РОДСТВЕННЫЕ ЗАЯВКИ
Данная заявка связана с предварительной заявкой на патент Индии IN 201721025857, поданной 20-го июля 2017 г., включенной в данный документ во всей своей полноте.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к твердой композиции на основе бетагистина для перорального применения с непульсирующим пролонгированным высвобождением. В частности, к твердой композиции для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов, содержащей некоторое количество бетагистина или его фармацевтически приемлемой соли, эквивалентное 48 мг бетагистина дигидрохлорида. Изобретение также относится к способу ее получения и применения в терапии, особенно в лечении заболевания или состояния вестибулярного аппарата, более конкретно, в лечении болезни Меньера.
УРОВЕНЬ ТЕХНИКИ
Бетагистин является международным непатентованным названием (INN) N-метил-2-(пиридин-2-ил)этанамина, которому соответствует номер CAS 5638-76-6. Структура бетагистина соответствует формуле (I):
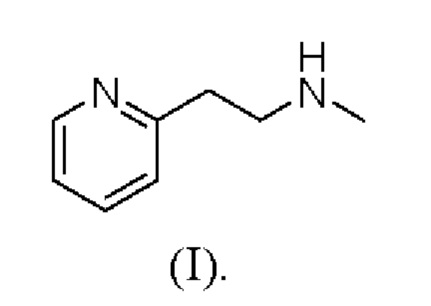
Бетагистин представляет собой сосудорасширяющее средство, и его обычно применяют в виде его дигидрохлоридной соли. После перорального введения бетагистина дигидрохлорид почти полностью всасывается во всех отделах желудочно-кишечного тракта, а после всасывания бетагистина дигидрохлорид быстро и почти полностью подвергается метаболическому превращению в свой метаболит 2-пиридилуксусную кислоту (2-РАА).
Первая лицензия на продажу бетагистина дигидрохлорида в Европе была выдана в 1970 г. для компенсирования или облегчения симптомов головокружения, шума в ушах, потери слуха и тошноты, ассоциированных с болезнью Меньера. Лечение болезни Меньера представляет собой долговременное лечение, которое охватывает первую начальную фазу перорального лечения с последующей второй фазой поддержания. В частности, первая начальная фаза лечения включает пероральное введение 8-16 мг бетагистина дигидрохлорида три раза в день; при этом вторая фаза поддерживающего лечения включает пероральное введение 24-48 мг бетагистина дигидрохлорида без превышения общей суточной дозы 48 мг.
Бетагистина дигидрохлорид является коммерчески доступным в форме таблеток с немедленным высвобождением с дозировкой 8 мг, 16 мг или 24 мг или в форме раствора для перорального применения с дозировкой 8 мг/мл. Следовательно, указанный расчет дозы для лечения болезни Меньера предусматривает от двух до четырех приемов в сутки перорально в зависимости от лекарственной формы. Таким образом, наиболее важным фактором, который снижает эффективность лечения болезни Меньера с помощью бетагистина дигидрохлорида, в течение последних 40 лет является соблюдение пациентом режима приема лекарственного средства.
Следовательно, были сделаны различные попытки улучшения соблюдения режима приема лекарственного средства путем уменьшения частоты приемов в сутки при поддержании уровней активного ингредиента в плазме в течение длительного периода времени.
В уровне техники является общеизвестным, что применение композиций с матричной основой может определять поглощение активных ингредиентов и, следовательно, их высвобождение из композиции с матричной основой. В частности, в уровне техники были раскрыты цельные композиции с матричной основой с пролонгированным высвобождением, содержащие от 12 мг до 48 мг бетагистина гидрохлорида. В частности, Kovshel A. Yu. et al. раскрывает композиции с матричной основой с замедленным высвобождением, содержащие 48 мг бетагистина дигидрохлорида. Такие композиции преимущественно основаны на матрице, образованной повидоном и поливинилацетатом, в комбинации с микрокристаллической целлюлозой и гидрофосфатом кальция в качестве вспомогательных веществ. После введения единичной дозы таких композиций с замедленным высвобождением бетагистина гидрохлорид высвобождается из композиции в течение 12 часов. К сожалению, преждевременное и чрезмерное высвобождение активного ингредиента из таких композиций (т.е. эффект сброса дозы) является неизбежным. В частности, приблизительно 40% бетагистина дигидрохлорида высвобождалось из композиции через 1 час после введения и приблизительно 60% - через 2 часа. Данный эффект сброса дозы может обуславливать значительное увеличение концентрации активного ингредиента в организме, что вызывает колебания его концентрации в плазме и, следовательно, негативные эффекты, связанные с недостаточной и/или чрезмерной дозировкой. Таким образом, введение данных композиций не обеспечивает поддержание высвобождения в течение достаточно длительного времени, чтобы их вводили один раз в сутки, а это означает, что все еще необходимы два или более приемов таких композиций для обеспечения терапевтически эффективного количества бетагистина дигидрохлорида на протяжении 24 часов (см. Kovshel A. Yu. et al, «sustained release betahistine tablets: elaboration of their composition and technology)), Pharmacy, 2014, vol. 6, pp. 40).
В этом отношении в РСТ-заявке на патент WO 2014001267 раскрывается применение композиций с матричной основой с замедленным высвобождением, пригодных для контроля высвобождения таких активных ингредиентов, которые характеризуются низкой растворимостью и/или не являются гигроскопичными. Однако возникают значительные сложности в отношении контроля высвобождения гигроскопических активных ингредиентов, которые в то же время характеризуются высокой растворимостью. В РСТ-заявке на патент WO 2014001267 обращается внимание на то, что это относится к бетагистина дигидрохлориду, который можно считать высокорастворимым в воде (растворимость в воде составляет 49,3 мг/мл), а также гигроскопическим активным ингредиентом.
Кроме того, в WO 2014001267 также указывается, что дополнительные сложности возникают при модификации профиля высвобождения такого активного ингредиента вследствие его кислотных свойств. А именно, высокая кислотность бетагистина может приводить к распаду и/или расщеплению вспомогательных веществ или носителей в составе и затем может обуславливать модифицирование (изменение) высвобождения активного ингредиента.
Подводя итог, в РСТ-заявке на патент WO 2014001267 раскрывается, что вследствие высокой растворимости, гигроскопичности и кислотных свойств бетагистина, практически невозможно контролировать его высвобождение исключительно путем применения стандартного состава с матричной основой с непульсирующим замедленным высвобождением, определенного выше. Следовательно, в WO 2014001267 предусматривается более целесообразный подход к продлению высвобождения бетагистина дигидрохлорида и, следовательно, уменьшению частоты приемов дозы лекарственного средства. Данный подход ориентирован на применение композиции в виде множества частиц с матричной основой с пульсирующим пролонгированным высвобождением, которая обеспечивает сначала немедленное высвобождение первой порции активного ингредиента сразу же после приема лекарственного средства и замедленное высвобождение второй порции активного ингредиента. По сути, такие композиции с пульсирующим высвобождением симулируют (имитируют) прием двух лекарственных форм в сутки вместо обеспечения отдельной лекарственной формы с непульсирующим пролонгированным высвобождением активных ингредиентов.
В частности, в РСТ-заявке на патент WO 2014001267 раскрывается композиция типа ядро-оболочка с пульсирующим пролонгированным высвобождением, содержащая 24 мг бетагистина дигидрохлорида. Композиция предусматривает ядро с замедленным высвобождением, содержащее 12 мг бетагистина дигидрохлорида, и внешнюю оболочку с немедленным высвобождением, содержащую оставшееся количество активного ингредиента, составляющее 12 мг.
Однако, опять-таки вследствие высокой растворимости и гигроскопичности активного ингредиента, все так же трудно обеспечить достаточное разделение во времени первого импульса немедленного высвобождения бетагистина (из оболочки) и второго импульса замедленного высвобождения бетагистина (из ядра) исключительно путем применения ядра с матричной основой с замедленным высвобождением. Следовательно, обычно требуется промежуточное покрытие в виде пленки между ядром и оболочкой. В связи с этим данные композиции все так же обуславливают колебания концентрации активного ингредиента в плазме, и, следовательно, все так же сохраняются негативные эффекты, ассоциированные с недостаточной и/или чрезмерной дозировкой.
Поэтому, исходя из сведений, известных в данной области, пришли к выводу, что давно существует потребность в обеспечении устойчивых композиций для перорального применения с пролонгированным высвобождением в течение 24 часов, содержащих некоторое количество бетагистина или его фармацевтически приемлемой соли, эквивалентное 48 мг бетагистина дигидрохлорида, которые могут быть подходящими для введения один раз в сутки и не обладают побочными эффектами, ассоциированными с колебаниями концентрации активного ингредиента в плазме.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения, несмотря на противоречащие идеи из WO 2014001267, неожиданно обнаружили твердую композицию для перорального применения с непульсирующим пролонгированным высвобождением, содержащую некоторое количество бетагистина или его фармацевтически приемлемой соли, эквивалентное 48 мг бетагистина дигидрохлорида, характеризующуюся приемлемым профилем растворения, который может быть подходящим для поддержания терапевтически эффективной концентрации в плазме исходного лекарственного средства бетагистина и основного метаболита 2-РАА (абсолютная биологическая доступность приблизительно 99%) - в течение не более 24 часов.
Введение один раз в сутки твердой композиции для перорального применения по настоящему изобретению обеспечивает более низкий показатель максимальной концентрации в крови (Cmax) и более высокий показатель минимальной концентрации в крови (Cmin) бетагистина, а также метаболита бетагистина - 2-РАА - по сравнению с введением дважды в сутки твердой композиции для перорального применения с немедленным высвобождением, что уменьшает колебания его концентрации в плазме и, следовательно, исключает побочные эффекты недостаточной или чрезмерной дозировки, связанные с введением по меньшей мере двух отдельных лекарственных форм с немедленным высвобождением или отдельной лекарственной формы в виде множества частиц, содержащей лекарственные формы с немедленным и замедленным высвобождением.
Кроме того, авторы настоящего изобретения также неожиданно обнаружили, что общее воздействие лекарственного средства, достигаемое после введения один раз в сутки композиции, содержащей 48 мг бетагистина дигидрохлорида, по настоящему изобретению, является биоэквивалентным общему воздействию лекарственного средства, достигаемому после введения дважды в сутки композиции, содержащей 24 мг бетагистина дигидрохлорида, с немедленным высвобождением. Это означает, что композиция, содержащая 48 мг бетагистина дигидрохлорида по настоящему изобретению (раз в сутки), демонстрирует сравнительные фармакокинетические параметры биологической доступности (например, Cmax, AUC0-t, AUC0-∞, AUC0-τ,ss, Cmax, ss и Cmin, ss) бетагистина и метаболита бетагистина - 2-РАА - по сравнению с таковыми для состава на основе бетагистина дигидрохлорида с немедленным высвобождением (принимаемого дважды в сутки), как указано в документе Европейского агентства по лекарственным средствам (EMA) «Guideline on the pharmacokinetic and clinical evaluation of modified release dosage forms» (EMA/CPMP/EWP/280/96Corr1). Таким образом, композиции по настоящему изобретению можно считать эквивалентными введению дважды в сутки композиции с немедленным высвобождением в соответствии с рекомендациями Европейского агентства по лекарственным средствам (ЕМА) в отношении составов с модифицированным высвобождением.
Более того, при высокой дозировке композиция с пролонгированным высвобождением по настоящему изобретению также демонстрирует надлежащую стабильность активного ингредиента, несмотря на ее высокую растворимость в воде, высокую гигроскопичность и высокую кислотность. В частности, композиции по настоящему изобретению соответствуют строгим критериям норм в отношении граничного содержания примесей, которые предъявляют органы регулирования качества лекарственных препаратов.
Кроме того, композиция по настоящему изобретению, которая содержит бетагистин или его фармацевтически приемлемую соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, обеспечивает возможность упрощения расчета дозы за счет уменьшения числа приемов в сутки до введения один раз в сутки. Это является преимущественным, поскольку позволяет улучшить соблюдение режима лечения в результате обеспечения большего удобства для пациентов, что приводит к лучшему выполнению пациентом указаний и к лучшей эффективности лечения.
Профиль высвобождения композиций по настоящему изобретению также обеспечивает поддержание терапевтически эффективного количества бетагистина или его фармацевтически приемлемой соли, эквивалентного 48 мг бетагистина дигидрохлорида, в течение 24 часов и даже в течение более длительного периода времени через несколько дней после введения (т.е. устойчивый уровень при многократном приеме) при приеме дозы один раз в сутки без побочных эффектов, связанных с накоплением дозы. Также композиция по настоящему изобретению обеспечивает лучшую переносимость в результате меньших колебаний между максимальным и минимальным значениями концентрации в плазме вследствие устойчивого уровня высвобождения лекарственного средства в течение длительного периода времени.
