Притязание на приоритет
Настоящая заявка испрашивает приоритет на основании заявок на патент США №61/779908 и 61/779962, поданных 13 марта 2013 года, содержание которых включено в настоящую заявку во всей полноте посредством ссылок.
Область техники
Настоящее изобретение относится к соединениям и композиции для ингибирования синтазы жирных кислот («FASN»), способу их получения, применениям и антидоту. Указанная заявка не испрашивает приоритет на основании какой-либо другой заявки.
Уровень техники
Синтаза жирных кислот (далее «FASN»; также известная как «FAS») играет основополагающую роль при метаболизме клетки и в сигнальной клеточной системе. FASN катализирует образование длинноцепочечных жирных кислот из ацетил-CoA, малонил-CoA и никотинамидадениндинуклеотидфосфата (НАДФ) и тем самым задействована в выработке и хранении энергии, клеточной структуре и образовании промежуточных веществ биосинтеза гормонов и других биологически важных молекул.
Для изучения экспрессии, функции и регуляции генов, кодирующих FASN, и различных форм белков FASN проводился ряд углубленных исследований. В некоторых исследованиях было показано, что FASN задействована при онкогенезе и прогрессировании опухоли при различных раковых заболеваниях. Например, амплификацию гена FASN и повышенную экспрессию белка наблюдали в клеточных линиях рака груди человека (Hunt DA, Lane HM, Zygmont ME, Dervan PA, Hennigar RA (2007), MRNA stability and overexpression of fatty acid synthase in human breast cancer cell lines. Anticancer Res. 27 (1A): 27–34; Kuhaja FP, (2006) Fatty acid synthase and cancer: New application of an old pathway. Cancer Research, 66(12) 5977-5980; Menendez JA, Lupu R (2007) Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nature Review Cancer, 7, 763-777). Кроме того, в исследованиях, направленных на новообразования в яичниках, было показано, что повышенный уровень FASN является индикатором сокращения срока выживания субъекта (Gansler TS, Hardman W, Hunt DA, Schaffel S, Hennigar RA (June 1997). Increased expression of fatty acid synthase (OA-519) in ovarian neoplasms predicts shorter survival. Hum. Pathol. 28 (6): 686–92). Вкратце, связь повышенной экспрессии или активности FASN при опухолях тяжелой степени и распространенных стадиях первичного рака груди, простаты и колоректального рака определила внимание к указанному ферменту как к возможной мишени для лекарственного вмешательства и маркеру плохого прогноза.
Также было показано, что помимо участия в онкогенезе FASN является фактором, который может влиять на прогрессирование заболеваний, таких как диабет и лейомиомы матки. В частности, в одном из исследований обнаружили, что ингибитор FASN платенсимицин снижал фоновый уровень глюкозы в моделях диабета у мышей. Кроме того, было показано, что ингибиторы FASN могут быть эффективными для инициирования снижения веса (см., например, Европейский патент ЕР0869784-А). Аналогично, результаты общегеномного исследования позволяют предположить, что FASN может влиять на предрасположенность к лейомиомам матки.
Кроме того, было показано, что FASN является мишенью при лечении микробных инфекций. В частности, показано, что синтез жирных кислот и уровень жирных кислот очень важны при патогенезе вируса. Кроме того, FASN задействована в патогенезе цитомегаловируса человека (ЦМВЧ), вирусах гриппа А и гепатита С (см., например, Munger et al., Nature Biotechnology, 26: 1179-1186 (2008)). Также показано, что экспрессия FASN повышена в клетках, инфицированных вирусом Коксаки В3 (CVB3), пикорнавирусом, и репликация CVB3 блокируется ингибиторами FASN (см. Rassmann et al., Antiviral Research, 76: 150-158 (2007)). Кроме того, сообщалось, что FASN важна при литической репликации вируса Эпштейна-Барр (EBV) (Li et al., Journal of Virology, 78(8): 4197-4206 (2004)). FASN также участвует в репликации вируса денге (см., например, Heaton et al., Proc. Natl. Acad. Sci., 107(40): 17345-17350 (2010); и Samsa et al., PLoS Pathegens, 5(10): e1000632 (2009)). Кроме того, FASN играет важную роль при инфекции ВГС путем контролирования попадания вируса в клетку и его размножения (Yang W, Hood BL, Chadwick SL, Watkins, Luo G, Conrads TP, Wang T (2008), Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology, 48, 13967-1403).
Значительные усилия направлены на разработку ингибиторов FASN, которые могут обеспечивать способ лечения рака и других родственных заболеваний. Были определены и опубликованы ряд семейств ингибиторов, таких как азабензимидазолы (WO 2011/066211 и родственные публикации) и сульфонамидные производные (WO 2008/075070 и родственные публикации), полученные в AstraZeneca UK Ltd. Тем не менее вследствие важной роли FASN и недостатков соединений, приведенных в публикациях, сохраняется неудовлетворенная потребность в ингибиторах FASN с высокой активностью и специфичностью.
В настоящем изобретении предложена новая группа соединений, которые селективно ингибируют активность FASN и модулируют рост и пролиферацию раковых клеточных линий. Также включены способы синтеза новых соединений. Указанные соединения могут иметь большую фармацевтическую значимость при лечении рака, а также других заболеваний, таких как вирусные инфекции, ожирение и диабет.
Краткое описание изобретения
В одном из аспектов изобретения предложены соединения, композиции и наборы для ингибирования FASN, содержащие соединение формулы I:

где
R1 представляет собой С1-С3 гидроксил-алкил, который является незамещенным или замещен -СН3 или -CHzF3-z, 5-членный циклоалкил, который является незамещенным или замещен заместителями, выбранными из группы, состоящей из дейтерия, -Rp, -ORp, -NHRp и -NRpRp1, или 3- или 4-членный циклоалкил или гетероциклоалкил, где (i) гетероатом, являющийся членом кольца 3- или 4-членного гетероциклоалкила, независимо выбран из O, S или N, и (ii) каждый указанный 3- или 4-членный циклоалкил или гетероциклоалкил является незамещенным или необязательно замещен заместителями, выбранными из группы, состоящей из дейтерия, -Ra, -ORa, -NHRa и -NRaRa1;
L представляет собой 5-10-членный моноциклический или бициклический алкил или гетероалкил, где (i) гетероатомы, являющиеся членами кольца 5-10-членного моноциклического или бициклического гетероалкила, независимо выбраны из O, S или N, и (ii) каждый 5-10-членный моноциклический или бициклический алкил или гетероалкил является незамещенным или необязательно замещен заместителями, выбранными из группы, состоящей из дейтерия и -Rb;
А и В независимо представляют собой О или S;
Ar1 представляет собой 4-10-членный моноциклический или бициклический арил, гетероарил или гетероциклоалкил, где (i) указанный 4-10-членный моноциклический или бициклический гетероарил или гетероциклоалкил содержит 1, 2, 3 или 4 гетероатома, независимо выбранные из N, S или О, и (ii) каждый указанный 4-10-членный моноциклический или бициклический арил, гетероарил или гетероциклоалкил является незамещенным или необязательно независимо замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, алкила, -CHzF3-z, циано, гидроксила, гидроксиалкила, амино, аминоалкила-, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -OCHzF3-z, -алкила, -алкенила, -алкинила, -алкокси или (алкоксиалкил)амино-, -N(Rc)-C(O)-алкила, -N(Rc)-C(O)-арила, -циклоалкила, -гетероциклоалкила, -арила и -гетероарила при условии, что никакие два расположенные по соседству гетероатома одновременно не являются S или О;
R2 представляет собой Н или 4-15-членный моноциклический, бициклический или трициклический арил, гетероарил, циклоалкил или гетероциклоалкил, где (i) 4-15-членный моноциклический, бициклический или трициклический гетероарил или гетероциклоалкил содержит 1, 2, 3, 4, 5, 6, 7 или 8 гетероатомов, независимо выбранных из N, S или О, и (ii) каждый указанный арил, гетероарил, циклоалкил и гетероциклоалкил является незамещенным или необязательно замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, циано, гидроксила, гидроксил-алкила-, гидроксилциклоалкила-, гидроксил-гетероциклоалкила-, гидроксил-арила-, гидроксил-гетероарила-, амино, аминоалкила, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -CHzF3-z, -OCHzF3-z, -алкила, алкокси-, -алкенила, -алкинила, арилокси-, (алкоксиалкил)амино-, -циклоалкила, -гетероциклоалкила, (гетероциклоалкил)алкила-, -арила, -гетероарила, -O(алкил), -O(циклоалкил), -O(гетероциклоалкил), -O(арил), -O(гетероарил), ONH2, -C(O)NH(алкил), -C(O)N(арил)2, -C(O)NH(циклоалкил), -NH(CO)циклоалкила, -NH(SO2), -NH(SO2)алкила, -NH(SO2)арила, -NH(SO2)гетероарила, -N(SO2)циклоалкила, -C(O)N(алкил)2, (арил)алкила-, -гетероарила, (гетероарил)алкила-, -S(O)2-алкила, -S(O)2-арила, -S(O)2-циклоалкила, -C(O)N(алкил)2, -C(O)алкила, -NH-C(O)-алкила, -NH-C(O)-циклоалкила, NH-C(O)-гетероциклоалкила, NH-C(O)-гетероциклоалкил-Rd, -NH-C(O)-Rd-(O)алкила, -NH-C(O)-арила, -NH-C(O)-NH-алкила, NH-C(O)-NH-циклоалкила, NH2(CO)циклоалкила-, NH-C(O)-NH-арила, -NH-C(O)-O-алкила, NH-C(O)-NH-циклоалкила, -NH-C(O)-O-циклоалкила, -NH(Rd)-C(O)-алкила, -NH(Rd)-C(O)-арила, -NH(Rd)-S(O2)циклоалкила, -S(O2)NH2, -S(O2)NH(алкил), -S(O2)N(Rd)циклоалкила, -S(O2)N(алкил)2, -C(O)N(H)(алкил), -C(O)N(Rd)(циклоалкил), метилендиокси, -CHzF3-z, -OCHzF3-z и -алкокси;
Rp и Rp1 независимо представляют собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Ra и Ra1 независимо представляют собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Rb представляет собой Н, галоген, С1-С4 алкил, С1-С3 гидроксил-алкил или С3-С4 циклоалкил;
Rc представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Rd представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
и z равен 0, 1 или 2;
и его фармацевтически приемлемые соли, сольваты, сложные эфиры, пролекарства и изомеры.
В другом аспекте настоящего изобретения предложены соединения, композиции и наборы для ингибирования FASN, содержащие соединение формулы I-A:

(I-A)
где:
R1 представляет собой С1-С3 гидроксил-алкил, который является незамещенным или замещен -СН3 или -CHzF3-z, 5-членный циклоалкил, который является незамещенным или замещен заместителями, выбранными из группы, состоящей из дейтерия, -Rp, -ORp, -NHRp и -NRpRp1, или 3- или 4-членный циклоалкил или гетероциклоалкил, где (i) гетероатом, являющийся членом кольца 3- или 4-членного гетероциклоалкила, независимо выбран из O, S или N, и (ii) каждый указанный 3- или 4-членный циклоалкил или гетероциклоалкил является незамещенным или необязательно замещен заместителями, выбранными из группы, состоящей из дейтерия, -Ra, -ORa, -NHRa и -NRaRa1;
Ar1 представляет собой 4-10-членный моноциклический или бициклический арил, гетероарил или гетероциклоалкил, где (i) указанный 4-10-членный моноциклический или бициклический гетероарил или гетероциклоалкил содержит 1, 2, 3 или 4 гетероатома, независимо выбранные из N, S или О, и (ii) каждый указанный 4-10-членный моноциклический или бициклический арил, гетероарил или гетероциклоалкил является незамещенным или необязательно независимо замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, алкила, -CHzF3-z, циано, гидроксила, гидроксиалкила, амино, аминоалкила-, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -OCHzF3-z, -алкила, -алкенила, -алкинила, -алкокси или (алкоксиалкил)амино-, -N(Rc)-C(O)-алкила, -N(Rc)-C(O)-арила, -циклоалкила, -гетероциклоалкила, -арила и -гетероарила, при условии, что никакие два расположенные по соседству гетероатома одновременно не являются S или О;
R2 представляет собой Н или 4-15-членный моноциклический, бициклический или трициклический арил, гетероарил, циклоалкил или гетероциклоалкил, где (i) 4-15-членный моноциклический, бициклический или трициклический гетероарил или гетероциклоалкил содержит 1, 2, 3, 4, 5, 6, 7 или 8 гетероатомов, независимо выбранных из N, S или О, и (ii) каждый указанный арил, гетероарил, циклоалкил и гетероциклоалкил является незамещенным или необязательно замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, циано, гидроксила, гидроксил-алкила-, гидроксилциклоалкила-, гидроксил-гетероциклоалкила-, гидроксил-арила-, гидроксил-гетероарила-, амино, аминоалкила, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -CHzF3-z, -OCHzF3-z, -алкила, алкокси-, -алкенила, -алкинила, арилокси-, (алкоксиалкил)амино-, -циклоалкила, -гетероциклоалкила, (гетероциклоалкил)алкила-, -арила, -гетероарила, -O(алкил), -O(циклоалкил), -O(гетероциклоалкил), -O(арил), -O(гетероарил), ONH2, -C(O)NH(алкил), -C(O)N(арил)2, -C(O)NH(циклоалкил), -NH(CO)циклоалкила, -NH(SO2), -NH(SO2)алкила, -NH(SO2)арила, -NH(SO2)гетероарила, -N(SO2)циклоалкила, -C(O)N(алкил)2, (арил)алкила-, -гетероарила, (гетероарил)алкила-, -S(O)2-алкила, -S(O)2-арила, -S(O)2-циклоалкила, -C(O)N(алкил)2, -C(O)алкила, -NH-C(O)-алкила, -NH-C(O)-циклоалкила, NH-C(O)-гетероциклоалкила, NH-C(O)-гетероциклоалкил-Rd, -NH-C(O)-Rd-(O)алкила, -NH-C(O)-арила, -NH-C(O)-NH-алкила, NH-C(O)-NH-циклоалкила, NH2(CO)циклоалкила-, NH-C(O)-NH-арила, -NH-C(O)-O-алкила, NH-C(O)-NH-циклоалкила, -NH-C(O)-O-циклоалкила, -NH(Rd)-C(O)-алкила, -NH(Rd)-C(O)-арила, -NH(Rd)-S(O2)циклоалкила, -S(O2)NH2, -S(O2)NH(алкил), -S(O2)N(Rd)циклоалкила, -S(O2)N(алкил)2, -C(O)N(H)(алкил), -C(O)N(Rd)(циклоалкил), метилендиокси, -CHzF3-z, -OCHzF3-z и -алкокси;
Rp и Rp1 независимо представляют собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Ra и Ra1 независимо представляют собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Rc представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Rd представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
и z равен 0, 1 или 2;
и его фармацевтически приемлемые соли, сольваты, сложные эфиры, пролекарства и изомеры.
В другом аспекте настоящего изобретения предложены соединения, композиции и наборы для ингибирования FASN, содержащие соединение формулы I-B:
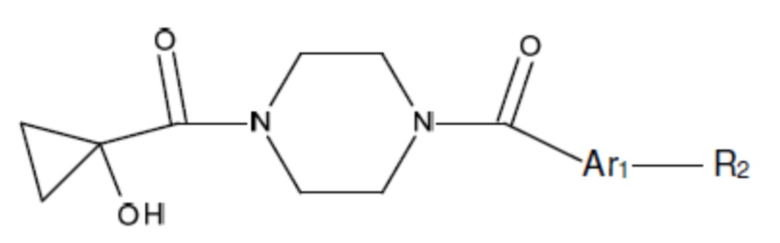
(I-B)
где:
Ar1 представляет собой 4-10-членный моноциклический или бициклический арил, гетероарил или гетероциклоалкил, где (i) указанный 4-10-членный моноциклический или бициклический гетероарил или гетероциклоалкил содержит 1, 2, 3 или 4 гетероатома, независимо выбранные из N, S или О, и (ii) каждый указанный 4-10-членный моноциклический или бициклический арил, гетероарил или гетероциклоалкил является незамещенным или необязательно независимо замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, алкила, -CHzF3-z, циано, гидроксила, гидроксиалкила, амино, аминоалкила-, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -OCHzF3-z, -алкила, -алкенила, -алкинила, -алкокси или (алкоксиалкил)амино-, -N(Rc)-C(O)-алкила, -N(Rc)-C(O)-арила, -циклоалкила, -гетероциклоалкила, -арила и -гетероарила, при условии, что никакие два расположенные по соседству гетероатома одновременно не являются S или О;
R2 представляет собой Н или 4-15-членный моноциклический, бициклический или трициклический арил, гетероарил, циклоалкил или гетероциклоалкил, где (i) 4-15-членный моноциклический, бициклический или трициклический гетероарил или гетероциклоалкил содержит 1, 2, 3, 4, 5, 6, 7 или 8 гетероатомов, независимо выбранных из N, S или О, и (ii) каждый указанный арил, гетероарил, циклоалкил и гетероциклоалкил является незамещенным или необязательно замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, циано, гидроксила, гидроксил-алкила-, гидроксилциклоалкила-, гидроксил-гетероциклоалкила-, гидроксил-арила-, гидроксил-гетероарила-, амино, аминоалкила, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -CHzF3-z, -OCHzF3-z, -алкила, алкокси-, -алкенила, -алкинила, арилокси-, (алкоксиалкил)амино-, -циклоалкила, -гетероциклоалкила, (гетероциклоалкил)алкила-, -арила, -гетероарила, -O(алкил), -O(циклоалкил), -O(гетероциклоалкил), -O(арил), -O(гетероарил), ONH2, -C(O)NH(алкил), -C(O)N(арил)2, -C(O)NH(циклоалкил), -NH(CO)циклоалкила, -NH(SO2), -NH(SO2)алкила, -NH(SO2)арила, -NH(SO2)гетероарила, -N(SO2)циклоалкила, -C(O)N(алкил)2, (арил)алкила-, -гетероарила, (гетероарил)алкила-, -S(O)2-алкила, -S(O)2-арила, -S(O)2-циклоалкила, -C(O)N(алкил)2, -C(O)алкила, -NH-C(O)-алкила, -NH-C(O)-циклоалкила, NH-C(O)-гетероциклоалкила, NH-C(O)-гетероциклоалкил-Rd, -NH-C(O)-Rd-(O)алкила, -NH-C(O)-арила, -NH-C(O)-NH-алкила, NH-C(O)-NH-циклоалкила, NH2(CO)циклоалкила-, NH-C(O)-NH-арила, -NH-C(O)-O-алкила, NH-C(O)-NH-циклоалкила, -NH-C(O)-O-циклоалкила, -NH(Rd)-C(O)-алкила, -NH(Rd)-C(O)-арила, -NH(Rd)-S(O2)циклоалкила, -S(O2)NH2, -S(O2)NH(алкил), -S(O2)N(Rd)циклоалкила, -S(O2)N(алкил)2, -C(O)N(H)(алкил), -C(O)N(Rd)(циклоалкил), метилендиокси, -CHzF3-z, -OCHzF3-z и -алкокси;
Rc представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Rd представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
и z равен 0, 1 или 2;
и его фармацевтически приемлемые соли, сольваты, сложные эфиры, пролекарства и изомеры.
В другом аспекте настоящего изобретения предложены соединения, композиции и наборы для ингибирования FASN, содержащие соединение формулы I-C:

(I-C)
где:
R1 представляет собой С1-С3 гидроксил-алкил, который является незамещенным или замещен -СН3 или -CHzF3-z, 5-членный циклоалкил, который является незамещенным или замещен заместителями, выбранными из группы, состоящей из дейтерия, -Rp, -ORp, -NHRp и -NRpRp1, или 3- или 4-членный циклоалкил или гетероциклоалкил, где (i) гетероатом, являющийся членом кольца 3- или 4-членного гетероциклоалкила, независимо выбран из O, S или N, и (ii) каждый указанный 3- или 4-членный циклоалкил или гетероциклоалкил является незамещенным или необязательно замещен заместителями, выбранными из группы, состоящей из дейтерия, -Ra, -ORa, -NHRa и -NRaRa1;
R2 представляет собой Н или 4-15-членный моноциклический, бициклический или трициклический арил, гетероарил, циклоалкил или гетероциклоалкил, где (i) 4-15-членный моноциклический, бициклический или трициклический гетероарил или гетероциклоалкил содержит 1, 2, 3, 4, 5, 6, 7 или 8 гетероатомов, независимо выбранных из N, S или О, и (ii) каждый указанный арил, гетероарил, циклоалкил и гетероциклоалкил является незамещенным или необязательно замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, циано, гидроксила, гидроксил-алкила-, гидроксилциклоалкила-, гидроксил-гетероциклоалкила-, гидроксил-арила-, гидроксил-гетероарила-, амино, аминоалкила, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -CHzF3-z, -OCHzF3-z, -алкила, алкокси-, -алкенила, -алкинила, арилокси-, (алкоксиалкил)амино-, -циклоалкила, -гетероциклоалкила, (гетероциклоалкил)алкила-, -арила, -гетероарила, -O(алкил), -O(циклоалкил), -O(гетероциклоалкил), -O(арил), -O(гетероарил), ONH2, -C(O)NH(алкил), -C(O)N(арил)2, -C(O)NH(циклоалкил), -NH(CO)циклоалкила, -NH(SO2), -NH(SO2)алкила, -NH(SO2)арила, -NH(SO2)гетероарила, -N(SO2)циклоалкила, -C(O)N(алкил)2, (арил)алкила-, -гетероарила, (гетероарил)алкила-, -S(O)2-алкила, -S(O)2-арила, -S(O)2-циклоалкила, -C(O)N(алкил)2, -C(O)алкила, -NH-C(O)-алкила, -NH-C(O)-циклоалкила, NH-C(O)-гетероциклоалкила, NH-C(O)-гетероциклоалкил-Rd, -NH-C(O)-Rd-(O)алкила, -NH-C(O)-арила, -NH-C(O)-NH-алкила, NH-C(O)-NH-циклоалкила, NH2(CO)циклоалкила-, NH-C(O)-NH-арила, -NH-C(O)-O-алкила, NH-C(O)-NH-циклоалкила, -NH-C(O)-O-циклоалкила, -NH(Rd)-C(O)-алкила, -NH(Rd)-C(O)-арила, -NH(Rd)-S(O2)циклоалкила, -S(O2)NH2, -S(O2)NH(алкил), -S(O2)N(Rd)циклоалкила, -S(O2)N(алкил)2, -C(O)N(H)(алкил), -C(O)N(Rd)(циклоалкил), метилендиокси, -CHzF3-z, -OCHzF3-z и -алкокси;
Rp и Rp1 независимо представляют собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Ra и Ra1 независимо представляют собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Rd представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Rq представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
и z равен 0, 1 или 2;
и его фармацевтически приемлемые соли, сольваты, сложные эфиры, пролекарства и изомеры.
В другом аспекте настоящего изобретения предложены соединения, композиции и наборы для ингибирования FASN, содержащие соединение формулы I-D:
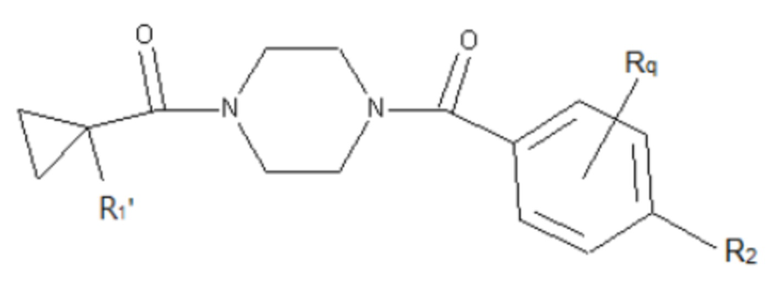
(I-D)
где:
R1' представляет собой ОН или NH2;
R2 представляет собой Н или 4-15-членный моноциклический, бициклический или трициклический арил, гетероарил, циклоалкил или гетероциклоалкил, где (i) 4-15-членный моноциклический, бициклический или трициклический гетероарил или гетероциклоалкил содержит 1, 2, 3, 4, 5, 6, 7 или 8 гетероатомов, независимо выбранных из N, S или О, и (ii) каждый указанный арил, гетероарил, циклоалкил и гетероциклоалкил является незамещенным или необязательно замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, циано, гидроксила, гидроксил-алкила-, гидроксилциклоалкила-, гидроксил-гетероциклоалкила-, гидроксил-арила-, гидроксил-гетероарила-, амино, аминоалкила, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -CHzF3-z, -OCHzF3-z, -алкила, алкокси-, -алкенила, -алкинила, арилокси-, (алкоксиалкил)амино-, -циклоалкила, -гетероциклоалкила, (гетероциклоалкил)алкила-, -арила, -гетероарила, -O(алкил), -O(циклоалкил), -O(гетероциклоалкил), -O(арил), -O(гетероарил), ONH2, -C(O)NH(алкил), -C(O)N(арил)2, -C(O)NH(циклоалкил), -NH(CO)циклоалкила, -NH(SO2), -NH(SO2)алкила, -NH(SO2)арила, -NH(SO2)гетероарила, -N(SO2)циклоалкила, -C(O)N(алкил)2, (арил)алкила-, -гетероарила, (гетероарил)алкила-, -S(O)2-алкила, -S(O)2-арила, -S(O)2-циклоалкила, -C(O)N(алкил)2, -C(O)алкила, -NH-C(O)-алкила, -NH-C(O)-циклоалкила, NH-C(O)-гетероциклоалкила, NH-C(O)-гетероциклоалкил-Rd, -NH-C(O)-Rd-(O)алкила, -NH-C(O)-арила, -NH-C(O)-NH-алкила, NH-C(O)-NH-циклоалкила, NH2(CO)циклоалкила-, NH-C(O)-NH-арила, -NH-C(O)-O-алкила, NH-C(O)-NH-циклоалкила, -NH-C(O)-O-циклоалкила, -NH(Rd)-C(O)-алкила, -NH(Rd)-C(O)-арила, -NH(Rd)-S(O2)циклоалкила, -S(O2)NH2, -S(O2)NH(алкил), -S(O2)N(Rd)циклоалкила, -S(O2)N(алкил)2, -C(O)N(H)(алкил), -C(O)N(Rd)(циклоалкил), метилендиокси, -CHzF3-z, -OCHzF3-z и -алкокси;
Rd представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
и z равен 0, 1 или 2;
и его фармацевтически приемлемые соли, сольваты, сложные эфиры, пролекарства и изомеры.
В другом аспекте настоящего изобретения предложены способы лечения заболевания посредством ингибирования FASN у субъекта (например, человека), нуждающегося в этом, путем введения субъекту эффективного количества соединения или фармацевтического состава согласно настоящему изобретению.
Другой предпочтительный вариант реализации представляет собой фармацевтический состав, содержащий фармацевтически приемлемое соединение согласно настоящему изобретению, которое при введении человеку обеспечивает снижение опухолевой нагрузки и/или метастазов. Фармацевтический состав можно вводить перорально или при помощи других способов, например, внутривенно или путем инъекции.
Другой вариант реализации представляет собой способ лечения рака яичников у субъекта (например, человека), нуждающегося в этом, путем введения субъекту терапевтически эффективного количества соединения или фармацевтического состава согласно настоящему изобретению.
Другой вариант реализации представляет собой способ лечения рака толстой кишки у субъекта (например, человека), нуждающегося в этом, путем введения субъекту эффективного количества соединения или фармацевтического состава согласно настоящему изобретению.
Другой вариант реализации представляет собой способ лечения рака груди у субъекта (например, человека), нуждающегося в этом, путем введения субъекту эффективного количества фармацевтического состава согласно настоящему изобретению.
Другой вариант реализации представляет собой способ лечения лейкоза у субъекта (например, человека), нуждающегося в этом, путем введения субъекту эффективного количества соединения или фармацевтического состава согласно настоящему изобретению.
Другой вариант реализации представляет собой способ лечения рака толстой кишки до или после хирургической резекции и/или радиотерапии у субъекта (например, человека), нуждающегося в этом, путем введения субъекту эффективного количества соединения или фармацевтического состава согласно настоящему изобретению.
Другой вариант реализации представляет собой способ лечения рака до или после хирургической резекции и/или радиотерапии у субъекта (например, человека), нуждающегося в этом, путем введения субъекту эффективного количества соединения или фармацевтического состава согласно настоящему изобретению совместно с проведением дополнительной терапии для лечения тошноты с использованием дексаметазона или без него.
Другой вариант реализации представляет собой способ лечения рака до или после хирургической резекции и/или радиотерапии у субъекта (например, человека), нуждающегося в этом, путем введения субъекту эффективного количества соединения или фармацевтического состава согласно настоящему изобретению совместно с проведением дополнительной терапии с использованием одного или более дополнительных терапевтических агентов или их фармацевтически приемлемых солей. Неограничивающие примеры указанных дополнительных терапевтических агентов включают цитотоксические агенты (такие как, например, без ограничений агенты, воздействующие на ДНК (такие как цисплатин или доксорубицин)); таксаны (например, таксотер, таксол); ингибиторы топоизомеразы II (такие как этопозид); ингибиторы топоизомеразы II (такие как иринотекан (или CPT-11), камптостар или топотекан); агенты, воздействующие на тубулин (такие как паклитаксел, доцетаксел или эпотилоны); гормональные агенты (такие как тамоксифен); ингибиторы тимидилатсинтазы (такие как 5-фторурацил или 5-FU); антиметаболиты (такие как метотрексат); алкилирующие агенты (такие как темозоломид, циклофосфамид); ингибиторы фарнезилпротеинтрансферазы (такие как SARASAR™ (4-[2-[4-[(11R)-3,10-дибром-8-хлор-6,11-дигидро-5H-бензо[5,6]циклогепта[1,2-b]пиридин-11-ил]-1-пиперидинил]-2-оксоэтил]-1-пиперидинкарбоксамид или SCH 66336), типифарниб (Zarnestra® или R115777 производства Janssen Pharmaceuticals), L778,123 (ингибитор фарнезилпротеинтрансферазы производства Merck & Company, Whitehouse Station, N.J.), BMS 214662 (ингибитор фарнезилпротеинтрансферазы производства Bristol-Myers Squibb Pharmaceuticals, Princeton, N.J.)); ингибиторы сигнальной трансдукции (такие как Iressa® (производства Astra Zeneca Pharmaceuticals, England), Tarceva® (ингибиторы EGFR киназы), антитела к EGFR (например, С225), GLEEVEC® (ингибитор C-abl киназы производства Novartis Pharmaceuticals, East Hanover, N.J.)); интерфероны, такие как, например, Intron® (производства Merck & Company), Peg-Intron® (производства Merck & Company); комбинированные гормональные терапии; комбинации ароматаз; ara-C, адриамицин, цитоксан и гемцитабин.
Другие агенты против рака (также известные как антинеопластические агенты) включают, но не ограничиваются ими, урациловый иприт, хлорметин, ифосфамид, мелфалан, хлорамбуцил, пипоброман, триэтиленмеламин, триэтилентиофосфорамид, бусульфан, кармустин, ломустин, стрептозоцин, дакарбазин, флоксуридин, цитарабин, 6-меркаптопурин, 6-тиогуанин, флударабина фосфат, оксалиплатин, лейковорин, оксалиплатин (ELOXATIN® производства Sanofi-Synthelabo Pharmaceuticals, France), пентостатин, винбластин, винкристин, виндесин, блеомицин, дактиномицин, даунорубицин, доксорубицин, эпирубицин, идарубицин, митрамицин, деоксикоформицин, митомицин-С, L-аспарагиназу, тенипозид, 17α-этинилэстрадиол, диэтилстилбестрол, тестостерон, преднизон, флуоксиместерон, дромостанолона пропионат, тестолактон, мегестрола ацетат, метилпреднизолон, метилтестостерон, преднизолон, триамцинолон, хлортрианизен, гидроксипрогестерон, аминоглутетимид, эстрамустин, медроксипрогестерона ацетат, леупролид, флутамид, торемифен, госерелин, цисплатин, карбоплатин, гидроксимочевину, амсакрин, прокарбазин, митотан, митоксантрон, левамизол, навелбен, анастрозол, летрозол, капецитабин, релоксафин, дролоксафин, гексаметилмеламин, авастин, герцептин, бексар, Velcade®, зевалин, трисенокс, кселода, винорелбин, порфимер, эрбитукс, липосомальный, тиотепа, алтретамин, мелфалан, трастузумаб, лерозол, фулвестрант, эксеместан, ифосфамид, ритуксимаб, С225 и кампат, 5-фторурацил и лейковорин совместно с ингибитором рецептора 5-HT3 (например, долансетроном, гранисетроном, ондансетроном) или без него совместно с дексаметазоном или без него.
Другой вариант реализации представляет собой способ лечения диабета у субъекта (например, человека), нуждающегося в этом, путем введения субъекту эффективного количества соединения или фармацевтического состава согласно настоящему изобретению.
Другой вариант реализации представляет собой способ лечения ожирения или избыточного веса у субъекта (например, человека), нуждающегося в этом, путем введения субъекту эффективного количества соединения или фармацевтического состава согласно настоящему изобретению.
Другой вариант реализации представляет собой способ лечения лейомиом матки у субъекта (например, человека), нуждающегося в этом, путем введения субъекту эффективного количества соединения или фармацевтического состава согласно настоящему изобретению.
Другой вариант реализации представляет собой способ лечения микробных инфекций у субъекта (например, человека), нуждающегося в этом, путем введения субъекту эффективного количества соединения или фармацевтического состава согласно настоящему изобретению.
Другой вариант реализации представляет собой способ лечения вирусных инфекций, включая, но не ограничиваясь ими, инфекции, вызванные ЦМВЧ, вирусом гриппа А, вирусом гепатита С, CVB3, пикорнавирусом, EBC и вирусом денге, у субъекта (например, человека), нуждающегося в этом, путем введения субъекту эффективного количества соединения или фармацевтического состава согласно настоящему изобретению.
В случае фиксированной дозы соединения согласно настоящему изобретению используют в указанных комбинированных продуктах в диапазоне дозировок, описанном в настоящем описании (или известном специалистам в данной области техники), и другой фармацевтически активный агент или способ лечения применяют в соответствующем диапазоне дозировок. Например, было обнаружено, что ингибитор CDC2 оломуцин обладает синергическим действием с известными цитотоксическими агентами в отношении инициирования апоптоза (J. Cell Sci., (1995) 108, 2897). Соединения согласно настоящему изобретению также можно вводить последовательно совместно с известными противораковыми или цитотоксическими агентами, если использование комбинированного состава неуместно. При любом комбинированном способе лечения изобретение не ограничено последовательностью введения; соединения описанных формул можно вводить до или после введения известного противоракового или цитотоксического агента. Например, последовательность введения противораковых агентов влияет на цитотоксическую активность ингибитора циклин-зависимой киназы флавопиридола (Cancer Research, (1997) 57, 3375). Указанные способы находятся в рамках компетенции специалистов в данной области техники, а также лечащих врачей.
Любой из указанных выше способов может быть дополнен путем введения жидких сред (таких как вода), петлевых диуретиков, одного или более химиотерапевтических или антинеопластических агентов, таких как лейковорин и фторурацил, и вспомогательных химиотерапевтических агентов, таких как (филграстим и эритропоэтин) или любой комбинации указанных выше агентов.
Другой вариант реализации представляет собой способ введения соединения согласно настоящему изобретению субъекту (например, человеку), нуждающемуся в этом, путем введения субъекту фармацевтического состава согласно настоящему изобретению.
Другой вариант реализации представляет собой способ получения фармацевтического состава согласно настоящему изобретению путем смешения по меньшей мере одного фармацевтически приемлемого соединения согласно настоящему изобретению и необязательно одной или более фармацевтически приемлемых добавок или вспомогательных веществ.
Инертные фармацевтически приемлемые носители, используемые для получения фармацевтических композиций из соединений, описанных в настоящем изобретении, могут быть твердыми или жидкими. Твердые препараты включают порошки, таблетки, диспергируемые гранулы, капсулы, крахмальные капсулы и суппозитории. Порошки и таблетки могут содержать от примерно 5 до примерно 95 процентов активного ингредиента. Подходящие твердые носители известны в данной области техники и включают, например, карбонат магния, стеарат магния, тальк, сахар или лактозу. Таблетки, порошки, капсулы и крахмальные капсулы можно применять в качестве твердых лекарственных форм, подходящих для перорального введения. Примеры фармацевтически приемлемых носителей и способов получения различных композиций можно найти в A. Gennaro (ed.), Remington's Pharmaceutical Sciences, 18th Edition, (1990), Mack Publishing Co., Easton, Pa.
Жидкие препараты включают растворы, суспензии и эмульсии. В качестве примера можно отметить воду или растворы вода-пропиленгликоль для парентеральной инъекции или добавки подсластителей и замутняющих агентов для пероральных растворов, суспензий и эмульсий. Жидкие препараты также могут включать растворы для интраназального введения.
Препараты в виде аэрозолей, подходящие для ингаляции, могут включать растворы и твердые вещества в порошковой форме, которые можно объединять с фармацевтически приемлемым носителем, таким как инертный сжатый газ, например, азот.
Также включены твердые препараты, которые незадолго до применения необходимо превращать в жидкие препараты для перорального или парентерального введения. Указанные жидкие формы включают растворы, суспензии и эмульсии.
Соединения согласно настоящему изобретению также можно доставлять чрескожно. Чрескожные композиции могут иметь форму кремов, лосьонов, аэрозолей и/или эмульсий и могут содержаться в чрескожном пластыре, имеющем матрицу или резервуар, традиционно используемые в данной области техники.
Соединения согласно настоящему изобретению также можно доставлять подкожно.
Предпочтительно соединение вводят перорально или внутривенно.
Предпочтительно фармацевтический препарат содержится в стандартной лекарственной форме. В указанной форме препарат разделен на стандартные дозы подходящего размера, содержащие соответствующие количества активного компонента, например, эффективное количество, обеспечивающее исполнение желаемой задачи.
Количество активного соединения в стандартной дозе препарата можно изменять или регулировать от примерно 1 мг до примерно 1000 мг, предпочтительно от примерно 1 мг до примерно 500 мг, более предпочтительно от примерно 1 мг до примерно 250 мг, еще более предпочтительно от примерно 1 мг до примерно 25 мг в зависимости от конкретного применения.
Фактическая используемая дозировка может быть различной в зависимости от требований пациента и тяжести состояния, подвергающегося лечению. Определение подходящей схемы лечения для конкретной ситуации входит в рамки компетенции специалистов в данной области техники. Для удобства общая ежедневная дозировка может быть разделена, и при необходимости ее можно вводить по частям в течение дня.
Количество и частоту введения соединений согласно настоящему изобретению и/или их фармацевтически приемлемых солей можно регулировать в зависимости от выбора лечащего врача с учетом таких факторов, как возраст, состояние и размер пациента, а также тяжесть симптомов, подвергающихся лечению. Типовая рекомендованная дневная дозировка при пероральном введении может находиться в диапазоне от примерно 1 мг/день до примерно 500 мг/день, предпочтительно от 1 мг/день до 200 мг/день в составе двух – четырех отдельных доз.
Определения
Следует понимать, что следующие термины, используемые выше и далее в настоящем описании, если не указано иное, имеют приведенные далее значения. Если определение отсутствует, то следует использовать традиционные определения, известные специалистам в данной области техники.
«Пациент» включает человека и животных.
«Млекопитающее» обозначает человека и других млекопитающих.
Термин «FASN» относится ко всем классам, типам, подтипам, изотипам, сегментам, вариантам и мутантным формам синтазы жирных кислот.
Термин «ингибитор» относится к молекуле, например, соединения, лекарственного средства, активатора фермента или гормона, которая блокирует или иным образом нарушает конкретную биологическую активность.
Термины «эффективное количество» или «терапевтически эффективное количество» относятся к достаточному количеству агента, обеспечивающему желаемый биологический результат. Указанный результат может представлять собой снижение и/или ослабление признаков, симптомов или причин заболевания или любое другое желаемое изменение биологической системы. Например, «эффективное количество» для терапевтического применения представляет собой количество композиции, содержащей соединение, предложенное в настоящем описании, требуемое для обеспечения клинически значимого облегчения заболевания. Специалисты в данной области техники в каждом отдельном случае могут определять соответствующее «эффективное» количество при помощи стандартной экспериментальной работы. Таким образом, выражение «эффективное количество» в общем случае относится к количеству, в котором активное вещество обладает терапевтическим действием. В указанном случае активное вещество представляет собой ингибитор синтазы жирных кислот (FASN).
Согласно настоящему описанию термины «лечить» или «лечение» являются синонимами термина «предотвращать» и обозначают отсрочку развития заболеваний, предотвращение развития заболеваний и/или снижение тяжести указанных симптомов, развитие которых возможно или ожидается в будущем. Таким образом, указанные термины включают ослабление существующих симптомов заболевания, предотвращение возникновения дополнительных симптомов, ослабление или предотвращение метаболических причин, вызывающих симптомы, подавление нарушения или заболевания, например, блокаду развития нарушения или заболевания, облегчение нарушения или заболевания, обращение вспять нарушения или заболевания, облегчение состояния, вызванного заболеванием или нарушением, или устранение или ослабление симптомов заболевания или нарушения.
Под «фармацевтически приемлемым» или «фармакологически приемлемым» понимают вещество, которое не является нежелательным с точки зрения биологии или по иным причинам, вещество можно вводить индивидууму в отсутствие каких-либо нежелательных биологических эффектов или отрицательных взаимодействий с какими-либо компонентами композиции, в которой содержится указанное вещество.
«Вещества-носители», которые также называют «вспомогательными веществами», включают любые вспомогательные вещества, традиционно используемые в фармацевтике, которые следует выбирать с учетом совместимости и профиля высвобождения из целевой лекарственной формы. Типовые вещества-носители включают, например, связывающие вещества, суспендирующие агенты, разрыхлители, наполнители, поверхностно-активные вещества, вещества, увеличивающие растворимость, стабилизаторы, смазывающие вещества, увлажнители, разбавители и т.д. «Фармацевтически совместимые вещества-носители» могут содержать, например, аравийскую камедь, желатин, коллоидный диоксид кремния, глицерофосфат кальция, лактат кальция, мальтодекстрин, глицерин, силикат магния, казеинат натрия, соевый лецитин, хлорид натрия, фосфат трикальция, фосфат дикалия, стеароиллактилат натрия, каррагинан, моноглицерид, диглицерид, прежелатинизированный крахмал и т.д. См., например, Hoover, John E., Remington’s Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa. 1975.
Согласно настоящему описанию термин «субъект» охватывает млекопитающих и животных, отличных от млекопитающих. Примеры млекопитающих включают, но не ограничиваются ими, любого члена класса млекопитающих: человека, приматов, отличных от человека, таких как шимпанзе, и другие виды обезьян и мартышек; сельскохозяйственных животных, таких как крупный рогатый скот, лошади, овцы, козы, свиньи; домашних животных, таких как кролики, собаки и кошки; лабораторных животных, включая грызунов, таких как крысы, мыши и морские свинки и т.д. Примеры животных, отличных от млекопитающих, включают, но не ограничиваются ими, птиц, рыб и т.д. В одном из вариантов реализации настоящего изобретения млекопитающее представляет собой человека.
Согласно настоящему описанию «алкил» обозначает линейную или разветвленную цепь, содержащую от 1 до 10 атомов углерода. Типовые насыщенные алкильные группы включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, 2-метил-1-пропил, 2-метил-2-пропил, 2-метил-1-бутил, 3-метил-1-бутил, 2-метил-3-бутил, 2,2-диметил-1-пропил, 2-метил-1-пентил, 3-метил-1-пентил, 4-метил-1-пентил, 2-метил-2-пентил, 3-метил-2-пентил, 4-метил-2-пентил, 2,2-диметил-1-бутил, 3,3-диметил-1-бутил, 2-этил-1-бутил, бутил, изобутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил и более длинные алкильные группы, такие как гептил и октил и т.д. Алкильная группа может быть незамещенной или замещенной. Алкильные группы, содержащие три или более атомов углерода, могут быть линейными, разветвленными или циклизованными. Согласно настоящему описанию «низший алкил» обозначает алкил, содержащий от 1 до 6 атомов углерода.
Согласно настоящему описанию «алкенил» включает неразветвленную или разветвленную углеводородную цепь, содержащую одну или более двойных связей. Двойная связь в алкенильной группе может быть изолированной или сопряжена с другой ненасыщенной группой. Иллюстративные алкенильные группы включают, но не ограничиваются ими, (С2-С8) алкенильные группы, такие как, этиленил, винил, аллил, бутенил, пентенил, гексенил, бутадиенил, пентадиенил, гексадиенил, 2-этилгексенил, 2-пропил-2-бутенил, 4-(2-метил-3-бутен)пентенил и т.д. Алкенильная группа может быть незамещенной или замещенной.
Согласно настоящему описанию «алкинил» включает неразветвленную или разветвленную углеводородную цепь, содержащую одну или более тройных связей. Тройная связь в алкинильной группе может быть изолированной или сопряжена с другой ненасыщенной группой. Подходящие алкинильные группы включают, но не ограничиваются ими, (С2-С6) алкинильные группы, такие как этинил, пропинил, бутинил, пентинил, гексинил, метилпропинил, 4-метил-1-бутинил, 4-пропил-2-пентинил, 4-бутил-2-гексинил и т.д. Алкинильная группа может быть незамещенной или замещенной.
Термины «трифторметил», «сульфонил» и «карбоксил» включают CF3, SO2 и СО2Н, соответственно.
Термин «гидроксил» обозначает группу ОН.
Термин алкилгидроксил или гидроксиалкил обозначает алкильную группу, такую как определено выше, где алкильная группа содержит группу ОН.
Термин «алкокси», используемый в настоящем описании, включает -О-(алкил), где алкил такой, как определено выше.
Термин «аминоалкил», используемый в настоящем описании, обозначает группу, содержащую один или более атомов азота и одну или более алкильных групп, таких как определено выше, присоединенных к атому азота.
«Аралкил» или «арилалкил» обозначает группу арил-алкил-, в которой арил и алкил, такие как описано выше. Предпочтительные аралкилы содержат низшую алкильную группу. Неограничивающие примеры подходящих аралкильных групп включают бензил, 2-фенэтил и нафталинилметил. Группа соединена с исходным фрагментом через алкил.
«Гетероарилалкил» обозначает гетероарильный фрагмент, определенный в настоящем описании, связанный посредством алкильного фрагмента (определенного выше) с исходным ядром. Неограничивающие примеры подходящих гетероарилов включают 2-пиридинилметил, хинолинилметил и т.д.
«Гетероциклилалкил» обозначает гетероциклильный фрагмент, определенный в настоящем описании, связанный посредством алкильного фрагмента (определенного выше) с исходным ядром. Неограничивающие примеры подходящих гетероциклилалкилов включают пиперидинилметил, пиперазинилметил и т.д.
Также следует понимать, что если в тексте, на схемах, в примерах и таблицах, приведенных в настоящем описании, встречается какой-либо атом углерода, а также гетероатом, имеющий незанятые валентности, то он содержит достаточное количество атомов водорода, занимающих недостающие валентности.
Если какая-либо переменная (например, арил, гетероцикл, R2 и т.д.) встречается более одного раза в составе какого-либо компонента формулы, то в каждом случае ее определение не зависит от определения во всех других случаях.
Согласно настоящему описанию термин «композиция» охватывает продукт, содержащий указанные ингредиенты в указанных количествах, а также любые продукты, которые образуются, напрямую или косвенно, из комбинации указанных ингредиентов в указанных количествах.
Термин «дейтерий», используемый в настоящем описании, обозначает стабильный изотоп водорода, содержащий равное количество протонов и нейтронов.
Термин «галоген», используемый в настоящем описании, обозначает заместитель, содержащий по меньшей мере один атом галогена, выбранного из фтора, хлора, брома и йода.
Термин «циано», используемый в настоящем описании, обозначает заместитель, содержащий атом углерода, связанный с атомом азота посредством тройной связи.
Термин «амино», используемый в настоящем описании, обозначает заместитель, содержащий по меньшей мере один атом азота.
Термин «(амино)алкокси», используемый в настоящем описании, обозначает заместитель, содержащий по меньшей мере одну аминогруппу и по меньшей мере одну алкоксигруппу.
Термин «арилокси», используемый в настоящем описании, обозначает заместитель вида Ar-O-, где Ar представляет собой арильную группу, такую как определено в настоящем описании.
Термин «метилендиокси», используемый в настоящем описании, обозначает функциональную группу структурной формулы -O-CH2-O-, связанную с молекулой при помощи двух химических связей через атомы кислорода.
Согласно настоящему описанию «алкоксиалкил» обозначает -(алкил)-О-(алкил), где каждый «алкил» независимо представляет собой алкильную группу, определенную выше.
Термин «(алкоксиалкил)амино», используемый в настоящем описании, обозначает заместитель, содержащий по меньшей мере одну алкоксиалкильную группу, определенную выше, и по меньшей мере одну аминогруппу, определенную выше.
Согласно настоящему описанию термин «арил» относится к моноциклическому или конденсированному полициклическому ароматическому карбоциклу (кольцевой структуре, в которой все атомы кольца представляют собой атомы углерода), содержащему от 3 до 24 атомов в кольце. Иллюстративные примеры арильных групп включают, но не ограничиваются ими, следующие фрагменты:
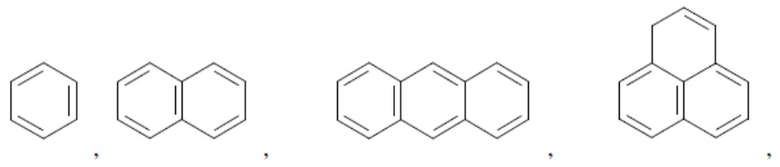
 и т.д.
и т.д.
Иллюстративные замещенные арилы включают:

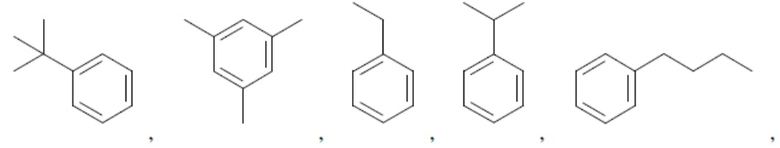
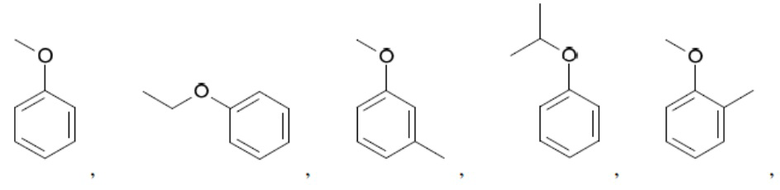
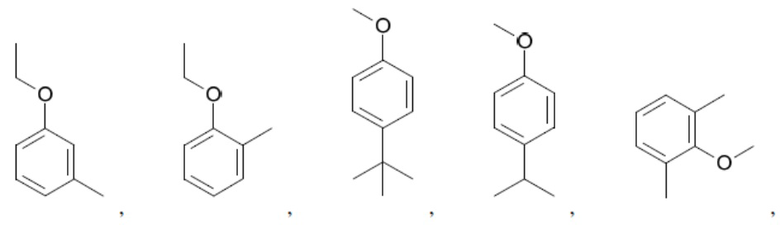
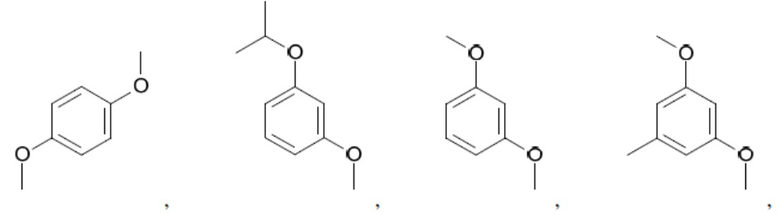
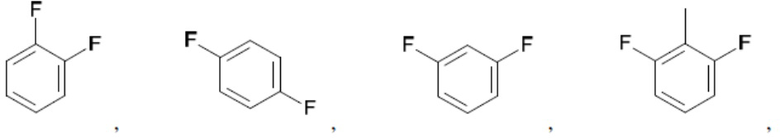
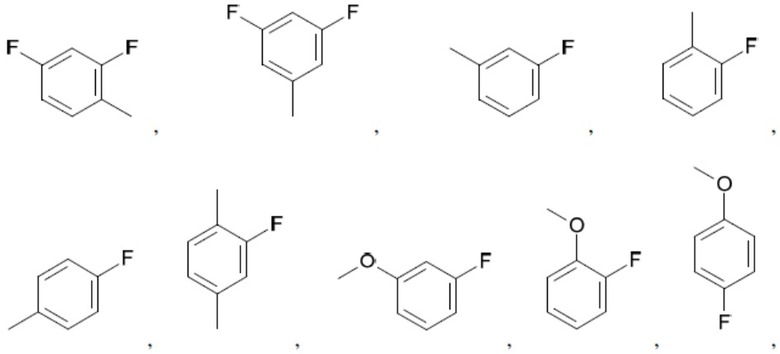
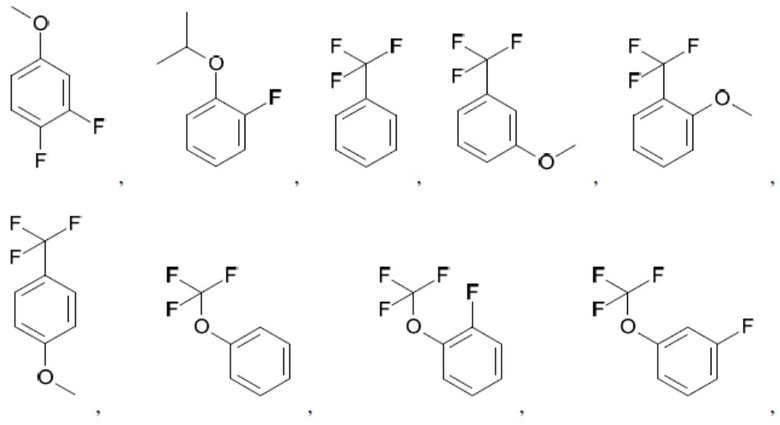
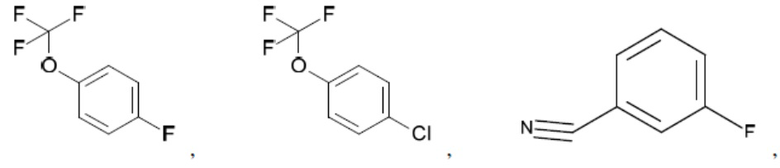
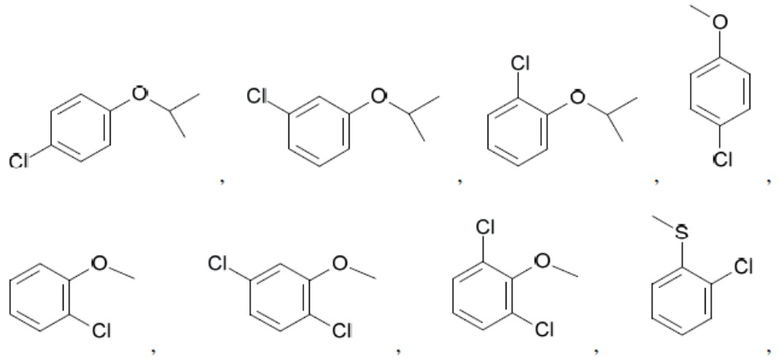

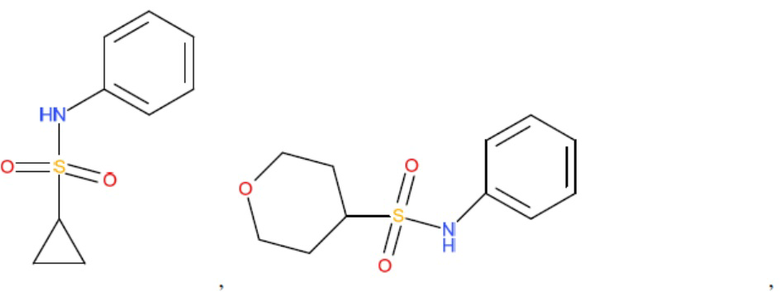
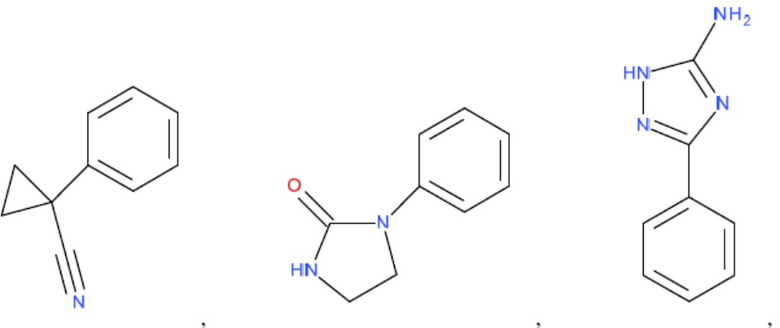
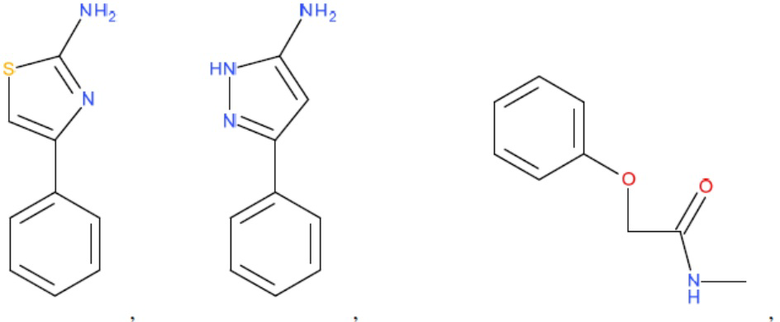

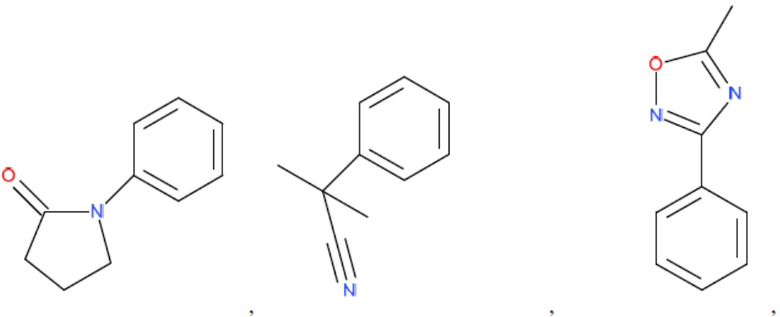
 и т.д.
и т.д.
Согласно настоящему описанию термин «гетероарил» относится к моноциклическому или конденсированному полициклическому гетероциклу (кольцевой структуре, в которой атомы в кольце выбраны из атомов углерода, а также гетероатомов азота, кислорода и серы), содержащему от 3 до 24 атомов в кольце. Иллюстративные примеры гетероарильных и замещенных гетероарильных групп включают, но не ограничиваются ими, следующие фрагменты:
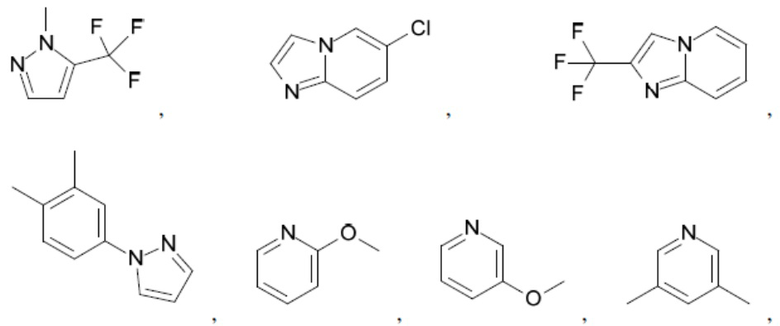
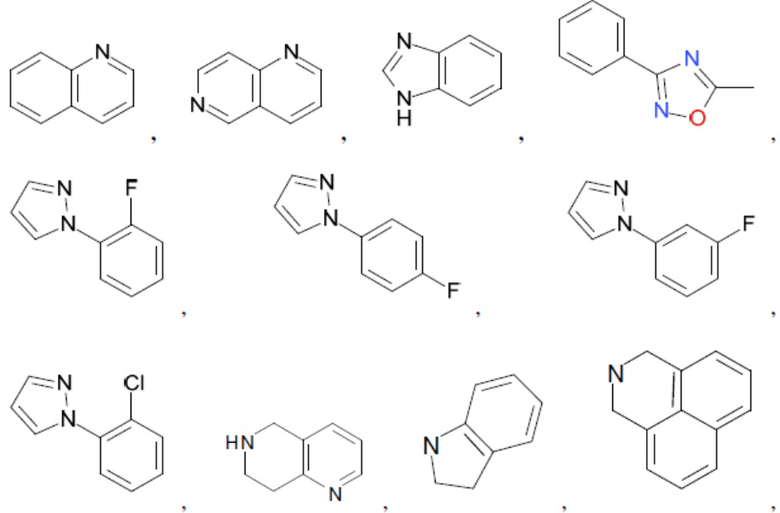
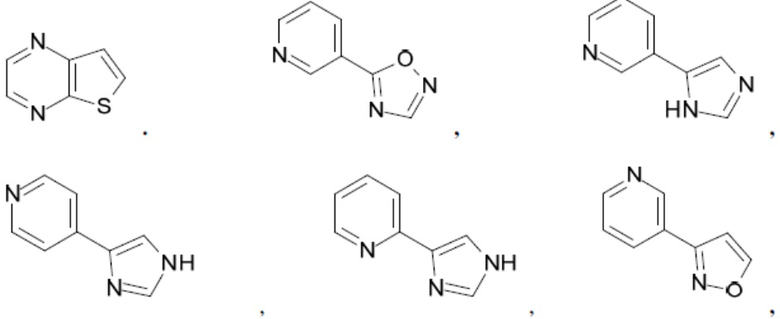
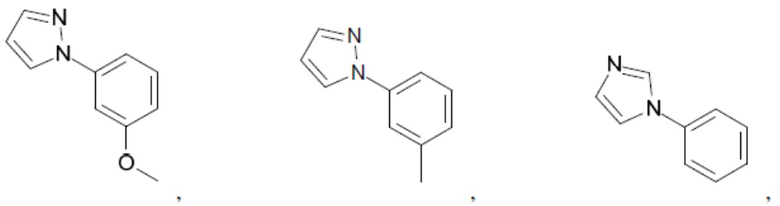
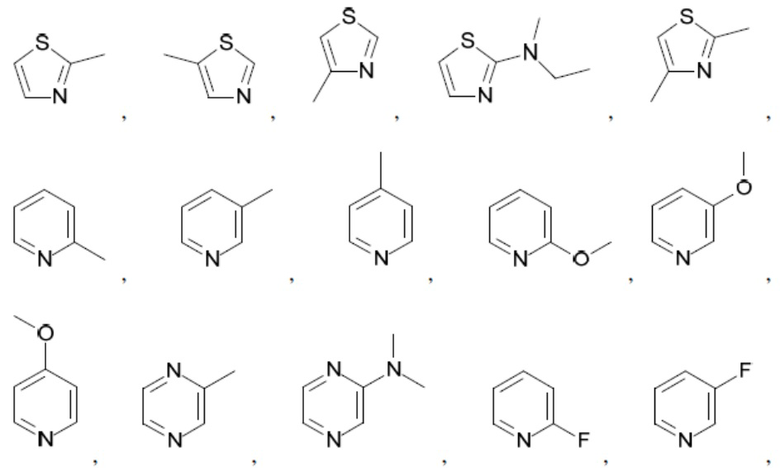

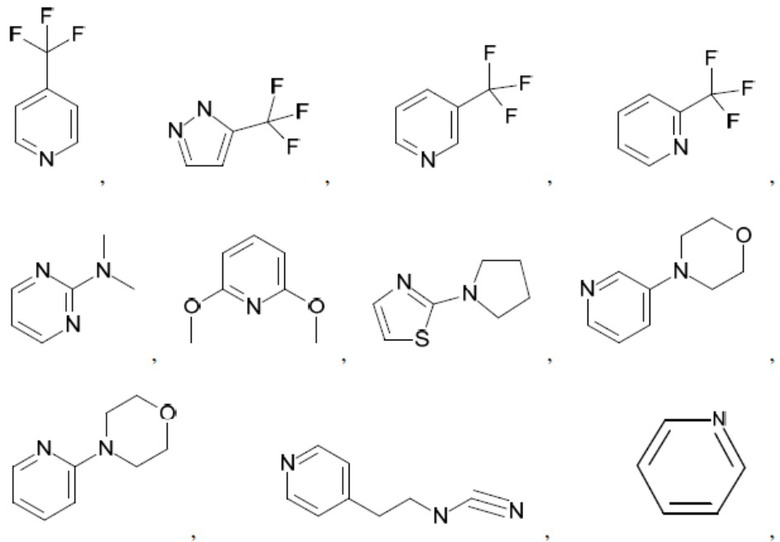
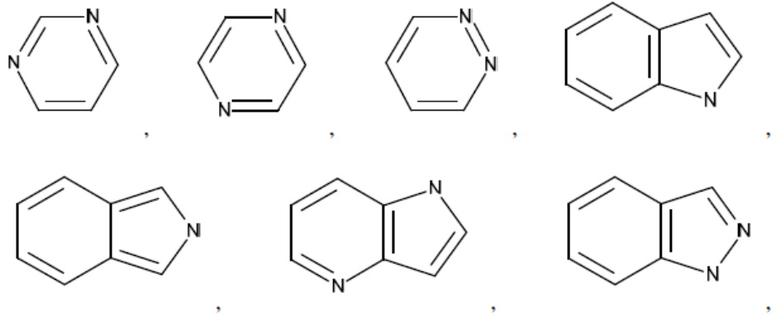
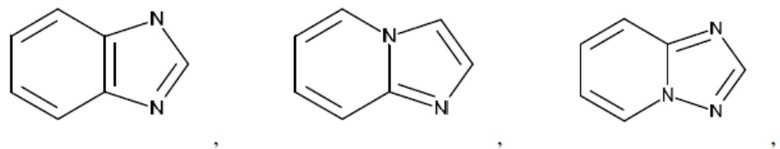

 и т.д.
и т.д.
Согласно настоящему описанию термин «циклоалкил» относится к насыщенному или частично насыщенному моноциклическому или конденсированному или спиро-полициклическому карбоциклу, содержащему от 3 до 24 атомов в кольце. Иллюстративные примеры циклоалкильных групп включают, но не ограничиваются ими, следующие фрагменты:
 и т.д.
и т.д.
Согласно настоящему описанию термин «гетероциклоалкил» относится к моноциклической или конденсированной или спиро-полициклической кольцевой структуре, которая является насыщенной или частично насыщенной и содержит от 3 до 24 атомов в кольце, выбранных из атомов С и гетероатомов N, O и S. Иллюстративные примеры гетероциклоалкильных и замещенных гетероциклоалкильных групп включают, но не ограничиваются ими:
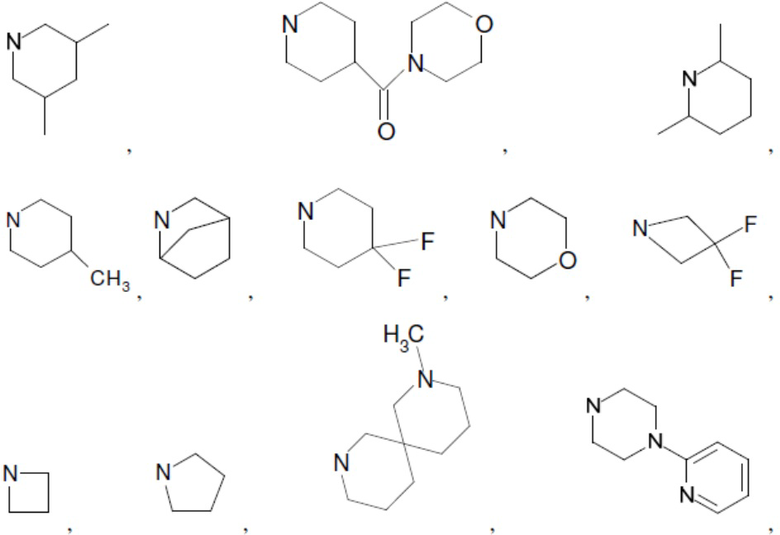

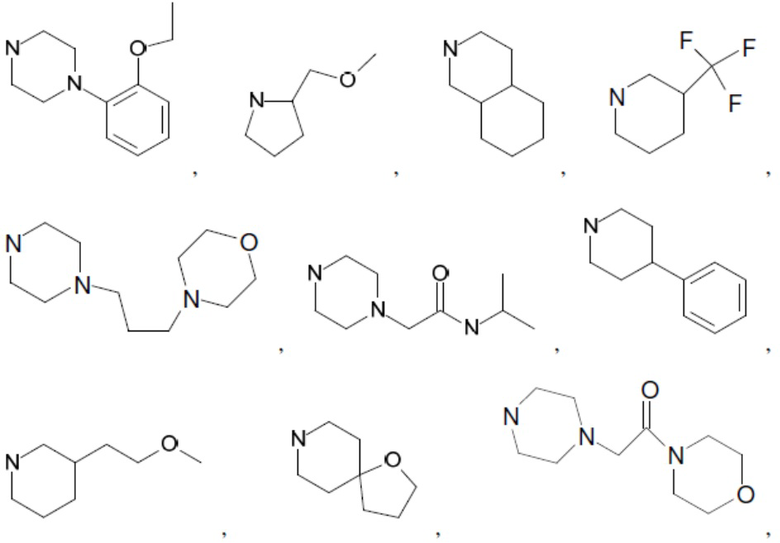
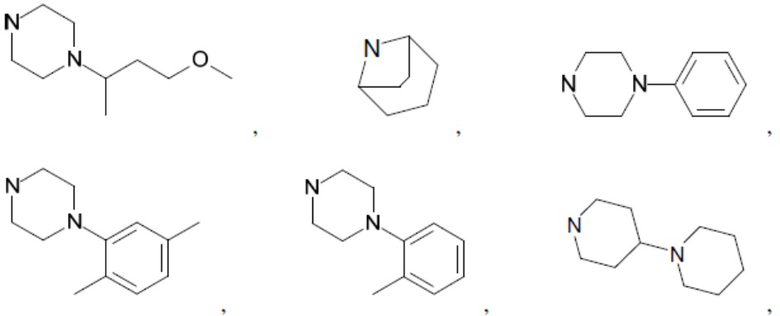
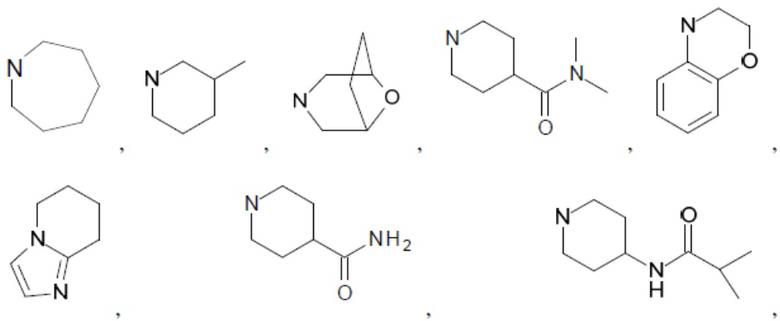
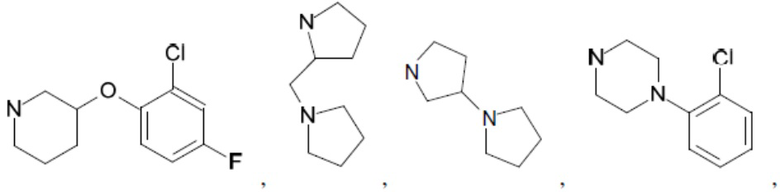

