ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННУЮ ЗАЯВКУ
Настоящая заявка испрашивает приоритет на основании предварительной заявки на патент США №61/566879, поданной 5 декабря 2011 года. Полное содержание указанной заявки включено в настоящее описание посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к способам и составам для доставки силденафила и его производных в кровеносную систему путем введения с помощью состава в форме спрея для перорального введения для лечения таких заболеваний, как легочная артериальная гипертензия и индуцированная селективными ингибиторами обратного захвата серотонина (СИОЗС-индуцированная) сексуальная дисфункция.
УРОВЕНЬ ТЕХНИКИ
Федеральным управлением по контролю качества пищевых продуктов и лекарственных средств США (FDA) были одобрены таблетки силденафила цитрата для лечения легочной артериальной гипертензии (функциональная группа I по классификации ВОЗ) под торговым названием Ревацио (REVATIO®). Рекомендуемая доза препарата Ревацио составляет 20 мг три раза в сутки.
Силденафила цитрат был впервые одобрен FDA для лечения эректильной дисфункции у мужчин под торговым названием Виагра (VIAGRA®).
Силденафил, как сообщают, является селективным ингибитором цГМФ-специфической фосфодиэстеразы типа 5 (ФДЭ-5). Его действие при лечении легочной артериальной гипертензии (и эректильной дисфункции) осуществляется за счет усиления последующих эффектов снижения тонуса сосудов, опосредованного оксидом азота (NO). ФДЭ-5 обнаружена в гладкой мускулатуре легочных сосудов и пещеристом теле, а также в тканях, таких как гладкая мускулатура сосудов и висцеральные гладкие мышцы, а также в тромбоцитах. Силденафил повышает уровень циклического гуанозинмонофосфата (цГМФ) за счет ингибирования ФДЭ-5. ФДЭ-5 ответственна за расщепление цГМФ. В результате силденафил повышает уровень цГМФ в гладкомышечных клетках легочных сосудов. У пациентов, страдающих легочной гипертензией, это может привести к расширению легочного сосудистого ложа и, в меньшей степени, к сосудорасширяющему эффекту в системном кровотоке (см., например, информацию по препарату Ревацио).
В дополнение к терапевтическому эффекту при таких заболеваниях, как легочная артериальная гипертензия (ЛАГ) и эректильная дисфункция, силденафил, как сообщают, эффективен для лечения сексуальных дисфункций, ассоциированных с приемом СИОЗС. См., например, Stimmel, GL, "Sexual dysfunction and psychotropic medications" CNS Spectr. (2006) 11: 8 (Suppl 9): 24-30 и Wang, W-F et al. "Selective serotonin reuptake inhibitors in the treatment of premature ejaculation" Chin Med J (2007) 120(11): 1000-1006. Сексуальные дисфункции представляют собой основной побочный эффект СИОЗС и включают снижение либидо, возбуждения, задержку или отсутствие оргазма (например, аноргазмию у женщин) и задержку эякуляции, анэякуляцию или эректильную дисфункцию у мужчин. Там же. У пациентов, страдающих сексуальной дисфункцией в результате лечения СИОЗС, введение силденафила в качестве вспомогательной терапии может, например, улучшить и поддерживать возбуждение у пациента благодаря усилению кровотока, и силденафил, как полагают, оказывает косвенное благоприятное воздействие на другие аспекты сексуальной реакции посредством того же механизма. См., например, Stimmel, GL. CNS Spectr. (2006) 11: 8(Suppl 9): 24-30.
Силденафил был также предложен в качестве потенциального лекарственного средства для лечения ряда других состояний. Эти состояния включают сексуальную дисфункцию у женщин, включая ассоциированную с менопаузой сексуальную дисфункцию, такую как снижение сексуального возбуждения. См., например, Mattar, CN et al. "Care of women in menopause: sexual function, dysfunction and therapeutic modalities" Ann Acad Med Singapore (2008) 37: 215-223. Другие состояния, при которых силденафил может обеспечить терапевтический эффект, включают высотную болезнь (см., например, Luks, AM et al CHEST (2008) 133: 744-755), боль, инсульт, рассеянный склероз и синдром раздраженного кишечника (см., например, Sharma, R Indian J Med Sci (2007) 61: 667-679 и Uthayathas, S et al. Pharmacological Reports (2007) 59: 150-163).
Несмотря на эффективность силденафила при лечении таких заболеваний, как ЛАГ и эректильная дисфункция, пероральное введение его твердых лекарственных форм может также вызывать нежелательные побочные эффекты. При высоких дозах частота возникновения таких побочных эффектов возрастает - например, искажения зрительного восприятия (от синего или зеленого ореола до затуманенного зрения), диспепсии, заложенности носа, мигреней, приливов, покраснения, диареи, головокружения, сыпи и инфекции мочевыводящих путей. В некоторых случаях могут возникать другие более серьезные побочные эффекты в результате физиологической предрасположенности, неблагоприятного взаимодействия или усиления действия лекарственного средства или злоупотребления лекарственным средством. Такие побочные эффекты включают обморок (потерю сознания), приапизм (эрекцию продолжительностью 4 или более часов) и повышенный риск сердечно-сосудистых осложнений (коитальный инфаркт). В частности, гипотонический криз может возникать в результате сочетания силденафила цитрата и органических нитратов, в некоторых случаях вызывая смерть. Следовательно, введение его пациентам, одновременно принимающим органические нитраты (такие как нитроглицерин) в любой форме, противопоказано. Кроме того, долгосрочные эффекты приема больших доз силденафилсодержащих лекарственных средств неизвестны (Handy В., "The Viagra® Craze," Time, pp.50-57 (May 4, 1998)).
Многие лекарственные средства демонстрируют низкую биодоступность по причине экстенсивного пресистемного метаболизма. Различия в биодоступности могут иметь важное клиническое значение. Например, силденафила цитрат обладает лишь 40% биодоступностью после перорального введения в форме таблеток, в которых активный метаболит составляет примерно половину.
По сравнению с пероральным введением твердых лекарственных форм, всасываемых в желудочно-кишечном тракте, введение через полость рта обеспечивает возможность более быстрой и эффективной доставки лекарственного средства благодаря ее обильному кровоснабжению. Полость рта характеризуется минимальными барьерными свойствами с точки зрения транспорта лекарственных средств, что может приводить к быстрому возрастанию концентрации лекарственных средств в сыворотке крови.
Однако приготовление лекарственных форм для введения через слизистую оболочку полости рта часто ставит нетривиальные задачи. Для обеспечения существенного всасывания лекарственного средства через слизистую оболочку полости рта требуется пролонгированное воздействие лекарственного средства на поверхность слизистой оболочки, и состав должен быть как химически стабильным, так и фармацевтически приемлемым для пациентов.
В WO 01/35926 предложены лекарственные формы силденафила для введения посредством пути, отличного от перорального, такого как назальная доставка. Однако в настоящее время не существует коммерчески доступной лекарственной формы для пероральной доставки силденафила путем распыления.
Таким образом, существует насущная потребность и интерес в отношении компактных и удобных лекарственных форм силденафила для лечения таких заболеваний, как легочная артериальная гипертензия и СИОЗС-индуцированная сексуальная дисфункция, при этом указанные лекарственные формы должны быть как химически стабильными, так и фармацевтически приемлемыми.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам и составам для лечения таких заболеваний, как легочная артериальная гипертензия и/или СИОЗС-индуцированная сексуальная дисфункция. В частности, настоящее изобретение относится к пероральному введению силденафила или силденафила цитрата.
Изобретение, описанное в настоящем документе, относится к составу в форме спрея для перорального введения для доставки силденафила с целью лечения таких заболеваний, как легочная артериальная гипертензии и/или СИОЗС-индуцированная сексуальная дисфункция. Состав на основе силденафила в форме спрея для перорального введения может представлять собой прозрачный бесцветный или светло-желтый раствор, предназначенный для распыления непосредственно в полость рта, области над или под языком.
В одном из аспектов состав в форме спрея для перорального введения может содержать силденафил или его фармацевтически приемлемую соль. В другом аспекте состав в форме спрея для перорального введения может содержать силденафила цитрат.
В другом аспекте состав в форме спрея для перорального введения, содержащий силденафил или его фармацевтически приемлемую соль, может иметь рН от примерно 1,5 до менее чем 3,0. В некоторых вариантах реализации рН состава составляет примерно 2,2±0,5. В конкретном варианте реализации рН состава составляет примерно 2,2.
В другом аспекте состав в форме спрея для перорального введения может содержать полярный растворитель. Полярный растворитель может содержать пропиленгликоль и этиловый спирт. В некоторых вариантах реализации соотношение пропиленгликоль:этиловый спирт составляет примерно 62,5:37,5% об./об.
В одном из аспектов указанный полярный растворитель может содержать один или. более регуляторов рН. Регуляторы рН могут представлять собой подкисляющие агенты, подщелачивающие агенты или их комбинацию. В одном из аспектов подкисляющий агент может представлять собой соляную кислоту (HCl). В конкретном варианте реализации HCl присутствует в количестве примерно 10% об./об. из расчета на общий объем состава. В другом аспекте подщелачивающий агент может представлять собой гидроксид натрия (NaOH). В конкретном варианте реализации NaOH присутствует в количестве примерно 2,1% об./об. из расчета на общий объем состава.
В одном из аспектов состав в форме спрея для перорального введения, содержащий силденафил или его фармацевтически приемлемую соль, может дополнительно содержать агент, маскирующий вкус. В некоторых вариантах реализации агент, маскирующий вкус, содержит мяту, В некоторых вариантах реализации мята представляет собой мяту перечную или мяту колосистую. В других вариантах реализации состав в форме спрея для перорального введения дополнительно содержит фруктовый и/или шоколадный ароматизатор. В некоторых вариантах реализации состав в форме спрея для перорального введения содержит подсластитель. В конкретном варианте реализации подсластитель представляет собой сукралозу.
В другом аспекте состав в форме спрея для перорального введения может дополнительно содержать одно или более фармацевтически приемлемых вспомогательных веществ, носителей или их комбинацию.
Предложенное в настоящей заявке изобретение также относится к способу лечения заболевания, такого как ЛАГ или СИОЗС-индуцированная сексуальная дисфункция, который может включать введение пациенту, нуждающемуся в этом, состава на основе силденафила в форме спрея для перорального введения. В некоторых вариантах реализации вводимое пациенту количество силденафила обеспечивает среднее геометрическое значение логарифмически преобразованной и приведенной к дозе 25 мг AUC0-t (нг⋅ч/мл), составляющее приблизительно 299,36. В других вариантах реализации вводимое пациенту количество силденафила обеспечивает среднее геометрическое значение логарифмически преобразованной и приведенной к дозе 25 мг AUC0-t (нг⋅ч/мл), составляющее приблизительно 323,16. В других вариантах реализации вводимое пациенту количество силденафила обеспечивает среднее геометрическое значение логарифмически преобразованной и приведенной к дозе 25 мг AUC0-t (нг⋅ч/мл), составляющее приблизительно 304,98. В некоторых вариантах реализации вводимое пациенту количество силденафила обеспечивает среднее геометрическое значение логарифмически преобразованной и приведенной к дозе 25 мг AUC0-∞ (нг⋅ч/мл), составляющее приблизительно 310,60. В других вариантах реализации вводимое пациенту количество силденафила обеспечивает среднее геометрическое значение логарифмически преобразованной и приведенной к дозе 25 мг AUC0-∞ (нг⋅ч/мл), составляющее приблизительно 331,22. В других вариантах реализации вводимое пациенту количество силденафила. обеспечивает среднее геометрическое значение логарифмически преобразованной и приведенной к дозе 25 мг AUC0-∞ (нг⋅ч/мл), составляющее приблизительно 311,05.
В некоторых вариантах реализации силденафил присутствует в составе в форме спрея для перорального введения согласно изобретению в количестве от примерно 7 до примерно 9% масс./об. из расчета на общий объем состава. В некоторых вариантах реализации он присутствует в количестве примерно 8,31% масс./об. из расчета на общий объем состава. В других вариантах реализации соль силденафила присутствует в количестве от примерно 10 до примерно 12% масс./об. из расчета на общий объем состава. В конкретном варианте реализации соль силденафила присутствует в количестве примерно 11,67% масс./об. из расчета на общий объем состава.
В некоторых вариантах реализации количество силденафила в составе в форме спрея для перорального введения согласно изобретению, вводимое в одной дозе спрея, составляет 10 мг. В других вариантах реализации количество силденафила, вводимое в одной дозе спрея, составляет 20 мг. В других вариантах реализации количество силденафила, вводимое в одной дозе спрея, составляет 30 мг.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фигура 1 (фигура 7.2.6.1-1) представляет собой график, отображающий не приведенную к дозе среднюю концентрацию силденафила в плазме (0-24 часа), N=24.
Фигура 2 (фигура 7.2.6.2-1) представляет собой график, отображающий приведенную к дозе среднюю концентрацию силденафила в плазме (0-24 часа), N=24.
Фигура 3 (фигура 7.2.6.3-1) представляет собой график, отображающий не приведенную к дозе среднюю концентрацию Ν-десметилсилденафила в плазме (0-24 часа), N=24.
Фигура 4 (фигура 7.2.6.4-1) представляет собой график, отображающий приведенную к дозе среднюю концентрацию Ν-десметилсилденафила в плазме (0-24 часа), N=24.
Фигура 5 (фигура 7.2.6.6-1) представляет собой график, отображающий отношение средних концентраций Ν-десметилсилденафил/силденафил в плазме (0-4 часа), N=24.
ПОДРОБНОЕ ОПИСАНИЕ
Силденафил имеет следующее химическое название: 1-[[3-(6,7-дигидро-1-метил-7-оксо-3-пропил-1Н-пиразол[4,3-d]пиримидин-5-ил)-4-этоксифенил]сульфонил]-4-метилпиперазин.
Предложенный в настоящей заявке состав в форме спрея для перорального введения. содержит силденафил или его фармацевтически приемлемую соль. Термин фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных кислот, включая органические и неорганические кислоты. Такие кислоты включают уксусную, бензолсульфокислоту, бензойную, камфорсульфоновую, лимонную, этансульфокислоту, фумаровую, глюконовую, глутаминовую, бромоводородную, соляную, изэтионовую, молочную, малеиновую, миндальную, метансульфокислоту, слизевую, азотную, памоевую, пантотеновую, фосфорную, янтарную, серную, винную кислоты и п-толуолсульфокислоту.
Ν-десметилсилденафил является одним из известных активных метаболитов силденафила.
Силденафил, вводимый в виде коммерчески доступного состава Ревацио для лечения легочной артериальной гипертензии (функциональная группа I по классификации ВОЗ) с целью улучшения физических возможностей и замедления усугубления клинических симптомов, всасывается в желудочно-кишечном тракте после перорального введения с абсолютной биодоступностью, составляющей примерно 40%. Как следует из предоставленной производителем информации по препарату Ревацио, максимальные наблюдаемые концентрации в плазме достигаются за период времени от 30 до 120 минут (в среднем 60 минут) при пероральном введении натощак. В случае, когда препарат Ревацио принимают вместе с пищей с высоким содержанием жира, скорость всасывания уменьшается, при этом наблюдают увеличение времени достижения максимальной концентрации Tmax в среднем на 60 минут и снижение максимальной концентрации Cmax в плазме в среднем на 29%. Средний объем распределения в равновесном состоянии (Vss) для силденафила, по имеющимся данным, составляет 105 л, что указывает на распределение лекарственного средства по тканям.
Предложенный в настоящей заявке состав в форме спрея для перорального введения приготовлен для доставки через полость рта. Доставка состава на основе силденафила в форме спрея для перорального введения может быть осуществлена посредством трансмукозального пути, сублингвального пути, через полость рта, через мембраны слизистой оболочки и/или через желудочно-кишечный тракт.
Состав в форме спрея для перорального введения согласно настоящей заявке подходит для применения при лечении одного или более заболеваний, в случае которых введение силденафила считают эффективным. Примеры таких заболеваний включают ЛАГ, СИОЗС-индуцированную сексуальную дисфункцию и эректильную дисфункцию. В некоторых вариантах реализации состав на основе силденафила в форме спрея для перорального введения согласно настоящей заявке подходит для применения при лечении ВИЧ-ассоциированной ЛАГ; ассоциированной с гемолитическими заболеваниями ЛАГ; портопульмональной гипертензии; ЛАГ у детей (например, идиопатической ЛАГ, ЛАГ после хирургического устранения врожденного порока сердца и синдрома Эйзенменгера); легочной гипертензии, ассоциированной с сердечной недостаточностью, хирургическими вмешательствами на сердце и пересадкой сердца; тромбоэмболии легочной артерии; ассоциированной с фиброзом легких легочной гипертензии и/или высотной легочной гипертензии. См., например, Barnett, CF et al. Vascular Health and Risk Management (2006) 2(4): 411422. В других вариантах реализации состав на основе силденафила в форме спрея для перорального введения согласно настоящей заявке подходит для применения при лечении сексуальной дисфункции у женщин, включая ассоциированную с менопаузой сексуальную дисфункцию, такую как снижение сексуального возбуждения. В других вариантах реализации состав на основе силденафила в форме спрея для перорального введения согласно настоящей заявке подходит для применения при лечении высотной болезни, боли, инсульта, рассеянного склероза и/или синдрома раздраженного кишечника.
Предложенный в настоящей заявке состав в форме спрея для перорального введения может содержать силденафил в форме его свободного основания или его фармацевтически приемлемую соль в количестве от примерно 6 до примерно 14% масс./об., от примерно 7 до примерно 13% масс./об. или от примерно 8 до примерно 12% масс./об. из расчета на общий объем состава. В одном из аспектов состав в форме спрея для перорального введения может содержать силденафил или его фармацевтически приемлемую соль в количестве от примерно 8 до примерно 10% масс./об. из расчета на общий объем состава.
В другом аспекте предложенный в настоящем изобретении состав в форме спрея для перорального введения может содержать силденафил в форме его свободного основания в количестве от примерно 6 до примерно 10% масс./об., от примерно 7 до примерно 9% масс./об. или примерно 8% масс./об. из расчета на общий объем состава. В другом аспекте состав в форме спрея для перорального введения может содержать силденафил в форме его свободного основания в количестве примерно 8,31% масс./об. из расчета на общий объем состава.
В одном из аспектов предложенный в настоящем изобретении состав в форме спрея для перорального введения может содержать силденафила цитрат в количестве от примерно 10 до примерно 14% масс./об., от примерно 11 до примерно 13% масс./об. или примерно 12% масс./об. из расчета на общий объем состава. В другом аспекте состав в форме спрея для перорального введения может содержать силденафила цитрат в количестве примерно 11,67% масс./об. из расчета на общий объем состава.
Предложенный в настоящем изобретении состав в форме спрея для перорального введения может обеспечить доставку силденафила в форме его свободного основания или его фармацевтически приемлемой соли в количестве (на одну дозу спрея) от примерно 3 до примерно 25 мг, от примерно 6 до примерно 20 мг, от примерно 8 до примерно 18 мг, от примерно от 10 до примерно 16 мг или от примерно 12 до примерно 14 мг.
В одном из аспектов состав в форме спрея для перорального введения может обеспечить доставку силденафила в форме его свободного основания в количестве (на одну дозу спрея) от примерно 6 до примерно 14 мг или от примерно 8 до примерно 12 мг. В другом аспекте состав в форме спрея для перорального введения может обеспечить доставку примерно 10 мг на одну дозу спрея.
В другом аспекте для состав в форме спрея для перорального введения может обеспечить доставку силденафила цитрата в количестве (на одну дозу спрея) от примерно 10 до примерно 18 мг или от примерно 12 до примерно 16 мг. В другом аспекте состав в форме спрея для перорального введения может обеспечить доставку примерно 14 мг на одну дозу спрея.
В одном из аспектов пациенту могут быть введены от одной до пяти доз спрея состава в форме спрея для перорального введения. В другом аспекте в организм пациента осуществляют доставку от одной до трех доз спрея состава на основе силденафила. В другом аспекте одна доза спрея может обеспечить доставку от примерно 100 до примерно 120 мкл (от примерно 0,1 до примерно 0,12 мл) состава. В другом аспекте одна доза спрея может обеспечить доставку примерно 120 мкл состава. В некоторых вариантах реализации одна доза спрея обеспечивает доставку от примерно 200 до примерно 250 мкл состава. В некоторых вариантах реализации одна доза спрея обеспечивает доставку примерно 250 мкл состава.
