Область техники
Настоящее изобретение относится к соединениям бензимидазола и к их применению для лечения или предотвращения респираторно-синцитиальной вирусной (РСВ) инфекции.
Уровень техники
РСВ представляет собой вирус семейства парамиксовирусов (Paramyxoviridae), содержащий отрицательно-полярную одноцепочечную РНК. РСВ легко передается через выделения от инфицированного человека через поверхности или при непосредственном контакте. В отличие от гриппа, он не передается через аэрозоли мелких частиц. После успешной инокуляции инкубационный период продолжается от четырех до шести дней, и за это время вирус распространяется от носоглотки до нижних дыхательных путей посредством слияния инфицированных клеток с неинфицированными и путем отторжения некротического эпителия. В сочетании с повышенной секрецией слизи и отеком у младенцев это может привести к закупорке слизью, что вызывает чрезмерное раздувание и коллапс дистальной легочной ткани, что указывает на бронхиолит. При этом обычно наблюдается гипоксия, и из-за нарушения дыхания часто затрудняется возможность кормления. При РСВ пневмонии воспалительная инфильтрация дыхательных путей состоит из одноядерных клеток и является более генерализованной с вовлечением в патологический процесс бронхиол, бронхов и альвеол. Было обнаружено, что продолжительность и степень выделения вируса в среду коррелируют с клиническими признаками и тяжестью заболевания.
РСВ является главной причиной тяжелых инфекций дыхательных путей у младенцев и детей младшего возраста во всем мире. Самая высокая заболеваемость и смертность наблюдается среди тех детей, кто родился раньше срока, и тех, кто страдает от хронической болезни легких или сердца, хотя многие младенцы, которых госпитализируют по поводу РСВ инфекции, не имеют других заболеваний. Тяжелая РСВ инфекция в младенческом возрасте может привести к возникновению рецидивирующей бронхиальной обструкции в течение нескольких лет и связана с развитием впоследствии астмы.
РСВ также является основной причиной заболеваемости и смертности у пожилых людей и у детей и взрослых с ослабленным иммунитетом, а также тех, кто страдает от хронической обструктивной болезни легких (ХОБЛ) и застойной сердечной недостаточности (ЗСН).
Заболеваемость РСВ имеет сезонный характер; ее можно очень точно прогнозировать, и РСВ наблюдается зимой в обоих полушариях, с сентября по май в Европе и Северной Америке, при этом пик заболеваемости приходится на декабрь и январь, а в тропических странах РСВ может наблюдаться в течение всего года. РСВ поражает >90% младенцев и детей младшего возраста до двух лет, и, поскольку естественный иммунитет является нестойким, многие будут повторно инфицироваться каждый год. Как и в случае гриппа, у пожилых людей РСВ является причиной примерно 10% госпитализаций зимой, при этом связанная с ним смертность составляет 10%.
Существующее лечение, направленное против РСВ, включает применение моноклонального антитела к РСВ, называемого паливизумабом. Такое применение паливизумаба является профилактическим, а не терапевтическим лечением РСВ. Хотя данное антитело часто является эффективным, его применение ограничивается применением у недоношенных детей и младенцев группы высокого риска. Действительно, его ограниченная применимость означает, что оно недоступно для многих людей, нуждающихся в лечении РСВ. Соответственно, существует острая необходимость в разработке эффективных альтернатив существующему лечению РСВ.
Кроме того, в качестве ингибиторов РСВ были предложены некоторые соединения, включая соединения на основе бензимидазола. Например, в K D Combrink et al., Bioorganic & Medicinal Chemistry Letters, 17 (2007), 4784-4790 описано соединение BMS-433771 и его варианты. Другие соединения на основе бензимидазола описаны в WO-02/062290, WO-03/053344 и WO-10/103306.
Краткое описание изобретения
Недавно было обнаружено, что новая группа бензимидазолов представляет собой соединения, проявляющие активность в качестве ингибиторов РСВ. Указанные соединения обладают уменьшенными липофильными свойствами, благоприятными фармакокинетическими и токсикологическими свойствами и могут быть легко приготовлены в виде составов для применения в фармацевтике.
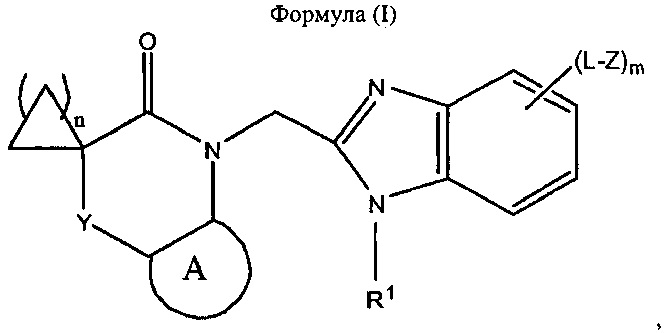
Соответственно, согласно настоящему изобретению предложено соединение, представляющее собой бензимидазол формулы (I):
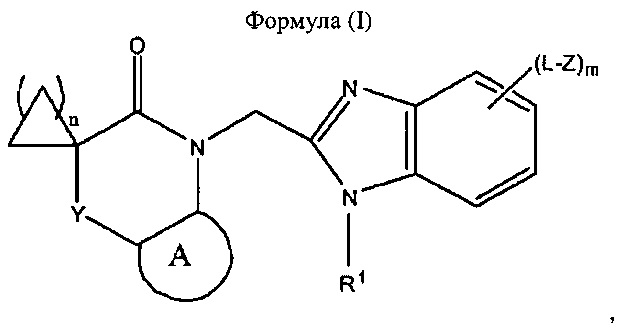
где:
- А представляет собой 5-12-членный арил или 5-12-членный гетероарил, каждый из которых является не содержит заместителей или содержит заместители;
- Y представляет собой одинарную связь, -(СН2)p-, -Х-, -СН2-Х- или -Х-СН2-;
- X представляет собой -О-, -S-, -N(R2)-, >С=O, >S(=O)2, >S(=O)2, -O-С(=O)-, -С(=O)-O-, -N(R2)-C(=O)- или -C(=O)-N(R2)-;
- каждый L независимо представляет собой одинарную связь, С1-3алкилен, С2-3алкенилен или С2-3алкинилен;
- R1 представляет собой C1-6алкил, С2-6алкенил или С2-6алкинил, каждый из которых не содержит заместителей или содержит заместители;
- каждый Z независимо представляет собой -N(R2)2, -OR2, -SR2, -S(=O)R2, -S(=O)2R2;
- каждый R2 независимо представляет собой водород, C1-6алкил, С2-6алкенил или С2-6алкинил, при этом указанная алкильная, алкенильная и алкинильная группы не содержат заместителей или содержат заместители;
- m равен 0, 1, 2 или 3;
- n равен 1, 2 или 3; и
- p равен 1, 2 или 3;
или его фармацевтически приемлемая соль.
Подробное описание изобретения
Когда какая-либо группа, кольцо, заместитель или фрагмент, определенные в настоящем описании, содержат заместители, они, как правило, содержат в качестве заместителя Q, как определено ниже.
С1-6 алкильная группа или фрагмент является линейной или разветвленной. С1-6 алкильная группа, как правило, представляет собой С1-4 алкильную группу или С4-6 алкильную группу. Примеры C1-6 алкильных групп и фрагментов включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-пентил, изопентил (т.е. 3-метилбут-1-ил), трет-пентил (т.е. 2-метилбут-2-ил), неопентил (т.е. 2,2-диметилпропан-1-ил), н-гексил, изогексил (т.е. 4-метилпентан-1-ил), трет-гексил (т.е. 3-метилпентан-3-ил) и неопентил (т.е. 3,3-диметилбутан-1-ил). Во избежание неопределенности, в случае когда два алкильных фрагмента присутствуют в группе, данные алкильные фрагменты могут быть одинаковыми или разными. C1-6 алкильная группа не содержит заместителей или содержит в качестве заместителей, как правило, одну или более групп Q, определенных ниже. Например, C1-6 алкильная группа не содержит заместителей или содержит в качестве заместителей 1, 2 или 3 группы Q, определенные ниже.
Q представляет собой галоген, нитро, -CN, ОН, C1-6алкокси, С1-6гидроксиалкил, C1-6алкилтио, C1-6галогеналкил, С1-4галогеналкокси, -CO2R''', -NR'''2, -SR''', -S(=O)R''', -S(=O)2R''', С3-С10циклоалкил, 5-10-членный гетероциклил, 5-12-членный арил или 5-12-членный гетероарил, где каждый R''' независимо выбран из Н, С1-6алкила, С3-10циклоалкила, 5-10-членного гетероциклила, 5-12-членного арила и 5-12-членного гетероарила.
С1-3 алкиленовая группа или фрагмент представляет собой не содержащую заместителей или содержащую заместители, линейную или разветвленную насыщенную двухвалентную алифатическую углеводородную группу или фрагмент, содержащий от 1 до 3 атомов углерода. Примеры включают метиленовые, этиленовые, н-пропиленовые и изопропиленовые группы и фрагменты. Когда алкиленовая группа содержит заместители, она, как правило, содержит в качестве заместителей группу Q, определенную выше.
С2-6 алкенильная группа представляет собой не содержащий заместителей или содержащий заместители, линейный или разветвленный углеводородный радикал из двух-шести атомов углерода с по меньшей мере одним ненасыщенным участком, т.е. двойной связью углерод-углерод sp2. Алкенильная группа может иметь «цис»- или «транс»-ориентацию, или в качестве альтернативы «Е»- или «Z»-ориентацию. Как правило, она представляет собой С2-4алкенильную группу или С4-6алкенильную группу. Примеры включают этиленил или винил (-СН=СН2) и аллил (-СН2СН=СН2). Когда алкенильная группа содержит заместители, она, как правило, содержит в качестве заместителей группу Q, определенную выше.
С2-3 алкениленовая группа или фрагмент представляет собой линейную или разветвленную, ненасыщенную двухвалентную алифатическую углеводородную группу или фрагмент, содержащий два или три атома углерода, с по меньшей мере одной двойной связью углерод-углерод sp2. Алкениленовая группа может иметь «цис»- или «транс»-ориентацию, или в качестве альтернативы «Е»- или «Z»-ориентацию. Примеры включают группы и фрагменты -СН=СН-, -СН=СНСН2- и -СН2СН=СН-.
С2-6 алкинильная группа представляет собой не содержащий заместителей или содержащий заместители, линейный или разветвленный углеводородный радикал из двух-шести атомов углерода с по меньшей мере одним ненасыщенным участком, т.е. тройной связью углерод-углерод sp. Как правило, она представляет собой С2-4алкинильную группу или С4-6алкинильную группу. Алкинильная группа может иметь «цис»- или «транс»-ориентацию, или в качестве альтернативы «Е»- или «Z»-ориентацию. Примеры включают этинил (-C≡СН) или пропинил (пропаргил, -СН2С≡СН). Когда алкинильная группа содержит заместители, она, как правило, содержит в качестве заместителей одну или более групп Q, определенных выше.
С2-3 алкиниленовая группа представляет собой линейную ненасыщенную двухвалентную алифатическую углеводородную группу или фрагмент, содержащий два или три атома углерода, с одной тройной связью углерод-углерод sp. Алкиниленовая группа может иметь «цис»- или «транс»-ориентацию, или в качестве альтернативы «Е»- или «Z»-ориентацию. Примеры включают группы и фрагменты -С≡С-, -С≡ССН2- и -СН2С≡С-.
C1-6 алкоксигруппа является линейной или разветвленной. Как правило, она представляет собой С1-4 алкоксигруппу, например, метокси-, этокси-, пропокси-, изопропокси-, н-пропокси-, н-бутокси-, втор-бутокси- или трет-бутоксигруппу. C1-6 алкоксигруппа не содержит заместителей или содержит в качестве заместителей, как правило, одну или более групп Q, как определено.
C1-6 алкилтиогруппа является линейной или разветвленной. Как правило, она представляет собой С1-4 алкилтиогруппу, например, метилтио-, этилтио-, пропилтио-, изопропилтио-, н-пропилтио-, н-бутилтио-, втор-бутилтио- или трет-бутилтиогруппу. C1-6 алкилтиогруппа не содержит заместителей или содержит в качестве заместителей, как правило, одну или более групп Q, как определено.