Таким образом, первый аспект настоящего изобретения относится к твердой композиции для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов, содержащей некоторое количество бетагистина или его фармацевтически приемлемой соли, эквивалентное 48 мг бетагистина дигидрохлорида, вместе с одним или несколькими фармацевтически приемлемыми вспомогательными веществами или носителями, где композиция характеризуется профилем растворения, в соответствии с которым: не более 30% по весу бетагистина растворяется за 1 час; от 35% до 45% по весу бетагистина растворяется за 2 часа; от 46% до 60% по весу бетагистина растворяется за 4 часа; от 61% до 80% по весу бетагистина растворяется за 8 часов; от 81% до 97% по весу бетагистина растворяется за 16 часов и от 98% до 100% по весу бетагистина растворяется за 24 часа, где профиль растворения измеряют с применением аппарата I типа (вращающаяся корзинка) в соответствии с USP путем помещения композиции в 900 мл фосфатного буфера с рН 6,8 при 37°С и 100 об./мин. Указанная твердая композиция для перорального применения по настоящему изобретению, как было доказано, характеризуется по сути подобным профилем растворения при физиологических условиях с различным рН. А именно, она характеризуется по сути таким же профилем растворения, как описанный выше, в случае измерения при тех же условиях, что и выше, но в 0,1 н. хлористоводородной кислоте или в ацетатном буфере с рН 4,5 вместо фосфатного буфера.
Второй аспект настоящего изобретения относится к способу получения композиции, определенной в первом аспекте настоящего изобретения, который предусматривает: (а) получение раствора бетагистина или его фармацевтически приемлемой соли в полярном растворителе, выбранном из группы (С1-С4)алкил-СО-(С1-С4)алкила, (С1-С4)алкил)-СОО-(С1-С4)алкила, воды и их смеси; (b) просеивание и смешивание одного или нескольких разбавителей, одного или нескольких стабилизаторов и необязательно одного или нескольких связующих с получением смеси; (с) гранулирование смеси со стадии (b) путем добавления раствора со стадии (а) с получением влажных гранул; (d) высушивание влажных гранул, полученных на стадии (с), с получением высушенных гранул; (е) смешивание высушенных гранул, полученных на стадии (d), с одним или несколькими средствами, образующими гидрофильную матрицу, с получением смеси; (f) необязательно смешивание смеси, полученной на стадии (е), с одним или несколькими вспомогательными веществами или носителями, выбранными из группы, состоящей из вещества, способствующего скольжению, и смазывающего вещества, с получением смеси; (g) прессование смеси, полученной на стадии (f), с образованием таблеток и (h) необязательно нанесение покрытия на таблетки, полученные на стадии (g).
Также частью настоящего изобретения является композиция в соответствии с первым аспектом настоящего изобретения для применения в терапии; и композиция в соответствии с первым аспектом настоящего изобретения для применения в лечении заболевания или состояния вестибулярного аппарата, предпочтительно для применения в лечении головокружения, шума в ушах, потери слуха и тошноты, ассоциированных с синдромом Меньера.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг. 1 показана средняя концентрация в плазме (МРС) в течение 24 часов после единственного перорального введения композиции из примера 1 настоящего изобретения, содержащей 48 мг бетагистина дигидрохлорида. МРС означает среднюю концентрацию в плазме (нг/мл), и Т означает время, выраженное в часах.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Все термины, используемые в данной заявке, если не указано иное, следует понимать в их обычном значении, известном в данной области техники. Другие, более конкретные определения некоторых терминов, используемых в данной заявке, изложены ниже, при этом подразумевается, что они применяются единообразно по всему описанию и формуле изобретения, если только иное явно изложенное определение не предоставляет более широкое определение.
Для целей настоящего изобретения любые приведенные диапазоны включают как нижние, так и верхние граничные значения диапазона. Приведенные диапазоны, такие как значения температуры, времени, веса и т.п., следует считать приблизительными, если конкретно не указано иное.
Термины «доля в процентах (%) по весу», «вес/вес, %» и «вес./вес. %» имеют одно и то же значение и используются взаимозаменяемо. Они означают долю в процентах каждого ингредиента композиции по отношению к общему весу композиции.
Термин «весовое соотношение» означает соотношение по весу заданного соединения и другого заданного соединения, например, гидроксипропилметилцеллюлози и каррагенана.
Термин «комнатная температура» означает температуру окружающей среды, без нагревания или охлаждения, и обычно она составляет от 20°С до 25°С.
Термины «в условиях приема пищи» или «после приема пищи», используемые в данном документе, означают, что фармакокинетические параметры были измерены, когда композицию бетагистина или его фармацевтически приемлемой соли по настоящему изобретению вводят перорально человеку одновременно с едой или сразу после приема еды, в отличие от «натощак» или «в условиях воздержания от пищи», что означает, что фармакокинетические параметры были измерены, когда композицию бетагистина по настоящему изобретению вводят перорально человеку, лишенному еды.
Термин «алкил» означает насыщенную прямую или разветвленную углеводородную цепь, содержащую такое число атомов углерода, которое указано в описании или формуле изобретения. Примеры включают, среди прочих, следующие группы: метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил.
Как указано выше, первый аспект настоящего изобретения относится к композиции с непульсирующим пролонгированным высвобождением в течение 24 часов, содержащей некоторое количество бетагистина или его фармацевтически приемлемой соли, эквивалентное 48 мг бетагистина дигидрохлорида, вместе с одним или несколькими фармацевтически приемлемыми вспомогательными веществами или носителями, где композиция характеризуется вышеуказанным целевым профилем растворения.
Термины «пульсирующий», «пульсирующее высвобождение» или «импульсный» имеют одно и то же значение и используются взаимозаменяемо. Они означают профиль высвобождения, в котором по меньшей мере два дискретных количества активного ингредиента высвобождаются с временными интервалами, что обуславливает по меньшей мере два отчетливых пика концентрации в плазме. Также, термин «непульсирующее» высвобождение означает профиль высвобождения, в котором общее количество активного ингредиента высвобождается отдельным импульсом, обуславливающим только один пик концентрации в плазме.
В контексте настоящего изобретения термин «пролонгированное высвобождение» означает композицию, в которой скорость высвобождения активного ингредиента из состава после введения была снижена. Другими словами, это композиция, которая характеризуется более медленным высвобождением активного средства, чем таковое у традиционной фармацевтической композиции с немедленным высвобождением, вводимой тем же путем. В целом термин «лекарственная форма с пролонгированным высвобождением» означает, что активный ингредиент высвобождается из фармацевтической лекарственной формы в течение длительного периода времени, а не путем немедленного высвобождения (см. Quality of prolonged release oral solid dosage forms Directive 75/318/EEC, октябрь 1992 г., раздел 3AQ19a, стр. 167-174). В контексте настоящего изобретения «пролонгированное высвобождение» означает, что бетагистин или его фармацевтически приемлемая соль высвобождаются из композиции в течение длительного периода времени, составляющего 24 часа. В частности, для целей настоящего изобретения термин «пролонгированное высвобождение» означает композицию, которая характеризуется профилем растворения, в соответствии с которым: не более 30% по весу бетагистина растворяется за 1 час; от 35% до 45% по весу бетагистина растворяется за 2 часа; от 46% до 60% по весу бетагистина растворяется за 4 часа; от 61% до 80% по весу бетагистина растворяется за 8 часов; от 81% до 97% по весу бетагистина растворяется за 16 часов и от 98% до 100% по весу бетагистина растворяется за 24 часа, где профиль растворения измеряют с применением аппарата I типа (вращающаяся корзинка) в соответствии с USP путем помещения композиции в 900 мл 0,1 н. хлористоводородной кислоты, или ацетатного буфера с рН 4,5, или фосфатного буфера с рН 6,8 при 37°С и 100 об./мин.
Композиция по настоящему изобретению содержит бетагистин или его фармацевтически приемлемую соль. В одном варианте осуществления композиция по настоящему изобретению содержит свободное основание бетагистина в количестве, эквивалентном 48 мг бетагистина дигидрохлорида. В одном варианте осуществления композиция по настоящему изобретению является такой, где бетагистин представлен в форме его фармацевтически приемлемой соли, при этом в количестве, эквивалентном 48 мг бетагистина дигидрохлорида. Термин «фармацевтически приемлемая соль», используемый в данном документе, охватывает соль, образованную из фармацевтически приемлемых нетоксичных кислот, в том числе неорганических или органических кислот. В отношении солей отсутствуют какие-либо ограничения, за исключением того, что в случае применения для терапевтический целей они должны быть фармацевтически приемлемыми. Соли бетагистина можно получать из фармацевтически приемлемых нетоксичных кислот, в том числе неорганических и органических кислот. Такие кислоты включают, среди прочих, уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромистоводородную, хлористоводородную, молочную, малеиновую, яблочную, миндальную, метансульфоновую, фосфорную, янтарную, серную, винную и п-толуолсульфоновую кислоты. В одном варианте осуществления бетагистин представлен в форме соли дигидрохлористой кислоты и бетагистина, т.е. бетагистина дигидрохлорида.
Композиция в соответствии с первым аспектом настоящего изобретения содержит некоторое количество бетагистина или его фармацевтически приемлемой соли, эквивалентное 48 мг бетагистина дигидрохлорида; предпочтительно содержит 48 мг бетагистина дигидрохлорида. Если приведено значение количества бетагистина или фармацевтически приемлемой соли, подразумевается, что это «приблизительное» значение с учетом погрешности измерения. Следует понимать, что если указано количество 48 мг, оно соответствует количеству ± 0,5, что означает от 47,5 мг до 48,5 мг. Вариабельность результатов обусловлена характерной точностью используемого аналитического оборудования для анализа.
В одном варианте осуществления бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, характеризуются степенью чистоты, требуемой регулирующими органами для применения в качестве активного ингредиента. В одном варианте осуществления бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, характеризуются химической чистотой, равной 98% пощади пиков или больше, измеренной с помощью HPLC; предпочтительно равной 99% площади пиков или больше, измеренной с помощью HPLC.
Условия проведения анализа методом HPLC:
- расход: 1,5 мл/мин.,
- колонка: Zorbax Eclipse XDB С18 (150 мм × 4,6 мм) 5 мкм, производитель: Agilent,
- размер пор колонки: 5 мкм,
- фазы: буфер:ACN (60:40),
- температура колонки: 35°С,
- объем вводимой пробы: 15 мкл,
- длина волны детектора: 260 нм,
- градиент: отсутствует,
- изократический режим: буфер:ACN (60:40).
В одном варианте осуществления бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, представлены в кристаллической форме.
В одном варианте осуществления твердая композиция для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов по настоящему изобретению характеризуется профилем растворения, в соответствии с которым: не более 30% по весу бетагистина растворяется за 1 час; от 35% до 45% по весу бетагистина растворяется за 2 часа; от 46% до 60% по весу бетагистина растворяется за 4 часа; от 61% до 80% по весу бетагистина растворяется за 8 часов; от 81% до 95% по весу бетагистина растворяется за 16 часов и от 98% до 100% по весу бетагистина растворяется за 24 часа, где профиль растворения измеряют с применением аппарата I типа (вращающаяся корзинка) в соответствии с USP путем помещения композиции в 900 мл 0,1 н. хлористоводородной кислоты, или ацетатного буфера с рН 4,5, или фосфатного буфера с рН 6,8 при 37°С и 100 об./мин.
В одном варианте осуществления твердая композиция для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов по настоящему изобретению характеризуется профилем растворения, в соответствии с которым: не более 30% по весу бетагистина растворяется за 1 час; от 35% до 40% по весу бетагистина растворяется за 2 часа; от 46% до 58% по весу бетагистина растворяется за 4 часа; от 61% до 78% по весу бетагистина растворяется за 8 часов; от 81% до 95% по весу бетагистина растворяется за 16 часов и от 98% до 100% по весу бетагистина растворяется за 24 часа, где профиль растворения измеряют с применением аппарата I типа (вращающаяся корзинка) в соответствии с USP путем помещения композиции в 900 мл 0,1 н. хлористоводородной кислоты, или ацетатного буфера с рН 4,5, или фосфатного буфера с рН 6,8 при 37°С и 100 об./мин.
В одном варианте осуществления после введения единичной пероральной дозы композиции в условиях приема пищи бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, демонстрируют максимальную концентрацию в плазме (Cmax), составляющую от 400 нг/мл до 700 нг/мл, в пересчете на концентрацию в плазме их метаболита 2-пиридилуксусной кислоты (2-РАА). В предпочтительном варианте осуществления после введения единичной пероральной дозы композиции в условиях приема пищи бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, демонстрируют максимальную концентрацию в плазме (Cmax), составляющую от 500 нг/мл до 600 нг/мл, в пересчете на концентрацию в плазме их метаболита - 2-пиридилуксусной кислоты. В более предпочтительном варианте осуществления после перорального введения единичной дозы композиции в условиях приема пищи бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, демонстрируют максимальную концентрацию в плазме (Cmax), составляющую от 530 нг/мл до 570 нг/мл, в пересчете на концентрацию в плазме их метаболита 2-пиридилуксусной кислоты. Термин «Cmax» означает максимальную концентрацию метаболита бетагистина - 2-РАА - в крови после введения единичной пероральной дозы композиции в условиях приема пищи.
