[0001] Область техники
[0002] Данная заявка относится к аналогам диазонамида формулы (I) и к их солям, фармацевтическим композициям и конъюгатам, которые применимы в качестве антипролиферативных средств.
[0003] Предшествующий уровень техники
[0004] Диазонамид А представляет собой разрушающее митотическое веретено средство, впервые выделенное из морского организма Diazona angulata, имеющее структуру:
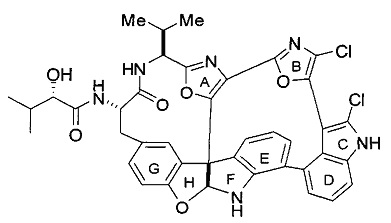 .
.
[0005] Получение аналогов диазонамида посредством макроциклических индолиновых промежуточных соединений, несущих карбобензилокси (Cbz) или о-нитрофенилсульфонильную защищенную аминогруппу, было описано ранее. В патентных документах США 7022720 и 7517895 точно раскрыта структура диазонамида А и описан синтез некоторых из его аналогов. В патентном документе США 7851620 (продолженном патентным документом США с серийным номером 12/896898) описаны синтетические способы получения аналогов диазонамида посредством промежуточных индолиновых соединений. В патентном документе США 7538129 описаны аналоги диазонамида А. Патентный документ США с серийным номером 12/432615 является родственной заявкой, находящейся на рассмотрении, где раскрыт индолин, который не имеет жесткой макроциклической структуры, образующей мостик между А- и Е-кольцами диазонамидного скелета. В этом документе раскрыты соединения формулы (I) и дополнительные новые аналоги диазонамида, которые обладают сильной цитотоксической активностью и пригодны для лечения клеточных пролиферативных нарушений.
[0006] Краткое описание изобретения
[0007] Настоящее изобретение направлено на соединения формулы (I) и их фармацевтически приемлемые соли и конъюгаты, фармацевтические композиции, содержащие соединение формулы (I) и/или его соль или конъюгат, модифицированные формы таких соединений, конъюгированных со стабилизирующими или нацеливающими средствами, и на способы получения и применения таких соединений и составов, где формула (I) представляет собой:
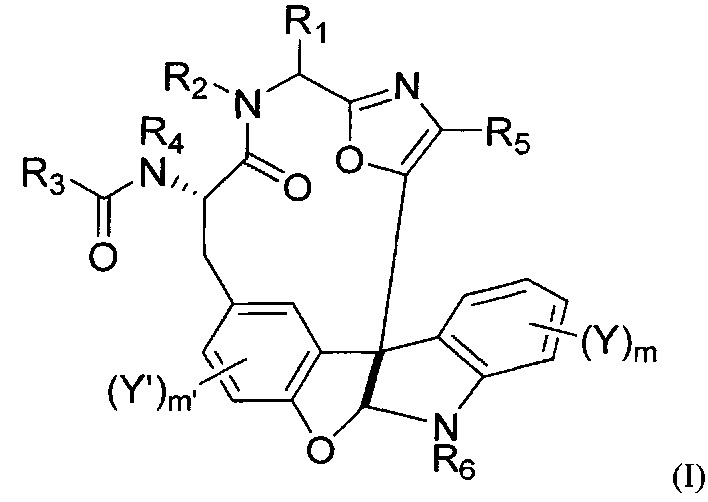 ,
,
или их фармацевтически приемлемой соли или конъюгата;
где:
R1 представляет собой необязательно замещенный C1-C4 алкил;
R2 представляет собой Н или необязательно замещенный C1-C4 алкил;
R3 представляет собой C1-C12 алкил, C1-C12 гетероалкил, C2-C12 алкенил, C2-C12 гетероалкенил, C3-C8 циклоалкил, C3-C8 гетероциклил, C4-C12 циклоалкилалкил, C4-C12 гетероциклилалкил, C6-C12 арил, C5-C12 гетероарил, C7-C14 арилалкил или C6-C14 гетероарилалкил, каждый из которых может быть необязательно замещенным;
R4 представляет собой H или необязательно замещенный C1-C4 алкил;
R5 представляет собой необязательно замещенный C6-C12 арил или необязательно замещенный C5-C12 гетероарил;
R6 представляет собой H или необязательно замещенный C1-C4алкил;
каждый из Y и Y' независимо представляет собой галоген, OH, C1-C4 алкокси, или C1-C8 алкил, C2-C8 алкенил, C2-C8 алкинил, C6-C12 арил, или C7-C14 арилалкил, или гетероформу одного из них, каждый из которых может быть необязательно замещенным;
m равняется 0-4; и
m' равняется 0-3.
[0008] Настоящее изобретение охватывает все комбинации различных предпочтительных вариантов осуществления/замен формулы (I), описанной в данном документе.
[0009] В следующем аспекте настоящее изобретение предусматривает фармацевтическую композицию, содержащую по меньшей мере одно соединение формулы (I) или его раскрытый вариант осуществления и фармацевтически приемлемый наполнитель.
[0010] В некоторых вариантах осуществления соединение формулы (I) или раскрытый его вариант осуществления представляет собой соединение одной из таблиц, предоставленных в данном документе, или фармацевтически приемлемую соль или конъюгат одного из таких соединений.
[0011] В другом аспекте настоящее изобретение предусматривает способ лечения или уменьшения интенсивности клеточного пролиферативного нарушения, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества по меньшей мере одного соединения формулы (I) или его раскрытого варианта осуществления, или его соли, конъюгата или фармацевтической композиции. В некоторых вариантах осуществления введенное количество является достаточным для ингибирования клеточной пролиферации. В других вариантах осуществления количество является достаточным для замедления роста опухоли или уменьшения размера опухоли. В некоторых вариантах осуществления соединение формулы (I) или его раскрытый вариант осуществления применяют в комбинации с другим химиопрепаратом или подходом.
[0012] Также предусмотрены способы ингибирования клеточной пролиферации в клетке, включающие приведение в контакт клетки с соединением одной из формул, описанных в данном документе, или его солью, или конъюгатом в количестве, эффективном для ингибирования клеточной пролиферации. В некоторых вариантах осуществления клетки являются клеточной линией, такой как клеточная линия рака (например, клеточная линия, полученная из рака молочной железы, предстательной железы, поджелудочной железы, легких или рака крови и кроветворной ткани и т.д.). В некоторых вариантах осуществления клетки находятся в ткани, в некоторых таких вариантах осуществления, при этом ткань может быть у субъекта. В других вариантах осуществления клетки находятся в ткани, а иногда находятся в ткани у субъекта.
[0013] Также предусмотрены способы лечения рака у субъекта, нуждающегося в таком лечении, включающие введение субъекту терапевтически эффективного количества соединения формулы (I), или его раскрытого варианта осуществления, или его соли, или конъюгата, как описано в данном документе, в количестве, эффективном для лечения или уменьшения интенсивности указанного рака.
[0014] Настоящее изобретение также предусматривает способы лечения или уменьшения интенсивности состояния, связанного с нарушенной клеточной пролиферацией. Например, предусмотрены способы лечения или уменьшения интенсивности нарушения клеточной пролиферации у субъекта, включающие введение соединения формулы (I), или его раскрытого варианта осуществления, или его соли, или конъюгата, как описано в данном документе, субъекту, нуждающемуся в этом, в количестве, эффективном для лечения или уменьшения интенсивности состояния.
[0015] В описанных в данном документе способах субъектом может быть экспериментальное животное (например, грызун, собака, кот, обезьяна), необязательно содержащее опухоль, такую как, например, привитая опухоль (например, опухоль человека), или им может быть человек.
[0016] Краткое описание графических материалов
[0017] На фигуре 1 показаны данные для исследуемых соединений на модели привитого рака легких человека НСС461 на мышах.
[0018] На фигуре 2 показаны данные для исследуемых соединений на модели привитого рака поджелудочной железы Miapaca на мышах.
[0019] Подробное описание отдельных вариантов осуществления
[0020] Настоящее изобретение может быть легко понято посредством ссылки на нижеследующее подробное описание предпочтительных вариантов осуществления настоящего изобретения и примеры, включенные в данный документ. Должно быть понятно, что терминология, используемая в данном документе, служит только с целью описания конкретных вариантов осуществления и не предназначена быть ограничивающей. Также следует понимать, кроме случаев, если в данном документе определено конкретно, что терминология, используемая в данном документе, приведена в ее общепринятом значении, известном в соответствующей области.
[0021] Используемые в данном документе формы единственного числа включают ссылки на множественное число, если не указано иное.
[0022] Используемое в данном документе выражение "субъект" относится к субъекту, являющемуся человеком или животным. В предпочтительных вариантах осуществления субъектом является человек.
[0023] Выражения "лечить", "лечение" или "обработка" в отношении отдельного заболевания или нарушения включают профилактику заболевания или нарушения и/или снижение, улучшение, уменьшение интенсивности, облегчение или устранение симптомов и/или патологии заболевания или нарушения.
[0024] Выражение "терапевтически эффективное количество" или "эффективное количество" предназначено означать такое количество лекарственного средства или фармацевтического средства, с помощью которого можно добиться биологического или медицинского ответа клетки, ткани, системы, животного или человека, находящихся под наблюдением исследователя, ветеринара, врача или другого клинициста. Выражения также могут относиться к уменьшению степени или прекращению клеточной пролиферации (например, замедлению или остановке роста опухоли) или уменьшению количества пролиферирующих раковых клеток (например, устранению части или всей опухоли). Иногда степень клеточной пролиферации уменьшается на 10%, 20%, 30%, 40%, 50%, 60% или 70% или больше. Иногда количество пролиферирующих клеток уменьшается на 10%, 20%, 30%, 40%, 50%, 60% или 70% или больше.
[0025] Используемые в данном документе выражения "алкил", "алкенил" и "алкинил" включают одновалентные углеводородные радикалы с неразветвленной цепью, с разветвленной цепью и циклической структуры и их комбинации, которые содержат только C и H, если они незамещенные. Примеры включают метил, этил, изопропил, изобутил, трет-бутил, циклогексил, циклопентилэтил, 2-пропенил, 3-бутинил и т.п. Иногда в каждой такой группе в данном документе указано общее число атомов углерода, что в ряде случаев, например, если группа может содержать до двенадцати атомов углерода, может быть представлено как 1-12C, или как C1-C12, или как C1-12, или как C1-12. Если гетероатомы (как правило, N, О и S) замещают атомы углерода алкильной, алкенильной или алкинильной группы, как, например, в гетероалкильных группах, то числа, описывающие группу, оставаясь все еще записанными как, например, C1-C6, представляют сумму числа атомов углерода в группе плюс число таких гетероатомов, которые включены как замены атомов углерода в описанном кольце или цепи.
[0026] Как правило, алкильные, алкенильные и алкинильные заместители по настоящему изобретению включают 1-12C (алкил) или 2-12C (алкенил или алкинил). Предпочтительно они включают 1-8C (алкил) или 2-8C (алкенил или алкинил). Иногда они включают 1-4C (алкил) или 2-4C (алкенил или алкинил). Отдельная группа может включать больше одного типа многократной связи или больше одной многократной связи; такие группы охватываются определением выражения "алкенил", если они содержат по меньшей мере одну двойную связь углерод-углерод, и они охватываются выражением "алкинил", если они содержат по меньшей мере одну тройную связь углерод-углерод.
[0027] "Гетероалкил", "гетероалкенил" и "гетероалкинил" и т.п. определены аналогично соответствующим углеводородным (алкильной, алкенильной и алкинильной) группам, но выражения ‘гетеро’ относятся к группам, которые содержат один или несколько гетероатомов, выбранных из O, S и N и их комбинации, в пределах остатка главной цепи; таким образом, по меньшей мере один атом углерода соответствующей алкильной, алкенильной или алкинильной группы замещается одним из указанных гетероатомов с образованием гетероалкильной, гетероалкенильной или гетероалкинильной группы. Предпочтительно, каждая гетероалкильная, гетероалкенильная и гетероалкинильная группа содержит только 1-2 гетероатома как часть скелета главной цепи гетероалкильной группы, т.е. не включая заместителей, которые могут присутствовать. Поэтому, гетероалкилы включают алкоксилы, такие как О-алкил, алкиловые эфиры, вторичные и третичные алкиламины, алкилсульфиды, алкилсульфонилы и т.п.
[0028] Характерные и предпочтительные размеры гетероформ алкильной, алкенильной и алкинильной групп обычно являются такими же, как для соответствующих углеводородных групп, а заместители, которые могут присутствовать на гетероформах, являются такими же как, как они описаны выше для углеводородных групп. Если такие группы содержат N, атом азота может присутствовать как NH или он может быть замещенным, если гетероалкильная или подобная группа описана как необязательно замещенная. Если такие группы содержат S, атом серы необязательно может быть окисленным до SO или SO2, если не указано иное. Ввиду химической стабильности также очевидно, если не указано иное, что такие группы не включают больше трех соседних гетероатомов как часть гетероалкильной цепи, несмотря на то, что при N или S может присутствовать оксогруппа, как в нитро или сульфонильной группе. Таким образом, -C(O)NH2 может быть C2 гетероалкильной группой, замещенной =O; и -SO2NH- может быть C2 гетероалкиленом, где S заменяет один углерод, N заменяет один углерод, и S замещен двумя =O группами.
[0029] Несмотря на то, что используемый в данном документе "алкил" включает циклоалкильные и циклоалкилалкильные группы, выражение "циклоалкил" может использоваться в данном документе для описания главным образом насыщенного или частично насыщенного, моноциклического, или конденсированного, или спирополициклического карбоцикла, который присоединен через атом углерода кольца, и "циклоалкилалкил" может использоваться для описания карбоциклической неароматической группы, которая присоединена к основной молекуле через алкильный линкер. Аналогично, "гетероциклил" может использоваться для описания неароматической циклической группы, которая содержит по меньшей мере один гетероатом в качестве члена кольца и которая присоединена к молекуле через атом кольца циклической группы, который может представлять собой С или N; и "гетероциклилалкил" может использоваться для описания такой группы, которая присоединена к другой молекуле через алкильный линкер. Размеры и заместители, которые являются подходящими для циклоалкильных, циклоалкилалкильных, гетероциклильных и гетероциклилалкильных групп, являются такими же, как описаны выше для алкильных групп. Зачастую циклоалкильные и гетероциклильные группы представляют собой C3-C8, а циклоалкилалкильные или гетероциклилалкильные группы представляют собой C4-C12. Размер циклоалкилалкильной или гетероциклилалкильной группы описывает общее число атомов углерода или атомов углерода плюс гетероатомов, которые заменяют атомы углерода алкильной, алкенильной, алкинильной, циклоалкильной или циклоалкилалкильной части. Используемые в данном документе такие выражения также включают кольца, которые содержат двойную связь или две, при условии, что кольцо не является ароматическим.
[0030] Используемый в данном документе "ацил" охватывает группы, содержащие алкильный, алкенильный, алкинильный, арильный или арилалкильный радикал, присоединенный в одном из двух доступных по валентности положений атома углерода карбонила (что может быть отображено в данном документе как -C(=O)R, -C(O)R или COR), где R представляет собой алкильную, алкенильную, алкинильную, арильную или арилалкильную группу, и гетероацил относится к соответствующим группам, где по меньшей мере один углерод, отличный от углерода карбонила, заменен гетероатомом, выбранным из N, O и S. Таким образом, гетероацил включает, например, -C(=O)OR и -C(=O)NR2, а также -C(=O)-гетероарил. Также охваченными определением гетероацильных групп являются тиоацильные заместители, например, -C(=S)R, и имино группы, например, -C(=NH)R.
[0031] Ацильные и гетероацильные группы связаны с любой группой или молекулой, к которой они присоединены, посредством свободной валентности атома углерода карбонила. Как правило, они представляют собой C1-C8 ацильные группы, которые включают формил, ацетил, трифторацетил, пивалоил и бензоил, и C2-C8 гетероацильные группы, которые включают метоксиацетил, этоксикарбонил и 4-пиридиноил. Углеводородные группы, арильные группы и гетероформы таких групп, которые включают ацильную или гетероацильную группу, могут быть замещены заместителями, описанными в данном документе, как правило, подходящими заместителями для каждого соответствующего компонента ацильной или гетероацильной группы.
[0032] "Ароматический" фрагмент или "арильный" фрагмент относится к моноциклическому или конденсированному бициклическому фрагменту, обладающему хорошо известными признаками ароматичности; примеры включают фенил и нафтил. Карбоциклические арильные кольца и кольцевые системы, как правило, являются кольцом из 6-12 атомов углерода и могут включать насыщенное или частично ненасыщенное карбоциклическое кольцо, конденсированное до ароматического кольца, например, тетрагидронафталеновой, индановой или инденовой кольцевой системы. Аналогично, "гетероароматический" и "гетероарил" относится к таким моноциклическим или конденсированным бициклическим кольцевым системам, которые содержат в качестве кольцевых членов один или несколько гетероатомов, выбранных из О, S и N. Включение гетероатома обеспечивает ароматичность в 5-членных кольцах, а также 6-членных кольцах. Обычные гетероароматические системы включают моноциклические C5-C6 ароматические группы, такие как пиридильные, примидильные, пиразинильные, пиридазинильные, триазинильные, тиенильные, фуранильные, пирролильные, пиразолильные, тиазолильные, изотиазолильные, оксазолильные, изоксазолильные, имидазолильные, триазолильные, тиадиазолильные, оксадиазолильные и тетразолильные кольца, и конденсированные бициклические фрагменты, полученные путем конденсации одной из таких моноциклических групп с фенильным кольцом или с любой из гетероароматических моноциклических групп, с образованием C8-C10 бициклической группы, например, индолил, бензимидазолил, индазолил, бензотриазолил, изохинолинил, хинолинил, бензотиазолил, бензофуранил, бензотиенил, бензизоксазолил, пиразолопиридил, хиназолинил, хиноксалинил, циннолинил и т.п. Этим определением охватывается любая моноциклическая или конденсированная кольцевая бициклическая система, которая имеет признаки ароматичности в отношении распределения электронов по всей кольцевой системе. Определение также охватывает бициклические группы, где по меньшей мере одно кольцо имеет признаки ароматичности, даже если оно может быть конденсированным до неароматического кольца. Как правило, кольцевые системы содержат 5-12 атомов кольца. Предпочтительно моноциклические арильные и гетероарильные группы содержат 5-6 членов кольца, а бициклические арильные и гетероарильные группы содержат 8-10 членов кольца.
[0033] Аналогично, "арилалкил" и "гетероарилалкил" относятся к ароматическим и гетероароматическим кольцевым системам, которые связаны с точкой их прикрепления посредством связывающей группы, такой как алкилен, включая замещенные или незамещенные, насыщенные или ненасыщенные, циклические или ациклические линкеры. Как правило, линкер представляет собой C1-C8 алкил или его гетероформу, предпочтительно C1-C4 алкил. Такие линкеры могут также включать карбонильную группу, что тем самым делает их способными обеспечить заместители в виде ацильных или гетероацильных фрагментов.
[0034] "Арилалкильные" группы, используемые в данном документе, представляют собой углеводородные группы, если они являются незамещенными, и описаны посредством общего числа атомов углерода в кольце и алкилене или подобном линкере. Таким образом, бензильная группа представляет собой C7-арилалкильную группу, и фенилэтил представляет собой C8-арилалкил. Предпочтительно, арилалкильная группа включает одно или два необязательно замещенных фенильных кольца и C1-C4 алкилен, незамещенный или замещенный одной или двумя C1-C4 алкильными группами или C1-C4 гетероалкильными группами, где алкильные или гетероалкильные группы необязательно могут замыкаться в цикл с получением кольца, такого как циклопропан, диоксолан или оксациклопентан, и где алкильные или гетероалкильные группы необязательно могут быть фторированными. Примеры арилалкильных групп необязательно включают замещенные бензильные, фенилэтильные, дифенилметильные и трифенилметильные группы. Необязательные заместители, если присутствуют на арильном кольце арилалкильной группы, являются такими же, как описанные в данном документе для арильного кольца. Арилалкильные группы, как правило, содержат 7-20 атомов, предпочтительно 7-14 атомов.
[0035] "Гетероарилалкил", описанный выше, относится к фрагменту, содержащему арильную группу, присоединенную через связывающую группу, и отличается от "арилалкила" тем, что по меньшей мере один атом кольца арильного фрагмента или один атом в связывающей группе является гетероатомом, выбранным из N, O и S. Гетероарилалкильные группы описаны в данном документе в соответствии с общим числом атомов в кольце и комбинированном линкере, и они включают арильные группы, связанные через гетероалкильный линкер; гетероарильные группы, связанные через углеводородный линкер, такой как алкилен; и гетероарильные группы, связанные через гетероалкильный линкер. Например, гетероарильные группы включают пиридилметил, пиридилэтил, -O-бензил и т.п. Гетероарилалкильные группы, как правило, содержат 6-20 атомов, предпочтительно 6-14 атомов.
[0036] "Алкилен", используемый в данном документе, относится к двухвалентной углеводородной группе; поскольку он двухвалентный, то может связывать две другие группы вместе. Как правило, это относится к -(CH2)n-, где n равняется 1-8 и предпочтительно n равняется 1-4, тем не менее, если указано, алкилен также может быть замещенным другими группами и может быть другой длины, а свободные валентности не обязательно должны быть на противоположных концах цепи. Таким образом, -CH(Me)- и -C(Me)2- также могут быть названы алкиленами, равно как и циклическая группа, такая как циклопропан-1,1-диил. Однако, для ясности, трех-атомный линкер, который представляет собой, например, алкиленовую группу, относится к двухвалентной группе, в которой доступные валентности для присоединения к другим группам разделены тремя атомами, а именно -(CH2)3-, т.е. указанная длинна отображает число атомов, связывающих точки присоединения, а не общее число атомов в углеводородной группе: -C(Me)2-, таким образом, будет одноатомным линкером, поскольку доступные валентности разделены только одним атомом. Если алкиленовая группа замещенная, то заместители включают те, которые обычно присутствуют на алкильных группах, описанных в данном документе, таким образом, -C(=O)- является примером алкилена, замещенного по одному углероду. В случаях, когда алкилен описан ненасыщенным, он может содержать одну или несколько двойных или тройных связей.
[0037] "Гетероалкилен", используемый в данном документе, определен аналогично соответствующим алкиленовым группам, но выражения ‘гетеро’ относятся к группам, которые содержат один или несколько гетероатомов, выбранных из О, S и N и их комбинаций, в пределах остатка главной цепи; таким образом по меньшей мере один атом углерода соответствующей алкиленовой группы замещается одним из указанных гетероатомов с образованием гетероалкиленовой группы. Таким образом, -C(=O)NH- является примером гетероалкилена, замещенного по двум углеродам, где один углерод заменяет N, и C замещается =O группой.
[0038] "Гетероформа", используемая в данном документе, относится к производному группы, такой как алкил, арил или ацил, где по меньшей мере один атом углерода предусмотренной карбоциклической группы заменен гетероатомом, выбранным из N, О и S. Таким образом, гетероформы алкила, алкенила, циклоалкила, алкинила, ацила, арила и арилалкила представляют собой гетероалкил, гетероалкенил, гетероциклил, гетероалкинил, гетероацил, гетероарил и гетероарилалкил, соответственно. Следует иметь ввиду, что гетероформа арильного или арилалкильного фрагмента может содержать на один атом "C" меньше, чем вся соответствующая углеродная система, так как включение гетероатома обеспечивает ароматичность в 5-членных кольцах. Например, гетероформа C6-C12 арила представляет собой C5-C12 гетероарил, а гетероформа C7-C20 арилалкила представляет собой C6-C20 гетероарилалкил. Известно, что обычно присоединены последовательно не больше двух атомов N, O или S, за исключением случаев, когда оксогруппа прикрепляется к N или S с образованием нитро или сульфонильной группы, или в случае некоторых гетероароматических колец, таких как триазин, триазол, тетразол, оксадиазол, тиадиазол и т.п.
[0039] Если не указано иное, выражение "оксо" относится к =O.
[0040] "Галоген", используемый в данном документе, включает фтор, хлор, бром и иод. Зачастую предпочтительны фтор, хлор и бром.
[0041] "Амино", используемый в данном документе, относится к NH2, но там, где амино описан как "замещенный" или "необязательно замещенный", выражение включает NR2, где каждый R независимо представляет собой H, или представляет собой алкильную, алкенильную, алкинильную, ацильную, арильную или арилалкильную группу, или гетероформу одной из этих групп, как дополнительно определено в данном документе, каждый из которых может быть необязательно замещенным заместителями, описанными в данном документе, по мере того, как они подходят к соответствующему типу группы. Выражение также включает формы, где две группы R на одном атоме азота (т.е. NR2) связываются вместе с образованием 3-8-членного моноциклического азациклического кольца или 8-12-членной бициклической конденсированной азациклической кольцевой системы, каждый из которых может быть насыщенным, ненасыщенным или ароматическим и может содержать 1-3 гетероатома, включая атом азота азациклического кольца, независимо выбранных из N, O и S в качестве членов кольца (т.е. 0-2 гетероатома, выбранные из N, O и S, дополнительно к атому азота азациклического кольца), и которые необязательно могут быть замещенными заместителями, описанными как подходящие для алкильных групп, или, если NR2 включает ароматическую группу, то необязательно могут быть замещенными заместителями, описанными как типичные для арильных или гетероарильных групп. Такие предпочтительные азациклические кольца включают пирролидин, пиперидин, гомопиперидин, морфолин, тиоморфолин, пиперазин и гомопиперазин.
[0042] Аминогруппы необязательно могут быть в защищенной или незащищенной форме. Специалисту в данной области будет понятно, что подходящие амин-защитные группы могут изменяться в зависимости от функциональности, присутствующей в отдельной молекуле, и природы аминогруппы. Подходяще защищенные амины могут включать, например, амины, защищенные как карбаматы (например, трет-бутоксикарбонил (Boc), бензилоксикарбонил (Cbz), флуоренилметилоксикарбонил (Fmoc), аллилоксикарбонил (Alloc) или (триалкилсилил)этоксикарбонил), карбоксамиды (например, формил, ацил или трифторацетил, бензоил), сульфонамиды, фталимиды, сукцинимиды, производные основания Шиффа и т.п. Также включены алкил или аллиламины, а также триалкилсилил-защищенные амины.
