Уровень техники
Изобретение относится к способу применения ингибиторов белка-переносчика эфиров холестерина (CETP) у субъектов с высоким уровнем триглицеридов для повышения уровней некоторых липидов в плазме, включая липопротеин высокой плотности (ЛВП)-холестерин, и для снижения уровней других липидов в плазме, таких как липопротеин низкой плотности (ЛНП)-холестерин, и, таким образом, для лечения заболеваний, на которые влияют низкие уровни ЛВП-холестерина и/или высокие уровни ЛНП-холестерина, таких как атеросклероз, дислипидемия и сердечно-сосудистые заболевания.
Атеросклероз и его клинические последствия, ишемическая болезнь сердца, являются основной причиной смертности в промышленно развитых странах. Давно признано, что изменения в характере циркулирующих липопротеинов коррелируют с риском развития атеросклероза и ишемической болезни сердца. Тогда как повышенный ЛНП-C может быть наиболее признанной формой дислипидемии, низкий ЛВП-C также является известным фактором риска развития ишемической болезни сердца.
Метаболический контроль уровней липопротеинов представляет собой сложный и динамический процесс, включающий множество факторов. Среди многих факторов одним из наиболее важных является метаболический контроль у человека белка-переносчика эфиров холестерина (CETP).
CETP является гликопептидом массой 74 кДа, он секретируется печенью и играет ключевую роль, способствуя переносу липидов между различными липопротеинами в плазме. Основная функция CETP заключается в перераспределении сложных эфиров холестерина (CE) и триглицеридов между частицами липопротеинов, включая липопротеины высокой плотности (ЛВП), липопротеины низкой плотности (ЛНП), липопротеины очень низкой плотности (ЛОНП) и хиломикроны (см. Assmann, G et al., "HDL cholesterol and protective factors in atherosclerosis", Circulation, 109:1118-1114 (2004)). Общим результатом активности CETP является понижение ЛВП-холестерина и повышение ЛНП-холестерина. Было показано, что такое влияние на профиль липопротеинов является проатерогенным, особенно у субъектов, липидный профиль которых представляет повышенный риск развития ишемической болезни сердца. Были проведены многочисленные эпидемиологические исследования, связывающие эффекты естественного изменения активности CETP с риском развития ишемической болезни сердца (См. Hirano, K.I. et al. (2000), "Pros and Cons of inhibiting cholesteryl ester transfer protein", Curr. Opin. Lipidol. 11(6), 589-596). Эти исследования четко продемонстрировали обратную корреляцию между концентрацией ЛВП-C в плазме и активностью CETP (см. Inazu A., et al. (2000), "Cholesteryl ester transfer protein and atherosclerosis", Curr. Opin. Lipidol. 11(4), 389-396), что привело к предположению, что фармакологическое ингибирование активности CETP в переносе липидов выгодно для людей при повышении уровней ЛВП-C, с понижением уровней ЛНП.
Анацетрапиб, ингибитор CETP, разработанный компанией Merck, в настоящее время находится в III фазе клинических исследований для лечения дислипидемии и ишемической болезни сердца. Данные in vitro указывают, что анацетрапиб не сохраняет свою активность ингибирования CETP в плазме, полученной от субъектов с повышенными уровнями триглицеридов. Это может привести к снижению эффективности анацетрапиба у пациентов с высокими уровнями триглицеридов, которые составляют значительную часть субъектов с дислипидемией. В целом, у 31% взрослого населения США концентрация триглицеридов в плазме повышена (≥150 мг/дл) (Carroll MD. Trends in serum lipids and lipoproteins of adults, 1960-2002. Journal of the American Medical Association 2005;294:1773-81). Высокие (≥200 мг/дл) и очень высокие (≥500 мг/дл) уровни триглицеридов натощак были обнаружены у 16,2% и 1,1% взрослых, соответственно. Таким образом, существует явная потребность в улучшенной терапии для лечения и предотвращения заболеваний и состояний, связанных с активностью CETP, у пациента с высокими уровнями триглицеридов в плазме. До настоящего времени, Merck не предоставила информации об эффективности анацетрапиба у пациентов, разделенных по концентрации триглицеридов в плазме.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Цель настоящего изобретения состоит в предоставлении способа лечения, предотвращения или уменьшения атеросклероза или дислипидемии, или способа повышения ЛВП-C и/или понижения ЛНП-C, у субъекта с высоким уровнем триглицеридов, включающего введение субъекту терапевтически эффективного количества соединения следующей формулы I:
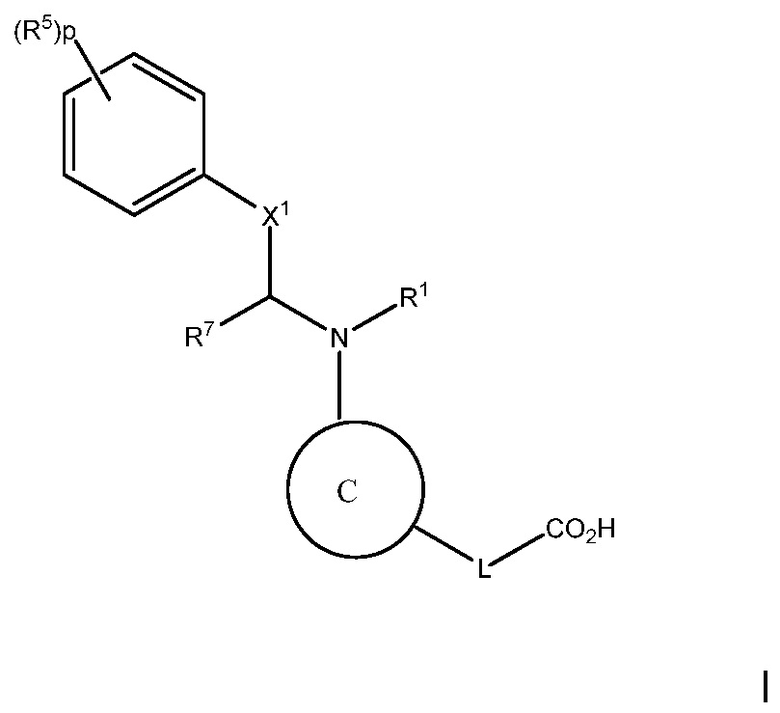
или его фармацевтически приемлемой соли, в которой:
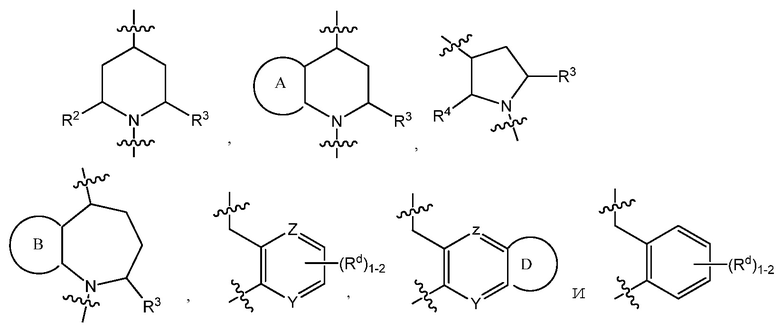
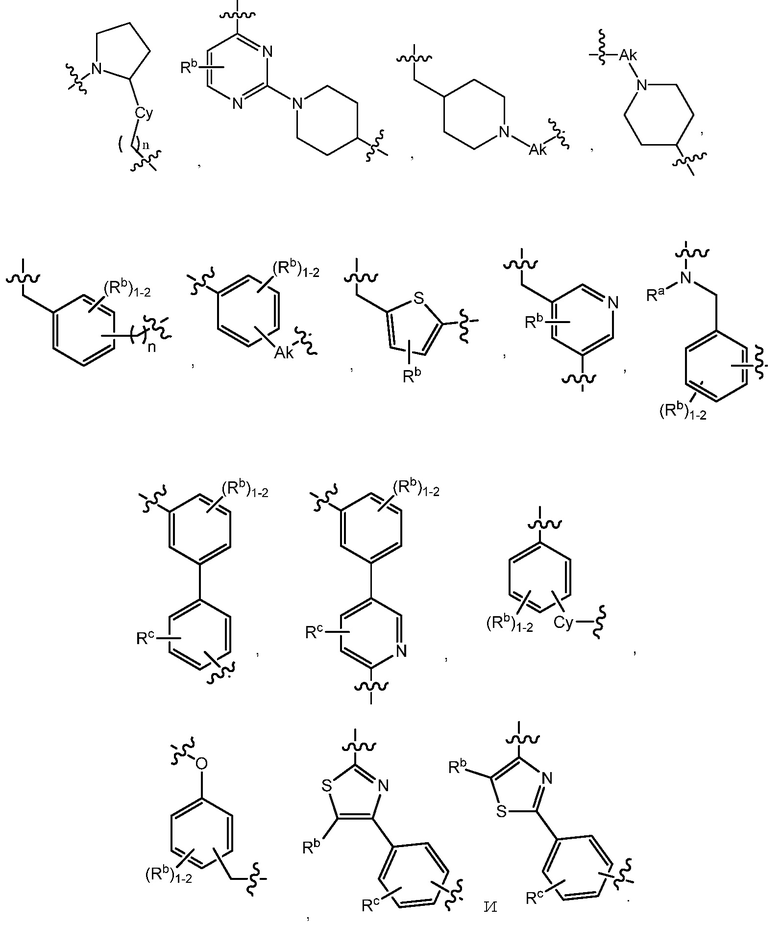
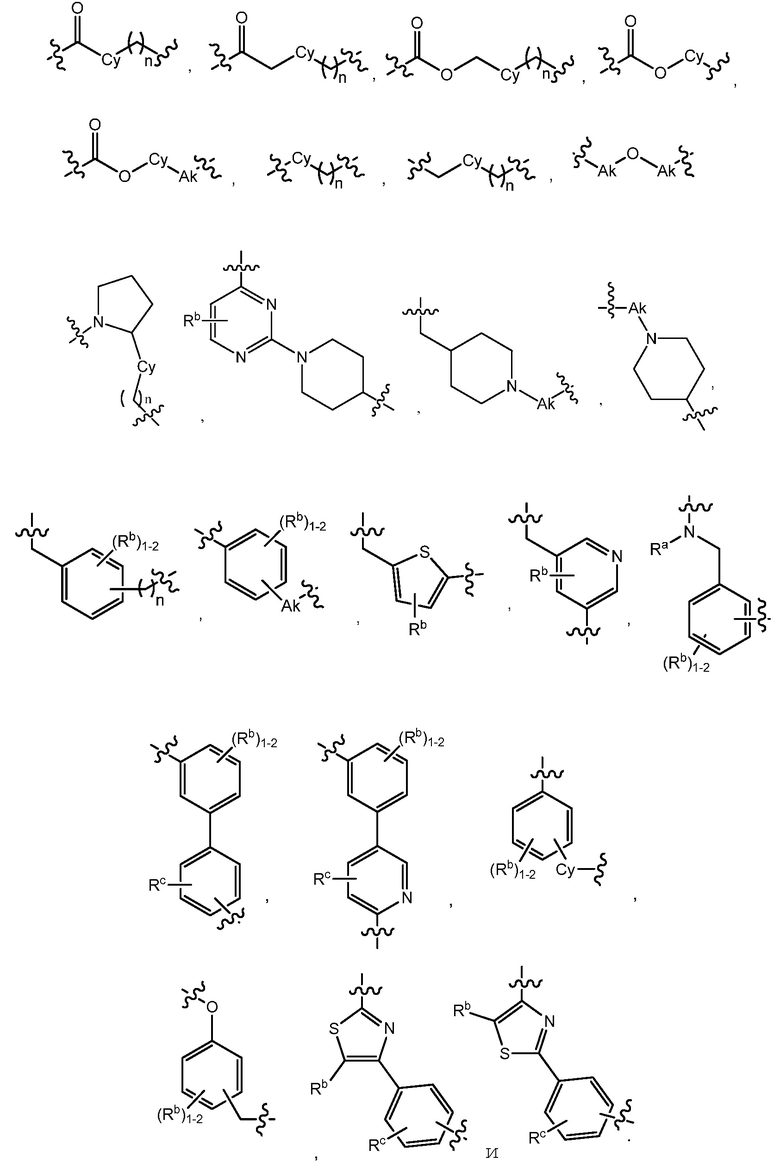
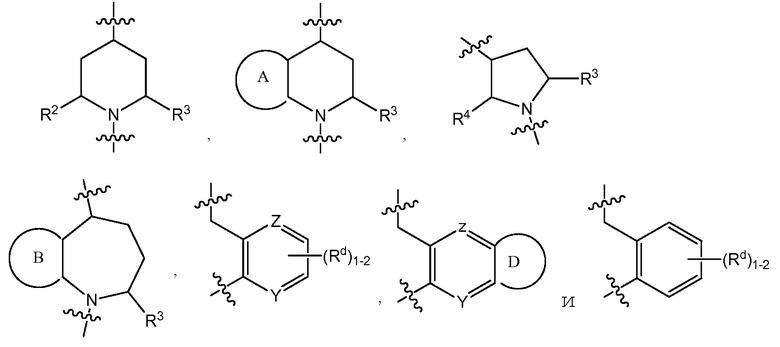
C представляет собой центральную структуру, выбранную из:
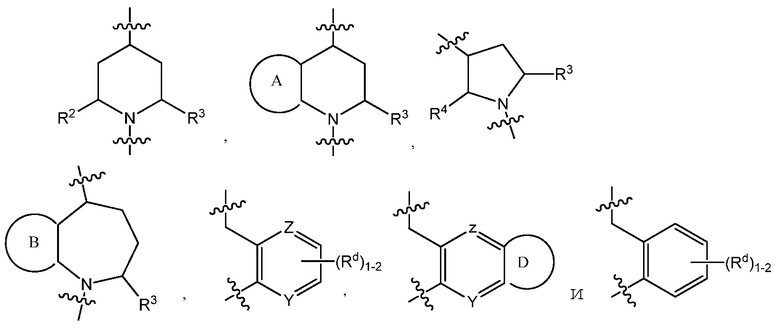
Y и Z независимо являются CH или N;
кольца A и B независимо являются фенилом или 5- или 6-членным гетероарилом, где фенил и гетероарил необязательно замещены 1-3 заместителями, независимо выбранными из C1-7алкила, C1-7алкокси, галогена, галоген-C1-7алкила, галоген-C1-7алкокси, амино, C1-7алкиламино, ди-C1-7алкиламино, C3-7циклоалкила и C3-7циклоалкокси;
D является C3-7циклоалкилом, фенилом, 5- или 6-членным гетероарилом, где фенил и гетероарил необязательно замещены 1-3 заместителями, независимо выбранными из 1-3 заместителей, независимо выбранных из C1-7алкила, C1-7алкокси, CN, NO2, C3-7циклоалкил-C1-7алкила, галогена, галоген-C1-7алкила, галоген-C1-7алкокси, амино, C1-7алкиламино, ди-C1-7алкиламино, C3-7циклоалкила и C3-7циклоалкокси;
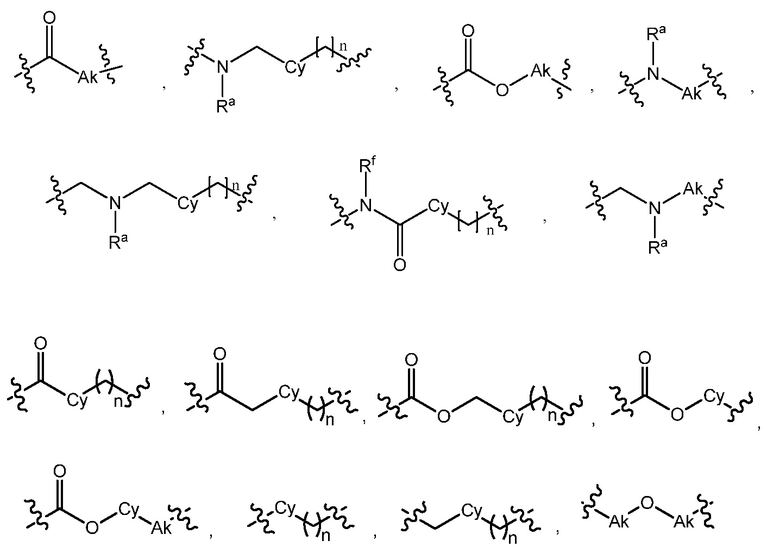
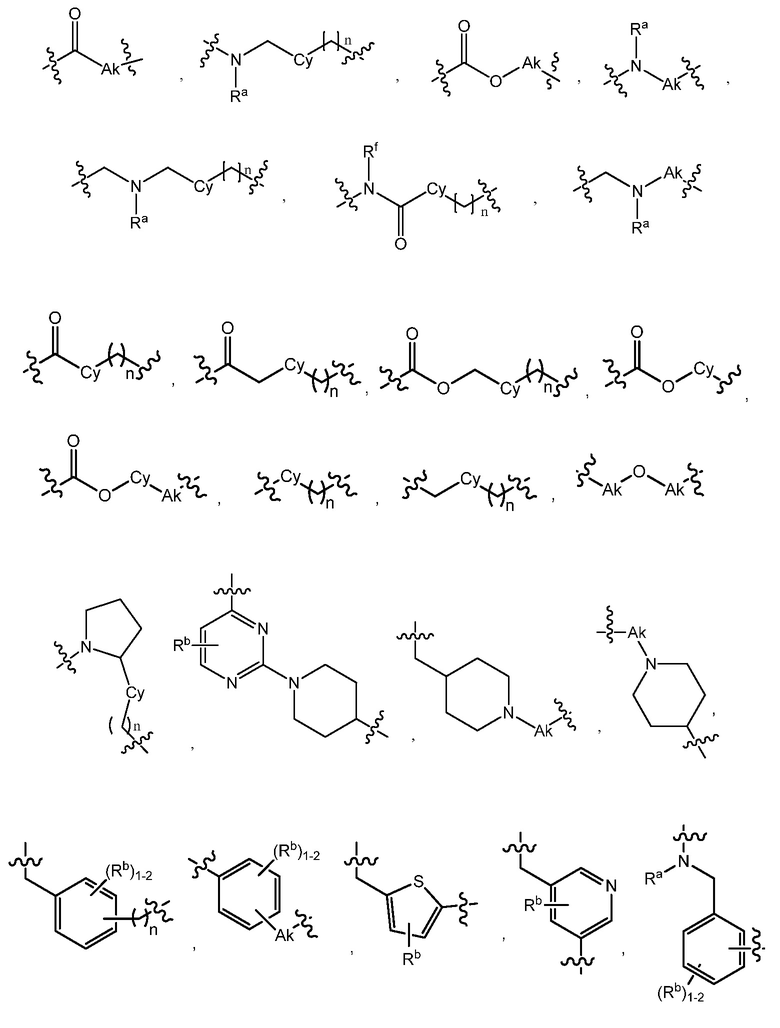
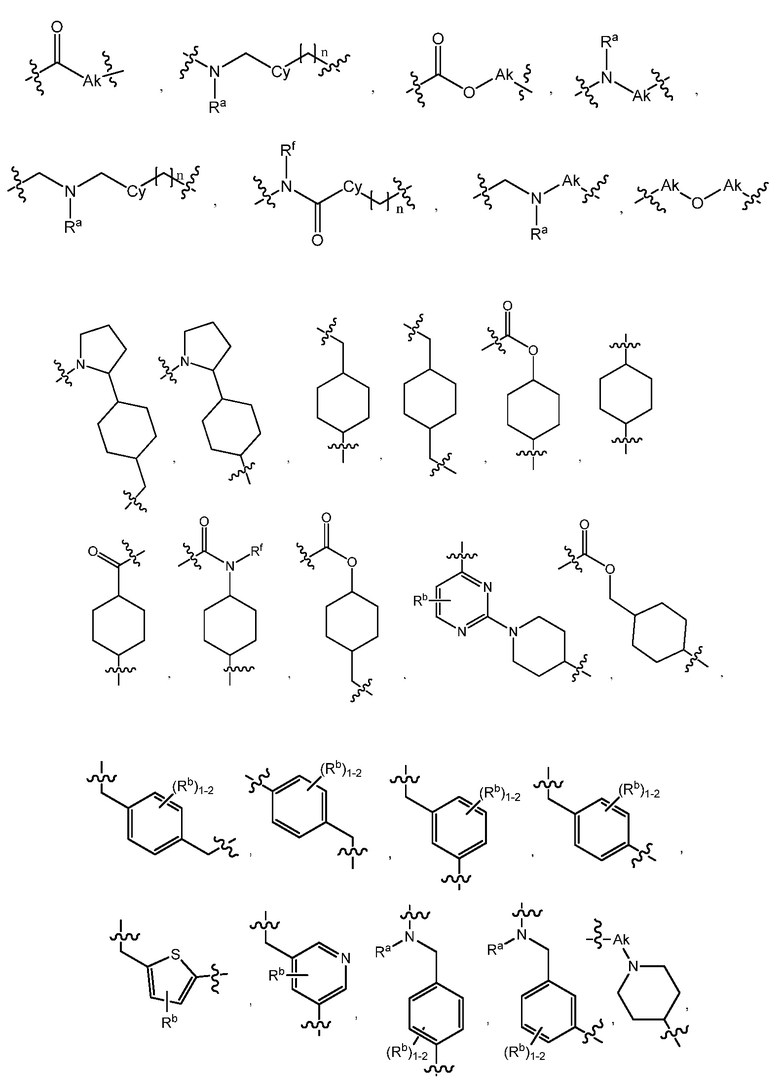
L является линкером, выбранным из C1-7алкила (прямого или разветвленного), или линкером, выбранным из:
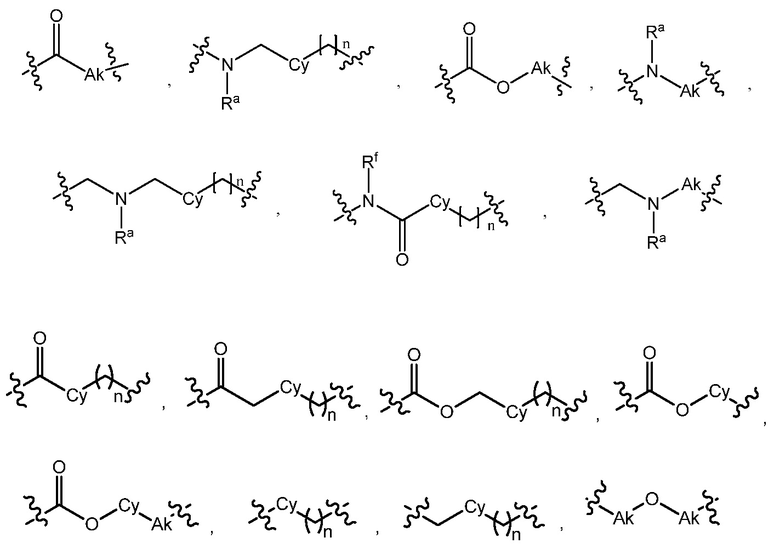
при условии, что центральная структура C и L не образуют N-N связь или N-O связь;
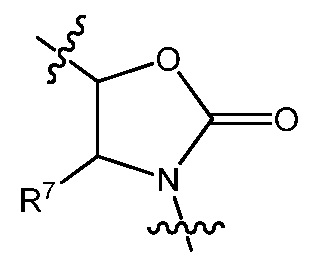
X1 отсутствует или является CR6, где R6 образует с R1 следующее кольцо:
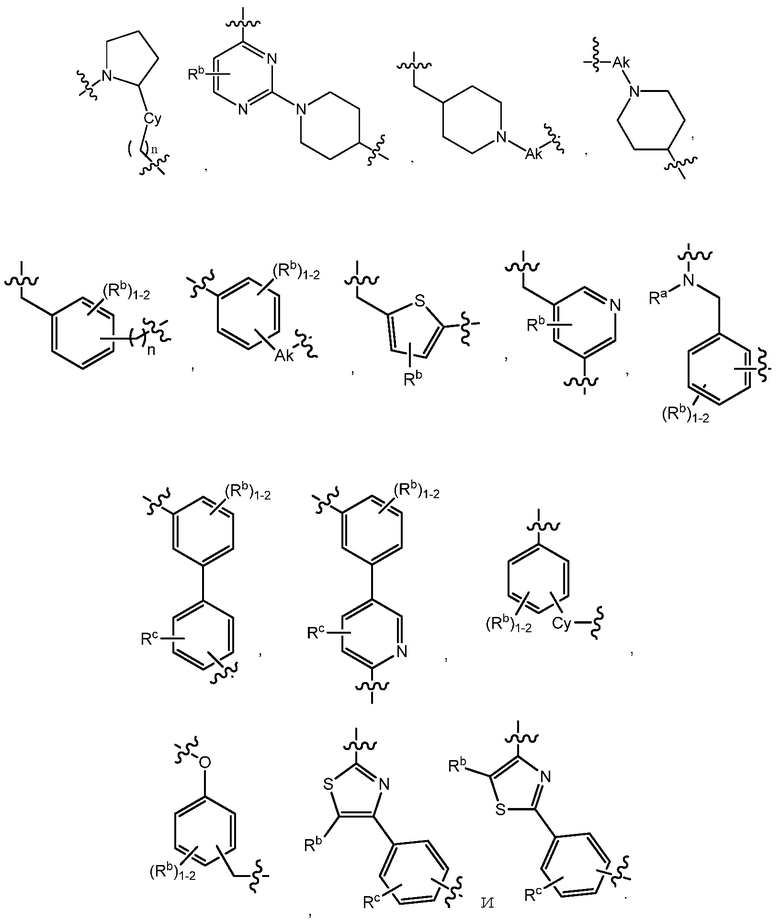
R1 является C(O)O-алкилом, C(O)-алкилом или 5- или 6-членным гетероарилом, необязательно замещенным 1-3 заместителями, независимо выбранными из C1-7алкила, гидрокси-C1-7алкила, C1-7алкокси, который необязательно замещен C1-7алкокси, гидрокси, галогена или -S(O)2C1-4алкила, амино, C1-7алкиламино, ди-C1-7алкиламино, амино-C1-7алкила, C1-7алкиламино-C1-7алкила, ди-C1-7алкиламино-C1-7алкила, (гидроксиС1-7алкил)амино, галогена, бензилокси, 5- или 6-членного гетероциклила или 5- или 6-членного гетероарила, где каждый гетероарил и гетероциклил необязательно замещены 1-3 заместителями, независимо выбранными из оксо, C1-7алкила, C1-7алканоила и гидрокси;
R5 в каждом случае независимо является галогеном, галоген-C1-7алкилом, NO2 или CN;
p является 0, 1 или 2;
n является 0, 1 или 2;
R2, R3 и R4 независимо являются H, C1-7алкилом, галоген-C1-7алкилом, C1-7алкокси-C1-7алкилом или C6-10арил-C1-7алкилом;
R7 является H или C1-7алкилом;
Ra является H, C1-7алкилом или C3-7циклоалкилом, C(O)-C1-7алкилом, C(O)O-C1-7алкилом;
Rb, Rc и Rd независимо являются H, C1-7алкилом, C3-7циклоалкилом, CN, галоген-C1-7алкилом, C1-7алкокси, галогеном, галоген-C1-7алкокси, C3-7циклоалкилокси, фенилом, 5- или 6-членным гетероарильным кольцом или гидрокси;
Rf является H, C1-7алкилом или C3-7циклоалкилом;
Ak является C1-6 линейным или разветвленным алкилом;
Cy является C3-7 циклоалкилом.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
В целях пояснения настоящего описания будут применяться следующие определения, если не определено иное и в любом соответствующем случае, при этом термины, используемые в единственном числе, будут также включать множественное число, и наоборот.
Используемый в настоящем описании термин "алкил" относится к полностью насыщенной, разветвленной или неразветвленной (или прямой, или линейной цепью) углеводородной группе, включающей 1-20 атомов углерода. Предпочтительно алкил включает 1-7 атомов углерода, и более предпочтительно 1-4 атома углерода. Репрезентативные примеры алкила включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил, 3-метилгексил, 2,2-диметилпентил, 2,3-диметилпентил, н-гептил. Термин "C1-7алкил" относится к углеводороду, содержащему от одного до семи атомов углерода. Термин "алкилен" относится к двухвалентному алкильному радикалу, где алкил является таким, как определено выше.
Используемый в настоящем описании термин "галогеналкил" относится к алкилу, определенному в настоящем описании, который замещен одной или более группами галогенов, как определено в настоящем описании. Предпочтительно галогеналкил может быть моногалогеналкилом, дигалогеналкилом или полигалогеналкилом, включая пергалогеналкил. Моногалогеналкил может иметь один атом иода, брома, хлора или фтора в алкильной группе. Дигалогеналкильные и полигалогеналкильные группы могут иметь два или более из тех же атомов галогенов или комбинацию различных групп галогенов в алкиле. Предпочтительно, полигалогеналкил содержит до 12, или 10, или 8, или 6, или 4, или 3, или 2 группы галогенов. Более предпочтительно, полигалогеналкилом является CF3. Репрезентативными примерами галогеналкила являются фторметил, дифторметил, трифторметил, хлорметил, дихлорметил, трихлорметил, пентафторэтил, гептафторпропил, дифторхлорметил, дихлорфторметил, дифторэтил, дифторпропил, дихлорэтил и дихлорпропил. Пергалогеналкил относится к алкилу, все атомы водорода в котором заменены атомами галогена. Термин "галоген-C1-7алкил" относится к углеводороду, содержащему от одного до семи атомов углерода и замещенному одной или более группами галогенов.
Используемый в настоящем описании термин "гидроксиалкил" обозначает алкил, определенный в настоящем описании, который замещен гидроксигруппой.
Используемый в настоящем описании термин "аминоалкил" относится к алкилу, как определено в настоящем описании, замещенному аминогруппой (-NH2). Аналогично, термин "алкиламиноалкил" относится к алкилу, замещенному алкиламиногруппой (алкил-NH-).
Используемый в настоящем описании термин "алкокси" относится к алкил-O-, где алкил определен выше. Репрезентативные примеры алкокси включают, без ограничения перечисленными, метокси, этокси, пропокси, 2-пропокси, бутокси, трет-бутокси, пентилокси, гексилокси, циклопропилокси-, циклогексилокси- и т.п. Предпочтительно алкоксигруппы содержат приблизительно 1-7, более предпочтительно приблизительно 1-4 атомов углерода.
Используемый в настоящем описании термин "галогеналкокси" относится к алкокси, как определено в настоящем описании, который замещен одной или более группами галогенов, как определено в настоящем описании. Предпочтительно галогеналкокси может представлять собой моногалогеналкокси, дигалогеналкокси или полигалогеналкокси, включая пергалогеналкокси.
Используемый в настоящем описании термин "алкоксиалкил" относится к алкилу, определенному в настоящем описании, который замещен алкокси, как определено в настоящем описании. Термин "C1-7алкокси-C1-7алкил" относится к углеводороду, который содержит от одного до семи атомов углерода и замещен алкоксигруппой, содержащей 1-7 атомов углерода.
Используемый в настоящем описании термин "гидроксиалкокси" относится к алкоксигруппе, как определено в настоящем описании, замещенной гидроксигруппой. Термин "гидрокси C1-7алкокси" относится к алкоксигруппе, содержащей 1-7 атомов углерода и замещенной гидроксигруппой.
Используемый в настоящем описании термин "амино" относится к -NH2. Термин "алкиламино" относится к алкил-NH- и термин "диалкиламино" относится к (алкил)2N-, где алкил определен выше.
Используемый в настоящем описании термин "циклоалкил" относится к насыщенной или частично ненасыщенной моноциклической углеводородной группе. Термин "C3-7 циклоалкил" относится к насыщенной или частично ненасыщенной моноциклической углеводородной группе, содержащей 3-7 атомов углерода. Примеры моноциклических углеводородных групп включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил и циклогексенил.
Используемый в настоящем описании термин "циклоалкилалкил" относится к алкилу, определенному в настоящем описании, который замещен циклоалкилом. Термин "C3-7циклоалкил C1-7алкил" относится к углеводороду, содержащему 1-7 атомов углерода, замещенному циклоалкилом, содержащим 3-7 атомов углерода в кольце.
Используемый в настоящем описании термин "циклоалкокси" или "циклоалкилокси" относится попеременно к циклоалкил-O-, где циклоалкил определен в настоящем описании.