Предложенный в настоящем изобретении состав в форме спрея для перорального введения может иметь рН от примерно 1,5 до менее чем 3,0. В одном из аспектов рН состава в форме спрея для перорального введения может составлять 1,5, 1,6, 1,7, 1,8, 1,9, 2,0, 2,1, 2,2, 2,3, 2,4, 2,5, 2,6, 2,7, 2,8 или 2,9.. В другом аспекте рН состава в форме спрея для перорального введения может составлять примерно 2,2±0,5 или примерно 2,3±0,5.
Полярные растворители, содержащиеся в предложенном в настоящем изобретении составе в форме спрея для перорального введения, включают, но не ограничиваются перечисленными, этиловый спирт, пропиленгликоль, глицерин и полиэтиленгликоли, имеющие номинальную молярную массу 200-600 г/моль, N-метил-2-пирролидон или их комбинации. В одном из аспектов полярный растворитель может содержать пропиленгликоль и дегидратированный спирт (например, этиловый спирт). Пропиленгликоль и дегидратированный спирт могут содержаться в составе в форме спрея для перорального введения в соотношении пропиленгликоль:спирт, составляющем примерно 70:30% об./об., примерно 65:35% об./об., примерно 60:40% об./об., примерно 55:45% об./об. или примерно 50:50% об./об. В другом аспекте соотношение пропиленгликоль:спирт может составлять примерно 62,5:37,5% об./об.
В одном из аспектов пропиленгликоль присутствует в количестве примерно 55% об./об., и этиловый спирт присутствует в количестве примерно 33% об./об. из расчета на конечный объем состава в форме спрея для перорального введения.
Предложенные в настоящем изобретении составы могут содержать один или более регуляторов рН. Регуляторы рН могут включать подкисляющие агенты или подщелачивающие агенты. Подкисляющие агенты согласно настоящему изобретению могут включать, но не ограничиваются перечисленными, соляную кислоту, лимонную кислоту, молочную кислоту, гликолевую кислоту, уксусную кислоту, ледяную уксусную кислоту, яблочную кислоту и пропионовую кислоту. Подщелачивающие агенты согласно настоящему изобретению могут включать, но не ограничиваются перечисленными, гидроксид натрия, тетрагидроксипропилэтилендиамин (эдетол), карбонат калия, гидроксид калия, борат натрия, карбонат натрия, цитрат натрия, лактат натрия и гликолят натрия. В одном из аспектов состав в форме спрея для перорального введения может содержать подкисляющий агент, подщелачивающий агент или их комбинацию. Подкисляющий агент может присутствовать в растворителе в количестве от примерно 5 до примерно 15% об./об. из расчета на общий объем растворителя или примерно 10% об./об. из расчета на общий объем растворителя. Подщелачивающий агент может присутствовать в растворителе в количестве от примерно 1,5 до примерно 4% об./об. из расчета на общий объем растворителя или примерно 2,1% об./об. из расчета на общий объем растворителя.
Cmax, получаемая после введения предложенного в настоящем изобретении состава в форме спрея для перорального введения, может составлять, но без ограничения, от примерно 50 до примерно 150% от Cmax для эталонного препарата. В одном из аспектов Cmax для эталонного препарата может представлять собой Cmax, получаемую после введения таблеток силденафила цитрата. В другом аспекте Cmax, получаемая после введения предложенного в настоящем изобретении состава в форме спрея для перорального введения, может соответствовать разработанным FDA стандартам биоэквивалентности, включая то, что 90% доверительный интервал отношения средних геометрических значений Cmax для исследуемого и эталонного препаратов находится в диапазоне 80-125%.
AUC, получаемая после введения предложенного в настоящем изобретении состава в форме спрея для перорального введения, может составлять, но без ограничения, от примерно 50 до примерно 150% от AUC для эталонного препарата. В одном из аспектов AUC для эталонного препарата может представлять собой AUC, получаемую после введения таблеток силденафила цитрата. В другом аспекте AUC, получаемая после введения предложенного в настоящем изобретении состава в форме спрея для перорального введения, может соответствовать разработанным FDA стандартам биоэквивалентности, включая то, что 90% доверительный интервал отношения средних геометрических значений AUC для исследуемого и эталонного препаратов находится в диапазоне 80-125%.
Tmax, получаемое после введения предложенного в настоящем изобретении состава в форме спрея для перорального введения, может составлять, но без ограничения, от примерно 50 до примерно 150% от Tmax для эталонного препарата. В одном из аспектов Tmax для эталонного препарата может представлять собой Tmax, получаемое после введения таблеток силденафила цитрата. В другом аспекте Tmax, получаемое после введения предложенного в настоящем изобретении состава в форме спрея для перорального введения, может соответствовать разработанным FDA стандартам биоэквивалентности, включая то, что 90% доверительный интервал отношения средних геометрических значений Tmax для исследуемого и эталонного препаратов находится в диапазоне 80-125%. Предложенный в настоящем изобретении состав в форме спрея для перорального введения может дополнительно содержать одно или несколько фармацевтически приемлемых вспомогательных веществ, носителей или их комбинацию.
Вспомогательные вещества могут включать, но не ограничиваются перечисленными, сорастворители и/или солюбилизирующие агенты, способствующие проникновению агенты, стабилизаторы, буферные агенты, регуляторы тоничности, противомикробные агенты и модификаторы вязкости.
Сорастворители могут включать, но не ограничиваются перечисленными, этиловый спирт, пропиленгликоль, глицерин и полиэтиленгликоли, имеющие номинальную молярную массу 200-600 г/моль, и N-метил-2-пирролидон.
Солюбилизирующие агенты могут включать, но не ограничиваются перечисленными, очищенный моноэтиловый простой эфир диэтиленгликоля, циклодекстрины, глицерина моностеарат, лецитин, полоксамер, полиэтиленалкиловые простые эфиры, полиоксиэтиленовые производные касторового масла, полиоксиэтиленсорбитановые сложные эфиры жирных кислот, полиоксиэтиленстеараты, стеариновую кислоту, лимонную кислоту и аскорбиновую кислоту и т.п.; поверхностно-активные агенты, такие как полисорбаты, сложные эфиры сорбитана, поливиниловый спирт, бензалкония хлорид, бензетония хлорид, цетримид, докузат натрия, лаурилсульфат натрия и октоксинол.
Усилители растворимости могут включать, но не ограничиваются перечисленными, DL-метионин, кофеин, никотинамид, ванилин, бензиловый спирт, этанол и Транскутол (Transcutol®) (моноэтиловый простой эфир диэтиленгликоля). В некоторых вариантах реализации состав согласно изобретению содержит кофеин в качестве усилителя растворимости. В других вариантах реализации кофеин присутствует в составе в количестве примерно 25 мг/мл. В некоторых вариантах реализации состав согласно изобретению содержит никотинамид в качестве усилителя растворимости. В других вариантах реализации никотинамид присутствует в составе в количестве примерно 5% масс./масс.
Буферные агенты могут включать, но не ограничиваются перечисленными, соляную кислоту, ацетат натрия, ледяную уксусную кислоту, ортофосфорную кислоту и дигидроортофосфат калия.
Стабилизаторы могут включать, но не ограничиваются перечисленными, метабисульфит натрия, бисульфит натрия, динатриевую соль этилендиаминтетрауксусной кислоты (ЭДТА) и аскорбиновую кислоту.
Противомикробные агенты могут включать, но не ограничиваются перечисленными, бензиловый спирт, бензалкония хлорид, фенилмеркурацетат и фенилэтиловый спирт.
Модификаторы вязкости могут включать, но не ограничиваются перечисленными, гидроксипропилметилцеллюлозу и полиакриловую кислоту или водорастворимые полимеры, такие как карбопол, натрий-карбоксиметилцеллюлозу, гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу и поливинилпирролидоны различных марок (например, K-15, K-30, K-60 и K-90).
В некоторых вариантах реализации может возникнуть необходимость в увеличении времени удержания силденафила, вводимого пациенту в составе предложенного в настоящем изобретении спрея для перорального введения, в слизистой оболочке (например, слизистой оболочке полости рта). Соответственно, в некоторых вариантах реализации состав в форме спрея для перорального введения согласно настоящем изобретении может содержать один или более способствующих проникновению агентов и/или мукоадгезивных агентов. Примеры подходящих способствующих проникновению агентов включают никотинамид, кофеин, масло мяты перечной, гликохолат натрия, фосфолипиды, алкилсахариды, апротинин, бензалкония хлорид, щерамиды, цетилпиридиния хлорид, хитозан, хитоза-4-иобутиламидин, циклодекстрины, декстрансульфат, додецилазациклогептил-2-кетон, липиды с простой эфирной связью (плазмалогены), глицерин, гликозилированные сфингозины, лауриновую кислоту, 23-лауриловый простой эфир, лизофосфатидилхолин, ментол, метоксисалицилат, фосфатидилхолин, 1-пальмитоил-2-глутароил-sn-глицеро-3-фосфохолин, поликарбофил-цистеин, поли-L-аргинин, полиоксиэтилен, полиоксиэтилен-9-лауриловый простой эфир, полисорбат 80, пропиленгликоль, ЭДТА, дезоксихолат натрия, гликохолат натрия, гликодезоксихолат натрия, лаурилсульфат натрия, салицилат натрия, таурохолат натрия, тауродезоксихолат натрия, тауродигидрофусидат натрия, сфинголипиды и стерины. Примеры подходящих мукоадгезивных агентов включают гидроксипропилцеллюлозу, желатин, поперечносшитую полиакриловую кислоту, полиметакриловую кислоту, полигидроксиэтилметакриловую кислоту, гидроксипропилметилцеллюлозу, полиэтиленгликоль, натрий-карбоксиметилцеллюлозу, гиалуроновую кислоту, хитозан, поликарбофил, пектин, ксантановую камедь, альгинат, сополимеры декстрана, полиакриламид, гуммиарабик, сополимер капролактона и этиленоксида, карбопол 934, трагакант и Эудрагит (Eudragit®).
Предложенный в настоящем изобретении состав в форме спрея для перорального введения может содержать агенты, маскирующие вкус, или ароматизаторы. В настоящем описании агенты, маскирующие вкус, или ароматизаторы представляют собой агенты, которые могут скрывать или минимизировать нежелательный привкус, такой как горький или кислый привкус. Примеры агентов, маскирующих вкус, или ароматизаторов, подходящих для применения в составах согласно настоящему изобретению, включают, но не ограничиваются перечисленными, синтетическое или натуральное масло мяты перечной, масло мяты колосистой, цитрусовые масла, фруктовые ароматизаторы (например, цитрусовые, такие как апельсин, лимон, лайм; клубника; вишня; виноград; дыня и их смеси), ароматизатор "шоколад", специи (например, анис, корицу), ваниль, ароматизатор "жевательная резинка" и подсластители (например, сахара, аспартам, сахарин и сукралозу). В некоторых вариантах реализации состав в форме спрея для перорального введения содержит подсластитель, представляющий собой сукралозу. В некоторых вариантах реализации агент, маскирующий вкус, или ароматизатор представляет собой мяту. В некоторых вариантах реализации мята представляет собой мяту перечную. В других вариантах реализации мята представляет собой сильную мяту, например, нефракционированное масло мяты, перечной. В других вариантах реализации состав согласно изобретению содержит агент, маскирующий вкус, или ароматизатор, обеспечивающий вкус "шоколад с мятой".
Агенты, маскирующие вкус, и/или ароматизаторы в составе согласно изобретению могут быть оценены в соответствии со способом определения вкусовых характеристик (см. Keane, P. The Flavor Profile Method. In С. Hootman (Ed.), Manual on Descriptive Analysis Testing for Sensory Evaluation ASTM Manual Series: MNL 13. Baltimore, MD (1992)), например, для идентификации, характеристики и/или количественной оценки органолептических свойств продуктов, например, основных вкусовых ощущений, аромата, консистенции и вкусового впечатления.
В некоторых вариантах реализации состав согласно настоящему изобретению содержит агент, маскирующий вкус, и/или ароматизатор, который достигает интенсивности, аналогичной таковой для обычного мятного спрея для освежения дыхания. В некоторых вариантах реализации буферная сила состава уменьшена (например, за счет снижения количества NaOH) для достижения требуемого сбалансированного вкуса состава (например, сбалансированных основных вкусовых ощущений).
Предложенный в настоящем изобретении состав в форме спрея для перорального введения может быть упакован во флаконы Schott 1 типа из оранжевого стекла, оснащенные нажимными помповыми дозирующими устройствами Pfeiffer на объем 100 или 120 мкл, или в фармацевтически приемлемую упаковку, емкость, упаковку с помповым дозирующим устройством или флакон любого типа.
В одном из аспектов предложенный в настоящем изобретении состав в форме спрея для перорального введения состоит из силденафила цитрата, пропиленгликоля, этилового спирта, соляной кислоты и гидроксида натрия.
В другом аспекте состав в форме спрея для перорального введения состоит из силденафила цитрата, пропиленгликоля, этилового спирта, соляной кислоты, гидроксида натрия и агента, маскирующего вкус.
В другом аспекте состав в форме спрея для перорального введения состоит по существу из силденафила цитрата, пропиленгликоля, этилового спирта, соляной кислоты и гидроксида натрия.
В другом аспекте состав в форме спрея для перорального введения состоит по существу из силденафила цитрата, пропиленгликоля, этилового спирта, соляной кислоты, гидроксида натрия и агента, маскирующего вкус.
Предложенный в настоящем изобретении состав в форме спрея для перорального введения далее будет описан в следующих неограничивающих примерах.
ПРИМЕР 1
Коммерчески доступная цитратная солевая форма силденафила была выбрана в качестве активного фармацевтического ингредиента для разработки состава в форме спрея для перорального введения.
Конечная система растворителей содержала комбинацию 62,5% об./об. пропиленгликоля и 37,5% об./об. смеси спиртов, подкисленной с помощью 0,5 мл разбавленной HCl с доведением рН до 2,0 или ниже с применением 5 н. NaOH. Эта комбинация обеспечивала растворение от 12 до 14% масс./об. силденафила цитрата и сохранения активных фармацевтических ингредиентов (АФИ) в растворе.

Раствор силденафила цитрата (с концентрацией соли 11,67% масс./об. (11,67 мг/мл)) может быть введен пациенту с помощью коммерчески доступного помпового распыляющего устройства, предназначенного для доставки 14 мг соли/120 мкл спрея (или 10 мг свободного основания/120 мкл спрея).
Переносящая среда для состава оставалась стабильной при хранении при 25°С/60% относительной влажности (ОВ) и 40°С/75% ОВ в течение шести месяцев.
ПРИМЕР 2
Субъекты получали силденафила цитрат в лекарственной форме, представляющей собой спрей для перорального введения, в виде части исследования с однократным введением. Одна подгруппа субъектов получала одну дозу спрея состава на основе силденафила в форме спрея для перорального введения, содержащего 14 мг силденафила цитрата на дозу (с обеспечением общего вводимого количества, эквивалентного 10 мг силденафила в форме его свободного основания). Вторая подгруппа субъектов получала две дозы спрея состава на основе силденафила в форме спрея для перорального введения, содержащего 14 мг силденафила цитрата на дозу (с обеспечением общего вводимого количества, эквивалентного 20 мг силденафила в форме его свободного основания). Третья подгруппа субъектов получала три дозы спрея состава на основе силденафила в форме спрея для перорального введения, содержащего 14 мг силденафила цитрата на дозу (с обеспечением общего вводимого количества, эквивалентного 30 мг силденафила в форме его свободного основания).
ПРИМЕР 3
Исследование относительной биодоступности натощак силденафила в форме спрея для перорального введения в дозах 10 мг, 20 мг и 30 мг по сравнению с таблетками Виагра, содержащими 25 мг силденафила
Цель
В данном исследовании оценивали относительную биодоступность силденафила в форме спрея для перорального введения по сравнению с таковой для таблеток Виагра (VIAGRA®) от Pfizer Labs после однократного перорального введения [1×14 мг/0,12 мл] силденафила цитрата в форме спрея (эквивалентных 10 мг силденафила в форме его свободного основания), 2×14 мг/0,12 мл силденафила цитрата в форме спрея (эквивалентных 20 мг силденафила в форме его свободного основания), 3×14 мг/0,12 мл силденафила цитрата в форме спрея (эквивалентных 30 мг силденафила в форме его свободного основания) или таблетки 1×25 мг здоровым взрослым субъектам, при этом введение осуществляли натощак.
Дополнительные цели заключались в оценке относительной безопасности силденафила в форме спрея для перорального введения после перорального введения однократной дозы по сравнению с таковой для таблеток Виагра. Оценки включали определение изменений состояния ортостатической гипотензии, степени раздражения полости рта, жизненно важных показателей, регистрацию электрокардиограммы (ЭКГ) в 12 отведениях и клинические анализы.
ДИЗАЙН ИССЛЕДОВАНИЯ
Исследование представляло собой открытое, рандомизированное, четырехпериодное перекрестное исследование с однократным введением натощак с применением четырех видов терапии. Общее число здоровых взрослых субъектов мужского пола, включенных в исследование, составляло двадцать четыре (24). Общая продолжительность исследования, скрининга вплоть до завершения исследования составляла приблизительно девять недель с по меньшей мере трехдневным периодом отмывки между введениями доз. При регистрации для участия в исследовании субъекты прибывали в клинический центр по меньшей мере за 36 часов до Дня 1 введения препаратов для Периода I и по меньшей мере за 12 часов до Дня 1 введения препаратов для Периодов II, III и IV. Субъекты должны были оставаться в центре в течение по меньшей мере 24 часов после каждого периода введения исследуемых препаратов.
После каждого периода отмывки субъекты возвращались в клинический центр для получения альтернативной терапии в соответствии с рандомизацией. Забор образцов крови осуществляли в течение 90-минутного периода, но не позднее, чем за 30 минут, до введения (0 часов) и через 0,083, 0,167, 0,25, 0,33, 0,50, 0,75, 1, 1,5, 2, 3, 4, 6, 8, 10, 12, 16 и 24 часа после введения дозы. Фиксировали фактическое время забора образцов. Во время каждого периода исследования в общей сложности забирали 18 образцов крови с получением в совокупности 72 образцов и общего объема образцов, составляющего 432 мл. Фиксировали фактическое время забора образцов.
Анализ образцов посредством биоаналитических методов
Концентрации силденафила и Ν-десметилсилденафила в плазме измеряли с применением валидированного биоаналитического метода и в соответствии со стандартными операционными процедурами и руководствами FDA. Валидированный диапазон обнаружения силденафила в плазме человека составлял от 1 до 300 нг/мл. Валидированный диапазон обнаружения Ν-десметилсилденафила в плазме человека составлял от 0,25 до 75 нг/мл.
Анализы фармакокинетических показателей
Фармакокинетические параметры для концентрации силденафила и Ν-десметилсилденафила в плазме рассчитывали с применением стандартных некомпартментных подходов, как указано ниже:

Значения Kel, AUC0-∞ или Т1/2 не указаны для случаев, в которых не была обнаружена терминальная логарифмически линейная фаза для профиля концентрация-время. При необходимости могут быть рассчитаны другие фармакокинетические параметры.
Статистический анализ
Средние арифметические значения, стандартные отклонения и коэффициенты вариации были рассчитаны для вышеперечисленных параметров. Кроме того, были рассчитаны средние геометрические значения для AUC0-t, AUC0-∞ и Cmax.
Дисперсионный анализ (analysis of variance, ANOVA) выполняли для логарифмически преобразованных фармакокинетических параметров AUC0-t, AUC0-∞ и Cmax. Модель ANOVA включала последовательность, состав и период в качестве фиксированных эффектов и субъектов, сгруппированных внутри последовательности, в качестве случайных эффектов. Проводили анализ последовательности с применением субъектов, сгруппированных внутри последовательности, в качестве параметра ошибки. Для определения эффекта последовательности применяли 10% уровень значимости. Каждый дисперсионный анализ включал расчет средних значений методом наименьших квадратов, разности между приведенными средними значениями для состава и связанной с этой разностью стандартной ошибки. Вышеописанные статистические анализы выполняли с применением соответствующей процедуры в программе SAS®.
В соответствии с двумя односторонними критериями для биоэквивалентности, 90% доверительные интервалы для разности между средними значениями, полученными методом наименьших квадратов (LSM), для содержащих лекарственное средство составов рассчитывали для параметров AUC0-t, AUC0-∞ и Cmax с применением логарифмически преобразованных данных. Доверительные интервалы выражены в процентах относительно LSM для эталонного состава.
Были рассчитаны отношения средних значений с применением LSM для логарифмически преобразованных AUC0-t, AUC0-∞ и Cmax. Представлены средние геометрические значения. Отношения средних значений выражены в процентах относительно LSM для эталонного состава.