[0043] Если амин присутствует в защищенной форме, то иногда желательно снять защитную группу. Таким образом, способы по настоящему изобретению также необязательно включают этап снятия любых защитных групп на аминогруппе или аминоалкильной группе.
[0044] Выражения "алкилсульфонил" и "арилсульфонил", используемая в данном документе, относятся к фрагментам формы -SO2 алкил или -SO2 арил, где алкил и арил определены выше. Необязательно фторированный С1-4алкил и необязательно замещенные фенильные группы предпочтительны для сульфонильных фрагментов. Фенильные группы арилсульфонильного фрагмента необязательно могут быть замещенными одним или несколькими заместителями, подходящими для арильного кольца; например, они может быть замещены галогеном, метилом, нитро, алкокси, амино или т.п. Такие сульфонильные фрагменты, если присутствуют на кислороде, образуют сульфонаты. Такие сульфонильные фрагменты образуют сульфонамиды, если присутствуют на азоте, и сульфоны, если присутствуют на углероде. Типичные сульфонаты включают, например, -OSO2Me (мезилат), -OSO2CF3 (трифлат), -OSO2 толил (тозилат) и т.п.
[0045] Выражение "алкоксикарбонил", используемое в данном документе, относится к фрагменту формы -COOR', где R' представляет собой C1-C8 алкил, C2-C8 алкенил, C5-C6 арил или C7-C14 арилалкил, триалкилсилил или т.п, каждый из которых может быть необязательно замещенным. Присутствуя на азоте, такие алкоксикарбонильные фрагменты образуют карбаматы, которые часто используют в качестве азот-защитных групп. В некоторых таких вариантах осуществления R' необязательно может быть галогенированным C1-C4 алкилом (например, трет-бутилом, метилом, этилом, 2,2,2-трихлорэтилом, 1,1-диметил-2,2,2-трихлорэтилом), аллилом, необязательно замещенным бензилом, флуоренилметилом или триалкилсилилом (например, триизопропилсилилом, триэтилсилилом, трет-бутилдиметилсилилом). Присутствуя на углероде такие фрагменты также могут называться карбоксилатными сложными эфирами, карбоалкокси группами или т.п. В некоторых вариантах осуществления, включающих функциональную группу карбоксилатного сложного эфира, R' предпочтительно представляет собой C1-4 алкильную группу. В некоторых таких вариантах осуществления R' представляет собой метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил.
[0046] Выражение "замещенный" означает, что указанная группа или фрагмент несет один или несколько заместителей, не являющихся водородом. Выражение "незамещенный" означает, что указанная группа не несет таких заместителей.
[0047] "Необязательно замещенный", используемый в данном документе, указывает на то, что отдельная описываемая группа или группы могут не иметь заместителей, не являющихся водородом, или группа или группы могут иметь один или несколько заместителей, не являющихся водородом (т.е. группа может быть замещенной или незамещенной). Если не указано иное, общее число таких заместителей, которые могут присутствовать, равно числу атомов Н, присутствующих в незамещенной форме описываемой группы. Если необязательный заместитель присоединяется посредством двойной связи, такой как кислород карбонила (=O), группа занимает две доступные валентности, так что общее число заместителей, которые могут быть включены, уменьшается согласно числу доступных валентностей.
[0048] Алкильные, алкенильные и алкинильные группы часто являются замещенными в той мере, в которой такое замещение целесообразно химически. Обычные заместители включают без ограничения галоген, OH, =O, =N-CN, =N-OR, =NR, OR, NR2, SR, SOR, SO2R, SO2NR2, NRSO2R, NRCONR2, NRCOOR, NRCOR, CN, COOR, CONR2, OOCR, COR и NO2, где каждый R независимо представляет собой H, необязательно фторированный C1-C8 алкил, C2-C8 гетероалкил, C1-C8 ацил, C2-C8 гетероацил, C2-C8 алкенил, C2-C8 гетероалкенил, C2-C8 алкинил, C2-C8 гетероалкинил, C6-C12 арил, C5-C12 гетероарил, C5-C20 арилалкил или C5-C20 гетероарилалкил, и каждый R является необязательно замещенным одной или несколькими группами, выбранными из галогена, ОН, =O, =N-CN, =N-OR', =NR', OR', 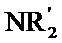
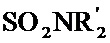
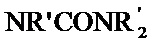
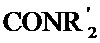
[0049] Предпочтительные заместители, если присутствуют на алкильной, алкенильной или алкинильной группе, или гетероформа одной из них включают галоген, OH, =O, OR, SR и NR2, где R определен выше; иногда, R представляет собой H, необязательно фторированный C1-C4 алкил или необязательно фторированный C1-C4ацил. Особенно предпочтительные заместители, если присутствуют на R3, включают OH, =O, C1-C4 алкокси, OAc, NHAc, NH2 и NHMe. Иногда, необязательные заместители, присутствующие на алкильной, алкенильной или алкинильной группе или гетероформе одной из них, включают NRSO2R, NRCONR2, COOR или CONR2, где R определен выше; предпочтительно, чтобы каждый R независимо представлял собой H, необязательно фторированный C1-C4 алкил или представлял собой C6-C12 арил, C5-C12 гетероарил, C7-C20 арилалкил или C6-C20 гетероарилалкил, каждый из которых может быть необязательно замещенным.
[0050] Арильные, гетероарильные и гетероциклильные фрагменты могут быть замещены различными заместителями, включающими необязательно фторированный C1-C8 алкил, C2-C8 алкенил, C2-C8 алкинил, C1-C8 ацил и их гетероформы, C6-C12 арил, C5-C12 для гетероарила, C6-20 арилалкил (C5-20 для гетероарилалкила), каждый из которых сам по себе может быть дополнительно замещенным; другие заместители для арильных и гетероарильных фрагментов включают галоген, OH, OR, СН2ОН, CH2OR, CH2NR2, NR2, SR, SOR, SO2R, SO2NR2, NRSO2R, NRCONR2, NRCOOR, NRCOR, CN, COOR, CONR2, OOCR, C(O)R и NO2, где каждый R независимо представляет собой H, необязательно фторированный C1-C8 алкил, C2-C8 гетероалкил, C2-C8 алкенил, C2-C8 гетероалкенил, C2-C8 алкинил, C2-C8 гетероалкинил, C6-C12 арил, C5-C12 гетероарил, C7-C20 арилалкил или C6-C20 гетероарилалкил, и каждый R является необязательно замещенным, как описано выше касательно алкильных групп. Замещающие группы на арильной или гетероарильной группе могут быть, разумеется, дополнительно замещены группами, описанными в данном документе, подходящими для каждого типа группы, которая включает заместитель. Предпочтительные заместители, если присутствуют на арильных, гетероарильных и гетероциклильных фрагментах, включают галоген, OH, OR, CH2OH, CH2OR, CH2NR2, SR, NR2, CN, COOR, CONR2 и NO2, где R определен выше, или необязательно замещенное C6-C12 арильное или C5-C12 гетероарильное кольцо.
[0051] Если арилалкильная или гетероарилалкильная группа описана как необязательно замещенная, то заместители могут находиться либо на алкильной или на гетероалкильной части, либо на арильной или гетероарильной части группы. Заместители, необязательно присутствующие на алкильной или гетероалкильной части, являются такими же, как описанные выше в основном для алкильных групп; заместители, необязательно присутствующие на арильной или гетероарильной части, являются такими же как, как описанные выше в основном для арильных групп.
[0052] Настоящее изобретение охватывает изомеры исследуемых соединений, в частности, стереоизомеры, такие как те, где атом углерода, несущий заместитель R1 в формуле (I), или соответствующий атом в раскрытых вариантах осуществления формулы (I) имеет (S)-конфигурацию.
[0053] Настоящее изобретение предусматривает новые индолиновые аналоги формулы (I), которые пригодны для обработки или уменьшения интенсивности пролиферативных нарушений, в частности, рака.
[0054] Настоящее изобретение охватывает все комбинации предпочтительных вариантов осуществления и предпочтительных заместителей, описанных в данном документе.
[0055] Предпочтительно, R1 представляет собой необязательно замещенный C2-C4 алкил, предпочтительно C2-C4 алкил, предпочтительно пропил или бутил, предпочтительно изопропил или трет-бутил.
[0056] Предпочтительно, R2, R4 и R6 независимо представляют собой H или метил, предпочтительно H. Заместитель R4 может выполнять функцию защитной группы, а способы, описанные в данном документе, включают необязательный этап снятия защиты, чтобы убрать любые защитные группы, присутствующие на молекуле.
[0057] Предпочтительно, R3 представляет собой замещенный метил общей формулы (-CRaRbRc), где Ra представляет собой OH, OR, CH2OR, SR и NR2, где каждый R независимо представляет собой H, необязательно галогенированный (предпочтительно фторированный или хлорированный) C1-C4 алкил или необязательно галогенированный C1-C4 ацил и предпочтительно ОН; и каждый из Rb и Rc независимо представляет собой H, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C3-C8 циклоалкилалкил, C6-C12 арил, C7-C14 арилалкил или гетероформу одного из них, каждый из которых может быть необязательно замещенным, и предпочтительно H или низший алкил C1-C4, более предпочтительно H и изопропил или трет-бутил, соответственно; или Rb и Rc вместе с углеродом, к которому они присоединены, могут образовывать C3-C8 циклоалкил или C3-C8 гетероциклильное кольцо, которые необязательно могут быть замещенными. Например, Rb и Rc могут быть взяты вместе с образованием необязательно замещенного циклопропилового, циклобутилового, циклопентилового, циклопентенилового, циклогексилового, циклогексенилового, тетрагидрофуранового, тетрагидропиранового, тетрагидротиофуранового, тетрагидротиопиранового, пирролидинового или пиперидинового кольца и т.п. В предпочтительном варианте осуществления Rb и Rc взяты вместе с образованием циклогексилового или циклопентилового кольца. В некоторых вариантах осуществления кольцо, образованное Rb и Rc, может быть конденсированным до замещенного или незамещенного фенильного кольца с обеспечением, например, индениловой или тетрагидронафтиловой кольцевой системы.
[0058] В других предпочтительных вариантах осуществления R3 представляет собой C1-C4 алкил, C3-C6 циклоалкил, C4-C8циклоалкилалкил или C6-C8 арилалкил, каждый из которых может быть необязательно замещенным. В предпочтительных вариантах осуществления алкильная группа, содержащая часть R3, является замещенной по меньшей мере одним заместителем, выбранным из группы, состоящей из OH, OR, CH2OR, SR и NR2, где каждый R независимо представляет собой H, необязательно фторированный C1-C4 алкил или необязательно фторированный C1-C4 ацил. Предпочтительно R3 является замещенным по меньшей мере одним заместителем, выбранный из группы, состоящей из OH, OMe, OAc, NH2, NHMe, СН2ОН и NHAc. В более конкретных вариантах осуществления R3 представляет собой C1-C8, предпочтительно C1-C4 , более предпочтительно C2-C3, наиболее предпочтительно С2-неразветвленную цепь, разветвленную, или циклоалкильную группу, каждая из которых является замещенной на атоме углерода, прилегающем к карбонильной группе, которая является частью R5 с OH, OMe, OAc, NH2, NHMe, CH2OH или NHAc, предпочтительно OH.
[0059] Предпочтительно R5 представляет собой необязательно замещенное фенильное, нафтильное, бензимидазольное, бензоксазольное, бензтиазольное, пиридинильное, пиримидинильное, пиразинильное или пиридазинильное кольцо и более предпочтительно R5 представляет собой необязательно замещенное оксазольное, оксазолиновое, тиазольное, тиазолиновое, пиразольное, пиразолиновое, имидазольное, имидазолиновое, пиррольное, пирролиновое, изоксазольное, изоксазолиновое, изотиазольное, изотиазолиновое, оксадиазольное, тиадиазольное, триазольное или тетразольное кольцо.
[0060] Предпочтительные заместители включают галоген, нитро, циано или необязательно фторированный C1-C4 алкил, необязательно фторированный C1-C4 алкокси, COOR8, 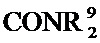
[0061] В конкретных предпочтительных вариантах осуществления R5 представляет собой необязательно замещенное оксазольное или тиазольное кольцо. В некоторых таких вариантах осуществления R5 представляет собой оксазольное кольцо, замещенное необязательно замещенным C6-C12 арильным или C5-C12 гетероарильным кольцом. В некоторых вариантах осуществления R5 представляет собой оксазольное кольцо, замещенное одним или несколькими заместителями, представляющими собой алкил, галоген, карбоновую кислоту, сложный эфир или амид.
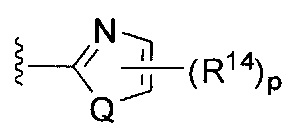
[0062] В конкретных вариантах осуществления формулы (I) R5 представляет собой необязательно замещенное гетероциклическое или гетероароматическое кольцо формулы:
 ;
; 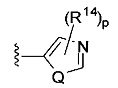 ;
; 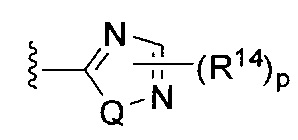 ;
; 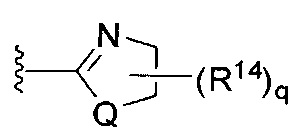
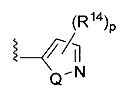 ;
; 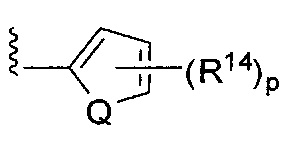 ;
; 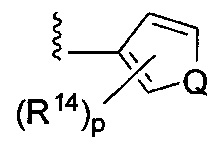 ; или
; или 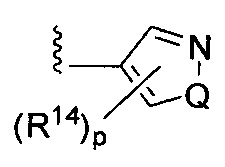 ,
,
[0063] где Q представляет собой О, S или NR13, где R13 представляет собой H или C1-C4 алкил; каждый R14 независимо представляет собой галоген, нитро, циано или необязательно фторированный C1-C4 алкил, необязательно фторированный C1-C4 алкокси, COOR8, 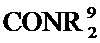
[0064] В конкретных предпочтительных вариантах осуществления формулы (I) R5 представляет собой
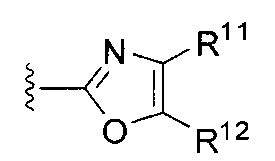 или
или 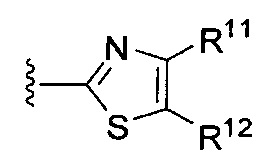 ,
,
[0065] где каждый из R11 и R12 независимо представляет собой H, галоген, нитро, циано или необязательно фторированный C1-C4 алкил, необязательно фторированный C1-C4 алкокси, COOR8, 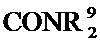
[0066] каждый R9 независимо представляет собой H, или C1-C12 алкил, C1-C12 гетероалкил, C2-C12 алкенил, C2-C12 гетероалкенил, C3-C8 циклоалкил, C3-C8 гетероциклил, C4-C12 циклоалкилалкил, C4-C12 гетероциклилалкил, C6-C12 арил, C5-C12 гетероарил, C7-C14 арилалкил, или C6-C14 гетероарилалкил, каждый из которых может быть необязательно замещенным; или два R9 на одном и том же N могут замыкать цикл с образованием необязательно замещенного 3- - 8-членного азациклического кольца, необязательно включая дополнительный гетероатом, выбранный из N, О и S, в качестве члена кольца. Такие азациклические кольца могут быть насыщенными, ненасыщенными или ароматическими; предпочтительные такие азациклические кольца включают пирролидин, пиперидин, гомопиперидин, морфолин, тиоморфолин, пиперазин и гомопиперазин.
[0067] В некоторых таких вариантах осуществления каждый из R11 и R12 независимо представляет собой H, галоген, нитро, циано, C1-C4 алкил, C1-C4 алкокси, COOR8, или 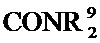
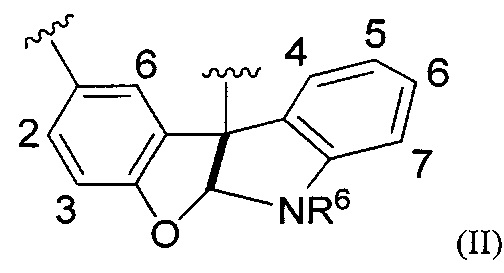
[0068] Заместители на индольних и тирозиновых компонентах макроциклического кольца формулы (I), Y и Y', соответственно, расположены в соответствующих положениях кольца, как показано в формуле II:
 ,
,
где R6 является таким, как определено в формуле (I); поэтому Y может находиться в одном или нескольких положениях 4, 5, 6 и 7 индольного фрагмента, a Y' может находиться в одном или нескольких положениях 2, 3 и 5 тирозинового фрагмента.
[0069] Предпочтительно каждый из Y и Y' независимо представляет собой галоген (F, О, Br или I), OH, C1-C4 алкокси, предпочтительно галоген, в частности, Cl или F; предпочтительно m равняется 3, 2, 1 или предпочтительно 0; и предпочтительно m' равняется 2, 1 или предпочтительно 0.
[0070] В предпочтительных вариантах осуществления Y находится в одном или нескольких положениях 5, 6 и 7, одном или нескольких положениях 5 и 7, одном из положений 5, 6 и 7, одном из положений 5 и 7, только в положении 5 или только в положении 7. В предпочтительных вариантах осуществления Y' находится в одном или нескольких положениях 2 и 3, одном из положений 2 и 3, только в положении 2 или только в положении 3. В отдельных вариантах осуществления одно или оба кольца являются замещенными.
[0071] Настоящее изобретение охватывает все комбинации предпочтительных вариантов осуществления и предпочтительных заместителей, как если бы каждая была подробно изложена, т.е. предпочтительные заместители в R1, объединенные с каждым предпочтительным заместителем в одном или нескольких из R2-R6 и Y/Y'/m/m' и т.д. Отдельные примеры таких комбинаций включают:
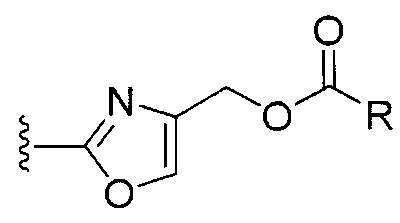
[0072] Ia. Оксазол, производные 4-оксазоила со сложными эфирами, отличными от сложного метилового эфира, в положении 4:
R1 представляет собой C1-C4 алкил, в частности, изопропил или трет-бутил,
R2, R4 и R6 представляют собой H,
R3 представляет собой замещенный метил формулы (-CRaRbRc), где Ra представляет собой OH, Rb представляет собой H, и Rc представляет собой изопропил или трет-бутил,
R5 представляет собой
 ,
,
где R представляет собой H, C1-C4 алкил или C1-C4 алкилокси, в частности, метил, H или метокси, и
Y представляет собой F и/или Cl, предпочтительно F, в положении 5 и/или 7, предпочтительно 5,
m равняется 0, 1 или 2, предпочтительно 0 или 1, и
m' равняется 0.
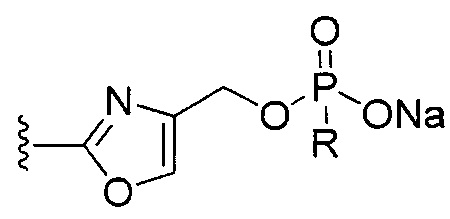
[0073] Ib. Оксазол, производные 4-оксазоила с фосфатными сложными эфирами в положении 4:
R1 представляет собой C1-C4 алкил, в частности, изопропил или трет-бутил,
R2, R4 и R6 представляют собой H,
R3 представляет собой замещенный метил формулы (-CRaRbRc), где Ra представляет собой OH, Rb представляет собой H, и Rc представляет собой изопропил или трет-бутил,
R5 представляет собой
 ,
,
где R представляет собой H, C1-C4 алкил, в частности, метил или H, и
Y представляет собой F и/или O, предпочтительно F, в положении 5 и/или 7, предпочтительно 5,
m равняется 0, 1 или 2, предпочтительно 0 или 1, и
m' равняется 0.
[0074] II. Оксазол, производные 4-оксазоила со спиртом или кетоном в положении 4:
R1 представляет собой C1-C4 алкил, в частности, изопропил или трет-бутил,
R2, R4 и R6 представляют собой H,
R3 представляет собой замещенный метил формулы (-CRaRbRc), где Ra представляет собой OH, Rb представляет собой H, и Rc представляет собой изопропил или трет-бутил,
R5 представляет собой
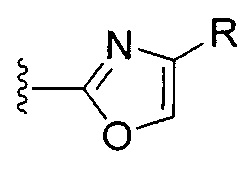 ,
,
где R представляет собой гидроксил, или C1-C4 спирт, или C1-C4 кетон, в частности, гидроксил, гидроксиметил, 1-гидроксиэтил или 1-гидроксиизопропил, и
Y представляет собой F и/или Cl, предпочтительно F, в положении 5 и/или 7, предпочтительно 5,
m равняется 0, 1 или 2, предпочтительно 0 или 1, и
m' равняется 0.
[0075] III. Оксазол, производные 4-оксазоила с амидом, амином, карбаматом или сульфонамидом в положении 4:
R1 представляет собой C1-C4 алкил, в частности, изопропил или трет-бутил,
R2, R4 и R6 представляют собой H,
R3 представляет собой замещенный метил формулы (-CRaRbRc), где Ra представляет собой OH, Rb представляет собой H, и Rc представляет собой изопропил или трет-бутил,
R5 представляет собой
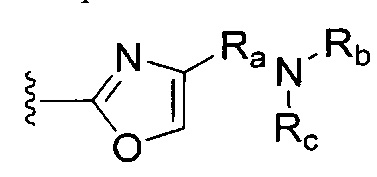 ,
,
где Ra представляет собой необязательно замещенный C0-C4 алкил, Rb и Rc независимо представляют собой H, C1-C8 алкил, C2-C8 алкенил, C6-C12 арил или гетероформу одного из них, каждый из которых может быть необязательно замещенным, в частности, где Ra представляет собой C0- или C1 алкил, Rb представляет собой H, и Rc представляет собой H, метил, сложный метиловый эфир, метилсульфонил или фенилсульфонил, и
Y представляет собой F и/или Cl, предпочтительно F, в 5 и/или 7, предпочтительно 5, m равняется 0, 1 или 2, предпочтительно 0 или 1, и
m' равняется 0.
[0076] IV. Оксазольное, производные 4-оксазоила с циано в положении 4:
R1 представляет собой C1-C4 алкил, в частности, изопропил или трет-бутил,
R2, R4 и R6 представляют собой H,
R3 представляет собой замещенный метил формулы (-CRaRbRc), где Ra представляет собой OH, Rb представляет собой H, и Rc представляет собой изопропил или трет-бутил,
R5 представляет собой
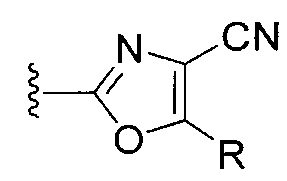 ,
,
где R представляет собой H, C1-C8алкил, C2-C8алкенил, C6-C12 арил или гетероформу одного из них, каждый из которых может быть необязательно замещенным, в частности, где R представляет собой H, метил, или NHAc, и
Y представляет собой F и/или Cl, предпочтительно F, в положении 5 и/или 7, предпочтительно 5,
m равняется 0, 1 или 2, предпочтительно 0 или 1, и
m' равняется 0.
[0077] V. Оксазол, производные 4-оксазоила с гетероциклом в положении 4:
R1 представляет собой C1-C4 алкил, в частности, изопропил или трет-бутил,
R2, R4 и R6 представляют собой H,
R3 представляет собой замещенный метил формулы (-CRaRbRc), где Ra представляет собой OH, Rb представляет собой H, и Rc представляет собой изопропил или трет-бутил,
R5 представляет собой
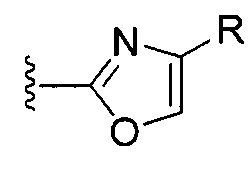 ,
,
где R представляет собой C3-C8 гетероциклил, C4-C12 гетероциклилалкил, C5-C12 гетероарил или C6-C14 гетероарилалкил, каждый из которых может быть необязательно замещенным, в частности, необязательно замещенное оксазольное, оксазолиновое, тиазольное, тиазолиновое, пиразольное, пиразолиновое, имидазольное, имидазолиновое, пиррольное, пирролиновое, изоксазольное, изоксазолиновое, изотиазольное, изотиазолиновое, оксадиазольное, тиадиазольное, триазольное или тетразольное кольцо, где предпочтительными заместителями являются галоген, нитро, циано, или необязательно фторированный C1-C4 алкил, необязательно фторированный C1-C4 алкокси, COOR8,
Y представляет собой F и/или О, предпочтительно F, в положении 5 и/или 7, предпочтительно 5,
m равняется 0, 1 или 2, предпочтительно 0 или 1, и
m' равняется 0.
[0078] Если в химические структуры включены хиральные углероды, кроме случаев, когда отображена конкретная ориентация, подразумевается, что охватываются обе стереоизомерные формы. Соединения формулы (I) и их раскрытые варианты осуществления могут, например, иметь два или больше центров асимметрии и поэтому существуют в разных энантиомерных и/или диастереоизомерных формах. Все оптические изомеры и стереоизомеры соединений, описанных в данном документе, и их смеси рассматриваются охваченными объемом настоящего изобретения, включая рацемат, одну или несколько энантиомерных форм, одну или несколько диастереоизомерных форм или их смеси. В частности, рацемические смеси отдельных диастереомеров, которые описаны, диастереомеров, характеризующихся диастереомерным избытком (d.e.) больше 90% или больше примерно 95%, и энантиомеров, характеризующихся энантиомерным избытком (e.e.) больше 90% или больше примерно 95%. Аналогично, если присутствуют двойные связи, соединения в некоторых случаях могут находиться в виде либо цис-, либо транс-изомеров; настоящее изобретение включает каждый изомер отдельно, а также смеси изомеров. Если описанные соединения могут также находиться в таутомерных формах, настоящее изобретение относится к применению всех таких таутомеров и их смесей.
