ОБЛАСТЬ ИЗОБРЕТЕНИЯ
В этой заявке описываются способы анализа сиалирования белков.
УРОВЕНЬ ТЕХНИКИ
Многие белки требуют гликозилирования для их биологической функции. Часто конечные, "кэппирующие" углеводы гликозильных цепей являются остатками сиаловой кислоты. К сиаловым кислотам относится группа N- и O-связанных нейраминовых кислот. N-связанные сиаловые кислоты образуются путем связывания ацетильных или гликозильных компонентов с амино-остатком нейраминовой кислоты, образуя N-ацетилнейраминовую кислоту (Neu5Ac) и N-гликозилнейраминовую кислоту (Neu5Gc), соответственно. Если амино-группа нейраминовой кислоты замещена гидроксильным компонентом, образуется 3-дезокси-D-глицеро-D-галакто-2-нонулосоновая кислота (KDN). O-связанные сиаловые кислоты образуются путем замещения одной или нескольких гидроксильных групп Neu5Ac, Neu5Gc или KDN метальными, ацетильными, лактоильными, сульфатными или фосфатными группами. Соответственно, существует большая и разнообразная популяция сиаловых кислот.
Кроме того, существует значительный интерес к анализу сиалирования белков в целом, благодаря многочисленным биологическим функциям, приписываемым этим модификациям. Сиалирование может быть важным для фармакокинетики и эффективности белковых биотерапевтических средств. Таким образом, были разработаны способы анализа с целью определения содержания сиаловой кислоты гликопротеинов. Например, могут применяться анализы на основе антител для распознавания конкретных углеводных компонентов. Конечные сиаловокислотные остатки могут ферментативным путем отсоединяться от соответствующего гликопротеина и подвергаться анализу при помощи HPLC. Однако каждый из этих способов имеет недостатки и, как правило, требует чистых образцов или высокой концентрации. Традиционные способы, применяемые в биофармацевтической отрасли, страдают недостатком точности, высокой изменчивостью данных и не могут применяться со сложными культуральными средами из-за помехи матрикса. Описанный авторами способ позволяет преодолеть такие недостатки и обеспечивает точный и воспроизводимый количественный анализ сиалирования белков.
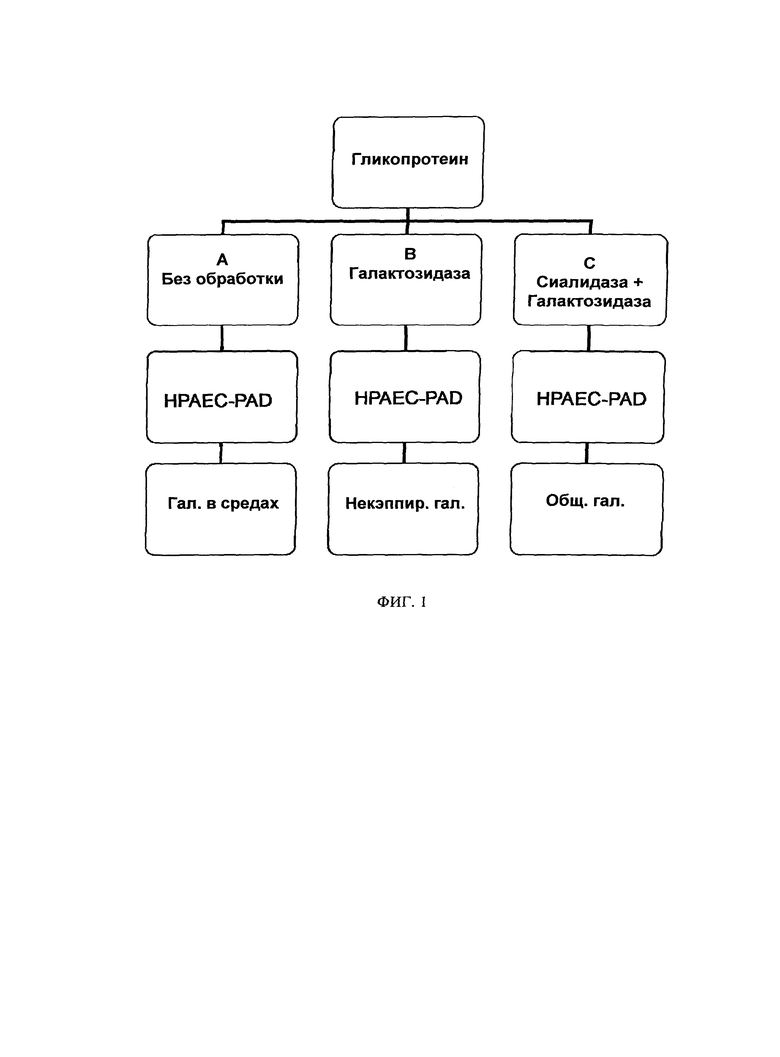
В целом описанный авторами способ включает два этапа: (1) ферментативную реакцию применяют для гидролиза галактозы и остатков сиаловой кислоты из гликопротеинов; и (2) способ ионообменной хроматографии применяют для отделения и подсчета галактозных остатков. Ферментная часть способа включает отцепление открытых (некэппированных) конечных галактозных остатков специфической экзогликозидазой, β-(1-4)-галактозидазой (β-галактозидазой), тогда, как конечные остатки сиаловой кислоты отцепляются α-(2-3,6,8,9)-сиалидазой (α-сиалидазой). Перед расщеплением образец распределяют как минимум по трем пробиркам. Содержимое первой пробирки. Реакция А представляет образец согласно существующему уровню техники и включает только буферы для ферментативной реакции. Содержимое второй пробирки. Реакцию В приводят в реакцию с β-галактозидазой, которая расщепляет все галактозные остатки, не кэппированные сиаловыми кислотами. Содержимое третьей пробирки. Реакция С совместно расщепляется нейраминидазой и β-галактозидазой. Фермент нейраминидаза удаляет кэппирующие сиаловые кислоты и позволяет β-галактозидазе расщеплять все открытые галактозные остатки. Затем применяют высокоэффективную анионообменную хроматографию с импульсным амперометрическим обнаружением (НРАЕС PAD) для определения количества галактозы, присутствующей в трех образцах. Соотношение некэппированной галактозы (т.е. Реакции В) с общим количеством галактозы (т.е. Реакцией С) применяют для расчета процента кэппинга галактозных остатков, при этом также учитывая любую свободную галактозу, присутствующую в среде (Реакция А).
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторами описываются способы анализа сиалирования белка. Также описывается способ определения содержания сиалированного белка, включающий: (а) подготовку белка к анализу; (b) ферментативную обработку подготовленного белка, включающую: разделение подготовленного белка на множество образцов белка, включающих (i) как минимум один образец белка в качестве образца среды (Реакция A); (ii) добавление как минимум β-галактозидазы к как минимум одному образцу белка (Реакция В); (iii) добавление как минимум β-галактозидазы и α-сиалидазы к как минимум еще одному образцу белка (Реакция С); и инкубацию множества образцов белка; и (с) анализ множества образцов белка с применением HPAEC-PAD-хроматографии; (d) определение содержания углеводов для множества образцов белка; и (е) расчет процента сиалирования для белка.