Галоген или галогеногруппа представляет собой F, Cl, Br или I. Предпочтительно, она представляет собой F, Cl или Br. C1-6 алкильная группа, содержащая в качестве заместителей галоген, может быть обозначена «C1-6галогеналкил», что означает C1-6 алкильную группу, определенную выше, в которой один или более атомов водорода замещены на галоген. Подобным образом, C1-6 алкоксигруппа, содержащая в качестве заместителей галоген, может быть обозначена «C1-6галогеналкокси», что означает C1-6 алкоксигруппу, определенную выше, в которой один или более атомов водорода замещены на галоген. Как правило, C1-6 галогеналкил или С1-6галогеналкокси содержит в качестве заместителей 1, 2 или 3 указанных атома галогена. Галогеналкильные и галогеналкоксигруппы включают пергалогеналкильные и пергалогеналкоксигруппы, такие как -СХ3 и -OCX3, где X представляет собой галоген, например, -CF3 -CCl3 -OCF3 и -OCCl3.
C1-6 гидроксиалкильная группа представляет собой C1-6 алкильную группу, определенную выше, которая содержит в качестве заместителей одну или более ОН-групп. Как правило, она содержит в качестве заместителей одну, две или три ОН-группы. Предпочтительно, она содержит в качестве заместителей одну ОН-группу.
5-12-членная арильная группа представляет собой ароматическую карбоциклическую группу, содержащую от 5 до 12 атомов углерода, например, от 6 до 10 атомов углерода, например, 6 или 10 атомов углерода. Она представляет собой моноциклическую или конденсированную бициклическую кольцевую систему, в которой ароматическое кольцо конденсировано с другим ароматическим карбоциклическим кольцом. Примеры 5-12-членной арильной группы включают фенил и нафталенил. Когда арильная группа содержит заместители, она, как правило, содержит в качестве заместителей С1-4алкил или группу Q, определенную выше, например, 1, 2 или 3 группы, выбранные из С1-4 алкильной группы и группы Q, определенной выше.
Аралкильная группа представляет собой арильную группу, определенную выше, присоединенную к алкильной группе, определенной выше. Примеры включают бензил.
С3-10 циклоалкильная группа представляет собой насыщенное углеводородное кольцо, содержащее от 3 до 10 атомов углерода. С3-10 циклоалкильная группа может представлять собой, например, С3-С7циклоалкил, такой как циклопропил, циклобутил, циклопентил, циклогексил, или циклогептил. Как правило, он представляет собой С3-С6циклоалкил, например, циклопропил, циклобутил или циклопентил. В одном из вариантов реализации она представляет собой циклопропил. С3-10 циклоалкильная группа не содержит заместителей или содержит в качестве заместителей, как правило, одну или более групп Q, определенных выше.
5-12-членная гетероарильная группа или фрагмент представляет собой 5-12-членную ароматическую гетероциклическую группу, которая содержит 1, 2, 3 или 4 гетероатома, выбранных из О, N и S. Она является моноциклической или бициклической. Как правило, она содержит один атом N и 0, 1, 2 или 3 дополнительных гетероатома, выбранных из О, S и N. Она может представлять собой, например, 5-7-членную гетероарильную группу, например, 5- или 6-членную N-содержащую гетероарильную группу. Примеры включают пиридильную, пиразинильную, пиримидинильную, пиридазинильную, фуранильную, тиенильную, пиразолидинильную, пирролильную, оксадиазолильную, оксазолильную, изоксазолильную, тиазолильную, тиадиазолильную, имидазолильную и пиразолильную группы. Фуранильная, тиенильная, пиридильная и пиримидильная группы являются предпочтительными. Когда гетероарильная группа содержит заместители, она, как правило, содержит в качестве заместителей одну или более, например, 1, 2 или 3 группы, выбранные из С1-4 алкила и группы Q, определенной выше.
5-10-членный гетероциклильный фрагмент представляет собой моноциклическое или бициклическое неароматическое насыщенное или ненасыщенное С5-10 карбоциклическое кольцо, в котором по меньшей мере один, например, 1, 2 или 3 атома углерода в кольце замещены на атом или группу, выбранную из О, S, SO, SO2, СО и N. Как правило, он представляет собой насыщенное С5-10 кольцо, в котором 1, 2 или 3 атома углерода в кольце замещены на атом или группу, выбранную из О, S, SO2, СО и NH. Чаще он представляет собой моноциклическое кольцо, предпочтительно моноциклическое C5-С6 кольцо. Примеры включают пиперидильный, пиперидин-2,6-дионильный, пиперидин-2-онильный, пиперазинильный, морфолинильный, тиоморфолинильный, S,S-диоксотиоморфолинильный, 1,3-диоксоланильный, пирролидинильный, имидазол-2-онильный, пирролидин-2-онильный, тетрагидрофуранильный и тетрагидропиранильный фрагменты.
Во избежание неопределенности, хотя приведенные выше определения гетероарильных и гетероциклильных групп относятся к атому «N», который может присутствовать в кольце, как будет понятно специалисту в области химии, указанный атом N будет протонированным (или будет иметь заместитель, определенный выше), если он присоединен к каждому из смежных атомов кольца через одинарную связь. Такие протонированные формы включены в определения гетероарильных групп и гетероциклильных групп, приведенные в настоящем описании.
В формуле (I), определенной выше, А, как правило, не содержит заместителей. В одном из вариантов реализации А представляет собой фенил или 5- или 6-членную N-содержащую гетероарильную группу. Например, А представляет собой фенил или пиридил.
В формуле (I), определенной выше, Y, как правило, представляет собой одинарную связь, -О-, -C(=O)-N(R2)- или -(СН2)p-. Предпочтительно, Y представляет собой одинарную связь, -О-, -C(=O)-NH или -СН2-. Во избежание неопределенности, левая часть двухвалентных фрагментов Y, изображенных в настоящем описании, присоединена к кольцу А, а правая часть присоединена к атому углерода в «голове» мостикового фрагмента спиро-фрагмента.
В формуле (I) каждый L, как правило, представляет собой С1-3алкилен. В одном из вариантов реализации каждый L представляет собой -СН2-.
R1, как правило, не содержит заместителей. В одном из вариантов реализации R1 представляет собой C1-6алкил. В другом варианте реализации R1 представляет собой разветвленный С3-6алкил, разветвленный С3-6алкенил или разветвленный С4-6алкинил. Как правило, R1 представляет собой разветвленный С4-6алкил. Предпочтительно, R1 представляет собой изопентил.
Каждый R2, как правило, представляет собой Н или С1-4алкил. Чаще каждый R2 представляет собой Н или метил. Предпочтительно, каждый R2 представляет собой Н.
Каждый Z, как правило, представляет собой -N(R2)2 или -OR2. Чаще каждый Z представляет собой -N(R2)2. В одном из вариантов реализации каждый Z независимо представляет собой -NHCH3, -N(CH3)2 или -NH2. Более предпочтительно, каждый Z представляет собой -NH2.
Когда m равен 1, 2 или 3, фрагмент -L-Z, как правило, присутствует в положении 5 бензимидазольного фрагмента. В одном из вариантов реализации m равен 0 или 1. Когда m равен 1, фрагмент -L-Z, как правило, присутствует в положении 5 бензимидазольного фрагмента. Предпочтительно, в данном варианте реализации -L-Z представляет собой фрагмент -CH2NH2.
В формуле (I) n равен 1, 2 или 3. Например, он равен 1 или 2, или он равен 2 или 3.
В формуле (I) p равен 1, 2 или 3. Например, он равен 1 или 2. Как правило, p равен 1.
В одном из вариантов формулы (I):
- А представляет собой не содержащую заместителей фенильную группу или не содержащую заместителей пиридильную группу;
- Y представляет собой одинарную связь, -О-, -C(=O)-NH- или -СН2-;
- L представляет собой -СН2-;
- R1 представляет собой разветвленную не содержащую заместителей С4-6 алкильную группу;
- Z представляет собой -NH2; и
- m равен 0 или 1; и
- n равен 1, 2 или 3.
В предпочтительном варианте реализации бензимидазол формулы (I) имеет следующую формулу (Ia):
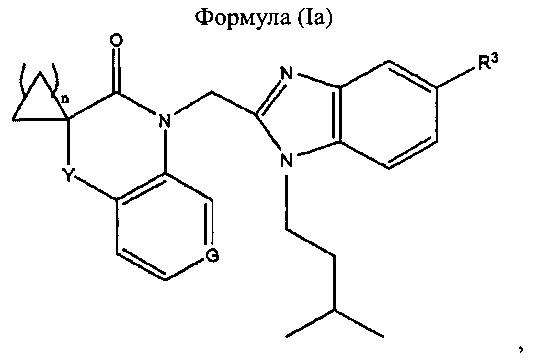
где
- G представляет собой N или СН
- Y представляет собой одинарную связь, -О-, -C(=O)-NH- или -СН2-;
- n равен 1, 2 или 3; и
- R3 представляет собой Н или -CH2NH2.
Конкретные примеры соединений согласно настоящему изобретению включают:
1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклопентан-1,3'-пирроло[2,3-с]пиридин]-2'(1'H)-он;
1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклопропан-1,3'-индолин]-2'-он;
1'-((1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклопропан-1,3'-индолин]-2'-он;
1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклопентан-1,3'-индолин]-2'-он;
1'-((1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклопентан-1,3'-индолин]-2'-он;
1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклобутан-1,3'-индолин]-2'-он;
1'-((1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклобутан-1,3'-индолин]-2'-он;
4-((5-(аминометил)-1-изопентил-1Н-бензо[d]имидазол-2-ил)метил)спиро[бензо[b][1,4]-оксазин-2,1'-циклопропан]-3(4Н)-он;
4-((1-изопентил-1Н-бензо[d]имидазол-2-ил)метил)спиро[бензо[b][1,4]оксазин-2,1'-циклопропан]-3(4Н)-он;
1'-((5-(аминометил)-1-изопентил-1Н-бензо[d]имидазол-2-ил)метил)-1'H-спиро[циклопропан-1,3'-хинолин]-2'(4'Н)-он;
1'-((1-изопентил-1Н-бензо[d]имидазол-2-ил)метил)-1'H-спиро[циклопропан-1,3'-хинолин]-2'(4'H)-он;
1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)-1'H-спиро[цикло-пентан-1,3'-хинолин]-2'(4'Н)-он;
1'-((1-изопентил-1H-бензо[d]имидазол-2-ил)метил)-1'H-спиро[циклопентан-1,3'-хинолин]-2'(4'H)-он; и
1-((1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[бензо[е][1,4]диазепин-3,1'-цикло-пропан]-2,5(1Н,4Н)-дион;
и их фармацевтически приемлемые соли.
Предпочтительные соединения согласно настоящему изобретению включают:
1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклопентан-1,3'-пирроло[2,3-с]пиридин]-2'(1'H)-он;
1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклопропан-1,3'-индолин]-2'-он;
1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклопентан-1,3'-индолин]-2'-он; и
1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклобутан-1,3'-индолин]-2'-он.
Соединения согласно настоящему изобретению могут содержать асимметрические или хиральные центры и, следовательно, существовать в разных стереоизомерных формах. Предполагается, что все стереоизомерные формы соединений согласно настоящему изобретению, включая, но не ограничиваясь ими, диастереомеры, энантиомеры и атропоизомеры, а также их смеси, такие как рацемические смеси, составляют часть настоящего изобретения. Соединения Формулы (I), содержащие один или более хиральных центров, можно использовать в энантиомерно или диастереоизомерно чистой форме, или в виде смеси изомеров.
Настоящее изобретение включает все геометрические и позиционные изомеры соединений согласно настоящему изобретению, определенных выше. Например, если соединение согласно настоящему изобретению содержит двойную связь или конденсированное кольцо, цис- и транс-формы, а также их смеси включены в объем настоящего изобретения. В объем настоящего изобретения также включены как отдельные позиционные изомеры, так и смеси позиционных изомеров.
Соединения согласно настоящему изобретению могут существовать в несольватированных, а также в сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этиловый спирт и т.п., и предполагается, что настоящее изобретение включает как сольватированные, так и несольватированные формы.
Соединения согласно настоящему изобретению могут существовать в разных таутомерных формах, и все такие формы включены в объем настоящего изобретения. Термин «таутомер» или «таутомерная форма» относится к структурным изомерам с разной энергией, которые подвергаются взаимным превращениям через низкий энергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимопревращения за счет миграции протона, такие как кето-енольная таутомеризация. Валентные таутомеры включают взаимопревращения путем перестройки некоторых связывающих электронов.
Соединения согласно настоящему изобретению могут быть получены в соответствии со следующими схемами реакций 1 и 2, на которых A, Y, L, R1, m и n в формулах (IPro), (II) и (III) являются такими, как определено выше для формулы (I).
