Настоящее изобретение касается способа получения акриловой кислоты из этанола и формальдегида. Кроме того, настоящее изобретение касается получения последующих продуктов из полученной таким образом акриловой кислоты.
В настоящее время промышленной получение акриловой кислоты в основном осуществляется исключительно при помощи гетерогенно катализируемого двухступенчатого частичного окисления пропилена (смотрите, например, немецкую заявку на патент DE-A 103 36 386).
Преимущество этого способа проведения процесса состоит в том, что он имеет сравнительно высокую селективность по целевому продукту в пересчете на вступивший во взаимодействие пропилен, что в случае циклического технологического режима для пропилена, не вступившего в реакцию при однократном прохождении, создает возможность высоких выходов акриловой кислоты из использованного пропилена. Кроме того, пропилен обладает исключительно экономичной обратной интеграцией для ископаемого основного нефтяного сырья (то есть, пропилен может получаться из нефти со сравнительно низкими затратами на производство), что в общей сложности дает возможность экономически благоприятного получения акриловой кислоты.
Однако с учетом предсказуемой нехватки ископаемых нефтяных ресурсов на будущее существует потребность в способах получения акриловой кислоты из сырья, которые также могут сравнительно экономически выгодно проводиться без обратной интеграции того же сырья с ископаемым основным нефтяным сырьем и которые одновременно имеют обратную интеграцию своего сырья с основным сырьем, чей диапазон использования во времени превосходит тот же диапазон для нефти.
Международная заявка WO 2005/093010 рассматривает сам пропилен в качестве такого сырья. Она предлагает также на будущее придерживаться двухступенчатого гетерогенно катализируемого газофазного окисления пропилена до акриловой кислоты, однако необходимый при этом пропилен получать, исходя из метанола. Преимущество такого способа проведения процесса состоит в том, что метанол может быть доступен как исходя из ископаемого основного сырья, такого как уголь (например, бурый уголь и каменный уголь; сравните, например, с международной заявкой WO 2010/072424) и природный газ (сравните, например, с международной заявкой WO 2010/067945), оба из которых обладают более значительным диапазоном использования во времени, чем нефть, так и исходя из возобновляемого основного сырья - биомассы, а также и непосредственно из диоксида углерода, содержащегося в атмосфере Земли (соответственно при необходимости при совместном использовании водяного пара или молекулярного водорода) (сравните, например, с G.A.Olah с соавт., Beyond Oil and Gas; The Methanol Economy, Wiley-VCH, 2009).
Однако недостаток способа проведения процесса, предложенного в международной заявке WO 2005/093010 состоит в том, что селективность получения пропилена, исходя из метанола, с помощью известных на сегодняшний день способов получения, в пересчете на вступивший в реакцию метанол, составляет менее 70% мольн., что не может удовлетворять (помимо пропилена, осуществляется, например, также образование этилена и бутилена).
Под ископаемым основным сырьем в данной публикации должно пониматься основное сырье, которое, как, например, бурый уголь, каменный уголь, природный газ и нефть, возникло в геологическом прошлом из продуктов разложения мертвых растений и мертвых животных.
В отличие от этого, под возобновляемым сырьем в данной публикации должно пониматься такое сырье, которое добывается из свежей биомассы, то есть, из выросшего вновь (в настоящее время) и в будущем (современном) растительного и животного материала.
Также уже было предложено (например, в международной заявке WO 2008/023040) получать акриловую кислоту и продукты ее превращения, исходя из возобновляемого сырья - глицерина. Однако недостатком такого способа проведения процесса является то, что глицерин экономически рентабельно доступен в качестве возобновляемого сырья в основном только как сопряженный продукт получения биодизельного топлива. Это является недостатком, поскольку современный энергетический баланс получения биодизельного топлива не может удовлетворять.
Кроме того, в уровне техники предлагается получение акриловой кислоты из пропана (например, в немецкой заявке на патент DE-A 102006024901), который образует компонент сырья природного газа. Однако недостатком такого способа получения акриловой кислоты является, с одной стороны, сравнительно высокая реакционная инертность пропана, а также то обстоятельство, что пропан также образует обладающий хорошим удобством использования, востребованный источник энергии.
Следовательно, задача настоящего изобретения состояла в том, чтобы предоставить альтернативный способ получения акриловой кислоты, который не имеет описанных недостатков способов из уровня техники и особенно обладает удовлетворительной селективностью в отношении образования целевого продукта, исходя из сырья, использованного для его получения.
В соответствии с этим предоставляется способ получения акриловой кислоты из этанола и формальдегида.
Притягательность такого способа проведения процесса, с одной стороны, состоит в том, что этанол представляет собой наиболее стабильное возобновляемое сырье (этанол образуется естественным путем при брожении биомассы, содержащей глюкозу; но можно также исходить из биомассы, содержащей крахмал и целлюлозу, путем того, что предварительно проводят процесс ферментативного или кислотного превращения, который переводит разновидности крахмала и целлюлозы в глюкозу; сравните, например, с международной заявкой WO 2010/092819).
Однако, как правило, этанол также является доступным в промышленности в результате реакции воды с этиленом при добавлении катализаторов, таких как серная кислота или нанесенная на крахмал фосфорная кислота, при температурах примерно 300°C и давлениях около 70 бар, с тем преимуществом, что этилен имеет тесную обратную интеграцию с ископаемыми ресурсами, такими как природный газ и уголь, которые обладают более значительным диапазоном использования во времени, чем нефть (например, Chemie Ingenieur Technik - CIT, Volume 82, страницы 201-213, Issue 3, опубликовано в интернете 9-го февраля 2010, Wiley - VCH Verlag Weinheim, «Alternative Synthesewege zum Ethylen», A.Behr, A.Kleyentreiber и H.Hertge).
Другое преимущество способа проведения процесса согласно изобретению обосновано тем, что формальдегид доступен в результате частичного окисления метанола (например, Catalysis Review 47, страницы с 125 по 174, 2004; европейская заявка на патент ЕР-А 2213370; международная заявка WO 2010/022923; немецкие заявки на патент DE-A 2334981; DE-A 1618413; международная заявка WO 2009/149809; немецкая заявка на патент DE-A 2145851; международные заявки WO 2005/063375; WO 03/053556; WO 2010/034480; WO 2007/059974; немецкая заявка на патент DE-A 102008059701 и издание Ullmann's Encyclopedia of Industrial Chemistry, Fifth, Completely Revised Edition, Volume A 11, 1988, страницы с 624 по 627), и метанол может получаться посредством синтез-газа (газовой смеси из монооксида угдерода и молекулярного водорода), в принципе, из всего углеродсодержащего ископаемого основного сырья и всего углеродсодержащего возобновляемого сырья (необходимый молекулярный водород, как в случае метана (способ получения метана из биогаза или соответственно биомассы, описанный, например, в немецкой заявке на патент DE-A 102008060310 или соответственно европейской заявке на патент EP-А 2220004), уже может содержаться в носителе углерода; как альтернатива, в качестве источника водорода имеется в распоряжении вода, из которой молекулярный водород может добываться, например, при помощи электролиза; источником кислорода, как правило, является воздух; (сравните, например, международные заявки WO 10-060236 и WO 10-060279). В качестве возобновляемого углеродсодержащего сырья для производства синтез-газа подходит, например, лигноцеллюлоза (сравните, например, с международной заявкой WO 10-062936). Также для получения синтез-газа пиролиз биомассы может непосредственно комбинироваться с риформингом с водяным паром.
Таким образом, объектом настоящего изобретения является способ получения акриловой кислоты из этанола и формальдегида, который включает следующие операции:
- через первую реакционную зону A, в которую загружен по меньшей мере один катализатор окисления A, пропускают поток поступающей реакционной газовой смеси A, содержащей реагенты - этанол и молекулярный кислород, а также по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и при прохождении этой реакционной зоны A этанол, содержащийся в поступающей реакционной газовой смеси A, в условиях гетерогенного катализа окисляется до уксусной кислоты и водяного пара, так что возникает газообразная смесь продуктов A, содержащая уксусную кислоту, водяной пар, молекулярный кислород, а также по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и поток газообразной смеси продуктов A покидает реакционную зону A, причем к проходящей сквозь реакционную зону A реакционной газовой смеси A на ее пути через эту реакционную зону A, на выбор, может подаваться дополнительный молекулярный кислород и/или дополнительный инертный газ-разбавитель,
- из потока газообразной смеси продуктов A, покидающего реакционную зону A (без проведения предварительно процесса его разделения), и по меньшей мере одного другого потока веществ, который содержит по меньшей мере один источник формальдегида, получается поток поступающей реакционной газовой смеси B, содержащей уксусную кислоту, водяной пар, молекулярный кислород, по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и формальдегид, в котором содержащееся молярное количество уксусной кислоты nНАс больше, чем содержащееся в нем молярное количество формальдегида nFd,
- через вторую реакционную зону B, в которую загружен по меньшей мере один катализатор альдольной конденсации B, пропускается поток поступающей реакционной газовой смеси B, и при прохождении этой реакционной зоны B формальдегид, содержащийся в поступающей реакционной газовой смеси B, вместе с уксусной кислотой, содержащейся в поступающей реакционной газовой смеси B, в условиях гетерогенного катализа конденсируется до акриловой кислоты и Н2O, так что возникает газообразная смесь продуктов B, содержащая акриловую кислоту, уксусную кислоту, водяной пар, молекулярный кислород, а также по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и поток газообразной смеси продуктов B покидает реакционную зону B, причем к проходящей сквозь реакционную зону B реакционной газовой смеси B на ее пути через эту реакционную зону B, на выбор, может подаваться дополнительный молекулярный кислород и/или дополнительный инертный газ-разбавитель,
- поток газообразной смеси продуктов B, покидающий реакционную зону B, подают в зону разделения T, и в этой зоне разделения T разделяется по меньшей мере на три потока веществ - X, Y и Z, причем
- поток акриловой кислоты, содержащийся в потоке вещества X, больше, чем потоки акриловой кислоты, содержащиеся в потоках веществ Y и Z, вместе взятые,
- поток уксусной кислоты, содержащийся в потоке вещества Y, больше, чем потоки уксусной кислоты, содержащиеся в потоках веществ X и Z, вместе взятые,
- поток инертного газа-разбавителя, отличающегося от водяного пара, содержащийся в потоке вещества Z, больше, чем потоки инертного газа-разбавителя, отличающегося от водяного пара, содержащиеся в потоках веществ X и Y, вместе взятые,
и
- поток вещества Y подается обратно в реакционную зону B и применяется дополнительно для получения поступающей реакционной газовой смеси B.
Согласно изобретению реакционная зона A предпочтительно заполнена по меньшей мере одним катализатором окисления A, содержащим по меньшей мере один оксид ванадия.
Особенно предпочтительно реакционная зона A заполнена по меньшей мере одним катализатором окисления A, содержащим по меньшей мере один оксид ванадия, который помимо по меньшей мере одного оксида ванадия дополнительно содержит по меньшей мере один оксид из группы оксида титана, алюминия, циркония и олова.
Наиболее предпочтительно реакционная зона A заполнена по меньшей мере одним катализатором окисления A, содержащим по меньшей мере один оксид ванадия, который помимо по меньшей мере одного оксида ванадия дополнительно содержит по меньшей мере один оксид титана.
Согласно изобретению катализаторы окисления A содержат ванадий в степени окисления +5. То есть, катализаторы окисления A, содержащие по меньшей мере один оксид ванадия, согласно изобретению целесообразно содержат действующую единицу (оксид ванадия) V2O5 (пентаоксид ванадия).
В отличие от этого, вышеуказанные катализаторы окисления A в случае элементов титана (Ti), циркония (Zr) и олова (Sn) преимущественно содержат эти элементы в степени окисления +4. То есть, катализаторы окисления, помимо оксида ванадия дополнительно содержащие по меньшей мере один оксид из группы оксида титана, оксида циркония и оксида олова, согласно изобретению целесообразно содержат по меньшей мере одну действующую единицу (диоксид элемента) из группы TiO2, ZrO2 и SnO2, причем внутри этой группы действующая единица TiO2 является наиболее предпочтительной для целей изобретения, особенно тогда, когда она присутствует в анатазной модификации.
В целом, в качестве катализаторов окисления A согласно изобретению предпочитают по меньшей мере один смешанный оксидный катализатор A, содержащий по меньшей мере один оксид ванадия, причем определение «смешанный оксид» проявляется в том, что каталитически активный оксид содержит по меньшей мере два отличающихся друг от друга элемента металлов.
В качестве катализатора окисления A, содержащего по меньшей мере один оксид ванадия, согласно изобретению подходят, например, все катализаторы окисления, раскрытые в европейской заявке на патент EP-А 294846. Особенно это те оксидные катализаторы из европейской заявки на патент EP-А 294846, которые, если сбросить со счетов содержащийся кислород, имеют стехиометрию MoxVyZz, где Z может отсутствовать или представлять собой по меньшей мере один определенный элемент металла.
Другими подходящими согласно изобретению катализаторами окисления A, содержащими по меньшей мере один оксид ванадия, являются смешанные оксидные катализаторы, раскрытые в патентной заявке патент US-A 5840971, активная масса которых состоит из элементов ванадия, титана и кислорода.
Также согласно изобретению в качестве катализаторов окисления A подходят катализаторы на носителе, полученные в немецкой заявке на патент DE-A 1642938 для применения при частичном окислении о-ксилола в ангидрид фталевой кислоты, содержащие пентаоксид ванадия и диоксид титана.
Наиболее предпочтительно для способа согласно изобретению в качестве катализаторов окисления A, содержащих по меньшей мере один оксид ванадия, используются те, которые рекомендуются в связи с этим в обосновывающем приоритет европейском патенте с номером заявки 09178015.5.
Подходящие согласно изобретению смешанные оксидные катализаторы A, содержащие по меньшей мере один оксид ванадия, могут получаться, например, в соответствии со способом получения, описанном в патентной заявке США US-A 4048112. При этом исходят из пористого оксида по меньшей мере одного из элементов Ti, алюминия (Al), Zr и Sn. Этот оксид пропитывается раствором соединения ванадия. Далее предпочтительно в значительной мере удаляется растворитель, использованный для получения раствора (как правило, в результате воздействия тепла и/или пониженного давления), а получающийся в результате при этом предшественник катализатора затем прокаливается.
При этом соединение ванадия, как правило, в атмосфере, содержащей молекулярный кислород, разлагается до оксида ванадия. При этом пористый оксид, который следует пропитывать, может иметь любую геометрическое пространственное строение. С точки зрения технологической целесообразности для способа согласно изобретению рассматриваются, прежде всего, шарики, кольца (полые цилиндры), экструдированные профили, таблетированные гранулы, а также монолитные формы. Предпочтительно наибольший размер вышеуказанных геометрических формованных изделий составляет от 1 или 2 до 10 мм (под наибольшим размером формованного изделия в настоящей публикации понимают, как правило, наиболее длинную прямую соединяющую линию между двумя точками, находящимися на поверхности этого формованного изделия). Для растворов, содержащих по меньшей мере одно соединение ванадия, которые следует применять для пропитывания, в качестве соединений ванадия подходят, например, пентаоксид ванадия, ванадилхлорид, ванадилсульфат и метаванадат аммония. В качестве растворителя предпочтительно применяется вода, к которой предпочтительно в качестве активаторов растворения добавлены комплексообразователи, такие как, например, щавелевая кислота. Удаление растворителя, которое следует предпринять после пропитки, а также прокаливание, которое следует провести, могут представлять собой плавно перетекающие друг в друга или также перекрывающиеся процессы.
Однако в предпочтительном варианте сначала удаляют растворитель при температуре от 100 до 200°C. Потом, после этого прокаливают при температуре от 400 до 800°C, или от 500 до 700°C. Прокаливание может осуществляться в атмосфере, содержащей молекулярный кислород, например, на воздухе, или в инертном газе. При этом атмосфера в процессе прокаливания может находиться над массой предшественника, которую следует прокалить, или протекать сквозь эту массу предшественника. Продолжительность прокаливания колеблется, как правило, в интервале от 0,5 часа до 10 часов. Обычно более высокие температуры прокаливания сопровождаются более короткой продолжительностью прокаливания. Если применяются сравнительно низкие температуры прокаливания, то прокаливание продолжается, как правило, в течение более длительного промежутка времени.
Выборочно процедура пропитки-сушки-прокаливания также может повторяться многократно, чтобы достичь желаемого насыщения оксидом ванадия.
В случае смешанных оксидных катализаторов А согласно изобретению, включающих оксид ванадия, в соответствии с изобретением в целом на действующую единицу V2O5 приходится предпочтительно от 0,1 до 60% масс, предпочтительно от 1 до 50% масс, или от 3 до 40% масс, особенно предпочтительно от 5 до 30% масс, соответственно в пересчете на общий вес активной массы.
Кроме того, в случае смешанных оксидных катализаторов A согласно изобретению, включающих оксид ванадия и оксид титана, на действующую единицу TiO2 приходится предпочтительно от 40 до 99,9% масс, предпочтительно от 50 до 99% масс, или от 60 до 97% масс, особенно предпочтительно от 70 до 95% масс, соответственно в пересчете на общий вес активной массы.
Альтернативный способ получения смешанных оксидных катализаторов A, содержащих оксид ванадия и диоксид титана, описывает патентная заявка США US-A 3464930. При этом сначала тонко измельченный диоксид титана обрабатывают соединением ванадия. Потом полученная в результате композиция может как до, так и после ее прокаливания формоваться с получением соответствующей геометрии катализатора. Однако в основном прокаленная композиция также может использоваться в качестве катализатора для соответствующего частичного окисления в форме порошка. Придание формы может, например, осуществляться таким образом, что из порошкообразной активной массы или непрокаленной массы ее предшественника в результате уплотнения (например, при помощи таблетирования или экструдирования или штранг-прессования) до желаемой геометрии катализатора получаются сплошные катализаторы или предшественники сплошного катализатора, причем перед приданием формы на выбор могут добавляться вспомогательные средства, такие как, например, графит или стеариновая кислота, в качестве смазочных средств и/или вспомогательных средств для формования, а также упрочняющие средства, такие как микроволокна из стекла, асбеста, карбида кремния или титаната калия. При этом подходящими согласно изобретению геометриями сплошных катализаторов являются (и в целом, в случае катализаторов окисления A (особенно в случае соответствующих сплошных катализаторов A (они состоят только из активной массы))), например, сплошные цилиндры и полые цилиндры с внешним диаметром и длиной от 1 или от 2 до 10 мм. В случае полых цилиндров целесообразной с технологической точки зрения является толщина стенки от 1 до 3 мм. В остальном, таблетирование предпочтительно может проводиться, как описано в публикациях международных заявок WO 2008/152079, WO 2008/087116, немецких заявок на патент DE-A 102008040094, DE-A 102008040093 и международной заявки WO 2010/000720. Также все геометрии, приведенные в указанных выше публикациях, подходят для катализаторов окисления A согласно изобретению.
Обработка тонко измельченного диоксида титана соединением ванадия может осуществляться, например, с помощью плохорастворимого соединения ванадия, такого как V2O5, в гидротермальных условиях. Однако, как правило, она проводится с помощью растворителя, содержащего растворенное соединение ванадия (например, в воде или в органическом растворителе (к примеру, формамиде или одно- или многоатомном спирте)). При этом в качестве соединений ванадия могут использоваться пентаоксид ванадия, ванадилхлорид, ванадилсульфат и метаванадат аммония. В качестве активаторов растворения могут быть добавлены комплексообразователи, такие как, например, щавелевая кислота.
Однако в качестве альтернативы сплошным катализаторам может также проводиться формование в виде слоистых катализаторов. При этом с помощью полученной порошкообразной активной массы или с помощью порошкообразной, еще не прокаленной массы предшественника при совместном использовании жидкого связующего средства покрывается поверхность инертного формованного изделия носителя (если покрывают еще не прокаленной массой предшественника, то прокаливание осуществляется после нанесения покрытия и, как правило, сушки). Обычно инертные формованные изделия носителя отличаются от каталитической активной массы (для этого в этой публикации в целом применяется также синоним «каталитически активная масса») тем, что они имеют существенно меньшую удельную поверхность. Как правило, их удельная поверхность составляет менее 3 м2/г формованного изделия носителя. Здесь следует придерживаться того, что все данные в этой публикации в отношении удельных поверхностей относятся к определениям согласно стандарту DIN 66131 (определение удельной поверхности твердых веществ с помощью газовой адсорбции (N2) по Брунауеру-Эммету-Теллеру (метод БЭТ)).
В качестве материалов для вышеуказанных инертных формованных изделий носителя подходят, например, кварц, кварцевое стекло, спеченная кремниевая кислота, спеченный или сплавленный глинозем, фарфор, спеченные или сплавленные силикаты, такие как силикат алюминия, силикат магния, силикат цинка, силикат циркония и особенно стеатит (например, стеатит С 220 фирмы CeramTec). Геометрия инертных формованных изделий носителя в основном может иметь нерегулярную форму, причем согласно изобретению предпочтительными являются имеющие регулярную форму формованные изделия носителя, такие как, например, шарики или полые цилиндры. С технологической точки зрения целесообразно наибольший размер вышеуказанных инертных формованных изделий носителя для целей изобретения составляет от 1 до 10 мм.
Покрытие инертных формованных изделий носителя соответствующим тонко измельченным порошком, как правило, осуществляется в подходящем вращающемся резервуаре, например, в дражирующем барабане. Жидкое связующее средство с точки зрения технологической целесообразности распыляется на инертные формованные изделия носителя, и увлаженную связующим средством поверхность инертных формованных изделий, перемешиваемых в дражирующем барабане, посыпают соответствующим порошком (сравните, например, с европейской заявкой на патент EP-A 714 700). Затем адгезионная жидкость, как правило, по меньшей мере частично удаляют из формованных изделий носителя с нанесенным покрытием (например, с помощью пропускания горячего газа сквозь эти формованные изделия носителя с нанесенным покрытием, как это описывает международная заявка WO 2006/094766). Однако в основном, для изготовления соответствующих слоистых катализаторов могут применяться все другие способы нанесения, отмеченные в европейской заявке EP-A 714700 в качестве уровня техники. В качестве жидких связующих средств рассматривают, например, воду и водные растворы (к примеру, глицерина в воде). Например, покрытие формованного изделия носителя также может проводиться путем того, что суспензию порошкообразной массы, которую следует нанести, в жидком связующем средстве (например, воде) распыляют на поверхность инертного формованного изделия носителя (как правило, при воздействии нагревания и высушивающего газа-носителя). В основном, нанесение покрытия может проводиться также в установке с псевдоожиженным слоем или установке для нанесения порошковых покрытий.
Толщину слоя активной массы, нанесенной на поверхность инертного формованного изделия носителя, с точки зрения технологической целесообразности выбирают лежащей в диапазоне от 10 до 2000 мкм или от 10 до 500 мкм, или от 100 до 500 мкм, или от 200 до 300 мкм. Согласно изобретению подходящими слоистыми катализаторами описанного типа для заполнения реакционной зоны A, среди прочих, являются те, у которых инертное формованное изделие носителя представляет собой полый цилиндр с длиной в диапазоне от 3 до 6 мм, внешним диаметром в диапазоне от 4 до 8 мм и толщиной стенки в диапазоне от 1 до 2 мм. Кроме того, для цели изобретения в реакционной зоне A для возможных инертных формованных изделий носителя кольцевидных слоистых катализаторов окисления A являются подходящими все предложенные в немецких заявках на патент DE-A 102010028328 и в DE-A 102010023312, а также в европейской заявке на патент EP-A 714700, геометрии кольца.
Другие способы получения подходящих согласно изобретению смешанных оксидных катализаторов A, содержащих оксид ванадия и диоксид титана, раскрывают патентные заявки США US-A 4228038 и US-A 3954857. Основой способа проведения процесса патентной заявки США US-A 4228038 является обработка диоксида титана водой и оксихлоридом ванадия, пока не будет достигнуто желаемое содержание ванадия. Основой способа проведения процесса заявки на патент США US-A 3954857 является нейтрализация солянокислого раствора пентаоксида ванадия и тетрахлорида титана, приводящая к реакции осаждения. Из получающихся в результате при обоих способах получения оксидных активных масс или из их масс предшественников, как описано выше, могут получаться сплошные и слоистые катализаторы, подходящие для реакционной зоны A.
Другой подходящий согласно изобретению способ проведения процесса для получения активных масс смешанных оксидов, содержащих оксид ванадия и диоксид титана, для катализаторов окисления A раскрывает патент США US 4448897. Основой этого способа проведения процесса является смешивание ванадилалкоксида с алкоксидом титана в водном растворе и последующая сушка, а также прокаливание образующихся при этом осадков. Формование до сплошных или слоистых катализаторов может осуществляться, исходя из соответствующего порошка активной массы или порошка массы ее предшественника, уже описанным (выше) способом. В заключение еще следует уточнить, что катализаторы, содержащие оксид ванадия, предложенные в международной заявке WO 2008/110468 и в немецкой заявке на патент DE-A 19649426, также являются подходящими в качестве катализаторов окисления A для реакционной зоны A способа согласно изобретению. Кроме того, инертные формованные изделия носителя для слоистых катализаторов окисления A (в отличие от катализаторов на носителе A) предпочтительно являются непористыми или соответственно имеют мало пор. В случае катализаторов на носителе A активная масса помещена в пористую структуру формованного изделия носителя.
Помимо уже указанных оксидов, активные массы катализаторов окисления A, подходящих согласно изобретению, дополнительно (к уже указанным оксидам элементов) могут еще содержать один или несколько оксидов металлов из группы бора, кремния, гафния, ниобия, вольфрама, лантана, церия, молибдена, хрома, сурьмы, щелочных металлов и щелочноземельных металлов, а также элементов 5-ой и 6-ой главной подгрупп периодической системы и других переходных металлов. Во многих случаях общее содержание вышеуказанных оксидов, в пересчете на общий вес активной массы, составляет от 1 до 15% масс. Согласно изобретению термин оксид элемента в основном включает также металлаты. Это отрицательно заряженные анионы, которые образованы исключительно из металла и кислорода.
Содержание в поступающей реакционной газовой смеси A этанола в случае способа согласно изобретению составляет, как правило, от 0,3 до 20% объемн., технологически предпочтительно от 0,5 до 15% объемн., особенно предпочтительно от 0,75 до 10% объемн. и наиболее предпочтительно от 1 до 5% объемн.
Содержащееся в поступающей реакционной газовой смеси A молярное количество молекулярного кислорода по в случае способа согласно изобретению с точки зрения технологической целесообразности отмеряется так, что оно больше, чем содержащееся в поступающей реакционной газовой смеси A молярное количество этанола nEt. Как правило, соотношение no: nEt в случае способа согласно изобретению будет составлять по меньшей мере 1,3, лучше по меньшей мере 1,5, предпочтительно по меньшей мере 1,75 и особенно предпочтительно по меньшей мере 2. В обычном случае это соотношение no: nEt, однако, будет составлять не более 10 и в большинстве случаев не более 5.
Вышеприведенные соотношения для способа согласно изобретению являются важными особенно тогда, когда они относятся к nо*: nEt*, причем no* представляет собой молярное количество молекулярного кислорода, в общей сложности подаваемого в реакционную зону A за промежуток времени t, а nEt* молярное количество этанола, в общей сложности подаваемого в реакционную зону A за тот же промежуток времени t в качестве компонента поступающей реакционной газовой смеси A.
Избыток молекулярного кислорода по отношению к реагенту этанолу, рассчитанный на протяжении реакционной зоны A, оказывается предпочтительным для способа согласно изобретению, как для срока службы загруженного катализатора в реакционной зоне A, так и для срока службы загруженного катализатора в реакционной зоне B, поскольку этот избыточный молекулярный кислород в случае способа согласно изобретению вносится в поступающую реакционную газовую смесь B.
В качестве инертного газа-разбавителя в настоящей публикации следует понимать компонент поступающей реакционной газовой смеси, который при условиях в каждой из обеих реакционных зон - A и B ведет себя как инертный и - при самостоятельном рассмотрении для каждой инертной составляющей реакционных газов - в соответствующей реакционной зоне остается химически неизменным более чем на 95% мольн., предпочтительно более чем на 97, или более чем на 98, или более чем на 99% мольн. Указанное выше определение соответствующим образом справедливо также для инертных газов-разбавителей в поступающей реакционной газовой смеси C и относительно реакционной зоны C, которые приводятся в дальнейшей части настоящей публикации.
Примерами инертных газов-разбавителей как для реакционной зоны A, так и для реакционных зон B и C являются Н2O, СO2, N2 и благородные газы, такие как аргон (Ar), а также смеси из вышеупомянутых газов. Инертный газ-разбавитель, среди прочего, сопутствует задаче поглощать теплоту реакции, выделяющуюся в реакционной зоне A, и тем самым ограничивать так называемую температуру горячей точки в реакционной зоне A, а также благоприятным образом регулировать характеристики воспламенения реакционной газовой смеси A. При этом под температурой горячей точки понимают максимальную температуру реакционной газовой смеси A на ее пути через реакционную зону A.
Водяной пар в качестве инертного газа-разбавителя в обеих реакционных зонах A и B занимает особенное место в сравнении с другими возможными инертными газами-разбавителями. Это можно объяснить тем, что присутствие водяного пара в поступающей реакционной газовой смеси A в реакционной зоне A облегчает десорбцию желаемого продукта частичного окисления с поверхности катализатора, что позитивно влияет на селективность образования уксусной кислоты. Кроме того, водяной пар по сравнению, например, с N2 и благородными газами имеет повышенную молярную теплоемкость.
Таким образом, согласно изобретению предпочтительно поступающая реакционная газовая смесь A может содержать от 1 до 40% объемн. Н2О. Однако поскольку присутствие водяного пара в реакционной зоне B, как правило, в определенном объеме уменьшает желаемую альдольную конденсацию и, кроме того, повышает необходимые энергетические затраты для отделения в зоне разделения T от газообразной смеси продуктов В потока вещества X, обогащенного по содержанию акриловой кислоты (акриловая кислота имеет повышенное сродство к H2O), то согласно изобретению предпочтительными являются величины содержания водяного пара в поступающей реакционной газовой смеси A от 1 до 20% объемн. При этом также принимают во внимание тот факт, что как в реакционной зоне A, так и в реакционной зоне B в качестве побочного продукта образуется H2O. Согласно изобретению особенно предпочтительно содержание водяного пара в поступающей реакционной газовой смеси A устанавливается от 5 до 15% объемн. или от 7,5 до 12,5% объемн.