Термин "арил" относится к моноциклическим или бициклическим ароматическим углеводородным группам, содержащим 6-10 атомов углерода в кольцевой части. Термин "арил" также относится к группе, в которой ароматическое кольцо конденсировано с циклоалкильным кольцом, где присоединяемый радикал находится на ароматическом кольце или на конденсированном циклоалкильном кольце. Репрезентативными примерами арила является фенил, нафтил, гексагидроиндил, инданил или тетрагидронафтил. Термин "C6-10 арил" относится к ароматическим углеводородным группам, содержащим 6-10 атомов углерода в кольцевой части.
Термин "арилалкил" относится к алкилу, замещенному арилом. Репрезентативными примерами арилалкила является бензил или фенил-CH2CH2-. Термин "C6-10арил-C1-7алкил" относится к углеводороду, содержащему от одного до семи атомов углерода и замещенному арилом, содержащим 6-10 атомов углерода.
Используемый в настоящем описании термин "бензилокси" относится к бензил-O-, где бензил представляет собой фенилСН2-.
Используемый в настоящем описании термин "алканоил" относится к алкил-C(O)-, где алкил определен в настоящем описании.
Термин "гетероарил" включает моноциклический или бициклический гетероарил, содержащий от 5-10 атомов в кольце, выбранных из атомов углерода и 1-5 гетероатомов, при этом каждый гетероатом независимо выбран из O, N или S, где S и N могут быть окислены до различных степеней окисления. Моноциклический гетероарил включает 5- или 6-членный гетероарил, содержащий 1-5 гетероатомов, независимо выбранных из O, N или S, где S и N могут быть окислены до различных степеней окисления. Что касается бициклической гетероарильной системы, система является полностью ароматической (то есть все кольца являются ароматическими).
Типичные моноциклические 5- или 6-членные гетероарильные группы включают тиенил, фурил, пирролил, имидазолил, пиразолил, тиазолил, изотиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, изотиазол-3-ил, изотиазол-4-ил, изотиазол-5-ил, оксазол-2-ил, оксазол-4-ил, оксазол-5-ил, изоксазол-3-ил, изоксазол-4-ил, изоксазол-5-ил, 1,2,4-триазол-3-ил, 1,2,4-триазол-5-ил, 1,2,3-триазол-4-ил, 1,2,3-триазол-5-ил, тетразолил, пирид-2-ил, пирид-3-ил или пиридил-4-ил, пиридазин-3-ил, пиридазин-4-ил, пиразин-3-ил, 2-пиразин-2-ил, пиразин-4-ил, пиразин-5-ил, 2-, 4- или 5-пиримидин-2-ил, пиримидин-4-ил, пиримидин-5-ил. Термин "гетероарил" также относится к группе, в которой гетероароматическое кольцо конденсировано с одним или более арильными кольцами, где радикал или положение присоединения находятся на гетероароматическом кольце или на конденсированном арильном кольце. Репрезентативными примерами бициклического гетероарила являются индолил, изоиндолил, индазолил, индолизинил, пиринил, хинолизинил, хинолинил, изохинолинил, циннолинил, фталазинил, нафтиридинил, хиназолинил, хинаксалинил, фенантридинил, фенантролинил, феназинил, фенотиазинил, феноксазинил, бензизохинолинил, тиено[2,3-b]фуранил, фуро[3,2-b]пиранил, 5H-пиридо[2,3-d]-o-оксазинил, 1H-пиразоло[4,3-d]оксазолил, 4H-имидазо[4,5-d]тиазолил, пиразино[2,3-d]пиридазинил, имидазо[2,1-b]тиазолил, имидазо[1,2-b][1,2,4]триазинил, 7-бензо[b]тиенил, бензоксазолил, бензимидазолил, бензотиазолил, бензоксапинил, бензоксазинил, 1H-пирроло[1,2-b][2]бензазапинил, бензофурил, бензотиофенил, бензотриазолил, пирроло[2,3-b]пиридинил, пирроло[3,2-c]пиридинил, пирроло[3,2-c]пиридинил, пирроло[3,2-b]пиридинил, имидазо[4,5-b]пиридинил, имидазо[4,5-c]пиридинил, пиразоло[4,3-d]пиридинил, пиразоло[4,3-c]пиридинил, пиразоло[3,4-c]пиридинил, пиразоло[3,4-d]пиридинил, пиразоло[3,4-b]пиридинил, имидазо[1,2-a]пиридинил, пиразоло[1,5-a]пиридинил, пирроло[1,2-b]пиридазинил, имидазо[1,2-c]пиримидинил, пиридо[3,2-d]пиримидинил, пиридо[4,3-d]пиримидинил, пиридо[3,4-d]пиримидинил, пиридо[2,3-d]пиримидинил, пиридо[2,3-b]пиразинил, пиридо[3,4-b]пиразинил, пиримидо[5,4-d]пиримидинил, пиразино[2,3-b]пиразинил или пиримидо[4,5-d]пиримидинил.
Используемый в настоящем описании термин "гетероциклил" или "гетероцикло" относится к необязательно замещенному, насыщенному или ненасыщенному неароматическому (частично ненасыщенному) кольцу, которое является 4-, 5-, 6- или 7-членным, моноциклическим и содержит по меньшей мере один гетероатом, выбранный из O, S и N, где N и S также могут быть необязательно окислены до различных степеней окисления. В случае бициклической и трициклической гетероциклильной кольцевой системы, неароматическая кольцевая система определена как неполностью или частично ненасыщенная кольцевая система. Поэтому бициклические и трициклические гетероциклильные кольцевые системы включают гетероциклильные кольцевые системы, в которых одно из конденсированных ядер является ароматическим, а другое(-ие) является неароматическим. В одном варианте осуществления, гетероциклильная группа представляет собой насыщенное моноциклическое ядро, содержащее от 5 до 7 атомов в кольце и необязательно содержащее дополнительный гетероатом, выбранный из O, S или N. Гетероциклическая группа может быть присоединена по гетероатому или атому углерода. Примеры гетероциклов включают дигидрофуранил, диоксоланил, диоксанил, дитианил, пиперазинил, пирролидин, дигидропиранил, оксатиоланил, дитиолан, оксатианил, тиоморфолино, оксиранил, азиридинил, оксетанил, оксепанил, азетидинил, тетрагидрофуранил, тетрагидротиофенил, пирролидинил, тетрагидропиранил, пиперидинил, морфолино, пиперазинил, азепинил, оксапинил, оксаазепанил, оксатианил, тиепанил, азепанил, диоксепанил и диазепанил.
Термин "галоген" или "гало" включает фтор, бром, хлор и иод. Термин "пергалогенированный" обычно относится к группе, в которой все водороды замещены атомами галогена.
Термин "гетероатом" включает атомы любого элемента кроме углерода или водорода. Предпочтительными гетероатомами являются азот, кислород, сера и фосфор. В одном варианте осуществления гетероатомы выбраны из N, O и S.
R1-2 обозначает 1 или 2 группы R. Таким образом, например, (Rb)1-2 обозначает 1 или 2 группы Rb, и, аналогично, (Rd)1-2 обозначает 1 или 2 Rd.
Соединения для способа по изобретению:
В настоящей заявке описаны различные варианты осуществления изобретения. Следует понимать, что признаки, указанные в каждом варианте осуществления, могут быть объединены с другими указанными признаками, с получением дополнительных вариантов осуществления.
В варианте осуществления 1, изобретение относится к соединению следующей формулы I:
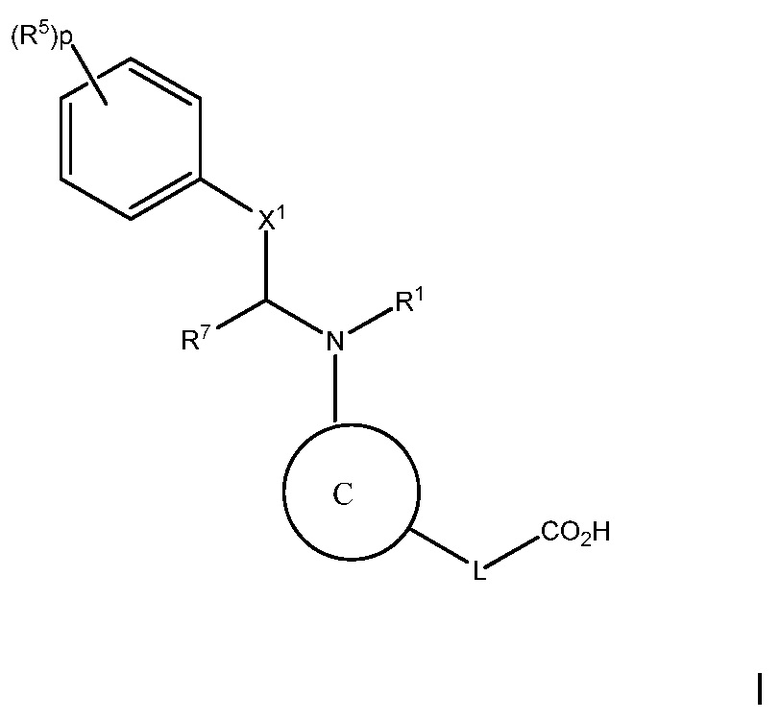
или его фармацевтически приемлемой соли, где:
C представляет собой центральную структуру, выбранную из:
Y и Z независимо представляют собой CH или N;
кольца A и B независимо являются фенилом или 5- или 6-членным гетероарилом, где фенил и гетероарил необязательно замещены 1-3 заместителями, независимо выбранными из C1-7алкила, C1-7алкокси, галогена, галоген-C1-7алкила, галоген-C1-7алкокси, амино, C1-7алкиламино, ди-C1-7алкиламино, C3-7циклоалкила и C3-7циклоалкокси;
D является C3-7циклоалкилом, фенилом, 5- или 6-членным гетероарилом, где фенил и гетероарил необязательно замещены 1-3 заместителями, независимо выбранными из 1-3 заместителей, независимо выбранных из C1-7алкила, C1-7алкокси, CN, NO2, C3-7циклоалкил-C1-7алкила, галогена, галоген-C1-7алкила, галоген-C1-7алкокси, амино, C1-7алкиламино, ди-C1-7алкиламино, C3-7циклоалкила и C3-7циклоалкокси;
L является линкером, выбранным из C1-7алкила (прямого или разветвленного), или линкером, выбранным из:
при условии, что центральная структура C и L не образуют N-N связь или N-O связь;
X1 отсутствует или является CR6, где R6 образует с R1 следующее кольцо:
R1 является C(O)O-алкилом, C(O)-алкилом или 5- или 6-членным гетероарилом, необязательно замещенным 1-3 заместителями, независимо выбранными из C1-7алкила, гидрокси-C1-7алкила, C1-7алкокси, амино, C1-7алкиламино, ди-C1-7алкиламино, амино-C1-7алкила, C1-7алкиламино-C1-7алкила, ди-C1-7алкиламино-C1-7алкила, (гидроксиС1-7алкил)амино, гидрокси-C1-7алкокси, галогена, бензилокси, 5- или 6-членного гетероциклила или 5- или 6-членного гетероарила, где каждый гетероарил и гетероциклил необязательно замещены 1-3 заместителями, независимо выбранными из оксо, C1-7алкила, C1-7алканоила и гидрокси;
R5 в каждом случае независимо является галогеном, галоген-C1-7алкилом, NO2 или CN;
p является 0, 1 или 2;
n является 0, 1 или 2;
R2, R3 и R4 независимо являются H, C1-7алкилом, галоген-C1-7алкилом, C1-7алкокси-C1-7алкилом или C6-10арил-C1-7алкилом;
R7 является H или C1-7алкилом;
Ra является H, C1-7алкилом или C3-7циклоалкилом, C(O)-C1-7алкилом, C(O)O-C1-7алкилом;
Rb, Rc и Rd независимо являются H, C1-7алкилом, C3-7циклоалкилом, CN, галоген-C1-7алкилом, C1-7алкокси, галогеном, галоген-C1-7алкокси, C3-7циклоалкилокси, фенилом, 5- или 6-членным гетероарильным кольцом или гидрокси;
Rf является H, C1-7алкилом или C3-7циклоалкилом;
Ak является C1-6линейным или разветвленным алкилом;
Cy является C3-7 циклоалкилом; для применения в лечении, предотвращении и/или уменьшении атеросклероза или дислипидемии, или для применения в повышении ЛВП-C и/или в снижении ЛНП-C, у субъекта с высоким уровнем триглицеридов.
В варианте осуществления 1A изобретения предложен способ лечения, предотвращения или уменьшении атеросклероза или дислипидемии, или способ повышения ЛВП-C и/или снижения ЛНП-C, у субъекта с высоким уровнем триглицеридов, включающий введение субъекту терапевтически эффективного количества соединения следующей формулы I:
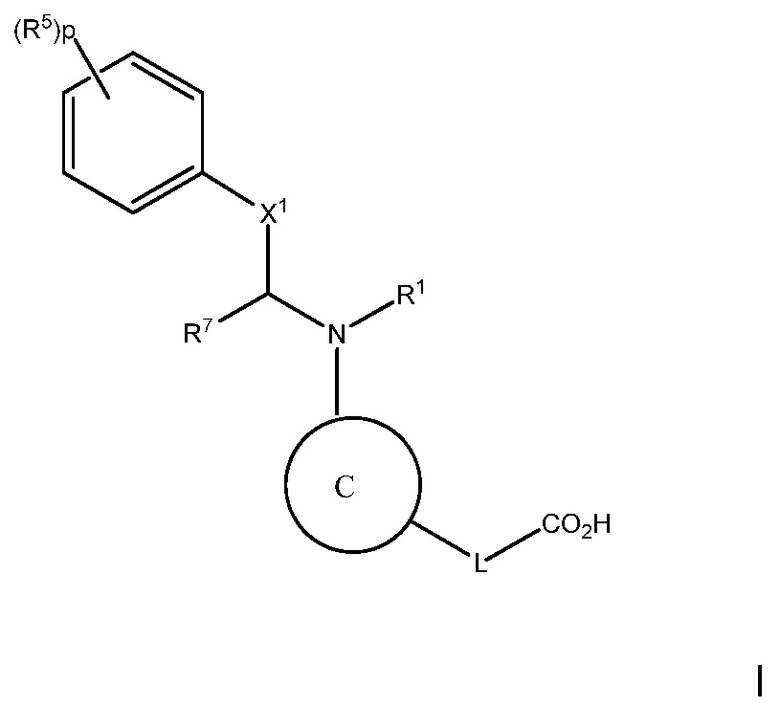
или его фармацевтически приемлемой соли, где:
C представляет собой центральную структуру, выбранную из:
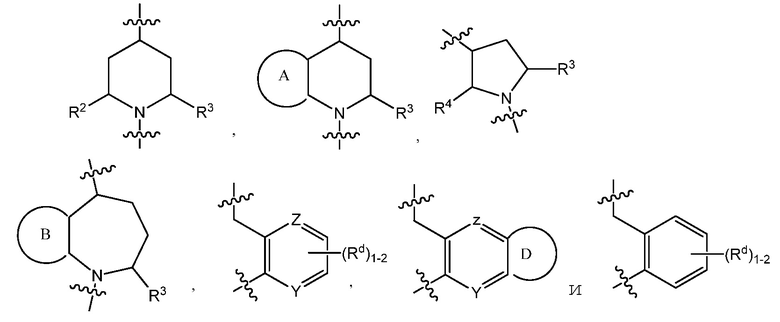
Y и Z независимо представляют собой CH или N;
кольца A и B независимо являются фенилом или 5- или 6-членным гетероарилом, где фенил и гетероарил необязательно замещены 1-3 заместителями, независимо выбранными из C1-7алкила, C1-7алкокси, галогена, галоген-C1-7алкила, галоген-C1-7алкокси, амино, C1-7алкиламино, ди-C1-7алкиламино, C3-7циклоалкила и C3-7циклоалкокси;
D является C3-7циклоалкилом, фенилом, 5- или 6-членным гетероарилом, где фенил и гетероарил необязательно замещены 1-3 заместителями, независимо выбранными из 1-3 заместителей, независимо выбранных из C1-7алкила, C1-7алкокси, CN, NO2, C3-7циклоалкил-C1-7алкила, галогена, галоген-C1-7алкила, галоген-C1-7алкокси, амино, C1-7алкиламино, ди-C1-7алкиламино, C3-7циклоалкила и C3-7циклоалкокси;
L является линкером, выбранным из C1-7алкила (прямого или разветвленного), или линкером, выбранным из:
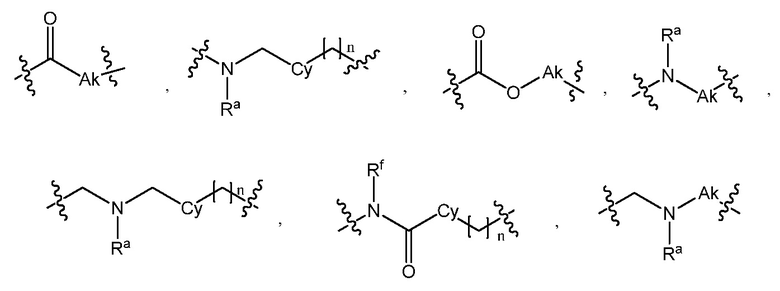
при условии, что центральная структура C и L не образуют N-N связь или N-O связь;
X1 отсутствует или является CR6, где R6 образует с R1 следующее кольцо:
R1 является C(O)O-алкилом, C(O)-алкилом или 5- или 6-членным гетероарилом, необязательно замещенным 1-3 заместителями, независимо выбранными из C1-7алкила, гидрокси-C1-7алкила, C1-7алкокси, амино, C1-7алкиламино, ди-C1-7алкиламино, амино-C1-7алкила, C1-7алкиламино-C1-7алкила, ди-C1-7алкиламино-C1-7алкила, (гидроксиС1-7алкил)амино, гидрокси-C1-7алкокси, галогена, бензилокси, 5- или 6-членного гетероциклила или 5- или 6-членного гетероарила, где каждый гетероарил и гетероциклил необязательно замещены 1-3 заместителями, независимо выбранными из оксо, C1-7алкила, C1-7алканоила и гидрокси;
R5 в каждом случае независимо является галогеном, галоген-C1-7алкилом, NO2 или CN;
p является 0, 1 или 2;
n является 0, 1 или 2;
R2, R3 и R4 независимо являются H, C1-7алкилом, галоген-C1-7алкилом, C1-7алкокси-C1-7алкилом или C6-10арил-C1-7алкилом;
R7 является H или C1-7алкилом;
Ra является H, C1-7алкилом или C3-7циклоалкилом, C(O)-C1-7алкилом, C(O)O-C1-7алкилом;
Rb, Rc и Rd независимо являются H, C1-7алкилом, C3-7циклоалкилом, CN, галоген-C1-7алкилом, C1-7алкокси, галогеном, галоген-C1-7алкокси, C3-7циклоалкилокси, фенилом, 5- или 6-членным гетероарильным кольцом или гидрокси;
Rf является H, C1-7алкилом или C3-7циклоалкилом;
Ak является C1-6линейным или разветвленным алкилом;
Cy является C3-7циклоалкилом.
В варианте осуществления 2 изобретения предложен способ лечения атеросклероза или дислипидемии, или способ повышения ЛВП-C и/или снижения ЛНП-C, включающий:
1. Отбор субъекта с высоким уровнем триглицеридов; и
2. Введение указанному субъекту терапевтически эффективного количества соединения формулы I:
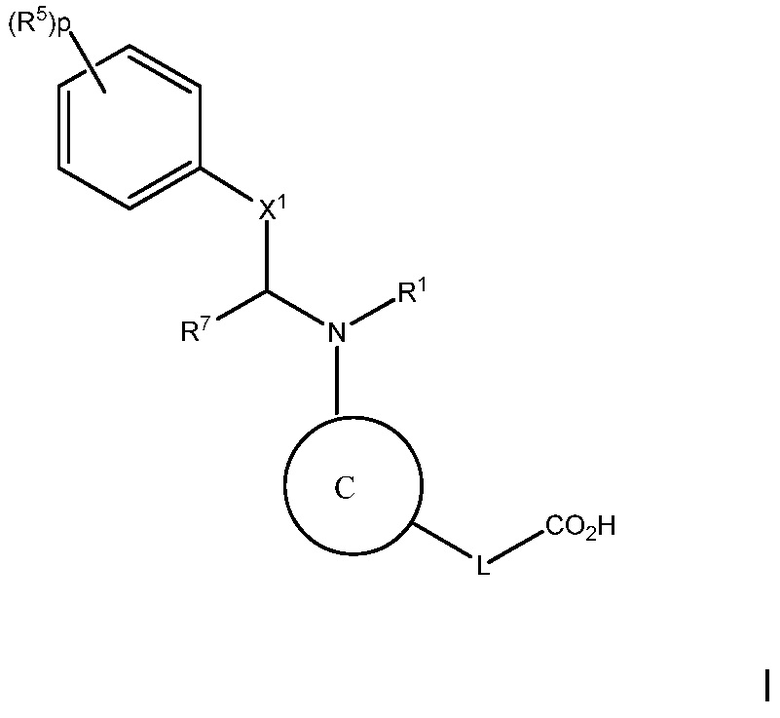
или его фармацевтически приемлемой соли, где:
C представляет собой центральную структуру, выбранную из:
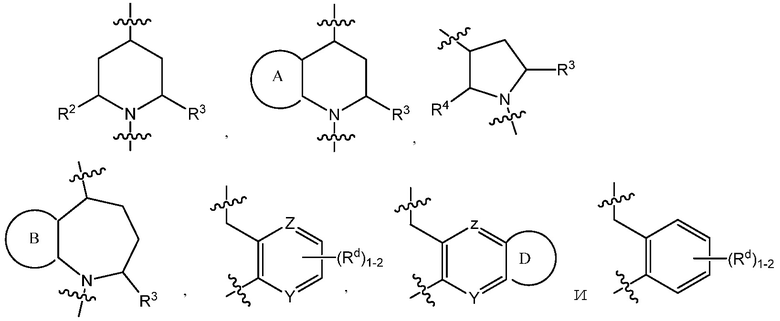
Y и Z независимо представляют собой CH или N;
кольца A и B независимо являются фенилом или 5- или 6-членным гетероарилом, где фенил и гетероарил необязательно замещены 1-3 заместителями, независимо выбранными из C1-7алкила, C1-7алкокси, галогена, галоген-C1-7алкил, галоген-C1-7алкокси, амино, C1-7алкиламино, ди-C1-7алкиламино, C3-7циклоалкила и C3-7циклоалкокси;
D является C3-7циклоалкилом, фенилом, 5- или 6-членным гетероарилом, где фенил и гетероарил необязательно замещены 1-3 заместителями, независимо выбранными из 1-3 заместителей, независимо выбранных из C1-7алкила, C1-7алкокси, CN, NO2, C3-7циклоалкил-C1-7алкила, галогена, галоген-C1-7алкила, галоген-C1-7алкокси, амино, C1-7алкиламино, ди-C1-7алкиламино, C3-7циклоалкила и C3-7циклоалкокси;
L является линкером, выбранным из C1-7алкила (прямого или разветвленного), или линкером, выбранным из:
при условии, что центральная структура C и L не образуют N-N связь или N-O связь;
X1 отсутствует или является CR6, где R6 образует с R1 следующее кольцо:
R1 является C(O)O-алкилом, C(O)-алкилом или 5- или 6-членным гетероарилом, необязательно замещенным 1-3 заместителями, независимо выбранными из C1-7алкила, гидрокси-C1-7алкила, C1-7алкокси, амино, C1-7алкиламино, ди-C1-7алкиламино, амино-C1-7алкила, C1-7алкиламино-C1-7алкила, ди-C1-7алкиламино-C1-7алкила, (гидроксиС1-7алкил)амино, гидрокси-C1-7алкокси, галогена, бензилокси, 5- или 6-членного гетероциклила или 5- или 6-членного гетероарила, где каждый гетероарил и гетероциклил необязательно замещены 1-3 заместителями, независимо выбранными из оксо, C1-7алкила, C1-7алканоила и гидрокси;
R5 в каждом случае независимо является галогеном, галоген-C1-7алкилом, NO2 или CN;
p является 0, 1 или 2;
n является 0, 1 или 2;
R2, R3 и R4 независимо являются H, C1-7алкилом, галоген-C1-7алкилом, C1-7алкокси-C1-7алкилом или C6-10арил-C1-7алкилом;
R7 является H или C1-7алкилом;
Ra является H, C1-7алкилом или C3-7циклоалкилом, C(O)-C1-7алкилом, C(O)O-C1-7алкилом;
Rb, Rc и Rd независимо являются H, C1-7алкилом, C3-7циклоалкилом, CN, галоген-C1-7алкилом, C1-7алкокси, галогеном, галоген-C1-7алкокси, C3-7циклоалкилокси, фенилом, 5- или 6-членным гетероарильным кольцом или гидрокси;
Rf является H, C1-7алкилом или C3-7циклоалкилом;
Ak является C1-6 линейным или разветвленным алкилом;
Cy является C3-7циклоалкилом.
В варианте осуществления 3 изобретение относится к способу или применению согласно варианту осуществления 1, 1A или 2, где C представляет собой центральную структуру, выбранную из:
Y и Z независимо представляют собой CH или N;
кольца A и B независимо являются фенилом или 5- или 6-членным гетероарилом, где фенил и гетероарил необязательно замещены 1-3 заместителями, независимо выбранными из C1-7алкила, C1-7алкокси, галогена, галоген-C1-7алкила, галоген-C1-7алкокси, амино, C1-7алкиламино, ди-C1-7алкиламино, C3-7циклоалкила и C3-7циклоалкокси;
D является C3-7циклоалкилом, фенилом, 5- или 6-членным гетероарилом, где фенил и гетероарил необязательно замещены 1-3 заместителями, независимо выбранными из 1-3 заместителей, независимо выбранных из C1-7алкила, C1-7алкокси, CN, NO2, C3-7циклоалкил-C1-7алкила, галогена, галоген-C1-7алкила, галоген-C1-7алкокси, амино, C1-7алкиламино, ди-C1-7алкиламино, C3-7циклоалкила и C3-7циклоалкокси;
L является линкером, выбранным из прямого или разветвленного C1-7алкила, или линкером, выбранным из:
при условии, что центральная структура C и L не образуют N-N связь или N-O связь;
X1 отсутствует или является CR6, где R6 образует с R1 следующее кольцо:
R1 является C(O)O-алкилом, C(O)-алкилом или 5- или 6-членным гетероарилом, необязательно замещенным 1-3 заместителями, независимо выбранными из C1-7алкила, гидрокси-C1-7алкила, C1-7алкокси, амино, C1-7алкиламино, ди-C1-7алкиламино, амино-C1-7алкила, C1-7алкиламино-C1-7алкила, ди-C1-7алкиламино-C1-7алкила, (гидроксиС1-7алкил)амино, гидрокси-C1-7алкокси, галогена, бензилокси, 5- или 6-членного гетероциклила или 5- или 6-членного гетероарила, где каждый гетероарил и гетероциклил необязательно замещены 1-3 заместителями, независимо выбранными из оксо, C1-7алкила, C1-7алканоила и гидрокси;
R5 в каждом случае независимо является галогеном, галоген-C1-7алкилом, NO2 или CN;
p является 0, 1 или 2;
n является 0, 1 или 2;
R2, R3 и R4 независимо являются H, C1-7алкилом, галоген-C1-7алкилом, C1-7алкокси-C1-7алкилом или C6-10арил-C1-7алкилом;
R7 является H или C1-7алкилом;
Ra является H, C1-7алкилом или C3-7циклоалкилом, C(O)-C1-7алкилом, C(O)O-C1-7алкилом;
Rb, Rc и Rd независимо являются H, C1-7алкилом, C3-7циклоалкилом, CN, галоген-C1-7алкилом, C1-7алкокси, галогеном, галоген-C1-7алкокси, C3-7циклоалкилокси, фенилом, 5- или 6-членным гетероарильным кольцом или гидрокси;
Rf является H, C1-7алкилом или C3-7циклоалкилом;
Ak является C1-6линейным или разветвленным алкилом;
Cy является C3-7циклоалкилом; или его фармацевтически приемлемая соль.
В варианте осуществления 4 изобретение относится к способу или применению согласно любому из вариантов осуществления 1, 1A, 2 и 3, где соединение имеет Формулу II:
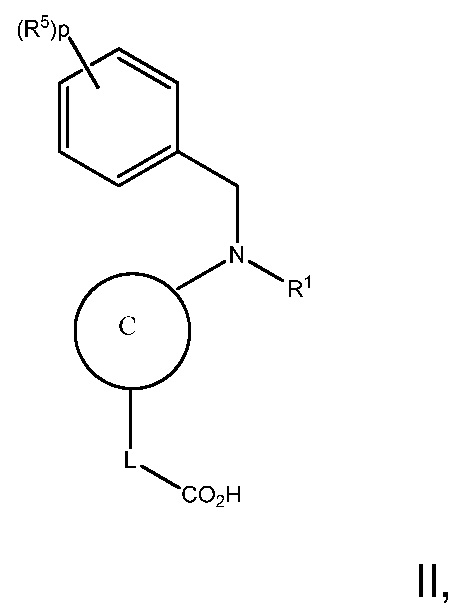
или его фармацевтически приемлемая соль, где R1, R5, C, L и p определены в вариантах осуществления 1, 1A, 2 или 3, выше.
В варианте осуществления 5 изобретение относится к способу или применению согласно любому из вариантов осуществления 1, 1A, 2, 3 и 4, где соединение имеет Формулу III:
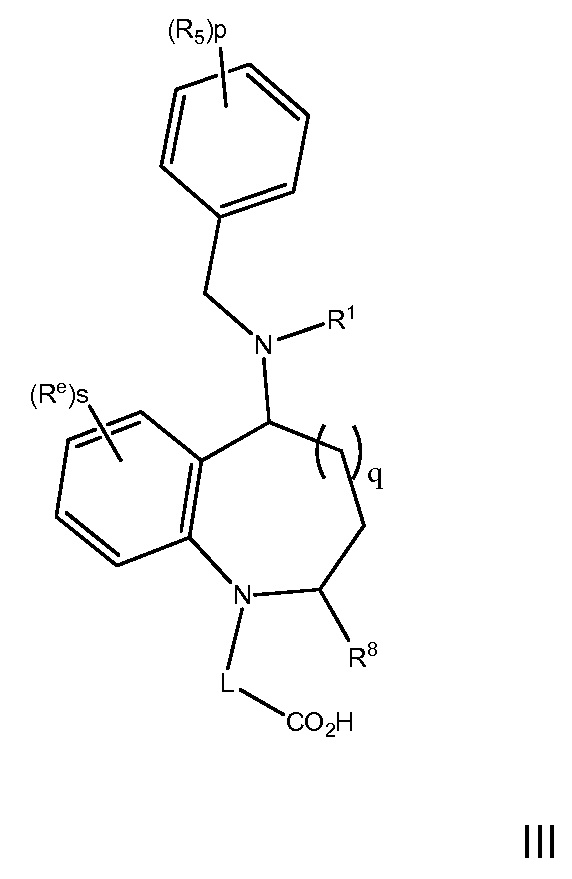
где R1, R5, L и p определены в варианте осуществления 1, 1A, 2 или 3, выше; и Re является C1-7алкилом, C1-7алкокси, галоген-C1-7алкокси, галоген-C1-7алкилом, гидрокси, q является 0 или 1; s является 0, 1, 2 или 3, и R8 является H или C1-7алкилом, или его фармацевтически приемлемая соль.
В варианте осуществления 5A изобретение относится к способу или применению согласно варианту осуществления 5, где соединения являются соединениями Формулы III, в которой q является 1, раскрытыми в заявках на патент США US 2007/244095 и US 2008/269284 (соответствующей WO 2006/002342) и заявке PCT WO 2011/002696, которые включены в настоящую заявку посредством ссылки. Примерами соединений Формулы III, которые раскрыты в WO 2006/002342, являются соединения примеров 31, 32, 89, 91, 92, 94, 101, 111, 122, 123, 125, 130, 134, 141-143, 153, 154, 175, 176, 197, 198 и 201-204, или их фармацевтически приемлемая соль.
