ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новым производным пиридона, способу их получения, содержащим их фармацевтическим композициям и их применению в качестве ингибиторов МЕК, и особенно в качестве терапевтических средств для лечения рака.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Статистические данные из Министерства здравоохранения Китая в 2008 году показали, что в Китае каждый год имеется приблизительно 2,127 миллиона новых случаев новообразований, из которых приблизительно 1,06 миллиона составляют новые случаи злокачественных новообразований; в то же время было зарегистрировано около 2,685 миллионов существующих пациентов с новообразованиями, из которых насчитывалось около 1,485 миллионов существующих пациентов со злокачественными новообразованиями. Министр здравоохранения Чэнь Чжу отметил на 21-м Всемирном конгрессе против рака, что за последние 30 лет смертность от рака в Китае увеличилась на 80%, ежегодное число смертей, вызванных раком, составило 1,8 миллиона, и рак стал первой причиной смерти для жителей Китая. По данным опроса "China Health Statistics Yearbook 2012", уровень смертности от злокачественных новообразований увеличивался, основными пятью типами злокачественных новообразований были рак легких, рак печени, рак желудка, рак пищевода и колоректальный рак соответственно, при этом быстрее всего возрастала смертность от рака легких и рака печени, и эти два рака занимают первое место по смертности среди заболеваний, вызванных злокачественными новообразованиями.
За последние полвека в области терапии опухолей были сделаны многие достижения. При тщательном изучении генетики и биологии опухолей было найдено множество ключевых внутриклеточных сигнальных путей, связанных с опухолью. Через эти внутриклеточные пути раковые клетки проводят внеклеточный сигнал до внутриклеточной трансдукции и регулируют активности, такие как непрерывная пролиферация и апоптоз, для поддержания злокачественноых фенотипов, и, с другой стороны, для создания устойчивости к лечению путем регулирования специфических генов и их белковых продуктов. Аномалия МАРК-киназного пути, приводящая к неконтролируемой клеточной пролиферации и замедлению дифференцировки, тесно связана с туморогенезом, в результате, МАРК-киназный сигнальный путь стал предпочтительной мишенью для разработки лекарств для лечения рака.
Серин/треонин митоген-активируемые протеинкиназы (МАРК, также называемые киназами, регулируемыми внеклеточными сигналами, ERK (от англ. extracellular signal-regulated)) активируются рецептором тирозинкиназы (например, рецептором ЭФР (эпидермального фактора роста)) и/или цитокиновым рецептором, связанным с гетеротримером G белка. МАРК могут взаимодействовать с внутриклеточными сигналами, инициированными различными вторичными мессенджерами, затем фосфорилироваться и регулировать активность различных ферментов и транскрипционных факторов (таких как NF-кВ, Rsk 90, фосфолипаза А2, с-Мус, CREB, Ets-1, АР-1 и c-jun, и т.д.). Среди путей МАРК, участвующих в нормальном и аномальном росте клеток, Ras/Raf/МЕК/Erk киназный путь является одним из наиболее хорошо изученных и наиболее важных путей. Более десяти лет назад ученые обнаружили, что белок семейства Erk-киназ участвует в промотировании пролиферации. В последующих исследованиях было быстро выявлено семейство МЕК, стоящее выше киназы ERK, после чего было установлено, что Raf может активировать МЕК, вышестоящим для Raf является Ras, относящийся к G-белку и связывающийся с активированной ГТФ, что может непрямым образом активировать Raf. Мутация гена Ras обнаружена приблизительно в 30% злокачественных новообразований у пациентов, частота мутаций Ras гена составляет даже до 90% при раке поджелудочной железы. Частота мутаций B-Raf составляет 50-70% при меланоме, 35% при раке яичников, 30% при раке щитовидной железы, и 10% при раке толстой кишки. Аналогично, МЕК могут быть активированы с помощью МЕК-киназы (также известной как МЕКК), не зависящей от Raf.
МЕК, также известные как киназы МАР-киназ (МАРКК или Erk-киназы), принадлежат к биспецифическим киназам, МЕК могут фосфорилировать серин/треониновые остатки и тирозиновые остатки МАРК (p44MAPK(Erk 1) и p42MAPK(Erk 2)) (сайтами фосфорилирования Erk1 являются Т202 и Y204, сайтами фосфорилирования Erk2 являются Т183 и Y185). Семейство МЕК включает пять генов: МЕК1, МЕК2, МЕКЗ, МЕК4 и МЕК5. N-терминальный конец МЕК является негативной регуляторной областью; С-концевой каталитический домен имеет функции связывания с Erk и активирования Erk. Тесты показали, что нокаут регуляторных областей МЕК1 приведет к ингибированию внутренней активности МЕК1 и Erk.
МЕК1, обладающий молекулярной массой около 44 кДа, имеющий в совокупности 393 аминокислоты, в основном экспрессируется в тканях взрослого организма, в особенности в ткани головного мозга. След экспрессии МЕК1 также может быть обнаружен в ходе эмбрионального развития. Активность МЕК1 инициируется фосфорилированием S218 и S222. Исследования показали, что в клетках NIH3T3 активность МЕК1 возрастает, когда фосфорилируются остатки аспарагиновой или глутаминовой кислот, и также увеличивается образование колоний. Внутренняя активность МЕК1 способствует старению клеток и экспрессии р53 и p16INK4a в первичной культуре клеток. Тем не менее, роль МЕК1 противоположна в иммортализованных клетках и p16INK4a или р53-дефицитных клетках. МЕК2, обладающий молекулярной массой около 45 кДа, имеет 79% сходство последовательностей с МЕК1, а его активность инициируется фосфорилированием S226 и S222. Каталитические активности при фосфорилировании МЕК1 и МЕК2 различны для различных изоформ МАРК, Erk1 и Erk2. МЕК 3, МЕК 4 и МЕК5 не играют роли посредством воздействия на Erks.
В настоящее время существует множество соединений для специфического ингибирования Raf и МЕК посредством сигнального пути МАРК, проходящих клинические испытания и находящихся на пострегистрационной стадии. При этом сорафениб (Bay 43-9006), выставленный на рынок в 2006 году, является неспецифическим серин/треонин и тирозин киназным ингибитором нацеленным на Raf, МЕК, VEGFR2/3, Flt-3, PDGFR, c-Kit и т.д. Специфические ингибиторы B-Raf, такие как дабрафениб (GSK2118436) и вемурафениб (PLX4032), показали хорошие клинические результаты, но их продолжительность не была достаточно длительной, в то же время клинические исследования показали, что симптомы большинства пациентов, получавших эффективное лечение PLX4032, рецидивировали, и было высказано предположение о том, что долговременное лечение ингибиторами B-Raf может вызвать приобретенную устойчивость к лекарству и сделать пациентов нечувствительными к ингибиторам B-Raf. Для того чтобы преодолеть устойчивость пациентов, в клинической терапии ингибиторы МЕК часто комбинируют с ингибиторами B-Raf. Специфический МЕК1/2 ингибитор траметиниб (GSK-1120212), разработанный GSK, уже вступил в стадию предварительной регистрации, другие ингибиторы МЕК1/2, такие как селуметиниб (AZD-6422), пимасертиба гидрохлорид (AS-703026) и TAK-733 и другие, достигли клинической стадии испытаний. Тем не менее, никаких данных по взаимодействию между этими ингибиторами МЕК и Erk1 или Erk2 не было раскрыто.
Был раскрыт ряд патентных заявок на ингибиторы МЕК, в том числе WO 2007096259, WO 2010003022 и WO 2012162293 и т.д.
Для достижения лучших онкотерапевтических целей, а также для более полного удовлетворения потребностей рынка, авторы надеятся разработать новое поколение ингибиторов сигнальных путей МАРК, в частности, ингибиторов МЕК, обладающих высокой эффективностью и низкой токсичностью. Настоящее изобретение относится к новым структурным ингибиторам МЕК, и было обнаружено, что соединения, обладающие такими структурами, имеют низкое ингибирование CYP450, хорошую активность, и обладают превосходной антипролиферативной активностью в отношении раковых клеток.
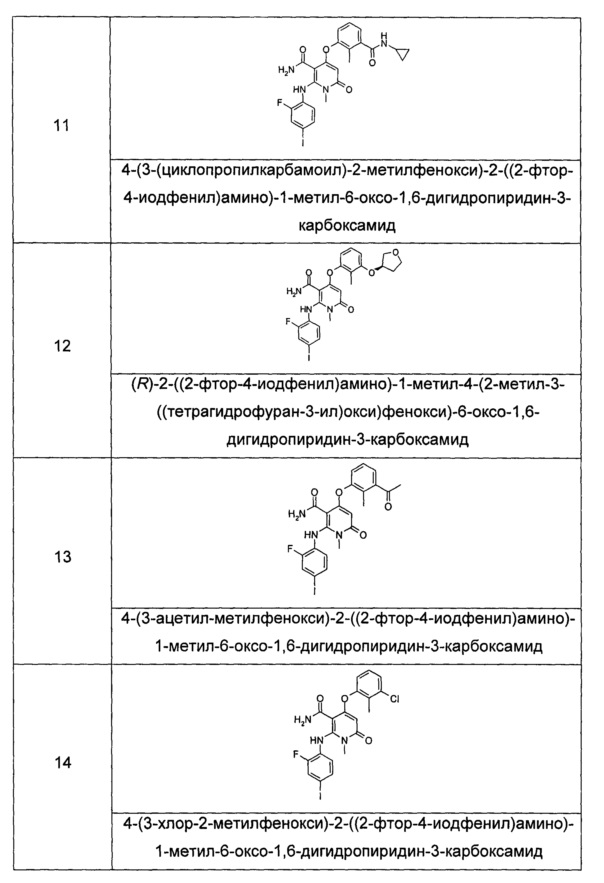
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению формулы (I), его таутомеру, мезомеру, рацемату, энантиомеру или диастереомеру, а также их смеси, и его фармацевтически приемлемой соли:

где:
R1 выбран из группы, состоящей из циклоалкила, гетероциклила, арила и гетероарила, где независимо друг от друга каждый из циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, циано-, нитро-, алкила, галогеналкила, гидроксиалкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, -OR7, -C(O)OR7, -OC(O)R7, -O(CH2)nC(O)OR7, -C(O)R7, -C(O)NHR7, -NHC(O)R7, -NHC(O)OR7, -NHS(O)mR7, -NR8R9, -OC(O)NR8R9 и -C(O)NR8R9;
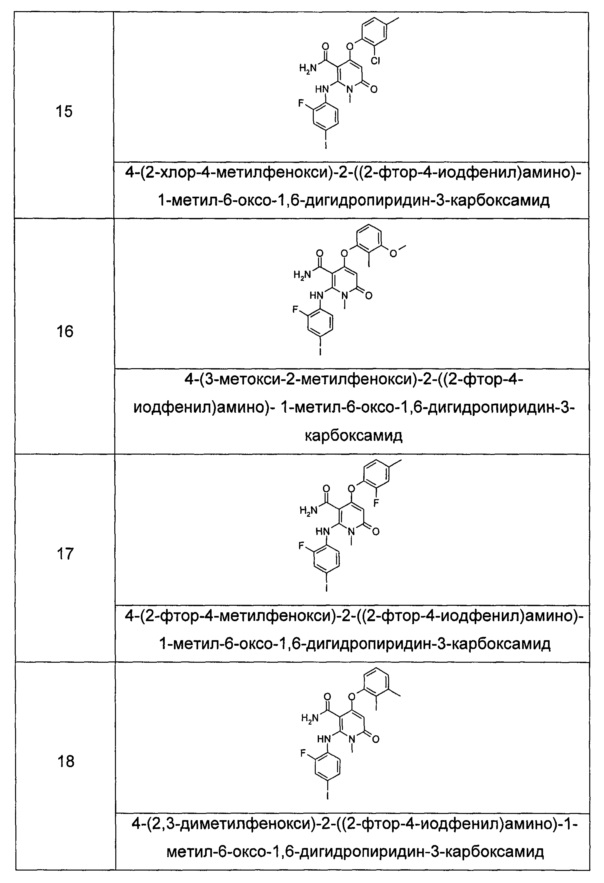
независимо друг от друга каждый из R2 и R3 выбран из группы, состоящей из водорода и алкила, где алкил необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, циано-, нитро-, алкенила, алкинила, гетероциклила, арила, гетероарила, -OR7, -C(O)OR7, -OC(O)R7, -O(CH2)nC(O)OR7, -C(O)R7, -NHC(O)R7, -NHC(O)OR7, -NHS(O)mR7 -NR8R9, -OC(O)NR8R9 и -C(O)NR8R9;
R4 выбран из группы, состоящей из арила и гетероарила, где независимо друг от друга каждый из арила и гетероарила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, циано-, гидрокси-, нитро-, алкила, галогеналкила, гидроксиалкила, алкенила, алкинила, гетероциклила, арила, гетероарила, -OR7, -C(O)OR7, -OC(O)R7, -O(CH2)nC(O)OR7, -C(O)R7, -NHC(O)R7, -NHC(O)OR7, -NHS(O)mR7, -NR8R9, -OC(O)NR8R9 и -C(O)NR8R9;
R5 выбран из группы, состоящей из водорода, алкила, алкенила и алкинила, где независимо друг от друга каждый из алкила, алкенила и алкинила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, гидрокси-, алкокси-, циано- и галогеналкила;
R6 выбран из группы, состоящей из водорода, галогена и алкила, где алкил необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, гидрокси-, циано-, нитро-, алкокси-, циклоалкила, гетероциклила, арила и гетероарила;
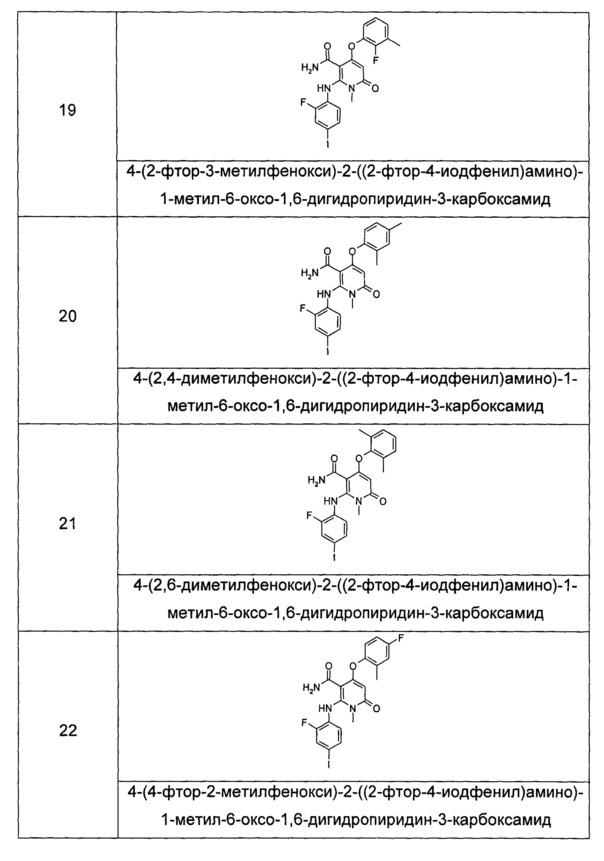
R7 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где независимо друг от друга каждый из алкила, циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из алкила, галогена, гидрокси-, алкокси-, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и алкоксикарбонила;
независимо друг от друга каждый из R8 и R9 выбраны из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где независимо друг от друга каждый из алкила, циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из алкила, галогена, гидрокси-, алкокси-, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и алкоксикарбонила;
альтернативно, R8 и R9 вместе с атомом азота, к которому они присоединены, образуют гетероциклил, где гетероциклил содержит один или несколько гетероатомов, выбранных из группы, состоящей из N, О или S(O)m, и гетероциклил необязательно дополнительно замещен одной или несколькими группами, выбранными из группы, состоящей из алкила, галогена, гидрокси-, алкокси-, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и алкоксикарбонила;
m равно 0, 1 или 2; и
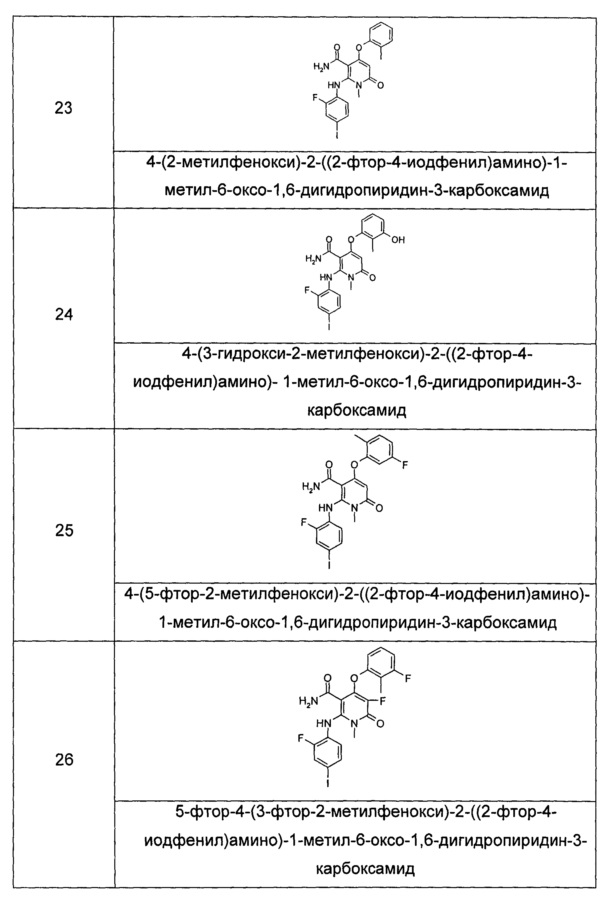
n равно 0, 1 или 2.
В предпочтительном воплощении настоящего изобретения, в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере или диастереомере, или их смеси, или его фармацевтически приемлемой соли, где R1 выбран из группы, состоящей из арила и гетероарила, где каждый из арила и гетероарила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, циано-, нитро-, алкила, галогеналкила, гидроксиалкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, -OR7, -C(O)OR7, -OC(O)R7, O(CH2)nC(O)OR7, -C(O)R7, -C(O)NHR7, -NHC(O)R7, -NHC(O)OR7, -NHS(O)mR7, -NR8R9, -OC(O)NR8R9 и -C(O)NR8R9, и R7, R8, R9, m и n являются такими, как определено в формуле (I).
В другом предпочтительном воплощении настоящего изобретения, в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере или диастереомере, или их смеси, или его фармацевтически приемлемой соли, где R1 выбран из группы, состоящей из фенила и пиридила, где каждый из фенила и пиридила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из алкила, галогена, галогеналкила, -OR7, -C(O)NHR7, -NHC(O)R7, -NHC(O)OR7 и -NHS(O)mR7, и R7 и m являются такими, как определено в формуле (I).
В другом предпочтительном воплощении настоящего изобретения, в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере или диастереомере, или их смеси, или его фармацевтически приемлемой соли, где R2 представляет собой водород, R3 выбран из группы, состоящей из водорода и алкила, где алкил необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, циано-, нитро-, алкила, алкенила, алкинила, гетероциклила, арила, гетероарила, -OR7, -C(O)OR7, -OC(O)R7, -O(CH2)nC(O)OR7, -C(O)R7, -NHC(O)R7, -NHC(O)OR7, -NHS(O)mR7, -NR8R9, -OC(O)NR8R9 и -C(O)NR8R9, и R7, R8, R9, m и n являются такими, как определено в формуле (I).
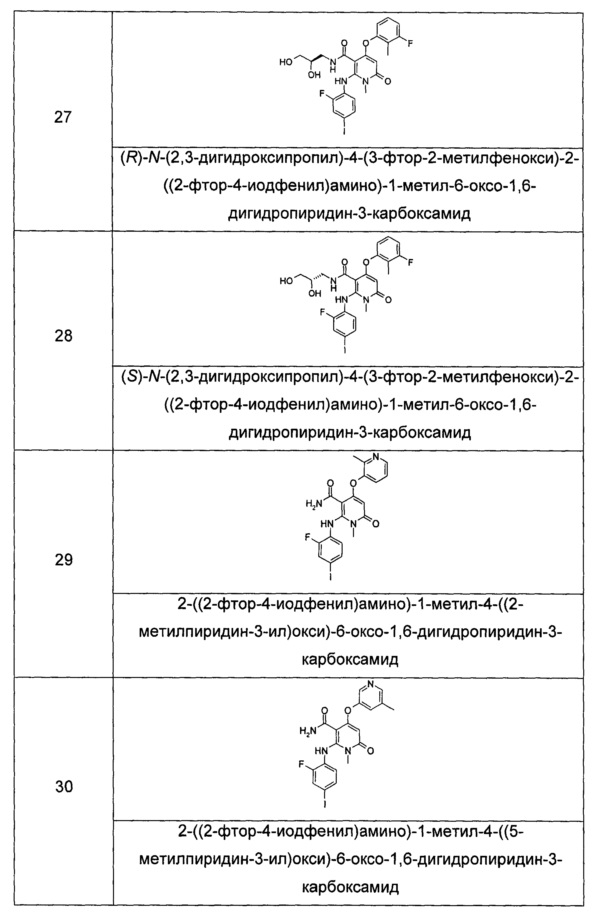
В еще одном предпочтительном воплощении настоящего изобретения, в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере или диастереомере, или их смеси, или его фармацевтически приемлемой соли, где R4 представляет собой арил, где арил необязательно замещен одним или несколькими атомами галогена.
В другом предпочтительном воплощении настоящего изобретения, в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере или диастереомере, или их смеси, или его фармацевтически приемлемой соли, где R5 представляет собой алкил, где алкил необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, гидрокси-, алкокси-, циано- и галогеналкила.
В другом предпочтительном воплощении настоящего изобретения, в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере или диастереомере, или их смеси, или его фармацевтически приемлемой соли, где R6 выбран из группы, состоящей из водорода или галогена.
В другом предпочтительном варианте осуществления настоящего изобретения, соединение формулы (I) или его таутомер, изомер, рацемат, энантиомер или диастереомер, или их смесь, или его фармацевтически приемлемая соль, является соединением формулы (II), или его таутомером, изомером, рацематом, энантиомером или диастереомером, или их смесью, или его фармацевтически приемлемой солью:
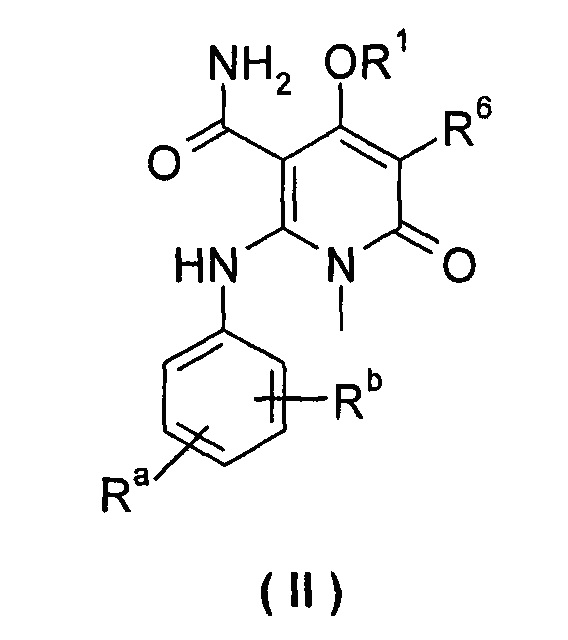
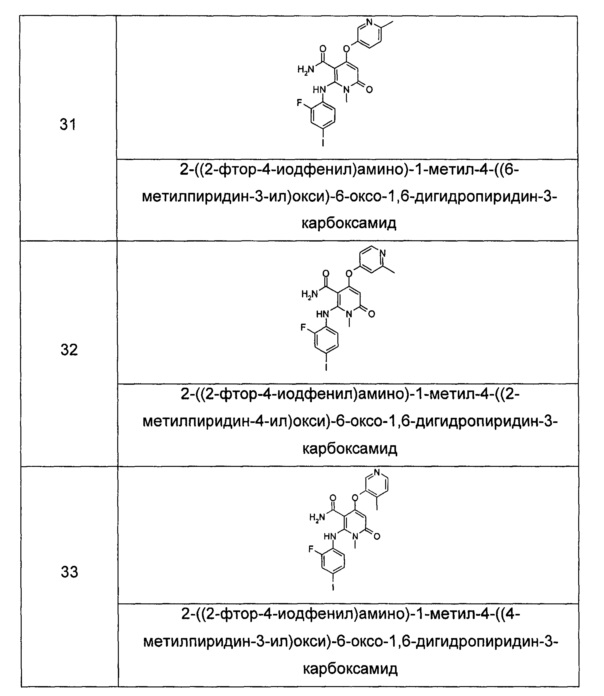
каждый из Ra и Rb выбран из группы, состоящей из водорода, галогена, алкила или галогеналкила;
R1 выбран из группы, состоящей из фенила и пиридинила, где каждый из фенила и пиридила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из алкила, галогена, галогеналкила, -OR7, -C(O)NHR7, -NHC(O)R7, -NHC(O)OR7 and -NHS(O)mR7;
R6 выбран из группы, состоящей из водорода, галогена и алкила, где алкил необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, гидрокси-, циано-, нитро-, алкокси-, циклоалкила, гетероциклила, арила и гетероарила; и
R7 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из алкила, циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из алкила, галогена, гидрокси-, алкокси-, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и алкоксикарбонила.
В другом предпочтительном воплощении настоящего изобретения, в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере или диастереомере, или их смеси, или его фармацевтически приемлемой соли, где R7 выбран из группы, состоящей из водорода, алкила, циклоалкила и гетероциклила, где алкил необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, гидрокси- и алкокси-.
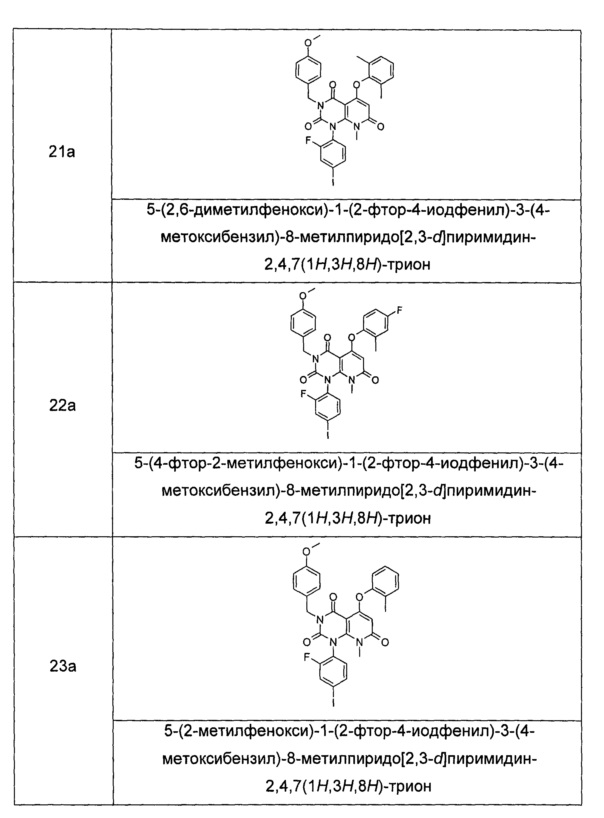
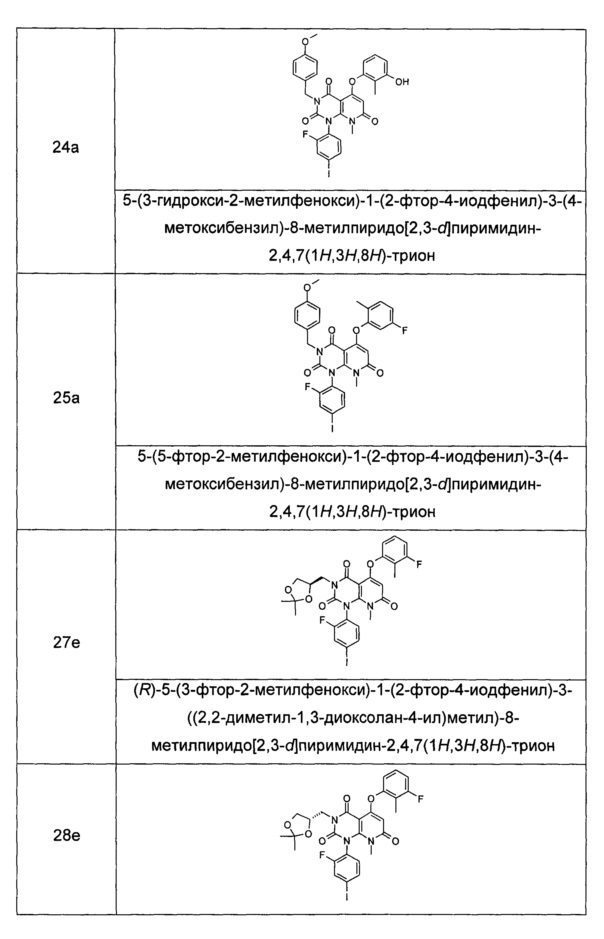
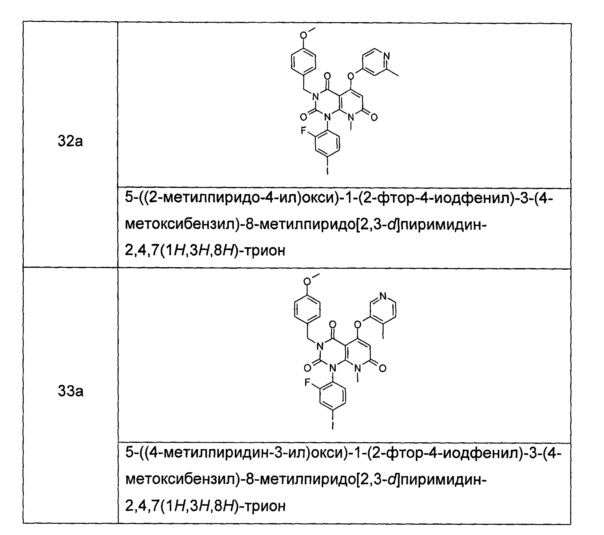
Типичные соединения по настоящему изобретению включают следующие, но не ограничиваются ими:
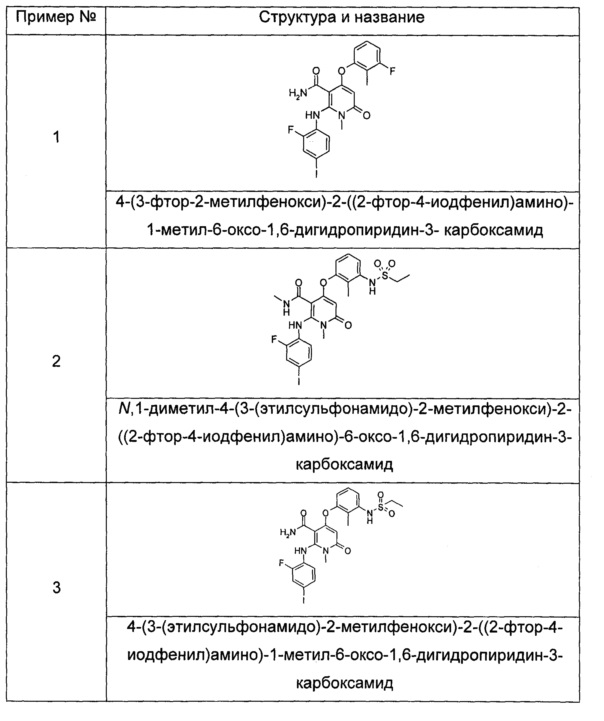
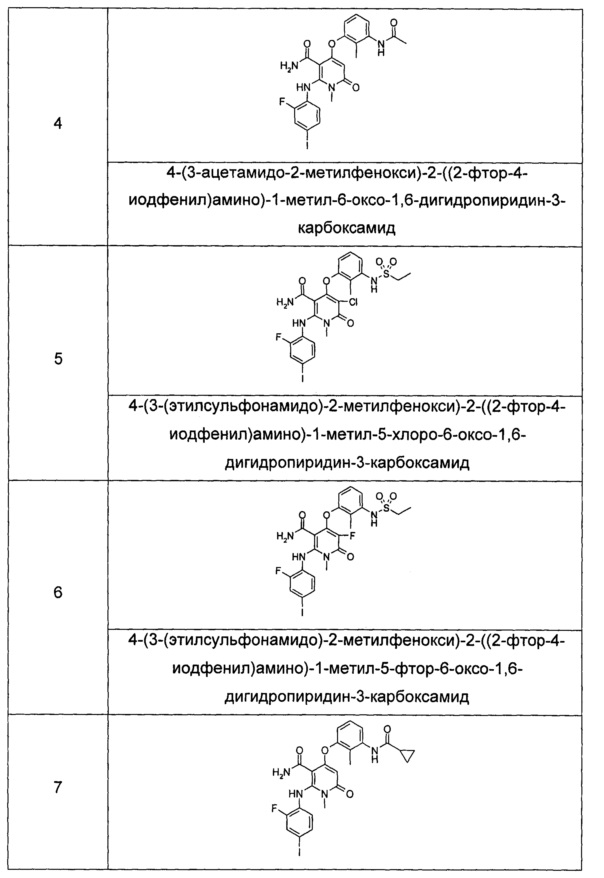
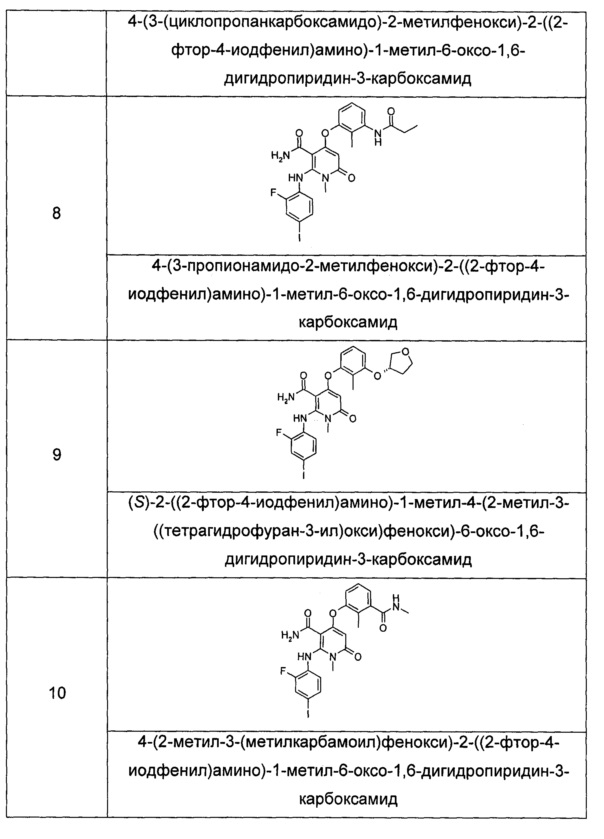
или их таутомером, мезомером, рацематом, энантиомером или диастереоизомером, или их смесью, или их фармацевтически приемлемой солью.