В одном варианте осуществления после введения единичной пероральной дозы композиции в условиях приема пищи бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, демонстрируют время максимальной концентрации в плазме (Tmax), составляющее от 5 ч. до 7 ч., в пересчете на концентрацию в плазме их метаболита 2-пиридилуксусной кислоты (2-РАА). В предпочтительном варианте осуществления после введения единичной пероральной дозы композиции в условиях приема пищи бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, демонстрируют время максимальной концентрации в плазме (Tmax), составляющее от 5 ч. до 6 ч., в пересчете на концентрацию в плазме их метаболита - 2-пиридилуксусной кислоты. В особенно предпочтительном варианте осуществления после введения единичной пероральной дозы композиции в условиях приема пищи бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, демонстрируют время максимальной концентрации в плазме (Tmax), составляющее от 5,2 ч. до 5,6 ч., в пересчете на концентрацию в плазме их метаболита - 2-пиридилуксусной кислоты. Термин «Tmax» означает время в часах, когда достигается Cmax после введения единичной пероральной дозы композиции по настоящему изобретению в условиях приема пищи.
В одном варианте осуществления после введения единичной пероральной дозы композиции в условиях приема пищи бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, демонстрируют площадь под кривой зависимости концентрации в плазме от времени во временном интервале от 0 до 24 часов (AUC (0-24)), составляющую от 5000 нг⋅ч./мл до 10000 нг⋅ч/мл, в пересчете на концентрацию в плазме их метаболита 2-пиридилуксусной кислоты. В предпочтительном варианте осуществления после введения единичной пероральной дозы композиции в условиях приема пищи бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, демонстрируют площадь под кривой зависимости концентрации в плазме от времени во временном интервале от 0 до 24 часов (AUC (0-24)), составляющую от 6000 нг⋅ч./мл до 8000 нг⋅ч/мл, в пересчете на концентрацию в плазме их метаболита 2-пиридилуксусной кислоты. В более предпочтительном варианте осуществления после введения единичной пероральной дозы композиции в условиях приема пищи бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, демонстрируют площадь под кривой зависимости концентрации в плазме от времени во временном интервале от 0 до 24 часов (AUC (0-24)), составляющую от 6500 нг⋅ч./мл до 7500 нг⋅ч/мл, в пересчете на концентрацию в плазме их метаболита - 2-пиридилуксусной кислоты. Термин «AUC» означает площадь под кривой зависимости концентрации в плазме от времени после введения единичной пероральной дозы композиции по настоящему изобретению. AUC 0-бесконечность обозначает площадь под кривой зависимости концентрации в плазме от времени во временном интервале от 0 до бесконечности, и AUC0-t обозначает площадь под кривой зависимости концентрации в плазме от времени во временном интервале от 0 до момента времени t.
В одном варианте осуществления после введения единичной пероральной дозы композиции в условиях приема пищи бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, демонстрируют период полувыведения (Т1/2) от 8 ч. до 10 ч, в пересчете на концентрацию в плазме их метаболита - 2-пиридилуксусной кислоты. В предпочтительном варианте осуществления после введения единичной пероральной дозы композиции в условиях приема пищи бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, демонстрируют период полувыведения (Т1/2) от 8,5 ч. до 9,5 ч, в пересчете на концентрацию в плазме их метаболита 2-пиридилуксусной кислоты. Термин «период полувыведения (Т1/2)» означает время, за которое метаболит бетагистина - 2-пиридилуксусная кислота (2-РАА) потеряет половину своей фармакологической активности.
Значения фармакокинетических параметров после введения отдельной единичной дозы композиции по настоящему изобретению, указанных выше (т.е. Cmax, Cmin, Tmax, AUC0-t, AUC0-∞), а также моделируемых фармакокинетических параметров после схемы из многочисленных отдельных единичных доз композиции по настоящему изобретению при устойчивом уровне (ср. значения AUC0-τ,ss, Cmax, ss и Cmin, ss в примере 5) указывают, что при введении либо в виде единичной дозы, либо в виде многократной дозы (устойчивый уровень) в условиях приема пищи введение один раз в сутки твердой композиции для перорального применения по настоящему изобретению обеспечивает более низкий показатель максимальной концентрации в крови (Cmin) и более высокий показатель минимальной концентрации в крови (Cmax) метаболита бетагистина - 2-РАА - по сравнению с введением два раза в сутки твердой композиции для перорального применения с немедленным высвобождением. Подобным образом, похожие фармакокинетические параметры (например, Cmax и Cmin) будут ожидаться для концентрации в крови исходного лекарственного средства бетагистина, которое демонстрирует примерно 1% абсолютной биологической доступности. Кроме того, значение AUC, которое представляет собой общее воздействие лекарственного средства, достигаемое после введения один раз в сутки композиции по настоящему изобретению, является биоэквивалентным общему воздействию лекарственного средства, получаемому после введения два раза в сутки композиции с немедленным высвобождением. Тем самым, композиции по настоящему изобретению можно считать эквивалентными введению дважды в сутки композиции с немедленным высвобождением в соответствии с документом Европейского агентства по лекарственным средствам (EMA) «Guideline on the pharmacokinetic and clinical evaluation of modified release dosage forms» (см. EMA/CPMP/EWP/280/96Corr1). Доверительный интервал 90% для соотношения фармакокинетических параметров композиции по настоящему изобретению и сравнительной композиции, которая не охватывается объемом настоящего изобретения, попадает в допустимый интервал 80,00-125,00% в соответствии с рекомендациями ЕМА в отношении исследования биоэквивалентности (СРМР/EWP/QWP/1401/98 Rev. 1/ Corr, 2010).
В одном варианте осуществления композиция по настоящему изобретению является таковой, где фармацевтически приемлемые вспомогательные вещества или носители предусматривают одно или несколько средств, образующих гидрофильную матрицу. Термин «средство, образующее гидрофильную матрицу» означает фармацевтический приемлемое вспомогательное вещество или носитель, которые образуют гель при контакте с водой и, следовательно, выполняют функцию гидрофильного полимера, контролирующего скорость диспергирования в воде.
В одном варианте осуществления композиция по настоящему изобретению является таковой, где одно или несколько средств, образующих гидрофильную матрицу, присутствуют в количестве от 20% до 85% по весу относительно общего веса композиции. В одном варианте осуществления композиция по настоящему изобретению является таковой, где одно или несколько средств, образующих гидрофильную матрицу, присутствуют в количестве от 35% до 65% по весу относительно общего веса композиции. В конкретном варианте осуществления композиция по настоящему изобретению является таковой, где одно или несколько средств, образующих гидрофильную матрицу, присутствуют в количестве от 40% до 60%, предпочтительно от 45% до 55% и более предпочтительно от 50% до 55% по весу относительно общего веса композиции.
В одном варианте осуществления композиция по настоящему изобретению является таковой, где фармацевтически приемлемые вспомогательные вещества или носители предусматривают одно или несколько средств, образующих гидрофильную матрицу, выбранных из группы, состоящей из производных целлюлозы, полисахаридов, отличных от целлюлозы; поливинилпирролидона; поливинилацетата; поливинилового спирта; полиакриловой кислоты; гиалуроновой кислоты, соли гиалуроновой кислоты; полиэтиленоксида и их смесей.
В одном варианте осуществления композиция по настоящему изобретению является таковой, где фармацевтически приемлемые вспомогательные вещества или носители предусматривают одно или несколько производных целлюлозы, являющихся простым эфиром целлюлозы, выбранным из группы, состоящей из метилцеллюлозы, этилцеллюлозы, гидроксипропилцеллюлозы, гидроксиэтилцеллюлозы, карбоксиметилцеллюлозы, гидроксипропилметилцеллюлозы и их смесей.
В одном варианте осуществления композиция по настоящему изобретению является таковой, где средство, образующее гидрофильную матрицу, предусматривает гидроксипропилметилцеллюлозу. В одном варианте осуществления композиция по настоящему изобретению является таковой, где средство, образующее гидрофильную матрицу, предусматривает гидроксипропилметилцеллюлозу, характеризующуюся содержанием метокси-фрагмента от 15% до 30% по весу относительно веса гидроксипропилметилцеллюлозы; предпочтительно от 19% до 24% по весу относительно веса гидроксипропилметилцеллюлозы. В одном варианте осуществления композиция по настоящему изобретению является таковой, где средство, образующее гидрофильную матрицу, предусматривает гидроксипропилметилцеллюлозу, характеризующуюся содержанием гидроксипропильного фрагмента от 5% до 15% по весу относительно веса гидроксипропилметилцеллюлозы; предпочтительно от 7% до 12% по весу относительно веса гидроксипропилметилцеллюлозы. Измерение содержания метокси-фрагмента и гидроксипропильного фрагмента можно проводить с помощью любого способа, известного в уровне техники. В конкретном варианте осуществления средство, образующее гидрофильную матрицу, представляет собой гидроксипропилметилцеллюлозу K100M.
В одном варианте осуществления композиция по настоящему изобретению является таковой, где средство, образующее гидрофильную матрицу, предусматривает гидроксипропилметилцеллюлозу, характеризующуюся структурной вязкостью от 65000 мПа⋅с до 150000 мПа⋅с; предпочтительно от 75000 мПа⋅с до 140000 мПа⋅с. Термин «структурная вязкость» означает сопротивление текучей среды усилию сдвига, при этом также называется динамической вязкостью или вязкостью при сдвиге и определяется математически как коэффициент отношения стресса к скорости сдвига. Измерение структурной вязкости можно проводить с помощью любого способа, известного в уровне техники. Обычно вязкость измеряют с применением таких устройств, как ротационные вискозиметры или реометры, с помощью которых измеряет крутящий момент, приложенный к оси, находящейся в контакте с образцом, вращаемым при точно контролируемой угловой скорости. Преобразование крутящего момента и скорости в стресс и скорость сдвига соответственно проводят путем прямого умножения на постоянные преобразования. В настоящем изобретении измерения вязкости проводят с применением раствора 2% по весу гидроксипропилметилцеллюлозы в воде при 20°С с использованием реометра от ТА Instraments, оснащенного системой измерения «конус-плита», где проводят цикл последовательного увеличения скорости сдвига от 0 до 150 с 1 и затем уменьшения от 150 до 0 с-1.
В одном варианте осуществления композиция по настоящему изобретению является таковой, где средство, образующее гидрофильную матрицу, содержит один или несколько из полисахаридов, отличных от целлюлозы, выбранных из группы, состоящей из каррагенана; сульфата амилозы; сульфата ксилана; галактоманнана; гуаровой камеди; камеди рожкового дерева; аравийской камеди; камеди стеркулии; агара; альгиновой кислоты, соли альгиновой кислоты и их смеси. В одном варианте осуществления композиция по настоящему изобретению является таковой, где средство, образующее гидрофильную матрицу, содержит каррагенан, предпочтительно каррагенан, выбранный из группы, состоящей из лямбда-каррагенана, йота-каррагенана, каппа-каррагенана и их смесей. В одном варианте осуществления композиция по настоящему изобретению является таковой, где одно или несколько средств, образующих гидрофильную матрицу, представляют собой лямбда-каррагенан. В частности, композиция по настоящему изобретению является таковой, где одно или несколько средств, образующих гидрофильную матрицу, представляют собой лямбда-каррагенан Viscarin РН 209.
В одном варианте осуществления композиция по настоящему изобретению содержит два или более средств, образующих гидрофильную матрицу. В конкретном варианте осуществления композиция по настоящему изобретению содержит два средства, образующих гидрофильную матрицу. В одном варианте осуществления композиция по настоящему изобретению является таковой, где одно или несколько средств, образующих гидрофильную матрицу, предусматривают смесь гидроксипропилметилцеллюлозы и каррагенана.
В одном варианте осуществления композиция по настоящему изобретению является таковой, где средства, образующие гидрофильную матрицу, предусматривают смесь гидроксипропилметилцеллюлозы и каррагенана, при этом весовое соотношение гидроксипропилметилцеллюлозы и каррагенана составляет от 3:1 до 12:1; предпочтительно от 5:1 до 11:1; более предпочтительно от 6:1 до 10:1. В конкретном варианте осуществления композиция по настоящему изобретению является таковой, где средства, образующие гидрофильную матрицу, предусматривают смесь гидроксипропилметилцеллюлозы и каррагенана, при этом весовое соотношение гидроксипропилметилцеллюлозы и каррагенана составляет 9:1.
