Область техники изобретения
Настоящее изобретение относится к пероральному твердому препарату комбинированного противотуберкулезного лекарственного средства и способу его получения, в частности к пероральному твердому препарату четырехкомпонентного противотуберкулезного лекарственного средства, содержащему в качестве активных ингредиентов рифампицин, изониазид, пиразинамид и этамбутола гидрохлорид.
Уровень техники
Туберкулез относится к типу хронических инфекционных заболеваний, вызванных туберкулезной бациллой, которые могут затрагивать различные ткани и органы в организме, например, легкие, почки, кишечник, кости и так далее. Среди прочих, наиболее часто встречающимся типом является туберкулез легких. Туберкулез является одной из основных проблем, влияющих на здоровье людей в развивающихся странах. Недавно возникшая проблема микобактериальных инфекций у людей, инфицированных ВИЧ, продолжает разрастаться и становится трудноразрешимой проблемой в развитых странах. Согласно официальным статистическим данным, опубликованным Всемирной Организацией Здравоохранения (ВОЗ), количество пациентов с подтвержденным диагнозом, страдающих от активного туберкулеза, в мире превышает 20 миллионов, при том, что число новых заболевших составляет до 8,7 миллионов в год, среди которых случаи легочного туберкулеза в Азии составляют 70% от общего числа в мире. Индия и Китай занимают первое и второе места по заболеваемости туберкулезом легких в Азии. Обычно в клинической практике для лечения туберкулеза используют антибиотики. Однако, в отличие от лечения типичных бактериальных инфекций, для полного исцеления пациентов с туберкулезом требуется больше времени и, как правило, лечение продолжается в течение примерно 6-12 месяцев. С точки зрения состояния клинического лечения, в результате длительного периода приема таблеток легко развивается лекарственная устойчивость. В определенной степени феномен лекарственной устойчивости становится все более распространенным в мире, главным образом, в результате неправильно проведенного лечения, особенно вследствие однократного использования одного конкретного вида антибиотиков, либо пациент обычно не принимает таблетки согласно предписанию, например, пропускает прием доз или принимает недостаточную дозу. Согласно документу «Multidrug and extensively drug-resistant tuberculosis: 2010 global report on surveillance and response. Geneva, Switzerland: World Health Organization, 2010», недавно вышедшему в свет, по оценкам, 440 тысяч человек в мире страдают от множественной лекарственной устойчивости, при этом каждый третий умирает. В связи с постоянно возникающей проблемой лекарственной устойчивости ВОЗ и Международный союз борьбы с туберкулезом и болезнями легких объявили, что имеет место чрезвычайная ситуация в области лечения туберкулеза легких. В связи с постоянно возникающим явлением лекарственной устойчивости для лечения туберкулеза стали широко использовать метод фиксированной комбинации доз (метод ФКД (FDC)). Одним из наиболее часто используемых комбинированных лекарственных средств в методе ФКД является четырехкомпонентное комбинированное лекарственное средство, то есть, помимо рифампицина (R) и изониазида (H), которые являются наиболее эффективными для лечения туберкулеза, оно дополнительно содержит пиразинамид (Z) и этамбутола гидрохлорид (E). Такое четырехкомпонентное комбинированное лекарственное средство может представлять собой таблетку и капсулу, и так далее, и оно было официально одобрено и принято ВОЗ для лечения туберкулеза. Такое четырехкомпонентное лекарственное средство состоит из комбинированного препарата, который способствует лучшему соблюдению пациентами режима и схемы лечения и может в некоторой степени снизить частоту возникновения лекарственной устойчивости.
Рифампицин (R), также известный как рифампин, является полусинтетическим производным рифамицина. Рифампицин представляет собой кристаллы кирпично-красного цвета и имеет температуру плавления 183°С. Рифампицин плохо растворим в воде (растворимость: 1,3 мг/мл в воде, рН 4,3; 2,5 мг/мл в воде, рН 7,3; 100 мг/мл в воде, ДМСО) и неустойчив в кислой среде. Рифампицин может легко вступать в реакцию с изониазидом, а также может окисляться под воздействием воздуха, освещенности и так далее. Молекулярная структура рифампицина приведена ниже:
Рифампицин является эффективным антибактериальным лекарственным средством широкого спектра действия и высокоэффективен для ингибирования или уничтожения туберкулезных бацилл. Противотуберкулезный эффект лишь немного слабее, чем у изониазида, но сильнее, чем у стрептомицина. Минимальная ингибирующая концентрация составляет примерно 0,02-0,05 мкг/мл. Рифампицин воздействует на бактерии не только в стадии размножения, но также и в стадии покоя. Рифампицин эффективен также в отношении штаммов, проявляющих лекарственную устойчивость к другим противотуберкулезным препаратам. Рифампицин может уничтожать туберкулезную бациллу в макрофагах, фиброзной полости, казеозном фокусе, и также имеет сильный ингибирующий эффект на грамположительные кокки, такие как золотистый стафилококк, стрептококк и пневмококк. В отношении грамотрицательных кокков, таких как менингококк, гонококк, и бациллы проказы рифампицин также демонстрирует сильный ингибирующий эффект. Высокая концентрация рифампицина ингибирует вирус varida и Chlamydia trachomatis. Однократное применение рифампицина имеет тенденцию приводить к лекарственной устойчивости и, следовательно, его обычно комбинируют с другими фармацевтическими препаратами первой линии для начального и повторного лечения пациентов, страдающих от серьезных заболеваний, для того, чтобы повысить эффективность и замедлить развитие лекарственной устойчивости. Не существует перекрестной лекарственной устойчивости между рифампицином и другими противотуберкулезными лекарственными средствами. Рифампицин может избирательно ингибировать бактериальную ДНК-зависимую РНК-полимеразу и блокирует синтез мРНК, но не оказывает влияния на молекулы животной РНК-полимеразы.
Изониазид (H), также известный как римифон, имеет химическое название 4-пиридилкарбонилгидразин. Изониазид легко растворим в воде, слабо растворим в этаноле и очень слабо растворим в этиловом эфире. Структура карбонилгидразина нестабильна, и в кислых или основных условиях он может гидролизоваться с образованием изоникотиновой кислоты и гидразина. Свободный гидразин увеличивает токсичность, а свет, тяжелые металлы, температура, pH и так далее могут усиливать его гидролиз.
Молекулярная структура изониазида приведена ниже:

Изониазид проявляет хороший ингибирующий бактерии эффект в отношении туберкулезной бациллы и отличается высокой эффективностью. Изониазид используют в узком диапазоне доз, и он проявляет относительно низкую токсичность, которая легко переносится пациентами. Степень абсорбции при пероральном приеме составляет 90%, и сывороточная концентрация лекарственного средства достигает пика через 1-2 часа после приема. Vd составляет 0,61±0,11 л/кг, а степень связывания с белками очень низка. Изониазид используют в основном во время прогрессирующей стадии, стадии растворения и распространения, стадии поглощения при туберкулезе легких, и его также можно использовать при туберкулезном менингите и других видах внелегочного туберкулеза, и так далее. Изониазид, как правило, используют в сочетании с другими противотуберкулезными лекарственными средствами в целях повышения эффективности и преодоления лекарственной устойчивости бактерий.
Пиразинамид (Z) слабо растворяется в воде и этаноле, и очень слабо растворяется в этиловом эфире. Пиразинамид оказывает влияние на ускорение реакции рифампицина с изониазидом. Молекулярная структура пиразинамида приведена ниже:
Пиразинамид проявляет хороший ингибирующий бактерии эффект в отношении человеческой туберкулезной бациллы, и наибольший бактерицидный эффект имеет место при pH 5-5,5. В частности, в настоящее время он представляет собой лучшее бактерицидное лекарственное средство против туберкулезной бациллы, медленно растущей в фагоцитах в кислых условиях. Минимальная ингибирующая концентрация пиразинамида составляет 12,5 мкг/мл. При достижении концентрации 50 мкг/мл пиразинамид может уничтожать туберкулезную бациллу. Внутриклеточная концентрация лекарственного средства для ингибирования туберкулезной бациллы в 10 раз ниже внеклеточной концентрации. Он почти не оказывает ингибирующего действия в нейтральной и щелочной среде. Механизм действия может быть связан с пиразиноевой кислотой. Когда пиразинамид проникает в фагоциты и в тело туберкулезной бациллы, амидаза в теле бактерии дезамидирует амидную группу пиразинамида, преобразует его в пиразиноевую кислоту, тем самым проявляется ингибирующий бактерии эффект. Кроме того, поскольку пиразинамид сходен по химической структуре с никотинамидом, пиразинамид прерывает действие дегидрогеназы, заменяя никотинамид, предотвращает эффект дегидрогенизации и предотвращает использование кислорода туберкулезной бациллой, таким образом влияя на обычный метаболизм бактерий и вызывая их гибель.
Этамбутола гидрохлорид (E) легко растворим в воде, легко поглощает влагу и обеспечивает условия для реакции между рифампицином и изониазидом. Молекулярная структура этамбутола гидрохлорида приведена ниже:
Этамбутола гидрохлорид подходит для лечения туберкулеза в сочетании с другими противотуберкулезными лекарственными средствами. Однократное использование этамбутола гидрохлорида имеет тенденцию приводить к развитию лекарственной устойчивости. Этамбутола гидрохлорид проявляет сильную активность в отношении бактерий в стадии роста и размножения, но почти не оказывает влияния на бактерии в стадии покоя.
Комбинация RHZE имеет важное значение в клинической практике, однако не обходится без проблем. Во-первых, при непосредственном контакте рифампицина с изониазидом они имеют тенденцию реагировать друг с другом, особенно в кислой среде желудка. Это делает биодоступность рифампицина в комбинированном препарате ниже, чем в случае использования только рифампицина, и, таким образом, у пациентов может не достигаться лечебный эффект или возникать лекарственная устойчивость. В литературных источниках сообщается (например, смотри Sosa et al., 2005, Ars Pharm, 46: 353-364), что в условиях кислой среды желудка (pH 1-3) прямой контакт между рифампицином и изониазидом легко приводит к реакции и образованию изониазона. Эксперименты, проведенные Singh et al. (смотри Singh et al., 2000, Pharm. Pharmacol. Commun. 6: 405-410), также продемонстрировали этот факт. Исследование показало, что в условиях кислой среды желудка рифампицин разлагается до 3-формилрифамицина в отсутствие изониазида. В присутствии изониазида полученный 3-формилрифамицин быстро реагирует с изониазидом и образует изониазон посредством реакции второго порядка. Поскольку изониазон нестабилен при кислых условиях, он обратимым образом вновь образует 3-формилрифамицин и изониазид посредством медленной реакции первого порядка. В такой сложной реакции рифампицин продолжает разлагаться, тогда как изониазид восстанавливается. Хотя изониазон обладает некоторой антибактериальной активностью, эта антибактериальная активность ниже, чем таковая у рифампицина. При этом, в комбинации RHZE пиразинамид оказывает каталитический эффект на реакцию между рифампицином и изониазидом, а этамбутола гидрохлорид, который легко поглощает влагу, создает условия для реакции между рифампицином и изониазидом. Это некоторые из основных факторов, которые делают препарат нестабильным, и биодоступность комбинированного препарата ниже, чем таковая в случае использования одного рифампицина. Во-вторых, растворимость рифампицина также является проблемой. Рифампицин представляет собой лекарственное средство с низкой растворимостью, высокой гиперосмотичностью и легко диссоциирует. Его растворимость зависит от рН, и он имеет сильно различающуюся растворимость при различных условиях pH в пищеварительном тракте. Некоторые эксперименты показали, что при рН 1,4 растворимость достигает примерно 125 мг/мл, и 80-90% растворяется за 10 минут. Однако при рН>3, растворимость составляет менее чем 6 мг/мл. Если применить простой метод с энтеросолюбильным покрытием, чтобы предотвратить высвобождение рифампицина в желудке, но вместо этого обеспечить его высвобождение в кишечнике, нерастворимость рифампицина в кишечнике также приведет непосредственно к снижению биодоступности рифампицина.
Таким образом, проблема, которую предстоит решить многим научно-исследовательским учреждениям и производителям лекарственных средств, заключается в том, каким образом улучшить биодоступность рифампицина в четырехкомпонентных (RHZE) комбинированных препаратах. Что касается этого, сообщалось о некоторых относящихся к делу исследованиях. Например, в WO02/11728 раскрыто, что рифампицин и рН-зависимый носитель растворяют в средах для получения твердых дисперсий с целью увеличения растворимости рифампицина. Однако авторы настоящего изобретения обнаружили в своих экспериментах, что, поскольку количество используемого pH-зависимого носителя относительно невелико, трудно контролировать высвобождение рифампицина в больших количествах в кислой среде. В приведенном патенте не удалось достичь такого же эффекта твердого препарата, как в настоящем изобретении, с точки зрения биодоступности рифампицина в комбинированном лекарственном средстве RHZE. Твердая дисперсия рифампицина в предлагаемом по изобретению твердом препарате не только повышает растворимость и скорость растворения рифампицина, но и обеспечивает меньшее высвобождение или отсутствие высвобождения рифампицина в кислой среде желудка и уменьшение реакции между рифампицином и изониазидом в организме. Кроме того, тот факт, что рифампицин и изониазид не контактируют друг с другом в препарате, обеспечивает стабильность препарата в процессе хранения. Стабильность рифампицина в in vivo и in vitro условиях в комбинированном лекарственном средстве RHZE способствует улучшению биодоступности рифампицина в комбинированном лекарственном средстве, обеспечивает эффективность лечения и снижает вероятность возникновения лекарственной устойчивости.