Выводы о биоэквивалентности были сделаны на основании 90% доверительных интервалов для силденафила.
Подготовка полости рта
В период времени между 1,25 часа (временная точка исследования -1,25 ч) и 1 часом (временная точка исследования-1 ч) до введения, включая День -1 Периода I введения плацебо, субъекты чистили зубы и язык зубной пастой, полученной в клиническом центре. После завершения чистки зубов и языка субъекты сразу три раза полоскали рот приблизительно 80 мл воды комнатной температуры до общего объема 240 мл (8 жидких унций).
Оценка рН слюны
рН слюны измеряли за 2300 часов (±30 минут) в День -1, а в День 1 в течение 30 минут до чистки зубов и языка, в течение 30 минут после завершения чистки зубов и языка и в течение 15 минут до введения препаратов. В случае введения плацебо (День -1 Периода I) рН слюны измеряли за 2300 часов (±30 минут) в День -2, а в День -1 в течение 30 минут до чистки зубов и языка, в течение 30 минут после завершения чистки зубов и языка и в течение 15 минут до введения плацебо. рН измеряли с применением тест-полосок для определения рН в диапазоне 5-9. Каждого субъекта, рН слюны которого составлял 5, дополнительно подвергали анализу с применением тест-полосок для определения рН в диапазоне 0-6.
Пероральное введение спрея
В течение 30 минут до введения флаконы с препаратом в форме спрея были подготовлены к применению путем выполнения пяти распылений в соответствии с предоставленными спонсором указаниями по подготовке флаконов. Для каждого субъекта и для каждой дозы применяли новый флакон со спреем.
В момент введения субъекты должны были открыть рот и поместить расслабленный язык напротив нижнего ряда зубов. Затем участвующий в проведении исследования персонал осуществлял введение требуемого количества доз спрея в соответствии со схемой рандомизации.
После введения субъекты должны были закрыть рот и поместить язык напротив нижнего ряда зубов на две минуты. Субъектам не следовало глотать или удалять препарат с.помощью языка в течение двух минут. Через две минуты субъекты должны были совершить глотательное движение, и фиксировали любое отклонение от этого времени. После совершения глотательного движения проводили объективную оценку раздражения полости рта (раздражения мягких тканей слизистой оболочки губ или щек и языка).
Введение эталонного препарата
Однократную дозу таблеток Виагра, содержащую 25 мг силденафила (1×25 мг), вводили в День 1 введения препаратов с приблизительно 240 мл (8 жидкими унциями) воды комнатной температуры. Персонал центра следил за тем, чтобы таблетка и жидкость были полностью проглочены субъектами.
Опросник после введения препарата
Непосредственно после завершения введения препаратов и через один час после введения субъектам были заданы следующие вопросы, относящиеся к исследуемому препарату:
- вкус препарата;
- готовность принять препарат снова;
- сколько-нибудь значимое избыточное слюноотделение.
Если раздражение полости рта продолжалось более одного часа, субъектов опрашивали по всем пунктам оценки степени раздражения и после одного часа с последующим вопросом: "Каковы ваши ощущения во рту?"
МЕТОДЫ ИССЛЕДОВАНИЯ
Регистрация для участия в исследовании (День -2 Периода I)
При регистрации для участия в исследовании субъекты прибывали в клинический центр по меньшей мере за 36 часов до Дня 1 введения препаратов и должны были оставаться в центре в течение 24 часов после Дня 1 введения. Субъектов оценивали с точки зрения того, продолжали ли они соответствовать критериям включения в исследование/исключения из исследования и ограничения. В течение периода голодания было разрешено неограниченное употребление воды. Измеряли артериальное давление в положении лежа на спине и стоя и частоту сердечных сокращений и регистрировали ЭКГ. Забирали образцы мочи для скрининга на злоупотребление лекарственным средством.
Субъекты прекращали прием пищи по меньшей мере за десять часов до введения в День -1. В течение исследования субъекты получали стандартизованную пищу и напитки. Состав и количество пищи оставались неизменными в течение каждого периода пребывания субъектов в центре. В момент времени 2300 часов (±30 минут) измеряли рН слюны до чистки зубов на ночь. Субъекты должны были лечь спать после завершения этого измерения.
Исключительно в День -1 Периода I пациенты получали три дозы спрея плацебо за 24 часа до введения препаратов в День 1 Периода I. Подготовка и введение дозы плацебо были идентичными таковым для исследуемых препаратов.
Регистрация для участия в исследовании (День 1 Периодов II, III и IV)
При регистрации для участия в исследовании субъекты прибывали в клинический центр по меньшей мере за 12 часов до Дня 1 введения препаратов и должны были оставаться в центре в течение 24 часов после Дня 1 введения. Субъектов быстро оценивали с точки зрения того, продолжали ли они соответствовать критериям включения в исследование/исключения из исследования и ограничения. В течение периода голодания было разрешено неограниченное употребление воды. Измеряли артериальное давление в положении лежа на спине и стоя и частоту сердечных сокращений и регистрировали ЭКГ. Забирали образцы мочи для скрининга на злоупотребление лекарственным средством.
Субъекты прекращали прием пищи по меньшей мере за десять часов до введения препаратов в День 1. В течение исследования субъекты получали стандартизованную пищу и напитки. Состав и количество пищи оставались неизменными в течение каждого периода пребывания субъектов в центре. За 2300 часов (±30 минут) измеряли рН слюны до чистки зубов на ночь. Субъекты должны были лечь спать после завершения этого измерения.
Процедуры в дни проведения исследования (День -1 Периода I)
Перед введением плацебо были выполнены следующие процедуры:
- Измерение рН слюны до чистки субъектами зубов и языка.
- Чистка субъектами зубов и языка до введения дозы.
- Измерение рН слюны после чистки субъектами зубов и языка.
- Измерение рН слюны в течение 15 минут до введения дозы.
- Оценка состояния полости рта для определения исходной степени раздражения полости рта до введения плацебо.
- Запрет на употребление жидкости с одного часа до введения дозы до одного часа после введения.
За 24 часа до Дня 1 введения препаратов каждому субъекту вводили последовательно одну дозу (три дозы спрея для перорального введения) плацебо в течение Периода I при общем количестве, составляющем одну дозу на каждого субъекта.
После введенния плацебо были выполнены следующие процедуры:
- Проведение как объективной, так и субъективной оценки степени раздражения полости рта непосредственно после введения и через один час после введения дозы плацебо.
- Поддержание режима голодания в течение по меньшей мере четырех часов после введения плацебо. В период исследования с пребыванием субъектов в центре употребление жидкости было возобновлено не ранее чем через один час после введения дозы. Когда употребление жидкостей не было запрещено, последние были доступны в неограниченном количестве.
- Запрет на чистку зубов и языка в течение первых двух часов после введения.
- Обед во временной точке исследования 4,25 часа после введения дозы плацебо.
- Ужин во временной точке исследования 10,25 часа после введения дозы плацебо.
- Вечерний легкий прием пищи за 10,5 часа до введения дозы в День 1.
- Измерение рН слюны за 2300 часов (±30 минут) до чистки зубов и языка на ночь. Субъекты должны были лечь спать после завершения этого измерения.
- Прекращение субъектами приема пищи по меньшей мере за десять часов до введения препаратов в День 1.
День 1
Перед введением препаратов были выполнены следующие процедуры:
- Измерение артериального давления в положении лежа на спине и стоя и частоты сердечных сокращений.
- Измерение рН слюны до чистки субъектами зубов и языка.
- Чистка субъектами зубов и языка до введения дозы.
- Измерение рН слюны после чистки субъектами зубов и языка.
- Забор образцов крови до введения дозы во временной точке 0 часов для проведения анализа фармакокинетических показателей.
- Измерение рН слюны в течение 15 минут до введения дозы.
- Оценка состояния полости рта для определения исходной степени раздражения полости рта.
- Запрет на употребление жидкости, за исключением жидкости, получаемой субъектами при введении эталонного препарата, с одного часа до введения дозы до одного часа после введения.
Каждому субъекту вводили последовательно одну дозу (1×14 мг/0,12 мл спрея для перорального введения, 2×14 мг/0,12 мл спрея для перорального введения, 3×14 мг/0,12 мл спрея для перорального введения или 1×25 мг в составе таблетки) в каждом из четырех периодов введения препаратов при общем количестве, составляющем четыре дозы на каждого субъекта.
После введения препаратов были выполнены следующие процедуры:
- Проведение как объективной, так и субъективной оценки степени раздражения полости рта непосредственно после введения и через один час после введения дозы.
- Поддержание режима голодания в течение по меньшей мере четырех часов после введения. В период исследования с пребыванием субъектов в центре употребление жидкости было возобновлено не ранее чем через один час после введения дозы. Когда употребление жидкостей не было запрещено, последние были доступны в неограниченном количестве.
- Запрет на чистку зубов и языка в течение первых двух часов после введения.
- Пристальное наблюдение участвующего в проведении исследования персонала за субъектами и нахождение субъектов в пределах видимости персонала в течение четырех часов после введения начальной дозы.
- Нахождение субъектов в положении сидя прямо в течение первых четырех часов после введения препаратов за исключением случаев, когда требовалось иное положение в связи с процедурами исследования или личными потребностями субъектов. Субъектам не разрешали ложиться (за исключением случаев возникновения последующих нежелательных эффектов, относительно которых были сделаны указания персоналом центра) в течение первых четырех часов после введения. Субъекты не занимались какой-либо физической активностью во время нахождения в клиническом центре и следовали правилам, регулирующим их действия, в соответствии с указаниями персонала клинического центра.
- Обед во временной точке исследования 4,25 часа.
- Ужин во временной точке исследования 10,25 часа.
- Вечерний легкий прием пищи во временной точке исследования 14,25 часа.
- Забор образцов крови для оценки фармакокинетического профиля.
- Измерение артериального давления в положении лежа на спине и стоя и частоты сердечных сокращений приблизительно через один час после введения каждой дозы.
- Регистрация ЭКГ приблизительно через один час после введения каждой дозы.
- Обследование всех субъектов для установления наличия каких-либо нежелательных эффектов.
День 2
- Забор образцов крови для оценки фармакокинетического профиля.
- Измерение артериального давления в положении лежа на спине и стоя и частоты сердечных сокращений приблизительно через 24 часа после введения каждой дозы.
- Обследование всех субъектов для установления наличия каких-либо нежелательных эффектов до покидания субъектами клинического центра.
- Обследование субъектов до покидания ими клинического центра для гарантии того, что отъезд субъектов из центра являлся для них безопасным.
- Покидание субъектами клинического центра через приблизительно 24 часа после введения дозы.
ОЦЕНКА СТЕПЕНИ РАЗДРАЖЕНИЯ ПОЛОСТИ РТА
Непосредственно перед введением первой дозы (0 часов) для каждого вида терапии с применением спрея для перорального введения, включая введение плацебо, и через указанные ниже интервалы времени оценивали состояние полости рта каждого добровольца на наличие местного раздражения. Местное раздражение оценивали относительно наблюдения во временной точке 0 часов. По возможности, все оценки для каждого индивидуального субъекта были выполнены одним и тем же экспертом в течение всех периодов исследования. В случае обнаружения раздражения было дано конкретное описание, определяющее локализацию, степень и масштабы раздражения. Информация по оценке раздражения и время исчезновения раздражения было зафиксировано в индивидуальных регистрационных картах (ИРК).
Схема оценки степени раздражения
Оценку степени раздражения выполняли в следующих временных точках: до каждого перорального введения спрея, сразу после совершения глотательного движения (2 минуты) и через 1 час после введения.
Если сколько-нибудь значимое раздражение сохранялось более одного часа периода оценки, проведение оценки продолжали каждые два часа до исчезновения раздражения вплоть до десяти часов после введения препаратов. Если раздражение все еще наблюдалось через десять часов, исследователь оценивал состояние субъекта с точки зрения того, следует ли субъекту прекратить свое участие в исследовании. Все оценки через один час также включали вопросы, позволяющие получить субъективную оценку.
Оценка степени раздражения
Обследование состояло из визуального осмотра/оценки состояния мягких тканей на присутствие раздражения слизистой оболочки губ и щек и языка. Были зафиксированы время и дата этих обследований. С целью облегчения обследования применяли небольшой фонарик или другое подобное устройство. Поверхности слизистой оболочки полости рта, с правой и с левой стороны, и места соединения губ и язык обследовали на присутствие раздражения и оценивали в соответствии со следующей системой классификации.
Шкала оценки степени раздражения
Нумерационный класс эритемы - слизистая оболочка полости рта (левая и правая стороны) и язык
0 - Отсутствие эритемы
1 - Очень легкая форма эритемы (едва заметная)
2 - Легкая форма эритемы (края области, хорошо определяемые по четко выраженным элементам)
3 - Умеренная форма эритемы (элементы размером 1,0 мм)
4 - Тяжелая форма эритемы (элементы размером более 1,0 мм, выходящие за область поражения)
СУБЪЕКТЫ ИССЛЕДОВАНИЯ
Распределение участников клинического исследования
Для участия в Периоде I были зарегистрированы двадцать восемь (28) добровольцев, а общее число субъектов составило 24 человека. Исследование проводили с участием 24 здоровых взрослых мужчин (23 продолжали участие вплоть до завершения исследования).
Регистрацию субъектов осуществляли в клиническом центре 30 июля 2010 года для Периода I, 6 августа 2010 года для Периода II, 13 августа 2010 года для Периода III и 20 августа 2010 года для Периода IV. Регистрацию проводили за два дня до введения дозы для Периода I и за один день до введения дозы для Периодов II, III и IV. Субъектов оставляли в клиническом центре на 24 часа после забора образцов крови во время каждого периода.,
В таблице 6.1-1 представлены все субъекты, досрочно прекратившие участие в исследовании, причина (причины) досрочного выбывания и временные точки в ходе исследования, представляющие собой моменты выбывания каждого субъекта.

В таблице 6.1-2 показано число субъектов, рандомизированных в соответствии с каждой последовательностью приема препаратов и выбывших после рандомизации в процессе проведения исследования.
В таблице 6.1-3 кратко представлено распределение субъектов по периодам исследования.
ОЦЕНКА ФАРМАКОКИНЕТИЧЕСКИХ ПАРАМЕТРОВ
Анализируемые совокупности данных
В исследование были включены двадцать четыре (24) субъекта, и все субъекты представляли собой здоровых взрослых.
Двадцать четыре (24) субъекта начинали исследование, и 23 субъекта завершили клиническую часть исследования в полном объеме.
Данные по всем 24 субъектам применяли в статистическом анализе для силденафила и Ν-десметилсилденафила для сравнений исследуемого препарата А (1 доза спрея; 10 мг) с эталонным препаратом D (1 таблетка, 25 мг) и сравнений исследуемого препарата С (3 дозы спрея; 30 мг) с эталонным препаратом D (1 таблетка, 25 мг). Субъект 24 отозвал информированное согласие до регистрации для участия в Периоде IV из-за невозможности следовать режиму дозирования. Данные по 23 из 24 субъектов применяли в статистическом анализе для силденафила и Ν-десметилсилденафила для сравнений исследуемого препарата В (2 дозы; 20 мг) с эталонным препаратом D (1 таблетка, 25 мг). Для расчета фармакокинетических параметров использовали значения фактического времени.
Краткое описание биоаналитического метода
Определение концентраций силденафила и Ν-десметилсилденафила в плазме выполняли в биоаналитической лаборатории института Cetero Research (Торонто). Анализ проводили с применением системы для ЖХ/МС/МС-анализа Micromass Quattro Ultima, оснащенной устройством для электрораспыления с Ζ-образной геометрией. Детекцию положительных ионов осуществляли в режиме мониторинга множественных реакций (MRM). Количественное определение аналитов выполняли с применением методики жидкость-жидкостной экстракции. После экстракции аликвоты объемом 20 мкл вводили в систему для ЖХ/МС/МС. Данные получали и обрабатывали с применением программного обеспечения Micromass "Mass lynx" версии 4.1. Для обеспечения наилучшего подбора калибровочной кривой к имеющимся данным применяли линейную регрессию с взвешиванием 1/х. Нижний предел количественного определения (НПКО) составлял 1,000/0,2500 нг/мл, а верхний предел количественного определения (ВПКО) составлял 300,0/75,00 нг/мл для сиденафила и Ν-десметилсилденафила. Стандартные соединения для построения калибровочной кривой и образцы для контроля качества (КК) для силденафила и Ν-десметилсилденафила соответствовали критериям приемлемости для серий анализов, проводимых при получении окончательных данных, что указывало на удовлетворительные рабочие характеристики метода в процессе анализа полученных от субъектов исследуемых образцов.
Результаты фармакокинетических исследований и представление данных по индивидуальным субъектам в виде таблиц
Данные статистического анализа
Фармакокинетические параметры рассчитывали с применением программного обеспечения WinNonlin® версии 5.0.1, разработанного специально для анализа фармакокинетических данных. Использовали модель расчетов Model 200 в программе WinNonlin® для путей введения лекарственных средств, отличных от введения в кровоток.
Для каждого фармакокинетического параметра выполняли дисперсионный анализ (ANOVA) с применением программного обеспечения SAS®. При сравнении эффектов исследуемого и эталонного препаратов использовали модель ANOVA, включающую факторы для последовательности введения препаратов, субъектов внутри последовательности, периодов и препаратов.
Обработка данных по выбывшим из исследования субъектам или отсутствующих данных
Данные по уровням соединений в плазме субъектов, завершивших исследование, были включены в окончательный анализ данных. Данные по уровням соединений в плазме субъектов, отозвавших информированное согласие или прекративших участие в исследовании по какой-либо причине, не применяли для окончательного анализа. Данные по субъектам, для которых отсутствовали некоторые значения концентраций (например, по причине пропуска субъектами забора крови, потери образцов, невозможности количественного определения соединений в образцах) применяли, если оценка фармакокинетических параметров с помощью данных для оставшихся временных точек представлялась возможной; в противном случае данные по этим субъектам были исключены из окончательного анализа.
Результаты фармакокинетических исследований
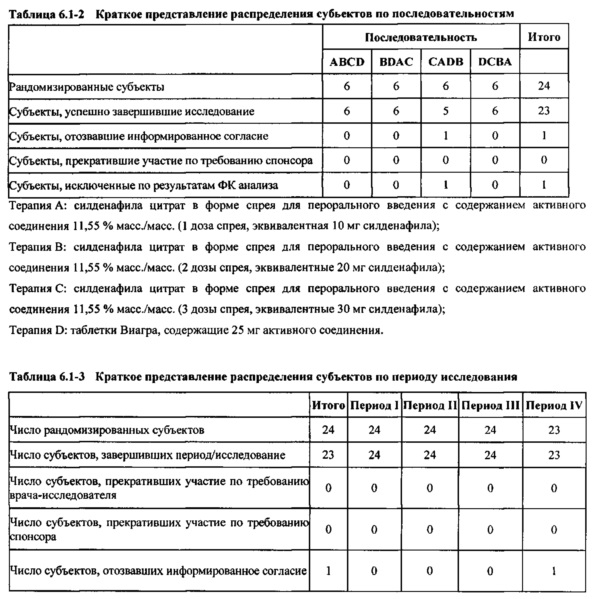
Силденафил (без приведения к дозе)
В таблице 7.2.6.1-1 кратко представлены не приведенные к дозе фармакокинетические параметры для каждого препарата силденафила, полученные с применением некомпартментного анализа.
В целом, концентрации силденафила в плазме были хорошо охарактеризованы при введенных перорально дозах 10 мг, 20 мг и 30 мг в форме спрея и дозе 25 мг в форме таблетки, и они снижались в соответствии с моноэкспоненциальной зависимостью после перорального введения доз спрея и таблетки у здоровых взрослых мужчин-добровольцев.
В общем, при пероральном введении доз 10 мг, 20 мг и 30 мг силденафила в форме спрея здоровым взрослым мужчинам тотальная длительность экспозиции и максимальные концентрации силденафила в плазме, как было оценено по средним значениям AUC и Cmax, соответственно, возрастали пропорционально увеличению дозы от 10 мг до 30 мг. Для всех видов терапии наблюдали тенденцию к быстрому достижению максимальных концентраций силденафила за приблизительно 1,00 час после введения дозы. Средние значения терминальных периодов полувыведения были, в общем, сходными для всех четырех видов терапии, при этом средние периоды полувыведения составляли, по оценкам, от 2,25 до 3,14 часа.