В качестве инертного газа-разбавителя, отличающегося от водяного пара, в случае способа согласно изобретению, как в реакционной зоне A, так и в реакционной зоне B предпочтительно используют молекулярный азот. Это благоприятно не в последнюю очередь потому, что молекулярный азот в воздухе выступает как природное сопутствующее вещество молекулярного кислорода, что относит воздух к предпочтительному источнику молекулярного кислорода, необходимого в реакционной зоне A. Однако, само собой разумеется, согласно изобретению в качестве источника кислорода также могут применяться чистый молекулярный кислород или воздух, обогащенный молекулярным кислородом, или другие газовые смеси из молекулярного кислорода и инертного газа-разбавителя.
Согласно изобретению по меньшей мере 80% объемн., предпочтительно по меньшей мере 90% объемн., часто по меньшей мере 95% объемн. и иногда 100% объемн. содержащегося в поступающей реакционной газовой смеси A инертного газа-разбавителя, отличающегося от водяного пара, приходится на молекулярный азот. То же самое относится и к характеристикам инертного газа в поступающей реакционной газовой смеси B. Как правило, поступающая реакционная газовая смесь A, помимо инертного газа-разбавителя, отличающегося от водяного пара, дополнительно еще содержит водяной пар в качестве инертного газа-разбавителя. Обычно содержание в поступающей реакционной газовой смеси A инертных газов-разбавителей, отличающихся от водяного пара, будет составлять по меньшей мере 5% объемн., однако, как правило, не более чем 95% объемн. Обычное содержание в поступающей реакционной газовой смеси A инертного газа-разбавителя, отличающегося от водяного пара, составляет от 10 до 90% объемн., предпочтительно от 30 до 90% объемн., особенно предпочтительно от 50 до 90% объемн. и наиболее предпочтительно от 60 до 90% объемн., или от 70 до 90% объемн., и прежде всего, от 75 до 85% объемн.
Таким образом, содержание в поступающей реакционной газовой смеси A молекулярного азота может составлять по меньшей мере 5% объемн., предпочтительно по меньшей мере 10% объемн., особенно предпочтительно по меньшей мере 20 или по меньшей мере 30% объемн. или соответственно по меньшей мере 40% объемн., однако, как правило, не более чем 95% объемн. Обычное содержание в поступающей реакционной газовой смеси A молекулярного азота может составлять от 10 до 90% объемн., предпочтительно от 30 до 90% объемн., особенно предпочтительно от 50 до 90% объемн. и наиболее предпочтительно от 60 до 90% объемн., или от 70 до 90% объемн., и прежде всего от 75 до 85% объемн.
Температура кипения инертного газа-разбавителя, отличающегося от водяного пара (в пересчете на давление 105 Па=1 бар), обычно находится заметно ниже той же температуры для водяного пара (в пересчете на то же давление), отчего в случае способа согласно изобретению поток вещества Z, как правило, обогащен по содержанию инертных газов-разбавителей, отличающихся от водяного пара (например, N2 и СO2).
Следовательно, с точки зрения технологии целесообразно также в качестве источника инертного газа-разбавителя, отличающегося от водяного пара, подавать обратно в реакционную зону A частичный поток из потока вещества Z для формирования поступающей реакционной газовой смеси A (технологический процесс с использованием газа в цикле). С точки зрения технологии благоприятно, когда разделение газообразной смеси продуктов B в зоне разделения T проводят таким образом, что поток вещества Z также имеет соответствующую долю водяного пара, а, следовательно, при использовании описанного выше технологического процесса с использованием газа в цикле также может исполнять роль источника для водяного пара, предпочтительно применяемого совместно в поступающей реакционной газовой смеси A (или соответственно C). Разумеется, частичные потоки из потока вещества Z могут подаваться обратно не только в реакционную зону A, но и в реакционную зону B (а также C).
Температура реакционной газовой смеси A (понятие реакционной газовой смеси A в настоящей заявке включает все встречающиеся в реакционной зоне A газовые смеси, которые находятся между поступающей реакционной газовой смесью A и газообразной смесью продуктов A; в полном соответствии с этим, понятие реакционной газовой смеси B включает все встречающиеся в реакционной зоне B газовые смеси, которые находятся между поступающей реакционной газовой смесью B и газообразной смесью продуктов B) в случае способа согласно изобретению внутри реакционной зоны A обычно будет находиться в диапазоне от 100°C до 450°C, предпочтительно в диапазоне от 150°C до 400°C и особенно предпочтительно в диапазоне от 150°C до 350°C или от 150°C до 300°C. Но, безусловно, вышеуказанный температурный диапазон может также составлять от 200°C до 300°C. Понятие температуры реакционной газовой смеси A (в данной публикации также обозначаемой как температура реакции в реакционной зоне A) при этом подразумевает, в первую очередь, ту температуру, которую реакционная газовая смесь A имеет от достижения степени превращения этанола, содержащегося в поступающей реакционной газовой смеси A, по меньшей мере 5% мольн. до достижения соответствующей конечной степени превращения этанола внутри реакционной зоны A.
Согласно изобретению температура реакционной газовой смеси A предпочтительно на протяжении всей реакционной зоны A лежит в вышеуказанном диапазоне температур. Предпочтительно также поступающую реакционную газовую смесь A подают в реакционную зону A уже с температурой, находящейся в диапазоне от 100°C до 350°C. Однако часто на входе в реакционную зону A в направлении движения потока до собственно каталитически активного заполнения катализатором в реакционной зоне A находится заполнение реакционной зоны A твердым инертным материалом или заполнение каталитически активным катализатором в высоком разбавлении инертным материалом такого типа. При прохождении такого предварительного заполнения реакционной зоны A температура поступающей реакционной газовой смеси A, подаваемой в реакционную зону A, может сравнительно просто устанавливаться на величину, с которой реакционная газовая смесь A должна поступать непосредственно в каталитически активное заполнение катализатором в этой реакционной зоне A. В основном заполнение реакционной зоны A по меньшей мере одним катализатором окисления A может выполняться в виде псевдоожиженного слоя. Однако с точки зрения технологии это заполнение реакционной зоны A катализатором окисления A предпочтительно выполнено в виде неподвижного слоя.
В принципе реакционная газовая смесь A может как продавливаться, так и просасываться сквозь реакционную зону A. В соответствии с этим рабочее давление (=абсолютному давлению) внутри реакционной зоны A может составлять как ≥105 Па, так и <105 Па. С точки зрения технологической целесообразности рабочее давление в реакционной зоне A будет составлять ≥105 Па. Как правило, рабочее давление в реакционной зоне A будет лежать в диапазоне от 1,2⋅105 Па до 50⋅105 Па, предпочтительно в диапазоне от 1,5⋅105 до 20⋅105 Па и особенно предпочтительно в диапазоне от 2⋅105 до 106 Па или в диапазоне от 2⋅105 до 6⋅105 Па.
Исполнение реакционной зоны A с точки зрения технологической целесообразности может производиться в так называемом «реакторе с теплообменом». Этот реактор имеет по меньшей мере один первичный объем и по меньшей мере один вторичный объем, оба из которых отделены друг от друга с помощью разделительной стенки. По меньшей мере в одном первичном объеме находится засыпанный катализатор, который включает по меньшей мере один катализатор окисления A, и через который протекает реакционная газовая смесь A. Одновременно через вторичный объем протекает жидкий теплоноситель, и через разделительную стенку между обоими объемами имеет место теплообмен, который преследует цель контролировать и регулировать температуру реакционной газовой смеси A на ее пути сквозь слой катализатора (поддерживать температурный режим в реакционной зоне A).
В качестве примеров, как подходящие согласно изобретению реакторы с теплообменом для реализации реакционной зоны A, следует назвать кожухотрубчатый реактор (как он предлагается, например, в европейской заявке на патент EP-A 700714 и в цитируемом в той публикации уровне техники) и пластинчатый реактор с теплообменом (как он предлагается, например, в публикациях европейской заявки на патент EP-A 1651344, немецких заявок на патент DE-A 10361456, DE-A 102004017150 и в отмеченном в этих публикациях уровне техники). В случае кожухотрубчатого реактора слой катализатора, через который протекает реакционная газовая смесь A, предпочтительно находится в его трубках (первичном объеме), а через объем, окружающий реакционные трубки (вторичный объем), подают по меньшей мере один теплоноситель. В качестве теплоносителей для реакторов с теплообменом принимают во внимание, например, расплавы солей, теплонесущие масла, ионные жидкости и водяной пар. Как правило, используемые в промышленности кожухотрубчатые реакторы содержат по меньшей мере от трех тысяч до нескольких десятков тысяч параллельно расположенных реакционных трубок (трубок реактора). Но, разумеется, исполнение реакционной зоны A также может осуществляться в реакторе с псевдоожиженным слоем или в микрореакторе.
Традиционные реакторы и микрореакторы отличаются своими характеристическими размерами и особенно характеристическими размерами реакционного объема, заключающего в себе слой катализатора, через который проходит реакционная газовая смесь.
Частичное окисление этанола до уксусной кислоты согласно изобретению протекает за две следующие друг за другом стадии. На первой стадии этанол частично окисляется до ацетальдегида, а на второй (следующей после первой) стадии этот ацетальдегид частично окисляется до уксусной кислоты.
Так, неожиданным образом было обнаружено, что для образования целевого продукта в реакционной зоне A является предпочтительным использовать для второй стадии реакции катализаторы окисления A, активная масса которых помимо оксида ванадия еще содержит оксид молибдена. То есть, для второй стадии частичного окисления этанола до уксусной кислоты согласно изобретению предпочтительно применяются полиметаллические оксидные катализаторы A, активная масса которых помимо кислорода содержит еще по меньшей мере элементы молибден (Мо) и ванадий (V).
Катализаторы окисления этого типа известны из литературы как катализаторы для гетерогенно катализируемого частичного газофазного окисления акролеина до акриловой кислоты. Так, неожиданным образом было обнаружено, что все полиметаллические катализаторы окисления вышеуказанного типа, которые известны из уровня техники как катализаторы для гетерогенно катализируемого частичного газофазного окисления акролеина до акриловой кислоты, также предпочтительно подходят в качестве катализаторов окисления для второй стадии частичного окисления этанола до уксусной кислоты - частичного окисления ацетальдегида до уксусной кислоты в реакционной зоне A.
Такие активные массы полиметаллических оксидов, содержащие Мо и V, включая катализаторы, содержащие их, могут быть взяты, например, из публикаций патентных заявок США US-A 3775474, US-A 3954855, US-A 3893951, US-A 4339355, европейских заявок на патент EP-A 614872, EP-A 1041062, международных заявок WO 03/055835 и WO 03/057653.
Однако особенно подходят для второй стадии в реакционной зоне A настоящего изобретения также содержащие Мо и V активные массы полиметаллических оксидов, включая катализаторы, содержащие их, такие как предложены в публикациях немецких заявок на патент DE-A 10325487, DE-A 10325488, европейской заявки на патент EP-A 427508, немецких заявок на патент DE-A 2909671, DE-C 3151805, DE-AS 2626887, DE-A 4302991, европейских заявок на патент EP-A 700893, EP-A 714700, немецких заявок на патент DE-A 19736105, DE-A 19927624, DE-A 102010028328 и DE-A 10360057. Это особенно относится к приведенным в качестве примеров вариантам исполнения европейской заявки на патент EP-A 714700, немецких заявок на патент DE-A 19736105, DE-A 19927624, DE-A 10360057, а также DE-A 102010028328.
При этом для соответствующей второй реакционной стадии в реакционной зоне A особенно хорошо подходят те катализаторы окисления A, активная масса которых представляет собой по меньшей мере один полиметаллический оксид, который помимо V и Мо дополнительно содержит по меньшей мере один из следующих элементов - вольфрам (W), ниобий (Nb), тантал (Та), хром (Cr) и церий (Се), а также по меньшей мере один из следующих элементов - медь (Cu), никель (Ni), кобальт (Со), железо (Fe), марганец (Mn) и цинк (Zn).
Среди этих катализаторов для катализа второй стадии частичного окисления этанола до уксусной кислоты прежде всего подходят активные массы полиметаллических оксидов общей формулы I
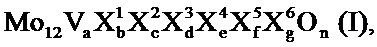
в которой переменные имеют следующие значения:
X1=W, Nb, Та, Cr и/или Ce,
X2=Cu, Ni, Со, Fe, Mn и/или Zn,
X3=Sb и/или Bi,
X4=один или несколько щелочных металлов,
X5=один или несколько щелочноземельных металлов,
X6=Si, Al, Ti и/или Zr,
A=от 1 до 6,
b=от 0,2 до 4,
c=от 0,5 до 1,8
d=от 0 до 40
e=от 0 до 2,
f=от 0 до 4,
g=от 0 до 40, и
n=стехиометрический коэффициент элементарного кислорода, который определяется с помощью стехиометрических коэффициентов элементов, отличных от кислорода, а также числом их зарядов в I.
В наиболее предпочтительных вариантах исполнения согласно изобретению переменные общей формулы I имеют следующие значения:
X1=W, Nb и/или Cr,
X2=Cu, Ni, Co, Fe, Mn и/или Fe,
X3=Sb,
X4=натрий (Na) и/или калий (K),
X5=кальций (Ca), стронций (Sr) и/или барий (Ba),
X6=кремний (Si), алюминий (Al) и/или титан (Ti),
a=от 1,5 до 5,
b=от 0,5 до 2,
c=от 0,5 до 3,
d=от 0 до 2,
e=от 0 до 0,2,
f=от 0 до 1,
g=от 0 до 1, и
n=стехиометрический коэффициент элементарного кислорода, который определяется с помощью стехиометрических коэффициентов элементов, отличных от кислорода, а также числом их зарядов в I.
Активные массы полиметаллических оксидов, содержащие V и Мо, особенно этих оксидов общей формулы I, могут использоваться для катализа частичного окисления ацетальдегида до уксусной кислоты как в порошкообразной форме, так и в виде сплошных катализаторов, формованными с получением определенной геометрии катализатора. В отношении подходящих при этом согласно изобретению геометрических характеристик катализатора соответствующим образом справедливы уже сделанные в этой публикации высказывания в отношении возможных геометрических характеристик сплошных катализаторов окисления A.
Однако при катализе соответствующей второй стадии реакции согласно изобретению описанные активные массы полиметаллических оксидов, содержащие V и Мо, предпочтительно применяются в форме слоистых катализаторов (то есть, нанесенными на внешнюю поверхность предварительно сформованных инертных носителей катализатора (формованных изделий носителя)). В отношении подходящих при этом согласно изобретению геометрических характеристик формованных изделий носителя соответствующим образом справедливы уже сделанные в этой публикации высказывания в связи со слоистыми катализаторами окисления A. Здесь также предпочтительными геометриями формованных изделий носителя являются шарики и кольца, наибольший размер которых может составлять от 1 до 10 мм, часто от 2 до 8 мм или от 3 до 6 мм. Согласно изобретению оптимальными геометрическими характеристиками кольца являются полые цилиндрические формованные изделия носителя с длиной от 2 до 10 мм, внешним диаметром от 4 до 10 мм и толщиной стенки от 1 до 4 мм. Предпочтительно эти полые цилиндрические формованные изделия носителя имеют длину от 3 до 6 мм, внешний диаметр от 4 до 8 мм и толщину стенки от 1 до 2 мм. В качестве примерной геометрии следует привести геометрию кольца 7 мм×3 мм×4 мм (внешний диаметр×длина×внутренний диаметр). Толщина слоя из каталитически активной оксидной массы, нанесенной на формованное изделие носителя, с точки зрения технологической целесообразности, как правило, составляет от 10 до 1000 мкм. Предпочтительно эта толщина слоя составляет от 10 до 500 мкм, особенно предпочтительно от 100 до 500 мкм и наиболее предпочтительно от 200 до 300 мкм. Предпочтительно эта толщина слоя при рассмотрении отдельного слоистого катализатора является насколько возможно одинаковой. При изготовлении большей партии продукции слоистых катализаторов толщина слоя при рассмотрении нескольких формованных изделий слоистых катализаторов также является насколько возможно одинаковой. В качестве материалов для инертных формованных изделий носителя рассматривают уже указанные в этой публикации инертные материалы. В качестве таких возможных инертных материалов следует еще раз упомянуть оксид алюминия, диоксид кремния, силикаты, такие как глина, каолин, стеатит, пемза, силикат алюминия и силикат магния, карбид кремния, диоксид циркония и диоксид тория (согласно изобретению особенно предпочтительным инертным материалом для формованных изделий носителя является стеатит типа C 220 фирмы CeramTec). Формованные изделия носителя с заметно сформированной неровностью поверхности (например, полые цилиндры с мелкозернистым покрытием, такие как описаны в базе данных раскрываемой информации Research Disclosure с номером 532036, опубликованной в августе 2008)) являются предпочтительными для получения формованных изделий слоистых катализаторов. В остальном, формованные изделия носителя предпочтительно являются насколько возможно непористыми.
Для изготовления формованных изделий слоистого катализатора сначала может как таковая получаться каталитически активная оксидная масса общей формулы I. Это обычно осуществляется путем того, что из источников элементарных составляющих каталитически активной оксидной массы получают как можно более плотную, предпочтительно тонко измельченную, составленную в соответствии с их стехиометрией сухую смесь (массу предшественника), и эту массу прокаливают (термически обрабатывают) при температурах от 350 до 600°C. Это прокаливание может проводиться как в атмосфере инертного газа, так и в окислительной атмосфере, такой как, например, воздух (или другая смесь из инертного газа и кислорода), а также в восстановительной атмосфере (например, смеси из инертного газа и восстанавливающих газов, таких как H2, NH3, CO, метан и/или акролеин, или соответственно по существу в указанных газах с восстанавливающим действием). Продолжительность прокаливания может составлять от нескольких минут до нескольких часов и обычно убывает с подъемом температуры прокаливания. Хорошо подходящий согласно изобретению способ прокаливания описывает, например, международная заявка WO 95/11081.
В качестве источников для элементарных составляющих каталитически активной оксидной массы общей формулы I рассматривают (как в общем в случае катализаторов окисления A), в частности, такие соединения, которые уже представляют собой оксиды, и/или такие соединения, которые могут переводиться в оксиды в результате нагревания, по меньшей мере в присутствии кислорода. Тщательное смешивание исходных соединений (источников) может осуществляться в сухом или во влажном виде. Если оно осуществляется в сухом виде, то эти исходные соединения целесообразным образом используются в виде тонко измельченного порошка и после смешивания и при необходимости уплотнения подвергаются прокаливанию. Однако предпочтительно это тщательное смешивание осуществляется во влажном виде. Обычно при этом исходные соединения смешивают друг с другом в форме водного раствора и/или суспензии.
Особенно плотные сухие смеси получаются при описанном способе смешивания тогда, когда исходят исключительно из источников элементарных составляющих, присутствующих в растворенной форме.
В качестве растворителя предпочтительно используют воду. Затем полученная жидкая (например, водная, масса высушивается, причем процесс сушки предпочтительно осуществляется путем распылительной сушки жидкой (например, водной) смеси с температурами на выходе от 100 до 150°C. Осушающий поток газа с точки зрения технологической целесообразности представляет собой воздух или молекулярный азот.
Полученная после прокаливания каталитически активная оксидная масса затем, например, с помощью размалывания переводится в тонко измельченный порошок, который потом обычно с помощью жидкого связующего средства может наноситься на внешнюю поверхность формованного изделия носителя. При этом тонкость измельчения каталитически активной оксидной массы, которую надлежит нанести на формованное изделие носителя, разумеется, как описано в уровне техники (сравните, например, с европейской заявкой на патент EP-A 714700), подбирается к желаемой толщине слоя.
Например, формованные изделия носителя контролируемо увлажняются, к примеру, с помощью распыления, жидким связующим средством, и увлаженные таким образом формованные изделия носителя посыпают тонко измельченной каталитически активной оксидной массой (сравните, например, европейскую заявку на патент EP-A 714 700 и немецкую заявку на патент DE-A102010023312). Затем адгезионную жидкость по меньшей мере частично удаляют из увлажненных формованных изделий носителя, покрытых активной оксидной массой (например, пропусканием горячего газа; сравните с международной заявкой WO 2006/094766). Однако для изготовления подходящих согласно изобретению слоистых катализаторов с активной массой из полиметаллического оксида I могут также применяться все другие способы нанесения, отмеченные в европейской заявке EP-A 714700 в качестве уровня техники. В качестве жидких связующих средств рассматривают, например, воду и водные растворы.
Однако в основном для изготовления слоистых катализаторов согласно изобретению можно также поступать таким образом, что на поверхность изделия носителя сначала наносят тонко измельченную массу предшественника, а прокаливание этой массы предшественника до каталитически активной оксидной массы общей формулы I проводят только потом, то есть, уже при нахождении на поверхности формованного изделия носителя.
В качестве источников элементов для получения каталитически активной оксидной массы общей формулы I помимо оксидов элементов в целом, прежде всего, рассматривают галогениды, нитраты, формиаты, оксалаты, ацетаты, карбонаты и гидроксиды. В качестве источника молибдена с точки зрения технологической целесообразности применяется тетрагидрат гептамолибдата аммония. Предпочтительным источником ванадия является метаванадат аммония, а в случае совместного применения элемента вольфрама, предпочтительным источником элемента является гептагидрат паравольфрамата аммония. В качестве источников элементарных составляющих меди для получения каталитически активной оксидной массы общей формулы I рассматривают, в частности, пентагидрат сульфата меди(II), гидрат нитрата меди(II) (содержание Cu=21,6% масс.) и моногидрат ацетата меди(II), среди которых предпочтительным является последний. Процесс термической обработки массы предшественника каталитически активной оксидной массы общей формулы I проводят согласно изобретению предпочтительно в соответствии со способом проведения процесса, описанным и приводимым в качестве примера в немецкой заявке на патент DE-A 10360057. В случае кольцевидных формованных изделий носителя предпочтительный способ нанесения каталитически активной оксидной массы общей формулы I на их поверхность представляет собой способ, описанный в европейской заявке на патент EP-A 714700. Тогда предпочтительным связующим средством является водный раствор из 75% масс, воды и 25% масс, глицерина. В остальном, можно поступать, как описано в примерах исполнения 1 и 2 немецкой заявки на патент DE-A 10360057. В случае шарообразных формованных изделий носителя предпочтительным связующим средством является вода.
Если в случае способа согласно изобретению в реакционной зоне A для второй стадии гетерогенно катализируемого частичного окисления этанола до уксусной кислоты используют по меньшей мере один катализатор окисления A, активная масса которого представляет собой полиметаллический оксид (например, такой оксид общей формулы I), который помимо кислорода содержит еще по меньшей мере элементы Mo и V, то согласно изобретению предпочтительно для первой стадии частичного окисления этанола до уксусной кислоты используется по меньшей мере один катализатор окисления A, активная масса которого представляет собой смешанный оксид, который помимо кислорода и V еще содержит по меньшей мере один из элементов Ti (предпочтительно), Zr и Al (и, как правило, не содержит Mo), (то есть, по меньшей мере один катализатор окисления A, который помимо оксида ванадия (предпочтительно V2O5) еще включает в себя по меньшей мере один оксид Ti (предпочтительно; предпочтительно TiO2), Zr и Al).
Такое заполнение реакционной зоны A катализатором окисления A согласно изобретению является предпочтительным, поскольку при высоких, рассчитанных на однократное прохождение реакционной смеси A через реакционную зону A степенях превращения этанола оно обеспечивает особенно высокие селективности по целевому продукту - уксусной кислоте.
То есть, согласно изобретению заполнение реакционной зоны A по меньшей мере одним катализатором окисления A предпочтительно включает в себя в направлении движения потока реакционной смеси A два следующих друг за другом в пространстве (в указанной числовой последовательности) участка 1 и 2 (между ними обоими, на выбор, может находиться участок, заполненный исключительно инертными формованными изделиями, что, однако, согласно изобретению является менее предпочтительным).
Активная масса по меньшей мере одного катализатора окисления A участка 1 (на участке 1), которая здесь также обозначается как каталитически активная масса 1, при этом представляет собой смешанный оксид, который помимо кислорода и ванадия содержит еще по меньшей мере один из элементов Ti (предпочтительно), Zr и Al (и, как правило, не содержит Mo), в то время как активная масса по меньшей мере одного катализатора окисления A участка 2 (на участке 2), которая здесь также обозначается как каталитически активная масса 2 (и отличается от каталитически активной массы 1), представляет собой смешанный оксид, содержащий по меньшей мере один из элементов V и Mo (предпочтительно такой оксид общей формулы I). То есть, по меньшей мере один катализатор окисления A участка 1 (первого участка в направлении движения потока реакционной газовой смеси A) помимо оксида ванадия (предпочтительно V2O5) содержит еще по меньшей мере один оксид Ti (предпочтительно; предпочтительно TiO2), Zr и Al (и, как правило, не содержит оксида Мо), в то время как по меньшей мере один катализатор окисления A участка 2 помимо оксида ванадия содержит еще по меньшей мере один оксид молибдена.
Особенно предпочтительно активная масса по меньшей мере одного катализатора окисления A участка 1 содержит, или эта активная масса состоит из количеств от 1 до 50% масс. V2O5 и от 50 до 99% масс. TiO2, предпочтительно от 3 до 40% масс. V2O5 и от 60 до 97% масс. TiO2, а также наиболее предпочтительно от 5 до 30% масс.V2O5 и от 70 до 95% масс. TiO2. Предпочтительно в вышеупомянутых случаях TiO2 присутствует в анатазной модификации.
Согласно изобретению поддержание температурного режима на участке 1 предпочтительно осуществляется независимо от поддержания температурного режима на участке 2. Технологически предпочтительно поддержание температурного режима на участках 1 и 2 осуществляется так, что на протяжении длины (каталитически активного) участка 1 средняя арифметическая температура реакционной газовой смеси A (это температура 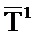
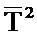
При этом предпочтительно 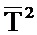
Длина обоих участков заполнения 1 и 2 согласно изобретению обычно выбирается таким образом, что степень превращения этанола, достигнутая за однократное прохождение реакционной газовой смеси A сквозь участок 1, составляет по меньшей мере 90% мольн., регулярно даже по меньшей мере 95% мольн., а степень превращения ацетальдегида, достигнутая на участке 2, также по меньшей мере 90% мольн. и регулярно даже по меньшей мере 95% мольн. Как правило, степень превращения этанола, достигнутая за однократное прохождение реакционной газовой смеси A сквозь участки 1 и 2, составляет ≥97% мольн., во многих случаях ≥98% мольн. и часто ≥99% мольн. Селективность сопровождающего это образования уксусной кислоты обычно составляет ≥85% мольн., часто ≥86% мольн. или ≥87% мольн., а во многих случаях даже ≥88% мольн. или ≥90% мольн.
Исполнение обоих участков 1 и 2 реакционной зоны A в простом способе возможно, например, в двух расположенных друг за другом реакторах с теплообменом (например, двух кожухотрубчатых реакторах), в соответствующих вторичных объемах которых соответственно протекает независимый жидкий теплоноситель. По меньшей мере один первичный объем первого из двух реакторов по направлению движения потока заключает в себе участок 1, в то время как по меньшей мере один первичный объем второго из двух реакторов по направлению движения потока заключает в себе участок 2.
Однако исполнение обоих участков 1 и 2 реакционной зоны A также может осуществляться, например, в так называемом двухзонном реакторе, таком как предлагает в качестве примера немецкая заявка на патент DE-A 2830765. При этом оба участка 1 и 2 размещены в пространстве друг за другом в одном первичном объеме, а граничащий с первичным объемом вторичный объем разделен на два частичных объема, из которых один распространяется на участок 1, а другой на участок 2, и через оба независимо протекают теплоносители, имеющие различные температуры на входе. Двухзонные реакторы относятся также к публикациям немецких заявок на патент DE-A 10313210, DE-A 10313209, DE-A 19948523, DE-A 19948241, DE-A 10313208 и международной заявки WO 2007/082827. Согласно изобретению предпочтительно используют двухзонные кожухотрубчатые реакторы.
Если оба участка 1 и 2 реализованы в одном двухзонном реакторе, поступающая реакционная газовая смесь A уже должна иметь (включать в себя) все компоненты, необходимые для частичного окисления этанола до уксусной кислоты в объеме необходимом для реакции. Если исполнение осуществляется в двух подключенных друг за другом реакторах с теплообменом, к реакционной газовой смеси A между двумя реакторами еще может добавляться, например, молекулярный кислород и/или инертный газ.
Согласно изобретению катализаторы окисления A, активная масса которых содержит по меньшей мере один оксид ванадия (также такие массы общей формулы I), предпочтительно при применении для гетерогенно катализируемого частичного окисления этанола до уксусной кислоты имеют также полностью удовлетворительный срок службы тогда, когда для получения поступающей реакционной газовой смеси A используют биоэтанол, то есть, этанол, который получается из возобновляемого основного сырья - биомассы. Биоэтанол в силу производственных причин в качестве загрязняющих примесей, как правило, содержит по меньшей мере одно химическое соединение, которое в химически связанном виде имеет элемент серу. В пересчете на массу этанола, содержащегося в биоэтаноле, и при выражении через массу серы, содержащейся в таких соединениях серы, содержание в биоэтаноле таких серусодержащих соединений, как правило, составляет ≥1 масс. частей на млн часто ≥2 масс. частей на млн или ≥3 масс. частей на млн. Как правило, вышеупомянутое содержание серы в биоэтаноле составляет ≤200 масс. частей на млн или ≤150 масс. частей на млн или соответственно частью ≤100 масс. частей на млн.