В другом аспекте настоящее изобретение относится к соединению формулы (IA) или его таутомеру, мезомеру, рацемату, энантиомеру или диастереомеру, или его фармацевтически приемлемой соли, которые могут быть использованы в качестве промежуточного продукта для получения соединения формулы (I), или его таутомера, изомера, рацемата, энантиомера, или его диастереомера,
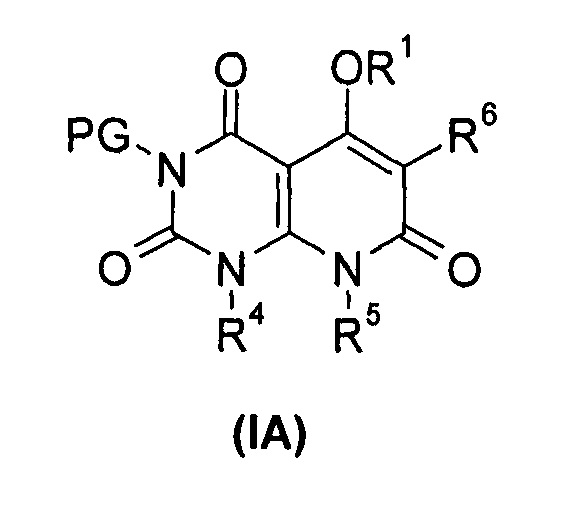
где:
R1, R4 до R6 представляют собой такие, как определено в формуле (I);
PG выбран из группы, состоящей из алкила и амино-защитной группы, где амино-защитной группой предпочтительно является бензил; каждый из алкила и бензила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, циано-, нитро-, алкила, циклоалкила, гетероциклила, гетероарила и -OR7; и
R7 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из алкила, циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из алкила, галогена, гидрокси-, алкокси-, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и алкоксикарбонила.
Типичные соединения формулы (IA) по настоящему изобретению включают следующие, но не ограничиваются ими:
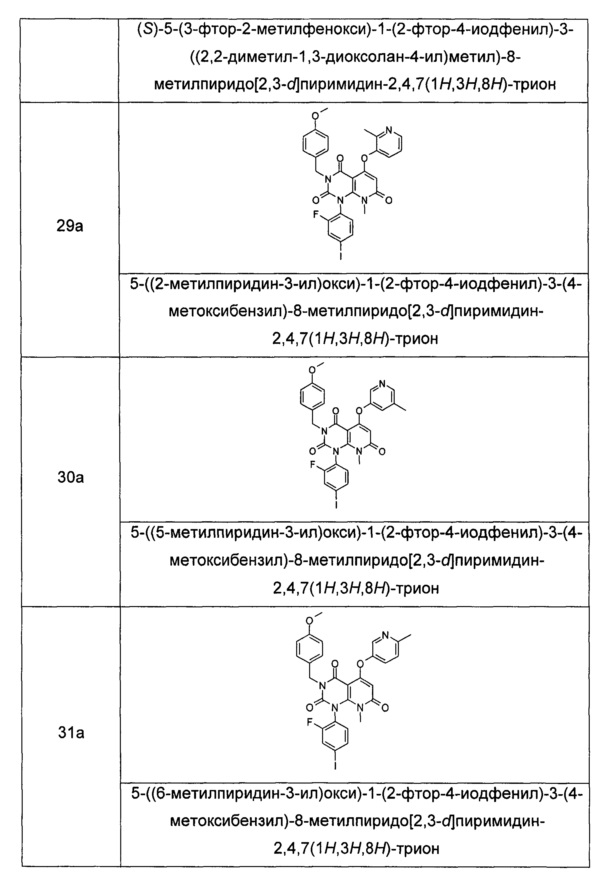
или их таутомер, мезомер, рацемат, энантиомер или диастереоизомер, или их смесь или их фармацевтически приемлемая соль.
В другом аспекте настоящее изобретение относится к способу получения соединения формулы (IA) или его таутомера, мезомера, рацемата, энантиомера или диастереомера, или их смеси, или его фармацевтически приемлемой соли, включающий стадию:

взаимодействия соединения формулы (Ii) с нуклеофилом R1H с получением соединения формулы (IA);
где: R1, R4 до R6 представляют собой такие, как определено в формуле (I);
-OG является уходящей группой, предпочтительно сульфонилокси-;
PG выбран из группы, состоящей из алкила и амино-защитной группы, где амино-защитная группа предпочтительно представляет собой бензил; каждый из алкила и бензила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, циано-, нитро-, алкила, циклоалкила, гетероциклила, арила, гетероарила и - OR7; и
R7 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из алкила, циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из алкила, галогена, гидрокси-, алкокси-, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и алкоксикарбонила.
В вышеуказанном техническом решении щелочные условия обеспечивают с помощью реагента, включающего органическую щелочь и неорганическую щелочь, где органическая щелочь включает, не ограничиваясь, триэтиламин, пиридин, 2,6-лутидин, метоксид натрия, гексаметилдисилазид лития, гексаметилдисилазид натрия, n-бутиллитий, трет-бутоксид калия и бромид тетрабутиламмония; и неорганическая щелочь включает, не ограничиваясь, гидрид натрия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия, карбонат цезия, гидроксид лития, гидроксид натрия и гидроксид калия; щелочной реагент предпочтительно представляет собой неорганическую щелочь, более предпочтительно гидрид натрия или карбонат цезия.
В другом аспекте настоящее изобретение относится к способу получения соединения формулы (I), или его таутомера, мезомера, рацемата, энантиомера или диастереомера, или их смеси, или его фармацевтически приемлемой соли, включающему стадию:
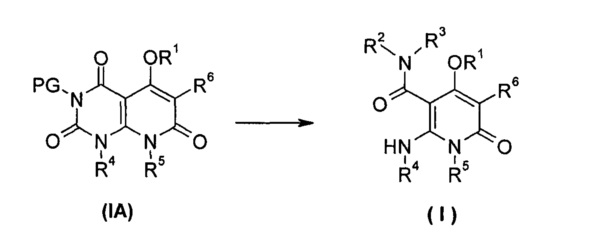
раскрытия кольца соединения формулы (IA) в щелочных условиях, необязательного удаления амино-защитной группы PG, с получением соединения формулы (I);
где:
R1-R6 являются такими, как определено в формуле (I);
PG выбран из группы, состоящей из алкила и амино-защитной группы, где амино-защитная группа предпочтительно представляет собой бензил; каждый из алкила и бензила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, циано-, нитро-, алкила, циклоалкила, гетероциклила, арила, гетероарила и -OR7; и
R7 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из алкила, циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из алкила, галогена, гидрокси-, алкокси-, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и алкоксикарбонила.
В вышеуказанном техническом решении щелочные условия обеспечивают с помощью реагента, включющего органическую щелочь и неорганическую щелочь, где указанная органическая щелочь включает, не ограничиваясь, триэтиламин, пиридин, 2,6-лутидин, метоксид натрия, гексаметилдисилазид лития, гексаметилдисилазид натрия, n-бутиллитий, трет-бутоксид калия и бромид тетрабутиламмония; и неорганическая щелочь включает, не ограничиваясь, гидрид натрия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия, карбонат цезия, гидроксид лития, гидроксид натрия и гидроксид калия; щелочной реагент в реакции с раскрытием колца в способе по настоящему изобретению предпочтительно представляет собой неорганическую щелочь, более предпочтительно гидроксид лития, гидроксид натрия или метилат натрия.
Настоящее изобретение также относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера или диастереомера, или их смеси, или его фармацевтически приемлемой соли, и фармацевтически приемлемый носитель, разбавитель или наполнитель.
Настоящее изобретение также относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера или диастереомера или их смеси, или его фармацевтически приемлемой соли, или содержащей его фармацевтической композиции, для получения лекарственного средства для ингибирования МЕК.
Настоящее изобретение также относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера или диастереомера или их смеси, или их фармацевтически приемлемой соли, или содержащей его фармацевтической композиции, для получения лекарственного средства для лечения воспалительного расстройства, аутоиммунного заболевания, сердечно-сосудистого расстройства, пролиферативного заболевания или ноцицептивного расстройства, где пролиферативное заболевание может представлять собой рак (как определено ниже).
Настоящее изобретение также относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера или диастереомера или их смеси, или его фармацевтически приемлемой соли, или содержащей его фармацевтической композиции, для получения лекарственного средства для лечения рака, где рак выбран из группы, состоящей из меланомы, опухоли головного мозга (глиомы, включая астроцитому и олигодендроглиому и т.д.), рака пищевода, рака желудка, рака печени, рака поджелудочной железы, колоректального рака (рака толстой кишки, рака прямой кишки, и т.д.), рака легкого (немелкоклеточного рака легкого, мелкоклеточного рака легкого, первичного или метастатического плоскоклеточного рака и т.д.), рака почки, рака молочной железы, рака яичников, рака предстательной железы, рака кожи, нейробластомы, саркомы, остеохондромы, остеомы, остеосаркомы, семиномы, рака яичка, рака матки (рака шейки матки, рака эндометрия, и т.д.), рака головы и шеи (рака костей верхней челюсти, рака гортани, рака носоглотки, рака языка, рака полости рта и т.д.), множественной миеломы, злокачественной лимфомы (ретикулярно-клеточной саркомы, лимфосаркомы, лимфомы Ходжкина и т.д.), истинной полицитемии, лейкемии (острого миелоидного лейкоза, хронического миелоидного лейкоза, острого лимфобластного лейкоза, хронического лимфоцитарного лейкоза и т.д.), рака щитовидной железы, рака мочеточника, рака мочевого пузыря, рака желчного пузыря, холангиокарциномы, хориокарциномы и педиатрических опухолей (саркомы Юинга, саркомы Вильмса, рабдомиосаркомы, ангиосаркомы, фетального рака яичка, нейробластомы, ретинобластомы, гепатобластомы, нефробластомы и т.д.).
Настоящее изобретение также относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера или диастереомера или их смеси, или его фармацевтически приемлемой соли, или содержащей его фармацевтической композиции, для получения лекарственного средства для лечения рака, где рак предпочтительно представляет собой колоректальный рак или рак легких.
Настоящее изобретение также относится к способу ингибирования активности МЕК, включающему стадию введения субъекту, нуждающемуся в таком введении, терапевтически эффективного количества соединения формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или его фармацевтически приемлемой соли, или содержащей его фармацевтической композиции.
Дополнительно, настоящее изобретение относится к способу лечения воспалительного расстройства, аутоиммунного заболевания, сердечнососудистого расстройства, пролиферативного заболевания или ноцицептивного расстройства, включающему стадию введения субъекту, нуждающемуся в таком введении, терапевтически эффективного количества соединения формулы (I), или его таутомера, мезомера, рацемата, энантиомера, или диастереомера, или их смеси, или его фармацевтически приемлемой соли, или содержащей его фармацевтической композиции, где пролиферативное расстройство может представлять собой рак (как определено ниже).
Настоящее изобретение дополнительно относится к способу лечения рака, включающему стадию введения субъекту, нуждающемуся в таком введении, терапевтически эффективного количества соединения формулы (I), или его таутомера, мезомера, рацемата, энантиомера или диастереомера, или их смеси, или его фармацевтически приемлемой соли, или содержащей его фармацевтической композиции, где рак выбран из группы, состоящей из меланомы, опухоли головного мозга (глиомы, включая астроцитому и олигодендроглиому, и т.д.), рака пищевода, рака желудка, рака печени, рака поджелудочной железы, колоректального рака (рака толстой кишки, рака прямой кишки, и т.д.), рака легкого (немелкоклеточного рака легкого, мелкоклеточного рака легкого, первичного или метастатического плоскоклеточного рака и т.д.), рака почки, рака молочной железы, рака яичников, рака предстательной железы, рака кожи, нейробластомы, саркомы, остеохондромы, остеомы, остеосаркомы, семиномы, рака яичка, рака матки (рака шейки матки, рака эндометрия, и т.д.), рака головы и шеи (рака костей верхней челюсти, рака гортани, рака носоглотки, рака языка, рака полости рта и т.д.), множественной миеломы, злокачественной лимфомы (ретикулярно-клеточной саркомы, лимфосаркомы, лимфомы Ходжкина и т.д.), истинной полицитемии, лейкемии (острого миелоидного лейкоза, хронического миелоидного лейкоза, острого лимфобластного лейкоза, хронического лимфоцитарного лейкоза и т.д.), рака щитовидной железы, рака мочеточника, рака мочевого пузыря, рака желчного пузыря, холангиокарциномы, хориокарциномы и педиатрических опухолей (саркомы Юинга, саркомы Вильмса, рабдомиосаркомы, ангиосаркомы, фетального рак яичка, нейробластомы, ретинобластомы, злокачественной опухоли печени, нефробластомы и т.д.).
Настоящее изобретение также относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли, или содержащей его фармацевтической композиции, для применения в качестве лекарственного средства для ингибирования активности МЕК.
Настоящее изобретение также относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру или диастереомеру или их смеси, или его фармацевтически приемлемой соли, или содержащей его фармацевтической композиции, для применения в качестве лекарственного средства для лечения воспалительного расстройства, аутоиммунного заболевания, сердечно-сосудистого расстройства, пролиферативного заболевания или ноцицептивного расстройства, где пролиферативное заболевание может представлять собой рак.
Настоящее изобретение также относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру или их диастереомеру или их смеси, или его фармацевтически приемлемой соли, или содержащей его фармацевтической композиции, для применения в качестве лекарственного средства для лечения рака, где рак выбран из группы, состоящей из меланомы, опухоли головного мозга (глиомы, включая астроцитому и олигодендроглиому и т.д.), рака пищевода, рака желудка, рака печени, рака поджелудочной железы, рака прямой кишки (ракы толстой кишки, колоректальныого рака, и т.д.), рака легкого (немелкоклеточного рака легкого, мелкоклеточного рака легкого, первичного или метастатического плоскоклеточного рака и т.д.), рака почки, рака молочной железы, рака яичников, рака предстательной железы, рака кожи, нейробластомы, саркомы, остеохондромы, остеомы, остеосаркомы, семиномы, рака яичка, рака матки (рака шейки матки, рака эндометрия, и т.д.), рака головы и шеи (рака костей верхней челюсти, рак гортани, рака носоглотки, рака языка, рака полости рта и т.д.), множественной миеломы, злокачественной лимфомы (ретикулярно-клеточной саркомы, лимфосаркомы, лимфомы Ходжкина и т.д.), истинной полицитемии, лейкемии (острого миелоидного лейкоза, хронического миелоидного лейкоза, острого лимфобластного лейкоза, хронического лимфоцитарного лейкоза и т.д.), рака щитовидной железы, рака мочеточника, рака мочевого пузыря, рака желчного пузыря, холангиокарциномы, хориокарциномы и педиатрических опухолей (саркомы Юинга, саркомы Вильмса, рабдомиосаркомы, ангиосаркомы, фетального рака яичка, нейробластомы, ретинобластомы, гепатобластомы, нефробластомы и т.д.).
Фармацевтическая композиция, содержащая активный ингредиент, может находиться в форме, подходящей для перорального введения, например, в форме таблеток, пастилок, таблеток для рассасывания, водных или масляных суспензий, дисперсных порошков или гранул, эмульсий, твердых или мягких капсул, или сиропов, или эликсиров. Композиции, предназначенные для перорального применения, необязательно получают в соответствии с известными способами, и такие композиции могут содержать один или несколько агентов, выбранных из группы, состоящей из подсластителей, ароматизаторов, красителей и консервантов, добавляемых для получения фармацевтически лучших и приятных на вкус препаратов. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, пригодными для изготовления таблеток. Эти наполнители могут представлять собой инертные наполнители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и дезинтегрирующе агенты, такие как микрокристаллическая целлюлоза, кроскармеллоза натрия, кукурузный крахмал или альгиновая кислота; связующие агенты, такие как крахмал, желатин, поливинилпирролидон или аравийская камедь; и лубриканты, такие как стеарат магния, стеариновая кислота или тальк. Таблетки могут быть непокрытыми оболочкой или покрытыми известными способами для маскировки вкуса лекарственного средства или задержки дезинтеграции и абсорбции в желудочно-кишечном тракте, обеспечивая тем самым пролонгированное высвобождение в течение длительного периода. Например, может быть использован водорастворимый маскирующий вкус материал, такой как гидроксипропилметилцеллюлоза или гидроксипропилцеллюлоза, или материал для увеличения времени, такой как этил целлюлоза или бутират ацетатцеллюлозы.
Пероральные композиции также могут быть представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, таким как карбонат кальция, фосфат кальция или каолин, или в виде мягких желатиновых капсул, в которых активный ингредиент смешан с водорастворимым носителем, таким как полиэтиленгликоль, или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом.
Водные суспензии содержат активное вещество с добавкой наполнителей, подходящих для получения водных суспензий. Такими наполнителями являются суспендирующие агенты, такие как натрий-карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон и аравийская камедь; диспергирующие или смачивающие агенты, которые могут представлять собой природные фосфатиды, такие как лецитин, или продукты конденсации алкилен оксида с жирными кислотами, такие как стеарат полиоксиэтилена, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, такими как гептадекаэтиленокси цетанол, или продукты конденсации этилен оксида с неполными сложными эфирами, полученными из жирных кислот и гекситов, такие как моноолеат полиоксиэтиленсорбитана, или продукты конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и ангидридов гексита, такие как моноолеат полиэтиленсорбитана. Водные суспензии могут также содержать один или несколько консервантов, таких как этилпарабен или н-пропилпарабен, один или несколько красителей, один или несколько вкусовых добавок и один или несколько подсластителей, таких как сахароза, сахарин или аспартам.
Масляные суспензии могут быть получены путем суспендирования активного ингредиента в растительном масле, таком как арахисовое масло, оливковое масло, кунжутное масло или кокосовое масло, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, такой как пчелиный воск, твердый парафин или цетиловый спирт. Указанные выше подсластители и вкусовые добавки могут быть добавлены для получения приятного на вкус препарата. Эти композиции могут быть презервированы путем добавления антиоксиданта, такого как бутилированный гидроксианизол или альфа-токоферол.
Дисперсные порошки и гранулы, пригодные для получения водной суспензии путем добавления воды, обеспечивают получение активного ингредиента с добавкой диспергирующего или увлажняющего агента, суспендирующего агента и одного или нескольких консервантов. Примеры подходящих диспергирующих или смачивающих агентов и суспендирующих агентов упомянуты выше. Также могут присутствовать дополнительные наполнители, такие как подсластители, вкусовые добавки и красители. Эти композиции могут быть презервированы путем добавления антиоксиданта, такого как аскорбиновая кислота.
Фармацевтические композиции могут также быть в форме эмульсий «масло в воде». Масляная фаза может представлять собой растительное масло, такое как оливковое масло или арахисовое масло, или минеральное масло, такое как жидкий парафин, или их смеси. Подходящими эмульгаторами могут быть природные фосфатиды, такие как соевый лецитин, а также эфиры или неполные эфиры, полученные из жирных кислот и ангидридов гексита, такие как моноолеат сорбитана, и продукты конденсации неполных эфиров с оксидом этилена, например, моноолеат полиоксиэтиленсорбитана. Эмульсии могут также содержать подсластители, вкусовые добавки, консерванты и антиоксиданты. Сиропы и эликсиры могут быть приготовлены с использованием подсластителей, таких как глицерин, пропиленгликоль, сорбит или сахароза. Такие препараты могут также содержать средство, уменьшающее раздражение, консервант, краситель и антиоксидант.
Фармацевтические композиции могут быть в форме стерильных инъецируемых водных растворов. Среди приемлемых носителей и растворителей, которые могут быть использованы, находятся вода, раствор Рингера и изотонический раствор хлорида натрия. Стерильный инъекционный препарат может также быть стерильной инъецируемой микроэмульсией типа «масло в воде», в которой активный ингредиент растворен в масляной фазе. Например, активный ингредиент может быть сначала растворен в смеси соевого масла и лецитина, затем масляный раствор вводят в смесь воды и глицерина и обрабатывают с образованием микроэмульсии. Инъекционные растворы или микроэмульсии могут быть введены в кровоток индивидуума путем местного введения болюса. Альтернативно, может быть целесообразно введение раствора или микроэмульсии таким образом, чтобы поддерживать постоянную циркулирующую концентрацию соединения по настоящему изобретению. Для того чтобы поддерживать такую постоянную концентрацию, может быть использовано устройство непрерывного внутривенного введения. Примером такого устройства является внутривенный насос Deltec CADD-PLUS™ модель 5400.
Фармацевтические композиции могут быть в форме стерильных инъекционных водных или масляных суспензий для внутримышечного и подкожного введения. Суспензии могут быть приготовлены в соответствии с известными в данной области техники способами, путем использования вышеупомянутых подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный инъекционный препарат может также быть стерильным инъекционным раствором или суспензией в нетоксичном парентерально приемлемом разбавителе или растворителе, например, раствором в 1,3-бутандиоле. Кроме того, стерильное нелетучее масло может быть обычным способом использовано в качестве растворителя или суспендирующей среды. Для этой цели может быть использована любая смесь нелетучего масла для синтеза моно- или диглицеридов. Кроме того, жирные кислоты, такие как олеиновая кислота, могут быть использованы при приготовлении инъекций.
Соединения по настоящему изобретению также могут быть введены в форме суппозиториев для ректального введения. Композиции могут быть получены путем смешивания активного ингредиента с подходящим не раздражающим наполнителем, который является твердым при обычных температурах, но жидким при ректальной температуре и, следовательно, будет плавиться в прямой кишке с высвобождением лекарственного средства. Такие материалы включают масло какао, глицинозированный желатин, гидрированные растительные масла, смеси полиэтиленгликолей различной молекулярной массы и эфиров жирных кислот полиэтиленгликоля.
Специалистам в данной области техники известно, что доза лекарственного препарата зависит от множества факторов, включающих следующие факторы, но не ограничивающимися ими: активность конкретного соединения, возраст пациента, масса пациента, общее состояние здоровья пациента, поведение пациента, диета пациента, время введения, путь введения, скорость выведения, сочетание лекарственных средств и т.д. Кроме того, лучшее лечение, такое как модель лечения, суточная доза соединения формулы (I) или тип его фармацевтически приемлемой соли, может быть установлено с помощью общепринятых программ лечения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, то термины, используемые в описании и формуле изобретения, имеют значения, описанные ниже.
"Алкил" относится к насыщенной алифатической углеводородной группе, включающей группы С1-С20 с неразветвленной цепью и с разветвленной цепью. Предпочтительно, алкильная группа представляет собой алкил, имеющий от 1 до 10 атомов углерода, и более предпочтительно алкил, имеющий от 1 до 6 атомов углерода, еще более предпочтительно алкил, имеющий от 1 до 4 атомов углерода, и наиболее предпочтительно метил. Типичные примеры включают, без ограничения, метил, этил, n-пропил, изопропил, n-бутил, изобутил, трет-бутил, втор-бутил, n-пентил, 1,1-диметил пропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, n-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилфенил, 3-метилфенил, 4-метилфенил, 2,3-диметилбутил, n-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, n-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, n-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, n-децил, 3,3-диэтилгексил, 2,2-диэтилгексил, и их различные изомеры с разветвленными цепями. Более предпочтительно, алкильная группа является низшим алкилом, имеющим от 1 до 6 атомов углерода. Типичные примеры включают, без ограничения, метил, этил, n-пропил, изопропил, n-бутил, изобутил, трет-бутил, втор-бутил, n-пентил, 1,1-диметил пропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилфенил, 3-метилфенил, 4-метилфенил, 2,3-диметилбутил и т.д. Алкильная группа может быть замещенной или незамещенной. При замещении, замещающая группа(ы) может быть замещена в любом доступном месте соединения, и, предпочтительно, замещающая группа(ы) является одной или более группой, независимо выбранной из группы, состоящей из алкила, алкенила, алкинила, алкокси-, алкилсульфо-, алкиламино-, галогена, тиола, гидрокси-, нитро-, циано-, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси-, гетероциклоалкокси-, циклоалкилтио-, гетероциклоалкилтио-, оксогруппы, амино-, галогеналкила, гидроксиалкила, карбоксила, алкоксикарбонила, OR7, -C(O)OR7, -OC(O)R7, -O(CH2)nC(O)OR7, -C(O)R7, -C(O)NHR7, -NHC(O)R7, -NHC(O)OR7, -NHS(O)mR7, -NR8R9, -OC(O)NR8R9 и -C(O)NR8R9
"Алкенил" относится к алкилу, такому как определено выше, имеющему по меньшей мере два атома углерода и по меньшей мере одну углерод-углеродную двойную связь, например, винил, 1-пропенил, 2- пропенил, 1-, 2-, или 3-бутенил и т.д., предпочтительно С2-10 алкенил, более предпочтительно С2-6 алкенил, и наиболее предпочтительно С2-4 алкенил. Алкенильная группа может быть замещенной или незамещенной. При замещении замещающая группа(ы) предпочтительно является одной или более группой, независимо выбранной из группы, состоящей из алкила, алкенила, алкинила, алкокси-, алкилсульфо-, алкиламино-, галогена, тиола, гидрокси-, нитро-, циано-, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси-, гетероциклоалкокси-, циклоалкилтио-, гетероциклоалкилтио-, оксогруппы, амино-, галогеналкила, гидроксиалкила, карбоксила, алкоксикарбонила, -OR7, -C(O)OR7, -OC(O)R7, -O(CH2)nC(O)OR7, -C(O)R7, -C(O)NHR7, -NHC(O)R7, -NHC(O)OR7, -NHS(O)mR7, -NR8R9, -OC(O)NR8R9 и -C(O)NR8R9. "Алкинил" относится к алкилу, такому как определено выше, имеющему по меньшей мере два атома углерода и по меньшей мере одну углерод-углеродную тройную связь, например, этинилу, 1-пропинилу, 2-пропинилу, 1-, 2-, или 3-бутинилу и т.д., предпочтительно С2-10 алкинилу, более предпочтительно С2-6 алкинилу, и наиболее предпочтительно С2-4 алкинилу. Алкинильная группа может быть замещенной или незамещенной. При замещении замещающая группа(ы) предпочтительно является одной или более группами, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкокси-, алкилсульфо-, алкиламино-, галогена, тиола, гидрокси-, нитро-, циано-, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси-, гетероциклоалкокси-, циклоалкилтио-, гетероциклоалкилтио-, оксогруппы, амино-, галогеналкила, гидроксиалкила, карбоксила и алкоксикарбонила.
"Циклоалкил" относится к насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 10 атомов углерода, еще более предпочтительно от 3 до 6 атомов углерода, и наиболее предпочтительно к циклопропилу. Характерные примеры моноциклических циклоалкилов включают, без ограничения, циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.д., предпочтительно циклопропил или циклогексенил. Полициклический циклоалкил включает циклоалкил, имеющий спиральное кольцо, конденсированное кольцо или мостиковое кольцо.
Циклоалкильная группа может быть замещенной или незамещенной. При замещении предпочтительно замещающая группа(ы) является одной или более группами, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкокси-, алкилсульфо-, алкиламино-, галогена, тиола, гидрокси-, нитро-, циано-, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси-, гетероциклоалкокси-, циклоалкилтио-, гетероциклоалкилтио-, оксогруппы, амино-, галогеналкила, гидроксиалкила, карбоксила, алкоксикарбонила, OR7, -C(O)OR7, -OC(O)R7, -O(CH2)nC(O)OR7, -C(O)R7, -C(O)NHR7, -NHC(O)R7, -NHC(O)OR7, -NHS(O)mR7, -NR8R9, -OC(O)NR8R9 and -C(O)NR8R9.
"Гетероциклил" относится к от 3 до 20-членной насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей один или несколько гетероатомов, выбранных из группы, состоящей из N, О, и S(O)m (где m представляет собой целое число, выбранное из группы, состоящей из 0, 1 и 2) в качестве кольцевых атомов, за исключением -O-O-, -О-S- или -S-S- в кольце, при этом остальные кольцевые атомы являются С. Предпочтительно гетероциклил имеет от 3 до 12 атомов, где от 1 до 4 атомов представляют собой гетероатомы; более предпочтительно от 3 до 10 атомов и наиболее предпочтительно от 5 до 6 атомов. Характерные примеры моноциклических гетероциклов включают, без ограничения, пирролидил, пиперидил, пиперазинил, морфолинил, сульфо-морфолинил, гомопиперазинил, пиранил, тетрагидрофуранил и т.д. Полициклический гетероциклил включает гетероциклил, имеющий спиральное кольцо, конденсированное кольцо или мостиковое кольцо. Гетероциклильная группа может быть замещенной или незамещенной. В случае замещения предпочтительно замещающая группа(ы) является одной или более группами, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкокси-, алкилсульфо-, алкиламино-, галогена, тиола, гидрокси-, нитро-, циано-, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси-, гетероциклоалкокси-, циклоалкилтио-, гетероциклоалкилтио-, оксогруппы, амино-, галогеналкила, гидроксиалкила, карбоксила, алкоксикарбонила, OR7, -C(O)OR7, -OC(O)R7, -O(CH2)nC(O)OR7, -C(O)R7, -C(O)NHR7, -NHC(O)R7, -NHC(O)OR7, -NHS(O)mR7, -NR8R9, -OC(O)NR8R9 и -C(O)NR8R9.