В таблицах 7.2.6.1-2, 7.2.6.1-3 и 7.2.6.1-4 кратко представлены результаты анализов, выполненных с целью определения не приведенных к дозе фармакокинетических параметров для силденафила.
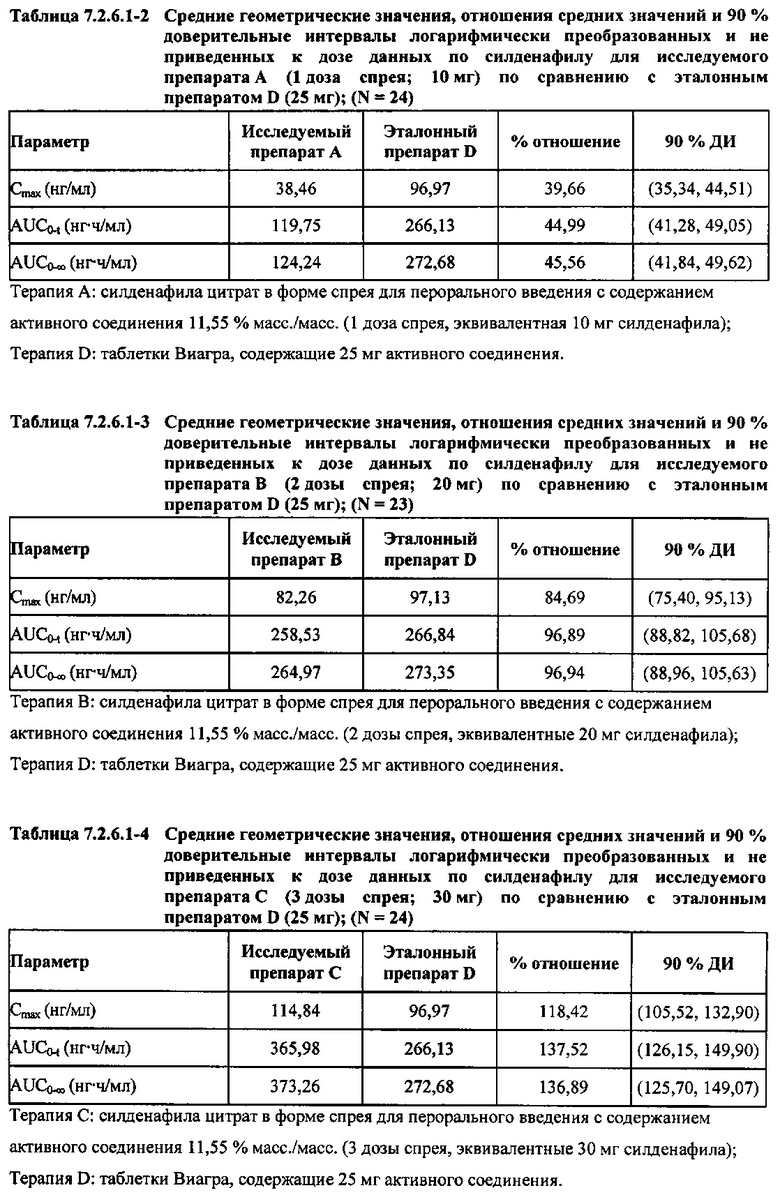
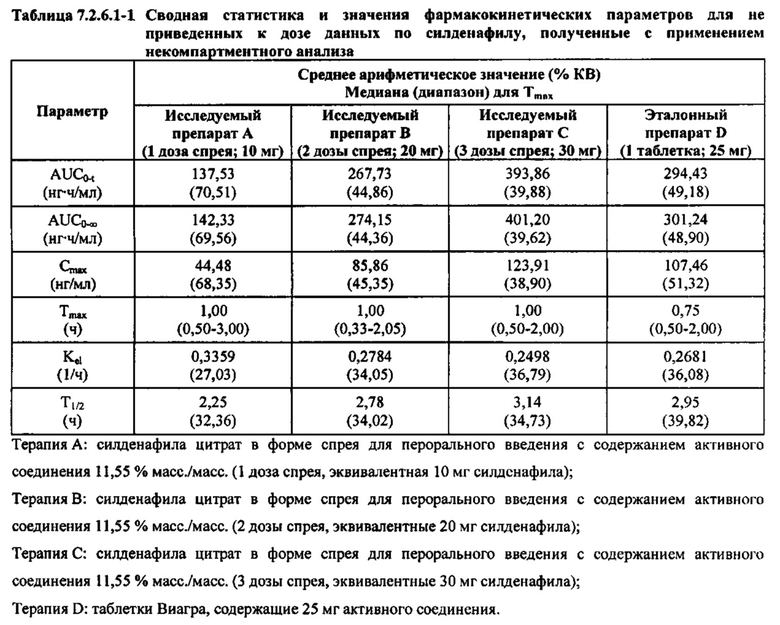
В случае, когда ФК параметры AUC и Cmax для исследуемых препаратов А (10 мг), В (20 мг) и С (30 мг) в форме спрея для перорального введения не были приведены к дозе (25 мг) эталонной таблетки D, относительная биодоступность силденафила в составе исследуемых препаратов в форме спрея для перорального введения по сравнению с таковой для эталонного препарата в форме таблетки характеризовалась тем, что:
- Не приведенные к дозе значения AUC для силденафила, вводимого в 2 дозах исследуемого препарата B в форме спрея (доза силденафила 20 мг), были сопоставимы с таковыми для эталонного препарата D в форме таблетки 25 мг. Точечная оценка для Cmax была приблизительно на 15% ниже, чем для эталонного препарата D в форме таблетки 25 мг, и нижняя граница 90% ДИ для Cmax, составляющая 75,40%, была лишь немного меньше нижней границы диапазона приемлемых значений.
- Как и ожидали, значения AUC и Cmax для 1 дозы исследуемого препарата А в форме спрея (доза силденафила 10 мг) были значительно ниже, а для 3 доз исследуемого препарата С в форме спрея (доза силденафила 30 мг) были значительно выше таковых для эталонного препарата D (таблетки 25 мг).
Силденафил (с приведением к дозе)
В таблице 7.2.6.2-1 кратко представлены приведенные к дозе фармакокинетические параметры для каждого препарата силденафила, полученные с применением некомпартментного анализа.
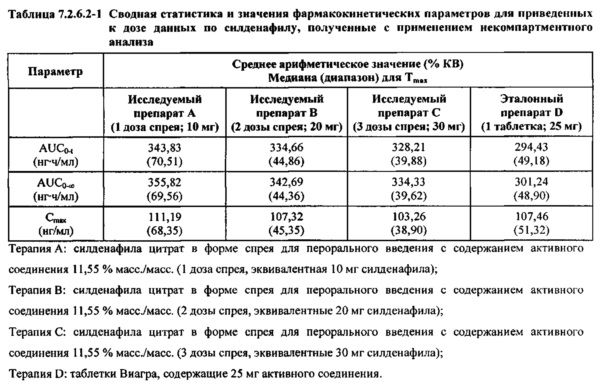
Приведенная к дозе Cmax для исследуемых препаратов на основе силденафила А (10 мг), В (20 мг) и С (30 мг) в форме спрея для перорального введения была сопоставима с таковой для эталонной таблетки D (25 мг), в то время как тотальная длительность экспозиции силденафила, как было оценено по приведенным к дозе значениям AUC, неизменно превышала таковую для эталонной таблетки D (25 мг), при этом значения AUC0-t находились в диапазоне от 328 до 344 нг⋅ч/мл в случае препаратов в форме спрея для перорального введения по сравнению с 294 нг⋅ч/мл для эталонной таблетки 25 мг, и значения AUC0-∞ находились в диапазоне от 344 до 356 нг⋅ч/мл в случае препаратов в форме спрея для перорального, (введения по сравнению с 301 нг⋅ч/мл для эталонной таблетки 25 мг.
Максимальные концентрации в плазме, как было оценено по значениям Cmax, для всех доз исследуемых препаратов (10 мг, 20 мг и 30 мг силденафила в форме спрея для перорального введения) были сопоставимы с дозой эталонного препарата (25 мг силденафила в форме таблетки для перорального введения). Для всех видов терапии максимальные концентрации в плазме находились в диапазоне от 103,26 нг/мл до 111,19 нг/мл.
В таблицах 7.2.6.2-2, 7.2.6.2-3 и 7.2.6.2-4 кратко представлены результаты анализов, выполненных с целью определения приведенных к дозе фармакокинетических параметров для силденафила.
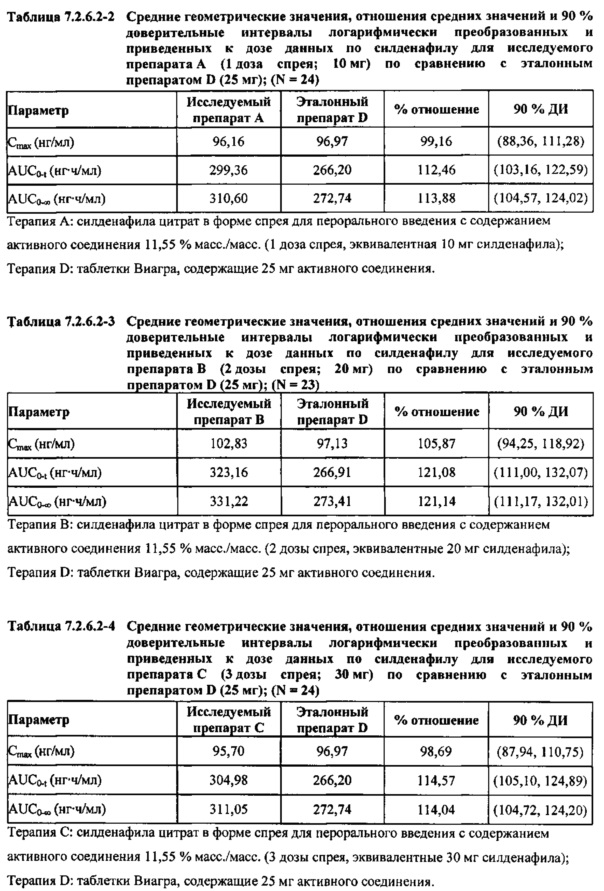
В случае, когда ФК параметры AUC и Cmax для исследуемых препаратов А (10 мг), В (20 мг) и С (30 мг) в форме спрея для перорального введения были приведены к дозе (25 мг) эталонной таблетки D, относительная биодоступность силденафила в составе исследуемых препаратов в форме спрея для перорального введения по сравнению с таковой в составе эталонного препарата в форме таблетки характеризовалась тем, что:
- Приведенные к дозе значения Cmax для доз 10 мг, 20 мг и 30 мг силденафила в составе препаратов в форме спрея для перорального введения были сопоставимы с таковыми для эталонного препарата D (таблетки 25 мг), и все они находились в пределах 90% ДИ.
- Точечные оценки соотношения исследуемый препарат/эталонный препарат для приведенных к дозе AUC0-∞ для всех доз силденафила в форме спрея для перорального введения составляли приблизительно от 114% до 121%. Верхняя граница 90% ДИ составляла 124% для доз исследуемых препаратов А (10 мг) и С (30 мг), т.е. чуть ниже верхней границы 125%. Точечные оценки соотношения исследуемый препарат/эталонный препарат для приведенных к дозе AUC0-t для всех доз силденафила в форме спрея для перорального введения составляли приблизительно от 112% до 121%. Верхняя граница 90% ДИ составляла 123% для дозы исследуемого препарата А (10 мг) и 125% для дозы исследуемого препарата С (30 мг). Для исследуемого препарата В (20 мг) в форме спрея для перорального введения точечные оценки для значений AUC были выше примерно на 21%, а граница 90% ДИ для AUC, составляющая 132%, была выше верхней границы диапазона приемлемых значений.
Эти данные указывают на то, что тотальная системная экспозиция силденафила в составе исследуемых препаратов в форме спрея для перорального введения обеспечивает большую системную биодоступность по сравнению с эталонным препаратом в форме таблетки 25 мг для перорального введения, вероятно, благодаря тому, что силденафил не подвергается пресистемному метаболизму при всасывании через слизистую оболочку.
Ν-десметилсилденафил (без приведения к дозе)
В таблице 7.2.6.3-1 кратко представлены не приведенные к дозе фармакокинетические параметры для каждого препарата Ν-десметилсилденафила, полученные с применением некомпартментного анализа.
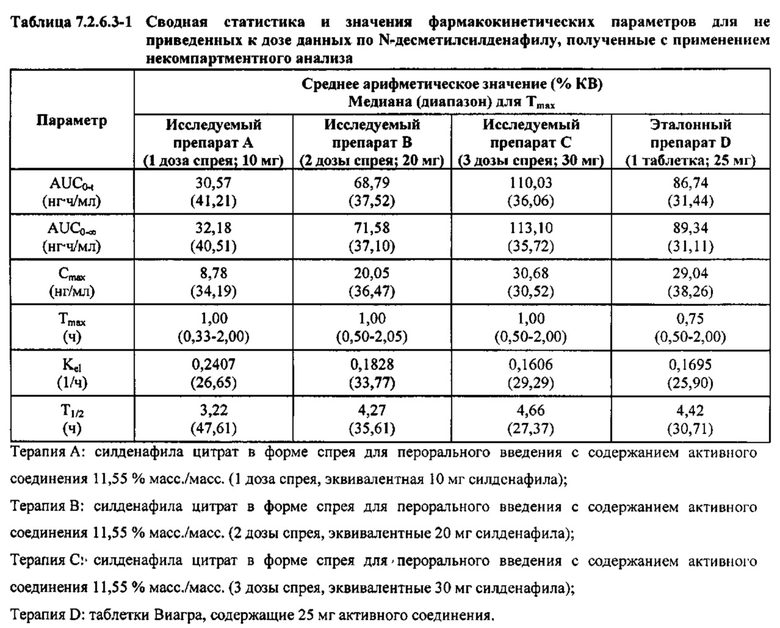
В общем, тотальная длительность экспозиции и максимальные концентрации Ν-десметилсилденафила в плазме для исследуемых препаратов А (10 мг), В (20 мг) и С (30 мг) в форме спрея для перорального введения, как было оценено по средним значениям AUC и Cmax, возрастали пропорционально увеличению дозы от 10 мг до 30 мг. Для всех доз силденафила в форме спрея для перорального введения время достижения максимальных концентраций Ν-десметилсилденафила было немного меньше по сравнению с Tmax для эталонного препарата в форме таблетки для перорального введения, составляющим 0,75 часа; однако диапазоны Tmax для всех исследуемых препаратов силденафила в форме спрея для перорального введения и эталонной таблетки для перорального введения были сходными. Средние значения терминального периода полувыведения были, в общем, сходными для всех четырех видов терапии.
В таблицах 7.2.6.3-2, 7.2.6.3-3 и 7.2.6.3-4 кратко представлены результаты анализов, выполненных с целью определения не приведенных к дозе фармакокинетических параметров для Ν-десметилсилденафила.
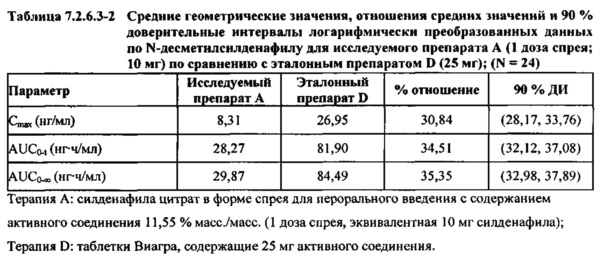
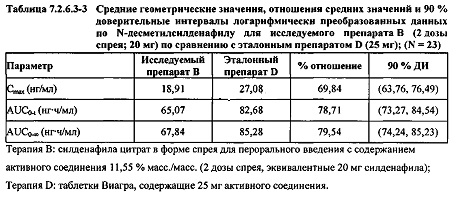
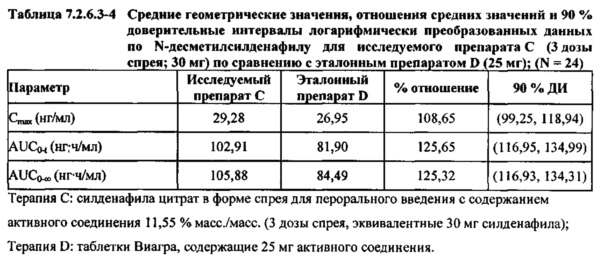
В случае, когда ФК параметры AUC и Cmax для Ν-десметилсилденафила для исследуемых препаратов А (10 мг), В (20 мг) и С (30 мг) в форме спрея для перорального введения не были приведены к дозе (25 мг) эталонной таблетки D, относительная биодоступность Ν-десметилсилденафила, образующегося в результате метаболизации исследуемых препаратов в форме спрея для перорального введения, по сравнению с таковой для эталонного препарата в форме таблетки характеризовалась тем, что:
- Точечные оценки для не приведенных к дозе AUC для Ν-десметилсилденафила в диапазоне доз 10 мг, 20 мг и 30 мг, обеспечиваемых пероральным введением исследуемых препаратов в форме спрея, были, в общем, сопоставимыми с разностью по отношению к эталонной таблетке 25 мг.
- Точечные оценки для Cmax в диапазоне доз 10 мг, 20 мг и 30 мг, обеспечиваемых пероральным введением исследуемых препаратов в форме спрея, были, в общем, меньше, чем разность по отношению к эталонной таблетке 25 мг.
Ν-десметилсилденафил (с приведением к дозе)
В таблице 7.2.6.4-1 кратко представлены приведенные к дозе фармакокинетические параметры для каждого препарата Ν-десметилсилденафила, полученные с применением некомпартментного анализа.
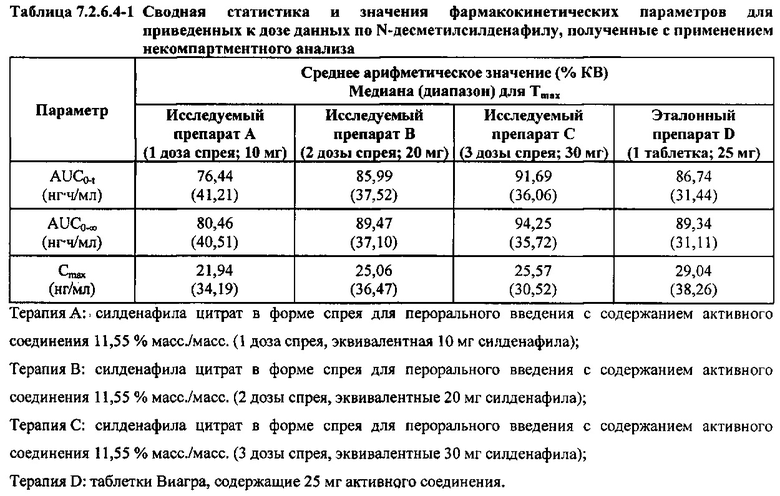
После приведения ФК параметров AUC и Cmax для Ν-десметилсилденафила для исследуемых препаратов А (10 мг), В (20 мг) и С (30 мг) в форме спрея для перорального введения к дозе (25 мг) эталонной таблетки D, тотальная длительность экспозиции Ν-десметилсилденафила, как было оценено по значениям AUC, демонстрировала тенденцию к небольшому увеличению с возрастанием дозы от 10 мг до 30 мг. Однако приведенные к дозе максимальные концентрации Ν-десметилсилденафила в плазме, как было оценено по значениям Cmax, были сходными для доз 20 мг и 30 мг силденафила в составе исследуемых препаратов в форме спрея для перорального введения, но немного меньше для дозы 10 мг в составе исследуемого препарата в форме спрея для перорального введения. Для всех четырех препаратов максимальные концентрации в плазме находились в диапазоне от 21,94 нг/мл до 29,04 нг/мл.
В таблицах 7.2.6.4-2, 7.2.6.4-3 и 7.2.6.4-4 кратко представлены результаты анализов, выполненных с целью определения приведенных к дозе фармакокинетических параметров для Ν-десметилсилденафила.