В качестве таких загрязнений, содержащих серу, рассматриваются, например, диметилсульфат и диметилсульфоксид. Определение содержания соединений серы может осуществляться с помощью газовой хроматографии. Следует отметить, что катализаторы окисления A, активная масса которых содержит по меньшей мере один оксид ванадия, являются очевидно в значительной мере устойчивыми по отношению к соединениям серы такого типа в качестве компонентов поступающей реакционной газовой смеси A, так что соответствующие содержания соединений серы в поступающей реакционной газовой смеси A, пересчитанные на содержание этанола в этой поступающей реакционной газовой смеси A, в случае способа согласно изобретению могут иметь отклонения. Однако, разумеется, для способа согласно изобретению в качестве источника этанола также рассматривается биоэтанол, у которого соответствующим образом измеренное содержание соединений серы было понижено до величины <1 масс. части на млн.
Например, для способа согласно изобретению может использоваться биоэтанол, который удовлетворяет следующей спецификации:
Согласно изобретению важным является то, что сера, содержащаяся в поступающей реакционной газовой смеси A, находящаяся химически связанной в соответствующих загрязняющих примесях, в случае способа согласно изобретению в качестве компонента поступающей реакционной газовой смеси B вносится в реакционную зону B. При этом удивляет то, что катализаторы альдольной конденсации, которые согласно изобретению следует использовать в реакционной зоне B, особенно предпочтительные согласно изобретению, имеют в полном объеме удовлетворительную невосприимчивость по отношению к соединениям, содержащим химически связанную серу.
Для получения поступающей реакционной газовой смеси A биоэтанол, используемый в качестве исходного сырья как таковой, переводится в паровую фазу и вводится в поступающую реакционную газовую смесь A. Разумеется, в случае способа согласно изобретению таким образом могут также использоваться водные растворы биоэтанола. В основном для способа согласно изобретению в качестве источника этанола также может использоваться получающаяся при выработке биоэтанола, содержащая растворенный биоэтанол водная плодово-ягодная мезга. Эту мезгу подвергают фильтрации и отфильтровывают содержащиеся в ней твердые материалы. Фильтрат переводится в паровую фазу и подается на получение поступающей реакционной газовой смеси A.
В остальном, нагрузка на находящийся в реакционной зоне A, включающий по меньшей мере один катализатор окисления A неподвижный слой катализатора по этанолу, содержащемуся в поступающей реакционной газообразной смеси A, в случае способа согласно изобретению составляет, например, от 20 до 500, предпочтительно от 30 до 100 и особенно предпочтительно от 50 до 100 нл/л⋅ч. Термин «нагрузка» при этом используется так, как определено в немецкой заявке на патент DE-A 19927624.
В качестве источника формальдегида, необходимого для поступающей реакционной газовой смеси B, для способа согласно изобретению рассматривают различное исходное сырье. Одним возможным источником являются водные растворы формальдегида (сравните, например, с немецкой заявкой на патент DE-A 102008059701), которые могут быть в продаже, например, с содержанием формальдегида от 35 до 50% масс, как формалин (например, формальдегид 49-2015 фирмы BASF SE). Обычно формалин в качестве стабилизатора еще содержит незначительные количества метанола. Эти количества могут составлять, в пересчете на массу формалина, от 0,5 до 20% масс, предпочтительно от 0,5 до 5% масс, и особенно предпочтительно 0,5 до 2% масс. Будучи переведенным в паровую фазу, этот формалин может непосредственно использоваться для получения поступающей реакционной газовой смеси B. Однако недостатком формалина как источника формальдегида является то, что помимо формалина он также содержит воду, что оказывает негативное влияние на положение равновесия реакции в реакционной зоне B.
В качестве альтернативного источника формальдегида рассматривают триоксан. Триоксан представляет собой гетероциклическое соединение из группы веществ - ацеталей, которое образуется в результате тримеризации формальдегида. При нормальном давлении (105 Па) и 25°C оно является твердым, плавится при 62°C и кипит при 115°C.При нагревании до температуры от 150 до 200°C оно снова деполимеризуется до мономерного формальдегида. Поскольку температура реакции в реакционной зоне B обычно лежит выше 250°C, то, таким образом, триоксан представляет собой благоприятный согласно изобретению источник формальдегида для получения поступающей реакционной газовой смеси B. Поскольку триоксан также сравнительно хорошо растворим в воде и спиртах, таких как метанол, то для способа согласно изобретению в качестве подходящих источников формальдегида согласно изобретению также могут применяться соответствующие растворы триоксана. Присутствие от 0,25 до 0,50% масс, серной кислоты в растворах триоксана способствует обратному расщеплению до формальдегида. В качестве альтернативы триоксан также может растворяться в жидком, состоящем главным образом из уксусной кислоты потоке вещества Y, а получающийся в результате раствор с целью получения поступающей реакционной газовой смеси B испаряется, и содержащийся в нем триоксан при повышенной температуре расщепляется обратно в формальдегид.
Кроме того, в качестве источника формальдегида для способа согласно изобретению может использоваться параформальдегид.
Параформальдегид представляет собой короткоцепочечный полимер формальдегида, степень полимеризации которого обычно составляет от 8 до 100. Он представляет собой белый порошок, который при низких значениях pH или при нагревании снова расщепляется в формальдегид.
При нагревании параформальдегида в воде он распадается, и получают водный раствор формальдегида, который также является подходящим источником согласно изобретению. Иногда он обозначается как водный «раствор параформальдегида», чтобы терминологически разграничить его с водными растворами формальдегида, которые получаются в результате разбавления формалина. Однако фактически параформальдегид как таковой в основном не растворим в воде.
Другим источником формальдегида, подходящим для способа согласно изобретению, является метилаль (диметоксиметан). Речь идет о продукте реакции формальдегида с метанолом, который при нормальном давлении и 25°C образует бесцветную жидкость. В водных кислотах он гидролизуется, и снова получается формальдегид и метанол. Будучи переведенным в паровую фазу, он подходит для получения поступающей реакционной газовой смеси B.
Также могут применяться способы непрерывного добавления формальдегида, раскрытые в Chemie Ingenieur Technik-CIT, Volume 66, Issue 4, на страницах с 498 по 502, опубликованных в сети Интернет в 2004 г.
В промышленности формальдегид получают при помощи гетерогенно катализируемого частичного газофазного окисления метанола. Поэтому особенно предпочтительным источником формальдегида согласно изобретению для образования поступающей реакционной газовой смеси B является газообразная смесь продуктов гетерогенно катализируемого частичного газофазного окисления метанола до формальдегида, на выбор - после того как было отделено частичное или полное количество содержащегося в ней в зависимости от обстоятельств непрореагировавшего метанола.
Из потока газообразной смеси продуктов A, выходящего из реакционной зоны A, и переведенного в паровую фазу источника формальдегида в качестве по меньшей мере одного дополнительного потока вещества, а также потока вещества Y, и на выбор - других потоков веществ, таких как, например, дополнительный водяной пар или дополнительный инертный газ, отличающийся от водяного пара (=инертный газ-разбавитель) может получаться поступающая реакционная газовая смесь B.
Температура реакционной газовой смеси B в случае способе согласно изобретению внутри реакционной зоны B обычно будет находиться в диапазоне от 260 до 400°C, предпочтительно в диапазоне от 270 до 390°C, особенно предпочтительно в диапазоне от 280 до 380°C, предпочтительнее в диапазоне от 300 до 370°C и в особенно предпочтительном варианте в диапазоне от 300 до 340°C.
Понятие температуры реакционной газовой смеси B (в этой публикации также обозначенной как температура реакции в реакционной зоне B) при этом подразумевает, в первую очередь, ту температуру, которую реакционная газовая смесь B имеет от достижения степени превращения формальдегида, содержащегося в поступающей реакционной газовой смеси B, по меньшей мере 5% мольн. до достижении соответствующей конечной степени превращения формальдегида внутри реакционной зоны B. Согласно изобретению температура реакционной газовой смеси B на протяжении всей реакционной зоны B предпочтительно лежит в вышеуказанном диапазоне температур. Предпочтительно также поступающая реакционная газовая смесь B подается в реакционную зону B уже с температурой, находящейся в диапазоне от 260°C до 400°C. Однако часто на входе в реакционную зону B в направлении движения потока до собственно каталитически активного заполнения катализатором в реакционной зоне B находится заполнение реакционной зоны B твердым инертным материалом или заполнение каталитически активным катализатором в высоком разбавлении инертным материалом такого типа. При прохождении такого предварительного заполнения реакционной зоны B температура поступающей реакционной газовой смеси B, подаваемой в реакционную зону B, может сравнительно просто устанавливаться на величину, с которой реакционная газовая смесь B должна поступать непосредственно в каталитически активное заполнение катализатором в этой реакционной зоне B. Как правило, температура газообразной смеси продуктов A, покидающей реакционную зону A, отличается от этой температуры. По этой причине поток газообразной смеси продуктов A на своем пути из реакционной зоны A в реакционную зону B может проходить через теплообменник с непрямым теплообменом, чтобы приблизить его температуру к предусмотренной температуре поступающей реакционной газовой смеси B на входе в реакционную зону B, или довести его до этой температуры.
В основном заполнение реакционной зоны B по меньшей мере одним катализатором альдольной конденсации B может выполняться в виде псевдоожиженного слоя. Однако с точки зрения технологии это заполнение реакционной зоны B катализатором альдольной конденсации B предпочтительно выполнено в виде неподвижного слоя.
В отношении имеющего места в реакционной зоне B рабочего давления соответствующим образом справедливо уже сказанное в отношении рабочего давления, имеющего место в реакционной зоне A. Как правило, рабочее давление в реакционной зоне B по причине возникающих при прохождении реакционной газовой смеси A сквозь реакционную зону A потерь давления меньше, чем рабочее давление в реакционной зоне A. Исполнение реакционной зоны B также может осуществляться в соответствующих реакторах с теплообменом, как и реакционной зоны A, причем справедливы аналогичные принципы предпочтения.
В случае способа согласно изобретению содержание в поступающей реакционной газовой смеси B формальдегида, как правило, составляет от 0,5 до 10% объемн., предпочтительно от 0,5 до 7% объемн. и особенно предпочтительно от 1 до 5% объемн.
Соотношение nHAс: nFd из молярного количества уксусной кислоты, содержащегося в поступающей реакционной газовой смеси B (nHAс), и содержащегося в ней молярного количества формальдегида (nFd) в случае способа согласно изобретению больше 1 и может составлять до 10 (при этом под nFd понимают сумму из формальдегидных структурных единиц, присутствующих в поступающей реакционной газовой смеси B как мономеры (предпочтительно) и олигомеры, а также как полимеры, поскольку в значительной мере расщепление до мономерного формальдегида также может устанавливаться только при прохождении реакционной газовой смеси B через заполнение катализатором реакционной зоны B). Согласно изобретению предпочтительно это соотношение nHAс:nFd в поступающей реакционной газовой смеси B составляет от 1,1 до 5 и особенно предпочтительно от 1,5 до 3,5. Часто содержание уксусной кислоты в поступающей реакционной газовой смеси B варьируется в диапазоне от 1 или от 1,5 до 20% объемн., преимущественно в диапазоне от 2 до 15% объемн. и особенно предпочтительно в диапазоне от 3 до 10% объемн. Содержание молекулярного кислорода в поступающей реакционной газовой смеси B в случае способа согласно изобретению с точки зрения технологической целесообразности варьируется в диапазоне от 0,5 до 5% объемн., предпочтительно в диапазоне от 1 до 5% объемн. и особенно предпочтительно в диапазоне от 2 или от 3 до 5% объемн. Присутствие молекулярного кислорода в поступающей реакционной газовой смеси B благоприятно сказывается на сроке службы засыпанного катализатора реакцонной зоны B. Однако если содержание кислорода в реакционной газовой смеси B слишком высоко, то в реакционной зоне B доходит до нежелательного образования оксида углерода.
Содержание водяного пара в поступающей реакционной газовой смеси B в случае способа согласно изобретению не должно бы превышать 30% объемн., поскольку присутствие водяного пара в реакционной газовой смеси B неблагоприятно сказывается на положении равновесия альдольной конденсации. Поэтому с точки зрения технологической целесообразности содержание водяного пара в поступающей реакционной газовой смеси B, как правило, не будет превышать 25% объемн. и предпочтительно 20% объемн. Как правило, содержание водяного пара в поступающей реакционной газовой смеси B будет составлять по меньшей мере 1,5 или по меньшей мере 2% объемн. Предпочтительно содержание водяного пара в поступающей реакционной газовой смеси B составляет от 5 до 15% объемн., а с учетом его действия и образования в реакционной зоне A, прежде всего, от 10 до 15% объемн. Объемная доля инертных газов-разбавителей, отличающихся от водяного пара, в поступающей реакционной газовой смеси B обычно будет составлять по меньшей мере 30% объемн. Предпочтительно вышеупомянутая доля инертных газов составляет по меньшей мере 40% объемн. или по меньшей мере 50% объемн. Как правило, доля инертных газов-разбавителей, отличающихся от водяного пара, в поступающей реакционной газовой смеси B не будет превышать 95% объемн. или в большинстве случаев 90% объемн. Технологически особенно предпочтительно поступающая реакционная газовая смесь B содержит от 60 до 90% объемн., особенно предпочтительно от 70 до 80% объемн. инертного газа-разбавителя, отличающегося от водяного пара. Согласно изобретению предпочтительным инертным газом-разбавителем, отличающимся от водяного пара, в поступающей реакционной газовой смеси B также является молекулярный азот (N2).
Таким образом, содержание молекулярного азота в поступающей реакционной газовой смеси B может составлять по меньшей мере 30% объемн., предпочтительно по меньшей мере 40% объемн. или по меньшей мере 50% объемн. Как правило, поступающая реакционная газовая смесь B содержит не более чем 95% объемн. и в большинстве случаев не более чем 90% объемн. молекулярного азота. Предпочтительно поступающая реакционная газовая смесь B содержит от 60 до 90% объемн., особенно предпочтительно от 70 до 80% объемн. молекулярного азота.
В качестве катализаторов для заполнения реакционной зоны B рассматривают, например, те, которые предложены в издании I & ЕС PRODUCT RESEARCH AND DEVELOPMENT, Vol.5, No. 1, March 1966, на страницах с 50 по 53. Эта группа основных катализаторов включает в себя, с одной стороны, цеолиты (алюмосиликаты) с анионным зарядом каркаса, у которых на внутренней и внешней поверхности находится по меньшей мере один тип катионов из группы ионов щелочных и щелочноземельных металлов (предпочтительно Na+, К+, Са2+ и/или Mg2+), чтобы уравновешивать (нейтрализовывать) отрицательный заряд каркаса. Но она включает также нанесенный на инертный носитель (например, аморфный диоксид кремния (силикагель)) гидроксид из группы, состоящей из гидроксидов щелочных металлов, гидроксидов щелочноземельных металлов и гидроксида алюминия (предпочтительно KOH, NaOH, Ca(OH)2 и Mg(OH)2).
Однако для заполнения реакционной зоны В также подходят кислотные катализаторы, которые предлагаются в европейской заявке на патент EP-A 164614.
При этом речь идет о катализаторах, которые включают в себя
- в качестве компонента а) по меньшей мере один оксид по меньшей мере одного из элементов кремния (Si), алюминия (Al), титана (Ti), циркония (Zr), кадмия (Cd), олова (Sn), галлия (Ga), иттрия (Y) и лантана (La) и/или цеолит,
и
- в качестве компонента b) по меньшей мере один оксид, выбираемый из оксида бора и оксида фосфора,
а также на выбор
- в качестве компонента c) один или более одного оксида по меньшей мере одного из элементов ванадия (V), хрома (Cr), кобальта (Co), никеля (Ni), молибдена (Mo) и свинца (Pb) и/или одну или более чем одну гетерополикислоту по меньшей мере с одним полиатомом, выбираемым из V, Mo и вольфрама (W).
При этом в качестве оксида бора будет предпочтительным B2O3, а в качестве оксида фосфора P2O5.
При этом предпочитают катализаторы, у которых содержание оксида бора (рассчитанное как B2O3 (в пересчете на содержащееся количество бора (B))) составляет от 1 до 50% масс. Однако благоприятными согласно изобретению катализаторами являются также такие, у которых содержание оксида фосфора (рассчитанное как P2O5 (в пересчете на содержащееся количество фосфора (P))) составляет от 1 до 50% масс. Однако для способа согласно изобретению в качестве катализаторов альдольной конденсации B рассматриваются также такие из вышеназванных катализаторов, у которых общее содержание оксида фосфора (рассчитанное как P2O5) и оксида бора (рассчитанное как B2O3) составляет от 1 до 50% масс. Предпочтительно указанные выше содержания оксида фосфора и/или оксида бора составляют от 5 до 30% масс.
Кроме того, в случае компонента a) речь предпочтительно идет по меньшей мере об одном оксиде по меньшей мере одного из элементов Si, Al, Ti и Zr.
Согласно изобретению особенно благоприятными являются комбинации оксида титана в качестве компонента a), а также оксида бора и фосфора в качестве компонента b), или соответственно диоксида кремния-оксида алюминия в качестве компонента a), а также оксида бора в качестве компонента b), или оксида алюминия в качестве компонента a), а также оксида бора или оксида фосфора в качестве компонента b). Если вышеуказанные катализаторы включают дополнительно гетерополикислоту, то эта кислота в качестве гетероатома содержит предпочтительно по меньшей мере один из элементов P, B и Si. Если вышеуказанные катализаторы включают компонент c), то его количество составляет обычно от 0,01 до 10 ммоль на грамм катализатора, а часто от 0,03 до 5 ммоль на грамм катализатора. При этом благоприятно, если катализаторы в качестве компонента c) содержат как по меньшей мере один из оксидов, так и по меньшей мере одну из гетерополикислот.
Однако согласно изобретению особенно предпочтительно реакционная зона B заполнена катализаторами альдольной конденсации B, активная масса которых представляет собой оксид ванадия-фосфора и/или оксид ванадия-фосфора, дотированный элементами, отличающимися от ванадия и фосфора (обобщенно в литературе обозначающиеся также как V-P-O-катализаторы).
Катализаторы такого типа уже описаны в литературе и рекомендуются там особенно в качестве катализаторов для гетерогенно катализируемого газофазного окисления углеводородов по меньшей мере с четырьмя атомами углерода (особенно н-бутана, н-бутена и/или бензола) до ангидрида малеиновой кислоты.
Неожиданным образом эти катализаторы, известные из уровня техники для вышеуказанного частичного окисления, в общем подходят как катализаторы альдольной конденсации B для заполнения реакционной зоны B. Они отличаются особенно высокими селективностями по образованию целевого продукта (образованию акриловой кислоты) (при одновременных высоких степенях превращения формальдегида).
В соответствии с этим, в случае способа согласно изобретению в качестве катализаторов альдольной конденсации B могут использоваться, например, все те, которые предложены в публикациях патентных заявок США US-A 5,275,996, US-A 5,641,722, US-A 5,137,860, US-A 5,095,125, немецкого патента DE-69702728 Т2, международных заявок WO 2007/012620, WO 2010/072721, WO 2001/68245, патентных заявок США US-А 4,933,312, международной заявки WO 2003/078310, издания Journal of Catalysis 107, страницы 201-208 (1987), немецкой заявки на патент DE-A 102008040094, международной заявки WO 97/12674, диссертационной работы «Neuartige Vanadium (IV)-phosphate fur die Partialoxidation von kurzkettigen Kohlenwasserstoffen-Synthesen, Kristallstrukturen, Redox-Verhalten и katalytische Eigenschaften», Dissertation von Dipl.Chem. Ernst Benser, 2007, Rheinische Friedrichs-Wilhelms-Universitat Bonn», международной заявки WO 2010/072723, диссертационной работы «Untersuchung von V-P-O-Katalysatoren fur die partielle Oxidation von Propan zu Acrylsaure, Dissertation von Dipl.Chem. Thomas Quandt, 1999, Ruhr-Universitat Bochum», международных заявок WO 2010/000720, WO 2008/152079, WO 2008/087116, немецких заявок на патент DE-A 102008040093, DE-A 102005035978 и DE-A 102007005602, а также в приведенном в этих публикациях уровне техники. В частности, это относится ко всем вариантам исполнения вышеуказанного уровня техники, приведенным в качестве примеров, особенно к таким вариантам международной заявки WO 2007/012620.
Соотношение атомов фосфора/ванадия в недотированных или дотированных оксидах ванадия-фосфора согласно изобретению предпочтительно составляет от 0,9 до 2,0, предпочтительно 0,9 до 1,5, особенно предпочтительно 0,9 до 1,2 и наиболее предпочтительно от 1,0 до 1,1. Средняя арифметическая степень окисления ванадия в них предпочтительно составляет от +3,9 до +4,4 и особенно предпочтительно от 4,0 до 4,3. Кроме того, эти активные массы предпочтительно обладают удельной поверхностью, измеренной по методу БЭТ, ≥15 м2/г, предпочтительно от ≥15 до 50 м2/г и наиболее предпочтительно от ≥15 до 40 м2/г. Предпочтительно они имеют общий объем пор ≥0,1 мл/г, предпочтительно от 0,15 до 0,5 мл/г и наиболее предпочтительно от 0,15 до 0,4 мл/г. Данные об общем объеме пор в данной публикации относятся к определению с помощью метода ртутной порометрии с применением измерительного прибора Auto Роге 9220 фирмы Micromeritics GmbH, DE-4040 Neuss (ширина диапазона от 30 А до 0,3 мм). Как уже излагалось, активные массы оксида ванадия-фосфора могут быть дотированы элементами промоторов, отличающимися от ванадия и фосфора. В качестве элементов промоторов такого типа рассматриваются элементы с 1-ой по 15-ю групп периодической системы, отличающиеся от Р и V. Дотированные оксиды ванадия-фосфора предлагают, например, международные заявки WO 97/12674, WO 95/26817, патентные заявки США US-A 5,137,860, US-A 5,296,436, US-А 5,158,923, US-A 4,795,818 и международная заявка WO 2007/012620.
Согласно изобретению предпочтительными промоторами являются элементы литий, калий, натрий, рубидий, цезий, таллий, молибден, цинк, гафний, цирконий, титан, хром, марганец, никель, медь, железо, бор, кремний, олово, ниобий, кобальт и висмут, среди которых, помимо железа, особенно предпочтительными являются ниобий, молибден, цинк и висмут. Активные массы оксида ванадия-фосфора могут содержать один или несколько элементов промоторов. Общее содержание промоторов в каталитически активной массе составляет, в пересчете на их массу, как правило, не более 5% масс, (отдельный элемент промотора, соответственно рассчитанный как электрически нейтральный оксид, в котором этот элемент промотора имеет такое же число зарядов (степень окисления), как в активной массе).
Следовательно, в качестве катализаторов альдольной конденсации B для заполнения реакционной зоны B особенно рассматриваются активные массы полиэлементных оксидов общей формулы II
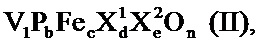
в которой переменные имеют следующее значение:
X1=Mo, Bi, Co, Ni, Si, Zn, Hf, Zr, Ti, Cr, Mn, Cu, B, Sn и/или Nb, предпочтительно Nb, Mo, Zn и/или Hf,
X2=Li, K, Na, Rb, Cs и/или TI,
b=от 0,9 до 2,0, предпочтительно от 0,9 до 1,5, особенно предпочтительно от 0,9 до 1,2 и наиболее предпочтительно от 1,0 до 1,1
c=от ≥0 до 0,1,
d=от ≥0 до 0,1,
e=от ≥0 до 0,1, и
n=стехиометрический коэффициент элемента кислорода, который определяется с помощью стехиометрических коэффициентов элементов, отличных от кислорода, а также числом их зарядов в II.
Независимо от стехиометрических коэффициентов d, е и b стехиометрический коэффициент с согласно изобретению предпочтительно составляет в активной массе общей формулы II от 0,005 до 0,1, предпочтительно от 0,005 до 0,05 и особенно предпочтительно от 0,005 до 0,02.
Катализаторы альдольной конденсации B могут содержать активные массы полиметаллического оксида общей формулы II, например, в чистой, неразбавленной форме или при разбавлении оксидным, в основном инертным материалом разбавителя как так называемые сплошные катализаторы. В качестве подходящих согласно изобретению инертных материалов разбавителя следует назвать, например, тонко измельченный оксид алюминия, диоксид кремния, алюмосиликаты, диоксид циркония, диоксид титана или их смеси. Неразбавленные сплошные катализаторы согласно изобретению являются предпочтительными. Эти сплошные катализаторы в основном могут иметь любую форму. Предпочтительными сплошными катализаторами являются шарики, сплошные цилиндры, полые цилиндры и трехдольчатые гранулы, наибольший размер которых во всех случаях составляет предпочтительно от 1 до 10 мм.
В случае формованных изделий сплошных катализаторов формование осуществляется предпочтительно с помощью порошка предшественника, который прокаливается только после придания формы. Это формование обычно осуществляется с добавлением вспомогательных средств для формования, таких как, например, графит (смазывающее средство), или минеральных волокон (усиливающих средств). Подходящими способами формования являются таблетирование, штранг-прессование и экструдирование.
С точки зрения технологической целесообразности внешний диаметр цилиндрических сплошных катализаторов составляет от 3 до 10 мм, предпочтительно от 4 до 8 мм и прежде всего от 5 до 7 мм. Их высота предпочтительно составляет от 1 до 10 мм, предпочтительно от 2 до 6 мм и прежде всего от 3 до 5 мм. В случае полых цилиндров справедливо то же самое. Дополнительно внутренний диаметр отверстия, проходящего сверху вниз, предпочтительно составляет от 1 до 8 мм, предпочтительно от 2 до 6 мм и наиболее предпочтительно от 2 до 4 мм. Толщина стенки от 1 до 3 мм в случае полых цилиндров технологически целесообразна. Разумеется, в качестве катализаторов альдольной конденсации B в реакционной зоне B также могут использоваться дотированные и недотированные активные массы оксидов ванадия-фосфора в форме порошка или как слоистые катализаторы со слоем активной массы, нанесенным на поверхность инертного формованного изделия носителя. При этом получение этих слоистых катализаторов, толщина слоя и геометрия инертных формованных изделий носителя может выбираться, как описано в случае слоистых катализаторов для реакционной зоны A.
В остальном, получение дотированных или недотированных активных масс оксидов ванадия-фосфора, а также сплошных катализаторов, изготовленных из этих масс, может осуществляться, как описано в публикациях уровня техники, на которые в настоящей патентной заявке делается ссылка.
Это, в частности, публикации международных заявок WO 2007/012620, WO 2010/07273, WO 2010/000720 и WO 2010/000764.
Например, можно поступать следующим образом:
a) взаимодействие соединения пятивалентного ванадия (например, V2O5) с органическим восстанавливающим растворителем (например, изобутанолом) в присутствии соединения пятивалентного фосфора (например, орто- и/или пирофосфорной кислоты) при нагревании от 75 до 205°C, предпочтительно от 100 до 120°C;
b) охлаждение реакционной смеси предпочтительно до температуры от 40 до 90°C;
c) на выбор, добавление соединений, содержащих дотирующие элементы, таких как, например, фосфат железа(III);
d) повторное нагревание от 75 до 205°C, предпочтительно от 100 до 120°C;
e) выделение образовавшейся твердой массы предшественника, содержащей V, Р, О и, например, Fe (к примеру, с помощью фильтрования);
f) сушка и/или предварительная термическая обработка этой массы предшественника (на выбор, до начинающегося предварительного формирования в результате отщепления воды из массы предшественника);
g) добавление вспомогательного средства для формования, такого как, например, графит или минеральные волокна, и последующее формование с получением формованных изделий предшественника сплошного катализатора, например, при помощи таблетирования;
h) после этого термическая обработка образовавшихся формованных изделий предшественника сплошного катализатора с помощью нагревания в атмосфере, которая содержит кислород, азот, благородные газы, диоксид углерода, монооксид углерода и/или водяной пар (например, как описано в международной заявке WO 2003/078310 на странице 20, строке 16 до страницы 21, строки 35). Температура этой термической обработки, как правило, превышает 250°C, часто 300°C или 350°C, однако обычно не превышает 600°C, предпочтительно не превышает 550°C и наиболее предпочтительно не превышает 500°C.
Нагрузка на заполнение катализатором реакционной зоны B по формальдегиду, содержащемуся в поступающей реакционной газовой смеси B, согласно изобретению может составлять, например, от 1 до 100, предпочтительно от 2 до 50 и особенно предпочтительно от 3 до 30 или от 4 до 10 нл/лч. Термин «нагрузка» при этом применяется так, как определено в немецкой заявке на патент DE-A 19927624. Как в реакционной зоне A, так и в реакционной зоне B соответствующий неподвижный слой катализатора (также в случае разделения на участок 1 / участок 2 в реакционной зоне A) может состоять как только из катализатора, содержащего активную массу, так и из смеси катализатора, содержащего активную массу, и инертных формованных изделий.
В частности, при применении V-P-O-катализаторов в качестве катализаторов альдольной конденсации в реакционной зоне B в случае способа согласно изобретению, в пересчете на однократное прохождение реакционной газовой смеси B сквозь реакционную зону B, подвергается превращению по меньшей мере 95% мольн., в большинстве случаев по меньшей мере 98% мольн. формальдегида, содержащегося в поступающей реакционной газовой смеси B. При этом селективность образования акриловой кислоты в пересчете на вступивший в реакцию формальдегид составляет, как правило, ≥95% мольн., часто ≥98% мольн.
Для исполнения реакционной зоны B согласно изобретению подходят те же реакторы с теплообменом, которые уже рекомендовались для исполнения реакционной зоны A.
Как уже упоминалось, формальдегид в промышленности получается в результате гетерогенно катализируемого частичного газофазного окисления метанола. Таким образом, особенно предпочтительным согласно изобретению источником формальдегида для образования поступающей реакционной газовой смеси B является газообразная смесь продуктов гетерогенно катализируемого частичного газофазного окисления метанола до формальдегида.