[0079] Соединения формулы (I) и их раскрытые варианты осуществления могут поставляться в форме свободного основания, или поставляться как фармацевтически приемлемая соль, или как смесь формы свободного основания и соответствующей соли. Соединения по настоящему изобретению могут быть выделены как соли, где присутствует ионизируемая группа, такая как основный амин или карбоновая кислота. Настоящее изобретение включает соли таких соединений, которые имеют фармацевтически приемлемые противоионы. Такие соли хорошо известны в области техники и включают, например, соли кислотных групп, что образованы путем реакции с органическими или неорганическими основаниями, и соли основных групп, что образованы путем реакции с органическими или неорганическими кислотами, при условии, что противоионы, введенные путем реакции, приемлемы для фармацевтических применений. Примеры неорганических оснований включают гидроксиды щелочных металлов (например, гидроксид натрия, гидроксид калия и т.д.), гидроксиды щелочноземельных металлов (например, кальция, магния и т.д.) и гидроксиды алюминия, аммония и т.д. Примеры органических оснований, которые можно применять, включают триметиламин, триэтиламин, пиридин, пиколин, этаноламин, диэтаноламин, триэтаноламин, дициклогексиламин, N,N'-дибензилэтилендиамин и т.д.
[0080] Подходящие соли включают соли неорганических кислот, такие как гидрохлориды, гидробромиды, сульфаты, гидросульфаты и т.п., или соли присоединения органических кислот. Примеры неорганических кислот, которые можно применять, включают соляную кислоту, бромистоводородную кислоту, азотную кислоту, серную кислоту, фосфорную кислоту и т.д. Примеры органических кислот включают муравьиную кислоту, щавелевую кислоту, уксусную кислоту, винную кислоту, метансульфоновую кислоту, бензолсульфоновую кислоту, яблочную кислоту, метансульфоновую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту и т.д. Также включены соли с основными аминокислотами, такими как аргинин, лизин, орнитин и т.д., и соли с кислотными аминокислотами, такими как аспарагиновая кислота, глутаминовая кислота и т.д. Фармацевтически приемлемые соли могут быть получены путем смешивания соединений по изобретению с органическим или неорганическим основанием либо с органической или неорганической кислотой любым способом, известным в области техники.
[0081] Кроме того, соединения формулы (I) и их раскрытые варианты осуществления могут быть связаны или конъюгированы с фрагментами, такими как нацеливающее средство. К таким нацеливающим средствам относятся антитела или их иммунологически активные части, в том числе формы одноцепочечных антител, направленные против антигенов опухоли или против рецепторов или интегринов, ассоциированных с опухолями, пептидомиметики, направленные против таких фрагментов, и т.п. Кроме того, соединения формулы (I) и их раскрытые варианты осуществления могут быть связаны или конъюгированы с наполнителем, например полимерным наполнителем, таким как полиэтиленгликоль, для изменения фармакокинетики, как описано в теме номера Advanced Drug Delivery Reviews (выпуск 61, ноябрь 2009 года) под названием Polymer Therapeutics: Clinical Applications and Challenges for Development, including Pasut and Veronese, Adv Drug Delivery Rev 61 (13): 1177-1188, 2009. Выбранный PEG может быть с любой подходящей молекулярной массой, и может быть линейным или разветвленным, и необязательно может быть конъюгированным посредством линкера. Средняя молекулярная масса PEG будет предпочтительно находиться в диапазоне от примерно 2 килодальтон (кДа) до примерно 100 кДа, более предпочтительно от примерно 5 кДа до примерно 40 кДа.
[0082] Соединения формулы (I) и их раскрытые варианты осуществления пригодны для лечения или уменьшения интенсивности клеточных пролиферативных заболеваний. В частности, соединения и способы, описанные в данном документе, пригодны для обработки или уменьшения интенсивности опухолей и злокачественных новообразований, ассоциированных с молочной железой, яичником, легким (SCLC и NSCLC), толстой кишкой, прямой кишкой, предстательной железой, яичками, кожей (например, меланома, базальноклеточная карцинома и плоскоклеточная карцинома), поджелудочной железой, печенью, почкой, головным мозгом (например, глиома, менингиома, невринома и гранулобластома), кровью и кроветворной системой, включая, например, лейкоз, неходжкинскую лимфому и множественную миелому.
[0083] В описанных в данном документе способах, например, может быть уменьшена клеточная пролиферация, и/или может быть вызвана гибель клеток, такая как апоптоз или апоптическая гибель клеток. Клеточным пролиферативным нарушением у субъекта, являющегося человеком или животным, может быть опухоль или рак без образования опухоли.
[0084] Соединения и способы, предусмотренные в данном документе для уменьшения клеточной пролиферации, и/или чтобы вызвать гибель клеток, могут применяться отдельно, или совместно с, или в комбинации с хирургическими, лучевыми, химиотерапевтическими способами, иммунотерапией и способами трансплантации костного мозга и/или стволовых клеток или с другими паллиативными средствами, такими как соединения, которые помогают при лечебном питании или улучшают общее состояние здоровья, противорвотные вещества и т.п.
[0085] В некоторых вариантах осуществления соединения по настоящему изобретению вводят в комбинации с химиопрепаратом и применяют для уменьшения клеточной пролиферации, чтобы вызвать гибель клеток и/или для лечения или уменьшения интенсивности клеточных пролиферативных нарушений.
[0086] Описанные в данном документе соединения также пригодны против устойчивых к конкретным лекарственным средствам опухолей и линий раковых клеток, в частности, против видов рака, которые устойчивы к TAXOL® и/или противораковых средств на основе алкалоида барвинка.
[0087] Если вводят дополнительное химиотерапевтическое лекарственное средство, оно, как известно, обладает цитостатической, цитотоксической или противоопухолевой активностью. Такие средства включают без ограничения антиметаболиты (например, цитарабин, флударагин, 5-фтор-2'-дезоксиуридин, гемцитабин, гидроксимочевину, метотрексат); средства, активные в отношении ДНК (например, блеомицин, хлорамбуцил, цисплатин, циклофосфамид); интеркалирующие средства (например, адриамицин и митоксантрон); ингибиторы белкового синтеза (например, L-аспарагиназу, циклогексимид, пуромицин); ингибиторы топоизомеразы I типа (например, камптотецин, топотекан или иринотекан); ингибиторы топоизомеразы II типа {например, этопозид, тенипозид, антрахиноны, антрациклины и подофиллотоксин); ингибиторы микротрубочек (например, таксаны, такие как паклитаксел и доцетаксел, колцемид, колхицины или алкалоиды барвинка, такие как винбластин и винкристин); ингибиторы киназ (например, флавопиридол, стауроспорин и гидроксистауроспорин), лекарственные средства, действующие на Hsp90 {например, гелданомицин и производные гелданомицина, радицикол, производные пурина и антитела или фрагменты антител, которые селективно связываются с Hsp90), TRAIL, антитело к рецептору TRAIL, TNF-α или TNF-β и/или лучевую терапию.
[0088] В некоторых предпочтительных вариантах осуществления дополнительное терапевтическое средство против рака представляет собой TRAIL, антитело к рецептору TRAIL, TNF-α или TNF-β. В других предпочтительных вариантах осуществления дополнительные лекарственные средства для совместного ведения с соединениями по настоящему изобретению действуют на Hsp90 (белок теплового шока 90).
[0089] Подходящие ингибиторы Hsp90 включают производные анзамицина, такие как гелданомицин и производные гелданомицина, в том числе 17-(аллиламино)-17-дезметоксигелданамицин (17-AAG), его дигидро-производное, 17-AAGH2 и 17-амино-производные гелданамицина, такие как 17-диметиламиноэтиламино-17-деметокси-гелданамицин (17-DMAG), 11-оксогелданамицин и 5,6-дигидрогелданамицин, которые раскрыты в патентах США №4261989; 5387584 и 5932566, каждый из которых включен в данный документ посредством ссылки. Другие подходящие ингибиторы Hsp90 включают радицикол, и оксимы, и другие их аналоги, раскрытые в Soga, et al., Curr. Cancer Drug Targets, 3, 359-69 (2003), и в Yamamoto, et al., Angew. Chem., 42, 1280-84 (2003); и в Moulin, et al., J. Amer. Chem. Soc., vol 127, 6999-7004 (2005); производные пурина, такие как PU3, PU24FCI и PUH64 (смотри Chiosis et al., ACS Chem. Biol. Vol.1(5), 279-284 (2006) и раскрытые в PCT заявке № WO 2002/0236075; родственные гетероциклические производные, раскрытые в РСТ заявке № WO 2005/028434; и 3,4-диарилпиразольные соединения, раскрытые в Cheung, et al., Bioorg. Med. Chem. Lett., vol.15, 3338-43 (2005). Антитела или части антител, которые селективно связываются с Hsp90, также могут вводиться в качестве лекарственных средств, чтобы вызвать ингибирование Hsp90, и могут применяться в комбинации с соединениями по настоящему изобретению.
[0090] Если описанное в данном документе соединение используют совместно с, или в комбинации с другим терапевтическим средством, два средства могут быть введены совместно или они могут быть введены раздельно, если их введение выдержано по времени так, чтобы два средства действовали одновременно или последовательно.
[0091] Соответственно, композиции, применяемые в описанных в данном документе способах, включают по меньшей мере одно соединение по настоящему изобретению и могут необязательно включать одно или несколько дополнительных цитотоксических или цитостатических терапевтических средств, без ограничения таких как те, что раскрыты выше. Аналогично, способы по настоящему изобретению включают способы, где субъекта, у которого установили необходимость лечения рака, лечат с помощью по меньшей мере одного соединения или композиции по настоящему изобретению, и одновременно или параллельно с этим лечат с помощью одного или нескольких дополнительных терапевтических средств, описанных выше.
[0092] Составление и введение
[0093] Составы, пригодные в настоящем изобретении, включают стандартные составы, такие как изложенные в Remington’s Pharmaceutical Sciences, последнем издании, Mack Publishing Co., Easton, PA, включенном в данный документ посредством ссылки. Такие составы включают разработанные для пероральной доставки, замедленного высвобождения, местного введение, парентерального введения или любого другого подходящего пути, что определяет лечащий врач или ветеринар. Таким образом, введение может быть системным или локальным. Подходящие среды или наполнители включают липосомы, мицеллы, наночастицы, полимерные матрицы, буферы и весь ассортимент составов, известных практикующим врачам.
[0094] Системные составы включают те, которые разработаны для введения путем инъекции (например, внутримышечной, внутривенной или подкожной инъекции) и полученные для трансдермального, чресслизистого или пероального введения. Состав обычно будет включать разбавитель, а также, в некоторых случаях, адъюванты, буферы, консерванты и т.п. Соединения могут быть введены также в липосомальных композициях или в виде микроэмульсий.
[0095] Способы введения путем инъекции иногда являются подходящими путями введения соединений для системных обработок, а иногда также для локализованных обработок. Такие включают способы внутривенной, внутримышечной, подкожной и другие способы внутренней доставки, с помощью которых проходят слизистый и кожный барьеры для доставки композиции непосредственно в живые ткани субъекта.
[0096] Для инъекции составы могут быть получены в общепринятых формах в виде жидких растворов или суспензий, или в виде твердых форм, подходящих для раствора или суспензии в жидкости перед инъекцией, или в виде эмульсий. Подходящие наполнители включают, например, воду, солевой раствор, декстрозу, глицерин и т.п. Такие композиции могут также включать некоторые количества нетоксических вспомагательных веществ, таких как смачивающие или эмульгирующие средства, влияющие на pH буферные средства и т.п., такие как, например, ацетат натрия, сорбитанмонолаурат и прочие.
[0097] Также были разработаны различные системы с замедленным высвобождением для лекарственных средств и могут использоваться с соединениями по настоящему изобретению. Смотри, например, патент США №5624677. При необходимости имеющиеся композиции могут быть использованы в таких системах доставки с контролируемым высвобождением.
[0098] Системное введение может также включать сравнительно неинвазивные способы, такие как применение суппозиториев, трансдермальных пластырей, чресслизистой доставки и интраназального введения. Пероральное введение также подходит для соединений по настоящему изобретению. Подходящие формы включают сиропы, капсулы, таблетки и т.п., как известно в данной области техники.
[0099] Отбор конкретного пути введения для данного субъекта и индикации находится в пределах квалификации простого специалиста в данной области. Например, часто целесообразна ректальная доставка в виде суппозитория, если субъект испытывает тошноту и рвоту, что исключает возможность эффективной пероральной доставки. Трансдермальные пластыри обычно способны осуществлять доставку дозы с контролируемым высвобождением в течение нескольких дней или в конкретное место и, поэтому, подходят для субъектов, для которых желательны такие эффекты.
[00100] Чресслизистая доставка также подходит для некоторых композиций и способов по настоящему изобретению. Таким образом, композиции по настоящему изобретению могут быть введены чресслизисто с применением методики и способов составления, известных в данной области техники.
[00101] Независимо от выбранного пути введения, описанные в данном документе соединения, которые могут применяться в подходящей гидратной форме, и/или фармацевтические композиции по настоящему изобретению составляют в фармацевтически приемлемые лекарственные формы посредством общепринятых способов, известных специалистам в данной области.
[00102] Фактические уровни дозировки активных ингредиентов в фармацевтических композициях по настоящему изобретению могут изменяться так, чтобы получить количество активного ингредиента, которое является эффективным для достижения необходимого терапевтического ответа у конкретного пациента, композицию и способ введения, не оказывающих токсичности на пациента.
[00103] Выбранный уровень дозы будет зависеть от ряда факторов, включая активность конкретного используемого соединения по настоящему изобретению или его сложного эфира, соли или амида, путь введения, время введения, скорость экскреции или метаболизма конкретного используемого соединения, скорость и степень абсорбции, продолжительность обработки, другие лекарственные средства, соединения и/или материалы, применяемые в комбинации с конкретным используемым соединением, возраст, пол, вес, состояние, общее состояние здоровья, и прежнюю историю болезни подлежащего лечению пациента, и подобные факторы, хорошо известные в областях медицины.
[00104] Для введения субъектам, являющимся животным или человеком, доза соединения по настоящему изобретению составляет, как правило, 10-2400 мг за введение. Однако уровни дозы очень зависят от природы состояния, состояния пациента, принимаемого практикующим врачом решения и кратности и способа введения. Выбор дозы таких соединений находится в пределах квалификации обычного специалиста и может быть осуществлен, начиная с довольно низкой дозы и увеличивая дозу, пока не будет достигнут приемлемый эффект.
[00105] Кратность введения соединений по настоящему изобретению также может быть легко определена специалистом в данной области с применением хорошо известных методик. Например, пациенту может быть введена низкая доза соединения или композиции по настоящему изобретению при малой повторяемости, такой как один раз в день или реже; и доза и/или кратность введения могут систематично увеличиваться, пока у пациента не будет достигнут требуемый эффект.
[00106] Способы синтеза
[00107] Исследуемые соединения были получены посредством эффективного многостадийного способа, как показано на схеме 1. Ключевой этап в способе включает электрохимическое окислительное замыкание кольца фенольного промежуточного соединения с получением индолинового соединения формулы (I), которое может быть дополнительно функционализированно на примере описанных в данном документе соединений. Окислительное замыкание кольца было описано в заявке США с серийным №12/134984, поданной 6 июня, 2008 года, и опубликованной как US 2009/0005572.
[00108] Как показано на схеме 1, дипептидные исходные материалы получали при стандартных условиях, известных в данной области техники, например, путем присоединения N-гидроксисукцинимидного сложного эфира или другого активированного сложного эфира защищенной аминокислоты к серину. Специалисту в данной области будет понятно, что для образования дипептидных исходных материалов можно применять широкое разнообразие подходящих условий, включая обширную совокупность литературы, описывающей синтез пептидов и пептидомиметиков.
[00109] Дипептид приводили в реакцию с необязательно замещенным индолом и активирующим реагентом, необязательно в присутствии протонсодержащей кислоты, для обеспечения индолсодержащего дипептида. Подходящие активирующие реагенты включают, например, ангидриды карбоновых кислот, смешанные ангидриды, или ацилгалогениды (например, уксусный ангидрид, трифторуксусный ангидрид, ацетилхлорид, оксалилхлорид), ангидриды сульфоновой кислоты или галогениды (например, метансульфоновый ангидрид, трифторметансульфоновый ангидрид, метансульфонилхлорид), галогениды минеральных кислот (например, тионилхлорид или фосфорилхлорид) и т.п.
[00110] В предпочтительном варианте осуществления активирующее средство представляло собой уксусный ангидрид, и реакцию проводили в уксусной кислоте в качестве протонного растворителя. В особенно предпочтительном варианте осуществления дипептид и необязательно замещенный индол приводили в реакцию с уксусным ангидридом в уксусной кислоте при примерно 80°C с получением требуемого соединения.
[00111] О получении N-ацетилтриптофановых производных путем реакции серина или N-ацетилсерина и необязательно замещенного индола в уксусном ангидриде и уксусной кислоте сообщалось ранее Y. Yokoyama, et al., Tetrahedron Letters (1999), 40: 7803; Y. Yokoyama, et al., Eur. J. Org. Chem. (2004), 1244; Y. Konda-Yamada, et al., Tetrahedron (2002), 58: 7851; M.W. Orme, et al., US 6,872,721. Однако получение других ацилированных триптофановых производных при этих условиях, таких как дипептидные аналоги по настоящему изобретению, насколько известно, ранее описаны не были.
[00112] Эстерификация свободной карбоновой кислоты с последующим окислительным замыканием цикла дипептидного промежуточного соединения с окисляющим средством, например, DDQ, предоставила оксазольное промежуточное соединение. Специалистам в данной области будет понятно, что могут использоваться другие окислительные условия, такие как, например, применение 7,7,8,8-тетрацианохинодиметана (TCNQ), цериевого нитрата аммония, гипервалентных йодистых реагентов и т.п.
[00113] Снятия защиты с защищенной аминогруппы, если присутствует, и образование амидной связи предусматривает фенольное промежуточное соединение. Электрохимическим окислительным замыканием цикла фенольного промежуточного соединения предусмотрено макроциклическое индолиновое соединение. Такие соединения дополнительно освещены для соединений формулы (I) посредством серии несложных химических превращений. Например, удаление Cbz-группы и ацилирование или образование амидной связи применяли для получения соединений формулы (I), где R5 представляет собой заместитель, являющийся ацилом, например -C(O)R3. Специалисту в данной области будет понятно, что порядок таких этапов может быть изменен на обратный в зависимости от природы вводимых функциональных групп и используемых защитных групп.
[00114] Схема 1 предусматривает общий путь синтеза, пригодный для получения макроциклического индолинового соединения формулы (I). Специалистам в данной области должно быть понятно, что конкретные условия реакции могут варьировать без изменения сущности настоящего изобретения. Например, реакции присоединения могут быть выполнены с множеством активированных сложных эфиров, а именно, только в качестве примера, N-гидроксибензотриазольным сложным эфиром, перфторфенильным сложным эфиром, N-гидроксифталимидным сложным эфиром, активированными сложными эфирами, образованными путем реакции карбоновой кислоты с карбодиимидом, и другими активированными сложными эфирами, обычно применяемыми для ацилирования амина с образованием амидных связей. Кроме того, специалисту в данной области будет понятно, что пока аминогруппы подходящим образом защищены как карбобензилокси (Cbz) группа, то могут использоваться другие подходящие защитные группы. Подходящие защитные группы и способы их присоединения и снятия хорошо известны в области техники и описаны, например, в Т.Н. Greene, Protective Groups in Organic Synthesis, 2-е изд.
Схема 1.
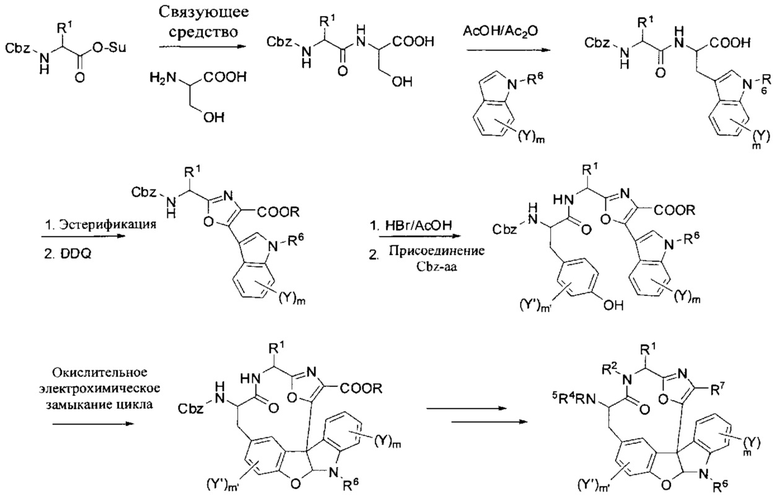
[00115] Описанный на схеме 1 способ применим для получения индолинов формулы (I) с высоким выходом и чистотой. В частности, соединения настоящего изобретения доступны с хорошим выходом и с высокой диастереоизомерной чистотой, предпочтительно с диастереомерным избытком больше 95%, иногда 98% диастереомерным избытком.
[00116] Нижеследующие примеры представлены для пояснения, а не ограничения настоящего изобретения.
[00117] Примеры
[00118] Синтез соединения 57
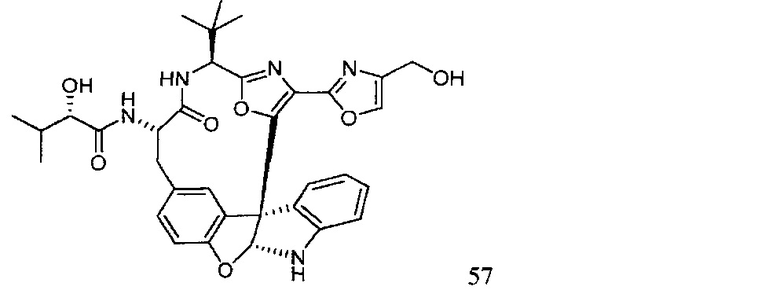
[00119] Этап 1
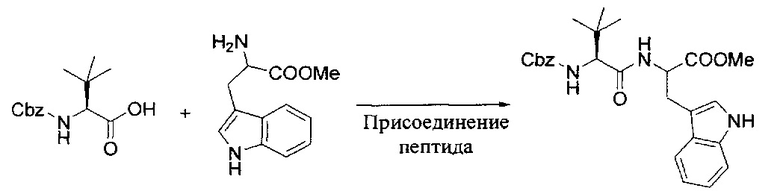
[00120] В сухую 100-мл колбу с магнитной мешалкой добавляли Cbz-L-α-трет-бутилглициновую DCHA соль (5,0 г, 11,2 ммоль), гидрохлорид сложного метилового эфира L-триптофана (3,14 г, 12,3 ммоль, 1,1 экв.), HOBt (1,76 г, 13,4 ммоль, 1,2 экв.), безводный DMF (30 мл) и N,N-диизопропилэтиламин (2,93 мл, 16,8 ммоль, 1,5 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (2,58 г, 13,4 ммоль, 1,2 экв.). Полученную в результате реакционную смесь перемешивали при RT в течение 16 часов. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (300 мл)/вода (100 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×50 мл). Объединенные органические слои промывали водой (100 мл), 10% водным NaHSO4 (100 мл), водой (100 мл), насыщенным NaHCO3 (100 мл) и солевым раствором (2×100 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00121] Этап 2
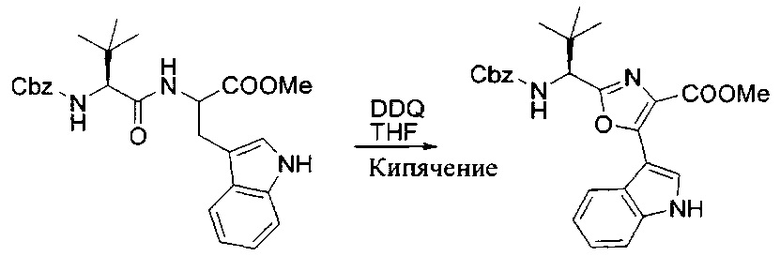
[00122] Раствор DDQ (6,2 г, 27,3 ммоль, 2,4 экв.) в THF (100 мл) добавляли к кипящему с обратным холодильником раствору соединения, синтезированного на этапе 1 выше (11,2 ммоль), в THF (200 мл) и темный раствор кипятили на масляной бане при 85°C в течение 1 часа. После охлаждения растворитель извлекали на роторном испарителе. Остаток растворяли в этилацетате (500 мл), который промывали водой (200 мл), водным насыщенным NaHCO3 (2×200 мл), водой (2×200 мл), солевым раствором (100 мл) и сушили над Na2SO4. После концентрирования смесь очищали с помощью колоночной флэш-хроматографии (20% EtOAc в CH2Cl2). Это дало 3,24 г (64% выход) продукта.