В варианте осуществления 5B изобретение относится к способу или применению согласно варианту осуществления 5, где соединения являются соединениями Формулы III, в которой q является 0, раскрытыми в US 2006/0063803, которая включена в настоящее описание посредством ссылки. Примерами соединений Формулы III, которые раскрыты в US 2006/0063803, являются соединения примеров 5, 6, 7, 10, 18, 26, 69, 70-74, 78 и 82-84, или их фармацевтически приемлемая соль.
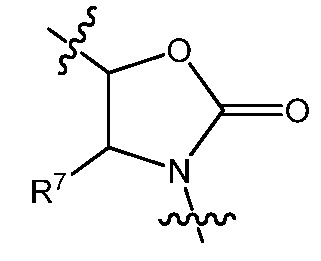
В варианте осуществления 5C изобретение относится к способу или применению согласно варианту осуществления 5, где q является 1, и R8 является H, представленному Формулой IIIA:
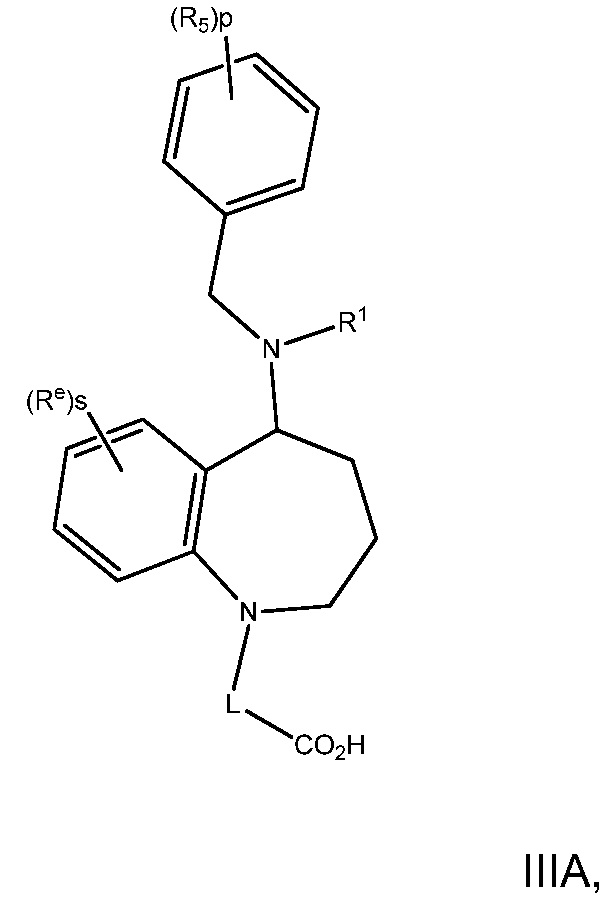
или его фармацевтически приемлемой соли, где R5, R1, L, Re, p и s определены в вариантах осуществления 1, 1A, 2, 3, 4 и 5, выше.
В варианте осуществления 6 изобретение относится к варианту осуществления 5 или 5C, где линкер L является C1-6алкилом или линкером, выбранным из:
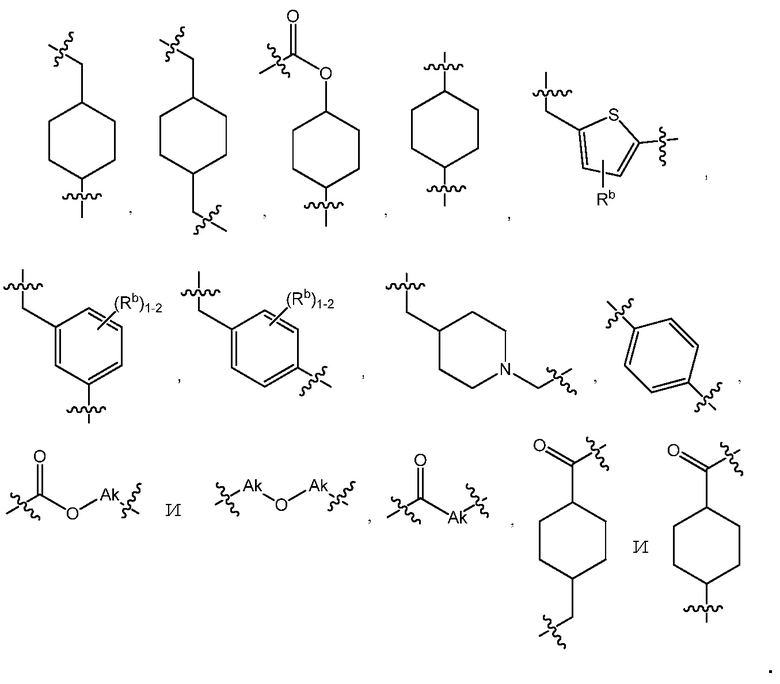
В варианте осуществления 7 изобретение относится к способу или применению согласно варианту осуществления 5, 5C или 6, где L выбран из:
В варианте осуществления 8 изобретение относится к способу или применению согласно любому из вариантов осуществления 4-7, где R1 представляет собой тетразол, необязательно замещенный C1-4 алкилом; или их фармацевтически приемлемой соли.
В варианте осуществления 8A изобретение относится к способу или применению варианта осуществления 8, где соединением Формулы III является:
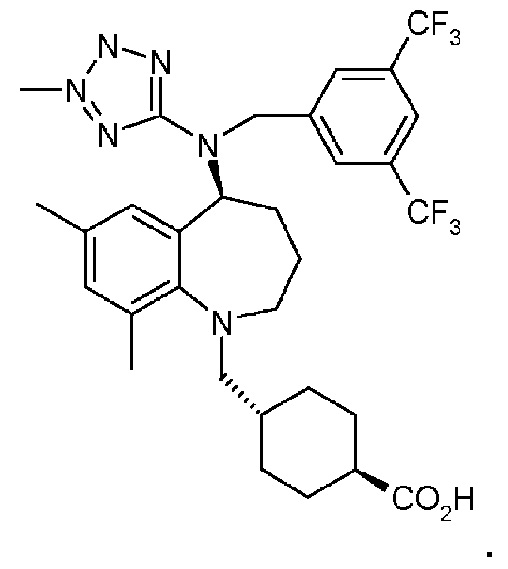
В варианте осуществления 9 изобретение относится к способу или применению согласно любому из вариантов осуществления 1, 1A, 2, 3 и 4, где соединение имеет Формулу IV:
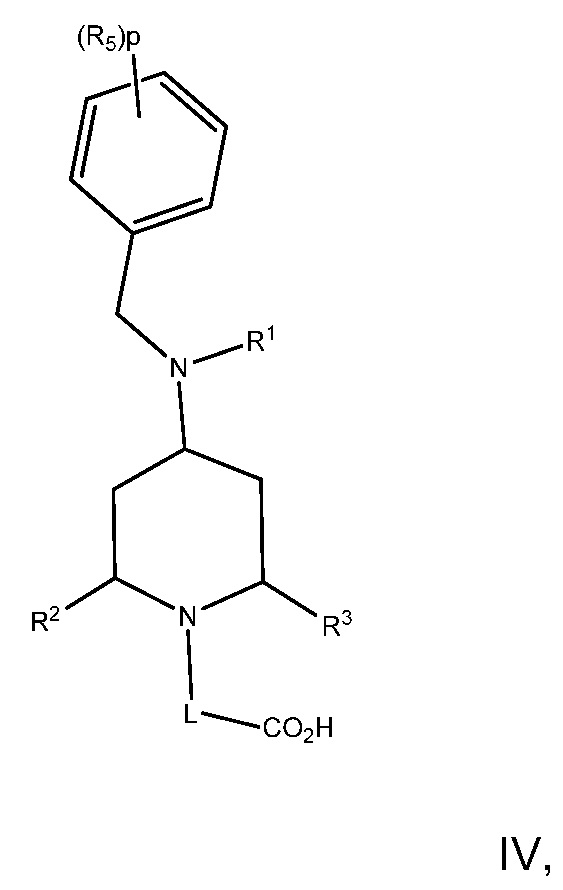
или его фармацевтически приемлемой соли, где R1, R2, R3, R5, L и p определены в варианте осуществления 1, 1A или 2, выше.
В варианте осуществления 10 изобретение относится к способу или применению согласно варианту осуществления 9, где соединение имеет Формулу IVA:
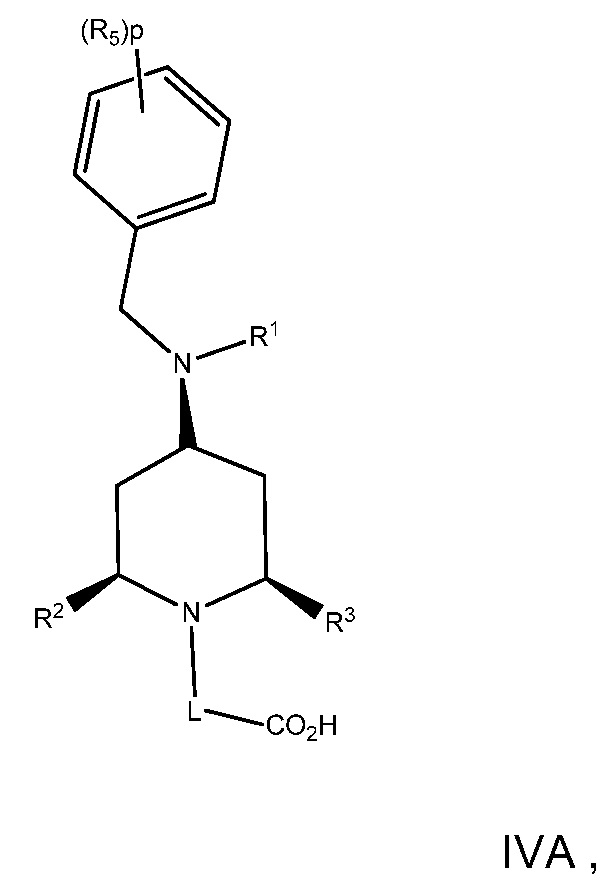
или его фармацевтически приемлемой соли.
В варианте осуществления 10A изобретение относится к способу или применению согласно варианту осуществления 10, где соединением Формулы IV или IVA являются соединения, раскрытые в US 2009/0118287 (WO 2009/059943), которая включена в настоящее описание посредством ссылки.
В варианте осуществления 11 изобретение относится к способу или применению согласно варианту осуществления 9 или 10, где L выбран из:
В варианте осуществления 12 изобретение относится к способу или применению согласно варианту осуществления 11, где L выбран из:
В варианте осуществления 13 изобретение относится к способу или применению согласно любому из вариантов осуществления 9-12, где R1 является 5- или 6-членным гетероарилом, где указанный гетороарил необязательно замещен одним-тремя заместителями, выбранными из галогена, C1-7алкила, гидрокси-C1-7алкила, ди-C1-7алкиламино, C1-7алкокси, 5- или 6-членного гетероциклила или 5- или 6-членного гетероарила, где указанные гетероциклил и гетероарил также необязательно замещены одним-тремя заместителями, выбранными из C1-7алкила, C1-7алканоила или гидрокси.
В варианте осуществления 14 изобретение относится к способу или применению согласно варианту осуществления 13, где R1 является пиримидином, замещенным морфолино, имидазолилом, пиразоилом или тетразолилом, где имидазолил, пиразоил и тетразолил необязательно замещены C1-7алкилом.
В варианте осуществления 15 изобретение относится к способу или применению согласно любому из вариантов осуществления 9-14, где R2 и R3 независимо являются C1-4алкилом.
В варианте осуществления 16 изобретение относится к способу или применению согласно любому из вариантов осуществления 1, 1A, 2, 3 и 4, где соединение имеет Формулу V:
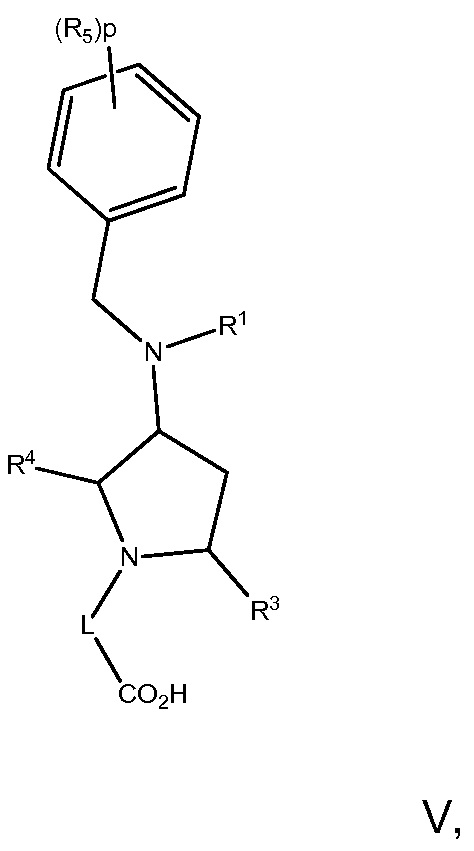
или его фармацевтически приемлемой соли, где R1, R3, R4, R5, L и p определены в варианте осуществления 1, 1A или 2, выше.
В варианте осуществления 17 изобретение относится к способу или применению согласно варианту осуществления 16, где соединение имеет Формулу VA:
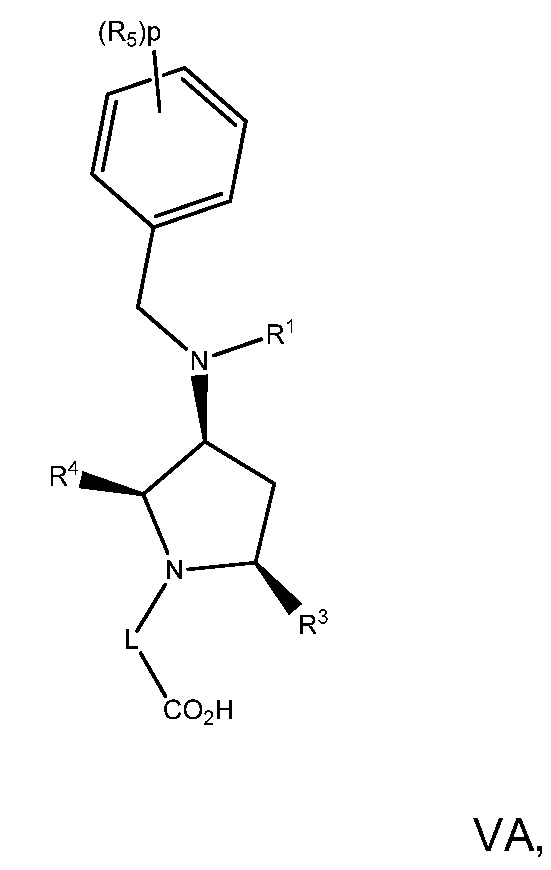
или его фармацевтически приемлемой соли.
В варианте осуществления 17A изобретение относится к способу или применению согласно варианту осуществления 17, где соединение является соединением Формулы V или VA, раскрытым в US 2010/0311750 (WO 2009/071509), которая включена в настоящее описание посредством ссылки.
В варианте осуществления 18 изобретение относится к способу или применению согласно варианту осуществления 16 или 17, где L выбран из:
В варианте осуществления 19 изобретение относится к способу или применению согласно варианту осуществления 18, где L выбран из:
В варианте осуществления 20 изобретение относится к способу или применению согласно любому из вариантов осуществления 16-19, где R3 является C1-7алкилом, и R4 является H.
В варианте осуществления 21 изобретение относится к способу или применению согласно любому из вариантов осуществления 16-20, где R1 является 5- или 6-членным гетероарилом, где указанный гетероарил необязательно замещен одним-тремя заместителями, выбранными из галогена, C1-7алкила, гидрокси-C1-7алкила, ди-C1-7алкиламино, C1-7алкокси, 5- или 6-членного гетероциклила или 5- или 6-членного гетероарила, где указанный гетероциклил и гетероарил также необязательно замещены одним-тремя заместителями, выбранными из C1-7алкила, C1-7алканоила или гидрокси.
В варианте осуществления 22 изобретение относится к способу или применению согласно варианту осуществления 21, где R1 является пиримидином, замещенным морфолино, имидазолилом, пиразоилом или тетразолилом, где имидазолил, пиразоил и тетразолил необязательно замещены C1-7алкилом.
В варианте осуществления 23 изобретение относится к способу или применению согласно варианту осуществления 1, 1A, 2, 3 или 4, где соединение имеет Формулу VI:
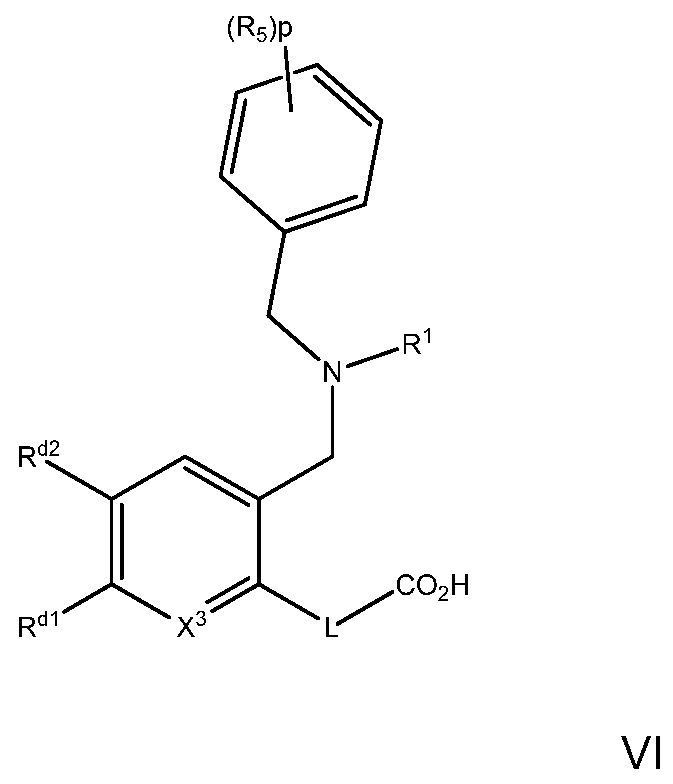
где R5, R1, L и p определены в варианте осуществления 1, 1A или 2, выше; и
X3 представляет собой CH или N; и
Rd1 и Rd2 независимо выбраны из H, C1-7алкила, галогена, галоген-C1-7алкила; или Rd1 и Rd2, вместе с атомами, к которым они присоединены, образуют фенил, необязательно замещенный 1-3 заместителями, независимо выбранными из галогена, C1-7алкила, галоген-C1-7алкила; или
его фармацевтически приемлемой соли.
В варианте осуществления 23A изобретение относится к способу или применению согласно варианту осуществления 23, где соединения являются соединениями Формулы VI, которые раскрыты в US 2009/0075968 (WO 2007/073934); в US 2009/0227580 (WO 2007/128568) и в WO 2004/020393, которые включены в настоящее описание посредством ссылки.
В варианте осуществления 24 изобретение относится к способу или применению согласно варианту осуществления 23, где L выбран из:
В варианте осуществления 25 изобретение относится к способу или применению согласно варианту осуществления 24, где L выбран из:
В варианте осуществления 26 изобретение относится к способу или применению согласно любому из вариантов осуществления 23-25, где R1 является тетразолом, который необязательно замещен C1-4алкилом.
В варианте осуществления 27 изобретение относится к способу или применению согласно любому из вариантов осуществления 23-26, где X3 представляет собой N, Rd1 представляет собой H, и Rd2 представляет собой галоген-C1-7алкил; или Rd1 и Rd2, вместе с атомами, к которым они присоединены, образуют фенил, который необязательно замещен 1-2 заместителями, независимо выбранными из галогена, C1-7алкила, галоген-C1-7алкила.
В варианте осуществления 27A изобретение относится к способу или применению согласно любому из вариантов осуществления 23-26, где X3 представляет собой CH, Rd1 является H или C1-7алкилом, и Rd2 является галогеном или галологен-C1-7алкилом. В другом аспекте варианта осуществления 27A изобретение относится к способу или применению, где соединения являются соединениями Формулы VI, как раскрыто в WO 2004/020393.
В варианте осуществления 27B изобретение относится к способу или применению согласно варианту осуществления 1, 1A, 2 или 3, где соединение имеет Формулу VIA:
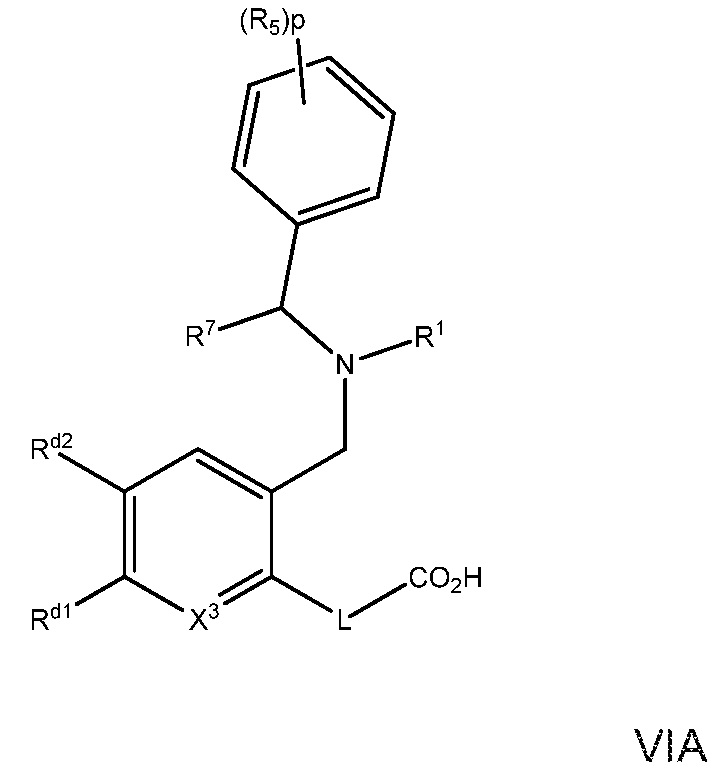
где R5, R7, R1, L и p определены в варианте осуществления 1, 1A или 2, выше; и
X3 представляет собой CH или N; и
Rd1 и Rd2 независимо выбраны из H, C1-7алкила, галогена, галоген-C1-7алкила; или
его фармацевтически приемлемая соль.
В варианте осуществления 27C изобретение относится к способу или применению согласно варианту осуществления 27B, где L выбран из:
В варианте осуществления 27D изобретение относится к способу или применению, где соединения являются соединениями Формулы VIA, которые раскрыты в US 2009/082352, которая включена в настоящее описание посредством ссылки.
В варианте осуществления 27E изобретение относится к способу или применению согласно варианту осуществления 27B, 27C или 27D, где R7 является алкилом, и R1 является пиримидином, который необязательно замещен C1-7алкокси, где алкокси также необязательно замещен -S(O)2-C1-4алкилом.
В варианте осуществления 27F изобретение относится к способу или применению согласно варианту осуществления 27B, 27C, 27D или 27E, где X3 представляет собой CH, Rd1 является H или C1-7алкилом, и Rd2 является галоген-C1-7алкилом.
В варианте осуществления 28 изобретение относится к способу или применению согласно варианту осуществления 1, 1A, 2 или 3, имеющему Формулу VII:
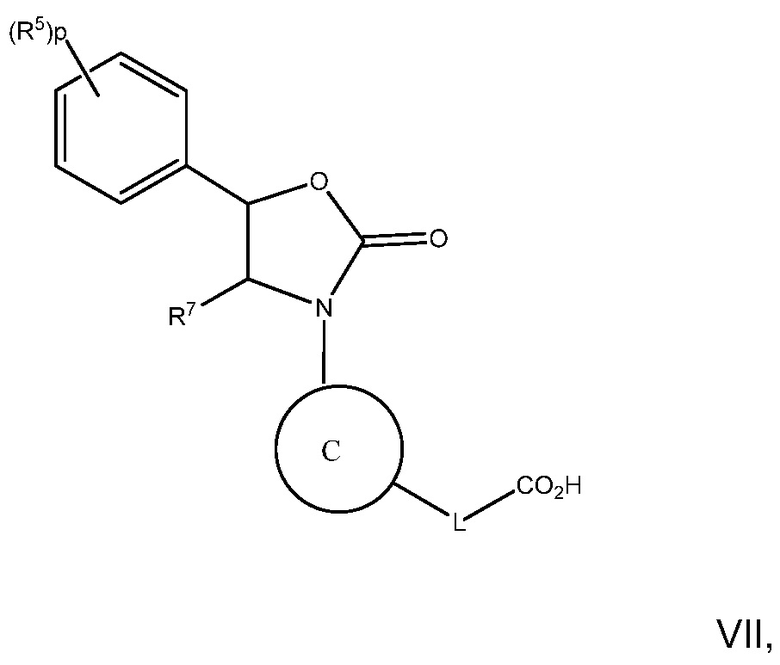
где R5, R7, L и p определены в варианте осуществления 1, 1A или 2; выше;
или его фармацевтически приемлемой соли.
В варианте осуществления 29 изобретение относится к способу или применению согласно варианту осуществления 28, имеющему Формулу VIIA:
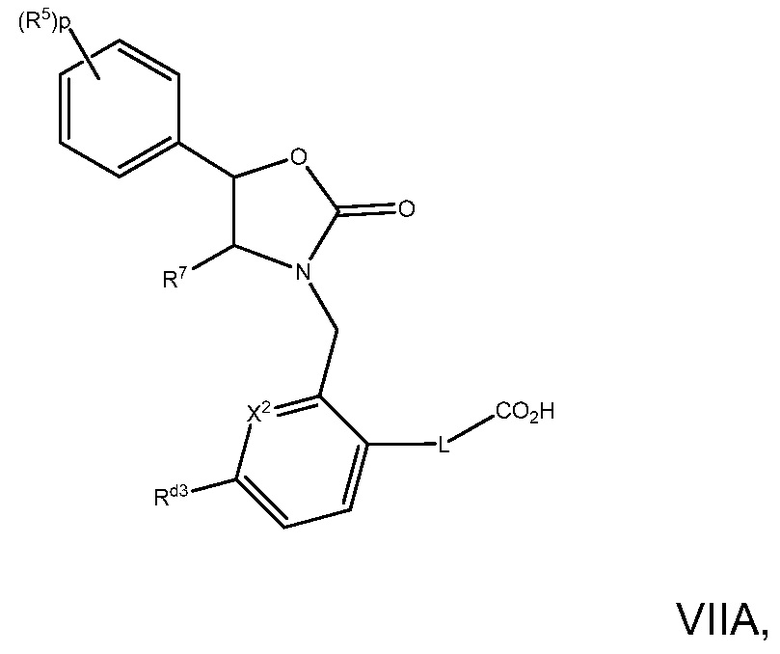
где X2 представляет собой CRd4 или N, Rd3 является C1-7алкокси, C1-7алкилом, галоген-C1-7алкилом, галоген-C1-7алкокси, Rd4 представляет собой H, или Rd3 и Rd4 вместе с 5- или 6-членным гетероциклилом или гетероарилом могут образовывать фенил или циклоалкил; или его фармацевтически приемлемой соли.
В варианте осуществления 29A изобретение относится к способу или применению согласно варианту осуществления 29, где соединения являются соединениями Формулы VII, которые раскрыты в заявках США US 2009/075979 (соответствующий WO 2007/081571) и US 2009/042892 (соответствующей WO 2007/081569), которые включены в настоящее описание посредством ссылки. Примерами соединений Формулы VII, которые раскрыты в WO 2007/081571, являются соединения примеров 13-15, 63-67, 69-77, 79-82, 84, 87 и 89, или их фармацевтически приемлемая соль. Примерами соединений Формулы VII, которые раскрыты в WO 2007/081569, являются соединения примеров 62, 64, 108-111, 113-118, 120, 121, 123, 125 и 127, или их фармацевтически приемлемая соль.
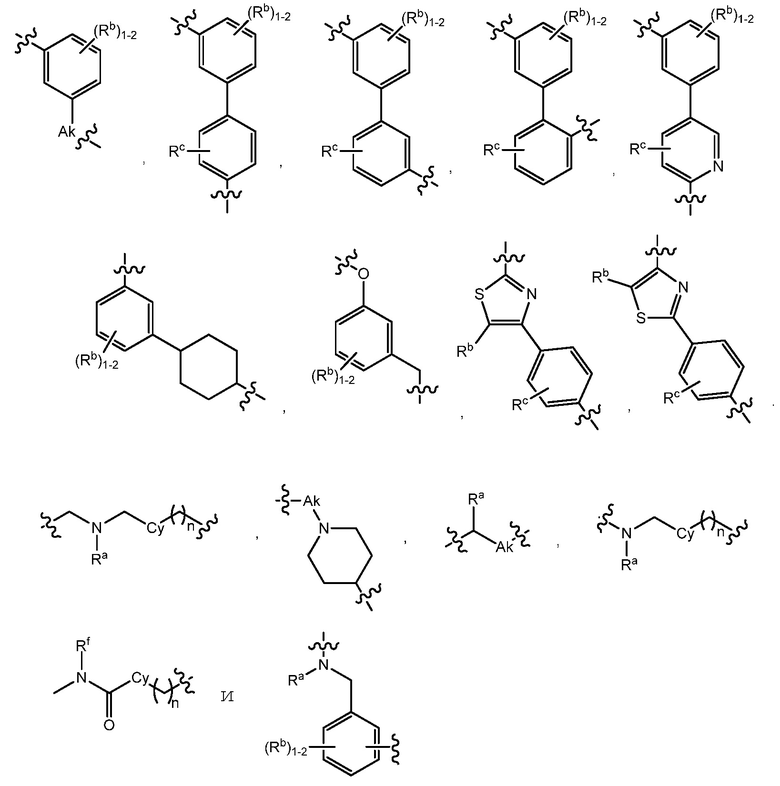
В варианте осуществления 30 изобретение относится к способу или применению согласно пункту 28 или 29, где L выбран из:
В варианте осуществления 31 изобретение относится к способу или применению согласно любому из вариантов осуществления 28-30, где L представляет собой:
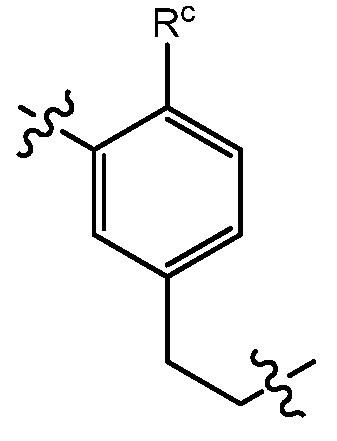
где Rc представляет собой C1-7алкокси или галоген-C1-7алкокси.