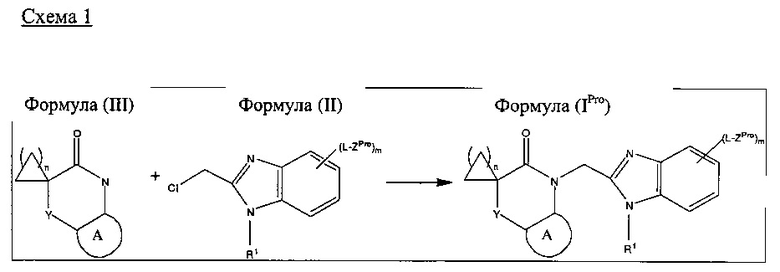
Соединения Формулы (I), в которых m равен 1, 2 или 3, могут быть получены в виде соединения Формулы (IPro), в котором ZPro представляет собой Z, или через соединение Формулы (IPro), в котором ZPro представляет собой защищенное производное Z, определенного выше. Подходящее защищенное производное Z для любого конкретного Z хорошо известно в данной области техники и может быть выбрано специалистом в области химии, например, ZPro может представлять собой BOC-защищенную аминогруппу, когда Z представляет собой -NH2. Соединение Формулы (IPro) может быть получено путем проведения реакции соединения Формулы (III) с соединением Формулы (II) в соответствующих условиях, например, условиях, используемых в Примерах 1 и 3 ниже.
Соединения Формулы (I), в которых m равен нулю, могут быть получены в соответствии с процедурой, описанной на схеме 1, без какого-либо дополнительного этапа удаления защитных групп. Подобным образом, соединения формулы (IPro), в которых m равен 1, 2 или 3, и ZPro представляет собой Z, соответствуют соединению Формулы (I), и удаление защитных групп не требуется.
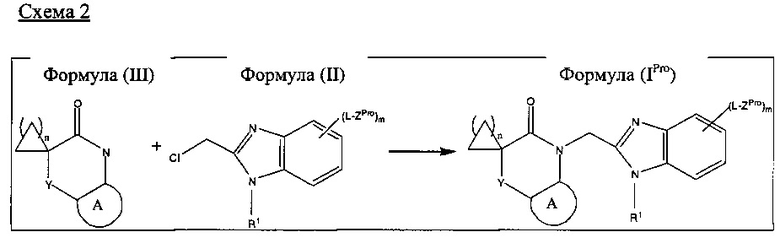
В соответствии с другим аспектом настоящего изобретения предложен способ получения соединения согласно настоящему изобретению, определенного выше, который включает обработку соединения формулы (III) соединением Формулы (II):
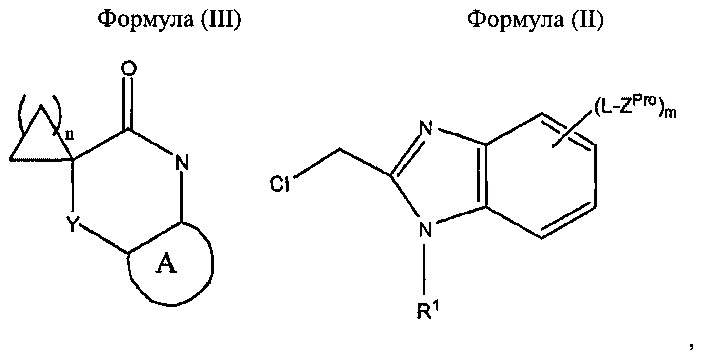
где A, Y, L, R1, m и n являются такими, как определено выше, и ZPro представляет собой Z, определенный выше, или защищенное производное Z; и, когда m равен 1, 2 или 3, и ZPro представляет собой защищенное производное Z, удаление защитных групп из полученного продукта.
Бензимидазол формулы (I) может быть получен путем удаления защитных групп из соединения формулы (IPro), определенного выше, в котором m равен 1, 2 или 3, и ZPro представляет собой защищенное производное Z, с использованием соответствующих реагентов и условий, которые могут быть легко определены специалистом в данной области техники в соответствии с природой ZPro. Например, когда ZPro представляет собой BOC-защищенную аминогруппу, защитная группа может быть удалена из соединения Формулы (IPro) с помощью концентрированной HCl.
A, Y, L, R1, m и n в Формулах (IPro), (II) и (III) являются такими, как определено выше для соединений Формулы (I). Соединения Формул (II) и (III) представляют собой известные соединения или могут быть получены по аналогии с известными способами.
Бензимидазол формулы (I) может быть переведен в его фармацевтически приемлемую соль, и соль может быть переведен в свободное соединение обычными способами. Например, бензимидазол формулы (I) может быть приведен в контакт с фармацевтически приемлемой кислотой с образованием фармацевтически приемлемой соли. Фармацевтически приемлемая соль представляет собой соль, образованную фармацевтически приемлемой кислотой или основанием.
Фармацевтически приемлемые кислоты включают как неорганические кислоты, такие как хлороводородная, серная, фосфорная, дифосфорная, бромистоводородная или азотная кислота, так и органические кислоты, такие как лимонная, фумаровая, малеиновая, яблочная, аскорбиновая, янтарная, винная, бензойная, уксусная, метансульфоновая, этансульфоновая, бензолсульфоновая или п-толуолсульфоновая кислота. Фармацевтически приемлемые основания включают гидроксиды щелочных металлов (например, натрия или калия) и щелочноземельных металлов (например, кальция или магния), и органические основания, такие как алкиламины, аралкиламины и гетероциклические амины.
В биологических тестах было обнаружено, что соединения согласно настоящему изобретению являются ингибиторами респираторно-синцитиального вируса (РСВ). Следовательно, данные соединения являются терапевтически полезными. Соответственно, согласно настоящему изобретению также предложено соединение, которое представляет собой бензимидазол формулы (I), определенной выше, или его фармацевтически приемлемую соль, для применения в способе лечения организма человека или животного посредством терапии. В соответствии с настоящим изобретением также предложено соединение согласно настоящему изобретению, определенное выше, для применения в способе лечения или предотвращения РСВ инфекции. В соответствии с настоящим изобретением также предложено применение соединения согласно настоящему изобретению, определенного выше, для изготовления лекарственного средства для применения для лечения или предотвращения РСВ инфекции. Таким образом, субъекта, страдающего от или подверженного РСВ инфекции, можно лечить с помощью способа, включающего введение указанному субъекту соединения согласно настоящему изобретению, определенного выше. Тем самым можно улучшать или облегчать состояние субъекта.
РСВ инфекция, как правило, представляет собой инфекцию дыхательных путей. РСВ инфекция может представлять собой инфекцию у ребенка, например ребенка в возрасте до десяти лет или младенца в возрасте до двух лет. В одном из вариантов реализации настоящего изобретения предложено соединение, определенное выше, для применения для лечения или предотвращения РСВ инфекции у пациентов детского возраста. В качестве альтернативы указанная инфекция может представлять собой инфекцию у взрослого зрелого возраста или пожилого взрослого, например, взрослого старше 60 лет, взрослого старше 70 лет или взрослого старше 80 лет. Согласно настоящему изобретению также предложено соединение для применения для лечения или предотвращения РСВ инфекции у пожилых пациентов.
РСВ инфекция может представлять собой инфекцию у индивидуума с ослабленным иммунитетом или индивидуума, страдающего от ХОБЛ или ЗСН. В другом варианте реализации РСВ инфекция представляет собой инфекцию у индивидуума без нарушений функций организма, например, индивидуума, который не имеет других заболеваний.
Соединение согласно настоящему изобретению можно вводить в различных лекарственных формах, например, перорально, как например, в форме таблеток, капсул, таблеток с сахарным или пленочным покрытием, жидких растворов или суспензий, или парентерально, например, внутримышечно, внутривенно или подкожно. Следовательно, соединение может быть введено путем инъекции, инфузии или путем ингаляции или распыления.
Доза зависит от различных факторов, включая возраст, массу тела и состояние пациента, и путь введения. Суточные дозы могут варьироваться в широких пределах и будут корректироваться в соответствии с потребностями индивидуума в каждом конкретном случае. Однако, как правило, доза, принятая для каждого пути введения, когда соединение вводят отдельно взрослым людям, составляет от 0,0001 до 650 мг/кг, чаще всего находится в диапазоне от 0,001 до 10 мг/кг массы тела, например, от 0,01 до 1 мг/кг. Такая доза может быть введена, например, от 1 до 5 раз в сутки. Для внутривенной инъекции подходящая суточная доза составляет от 0,0001 до 1 мг/кг массы тела, предпочтительно от 0,0001 до 0,1 мг/кг массы тела. Суточная доза может быть введена в виде однократной дозы или в соответствии с режимом введения разделенной дозы.
Единичная лекарственная форма, такая как таблетка или капсула, обычно будет содержать 1-250 мг активного ингредиента. Например, соединение формулы (I) может быть введено пациенту, представляющему собой человека, в дозе, составляющей 100-250 мг, либо один раз в сутки, два раза, либо три раза в сутки. Например, соединение формулы (I) может быть введено пациенту, представляющему собой человека, в дозе, составляющей 100-250 мг, либо один раз в сутки, два раза, либо три раза в сутки.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы сами по себе. В качестве альтернативы, они могут быть введены в форме фармацевтической композиции. Следовательно, согласно настоящему изобретению также предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, определенную выше, совместно с фармацевтически приемлемым адъювантом, разбавителем или носителем. Традиционные процедуры выбора и получения подходящих фармацевтических составов описаны, например, в "Pharmaceuticals - The Science of Dosage Form Designs", M.E. Aulton, Churchill Livingstone, 1988.
В зависимости от способа введения фармацевтическая композиция будет предпочтительно содержать от 0,05 до 99 масс. % (массовых процентов), более предпочтительно от 0,05 до 80 масс. %, более предпочтительно от 0,10 до 70 масс. % и еще более предпочтительно от 0,10 до 50 масс. % активного ингредиента; все массовые проценты приведены из расчета на общую массу композиции.
В соответствии с настоящим изобретением также предложен способ получения фармацевтической композиции согласно настоящему изобретению, включающий смешивание соединения формулы (I) или его фармацевтически приемлемой соли, определенной выше, с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Соединения согласно настоящему изобретению могут быть введены в различных лекарственных формах. Таким образом, они могут быть введены перорально, например, в виде таблеток, лепешек, пастилок, водных или масляных суспензий, растворов, диспергируемых порошков или гранул. Соединения согласно настоящему изобретению также могут быть введены парентерально либо подкожно, внутривенно, внутримышечно, внутригрудинно, трансдермально, способами инфузии, либо путем ингаляции или распыления. Соединения также могут быть введены в виде суппозиториев.
Твердые пероральные формы фармацевтической композиции согласно настоящему изобретению могут содержать наряду с активным соединением разбавители, например, лактозу, декстрозу, сахарозу, целлюлозу, кукурузный крахмал или картофельный крахмал; смазывающие вещества, например, диоксид кремния, тальк, стеариновую кислоту, стеарат магния или кальция, и/или полиэтиленгликоли; связующие вещества, например, крахмалы, аравийскую камедь, желатин, метилцеллюлозу, карбоксиметилцеллюлозу или поливинилпирролидон; дезагрегирующие агенты, например, крахмал, альгиновую кислоту, альгинаты или крахмалгликолят натрия; вспенивающиеся смеси; красители; подсластители; смачивающие агенты, такие как лецитин, полисорбаты, лаурилсульфаты; и, в целом, нетоксичные и фармакологически неактивные вещества, используемые в фармацевтических составах. Такие фармацевтические препараты могут быть изготовлены известным способом, например, способами смешивания, гранулирования, таблетирования, нанесения сахарного покрытия или нанесения пленочного покрытия.
Жидкие дисперсии для перорального введения могут представлять собой сиропы, эмульсии и суспензии. Сиропы могут содержать в качестве носителей, например, сахарозу или сахарозу с глицерином и/или маннит, и/или сорбит.
Суспензии и эмульсии могут содержать в качестве носителя, например, природную камедь, агар, альгинат натрия, пектин, метилцеллюлозу, карбоксиметилцеллюлозу или поливиниловый спирт. Суспензии или растворы для внутримышечных инъекций могут содержать наряду с активным соединением фармацевтически приемлемый носитель, например, стерильную воду, оливковое масло, этилолеат, гликоли, например, пропиленгликоль и при необходимости подходящее количество гидрохлорида лидокаина. Другие подходящие носители для суспензий включают стерильную воду, гидроксипропилметилцеллюлозу (ГПМЦ), полисорбат 80, поливинилпирролидон (ПВП), аэрозоль АОТ (т.е. натрия 1,2-бис(2-этилгексоксикарбонил)этансульфонат), плюроник F127 и/или каптизол (т.е. простой сульфобутиловый эфир-бета-циклодекстрин).