Также описывается способ, также включающий (f) анализ множества положительных и отрицательных контрольных образцов с применением НРАЕС-PAD- хроматографии; (g) анализ множества стандартных образцов с применением HPAEC-PAD-хроматографии; и (h) сравнение множества образцов белка с множеством стандартных и контрольных образцов.
Также описывается применение HPAEC-PAD-хроматографии для определения содержания сиалированного белка, включающее: (а) подготовку белка к анализу; (b) ферментативную обработку подготовленного белка, включающую: разделение подготовленного белка на множество образцов белка, включающих (i) как минимум один образец белка в качестве образца среды (Реакция А); (ii) добавление, как минимум, β-галактозидазы к как минимум одному образцу белка (Реакция В); (iii) добавление, как минимум, β-галактозидазы и α-сиалидазы к как минимум еще одному образцу белка (Реакция С); и инкубацию множества образцов белка; и (с) анализ множества образцов белка с применением HPAEC-PAD-хроматографии; (d) определение содержания углеводов для множества образцов белка; и (е) расчет процента сиалирования для белка.
Также описывается применение, также включающее: (f) анализ множества положительных и отрицательных контрольных образцов с применением НРАЕС-PAD-хроматографии; (g) анализ множества стандартных образцов с применением HPAEC-PAD-хроматографии; и (h) сравнение множества образцов белка с множеством стандартных и контрольных образцов.
Также описывается комплект, определяющий содержание любого сиалированного белка, включающий: как минимум одно вместилище, включающее множество вместилищ, включающих предварительно отмеренное количество галактозидазы и сиалидазы; вместилища необязательно содержат как минимум одну буферную композицию, положительный контрольный образец, отрицательный контрольный образец и стандартные углеводные образцы, а также инструкции, в которых описывается способ определения содержания сиалированного белка, включая описания: (а) подготовки белка к анализу; (b) ферментативной обработки подготовленного белка, включающую: разделение подготовленного белка на множество образцов белка, включающих (i) как минимум один образец белка в качестве образца среды (Реакция А); (ii) добавление, как минимум, β-галактозидазы к как минимум одному образцу белка (Реакция В); (iii) добавление, как минимум, β-галактозидазы и α-сиалидазы к как минимум еще одному образцу белка (Реакция С); и инкубацию множества образцов белка; и (с) анализа множества образцов белка с применением НРАЕС-PAD-хроматографии; (d) определения содержания углеводов для множества образцов белка; и (е) расчета процента сиалирования для белка.
Также описывается комплект, также включающий: (f) анализ положительного и отрицательного контрольных образцов с применением HPAEC-PAD-хроматографии; (g) анализ множества стандартных образцов с применением HPAEC-PAD-хроматографии; и (h) сравнение результатов множества образцов белка с результатами множества стандартных образцов.
КРАТКОЕ ОПИСАНИЕ ФИГУР
ФИГУРА 1 схематически показывает Реакции А, В и С и данные, полученные в результате каждой соответствующей реакции. Процент сиалирующего кэппинга определяют по частному разностей в Реакциях С и В и Реакциях С и А, соответственно. См. Уравнение 1.
ФИГУРА 2 показывает типичную хроматограмму для расщепленного образца recAlpha-1 в Реакции С. Время элюирования для галактозы составляет приблизительно от 14 до 16 минут.
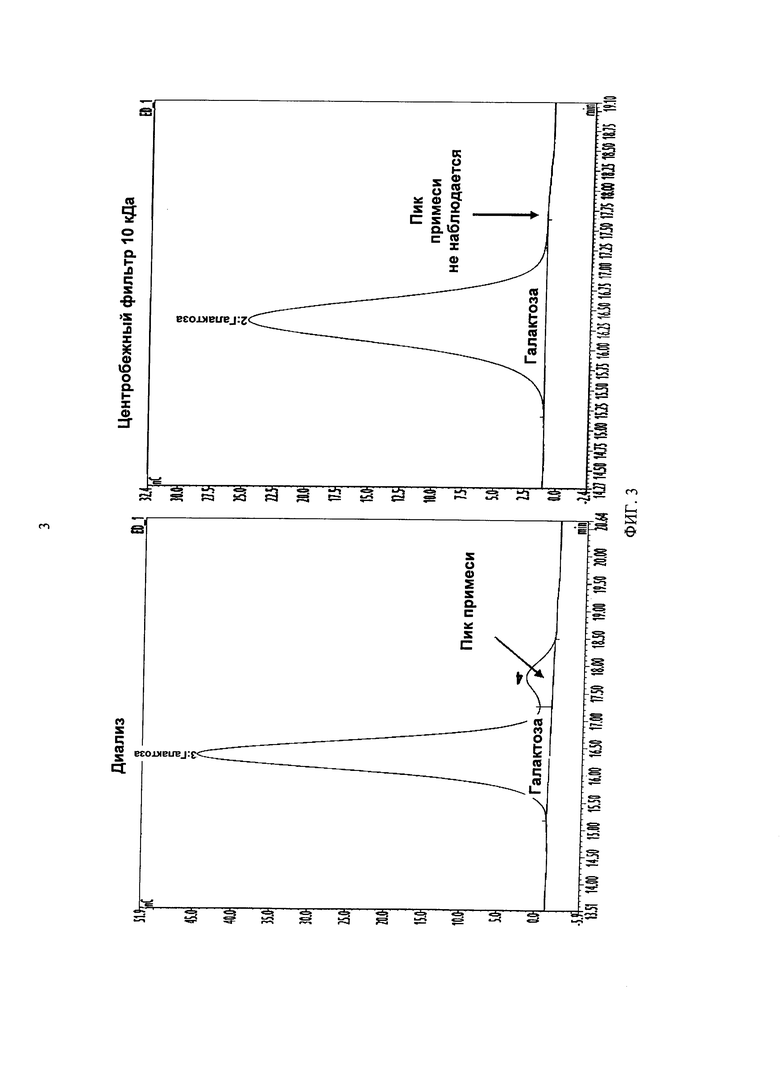
ФИГУРА 3 показывает сравнение предшествующего образца recAlpha-1, который был подвергнут либо диализу на деионизированной воде, либо центрифугированию 10 кДа в центробежном фильтре. В диализированном образце оставались примеси, но они почти полностью удалялись при помощи 10 кДа центробежного фильтра.
ФИГУРА 4 демонстрирует, что предыдущие вспомогательные вещества из культуральных сред recAlpha-1 могут обнаруживаться и подвергаться совместному элюированию поблизости от пика галактозы. На графике А показан пик вспомогательного вещества, который в некоторых образцах элюируется после пика галактозы после очистки центробежного фильтра. Для образцов Реакции В пик примесей представляет базовый показатель, полученный на основе пика галактозы, и не интегрируется. Как показано на графике В, пик примесей процесса совместно элюируется с образцом Реакции С. В этом случае пик примесей должен быть расщеплен, как показано выше, для исключения его интеграции с площадью под пиком галактозы.