В варианте осуществления 32 изобретение относится к способу или применению согласно вариантам осуществления 1, 1A или 2, где соединение выбрано из:
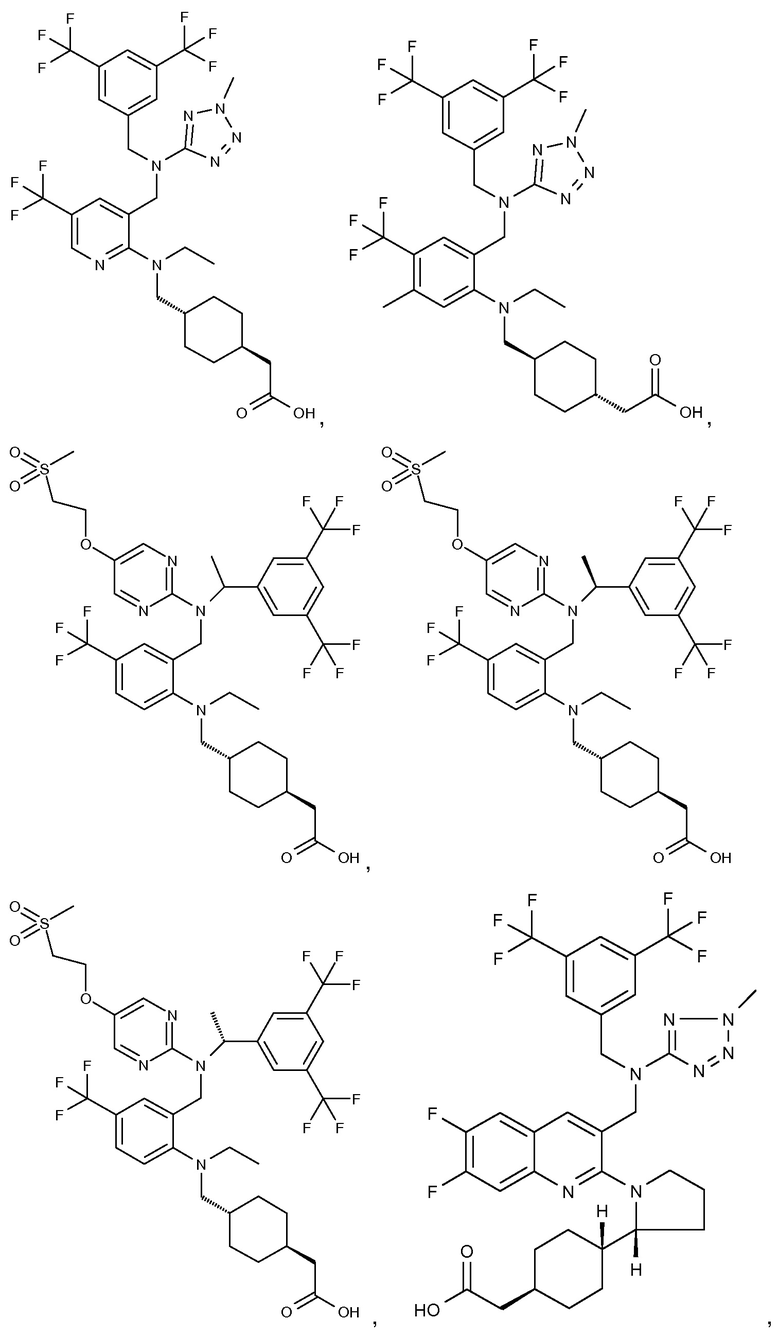
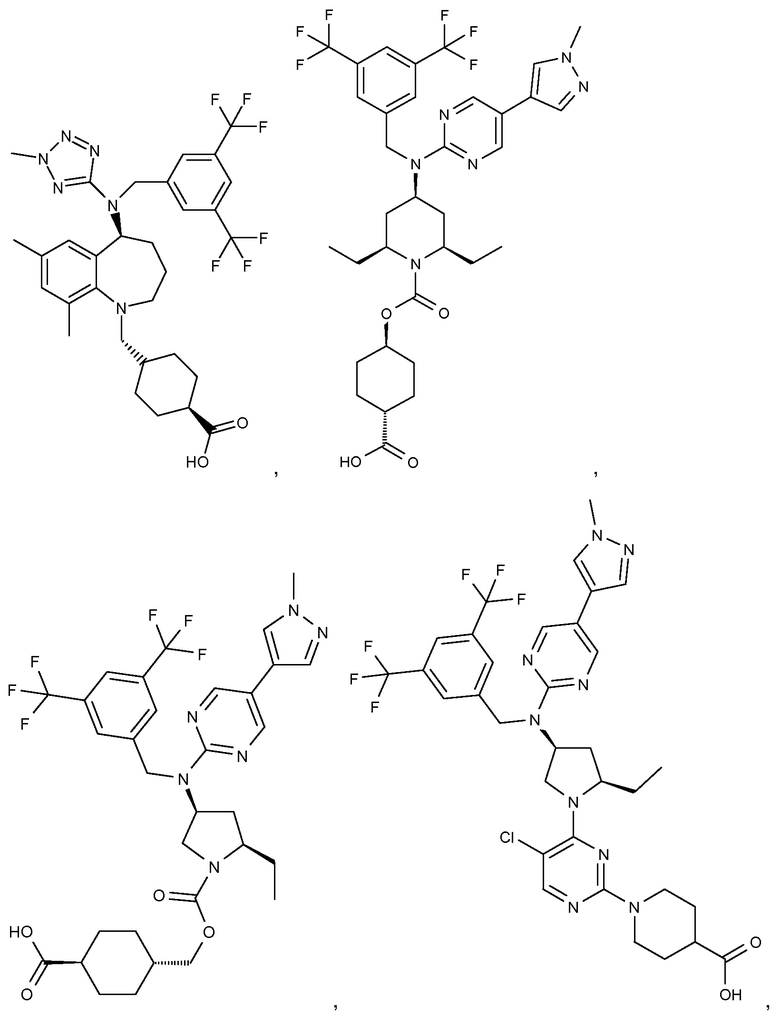
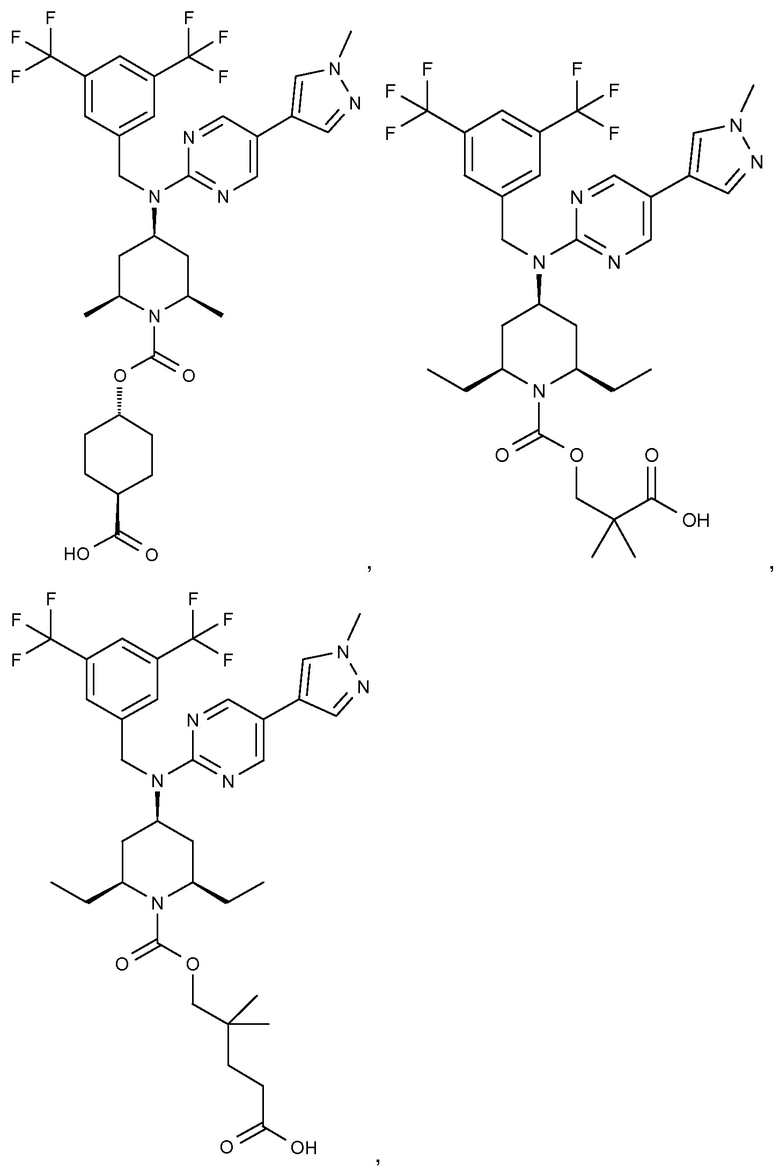
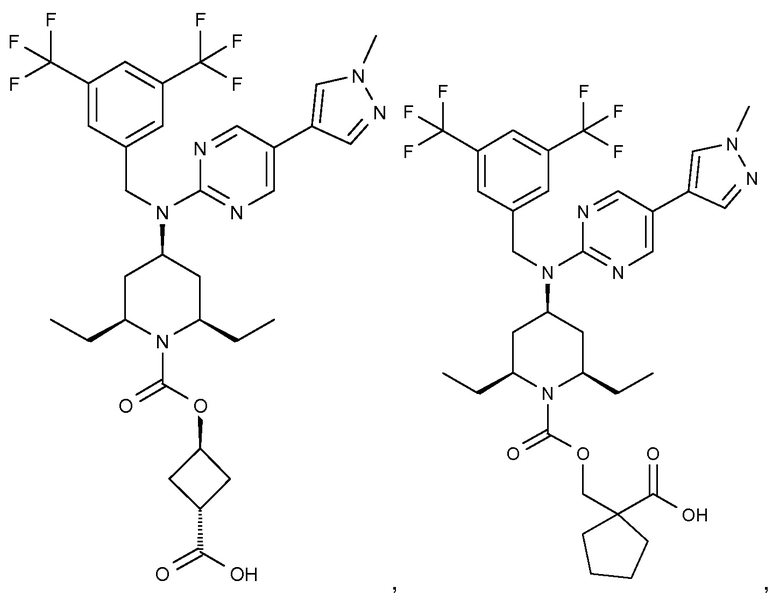
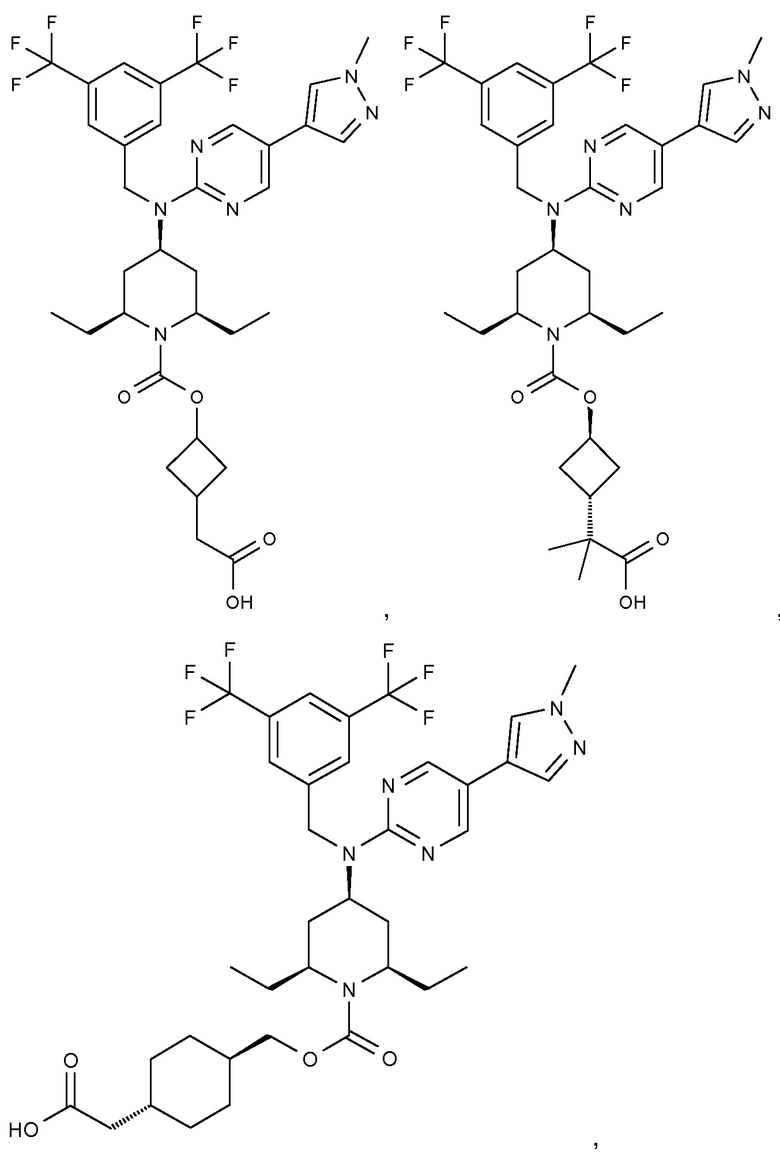
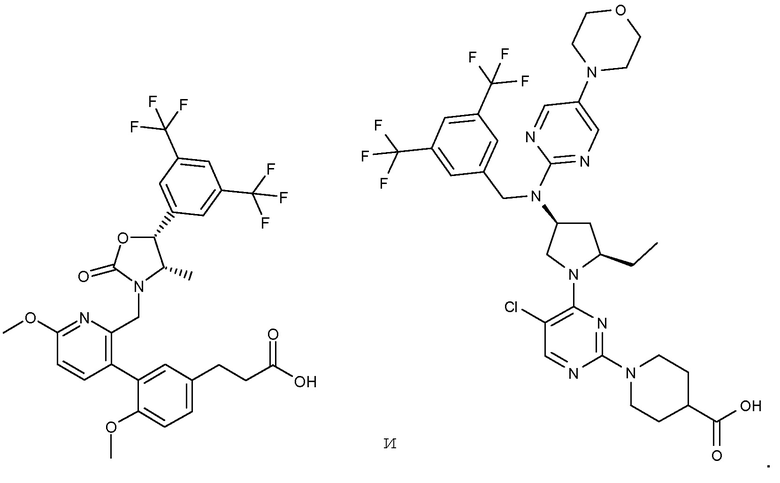
В варианте осуществления 34 изобретение относится к способу или применению согласно варианту осуществления 1, 1A или 2, где соединение выбрано из:
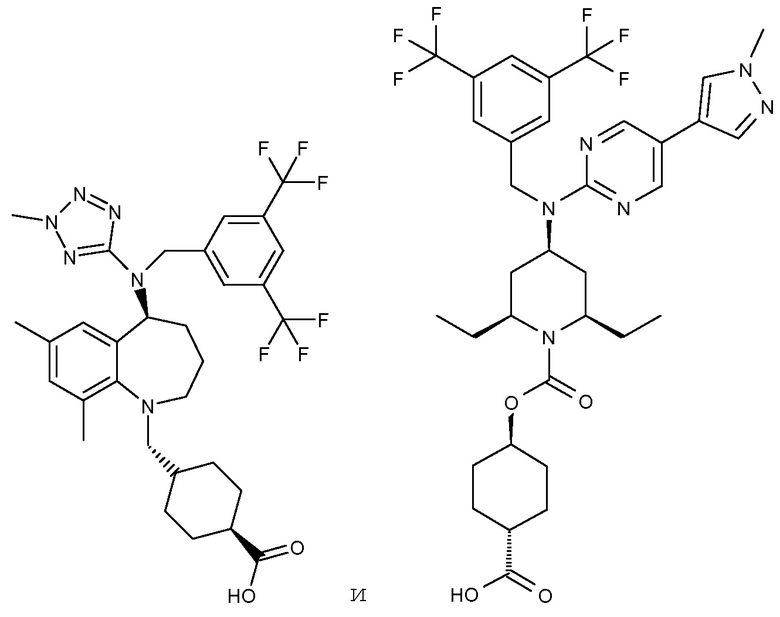 ,
,
или их фармацевтически приемлемой соли.
В варианте осуществления 34A изобретение относится к способу или применению согласно варианту осуществления 1, 1A или 2, где соединение представляет собой:
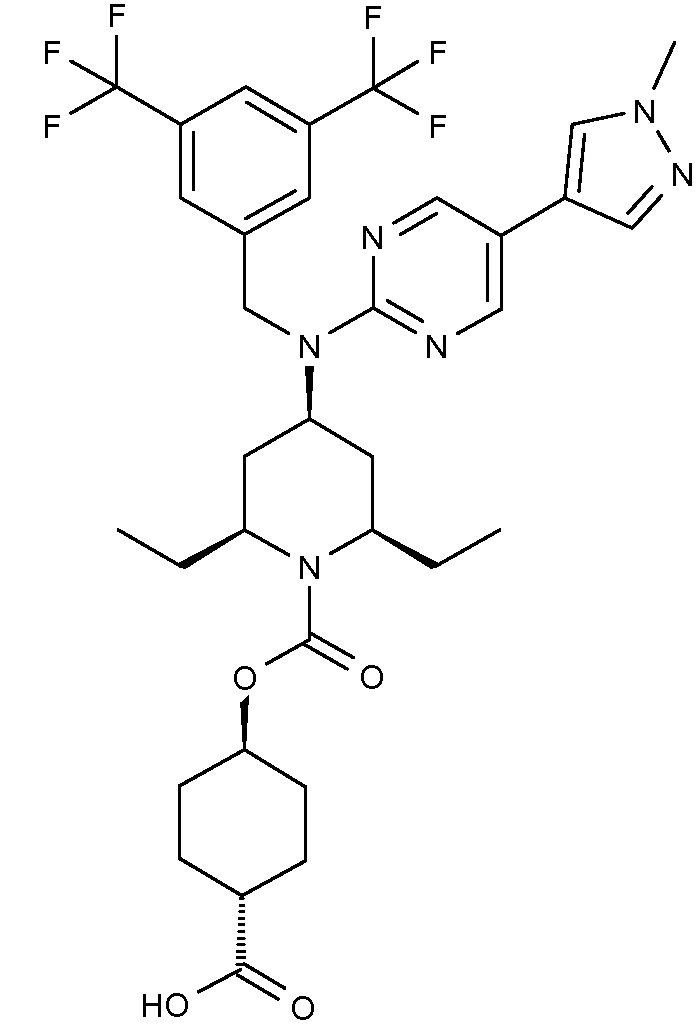
или его фармацевтически приемлемой соли.
В варианте осуществления 35 изобретение относится к способу или применению соединения согласно любому из предыдущих вариантов осуществления, в комбинации по меньшей мере с одним другим терапевтическим средством, в качестве комбинированного препарата для одновременного, раздельного или последовательного применения в терапии.
В варианте осуществления 36 изобретение относится к способу или применению согласно варианту осуществления 35, где другое терапевтическое средство выбрано из статина, ингибитора абсорбции холестерина, апрегулятора/индуктора АпоA-1, пре-бета ЛВП миметика, стабилизатора или индуктора ABCA1, агониста LXR, агониста FXR, ингибитора белка-переносчика фосфолипидов (PLTP), ингибитора альдостерон-синтазы (ASI), производного фибровой кислоты, рыбьего жира, ингибитора DGAT1 и ингибитора эндотелиальной липазы, или их фармацевтически приемлемой соли.
В варианте осуществления 37 изобретение относится к способу или применению согласно любому из предыдущих вариантов осуществления, где уровень триглицеридов является уровнем триглицеридов натощак >150 мг/дл.
В варианте осуществления 38 изобретение относится к способу или применению согласно любому из предыдущих вариантов осуществления, где уровень триглицеридов является уровнем триглицеридов натощак >200 мг/дл.
В варианте осуществления 39 изобретение относится к способу или применению согласно любому из предыдущих вариантов осуществления, где уровень триглицеридов является уровнем триглицеридов натощак >500 мг/дл.
В варианте осуществления 40 изобретение относится к способу или применению согласно любому из предыдущих вариантов осуществления повышения ЛВП-C до 100%.
Следует отметить, что структура некоторых соединений для применения в настоящем изобретении включает асимметричные атомы углерода. Таким образом, необходимо понимать, что изомеры, являющиеся результатом такой асимметрии (например, все энантиомеры и диастереомеры), включены в объем настоящего изобретения, если не указано иное. Такие изомеры могут быть получены в практически чистой форме с помощью классических методик разделения и стереохимически контролируемого синтеза. Кроме того, структуры и другие соединения и молекулы, обсуждаемые в настоящей заявке, также включают все соответствующие таутомеры.
Используемый в настоящем описании термин "изомеры" относится к различным соединениям, которые имеют одну и ту же молекулярную формулу, но отличаются расположением и конфигурацией атомов. Используемый в настоящем описании термин "оптический изомер" или "стереоизомер" также относится к любой из различных стереоизомерных конфигураций, которые могут существовать для данного соединения по настоящему изобретению и включают геометрические изомеры. Подразумевается, что заместитель может быть присоединен в хиральном центре атома углерода. Таким образом, изобретение включает энантиомеры, диастереомеры или рацематы соединения. "Энантиомеры" представляют собой пару стереоизомеров, которые являются несовпадающими зеркальными изображениями друг друга. Смесь 1:1 пары энантиомеров является "рацемической" смесью. Термин используется для обозначения рацемической смеси в соответствующих случаях. "Диастереоизомеры" являются стереоизомерами, которые содержат по меньшей мере два асимметричных атома, но которые не являются зеркальными отображениями друг друга. Абсолютную стереохимию определяют согласно R-S системе Кана-Ингольда-Прелога. Если соединение является чистым энантиомером, стереохимия на каждом хиральном углероде может быть обозначена либо индексом R, либо S. Разделенные соединения, абсолютная конфигурация которых неизвестна, могут быть обозначены (+) или (-), в зависимости от направления (право- или левовращающего), в котором они вращают плоскость поляризованного света при длине волны D-линии натрия. Некоторые соединения, описанные в настоящей заявке, содержат один или более центров или осей асимметрии и, таким образом, могут образовывать энантиомеры, диастереомеры и другие стереоизомерные формы, которые могут быть определены, с точки зрения абсолютной стереохимии, как (R)- или (S)-. Подразумевается, что настоящее изобретение включает все такие возможные изомеры, включая рацемические смеси, оптически чистые формы и смеси промежуточных соединений. Оптически активные (R)- и (S)-изомеры могут быть получены при использовании хиральных синтонов или хиральных реагентов, или разделены с использованием стандартных методик. Если соединение содержит двойную связь, заместитель может иметь E или Z конфигурацию. Если соединение содержит двузамещенный циклоалкил, циклоалкильный заместитель может иметь цис- или транс-конфигурацию. Предполагается, что включены также все таутомерные формы.
Любой асимметричный атом (например, атом углерода или подобный) соединения(-ий) по настоящему изобретению может присутствовать в рацемической или энантиомерно обогащенной форме, например, (R)-, (S)- или (R,S)-конфигурации. В некоторых вариантах осуществления каждый асимметричный атом имеет по меньшей мере 50% энантиомерный избыток, по меньшей мере 60% энантиомерный избыток, по меньшей мере 70% энантиомерный избыток, по меньшей мере 80% энантиомерный избыток, по меньшей мере 90% энантиомерный избыток, по меньшей мере 95% энантиомерный избыток или по меньшей мере 99% энантиомерный избыток в (R)- или (S)-конфигурации. Заместители в атомах с ненасыщенными связями могут, при наличии возможности, присутствовать в цис-(Z)- или транс-(E)-форме.
Таким образом, в данном контексте соединение по настоящему изобретению может находиться в форме одного из возможных изомеров, ротамеров, атропоизомеров, таутомеров или их смесей, например, в виде по существу чистых геометрических (цис или транс) изомеров, диастереомеров, оптических изомеров (антиподов), рацематов или их смесей.
Любые полученные смеси изомеров могут быть разделены с учетом физико-химических различий компонентов на чистые или по существу чистые геометрические или оптические изомеры, диастереомеры, рацематы, например, с помощью хроматографии и/или фракционной кристаллизации.
Любые полученные рацематы конечных продуктов или промежуточных соединений могут быть разделены на оптические антиподы с помощью известных методов, например, при разделении соответствующих диастереомерных солей, полученных с оптически активной кислотой или основанием, и высвобождением оптически активного кислотного или основного соединения. В частности, основная группа может использоваться, таким образом, для разделения соединений по настоящему изобретению на их оптические антиподы, например, с помощью фракционной кристаллизации соли, полученной с оптически активной кислотой, например, винной кислотой, дибензоилвинной кислотой, диацетилвинной кислотой, ди-O,O'-п-толуоил-винной кислотой, миндальной кислотой, яблочной кислотой или камфор-10-сульфоновой кислотой. Рацемические продукты также могут быть разделены с помощью хиральной хроматографии, например, высокоэффективной жидкостной хроматографии (ВЭЖХ) с использованием хирального адсорбента.
Используемый в настоящем описании термин "фармацевтически приемлемые соли" относится к солям, которые сохраняют биологическую эффективность и свойства соединений по настоящему изобретению и которые обычно не являются биологически или иным образом нежелательными. Во многих случаях, соединения по настоящему изобретению способны образовывать кислотные и/или основные соли благодаря присутствию амино и/или карбоксильных групп или подобных им групп.
Фармацевтически приемлемые соли присоединения кислоты, для применения в изобретении, могут быть образованы с минеральными кислотами и органическими кислотами, например, ацетат, аспартат, бензоат, безилат, бромид/гидробромид, бикарбонат/карбонат, бисульфат/сульфат, камфорсульфонат, хлорид/гидрохлорид, хлортеофиллонат, цитрат, этандисульфонат, фумарат, глюцептат, глюконат, глюкуронат, гиппурат, гидроиодид/иодид, изетионат, лактат, лактобионат, лаурилсульфат, малат, малеат, малонат, манделат, мезилат, метилсульфат, нафтоат, напсилат, никотинат, нитрат, октадеканоат, олеат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, полигалактуронат, пропионат, стеарат, сукцинат, сульфосалицилат, тартрат, тозилат и трифторацетат.
Минеральные кислоты, из которых могут быть получены соли, включают, например, соляную кислоту, бромоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и т.п.
Органические кислоты, из которых могут быть получены соли, включают, например, уксусную кислоту, пропионовую кислоту, гликолевую кислоту, щавелевую кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту, миндальную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, толуолсульфоновую кислоту, сульфосалициловую кислоту и т.п. Фармацевтически приемлемые соли присоединения основания могут быть получены с неорганическими и органическими основаниями. Неорганические основания, из которых могут быть получены соли, включают, например, соли аммония и металлов из I-XII колонок Периодической таблицы Менделеева. В некоторых вариантах осуществления соли получены из натрия, калия, аммония, кальция, магния, железа, серебра, цинка и меди; наиболее подходящие соли включают соли аммония, калия, натрия, кальция и магния.
Органические основания, из которых могут быть получены соли, включают, например, первичные, вторичные и третичные амины, замещенные амины, включая природные замещенные амины, циклические амины, основные ионообменные смолы и т.п. Некоторые органические амины включают изопропиламин, бензатин, холинат, диэтаноламин, диэтиламин, лизин, меглумин, пиперазин и трометамин.
Фармацевтически приемлемые соли для применения в настоящем изобретении могут быть синтезированы из исходного соединения, основной или кислотной молекулы, стандартными химическими методами. Как правило, такие соли могут быть получены при взаимодействии свободных кислотных форм таких соединений со стехиометрическим количеством соответствующего основания (такого как гидроксид, карбонат, бикарбонат Na, Ca, Mg или K, или подобного), или при взаимодействии свободных основных форм таких соединений со стехиометрическим количеством соответствующей кислоты. Такие реакции обычно проводят в воде или в органическом растворителе, или в их смеси. Обычно желательно использовать неводную среду, такую как эфир, этилацетат, этанол, изопропанол или ацетонитрил, при наличии возможности. Перечень дополнительных подходящих солей можно найти, например, в "Remington's Pharmaceutical Sciences", 20th ed., Mack Publishing Company, Easton, Pa., (1985); и в "Handbook of Pharmaceutical Salts: Properties, Selection, and Use", Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
Кроме того, любая формула, приведенная в настоящем описании, представляет немеченые формы, а также изотопно меченные формы соединений. Например, любой водород, обозначенный "H" в любой из формул в настоящем описании, представляет все изотопные формы водорода (например, 1H, 2H или D, 3H); любой углерод, обозначенный "C" в любой из формул в настоящем описании, представляет все изотопные формы углерода (например, 11C, 12C, 13C, 14C); любой азот, обозначенный "N", представляет все изотопные формы азота (например, 14N, 15N). Другие примеры изотопов, которые включены в изобретение, включают изотопы кислорода, серы, фосфора, фтора, иода и хлора, такие как 18F 31P, 32P, 35S, 36Cl, 125I. Изобретение включает различные изотопно меченные соединения, как определено в настоящей заявке, например, такие соединения, в которых присутствуют радиоактивные изотопы, такие как 3H, 13C и 14C. В одном варианте осуществления атомы в формулах в настоящем описании присутствуют с их природной распространенностью. В другом варианте осуществления один или более атомов водорода могут быть обогащены 2H; или/и один или более атомов углерода могут быть обогащены 11C, 13C или 14C; или/и один или более атомов азота могут быть обогащены 14N. Такие изотопно меченные соединения могут применяться в метаболических исследованиях (14C), исследованиях кинетики реакций (например, 2H или 3H), методиках обнаружения или визуализации, таких как позитронно-эмиссионная томография (ПЭТ), или однофотонная эмиссионная компьютерная томография (ОФЭКТ), включая анализы распределения лекарственного средства или субстрата в ткани, или в лучевой терапии пациентов. В частности, 18F или меченое соединение может особенно подходить для ПЭТ или ОФЭКТ исследований. Изотопно меченные соединения по настоящему изобретению и соответствующие пролекарства могут быть получены при выполнении методик, раскрытых в схемах или в примерах, и способов получения, описанных ниже, при замене реагента, не содержащего изотопную метку, общедоступным изотопно меченным реагентом.
Кроме того, обогащение более тяжелыми изотопами, в частности дейтерием (то есть 2H или D), может предоставить некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенным полупериодом выведения in vivo или требованием сниженной дозировки, или улучшением терапевтического индекса. Следует понимать, что дейтерий в данном контексте рассматривается как заместитель соединения согласно любой из формул I-VIIA. Концентрация такого более тяжелого изотопа, в особенности дейтерия, может определяться коэффициентом изотопного обогащения. Термин "коэффициент изотопного обогащения", используемый в настоящем описании, означает отношение между содержанием изотопа и распространенностью указанного изотопа в природе. Если заместитель в соединении по настоящему изобретению является указанным дейтерием, такое соединение имеет коэффициент изотопного обогащения для каждого обозначенного атома дейтерия по меньшей мере 3500 (включение 52,5% дейтерия в каждом обозначенном атоме дейтерия), по меньшей мере 4000 (включение 60% дейтерия), по меньшей мере 4500 (включение 67,5% дейтерия), по меньшей мере 5000 (включение 75% дейтерия), по меньшей мере 5500 (включение 82,5% дейтерия), по меньшей мере 6000 (включение 90% дейтерия), по меньшей мере 6333,3 (включение 95% дейтерия), по меньшей мере 6466,7 (включение 97% дейтерия), по меньшей мере 6600 (включение 99% дейтерия) или по меньшей мере 6633,3 (включение 99,5% дейтерия).
Изотопно обогащенные соединения согласно любой из формул I-VIIA в большинстве случаев могут быть получены при использовании стандартных методик, известных квалифицированным специалистам, или способов, аналогичных описанным в сопутствующих Примерах и Примерах получения, при использовании подходящего изотопнообогащенного реагента вместо необогащенного реагента, используемого ранее.
Фармацевтически приемлемые сольваты в соответствии с изобретением включают такие сольваты, в которых растворитель кристаллизации может быть изотопно замещенным, например, D2O, d6-ацетоном, d6-ДМСО.
Соединения для применения в изобретении, то есть соединения согласно любой из формул I-VIIA, которые содержат группы, способные действовать в качестве доноров и/или акцепторов водородных связей, могут быть способны к образованию сокристаллов с подходящими сокристаллическими формами. Указанные сокристаллы могут быть получены из соединений согласно любой из формул I-VIIA известными методиками образования сокристаллов. Такие методики включают размол, нагревание, совместную возгонку, совместное плавление или контакт в растворе соединений согласно любой из формул I-VIIA с сокристаллообразователем в условиях кристаллизации и выделение сокристаллов, сформированных таким образом. Подходящие сокристаллические формы включают описанные в WO 2004/078163. Следовательно, в изобретении также предложены сокристаллы, включающие соединение согласно любой из формул I-VIIA или их фармацевтически приемлемую соль.
Используемый в настоящем описании термин "фармацевтически приемлемый носитель" включает любые и все возможные растворители, дисперсионные среды, покрытия, поверхностно-активные вещества, антиоксиданты, консерванты (например, противобактериальные агенты, противогрибковые агенты), изотонические вещества, замедлители абсорбции, соли, консерванты, лекарственные средства, стабилизаторы лекарственных средств, связующие вещества, вспомогательные вещества, разрыхлители, смазывающие вещества, подсластители, ароматизаторы, красители и т.п., и их комбинации, известные специалистам в данной области техники (см., например, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289-1329). За исключением тех случаев, когда какой-либо стандартный носитель несовместим с действующим веществом, предполагается его применение в терапевтических или фармацевтических композициях.