Соединения согласно настоящему изобретению могут, например, быть приготовлены в виде водных суспензий в носителе, выбранном из:
(i) 0,5% масс./об. гидроксипропилметилцеллюлозы (ГПМЦ)/0,1% масс./об. полисорбата 80;
(ii) 0,67% масс./об. поливинилпирролидона (ПВП)/0,33% масс./об. аэрозоля АОТ (натрия 1,2-бис(2-этилгексоксикарбонил)этансульфоната);
(iii) 1% масс./об. плюроника F 127; и
(iv) 0,5% масс./об. полисорбата 80.
Носители могут быть получены с использованием стандартных процедур, известных специалисту в данной области техники. Например, каждый из носителей (i)-(iv) может быть получен путем взвешивания необходимого количества вспомогательного вещества в подходящий сосуд, добавления приблизительно 80% от конечного объема воды и перемешивания с помощью магнитной мешалки до образования раствора. Затем носитель доводят до объема водой. Водные суспензии соединений формулы I могут быть получены путем взвешивания необходимого количества соединения формулы I в подходящий сосуд, добавления 100% от необходимого объема носителя и перемешивания с помощью магнитной мешалки.
Растворы для инъекции или инфузии могут содержать в качестве носителя, например, стерильную воду или предпочтительно они могут находиться в форме стерильных водных изотонических солевых растворов.
Соединения согласно настоящему изобретению также могут быть введены в сочетании с другими соединениями, применяемыми для лечения вирусных инфекций. Таким образом, настоящее изобретение также относится к комбинированной терапии, при которой соединение согласно настоящему изобретению или его фармацевтически приемлемую соль, или фармацевтическую композицию или состав, содержащий соединение согласно настоящему изобретению, вводят одновременно или последовательно, или в виде комбинированного препарата с другим терапевтическим агентом или агентами для лечения или предотвращения вирусной инфекции, в частности, инфекции, вызываемой РСВ.
В настоящем описании, когда употребляется термин «комбинация», следует понимать, что он относится к одновременному, раздельному или последовательному введению. В соответствии с одним из аспектов настоящего изобретения термин «комбинация» относится к одновременному введению. В соответствии с другим аспектом настоящего изобретения термин «комбинация» относится к раздельному введению. В соответствии с другим аспектом настоящего изобретения термин «комбинация» относится к последовательному введению. В случае, когда введение является последовательным или раздельным, отсрочка введения второго компонента не должна приводить к снижению полезного эффекта комбинации.
Подходящие терапевтические агенты для применения в комбинированной терапии включают
(i) ингибиторы нуклеокапсидного (N)-белка РСВ;
(ii) другие ингибиторы белка РСВ, такие как те, которые ингибируют белок фосфопротеин (Р) и крупный белок (L);
(iii) моноклональные антитела против РСВ, такие как антитела к F-белку;
(iv) иммуномодулирующие соединения, представляющие собой toll-подобные рецепторы;
(v) другие противовирусные средства для лечения респираторного вируса, такие как противогриппозные и противориновирусные соединения; и/или
(vi) противовоспалительные соединения.
Нуклеокапсидный (N)-белок РСВ играет ключевую роль в транскрипции и репликации вируса, опосредуя взаимодействие между геномной РНК и кодируемой вирусом РНК-зависимой РНК-полимеразой. Р- и L-белки РСВ являются компонентами кодируемой вирусом РНК-зависимой РНК-полимеразы РСВ.
В соответствии с другим аспектом настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, определенная выше, в комбинации с одним или более терапевтическими агентами, перечисленными в пп. (i)-(vi) выше, для применения для лечения РСВ.
Следующие Примеры иллюстрируют настоящее изобретение. Однако они никоим образом не ограничивают настоящее изобретение.
Примеры
Все температуры приведены в °С. Тонкослойную хроматографию (ТСХ) проводили на А1 пластинах, покрытых Si 60G, с УФ-индикатором uv254 (Polygram). Все спектры ЯМР получали при 400 МГц в CDCl3, если не указано иное.
Условия анализа ЖХ-МС
Образцы анализировали на MicroMass Quattro Ultima с использованием электрораспыления с одновременным детектированием положительных-отрицательных ионов.
Колонка: Phenomenex Luna RP 50×3 мм, 3 мкМ.
Элюенты: А - Н2О, 0,1% муравьиная кислота; В - МеОН, 0,1% муравьиная кислота
Детектирование: НР1100 210-400 нм
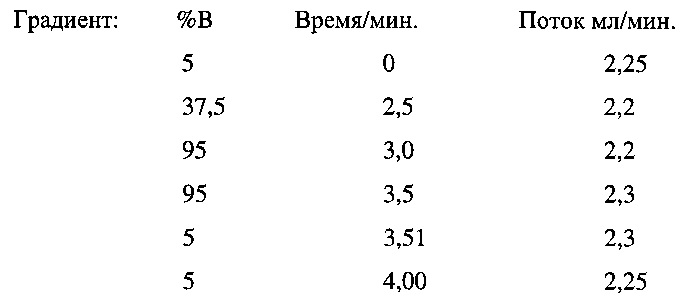
Условия препаративной ВЭЖХ
Градиент выбран в соответствии со временем удерживания аналитической ВЭЖХ,
т.е. для времени удерживания = 3,4 мин.
Преп. колонка: Phenomenex Luna RP 100×21,2 мм, 5 мкМ
Растворители: А - Вода со степенью чистоты, соответствующей требованиям ВЭЖХ + 0,1% муравьиная кислота
В - ацетонитрил

Сокращения
Пример получения 1: Этил-2-(3-нитропиридин-4-ил)ацетат
В сухой колбе в атмосфере азота при комнатной температуре быстро перемешивали трет-бутоксид калия (22,8 г, 203,4 ммоль) в ТГФ (68 мл) и в то же время по каплям добавляли раствор 3-нитропиридина (2,1 г, 16,95 ммоль) и метилхлорацетата (2,46 мл, 28,32 ммоль) в ТГФ (68 мл). Через 1 час добавляли 25% водный раствор хлорида аммония и смесь экстрагировали EtOAc. Объединенные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (20-50% EtOAc в изогексане) позволяла выделить целевое соединение (1,5 г, 7,63 ммоль, 45%).
1Н ЯМР (400 МГц): δ 3,75 (s, 3Н), 4,09 (s, 2Н), 7,36 (dd, 1H), 8,80 (dd, 1H), 9,32 (d, 1H). ЖХ/МС 197 (МН+).
Пример получения 2: Этил-1-(3-нитропиридин-4-ил)циклопентанкарбоксилат
В сухой колбе в атмосфере азота растворяли этил-2-(3-нитропиридин-4-ил)ацетат (265 мг, 1,35 ммоль, Пример получения 1) в MeOH (2,7 мл), обрабатывали 1,5-дийодбутаном (0,9 мл, 6,75 ммоль) при комнатной температуре. По каплям добавляли метоксид натрия (6,6 мл 0,5 М раствора в MeOH) при комнатной температуре. Полученный темно-фиолетовый раствор перемешивали при комнатной температуре в течение 16 часов. К смеси добавляли воду, а затем концентрировали в вакууме. Разделяли остаток между EtOAc и водой с последующей экстракцией водной фазы EtOAc. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (100% изогексан и 25% EtOAc в изогексане) позволяла выделить целевое соединение (110 мг, 0,44 ммоль, 32%).
1Н ЯМР (400 МГц): δ 1,75 (m, 2Н), 1,975 (m, 2Н), 2,05 (m, 2Н), 2,575 (m, 2Н), 3,67 (s, 3Н), 7,49 (d, 1Н), 8,79 (d, 1Н), 9,08 (s, 1H). ЖХ/МС 251 (МН+).
Пример получения 3: Спиро[циклопентан-1,3'-пирроло[2,3-с]пиридин]-2'(1'Н)-он
Этил-1-(3-нитропиридин-4-ил)циклопентанкарбоксилат (110 мг, 0,44 ммоль, Пример получения 2) в EtOH (12 мл) добавляли в сухую колбу в атмосфере азота, в которую помещен 10% палладий на угле (20 мг). Колбу продували водородом и перемешивали в течение 6 часов в данной атмосфере водорода. После продувания колбы азотом осуществляли фильтрацию через Целит и концентрирование в вакууме. Остаток переносили в Et2O (10 мл) и обрабатывали 5 мл 2 М водной соляной кислоты и перемешивали в течение 16 часов. Водную фазу отделяли и концентрировали в вакууме.
Остаток обрабатывали трихлоридом титана (2 мл 40% раствора в 20-30% соляной кислоте) в течение 6 часов. Добавляли твердый NaHCO3 для нейтрализации смеси и водную фазу экстрагировали EtOAc. Объединенные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Целевой продукт выделяли в виде белого твердого вещества (75 мг, 91%).
1Н ЯМР (400 МГц): δ 1,77 (m, 2Н), 1,95 (m, 6Н), 7,35 (d, 1H), 8,05 (s, 1H), 8,21 (d, 1Н), 10,5 (bs, 1H). ЖХ/МС 189 (МН+).
Пример получения 4: Спиро[циклопропан-1,3'-индолин]-2'-он
N-4-метоксибензил-3-спироциклопропилоксиндол (837 мг, 3 ммоль) растворяли в ТФУ (4,6 мл) в атмосфере азота и добавляли анизол (0,66 мл, 6 ммоль). Смесь нагревали при 60°С в течение 16 часов. Смесь охлаждали до комнатной температуры и концентрировали в вакууме. Остаток переносили в дихлорметан (12 мл). Добавляли солевой раствор и триэтиламин (2,5 мл). Водную фазу экстрагировали ДХМ. Объединенные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (20-40% EtOAc в изогексане) позволяла выделить целевой спироциклопропилоксиндол в виде бледно-розового твердого вещества (318 мг, 2 ммоль, 67%).
1Н ЯМР (400 МГц): δ 1,57 (m, 2Н), 1,79 (m, 2Н), 6,83 (d, 1H), 7,01 (m, 2Н), 7,21 (m, 1H), 9,18 (bs, 1Н). ЖХ/МС 160 (МН+).
Пример получения 5: 1-(4-метоксибензил)индолин-2-он
N-(4-метоксибензил)изатин (2,67 г) порциями добавляли к гидразина гидрату (20 мл) при комнатной температуре. Смесь нагревали при 95°С в течение 40 часов. Смесь охлаждали, разделяли между EtOAc и водой. Органическую фазу промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с выделением целевого соединения в виде оранжевого твердого вещества (1,82 г, 7,2 ммоль, 72%).
1Н ЯМР (400 МГц): δ 3,53 (s, 2Н), 3,70 (s, 3Н), 4,78 (s, 2Н), 6,66 (d, 1Н), 6,76 (d, 2Н), 6,95 (dd, 1H), 7,10 (dd, 1H), 7,37 (m, 3Н). ЖХ/МС 254 (МН+).
Пример получения 6: 1'-(4-метоксибензил)спиро[циклопропан-1,3'-индолин]-2'-он
В сухой колбе в атмосфере азота 1-(4-метоксибензил)индолин-2-он (264 мг, 1,04 ммоль, Пример получения 5) в ДМФА (1,5 мл) обрабатывали 1,2-дибромэтаном (0,1 мл, 1,18 ммоль) и данную смесь охлаждали до 0°С. При этой температуре порциями добавляли гидрид натрия (86 мг, 2,14 ммоль). Убирали охлаждающую баню и добавляли дополнительное количество гидрида натрия (43 мг, 1,07 ммоль), как только смесь нагревалась до комнатной температуры. Смесь перемешивали при комнатной температуре в течение 16 часов. Осторожно добавляли лед при 0°С и полученную суспензию разделяли между EtOAc и водой. Органическую фазу отделяли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (от изогексана до 50% EtOAc в изогексане) позволяла выделить целевой продукт в виде бесцветного твердого вещества (172 мг, 0,61 ммоль, 61%)
1Н ЯМР (400 МГц): δ 1,56 (m, 2Н), 1,82 (m, 2Н), 3,79 (s, 3Н), 4,95 (s, 2Н), 6,86 (m, 4Н), 7,01 (m, 1Н), 7,16 (m, 1Н), 7,28 (m, 1H). ЖХ/МС 280 (МН+).