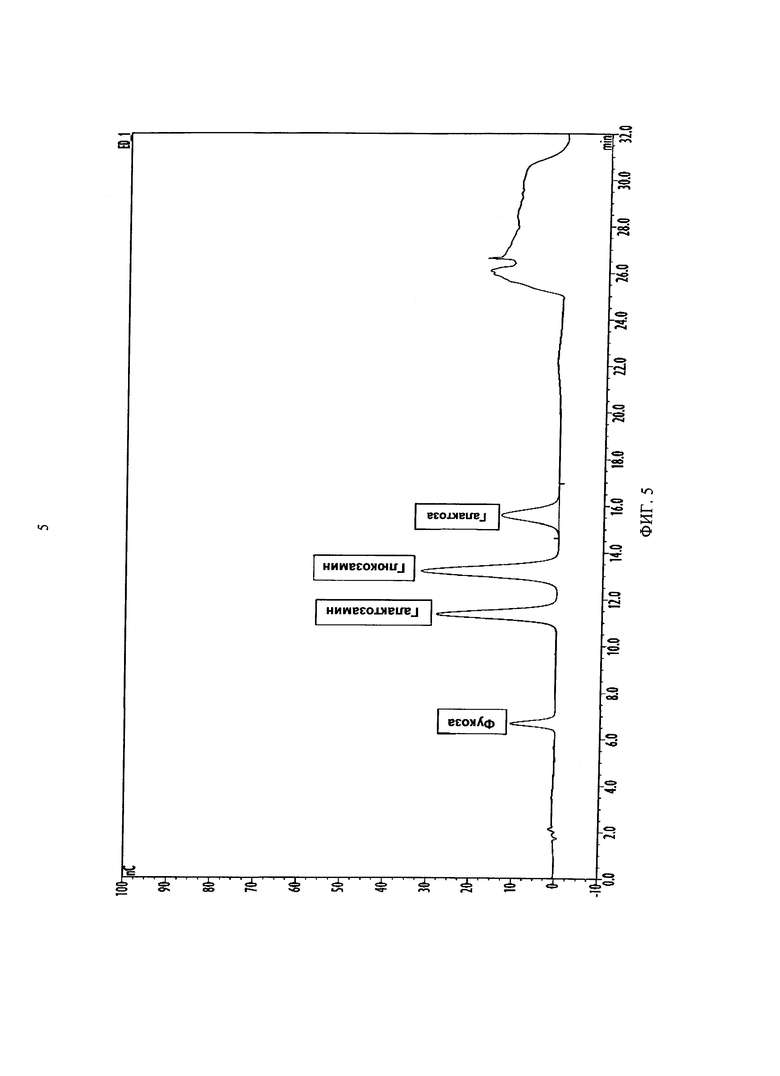
ФИГУРА 5. Специфичность способа подтверждалась анализом смеси нейтральных и аминомоносахаридов, производных от гликопротеинов.
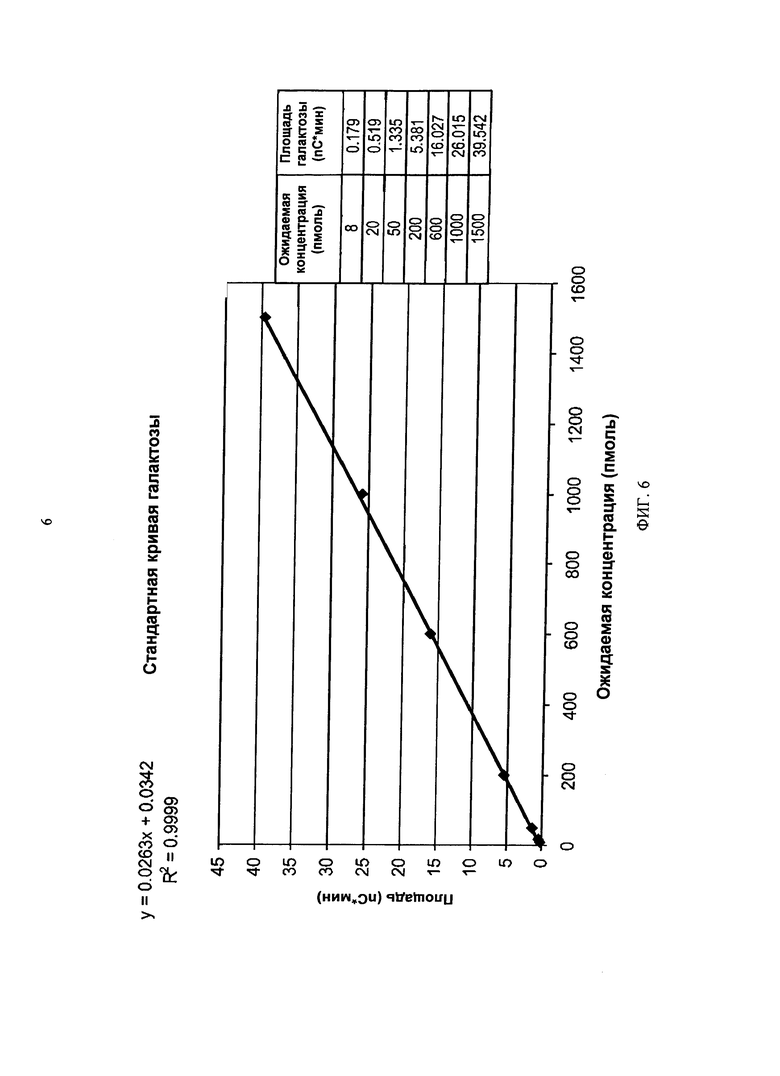
ФИГУРА 6. Пример типичной стандартной кривой галактозы.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Примером белка, поддающегося анализу с применением описанного авторами способа, является ингибитор альфа-1-протеиназы (также известный как альфа-1-антитрипсин). Ингибитор альфа-1-протеиназы является природным серпиновым гликопротеином, задействованным в защите клеток от протеазных ферментов, задействованных в свертывании и воспалении. Отсутствие ингибитора альфа-1-протеазы, дефицит альфа-1-антитрипсина, ведет к респираторным нарушениям, таким как эмфизема и хроническое обструктивное заболевание легких (COPD). Соответственно, существует интерес к использованию ингибитора альфа-1-протеазы в качестве биотерапевтического средства для лечения связанных с дефицитом ингибитора альфа-1-протеазы болезней. Ингибитор рекомбинантной альфа-1 протеиназы (recAlpha-1) подвергали инженерии для секреции клеточной линией PerC6 с N-связанными структурами углевода гликана, частично или полностью кэппированными конечными сиаловыми кислотами (N-ацетилнейраминовыми кислотами). Было продемонстрировано, что уменьшение количества конечных сиаловых кислот сокращает период полувыведения recAlpha-1 из сыворотки. Таким образом, важно знать процент галактозных остатков, кэппированных сиаловыми кислотами в recAlpha-1, при исследовании ее функции или эффективности в качестве терапевтического средства.
ПРИМЕРЫ
Пример 1: Приготовление образца для анализа и ферментативного расщепления
Авторами описывается способ определения содержания сиалирования гликопротеина. Способ начинается с приготовления образца белка для ферментативного гидролиза углеводных компонентов. Соответственно, белок должен быть приведен в состояние, совместимое с ферментативными реакциями, включая регулирование концентрации белка, удаление компонентов раствора, таких как растворенные соли, буферы, другие белки, углеводы, вспомогательные вещества и т.п., которые могут препятствовать действию фермента(ов). В контексте данного описания термин "подготовленный" или фраза "подготовка белка к анализу" означают процесс удаления компонентов раствора, которые могут препятствовать ферментативному гидролизу и разбавлению белкового раствора до оптимальной для анализа концентрации деионизированной водой.