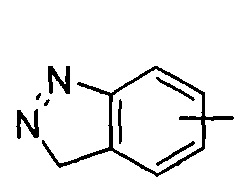
"Арил" относится к от 6 до 14-членному полностью углеродному моноциклическому кольцу или полициклическому конденсированному кольцу ("конденсированная" кольцевая система означает, что каждое кольцо в системе делит смежную пару атомов углерода с другим кольцом в системе), которое имеет полностью сопряженную pi-электронную систему. Предпочтительно арил является от 6 до 10-членным, более предпочтительно фенилом и нафтилом, и наиболее предпочтительно фенилом. Арил может быть конденсирован с кольцом гетероарила, гетероциклила или циклоалкила, где кольцом, связанным с исходной структурой, является арил. Типичные примеры включают, без ограничения, следующие группы:

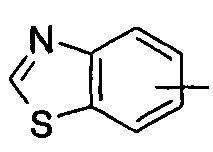
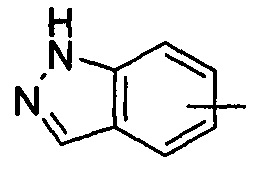
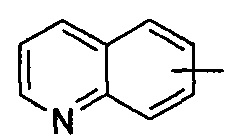
 и
и 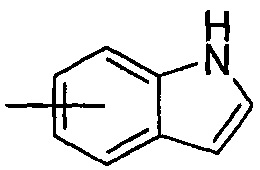 .
.
Арильная группа может быть замещенной или незамещенной. При замещении замещающая группа(ы) предпочтительно являются одной или более группами, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкокси-, алкилсульфо-, алкиламино-, галогена, тиола, гидрокси-, нитро-, циано-, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси-, гетероциклоалкокси-, циклоалкилтио-, гетероциклоалкилтио-, -OR7, -C(O)OR7, -OC(O)R7, -O(CH2)nC(O)OR7, -C(O)R7, -C(O)NHR7, -NHC(O)R7, -NHC(O)OR7, -NHS(O)mR7, -NR8R9, -OC(O)NR8R9 and -C(O)NR8R9.
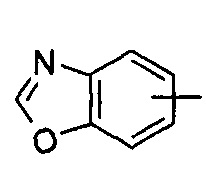
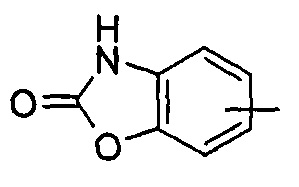
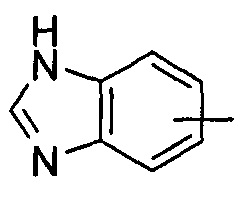
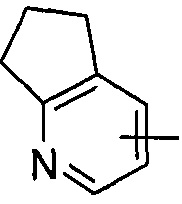
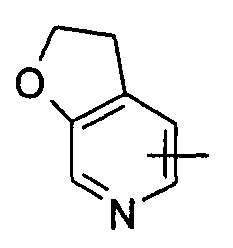
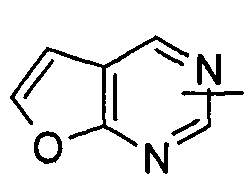
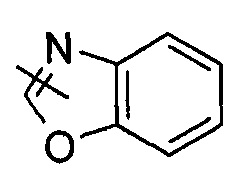
"Гетероарил" относится к от 5 до 14-членному моноциклическому кольцу или полициклическому конденсированному кольцу, имеющему полностью конъюгированную pi-электронную систему и дополнительно содержащему от 1 до 4 гетероатомов, выбранных из группы, состоящей из кислорода, серы или азота. Предпочтительно гетероарил имеет от 5 до 10 членов, более предпочтительно от 5 до 6 членов и наиболее предпочтительно является фурилом, тиенилом, пиридином, пирролом, N-алкилпирролилом, пиримидинилом, пиразинилом, имидазолилом или тетразолилом и т.д. Гетероарил может быть конденсирован с арильным, гетероциклильным или циклоалкильным кольцом, где кольцом, связанным с исходной структурой, является гетероарил. Типичные примеры включают, без ограничения, следующие группы:

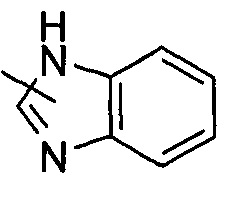

 и
и 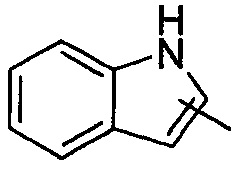 .
.
Гетероарильная группа может быть замещенной или незамещенной. При замещении замещающая группа(ы) предпочтительно является одной или более группами, независимо выбранными из группы, состоящей из алкил,а алкенила, алкинила, алкокси-, алкилсульфо-, алкиламино-, галогена, тиола, гидрокси-, нитро-, циано-, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси-, гетероциклоалкокси-, цикло-алкилтио-, гетероциклоалкилтио-, -OR7, -C(O)OR7, -OC(O)R7, -O(CH2)nC(O)OR7, -C(O)R7, -C(O)NHR7, -NHC(O)R7, -NHC(O)OR7, -NHS(O)mR7, -NR8R9, -OC(O)NR8R9 и -C(O)NR8R9.
"Алкокси" относится как к -О-(алкил), так и к -О-(незамещенный циклоалкил) группе, где алкил и циклоалкил являются такими, как определено выше. Типичные примеры включают, без ограничения, метокси-, этокси-, пропокси-, бутокси-, циклопропилокси-, циклобутилокси-, циклопентилокси-, циклогексилокси- и тому подобное. Алкокси- может быть замещенной или незамещенной. При замещении заместитель предпочтительно является одной или более группами, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкокси-, алкилсульфо-, алкиламино-, галогена, тиола, гидрокси-, нитро-, циано-, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси-, гетероциклоалкокси, циклоалкилтио-, гетероциклоалкилтио, амино-, галогеналкила, гидроксиалкила, карбоксила, алкоксикарбонила, -OR7, -C(O)OR7, -OC(O)R7, -O(CH2)nC(O)OR7, -C(O)R7, -C(O)NHR7, -NHC(O)R7, -NHC(O)OR7, -NHS(O)mR7, -NR8R9, -OC(O)NR8R9 и -C(O)NR8R9.
Талогеналкил" относится к алкильной группе, замещенной одним или несколькими атомами галогена, где котором алкил является таким, как определено выше.
"Гидрокси" относится к группе -ОН.
"Гидроксиалкил" относится к алкильной группе, замещенной гидроксильной группой, где алкил является таким, как определено выше. "Талоген" относится к атомам фтора, хлора, брома или иода.
"Амино" относится к группе -NH2.
"Циано" относится к группе -CN.
"Нитро" относится к группе -NO2.
"Оксогруппа " относится к группе =O.
"Карбоксил" относится к -С(O)ОН группе.
"Алкоксикарбонил" относится к -С(O)O(алкил) или (циклоалкил) группе, где алкил и циклоалкил являются такими, как определено выше.
"Необязательный" или "необязательно" означает, что событие или обстоятельство, описанное далее, может, но не обязательно должно иметь место, и описание включает случаи, в которых происходит или не происходит событие или обстоятельство. Например, "гетероциклическая группа, необязательно замещенная алкилом" означает, что алкильная группа может, но не обязательно должна присутствовать, и описание включает случай гетероциклической группы, замещенной алкилом и гетероциклической группы, не замещенной алкилом.
"Замещенный" относится к одному или более атомов водорода в группе, предпочтительно вплоть до 5, более предпочтительно от 1 до 3 атомов водорода, каждый из которых независимо замещен соответствующим числом заместителей. Само собой разумеется, что заместители существуют в единственной возможной химической позиции. Специалист в данной области может определить, является ли замена возможной или невозможной без приложения чрезмерных усилий путем эксперимента или теории. Например, сочетание амино- или гидроксильной группы, имеющей свободный водород и углеродных атомов, содержащих ненасыщенные связи (например, олефиновые), может быть неустойчивым.
"Фармацевтическая композиция" относится к смеси одного или более соединений, описанных в настоящем изобретении, или их физиологически /фармацевтически приемлемых солей или их пролекарств и других химических компонентов, таких как физиологически/фармацевтически приемлемые носители и наполнители. Целью фармацевтической композиции является облегчение введения соединения в организм, что способствует абсорбции активного ингредиента, тем самым проявляя биологическую активность.
От R7 до R9, m и n являются такими, как определено в приведенной выше формуле (I).
СПОСОБ СИНТЕЗА СОЕДИНЕНИЯ ПО ИЗОБРЕТЕНИЮ
Для того чтобы завершить цель настоящего изобретения, настоящее изобретение применяет следующее техническое решение:
Способ получения соединения формулы (I) по настоящему изобретению, или его таутомера, мезомера, рацемата, энантиомера или диастереомера, или их смеси, или его фармацевтически приемлемой соли, включает следующие стадии:
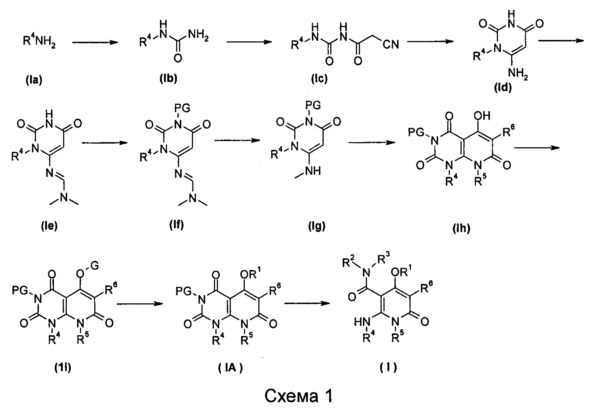
На ледяной бане проводят взаимодействие соединения формулы (Ia) с N,N'-карбонилдиимидазолом и аммиачной водой в щелочных условиях с получением соединения формулы (Ib); взаимодействие соединения формулы (Ib) с 2-циануксусной кислотой в присутствии метансульфонилхлорида с получением соединения формулы (Ic); циклизацию соединения формулы (Ic) в щелочных условиях с получением соединения формулы (Id); взаимодействие соединения формулы (Id) с ацеталем для получения соединения формулы (Ie); взаимодействие соединения формулы (Ie) с аминозащитным реагентом с получением соединения формулы (If); восстановление соединения формулы (If) в присутствии борогидрида натрия с получением соединения формулы (Ig); нагревание соединения формулы (Ig) и диэтил малоната через циклизацию с получением соединения формулы (1h); взаимодействие соединения формулы (1h) с гидроксизащитным реагентом с получением соединения формулы (1i); взаимодействие соединения формулы (1i), с нуклеофилом R1H с получением соединения формулы (IA); раскрытие кольца соединения формулы (IA) в щелочных условиях, необязательно удаление аминозащитной группы PG (защитная группа, от англ. protective group) с получением соединения формулы (I);
где от R1 до R6 являются такими, как определено в формуле (I);
-OG является уходящей группой, предпочтительно сульфонилокси-;
PG выбран из группы, состоящей из алкила и аминозащитной группы, где аминозащитная группа предпочтительно представляет собой бензил; каждый из алкила и бензила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, циано-, нитро- алкила, гетероциклила, гетероарила и -OR7; и
R7 является таким, как определено в формуле (I).
Способ получения соединения формулы (II) по изобретению, или его таутомера, мезомера, рацемата, энантиомера или диастереомера или их смеси, или его фармацевтически приемлемой соли, включает следующие стадии:
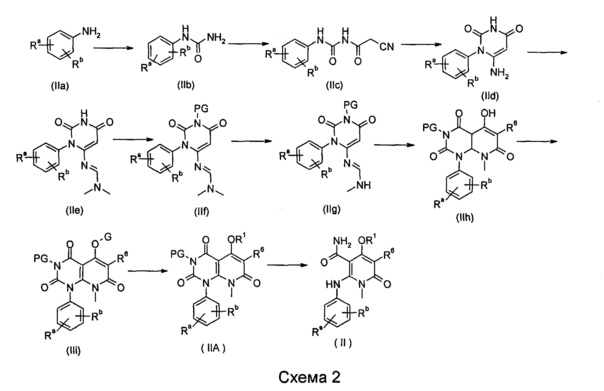
На ледяной бане проводят взаимодействие соединения формулы (IIa), с N,N'-карбонилдиимидазолом в щелочных условиях с получением соединения формулы (lib); взаимодействие соединения формулы (IIb) с 2-циануксусной кислотой в присутствии метансульфонилхлорида а получением соединения формулы (IIc); циклизация соединения формулы (IIc) в щелочных условиях с получением соединения формулы (IId); взаимодействие соединения формулы (IId) с ацеталем с получением соединения формулы (IIe); взаимодействие соединения формулы (IIe) с аминозащитным реагентом с получением соединения формулы (IIf); восстановление соединения формулы (If) в присутствии борогидрида натрия с получением соединения формулы (IIg); нагревание соединения формулы (IIg) и диэтилмалоната через циклизацию с получением соединения формулы (IIh); взаимодействие соединения формулы (IIh) с гидроксизащитным реагентом с получением соединения формулы (IIi); взаимодействие соединения формулы (IIi), с нуклеофилом R1H с получением соединения формулы (НА); раскрытие кольца соединения формулы (НА) в щелочных условиях, необязательно удаление аминозащитной группы PG с получением соединения формулы (II);
где Ra, Rb, R1, R6 являются такими, как определено в формуле (II);
-OG является уходящей группой, предпочтительно сульфонилокси-;
PG выбран из группы, состоящей из алкила и аминозащитной группы, где аминозащитная группа предпочтительно представляет собой бензил; каждый из алкила и бензила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, циано-, нитро-, алкила, гетероциклила, гетероарила и -OR7; и
R7 является таким, как определено в формуле (I).
В вышеуказанных схемах щелочное состояние обеспечивают с помощью реагента, вклющего органическую щелочь и неорганическую щелочь, где органическая щелочь включает, без ограничения, триэтиламин, пиридин, 2,6-лутидин, метоксид натрия, гексаметилдисилазид лития, гексаметилдисилазид натрия, n-бутиллитий, трет-бутоксид калия и тетрабутилбромид аммония; и неорганическая щелочь включает, без ограничения, гидрид натрия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия, карбонат цезия, гидроксид лития, гидроксид натрия и гидроксид калия; щелочным реагентом в реакции нуклеофильного замещения в способе по настоящему изобретению является предпочтительно неорганическая щелочь, более предпочтительно гидрид натрия или карбонат цезия; щелочным реагентом в реакции раскрытия кольца в способе по настоящему изобретению является предпочтительно неорганическая щелочь, более предпочтительно гидроксид лития или гидрид натрия.
ПРЕДПОЧТИТЕЛЬНЫЕ ВОПЛОЩЕНИЯ
Изобретение будет далее проиллюстрировано со ссылкой на следующие конкретные примеры. Следует понимать, что эти примеры предназначены только для демонстрации изобретения, без ограничения объема изобретения. Экспериментальные методы в следующих примерах, для которых не указано никаких конкретных условий, будут осуществляться в соответствии с традиционными условиями или рекомендуемыми условий для сырья и производителей продукции. Экспериментальные реагенты, для которых не указаны конкретные источники, будут представлять собой обычные реагенты, как правило, приобретенные коммерчески.
Примеры
Структуры соединений были идентифицированы с помощью ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). ЯМР химические сдвиги (6) были приведены в 10-6 (частей на миллион). ЯМР определяли с помощью прибора Bruker AVANCE-400. Растворители представляли собой дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD) с тетраметилсиланом (TMS) в качестве внутреннего стандарта.
МС определяли с помощью масс-спектрометра FINNIGAN LCQAd (ESI) (производитель: Thermo, тип: Finnigan LCQ advantage MAX).
Высокоэффективную жидкостную хроматографию (ВЭЖХ) проводили на спектрометре Agilent 1200 DAD для жидкостной хроматографии высокого давления (хроматографическая колонка Sunfire С18 150×4,6 мм) и спектрометре Waters 2695-2996 для жидкостной хроматографии высокого давления (хроматографическая KonoHKaGimini С18 150×4,6 мм),
Среднюю скорость ингибирования киназы и IC50 определяли с помощью Novostar ELIASA (BMG Co., Германия).
Для тонкослойной хроматографии на силикагеле (ТСХ) использовали пластины силикагеля Yantai Huanghai HSGF254 или Qingdao GF254. Размер пластин, использованных в ТСХ, составлял от 0,15 мм до 0,2 мм, а размер пластин, используемых при очистке продукта, составлял от 0,4 мм до 0,5 мм.
В колоночной хроматографии обычно использовали силикагель Yantai Huanghai с размером ячеек от 200 до 300 меш в качестве носителя.
Известные исходные материалы по изобретению могут быть получены с помощью традиционных способов синтеза из уровня техники, или могут быть приобретены в ABCR GmbH & Co. KG, Acros Organics, Aldrich Chemical Company, Inc. Accela ChemBio, или Dari Chemical Company, и т.д.
Если не указано иное, следующие реакции проводили в атмосфере азота или аргона.
Термин "в атмосфере аргона" или "в атмосфере азота" означает, что реакционная колба оснащена 1 л баллоном аргона или азота.
Если не указано иное, то раствор, используемый в примерах, относится к водному раствору.
Если не указано иное, то температура реакции в примерах представляла собой комнатную температуру в диапазоне от 20°С до 30°С.
Протекание реакции контролировали с помощью тонкослойной хроматографии (ТСХ), и система проявляющего растворителя включала: А: систему дихлорметана и метанола, В: систему n-гексана и этилацетата, С: систему петролейного эфира и этилацетата, D: ацетон, Отношение объема растворителя подбирали в соответствии с полярностью соединений.
Система элюирования для очистки соединений с помощью колоночной хроматографии и тонкослойной хроматографии включала: А: систему дихлорметана и метанола, В: систему n-гексана и этилацетата, С: систему дихлорметана и ацетона, D: систему этилацетата и дихлорметана. Объем растворителя подбирали в соответствии с полярностью соединений, а иногда также добавляли немного щелочного реагента, такого как триэтиламин или кислотный реагент.
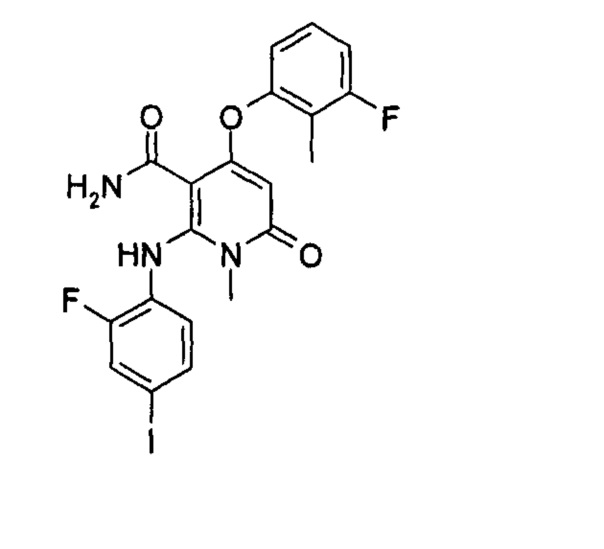
Пример 1
4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
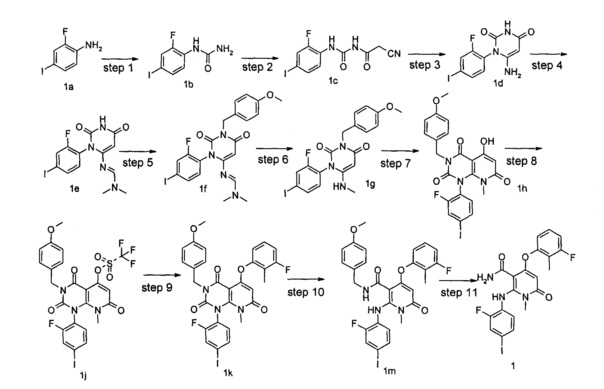
Стадия 1
1-(2-фтор-4-иодфенил)мочевина 2-фтор-4-иоданилин 1а (50,80 г, 214 ммоль) растворяли в 254 мл трихлорметана, с последующим добавлением триэтиламина (60 мл, 429 ммоль). Реакционный раствор охлаждали до 0°С и добавляли N,N'-карбонилдиимидазол (69,50 г, 429 ммоль). После перемешивания в течение 15 мин реакционный раствор нагревали до комнатной температуры и перемешивали в течение 4 ч. Реакционный раствор охлаждали до 0°С, затем добавляли 254 мл аммиачной воды и фильтровали. Осадок на фильтре промывали последовательно водой (50 мл×2), трихлорметаном (20 мл×2) и этилацетатом (50 мл×2) и сушили с получением неочищенного указанного в заголовке соединения 1-(2-фтор-4-иодфенил) мочевина 1b (53 г, белое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI - ионизация распылением в электрическом поле, от англ. electrospray ionization): 281.0 [М+1]
Стадия 2
2-циано-N-((2-фтор-4-иодфенил)карбамоил)ацетамид
Неочищенную 1-(2-фтор-4-йодфенил) мочевину 1b (113 г, 404 ммоль) растворяли в 450 мл N,N-диметилформамида, с последующим добавлением 2-цианоуксусной кислоты (41 г, 488 ммоль). После охлаждения до 0°С к реакционной смеси добавляли метансульфонилхлорид (55,44 г, 484 ммоль), а затем нагревали до комнатной температуры и перемешивали в течение 2 часов. К реакционному раствору добавляли 780 мл смеси воды и изопропанола (V:V=1:2), перемешивали в течение 1 часа и фильтровали. Осадок на фильтре промывали последовательно водой (200 мл×2) и этилацетатом (50 мл) и сушили с получением неочищенного указанного в заголовке соединения 2-циано-N-((2-фтор-4-иодфенил)карбамоил)ацетамид 1 с (143 г, белое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки. МС m/z (ESI): 345.9 [М-1]
Стадия 3
6-амино-1-(2-фтор-4-иодфенил)пиримидин-2,4(1H,3H)-дион
Неочищенный 2-циано-N-((2-фтор-4-йодфенол)карбамоил)ацетамид 1 с (156 г, 430 ммоль) растворяли в 628 мл воды с последующим добавлением 2М раствора гидроксида натрия (22,6 мл, 42 ммоль). Реакционный раствор нагревали до 85°С и перемешивали в течение 1 часа. После охлаждения до 0°С, в реакционный раствор добавляли по каплям 2 М соляную кислоту, чтобы довести значение рН до 3, с последующим добавлением 300 мл изопропанола, и фильтровали. Осадок на фильтре промывали последовательно водой (200 мл×2) и изопропиловым спиртом (100 мл×3) и сушили с получением неочищенного указанного в заголовке соединения 6-амино-1-(2-фтор-4-иодфенил) пиримидин-2,4(1H,3H)-дион 1d (128 г, белое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 348.0 [М+1]
Стадия 4
(Е)-N'-(3-(2-фтор-4-иодфенил)-2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-ил)-N,N-диметилформимидамид
Неочищенный 6-амино-1-(2-фтор-4-йодфенил)пиримидин-2,4(1Н,3H)-дион 1d (128 г, 368,80 ммоль) растворяли в 250 мл N,N-диметилформамида с последующим добавлением N,N-диметилформамида диметилацеталя (124 мл, 935 ммоль), и перемешивали в течение 4,5 часов. В реакционный раствор добавляли 720 мл смеси воды и изопропанола (V:V=5:1), перемешивали в течение 1 часа и фильтровали. Осадок на фильтре последовательно промывали последовательно водой (200 мл×2) и изопропиловым спиртом (50 мл×2) и сушили с получением неочищенного указанного в заголовке соединения (E)-N'-(3-(2-фтор-4-иодфенил)-2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-ил)-N,N-диметилформимидамид 1е (132 г, белое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 403.0 [М+1]
Стадия 5
(Е)-N'-(3-(2-фтор-4-иодфенил)-1-(4-метоксибензил)-2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-ил)-N,N-диметилформимидамид
Неочищенный (Е)-N'-(3-(2-фтор-4-йодфенил)-2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-ил)-N,N-диметилформимидамид 1е (20 г, 50 ммоль) растворяли в 150 мл N,N-диметилформамида с последующим добавлением 1,8-диазабицикло[5.4.0]ундец-7-ена (22,4 мл, 150 ммоль) и 4-метоксибензил хлорида (14,1 мл, 104,30 ммоль). Реакционный раствор нагревали до 75°С и перемешивали в течение 3 часов. После охлаждения до комнатной температуры к реакционному раствору добавляли 675 мл смеси воды и изопропилового спирта (V:V=2:1), перемешивали в течение 1 часа и фильтровали. Осадок на фильтре последовательно промывали водой (200 мл×2) и изопропиловым спиртом (50 мл×2) и сушили с получением неочищенного указанного в заголовке соединения (Е)-N'-(3-(2-фтор-4-иодфенил)-1-(4-метоксибензил)-2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-ил)-N,N-диметилформимидамид 1f (35 г, белое твердое вещество), которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ESI): 523.0 [М+1]
Стадия 6
1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-6-(метиламино)пиримидин-2,4(1Н,3Н)-дион
Борогидрид натрия (3,80 г, 100 ммоль) растворяли в 210 мл смеси этанола и трет-бутанола (V:V=1:2) с последующим добавлением неочищенного (Е)-N'-(3-(2-фтор-4-иодфенил)-1-(4-метоксибензил)-2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-ил)-N,N-диметилформимидамида 1f (35 г, 67 ммоль). Реакционный раствор нагревали до 65°С и перемешивали в течение 1 часа. После охлаждения до 0°С в реакционный раствор добавляли последовательно 175 мл воды и 140 мл 10% лимонной кислоты, и фильтровали. Осадок на фильтре последовательно промывали водой (200 мл×2) и изопропанолом (50 мл×2), и сушили с получением неочищенного указанного в заголовке соединения 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-6-(метиламино)пиримидин-2,4(1Н,3Н)-дион 1g (33 г, белое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 482.0 [М+1]
Стадия 7
1-(2-фтор-4-иодфенил)-5-гидрокси-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион
Неочищенный 1-(2-фтор-4-йод фенил)-3-(4-метокси бензил)-6-(метиламино)пиримидин-2,4(1Н,3Н)-дион 1g (10,80 г, 22,44 ммоль) и диэтил малонат (21,20 г, 157,09 ммоль) растворяли в 100 мл фенилового эфира. Реакционный раствор нагревали до 230°С и перемешивали в течение 1 часа. После охлаждения до комнатной температуры реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии с силикагелем с системой элюции В с получением указанного в заголовке соединения 1-(2-фтор-4-иодфенил)-5-гидрокси-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1H,3Н,8Н)-трион 1h (8,97 г, твердое вещество оранжевого цвета), выход: 72,9%.
МС m/z (ESI): 550.0 [М+1]
Стадия 8
1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат
1-(2-фтор-4-йодфенил)-5-гидрокси-3-(4-метоксибензоил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 1h (8,97 г, 16,33 ммоль) растворяли в 100 мл дихлорметана с последующим добавлением триэтиламина (7,00 г, 65,32 ммоль). После охлаждения до 0°С в реакционный раствор добавляли трифторметансульфоновый ангидрид (9,21 г, 32,66 ммоль), затем нагревали до комнатной температуры и перемешивали в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали с помощью хроматографии на колонке с силикагелем с системой элюции В с получением указанного в заголовке соединения 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]иримидин-5-ил трифторметансульфонат 1j (4,13 г, желтое твердое вещество), выход: 37,1%.
МС m/z (ESI): 682.0 [М+1]
Стадия 9
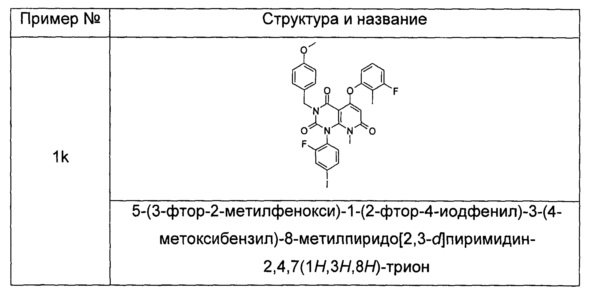
5-(3-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1H,3H,8H)-трион
3-фтор-2-метилфенол (30 мг, 0,24 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (12 мг, 0,30 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (136 мг, 0,20 ммоль), нагревали до 60°С и перемешивали в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении с получением указанного в заголовке соединения 5-(3-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d] пиримидин-2,4,7(1Н,3H,8Н)-трион 1k (131 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
МС m/z (ESI): 658.1 [М+1]
Стадия 10
4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-(3-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 1k (131 мг, 0,20 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=2:1) с последующим добавлением гидроксида лития (168 мг, 4 ммоль). Реакционный раствор нагревали до 40°С и перемешивали в течение 0,5 часа. После охлаждения до комнатной температуры реакционный раствор перемешивали в течение 12 часов и добавляли 50 мл дихлорметана. Органическую фазу промывали насыщенным раствором бикарбоната натрия (25 мл×3), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке неочищенного соединения 4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 1m (126 мг, коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 632.1 [М+1]
Стадия 11
4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(3-фтор-2-метил фенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 1m (126 мг, 0,20 ммоль) растворяли в 5 мл анизола, с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 3,5 ч, добавляли 10 мл воды и 1 мл 1М соляной кислоты и экстрагировали этилацетатом (20 мл×3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, а полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 1 (33 мг, светло-коричневого твердого вещества), выход: 32,3%.
МС m/z (ESI): 512.0 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.78 (s, 1Н), 7.60-7.66 (m, 3Н), 7.34-7.44 (m, 2Н), 7.18 (t, 1Н), 7.10 (d, 1Н), 6.67 (t, 1Н), 5.04 (s, 1Н), 3.15 (s, 3Н), 2.06 (s, 3Н).
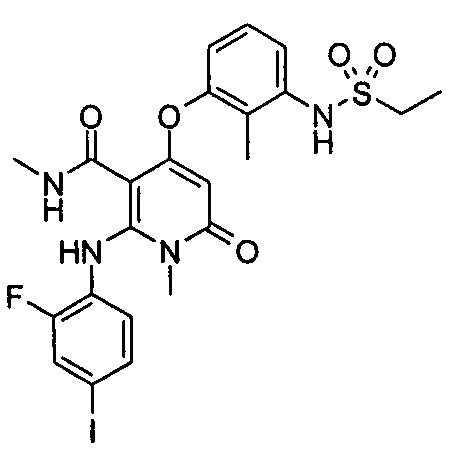
Пример 2
N,1-диметил-4-(3-(этилсульфонамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-6-оксо-1,6-дигидропиридин-3-карбоксамид
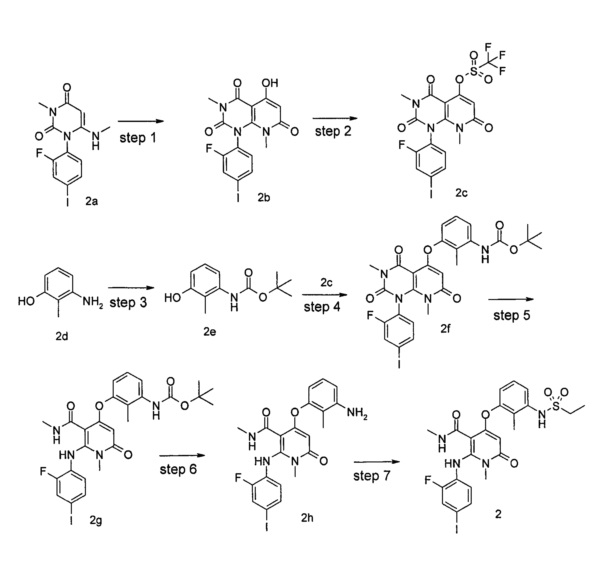
Стадия 1
3-метил-1-(2-фтор-4-иодфенил)-5-гидрокси-8-метилпиридо[2,3-d]пиримидин-2,4,7(7Н,3Н,8Н)-трион
3-метил-1-(2-фтор-4-иодфенил)-6-(метиламино)пиримидин-2,4(1H,3H)-диона 2a (2,70 г, 7,19 ммоль, полученного способом, раскрытым в патентной заявке WO 2005/121142 А1) и диэтиловый эфир малоновой кислоты (8,07 г, 50,38 ммоль) растворяли в 24 мл фенилового эфира. Реакционный раствор нагревали до 230°С и перемешивали в течение 2 часов, а затем концентрировали при пониженном давлении, и полученный остаток очищали с помощью хроматографии на колонке с силикагелем с системой элюции В с получением указанного в заголовке соединения 3-метил-1-(2-фтор-4-иодфенил)-5-гидрокси-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8H)-трион 2b (2,20 г, коричнево-красное твердое вещество), выход: 68,9%.