Сущность изобретения
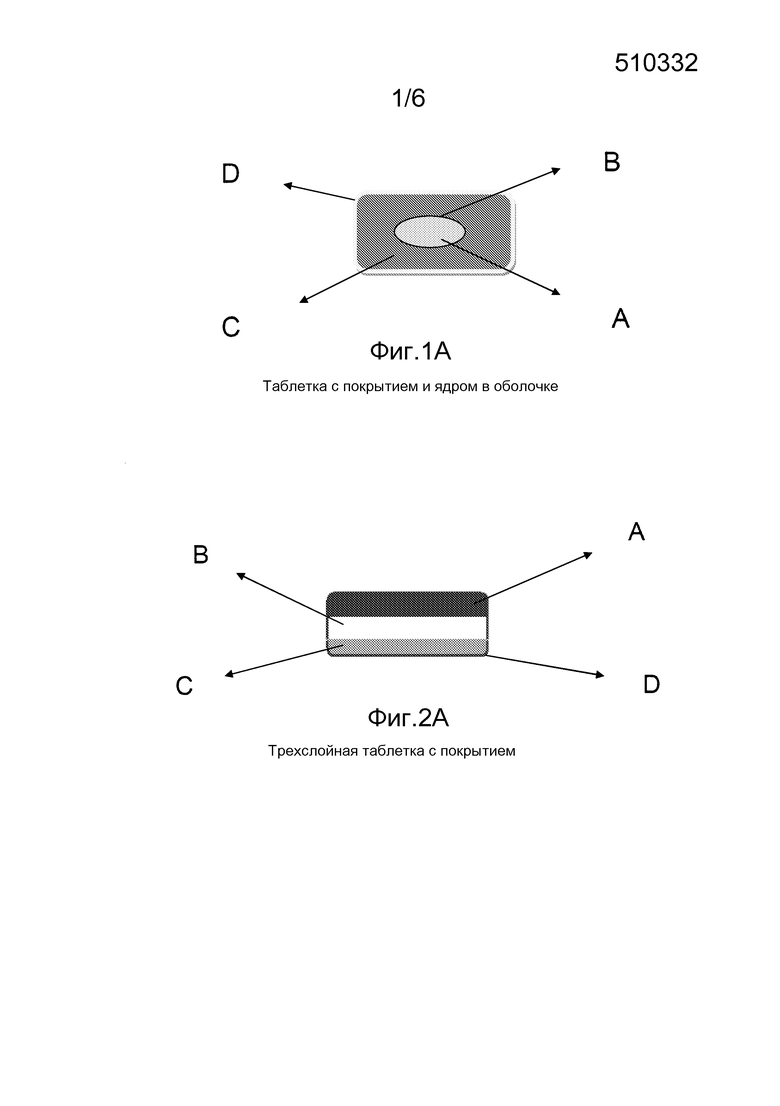
В первом варианте осуществления комбинированный пероральный твердый препарат с ядром в оболочке находится в форме таблетки с покрытием и ядром в оболочке, содержащей в качестве активных ингредиентов рифампицин, изониазид, пиразинамид и этамбутола гидрохлорид, в которой ядро внутреннего слоя содержит изониазид или рифампицин и покрыто нерастворимым в воде и не зависимым от рН пленочным покрытием, внешний слой твердого препарата с ядром в оболочке покрыт водорастворимым и не зависимым от рН пленочным покрытием, которое является влагоустойчивым и светонепроницаемым, после введения по меньшей мере один активный ингредиент из рифампицина и изониазида в меньшей степени высвобождается или не высвобождается в желудке.
Во втором варианте осуществления комбинированный пероральный твердый препарат с ядром в оболочке представляет собой трехслойную таблетку с покрытием, содержащую в качестве активных ингредиентов рифампицин, изониазид, пиразинамид и этамбутола гидрохлорид, в которой верхний слой и нижний слой независимо содержат один ингредиент, выбранный из изониазида и рифампицина, центральный слой содержит по меньшей мере один из пиразинамида и этамбутола гидрохлорида, сверху три слоя покрыты нерастворимым в воде и не зависимым от рН пленочным покрытием, после введения по меньшей мере один активный ингредиент из рифампицина и изониазида в меньшей степени высвобождается или не высвобождается в желудке.
В третьем варианте осуществления комбинированный пероральный твердый препарат с ядром в оболочке находится в форме таблетки с покрытием и ядром в оболочке, содержащей в качестве активных ингредиентов рифампицин, изониазид, пиразинамид и этамбутола гидрохлорид, в которой ядро внутреннего слоя содержит изониазид или рифампицин и покрыто водорастворимой и не зависимой от рН разделительной пленкой, рифампицин находится в форме энтеросолюбильной твердой дисперсии рифампицина, диспергированной в энтеросолюбильных твердых носителях, внешний слой твердого препарата с ядром в оболочке покрыт водорастворимым и не зависимым от рН пленочным покрытием, которое является влагоустойчивым и светонепроницаемым.
В четвертом варианте осуществления комбинированный пероральный твердый препарат с ядром в оболочке находится в форме трехслойной таблетки с покрытием, содержащей в качестве активных ингредиентов рифампицин, изониазид, пиразинамид и этамбутола гидрохлорид, в которой верхний слой и нижний слой независимо содержат один ингредиент, выбранный из изониазида и рифампицина, в слое, содержащем рифампицин, рифампицин находится в форме энтеросолюбильной твердой дисперсии рифампицина, диспергированной в энтеросолюбильных твердых носителях, центральный слой содержит по меньшей мере один из пиразинамида и этамбутола гидрохлорида, сверху три слоя покрыты водорастворимым и не зависимым от рН пленочным покрытием, которое является влагоустойчивым и светонепроницаемым.
В другом варианте осуществления нерастворимое в воде и не зависимое от рН пленочное покрытие в первом и втором вариантах осуществления содержит нерастворимый в воде и не зависимый от рН полимерный пленкообразующий материал и пластификатор, необязательно дополнительно содержит по меньшей мере одно из порообразующего вещества и антиадгезионного вещества.
В еще одном варианте осуществления нерастворимое в воде и не зависимое от рН пленочное покрытие во втором варианте осуществления дополнительно содержит замутняющее вещество.
В еще одном варианте осуществления нерастворимое в воде и не зависимое от рН пленочное покрытие в первом варианте осуществления дополнительно содержит полимер, который разрушается и быстро увеличивается в объеме при поглощении воды, и/или низкомолекулярный стимулятор проницаемости в ядре внутреннего слоя.
В еще одном варианте осуществления слой, содержащий рифампицин, во втором варианте осуществления дополнительно содержит полимер, который разрушается и быстро увеличивается в объеме при поглощении воды, а центральный слой дополнительно содержит замедлитель.
Без привязки к какой-либо теории, предложен дизайн комбинированного перорального твердого препарата, позволяющий двум активным ингредиентам в препарате, рифампицину и изониазиду, не вступать в прямой контакт друг с другом. После введения по меньшей мере один из активных ингредиентов, рифампицина и изониазида, контролируется так, чтобы в меньшей степени высвобождаться или не высвобождаться в желудке. Предпочтительное высвобождаемое количество составляет не более чем 15% от указанного содержания, более предпочтительно не более чем 10%, и еще более предпочтительно не более чем 5%, чтобы таким образом повысить стабильность комбинированного препарата и биодоступность рифампицина в комбинированном препарате.
В настоящем изобретении термин «стимулятор проницаемости» означает вещество, обладающее определенным осмотическим давлением и, таким образом, облегчающее проникновение молекул воды. В настоящем изобретении в отношении данного термина не существует конкретных ограничений, при условии, что стимулятор проницаемости подходит для использования в области создания фармацевтических препаратов по настоящему изобретению.
В настоящем изобретении термин «не зависимое от рН пленочное покрытие» относится к пленке, содержащей не зависимое от pH вещество. В настоящем изобретении термин «не зависимое от рН вещество» означает вещество, которое можно растворять в воде, и на него не влияет величина pH. В настоящем изобретении в отношении данного термина не существует конкретных ограничений, при условии, что не зависимое от рН пленочное покрытие подходит для использования в области создания фармацевтических препаратов по настоящему изобретению.
В настоящем изобретении термин «энтеросолюбильный носитель» означает вещество, которое практически нерастворимо в кислой среде желудка, но может растворяться в условиях pH кишечника. В настоящем изобретении в отношении данного термина не существует конкретных ограничений, при условии, что это энтеросолюбильный носитель, обычно используемый в области создания фармацевтических препаратов по настоящему изобретению.
В настоящем изобретении термин «связующее вещество» означает клейкое вещество, и оно способно связывать рассыпчатые порошки склеиванием. В настоящем изобретении в отношении данного термина не существует конкретных ограничений, при условии, что это связующее вещество, обычно используемое в области создания фармацевтических препаратов по настоящему изобретению.
В настоящем изобретении термин «смазывающее вещество» означает вещество, обладающее способностью уменьшать истирание и трение, вызванное контактом, если оно помещено между двумя противоположно движущимися объектами. В настоящем изобретении в отношении данного термина не существует конкретных ограничений, при условии, что это смазывающее вещество, обычно используемое в области создания фармацевтических препаратов по настоящему изобретению.
Настоящее изобретение относится к комбинированному пероральному твердому препарату с покрытием, содержащему в качестве активных ингредиентов рифампицин, изониазид, пиразинамид и этамбутола гидрохлорид, в котором активные ингредиенты составляют комбинацию с соотношением 200-300 мг: 75-300 мг: 250-500 мг: 250-275 мг, предпочтительно 150 мг: 75 мг: 400 мг: 275 мг.
В одном аспекте одна из технических характеристик изобретения заключается в том, что один или более из активных ингредиентов в комбинированном пероральном твердом препарате с покрытием по настоящему изобретению покрыт нерастворимым в воде и не зависимым от рН пленочным покрытием. Пленочное покрытие является водопроницаемым и используется для контроля замедленного высвобождения изониазида или рифампицина, и один из рифампицина или изониазида и других активных ингредиентов быстро высвобождается после введения. Характерная особенность дизайна также гарантирует, что рифампицин и изониазид не контактируют напрямую. Комбинированный пероральный твердый препарат с покрытием, обладающий характерной особенностью дизайна, может быть в форме таблетки с покрытием и ядром в оболочке, либо трехслойной таблетки с покрытием.
В одном предпочтительном варианте осуществления настоящего изобретения комбинированный пероральный твердый препарат по настоящему изобретению находится в форме таблетки с покрытием и ядром в оболочке, в которой внутренний слой представляет собой слой изониазида, который может находиться в виде одной или более отдельных гранул, драже или хлопьев, покрытых нерастворимым в воде и не зависимым от рН пленочным покрытием. Ядро внутреннего слоя с покрытием покрыто внешним слоем, содержащим рифампицин, пиразинамид и этамбутола гидрохлорид, и таким образом получается таблетка с покрытием и ядром в оболочке. Таблетка с покрытием и ядром в оболочке покрыта водорастворимым и не зависимым от рН пленочным покрытием, которое является влагоустойчивым и светонепроницаемым.
В таблетке с покрытием и ядром в оболочке, в дополнение к активным ингредиентам, ядро внутреннего слоя с нерастворимым в воде и не зависимым от рН пленочным покрытием может также содержать полимер, который разрушается и быстро увеличивается в объеме при поглощении воды, и необязательно содержит один или более носителей, выбранных из низкомолекулярного стимулятора проницаемости, связующего вещества и смазывающего вещества. Полимер, который разрушается и быстро увеличивается в объеме при поглощении воды, представляет собой один или более, выбранные из группы, состоящей из сшитой карбоксиметилцеллюлозы натрия (CCMC-Na), низкозамещенной гидроксипропилцеллюлозы (L-HPC), карбоксиметилцеллюлозы натрия (CMC-Na), карбоксиметилкрахмала натрия (CMS-Na), поливинилполипирролидона (PVPP) и микрокристаллической целлюлозы (MCC). Низкомолекулярный стимулятор проницаемости представляет собой вещество, выбранное из группы, состоящей из хлорида натрия и хлорида калия.
В таблетке с покрытием и ядром в оболочке нерастворимое в воде и не зависимое от рН пленочное покрытие, покрывающее сверху ядро внутреннего слоя изониазида, составляет от 6 до 30% по массе от ядра внутреннего слоя изониазида. Пленка проницаема для воды и содержит нерастворимый в воде и не зависимый от pH полимерный пленкообразующий материал, пластификатор и, необязательно, содержит порообразующее вещество и/или антиадгезионное вещество. Пленкообразующий материал в нерастворимом в воде и не зависимом от рН пленочном покрытии составляет от 70 до 90%, предпочтительно от 70 до 80% по массе от общей массы пленочного покрытия. Пленкообразующий материал представляет собой вещество, предпочтительно выбранное из группы, состоящей из Aquacoat ECD, этилцеллюлозы (EC), ацетата целлюлозы (CA), поливинилхлорида, поликарбоната, винилового спирта-винилацетата, а также их сочетаний. Пластификатор составляет от 10 до 30%, предпочтительно от 13 до 25% по массе от общей массы пленочного покрытия. Пластификатор представляет собой вещество, предпочтительно выбранное из группы, состоящей из триэтилцитрата (TEC), трибутилцитрата (TBC), ацетилтриэтилцитрата (ATEC), диметилсебацата (DMS), дибутилсебацата (DBS), дибутилфталата (DBP), а также их сочетаний. Порообразующее вещество составляет от 0 до 15%, предпочтительно от 4 до 13% по массе от общей массы пленочного покрытия. Порообразующее вещество представляет собой вещество, предпочтительно выбранное из группы, состоящей из полиэтиленгликолей, гидроксипропилметилцеллюлозы, Kollicoat®IR, поливинилового спирта, мочевины, а также их сочетаний. Антиадгезионное вещество составляет от 0 до 25%, предпочтительно от 0 до 15% по массе от общей массы пленочного покрытия. Антиадгезионное вещество представляет собой вещество, предпочтительно выбранное из группы, состоящей из порошка талька, легкого тонкоизмельченного силикагеля, а также их сочетаний.
Способ получения таблетки с покрытием и ядром в оболочке приведен ниже:
1) Получение ядра внутреннего слоя:
Изониазид и полимер, который разрушается и быстро увеличивается в объеме при поглощении воды, и/или низкомолекулярный стимулятор проницаемости смешивают, измельчают, гранулируют или дражируют, а затем высушивают и покрывают нерастворимым в воде и независимым от pH пленочным покрытием. При необходимости изготовления в виде внутреннего ядра таблетки с покрытием, к полученным гранулам необходимо добавлять глидант или смазывающее вещество, гомогенизировать, просеивать, а затем прессовать, получая таблетку, которую затем покрывать нерастворимым в воде и не зависимым от рН пленочным покрытием.
2) Получение гранул внешнего слоя:
Рифампицин, пиразинамид и этамбутола гидрохлорид смешивают с наполнителем и измельчают, а затем гранулируют со связующим веществом, высушивают, добавляют к смеси глидант или смазывающее вещество, гомогенизируют, просеивают, после чего они готовы к использованию.
3) Прессование таблетки с покрытием и ядром в оболочке:
Ядро таблетки с покрытием и ядром в оболочке из этапа 1) используют в качестве внутреннего ядра, а гранулы из этапа 2) используют в качестве внешнего слоя. Таблетку с покрытием и ядром в оболочке получают прессованием, либо гранулы с покрытием или драже с покрытием из этапа 1) смешивают с гранулами из этапа 2) и затем прессуют в форме таблетки.