Могут применяться как минимум два неограничивающих типичных способа для подготовки белковых компонентов раствора из белков для анализа: (1) диализ на деионизированной воде или (2) центробежное фильтрование (также известно как спиновая фильтрация). В обоих случаях использовали полупроницаемую мембрану с указанной границей отсечки по молекулярной массе (MWCO) для удаления более низкомолекулярных разновидностей при удерживании анализируемого белка. Диапазон значений MWCO включает 1 кДа, 2.5 кДа, 5 кДа, 10 кДа, 20 кДа, 50 кДа, 100 кДа, 250 кДа и 500 кДа. При процедуре диализа белок вставляли в диализную мембрану и диализировали на избыточном количестве деионизированной воды или подходящего буфера и/или солевого раствора в течение как минимум 4 часов при 4°С.
В альтернативном варианте вместо диализа образец белка может быть подготовлен к ферментативному расщеплению путем центробежного фильтрования. Во время центробежного фильтрования белковый раствор центрифугировали на полупроницаемую мембрану с указанным MWCO. Компоненты раствора с молекулярной массой, меньшей, чем MWCO, во время центрифугирования проходят сквозь фильтр, в то время как белок и более высокомолекулярные разновидности удерживаются. Как правило, белковый раствор концентрировали во время спиновой фильтрации за счет прохождения воды сквозь полупроницаемую мембрану. В качестве типичного неограничивающего примера для приготовления образца белка использовали 10 кДа центробежный фильтр в соответствии с инструкциями производителя.
Перед ферментативным расщеплением образцы белков также разбавляли до концентрации от 1,0 до 1,5 мг/мл деионизированной водой, таким образом, чтобы они пребывали в пределах линейного диапазона анализа. Сразу после разбавления образцов составляли как минимум одну реакцию для каждого состояния (т.е. А, В и С). Приготовленный белковый раствор разделяли как минимум на три образца для ферментативного дегликозилирования. См. Фигуру 1. Реакция А представляла собой контрольный образец существующего уровня техники (для контроля экзогенной галактозы). Эта реакция состоит только из буферов для ферментативных реакций и служит в качестве контроля для углеводов, которые могут существовать в среде, включающей анализируемый белок. Реакция В содержала β-галактозидазу, которая гидролизировала несиалированные углеводные группы, в отличие от "кэппированных" сиалильными группами. Реакция С содержала α-сиалидазу и β-галактозидазу. В этой реакции α-сиалидаза гидролизировала кэппирующие сиалильные компоненты, которые затем позволяли β-галактозидазе гидролизировать все углеводные группы. В этой комбинированной реакции α-сиалидазы и β-галактозидазы, все гликозилирования (т.е., сиалированные и несиалированные) удаляли из белка, в то время как в реакции β-галактозидазы удаляли только некэппированные (несиалированные) углеводные группы. Компоненты трех условий реакции показаны в Таблице 1.
Отдельные реакции подготавливали в нескольких экземплярах в зависимости от количества образцов, подлежащих расщеплению. Для каждого расщепления 45 мкл образца добавляли в три отдельные пробирки. Затем 20 мкл соответствующей ферментативной реакции А, В или С добавляли в пробирку, предназначенную для этого типа реакции, и нагревали на термомиксере при 37°С с умеренным встряхиванием в течение 2,5-4 часов. Реакцию гасили путем инкубации при 90°С в течение 5 минут.
Сразу после завершения расщепления образцы подготавливали для хроматографического анализа путем комбинирования 30 мкл образца и 20 мкл 0,02 мг/мл дезоксирибозного стандарта в флаконе и тщательного перемешивания. Затем образцы помещали на автодозатор для HPLC при 10°С и прогоняли, применяя способ хроматографии, описанный в Примере 2.
Пример 2: Способы хроматографии
Анализ при помощи высокоэффективной анионообменной хроматографии с импульсным амперометрическим обнаружением (HPAE-PAD) выполняли при помощи систем для ионной хроматографии Dionex ICS-3000 с одним насосом, термостатированными автодозаторами, установленными на 10°С, и электрохимическими детекторами (Dionex, Sunnyvale, CA). Используют золотые (Au) электроды для импульсного амперометрического обнаружения (PAD) (Dionex Prod. No.060139). Форма волны, используемая для анализа образца, была указана в технической записке 21 от Dionex, в которой описываются оптимальные настройки для PAD углеводов, как показано в Таблице 2. См. Dionex Technical Note 21, Optimal Settings for Pulsed Amperometric Detection of Carbohydrates Using the Dionex ED40 Electrochemical Detector, Dionex (1998).
Хроматографию выполняли с применением анионообменной колонки Dionex CarboPac РА10 4×250 мм (Dionex Prod. No.046110) с встроенной предколонкой CarboPac РА10 4×50 мм (Dionex Prod. No.046115) и 4×50 мм аминоулавливающей колонкой (Dionex Prod. No.046112) для связывания десиалированного белка и предотвращения его адсорбции в колонку.
Первую мобильную фазу (А), содержащую 20 мМ NaOH, использовали для изократического разделения при скорости потока 1 мл/мин в течение 30 минут (время первичного прогона составляло 28 минут, но продлевалось до 30 минут во время проявления, что давало больше времени на повторное уравновешивание в 100% мобильной фазе А). Вторую мобильную фазу (В), содержащую 500 мМ NaOH, использовали для элюирования колонки, очистки колонки и очистки электродов. Способ хроматографического элюирования, включающий функцию ramp wash с применением мобильных фаз А и В, показан в Таблице 3. Вводимый объем образца составлял 20 мкл. Анализы данных выполняли с использованием программы для анализа данных хроматографии Chromeleon® 6.8 (Dionex).
Пример 3: Стандарты, контрольные образцы, градуирование и приемлемость системы
Концентрацию галактозы определяют относительно известного количества 10 мМ стандарта галактозы (BioAssay Systems, EGAL-100), который последовательно разбавляли в линейном диапазоне от 8 пмоль до 1,5 нмоль. Во время разработки способа для каждой точки осуществляли одно введение перед введением образцов для обеспечения функционирования колонки и системы. См. Фигуру 6.
Моносахарид 2-дезокси-D-рибозу (дезоксирибозу) использовали в качестве внутреннего стандарта. Равноценное количество дезоксирибозы добавляли ко всем образцам белков, стандартным и контрольным образцам для наблюдения за функционированием электрода. Характер PAD требует применения внутреннего стандарта для исправления различий в показаниях детектора, которые для разных вводимых образцов могут быть разными. Площадь под пиком галактозы для всех образцов корректировали путем деления площади галактозы на площадь дезоксирибозы.
Положительный контроль использовали для обеспечения точности анализа при каждом его выполнении. Она требуется для наблюдения за функцией фермента и обеспечения надлежащего обращения с образцом. Контрольная реакция представляла собой смесь 1:1 серийно выпускаемого сиалированного фетуина крупного рогатого скота и асиалофетуинового стандарта (Sigma F3004 и А4781, соответственно). Отдельно сиалированный фетуин имеет процент кэппинга свыше 99%, а асиалофетуин имеет процент кэппинга 0%. При смешивании в одинаковых пропорциях соотношение кэппинга (сиалирования) для фетуина должно составлять 50%±3%. Фетуиновый контроль изготавливали большой партией, которую делили на аликвоты, замораживали при -70°С и отдельный образец размораживали и использовали в качестве контроля каждый раз при расщеплении и прогонке набора образцов с применением описанных авторами способов.