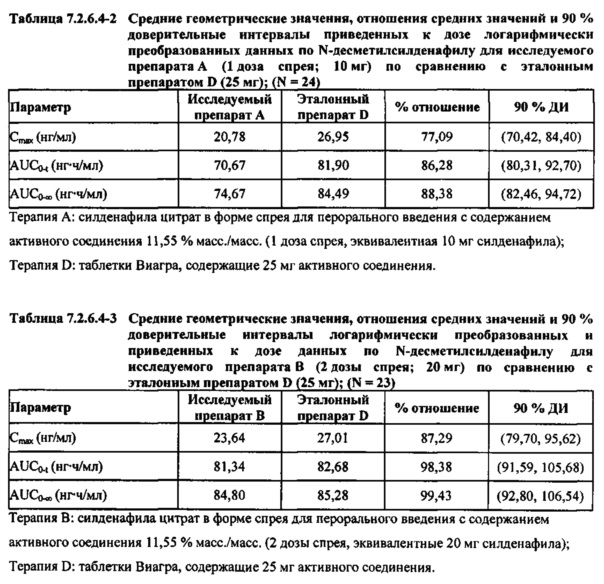
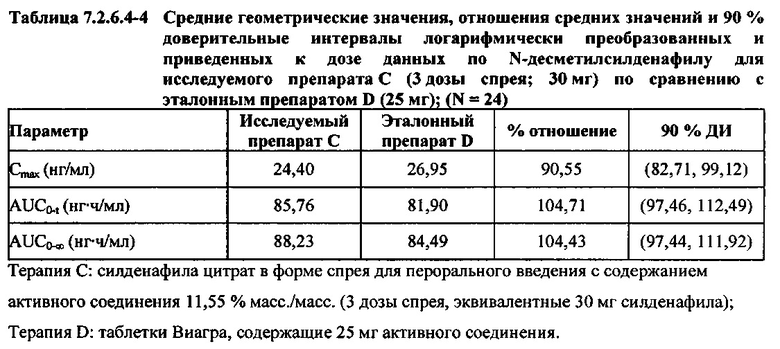
После приведения ФК параметров AUC и Cmax для Ν-десметилсилденафила для исследуемых препаратов А (10 мг), В (20 мг) и С (30 мг) в форме спрея для перорального введения к дозе (25 мг) эталонной таблетки D, результаты ФК и статистического анализов демонстрировали, что, за исключением Cmax для доз 10 мг и 20 мг силденафила в составе исследуемых препаратов в форме спрея для перорального введения, все значения AUC и Cmax соответствовали критерию 80%-125% для 90% ДИ.
Пропорциональность дозы
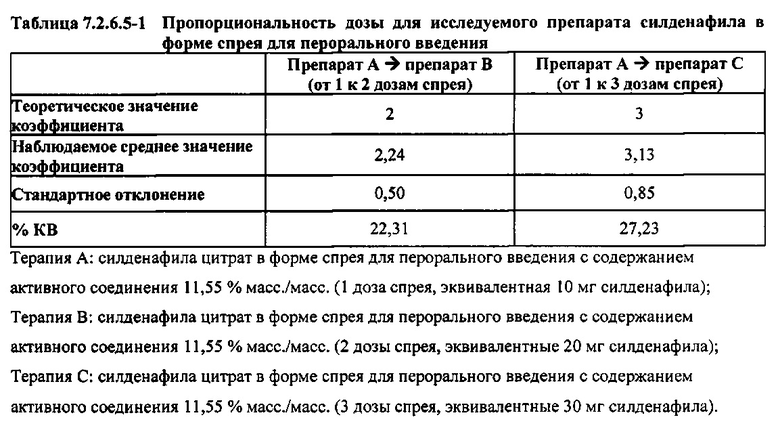
Результаты исследования пропорциональности дозы при однократных пероральных введениях доз 10 мг, 20 мг и 30 мг силденафила в форме спрея демонстрируют, что, в то время как AUC∞ возрастала линейно с увеличением дозы, она не возрастала в точности пропорционально дозе; в частности, наблюдали несколько большее, чем пропорциональное дозе, возрастание AUC∞. Как показывают представленные в таблице 7.2.6.5-1 результаты, в среднем было обнаружено 2,24- и 3,13-кратное возрастание AUC∞ при переходе от 1 к 2 дозам спрея и от 1 к 3 дозам спрея, соответственно. Николе с соавт. (Nichols et al.) сообщают об аналогичных превышающих непропорциональную дозу результатах, полученных после однократного перорального введения дозы силденафила в форме таблеток здоровым мужчинам-добровольцам, и, как отмечено в упомянутом исследовании и в настоящем исследовании, степень этой непропорциональности, по-видимому, не является клинически значимой.
Соотношение метаболит/исходное соединение (соотношение Ν-десметилсилденафил/силденафил)
В таблице 7.2.6.6-1 кратко представлены фармакокинетические параметры для соотношения метаболит/исходное соединение (Ν-десметилсилденафил/силденафил) для соответствующих каждому виду терапии исследуемых препаратов.
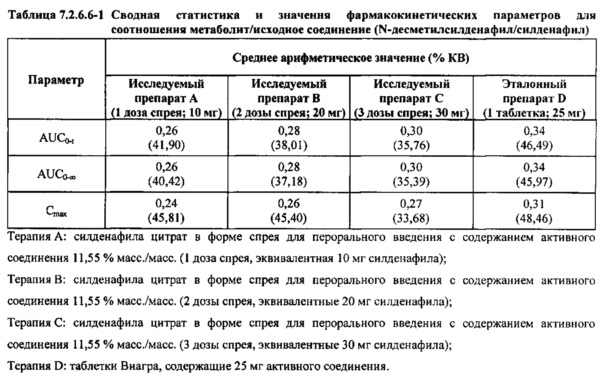
В таблицах 7.2.6.6-2, 7.2.6.6-3 и 7.2.6.6-4 кратко представлены результаты анализов, выполненных с целью определения фармакокинетических параметров для соотношения Ν-десметилсилденафил/силденафил.
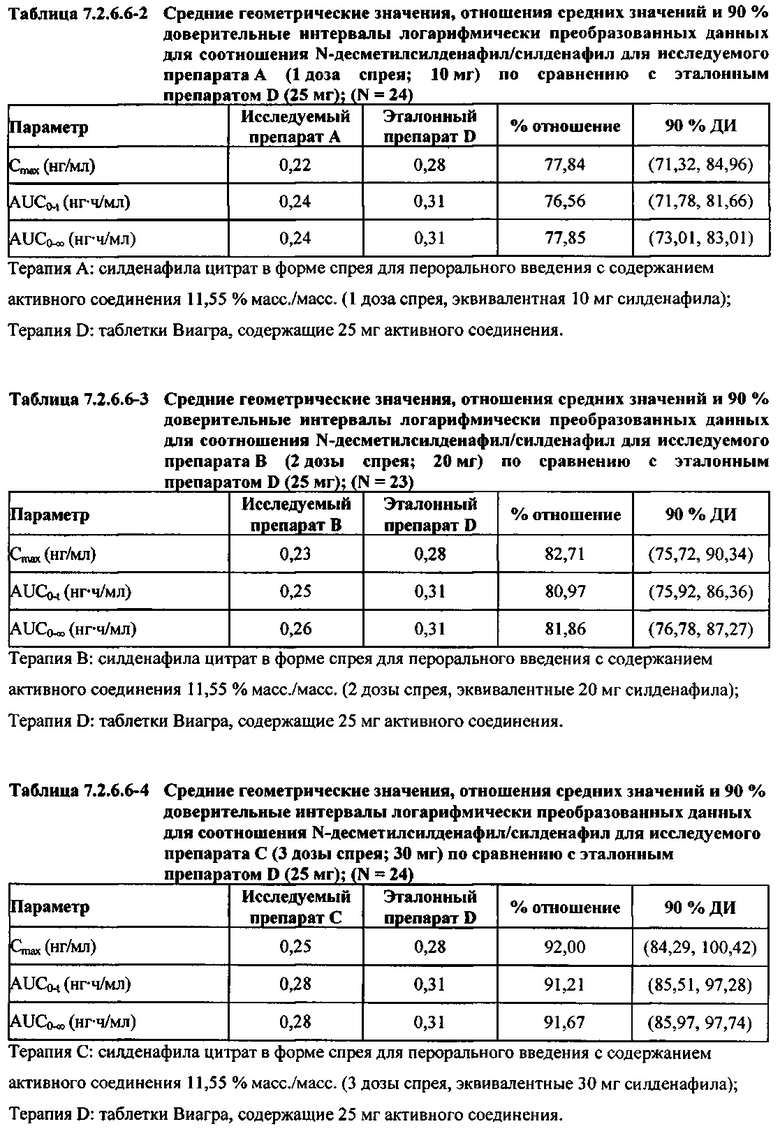
В целом, результаты анализа соотношений метаболит/исходное соединение (М/И) показали, что образование метаболита, как было оценено по соотношениям М/И для доз исследуемых препаратов А (10 мг) и В (20 мг) в форме спрея для перорального введения, происходило с меньшей интенсивностью по сравнению с соотношением М/И для эталонной таблетки D (25 мг), в то время как соотношения М/И для дозы исследуемого препарата С в форме спрея для перорального введения были сопоставимы с соотношениями М/И, наблюдаемыми для эталонной таблетки D (25 мг).
Соотношение М/И для каждой временной точки определения концентрации рассчитывали для каждого вида терапии и наносили на график зависимости от времени с целью визуальной оценки скорости образования метаболита. Эту процедуру выполняли до временной точки четыре часа после введения дозы. Данный график показан на фигуре 7.2.6.6-1. Можно отметить присутствие на графике быстро появляющегося пика на кривой соотношения М/И для эталонной таблетки для перорального введения, в то время как увеличение соотношения М/И с возрастанием времени для всех доз исследуемых препаратов в форме спрея для перорального введения происходило значительно медленнее, чем для эталонной таблетки. Это наблюдение указывает на то, что существует заметное снижение скорости образования метаболита в случае исследуемых препаратов в форме спрея для перорального введения по сравнению с эталонным препаратом в форме таблетки для перорального введения, и свидетельствует о том, что активное соединение в составе исследуемого спрея для перорального введения по большей части не подвергается пресистемному метаболизму за счет всасывания через слизистую оболочку.
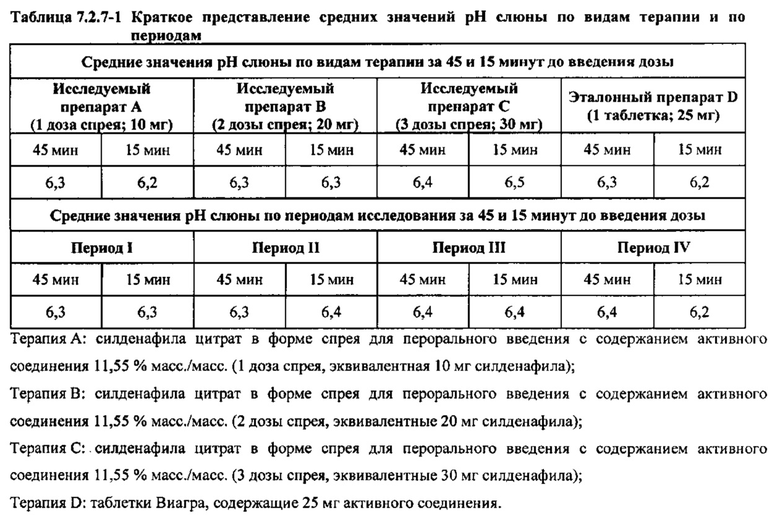
Оценка рН слюны
Всасывание лекарственных средств при введении их пациентам через слизистую оболочку может зависеть от рН полости рта. Таким образом, в настоящем исследовании измеряли рН слюны здоровых взрослых мужчин-добровольцев за 2300 часов (±30 минут) в День -1 и в течение 30 минут до чистки зубов и языка в День 1. Наблюдали минимальное изменение рН слюны до и после чистки зубов и языка. Затем рН слюны измеряли в течение 30 минут после завершения чистки зубов и языка (~ за 45 минут до введения дозы) и в течение 15 минут до введения дозы. рН измеряли с применением тест-полосок для определения рН в диапазоне 5-9. Ни у одного из субъектов рН не был равен 5, соответственно, не проводили дополнительного определения рН с применением тест-полосок для определения рН в диапазоне 0-6. Не было обнаружено каких-либо существенных различий между значениями рН слюны, соответствующими видам терапии или периодам исследования, за 45 минут и 15 минут до введения дозы, как показано в таблице 7.2.7-1.
Опросник после введения препарата
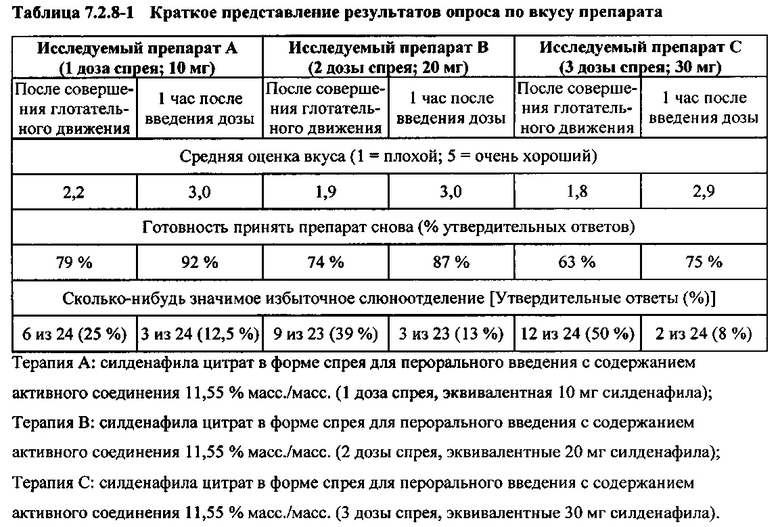
Непосредственно после завершения введения препаратов (2 минуты) и через один час после введения субъектам задавали следующие вопросы, относящиеся к исследуемому препарату (в форме спрея для перорального введения):
- Вкус препарата
- Готовность принять препарат снова
- Сколько-нибудь значимое избыточное слюноотделение
Оценка степени раздражения полости рта
Обследование состояло из визуального осмотра/оценки состояния мягких тканей на присутствие раздражения слизистой оболочки губ и щек и языка. Поверхности слизистой оболочки полости рта, с правой и с левой стороны, и места соединения губ и язык обследовали на присутствие раздражения и оценивали в соответствии со следующей системой классификации, описанной ниже.
Нумерационный класс эритемы - слизистая оболочка полости рта (левая и правая стороны) и язык
0 - Отсутствие эритемы
1 - Очень легкая форма эритемы (едва заметная)
2 - Легкая форма эритемы (края области, хорошо определяемые по четко выраженным элементам)
3 - Умеренная форма эритемы (элементы размером 1,0 мм)
4 - Тяжелая форма эритемы (элементы размером более 1,0 мм, выходящие за область поражения)
Оценку степени раздражения мягких тканей слизистой оболочки губ и щек и языка проводили после введения как активного соединения, так и плацебо. Оценки степени раздражения регистрировали в следующие временные точки: до каждого перорального введения спрея, сразу после совершения глотательного движения (2 минуты) и через 1 час после введения. Если сколько-нибудь значимое раздражение сохранялось более одного часа периода оценки, проведение оценки продолжали каждые два часа до исчезновения раздражения вплоть до десяти часов после введения препаратов. Если раздражение все еще наблюдалось через десять часов, исследователь оценивал состояние субъекта с точки зрения того, следует ли субъекту прекратить свое участие в исследовании. Все оценки через один час также включали вопросы, позволяющие получить субъективную оценку.
Результаты оценки раздражения полости рта свидетельствовали об отсутствии раздражения полости рта после введения плацебо (3 дозы спрея). Кроме того, не было обнаружено раздражения полости рта после введения исследуемого препарата А (1 доза спрея), исследуемого препарата В (2 дозы спрея) или исследуемого препарата С (3 дозы спрея) при обследовании сразу после введения (удерживания препарата во рту в течение двух минут, затем совершения глотательного движения) и при обследовании через 1 час после введения дозы.
ОБСУЖДЕНИЕ
Приведенные к дозе значения Cmax для силденафила в составе исследуемых препаратов А (10 мг), В (20 мг) С (30 мг) в форме спрея для перорального введения были, в общем, сопоставимы с таковыми для эталонной таблетки D (25 мг) для перорального введения. Точечные оценки соотношения исследуемый препарат/эталонный препарат для приведенных к дозе AUC для всех доз силденафила в форме спрея для перорального введения составляли приблизительно от 112% до 121%, и 90% ДИ для значений AUC составляли от 124% до 132% при верхней границе диапазона приемлемых значений 125%. Кроме того, не приведенные к дозе точечные оценки для значений AUC для 2 вводимых перорально доз спрея (20 мг) исследуемого препарата В составляли приблизительно 97%, что указывало на биодоступность, соспоставимую с таковой для эталонной таблетки D (25 мг) для перорального введения. Эти данные указывают на то, что тотальная экспозиция силденафила в составе исследуемого препарата в форме спрея для перорального введения обеспечивает большую системную биодоступность по сравнению с эталонной таблеткой для перорального введения, вероятно, благодаря тому, что силденафил не подвергается пресистемному метаболизму при всасывании через слизистую оболочку.
Силденафил, как известно, метаболизируется до его основного метаболита, Ν-десметилсилденафила, посредством пресистемного метаболизма на более чем 50% [абсолютная биодоступность составляет 41% (диапазон 25-63%)]. Фармакокинетические и статистические результаты, полученные после перорального введения силденафила в форме спрея здоровым взрослым субъектам мужского пола, демонстрировали повышенную системную экспозицию, которую можно обоснованно объяснить процессами всасывания через слизистую оболочку полости рта.
Оценка соотношения М/И (отношения метаболита (Ν-десметилсилденафила) к исходному соединению (силденафилу)) для исследуемых препаратов А (10 мг), В (20 мг) и С (30 мг) в форме спрея для перорального введения по сравнению с эталонной таблеткой D (25 мг) для перорального введения демонстрировала различия в скорости образования метаболита, свидетельствующие о всасывании препаратов через слизистую оболочку.
Было обнаружено минимальное изменение средних значений рН слюны при сравнении по видам терапии и периодам настоящего исследования, таким образом, рН слюны субъектов, вероятно, не являлся фактором, влияющим на кажущееся поглощение через слизистую оболочку, наблюдаемое после перорального введения спрея.
Ни у одного из субъектов не было обнаружено раздражения полости рта после перорального введения доз плацебо или активного соединения в форме спрея (вплоть до 3 доз спрея, доза силденафила 30 мг).
В целом, силденафила цитрат хорошо переносился в дозах 10 мг, 20 мг, 30 мг и хорошо переносился в однократной дозе 25 мг (таблетка 1×25 мг для перорального введения), вводимых здоровым взрослым субъектам в условиях наличия приемов пищи.
ПРИМЕР 4
Цель
Цель следующего исследования заключалась в оценке частных значений AUC экспозиции силденафила во временных точках один, четыре, двенадцать и двадцать четыре часа (AUC0-1, AUC0-4, AUC0-12 и AUC0-24) после перорального введения доз 10, 20 и 30 мг силденафила в составе исследуемых препаратов в форме спрея по сравнению с аналогичными значениями для дозы 25 мг силденафила в составе эталонного препарата в форме таблетки.
ДИЗАЙН ИССЛЕДОВАНИЯ
Настоящее исследование представляло собой рандомизированное, четырехпериодное перекрестное исследование, направленное на сравнение относительной биодоступности (скорости и степени абсорбции) одного исследуемого препарата в трех различных дозах (10 мг, 20 мг и 30 мг) с биодоступностью одного эталонного препарата при введении натощак. В данных экспериментах один исследуемый состав в трех различных дозах (10 мг, 20 мг и 30 мг в составе спрея на основе силденафила цитрата для перорального введения, обеспечивающего доставку 14 мг/0,12 мкл на дозу спрея) сопоставляли с таблетками Виагра 25 мг.
Двадцать четыре здоровых взрослых субъекта мужского пола, для которых не было заместителей, распределяли случайным образом по группам, соответствующим одной из четырех последовательностей введения следующих препаратов:

Четыре последовательности введения препаратов представляли собой следующие:
Последовательность 1 = ABCD
Последовательность 2 = BDAC
Последовательность 3 = CADB
Последовательность 4 = DCBA
Добровольцам вводили перорально натощак однократную дозу исследуемого или эталонного препарата в виде четырех раздельных процедур с по меньшей мере трехдневным периодом отмывки между введениями доз. В течение каждого периода пребываня субъектов в клиническом центре контролировали потребление ими пищи и жидкости.
Фармакокинетические параметры и для не приведенных к дозе, и для приведенных к дозе AUC0-1, AUC0-4, AUC0-12 и AUC0-24 для силденафила и его метаболита, Ν-десметилсилденафила, рассчитывали. с применением программного обеспечения WinNonlin® версии 5.0.1, разработанного специально для анализа фармакокинетических данных. Использовали модель расчетов Model 200 в программе WinNonlin® для путей введения лекарственных средств, отличных от введения в кровоток.