Пример получения 7: Спиро[циклопентан-1,3'-индолин]-2'-он
н-Бутиллитий (2,5 М в гексанах, 4,2 мл, 0,5 ммоль) по каплям добавляли к суспензии индолинона (0,665 мг, 5 ммоль) и TMEDA (1,5 мл, 10 ммоль) в ТГФ (20 мл) при -78°С в сухой колбе в атмосфере азота. Через 1 час при -78°С по каплям добавляли 1,4-дийодбутан (3,3 мл, 25 ммоль), и смесь оставляли медленно нагреваться до комнатной температуры. Через 12 часов при комнатной температуре, к смеси добавляли насыщенный водный раствор хлорида аммония и данную смесь экстрагировали EtOAc. Объединенные органические фазы промывали водой, солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (10-30% EtOAc в изогексане) позволяла выделить целевое соединение в виде бледно-розового твердого вещества (393 мг, 2,1 ммоль, 42%).
1Н ЯМР (400 МГц): δ 1,90 (m, 2Н), 2,03 (m, 2Н), 2,10 (m, 2Н), 2,21 (m, 2Н), 6,94 (m, 1H), 7,04 (m, 1Н), 7,20 (m, 2Н), 8,75 (bs, 1Н). ЖХ/МС 188 (МН+).
Пример получения 8: N'-фенилциклобутанкарбогидразид
В сухой колбе в атмосфере азота к смеси гидрохлорида фенилгидразина (3,92 мл, 40 ммоль) в ДХМ (80 мл) при комнатной температуре по каплям добавляли триэтиламин (5,4 мл, 48 ммоль). Смесь охлаждали на бане лед/соль и по каплям добавляли циклобутанкарбонилхлорид (4,8 мл, 42 ммоль) с такой скоростью, чтобы поддерживать внутреннюю температуру ниже -10°С. После 1,5 часов при -10°С, смеси давали нагреться до комнатной температуры и перемешивали при данной температуре в течение еще 16 часов. Смесь концентрировали в вакууме и добавляли разбавленный водный раствор NaHCO3. Смесь фильтровали, и твердое вещество промывали NaHCO3, Et2O и сушили с помощью насоса с получением целевого соединения в виде смеси с бис-циклобутанкарбогидразидом в соотношении 2,3-1 (5,33 г).
ЖХ/МС 191 (МН+)
Пример получения 9: Спиро[циклобутан-1,3'-индолин]-2'-он
К перемешиваемой суспензии оксида кальция (12,6 г, 124 ммоль) в хинолине (26 мл) добавляли N'-фенилциклобутанкарбогидразид (4,88 г, 25,6 ммоль, Пример получения 8). Смесь нагревали до 270-310°С и выдерживали при данной температуре в течение 75 минут. Смесь охлаждали до комнатной температуры и добавляли 2 М водный раствор соляной кислоты. Смесь экстрагировали EtOAc и объединенные экстракты промывали 2 М соляной кислотой, солевым раствором и сушили над Na2SO4. После фильтрации, а затем концентрирования в вакууме осуществляли хроматографию на силикагеле (40-60% EtOAc в гексане) с выделением целевого продукта в виде оранжевого твердого вещества (1,92 г, 11,1 ммоль, 43%).
1Н ЯМР (400 МГц): δ 2,18 (m, 1H), 2,28 (m, 3Н), 2,61 (m, 2Н), 6,79 (d, 1H), 7,01 (dd, 1H), 7,12 (dd, 1H), 7,41 (d, 1Н), 7,95 (bs, 1H). ЖХ/МС 174 (МН+):
Пример получения 10: 1'-(4-метоксибензил)-1'H-спиро[циклопропан-1,3'-хинолин]-2'(4'Н)-он
Этап 1: 1-(4-метоксибензил)-3,4-дигидрохинолин-2(1Н)-он (2,678 г, 10 ммоль) в ТГФ (10 мл) добавляли к свежеприготовленному раствору LiHMDS (полученному с использованием 4,8 мл 2,5 М бутиллития в гексанах и 2,1 мл HMDS) в ТГФ (10 мл) при -78°С. После 1 часа при -78°С данный анионный раствор по каплям добавляли через канюлю к перемешиваемому раствору 1-бром-2-хлорэтана (3 экв., 2,5 мл) в ТГФ (10 мл) при -78°С. Реакционной смеси давали нагреться до комнатной температуры в течение ночи, затем гасили путем добавления NH4Cl (водн.) и экстрагировали EtOAc. Органическую фазу сушили, фильтровали, концентрировали и очищали путем хроматографии на Si с использованием в качестве элюента смеси гексан/EtOAc в соотношении от 9:1 до 4:01. Это позволяло получить хлорэтильное соединение в виде оранжевого масла (1,65 г, чистота приблизительно 70%).
Этап 2: Раствор неочищенного 3-(2-хлорэтил)-1-(4-метоксибензил)-3,4-дигидрохинолин-2(1Н)-она (1,65 г) в ацетоне (20 мл) обрабатывали NaI (3,0 г, 4 экв.) и кипятили с обратным холодильником в течение 12 часов. Охлажденную реакционную смесь концентрировали и разделяли между водой и EtOAc. Добавляли тиосульфат натрия и отделяли органическую фазу. Концентрирование позволяло получить йодсодержащее соединение в виде оранжевого масла (2,05 г, чистота приблизительно 70%).
Этап 3: Раствор 3-(2-йодэтил)-1-(4-метоксибензил)-3,4-дигидрохинолин-2(1Н)-она (2,05 г, 4,8 ммоль) в ТГФ (10 мл) по каплям добавляли к свежеприготовленному раствору LiHMDS (полученному из BuLi 1 экв., 1,95 мл 2,5 М и HMDS 1 экв., 1,02 мл) в ТГФ (10 мл) при -78°С. Смесь перемешивали в течение 1 часа, затем давали нагреться до комнатной температуры в течение ночи. Добавляли солевой раствор, экстрагировали в EtOAc (2×25 мл), сушили, фильтровали и концентрировали с получением коричневого масла (1,05 г, 75%).
1Н ЯМР (400 МГц): δ 7,26 (s, 1H), 7,11 (m, 5Н), 6,90 (m, 5Н), 5,30 (d, J=1,0 Гц, 1H), 5,12 (s, 2Н), 5,02 (s, 2Н), 3,76 (s, 3Н), 2,87 (s, 2Н), 1,32 (m, 2Н), 0,77 (m, 2Н). ЖХ/МС 294,5 (МН+).
1-(4-метоксибензил)-3,4-дигидрохинолин-2(1Н)-он получали способом из Winter, D.K. et al, Journal of Organic Chemistry, 75(8), 2610-2618; 2010.
1H ЯМР (400 МГц): δ 7,07 (m, 4H), 6,83 (m, 4H), 5,04 (s, 2H), 3,70 (s, 3H), 2,89 (dd, J=8,7, 6,0 Гц, 2H), 2,70 (m, 2H).
Пример получения 11: 1'Н-спиро[циклопропан-1,3'-хинолин]-2'(4'Н)-он
1'-(4-метоксибензил)-1'Н-спиро[циклопропан-1,3'-хинолин]-2'(4'Н)-он (1,05 г, Пример получения 10) обрабатывали анизолом (1 экв.) и ТФУ (3 мл) при 65°С в течение 3 часов. Охлажденную смесь концентрировали, переносили в смесь солевой раствор/ДХМ и добавляли Et3N (3 мл). Водную фазу экстрагировали ДХМ (3×10 мл) и объединенные органические фазы сушили и концентрировали с получением желтой смолы (1,5 г). Очистка путем хроматографии на Si с использованием в качестве элюента смеси гексан/EtOAc (от 9:1 до 4:1) позволяла получить соединение, указанное в названии, в виде белого твердого вещества (465 мг), которое при растирании с изогексаном давало в результате фильтрации соединение, указанное в названии, в виде белого твердого вещества (301 мг, 48%).
1Н ЯМР (400 МГц): δ 8,20 (br s, 1H), 7,21 (m, 1H), 7,11 (m, 1Н), 7,01 (td, J=7,5, 1,1 гц, 1H), 6,78 (dd, J=7,8, 1,2 Гц, 1H), 2,89 (s, 2Н), 1,39 (m, 2Н), 0,79 (m, 2Н). ЖХ/МС 173,95 (М+), 205,95.
Пример получения 12: 3-(4-Йодбутил)-1-(4-метоксибензил)-3,4-дигидрохинолин-2(1Н)-он
1-(4-метоксибензил)-3,4-дигидрохинолин-2(1Н)-он (1,995 г, 7,4 ммоль) в ТГФ (5 мл) добавляли к свежеприготовленному раствору LDA (полученному с использованием 3,58 мл 2,5 М бутиллития в гексанах и 1,25 мл диизопропиламина) в ТГФ (5 мл) при -78°С. После 1 часа при -78°С данный анионный раствор по каплям добавляли через канюлю к перемешиваемому раствору 1,4-дийодбутана (2,95 мл, 22,2 ммоль, 3 экв.) в ТГФ (5 мл) при -78°С. Реакционной смеси давали нагреться до комнатной температуры в течение ночи, затем гасили путем добавления NH4Cl (водн.) и экстрагировали EtOAc. Органическую фазу сушили, фильтровали, концентрировали и очищали путем хроматографии на Si с использованием в качестве элюента смеси гексан/EtOAc (от 9:1 до 4:1). Это позволяло получить соединение, указанное в названии, в виде бледного масла (2,01 г, 60%).
1Н ЯМР (400 МГц): δ 7,16 (4Н, m), 6,98 (1H, t), 6,93 (1Н, d), 6,86 (2Н, d), 5,12 (2Н, s), 3,79 (3Н, s), 3,22 (2Н, m), 3,08 (1H, dd), 2,78 (1Н, dd), 2,68 (1H, m), 1,9 (3Н, m), 1,62 (5Н, m). ЖХ/МС 450,3.
Пример получения 13: 1'-(4-Метоксибензил)-1'Н-спиро[циклопентан-1,3'-хинолин]-2'(4'Н)-он
Раствор 3-(4-йодбутил)-1-(4-метоксибензил)-3,4-дигидрохинолин-2(1Н)-она (2 г, 4,4 ммоль, Пример получения 12) в ТГФ (10 мл) по каплям добавляли к раствору LiHMDS (полученному из 1,1 экв. бутиллития и 1,1 экв. HMDS) в ТГФ (10 мл) при -78°С. Убирали холодную баню и смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли солевой раствор и экстрагировали EtOAc. Объединенные органические фазы сушили и концентрировали с получением оранжевого масла, которое очищали путем хроматографии на Si с использованием в качестве элюента смеси гексан/Et2O (от 3:1 до 1:1). Это позволяло получить соединение, указанное в названии, в виде желтого масла (812 мг, 57%), содержащего некоторое количество примесей.
1H ЯМР (400 МГц): δ 7,10 (m 3Н), 6,94 (t, 1Н), 6,84 (m, 3Н), 5,09 (s, 2Н), 3,76 (s, 3Н), 2,85 (s, 2Н), 2,13 (m, 2Н), 1,75 (m, 5Н), 1,52 (m, 2Н). ЖХ/МС 322 (МН+)
Пример получения 14: 1'Н-спиро[циклопентан-1,3'-хинолин]-2'(4'Н)-он
Смесь 1'-(4-метоксибензил)-1'H-спиро[циклопентан-1,3'-хинолин]-2'(4'Н)-она (810 мг, 2,5 ммоль, Пример получения 13) и анизола (1 экв.) в ТФУ (1,9 мл) нагревали при 65°С в течение 3 часов. Смесь охлаждали, концентрировали в вакууме и переносили в ДХМ (10 мл). Добавляли Et3N (2 мл) и смесь промывали солевым раствором. Водную фазу экстрагировали дополнительным количеством ДХМ (2×10 мл) и объединенные органические фазы сушили и концентрировали с получением светло-коричневого твердого вещества. Хроматография на Si смесью гексан/Et2O в соотношении от 3:1 до 1:1 позволяла получить соединение, указанное в названии, в виде твердого вещества кремового цвета (373 мг, 74%).