Эксперимент на приемлемость системы для обеспечения эффективности аналитического способа наблюдали с использованием смеси галактозы и дезоксирибозы, которую вводили в начале прогона, с "заключением в скобки" после каждых введенных 12 образцов (т.е., 2 образца в двух экземплярах), и в конце прогона для наблюдения за функционированием электрода и колонки в течение всего прогона. Серия экспериментов для типичного хроматографического прогона представлена в Таблице 4.
Пример 4: Корректировка и расчет результатов
Репрезентативная хроматограмма показана на Фигуре 2. Площади всех введенных образцов корректировались площадью дезоксирибозы. Корректировку выполняли путем деления пика галактозы на площадь пика дезоксирибозы. Это скорректированное число использовали для расчета процента сиалирования. Для расчета этого процента определяли соотношение некэппированной галактозы с общей галактозой при помощи Уравнения 1:

Для расчета процента кэппирования площадь галактозы делили на площадь дезоксирибозы для получения скорректированных показателей площади галактозы. Процент кэппирования рассчитывали для каждого из введенных в двух экземплярах образцов с использованием скорректированных площадей. Скорректированную площадь галактозы, измеренную для Реакции В, вычитали из скорректированной площади галактозы, определенной на основе Реакции С; это значение соответствует количеству кэппированной сиалилом галактозы. Скорректированную площадь галактозы, определенную для Реакции А, затем вычитали из скорректированной площади галактозы, измеренной для Реакции С; это значение соответствует общему показателю галактозы (т.е., кэппированной и некэппированной). Показатель кэппированной галактозы (С-В) делили на общий показатель галактозы (С-А) и умножали на 100 для получения процента сиалилового кэппинга. Репрезентативные данные показаны в Таблице 5.
Приемлемость системы наблюдали с использованием смеси галактозы и дезоксирибоза, которую вводили в начале прогона и в конце прогона для наблюдения за функционированием электрода и колонки в течение всего прогона. Указанный средний процент кэппирования определяли на основе скорректированных площадей двукратных введений. Если площадь под пиком галактозы в Реакции В была меньшей, чем площадь под пиком галактозы в стандарте 8 пмоль, процент кэппирования указывали как ">[кэппинг]%" (т.е., "более"), рассчитанный на основе площади под пиком галактозы стандарта 8 пмоль в числителе расчета. Коэффициент асимметрии пика (называемый "асимметрией" в программе Chromeleon®), теоретических тарелок и разделение рассчитывали и определяли при помощи программы сбора данных Dionex. Расчеты остальных критериев приемлемости системы производили вручную. Репрезентативные параметры приемлемости системы показаны в Таблице 6.
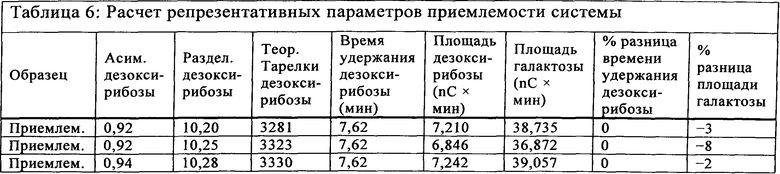
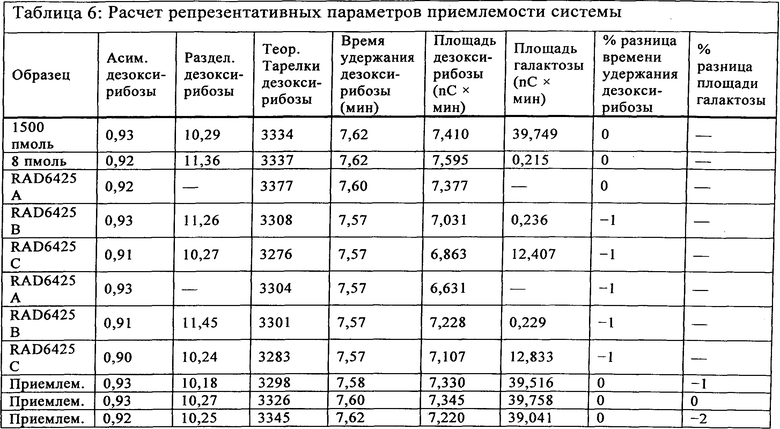
Критерии допустимости анализа приемлемости системы показаны в Таблице 7.
Пример 5: Стехиометрия образца/ферментативных реакций и время расщепления
С целью оптимизации скорости ферментативной реакции анализировали соотношение фермента с белком. Фермент β-галактозидаза обеспечивался производителем с активностью >3 ед./мл (удельная активность >6 ед./мг), в то время как α-Фермент сиалидаза обладает активностью 5 ед./мл (удельная активность при 135 ед./мг). Количество каждого фермента поддерживали на постоянном уровне по 4 мкл, что соответствует 0,012 единицы β-галактозидазы и 0,02 единицы α-сиалидазы в реакции, в то время как количество белка колебалось от 540 до 2160 пмоль. Анализируемые образцы представляли собой подвергнутый буферному обмену и фильтрованный recAlpha-1 в супернатанте клеточной культуры в концентрации 1,4 мг/мл. Образцы подготавливали, как показано в Таблице 8.
Для каждого показателя стехиометрии реакции и момента времени расщепления recAlpha-1 анализировали площадь под хроматографическим пиком галактозы и определяли процент кэппирования. В Таблице 9 показаны сводные результаты, демонстрирующие, что показатель кэппинга не меняется ни при каких стехиометрических условиях. Начальный показатель стехиометрии реакции (например, 2,2×10-5 ед./пмоль белка) определяли в соответствии с рекомендациями производителя. Однако эти результаты указывают, что ферменты могут быть избыточными в рекомендуемых поставщиком условиях, даже при повышении концентрации белка в 4 раза. При таком широком динамическом диапазоне стехиометрии выбирали "средние" стехиометрические условия (т.е., приблизительно 1,1×10-5 ед./пмоль белка) с целью упрощения процедуры, минимизации стоимости фермента и обеспечения возможности достаточной устойчивости в приготовлении образца.
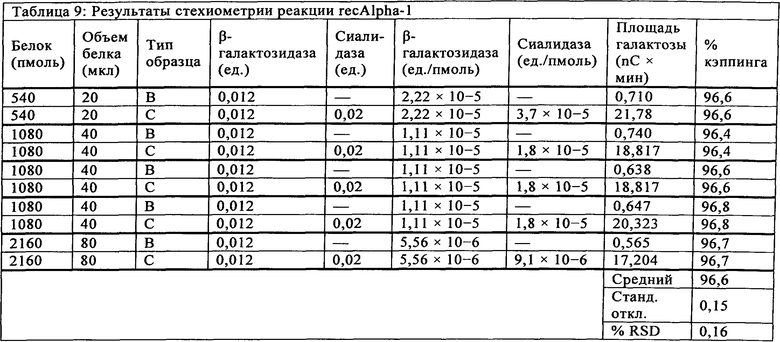
Стехиометрию реакции также исследовали путем сравнения результатов кэппинга для предыдущего образца recAlpha-1 (RAD-0637), изготовленного в 1-й день с использованием 4 мкл фермента в реакции и 2-й день (через 14 дней) с использованием 2 мкл фермента. Результаты эксперимента показывают одинаковый показатель кэппинга для количества фермента 2 мкл и 4 мкл, что свидетельствует о том, что количество 2 мкл фермента было достаточным для доведения реакции до завершения, в соответствии с вышеуказанным наблюдением (Таблица 10).