Для каждого частного фармакокинетического параметра AUC0-1, AUC0-4, AUC0-12 и AUC0-24 выполняли дисперсионный анализ (ANOVA) с применением программного обеспечения SAS®. При сравнении эффектов исследуемого и эталонного препаратов использовали модель ANOVA, включающую факторы для последовательности введения препаратов, субъектов внутри последовательности, периодов и препаратов. Для большинства субъектов расчет AUC0-24 не представлялся возможным в связи с тем, что значения концентрации во временной точке 24 часа были ниже предела количественного определения.
РЕЗУЛЬТАТЫ АНАЛИЗА ФАРМАКОКИНЕТИЧЕСКИХ ПАРАМЕТРОВ И ИХ ОБСУЖДЕНИЕ
В таблице 1 кратко представлены фармакокинетические параметры для каждого препарата силденафила, полученные с применением некомпартментного анализа.
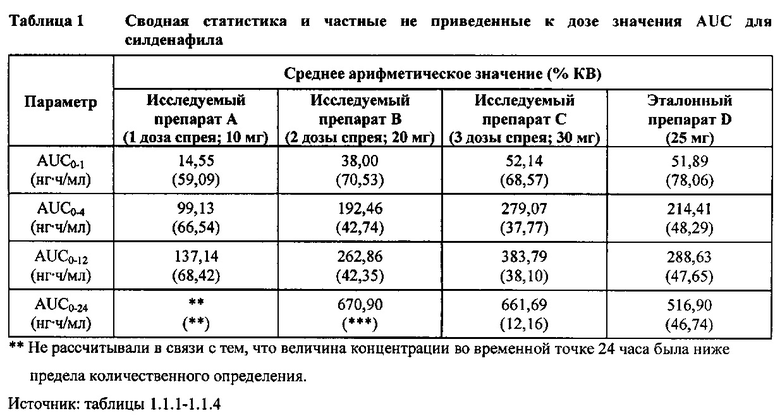
В общем случае, при пероральном введении здоровым взрослым мужчинам 10, 20 и 30 мг силденафила в составе исследуемых препаратов в форме спрея частная длительность экспозиции силденафила, как было оценено по не приведенным к дозе AUC0-1, AUC0-4 и AUC0-12, возрастала пропорционально увеличению дозы.
Представленные в таблице 1 не приведенные к дозе частные AUC для силденафила в составе спрея для перорального введения и эталонной таблетки показывают, что частные AUC0-1, AUC0-4, AUC0-12 и AUC0-24 находились в соответствии с разностью AUC0-t и AUC0-∞ (таблица 7.2.6.1-1), обнаруженной между спреем для перорального введения и эталонной таблеткой, причем, 2 дозы спрея, содержащие 20 мг силденафила, и эталонная таблетка 25 мг характеризовались по существу одними и теми же значениями AUC.
В таблицах 2, 3 и 4 кратко представлены результаты статистического анализа, проведенного для интервалов частных AUC для силденафила без приведения к дозе.
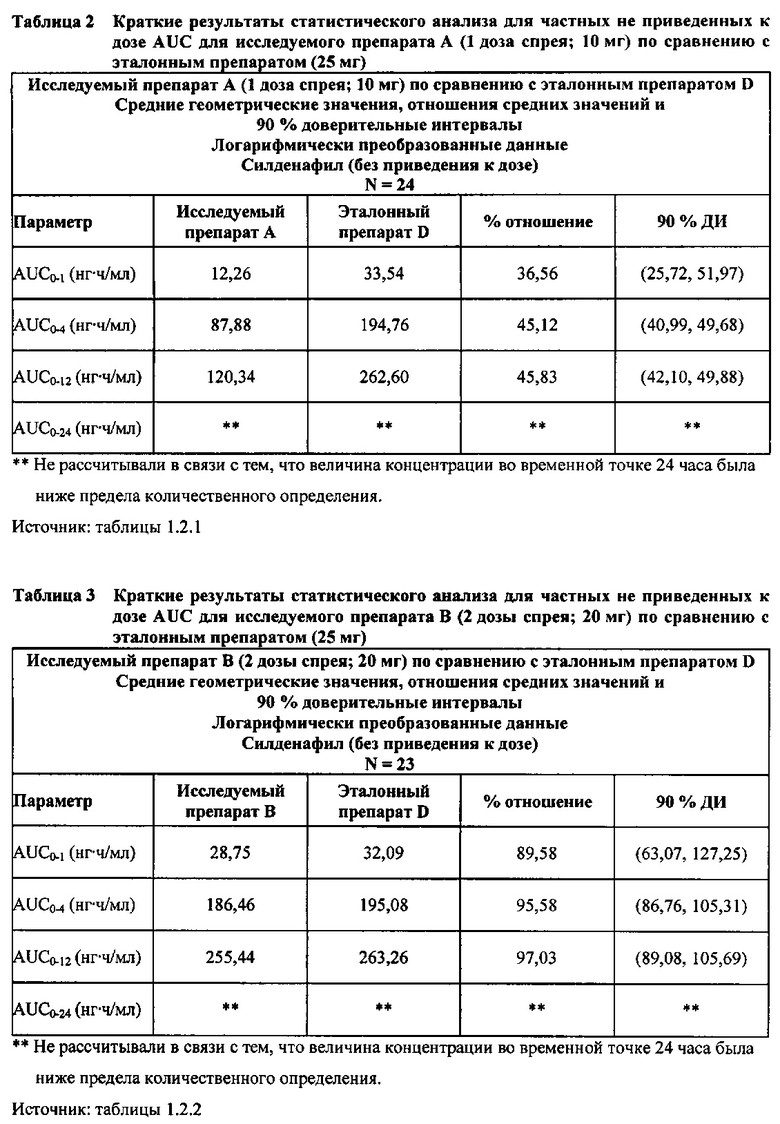
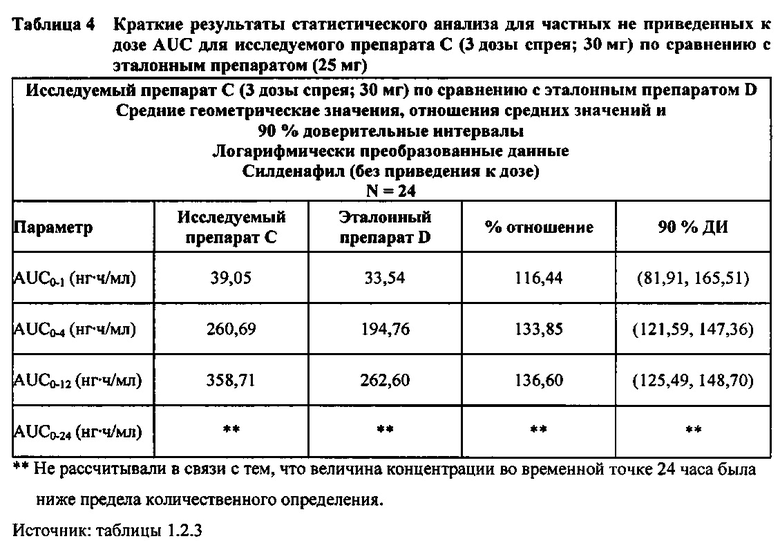
В случае, когда ФК параметры AUC0-1, AUC0-4, AUC0-12 для исследуемых препаратов А (10 мг), В (20 мг) и С (30 мг) в форме спрея для перорального введения не были приведены к дозе (25 мг) эталонной таблетки D, статистические результаты для частной длительности экспозиции силденафила во временных точках от 0 до 1, 4, 12 и 24 часов в составе исследуемых препаратов в форме спрея для перорального введения по сравнению с результатами для эталонного препарата в форме таблетки демонстрировали, что:
- существовали статистически значимые различия для частных не приведенных к дозе AUC0-1, AUC0-4, AUC0-12 в случае исследуемого препарата А силденафила (1 доза спрея, доза силденафила 10 мг) по сравнению с эталонным препаратом D в форме таблетки 25 мг. Длительность экспозиции силденафила, рассчитанная для временной точки один час (AUC0-1) была приблизительно на 63% ниже, в то время как AUC, рассчитанные для временных точек 4 и 12 часов были приблизительно на 55% выше, соответственно, чем аналогичные значения для эталонного препарата в форме таблетки в соответствии с разностью доз (см. таблицу 2).
- длительность экспозиции силденафила, рассчитанная для временных точек 4 и 12 часов (для не приведенных к дозе AUC0-4, AUC0-12), в составе исследуемого препарата В (2 дозы спрея, доза силденафила 20 мг) была сопоставима с таковой для эталонного препарата D в форме таблетки 25 мг, поскольку эти значения находились в пределах 90% ДИ. Однако частная AUC, рассчитанная для временной точки 1 час, не попадала в 90% ДИ, хотя точечная оценка была лишь немного ниже, приблизительно на 10% (как представлено в таблице 3).
- для исследуемого препарата С в форме спрея для перорального введения, содержащего дозу 30 мг силденафила, не приведенные к дозе частные AUC0-1, AUC0-4, AUC0-12 для силденафила отражали, в общем, разницу в дозе с эталонным препаратом D в форме таблетки 25 мг (и были приблизительно на 16, 34 и 37% выше, соответственно), и все значения, рассчитанные для частных AUC во временных точках от 0 до 1, 4 и 12 часов, не попадали в 90% ДИ (см. таблицу 4).
Силденафил (с приведением к дозе)
В таблице 5 кратко представлены приведенные к дозе фармакокинетические параметры для каждого препарата силденафила, полученные с применением некомпартментного анализа.
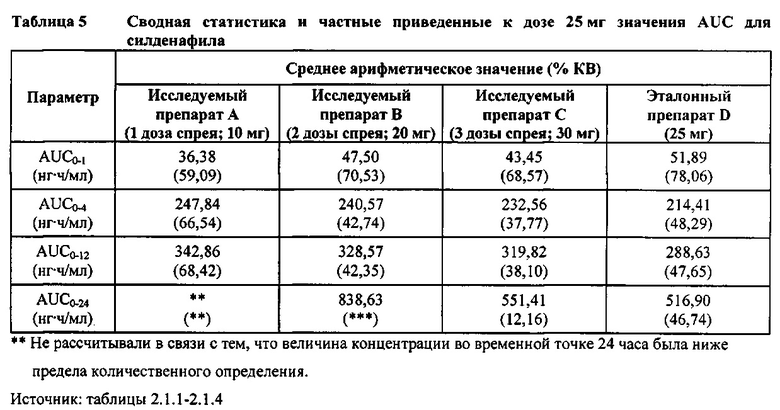
Представленные в таблице 5 результаты статистического анализа для приведенных к дозе частных ФК параметров AUC0-1, AUC0-4, AUC0-12 и AUC0-24 демонстрируют, что:
- приведенная к дозе AUC0-1 для 10, 20 и 30 мг силденафила в составе спрея для перорального введения была, в общем, сопоставима с таковой для 25 мг силденафила в составе эталонной таблетки.
- в общем, частные AUC, рассчитанные в интервале от 0 до 12 часов для всех 3 исследуемых препаратов силденафила в форме спрея для перорального введения, немного превышали таковые для эталонного препарата в форме таблетки 25 мг (на приблизительно 11-19%, соответственно).
- эти результаты указывают на то, что частная длительность экспозиции силденафила, рассчитанная для временной точки один час (AUC0-1), была меньше, а для следующей точки 4 часа (AUC0-4) была сопоставимой, но затем была больше при расчете для точки 12 часов (AUC0-12) для всех исследуемых препаратов в форме спрея для перорального введения по сравнению с эталонным препаратом в форме таблетки для перорального введения.
В таблицах 6, 7 и 8 кратко представлены результаты анализа, проведенного для фармакокинетических параметров для силденафила с приведением к дозе.
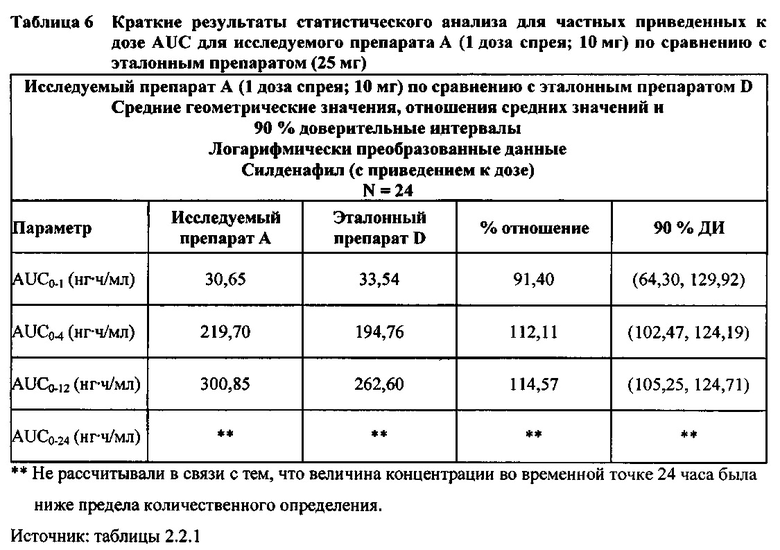
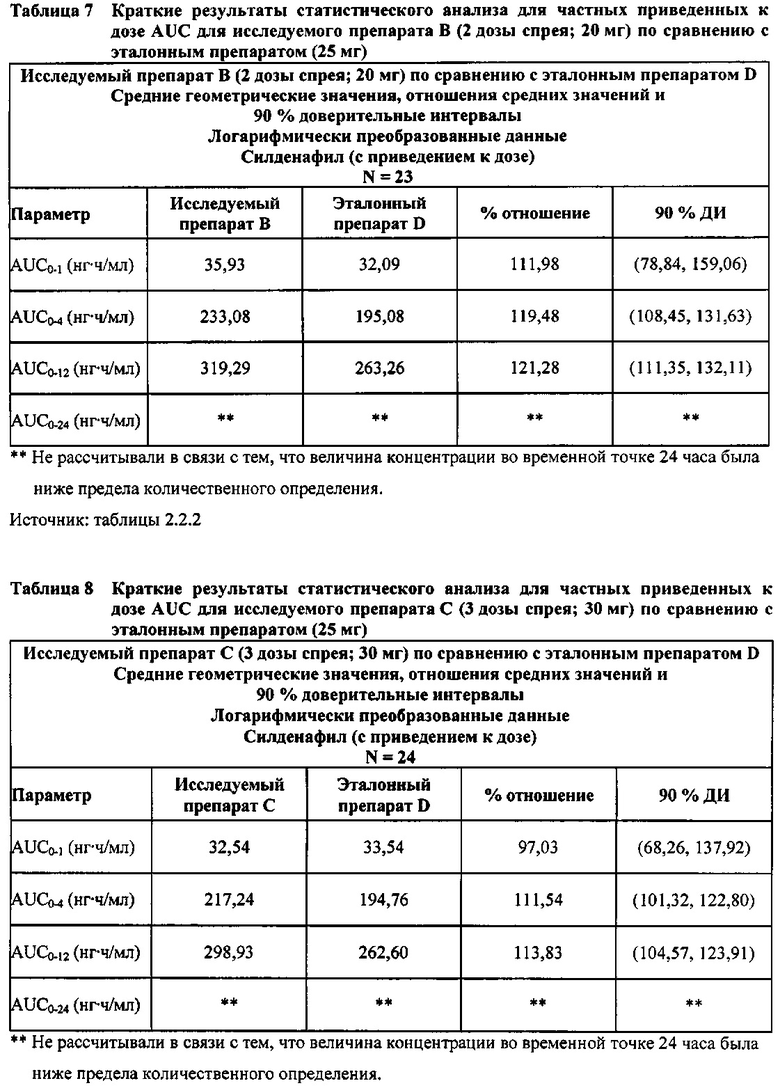
В случае, когда частные ФК параметры AUC0-1, AUC0-4, AUC0-12 для силденафила были приведены к дозе 25 мг эталонного состава в форме таблетки, частная системная экспозиция силденафила в составе препаратов в форме спрея для перорального введения по сравнению с таковой для эталонного состава в форме таблетки 25 мг характеризовалась тем, что (см. таблицы 6-8):
- значения приведенных к дозе AUC, рассчитанные во временных точках от 0 до 4 и до 12 часов для доз 10 мг и 30 мг силденафила в составе препаратов в форме спрея для перорального введения, были, в общем, сопоставимы с таковыми для эталонного препарата в форме таблетки 25 мг и находились в пределах 90% ДИ. Значения AUC для дозы 20 мг в составе исследуемого препарата В были немного выше по сравнению с таковыми для эталонного препарата в форме таблетки 25 мг и не попадали в 90% ДИ.
- значения, полученные для частных AUC, рассчитанных в интервале от 0 до 1 часа, в общем, не попадали в 90% ДИ; этот факт указывал на то, что частная длительность экспозиции силденафила, рассчитанная для временной точки 1 час для всех исследуемых препаратов в форме спрея для перорального введения, не была сопоставимой с аналогичным параметром для эталонного препарата в форме таблетки 25 мг.
- как указывают точечные оценки % исследуемый препарат/эталонный препарат, частная системная экспозиция силденафила, соответствующая AUC0-1, AUC0-4, AUC0-12 для дозы 20 мг в составе спрея для перорального введения (исследуемого препарата В), обеспечивала немного большую системную биодоступность по сравнению с эталонной таблеткой 25 мг для перорального введения (112% по сравнению с 119,5% по сравнению с 121,3%, соответственно).
Частные AUC для Ν-десметилсилденафила (без приведения к дозе)
В таблице 9 кратко представлены частные AUC для Ν-десметилсилденафила для каждого препарата, полученные с применением некомпартментного анализа.
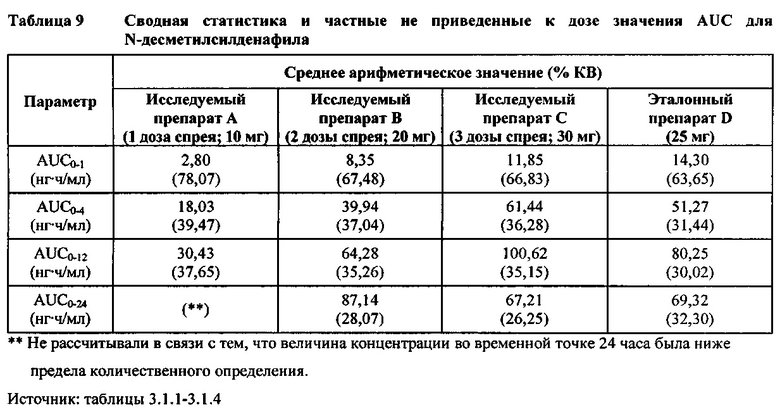
Представленные в таблице 9 результаты статистического анализа для не приведенных к дозе частных параметров AUC0-1, AUC0-4, AUC0-12 и AUC0-24 для метаболита, Ν-десметилсилденафила, демонстрируют, что:
- в общем, для всех доз силденафила в составе препарата в форме спрея для перорального введения длительность экспозиции Ν-десметилсилденафила, как было оценено по частным AUC, рассчитанным в интервалах от 0 до 4 и 12 часов, возрастала пропорционально увеличению дозы от 10 до 30 мг.
- в общем, разности, обнаруженные между частными AUC для Ν-десметилсилденафила, находились в соответствии с разницей доз в случаях спрея для перорального введения и эталонной таблетки 25 мг.
В таблицах 10, 11 и 12 кратко представлены результаты анализа, проведенного для частных параметров AUC для Ν-десметилсилденафила без приведения к дозе.