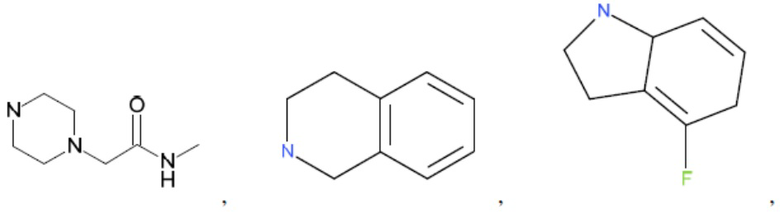
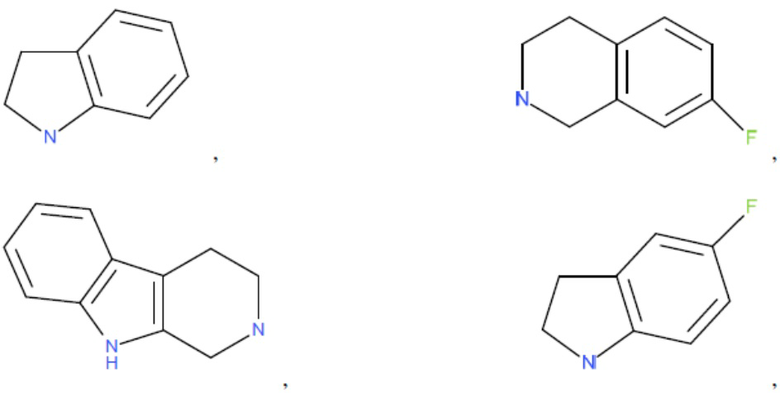
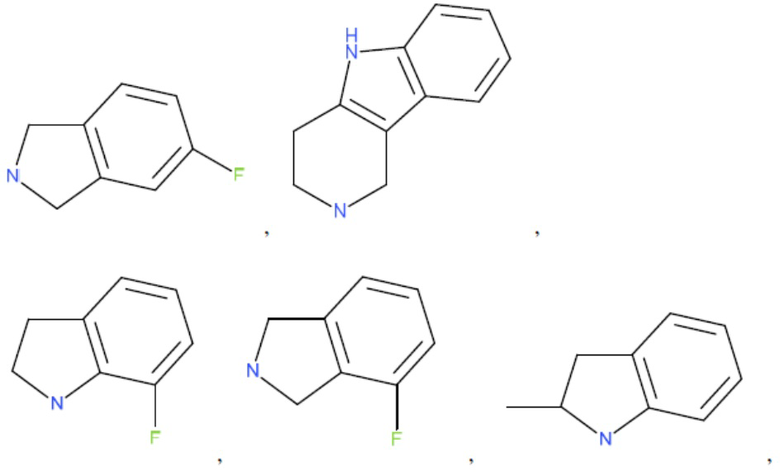
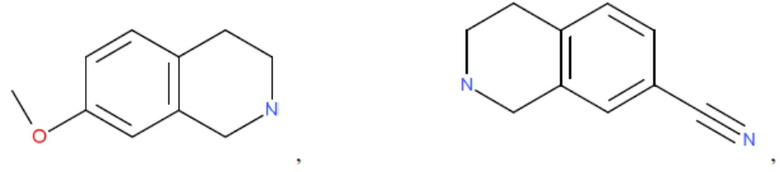
 и т.д.
и т.д.
Числовые диапазоны, используемые в настоящем описании, включают последовательные целые числа. Например, диапазон, выраженный как «от 0 до 4» включает 0, 1, 2, 3 и 4.
Согласно настоящему описанию термины «моноциклический, бициклический или трициклический арил, гетероарил, циклоалкил или гетероциклоалкил» означает, что любое кольцо в бициклической или трициклической структуре может независимо представлять собой арил, гетероарил, циклоалкил или гетероциклоалкил, распределенные в орто- или орто- и пери-конденсированной системе.
Согласно настоящему описанию термин «замещенный» означает, что указанная(-ый) группа или фрагмент содержит один или более подходящих заместителей, где заместители могут быть присоединены к указанной группе или фрагменту через одно или более положений. Например, арил, замещенный циклоалкилом, может означать, что циклоалкил связан с одним атомом арила посредством связи или конденсирован с арилом через два или более общих атомов.
Согласно настоящему описанию термин «незамещенный» означает, что указанная группа не содержит заместители.
Согласно настоящему описанию термин «возможно замещенный» означает, что указанная группа является незамещенной или замещена одним или более заместителями.
Если показан многофункциональный фрагмент, то место присоединения к ядру может быть обозначено линией. Например, (циклоалкилокси)алкил- относится к группе, в которой алкил является местом присоединения к ядру, тогда как циклоалкил присоединен к алкилу посредством оксигруппы. Если линия отсутствует, то подразумевается присоединение по любому положению.
Выражение «вспомогательный химиотерапевтический агент» в общем случае относится к агентам, которые излечивают, ослабляют, облегчают или снижают побочные эффекты химиотерапевтических агентов. Указанные агенты включают агенты, которые модифицируют рост и созревание клеток крови. Примеры вспомогательных химиотерапевтических агентов включают, но не ограничиваются ими, филграстим и эритропоэтин. Другие вспомогательные химиотерапевтические агенты включают агенты, которые подавляют тошноту, связанную с введение химиотерапевтических агентов, такие как ингибитор рецептора 5-HT3 (например, долансетрон, гранисетрон или ондансетрон) совместно с дексаметазоном или без него.
Термины «химиотерапевтический агент» и «антинеопластический агент» в общем случае относятся к агентам, которые лечат, предотвращают, подвергают лечению, излечивают, ослабляют, облегчают, изменяют, вылечивают, снижают, улучшают или воздействуют на злокачественные образования и их метастазы. Примеры указанных агентов (также известных как «антинеопластические агенты») включают, но не ограничиваются ими, преднизон, фторурацил (например, 5-фторурацил (5-FU)), анастрозол, бикалутамид, карбоплатин, цисплатин, хлорамбуцил, цисплатин, карбоплатин, доцетаксел, доксорубицин, флутамид, интерферон-альфа, летрозол, леупролид, мегестрол, митомицин, оксалиплатин, паклитаксел, пликамицин (Mithracin™), тамоксифен, тиотепа, топотекан, валрубицин, винбластин, винкристин и любую комбинацию каких-либо приведенных выше агентов. Указанные дополнительные агенты описаны далее.
Необходимо отметить, что в настоящем описании и прилагаемой формуле изобретения формы единственного числа включают ссылки на множество объектов, если по контексту явным образом не требуется иное.
При использовании в качестве терапевтических агентов ингибиторы FASN (FASN), описанные в настоящей заявке, можно вводить совместно с одним или более физиологически приемлемыми вспомогательными веществами. Физиологически приемлемый(-ое) носитель или вспомогательное вещество представляет собой состав, в который можно добавлять соединение для его растворения или упрощения введения иным образом.
Лекарственные формы согласно настоящему изобретению могут содержать смесь одного или более соединений согласно настоящему изобретению и могут включать дополнительные вещества, известные специалистам в данной области техники как фармацевтические вспомогательные вещества. В раствор для доставки агента можно включать стабилизирующие добавки. В случае некоторых лекарственных средств наличие таких добавок улучшает стабильность и распределение агента в растворе. Стабилизирующие добавки можно применять в концентрации в диапазоне от примерно 0,1 до 5% (масс./об.), предпочтительно примерно 0,5% (масс./об.). Подходящие, но не ограничивающие примеры стабилизирующих добавок включают аравийскую камедь, желатин, метилцеллюлозу, полиэтиленгликоль, карбоновые кислоты и их соли и полилизин. Предпочтительными стабилизирующими добавками являются аравийская камедь, желатин и метилцеллюлоза.
В качестве вспомогательных веществ можно применять подкисляющие агенты (уксусную кислоту, ледяную уксусную кислоту, лимонную кислоту, фумаровую кислоту, хлороводородную кислоту, разбавленную хлороводородную кислоту, яблочную кислоту, азотную кислоту, фосфорную кислоту, разбавленную фосфорную кислоту, серную кислоту, винную кислоту); газы-вытеснители для аэрозолей (бутан, дихлордифторметан, дихлортетрафторэтан, изобутан, пропан, трихлормонофторметан); вытеснители воздуха (диоксид углерода, азот); агенты для денатурации спирта (бензоат денатония, метилизобутилкетон, октацетат сахарозы); подщелачивающие агенты (концентрированный раствор аммиака, карбонат аммония, диэтаноламин, диизопропаноламин, гидроксид калия, бикарбонат натрия, борат натрия, карбонат натрия, гидроксид натрия, троламин); агенты, предотвращающие спекание (см. глиданты); агенты, предотвращающие вспенивание (диметикон, симетикон); противомикробные консерванты (хлорид бензалкония, раствор хлорида бензалкония, хлорид бензэтония, бензойную кислоту, бензиловый спирт, бутилпарабен, хлорид цетилпиридиния, хлорбутанол, хлоркрезол, крезол, дегидроуксусную кислоту, этилпарабен, метилпарабен, метилпарабен натрия, фенол, фенилэтиловый спирт, ацетат фенилртути, нитрат фенилртути, бензоат калия, сорбат калия, пропилпарабен, пропилпарабен натрия, бензоат натрия, дегидроацетат натрия, пропионат натрия, сорбиновую кислоту, тимеросал, тимол); антиоксиданты (аскорбиновую кислоту, аскорбилпальмитат, бутилированный гидроксианизол, бутилированный гидрокситолуол, гипофосфорную кислоту, монотиоглицерин, пропилгаллат, формальдегид-сульфоксилат натрия, метабисульфит натрия, тиосульфат натрия, диоксид серы, токоферол, вспомогательные токоферолы); буферные агенты (уксусную кислоту, карбонат аммония, фосфат аммония, борную кислоту, лимонную кислоту, молочную кислоту, фосфорную кислоту, цитрат калия, метафосфат калия, одноосновный фосфат калия, ацетат натрия, цитрат натрия, раствор лактата натрия, двухосновный фосфат натрия, одноосновный фосфат натрия); смазывающие агенты для капсул (см. смазывающие агенты для таблеток и капсул); хелатообразующие агенты (эдетат динатрия, этилендиаминтетрауксусную кислоту и ее соли, эдетовую кислоту); агенты, образующие покрытие (карбоксиметилцеллюлозу натрия, ацетат целлюлозы, ацетат-фталат целлюлозы, этилцеллюлозу, желатин, фармацевтическую глазурь, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, фталат гидроксипропилметилцеллюлозы, сополимер метакриловой кислоты, метилцеллюлозу, полиэтиленгликоль, поливинил-ацетат-фталат, шеллак, сахарозу, диоксид титана, воск карнауба, микрокристаллический воск, зеин); красители (карамельный, красный, желтый, черный или смеси, оксид железа (III)); комплексообразующие агенты (этилендиаминтетрауксусную кислоту и ее соли (ЭДТА), эдетовую кислоту, этаноламид гентизиновой кислоты, сульфат оксихинолина); поглотители влаги (хлорид кальция, сульфат кальция, диоксид кремния); эмульгаторы и/или агенты, увеличивающие растворимость (аравийскую камедь, холестерин, диэтаноламин (вспомогательный), глицерилмоностеарат, ланолиновые спирты, лецитин, моно- и ди-глицериды, моноэтаноламин (вспомогательный), олеиновую кислоту (вспомогательная), олеиловый спирт (стабилизатор), полоксамер, полиоксиэтилен-50-стеарат, полиоксил-35-касторовое масло, полиоксил-40-гидрированное касторовое масло, полиоксил-10-олеиловый простой эфир, полиоксил-20-цетостеариловый простой эфир, полиоксил-40-стеарат, полисорбат 20, полисорбат 40, полисорбат 60, полисорбат 80, диацетат пропиленгликоля, моностеарат пропиленгликоля, лаурилсульфат натрия, стеарат натрия, сорбитанмонолаурат, сорбитанмоноолеат, сорбитанмонопальмитат, сорбитанмоностеарат, стеариновую кислоту, троламин, эмульгирующий воск); фильтрующие добавки (порошковую целлюлозу, очищенную кремнистую землю); вкусоароматические добавки и ароматизаторы (анетол, бензальдегид, этилванилин, ментол, метилсалицилат, глутамат мононатрия, масло из цветков померанца, перечную мяту, масло перечной мяты, спиртовой раствор перечной мяты, розовое масло, концентрированную розовую воду, тимол, настойку тилуанского бальзама, ваниль, настойку ванили, ванилин); глиданты и/или агенты, предотвращающие спекание (силикат кальция, силикат магния, коллоидный диоксид кремния, тальк), увлажнители (глицерин, гексиленгликоль, пропиленгликоль, сорбит); пластификаторы (касторовое масло, диацетилированные моноглицериды, диэтилфталат, глицерин, моно- и ди-ацетилированные моноглицериды, полиэтиленгликоль, пропиленгликоль, триацетин, триэтилцитрат); полимеры (например, ацетат целлюлозы, алкилцеллюлозы, гидроксиалкилцеллюлозы, акриловые полимеры и сополимеры); растворители (ацетон, спирт, разбавленный спирт, гидрат амилена, бензилбензоат, бутиловый спирт, тетрахлорид углерода, хлороформ, кукурузное масло, хлопковое масло, этилацетат, глицерин, гексиленгликоль, изопропиловый спирт, метиловый спирт, метиленхлорид, метилизобутилкетон, минеральное масло, арахисовое масло, полиэтиленгликоль, пропиленкарбонат, пропиленгликоль, кунжутное масло, воду для инъекций, стерильную воду для инъекций, стерильную воду для промывания, очищенную воду); сорбенты (порошковую целлюлозу, активированный уголь, очищенную кремнистую землю); сорбенты диоксида углерода (бариевую известь, натронную известь); загустители (гидрированное касторовое масло, цетостеариловый спирт, цетиловый спирт, воск цетиловых эфиров, твердый жир, парафин, вспомогательный полиэтилен, стеариловый спирт, эмульгирующий воск, белый воск, желтый воск); суспендирующие агенты и/или агенты, повышающие вязкость (аравийскую камедь, агар, альгиновую кислоту, моностеарат алюминия, бентонит, очищенный бентонит, густую суспензию бентонита, карбомер 934р, карбоксиметилцеллюлозу кальция, карбоксиметилцеллюлозу натрия, карбоксиметилцеллюлозу натрия 12, каррагинан, микрокристаллическую целлюлозу и карбоксиметилцеллюлозу натрия, декстрин, желатин, гуаровую камедь, гидроксиэтилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, алюмосиликат магния, метилцеллюлозу, пектин, полиэтиленоксид, поливиниловый спирт, повидон, альгинат пропиленгликоля, диоксид кремния, коллоидный диоксид кремния, альгинат натрия, трагакантовую камедь, ксантановую камедь); подсластители (аспартам, декстраты, декстрозу, вспомогательную декстрозу, фруктозу, маннит, сахарин, сахарин кальция, сахарин натрия, сорбит, раствор сорбита, сахарозу, прессованный сахар, сахарную пудру, сироп); связывающие вещества для таблеток (аравийскую камедь, альгиновую кислоту, карбоксиметилцеллюлозу натрия, микрокристаллическую целлюлозу, декстрин, этилцеллюлозу, желатин, жидкую глюкозу, гуаровую камедь, гидроксипропилметилцеллюлозу, метилцеллюлозу, полиэтиленоксид, повидон, прежелатинизированный крахмал, сироп); разбавители для таблеток и/или капсул (карбонат кальция, двухосновный фосфат кальция, трехосновный фосфат кальция, сульфат кальция, микрокристаллическую целлюлозу, порошковую целлюлозу, декстраты, декстрин, вспомогательную декстрозу, фруктозу, каолин, лактозу, маннит, сорбит, крахмал, прежелатинизированный крахмал, сахарозу, прессованный сахар, сахарную пудру), разрыхлители для таблеток (альгиновую кислоту, микрокристаллическую целлюлозу, кроскармеллозу натрия, кросповидон, полакрилин калия, крахмал гликолят натрия, крахмал, прежелатинизированный крахмал); смазывающие агенты для таблеток и/или капсул (стеарат кальция, глицерилбегенат, стеарат магния, легкое минеральное масло, полиэтиленгликоль, стеарилфумарат натрия, стеариновую кислоту, очищенную стеариновую кислоту, тальк, гидрированное растительное масло, стеарат цинка); тонический агент (декстрозу, глицерин, маннит, хлорид калия, хлорид натрия); носитель: ароматизированный и/или с подсластителем (ароматический эликсир, смешанный эликсир бензальдегида, изоспиртовой эликсир, воду перечной мяты, раствор сорбита, сироп, сироп толуанского бальзама); носитель: масляный (миндальное масло, кукурузное масло, хлопковое масло, этилолеат, изопропилмиристат, изопропилпальмитат, минеральное масло, легкое минеральное масло, миристиловый спирт, октилдодеканол, оливковое масло, арахисовое масло, персидское масло, кунжутное масло, соевое масло, сквалан); носитель: твердый носитель (сахарные сферы); носитель: стерильный (бактериостатическую воду для инъекций, бактериостатический хлорид натрия для инъекций); агент, увеличивающий вязкость (см. суспендирующий агент); водоотталкивающий агент (циклометикон, диметикон, симетикон); и увлажняющий агент и/или агент, увеличивающий растворимость (хлорид бензалкония, хлорид бензэтония, хлорид цетилпиридиния, докузат натрия, ноноксинол 9, ноноксинол 10, октоксинол 9, полоксамер, полиоксил-35-касторовое масло, полиоксил 40, гидрированное касторовое масло, полиоксил-50-стеарат, полиоксил-10-олеиловый простой эфир, полиоксил 20, цетостеариловый простой эфир, полиоксил-40-стеарат, полисорбат 20, полисорбат 40, полисорбат 60, полисорбат 80, лаурилсульфат натрия, сорбитана монолаурат, сорбитана моноолеат, сорбитана монопальмитат, сорбитана моностеарат, тилоксапол). Указанный перечень не является исключающим, но включает только типовые классы вспомогательных веществ и конкретных вспомогательных веществ, которые можно применять в лекарственных формах согласно настоящему изобретению.
Соединения формулы I, I-A, I-B, I-C и I-D могут образовывать соли, которые также включены в объем настоящего изобретения. Следует понимать, что указание на соединение, имеющее формулу, описанную в настоящей заявке, включает его соли, если не указано иное. Термин «соль(-и)», используемый в настоящем описании, обозначает соли кислот, полученные из неорганических и/или органических кислот, а также соли оснований, полученные из неорганических и/или органических оснований. Кроме того, если соединение, имеющее формулу, содержит основный фрагмент, такой как без ограничений пиридин или имидазол, и кислотный фрагмент, такой как без ограничений карбоновая кислота, то могут образовываться цвиттер-ионы («внутренние соли»), которые включены в объем термина «соль(-и)», используемого в настоящем описании. Фармацевтически приемлемые (т.е. нетоксичные, физиологически приемлемые) соли являются предпочтительными, в то же время можно применять и другие соли. Соли соединений, имеющих формулу, можно получать, например, путем взаимодействия соединения, имеющего формулу, с количеством кислоты или основания, таким как эквивалентное количество, в среде, такой как среда, в которой происходит осаждение соли, или водная среда, и последующей лиофилизации.
Типовые соли присоединения кислот включают ацетаты, аскорбаты, бензоаты, бензолсульфонаты, бисульфаты, бораты, бутираты, цитраты, камфораты, камфорсульфонаты, фумараты, гидрохлориды, гидробромиды, гидроиодиды, лактаты, малеаты, метансульфонаты, нафталинсульфонаты, нитраты, оксалаты, фосфаты, пропионаты, салицилаты, сукцинаты, сульфаты, тартраты, тиоцианаты, толуолсульфонаты (также известные как тозилаты) и т.д. Кроме того, обсуждение кислот, которые в общем случае рассматривают как подходящие для получения фармацевтически приемлемых солей из основных фармацевтических соединений, приведено, например, в P. Stahl et al, Camille G. (ред.) Handbook of Pharmaceutical Salts. Properties, Selection and Use. (2002) Zurich: Wiley-VCH; S. Berge et al., Journal of Pharmaceutical Sciences (1977) 66(1) 1-19; P. Gould, International J. of Pharmaceutics (1986) 33 201-217; Anderson et al., The Practice of Medicinal Chemistry (1996), Academic Press, New York; и The Orange Book (Управление по контролю качества пищевых продуктов и лекарственных средств, Washington, D.C., электронный сайт). Содержание указанных работ включено в настоящую заявку посредством ссылок.
Типовые основные соли включают соли аммония, соли щелочных металлов, такие как соли натрия, лития и калия, соли щелочноземельных металлов, такие как соли кальция и магния, соли органических оснований (например, органических аминов), таких как дициклогексиламины, трет-бутиламины, и соли аминокислот, таких как аргинин, лизин и т.д. Основные азотсодержащие группы можно превращать в четвертичные с использованием агентов, таких как низшие алкилгалогениды (например, метил-, этил- и бутил-хлориды, -бромиды и -йодиды), диалкилсульфаты (например, диметил-, диэтил- и дибутилсульфаты), длинноцепочечные галогениды (например, децил-, лаурил- и стеарил-хлориды, -бромиды и -йодиды), аралкилгалогениды (например, бензил- и фенэтилбромиды) и т.д.
Предполагается, что все указанные соли присоединения кислот и оснований представляют собой фармацевтически приемлемые соли, включенные в объем настоящего изобретения, и для задач настоящего изобретения все соли присоединения кислот и оснований рассматривают как эквивалентные свободным формам соответствующих соединений.
Соединения, имеющие различные формулы, и их соли, сольваты, сложные эфиры и пролекарства могут существовать в таутомерной форме (например, амида или простого иминоэфира). Все указанные таутомерные формы рассматривают в настоящем описании как часть настоящего изобретения.
Соединения, имеющие различные формулы, могут содержать асимметрические центры и, таким образом, существуют в различных стереоизомерных формах. Предполагается, что все стереоизомерные формы соединений, имеющих различные формулы, а также их смеси, включая рацемические смеси, составляют часть настоящего изобретения. Кроме того, настоящее изобретение охватывает все геометрические и позиционные изомеры. Например, если соединение, имеющее различные формулы, содержит двойную связь или конденсированное кольцо, то в объем настоящего изобретения включены цис- и транс-формы, а также их смеси. Каждое соединение, предложенное в настоящем описании, включает все энантиомеры, удовлетворяющие общей структуре соединения. Соединения могут иметь рацемическую или энантиомерно-чистую форму или любую другую форму, если рассматривать стереохимию. В результатах исследования могут быть отражены данные, собранные для рацемической формы, энантиомерно-чистой формы или любой другой формы, если рассматривать стереохимию.
Диастереомерные смеси можно разделять на отдельные диастереомеры на основании физических и химических различий при помощи способов, хорошо известных специалистам в данной области техники, таких как, например, хроматография и/или фракционная кристаллизация. Энантиомеры можно разделять путем превращения энантиомерной смеси в диастереомерную смесь путем взаимодействия с соответствующим оптически активным соединением (например, хиральным вспомогательным веществом, таким как хиральный спирт или хлорангидрид кислоты Мошера), разделения диастереомеров и превращения (например, гидролиза) отдельных диастереомеров в соответствующие чистые энантиомеры. Также, некоторые соединения, имеющие различные формулы, могут представлять собой атропизомеры (например, замещенные биарилы), которые рассматривают как часть настоящего изобретения. Энантиомеры также можно разделять с использованием колонки для хиральной ВЭЖХ.
Соединения, имеющие различные формулы, также могут существовать в различных таутомерных формах, и все указанные формы включены в объем настоящего изобретения. Также, например, кето-енольные и имин-енаминовые формы соединений включены в объем изобретения.
Все стереоизомеры (например, геометрические изомеры, оптические изомеры и т.д.) соединений согласно настоящему изобретению (включая соли, сольваты, сложные эфиры и пролекарства соединений, а также соли, сольваты и сложные эфиры пролекарств), такие как стереоизомеры, которые могут существовать благодаря наличию асимметрических атомов углерода в различных заместителях, включая энантиомерные формы (которые могут существовать даже в отсутствие асимметрических атомов углерода), ротамерные формы, атропизомеры и диастереомерные формы, включены в объем настоящего изобретения наряду с позиционными изомерами (такими как, например, 4-пиридил и 3-пиридил). (Например, если соединение, имеющее различные формулы, содержит двойную связь или конденсированное кольцо, то в объем настоящего изобретения включены цис- и транс-формы, а также их смеси. Также, например, в объем изобретения включены все кето-енольные и имин-енаминовые формы соединений.) Отдельные стереоизомеры соединений согласно настоящему изобретению могут, например, по существу не содержать другие изомеры, или они могут быть смешаны, например, в виде рацематов или совместно со всеми другими или отдельными другими стереоизомерами. Хиральные центры согласно настоящему изобретению могут иметь S- или R-конфигурацию, определенную в соответствии с рекомендациями IUPAC от 1974 года. Предполагается, что использование терминов «соль», «сольват», «сложный эфир», «пролекарство» и т.д. в равной степени относится к соли, сольвату, сложному эфиру или пролекарству энантиомеров, стереоизомеров, ротамеров, таутомеров, позиционных изомеров, рацематов или пролекарств соединений согласно настоящему изобретению.
Настоящее изобретение также охватывает изотопно-меченные соединения согласно настоящему изобретению, идентичные тем, что указаны в настоящем описании, за исключением того, что один или более атомов заменены на атом, имеющий атомную массу или массовое число, отличающееся от атомной массы или массового числа атома, распространенного в природе. Примеры изотопов, которые могут содержаться в соединениях согласно настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2Н, 3Н, 13С, 14С, 15N, 18O, 17O, 31P, 32P, 35S, 18F и 36Cl, соответственно.
Определенные изотопно-меченные соединения, имеющие различные формулы, (например, меченные 3Н и 14С) подходят для исследований распределения соединения и/или субстрата в тканях. Изотопы трития (т.е. 3Н) и углерода-14 (т.е. 14С) особенно предпочтительны благодаря простоте получения и обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий (т.е. 2Н), может обеспечивать определенные терапевтические преимущества, определяемые повышенной метаболической стабильностью (например, увеличение периода полувыведения in vivo или пониженную требуемую дозировку), и тем самым может быть предпочтительным в определенных случаях. Изотопно-меченные соединения, имеющие различные формулы, в общем случае можно получать при помощи способов, аналогичных тем, что предложены далее на схемах и/или в примерах, путем замещения реагента, не меченного изотопами, на соответствующий изотопно-меченный реагент.
Предполагается, что полиморфные формы соединений, имеющие различные формулы, и солей, сольватов, сложных эфиров и пролекарств соединений, имеющих различный формулы, включены в объем настоящего изобретения.
Преимущества настоящего изобретения включают пероральное введение оптимального количества ингибитора FASN.
Преимущества настоящего изобретения включают внутривенное введение оптимального количества ингибитора FASN.
Преимущества настоящего изобретения включают интраперитонеальное введение оптимального количества ингибитора FASN.
Преимущества настоящего изобретения включают внутристеночное введение оптимального количества ингибитора FASN.
Преимущества настоящего изобретения включают внутримышечное введение оптимального количества ингибитора FASN.
Преимущества настоящего изобретения включают подкожное введение оптимального количества ингибитора FASN.
Преимущества настоящего изобретения включают внутриопухолевое введение оптимального количества ингибитора FASN.
Преимущества настоящего изобретения включают интратекальное введение оптимального количества ингибитора FASN.
Преимущества настоящего изобретения включают субдуральное введение оптимального количества ингибитора FASN.
Преимущества настоящего изобретения включают периорбитальное введение оптимального количества ингибитора FASN.
С учетом указанных результатов настоящее изобретение имеет важное значение для разработки новых стратегий лечения пациентов, страдающих от рака, включая лейкозы и солидные опухоли, воспалительных заболеваний, вирусных инфекций, остеопороза, атеросклероза; синдрома раздраженного или воспаленного кишечника; диабета, ожирения и других состояний, описанных в настоящей заявке или известных специалистам в данной области техники.
Описание предпочтительных вариантов реализации
Один из аспектов настоящего изобретения относится к соединениям, предложенным в настоящем описании.
Один из аспектов настоящего изобретения относится к соединениям, которые представляют собой или могут являться ингибиторами FASN.
Один из аспектов настоящего изобретения относится к применению ингибитора FASN для получения лекарственного средства, используемого для лечения, предотвращения, подавления или уничтожения опухолей.
Один из аспектов настоящего изобретения относится к применению ингибитора FASN для получения лекарственного средства, используемого для лечения, предотвращения, подавления или устранения рака.
Один из аспектов настоящего изобретения относится к применению ингибитора FASN для получения лекарственного средства, используемого для лечения, предотвращения, подавления или устранения рака, где раковое заболевание выбрано из лейкоза, лимфомы, рака яичников, рака груди, рака матки, рака толстой кишки, рака шейки матки, рака легкого, рака простаты, рака кожи, рака ЦНС, рака мочевого пузыря, рака поджелудочной железы и болезни Ходжкина.
Один из аспектов настоящего изобретения относится к применению ингибитора FASN для получения лекарственного средства, используемого для лечения, предотвращения, подавления или устранения диабета.
Один из аспектов настоящего изобретения относится к применению ингибитора FASN для получения лекарственного средства, используемого для лечения, предотвращения, подавления или устранения вирусных инфекционных заболеваний.
Один из аспектов настоящего изобретения относится к применению ингибитора FASN для получения лекарственного средства, используемого для лечения, предотвращения, подавления или устранения избыточного веса или ожирения.
Один из аспектов настоящего изобретения относится к применению ингибитора FASN для получения лекарственного средства, используемого для лечения, предотвращения, подавления или устранения лейомиом матки.
В настоящем изобретении также описаны один или более способов синтеза соединений согласно настоящему изобретению.
В изобретении также описаны одно или более применений соединений согласно настоящему изобретению.
В изобретении также описаны одно или более применений соединений согласно настоящему изобретению совместно со вспомогательным агентом, таких как применение совместно с ФНО, ГКСФ или другими химиотерапевтическими агентами.
В изобретении также описаны одно или более применений фармацевтических композиций согласно настоящему изобретению.
Один из аспектов настоящего изобретения относится к применению ингибитора FASN для получения лекарственного средства, используемого для лечения воспалительных заболеваний.
Один из аспектов настоящего изобретения относится к применению ингибитора FASN для получения лекарственного средства, используемого для лечения воспалительных заболеваний, таких как синдром раздраженного кишечника или воспалительная болезнь кишечника.
Один из аспектов настоящего изобретения относится к применению ингибитора FASN для получения лекарственного средства, используемого для лечения заболевания кости, такого как остеопороз.
Один из аспектов настоящего изобретения относится к применению ингибитора FASN для получения лекарственного средства, используемого для лечения заболевания сердечнососудистой системы, такого как атеросклероз.
Один из аспектов настоящего изобретения относится к применению ингибитора FASN для получения лекарственного средства, используемого для лечения заболевания или состояния, вызванного повышенным уровнем FASN.
Указанное заболевание или состояние представляет собой одно или более заболеваний или состояний, выбранных из группы, состоящей из рака яичников, рака груди, рака матки, рака толстой кишки, рака шейки матки, рака легкого, рака простаты, рака кожи, рака мочевого пузыря, рака поджелудочной железы, лейкоза, лимфомы, болезни Ходжкина, вирусных инфекций, вируса иммунодефицита человека, вируса гепатита, герпесвируса, простого герпеса, воспалительных нарушений, синдрома раздраженного кишечника, воспалительной болезни кишечника, ревматоидного артрита, астмы, хронической обструктивной болезни легких, остеоартрита, остеопороза, дерматита, атопического дерматита, псориаза, системной красной волчанки, рассеянного склероза, псориатического артрита, анкилозирующего спондилита, болезни трансплантат-против-хозяина, нарушения мозгового кровообращения, атеросклероза, диабета, гломерулонефрита, метаболического синдрома, немелкоклеточного рака легкого, мелкоклеточного рака легкого, множественной миеломы, лимфом, плоскоклеточных раковых заболеваний, рака почки, рака мочеиспускательного канала и мочевого пузыря, раковых заболеваний области головы и шеи, раковых заболеваний мозга и центральной нервной системы (ЦНС).
Соединения согласно настоящему изобретению могут подходить для лечения пролиферативных заболеваний, таких как рак, аутоиммунные заболевания, вирусные заболевания, грибковые заболевания, неврологические/нейродегенеративные нарушения, артрита, воспаления, антипролиферативного заболевания (например, глазной ретинопатии), заболевания нейронов, облысения и сердечнососудистого заболевания. Множество указанных заболеваний и нарушений перечислены в патенте США №6413974, содержание которого включено в настоящую заявку посредством ссылки.
Более конкретно, соединения могут подходить для лечения различных раковых заболеваний, включая (но не ограничиваясь ими) следующие: карциному, включая карциному мочевого пузыря, груди, толстой кишки, почки, печени, легкого, включая мелкоклеточный рак легкого, немелкоклеточный рак легкого, области головы и шеи, пищевода, желчного пузыря, яичников, поджелудочной железы, желудка, шейки матки, щитовидной железы, простаты и кожи, включая плоскоклеточную карциному; гематопоэтические опухоли лимфоидного происхождения, включая лейкоз, острый лимфоцитарный лейкоз, острый лимфобластный лейкоз, В-клеточную лимфому, Т-клеточную лимфому, лимфому Ходжкина, неходжкинскую лимфому, волосатоклеточную лимфому, лимфому из клеток мантии, миелому и лимфому Беркитта; гематопоэтические опухоли миелоидного происхождения, включая острый и хронический миелогенный лейкоз, миелодиспластический синдром и промиелоцитарный лейкоз; опухоли мезенхимального происхождения, включая фибросаркому и рабдомиосаркому; опухоли центральной и периферической нервной системы, включая астроцитому, нейробластому, глиому и шванномы; и другие опухоли, включая меланому, семиному, тератокарциному, остеосаркому, пигментную ксеродерму, кератоакантому, фолликулярный рак щитовидной железы и саркому Капоши.
Соединения согласно настоящему изобретению могут инициировать или подавлять апоптоз.
Соединения согласно настоящему изобретению также могут подходить для хемопревенции рака. Хемопревенция определена как подавление развития инвазивного рака путем блокировки начального мутагенного явления или блокировки прогрессирования уже пораженных предшественников злокачественных клеток или подавления обострения опухоли.
Дополнительный аспект изобретения представляет собой способ ингибирования FASN у животного, включающий введение указанному животному, нуждающемуся в этом, фармацевтически приемлемого количества соединения согласно настоящему изобретению.
Дополнительный аспект изобретения представляет собой фармацевтический состав, содержащий соединение согласно настоящему изобретению.
Другой вариант реализации изобретения включает фармацевтический состав согласно настоящему изобретению, где фармацевтический состав при введении человеку обеспечивает снижение опухолевой нагрузки.
Другой вариант реализации изобретения представляет собой фармацевтический состав, дополнительно содержащий один или более антинеопластических агентов, химиотерапевтических агентов или вспомогательных химиотерапевтических агентов.
Фармацевтические составы согласно настоящему изобретению могут дополнительно содержать терапевтически эффективное количество вспомогательного химиотерапевтического агента.
Вспомогательный химиотерапевтический агент может представлять собой агент, модифицирующий рост и созревание клеток крови. Неограничивающими примерами вспомогательных химиотерапевтических агентов являются филграстим, пегфилграстим и эритропоэтин.
Изобретение также относится к способу лечения или предотвращения нарушения, связанного с избыточной скоростью роста клеток, у млекопитающего, включающему введение млекопитающему эффективного количества фармацевтического состава согласно настоящему изобретению. Неограничивающие примеры нарушений включают рак или метастазы злокачественных опухолей.
Другой аспект изобретения представляет собой способ подавления скорости роста и деления опухолевых клеток у млекопитающего, страдающего от рака или другого нарушения, связанного с нарушенным делением клеток, включающий введение млекопитающему эффективного количества фармацевтического состава согласно настоящему изобретению.
Другой вариант реализации изобретения представляет собой способ лечения боли в кости, вызванной избыточным ростом опухоли или метастазами в кости, у млекопитающего, нуждающегося в этом, включающий введение млекопитающему эффективного количества фармацевтического состава согласно настоящему изобретению.
Другой вариант реализации изобретения представляет собой способ введения соединения, содержащего ингибитор FASN, млекопитающему, нуждающемуся в этом, включающий введение млекопитающему фармацевтического состава согласно настоящему изобретению. В одном из вариантов реализации млекопитающее представляет собой человека.
Дополнительный вариант реализации изобретения представляет собой способ получения фармацевтического состава, включающий смешение по меньшей мере одного фармацевтически приемлемого соединения согласно настоящему изобретению и необязательно одного или более фармацевтически приемлемых вспомогательных веществ или добавок.
Изобретение также относится к способам синтеза соединений согласно настоящему изобретению.
Соединения согласно настоящему изобретению
Настоящее изобретение относится к конкретным молекулам и их фармацевтически приемлемым солям или изомерам. Изобретение дополнительно относится к молекулам, которые подходят для ингибирования фермента синтазы жирных кислот (FASN), и к их фармацевтически приемлемым солям или изомерам.
Изобретение относится к соединениям, описанным в настоящей заявке, и к их фармацевтически приемлемым солям или изомерам и к фармацевтическим композициям, содержащим одно или более соединений, описанных в настоящей заявке, и их фармацевтически приемлемые соли или изомеры. В одном из аспектов настоящего изобретения предложены соединения, композиции и наборы для ингибирования FASN, содержащие соединение формулы I:
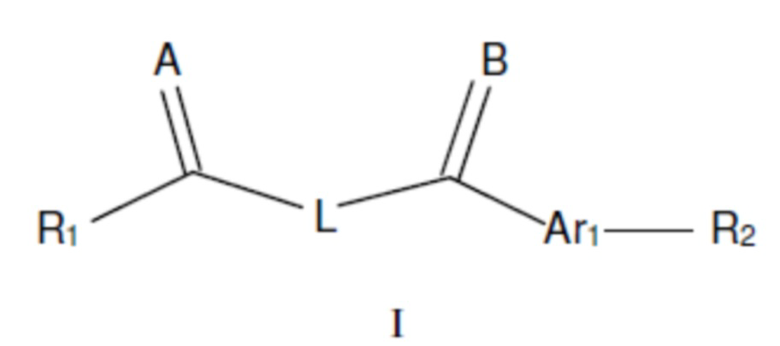
где
R1 представляет собой С1-С3 гидроксил-алкил, который является незамещенным или замещен -СН3 или -CHzF3-z, 5-членный циклоалкил, который является незамещенным или замещен заместителями, выбранными из группы, состоящей из дейтерия, -Rp, -ORp, -NHRp и -NRpRp1, или 3- или 4-членный циклоалкил или гетероциклоалкил, где (i) гетероатом, являющийся членом кольца 3- или 4-членного гетероциклоалкила, независимо выбран из O, S или N, и (ii) каждый указанный 3- или 4-членный циклоалкил или гетероциклоалкил является незамещенным или необязательно замещен заместителями, выбранными из группы, состоящей из дейтерия, -Ra, -ORa, -NHRa и -NRaRa1;
L представляет собой 5-10-членный моноциклический или бициклический алкил или гетероалкил, где (i) гетероатомы, являющиеся членами кольца 5-10-членного моноциклического или бициклического гетероалкила, независимо выбраны из O, S или N, и (ii) каждый 5-10-членный моноциклический или бициклический алкил или гетероалкил является незамещенным или необязательно замещен заместителями, выбранными из группы, состоящей из дейтерия и -Rb;
А и В независимо представляют собой О или S;
Ar1 представляет собой 4-10-членный моноциклический или бициклический арил, гетероарил или гетероциклоалкил, где (i) указанный 4-10-членный моноциклический или бициклический гетероарил или гетероциклоалкил содержит 1, 2, 3 или 4 гетероатома, независимо выбранные из N, S или О, и (ii) каждый указанный 4-10-членный моноциклический или бициклический арил, гетероарил или гетероциклоалкил является незамещенным или необязательно независимо замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, алкила, -CHzF3-z, циано, гидроксила, гидроксиалкила, амино, аминоалкила-, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -OCHzF3-z, -алкила, -алкенила, -алкинила, -алкокси или (алкоксиалкил)амино-, -N(Rc)-C(O)-алкила, -N(Rc)-C(O)-арила, -циклоалкила, -гетероциклоалкила, -арила и -гетероарила, при условии, что никакие два расположенные по соседству гетероатома одновременно не являются S или О;
R2 представляет собой Н или 4-15-членный моноциклический, бициклический или трициклический арил, гетероарил, циклоалкил или гетероциклоалкил, где (i) 4-15-членный моноциклический, бициклический или трициклический гетероарил или гетероциклоалкил содержит 1, 2, 3, 4, 5, 6, 7 или 8 гетероатомов, независимо выбранных из N, S или О, и (ii) каждый указанный арил, гетероарил, циклоалкил и гетероциклоалкил является незамещенным или необязательно замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, циано, гидроксила, гидроксил-алкила-, гидроксилциклоалкила-, гидроксил-гетероциклоалкила-, гидроксил-арила-, гидроксил-гетероарила-, амино, аминоалкила, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -CHzF3-z, -OCHzF3-z, -алкила, алкокси-, -алкенила, -алкинила, арилокси-, (алкоксиалкил)амино-, -циклоалкила, -гетероциклоалкила, (гетероциклоалкил)алкила-, -арила, -гетероарила, -O(алкил), -O(циклоалкил), -O(гетероциклоалкил), -O(арил), -O(гетероарил), ONH2, -C(O)NH(алкил), -C(O)N(арил)2, -C(O)NH(циклоалкил), -NH(CO)циклоалкила, -NH(SO2), -NH(SO2)алкила, -NH(SO2)арила, -NH(SO2)гетероарила, -N(SO2)циклоалкила, -C(O)N(алкил)2, (арил)алкила-, -гетероарила, (гетероарил)алкила-, -S(O)2-алкила, -S(O)2-арила, -S(O)2-циклоалкила, -C(O)N(алкил)2, -C(O)алкила, -NH-C(O)-алкила, -NH-C(O)-циклоалкила, NH-C(O)-гетероциклоалкила, NH-C(O)-гетероциклоалкил-Rd, -NH-C(O)-Rd-(O)алкила, -NH-C(O)-арила, -NH-C(O)-NH-алкила, NH-C(O)-NH-циклоалкила, NH2(CO)циклоалкила-, NH-C(O)-NH-арила, -NH-C(O)-O-алкила, NH-C(O)-NH-циклоалкила, -NH-C(O)-O-циклоалкила, -NH(Rd)-C(O)-алкила, -NH(Rd)-C(O)-арила, -NH(Rd)-S(O2)циклоалкила, -S(O2)NH2, -S(O2)NH(алкил), -S(O2)N(Rd)циклоалкила, -S(O2)N(алкил)2, -C(O)N(H)(алкил), -C(O)N(Rd)(циклоалкил), метилендиокси, -CHzF3-z, -OCHzF3-z и -алкокси;
Rp и Rp1 независимо представляют собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Ra и Ra1 независимо представляют собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Rb представляет собой Н, галоген, С1-С4 алкил, С1-С3 гидроксил-алкил или С3-С4 циклоалкил;
Rc представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Rd представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
и z равен 0, 1 или 2;
и его фармацевтически приемлемые соли, сольваты, сложные эфиры, пролекарства и изомеры.
В другом варианте реализации соединение формулы I представлено соединением формулы I-A:

( I - A)
где:
R1 представляет собой С1-С3 гидроксил-алкил, который является незамещенным или замещен -СН3 или -CHzF3-z, 5-членный циклоалкил, который является незамещенным или замещен заместителями, выбранными из группы, состоящей из дейтерия, -Rp, -ORp, -NHRp и -NRpRp1, или 3- или 4-членный циклоалкил или гетероциклоалкил, где (i) гетероатом, являющийся членом кольца 3- или 4-членного гетероциклоалкила, независимо выбран из O, S или N, и (ii) каждый указанный 3- или 4-членный циклоалкил или гетероциклоалкил является незамещенным или необязательно замещен заместителями, выбранными из группы, состоящей из дейтерия, -Ra, -ORa, -NHRa и -NRaRa1;
Ar1 представляет собой 4-10-членный моноциклический или бициклический арил, гетероарил или гетероциклоалкил, где (i) указанный 4-10-членный моноциклический или бициклический гетероарил или гетероциклоалкил содержит 1, 2, 3 или 4 гетероатома, независимо выбранные из N, S или О, и (ii) каждый указанный 4-10-членный моноциклический или бициклический арил, гетероарил или гетероциклоалкил является незамещенным или необязательно независимо замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, алкила, -CHzF3-z, циано, гидроксила, гидроксиалкила, амино, аминоалкила-, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -OCHzF3-z, -алкила, -алкенила, -алкинила, -алкокси или (алкоксиалкил)амино-, -N(Rc)-C(O)-алкила, -N(Rc)-C(O)-арила, -циклоалкила, -гетероциклоалкила, -арила и -гетероарила, при условии, что никакие два расположенные по соседству гетероатома одновременно не являются S или О;
R2 представляет собой Н или 4-15-членный моноциклический, бициклический или трициклический арил, гетероарил, циклоалкил или гетероциклоалкил, где (i) 4-15-членный моноциклический, бициклический или трициклический гетероарил или гетероциклоалкил содержит 1, 2, 3, 4, 5, 6, 7 или 8 гетероатомов, независимо выбранных из N, S или О, и (ii) каждый указанный арил, гетероарил, циклоалкил и гетероциклоалкил является незамещенным или необязательно замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, циано, гидроксила, гидроксил-алкила-, гидроксилциклоалкила-, гидроксил-гетероциклоалкила-, гидроксил-арила-, гидроксил-гетероарила-, амино, аминоалкила, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -CHzF3-z, -OCHzF3-z, -алкила, алкокси-, -алкенила, -алкинила, арилокси-, (алкоксиалкил)амино-, -циклоалкила, -гетероциклоалкила, (гетероциклоалкил)алкила-, -арила, -гетероарила, -O(алкил), -O(циклоалкил), -O(гетероциклоалкил), -O(арил), -O(гетероарил), ONH2, -C(O)NH(алкил), -C(O)N(арил)2, -C(O)NH(циклоалкил), -NH(CO)циклоалкила, -NH(SO2), -NH(SO2)алкила, -NH(SO2)арила, -NH(SO2)гетероарила, -N(SO2)циклоалкила, -C(O)N(алкил)2, (арил)алкила-, -гетероарила, (гетероарил)алкила-, -S(O)2-алкила, -S(O)2-арила, -S(O)2-циклоалкила, -C(O)N(алкил)2, -C(O)алкила, -NH-C(O)-алкила, -NH-C(O)-циклоалкила, NH-C(O)-гетероциклоалкила, NH-C(O)-гетероциклоалкил-Rd, -NH-C(O)-Rd-(O)алкила, -NH-C(O)-арила, -NH-C(O)-NH-алкила, NH-C(O)-NH-циклоалкила, NH2(CO)циклоалкила-, NH-C(O)-NH-арила, -NH-C(O)-O-алкила, NH-C(O)-NH-циклоалкила, -NH-C(O)-O-циклоалкила, -NH(Rd)-C(O)-алкила, -NH(Rd)-C(O)-арила, -NH(Rd)-S(O2)циклоалкила, -S(O2)NH2, -S(O2)NH(алкил), -S(O2)N(Rd)циклоалкила, -S(O2)N(алкил)2, -C(O)N(H)(алкил), -C(O)N(Rd)(циклоалкил), метилендиокси, -CHzF3-z, -OCHzF3-z и -алкокси;
Rp и Rp1 независимо представляют собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Ra и Ra1 независимо представляют собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Rc представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Rd представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
и z равен 0, 1 или 2;
и его фармацевтически приемлемыми солями, сольватами, сложными эфирами, пролекарствами и изомерами.
В другом варианте реализации соединение формулы I представлено соединением формулы I-B:
(I-B)
где:
Ar1 представляет собой 4-10-членный моноциклический или бициклический арил, гетероарил или гетероциклоалкил, где (i) указанный 4-10-членный моноциклический или бициклический гетероарил или гетероциклоалкил содержит 1, 2, 3 или 4 гетероатома, независимо выбранные из N, S или О, и (ii) каждый указанный 4-10-членный моноциклический или бициклический арил, гетероарил или гетероциклоалкил является незамещенным или необязательно независимо замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, алкила, -CHzF3-z, циано, гидроксила, гидроксиалкила, амино, аминоалкила-, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -OCHzF3-z, -алкила, -алкенила, -алкинила, -алкокси или (алкоксиалкил)амино-, -N(Rc)-C(O)-алкила, -N(Rc)-C(O)-арила, -циклоалкила, -гетероциклоалкила, -арила и -гетероарила, при условии, что никакие два расположенные по соседству гетероатома одновременно не являются S или О;
R2 представляет собой Н или 4-15-членный моноциклический, бициклический или трициклический арил, гетероарил, циклоалкил или гетероциклоалкил, где (i) 4-15-членный моноциклический, бициклический или трициклический гетероарил или гетероциклоалкил содержит 1, 2, 3, 4, 5, 6, 7 или 8 гетероатомов, независимо выбранных из N, S или О, и (ii) каждый указанный арил, гетероарил, циклоалкил и гетероциклоалкил является незамещенным или необязательно замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, циано, гидроксила, гидроксил-алкила-, гидроксилциклоалкила-, гидроксил-гетероциклоалкила-, гидроксил-арила-, гидроксил-гетероарила-, амино, аминоалкила, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -CHzF3-z, -OCHzF3-z, -алкила, алкокси-, -алкенила, -алкинила, арилокси-, (алкоксиалкил)амино-, -циклоалкила, -гетероциклоалкила, (гетероциклоалкил)алкила-, -арила, -гетероарила, -O(алкил), -O(циклоалкил), -O(гетероциклоалкил), -O(арил), -O(гетероарил), ONH2, -C(O)NH(алкил), -C(O)N(арил)2, -C(O)NH(циклоалкил), -NH(CO)циклоалкила, -NH(SO2), -NH(SO2)алкила, -NH(SO2)арила, -NH(SO2)гетероарила, -N(SO2)циклоалкила, -C(O)N(алкил)2, (арил)алкила-, -гетероарила, (гетероарил)алкила-, -S(O)2-алкила, -S(O)2-арила, -S(O)2-циклоалкила, -C(O)N(алкил)2, -C(O)алкила, -NH-C(O)-алкила, -NH-C(O)-циклоалкила, NH-C(O)-гетероциклоалкила, NH-C(O)-гетероциклоалкил-Rd, -NH-C(O)-Rd-(O)алкила, -NH-C(O)-арила, -NH-C(O)-NH-алкила, NH-C(O)-NH-циклоалкила, NH2(CO)циклоалкила-, NH-C(O)-NH-арила, -NH-C(O)-O-алкила, NH-C(O)-NH-циклоалкила, -NH-C(O)-O-циклоалкила, -NH(Rd)-C(O)-алкила, -NH(Rd)-C(O)-арила, -NH(Rd)-S(O2)циклоалкила, -S(O2)NH2, -S(O2)NH(алкил), -S(O2)N(Rd)циклоалкила, -S(O2)N(алкил)2, -C(O)N(H)(алкил), -C(O)N(Rd)(циклоалкил), метилендиокси, -CHzF3-z, -OCHzF3-z и -алкокси;
Rc представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Rd представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
и z равен 0, 1 или 2;
и его фармацевтически приемлемыми солями, сольватами, сложными эфирами, пролекарствами и изомерами.
В другом варианте реализации соединение формулы I представлено соединением формулы I-C:
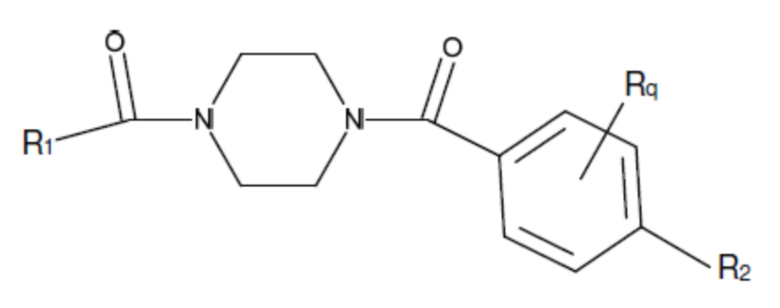
(I-C)
где:
R1 представляет собой С1-С3 гидроксил-алкил, который является незамещенным или замещен -СН3 или -CHzF3-z, 5-членный циклоалкил, который является незамещенным или замещен заместителями, выбранными из группы, состоящей из дейтерия, -Rp, -ORp, -NHRp и -NRpRp1, или 3- или 4-членный циклоалкил или гетероциклоалкил, где (i) гетероатом, являющийся членом кольца 3- или 4-членного гетероциклоалкила, независимо выбран из O, S или N, и (ii) каждый указанный 3- или 4-членный циклоалкил или гетероциклоалкил является незамещенным или необязательно замещен заместителями, выбранными из группы, состоящей из дейтерия, -Ra, -ORa, -NHRa и -NRaRa1;
R2 представляет собой Н или 4-15-членный моноциклический, бициклический или трициклический арил, гетероарил, циклоалкил или гетероциклоалкил, где (i) 4-15-членный моноциклический, бициклический или трициклический гетероарил или гетероциклоалкил содержит 1, 2, 3, 4, 5, 6, 7 или 8 гетероатомов, независимо выбранных из N, S или О, и (ii) каждый указанный арил, гетероарил, циклоалкил и гетероциклоалкил является незамещенным или необязательно замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, циано, гидроксила, гидроксил-алкила-, гидроксилциклоалкила-, гидроксил-гетероциклоалкила-, гидроксил-арила-, гидроксил-гетероарила-, амино, аминоалкила, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -CHzF3-z, -OCHzF3-z, -алкила, алкокси-, -алкенила, -алкинила, арилокси-, (алкоксиалкил)амино-, -циклоалкила, -гетероциклоалкила, (гетероциклоалкил)алкила-, -арила, -гетероарила, -O(алкил), -O(циклоалкил), -O(гетероциклоалкил), -O(арил), -O(гетероарил), ONH2, -C(O)NH(алкил), -C(O)N(арил)2, -C(O)NH(циклоалкил), -NH(CO)циклоалкила, -NH(SO2), -NH(SO2)алкила, -NH(SO2)арила, -NH(SO2)гетероарила, -N(SO2)циклоалкила, -C(O)N(алкил)2, (арил)алкила-, -гетероарила, (гетероарил)алкила-, -S(O)2-алкила, -S(O)2-арила, -S(O)2-циклоалкила, -C(O)N(алкил)2, -C(O)алкила, -NH-C(O)-алкила, -NH-C(O)-циклоалкила, NH-C(O)-гетероциклоалкила, NH-C(O)-гетероциклоалкил-Rd, -NH-C(O)-Rd-(O)алкила, -NH-C(O)-арила, -NH-C(O)-NH-алкила, NH-C(O)-NH-циклоалкила, NH2(CO)циклоалкила-, NH-C(O)-NH-арила, -NH-C(O)-O-алкила, NH-C(O)-NH-циклоалкила, -NH-C(O)-O-циклоалкила, -NH(Rd)-C(O)-алкила, -NH(Rd)-C(O)-арила, -NH(Rd)-S(O2)циклоалкила, -S(O2)NH2, -S(O2)NH(алкил), -S(O2)N(Rd)циклоалкила, -S(O2)N(алкил)2, -C(O)N(H)(алкил), -C(O)N(Rd)(циклоалкил), метилендиокси, -CHzF3-z, -OCHzF3-z и -алкокси;
Rp и Rp1 независимо представляют собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Ra и Ra1 независимо представляют собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Rd представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
Rq представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
и z равен 0, 1 или 2;
и его фармацевтически приемлемыми солями, сольватами, сложными эфирами, пролекарствами и изомерами.
В другом варианте реализации соединение формулы I представлено соединением формулы I-D:
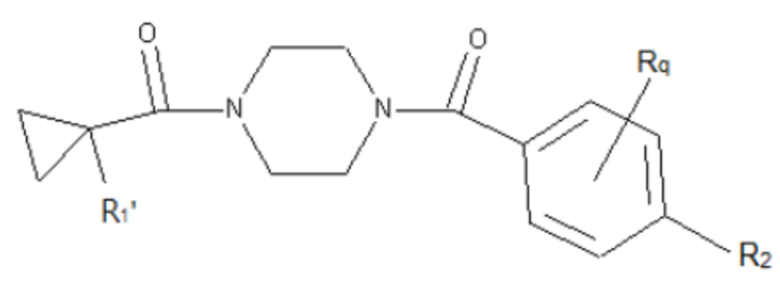
(I-D)
где:
R1' представляет собой ОН или NH2;
R2 представляет собой Н или 4-15-членный моноциклический, бициклический или трициклический арил, гетероарил, циклоалкил или гетероциклоалкил, где (i) 4-15-членный моноциклический, бициклический или трициклический гетероарил или гетероциклоалкил содержит 1, 2, 3, 4, 5, 6, 7 или 8 гетероатомов, независимо выбранных из N, S или О, и (ii) каждый указанный арил, гетероарил, циклоалкил и гетероциклоалкил является незамещенным или необязательно замещен 1 или более заместителями, которые могут быть одинаковыми или различными и независимо выбраны из группы, состоящей из дейтерия, галогена, циано, гидроксила, гидроксил-алкила-, гидроксилциклоалкила-, гидроксил-гетероциклоалкила-, гидроксил-арила-, гидроксил-гетероарила-, амино, аминоалкила, (амино)алкокси-, -CONH2, -C(O)NH(алкил), -C(O)N(алкил)2, -C(O)NH(арил), -C(O)N(арил)2, -CHzF3-z, -OCHzF3-z, -алкила, алкокси-, -алкенила, -алкинила, арилокси-, (алкоксиалкил)амино-, -циклоалкила, -гетероциклоалкила, (гетероциклоалкил)алкила-, -арила, -гетероарила, -O(алкил), -O(циклоалкил), -O(гетероциклоалкил), -O(арил), -O(гетероарил), ONH2, -C(O)NH(алкил), -C(O)N(арил)2, -C(O)NH(циклоалкил), -NH(CO)циклоалкила, -NH(SO2), -NH(SO2)алкила, -NH(SO2)арила, -NH(SO2)гетероарила, -N(SO2)циклоалкила, -C(O)N(алкил)2, (арил)алкила-, -гетероарила, (гетероарил)алкила-, -S(O)2-алкила, -S(O)2-арила, -S(O)2-циклоалкила, -C(O)N(алкил)2, -C(O)алкила, -NH-C(O)-алкила, -NH-C(O)-циклоалкила, NH-C(O)-гетероциклоалкила, NH-C(O)-гетероциклоалкил-Rd, -NH-C(O)-Rd-(O)алкила, -NH-C(O)-арила, -NH-C(O)-NH-алкила, NH-C(O)-NH-циклоалкила, NH2(CO)циклоалкила-, NH-C(O)-NH-арила, -NH-C(O)-O-алкила, NH-C(O)-NH-циклоалкила, -NH-C(O)-O-циклоалкила, -NH(Rd)-C(O)-алкила, -NH(Rd)-C(O)-арила, -NH(Rd)-S(O2)циклоалкила, -S(O2)NH2, -S(O2)NH(алкил), -S(O2)N(Rd)циклоалкила, -S(O2)N(алкил)2, -C(O)N(H)(алкил), -C(O)N(Rd)(циклоалкил), метилендиокси, -CHzF3-z, -OCHzF3-z и алкокси;
Rd представляет собой Н, галоген, С1-С4 алкил или С3-С4 циклоалкил;
и z равен 0, 1 или 2;
и его фармацевтически приемлемыми солями, сольватами, сложными эфирами, пролекарствами и изомерами.
В соединениях формулы I, I-A, I-B, I-C и I-D различные фрагменты выбраны независимо.
Следующие варианты реализации относятся к соединениям формулы I, I-A, I-B, I-C и I-D, если это возможно. Если для каких-либо фрагментов отсутствуют конкретные определения, то следует учитывать предыдущие определения. Кроме того, арильный, гетероарильный и гетероциклоалкильный фрагменты в указанных вариантах реализации независимо могут быть незамещенными или необязательно замещены или необязательно конденсированы, как описано выше.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R1 представляет собой С1-С3 гидроксил-алкил, который является незамещенным или замещен -СН3 или -CHzF3-z, и А, В, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R1 представляет собой 5-членный циклоалкил, который является незамещенным или замещен гидроксилом, и А, В, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R1 представляет собой 3- или 4-членный циклоалкил, и А, В, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R1 представляет собой 3- или 4-членный гетероциклоалкил, и А, В, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R1 представляет собой  ,
,  ,
, 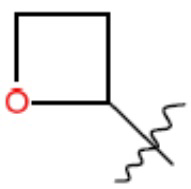 ,
, 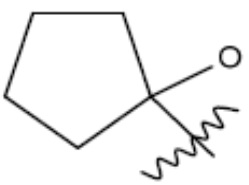 ,
, 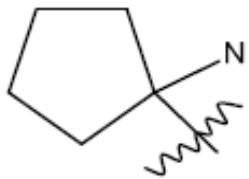 ,
, 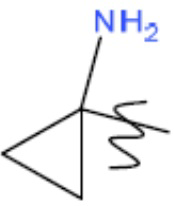 или
или 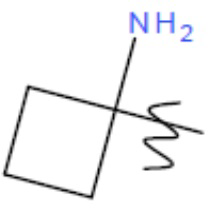 , и А, В, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, и А, В, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R1 представляет собой 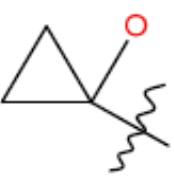 ,
, 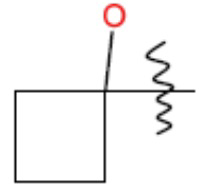 ,
,  ,
, 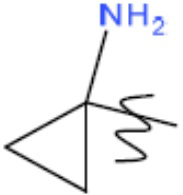 или
или  , и А, В, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, и А, В, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R1 представляет собой 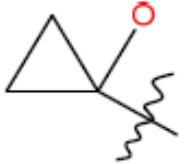 , и А, В, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, и А, В, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R1 представляет собой 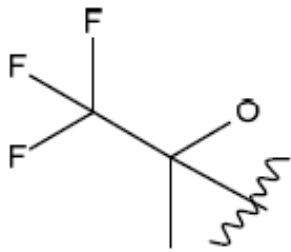 ,
, 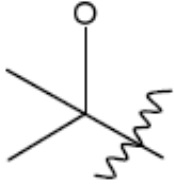 ,
, 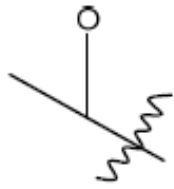 ,
,  или
или  , и А, В, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, и А, В, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, А и В представляют собой О, и R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, А и В представляют собой S, и R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, один из А или В представляет собой О, другой представляет собой S, и R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, L представляет собой 5-10-членный моноциклический алкил, и А, В, R1, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, L представляет собой 5-10-членный бициклический алкил, и А, В, R1, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, L представляет собой 5-10-членный моноциклический гетероалкил, и А, В, R1, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, L представляет собой 5-10-членный бициклический гетероалкил, и А, В, R1, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, L представляет собой 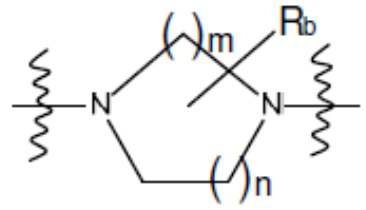 , m равен 1, 2 или 3, n равен 0, 1, 2 или 3, и А, В, R1, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, m равен 1, 2 или 3, n равен 0, 1, 2 или 3, и А, В, R1, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, L представляет собой  , и А, В, R1, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, и А, В, R1, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, L представляет собой  , и А, В, R1, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, и А, В, R1, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой арил, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой гетероарил, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой 5-10-членный моноциклический арил, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой 5-10-членный бициклический арил, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой 5-10-членный моноциклический гетероарил, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой 5-10-членный бициклический гетероарил, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой замещенный или незамещенный 5-членный моноциклический арил или гетероарил, и указанный гетероарил содержит 1 или 2 гетероатома, независимо представляющие собой S или N, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой замещенный или незамещенный 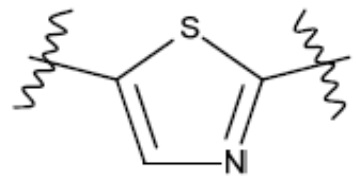 , и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой замещенный или незамещенный 6-членный моноциклический арил или гетероарил, и указанный гетероарил содержит 1 или 2 гетероатома, представляющие собой N, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой замещенный или незамещенный 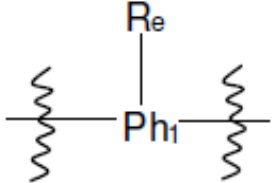 , Ph1 представляет собой фенил, пиридинил, пиразинил или пиримидинил, Re представляет собой Н, галоген или С1-С3 алкил, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, Ph1 представляет собой фенил, пиридинил, пиразинил или пиримидинил, Re представляет собой Н, галоген или С1-С3 алкил, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой замещенный или незамещенный  ,
, 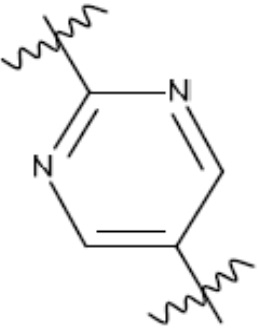 ,
, 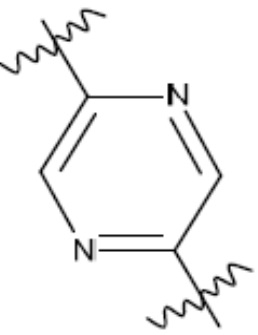 или
или 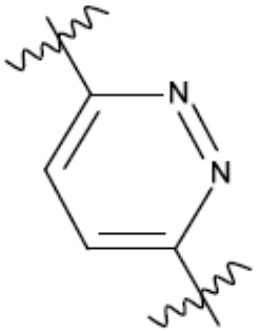 , и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой замещенный или незамещенный 6-членный моноциклический арил, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой 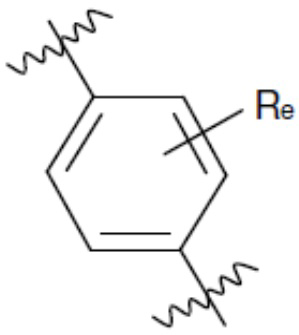 , Re представляет собой Н, галоген или С1-С3 алкил, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, Re представляет собой Н, галоген или С1-С3 алкил, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой 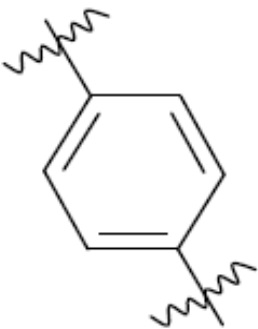 ,
, 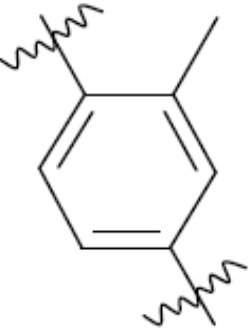 ,
,  или
или  , и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой замещенный или незамещенный 9-членный 6,5-бициклический гетероарил, и указанный гетероарил содержит 1, 2 или 3 гетероатома, независимо представляющие собой О, S или N, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ar1 представляет собой замещенный или незамещенный  ,
,  ,
,  или
или 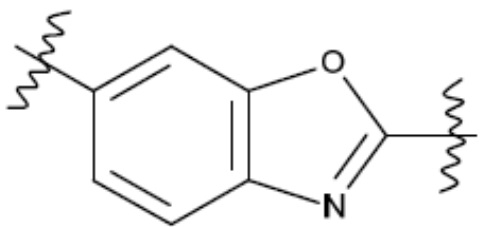 , и указанный гетероарил содержит 1, 2 или 3 гетероатома, независимо представляющие собой S или N, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, и указанный гетероарил содержит 1, 2 или 3 гетероатома, независимо представляющие собой S или N, и А, В, R1, L, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R2 представляет собой замещенный или незамещенный арил, и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R2 представляет собой замещенный или незамещенный гетероарил, и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R2 представляет собой замещенный или незамещенный циклоалкил, и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R2 представляет собой замещенный или незамещенный гетероциклоалкил, и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R2 представляет собой замещенный или незамещенный моноциклический или бициклический 5-10-членный арил или гетероарил, и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R2 представляет собой замещенный или незамещенный моноциклический 6-членный арил, и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R2 представляет собой  ,
, 
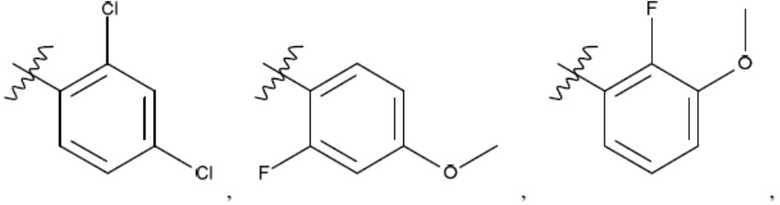
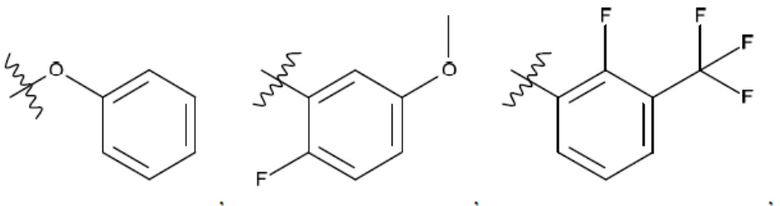
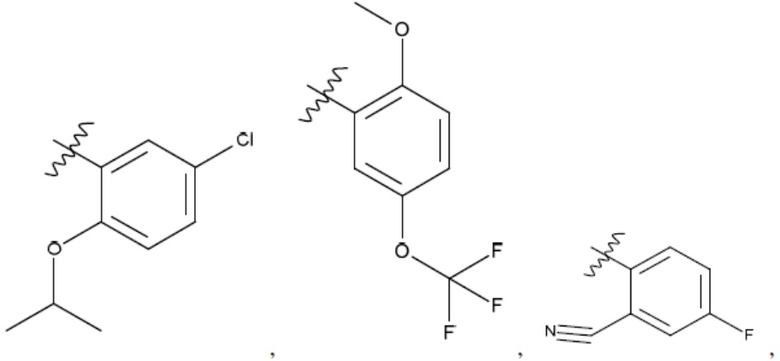
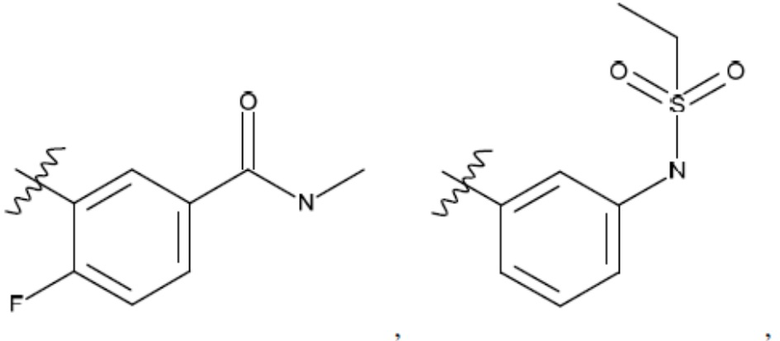