Пример 6: Внутренний стандарт
При этом способе HPAEC-PAD применяли внутренний стандарт для нормализации площади под пиком галактозы вследствие естественной изменчивости амперометрического обнаружения каждого вводимого образца. Сначала испытывали два внутренних стандарта: галактозамин и дезоксирибозу.
Оба внутренних стандарта функционировали надлежащим образом, и время элюирования обоих достаточно отличалось от времени галактозы и не препятствовало количественному определению. Хотя галактозамин функционировал надлежащим образом, наблюдались некоторые несоответствия показателей площади под пиком, которые требовали более широких критериев допустимости для наблюдения за функционированием системы. Таким образом, в качестве внутреннего стандарта выбирали дезоксирибозу. Кроме того, дезоксирибозу обычно используют в качестве промышленного стандарта для осуществления способов амперометрического обнаружения. Площади под пиками дезоксирибозы демонстрируют меньшую изменчивость по всей последовательности введений и могут использоваться при более жестких критериях допустимости.
Во время проявки наблюдались некоторые вещества с необъяснимым увеличением или уменьшением площади дезоксирибозы. Однако в этих же образцах за увеличением или уменьшением площади дезоксирибозы следовало противоположное изменение пика галактозы (Таблица 11).
Как правило, колебания площади под пиком могли объясняться как различия в реакции со стороны электрода. В таком случае можно было бы ожидать, что оба пика должны повышаться или понижаться. Был проведен эксперимент, в котором дезоксирибозу добавляли к трем образцам, но не смешивали. Образцы анализировали путем HPAEC-PAD, затем удаляли и вихревали для обеспечения смешивания образца и внутреннего стандарта. Затем образцы вводили повторно. Результаты этих экспериментов показывают, что колебания площади являются результатом недостаточного смешивания образца и внутреннего стандарта. Это указывает на возможность расслоения между раствором внутреннего стандарта и образцом, и пользователи должны в достаточной мере смешивать реакцию перед помещением на поддон автодозатора. Результаты этих экспериментов сведены в Таблице 12.
Пример 7: Сравнение ферментов от разных поставщиков
С учетом критичности качества ферментов β-галактозидазы и сиалидазы для скорости реакции и количественного определения галактозы сравнивали ферменты β-галактозидазу и α-сиалидазу от четырех разных поставщиков. Цель состояла в измерении достаточности скорости реакции, которая бы обеспечивала точную реакцию, и определении запасного поставщика на случай недоступности ферментов от основного поставщика. Выбранными поставщиками были Sigma, Glyko-Prozyme, New England BioLabs и QA Bio. Ферменты от QA Bio использовали в большинстве описанных авторами экспериментов. Ферменты от Sigma были исключены, когда ферментативная реакция не дала ответа. Ферменты от Glyko-Prozyme также были исключены как вариант, когда показатели площади под пиком галактозы для образцов, обработанных β-галактозидазой, оказались такими же, как для образцов, обработанных β-галактозидазой и α-сиалидазой в двух отдельных экспериментах (т.е., активность α-сиалидаза не обнаруживалась). Эксперименты с прямым сравнением ферментов от New England BioLabs и QA Bio подготавливали и проводили в один день. Результаты также сравнивали с данными предыдущих дней с теми же образцами, расщепленными ферментами от QA Bio. Процентные различия между двумя ферментами были пренебрежимыми, и относительное стандартное отклонение (% RSD) для нескольких экспериментальных прогонов указывало, что ферменты от New England BioLabs β-Галактозидаза и α-сиалидаза были сравнимы с QA Bio, и этот поставщик может использоваться в качестве запасного на случай недоступности ферментов от QA Bio. Результаты экспериментов сведены в Таблице 13.
Пример 8: Типы и приготовление образцов
С учетом того, что предыдущие образцы белка могут содержать 5 мг/мл галактозы из среды для культур клеток, способ приготовления образца разрабатывался для удаления из среды большей части избыточной галактозы, а также других потенциально создающих помехи вспомогательных веществ. Сама по себе такая очистка была недостаточной для удаления всех технологических примесей, поэтому должна выполняться дополнительная очистка в рамках подготовки способа кэппинга. Оценивали два способа быстрой очистки: диализ на деионизированной воде и центробежное фильтрование 10 кДа.
Для эксперимента использовали подвергнутый буферному обмену образец recAlpha-1. В этом эксперименте образец подвергали диализу на деионизированной воде в течение 4 часов при одновременной очистке другого образца с использованием центробежного фильтра 10 кДа. Затем оба образца анализировали с применением способа кэппинга. Согласно выводу по результатам эксперимента, центробежные фильтры 10 кДа более эффективны при удалении примесей по сравнению с диализом и позволяют приготавливать до 30 образцов за раз, при значительном продлении времени цикла. Хроматограммы обеих процедур очистки, включавших расщепление Реакцией С (β-галактозидаза и α-сиалидаза), показаны на Фигуре 3. Подвергнутый центробежному фильтрованию образец восстанавливали деионизированной водой, и, следовательно он был менее концентрированным, чем диализированный образец. Хотя следовое количество галактозы может сохраняться после очистки, все фоновые пики (т.е., Реакция А) впоследствии учитывались при расчете процента кэппирования путем вычитания площади под пиком примеси.
Также наблюдалось, что некоторые вспомогательные вещества из супернатантов культуры клеток элюируются вскоре после пика галактозы и в некоторых случаях имеют форму плеча пика на пике галактозы (см. Фигуру 4). В таких случаях, когда некоторые остаточные вспомогательные вещества на могут не могут быть удалены при помощи центробежного фильтра 10 кДа, присутствие этой примеси исключали из площади под пиком галактозы путем выполнения "спадающей" интеграции без интеграции пика примесей.
Пример 9: Специфичность
Специфичность означает способность способа к оценке аналита в присутствии компонентов, наличие которых может ожидаться, таких, как примеси, продукты распада, матриксы и т.п. Специфичность способа определяли путем приготовления смеси нейтральных и аминомоносахаридов промышленного производства и анализа смеси при помощи HPAEC-PAD. Оцениваемыми сахарами были фукоза, галактозамин, глюкозамин и галактоза. Сахара анализировали отдельно для подтверждения времени удержания, а затем анализировали как смесь для определения специфичности (Фигура 5). Отделение моносахаридов было сравнимым с описанным в технической записке 20 от Dionex, согласно которой галактоза элюируется после всех трех других моносахаридов. См. Dionex Technical Note 20, Analysis of Carbohydrates by High Performance Anion Exchange Chromatography with Pulsed Amperometric Detection (HPAE-PAD), Dionex Corp. (2000). Кроме того, для образцов с высоким содержанием примесей, т.е., супернатантов культур клеток, фоновые уровни анализировали для определения присутствия каких-либо создающих помехи вспомогательных веществ и их удаления при приготовлении образца.