Таким образом, с точки зрения технологической целесообразности способ согласно изобретению включает в себя еще одну, третью реакционную зону C, которая заполнена по меньшей мере одним катализатором окисления C и предпочтительно включает следующие дополнительные операции:
- через третью реакционную зону C, которая заполнена по меньшей мере одним катализатором окисления C, пропускают поток поступающей реакционной газовой смеси C, содержащей реагенты - метанол и молекулярный кислород, а также по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и при прохождении реакционной зоны C метанол, содержащийся в поступающей реакционной газовой смеси C, окисляют в условиях гетерогенного катализа до формальдегида и водяного пара, так что получается газообразная смесь продуктов C, содержащая формальдегид, водяной пар и по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и поток газообразной смеси продуктов C покидает реакционную зону C, причем к проходящей сквозь реакционную зону C реакционной газовой смеси C на ее пути через эту реакционную зону С на выбор может подаваться дополнительный молекулярный кислород и/или дополнительный инертный газ-разбавитель.
Тогда выходящий из реакционной зоны C поток газообразной смеси продуктов C, содержащий формальдегид, может применяться как таковой (то есть, без того, чтобы проводить для него предварительно процесс разделения), чтобы получать поступающую реакционную газовую смесь B. Как правило, для этой цели газообразную смесь продуктов C при покидании реакционной зоны C сначала охлаждают (быстрое охлаждение), чтобы уменьшить нежелательные побочные реакции в газообразной смеси продуктов C перед ее введением в поступающую реакционную газовую смесь B. Обычно ее как можно быстрее охлаждают до температур от 150 до 350°C или от 200 до 250°C.
Однако на выбор, из газообразной смеси продуктов C также сначала в зоне разделения Т* может отделяться частичное или все количество при необходимости еще содержащегося в ней, не вступившего в реакцию в реакционной зоне C метанола, а затем остающаяся при этом газообразная смесь продуктов C*, содержащая формальдегид (которая может в рамках этого отделения проходить через жидкое агрегатное состояние), используется для получения поступающей реакционной газовой смеси B. С технологической точки зрения предпочтительно это разделение проводят ректификацией. Для этой цели газообразная смесь продуктов C, на выбор - после предварительно произведенного прямого или непрямого охлаждения, в газообразном виде подается в соответствующую ректификационную колонну, снабженную охлаждающим контуром. Однако, разумеется, из газообразной смеси продуктов C сначала могут переводиться в жидкую фазу те компоненты, температура кипения которых при нормальном давлении (105 Па) меньше или такая же, как температура кипения формальдегида (например, с помощью конденсации), и ректификацию проводят из жидкой фазы. Как правило, такое отделение метанола также сопровождает отделение водяного пара, содержащегося в газообразной смеси продуктов C. Для целей упомянутого выше прямого охлаждения в ректификационную колонну может, например, вводиться отбираемая из ее кубовой части и при необходимости с помощью непрямого теплообмена дополнительно охлажденная жидкая фаза, которая через соответствующие сопла распыляется на мелкие капельки, которые предоставляют необходимую большую поверхность теплообмена для горячей газообразной смеси продуктов C. Согласно изобретению отделенный метанол целесообразно подавать обратно в реакционную зону C и применять дополнительно для получения поступающей реакционной газовой смеси C (сравните с немецкой заявкой DE-A 1618413). Отделение метанола от газообразной смеси продуктов C до его использования для получения поступающей реакционной газовой смеси B, как правило, производится тогда, когда реакционная зона C оформлена таким образом, что результирующая степень превращения метанола в реакционной зоне C, в пересчете на однократное прохождение газообразной смеси продуктов C через реакционную зону C, составляет не более 90% мольн. Однако, разумеется, такое отделение метанола может также применяться при соответствующих степенях превращения метанола не более 95% мольн. Например, такое отделение метанола может проводиться, как описано в издании Ullmann's Encyclopedia of Industrial Chemistry, Vol.A11, 5th Ed., VCH Weinheim на странице 626 и далее.
Особенно подходящие для заполнения реакционной зоны C катализаторы окисления C в основном могут подразделяться на две группы.
Первая из двух групп включает в себя так называемые серебряные катализаторы (катализаторы на основе серебра), которые в качестве активной массы содержат элементарное серебро, чистота которого предпочтительно составляет ≥99,7% масс, преимущественно ≥99,8% масс, предпочтительно ≥99,9% масс, и наиболее предпочтительно ≥99,99% масс. Относящиеся к этому способы гетерогенно катализируемого частичного газофазного окисления метанола до формальдегида на этих «серебряных катализаторах» обозначаются в уровне техники как способы с серебром (сравните, например, с «А.Nagy, G.Mestl: High temperature partial oxidation reactions over silver catalysts, Appl. Catal. 188 (1999), стр. с 337 по 353», «H. Schubert, U. Tegtmayr, R. Schlogl: On the mechanism of the selective oxidation of methanol over elemental silver, Catalyst Letters, 28 (1994), стр. с 383 по 395», «L. Lefferts, Factors controlling the selectivity of silver catalysts for methanol oxidation, Dissertation, Universitat Twente (1987)» и немецкой заявкой DE-A 2334981).
Предпочтительные согласно изобретению серебряные катализаторы окисления C для заполнения реакционной зоны C предложены, например, в Ullmann's Encyclopedia of Industrial Chemistry, Vol.A11, 5th Ed., VCH, Weinheim, стр. с 619 по 652, или в издании Encyclopedia of Chemical Technology, Vol.11, 4th Ed., Wiley & Sons, New York, стр. с 929 по 949, в немецких заявках на патент DE-AS 1231229, DE-AS 1294360, DE-A 1903197 и в бельгийской патентной публикации BE 683130. Обычно речь идет о кристаллах элементарного серебра, осажденных в результате электролиза водных растворов солей серебра (их форма также может быть округлой) (предпочтительно указанной выше чистоты), которые засыпаются на перфорированную подложку (например, пластинку с отверстиями, сито или ячеистую сетку (предпочтительно также изготовленную из серебра)) в качестве неподвижного слоя катализатора (обычная высота насыпанного слоя составляет от 10 до 50 мм, часто от 15 до 30 мм). Общее содержание в каталитически активном серебре металлов, присутствующих в элементарном виде, отличающихся от серебра (Ag) (например, меди (Cu), палладия (Pd), свинца (Pb), висмута (Bi), железа (Fe), платины (Pt) и золота (Au)) предпочтительно составляет ≤2000 масс. частей на млн, лучше ≤1000 масс. частей на млн, предпочтительно ≤100 масс.частей на млн и особенно предпочтительно ≤50 масс. частей на млн или ≤30 масс. частей на млн. Наибольший размер кристаллов серебра обычно находится в области от 0,1 до 5 мм и предпочтительно увеличивается в направлении движения потока реакционной газовой смеси C. Предпочтительно неподвижный слой серебра выполняется как двухслойный, причем нижний слой имеет толщину, например, от 15 до 40 мм, предпочтительно от 20 до 30 мм, и состоит по меньшей мере на 50% масс, из кристаллов серебра с размером зерна от 1 до 4 мм, предпочтительно от 1 до 2,5 мм. Верхний слой может иметь толщину (толщину слоя), например от 0,75 до 3 мм, предпочтительно от 1 до 2 мм, и состоять из кристаллов с размером зерна (наибольшим размером) от 0,1 до 1 мм, предпочтительно от 0,2 до 0,75 мм. Обтекание потоком поступающей реакционной газовой смеси C в этом случае осуществляется сверху вниз.
Чтобы противодействовать спеканию кристаллов серебра, снижающему эффективность неподвижного слоя катализатора, с возрастающим сроком службы (при сравнительно высоких температурах реакции) международная заявка WO 2010/022923 рекомендует покрывать эти кристаллы серебра тонким пористым слоем из оксидного материала по меньшей мере одного из элементов Al, Si, Zr и Ti (толщина этого слоя может составлять от 0,3 до 10 мкм, предпочтительно от 1,0 до 5,0 мкм, особенно предпочтительно от 2,0 до 4,0 мкм и лучше всего примерно 3 мкм), и таким образом достигать продления срока службы неподвижного слоя катализатора.
В случае способа с серебром содержание метанола в поступающей реакционной газовой смеси C обычно составляет по меньшей мере 5% объемн., большей частью по меньшей мере 10% объемн. и может распространяться до 60% объемн. Предпочтительно вышеупомянутое содержание метанола в случае способа с серебром составляет от 15 до 50% объемн. и особенно предпочтительно от 20 до 40 или до 30% объемн.
Кроме того, соотношение молярного количества молекулярного кислорода (no), содержащегося в поступающей реакционной газовой смеси С, и молярного количество метанола (nМе), содержащегося в поступающей реакционной газовой смеси C, nо: nМе, в случае способа с серебром составляет обычно 1 (<1), предпочтительно ≤0,8. Особенно предпочтительно оно будет составлять от 0,2 до 0,6 и наиболее предпочтительно от 0,3 до 0,5 или от 0,4 до 0,5. Как правило, no: nМе в случае способа с серебром будет составлять не менее чем 0,1.
Сказанное в отношении инертных газов-разбавителей для реакционной зоны A, в случае способа с серебром также в основном справедливо и для реакционной зоны C. Примерами инертных газов-разбавителей, которые можно применять в случае способа с серебром являются H2O, CO2, N2 и благородные газы, такие как аргон, а также смеси из вышеупомянутых газов.
В качестве инертного газа-разбавителя, отличающегося от водяного пара, в случае способа с серебром для поступающей реакционной газовой смеси C также предпочитают молекулярный азот. Его преимущество не в последнюю очередь основывается на том, что молекулярный азот в воздухе выступает как природное сопутствующее вещество молекулярного кислорода, что делает воздух предпочтительным источником молекулярного кислорода, необходимого в реакционной зоне C. Однако, само собой разумеется, в случае способа с серебром согласно изобретению в качестве источника кислорода также может применяться чистый молекулярный кислород или воздух, обогащенный молекулярным кислородом, или другая смесь из молекулярного кислорода и инертного газа-разбавителя.
Обычно поступающая реакционная газовая смесь C в случае способа с серебром содержит от 20 до 80% объемн., или от 30 до 70% объемн., или от 40 до 60% объемн. инертного газа-разбавителя. Этот же газ может совершенно не содержать водяного пара. То есть, поступающая реакционная газовая смесь C в случае способа с серебром может содержать от 20 до 80% объемн., или от 30 до 70% объемн., или от 40 до 60% объемн. молекулярного азота. Однако из таких же соображений, как и для поступающей реакционной газовой смеси A, в случае способа с серебром в поступающей реакционной газовой смеси C также, как правило, совместно применяется водяной пар в качестве инертного газа-разбавителя.
Как правило, поступающая реакционная газовая смесь C в случае способа с серебром может содержать от >0 до 50% объемн. Н2O. Однако из сравнимых соображений, как и в случае поступающей реакционной газовой смеси A, согласно изобретению предпочтительно в случае способа с серебром сравнительно ограничивать в поступающей реакционной газовой смеси C содержание Н2O.
То есть, предпочтительно поступающая реакционная газовая смесь C в случае способа с серебром содержит от ≥5 до 45% объемн. Н2O, предпочтительно от ≥10 до 40% объемн. и особенно предпочтительно от 15 до 35% объемн., или от 20 до 30% объемн. Н2O. В качестве источника инертного газа в случае способа с серебром для поступающей реакционной газовой смеси C также может применяться поток вещества Z, образующийся в зоне разделения T. Таким образом, в случае способа с серебром с технологической точки зрения частичный поток потока вещества Z целесообразно подается обратно в реакционную зону C для получения поступающей реакционной газовой смеси C.
То есть, согласно изобретению подходящие поступающие реакционные газовые смеси C в случае способа с серебром могут содержать, например, от 10 до 50% объемн. Н2O и от 20 до 60% объемн. инертного газа-разбавителя, отличающегося от водяного пара (например, N2 или N2+CO2, или N2+благородный газ (например, Ar), или N2+CO2+благородный газ (например, Ar).
Само собой разумеется, поступающие реакционные газовые смеси C в случае способа с серебром могут содержать также от 10 до 40% объемн. Н2O и от 30 до 60% объемн. инертных газов-разбавителей, отличающихся от водяного пара (например, вышеуказанных).
Однако, естественно, поступающая реакционная газовая смесь C в случае способа с серебром может содержать также от 20 до 40% объемн. Н2O и от 30 до 50% объемн. инертных газов-разбавителей, отличающихся от водяного пара (например, вышеуказанных).
В принципе, в случае способа с серебром реакционная газовая смесь C может как продавливаться, так и просасываться сквозь реакционную зону C. В соответствии с этим в случае способа с серебром рабочее давление внутри реакционной зоны C может составлять как ≥105 Па, так и <105 Па. С точки зрения технологической целесообразности в случае способа с серебром рабочее давление в реакционной зоне C будет составлять от 103 до 106 Па, предпочтительно от 104 до 5⋅105 Па, особенно предпочтительно от 104 до 2⋅105 Па и особенно предпочтительно от 0,5⋅105 Па до 1,8⋅105 Па.
Температура реакционной газовой смеси C (понятие реакционной газовой смеси C в настоящей заявке включает все встречающиеся в реакционной зоне C газовые смеси, которые находятся между поступающей реакционной газовой смесью C и газообразной смесью продуктов C) в случае способа с серебром внутри реакционной зоны C обычно будет находиться в диапазоне от 400 до 800°C, предпочтительно в диапазоне от 450 до 800°C и особенно предпочтительно в диапазоне от 500 до 800°C. Понятие температуры реакционной газовой смеси C (в данной публикации также обозначаемой как температура реакции в реакционной зоне C) при этом подразумевает в первую очередь ту температуру, которую реакционная газовая смесь C имеет от достижения степени превращения метанола, содержащегося в поступающей реакционной газовой смеси C, по меньшей мере 5% мольн. до достижения соответствующей конечной степени превращения метанола внутри реакционной зоны C.
Согласно изобретению температура поступающей реакционной газовой смеси C в случае способа с серебром предпочтительно на протяжении всей реакционной зоны C лежит в вышеуказанном диапазоне температур.
Предпочтительно в случае способа с серебром поступающая реакционная газовая смесь C также подается в реакционную зону C уже с температурой, находящейся в указанном диапазоне. Часто в случае способа с серебром на входе в реакционную зону C в направлении движения потока до собственно каталитически активного заполнения катализатором (который также может быть разбавлен инертными формованными изделиями) находится заполнение реакционной зоны C твердым инертным материалом или заполнение каталитически активным катализатором в высоком разбавлении инертным материалом такого типа. При прохождении такого предварительного заполнения реакционной зоны C температура поступающей реакционной газовой смеси C, подаваемой в случае способа с серебром в реакционную зону C, может сравнительно просто устанавливаться на величину, с которой реакционная газовая смесь C в случае способа с серебром должна поступать непосредственно в каталитически активное заполнение катализатором в этой реакционной зоне C.
Если температура реакционной газовой смеси C в случае способа с серебром внутри реакционной зоны С ограничивается величиной от 450 до 650°C, предпочтительно от 500 до 600°C, то степень превращения метанола, как правило, составляет ≤90% мольн., часто ≤85% мольн. или ≤80% мольн., в то время как селективность образования формальдегида находится в области значений ≥90% мольн., часто ≥93% мольн. или ≥95% мольн. В этом случае (при котором содержание водяного пара в поступающей реакционной газовой смеси составляет предпочтительно <10% объемн.) согласно изобретению целесообразно от газообразной смеси продуктов C перед ее использованием для получения поступающей реакционной газовой смеси B отделять по меньшей мере частичное количество не вступившего в реакцию метанола и подавать обратно на получение поступающей реакционной газовой смеси C.
Следовательно, температура реакционной газовой смеси C в случае способа с серебром внутри реакционной зоны C предпочтительно будет составлять от 550 до 800°C, более предпочтительно от 600 до 750°C и особенно предпочтительно от 650 до 750°C.
Одновременно содержание водяного пара в поступающей реакционной газовой смеси C в случае способа с серебром предпочтительно устанавливается на величину ≥10% объемн., предпочтительно ≥15% объемн. и особенно предпочтительно ≥20% объемн. Как повышенная температура, так и повышенное содержание водяного пара в поступающей реакционной газовой смеси C в случае способа с серебром благоприятно сказываются на степени превращения метанола (в пересчете на однократное прохождение реакционной газовой смеси C через реакционную зону C). Как правило, эта степень превращения будет составлять >90% мольн., часто ≥92% мольн., или ≥95% мольн. и даже часто ≥97% мольн. (сравните, например, с Ullmann's Encylopedia of Industrial Chemistry, Vol.A 11, 5th Ed., VCH Weinheim на странице 625 и далее) (высокие степени превращения метанола, которые можно достичь несмотря на незначительные соотношения no: nМе в поступающей реакционной газовой смеси C в случае способа с серебром, прежде всего, могут объясняться тем, что с увеличивающейся температурой реакционной газовой смеси C в реакционной зоне C экзотермическое частичное окисление CH3OH+0,5 O2→HCHO+H2O все больше сопровождается эндотермическим дегидрированием CH3OH↔HCHO+H2).
Таким образом в случае способа с серебром регулярно могут достигаться пересчитанные на однократное прохождение реакционной газовой смеси С через реакционную зону C, а также на вступившее при этом во взаимодействие молярное количество метанола выходы формальдегида ≥85% мольн., чаще всего ≥87% мольн. и часто ≥89% мольн. В остальном, способ с серебром можно проводить, как описано в уже упомянутых касательно этого публикациях уровня техники, или в публикациях заявок на патенты США US-A 4080383, US-A 3994977, US-A 3987107, US-A 4584412 и US-A 4343954. Разумеется, при описанном способе с серебром в качестве исходного сырья (источника) может использоваться не только сравнительно чистый метанол. Согласно изобретению подходящим в этом отношении метанольным исходным сырьем являются также водные растворы метанола и технический метанол, которые после соответствующего выпаривания могут применяться для получения поступающей реакционной газовой смеси C.
Для исполнения способа с серебром в реакционной зоне C помимо реакторов, рекомендуемых в указанном выше уровне техники, среди прочего, подходят также те реакторы с теплообменом, которые уже рекомендовались для исполнения реакционной зоны A. Нагрузка реактора, заполненного кристаллами серебра, по метанолу, содержащемуся в поступающей реакционной газовой смеси C, как правило, будет составлять (от 0,5 до 6)⋅103 кг метанола на м2 поперечного сечения реактора или соответственно поперечного сечения неподвижного слоя катализатора.
Однако согласно изобретению гетерогенно катализируемое частичное газофазное окисление метанола до формальдегида предпочтительно проводится в соответствии с ФОРМОКС-процессом.
В отличие от способа с серебром ФОРМОКС-процесс проводится на катализаторах окисления С, активная масса которых представляет собой смешанный оксид, который содержит по меньшей мере один переходный металл в окисленном состоянии (сравните, например, с международной заявкой WO 03/053556 и европейской заявкой на патент EP-A 2213370). При этом понятие «переходные металлы» подразумевает химические элементы периодической системы с порядковыми номерами от 21 до 30, от 39 до 48 и от 57 до 80.
Согласно изобретению вышеуказанные активные массы смешанных оксидов содержат предпочтительно по меньшей мере один из переходных металлов Мо и V в окисленном состоянии. Согласно изобретению наиболее предпочтительно вышеупомянутые активные массы представляют собой смешанные оксиды, содержащие по меньшей мере элементы Fe и Мо в окисленном состоянии (сравните, например, с заявками на патенты США US-A 3983073, US-A 3978136, US-A 3975302, US-A 3846341, US-A 3716497, US-A 4829042, европейской заявкой на патент EP-A 2213370, а также международной заявкой WO 2005/063375, патентом США US 3408309, заявками на патенты США US-A 3198753, US-А 3152997, международной заявкой WO 2009/1489809, немецкой заявкой на патент DE-A 2145851, международными заявками WO 2010/034480, WO 2007/059974 и публикацией «Methanol Selective Oxidation to Formaldehyde over Iron-Molybdate Catalysts, Ana Paula Vieira Soares and Manuel Farinha Portela and Alain Kiennemann in Catalysis Review 47, страницы с 125 no 174 (2004)» и уровнем техники, приведенным в этих публикациях).
Другое различие между способом с серебром и ФОРМОКС-процессом состоит в том, что соотношение содержащегося в поступающей реакционной газовой смеси C молярного количества молекулярного кислорода (no) и содержащегося в поступающей реакционной газовой смеси C молярного количества метанола (nМе), no: nМе, обычно составляет по меньшей мере 1 или больше 1 (≥1), предпочтительно ≥1,1. Однако, как правило, это соотношение nо: nMе в поступающей реакционной газовой смеси C при ФОРМОКС-процессе составляет не более 5, часто не более 4. Согласно изобретению предпочтительные соотношения nо: nМе в поступающей реакционной газовой смеси C составляют от 1,5 до 3,5, предпочтительно от 2 до 3. Кроме того, содержание в поступающей реакционной газовой смеси C метанола при ФОРМОКС-процесс обычно будет составлять не более 15% объемн., в большинстве случаев не больше 11% объемн. Это может объясняться тем, что газовые смеси из молекулярного азота, молекулярного кислорода и метанола с содержанием молекулярного кислорода не более чем приблизительно 11% объемн. находятся за пределами области воспламенения. Обычно содержание метанола в поступающей реакционной газовой смеси C при ФОРМОКС-процессе составляет ≥2% объемн., предпочтительно от 4 до 10% объемн. и особенно предпочтительно от 6 до 9% объемн. или от 5 до 7% объемн. Газовые смеси из молекулярного азота, молекулярного кислорода и метанола, содержание метанола в которых составляет ≤6,7% объемн., независимо от их содержания молекулярного кислорода находятся за пределами области воспламенения, отчего в этой области концентраций могут применяться особенно высокие соотношения no: nMе в поступающей реакционной газовой смеси C.
Однако ФОРМОКС-процесс также отличается от способа с серебром тем, что с помощью этого процесса в пересчете на однократное прохождение реакционной газовой смеси C через реакционную зону C достигаемые степени превращения метанола в основном независимо от совместно применяемого в поступающей реакционной газовой смеси C инертного газа-разбавителя регулярно составляют >90% мольн., обычно ≥92% мольн., в большинстве случаев ≥95% мольн. и часто даже ≥97% мольн. или ≥98% мольн., или соответственно ≥99% мольн. Сопровождающие это селективности образования формальдегида составляют регулярно ≥90% мольн., в большинстве случаев ≥92% мольн. и часто ≥94% мольн., а часто даже ≥96% мольн.
В качестве инертных газов-разбавителей в поступающей реакционной газовой смеси C согласно изобретению для ФОРМОКС-процесса в реакционной зоне C также рассматриваются такие газы, как H2O, N2, CO2 и благородные газы, такие как Ar, а также смеси из вышеназванных газов. Предпочтительным инертным газом-разбавителем, отличающимся от водяного пара, в поступающей реакционной газовой смеси C при ФОРМОКС-процессе также является молекулярный азот.
Содержание в поступающей реакционной газовой смеси C инертного газа-разбавителя (определение инертного газа-разбавителя для реакционной зоны C аналогично тому же для реакционных зон A и B) при ФОРМОКС-процессе может составлять от 70 до 95% объемн., часто от 70 до 90% объемн. и предпочтительно от 70 до 85% объемн. То есть, содержание в поступающей реакционной газовой смеси C молекулярного азота при использовании ФОРМОКС-процесса может составлять от 70 до 95% объемн., или от 70 до 90% объемн., или от 70 до 85% объемн. Согласно изобретению предпочтительно поступающая реакционная газовая смесь C при ФОРМОКС-процессе может быть не содержащей водяного пара. С точки зрения технологической целесообразности поступающая реакционная газовая смесь C при использовании ФОРМОКС-процесса в реакционной зоне C из таких же соображений, как и в случае поступающей реакционной газовой смеси A, будет иметь незначительное содержание водяного пара. Как правило, это содержание водяного пара в поступающей реакционной газовой смеси C при ФОРМОКС-процессе составляет в реакционной зоне C ≥0,1% объемн. и ≤20% объемн. или соответственно ≤10% объемн., предпочтительно ≥0,2% объемн. и ≤7% объемн., более предпочтительно ≥0,5 и ≤5% объемн. Другое преимущество применения ФОРМОКС-процесса в реакционной зоне C согласно изобретению обосновывается тем, что описываемые высокие степени превращения метанола устанавливаются в основном при более низких температурах реакции по сравнению с применением способа с серебром.
Температура реакционной газовой смеси C при ФОРМОКС-процессе в реакционной зоне C обычно будет лежать в диапазоне от 250 до 500°C, предпочтительно в диапазоне от 300 до 450°C и часто в диапазоне от 270 до 400°C. При этом значение термина «температура реакционной газовой смеси C» при ФОРМОКС-процессе соответствует тому, которое уже было дано в данной публикации для способа с серебром.
Согласно изобретению температура реакционной газовой смеси C (в этой публикации также обозначенной как температура реакции в реакционной зоне C) предпочтительно на протяжении всей реакционной зоны C лежит в указанных выше температурных диапазонах. Также при ФОРМОКС-процессе предпочтительно подаваемая реакционная газовая смесь C подается в реакционную зону C уже с температурой, лежащей в вышеуказанной области. Часто при ФОРМОКС-процессе на входе в реакционную зону C в направлении движения потока до собственно каталитически активного заполнения катализатором (который также может быть разбавлен инертными формованными изделиями) находится заполнение реакционной зоны C твердым инертным материалом или заполнение каталитически активным катализатором в высоком разбавлении инертным материалом такого типа. При прохождении такого предварительного заполнения реакционной зоны C температура поступающей реакционной газовой смеси C, подаваемой в реакционную зону C, при ФОРМОКС-процессе может сравнительно просто устанавливаться на величину, с которой реакционная газовая смесь C при ФОРМОКС-процессе должна поступать непосредственно в каталитически активное заполнение катализатором в этой реакционной зоне C.
В отношении рабочего давления в реакционной зоне C для ФОРМОКС-процесса соответствующим образом также справедливо сказанное для способа с серебром.
Особенно подходящими для ФОРМОКС-процесса активными массами смешанных оксидов являются такие массы общей формулы III,
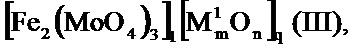
в которой переменные имеют следующие значения:
М1=Мо и/или Fe, или
Mo и/или Fe, а также в пересчете на общее молярное количество из Mo и Fe общее молярное количество вплоть до 10% мольн. (например, от 0,01 до 10% мольн., или от 0,1 до 10% мольн.), предпочтительно не более чем 5% мольн., одного или более чем одного элемента из группы, состоящей из Ti, Sb, Sn, Ni, Cr, Се, Al, Ca, Mg, V, Nb, Ag, Mn, Cu, Co, Si, Na, K, Tl, Zr, W, Ir, Та, As, P и В,
q=от 0 до 5, или от 0,5 до 3, или от 1 до 2,
m=от 1 до 3, и
n=от 1 до 6,
при условии, что содержимое обеих квадратных скобок является электрически нейтральным, то есть, не имеет электрического заряда.
Согласно изобретению предпочтительно активные массы смешанных оксидов III предпочтительно содержат менее чем 50% мольн., особенно предпочтительно менее чем 20% мольн. и особенно предпочтительно менее чем 10% мольн. железа (Fe), содержащегося в этой активной массе смешанных оксидов III, в степени окисления +2, и соответственно остальное количество содержащегося в ней Fe в степени окисления +3. Наиболее предпочтительно активная масса смешанных оксидов III содержит все содержащееся в ней железо в степени окисления +3.
Соотношение nMo: nFe содержащегося в активной массе смешанных оксидов III молярного количества Мо (nМо) и содержащегося в той же активной массе смешанных оксидов молярного количества Fe (nFe) предпочтительно составляет от 1:1 до 5:1.
Кроме того, согласно изобретению предпочтительно, когда М1=Мо, и m=1, а также n=3. Предпочтительные массы смешанных оксидов согласно изобретению также получаются тогда, когда М1=Fe, и m=2, а также n=3.
Кроме того, согласно изобретению благоприятными активными массами смешанных оксидов III являются такие, стехиометрия которых составлена так, что формально они могут рассматриваться (представляться) как смесь из MoO3 и Fe2O3, и содержание MoO3 в этой смеси составляет от 65 до 95% масс, а содержание Fe2O3 в этой смеси от 5 до 35% масс.
Получение активных масс смешанных оксидов III может осуществляться, как описано в приведенных публикациях уровня техники.
Как правило, при этом поступают таким образом, что из источников каталитически активной оксидной массы III получают как можно более плотную, предпочтительно тонко измельченную, составленную в соответствии со стехиометрией желаемой оксидной массы III сухую смесь (массу предшественника), и эту массу прокаливают (термически обрабатывают) при температурах от 300 до 600°C, предпочтительно от 400 до 550°C. Это прокаливание может проводиться как в атмосфере инертного газа, так и в окислительной атмосфере, такой как, например, воздух (или другая смесь из инертного газа и кислорода), а также в восстановительной атмосфере (например, смеси из инертного газа и восстанавливающих газов, таких как NH3 и СО). Продолжительность прокаливания, как правило, будет составлять несколько часов и обычно убывает с подъемом температуры прокаливания.
В качестве источников для элементарных составляющих каталитически активной оксидной массы III рассматривают особенно такие соединения, которые уже представляют собой оксиды, и/или такие соединения, которые могут переводиться в оксиды в результате нагревания, по меньшей мере в присутствии кислорода. Тщательное смешивание исходных соединений (источников) может осуществляться в сухой или во влажной форме. Если оно осуществляется в сухой форме, то эти исходные соединения целесообразным образом используются в виде тонко измельченного порошка и после смешивания и при необходимости уплотнения подвергаются прокаливанию. Однако предпочтительно это тщательное перемешивание осуществляется во влажной форме. Обычно при этом исходные соединения смешивают друг с другом в форме водных суспензий и/или растворов. Особенно тщательно смешанные сухие смеси получаются при описанном способе смешивания тогда, когда исходят исключительно из источников элементарных составляющих, присутствующих в растворенной форме.