Термин "терапевтически эффективное количество" соединения настоящего по изобретению относится к такому количеству соединения по настоящему изобретению, которое вызывает биологический или медицинский ответ субъекта, например, снижение или ингибирование активности фермента или белка, или уменьшение интенсивности симптома, облегчение состояния, замедление или задержку развития болезни, или предотвращение болезни и т.д. В одном неограничивающем варианте осуществления, термин "терапевтически эффективное количество" относится к такому количеству соединения по настоящему изобретению, которое при введении субъекту является эффективным (1) для по меньшей мере частичного облегчения, замедления, предотвращения и/или уменьшения тяжести состояния, нарушения или болезни, или ее симптома, (i) интенсивность которых уменьшается при ингибировании CETP, или (ii) связанных с активностью CETP, или (iii) характеризуемых аномальной активностью CETP; или (2) уменьшения или ингибирования активности CETP; или (3) уменьшения или ингибирования экспрессии CETP. В другом неограничивающем варианте осуществления термин "терапевтически эффективное количество" относится к такому количеству соединения по настоящему изобретению, которое при введении в клетку или ткань, или в бесклеточный биологический материал или среду, является эффективным для по меньшей мере частичного уменьшения или ингибирования активности CETP; или по меньшей мере частичного уменьшения или ингибирования экспрессии CETP.
Используемый в настоящем описании термин "субъект" относится к животному. Обычно животное является млекопитающим. Субъект также относится например, к приматам (например, людям), коровам, овцам, козам, лошадям, собакам, кошкам, кроликам, крысам, мышам, рыбам, птицам и т.п. В некоторых вариантах осуществления субъектом является примат. В других вариантах осуществления субъектом является человек.
Используемый в настоящем описании термин "ингибировать", "ингибирующий" или "ингибирование" относится к уменьшению или подавлению данного состояния, симптома или нарушения, или болезни, или значительному уменьшению исходной биологической активности или активности процесса.
Используемый в настоящем описании термин "лечить", "подвергаемый лечению" или "лечение" любого заболевания или нарушения относится в одном варианте осуществления к уменьшению тяжести заболевания или нарушения (то есть замедлению или прекращению, или уменьшению развития заболевания или по меньшей мере одного из его клинических симптомов). В другом варианте осуществления "лечить", "подвергаемый лечению" или "лечение" относятся к облегчению или уменьшению тяжести по меньшей мере одного физического параметра, включая параметры, которые могут быть незаметными для пациента. В еще одном варианте осуществления "лечить", "подвергаемый лечению" или "лечение" относятся к модуляции заболевания или нарушения, либо физически (например, путем стабилизации заметного симптома), физиологически (например, путем стабилизации физического параметра), либо и тому, и другому. В еще одном варианте осуществления "лечить", "подвергаемый лечению" или "лечение" относятся к предотвращению или задержке начала развития или прогрессии заболевания или нарушения.
Используемый в настоящем описании субъект "испытывает потребность" в лечении, если для такого субъекта подобное лечение будет благоприятным биологически, с медицинской точки зрения или в отношении качества жизни.
Используемый в оригинальном тексте настоящего описания термины "какой-либо", "этот" и подобные термины, используемые в контексте настоящего изобретения (особенно в контексте формулы изобретения), следует считать охватывающими как единственное, так и множественное число, если не указано иное или если иное прямо не противоречит контексту.
Все способы, описанные в настоящей заявке, могут быть выполнены в любом подходящем порядке, если не указано иное не или если иное прямо не противоречит контексту. Применение любых возможных примеров или примерного выражения (например, "такой как"), приводимого в настоящем описании, предназначено исключительно для более полного объяснения изобретения и не накладывает ограничений на объем заявленного изобретения.
Соединения для применения в настоящем изобретении используются либо в свободной форме, либо в виде соответствующей соли.
В случае, когда основная группа и кислотная группа присутствуют в одной молекуле, соединения по настоящему изобретению могут также образовывать внутренние соли, например, цвиттер-ионные молекулы.
Кроме того, соединения по настоящему изобретению, включая их соли, также могут быть получены в форме гидратов или могут включать другие растворители, используемые для их кристаллизации.
В другом аспекте настоящего изобретения предложена фармацевтическая композиция, включающая соединение по настоящему изобретению или его фармацевтически приемлемую соль и один или более фармацевтически приемлемых носителей для применения в предотвращении, уменьшении или лечении атеросклероза или дислипидемии, или для повышения ЛВП-C и/или снижения ЛНП-C, у субъекта с высоким уровнем триглицеридов. Фармацевтическая композиция может быть составлена для конкретных путей введения, таких как пероральное введение, перентеральное введение и ректальное введение, и т.д. Кроме того, фармацевтические композиции по настоящему изобретению могут быть приготовлены в твердой форме (включая, без ограничения, капсулы, таблетки, пилюли, гранулы, порошки или суппозитории) или в жидкой форме (включая, без ограничения, растворы, суспензии или эмульсии). Фармацевтические композиции могут быть подвергнуты стандартным фармацевтическим операциям, таким как стерилизация, и/или могут содержать стандартные инертные растворители, смазывающие вещества или буферные вещества, а также вспомогательные добавки, такие как консерванты, стабилизаторы, смачивающие вещества, эмульгаторы и буферы, и т.д.
Как правило, фармацевтические композиции представляют собой таблетки или желатиновые капсулы, включающие действующее вещество вместе с:
a) разбавителями, например, лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином;
b) смазывающими веществами, например, диоксидом кремния, тальком, стеариновой кислотой, ее магниевой или кальциевой солью и/или полиэтиленгликолем; для таблеток также
c) связующими компонентами, например, силикатом алюминия магния, крахмальной пастой, желатином, трагакантом, метилцеллюлозой, натрий-карбоксиметилцеллюлозой и/или поливинилпирролидоном; при необходимости
d) разрыхлителями, например, крахмалами, агаром, альгиновой кислотой или ее натриевой солью, или шипучими смесями; и/или
e) абсорбентами, пигментами, ароматизаторами и подсластителями.
Таблетки могут быть покрыты пленкой или энтеросолюбильным покрытием согласно способам, известным в уровне техники.
Подходящие соединения для перорального введения включают эффективное количество соединения по изобретению в форме таблеток, леденцов, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсии, твердых или мягких капсул, или сиропов или эликсиров. Композиции, предназначенные для перорального применения, получают согласно любому способу, известному в уровне техники для производства фармацевтических соединений, и такие композиции могут содержать одно или более веществ, выбранных из группы, состоящей из подсластителей, ароматизаторов, пигментов и консервантов, для получения фармацевтически изящных и приемлемых на вкус препаратов. Таблетки могут содержать действующее вещество в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, которые являются подходящими для производства таблеток. Указанные эксципиенты являются, например, инертными разбавителями, такими как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующими и разрыхляющими добавками, например, кукурузным крахмалом или альгиновой кислотой; связующими веществами, например, крахмалом, желатином или гуммиарабиком; и смазывающими веществами, например, стеаратом магния, стеариновой кислотой или тальком. Таблетки не имеют покрытия, или покрытие наносят на них известными методиками, чтобы задержать разрыхление и абсорбцию в желудочно-кишечном тракте и таким образом обеспечить пролонгированное действие в течение более длительного периода. Например, может использоваться материал, задерживающий высвобождение, такой как глицерилмоностеарат или глицерилдистеарат. Композиции для перорального применения могут быть представлены в виде твердых желатиновых капсул, в которых действующее вещество смешано с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых действующее вещество смешано с водой или масляной средой, например, арахисовым маслом, жидким вазелиновым маслом или оливковым маслом.
Некоторые композиции для инъекций являются водными изотоническими растворами или суспензиями, а суппозитории предпочтительно приготавливают из жировых эмульсий или суспензий. Указанные композиции могут быть стерилизованы и/или содержать адъюванты, такие как консервирующие, стабилизирующие, смачивающие или эмульгирующие вещества, ускорители растворения, соли для регуляции осмотического давления и/или буферы. Кроме того, они могут также содержать другие терапевтически ценные вещества. Указанные композиции получают согласно стандартным методам смешивания, гранулирования или нанесения покрытий, соответственно, при этом они содержат приблизительно 0,1-75% или содержат приблизительно 1-50% действующего вещества.
Подходящие композиции для трансдермального применения включают эффективное количество соединения по изобретению с подходящим носителем. Носители, подходящие для трансдермальной доставки, включают абсорбируемые фармакологически приемлемые растворители, способствующие проникновению через кожу хозяина. Например, трансдермальные устройства находятся в форме бандажа, включающего поддерживающий элемент, резервуар, содержащий соединение, необязательно с носителями, необязательно барьер, регулирующий скорость, для доставки соединения в кожу реципиента с регулируемой и установленной скоростью в течение длительного периода времени, и приспособления для закрепления устройства на коже.
Подходящие композиции для местного применения, например, на коже и глазах, включают водные растворы, суспензии, мази, кремы, гели или распыляемые композиции, например, для доставки аэрозолем или т.п. Такие системы местной доставки, в частности, подходят для нанесения на кожу. Таким образом, они особенно подходят для применения в качестве препаратов для местного применения, в том числе косметических, которые известны в уровне техники. Такие препараты могут содержать солюбилизаторы, стабилизаторы, вещества, регулирующие тоничность, буферы и консерванты.
Используемое в настоящем описании местное применение может также относится к ингаляции или интраназальному применению. Они могут быть удобно доставлены в форме сухого порошка (либо отдельно, в виде смеси, например, сухой смеси с лактозой, либо в виде частиц смешанного состава, например, с фосфолипидами) из ингалятора сухого порошка, или в форме аэрозольного спрея из баллона под давлением, насоса, спрея, распылителя или небулайзера, с или без использования подходящего пропеллента.
В настоящем изобретении также предложены безводные фармацевтические композиции и лекарственные формы, включающие соединения по настоящему изобретению в качестве действующих веществ, поскольку вода может способствовать разложению некоторых соединений, для применения в изобретении.
Безводные фармацевтические композиции и лекарственные формы для применения в изобретении могут быть получены с использованием безводных и имеющих малое содержание влаги компонентов или условий низкой влажности. Безводную фармацевтическую композицию можно приготовить и хранить таким образом, чтобы сохранялась ее безводная природа. Таким образом, безводные соединения упаковывают, используя материалы, известные для предотвращения контакта с водой, при этом они могут быть включены в подходящие формулярные наборы. Примеры подходящей упаковки включают, без ограничения перечисленными, герметично запаяные упаковки из фольги, пластмассы, контейнеры с единичной дозой (например, флаконы), блистерные упаковки и контурные упаковки.
В изобретении также предложены фармацевтические композиции и лекарственные формы, которые содержат одно или более веществ, которые снижают скорость, с которой соединение по настоящему изобретению, в качестве действующего вещества, разлагается. Такие вещества, которые указаны в настоящем описании как "стабилизаторы", включают, без ограничения перечисленными, антиоксиданты, такие как аскорбиновую кислоту, pH буферы или солевые буфера и т.д.
Подходящая фармацевтическая композиция включает композицию, содержащую твердую аморфную дисперсию, которую приготавливают с помощью процессов экструзии из горячего расплава. Примеры твердой аморфной дисперсии представлены ниже.
Пример состава 1: Твердая аморфная дисперсия соединения Примера 8
Пример состава 2: Твердая аморфная дисперсия соединения Примера 17
Соединения для применения в изобретении:
Соединения по изобретению могут быть синтезированы при использовании способов, описанных в следующих заявках: US 2009/0075968, US 2009/0227580, US 2006/0063803, US 2009/075979, US 2009/042892, US 2007/244095, WO 2011/028395, WO 2009/027785 и US 2008/269284, которые включены в настоящую заявку посредством ссылки.
Соединения согласно любой из формул I-VIIA для применения в способе по изобретению, или их фармацевтически приемлемая соль, в свободной форме или в форме фармацевтически приемлемой соли, демонстрируют ценные фармакологические свойства, например, CETP-ингибирующие свойства, например, как показано в in vitro и in vivo тестах, представленных в следующих разделах, и поэтому показаны для лечения, уменьшения и/или предотвращения атеросклероза или дислипидемии, или для повышения ЛВП-C и/или снижения ЛНП-C у субъекта с высоким уровнем триглицеридов.
Фармацевтическая композиция или комбинация по изобретению для применения в способе по изобретению может находиться в форме единичной дозировки, соответствующей приблизительно 1-1000 мг действующего вещества (веществ) для субъекта весом приблизительно 50-70 кг, или приблизительно 1-500 мг или приблизительно 1-250 мг или приблизительно 1-150 мг или приблизительно 0,5-100 мг, или приблизительно 1-50 мг действующих веществ. Терапевтически эффективная дозировка соединения, фармацевтической композиции или их комбинаций зависит от вида субъекта, массы тела, возраста и индивидуального состояния, тяжести нарушения. Врач, клинический врач или ветеринарный врач средней квалификации сумеют с легкостью определить эффективное количество каждого из действующих веществ, необходимое для предотвращения, лечения или замедления прогрессии нарушения или заболевания.
Вышеуказанные свойства дозировки подтверждаются in vitro и in vivo тестами с использованием предпочтительно млекопитающих, например, мышей, крыс, собак, обезьян, или изолированных органов, тканей и их препаратов. Соединения по настоящему изобретению могут быть применены in vitro в форме растворов, например, водных растворов, и in vivo или энтерально, перентерально, предпочтительно внутривенно, например, в виде суспензии или в водном растворе. Дозировка in vitro может изменяться в диапазоне молярных концентраций от приблизительно 10-3 до 10-9 моль. Терапевтически эффективное in vivo количество может изменяться, в зависимости от пути введения, в пределах приблизительно 0,1-500 мг/кг, или в пределах приблизительно 1-100 мг/кг.
Способ по изобретению:
In vitro и in vivo анализы:
Получение человеческого про-аполипопротеина A-1 (про-апоA-1)
кДНК человеческого про-апоA1 (регистрационный номер NCBI NM_000039) клонировали из кДНК печени человека Quick-CloneTM (Clontech, CA, каталожный номер 7113-1) и встраивали в вектор pET28a (Novagen, Germany) для бактериальной экспрессии. Экспрессированный белок в виде слитого белка с 6×His-меткой на N-конце в BL-21 Gold (DE3) (Strategene, CA) очищали при использовании HiTrap Chelating (GE Healthcare, CT).
Приготовление микроэмульсии доноров
Микроэмульсию, содержащую про-апоA-1, в качестве донорной частицы приготавливали согласно предыдущему сообщению (J. Biol. Chem., 280:14918-22). Глицерилтриолеат (500 мкг, Sigma-Aldrich, каталожный номер T7140), 3-sn-фосфатидилхолин (4680 мкг, Sigma, каталожный номер P2772) и холестерил BODIPY FL C12 (2 мг, Invitrogen, CA, каталожный номер C-3927MP) растворяли в 1,5 мл хлороформа. Раствор выпаривали, и удаляли остатки растворителя в вакууме более 1 часа при комнатной температуре. Высушенную смесь липидов растворяли в 7 мл буфера для анализа (50 мМ Трис-HCl (pH 7,4), содержащего 150 мМ NaCl и 2 мМ ЭДТА), и обрабатывали ультразвуком в водяной бане в течение 1 часа. Раствор делили на две пробирки по 3,5 мл каждая. Каждую пробирку обрабатывали ультразвуком при 50°C с микронаконечника (MICROSONTM ULTRASONIC CELL DISRUPTOR, Misonix, Farmingdale, NY) при выходной мощности 006 в течение 10 минут (каждые 25 секунд). Раствор охлаждали до 40°C и в каждую пробирку добавляли 400 мкг про-апоA-1 (общий объем 4 мл). Затем раствор обрабатывали ультразвуком при выходной мощности 004 в течение 20 минут (каждые 30 секунд). Конечная концентрация: глицерилтриолеат: 62,5 мкг/мл, ФХ: 585 мкг/мл, Bodipy CE: 250 мкг/мл, про-апоA-1: 100 мкг/мл. Раствор центрифугировали при 5200×g в течение 5 минут, и супернатант хранили при 4°C.
(1) In vitro эксперименты:
Образцы человеческой сыворотки
Образцы человеческой сыворотки получали из центра биомедицинских исследований Новартис (Novartis Institutes for BioMedical Research) по программе доноров (нормальные образцы триглицеридов) или приобретали у Bioreclamation (Westbury, NY) или Uniglobe (Reseda, CA). Общие требования к донорам человеческой сыворотки включали: мужской пол, возраст 20-55 лет, в настоящее время не проходит лечение средствами, изменяющими липидный состав, не воздерживается от приема пищи на момент забора крови, и общий уровень холестерина в пределах 200-300 мг/дл. Образцы доноров отбирали прежде всего на основе уровня триглицеридов в сыворотке, используя следующие классификации: нормальные триглицериды (<150 мг/дл); высокие триглицериды (300-500 мг/дл); и очень высокие триглицериды (750-1200 мг/дл). В данных экспериментах использовали в общей сложности 4 отдельных образца сыворотки из каждой классификации. Для каждой из 3 классификаций триглицеридов подготавливали два пула образцов сыворотки. Средние концентрации триглицеридов в плазме (мг/дл) для пулов составили: нормальная: 101,35; высокая: 347,5; очень высокая 1, 868,45; очень высокая 2, 1269,35.
Анализ активности CETP in vitro в человеческой плазме
Раствор донора приготавливали путем разведения микроэмульсии донора буфером для анализа (1,7 мкл донора + 8,3 мкл буфера). Человеческую сыворотку (25 мкл), буфер для анализа (14 мкл) и 50× тестируемое соединение, растворенное в 100% диметилсульфоксиде (1 мкл), добавляли в каждую лунку 384-луночного черного планшета с прозрачным дном (NUNC. Thermo Fisher # 242764). Реакцию инициировали добавлением раствора донора (10 мкл) в каждую лунку. Интенсивность флуоресценции измеряли каждые 10 мин при 37°C при длине волны возбуждения 485 нм и длине волны эмиссии 515 нм в течение 80-минутного периода, используя SpectraMax M5 производства Molecular Devices (Sunnyvale, CA). Активность CETP (ОЕФ/мин) определяли как изменение интенсивности флуоресценции от 40 до 70 мин. Процент ингибирования CETP вычисляли следующим образом: ((100%Act-Wellcmpnd)/100%Act))*100. Значения IC50 вычисляли при использовании программы GraphPad Prism 5.
При использовании анализа (описанного выше), соединения по изобретению показали ингибирующую эффективность в соответствии с Таблицей 1, представленной ниже.
Ингибирующая активность соединений
Эти данные демонстрируют, что соединения по настоящему изобретению сохраняют ингибирующую активность CETP в плазме, выделенной у гипертриглицеридемических пациентов в сравнении с анацетрапибом (разрабатываемым в настоящее время Merck). Данные показывают, что смещение IC50 от нормальной триглицеридемической плазмы до высокой триглицеридемической плазмы является небольшим по сравнению с соответствующим смещением IC50 для анацетрапиба. Смещение IC50 определяется как:

Смещение IC50 для соединений Формулы I меньше 5, меньше 10, меньше 20 или меньше 30. Предпочтительно смещение IC50 для соединений по настоящему изобретению меньше 5.
(2) In vivo эксперименты
Животные и условия эксперимента
Самцов золотистых сирийских хомяков возрастом девять-десять недель приобрели в Japan SLC Inc. (Shizuoka, Japan). Животных подвергали акклиматизации в течение 1 недели в следующих условиях: световой цикл 8:00/20:00 свет/темнота, комнатная температура 23±2°C (диапазон), влажность 55±15% (диапазон). В это время животные получали фильтрованную (0,5 мкм) воду и стандартный корм без ограничений.
Для индукции острого повышения уровней триглицеридов в плазме, тритон WR-1339 (каталожный номер 35418-12, Nacalai Tesque Inc, Kyoto, Japan) растворяли в физиологическом солевом растворе (Otsuka Normal Saline, Otsuka Pharmaceuticals, Tokushima, Japan) при концентрации 2,5-10% (вес/вес) и вводили внутривенно в объеме 2 мл/кг из локтевой подкожной вены под ингаляционной анестезией изофлураном (FORANE®, Abbott Japan Co. Ltd., Tokyo, Japan). Пробы цельной крови забирали при использовании промытого гепарином шприца (гепарин натрий для инъекций "Ajinomoto"; Ajinomoto, Tokyo, Japan) из брюшной вены при венепункции под ингаляционной анестезией изофлураном (Abbott Japan Co. Ltd.). После центрифугирования при 15000 об/мин в течение 10 минут при 4°C получали гепаринизированную плазму и хранили при -80°C до использования.
Соединения (ингибиторы CETP) суспендировали в 0,5% растворе метилцеллюлозы (метилцеллюлоза 400cP, каталожный номер 138-05072, Wako Pure Chemical Industries, Osaka, Japan), который использовали в качестве среды. Соединение или среду (10 мл/кг) вводили перорально через зонд за 5 ч перед введением Тритона WR-1339.
Определение концентраций общего холестерина и триглицеридов в плазме
Концентрации холестерина и триглицеридов в плазме и сыворотке измеряли вручную с помощью наборов (Cholesterol E-test, каталожный номер 439-17501 и Triglyceride E-test, каталожный номер 432-40201, Wako Pure Chemical, Osaka, Japan).
Ex vivo активность CETP в плазме
Активность CETP в плазме ex vivo определяли следующим образом: 50 мкл плазмы хомяка (конечная 80% плазма) смешивали с 10 мкл раствора донора, разведенного буфером для анализа, состоящим из 50 мМ Трис-HCl (pH 7,4), содержащего 150 мМ NaCl и 2 мМ ЭДТА, в 96-луночном черном плоскодонном планшете с половинной площадью лунок (каталожный номер 3686, Corning, Corning, NY). Интенсивность флуоресценции измеряли каждые 15 минут при длине волны возбуждения 485 нм и длине волны эмиссии 535 нм в течение 120 мин при 37°C, используя ARVO SX+L (PerkinElmer, Wellesley, MA). Активность CETP (ОЕФ/мин) определяли как изменение интенсивности флуоресценции от 30 до 90 мин.
Концентрации CETP в плазме
Концентрации CETP в плазме измеряли с использованием набора для иммуноферментного анализа (ELISA) (каталожный номер 278181, Daiichi Pure Chemicals, Tokyo, Japan). Рекомбинантный человеческий белок CETP использовали в качестве стандарта. Плазму хомяка разводили в 250 раз буфером для разведения из набора, после чего концентрацию CETP измеряли согласно методике производителя.
Система высокоэффективной жидкостной хроматографии (ВЭЖХ) для анализа липопротеинов в плазме хомяка
Систему ВЭЖХ-эксклюзионной хроматографии с поточной системой ферментной двойной детекции липидов использовали для анализа липопротеинов. Образцы плазмы разбавляли (в 5 раз) солевым раствором после фильтрации (0,45 мкм, Millipore Co, каталожный номер UFC30HV00) и вводили в систему ВЭЖХ в объеме 100 мкл (20 мкл плазмы) каждые 95 минут, используя автодозатор. Липопротеины плазмы разделяли при использовании одной колонки Superose 6 и фильтрованного фосфатно-солевого буфера (0,45 мкм, Millipore Co., Bedford, MA, каталожный номер SJHVM4710; PBS, Dainippon Pharmaceutical, Osaka, Japan, каталожный номер 28-103-05 FN) при расходе 0,5 мл/мин. Каждый ферментативный реактив подавали насосом при расходе 0,25 мл/мин. Обе ферментативных реакции протекали при 37°C в реакторе (тефлоновая трубка, 15 м × 0,4 мм (внутренний диаметр)) в термостате колонки. Окрашивание, развивающееся после ферментативной реакции, измеряли при 580 нм, и при этом регистрировали электрический сигнал (каждые 0,5 секунды).
Контрольный смешанный образец ЭДТА-плазмы использовали для стандартизации уровней холестерина и фосфолипидов по липопротеинам. Уровни холестерина и фосфолипидов в контрольной плазме измеряли вручную с использованием наборов (Cholesterol E-test, каталожный номер 439-17501, и Phospholipid C-test, каталожный номер 433-36201, соответственно, Wako Pure Chemical Industries), при этом концентрации составили 121,8 и 216,8 мг/дл, соответственно. Концентрации ЛОНП, ЛНП, а также уровни ЛВП-холестерина и -фосфолипидов вычисляли, используя площади хроматограмм, наблюдаемые для контрольных образцов плазмы хомяка. Согдасно данной методике значения времени удержания контрольных ЛОНП, ЛНП и ЛВП хомяка составили 18,5-24,0, 24,0-30,5 и 31,5-42,0 мин, соответственно.
Анализ данных
Данные выражены как среднее и стандартная ошибка среднего (SEM). Для оценки статистической значимости множества групп использовали критерий множественных сравнений Даннетта, после однофакторного дисперсионного анализа (ANOVA). Для статистического анализа между двумя группами использовали t-критерий Стьюдента. Статистическую значимость определяли как P<0,05.
Острая гипертриглицеридемия у хомяков
Чтобы определить эффекты резких изменений уровней триглицеридов в плазме на активность CETP in vivo, Тритон WR-1339 внутривенно вводили хомякам, получавшим стандартный кормовой рацион. Тритон WR-1339 повышает уровни триглицеридов в плазме посредством ингибирования липопротеинлипазного пути и, в свою очередь, липолиза ЛОНП (Borensztajn, Rone, and Kotlar, "The inhibition in vivo of lipoprotein lipase (clearing-factor lipase) activity by triton WR-1339", Biochem J. 1976; 156:539-543).
Внутривенное введение Тритона WR-1339 в дозах 50, 100 или 200 мг/кг приводило к дозозависимому увеличению уровней триглицеридов в плазме по сравнению с группой, обработанной солевым раствором. Максимальное повышение уровней триглицеридов наблюдалось через 8 часов после введения Тритона. В соответствии с повышением уровней триглицеридов, активность CETP в плазме также возрастала в зависимости от дозы. На основе данных результатов, фармакодинамические параметры определяли через 3 ч после введения Тритона WR-1339 в последующих исследованиях.
Через 3 ч после введения, Тритон WR-1339 в дозах 50 и 100 мг/кг повышал концентрации триглицеридов в плазме (в 2,0 и 4,0 раза), общий холестерин (в 1,2 и 1,4 раза) и активность CETP (в 1,7 и 3,7 раза) со статистической значимостью. Повышение активности CETP было в большей степени связано с изменением уровней триглицеридов, нежели с изменением уровней общего холестерина. Примечательно, что Тритон WR-1339 не оказывал значительного влияния на концентрации CETP в плазме при любой дозе, что указывает на то, что повышение активности CETP под действием Тритона WR-1339 обусловлено резким увеличением акцептора в процессе переноса холестерина, а не увеличением продукции белка CETP.
Эффект от внутривенного введения Тритона WR-1339 на профиле липопротеинов плазмы определяли с помощью ВЭЖХ анализа. По сравнению с группой, получавшей солевой раствор, Тритон WR-1339 повышал содержание холестерина в ЛОНП и увеличивал размер частиц ЛОНП. В частности, ЛВП-холестерин не изменялся при обработке дозой 50 мг/кг, но снижался в группе 100 мг/кг. ЛНП-холестерин снижался при введении Тритона WR-1339. Приведенные результаты указывают, что обработка Тритоном WR-1339 повышала активность CETP прежде всего посредством увеличения богатых триглицеридами частиц.
Влияние обработки Тритоном WR-1339 на ингибирование активности CETP торцетрапибом
Исследовали влияние резкого увеличения триглицеридов плазмы in vivo (резкого увеличения богатых триглицеридами частиц) на способность торцетрапиба изменять активность CETP в плазме, уровни триглицеридов и общего холестерина. Торцетрапиб в дозах 3, 10 или 30 мг/кг или среде вводили однократно с кормом хомякам, получавшим стандартный кормовой рацион. Тритон WR-1339 (50 или 100 мг/кг) или солевой раствор вводили внутривенно через 5 ч после введения торцетрапиба. Параметры плазмы измеряли через 3 ч после инъекции Тритона WR-1339.
Торцетрапиб вызывал дозозависимое и значительное ингибирование активности CETP в плазме в группе, получавшей солевой раствор. Ингибиторные эффекты в дозах 3, 10 и 30 мг/кг составили 38, 74 и 88%, соответственно. По сравнению с группой, получавшей солевой раствор, способность торцетрапиба ингибировать активность CETP уменьшалась у животных, обработанных WR-1339. Различия между группами, получавшими солевой раствор и W-1339, по степени ингибирования, наблюдаемого при дозах 3, 10 и 30 мг/кг торцетрапиба, составляли 5,5%, 13%, 35% (P<0,01), соответственно. Кроме того, ингибиторное действие торцетрапиба при дозе 10 мг/кг уменьшалось при обработке Тритоном WR-1339. В отношении группы, получавшей солевой раствор (74% ингибирование), степень ингибирования снижалась на 44 и 13% при дозах 50 и 100 мг/кг, соответственно.
Также определяли эффекты торцетрапиба в отношении уровней триглицеридов и общего холестерина в плазме в группах, получавших солевой раствор и Тритон WR-1339. Обработка торцетрапибом приводила к небольшому, но значительному, снижению уровней триглицеридов в плазме в группе, получавшей солевой раствор, при дозах 3 и 30 мг/кг. С другой стороны, торцетрапиб не влиял на триглицериды плазмы в группе, обработанной Тритоном WR-1339. Уровни общего холестерина в группах, получавших солевой раствор или Тритон WR-1339, при обработке торцетрапибом не изменялись.
С учетом представленных результатов, дозу 100 мг/кг Тритона WR-1339 выбирали для дальнейших сравнительных исследований ингибиторов CETP.
Влияние Тритона WR-1339 на ингибирование активности CETP, вызванной анацетрапибом, соединением Примера 49 и соединением Примера 8
Исследовали влияние резкого повышения триглицеридов плазмы in vivo на степень ингибирования CETP, вызываемого торцетрапибом, анацетрапибом, соединением Примера 49 или соединением Примера 8. Соединение или среду вводили с кормом хомякам, получавшим стандартный кормовой рацион. Тритон WR-1339 (100 мг/кг) или солевой раствор вводили внутривенно через 5 ч после введения соединения. Параметры плазмы измеряли через 3 ч после инъекции Тритона WR-1339. Были выбраны две дозы для каждого ингибитора: около 55-75% (низкая доза) или 75-80% (высокая доза) ингибирования активности CETP в группе, получавшей солевой раствор.
В группе низкой дозы, торцетрапиб (5 мг/кг), анацетрапиб (1,5 мг/кг), соединение Примера 49 (1,5 мг/кг) и соединение Примера 8 (0,75 мг/кг) значительно ингибировали активность CETP на 60, 56, 63 и 72% в группе, получавшей солевой раствор, соответственно. Тритон WR-1339 снижал ингибиторные эффекты всех соединений до 13, 8, 26 и 20%, соответственно, без статистической значимости по сравнению с группой, получавшей среду.
В противоположность группе низкой дозы, в группе высокой дозы, торцетрапиб (10 мг/кг), анацетрапиб (3 мг/кг), соединение Примера 49 (3 мг/кг) и соединение Примера 8 (1,5 мг/кг) значительно ингибировали активность CETP на 75, 78, 77 и 79% в группе, получавшей солевой раствор, соответственно. Тритон WR-1339 заметно снижал ингибиторные эффекты торцетрапиба и анацетрапиба в отношении активности CETP до 8 и 13%, соответственно, без статистической значимости по сравнению с группой, получавшей среду. Однако, соединение Примера 49 и соединение Примера 8 значительно ингибировали активность CETP на 38 и 47% при введении Тритона WR-1339, умеренно снижающего их ингибиторные эффекты.
Представленные результаты указывают, что ингибирование активности CETP торцетрапибом и анацетрапибом, даже при высоких дозах, уменьшалось при резком повышении триглицеридов плазмы у хомяков, обработанных Тритоном WR-1339. По сравнению с торцетрапибом и анацетрапибом, гипертриглицеридемия оказывала меньшее влияние на активность соединения Примера 49 и соединения Примера 8 в отношении ингибирования CETP.
Для определения максимального ингибирования в анализе ex vivo, торцетрапиб или соединение примера 49 добавляли извне (поскольку различие в условии анализа заключалось между in vitro (50% плазмы) и ex vivo (80% плазмы) анализом активности CETP в плазме). Оба соединения при концентрации 10 мкМ вызывали практически 100% ингибирование в группе, получавшей солевой раствор. В группе, получавшей Тритон WR-1339, соединение Примера 49 вызывало практически 100% ингибирование, а ингибирование под действием торцетрапиба составляло приблизительно 75%. Представленные результаты указывают, что селективное ингибирование CETP соединениями в исследовании in vivo вероятно вызвано другим механизмом, а не сниженным ингибированием активности CETP в зависимости от соединения.