Пример 10: Линейность
Линейность анализа НРАЕС PAD сиалирования означает его способность к обеспечению результатов испытаний, которые являются прямо пропорциональными концентрации аналита или содержанию в данном диапазоне. Кроме того, диапазон, выведенный в результате исследования линейности, использовали для подтверждения степени приемлемости линейности, точности и точности, достигаемой при использовании процедуры. Линейность для способа оценивали путем построения стандартной кривой градуирования для галактозы при оптимальном диапазоне от 8 пмоль до 1,5 нмоль (Фигура 6). Коэффициент определения (коэффициент регрессии) составлял 0,99 или более для диапазона от 8 пмоль до 1,5 нмоль. Остатки регрессии также анализировали, и они не проявляли сдвига в этом диапазоне. Кривая градуирования может продлеваться до 2 нмоль и сохранять приемлемую линейность, но с потерей точности на нижнем конце кривой градуирования. Оптимальный диапазон кривой градуирования наблюдали в разные дни, с использованием разных систем, колонок и одноразовых золотых электродов Dionex ICS-3000. Эти результаты сведены в Таблице 14.
Пример 11: Граница количественного определения
Граница количественного определения (LOQ) НРАЕС PAD анализа сиалирования указывает наименьшее количество аналита в образце, которое поддается количественному определению с надлежащей точностью. Существует несколько способов расчета LOQ количественного способа. Один способ основывается на стандартном отклонении точки пересечения с осью у, деленном на средний уклон. Другой способ основывается на визуальном анализе хроматограммы. Сначала рассчитывали LOQ пробы на основе отношения сигнал/шум типичной хроматограммы. В частности, измеряли уровень шума в 1-минутной горизонтальной области хроматограммы и умножали на 10 для получения LOQ как высоты пика. Высоту пика переводили в пикомольное количество на основе высоты пика по стандартам градуирования. Результаты уровня шума сравнивали со стандартом градуирования, который обеспечивал подобный ответ (см. Таблицу 15). Уровень шума определяли в шесть разных дней на двух разных системах ICS-3000. Эти результаты указывают, что уровень шума в разные дни давал сходные результаты (±0,02) и соответствовал стандартному уровню приблизительно 8 пмоль (разница 0,12 nC × мин, которая была незначительной по сравнению с высотой стандарта 1500 пмоль приблизительно при 70 nC × мин).
Поскольку уровень шума хроматограммы может изменяться в зависимости от инструмента, LOQ также рассчитывали другим способом. На основе значений из Таблицы 14 и, в частности, на основе стандартного отклонения точки пересечения с осью у (0,0577), деленного на средний уклон (0,0283) и умноженного на 10, рассчитывали показатель LOQ, который составлял 20 пмоль. Два способа расчета границы количественного определения указывают, что показатель LOQ составляет приблизительно от 8 до 20 пмоль.
Пример 12: Точность способа
Точность способа определяли по согласованию между известным стандартом и экспериментально измеренным результатам. Поскольку не существует "золотого стандарта", который служил бы эталоном, точность способа определяли, используя смесь сиалированного и асиалофетуинового стандартов промышленного производства в равных пропорциях. Сиалированный фетуин крупного рогатого скота и асиалофетуин в качестве стандартных образцов приготавливали в равных концентрациях, как определялось путем УФ-поглощения при 280 нм (т.е., А280). Стандартные образцы анализировали отдельно, затем смешивали в соотношении 1:1 и анализировали. Сиалированный фетуиновый стандарт имел процент кэппирования 99,4%, а асиалофетуиновый стандарт имел процент кэппирования 1,2%, что объяснялось тем, что количество галактозы в Реакции В было несколько выше по сравнению с Реакцией С. Согласно этим данным, ожидаемый процент кэппинга для контрольной фетуиновой смеси должен был составлять приблизительно 50%. Фактический результат для фетуиновой смеси был определен как 49,3% на основе среднего показателя нескольких прогонов. Результаты эксперимента сведены в Таблице 16. Хотя точную производную изменчивости или точности от измерения А280 получить невозможно, эти результаты указывают, что этот способ был точным в пределах нескольких процентов. Кроме того, этот фетуиновый контроль анализировали в рамках исследований внутрилабораторной воспроизводимости в разные дни (см. Таблицу 18 и Таблицу 19 ниже), и он продемонстрировал относительное стандартное отклонение (% RSD) 3%. Применение такого контроля рекомендуется каждый раз при выполнении анализа.
Пример 13: Воспроизводимость
Воспроизводимость анализа оценивали по постоянству результатов, полученных при выполнении способа в течение коротких интервалов времени в указанных условиях. Воспроизводимость способа определяли с использованием образца супернатанта культуры клеток recAlpha-1, который вводили в шести экземплярах. Образец согласно существующему уровню техники (т.е., Реакция А) не анализировали, поскольку этот ранее анализировался и не обнаруживал содержания создающей помехи галактозы. Относительное стандартное отклонение определяли для площади галактозамина, площади галактозы и процента кэппирования для шести введенных экземпляров. Результаты сведены в Таблице 17 и показываются как скорректированные площадью галактозамина и без коррекции. Данные показывают воспроизводимость около 0,20%.
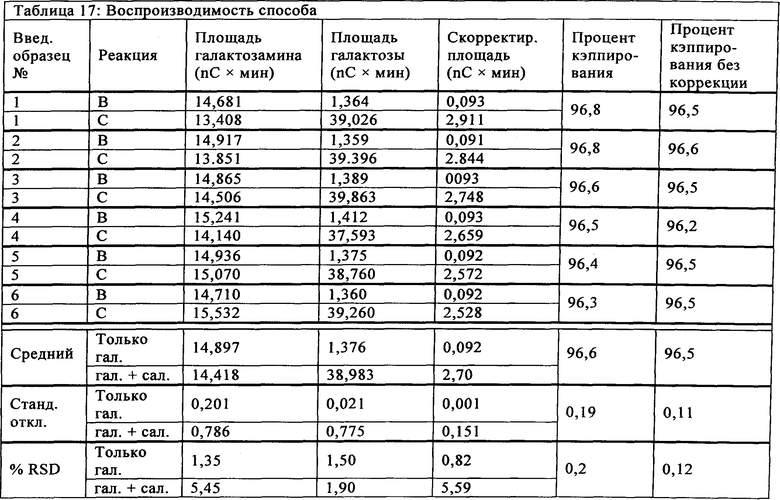
Пример 14: Внутрилабораторная воспроизводимость
Помимо исследования воспроизводимости, анализ внутрилабораторной воспроизводимости включал несколько дополнительных факторов: разные дни, разные настройки инструментов и разные препараты образцов. Внутрилабораторную воспроизводимость способа исследовали путем приготовления разработанного для дальнейшего процесса образца recAlpha-1 (RAD-5904) для анализа кэппинга в три разных дня и в трех разных концентрациях. Для анализов использовали разные хроматографические системы Dionex ICS-3000, одноразовые электроды, аминоулавливающие колонки, предколонки и аналитические колонки. Результаты показывают относительное стандартное отклонение (RSD) образца 0,25%. Кроме того, также сравнивали процент кэппинга фетуинового контроля, приготавливаемого в течение трех дней анализа. Показатель RSD фетуинового контроля составлял 2,99%. Результаты внутрилабораторной воспроизводимости образцов recAlpha-1 и фетуинового контроля сведены в Таблице 18 и Таблице 19, соответственно. Показатели площади представляли среднее значение двух прогонов и не корректировались.