Композиции по настоящему изобретению содержат одно или несколько фармацевтически приемлемых вспомогательных веществ или носителей. Термин фармацевтически приемлемые вспомогательные вещества или носители» означает такие вспомогательные вещества или носители, которые являются подходящими для применения в фармацевтической технологии с целью получения композиций, предусматривающих применение в медицине.
Твердые композиции для перорального применения по настоящему изобретению можно составлять в любой форме, которая включает любую единичную стандартную лекарственную форму и любые лекарственные формы в виде множественных структурно обособленных единиц. Термин «отдельная единица» охватывает один объект, такой как отдельная таблетка, отдельная гранула и отдельная пеллета. Термин «единичная стандартная лекарственная форма» определяет лекарственную форму, которая состоит только из одной единицы, содержащей эффективное количество бетагистина. Термин «лекарственная форма в виде множественных структурно обособленных единиц» определяет лекарственную форму, которая состоит из более чем одной единицы, при этом содержит эффективное количество бетагистина. Обычно лекарственные формы в виде множественных структурно обособленных единиц основаны на субъединицах, таких как гранулы, пеллеты или минитаблетки. Они обычно доставляются в твердых желатиновых капсулах или преобразованы в таблетки. Таким образом, частью настоящего изобретения также является стандартная лекарственная форма, которая содержит композицию по настоящему изобретению. В одном варианте осуществления стандартная лекарственная форма, которая содержит композицию по настоящему изобретению, представляет собой единичную стандартную лекарственную форму. В одном варианте осуществления стандартная лекарственная форма, которая содержит композицию по настоящему изобретению, представляет собой лекарственную форму в виде множественных структурно обособленных единиц.
Подходящие вспомогательные вещества и/или носители, а также их количества, могут быть легко определены специалистами в данной области в зависимости от типа состава, который получают.
В одном варианте осуществления композиция по настоящему изобретению содержит: некоторое количество бетагистина или его фармацевтически приемлемой соли, эквивалентное 48 мг бетагистина дигидрохлорида; от 20 до 85% по весу одного или нескольких средств, образующих гидрофильную матрицу, определенных выше; от 20 до 40% по весу одного или нескольких разбавителей; от 0,1 до 15% по весу одного или нескольких стабилизаторов; необязательно от 0,1 до 15% по весу одного или нескольких связующих; необязательно от 0,1 до 10% по весу одного или нескольких веществ, способствующих скольжению; и необязательно от 0,1 до 10% по весу одного или нескольких смазывающих веществ; при этом сумма ингредиентов составляет 100% по весу.
В одном варианте осуществления композиция по настоящему изобретению содержит: некоторое количество бетагистина или его фармацевтически приемлемой соли, эквивалентное 48 мг бетагистина дигидрохлорида; от 35 до 65%, предпочтительно от 40 до 60%, более предпочтительно от 45 до 55%, еще более предпочтительно от 50 до 55% по весу одного или нескольких средств, образующих гидрофильную матрицу, определенных выше; от 25 до 35%, предпочтительно от 28 до 30% по весу одного или нескольких разбавителей; необязательно от 0,5 до 5%, предпочтительно от 1 до 2% по весу одного или нескольких связующих; предпочтительно от 1 до 10%, более предпочтительно от 5 до 7% по весу одного или нескольких стабилизаторов; необязательно от 0,5 до 5%, предпочтительно от 1 до 3% по весу одного или нескольких веществ, способствующих скольжению; и необязательно от 0,5 до 5%, предпочтительно от 1 до 3% по весу одного или нескольких смазывающих веществ; при этом сумма ингредиентов составляет 100% по весу.
Термины «наполнитель» и «разбавитель» имеют одно и то же значение и используются взаимозаменяемо. Они означают любое фармацевтически приемлемое вспомогательное вещество или носитель (материал), за счет которых формируется объем композиции, что обеспечивает практичность ее изготовления и удобство ее применения потребителем. Материалы, обычно применяемые в качестве наполнителей, включают карбонат кальция, фосфат кальция, двухосновный фосфат кальция, трехосновный сульфат кальция, карбоксиметилцеллюлозу кальция, целлюлозу, продукты на основе целлюлозы, такие как микрокристаллическая целлюлоза и ее соли, производные декстрина, декстрин, декстрозу, фруктозу, лактит, лактозу, крахмалы или модифицированные крахмалы, карбонат магния, оксид магния, мальтит, мальтодекстрины, мальтозу, маннит, сорбит, крахмал, сахарозу, сахар, ксилит, эритрит и их смеси. В одном варианте осуществления композиция по настоящему изобретению является таковой, где фармацевтически приемлемые вспомогательные вещества или носители содержат один или несколько наполнителей; предпочтительно содержат микрокристаллическую целлюлозу. В одном варианте осуществления композиция по настоящему изобретению является таковой, где фармацевтически приемлемые вспомогательные вещества или носители содержат один или несколько наполнителей, предпочтительно микрокристаллическую целлюлозу, в количестве от 20 до 40%, предпочтительно от 25 до 35%, более предпочтительно от 28 до 30% по весу композиции.
Термин «стабилизатор» означает любое вещество, которое замедляет или задерживает разложение или изменение активного ингредиента. Примеры подходящих стабилизирующих средств в соответствии с настоящим изобретением включают без ограничения альфа-гидроксилкарбоновую кислоту, такую как молочная кислота, винная кислота или лимонная кислота. В одном варианте осуществления композиция по настоящему изобретению является таковой, где фармацевтически приемлемые вспомогательные вещества или носители содержат один или несколько стабилизаторов; предпочтительно предусматривающих лимонную кислоту. Композиции по настоящему изобретению, которые содержат лимонную кислоту в качестве стабилизирующего средства, являются особенно преимущественными, поскольку они характеризуются более высокой стабильностью. В одном варианте осуществления композиция по настоящему изобретению является таковой, где фармацевтически приемлемые вспомогательные вещества или носители содержат один или несколько стабилизаторов в количестве от 0,1 до 15%, предпочтительно от 1 до 10%, более предпочтительно от 5 до 7% по весу композиции.
Термин «связующее» означает любое фармацевтически приемлемое соединение, обладающее связующими свойствами. Материалы, обычно применяемые в качестве связующих, включают повидон, такой как поливинилпирролидон K30, полимеры на основе метилцеллюлозы, гидроксиэтилцеллюлозу, гидроксипропилцеллюлозу, L-гидроксипропилцеллюлозу (низкозамещеиную), гидроксипропилметилцеллюлозу (НРМС), карбоксиметилцеллюлозу натрия, карбоксиметилен, карбоксиметилгидроксиэтилцеллюлозу и другие производные целлюлозы, крахмалы или модифицированные крахмалы и их смесь. В одном варианте осуществления композиция по настоящему изобретению является таковой, где фармацевтически приемлемые вспомогательные вещества или носители предусматривают одно или несколько связующих, предпочтительно предусматривают поливинилпирролидон, например поливинилпирролидон K30. В одном варианте осуществления композиция по настоящему изобретению является таковой, где фармацевтически приемлемые вспомогательные вещества или носители содержат одно или несколько связующих в количестве от 0,1 до 15%, предпочтительно от 0,5 до 5%, более предпочтительно от 1 до 2% по весу композиции.
Термин «вещество, способствующее скольжению» означает вещество, которое улучшает характеристики сыпучести порошкообразных смесей в сухом состоянии. Материалы, обычно применяемые в качестве вещества, способствующего скольжению, включают стеарат магния, коллоидный диоксид кремния или тальк. В одном варианте осуществления композиция по настоящему изобретению является таковой, где фармацевтически приемлемые вспомогательные вещества или носители содержат одно или несколько веществ, способствующих скольжению; предпочтительно содержат коллоидный диоксид кремния. В одном варианте осуществления композиция по настоящему изобретению является таковой, где фармацевтически приемлемые вспомогательные вещества или носители предусматривают одно или несколько веществ, способствующих скольжению, в количестве от 0,1 до 10%, предпочтительно от 0,5 до 5%, более предпочтительно от 1 до 3% по весу композиции.
Термин «смазывающее вещество» означает вещество, которое предотвращает комкование ингредиентов композиции вместе и их прилипание к таблеточным формам или к устройству для заполнения капсул и улучшает текучесть смеси композиции. Материалы, обычно применяемые в качестве смазывающего вещества, включают олеат натрия, стеарат натрия, бензоат натрия, стеарат натрия, хлорид натрия, стеариновую кислоту, стеарилфумарат натрия, стеарат кальция, стеарат магния, лаурилсульфат магния, стеарилфумарат натрия, сложные эфиры или жирную кислоту сахарозы, цинк, полиэтиленгликоль, тальк и их смеси. Присутствие смазывающего вещества является особенно предпочтительным, если композиция представляет собой таблетку, для улучшения процесса таблетирования.
В одном варианте осуществления композиция по настоящему изобретению является таковой, где фармацевтически приемлемые вспомогательные вещества или носители предусматривают одно или несколько смазывающих веществ; предпочтительно содержат смесь стеарилфумарата натрия и талька, предпочтительно в соотношении 1:1. В одном варианте осуществления композиция по настоящему изобретению является таковой, где фармацевтически приемлемые вспомогательные вещества или носители предусматривают одно или несколько смазывающих веществ в количестве от 0,1 до 10%, предпочтительно от 0,5 до 5%, более предпочтительно от 1 до 3% по весу композиции.
Дополнительно композиции по настоящему изобретению могут содержать другие ингредиенты, такие как красящие вещества, разрыхлители и другие компоненты, известные в данной области техники для применения в твердых составах для перорального применения.
В одном варианте осуществления композиция по настоящему изобретению содержит: некоторое количество бетагистина или его фармацевтически приемлемой соли, эквивалентное 48 мг бетагистина дигидрохлорида; от 20 до 85% по весу смеси гидроксипропилметилцеллюлозы и каррагенана, определенной выше, предпочтительно в весовом соотношении от 3:1 до 12:1; от 20 до 40% по весу микрокристаллической целлюлозы; от 0,1 до 15% по весу лимонной кислоты; необязательно от 0,1 до 15% по весу одного или нескольких видов поливинилпирролидона; необязательно от 0,1 до 10% по весу коллоидного диоксида кремния, предпочтительно в весовом соотношении от 1:2 до 2:1; и необязательно от 0,1 до 10% по весу смеси стеарилфумарата натрия и талька, определенных выше, предпочтительно в весовом соотношении 1:1; при этом сумма ингредиентов составляет 100% по весу.
В одном варианте осуществления композиция по настоящему изобретению содержит: некоторое количество бетагистина или его фармацевтически приемлемой соли, эквивалентное 48 мг бетагистина дигидрохлорида; от 35 до 65%, более предпочтительно от 40 до 60%, более предпочтительно от 45 до 55%, еще более предпочтительно от 50 до 55% по весу смеси гидроксипропилметилцеллюлозы и каррагенана, определенной выше, предпочтительно в весовом соотношении от 3:1 до 12:1; предпочтительно от 5:1 до 11:1; более предпочтительно от 6:1 до 10:1 и еще более предпочтительно 9:1; от 25 до 35%, более предпочтительно от 28 до 30% по весу микрокристаллической целлюлозы; необязательно от 0,5 до 5%, предпочтительно от 1 до 3% по весу поливинилпирролидона; от 1 до 10%, предпочтительно от 5 до 7% по весу лимонной кислоты; необязательно от 0,5 до 5%, предпочтительно от 1 до 3% по весу коллоидного диоксида кремния; и необязательно от 0,5 до 5%, предпочтительно от 1 до 3% по весу смеси стеарилфумарата натрия и талька, определенной выше, предпочтительно в весовом соотношении 1:1; где сумма ингредиентов составляет 100% по весу.
В предпочтительном варианте осуществления композиция по настоящему изобретению содержит: 48 мг бетагистина дигидрохлорида; от 50 до 55% по весу смеси гидроксипропилметилцеллюлозы и каррагенана в весовом соотношении 9:1; от 28 до 30% по весу микрокристаллической целлюлозы; от 1 до 2% по весу поливинилпирролидона; от 5 до 7% по весу лимонной кислоты; от 1 до 3% по весу коллоидного диоксида кремния; и от 1 до 3% по весу смеси стеарилфумарата натрия и талька в весовом соотношении 1:1; где сумма ингредиентов составляет 100% по весу.
Другой аспект настоящего изобретения представляет собой способ получения композиций по настоящему изобретению, определенных выше. Композиции по настоящему изобретению можно получать в соответствии со способами, широко известными в данной области техники. Подходящие способ и условия могут быть легко определены специалистами в данной области в зависимости от типа состава, который получают.