[00123] Этап 3
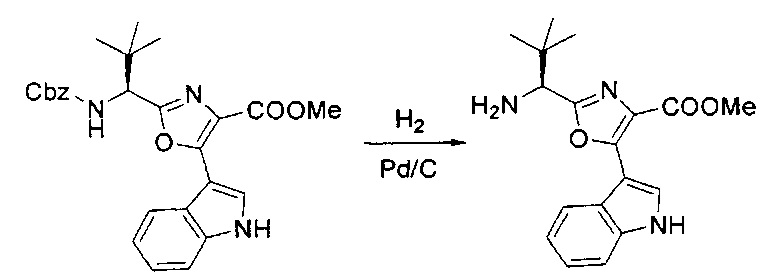
[00124] В 100-мл колбу, содержащую материал, синтезированный на этапе 2 выше (3,24 г, 7,02 ммоль), добавляли метанол (30 мл) и Pd/C (10%) (650 мг, 0,61 ммоль, 0,09 экв.) под N2. Подводили баллон с H2 и колбу продували H2 4 раза. Затем H2 баллон открывали в реакционную систему. После 3 часов перемешивания исходного материала практически не оставалось. Реакцию останавливали. Реакционную смесь фильтровали через подушку из целита и черный осадок промывали метанолом (3×10 мл). Фильтрат концентрировали, а остаток применяли непосредственно на следующем этапе без дополнительной очистки.
[00125] Этап 4
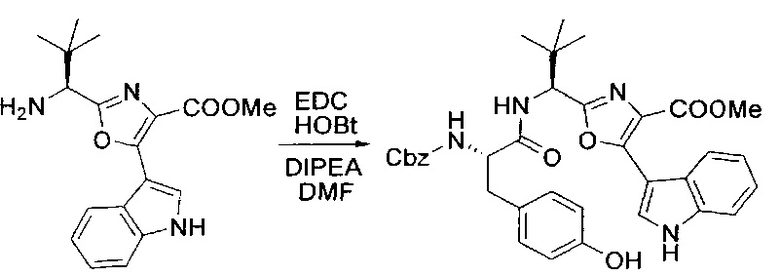
[00126] В сухую 100-мл колбу с магнитной мешалкой добавляли амин, синтезированный на этапе 3 (2,06 г, 6,29 ммоль), Cbz-L-тирозин (1,98 г, 6,91 ммоль, 1,1 экв.), HOBt (0,94 г, 6,91 ммоль, 1,1 экв.), безводный DMF (30 мл) и N,N-диизопропилэтиламин (1,31 мл, 7,54 ммоль, 1,2 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (1,33 г, 6,91 ммоль, 1,1 экв.). Полученную в результате реакционную смесь перемешивали при RT в течение 16 часов. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (300 мл Увода (100 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×50 мл). Объединенные органические слои промывали водой (100 мл), 10% водным NaHSO4 (100 мл), водой (100 мл), насыщенным NaHCO3 (100 мл) и солевым раствором (2×100 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00127] Этап 5
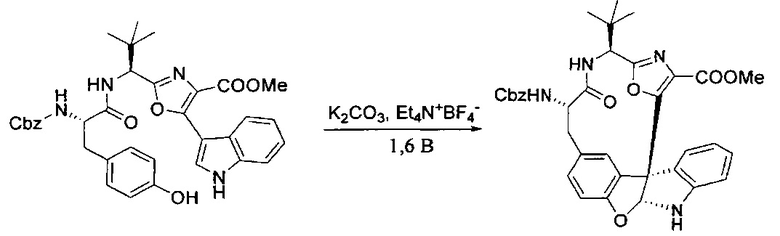
[00128] Электрохимическую ячейку компоновали с применением стеклянного цилиндра (диаметр 6 см × высота 11 см) и обычной подвески (полипропилен и нейлон), что удерживала 9 вертикальных графитовых стержней (диаметр 6,15 мм × длина 12 см). Стержни располагали в модель кольца из 6 анодов и 3 катодов. Электроды погружали на глубину 6,5 см. Фенольный материал, синтезированный на этапе 4 выше (5,00 г, 8,0 ммоль), Et4NBF4 (4,00 г, 18,4 ммоль, 2,9 экв.) и (NH4)2CO3 (1,0 г, 10,4 ммоль, 1,3 экв.) и ID воду (4 мл) добавляли в DMF (200 мл). Раствор энергично перемешивали на пластине для смешивания (приблиз. 600 rpm). Электрохимическую реакцию осуществляли при электрическом потенциале 1,5-1,6 вольт. Через 3 дня большая часть первоначального SM была поглощена, что определяли с помощью интегрирования ВЭЖХ при 220 нМ. Реакционную смесь концентрировали на роторном испарителе (температура бани ≤35°C) и дополнительно сушили на вакуумном коллекторе. Остаток разделяли между EtOAc (200 мл) и 0,5 н водной HCl (60 мл). Органический слой промывали насыщенным водным NaHCO3 (50 мл), а затем насыщенным водным NaCl (50 мл). Водные слои экстрагировали последовательно EtOAc (2×50 мл). Объединенные органические слои сушили (Na2SO4), декантировали и испаряли. Этот материал очищали с помощью колоночной флэш-хроматографии с 20% EtOAc в CH2Cl2. Это дало 1,24 г (24,8% выход) продукта в виде смеси стереоизомеров (71:29, как измерено с помощью интегрирования ВЭЖХ при 254 нМ).
[00129] Этап 6
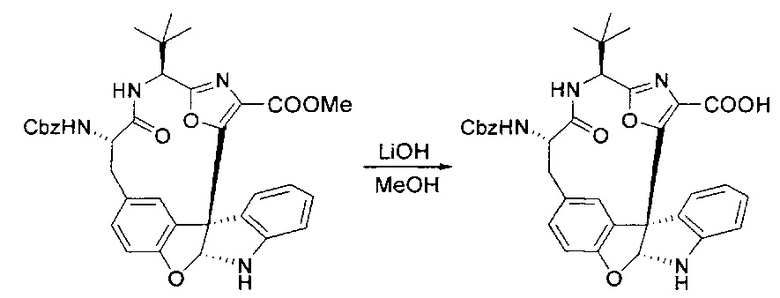
[00130] Соединение, синтезированное на этапе 6 (725 мг, 2,33 ммоль), растворяли в метаноле (22 мл) и раствор охлаждали на ледяной бане. В течение 5 минут добавляли раствор LiOH (558 мг, 23,3 ммоль, 10 экв.) в воде (7,0 мл). Ледяную баню убирали и смесь перемешивали в течение 18 часов. Смесь охлаждали на ледяной бане, добавляли воду (30 мл), а затем 1 н водную HCl (24 мл), поддерживая температуру реакционной смеси ниже 10°C. Смесь разделяли между водой (15 мл) и EtOAc (100 мл) и промывали органический слой насыщенным водным NaCl. Водные слои экстрагировали последовательно EtOAc (30 мл). Объединенные органические слои сушили (Na2SO4), декантировали и испаряли с получением продукта, представляющего собой кислоту, в виде чисто белых кристаллов.
[00131] Этап 7
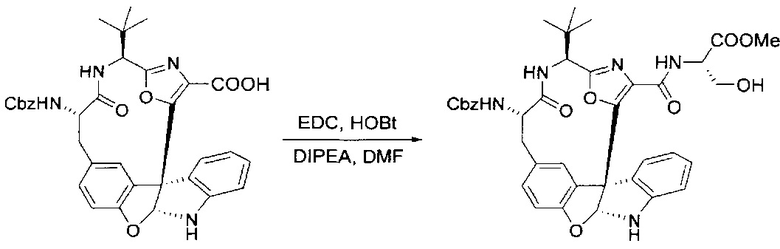
[00132] В сухую 100-мл колбу с магнитной мешалкой добавляли карбоновую кислоту, синтезированную на этапе 6 выше (2,33 ммоль), гидрохлорид сложного метилового эфира L-серина (435 мг, 2,8 ммоль, 1,2 экв.), HOBt (378 мг, 2,8 ммоль, 1,2 экв.), безводный DMF (25 мл) и N,N-диизопропилэтиламин (1,01 мл, 5,83 ммоль, 2,5 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (537 мг, 2,8 ммоль, 1,2 экв.). Полученную в результате реакционную смесь перемешивали при RT в течение 16 часов. Реакцию контролировали с помощью LCMS. Большую часть растворителей испаряли при пониженном давлении. Остаток разбавляли EtOAc (100 мл)/вода (30 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×20 мл). Объединенные органические слои промывали водой (40 мл), 10% водным NaHSO4 (40 мл), водой (40 мл), насыщенным NaHCO3 (40 мл) и солевым раствором (2×40 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00133] Этап 8
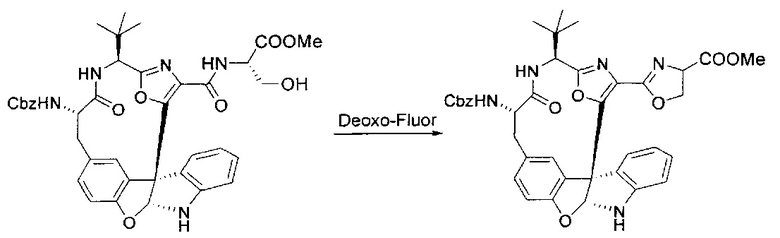
[00134] В сухую колбу добавляли неочищенный продукт этапа 8 выше (2,33 ммоль) и безводный CH2Cl2 (40 мл). Реакционный раствор становился мутным по мере того, как его охлаждали до -20°C на бане сухой лед/ацетон/вода. Свежеполученный основной раствор бис(2-метоксиэтил)аминосеры трифторида (0,644 мл, 0,022 ммоль, 2,8 экв.) в CH2Cl2 (4 мл) добавляли по каплям. Полученную в результате реакционную смесь перемешивали при -20°C в течение 1 часа и нагревали до комнатной температуры. Реакционную смесь гасили посредством добавления насыщенного водного NaHCO3 (20 мл), разбавляли EtOAc (100 мл), промывали водой (2×30 мл), а также солевым раствором (30 мл), и сушили над Na2SO4. После концентрирования остаток применяли на следующем этапе.
[00135] Этап 9
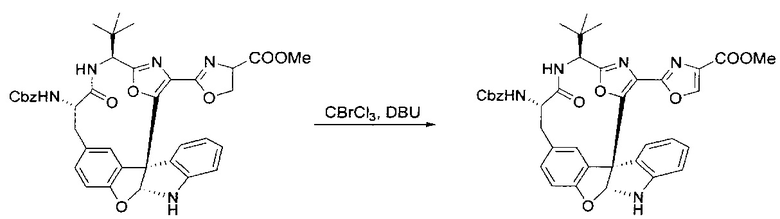
[00136] В сухую колбу, содержащую неочищенный продукт этапа 8 выше (2,33 ммоль), добавляли безводный CH2Cl2 (40 мл). Смесь охлаждали до 0°C. Затем добавляли CBrCl3 (0,345 мл, 3,5 ммоль, 1,5 экв.) и DBU (0,523 мл, 3,5 ммоль, 1,5 экв.), соответственно. Полученной в результате смеси давали возможность нагреться до комнатной температуры и перемешивали ее в течение 1 часа. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (100 мл), промывали 10% NaHSO4 (30 мл), водой (2×30 мл), насыщенным водным NaHCO3 (30 мл), водой (30 мл) и солевым раствором (30 мл), сушили над Na2SO4. После концентрирования остаток применяли на следующем этапе.
[00137] Этап 10
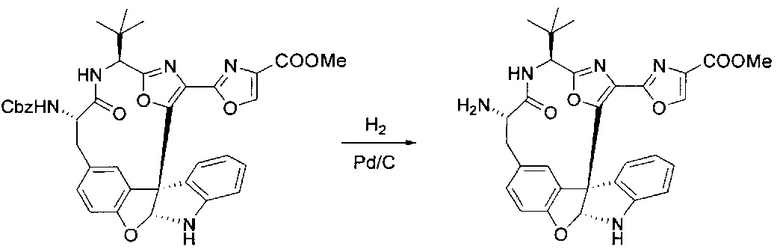
[00138] В 50-мл колбу, содержащую материал, синтезированный на этапе 9 выше (400 мг, 0,58 ммоль), добавляли метанол (15 мл), трет-бутиламин (0,086 мл, 0,87 ммоль, 1,5 экв.) и Pd/C (10%) (62 мг, 0,058 ммоль, 0,1 экв.) под N2. Подводили баллон с H2 и продували колбу H2 4 раза. Затем баллон с H2 открывали в реакционную систему. После 4 часов перемешивания исходного материала практически не оставалось. Реакцию останавливали. Реакционную смесь фильтровали через подушку из целита и черный осадок промывали метанолом (3×10 мл). Фильтрат концентрировали, а остаток применяли непосредственно на следующем этапе без дополнительной очистки.
[00139] Этап 11
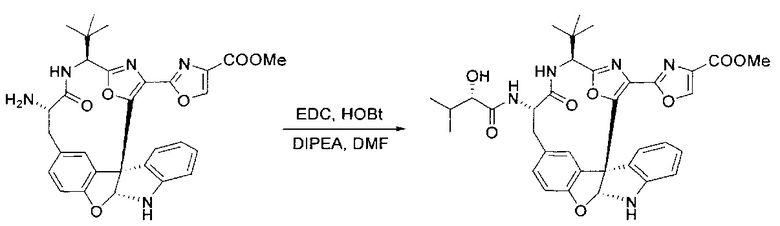
[00140] В сухую 25-мл колбу, содержащую амин, синтезированный на этапе 10 выше (0,58 ммоль), добавляли (S)-(+)-2-гидрокси-3-метилбутановую кислоту (82 мг, 0,696 ммоль, 1,2 экв.), HOBt (94 мг, 0,696 ммоль, 1,2 экв.), безводный DMF (8 мл) и N,N-диизопропилэтиламин (0,152 мл, 0,87 ммоль, 1,5 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (133 мг, 0,696 ммоль, 1,2 экв.). Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (80 мл)/вода (30 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×20 мл). Объединенные органические слои промывали водой (30 мл), 10% водным NaHSO4 (30 мл), водой (30 мл), насыщенным NaHCO3 (30 мл) и солевым раствором (2×30 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00141] Этап 12
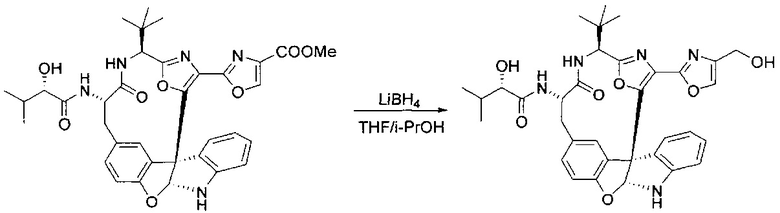
[00142] В сухую колбу добавляли неочищенный материал, синтезированный на этапе 11 (0,58 ммоль), THF (4 мл) и 2-пропанол (12 мл). Этот раствор охлаждали до 0°C с последующим добавлением твердого борогидрида лития (152 мг, 6,96 ммоль, 12 экв.). Полученной в результате смеси давали возможность нагреться до комнатной температуры и перемешивали в течении 22 часов. Реакцию контролировали с помощью LCMS. Исходного материала практически не оставалось. Реакционную смесь охлаждали до 0°C. Добавляли 2-пропанол (24 мл) и воду (40 мл) с последующим добавлением NH4Cl (3,1 г, 58 ммоль, 100 экв.). Реакционную смесь перемешивали в течение 1 часа и разбавляли EtOAc (250 мл)/вода (50 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×50 мл). Объединенные органические слои промывали водой (3×50 мл), 10% NaHSO4 (2×50 мл), водой (2×50 мл), насыщенным NaHCO3 (50 мл) и солевым раствором (2×50 мл), а затем сушили над Na2SO4. После концентрирования остаток очищали с помощью колоночной флэш-хроматографии (EtOAc до 10% EtOAc/MeOH) с получением целевого продукта в виде грязнобелого твердого вещества (188 мг, 0,108 ммоль, 52% за три этапа). MS: масса/заряд = 627,9 (М+1).
[00143] Синтез соединения 81
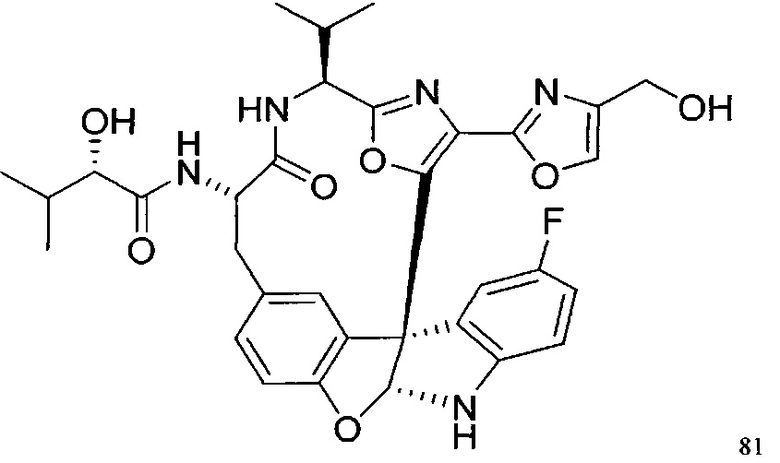
[00144] Этап 1
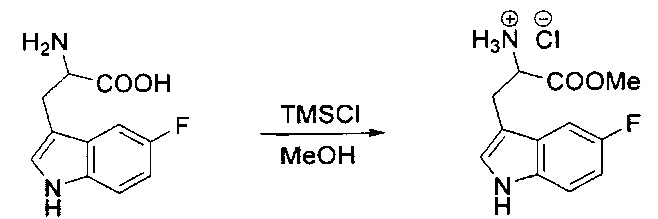
[00145] В сухую 250-мл колбу добавляли 5-фтор-DL-триптофан (5,0 г, 22,5 ммоль) и безводный метанол (120 мл). Суспензию охлаждали до 0°C с последующим добавлением хлортриметилсилана (12,8 мл, 101,3 ммоль, 4,5 экв.) с такой скоростью, чтобы поддерживать температуру реакционной смеси ниже 6°C. Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Реакцию контролировали с помощью TLC. Большую часть летучих веществ испаряли при пониженном давлении. Неочищенный продукт применяли на следующем этапе.
[00146] Этап 2
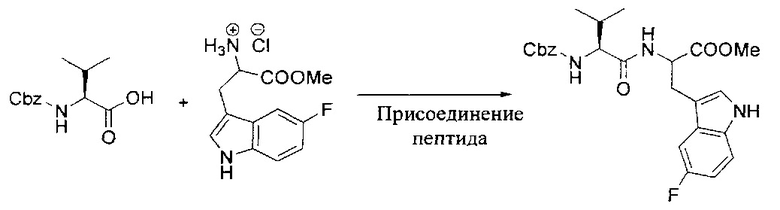
[00147] В сухую 250-мл колбу с магнитной мешалкой добавляли соль амина, синтезированную на этапе 1 выше (22,5 ммоль.), Cbz-L-валин (6,22 г, 24,75 ммоль, 1,1 экв.), HOBt (3,34 г, 24,75 ммоль, 1,1 экв.), безводный DMF (80 мл) и N,N-диизопропилэтиламин (11,8 мл, 67,5 ммоль, 3,0 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (4,74 г, 24,75 ммоль, 1,1 экв.). Полученную в результате реакционную смесь перемешивали при RT в течение 16 часов. Реакцию контролировали с помощью LCMS. Большую часть растворителей испаряли при пониженном давлении. Затем остаток разбавляли EtOAc (600 мл)/вода (200 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×50 мл). Объединенные органические слои промывали водой (100 мл), 10% водным NaHSO4 (100 мл), водой (100 мл), насыщенным NaHCO3 (100 мл) и солевым раствором (2×100 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00148] Этап 3
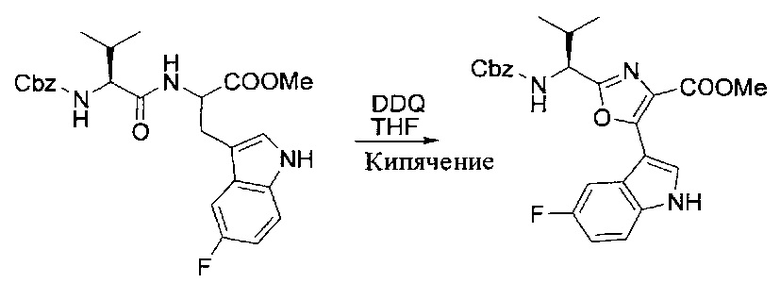
[00149] Раствор DDQ (12,8 г, 56,25 ммоль, 2,5 экв.) в THF (500 мл) добавляли к кипящему с обратным холодильником раствору соединения, синтезированного на этапе 2 выше (22,5 ммоль), в THF (250 мл) и темный раствор выдерживали при кипении на масляной бане при 85°C в течение 1 часа. После охлаждения растворитель извлекали на роторном испарителе. Остаток растворяли в этилацетате (600 мл) и добавляли NaHCO3 (13 г). Смесь перемешивали в течение 1 часа с последующим фильтрованием через воронку с пористым фильтром. Фильтрат промывали водой (200 мл), водным насыщенным NaHCO3 (2×200 мл), водой (2×200 мл), солевым раствором (100 мл) и сушили над Na2SO4. После концентрирования смесь очищали с помощью колоночной флэш-хроматографии (5% EtOAc в CH2Cl2). Это дало 4,63 г (44,2% выход) продукта.
[00150] Этап 4
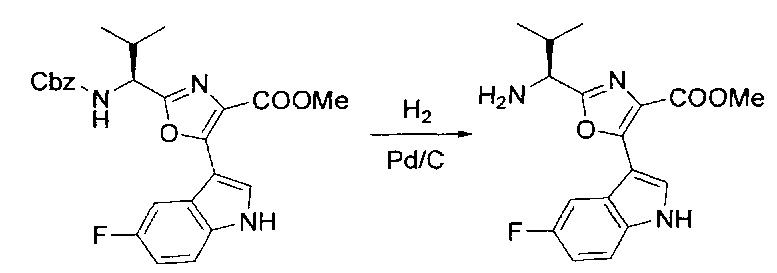
[00151] В 250-мл колбу, содержащую материал, синтезированный на этапе 3 выше (4,63 г, 9,94 ммоль), добавляли метанол (50 мл) и Pd/C (10%) (530 мг, 0,497 ммоль, 0,05 экв.) под N2. Подводили баллон с Н2 и продували колбу Н2 4 раза. Затем баллон с H2 открывали в реакционную систему. После 1 часа перемешивания исходного материала практически не оставалось. Реакцию останавливали. Реакционную смесь фильтровали через подушку из целита и черный осадок промывали метанолом (3×15 мл). Фильтрат концентрировали, а остаток применяли непосредственно на следующем этапе без дополнительной очистки.
[00152] Этап 5
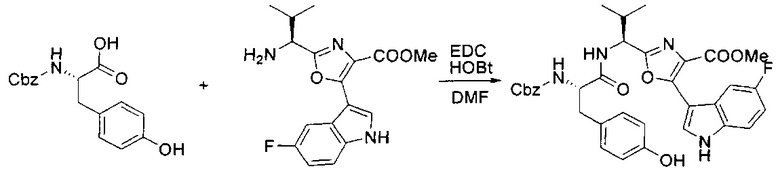
[00153] В сухую 100-мл колбу с магнитной мешалкой добавляли амин, синтезированный на этапе 4 (9,94 ммоль), Cbz-L-тирозин (3,45 г, 10,93 ммоль, 1,1 экв.), HOBt (1,48 г, 10,93 ммоль, 1,1 экв.), безводный DMF (30 мл). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (2,10 г, 10,93 ммоль, 1,1 экв.). Полученную в результате реакционную смесь перемешивали при RT в течение 16 часов. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (400 мл)/вода (150 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×100 мл). Объединенные органические слои промывали водой (200 мл), 10% водным NaHSO4 (150 мл), водой (150 мл), насыщенным NaHCO3 (150 мл) и солевым раствором (2×100 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт (6,58 г) применяли непосредственно на следующем этапе.
[00154] Этап 6
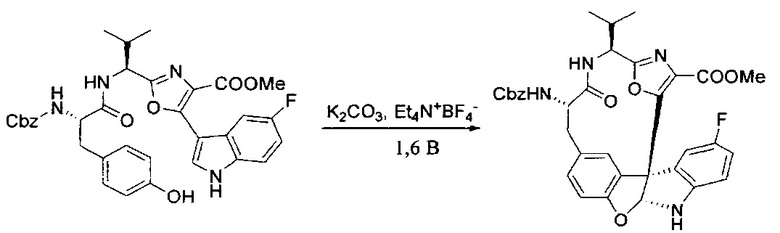
[00155] Электрохимическую ячейку компоновали с применением стеклянного цилиндра (диаметр 6 см × высота 11 см) и обычной подвески (полипропилен и нейлон), что удерживала 9 вертикальных графитовых стержней (диаметр 6,15 мм × длина 12 см). Стержни располагали в модель кольца из 6 анодов и 3 катодов. Электроды погружали на глубину 6,5 см. Фенольный материал, синтезированный на этапе 5 выше (2,00 г, 3,18 ммоль), Et4NBF4 (2,00 г, 9,2 ммоль, 3 экв.), K2CO3 (0,44 г, 3,18 ммоль, 1,0 экв.) и ID воду (4 мл) добавляли в DMF (200 мл). Раствор энергично перемешивали на пластине для смешивания (приблиз. 600 rpm). Электрохимическую реакцию осуществляли при электрическом потенциале 1,5-1,6 вольт. Через 3 дня большая часть первоначального SM была поглощена, что определяли с помощью интегрирования ВЭЖХ при 220 нМ. Электрохимическую реакцию повторяли 4 раза, чтобы поглотился весь фенольный материал, синтезированный на этапе 5. Объединенные реакционные смеси концентрировали на роторном испарителе (температура бани ≤35°C) и дополнительно сушили на вакуумном коллекторе. Остаток разбавляли EtOAc (500 мл) с последующим фильтрованием через воронку с пористым фильтром. Фильтрат промывали водой (2×200 мл), солевым раствором (200 мл). Водные слои экстрагировали последовательно EtOAc (2×50 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали. Этот материал очищали с помощью колоночной флэш-хроматографии с 15% MeCN в CH2Cl2. Это дало 900 мг целевого продукта при 14% выходе за три этапа.