МС m/z (ESI): 444.1 [М+1]
Стадия 2
3,8-диметил-1-(2-фтор-4-иодфенил)-2,4,7-гтриоксо-1,2,3,4,7,8-гексагидропиридо[2,3-d]пиримидин-5-ил трифторметансульфонат
3-метил-1-(2-фтор-4-йодфенил)-5-гидрокси-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3H,8Н)-трион 2b (2,20 г, 5 ммоль) растворяли в 20 мл дихлорметана с последующим добавлением триэтиламина (2,14 г, 20 ммоль). Реакционный раствор охлаждали до 0°С, добавляли трифторметансульфоновый ангидрид (2,82 г, 10 ммоль), затем нагревали до комнатной температуры и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали с помощью хроматографии на колонке с силикагелем с системой элюции В с получением указанного в заголовке соединения 3,8-диметил-1-(2-фтор-4-иодфенил)-2,4,7-триоксо-1,2,3,4,7,8-гексагидропиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 2с (1,50 г, желтое твердое вещество), выход: 52,1%.
МС m/z (ESI): 576.0 [М+1]
Стадия 3
трет-бутил (3-гидрокси-2-метилфенил)карбамат
3-амино-2-метилфенол 2d (2 г, 16,20 ммоль) растворяли в 300 мл дихлорметана с последующим добавлением ди-трет-бутилдикарбоната (4,25 г, 19,50 ммоль). Реакционный раствор нагревали до 70°С и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали с помощью хроматографии на колонке с силикагелем с системой элюции В, получая указанное в заголовке соединение трет-бутил(3-гидрокси-2-метилфенил)карбамат 2е (3,0 г, белое твердое вещество) выход: 83,1%.
МС m/z (ESI): 222.2 [М-1]
Стадия 4
трет-бутил (3-(3,8-диметил-(1-(2-фтор-4-иодфенил)-2,4,7-триоксо-1,2,3,4,7,8-гексагидропиридо[2,3-d]пиримидин-5-ил)окси)-2-метилфенил)карбамат
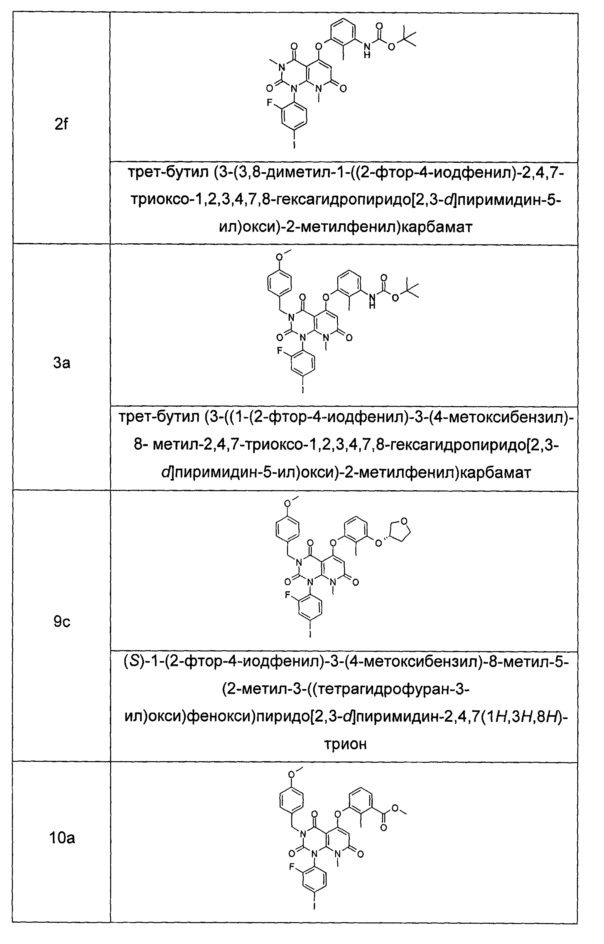
Трет-бутил (3-гидрокси-2-метилфенил)карбамат 2е (120 мг, 0,52 ммоль) растворяли в 10 мл тетрагидрофурана с последующим добавлением гидрида натрия (25 мг, 0,63 ммоль). Реакционный раствор перемешивали в течение 2 часов с последующим добавлением 3,8-диметил-1-(2-фтор-4-иодфенил)-2,4,7-триоксо-1,2,3,4,7,8-гексагидропиридо[2,3-d]пиримидин-5-ил трифторметансульфоната 2 с (300 мг, 0,53 ммоль), а затем нагревали до 70°С и перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении, добавляли 50 мл этилацетата и 20 мл воды. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения трет-бутил(3-(3,8-диметил-(1-(2-фтор-4-иодфенил)-2,4,7-триоксо-1,2,3,4,7,8-гексагидропиридо[2,3-d]пиримидин-5-ил)окси)-2-метилфенил)карбамат 2f (339 мг, коричневая жидкость), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 649.1 [М+1]
Стадия 5
трет-бутил (3-(5-(метилкарбамоил)-(6-((2-фтор-4-иодфенил)амино)-1-метил-2-оксо-1,2-дигидропиридин-4-ил)окси)-2-метилфенил)карбамат
Неочищенный трет-бутил (3-(3,8-метил-(1-(2-фтор-4-иодфенил)2,4,7-триоксо-1,2,3,4,7,8-гексагидропиридо[2,3-d]пиримидин-5-ил)окси)-2-метилфенил)карбамат 2f (339 мг, 0,52 ммоль) растворяли в 10 мл тетрагидрофурана и добавляли 2 мл воды с последующим добавлением гидроксида лития (218 мг, 5,20 ммоль). Реакционный раствор нагревали до 40°С и перемешивали в течение 1,5 часов. Реакционный раствор концентрировали при пониженном давлении, добавляли 50 мл этилацетата, промывали водой (10 мл). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения трет-бутил (3-(5-(метилкарбамоил)-(6-((2-фтор-4-иодфенил) амино)-1-метил-2-оксо-1,2-дигидропиридин-4-ил) окси)-2-метилфенил)карбамат 2g (325 мг, коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z(ESI): 623.0 [М+1]
Стадия 6
N,1-диметил-4-(3-амино-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный трет-бутил (3-(5-(метил карбамоил)-(6-((2-фтор-4-иодфенил)амино)-1-метил-2-оксо-1,2-дигидропиридин-4-ил)окси)-2-метилфенил)карбамата 2g (325 мг, 0,52 ммоль) растворяли в 10 мл дихлорметана с последующим добавлением 3 мл трифторуксусной кислоты и перемешивали в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении, добавляли 100 мл этилацетата. Органическую фазу промывали насыщенным раствором бикарбоната натрия (30 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения N,1-диметил-4-(3-амино-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-6-оксо-1,6-дигидропиридин-3-карбоксамид 2h (253 мг, коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 522.9 [М+1]
Стадия 7
N,1-диметил-4-(3-(этилсульфонамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный N,1-диметил-4-(3-амино-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-6-оксо-1,6-дигидропиридин-3-карбоксамид 2h (175 мг, 0,34 ммоль) растворяли в 10 мл дихлорметана с последующим добавлением 2,5 мл пиридина. После охлаждения до 0°С в реакционный раствор добавляли этансульфонилхлорид (65 мг, 0,50 ммоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 12 часов, затем добавляли 100 мл этилацетата и 30 мл воды. Органическую фазу промывали водой (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения N,1-диметил-4-(3-(этилсульфонамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-6-оксо-1,6-дигидропиридин-3-карбоксамид 2 (85 мг, желтое твердое вещество), выход: 41,4%.
МС m/z (ESI): 615.1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.26 (s, 1Н), 9.23 (s, 1Н), 8.03-8.07 (m, 1Н), 7.59-7.64 (dd, 1Н), 7.40-7.44 (dd, 1Н), 7.25-7.35 (m, 2Н), 7.07-7.10 (dd, 1Н), 6.67-6.72 (t, 1Н), 4.95 (s, 1Н), 3.22 (s, 3Н), 3.10-3.17 (q, 2Н), 2.50 (s, 3Н), 2.11 (s, 3Н), 1.25-1.29 (t, 3Н).
Пример 3
4-(3-(этилсульфонамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
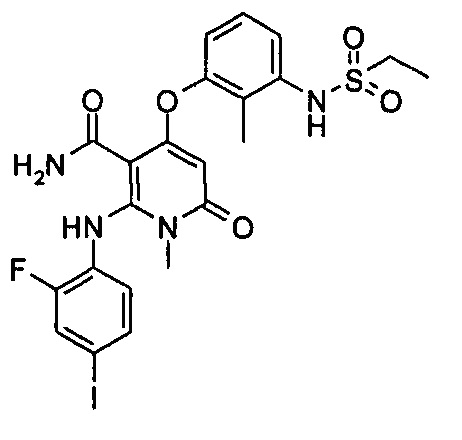
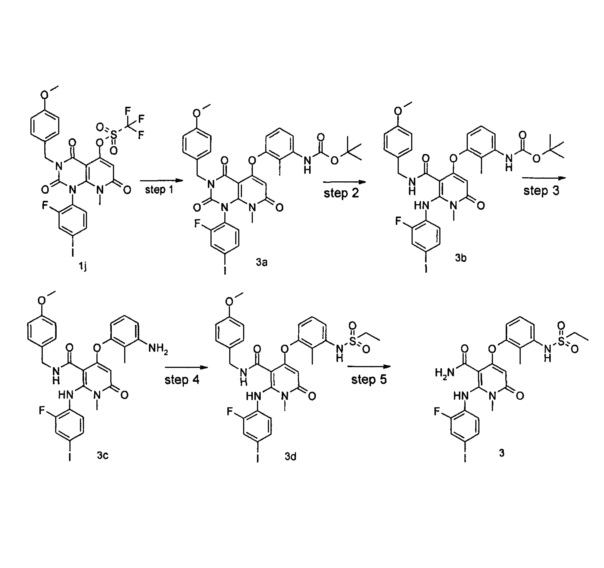
Стадия 1
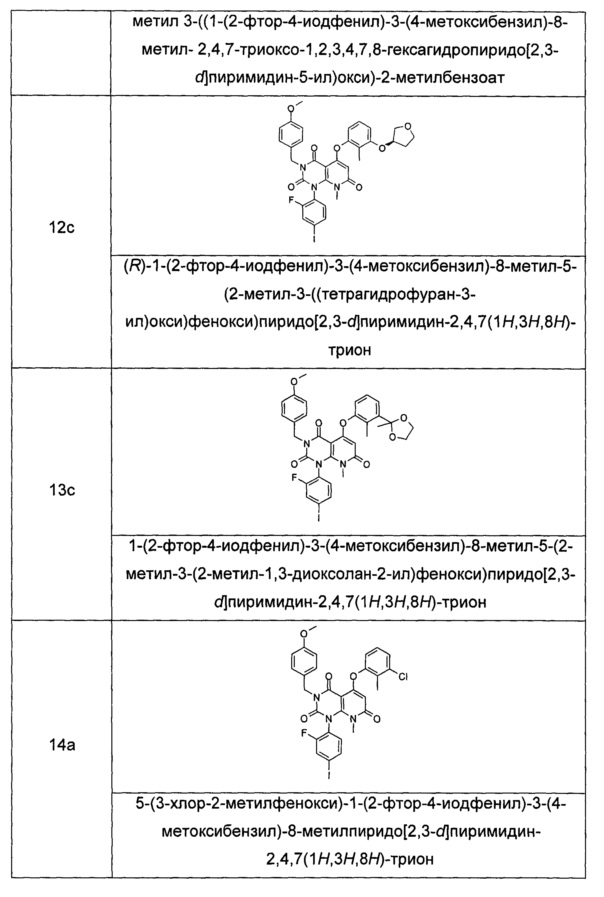
трет-бутил (3-((1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-1,2,3,4,7,8-гексагидропиридо[2,3-d]пиримидин-5-ил)окси)-2-метилфенил)карбамат
Трет-бутил (3-гидрокси-2-метил фенил)карбамат (165 мг, 0,74 ммоль) растворяли в 10 мл тетрагидрофурана с последующим добавлением гидрида натрия (37 мг, 0,93 ммоль). Реакционный раствор перемешивали в течение 2 часов с последующим добавлением 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфоната 1j (420 мг, 0,62 ммоль), затем нагревали до 60°С. После перемешивания в течение 0,5 ч реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения трет-бутил(3-((1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-1,2,3,4,7,8-гексагидропиридо[2,3-d]пиримидин-5-ил)окси)-2-метилфенил)карбамат 3а (465 мг, светло-желтая жидкость), которую использовали непосредственно на следующей стадии без дополнительной очистки.
МС m/z (ESI): 755.1 [М+1]
Стадия 2
трет-бутил (3-((6-((2-фтор-4-иодфенил)амино)-5-((4-метоксибензил)карбамоил)-1-метил-2-оксо-1,2-дигидропиридин-4-ил)окси)-2-метилфенил)карбамат
Неочищенный трет-бутил (3-((1-(2-фтор-4-йодфенил)-3-(4-метоксибензил)-в-метил-2,4,7-триоксо-1,2,3,4,7,8-гексагидропиридо[2,3-d]пиримидин-5-ил)окси-2-метилфенил)карбамат За (465 мг, 0,62 ммоль) растворяли в 12,5 мл смеси тетрагидрофурана и воды (V:V=4:1) с последующим добавлением гидроксида лития (517 мг, 12,32 ммоль) Реакционный раствор подогревали до 40°С и перемешивали в течение 1 ч. После охлаждения до комнатной температуры реакционный раствор перемешивали в течение 12 ч с последующим добавлением 15 мл воды и 50 мл дихлорметана. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения трет-бутил (3-((6-((2-фтор-4-иодфенил)амино)-5-((4-метоксибензил)карбамоил)-1-метил-2-оксо-1,2-дигидропиридин-4-ил)окси)-2-метилфенил)карбамат 3b (449 мг, коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 729.2 [М+1]
Стадия 3
4-(3-амино-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный трет-бутил (3-((6-((2-фтор-4-йодфенил)амино)-5-((4-метоксибензил)карбамоил)-1-метил-2-оксо-1,2-дигидропиридин-4-ил)окси)-2-метилфенил)карбамат 3b (449 мг, 0,62 ммоль) растворяли в 10 мл дихлорметана с последующим добавлением трифторуксусной кислоты (1,40 г, 12,32 ммоль). После перемешивания в течение 2 часов реакционный раствор концентрировали при пониженном давлении, добавляли 10 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Реакционный раствор перемешивали в течение 12 ч и экстрагировали дихлорметаном (30 мл×2). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(3-амино-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 3с (387 мг, светло-коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 629.0 [М+1]
Стадия 4
4-(3-(этилсульфонамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(3-амино-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 3с (190 мг, 0,30 ммоль) растворяли в 10 мл смеси дихлорметана и пиридина (V:V=1:1). После охлаждения до 0°С в реакционный раствор добавляли этансульфонилхлорид (42 мг, 0,33 ммоль), нагревали до комнатной температуры и перемешивали в течение 12 часов. Добавляли 50 мл дихлорметана, органическую фазу промывали водой (20 мл), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(3-(этилсульфонамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 3d (216 мг, коричневато-черное твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 720.9 [М+1]
Стадия 5
4-(3-(этилсульфонамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(3-(этилсульфонамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 3d (200 мг, 0,28 ммоль) растворяли в 10 мл анизола с последующим добавлением хлорида алюминия (185 мг, 1,39 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 1 часа с последующим добавлением 10 мл воды и 1 мл 1М соляной кислоты и экстрагировали этилацетатом (20 мл×3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(3-(этилсульфонамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 3 (35 мг, светло-коричневое твердое вещество), выход: 20,8%.
МС m/z (ESI): 601.0 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.88 (s, 1Н), 9.27 (s, 1Н), 7.58-7.68 (m, 3Н), 7.43 (d, 1Н), 7.27-7.35 (m, 2Н), 7.14 (d, 1Н), 6.67 (t, 1Н), 4.96 (s, 1Н), 3.14 (s, 3Н), 3.13 (q, 2Н), 2.12 (s, 3Н), 1.26 (t, 3Н).
Пример 4
4-(3-ацетамидо-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
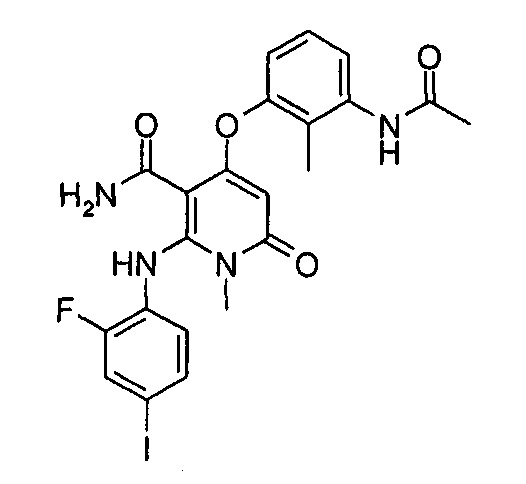
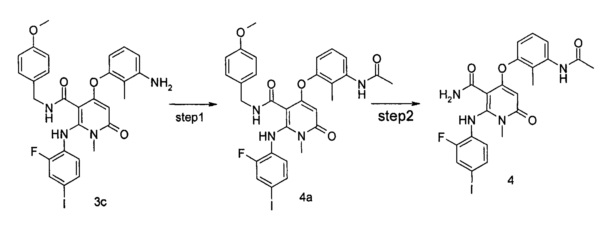
Стадия 1
4-(3-ацетамидо-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(3-амино-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 3с (190 мг, 0,30 ммоль) растворяли в 10 мл смеси дихлорметана и пиридина (V:V=1:1). После охлаждения до 0°С в реакционный раствор добавляли ацетилхлорид (26 мг, 0,33 ммоль), затем нагревали до комнатной температуры и перемешивали в течение 12 ч с последующим добавлением 50 мл дихлорметана. Органическую фазу промывали водой (20 мл), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(3-ацетамидо-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 4а (201 мг, светло-коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 671.1 [М+1]
Стадия 2
4-(3-ацетамидо-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(3-ацетамидо-2-метилфенокси)-2-((2-фтор-4-иодфенил) амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 4а (201 мг, 0,30 ммоль) растворяли в 10 мл анизола с последующим добавлением хлорида алюминия (200 мг, 1,50 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 5 часов, затем концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(3-ацетамидо-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 4 (30 мг, коричневое твердое вещество), выход: 18,2%.
МС m/z(ESI): 551.0 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.89 (s, 1Н), 9.47 (s, 1Н), 7.58-7.68 (m, 3Н), 7.42 (t, 2Н), 7.29 (t, 1Н), 7.06 (d, 1Н), 6.67 (t, 1Н), 4.96 (s, 1Н), 3.14 (s, 3Н), 2.07 (s, 3Н), 2.02 (s, 3Н).
Пример 5
4-(3-(этилсульфонамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-5-хлоро-6-оксо-1,6-дигидропиридин-3-карбоксамид
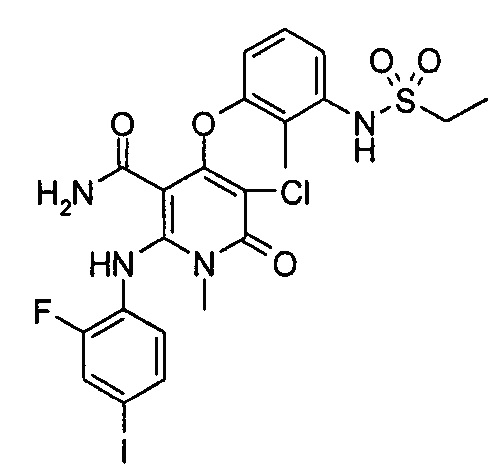
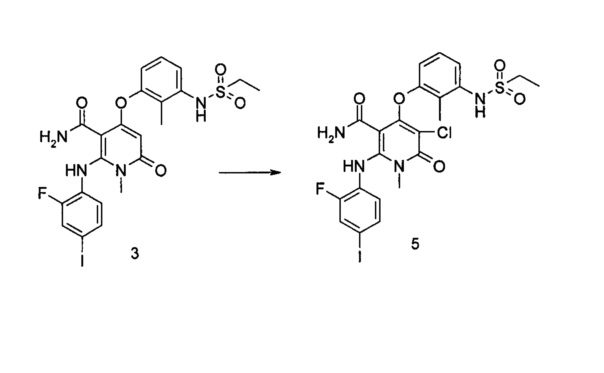
4-(3-(этилсульфонамидо)-2-метилфенокси)-2-((2-фтор-4-йодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 3 (30 мг, 0,05 ммоль) растворяли в 9 мл дихлорметана. После охлаждения до 0°С в реакционный раствор добавляли 3 мл раствора N-хлорсукцинимида (7 мг, 0,05 ммоль) в дихлорметане, затем нагревали до 40°С и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(3-(этилсульфонамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-5-хлор-6-оксо-1,6-дигидропиридин-3-карбоксамид 5 (19 мг, твердое вещество белого цвета), выход: 59,4%.
МС m/z (ESI): 635.0 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.17 (s, 1Н), 8.96 (s, 1Н), 7.60 (dd, 1Н), 7.51 (s, 1Н), 7.36-7.46 (m, 2Н), 6.99-7.09 (m, 2Н), 6.74 (t, 1Н), 6.58 (d, 1Н), 3.36 (s, 3Н), 3.09 (q, 2Н), 2.30 (s, 3Н), 1.26 (t, 3Н).
Пример 6
4-(3-(этилсульфонамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-5-фтор-6-оксо-1,6-дигидропиридин-3-карбоксамид
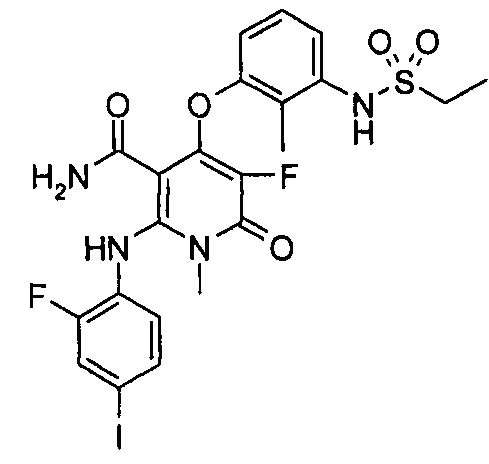
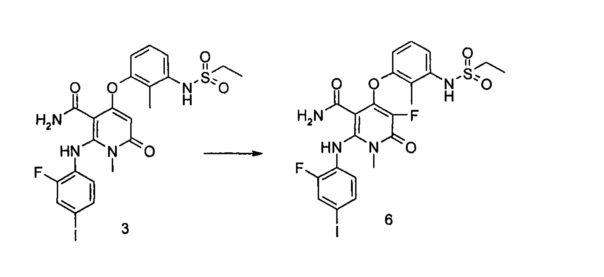
4-(3-(этилсульфонамидо)-2-метилфенокси)-2-((2-фтор-4-йодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 3 (12 мг, 0,02 ммоль) растворяли в 10 мл дихлорметана. После охлаждения до 0°С в реакционный раствор добавляли 3 мл раствора 1-хлорметил-4-фтор-1,4-диазониабицикло[2.2.2]октана бис(тетрафторбората) (7 мг, 0,02 ммоль) в дихлорметане, затем смесь нагревали до комнатной температуры и перемешивали в течение 12 ч. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(3-(этилсульфонамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-5-фтор-6-оксо-1,6-дигидропиридин-3-карбоксамид 6 (6 мг, светло-коричневое твердое вещество), выход: 31,5%.
МС m/z (ESI): 619.0 [М+1]
1H ЯМР (400 МГц, ДМСО-d6): δ 9.20 (s, 1Н), 8.68 (s, 1Н), 7.42-7.66 (m, 3Н), 7.30-7.40 (m, 1Н), 7.00-7.22 (m, 2Н), 6.78-7.88 (m, 1Н), 6.58-6.70 (m, 1Н), 3.33 (s, 3Н), 3.10(q, 2Н), 2.28 (s, 3Н), 1.25 (t, 3Н).
Пример 7
4-(3-(циклопропанкарбоксамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
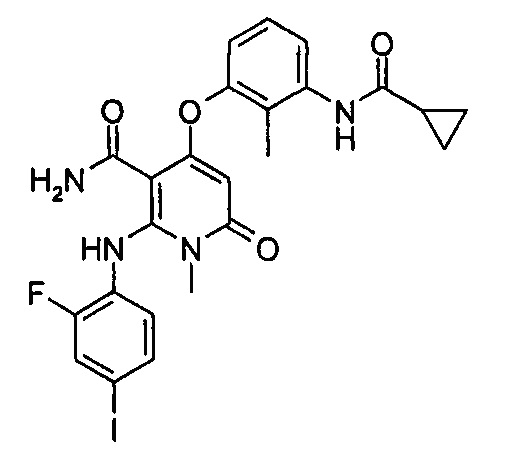
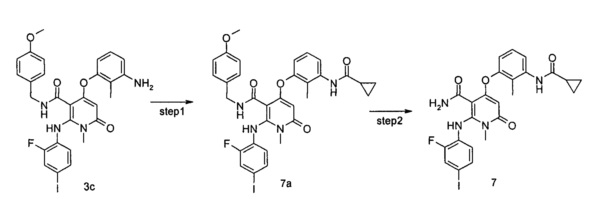
Стадия 1
4-(3-(циклопропанкарбоксамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(3-амино-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 3с (126 мг, 0,20 ммоль) растворяли в 2 мл смеси дихлорметана и пиридина (V:V=1:1). После охлаждения до 0°С в реакционный раствор добавляли циклопропанкарбонил хлорид (23 мг, 0,22 ммоль), затем нагревали до комнатной температуры и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении и добавляли 50 мл дихлорметана. Органическую фазу промывали водой (20 мл), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(3-(циклопропанкарбоксамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 7а (139 мг, светло-коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 697.1 [М+1]
Стадия 2
4-(3-(циклопропанкарбоксамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(3-(циклопропанкарбоксамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 7а (139 мг, 0,20 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 1,5 ч с последующим добавлением 10 мл воды и 1 мл 1М соляной кислоты, затем экстрагировали этилацетатом (20 мл×3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(3-(циклопропанкарбоксамидо)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 7 (49 мг, светло-коричневое твердое веществ), выход: 42,6%. МС m/z (ESI): 577.1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.88 (s, 1Н), 9.70 (s, 1Н), 7.59-7.69 (m, 3Н), 7.38-7.46 (m, 2Н), 7.28 (t, 1Н), 7.05 (d, 1Н), 6.66 (t, 1Н), 4.96 (s, 1Н), 3.14 (s, 3Н), 2.01 (s, 3Н), 1.83-1.94 (m, 1Н), 0.75-0.83 (m, 4Н).
Пример 8
4-(3-пропионамидо-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
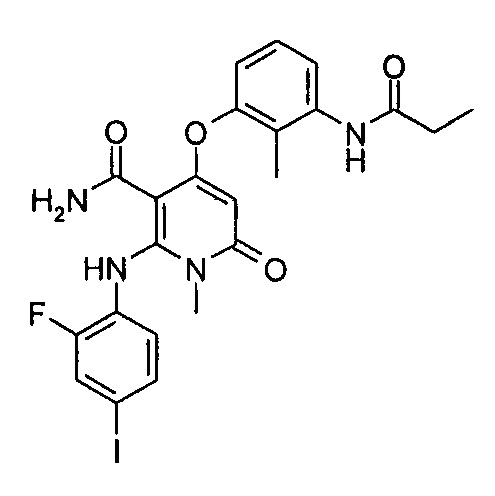
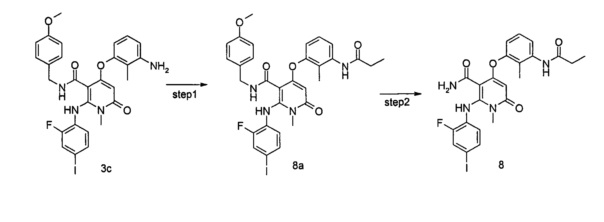
Стадия 1
4-(3-пропионамидо-2-метилфенокси-)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(3-амино-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино) -N-(4-метоксибензил)-1 -метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 3с (126 мг, 0,20 ммоль) растворяли в 2 мл смеси дихлорметана и пиридина (V:V=1:1) с последующим добавлением пропионилхлорида (20 мг, 0,22 ммоль). После перемешивания в течение 12 ч реакционный раствор концентрировали при пониженном давлении, добавляли 30 мл дихлорметана. Органическую фазу промывали водой (20 мл), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(3-пропионамидо-2-метилфенокси-)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 8а (137 мг, коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
MS m/z (ESI): 685.1 [М+1]
Стадия 2
4-(3-пропионамидо-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(3-пропионамидо-2-метил фенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 8а (137 мг, 0,20 ммоль) растворяли в 5 мл анизола, с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 1,5 ч с последующим добавлением 10 мл воды и 1 мл 1 М соляной кислоты, а затем экстрагировали этилацетатом (20 мл×3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(3-пропионамидо-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 8 (55 мг, светло-коричневого твердого вещества), выход: 48,7%.
МС m/z (ESI): 565.1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.89 (s, 1Н), 9.39 (s, 1Н), 7.59-7.69 (m, 3Н), 7.35-7.46 (m, 2Н), 7.29 (t, 1Н), 7.06 (d, 1Н), 6.66 (t, 1Н), 4.96 (s, 1Н), 3.14 (s, 3Н), 2.36 (m, 2Н), 1.99 (s, 3Н), 1.10 (t, 3Н).