1Н ЯМР (400 МГц): δ 7,75 (br s, 1H), 7,04 (m, 4Н), 3,80 (m, 2Н), 2,86 (s, 2Н), 2,12 (m, 2Н), 1,75 (m, 4Н), 1,54 (m, 1H). ЖХ/МС 202 (МН+)
Пример получения 15: 1-(2-(((1-изопентил-1Н-бензо[d]имидазол-2-ил)метил)амино)-бензамидо)циклопропанкарбоновая кислота
DIAD (0,9 мл, 1 экв.) в ТГФ (2 мл) по каплям добавляли к перемешиваемому раствору, содержащему (1-изопентил-1Н-бензо[d]имидазол-2-ил)метанол (1,0 г), PPh3 (1,2 г, 1 экв.) и изатоевый ангидрид (748 мг, 1 экв.) в ТГФ (30 мл), и перемешивали в течение ночи. Реакционную смесь концентрировали с получением светло-коричневого остатка, который переносили в ДМФА (20 мл) и обрабатывали этил-1-аминоциклопропанкарбоксилата гидрохлоридом (1,05 г, 1,5 экв.), и нагревали до 75°С в течение 48 часов. В результате водной обработки между Et2O и водой получали коричневую пену (| 1,2 г). Данный неочищенный сложный эфир растворяли в 2 М NaOH/EtOH (4:1, 10 мл) и нагревали до 70°С в течение 5 часов. Охлажденную реакционную смесь концентрировали, переносили в 2 М NaOH и промывали Et2O. Водную фазу подкисляли 2 М HCl до pH 4 и экстрагировали в EtOAc (2×25 мл). Это позволяло получить неочищенную коричневую пену, которую очищали на силикагеле с использованием в качестве элюента смеси изогексан/EtOAc (от 3:1 до 1:1). Это позволяло получить целевую кислоту в качестве более полярного компонента в виде твердого вещества кремового цвета (279 мг, 15%).
1Н ЯМР (400 МГц, ДМСО): δ 8,85 (1H, s), 8,8 (1H, t), 7,58 (1Н, d), 7,48 (1H, d), 7,349 (1H, dd), 7,18 (3Н, m), 6,93 (1H, d), 6,55 (1H, t), 4,68 (2Н, d), 4,29 (2Н, m), 1,67 (1H, септ), 1,578 (2Н, m), 1,085 (2Н, m), 0,94 (8Н, d). ЖХ/МС 421,5 (МН+).
Пример 1: 1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро-[циклопентан-1,3'-пирроло[2,3-с]пиридин]-2'(1'H)-он
В сухую колбу в атмосфере азота помещали гидрид натрия (42 мг, 1,05 ммоль) и ДМФА (0,5 мл). Данную быстро перемешиваемую суспензию охлаждали до 0°C с использованием бани лед/вода, и по каплям добавляли спиро[циклопентан-1,3'-пирроло[2,3-с]пиридин]-2'(1'H)-он (66 мг, 0,35 ммоль, Пример получения 1) в виде раствора в ДМФА (1,2 мл). Баню лед/вода убирали за 15 минут до повторного охлаждения смеси до 0°С, и по каплям добавляли трет-бутил-((2-(хлорметил)-1-изопентил-1H-бензо[d]имидазол-5-ил)метил)карбамата гидрохлорид (143 мг, 0,39 ммоль) в виде раствора в ДМФА (1,3 мл). Смеси давали нагреться до комнатной температуры и перемешивали в течение 1,5 часов.
Смесь охлаждали до 0°С и осторожно добавляли лед. После разделения между EtOAc и водой осуществляли разделение, сушку и концентрирование в вакууме. Полученный остаток обрабатывали 4 М соляной кислотой в диоксане (8 мл) при комнатной температуре в течение 16 часов. Смесь разделяли между этилацетатом и водой, и водную фазу промывали EtOAc. После нейтрализации водной фазы твердым Na2CO3 осуществляли экстракцию EtOAc, сушку объединенных экстрактов (Na2SO4) и концентрирование в вакууме. Хроматография на силикагеле (ДХМ/EtOH/водный раствор NH3, от 100/8/1 до 50/8/1) позволяла выделить соединение, указанное в названии, в виде бесцветного масла. В результате последующей сушки методом лиофилизации из MeCN/Н2О (2 мл/1 мл) получали бесцветное твердое вещество (75 мг, 51%).
1Н ЯМР (400 МГц): δ 1,01 (d, 6Н), 1,58 (m, 2Н), 1,71 (m, 1H), 1,90 (m, 2Н), 2,02 (m, 2Н), 2,15 (m, 2Н), 2,16 (m, 2Н), 3,99 (s, 2Н), 4,26 (m, 2Н), 5,26 (s, 2Н), 7,13 (d, 2Н), 7,27 (m, 2Н), 7,70 (d, 1H), 8,37 (d, 1Н), 8,77 (s, 1H). ЖХ/МС 418 (МН+).
Пример 2: 1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро-[циклопропан-1,3'-индолин]-2'-он
В способе, аналогичном описанному в Примере 1, проводили реакцию спиро[циклопропан-1,3'-индолин]-2'-она (160 мг, 1 ммоль, Пример получения 4) с трет-бутил-((2-(хлорметил)-1-изопентил-1H-бензо[d]имидазол-5-ил)метил)карбамата гидрохлоридом (366 мг, 1 ммоль) с получением 85 мг (0,22 ммоль, 22%) целевого соединения в виде бесцветного твердого вещества.
1Н ЯМР (400 МГц): δ 0,90 (d, 6Н), 1,46 (m, 2Н), 1,59 (m, 2Н), 1,69 (m, 2Н), 1,82 (m, 2Н), 3,99 (s, 2Н), 4,24 (m, 2Н), 5,34 (s, 2Н), 6,83 (d, 1H), 7,01 (d, 1H), 7,26 (m, 2Н), 7,45 (d, 1H), 7,73 (s, 1Н). ЖХ/МС 389 (МН+).
Пример 3: 1'-((1-изопентил)-1H-бензо[d]имидазол-2-ил)метил)спиро[циклопропан-1,3'-индолин]-2'-он
В сухую колбу в атмосфере азота помещали гидрид натрия (120 мг, 3,0 ммоль) и ДМФА (1 мл). Данную быстро перемешиваемую суспензию охлаждали до 0°C с использованием бани лед/вода, и по каплям добавляли спиро[циклопропан-1,3'-индолин]-2'-он (160 мг, 1,0 ммоль, Пример получения 4) в виде раствора в ДМФА (2 мл). Баню лед/вода убирали за 15 минут до повторного охлаждения смеси до 0°С, и по каплям добавляли 2-(хлорметил)-1-изопентил-1Н-бензо[d]имидазола гидрохлорид (236 мг, 1,0 ммоль) в виде раствора в ДМФА (2,1 мл). Смеси давали нагреться до комнатной температуры и перемешивали при данной температуре в течение 16 часов.
Смесь охлаждали до 0°С и осторожно добавляли лед. После разделения между EtOAc и водой осуществляли разделение, сушку и концентрирование в вакууме. Препаративная ВЭЖХ позволяла выделить целевое соединение (110 мг, 0,31 ммоль, 31%).
1Н ЯМР (400 МГц): δ 0,96 (d, 6Н), 1,45 (m, 2Н), 1,60 (m, 2Н), 1,69 (m, 1H), 1,82 (m, 2Н), 4,26 (m, 2Н), 5,35 (s, 2Н), 6,83 (d, 1H), 7,02 (dd, 1H), 7,19 (dd, 1H), 7,29 (m, 3Н), 7,47 (d, 1H), 7,81 (m, 1H). ЖХ/МС 360 (МН+).
Пример 4: 1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро-[циклопентан-1,3'-индолин]-2'-он
В способе, аналогичном описанному в Примере 1, проводили реакцию спиро[циклопентан-1,3'-индолин]-2'-она (187 мг, 1 ммоль) с трет-бутил-((2-(хлорметил)-1-изопентил-1H-бензо[d]имидазол-5-ил)метил)карбамата гидрохлоридом (366 мг, 1 ммоль) с получением 65 мг (0,22 ммоль, 22%) целевого соединения в виде бесцветного твердого вещества.
1Н ЯМР (400 МГц): δ 1,00 (d, 6Н), 1,55 (m, 2Н), 1,70 (bs, 2Н), 1,74 (m, 1Н), 1,88 (m, 2Н), 2,03 (m, 2Н), 2,13 (m, 2Н), 2,22 (m, 2Н), 3,99 (s, 2Н), 4,25 (m, 2Н), 5,25 (s, 2Н), 7,03 (m, 1H), 7,19 (m, 2Н), 7,28 (m, 2Н), 7,46 (d, 1Н), 7,72 (s, 1H). ЖХ/МС 417 (МН+).
Пример 5: 1'-((1-изопентил)-1H-бензо[d]имидазол-2-ил)метил)спиро[циклопентан-1,3'-индолин]-2'-он
В способе, аналогичном описанному в Примере 3, проводили реакцию спиро[циклопентан-1,3'-индолин]-2'-она (187 мг, 1 ммоль, Пример получения 7) с 2-(хлорметил)-1-изопентил-1H-бензо[d]имидазола гидрохлоридом (238 мг, 1 ммоль) с получением 60 мг (0,22 ммоль, 15%) целевого соединения в виде бесцветного твердого вещества.
1Н ЯМР (400 МГц): δ 0,91 (d, 6Н), 1,58 (m, 2Н), 1,65 (m, 1Н), 1,9-2,15 (m, 6Н), 4,18 (m, 2Н), 5,15 (s, 2Н), 6,93 (m, 1H), 7,10 (m, 2Н), 7,19 (m, 2Н), 7,21 (m, 1H), 7,38 (m, 1H), 7,71 (m, 1Н). ЖХ/МС 388 (МН+).
Пример 6: 1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро-[циклобутан-1,3'-индолин]-2'-он
В способе, аналогичном описанному в Примере 1, проводили реакцию спиро[циклобутан-1,3'-индолин]-2'-она (173 мг, 1 ммоль, Пример получения 9) с трет-бутил-((2-(хлорметил)-1-изопентил-1H-бензо[d]имидазол-5-ил)метил)карбамата гидрохлоридом (366 мг, 1 ммоль) с получением 72 мг (0,22 ммоль, 18%) целевого соединения в виде бесцветного твердого вещества.
1Н ЯМР (400 МГц): δ 0,98 (d, 6Н), 1,49 (m, 2Н), 1,72 (m, 1H), 1,75 (bs, 2Н), 2,35 (m, 4Н), 2,74 (m, 2Н), 3,99 (s, 2Н), 4,25 (m, 2Н), 5,22 (s, 2Н), 7,08 (m, 1Н), 7,20 (m, 1Н), 7,26 (m, 2Н), 7,40 (d, 1H), 7,51 (dd, 1H), 7,71 (s, 1H). ЖХ/МС 403 (МН+).
Пример 7: 1'-((1-изопентил)-1H-бензо[d]имидазол-2-ил)метил)спиро[циклобутан-1,3'-индолин]-2'-он
В способе, аналогичном описанному в Примере 3, проводили реакцию спиро[циклобутан-1,3'-индолин]-2'-она (126 мг, 0,58 ммоль, Пример получения 9) с 2-(хлорметил)-1-изопентил-1H-бензо[d]имидазола гидрохлоридом (122 мг, 1 ммоль) с получением 69 мг (0,18 ммоль, 32%) желаемого соединения в виде бесцветного твердого вещества.
1Н ЯМР (400 МГц): δ 0,99 (d, 6Н), 1,52 (m, 2Н), 1,72 (m, 1Н), 2,39 (m, 4Н), 2,74 (m, 2Н), 4,28 (m, 2Н), 5,24 (s, 2Н), 7,09 (dd, 1H), 7,21 (dd, 2Н), 7,28 (m, 3Н), 7,51 (d, 1H), 7,52 (d, 1H), 7,80 (m, 1H). ЖХ/МС 374 (МН+).
Пример 8: 4-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро-[бензо[b][1,4]оксазин-2,1'-циклопропан]-3(4Н)-он
Способом, аналогичным описанному в Примере 1, проводили реакцию спиро[бензо[b][1,4]оксазин-2,1'-циклопропан]-3(4H)-она (125 мг, 0,71 ммоль) с трет-бутил-((2-(хлорметил)-1-изопентил-1H-бензо[d]имидазол-5-ил)метил)карбамата гидрохлоридом (0,7 ммоль) с получением соединения, указанного в названии, в виде твердого вещества кремового цвета (149 мг, 49%).
1Н ЯМР (400 МГц): δ 7,65 (m, 2Н), 7,27 (m, 2Н), 6,99 (dt, J=7,6, 1,5 Гц, 2Н), 6,86 (dd, J=7,8, 1,6 Гц, 1Н), 5,55 (s, 2Н), 4,19 (m, 2Н), 3,98 (s, 2Н), 1,75 (dp, J=13,2, 6,6 Гц, 1Н), 1,59 (m, 6Н), 1,47 (m, 2Н), 1,29 (m, 2Н), 1,02 (d, J=6,6 Гц, 6Н). ЖХ/МС 405 (МН+).
Спиро[бензо[b][1,4]оксазин-2,1'-циклопропан]-3(4H)-он получали способом из Powell et al, J.Med.Chem 15 (2007) 5912.
1H ЯМР (400 МГц): δ 8,53 (s, 1H), 6,91 (m, 4Н), 1,46 (m, 2Н), 1,26 (m, 2Н).