Кроме того, эксперименты in vivo проводили для оценки экспозиции и эффективности Примера 8 и анацетрапиба (Merck) на нормальных обезьянах, получавших корм с высоким содержанием углеводов.
Животные и рацион
Нормальных самцов яванских макак (n = 17) весом 8-14 кг содержали на рационе с высоким содержанием углеводов и низким содержанием жиров. Калорийный состав рациона включал приблизительно 13,4% белков, 79,1% углеводов (с соотношением сахара и крахмала 50:50) и 7,5% жиров. Калории поступали со стандартным кормом для обезьян (LabDiet® 5047, PMI, Richmond, IN или LabDiet® 5000, Balanced Fiber diet, PMI, Richmond, IN) в количествах, необходимых для удовлетворения потребностей в белках и жирах, а остальные калории поступали с подслащенными сахаром злаками, леденцами и фруктами (банан, апельсин, канталупа, дыня, ананас). Рацион с высоким содержанием углеводов (CHO) поддерживали в течение 18 дней (4 дня перед введением лекарственного средства, 14 дней введения лекарственного средства), после чего животных возвращали на их стандартные рационы в течение 14-дневного периода вымывания, сопровождаемого второй 14-дневной фазой исследования с введением доз (нормальный рацион).
Распределение в группы обработки соединениями будет основано на триглицеридных ответах после 3 дней на рационе с высоким содержанием углеводов. Животные будут выступать в качестве собственного контроля в течение фазы введения лекарственного средства с высоким CHO и фазы введения лекарственного средства с нормальным рационом.
Приготовление лекарственной формы соединения
Соединение Примера 8 растворяли в концентрации 30 мг/мл в 1 М NaOH + этанол:глицерид кукурузного масла:пропиленгликоль: кремофор RH40 (10:36:9:45) и разводили 1:10 в ddH2O (бидист.). Анацетрапиб растворяли в концентрации 85 мг/мл в кремофоре EL: Span80:кукурузном масле (1:1:1) и разводили до 10 мг/мл в ddH2O. Лекарственные формы приготавливали каждый день (дозы для введения на выходных приготавливали в пятницу) и хранили при перемешивании и с защитой от света при комнатной температуре до введения. При получении дозы в нее добавляли искусственные ароматизаторы и красители.
Введение соединения
Соединения (1 мл/кг) дают животным внутрь без принуждения из пластмассового шприца через ограждение клетки, в которой их содержат, приблизительно в 8:00 ежедневно. Животным дают корм через 60 минут после введения дозы.
Забор крови
Забор крови производят еженедельно, в течение каждой фазы введения доз. До забора крови животным не дают корм (приблизительно в течение 16 часов). Пробы крови забирают из подкожной вены с помощью иглы и шприца в покрытые ЭДТА пробирки объемом 2 мл, которые сразу помещают в лед до центрифугирования. Пробы центрифугируют при 3000 об/мин в течение 20 минут. Плазму собирают, разделяют на аликвоты (указанные ниже) и до анализа хранят при -70°C.
Разделение пробы
Масса тела
Массу тела (МТ) определяют натощак и регистрируют раз в неделю в Lab Animal Services.
Результаты представлены в таблице 2 ниже:
% Δ в сравнении с днем 0
% Δ в сравнении с днем 0
Результаты показывают, что у обезьян с индуцированной рационом гипертриглицеридемией (TG>500 мг/дл), соединение примера 8 сохраняет CETP-ингибирующую активность и демонстрирует более высокое максимальное ингибирование активности CETP по сравнению с анацетрапибом.
В заключение, данные in vivo и in vitro подтверждают, что соединения по изобретению сохраняют ингибирующую активность в противоположность другим соединениям-ингибиторам CETP (анацетрапибу и торцетрапибу), разрабатываемым в настоящее время или созданным ранее.
Клинический эксперимент:
В первоначальное исследование проверки концепции (PoC) включали субъектов со смешанной дислипидемией и тяжелой гипертриглицеридемией, отсутствием в анамнезе острого коронарного синдрома (ACS), болезни коронарных артерией (CAD), инфаркта миокарда (MI) или инсульта. Параметры липидов натощак у этих пациентов должны соответствовать следующим критериям: ЛНП-C 100-150 мг/дл, ЛВП-C <40 мг/дл, TG >500 мг/дл. Исследование PoC планировали как рандомизированное, двойное слепое, плацебо-контролируемое перекрестное исследование с 2 периодами (терапия в течение 14 дней в каждый период и вымывание в течение 14 дней между периодами), для оценки фармакокинетических и фармакодинамических параметров у гипертриглицеридемических (TG >500 мг/дл) пациентов, наряду с сопутствующим контролем артериального давления в амбулаторных условиях (ABPM) для оценки влияния на артериальное давление, концентрации альдостерона и кортизола в плазме.
После обработки ингибитором CETP Формулы I параметры липидов определяют и сравнивают у пациентов с нормальным уровнем триглицеридов натощак и у лиц с высоким уровнем триглицеридов натощак. С учетом данных in vitro и in vivo, представленных в настоящей заявке, фармакодинамический эффект соединений Формулы I у пациентов с высокими или опасно высокими уровнями триглицеридов, как ожидают, будет аналогичен в ингибировании CETP и увеличении ЛВП, чем у пациентов с нормальным уровнем триглицеридов.
Исследование направлено на демонстрацию остаточной CETP-ингибирующей эффективности соединений Формулы I у пациентов с высокими уровнями триглицеридов, а также безопасности (отсутствие повышения артериального давления или уровня альдостерона) в целевой популяции пациентов. Остаточная CETP-ингибирующая эффективность может быть определена как способность соединения Формулы I сохранять ≥50%, предпочтительно ≥65% или более предпочтительно ≥75% ингибирования CETP-ингибирующей активности у субъектов с высокими уровнями триглицеридов в плазме по сравнению с субъектом, имеющим нормальные уровни триглицеридов в плазме. В предпочтительном варианте осуществления остаточная CETP-ингибирующая эффективность может быть определена как способность соединения Формулы I сохранять ≥50%, предпочтительно ≥65% или более предпочтительно ≥75% ингибирования CETP-ингибирующей активности без необходимости в повышении дозы у субъектов с высокими уровнями триглицеридов в плазме по сравнению с субъектом, имеющим нормальные уровни триглицеридов в плазме. Исследование также направлено на демонстрацию того, что соединение Формулы I повышает уровень ЛВП-C на 80%, 90% или 100% у пациентов с высоким уровнем триглицеридов (>300 мг/дл) и наиболее предпочтительно у пациентов с очень высокими уровнями триглицеридов (>750 мг/дл).
Субъект с повышенным уровнем триглицеридов включает пациента, у которого уровень триглицеридов в сыворотке натощак превышает 150 мг/дл (миллиграммов на децилитр). Высокие и очень высокие уровни триглицеридов натощак определяют как ≥200 мг/дл и ≥500 мг/дл, соответственно.
Текущие обозначения для уровней триглицеридов натощак согласно NCEP (Национальная образовательная программа по холестерину) являются следующими: 150-199 мг/дл соответствует границе высокого уровня; 200-499 мг/дл соответствует высокому уровню, и ≥500 мг/дл считают очень высоким уровнем (Triglyceride and cardiovascular diseases, Journal of the American Heart Association, Circulation, 2011, 123, 2293-2333).
Субъект с высокими уровнями триглицеридов включает пациента, у которого уровень триглицеридов в сыворотки натощак превышает 150 мг/дл (миллиграммов на децилитр). В одном варианте осуществления субъект с высокими уровнями триглицеридов включает пациента, у которого уровень триглицеридов натощак выше чем 200 мг/дл или выше чем 300 мг/дл, или выше чем 750 мг/дл.
Популяция пациентов:
Доказательство из эпидемиологических и контролируемых клинических испытаний продемонстрировало, что на уровни триглицеридов заметно влияют статус массы тела и распределение жира в теле (Ford ES, Li C, Zhao G, Pearson WS, Mokdad AH. Hyperglyceridemia and its pharmacologic treatment among US adults. Arch Intern Med. 2009; 169: 572-578). В данном исследовании было продемонстрировано, что у 80% участников, классифицированных как имеющих избыточный вес (BMI 25-30 кг/м2) и ожирение (BMI >30 кг/м2), уровни триглицеридов превышали 150 мг/дл, и с границей уровня триглицеридов >200 мг/дл, 83% участников были классифицированы как имеющие избыточный вес.
Гипертриглицеридемию наблюдали у многих пациентов с липодистрофическими нарушениями, редким генетическим нарушением, часто ассоциируемым с низким ЛВП-C (Simha V, Gard A. Lipodystrophy: lessons in lipid and energy metabolism. Curr Opin Lipidol. 2006; 77:162-169).
Высокие уровни триглицеридов также, как известно, сопровождают нормальную или нарушенную глюкозу натощак. У 35% взрослых с сахарным диабетом 2 типа уровни триглицеридов натощак превышают 200 мг/дл, что связано с пониженным ЛВП-C (Resnick HE, Foster GL, Bardsley J, Ratner RE. Achievement of American Diabetes Association clinical pratice recommendations among US adults with diabetes, 1999-2002: The National Health and Nutrition Examination Survey. Diabetes Care, 2006; 29:531-537). Пациенты с плохо контролируемым сахарным диабетом 1 типа могут демонстрировать подобный характер дислипидемии. Причины гипертриглицеридемии у пациентов с сахарным диабетом включают повышенный синтез ЛОНП печени (липопротеинов очень низкой плотности) и нарушение удаления хиломикронов и CMR (остатков хиломикронов) (Kreisberg RA. Diabetic dyslipidemia Am. J. Cardiol. 1998; 82:67U-73U).
Повышенные уровни триглицеридов, наряду с увеличенной окружностью талии, повышенный уровень глюкозы натощак, повышенное артериальное давление или пониженные уровни ЛВП-C, как известно, являются факторами риска развития метаболического синдрома (Ninomiya JK, L'ltalien G, Criqui MH, Whyte JL, Gamst A, Chen RS. Association of the metabolic syndrome with history of myocardial infarction and stroke in the third national health and nutrition examination Survey. Circulation. 2004; 109:42-46). Распространенность повышенных уровней триглицеридов у субъектов с метаболическим синдромом почти в два раза выше, чем у субъектов без метаболического синдрома (Schwartz GG, Olson AG, Szarek M, Sasiela WJ. Relation of characteristics of metabolic syndrome to short-term prognosis and effects of intensive statin therapy after acute coronary syndrome: and analysis of the myocardial ischemia reduction with aggressive cholesterol lowering (MIRACL) trial. Diabetes Care. 2005; 28: 2508-2513). Среди лиц с элементами метаболического синдрома высокие уровни триглицеридов были вторым наиболее часто встречаемым фактором после повышенного артериального давления (Kasai T, Miyauchi K, Kurata T, Ohta H, Okazaki S, Miyazaki T, Kajimoto K, Kubota N, Daida H. Prognostic value of the metabolic syndrome for long-term outcomes in patients undergoing percutaneous coronary intervention Circ. J. 2006; 70:1531-1537).
Заметно повышенные уровни триглицеридов также наблюдали у пациентов с генетическими синдромами метаболизма триглицеридов. Указанные генетические синдромы включают синдром хиломикронемии, обусловленный дефицитом липопротеинлипазы или дефицитом аполипопротеина C-II, или мутациями APOA5 и GP1 HBP1 с потерей функции (Brunzell JD. Familial lipoprotein lipase deficiency and other causes of chylomicronemia syndrome. In: Scriver CR et al. The metabolic and molecular base of inherited diseases. New York, NY: McGraw-Hill; 1995; 1913-1932; Priore Oliva C. et al. inherited apolipoprotein A-V deficiency in severe hypertriglyceridemia. Arterioscler Thromb Vasc Biol. 2005; 25:41 1-417; Olivecrona G et al. Mutation of conserved cysteines in Ly6 domain of GPIHBP1 in familial chylomicronemia. J. Lipid Res. 2010; 51 :1535-1545; Beigneux AP et al. Chylmicronemia with a mutant GPIHBP1 (Q115P) which can not bind lipoprotein lipase. Arterioscler Thromb Vase. Biol. 2009; 29:956-962). Дополнительные генетические синдромы, ассоциируемые с гипертриглицеридемией, включают семейную гипертриглицеридемию, семейную комбинированную гиперлипидемию или дисбеталипопротеинемию III типа (Goldstein JL et al. genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J. Clin. Invest. 1973; 52: 1544-1568; Brunzell JD et al. Myocardial infraction in the familial forms of hypertriglyceridemia. Metabolism. 1976; 25: 313-320).
Таким образом, субъект с высокими уровнями триглицеридов может включать пациента с семейным синдромом хиломикронемии, обусловленным дефицитом липопротеинлипазы или дефицитом аполипопротеина C-II, или мутацией APOA5 и GP1 HBP1 с потерей функции, пациентов с коронарной недостаточностью, пациентов с острым коронарным синдромом, пациентов с инфарктом миокарда, пациентов с диабетом, пациентов с ожирением, пациентов с липодистрофическими нарушениями, пациентов с хроническим заболеванием почек, пациентов с метаболическим синдромом, пациентов с другими генетическими синдромами, при которых обычно наблюдают приобретенное повышение триглицеридов, такие как семейная гипертриглицеридемия, семейная комбинированная гиперлипидемия или дисбеталипопротеинемия III типа.
Соединение для применения в настоящем изобретении можно вводить одновременно, либо до, либо после, с одним или более другими терапевтическими средствами. Соединение по настоящему изобретению можно вводить отдельно, одними и теми же или разными путями введения, или вместе, в одной фармацевтической композиции, как другие средства.
В одном варианте осуществления изобретение относится к способу или применению согласно любому из вариантов осуществления 1-34A, включающему введение субъекту продукта, включающего соединение согласно любой из формул I-VIIA, или его фармацевтически приемлемую соль, и по меньшей мере одно другое терапевтическое средство, в виде комбинированного препарата для одновременного, раздельного или последовательного применения в терапии.
Продукты, предложенные в виде комбинированного препарата для применения в способе по изобретению, включают композицию, включающую соединение согласно любой из формул I-VIIA, или его фармацевтически приемлемую соль, и другое терапевтическое средство(-а) вместе в одной фармацевтической композиции, или соединение согласно любой из формул I-VIIA, или его фармацевтически приемлемую соль, и другое терапевтическое средство(-а) в раздельной форме, например, в форме набора.
В одном варианте осуществления изобретение относится к способу или применению согласно любому из вариантов осуществления 1-34A, включающему введение субъекту фармацевтической композиции, включающей соединение согласно любой из формул I-VIIA, или его фармацевтически приемлемую соль, и по меньшей мере одно другое терапевтическое средство, в виде комбинированного препарата для одновременного, раздельного или последовательного применения в терапии. Необязательно, фармацевтическая композиция для применения в способе по изобретению может включать фармацевтически приемлемый эксципиент, как описано выше.
В одном варианте осуществления изобретения предложен набор для применения в способе по изобретению, включающий две или более отдельных фармацевтических композиции, из которых по меньшей мере одна композиция содержит соединение согласно любой из формул I-VIIA или его фармацевтически приемлемую соль. В одном варианте осуществления набор включает средства для отдельного хранения указанных композиций, такие как емкость, раздельный флакон или раздельный пакет из фольги. Примером такого набора является блистерная упаковка, какую обычно используют для упаковки таблеток, капсул и т.п.
Набор по изобретению может применяться для введения различных лекарственных форм, например, пероральных и парентеральных, для введения отдельных композиций в различных интервалах дозировки, или для титрованиях отдельных композиций друг против друга. Чтобы способствовать соблюдению режима терапии, набор по изобретению обычно включает указания по применению.
В комбинированных терапиях согласно изобретению соединение по изобретению и другое терапевтическое средство может быть получено и/или составлено одним или разными производителями. Кроме того, соединение по изобретению и другое терапевтическое средство могут быть объединены в комбинированной терапии: (i) перед передачей комбинированного продукта врачам (например, в случае набора, включающего соединение по изобретению и другое терапевтическое средство); (ii) врачом самостоятельно (или под руководством врача) незадолго до введения; (iii) в организме пациента, например, во время последовательного введения соединения по изобретению и другого терапевтического средства.
Таким образом, в изобретении предложен способ или применение согласно любому из вариантов осуществления 1-34A, включающие введение соединения согласно любой из формул I-VIIA, или его фармацевтически приемлемой соли, где лекарственное средство приготовлено для введения с другим терапевтическим средством.
В изобретении также предложено соединение согласно любой из формул I-VIIA, или его фармацевтически приемлемая соль, для применения в способе по изобретению, где соединение согласно любой из формул I-VIIA или его фармацевтически приемлемая соль приготовлены для введения с другим терапевтическим средством. В изобретении также предложено другое терапевтическое средство для применения в способе по изобретению, где другое терапевтическое средство приготовлено для введения с соединением согласно любой из формул I-VIIA или его фармацевтически приемлемой солью.
В изобретении также предложено применение соединения согласно любой из формул I-VIIA или его фармацевтически приемлемой соли для лечения, предотвращения и/или уменьшения интенсивности атеросклероза или дислипидемии, или для повышения ЛВП-C и/или снижения ЛНП-C, у субъекта с высоким уровнем триглицеридов, где пациент ранее (например, в течение 24 часов) получал другое терапевтическое средство. В изобретение также предложено применение другого терапевтического средства при лечении, предотвращении и/или уменьшении интенсивности атеросклероза или дислипидемии, или для повышения ЛВП-C и/или снижения ЛНП-C, у пациента с высоким уровнем триглицеридов, где пациент ранее (например, в течение 24 часов) получал соединение согласно любой из формул I-VIIA или его фармацевтически приемлемую соль.
В одном варианте осуществления другое терапевтическое средство выбрано из: статина, ингибитора абсорбции холестерина, апрегулятора/индуктора АпоA-1, пре-бета ЛВП миметика, стабилизатора или индуктора ABCA1, агониста LXR, агониста FXR, ингибитора белка-переносчика фосфолипидов (PLTP), ингибитора альдостерон-синтазы (ASI), производного фибровой кислоты, рыбьего жира, ингибитора DGAT1 и ингибитора эндотелиальной липазы.
Термин "в комбинации с" вторым средством или терапией включает совместное введение соединения по изобретению (например, соединения согласно любой из Формул I-VIIA или соединения, описанного в настоящей заявке иным образом) со вторым средством или терапией, введение соединения по изобретению вначале с последующим введением второго средства или терапии, и введения второго средства или терапии вначале с последующим введением соединения по изобретению.
Термин "второе средство" включает любое средство, которое известно в уровне техники для лечения, предотвращения или уменьшения симптомов атеросклероза или дислипидемии.
Примеры вторых средств включают:
i) статин (также известный как ингибитор HMG-CoA редуктазы);
ii) ингибитор абсорбции холестерина;
iii) апрегулятор/индуктор АпоA-1;
iv) пре-бета ЛВП миметик;
v) стабилизатор или индуктор ABCA1;
vi) агонист LXR;
vii) агонист FXR;
viii) ингибитор белка-переносчика фосфолипидов (PLTP);
ix) ингибитор альдостерон-синтазы;
x) производное фибровой кислоты;
xi) рыбий жир;
xii) ингибитор DGAT1;
xiii) ингибитор эндотелиальной липазы;
или их фармацевтически приемлемую соль.
i) Статины (или ингибиторы HMG-CoA редуктазы) являются классом лекарственных средств, которые используются для снижения уровней холестерина путем ингибирования фермента HMG-CoA редуктазы, который играет центральную роль в синтезе холестерина в печени. Повышенные уровни холестерина были связаны с сердечно-сосудистыми заболеваниями, и поэтому статины применяются в профилактике таких заболеваний. Примеры включают аторвастатин, церивастатин, компактин, далвастатин, дигидрокомпактин, флуиндостатин, флувастатин, ловастатин, питавастатин, мевастатин, правастатин, ривастатин, симвастатин и велостатин или их фармацевтически приемлемые соли.
ii) Ингибиторы абсорбции холестерина являются классом соединений, которые предотвращают поглощение холестерина из тонкой кишки в кровеносную систему и, в свою очередь, понижают концентрации ЛНП-C в плазме. Повышенные уровни холестерина связаны с увеличенным риском CVD; таким образом, ингибиторы абсорбции холестерина применяются с целью снижения риска CVD. Примером ингибитора абсорбции холестерина является эзетимиб, ранее известный как "Sch-58235". Другим примером является Sch-48461. Оба соединения разработаны компанией Schering-Plough.
iii) Аполипопротеин A-1 является белком, который у людей кодируется геном APOA1. Он играет определенную роль в липидном обмене. Аполипопротеин A-1 является главным белковым компонентом липопротеина высокой плотности (ЛВП) в плазме. Хиломикроны, секретируемые энтероцитами кишечника, также содержат ApoA1, но он быстро переносится к ЛВП в кровотоке. Белок способствует выходу холестерина из тканей в печени для выделения. Он является кофактором лецитинхолестеринацилтрансферазы (LCAT), которая ответственна за формирование большинства эфиров холестерина плазмы. Было показано, что инфузия варианта АпоA-1 людям вызывает регресс атеросклеротических бляшек согласно оценке с помощью внутрисосудистого ультразвукового исследования; таким образом, АпоA-1 понижает риск CVD и обладает способностью одновременно замедлять прогрессию и вызвать регрессию атеросклероза. Примером апрегулятора/индуктора АпоA-1 является RVX208.
iv) Примером пре-бета ЛВП миметика является CER-001. CER-001 представляет собой инновационный комплекс рекомбинантного человеческого АпоA-1, главного структурного белка ЛВП, и фосфолипидов. Он был разработан с целью имитации структуры и функций природных, формирующихся ЛВП, также известных как пре-бета ЛВП, которые, как полагают, обеспечивают защиту против атеросклероза. Предполагается, что CER-001 также будет снижать частоту сердечно-сосудистых явлений у пациентов с высоким риском, способствуя удалению холестерина из стенок сосудов.
v) Переносчик АТФ-связывающих кассет ABCA1 (член 1 подсемейства переносчиков ABCA человека), также известный как белок регуляции выведения холестерина (CERP), является белком, который у людей кодируется геном ABCA1. Указанный переносчик является главным регулятором гомеостаза клеточного холестерина и фосфолипидов. Примером регулятора ABCA1 является пробукол. Пробукол снижает уровень холестерина в крови путем повышения скорости катаболизма ЛНП. Кроме того, пробукол может ингибировать синтез холестерина и задерживать абсорбцию холестерина. Пробукол является сильным антиоксидантом, ингибирующим окисление холестерина в ЛНП; он замедляет формирование пенистых клеток, которые способствуют образованию атеросклеротических бляшек.
vi) X-рецептор печени (LXR) является представителем семейства ядерных рецепторов факторов транскрипции и близко связаны с такими ядерными рецепторами, как PPAR, FXR и RXR. X-рецепторы печени (LXR) являются важными регуляторами гомеостаза холестерина, жирных кислот и глюкозы. Агонисты LXR эффективны при лечении атеросклероза, диабета, воспаления и болезни Альцгеймера в моделях на мышах. Лечение с применением агонистов LXR (гипохоламид, T0901317, GW3965 или N,N-диметил-3бета-гидрокси-холенамид (DMHCA)) снижает уровень холестерина в сыворотке и печени и ингибирует развитие атеросклероза в моделях болезни на мышах. Примерами агонистов LXR являются GW3965 (синтетический нестероидный агонист/активатор X-рецепторов печени (LXR)) и T0901317 (двойной агонист LXR/FXR).
vii) фарнезоидный X-рецептор (FXR), также известный как NR1H4 (член 4 группы H подсемейства ядерных рецепторов 1), является ядерным рецептором гормонов с активностью, аналогичной наблюдаемой в других рецепторах стероидов, таких как эстроген или прогестерон, но более сходным по форме с PPAR, LXR и RXR. Активация ядерного рецептора FXR, как известно, снижает гипергликемию и гиперлипидемию. Примером агониста FXR является GW4064 (3-(2,6-дихлорфенил)-4-(3'-карбокси-2-хлорстильбен-4-ил)-оксиметил-5-изопропилизоксазол).
viii) Белок-переносчик фосфолипидов (PLTP) является белком, который у людей кодируется геном PLTP. Белок, кодируемый указанным геном, является одним из по меньшей мере двух белков-переносчиков липидов, обнаруженных в плазме человека, а другим является CETP. Кодируемый белок переносит фосфолипиды от богатых триглицеридами липопротеинов к липопротеинам высокой плотности (ЛВП). В дополнение к регуляции размера частиц ЛВП данный белок может быть включен в метаболизм холестерина. Для этого гена были обнаружены по меньшей мере два варианта транскриптов, кодирующих различные изоформы. Поскольку PLTP влияет на метаболизм богатых триглицеридами липопротеинов и ЛВП, модуляция данного белка-переносчика потенциально может изменять риск возникновения сердечно-сосудистого заболевания.
ix) Класс ингибиторов альдостеронсинтазы включает стероидные и нестероидные ингибиторы альдостеронсинтазы, причем последние являются наиболее предпочтительными.
Предпочтение отдают коммерчески доступным ингибиторам альдостеронсинтазы или таким ингибиторам альдостеронсинтазы, которые были одобрены органами здравоохранения.
Класс ингибиторов альдостеронсинтазы включает соединения, обладающие различными структурными особенностями. Наиболее предпочтительным нестероидным ингибитором альдостеронсинтазы является (+)-энантиомер гидрохлорида фадрозола (патенты США 4617307 и 4889861) формулы
или, в соответствующем случае, его фармацевтически приемлемая соль.
Ингибиторы альдостеронсинтазы, которые могут применяться в указанной комбинации, являются соединениями и аналогами, которые, в общем и в частности, раскрыты, например, в US 2007/0049616, в особенности в пунктах формулы, в которых заявлены соединения, при этом конечные продукты рабочих примеров, объект конечных продуктов, фармацевтические препараты и пункты формулы включены в настоящую заявку посредством ссылки на данную публикацию. Предпочтительные ингибиторы альдостеронсинтазы, подходящие для применения в настоящем изобретении, включают, без ограничения, 4-(6,7-дигидро-5H-пирроло[1,2-c]имидазол-5-ил)-3-метилбензонитрил; (4-метоксибензил)метиламид 5-(2-хлор-4-цианофенил)-6,7-дигидро-5H-пирроло[1,2-c]имидазол-5-карбоновой кислоты; 4'-фтор-6-(6,7,8,9-тетрагидро-5H-имидазо[1,5-a]азепин-5-ил)бифенил-3-карбонитрил; бутиловый эфир 5-(4-циано-2-метоксифенил)-6,7-дигидро-5H-пирроло[1,2-c]имидазол-5-карбоновой кислоты; 4-(6,7-дигидро-5H-пирроло[1,2-c]имидазол-5-ил)-2-метоксибензонитрил; 4-фторбензиловый эфир 5-(2-хлор-4-цианофенил)-6,7-дигидро-5H-пирроло[1,2-c]имидазол-5-карбоновой кислоты; метиловый эфир 5-(4-циано-2-трифторметоксифенил)-6,7-дигидро-5H-пирроло[1,2-c]имидазол-5-карбоновой кислоты; 2-изопропоксиэтиловый эфир 5-(4-циано-2-метоксифенил)-6,7-дигидро-5H-пирроло[1,2-c]имидазол-5-карбоновой кислоты; 4-(6,7-дигидро-5H-пирроло[1,2-c]имидазол-5-ил)-2-метилбензонитрил; 4-(6,7-дигидро-5H-пирроло[1,2-c]имидазол-5-ил)-3-фторбензонитрил; 4-(6,7-дигидро-5H-пирроло[1,2-c]имидазол-5-ил)-2-метоксибензонитрил; 3-фтор-4-(7-метилен-6,7-дигидро-5H-пирроло[1,2-c]имидазол-5-ил)бензонитрил; цис-3-фтор-4-[7-(4-фторбензил)-5,6,7,8-тетрагидро-имидазо[1,5-a]пиридин-5-ил]бензонитрил; 4'-фтор-6-(9-метил-6,7,8,9-тетрагидро-5H-имидазо[1,5-a]азепин-5-ил)бифенил-3-карбонитрил; 4'-фтор-6-(9-метил-6,7,8,9-тетрагидро-5H-имидазо[1,5-a]азепин-5-ил)бифенил-3-карбонитрил или, в каждом случае, их (R) или (S) энантиомер; или, в соответствующем случае, их фармацевтически приемлемую соль.
Термин "ингибиторы альдостеронсинтазы" также включают соединения и аналоги, раскрытые в WO 2008/076860, WO 2008/076336, WO 2008/076862, WO 2008/027284, WO 2004/046145, WO 2004/014914, WO 2001/076574.
Кроме того, ингибиторы альдостеронсинтазы также включают соединения и аналоги, раскрытые в заявках на патенты США US 2007/0225232, US 2007/0208035, US 2008/0318978, US 2008/0076794, US 2009/0012068, US 2009/0048241 и в заявках PCT WO 2006/005726, WO 2006/128853, WO 2006/128851, WO 2006/128852, WO 2007/065942, WO 2007/116099, WO 2007/116908, WO 2008/119744 и в европейской заявке на патент EP 1886695. Предпочтительные ингибиторы альдостеронсинтазы, подходящие для применения в настоящем изобретении, включают, без ограничения, 8-(4-фторфенил)-5,6-дигидро-8H-имидазо[5,1-c][1,4]оксазин; 4-(5,6-дигидро-8H-имидазо[5,1-c][1,4]оксазин-8-ил)-2-фторбензонитрил; 4-(5,6-дигидро-8H-имидазо[5,1-c][1,4]оксазин-8-ил)-2,6-дифторбензонитрил; 4-(5,6-дигидро-8H-имидазо[5,1-c][1,4]оксазин-8-ил)-2-метоксибензонитрил; 3-(5,6-дигидро-8H-имидазо[5,1-c][1,4]оксазин-8-ил)бензонитрил; 4-(5,6-дигидро-8H-имидазо[5,1-c][1,4]оксазин-8-ил)фталонитрил; 4-(8-(4-цианофенил)-5,6-дигидро-8H-имидазо[5,1-c][1,4]оксазин-8-ил)бензонитрил; 4-(5,6-дигидро-8H-имидазо[5,1-c][1,4]оксазин-8-ил)бензонитрил; 4-(5,6-дигидро-8H-имидазо[5,1-c][1,4]оксазин-8-ил)нафталин-1-карбонитрил; 8-[4-(1H-тетразол-5-ил)фенил]-5,6-дигидро-8H-имидазо[5,1-c][1,4]оксазин, разработанный компанией Speedel или, в каждом случае, их (R) или (S) энантиомер; или, в соответствующем случае, их фармацевтически приемлемую соль.