4) Нанесение покрытия:
Таблетку с покрытием и ядром в оболочке из этапа 3) покрывают водорастворимым и не зависимым от рН пленочным покрытием, которое является влагоустойчивым и светонепроницаемым.
В другом предпочтительном варианте осуществления комбинированный пероральный твердый препарат по настоящему изобретению находится в форме трехслойной таблетки с покрытием, состоящий из верхнего слоя, центрального слоя и нижнего слоя, в которой верхний слой и нижний слой, соответственно, представляют собой слой рифампицина и слой изониазида/пиразинамида, а центральный слой представляет собой слой этамбутола гидрохлорида. Слой рифампицина содержит полимер, который разрушается и быстро увеличивается в объеме при поглощении воды, и другие фармацевтические носители. Слой этамбутола гидрохлорида содержит замедлитель, который задерживает быстрый распад или высвобождение этамбутола гидрохлорида и других фармацевтических носителей. Слой изониазида/пиразинамида содержит другие фармацевтические носители. Трехслойная таблетка покрыта нерастворимым в воде и не зависимым от рН пленочным покрытием.
Нерастворимое в воде и не зависимое от рН пленочное покрытие, покрывающее сверху трехслойную таблетку, проницаемо для воды, и пленочное покрытие составляет от 5 до 12% по массе от непокрытого ядра трехслойной таблетки. Нерастворимое в воде и не зависимое от рН пленочное покрытие содержит нерастворимый в воде и не зависимый от рН полимерный пленкообразующий материал, пластификатор, необязательно дополнительно содержит по меньшей мере одно из порообразующего вещества, замутняющего вещества, антиадгезионного вещества. Нерастворимый в воде и не зависимый от рН полимерный пленкообразующий материал представляет собой вещество, выбранное из группы, состоящей из Aquacoat ECD, этилцеллюлозы (EC), ацетата целлюлозы (CA), поливинилхлорида, поликарбоната, винилового спирта-винилацетата, а также их сочетаний. Нерастворимый в воде и не зависимый от рН полимерный пленкообразующий материал составляет от 50 до 80% по массе от общей массы пленочного покрытия. Пластификатор представляет собой вещество, выбранное из группы, состоящей из триэтилцитрата (TEC), трибутилцитрата (TBC), ацетилтриэтилцитрата (ATEC), диметилсебацата (DMS), дибутилсебацата (DBS), дибутилфталата (DBP), а также их сочетаний. Пластификатор составляет от 20 до 45% по массе от общей массы пленочного покрытия. Порообразующее вещество представляет собой вещество, выбранное из группы, состоящей из полиэтиленгликолей, гидроксипропилметилцеллюлозы, Kollicoat®IR, поливинилового спирта, мочевины, а также их сочетаний. Порообразующее вещество составляет от 0 до 15% по массе от общей массы пленочного покрытия. Антиадгезионное вещество представляет собой вещество, предпочтительно выбранное из группы, состоящей из порошка талька, легкого тонкоизмельченного силикагеля, а также их сочетаний. Антиадгезионное вещество составляет от 0 до 20% по массе от общей массы пленочного покрытия. Замутняющее вещество представляет собой диоксид титана. Замутняющее вещество составляет от 0 до 20% по массе от общей массы пленочного покрытия.
Слой рифампицина трехслойной таблетки с покрытием содержит полимер, который разрушается и быстро увеличивается в объеме при поглощении воды, и полимер представляет собой один или более, выбранные из группы, состоящей из сшитой карбоксиметилцеллюлозы натрия (CCMC-Na), низкозамещенной гидроксипропилцеллюлозы (L-HPC), карбоксиметилцеллюлозы натрия (CMC-Na), карбоксиметилкрахмала натрия (CMS-Na), поливинилполипирролидона (PVPP) и микрокристаллической целлюлозы (MCC).
Слой этамбутола гидрохлорида трехслойной таблетки с покрытием содержит замедлитель, представляющий собой один или более, выбранные из группы, состоящей из нерастворимого в воде каркасного материала, такого как этилцеллюлоза, полиэтилены, акриловые смолы; эрозивного каркасного материала, такого как пчелиный воск, гидрогенизированное растительное масло, стеариновая кислота, полиэтиленгликоль, карнаубский воск, стеарат глицерина, стеарат пропиленгликоля и стеариловый спирт; и водорастворимого каркасного материала, такого как гидроксипропилметилцеллюлоза, полиэтиленоксид (PEO).
Способ получения трехслойной таблетки с покрытием приведен ниже:
1) Получение слоя рифампицина:
Рифампицин и полимер, который разрушается и быстро увеличивается в объеме при поглощении воды, смешивают, измельчают, просеивают, гранулируют со связующим веществом, а затем высушивают. Добавляют глидант или смазывающее вещество, гомогенизируют, просеивают, после чего смесь готова для прессования в таблетку.
2) Получение слоя этамбутола гидрохлорида:
Этамбутола гидрохлорид, замедлитель и наполнитель смешивают, измельчают, просеивают, гранулируют со связующим веществом, а затем высушивают. Добавляют глидант или смазывающее вещество, гомогенизируют, просеивают, после чего смесь готова для прессования в таблетку.
3) Получение слоя изониазида/пиразинамида:
Изониазид, пиразинамид и наполнитель смешивают, измельчают, просеивают, гранулируют со связующим веществом, а затем высушивают. Добавляют глидант или смазывающее вещество, гомогенизируют, просеивают, после чего смесь готова для прессования в таблетку.
4) Прессование трехслойной таблетки:
Гранулы из этапа 1), 2) и 3) прессуют, соответственно, как верхний, центральный и нижний слои.
5) Нанесение покрытия:
Трехслойную таблетку из этапа 4) покрывают нерастворимым в воде и не зависимым от рН пленочным покрытием.
В другом аспекте другая характерная особенность дизайна по настоящему изобретению заключается в том, что, поскольку рифампицин является практически нерастворимым лекарственным средством, стабильность комбинированного лекарственного препарата по настоящему изобретению и биодоступность рифампицина в препарате улучшают путем увеличения растворимости и скорости растворения рифампицина при помощи технологии энтеросолюбильной твердой дисперсии. Характерная особенность дизайна заключается в том, что комбинированный пероральный твердый препарат представляет собой таблетку с покрытием и ядром в оболочке, либо трехслойную таблетку с покрытием.
В одном предпочтительном варианте осуществления настоящего изобретения комбинированный пероральный твердый препарат с ядром в оболочке находится в форме таблетки с покрытием и ядром в оболочке, в которой ядро внутреннего слоя содержит изониазид или рифампицин и покрыто водорастворимой и не зависимой от рН разделительной пленкой. Кроме того, рифампицин находится в форме энтеросолюбильной твердой дисперсии рифампицина, диспергированной в энтеросолюбильных твердых носителях. Внешний слой твердого препарата с ядром в оболочке покрыт водорастворимым и не зависимым от рН пленочным покрытием, которое является влагоустойчивым и светонепроницаемым.
Весовое соотношение рифампицина и энтеросолюбильного носителя в твердых дисперсиях рифампицина по настоящему изобретению находится в диапазоне от 2:1 до 1:3, при этом носитель энтеросолюбильных твердых дисперсий представляет собой один или более, выбранные из группы, состоящей из: фталата поливинилацетата (PVAP), сополимера метакриловой кислоты/метилметакрилата, такого как Eudragit L30D-55, Eudragit L100, Eudragit S100; целлюлозы и ее производных, таких как ацетатфталат целлюлозы (CAP), ацетаттримеллитат целлюлозы (CAT), фталат гидроксипропилметилцеллюлозы (HPMCP), тримеллитат гидроксипропилметилцеллюлозы (HPMCT); ацетатсукцинат целлюлозы (CAS) и ацетатсукцинат гидроксипропилметилцеллюлозы (HPMCAS), ацетатфталат гидроксипропилметилцеллюлозы (HPMCAP), и ацетатсукцинат гидроксипропилметилцеллюлозы (HPMCAS) является предпочтительным.
Водорастворимый и не зависимый от рН полимерный пленкообразующий материал представляет собой вещество, выбранное из группы, состоящей из гидроксипропилметилцеллюлозы (HPMC), Kollicoat®IR, Opadry II, а также их сочетаний.
Метод получения твердой дисперсии рифампицина по настоящему изобретению может быть общепринятым методом получения твердых дисперсий, таким как метод горячего плавления, метод сушки распылением, одноэтапный метод псевдоожиженного слоя или метод с испарением растворителя.
Способ получения таблетки с покрытием и ядром в оболочке приведен ниже:
1) Получение ядра внутреннего слоя:
Изониазид и наполнитель смешивают, измельчают, гранулируют или дражируют со связующим веществом, а затем высушивают и покрывают водорастворимым и не зависимым от pH разделительным пленочным покрытием; при необходимости изготовления в виде внутреннего ядра таблетки с покрытием, к полученным гранулам необходимо добавлять глидант или смазывающее вещество, гомогенизировать, просеивать, а затем прессовать, получая таблетку, которую затем покрывать водорастворимым и не зависимым от pH пленочным покрытием в качестве разделительного пленочного покрытия.
2) Получение гранул внешнего слоя:
Во-первых, из рифампицина получают энтеросолюбильную твердую дисперсию, а затем полученную твердую дисперсию рифампицина, пиразинамид и этамбутола гидрохлорид смешивают с наполнителем, разрыхлителем, и гранулы, полученные при помощи связующего вещества, с добавленным к ним глидантом или смазывающим веществом гомогенизируют, просеивают, после чего они готовы к использованию.
3) Прессование таблетки с покрытием и ядром в оболочке:
Ядро таблетки с покрытием и ядром в оболочке из этапа 1) используют в качестве внутреннего ядра, а гранулы из этапа 2) используют в качестве внешнего слоя. Таблетку с покрытием и ядром в оболочке получают путем прессования, либо гранулы с покрытием или драже с покрытием из этапа 1) смешивают с гранулами из этапа 2), а затем прессуют, получая таблетку.
4) Нанесение покрытия:
Таблетку с покрытием и ядром в оболочке из этапа 3) покрывают водорастворимым и не зависимым от рН пленочным покрытием, которое является влагоустойчивым и светонепроницаемым.
В другом предпочтительном варианте осуществления комбинированный пероральный твердый препарат представляет собой трехслойную таблетку с покрытием, верхний слой и нижний слой которой независимо содержат любой из изониазида и рифампицина. В слое, содержащем рифампицин, рифампицин находится в форме энтеросолюбильной твердой дисперсии рифампицина, диспергированной в энтеросолюбильных твердых носителях. Центральный слой содержит по меньшей мере один из пиразинамида и этамбутола гидрохлорида. Сверху три слоя покрыты водорастворимым и не зависимым от рН пленочным покрытием, которое является влагоустойчивым и светонепроницаемым. Диапазон массового соотношения рифампицина и энтеросолюбильного носителя, способы получения носителя энтеросолюбильной твердой дисперсии и твердой дисперсии рифампицина описаны выше.
Способ получения трехслойной таблетки с покрытием приведен ниже:
1) Получение слоя рифампицина:
Во-первых, из рифампицина получают энтеросолюбильную твердую дисперсию, а затем полученную твердую дисперсию рифампицина и другие фармацевтические носители смешивают, гранулируют или непосредственно используют для прессования в таблетку;
2) Получение слоя этамбутола гидрохлорида:
Этамбутола гидрохлорид и другие фармацевтические носители смешивают, измельчают, просеивают, затем гранулируют со связующим веществом, высушивают, добавляют к смеси глидант или смазывающее вещество, гомогенизируют, просеивают, после чего смесь готова к использованию;
3) Получение слоя изониазида/пиразинамида:
Изониазид, пиразинамид и другие фармацевтические носители смешивают, измельчают, просеивают, затем гранулируют со связующим веществом, высушивают, добавляют к смеси глидант или смазывающее вещество, гомогенизируют, просеивают, после чего смесь готова к использованию;
4) Прессование трехслойной таблетки:
Гранулы из этапа 1), 2) и 3) прессуют, соответственно, как верхний, центральный и нижний слои.
5) Нанесение покрытия:
Трехслойную таблетку из этапа 4) покрывают нерастворимым в воде и не зависимым от рН пленочным покрытием.
Описанное в вышеприведенных вариантах осуществления водорастворимое и не зависимое от рН пленочное покрытие, которое является влагоустойчивым и светонепроницаемым, содержит вещество, выбранное из группы, состоящей из Kollicoat®IR, гидроксипропилметилцеллюлозы (HPMC), сочетания диоксида титана и порошка талька, а также Opadry II, содержащего сочетание гидроксипропилметилцеллюлозы (HPMC), диоксида титана и порошка талька.
Комбинированный пероральный твердый препарат с ядром в оболочке по настоящему изобретению может также содержать другие фармацевтические носители, выбранные из группы, состоящей из наполнителя, связующего вещества, смазывающего вещества или глиданта и/или разрыхлителя.
Краткое описание чертежей
На фигуре 1 схематически изображена таблетка с покрытием и ядром в оболочке, A: ядро внутреннего слоя; B: пленочное покрытие внутреннего слоя; C: внешний слой; D: пленочное покрытие.
На фигуре 2 схематически изображена трехслойная таблетка с покрытием; A: верхний слой; B: центральный слой; C: нижний слой; D: пленочное покрытие.