Пример 9: 4-((1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[бензо[b][1,4]оксазин-2,1'-циклопропан]-3(4H)-он
Способом, аналогичным описанному в Примере 3, проводили реакцию спиро[бензо[b][1,4]оксазин-2,1'-циклопропан]-3(4H)-она (125 мг, 0,71 ммоль) с 2-(хлорметил)-1-изопентил-1H-бензо[d]имидазола гидрохлоридом (0,7 ммоль) с получением после очистки путем обращенно-фазовой ВЭЖХ соединения, указанного в названии, в виде белого твердого вещества (46 мг, 17%).
1Н ЯМР (400 МГц): δ 7,74 (m, 1Н), 7,64 (dd, J=8,0, 1,6 Гц, 1H), 7,26 (m, 3Н), 6,98 (dtd, J=27,6, 7,6, 1,6 Гц, 2Н), 6,84 (dd, J=7,9, 1,6 Гц, 1H), 5,54 (s, 2Н), 4,19 (m, 2Н), 1,73 (m, 2Н), 1,58 (m, 1H), 1,47 (q, J=5,2 Гц, 2Н), 1,27 (q, J=5,1 Гц, 2Н), 1,00 (d, J=6,6 Гц, 6Н). ЖХ/МС 376 (МН+). Спиро[бензо[b][1,4]оксазин-2,1'-циклопропан]-3(4H)-он получали способом из Powell et al, J.Med.Chem 15 (2007) 5912.
1H ЯМР (400 МГц): δ 8,53 (s, 1H), 6,91 (m, 4Н), 1,46 (m, 2Н), 1,26 (m, 2Н).
Пример 10: 1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)-1'H-спиро[циклопропан-1,3'-хинолин]-2'(4'Н)-он
Способом, аналогичным описанному в Примере 1, проводили реакцию 1'H-спиро[циклопропан-1,3'-хинолин]-2'(4'Н)-она (150 мг, 0,85 ммоль, Пример получения 11) с трет-бутил-((2-(хлорметил)-1-изопентил-1H-бензо[d]имидазол-5-ил)метил)карбамата гидрохлоридом (348 мг) с получением соединения, указанного в названии, в виде пены кремового цвета (93 мг, 27%).
1Н ЯМР (400 МГц): δ 7,65 (m, 2Н), 7,25 (m, 3Н), 7,04 (m, 2Н), 5,54 (s, 2Н), 4,21 (m, 2Н), 3,96 (s, 2Н), 2,86 (s, 2Н), 1,85 (br m, 4Н), 1,70 (m, 2Н), 1,41 (q, J=4,0 Гц, 2Н), 1,02 (d, J=6,5 Гц, 6Н), 0,82 (m, 3Н). ЖХ/МС 403 (МН+).
Пример 11: 1'-((1-изопентил)-1Н-бензо[d]имидазол-2-ил)метил)-1'Н-спиро[циклопропан-1,3'-хинолин]-2'(4'Н)-он
Способом, аналогичным описанному в Примере 3, проводили реакцию 1'H-спиро[циклопропан-1,3'-хинолин]-2'(4'H)-она (154 мг, 0,8 ммоль, Пример получения 11) с 2-(хлорметил)-1-изопентил-1H-бензо[d]имидазола гидрохлоридом (0,8 ммоль) с получением после очистки путем обращенно-фазовой ВЭЖХ соединения, указанного в названии, в виде белого твердого вещества (52 мг, 17%).
1H ЯМР (400 МГц): δ 7,74 (1Н, m), 7,67 (1H, d), 7,278 (4Н, m), 7,074 (1H, d), 7,017 (1H, t), 5,56 (2Н, s), 4,23 (2Н, m), 2,86 (2Н, s), 1,77 (1H, септ), 1,65 (|4H, m), 1,41 (2Н, m), 1,03 (6Н, d), 0,82 (2Н, m). ЖХ/МС 374 (МН+).
Пример 12: 1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)-1'Н-спиро[циклопентан-1,3'-хинолин]-2'(4'Н)-он
Способом, аналогичным описанному в Примере 1, проводили реакцию 1'H-спиро[циклопентан-1,3'-хинолин]-2'(4'Н)-она (125 мг, 0,62 ммоль, Пример получения 14) с трет-бутил-((2-(хлорметил)-1-изопентил-1H-бензо[d]имидазол-5-ил)метил)карбамата гидрохлоридом (1 экв.) с получением соединения, указанного в названии, в виде пены бледного цвета (101 мг, 38%).
1H ЯМР (400 МГц): δ 7,65 (s, 1H), 7,58 (d, 1H), 7,25 (m, 4Н), 7,12 (dd, J=7,4, 1,5 Гц, 1H), 6,98 (td, J=7,4, 1,0 Гц, 1H), 5,52 (s, 2Н), 4,24 (m, 2Н), 3,96 (s, 2Н), 2,86 (s, 2Н), 2,16 (m, 2Н), 1,76 (m, 5Н), 1,63 (m, 8Н), 1,53 (m, 2Н), 1,02 (d, J=6,5 Гц, 6Н). ЖХ/МС 431,5(МН+)
Пример 13: 1'-((1-изопентил)-1H-бензо[d]имидазол-2-ил)метил)-1'H-спиро[циклопентан-1,3'-хинолин]-2'(4'Н)-он
Способом, аналогичным описанному в Примере 3, проводили реакцию 1'H-спиро[циклопентан-1,3'-хинолин]-2'(4'Н)-она (125 мг, 0,62 ммоль) с 2-(хлорметил)-1-изопентил-1H-бензо[d]имидазола гидрохлоридом (0,62 ммоль) и очищали путем обращенно-фазовой ВЭЖХ с получением соединения, указанного в названии, в виде пены бледного цвета (80 мг, 32%).
1H ЯМР (400 МГц): δ 7,64 (m, 1H), 7,51 (d, J=8,2 Гц, 1H), 7,16 (m, 4Н), 7,02 (dd, J=7,3, 1,6 Гц, 1Н), 6,89 (td, J=7,4, 1,1 Гц, 1H), 5,44 (s, 2Н), 4,15 (m, 2Н), 2,76 (s, 2Н), 2,06 (m, 2Н), 1,58 (m, 9Н), 0,93 (d, J=6,5 Гц, 6Н). ЖХ/МС 402,4 (МН+).
Пример 14: 1-((1-изопентил)-1H-бензо[d]имидазол-2-ил)метил)спиро-[бензо[е][1,4]диазепин-3,1'-циклопропан]-2,5(1Н,4Н)-дион
Раствор 1-(2-(((1-изопентил-1H-бензо[d]имидазол-2-ил)метил)амино)бензамидо)-циклопропанкарбоновой кислоты (279 мг, Пример получения 15) в ДМФА (5 мл) обрабатывали TBTU (284 мг, 1,2 экв.) и DIPEA (190 мкл, 1,3 экв.), затем перемешивали в течение 12 часов при комнатной температуре. После концентрирования в вакууме остаток разделяли между Et2O и 2М NaOH. Фазу Et2O отделяли, сушили и концентрировали до очистки путем хроматографии на Si геле с использованием в качестве элюента смеси изогексан/EtOAc (4:1). Это позволяло получить соединение, указанное в названии, в виде смолы, которую сушили методом лиофилизации из водного раствора MeCN с получением порошка серого цвета (42 мг, 16%). 1Н ЯМР (400 МГц) δ 8,3 (1Н, t), 7,2 (1Н, dd), 7,68 (1H, m), 7,24 (4Н, m), 6,91 (1H, dm), 6,67 (1Н, ddd), 4,68 (2Н, d), 4,11 (2Н, m), 1,75 (2Н, m), 1,72 (2Н, m), 1,58 (2Н, m), 0,88 (6Н, d). ЖХ/МС 403,2 (МН+).
Пример 15: Биологическое тестирование
Соединения согласно настоящему изобретению, полученные, как описано в предыдущих Примерах, подвергали следующему биологическому анализу:
Тест нейтрализации бляшкообразования
Соединения из Примеров 1-13 представляли для анализа в виде предварительно взвешенного количества, эквивалентного сотой части их молекулярной массы. В результате добавления 1,00 мл 100% диметилсульфоксида (ДМСО) соответствующего качества для выращивания культуры клеток получали исходный раствор с концентрацией 10 мМ. Растворение при необходимости облегчали путем обработки ультразвуком при комнатной температуре или путем слабого нагревания (<40°С), и аспирацию осуществляли пипеткой.
Затем из исходного раствора в ДМСО брали аликвоты и разводили до необходимой концентрации путем добавления буфера, подходящего для анализа, до конечной концентрации, содержащей не менее 0,5% ДМСО. 100% исходный раствор в ДМСО хранили при 4°С в соответствии с требованиями с защитой от света и внешней влаги. Его оставляли мягко оттаивать в течение ночи и, если было видно твердое вещество, аспирировали или иным образом повторно солюбилизировали перед забором аликвоты.
Реакция нейтрализации бляшкообразования:
Клетки Vero высевали в 96-луночные планшеты в объеме 100 мкл Optimem с добавлением 3% FCS в концентрации 4×104 клеток на лунку. После инкубации в течение ночи при 37°С в увлажненной атмосфере, содержащей 5% CO2, монослой клеток должен демонстрировать приблизительно 90% конфлюэнтность. Противовирусные соединения титровали в предварительно нагретой бессывороточной (SF) среде Optimem в планшете на 96 лунок с U-образным дном. Для соединений в растворе ДМСО сначала выполняли титрование в 100% ДМСО, и каждую концентрацию добавляли по отдельности до конечной концентрации, разбавленной в 2 раза (2 x), в 4% ДМСО в бессывороточных средах перед смешиванием с вирусом (2% конечный ДМСО с вирусом). Затем с клеток удаляли среды и заменяли на ФБР (100 мкл/лунка). Исходный раствор РСВ размораживали и разбавляли в бессывороточных средах Optimem до 4000 БОЕ/мл. Равный объем вируса добавляли к соединениям в планшете для титрования. Удаляли ФБР с клеток, которые затем инокулировали раствором вируса/соединения (50 мкл/лунка). Клетки инкубировали в течение 2 часов в увлажненном инкубаторе 37°С + 5% CO2 для того, чтобы произошло инфицирование. Инокулят удаляли, и к клеткам добавляли среды (Optimem + 1% FCS) (100 мкл/лунка). Затем клетки инкубировали в течение 48 часов при 37°С + 5% CO2 в увлажненном инкубаторе.
Процедура иммуноокрашивания:
Среды удаляли с клеток, и монослой промывали ФБР. Клетки фиксировали ледяным 80% ацетоном в ФБР (100 мкл/лунка) в течение 20 минут при -20°С. Фиксатор удаляли, и клетки сушили в течение 30 минут в перевернутых планшетах. К клеткам добавляли блокирующий раствор (5% сухое обезжиренное молоко в ФБР-Т) (150 мкл/лунка) и планшеты инкубировали в течение 30 минут при комнатной температуре. Блокирующий раствор удаляли и планшеты один раз промывали ФБР-Т. В планшеты добавляли первичное антитело в блокирующем растворе (50 мкл/лунка) и инкубировали в течение 1 часа при 37°С. Затем планшеты 3 раза промывали ФБР-Т. В планшеты добавляли вторичное антитело в блокирующем растворе (50 мкл/лунка) и инкубировали в течение 1 часа при 37°С в темноте. Планшеты промывали, как описано выше, а затем сушили в течение 10 минут. Планшеты сканировали на Odyssey Imager (Li-Cor Biosciences) с разрешением 42 мкМ, средним качеством и интенсивностью 5 уровня в канале 800 нМ.
Анализ данных:
Полученные изображения сохраняли, и количество бляшек подсчитывали с помощью компьютерного программного обеспечения для обработки изображений. Значения ЕС50 для соединений получали из кривых зависимости «доза-ответ» [график зависимости ответа от логарифма концентрации ингибитора (log(ингибитор)) для трех разных значений концентрации], полученных с использованием программного обеспечения Graphpad Prism.
Результаты:
Было обнаружено, что все тестируемые соединения имеют ЕС50, составляющую 80 мкМ или меньше.
Пример 16: Водный состав
Готовили соста соединения из Примера 10 в виде раствора в 30% масс./об. каптизоле (т.е. простом сульфобутиловом эфире-бета-циклодекстрине) при pH4 в соответствии со следующей процедурой.
Носитель, представляющий собой 30% масс./об. каптизол (т.е. простой сульфобутиловый эфир-бета-циклодекстрин) получали путем взвешивания необходимого количества каптизола в подходящий сосуд, добавления приблизительно 80% от конечного объема воды и перемешивания с помощью магнитной мешалки до образования раствора. Затем носитель доводили до объема водой.