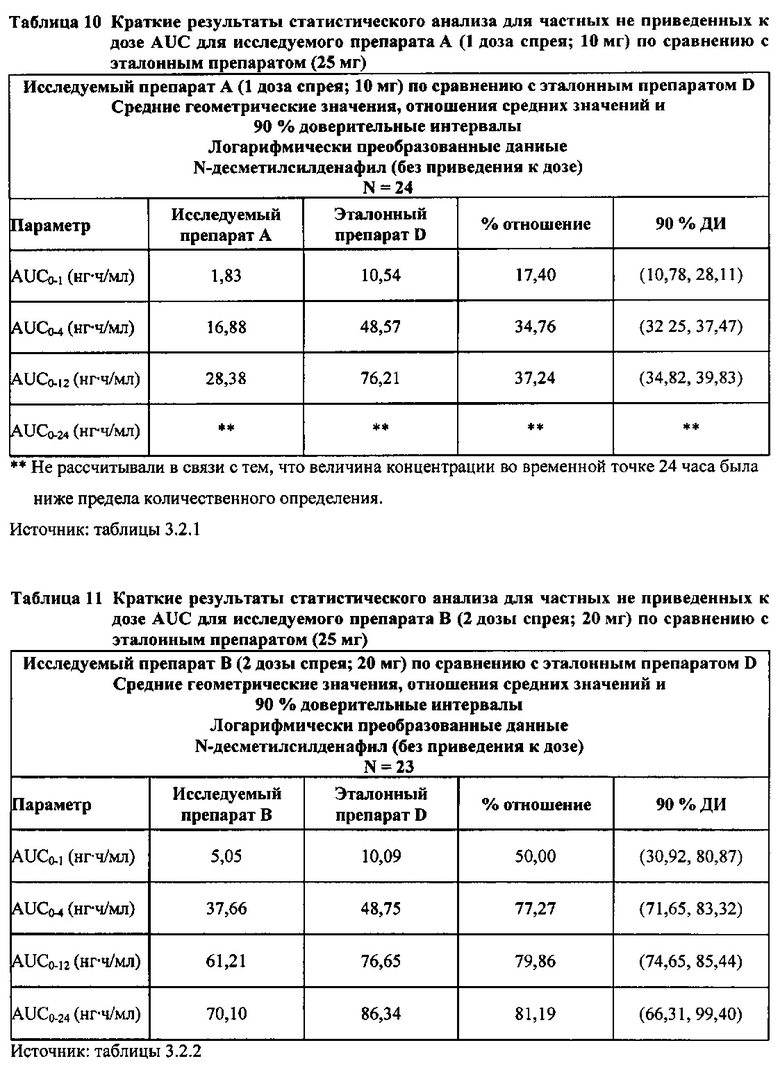
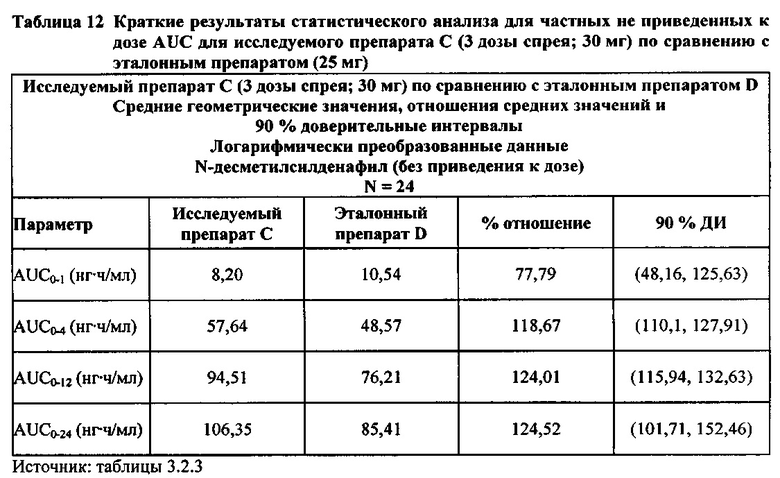
Не приведенные к дозе 25 мг эталонной таблетки частные AUC0-1, AUC0-4, AUC0-12 и AUC0-24 для Ν-десметилсилденафила, обеспечиваемые пероральным введением доз силденафила в форме спрея, по сравнению с эталонным составом в форме таблетки 25 мг характеризовались тем, что:
- точечные оценки для не приведенных к дозе частных AUC, рассчитанных для Ν-десметилсилденафила в интервале от 0 до 1 часа (AUC0-1) для вводимых перорально в составе спрея доз 10, 20 и 30 мг силденафила, были, в общем, ниже с точностью до разницы в дозах относительно эталонной таблетки 25 мг.
- Точечные оценки для частных AUC, рассчитанные для временных точек 4, 12 и 24 часа для вводимых перорально в составе спрея доз 10 мг и 20 мг, в целом находились в соответствии с разницей в мг дозы относительно эталонной таблетки 25 мг.
Частные AUC для Ν-десметилсилденафила (с приведением к дозе)
В таблице 13 кратко представлены приведенные к дозе фармакокинетические параметры для Ν-десметилсилденафила для каждого препарата, полученные с применением некомпартментного анализа.
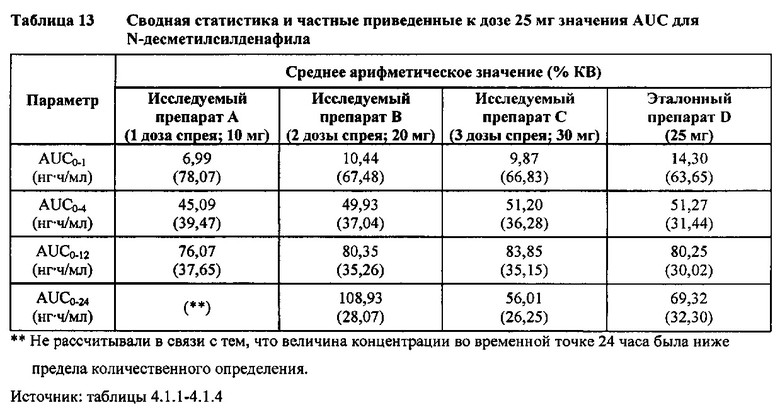
После приведения к дозе частные AUC0-1 для вводимых перорально в составе спрея доз 10, 20 и 30 мг оказались меньше по сравнению с вводимой перорально в составе таблетки дозой 25 мг.
Приведенные к дозе AUC, рассчитанные в интервалах от 0 до 4 и 12 часов для метаболита исследуемых препаратов А, В и С, были, в общем, сопоставимы, в то время как не наблюдали какой-либо конкретной динамики для AUC0-24 для всех исследуемых препаратов в форме спрея для перорального введения относительно эталонного препарата в форме таблетки.
В таблицах 14, 15 и 16 кратко представлены результаты анализа, проведенного для частных AUC для Ν-десметилсилденафила с приведением к дозе.
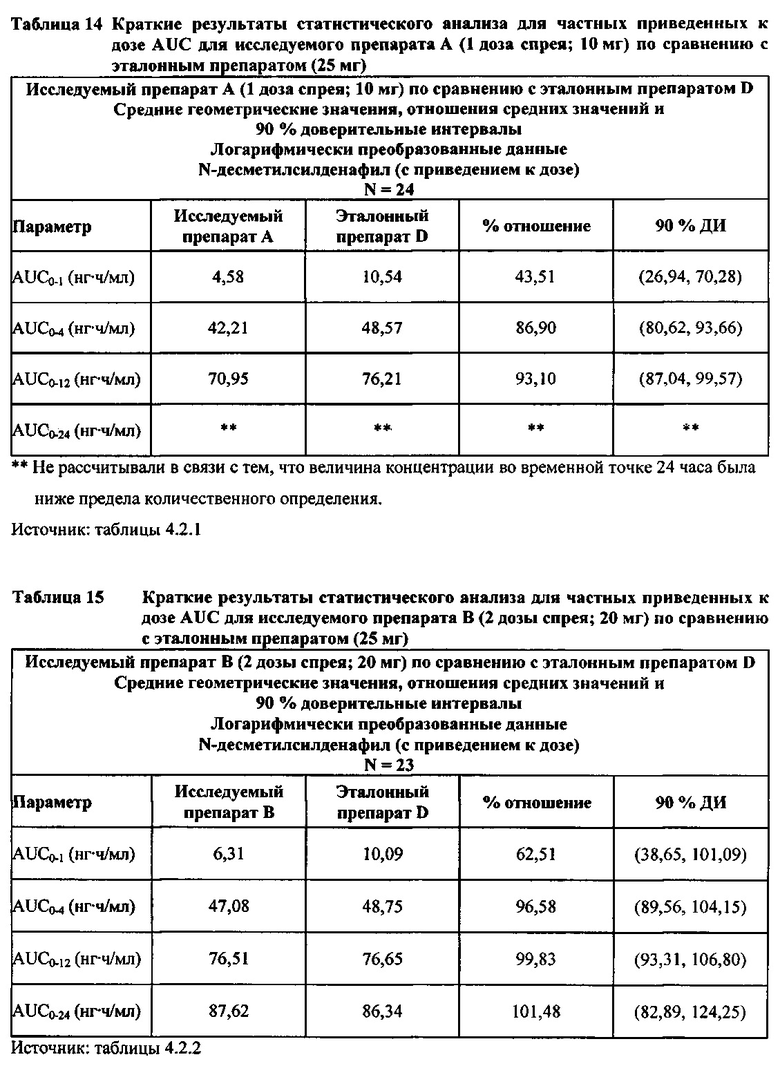
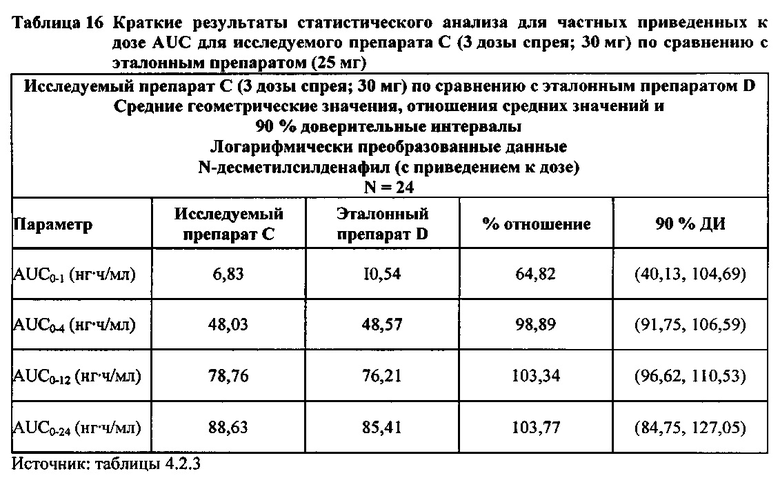
При сравнении приведенных к дозечастных AUC0-1, AUC0-4, AUC0-12 и AUC0-24 для Ν-десметилсилденафила для вводимых перорально в составе спрея доз результаты фармакокинетического и статистического анализа для Ν-десметилсилденафила демонстрировали, что:
- для 1 дозы спрея (доза силденафила 10 мг), 2 доз спрея (доза 20 мг) и 3 доз спрея (доза 30 мг) все значения AUC для метаболита, рассчитанные для временных точек 4 и 12 часов, находились, в общем, в пределах 90% ДИ, соответствующего критерию биоэквивалентности 80-125%, и, следовательно, их считали сопоставимыми с аналогичными значениями для эталонного препарата в форме таблетки 25 мг.
- значения, полученные для частных AUC, рассчитанных для временной точки один час, демонстрировали, что, в общем, для всех исследуемых препаратов в форме спрея для перорального введения значения для AUC0-1 были по существу меньше таковых для эталонного препарата в форме таблетки.
Частные AUC для соотношения метаболит/исходное соединение (соотношения Ν-десметилсилденафил/силденафил)
В таблице 17 кратко представлены фармакокинетические параметры для соотношения метаболит/исходное соединение (N-десметилсилденафил/силденафил) для каждого препарата.
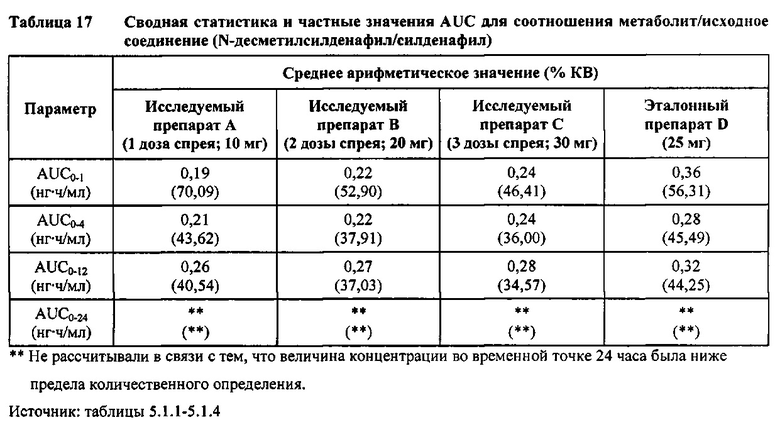
В таблицах 18, 19 и 20 кратко представлены результаты анализа, проведенного для фармакокинетических параметров для соотношения N-десметилсилденафил/силденафил.
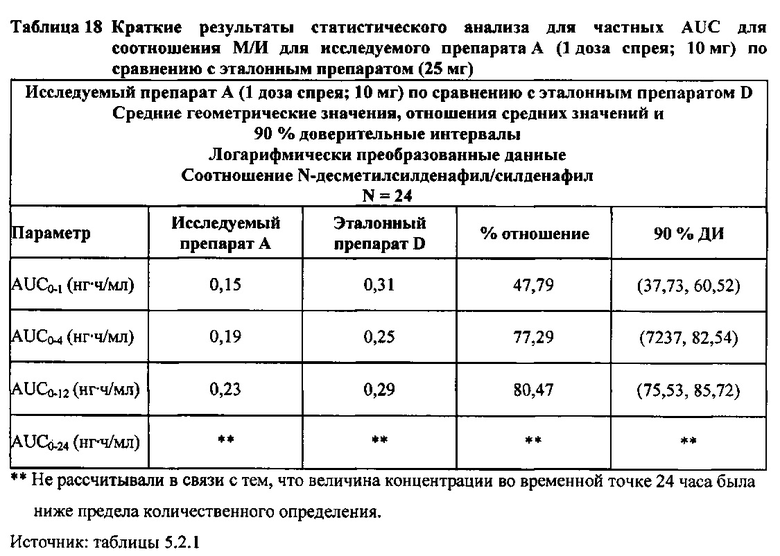
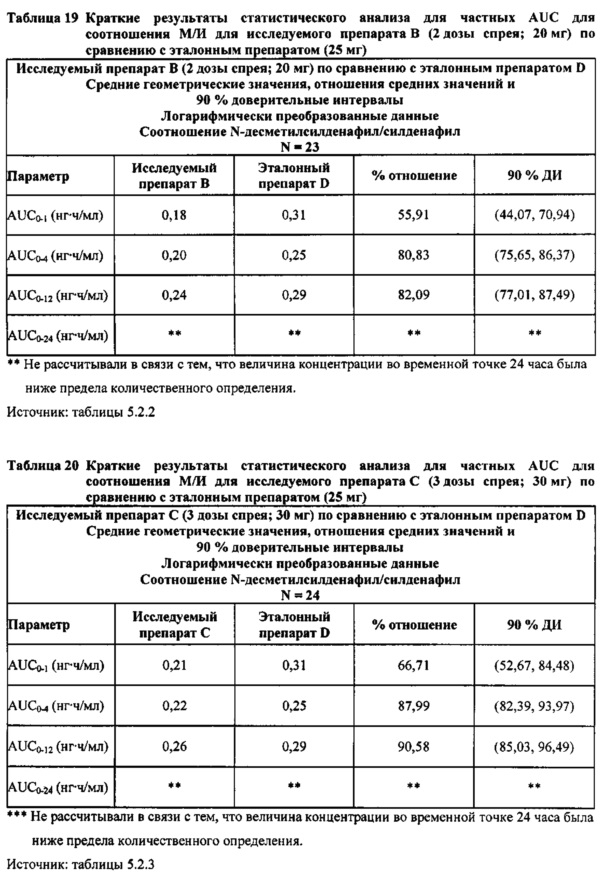
В целом, процесс образования метаболита, как было оценено по соотношению М/И для вводимых перорально в составе спрея доз 10, 20 и 30 мг, характеризовался тем, что наиболее существенное различие наблюдали в течение соответствующего AUC0-1 интервала. Эти различия также по существу не зависели от дозы исследуемого препарата (1 доза спрея, 2 дозы спрея и 3 дозы спрея), поскольку были обнаружены лишь минимальные различия между количеством доз спрея. По существу не было замечено различий в частных AUC для соотношения М/И в течение интервалов времени, соответствующих AUC0-4 и AUC0-12.
ОБСУЖДЕНИЕ
Оценка частных AUC указывает на то, что наиболее существенное различие между пероральным введением силденафила в составе спрея и в составе эталонной таблетки Виагра возникало в интервале, соответствующем AUC0-1. Далее не было обнаружено существенных различий в течение интервалов времени 0-4, 0-12 и 0-24 часа, соответствующих частным AUC, между исследуемым препаратом в форме спрея для перорального введения и эталонной таблеткой. Это было снова продемонстрировано при анализе параметров для соотношения метаболит/исходное соединение, когда наиболее существенное различие наблюдали в течение соответствующего AUC0-1 интервала. Данные различия также по существу не зависели от дозы исследуемого препарата (1 доза спрея, 2 дозы спрея и 3 дозы спрея).
ССЫЛКИ
Food and Drug Administration. Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products-General Considerations. Center for Drug Evaluation and Research, March 2003.
Chow, S.C. and. J.P. Liu. Design and Analysis of Bioavailability and Bioequivalence Studies, Second Edition, Revised and Expanded. New York: Marcel Dekker, Inc., 2000.
Pharsight Corporation. WinNonlin® User's Guide. Pharsight Corporation, 1998-2005.
SAS Institute, Inc. SAS OnlineDoc® Version 9.1. Cary, NC: SAS Institute, Inc., 2002-2006.
Pharmacokinetics of sildenafil after single oral doses in healthy male subjects: absolute bioavailability, food effects and dose proportionality. Donald J. Nichols, Gary J. Muirhead, Jane A. Harness Br. J. Clin Pharmaco, Vol 53 supp. Pages: 5S-12S, Feb 2002.
Comparative human pharmacokinetics and metabolism of single-dose oral and intravenous sildenafil authors: Gary J. Muirhead, David J. Ranee, Br. J. Clin Pharmaco, Vol 53 supp. Pages: 13S-20S, Mar 2002.
Таким образом, имея настоящее подробное описание предпочтительных вариантов реализации настоящего изобретения, следует понимать, что определенное выше изобретение не ограничивается изложенными конкретными подробностями, поскольку возможно большое количество его очевидных вариаций, не выходящих за рамки сущности и объема настоящего изобретения.
Следует считать, что термины "включают", "включает" или "включая" сопровождаются словами "без ограничения", если не указано иное.