Пример 9
(S)-2-((2-фтор-4-иодфенил)амино)-1-метил-4-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
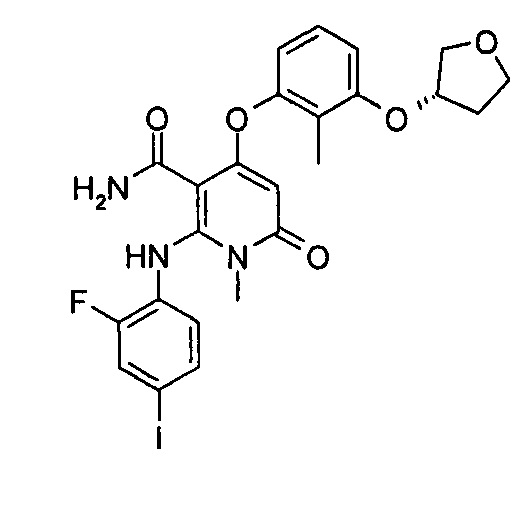
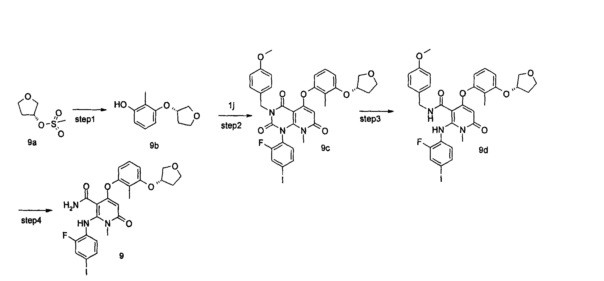
Стадия 1
(S)-2-метил-3-((тетрагидрофуран-3-ил)окси)фенол
(R)-тетра гидрофура н-3-ил метансульфонат 9а (373 мг, 3 ммоль, полученный с помощью хорошо известного способа, раскрытого в "Journal of Organic Chemistry, 73(14), 5397-5409; 2008"), 2-метилбензол-1,3-диол (500 мг, 3 ммоль) и карбонат цезия (977 мг, 3 ммоль) растворяли в 10 мл диметилформамида. Реакционный раствор нагревали до 80°С и перемешивали в течение 12 часов с последующим добавлением 10 мл воды и 50 мл этилацетата, по каплям добавляли 1М соляную кислоту для доведения значения рН до 3, затем экстрагировали дихлорметаном (20 мл×3). Органические фазы объединяли, промывали водой (20 мл×3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюента В с получением указанного в заголовке соединения (S)-2-метил-3-((тетрагидрофуран-3-ил)окси)фенол 9b (290 мг, коричневое твердое вещество), выход: 49,8%.
МС m/z (ESI): 195.1 [М+1]
Стадия 2
(S)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-5-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)пиридо[2,3-d]пиримидин-2,4,7(1Н,3H,8А7)-трион
(S)-2-метил-3-((тетрагидрофуран-3-ил)окси)фенол 9b (290 мг, 1,49 ммоль) растворяли в 10 мл тетрагидрофурана, и добавляли гидрид натрия (119 мг, 2,98 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (300 мг, 0,44 ммоль), затем нагревали до 60°С и перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (S)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-5-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)пиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 9 с (319 мг, светло-желтое масло), которое непосредственно использовали на следующей стадии без дополнительной очистки. Стадия 3 (S)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-4-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный (S)-1-(2-фтор-4-йодфенил)-3-(4-метоксибензил)-8-метил-5-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)пиридо[2,3-с!]пиримидин-2,4,7(1Н,3Н,8Н)-трион 9 с (319 мг, 0,44 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=5:1), с последующим добавлением гидроксида лития (185 мг, 4,40 ммоль). После перемешивания в течение 12 часов реакционный раствор концентрировали при пониженном давлении, добавляли 50 мл этилацетата и 10 мл воды. Органическую фазу сушили над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (S)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-4-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)-6-оксо-1,6-дигидропиридин-3-карбоксамид 9d (130 мг, твердое вещество серого цвета), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 700.1 [М+1]
Стадия 4
(S)-2-((2-фтор-4-иодфенил)амино)-1-метил-4-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный (S)-2-((2-фтор-4-йодфенил)амино)-N-(4-метоксибензил)-1-метил-4-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)-6-оксо-1,6-дигидропиридин-3-карбоксамид 9d (130 мг, 0,20 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (124 мг, 0,93 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 4-х часов, затем концентрировали при пониженном давлении и добавляли 50 мл этилацетата и 10 мл воды. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения (S)-2-((2-фтор-4-иодфенил)амино)-1-метил-4-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)-6-оксо-1,6-дигидропиридин-3-карбоксамид 9 (50 мг, светло-коричневое твердое вещество), выход: 46,5%.
МС m/z (ESI): 580.1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.90 (s, 1Н), 7.62-7.67 (m, 3Н), 7.42-7.44 (d, 1Н), 7.26-7.31 (t, 1Н), 6.93-6.96 (d, 1Н), 6.83-6.85 (d, 1Н), 6.64-6.69 (t, 1Н), 5.10 (br, 1Н), 5.00 (s, 1Н), 3.76-3.95 (m, 4Н), 3.15 (s, 3Н), 2.22-2.27 (m, 1Н), 2.19-2.03 (m, 4Н).
Пример 10
4-(2-метил-3-(метилкарбамоил)фенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
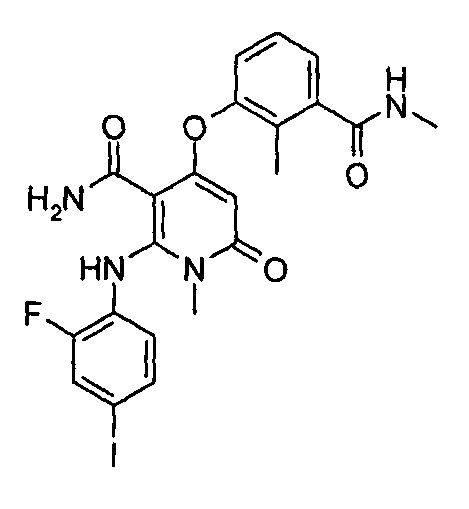
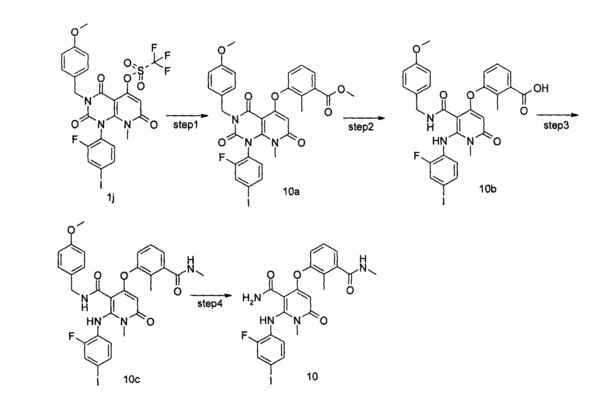
Стадия 1
метил 3-((1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-1,2,3,4,718-гексагидропиридо[2,3-d]пиримидин-5-ил)окси)-2-метилбензоат
Метил 3-гидрокси-2-метилбензоат (122 мг, 0,73 ммоль, полученный известным способом, раскрытым в "Tetrahedron Letters, 48 (31), 5465-5469; 2007") растворяли в 10 мл тетрагидрофурана с последующим добавлением гидрида натрия (60 мг, 1,50 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-уил трифторметансульфонат 1j (500 мг, 0,73 ммоль), нагревали до 70°С и перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения метил 3-((1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-1,2,3,4,7,8-гексагидропиридо[2,3-d]пиримидин-5-ил)окси)-2-метилбензоат 10а (509 мг, светло-желтая жидкость), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 698.1 [М+1]
Стадия 2
3-((6-((2-фтор-4-иодфенил)амино)-5-((4-метоксибензил)карбамоил)-1-метил-2-оксо-1,2-дигидропиридин-4-ил)окси)-2-метилбензойная кислота
Неочищенный метил 3-((1-(2-фтор-4-йодфенол)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-1,2,3,4,7,8-гексагидропиридо [2,3-d]пиримидин-5-ил)окси)-2-метилбензоат 10а (509 мг, 0,73 ммоль) растворяли в 24 мл смеси тетрагидрофурана и воды (V:V=5:1) с последующим добавлением гидроксида лития (308 мг, 7,34 ммоль). После перемешивания в течение 12 часов в реакционную смесь добавляли 10 мл воды и 50 мл этилацетата. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения неочищенной 3-((6-((2-фтор-4-иодфенил)амино)-5-((4-метоксибензил)карбамоил)-1-метил-2-оксо-1,2-дигидропиридин-4-ил)окси)-2-метилбензойную кислоту 10b (500 мг, желтое твердое вещество), которую непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 658.1 [М+1]
Стадия 3
4-(2-метил-3-(метилкарбамоил)фенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенную 3-((6-((2-фтор-4-иодфенил)амино)-5-((4-метоксибензил) карбамоил)-1-метил-2-оксо-1,2-дигидропиридин-4-ил)окси)-2-метилбензойную кислоту 10b (250 мг, 0,38 ммоль) растворяли в 5 мл диметилформамида с последующим добавлением последовательно 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорида (146 мг, 0,76 ммоль), 1-гидроксибензотриазола (103 мг, 0,76 ммоль) и 0,5 мл N,N-диизопропилэтиламина. После перемешивания в течение 10 минут в реакционный раствор добавляли метиламин (0,2 мл, 0,38 ммоль) и перемешивали в течение 12 ч с последующим добавлением 20 мл 10% хлорида лития и 50 мл этилацетата. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения неочищенного 4-(2-метил-3-(метилкарбамоил)фенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 10с (200 мг, коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 4
4-(2-метил-3-(метилкарбамоил)фенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(2-метил-3-(метилкарбамоил)фенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 10 с (200 мг, 0,30 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (200 мг, 1,50 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 3 часов, затем концентрировали при пониженном давлении и добавляли 50 мл этилацетата и 15 мл воды. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(2-метил-3-(метилкарбамоил)фенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 10 (35 мг, белое твердое вещество), выход: 21,3%.
МС m/z (ESI): 551.1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.90 (s, 1Н), 8.31-8.33 (m, 1Н), 7.63-7.67 (m, 3Н), 7.27-7.45 (m, 4Н), 6.65-6.70 (t, 1Н), 5.01 (s, 1Н), 3.15 (s, 3Н), 2.76-2.78 (d, 3Н), 2.12 (s, 3Н).
Пример 11
4-(3-(циклопропилкарбамоил)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
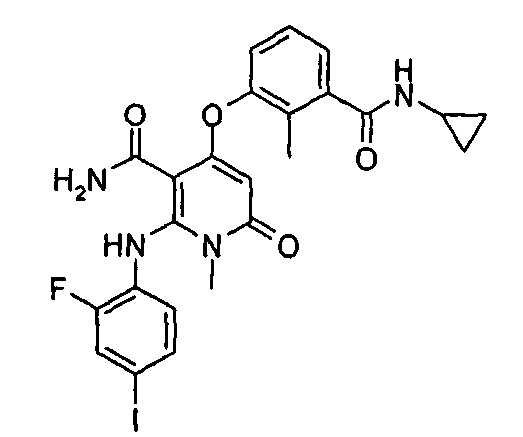
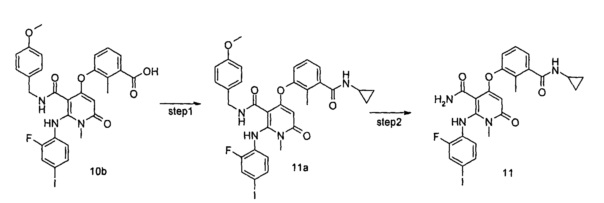
Стадия 1
4-(3-(циклопропилкарбамоил)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенную 3-((6-((2-фтор-4-йодфенил)амино)-5-((4-метоксибензил) карбамоил)-1-метил-2-оксо-1,2-дигидропиридин-4-ил)окси)-2-метилбензойную кислоту 10b (250 мг, 0,38 ммоль) растворяли в 5 мл диметилформамида с последующим добавлением последовательно 1-этил-3-(3-диметиламинопропил) карбодиимид гидрохлорида (146 мг, 0,76 ммоль), 1-гидроксибензотриазола (103 мг, 0,76 ммоль) и 0,5 мл N',N'-диизопропилэтиламина. После перемешивания в течение 10 минут в реакционный раствор добавляли циклопропанметиламин (22 мг, 0,38 ммоль) и перемешивали в течение 12 ч с последующим добавлением 20 мл 10% хлорида лития и 50 мл этилацетата. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(3-(циклопропилкарбамоил)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 11а (130 мг, коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 2
4-(3-(циклопропилкарбамоил)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1 -метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(3-(циклопропилкарбамоил)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 11а (130 мг, 0,20 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 3 часов, затем концентрировали при пониженном давлении и добавляли 50 мл этилацетата и 15 мл воды. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(3-(циклопропилкарбамоил)-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 11 (40 мг, коричневое твердое вещество), выход: 35,4%.
МС m/z (ESI): 577.1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.90 (s, 1Н), 8.43-8.45 (d, 1Н), 7.63-7.67 (m, 3Н), 7.24-7.45 (m, 4Н), 6.65-6.70 (t, 1Н), 5.01 (s, 1Н), 3.15 (s, 3Н), 2.83-2.84 (m, 1Н), 2.11 (s, 3Н), 0.68-0.71 (m, 2Н), 0.52-0.54 (m, 2Н).
Пример 12
(R)-2-((2-фтор-4-иодфенил)амино)-1-метил-4-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
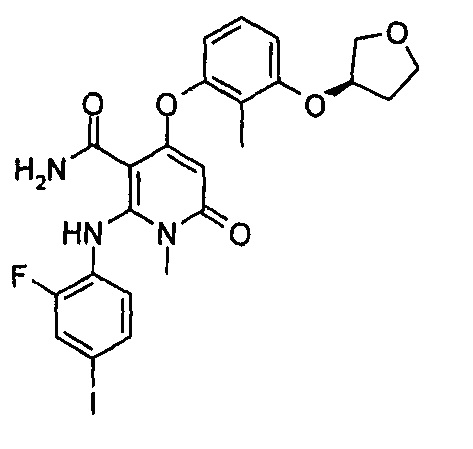
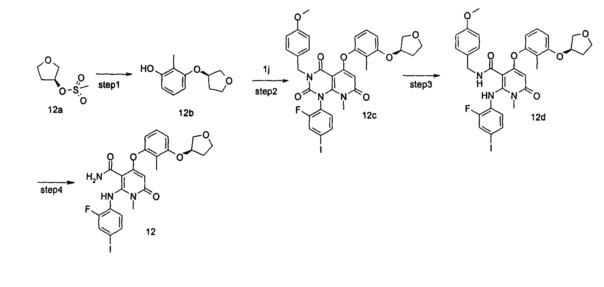
Стадия 1
(R)-2-метил-3-((тетрагидрофуран-3-ил)окси)фенол
(S)-тетрагидрофуран-3-ил метансульфонат 12а (1,65 г, 9,93 ммоль, полученный с помощью хорошо известного способа, раскрытого в "Journal of Organic Chemistry, 73 (14), 5397-5409; 2008"), 2-метилбензол-1,3-диол (2,46 г, 19,90 ммоль) и карбонат цезия (3,23 г, 9,93 ммоль) растворяли в 250 мл диметилформамида. Реакционный раствор нагревали до 80°С и перемешивали в течение 12 ч, по каплям добавляли 1М соляную кислоту для доведения значения рН до 7, и экстрагировали дихлорметаном (100 мл×3). Органические фазы объединяли, промывали водой (100 мл×3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюента В с получением указанного в заголовке соединения (R)-2-метил-3-((тетрагидрофуран-3-ил)окси)фенол 12b (1,03 г, беловатое твердое вещество), выход: 53,3%.
МС m/z (ESI): 195.1 [М+1]
Стадия 2
(R)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-5-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)пиридо[2,3-d]пиримидин-2,4,7(1Н,3H,8H)-трион
(R)-2-метил-3-((тетрагидрофуран-3-ил)окси)фенол 12b (47 мг, 0,24 ммоль) растворяли в 5 мл тетрагидрофурана и добавляли гидрид натрия (12 мг, 0,30 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d)пиримидин-5-ил трифторметансульфонат 1j (136 мг, 0,20 ммоль), затем нагревали до 60°С и перемешивали в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (R)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-5-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)пиридо[2,3-d]пиримидин-2,4,7(1H,3Н,8Н)-трион 12 с (145 мг, светло-желтое масло), которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 3
(R)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-4-(2-метил-3-((тетрагидрофуран -3-ил)окси)фенокси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный (R)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-5-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)пиридо[2,3-d]пиримидин-2,4,7(1Н,3H,8Н)-трион 12 с (145 мг, 0,20 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=4:1) с последующим добавлением гидроксида лития (168 мг, 4 ммоль) Реакционный раствор подогревали до 40°С и перемешивали в течение 1 ч, затем добавляли 10 мл воды, экстрагировали дихлорметаном (10 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (R)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-4-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)-6-оксо-1,6-дигидропиридин-3-карбоксамид 12d (140 мг, светло-коричневое твердое вещество), который непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 700.0 [М+1]
Стадия 4
(R)-2-((2-фтор-4-иодфенил)амино)-1-метил-4-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный (R)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-4-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)-6-оксо-1,6-дигидропиридин-3-карбоксамид 12d (140 мг, 0,20 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 3,5 ч, добавляли 10 мл воды и 1 мл 1М соляной кислоты, и экстрагировали этилацетатом (20 мл×3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, а полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения (R)-2-((2-фтор-4-иодфенил)амино)-1-метил-4-(2-метил-3-((тетрагидрофуран-3-ил)окси)фенокси)-6-оксо-1,6-дигидропиридин-3-карбоксамид 12 (35 мг, светло-коричневое твердое вещество), выход: 30,1%.
МС m/z (ESI): 580.1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.89 (s, 1Н), 7.59-7.69 (m, 3Н), 7.40-7.47 (m, 1Н), 7.29 (t, 1Н), 6.95 (d, 1Н), 6.84 (d, 1Н), 6.67 (t, 1Н), 5.07-5.13(т, 1Н), 4.99 (s, 1Н), 3.75-3.95 (m, 4Н), 3.13 (s, 3Н), 2.19-2.29 (m, 1Н), 1.97-2.04 (m, 1Н), 1.95 (s, 3Н).
Пример 13
4-(3-ацетил-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
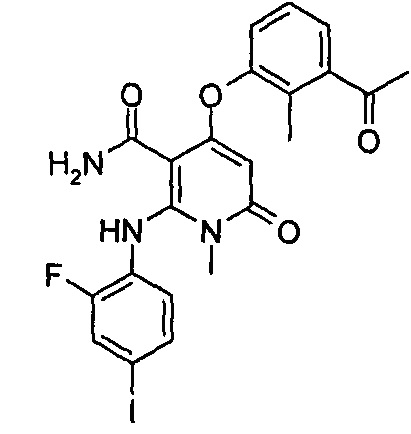
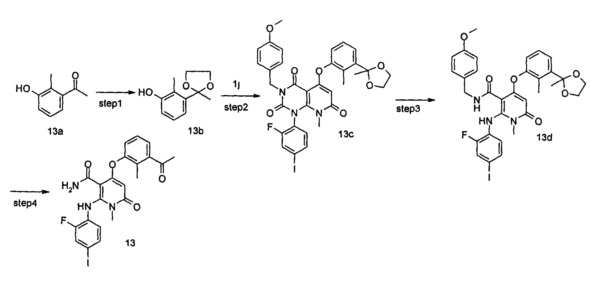
Стадия 1
2-метил-3-(2-метил-1,3-диоксолан-2-ил)фенол
1-(3-гидрокси-2-метилфенил)этанон 13а (3 г, 20 ммоль, полученный с помощью хорошо известного способа, раскрытого в "Journal of Medicinal Chemistry, 52 (20), 6433-6446; 2009") и 30 мл этиленгликоля растворяли в 30 мл толуола. После перемешивания в течение 3,5 часов с обратным холодильником, в реакционный раствор добавляли 200 мл этилацетата, промывали водой (100 мл×3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, а полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюента В, получая указанное в заголовке соединение 2-метил-3-(2-метил-1,3-диоксолан-2-ил)фенол 13b (1,03 г, твердое вещество белого цвета), выход: 54,1%.
МС m/z (ESI): 195.1 [М+1]
Стадия 2
1-(2-фтор-4-йодфенил)-3-(4-метоксибензил)-8-метил-5-(2-метил-3-(2-метил-1,3-диоксолан-2-ил)фенокси)пиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8H)-трион
2-метил-3-(2-метил-1,3-диоксолан-2-ил)фенол 13b (47 мг, 0,24 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (12 мг, 0,30 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (136 мг, 0,20 ммоль), затем нагревали до 60°С и перемешивали в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-5-(2-метил-3-(2-метил-1,3-диоксолан-2-ил)фенокси)пиридо[2,3-d]пиримидин-2,4,7(1Н,3H,8Н)-трион 13 с (145 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
МС m/z (ESI): 726.2 [М+1]
Стадия 3
2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-4-(2-метил-3-(2-метил-1,3-диоксолан-2-ил)фенокси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 1-(2-фтор-4-йодфенил)-3-(4-метоксибензил)-8-метил-5-(2-метил-3-(2-метил-1,3-диоксолан-2-ил)фенокси)пиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8H)-трион 13 с (145 мг, 0,20 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=4:1) с последующим добавлением гидроксида лития (168 мг, 4 ммоль) Реакционный раствор подогревали до 40°С и перемешивали в течение 1 часа. К реакционному раствору добавляли 50 мл этилацетата, промывали водой (25 мл×3), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-4-(2-метил-3-(2-метил-1,3-диоксолан-2-ил)фенокси)-6-оксо-1,6-дигидропиридин-3-карбоксамид 13d (140 мг, светло-коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 700.1 [М+1]
Стадия 4
4-(3-ацетил-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 2-((2-фтор-4-йодфенил)амино)-N-(4-метоксибензил)-1-метил-4-(2-метил-3-(2-метил-1,3-диоксолан-2-ил)фенокси)-6-оксо-1,6-дигидропиридин-3-карбоксамид 13d (140 мг, 0,20 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 3,5 часов, добавляли 10 мл воды и 1 мл 1М соляной кислоты и экстрагировали этилацетатом (20 мл×3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(3-ацетил-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 13 (7 мг, светло-коричневое твердое вещество), выход: 6,5%.
МС m/z (ESI): 536.1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.87 (s, 1Н), 7.74-7.78 (m, 1Н), 7.59-7.69 (m, 3Н), 7.39-7.49 (m, 3Н), 6.67 (t, 1Н), 4.95 (s, 1Н), 3.14 (s, 3Н), 2.60 (s, 3Н), 2.19 (s, 3Н).
Пример 14
4-(3-хлор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
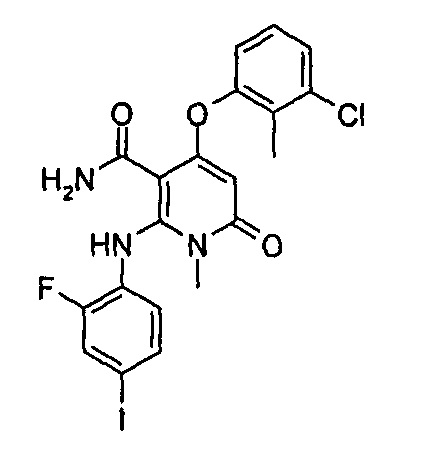
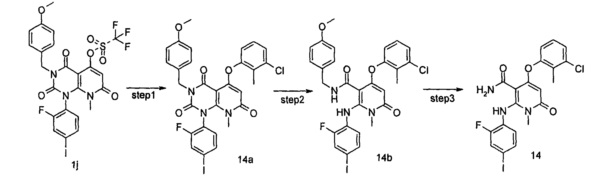
Стадия 1
5-(3-хлор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион
3-хлор-2-метил-фенол (26 мг, 0,18 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (9 мг, 0,23 ммоль). После перемешивания в течение 1 часа в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (102 мг, 0,15 ммоль) и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-(3-хлор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-с!]пиримидин-2,4,7(1Н,3Н,8Н)-трион 14а (100 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
МС m/z (ESI): 674.0 [М+1]
Стадия 2
4-(3-хлор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-(3-хлор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d] пиримидин-2,4, 7(1Н,3Н,8Н)-трион 14а (100 мг, 0,15 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=5:1) с последующим добавлением гидроксида лития (31 мг, 0,75 ммоль). Реакционную смесь перемешивали в течение 12 ч, добавляли 10 мл воды и 50 мл этилацетата. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(3-хлор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 14b (106 мг, желто-коричневое масло), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 648.0 [М+1]
Стадия 3
4-(3-хлор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(3-хлор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 14b (106 мг, 0,15 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (100 мг, 0,75 ммоль). После перемешивания в течение 3 ч при 120°С реакционный раствор концентрировали при пониженном давлении, добавляли 50 мл этилацетата и 15 мл воды. Органическую фазу промывали 1М соляной кислотой (25 мл×3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью препаративного разделения с получением указанного в заголовке соединения 4-(3-хлор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 14 (21 мг, твердое вещество белого цвета), выход: 26,5%.
МС m/z (ESI): 528.0 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.79 (s, 1Н), 7.60-7.68 (m, 3Н), 7.41-7.47 (m, 2Н), 7.36 (t, 1Н), 7.23 (d, 1Н), 6.67 (t, 1Н), 5.00 (s, 1Н), 3.15 (s, 3Н), 2.18 (s, 3Н).
Пример 15
4-(2-хлор-4-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
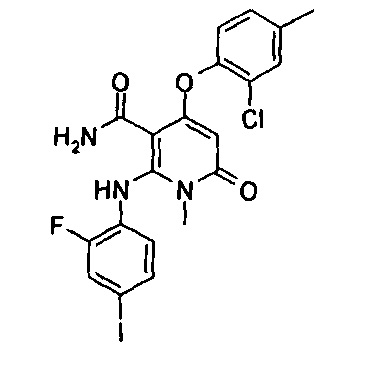
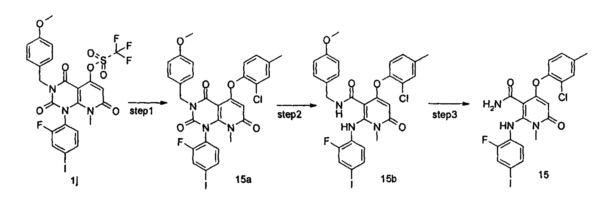
Стадия 1
5-(2-хлор-4-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1H,3Н,8Н)-трион
2-хлор-4-метил-фенол (34 мг, 0,24 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (12 мг, 0,30 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (136 мг, 0,20 ммоль) и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-(2-хлор-4-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 15а (135 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
МС m/z (ESI): 674.0 [М+1]
Стадия 2
4-(2-хлор-4-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-(2-хлор-4-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 15а (135 мг, 0,20 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=4:1) с последующим добавлением гидроксида лития (168 мг, 4 ммоль). Реакционный раствор нагревали до 40°С и перемешивали в течение 1 часа с последующим добавлением 50 мл этилацетата. Органическую фазу промывали с помощью 1М раствора гидроксида натрия (25 мл×3), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(2-хлор-4-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 15b (130 мг, желто-коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 648.1 [М+1]
Стадия 3
4-(2-хлор-4-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(2-хлор-4-метилендиокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 15b (130 мг, 0,20 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 1,5 часов с последующим добавлением 10 мл воды и 1 мл 1М соляной кислоты, и экстрагировали этилацетатом (20 мл×3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(2-хлор-4-метилфенокси)-2-((2-фтор-4-иодфенил) амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 15 (50 мг, серо-белое твердое вещество), выход: 47,2%.
МС m/z (ESI): 528.0 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.95 (s, 1Н), 7.62-7.71 (m, 2Н), 7.52-7.58 (m, 1Н), 7.48-7.52 (m, 1Н), 7.44 (d, 1Н), 7.87 (d, 1Н), 7.26-7.34 (m, 1Н), 6.67 (t, 1Н), 4.97 (s, 1Н), 3.14 (s, 3Н), 2.35 (s, 3Н).
Пример 16
4-(3-метокси-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
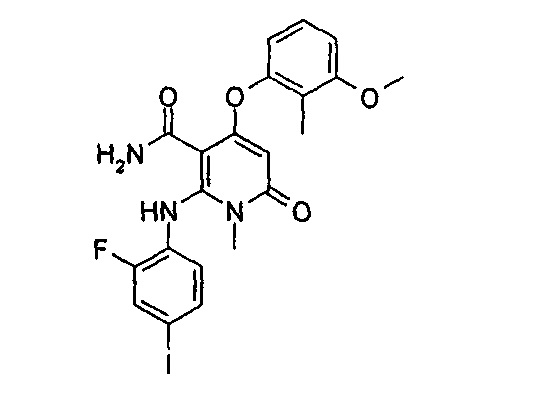
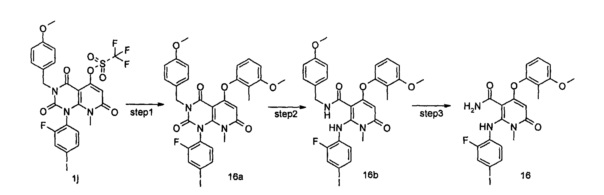
Стадия 1
5-(3-метокси-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион
3-метокси-2-метил-фенол (27 мг, 0,19 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (7 мг, 0,29 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (130 мг, 0,19 ммоль), нагревали до 60°С и перемешивали в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-(3-метокси-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8H)-трион 16а (127 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2
4-(3-метокси-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-(3-метокси-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8H)-трион 16а (127 мг, 0,19 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=4:1) с последующим добавлением гидроксида лития (80 мг, 1,91 ммоль). После перемешивания в течение 12 часов в реакционный раствор добавляли 50 мл этилацетата. Органическую фазу промывали с помощью 1М раствора гидроксида натрия (25 мл×3), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(3-метокси-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 16b (170 мг, коричневое масло), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 644.1 [М+1]
Стадия 3
4-(3-метокси-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(3-метокси-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 16b (170 мг, 0,19 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (127 мг, 0,95 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 1,5 часов, добавляли 10 мл воды и 1 мл 1М соляной кислоты и экстрагировали этилацетатом (20 мл×3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, а полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(3-метокси-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 16 (28 мг, серо-белое твердое вещество), выход: 28,0%.
МС m/z (ESI): 524.1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.83 (s, 1Н), 7.61-7.67 (m, 3Н), 7.42-7.44 (d, 1Н), 7.28-7.33 (t, 1Н), 6.95-6.98 (d, 1Н), 6.82-6.85 (d, 1Н), 6.64-6.69 (t, 1Н), 4.99 (s, 1Н), 3.85 (s, 3Н), 3.14 (s, 3Н), 1.97 (s, 3Н).
Пример 17
4-(2-фтор-4-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
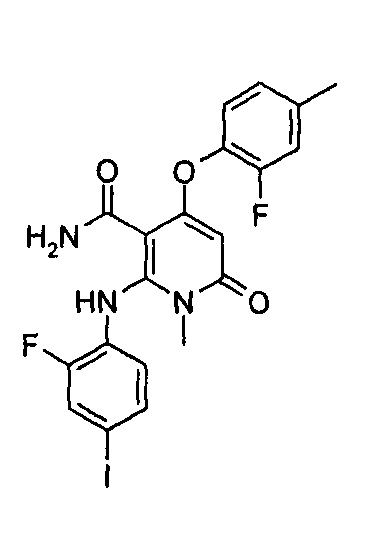

Стадия 1
5-(2-фтор-4-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3H,8Н)-трион
2-нитро-4-метил-фенол (33 мг, 0,26 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (16 мг, 0,40 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (136 мг, 0,20 ммоль) и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-(2-фтор-4-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 17а (131 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
МС m/z (ESI): 658.0 [М+1]
Стадия 2
4-(2-фтор-4-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-(2-фтор-4-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3H,8Н)-трион 17а (131 мг, 0,20 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=4:1), с последующим добавлением гидроксида лития (42 мг, 1 ммоль). После перемешивания в течение 4 часов реакционный раствор добавляли 50 мл этилацетата. Органическую фазу промывали последовательно водой (30 мл×1), 0,5М соляной кислотой (30 мл×1) и насыщенным раствором хлорида натрия (30 мл×1), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(2-фтор-4-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 17b (126 мг, коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 632.0 [М+1]
Стадия 3
4-(2-фтор-4-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(2-фтор-4-метилендиокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 17b (126 мг, 0,20 ммоль) растворяли в 3 мл анизола с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 100°С и перемешивали в течение 1 часа с последующим добавлением 60 мл дихлорметана. Органическую фазу промывали последовательно водой (50 мл×2) и насыщенным раствором хлорида натрия (50 мл×1), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(2-фтор-4-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 17 (32 мг, почти белого твердого вещества), выход: 31,4%.