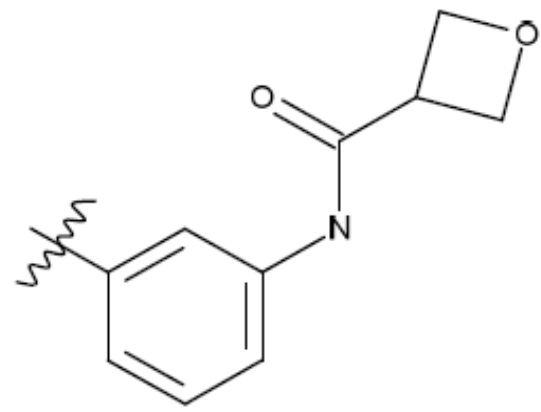 или
или 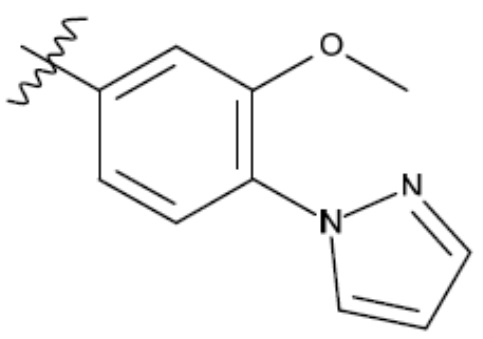 , и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R2 представляет собой замещенный или незамещенный бициклический 8-10-членный арил или 8-10-членный гетероарил, и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R2 представляет собой замещенный или незамещенный 8-членный 5,5-бициклический гетероарил, и указанный гетероарил содержит 1, 2, 3 или 4 гетероатома, и указанные гетероатомы независимо представляют собой О, S или N, и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R2 представляет собой замещенный или незамещенный 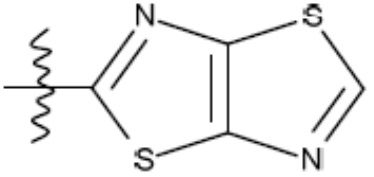 ,
, 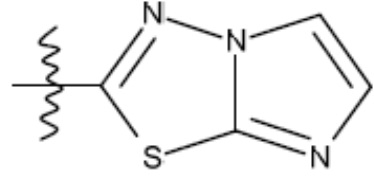 ,
, 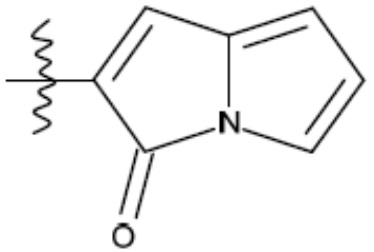 или
или 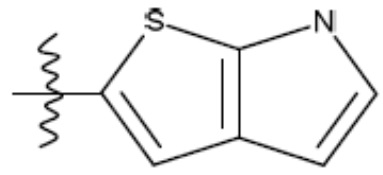 , и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R2 представляет собой замещенный или незамещенный 9-членный 6,5-бициклический гетероарил, и указанный гетероарил содержит 1, 2, 3 или 4 гетероатома, и указанные гетероатомы независимо представляют собой О, S или N, и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R2 представляет собой замещенный или незамещенный  ,
, 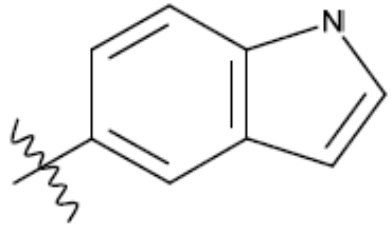 ,
, 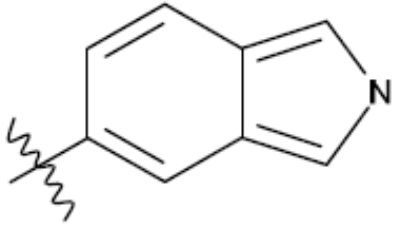 ,
, 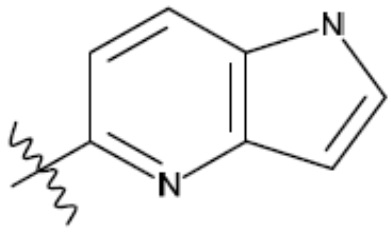 ,
, 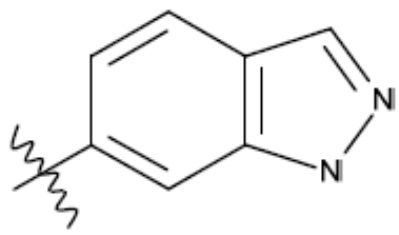 ,
, 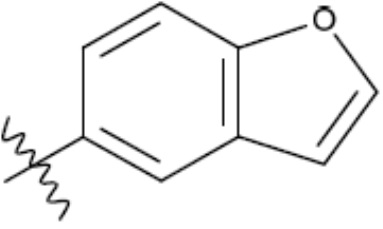 ,
, 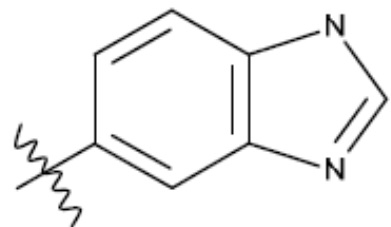 ,
,  ,
, 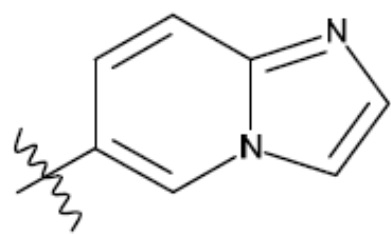 ,
, 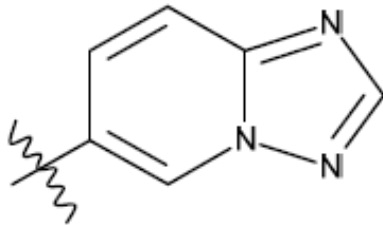 ,
, 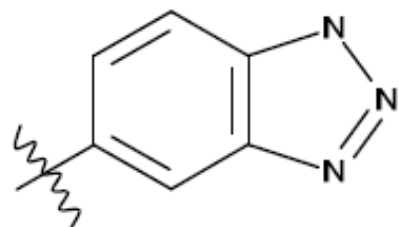 ,
, 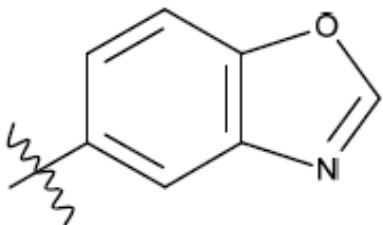 ,
, 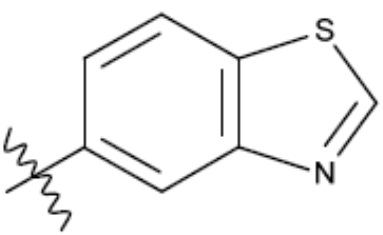 ,
, 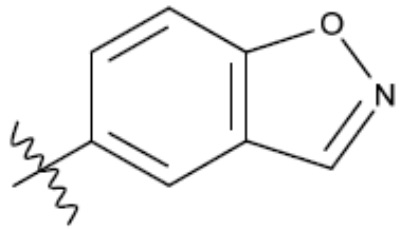 ,
,  ,
, 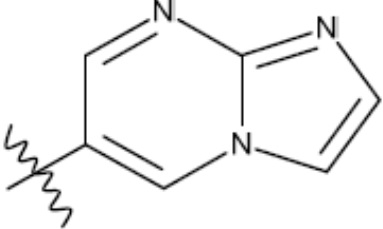 ,
,  ,
, 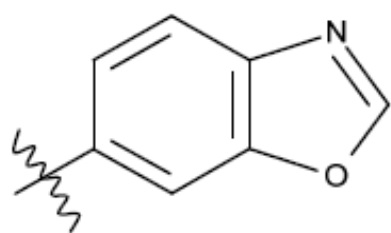 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 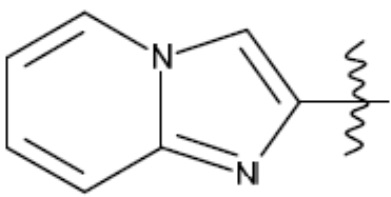 ,
, 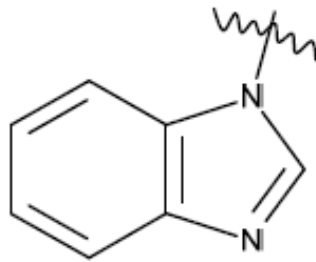 или
или 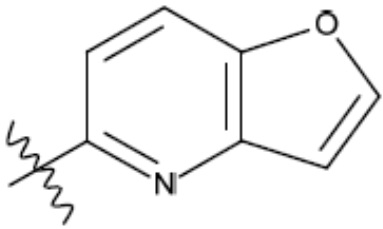 , и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R2 представляет собой замещенный или незамещенный 10-членный 6,6-бициклический арил или гетероарил, и указанный гетероарил содержит 1, 2, 3 или 4 гетероатома, и указанные гетероатомы представляют собой О, S или N, и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, R2 представляет собой замещенный или незамещенный 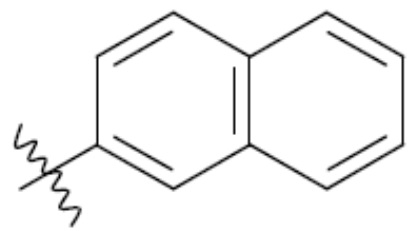 ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 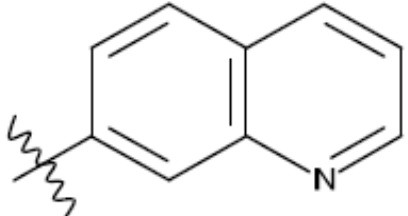 ,
, 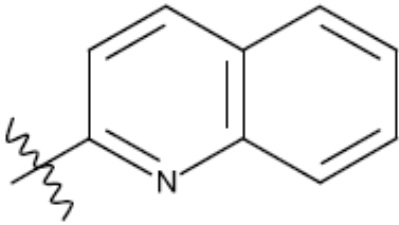 ,
, 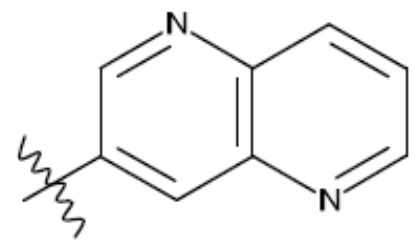 или
или  , и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
, и А, В, R1, L, Ar1, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rp представляет собой Н, и А, В, R1, L, Ar1, R2, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rp представляет собой галоген, и А, В, R1, L, Ar1, R2, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rp представляет собой С1-С4 алкил, и А, В, R1, L, Ar1, R2, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rp представляет собой С3-С4 циклоалкил, и А, В, R1, L, Ar1, R2, Rp1, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rp1 представляет собой Н, и А, В, R1, L, Ar1, R2, Rp, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rp1 представляет собой галоген, и А, В, R1, L, Ar1, R2, Rp, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rp1 представляет собой С1-С4 алкил, и А, В, R1, L, Ar1, R2, Rp, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rp1 представляет собой С3-С4 циклоалкил, и А, В, R1, L, Ar1, R2, Rp, Ra, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ra представляет собой Н, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ra представляет собой галоген, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ra представляет собой С1-С4 алкил, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ra представляет собой С3-С4 циклоалкил, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra1, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ra1 представляет собой Н, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ra1 представляет собой галоген, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ra1 представляет собой С1-С4 алкил, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Ra1 представляет собой С3-С4 циклоалкил, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Rb, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rb представляет собой Н, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rb представляет собой галоген, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rb представляет собой С1-С4 алкил, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rb представляет собой С1-С3 гидроксил-алкил, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rb представляет собой С3-С4 циклоалкил, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rc, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rc представляет собой Н, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rc представляет собой галоген, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rc представляет собой С1-С4 алкил, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rc представляет собой С3-С4 циклоалкил, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rd, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rd представляет собой Н, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rd представляет собой галоген, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rd представляет собой С1-С4 алкил, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rd представляет собой С3-С4 циклоалкил, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rq и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rq представляет собой Н, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rq представляет собой галоген, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rq представляет собой С1-С4 алкил, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, Rq представляет собой С3-С4 циклоалкил, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd и z такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, z равен 0, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd и Rq такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, z равен 1, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd и Rq такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, где различные фрагменты выбраны независимо, z равен 2, и А, В, R1, L, Ar1, R2, Rp, Rp1, Ra, Ra1, Rb, Rc, Rd и Rq такие, как определено в настоящем описании.
В одном из вариантов реализации изобретения предложено соединение, описанное формулами I, I-A, I-B, I-C или I-D, где R2 не является замещенным или незамещенным 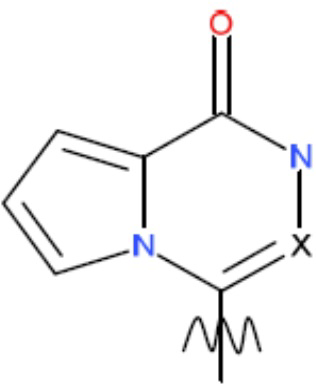 , в котором Х представляет собой N или CH.
, в котором Х представляет собой N или CH.
В одном из вариантов реализации изобретения предложено соединение, описанное формулами I, I-A, I-B, I-C или I-D, где если Ar1 представляет собой 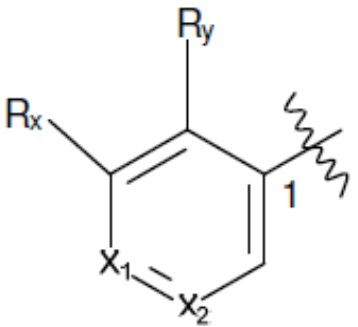 , соединенный с
, соединенный с 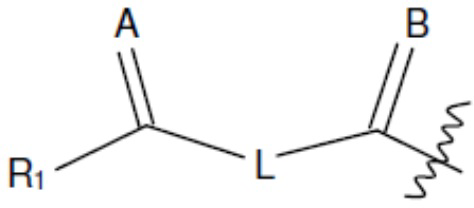 через положение 1, и X1 и X2 независимо представляют собой N или C-Rz, и Ry и Rz представляют собой какой-либо заместитель, то Rx не включает алкинил, алкенил, арил, 5-14-членный гетероцикл, 5-14-членную гетероароматическую группу или 4-9-членный карбоцикл.
через положение 1, и X1 и X2 независимо представляют собой N или C-Rz, и Ry и Rz представляют собой какой-либо заместитель, то Rx не включает алкинил, алкенил, арил, 5-14-членный гетероцикл, 5-14-членную гетероароматическую группу или 4-9-членный карбоцикл.
В одном из вариантов реализации изобретения предложено соединение, описанное формулами I, I-A, I-B, I-C или I-D, где если R2 представляет собой  , то Ar1 не является замещенным или незамещенным
, то Ar1 не является замещенным или незамещенным  ,
, 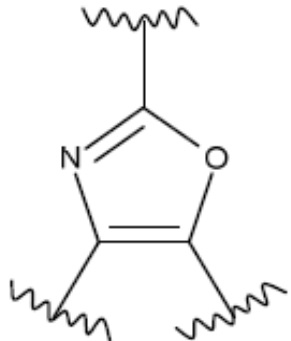 ,
, 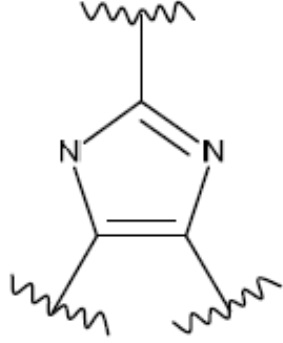 ,
, 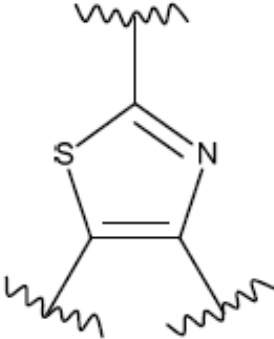 ,
, 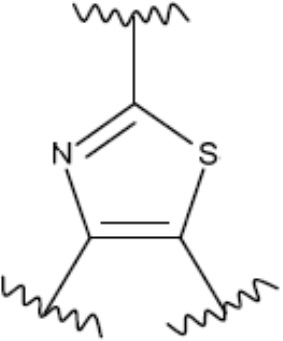 ,
,  или
или  .
.
В одном из вариантов реализации изобретения предложено соединение, описанное формулами I, I-A, I-B, I-C или I-D, где если Ar1 представляет собой замещенный или незамещенный 5-членный гетероарил, то Ar1 представляет собой  .
.
В другом варианте реализации изобретение дополнительно проиллюстрировано соединениями, приведенными в таблице 1, в которой перечислены названия согласно IUPAC и структуры соединений.
Таблица 1
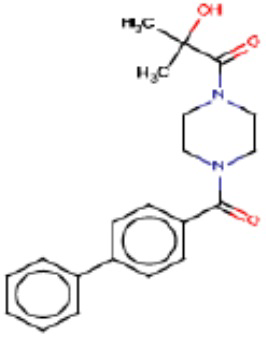
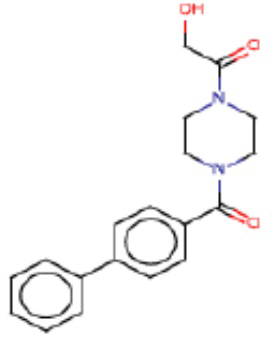
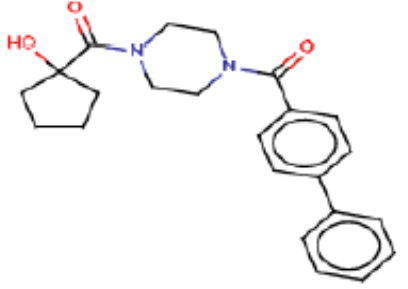
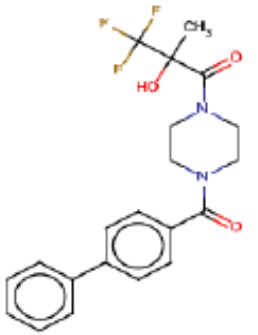
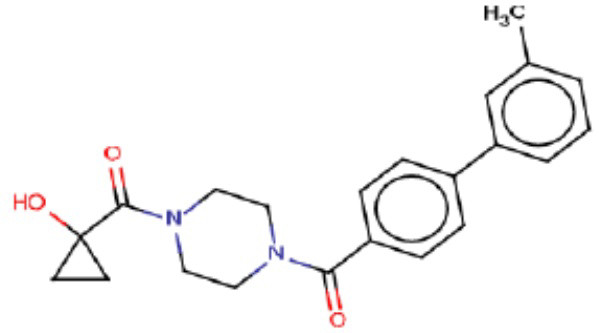
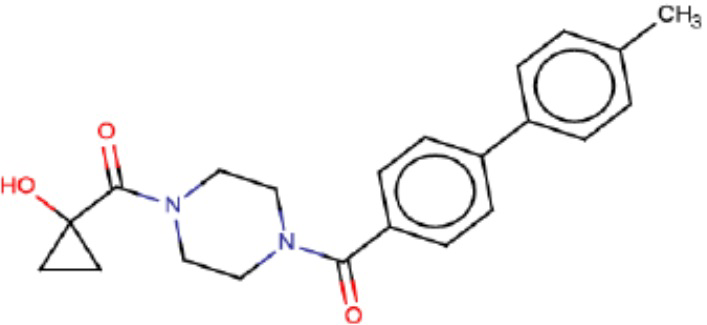

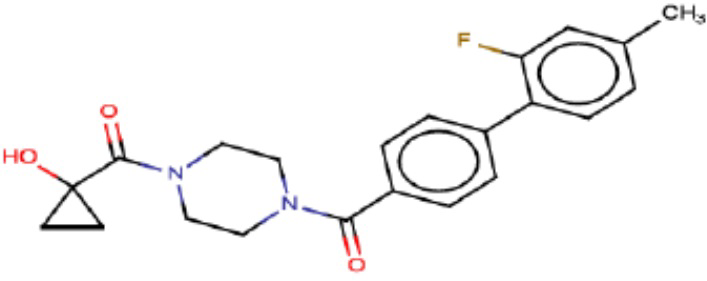
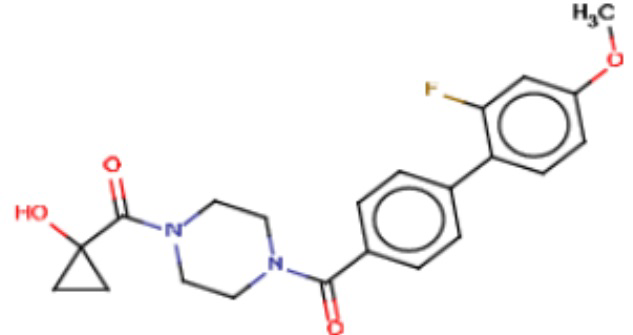
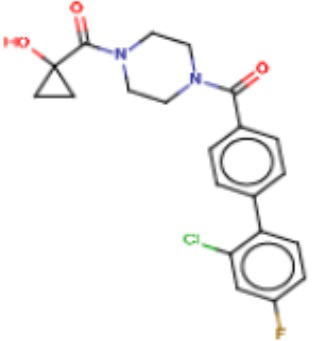
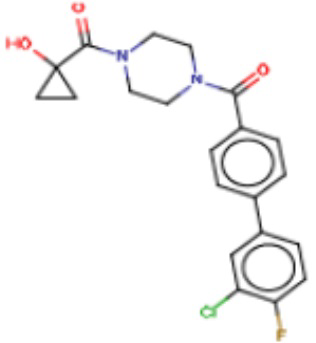
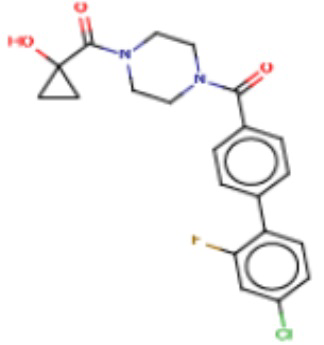
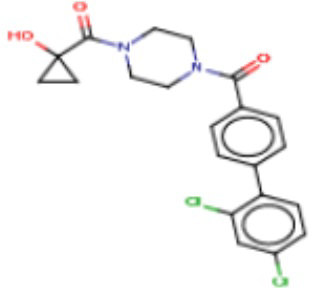
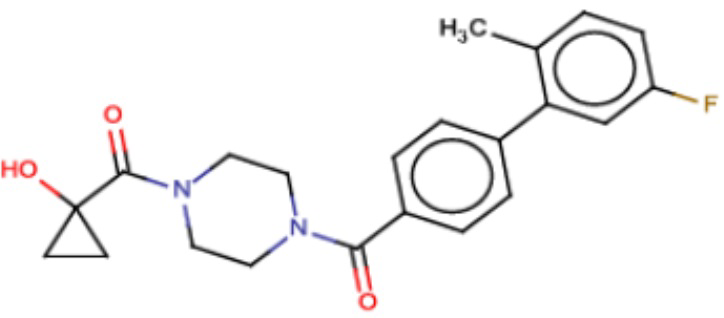
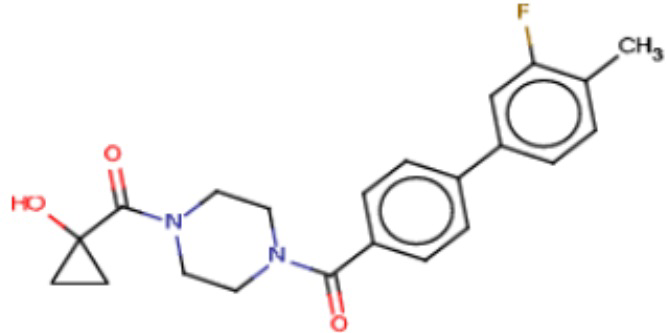
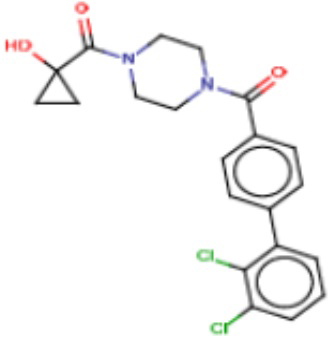
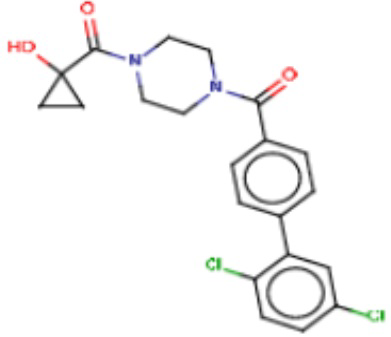
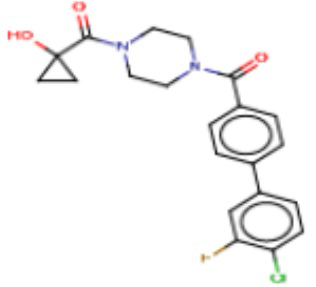
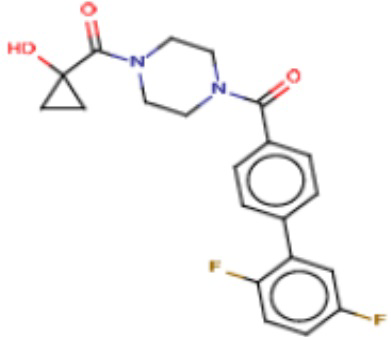
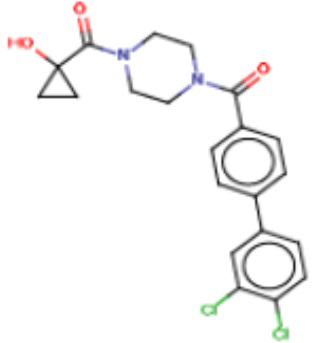
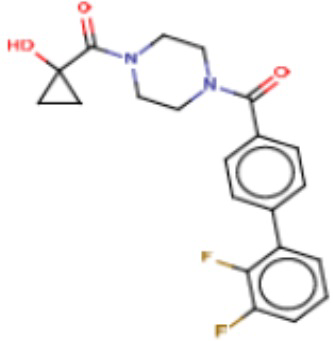
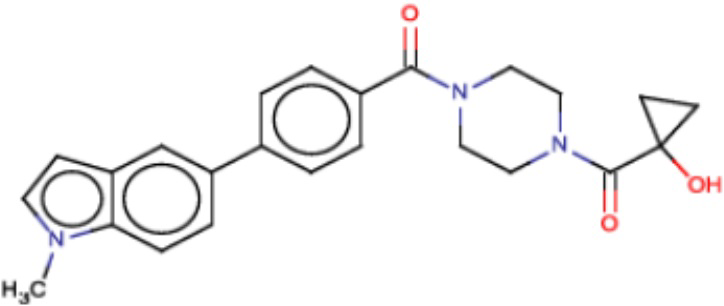
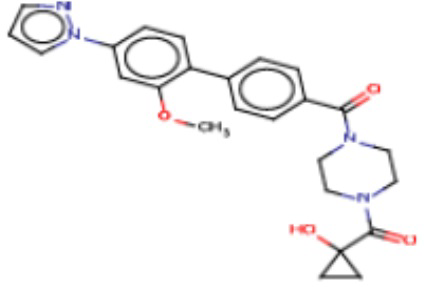
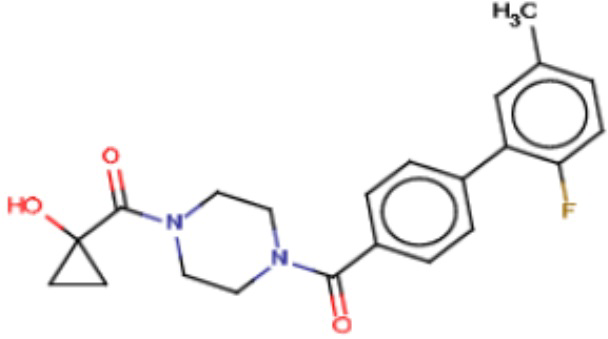
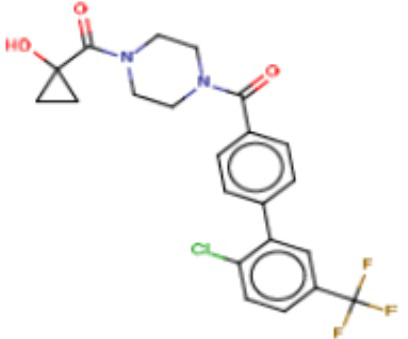
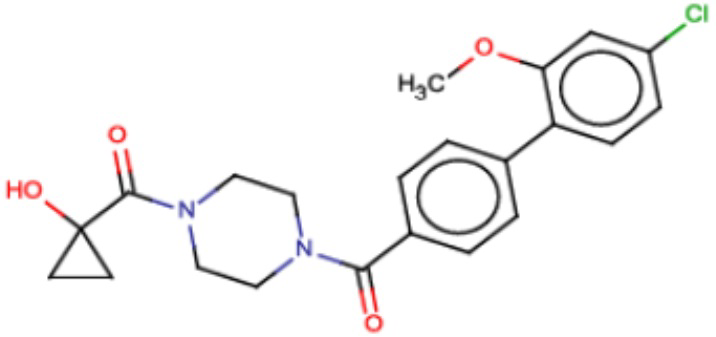

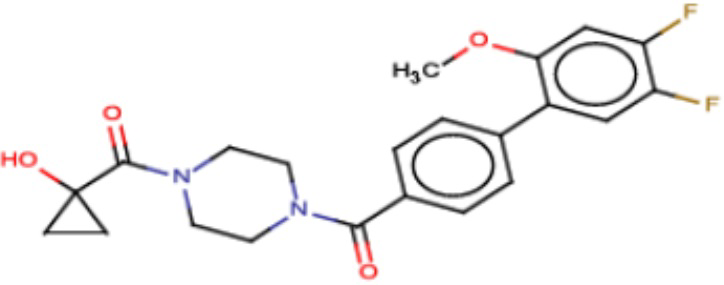
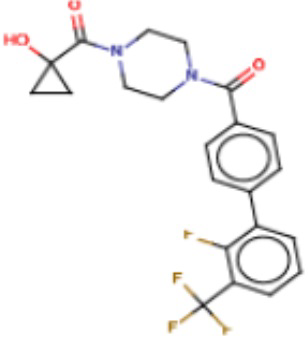
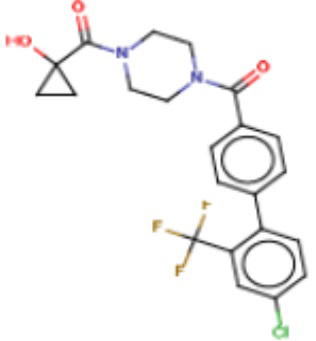
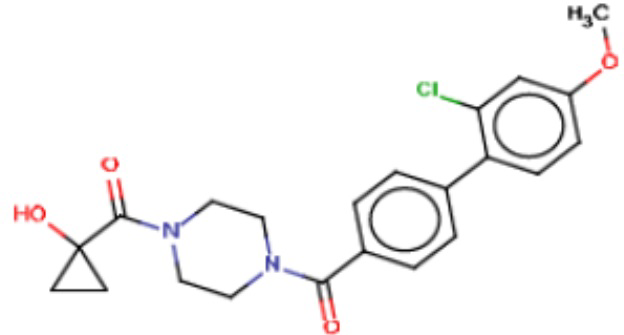
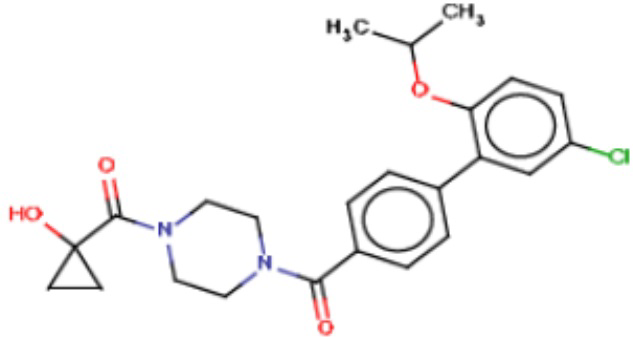
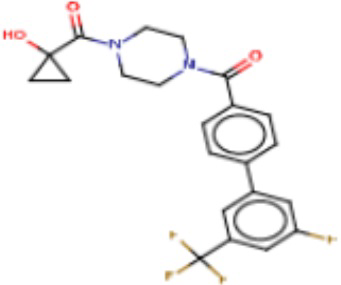
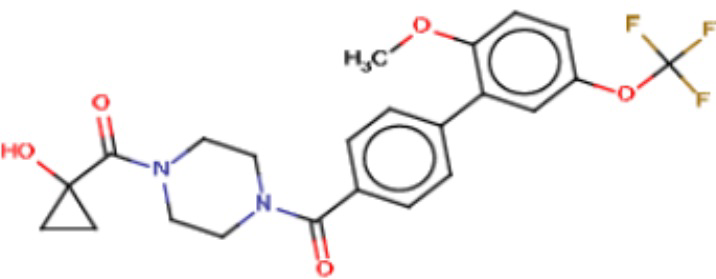
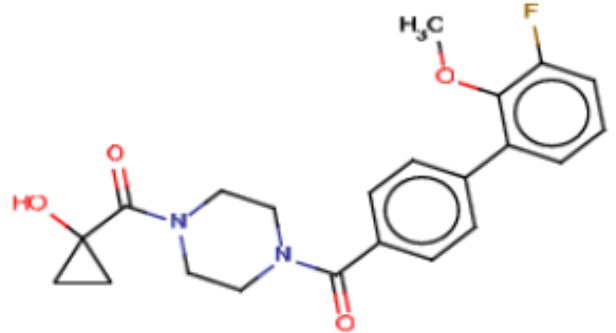

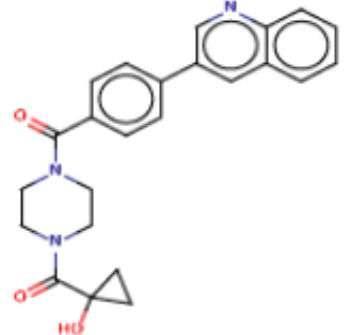
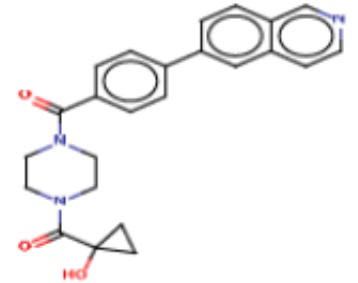
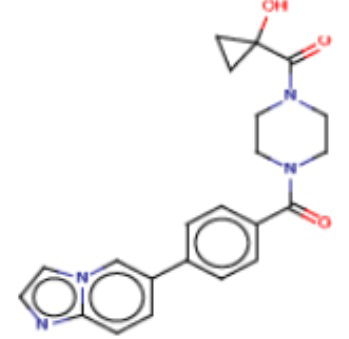
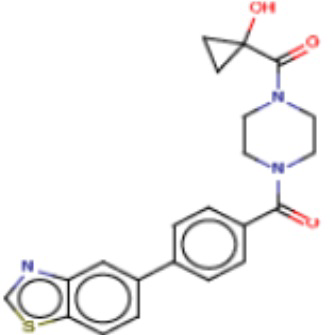
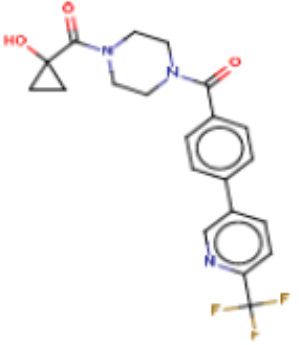

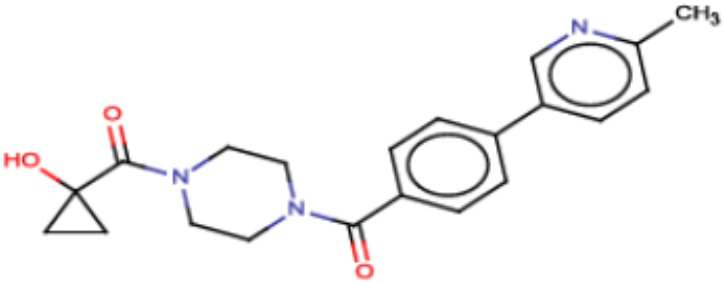
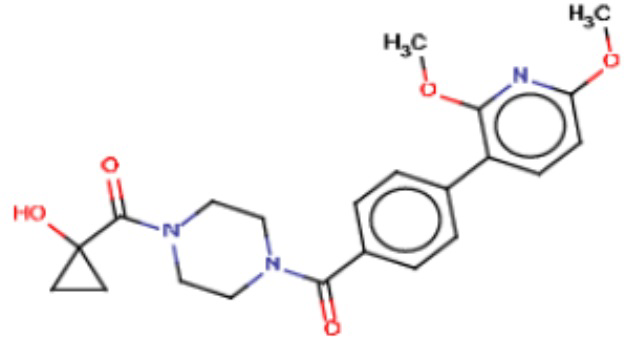

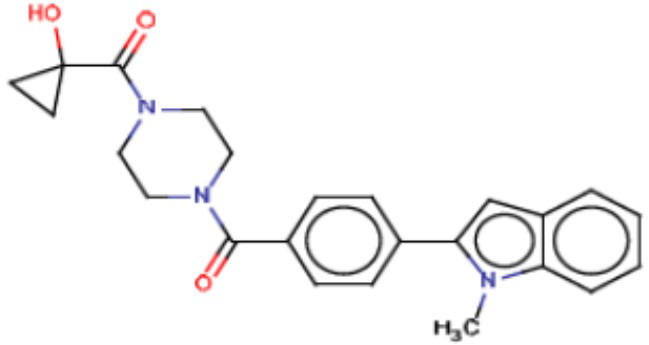
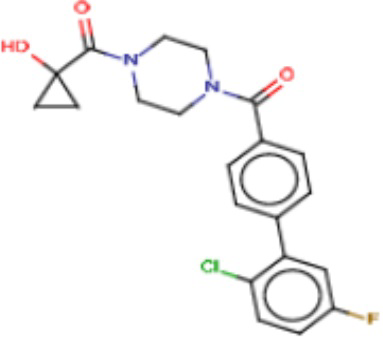
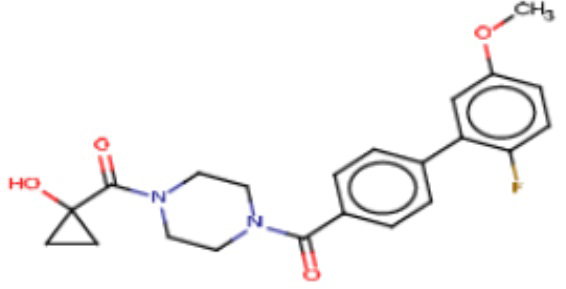
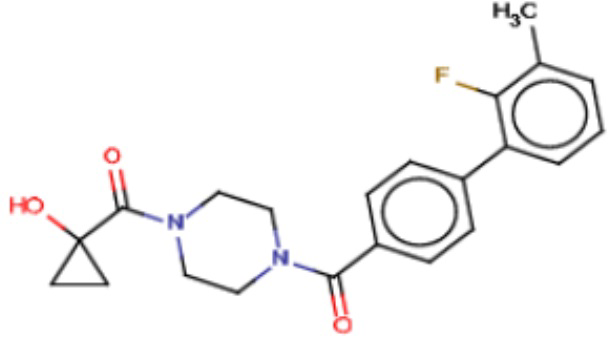


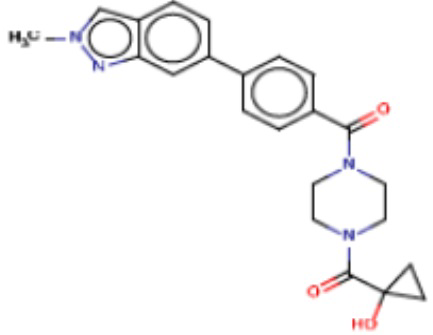
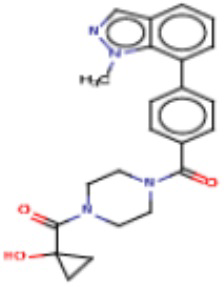
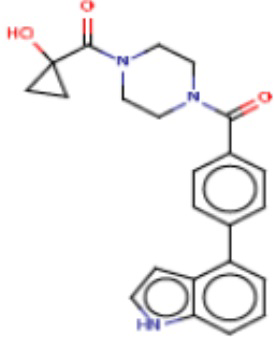
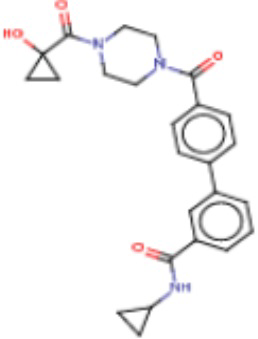
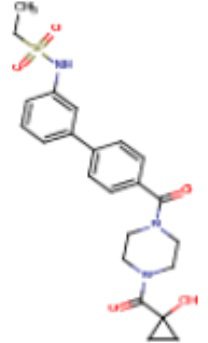
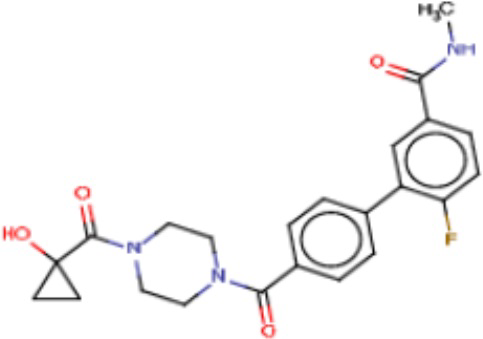
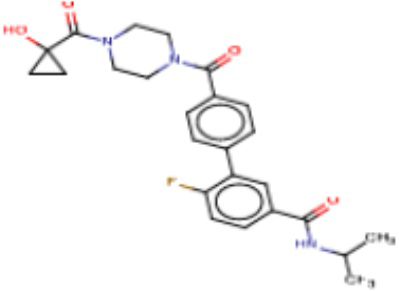
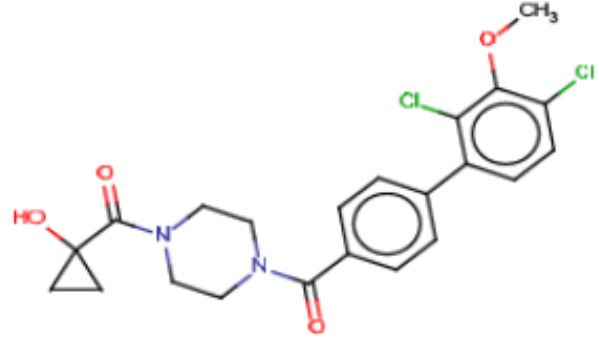
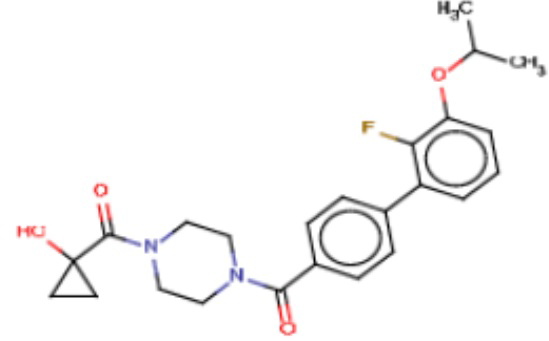
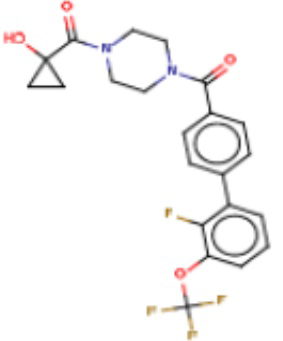

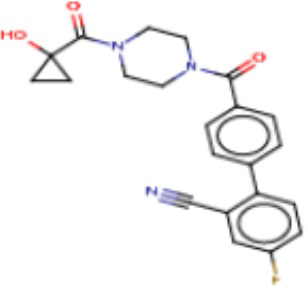
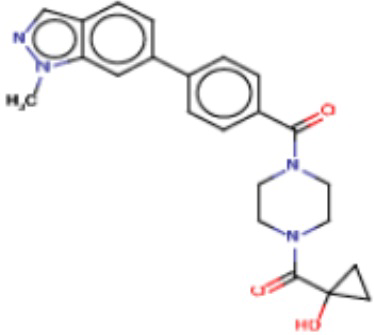
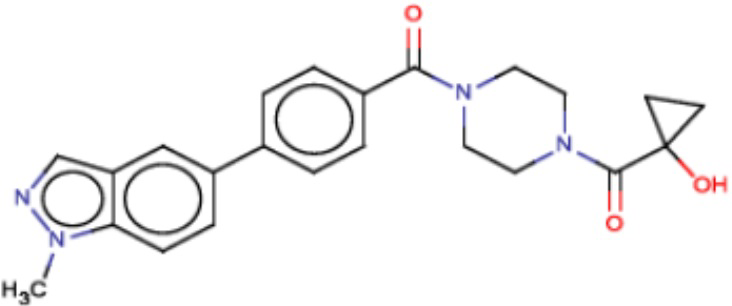
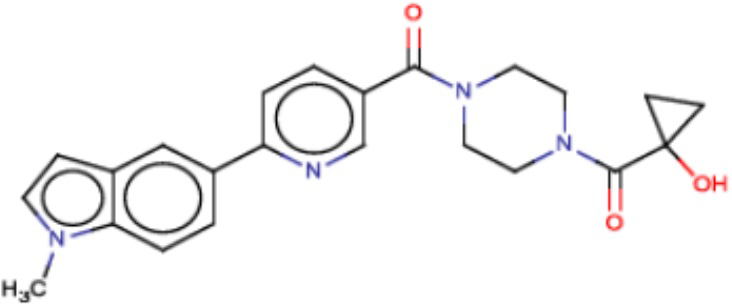
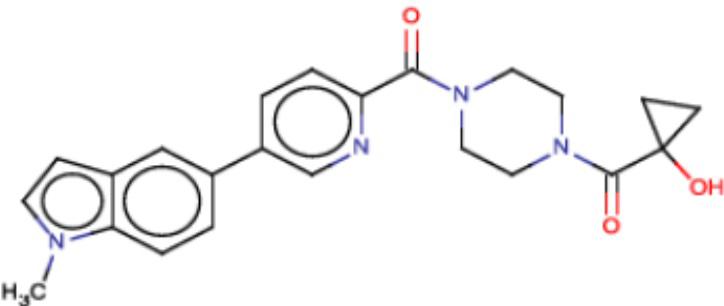
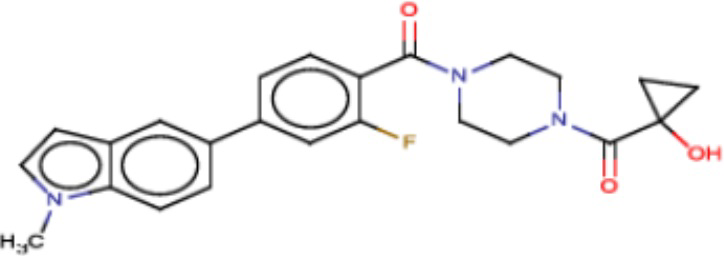

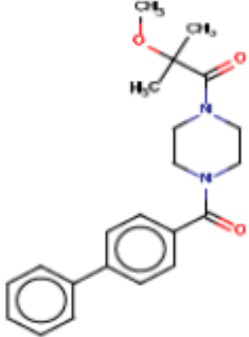
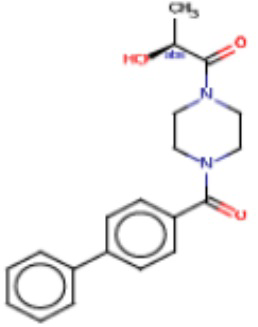

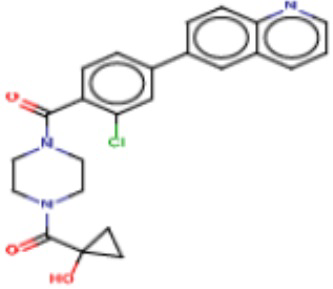
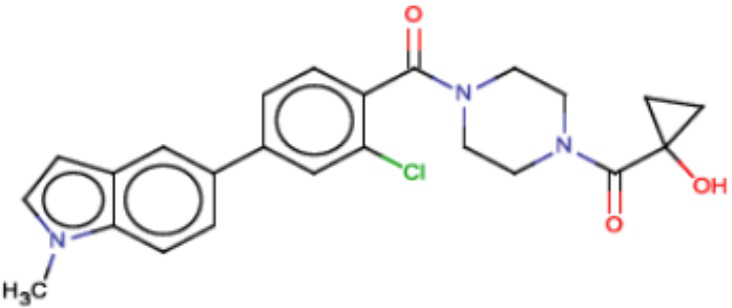
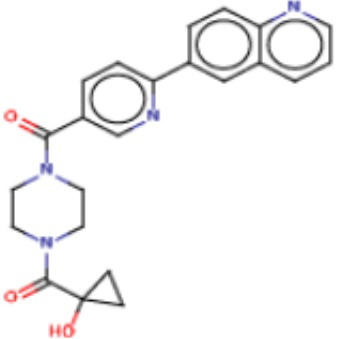
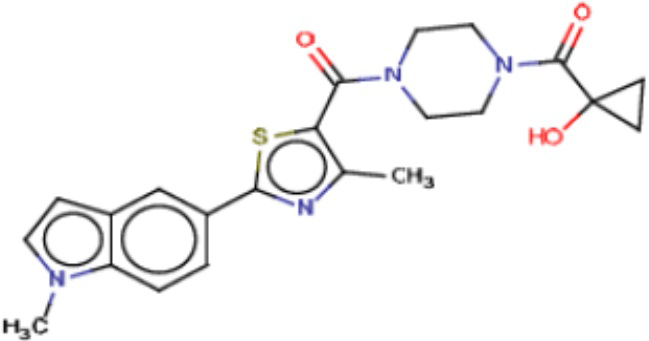
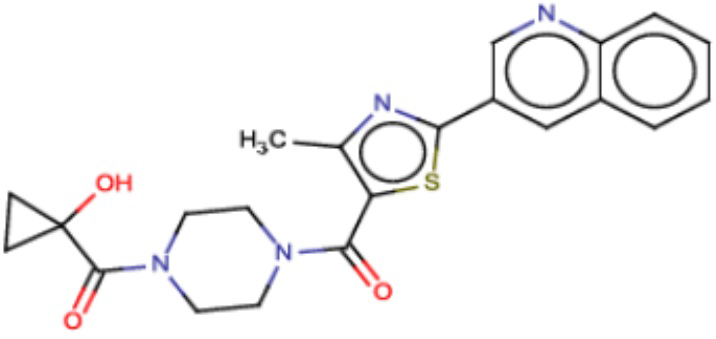
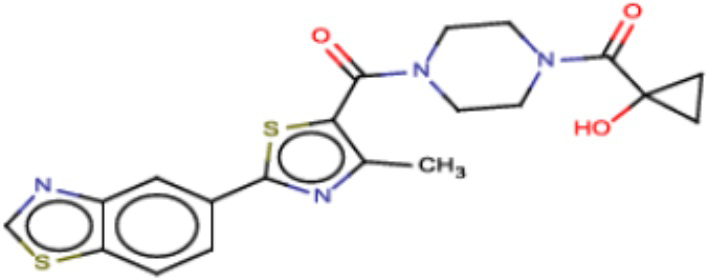
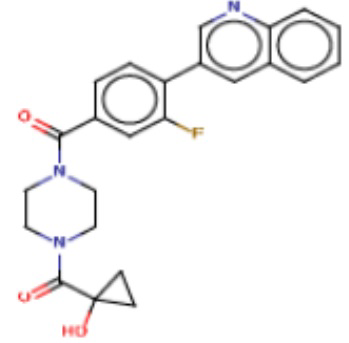
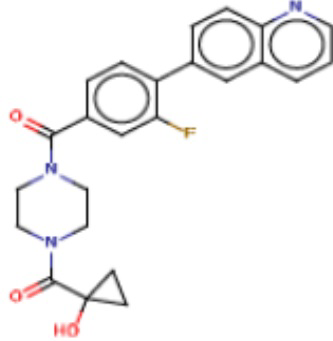
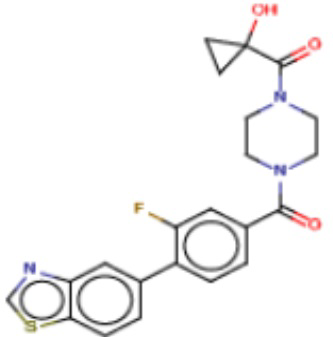
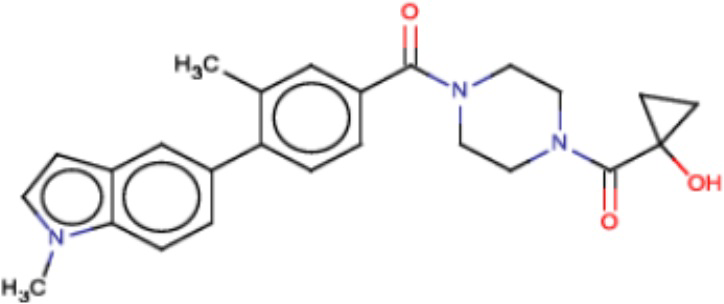
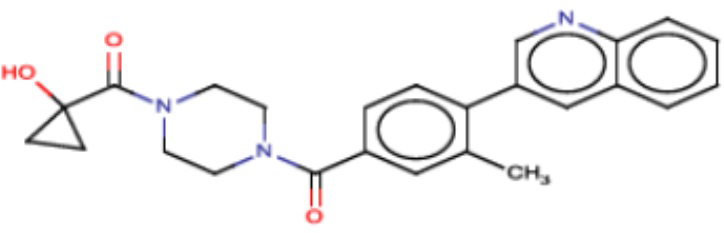
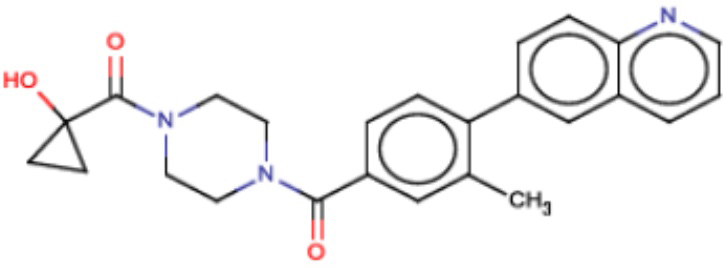
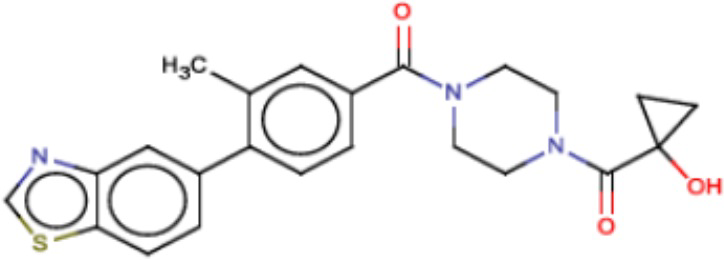
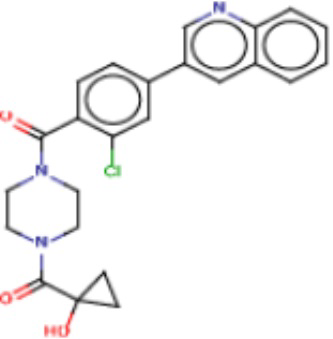
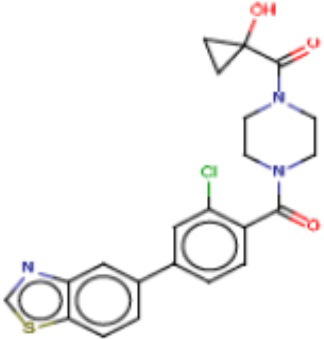
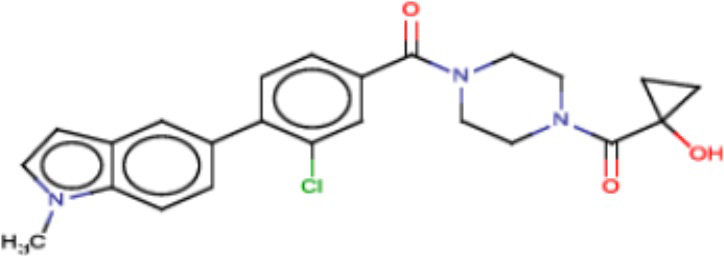
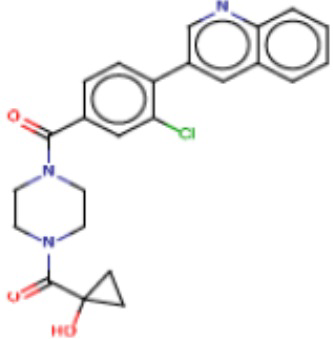
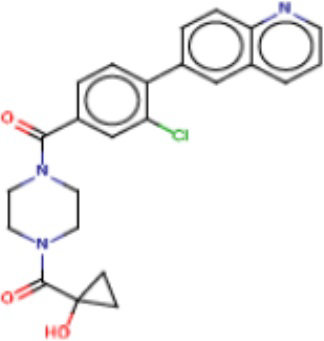
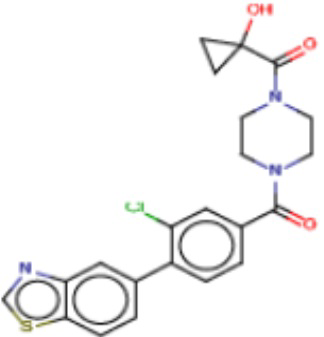


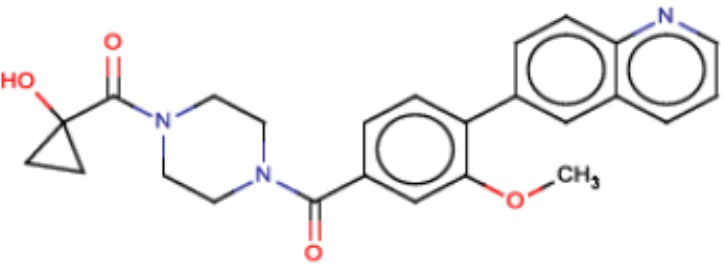
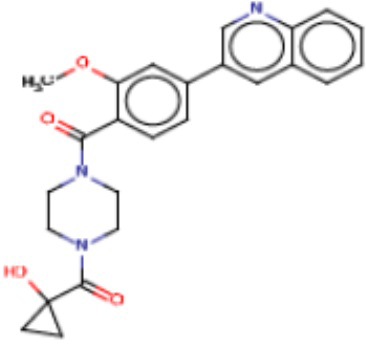
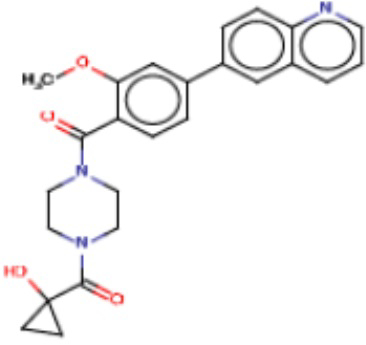
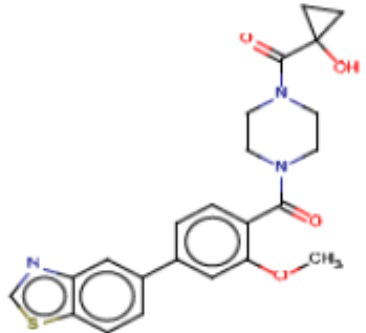
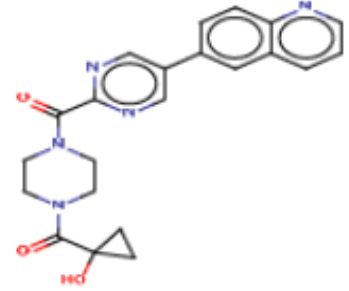
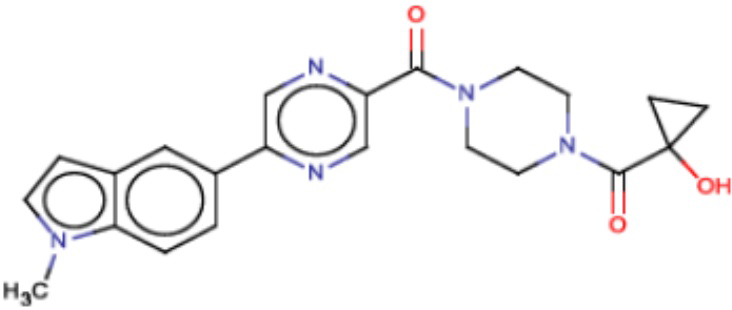
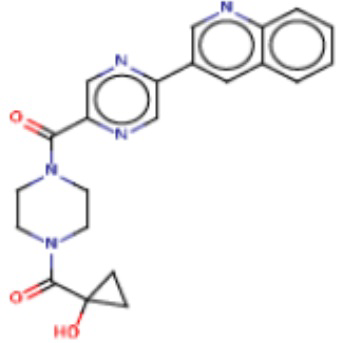
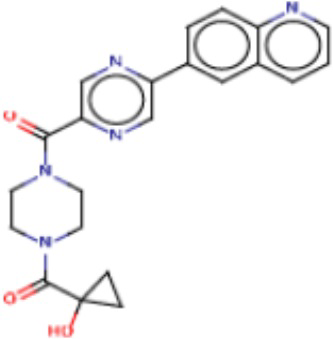
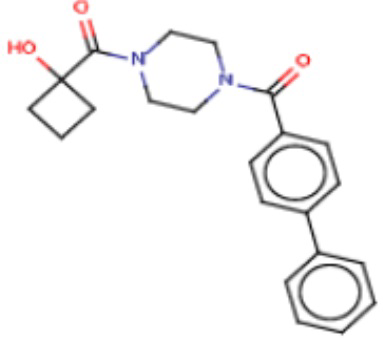
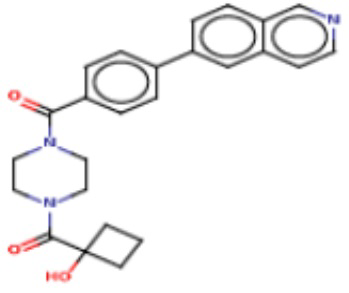

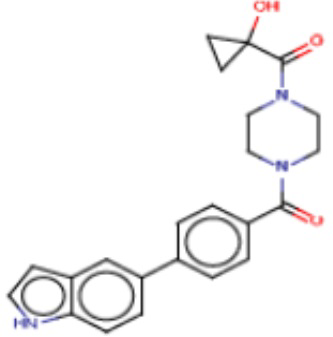
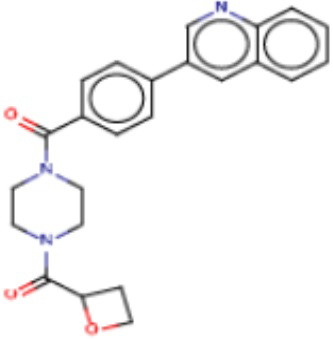
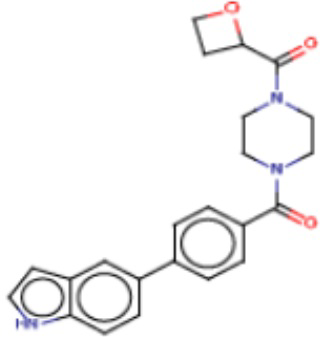
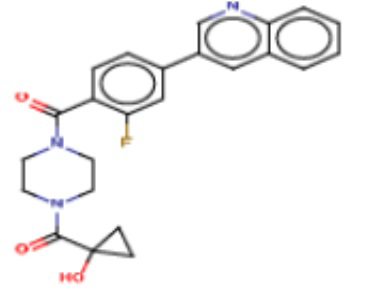
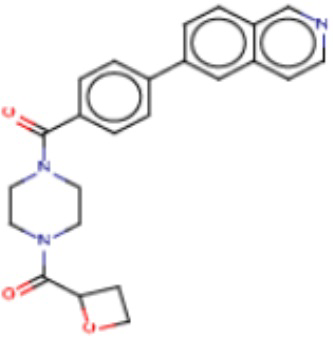
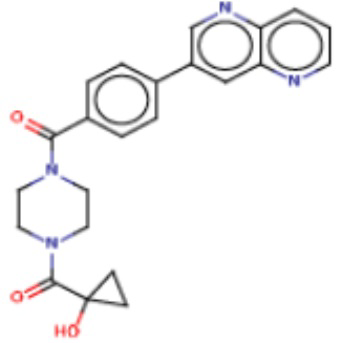
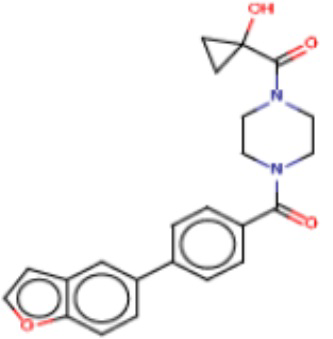
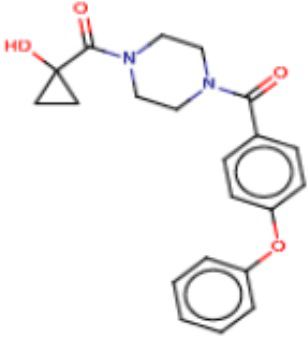
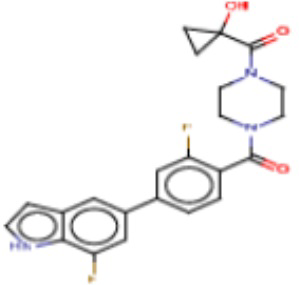
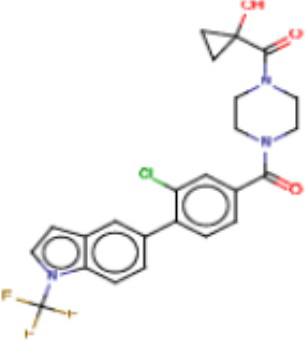
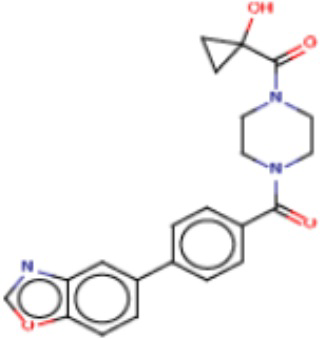
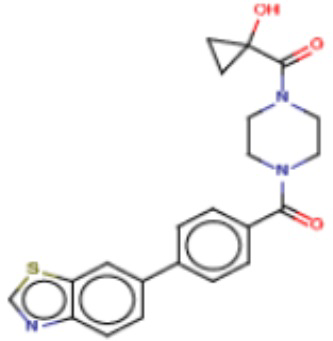
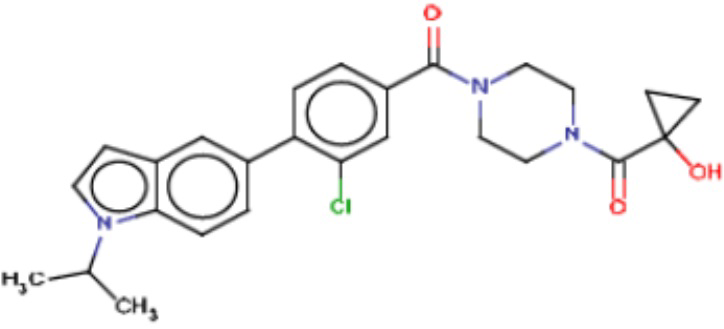
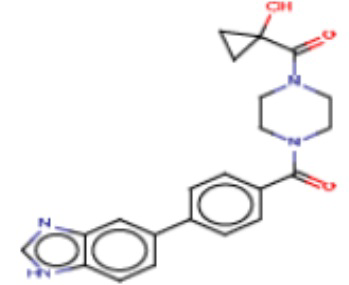
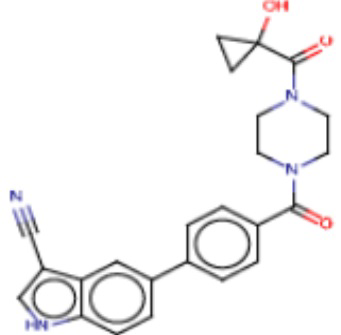
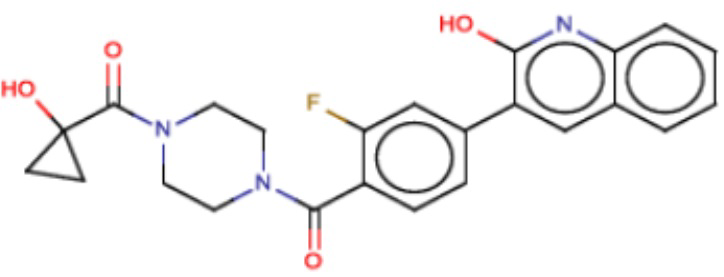
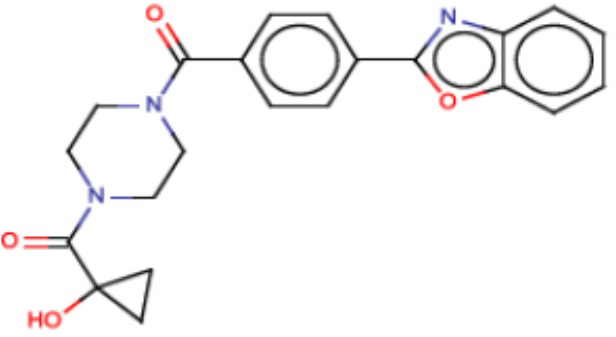
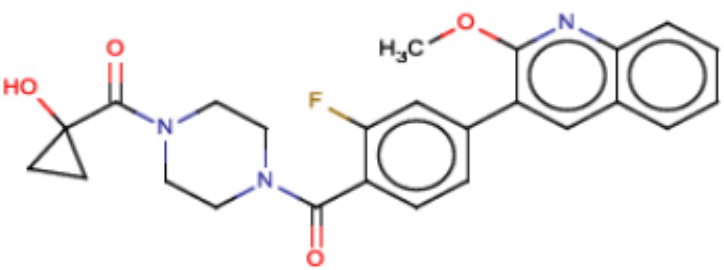
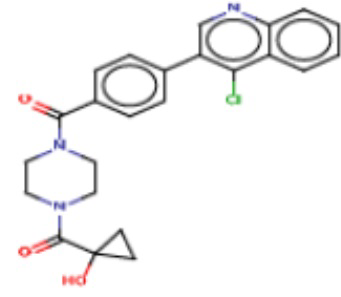
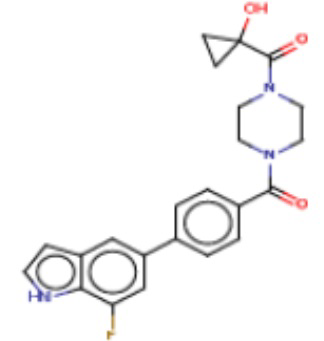
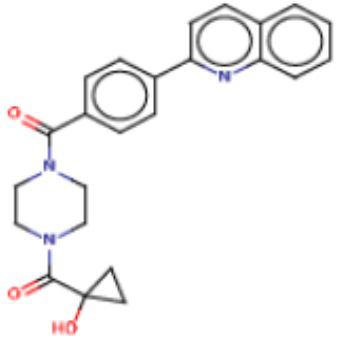

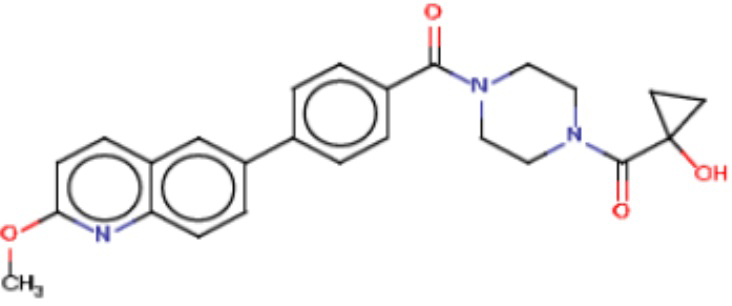
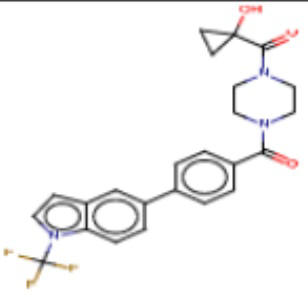
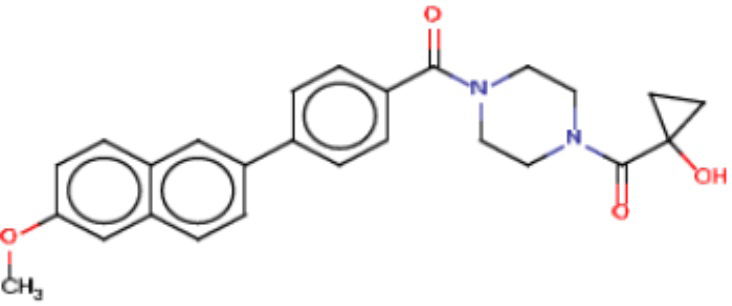




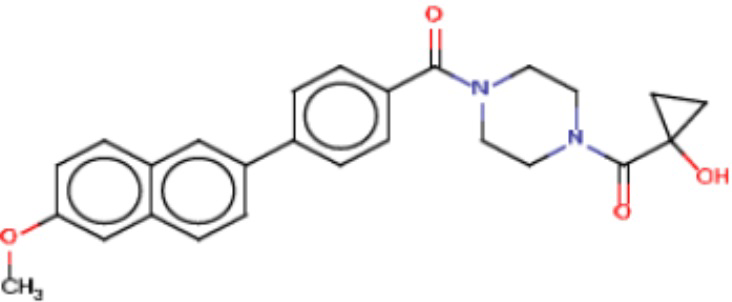
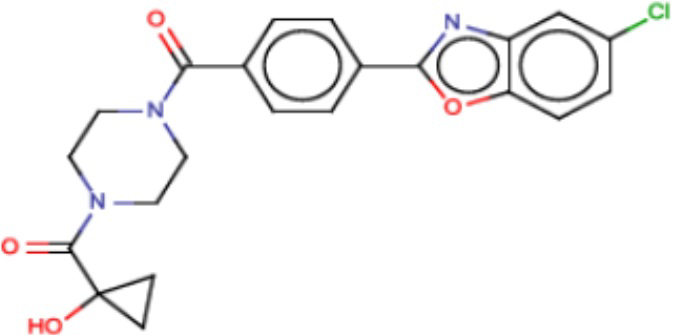
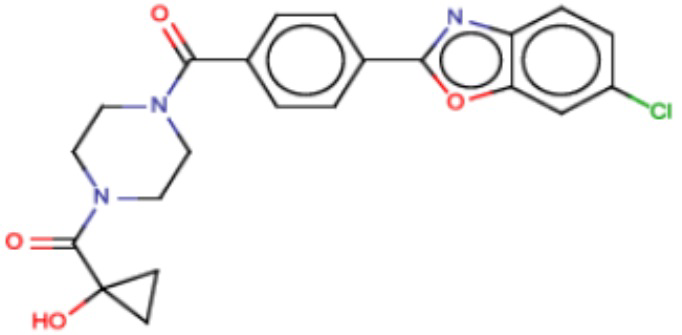

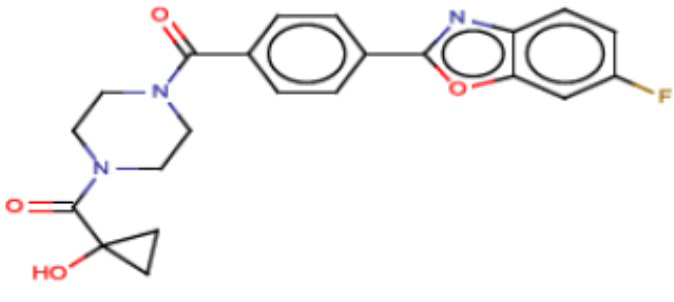
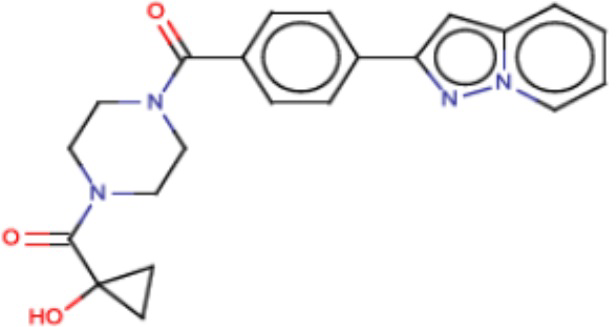

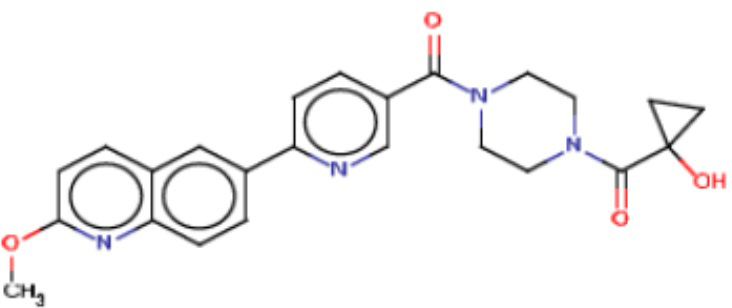
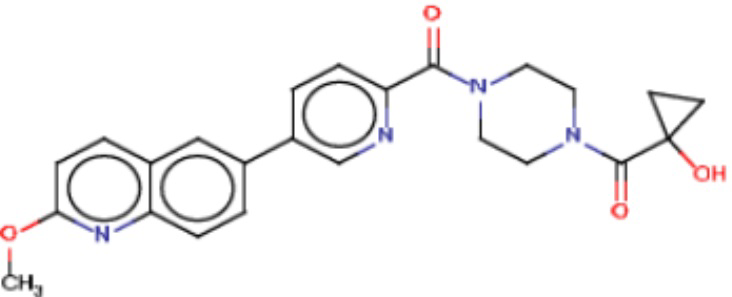