[00156] Этап 7
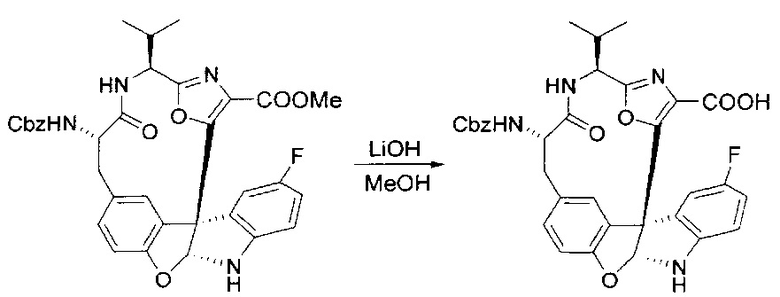
[00157] Соединение, синтезированное на этапе 6 (900 мг, 1,43 ммоль), растворяли в метаноле (28 мл) и раствор охлаждали на ледяной бане. В течение 5 минут добавляли раствор LiOH (344 мг, 14,3 ммоль, 10 экв.) в воде (4,5 мл). Ледяную баню убирали и смесь перемешивали при RT в течение 18 часов. Смесь охлаждали на ледяной бане, добавляли воду (40 мл), а затем 1 н водную HCl (14,5 мл), поддерживая температуру реакционной смеси ниже 10°C. Смесь разделяли между водой (25 мл) и EtOAc (200 мл) и промывали органический слой насыщенным водным NaCl. Водные слои экстрагировали последовательно EtOAc (50 мл). Объединенные органические слои сушили (Na2SO4), декантировали и испаряли с получением продукта, представляющего собой кислоту, в виде чисто белых кристаллов.
[00158] Этап 8
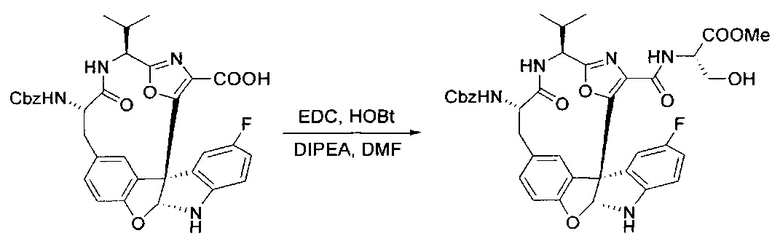
[00159] В сухую 50-мл колбу с магнитной мешалкой добавляли карбоновую кислоту, синтезированную на этапе 7 выше (1,43 ммоль), гидрохлорид сложного метилового эфира L-серина (268 мг, 1,72 ммоль, 1,2 экв.), HOBt (232 мг, 1,72 ммоль, 1,2 экв.), безводный DMF (15 мл) и N,N-диизопропилэтиламин (0,624 мл, 3,58 ммоль, 2,5 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (330 мг, 1,72 ммоль, 1,2 экв.). Полученную в результате реакционную смесь перемешивали при RT в течение 16 часов. Реакцию контролировали с помощью LCMS. Большую часть растворителей испаряли при пониженном давлении. Остаток разбавляли EtOAc (150 мл)/вода (50 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×30 мл). Объединенные органические слои промывали водой (60 мл), 10% водным NaHSO4 (60 мл), водой (60 мл), насыщенным NaHCO3 (60 мл) и солевым раствором (2×60 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00160] Этап 9
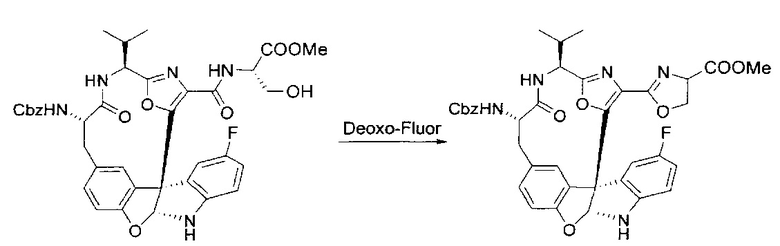
[00161] В сухую колбу добавляли неочищенный продукт этапа 8 выше (1,43 ммоль) и безводный CH2Cl2 (25 мл). Реакционный раствор становился мутным по мере того, как его охлаждали до -20°C на бане сухой лед/ацетон/вода. Свежеполученный основной раствор бис(2-метоксиэтил)аминосеры трифторида (0,395 мл, 2,15 ммоль, 1,5 экв.) в CH2Cl2 (4 мл) добавляли по каплям. Полученную в результате реакционную смесь перемешивали при -20°C в течение 1 часа и нагревали до комнатной температуры. Реакционную смесь гасили посредством добавления насыщенного водным NaHCOs (15 мл), разбавляли EtOAc (100 мл), промывали водой (2×20 мл), а также солевым раствором (30 мл), и сушили над Na2SO4. После концентрирования остаток применяли на следующем этапе.
[00162] Этап 10
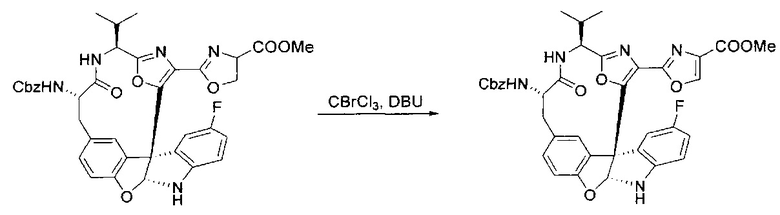
[00163] В сухую колбу, содержащую неочищенный продукт этапа 9 выше (1,43 ммоль), добавляли безводный CH2Cl2 (25 мл). Смесь охлаждали до 0°C. Затем добавляли CH2Cl3 (0,211 мл, 2,15 ммоль, 1,5 экв.) и DBU (0,321 мл, 2,15 ммоль, 1,5 экв.), соответственно. Полученной в результате смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 1 часа. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (100 мл), промывали 10% NaHSO4 (30 мл), водой (2×30 мл), насыщенным водным NaHCO3 (15 мл), водой (30 мл) и солевым раствором (30 мл), сушили над Na2SO4. После концентрирования остаток применяли на следующем этапе.
[00164] Этап 11
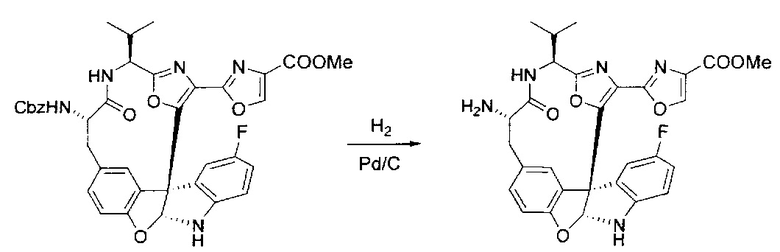
[00165] В 100-мл колбу, содержащую материал, синтезированный на этапе 10 выше (1,43 ммоль), добавляли метанол (20 мл), трет-бутиламин (0,226 мл, 2,15 ммоль, 1,5 экв.) и PaVC (10%) (152 мг, 0,143 ммоль, 0,1 экв.) под N2. Подводили баллон с H2 и продували колбу H2 4 раза. Затем баллон с H2 открывали в реакционную систему. После 4 часов перемешивания исходного материала практически не оставалось. Реакцию останавливали. Реакционную смесь фильтровали через подушку из целита и черный осадок промывали метанолом (3×15 мл). Фильтрат концентрировали, а остаток применяли непосредственно на следующем этапе без дополнительной очистки.
[00166] Этап 12
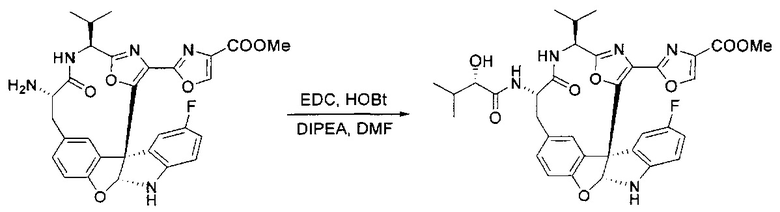
[00167] В сухую 25-мл колбу, содержащую амин, синтезированный на этапе 11 выше (1,43 ммоль), добавляли (S)-(+)-2-гидрокси-3-метилбутановую кислоту (203 мг, 1,72 ммоль, 1,2 экв.), HOBt (232 мг, 1,72 ммоль, 1,2 экв.), безводный DMF (15 мл) и N,N-диизопропилэтиламин (0,374 мл, 2,15 ммоль, 1,5 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (330 мг, 1,72 ммоль, 1,2 экв.). Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (150 мл)/вода (50 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×30 мл). Объединенные органические слои промывали водой (50 мл), 10% водным NaHSO4 (50 мл), водой (30 мл), насыщенным NaHCO3 (50 мл) и солевым раствором (2×50 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00168] Этап 13
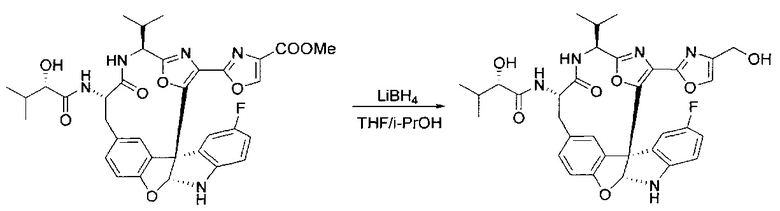
[00169] В сухую колбу добавляли неочищенный материал, синтезированный на этапе 12 (1,43 ммоль), THF (13 мл) и 2-пропанол (40 мл). Этот раствор охлаждали до 0°C с последующим добавлением твердого борогидрида лития (467 мг, 21,45 ммоль, 15 экв.). Полученной в результате смеси давали возможность нагреться до комнатной температуры и перемешивали в течении 18 часов. Реакцию контролировали с помощью LCMS. Исходного материала практически не оставалось. Реакционную смесь охлаждали до 0°C. Добавляли 2-пропанол (40 мл) и воду (80 мл) с последующим добавлением NH4Cl (7,6 г, 143 ммоль, 100 экв.). Реакционную смесь перемешивали в течение 1 часа и разбавляли EtOAc (400 мл)/вода (100 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×100 мл). Объединенные органические слои промывали водой (3×100 мл), 10% NaHSO4 (2×100 мл), водой (2×100 мл), насыщенным NaHCO3 (100 мл) и солевым раствором (2×100 мл), а затем сушили над Na2SO4. После концентрирования остаток очищали с помощью колоночной флэш-хроматографии (чистый EtOAc до 7% MeCN/EtOAc) с получением целевого продукта в виде грязнобелого твердого вещества (215 мг, 0,340 ммоль, 24% за семь этапов).
[00170] Синтез соединения 85
[00171]
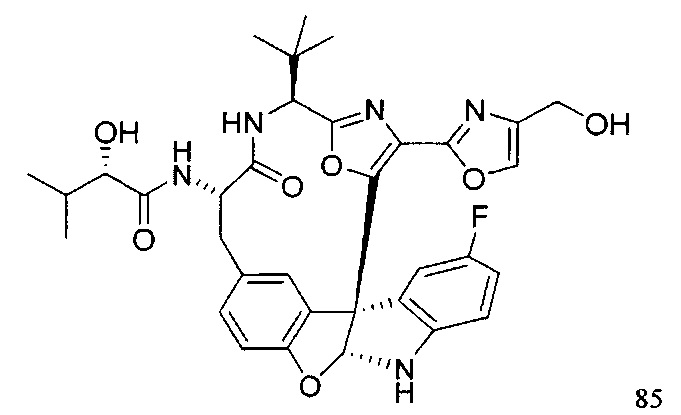
[00172] Этап 1
[00173] В сухую 250-мл колбу добавляли 5-фтор-DL-триптофан (6,0 г, 27,0 ммоль) и безводный метанол (120 мл). Суспензию охлаждали до 0°C с последующим добавлением хлортриметилсилана (15,4 мл, 121,5 ммоль, 4,5 экв.) с такой скоростью, чтобы поддерживать температуру реакционной смеси ниже 6°C. Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Реакцию контролировали с помощью TLC. Большую часть летучих веществ испаряли при пониженном давлении. Неочищенный продукт применяли на следующем этапе.
[00174] Этап 2
[00175] В сухую 250-мл колбу с магнитной мешалкой добавляли соль амина, синтезированную на этапе 1 выше (27 ммоль.), Cbz-L-α-трет-бутилглициновую DCHA соль (13,26 г, 29,7 ммоль, 1,1 экв.), HOBt (4,01 г, 29,7 ммоль, 1,1 экв.), безводный DMF (100 мл) и N,N-диизопропилэтиламин (14,1 мл, 81 ммоль, 3,0 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (5,69 г, 29,7 ммоль, 1,1 экв.). Полученную в результате реакционную смесь перемешивали при RT в течение 16 часов. Реакцию контролировали с помощью LCMS. Большую часть растворителей испаряли при пониженном давлении. Затем остаток разбавляли EtOAc (700 мл)/вода (200 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×50 мл). Объединенные органические слои промывали водой (100 мл), 10% водным NaHSO4 (100 мл), водой (100 мл), насыщенным NaHCO3 (100 мл) и солевым раствором (2×100 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00176] Этап 3
[00177] Раствор DDQ (15,32 г, 67,5 ммоль, 2,5 экв.) в THF (100 мл) добавляли к кипящему с обратным холодильником раствору соединения, синтезированного на этапе 2 выше (27 ммоль), в THF (300 мл) и темный раствор выдерживали при кипении на масляной бане при 85°C в течение 1 часа. После охлаждения растворитель извлекали на роторном испарителе. Остаток растворяли в этилацетате (700 мл) и добавляли NaHCO3 (15 г). Смесь перемешивали в течение 1 часа с последующим фильтрованием через воронку с пористым фильтром. Фильтрат промывали водой (200 мл), водным насыщенным NaHCO3 (2×200 мл), водой (2×200 мл), солевым раствором (100 мл) и сушили над Na2SO4. После концентрирования смесь очищали с помощью колоночной флэш-хроматографии (5% EtOAc в CH2Cl2). Это дало 6,42 г (50% выход) продукта.
[00178] Этап 4
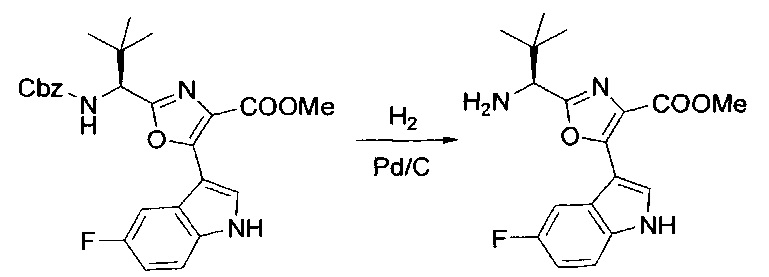
[00179] В 250-мл колбу, содержащую материал, синтезированный на этапе 3 выше (6,42 г, 13,4 ммоль), добавляли метанол (60 мл) и Pd/C (10%) (1,43 г, 1,34 ммоль, 0,1 экв.) под N2. Подводили баллон с H2 и продували колбу H2 4 раза. Затем баллон H2 открывали в реакционную систему. После 1 часа перемешивания исходного материала практически не оставалось. Реакцию останавливали. Реакционную смесь фильтровали через подушку из целита и черный осадок промывали метанолом (3×15 мл). Фильтрат концентрировали, а остаток применяли непосредственно на следующем этапе без дополнительной очистки.
[00180] Этап 5
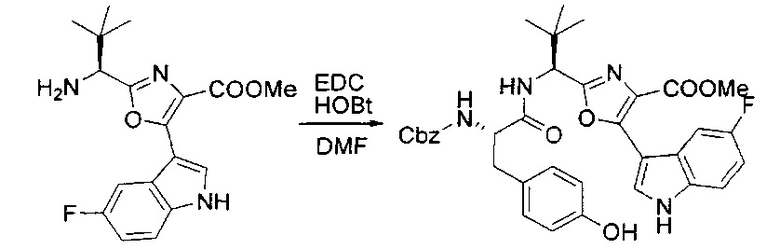
[00181] В сухую 100-мл колбу с магнитной мешалкой добавляли амин, синтезированный на этапе 4 (13,4 ммоль), Cbz-L-тирозин (4,65 г, 14,74 ммоль, 1,1 экв.), HOBt (2,0 г, 14,74 ммоль, 1,1 экв.), безводный DMF (40 мл). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (2,83 г, 14,74 ммоль, 1,1 экв.). Полученную в результате реакционную смесь перемешивали при RT в течение 16 часов. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (500 мл)/вода (150 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×100 мл). Объединенные органические слои промывали водой (200 мл), 10% водным NaHSO4 (150 мл), водой (150 мл), насыщенным NaHCO3 (150 мл) и солевым раствором (2×100 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00182] Этап 6
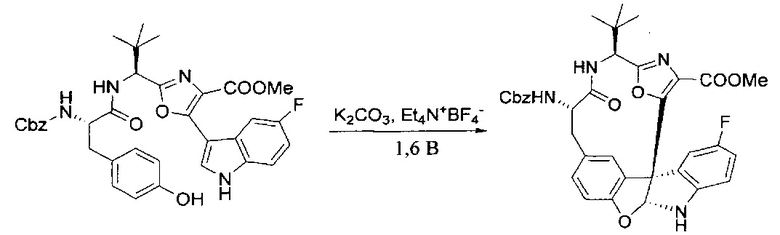
[00183] Электрохимическую ячейку компоновали с применением стеклянного цилиндра (диаметр 6 см × высота 11 см) и обычной подвески (полипропилен и нейлон), что удерживала 9 вертикальных графитовых стержней (диаметр 6,15 мм × длина 12 см). Стержни располагали в модель кольца из 6 анодов и 3 катодов. Электроды погружали на глубину 6,5 см. Фенольный материал, синтезированный на этапе 5 выше (2,00 г, 3,11 ммоль), Et4NBF4 (2,00 г, 9,2 ммоль, 3 экв.), K2CO3 (0,409 г, 2,96 ммоль, 0,95 экв.) и ID воду (4 мл) добавляли в DMF (200 мл). Раствор энергично перемешивали на пластине для смешивания (приблиз. 600 rpm). Электрохимическую реакцию осуществляли при электрическом потенциале 1,5-1,6 вольт. Через 3 дня большая часть первоначального SM была поглощена, что определяли с помощью интегрирования ВЭЖХ при 220 нМ. Электрохимическую реакцию повторяли 4 раза, чтобы поглотился весь фенольный материал, синтезированный на этапе 5. Объединенные реакционные смеси концентрировали на роторном испарителе (температура бани ≤35°C) и дополнительно сушили на вакуумном коллекторе. Остаток разбавляли EtOAc (500 мл) с последующим фильтрованием через воронку с пористым фильтром. Фильтрат промывали водой (2×200 мл), солевым раствором (200 мл). Водные слои экстрагировали последовательно EtOAc (2×50 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали. Этот материал очищали с помощью колоночной флэш-хроматографии с 15% MeCN в CH2Cl2. Это дало 553 мг целевого продукта при 6,4% выходе за три этапа.
[00184] Этап 7
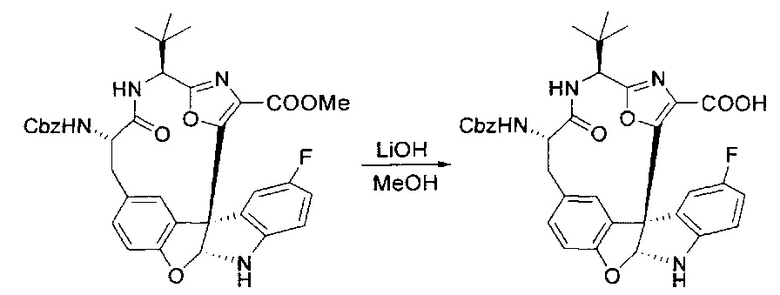
[00185] Соединение, синтезированное на этапе 6 (553 мг, 0,863 ммоль), растворяли в метаноле (17 мл) и раствор охлаждали на ледяной бане. Раствор LiOH (207 мг, 8,63 ммоль, 10 экв.) в воде (2,7 мл) добавляли в течение 5 минут. Ледяную баню убирали и смесь перемешивали при RT в течение 18 часов. Смесь охлаждали на ледяной бане и добавляли воду (20 мл), а затем 1 н водную HCl (8,8 мл), поддерживая температуру реакционной смеси ниже 10°C. Смесь разделяли между водой (15 мл) и EtOAc (100 мл) и промывали органический слой насыщенным водным NaCl. Водные слои экстрагировали последовательно EtOAc (30 мл). Объединенные органические слои сушили (Na2SO4), декантировали и испаряли с получением продукта, представляющего собой кислоту, в виде чисто белых кристаллов.
[00186] Этап 8
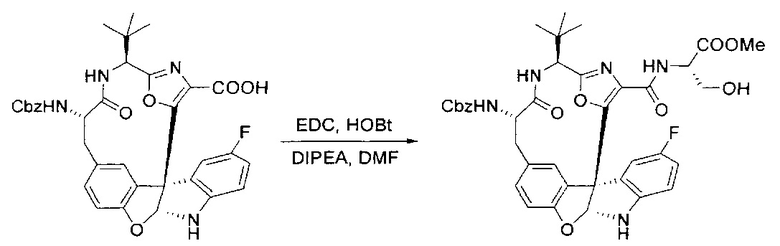
[00187] В сухую 50-мл колбу с магнитной мешалкой добавляли карбоновую кислоту, синтезированную на этапе 7 выше (0,863 ммоль), гидрохлорид сложного метилового эфира L-серина (161 мг, 1,036 ммоль, 1,2 экв.), HOBt (140 мг, 1,036 ммоль, 1.2 экв.), безводный DMF (15 мл) и N,N-диизопропилэтиламин (0,346 мл, 1,99 ммоль, 2.3 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (199 мг, 1,036 ммоль, 1,3 экв.). Полученную в результате реакционную смесь перемешивали при RT в течение 16 часов. Реакцию контролировали с помощью LCMS. Большую часть растворителей испаряли при пониженном давлении. Остаток разбавляли EtOAc (100 мл)/вода (30 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×20 мл). Объединенные органические слои промывали водой (40 мл), 10% водным NaHSO4 (40 мл), водой (40 мл), насыщенным NaHCO3 (40 мл) и солевым раствором (2×40 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00188] Этап 9
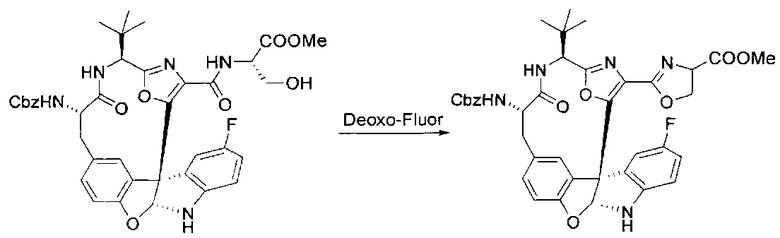
[00189] В сухую колбу добавляли неочищенный продукт этапа 8 выше (0,863 ммоль) и безводный CH2Cl2 (15 мл). Реакционный раствор становился мутным по мере того, как его охлаждали до -20°C на бане сухой лед/ацетон/вода. Свежеполученный основной раствор бис(2-метоксиэтил)аминоаминосеры трифторида (0,239 мл, 1,29 ммоль, 1,5 экв.) в CH2Cl2 (2 мл) добавляли по каплям. Полученную в результате реакционную смесь перемешивали при -20°C в течение 1 часа и нагревали до комнатной температуры. Реакционную смесь гасили посредством добавления насыщенного водного NaHCO3 (10 мл), разбавляли EtOAc (50 мл), промывали водой (2×15 мл), а также солевым раствором (20 мл), и сушили над Na2SO4. После концентрирования остаток применяли на следующем этапе.
[00190] Этап 10
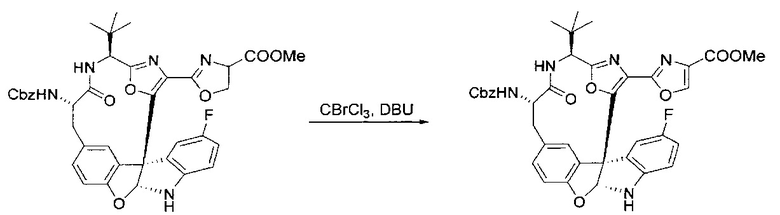
[00191] В сухую колбу, содержащую неочищенный продукт этапа 9 выше (0,866 ммоль), добавляли безводный CH2Cl2 (15 мл). Смесь охлаждали до 0°C. Затем добавляли CBrCl3 (0,128 мл, 1,29 ммоль, 1,5 экв.) и DBU (0,193 мл, 1,29 ммоль, 1,5 экв.), соответственно. Полученной в результате смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 1 часа. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (50 мл), промывали 10% NaHSO4 (15 мл), водой (2×15 мл), насыщенным водным NaHCO3 (15 мл), водой (15 мл) и солевым раствором (15 мл), сушили над Na2SO4. После концентрирования остаток применяли на следующем этапе.
[00192] Этап 11
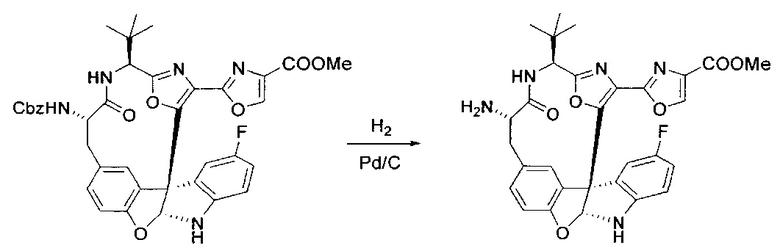
[00193] В 50-мл колбу, содержащую материал, синтезированный на этапе 10 выше (0,863 ммоль), добавляли метанол (12 мл), трет-бутиламин (0,137 мл, 1,3 ммоль, 1,5 экв.) и Pd/C (10%) (91 мг, 0,0863 ммоль, 0,1 экв.) под N2. Подводили баллон с H2 и продували колбу H2 4 раза. Затем баллон с H2 открывали в реакционную систему. После 4 часов перемешивания исходного материала практически не оставалось. Реакцию останавливали. Реакционную смесь фильтровали через подушку из целита и черный осадок промывали метанолом (3×10 мл). Фильтрат концентрировали, а остаток применяли непосредственно на следующем этапе без дополнительной очистки.