В одном варианте осуществления композиция представлена в форме таблетки. В одном варианте осуществления способ получения композиции по настоящему изобретению в форме таблетки, определенной выше, предусматривает таблетку, полученную в результате влажной грануляции. Способ изготовления таких композиций предусматривает любой способ, известный в уровне техники, который предусматривает стадию влажной грануляции. В одном варианте осуществления способ получения композиции, определенной выше, предусматривает: (а) получение раствора бетагистина или его фармацевтически приемлемой соли в полярном растворителе, выбранном из группы (С1-С4)алкил-СО-(С1-С4)алкила, (С1-С4)алкил)-СОО-(С1-С4)алкила, воды и их смеси; (b) просеивание и смешивание одного или нескольких разбавителей, одного или нескольких стабилизаторов и необязательно одного или нескольких связующих с получением смеси; (с) гранулирование смеси со стадии (b) путем добавления раствора со стадии (а) с получением влажных гранул; (d) высушивание влажных гранул, полученных на стадии (с), с получением высушенных гранул; (е) смешивание высушенных гранул, полученных на стадии (d), с одним или несколькими средствами, образующими гидрофильную матрицу, с получением смеси; (f) необязательно смешивание смеси, полученной на стадии (е), с одним или несколькими вспомогательными веществами или носителями, выбранными из группы, состоящей из вещества, способствующего скольжению, и смазывающего вещества, с получением смеси; (g) прессование смеси, полученной на стадии (f), с образованием таблеток и (h) необязательно нанесение покрытия на таблетки, полученные на стадии (g). Данный способ является преимущественным, поскольку он обеспечивает получение устойчивой композиции, содержащей некоторое количество бетагистина или его фармацевтически приемлемой соли, эквивалентное 48 мг бетагистина дигидрохлорида, вместе с одним или несколькими фармацевтически приемлемыми вспомогательными веществами или носителями, которая характеризуется целевым профилем растворения. В частности, данный способ предусматривает стадию влажной грануляции, которая обеспечивает получение промежуточных гранул, содержащих активный ингредиент бетагистин вместе с одним или несколькими разбавителями, одним или несколькими стабилизаторами и необязательно одним или несколькими связующими. Данная стадия способа увеличивает стабильность активного ингредиента и исключает абсорбцию или впитывание влаги или молекул воды из воздуха. Данный способ является особенно преимущественным в данном случае, поскольку бетагистин, особенно бетагистина дигидрохлорид, является активным ингредиентом, который характеризуется высокой растворимостью в воде и гигроскопичностью.
В одном варианте осуществления стадию (а) способа получения композиции по настоящему изобретению проводят при комнатной температуре. В одном варианте осуществления стадию (а) способа получения композиции по настоящему изобретению проводят в высокоскоростном смесителе-грануляторе.
В одном варианте осуществления стадию (b) способа получения композиции по настоящему изобретению проводят при комнатной температуре. В одном варианте осуществления стадию (а) способа получения композиции по настоящему изобретению проводят в высокоскоростном смесителе-грануляторе.
В одном варианте осуществления стадию (с) способа получения композиции по настоящему изобретению проводят при комнатной температуре, предпочтительно при температуре от 20°С до 25°С. В одном варианте осуществления стадию (с) способа получения композиции по настоящему изобретению проводят в любом оборудовании, которое представляет собой смеситель-гранулятор; предпочтительно в высокоскоростном смесителе-грануляторе.
В одном варианте осуществления стадию (d) способа получения композиции по настоящему изобретению проводят при температуре от 45°С до 70°С; предпочтительно от 50°С до 60°С. В одном варианте осуществления стадию (d) способа получения композиции по настоящему изобретению проводят, пока содержание воды (LOD) не станет равным 2% по весу или меньше.
В одном варианте осуществления стадии (е) и (f) способа получения композиции по настоящему изобретению проводят при комнатной температуре, предпочтительно проводят при температуре от 20°С до 25°С.
В одном варианте осуществления стадию прессования (g) способа получения композиции по настоящему изобретению проводят с применением оборудования, которое представляет собой таблеточный пресс. В одном варианте осуществления стадию прессования (g) способа получения композиции по настоящему изобретению проводят при усилии сжатия от 150 Н до 220 Н; в частности, при 180 Н получают таблетки с весом от приблизительно 700 до 900 мг, предпочтительно с весом от приблизительно 750 до 800 мг, более предпочтительно с весом приблизительно 785 мг.
Все варианты осуществления, раскрытые выше в отношении твердой композиции для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов по настоящему изобретению, также применяются для способа ее получения.
В одном варианте осуществления композиция представлена в форме твердых желатиновых капсул. В одном варианте осуществления способ получения композиции по настоящему изобретению можно проводить любым способом, известным из уровня техники, для получения твердых желатиновых капсул. Как правило, способ предусматривает смешивание бетагистина или его фармацевтически эффективного количества с подходящими фармацевтически приемлемыми вспомогательными веществами или носителями с последующим заполнением твердых желатиновых капсулу вручную или с применением полуавтоматических или автоматических устройств.
В одном варианте осуществления композиция представлена в форме пеллет. В одном варианте осуществления способ получения композиции по настоящему изобретению можно проводить любым способом, известным из уровня техники, для получения пеллет. Как правило, пеллеты можно получать с помощью способа экструзии-сферонизации или путем нанесения слоев на внутреннее ядро.
Твердая композиция для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов может быть определена посредством способа ее получения, определенному выше, и, следовательно, твердая композиция для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов по настоящему изобретению, получаемая с помощью способа по настоящему изобретению, считается частью настоящего изобретения. Для целей настоящего изобретения выражения «получаемый», «полученный» и эквивалентные выражения используются взаимозаменяемо, и в любом случае выражение «получаемый» охватывает выражение «полученный».
Все варианты осуществления, раскрытые выше в отношении твердой композиции для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов по настоящему изобретению, а также в отношении способа ее получения, также применяются к твердой композиции для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов, получаемой с помощью данного способа получения.
Третий аспект настоящего изобретения относится к твердой композиции для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов по настоящему изобретению, определенной выше, для применения в качестве лекарственного препарата. В одном варианте осуществления настоящее изобретение относится к твердой композиции для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов по настоящему изобретению, определенной выше, для применения в качестве лекарственного препарата, где лекарственный препарат вводят один раз в сутки.
Четвертый аспект настоящего изобретения относится к твердой композиции для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов по настоящему изобретению, определенной выше, для применения в лечении заболевания или состояния вестибулярного аппарата (внутреннего уха). Данный аспект также можно сформулировать как применение твердой композиции для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов по настоящему изобретению, определенной выше, с целью получения лекарственного препарата для профилактики и/или лечения заболевания или состояния вестибулярного аппарата. Он также относится к способу профилактики и/или лечения млекопитающих, страдающих от заболевания или состояния вестибулярного аппарата или подверженных риску их возникновения, где способ предусматривает введение указанному млекопитающему твердой композиции для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов по настоящему изобретению, определенной выше. В одном варианте осуществления заболевание или состояние вестибулярного аппарата означает болезнь Меньера или нарушение, симптомы которых могут включать головокружение, шум в ушах, потерю слуха и тошноту.
В одном варианте осуществления предусматривается твердая композиция для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов по настоящему изобретению, определенная выше, для применения в лечении заболевания или состояния вестибулярного аппарата, предпочтительно для применения в лечении болезни Меньера, где лечение предусматривает введение один раз в сутки твердой композиции для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов.
Все варианты осуществления, раскрытые выше в отношении твердой композиции для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов по настоящему изобретению, также применяются к твердой композиции для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов, ограниченной ее применением.
По всему описанию и формуле изобретения слово «содержит» и вариации слова не предназначены исключать иные технические признаки, добавки, компоненты или стадии. Кроме того, слово «содержит» охватывает случай «состоит из». Дополнительные цели, преимущества и признаки настоящего изобретения будут очевидны специалистам в данной области после ознакомления с описанием или могут быть изучены при применении на практике настоящего изобретения. Следующие примеры и графические материалы представлены в качестве иллюстрации, и они не предназначены для ограничения настоящего изобретения. Ссылочные позиции, связанные с графическими материалами и помещенные в круглые скобки в пункте формулы изобретения, предназначены исключительно для улучшения ясности пункта формулы изобретения и не должны рассматриваться как ограничивающие объем пункта формулы изобретения. Кроме того, настоящее изобретение охватывает все возможные комбинации конкретных и предпочтительных вариантов осуществления, описанных в данном документе.
ПРИМЕРЫ
Общие положения
Молекула, для которой измеряют фармакокинетические показатели: метаболит бетагистина 2-РАА. В соответствии с краткой характеристикой лекарственного препарата, а именно таблеток бетагистина дигидрохлорида (таблетки Betaserc®), выпущенной Европейским агентством по лекарственным средствам, перорально вводимый бетагистин легко и почти полностью всасывается во всех отделах желудочно-кишечного тракта. После всасывания лекарственное средство быстро и почти полностью подвергается метаболическому превращению в 2-пиридилуксусную кислоту (2-РАА). Абсолютная биологическая доступность бетагистина, дозированного в виде таблетки с немедленным высвобождением или таблетки, распадающейся в полости рта, по расчетам составляет около 1% вследствие его очень высокого пресистемного метаболизма. Уровни бетагистина в плазме являются очень низкими. Следовательно, большинство фармакокинетических анализов основаны на измерениях 2-РАА в плазме и моче, что предполагается как суррогатный маркер в биологическом анализе бетагистина. Следовательно, биоэквивалентность на основе метаболита 2-пиридилуксусной кислоты (2-РАА) - считается приемлемой органами государственного регулирования лекарственных препаратов. Подобным образом, ожидается, что фармакокинетические анализы на основе уровней в плазме малодоступного исходного лекарственного средства бетагистина будут обеспечивать подобные результаты биоэквивалентности.
Пример 1. Композиции
Компоненты примера композиции по настоящему изобретению с пролонгированным высвобождением в форме таблетки показаны в таблице 1, где количества компонентов выражены в процентах по весу.
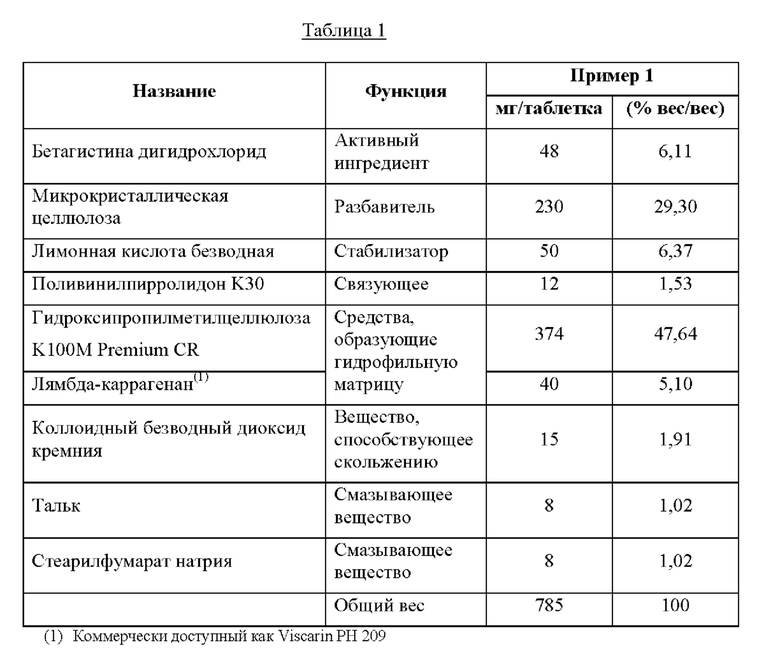
Композицию по настоящему изобретению из примера 1, определенную в таблице 1, получали с помощью способа, раскрытого ниже, с применением количеств ингредиентов, указанных в таблице выше.
(a) Бетагистин растворяли в достаточном количестве очищенной воды для полного растворения бетагистина при непрерывном перемешивании до получения прозрачного раствора.
(b) Смесь разбавителя, стабилизатора и связующего перемешивали и просеивали через сито 40 меш и загружали в высокоскоростной смеситель-гранулятор (RMG).
(c) Гранулировали смесь, полученную на стадии (b), путем добавления раствора, полученного на стадии (а). Полученную смесь перемешивали в течение 5 минут с получением влажных гранул.
(d) Влажные гранулы, полученные на стадии (с), высушивали в сушилке при температуре 55±10°С, пока содержание воды (LOD потеря при высушивании) не достигало 2,0%. Затем высушенные гранулы просеивали через сито 40 меш и перемешивали в течение 5 минут при 16 об./мин.
(e) Смесь средств, образующих гидрофильную матрицу, получали и просеивали через сито 40 меш; и данную смесь добавляли к высушенным гранулам, полученным на стадии (d), и смешивали в смесителе в течение 20 минут при 16 об./мин.