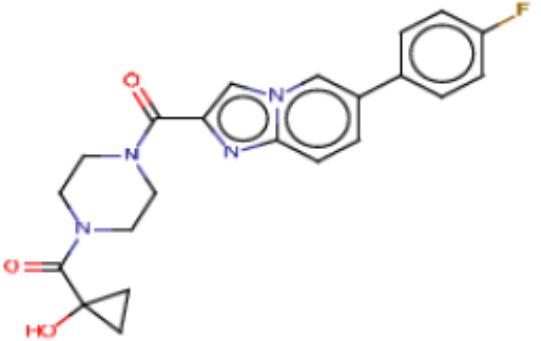
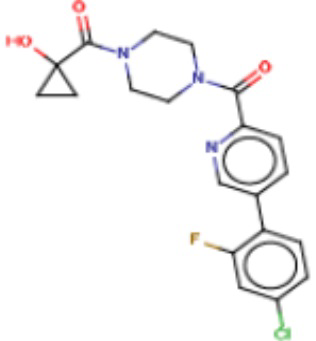
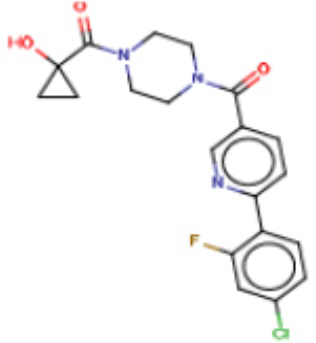
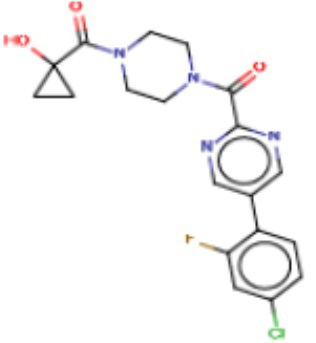
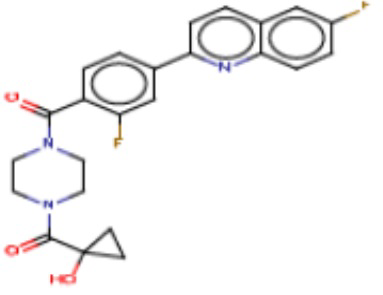
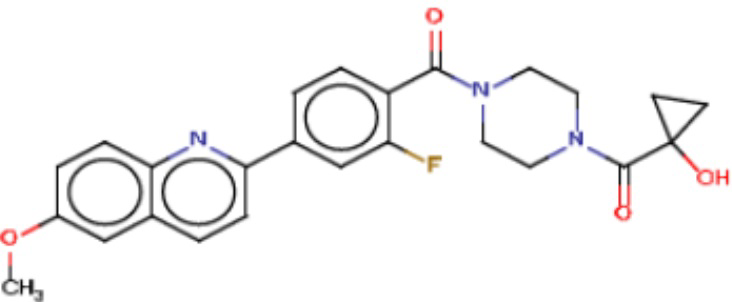
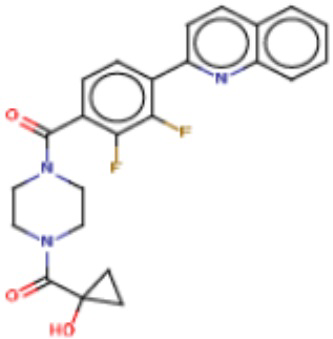
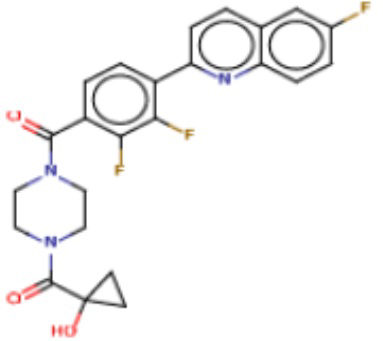
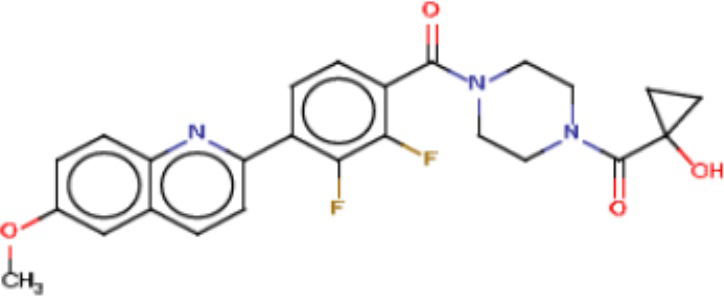
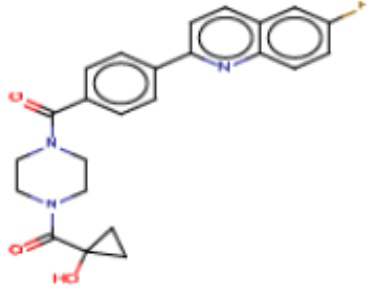
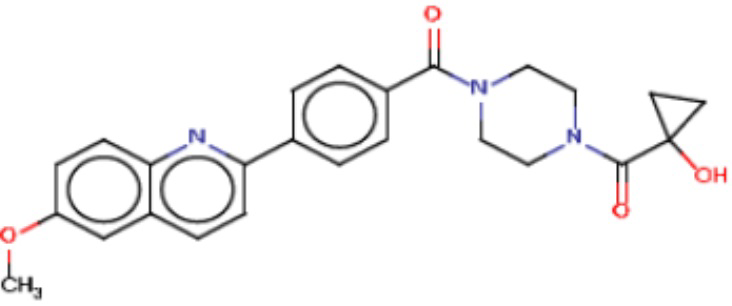
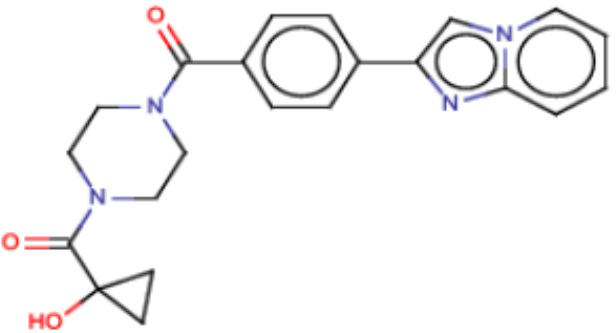
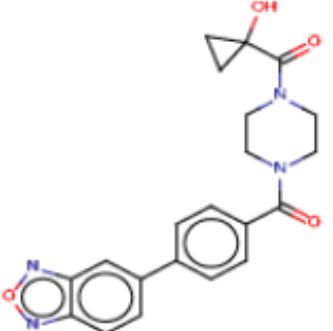
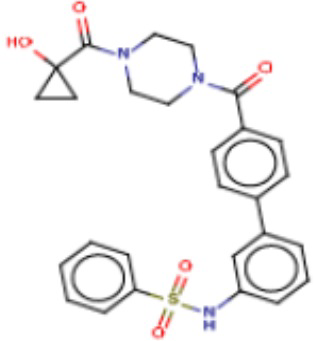
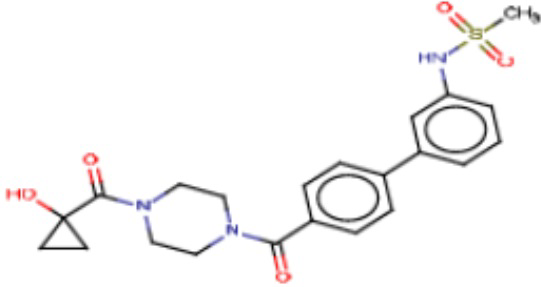
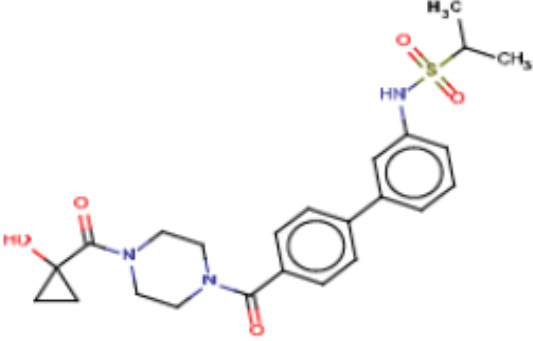
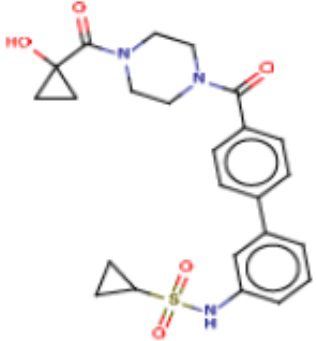
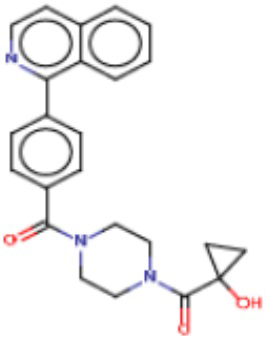
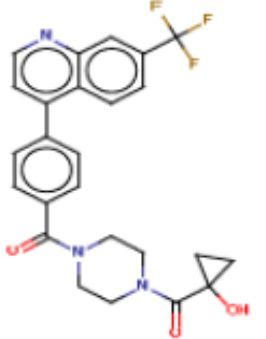
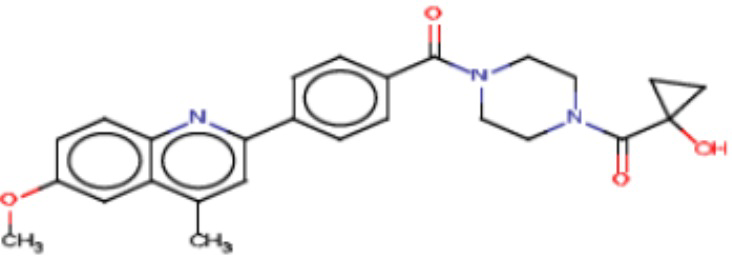
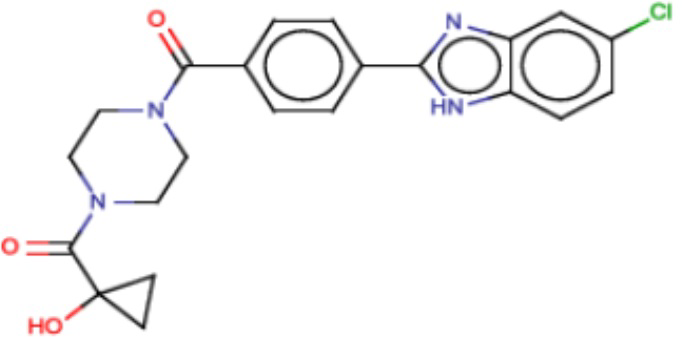
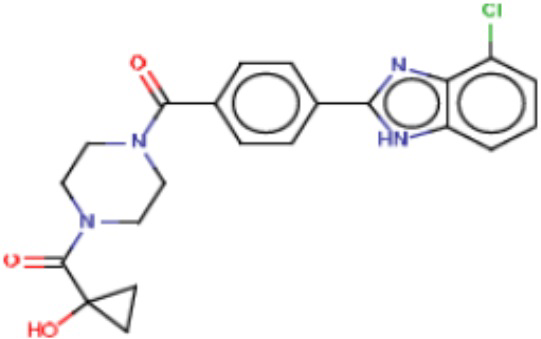
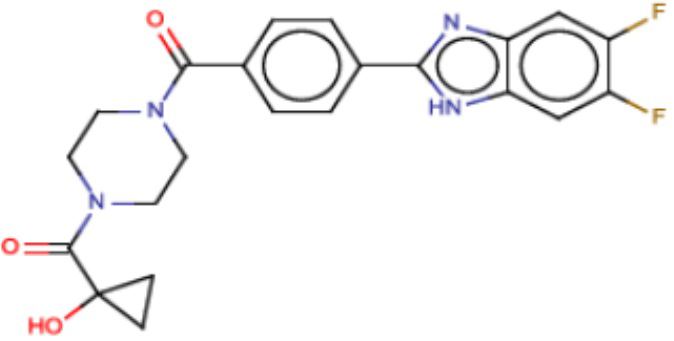
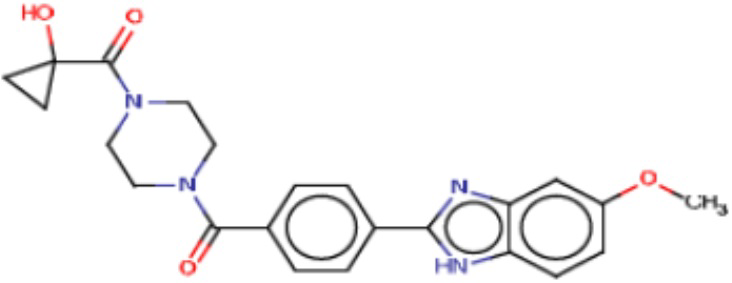
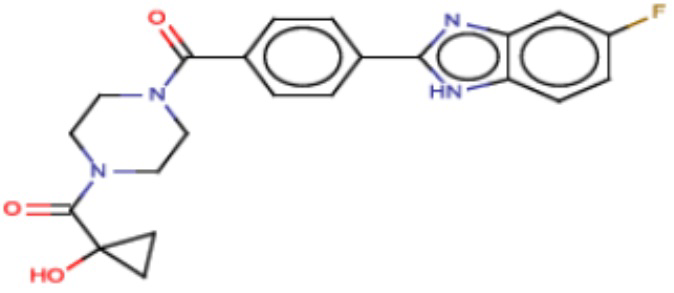
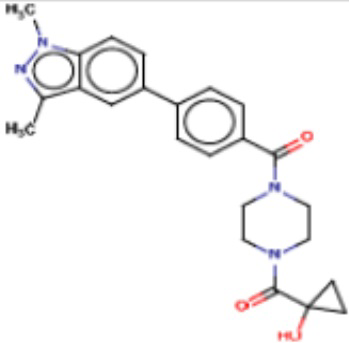
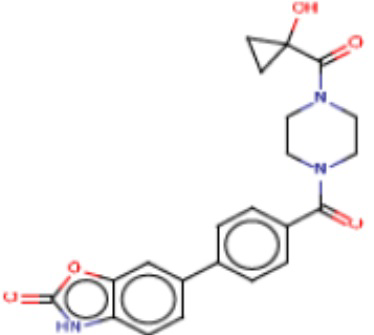
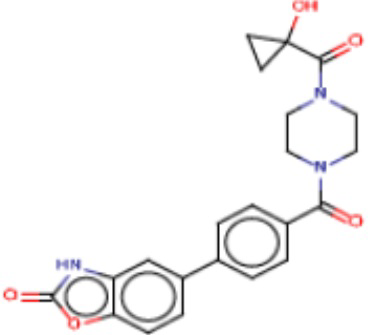
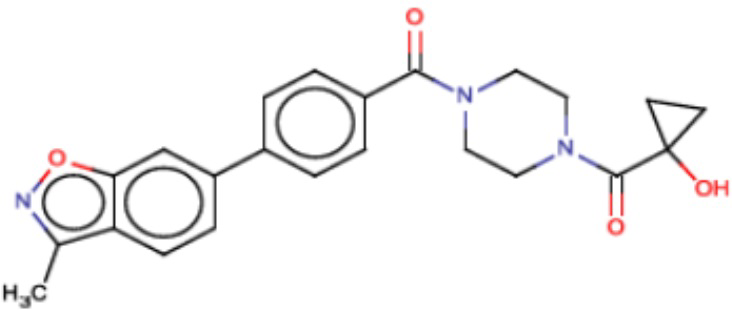
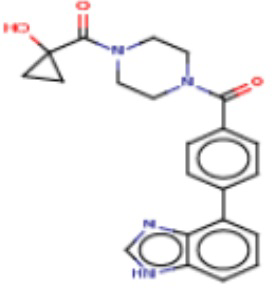
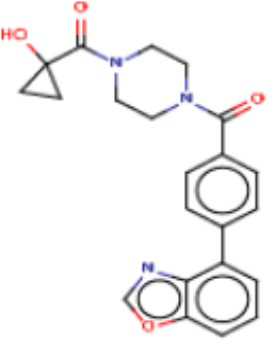
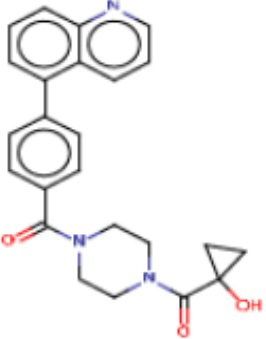
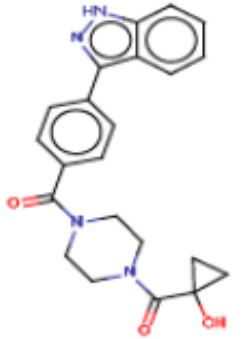

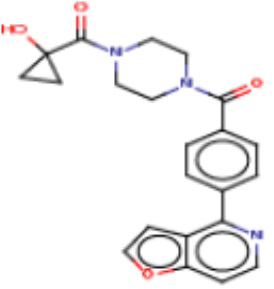
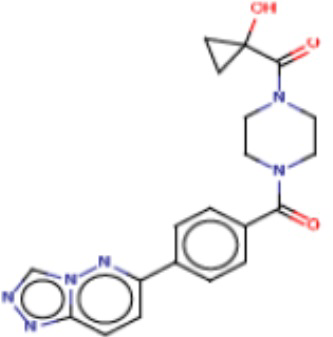
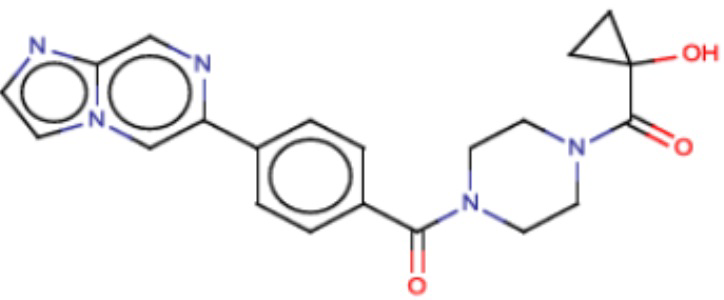

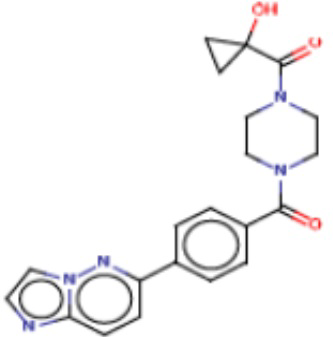
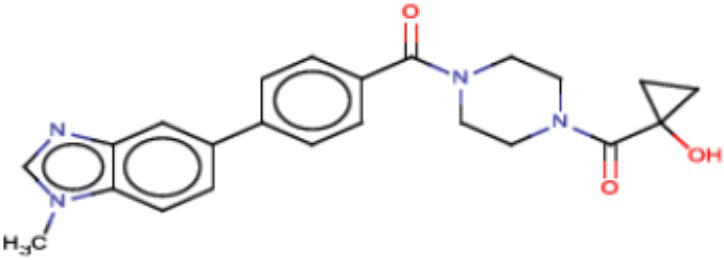
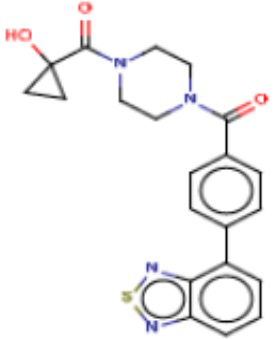
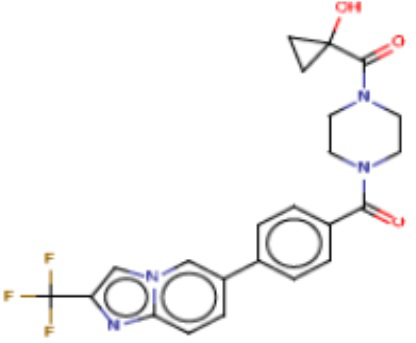
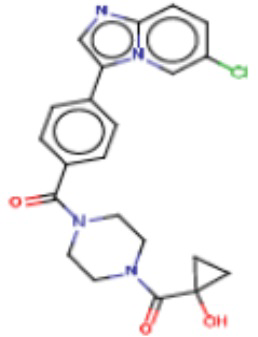
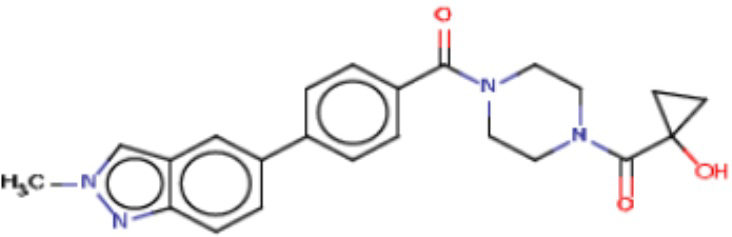
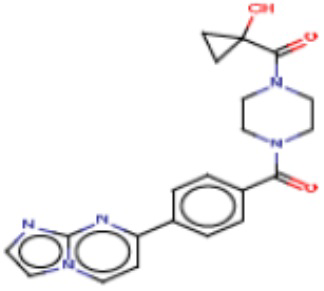
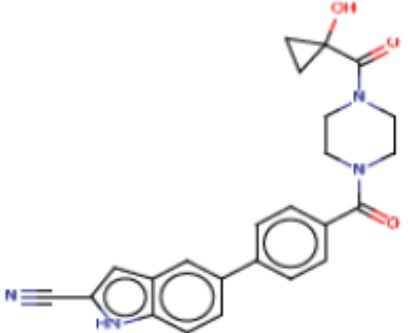
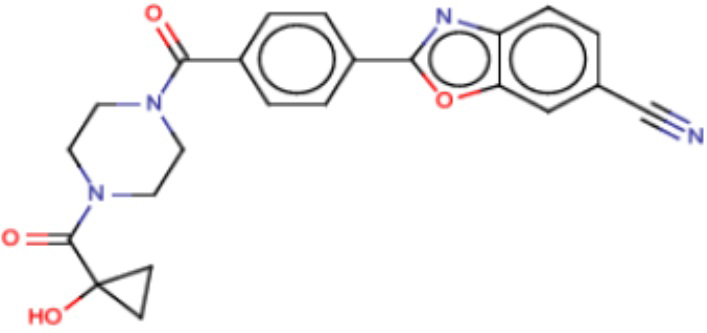
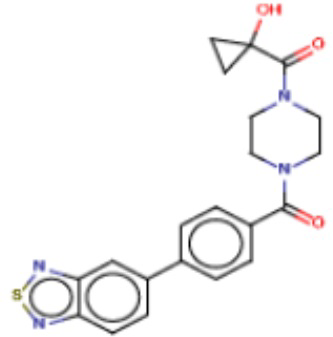
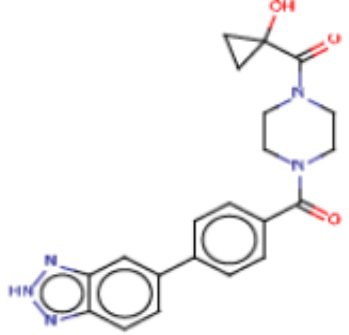
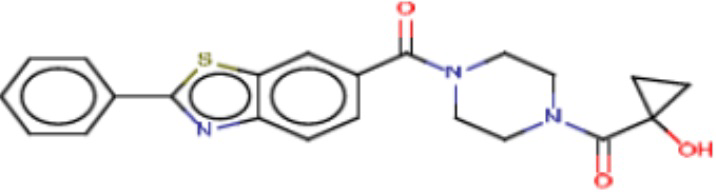
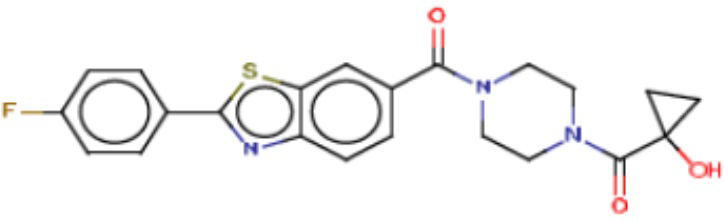
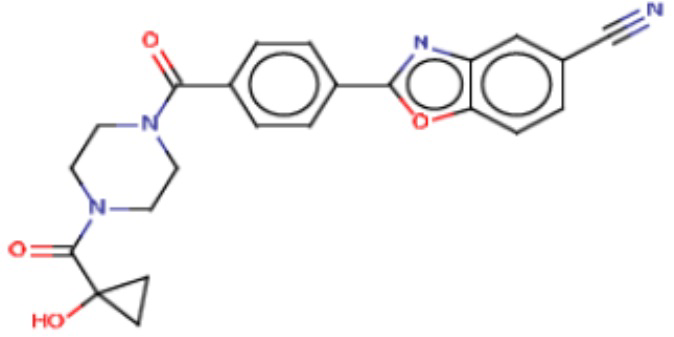
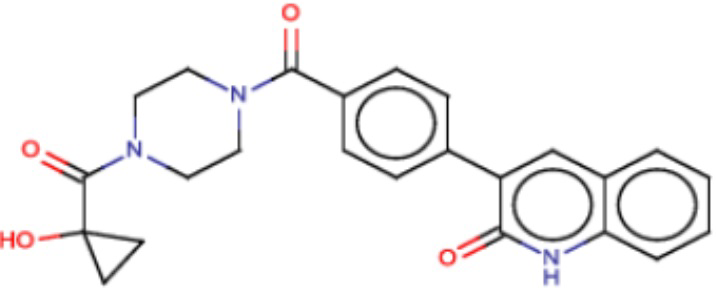
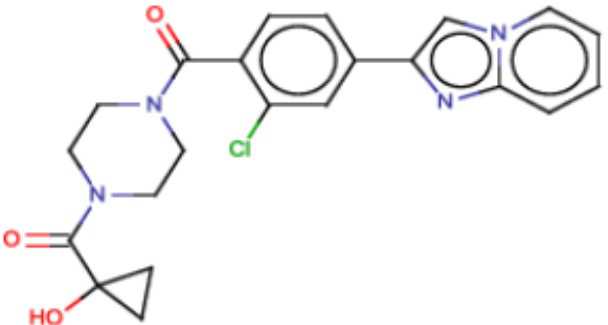
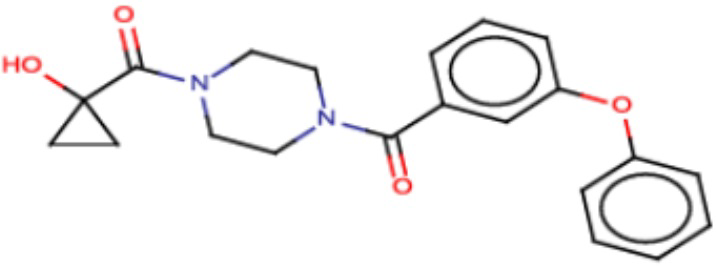
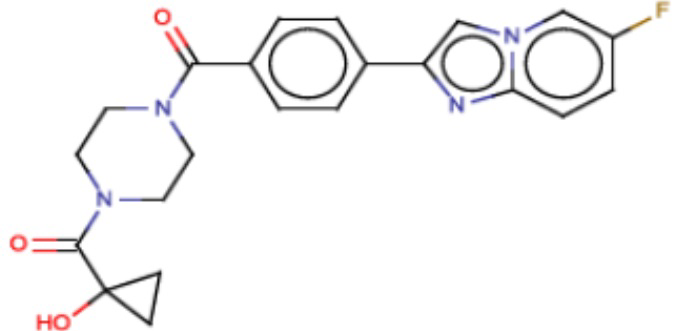
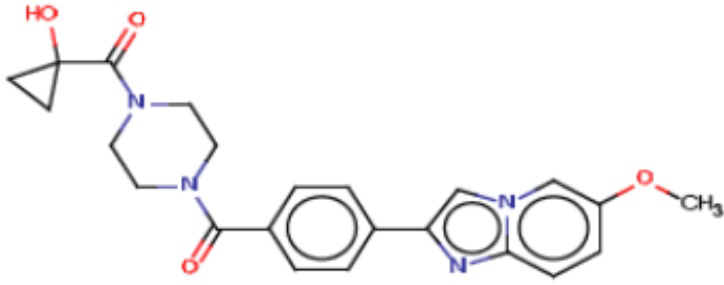
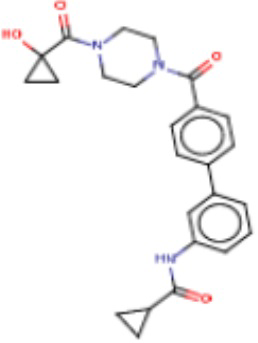
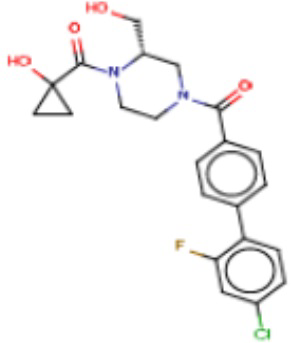
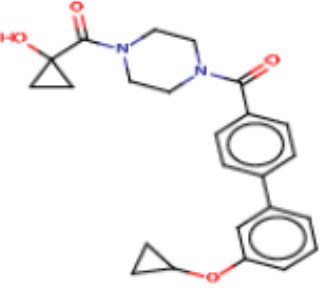
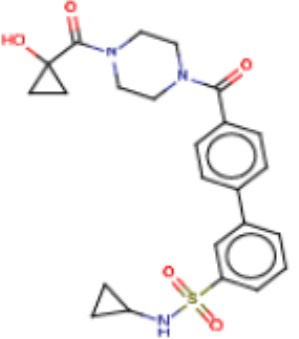
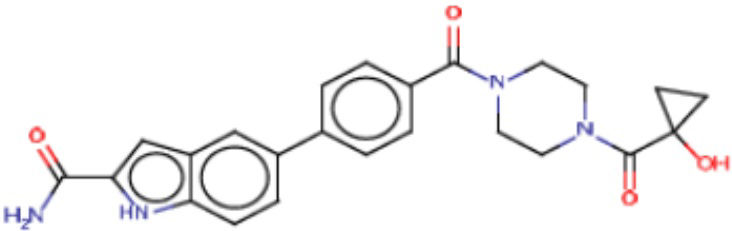
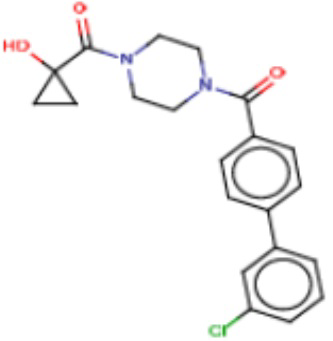

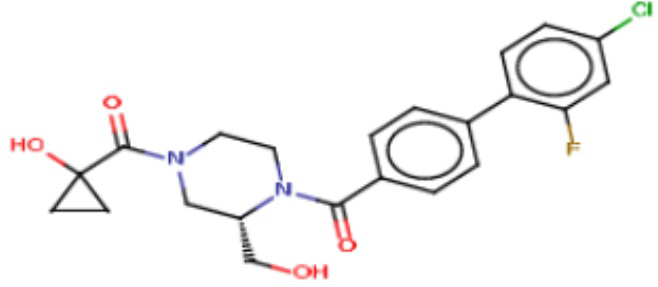


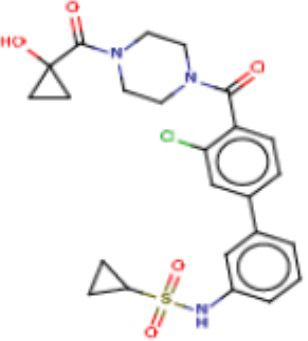
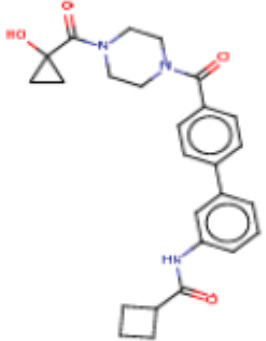
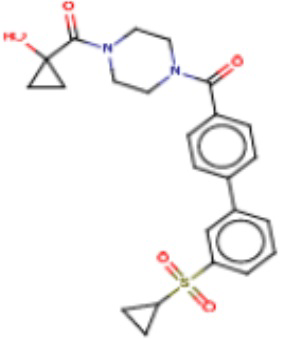
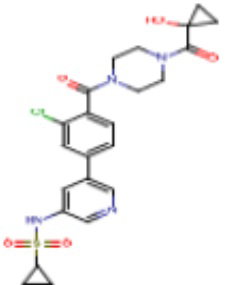
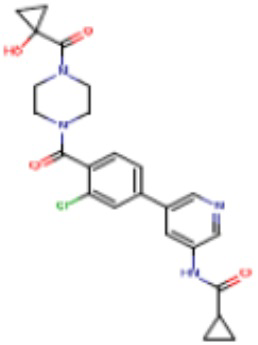

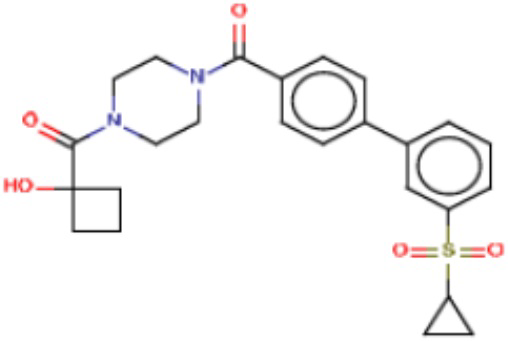
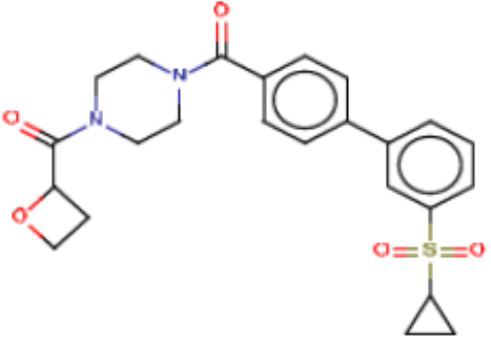

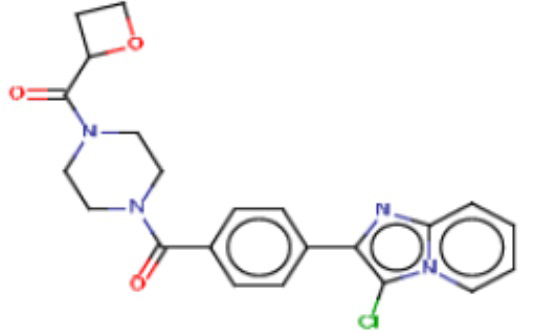
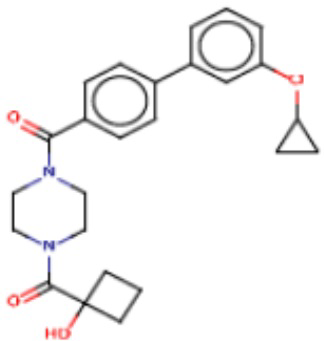
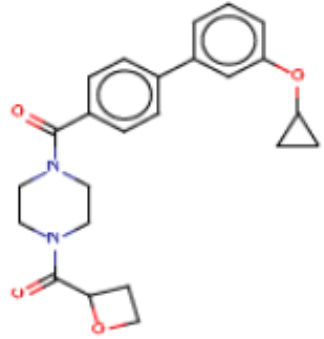
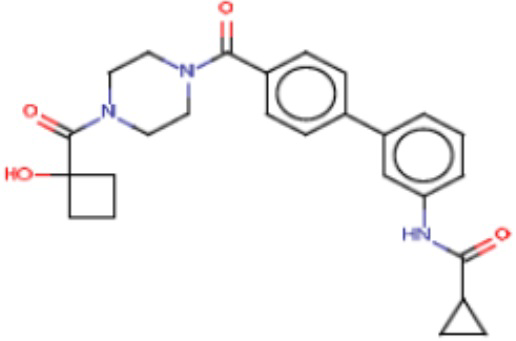
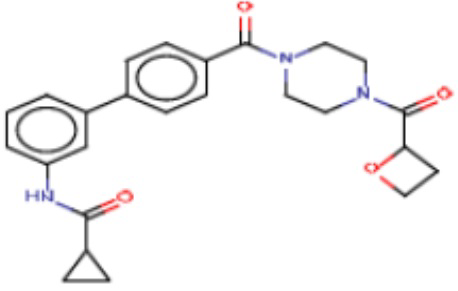
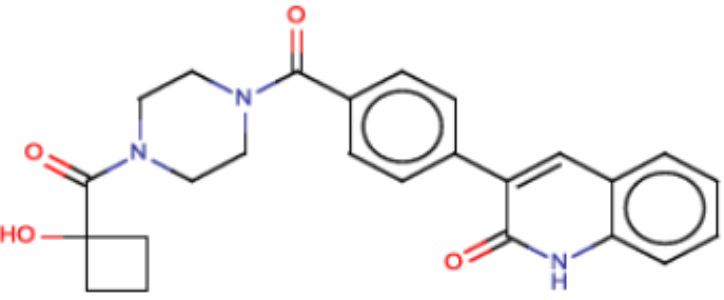
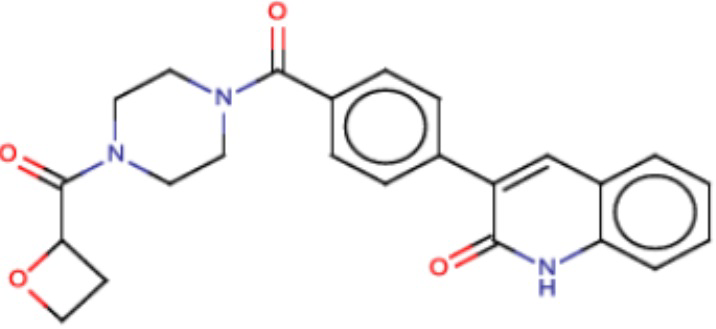
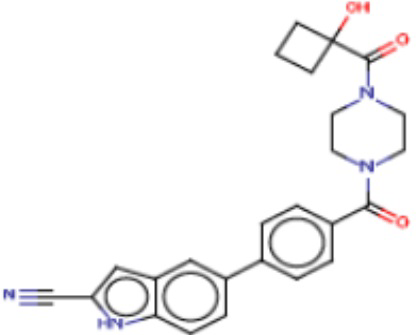
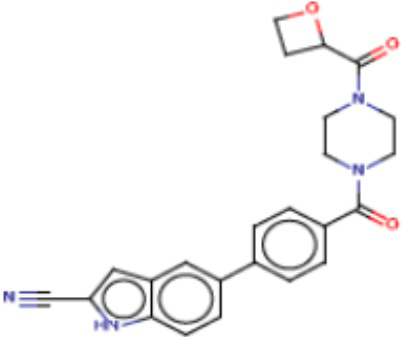
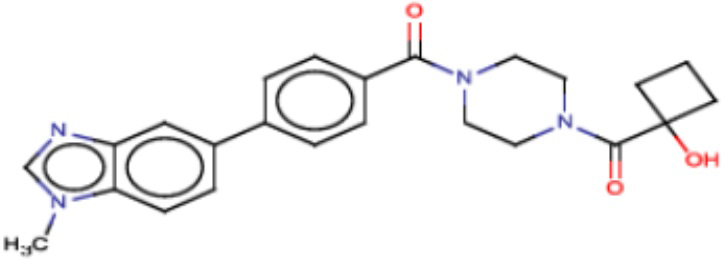
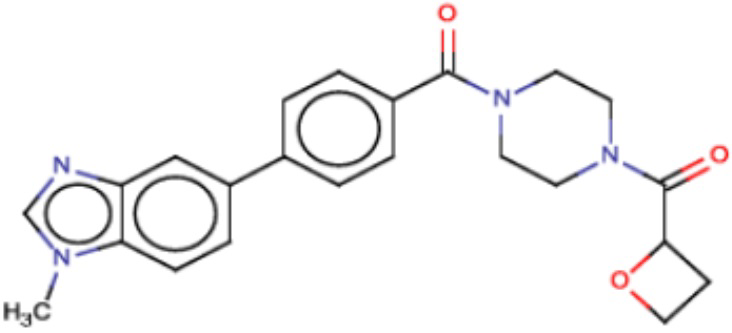

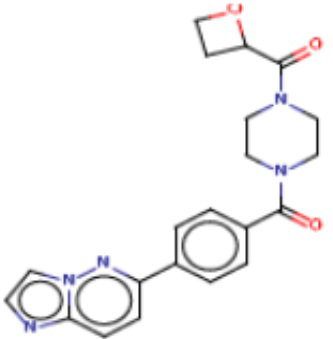
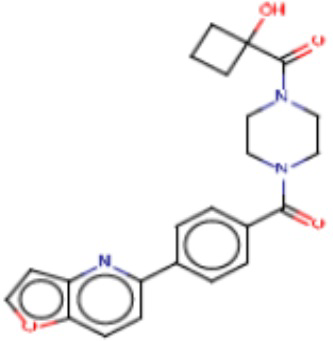

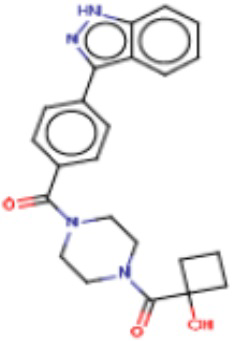
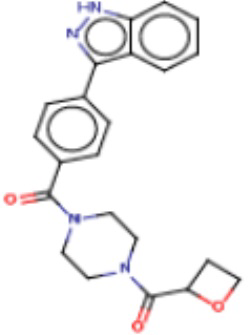
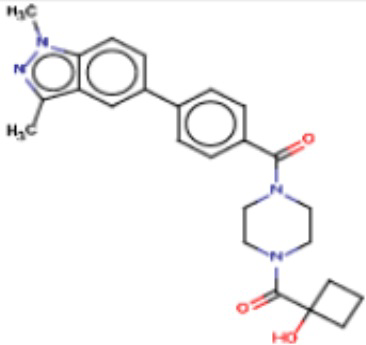
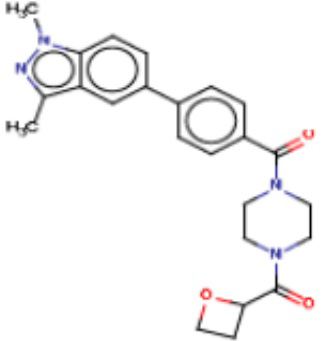
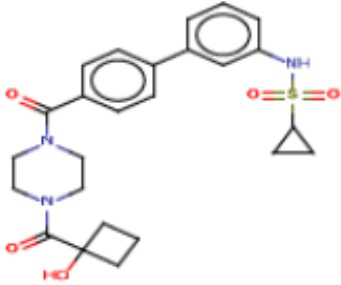
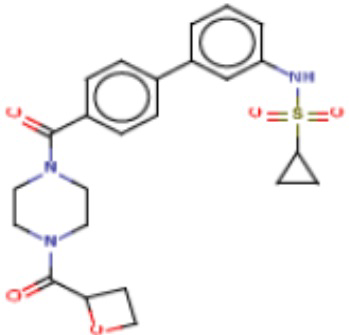
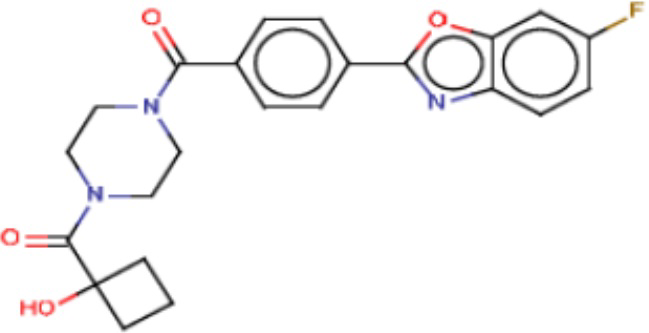


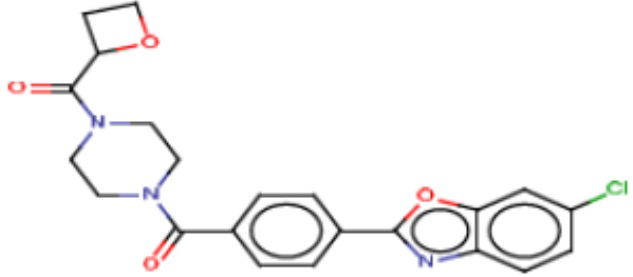
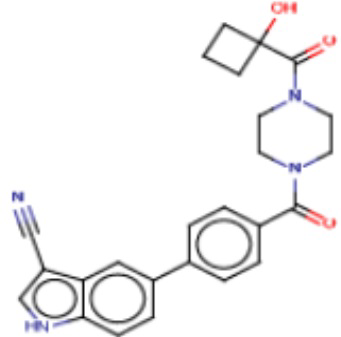

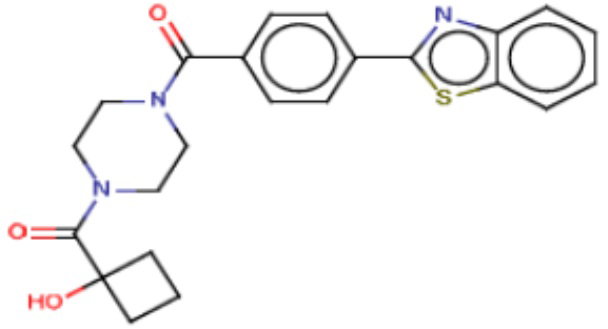

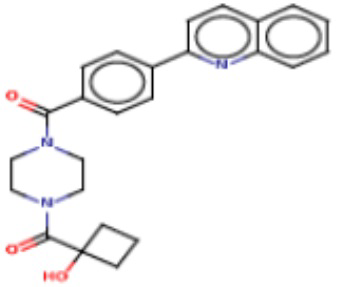

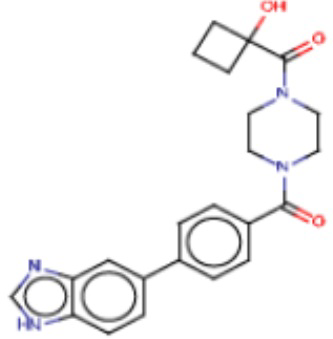
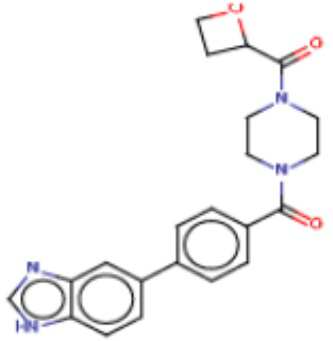
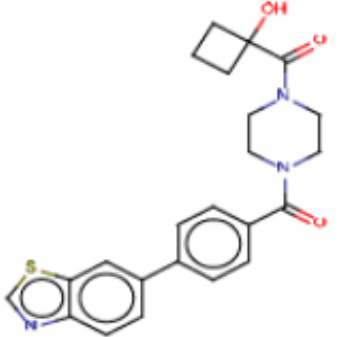
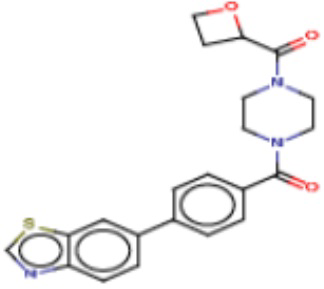
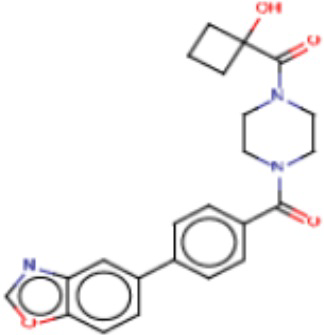
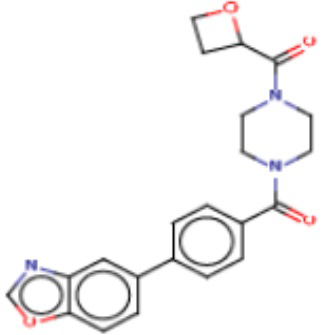

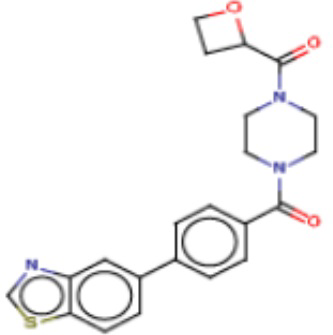
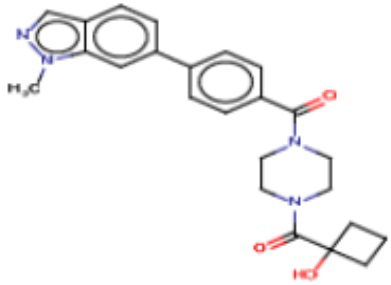
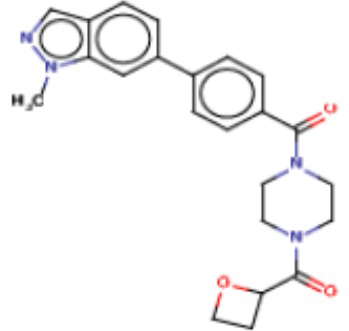
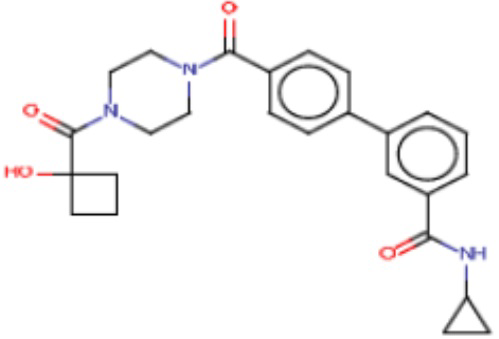
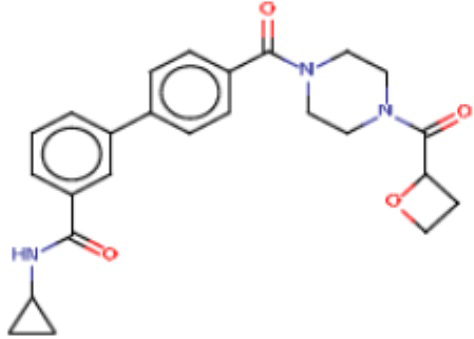
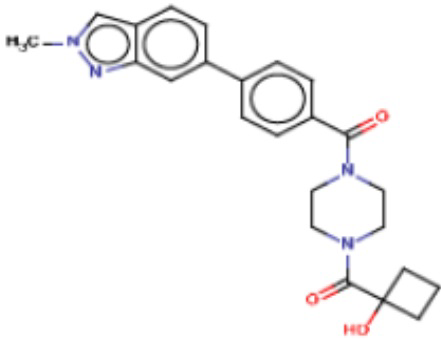
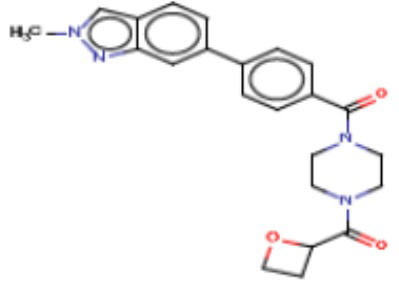
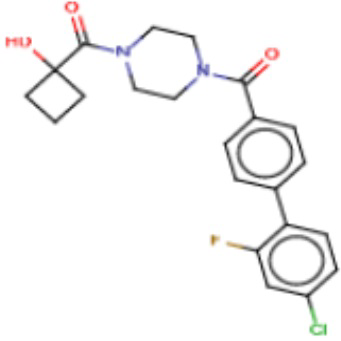
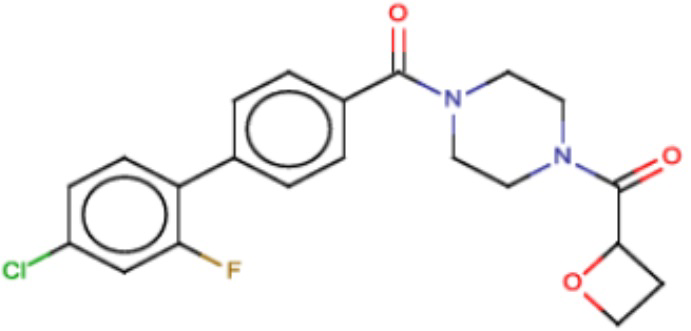
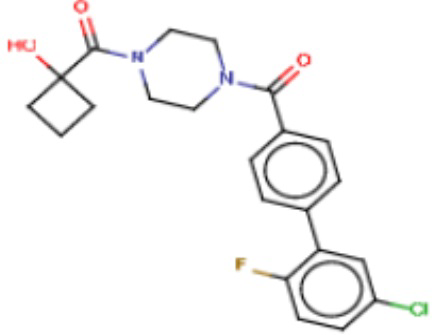
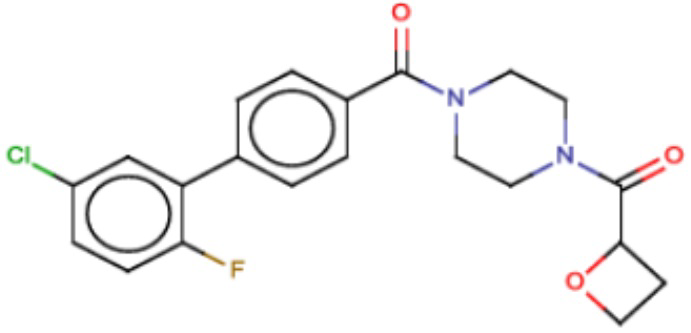

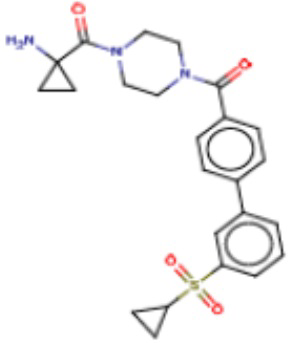
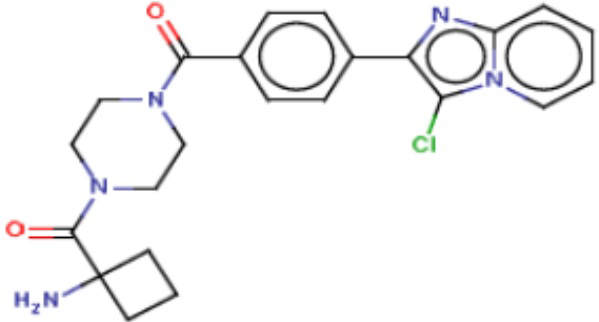
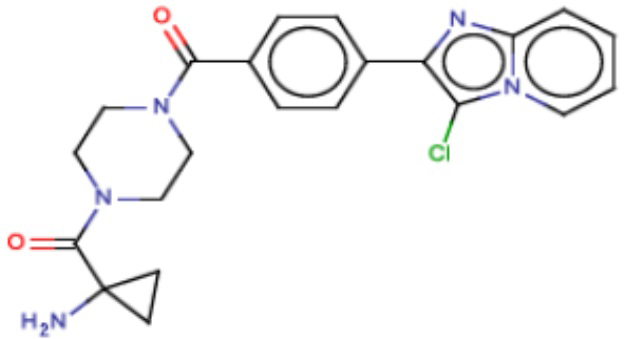
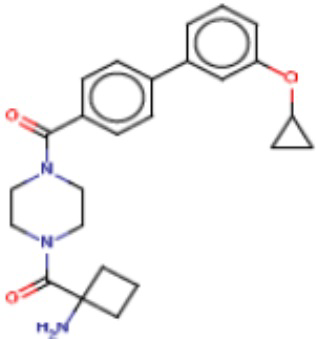
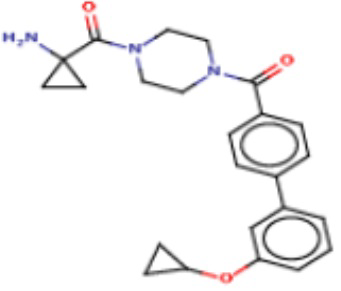
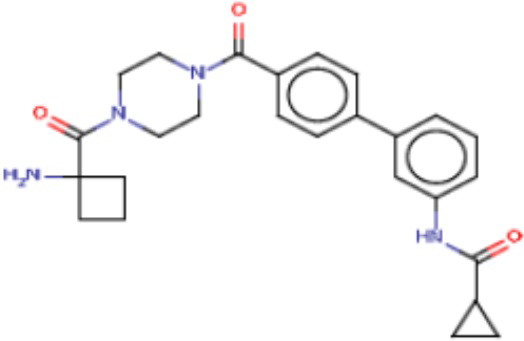
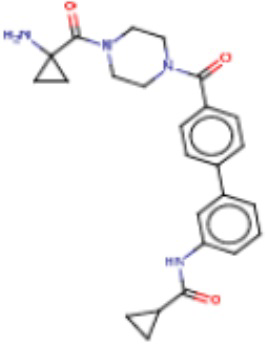
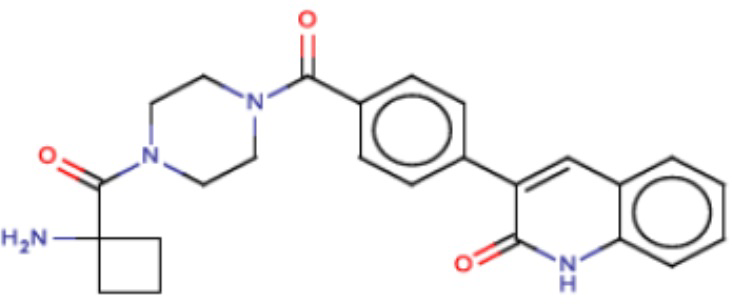
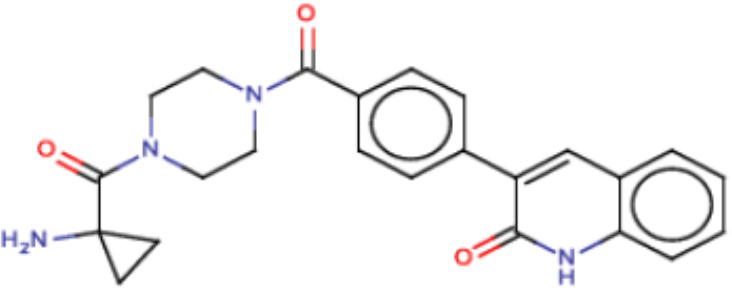
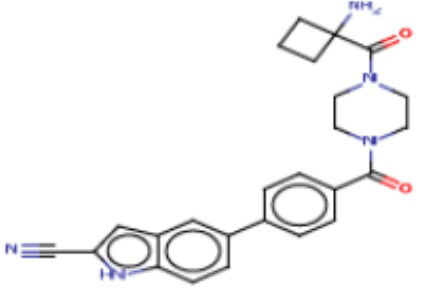
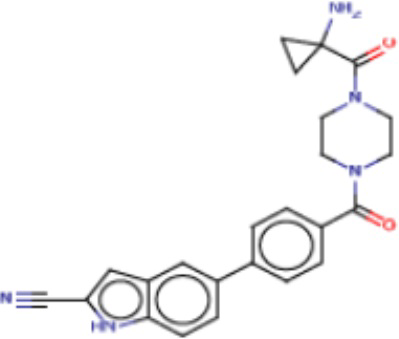
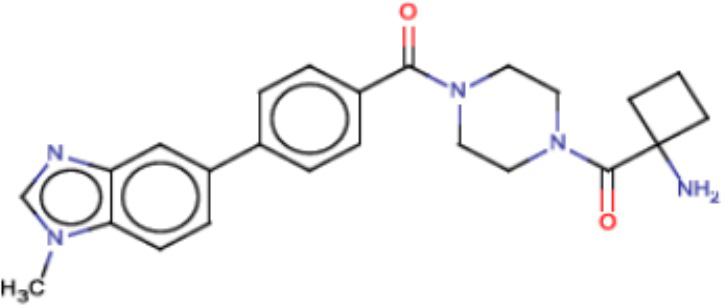

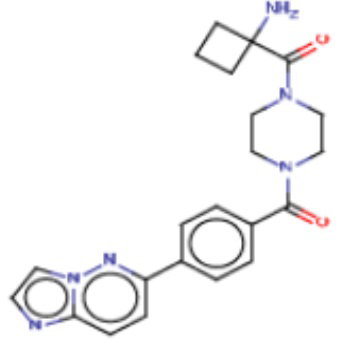
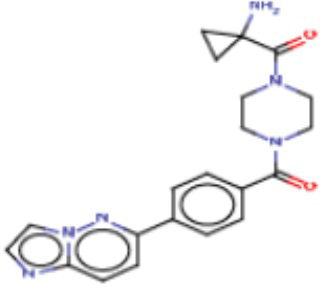
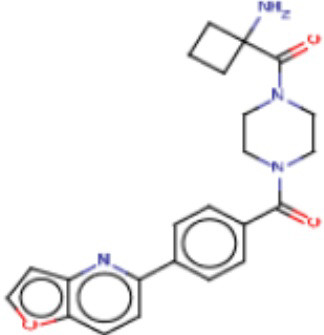

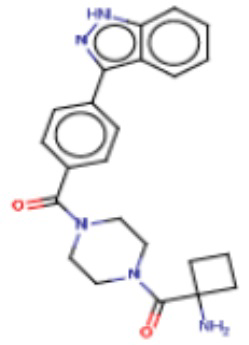

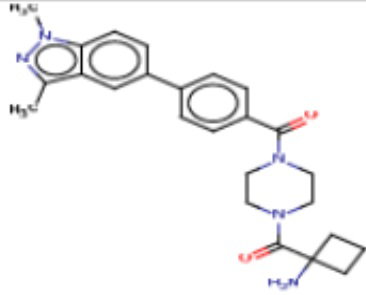
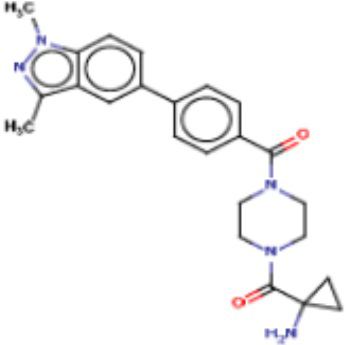
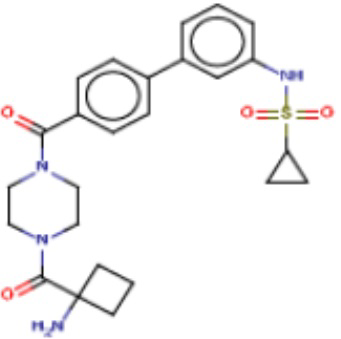
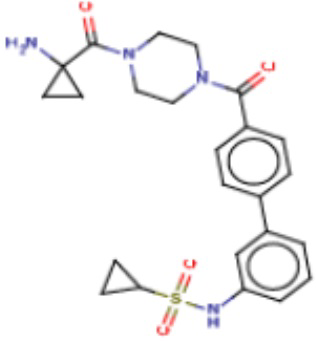
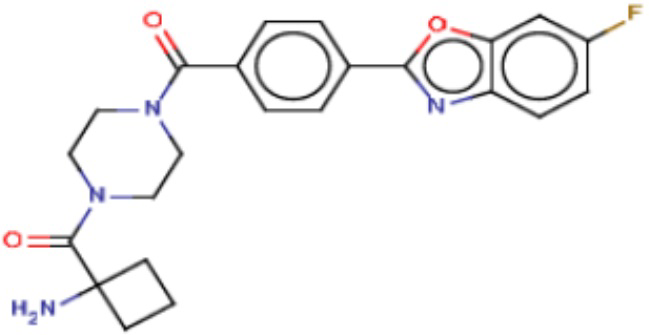
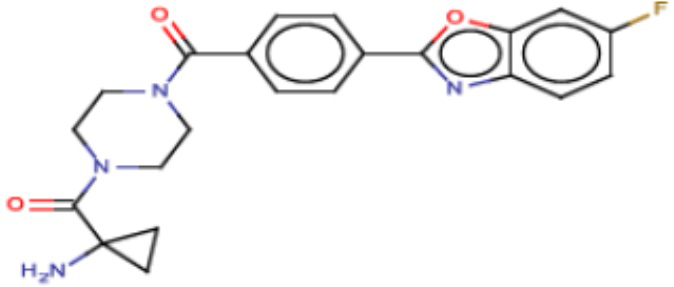
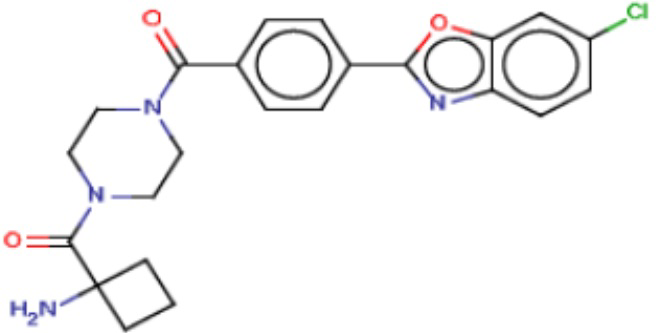
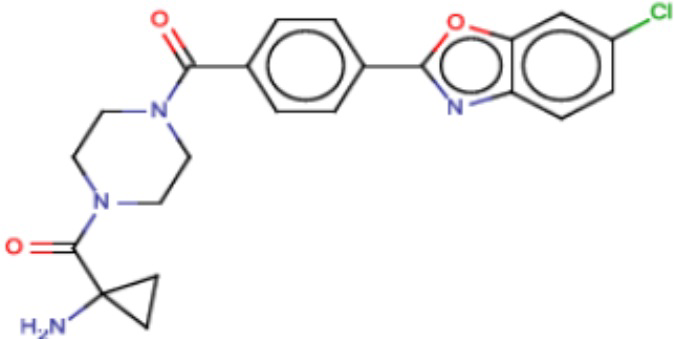
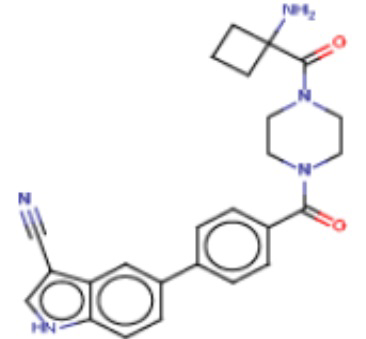
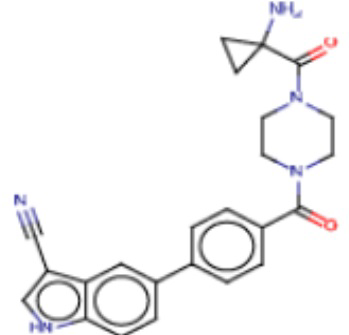
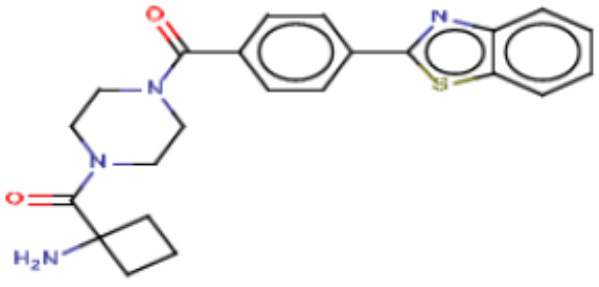
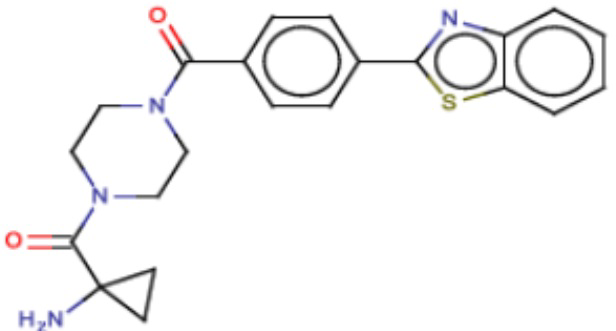

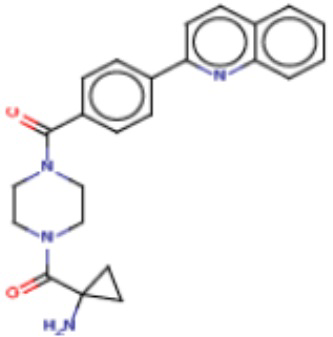
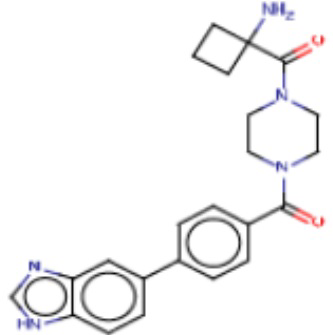
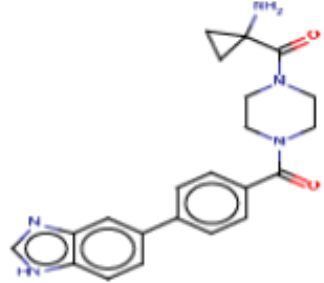
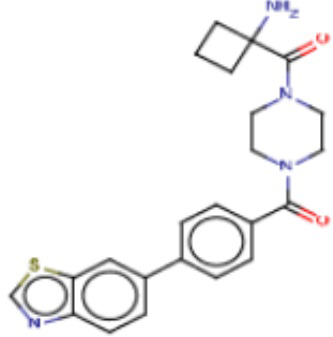
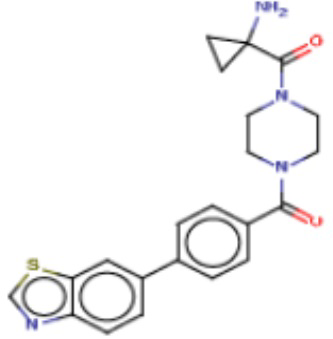
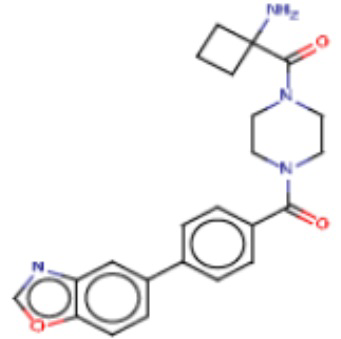
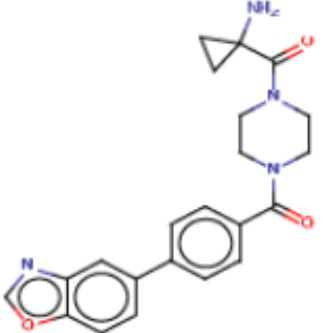
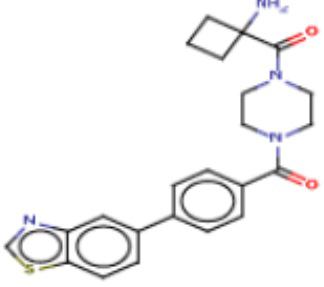
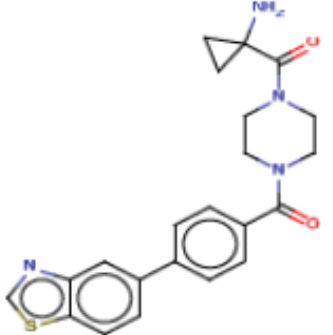
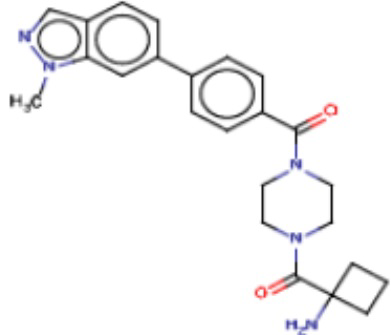
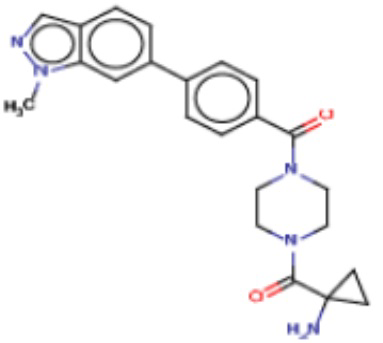
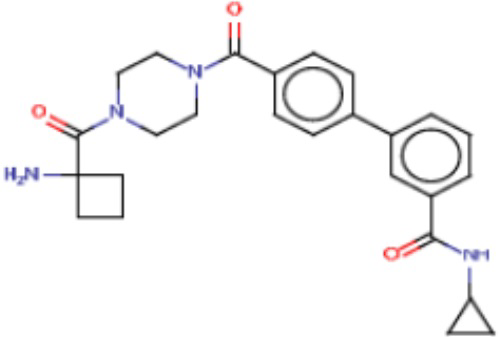
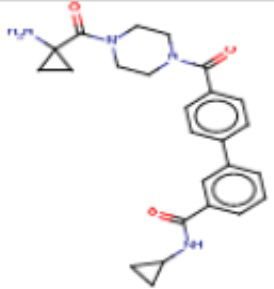

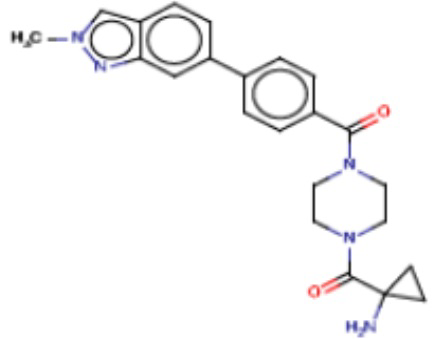
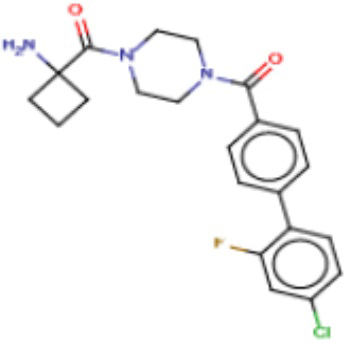
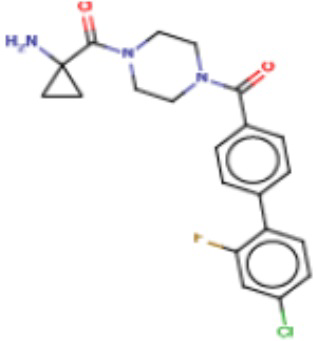
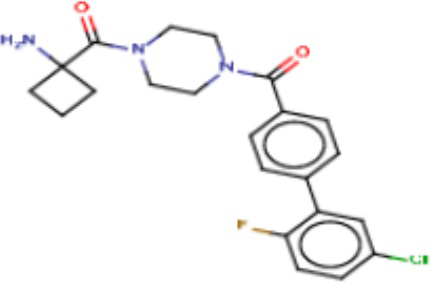
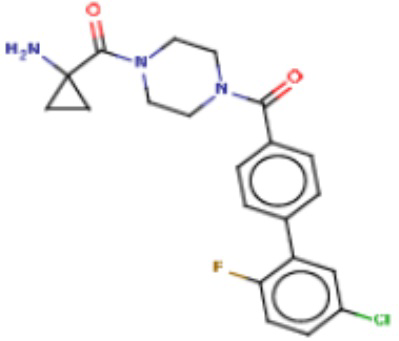