[00194] Этап 12
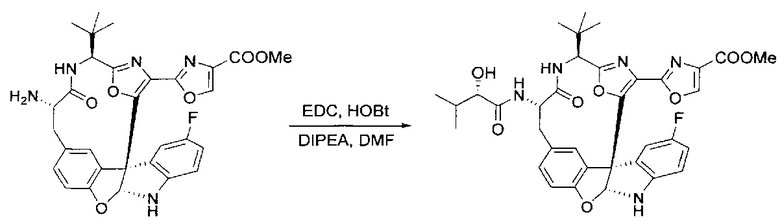
[00195] В сухую 25-мл колбу, содержащую амин, синтезированный на этапе 11 выше (0,863 ммоль), добавляли (S)-(+)-2-гидрокси-3-метилбутановую кислоту (122 мг, 1,036 ммоль, 1,2 экв.), HOBt (140 мг, 1,036 ммоль, 1,2 экв.), безводный DMF (10 мл) и N,N-диизопропилэтиламин (0,225 мл, 1,29 ммоль, 1,5 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (199 мг, 1,036 ммоль, 1,2 экв). Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (100 мл)/вода (30 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×20 мл). Объединенные органические слои промывали водой (30 мл), 10% водным NaHSO4 (30 мл), водой (30 мл), насыщенным NaHCO3 (30 мл) и солевым раствором (2×30 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00196] Этап 13
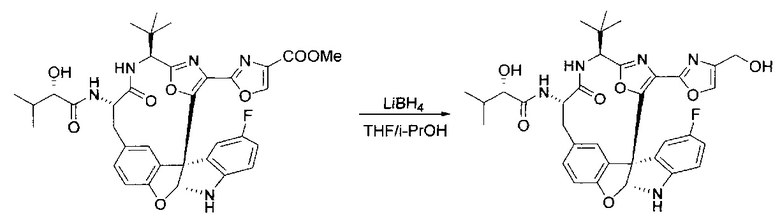
[00197] В сухую колбу добавляли неочищенный материал, синтезированный на этапе 12 (0,863 ммоль), THF (10 мл) и 2-пропанол (30 мл). Этот раствор охлаждали до 0°C с последующим добавлением твердого борогидрида лития (282 мг, 12,95 ммоль, 15 экв.). Полученной в результате смеси давали возможность нагреться до комнатной температуры и перемешивали в течении 22 часов. Реакцию контролировали с помощью LCMS. Исходного материала практически не оставалось. Реакционную смесь охлаждали до 0°C. Добавляли 2-пропанол (24 мл) и воду (40 мл) с последующим добавлением NH4Cl (4,6 г, 86,3 ммоль, 100 экв.). Реакционную смесь перемешивали в течение 1 часа и разбавляли EtOAc (250 мл)/вода (50 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×50 мл). Объединенные органические слои промывали водой (3×100 мл), 10% NaHSO4 (2×100 мл), водой (2×100 мл), насыщенным NaHCO3 (100 мл) и солевым раствором (2×100 мл), а затем сушили над Na2SO4. После концентрирования остаток очищали с помощью колоночной флэш-хроматографии (70% EtOAc/DCM) с получением целевого продукта в виде грязнобелого твердого вещества (124 мг, 0,192 ммоль, 22% за семь этапов).
[00198] Синтез соединения 86
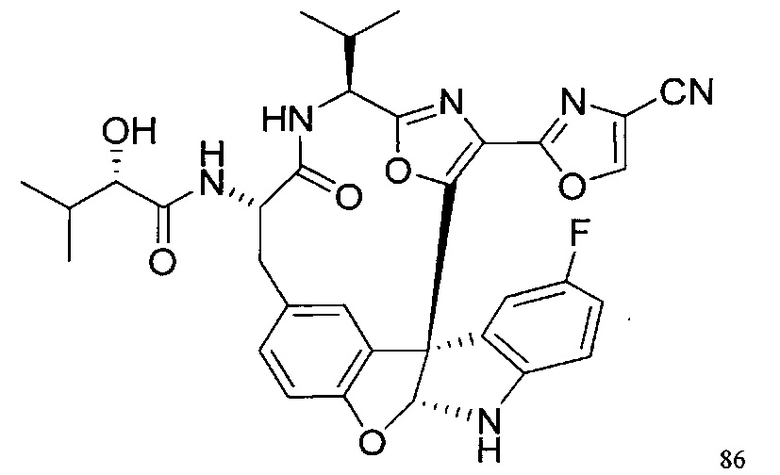
[00199] Этап 1
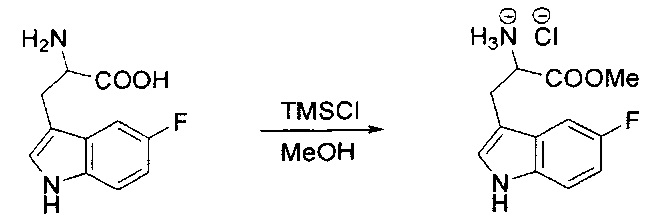
[00200] В сухую 250-мл колбу добавляли 5-фтор-DL-триптофан (5,0 г, 22,5 ммоль) и безводный метанол (120 мл). Суспензию охлаждали до 0°C с последующим добавлением хлортриметилсилана (12,8 мл, 101,3 ммоль, 4,5 экв.) с такой скоростью, чтобы поддерживать температуру реакционной смеси ниже 6°C. Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Реакцию контролировали с помощью TLC. Большую часть летучих веществ испаряли при пониженном давлении. Неочищенный продукт применяли на следующем этапе.
[00201] Этап 2
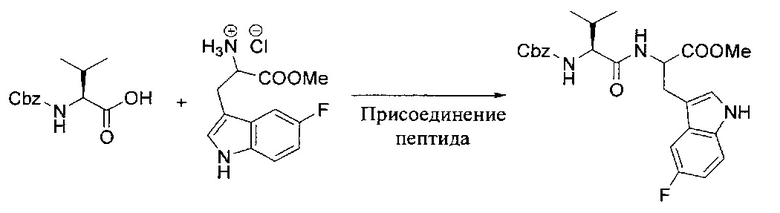
[00202] В сухую 250-мл колбу с магнитной мешалкой добавляли соль амина, синтезированную на этапе 1 выше (22,5 ммоль.), Cbz-L-валин (6,22 г, 24,75 ммоль, 1,1 экв.), HOBt (3,34 г, 24,75 ммоль, 1,1 экв.), безводный DMF (80 мл) и N,N-диизопропилэтиламин (11,8 мл, 67,5 ммоль, 3,0 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (4,74 г, 24,75 ммоль, 1,1 экв.). Полученную в результате реакционную смесь перемешивали при RT в течение 16 часов. Реакцию контролировали с помощью LCMS. Большую часть растворителей испаряли при пониженном давлении. Затем остаток разбавляли EtOAc (600 мл)/вода (200 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×50 мл). Объединенные органические слои промывали водой (100 мл), 10% водным NaHSO4 (100 мл), водой (100 мл), насыщенным NaHCO3 (100 мл) и солевым раствором (2×100 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00203] Этап 3
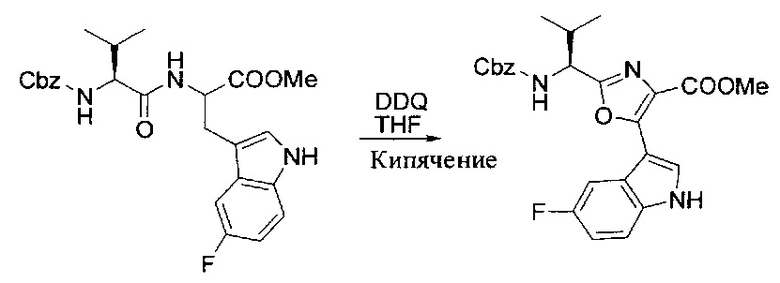
[00204] Раствор DDQ (12,8 г, 56,25 ммоль, 2,5 экв.) в THF (500 мл) добавляли к кипящему с обратным холодильником раствору соединения, синтезированного на этапе 2 выше (22,5 ммоль), в THF (250 мл) и темный раствор выдерживали при кипении на масляной бане при 85°C в течение 1 часа. После охлаждения растворитель извлекали на роторном испарителе. Остаток растворяли в этилацетате (600 мл) и добавляли NaHCO3 (13 г). Смесь перемешивали в течение 1 часа с последующим фильтрованием через воронку с пористым фильтром. Фильтрат промывали водой (200 мл), водным насыщенным NaHCO3 (2×200 мл), водой (2×200 мл), солевым раствором (100 мл) и сушили над Na2SO4. После концентрирования смесь очищали с помощью колоночной флэш-хроматографии (5% EtOAc в CH2Cl2). Это дало 4,63 г (44,2% выход) продукта.
[00205] Этап 4
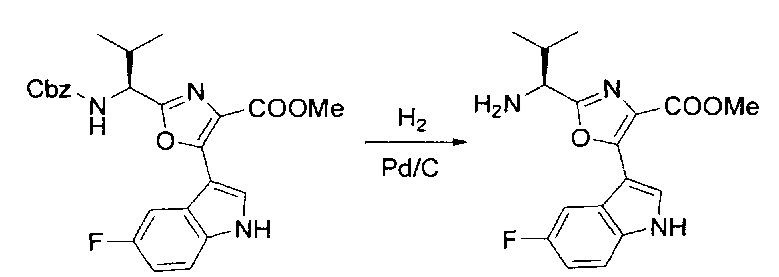
[00206] В 250-мл колбу, содержащую материал, синтезированный на этапе 3 выше (4,63 г, 9,94 ммоль), добавляли метанол (50 мл) и Pd/C (10%) (530 мг, 0,497 ммоль, 0,05 экв.) под N2. Подводили баллон с H2 и продували колбу H2 4 раза. Затем баллон с H2 открывали в реакционную систему. После 1 часа перемешивания исходного материала практически не оставалось. Реакцию останавливали. Реакционную смесь фильтровали через подушку из целита и черный осадок промывали метанолом (3×15 мл). Фильтрат концентрировали, а остаток применяли непосредственно на следующем этапе без дополнительной очистки.
[00207] Этап 5
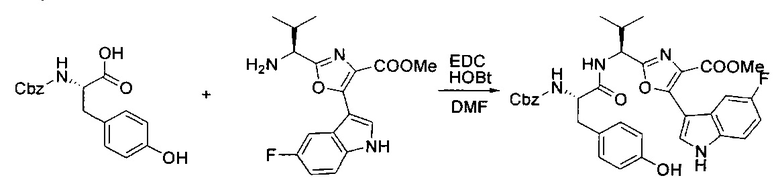
[00208] В сухую 100-мл колбу с магнитной мешалкой добавляли амин, синтезированный на этапе 4 (9,94 ммоль), Cbz-L-тирозин (3,45 г, 10,93 ммоль, 1,1 экв.), HOBt (1,48 г, 10,93 ммоль, 1,1 экв.), безводный DMF (30 мл). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (2,10 г, 10,93 ммоль, 1,1 экв.). Полученную в результате реакционную смесь перемешивали при RT в течение 16 часов. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (400 мл)/вода (150 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×100 мл). Объединенные органические слои промывали водой (200 мл), 10% водным NaHSO4 (150 мл), водой (150 мл), насыщенным NaHCO3 (150 мл) и солевым раствором (2×100 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт (6,58 г) применяли непосредственно на следующем этапе.
[00209] Этап 6
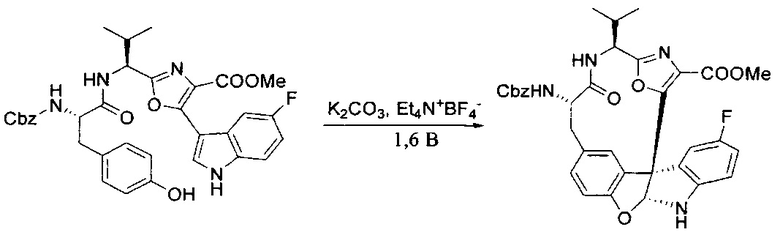
[00210] Электрохимическую ячейку компоновали с применением стеклянного цилиндра (диаметр 6 см × высота 11 см) и обычной подвески (полипропилен и нейлон), что удерживала 9 вертикальных графитовых стержней (диаметр 6,15 мм × длина 12 см). Стержни располагали в модель кольца из 6 анодов и 3 катодов. Электроды погружали на глубину 6,5 см. Фенольный материал, синтезированный на этапе 5 выше (2,00 г, 3,18 ммоль), Et4NBF4 (2,00 г, 9,2 ммоль, 3 экв.), K2CO3 (0,44 г, 3,18 ммоль, 1,0 экв.) и ID воду (4 мл) добавляли в DMF (200 мл). Раствор энергично перемешивали на пластине для смешивания (приблиз. 600 rpm). Электрохимическую реакцию осуществляли при электрическом потенциале 1,5-1,6 вольт. Через 3 дня большая часть первоначального SM была поглощена, что определяли с помощью интегрирования ВЭЖХ при 220 нМ. Электрохимическую реакцию повторяли 4 раза, чтобы поглотился весь фенольный материал, синтезированный на этапе 5. Объединенные реакционные смеси концентрировали на роторном испарителе (температура бани ≤35°C) и дополнительно сушили на вакуумном коллекторе. Остаток разбавляли EtOAc (500 мл) с последующим фильтрованием через воронку с пористым фильтром. Фильтрат промывали водой (2×200 мл), солевым раствором (200 мл). Водные слои экстрагировали последовательно EtOAc (2×50 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали. Этот материал очищали с помощью колоночной флэш-хроматографии с 15% MeCN в CH2Cl2. Это дало 900 мг целевого продукта при 14% выходе за три этапа.
[00211] Этап 7
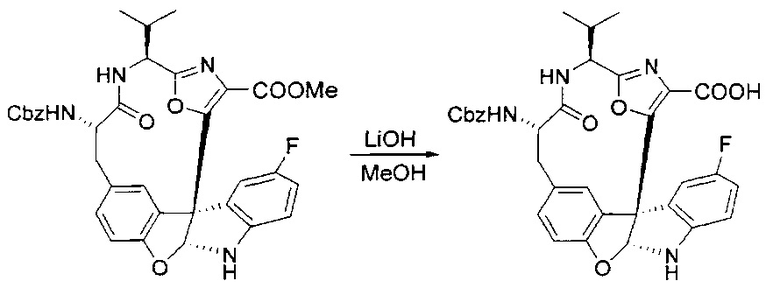
[00212] Соединение, синтезированное на этапе 6 (900 мг, 1,43 ммоль), растворяли в метаноле (28 мл) и раствор охлаждали на ледяной бане. Раствор LiOH (344 мг, 14,3 ммоль, 10 экв.) в воде (4,5 мл) добавляли в течение 5 минут. Ледяную баню убирали и смесь перемешивали при RT в течение 18 часов. Смесь охлаждали на ледяной бане и добавляли воду (40 мл), а затем 1 н водную HCl (14,5 мл), поддерживая температуру реакционной смеси ниже 10°C. Смесь разделяли между водой (25 мл) и EtOAc (200 мл) и промывали органический слой насыщенным водным NaCl. Водные слои экстрагировали последовательно EtOAc (50 мл). Объединенные органические слои сушили (Na2SO4), декантировали и испаряли с получением продукта, представляющего собой кислоту, в виде чисто белых кристаллов.
[00213] Этап 8
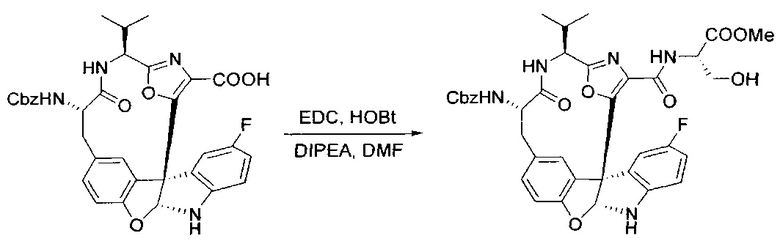
[00214] В сухую 50-мл колбу с магнитной мешалкой добавляли карбоновую кислоту, синтезированную на этапе 7 выше (1,43 ммоль), гидрохлорид сложного метилового эфира L-серина (268 мг, 1,72 ммоль, 1,2 экв.), HOBt (232 мг, 1,72 ммоль, 1,2 экв.), безводный DMF (15 мл) и N,N-диизопропилэтиламин (0,624 мл, 3,58 ммоль, 2,5 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (330 мг, 1,72 ммоль, 1,2 экв.). Полученную в результате реакционную смесь перемешивали при RT в течение 16 часов. Реакцию контролировали с помощью LCMS. Большую часть растворителей испаряли при пониженном давлении. Остаток разбавляли EtOAc (150 мл)/вода (50 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×30 мл). Объединенные органические слои промывали водой (60 мл), 10% водным NaHSO4 (60 мл), водой (60 мл), насыщенным NaHCO3 (60 мл) и солевым раствором (2×60 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00215] Этап 9
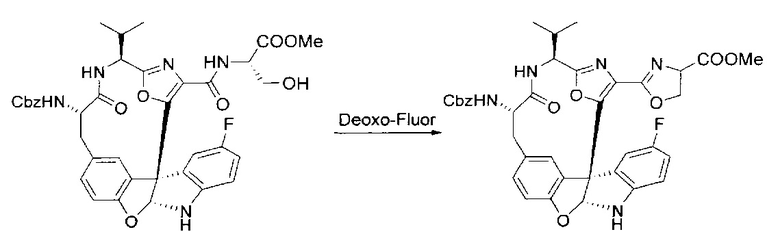
[00216] В сухую колбу добавляли неочищенный продукт этапа 8 выше (1,43 ммоль) и безводный CH2Cl2 (25 мл). Реакционный раствор становился мутным по мере того, как его охлаждали до -20°C на бане сухой лед/ацетон/вода. Свежеполученный основной раствор бис(2-метоксиэтил)аминоаминосеры трифторида (0,395 мл, 2,15 ммоль, 1,5 экв.) в CH2Cl2 (4 мл) добавляли по каплям. Полученную в результате реакционную смесь перемешивали при -20°C в течение 1 часа и нагревали до комнатной температуры. Реакционную смесь гасили посредством добавления насыщенного водного NaHCO3 (15 мл), разбавляли EtOAc (100 мл), промывали водой (2×20 мл), а также солевым раствором (30 мл), и сушили над Na2SO4. После концентрирования остаток применяли на следующем этапе.
[00217] Этап 10
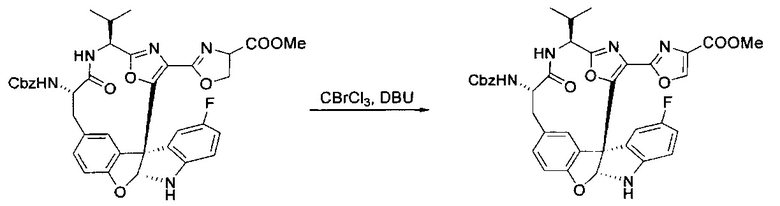
[00218] В сухую колбу, содержащую неочищенный продукт этапа 9 выше (1,43 ммоль), добавляли безводный CH2Cl2 (25 мл). Смесь охлаждали до 0°C. Затем добавляли CBrO3 (0,211 мл, 2,15 ммоль, 1,5 экв.) и DBU (0,321 мл, 2,15 ммоль, 1,5 экв.), соответственно. Полученной в результате смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 1 часа. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (100 мл), промывали 10% NaHSO4 (30 мл), водой (2×30 мл), насыщенным водным NaHCO3 (15 мл), водой (30 мл) и солевым раствором (30 мл), сушили над Na2SO4. После концентрирования остаток применяли на следующем этапе.
[00219] Этап 11
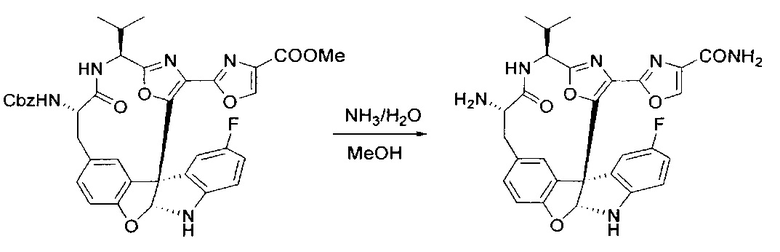
[00220] В колбу добавляли продукт этапа 10 (0,735 ммоль), метанол (30 мл), водный аммиачный раствор (28%, 15 мл). Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Реакцию контролировали с помощью TLC. Большую часть метанола испаряли под уменьшенным давлением. Остаток экстрагировали этилацетатом (3×30 мл), промывали 2% NaHSO4 (30 мл), водой (30 мл), 5% KaHCO3 (30 мл), солевым раствором (30 мл). Органическую фазу сушили над Na2SO4. После концентрирования неочищенный продукт применяли на следующем этапе.
[00221] Этап 12
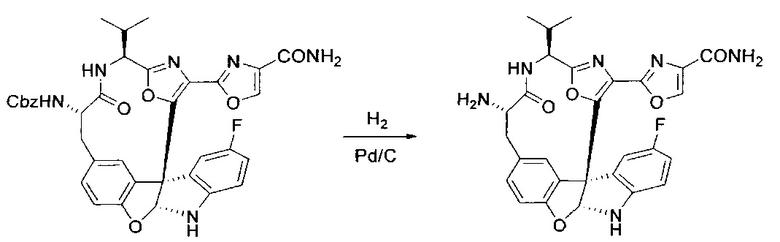
[00222] В 100-мл колбу, содержащую материал, синтезированный на этапе 11 выше (0,735 ммоль), добавляли метанол (20 мл), трет-бутиламин (0,116 мл, 1,1 ммоль, 1,5 экв.) и Pd/C (10%) (78 мг, 0,052 ммоль, 0,1 экв.) под N2. Подводили баллон с H2 и продували колбу H2 4 раза. Затем баллон с H2 открывали в реакционную систему. Через 4 часа перемешивания исходного материала практически не оставалось. Реакцию останавливали. Реакционную смесь фильтровали через подушку из целита и черный осадок промывали метанолом (3×15 мл). Фильтрат концентрировали, а остаток применяли непосредственно на следующем этапе без дополнительной очистки.
[00223] Этап 13
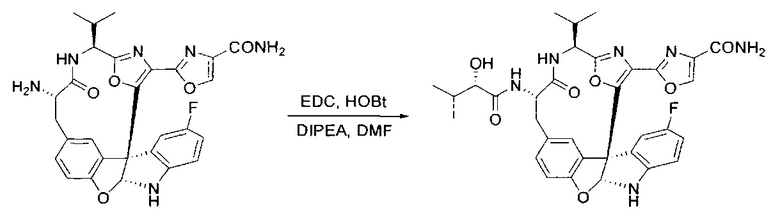
[00224] В сухую 25-мл колбу, содержащую амин, синтезированный на этапе 12 выше (0,735 ммоль), добавляли (S)-(+)-2-гидрокси-3-метилбутановую кислоту (104 мг, 0,882 ммоль, 1,2 экв.), HOBt (119 мг, 0,882 ммоль, 1,2 экв.), безводный DMF (10 мл) и N,N-диизопропилэтиламин (0,192 мл, 1,1 ммоль, 1,5 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (169 мг, 0,882 ммоль, 1,2 экв.). Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (100 мл)/вода (30 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×30 мл). Объединенные органические слои промывали водой (50 мл), 10% водным NaHSO4 (50 мл), водой (30 мл), насыщенным NaHCO3 (50 мл) и солевым раствором (2×50 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00225] Этап 14
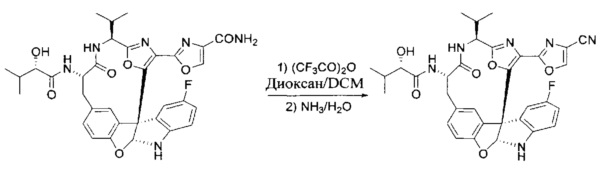
[00226] В сухую колбу добавляли неочищенный материал, синтезированный на этапе 13 (0,735 ммоль), диоксан (10 мл), и CH2Cl2 (10 мл), и пиридин (1,2 мл, 14,7 ммоль). Этот раствор охлаждали до -17°C с последующим добавлением трифторуксусного ангидрида (1,5 мл, 11 ммоль) при температуре от -10 до -17°C. После добавления полученную в результате смесь перемешивали при -15°C в течение 1 часа. Затем по каплям добавляли водный раствор NH3 (28%, 10 мл) при -15°C с последующим нагреванием до комнатной температуры и перемешивали в течение 1 часа. Реакцию контролировали с помощью LCMS. Большую часть растворителя переносили при уменьшенном давлении. Остаток экстрагировали этилацетатом (3×20 мл), промывали водой (2×20 мл), 5% NaHSO4 (20 мл), водой (20 мл), насыщ. NaHCO3 (20 мл), водой (20 мл) и солевым раствором (20 мл). Органическую фазу сушили над Na2SO4. После концентрирования остаток очищали с помощью колоночной флэш-хроматографии (30% MeCN/DCM до 40% MeCN/DCM) с получением целевого продукта в виде грязнобелого твердого вещества (115 мг, 0,184 ммоль, 25% за восемь этапов).