МС m/z (ESI): 512.0 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.84(8, 1Н), 7.61-7.66 (m, 3Н), 7.41-7.43 (m, 1Н), 7.28-7.34 (m, 2Н), 7.12-7.15 (m, 1Н), 6.64-6.68 (m, 1Н), 5.10 (s, 1Н), 3.15 (s, 3Н), 2.36 (s, 3Н).
Пример 18
4-(2,3-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
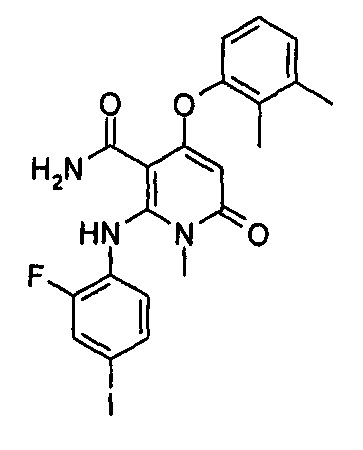
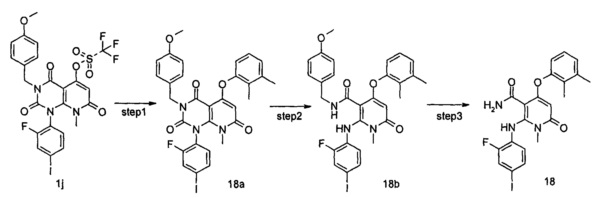
Стадия 1
5-(2,3-диметилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион
2,3-диметил-фенол (24 мг, 0,19 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (7 мг, 0,29 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (130 мг, 0,19 ммоль) и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-(2,3-диметилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8H)-трион 18а (125 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2
4-(2,3-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-(2,3-диметилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1H,3Н,8Н)-трион 18а (125 мг, 0,19 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=4:1) с последующим добавлением гидроксида лития (80 мг, 1,91 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 50 мл этилацетата. Органическую фазу промывали 1М раствором гидроксида натрия (25 мл×3), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(2,3-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 18b (130 мг, желтое масло), которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 3
4-(2,3-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(2,3-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 18b (130 мг, 0,19 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (127 мг, 0,95 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 1,5 часа с последующим добавлением 10 мл воды и 1 мл 1М соляной кислоты, и экстрагировали этилацетатом (20 мл×3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(2,3-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 18 (26 мг, твердое вещество белого цвета), выход: 28,9%.
МС m/z (ESI): 506.0 [М-1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.91 (s, 1Н), 7.63-7.67 (m, 3Н), 7.42-7.44 (d, 1Н), 7.15-7.22 (m, 2Н), 7.04-7.06 (d, 1Н), 6.64-6.69 (t, 1Н), 4.95 (s, 1Н), 3.14 (s, 3Н), 2.31 (s, 3Н), 2.04 (s, 3Н).
Пример 19
4-(2-фтор-3-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид

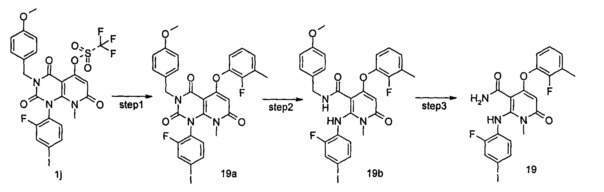
Стадия 1
5-(2-фтор-3-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион
2-фтор-3-метилфенол (30 мг, 0,24 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (12 мг, 0,30 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (136 мг, 0,20 ммоль), нагревали до 60°С и перемешивали в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-(2-фтор-3-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1H,3Н,8Н)-трион 19а (131 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
МС m/z (ESI): 658.1 [М+1]
Стадия 2
4-(2-фтор-3-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-(2-фтор-3-метилендиокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4)7(1Н,3Н,8Н)-трион 19а (131 мг, 0,20 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=4:1) с последующим добавлением гидроксида лития (168 мг, 4 ммоль). Реакционный раствор нагревали до 40°С и перемешивали в течение 1 часа с последующим добавлением 50 мл этилацетата. Органическую фазу промывали с помощью 1М раствора гидроксида натрия (25 мл×3), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(2-фтор-3-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 19b (126 мг, светло-коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 632.1 [М+1]
Стадия 3
4-(2-фтор-3-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(2-фтор-3-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 19b (126 мг, 0,20 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 1,5 часа с последующим добавлением 10 мл воды и 1 мл 1М соляной кислоты, и экстрагировали этилацетатом (20 мл×3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(2-фтор-3-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 19 (35 мг, серо-белое твердое вещество), выход: 34,3%.
МС m/z (ESI): 512.1 [М+1]
1H ЯМР (400 МГц, ДМСО-d6): δ 9.88 (s, 1Н), 7.60-7.66 (m, 3Н), 7.40-7.46 (m, 1Н), 7.18-7.29 (m, 3Н), 6.67 (t, 1Н), 5.11 (s, 1Н), 3.14 (s, 3Н), 2.30 (s, 3Н).
Пример 20
4-(2,4-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
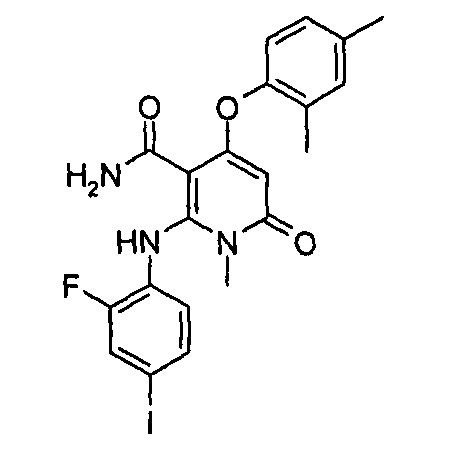
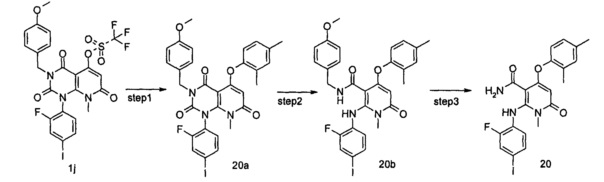
Стадия 1
5-(2,4-диметилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион
2,4-диметил-фенол (32 мг, 0,26 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (16 мг, 0,40 ммоль). После перемешивания в течение 2 часов к реакционному раствору добавляли 1-(2-Фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (136 мг, 0,20 ммоль), и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-(2,4-диметилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 20а (130 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2
4-(2,4-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-(2,4-диметилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1 Н,3Н,8Н)-трион 20а (130 мг, 0,20 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=4:1) с последующим добавлением гидроксида лития (42 мг, 1 ммоль). После перемешивания в течение 4 часов в реакционный раствор добавляли 50 мл этилацетата. Органическую фазу промывали последовательно водой (30 мл×1), 0,5М соляной кислотой (30 мл×1) и насыщенным раствором хлорида натрия (30 мл×1), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения неочищенного 4-(2,4-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 20b (124 мг, рыжевато-коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 628.1 [М+1]
Стадия 3
4-(2,4-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(2,4-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 20b (124 мг, 0,20 ммоль) растворяли в 3 мл анизола с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 100°С и перемешивали в течение 1 часа, с последующим добавлением 60 мл дихлорметана. Органическую фазу промывали последовательно водой (50 мл×2) и насыщенным раствором хлорида натрия (50 мл×1), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(2,4-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 20 (15 мг, белое твердое вещество), выход: 31,4%.
МС m/z (ESI): 508.1 [M+1]
1H ЯМР (400 МГц, ДМСО-d6): δ 10.0 (s, 1H), 7.61-7.66 (m, 3H), 7.41-7.44 (m, 1H), 7.17-7.18 (m, 1H), 7.07-7.14 (m, 2H), 6,64-6.68 (m, 1H), 4.96 (s, 1H), 3.13 (s, 3H), 2.31 (s, 3H), 2.10 (s, 3H).
Пример 21
4-(2,6-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
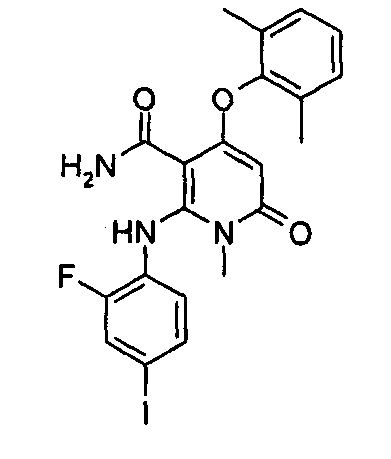
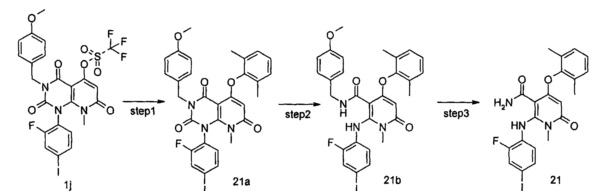
Стадия 1
5-(2,6-диметилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3H,8Н)-трион
2,6-диметил-фенол (32 мг, 0,26 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (16 мг, 0,40 ммоль). После перемешивания в течение 1,5 часов к реакционному раствору добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (136 мг, 0,20 ммоль) и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-(2,6-диметилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 21а (130 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2
4-(2,6-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-(2,6-диметилендиокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 21а (130 мг, 0,20 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (1:V=4:1) с последующим добавлением гидроксида лития (42 мг, 1 ммоль). После перемешивания в течение 4 часов к реакционной смеси добавляли 70 мл дихлорметана. Органическую фазу промывали последовательно водой (30 мл×1) и насыщенным раствором хлорида натрия (30 мл×1), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(2,6-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 21 b (125 мг, рыжевато-коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 628.1 [М+1]
Стадия 3
4-(2,6-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(2,6-диметилендиокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 21 b (125 мг, 0,20 ммоль) растворяли в 3 мл анизола с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 100°С и перемешивали в течение 1,5 часов с последующим добавлением 60 мл дихлорметана. Органическую фазу промывали последовательно водой (50 мл×2) и насыщенным раствором хлорида натрия (50 мл×1), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(2,6-диметилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 21 (10 мг, белое твердое вещество), выход: 9,9%.
МС m/z (ESI): 508.1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 10.33 (s, 1Н), 7.62-7.67 (m, 3Н), 7.42-7.44 (m, 1Н), 7.16-7.21 (m, 3Н), 6.69-6.74 (m, 1Н), 4.88 (s, 1Н), 3.12 (s, 3Н), 2.13 (s, 6Н).
Пример 22
4-(4-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
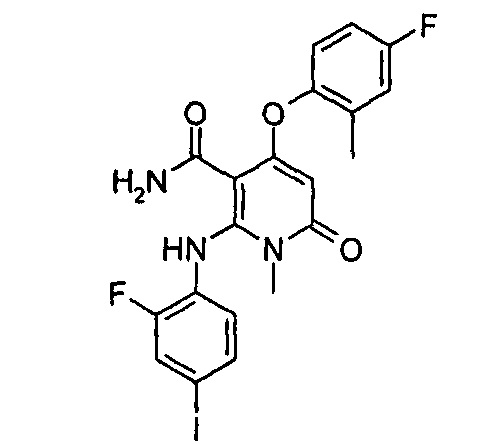
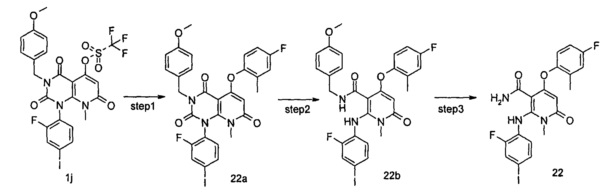
Стадия 1
5-(4-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3H,8H)-трион
4-фтор-2-метил фенол (24 мг, 0,19 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (7 мг, 0,29 ммоль). После перемешивания в течение 2 часов к реакционному раствору добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (130 мг, 0,19 ммоль) и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-(4-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1H,3Н,8H)-трион 22а (124 мг, светло-желтая жидкость), которую использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2
4-(4-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-(4-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]птримидин-2,4,7(1H,3H,8H)-трион 22а (124 мг, 0,19 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=4:1) с последующим добавлением гидроксида лития (80 мг, 1,91 ммоль). После перемешивания в течение 2 часов к реакционному раствору добавляли 50 мл этилацетата и 10 мл воды, сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(4-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 22b (150 мг, желтое масло), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 630.1 [М-1]
Стадия 3
4-(4-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(4-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 22b (150 мг, 0,19 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (127 мг, 0,95 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 1,5 часов с последующим добавлением 20 мл дихлорметана и 5 мл воды. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(4-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 22 (26 мг, белое твердое вещество), выход: 26,6%.
МС m/z (ESI): 510.0 [М-1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.94 (s, 1Н), 7.63-7.67 (m, 3Н), 7.42-7.45 (d, 1Н), 7.26-7.30 (m, 2Н), 7.16-7.20 (m, 1Н), 6.64-6.70 (t, 1Н), 4.99 (s, 1Н), 3.15 (s, 3Н), 2.15 (s, 3Н).
Пример 23
4-(2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Стадия 1
5-(2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион
2-метил-фенол (21 мг, 0,19 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (7 мг, 0,29 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (130 мг, 0,19 ммоль) и перемешивали в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-(2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 23а (122 мг, светло-желтая жидкость), которое непосредственно использовали на следующей стадии.
Стадия 2
4-(2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-(2-метилфенокси)-1-(2-фтор-4-йодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1H,3Н,8Н)-трион 23а (122 мг, 0,19 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=4:1) с последующим добавлением гидроксида лития (80 мг, 1,91 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 50 мл этилацетата и 10 мл воды, сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 23b (140 мг, желтое масло), которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 3
4-(2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(2-метилфенокси)-2-((2-фтор-4-йодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 23b (150 мг, 0,19 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (127 мг, 0,95 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 1,5 часов с последующим добавлением 20 мл дихлорметана и 5 мл воды. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 23 (29 мг, белое твердое вещество), выход: 30,9%.
МС m/z (ESI): 492.0 [М-1]
1H ЯМР (400 МГц, ДМСО-d6) δ 10.00 (s, 1Н), 7.64-7.67 (m, 3Н), 7.21-7.45 (m, 5Н), 6.65-6.70 (t, 1Н), 4.97 (s, 1Н), 3.14 (s, 3Н), 2.15 (s, 3Н).
Пример 24
4-(3-гидрокси-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
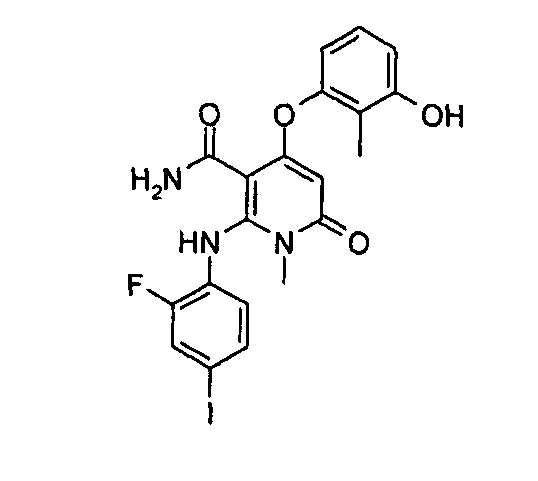
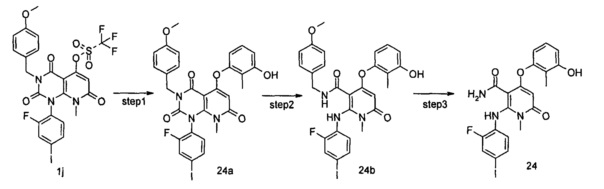
Стадия 1
5-(3-гидрокси-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион
3-гидрокси-2-метилфенол (24 мг, 0,19 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (7 мг, 0,29 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (130 мг, 0,19 ммоль) и перемешивали в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-(3-гидрокси-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 24а (125 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
МС m/z (ESI): 656.0 [М+1]
Стадия 2
4-(3-гидрокси-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-(3-гидрокси-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(7Н,3Н,8Н)-трион 24а (125 мг, 0,19 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=4:1) с последующим добавлением гидроксида лития (80 мг, 1,91 ммоль). После перемешивания в течение 12 часов в реакционный раствор добавляли 50 мл этилацетата и 10 мл воды, сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(3-гидрокси-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 24b (140 мг, коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 629.9 [М+1]
Стадия 3
4-(3-гидрокси-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(3-гидрокси-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 24b (140 мг, 0,19 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (127 мг, 0,95 ммоль). Реакционный раствор нагревали до 100°С и перемешивали в течение 3 часов с последующим добавлением 20 мл этилацетата и 5 мл воды. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(3-гидрокси-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 24 (15 мг, твердое вещество серого цвета), выход: 15,4%.
МС m/z (ESI): 508.1 [М-1]
1H ЯМР (400 МГц, ДМСО-d6) δ 9.92 (s, 1Н), 9.80 (s, 1Н),7.59-7.67 (m, 3Н), 7.42-7.44 (d, 1Н), 7.09-7.14 (t, 1Н), 6.78-6.81 (d, 1Н), 6.64-6.69 (m, 2Н), 5.01 (s, 1Н), 3.14 (s, 3Н), 1.93 (s, 3Н).
Пример 25
4-(5-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
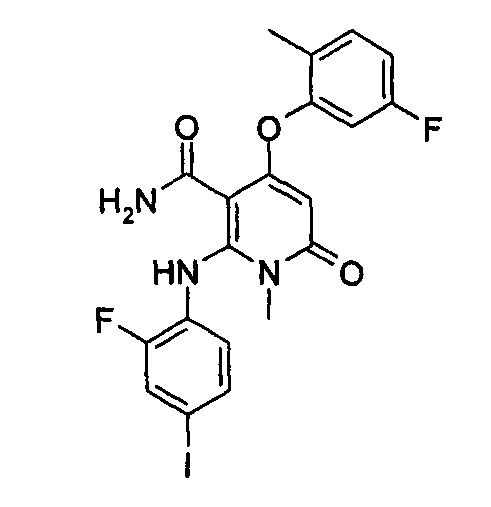
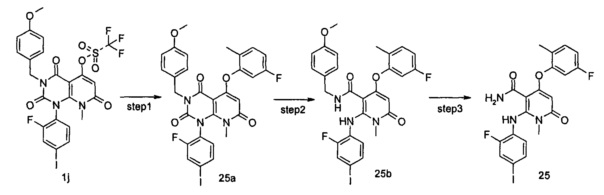
Стадия 1
5-(5-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион
5-фтор-2-метил-фенол (33 мг, 0,26 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (16 мг, 0,40 ммоль). После перемешивания в течение 1 часа в реакционную смесь добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (136 мг, 0,20 ммоль) и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-(5-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1H,3Н,8Н)-трион 25а (131 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2
4-(5-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-(5-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1H,3Н,8Н)-трион 25а (131 мг, 0,20 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=4:1) с последующим добавлением гидроксида лития (42 мг, 1 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 60 мл этилацетата. Органическую фазу промывали последовательно водой (30 мл×1) и насыщенным раствором хлорида натрия (30 мл×1), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-(5-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 25b (126 мг, коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 632.1 [М+1]
Стадия 3
4-(5-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-(5-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 25b (126 мг, 0,20 ммоль) растворяли в 3 мл анизола с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 100°С и перемешивали в течение 1 часа с последующим добавлением 40 мл дихлорметана. Органическую фазу промывали последовательно водой (50 мл×2) и насыщенным раствором хлорида натрия (50 мл×1), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 4-(5-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 25 (10 мг, светло-коричневое твердое вещество), выход: 23,5%.
МС m/z (ESI): 512.1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.88 (s, 1Н), 7.61-7.66 (m, 3Н), 7.40-7.44 (m, 2Н), 7.18-7.21 (m, 1Н), 7.10-7.15 (m, 1Н), 6.64-6.69 (m, 1Н), 5.03 (s, 1Н), 3.15 (s, 3Н), 2.11 (s, 3Н).
Пример 26
5-фтор-4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
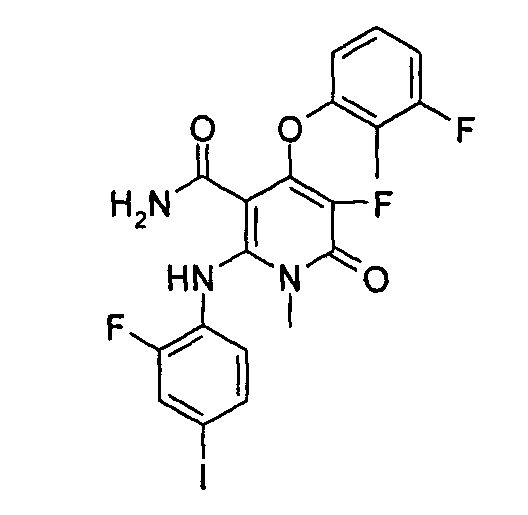
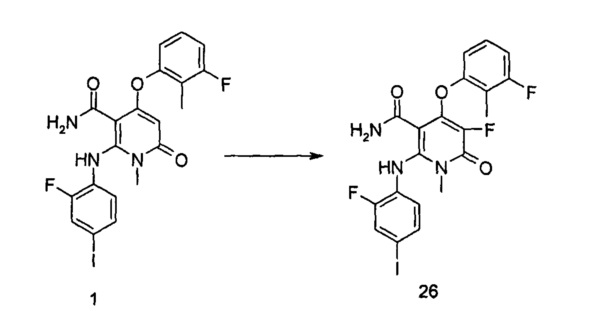
4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-йодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 1 (10 мг, 0,02 ммоль) растворяли в 6 мл дихлорметана. После охлаждения до 0°С в реакционный раствор добавляли 5 мл раствора 1-хлорметил-4-фтор-1,4-диазониабицикло[2.2.2]октана бис(тетрафторбората) (11 мг, 0,03 ммоль) в дихлорметане, нагревали до комнатной температуры и перемешивали в течение 12 ч. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 5-фтор-4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,б-дигидропиридин-3-карбоксамид 26 (3 мг, красновато-коричневое твердое вещество), выход: 30,0%.
МС m/z (ESI): 530.1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 8.68 (s, 1Н), 7.54-7.60 (m, 2Н), 7.44-7.46 (m, 1Н), 7.35-7.37 (m, 1Н), 7.14-7.20 (m, 1Н), 6.93-6.97 (m, 1Н), 6.76-6.78 (m, 1Н), 6.62-6.66(т, 1Н), 3.33 (s, 3Н), 2.21 (s, 3Н).
Пример 27
(R)-N-(2,3-дигидроксипропил)-4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид

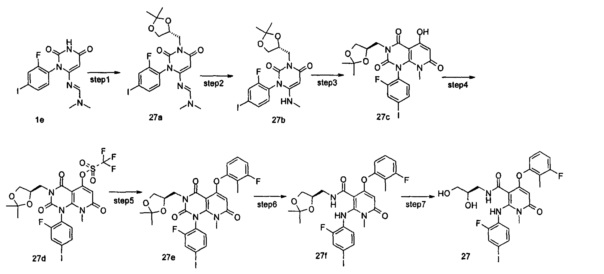
Стадия 1
(R,E)-N'-3-(2-фтор-4-иодфенил)-(1-((2,2-диметил-1,3-диоксолан-4-ил)метил)-2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-ил)-N,N-диметилформимидамид
Неочищенный (Е)-N'-(3-(2-фтор-4-йодфенил)-2,6-диоксо-1,2,3,6-тетрагидро пиримидин-4-ил)-N,N-диметилформимидамид 1е, (3,30 г, 8,20 ммоль), (R)-4-хлорметил-2,2-диметил-1,3-диоксолан (2,47 г, 16,40 ммоль), карбонат цезия (5,34 г, 16,40 ммоль) и йодид калия (3,30 г, 8,20 ммоль) (200 мг, 1,20 ммоль) растворяли в 33 мл N,N-диметилформамида. Реакционный раствор нагревали до 100°С и перемешивали в течение 12 часов. После охлаждения до комнатной температуры в реакционный раствор добавляли 200 мл этилацетата. Органическую фазу промывали водой (100 мл×3), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (R,E)-N'-3-(2-фтор-4-иодфенил)-(1-((2,2-диметил-1,3-диоксолан-4-ил)метил)-2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-ил)-N,N-диметилформимидамид 27а (3,56 г, темно-коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 517.0 [М+1]
Стадия 2
(R)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-(2-фтор-4-иодфенил)-6-(метиламино)пиримидин-2,4(1Н,3Н)-дион
Борогидрид натрия (391 мг, 10,34 ммоль) растворяли в 15 мл смеси этанола и трет-бутанола (V:V=1:2). После охлаждения до 0°С в реакционный раствор добавляли 15 мл неочищенного (R,Е)-N'-3-(2-фтор-4-иодфенил)-(1-((2,2-диметил-1,3-диоксолан-4-ил)метил)-2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-ил)-N,N-диметилформимидамида 27а (3,56 г, 6,90 ммоль) в метаноле и перемешивали в течение 1 часа. Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 12 часов. После охлаждения до 0°С в реакционный раствор добавляли 100 мл воды, и органическую фазу промывали водой (50 мл×3), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (R)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-(2-фтор-4-иодфенил)-6-(метиламино)пиримидин-2,4(1Н,3Н)-дион 27b (3,16 г, коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 476.0 [М+1]
Стадия 3
(R)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-(2-фтор-4-иодфенил)-5-гидрокси-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион
Неочищенный (R)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-(2-фтор-4-иодфенил)-6-(метиламино)пиримидин-2,4(7Н,3Н)-дион 27b (3,16 г, 6,65 ммоль) растворяли в 35 мл диэтилового эфира малоновой кислоты. Реакционный раствор перемешивали в течение 1 часа с обратным холодильником. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюента В с получением указанного в заголовке соединения (R)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-(2-фтор-4-иодфенил)-5-гидрокси-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 27с (356 мг, желтое твердое вещество), выход: 9,86%.
МС m/z (ESI): 544.0 [М+1]
Стадия 4
(Р)-1-(2-фтор-4-иодфенил)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат
(R)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-(2-фтор-4-иодфенил)-5-гидрокси-8-метилпиридо[2,3-d]пиримидин-2,4)7(1Н,3Н,8H)-трион 27 с (356 мг, 0,66 ммоль) растворяли в 5 мл дихлорметана с последующим добавлением триэтиламина (281 мг, 2,62 ммоль). После охлаждения до 0°С в реакционный раствор добавляли трифторметансульфоновый ангидрид (370 мг, 1,31 ммоль), нагревали до комнатной температуры и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюента В с получением указанного в заголовке соединения (R)-1-(2-фтор-4-иодфенил)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 27d (150 мг, светло-желтое твердое вещество), выход: 33,9%.
МС m/z (ESI): 693.1 [М+18]
Стадия 5
(R)-5-(3-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион
3-фтор-2-метилфенол (34 мг, 0,27 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (13 мг, 0,33 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли (R)-5-(3-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(7Н,3Н,8Н)-трион 27d (150 мг, 0,22 ммоль), и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (R)-5-(3-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 27е (145 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 6
(R)-4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный (R)-5-(3-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 27е (145 мг, 0,22 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=2:1) с последующим добавлением гидроксида лития (186 мг, 4,44 ммоль). Реакционный раствор нагревали до 40°С и перемешивали в течение 1 часа с последующим добавлением 20 мл этилацетата. Органическую фазу промывали последовательно насыщенным раствором бикарбоната натрия (10 мл×3) и водой (20 мл×1), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (R)-4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 27f (139 мг, красновато-коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 626.1 [М+1]
Стадия 7
(R)-N-(2,3-дигидроксипропил)-4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный (R)-4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 27f (139 мг, 0,22 ммоль) растворяли в 10 мл тетрагидрофурана с последующим добавлением 5 мл 1М соляной кислоты. После перемешивания в течение 4 часов реакционный раствор концентрировали при пониженном давлении, а полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения (R)-N-(2,3-дигидроксипропил)-4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 27 (70 мг, светло-коричневое твердое вещество), выход: 53,8%.
МС m/z (ESI): 586.2 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.33 (s, 1Н), 8.09 (t, 1Н), 7.61 (d, 1Н), 7.33-7.42 (m, 2Н), 7.17 (t, 1Н), 7.06 (d, 1Н), 6.69 (t, 1Н), 5.04 (s, 1Н), 3.39-3.46 (m, 1Н), 3.11-3.24 (m, 3Н), 3.19 (s, 3Н), 2.93-3.00 (m, 1Н), 2.06 (s, 3Н).
Пример 28
(S)-N-(2,3-дигидроксипропил)-4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
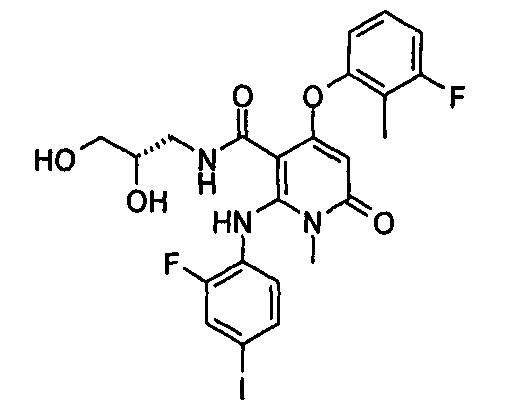
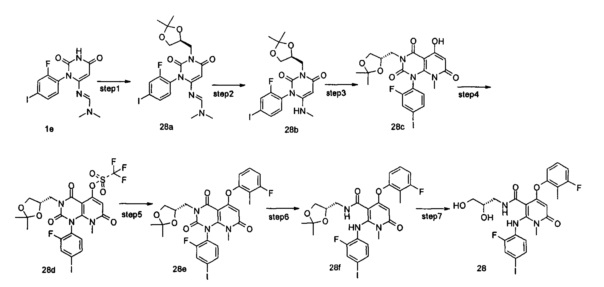
Стадия 1
(S,E)-N'-(3-(2-фтор-4-иодфенил)-1-((2,2-диметил-1,3-диоксолан-4-ил)метил)-2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-ил)-N,N-диметилформимидамид
Неочищенный (E)-N'-(3-(2-фтор-4-йодфенил)-2,6-диоксо-1,2,3,6-тетрагидро пиримидин-4-ил)-N,N-диметилформимидамид 1е (3,30 г, 8,20 ммоль), (S)-4-хлор-2,2-диметил-1,3-диоксолан (2,47 г, 16,40 ммоль), карбонат цезия (5,34 г, 16,40 ммоль) и йодид калия (200 мг, 1,20 ммоль) растворяли в 33 мл N,N-диметилформамида. Реакционный раствор нагревали до 100°С и перемешивали в течение 12 часов. После охлаждения до комнатной температуры в реакционный раствор добавляли 200 мл этилцетата. Органическую фазу промывали водой (100 мл×3), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (S,Е)-N'-(3-(2-фтор-4-иодфенил)-1-((2,2-диметил-1,3-диоксолан-4-ил)метил)-2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-ил)-N,N-диметилформимидамид 28а (3,80 г, темно-коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 517.0 [М+1]
Стадия 2
(S)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-(2-фтор-4-иодфенил)-6-(метиламино)пиримидин-2,4(1Н,3Н)-дион
Борогидрид натрия (418 мг, 11,04 ммоль) растворяли в 15 мл смеси этанола и трет-бутанола (V:V=1:2). После охлаждения до 0°С в реакционный раствор добавляли неочищенный (S,E)-N'-(3-(2-фтор-4-иодфенил)-1-((2,2-диметил-1,3-диоксолан-4-ил)метил)-2,6-диоксо-1,213,6-тетрагидропиримидин-4-ил)-N,N-диметилформимидамид 28а (3,80 г, 7,36 ммоль), и перемешивали в течение 1 часа. После нагревания до комнатной температуры реакционный раствор перемешивали в течение 12 часов, затем охлаждали до 0°С с последующим добавлением 100 мл воды. Органическую фазу промывали водой (50 мл×3), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (S)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-(2-фтор-4-иодфенил)-6-(метиламино)пиримидин-2,4(1Н,3Н)-дион 28b (3,35 г, коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 476.1 [М+1]
Стадия 3
(S)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-(2-фтор-4-иодфенил)-5-гидрокси-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион
Неочищенный (S)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-(2-фтор-4-иодфенил)-6-(метиламино)пиримидин-2,4(1Н,3Н)-дион 28b (3,35 г, 7,05 ммоль) растворяли в 35 мл диэтилового эфира малоновой кислоты. Реакционный раствор перемешивали в течение 1 часа с обратным холодильником, а затем концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюента В с получением указанного в заголовке соединения (S)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-(2-фтор-4-иодфенил)-5-гидрокси-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 28 с (300 мг, желтое твердое вещество), выход: 7,83%.