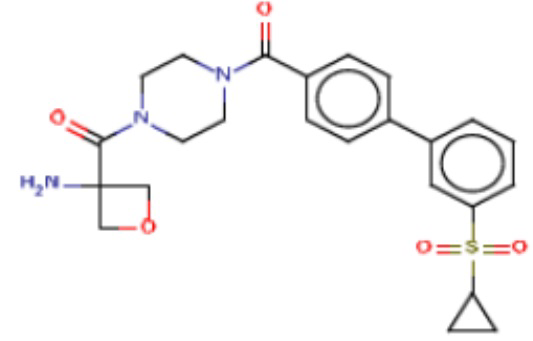
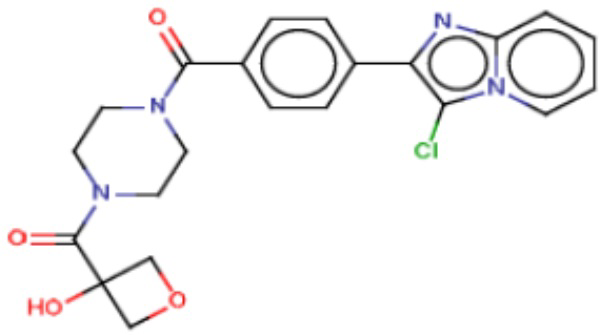
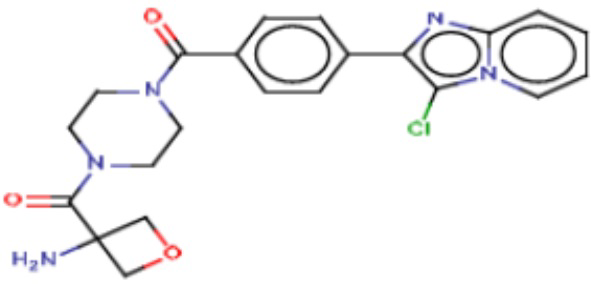
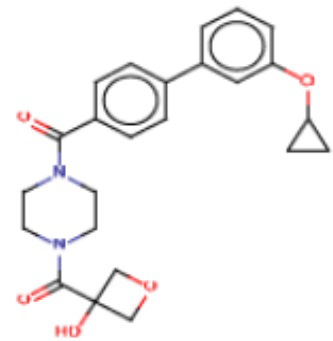
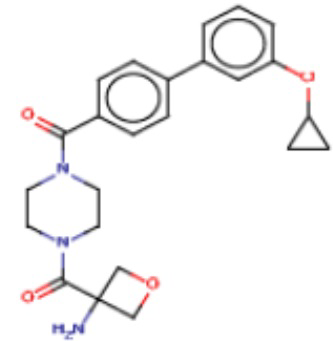
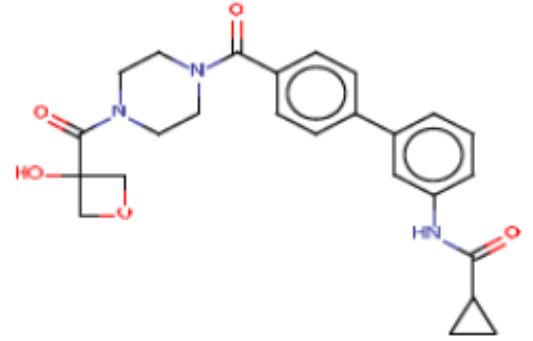
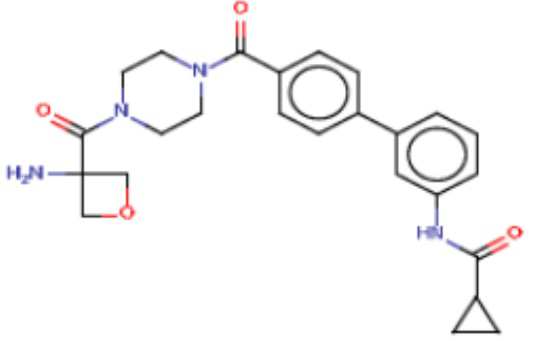
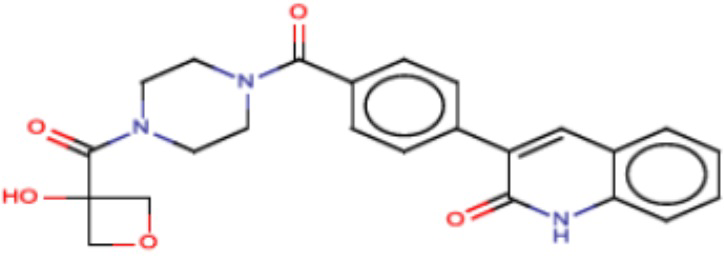
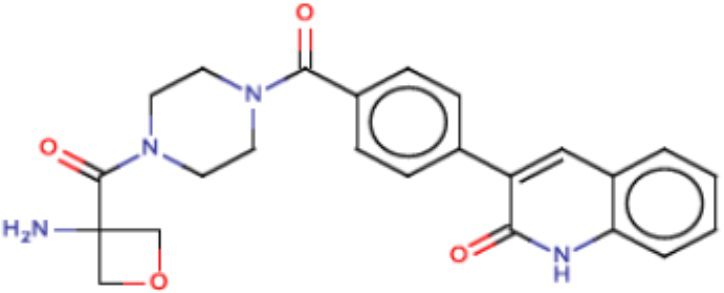
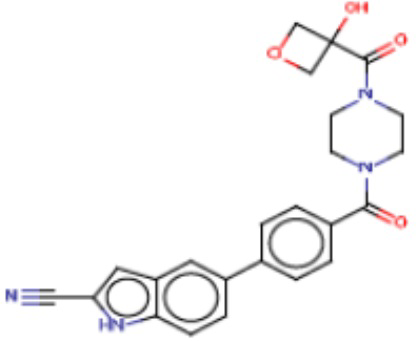

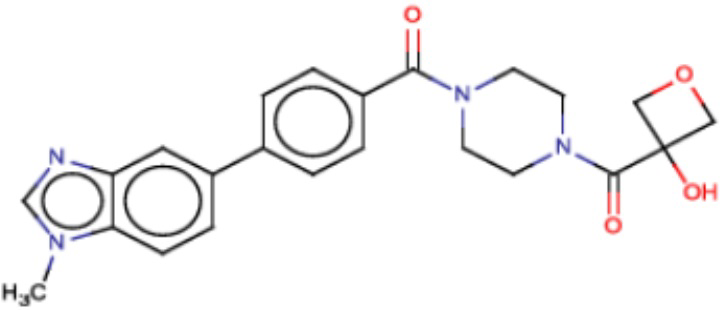
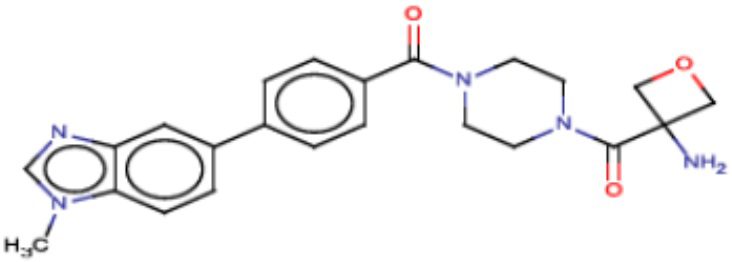
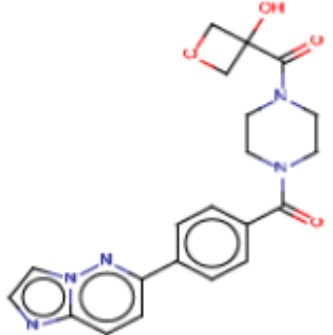
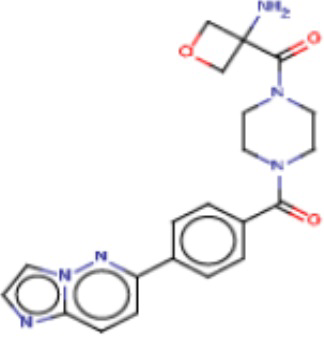
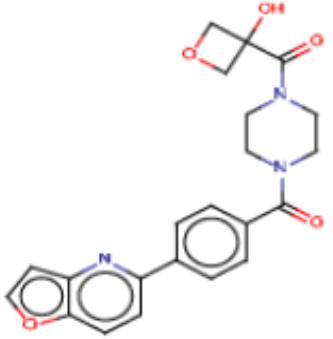
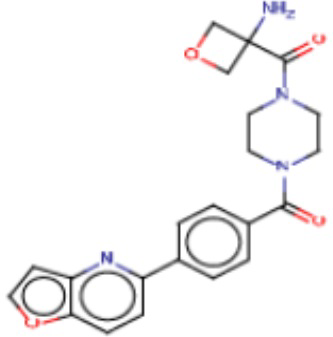
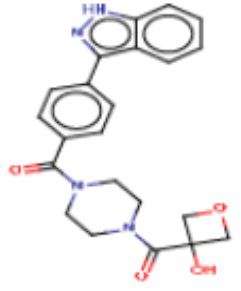
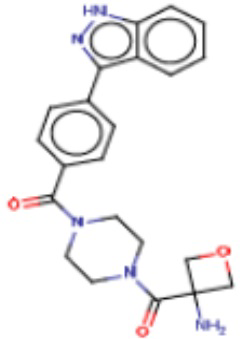
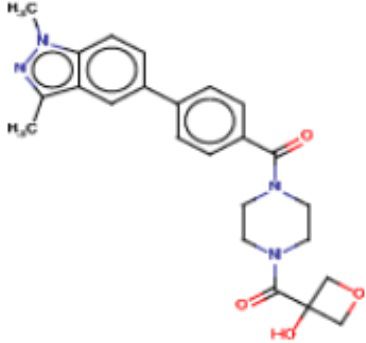

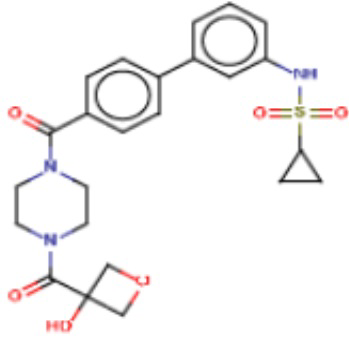
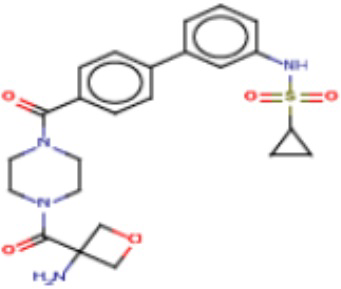
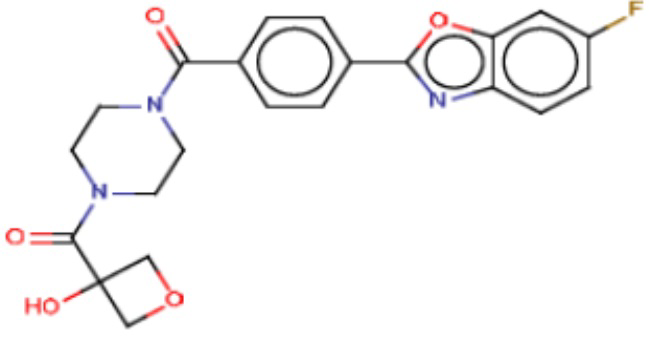
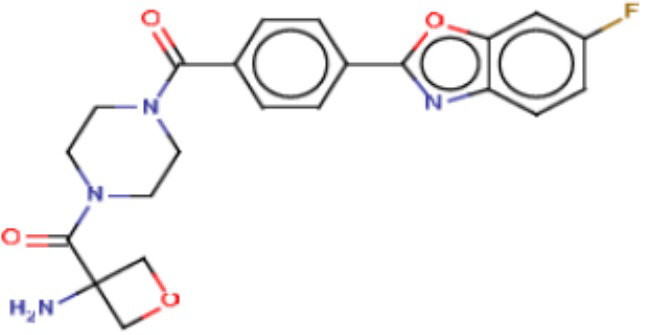


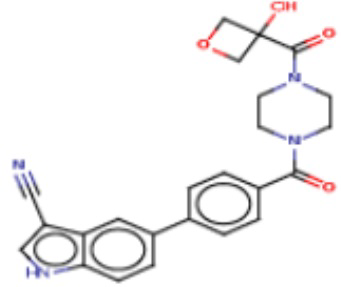
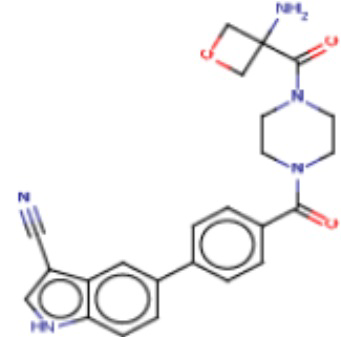
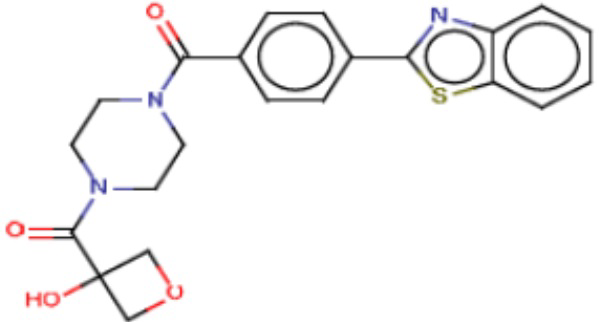
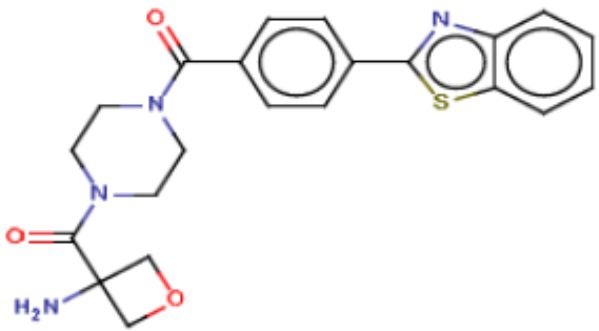
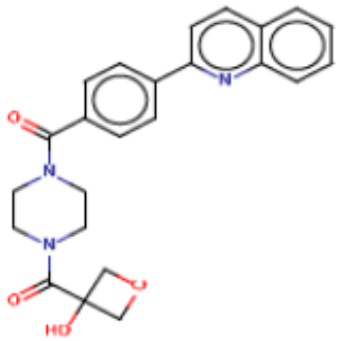
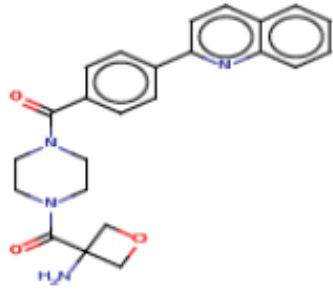
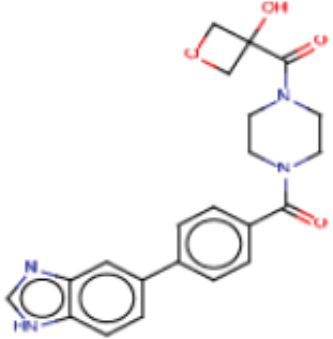


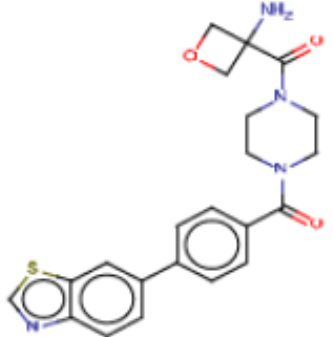
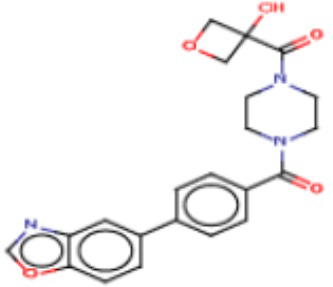
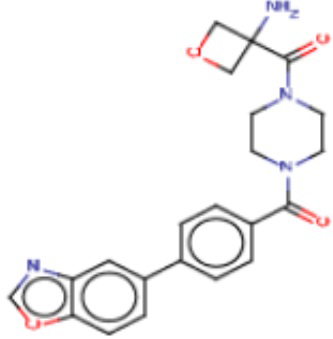
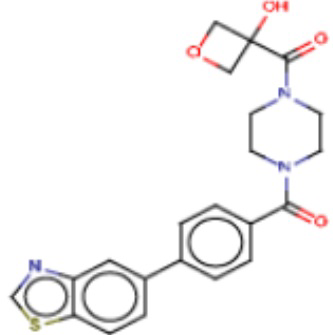
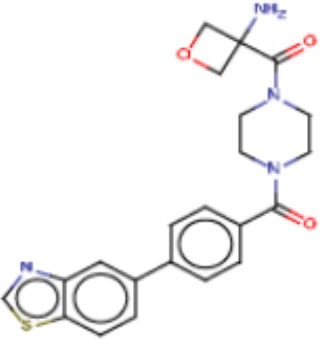
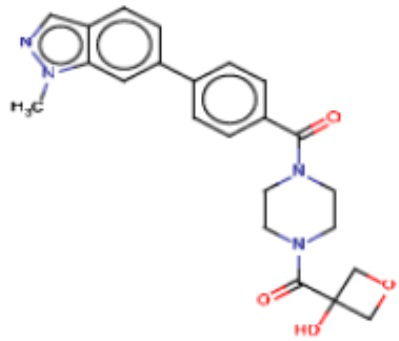
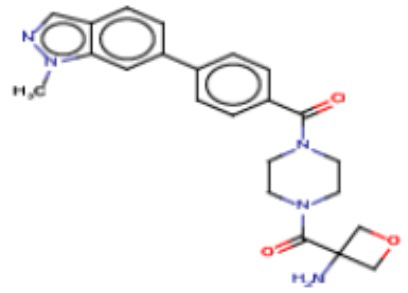
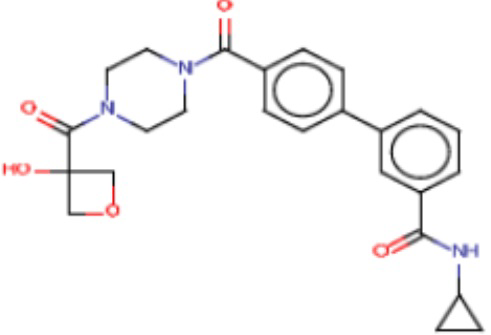
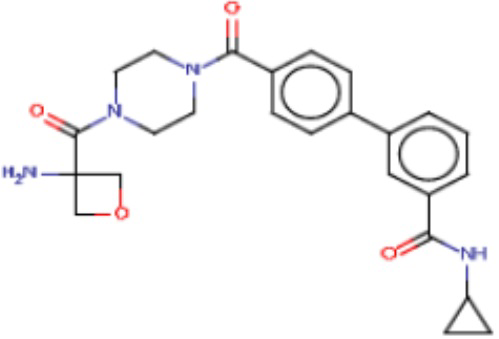
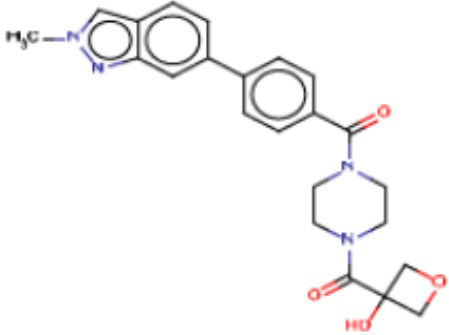
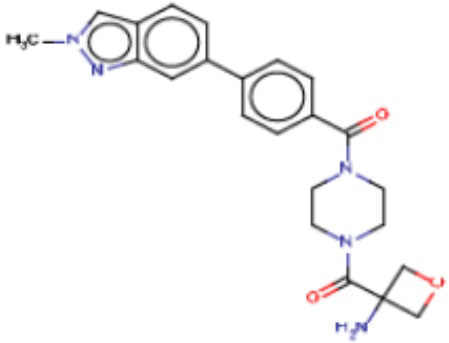

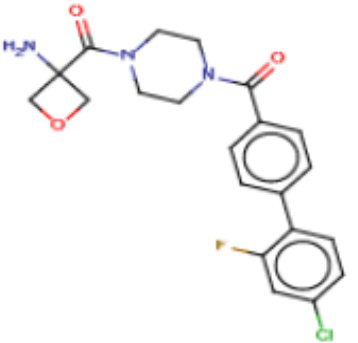
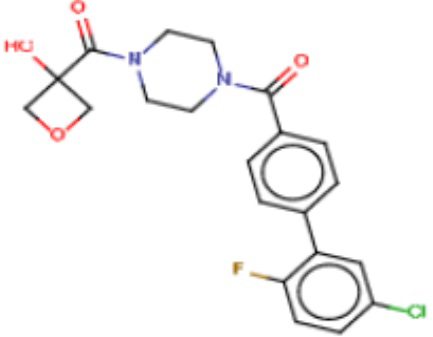
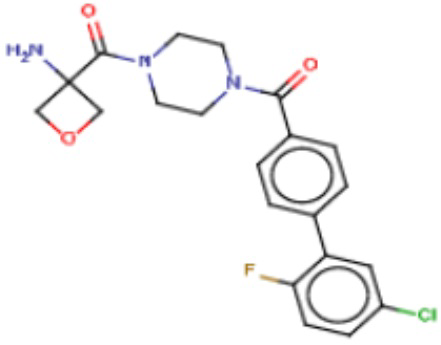
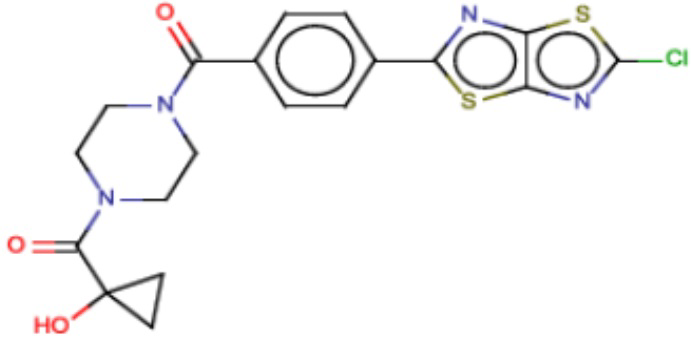
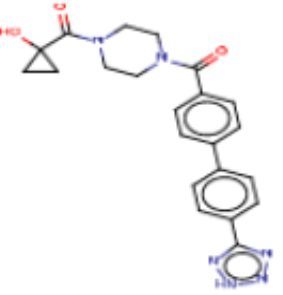
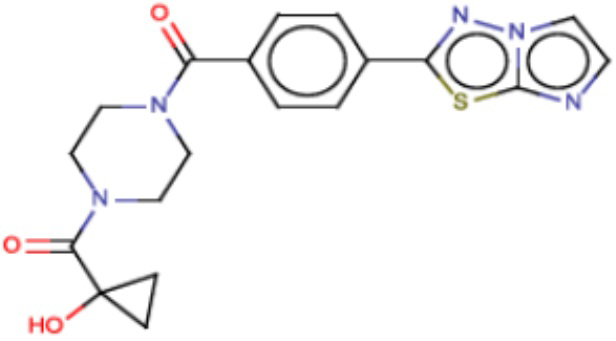
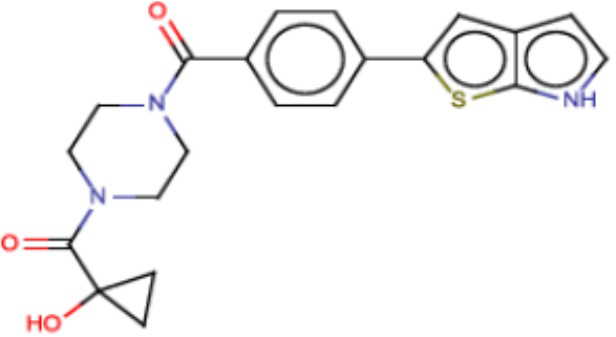
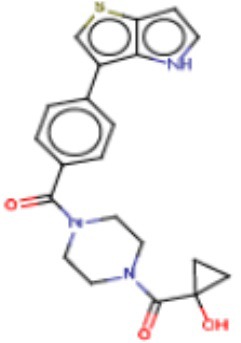
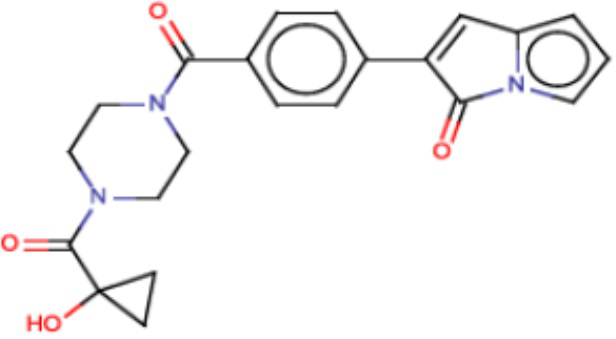
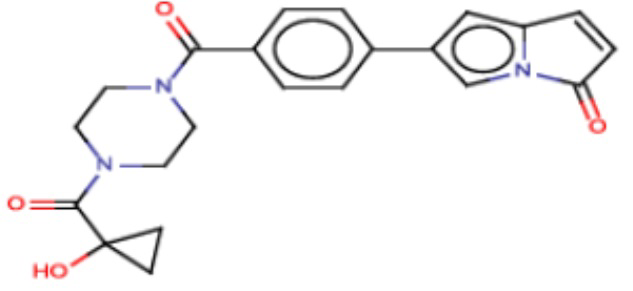
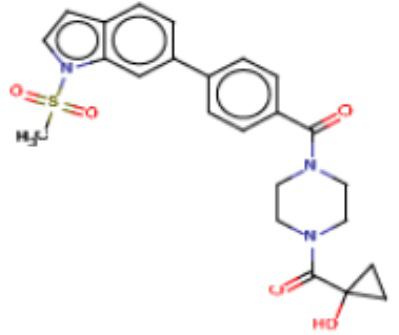
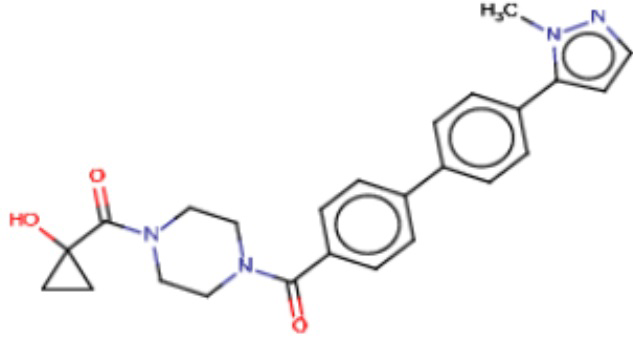
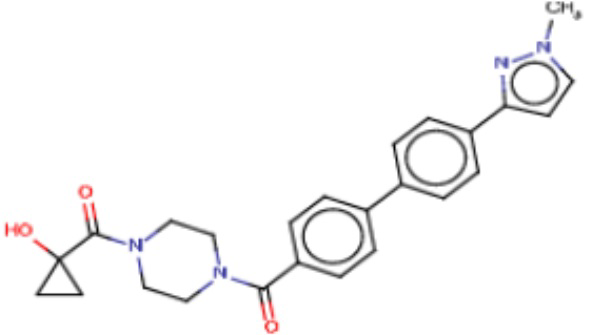
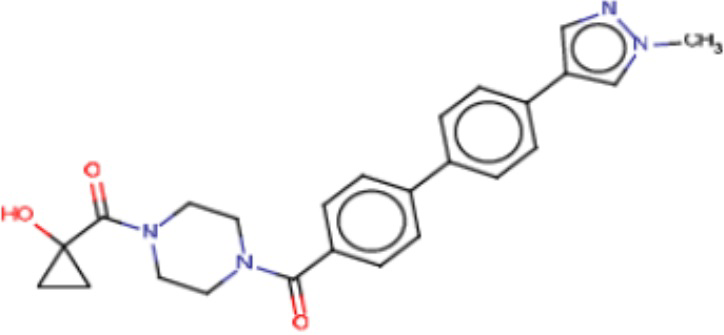
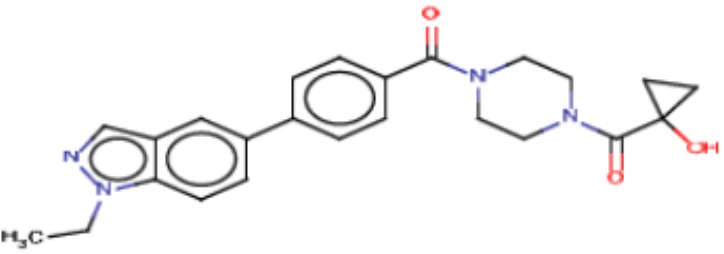
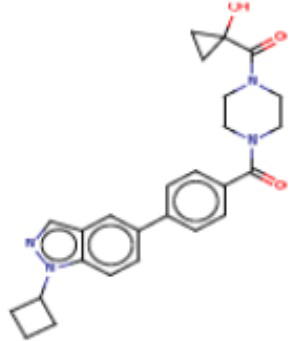
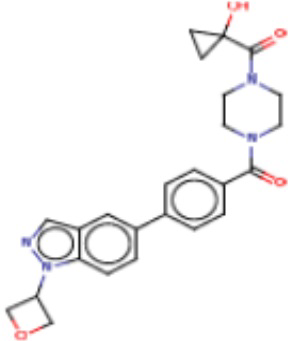
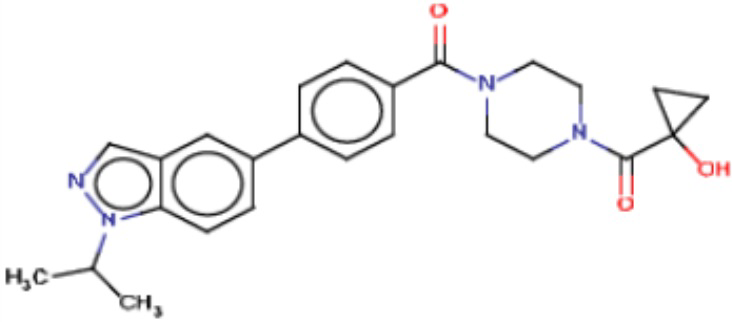
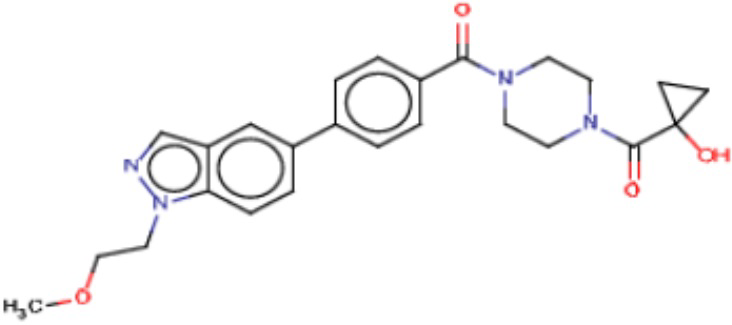
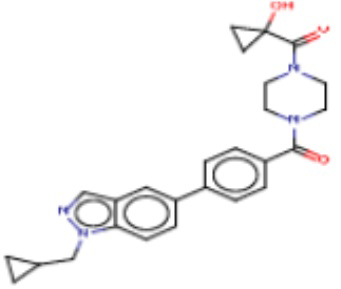
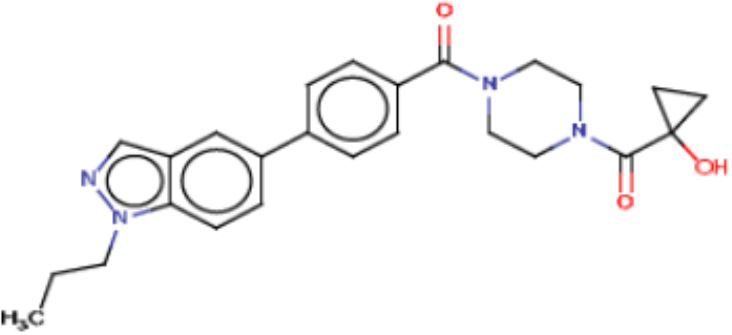
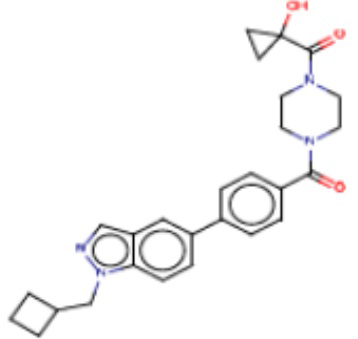

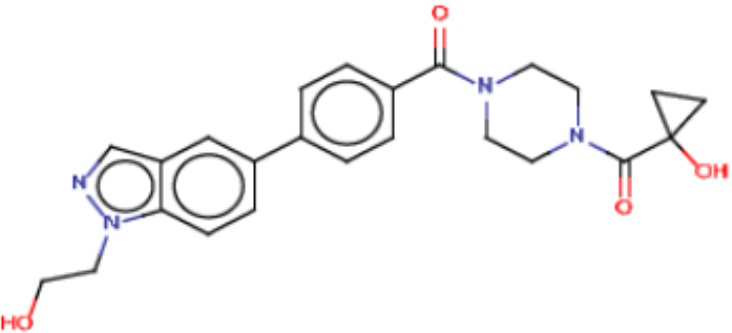
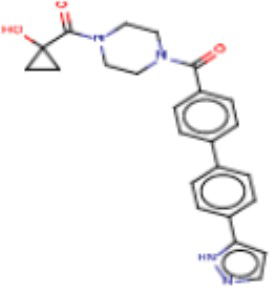
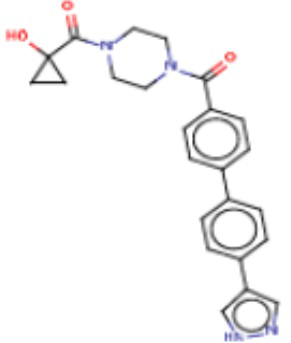
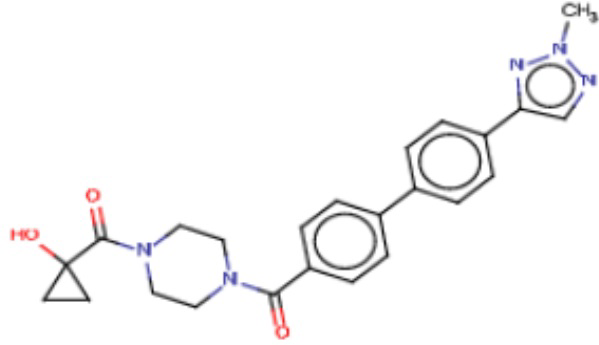
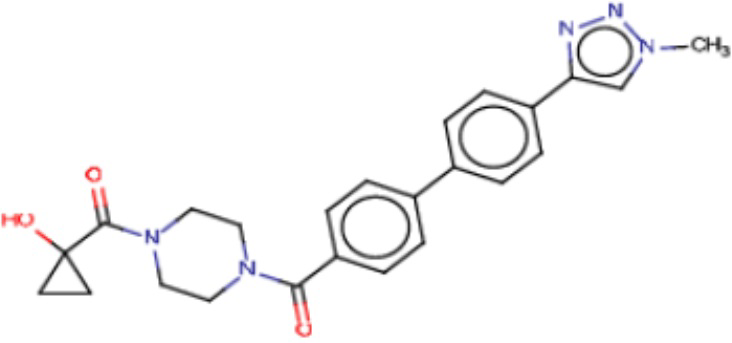
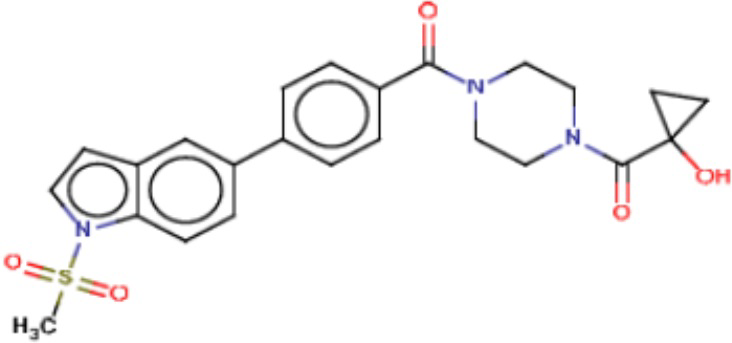
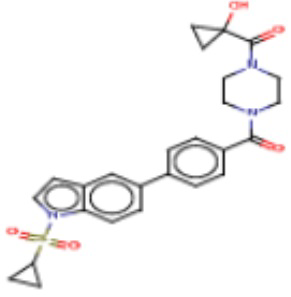

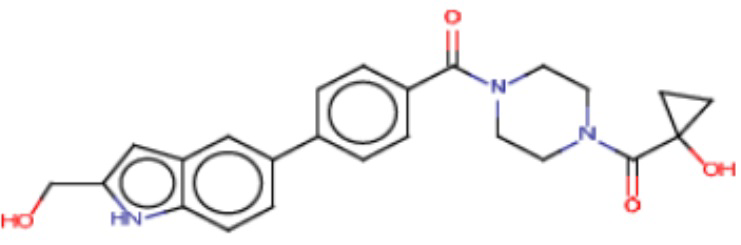
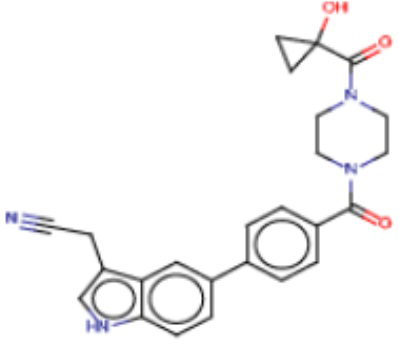
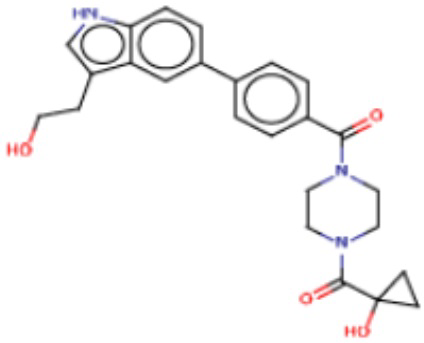
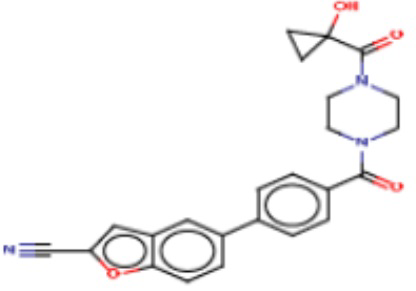
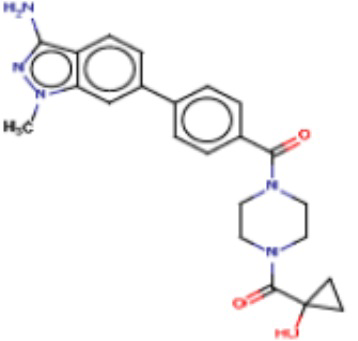
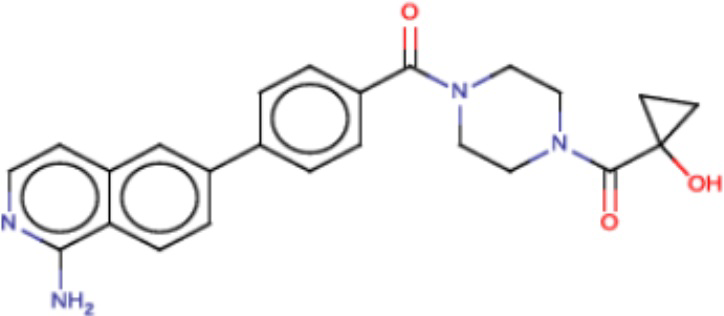
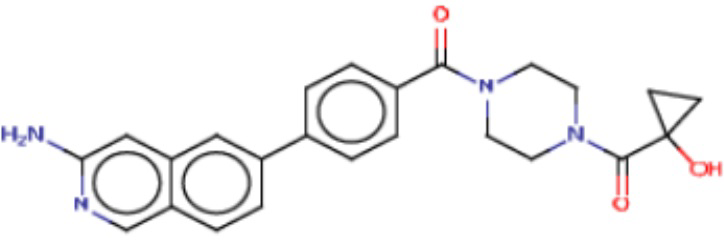
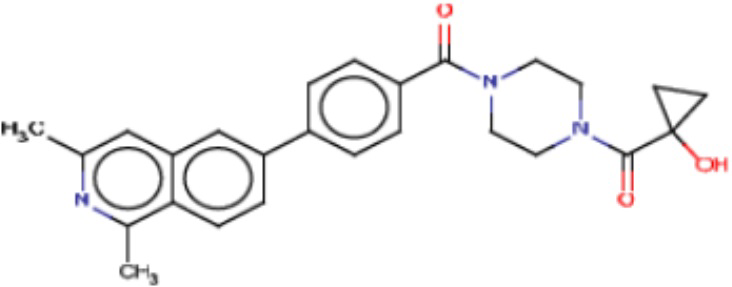
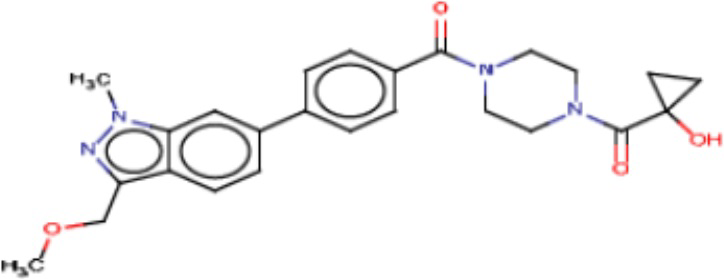
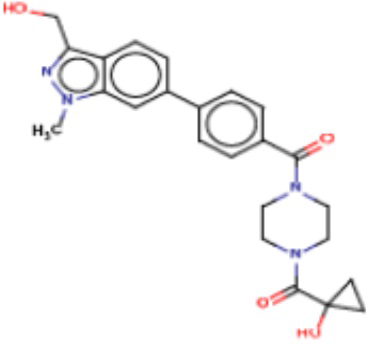
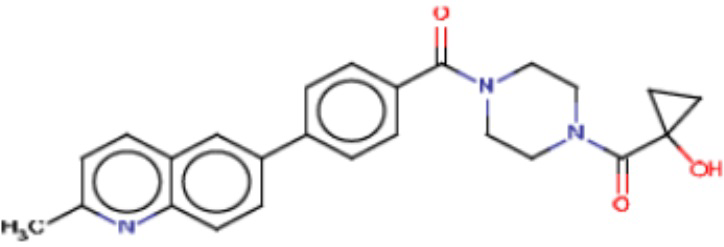
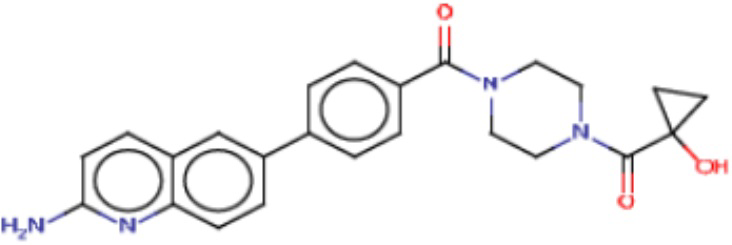
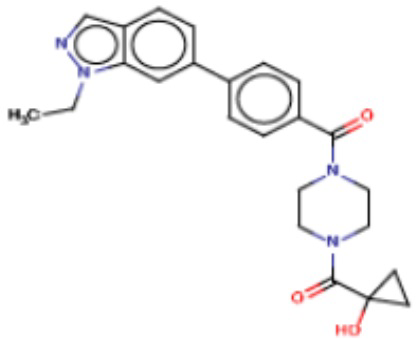
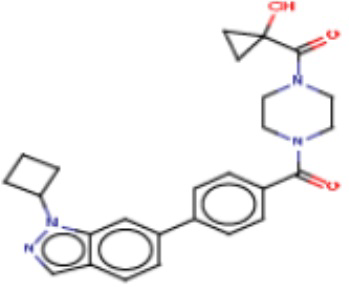
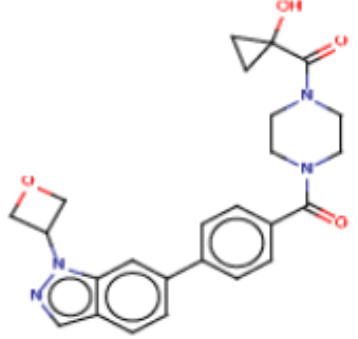
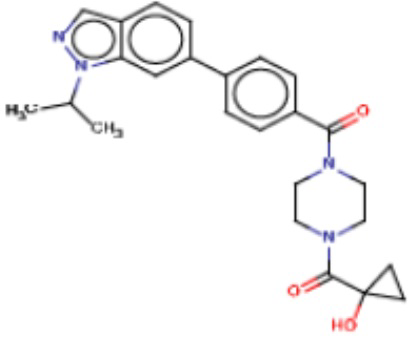
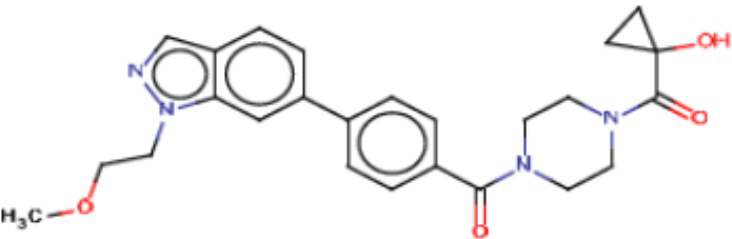
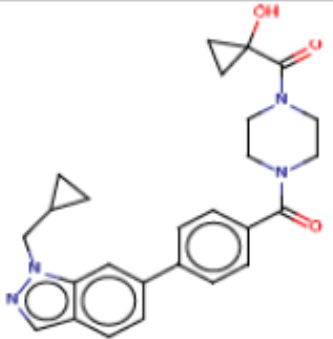
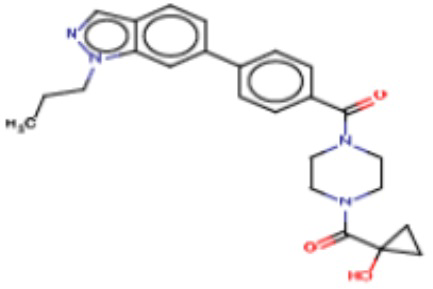
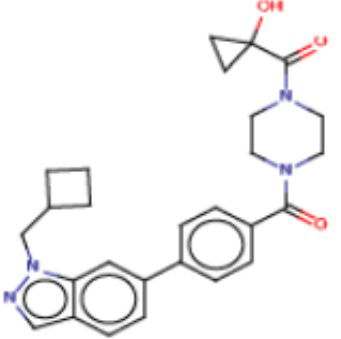
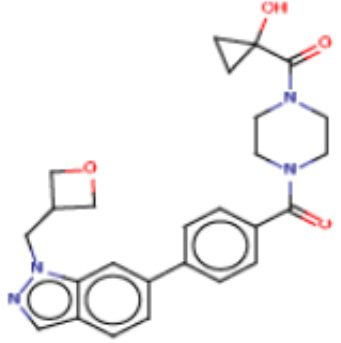


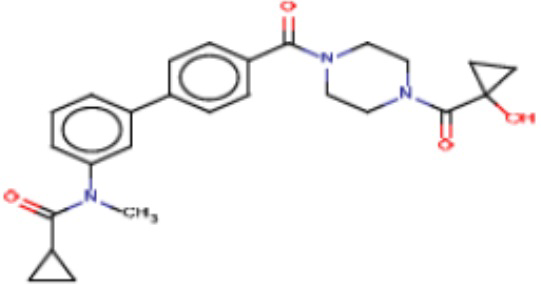
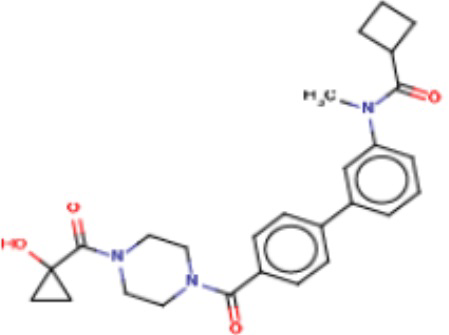
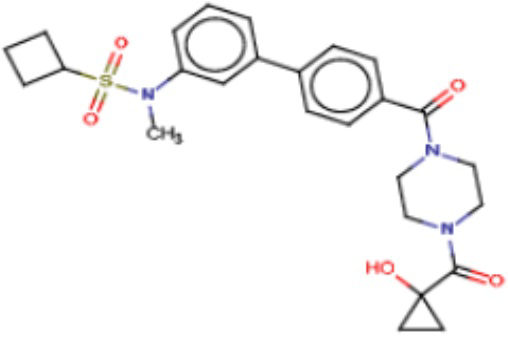

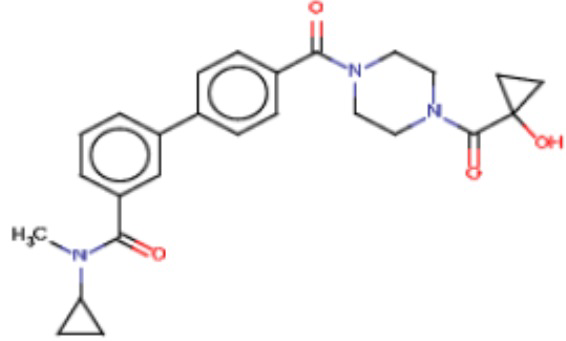
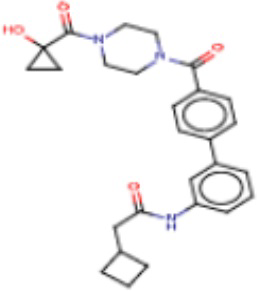

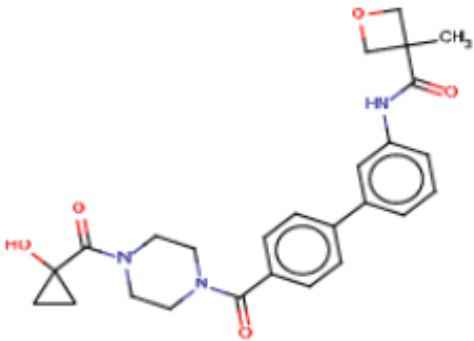
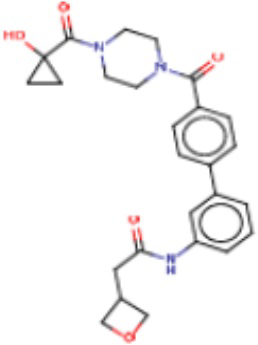
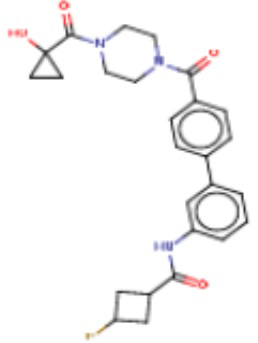
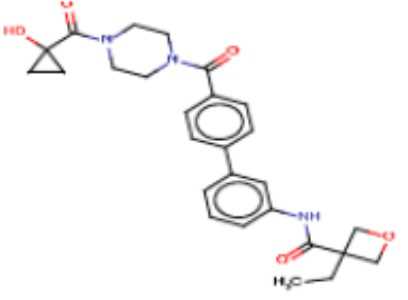
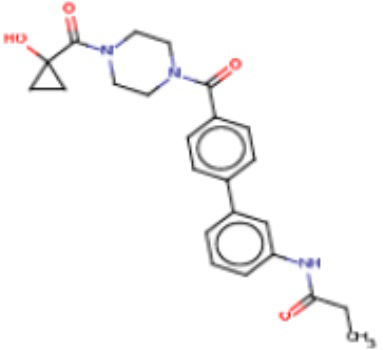
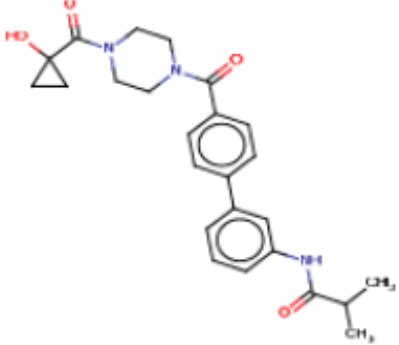


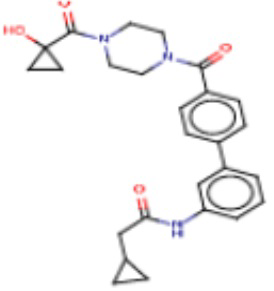
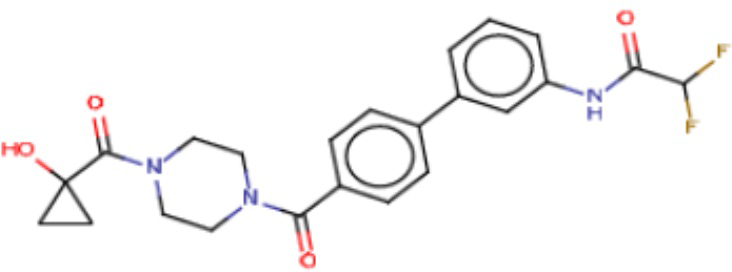
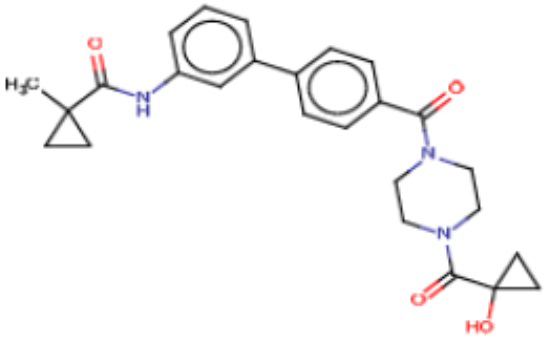
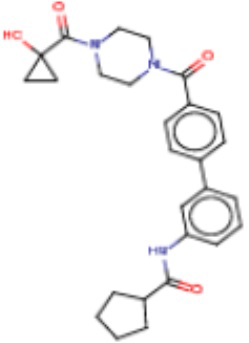
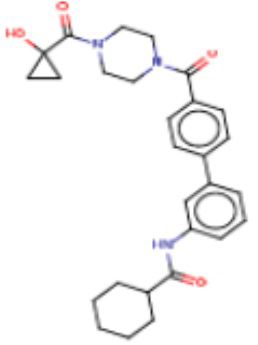
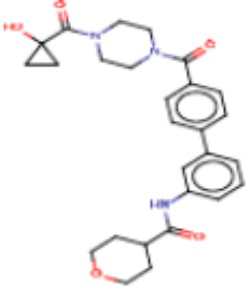

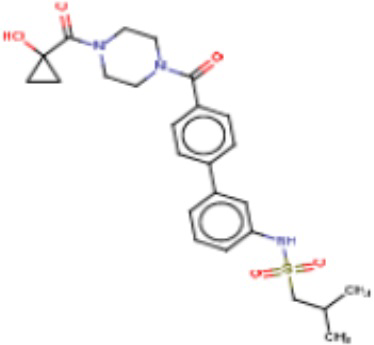
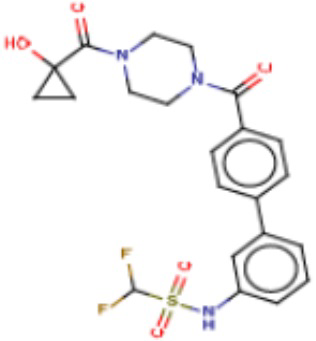
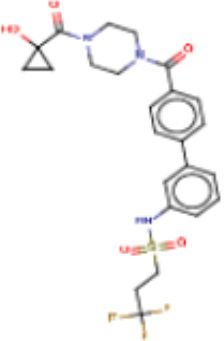

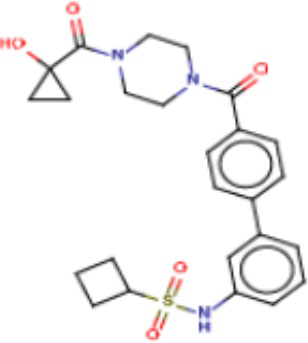
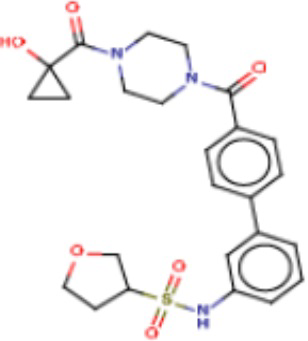
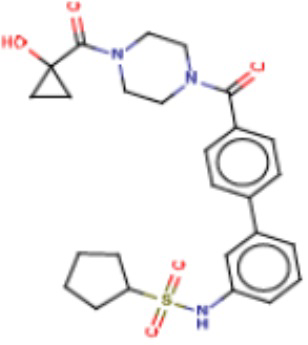
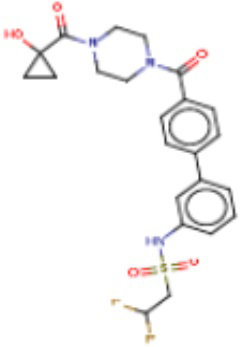
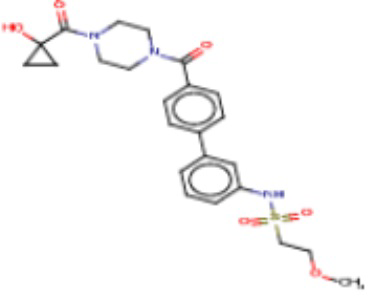
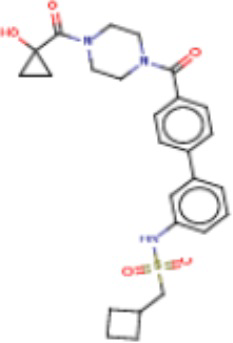
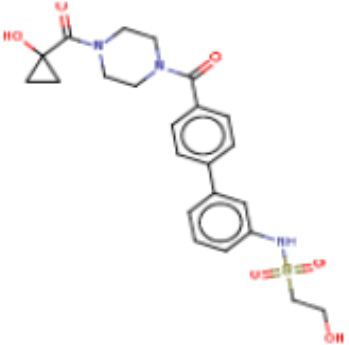
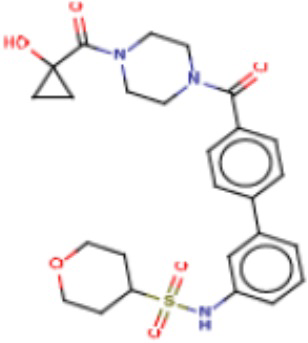
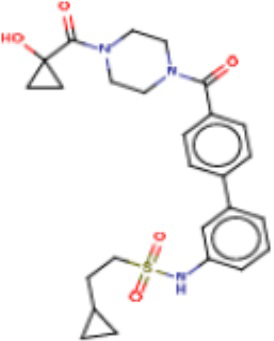
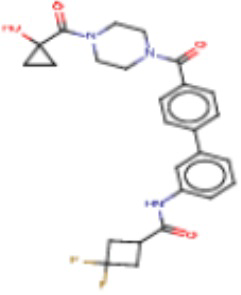
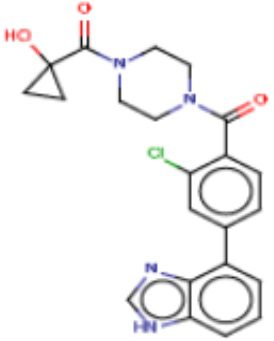
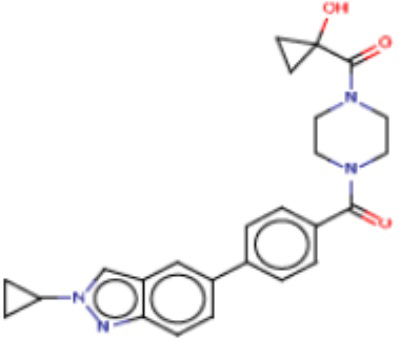
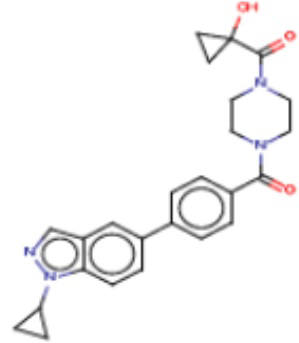
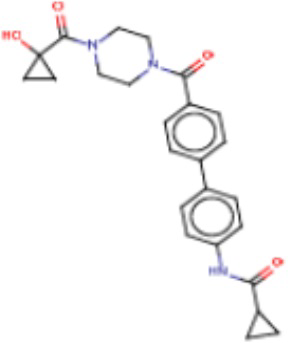
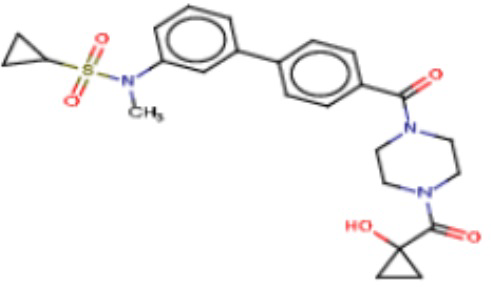
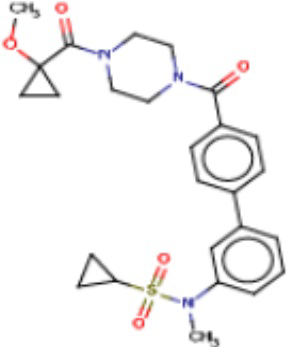
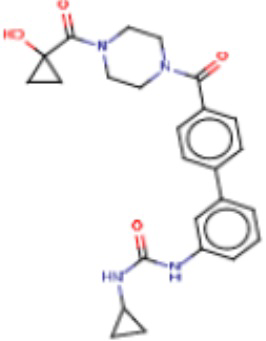

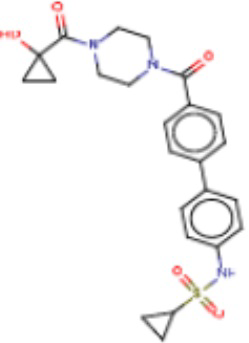
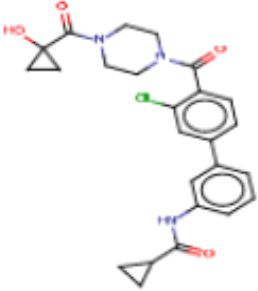
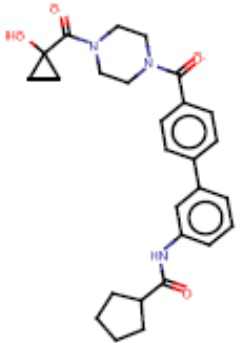
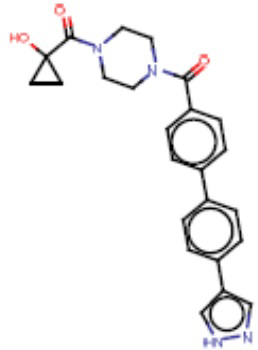
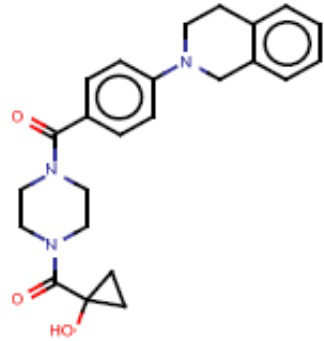
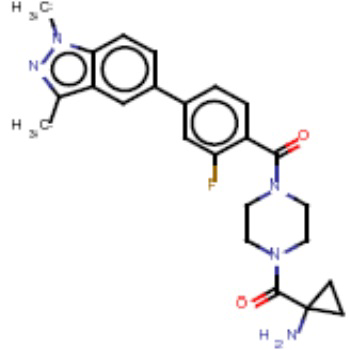

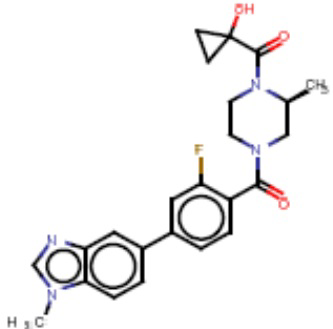
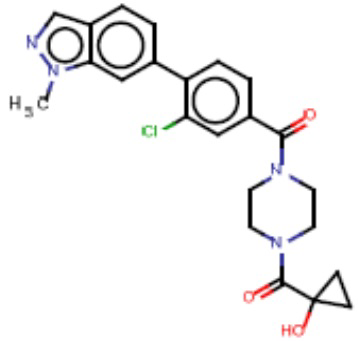
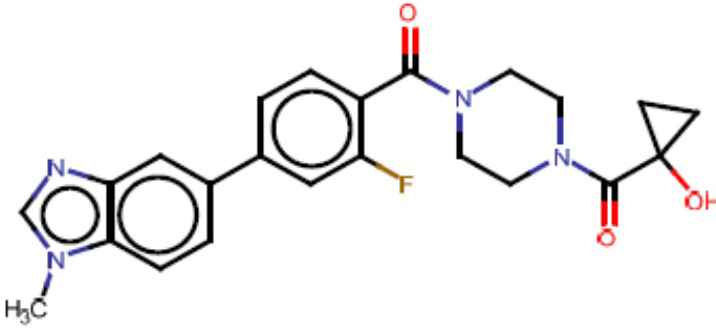
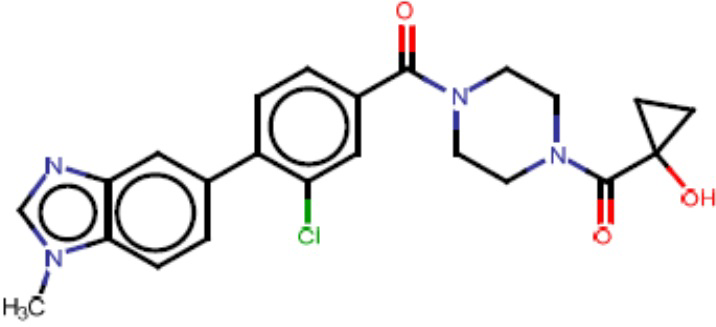
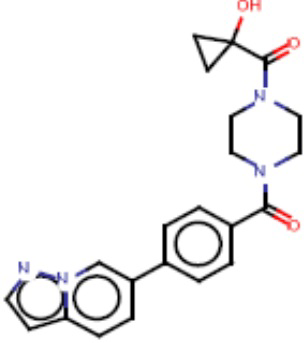
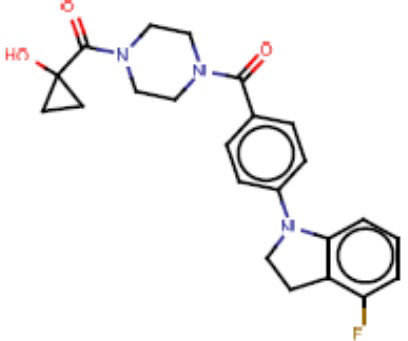



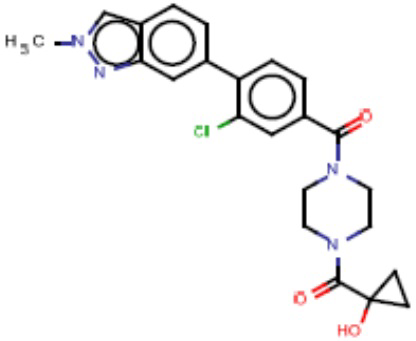
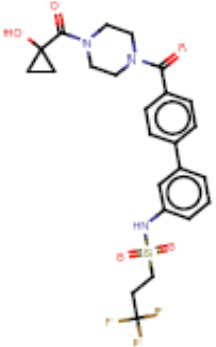
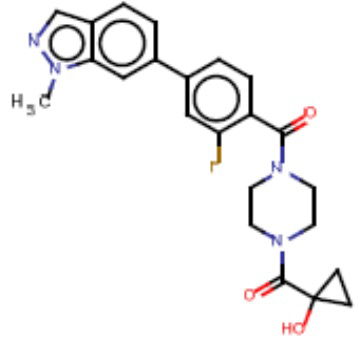
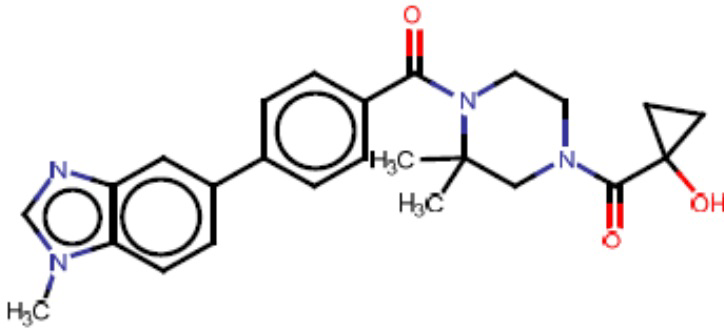
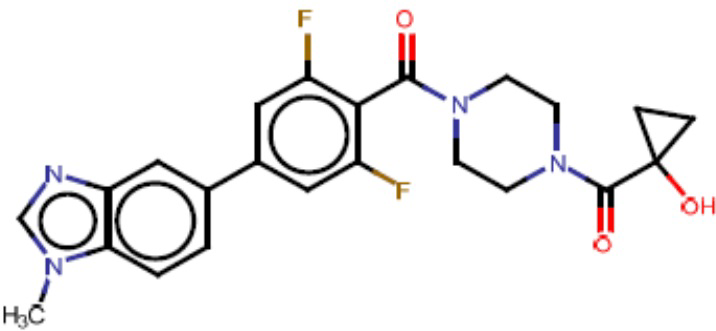
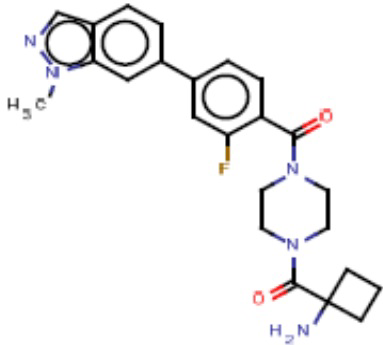
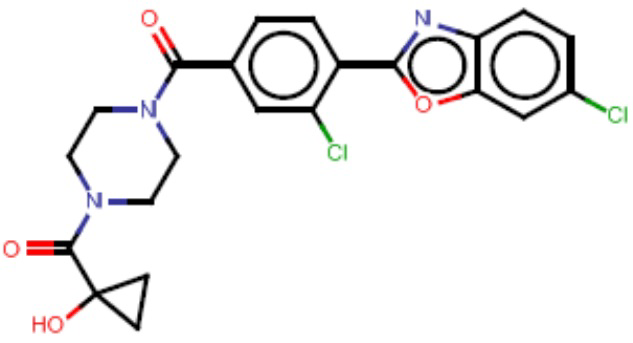

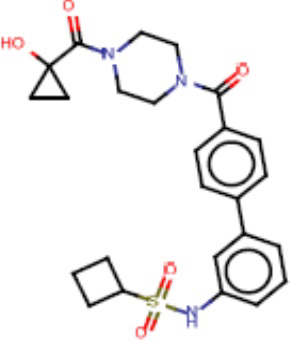
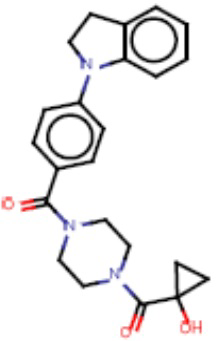
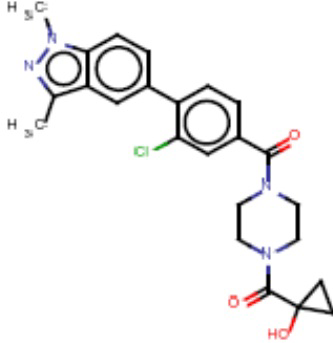

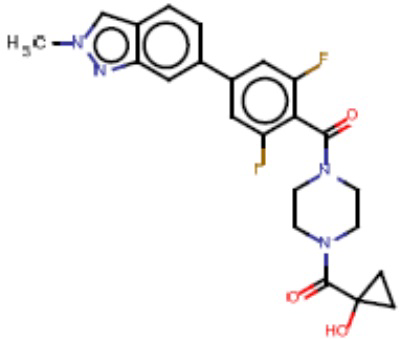

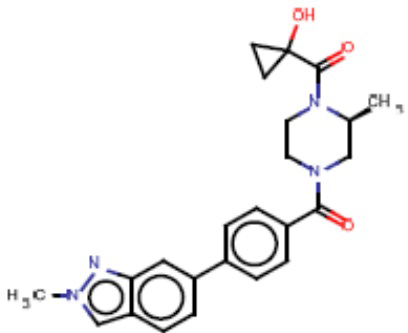

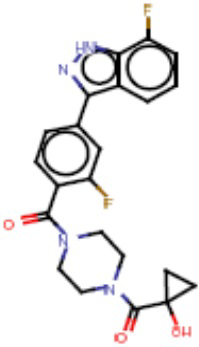
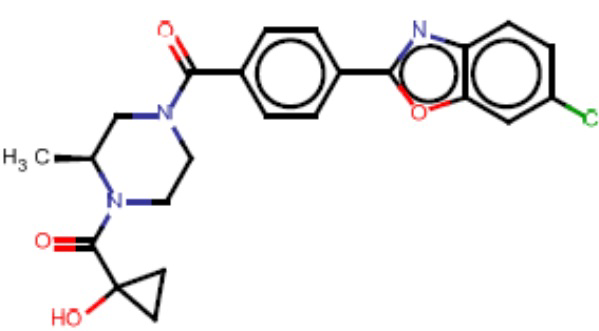
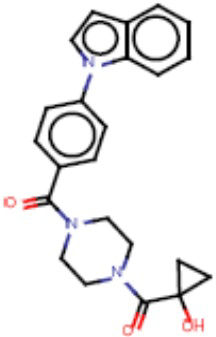
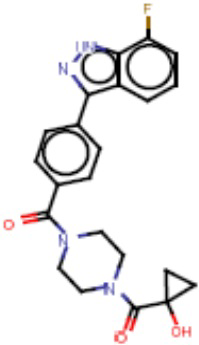
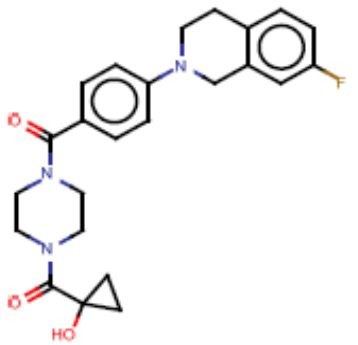
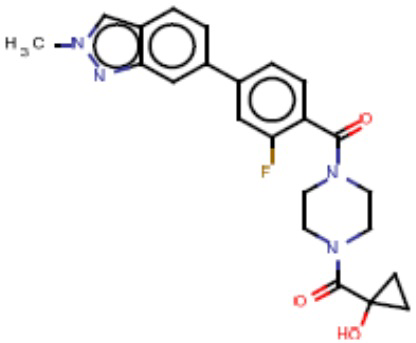

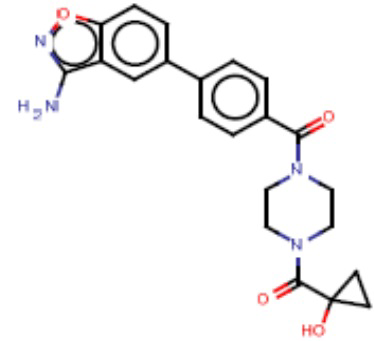
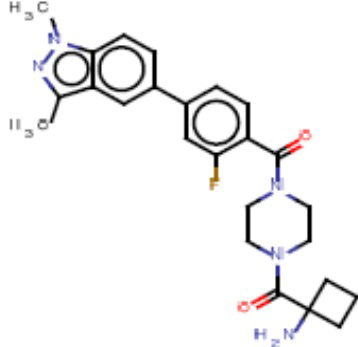