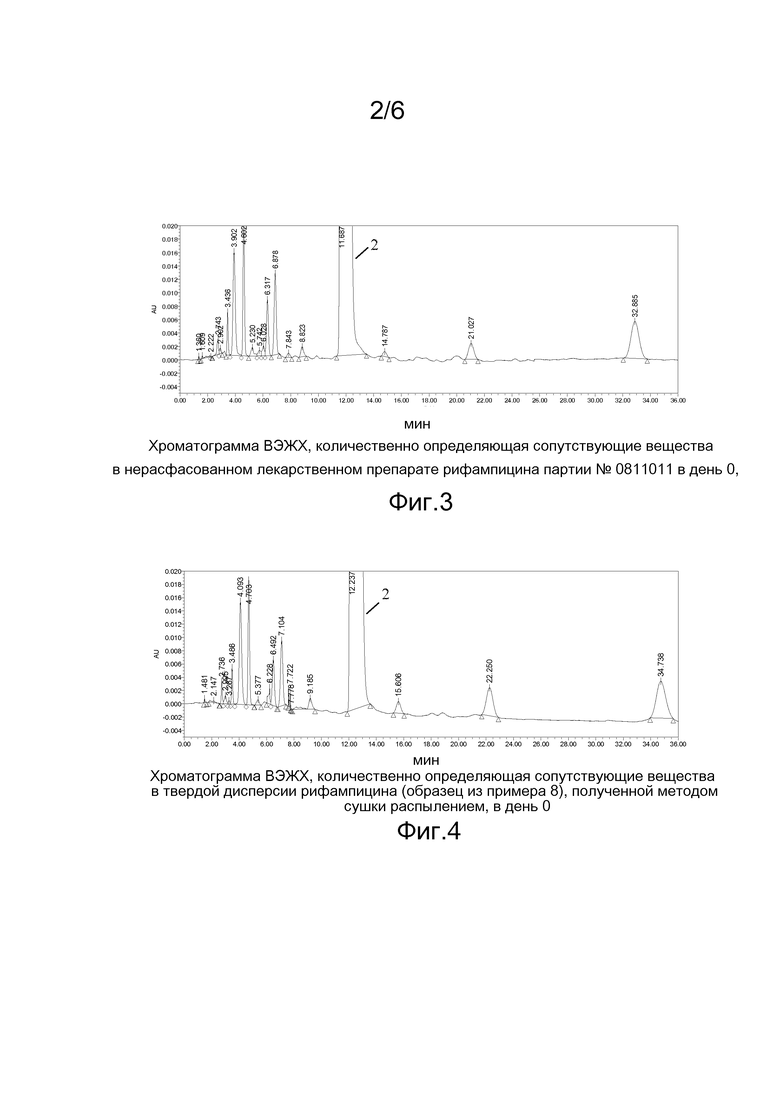
На фигуре 3 приведена хроматограмма ВЭЖХ, количественно определяющая сопутствующие вещества в нерасфасованном лекарственном препарате рифампицина партии № 0811011 в день 0;
На фигуре 4 приведена хроматограмма ВЭЖХ, количественно определяющая сопутствующие вещества в твердой дисперсии рифампицина (образец из примера 8), полученной методом сушки распылением, в день 0;
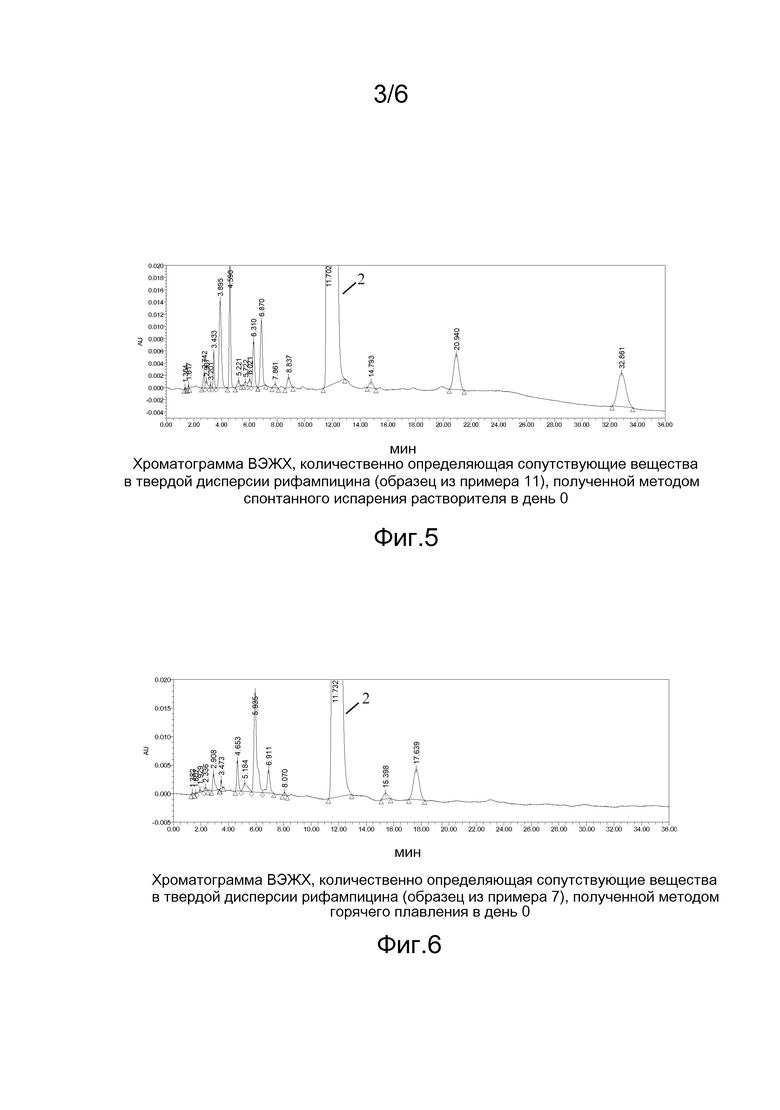
На фигуре 5 приведена хроматограмма ВЭЖХ, количественно определяющая сопутствующие вещества в твердой дисперсии рифампицина (образец из примера 11), полученной методом спонтанного испарения растворителя, в день 0;
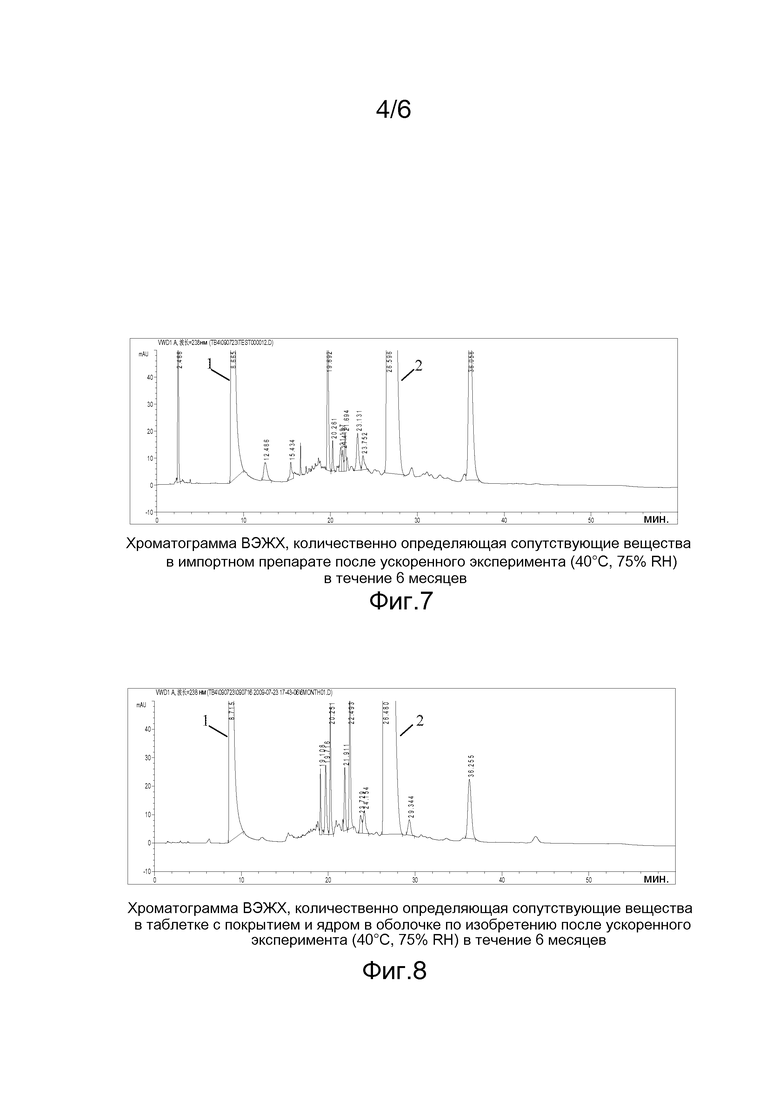
На фигуре 6 приведена хроматограмма ВЭЖХ, количественно определяющая сопутствующие вещества в твердой дисперсии рифампицина (образец из примера 7), полученной методом горячего плавления, в день 0;
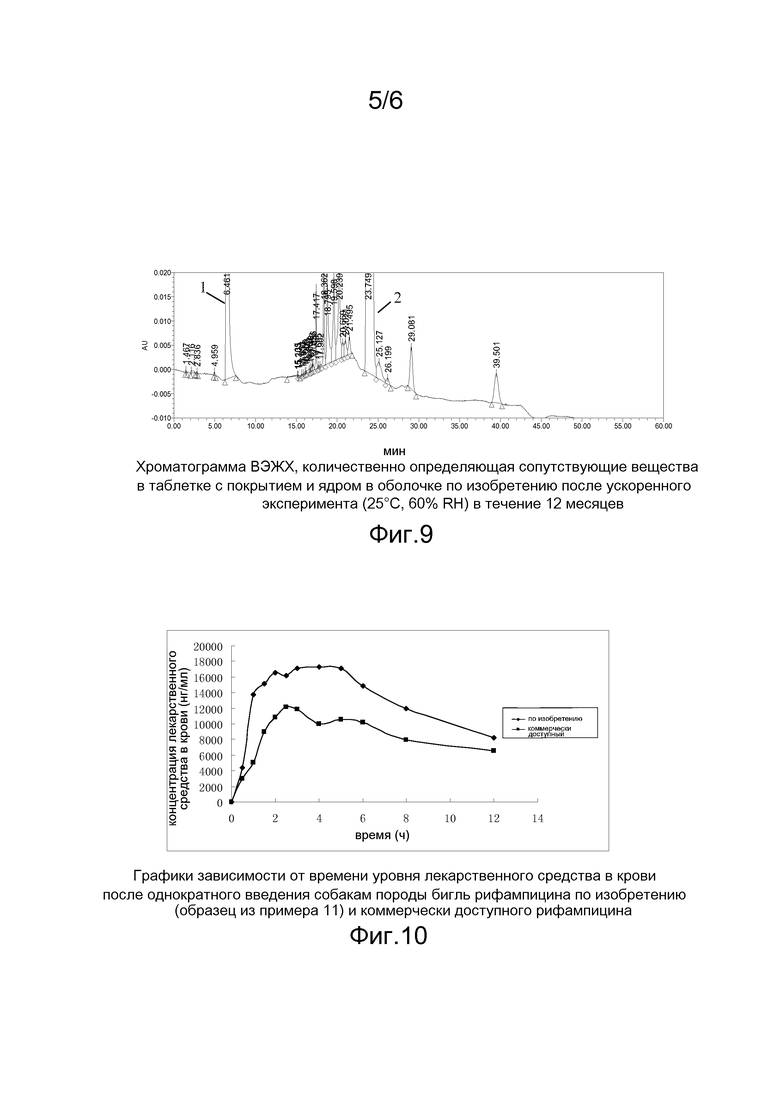
На фигуре 7 приведена хроматограмма ВЭЖХ, количественно определяющая сопутствующие вещества в импортном препарате после ускоренного эксперимента (40°C, относительная влажность (RH) 75%) в течение 6 месяцев;
На фигуре 8 приведена хроматограмма ВЭЖХ, количественно определяющая сопутствующие вещества в таблетке с покрытием и ядром в оболочке по изобретению после ускоренного эксперимента (40°C, 75% RH) в течение 6 месяцев;
На фигуре 9 приведена хроматограмма ВЭЖХ, количественно определяющая сопутствующие вещества в таблетке с покрытием и ядром в оболочке по изобретению после ускоренного эксперимента (25°C, 60% RH) в течение 12 месяцев;
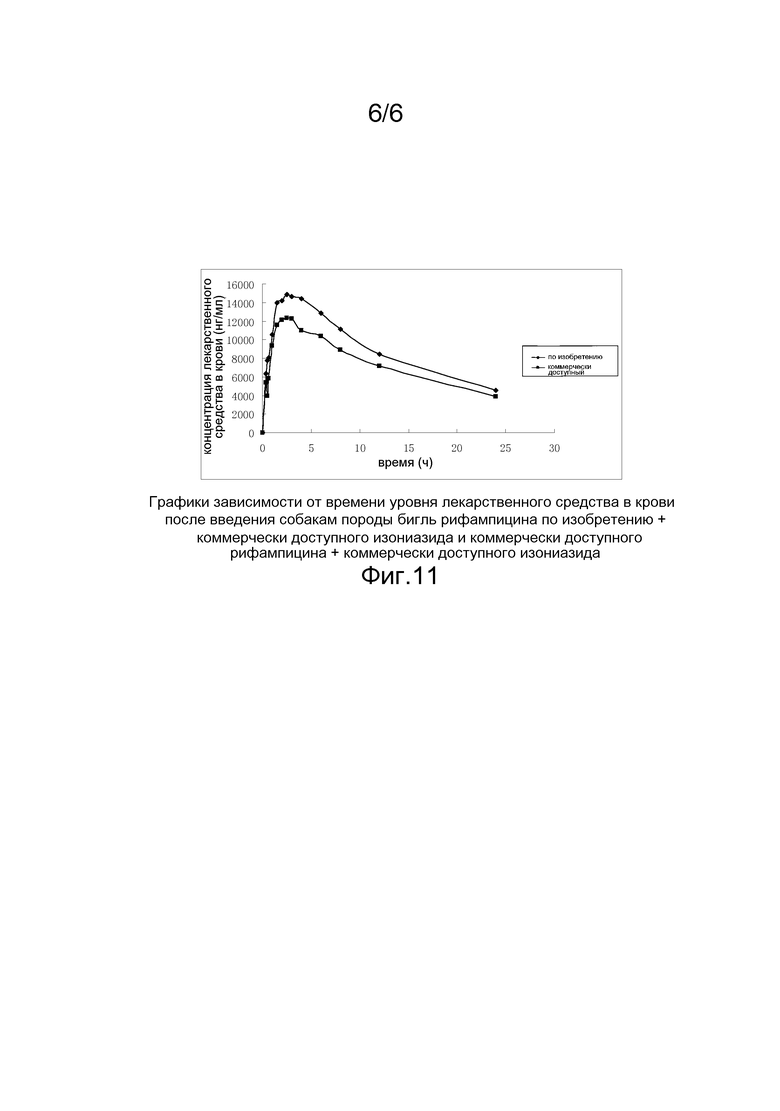
На фигуре 10 приведены графики зависимости от времени уровня лекарственного средства в крови после однократного введения собакам породы бигль рифампицина по изобретению (образец из примера 11) и коммерчески доступных рифампицинов;
На фигуре 11 приведены графики зависимости от времени уровня лекарственного средства в крови после введения собакам породы бигль рифампицина по изобретению + коммерчески доступного изониазида и коммерчески доступного рифампицина + коммерчески доступного изониазида.