(f) К полученной смеси, полученной на стадии (е), добавляли вещества, способствующие скольжению, и смазывающие вещества, ранее просеянные через сито 40 меш; и полученную смесь перемешивали в течение 5 минут при 16 об./мин.
(g) Полученную смесь, полученную на стадии (f), прессовали с применением овального вогнутого пуансона 18 × 10 мм с получением таблетки, характеризующейся прочностью на раздавливание от 150 Н до 220 Н.
Полученная таблетка характеризовалась общим содержанием примесей ниже 0,15%, измеренным с помощью способа HPLC.
Условия способа HPLC являются следующими:
- расход: 1,3 мл/мин.,
- колонка: Peerless basic С18 (250 мм × 4,6 мм), 4 мкм; производитель: Chromatopak,
- размер пор колонки: 4 мкм,
- фазы: подвижная фаза А: [(буфер:метанол:ацетонитрил): (45:40:15)],
подвижная фаза В: [(буфер:ацетонитрил): (45:55)].
- длина волны детектора: 260 нм,
- объем вводимой пробы: 50 мкл,
- температура колонки: 25°С,
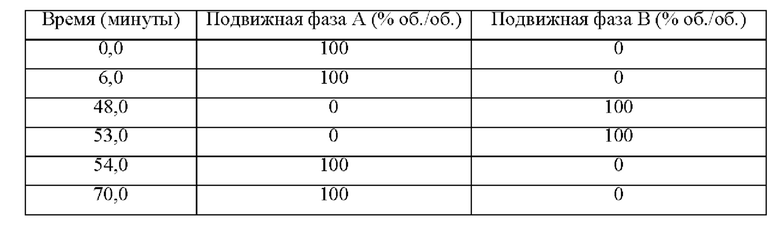
- градиент:
Пример 2. Тест на растворение
Профиль растворения композиции в форме таблетки с пролонгированным высвобождением из примера 1, содержащей 48 мг бетагистина дигидрохлорида, измеряли при различных условиях рН.
Условия, касающиеся бани для растворения
- Скорость вращения мешалки: 100 об./мин.
- Температура среды для растворения: 37°С±0,5°С.
- Среда для растворения: условия А: 0,1 н. HCl,
условия В: рН 4,5 с ацетатным буфером,
условия С: рН 6,8 с фосфатным буфером.
- Объем сосуда: 900 мл.
- Момент времени (часы): 0,5, 1, 4, 8, 16 и 24.
- Количество образцов: 6.
Проведение способа
В налаженном аппарате для проведения растворения, содержащем 900 мл среды для растворения, определенной выше, в каждый из шести стеклянных сосудов при указанных выше условиях добавляли таблетку тестируемого образца в каждую корзинку в вытяжном шкафу, при этом обеспечивали отсутствие пузырьков воздуха на поверхности таблетки, и сразу же включали аппарат.
После этого в каждый момент времени отбирали 10 мл тестируемого образца из каждого из шести стеклянных сосудов. Затем 2 мл аликвоты тестируемого образца переносили из собранного образца в отдельную пробирку, где ее перемешивали и фильтровали через фильтр из поливинилиденфторида (PVDF-фильтр) с размером пор 0,45 мкм. Кроме того, объем, который отбирали в каждый момент времени, заменяли путем добавления равного количества свежей среды для растворения при 37°С±0,5°С.
Условия хроматографического анализа
- Прибор: хроматограф для высокоэффективной жидкостной хроматографии, оснащенный автоматическим дозатором, УФ-детектором и участком колонки со встроенным термостатом.
- Колонка: Zorbax Eclipse XDB С18 (150 мм × 4,6 мм) 5 мкм, производитель: Agilent или аналог.
- Расход: 1,5 мл/мин.
- Температура колонки: 35°С.
- Объем вводимой пробы: 10 мкл.
- Длина волны: 260 нм.
- Время анализа: 3,5 мин.
- Время удерживания бетагистина: приблизительно 2,7 мин.
Подвижная фаза: смешивают 600 объемов буфера 1 и 400 объемов ацетонитрила, фильтруют через нейлоновый фильтр с размером пор 0,45 мкм и дегазируют с помощью ультразвуковой обработки.
Буфер 1: растворяют 4,6 г моногидрата однозамещенного ортофосфата натрия (NaH2PO4-H2O) в 900 мл воды; добавляют 0,66 г гексиламина и перемешивают. Затем рН регулируют до 2,7±0,05 путем добавления 10% об./об. ортофосфорной кислоты. Наконец добавляют воду до 1000 мл и перемешивают; и добавляют 2,7 г додецилсульфата натрия при осторожном перемешивании до растворения.
Получение исходного внутреннего эталонного стандарта бетагистина. Переносят 33,3 мг рабочего стандарта бетагистина дигидрохлорида в мерную колбу объемом 50 мл; добавляют 40 мл воды и подвергают воздействию ультразвука до растворения.
Наконец, разбавляют до необходимого объема водой и перемешивают с получением конечной концентрации 0,666 мг/мл.
Получение внутреннего стандарта бетагистина: Разбавляют 4,0 мл препарата исходного внутреннего эталонного стандарта бетагистина до 50 мл средой для растворения, определенной выше, и перемешивают до получения конечной концентрации 0,053 мг/мл.
Проведение способа
Определение пригодности системы: Уравновешивают колонку с помощью подвижной фазы, определенной выше, при указанных выше условиях до получения устойчивой нулевой линии. Вводят препарат стандарта в шести повторностях введений в жидкостный хроматограф и регистрируют хроматограммы. Измеряют ответ для пика бетагистина.
На хроматограмме, полученной для препарата стандарта:
• % Относительного стандартного отклонения для области пика бетагистина в шести повторностях введений должен составлять не более 2,00.
• Количество теоретических тарелок для пика бетагистина должно составлять не менее 1000.
Фактор асимметрии для пика бетагистина должен составлять от 0,80 до 2,00.
Проведение процедуры: Вводят отдельно по одному введению каждой среды для растворения в качестве холостого опыта и тестируемого образца в жидкостный хроматограф и регистрируют хроматограммы. Измеряют ответ для пика бетагистина. Рассчитывают количество растворенного бетагистина дигидрохлорида в процентах от заявленного содержания на основе средних площадей пиков препарата стандарта и площади пика тестируемого образца и степени разведения применяемого рабочего стандарта в процентах.
Расчет: % растворенного бетагистина дигидрохлорида
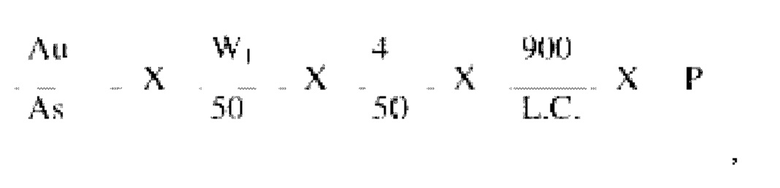
где
Au = Площадь пика бетагистина, полученного для препарата образца.
As = Средняя площадь пика бетагистина, полученного для препарата стандарта.
W1 = Вес рабочего стандарта бетагистина дигидрохлорида, выраженный в мг.
L.C. = Заявленное содержание бетагистина дигидрохлорида в мг/таблетка.
Р = Степень разведения рабочего стандарта бетагистина дигидрохлорида в процентах относительно исходного значения.
Рассчитывают поправочный коэффициент процента потери лекарственного средства в конце каждого момента времени. Добавляют данный поправочный коэффициент к значению % растворенного вещества, полученному в следующий момент времени.

Для момента 0,5 ч.: % растворенного вещества за 0,5 ч.
Для момента 1 ч.: поправочный коэффициент для 0,5 ч. + % растворенного вещества за 1 ч.
Для момента 4 ч.: поправочный коэффициент для (0,5 ч. + 1 ч.) + % растворенного вещества за 4 ч.
Для момента 8 ч.: поправочный коэффициент для (0,5 ч. + 1 ч. + 4 ч.) + % растворенного вещества за 8 ч.
Для момента 16 ч.: поправочный коэффициент для (0,5 ч. + 1 ч. + 4 ч. + 8 ч.) + % растворенного вещества за 16 ч.
Для момента 24 ч.: поправочный коэффициент для (0,5 ч. + 1 ч. + 4 ч. + 8 ч. + 16 ч.) + % растворенного вещества за 24 ч.
Результаты
Значения в процентах по весу бетагистина дигидрохлорида, высвобожденного из композиции из примера 1 настоящего изобретения, подверженного различным условиям растворения в моменты времени от 0,5 часов до 24 часов; а также относительное стандартное отклонение показаны таблице 2.
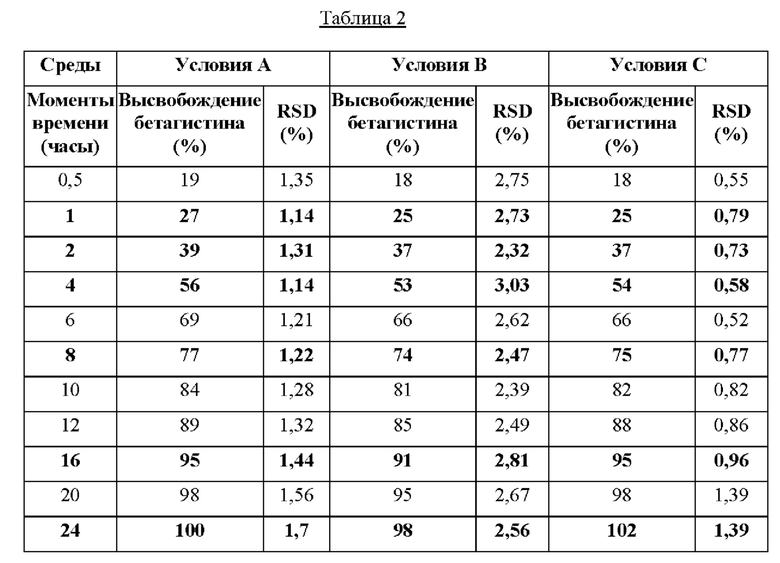
Результаты профилей растворения в таблице 2 показали, что композиция, содержащая 48 мг бетагистина дигидрохлорида, по настоящему изобретению характеризуется необходимым профилем растворения независимо от рН. Это значит, что не более 30% бетагистина растворяется за 1 час; от 35% до 45% бетагистина растворяется за 2 часа; от 46% до 60% бетагистина растворяется за 4 часа; от 61% до 80% бетагистина растворяется за 8 часов; от 81% до 97% бетагистина растворяется за 16 часов и от 98% до 100% по весу бетагистина растворяется за 24 часа.
Кроме того, целевой профиль растворения, показанный в таблице 2, поддерживался даже в том случае, когда тест на растворение проводили в присутствии до 40% этанола в среде 0,1 н. HCl в течение не более 2 часов.
Таким образом, композиции по настоящему изобретению являются преимущественными, поскольку они характеризуются растворимостью, не зависящей от рН, и они исключают эффект сброса дозы (даже в присутствии этанола) в первые часы (от 0 до 2 часов). Кроме того, композиции по настоящему изобретению также обеспечивают возможность контроля высвобождения бетагистина дигидрохлорида, даже если растворение бетагистина дигидрохлорида уже началось (т.е. от 4 до 16 часов), без необходимости в дополнительных слоях или покрытиях, обеспечивающих контролируемое высвобождение. Это также является преимущественным, поскольку композиция по настоящему изобретению обеспечивает возможность поддержания терапевтически эффективного количества бетагистина в течение 24 часов и даже в течение более длительного периода времени через несколько дней (устойчивый уровень при многократной дозе) при приеме дозы один раз в сутки. Также композиция по настоящему изобретению обеспечивает лучшую переносимость в результате меньших колебаний между максимальным и минимальным значениями концентрации в плазме вследствие устойчивого уровня высвобождения лекарственного средства в течение длительного периода времени.
Пример 3. Тест на биологическую доступность
Изучали биологическую доступность композиций с пролонгированным высвобождением по настоящему изобретению.
A. План исследования
Испытание проводили как открытое, сбалансированное, рандомизированное, перекрестное исследование сравнительной биологической доступности, предусматривающее две схемы лечения, две последовательности приема, два периода времени и пероральный прием дозы, с участием обычных, здоровых, взрослых субъектов-людей в условиях приема пищи.