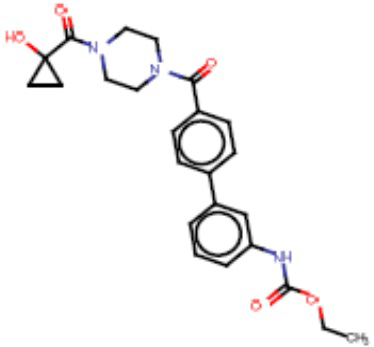
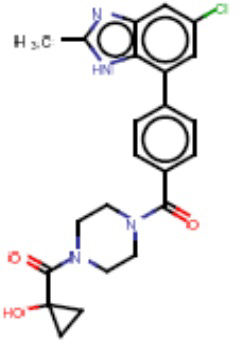
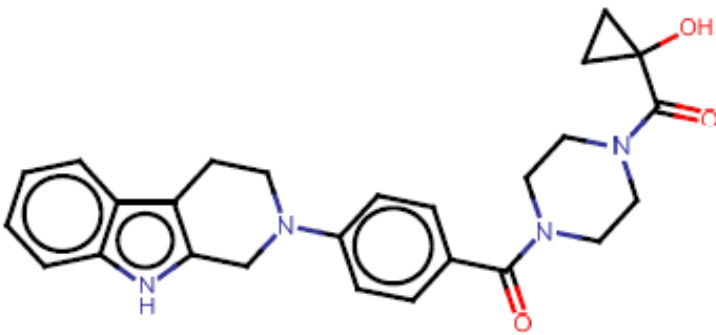
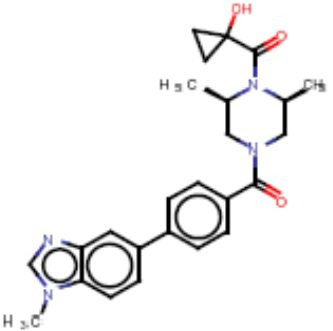
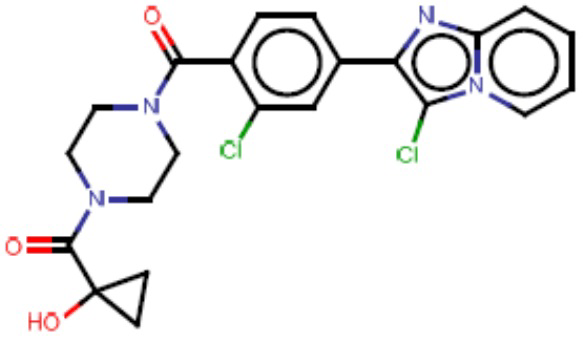
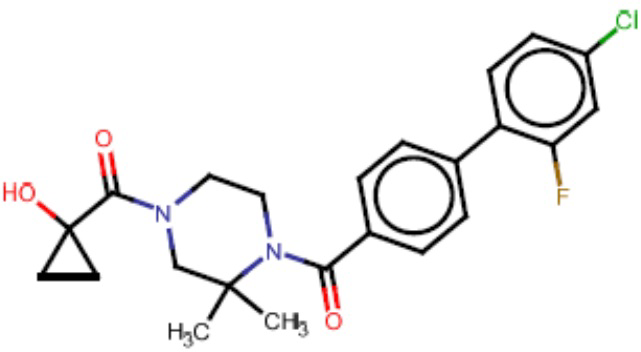
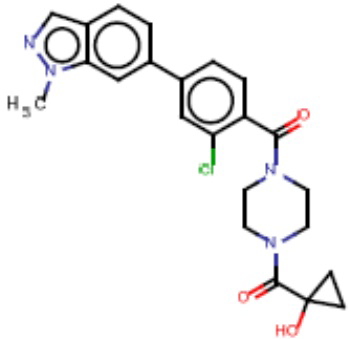
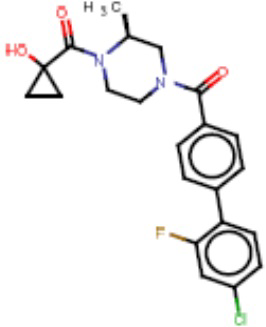
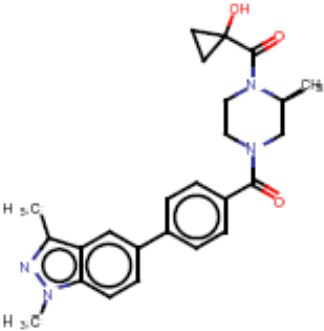

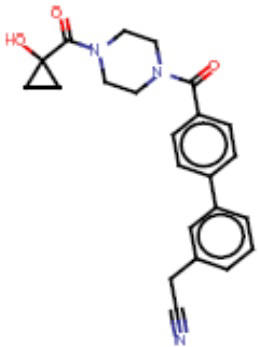
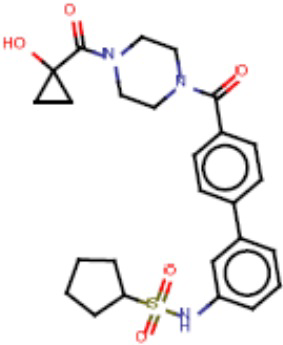
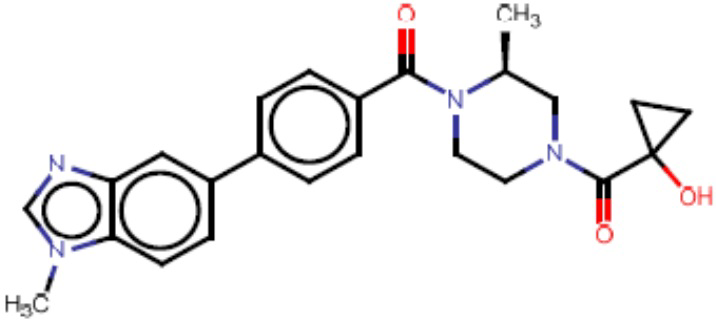
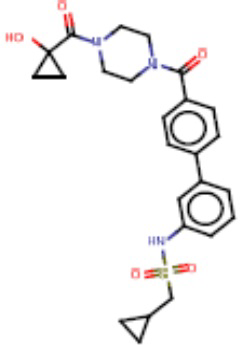
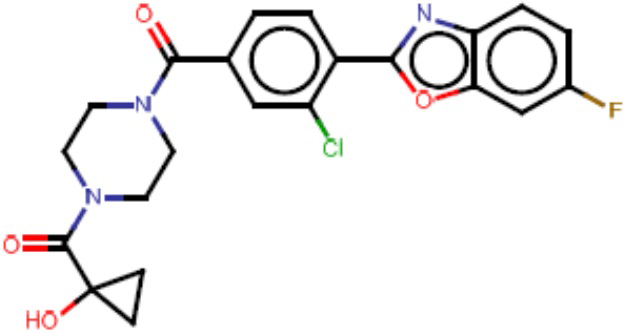
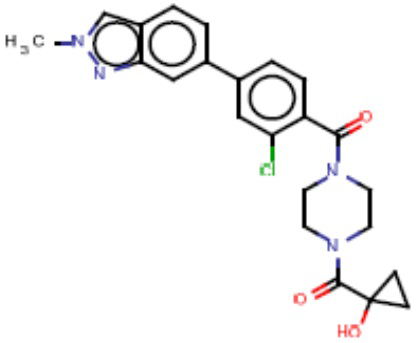
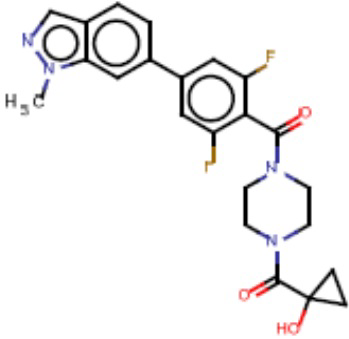
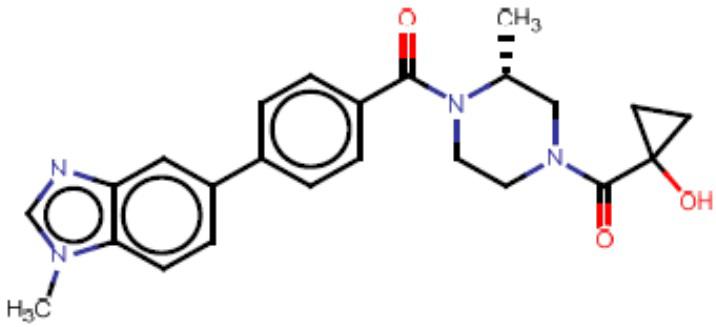
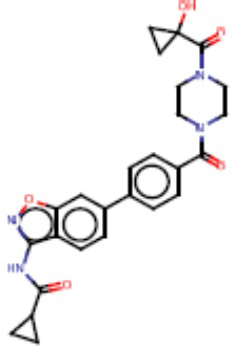
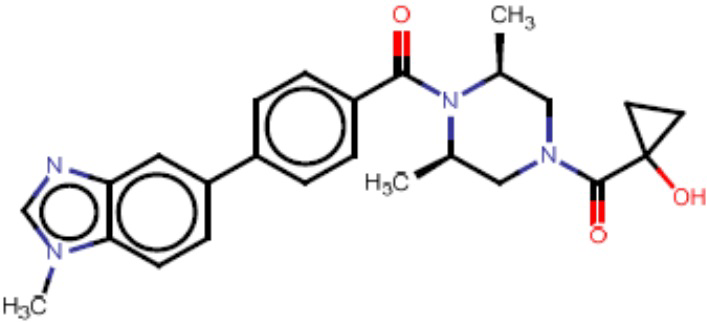
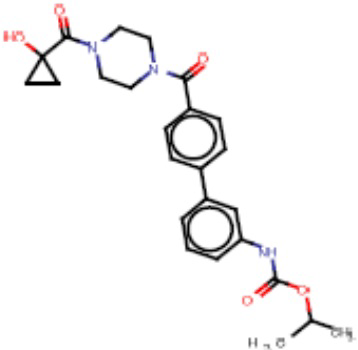
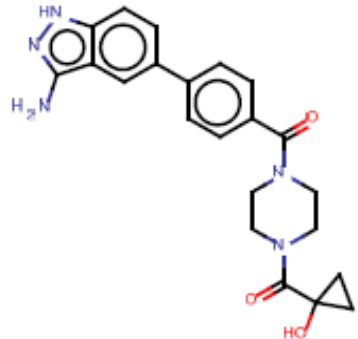
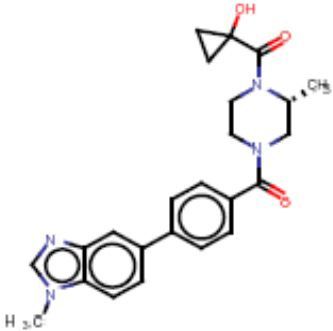
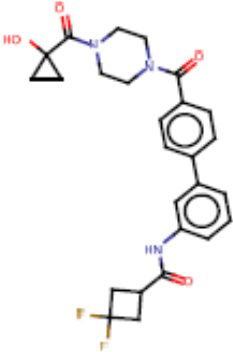
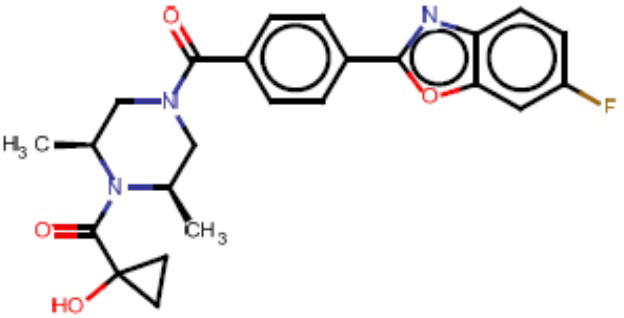
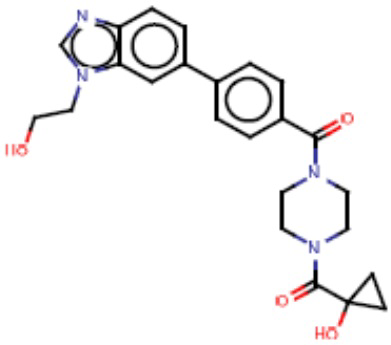
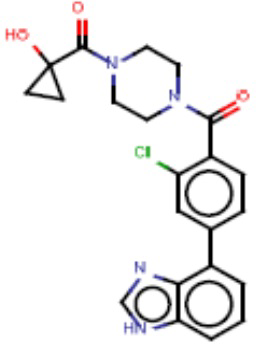


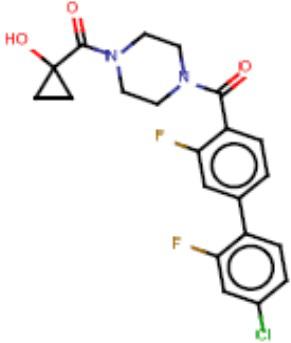
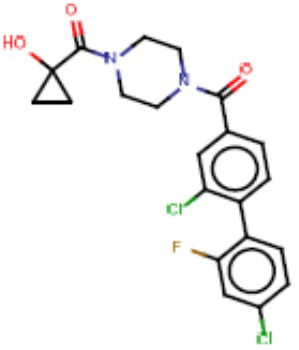
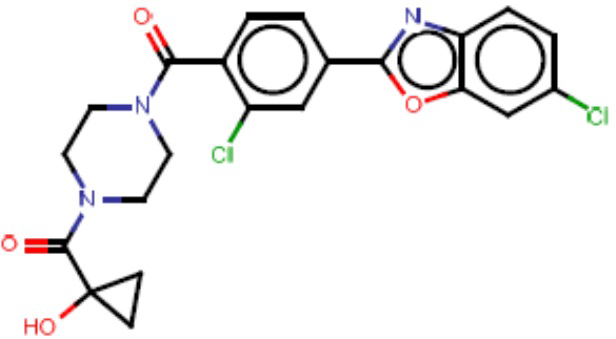
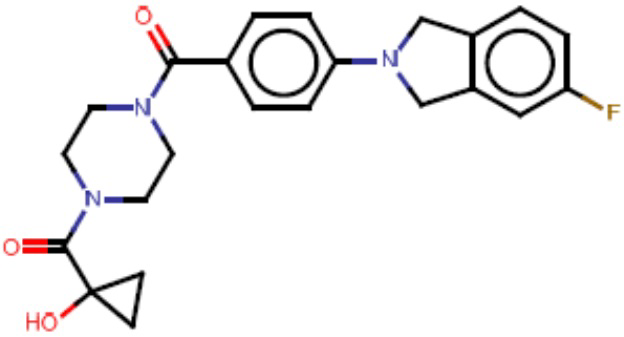
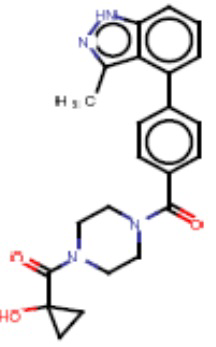
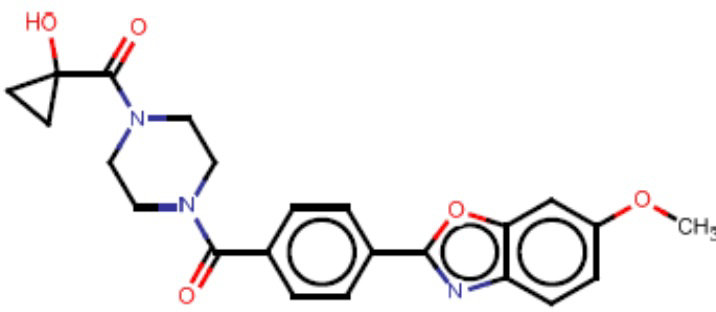
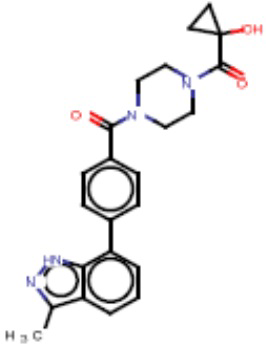
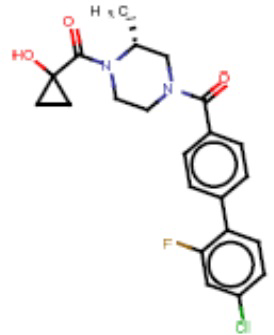


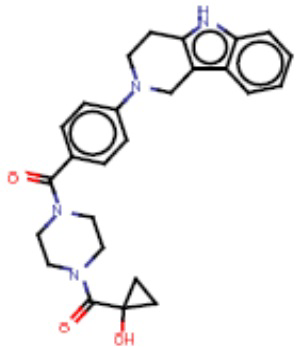
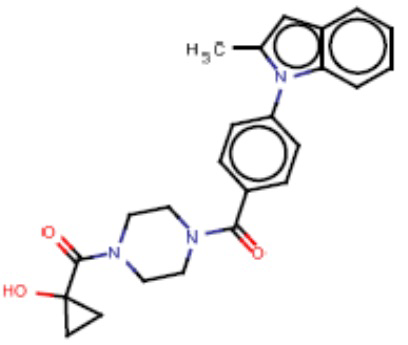

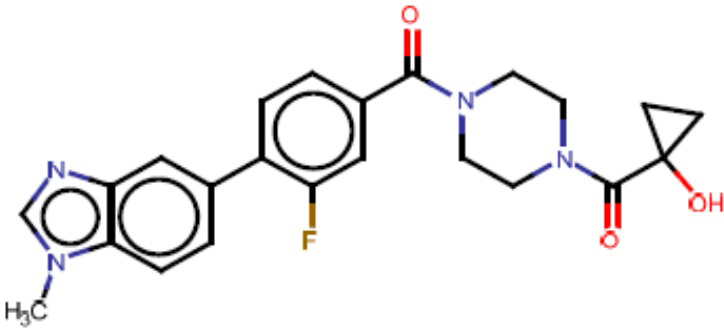
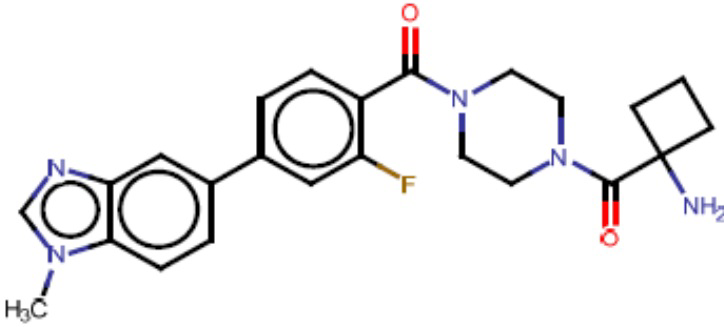
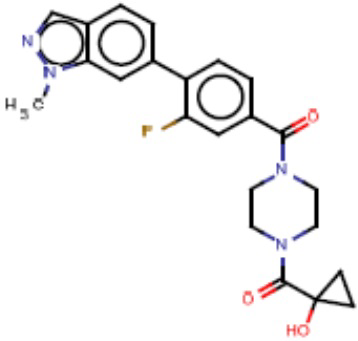
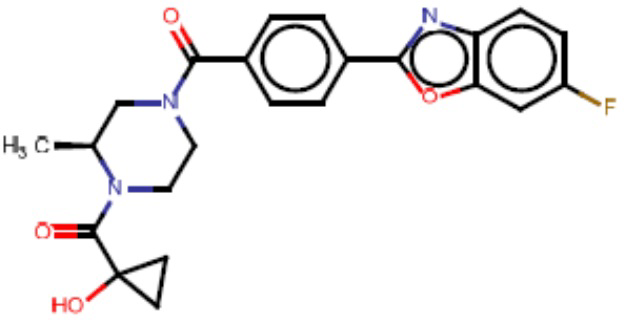
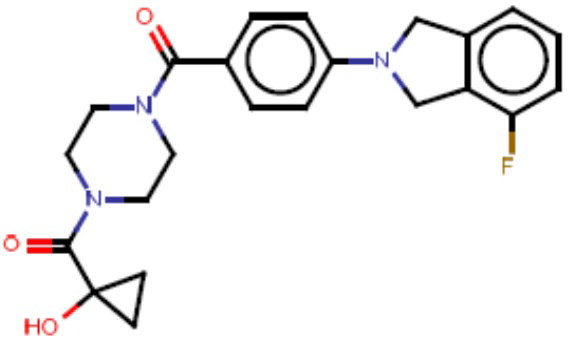
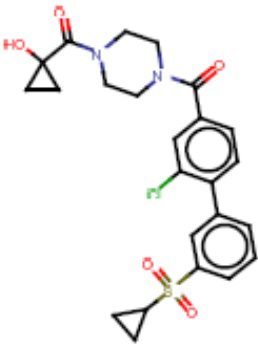

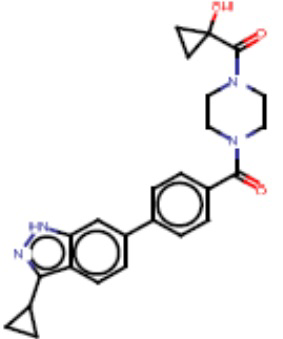
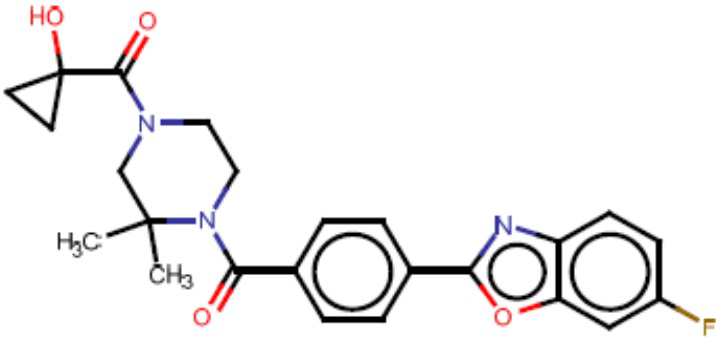
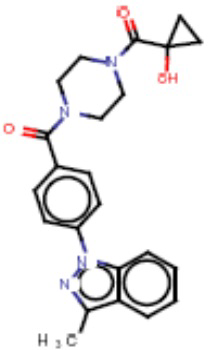
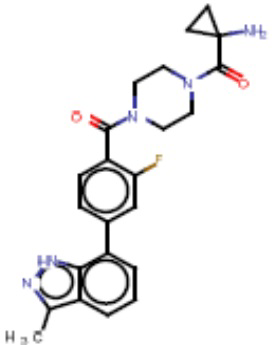

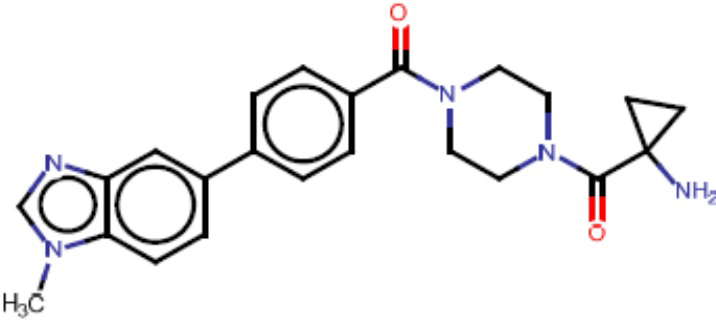
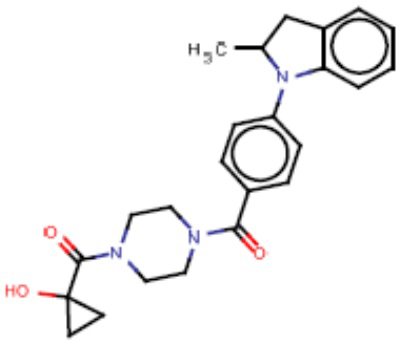
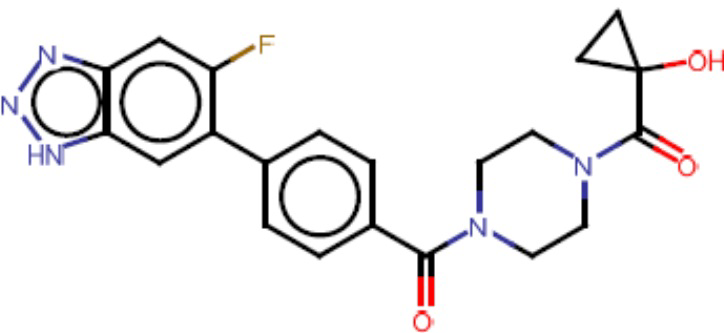
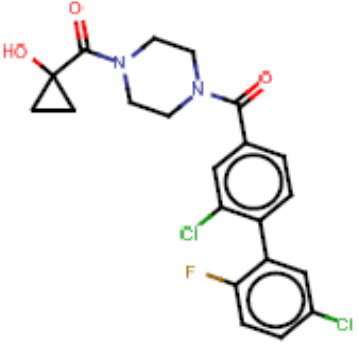
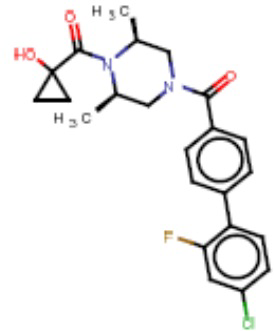
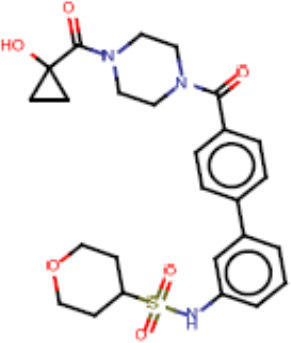

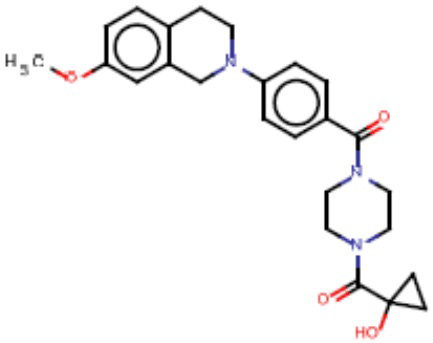
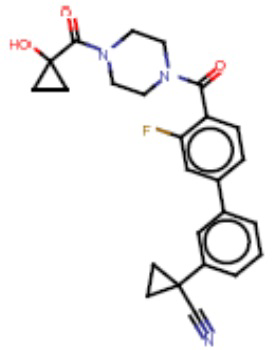
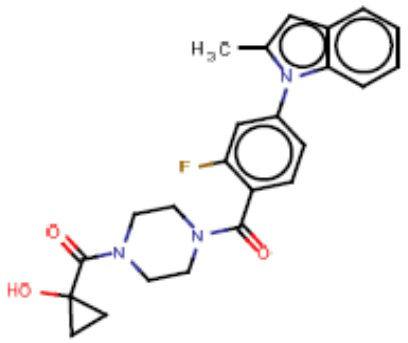



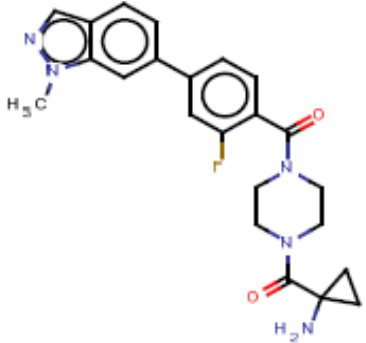
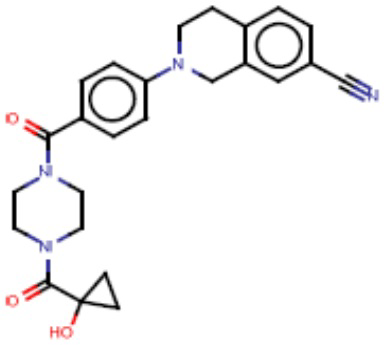
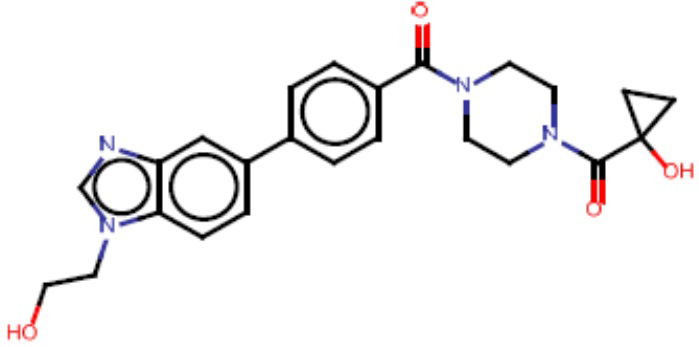


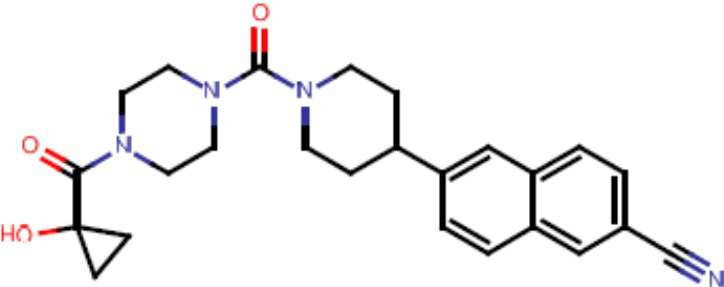
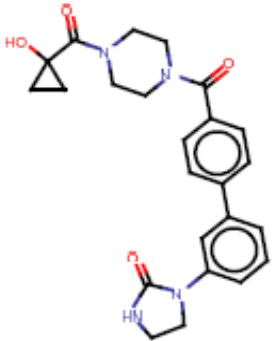
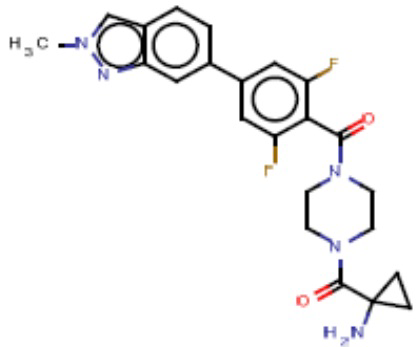
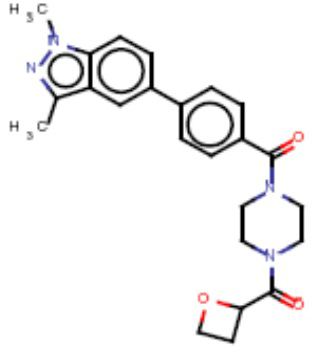
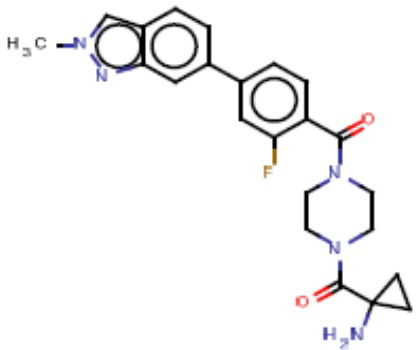

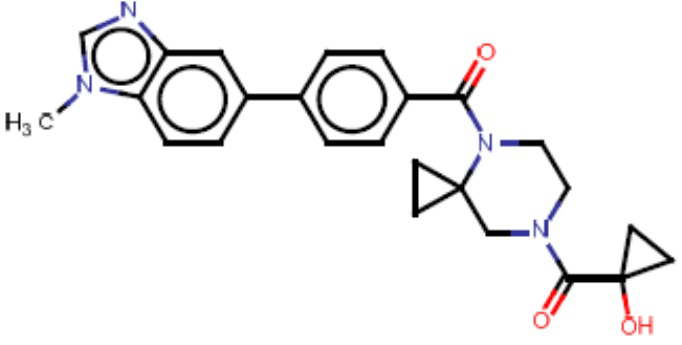
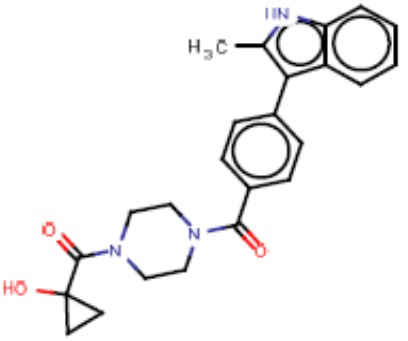

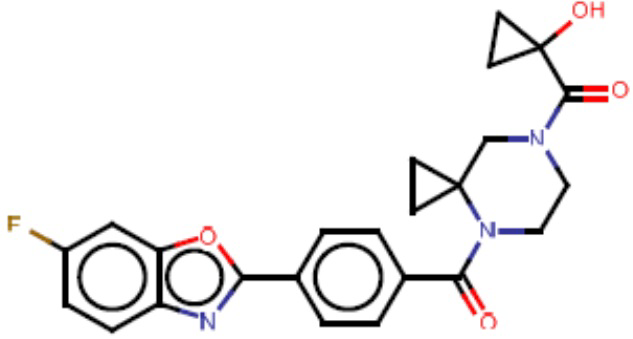
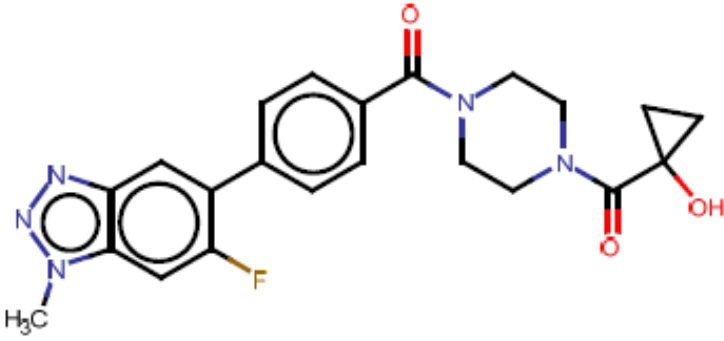
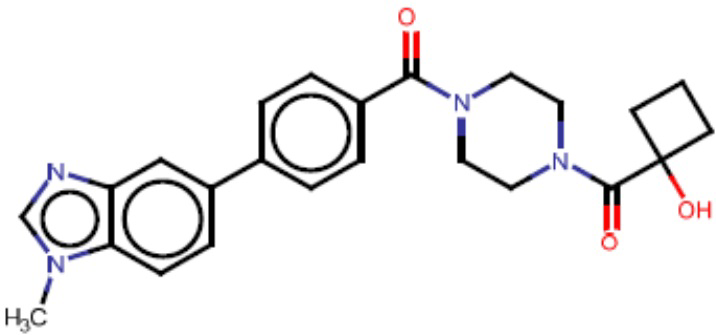
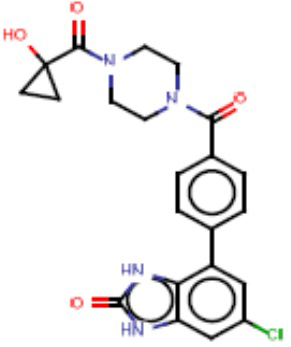
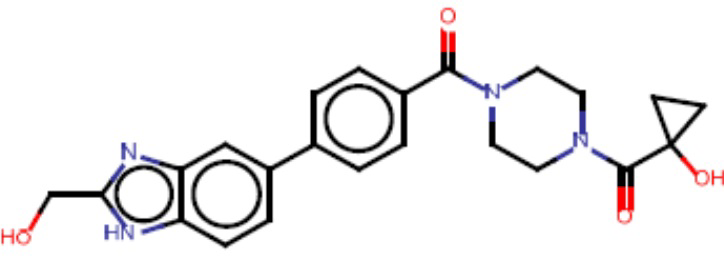
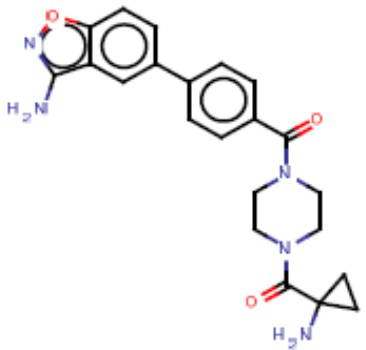
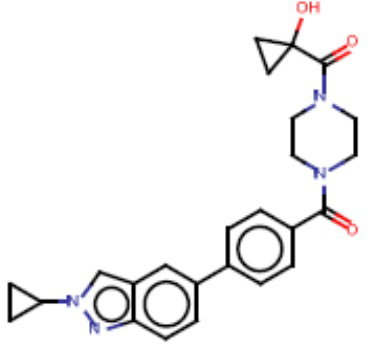
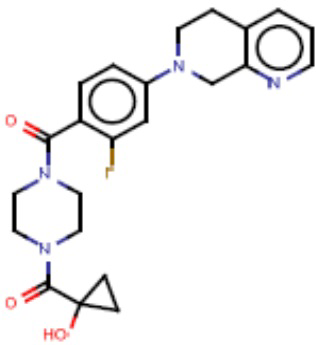
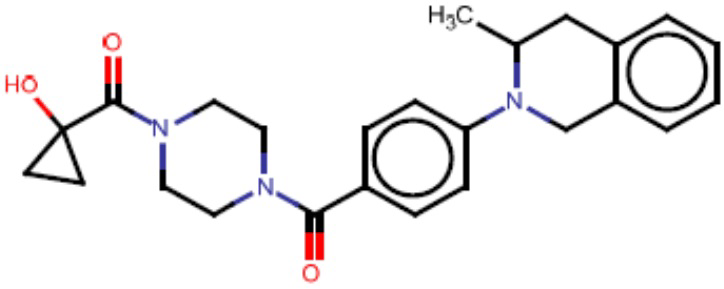
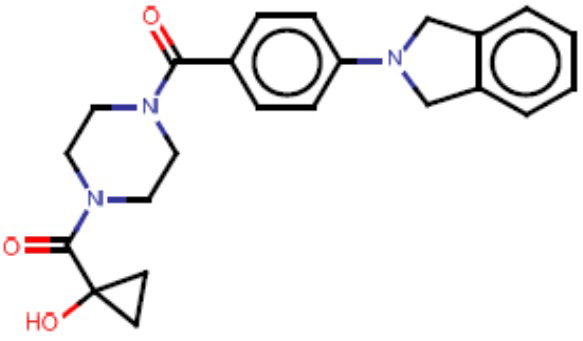
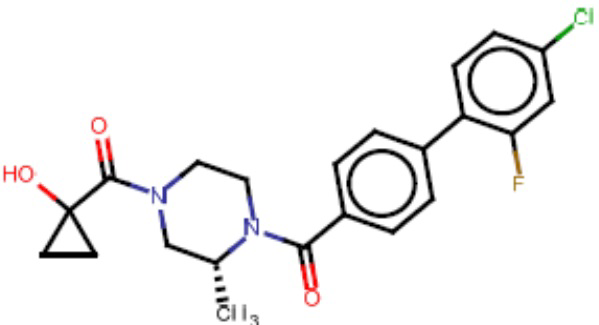
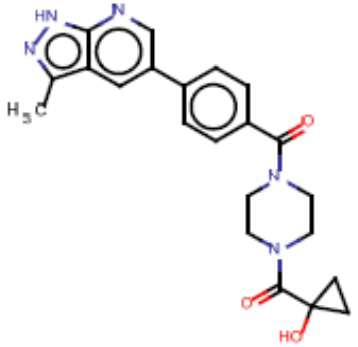

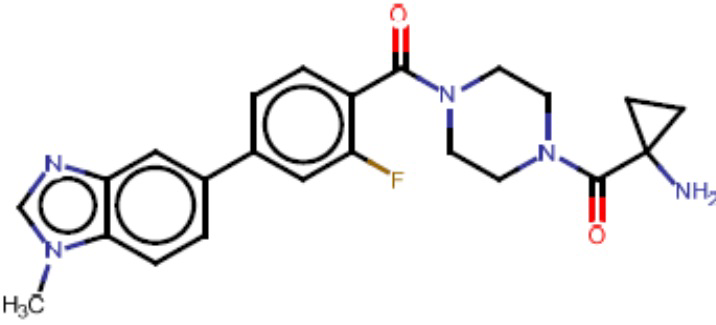
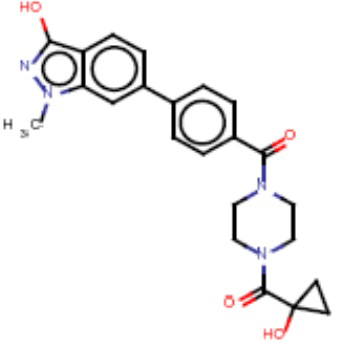


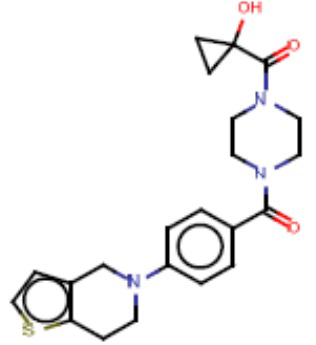
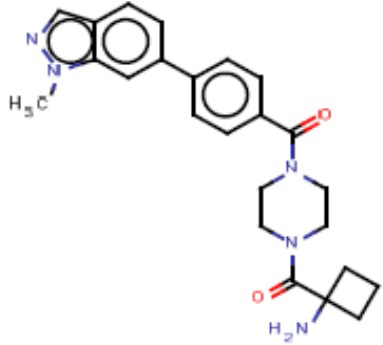
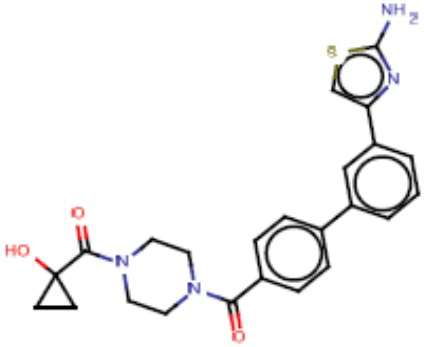
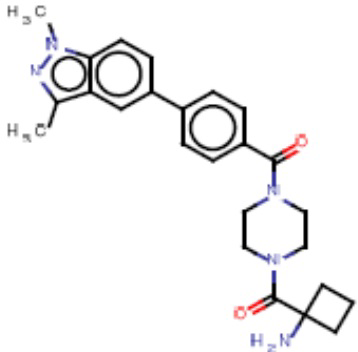

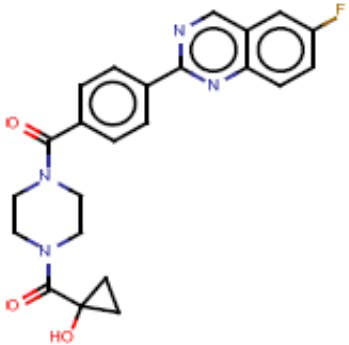
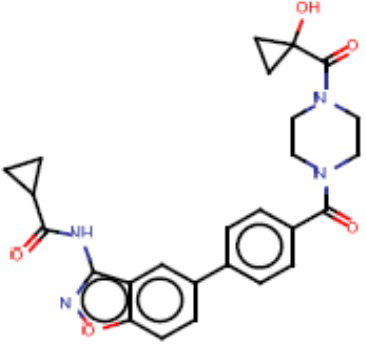
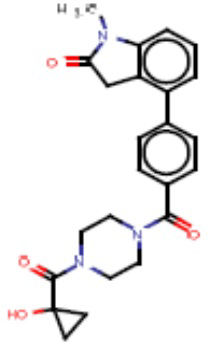

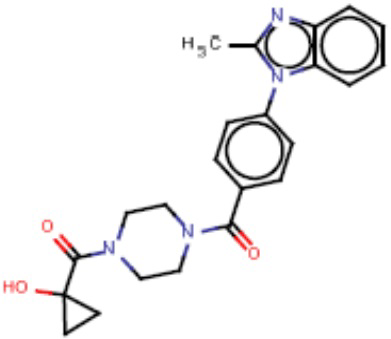
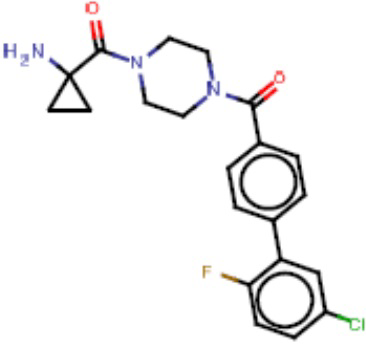

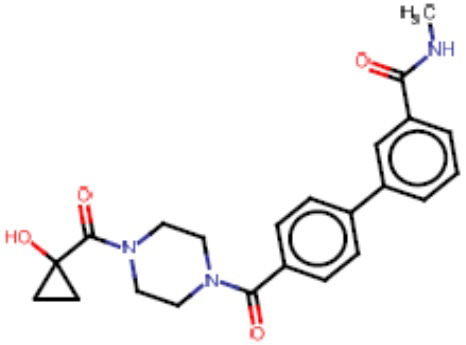
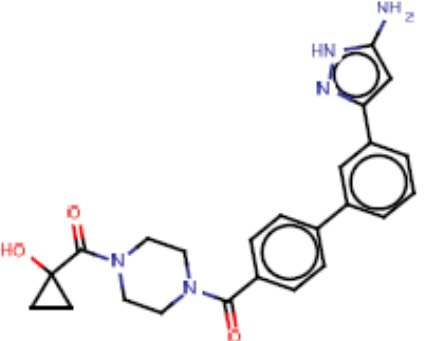
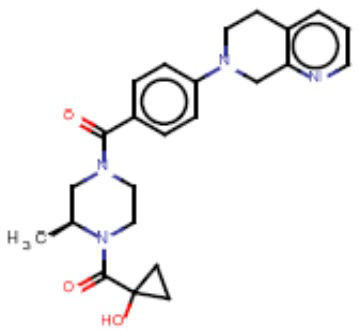
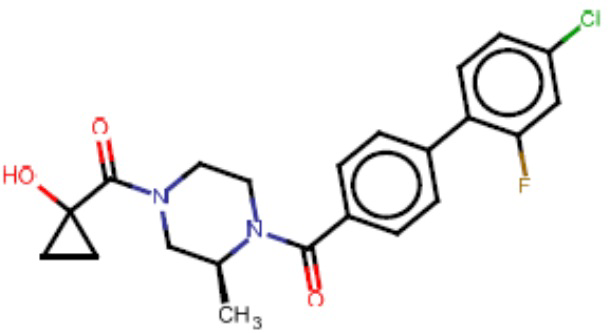
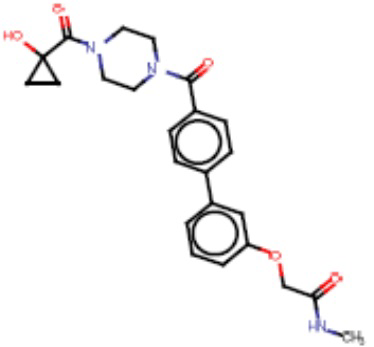

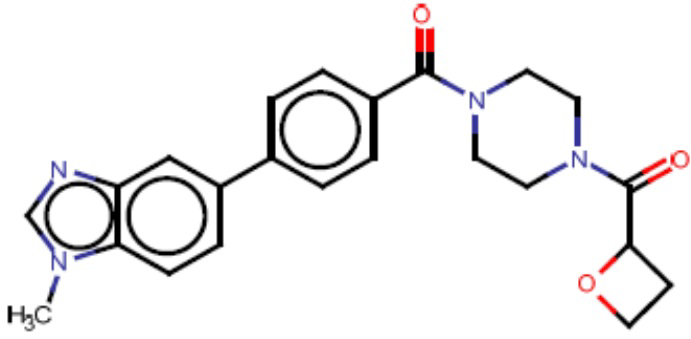
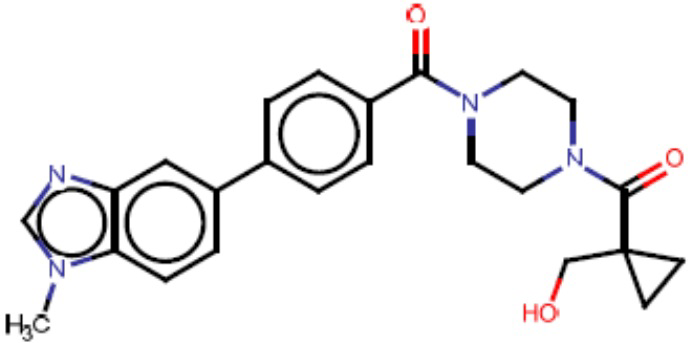
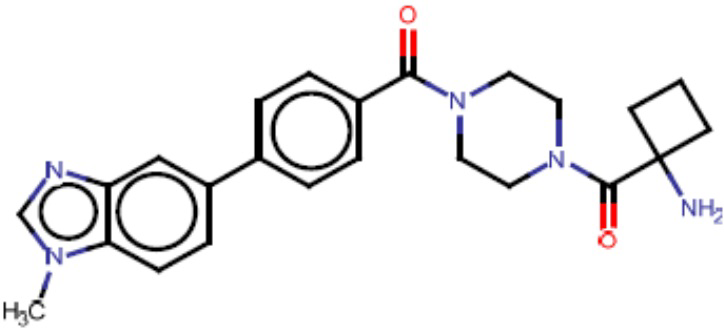
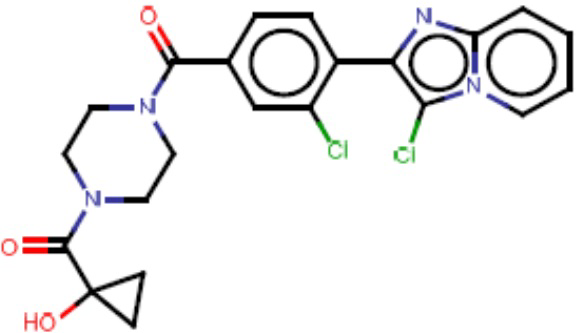
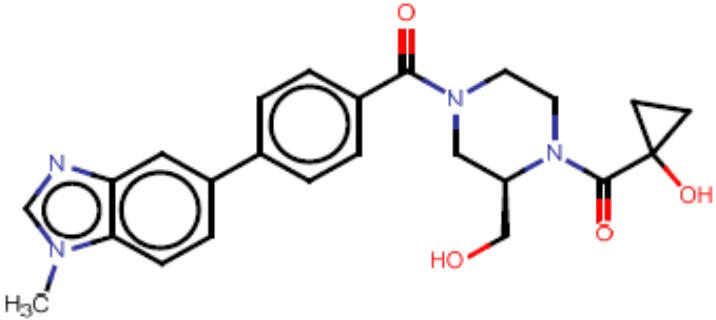
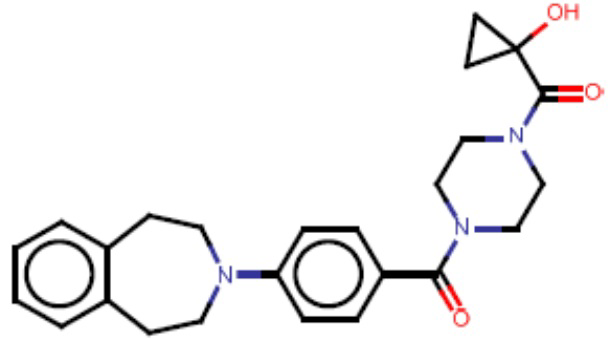
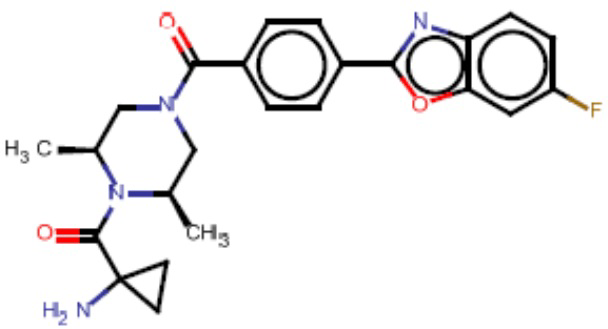
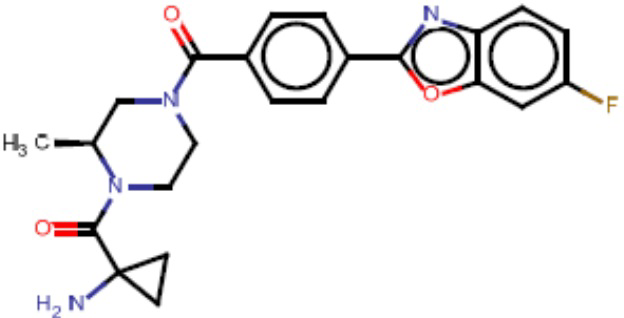
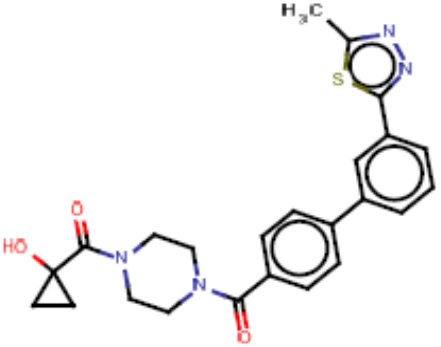
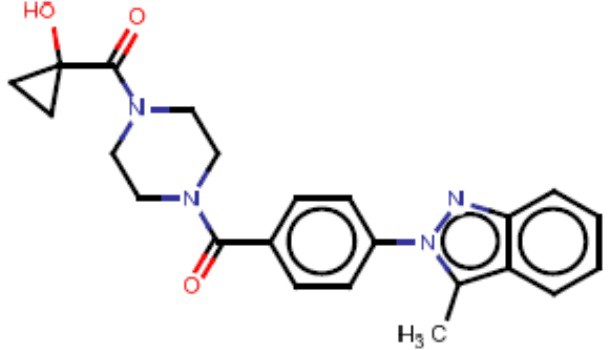
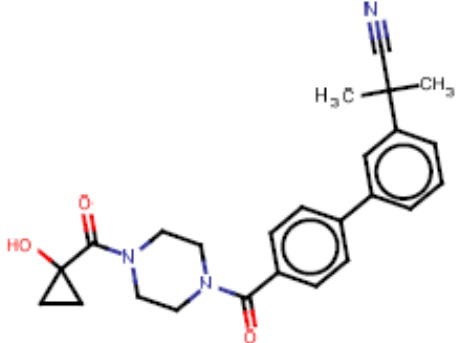
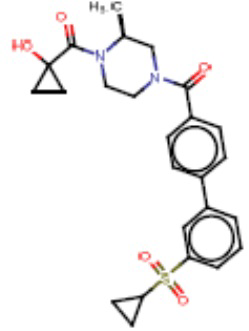
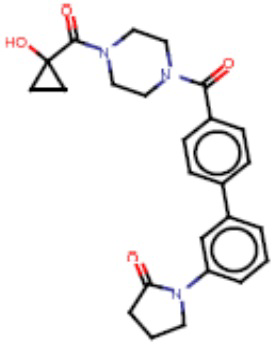
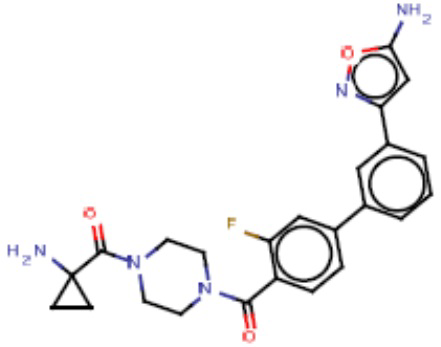
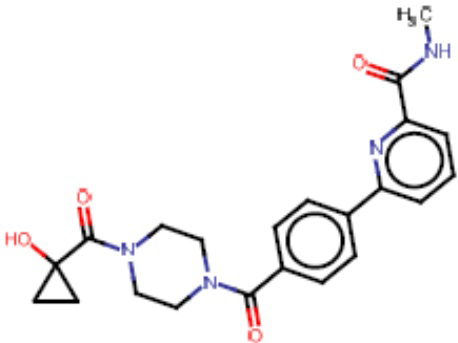

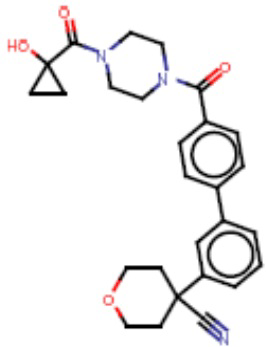
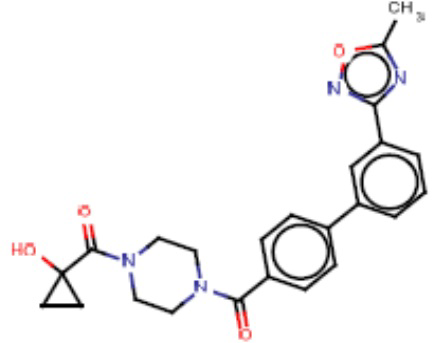
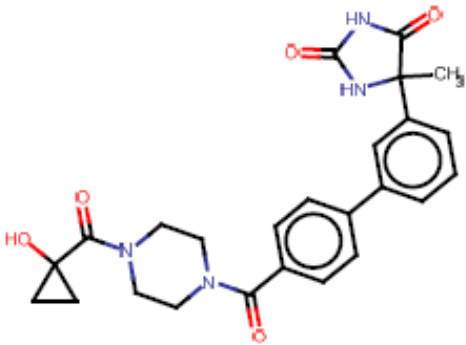
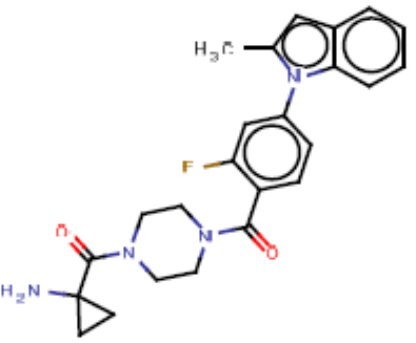

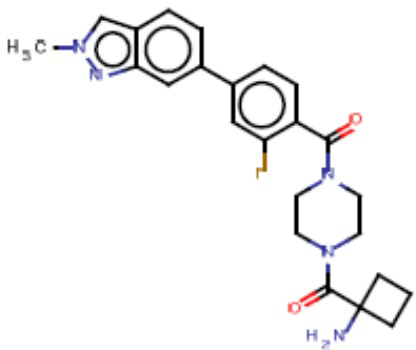
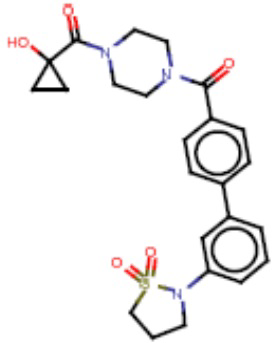
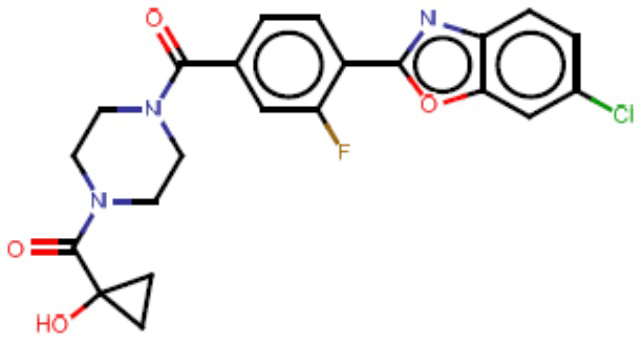
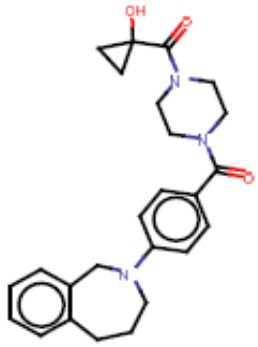
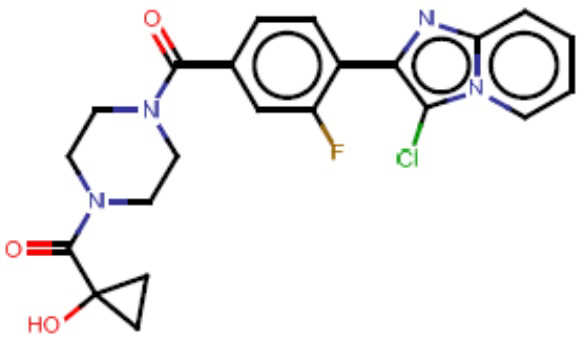
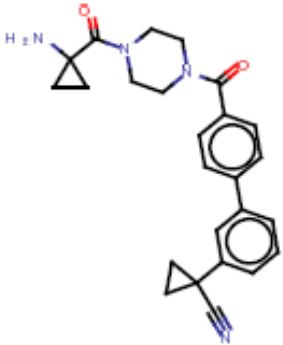