Пример 15: Устойчивость
Устойчивость анализа означает меру его способности оставаться неизменным при небольших, но преднамеренных изменениях параметров способа или обработки образца. В нескольких сериях экспериментов преднамеренно варьировали несколько разных факторов, таких, как устойчивость автодозатора, время ферментативной реакции, объем фермента и помеха матрикса.
Устойчивость образца в автодозаторе
Устойчивость определяли путем исследования устойчивости образца в течение 48 часов в автодозаторе для условий HPLC (т.е. 10°С), чтобы определить, может ли образец, ожидающий введения в автодозатор при 10°С, ухудшить качество результатов. Отдельно приготовленный образец, который держали в автодозаторе, вводили с разными интервалами и для каждого момента времени определяли максимальный отклик количества галактозы. Результаты сведены в Таблице 20 и показывают значение относительного стандартного отклонения, составляющее 0,23%, что согласуется с относительным стандартным отклонением, определяемым по внутрилабораторной воспроизводимости. Результаты показываются как скорректированные площадью галактозамина под пиком и без коррекции. Эти данные показывают, что образцы могут храниться в автодозаторе в течение 48 часов перед введение без ухудшения результатов кэппинга.
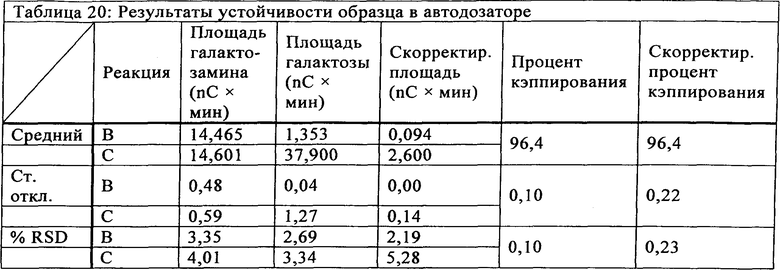
Время ферментативной реакции
Устойчивость также исследовали путем изменения времени ферментативной реакции с 1 до 4 часов в поисках объяснения изменчивости скорости реакции для разных партий фермента. Образец инкубировали в моменты времени 1, 2, 5 и 4 часа и каждый раз анализировали. Результаты сведены в Таблице 21 и показывают, что, несмотря на сходные показатели процента кэппирования для трех значений времени реакции, образец после одночасового расщепления демонстрировал более низкие показатели по сравнению с более длительными реакциями. На основе этих данных 2,5-часовое расщепление определяли как достаточное для доведения реакции до завершения. С учетом того, что способ зависит от скорости реакции двух ферментов, более низкие процентные результаты кэппирования для 1-часового времени реакции указывают, что фермент сиалидаза является ограничивающим скорость фактором.
Помехи матрикса
Хотя помехи со стороны вспомогательных веществ и матрикса рассматривались в разделе специфичности, выполняли исследование с введением матрикса для исключения сдвига результатов предыдущим матриксом. Полученный из плазмы образец Alpha-1 (PD Alpha-1), содержащий различные матриксы, добавляли к средам культуры клеток и процент кэппирования измеряли с применением описанного авторами способа. Среды, выбранные для эксперимента, содержали наибольшее количество добавок, применяемых в предыдущих экспериментах по разработке, таких как среды CDM4PERMAB™ (Hyclone), Pluronic® F-68 (BASF), Antifoam-C (Dow Corning®), и различные среды для культур клеток. Полученный из плазмы Alpha-1 добавляли к средам в концентрации 0,5 мг/мл. Затем образец приготавливали, используя типичную процедуру очистки обмена буфера на 20 мМ фосфата, pH 7, с последующим центробежным фильтрованием 10 кДа. Процент кэппинга PD Alpha-1, добавляемого к культуральным средам, сравнивали с результатами кэппинга для PD Alpha-1, который не добавляли к средам для культур клеток (см. Таблицу 22). Результаты показывают процент кэппирования 99,4% для PD Alpha-1, добавляемого к средам, и 99,2% (средний) для PD Alpha-1, который не добавляли к средам, что соответствовало внутрилабораторной воспроизводимости, связанной с этим способом. Эти результаты указывают, что среды для культур клеток не препятствуют анализу после применения к образцам соответствующих этапов очистки.
Выводы
Результаты, полученные в ходе исследовательских и предквалификационных экспериментов, сведены в Таблице 23. На основе вышеописанных исследований этот способ был оптимизирован для определения показателей кэппинга в образцах белков. Параметры линейности, LOQ, точности и устойчивости, определенные для этого анализа, показывают, что этот способ является последовательным, точным и надежным.


Изобретение относится к области биохимии. Описана группа изобретений, включающая способ определения процента сиалирования гликопротеина и набор для определения содержания сиаловой кислоты в гликопротеине. Способ включает в себя стадии подготовки белка к анализу, ферментативную обработку подготовленного белка, анализ образцов белка с применением HPAEC-PAD-хроматографии, определение содержания сиаловой кислоты в образцах белка, расчет процента сиалирования белка. Изобретение расширяет арсенал средств для определения процента сиалирования гликопротеинов. 2 н. и 2 з.п. ф-лы, 6 ил, 23 табл, 15 пр.
1. Способ определения процента сиалирования гликопротеина, включающий:
(a) подготовку белка к анализу;
(b) ферментативную обработку подготовленного белка, включающую: разделение подготовленного белка на три группы образцов, включающих
(i) группу образцов белка в качестве образца среды (Реакция А);
(ii) группу образцов белка для добавления β-галактозидазы (Реакция В);
(iii) группу образцов белка для добавления β-галактозидазы и α-сиалидазы (Реакция С); и
инкубацию образцов белка; и
(c) анализ образцов белка с применением HPAEC-PAD-хроматографии;
(d) определение содержания сиаловой кислоты в образцах белка;
(e) расчет процента сиалирования белка.
2. Способ по п. 1, отличающийся тем, что также включает:
(f) анализ положительных и отрицательных контрольных образцов с применением HPAEC-PAD-хроматографии;
(g) анализ стандартных образцов с применением HPAEC-PAD-хроматографии; и
(h) сравнение образцов белка со стандартными и контрольными образцами.
3. Набор для определения содержания сиаловой кислоты в гликопротеине, включающий:
контейнер, включающий емкости, который содержит предварительно отмеренное количество галактозидазы и сиалидазы, или отдельные контейнеры, содержащие указанные ферменты,
а также инструкции, в которых описывается способ определения содержания сиаловой кислоты в гликопротеине по п. 1.
4. Набор по п. 3, в котором контейнер включает дополнительные емкости, содержащие как минимум одну буферную композицию, положительный контрольный образец, отрицательный контрольный образец и стандартные углеводные образцы, или он дополнительно включает отдельные контейнеры с указанными веществами.
| WO2013011178 A1 24.01.2013 | |||
| V.P | |||
| BHAVANANDAN et al.: "Identification of the glycosidically bound sialic acid in mucin glycoproteins that reacts as ''free sialic acid'' in the Warren assay", GLYCOBIOLOGY, vol | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| Jeffrey S | |||
| Rohrer ";Analyzing Sialic Acids Using High-Performance Anion-Exchange Chromatography with Pulsed | |||