Примеры
Далее настоящее изобретение будет проиллюстрировано с помощью примеров, но настоящее изобретение не должно быть ограничено ими.
Пример 1
Получение таблетки с покрытием и ядром в оболочке
1) Получение ядра таблетки без оболочки
75,00 г изониазида, 2,00 г хлорида натрия, 18,70 г микрокристаллической целлюлозы и 10,30 г сшитой карбоксиметилцеллюлозы натрия смешивали до однородного состояния, а затем измельчали для пропускания через сито с размером пор 0,250 мм. Получали гибкий материал с использованием 2% гидроксипропилметилцеллюлозы, а затем пропускали через сито с размером пор 0,710 мм и гранулировали. Смесь высушивали при 50°С, пока содержание влаги в гранулах не становилось менее чем 3,0%. Добавляли 1% по массе стеарата магния, исходя из массы сухих гранул. Смесь дополнительно перемешивали и пропускали через сито с размером пор 0,85 мм, а затем смешивали до однородного состояния и прессовали, получая таблетку определенной формы при помощи 4*9 мм штампа для прессования. Давление поддерживали на уровне от 50 до 65 Н. Время распада ядра таблетки до нанесения покрытия, наиболее предпочтительно, поддерживалось в пределах 1 минуты.
2) Покрытие ядра внутреннего слоя таблетки
Раствор для нанесения покрытия с содержанием твердых веществ 15% готовили в соответствии с весовым соотношением твердых веществ Aquacoat ECD: триэтилцитрат (TEC): Kollicoat®IR = 8,05: 2,01: 0,50. Температуру покрывающего слоя поддерживали в диапазоне от 39°C до 42°C. Увеличение массы составило от 8 до 10%. Смесь отверждали при 60°С в течение 2 часов после нанесения покрытия, после чего она была готова к применению в качестве ядра внутреннего слоя таблетки с покрытием и ядром в оболочке. Профиль высвобождения изониазида в ядре внутреннего слоя таблетки с покрытием и ядром в оболочке приведен в таблице 1.
3) Подготовка к прессованию гранул внешнего слоя
150,00 г рифампицина, 400,00 г пиразинамида, 275,00 г этамбутола гидрохлорида, 160,00 г микрокристаллической целлюлозы и 54,00 г низкозамещенной гидроксипропилцеллюлозы смешивали до однородного состояния и затем формовали в порошок. Смесь пропускали через сито с размером пор 0,250 мм и гранулировали с использованием 5%-гидроксипропилметилцеллюлозы. Смесь высушивали при 50°C, и влажность гранул поддерживали на уровне менее чем 3%. Добавляли 0,5% тонкоизмельченного силикагеля, исходя из массы сухих гранул, и смесь пропускали через сито с размером пор 0,600 мм. Полученный продукт прессовали в таблетку с покрытием и ядром в оболочке.
4) Получение внешнего слоя
Готовили раствор для нанесения покрытия с 20% концентрацией Opadry II. Температуру покрывающих материалов поддерживали на уровне от 40 до 45°C. Увеличение массы составило примерно 3%.
Пример 2
Получение таблетки с покрытием и ядром в оболочке
1) Получение ядра таблетки без оболочки
75,00 г изониазида, 18,70 г микрокристаллической целлюлозы, 8,20 г сшитой карбоксиметилцеллюлозы натрия смешивали до однородного состояния, а затем измельчали для пропускания через сито с размером пор 0,250 мм. Получали гибкий материал с использованием 2% гидроксипропилметилцеллюлозы, а затем пропускали через сито с размером пор 0,710 мм и гранулировали. Смесь высушивали при 50°C, пока содержание влаги в гранулах не становилось менее чем 3,0%. Добавляли 1% стеарата магния, исходя из массы сухих гранул. Смесь перемешивали и пропускали через сито с размером пор 0,85 мм, а затем смешивали до однородного состояния и прессовали в таблетку, используя круглый штамп с углублением 6,5. Давление поддерживали на уровне от 50 до 65 Н. Время распада ядра таблетки до нанесения покрытия оптимально находилось в пределах 1 минуты.
2) Покрытие ядра внутреннего слоя таблетки
Раствор для нанесения покрытия с содержанием твердых веществ 15% готовили в соответствии с весовым соотношением твердых веществ Aquacoat ECD: триэтилцитрат (TEC): Kollicoat®IR = 8,05: 2,01: 0,50. Температуру покрывающего слоя поддерживали в диапазоне от 39°C до 42°C. Увеличение массы составило от 8 до 10%. Смесь отверждали при 60°C в течение 2 часов после нанесения покрытия, после чего она была готова к применению в качестве ядра внутреннего слоя таблетки с покрытием и ядром в оболочке. Профиль высвобождения изониазида в ядре внутреннего слоя таблетки с покрытием и ядром в оболочке приведен в таблице 2.
3) Прессование и нанесение покрытия на таблетку с покрытием и ядром в оболочке
Гранулы внешнего слоя из примера 1 использовали для прессования таблетки с покрытием и ядром в оболочке, полученную таблетку покрывали раствором для нанесения покрытия с 20% концентрацией Opadry II. Температуру покрывающих материалов поддерживали на уровне от 40 до 45°C. Увеличение массы составило примерно 3%.
Пример 3
Получение таблетки с покрытием и ядром в оболочке
1) Получение ядра таблетки без оболочки
75,00 г изониазида, 18,70 г микрокристаллической целлюлозы, 8,20 г сшитой карбоксиметилцеллюлозы натрия смешивали до однородного состояния, а затем измельчали для пропускания через сито с размером пор 0,250 мм. Получали гибкий материал с использованием 2% гидроксипропилметилцеллюлозы. Во-первых, изониазид измельчали и пропускали через сито с размером пор 0,250 мм. Смесь экструдировали через сито с размером пор 600 мкм. Смесь вращали при 700 об/мин, а затем получали драже. Драже высушивали при 50°C.
2) Покрытие ядра драже
Раствор для нанесения покрытия с содержанием твердых веществ 15% готовили в соответствии с весовым соотношением твердых веществ Aquacoat ECD: триэтилцитрат (TEC): Kollicoat®IR = 8,05: 2,01: 0,50. Температуру покрывающего слоя поддерживали в диапазоне от 39°C до 42°C. Увеличение массы при нанесении покрытия на драже составило от 22 до 25%. Смесь отверждали при 60°С в течение 2 часов после нанесения покрытия, после чего она была готова к применению в качестве ядра внутреннего слоя таблетки с покрытием и ядром в оболочке.
3) Прессование и нанесение покрытия на таблетку с покрытием и ядром в оболочке
Гранулы внешнего слоя из примера 1 использовали для прессования таблетки, имеющей соотношение изониазид: рифампицин: пиразинамид: этамбутола гидрохлорид, равное 75 мг: 150 мг: 400 мг: 275 мг, полученную таблетку покрывали раствором для нанесения покрытия с 20% концентрацией Opadry II. Температуру покрывающих материалов поддерживали на уровне от 40 до 45°C. Увеличение массы составило примерно 3%.
Пример 4
Получение трехслойной таблетки с покрытием
1) Получение гранул слоя рифампицина
30,03 г изониазида, 5,60 г микрокристаллической целлюлозы, 7,40 г сшитой карбоксиметилцеллюлозы натрия смешивали до однородного состояния, а затем измельчали для пропускания через сито с размером пор 0,250 мм. Смесь пропускали через сито с размером пор 0,710 мм с 2% раствором гидроксипропилметилцеллюлозы. Смесь высушивали при 50°С, пока содержание влаги в гранулах не становилось менее чем 3,0%. Добавляли 0,5% тонкоизмельченного силикагеля, исходя из массы сухих гранул, и смесь пропускали через сито с размером пор 0,850 мм. Полученный продукт был смешан до однородного состояния и готов для прессования в таблетку.
2) Получение гранул слоя этамбутола гидрохлорида
55,02 г этамбутола гидрохлорида и 5,02 г микрокристаллической целлюлозы смешивали, измельчали и пропускали через сито с размером пор 0,250 мм. Смесь пропускали через сито с размером пор 0,710 мм с 5% раствором этилцеллюлозы в смеси этанол/вода (этанол:дистиллированная вода = 90:10 об./об.) и гранулировали. Смесь высушивали при 80°C, пока содержание влаги в гранулах не становилось менее чем 2,0%. Добавляли 0,5% стеариновой кислоты, исходя из массы сухих гранул, и смесь пропускали через сито с размером пор 0,850 мм. Полученный продукт был смешан до однородного состояния и готов для прессования в таблетку.
3) Получение гранул слоя изониазида/пиразинамида
15,00 г этамбутола гидрохлорида, 80,00 г пиразинамида и 8,02 г микрокристаллической целлюлозы смешивали, измельчали и пропускали через сито с размером пор 0,250 мм. Смесь пропускали через сито с размером пор 0,710 мм с 2% водным раствором гидроксипропилметилцеллюлозы и гранулировали. Смесь высушивали при 80°C, пока содержание влаги в гранулах не становилось менее чем 3,0%. Добавляли 0,5% стеариновой кислоты, исходя из массы сухих гранул, и смесь пропускали через сито с размером пор 0,850 мм. Полученный продукт был смешан до однородного состояния и готов для прессования в таблетку.
4) Прессование таблетки
Слой рифампицина, слой этамбутола гидрохлорида и слой изониазида/пиразинамида прессовали в трехслойную таблетку, имеющую соотношение рифампицин: этамбутола гидрохлорид: изониазид: пиразинамид=150 мг: 275 мг: 75 мг: 400 мг.
5) Нанесение покрытия
15% раствор для нанесения покрытия готовили в соответствии с весовым соотношением твердых веществ Aquacoat ECD: DBS: TEC: диоксид титана = 60: 20: 10: 10. Увеличение массы при нанесении покрытия составило примерно 8%. Отверждение проводили при 60°C в течение 2 часов.
Пример 5
Получение трехслойной таблетки с покрытием
1) Получение гранул слоя рифампицина
30,13 г изониазида, 5,66 г микрокристаллической целлюлозы, 7,50 г сшитой карбоксиметилцеллюлозы натрия смешивали до однородного состояния, а затем измельчали для пропускания через сито с размером пор 0,250 мм. Смесь пропускали через сито с размером пор 0,710 мм с 2% раствором гидроксипропилметилцеллюлозы. Смесь высушивали при 50°C, пока содержание влаги в гранулах не становилось менее чем 3,0%. Добавляли 0,5% тонкоизмельченного силикагеля, исходя из массы сухих гранул, и смесь пропускали через сито с размером пор 0,850 мм. Полученный продукт был смешан до однородного состояния и готов для прессования в таблетку.
2) Получение гранул слоя этамбутола гидрохлорида
55,22 г этамбутола гидрохлорида, 5,99 г микрокристаллической целлюлозы и 3,01 г гидроксипропилцеллюлозы K100M смешивали, измельчали и пропускали через сито с размером пор 0,250 мм. Гибкий материал, полученный с использованием раствора 70% этанол/вода (об./об.), пропускали через сито с размером пор 0,710 мм и гранулировали. Смесь высушивали при 80°C, пока содержание влаги в гранулах не становилось менее чем 2,0%. Добавляли 0,5% стеариновой кислоты, исходя из массы сухих гранул, и смесь пропускали через сито с размером пор 0,850 мм. Полученный продукт был смешан до однородного состояния и готов для прессования в таблетку.
3) Получение гранул слоя изониазида/пиразинамида
15,08 г изониазида, 80,10 г пиразинамида и 8,10 г микрокристаллической целлюлозы смешивали и измельчали для пропускания через сито с размером пор 0,250 мм. Смесь пропускали через сито с размером пор 0,710 мм с 2% раствором гидроксипропилметилцеллюлозы. Смесь высушивали при 80°C, пока содержание влаги в гранулах не становилось менее чем 3,0%. Добавляли 0,5% стеариновой кислоты, исходя из массы сухих гранул, и смесь пропускали через сито с размером пор 0,850 мм. Полученный продукт был смешан до однородного состояния и готов для прессования в таблетку.
4) Прессование таблетки
Слой рифампицина, слой этамбутола гидрохлорида и слой изониазида/пиразинамида прессовали в трехслойную таблетку, имеющую соотношение рифампицин: этамбутола гидрохлорид: изониазид: пиразинамид = 150 мг: 275 мг: 75 мг: 400 мг.
5) Нанесение покрытия
15% раствор для нанесения покрытия готовили в соответствии с весовым соотношением твердых веществ Aquacoat ECD:DBS:TEC:диоксид титана=60:20:10:10. Увеличение массы при нанесении покрытия составило примерно 8%. Отверждение проводили при 60°C в течение 2 часов.
Пример 6
15% раствор для нанесения покрытия готовили в соответствии с весовым соотношением твердых веществ Aquacoat ECD:TEC:HPMC:диоксид титана=70,8:12,5:8,3:8,3. Увеличение массы при нанесении покрытия составило примерно 8%. Отверждение проводили при 60°C в течение 2 часов.
Пример 7
Получение таблетки с покрытием и ядром в оболочке
1) Получение ядра таблетки без оболочки
15,02 г изониазида и 2,80 г микрокристаллической целлюлозы смешивали до однородного состояния, а затем измельчали для пропускания через сито с размером пор 0,250 мм. Получали гибкий материал с использованием 2% гидроксипропилметилцеллюлозы, а затем пропускали через сито с размером пор 0,710 мм и гранулировали. Смесь высушивали при 50°C, пока содержание влаги в гранулах не становилось менее чем 3,0%. Добавляли 1% стеарата магния, исходя из массы сухих гранул. Смесь перемешивали и пропускали через сито с размером пор 0,85 мм, а затем смешивали до однородного состояния и прессовали, получая таблетку определенной формы при помощи 4*9 мм штампа для прессования. Давление поддерживали на уровне от 50 до 65 Н.
2) Покрытие ядра внутреннего слоя таблетки разделительным пленочным покрытием
Для нанесения покрытия использовали 10% раствор HPMC. Температуру покрывающего слоя поддерживали в диапазоне от 28°C до 32°C. Увеличение массы составило примерно 5%.
3) Получение твердой дисперсии рифампицина во внешнем слое
15,05 г рифампицина и 15,10 г ацетатсукцината гидроксипропилметилцеллюлозы смешивали до однородного состояния. Твердую дисперсию рифампицина получали методом горячего плавления, а затем измельчали и просеивали через сито с размером пор 0,250 мм.