В качестве растворителя предпочтительно используют воду. Предпочтительно из исходных соединений получают по меньшей мере два водных раствора, из которых по меньшей мере один представляет собой кислый раствор, и по меньшей мере один аммонийный щелочной (основный) раствор.
При соединении этих водных растворов, как правило, происходят реакции осаждения, при которых образуются соединения-предшественники активной массы смешанных оксидов III.
Затем полученную водную массу высушивают, причем процесс сушки может осуществляться, например, с помощью распылительной сушки.
Полученная после прокаливания сухой массы каталитически активная оксидная масса может в тонко измельченной форме как таковой или с помощью жидкого связующего средства будучи нанесенной на внешнюю поверхность формованного изделия носителя в качестве слоистого катализатора применяться для заполнения реакционной зоны C для ФОРМОКС-процесса. Однако изготовление слоистых катализаторов также может осуществляться таким образом, что с помощью жидкого связующего средства тонко измельченный порошок предшественника наносится на внешнюю поверхность формованного изделия носителя, а прокаливание вещества предшественника осуществляется только после проведенного нанесения и сушки.
Однако активные массы полиметаллического оксида III могут также использоваться в реакционной зоне C в чистой, неразбавленной форме или при разбавлении оксидным, в основном инертным материалом разбавителя в качестве так называемых сплошных катализаторов (согласно изобретению это предпочтительно). В качестве подходящих согласно изобретению инертных материалов разбавителя следует назвать, например, тонко измельченный оксид алюминия, диоксид кремния, алюмосиликаты, диоксид циркония, диоксид титана или их смеси. Неразбавленные сплошные катализаторы согласно изобретению являются предпочтительными.
В случае формованных изделий сплошных катализаторов формование осуществляется предпочтительно с помощью порошка предшественника, который прокаливается только после придания формы. Это формование обычно осуществляется с добавлением вспомогательных средств для формования, таких как, например, графит (смазывающих средств), или минеральных волокон (усиливающих средств). Подходящими способами формования являются таблетирование, штранг-прессование и экструдирование. Но, разумеется, это формование также может проводиться, например, со смесью из порошка активной массы и порошка предшественника, к которой перед формованием, в свою очередь, добавляется вспомогательное средство для формования, а также на выбор - порошок инертного разбавителя. После формования снова прокаливают. Как правило, формование с получением сплошных катализаторов может проводиться также только с уже предварительно полученным порошком активной массы, а также на выбор - с указанными вспомогательными средствами. Этот способ проведения процесса является менее предпочтительным. После формования в этом случае также, как правило, еще раз прокаливают.
Желательным источником Mo является, например, тетрагидрат гептамолибдата аммония (NH4)6(Mo7O24)⋅4H2O. Предпочтительными источниками железа являются, например, нитрат железа (III) [Fe(NO3)3], хлорид железа(III) [FeCl3] или гидраты нитрата железа(III), такие как, например, Fe(NO3)3⋅9H2O.
В отношении подходящих согласно изобретению геометрических характеристик и материалов формованных изделий носителя, а также способов нанесения покрытий для получения слоистых катализаторов из активной массы смешанных оксидов III соответствующим образом справедливы высказывания, уже сделанные в этой публикации в связи со слоистыми катализаторами - катализаторами окисления A. Предпочтительными геометриями формованных изделий носителя здесь также являются шарики и кольца, наибольший размер которых составляет от 1 до 10 мм, часто от 2 до 8 мм или от 3 до 6 мм. Желаемую согласно изобретению геометрию кольца имеют полые цилиндрические формованные изделия носителя с длиной от 2 до 10 мм, внешним диаметром от 4 до 10 мм и толщиной стенки от 1 до 4 мм. Предпочтительно эти полые цилиндрические формованные изделия носителя имеют длину от 3 до 6 мм, внешний диаметр от 4 до 8 мм и толщину стенки от 1 до 2 мм.
Толщина слоя из каталитически активной массы, нанесенного в случае вышеуказанных слоистых катализаторов на формованное изделие носителя, в случае активных масс смешанных оксидов III с точки зрения технологической целесообразности также, как правило, составляет от 10 до 1000 мкм. Предпочтительно эта толщина слоя составляет от 10 до 500 мкм, особенно предпочтительно от 100 до 500 мкм и наиболее предпочтительно от 200 до 300 мкм.
Предпочтительными формованными изделиями сплошных катализаторов, включающими активные массы смешанных оксидов III, являются сплошные цилиндры, полые цилиндры и трехдольчатые гранулы. Внешний диаметр цилиндрических сплошных катализаторов с точки зрения технологической целесообразности составляет от 3 до 10 мм, предпочтительно от 4 до 8 мм и прежде всего от 5 до 7 мм. Их высота предпочтительно составляет от 1 до 10 мм, предпочтительно от 2 до 6 мм и прежде всего от 3 до 5 мм. В случае полых цилиндров справедливо то же самое. Дополнительно, внутренний диаметр отверстия, проходящего насквозь сверху вниз, предпочтительно составляет от 1 до 8 мм, предпочтительно от 2 до 6 мм и наиболее предпочтительно от 2 до 4 мм. Технологически целесообразно толщина стенки полых цилиндров составляет от 1 до 3 мм.
Однако катализаторы окисления C на основе активных масс смешанных оксидов III в реакционной зоне C могут также применяться как катализаторы на носителе. В этом случае исходят из сравнительно пористых формованных изделий носителя, которые, например, друг за другом пропитываются по меньшей мере двумя растворами соединений предшественников. Описанная реакция осаждения осуществляется в порах формованного изделия носителя, а соединения предшественника, образующиеся при этом в этих порах, могут после этого переводиться в желаемые активные массы смешанных оксидов III при помощи прокаливания. В качестве альтернативы можно также пропитывать раствором, содержащим в растворенном виде все требуемые источники, высушивать и после этого кальцинировать (сравните, например, с немецкой заявкой на патент DE-A 2442311). В остальном, для получения катализаторов окисления C на основе активных масс смешанных оксидов III можно поступать, как в публикациях из уровня техники, на которые в настоящей заявке в этом отношении делается ссылка.
В частности, это публикации заявок на патенты США US-A 3716497, US-A 3846341, европейской заявки на патент EP-A 199359, немецкой заявки на патет-DE-A 2145851, заявки на патент США US-A 3983073, немецкой заявки на патент DE-A 2533209, европейской заявки на патент EP-A 2213370 и Catalysis Review, 47, страницы 125-174 (2004).
Разумеется, при ФОРМОКС-процессе для получения поступающей реакционной газовой смеси C также может использоваться не только сравнительно чистый метанол. Согласно изобретению подходящим в этом отношении метанольным исходным сырьем являются также водные растворы метанола, а также технический метанол, которые после соответствующего упаривания могут применяться для получения поступающей реакционной газовой смеси C.
Также реакционная зона C может заполняться неподвижным слоем катализатора, который содержит катализаторы окисления C для процесса ФОРМОКС в виде разбавленных инертными формованными изделиями.
Нагрузка неподвижного слоя катализатора, находящегося в реакционной зоне C, по поступающей реакционной газовой смеси C при применяемом согласно изобретению ФОРМОКС-процессе, как правило, будет составлять от 3500 нл/л⋅ч до 75000 нл/л⋅ч, предпочтительно от 25000 нл/л⋅ч до 35000 нл/л⋅ч. Понятие нагрузки при этом применяется так, как определено в немецкой заявке на патент DE-A 19927624.
Для исполнения ФОРМОКС-процесса в реакционной зоне C подходят, в частности, также реакторы с теплообменом, которые уже были рекомендованы для исполнения реакционной зоны A (сравните, например, с международной заявкой WO 2005/063375).
Согласно изобретению для получения акриловой кислоты особенно предпочтительны такие способы, при которых для получения поступающей реакционной газовой смеси B в качестве источника формальдегида применяется газообразная смесь продуктов C, выходящая из реакционной зоны C, которая представляет собой продукт ФОРМОКС-процесса, проводимого в реакционной зоне C. Это также потому, что такая газообразная смесь продуктов C в отличие от газообразной смеси продуктов C после способа с серебром не содержит молекулярного водорода.
То есть, газообразная смесь продуктов C гетерогенно катализируемого частичного газофазного окисления метанола до формальдегида согласно ФОРМОКС-процессу (без того, чтобы ее сначала следовало подвергать процессу разделения, без того, чтобы предварительно проводить для нее процесс разделения) представляет собой идеальный источник формальдегида для формальдегида, необходимого в поступающей реакционной газовой смеси B.
Часто газообразная смесь продуктов C при ФОРМОКС-процессе получается с температурой, с которой она может использоваться для получения поступающей реакционной газовой смеси B без дополнительной предварительной термической обработки. Однако часто температура газообразной смеси продуктов C, выходящей из реакционной зоны C, как в случае способа с серебром, так и при ФОРМОКС-процессе, отличается от той температуры, с которой она должна использоваться для получения поступающей реакционной газовой смеси B. По этим соображениям поток газообразной смеси продуктов C на своем пути из реакционной зоны C в реакционную зону B может проходить сквозь непрямой теплообменник, чтобы привести его температуру в соответствие с температурой смешивания, предусмотренной для получения поступающей реакционной газовой смеси B.
Как правило, заполнение реакционной зоны C по меньшей мере одним катализатором окисления C может выполняться в виде псевдоожиженного слоя. Однако с точки зрения технологии заполнение реакционной зоны C катализатором окисления C предпочтительно выполняется в виде неподвижного слоя.
Ради полноты еще следует добавить, что также при использовании ФОРМОКС-процесса в реакционной зоне C поток вещества Z, полученный в случае способа согласно изобретению в зоне разделения T, образует подходящий источник инертного газа для инертного газа, необходимого в поступающей реакционной газовой смеси C, и с точки зрения технологической целесообразности частичный поток потока вещества Z подается обратно в реакционную зону C для получения поступающей реакционной газовой смеси C.
Разделение газообразной смеси продуктов B, выходящей из реакционной зоны B, содержащей образовавшуюся акриловую кислоту, не вступившую в реакцию уксусную кислоту, по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, молекулярный кислород, а также водяной пар, в зоне разделения T по меньшей мере на три потока веществ X, Y и Z может осуществляться известным способом.
Например, это разделение может осуществляться с помощью фракционированной конденсации, как это рекомендуется в публикациях немецких заявок на патент DE-A 102007004960, DE-A 10 2007055086, DE-A 10243625, DE-A 10235847 и DE-A 19924532. При этом способе проведения процесса температура газообразной смеси продуктов B при необходимости сначала снижается при помощи прямого и/или непрямого охлаждения, и эта газообразная смесь продуктов B затем подается в конденсационную колонну, снабженную способствующими разделению встроенными элементами (например, тарелками для массообмена) и на выбор - оснащенную циклом охлаждения, и внутри этой конденсационной колонны, поднимаясь сама по себе, конденсируется по фракциям. С помощью соответствующего выбора числа теоретических ступеней разделения (теоретических тарелок) в конденсационной колонне потоки веществ X, Y и Z могут выводиться из этой конденсационной колонны как отделенные друг от друга фракции с соответствующей степенью обогащения.
С точки зрения технологической целесообразности поток вещества X, как правило, отделяется с содержанием акриловой кислоты ≥90% масс, предпочтительно ≥95% масс, и выводится из конденсационной колонны. При повышенной потребности в чистоте поток вещества X с точки зрения технологии предпочтительно может дополнительно очищаться при помощи кристаллизации (предпочтительно суспензионной кристаллизации) (сравните с указанными выше публикациями из уровня техники и международной заявкой WO 01/77056). Разумеется, поток вещества X, выведенный из конденсационной колонны, также может дополнительно очищаться перегонкой. Обоими путями таким образом со сравнительно небольшими затратами может достигаться чистота акриловой кислоты ≥99,9% масс, которая подходит для получения водопоглощающих смол с помощью радикальной полимеризации акриловой кислоты и/или смесей мономеров, включающих ее натриевую соль.
Получение водопоглощающих смол может осуществляться, например, как описано в публикациях международной заявки WO 2008/116840, немецких заявок DE-A 102005062929, DE-A 102004057874, DE-A 102004057868, DE-А 102004004496 и DE-A 19854575.
Соответствующим образом поток вещества Y также обычно выводится из конденсационной колонны с содержанием уксусной кислоты ≥90% масс, предпочтительно ≥95% масс. Отделенный таким образом поток вещества Y может как таковой подаваться обратно в реакционную зону В для получения поступающей реакционной газовой смеси B. Разумеется, также возможно перед подачей отделенного, как описано, потока вещества Y обратно в реакционную зону B с помощью перегонки и/или кристаллизации дополнительно обогащать его по содержанию уксусной кислоты (например, до содержания уксусной кислоты ≥99% масс), или при помощи увеличения числа теоретических ступеней разделения в конденсационной колонне непосредственно в ней самой отделять поток вещества Y с такой повышенной чистотой. Поток вещества Z обычно выходит из конденсационной колонны через головную часть.
В качестве альтернативы можно также проводить процесс, как рекомендуется в публикациях немецких заявок на патент DE-A 102009027401 и DE-A 10336386. После осуществленного предварительно при необходимости прямого и/или непрямого охлаждения газообразная смесь продуктов В при этом способе проведения процесса подается в абсорбционную колонну, предпочтительно оснащенную встроенными элементами с эффектом разделения, в противотоке с органическим растворителем, при нормальном давлении (105 Па) кипящем выше, чем акриловая кислота, (в качестве таковых рассматривают, например, органические растворители, указанные в немецких заявках на патент DE-A 102009027401 и DE-A 10336386) и уксусная кислота и акриловая кислота, содержащиеся в газообразной смеси продуктов B, абсорбируются в органический растворитель, в то время как поток вещества Z выходит из абсорбционной колонны через ее верх. Из продукта абсорбции, содержащего уксусную кислоту и акриловую кислоту, потоки вещества X и Y с соответствующей желаемой степенью обогащения могут выделяться с помощью соответствующего выбора числа теоретических ступеней разделения (теоретических тарелок) известным способом в результате ректификации (фракционной перегонки) в ректификационной колонне. Как правило, эта степень обогащения по акриловой кислоте или соответственно по уксусной кислоте будет составлять по меньшей мере 90% масс, предпочтительно по меньшей мере 95% масс. Последующая кристаллизационная дополнительная очистка отделенного потока вещества X (например, как предложено в международной заявке WO 01/77056) со сравнительно небольшими затратами ведет к чистоте акриловой кислоты ≥99,9% масс, которая подходит для получения водопоглощающих смол с помощью радикальной полимеризации акриловой кислоты и/или смесей мономеров, включающих ее натриевую соль. Отделенный, как описано, ректификацией поток вещества Y может как таковой или при необходимости после проведенной кристаллизационной и/или ректификационной дополнительной очистки (например, до содержания уксусной кислоты ≥99% масс.) подаваться обратно в реакционную зону B для получения поступающей реакционной газовой смеси B. С помощью соответствующего увеличения числа теоретических ступеней разделения поток вещества Y также может непосредственно с такой степенью обогащения отделяться от продукта абсорбции ректификацией.
Вместо того, чтобы применять органический абсорбирующий агент, руководствуясь техническим описанием европейских заявок на патент EP-A 551111 или EP-A 778255, можно акриловую кислоту, а также уксусную кислоту, содержащиеся в газообразной смеси продуктов B, поглощать из нее в абсорбционной колонне также водным абсорбирующим агентом, в то время как поток вещества Z выходит из этой абсорбционной колонны через ее верх. Последующее разделение ректификацией водного продукта абсорбции, при желании с привлечением азеотропных агентов, дает желаемые потоки веществ X и Y.
Переведение уксусной кислоты и акриловой кислоты, содержащихся в реакционной газовой смеси B, в конденсированную фазу с сохранением газообразного потока вещества Z также может осуществляться, например, в результате одноступенчатой конденсации тех компонентов, содержащихся в реакционной газовой смеси B, температура кипения которых при нормальном давлении лежит не выше такой температуры уксусной кислоты. После этого конденсат, содержащий акриловую кислоту и уксусную кислоту, снова с соответствующей степенью обогащения может разделяться на по меньшей мере один поток вещества Y и по меньшей мере один поток вещества X.
С точки зрения технологической целесообразности в случае способа согласно изобретению по меньшей мере 90% мольн., предпочтительно по меньшей мере 95% мольн., особенно предпочтительно по меньшей мере 98% мольн. или по меньшей мере 99% мольн. уксусной кислоты, содержащейся в газообразной смеси продуктов B, подается обратно в реакционную зону B для получения поступающей реакционной газовой смеси B.
Вместо того, чтобы за способом согласно изобретению следовал процесс, при котором акриловая кислота, содержащаяся в потоке вещества X, или смесь из акриловой кислоты, содержащейся в потоке вещества X, и одного или нескольких отличающихся от акриловой кислоты, по меньшей мере однократно ненасыщенных по этиленовому типу мономеров полимеризуются с получением полимеров (например, радикально; эта полимеризация может представлять собой, например, полимеризацию в растворе или водную эмульсионную полимеризацию, или суспензионную полимеризацию), за способом согласно изобретению может также следовать процесс, при котором акриловая кислота, содержащаяся в потоке вещества X, этерифицируется с помощью по меньшей мере одного спирта, содержащего, например, от 1 до 8 атомов углерода, (например, такого спирта как метанол, этанол, н-бутанол, трет-бутанол и 2-этилгексанол) до соответствующих сложных эфиров акриловой кислоты (акрилатов). Тогда за процессом получения сложных эфиров акриловой кислоты снова может следовать процесс, при котором полученный сложный эфир акриловой кислоты или смесь полученных сложных эфиров акриловой кислоты и одного или нескольких отличающихся от полученных сложных эфиров акриловой кислоты, по меньшей мере однократно ненасыщенных по этиленовому типу мономеров полимеризуется с получением полимеров (например, радикально; эта полимеризация может представлять собой, например, полимеризацию в растворе или водную эмульсионную полимеризацию, или суспензионную полимеризацию).
Для порядка еще следует уточнить, что деактивации различных катализаторов в различных реакционных зонах способа согласно изобретению можно препятствовать тем, что соответствующим образом поднимают температуру реакции в соответствующей реакционной зоне (чтобы поддерживать стабильной степень превращения реагентов, рассчитанную на однократное прохождение реакционной газовой смеси через слой катализатора). Также оксидные активные массы в реакционных зонах A, B и C соответствующим образом, как описано для сравнимых оксидных катализаторов в международной заявке WO 2005/042459, могут регенерироваться с помощью пропускания обладающего окислительным действием газа, содержащего кислород, при повышенной температуре.
Бесперебойная работа, особенно в реакционных зонах A и C, в случае способа согласно изобретению может обеспечиваться при помощи аналогичного применения способа проведения процесса, описанного в международной заявке WO 2004/007405.
Способ согласно изобретению подкупает, с одной стороны, его широкой и обширной по времени использования сырьевой базой. С другой стороны, речь идет о способе, который в отличие от способов из уровня техники, делает возможным при сохранении способа проведения процесса скользящий переход от «ископаемой акриловой кислоты» к «возобновляемой акриловой кислоте».
При этом под «ископаемой акриловой кислотой» понимают акриловую кислоту, для которой соотношение молярного количества ядер атомов 14C, содержащихся в этой акриловой кислоте, и молярного количества ядер атомов 12C, содержащихся в той же акриловой кислоте, n14C:n12C, сведено на нет.
При этом под «возобновляемой акриловой кислотой» понимают акриловую кислоту, для которой это соотношение n14C n12C соответствует присутствующему в CO2 в атмосфере Земли соотношению V* из n14C:n12C, причем определение этого соотношения n14C:n12C осуществляется согласно способу, разработанному Уиллардом Френком Либби (ссылка на страницу в сети Интернет: http://de.wikipedia.orgn/wiki/Radikohlenstoffdatierung).
Соответствующим образом в этой публикации применяются термины «возобновляемый углерод» и «ископаемый углерод».
Способ, разработанный Либби, основан на том, что по сравнению с обоими ядрами атомов углерода 12C и 13C, третье встречающееся в природе ядро атома углерода 14C является нестабильным и поэтому также называется радиоактивным углеродом (период полураспада=примерно 5700 лет).
В верхних слоях земной атмосферы 14C постоянно образуется снова в результате ядерной реакции. Одновременно 14C распадается с периодом полураспада 5700 лет путем β-распада. Между постоянным новым образованием и постоянным распадом в атмосфере на Земле возникает равновесие, так что доля ядер 14C для углерода в атмосфере на Земле постоянна на протяжении большого промежутка времени, в течение которого в атмосфере на Земле существует стабильное соотношение V*.
Сформировавшийся в атмосфере радиоактивный углерод связывается с кислородом воздуха с образованием CO2, который в результате фотосинтеза затем попадает в биосферу. Поскольку живые организмы (растения, животные, люди) при своем обмене веществ таким образом постоянно обмениваются углеродом с окружающей их атмосферой, в живых организмах устанавливается то же самое соотношение распределения изотопов углерода, а, следовательно, то же самое соотношение n14C:n12C, какое имеет место в окружающей их атмосфере. Если этот обмен в момент смерти живого организма прерывается, то соотношение между 14C и 12C в умершем организме изменяется, поскольку распадающиеся атомные ядра 14C больше не заменяются на новые (углерод, содержащийся в умерших организмах, становится ископаемым).
Если со смерти организма (живого организма) проходит более чем 50000 лет, его содержание 14C лежит ниже границы обнаружения. Сегодняшние и будущие биологические («возобновляемые») источники сырья и полученные из них химические вещества имеют соответствующие текущие концентрации 14C в CO2 атмосферы на Земле (это соотношение n14C:n12C=V*). Однако ископаемые источники углерода, такие как уголь, нефть и природный газ уже несколько миллионов лет хранятся на Земле «мертвыми», отчего они, как и полученные из них химические вещества, не содержат больше 14C.
Если в случае способа согласно изобретению используется ископаемый этанол (этанол, полученный из ископаемого сырья) и возобновляемый формальдегид (формальдегид, полученный из возобновляемого сырья), получается акриловая кислота, у которой соотношение n14C:n12C составляет только (1/3)×V*.
И наоборот, если в случае способа согласно изобретению используется этанол, полученный из возобновляемого сырья, и формальдегид, полученный из ископаемого сырья, получается акриловая кислота, у которой соотношение n14C:n12C=(2/3)×V*.
Если в случае способа согласно изобретению используется как ископаемый (возобновляемый) этанол, так и ископаемый (возобновляемый) формальдегид, получается акриловая кислота, у которой соотношение n14C:n12C=0(=V*).
Если случае способа согласно изобретению дополнительно привлекают возможность смешивания возобновляемых и ископаемых исходных веществ (сырья), то, таким образом, изготовитель акриловой кислоты при использовании способа проведения процесса согласно изобретению без изменения способа получения (то есть, с помощью одной и той же производственной установки) может на собственное усмотрение установить в соответствии с желанием потребителя (например, изготовителя суперабсорбентов (=абсорбирующих воду смол)) «степень возобновляемости» акриловой кислоты, которую следует произвести, (желаемое потребителем соотношение n14C:n12C для акриловой кислоты, которую следует произвести).
В результате этерификации акриловой кислоты, для которой имеет место V=V*, с помощью биометанола или биоэтанола, могут получаться сложные эфиры акриловой кислоты, для которых соотношение n14C и n12C также равно V*.
Другое преимущество способа проведения процесса согласно изобретению состоит в том, что ни целевой продукт реакционной зоны A, ни целевой продукт реакционной зоны C не требуют выделения из газообразной смеси продуктов A или соответственно C, чтобы их можно было привлечь для получения поступающей реакционной газовой смеси B. Это обеспечивает для способа согласно изобретению как высокую экономию, так и эффективный энергетический баланс. Кроме того, при конденсации уксусной кислоты с формальдегидом в качестве побочного продукта не образуются ни глиоксаль, ни пропионовая кислота, как это обязательно имеет место при гетерогенно катализируемом частичном окислении пропилена, пропана, акролеина, пропионового альдегида и/или глицерина до акриловой кислоты.
Кроме того, способ согласно изобретению обеспечивает высокий выход продукта за один проход в единицу времени на единицу объема при одновременно высокой селективности по целевому продукту, рассчитанной на вступившие в реакцию исходные вещества.
Таким образом, настоящая заявка включает в себя, в частности, следующие варианты исполнения согласно изобретению:
1. Способ получения акриловой кислоты из этанола и формальдегида, который включает следующие операции:
- через первую реакционную зону A, в которую загружен по меньшей мере один катализатор окисления A, пропускается поток поступающей реакционной газовой смеси A, содержащей реагенты - этанол и молекулярный кислород, а также по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и при прохождении этой реакционной зоны A этанол, содержащийся в поступающей реакционной газовой смеси A, в условиях гетерогенного катализа окисляется до уксусной кислоты и водяного пара, так что возникает газообразная смесь продуктов A, содержащая уксусную кислоту, водяной пар, молекулярный кислород, а также по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и поток газообразной смеси продуктов A покидает реакционную зону A, причем к проходящей сквозь реакционную зону A реакционной газовой смеси A на ее пути через эту реакционную зону A на выбор может подаваться дополнительный молекулярный кислород и/или дополнительный инертный газ-разбавитель,
- из потока газообразной смеси продуктов A, покидающего реакционную зону A, и по меньшей мере одного другого потока веществ, который содержит по меньшей мере один источник формальдегида, получается поток поступающей реакционной газовой смеси B, содержащей уксусную кислоту, водяной пар, молекулярный кислород, по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и формальдегид, в котором содержащееся молярное количество уксусной кислоты nHAc больше чем содержащееся в нем молярное количество формальдегида nFd,
- через вторую реакционную зону B, в которую загружен по меньшей мере один катализатор альдольной конденсации B, пропускается поток поступающей реакционной газовой смеси B, и при прохождении этой реакционной зоны B формальдегид, содержащийся в поступающей реакционной газовой смеси B, вместе с уксусной кислотой, содержащейся в поступающей реакционной газовой смеси B, в условиях гетерогенного катализа конденсируется до акриловой кислоты и H2O, так что возникает газообразная смесь продуктов B, содержащая акриловую кислоту, уксусную кислоту, водяной пар, молекулярный кислород, а также по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и поток газообразной смеси продуктов B покидает реакционную зону B, причем к проходящей сквозь реакционную зону B реакционной газовой смеси B на ее пути через эту реакционную зону B на выбор может подаваться дополнительный молекулярный кислород и/или дополнительный инертный газ-разбавитель,
- поток газообразной смеси продуктов B, покидающий реакционную зону B, подается в зону разделения T, и в этой зоне разделения T разделяется по меньшей мере на три потока веществ - X, Y и Z, причем
- поток акриловой кислоты, содержащийся в потоке вещества X, больше, чем потоки акриловой кислоты, содержащиеся в потоках веществ Y и Z, вместе взятые,
- поток уксусной кислоты, содержащийся в потоке вещества Y, больше, чем потоки уксусной кислоты, содержащиеся в потоках веществ X и Z, вместе взятые,
- поток инертного газа-разбавителя, отличающегося от водяного пара, содержащийся в потоке вещества Z, больше, чем потоки инертного газа-разбавителя, отличающегося от водяного пара, содержащиеся в потоках веществ X и Y, вместе взятые,
и
- поток вещества Y подается обратно в реакционную зону В и применяется дополнительно для получения поступающей реакционной газовой смеси В.
2. Способ согласно варианту исполнения 1, отличающийся тем, что по меньшей мере один катализатор окисления A содержит каталитически активную массу, которая содержит по меньшей мере один оксид ванадия.
3. Способ согласно вариантам исполнения 1 или 2, отличающийся тем, что по меньшей мере один катализатор окисления A содержит каталитически активную массу, которая содержит по меньшей мере один оксид ванадия и дополнительно по меньшей мере один оксид из группы оксидов титана, алюминия, циркония и олова.
4. Способ согласно вариантам исполнения 2 или 3, отличающийся тем, что по меньшей мере один оксид ванадия содержит ванадий в степени окисления +5.
5. Способ согласно варианту исполнения 3, отличающийся тем, что по меньшей мере один оксид ванадия содержит ванадий в степени окисления +5, и по меньшей мере оксид из группы оксидов титана, алюминия, циркония и олова содержит элемент из группы титана, алюминия, циркония и олова в степени окисления +4.
6. Способ согласно одному из вариантов исполнения 2-5, отличающийся тем, что каталитически активная масса в качестве по меньшей мере одного оксида ванадия содержит V2O5.
7. Способ согласно варианту исполнения 6, отличающийся тем, что каталитически активная масса содержит от 0,1 до 60% масс. V2O5.
8. Способ согласно варианту исполнения 6, отличающийся тем, что каталитически активная масса содержит от 1 до 50% масс. V2O5.
9. Способ согласно варианту исполнения 6, отличающийся тем, что каталитически активная масса содержит от 3 до 40% масс. V2O5.
10. Способ согласно варианту исполнения 6, отличающийся тем, что каталитически активная масса содержит от 5 до 30% масс. V2O5.
11. Способ согласно одному из вариантов исполнения 2-10, отличающийся тем, что каталитически активная масса содержит TiO2.
12. Способ согласно одному из вариантов исполнения 2-7, отличающийся тем, что каталитически активная масса содержит от 40 до 99,9% масс. TiO2.
13. Способ согласно варианту исполнения 8, отличающийся тем, что каталитически активная масса содержит от 50 до 99% масс. TiO2.
14. Способ согласно варианту исполнения 9, отличающийся тем, что каталитически активная масса содержит от 60 до 97% масс. TiO2.
15. Способ согласно варианту исполнения 10, отличающийся тем, что каталитически активная масса содержит от 70 до 95% масс. TiO2.
16. Способ согласно одному из вариантов исполнения 11-15, отличающийся тем, что каталитически активная масса состоит из V2O5 в качестве по меньшей мере одного оксида ванадия и из TiO2.
17. Способ согласно одному из вариантов исполнения 11-16, отличающийся тем, что по меньшей мере частичное количество TiO2 присутствует в анатазной модификации.