B. Добровольцы
Достаточное число здоровых, взрослых добровольцев-людей отбирали с их согласия для набора 16 субъектов для исследования, и они дополнительно соответствовали следующим критериям включения:
- здоровые, взрослые субъекты-люди возрастом от 18 до 45 лет (обе границы включительно);
- имеющие индекс массы тела (BMI) от 18,5 до 30,0 (обе границы включительно), который рассчитывается как вес в кг/рост в м2;
- не имеющие серьезных заболеваний или клинически значимых патологических отклонений во время отбора, в истории болезни, при клиническом обследовании, при лабораторных испытаниях, при ЭКГ в 12 отведениях и на рентгенограммах грудной клетки (вид спереди и сзади);
- способные понять процедуры исследования и следовать им по мнению исследователя;
- добровольцы, которые не являются курильщиками и не страдают алкоголизмом;
- способные предоставить добровольное письменное информированное согласие на участие в исследовании;
- в случае субъектов-женщин:
- стерилизованные хирургическим путем по меньшей мере за 6 месяцев до участия в исследовании; или
- если имеют способность к деторождению, согласны применять подходящий и эффективный двойной барьерный способ контрацепции или внутриматочную спираль во время исследования, и
- сывороточный тест на беременность должен быть отрицательным.
C. Образцы
Образцы, применяемые в тесте в отношении биологической доступности, являются следующими:
a) тестируемый образец: композиция с пролонгированным высвобождением из примера 1 в форме таблетки, упакованной в двухстороннюю блистерную упаковку из алюминиевой фольги, которая содержит 48 мг бетагистина дигидрохлорида; и
b) эталонный тест: Betaserc® 24 мг в форме таблеток.
D. Способ проведения теста
Тестируемые и эталонные образцы вводили субъектам в сидячем положении с 240±02 мл питьевой воды при температуре окружающей среды. Таблетку следует глотать целиком, без разжевывания или раздавливания.
В случае тестируемого образца:
После воздержания от пищи в течение ночи, по меньшей мере в течение 10 часов, субъектам обеспечивали стандартизированный высококалорийный вегетарианский завтрак с высоким содержанием жира, который они должны были съесть в течение 30 минут. Тестируемый образец подлежал введению один раз через 30 минут после обеспечения стандартизированного высококалорийного вегетарианского завтрака с высоким содержанием жира.
В случае эталонного образца:
После воздержания от пищи в течение ночи, по меньшей мере в течение 10 часов, субъектам обеспечивали стандартизированный высококалорийный вегетарианский завтрак с высоким содержанием жира, который они должны были съесть в течение 30 минут. Первую дозу продукта эталонного образца вводили через 30 минут после обеспечения стандартизированного высококалорийного вегетарианского завтрака с высоким содержанием жира. Кроме того, стандартизированный высококалорийный вегетарианский завтрак с высоким содержанием жира снова обеспечивали через 11,5 часов после первой дозы, который они должны были съесть в течение 30 минут. Вторую дозу вводили в 12,00 часов (интервал между двумя последовательными дозами составляет 12 часов ± 5 минут).
Отбор образцов
В исходное время и через каждый час (т.е. в каждый момент времени) после введения отбирали кровь.
Образцы крови отбирали через постоянный внутривенный катетер (Venflon), помещенный в вену предплечья субъектов. Постоянный внутривенный катетер поддерживали in situ как можно дольше путем введения 0,5 мл физиологического раствора для предотвращения закупорки катетера, препятствующей сбору всех образцов крови в течение установки.
Образцы крови центрифугировали при 3000-100 rcf в течение 5 минут при температуре ниже 10°С для отделения плазмы. Отделенную плазму переносили в предварительно меченую полипропиленовую пробирку и хранили в вертикальном положении в контейнере, содержащем сухой лед, или в морозильной камере при температуре -65±10°С для временного хранения в соответствии с требованиями.
Наконец, образцы переносили на хранение в контейнер с сухим льдом и хранили в биоаналитической морозильной камере при -65±10°С до завершения анализа.
Определения
Способ включал измерение концентрации в плазме метаболита бетагистина - 2-пиридилуксусной кислоты (2-РАА) - в течение 24 часов в предварительно определенные моменты времени.
Определяли среднюю концентрацию метаболита - 2-пиридилуксусной кислоты.
Значения концентрации 2-пиридилуксусной кислоты после введения один раз в сутки тестируемого образца по настоящему изобретению и после введения два раза в сутки эталонного образца кратко изложены в таблице 3. Кроме того, профиль зависимости концентрации в плазме от времени после введения два раза в сутки тестируемого образца показан на фиг. 1.
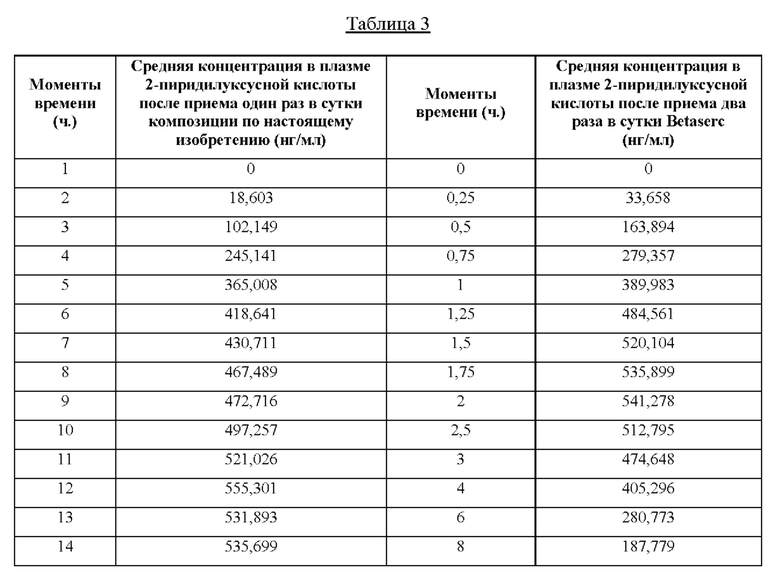
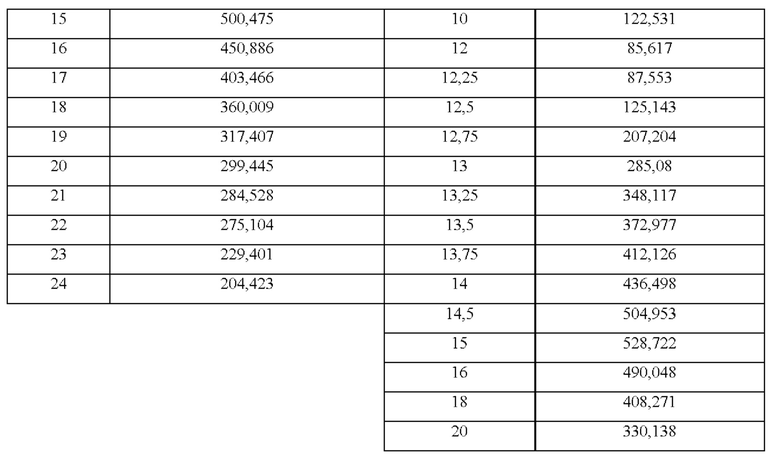
Кроме того, основные фармакокинетические параметры, проявляемые после введения один раз в сутки композиции по настоящему изобретению (пример 1) и введения два раза в сутки эталонного образца, измеряли с помощью способа LC-MS/MS.
В частности, значения Cmax, Tmax, AUC, Tlag, 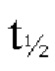 (период полувыведения) и λz (константа скорости выведения), а также стандартное отклонение (SD) тестируемого образца и эталонного образца кратко изложены в таблице 4.
(период полувыведения) и λz (константа скорости выведения), а также стандартное отклонение (SD) тестируемого образца и эталонного образца кратко изложены в таблице 4.
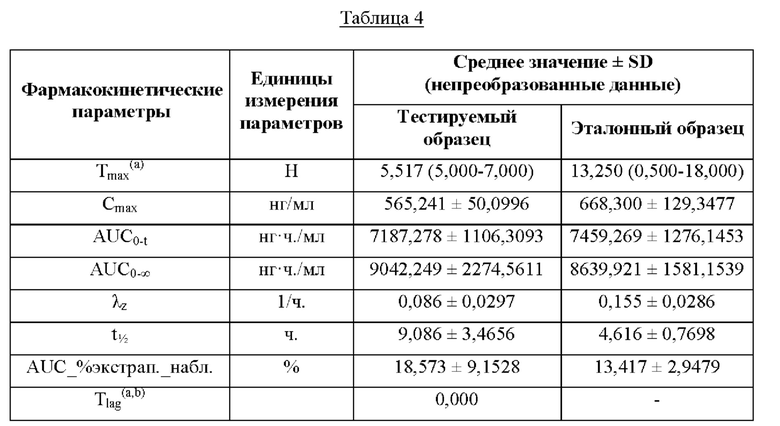
(a) Tmax и Tlag представлены медианным (min-max) значением.
(b) Рассчитано только для тестируемого состава.
Результаты анализов в отношении относительной биологической доступности (т.е. геометрический геометрические средние, рассчитанные методом наименьших квадратов, соотношение, 90% доверительный интервал, коэффициент вариации для одного субъекта и статистическая мощность) тестируемого образца по сравнению с эталонным образцом в отношении метаболита бетагистина - 2-пиридилуксусной кислоты - кратко изложены в таблице 5.
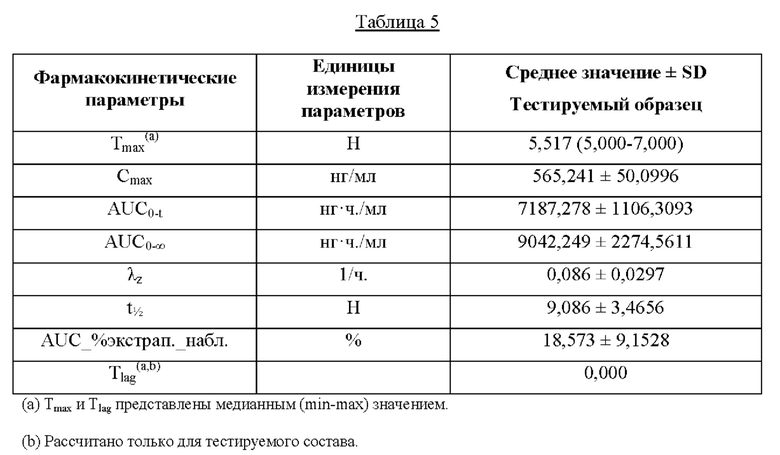
Указанные выше фармакокинетические параметры и значения средней концентрации в плазме метаболита бетагистина - 2-пиридилуксусной кислоты, приведенные в таблицах выше, показали, что введение один раз в сутки композиции по настоящему изобретению, которая содержит 48 мг, обеспечивает более низкий показатель максимальной концентрации в крови и более высокий показатель минимальной концентрации в крови, чем введение два раза в сутки эталонного соединения. Это является преимущественным, поскольку улучшается переносимость активного ингредиента, а также контроль эффективности.
Указанные выше значения также показали, что колебания концентрации в плазме уменьшались. Данное снижение колебаний содержания в плазме обеспечивает возможность поддержания терапевтически эффективного количества бетагистина в плазме в течение 24 часов после введения один раз в сутки и исключения побочных эффектов недостаточной или чрезмерной дозировки, связанных с введением два раз в сутки композиции с немедленным высвобождением, которая содержит 24 мг бетагистина дигидрохлорида (эталонный образец).
Кроме того, было неожиданно обнаружено, что значение AUC, которое представляет собой общее воздействие лекарственного средства, достигаемое после введения один раз в сутки композиции по настоящему изобретению, является биоэквивалентным общему воздействию лекарственного средства, получаемому после введения два раза в сутки эталонного образца. Это означает, что композиция по настоящему изобретению характеризуется сопоставимой биологической доступностью, поскольку скорость и степень того, как бетагистина дигидрохлорид всасывается из композиции по настоящему изобретению и становится доступным в месте действия лекарственного средства, являются такими же, как у эталонного образца. Следовательно, введение один раз в сутки композиции по настоящему изобретению может считаться эквивалентным введению два раза в сутки эталона с немедленным высвобождением в соответствии с рекомендациями в отношении составов с модифицированным высвобождением от Европейского агентства по лекарственным средствам (ЕМА).
В заключение, композиция по настоящему изобретению, которая содержит бетагистин или его фармацевтически приемлемую соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, обеспечивает возможность упрощения расчета дозы бетагистина для пациентов путем уменьшения числа приемов в сутки. Это является преимущественным, поскольку позволяет уменьшить процент пациентов, прекративших лечение, и, как следствие, увеличить эффективность лечения.
Список использованной литературы
1. Kovshel A. Yu. et al, «sustained release betahistine tablets: elaboration of their composition and technology», Pharmacy, 2014, vol. 6, pp. 40.
2. Quality of prolonged release oral solid dosage forms Directive 75/318/EEC October 1992. Section 3AQ19a pp. 167-174.
3. Public Assessment Report of the Medicines Evaluation Board in The Netherlands for betahistine hydrochloride (EU-procedure number: NL/H/1046/01-02/MR; Registration number in the Netherlands: RVG 34738, 34739; 25 March 2008).