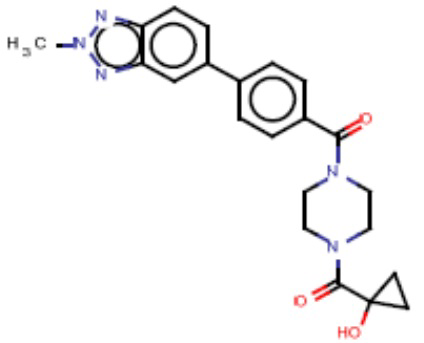
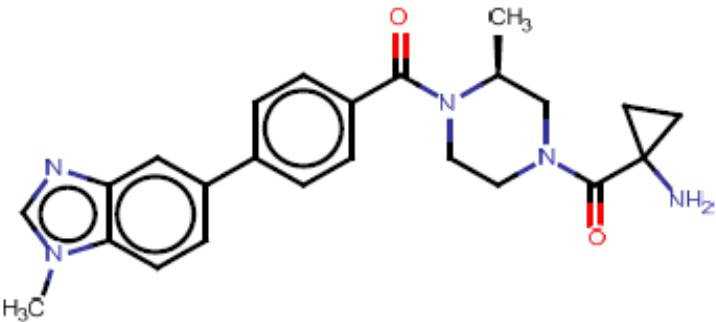
Примеры
Далее приведены иллюстративные, но неограничивающие примеры определенных вариантов реализации настоящего изобретения. Приведены схемы синтеза определенных соединений, предложенных в настоящем описании. Также описан способ и результаты исследования ингибирования FASN и действия в отношении пролиферации раковой клеточной линии.
Определения, используемые на приведенных далее схемах и в настоящем описании:
Материалы
Если не указано иное, все материалы получали у коммерческих поставщиков и использовали без дополнительной очистки. Безводные растворители получали в Sigma-Aldrich (Milwaukee, WI) и использовали без обработки. Все взаимодействия с использованием реагентов, чувствительных к воздуху или влаге, проводили в атмосфере азота. Данные чистоты и масс-спектрометрии с низким разрешением получали с использованием: (1) системы сверхэффективной жидкостной хроматографии (СВЭЖХ) Waters Acquity (система СВЭЖХ Waters Acquity, оборудованная устройством распределения образцов (Sample Organizer) и масс-спектрометром Waters Micromass ZQ), детектирование проводили в УФ диапазоне при 220 нм в режиме детектирования положительных ионов, получаемых путем ионизации распылением низкорезонансных электронов (ИЭР) (колонка: Acquity UPLC BEH C18, 1,7 мкм, 2,1 х 50 мм; градиент: 5-100% растворителя В (95/5/0,09%: ацетонитрил/вода/муравьиная кислота) в растворителе А (95/5/0,1%: 10 мМ формиат аммония/ацетонитрил/муравьиная кислота) в течение 2,2 мин, затем 100-5% растворителя В в растворителе А в течение 0,01 мин, затем выдерживали 5% растворителя В в растворителе А в течение 0,29 мин) или (2) системы высокоэффективной жидкостной хроматографии (ВЭЖХ) Waters HT2790 Alliance (Waters 996 PDA и масс-спектрометр Waters ZQ Single Quad), детектирование проводили в УФ диапазоне при 220 нм и 254 нм в режиме детектирования положительных/отрицательных ионов, получаемых путем ионизации распылением низкорезонансных электронов (ИЭР) (колонка: XBridge Phenyl или С18, 5 мкм, 4,6х50 мм; градиент: 5-95% растворителя В (95% метанола/5% воды, содержащая 0,1% муравьиной кислоты) в растворителе А (95% воды/5% метанола, содержащая 0,1% муравьиной кислоты) в течение 2,5 мин, затем выдерживали 95% растворителя В в растворителе А в течение 1 мин).
Общие способы синтеза соединения:
Способ 1.
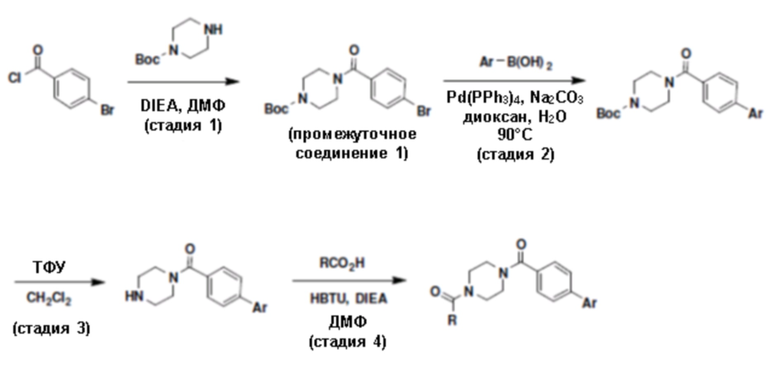
Стадия 1. Трет-бутил-4-(4-бромбензоил)пиперазин-1-карбоксилат (промежуточное соединение 1)
В выдерживаемый при 0°С раствор трет-бутил-пиперазин-1-карбоксилата (10,18 г, 54,7 ммоль) и 4-бромбензоилхлорида (12,0 г, 54,7 ммоль) в ДМФ (80 мл) добавляли N,N-диизопропилэтиламин (28,6 мл, 164 ммоль) и реакционную смесь перемешивали при КТ в течение 6 часов. Добавляли воду и полученную смесь перемешивали в течение ночи, а затем фильтровали и сушили с получением трет-бутил-4-(4-бромбензоил)пиперазин-1-карбоксилата (19,388 г, 52,5 ммоль, выход 96%) в виде беловатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 369, 371 (М+1).
Стадия 2. Трет-бутил-4-(4'-хлор-2'-фторбифенилкарбонил)пиперазин-1-карбоксилат
В смесь трет-бутил-4-(4-бромбензоил)пиперазин-1-карбоксилата (5,00 г, 13,54 ммоль), 4-хлор-2-фторфенилбороновой кислоты (2,95 г, 16,93 ммоль) и карбоната натрия (5,74 г, 54,2 ммоль) в 1,4-диоксане (50 мл) и воде (10 мл) добавляли тетракис(трифенилфосфин)-палладий (0) (1,565 г, 1,354 ммоль). Смесь перемешивали при 70°С в течение 5 часов. Фильтровали реакционную смесь через Celite и концентрировали с получением оранжевой маслянистой жидкости. Полученное вещество очищали путем колоночной хроматографии на силикагеле (100 г колонка Biotage, элюировали с градиентом смесями 0-50% этилацетат-гексан) с получением трет-бутил-4-(4'-хлор-2'-фторбифенилкарбонил)пиперазин-1-карбоксилата (5,508 г, 13,15 ммоль, выход 97%) в виде коричневатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 419 (М+1).
Стадия 3. 2,2,2-трифторацетат (4'-хлор-2'-фторбифенил-4-ил)(пиперазин-1-ил)метанона
В раствор трет-бутил-4-(4'-хлор-2'-фторбифенилкарбонил)пиперазин-1-карбоксилата (11,35 г, 27,1 ммоль) в дихлорметане (100 мл) добавляли трифторуксусную кислоту (20,0 мл, 260 ммоль) и раствор перемешивали при КТ в течение 1,5 часа. Концентрировали реакционную смесь и растирали остаток с диэтиловым эфиром с получением 2,2,2-трифторацетата (4'-хлор-2'-фторбифенил-4-ил)(пиперазин-1-ил)метанона (12,02 г, 27,8 ммоль, выход 100%) в виде коричневатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 319 (М+1).
Стадия 4. (4'-хлор-2'-фторбифенил-4-ил)(4-(1-гидроксициклопропанкарбонил)-пиперазин-1-ил)метанон
В раствор 2,2,2-трифторацетата (4'-хлор-2'-фторбифенил-4-ил)(пиперазин-1-ил)метанона (5,647 г, 13,05 ммоль), 1-гидроксициклопропанкарбоновой кислоты (1,332 г, 13,05 ммоль) и гексафторфосфата О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (7,42 г, 19,57 ммоль) в ДМФ (50,0 мл) добавляли N,N-диизопропилэтиламин (9,12 мл, 52,2 ммоль) и реакционную смесь перемешивали при КТ в течение 18 часов. Разделяли реакционную смесь в этилацетате и воде. Органическую фазу промывали водой и солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением желтой маслянистой жидкости. Полученное вещество очищали путем колоночной хроматографии на силикагеле (100 г колонка Biotage, элюировали с градиентом 0-5% смесями метанол-этилацетат) с получением беловатого твердого вещества, которое дополнительно очищали путем колоночной хроматографии на силикагеле (100 г колонка Biotage, элюировали с градиентом 0-10% смесями метанол-дихлорметан) с получением (4'-хлор-2'-фторбифенил-4-ил)(4-(1-гидроксициклопропанкарбонил)пиперазин-1-ил)метанона (1,554 г, 3,86 ммоль, выход 30%) в виде белого твердого вещества. МС (ИЭР, пол.ионы) m/z: 403 (М+1).
Способ 2.
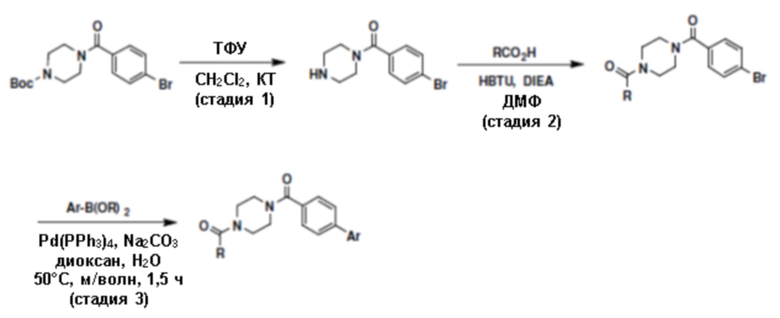
Стадия 1. 2,2,2-трифторацетат (4-бромфенил)(пиперазин-1-ил)метанона
В раствор трет-бутил-4-(4-бромбензоил)пиперазин-1-карбоксилата (2,00 г, 5,42 ммоль) в дихлорметане (25,0 мл) добавляли трифторуксусную кислоту (5,0 мл, 64,9 ммоль) и раствор перемешивали при КТ в течение 2 часов. Концентрировали реакционную смесь и растирали остаток с диэтиловым эфиром с получением 2,2,2-трифторацетата (4-бромфенил)(пиперазин-1-ил)метанона (1,992 г, 5,20 ммоль, выход 96%) в виде белого твердого вещества. МС (ИЭР, пол.ионы) m/z: 269, 271 (М+1).
Стадия 2. (4-(4-бромбензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанон
В раствор 2,2,2-трифторацетата (4-бромфенил)(пиперазин-1-ил)метанона (0,250 г, 0,652 ммоль), 1-гидроксициклопропанкарбоновой кислоты (0,067 г, 0,652 ммоль) и гексафторфосфата О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (0,371 г, 0,979 ммоль) в ДМФ (5,0 мл) добавляли N,N-диизопропилэтиламин (0,46 мл, 2,6 ммоль) и реакционную смесь перемешивали при КТ в течение 18 часов. Разделяли реакционную смесь в этилацетате и воде. Органическую фазу промывали солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением оранжевой маслянистой жидкости. Полученное вещество очищали путем колоночной хроматографии на силикагеле (25 г колонка Biotage, элюировали с градиентом 0-5% смесями метанол-дихлорметан) с получением (4-(4-бромбензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанона (0,195 г, 0,552 ммоль, выход 85%) в виде светло-оранжевого твердого вещества. МС (ИЭР, пол.ионы) m/z: 353, 355 (М+1).
Стадия 3. (4-(4-(бензо[d]тиазол-5-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)-метанон
В смесь (4-(4-бромбензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанона (0,050 г, 0,142 ммоль), 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензо[d]тиазола (0,055 г, 0,212 ммоль) и карбоната натрия (0,060 г, 0,566 ммоль) в диоксане (1,5 мл) и воде (0,30 мл) добавляли тетракис(трифенилфосфин)палладий (0) (0,016 г, 0,014 ммоль). Смесь перемешивали в микроволновом реакторе при 50°С в течение 1 часа. Реакционную смесь фильтровали через Celite и концентрировали с получением желтой маслянистой жидкости. Полученное вещество очищали путем колоночной хроматографии на силикагеле (25 г колонка Biotage, элюировали с градиентом 0-5% смесями метанол-этилацетат) с получением (4-(4-(бензо[d]тиазол-5-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанона (0,020 г, 0,049 ммоль, выход 35%) в виде беловатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 408 (М+1).
Способ 3.
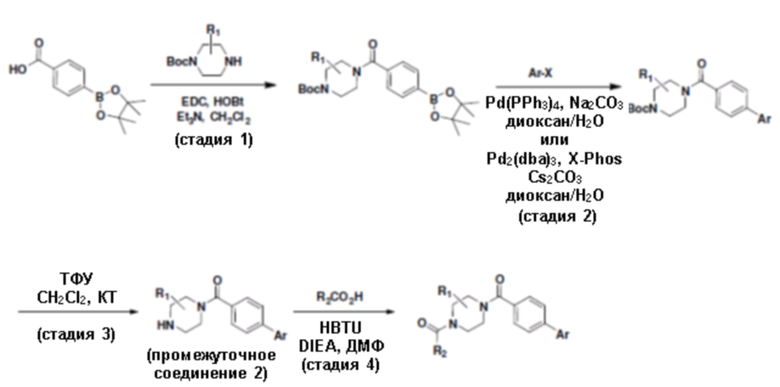
Стадия 1. Трет-бутил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоил)-пиперазин-1-карбоксилат
Смесь 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензойной кислоты (15 г, 60,5 ммоль), трет-бутил-пиперазин-1-карбоксилата (11,26 г, 60,5 ммоль), гидрата 1Н-бензо[d][1,2,3]триазол-1-ола (4,63 г, 30,2 ммоль), гидрохлорида N1-((этилимино)метилен)-N3,N3-диметилпропан-1,3-диамина (12,75 г, 66,5 ммоль) и триэтиламина (33,7 мл, 242 ммоль) в дихлорметане (180 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь промывали насыщенным водным раствором бикарбоната натрия (75 мл), 0,5М раствором HCl (75 мл) и снова насыщенным водным раствором бикарбоната натрия (75 мл). Концентрировали объединенные органические фазы и полученное твердое вещество суспендировали в растворе 1:1 метил-трет-бутиловый эфир/гексан (200 мл). Фильтровали и сушили полученное вещество с получением трет-бутил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоил)пиперазин-1-карбоксилата (20,03 г, 48,1 ммоль, 80%). МС (ИЭР, пол.ионы) m/z: 417 (М+1).
Стадия 2 (Pd(PPh3)4). Трет-бутил-4-(4-(1Н-бензо[d]имидазол-2-ил)бензоил)пиперазин-1-карбоксилат
В смесь трет-бутил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоил)пиперазин-1-карбоксилата (0,050 г, 0,120 ммоль), 2-бром-1Н-бензо[d]имидазола (0,035 г, 0,180 ммоль) и карбоната натрия (0,051 г, 0,480 ммоль) в диоксане (1,5 мл) и воде (0,30 мл) добавляли тетракис(трифенилфосфин)палладий (0) (0,028 г, 0,024 ммоль). Смесь перемешивали в микроволновом реакторе при 50°С в течение 3 часов. Реакционную смесь фильтровали через Celite и концентрировали. Очищали остаток путем колоночной хроматографии на силикагеле (10 г колонка Biotage, элюировали с градиентом 0-50% смесями этилацетат-гексан) с получением трет-бутил-4-(4-(1Н-бензо[d]имидазол-2-ил)бензоил)пиперазин-1-карбоксилата (0,027 г, 0,066 ммоль, выход 55%) в виде беловатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 407 (М+1).
Стадия 2 (X-Phos). Трет-бутил-4-(4-(пиразоло[1,5-a]пиридин-2-ил)бензоил)пиперазин-1-карбоксилат
Смесь трет-бутил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоил)пиперазин-1-карбоксилата (0,100 г, 0,240 ммоль), 2-хлорпиразоло[1,5-a]пиридина (0,046 г, 0,300 ммоль), трис(дибензилиденацетон)дипалладия (0,011 г, 0,012 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенила (X-Phos, 0,011 г, 0,024 ммоль) и карбоната цезия (0,235 г, 0,721 ммоль) в диоксане (2,5 мл) и воде (0,50 мл) перемешивали в микроволновом реакторе при 80°С в течение 1 часа. Фильтровали реакционную смесь через Celite и концентрировали с получением желто-коричневой маслянистой жидкости. Полученное вещество очищали путем колоночной хроматографии на силикагеле (25 г колонка Biotage, элюировали с градиентом 0-50-100% смесями этилацетат-гексан) с получением трет-бутил-4-(4-(пиразоло[1,5-a]пиридин-2-ил)бензоил)пиперазин-1-карбоксилата (0,056 г, 0,138 ммоль, выход 57%) в виде беловатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 407 (М+1).
Стадия 3. 2,2,2-трифторацетат (4-(1Н-бензо[d]имидазол-2-ил)фенил)(пиперазин-1-ил)метанона (промежуточное соединение 2)
В раствор трет-бутил-4-(4-(1Н-бензо[d]имидазол-2-ил)бензоил)пиперазин-1-карбоксилата (0,027 г, 0,066 ммоль) в дихлорметане (3,0 мл) добавляли трифторуксусную кислоту (1,0 мл, 12,98 ммоль) и раствор перемешивали при КТ в течение 1,5 часа. Концентрировали реакционную смесь и растирали остаток с диэтиловым эфиром и фильтровали с получением 2,2,2-трифторацетата (4-(1Н-бензо[d]имидазол-2-ил)фенил)(пиперазин-1-ил)метанона (0,028 г, 0,067 ммоль, выход 100%) в виде коричневой пленки. МС (ИЭР, пол.ионы) m/z: 307 (М+1).
Стадия 4. (4-(4-(1Н-бензо[d]имидазол-2-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанон
В раствор 2,2,2-трифторацетата (4-(1Н-бензо[d]имидазол-2-ил)фенил)(пиперазин-1-ил)метанона (0,028 г, 0,067 ммоль), 1-гидроксициклопропанкарбоновой кислоты (7,14 мг, 0,070 ммоль) и гексафторфосфата О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (0,038 г, 0,100 ммоль) в ДМФ (1,0 мл) добавляли DIEA (0,047 мл, 0,266 ммоль) и реакционную смесь перемешивали при КТ в течение 20 часов. Добавляли воду и разделяли смесь в этилацетате и воде. Отделяли водную фазу и экстрагировали дихлорметаном. Объединенные органические фазы промывали солевым раствором, сушили над безводным сульфатом натрия и фильтровали с получением коричневой маслянистой жидкости. Полученное вещество очищали путем колоночной хроматографии на силикагеле (10 г колонка Biotage, элюировали с градиентом 0-5% смесями метанол-этилацетат) с получением (4-(4-(1Н-бензо[d]имидазол-2-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанона (0,004 г, 10,24 мкмоль, выход 15%) в виде белого твердого вещества. МС (ИЭР, пол.ионы) m/z: 391 (М+1).
Способ 4.
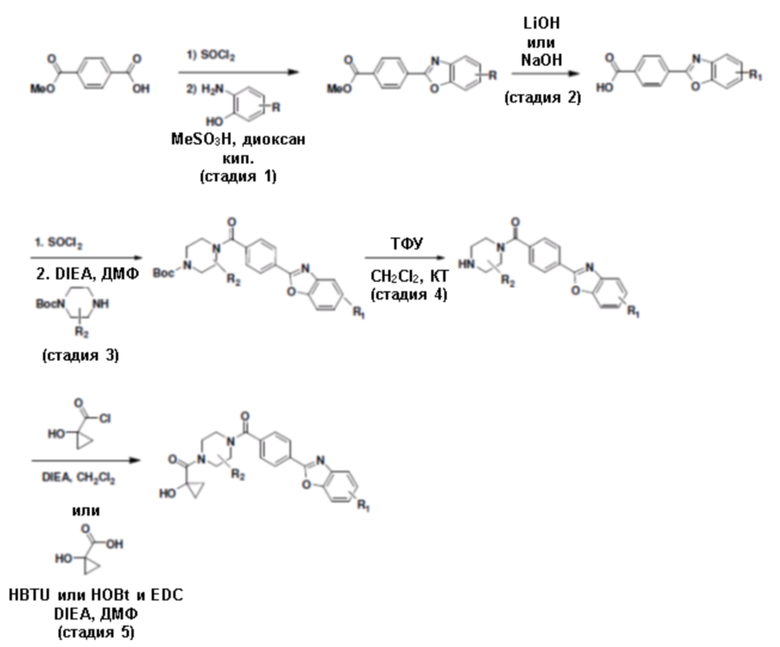
Стадия 1. Метил-4-(бензо[d]оксазол-2-ил)бензоат
К моно-метил-терефталату (5,00 г, 27,8 ммоль) добавляли тионилхлорид (10 мл, 137 ммоль). Добавляли каплю ДМФ и смесь грели при 80°С в течение 2 часов. Удаляли избыток тионилхлорида и обрабатывали остаток 2-аминофенолом (3,03 г, 27,8 ммоль) и диоксаном (60 мл). Добавляли метансульфокислоту (5,59 мл, 86 ммоль) и реакционную смесь грели при температуре обратной конденсации в течение 18 часов. Концентрировали раствор и разделяли остаток в дихлорметане и нас.водн. растворе бикарбоната натрия. Отделяли водную фазу и промывали дихлорметаном и промывали объединенные органические фазы солевым раствором, фильтровали и концентрировали с получением коричневого твердого вещества. Полученное вещество растирали с метанолом, фильтровали и сушили с получением метил-4-(бензо[d]оксазол-2-ил)бензоата (4,827 г, 19,06 ммоль, выход 69%) в виде беловатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 254 (М+1).
Стадия 2 (LiOH). 4-(бензо[d]оксазол-2-ил)бензойная кислота
В раствор метил-4-(бензо[d]оксазол-2-ил)бензоата (4,728 г, 18,67 ммоль) в ТГФ (25 мл), метаноле (25 мл) и воде (25 мл) добавляли гидроксид лития (0,894 г, 37,3 ммоль) и смесь перемешивали при КТ в течение 18 часов. Концентрировали реакционную смесь и подкисляли водный раствор до рН=5-6 при помощи 1н. водн. раствора HCl. Отфильтровывали полученный беловатый осадок и сушили с получением 4-(бензо[d]оксазол-2-ил)бензойной кислоты (3,846 г, 16,08 ммоль, выход 86%) в виде беловатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 240 (М+1).
Стадия 2 (NaOH). 4-(5-цианобензо[d]оксазол-2-ил)бензойная кислота
В раствор метил-4-(5-цианобензо[d]оксазол-2-ил)бензоата (0,233 г, 0,837 ммоль) в ТГФ (4,0 мл) и воде (2,00 мл) добавляли гидроксид натрия (0,067 г, 1,675 ммоль) и смесь перемешивали при КТ в течение 18 часов. Концентрировали реакционную смесь и подкисляли водный раствор до рН=5-6 при помощи 1н. водн. раствора HCl. Отфильтровывали полученный беловатый осадок и сушили с получением 4-(5-цианобензо[d]оксазол-2-ил)бензойной кислоты (0,176 г, 0,666 ммоль, выход 80%) в виде беловатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 265 (М+1).
Стадия 3. Трет-бутил-4-(4-(бензо[d]оксазол-2-ил)бензоил)пиперазин-1-карбоксилат
К 4-(бензо[d]оксазол-2-ил)бензойной кислоте (3,5 г, 14,63 ммоль) добавляли тионилхлорид (25,0 мл, 343 ммоль). Добавляли каплю ДМФ и смесь грели при 80°С в течение 1 часа. Удаляли избыток тионилхлорида и добавляли ДМФ (40 мл). Добавляли трет-бутил-пиперазин-1-карбоксилат (2,72 г, 14,63 ммоль) и N,N-диизопропилэтиламин (7,67 мл, 43,9 ммоль) и реакционную смесь перемешивали при КТ в течение 1,5 часа. Добавляли воду и перемешивали полученную смесь, а затем фильтровали и сушили с получением трет-бутил-4-(4-(бензо[d]оксазол-2-ил)бензоил)пиперазин-1-карбоксилата (5,006 г, 12,29 ммоль, выход 84%) в виде светло-коричневатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 408 (М+1).
Стадия 4. 2,2,2-трифторацетат (4-(бензо[d]оксазол-2-ил)фенил)(пиперазин-1-ил)метанона
В раствор трет-бутил-4-(4-(бензо[d]оксазол-2-ил)бензоил)пиперазин-1-карбоксилата (5,006 г, 12,29 ммоль) в дихлорметане (50 мл) добавляли трифторуксусную кислоту (10,0 мл, 130 ммоль) и раствор перемешивали при КТ в течение 5 часов. Концентрировали реакционную смесь и растирали остаток с диэтиловым эфиром и фильтровали с получением 2,2,2-трифторацетата (4-(бензо[d]оксазол-2-ил)фенил)(пиперазин-1-ил)метанона (5,501 г, 13,06 ммоль, выход 100%) в виде беловатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 308 (М+1).
Стадия 5 (сочетание с хлорангидридом кислоты). (4-(4-(бензо[d]оксазол-2-ил)бензоил)-пиперазин-1-ил)(1-гидроксициклопропил)метанон
К 1-гидроксициклопропанкарбоновой кислоте (0,115 г, 1,123 ммоль) добавляли тионилхлорид (2,0 мл, 27,4 ммоль). Добавляли каплю ДМФ и смесь грели при 80°С в течение 1 часа. Удаляли избыток тионилхлорида с получением коричневой маслянистой жидкости. Полученное вещество добавляли в раствор 2,2,2-трифторацетата (4-(бензо[d]оксазол-2-ил)фенил)(пиперазин-1-ил)метанона (0,338 г, 0,802 ммоль) и N,N-диизопропилэтиламина (0,70 мл, 4,01 ммоль) в дихлорметане (3,0 мл). Реакционную смесь перемешивали при КТ в течение 2 часов. Концентрировали смесь и очищали остаток путем колоночной хроматографии на силикагеле (25 г колонка Biotage, элюировали с градиентом 0-5% смесями метанол-этилацетат) с получением (4-(4-(бензо[d]оксазол-2-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанона (0,208 г, 0,531 ммоль, выход 66%) в виде розового твердого вещества. МС (ИЭР, пол.ионы) m/z: 392 (М+1).
Стадия 5 (сочетание с HBTU). (4-(4-(5-хлорбензо[d]оксазол-2-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанон
В раствор 2,2,2-трифторацетата (4-(5-хлорбензо[d]оксазол-2-ил)фенил)(пиперазин-1-ил)метанона (0,245 г, 0,537 ммоль), 1-гидроксициклопропанкарбоновой кислоты (0,058 г, 0,564 ммоль) и гексафторфосфата О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HBTU, 0,306 г, 0,806 ммоль) в ДМФ (5,0 мл) добавляли N,N-диизопропилэтиламин (0,376 мл, 2,150 ммоль) и реакционную смесь перемешивали при КТ в течение 20 часов. Добавляли воду и отфильтровывали и сушили полученный осадок. Полученное вещество очищали путем колоночной хроматографии на силикагеле (50 г колонка Biotage, элюировали с градиентом 0-5% смесями метанол-этилацетат) с получением (4-(4-(5-хлорбензо[d]оксазол-2-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанона (0,136 г, 0,319 ммоль, выход 59%) в виде беловатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 426 (М+1).
Способ 5.
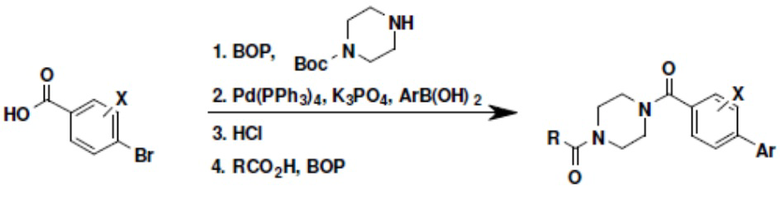
Получение (4-(2-хлор-4-(хинолин-6-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанона
В раствор трет-бутил-пиперазин-1-карбоксилата (0,2М раствор в 1,4-диоксане, содержащем 5% N,N-диизопропилэтиламина, 150 мкл, 0,03 ммоль) добавляли 4-бром-2-хлорбензойную кислоту (0,2М в 1,4-диоксане, 180 мкл, 0,036 ммоль). Добавляли гексафторфосфат (бензотриазол-1-илокси)трис(диметиламино)фосфония (BOP, 0,5М раствор в 1,2-дихлорэтане, 72 мкл, 0,036 ммоль) и полученную смесь помещали во встряхиватель при КТ на 2 часа. Добавляли хинолин-6-илбороновую кислоту (0,2М раствор в 1,4-диоксане, 225 мкл, 0,045 ммоль) и фосфат калия (1М водный, 150 мкл, 0,15 ммоль). Затем добавляли раствор тетракис(трифенилфосфин)палладия (0) (0,02М в толуоле, 75 мкл, 1,5 мкмоль) в атмосфере азота и полученную смесь помещали во встряхиватель в защитной перчаточной камере в атмосфере азота и грели при 80°С в течение ночи. После охлаждения до КТ разбавляли смесь солевым раствором (0,30 мл) и этилацетатом (0,5 мл). Отделяли органический слой и снова экстрагировали водный слой этилацетатом (0,6 мл). Сушили объединенные органические слои и перерастворяли остаток в метаноле (400 мкл). Добавляли раствор HCl (4н. в 1,4-диоксане, 75 мкл, 0,2 ммоль) и смесь грели на встряхивателе при 50°С в течение 1 часа. Реакционную смесь сушили в вакууме и перерастворяли остаток в 10% растворе N,N-диизопропилэтиламина в диметилацетамиде (200 мкл). Добавляли 1-гидроксициклопропанкарбоновую кислоту (0,2М в 1,4-диоксане, 225 мкл, 0,045 ммоль), затем раствор BOP (0,5М в 1,2-ДХЭ, 90 мкл, 0,045 ммоль). Смесь помещали во встряхиватель при КТ на 2 часа. Затем реакционную смесь разбавляли раствором гидроксида натрия (1н. в солевом растворе, 0,45 мл) и этилацетатом (0,5 мл). Отделяли органический слой и экстрагировали водный слой этилацетатом (0,6 мл). Концентрировали объединенные органические слои и очищали остаток путем высокоэффективной жидкостной хроматографии (фракции собирали на системе препаративной ВЭЖХ-МС Waters Autopurification в следующих условиях: колонка: Waters XBridge OBD С18, 5 мкм, 19х50 мм; расход: 20 мл/мин; мобильная фаза: вода, содержащая 0,1% гидроксида аммония, (А), и метанол, содержащий 0,1% гидроксида аммония, (В), использовали следующий градиент: от 0 до 2 минут (15%В), от 2 до 6 минут (15-100%В); детектор ZQ Mass Detector в режиме ионизации электронным распылением) с получением (4-(2-хлор-4-(хинолин-6-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанона (5,9 мг, 14 мкмоль, выход 45%). МС (ИЭР, пол.ионы) m/z: 436 (М+1).
Способ 6.
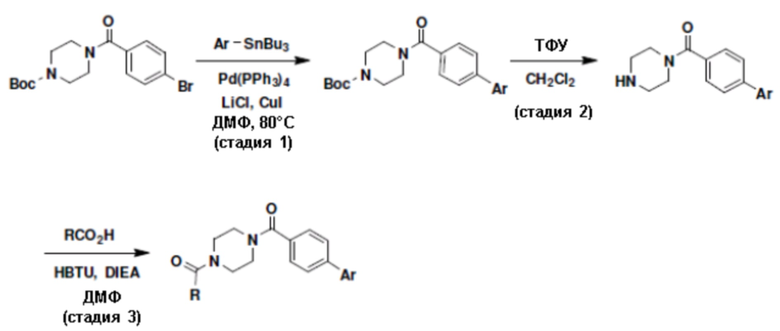
Стадия 1. Трет-бутил-4-(4-(бензо[d]тиазол-2-ил)бензоил)пиперазин-1-карбоксилат
В смесь трет-бутил-4-(4-бромбензоил)пиперазин-1-карбоксилата (1,00 г, 2,71 ммоль), 2-(трибутилстаннил)бензо[d]тиазола (1,00 мл, 2,84 ммоль), хлорида лития (0,230 г, 5,42 ммоль) и йодида меди (I) (0,026 г, 0,135 ммоль) в ДМФ (25 мл) добавляли тетракис(трифенилфосфин)палладий (0) (0,313 г, 0,271 ммоль). Смесь перемешивали при 80°С в течение 2 часов, а затем при 90°С в течение 8 часов. Фильтровали реакционную смесь через Celite и разделяли фильтрат в дихлорметане и воде. Отделяли органическую фазу и промывали солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением оранжевой маслянистой жидкости. Полученное вещество очищали путем колоночной хроматографии на силикагеле (50 г колонка Biotage, элюировали с градиентом 0-50% смесями этилацетат-гексан) с получением трет-бутил-4-(4-(бензо[d]тиазол-2-ил)бензоил)пиперазин-1-карбоксилата (0,586 г, 1,384 ммоль, выход 51%) в виде коричневого твердого вещества. МС (ИЭР, пол.ионы) m/z: 424 (М+1).
Стадия 2. 2,2,2-трифторацетат (4-(бензо[d]тиазол-2-ил)фенил)(пиперазин-1-ил)метанона
В раствор трет-бутил-4-(4-(бензо[d]тиазол-2-ил)бензоил)пиперазин-1-карбоксилата (0,586 г, 1,38 ммоль) в дихлорметане (5,0 мл) добавляли трифторуксусную кислоту (1,00 мл, 13,0 ммоль) и перемешивали раствор при КТ в течение 3 часов. Концентрировали реакционную смесь и растирали остаток с диэтиловым эфиром и фильтровали с получением 2,2,2-трифторацетата (4-(бензо[d]тиазол-2-ил)фенил)(пиперазин-1-ил)метанона (0,542 г, 1,239 ммоль, выход 90%) в виде коричневатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 324 (М+1).
Стадия 3. (4-(4-(бензо[d]тиазол-2-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)-метанон
В раствор 2,2,2-трифторацетата (4-(бензо[d]тиазол-2-ил)фенил)(пиперазин-1-ил)метанона (0,542 г, 1,239 ммоль), 1-гидроксициклопропанкарбоновой кислоты (0,133 г, 1,301 ммоль) и гексафторфосфата О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (0,705 г, 1,859 ммоль) в ДМФ (5,0 мл) добавляли N,N-диизопропилэтиламин (0,87 мл, 5,0 ммоль) и перемешивали реакционную смесь при КТ в течение 16 часов. Добавляли воду и отфильтровывали и сушили полученный осадок. Полученное вещество очищали путем колоночной хроматографии на силикагеле (50 г колонка Biotage, элюировали с градиентом 0-5% смесями метанол-этилацетат) с получением (4-(4-(бензо[d]тиазол-2-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанона (0,235 г, 0,577 ммоль, выход 47%) в виде беловатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 408 (М+1).
Способ 7.

Получение 5-(4-(4-(1-гидроксициклопропанкарбонил)пиперазин-1-карбонил)фенил)-1Н-индол-3-карбонитрила:
В раствор трет-бутил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоил)-пиперазин-1-карбоксилата (0,2М в 1,4-диоксане, 100 мкл, 0,02 ммоль) добавляли 5-бром-1Н-индол-3-карбонитрил (0,2М в 1,4-диоксане, 100 мкл, 0,02 ммоль) и раствор фосфата калия (1М водный, 100 мкл, 0,1 ммоль). Смесь барботировали азотом и добавляли тетракис(трифенилфосфин)палладий (0) (0,02М в толуоле, 50 мкл, 1 мкмоль). Полученную смесь помещали во встряхиватель в защитной перчаточной камере в атмосфере азота и грели при 80°С в течение ночи. После охлаждения до КТ разбавляли смесь 0,35 мл солевого раствора и 0,5 мл этилацетата. Отделяли органический слой и снова экстрагировали водный слой этилацетатом (0,6 мл). Концентрировали объединенные органические слои и растворяли остаток в 200 мкл метанола. Добавляли раствор HCl (50 мкл, 4н. в 1,4-диоксане, 0,2 ммоль). Смесь помещали во встряхиватель при 50°С на 1 час. Концентрировали реакционную смесь в вакууме и растворяли остаток в 10% растворе N,N-диизопропилэтиламина в диметилацетамиде (200 мкл). Добавляли 1-гидроксициклопропанкарбоновую кислоту (0,2М в 1,4-диоксане, 120 мкл, 0,024 ммоль), затем раствор BOP (0,5М в 1,2-дихлорэтане, 48 мкл, 0,024 ммоль). Смесь помещали во встряхиватель при КТ на 2 часа. Затем реакционную смесь разбавляли 0,45 мл 1н. NaOH в солевом растворе и 0,5 мл этилацетата. Отделяли органический слой и снова экстрагировали водный слой этилацетатом (0,6 мл). Концентрировали объединенные органические слои и очищали остаток путем ВЭЖХ: фракции собирали на системе препаративной ВЭЖХ-МС Waters Autopurification в следующих условиях: колонка, Waters XBridge OBD C18, 5 мкм, 19х50 мм; расход 20 мл/мин; мобильная фаза, вода, содержащая 0,1% гидроксида аммония, (А), и метанол, содержащий 0,1% гидроксида аммония, (В), использовали следующий градиент, от 0 до 2 минут (15%В), от 2 до 6 минут (15-100%В); детектор ZQ Mass Detector в режиме ионизации электронным распылением. Получали 5-(4-(4-(1-гидроксициклопропанкарбонил)пиперазин-1-карбонил)фенил)-1Н-индол-3-карбонитрил (3,5 мг, 8,4 мкмоль, выход 42%). МС (ИЭР, пол.ионы) m/z: 415 (М+1).
Способ 8.
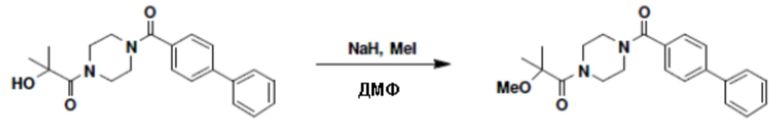
1-(4-(бифенилкарбонил)пиперазин-1-ил)-2-метокси-2-метилпропан-1-он
В раствор 1-(4-(бифенилкарбонил)пиперазин-1-ил)-2-гидрокси-2-метилпропан-1-она (32 мг, 0,091 ммоль) в ДМФ (4 мл) добавляли гидрид натрия (9,08 мг, 0,227 ммоль). После перемешивания при КТ в течение 30 минут добавляли метилиодид (0,028 мл, 0,454 ммоль). Смесь перемешивали при КТ в течение ночи. Концентрировали смесь и очищали остаток путем препаративной обращенно-фазовой ВЭЖХ (20 мл/мин, 10 мин, градиент 15%-85% CH3CN, 0,01% HCO2H, на колонке XTerra Prep MS С18 OBD, 5 мкм, 19х100 мм) с получением 1-(4-(бифенилкарбонил)пиперазин-1-ил)-2-метокси-2-метилпропан-1-она (12,2 мг, 37%). МС (ИЭР, пол.ионы) m/z: 367 (М+1).
Способ 9.
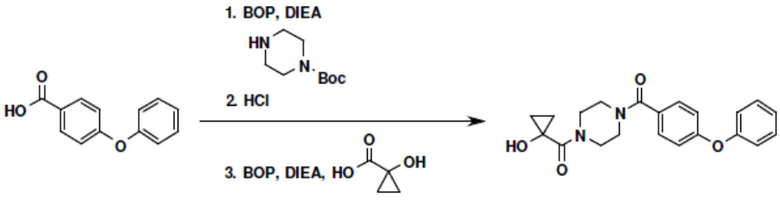
Получение (4-(2-хлор-4-(хинолин-6-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанона
В раствор трет-бутил-пиперазин-1-карбоксилата (0,2М в 1,4-диоксане, содержащем 5% N,N-диизопропилэтиламина, 150 мкл, 0,03 ммоль) добавляли 4-феноксибензойную кислоту (0,2М в 1,4-диоксане, 150 мкл, 0,03 ммоль), затем раствор гексафторфосфата (бензотриазол-1-илокси)трис(диметиламино)фосфония (BOP, 0,5М в 1,2-дихлорэтане, 66 мкл, 0,033 ммоль). Полученную смесь помещали во встряхиватель при комнатной температуре на 2 часа. Добавляли раствор хлороводородной кислоты (4н. в 1,4-диоксане, 75 мкл) и смесь помещали во встряхиватель при 50°С на 1 час. После охлаждения до комнатной температуры концентрировали смесь и перерастворяли остаток в 10% растворе диизопропилэтиламина в диметилацетамиде (200 мкл). В смесь добавляли 1-гидроксициклопропанкарбоновую кислоту (0,2М в 1,4-диоксане, 180 мкл, 0,036 ммоль), затем раствор BOP (0,5М в 1,2-дихлорэтане, 72 мкл, 0,036 ммоль). Смесь помещали во встряхиватель при комнатной температуре на 2 часа. Затем реакционную смесь разбавляли раствором гидроксида натрия (1н. в солевом растворе) и этилацетатом (0,5 мл). Отделяли органический слой и экстрагировали водный слой этилацетатом (0,6 мл). Концентрировали объединенные органические слои и очищали остаток путем высокоэффективной жидкостной хроматографии (фракции собирали на системе препаративной ВЭЖХ-МС Waters Autopurification в следующих условиях: колонка: Waters XBridge OBD C18, 5 мкм, 19х50 мм; расход 20 мл/мин; мобильная фаза: вода, содержащая 0,1% гидроксида аммония, (А), и метанол, содержащий 0,1% гидроксида аммония, (В), использовали следующий градиент: от 0 до 2 минут (15%В), от 2 до 6 минут (15-100%В); детектор ZQ Mass Detector в режиме ионизации электронным распылением) с получением (4-(2-хлор-4-(хинолин-6-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанона (4,1 мг, 11 мкмоль, выход 37%). МС (ИЭР, пол.ионы) m/z: 367 (М+1).
Способ 10.
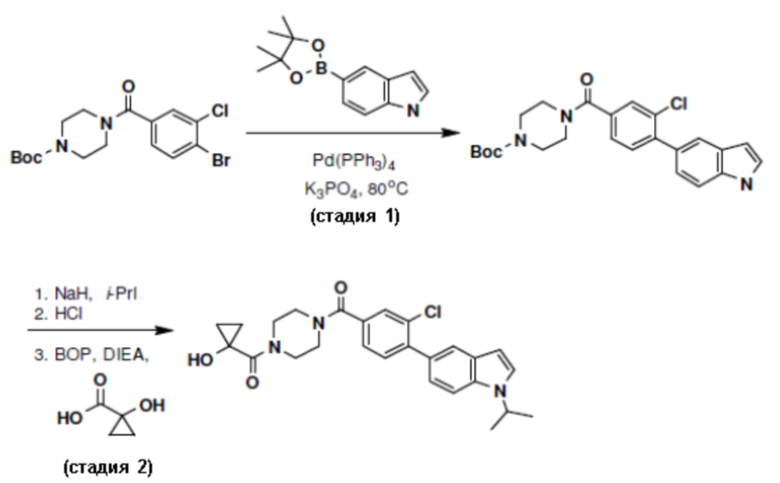
Стадия 1. Трет-бутил-4-(3-хлор-4-(1Н-индол-5-ил)бензоил)пиперазин-1-карбоксилат
В раствор трет-бутил-4-(4-бром-3-хлорбензоил)пиперазин-1-карбоксилата (505 мг, 1,25 ммоль) в 1,4-диоксане (9 мл) добавляли 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индол (365 мг, 1,5 ммоль), фосфат калия (1,06 г, 5 ммоль) и воду (3 мл). Смесь барботировали азотом и добавляли тетракис(трифенилфосфин)палладий (0) (72,2 мг, 0,063 ммоль). Закрывали смесь и грели при 80°С в течение ночи. После охлаждения до КТ разбавляли смесь водой (10 мл) и этилацетатом (20 мл). Отделяли органический слой и экстрагировали водный слой этилацетатом (20 мл). Концентрировали объединенные органические слои с получением трет-бутил-4-(3-хлор-4-(1Н-индол-5-ил)бензоил)-пиперазин-1-карбоксилата (578 мг, 1,31 ммоль, выход 100%). Неочищенный продукт использовали без дополнительной очистки. МС (ИЭР, пол.ионы) m/z: 440 (М+1).
Стадия 2. (4-(3-хлор-4-(1-изопропил-1Н-индол-5-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанон
В пробирку, содержащую гидрид натрия (60% в минеральном масле, 1,6 мг, 0,040 ммоль) и ДМФ (0,1 мл) добавляли трет-бутил-4-(3-хлор-4-(1Н-индол-5-ил)бензоил)-пиперазин-1-карбоксилат (0,2М в ДМФ, 0,1 мл, 0,02 ммоль), затем 2-йодпропан (0,2М в ДМФ, 0,2 мл, 0,04 ммоль). Полученную смесь помещали во встряхиватель на 30 минут при комнатной температуре. Затем смесь разбавляли водой и этилацетатом. Отделяли органический слой и экстрагировали водный слой этилацетатом. Концентрировали объединенные органические слои в вакууме. Остаток растворяли в метаноле (0,3 мл) и добавляли раствор HCl (4н. в 1,4-диоксане, 50 мкл). Смесь помещали во встряхиватель при 50°С на 1 час. После охлаждения до комнатной температуры концентрировали смесь. Остаток растворяли в 5% растворе N,N-диизопропилэтиламина в диметилацетамиде (0,2 мл) и добавляли 1-гидроксициклопропанкарбоновую кислоту (0,2М в 1,4-диоксане, 120 мкл, 0,024 ммоль), затем раствор BOP (0,5М в 1,2-дихлорэтане, 48 мкл, 0,024 ммоль). Смесь помещали во встряхиватель при комнатной температуре на 2 часа. Реакционную смесь разбавляли раствором гидроксида натрия (1н. в солевом растворе, 0,45 мл) и этилацетатом (0,5 мл). Отделяли органический слой и экстрагировали водный слой этилацетатом (0,6 мл). Концентрировали объединенные органические слои и очищали остаток путем высокоэффективной жидкостной хроматографии (фракции собирали на системе препаративной ВЭЖХ-МС Waters Autopurification в следующих условиях: колонка: Waters XBridge OBD C18, 5 мкм, 19х50 мм; расход 20 мл/мин; мобильная фаза: вода, содержащая 0,1% гидроксида аммония, (А), и метанол, содержащий 0,1% гидроксида аммония, (В), использовали следующий градиент: от 0 до 2 минут (15%В), от 2 до 6 минут (15-100%В); детектор ZQ Mass Detector в режиме ионизации электронным распылением) с получением (4-(3-хлор-4-(1-изопропил-1Н-индол-5-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)-метанона (2,2 мг, 4,7 мкмоль, выход 24%). МС (ИЭР, пол.ионы) m/z (М+1).
Способ 11.
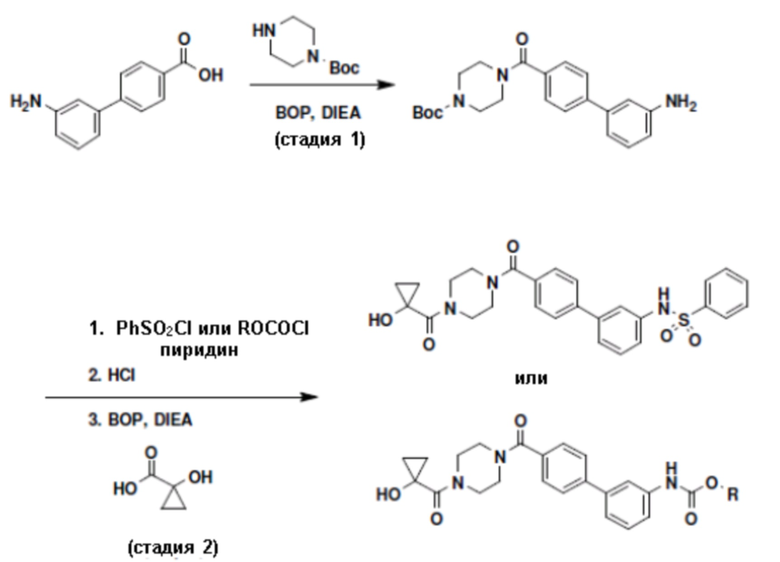
Стадия 1. Трет-бутил-4-(3'-амино-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилат
В раствор трет-бутил-пиперазин-1-карбоксилата (745 мг, 4 ммоль) в ДМФ (20 мл) добавляли N,N-диизопропилэтиламин (1,43 мл, 8 ммоль), затем 3'-аминобифенил-4-карбоновую кислоту (853 мг, 4 ммоль) и гексафторфосфат (бензотриазол-1-илокси)трис-(диметиламино)фосфония (BOP, 2,12 г, 4,8 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 часа. Затем смесь выливали в смесь лед-вода (40 мл) при перемешивании. Отфильтровывали осадок, промывали водой и сушили на воздухе с получением трет-бутил-4-(3'-аминобифенилкарбонил)пиперазин-1-карбоксилата (1,2 г, 3,15 ммоль, выход 79%). Продукт использовали на следующей стадии без дополнительной очистки. МС (ИЭР, пол.ионы) m/z: 382 (М+1).
Стадия 2. N-(4'-(4-(1-гидроксициклопропанкарбонил)пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)бензолсульфонамид или этил-(4'-(4-(1-гидроксициклопропан-1-карбонил)пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)карбамат
В раствор трет-бутил-4-(3'-аминобифенилкарбонил)пиперазин-1-карбоксилата (0,2М в 1,4-диоксане, 150 мкл, 0,03 ммоль) добавляли пиридин (9,7 мкл, 0,12 ммоль), затем бензолсульфонилхлорид (0,2М в 1,4-диоксане, 300 мкл, 0,06 ммоль) или этил-карбонохлоридат (0,2М в 1,4-диоксане, 300 мкл, 0,06 ммоль). Полученную смесь помещали во встряхиватель при 80°С на ночь. Реакционную смесь разбавляли раствором гидроксида натрия (1н. в солевом растворе, 0,45 мл) и этилацетатом (0,6 мл). Отделяли органический слой и экстрагировали водный слой этилацетатом (0,6 мл). Концентрировали объединенные органические слои. Остаток растворяли в метаноле (200 мкл) и добавляли раствор HCl (4н. в 1,4-диоксане, 75 мкл). Смесь помещали во встряхиватель при 50°С на 1 час. Концентрировали реакционную смесь в вакууме и растворяли остаток в растворе диметилацетамида, содержащем 10% N,N-диизопропилэтиламина (200 мкл). Добавляли 1-гидроксициклопропанкарбоновую кислоту (0,2М в 1,4-диоксане, 180 мкл, 0,036 ммоль), затем раствор BOP (0,5М в 1,2-дихлорэтане, 72 мкл, 0,036 ммоль). Смесь помещали во встряхиватель при КТ на 2 часа. Затем разбавляли реакционную смесь раствором гидроксида натрия (1н. в солевом растворе, 0,45 мл) и этилацетатом (0,5 мл). Отделяли органический слой и экстрагировали водный слой этилацетатом (0,6 мл). Концентрировали объединенные органические слои и очищали остаток путем высокоэффективной жидкостной хроматографии (фракции собирали на системе препаративной ВЭЖХ-МС Waters Autopurification в следующих условиях: колонка: Waters XBridge OBD C18, 5 мкм, 19х50 мм; расход 20 мл/мин; мобильная фаза: вода, содержащая 0,1% гидроксида аммония, (А), и метанол, содержащий 0,1% гидроксида аммония, (В), использовали следующий градиент: от 0 до 2 минут (15%В), от 2 до 6 минут (15-100%В); детектор ZQ Mass Detector в режиме ионизации электронным распылением) с получением N-(4'-(4-(1-гидроксициклопропанкарбонил)пиперазин-1-карбонил)бифенил-3-ил)бензолсульфонамида (7,8 мг, 15 мкмоль, выход 51%). МС (ИЭР, пол.ионы) m/z: 506 (М+1).
Способ 12.
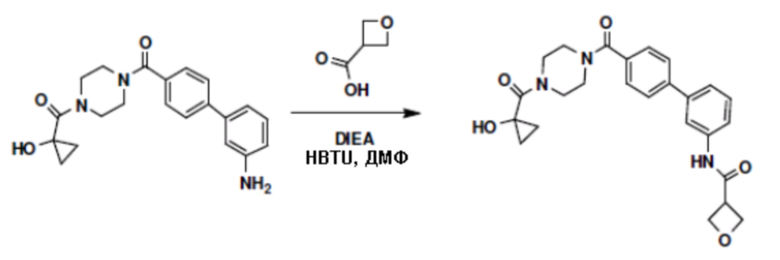
Получение N-(4'-(4-(1-гидроксициклопропанкарбонил)пиперазин-1-карбонил)бифенил-3-ил)оксетан-3-карбоксамида
(3'-аминобифенил-4-ил)(4-(1-гидроксициклопропанкарбонил)пиперазин-1-ил)метанон (0,168 г, 0,460 ммоль), N,N-диизопропилэтиламин (0,241 мл, 1,379 ммоль) и оксетан-3-карбоновую кислоту (0,055 г, 0,506 ммоль) объединяли в ДМФ (5 мл). Добавляли гексафторфосфат О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (0,244 г, 0,644 ммоль) с получением светло-желтого раствора, который перемешивали в течение 18 часов. Добавляли N,N-диизопропилэтиламин (0,120 мл, 0,687 ммоль), гексафторфосфат О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (0,18 г, 0,474 ммоль) и оксетан-3-карбоновую кислоту (0,025 г, 0,244 ммоль). Продолжали перемешивать в течение 5 часов. Разбавляли реакционную смесь 10 мл воды и перемешивали. Добавляли еще 15 мл воды и экстрагировали водную эмульсию 50 мл дихлорметана, а затем 20 мл дихлорметана. Объединенные органические слои промывали 20 мл воды, а затем 20 мл солевого раствора, сушили над MgSO4 и концентрировали. Полученное вещество очищали путем колоночной хроматографии на силикагеле (25 г колонка Biotage, элюировали с градиентом 0-6% смесями метанол/дихлорметан). Полученное вещество повторно очищали на второй колонке с силикагелем (25 г колонка Biotage, 4-6,5% метанол/дихлорметан). Концентрирование фракций приводило к получению желтой пленки. Полученное вещество помещали в 40 мл дихлорметана и дважды промывали 5 мл разбавленного NaHCO3 и один раз 5 мл солевого раствора. Раствор сушили над MgSO4 и концентрировали с получением желтой маслянистой жидкости. Полученную маслянистую жидкость помещали в дихлорметан и растирали с гексанами. Фильтрование приводило к получению N-(4'-(4-(1-гидроксициклопропанкарбонил)пиперазин-1-карбонил)бифенил-3-ил)оксетан-3-карбоксамида (0,036 г, 0,076 ммоль, 17%) в виде желтого твердого вещества. МС (ИЭР, пол.ионы) m/z: 450 (М+1).
Способ 13.
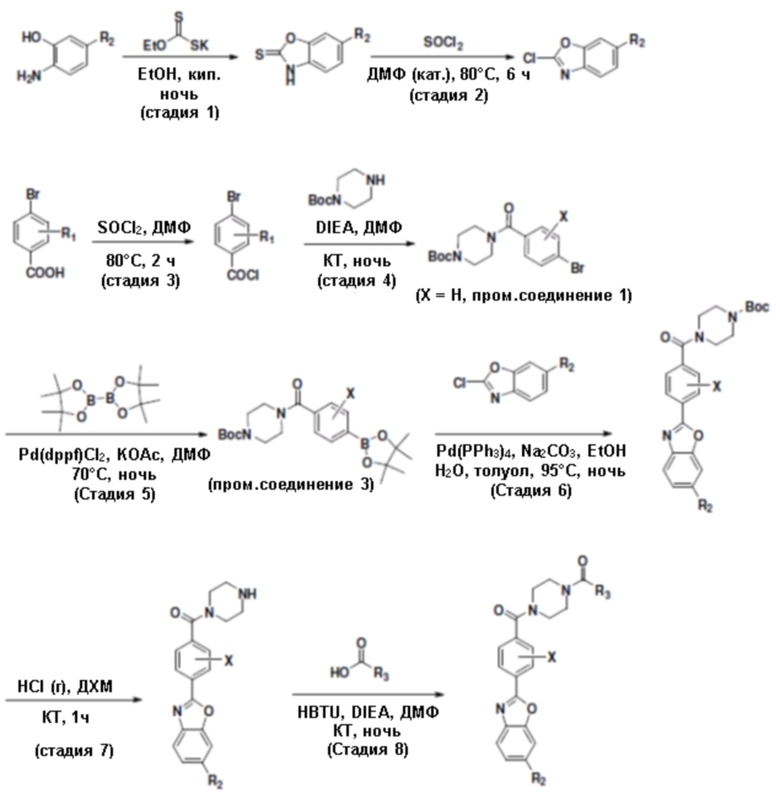
Стадия 1. 6-фторбензо[d]оксазол-2(3Н)-тион
В 250 мл круглодонную колбу помещали 2-амино-5-фторфенол (8 г, 62,93 ммоль, 1,00 экв.), этокси(калийсульфанил)метантион (11,1 г, 69,25 ммоль, 1,10 экв.), этанол (150 мл). Полученный раствор нагревали до температуры обратной конденсации в течение ночи на масляной бане. Охлаждали реакционную смесь до комнатной температуры на водяной бане, а затем концентрировали в вакууме. Полученную суспензию разбавляли 150 мл воды и доводили рН до 5 при помощи уксусной кислоты, образовывался осадок. Собирали твердые вещества путем фильтрования и сушили в печи. Это приводило к получению 9,2 г (86%) титульного соединения в виде серого твердого вещества. ЖХ-МС (ИЭР, m/z): 170 [М+Н]+.
Стадия 2. 2-хлор-6-фторбензо[d]оксазол
В 500 мл круглодонную колбу помещали 6-фтор-2,3-дигидро-1,3-бензоксазол-2-тион (9,2 г, 54,38 ммоль, 1,00 экв.), тионилхлорид (200 мл) и ДМФ (0,5 мл). Полученный раствор перемешивали в течение 6 часов при 80°С на масляной бане. После завершения взаимодействия удаляли избыток SOCl2 в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесями этилацетат/петролейный эфир (0/100~7/93). Собирали фракции и концентрировали в вакууме. Это приводило к получению 3,5 г (38%) титульного соединения в виде коричневой маслянистой жидкости. ЖХ-МС (ИЭР, m/z): 172 [М+Н]+.
Стадия 3. 4-бром-2-фторбензоилхлорид
В 250 мл круглодонную колбу помещали 4-бром-2-фторбензойную кислоту (10 г, 45,66 ммоль), тионилхлорид (50 мл) и N,N-диметилформамид (0,1 мл). Полученную смесь перемешивали в течение 2 часов при 80°С. После охлаждения до комнатной температуры концентрировали раствор в вакууме. Это приводило к получению 10 г (92%) титульного соединения в виде светло-желтой маслянистой жидкости. Продукт использовали на следующей стадии непосредственно без дополнительной очистки.
Стадия 4. Трет-бутил-4-(4-бром-2-фторбензоил)пиперазин-1-карбоксилат
В 250 мл круглодонную колбу помещали раствор 4-бром-2-фторбензоилхлорида (10 г, 42,11 ммоль, 1,00 экв.) в N,N-диметилформамиде (100 мл). После этого за несколько раз добавляли трет-бутил-пиперазин-1-карбоксилат (7,88 г, 42,31 ммоль, 1,00 экв.). Затем добавляли DIEA (16,25 г, 125,74 ммоль, 2,99 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Осаждали продукт путем добавления 400 мл H2O. Собирали твердые вещества путем фильтрования и сушили в печи. Это приводило к получению 14 г (86%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 387, 389 [М+Н]+.
Стадия 5. Трет-бутил-4-(2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоил)пиперазин-1-карбоксилат (промежуточное соединение 3)
В 2000 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор трет-бутил-4-[(4-бром-2-фторфенил)карбонил]пиперазин-1-карбоксилата (57 г, 147,19 ммоль, 1,00 экв.) в N,N-диметилформамиде (800 мл), 4,4,5,5-тетраметил-2-(тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксаборолан (76,2 г, 300,07 ммоль, 2,01 экв.), PdCl2(dppf) (11 г, 15,03 ммоль, 0,10 экв.), KOAc (44,1 г, 449,36 ммоль, 3,00 экв.). Полученную смесь перемешивали в течение ночи при 70°С. После охлаждения до комнатной температуры смесь выливали в 3 л H2O. Собирали твердые вещества путем фильтрования, дополнительно очищали на колонке с силикагелем с использованием смеси этилацетат/петролейный эфир (3:7). Объединяли собранные фракции и концентрировали в вакууме. Это приводило к получению 30 г (47%) титульного соединения в виде светло-коричневого твердого вещества. ЖХ-МС (ИЭР, m/z): 435 [М+Н]+.
Стадия 6. Трет-бутил-4-(2-фтор-4-(6-фторбензо[d]оксазол-2-ил)бензоил)пиперазин-1-карбоксилат
В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор трет-бутил-4-[[2-фтор-4-(тетраметил-1,3,2-диоксаборолан-2-ил)фенил]карбонил]пиперазин-1-карбоксилата (1,26 г, 2,90 ммоль, 1,00 экв.) в толуоле (24 мл), 2-хлор-6-фтор-1,3-бензоксазол (500 мг, 2,91 ммоль, 1,00 экв.), Pd(PPh3)4 (336 мг, 0,29 ммоль, 0,10 экв.), карбонат натрия (12 мл, 2М), этанол (3,3 мл). Полученную смесь перемешивали в течение ночи при 95°С. После охлаждения до комнатной температуры полученный раствор разбавляли 30 мл воды и экстрагировали 2х30 мл этилацетата. Объединяли органические слои и сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью ЭА/ДХМ (3/10). Это приводило к получению 1,1 г (86%) титульного соединения в виде серого твердого вещества. ЖХ-МС (ИЭР, m/z): 444 [М+Н]+.
Стадия 7. (2-фтор-4-(6-фторбензо[d]оксазол-2-ил)фенил)(пиперазин-1-ил)метанон
В 250 мл круглодонную колбу помещали раствор трет-бутил-4-[[2-фтор-4-(6-фтор-1,3-бензоксазол-2-ил)фенил]карбонил]пиперазин-1-карбоксилата (1,1 г, 2,48 ммоль, 1,00 экв.) в дихлорметане (100 мл). В полученную выше смесь добавляли газообразный хлороводород. Полученный раствор перемешивали в течение 1 часа при комнатной температуре. Собирали твердые вещества путем фильтрования, а затем растворяли в воде, затем доводили рН до 6~7 при помощи бикарбоната натрия. Затем собирали твердые вещества путем фильтрования и сушили в печи. Это приводило к получению 700 мг (82%) титульного соединения в виде серого твердого вещества. ЖХ-МС (ИЭР, m/z): 344 [М+Н]+.
Стадия 8. (4-(2-фтор-4-(6-фторбензо[d]оксазол-2-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанон
В 50 мл круглодонную колбу помещали раствор 6-фтор-2-[3-фтор-4-[(пиперазин-1-ил)карбонил]фенил]-1,3-бензоксазола (538 мг, 1,57 ммоль, 1,00 экв.) в N,N-диметилформамиде (15 мл), 1-гидроксициклопропан-1-карбоновую кислоту (160 мг, 1,57 ммоль, 1,00 экв.), HBTU (891 мг, 2,35 ммоль, 1,50 экв.), DIEA (809 мг, 6,26 ммоль, 4,00 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Затем реакционную смесь разбавляли 30 мл воды. Полученный раствор экстрагировали 2х20 мл дихлорметана и объединяли органические слои и сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью этилацетат/петролейный эфир (7:1). Собирали фракцию и концентрировали. Полученный продукт перекристаллизовывали из смеси ЭА/ПЭ с отношением 10/1. Это приводило к получению 206,7 мг (30%) титульного соединения в виде беловатого твердого вещества. ЖХ-МС (ИЭР, m/z): 428 [М+Н]+.
Способ 14.
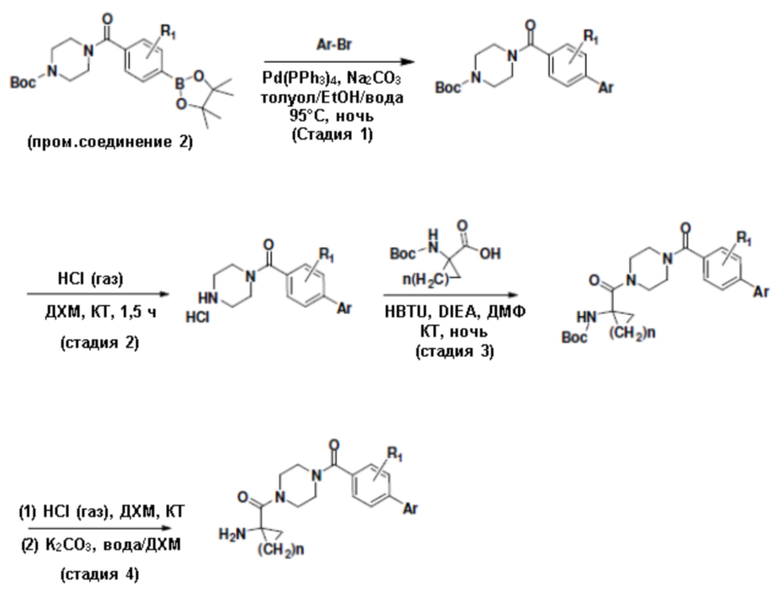
Стадия 1. Трет-бутил-4-(4-(1-метил-1Н-бензо[d]имидазол-5-ил)бензоил)пиперазин-1-карбоксилат
В 100 мл круглодонную колбу помещали трет-бутил-4-[[4-(тетраметил-1,3,2-диоксаборолан-2-ил)фенил]карбонил]пиперазин-1-карбоксилат (832 мг, 2,00 ммоль, 1,00 экв.), 5-бром-1-метил-1Н-1,3-бензотриазол (422 мг, 2,00 ммоль, 1,00 экв.), Pd(PPh3)4 (232 мг, 0,20 ммоль, 0,10 экв.), карбонат натрия (2н., 5 мл), толуол (10 мл), этанол (3 мл). Полученную смесь перемешивали в течение ночи при 95°С на масляной бане. После охлаждения до комнатной температуры полученный раствор разбавляли 10 мл воды, экстрагировали 3х20 мл этилацетата. Объединяли органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесями этилацетат/петролейный эфир (0/100~100/0). Объединяли собранные фракции и концентрировали в вакууме. Это приводило к получению 800 мг (95%) титульного соединения в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 421 [М+Н]+.
Стадия 2. Гидрохлорид (4-(1-метил-1Н-бензо[d]имидазол-5-ил)фенил)(пиперазин-1-ил)метанона
В 100 мл круглодонную колбу помещали трет-бутил-4-[[4-(1-метил-1Н-1,3-бензодиазол-5-ил)фенил]карбонил]пиперазин-1-карбоксилат (800 мг, 1,90 ммоль, 1,00 экв.), дихлорметан (20 мл). Затем вводили газообразный HCl. Полученный раствор перемешивали в течение 1,5 часа при комнатной температуре. Собирали твердые вещества путем фильтрования и сушили в вакууме. Это приводило к получению 580 мг (95%) титульного соединения в виде беловатого твердого вещества. ЖХ-МС (ИЭР, m/z): 321 [М+Н]+.
Стадия 3. Трет-бутил-(1-(4-(4-(1-метил-1Н-бензо[d]имидазол-5-ил)бензоил)пиперазин-1-карбонил)циклопропил)карбамат
В 50 мл круглодонную колбу помещали гидрохлорид 1-метил-5-[4-[(пиперазин-1-ил)карбонил]фенил]-1Н-1,3-бензодиазола (580 мг, 1,63 ммоль, 1,00 экв.), 1-[[(трет-бутокси)-карбонил]амино]циклопропан-1-карбоновую кислоту (360 мг, 1,79 ммоль, 1,10 экв.), HBTU (927 мг, 2,44 ммоль, 1,50 экв.), DIEA (841 мг, 6,51 ммоль, 4,00 экв.), N,N-диметилформамид (10 мл). Смесь перемешивали в течение ночи при комнатной температуре. Полученный раствор разбавляли 60 мл воды, экстрагировали 3х60 мл дихлорметана. Объединяли органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью дихлорметан/метанол (80/20). Объединяли собранные фракции и концентрировали в вакууме. Это приводило к получению 800 мг (количественный выход) титульного соединения в виде бесцветной маслянистой жидкости. ЖХ-МС (ИЭР, m/z): 504 [М+Н]+.
Стадия 4. (4-(1-аминоциклопропан-1-карбонил)пиперазин-1-ил)(4-(1-метил-1Н-бензо[d]имидазол-5-ил)фенил)метанон
В 100 мл круглодонную колбу помещали трет-бутил-N-[1-[(4-[[4-(1-метил-1Н-1,3-бензодиазол-5-ил)фенил]карбонил]пиперазин-1-ил)карбонил]циклопропил]карбамат (800 мг, 1,59 ммоль, 1,00 экв.), дихлорметан (50 мл). Затем барботировали газообразный HCl в течение 1,5 часа при комнатной температуре. Собирали твердые вещества путем фильтрования и промывали ДХМ (50 мл). Затем растворяли твердые вещества в 5 мл воды. рН раствора доводили до 8 при помощи карбоната калия (2М). Полученную смесь экстрагировали 2х50 мл дихлорметана и объединяли органические слои и концентрировали в вакууме. Неочищенный продукт очищали путем препаративной ВЭЖХ в следующих условиях (Waters III): колонка, Xbridge RP18, 19*150; мобильная фаза, вода (0,05% NH4HCO3) и MeCN (от 15% CH3CN до 75% за 10 минут); детектор, УФ 220 и 254 нм. Это приводило к получению 93,1 мг (15%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z):404 [М+Н]+.
Способ 15.
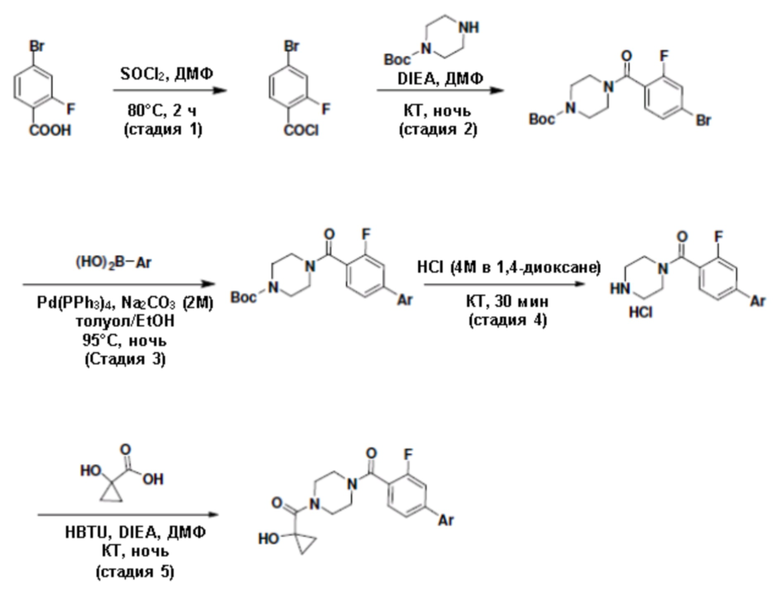
Стадия 1. 4-бром-2-фторбензоилхлорид
В 250 мл круглодонную колбу помещали 4-бром-2-фторбензойную кислоту (10 г, 45,66 ммоль, 1,00 экв.), тионилхлорид (50 мл), N,N-диметилформамид (0,1 мл). Полученный раствор перемешивали в течение 2 часов при 80°С. После охлаждения до комнатной температуры полученную смесь концентрировали в вакууме. Это приводило к получению 10 г (92%) титульного соединения в виде светло-желтой маслянистой жидкости. Продукт использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2. Трет-бутил-4-(4-бром-2-фторбензоил)пиперазин-1-карбоксилат
В 250 мл круглодонную колбу помещали раствор 4-бром-2-фторбензоилхлорида (10 г, 42,11 ммоль, 1,00 экв.) в N,N-диметилформамиде (100 мл). После этого за несколько раз добавляли трет-бутил-пиперазин-1-карбоксилат (7,88 г, 42,31 ммоль, 1,00 экв.). Затем в смесь добавляли DIEA (16,25 г, 125,74 ммоль, 2,99 экв.). Полученную смесь перемешивали в течение ночи при комнатной температуре. Смесь выливали в 300 мл воды. Собирали твердые вещества путем фильтрования и сушили в вакууме. Это приводило к получению 14 г (86%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 387, 389 [М+Н]+.
Стадия 3. Трет-бутил-4-(4'-хлор-2',3-дифтор-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилат
В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор трет-бутил-4-(4-бром-2-фторбензоил)пиперазин-1-карбоксилата (1 г, 2,58 ммоль, 1,00 экв.) в толуоле (10 мл), (4-хлор-2-фторфенил)бороновую кислоту (540 мг, 3,10 ммоль, 1,20 экв.), Pd(PPh3)4 (358 мг, 0,31 ммоль, 0,12 экв.), карбонат натрия (2М в воде, 5 мл), этанол (1,4 мл). Полученную смесь перемешивали в течение ночи при 105°С. После охлаждения до комнатной температуры полученный раствор разбавляли 20 мл H2O и экстрагировали 3х20 мл этилацетата. Объединяли органические слои, промывали 3х20 мл солевого раствора, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, использованием смесь этилацетат/петролейный эфир (3:7). Объединяли собранные фракции и концентрировали в вакууме. Это приводило к получению 1 г (89%) титульного соединения в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 437 [М+Н]+.
Стадия 4. Гидрохлорид (4'-хлор-2',3-дифтор-[1,1'-бифенил]-4-ил)(пиперазин-1-ил)метанона
В 100 мл круглодонную колбу помещали трет-бутил-4-(4'-хлор-2',3-дифтор-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилат (1 г, 2,28 ммоль, 1,00 экв.), хлороводородную кислоту в 1,4-диоксане (4М, 30 мл). Полученный раствор перемешивали в течение 30 минут при комнатной температуре. Полученную смесь концентрировали в вакууме. Это приводило к получению 600 мг (78%) титульного соединения в виде беловатого твердого вещества. ЖХ-МС (ИЭР, m/z): 337 [М+Н]+.
Стадия 5. (4-(4'-хлор-2',3-дифтор-[1,1'-бифенил]-4-карбонил)пиперазин-1-ил)(1-гидроксициклопропил)метанон
В 100 мл круглодонную колбу помещали раствор гидрохлорида (4'-хлор-2',3-дифтор-[1,1'-бифенил]-4-ил)(пиперазин-1-ил)метанона (500 мг, 1,48 ммоль, 1,00 экв.), N,N-диметилформамида (20 мл), 1-гидроксициклопропан-1-карбоновой кислоты (166 мг, 1,63 ммоль, 1,10 экв.), HBTU (841 мг, 2,22 ммоль, 1,49 экв.), DIEA (764 мг, 5,91 ммоль, 3,98 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Смесь выливали в 80 мл воды, образовывался осадок. Собирали твердые вещества путем фильтрования и помещали в колонку с силикагелем, элюировали смесью этилацетат/гексан (3:2). Объединяли собранные фракции и концентрировали в вакууме. Это приводило к получению 260 мг (42%) титульного соединения в виде беловатого твердого вещества. ЖХ-МС (ИЭР, m/z): 421 [М+Н]+.
Способ 16.
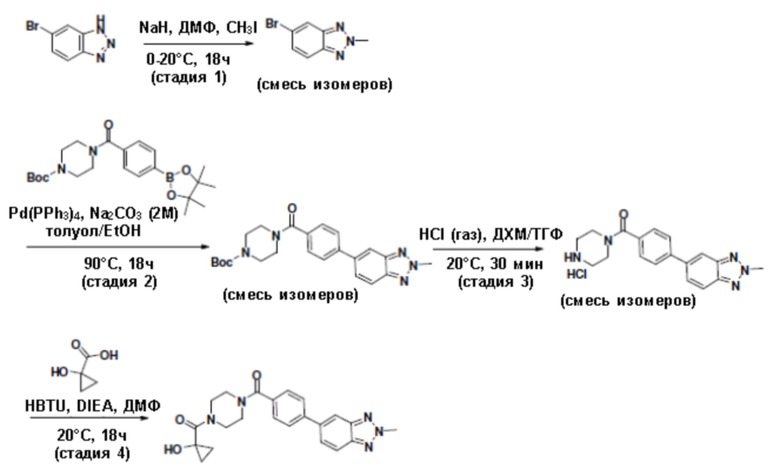
Стадия 1. 5-бром-N-метил-2Н-бензо[d][1,2,3]триазол
В 100 мл круглодонную колбу помещали раствор 6-бром-1Н-1,2,3-бензотриазола (600 мг, 3,03 ммоль, 1,00 экв.) в N,N-диметилформамиде (10 мл). После этого добавляли гидрид натрия (60% в масле, 303 мг, 7,58 ммоль, 2,50 экв.) по частям при 0°С. После перемешивания в течение 30 минут при 0°С по каплям добавляли CH3I (650 мг, 4,58 ммоль, 1,50 экв.). Полученный раствор оставляли для прохождения взаимодействия при перемешивании еще на 18 часов при 20°С. Затем реакционную смесь разбавляли 10 мл смеси лед/вода, экстрагировали 3х20 мл этилацетата. Объединяли органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесями ПЭ:ЭА (200:1~3:1). Это приводило к получению 700 мг (масса неочищенного вещества) смеси титульных соединений в виде желтой маслянистой жидкости. ЖХ-МС (ИЭР, m/z): 212, 214 [М+Н]+.
Стадия 2. Трет-бутил-4-(4-(N-метил-2Н-бензо[d][1,2,3]триазол-5-ил)бензоил)пиперазин-1-карбоксилат
В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали трет-бутил-4-[[4-(тетраметил-1,3,2-диоксаборолан-2-ил)фенил]карбонил]-пиперазин-1-карбоксилат (1,51 г, 3,63 ммоль, 1,10 экв.), смесь 5-бром-N-метил-2Н-бензо[d][1,2,3]триазолов (700 мг, 3,30 ммоль, 1,00 экв.), Pd(PPh3)4 (382 мг, 0,33 ммоль, 0,10 экв.) и толуол/EtOH (30/4,5 мл), карбонат натрия (15 мл, 2М). Полученную смесь перемешивали в течение 18 часов при 90°С. После охлаждения до комнатной температуры реакционную смесь разбавляли 50 мл воды, экстрагировали 3х30 мл этилацетата. Объединяли органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесями дихлорметан/метанол (200/1~20/1). Это приводило к получению 1,2 г (масса неочищенного вещества) смеси титульных соединений в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 422 [М+Н]+.
Стадия 3. Гидрохлорид (4-(N-метил-2Н-бензо[d][1,2,3]триазол-5-ил)фенил)(пиперазин-1-ил)метанона
В 100 мл круглодонную колбу помещали смесь трет-бутил-4-(4-(N-метил-2Н-бензо[d][1,2,3]триазол-5-ил)бензоил)пиперазин-1-карбоксилата (1,2 г, 2,85 ммоль, 1,00 экв.) и дихлорметана (50 мл). Затем вводили хлороводород (газ). Полученный раствор перемешивали в течение 30 минут при 20°С. Собирали полученные твердые вещества путем фильтрования и сушили при пониженном давлении. Это приводило к получению 800 мг (79%) смеси титульных соединений в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 322 [М+Н]+.
Стадия 4. (4-(1-гидроксициклопропан-1-карбонил)пиперазин-1-ил)(4-(2-метил-2Н-бензо[d][1,2,3]триазол-5-ил)фенил)метанон
В 100 мл круглодонную колбу помещали раствор 1-гидроксициклопропан-1-карбоновой кислоты (240 мг, 2,35 ммоль, 1,10 экв.) в N,N-диметилформамиде (10 мл), смесь гидрохлорида (4-(N-метил-2Н-бензо[d][1,2,3]триазол-5-ил)фенил)(пиперазин-1-ил)метанона (766 мг, 2,14 ммоль, 1,00 экв., 98%), HBTU (1,22 г, 3,22 ммоль, 1,50 экв.), DIEA (1,11 г, 8,59 ммоль, 4,00 экв.). Полученный раствор перемешивали в течение 18 часов при 20°С. Реакционную смесь разбавляли 20 мл воды, экстрагировали 3х20 мл этилацетата. Объединяли органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Очищали остаток путем препаративной ВЭЖХ в следующих условиях (Waters III): колонка XBridge Prep C18 OBD, 5 мкм, 19*150 мм; мобильная фаза, вода, содержащая 0,03% NH3.H2O, и MeCN (от 16% MeCN до 34% за 8 минут); детектор, 254 и 220 нм. Это приводило к получению 90 мг (10%) титульного соединения в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 406 [М+Н]+.
Способ 17.