МС m/z (ESI): 544.0 [М+1]
Стадия 4
(S)-1-(2-фтор-4-иодфенил)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат
(S)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-(2-фтор-4-иодфенил)-5-гидрокси-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 28 с (300 мг, 0,55 ммоль) растворяли в 5 мл дихлорметана с последующим добавлением 2,6-лутидина (237 мг, 2,21 ммоль). После охлаждения до 0°С в реакционный раствор добавляли трифторметансульфоновый ангидрид (311 мг, 1,10 ммоль), затем нагревали до комнатной температуры и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюента В с получением указанного в заголовке соединения (S)-1-(2-фтор-4-иодфенил)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 28d (150 мг, светло-желтое твердое вещество), выход: 40,2%.
МС m/z (ESI): 676.1 [М+1]
Стадия 5
(S)-5-(3-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион
3-фтор-2-метилфенол (34 мг, 0,27 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (13 мг, 0,33 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли (S)-1-(2-фтор-4-иодфенил)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 28d (150 мг, 0,22 ммоль), затем нагревали до 60°С и перемешивали в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (S)-5-(3-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 28е (145 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 6
(S)-4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный (S)-5-(3-фтор-2-метилфенокси)-1-(2-фтор-4-иодфенил)-3-((2,2-диметил-1,3-диоксолан-4-ил)метил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(7Н,3Н,8Н)-трион 28e (145 мг, 0,22 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=2:1) с последующим добавлением гидроксида лития (186 мг, 4,44 ммоль) Реакционный раствор нагревали до 40°С и перемешивали в течение 1 часа с последующим добавлением 20 мл этилацетата. Органическую фазу промывали последовательно насыщенным раствором бикарбоната натрия (10 мл×3) и водой (20 мл×1), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (S)-4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-N-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 28f (139 мг, светло-коричневое твердое вещество), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 626.2 [М+1]
Стадия 7
(S)-N-(2,3-дигидроксипропил)-4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный (S)-4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил) амино)-N-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 28f (139 мг, 0,22 ммоль) растворяли в 10 мл тетрагидрофурана с последующим добавлением 5 мл 1М соляной кислоты. После перемешивания в течение 2 часов реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения (S)-N-(2,3-дигидроксипропил)-4-(3-фтор-2-метилфенокси)-2-((2-фтор-4-иодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 28 (70 мг, светло-коричневое твердое вещество), выход: 53,8%.
МС m/z (ESI): 586.2 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.33 (s, 1Н), 8.09 (t, 1Н), 7.61 (dd, 1Н), 7.33-7.42 (m, 2Н), 7.17 (t, 1Н), 7.06 (d, 1Н), 6.69 (t, 1Н), 5.04 (s, 1Н), 3.39-3.46 (m, 1Н), 3.11-3.24 (m, 3Н), 3.19 (s, 3Н), 2.93-3.00 (m, 1Н), 2.06 (s, 3Н).
Пример 29
2-((2-фтор-4-иодфенил)амино)-1-метил-4-((2-метилпиридин-3-ил)окси)-6-оксо-1,6-дигидропиридин-3-карбоксамид

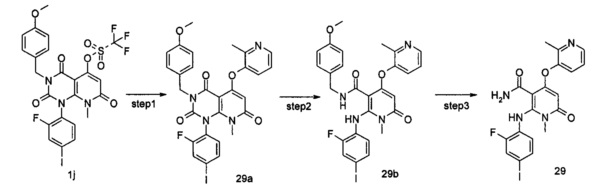
Стадия 1
5-((2-метилпиридин-3-ил)окси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 2-метил-3-гидроксипиридин (55 мг, 0,50 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (24 мг, 0,60 ммоль). После перемешивания в течение 1 часа в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (136 мг, 0,20 ммоль) и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-((2-метилпиридин-3-ил)окси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 29а (128 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2
4-((2-метилпиридин-3-ил)окси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-((2-метилпиридин-3-ил)окси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 29а (128 мг, 0,20 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=5:1) с последующим добавлением гидроксида лития (42 мг, 1 ммоль). После перемешивания в течение 4 часов в реакционный раствор добавляли 50 мл этилацетата и 10 мл воды. Водную фазу экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-((2-метилпиридин-3-ил)окси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 296 (123 г, красно-коричневое масло), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 615.1 [М+1]
Стадия 3
2-((2-фтор-4-иодфенил)амино)-1-метил-4-((2-метилпиридин-3-ил)окси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-((2-метилпиридин-3-ил)окси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 296 (123 мг, 0,20 ммоль) растворяли в 4 мл анизола с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 1 часа с последующим добавлением 50 мл этилацетата и 15 мл воды. Органическую фазу промывали 1М соляной кислотой (25 мл×3), сушили над безводным сульфатом натрия и фильтровали, фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 2-((2-фтор-4-иодфенил)амино)-1-метил-4-((2-метилпиридин-3-ил)окси)-6-оксо-1,6-дигидропиридин-3-карбоксамид 29 (9 мг, светло-коричневого твердого вещества), выход: 9,1%.
МС m/z (ESI): 495.1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.89 (s, 1Н), 8.43 (dd, 1Н), 7.59-7.70 (m, 4Н), 7.43 (dd, 1Н), 7.38 (dd, 1Н), 6.68 (t, 1Н), 5.00 (s, 1Н), 3.15 (s, 3Н), 2.35 (s, 3Н).
Пример 30
2-((2-фтор-4-иодфенил)амино)-1-метил-4-((5-метилпиридин-3-ил)окси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
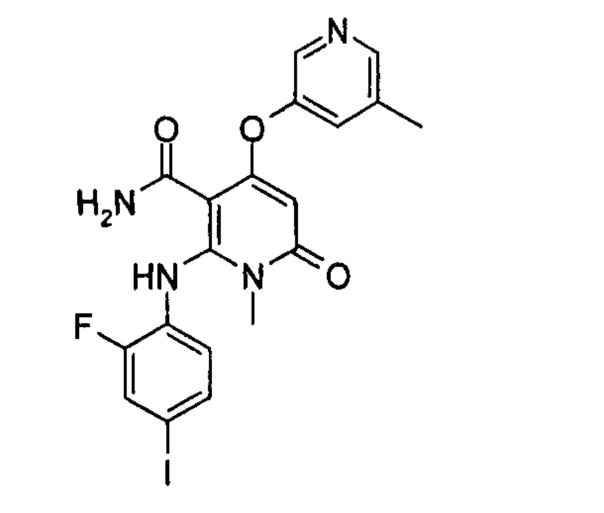
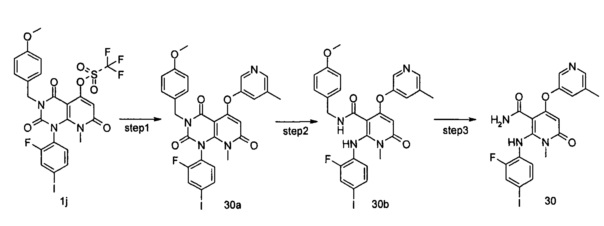
Стадия 1
5-((5-метилпиридин-3-ил)окси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 5-метил-3-гидроксипиридин (43 мг, 0,40 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (16 мг, 0,40 ммоль). После перемешивания в течение 1 часа в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (136 мг, 0,20 ммоль) и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-((5-метилпиридин-3-ил)окси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 30а (128 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2
4-((5-метилпиридин-3-ил)окси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-((5-метилпиридин-3-ил)окси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2|4,7(1Н,3Н,8Н)-трион 30а (128 мг, 0,20 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=4:1) с последующим добавлением гидроксида лития (42 мг, 1 ммоль). После перемешивания в течение 3 часов в реакционный раствор добавляли 50 мл этилацетата и 10 мл воды. Водную фазу экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл×3), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-((5-метилпиридин-3-ил)окси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 30b (122 мг, коричневое масло), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 615.0 [М+1]
Стадия 3
2-((2-фтор-4-иодфенил)амино)-1-метил-4-((5-метилпиридин-3-ил)окси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-((5-метилпиридин-3-ил)окси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 30b (122 мг, 0,20 ммоль) растворяли в 4 мл анизола с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 100°С, перемешивали в течение 4 часов с последующим добавлением 50 мл этилацетата и 15 мл воды. Органическую фазу промывали водой (25 мл×3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения для получения указанного в заголовке соединения 2-((2-фтор-4-иодфенил)амино)-1-метил-4-((5-метилпиридин-3-ил)окси)-6-оксо-1,6-дигидропиридин-3-карбоксамид 30 (18 мг, почти белого твердого вещества), выход: 18,2%.
МС m/z (ESI): 495.1 [М+1]
1H ЯМР (400 МГц, ДМСО-d6): δ 9.79 (s, 1Н), 8.36-8.38 (m, 2Н), 7.60-7.66 (m, 4Н), 7.42-7.44 (m, 1Н), 6.63-6.68 (m, 1Н), 5.15 (s, 1Н), 3.16 (s, 3Н), 2.36 (s, 3Н).
Пример 31
2-((2-фтор-4-иодфенил)амино)-1-метил-4-((6-метилпиридин-3-ил)окси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
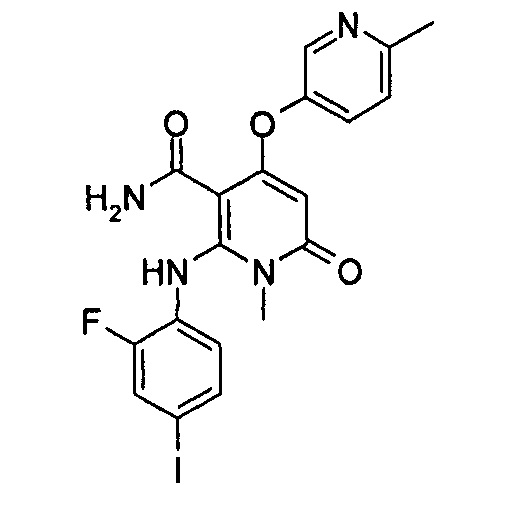
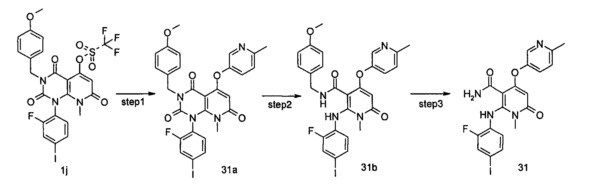
Стадия 1
5-((6-метилпиридин-3-ил)окси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1H,3Н,8H)-трион 6-метил-3-гидроксипиридин (26 мг, 0,24 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (12 мг, 0,30 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридол[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (136 мг, 0,20 ммоль), и нагревали до 60°С и перемешивали в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-((6-метилпиридин-3-ил)окси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 31а (128 мг, светло-желтая жидкость), которое использовали непосредственно на следующей стадии без дополнительной очистки.
МС m/z (ESI): 641.1 [М+1]
Стадия 2
4-((6-метилпиридин-3-ил)окси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-((6-метилпиридин-3-ил)окси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 31а (128 мг, 0,20 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=4:1), с последующим добавлением гидроксида лития (168 мг, 4 ммоль). Реакционный раствор нагревали до 40°С и перемешивали в течение 1 часа с последующим добавлением 50 мл этилацетата. Органическую фазу промывали 1М раствором гидроксида натрия (30 мл×3), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-((6-метилпиридин-3-ил)окси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 31b (123 мг, коричневое масло), которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 615.0 [М+1]
Стадия 3
2-((2-фтор-4-иодфенил)амино)-1-метил-4-((6-метилпиридин-3-ил)окси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-((6-метилпиридин-3-ил)окси)-2-((2-фтор-4-иодфенил) амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 31b (123 мг, 0,20 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 120°С и перемешивали в течение 4 часов с последующим добавлением 50 мл этилацетата и 15 мл воды. Органическую фазу промывали водой (25 мл×3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 2-((2-фтор-4-иодфенил)амино)-1-метил-4-((6-метилпиридин-3-ил)окси)-6-оксо-1,6-дигидропиридин-3-карбоксамид 31 (30 мг, светло-коричневое твердое вещество), выход: 30,3%.
МС m/z (ESI): 495.0 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.78 (s, 1Н), 8.38-8.44 (m, 1Н), 7.57-7.75 (m, 4Н), 7.35-7.49 (m, 2Н), 6.65 (t, 1Н), 5.09 (s, 1Н), 3.15 (s, 3Н), 2.51 (s, 3Н).
Пример 32
2-((2-фтор-4-иодфенил)амино)-1-метил-4-((2-метилпиридин-4-ил)окси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
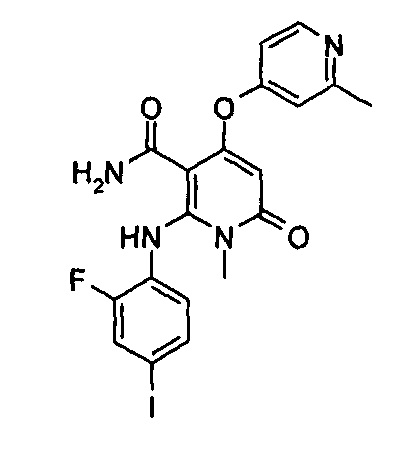
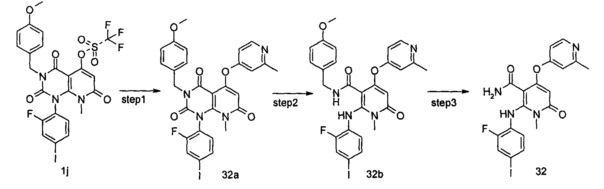
Стадия 1
5-((2-метилпиридин-4-ил)окси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 2-метил-4-гидроксипиридин (21 мг, 0,19 ммоль) растворяли в 10 мл тетрагидрофурана с последующим добавлением гидрида натрия (7 мг, 0,29 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (130 мг, 0,19 ммоль). После перемешивания в течение 12 часов реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-((2-метилпиридин-4-ил)окси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8H)-триона 32а (122 мг, светло-желтая жидкость), который непосредственно использовали на следующей стадии без дальнейшей очистки.
Стадия 2
4-((2-метилпиридин-4-ил)окси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-((2-метилпиридин-4-ил)окси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 32а (122 мг, 0,19 ммоль) растворяли в 6 мл смеси тетрагидрофурана и воды (V:V=4:1), с последующим добавлением гидроксида лития (80 мг, 1,91 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 50 мл этилацетата и 10 мл воды. Водную фазу экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл×3), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-((2-метилпиридин-4-ил)окси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамида 32b (140 г, желтое масло), которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 3
2-((2-фтор-4-иодфенил)амино)-1-метил-4-((2-метилпиридин-4-ил)окси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-((2-метилпиридин-4-ил)окси)-2-((2-фтор-4-иодфенил) амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 32b (140 мг, 0,19 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (127 мг, 0,96 ммоль). Реакционный раствор нагревали до 100°С и перемешивали в течение 3 ч с последующим добавлением 50 мл этилацетата и 15 мл воды. Органическую фазу промывали водой (25 мл×3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения с получением указанного в заголовке соединения 2-((2-фтор-4-иодфенил)амино)-1-метил-4-((2-метилпиридин-4-ил)окси)-6-оксо-1,6-дигидропиридин-3-карбоксамид 32 (5 мг, желтое твердое вещество), выход: 5,3%.
МС m/z (ESI): 495.1 [М+1]
Пример 33
2-((2-фтор-4-иодфенил)амино)-1-метил-4-((4-метилпиридин-3-ил)окси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
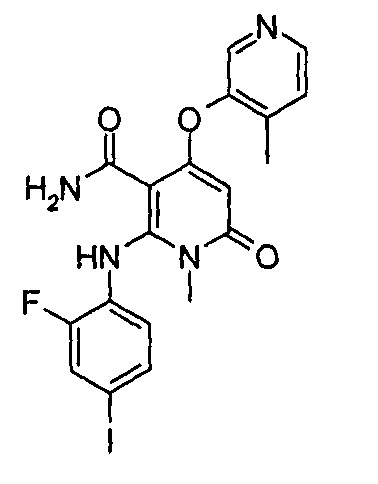
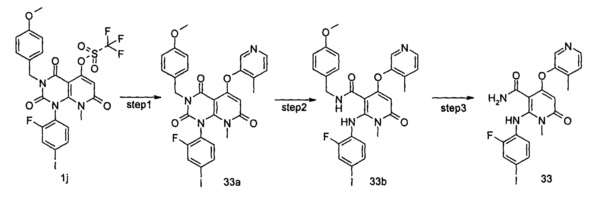
Стадия 1
5-((4-метилпиридин-3-ил)окси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1H,3Н,8Н)-трион
4-метил-3-гидроксипиридин (44 мг, 0,40 ммоль) и 1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метил-2,4,7-триоксо-пиридо[2,3-d]пиримидин-5-ил трифторметансульфонат 1j (140 мг, 0,20 ммоль) растворяли в 5 мл тетрагидрофурана с последующим добавлением гидрида натрия (24 мг, 0,60 ммоль). После перемешивания в течение 1 часа, в реакционную смесь добавляли и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-((4-метилпиридин-3-ил)окси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 33a (128 мг, желтая жидкость), которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 2
4-((4-метилпиридин-3-ил)окси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 5-((4-метилпиридин-3-ил)окси)-1-(2-фтор-4-иодфенил)-3-(4-метоксибензил)-8-метилпиридо[2,3-d]пиримидин-2,4,7(1Н,3Н,8Н)-трион 33а (128 мг, 0,20 ммоль) растворяли в 12 мл смеси тетрагидрофурана и воды (V:V=6:1) с последующим добавлением гидроксида лития (84 мг, 2 ммоль). После перемешивания в течение 2 часов в реакционный раствор добавляли 50 мл этилацетата и 10 мл воды. Водную фазу экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл×3), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4-((4-метилпиридин-3-ил)окси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 336 (122 г, желтое масло), которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 3
2-((2-фтор-4-иодфенил)амино)-1-метил-4-((4-метилпиридин-3-ил)окси)-6-оксо-1,6-дигидропиридин-3-карбоксамид
Неочищенный 4-((4-метилпиридин-3-ил)окси)-2-((2-фтор-4-иодфенил)амино)-N-(4-метоксибензил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид 336 (122 мг, 0,20 ммоль) растворяли в 5 мл анизола с последующим добавлением хлорида алюминия (133 мг, 1 ммоль). Реакционный раствор нагревали до 100°С и перемешивали в течение 12 ч с последующим добавлением 50 мл дихлорметана и 15 мл воды. Органическую фазу промывали водой (25 мл×3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью препаративного способа разделения для получения указанного в заголовке соединения 2-((2-фтор-4-иодфенил)амино)-1-метил-4-((4-метилпиридин-3-ил)окси)-6-оксо-1,6-дигидропиридин-3-карбоксамид 33 (31 мг, бледно-желтого твердого вещества),
выход: 31,3%.
МС m/z (ESI): 495.1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.88 (s, 1Н), 8.59 (s, 1Н), 8.51-8.53 (d, 1Н), 7.60-7.70 (m, 4Н), 7.43-7.46 (dd, 1Н), 6.68-6.73 (t, 1Н), 5.14 (s, 1Н), 3.17 (s, 3Н), 2.27 (s, 3Н).
ТЕСТОВЫЕ ПРИМЕРЫ:
Биологическая оценка Тестовый пример 1. Исследование для определения активности соединений по настоящему изобретению в отношении MEK1
Активность киназы MEK1 тестировали in vitro с помощью следующего способа.
Киназа MEK-, использованная в исследовании: MEK1 (Рекомбинантный белок человека, Invitrogen, каталожный №PV3093).
Набор, использованный в исследовании: Z'-LYTE™ Kinase Assay Kit-Ser/Thr 03 Peptide (Invitrogen, каталожный No. PV3176).
Следующее исследование in vitro проводили с целью определения активности соединений по настоящему изобретению в отношении ингибирования пролиферации киназы MEK. Исследуемое соединение растворяли в диметилсульфоксиде до желаемой концентрации. Был приготовлен 1×Буфер A (Invitrogen, каталожный №PV3189), АТФ (аденозинтрифосфат) разбавляли 1×Буфера А с получением 400 мкМ раствор АТФ, смешивали соответствующие количества пептида Z'-LYTE™ Ser/Thr 03 Peptide (Invitrogen, каталожный №PV3200), ферментов киназы MEK (MEK1) и 1×буфера А, и соответствующие количества субстрата Z'-LYTE™ Ser/Thr 03 phosphoPeptide (Invitrogen, каталожный №PV3215) и 1×буфера А приводили в смесь для испытания. Из 1×буфера и ДМСО раствора исследуемого соединения был приготовлен 4% раствор ДМСО исследуемого соединения. В реакционную лунку добавляли 2,5 мкл ДМСО раствора испытуемого соединения, и затем 1×Буфер А, 2,5 мкл 400 мкМ раствора АТФ, 5 мкл фермента и смесь субстратов добавляли таким образом, чтобы получить 10 мкл реакционной системы. Реакционную систему инкубировали в течение 4 часов при 37°С, и смесь готовили в соответствии с Реагентом А: Буфер В=1:1024. В реакционную лунку добавляли 5 мкл смеси Реагента A (Invitrogen, каталожный №PV3295) и Буфера В (Invitrogen, каталожный №Р3127), реакционную систему инкубировали в течение 60 мин при 25°С, и флуоресценцию считывали с помощью Novostar ELIASA с длиной волны возбуждения: 400 нм, длиной волны эмиссии: 445 нм и 520 нм.
Активность соединений по настоящему изобретению: Вышеуказанное исследование использовали для определения биохимической активности соединений по настоящему изобретению в отношении ингибирования киназы MEK (MEK1). Величины IC50 (концентрации полумаксимального ингибирования) приведены в таблице 1.
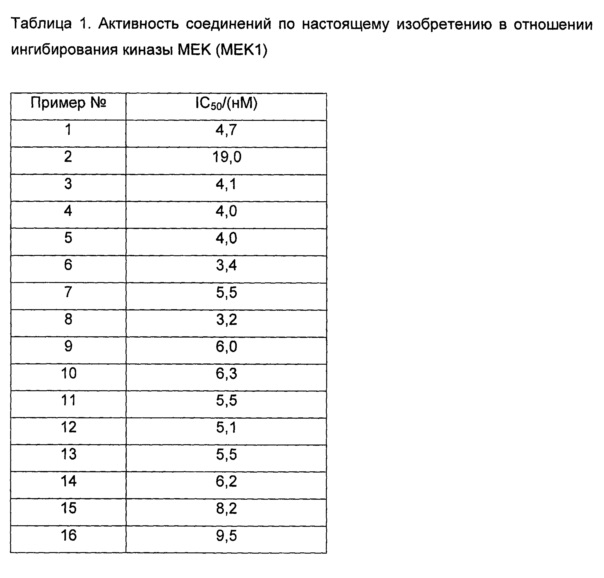
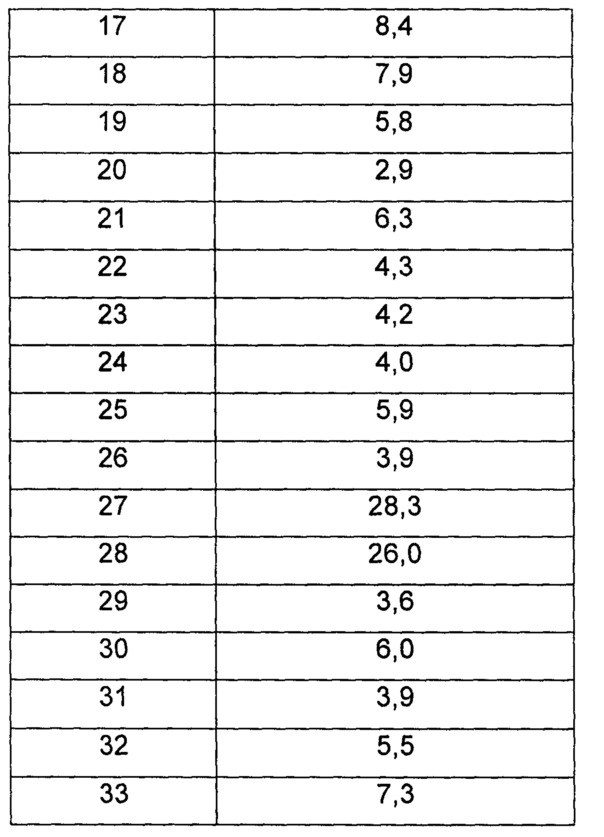
Заключение: Соединения по настоящему изобретению обладают значительной активностью в отношении ингибирования MEK1.
Тестовый пример 2. Исследование для определения активности соединений по настоящему изобретению в отношении ингибирования пролиферации MEK2
Активность киназы MEK2 исследовали in vitro с помощью следующего метода.
Киназа MEK-, использованная в анализе:
MAP2K2 (MEK2) Рекомбинантный белок человека (Invitrogen, каталожный №PV3615)
MAPK1 (ERK2) Рекомбинантный белок человека (Invitrogen, каталожный №PV3314).
Набор, использованный в исследовании: Z'-LYTE™ Kinase Assay Kit-Ser/Thr 03 Peptide (Invitrogen, каталожный №PV3176).
Следующий анализ in vitro проводили с целью определения активности соединений по настоящему изобретению в отношении ингибирования пролиферации киназы MEK. Исследуемое соединение растворяли в диметилсульфоксиде до желаемой концентрации. Готовили 1×буфер А (Invitrogen, каталожный №PV3189), АТФ разбавляли 1×буфером А для получения 400 мкМ раствора АТФ, смешивали соответствующее количество пептида Z'-LYTE™ Ser/Thr 03 Peptide (Invitrogen, каталожный №PV3200), ферментов киназы MEK (MEK 2), ферментов (ERK2) и 1×буфера А, и соответствующие количества субстрата Z'-LYTE™ Ser/Thr 03 phosphoPeptide (Invitrogen, каталожный №PV3215) и 1×буфера А приводили в смесь для испытания. Из 1×буфера и ДМСО раствора исследуемого соединения готовили 4% раствор ДМСО исследуемого соединения. В реакционную лунку добавляли 2,5 мкл ДМСО раствора исследуемого соединения, и затем 2,5 мкл 400 мкМ раствора АТФ, 5 мкл фермента и субстратной смеси для получения 10 мкл реакционной системы. Реакционную систему инкубировали в течение 1,5 ч при температуре 25°С, и смесь готовили в соответствии с реагентом А: Буфер В=1:1024. В реакционную лунку добавляли 5 мкл смеси реагента A (Invitrogen, каталожный №PV3295) и буфера В (Invitrogen, позиция №Р3127), реакционную систему смесь инкубировали в течение 60 мин при 25°С, и затем флуоресценцию считывали с помощью Novostar ELIASA с длиной волны возбуждения: 400 нм, длиной волны эмиссии: 445 нм и 520 нм.
Приведенный выше анализ использовали для определения биохимической активности соединений по настоящему изобретению в отношении ингибирования киназы MEK (MEK2). Величины IC50 приведены в таблице 3.
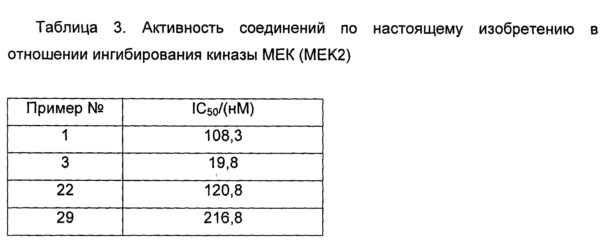
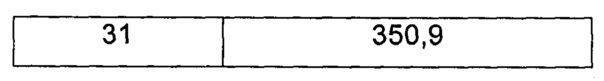
Заключение: Соединения по настоящему изобретению обладают значительной активностью в отношении ингибирования MEK2.
Тестовый пример 3. Исследование для определения активности соединений по настоящему изобретению в отношении ингибирования пролиферации Colo205
Клеточная линия, использованная в исследовании: Colo205 (Клеточный банк Китайской Академии наук, каталожный №. TCHu102).
Следующее in vitro исследование на клетках проводили для определения активности соединений по настоящему изобретению в отношении ингибирования пролиферации клеток рака толстой кишки человека, и активность может быть выражена в IC50. Общие программы такого рода исследования заключались в следующем: Во-первых, исследуемую клеточную линию (приобретенную в Клеточном банке Китайской Академии наук) засевали в 96-луночный культуральный планшет с подходящей концентрацией клеток, составляющей 4000 клеток/мл среды, и культивировали в инкубаторе с диоксидом углерода при 37°С, а затем растили в течение ночи. Среду заменяли новой средой с добавлением серии концентраций (10000 нМ, 1000 нМ, 100 нМ, 10 нМ, 1 нМ, 0,1 нМ) растворов исследуемого соединения. Планшеты снова помещали в инкубатор и непрерывно культивировали в течение 72 часов. Через 72 часов, способ CCK8 (Cell Counting Kit-8, Набор для подсчета клеток-8, каталожный №CK04, приобретен у коллег в химической компании) был использован для определения ингибирующей пролиферацию активности испытуемых соединений. Значения IC50 рассчитывали по данным по ингибированию для серии исследуемых соединений в различных концентрациях.
Приведенное выше исследование использовали для определения биохимической активности соединений по настоящему изобретению. Значения IC50 приведены в таблице 2.
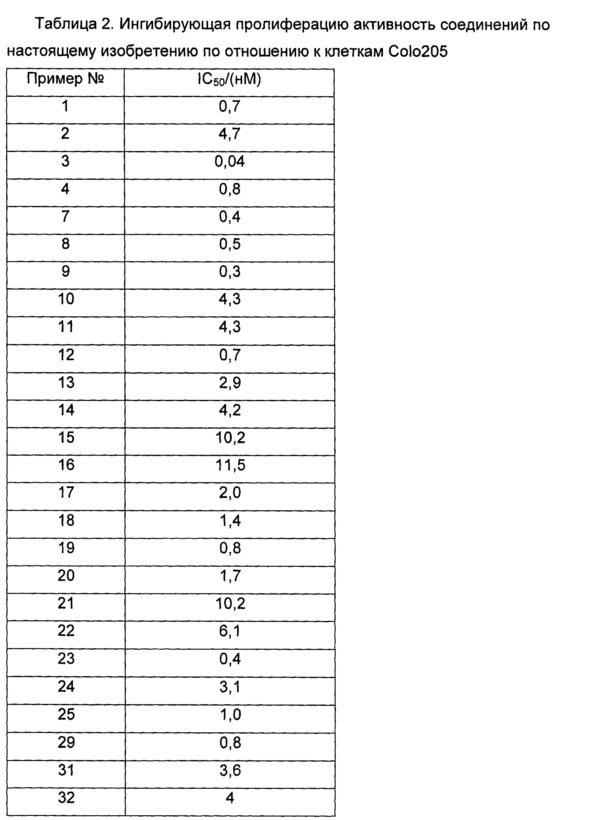
Заключение: Соединения по настоящему изобретению обладают значительной активностью в отношении ингибирования пролиферации клеток Colo205.
Тестовый пример 4. Исследование для определения активности соединений по настоящему изобретению в отношении ингибирования пролиферацииклеток рака толстой кишки человека НСТ116
Следующее in vitro исследование на клетках проводили с целью определения активности соединений по настоящему изобретению в отношении ингибирования пролиферации клеток рака толстой кишки человека, и активность может быть выражена с помощью IC50.