4) Получение гранул внешнего слоя пиразинамида/этамбутола
400,70 г рифампицина, 275,05 г этамбутола гидрохлорида, 161,04 г микрокристаллической целлюлозы и 54,50 г низкозамещенной гидроксипропилцеллюлозы смешивали до однородного состояния и затем формовали в порошок. Смесь пропускали через сито с размером пор 0,250 мм и гранулировали с использованием 5%-гидроксипропилметилцеллюлозы. Смесь высушивали, и влажность гранул поддерживали на уровне менее чем 3%.
5) Смешивание гранул внешнего слоя
Порошок твердой дисперсии рифампицина смешивали с высушенными гранулами пиразинамида/этамбутола гидрохлорида из этапа 4) с соотношением рифампицин: этамбутола гидрохлорид: пиразинамид = 150 мг: 275 мг: 400 мг. Добавляли 0,5% тонкоизмельченного силикагеля, исходя из массы смешанных гранул, и пропускали через сито с размером пор 0,600 мм. Полученный продукт был готов для прессования в таблетку.
6) Прессование таблетки с покрытием и ядром в оболочке
Таблетку с покрытием из этапа 2) использовали в качестве ядра таблетки, а гранулы, полученные на этапе 5), использовали в качестве внешнего слоя и прессованием получали таблетку с покрытием и ядром в оболочке.
7) Покрытие внешнего слоя
Готовили раствор для покрытия с 20% концентрацией Opadry II. Температуру покрывающих материалов поддерживали на уровне от 40 до 45°C. Увеличение массы составило примерно 3%.
Пример 8
Получение таблетки с покрытием и ядром в оболочке
1) Получение твердой дисперсии рифампицина во внешнем слое
150,00 г рифампицина и 150,00 г ацетатсукцината гидроксипропилметилцеллюлозы перемешивали до однородного состояния, затем растворяли в 1500,0 г дихлорметана и 1500,0 г ацетона, соответственно, полученные растворы смешивали до однородного состояния, а затем получали твердую дисперсию рифампицина методом сушки распылением. Температуру проточного воздуха поддерживали на уровне от 50 до 55°C.
2) Смешивание гранул внешнего слоя
Порошок твердой дисперсии рифампицина, полученный методом сушки распылением, смешивали с высушенными гранулами пиразинамида/этамбутола гидрохлорида из этапа 4) примера 7 с соотношением рифампицин: этамбутола гидрохлорид: пиразинамид = 150 мг: 275 мг: 400 мг. Добавляли 0,5% тонкоизмельченного силикагеля, исходя из массы смешанных гранул, и пропускали через сито с размером пор 0,600 мм. Полученный продукт был готов для прессования в таблетку.
3) Прессование таблетки с покрытием и ядром в оболочке
Таблетку с покрытием из этапа 2) примера 7 использовали в качестве ядра таблетки, а гранулы, полученные на этапе 2) примера 8 использовали в качестве внешнего слоя и прессованием получали таблетку с покрытием и ядром в оболочке.
4) Покрытие внешнего слоя
Готовили раствор для покрытия с 20% концентрацией Opadry II. Температуру покрывающих материалов поддерживали на уровне от 40 до 45°C. Увеличение массы составило примерно 3%.
Пример 9
Получение таблетки с покрытием и ядром в оболочке
1) Получение раствора рифампицина
150,00 г рифампицина и 150,00 г ацетатсукцината гидроксипропилметилцеллюлозы перемешивали до однородного состояния, затем растворяли в 1500,0 г дихлорметана и 1500,0 г ацетона, соответственно, полученные растворы смешивали до однородного состояния для последующего использования.
2) Получение гранул внешнего слоя
400,00 г пиразинамида, 275,00 г этамбутола гидрохлорида, 160,00 г микрокристаллической целлюлозы и 54,00 г низкозамещенной гидроксипропилцеллюлозы смешивали до однородного состояния и измельчали. Смесь пропускали через сито с размером пор 0,250 мм, а затем высыпали в псевдоожиженный слой. Раствор рифампицина из этапа 1) использовали в одностадийном методе гранулирования. Температуру материалов поддерживали на уровне от 35 до 40°C и содержание влаги в гранулах поддерживали на уровне менее чем 3%. Добавляли предписанное количество тонкоизмельченного силикагеля и пропускали через сито с размером пор 0,600 мм. Полученный продукт был готов к использованию.
3) Прессование таблетки с покрытием и ядром в оболочке
Таблетку с покрытием из этапа 2) примера 7 использовали в качестве ядра таблетки, а гранулы, полученные на этапе 2) примера 9, использовали в качестве внешнего слоя и прессованием получали таблетку с покрытием и ядром в оболочке.
4) Покрытие внешнего слоя
Готовили раствор для покрытия с 20% концентрацией Opadry II. Температуру покрывающих материалов поддерживали на уровне от 40 до 45°C. Увеличение массы составило примерно 3%.
Пример 10
Получение трехслойной таблетки с покрытием
1) Получение гранул слоя рифампицина
60,30 г твердой дисперсии рифампицина из примера 8, 5,60 г микрокристаллической целлюлозы, 7,32 г сшитой карбоксиметилцеллюлозы натрия смешивали до однородного состояния, а затем измельчали для пропускания через сито с размером пор 0,250 мм. Смесь пропускали через сито с размером пор 0,710 мм с 2% раствором гидроксипропилметилцеллюлозы. Смесь высушивали при 50°C, пока содержание влаги в гранулах не становилось менее чем 3,0%. Добавляли 0,5% стеариновой кислоты, исходя из массы сухих гранул, и смесь пропускали через сито с размером пор 0,850 мм. Полученный продукт был смешан до однородного состояния и готов для прессования в таблетку.
2) Получение гранул слоя этамбутола гидрохлорида
55,22 г этамбутола гидрохлорида и 6,03 г микрокристаллической целлюлозы смешивали, измельчали и пропускали через сито с размером пор 0,250 мм. Смесь пропускали через сито с размером пор 0,710 мм с 5% водным раствором гидроксипропилметилцеллюлозы и гранулировали. Смесь высушивали при 80°C, пока содержание влаги в гранулах не становилось менее чем 2,0%. Добавляли 0,5% стеариновой кислоты, исходя из массы сухих гранул, и смесь пропускали через сито с размером пор 0,850 мм. Полученный продукт был смешан до однородного состояния и готов для прессования в таблетку.
3) Прессование таблетки
1) Слой рифампицина, 2) слой этамбутола гидрохлорида и 3) слой изониазида/пиразинамида из примера 5 прессовали в трехслойную таблетку, имеющую соотношение рифампицин: этамбутола гидрохлорид: изониазид: пиразинамид = 150 мг: 275 мг: 75 мг: 400 мг.
4) Нанесение покрытия
Готовили раствор для нанесения покрытия с 20% концентрацией Opadry II. Температуру покрывающих материалов поддерживали на уровне от 40 до 45°C. Увеличение массы составило примерно 3%.
Пример 11
148,84 г твердой дисперсии рифампицина из примера 8, 20,75 г микрокристаллической целлюлозы, 8,94 г карбоксиметилцеллюлозы натрия и 0,90 г тонкоизмельченного силикагеля смешивали до однородного состояния и прессовали в таблетку, используя круглый штамп с углублением 12. Давление поддерживали на уровне от 50 до 90 Н. Полученную таблетку покрывали раствором для нанесения покрытия с 20% концентрацией Opadry II. Температуру покрывающих материалов поддерживали на уровне от 4 0 до 45°C. Увеличение массы составило примерно 3%. Экспериментальные in vivo данные по препарату, полученные в примере, приведены в таблицах 10-11.
Пример 12
Твердые дисперсии рифампицина с различными соотношениями получали методом спонтанного испарения растворителя. Каждую из навесок 9,0, 6,0, 3,0, 1,5 и 1,0 г HPMCAS растворяли в 30,0 г дихлорметана, получая растворы. Затем каждый из растворов рифампицина выливали в раствор HPMCAS. После смешивания до однородного состояния в течение 1 часа, каждую из смесей выливали на часовые стекла и помещали в вытяжной шкаф для выпаривания досуха. Получали твердые дисперсии рифампицина, а затем измельчали для использования. Результаты определения степени высвобождения рифампицина в препарате из данного примера приведены в таблице 5.
Материалы
Рифампицин (Shenyang Antibiotics Factory); изониазид (Zhejiang Xinsai Pharmaceuticals Co. Ltd.); пиразинамид (Jiangsu Sihuan Biotec Co. Ltd.); этамбутола гидрохлорид (Shanghai Wuzhou Pharmaceuticals Co. Ltd.); XEED® (Panacea Biotec Ltd); капсулы рифампицина, произведенные в Китае (Shenyang Hongqi pharmaceuticals Co. Ltd.); таблетки изониазида, произведенные в Китае (Shenyang Hongqi pharmaceuticals Co. Ltd.); микрокристаллическая целлюлоза (Asahi Kasei Chemicals Corporation); низкозамещенная гидроксипропилцеллюлоза (Shin-Etsu); гидроксипропилметилцеллюлоза E5 и K100M (Colorcon); стеариновая кислота (Hunan Huari Pharmaceuticals Co. Ltd.); тонкоизмельченный силикагель (Evonik); ацетатсукцинат гидроксипропилметилцеллюлозы (Shin-Etsu); сшитая карбоксиметилцеллюлоза натрия (Nichirin Chemical Industries Ltd.); Aquacoat ECD (FMC Biopolymer); триэтилцитрат (Shanghai Shenbao Flavours and Perfumes Co. Ltd.); Kollicoat®IR (BASF); стеарат магния (Anhui Shanhe Pharmaceutic Adjuvant Co. Ltd.); витамин С (Hebei Welcome Pharmaceutical Co.,Ltd ); лаурилсульфат натрия (Anhui Shanhe Pharmaceutic Adjuvant Co. Ltd.); ПЭГ 6000 (Chemical Reagents Co. Ltd., Sinopharm Group); Opadry II (Colorcon); дихлорметан (Chemical Reagents Co. Ltd., Sinopharm Group); ацетон (Chemical Reagents Co. Ltd., Sinopharm Group); хлорид натрия (Shanghai Qingxi Chemical Technology Co. Ltd.); ацетонитрил (хроматографически чистый, MERCK Co. Ltd., US); безводный дигидрофосфат натрия (аналитически чистый, Shanghai Lingfeng Chemical Reagent Co. Ltd.); фосфорная кислота (аналитически чистая, Chemical Reagents Co. Ltd., Sinopharm Group); гидроксид натрия (аналитически чистый, Shanghai Lingfeng Chemical Reagent Co. Ltd.); уксусная кислота (аналитически чистая, Shanghai Lingfeng Chemical Reagent Co. Ltd.).
Приборы и инструменты
Многофункциональное экспериментальное оборудование мини-типа (Chongqing Jinggong Pharmaceutical machine Co. Ltd.); таблеточный пресс с одним пуансоном (Shanghai Tianfan Pharmaceutical machinery factory); распылительная сушилка (BUCHI); прибор для определения влажности (Sartorius Co. Ltd., Beijing); интеллектуальный прибор для проверки растворимости (Tianda Tianfa Co. Ltd.); климатическая камера, типа 720 (BINDER); электронные весы BT224S (Sartorius Co. Ltd., Beijing); установка для нанесения покрытий OHHARA лабораторного типа; установка для ВЭЖХ Agilent 1200 (Agilent Technology, Co. Ltd.); pH-метр типа FE20 (METTLER TOLEDO Co. Ltd: Phenomenex Luna C18 100A (250×4,6 мм, 5 мкм, Phenomenex Co. Ltd., US); Agilent Eclipse XDB-C18 (150×4,6 мм, 5 мкм, Agilent Technology, Co. Ltd., US).
Экспериментальные примеры
(1) In-vitro тест
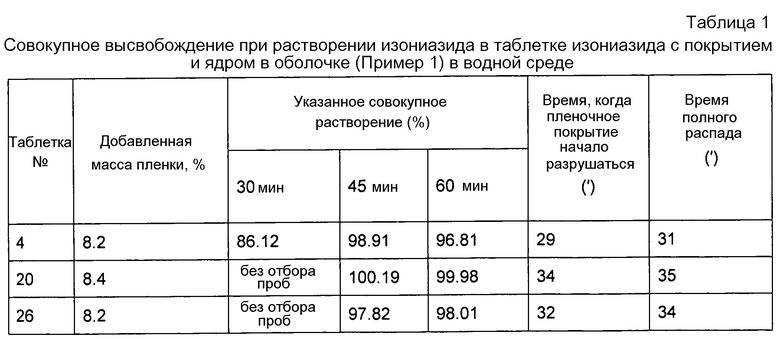
Примечание: Согласно опыту, полученному при проведении экспериментов, если пленочное покрытие таблетки не разрушалось, высвобождение изониазида было относительно невелико, то есть, менее чем 10%. Вследствие этого, образцы в случае таблеток № 20 и 26 не отбирали и не измеряли.
Как видно из таблицы 1, время разрушения пленочного покрытия внутреннего слоя практически может находиться в пределах необходимого контролируемого времени, то есть, от 30 до 60 минут. После разрушения пленочного покрытия лекарственные средства могут полностью высвобождаться в течение короткого времени.
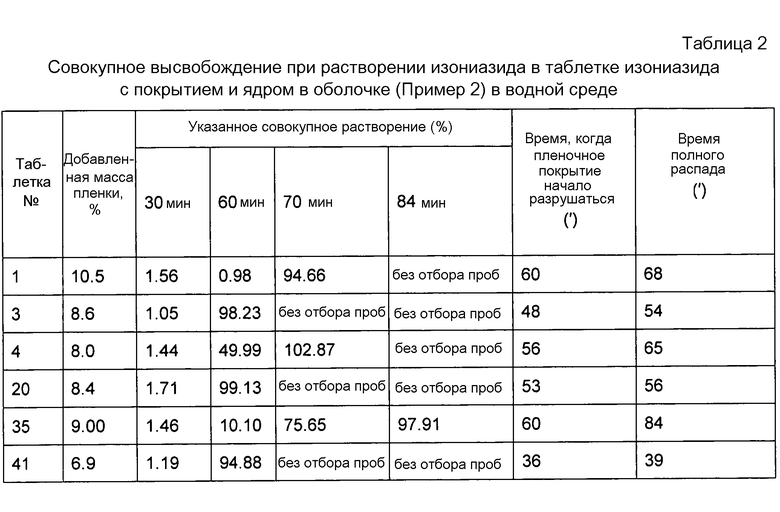
Как видно из таблицы 2, каждое из пленочных покрытий на центральном слое препарата разрушается в течение 30-60 минут. В течение 30 минут высвобождение изониазида составляет не более 2%, что значительно ниже, чем стандартные 10% в оптимизированном режиме для контроля качества. В течение 90 минут лекарственное средство практически полностью освобождается.