18. Способ согласно одному из вариантов исполнения 11-16, отличающийся тем, что от 50 до 100% масс. TiO2 присутствует в анатазной модификации.
19. Способ согласно одному из вариантов исполнения 2-18, отличающийся тем, что по меньшей мере один катализатор оксиления A представляет собой сплошной катализатор.
20. Способ согласно варианту исполнения 19, отличающийся тем, что сплошной катализатор представляет собой шарик, кольцо или сплошной цилиндр.
21. Способ согласно варианту исполнения 20, отличающийся тем, что наибольший размер сплошного катализатора составляет от 1 до 10 мм.
22. Способ согласно одному из вариантов исполнения 2-18, отличающийся тем, что по меньшей мере один катализатор оксиления A представляет собой слоистый катализатор, который содержит каталитически активную массу в качестве слоя на поверхности инертного формованного изделия носителя.
23. Способ согласно варианту исполнения 22, отличающийся тем, что формованное изделие носителя представляет собой шарик или кольцо.
24. Способ согласно варианту исполнения 22 или 23, отличающийся тем, что наибольший размер формованного изделия носителя составляет от 1 до 10 мм.
25. Способ согласно одному из вариантов исполнения 22-24, отличающийся тем, что инертное формованное изделие носителя состоит из стеатита.
26. Способ согласно одному из вариантов исполнения 22-25, отличающийся тем, что толщина оболочки из каталитически активной массы составляет от 10 до 2000 мкм, или от 10 до 500 мкм, или от 100 до 500 мкм, или от 200 до 300 мкм.
27. Способ согласно варианту исполнения 1, отличающийся тем, что заполнение реакционной зоны A по меньшей мере одним катализатором окисления A включает в себя в направлении движения потока поступающей реакционной газовой смеси A два следующих друг за другом в пространстве в указанной числовой последовательности участка 1 и 2, которые заполнены отличающимися друг от друга катализаторами окисления A, причем
- по меньшей мере один катализатор окисления A участка 1 содержит каталитически активную массу 1, которая содержит по меньшей мере один оксид ванадия, а также по меньшей мере один оксид из группы оксидов титана, алюминия, циркония и олова, и
- по меньшей мере один катализатор окисления A участка 2 содержит каталитически активную массу 2, которая представляет собой мультиметаллический оксид, который помимо ванадия (V) и молибдена (Мо) дополнительно содержит по меньшей мере один из элементов - вольфрам (W), молибден (Mo), тантал (Ta), хром (Cr) и церий (Ce), а также по меньшей мере один из элементов - медь (Cu), никель (Ni), кобальт (Co), железо (Fe), марганец (Mn) и цинк (Zn).
28. Способ согласно варианту исполнения 27, отличающийся тем, что каталитически активная масса 1 содержит от 1 до 50% масс. V2O5 в качестве по меньшей мере одного оксида ванадия и от 50 до 99% масс. TiO2 в качестве оксида титана (предпочтительно в анатазной модификации) или состоит из них.
29. Способ согласно варианту исполнения 27, отличающийся тем, что каталитически активная масса 1 содержит от 3 до 40% масс. V2O5 в качестве по меньшей мере одного оксида ванадия и от 60 до 97% масс. TiO2 в качестве оксида титана (предпочтительно в анатазной модификации) или состоит из них.
30. Способ согласно варианту исполнения 27, отличающийся тем, что каталитически активная масса 1 содержит от 5 до 30% масс. V2O5 в качестве по меньшей мере одного оксида ванадия и от 70 до 95% масс. TiO2 в качестве оксида титана (предпочтительно в анатазной модификации) или состоит из них.
31. Способ согласно одному из вариантов исполнения 27-30, отличающийся тем, что по меньшей мере один катализатор окисления A участка 1 представляет собой сплошной катализатор.
32. Способ согласно варианту исполнения 31, отличающийся тем, что геометрия сплошного катализатора выбирается из группы, состоящей из шариков, колец и сплошных цилиндров, а также имеет наибольший размер в интервале от 1 до 10 мм.
33. Способ согласно одному из вариантов исполнения 27-30, отличающийся тем, что по меньшей мере один катализатор окисления A участка 1 представляет собой слоистый катализатор, который содержит каталитически активную массу 1 в качестве оболочки, нанесенной на поверхность инертного формованного изделия носителя.
34. Способ согласно варианту исполнения 33, отличающийся тем, что формованное изделие носителя представляет собой шарик или кольцо.
35. Способ согласно варианту исполнения 33 или 34, отличающийся тем, что наибольший размер формованного изделия носителя составляет от 1 до 10 мм.
36. Способ согласно одному из вариантов исполнения 33-35, отличающийся тем, что инертное изделие носителя состоит из стеатита.
37. Способ согласно одному из вариантов исполнения 33-36, отличающийся тем, что толщина слоя из каталитически активной массы 1 составляет от 10 до 2000 мкм, или от 10 до 500 мкм, или от 100 до 500 мкм, или от 200 до 300 мкм.
38. Способ согласно одному из вариантов исполнения 27-37, отличающийся тем, что каталитически активная масса 1 не содержит молибдена (Mo).
39. Способ согласно одному из вариантов исполнения 27-38, отличающийся тем, что каталитически активная масса 2 представляет собой по меньшей мере один мультиметаллический оксид общей формулы I,
в которой переменные имеют следующее значение:
X1=W, Nb, Та, Cr и/или Се,
X2=Cu, Ni, Со, Fe, Mn и/или Zn,
X3=Sb и/или Bi,
X4=один или несколько щелочных металлов,
X5=один или несколько щелочноземельных металлов,
X6=Si, AI, Ti и/или Zr,
A=от 1 до 6,
b=от 0,2 до 4,
C=от 0,5 до 1,8
d=от 0 до 40,
e=от 0 до 2,
f=от 0 до 4,
g=от 0 до 40, и
n=стехиометрический коэффициент элементарного кислорода, который определяется с помощью стехиометрических коэффициентов элементов, отличных от кислорода, а также числом их зарядов в I.
40. Способ согласно одному из вариантов исполнения 27-38, отличающийся тем, что каталитически активная масса 2 представляет собой по меньшей мере один мультиметаллический оксид общей формулы I
в которой переменные имеют следующее значение:
X1=W, Nb и/или Cr,
X2=Cu, Ni, Co, Fe, Mn и/или Fe,
X3=Sb,
X4=натрий (Na) и/или калий (К),
X5=кальций (Са), стронций (Sr) и/или барий (Ва),
X6=Si, AI и/или Ti,
a=от 1,5 до 5,
b=от 0,5 до 2,
c=от 0,5 до 3,
d=от 0 до 2,
e=от 0 до 0,2,
f=от 0 до 1,
g=от 0 до 1, и
n=стехиометрический коэффициент элементарного кислорода, который определяется с помощью стехиометрических коэффициентов элементов, отличных от кислорода, а также числом их зарядов в I.
41. Способ согласно одному из вариантов исполнения 27-40, отличающийся тем, что по меньшей мере один катализатор окисления A участка 2 представляет собой слоистый катализатор, который содержит каталитически активную массу 2 в качестве оболочки, нанесенной на поверхность инертного формованного изделия носителя.
42. Способ согласно варианту исполнения 41, отличающийся тем, что формованное изделие носителя представляет собой шарик или кольцо.
43. Способ согласно варианту исполнения 41 или 42, отличающийся тем, что наибольший размер формованного изделия носителя составляет от 1 до 10 мм.
44. Способ согласно одному из вариантов исполнения 41-43, отличающийся тем, что инертное изделие носителя состоит из стеатита.
45. Способ согласно одному из вариантов исполнения 41-44, отличающийся тем, что толщина слоя из каталитически активной массы 2 составляет от 10 до 2000 мкм, или от 10 до 500 мкм, или от 100 до 500 мкм, или от 200 до 300 мкм.
46. Способ согласно одному из вариантов исполнения 1-45, отличающийся тем, что реакционная зона A, которая заполнена по меньшей мере одним катализатором окисления A, заполнена неподвижным слоем катализатора.
47. Способ согласно одному из вариантов исполнения 1-46, отличающийся тем, что температура реакции в реакционной зоне A лежит в диапазоне от 100 до 450°C.
48. Способ согласно одному из вариантов исполнения 1-46, отличающийся тем, что температура реакции в реакционной зоне A лежит в диапазоне от 100 до 350°C.
49. Способ согласно одному из вариантов исполнения 27-45, отличающийся тем, что на протяжении длины участка 1 средняя арифметическая температура реакции 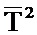
50. Способ согласно варианту исполнения 49, отличающийся тем, что 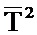
51. Способ согласно варианту исполнения 49, отличающийся тем, что
52. Способ согласно варианту исполнения 49, отличающийся тем, что
53. Способ согласно одному из вариантов исполнения 49-52, отличающийся тем, что
54. Способ согласно одному из вариантов исполнения 27-46 и 49-53, отличающийся тем, что этанол, содержащийся в поступающей реакционной газовой смеси A, при прохождении сквозь участок 1 подвергается взаимодействию по меньшей мере на 90% мольн.
55. Способ согласно одному из вариантов исполнения 27-46 и 49-53, отличающийся тем, что этанол, содержащийся в поступающей реакционной газовой смеси A, при прохождении сквозь участок 1 подвергается взаимодействию по меньшей мере на 95% мольн.
56. Способ согласно одному из вариантов исполнения 27-46 и 49-55, отличающийся тем, что этанол, содержащийся в поступающей реакционной газовой смеси A, при прохождении сквозь участки 1 и 2 подвергается взаимодействию по меньшей мере на 97% мольн.
57. Способ согласно одному из вариантов исполнения 27-46 и 49-56, отличающийся тем, что этанол, содержащийся в поступающей реакционной газовой смеси A при прохождении сквозь участки 1 и 2 подвергается взаимодействию по меньшей мере на 99% мольн.
58. Способ согласно одному из вариантов исполнения 1-57, отличающийся тем, что поступающая реакционная газовая смесь A содержит от 0,3 до 20% объемн. этанола.
59. Способ согласно одному из вариантов исполнения 1-57, отличающийся тем, что поступающая реакционная газовая смесь A содержит от 0,5 до 15% объемн. этанола.
60. Способ согласно одному из вариантов исполнения 1-57, отличающийся тем, что поступающая реакционная газовая смесь A содержит от 0,75 до 10% объемн. или от 1 до 5% объемн. этанола.
61. Способ согласно одному из вариантов исполнения 1-60, отличающийся тем, что поступающая реакционная газовая смесь A содержит молекулярный кислород в молярном количестве nо и этанол в молярном количестве nEt, и соотношение nо:nEt составляет по меньшей мере 1,3.
62. Способ согласно варианту исполнения 61, отличающийся тем, что nо: nEt составляет по меньшей мере 1,5.
63. Способ согласно варианту исполнения 61, отличающийся тем, что nо: nEt составляет по меньшей мере 1,75.
64. Способ согласно одному из вариантов исполнения 61-63, отличающийся тем, что no:nEt составляет не более 10.
65. Способ согласно одному из вариантов исполнения 61-63, отличающийся тем, что no:nEt составляет не более 5.
66. Способ согласно одному из вариантов исполнения 1-65, отличающийся тем, что поступающая реакционная газовая смесь A содержит от 1 до 40% объемн. Н2O.
67. Способ согласно одному из вариантов исполнения 1-65, отличающийся тем, что поступающая реакционная газовая смесь A содержит от 1 до 20% объемн. Н2O.
68. Способ согласно одному из вариантов исполнения 1-65, отличающийся тем, что поступающая реакционная газовая смесь A содержит от 5 до 15% объемн. Н2O.
69. Способ согласно одному из вариантов исполнения 1-65, отличающийся тем, что поступающая реакционная газовая смесь A содержит от 7,5 до 12,5% объемн. Н2O.
70. Способ согласно одному из вариантов исполнения 1-69, отличающийся тем, что по меньшей мере 80% объемн. содержащегося в поступающей реакционной газовой смеси A инертного газа-разбавителя, отличающегося от водяного пара, представляет собой молекулярный азот.
71. Способ согласно одному из вариантов исполнения 1-69, отличающийся тем, что по меньшей мере 90% объемн. содержащегося в поступающей реакционной газовой смеси A инертного газа-разбавителя, отличающегося от водяного пара, представляет собой молекулярный азот.
72. Способ согласно одному из вариантов исполнения 1-69, отличающийся тем, что по меньшей мере 95% объемн. содержащегося в поступающей реакционной газовой смеси A инертного газа-разбавителя, отличающегося от водяного пара, представляет собой молекулярный азот.
73. Способ согласно одному из вариантов исполнения 1-72, отличающийся тем, что поступающая реакционная газовая смесь A в качестве по меньшей мере одного инертного газа-разбавителя, отличающегося от водяного пара, содержит по меньшей мере 10% объемн. молекулярного азота.
74. Способ согласно одному из вариантов исполнения 1-72, отличающийся тем, что поступающая реакционная газовая смесь A в качестве по меньшей мере одного инертного газа-разбавителя, отличающегося от водяного пара, содержит по меньшей мере 30% объемн. молекулярного азота.
75. Способ согласно одному из вариантов исполнения 1-72, отличающийся тем, что поступающая реакционная газовая смесь A в качестве по меньшей мере одного инертного газа-разбавителя, отличающегося от водяного пара, содержит по меньшей мере 40% объемн. молекулярного азота.
76. Способ согласно одному из вариантов исполнения 1-72, отличающийся тем, что поступающая реакционная газовая смесь A в качестве по меньшей мере одного инертного газа-разбавителя, отличающегося от водяного пара, содержит не более 90% объемн. молекулярного азота.
77. Способ согласно одному из вариантов исполнения 1-76, отличающийся тем, что рабочее давление в реакционной зоне A составляет от 1,2⋅105 Па до 50⋅105 Па.
78. Способ согласно одному из вариантов исполнения 1-77, отличающийся тем, что в качестве источника этанола, содержащегося в поступающей реакционной газовой смеси A, используется биоэтанол.
79. Способ согласно варианту исполнения 78, отличающийся тем, что поступающая реакционная газовая смесь A, в пересчете на массу содержащегося в ней этанола, содержит по меньшей мере 1 масс, частей на млн химического соединения, содержащего элемент серу, в пересчете на количество этой содержащейся серы.
80. Способ согласно варианту исполнения 78, отличающийся тем, что поступающая реакционная газовая смесь A, в пересчете на массу содержащегося в ней этанола, содержит по меньшей мере от 2 до 200 масс, частей на млн химического соединения, содержащего элемент серу, в пересчете на количество этой содержащейся серы.
81. Способ согласно одному из вариантов исполнения 1-78, отличающийся тем, что поступающая реакционная газовая смесь A, в пересчете на массу содержащегося в ней этанола, содержит от 0 до <1 масс, частей на млн химического соединения, содержащего элемент серу, в пересчете на количество этой содержащейся серы.
82. Способ согласно одному из вариантов исполнения 78-81, отличающийся тем, что источник этанола, содержащегося в поступающей реакционной газовой смеси A, представляет собой водный раствор биоэтанола.
83. Способ согласно варианту исполнения 82, отличающийся тем, что в качестве источника этанола, содержащегося в поступающей реакционной газовой смеси A, используется фильтрат получающейся при выработке биоэтанола, содержащей растворенный биоэтанол, водной плодово-ягодной мезги.
84. Способ согласно одному из вариантов исполнения 1-83, отличающийся тем, что в качестве источника формальдегида для формальдегида, содержащегося в поступающей реакционной газовой смеси В, используется по меньшей мере один из источников - триоксан, параформальдегид, формалин, метилаль, водный раствор параформальдегида, водный раствор формальдегида и газообразная смесь продуктов гетерогенно катализируемого частичного газофазного окисления метанола до формальдегида, из которой, на выбор, при необходимости был отделен содержащийся в ней не вступивший в реакцию метанол.
85. Способ согласно одному из вариантов исполнения 1-84, отличающийся тем, что температура реакции в реакционной зоне B составляет от 260 до 400°C.
86. Способ согласно одному из вариантов исполнения 1-84, отличающийся тем, что температура реакции в реакционной зоне B составляет от 280 до 380°C.
87. Способ согласно одному из вариантов исполнения 1-84, отличающийся тем, что температура реакции в реакционной зоне B составляет от 300 до 370°C.
88. Способ согласно одному из вариантов исполнения 1-87, отличающийся тем, что рабочее давление в реакционной зоне B составляет от 1,2⋅105 Па до 50⋅105 Па.
89. Способ согласно одному из вариантов исполнения 1-88, отличающийся тем, что содержание формальдегида в поступающей реакционной газовой смеси B составляет от 0,5 до 10% объемн.
90. Способ согласно одному из вариантов исполнения 1-88, отличающийся тем, что содержание формальдегида в поступающей реакционной газовой смеси B составляет от 0,5 до 7% объемн.
91. Способ согласно одному из вариантов исполнения 1-88, отличающийся тем, что содержание формальдегида в поступающей реакционной газовой смеси B составляет от 1 до 5% объемн.
92. Способ согласно одному из вариантов исполнения 1-91, отличающийся тем, что поступающая реакционная газовая смесь B содержит уксусную кислоту в молярном количестве nHAc и формальдегид в молярном количестве nFd, и соотношение nHAс:nFd составляет больше 1 и ≤10.
93. Способ согласно варианту исполнения 92, отличающийся тем, что это соотношение nHAc: nFd составляет от 1,1 до 5.
94. Способ согласно варианту исполнения 92, отличающийся тем, что это соотношение nHAc:nFd составляет от 1,5 до 3,5.
95. Способ согласно одному из вариантов исполнения 1-94, отличающийся тем, что содержание уксусной кислоты в поступающей реакционной газовой смеси B составляет от 1,5 до 20% объемн.
96. Способ согласно одному из вариантов исполнения 1-94, отличающийся тем, что содержание уксусной кислоты в поступающей реакционной газовой смеси B составляет от 2 до 15% объемн.
97. Способ согласно одному из вариантов исполнения 1-94, отличающийся тем, что содержание уксусной кислоты в поступающей реакционной газовой смеси B составляет от 3 до 10% объемн.
98. Способ согласно одному из вариантов исполнения 1-97, отличающийся тем, что содержание молекулярного кислорода в поступающей реакционной газовой смеси B составляет от 0,5 до 5% объемн.
99. Способ согласно одному из вариантов исполнения 1-97, отличающийся тем, что содержание молекулярного кислорода в поступающей реакционной газовой смеси В составляет от 2 до 5% объемн.
100. Способ согласно одному из вариантов исполнения 1-99, отличающийся тем, что содержание водяного пара в поступающей реакционной газовой смеси В не превышает 30% объемн. и не опускается ниже 1,5% объемн.
101. Способ согласно одному из вариантов исполнения 1-99, отличающийся тем, что содержание водяного пара в поступающей реакционной газовой смеси B не превышает 20% объемн. и не опускается ниже 2% объемн.
102. Способ согласно одному из вариантов исполнения 1-99, отличающийся тем, что содержание водяного пара в поступающей реакционной газовой смеси B составляет от 5 до 15% объемн. или от 10 до 15% объемн.
103. Способ согласно одному из вариантов исполнения 1-102, отличающийся тем, что содержание в поступающей реакционной газовой смеси B инертного газа-разбавителя, отличающегося от водяного пара, составляет по меньшей мере 30% объемн. или по меньшей мере 40% объемн.
104. Способ согласно одному из вариантов исполнения 1-102, отличающийся тем, что содержание в поступающей реакционной газовой смеси B инертного газа-разбавителя, отличающегося от водяного пара, составляет по меньшей мере 50% объемн.
105. Способ согласно одному из вариантов исполнения 1-104, отличающийся тем, что поступающая реакционная газовая смесь B в качестве по меньшей мере одного инертного газа-разбавителя, отличающегося от водяного пара, содержит по меньшей мере 30% объемн. или по меньшей мере 40% объемн. N2.
106. Способ согласно одному из вариантов исполнения 1-104, отличающийся тем, что поступающая реакционная газовая смесь B в качестве по меньшей мере одного инертного газа-разбавителя, отличающегося от водяного пара, содержит по меньшей мере 50% объемн. N2.
107. Способ согласно одному из вариантов исполнения 1-106, отличающийся тем, что по меньшей мере один катализатор альдольной конденсации B представляет собой цеолит с анионным зарядом каркаса, у которого на внутренней и внешней поверхности находится по меньшей мере один тип катионов из группы ионов щелочных и щелочноземельных металлов, чтобы нейтрализовывать отрицательный заряд каркаса.
108. Способ согласно одному из вариантов исполнения 1-106, отличающийся тем, что по меньшей мере один катализатор альдольной конденсации B представляет собой гидроксид из группы, состоящей из гидроксидов щелочных металлов, гидроксидов щелочноземельных металлов и гидроксида алюминия, нанесенный на аморфный диоксид кремния.
109. Способ согласно варианту исполнения 108, отличающийся тем, что гидроксид, нанесенный на аморфный диоксид кремния, представляет собой KOH, NaOH, Ca(OH)2 или Mg(OH)2.
110. Способ согласно одному из вариантов исполнения 1-106, отличающийся тем, что по меньшей мере один катализатор альдольной конденсации B представляет собой катализатор, который включает
- в качестве компонента a) по меньшей мере один оксид по меньшей мере одного из элементов Si, AI, Ti, Zr, Cd, Sn, Ga, Y и La и/или цеолит,
и
- в качестве компонента b) по меньшей мере один оксид, выбираемый из оксида бора и оксида фосфора,
а также на выбор
- в качестве компонента c) один или более одного оксида по меньшей мере одного из элементов V, Cr, Co, Ni, Mo и Pb и/или более чем одну гетерополикислоту по меньшей мере с одним полиатомом, выбираемым из V, Mo и W.
111. Способ согласно варианту исполнения 110, отличающийся тем, что по меньшей мере один катализатор альдольной конденсации B содержит от 1 до 50% масс, оксида бора или от 1 до 50% масс, оксида фосфора, или от 1 до 50% масс, оксида бора и оксида фосфора, причем оксид бора рассчитывается в пересчете на содержащееся количество B, всегда как B2O3, и оксид фосфора, в пересчете на содержащееся количество P, всегда как P2O5.
112. Способ согласно одному из вариантов исполнения 1-106, отличающийся тем, что по меньшей мере один катализатор альдольной конденсации B содержит каталитически активную массу, которая представляет собой оксид ванадия-фосфора или оксид ванадия-фосфора, дотированный элементами, отличными от ванадия и фосфора.
113. Способ согласно варианту исполнения 112, отличающийся тем, что каталитически активная масса представляет собой каталитически активную массу мультиэлементных оксидов общей формулы II
в которой переменные имеют следующее значение:
X1=Mo, Bi, Co, Ni, Si, Zn, Hf, Zr, Ti, Cr, Mn, Cu, B, Sn и/или Nb, предпочтительно Nb, Mo, Zn и/или Hf,
X2=Li, K, Na, Rb, Cs и/или TI,
b=от 0,9 до 2,0,
c=от >0 до 0,1,
d=от >0 до 0,1,
e=от >0 до 0,1, и
n=стехиометрический коэффициент элемента кислорода, который определяется с помощью стехиометрических коэффициентов элементов, отличных от кислорода, а также числом их зарядов в II.
114. Способ согласно варианту исполнения 113, отличающийся тем, что X1=Nb, Mo, Zn и/или Hf.
115. Способ согласно варианту исполнения 113 или 114, отличающийся тем, что b составляет от 0,9 до 1,5.
116. Способ согласно варианту исполнения 113 или 114, отличающийся тем, что что b составляет от 0,9 до 1,2.
117. Способ согласно одному из вариантов исполнения 113-116, отличающийся тем, что X1=Mo.
118. Способ согласно одному из вариантов исполнения 113-117, отличающийся тем, что с составляет от 0,005 до 0,1.
119. Способ согласно одному из вариантов исполнения 113-117, отличающийся тем, что с составляет от 0,005 до 0,05 или 0,005 до 0,02.
120. Способ согласно варианту исполнения 112, отличающийся тем, что соотношение np:nv из содержащегося в каталитически активной массе молярного количества фосфора nр и содержащегося в каталитически активной массе молярного количества ванадия nv составляет от 0,09 до 2,0, предпочтительно от 0,9 до 1,5 и особенно предпочтительно от 0,9 до 1,2.
121. Способ согласно одному из вариантов исполнения 112 или 120, отличающийся тем, что содержащиеся в каталитически активной массе элементы, отличающиеся от ванадия и фосфора, представляют собой один или более одного элемента из группы, состоящей из лития, калия, натрия, рубидия, цезия, таллия, молибдена, цинка, гафния, циркония, титана, хрома, марганца, никеля, меди, железа, бора, кремния, олова, ниобия, кобальта и висмута.
122. Способ согласно варианту исполнения 121, отличающийся тем, что общее содержание в каталитически активной массе элементов, отличающихся от ванадия и фосфора, в пересчете на ее массу составляет не более 5% масс, причем этот соответствующий элемент, отличающийся от ванадия и фосфора, рассчитывается как электрически нейтральный оксид, в котором элемент имеет такое же число зарядов, как и в активной массе.
123. Способ согласно одному из вариантов исполнения 112-122, отличающийся тем, что средняя арифметическая степень окисления ванадия в каталитически активной массе составляет от +3,9 до +4,4 или от +4,0 до +4,3.
124. Способ согласно одному из вариантов исполнения 112-123, отличающийся тем, что удельная поверхность, определенная по методу БЭТ, каталитически активной массы составляет от ≥15 до 50 м2/г.
125. Способ согласно одному из вариантов исполнения 112-124, отличающийся тем, что общий объем пор каталитически активной массы составляет от 0,1 до 0,5 мл/г.
126. Способ согласно одному из вариантов исполнения 112-125, отличающийся тем, что общий объем пор каталитически активной массы составляет от 0,15 до 0,4 мл/г.
127. Способ согласно одному из вариантов исполнения 112-126, отличающийся тем, что по меньшей мере один катализатор окисления B представляет собой сплошной катализатор или катализатор на носителе.
128. Способ согласно варианту исполнения 127, отличающийся тем, что геометрия сплошного катализатора выбирается из группы, состоящей из шариков, колец и сплошных цилиндров, а также имеет наибольший размер в интервале от 1 до 10 мм.
129. Способ согласно варианту исполнения 127, отличающийся тем, что геометрия сплошного катализатора представляет собой кольцо (полый цилиндр) с внешним диметром в интервале от 3 до 10 мм, высотой от 1 до 10 мм, внутренним диаметром от 1 до 8 мм и толщиной стенки от 1 до 3 мм.
130. Способ согласно одному из вариантов исполнения 112-126, отличающийся тем, что по меньшей мере один катализатор альдольной конденсации B представляет собой слоистый катализатор, который содержит каталитически активную массу в качестве оболочки, нанесенной на поверхность инертного формованного изделия носителя.
131. Способ согласно варианту исполнения 130, отличающийся тем, что формованное изделие носителя представляет собой шарик или кольцо.
132. Способ согласно варианту исполнения 130 или 131, отличающийся тем, что наибольший размер формованного изделия носителя составляет от 1 до 10 мм.
133. Способ согласно одному из вариантов исполнения 130-132, отличающийся тем, что инертное формованное изделие носителя состоит из стеатита.
134. Способ согласно одному из вариантов исполнения 130-133, отличающийся тем, что толщина слоя из каталитически активной массы составляет от 10 до 2000 мкм, или от 10 до 500 мкм, или от 100 до 500 мкм, или от 200 до 300 мкм.
135. Способ согласно одному из вариантов исполнения 1-134, отличающийся тем, что он включает еще одну, третью реакционную зону C, которая заполнена по меньшей мере одним катализатором окисления C, и включает следующие дополнительные операции:
- через третью реакционную зону C, которая заполнена по меньшей мере одним катализатором окисления C, пропускается поток поступающей реакционной газовой смеси C, содержащей реагенты - метанол и молекулярный кислород, а также по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и при прохождении реакционной зоны C метанол, содержащийся в поступающей реакционной газовой смеси C, окисляется в условиях гетерогенного катализа до формальдегида и водяного пара, так что получается газообразная смесь продуктов C, содержащая формальдегид, водяной пар и по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и поток газообразной смеси продуктов C покидает реакционную зону C, причем к проходящей сквозь реакционную зону C реакционной газовой смеси C на ее пути через эту реакционную зону C на выбор может подаваться дополнительный молекулярный кислород и/или дополнительный инертный газ-разбавитель.
- на выбор, из газообразной смеси продуктов C в зоне разделения Т* от газообразной смеси продуктов C отделяется при необходимости еще содержащийся в газообразной смеси продуктов С, не вступивший в реакцию метанол, причем остается газообразная смесь продуктов C*, содержащая формальдегид, и
- газообразная смесь продуктов C или газообразная смесь продуктов С* подается в реакционную зону B для получения поступающей реакционной газовой смеси B.
136. Способ согласно варианту исполнения 135, отличающийся тем, что метанол, отделенный в в зоне разделения Т*, подается обратно в реакционную зону C для получения поступающей реакционной газовой смеси C.
137. Способ согласно вариантам исполнения 135 или 136, отличающийся тем, что отделение метанола в зоне разделения Т* осуществляется ректификацией.
138. Способ согласно одному из вариантов исполнения 135-137, отличающийся тем, что по меньшей мере один катализатор окисления C содержит каталитически активную массу, которая включает по меньшей мере элементарное серебро.
139. Способ согласно варианту исполнения 138, отличающийся тем, что чистота этого элементарного серебра составляет ≥99,7% масс.
140. Способ согласно варианту исполнения 138, отличающийся тем, что чистота этого элементарного серебра составляет ≥99,9 или ≥99,99% масс.
141. Способ согласно одному из вариантов исполнения 138-140, отличающийся тем, что по меньшей мере один катализатор окисления C включает кристаллы серебра, у которых наибольший размер находится в диапазоне от 0,1 до 5 мм.
142. Способ согласно варианту исполнения 141, отличающийся тем, что кристаллы серебра покрыты пористым слоем из оксидного материала по меньшей мере одного из элементов Al, Si, Zr и Ti, толщина которого лежит в диапазоне от 0,3 до 10 мкм.