Ингибиторы альдостеронсинтазы, которые могут применяться в указанной комбинации, являются соединениями и аналогами, в общем и в частности раскрытыми, например, в WO 2009/156462 и WO 2010/130796, в особенности, в пунктах формулы, в которых заявлены соединения, конечных продуктах рабочих примеров, объекте конечных продуктов, фармацевтических препаратах и формуле изобретения. Предпочтительные ингибиторы альдостеронсинтазы, подходящие для комбинации в настоящем изобретении, включают гидрохлорид 3-(6-фтор-3-метил-2-пиридин-3-ил-1H-индол-1-илметил)бензонитрила, 1-(4-метансульфонил-бензил)-3-метил-2-пиридин-3-ил-1H-индол, 2-(5-бензилокси-пиридин-3-ил)-6-хлор-1-метил-1H-индол, этиловый эфир 5-(3-циано-1-метил-1H-индол-2-ил)никотиновой кислоты, N-[5-(6-хлор-3-циано-1-метил-1H-индол-2-ил)пиридин-3-илметил]этансульфонамид, 5-(6-хлор-3-циано-1-метил-1H-индол-2-ил)пиридин-3-иловый эфир пирролидин-1-сульфоновой кислоты, N-метил-N-[5-(1-метил-1H-индол-2-ил)пиридин-3-илметил]метансульфонамид, 6-хлор-1-метил-2-{5-[(2-пирролидин-1-илэтиламино)метил]пиридин-3-ил}-1H-индол-3-карбонитрил, 6-хлор-2-[5-(4-метансульфонилпиперазин-1-илметил)пиридин-3-ил]-1-метил-1H-индол-3-карбонитрил, 6-хлор-1-метил-2-{5-[(1-метилпиперидин-4-иламино)метил]пиридин-3-ил}-1H-индол-3-карбонитрил, [5-(6-хлор-3-циано-1-метил-1H-индол-2-ил)пиридин-3-илметил]амид морфолин-4-карбоновой кислоты, N-[5-(6-хлор-1-метил-1H-индол-2-ил)пиридин-3-илметил]этансульфонамид, C,C,C-трифтор-N-[5-(1-метил-1H-индол-2-ил)пиридин-3-илметил]метансульфонамид, N-[5-(3-хлор-4-цианофенил)пиридин-3-ил]-4-трифторметилбензолсульфонамид, N-[5-(3-хлор-4-цианофенил)-пиридин-3-ил]-1-фенилметансульфонамид, N-(5-(3-хлор-4-цианофенил)пиридин-3-ил)бутан-1-сульфонамид, N-(1-(5-(4-циано-3-метоксифенил)пиридин-3-ил)этил)этансульфонамид, N-((5-(3-хлор-4-цианофенил)пиридин-3-ил)(циклопропил)метил)этансульфонамид, N-(циклопропил(5-(1H-индол-5-ил)пиридин-3-ил)метил)этансульфонамид, N-(циклопропил(5-нафталин-1-ил-пиридин-3-ил)метил)этансульфонамид, [5-(6-хлор-1-метил-1H-пирроло[2,3-b]пиридин-2-ил)пиридин-3-илметил]амид этансульфоновой кислоты и {[5-(3-хлор-4-цианофенил)пиридин-3-ил]циклопропилметил}этиламид этансульфоновой кислоты.
x) производные фибровой кислоты понижают триглицериды и повышают холестерин ЛВП. Они могут оказывать небольшой эффект на холестерин ЛНП. Например, гемфиброзил или фенофибрат назначают людям, которые имеют очень высокие уровни триглицеридов или имеют низкие уровни ЛВП и высокие уровни триглицеридов. Гемфиброзил может применяться для снижения риска сердечного приступа у людей с коронарной недостаточностью (CAD), которые имеют низкие уровни ЛВП и высокие уровни триглицеридов.
xi) рыбий жир - жир, полученный из тканей жирной рыбы. Рыбьи жиры содержат омега-3 жирные кислоты - эйкозапентаеновую кислоту (EPA) и докозагексаеновую кислоту (PHA), предшественников эйкозаноидов, которые, как известно, обладают многими полезными для здоровья свойствами. Рыбий жир и другие источники омега-3 более всего рекомендуются при следующих состояниях: гипертриглицеридемия, вторичное сердечно-сосудистое заболевание и предотвращение высокого артериального давления. Например, Ловаза используется в сочетании с диетой с низким содержанием жиров и холестерина для понижения очень высоких уровней триглицеридов (жиров) в крови.
xii) DGAT является ферментом, который катализирует последнюю стадию биосинтеза триацилглицеринов. DGAT катализирует конденсацию 1,2-диацилглицерина с ацил-КоА жирной кислоты с получением кофермента A и триацилглицерина. Были идентифицированы два фермента, которые проявляют активность DGAT: DGAT1 (ацил-КоА-диацилглицеринацилтрансфераза 1, см. Cases et al., Proc. Natl. Acad. Sci. 95:13018-13023, 1998) и DGAT2 (ацил-CoA-диацилглицеринацилтрансфераза 2, видят Cases et al., J. Biol. Chem. 276:38870-38876, 2001). DGAT1 и DGAT2 не имеют значительной общей гомологии белковой последовательности. Важно отметить, что мыши с нокаутом по DGAT1 защищены от набора веса, вызванного потреблением корма с высоким содержанием жиров, и инсулинорезистентности (Smith et al, Nature Genetics 25:87-90, 2000). Фенотип DGAT1 нокаутных мышей указывает, что ингибитор DGAT1 может применяться для лечения ожирения и связанных с ожирением осложнений. Ингибиторы DGAT1, которые полезны в указанной комбинации, являются соединениями и аналогами, в общем и в частности раскрытыми, например, в WO 2007/126957 и WO 2009/040410, в особенности в пунктах формулы, в которых заявлены соединения, а также конечных продуктах рабочих примеров, объекте конечных продуктов, фармацевтических препаратах и формуле изобретения. Предпочтительные ингибиторы DGAT1, подходящие для применения в настоящем изобретении, включают {4-[4-(3-метокси-5-фениламинопиридин-2-ил)фенил]циклогексил}уксусную кислоту, (4-{4-[5-(1-метил-1H-пиразол-3-иламино)пиридин-2-ил]фенил}циклогексил)уксусную кислоту, (4-{4-[5-(5-фтор-6-метоксипиридин-3-иламино)пиридин-2-ил]фенил}циклогексил)уксусную кислоту, (4-{5-[5-(6-трифторметилпиридин-3-иламино)пиридин-2-ил]спироциклогексилиденил-1,1'-инданил}уксусную кислоту, (4-{4-[5-(бензооксазол-2-иламино)пиридин-2-ил]фенил}циклогексил)уксусную кислоту, 4-(4-{4-[2-(3-хлорфениламино)оксазол-5-ил]фенил}циклогексил)масляную кислоту, (4-{4-[5-(6-трифторметилпиридин-3-иламино)пиридин-2-ил]фенил}циклогексил)уксусную кислоту, (6-{4-[4-(2H-тетразол-5-илметил)циклогексил]фенил}пиридазин-3-ил)-(6-трифторметилпиридин-3-ил)амин, 3-(4-{4-[6-(6-трифторметилпиридин-3-иламино)пиридазин-3-ил]фенил}циклогексилметил)-4H-[1,2,4]оксадиазол-5-он, (1-{4-[6-(3-трифторметилфениламино)пиридазин-3-ил]фенил}пиперидин-4-ил)уксусную кислоту, (4-{4-[4-метил-6-(6-трифторметилпиридин-3-иламино)пиридазин-3-ил]фенил}циклогексил)уксусную кислоту, (4-{4-[5-(6-трифторметилпиридин-3-иламино)пиразин-2-ил]фенил}циклогексил)уксусную кислоту, 6-[5-(4-хлорфенил)[1,3,4]оксадиазол-2-ил]-2-(2,6-дихлорфенил)-1H-бензоимидазол, 6-(5-циклогексил[1,3,4]оксадиазол-2-ил)-2-(2,6-дихлорфенил)-1H-бензоимидазол, 6-(5-бутил[1,3,4]оксадиазол-2-ил)-2-(2,6-дихлорфенил)-1H-бензоимидазол, 2-(2,6-дихлорфенил)-6-[5-(5-метилпиридин-3-ил)[1,3,4]оксадиазол-2-ил]-1H-бензоимидазол, 6-[5-(4-хлорфенил)[1,3,4]оксадиазол-2-ил]-2-(2,6-диметил-4-морфолин-4-илфенил)-1H-бензоимидазол, 6-[5-(4-хлорфенил)[1,3,4]оксадиазол-2-ил]-2-(3,5-дихлорпиридин-4-ил)-1H-бензоимидазол, 3-(4-{5-[5-(4-метоксифенил)[1,3,4]оксадиазол-2-ил]-1H-бензоимидазол-2-ил}-3,5-диметилфенил)-2,2-диметилпропионовую кислоту, 3-(4-{6-[5-(4-метоксифенил)[1,3,4]оксадиазол-2-ил]-1H-бензоимидазол-2-ил}-3,5-диметилфенил)пропионовую кислоту, 3-(4-{6-[5-(4-метоксифениламино)[1,3,4]оксадиазол-2-ил]-1H-бензимидазол-2-ил}-3,5-диметилфенил)пропионовую кислоту, [3-(4-{6-[5-(4-хлорфенил)[1,3,4]оксадиазол-2-ил]-1H-бензоимидазол-2-ил}-3,5-диметилфенил)пропил]фосфоновую кислоту, 2-(2,6-дихлорфенил)-6-(4,5-дифенилоксазол-2-ил)-1H-бензоимидазол, (4-{6-[5-(4-хлорфенил)[1,3,4]оксадиазол-2-ил]-1H-бензоимидазол-2-ил}-3,5-диметилфенокси)уксусную кислоту, 2-(2,6-дихлорфенил)-6-(5-пирролидин-1-ил[1,3,4]оксадиазол-2-ил)-1H-бензоимидазол и 3,5-диметил-4-{6-[5-(4-трифторметилфениламино)[1,3,4]оксадиазол-2-ил]-1H-бензоимидазол-2-ил}фенол.