Из данных в таблице 3 можно видеть, что высвобождение изониазида может достигать стандарта, необходимого для контроля, то есть, менее чем 15% высвобождение в течение 30 минут и более чем 80% высвобождение в течение 90 минут.
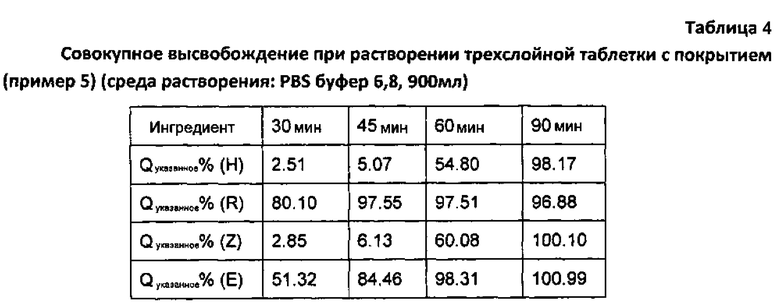
Из данных в таблице 4 можно видеть, что контролирующая высвобождение пленка и слой этамбутола гидрохлорида отлично зарекомендовали себя для контроля замедленного высвобождения изониазида.
Примечание: 1) Условия растворения  были такими, что кислота и основание (900 мл каждого) не содержались в одной емкости, и его проводили независимо методом с использованием лопастной мешалки со скоростью 100 об/мин при 37,0°C. Условия растворения
были такими, что кислота и основание (900 мл каждого) не содержались в одной емкости, и его проводили независимо методом с использованием лопастной мешалки со скоростью 100 об/мин при 37,0°C. Условия растворения  были такими, что кислота и основание содержались в одной и той же емкости. Другими словами, сначала добавляли 750 мл кислого вещества (0,1 моль/л соляной кислоты). Отбирали образец и добавляли раствор после того, как растворение продолжалось в течение 45 минут, при том, что добавляли 250 мл 0,2 моль/л раствора фосфата натрия и добавляли 4,5 мл 2 моль/л раствора гидроксида натрия для доведения pH раствора до 6,8±0,05 как у основной среды. Растворение проводили методом с использованием лопастной мешалки со скоростью 100 об/мин при 37,0°C.
были такими, что кислота и основание содержались в одной и той же емкости. Другими словами, сначала добавляли 750 мл кислого вещества (0,1 моль/л соляной кислоты). Отбирали образец и добавляли раствор после того, как растворение продолжалось в течение 45 минут, при том, что добавляли 250 мл 0,2 моль/л раствора фосфата натрия и добавляли 4,5 мл 2 моль/л раствора гидроксида натрия для доведения pH раствора до 6,8±0,05 как у основной среды. Растворение проводили методом с использованием лопастной мешалки со скоростью 100 об/мин при 37,0°C.
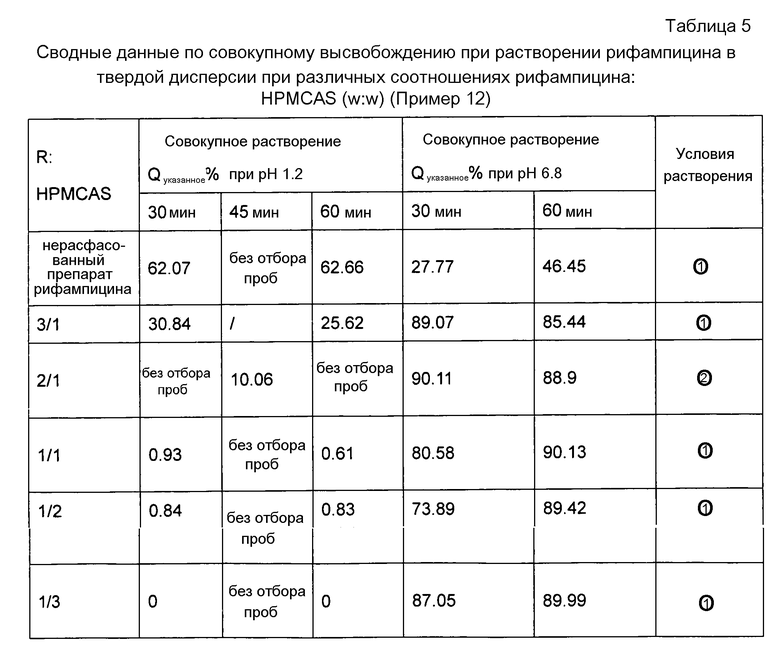
Как видно из таблицы 5, результаты эксперимента свидетельствуют о том, что в соответствии с настоящим изобретением лучший солюбилизирующий эффект для рифампицина достигается при переведении рифампицина в твердую суспензию (соотношение рифампицин: HPMCAS соответствует от 3:1 до 1:3). Однако при соотношении рифампицин: HPMCAS, равном 3:1, высвобождение рифампицина в кислых условиях желудка было вне контролируемого диапазона, и поэтому такое соотношение не было выбрано для препарата. Как можно видеть в таблице, наиболее подходящим для выбора является массовое соотношение рифампицин: HPMCAS, равное 1:1, поскольку происходило растворение, и высвобождение рифампицина в кислой среде можно было контролировать, используемое количество также было относительно невелико.
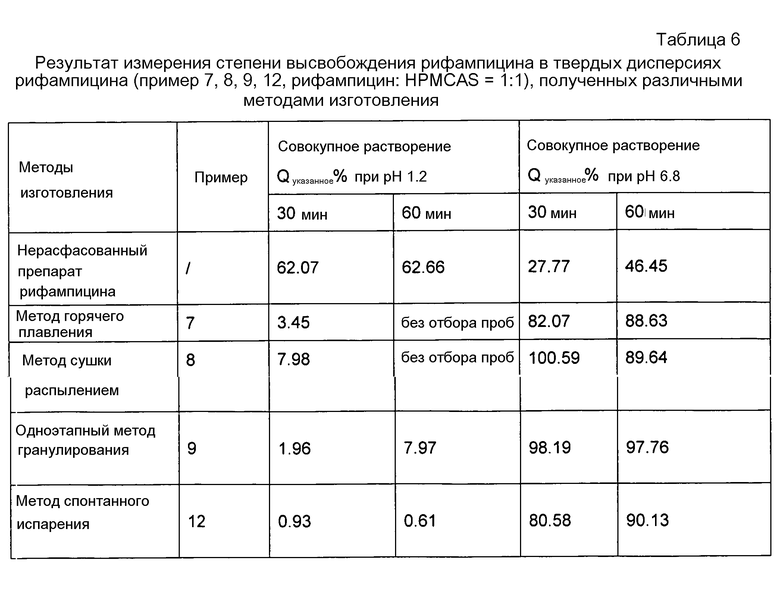
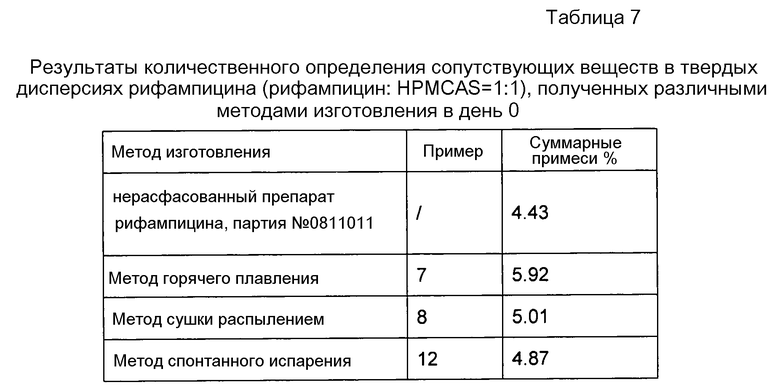
Данные в таблице 6 свидетельствуют, что методы изготовления не оказывают существенного солюбилизирующего эффекта на рифампицин.
Примечание: 1) Хроматограммы приведены на фигурах 1-4. Пик 2 на фигурах соответствует пику рифампицина.
2) Оборудование и условия хроматографии: ВЭЖХ хроматограф Waters e2695-2998, колонка Phenmenex luna C18 (250×4,6 мм, 5 мкм); скорость потока составляла 1,0 мл/мин, длина волны измерений составляла 238 нм, подвижная фаза: фаза A=pbs 6,8/ацетонитрил (96/4), фаза B=pbs 6,8/ацетонитрил (50/50), фаза A: фаза B=25:75.
Как видно из таблицы 7, показатели стабильности рифампицина при получении твердых дисперсий тремя методами располагаются в следующем порядке: метод спонтанного испарения > метод сушки распылением >метод горячего плавления. С учетом промышленного характера производства предпочтительным является метод сушки распылением.

Примечание: 1) импортная четырехкомпонентная комбинированная таблетка с покрытием и ядром в оболочке произведена компанией Panacea Biotec Ltd из Индии (партия №: 9988512). Хроматограммы приведены на фигурах 5-6, на них пик 1 соответствует пиразинамиду, а пик 2 соответствует рифампицину.
2) Оборудование и условия хроматографии: ВЭЖХ хроматограф Agilent 1200, колонка Phenmenex luna C18 (250×4,6 мм, 5 мкм), длина волны измерений составляла 238 нм, скорость потока составляла 1,0 мл/мин, подвижная фаза: фаза A = pbs 6,8/ацетонитрил (96/4), фаза B = pbs 6,8/ацетонитрил (40/60), и градиент был таким, как указано ниже:
Результаты изучения стабильности, приведенные в таблице 8, свидетельствуют о том, что препарат по изобретению более стабилен, чем импортный коммерчески доступный препарат. Таким образом, настоящее изобретение позволяет улучшить стабильность четырехкомпонентного комбинированного противотуберкулезного препарата.
Примечание: 1) Хроматограмма приведена на фигуре 7, на ней пик 1 соответствует пиразинамиду, а пик 2 соответствует рифампицину.
2) Оборудование и условия хроматографии: ВЭЖХ хроматограф Waters e2695-2998, колонка Phenmenex luna C18 (250×4,6 мм, 5 мкм), скорость потока составляла 1,0 мл/мин, длина волны измерений составляла 238 нм, подвижная фаза: фаза A=pbs 6,8/ацетонитрил (96/4), фаза B=pbs 6,8/ацетонитрил (50/50), и градиент был таким, как указано ниже:
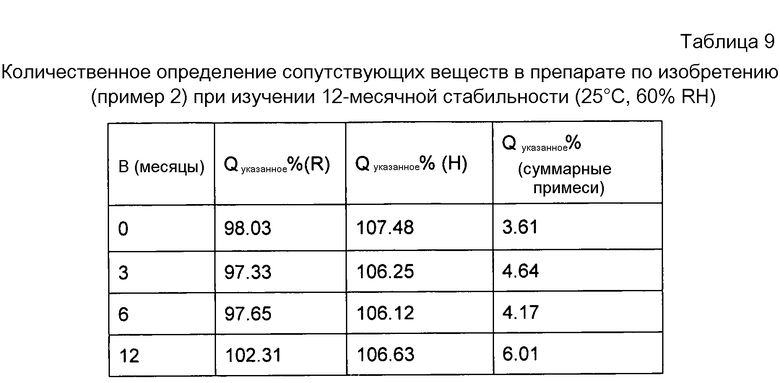
Как видно из таблицы 9, результат изучения стабильности в течение 12-месячного срока свидетельствует о том, что при хранении в условиях нормальной температуры количество примесей за счет рифампицина и изониазида возрастало в препаратах по изобретению в изучаемый период времени, однако препарат по-прежнему оставался относительно стабильным.
(2) Эксперимент in-vivo на собаках породы бигль
Процедура введения в группе введения одного рифампицина:
12 биглей были случайным образом разделены на группы А и В. Кормление в каждой группе проводили в 8 часов утра и введение проводили через 0,5 часа. Животным группы A вводили одну таблетку рифампицина по изобретению (150 мг, что соответствует 10 мг/кг, рифампицин сначала переведен в энтеросолюбильные твердые дисперсии методом сушки распылением, а затем спрессован с другими вспомогательными веществами для получения образца с покрытием (то есть, образец из примера 11). Животным группы B вводили одну капсулу рифампицина производства Shenyang Hongqi Pharmaceutical Co. Ltd. (150 мг, партия № 0907011).
Процедура введения сочетания рифампицин+изониазид
Через две недели после окончания описанного выше эксперимента животных каждой из групп A и B кормили и через 0,5 часа проводили введение. Животным группы A вводили одну таблетку рифампицина по изобретению (150 мг, что соответствует 10 мг/кг, образец был таким же, что и образец при введении одного рифампицина) и одну капсулу изониазида производства Shenyang Hongqi Pharmaceutical Co. Ltd. (75 мг/капсулу, полученную путем измельчения таблетки, содержащей 0,3 г/таблетку из партии № 0907011, и расфасовки в капсулы). Животным группы B вводили одну капсулу рифампицина (150 мг, партия № 0907011) и одну капсулу изониазида (такую же, как в группе A) производства Shenyang Hongqi Pharmaceutical Co. Ltd.


Примечание:
Относительная биодоступность 1(%)= среднее значение AUC 0-12 одной таблетки рифампицина по изобретению/среднее значение AUC 0-12 одной капсулы коммерчески доступного рифампицина × 100%;
Относительная биодоступность 2(%)=среднее значение AUC 0-12 таблетки комбинированного препарата рифампицина по изобретению/среднее значение AUC 0-12 капсулы коммерчески доступного комбинированного препарата рифампицина×100%.