[00227] Синтез соединения 87
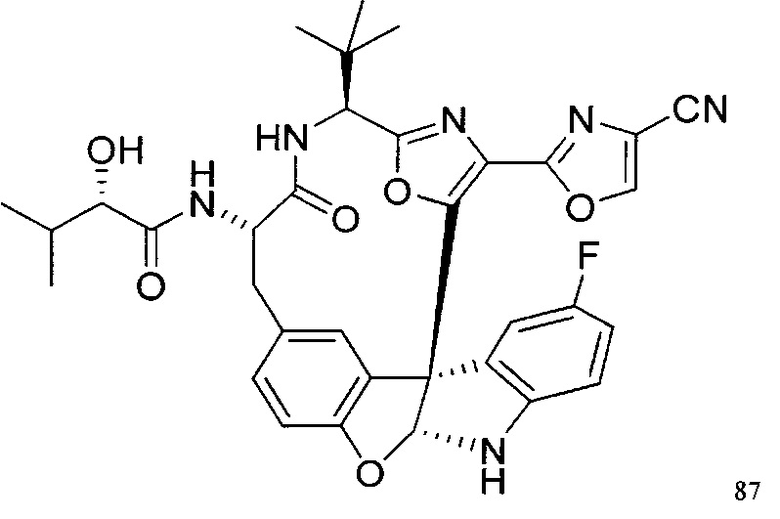
[00228] Этап 1

[00229] В сухую 250-мл колбу добавляли 5-фтор-DL-триптофан (6,0 г, 27,0 ммоль) и безводный метанол (120 мл). Суспензию охлаждали до 0°C с последующим добавлением хлортриметилсилана (15,4 мл, 121,5 ммоль, 4,5 экв.) с такой скоростью, чтобы поддерживать температуру реакционной смеси ниже 6°C. Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Реакцию контролировали с помощью TLC. Большую часть летучих веществ испаряли при пониженном давлении. Неочищенный продукт применяли на следующем этапе.
[00230] Этап 2
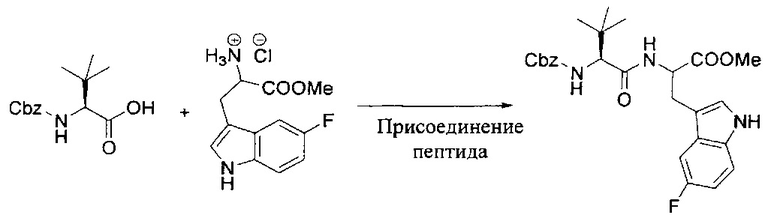
[00231] В сухую 250-мл колбу с магнитной мешалкой добавляли соль амина, синтезированную на этапе 1 выше (27 ммоль.), соль Cbz-L-α-трет-бутилглицина DCHA (13,26 г, 29,7 ммоль, 1,1 экв.), HOBt (4,01 г, 29,7 ммоль, 1,1 экв.), безводный DMF (100 мл) и N,N-диизопропилэтиламин (14,1 мл, 81 ммоль, 3,0 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (5,69 г, 29,7 ммоль, 1,1 экв.). Полученную в результате реакционную смесь перемешивали при RT в течение 16 часов. Реакцию контролировали с помощью LCMS. Большую часть растворителей испаряли при пониженном давлении. Затем остаток разбавляли EtOAc (700 мл)/вода (200 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×50 мл). Объединенные органические слои промывали водой (100 мл), 10% водным NaHSO4 (100 мл), водой (100 мл), насыщенным NaHCO3 (100 мл) и солевым раствором (2×100 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00232] Этап 3
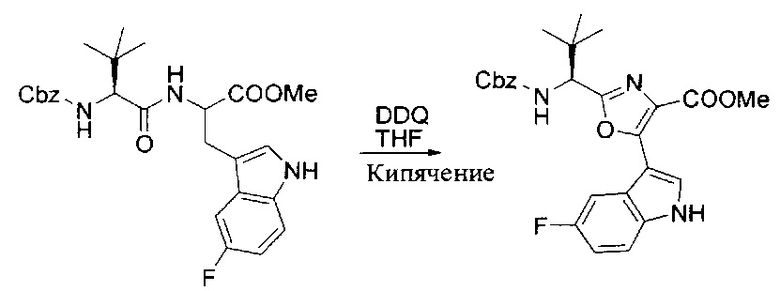
[00233] Раствор DDQ (15,32 г, 67,5 ммоль, 2,5 экв.) в THF (100 мл) добавляли к кипящему с обратным холодильником раствору соединения, синтезированного на этапе 2 выше (27 ммоль), в THF (300 мл) и темный раствор выдерживали при кипении на масляной бане при 85°C в течение 1 часа. После охлаждения растворитель извлекали на роторном испарителе. Остаток растворяли в этилацетате (700 мл) и добавляли NaHCO3 (15 г). Смесь перемешивали в течение 1 часа с последующим фильтрованием через воронку с пористым фильтром. Фильтрат промывали водой (200 мл), водным насыщенным NaHCO3 (2×200 мл), водой (2×200 мл), солевым раствором (100 мл) и сушили над Na2SO4. После концентрирования смесь очищали с помощью колоночной флэш-хроматографии (5% EtOAc в CH2Cl2). Это дало 6,42 г (50% выход) продукта.
[00234] Этап 4
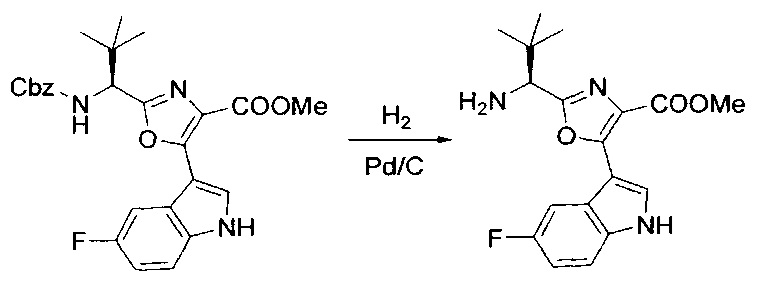
[00235] В 250-мл колбу, содержащую материал, синтезированный на этапе 3 выше (6,42 г, 13,4 ммоль), добавляли метанол (60 мл) и Pd/C (10%) (1,43 г, 1,34 ммоль, 0,1 экв.) под N2. Подводили баллон с H2 и продували колбу H2 4 раза. Затем баллон H2 открывали в реакционную систему. После 1 часа перемешивания исходного материала практически не оставалось. Реакцию останавливали. Реакционную смесь фильтровали через подушку из целита и черный осадок промывали метанолом (3×15 мл). Фильтрат концентрировали, а остаток применяли непосредственно на следующем этапе без дополнительной очистки.
[00236] Этап 5
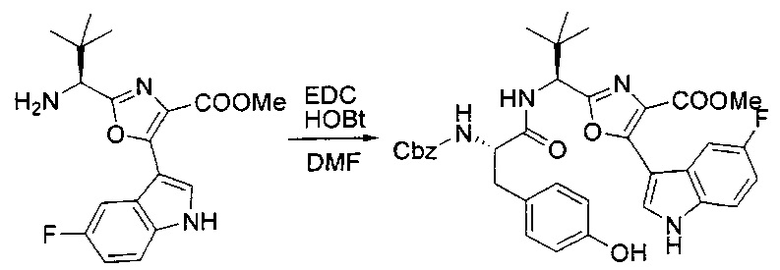
[00237] В сухую 100-мл колбу с магнитной мешалкой добавляли амин, синтезированный на этапе 4 (13,4 ммоль), Cbz-L-тирозин (4,65 г, 14,74 ммоль, 1,1 экв.), HOBt (2,0 г, 14,74 ммоль, 1,1 экв.), безводный DMF (40 мл). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (2,83 г, 14,74 ммоль, 1,1 экв.). Полученную в результате реакционную смесь перемешивали при RT в течение 16 часов. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (500 мл)/вода (150 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×100 мл). Объединенные органические слои промывали водой (200 мл), 10% водным NaHSO4 (150 мл), водой (150 мл), насыщенным NaHCO3 (150 мл) и солевым раствором (2×100 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00238] Этап 6
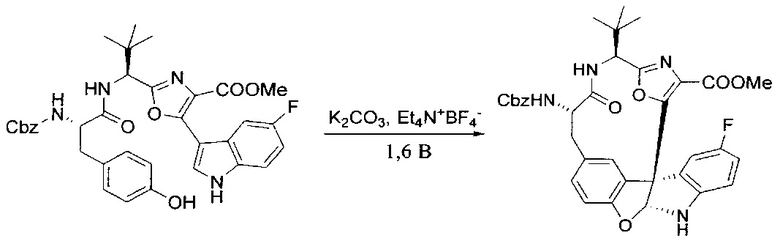
[00239] Электрохимическую ячейку компоновали с применением стеклянного цилиндра (диаметр 6 см × высота 11 см) и обычной подвески (полипропилен и нейлон), что удерживала 9 вертикальных графитовых стержней (диаметр 6,15 мм × длина 12 см). Стержни располагали в модель кольца из 6 анодов и 3 катодов. Электроды погружали на глубину 6,5 см. Фенольный материал, синтезированный на этапе 5 выше (2,00 г, 3,11 ммоль), Et4NBF4 (2,00 г, 9,2 ммоль, 3 экв.), K2CO3 (0,409 г, 2,96 ммоль, 0,95 экв.) и ID воду (4 мл) добавляли в DMF (200 мл). Раствор энергично перемешивали на пластине для смешивания (приблиз. 600 rpm). Электрохимическую реакцию осуществляли при электрическом потенциале 1,5-1,6 вольт. Через 3 дня большая часть первоначального SM была поглощена, что определяли с помощью интегрирования ВЭЖХ при 220 нМ. Электрохимическую реакцию повторяли 4 раза, чтобы поглотился весь фенольный материал, синтезированный на этапе 5. Объединенные реакционные смеси концентрировали на роторном испарителе (температура бани ≤35°C) и дополнительно сушили на вакуумном коллекторе. Остаток разбавляли EtOAc (500 мл) с последующим фильтрованием через воронку с пористым фильтром. Фильтрат промывали водой (2×200 мл), солевым раствором (200 мл). Водные слои экстрагировали последовательно EtOAc (2×50 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали. Этот материал очищали с помощью колоночной флэш-хроматографии с 15% MeCN в CH2Cl2. Это дало 553 мг целевого продукта при 6,4% выходе за три этапа.
[00240] Этап 7
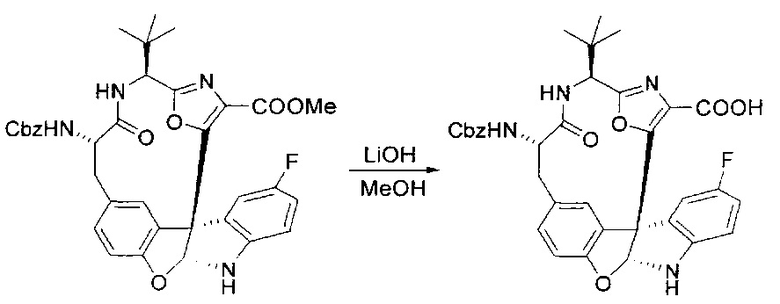
[00241] Соединение, синтезированное на этапе 6 (553 мг, 0,863 ммоль), растворяли в метаноле (17 мл) и раствор охлаждали на ледяной бане. Раствор LiOH (207 мг, 8,63 ммоль, 10 экв.) в воде (2,7 мл) добавляли в течение 5 минут. Ледяную баню убирали и смесь перемешивали при RT в течение 18 часов. Смесь охлаждали на ледяной бане и добавляли воду (20 мл), а затем 1 н водную HCl (8.8 мл), поддерживая температуру реакционной смеси ниже 10°C. Смесь разделяли между водой (15 мл) и EtOAc (100 мл) и промывали органический слой насыщенным водным NaCl. Водные слои экстрагировали последовательно EtOAc (30 мл). Объединенные органические слои сушили (Na1SO4), декантировали и испаряли с получением продукта, представляющего собой кислоту, в виде чисто белых кристаллов.
[00242] Этап 8
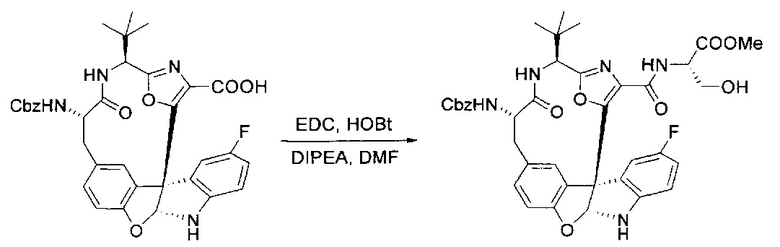
[00243] В сухую 50-мл колбу с магнитной мешалкой добавляли карбоновую кислоту, синтезированную на этапе 7 выше (0,863 ммоль), гидрохлорид сложного метилового эфира L-серина (161 мг, 1,036 ммоль, 1,2 экв.), HOBt (140 мг, 1,036 ммоль, 1,2 экв.), безводный DMF (15 мл) и N,N-диизопропилэтиламин (0,346 мл, 1,99 ммоль, 2,3 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (199 мг, 1,036 ммоль, 1,3 экв.). Полученную в результате реакционную смесь перемешивали при RT в течение 16 часов. Реакцию контролировали с помощью LCMS. Большую часть растворителей испаряли при пониженном давлении. Остаток разбавляли EtOAc (100 мл)/вода (30 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×20 мл). Объединенные органические слои промывали водой (40 мл), 10% водным NaHSO4 (40 мл), водой (40 мл), насыщенным NaHCO3 (40 мл) и солевым раствором (2×40 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00244] Этап 9
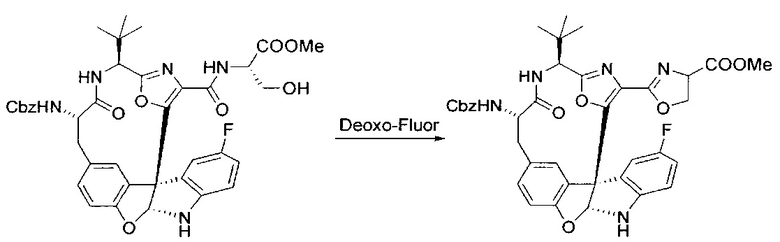
[00245] В сухую колбу добавляли неочищенный продукт этапа 8 выше (0,863 ммоль) и безводный CH2Cl2 (15 мл). Реакционный раствор становился мутным по мере того, как его охлаждали до -20°C на бане сухой лед/ацетон/вода. Свежеполученный основной раствор бис(2-метоксиэтил)аминоаминосеры трифторида (0,239 мл, 1,29 ммоль, 1,5 экв.) в CH2Cl2 (2 мл) добавляли по каплям. Полученную в результате реакционную смесь перемешивали при -20°C в течение 1 часа и нагревали до комнатной температуры. Реакционную смесь гасили посредством добавления насыщенного водного NaHCO3 (10 мл), разбавляли EtOAc (50 мл), промывали водой (2×15 мл), а также солевым раствором (20 мл), и сушили над Na2SO4. После концентрирования остаток применяли на следующем этапе.
[00246] Этап 10
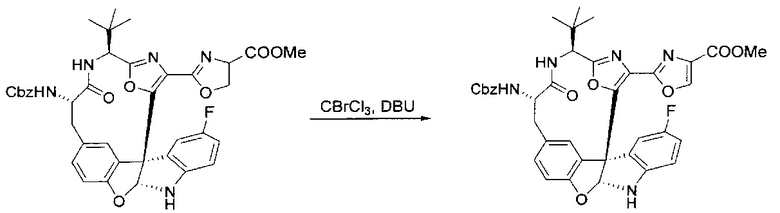
[00247] В сухую колбу, содержащую неочищенный продукт этапа 9 выше (0,866 ммоль), добавляли безводный CH2Cl2 (15 мл). Смесь охлаждали до 0°C. Затем добавляли CBrO3 (0,128 мл, 1,29 ммоль, 1,5 экв.) и DBU (0,193 мл, 1,29 ммоль, 1,5 экв.), соответственно. Полученной в результате смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 1 часа. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (50 мл), промывали 10% NaHSO4 (15 мл), водой (2×15 мл), насыщенным водным NaHCO3 (15 мл), водой (15 мл) и солевым раствором (15 мл), сушили над Na2SO4. После концентрирования остаток применяли на следующем этапе.
[00248] Этап 11
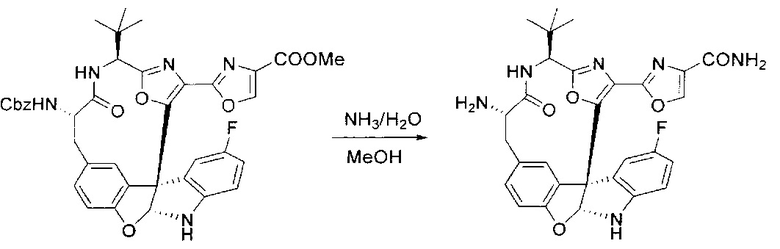
[00249] В колбу добавляли продукт этапа 10 (0,52 ммоль), метанол (25 мл), водный аммиачный раствор (28%, 10 мл). Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Реакцию контролировали с помощью TLC. Большую часть метанола испаряли при уменьшенном давлении. Остаток экстрагировали этилацетатом (3×30 мл), промывали 2% NaHSO4 (30 мл), водой (30 мл), 5% NaHCO3 (30 мл), солевым раствором (30 мл). Органическую фазу сушили над Na2SO4. После концентрирования неочищенный продукт применяли на следующем этапе.
[00250] Этап 12
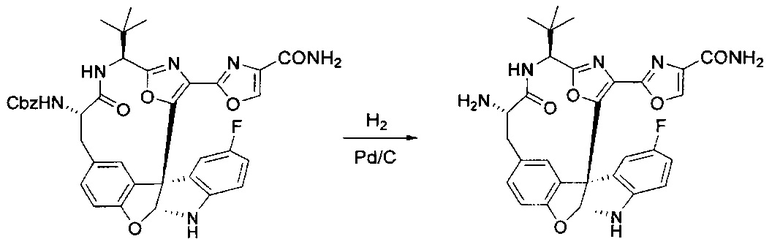
[00251] В 100-мл колбу, содержащую материал, синтезированный на этапе 11 выше (0,52 ммоль), добавляли метанол (10 мл), трет-бутиламин (0,082 мл, 0,78 ммоль, 1,5 экв.) и Pd/C (10%) (55 мг, 0,052 ммоль, 0,1 экв.) под N2. Подводили баллон с H2 и продували колбу H2 4 раза. Затем баллон с H2 открывали в реакционную систему. После 4 часов перемешивания исходного материала практически не оставалось. Реакцию останавливали. Реакционную смесь фильтровали через подушку из целита и черный осадок промывали метанолом (3×15 мл). Фильтрат концентрировали, а остаток применяли непосредственно на следующем этапе без дополнительной очистки.
[00252] Этап 13
[00253] 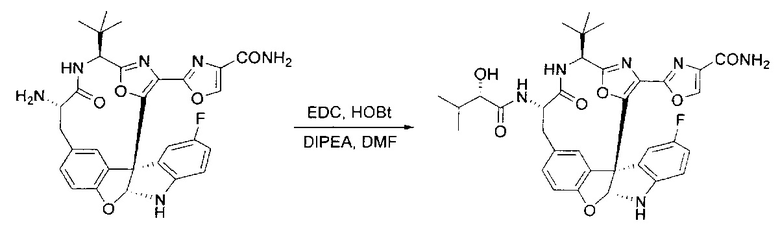
[00254] В сухую 25-мл колбу, содержащую амин, синтезированный на этапе 12 выше (0,52 ммоль), добавляли (S)-(+)-2-гидрокси-3-метилбутановую кислоту (74 мг, 0,624 ммоль, 1,2 экв.), HOBt (85 мг, 0,624 ммоль, 1,2 экв.), безводный DMF (10 мл) и 1 N,N-диизопропилэтиламин (0,136 мл, 0,78 ммоль, 1,5 экв.). Реакционную смесь охлаждали до 0°C с последующим добавлением EDC⋅HCl (120 мг, 0,624 ммоль, 1,2 экв.). Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию контролировали с помощью LCMS. Реакционную смесь разбавляли EtOAc (100 мл)/вода (30 мл). Органическую фазу отделяли, а водную фазу экстрагировали EtOAc (2×30 мл). Объединенные органические слои промывали водой (50 мл), 10% водным NaHSO4 (50 мл), водой (30 мл), насыщенным NaHCO3 (50 мл) и солевым раствором (2×50 мл), а затем сушили над Na2SO4. После концентрирования неочищенный продукт применяли непосредственно на следующем этапе.
[00255] Этап 14
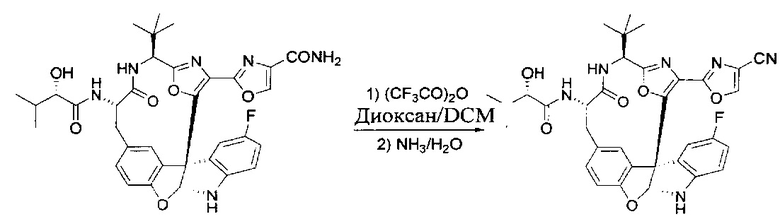
[00256] В сухую колбу добавляли неочищенный материал, синтезированный на этапе 13 (0,52 ммоль), диоксан (7 мл), и CH2Cl2 (7 мл), и пиридин (0,841 мл, 14,7 ммоль). Этот раствор охлаждали до -17°C с последующим добавлением трифторуксусного ангидрида (1,1 мл, 7,8 ммоль) при температуре от -10 до -17°C. После добавления полученную в результате смесь перемешивали при -15°C в течение 1 часа. Затем по каплям добавляли водный раствор NH3 (28%, 7 мл) при -15°C с последующим нагреванием до комнатной температуры и перемешивали в течение 1 часа. Реакцию контролировали с помощью LCMS. Большую часть растворителя переносили под уменьшенным давлением. Остаток экстрагировали этилацетатом (3×20 мл), промывали водой (2×20 мл), 5% NaHSO4 (20 мл), водой (20 мл), насыщ. NaHCO3 (20 мл), водой (20 мл) и солевым раствором (20 мл). Органическую фазу сушили над Na2S04. После концентрирования остаток очищали с помощью колоночной флэш-хроматографии (20% MeCN/DCM до 30% MeCN/DCM) с получением целевого продукта в виде грязнобелого твердого вещества (51 мг, 0,080 ммоль, 16% за восемь этапов).
[00257] Протокол анализа жизнеспособности клеток
[00258] Анализы жизнеспособности клеток проводили с применением стандартных протоколов, известных специалистам в данной области. Клетки высевали в 96-луночные планшеты при плотности 3000-10000 клеток на лунку. Через двадцать четыре часа клетки обрабатывали тестируемыми соединениями с увеличивающимися концентрациями (от 1 нМ до 1 мкМ). Еще через 48 часов измеряли выживаемость клеток с применением реагента Cell-Titer-Glo® (Promega), следуя протоколу, предоставленному производителем. Величину IC50 определяли как концентрацию тестируемого соединения, которая убивает 50% клеточной популяции.