xiii) Активность эндотелиальной липазы (EL) была задействована в катаболизме ЛВП, воспалении сосудов и атерогенезе. У EL нокаутных мышей присутствовало явное повышение холестерина ЛВП по сравнению с мышами дикого типа. Ингибиторы, как поэтому ожидают, будут применяться для лечения сердечно-сосудистого заболевания.
Второе средство, представляющее наибольший интерес, включает статин и ингибитор абсорбции холестерина.
Иллюстративные примеры изобретения:
Следующие примеры предназначены для иллюстрации изобретения и не должны рассматриваться как ограничивающие изобретения.
ИЛЛЮСТРАТИВНЫЕ ПРИМЕРЫ ИЗОБРЕТЕНИЯ:
Примеры 1-27 являются соединениями, раскрытыми в US 2009/01 18287 (WO 2009/059943):
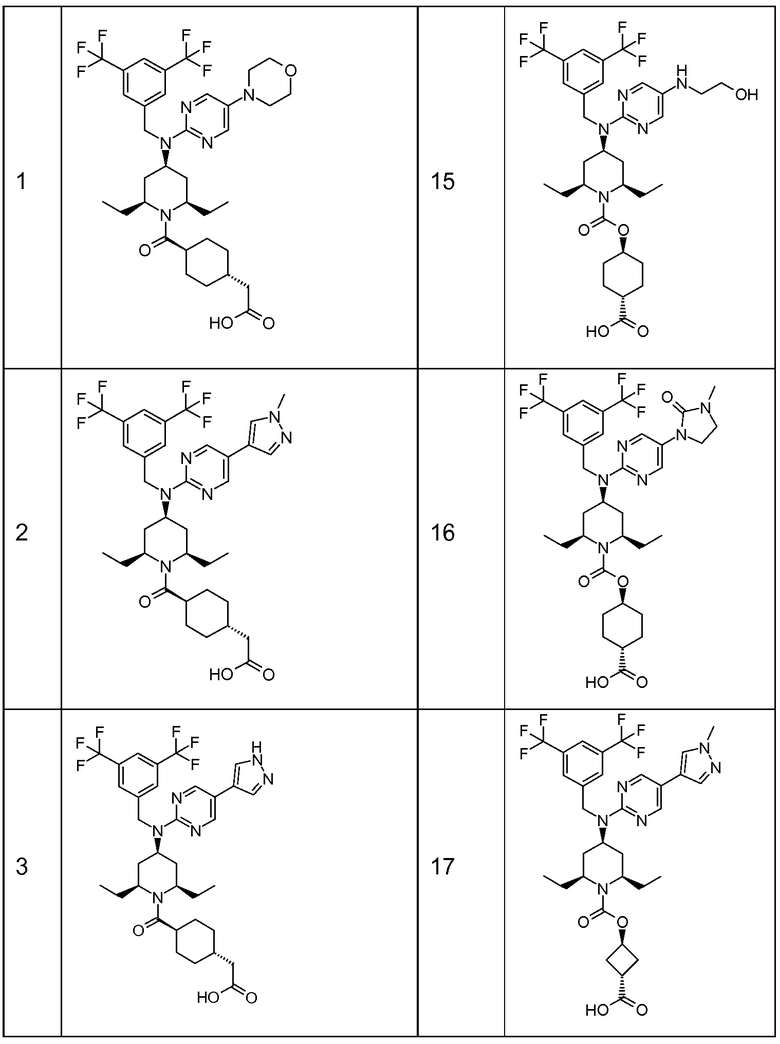
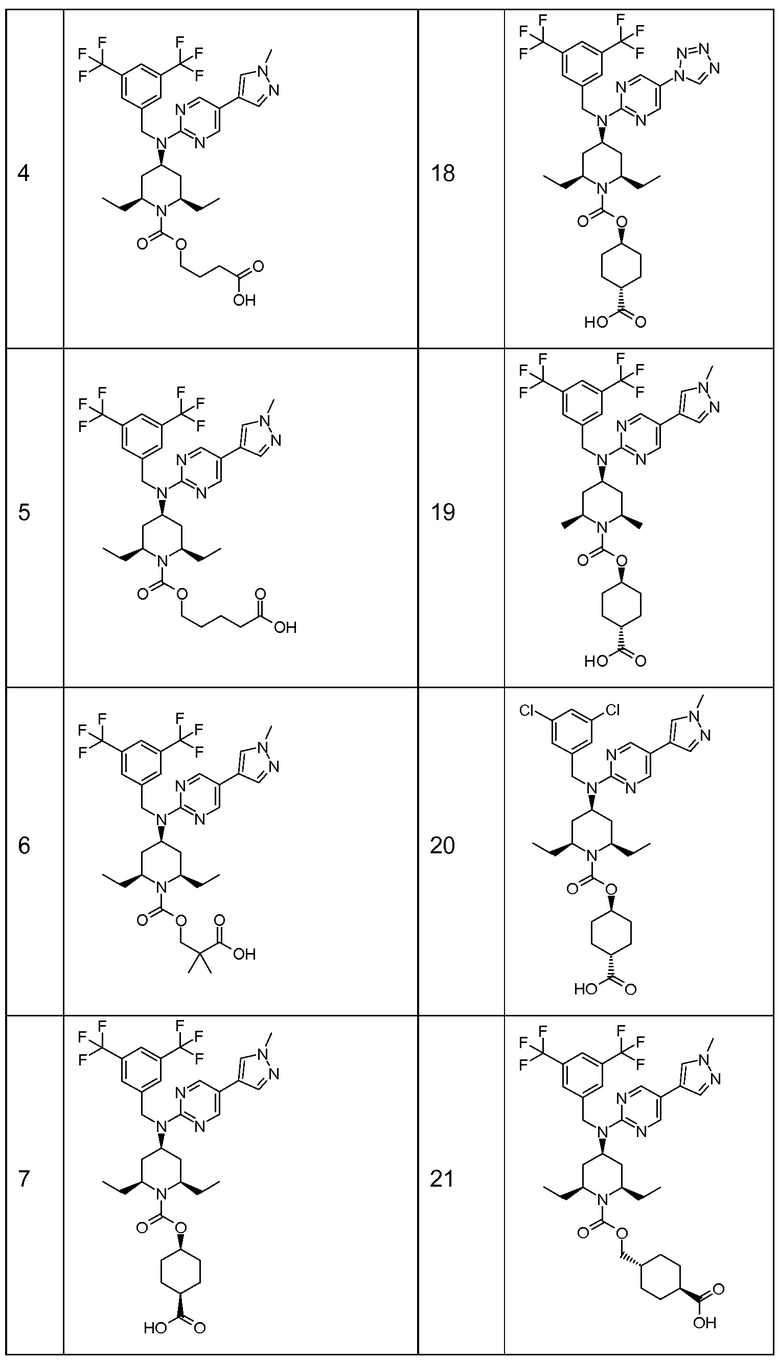
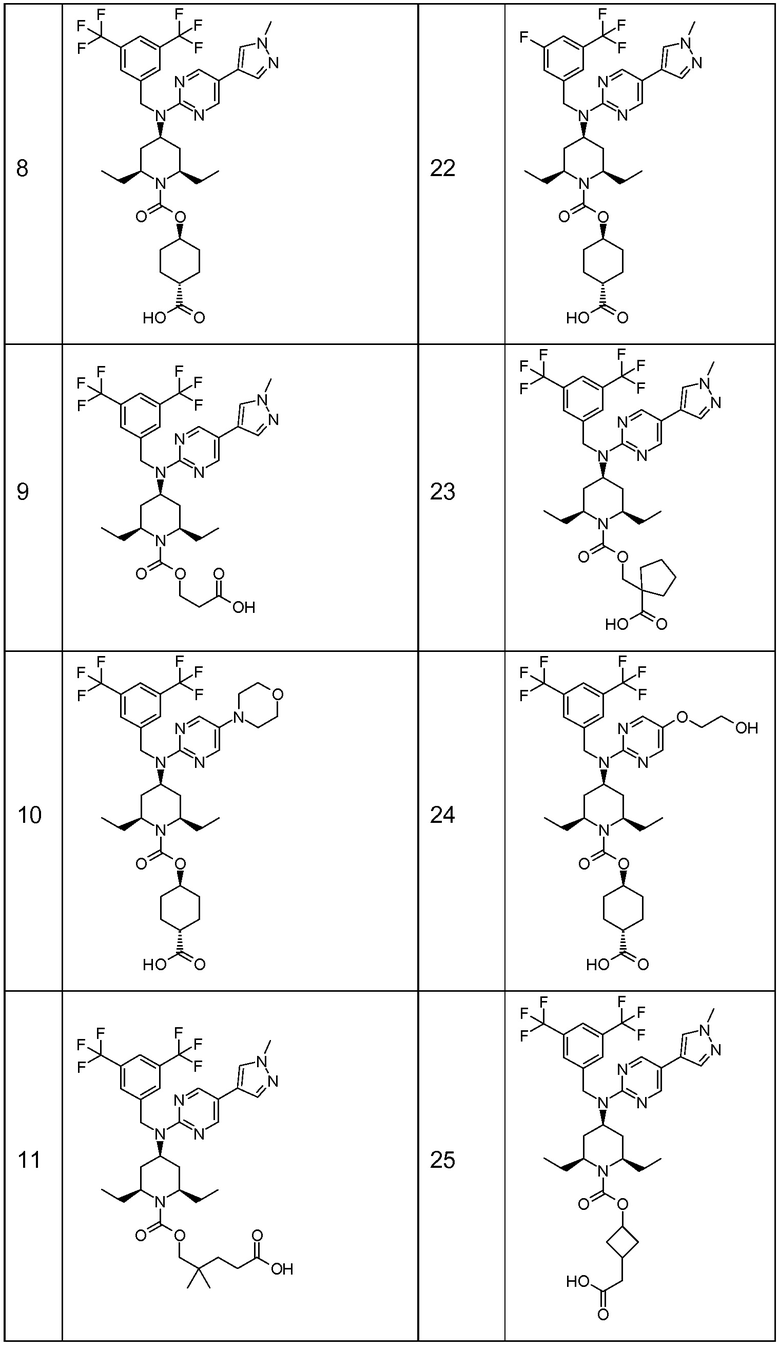
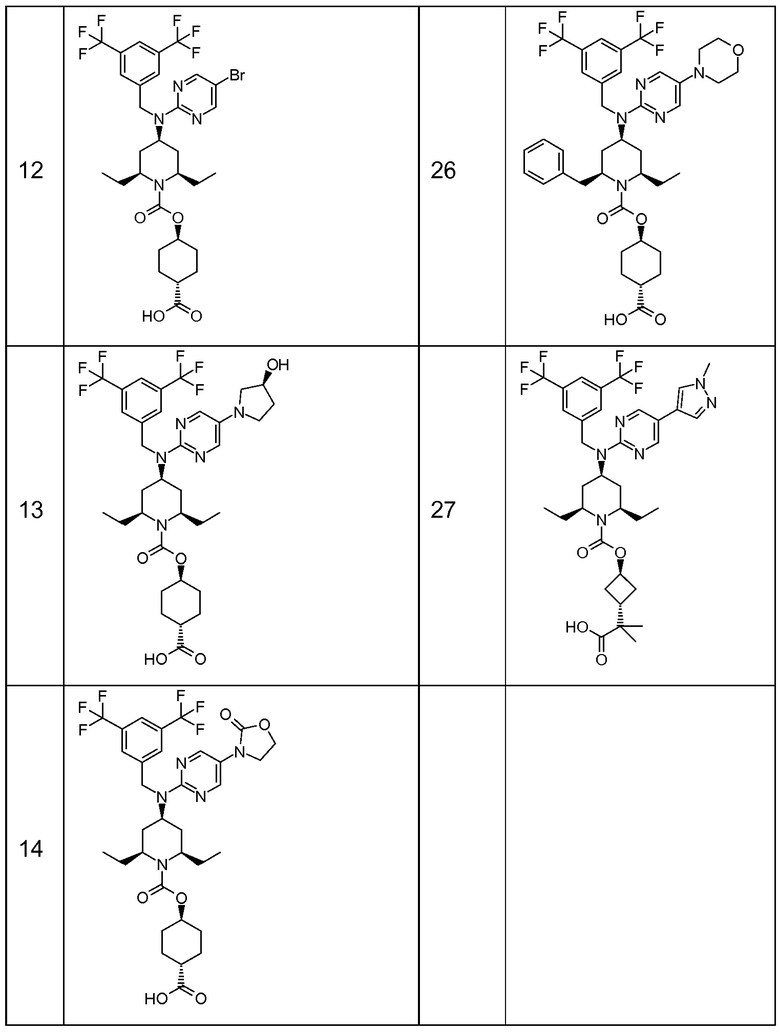
Примеры 28-41 являются соединениями, раскрытыми в US 2010/0311750 (WO 2009/071509).
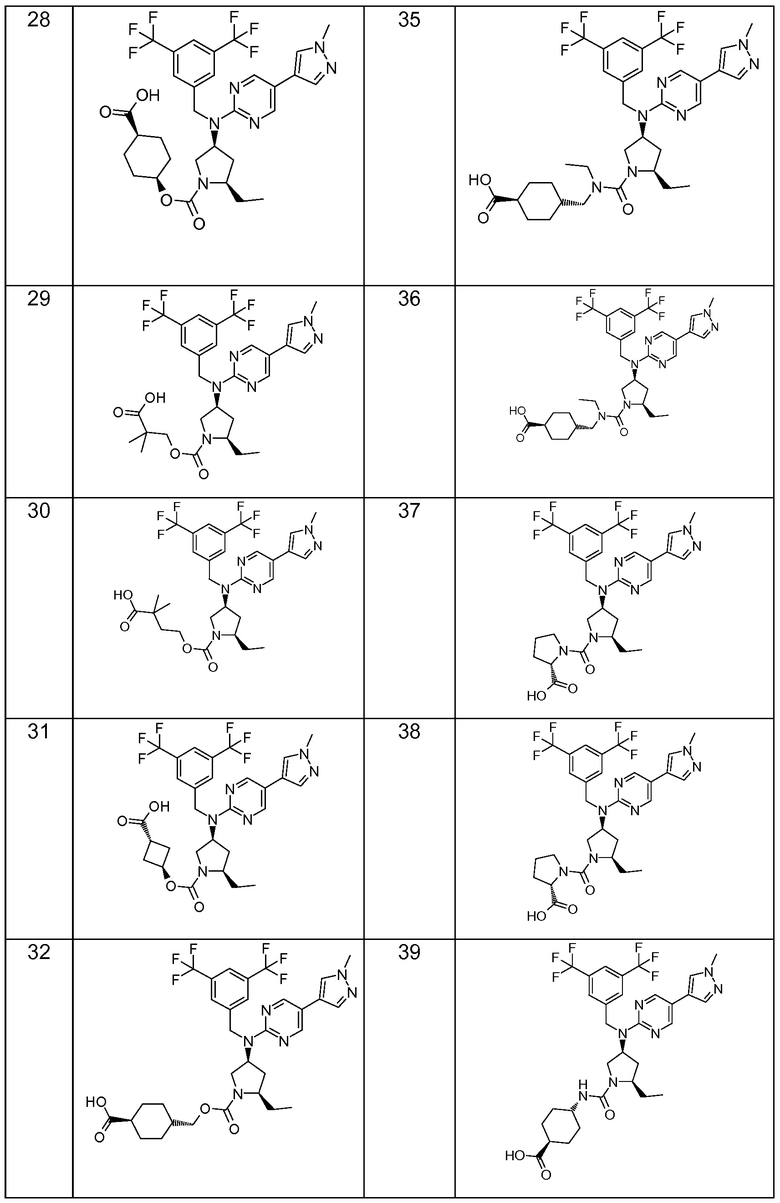
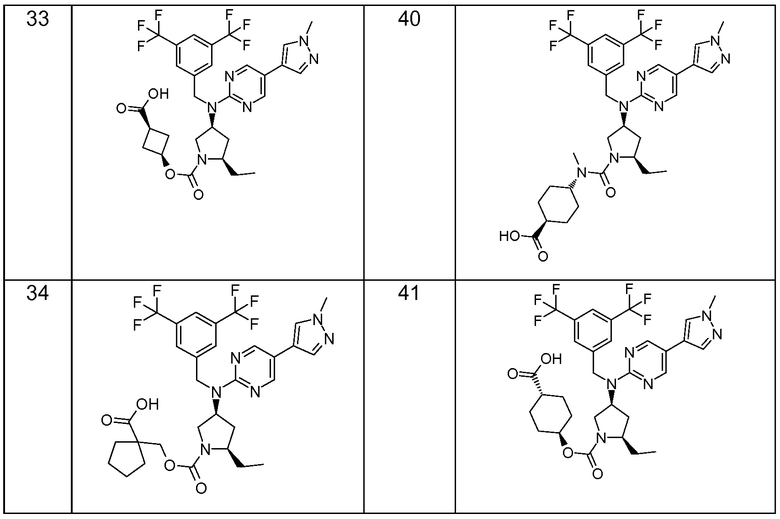
Примеры 42-46 являются соединениями, раскрытыми в US 2009/0075968 (WO 2007/073934):
Пример 42:
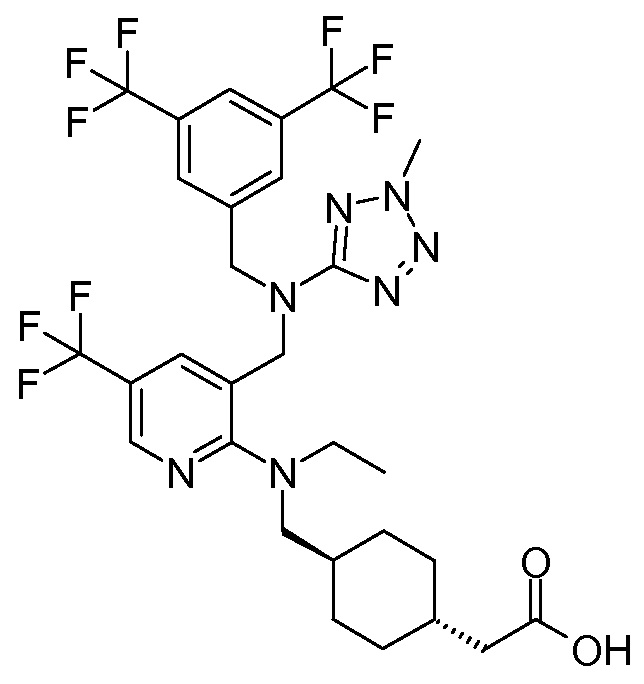
Пример 43:
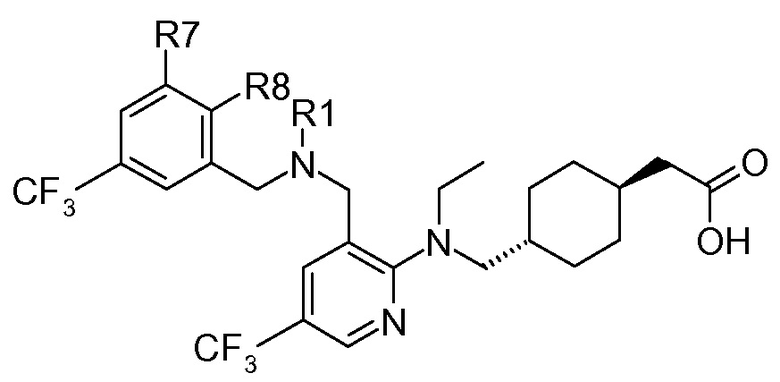
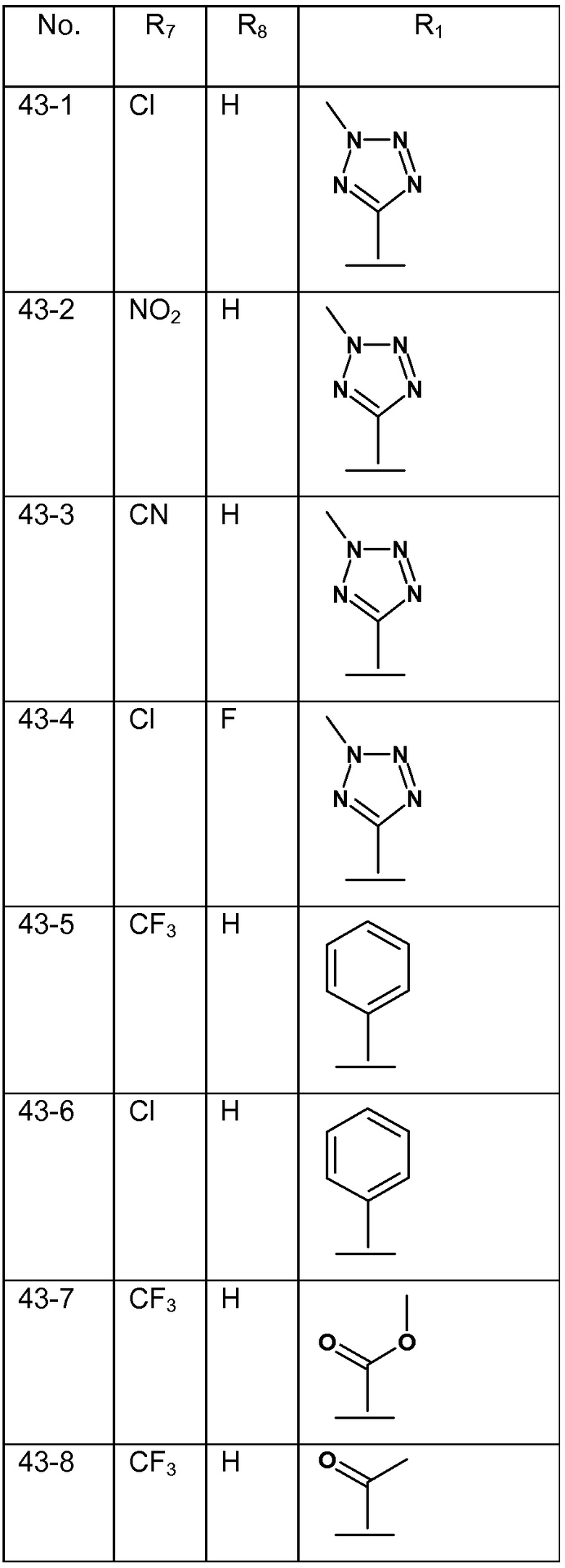
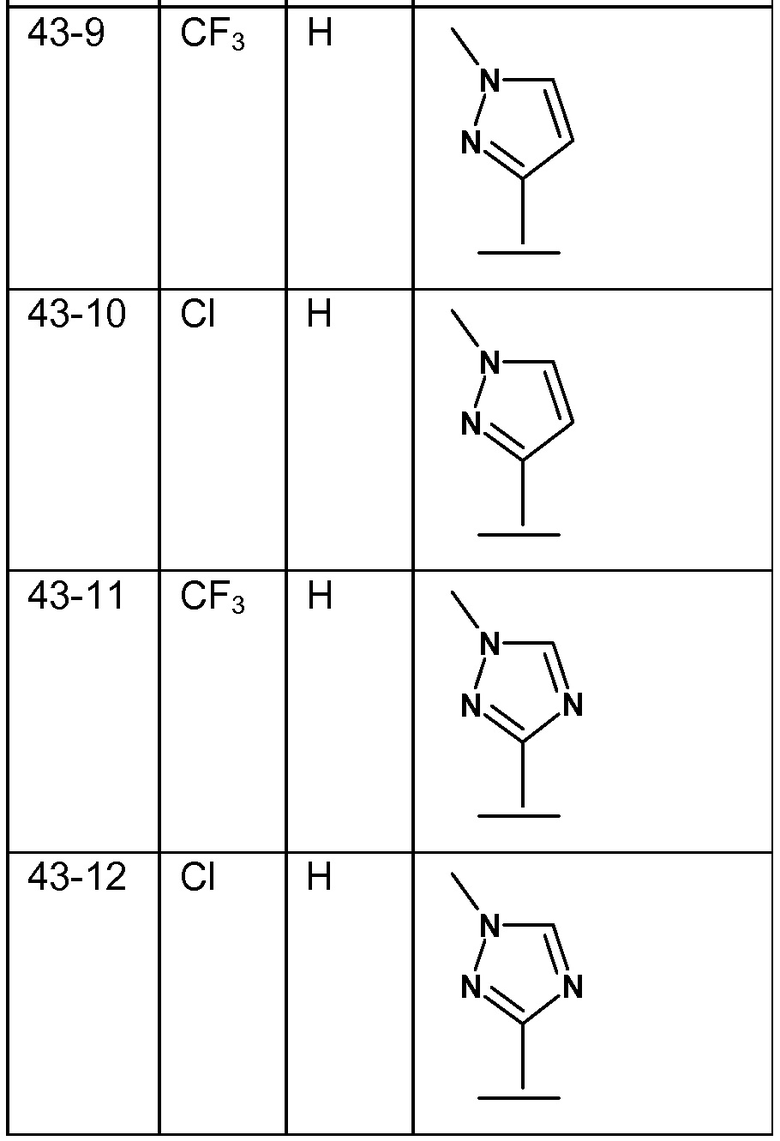
Пример 44:
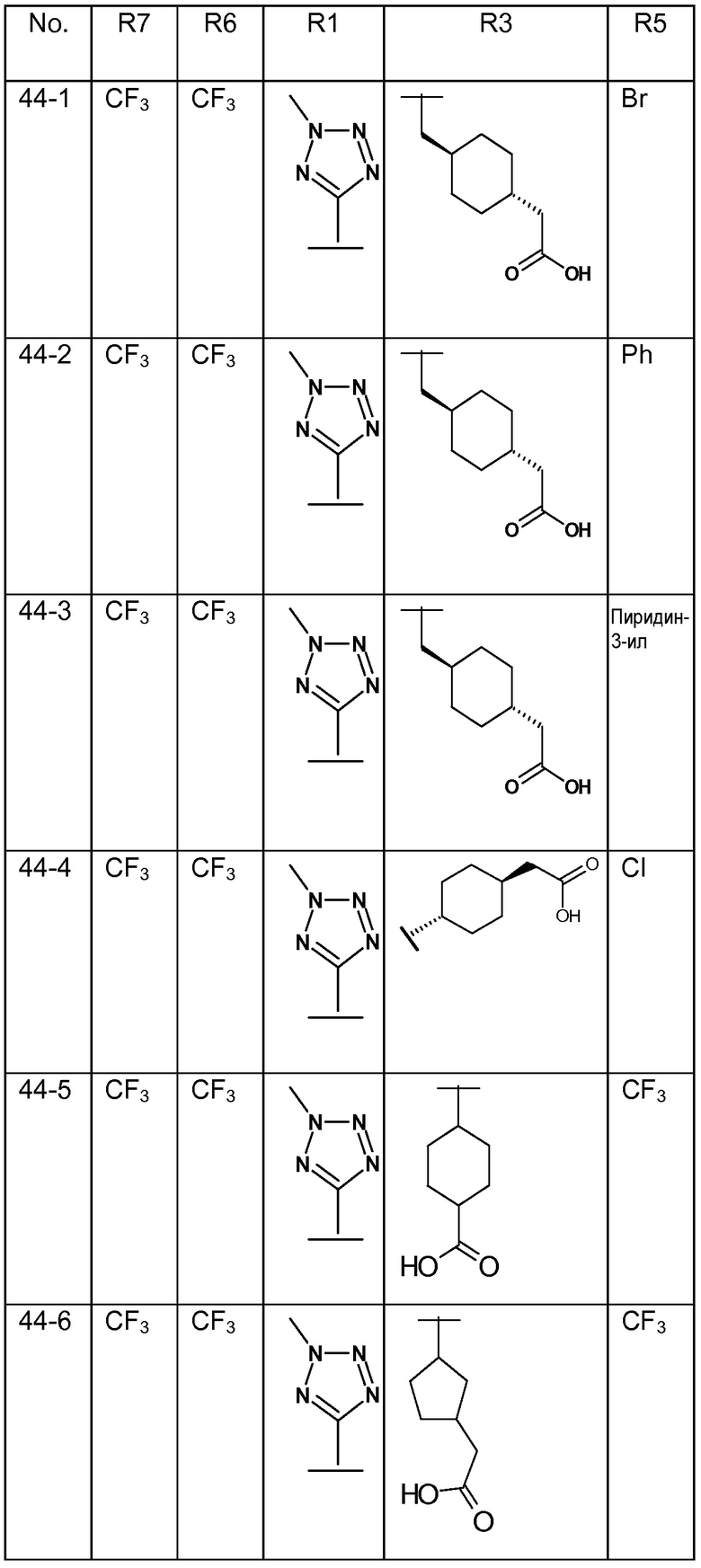
Пример 45:
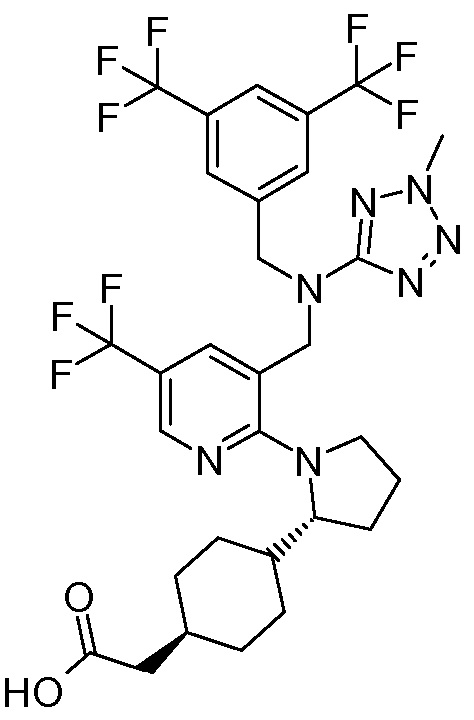
Пример 46:
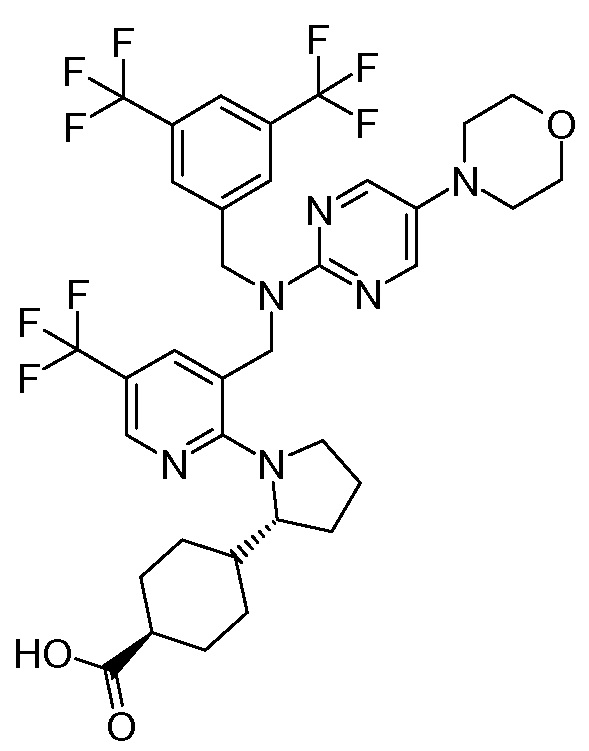
Примеры 47-52 являются соединениями, раскрытыми в US 2009/0227580 (WO 2007/128568)
Пример 47:
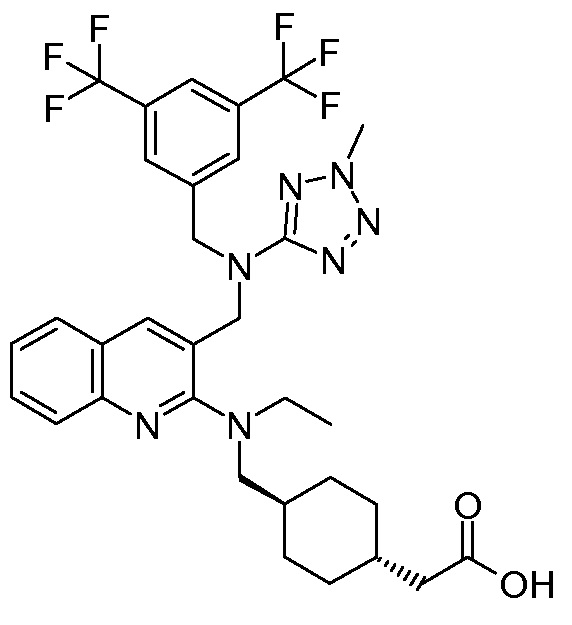
Пример 48:
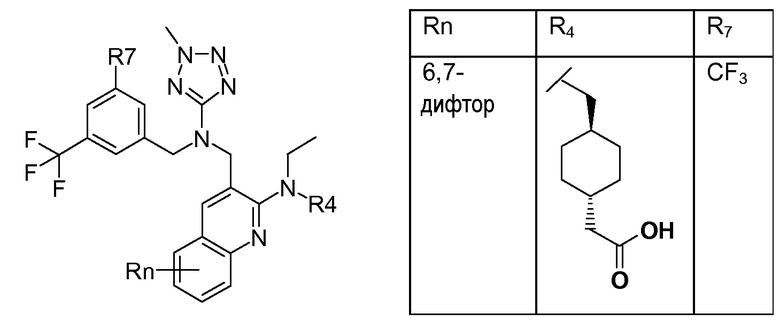
Пример 49:
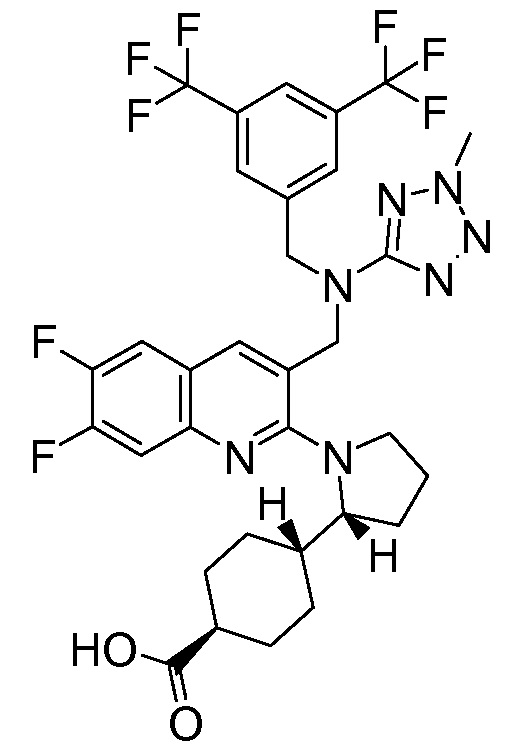
Пример 50:
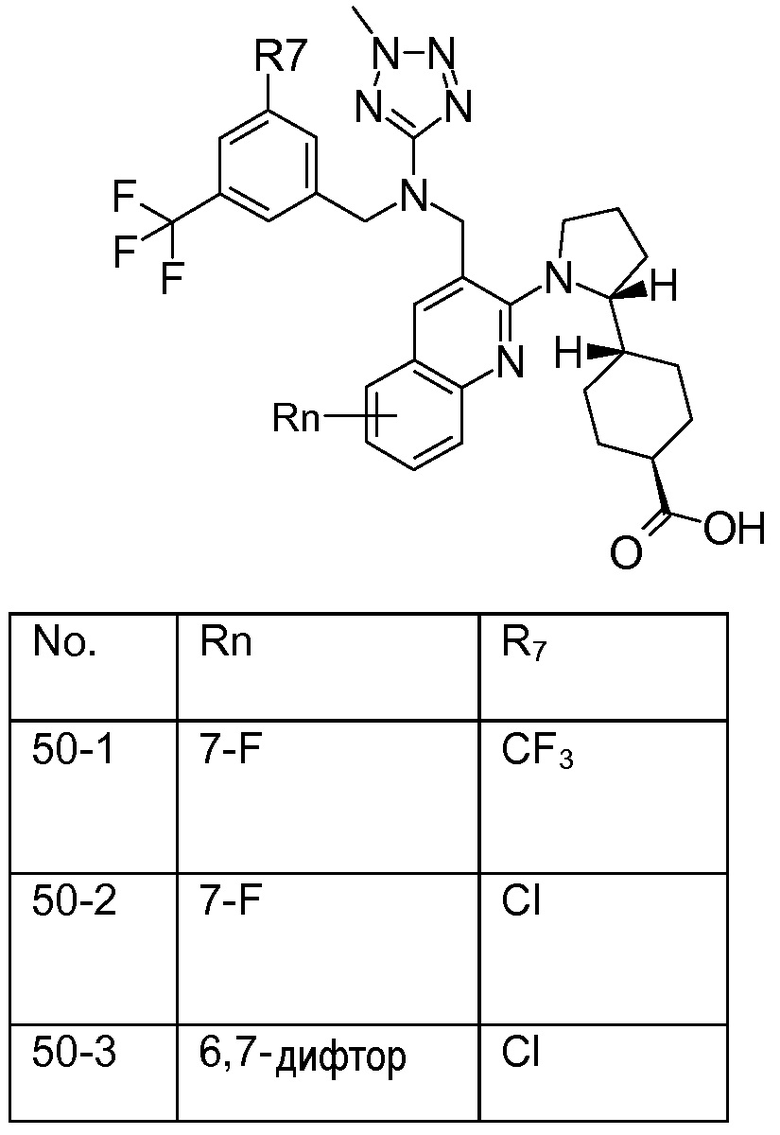
Пример 51:
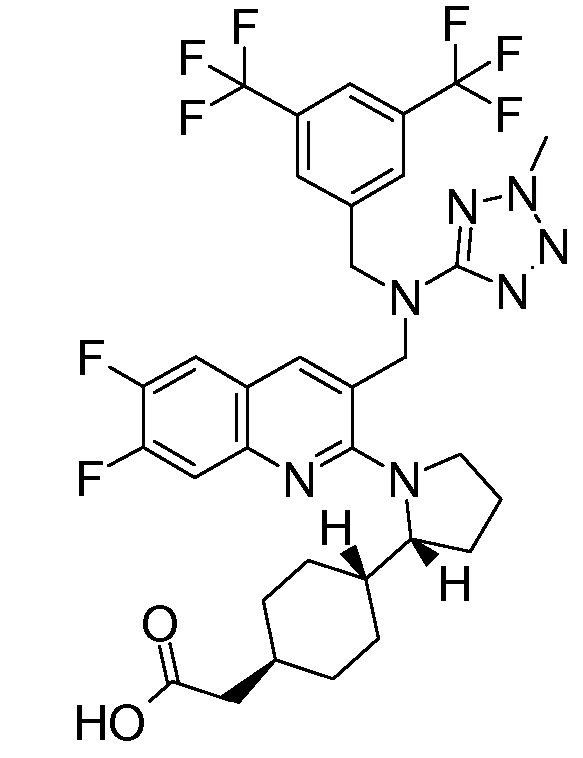
Пример 52:
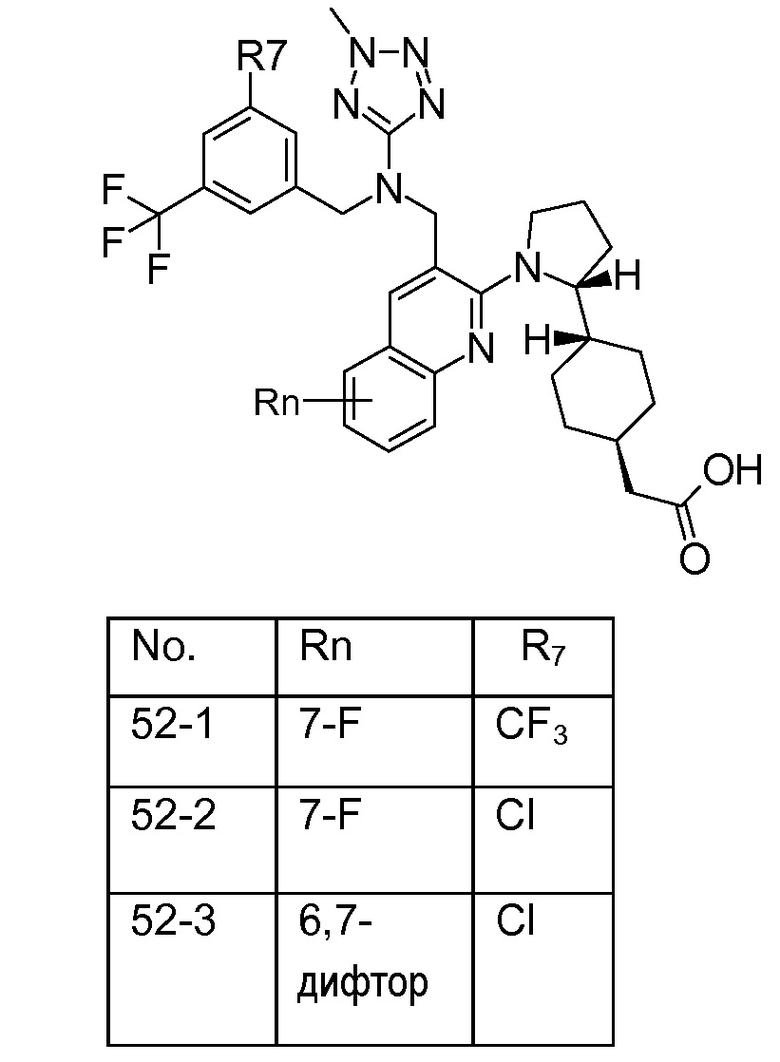
Пример 53: раскрыт в WO 2006/002342 (US 2008/269284)
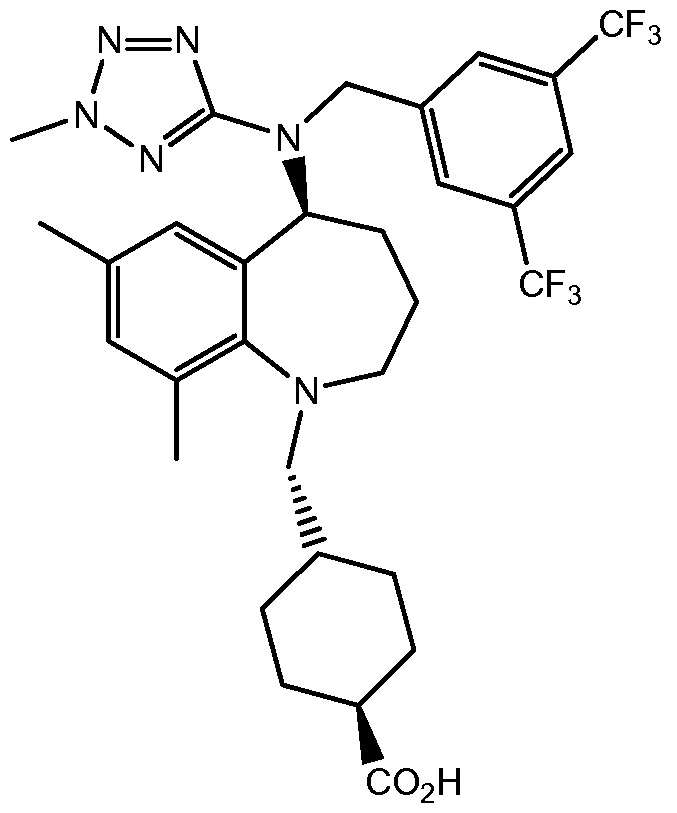
Пример 54: раскрыт в WO 2007/081569 (US 2009/042892)
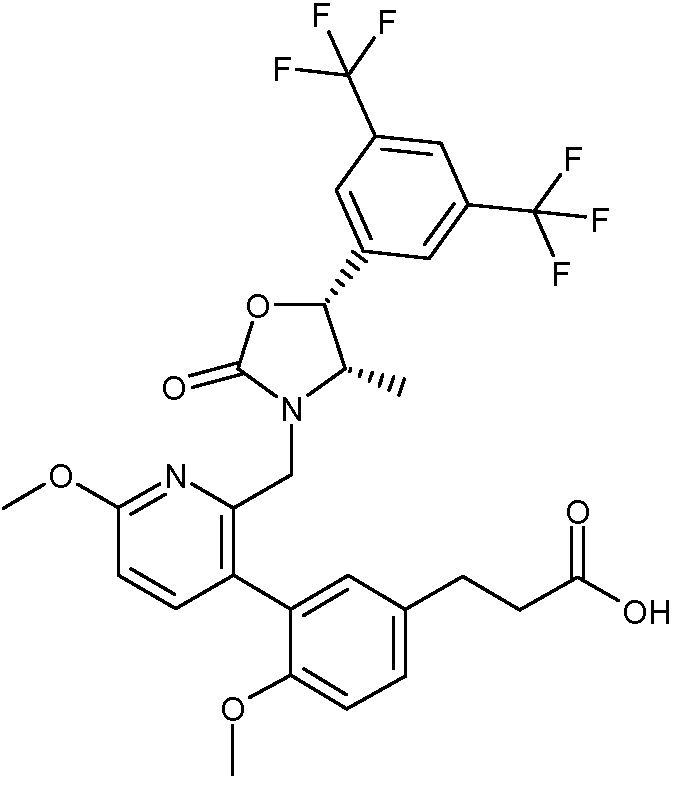
Пример 55: раскрыт в WO 2007/081571 (US 2009/075979) - Пример 64
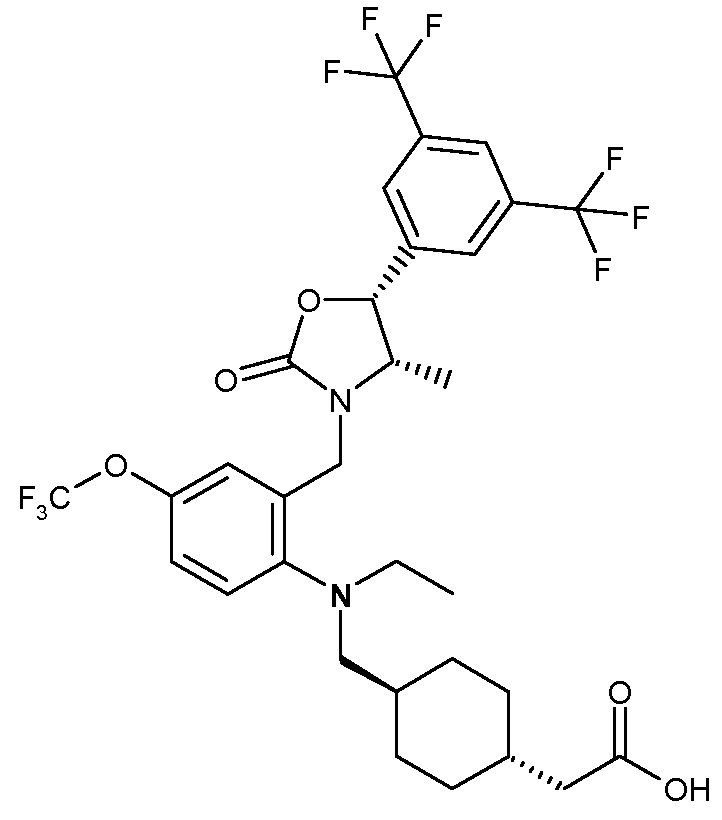
Пример 56: раскрыт в US 2009/082352 - Пример 45
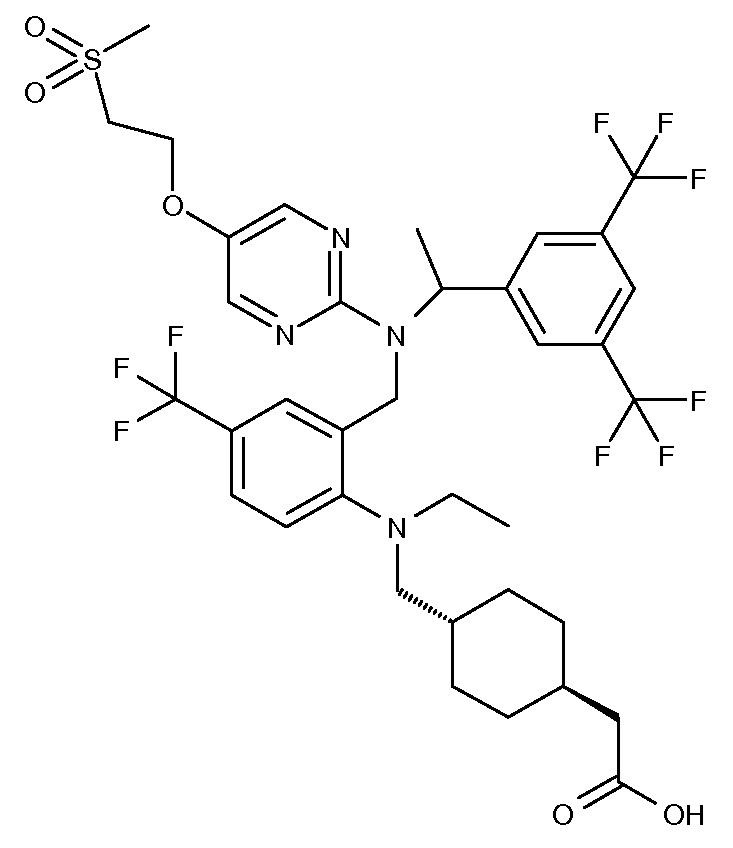
Пример 57:
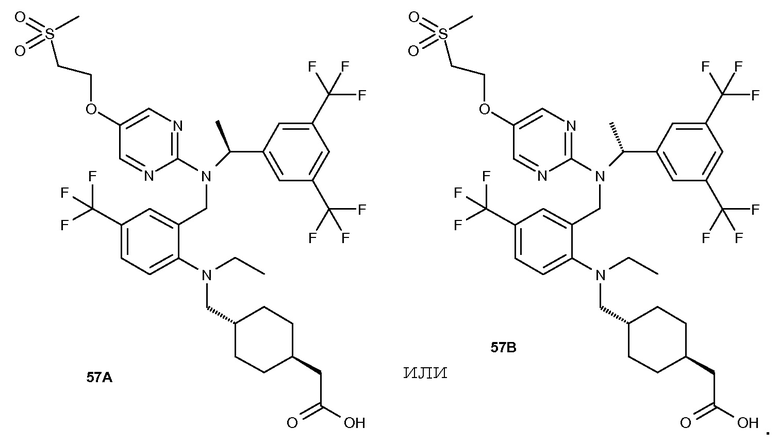
Пример 58: раскрыт в WO 2004/020393:
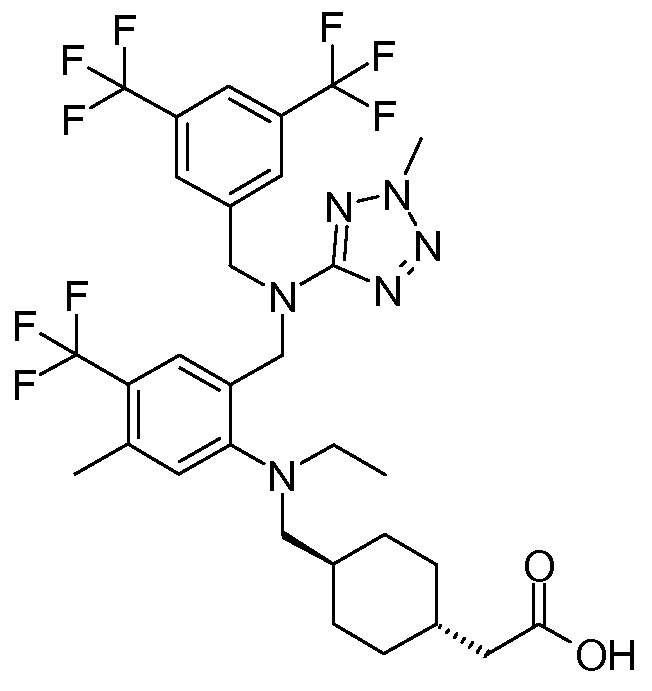
Можно заметить, что соединения по изобретению могут применяться в качестве ингибиторов CETP и в лечении заболеваний и состояний, связанных с активностью CETP, таких как заболевания, раскрытые в настоящей заявке, у субъекта с высоким уровнем триглицеридов.
Следует понимать, что изобретение было описано лишь в качестве примера, и модификации могут быть сделаны без выхода за рамки объема и сущности изобретения.
Настоящая группа изобретений относится к медицине, а именно к терапии, и касается лечения атеросклероза и гипертриглицеридемии. Для этого вводят соединение формулы
 или формулы
или формулы 
в эффективном количестве. Это обеспечивает лечение, уменьшение интенсивности и/или предотвращение таких заболеваний и патологических состояний, как атеросклероз и дислипидемия, у субъекта с высокими уровнями триглицеридов. 2 н. и 10 з.п. ф-лы, 2 табл., 58 пр.
1. Способ лечения, предотвращения или уменьшения интенсивности атеросклероза или дислипидемии у субъекта с высоким уровнем триглицеридов, включающий введение субъекту терапевтически эффективного количества соединения формулы
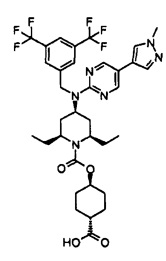 или соединения формулы
или соединения формулы 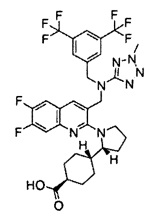
2. Способ по п. 1, включающий введение субъекту указанных соединений в комбинации с по меньшей мере одним другим терапевтическим средством.
3. Способ по п. 1 или 2, где уровень триглицеридов у субъекта с высоким уровнем триглицеридов является уровнем триглицеридов натощак >200 мг/дл.
4. Способ по п. 1 или 2, где уровень триглицеридов у субъекта с высоким уровнем триглицеридов является уровнем триглицеридов натощак >500 мг/дл.
5. Способ по п. 1 или 2, для повышения уровня ЛВП-С до 100%.
6. Способ по п. 1 или 2, где указанные соединения обладают способностью сохранять ≥50% ингибирования СЕТР-ингибирующей активности у субъектов с высокими уровнями триглицеридов в плазме по сравнению с субъектом, имеющим нормальные уровни триглицеридов в плазме.
7. Способ лечения атеросклероза или дислипидемии, включающий:
a. Отбор субъекта с высоким уровнем триглицеридов; и
b. Введение указанному субъекту терапевтически эффективного количества соединения формулы
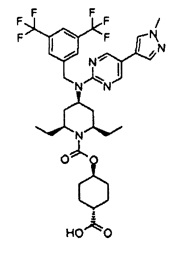 или соединения формулы
или соединения формулы 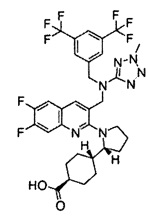
8. Способ по п. 7, включающий введение субъекту указанных соединений в комбинации с по меньшей мере одним другим терапевтическим средством.
9. Способ по п. 7 или 8, где уровень триглицеридов у субъекта с высоким уровнем триглицеридов является уровнем триглицеридов натощак >200 мг/дл.
10. Способ по п. 7 или 8, где уровень триглицеридов у субъекта с высоким уровнем триглицеридов является уровнем триглицеридов натощак >500 мг/дл.
11. Способ по п. 7 или 8 для повышения уровня ЛВП-С до 100%.
12. Способ по п. 7 или 8, где указанные соединения обладают способностью сохранять ≥50% ингибирования СЕТР-ингибирующей активности у субъектов с высокими уровнями триглицеридов в плазме по сравнению с субъектом, имеющим нормальные уровни триглицеридов в плазме.
| ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИНГИБИТОРОВ CETP И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2265010C2 |
| RU2008136198A 20.03.2010 | |||
| WO 2007128568 A1 15.11.2007 | |||
| WO2008009436 A1 24.01.2008 | |||
| БАБАК О.Я | |||
| и др | |||
| "Эффективность статинов в зависимости от плиморфизма гена СЕТР" | |||
| Украинский терапевтический журнал, 2010. | |||