Из данных по AUC 0-24 ч и AUC 0-∞ ч в таблице 10 и таблице 11 можно видеть, что после того, как рифампицин был переведен в твердую дисперсию, его биодоступность значительно увеличилась.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ПРОТИВОТУБЕРКУЛЕЗНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ И СПОСОБ ИХ ИЗГОТОВЛЕНИЯ | 2001 |
|
RU2240795C9 |
| ПРОТИВОТУБЕРКУЛЕЗНАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ РИФАМПИЦИН, ИЗОНИАЗИД, ЭТАМБУТОЛ И ПИРАЗИНАМИД, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2014 |
|
RU2672879C2 |
| КОМБИНИРОВАННЫЙ ПРОТИВОТУБЕРКУЛЕЗНЫЙ ПРЕПАРАТ | 2007 |
|
RU2354378C1 |
| КОМБИНИРОВАННАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С ПРОТИВОТУБЕРКУЛЁЗНЫМ ДЕЙСТВИЕМ | 2003 |
|
RU2247561C1 |
| ФАРМАЦЕВТИЧЕСКАЯ ПРОТИВОТУБЕРКУЛЕЗНАЯ КОМБИНИРОВАННАЯ КОМПОЗИЦИЯ | 2011 |
|
RU2478389C2 |
| ПРОТИВОТУБЕРКУЛЕЗНОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ОСНОВЕ 4-ТИОУРЕИДОИМИНОМЕТИЛПИРИДИНИЯ ПЕРХЛОРАТА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ЛЕЧЕНИЯ | 2010 |
|
RU2423977C1 |
| КОМБИНИРОВАННЫЙ СОСТАВ С ПРОТИВОТУБЕРКУЛЁЗНЫМ ДЕЙСТВИЕМ | 2003 |
|
RU2247560C1 |
| ПРОТИВОТУБЕРКУЛЁЗНОЕ СРЕДСТВО | 2003 |
|
RU2247559C1 |
| КОМБИНИРОВАННАЯ ПРОТИВОТУБЕРКУЛЕЗНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2009 |
|
RU2422145C2 |
| КОМБИНИРОВАННОЕ ПРОТИВОТУБЕРКУЛЕЗНОЕ СРЕДСТВО И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2008 |
|
RU2431487C2 |
Предложен пероральный твердый препарат комбинированного противотуберкулезного лекарственного средства, в котором активными ингредиентами являются рифампицин, изониазид, пиразинамид и этамбутола гидрохлорид в массовом соотношении 150:75:400:275, соответственно. Комбинированный пероральный твердый препарат представляет собой таблетку с покрытием и ядром в оболочке или трехслойную таблетку с покрытием, в которой два активных ингредиента рифампицин и изониазид не контактируют напрямую. В таблетке с покрытием и ядром в оболочке ядро внутреннего слоя содержит изониазид или рифампицин и полимер, который быстро разрушается и быстро увеличивается в объеме при поглощении воды, или рифампицин в ядре внутреннего слоя находится в форме энтеросолюбильной твердой дисперсии. В трехслойной таблетке с покрытием верхний слой и нижний слой отдельно и независимо представляют собой слой рифампицина, включающий полимер, который разрушается и быстро увеличивается в объеме при поглощении воды, и слой изониазида/пиразинамида, а центральный слой представляет собой слой этамбутола гидрохлорида, включающий замедлитель, который задерживает быстрый распад или высвобождение этамбутола гидрохлорида, или в слое, содержащем рифампицин, рифампицин находится в форме энтеросолюбильной твердой дисперсии, где массовое соотношение рифампицина и энтеросолюбильного твердого носителя составляет от 2:1 до 1:3. Комбинированный пероральный твердый препарат по изобретению характеризуется повышенной стабильностью, повышенной биодотупностью рифампицина, что усиливает эффективность лечения и снижает вероятность возникновения лекарственной устойчивости. 4 н. и 15 з.п. ф-лы, 11 ил., 11 табл., 11 пр.
1. Комбинированный пероральный твердый препарат с покрытием в форме таблетки с покрытием и ядром в оболочке, содержащей в качестве активных ингредиентов рифампицин, изониазид, пиразинамид и этамбутола гидрохлорид в массовом соотношении 200-300:75-300:250-500:250-275, соответственно, в которой ядро внутреннего слоя содержит изониазид или рифампицин и полимер, который разрушается и быстро увеличивается в объеме при поглощении воды, причем полимер представляет собой один или более выбранных из группы, состоящей из сшитой карбоксиметилцеллюлозы натрия (CCMC-Na), низкозамещенной гидроксипропилцеллюлозы (L-HPC), карбоксиметилцеллюлозы натрия (CMC-Na), карбоксиметилкрахмала натрия (CMS-Na), поливинилполипирролидона (PVPP) и микрокристаллической целлюлозы (МСС), причем ядро внутреннего слоя покрыто нерастворимым в воде не зависимым от pH пленочным покрытием, а внешний слой твердого препарата с покрытием покрыт водорастворимым и не зависимым от pH пленочным покрытием, которое является влагоустойчивым и светонепроницаемым, в которой после введения по меньшей мере один активный ингредиент из рифампицина и изониазида высвобождается не более чем на 15% от указанного процентного содержания или не высвобождается в желудке.
2. Комбинированный пероральный твердый препарат с покрытием в форме трехслойной таблетки с покрытием, содержащей в качестве активных ингредиентов рифампицин, изониазид, пиразинамид и этамбутола гидрохлорид в массовом соотношении 200-300:75-300:250-500:250-275, соответственно, в которой верхний слой и нижний слой отдельно и независимо представляют собой слой рифампицина, включающий полимер, который разрушается и быстро увеличивается в объеме при поглощении воды, причем полимер представляет собой один или более выбранных из группы, состоящей из сшитой карбоксиметилцеллюлозы натрия (CCMC-Na), низкозамещенной гидроксипропилцеллюлозы (L-HPC), карбоксиметилцеллюлозы натрия (CMC-Na), карбоксиметилкрахмала натрия (CMS-Na), поливинилполипирролидона (PVPP) и микрокристаллической целлюлозы (МСС) и слой изониазида/пиразинамида, а центральный слой представляет собой слой этамбутола гидрохлорида, включающий замедлитель, который задерживает быстрый распад или высвобождение этамбутола гидрохлорида, при этом сверху три слоя покрыты нерастворимым в воде не зависимым от pH пленочным покрытием, в которой после введения по меньшей мере один активный ингредиент из рифампицина и изониазида в меньшей степени высвобождается не более чем на 15% от указанного процентного содержания или не высвобождается в желудке.
3. Комбинированный пероральный твердый препарат с покрытием в форме таблетки с покрытием и ядром в оболочке, содержащей в качестве активных ингредиентов рифампицин, изониазид, пиразинамид и этамбутола гидрохлорид в массовом соотношении 200-300:75-300:250-500:250-275, соответственно, в которой ядро внутреннего слоя содержит изониазид или рифампицин и покрыто водорастворимой и не зависимой от pH разделительной пленкой, где рифампицин находится в форме энтеросолюбильной твердой дисперсии, в которой рифампицин диспергирован в энтеросолюбильном твердом носителе, причем энтеросолюбильный твердый носитель выбран из группы, состоящей из фталата поливинилацетата (PVAP), сополимера метакриловой кислоты/метилметакрилата, ацетатсукцината гидроксипропилметилцеллюлозы (HPMCAS), и где массовое соотношение рифампицина и энтеросолюбильного твердого носителя составляет от 2:1 до 1:3; а внешний слой твердого препарата с покрытием покрыт водорастворимым и не зависимым от pH пленочным покрытием, которое является влагоустойчивым и светонепроницаемым.
4. Комбинированный пероральный твердый препарат с покрытием в форме трехслойной таблетки с покрытием, содержащей в качестве активных ингредиентов рифампицин, изониазид, пиразинамид и этамбутола гидрохлорид в массовом соотношении 200-300:75-300:250-500:250-275, соответственно, в которой верхний слой и нижний слой отдельно и независимо представляют собой слой рифампицина и слой изониазида/пиразинамида, причем в слое, содержащем рифампицин, рифампицин находится в форме энтеросолюбильной твердой дисперсии, в которой рифампицин диспергирован в энтеросолюбильном твердом носителе, причем энтеросолюбильный твердый носитель выбран из группы, состоящей из фталата поливинилацетата (PVAP), сополимера метакриловой кислоты/метилметакрилата, ацетатсукцината гидроксипропилметилцеллюлозы (HPMCAS), и где массовое соотношение рифампицина и энтеросолюбильного твердого носителя составляет от 2:1 до 1:3; и центральный слой представляет собой слой этамбутола гидрохлорида, и сверху три слоя покрыты водорастворимым и не зависимым от pH пленочным покрытием, которое является влагоустойчивым и светонепроницаемым.
5. Комбинированный пероральный твердый препарат с покрытием по п.1 или 2, отличающийся тем, что указанное нерастворимое в воде и не зависимое от pH пленочное покрытие содержит нерастворимый в воде и не зависимый от pH полимерный пленкообразующий материал и пластификатор, необязательно дополнительно содержит по меньшей мере одно из порообразующего вещества и антиадгезионного вещества.
6. Комбинированный пероральный твердый препарат с покрытием по п.2, отличающийся тем, что указанное нерастворимое в воде и не зависимое от pH пленочное покрытие дополнительно содержит замутняющее вещество.
7. Комбинированный пероральный твердый препарат с покрытием по п.5, отличающийся тем, что указанное нерастворимое в воде и не зависимое от pH пленочное покрытие содержит 70-90% по массе пленкообразующего материала, 10-30% по массе пластификатора, 0-15% по массе порообразующего вещества и 0-25% по массе антиадгезионного вещества, и предпочтительно 70-80% по массе пленкообразующего материала, 13-25% по массе пластификатора и/или 4-13% по массе порообразующего вещества и/или 0-15% по массе антиадгезионного вещества, исходя из общей массы пленочного покрытия.
8. Комбинированный пероральный твердый препарат с покрытием по п.1, отличающийся тем, что увеличение массы за счет нерастворимого в воде и не зависимого от pH пленочного покрытия составляет 6-30% по массе от массы изониазидного ядра внутреннего слоя.
9. Комбинированный пероральный твердый препарат с покрытием по п.1, отличающийся тем, что более чем 80%, предпочтительно более чем 85% от указанного процентного содержания рифампицина, этамбутола гидрохлорида и пиразинамида в каждом случае высвобождается в течение 45 минут; нерастворимое в воде и не зависимое от pH пленочное покрытие, покрывающее сверху ядро внутреннего слоя, разрушается в течение 30-90 минут, не более чем 15% изониазида от указанного процентного содержания высвобождается в течение 30 минут, в то время как более чем 80%, предпочтительно более чем 85% от указанного процентного содержания изониазида высвобождается в течение 90 минут.
10. Комбинированный пероральный твердый препарат с покрытием по п.2, отличающийся тем, что после введения нерастворимое в воде и не зависимое от pH пленочное покрытие, покрывающее сверху слой рифампицина, сначала разрушается в течение 5-10 минут; полное время распада для центрального слоя составляет более чем 15 минут; более чем 80%, предпочтительно более чем 85% рифампицина от указанного процентного содержания высвобождается в течение 45 минут, и не более чем 15% изониазида высвобождается в течение 30 минут; и более чем 80%, предпочтительно более чем 85% изониазида от указанного процентного содержания высвобождается в течение 90 минут.
11. Комбинированный пероральный твердый препарат с покрытием по п.2, отличающийся тем, что указанное нерастворимое в воде и не зависимое от pH пленочное покрытие содержит 50-80% по массе пленкообразующего материала, 20-45% по массе пластификатора, 0-20% по массе замутняющего вещества и/или 0-15% по массе порообразующего вещества и/или 0-20% по массе антиадгезионного вещества, исходя из общей массы пленочного покрытия.
12. Комбинированный пероральный твердый препарат с покрытием по п.2, отличающийся тем, что увеличение массы за счет нерастворимого в воде и не зависимого от pH пленочного покрытия составляет 5-12% по массе от общей массы ядра трехслойной таблетки с покрытием.
13. Комбинированный пероральный твердый препарат с покрытием по п.2, отличающийся тем, что указанный замедлитель представляет собой один или более, выбранные из группы, состоящей из нерастворимого в воде каркасного материала, эрозивного каркасного материала и водорастворимого каркасного материала.
14. Комбинированный пероральный твердый препарат с покрытием по п.3 или 4, отличающийся тем, что после введения не более чем 15%, предпочтительно не более чем 10% рифампицина от указанного процентного содержания высвобождается в течение 30 минут в кислой среде желудка, и более чем 80%, предпочтительно более чем 85% рифампицина от указанного процентного содержания высвобождается в течение 45 минут в условиях pH кишечника.
15. Комбинированный пероральный твердый препарат с покрытием по п.3, отличающийся тем, что указанный пленкообразующий материал водорастворимой и не зависимой от pH разделительной пленки представляет собой один или более, выбранные из группы, состоящей из гидроксипропилметилцеллюлозы (НРМС), Kollicoat® IR и Opadry II.
16. Комбинированный пероральный твердый препарат с покрытием по любому из пп. 1, 3 или 4, отличающийся тем, что указанное водорастворимое и не зависимое от pH пленочное покрытие, которое является влагоустойчивым и светонепроницаемым, представляет собой покрытие, выбранное из группы, состоящей из Kollicoat® IR, гидроксипропилметилцеллюлозы (НРМС), сочетания диоксида титана и порошка талька, и Opadry II, содержащего сочетание гидроксипропилметилцеллюлозы (НРМС), диоксида титана и порошка талька.
17. Комбинированный пероральный твердый препарат с покрытием по любому из пп. 1-4, отличающийся тем, что после введения не более чем 15% от указанного процентного содержания по меньшей мере одного из активных ингредиентов, рифампицина и изониазида, высвобождается в течение 30 минут.
18. Комбинированный пероральный твердый препарат с покрытием по любому из пп. 1-4, дополнительно содержащий другие фармацевтические носители, выбранные из группы, состоящей из: наполнителя, связующего вещества, смазывающего вещества или глиданта и/или разрыхлителя.
19. Комбинированный пероральный твердый препарат с покрытием по любому из пп. 1-4, где массовое соотношение рифампицина, изониазида, пиразинамида и этамбутола гидрохлорида составляет 150:75:400:275, соответственно.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ПРОТИВОТУБЕРКУЛЕЗНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ И СПОСОБ ИХ ИЗГОТОВЛЕНИЯ | 2001 |
|
RU2240795C9 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ измерения коэффициента затухания упругих волн в материалах | 1983 |
|
SU1408354A1 |
| US 6372255 B1, 16.04.2002 | |||
| CN 101524355 A, 09.09.2009 | |||
| Устройство для обработки меховых шкур | 1988 |
|
SU1602872A1 |
| КОМБИНИРОВАННАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С ПРОТИВОТУБЕРКУЛЁЗНЫМ ДЕЙСТВИЕМ | 2003 |
|
RU2247561C1 |