4. «Guideline on the pharmacokinetic and clinical evaluation of modified release dosage forms» Европейского агентства по лекарственным средствам (ЕМА), доступно с июня 2017 г. на сайте EMA/CPMP/EWP/280/96Corr1.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТАБЛЕТКА, СОДЕРЖАЩАЯ ЦЕТИРИЗИН И ПСЕВДОЭФЕДРИН | 2002 |
|
RU2286784C2 |
| СОСТАВЫ НА ОСНОВЕ БЕТАГИСТИНА С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2000 |
|
RU2248791C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПРОЛОНГИРОВАННОГО ВЫСВОБОЖДЕНИЯ ФЕНИЛЭФРИНА | 2007 |
|
RU2450803C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ МЕТФОРМИНА С ДЛИТЕЛЬНЫМ ВЫСВОБОЖДЕНИЕМ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2006 |
|
RU2433821C2 |
| КОМПОЗИЦИИ ТРАМАДОЛА ПРОЛОНГИРОВАННОГО ВЫСВОБОЖДЕНИЯ С 24-ЧАСОВЫМ ДЕЙСТВИЕМ | 2003 |
|
RU2328275C2 |
| Фармацевтическая композиция пролонгированного действия на основе 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида и/или основания (Афобазола) | 2017 |
|
RU2694837C2 |
| СПОСОБ ЛЕЧЕНИЯ СЕРДЕЧНО-СОСУДИСТЫХ ЗАБОЛЕВАНИЙ | 1999 |
|
RU2207856C2 |
| ФАРМАЦЕВТИЧЕСКИЕ ЛЕКАРСТВЕННЫЕ ФОРМЫ И КОМПОЗИЦИИ ФЕНИЛЭФРИНА ДЛЯ АБСОРБЦИИ В ОБОДОЧНОЙ КИШКЕ | 2007 |
|
RU2454225C2 |
| ПРЕПАРАТЫ РАНОЛАЗИНА С ПРОЛОНГИРОВАННЫМ ДЕЙСТВИЕМ | 1999 |
|
RU2214233C2 |
| КОМПОЗИЦИИ ГИДРОКОДОНА С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2001 |
|
RU2253452C2 |
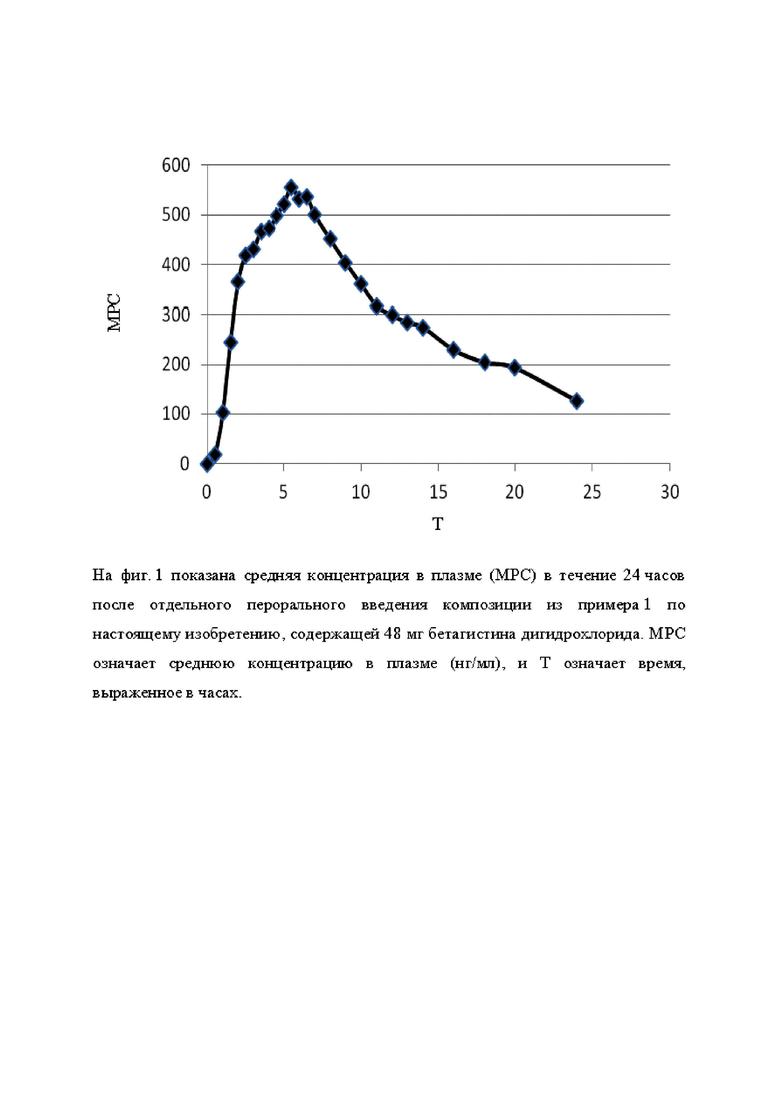
Группа изобретений относится к области медицины, а именно к твердой композиции для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов, содержащей некоторое количество бетагистина или его фармацевтически приемлемой соли, эквивалентное 48 мг бетагистина дигидрохлорида, вместе с одним или несколькими фармацевтически приемлемыми вспомогательными веществами или носителями, где композиция характеризуется профилем растворения, в соответствии с которым: не более 30% по весу бетагистина растворяется за 1 час; от 35 до 45% по весу бетагистина растворяется за 2 часа; от 46 до 60% по весу бетагистина растворяется за 4 часа; от 61 до 80% по весу бетагистина растворяется за 8 часов; от 81 до 97% по весу бетагистина растворяется за 16 часов и от 98 до 100% по весу бетагистина растворяется за 24 часа, где фармацевтически приемлемые вспомогательные вещества или носители предусматривают одно или несколько средств, образующих гидрофильную матрицу, где средство, образующее гидрофильную матрицу, представляет собой смесь гидроксипропилметилцеллюлозы и каррагенана в весовом соотношении от 3:1 до 12:1, и также относится к способу получения, который предусматривает: (a) получение раствора бетагистина или его фармацевтически приемлемой соли в полярном растворителе, выбранном из группы (C1-C4)алкил-CO-(C1-C4)алкила, (C1-C4)алкил)-COO-(C1-C4)алкила, воды и их смеси; (b) просеивание и смешивание одного или нескольких разбавителей, одного или нескольких стабилизаторов и необязательно одного или нескольких связующих с получением смеси; (c) гранулирование смеси со стадии (b) путем добавления раствора со стадии (a) с получением влажных гранул; (d) высушивание влажных гранул, полученных на стадии (c), с получением высушенных гранул; (e) смешивание высушенных гранул, полученных на стадии (d), с одним или несколькими средствами, образующими гидрофильную матрицу, с получением смеси; и (f) прессование смеси, полученной на стадии (e), с образованием таблеток. Группа изобретений обеспечивает предоставление твердой композиции для перорального применения, обеспечивающей более низкий показатель максимальной концентрации в крови (Cmax) и более высокий показатель минимальной концентрации в крови (Cmin) бетагистина, а также метаболита бетагистина – 2-PAA – по сравнению с введением дважды в сутки твердой композиции для перорального применения с немедленным высвобождением, что уменьшает колебания его концентрации в плазме и, следовательно, исключает побочные эффекты недостаточной или чрезмерной дозировки, связанные с введением по меньшей мере двух отдельных лекарственных форм с немедленным высвобождением или отдельной лекарственной формы в виде множества частиц, содержащей лекарственные формы с немедленным и замедленным высвобождением. 2 н. и 7 з.п. ф-лы, 1 ил., 5 табл., 3 пр.
1. Твердая композиция для перорального применения с непульсирующим пролонгированным высвобождением в течение 24 часов, содержащая некоторое количество бетагистина или его фармацевтически приемлемой соли, эквивалентное 48 мг бетагистина дигидрохлорида, вместе с одним или несколькими фармацевтически приемлемыми вспомогательными веществами или носителями, где композиция характеризуется профилем растворения, в соответствии с которым:
не более 30% по весу бетагистина растворяется за 1 час;
от 35 до 45% по весу бетагистина растворяется за 2 часа;
от 46 до 60% по весу бетагистина растворяется за 4 часа;
от 61 до 80% по весу бетагистина растворяется за 8 часов;
от 81 до 97% по весу бетагистина растворяется за 16 часов и
от 98 до 100% по весу бетагистина растворяется за 24 часа,
где фармацевтически приемлемые вспомогательные вещества или носители предусматривают одно или несколько средств, образующих гидрофильную матрицу,
где средство, образующее гидрофильную матрицу, представляет собой смесь гидроксипропилметилцеллюлозы и каррагенана в весовом соотношении от 3:1 до 12:1.
2. Композиция по п. 1, которая содержит дигидрохлоридную соль бетагистина.
3. Композиция по п. 1, где после перорального введения единичной дозы композиции в условиях приема пищи бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, демонстрируют максимальную концентрацию в плазме (Cmax), составляющую от 400 до 700 нг/мл, в пересчете на концентрацию в плазме их метаболита – 2-пиридилуксусной кислоты.
4. Композиция по п. 1, где после перорального введения единичной дозы композиции в условиях приема пищи бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, демонстрируют время до максимальной концентрации в плазме (Tmax), составляющее от 5 до 7 ч, в пересчете на концентрацию в плазме их метаболита – 2-пиридилуксусной кислоты.
5. Композиция по п. 1, где после перорального введения единичной дозы композиции в условиях приема пищи бетагистин или его фармацевтически приемлемая соль в количестве, эквивалентном 48 мг бетагистина дигидрохлорида, демонстрируют площадь под кривой зависимости концентрации в плазме от времени во временном интервале от 0 до 24 часов (AUC (0-24)), составляющую от 5000 до 10000 нг·ч./мл, в пересчете на концентрацию в плазме их метаболита – 2-пиридилуксусной кислоты.
6. Композиция по п. 1, где одно или несколько средств, образующих гидрофильную матрицу, присутствуют в количестве от 20 до 85%, предпочтительно в количестве от 35 до 65%, предпочтительно от 40 до 60%, более предпочтительно от 45 до 55% и еще более предпочтительно от 50 до 55% по весу относительно общего веса композиции.
7. Композиция по любому из пп. 1-6, которая содержит:
некоторое количество бетагистина или его фармацевтически приемлемой соли, эквивалентное 48 мг бетагистина дигидрохлорида;
от 20 до 85% по весу одного или нескольких средств, образующих гидрофильную матрицу, как определено в п. 6;
от 20 до 40% по весу одного или нескольких разбавителей;
от 0,1 до 15% по весу одного или нескольких стабилизаторов;
необязательно от 0,1 до 15% по весу одного или нескольких связующих;
необязательно от 0,1 до 10% по весу одного или нескольких веществ, способствующих скольжению,
и необязательно от 0,1 до 10% по весу одного или нескольких смазывающих веществ;
при этом сумма ингредиентов составляет 100% по весу.
8. Способ получения композиции по любому из пп. 1-7, который предусматривает:
(a) получение раствора бетагистина или его фармацевтически приемлемой соли в полярном растворителе, выбранном из группы (C1-C4)алкил-CO-(C1-C4)алкила, (C1-C4)алкил)-COO-(C1-C4)алкила, воды и их смеси;
(b) просеивание и смешивание одного или нескольких разбавителей, одного или нескольких стабилизаторов и необязательно одного или нескольких связующих с получением смеси;
(c) гранулирование смеси со стадии (b) путем добавления раствора со стадии (a) с получением влажных гранул;
(d) высушивание влажных гранул, полученных на стадии (c), с получением высушенных гранул;
(e) смешивание высушенных гранул, полученных на стадии (d), с одним или несколькими средствами, образующими гидрофильную матрицу, с получением смеси; и
(f) прессование смеси, полученной на стадии (e), с образованием таблеток.
9. Способ по п. 8, дополнительно включающий смешивание смеси, полученной на стадии (e), с одним или несколькими вспомогательными веществами или носителями, выбранными из группы, состоящей из вещества, способствующего скольжению, и смазывающего вещества, с получением смеси, и/или нанесение покрытия на таблетки, полученные на стадии (f).
| WO 2012131722 A1, 04.10.2012 | |||
| Стробоскопическое устройство для измерения фазовых углов горизонтальных составляющих посредством резонансного гармонического анализатора | 1935 |
|
SU53162A1 |
| Vladimir A | |||
| Parfenov et al | |||
| Приспособление для автоматической односторонней разгрузки железнодорожных платформ | 1921 |
|
SU48A1 |
| Устройство для сборки деталей запрессовкой | 1986 |
|
SU1493435A1 |