Клеточная линия, использованная в эксперименте: НСТ116 (Клеточный банк Китайской Академии наук, каталожный №TCHu99).
Общие программы исследования заключались в следующем: Во-первых, исследуемую клеточную линию засевали в 384-луночный культуральный планшет с подходящей концентрацией клеток, составляющей 1000 клеток/лунка, и культивировали в инкубаторе в условиях 37°С и 5% СО2, затем растили в течение ночи. Среду заменяли новой средой с добавлением серии концентраций (1000 нм, 250 нм, 62,50 нм, 15,63 нм, 3,91 нмоль, 0.98 nM, 0,24 нМ, 0,06 нМ, 0,015 нм, 0,004 нМ) растворов исследуемого соединения. Планшеты снова помещали в инкубатор и непрерывно культивировали в течение 72 часов. Через 72 часа способ CCK8 (Cell Counting Kit-8, набор для подсчета клеток-8, каталожный №:CK04, приобретенный у коллег в химической компании) использовали для определения ингибирующей пролиферацию активности исследуемых соединений. Значения IC50 рассчитывали из данных по ингибированию для серии исследуемых соединений в различных концентрациях.
Приведенное выше исследование использовали для определения биохимической активности соединений по настоящему изобретению. Величины IC50 приведены в таблице 4.

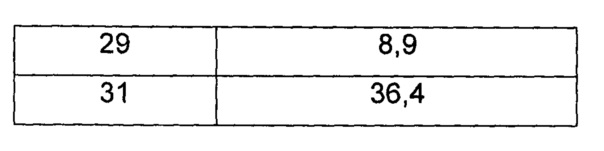
Заключение: Соединения по настоящему изобретению обладают значительной ингибирующей пролиферацию активностью в отношении клеток НСТ116.
In vitro оценка ингибирующей активности соединений по настоящему
изобретению в отношении фермента цитохрома Р450 Тестовый пример 5: In vitro исследование для определения ингибирующей активности соединений по Примеру 1, Примеру 3, Примеру 4, Примеру 22, Примеру 29, Примеру 30 и Примеру 31 настоящего изобретения в отношении фермента цитохрома Р450
1. Резюме
Система инкубации микросом печени человека была использована для отображения активности ферментов посредством количества продуцируемых метаболитов. Ферментное ингибирование испытуемых соединений исследовали на CYP1A2, CYP2C9, CYP2C6, CYP3A4m, CYP3A4t и CYP2C19, и измеряли величины IC50 (концентрации исследуемого соединения, необходимой для 50% ингибирования активности фермента).
2. Протокол
2.1 Образцы
Соединения по Примеру 1, Примеру 3, Примеру 4, Примеру 22, Примеру 29, Примеру 29, Примеру 30 и Примеру 31.
2.2 Материалы
2.2.1. Получение натрий-фосфатного буфера (PBS, от англ. phosphate buffered saline)
18,303 г K2HPO4, 2,695 г KH2PO4, 11,175 г KCl и 372,2 мг ЭДТА (этилендиаминтетрауксусной кислоты) взвешивали и разводили сверхчистой водой до 1000 мл для натрий-фосфатного буфера с рН 7,4 (ЭДТА 1 мМ, KCl 0,15 М), и PBS хранили в холодильнике при температуре 4°С.
2.2.2. Взвешивание и приготовление НАДФ (никотинамидадениндинуклеотидфосфата)
Приготовление раствора 40 мМ НАДФ: стандартный образец 100 мг НАДФ (молекулярная масса составляла 833,4 г/моль) взвешивали и растворяли в 3 мл PBS, а затем раствор равномерно смешивали.
2.2.3. Получение PBS раствора микросом печени
Приготовление 0,25 мг/мл раствора микросом печени: микросомы печени человека (20 мг/мл) разводили PBS до 0,25 мг/мл.
2.2.4. Приготовление реакционного раствора исследуемого соединения Соответствующее количество стандартного образца исследуемого
соединения взвешивали и разбавляли ДМСО до 50 мМ с получением исходного раствора I. Исходный раствор I разбавляли PBS до 100 мкМ с получением реакционной жидкости, которую использовали при инкубации.
2.2.5. Получение маркерного субстрата цитохрома Р450 и селективного ингибитора
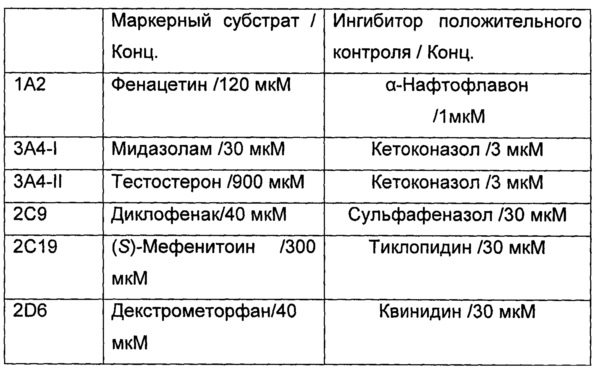
Конечную концентрацию маркерного субстрата и ингибиторы положительного контроля готовили с PBS.
3. Способ
Приготовленная реакционная смесь: 60 мкл

Исследуемое соединение/ ингибиторы положительного контроля 10 мкл
Вышеуказанную смесь предварительно инкубировали в течение 5 минут при 37°С с последующим добавлением 40 мкл НАДФ (2,5 мМ, раствор в PBS). Раствор смеси инкубировали в течение 20 минут при 37°С. Все инкубированные образцы были сделаны в двойных пробах. Для окончания реакции добавляли 300 мкл ледяного ацетонитрила. К реакционному раствору добавляли 100 мкл внутреннего стандарта, равномерно смешивали и центрифугировали в течение 10 минут при 3500 об./мин. Супернатант направляли на ЖХ(жидкостная хроматография)-МС/МС анализ.
4. Анализ данных
Активность ферментов отражалась посредством количества продуцируемых метаболитов. С применением метода одной точки формулу рассчитывали следующим образом:
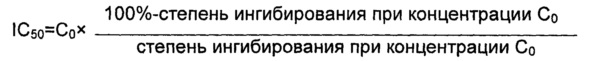
[предположительно коэффициент Хилла = 1] Со соответствует концентрации тестируемого соединения В соответствии с известными литературными данными: IC50>10 мкМ относится к слабому ингибированию, 1 мкМ<IC50<10 мкМ относится к умеренному ингибированию, IC50<1 мкМ относится к сильному ингибированию.
5. Результаты ингибирования фермента цитохрома Р450 in vitro Результаты ингибирования фермента цитохрома Р450 соединениями по изобретению in vitro показаны ниже.
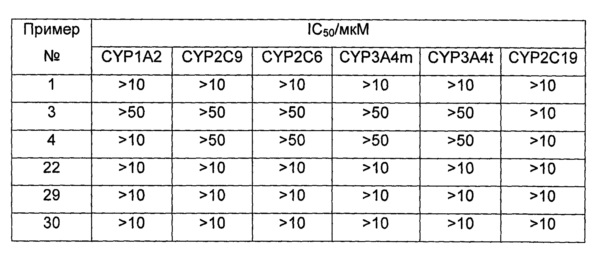

Предпочтительные соединения по настоящему изобретению имели более слабое ингибирование по отношению к ферментам CYP1A2, CYP2C9, CYP2C6, CYP3A4m, CYP3A4t и CYP2C19. Соединения по настоящему изобретению, таким образом, имели меньшую вероятность влияния лекарственных препаратов на метаболизм при клиническом введении.
Фармакокинетическое исследование Тестовый пример 6. Фармакокинетическое исследование соединений по Примеру 1, Примеру 22, Примеру 29 и Примеру 31 настоящего изобретения
1. Резюме
В качестве подопытных животных использовали крыс линии Спрег-Доули (SD, от англ. Sprague Dawley). Соединения по Примеру 1, Примеру 22, Примеру 29 и Примеру 31 крысам вводили внутрижелудочно, чтобы определить концентрацию лекарственного средства в плазме крови в различные моменты времени с помощью метода ЖХ/МС-МС (метод высокоэффективной жидкостной хроматографии с тандемной масс-спектрометрией). Фармакокинетическое поведение соединений по настоящему изобретению изучали и оценивали на крысах.
2. Протокол 2.1 Образцы
Соединения по Примеру 1, Примеру 22, Примеру 29 и Примеру 31. 2.2 Подопытные животные
16 здоровых взрослых крыс SD, половина самцов и половина самок, приобретенных у SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., СО Сертификат №: SCXK (Shanghai) 2008-0016, были разделены на четыре группы, по 4 крысы в каждой группе.
2.3 Приготовление исследуемых соединений
Соответствующие количества исследуемых соединений взвешивали и смешивали посредством ультразвукового метода с 40 мкл Tween 80 (полисорбат-80) и 0,5% Na-КМЦ (натриевая соль карбоксиметилцеллюлозы) с получением суспензии 1 мг/мл. 2.4 Введение
После голодания в течение ночи 16 крыс SD, половина самцы, половина самки, были разделены на 4 группы, по 4 крысы в каждой группе, и им вводили внутрижелудочно исследуемые соединения в дозе 10 мг/кг, и объем введения составлял 10 мл/кг.
3. Способ
Образцы крови (0,1 мл) отбирали из орбитального синуса перед введением и через 0,5 ч, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 11 ч, 24 ч и 48 ч после введения, хранили в пробирках с гепарином и центрифугировали в течение 10 минут при 3500 об./мин для отделения плазмы крови. Образцы плазмы хранили при -20°С. Крыс кормили через 2 часа после введения.
Концентрация исследуемых соединений в плазме крови крыс после внутрижелудочного введения исследуемых соединений анализировали способом ЖХ/МС-МС. Диапазон линейности метода составил 1.00-2000 нг/мл, а нижний предел количественного определения составил 1,00 нг/мл. Образцы плазмы анализировали после осаждения белка.
4. Полученные фармакокинетические параметры
Были получены следующие фармакокинетические параметры соединений по настоящему изобретению:

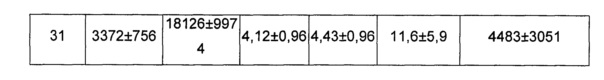
Заключение: Соединения по настоящему изобретению обладают хорошей фармакокинетической абсорбцией и значительным преимуществом фармакокинетических свойств.
Изобретение относится к производным пиридилкетона формулы (I) или его фармацевтически приемлемым солям:
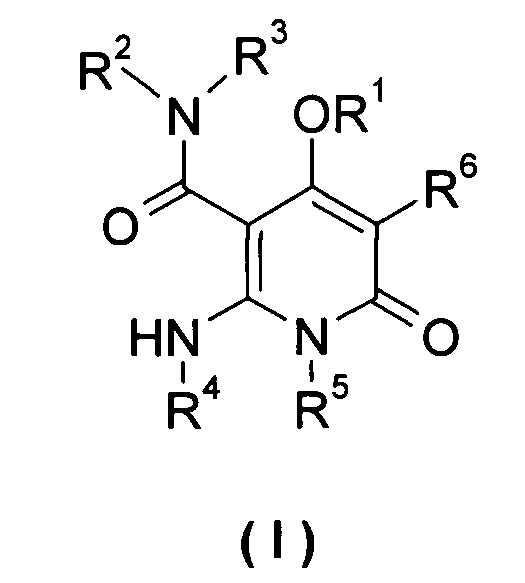
где: R1 выбран из группы, состоящей из фенила и пиридила, где независимо друг от друга каждый из фенила и пиридила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, алкила, галогеналкила, -OR7, -C(O)R7, -C(O)NHR7, -NHC(O)R7, -NHC(O)OR7 и -NHS(O)mR7; R2 представляет собой водород; R3 выбран из группы, состоящей из водорода и алкила, где алкил необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из -OR7; R4 представляет собой фенил, где фенил необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена и алкила; R5 выбран из группы, состоящей из водорода и алкила; R6 выбран из группы, состоящей из водорода, галогена и алкила; R7 выбран из группы, состоящей из водорода, алкила, С3-6циклоалкила, 5- или 6-членного гетероциклила, где независимо друг от друга каждый из указанных радикалов необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из алкила и галогена; где гетероциклил содержит один или два гетероатома, выбранные из группы, состоящей из N, О и S(O)m; независимо друг от друга каждый из R8 и R9 выбран из группы, состоящей из водорода или алкила; и m равно 2, и их применению в качестве ингибиторов MEK, и особенно в качестве терапевтических средств для лечения рака. 8 н. и 4 з.п. ф-лы, 4 табл., 33 пр.
1. Соединение формулы (I) или его фармацевтически приемлемая соль:
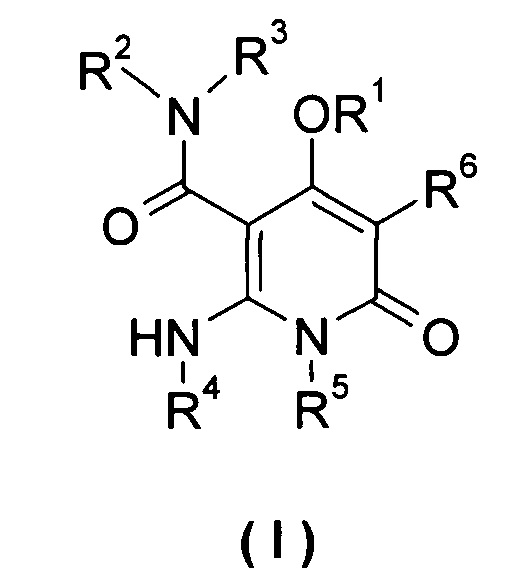
где:
R1 выбран из группы, состоящей из фенила и пиридила, где независимо друг от друга каждый из фенила и пиридила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, алкила, галогеналкила, -OR7, -C(O)R7, -C(O)NHR7, -NHC(O)R7, -NHC(O)OR7 и -NHS(O)mR7;
R2 представляет собой водород;
R3 выбран из группы, состоящей из водорода и алкила, где алкил необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из -OR7;
R4 представляет собой фенил, где фенил необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена и алкила;
R5 выбран из группы, состоящей из водорода и алкила;
R6 выбран из группы, состоящей из водорода, галогена и алкила;
R7 выбран из группы, состоящей из водорода, алкила, С3-6циклоалкила, 5- или 6-членного гетероциклила, где независимо друг от друга каждый из указанных радикалов необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из алкила и галогена;
где гетероциклил содержит один или два гетероатома, выбранные из группы, состоящей из N, О и S(O)m;
независимо друг от друга каждый из R8 и R9 выбран из группы, состоящей из водорода или алкила; и m равно 2.
2. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где R6 выбран из группы, состоящей из водорода и галогена.
3. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, представляющее собой соединение формулы (II):
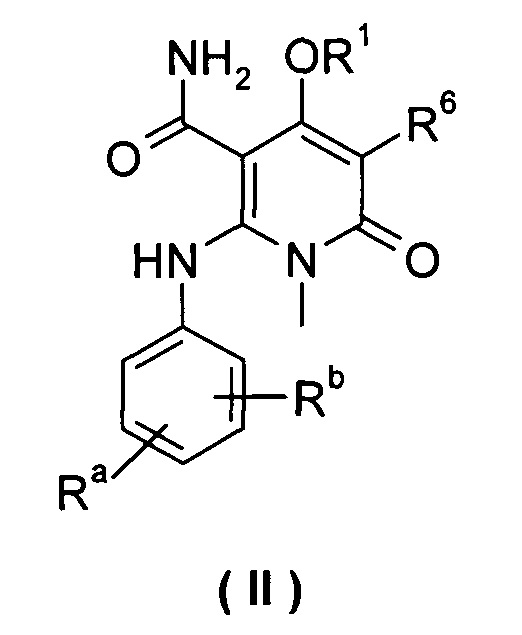
каждый из Ra и Rb выбран из группы, состоящей из водорода, галогена и алкила;
R1 выбран из группы, состоящей из фенила и пиридила, где каждый из фенила и пиридила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из алкила, галогена, галогеналкила, -OR7, -C(O)NHR7, -NHC(O)R7, - NHC(O)OR7 и -NHS(O)mR7;
R6 выбран из группы, состоящей из водорода, галогена и алкила; и
R7 выбран из группы, состоящей из водорода, алкила, С3-6циклоалкила, 5- или 6-членного гетероциклила, где независимо друг от друга каждый из указанных радикалов необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из алкила и галогена,
где гетероциклил содержит один или два гетероатома, выбранные из группы, состоящей из N, О и S(O)m.
4. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где соединение выбрано из группы, состоящей из:
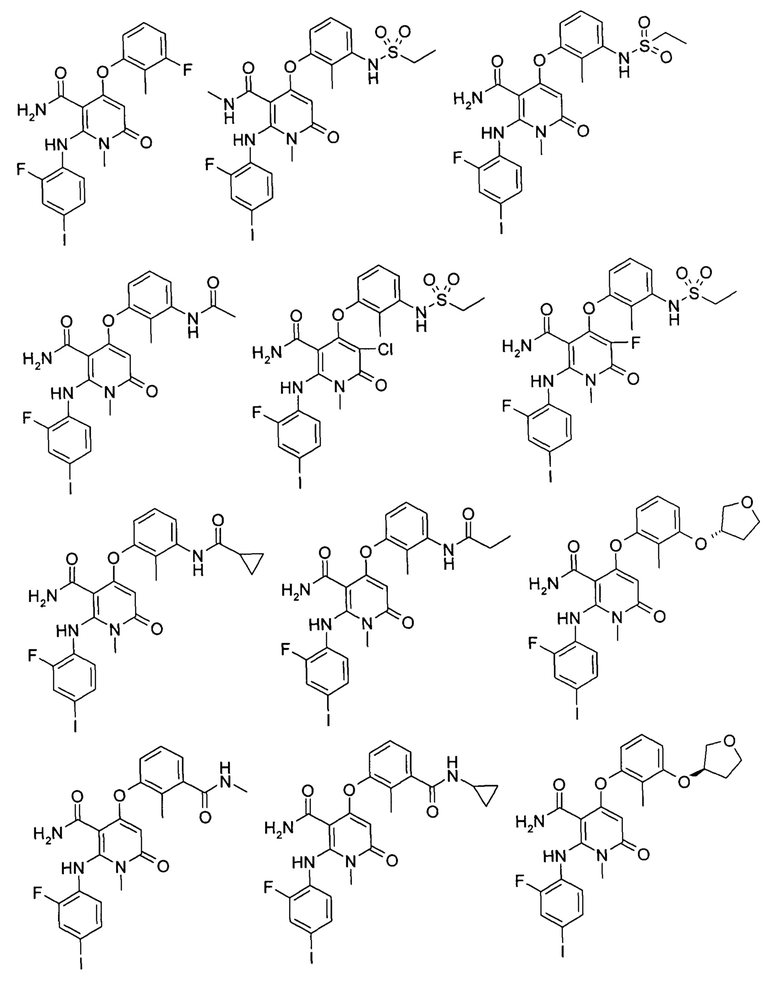
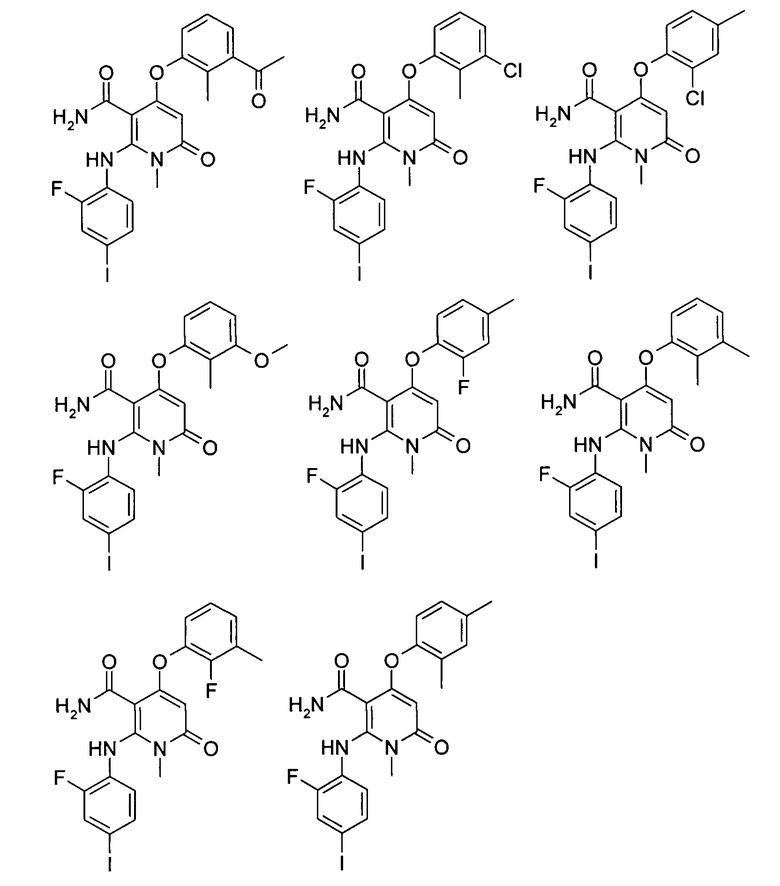

5. Соединение формулы (IA) или его фармацевтически приемлемая соль:
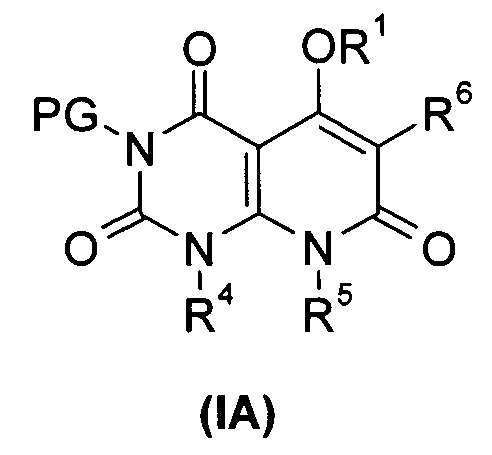
где:
R1, R4, R5, и R6 являются такими, как определено в п. 1;
PG выбран из группы, состоящей из алкила и бензила; каждый из алкила и бензила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, циано-, нитро-, алкила, циклоалкила, гетероциклила, гетероарила и -OR7; и
R7 выбран из группы, состоящей из водорода, алкила, С3-6циклоалкила, 5- или 6-членного гетероциклила, где независимо друг от друга каждый из указанных радикалов необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из алкила и галогена,
где гетероциклил содержит один или два гетероатома, выбранные из группы, состоящей из N, О и S(O)m.
6. Соединение формулы (IA) по п. 5, где соединение выбрано из группы, состоящей из:
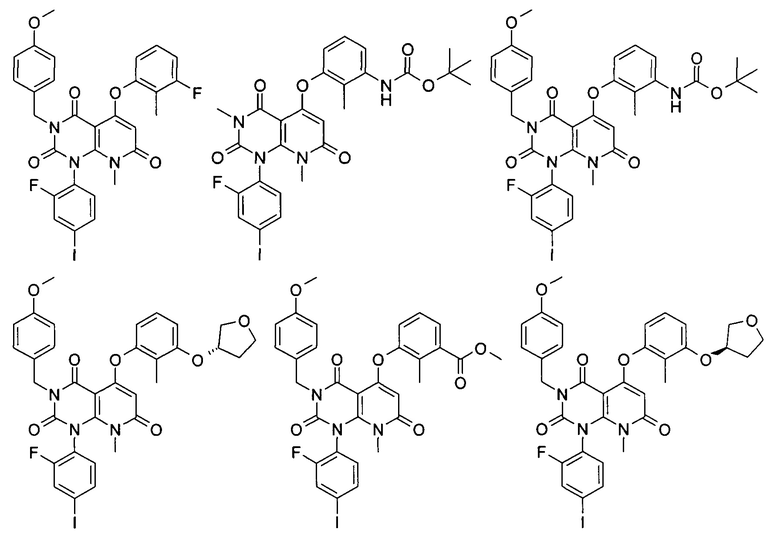
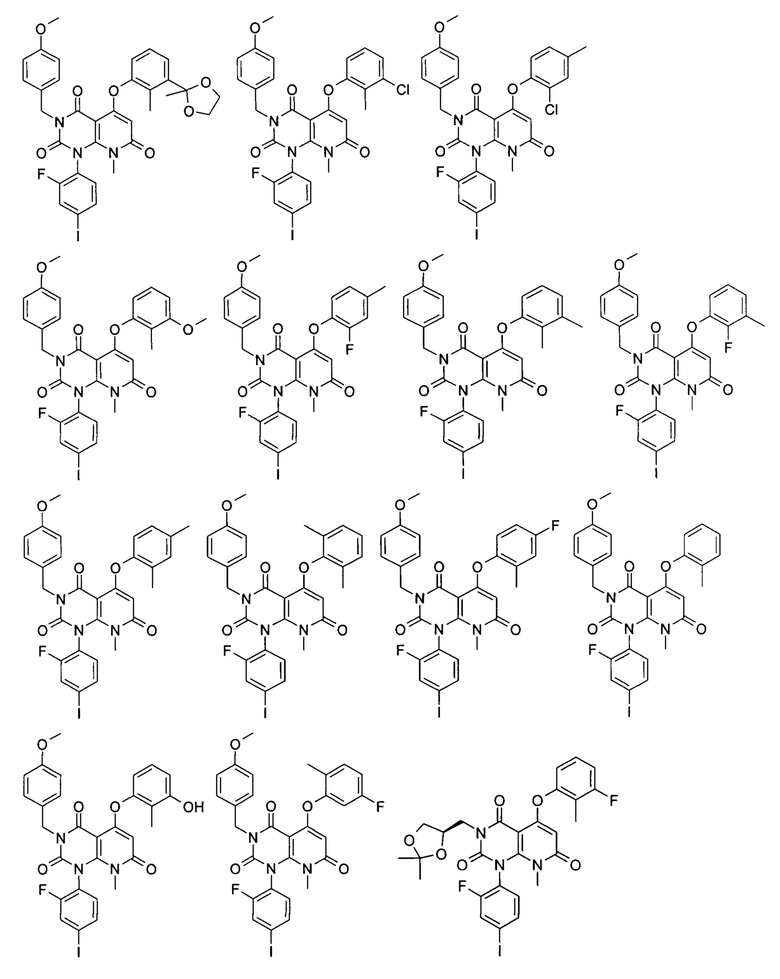
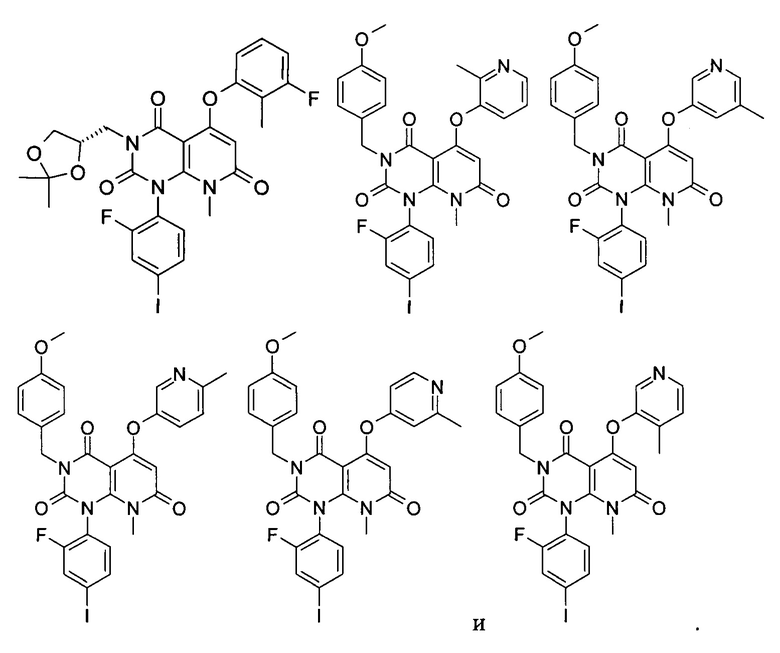
7. Способ получения соединения формулы (I) по п. 1 или его фармацевтически приемлемой соли, включающий стадию:
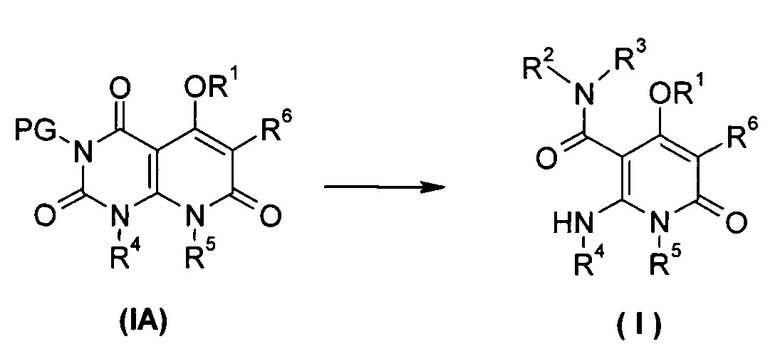
раскрытия кольца соединения формулы (IA) в щелочных условиях, необязательного удаления амино-защитной группы PG, с получением соединения формулы (I);
где: R1-R6 являются такими, как определено в п. 1;
PG выбран из группы, состоящей из алкила и бензила; каждый из алкила и бензила необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, циано-, нитро-, алкила, циклоалкила, гетероциклила, арила, гетероарила и -OR7; и
R7 выбран из группы, состоящей из водорода, алкила, С3-6циклоалкила, 5- или 6-членного гетероциклила, где независимо друг от друга каждый из указанных радикалов необязательно замещен одной или несколькими группами, выбранными из группы, состоящей из алкила и галогена, где гетероциклил содержит один или два гетероатома, выбранные из группы, состоящей из N, О и S(O)m.
8. Фармацевтическая композиция, обладающая активностью ингибитора MEK, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-4, и фармацевтически приемлемый носитель, разбавитель или наполнитель.
9. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-4, или фармацевтической композиции по п. 8 для получения лекарственного средства для ингибирования MEK.
10. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-4, или фармацевтической композиции по п. 8 для получения лекарственного средства для лечения рака, где рак выбран из группы, состоящей из меланомы, опухоли головного мозга, рака пищевода, рака желудка, рака печени, рака поджелудочной железы, колоректального рака, рака легкого, рака почки, рака молочной железы, рака яичников, рака предстательной железы, рака кожи, нейробластомы, саркомы, остеохондромы, остеомы, остеосаркомы, семиномы, рака яичка, рака матки, рака головы и шеи, множественной миеломы, злокачественной лимфомы, истинной полицитемии, лейкемии, рака щитовидной железы, рака мочеточника, рака мочевого пузыря, рака желчного пузыря, холангиокарциномы, хориокарциномы и педиатрической опухоли, предпочтительно колоректального рака или рака легкого.
11. Способ ингибирования активности MEK, включающий стадию введения субъекту, нуждающемуся в таком введении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-4, или фармацевтической композиции по п. 8.
12. Способ лечения рака, включающий стадию введения субъекту, нуждающемуся в таком введении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-4, или фармацевтической композиции по п. 8, где рак выбран из группы, состоящей из меланомы, опухоли головного мозга, рака пищевода, рака желудка, рака печени, рака поджелудочной железы, колоректального рака, рака легкого, рака почки, рака молочной железы, рака яичников, рака предстательной железы, рака кожи, нейробластомы, саркомы, остеохондромы, остеомы, остеосаркомы, семиномы, рака яичка, рака матки, рака головы и шеи, множественной миеломы, злокачественной лимфомы, истинной полицитемии, лейкемии, рака щитовидной железы, рака мочеточника, рака мочевого пузыря, рака желчного пузыря, холангиокарциномы, хориокарциномы и педиатрических опухолей, предпочтительно колоректального рака или рака легкого.
| US 2012238599 A1, 20.09.2012 | |||
| 2005 |
|
RU2364596C2 | |
| WO 2010145197 A1, 23.12.2010 | |||
| WO 2013136249 A1, 19.09.2013. | |||