143. Способ согласно одному из вариантов исполнения 138-142, отличающийся тем, что содержание метанола в поступающей реакционной газовой смеси C составляет по меньшей мере 5% объемн.
144. Способ согласно варианту исполнения 143, отличающийся тем, что содержание метанола в поступающей реакционной газовой смеси C составляет не более 60% объемн.
145. Способ согласно одному из вариантов исполнения 138-142, отличающийся тем, что содержание метанола в поступающей реакционной газовой смеси C составляет от 15 до 50% объемн.
146. Способ согласно одному из вариантов исполнения 138-142, отличающийся тем, что содержание метанола в поступающей реакционной газовой смеси C составляет от 20 до 40% объемн. или 20 до 30% объемн.
147. Способ согласно одному из вариантов исполнения 138-146, отличающийся тем, что поступающая реакционная газовая смесь C содержит молекулярный кислород в молярном количестве no и метанол в молярном количестве nMе, и соотношение nо:nМе составляет меньше 1.
148. Способ согласно варианту исполнения 147, отличающийся тем, что nо:nМе составляет от 0,1 до 0,8 или от 0,2 до 0,6.
149. Способ согласно одному из вариантов исполнения 138-148, отличающийся тем, что nо:nМе составляет от 0,3 до 0,5.
150. Способ согласно одному из вариантов исполнения 138-149, отличающийся тем, что поступающая реакционная газовая смесь C содержит от ≥0 до 50% объемн. H2O.
151. Способ согласно варианту исполнения 150, отличающийся тем, что поступающая реакционная газовая смесь С содержит от 15 до 35% объемн. или 20 до 30% объемн. H2O.
152. Способ согласно одному из вариантов исполнения 138-151, отличающийся тем, что поступающая реакционная газовая смесь C в качестве по меньшей мере одного инертного газа-разбавителя, отличающегося от водяного пара, содержит N2.
153. Способ согласно варианту исполнения 152, отличающийся тем, что поступающая реакционная газовая смесь C содержит от 20 до 80% объемн. N2.
154. Способ согласно вариантам исполнения 152 или 153, отличающийся тем, что поступающая реакционная газовая смесь C содержит от 30 до 70% объемн. N2.
155. Способ согласно одному из вариантов исполнения 152-154, отличающийся тем, что поступающая реакционная газовая смесь C содержит от 40 до 60% объемн. N2.
156. Способ согласно одному из вариантов исполнения 138-155, отличающийся тем, что метанол в реакционной зоне C при температуре реакции в интервале от 400 до 800°C окисляется до формальдегида и воды.
157. Способ согласно одному из вариантов исполнения 138-156, отличающийся тем, что метанол в реакционной зоне C при температуре реакции в интервале от 500 до 800°C окисляется до формальдегида и воды.
158. Способ согласно одному из вариантов исполнения 138-156, отличающийся тем, что метанол в реакционной зоне C при температуре реакции в интервале от 450 до 650°C или от 500 до 600°С окисляется до формальдегида и воды.
159. Способ согласно одному из вариантов исполнения 138-156, отличающийся тем, что метанол в реакционной зоне С при температуре реакции в интервале от 600 до 750°С окисляется до формальдегида и воды.
160. Способ согласно одному из вариантов исполнения 138-159, отличающийся тем, что метанол в реакционной зоне С при рабочем давлении в диапазоне от 103 до 106 Па или от 104 до 2⋅105 Па окисляется до формальдегида и воды.
161. Способ согласно одному из вариантов исполнения 135-137, отличающийся тем, что по меньшей мере один катализатор окисления С содержит каталитически активную массу, которая представляет собой смешанный оксид, который имеет по меньшей мере один переходный металл в окисленном состоянии.
162. Способ согласно варианту исполнения 161, отличающийся тем, что по меньшей мере один переходный металл включает в себя Mo и/или V.
163. Способ согласно варианту исполнения 161, отличающийся тем, что по меньшей мере один переходный металл включает в себя Mo и Fe.
164. Способ согласно варианту исполнения 161, отличающийся тем, что каталитически активная масса представляет собой смешанный оксид общей формулы III,
в которой переменные имеют следующие значения:
M1=Mo и/или Fe, или
Mo и/или Fe, а также в пересчете на общее молярное количество из Mo и Fe общее молярное количество вплоть до 10% мольн. (например, от 0,01 до 10% мольн., или от 0,1 до 10% мольн.), предпочтительно не более чем 5% мольн., одного или нескольких элементов из группы, состоящей из Ti, Sb, Sn, Ni, Cr, Се, AI, Ca, Mg, V, Nb, Ag, Mn, Cu, Co, Si, Na, K, Tl, Zr, W, Ir, Та, As, P и В,
q=от 0 до 5,
m=от 1 до 3,
n=от 1 до 6.
165. Способ согласно варианту исполнения 164, отличающийся тем, что q=от 0,5 до 3.
166. Способ согласно вариантам исполнения 164 или 165, отличающийся тем, что q=от 1 до 2.
167. Способ согласно одному из вариантов исполнения 164-166, отличающийся тем, что M1=Mo, m=1, и n=3.
168. Способ согласно одному из вариантов исполнения 164-166, отличающийся тем, что M1=Fe, m=2, и n=3.
169. Способ согласно одному из вариантов исполнения 164-168, отличающийся тем, что менее чем 50% мольн. железа, содержащегося в смешанном оксиде III, присутствует в степени окисления +2.
170. Способ согласно одному из вариантов исполнения 164-168, отличающийся тем, что менее чем 20% мольн. железа, содержащегося в смешанном оксиде III, присутствует в степени окисления +2.
171. Способ согласно одному из вариантов исполнения 164-168, отличающийся тем, что менее чем 10% мольн. железа, содержащегося в смешанном оксиде III, присутствует в степени окисления +2.
172. Способ согласно одному из вариантов исполнения 164-168, отличающийся тем, что все количество железа, содержащегося в смешанном оксиде III, присутствует в степени окисления +3.
173. Способ согласно одному из вариантов исполнения 164-172, отличающийся тем, что соотношение nMo:nFe, образованное из содержащегося с смешанном оксиде III молярного количества молибдена и содержащегося в том же смешанном оксиде III молярного количества железа, составляет от 1:1 до 5:1.
174. Способ согласно одному из вариантов исполнения 164-172, отличающийся тем, что каталитически активная масса формально может представляться как смесь из MoO3 и Fe2O3, причем содержание MoO3 в этой смеси составляет от 65 до 95% масс, а содержание Fe2O3 в этой смеси от 5 до 35% масс.
175. Способ согласно одному из вариантов исполнения 161-174, отличающийся тем, что по меньшей мере один катализатор окисления С представляет собой сплошной катализатор.
176. Способ согласно варианту исполнения 175, отличающийся тем, что геометрия сплошного катализатора выбирается из группы, состоящей из шариков, колец и сплошных цилиндров.
177. Способ согласно варианту исполнения 176, отличающийся тем, что наибольший размер сплошного катализатора составляет от 1 до 10 мм.
178. Способ согласно варианту исполнения 175, отличающийся тем, что сплошной катализатор имеет геометрию кольца с внешним диметром в интервале от 3 до 10 мм, высотой от 1 до 10 мм и внутренним диаметром от 1 до 8 мм.
179. Способ согласно варианту исполнения 178, отличающийся тем, что кольцо имеет толщину стенки от 1 до 3 мм.
180. Способ согласно одному из вариантов исполнения 161-174, отличающийся тем, что по меньшей мере один катализатор окисления С представляет собой слоистый катализатор, который содержит каталитически активный смешанный оксид в качестве оболочки, нанесенной на поверхность инертного формованного изделия носителя.
181. Способ согласно варианту исполнения 180, отличающийся тем, что формованное изделие носителя представляет собой шарик или кольцо.
182. Способ согласно варианту исполнения 181, отличающийся тем, что наибольший размер формованного изделия носителя составляет от 1 до 10 мм.
183. Способ согласно варианту исполнения 180, отличающийся тем, что инертное формованное изделие носителя представляет собой кольцо с длиной от 2 до 10 мм, внешним диаметром от 4 до 10 мм и толщиной стенки от 1 до 4 мм.
184. Способ согласно одному из вариантов исполнения 180-183 отличающийся тем, что инертное формованное изделие носителя состоит из стеатита.
185. Способ согласно одному из вариантов исполнения 180-184, отличающийся тем, что слой из каталитически активного смешанного оксида имеет толщину от 10 до 2000 мкм, или от 10 до 500 мкм, или от 100 до 500 мкм, или от 200 до 300 мкм.
186. Способ согласно одному из вариантов исполнения 161-185, отличающийся тем, что поступающая реакционная газовая смесь C содержит не более 15% объемн. метанола.
187. Способ согласно одному из вариантов исполнения 161-185, отличающийся тем, что поступающая реакционная газовая смесь C содержит не более 11% объемн. метанола.
188. Способ согласно одному из вариантов исполнения 161-187, отличающийся тем, что поступающая реакционная газовая смесь C содержит от 2 до 10% объемн. метанола.
189. Способ согласно одному из вариантов исполнения 161-188, отличающийся тем, что поступающая реакционная газовая смесь C содержит от 6 до 9% объемн. метанола.
190. Способ согласно одному из вариантов исполнения 161-189, отличающийся тем, что поступающая реакционная газовая смесь C содержит молекулярный кислород в молярном количестве no и метанол в молярном количестве nМе, и соотношение no:nМе составляет по меньшей мере 1 или больше 1.
191. Способ согласно варианту исполнения 190, отличающийся тем, что соотношение nо:nМе составляет от 1,1 до 5.
192. Способ согласно варианту исполнения 190 или 191, отличающийся тем, что соотношение nо:nMе составляет от 1,5 до 3,5.
193. Способ согласно одному из вариантов исполнения 161-192, отличающийся тем, что поступающая реакционная газовая смесь C в качестве по меньшей мере одного инертного газа-разбавителя, отличающегося от водяного пара, содержит N2.
194. Способ согласно варианту исполнения 193, отличающийся тем, что поступающая реакционная газовая смесь C содержит от 70 до 95% объемн. N2.
195. Способ согласно одному из вариантов исполнения 161-194, отличающийся тем, что поступающая реакционная газовая смесь C содержит от 0 до 20% объемн. Н2O.
196. Способ согласно варианту исполнения 195, отличающийся тем, что поступающая реакционная газовая смесь C содержит от 0,1 до 10% объемн. Н2O.
197. Способ согласно вариантам исполнения 195 или 196, отличающийся тем, что поступающая реакционная газовая смесь C содержит от 0,2 до 7% объемн. Н2O.
198. Способ согласно одному из вариантов исполнения 194-196, отличающийся тем, что поступающая реакционная газовая смесь C содержит от 0,5 до 5% объемн. H2O.
199. Способ согласно одному из вариантов исполнения 161-198, отличающийся тем, что метанол в реакционной зоне C при температуре реакции в диапазоне от 250 до 500°C окисляется до формальдегида и воды.
200. Способ согласно варианту исполнения 199, отличающийся тем, что метанол в реакционной зоне C при температуре реакции в диапазоне от 250 до 400°C окисляется до формальдегида и воды.
201. Способ согласно одному из вариантов исполнения 161-200, отличающийся тем, что метанол в реакционной зоне C при рабочем давлении в интервале от 103 до 106 Па или от 104 до 2⋅105 Па окисляется до формальдегида и воды.
202. Способ согласно одному из вариантов исполнения 1-201, отличающийся тем, что частичное количество потока вещества Y подается обратно в реакционную зону A для получения поступающей реакционной газовой смеси A.
203. Способ согласно одному из вариантов исполнения 135-202, отличающийся тем, что частичное количество потока вещества Y подается обратно в реакционную зону C для получения поступающей реакционной газовой смеси C.
204. Способ согласно одному из вариантов исполнения 1-203, отличающийся тем, что для разделения газообразной смеси продуктов B в зоне разделения T эта газообразная смесь продуктов B, на выбор - после прямого и/или непрямого охлаждения этой смеси, подается в конденсационную колонну, снабженную способствующими разделению встроенными элементами, и внутри этой конденсационной колонны конденсируется по фракциям, а потоки веществ X, Y и Z выводятся из этой конденсационной колонны как отделенные друг от друга фракции.
205. Способ согласно одному из вариантов исполнения 1-203, отличающийся тем, что для разделения газообразной смеси продуктов B в зоне разделения T эта газообразная смесь продуктов B, на выбор - после прямого и/или непрямого охлаждения этой смеси, подается в абсорбционную колонну, оснащенную встроенными элементами с эффектом разделения, в противотоке с органическим растворителем, при нормальном давлении кипящем выше, чем акриловая кислота, а уксусная кислота и акриловая кислота, содержащиеся в газообразной смеси продуктов B, абсорбируются в органический растворитель с получением продукта абсорбции, в то время как поток вещества Z выходит из абсорбционной колонны через ее верх, а затем из продукта абсорбции потоки вещества X и Y выделяются в качестве разделенных фракций с помощью фракционной перегонки этого продукта в ректификационной колонне.
206. Способ согласно одному из вариантов исполнения 1-203, отличающийся тем, что для разделения газообразной смеси продуктов B в зоне разделения T эта газообразная смесь продуктов B, на выбор - после прямого и/или непрямого охлаждения этой смеси, подается в абсорбционную колонну, оснащенную встроенными элементами с эффектом разделения, в противотоке с водным раствором в качестве абсорбирующего агента, и уксусная кислота и акриловая кислота, содержащиеся в газообразной смеси продуктов B, абсорбируются растворителем с получением продукта абсорбции, в то время как поток вещества Z выходит из абсорбционной колонны через ее верх, а затем из продукта абсорбции потоки вещества X и Y выделяются в качестве разделенных фракций с помощью фракционной перегонки этого продукта в ректификационной колонне.
207. Акриловая кислота, для которой соотношение V из молярного количества ядер атомов 14C, содержащихся в этой акриловой кислоте, n14C и молярного количества ядер атомов 12C, содержащихся в той же акриловой кислоте, n12C, V=n14C:n12C, больше 0 и меньше, чем соответствующее молярное соотношение V* ядер атомов 14C и ядер атомов 12C, присутствующее в диоксиде углерода в атмосфере Земли.
208. Акриловая кислота согласно варианту исполнения 207, отличающаяся тем, что V=(1/3)⋅V*.
209. Акриловая кислота согласно варианту исполнения 207, отличающаяся тем, что V=(2/3)⋅V*.
210. Жидкая фаза Р, содержащая по меньшей мере 1 кг акриловой кислоты, отличающаяся тем, что содержащаяся акриловая кислота представляет собой акриловую кислоту согласно одному из вариантов исполнения 207-209.
Примеры
I) Получение различных катализаторов
А) Получение катализатора из смешанного оксида для гетерогенно катализируемого частичного газофазного окисления метанола до формальдегида по ФОРМОКС-процессу
В имеющей температуру 60°C смеси из 800 мл воды и 250 г водного раствора аммиака с концентрацией 25% масс, при поддержании 60°C растворяли 530 г тетрагидрата гептамолибдата аммония ((NH4)6Mo7O24⋅4H2O). При этом получали раствор 1 с температурой 60°C.
В 1000 мл нагретой до 60°C воды растворяли 808 г нонагидрата нитрата железа-(III) (Fe(NO3)3⋅9H2O) при поддержании температуры 60°C. При этом получали раствор 2 с температурой 60°C.
В течение 20 минут раствор 2 при поддержании 60°C непрерывно вводили при перемешивании в раствор 1. Затем дополнительно перемешивали еще 5 минут при 60°C.После этого полученную водную суспензию подвергали распылительной сушке при температуре на входе 340°C и температуре на выходе 110°C в потоке воздуха в течение 1,5 часов (башня для распылительной сушки типа Mobile Minor 2000 (MM-I) фирмы Niro A/S, Gladsaxevej 305, 2860 Soborg, Дания, с центробежным распылителем типа F01A и разбрызгивающим колесом типа SL24-50). В процессе распылительной сушки соответственно еще не распыленную часть суспензии дополнительно перемешивали при поддержании 60°C.
Полученный таким образом порошок после распылительной сушки, в пересчете на его массу, гомогенно перемешивали с 1% масс, графита TIMREX® Т44 фирмы Timcal AG (сравните с международной заявкой WO 2008/087116) (в барабанно-кольцевом смесителе; диаметр колеса: 650 мм, объем барабана: 5 л, число оборотов: примерно 30 об/мин, продолжительность перемешивания: 30 минут). Потом полученная в результате смесь уплотнялась в роликовом прессе типа RCC 100×20 фирмы Powtec с 2 противоположно вращающимися стальными валиками при давлении прессования 12 бар, а затем продавливалась через сито с квадратными ячейками с размером ячеек 0,8 мм. Затем полученный в результате плотный продукт (он имел насыпную массу 1050 г/л и в основном однородный размер частиц составляющий ≥0,4 мм и ≤0,8 мм) в упомянутом выше барабанно-кольцевом смесителе при числе оборотов примерно 30 об/мин в течение 30 минут, перемешивали, в пересчете на его массу, с 3% масс, того же самого графита, а после этого, как описано в немецкой заявке DE-A 10 2008040093, спрессовывали с получением кольцевидных формованных изделий предшественника сплошного катализатора с геометрическими характеристиками 5 мм×3 мм×3 мм (внешний диаметр х высота х внутренний диаметр) с прочностью при боковом сдавливании 22±5 H и массой 130 мг (высота заполнения: 7,5-9 мм; прессующее усилие: 3,0-3,5 кН; роторный таблеточный пресс Kilian (таблетирующая установка на 9 гнезд) типа S100 (фирмы Kilian, D-50735 Кельн). Это таблетирование осуществлялось в атмосфере азота.
Для заключительной термической обработки формованные изделия предшественника сплошного катализатора равномерно распределяли на 4 расположенных рядом друг с другом сетчатых решетках с квадратной поверхностью основания соответственно 150 мм×150 мм (высота засыпки: примерно 40 мм) и подвергали обработке в конвекционной сушильной печи с подаваемым потоком воздуха (фирмы Heraeus Instruments GmbH, D-63450 Hanau, модель: К 750/2) следующим образом. Поток воздуха составлял 100 нл/ч и первоначально имел температуру 120°C. Сначала в течение 10 часов поддерживали эти 120°C. Потом в течение 10 часов температуру в основном линейно повышали до 460°C и затем поддерживали эти 460°C в течение дополнительных 4 часов. После этого в течение 5 часов охлаждали до 25°C.
Таким образом получали кольцевидный сплошной катализатор окисления C, у которого активная масса смешанных оксидов имела стехиометрию Fe2(MoO4)3. После этого кольца сплошного катализатора продавливали через сито с квадратными ячейками с размером ячеек 0,8 мм. Прошедший через сито продукт (катализатор окисления C в форме крошки) имел в основном однородный размер частиц (наибольший размер), составляющий ≥0,4 мм и ≤0,8 мм.
В) Получение катализатора из смешанных оксидов для заполнения участка 1 реакционной зоны A для гетерогенно катализируемого частичного газофазного окисления этанола до уксусной кислоты.
380 г воды загружали в химический стакан объемом 2 л и нагревали до 55°C. Потом добавляли 220 г дигидрата щавелевой кислоты и при поддержании 55°C растворяли при перемешивании. После полного растворения дигидрата щавелевой кислоты добавляли 116 г тонко измельченного V2O5, причем образовывался темно-синий ванадиевый комплекс. После завершенного добавления V2O5 смесь при перемешивании в течение 20 минут в основном линейно нагревалась до 80°C. Эту температуру при перемешивании поддерживали в течение дополнительных 10 минут. После этого образовавшийся водный раствор в течение 60 минут в основном линейно охлаждали до 25°C.
К 135 мл водного раствора, полученного, как описано, добавляли 97,5 г тонко измельченного диоксида титана (изготовитель: фирма Fuji; тип: ТА 100С; анатазная модификация; удельная поверхность (по методу БЭТ): 20 м2/г; 2200 частей на млн серы; 1600 частей на млн ниобия; распределение размеров частиц (1 г TiO2 перемешивали в 100 мл воды 10 минут с помощью магнитной мешалки, затем определяли распределение размеров частиц с помощью динамического светорассеяния в приборе Mastersizer S фирмы Malvern Instruments с маленьким модулем влажного диспергирования MS1): d10=0,6 мкм, d50=1,1 мкм, d90=2,1 мкм; dx при этом подразумевает, что X % от общего объема частиц состоит из частиц с данным или меньшим размером). После этого смесь в течение 3 минут гомогенизировали с помощью смесителя Ultra-Turax фирмы Janke & Kunkel GmbH и Co. KG - IKA Labortechnik (тип вала: S 50KR-G45 F fein, диаметр трубки вала: 25 мм, диаметр статора: 45 мм, диаметр ротора: 40 мм) при 8000 об/мин. Полученная в результате водная дисперсия применялась для нанесения покрытия на 150 г крошки из стеатита С220 фирмы CeramTec с наибольшим размером в диапазоне от 1 мм до 1,5 мм.
Для этой цели 150 г стеатитовой крошки вносили во вращающийся дражирующий барабан (с внутренним радиусом 15 см и внутренним объемом 1800 см3) в качестве установки для нанесения покрытий. Число оборотов этого дражирующего барабана составило 200 об/мин. Нанесение водной дисперсии на поверхность стеатитовой крошки осуществлялось путем распыления этой водной дисперсии при помощи двойного распылительного сопла.
В качестве распыляющей среды использовали 250 нл/ч сжатого воздуха (10 бар (изб.)), который имел температуру 25°C. Поток дисперсии составил 100 мл/ч при температуре водной дисперсии 25°C. Форма распыления представляла собой круглый сплошной конус (10° - 40°). Тип распыления представлял собой туман с размером капель от <50 до 150 мкм. Размер поперечного сечения потока для дисперсии составил 1 мм. Внутреннюю температуру в барабане для нанесения покрытий при процессе нанесения покрытия поддерживали при 120°C. После оконченного процесса распыления в остальном при неизменных условиях еще 30 минут сушили в этом дражирующем барабане. После этого частицы носителя с нанесенным покрытием помещали в фарфоровую чашку и при нахождении в этой чашке прокаливали в муфельной печи (внутренний объем 1,5 л) на воздухе (без движения). Для этого температуру в прокаливаемом продукте со скоростью нагрева 3°C/мин повышали от 100°C до 500°C, а затем выдерживали 3 часа при этой температуре. В заключение прокаливаемый продукт в течение 12 часов, находясь в муфельной печи, в основном линейно охлаждался до 25°C.
Таким образом, в качестве катализатора окисления A(1) получали слоистый катализатор, каталитически активная масса смешанных оксидов которого в среднем имела стехиометрию (V2O5)x (TiO2)y, где х=18,3% масс, и y=81,7% масс.
С) Получение катализатора из смешанных оксидов для заполнения участка 2 реакционной зоны A для гетерогенно катализируемого частичного газофазного окисления этанола до уксусной кислоты
В 2700 г воды, которая имела температуру 25°C, при поддержании этой температуры растворяли 127 г моногидрата ацетата меди(II) с получением раствора 1.
В 5500 г воды, которая имела температуру 95°C, при поддержании этой температуры в течение 5 минут растворяли 860 г тетрагидрата гептамолибдата аммония. Затем при поддержании 95°C добавляли 143 г метаванадата аммония и полученный в результате раствор при 95°C перемешивали еще 30 минут. После этого добавляли 126 г гептагидрата паравольфрамата аммония и полученный в результате раствор при 95°C перемешивали еще 40 минут. Затем полученный таким образом раствор 2 в течение 10 минут охлаждали до 80°C.
Имеющий температуру 25°C раствор 1 потом в течение 5 минут при поддержании 80°C вводили при перемешивании в раствор 2, имеющий температуру 80°C. После этого полученный в результате раствор подвергали распылительной сушке при температуре на входе 350°C и температуре на выходе 110°C в течение 3 часов в потоке воздуха (башня для распылительной сушки типа Mobile Minor 2000 (ММ-1) фирмы Niro A/S, Gladsaxevej 305, 2860 Soborg, Дания, с центробежным распылителем типа F01A и разбрызгивающим колесом типа SL24-50). В процессе распылительной сушки соответственно еще не распыленную часть суспензии дополнительно перемешивали при поддержании 80°C. Соответственно 1 кг полученного после распылительной сушки порошка смешивали с 0,15 кг воды, которая имела температуру 25°C, (смеситель фирмы Werner & Pfleiderer, модель ZS1-80; продолжительность смешивания: примерно 2 часа; температура смешивания: от 30 до 35°C). Затем продукт смешивания при толщине слоя примерно 2 см прокаливали в сушильном шкафу с циркуляцией воздуха, заполненном смесью кислород/азот. При этом содержание молекулярного кислорода в этой смеси кислород/азот устанавливали так, что на выходе их этого сушильного шкафа с циркуляцией воздуха содержание 02 составляло 1,5% объемн. В процессе прокаливания смешанная масса сначала со скоростью нагрева 10°C/мин нагревалась от 30°C до 300°C (соответствующей температуры прокаливаемого продукта) и затем в течение 6 часов выдерживалась при этой температуре. После этого продукт прокаливания со скоростью нагрева 10°C/мин нагревался до 400°C, и затем эту температуру поддерживали в течение 1 часа. После этого температуру продукта прокаливания в основном линейно снижают в течение 12 часов до 25°C. Нагрузка печи для прокаливания составляла 250 г/л (г продукта смешивания на л внутреннего объема сушильного шкафа с циркуляцией воздуха). Объемный поток поступающей смеси кислород/азот составлял 80 нл/ч, а время пребывания заполнения смесью кислород/азот (рассчитанное как соотношение из внутреннего объема сушильного шкафа с циркуляцией воздуха и объемного потока подаваемой при 25°C смеси кислород/азот) составляло 135 секунд. Внутренний объем сушильного шкафа с циркуляцией воздуха составлял 3 л.
Полученный в результате каталитически активный смешанный оксид имел стехиометрию Mo12V3W1,2Cu1,6On.
Каталитически активная оксидная масса затем размалывалась в мельнице ZM 200 фирмы Retsch до тонко измельченного порошка, размер частиц которого (наибольший размер) в основном составлял от 0,1 до 50 мкм (благоприятное для изобретения распределение размеров частиц демонстрирует Фиг.2 немецкой заявки DE-A 102010023312). Этим порошком активной массы во вращающемся барабане по аналогии со способом нанесения покрытия, описанным в европейской заявке EP-A 714 700, с добавлением воды в качестве связующего средства были покрыты шероховатые на поверхности шарики из стеатита С220 фирмы CeramTec с диаметром шариков от 2 до 3 мм. В заключение шарики с нанесенным покрытием сушили с помощью нагретого до 110°C воздуха. Доля активной массы полученного таким образом слоистого катализатора A(2) однородно устанавливалась как 20% масс. Остаточное содержание воды в активной массе после сушки составляло 0,2% масс.
D) Получение катализатора альдольной конденсации B, активная масса которого представляет собой оксид ванадия-фосфора, дотированный железом(III)
В заполненный молекулярным азотом (содержание N2 ≥99,99% объемн.) и обогреваемый снаружи водой под давлением сосуд с перемешиванием, имеющий внутренний объем 8 м3, эмалированный на внутренней поверхности и оснащенный отбойными перегородками, а также турбинной мешалкой, помещали 4602 кг изобутанола. После запуска трехступенчатой турбинной мешалки изобутанол при кипячении с обратным холодильником нагревался до 90°C. Теперь при поддержании этой температуры начинали непрерывно подавать через транспортирующий шнек 690 кг тонко измельченного пентаоксида ванадия. После того как в течение 20 минут добавили 460 кг пентаоксида ванадия (V2O5), при продолжении подачи пентаоксида ванадия начинали непрерывную подачу насосом 805 кг фосфорной кислоты с концентрацией 105% масс, (сравните с немецкой заявкой DE-A 3520053), причем температуру поддерживали при 90°C (скорость нагнетания=160 кг/ч).
После оконченного добавления фосфорной кислоты смесь при перемешивании и с обратным холодильником нагревали до температуры в интервале от 100 до 108°C и в течение 14 часов выдерживали в этом температурном интервале. Затем полученную горячую суспензию в течение 75 минут в основном линейно охлаждали до 60°C и добавляли 22,7 кг гидрата фосфата железа(III) (содержание Fe: 29,9% масс; производитель: фирма Dr. Paul Lohmann; не содержит примесей Fe(II)). Затем снова с обратным холодильником нагревали до температуры от 100 до 108°C и эту суспензию при дополнительном перемешивании выдерживали в течение 1 часа при этой температуре. Затем эту нагретую примерно до 105°C суспензию сливали в предварительно заполненный инертной атмосферой из азота, а также нагретый до 100°C нутч-фильтр и отфильтровывали под давлением над нутч-фильтром примерно 0,35 МПа по абсолютной величине. Образовавшийся остаток на фильтре продували в сухом состоянии при помощи постоянного пропускания азота при перемешивании и при 100°C в течение 1 часа. После сухой продувки нагревали до 155°C и создавали вакуум до абсолютного давления 15 кПа (150 мбар абс).
При этих условиях проводили сушку до остаточного содержания изобутанола в высушенной массе предшественника 2% масс. Эта масса содержала Fe и V в молярном соотношении Fe/V, составляющем 0,016.
Затем сухой порошок дополнительно обрабатывали в наклонной вращающейся трубе из благородной стали, через которую пропускали 100 м3/ч воздуха (его температура на входе составляла 150°C), и которая имела находящуюся внутри спиралевидную нарезку. Длина трубы составляла 6,5 м, внутренний диаметр 0,9 м, а толщина стенки вращающейся трубы была 2,5 см. Число оборотов этой вращающейся трубы составляло 0,4 об/мин. Сухой порошок подавался во внутреннюю часть вращающейся трубы в количестве 60 кг/ч у ее верхнего конца. Спиралевидная нарезка обеспечивала равномерное движение (вниз) осадка сухого порошка внутри этой вращающейся трубы. Длина вращающейся трубы была поделена на пять зон обогрева одинаковой длины, температура в которых поддерживалась снаружи. Температуры, измеренные на внешней стороне вращающейся трубы, сверху вниз составляли 250°C, 300°C, 345°C, 345°C и 345°C.