Полное содержание каждого патента, заявки на патент и публикации, цитируемых или описанных в настоящей заявке, включено в настоящее описание посредством ссылки, как если бы для каждого индивидуального патента, заявки на патент или публикации было конкретно и индивидуально указано, что его содержание включено в настоящее описание посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТВЕРДЫЕ ЛЕКАРСТВЕННЫЕ ФОРМЫ ОНДАНСЕТРОНА С ПРОЛОНГИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ ДЛЯ ЛЕЧЕНИЯ СИМПТОМОВ ТОШНОТЫ, РВОТЫ И ДИАРЕИ | 2015 |
|
RU2706708C2 |
| КИШЕЧНОРАСТВОРИМЫЕ ЛЕКАРСТВЕННЫЕ ФОРМЫ ДЛЯ ПЕРОРАЛЬНОГО ПРИМЕНЕНИЯ С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2018 |
|
RU2812901C2 |
| ТРАНСМУКОЗАЛЬНОЕ ВВЕДЕНИЕ 2,3-ДИМЕТОКСИ-5-МЕТИЛ-6-(10-ГИДРОКСИДЕЦИЛ)-1,4-БЕНЗОХИНОНА | 2007 |
|
RU2429830C2 |
| СОСТАВЫ, УСТРОЙСТВА И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ АЛКОГОЛЬНОЙ ЗАВИСИМОСТИ | 2017 |
|
RU2767062C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ И СПОСОБЫ НА ОСНОВЕ ЭСТРАДИОЛА ДЛЯ ИНТРАВАГИНАЛЬНОГО ВВЕДЕНИЯ | 2014 |
|
RU2713888C2 |
| НИЗКОДОЗОВАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ИЗОТРЕТИНОИНА ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ | 2015 |
|
RU2707753C2 |
| ЛЕКАРСТВЕННЫЕ СРЕДСТВА, СОДЕРЖАЩИЕ ДИАЦЕРЕИН, И СПОСОБЫ СНИЖЕНИЯ УРОВНЕЙ МОЧЕВОЙ КИСЛОТЫ В КРОВИ С ЕГО ИСПОЛЬЗОВАНИЕМ | 2015 |
|
RU2690372C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ чГР, ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ | 2007 |
|
RU2493868C2 |
| Композиции, содержащие триптановые соединения | 2010 |
|
RU2710372C2 |
| Твердые композиции на основе бетагистина для перорального применения с непульсирующим пролонгированным высвобождением | 2018 |
|
RU2774644C2 |
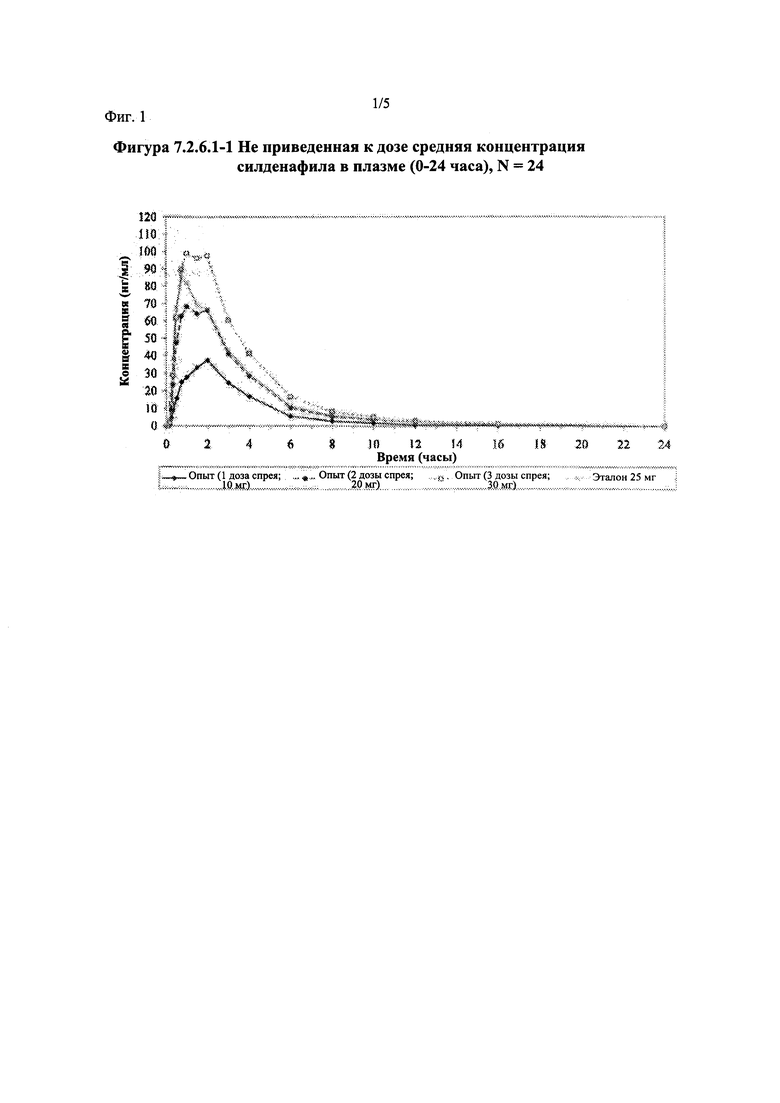
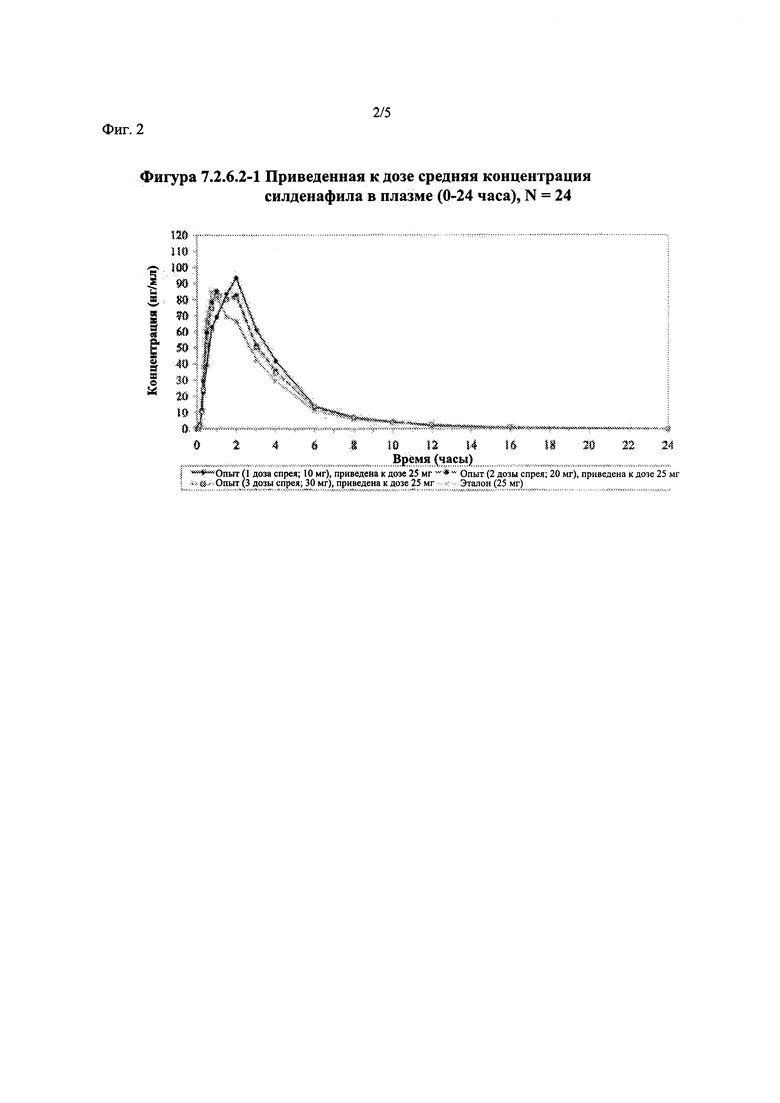
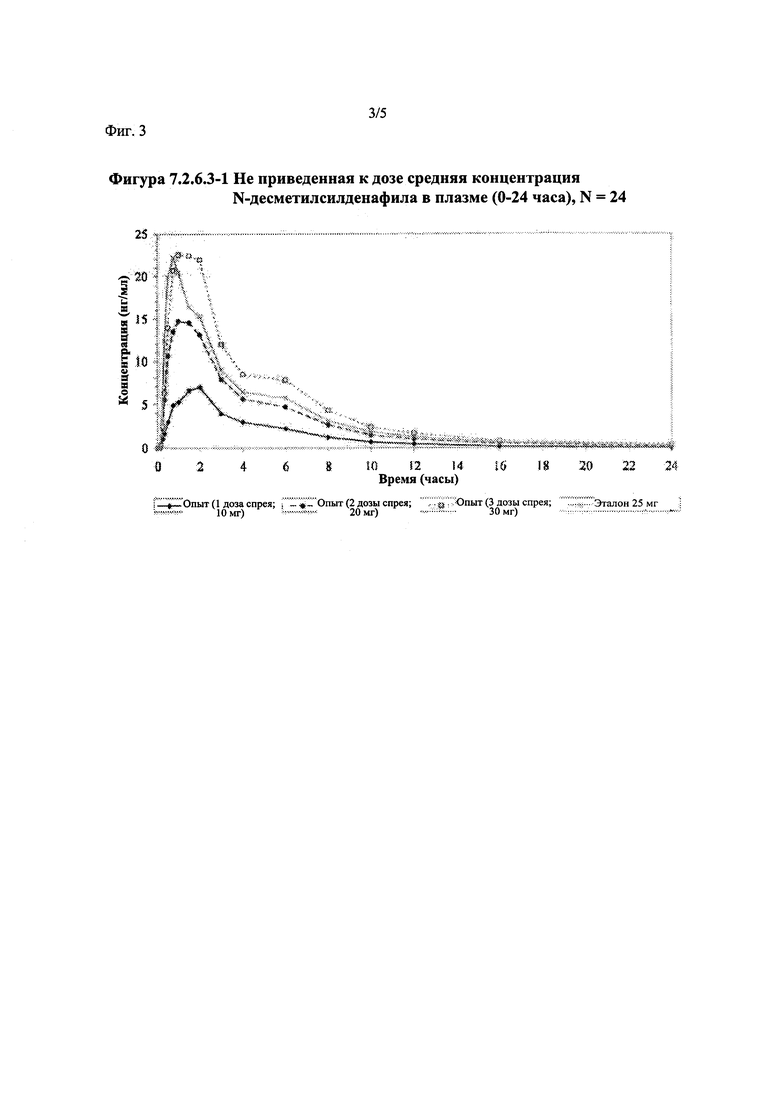
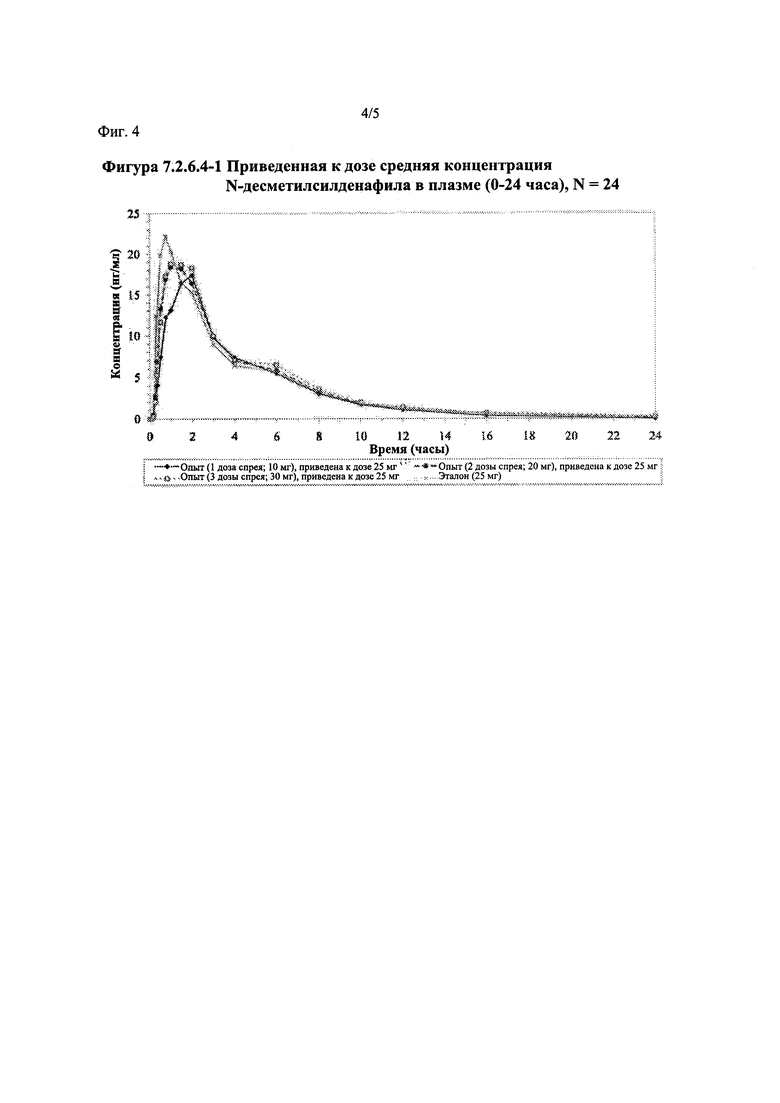
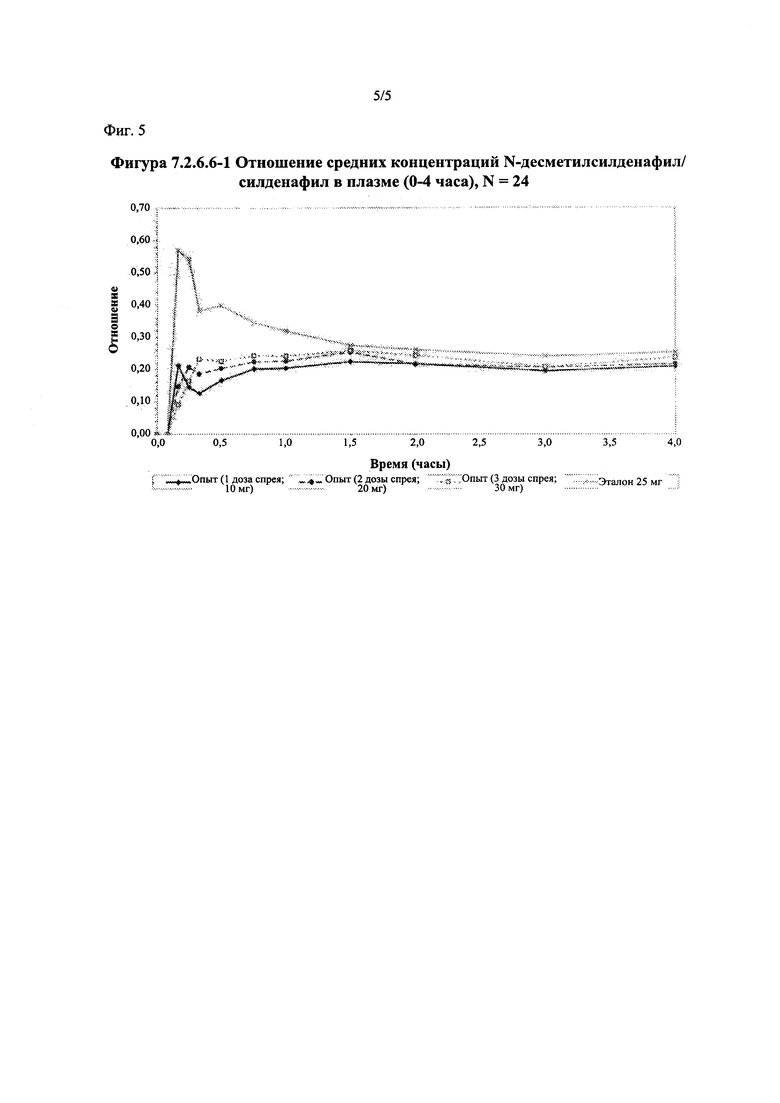
Настоящая группа изобретений относится к области фармацевтики. Описан биодоступный состав в форме спрея для перорального введения для лечения легочной артериальной гипертензии и/или индуцированной селективными ингибиторами обратного захвата серотонина (СИОЗС-индуцированной) сексуальной дисфункции. Указанный состав содержит силденафил в форме его свободного основания или его фармацевтически приемлемую соль, при этом рН состава составляет от 1,5 до 2,4. Описан также способ лечения легочной артериальной гипертензии и/или СИОЗС-индуцированной сексуальной дисфункции. Изобретение обеспечивает увеличение биодоступности активного вещества. 2 н. и 24 з.п. ф-лы, 20 табл., 4 пр., 5 ил.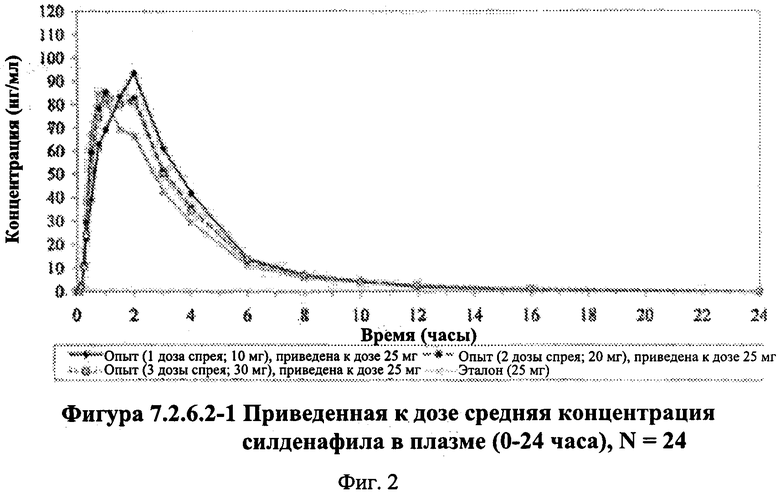
1. Биодоступный состав в форме спрея для перорального введения для лечения легочной артериальной гипертензии и/или индуцированной селективными ингибиторами обратного захвата серотонина (СИОЗС-индуцированной) сексуальной дисфункции, содержащий силденафил в форме его свободного основания или его фармацевтически приемлемую соль, при этом рН состава составляет от 1,5 до 2,4.
2. Состав в форме спрея для перорального введения по п. 1, отличающийся тем, что силденафил в форме его свободного основания присутствует в количестве от 7 до 9% мас./об. из расчета на общее количество состава.
3. Состав в форме спрея для перорального введения по п. 2, отличающийся тем, что силденафил в форме его свободного основания присутствует в количестве 8,31% мас./об. из расчета на общее количество состава.
4. Состав в форме спрея для перорального введения по п. 1, отличающийся тем, что указанная соль силденафила присутствует в количестве от 10 до 12% мас./об. из расчета на общее количество состава.
5. Состав в форме спрея для перорального введения по п. 4, отличающийся тем, что указанная соль силденафила присутствует в количестве 11,67% мас./об. из расчета на общее количество состава.
6. Состав в форме спрея для перорального введения по п. 4 или 5, отличающийся тем, что указанная соль силденафила представляет собой силденафила цитрат.
7. Состав в форме спрея для перорального введения по п. 1, отличающийся тем, что указанный состав содержит полярный растворитель.
8. Состав в форме спрея для перорального введения по п. 7, отличающийся тем, что указанный полярный растворитель содержит пропиленгликоль и этиловый спирт.
9. Состав в форме спрея для перорального введения по п. 8, отличающийся тем, что соотношение пропиленгликоль : этиловый спирт составляет 70:30% об./об., 65:35% об./об., 62,5:37,5% об./об., 60:40% об./об., 55:45% об./об. или 50:50% об./об.
10. Состав в форме спрея для перорального введения по п. 1, отличающийся тем, что указанный состав содержит один или более регуляторов рН, выбранных из списка, включающего подкисляющие агенты, подщелачивающие агенты или их комбинации.
11. Состав в форме спрея для перорального введения по п. 10, отличающийся тем, что указанный подкисляющий агент представляет собой соляную кислоту (HCl).
12. Состав в форме спрея для перорального введения по п. 11, отличающийся тем, что HCl присутствует в количестве 10% об./об. из расчета на общее количество состава.
13. Состав в форме спрея для перорального введения по п. 10, отличающийся тем, что указанный подщелачивающий агент представляет собой гидроксид натрия (NaOH).
14. Состав в форме спрея для перорального введения по п. 13, отличающийся тем, что NaOH присутствует в количестве 2,1% об./об. из расчета на общее количество состава.
15. Состав в форме спрея для перорального введения по п. 1, отличающийся тем, что указанный состав содержит агент, маскирующий вкус, и/или ароматизатор и/или подсластитель.
16. Состав в форме спрея для перорального введения по п. 15, отличающийся тем, что указанный агент, маскирующий вкус, представляет собой мяту, и/или указанный ароматизатор представляет собой фруктовый и/или шоколадный ароматизатор, и/или указанный подсластитель представляет собой сукралозу.
17. Состав в форме спрея для перорального введения по п. 1, отличающийся тем, что указанный состав содержит одно или более фармацевтически приемлемых вспомогательных веществ, носителей или их комбинацию.
18. Состав в форме спрея для перорального введения по п. 1, отличающийся тем, что рН указанного состава составляет 2,2.
19. Состав в форме спрея для перорального введения по п. 1, отличающийся тем, что количество силденафила, вводимое за одно распыление, составляет от 10 до 30 мг.
20. Способ лечения легочной артериальной гипертензии и/или СИОЗС-индуцированной сексуальной дисфункции, включающий введение пациенту состава в форме спрея для перорального введения, содержащего силденафил в форме его свободного основания или его фармацевтически приемлемую соль, при этом рН состава составляет от 1,5 до 2,4.
21. Способ по п. 20, отличающийся тем, что силденафил в форме его свободного основания присутствует в количестве от 7 до 9% мас./об. из расчета на общее количество состава, и/или указанная соль силденафила присутствует в количестве от 10 до 12% мас./об. из расчета на общее количество состава.
22. Способ по п. 20, отличающийся тем, что указанная соль силденафила представляет собой силденафила цитрат.
23. Способ по п. 20, отличающийся тем, что указанный состав содержит полярный растворитель.
24. Способ по п. 20, отличающийся тем, что указанный состав содержит один или более регуляторов рН, выбранных из списка, включающего подкисляющие агенты, подщелачивающие агенты или их комбинации.
25. Способ по п. 20, отличающийся тем, что количество силденафила, вводимое за одно распыление, составляет от 10 до 30 мг.
26. Способ по п. 20, отличающийся тем, что указанный состав содержит агент, маскирующий вкус, и/или ароматизатор, и/или подсластитель.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| ВАГОННЫЙ РАСПРЕДЕЛИТЕЛЬНЫЙ КРАН ДЛЯ АВТОМАТИЧЕСКОГО ВОЗДУШНОГО ТОРМОЗА | 1928 |
|
SU10692A1 |
| Телефон | 1923 |
|
SU6242A1 |