[00259] Примерные биологические данные
[00260] Данные о жизнеспособности клеток, полученные согласно описанному выше протоколу, были разработаны для примерных соединений в клетках А2058 и U937. Представленные в таблице 1 соединения были получены способами, описанными в данном документе для структурно подобных соединений. Эталонное соединение представляло собой искусственный аналог диазонамида, имеющий структуру:
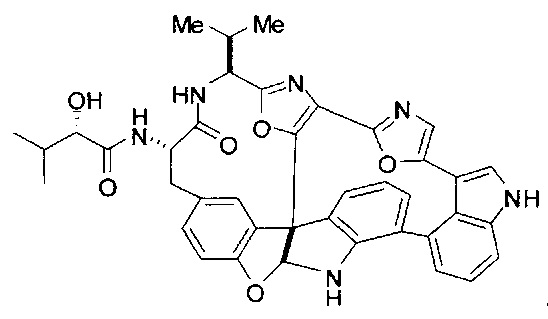
[00261]
анализа жизнеспособности клеток А2058
жизнеспособности клеток U937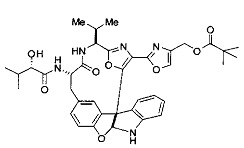
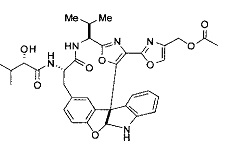
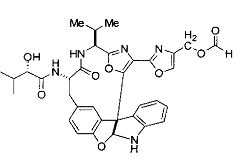
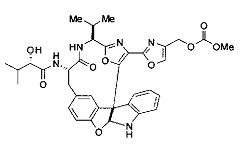
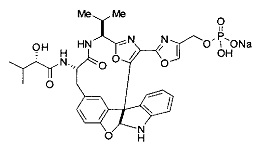
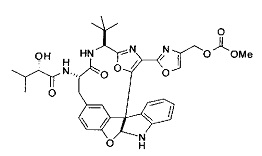
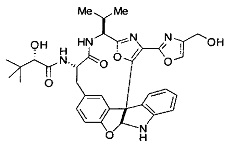
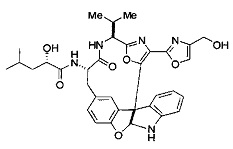
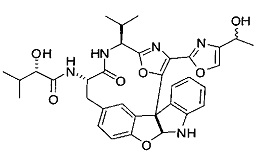
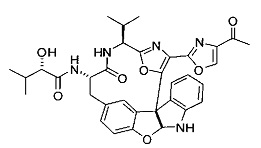
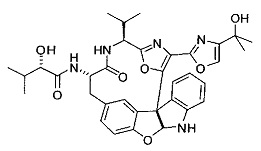
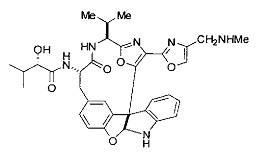
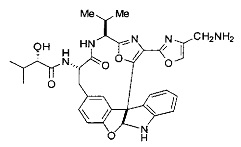
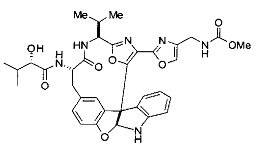
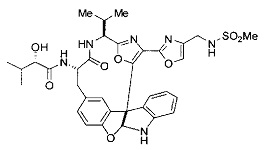
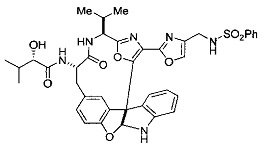
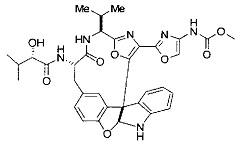
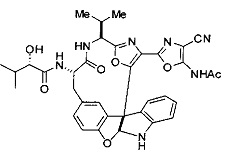
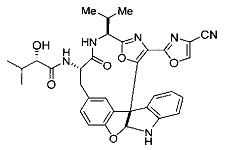
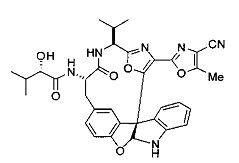
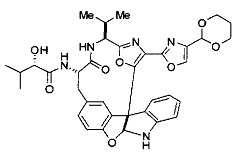
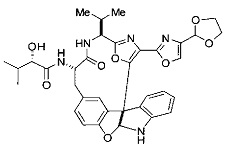
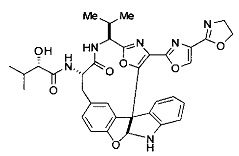
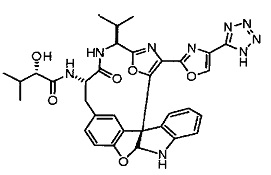
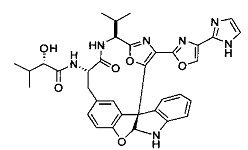
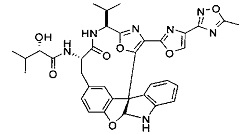
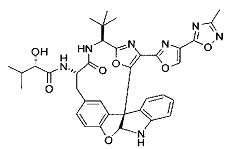
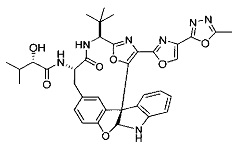
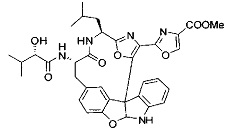
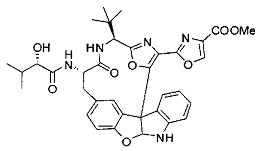
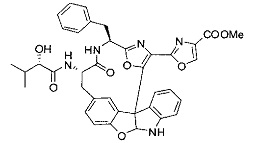
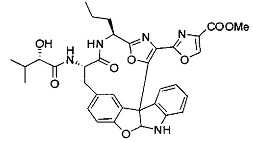
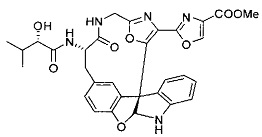
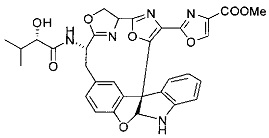
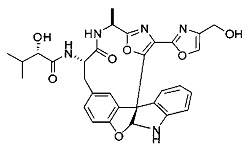
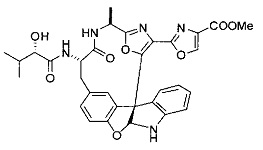
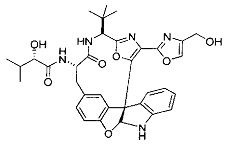
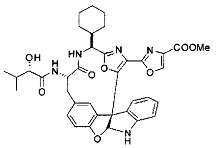
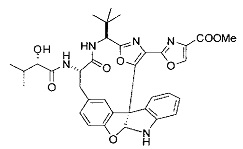
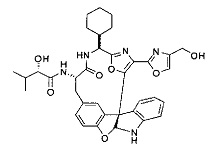
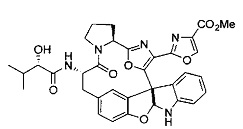
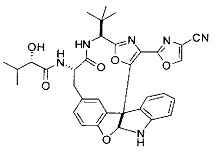
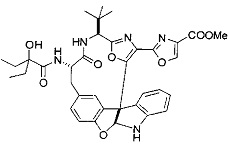
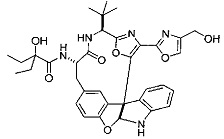
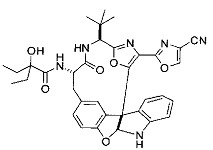
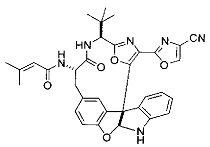
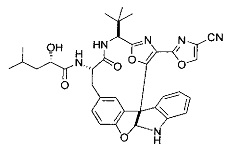
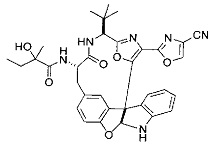
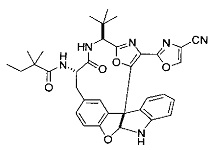
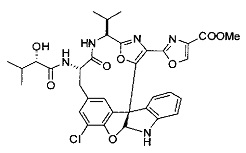
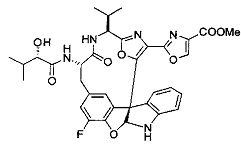
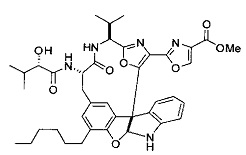
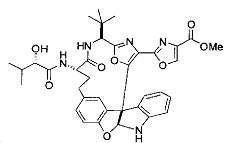
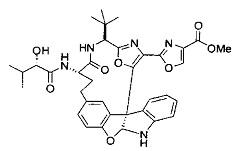
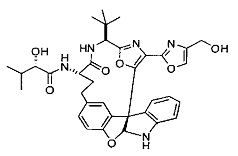
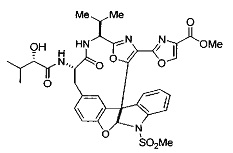
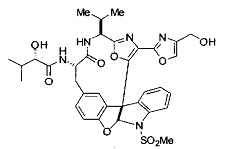
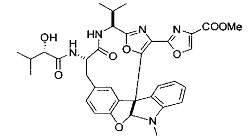
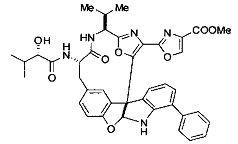
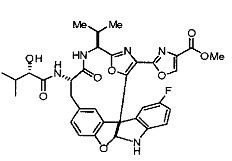
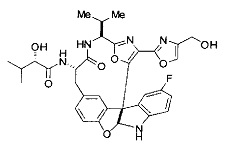
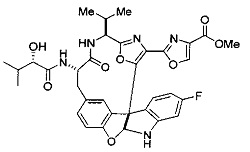
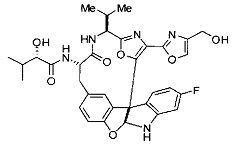
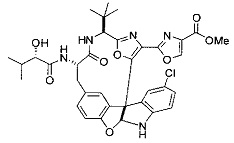
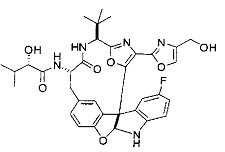
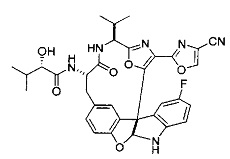
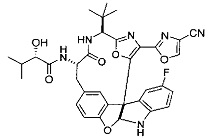
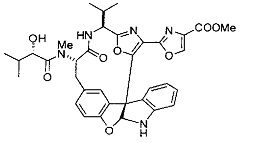
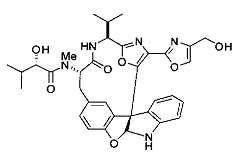
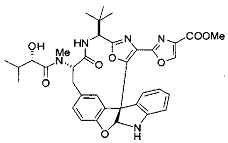
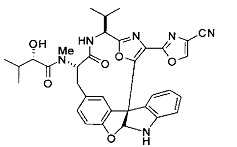
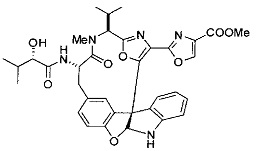
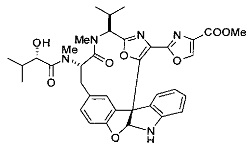
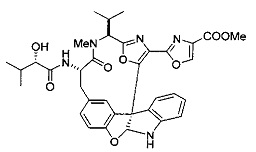
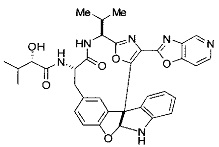
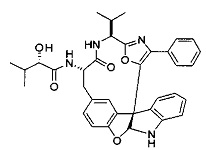
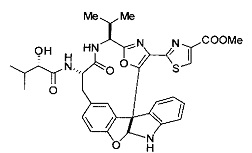
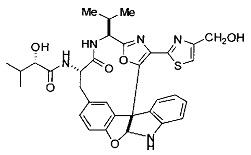
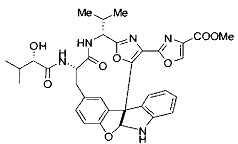
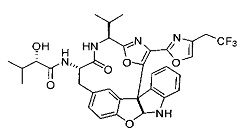
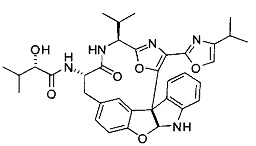
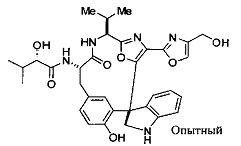
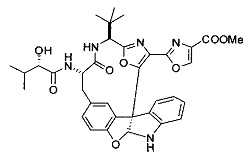
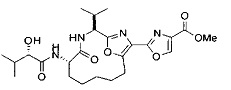
[00262] Модели привитых опухолей
[00263] Соединения тестировали на моделях привитого рака легких человека HCC461 и привитого рака поджелудочной железы Miapaca на бестимусных голых мышах Foxnlnu Harlan 5-6-недельного возраста.
Протокол:
[00264] Получение опухолевых клеток
[00265] Опухолевые клетки культивировали в полной среде RPMI и исключали любую контаминацию. Когда клетки обладали 70-80% конфлюентностью, среду извлекали и клетки промывали бессывороточной средой, трипсинизировали, собирали и промывали бессывороточной средой три раза посредством центрифугирования. После конечного промывания клетки подсчитывали и смешивали с матригелем в отношении 1:1 в объеме. Клетки суспендировали в объеме 200 мкл, что содержал необходимое количество клеток в расчете на инъекцию.
[00266] Подготовка к инъекции
[00267] Очищали и обеззараживали область заражения мышей с помощью растворов йода и этанола. Отбирали клетки шприцом 1-cc. Инъецировали опухолевые клетки (1×107) мышам подкожно (s.c.) в паховую область. Когда опухоли достигали размера 200-300 мм3, мышей отбирали в случайном порядке в группы, подвергаемые обработке, из пяти мышей на группу. Мышей взвешивали и измеряли опухоли с применением штангенциркулей два раза в неделю. Объем опухоли в мм3 рассчитывали по формуле: объем (мм3)=(длина × ширина2)/2.
[00268] Обработка
[00269] Соединения растворяли в кремофоре/этаноле (1:1) при 20 мг/мл в качестве основного раствора, а затем растворяли в солевом растворе до 2,5 мг/мл. Соединения и среду (6,25% кремофор/6,25% этанол в солевом растворе) вводили внутривенно в общем объеме 0,2 мл три раза в неделю для всего шести обработок.
[00270] В случае модели привитого рака легких человека HCC461 животным делали инъекцию в дни 7, 11, 14 и 18 после инъекции опухолевых клеток.
[00271] В случае модели привитого рака поджелудочной железы Miapaca животным делали инъекцию в дни 6, 13 и 20 после инъекции опухолевых клеток.
[00272] Результаты
[00273] Активности примерных соединений и дозы показаны на фигурах 1 и 2.
[00274] Полный объем каждого патента, патентной заявки, публикации и документа, упоминаемых в данном документе, тем самым включен посредством ссылки. Цитирование вышеуказанных патентов, патентных заявок, публикаций и документов не является признанием того, что любое из упомянутого выше непосредственно связано с уровнем техники, а также не представляет какого-либо допущения в отношении содержания или даты этих публикаций или документов.
[00275] В вышеизложенном могут быть сделаны модификации без отклонения от основных аспектов настоящего изобретения. Несмотря на то, что настоящее изобретение было очень подробно описано со ссылкой на один или несколько конкретных вариантов осуществления, специалистам в данной области будет понятно, что в конкретно описанных в данной заявке вариантах осуществления могут быть сделаны изменения, и все эти модификации и улучшения находятся в пределах объема и цели настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ЖЕЛЧНОЙ КИСЛОТЫ В КАЧЕСТВЕ АГОНИСТОВ FXR/TGR5 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2707280C2 |
| ПРОИЗВОДНЫЕ ЖЕЛЧНЫХ КИСЛОТ В КАЧЕСТВЕ АГОНИСТОВ FXR/TGR5 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2712099C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ИММУНОЛОГИЧЕСКИХ И ОНКОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2009 |
|
RU2545023C9 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2808166C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2684103C2 |
| ПРОИЗВОДНЫЕ ФУРО[3,2-В]ПИРАНА, ПРИМЕНИМЫЕ В СИНТЕЗЕ АНАЛОГОВ | 2011 |
|
RU2579511C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ, КОМПОЗИЦИИ И ИХ ИСПОЛЬЗОВАНИЕ | 2014 |
|
RU2754534C2 |
| ХИНОКСАЛИНСОДЕРЖАЩИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСА ГЕПАТИТА С | 2008 |
|
RU2493160C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ, КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2808165C2 |
| ИНГИБИТОРЫ MEK И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2812929C1 |
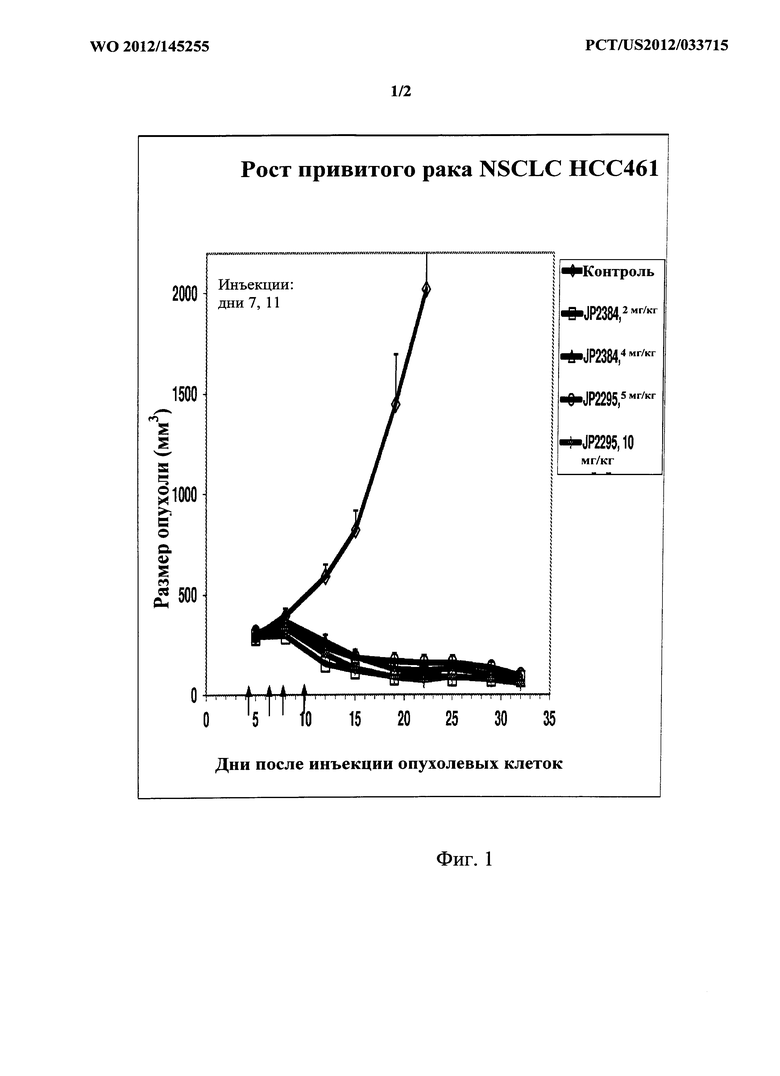
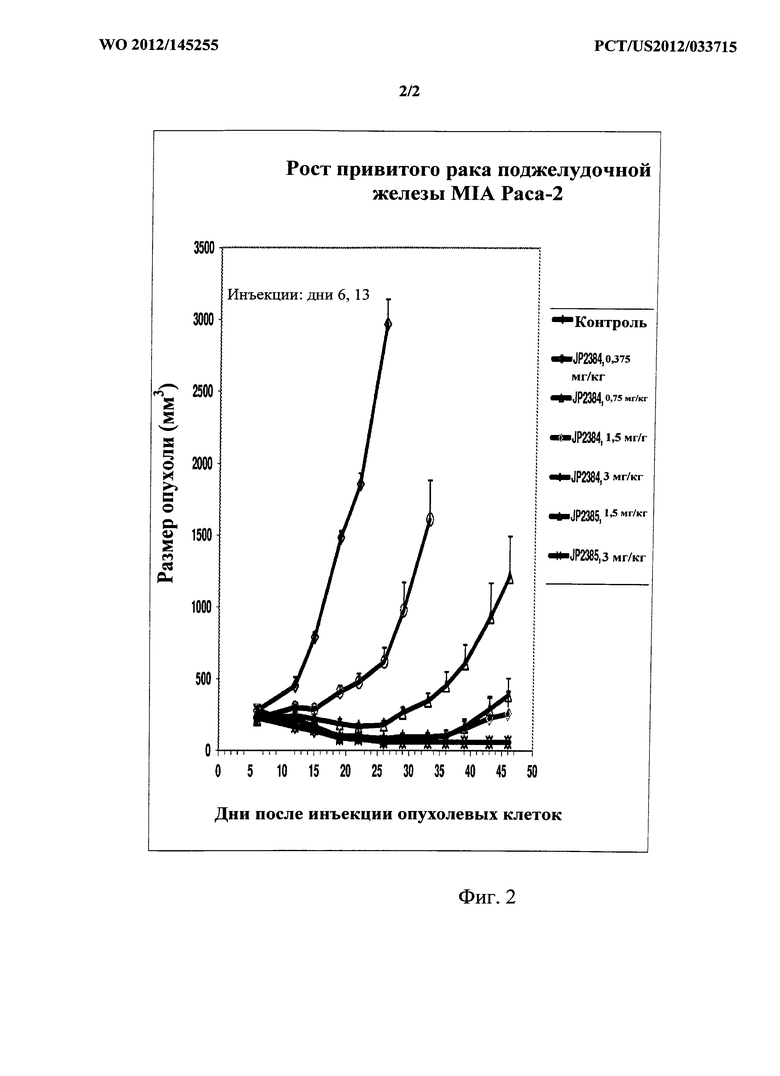
Изобретение относится к конкретным аналогам диазонамида, структуры которых приведены в формуле изобретения. Соединения по изобретению применяют для изготовления фармацевтической композиции, предназначенной для применения в качестве антипролиферативного средства, содержащей терапевтически эффективное количество соединения в единичной лекарственной форме по меньшей мере с одним фармацевтически приемлемым наполнителем. Также соединения применяют для уменьшения интенсивности или лечения клеточного пролиферативного нарушения у субъекта. Технический результат – новые аналоги диазонамида, обладающие антипролиферативным действием. 5 н.п. ф-лы, 2 ил., 1 табл.
1. Соединение, выбранное из группы, состоящей из:
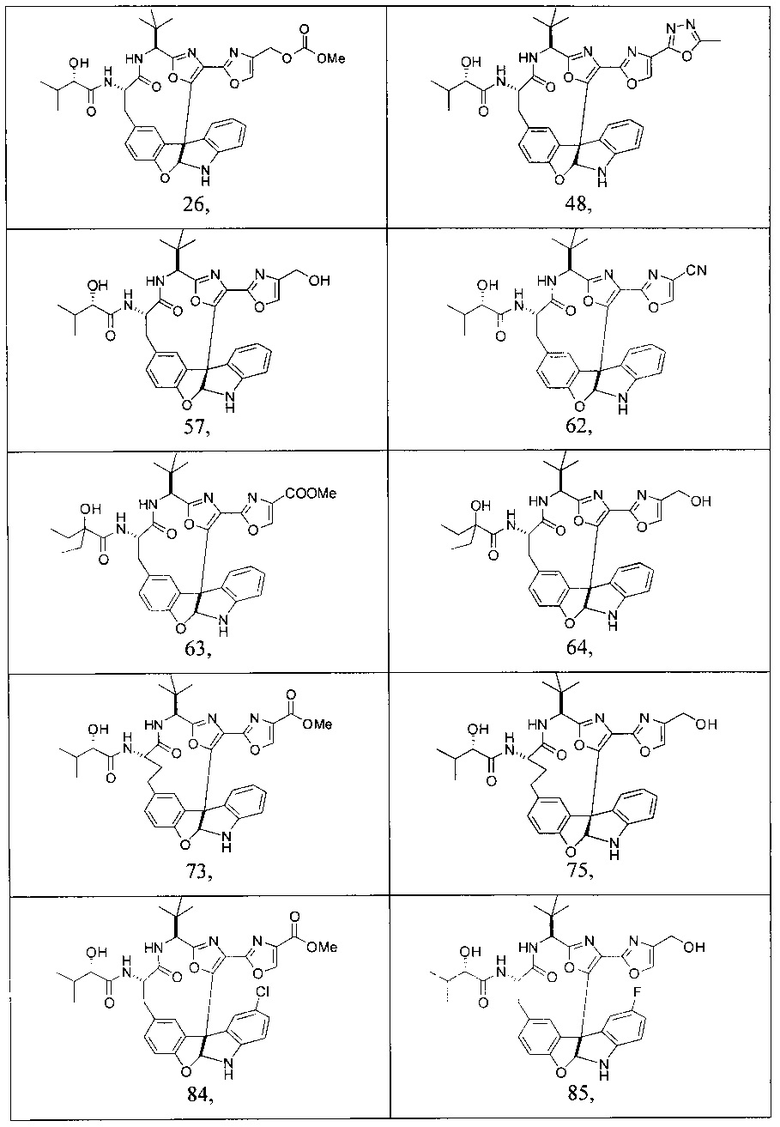
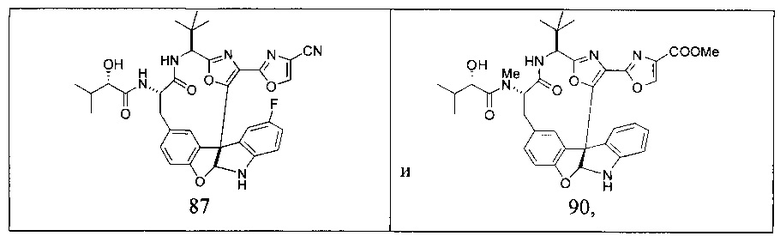
или его фармацевтически приемлемая соль.
2. Соединение, выбранное из группы, состоящей из:
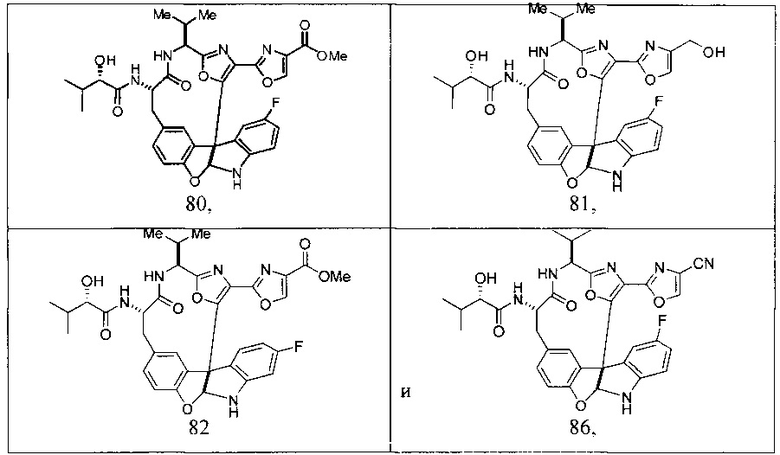
или его фармацевтически приемлемая соль.
3. Соединение, выбранное из группы, состоящей из:
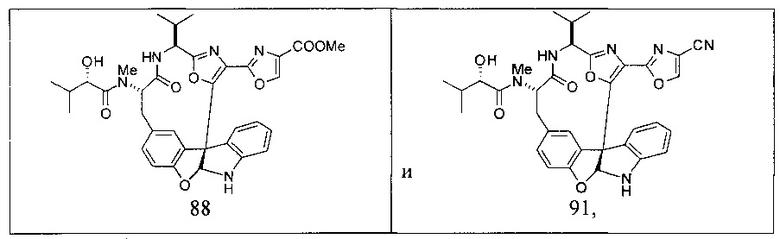
или его фармацевтически приемлемая соль.
4. Фармацевтическая композиция, предназначенная для применения в качестве антипролиферативного средства, содержащая терапевтически эффективное количество соединения по п. 1, или 2, или 3 в единичной лекарственной форме по меньшей мере с одним фармацевтически приемлемым наполнителем.
5. Способ применения соединения по п. 1, или 2, или 3, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения, где соединение применяют для уменьшения интенсивности или лечения клеточного пролиферативного нарушения у субъекта.
| Колосоуборка | 1923 |
|
SU2009A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| ANTHONY W.G.BURGETT et al.: "A concise and flexible total synthesis of (-)diazonamide A**";, ANGEW.CHEM.INT.ED., 2003, vol.42, no.40, p.4961-4966 | |||
| ОБТЮРАТОР ДЛЯ КИНОПРОЕКТОРА | 1929 |
|
SU13692A1 |
| US 7851620 B2, 14.12.2010 | |||
| US 7538129 B2, 26.05.2009. | |||