400 г порошка, выходящего из вращающейся трубы, гомогенно перемешивали в пересчете на его массу с 1% масс, графита (Asbury 3160, фирмы Timcal Ltd., сравните с международной заявкой WO 2008/087116) (в барабанно-кольцевом смесителе с диаметром колеса 650 мм, объемом барабана 5 л, числом оборотов 30 об/мин, продолжительность перемешивания: 30 минут). Потом полученная в результате смесь уплотнялась с помощью роликового пресса фирмы Powtec с 2 противоположно вращающимися стальными валиками при давлении прессования 9 бар, а затем продавливалась через сито с квадратными ячейками с размером 1 мм. Полученный в результате плотный продукт имел насыпную массу 1100 г/л и в основном однородный размер частиц, составляющий ≥0,7 мм и ≤0,8 мм. 30 мл насыпного объема этого гранулята помещали в вертикальную трубчатую печь (внутренний диаметр трубки: 26 мм; в центре этой трубки проходящая сверху вниз термогильза с внешним диаметром 4 мм для помещения термоэлемента). Через трубчатую печь пропускали 25 нл/ч воздуха с температурой на входе 160°C. Со скоростью нагрева 5°C/мин температура находящегося в трубчатой печи продукта прокаливания повышалась от 25°C до 250°C. После достижения температуры 250°C температуру продукта прокаливания поднимали со скоростью нагрева 2°C/мин до 330°C. Эту температуру поддерживали в течение промежутка времени 40 минут.
Потом при поддержании объемного потока 25 нл/ч переключали с прохождения воздуха на прохождение через трубчатую печь смеси из 50% объемн. N2 и 50% объемн. водяного пара (температура которой на входе составляла 160°C) и температуру продукта прокаливания со скоростью нагрева 3°C/мин поднимали до 425°C. Эту температуру поддерживали в течение промежутка времени 180 минут. Потом при поддержании объемного потока 25 нл/ч снова переключали на прохождение потока воздуха (температура потока воздуха на входе была 25°C). Потом температуру продукта прокаливания в течение 120 минут понижали до 25°C.
Стехиометрия полученного, как описано, дотированного Fe(III) сплошного катализатора альдольной конденсации В на основе оксида ванадия-фосфора была V1P1,05Fe0,016On.
II) Проведение способа согласно изобретению от А) до D) для получения акриловой кислоты из этанола и формальдегида с применением катализаторов, полученных в разделе I) (содержание компонентов во всех поступающих реакционных газовых смесях и исходных веществах определялись с помощью газовой хроматографии (ГХ))
А) 1. Оформление реакционной зоны A
Исполнение реакционной зоны A осуществлялось в трубчатом реакторе A (внутренний диаметр: 12 мм; толщина стенки: 1,5 мм; длина: 2000 мм; материал: благородная сталь, конструкционный материал 1.4541 по стандарту DIN), который с внешней стороны мог электрически обогреваться по зонам. Заполнение катализатором в трубчатом реакторе A было оформлено следующим образом:
Участки 1 и 2 непосредственно граничат друг с другом.
Температура трубчатого реактора A устанавливалась в области участка 1 на 185°C и в области участка 2 на 220°C (в каждом случае температуры с внешней стороны стенки трубчатого реактора A). 41,8 нл/ч поступающей реакционной газовой смеси A подавали в предварительную засыпку из стеатитовой крошки с температурой на входе 150°C. Абсолютное давление на входе в трубчатый реактор A составляло 2,0 бар.
Поступающая реакционная газовая смесь A имела следующее содержание компонентов:
Последние 1326 мм трубчатого реактора A не обогревались. Нагрузка заполнения катализатором по поступающей реакционной газовой смеси A составляла примерно 5000 ч-1. Нагрузка по этанолу составляла 80 ч-1 (нл/лч).
Газообразная смесь продуктов A, выходящая из трубчатого реактора A (41,9 нл/ч), имела следующее содержание компонентов (анализ с помощью ГХ в реальном времени):
2. Оформление реакционной зоны B
Исполнение реакционной зоны B осуществлялось в трубчатом реакторе B (внутренний диаметр: 15 мм; толщина стенки: 1,2 мм; длина: 2000 мм; материал: благородная сталь, конструкционный материал 1.4541 по стандарту DIN), который с внешней стороны мог электрически обогреваться. Заполнение катализатором в трубчатом реакторе B было оформлено следующим образом:
Из 41,9 нл/ч газообразной смеси продуктов A, 2,0 нл/ч водного раствора формальдегида в воде, переведенного в паровую фазу, и 1,3 нл/ч уксусной кислоты, переведенной в паровую фазу (которая изображала поток вещества Y), получали 45,1 нл/ч поступающей реакционной газовой смеси B.
Содержание компонентов в испаренном растворе формальдегида было следующим:
Содержание компонентов в испаренной уксусной кислоте было следующим:
99% объемн. уксусной кислоты.
Содержание компонентов в поступающей реакционной газовой смеси B было следующим:
Поступающая реакционная газовая смесь B подавалась в предварительную засыпку из стеатитовой крошки с температурой на входе 350°C. Абсолютное давление на входе в трубчатый реактор B составляло 1,5 бар. Температура трубчатого реактора B по длине его заполнения неподвижным слоем была установлена на 375°C (температура на внешней стенке трубчатого реактора B). Оставшаяся длина трубчатого реактора B не обогревалась. Нагрузка заполнения катализатором по поступающей реакционной газовой смеси B составляла 340 ч-1.
Газообразная смесь продуктов B, выходящая из трубчатого реактора B (45,1 нл/ч), имела следующее содержание компонентов (анализ с помощью ГХ в реальном времени):
В пересчете на поданное в реакционную зону A молярное количество этанола достигнутый выход акриловой кислоты составил 87,0% мольн.
В) Реакционная зона A и реакционная зона B были оформлены, как в разделе II)A), и заполнены тем же самым слоем катализатора. Однако длина засыпки каталитически активной части на участке 1 составила 62 мм (гомогенная смесь из 3,5 мл катализатора окисления A(1) и 3,5 мл стеатитовой крошки). Проведение гетерогенно катализируемого частичного окисления этанола до уксусной кислоты проводилось как в разделе II)A), но подводимый поток поступающей реакционной газовой смеси A составлял 43,3 нл/ч. Было получено 43,4 нл/ч газообразной смеси продуктов A. Состав газообразной смеси продуктов A соответствовал этой смеси в разделе II)А). Нагрузка заполнения катализатором в трубчатом реакторе A по поступающей реакционной газовой смеси А составляла примерно 5000 ч-1.
Из 43,4 нл/ч газообразной смеси продуктов A, потока 0,7 нл/ч формальдегида, полученного в результате испарения и расщепления триоксана, и 1,0 нл/ч уксусной кислоты, переведенной в паровую фазу (которая изображала поток вещества Y), получали 45,1 нл/ч поступающей реакционной газовой смеси B.
Содержание компонентов в потоке формальдегида было следующим:
100% объемн. формальдегида.
Содержание компонентов в испаренной уксусной кислоте было следующим:
99% объемн. уксусной кислоты.
Содержание компонентов в поступающей реакционной газовой смеси B было следующим:
Поступающая реакционная газовая смесь В подавалась в предварительную засыпку из стеатитовой крошки с температурой на входе 320°C. Абсолютное давление на входе в трубчатый реактор В составляло 1,5 бар. Температура трубчатого реактора B по длине его заполнения неподвижным слоем была установлена на 340°C (температура на внешней стенке трубчатого реактора B). Остальная длина трубчатого реактора B не обогревалась. Нагрузка заполнения катализатором по поступающей реакционной газовой смеси B составляла 340 ч-1 (нл/лч).
Газообразная смесь продуктов B, выходящая из трубчатого реактора B (45,1 нл/ч), имела следующее содержание компонентов (анализ с помощью ГХ в реальном времени):
В пересчете на поданное в реакционную зону A количество этанола достигнутый выход акриловой кислоты составил 88% мольн.
C) Реакционная зона A и реакционная зона B были оформлены, как в разделе II)А), и заполнены тем же самым слоем катализатора. Гетерогенно катализируемое частичное окисление этанола до уксусной кислоты проводилось как в разделе II)А), но поступающая реакционная газовая смесь A имела следующее содержание компонентов:
Газообразная смесь продуктов A, выходящая из трубчатого реактора A (41,9 нл/ч), имела следующее содержание компонентов (анализ с помощью ГХ в реальном времени):
Из 41,9 нл/ч газообразной смеси продуктов A, потока 1,2 нл/лч формальдегида, полученного в результате испарения и расщепления триоксана, и 2,0 нл/ч уксусной кислоты, переведенной в паровую фазу (которая изображала поток вещества Y), получали 45,1 нл/ч поступающей реакционной газовой смеси B.
Содержание компонентов в потоке формальдегида было следующим: 100% объемн. формальдегида.
Содержание компонентов в испаренной уксусной кислоте было следующим:
99% объемн. уксусной кислоты.
Содержание компонентов в поступающей реакционной газовой смеси B было следующим:
Поступающая реакционная газовая смесь B подавалась в предварительную засыпку из стеатитовой крошки с температурой на входе 320°C. Абсолютное давление на входе в трубчатый реактор В составляло 1,5 бар. Температура трубчатого реактора B по длине его заполнения неподвижным слоем была установлена на 340°C (температура на внешней стенке трубчатого реактора B). Остальная длина трубчатого реактора B не обогревалась. Нагрузка заполнения катализатором по поступающей реакционной газовой смеси B составляла 340 ч-1.
Газообразная смесь продуктов B, выходящая из трубчатого реактора В (45,1 нл/ч), имела следующее содержание компонентов (анализ с помощью ГХ в реальном времени):
В пересчете на поданное в реакционную зону A количество этанола достигнутый выход акриловой кислоты составил 88% мольн.
D) Реакционная зона A и реакционная зона B были оформлены как в разделе II)A) и заполнены тем же самым слоем катализатора. Однако длина засыпки каталитически активной части на участке 1 составила 43 мм (гомогенная смесь из 2,4 мл катализатора окисления A(1) и 2,4 мл стеатитовой крошки), а длина засыпки каталитически активной части на участке 2 составила только 11 мм (1,2 мл слоистого катализатора А(2)). Гетерогенно катализируемое частичное окисление этанола до уксусной кислоты проводилось как в разделе II)A), но поступающая реакционная газовая смесь A имела следующее содержание компонентов:
Кроме того, подаваемый поток поступающей реакционной газовой смеси A составлял только 30,6 нл/ч. Газообразная смесь продуктов А, выходящая из трубчатого реактора A (30,6 нл/ч), имела следующее содержание компонентов (анализ с помощью ГХ в реальном времени):
Из 30,6 нл/ч газообразной смеси продуктов А, 1,8 нл/ч уксусной кислоты, переведенной в паровую фазу (которая изображала поток вещества Y), и 12,7 нл/ч содержащей формальдегид газообразной смеси продуктов С, которая получалась в результате гетерогенно катализируемого частичного газофазного окисления метанола в ФОРМОКС-процессе в реакционной зоне С, получали 45,1 нл/ч поступающей реакционной газовой смеси В.
Содержание компонентов в испаренной уксусной кислоте было следующим:
99% объемн. уксусной кислоты.
Содержание компонентов в газообразной смеси продуктов C было следующим:
Содержание компонентов в поступающей реакционной газовой смеси B было следующим:
Поступающая реакционная газовая смесь B подавалась в предварительную засыпку из стеатитовой крошки с температурой на входе 320°C. Абсолютное давление на входе в трубчатый реактор В составляло 1,5 бар. Температура трубчатого реактора B по длине его заполнения неподвижным слоем была установлена на 340°C (температура на внешней стенке трубчатого реактора B). Остальная длина трубчатого реактора B не обогревалась. Нагрузка заполнения катализатором по поступающей реакционной газовой смеси B составляла 340 ч-1.
Газообразная смесь продуктов B, выходящая из трубчатого реактора И (45,1 нл/ч), имела следующее содержание компонентов:
В пересчете на поданное в реакционную зону A количество этанола достигнутый выход акриловой кислоты составил 88% мольн.
Исполнение реакционной зоны C осуществлялось в трубчатом реакторе C (внутренний диаметр: 8 мм; толщина стенки: 1 мм; длина: 100 мм; материал: благородная сталь, конструкционный материал 1.4541 по стандарту DIN), который с внешней стороны мог электрически обогреваться. Заполнение катализатором в трубчатом реакторе В было оформлено следующим образом: 50 мм предварительная засыпка из стеатитовой крошки (как в реакционной зоне A) на входе в реактор; и 37 мм 1,87 мл катализатора окисления C в виде крошки.
Содержание компонентов в поступающей реакционной газовой смеси C было следующим:
Поступающая реакционная газовая смесь C (12,2 нл/ч) подавалась в предварительную засыпку из стеатитовой крошки с температурой на входе 265°C. Абсолютное давление на входе в трубчатый реактор C составляло 2 бар. Температура трубчатого реактора C по длине его заполнения неподвижным слоем была установлена на 265°C (температура на внешней стенке трубчатого реактора C). Остальная длина трубчатого реактора C не обогревалась. Нагрузка заполнения катализатором по поступающей реакционной газовой смеси С составляла 6500 ч-1.
Предварительное описание заявки на патент США US Provisional Patent Application №61/383358, поданной 16 сентября 2010 г, включено в настоящую заявку путем литературной ссылки. Что касается вышеприведенного технического описания, то возможны многочисленные изменения и отклонения от настоящего изобретения. Поэтому можно исходить из того, что изобретение, в рамках приложенной Формулы изобретения может исполняться не так, как конкретно описано в данном документе.
Изобретение относится к способу получения акриловой кислоты из этанола и формальдегида, который включает следующие операции: через первую реакционную зону А пропускают поток поступающей реакционной газовой смеси А, содержащей реагенты - этанол и молекулярный кислород, а также инертный разбавляющий газ, отличающийся от водяного пара, и при прохождении этой реакционной зоны А этанол в условиях гетерогенного катализа окисляют до уксусной кислоты и водяного пара, так что образуется газообразная смесь продуктов А, содержащая уксусную кислоту, водяной пар, молекулярный кислород, а также инертный разбавляющий газ, отличающийся от водяного пара, и поток газообразной смеси продуктов А покидает реакционную зону А, причем к реакционной газовой смеси А на ее пути через реакционную зону А на выбор может подаваться дополнительный молекулярный кислород и/или дополнительный инертный газ-разбавитель, из потока газообразной смеси продуктов А, покидающего реакционную зону А, и по меньшей мере одного другого потока веществ, который содержит по меньшей мере один источник формальдегида, получают поток поступающей реакционной газовой смеси В, содержащей уксусную кислоту, водяной пар, молекулярный кислород, инертный газ-разбавитель, отличающийся от водяного пара, и формальдегид, в котором содержащееся молярное количество уксусной кислоты nНАс больше, чем содержащееся в нем молярное количество формальдегида nFd, через вторую реакционную зону В, в которую загружен катализатор альдольной конденсации В, пропускают поток поступающей реакционной газовой смеси В, и при прохождении этой реакционной зоны В формальдегид, содержащийся в поступающей реакционной газовой смеси В, вместе с уксусной кислотой, содержащейся в поступающей реакционной газовой смеси В, в условиях гетерогенного катализа конденсируют до акриловой кислоты и воды, так что образуется газообразная смесь продуктов В, содержащая акриловую кислоту, уксусную кислоту, водяной пар, молекулярный кислород, инертный газ-разбавитель, отличающийся от водяного пара, и поток газообразной смеси продуктов В покидает реакционную зону В, причем к реакционной газовой смеси В на ее пути через эту реакционную зону В на выбор может подаваться дополнительный молекулярный кислород и/или дополнительный инертный газ-разбавитель, поток газообразной смеси продуктов В, покидающий реакционную зону В, подают в зону разделения Т, и в этой зоне разделения Т разделяют по меньшей мере на три потока веществ - X, Y и Z, причем поток акриловой кислоты, содержащийся в потоке вещества X, больше, чем поток акриловой кислоты, содержащийся в потоках веществ Y и Z, вместе взятых, поток уксусной кислоты, содержащийся в потоке вещества Y, больше, чем поток уксусной кислоты, содержащийся в потоках веществ X и Z, вместе взятых, поток инертного газа-разбавителя, отличающегося от водяного пара, содержащийся в потоке вещества Z, больше, чем поток инертного газа-разбавителя, отличающегося от водяного пара, содержащийся в потоках веществ X и Y, вместе взятых, и поток вещества Y подают обратно в реакционную зону В и используют дополнительно для получения поступающей реакционной газовой смеси В. Способ позволяет получать целевой продукт с высокой селективностью. 20 з.п. ф-лы.
1. Способ получения акриловой кислоты из этанола и формальдегида, который включает следующие операции:
- через первую реакционную зону А, в которую загружен по меньшей мере один катализатор окисления А, пропускают поток поступающей реакционной газовой смеси А, содержащей реагенты - этанол и молекулярный кислород, а также по меньшей мере один инертный разбавляющий газ, отличающийся от водяного пара, и при прохождении этой реакционной зоны А этанол, содержащийся в поступающей реакционной газовой смеси А, в условиях гетерогенного катализа окисляют до уксусной кислоты и водяного пара, так что образуется газообразная смесь продуктов А, содержащая уксусную кислоту, водяной пар, молекулярный кислород, а также по меньшей мере один инертный разбавляющий газ, отличающийся от водяного пара, и поток газообразной смеси продуктов А покидает реакционную зону А, причем к проходящей сквозь реакционную зону А реакционной газовой смеси А на ее пути через эту реакционную зону А на выбор может подаваться дополнительный молекулярный кислород и/или дополнительный инертный газ-разбавитель,
- из потока газообразной смеси продуктов А, покидающего реакционную зону А, и по меньшей мере одного другого потока веществ, который содержит по меньшей мере один источник формальдегида, получают поток поступающей реакционной газовой смеси В, содержащей уксусную кислоту, водяной пар, молекулярный кислород, по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и формальдегид, в котором содержащееся молярное количество уксусной кислоты nНАс больше, чем содержащееся в нем молярное количество формальдегида nFd,
- через вторую реакционную зону В, в которую загружен по меньшей мере один катализатор альдольной конденсации В, пропускают поток поступающей реакционной газовой смеси В, и при прохождении этой реакционной зоны В формальдегид, содержащийся в поступающей реакционной газовой смеси В, вместе с уксусной кислотой, содержащейся в поступающей реакционной газовой смеси В, в условиях гетерогенного катализа конденсируют до акриловой кислоты и воды, так что образуется газообразная смесь продуктов В, содержащая акриловую кислоту, уксусную кислоту, водяной пар, молекулярный кислород, а также по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и поток газообразной смеси продуктов В покидает реакционную зону В, причем к проходящей сквозь реакционную зону В реакционной газовой смеси В на ее пути через эту реакционную зону В на выбор может подаваться дополнительный молекулярный кислород и/или дополнительный инертный газ-разбавитель,
- поток газообразной смеси продуктов В, покидающий реакционную зону В, подают в зону разделения Т, и в этой зоне разделения Т разделяют по меньшей мере на три потока веществ - X, Y и Z, причем
- поток акриловой кислоты, содержащийся в потоке вещества X, больше, чем поток акриловой кислоты, содержащийся в потоках веществ Y и Z, вместе взятых,
- поток уксусной кислоты, содержащийся в потоке вещества Y, больше, чем поток уксусной кислоты, содержащийся в потоках веществ X и Z, вместе взятых,
- поток инертного газа-разбавителя, отличающегося от водяного пара, содержащийся в потоке вещества Z, больше, чем поток инертного газа-разбавителя, отличающегося от водяного пара, содержащийся в потоках веществ X и Y, вместе взятых,
и
- поток вещества Y подают обратно в реакционную зону В и используют дополнительно для получения поступающей реакционной газовой смеси В.
2. Способ по п. 1, отличающийся тем, что по меньшей мере один катализатор окисления А содержит каталитически активную массу, которая содержит от 0,1 до 60% масс.V2O5 и от 40 до 99,9 мас.% TiO2.
3. Способ по п. 1, отличающийся тем, что заполнение реакционной зоны А по меньшей мере одним катализатором окисления А включает в себя в направлении движения потока поступающей реакционной газовой смеси А два следующих друг за другом в пространстве в указанной числовой последовательности участка 1 и 2, которые заполнены отличающимися друг от друга катализаторами окисления А, причем
- по меньшей мере один катализатор окисления А участка 1 содержит каталитически активную массу 1, которая содержит по меньшей мере один оксид ванадия, а также по меньшей мере один оксид из группы оксидов титана, алюминия, циркония и олова, и
- по меньшей мере один катализатор окисления А участка 2 содержит каталитически активную массу 2, которая представляет собой полиметаллический оксид, который помимо ванадия (V) и молибдена (Мо) дополнительно содержит по меньшей мере один из элементов -вольфрам (W), молибден (Мо), тантал (Та), хром (Cr) и церий (Се), а также по меньшей мере один из элементов - медь (Cu), никель (Ni), кобальт (Со), железо (Fe), марганец (Mn) и цинк (Zn).
4. Способ по п. 3, отличающийся тем, что каталитически активная масса 2 представляет собой по меньшей мере один полиметаллический оксид общей формулы I
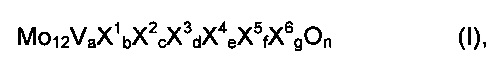
в которой переменные имеют следующее значение:
X1=W, Nb, Та, Cr и/или Се,
X2=Cu, Ni, Со, Fe, Mn и/или Zn,
X3=Sb и/или Bi,
X4=один или несколько щелочных металлов,
X5=один или несколько щелочноземельных металлов,
X6=Si, Al, Ti и/или Zr,
а=от 1 до 6,
b=от 0,2 до 4,
с=от 0,5 до 1,8,
d=от 0 до 40,
е=от 0 до 2,
f=от 0 до 4,
g=от 0 до 40, и
n=стехиометрический коэффициент элемента кислорода, который определяется с помощью стехиометрических коэффициентов элементов, отличных от кислорода, а также числом их зарядов в I.
5. Способ по п. 1, отличающийся тем, что поступающая реакционная газовая смесь А содержит от 0,75 до 10 об.% этанола.
6. Способ по п. 1, отличающийся тем, что поступающая реакционная газовая смесь А содержит от 1 до 40 об.% воды.
7. Способ по п. 1, отличающийся тем, что поступающая реакционная газовая смесь А, в пересчете на массу содержащегося в ней этанола, содержит по меньшей мере 1 мас.ч. на млн химического соединения, содержащего элемент серу, пересчитанное как количество этой содержащейся серы.
8. Способ по п. 1, отличающийся тем, что в качестве источника формальдегида для формальдегида, содержащегося в поступающей реакционной газовой смеси В, используют по меньшей мере один из источников - триоксан, параформальдегид, формалин, метилаль, водный раствор параформальдегида, водный раствор формальдегида и газообразную смесь продуктов гетерогенно катализируемого частичного газофазного окисления метанола до формальдегида, из которой, на выбор, был отделен содержащийся в ней при необходимости не вступивший в реакцию метанол.
9. Способ по п. 1, отличающийся тем, что содержание формальдегида в поступающей реакционной газовой смеси В составляет от 0,5 до 10 об.%.
10. Способ по п. 1, отличающийся тем, что поступающая реакционная газовая смесь В содержит уксусную кислоту в молярном количестве nНАс и формальдегид в молярном количестве nFd, и соотношение nНАс:nFd составляет >1 и ≤ 10.
11. Способ по п. 1, отличающийся тем, что содержание уксусной кислоты в поступающей реакционной газовой смеси В составляет от 1,5 до 20 об.%.
12. Способ по п. 1, отличающийся тем, что содержание водяного пара в поступающей реакционной газовой смеси В не превышает 30 об.% и не опускается ниже 1,5 об.%.
13. Способ по п. 1, отличающийся тем, что по меньшей мере один катализатор альдольной конденсации В содержит каталитически активную массу, которая представляет собой активную массу полиэлементных оксидов общей формулы II
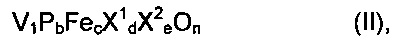
в которой переменные имеют следующее значение:
X1=Мо, Bi, Со, Ni, Si, Zn, Hf, Zr, Ti, Cr, Mn, Cu, B, Sn и/или Nb,
X2=Li, K, Na, Rb, Cs и/или TI,
b=от 0,9 до 2,0,
с=от ≥0 до 0,1,
d=от ≥ 0 до 0,1,
е=от ≥ 0 до 0,1, и
n=стехиометрический коэффициент элемента кислорода, который определяется с помощью стехиометрических коэффициентов элементов, отличных от кислорода, а также числом их зарядов в II.
14. Способ по п. 1, отличающийся тем, что он включает еще одну, третью реакционную зону С, которая заполнена по меньшей мере одним катализатором окисления С, и включает следующие дополнительные операции:
- через третью реакционную зону С, которая заполнена по меньшей мере одним катализатором окисления С, пропускают поток поступающей реакционной газовой смеси С, содержащей реагенты - метанол и молекулярный кислород, а также по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и при прохождении реакционной зоны С метанол, содержащийся в поступающей реакционной газовой смеси С, окисляют в условиях гетерогенного катализа до формальдегида и водяного пара, так что получают газообразную смесь продуктов С, содержащую формальдегид, водяной пар и по меньшей мере один инертный газ-разбавитель, отличающийся от водяного пара, и поток газообразной смеси продуктов С покидает реакционную зону С, причем к проходящей сквозь реакционную зону С реакционной газовой смеси С на ее пути через эту реакционную зону С на выбор может подаваться дополнительный молекулярный кислород и/или дополнительный инертный газ-разбавитель,
- на выбор, от газообразной смеси продуктов С в зоне разделения Т* отделяют при необходимости еще содержащийся в газообразной смеси продуктов С, не вступивший в реакцию метанол, причем остается газообразная смесь продуктов С*, содержащая формальдегид, и
- газообразную смесь продуктов С или газообразную смесь продуктов С* подают в реакционную зону В для получения поступающей реакционной газовой смеси В.
15. Способ по п. 14, отличающийся тем, что по меньшей мере один катализатор окисления С содержит каталитически активную массу, которая представляет собой смешанный оксид общей формулы III
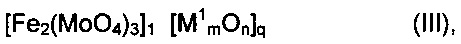
в которой переменные имеют следующие значения:
М1=Мо и/или Fe, или
Мо и/или Fe, а также, в пересчете на общее молярное количество из Мо и Fe, общее молярное количество вплоть до 10 мол.% одного или нескольких элементов из группы, состоящей из Ti, Sb, Sn, Ni, Cr, Се, Al, Ca, Mg, V, Nb, Ag, Mn, Cu, Co, Si, Na, K, TI, Zr, W, Ir, Та, As, P и В,
q=от 0 до 5,
m=от 1 до 3,
n=от 1 до 6.
16. Способ по п. 14, отличающийся тем, что поступающая реакционная газовая смесь С содержит от 2 до 15 об.% метанола.
17. Способ по п. 14, отличающийся тем, что поступающая реакционная газовая смесь С содержит от 0 до 20 об.% H2O.
18. Способ по п. 14, отличающийся тем, что частичное количество потока вещества Y подают обратно в реакционную зону С для получения поступающей реакционной газовой смеси С.
19. Способ по одному из пп. 1-18, отличающийся тем, что для разделения газообразной смеси продуктов В в зоне разделения Т эту газообразную смесь продуктов В, на выбор - после прямого и/или непрямого охлаждения этой смеси, подают в конденсационную колонну, снабженную способствующими разделению встроенными элементами, и внутри этой конденсационной колонны, конденсируют по фракциям, а потоки веществ X, Y и Z выводят из этой конденсационной колонны как отделенные друг от друга фракции.
20. Способ по одному из пп. 1-18, отличающийся тем, что для разделения газообразной смеси продуктов В в зоне разделения Т эту газообразную смесь продуктов В, на выбор - после прямого и/или непрямого охлаждения этой смеси, подают в абсорбционную колонну, оснащенную встроенными элементами с эффектом разделения, в противотоке с органическим растворителем, при нормальном давлении кипящем выше, чем акриловая кислота, а уксусную кислоту и акриловую кислоту, содержащиеся в газообразной смеси продуктов В, абсорбируют органическим растворителем с получением продукта абсорбции, в то время как поток вещества Z выходит из абсорбционной колонны через ее верх, а затем из продукта абсорбции потоки вещества X и Y выделяют в качестве разделенных фракций с помощью фракционной перегонки этого продукта в ректификационной колонне.
21. Способ по одному из пп. 1-18, отличающийся тем, что для разделения газообразной смеси продуктов В в зоне разделения Т эту газообразную смесь продуктов В, на выбор - после прямого и/или непрямого охлаждения этой смеси, подают в абсорбционную колонну, оснащенную встроенными элементами с эффектом разделения, в противотоке с водным раствором в качестве абсорбирующего агента, и уксусную кислоту и акриловую кислоту, содержащиеся в газообразной смеси продуктов В, абсорбируют растворителем с получением продукта абсорбции, в то время как поток вещества Z выходит из абсорбционной колонны через ее верх, а затем из продукта абсорбции потоки вещества X и Y выделяют в качестве разделенных фракций с помощью фракционной перегонки этого продукта в ректификационной колонне.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Питательное приспособление к трепальным машинам для лубовых растений | 1922 |
|
SU201A1 |
| US 5840971 A, 24.11.1998 | |||
| СN 101462069 A, 24.06.2009 | |||
| Xiaobo XU et al | |||
| "Advances in the Research and Development of Acrylic Acid Production from Biomass" Chinese Journal of Chemical Engineering, v | |||
| Паровоз для отопления неспекающейся каменноугольной мелочью | 1916 |
|
SU14A1 |
| Устройство для биологического очищения сточных вод | 1924 |
|
SU419A1 |
| US 2009239995 A1, 24.09.2009 | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| RU 2008135966 A, 20.03.2010. | |||