Водный раствор соединения из Примера 10 получали путем взвешивания 175 мг указанного соединения в подходящий сосуд и добавления приблизительно 80% от необходимого объема носителя. С использованием водного раствора хлороводородной кислоты pH доводили до pH2 и полученную смесь перемешивали с помощью магнитной мешалки до образования раствора. Затем состав доводили носителем до объема и pH доводили до pH4 с использованием водного раствора гидроксида натрия.
Пример 17: Композиция таблетки
Таблетки, каждую массой 0,15 г и содержащую 25 мг соединения согласно настоящему изобретению, изготавливали следующим образом:
Композиция для 10000 таблеток
Соединение согласно настоящему изобретению (250 г)
Лактоза (800 г)
Кукурузный крахмал (415 г)
Тальк, порошок (30 г)
Стеарат магния (5 г)
Смешивали соединение согласно настоящему изобретению, лактозу и половину кукурузного крахмала. Затем смесь продавливали через сито с размером отверстий 0,5 мм. Суспендировали кукурузный крахмал (10 г) в теплой воде (90 мл). Полученную пасту использовали для гранулирования порошка. Гранулят сушили и разбивали на мелкие фрагменты на сите с размером отверстий 1,4 мм. Добавляли оставшееся количество крахмала, тальк и магний, тщательно смешивали и перерабатывали в таблетки.
Пример 18: Состав для инъекций
Соединение согласно настоящему изобретению растворяли в большей части воды (35°С-40°С) и при необходимости pH доводили до 4,0-7,0 соляной кислотой или гидроксидом натрия. Затем партию доводили до объема водой и фильтровали через стерильный микропористый фильтр в стерильный 10 мл флакон из янтарного стекла (1 тип) и герметизировали стерильными укупорочными средствами и дополнительными укупорочными средствами.
Пример 19: Внутримышечная инъекция
Соединение согласно настоящему изобретению растворяли в гликофуроле. Затем добавляли и растворяли бензиловый спирт, и добавляли воду до 3 мл. Затем смесь фильтровали через стерильный микропористый фильтр и герметизировали в стерильных 3 мл стеклянных флаконах (1 типа).
Пример 20: Состав в форме сиропа
Соединение согласно настоящему изобретению растворяли в смеси глицерина и большей части очищенной воды. Затем к раствору добавляли водный раствор бензоата натрия с последующим добавлением раствора сорбита и, наконец, ароматизатора. Очищенной водой доводили до объема и хорошо перемешивали.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПИРОИНДОЛИНОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ РЕСПИРАТОРНО-СИНЦИТИАЛЬНОГО ВИРУСА (RSV) | 2015 |
|
RU2734248C2 |
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ FASN | 2014 |
|
RU2737434C2 |
| ПРОИЗВОДНЫЕ ИНДОЛИН-2-ОНА ИЛИ ПИРРОЛОПИРИДИН/ПИРИМИДИН-2-ОНА | 2014 |
|
RU2666532C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛОПИРИМИДИНЫ И ЗАМЕЩЕННЫЕ ПУРИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ УБИКВИТИН-СПЕЦИФИЧЕСКОЙ ПРОЦЕССИРУЮЩЕЙ ПРОТЕАЗЫ 1 (USP1) | 2019 |
|
RU2833222C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2667498C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2840815C2 |
| АНАЛОГИ НОЦИЦЕПТИНА | 2002 |
|
RU2426730C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЕСПЕЧИВАЮЩИЕ РАЗРУШЕНИЕ БЕЛКА IKAROS И БЕЛКА AIOLOS | 2020 |
|
RU2833608C2 |
| СОЕДИНЕНИЕ ИЗОИНДОЛИН, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2813232C2 |
Настоящее изобретение относится к производному бензимидазола формулы (I) или к его фармацевтически приемлемой соли, где А: незамещенный 6-членный арил или незамещенный 6-членный гетероарил, содержащий один атом азота; Y: одинарная связь, -(СН2)p- или -Х-; X: -О-, -N(R2)-C(=O)- или -C(=O)-N(R2)-; каждый L: C1-3 алкилен; R1: незамещенный C1-6 алкил; каждый Z: -N(R2)2; каждый R2: водород; m равен 0 или 1; n равен 1, 2 или 3; и p равен 1. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I), способу ингибирования РСВ, основанному на использовании соединения формулы (I), способу получения соединения формулы (I). Технический результат: получены новые производные бензимидазола, полезные в качестве ингибитора респираторно-синцитиального вируса (РСВ). 5 н. и 6 з.п. ф-лы, 35 пр.
1. Соединение, представляющее собой бензимидазол формулы (I): 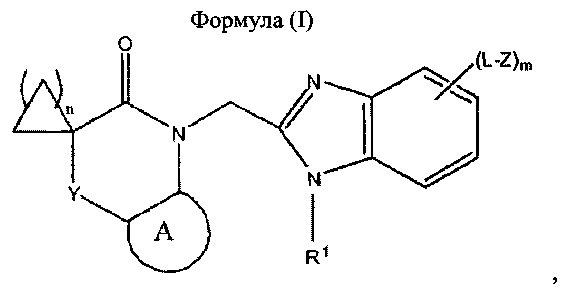
где:
- А представляет собой 6-членный арил или 6-членный гетероарил, содержащий один атом азота, каждый из которых не содержит заместителей;
- Y представляет собой одинарную связь, -(СН2)p- или -Х-;
- X представляет собой -О-, -N(R2)-C(=O)- или -C(=O)-N(R2)-;
- каждый L независимо представляет собой C1-3 алкилен;
- R1 представляет собой C1-6 алкил, который не содержит заместителей;
- каждый Z независимо представляет собой -N(R2)2;
- каждый R2 независимо представляет собой водород;
- m равен 0 или 1;
- n равен 1, 2 или 3; и
- p равен 1;
или его фармацевтически приемлемую соль.
2. Соединение по п.1, отличающееся тем, что А представляет собой фенил или 6-членный гетероарил.
3. Соединение по п.1 или 2, отличающееся тем, что Y представляет собой одинарную связь.
4. Соединение по п.1 или 2, отличающееся тем, что R1 представляет собой разветвленную С3-6 алкильную группу.
5. Соединение по п.1, отличающееся тем, что в формуле (I):
А представляет собой не содержащую заместителей фенильную группу или не содержащую заместителей пиридильную группу;
Y представляет собой одинарную связь, -О-, -C(=O)-NH- или -CH2-;
L представляет собой -СН2-;
R1 представляет собой разветвленную не содержащую заместителей С4-6 алкильную группу;
Z представляет собой -NH2;
m равен 0 или 1; и
n равен 1, 2 или 3.
6. Соединение по п.1, отличающееся тем, что бензимидазол формулы (I) имеет следующую формулу (Ia):
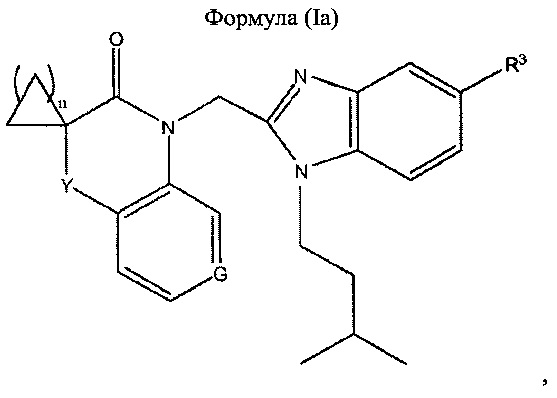
где
- G представляет собой N или СН;
- Y представляет собой одинарную связь, -О-, -C(=O)-NH- или -CH2-;
- n равен 1, 2 или 3; и
- R3 представляет собой Н или -CH2NH2.
7. Соединение по п.1, выбранное из
1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклопентан-1,3'-пирроло[2,3-с]пиридин]-2'(1'H)-она;
1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклопропан-1,3'-индолин]-2'-она;
1'-((1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклопропан-1,3'-индолин]-2'-она;
1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклопентан-1,3'-индолин]-2'-она;
1'-((1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклопентан-1,3'-индолин]-2'-она;
1'-((5-(аминометил)-1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклобутан-1,3'-индолин]-2'-она;
1'-((1-изопентил-1H-бензо[d]имидазол-2-ил)метил)спиро[циклобутан-1,3'-индолин]-2'-она;
4-((5-(аминометил)-1-изопентил-1Н-бензо[d]имидазол-2-ил)метил)спиро[бензо[b][1,4]-оксазин-2,1'-циклопропан]-3(4Н)-она;
4-((1-изопентил-1Н-бензо[d]имидазол-2-ил)метил)спиро[бензо[b][1,4]оксазин-2,1'-циклопропан]-3(4Н)-она;
1'-((5-(аминометил)-1-изопентил-1Н-бензо[d]имидазол-2-ил)метил)-1'Н-спиро[цикло-пропан-1,3'-хинолин]-2'(4'Н)-она;
1'-((1-изопентил-1Н-бензо[d]имидазол-2-ил)метил)-1'Н-спиро[циклопропан-1,3'-хинолин]-2'(4'Н)-она;
1'-((5-(аминометил)-1-изопентил-1Н-бензо[d]имидазол-2-ил)метил)-1'Н-спиро[цикло-пентан-1,3'-хинолин]-2'(4'Н)-она;
1'-((1-изопентил-1Н-бензо[d]имидазол-2-ил)метил)-1'Н-спиро[циклопентан-1,3'-хинолин]-2'(4'Н)-она; и
1-((1-изопентил-1Н-бензо[d]имидазол-2-ил)метил)спиро[бензо[e][1,4]диазепин-3,1'-циклопропан]-2,5(1Н,4Н)-диона;
и их фармацевтически приемлемых солей.
8. Фармацевтическая композиция для применения для лечения или предотвращения респираторно-синцитиальной вирусной (РСВ) инфекции, содержащая эффективное количество соединения по любому из пп.1-7 и фармацевтически приемлемый носитель или разбавитель.
9. Способ ингибирования РСВ у субъекта, включающий введение указанному субъекту эффективного количества соединения по любому из пп.1-7.
10. Фармацевтическая композиция для применения для лечения или предотвращения респираторно-синцитиальной вирусной (РСВ) инфекции, содержащая (а) соединение по любому из пп.1-7 и (b) один или более дополнительных терапевтических агентов, выбранных из:
(i) ингибитор нуклеокапсидного (N)-белка РСВ;
(ii) ингибитор другого белка, например ингибирующий белок фосфопротеин (Р) и/или крупный белок (L);
(iii) моноклональное антитело против РСВ, такое как антитело к F-белку;
(iv) иммуномодулирующее соединение, представляющее собой toll-подобный рецептор;
(v) другое противовирусное средство для лечения респираторного вируса, такое как противогриппозное и противориновирусное соединение; и/или
(vi) противовоспалительное соединение;
совместно с фармацевтически приемлемым носителем или разбавителем.
11. Способ получения соединения по любому из пп.1-7, включающий добавление раствора в ДМФА соединения формулы (III), определенного ниже, к гидриду натрия в ДМФА, и последующее добавление раствора в ДМФА соединения формулы (II), определенного ниже:
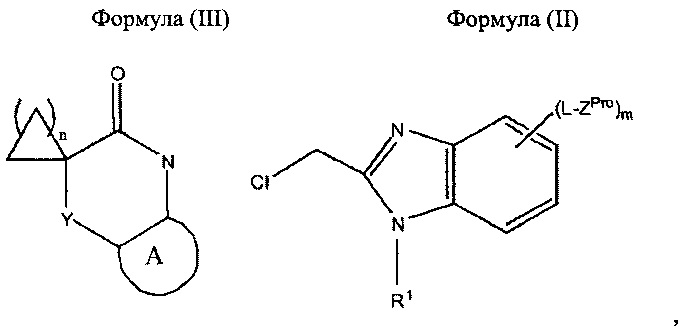
где A,Y, L, R1, m и n являются такими, как определено в п.1, и ZPro представляет собой Z, как определено в п.1, или защищенное производное Z; и, когда m равен 1, 2 или 3 и ZPro представляет собой защищенное производное Z; и удаление защитных групп из полученного соединения.
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| АВТОМАТИЧЕСКАЯ ТЕЛЕФОННАЯ СТАНЦИЯ | 1927 |
|
SU7157A1 |
| ПРОИЗВОДНЫЕ СПИРОПИПЕРИДИНА, ЛЕКАРСТВЕННОЕ СРЕДСТВО, СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ | 1999 |
|
RU2184735C2 |