Стадия 1. (4-(4-(трет-бутоксикарбонил)пиперазин-1-карбонил)фенил)бороновая кислота
В 1 л круглодонную колбу помещали промежуточное соединение 3 (12,5 г, 30,03 ммоль, 1,00 экв.), раствор периодата натрия (32,1 г, 150,00 ммоль, 5,00 экв.) в ацетоне (300 мл), ацетат аммония (150 мл, 5,00 экв., 1М). Полученный раствор перемешивали в течение 18 часов при КТ. Полученный раствор разбавляли 300 мл воды и экстрагировали 3х100 мл этилацетата. Объединяли органические слои и сушили над сульфатом натрия. Отфильтровывали твердые вещества и концентрировали раствор в вакууме. Это приводило к получению титульного соединения (10,1 г, 96%) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР, m/z):335 [М+Н]+.
Стадия 2. Трет-бутил-4-(4-(2-метил-1Н-бензо[d]имидазол-1-ил)бензоил)пиперазин-1-карбоксилат
В 250 мл круглодонную колбу помещали раствор (4-(4-(трет-бутоксикарбонил)пиперазин-1-карбонил)фенил)бороновой кислоты (3,34 г, 9,05 ммоль, 2,00 экв.) в дихлорметане (50 мл), 2-метил-1Н-бензо[d]имидазол (660 мг, 4,99 ммоль, 1,00 экв.), Cu(OAc)2 (1,23 мг, 1,50 экв.), пиридин (790 мг, 9,99 ммоль, 2,00 экв.), молекулярные сита 4Å (3 г). Полученную смесь перемешивали в течение 18 часов при 20°С. Отфильтровывали твердые вещества. Концентрировали фильтрат в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесями ДХМ/МеОН (200/1~20/1). Это приводило к получению 1 г (27%) титульного соединения в виде желтой маслянистой жидкости. ЖХ-МС (ИЭР, m/z):421 [М+Н]+.
Стадия 3. Гидрохлорид (4-(2-метил-1Н-бензо[d]имидазол-1-ил)фенил)(пиперазин-1-ил)метанона
В 100 мл круглодонную колбу помещали трет-бутил-4-(4-(2-метил-1Н-бензо[d]имидазол-1-ил)бензоил)пиперазин-1-карбоксилат (960 мг, 2,28 ммоль, 1,00 экв.), ДХМ (30 мл), ТГФ (30 мл). Затем вводили хлороводород (газ). Полученный раствор перемешивали в течение 30 минут при 20°С. Собирали твердые вещества путем фильтрования и сушили в вакууме. Это приводило к получению 600 мг титульного соединения в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР, m/z):321 [М+Н]+.
Стадия 4. (4-(1-гидроксициклопропан-1-карбонил)пиперазин-1-ил)(4-(2-метил-1Н-бензо[d]имидазол-1-ил)фенил)метанон
В 100 мл круглодонную колбу помещали 1-гидроксициклопропан-1-карбоновую кислоту (190 мг, 1,86 ммоль, 1,10 экв.), гидрохлорид (4-(2-метил-1Н-бензо[d]имидазол-1-ил)фенил)-(пиперазин-1-ил)метанона (600 мг, 1,68 ммоль, 1,00 экв.), HBTU (955 мг, 2,52 ммоль, 1,50 экв.), N,N-диметилформамид (10 мл), DIEA (867 мг, 6,71 ммоль, 4,00 экв.). Полученный раствор перемешивали в течение 18 часов при 20°С. Реакционную смесь разбавляли 20 мл воды, экстрагировали 3х20 мл этилацетата. Объединяли все органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Очищали остаток путем препаративной ВЭЖХ в следующих условиях (Waters I): колонка, Xbridge Prep C18 OBD, 5 мкм, 19*150 мм; мобильная фаза, вода (0,05% NH4HCO3) и CH3CN (от 16% CH3CN до 34% за 10 минут); детектор, УФ 220 и 254 нм. Это приводило к получению 130 мг (19%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z):405 [М+Н]+.
Способ 18.
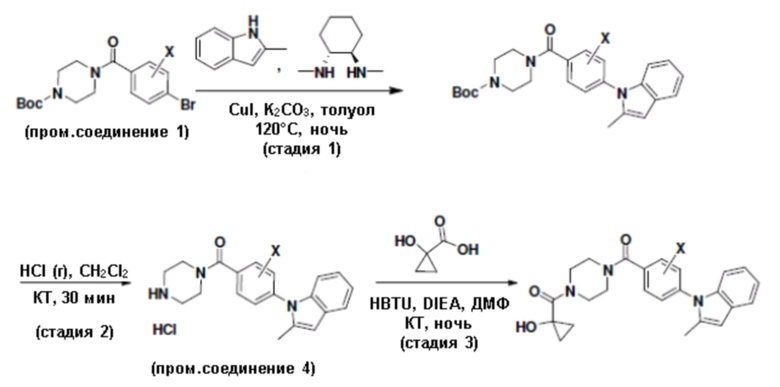
Стадия 1. Трет-бутил-4-(4-(2-метил-1Н-индол-1-ил)бензоил)пиперазин-1-карбоксилат
В 250 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор трет-бутил-4-[(4-бромфенил)карбонил]пиперазин-1-карбоксилата (промежуточное соединение 1 (X=H), 1,69 г, 4,58 ммоль, 1,00 экв.) в толуоле (50 мл), 2-метил-1Н-индол (600 мг, 4,57 ммоль, 1,00 экв.), CuI (87 мг, 0,46 ммоль, 0,10 экв.), карбонат калия (1,9 г, 13,75 ммоль, 3,00 экв.), (1R,2R)-1-N,2-N-диметилциклогексан-1,2-диамин (130 мг, 0,91 ммоль, 0,20 экв.). Полученную смесь перемешивали в течение ночи при 120°С. После охлаждения до комнатной температуры разбавляли реакционную смесь 50 мл H2O, экстрагировали 3х50 мл этилацетата. Объединяли органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью этилацетат/петролейный эфир (1:1). Объединяли собранные фракции и концентрировали в вакууме. Это приводило к получению 1,4 г (73%) титульного соединения в виде красного твердого вещества. ЖХ-МС (ИЭР, m/z):420 [М+Н]+.
Стадия 2. Гидрохлорид (4-(2-метил-1Н-индол-1-ил)фенил)(пиперазин-1-ил)метанона (промежуточное соединение 4)
В 100 мл круглодонную колбу помещали раствор трет-бутил-4-(4-(2-метил-1Н-индол-1-ил)бензоил)пиперазин-1-карбоксилата (1,4 г, 3,34 ммоль) в дихлорметане (30 мл). В полученную выше смесь вводили газообразный HCl. Полученный раствор перемешивали в течение 30 минут при комнатной температуре. Полученную смесь концентрировали в вакууме. Это приводило к получению 1,4 г (59%) титульного соединения в виде красного твердого вещества. ЖХ-МС (ИЭР, m/z):320 [М+Н]+.
Стадия 3. (4-(1-гидроксициклопропан-1-карбонил)пиперазин-1-ил)(4-(2-метил-1Н-индол-1-ил)фенил)метанон
В 100 мл круглодонную колбу помещали раствор гидрохлорида (4-(2-метил-1Н-индол-1-ил)фенил)(пиперазин-1-ил)метанона (658 мг, 1,85 ммоль, 1,00 экв.) в N,N-диметилформамиде (20 мл), 1-гидроксициклопропан-1-карбоновую кислоту (189 мг, 1,85 ммоль, 1,00 экв.), HBTU (1,05 г, 2,77 ммоль, 1,50 экв.), DIEA (955 мг, 7,39 ммоль, 4,00 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Разбавляли реакционную смесь 30 мл воды и экстрагировали 3х30 мл этилацетата. Объединяли органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью дихлорметан/метанол (10:1). Объединяли собранные фракции и концентрировали в вакууме. Это приводило к получению 112,3 мг (15%) титульного соединения в виде светло-коричневого твердого вещества. ЖХ-МС (ИЭР, m/z):404 [М+Н]+.
Способ 19.
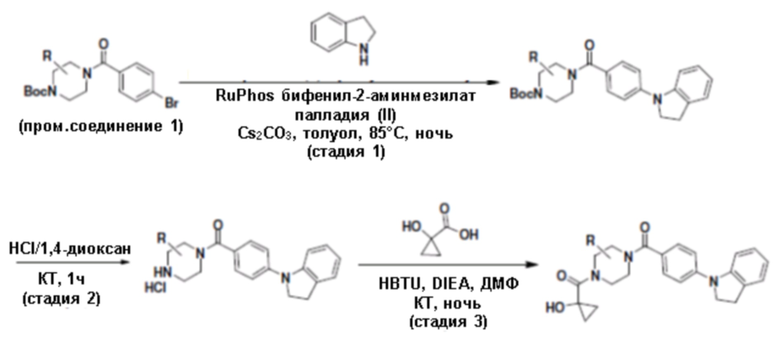
Стадия 1. Трет-бутил-4-(4-(индолин-1-ил)бензоил)пиперазин-1-карбоксилат
В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали 2,3-дигидро-1Н-индол (390 мг, 3,27 ммоль, 1,00 экв.), трет-бутил-4-[(4-бромфенил)карбонил]пиперазин-1-карбоксилат (промежуточное соединение 1 (R=H), 1,5 г, 3,60 ммоль, 1,10 экв.), Cs2CO3 (3,74 г, 11,48 ммоль, 3,51 экв.), толуол (50 мл), RuPhos бифенил-2-аминмезилат палладия (II) (276 мг). Полученную смесь перемешивали в течение ночи при 85°С. После охлаждения до комнатной температуры концентрировали полученную смесь в вакууме и перерастворяли в 200 мл ЭА. Полученную смесь промывали 3х50 мл солевого раствора, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью этилацетат/петролейный эфир (50:50). Это приводило к получению 1 г (75%) титульного соединения в виде желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 408 [М+Н]+.
Стадия 2. Гидрохлорид (4-(индолин-1-ил)фенил)(пиперазин-1-ил)метанона
В 100 мл круглодонную колбу помещали трет-бутил-4-(4-(индолин-1-ил)бензоил)пиперазин-1-карбоксилат (1 г, 2,45 ммоль, 1,00 экв.), раствор хлороводорода в 1,4-диоксане (4М, 20 мл). Полученный раствор перемешивали в течение 1 часа при комнатной температуре. Собирали твердые вещества путем фильтрования и сушили в вакууме. Это приводило к получению 800 мг (95%) титульного соединения в виде желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 308 [М+Н]+.
Стадия 3. (4-(1-гидроксициклопропан-1-карбонил)пиперазин-1-ил)(4-(индолин-1-ил)фенил)метанон
В 100 мл круглодонную колбу помещали гидрохлорид (4-(индолин-1-ил)фенил)(пиперазин-1-ил)метанона (540 мг, 1,57 ммоль, 1,00 экв.), 1-гидроксициклопропан-1-карбоновую кислоту (160 мг, 1,57 ммоль, 1,00 экв.), N,N-диметилформамид (20 мл), HBTU (892 мг, 2,35 ммоль, 1,50 экв.), DIEA (810 мг, 6,27 ммоль, 3,99 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли 50 мл ЭА, промывали 3х30 мл солевого раствора, сушили над безводным сульфатом натрия и концентрировали в вакууме. Очищали остаток путем препаративной ВЭЖХ в следующих условиях (Waters I): колонка SunFire Prep C18, 5 мкм, 19*100 мм; мобильная фаза, вода (0,05% NH4HCO3) и CH3CN (от 50% CH3CN до 100% за 10 минут); детектор, УФ 220 и 254 нм. Это приводило к получению 63,8 мг (10%) титульного соединения в виде беловатого твердого вещества. ЖХ-МС (ИЭР, m/z): 392 [М+Н]+.
Способ 20.
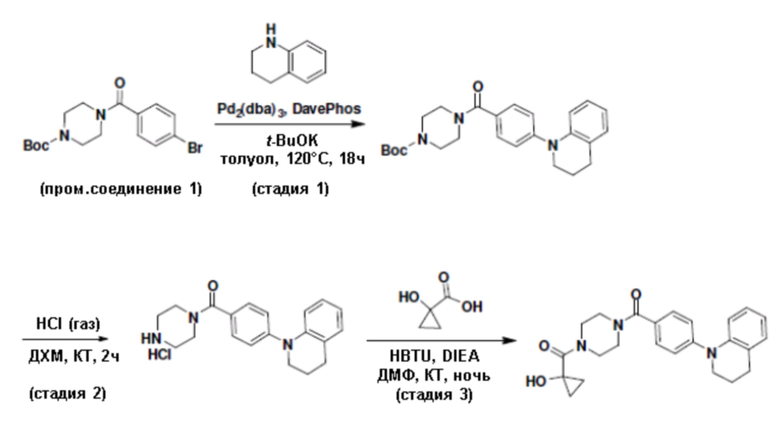
Стадия 1. Трет-бутил-4-(4-(3,4-дигидрохинолин-1(2Н)-ил)бензоил)пиперазин-1-карбоксилат
В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали Pd2(dba)4 (305 мг), DavePhos (627 мг), толуол (50 мл). Смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли 1,2,3,4-тетрагидрохинолин (700 мг, 5,26 ммоль, 1,00 экв.), трет-бутил-4-[(4-бромфенил)карбонил]пиперазин-1-карбоксилат (промежуточное соединение 1, 1,94 г, 5,25 ммоль, 1,00 экв.), t-BuOK (1,18 г, 10,52 ммоль, 2,00 экв.). Пять раз вакуумировали систему и повторно заполняли N2. Полученную смесь перемешивали в течение 18 часов при 120°С на масляной бане. После охлаждения до комнатной температуры концентрировали полученную смесь в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесями этилацетат/петролейный эфир (0/100~45/55). Объединяли собранные фракции и концентрировали в вакууме. Это приводило к получению 540 мг (24%) титульного соединения в виде желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 422 [М+Н]+.
Стадия 2. Гидрохлорид (4-(3,4-дигидрохинолин-1(2Н)-ил)фенил)(пиперазин-1-ил)метанона
В 100 мл круглодонную колбу помещали трет-бутил-4-(4-(3,4-дигидрохинолин-1(2Н)-ил)бензоил)пиперазин-1-карбоксилат (540 мг, 1,28 ммоль, 1,00 экв.), дихлорметан (40 мл). Затем барботировали газообразным хлороводородом. Полученный раствор перемешивали в течение 2 часов при комнатной температуре. Концентрировали полученную смесь в вакууме. Это приводило к получению 635 мг (количественный выход) титульного соединения в виде желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 322 [М+Н]+.
Стадия 3. (4-(4-(3,4-дигидрохинолин-1(2Н)-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанон
В 100 мл круглодонную колбу помещали 1-гидроксициклопропан-1-карбоновую кислоту (635 мг, 6,22 ммоль, 1,00 экв.), гидрохлорид (4-(3,4-дигидрохинолин-1(2Н)-ил)фенил)-(пиперазин-1-ил)метанона (181 мг, 0,51 ммоль, 0,08 экв.), HBTU (1,34 г, 3,53 ммоль, 0,57 экв.), N,N-диметилформамид (15 мл), DIEA (457 мг, 3,54 ммоль, 0,57 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Разбавляли реакционную смесь 60 мл ЭА, промывали 4х20 мл воды и 1х20 мл солевого раствора. Сушили органическую фазу над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесями этилацетат/петролейный эфир (0/100~100/0). Полученный неочищенный продукт дополнительно очищали путем препаративной ВЭЖХ в следующих условиях (Waters III): колонка Xbridge RP18, 19*150; мобильная фаза, вода (0,05% NH4HCO3) и MeCN (от 15% CH3CN до 75% за 10 минут); детектор, УФ 220 и 254 нм. Это приводило к получению 66,3 мг (3%) титульного соединения в виде пурпурного твердого вещества. ЖХ-МС (ИЭР, m/z): 406 [М+Н]+.
Способ 21.
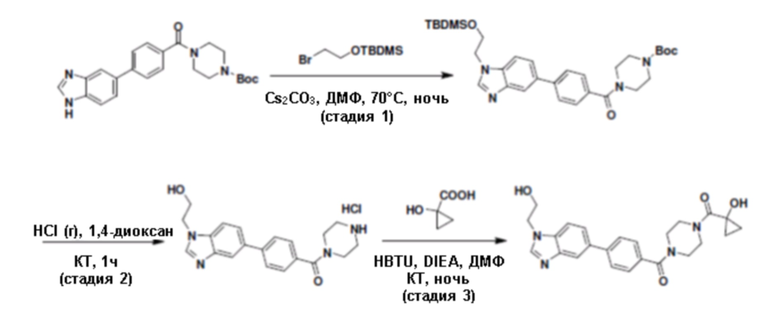
Стадия 1. Трет-бутил-4-(4-(1-(2-((трет-бутилдиметилсилил)окси)этил)-1Н-бензо[d]имидазол-5- и -6-ил)бензоил)пиперазин-1-карбоксилат
В 250 мл круглодонную колбу помещали трет-бутил-4-(4-(1Н-бензо[d]имидазол-5-ил)бензоил)пиперазин-1-карбоксилат (1 г, 2,46 ммоль, 1,00 экв.), (2-бромэтокси)(трет-бутил)-диметилсилан (883 мг, 3,69 ммоль, 1,50 экв.), N,N-диметилформамид (50 мл), Cs2CO3 (2,41 г, 7,40 ммоль, 3,01 экв.). Полученную смесь перемешивали в течение ночи при 70°С. После охлаждения до комнатной температуры разбавляли реакционную смесь 100 мл ЭА, промывали 3х50 мл воды и 50 мл солевого раствора. Органическую фазу сушили над безводным сульфатом натрия и концентрировали в вакууме. Очищали остаток путем препаративной ВЭЖХ в следующих условиях (Waters I): колонка SunFire Prep C18, 5 мкм, 19*100 мм; мобильная фаза, вода (0,05% NH4HCO3) и CH3CN (от 55% CH3CN до 75% за 7 минут); детектор, УФ 220 и 254 нм. Это приводило к получению 380 мг (27%) трет-бутил-4-(4-(1-(2-((трет-бутилдиметилсилил)окси)этил)-1Н-бензо[d]имидазол-5-ил)бензоил)-пиперазин-1-карбоксилата в виде желтого твердого вещества и 390 мг (28%) трет-бутил-4-(4-(1-(2-((трет-бутилдиметилсилил)окси)этил)-1Н-бензо[d]имидазол-6-ил)бензоил)пиперазин-1-карбоксилата в виде желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 565 [М+Н]+.
Стадия 2. Гидрохлорид (4-(1-(2-гидроксиэтил)-1Н-бензо[d]имидазол-6-ил)фенил)-пиперазин-1-метанона
В 100 мл круглодонную колбу помещали трет-бутил-4-(4-(1-(2-((трет-бутилдиметилсилил)-окси)этил)-1Н-бензо[d]имидазол-6-ил)бензоил)пиперазин-1-карбоксилат (380 мг, 0,67 ммоль, 1,00 экв.) и диоксан (40 мл). Затем вводили газообразный HCl. Полученный раствор перемешивали в течение 1 часа при комнатной температуре. Концентрировали полученную смесь в вакууме. Это приводило к получению 250 мг (96%) титульного соединения в виде желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 351 [М+Н]+.
Стадия 3. (4-(1-гидроксициклопропан-1-карбонил)пиперазин-1-ил)(4-(1-(2-гидроксиэтил)-1Н-бензо[d]имидазол-5-ил)фенил)метанон
В 100 мл круглодонную колбу помещали гидрохлорид (4-(1-(2-гидроксиэтил)-1Н-бензо[d]имидазол-6-ил)фенил)(пиперазин-1-ил)метанона (250 мг, 0,65 ммоль, 1,00 экв.), 1-гидроксициклопропан-1-карбоновую кислоту (66 мг, 0,65 ммоль, 1,00 экв.), N,N-диметилформамид (20 мл), HBTU (367 мг, 0,97 ммоль, 1,50 экв.), DIEA (333 мг, 2,58 ммоль, 3,99 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Реакционную смесь промывали водой, сушили над Na2SO4 и концентрировали в вакууме. Очищали остаток путем препаративной ВЭЖХ в следующих условиях (Waters I): колонка SunFire Prep C18, 5 мкм, 19*100 мм; мобильная фаза, вода (0,05% NH4HCO3) и CH3CN (от 55% CH3CN до 75% за 7 минут); детектор, УФ 220 и 254 нм. Это приводило к получению 75 мг (27%) титульного соединения в виде беловатого твердого вещества. ЖХ-МС (ИЭР, m/z): 435 [М+Н]+.
Способ 22.
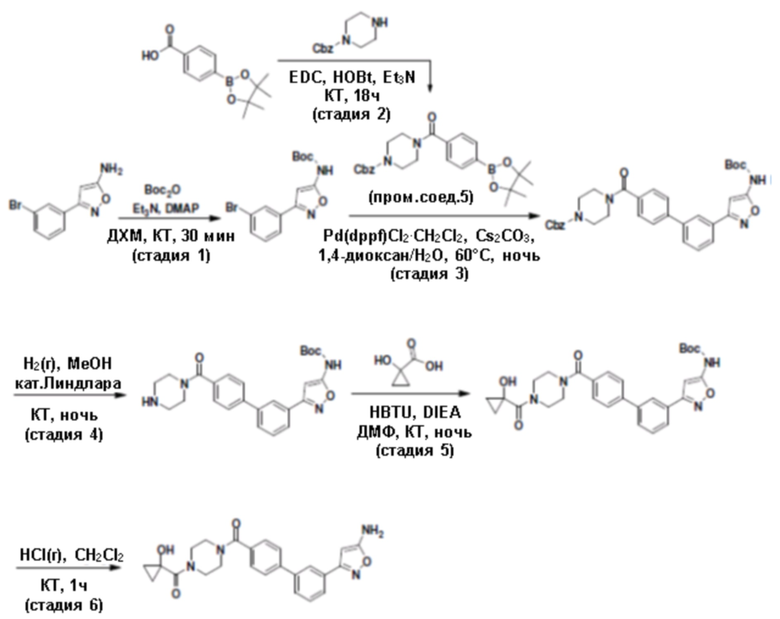
Стадия 1. Трет-бутил-(3-(3-бромфенил)изоксзаол-5-ил)карбамат
В 250 мл круглодонную колбу помещали раствор 3-(3-бромфенил)-1,2-оксазол-5-амина (1 г, 4,18 ммоль, 1,00 экв.) в дихлорметане (40 мл), Boc2O (1,83 г, 8,38 ммоль, 2,00 экв.), триэтиламин (1,27 г, 12,55 ммоль, 3,00 экв.), 4-диметиламинопиридин (51,24 мг, 0,42 ммоль, 0,10 экв.). Полученный раствор перемешивали в течение 1 часа при комнатной температуре. Полученную смесь промывали 2х20 мл хлороводородной кислоты (0,5М), сушили над безводным сульфатом натрия, концентрировали в вакууме. Это приводило к получению 1,42 г (масса неочищенного вещества) титульного соединения в виде беловатого твердого вещества. ЖХ-МС (ИЭР, m/z): 339 [М+Н]+.
Стадия 2. Бензил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоил)пиперазин-1-карбоксилат (промежуточное соединение 5)
В 1 л круглодонную колбу помещали бензил-пиперазин-1-карбоксилат (15 г, 68,10 ммоль, 1,00 экв.), 4-(тетраметил-1,3,2-диоксаборолан-2-ил)бензойную кислоту (16,9 г, 68,12 ммоль, 1,00 экв.), EDC (14,4 г, 75,12 ммоль, 1,10 экв.), HOBt (5,8 г, 42,92 ммоль, 0,50 экв.) и триэтиламин (27,6 г, 272,75 ммоль, 4,00 экв.) в дихлорметане (300 мл). Полученный раствор перемешивали в течение 18 часов при КТ. Затем смесь промывали 0,5М карбонатом натрия (водн., 75 мл). Затем полученную смесь промывали 0,5М HCl (водн., 75 мл), а после этого 0,5М карбонатом натрия (водн., 75 мл). Концентрировали раствор в вакууме и перекристаллизовывали неочищенный продукт из смеси МТБЭ/гексан (1:1). Это приводило к получению титульного соединения (26 г, 84%) в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 451 [М+Н]+.
Стадия 3. Бензил-4-(3'-(5-((трет-бутоксикарбонил)амино)изоксазол-3-ил)-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилат
В 250 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор трет-бутил-(3-(3-бромфенил)изоксазол-5-ил)карбамата (1,42 г, 4,19 ммоль, 1,00 экв.) в 1,4-диоксане (30 мл), бензил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоил)пиперазин-1-карбоксилат (1,89 г, 4,20 ммоль, 1,00 экв.), Pd(dppf)Cl2.CH2Cl2 (410 мг, 0,50 ммоль, 0,12 экв.), Cs2CO3 (4,11 г, 12,61 ммоль, 3,01 экв.), воду (10 мл). Полученный раствор перемешивали в течение ночи при 60°С. После охлаждения до комнатной температуры разбавляли реакционную смесь 30 мл H2O, экстрагировали 3х30 мл этилацетата. Объединяли органические слои, промывали 30 мл солевого раствора, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью этилацетат/петролейный эфир (1:1). Объединяли собранные фракции и концентрировали в вакууме. Это приводило к получению 780 мг (32%) титульного соединения в виде беловатого твердого вещества. ЖХ-МС (ИЭР, m/z): 583 [М+Н]+.
Стадия 4. Трет-бутил-(3-(4'-(пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)изоксазол-5-ил)карбамат
В 100 мл круглодонную колбу помещали бензил-4-(3'-(5-((трет-бутоксикарбонил)амино)-изоксазол-3-ил)-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилат (1,8 г, 3,09 ммоль, 1,00 экв.), метанол (40 мл) и катализатор Линдлара (1,8 г). Колбу 6 раз вакуумировали и повторно заполняли водородом. Полученный раствор перемешивали в течение ночи при комнатной температуре. Отфильтровывали твердые вещества. Концентрировали фильтрат в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью дихлорметан/метанол (90:10). Объединяли собранные фракции и концентрировали в вакууме. Это приводило к получению 850 мг (61%) титульного соединения в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 449 [М+Н]+.
Стадия 5. Трет-бутил-(3-(4'-(4-(1-гидроксициклопропан-1-карбонил)пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)изоксазол-5-ил)карбамат
В 100 мл круглодонную колбу помещали раствор трет-бутил-(3-(4'-(пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)изоксазол-5-ил)карбамата (850 мг, 1,90 ммоль, 1,00 экв.) в N,N-диметилформамиде (20 мл), 1-гидроксициклопропан-1-карбоновую кислоту (194 мг, 1,90 ммоль, 1,00 экв.), HBTU (1,08 г, 2,85 ммоль, 1,50 экв.), DIEA (980 мг, 7,58 ммоль, 4,00 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Разбавляли реакционную смесь 30 мл ЭА, промывали 3х30 мл солевого раствора, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью дихлорметан/метанол (90:10). Объединяли собранные фракции и концентрировали в вакууме. Это приводило к получению 600 мг (59%) титульного соединения в виде коричневого твердого вещества. ЖХ-МС (ИЭР, m/z): 533 [М+Н]+.
Стадия 6. (4-(3'-(5-аминоизоксазол-3-ил)-[1,1'-бифенил]-4-карбонил)пиперазин-1-ил)(1-гидроксициклопропил)метанон
В 100 мл круглодонную колбу помещали трет-бутил-(3-(4'-(4-(1-гидроксициклопропан-1-карбонил)пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)изоксазол-5-ил)карбамат (600 мг, 1,13 ммоль, 1,00 экв.), дихлорметан (30 мл). В полученную выше смесь вводили газообразный HCl. Полученный раствор перемешивали в течение 1 часа при комнатной температуре. Концентрировали смесь и растворяли в 5 мл метанола. рН раствора доводили до 8 при помощи насыщенного раствора бикарбоната натрия. Полученный раствор экстрагировали 2х30 мл дихлорметана. Объединяли органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Очищали остаток путем препаративной ВЭЖХ в следующих условиях (Waters I): колонка Xbridge Prep C18 OBD, 5 мкм, 19*150 мм; мобильная фаза, вода (0,05% NH4HCO3) и CH3CN (от 30% CH3CN до 70% за 9 минут); детектор, УФ 220 и 254 нм. Это приводило к получению 107,4 мг (22%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 433 [М+Н]+.
Способ 23.

Стадия 1. 4-(1-метил-1Н-бензо[d]имидазол-5-ил)бензойная кислота
В 250 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 4-(тетраметил-1,3,2-диоксаборолан-2-ил)бензойной кислоты (2,35 г, 9,47 ммоль, 1,00 экв.) в смеси CH3CN/толуол (70/12 мл), 5-бром-1-метил-1Н-бензо[d]имидазол (2 г, 9,48 ммоль, 1,00 экв.), Pd(PPh3)4 (548 мг, 0,47 ммоль, 0,05 экв.), карбонат натрия (0,4М, 50 мл, 2,00 экв.). Полученный раствор перемешивали в течение 18 часов при 90°С. После охлаждения до комнатной температуры реакционную смесь выливали в 50 мл воды. Смесь промывали 2х100 мл этилацетата и собирали водный слой. рН раствора доводили до 4 при помощи хлороводородной кислоты (1н.). Собирали твердые вещества путем фильтрования и сушили в вакууме. Это приводило к получению 2 г (84%) титульного соединения в виде белого твердого вещества, которое сушили в вакууме. ЖХ-МС (ИЭР, m/z): 253 [М+Н]+.
Стадия 2. Трет-бутил-(R)-2-метил-4-(4-(1-метил-1Н-бензо[d]имидазол-5-ил)бензоил)-пиперазин-1-карбоксилат
В 50 мл круглодонную колбу помещали раствор 4-(1-метил-1Н-бензо[d]имидазол-5-ил)бензойной кислоты (600 мг, 2,38 ммоль, 1,00 экв.) в N,N-диметилформамиде (15 мл), трет-бутил-(2R)-2-метилпиперазин-1-карбоксилат (476 мг, 2,38 ммоль, 1,00 экв.), HBTU (1,35 г, 3,56 ммоль, 1,50 экв.), DIEA (1,22 г, 9,44 ммоль, 4,00 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Реакционную смесь выливали в 100 мл воды, экстрагировали 3х100 мл дихлорметана. Объединяли органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью этилацетат/петролейный эфир (1/15). Это приводило к получению 650 мг (63%) титульного соединения в виде красной маслянистой жидкости. ЖХ-МС (ИЭР, m/z): 435 [М+Н]+.
Стадия 3. Гидрохлорид (R)-(4-(1-метил-1Н-бензо[d]имидазол-5-ил)фенил)(3-метилпиперазин-1-ил)метанона
В 50 мл круглодонную колбу помещали раствор трет-бутил-(R)-2-метил-4-(4-(1-метил-1Н-бензо[d]имидазол-5-ил)бензоил)пиперазин-1-карбоксилата (650 мг, 1,50 ммоль, 1,00 экв.) в дихлорметане (20 мл). В полученную выше смесь вводили хлороводород (газ). Полученный раствор перемешивали в течение 0,5 часа при комнатной температуре. Концентрировали полученную смесь в вакууме. Остаток растирали с 2х100 мл диэтилового эфира. Собирали твердые вещества путем фильтрования и сушили в вакууме. Это приводило к получению 500 мг (90%) титульного соединения в виде красного твердого вещества. ЖХ-МС (ИЭР, m/z): 335 [М+Н]+.
Стадия 4. (R)-(4-(1-гидроксициклопропан-1-карбонил)-3-метилпиперазин-1-ил)(4-(1-метил-1Н-бензо[d]имидазол-5-ил)фенил)метанон
В 50 мл круглодонную колбу помещали раствор гидрохлорида (R)-(4-(1-метил-1Н-бензо[d]имидазол-5-ил)фенил)(3-метилпиперазин-1-ил)метанона (500 мг, 1,35 ммоль, 1,00 экв.) в N,N-диметилформамиде (15 мл), 1-гидроксициклопропан-1-карбоновую кислоту (137 мг, 1,34 ммоль, 1,00 экв.), HBTU (766 мг, 2,02 ммоль, 1,50 экв.), DIEA (695 мг, 5,38 ммоль, 4,00 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Реакционную смесь выливали в 100 мл воды, экстрагировали 3х50 мл дихлорметана. Объединяли органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток очищали путем препаративной ВЭЖХ в следующих условиях (IntelFlash-1): колонка С18 силикагелем; мобильная фаза, CH3CN/вода, содержащая 0,05% NH4HCO3, = 20%, повышали до CH3CN/вода, содержащая 0,05% NH4HCO3, = 60% за 30 минут; детектор, УФ 254 и 220 нм. Это приводило к получению 200,3 мг (36%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 419 [М+Н]+.
Способ 24.
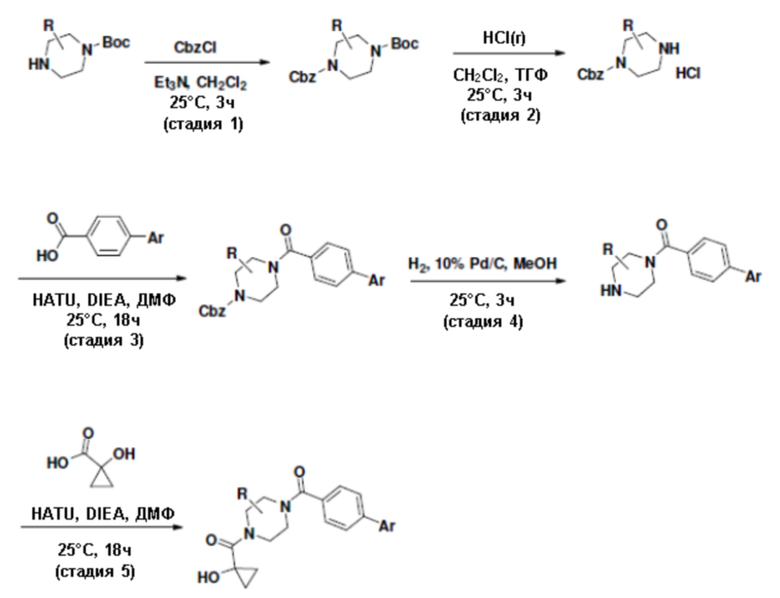
Стадия 1. 7-бензил-4-(трет-бутил)-4,7-диазаспиро[2.5-]октан-4,7-дикарбоксилат
В 100 мл круглодонную колбу помещали раствор трет-бутил-4,7-диазаспиро[2.5]октан-4-карбоксилата (3 г, 14,13 ммоль, 1,00 экв.) и ТЭА (4,25 г, 42,00 ммоль, 2,97 экв.) в дихлорметане (30 мл). После этого по каплям добавляли CbzCl (2,88 г, 16,88 ммоль, 1,19 экв.) при перемешивании при 0°С. Полученный раствор перемешивали в течение 3 часов при 25°С. Реакционную смесь разбавляли 50 мл воды, экстрагировали 3х50 мл дихлорметана. Объединяли органические слои, промывали 2х100 мл раствора карбоната натрия (2М), сушили над Na2SO4 и концентрировали в вакууме. Это приводило к получению 4 г (82%) титульного соединения в виде желтой маслянистой жидкости. ЖХ-МС (ИЭР, m/z): 347 [М+Н]+.
Стадия 2. Гидрохлорид бензил-4,7-диазаспиро[2.5]октан-7-карбоксилата
В 100 мл круглодонную колбу помещали раствор 7-бензил-4-(трет-бутил)-4,7-диазаспиро[2.5-]октан-4,7-дикарбоксилата (4 г, 11,55 ммоль, 1,00 экв.) в смеси дихлорметан/тетрагидрофуран (20/20 мл). В полученный выше раствор вводили хлороводород (газ). Полученный раствор перемешивали в течение 3 часов при 25°С. Собирали твердые вещества путем фильтрования. Это приводило к получению 3 г (масса неочищенного вещества) титульного соединения в виде желтой маслянистой жидкости. ЖХ-МС (ИЭР, m/z): 247 [М+Н]+.
Стадия 3. Бензил-4-(4-(6-фторбензо[d]оксазол-2-ил)бензоил)-4,7-диазаспиро[2.5]октан-7-карбоксилат
В 100 мл круглодонную колбу помещали HATU (1,55 г, 4,08 ммоль, 1,00 экв.), 4-(6-фтор-1,3-бензоксазол-2-ил)бензойную кислоту (951 мг, 3,70 ммоль, 0,91 экв.), гидрохлоридную соль бензил-4,7-диазаспиро[2.5]октан-7-карбоксилата (1 г, 4,06 ммоль, 1,00 экв.), DIEA (955 мг, 7,39 ммоль, 1,82 экв.) и N,N-диметилформамид (30 мл). Полученный раствор перемешивали в течение 18 часов при 25°С. Затем смесь разбавляли 50 мл ЭА, промывали 2х50 мл воды, сушили над Na2SO4 и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесями этилацетат/петролейный эфир (10%-90%). Это приводило к получению 1,2 г (масса неочищенного вещества) титульного соединения в виде желтой маслянистой жидкости. ЖХ-МС (ИЭР, m/z): 486 [М+Н]+.
Стадия 4. (4-(6-фторбензо[d]оксазол-2-ил)фенил)(4,7-диазаспиро[2.5]октан-4-ил)метанон
В 100 мл круглодонную колбу помещали бензил-4-[[4-(6-фтор-1,3-бензоксазол-2-ил)фенил]-карбонил]-4,7-диазаспиро[2.5]октан-7-карбоксилат (промежуточное соединение 2, 1,2 г, 2,47 ммоль, 1,00 экв.), палладий на подложке активированного угля (1,2 г) и метанол (30 мл). Затем колбу три раза вакуумировали и повторно заполняли водородом. Полученный раствор перемешивали в течение 3 часов при 25°С. Отфильтровывали твердые вещества. Концентрировали фильтрат в вакууме. Это приводило к получению 700 мг (масса неочищенного вещества) титульного соединения в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 352 [М+Н]+.
Стадия 5. (4-(4-(6-фторбензо[d]оксазол-2-ил)бензоил)-4,7-диазаспиро[2.5]октан-7-ил)(1-гидроксициклопропил)метанон
В 100 мл круглодонную колбу помещали HATU (658 мг, 1,73 ммоль, 1,00 экв.), 1-гидроксициклопропан-1-карбоновую кислоту (160 мг, 1,57 ммоль, 0,91 экв.), 2-[4-([4,7-диазаспиро[2.5]октан-4-ил]карбонил)фенил]-6-фтор-1,3-бензоксазол (608 мг, 1,73 ммоль, 1,00 экв.), DIEA (406 мг, 3,14 ммоль, 1,82 экв.) и N,N-диметилформамид (15 мл). Полученный раствор перемешивали в течение 18 часов при 25°С. Концентрировали полученную смесь в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесями этилацетат/петролейный эфир (10%-90%). Неочищенный продукт очищали путем препаративной ВЭЖХ в следующих условиях (Waters I): колонка, Xbridge Prep C18 OBD, 5 мкм, 19*150 мм; мобильная фаза, вода (0,05% NH4HCO3) и CH3CN (от 10% CH3CN до 40% за 9 минут); детектор, УФ 220 и 254 нм. Это приводило к получению 171,6 мг (23%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 436 [М+Н]+.
Способ 25.
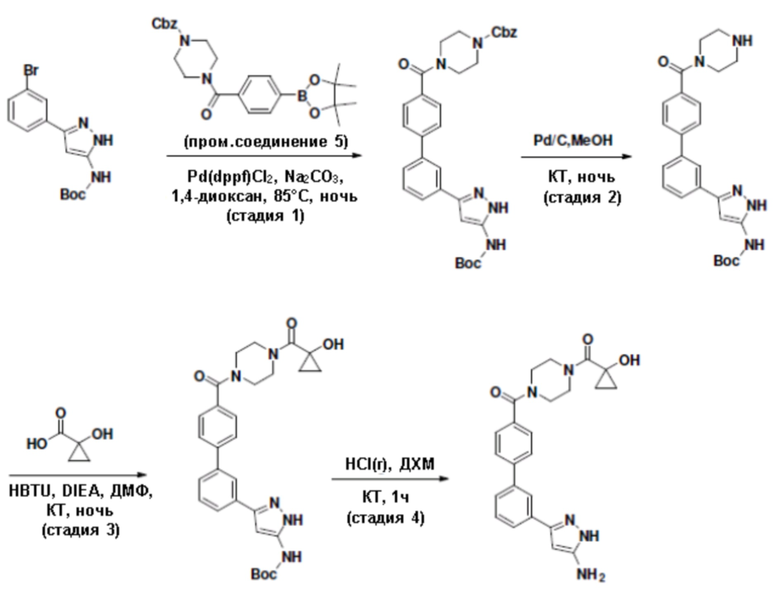
Стадия 1. Бензил-4-(3'-(5-((трет-бутоксикарбонил)амино)-1Н-пиразол-3-ил)-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилат
В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор бензил-4-[[4-(тетраметил-1,3,2-диоксаборолан-2-ил)фенил]карбонил]-пиперазин-1-карбоксилата (промежуточное соединение 5, 710 мг, 1,58 ммоль, 1,00 экв.) в 1,4-диоксане (20 мл), трет-бутил-N-[3-(3-бромфенил)-1Н-пиразол-5-ил]карбамат (533,2 мг, 1,58 ммоль, 1,00 экв.), Pd(dppf)Cl2 (115,5 мг, 0,16 ммоль, 0,10 экв.), карбонат натрия (2М, 5 мл). Полученную смесь перемешивали в течение ночи при 85°С. После охлаждения до комнатной температуры разбавляли реакционную смесь 30 мл воды, экстрагировали 2х100 мл этилацетата. Объединяли органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью этилацетат/петролейный эфир (4/1). Это приводило к получению 300 мг (33%) титульного соединения в виде желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 582 [М+Н]+.
Стадия 2. Трет-бутил-(3-(4'-(пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)-1Н-пиразол-5-ил)карбамат
В 50 мл круглодонную колбу помещали раствор бензил-4-(3'-(5-((трет-бутоксикарбонил)-амино)-1Н-пиразол-3-ил)-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилата (670 мг, 1,15 ммоль, 1,00 экв.) в метаноле (20 мл). После этого добавляли палладий на подложке углерода (670 мг). В полученную выше смесь вводили водород. Полученный раствор перемешивали в течение 5 часов при комнатной температуре. Отфильтровывали твердые вещества. Концентрировали фильтрат в вакууме. Это приводило к получению 515,7 мг (100%) титульного соединения в виде оранжевого твердого вещества. ЖХ-МС (ИЭР, m/z): 448 [М+Н]+.
Стадия 3. Трет-бутил-(3-(4'-(4-(1-гидроксициклопропан-1-карбонил)пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)-1Н-пиразол-5-ил)карбамат
В 250 мл круглодонную колбу помещали раствор трет-бутил-(3-(4'-(пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)-1Н-пиразол-5-ил)карбамата (515,7 мг, 1,15 ммоль, 1,00 экв.) в N,N-диметилформамиде (50 мл), 1-гидроксициклопропан-1-карбоновую кислоту (117 мг, 1,15 ммоль, 1,00 экв.), HBTU (654 мг, 1,72 ммоль, 1,50 экв.), DIEA (445,5 мг, 3,45 ммоль, 3,00 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Затем реакционную смесь разбавляли 50 мл воды, экстрагировали 2х150 мл дихлорметана. Объединяли органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью этилацетат/петролейный эфир (2/1). Это приводило к получению 400 мг (65%) титульного соединения в виде коричневого твердого вещества. ЖХ-МС (ИЭР, m/z): 532 [М+Н]+.
Стадия 4. (4-(3'-(5-амино-1Н-пиразол-3-ил)-[1,1'-бифенил]-4-карбонил)пиперазин-1-ил)(1-гидроксициклопропил)метанон
В 100 мл круглодонную колбу помещали раствор Трет-бутил-(3-(4'-(4-(1-гидроксициклопропан-1-карбонил)пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)-1Н-пиразол-5-ил)карбамата (400 мг, 0,75 ммоль, 1,00 экв.) в дихлорметане (30 мл). В полученную выше смесь вводили газообразный HCl. Полученный раствор перемешивали в течение 1 часа при комнатной температуре. Собирали твердые вещества путем фильтрования и растворяли в 10 мл воды. рН доводили до 6~7 при помощи насыщенного раствора бикарбоната натрия. Собирали твердые вещества путем фильтрования. Остаток очищали путем препаративной флэш-ВЭЖХ в следующих условиях (IntelFlash-1): колонка, С18 с силикагелем; мобильная фаза, CH3CN/вода, содержащая 0,1% NH4HCO3, = 1/5, повышали до CH3CN/вода, содержащая 0,1% NH4HCO3, = 3/2 за 20 минут; детектор, УФ 254 и 220 нм. Это приводило к получению 102,9 мг (32%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 432 [М+Н]+.
Способ 26.
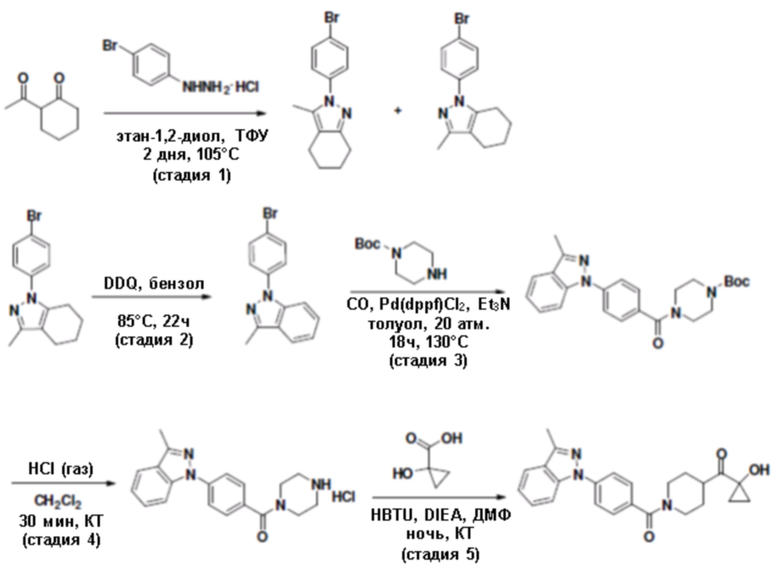
Стадия 1. N-(4-бромфенил)-3-метил-4,5,6,7-тетрагидро-1- и -2Н-индазол
В 250 мл круглодонную колбу (1 атм), продутую и выдерживаемую в инертной атмосфере азота, помещали 2-ацетилциклогексан-1-он (4,08 г, 29,11 ммоль, 1,00 экв.), гидрохлорид (4-бромфенил)гидразина (6,5 г, 29,08 ммоль, 1,00 экв.), TsOH (500 мг, 2,90 ммоль, 0,10 экв.), этан-1,2-диол (100 мл). Полученный раствор перемешивали в течение 2 дней при 105°С на масляной бане. Концентрировали полученную смесь в вакууме. Неочищенный продукт (4 г) очищали путем препаративной флэш-ВЭЖХ в следующих условиях (IntelFlash-1): колонка С18 с силикагелем; мобильная фаза, 0,05% раствор NH4HCO3, MeCN = 95/5, повышали до 0,05% раствор NH4HCO3, MeCN = 0/100 за 60 минут; детектор, УФ 254 нм. Получали 1,5 г продукта. Это приводило к получению 1,5 г (18%) 2-(4-бромфенил)-3-метил-4,5,6,7-тетрагидро-2Н-индазола в виде светло-желтого твердого вещества и 0,8 г (9%) 1-(4-бромфенил)-3-метил-4,5,6,7-тетрагидро-2Н-индазола в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 292 [М+Н]+.
Стадия 2. 1-(4-бромфенил)-3-метил-1Н-индазол
В 250 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали 1-(4-бромфенил)-3-метил-4,5,6,7-тетрагидро-1Н-индазол (3,5 г, 12,02 ммоль, 1,00 экв.), бензол (100 мл), DDQ (10,9 г, 48,02 ммоль, 3,99 экв.). Полученный раствор перемешивали в течение 22 часов при 85°С на масляной бане. После охлаждения до комнатной температуры концентрировали полученную смесь в вакууме и разбавляли 200 мл ЭА и промывали 1х250 мл воды и 3х100 мл солевого раствора. Органическую фазу сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесями этилацетат/петролейный эфир (0/100~10/90). Это приводило к получению 800 мг (23%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 287, 289 [М+Н]+.
Стадия 3. Трет-бутил-4-(4-(3-метил-1Н-индазол-1-ил)бензоил)пиперазин-1-карбоксилат
В 250 мл резервуар, стойкий к давлению, помещали 1-(4-бромфенил)-3-метил-1Н-индазол (800 мг, 2,79 ммоль, 1,00 экв.), трет-бутил-пиперазин-1-карбоксилат (770 мг, 4,13 ммоль, 1,48 экв.), Pd(dppf)Cl2.CH2Cl2 (113 мг, 0,14 ммоль, 0,05 экв.), толуол (120 мл), триэтиламин (837 мг, 8,27 ммоль, 2,97 экв.). Затем реактор три раза вакуумировали и повторно заполняли СО. Полученную смесь перемешивали в течение 24 часов при 130°С под давлением СО 20 атмосфер. После охлаждения до комнатной температуры концентрировали полученную смесь в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесями этилацетат/петролейный эфир (0/100~25/75). Это приводило к получению 1,0 г (85%) титульного соединения в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 421 [М+Н]+.
Стадия 4. Гидрохлорид (4-(3-метил-1Н-индазол-1-ил)фенил)(пиперазин-1-ил)метанона
В 250 мл круглодонную колбу помещали трет-бутил-4-(4-(3-метил-1Н-индазол-1-ил)бензоил)пиперазин-1-карбоксилат (1 г, 2,38 ммоль, 1,0 экв.), дихлорметан (100 мл). Затем вводили хлороводород (газ). Полученный раствор перемешивали в течение 2 часов при комнатной температуре. Собирали твердые вещества путем фильтрования. Это приводило к получению 930 мг (масса неочищенного вещества) титульного соединения в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 321 [М+Н]+.
Стадия 5. (4-(1-гидроксициклопропан-1-карбонил)пиперазин-1-ил)(4-(3-метил-1Н-индазол-1-ил)фенил)метанон
В 100 мл круглодонную колбу помещали 1-гидроксициклопропан-1-карбоновую кислоту (265 мг, 2,60 ммоль, 1,00 экв.), HBTU (1,086 г, 2,86 ммоль, 1,10 экв.), N,N-диметилформамид (20 мл), гидрохлорид (4-(3-метил-1Н-индазол-1-ил)фенил)(пиперазин-1-ил)метанона (930 мг, 2,61 ммоль, 1,00 экв.), DIEA (1,68 г, 13,00 ммоль, 5,01 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Полученный раствор разбавляли 100 мл ЭА, промывали 3х20 мл воды и 1х20 мл солевого раствора. Органическую фазу сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесями этилацетат/петролейный эфир (0/100~100/0). Объединяли собранные фракции и концентрировали в вакууме. Полученное твердое вещество перемешивали в 2х20 мл MeCN, собирали путем фильтрования и сушили при пониженном давлении. Это приводило к получению 228,5 мг (22%) титульного соединения в виде беловатого твердого вещества. ЖХ-МС (ИЭР, m/z): 405 [М+Н]+.
Способ 27.
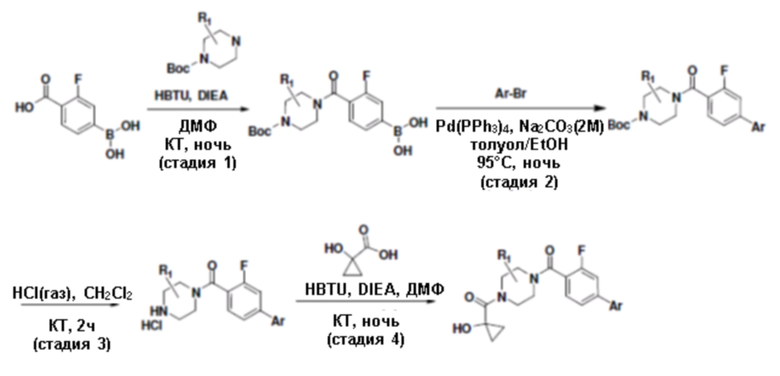
Стадия 1. (S)-(4-(4-(трет-бутоксикарбонил)-3-метилпиперазин-1-карбонил)-3-фторфенил)бороновая кислота
В 250 мл круглодонную колбу помещали раствор 4-(дигидроксиборанил)-2-фторбензойной кислоты (1,84 г, 10,00 ммоль, 1,00 экв.) в ДМФ (30 мл). Добавляли HBTU (15,16 г, 39,97 ммоль, 4,00 экв.), затем DIEA (1,94 г, 15,01 ммоль, 1,50 экв.). Полученный раствор перемешивали в течение 10 минут при КТ, затем добавляли раствор трет-бутил-(2R)-2-метилпиперазин-1-карбоксилата (2 г, 9,99 ммоль, 1,00 экв.) в ДМФ (5 мл). Полученный раствор оставляли перемешиваться на ночь. Затем раствор разбавляли EtOAc (50 мл) и промывали солевым раствором (3х50 мл). Затем органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очищали остаток путем FCC, элюируя смесью дихлорметан/метанол (90:10). Это приводило к получению титульного соединения (3,5 г, 96%) в виде коричневого твердого вещества. ЖХ-МС (ИЭР, m/z): 367 [М+Н]+.
Стадия 2. Трет-бутил-(S)-4-(2-фтор-4-(1-метил-1Н-бензо[d]имидазол-5-ил)бензоил)-2-метилпиперазин-1-карбоксилат
В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор (S)-(4-(4-(трет-бутоксикарбонил)-3-метилпиперазин-1-карбонил)-3-фторфенил)бороновой кислоты (837 мг, 2,29 ммоль, 1,00 экв.) в толуоле (20 мл), 5-бром-1-метил-1Н-1,3-бензодиазол (400 мг, 1,90 ммоль, 0,83 экв.), Pd(PPh3)4 (264 мг, 0,23 ммоль, 0,10 экв.), карбонат натрия (2М, 10 мл), этанол (2,8 мл). Полученную смесь перемешивали в течение ночи при 95°С. После охлаждения до комнатной температуры полученный раствор разбавляли 20 мл H2O, экстрагировали 3х30 мл этилацетата. Объединяли органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью дихлорметан/метанол (90:10). Объединяли собранные фракции и концентрировали в вакууме. Это приводило к получению 800 мг (77%) титульного соединения в виде коричневого твердого вещества. ЖХ-МС (ИЭР, m/z): 453 [М+Н]+.
Стадия 3. Гидрохлорид (S)-(2-фтор-4-(1-метил-1Н-бензо[d]имидазол-5-ил)фенил)(3-метилпиперазин-1-ил)метанона
В 100 мл круглодонную колбу помещали раствор трет-бутил-(S)-4-(2-фтор-4-(1-метил-1Н-бензо[d]имидазол-5-ил)бензоил)-2-метилпиперазин-1-карбоксилата (800 мг, 1,77 ммоль, 1,00 экв.) в дихлорметане (30 мл). В полученную выше смесь вводили HCl (газ). Полученный раствор перемешивали в течение 2 часов при комнатной температуре. Собирали твердые вещества путем фильтрования. Это приводило к получению 650 мг (95%) титульного соединения в виде коричневого твердого вещества. ЖХ-МС (ИЭР, m/z): 353 [М+Н]+.
Стадия 4. (S)-(4-(2-фтор-4-(1-метил-1Н-бензо[d]имидазол-5-ил)бензоил)-2-метилпиперазин-1-ил)(1-гидроксициклопропил)метанон
В 100 мл круглодонную колбу помещали раствор гидрохлорида (S)-(2-фтор-4-(1-метил-1Н-бензо[d]имидазол-5-ил)фенил)(3-метилпиперазин-1-ил)метанона (650 мг, 1,67 ммоль, 1,00 экв.) в N,N-диметилформамиде (20 мл), 1-гидроксициклопропан-1-карбоновую кислоту (180 мг, 1,76 ммоль, 1,05 экв.), HBTU (734 мг, 1,94 ммоль, 1,16 экв.), DIEA (908 мг, 7,03 ммоль, 4,20 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли 30 мл ЭА, промывали 3х30 мл воды и 30 мл солевого раствора. Органическую фазу сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью дихлорметан/метанол (90:10). Неочищенный продукт дополнительно очищали путем препаративной ВЭЖХ в следующих условиях (Waters III): колонка Xbridge RP18, 5 мкм, 19*150; мобильная фаза, вода (0,05% NH4HCO3) и MeCN (от 10% CH3CN до 70% за 9 минут); детектор, УФ 220 и 254 нм. Это приводило к получению 203,1 мг (28%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 437 [М+Н]+.
Способ 28.
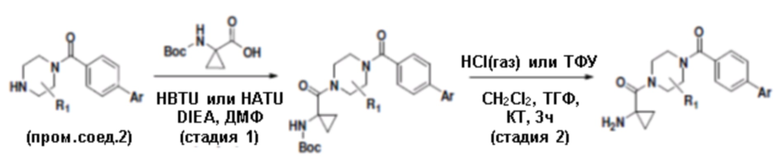
Стадия 1. Трет-бутил-(S)-(1-(4-(4-(6-фторбензо[d]оксазол-2-ил)бензоил)-2-метилпиперазин-1-карбонил)циклопропил)карбамат
В 100 мл круглодонную колбу помещали 1-[(трет-бутокси)карбонил]аминоциклопропан-1-карбоновую кислоту (промежуточное соединение 2, 400 мг, 1,99 ммоль, 0,90 экв.), HBTU (830 мг, 2,19 ммоль, 1,0 экв.), N,N-диметилформамид (30 мл), гидрохлорид (S)-(4-(6-фторбензо[d]оксазол-2-ил)фенил)(3-метилпиперазин-1-ил)метанона (750 мг, 2,21 ммоль, 1,00 экв.) и DIEA (514 мг, 3,98 ммоль, 1,80 экв.). Полученный раствор перемешивали в течение 18 часов при 25°С. Полученный раствор разбавляли 50 мл ЭА и промывали 2х50 мл воды. Органический слой сушили над Na2SO4 и концентрировали в вакууме. Это приводило к получению 800 мг (масса неочищенного вещества) титульного соединения в виде желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 523 [М+Н]+.
Стадия 2. (S)-(4-(1-аминоциклопропан-1-карбонил)-3-метилпиперазин-1-ил)(4-(6-фторбензо[d]оксазол-2-ил)фенил)метанон
В 100 мл круглодонную колбу помещали трет-бутил-(S)-(1-(4-(4-(6-фторбензо[d]оксазол-2-ил)бензоил)-2-метилпиперазин-1-карбонил)циклопропил)карбамат (800 мг, 1,53 ммоль, 1,00 экв.), дихлорметан (20 мл), тетрагидрофуран (20 мл). Затем смесь барботировали хлороводородом (газ). Полученный раствор перемешивали в течение 3 часов при 25°С. Концентрировали полученную смесь в вакууме и растворяли в 10 мл воды. рН раствора доводили до 8 при помощи насыщенного раствора карбоната натрия. Собирали твердые вещества путем фильтрования. Неочищенный продукт очищали путем препаративной ВЭЖХ в следующих условиях (Waters I): колонка Xbridge Prep C18 OBD, 5 мкм, 19*150 мм; мобильная фаза, вода (0,05% NH4HCO3) и CH3CN (от 7% CH3CN до 35% за 10 минут); детектор, УФ 220 и 254 нм. Это приводило к получению 163,6 мг (25%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 423 [М+Н]+.
Способ 29.
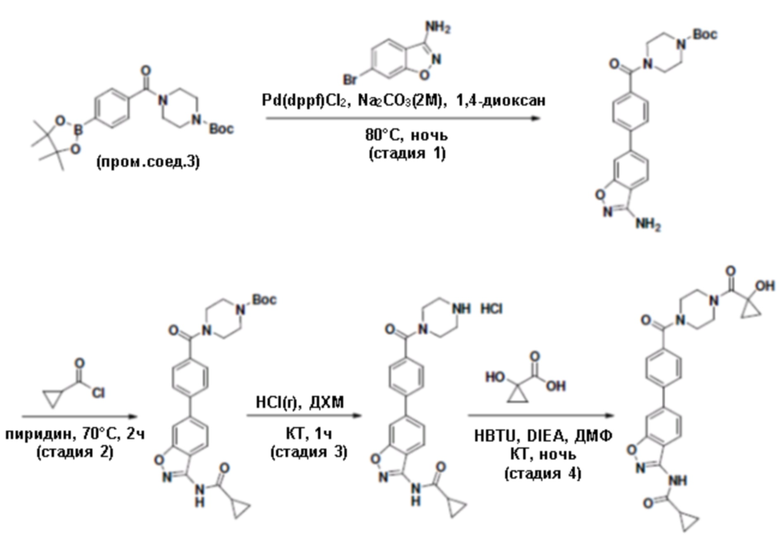
Стадия 1. Трет-бутил-4-(4-(3-аминобензо[d]изоксазол-6-ил)бензоил)пиперазин-1-карбоксилат
В 250 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор трет-бутил-4-[[4-(тетраметил-1,3,2-диоксаборолан-2-ил)фенил]карбонил]-пиперазин-1-карбоксилата (промежуточное соединение 3, 1,9 г, 4,56 ммоль, 1,00 экв.) в диоксане (100 мл), 6-бром-1,2-бензоксазол-3-амин (1 г, 4,69 ммоль, 1,00 экв.), Pd(dppf)Cl2 (344 мг, 0,47 ммоль, 0,10 экв.), карбонат натрия (20 мл, 2М). Полученный раствор перемешивали в течение ночи при 80°С. После охлаждения до комнатной температуры полученную смесь разбавляли ДХМ, промывали водой и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью дихлорметан/метанол (10/1). Это приводило к получению 1,6 г (83%) титульного соединения в виде серого твердого вещества. ЖХ-МС (ИЭР, m/z): 423 [М+Н]+.
Стадия 2. Трет-бутил-4-(4-(3-(циклопропанкарбоксамидо)бензо[d]изоксазол-6-ил)бензоил)пиперазин-1-карбоксилат
В 100 мл круглодонную колбу помещали раствор трет-бутил-4-[[4-(3-амино-1,2-бензоксазол-6-ил)фенил]карбонил]пиперазин-1-карбоксилата (800 мг, 1,89 ммоль, 1,00 экв.) в пиридине (20 мл), циклопропанкарбонилхлорид (199 мг, 1,90 ммоль, 1,00 экв.). Полученный раствор перемешивали в течение 2 часов при 70°С. После охлаждения до комнатной температуры реакционную смесь разбавляли EtOAc, промывали водой и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью этилацетат/петролейный эфир (1/1). Это приводило к получению 535 мг (58%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 491 [М+Н]+.
Стадия 3. Гидрохлорид N-(6-(4-(пиперазин-1-карбонил)фенил)бензо[d]изоксазол-3-ил)циклопропанкарбоксамида
В 100 мл круглодонную колбу помещали раствор трет-бутил-4-[[4-(3-циклопропанамидо-1,2-бензоксазол-6-ил)фенил]карбонил]пиперазин-1-карбоксилата (535 мг, 1,09 ммоль, 1,00 экв.) в дихлорметане (30 мл). В полученный выше раствор вводили хлороводород (газ). Полученный раствор перемешивали в течение 1 часа при комнатной температуре. Концентрировали полученную смесь в вакууме. Это приводило к получению 466 мг (количественный выход) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 391 [М+Н]+.
Стадия 4. N-(6-(4-(4-(1-гидроксициклопропан-1-карбонил)пиперазин-1-карбонил)-фенил)бензо[d]изоксазол-3-ил)циклопропанкарбоксамид
В 50 мл круглодонную колбу помещали раствор 1-гидроксициклопропан-1-карбоновой кислоты (111 мг, 1,09 ммоль, 1,00 экв.) в N,N-диметилформамиде (20 мл), HBTU (620 мг, 1,63 ммоль, 1,50 экв.), DIEA (563 мг, 4,36 ммоль, 4,00 экв.). Полученную выше смесь перемешивали в течение 1 часа при комнатной температуре. В полученную смесь добавляли гидрохлорид N-(6-[4-[(пиперазин-1-ил)карбонил]фенил]-1,2-бензоксазол-3-ил)циклопропанкарбоксамида (466 мг, 1,09 ммоль, 1,00 экв.). Полученную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли ДХМ, промывали водой и концентрировали в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью дихлорметан/метанол (10/1) и дополнительно очищали путем препаративной ВЭЖХ в следующих условиях (IntelFlash-1): колонка С18 с силикагелем; мобильная фаза, CH3CN/вода, содержащая 0,05% NH4HCO3, = 40%, повышали до CH3CN/вода, содержащая 0,05% NH4HCO3, = 60% за 15 минут; детектор, УФ 254 и 220 нм. Это приводило к получению 123,9 мг (24%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 475 [М+Н]+.
Способ 30.
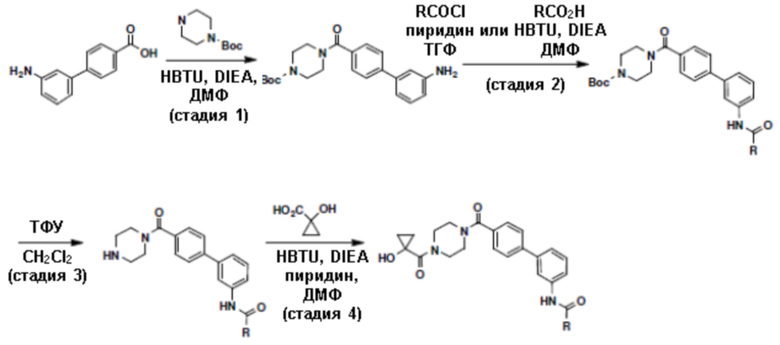
Стадия 1. Трет-бутил-4-(3'-амино-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилат
В 100 мл круглодонную колбу добавляли трет-бутил-пиперазин-1-карбоксилат (1,310 г, 7,03 ммоль), 3'-аминобифенил-4-карбоновую кислоту (1,5 г, 7,03 ммоль) и DIEA (2,457 мл, 14,07 ммоль) в ДМФ (25 мл), затем HBTU (3,20 г, 8,44 ммоль) с получением коричневой суспензии, которую перемешивали при КТ в течение 8 часов. Добавляли воду (50 мл) и дважды экстрагировали реакционную смесь метиленхлоридом (2х100 мл). Объединяли органические слои и промывали водой (25 мл) и солевым раствором (25 мл). Затем органический слой сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Неочищенный остаток очищали путем FCC (0-4% MeOH/CH2Cl2) с получением титульного соединения (1,70 г, 63%) в виде желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 382 [М+Н]+.
Стадия 2 (RCOCl). Трет-бутил-4-(3'-(циклопентанкарбоксамидо)-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилат
В 20 мл сцинтилляционную пробирку добавляли трет-бутил-4-(3'-аминобифенилкарбонил)-пиперазин-1-карбоксилат (150 мг, 0,393 ммоль), пиридин (0,095 мл, 1,180 ммоль) и циклопентанкарбонилхлорид (0,057 мл, 0,472 ммоль) в ТГФ (9 мл) с получением светло-желтой суспензии, которую перемешивали при КТ в течение 2 часов. Реакционную смесь помещали в 75 мл метиленхлорида. Органический раствор дважды промывали разбавленным раствором карбоната натрия (водн., 35 мл) и один раз солевым раствором (15 мл). Затем органический слой сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали путем FCC (0-60% EtOAc/гексаны) с получением титульного соединения (116 мг, 62%). ЖХ-МС (ИЭР, m/z): 478 [М+Н]+.
Стадия 2 (RCO2H). Трет-бутил-4-(3'-(3,3-дифторциклобутанкарбоксамидо)-бифенилкарбонил)пиперазин-1-карбоксилат
В 20 мл сцинтилляционную пробирку добавляли трет-бутил-4-(3'-аминобифенилкарбонил)-пиперазин-1-карбоксилат (200 мг, 0,524 ммоль), 3,3-дифторциклобутанкарбоновую кислоту (86 мг, 0,629 ммоль) и основание Хюнига (0,275 мл, 1,573 ммоль) в ДМФ (5 мл), затем HBTU (239 мг, 0,629 ммоль) с получением коричневой суспензии, которую перемешивали при КТ в течение ночи, затем реакционную смесь разбавляли водой (20 мл). Затем реакционную смесь экстрагировали метиленхлоридом (2х40 мл). Объединенные органические слои промывали водой (20 мл) и солевым раствором (20 мл). Органический слой сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали путем FCC (0-60% EtOAc/гексаны) с получением титульного соединения в виде вспененного стекла. ЖХ-МС (ИЭР, m/z): 500 [М+Н]+.
Стадия 3. 2,2,2-трифторацетат N-(4'-(пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)циклопентанкарбоксамида
В 100 мл круглодонную колбу добавляли трет-бутил-4-(3'-(циклопентанкарбоксамидо)-бифенилкарбонил)пиперазин-1-карбоксилат (114 мг, 0,239 ммоль) в метиленхлориде (15 мл) с получением желтой суспензии. Постепенно при помощи шприца добавляли трифторуксусную кислоту (0,552 мл, 7,16 ммоль). Раствор перемешивали при КТ в течение 2,5 часа. Концентрировали раствор и растирали неочищенный продукт с диэтиловым эфиром, затем декантировали. Полученное твердое вещество сушили в вакууме с получением титульного соединения (117 мг, 100%) в виде твердой пены.
Стадия 4. N-(4'-(4-(1-гидроксициклопропан-1-карбонил)пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)циклопентанкарбоксамид
В 100 мл круглодонную колбу добавляли 2,2,2-трифторацетат N-(4'-(пиперазин-1-карбонил)-бифенил-3-ил)циклопентанкарбоксамида (118 мг, 0,24 ммоль), 1-гидроксициклопропанкарбоновую кислоту (19,60 мг, 0,192 ммоль) и пиридин (0,058 мл, 0,720 ммоль) в ДМФ (5 мл), затем HBTU (127 мг, 0,336 ммоль) с получением желтой суспензии, которую перемешивали при КТ в течение 4,5 часа. Добавляли основание Хюнига (0,021 мл, 0,12 ммоль) и продолжали перемешивать. Через 18 часов дополнительно добавляли основание Хюнига (0,063 мл, 0,36 ммоль) и реакционный раствор перемешивали в течение ночи. Реакционную смесь разбавляли водой (20 мл) и экстрагировали метиленхлоридом (2х50 мл). Объединяли органические слои и промывали водой (10 мл) и солевым раствором (10 мл). Раствор сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали путем FCC (0-7% MeOH/CH2Cl2) с получением титульного соединения (35,5 мг, 29%) в виде беловатого твердого вещества. ЖХ-МС (ИЭР, m/z): 462 [М+Н]+.
Способ 31.

Стадия 1. Трет-бутил-4-(4'-бром-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилат
В суспензию 4'-бром-[1,1'-бифенил]-4-карбоновой кислоты (1,5 г, 5,4 ммоль) в ДМФ (35 мл) добавляли ТЭА (1,21 мл, 8,6 ммоль), затем трет-бутил-пиперазин-1-карбоксилат (1,11 г, 6,0 ммоль) и HATU (2,6 г, 6,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем разбавляли водой и экстрагировали EtOAc. Органическую фазу промывали водой, 5% уксусной кислотой, насыщенным NaHCO3, солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очищали путем хроматографии на колонке с силикагелем, элюируя с градиентом смесями EtOAc в CH2Cl2, с получением 2,0 г (выход 83%) титульного соединения.
Стадия 2. Трет-бутил-4-(4'-(1Н-пиразол-4-ил)-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилат
Трет-бутил-4-(4'-бром-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилат (1,0 г, 2,3 ммоль), (1Н-пиразол-4-ил)бороновую кислоту (соль HCl, 0,67 г, 4,5 ммоль) и 2М K3PO4 (водн.) (4,5 мл, 9,0 ммоль) смешивали с 30 мл ДМФ в пробирке для микроволнового реактора. Полученную смесь несколько раз вакуумировали и повторно заполняли аргоном, после чего добавляли Pd(PPh3)4 (0,39 г, 0,3 ммоль). Реакционную смесь облучали в микроволновом реакторе в течение 35 минут при 150°С. После охлаждения до комнатной температуры разбавляли реакционную смесь водой и насыщенным NaHCO3 и экстрагировали смесью растворителей MeOH/CHCl3. Органическую фазу сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный остаток дважды очищали на системе ISCO (24 г колонка с силикагелем), элюируя с градиентом смесями 0,7н. NH3/MeOH в CHCl3, с получением 160 мг (выход ~17%) целевого продукта.
Стадия 3. Соль HCl (4'-(1Н-пиразол-4-ил)-[1,1'-бифенил]-4-ил)(пиперазин-1-ил)метанона
Трет-бутил-4-(4'-(1Н-пиразол-4-ил)-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилат (160 мг, 0,37 ммоль) растворяли в 18 мл безводного MeOH, насыщенного HCl (газ), при 0°С и перемешивали при комнатной температуре в течение ~3 часов. Полученный раствор концентрировали досуха при пониженном давлении. Остаток растирали с 2% MeOH в Et2O, затем в Et2O и пентане с получением 150 мг (колич. выход) титульного соединения в виде гидрохлоридной соли, которую использовали на следующей стадии без дополнительной очистки.
Стадия 4. (4-(4'-(1Н-пиразол-4-ил)-[1,1'-бифенил]-4-карбонил)пиперазин-1-ил)(1-гидроксициклопропил)метанон
В раствор соли HCl (4'-(1Н-пиразол-4-ил)-[1,1'-бифенил]-4-ил)(пиперазин-1-ил)метанона (150 мг, 0,37 моль) и 1-гидроксициклопропан-1-карбоновой кислоты (69 мг, 0,56 ммоль) в 3 мл ДМФ добавляли HBTU (230 мг, 0,6 ммоль), затем добавляли DIEA (0,33 мл, 1,8 ммоль). Полученный раствор перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли водой и насыщенным NaHCO3 и несколько раз экстрагировали смесями растворителей EtOAc/MeOH и MeOH/CHCl3. Объединяли органические слои, промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный остаток дважды очищали на системе ISCO (24 г золотая колонка с силикагелем), элюируя с градиентом смесями 0,7н. NH3/MeOH в EtOAc и 0,7н. NH3/MeOH в CHCl3. Продукт, полученный после хроматографии, растирали в Et2O с получением 110 мг (выход 71%) чистого целевого соединения. ЖХ-МС (ИЭР, m/z): 417 [М+Н]+.
Способ 32.
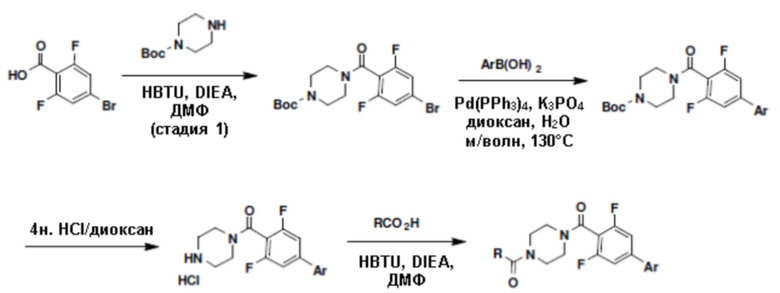
Стадия 1. Трет-бутил-4-(4-бром-2,6-дифторбензоил)пиперазин-1-карбоксилат
В раствор 4-бром-2,6-дифторбензойной кислоты (1,00 г, 4,22 ммоль), трет-бутил-пиперазин-1-карбоксилата (0,786 г, 4,22 ммоль) и HBTU (1,925 г, 5,06 ммоль) в ДМФ (10,0 мл) добавляли DIEA (2,95 мл, 16,88 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный бикарбонат натрия (водный, 10 мл), затем воду (10 мл). Затем реакционный раствор несколько раз экстрагировали EtOAc (3х20 мл). Объединяли органические экстракты и промывали водой (2х20 мл) и солевым раствором (20 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением неочищенного продукта в виде оранжевой маслянистой жидкости. Неочищенный продукт очищали путем FCC (Biotage SNAP 25; элюент: 15% - 25% EtOAc в гексанах за 10 объемов колонки (CV)). Это приводило к получению титульного соединения (1,44 г, 86%) в виде беловатого твердого вещества. ЖХ-МС (ИЭР, m/z): 405 [М+Н]+.
Стадия 2. Трет-бутил-4-(2,6-дифтор-4-(1-метил-1Н-индазол-6-ил)бензоил)пиперазин-1-карбоксилат
Трет-бутил-4-(4-бром-2,6-дифторбензоил)пиперазин-1-карбоксилат (383,2 мг, 0,946 ммоль), 1-метил-1Н-индазол-6-илбороновую кислоту (166 мг, 0,946 ммоль) и фосфат калия (1,00 г, 4,73 ммоль) суспендировали в растворе диоксана (6,0 мл) и воды (1,2 мл), продутом азотом. Реакционную смесь дополнительно продували азотом в течение 5 минут. Добавляли тетракис(трифенилфосфин)палладий (109 мг, 0,095 ммоль) и реакционный раствор продували азотом в течение еще 5 минут. Смесь облучали в микроволновом реакторе при 130°С в течение 30 минут, что приводило к получению желтого двухфазного раствора. Удаляли органический (верхний) слой, фильтровали через целит и концентрировали в вакууме с получением неочищенного продукта в виде светло-красного порошка. Неочищенный продукт очищали путем FCC (Biotage SNAP 25; элюировали с градиентом: 0-20% MeOH в EtOAc, содержащем 0,5% триэтиламина, за 15 CV). Это приводило к получению титульного соединения (327 мг, 76%) в виде светло-бежевого порошка. ЖХ-МС (ИЭР, m/z): 421 [М+Н]+.
Стадия 3. Гидрохлорид (2,6-дифтор-4-(1-метил-1Н-индазол-6-ил)фенил)(пиперазин-1-ил)метанона
В раствор трет-бутил-4-(2,6-дифтор-4-(1-метил-1Н-индазол-6-ил)бензоил)пиперазин-1-карбоксилата (320,0 мг, 0,701 ммоль) в диоксане (4,0 мл) добавляли HCl (4н. в 1,4-диоксане) (0,106 мл, 3,51 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 2 часов, что приводило к образованию белого осадка. В реакционную смесь добавляли диэтиловый эфир (10 мл), затем собирали осадок путем фильтрования. Осадок дополнительно промывали диэтиловым эфиром (10 мл), собирали и сушили в вакууме с получением титульного соединения (275 мг, 100%) в виде белого порошка. ЖХ-МС (ИЭР, m/z): 356 [М+Н]+.
Стадия 4. (4-(2,6-дифтор-4-(1-метил-1Н-индазол-6-ил)бензоил)пиперазин-1-ил)(1-гидроксициклопропил)метанон
В раствор гидрохлорида (2,6-дифтор-4-(1-метил-1Н-индазол-6-ил)фенил)(пиперазин-1-ил)метанона (218,3 мг, 0,556 ммоль), 1-гидроксициклопропанкарбоновой кислоты (56,7 мг, 0,556 ммоль) и HBTU (254 мг, 0,667 ммоль) в ДМФ (4,0 мл) добавляли DIEA (0,388 мл, 2,223 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный бикарбонат натрия (водный, 10 мл), затем воду (10 мл). Затем реакционный раствор несколько раз экстрагировали EtOAc (3х20 мл). Объединяли органические экстракты и промывали водой (2х20 мл) и солевым раствором (20 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением неочищенного продукта в виде оранжевой маслянистой жидкости. Неочищенный продукт очищали путем FCC (Biotage SNAP 25; элюент: 20-10% смеси гексанов в EtOAc за 10 CV). Затем продукт подвергали дополнительной очистке путем FCC (Biotage SNAP 25; элюировали с градиентом: 5-15% смесями МеОН в метиленхлориде). Это приводило к получению титульного соединения (105,2 мг, 43%) в виде белого порошка. ЖХ-МС (ИЭР, m/z): 441 [М+Н]+.
Способ 33.
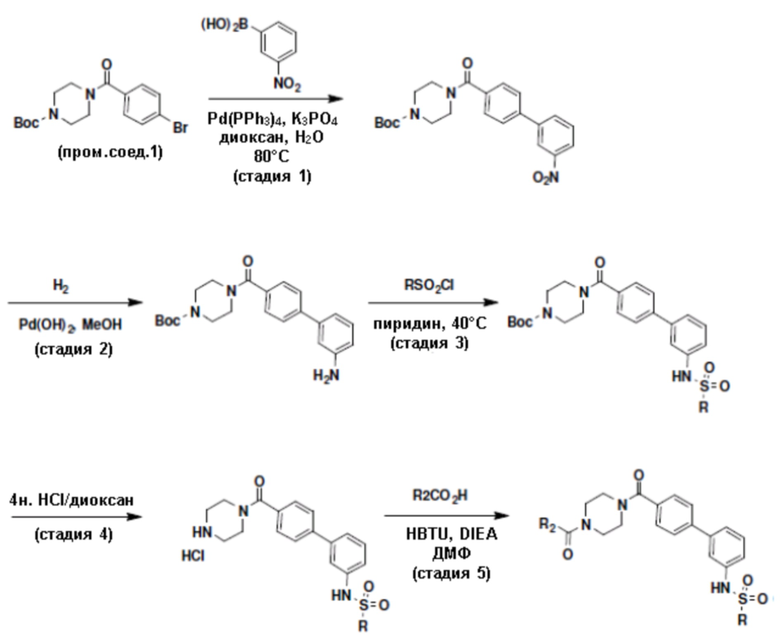
Стадия 1. Трет-бутил-4-(3'-нитробифенилкарбонил)пиперазин-1-карбоксилат
В раствор трет-бутил-4-(4-бромбензоил)пиперазин-1-карбоксилата (1,00 г, 2,71 ммоль), 3-нитрофенилбороновой кислоты (0,452 г, 2,71 ммоль) и фосфата калия (2,87 г, 13,54 ммоль) в диоксане, продутом азотом, (10,0 мл) и воде, продутой азотом, (2,0 мл) добавляли тетракис-(трифенилфосфин)палладий (0,313 г, 0,271 ммоль). Реакционный раствор дополнительно продували током азота в течение 15 минут. Затем проводили взаимодействие в течение ночи при 80°С. Реакционную смесь охлаждали до комнатной температуры, что приводило к образованию желтого осадка. Собирали осадок путем фильтрования. Затем промывали осадок метанолом (20 мл) и диэтиловым эфиром (10 мл). Это приводило к получению титульного соединения (1,0 г, 92%) в виде светло-желтого порошка. ЖХ-МС (ИЭР, m/z): 412 [М+Н]+.
Стадия 2. Трет-бутил-4-(3'-аминобифенилкарбонил)пиперазин-1-карбоксилат
Смесь трет-бутил-4-(3'-нитробифенилкарбонил)пиперазин-1-карбоксилата (1,00 г, 2,430 ммоль) и гидроксида палладия на активированном угле (0,341 г, 2,430 ммоль) суспендировали в МеОН (30 мл). Бомбу помещали во встряхиватель Парра и заполняли газообразным водородом при 30 psi. Реакционную смесь оставляли встряхиваться на 3 часа. Затем реакционную смесь продували азотом, фильтровали через целит и концентрировали в вакууме с получением титульного соединения (924 мг, 100%) в виде светло-коричневого твердого вещества. ЖХ-МС (ИЭР, m/z): 382 [М+Н]+.
Стадия 3. Трет-бутил-4-(3'-(циклобутансульфонамидо)бифенилкарбонил)пиперазин-1-карбоксилат
В раствор трет-бутил-4-(3'-аминобифенилкарбонил)пиперазин-1-карбоксилата (500,0 мг, 1,311 ммоль) в безводном пиридине (6,5 мл) добавляли циклобутансульфонилхлорид (203 мг, 1,311 ммоль) при 40°С. Реакционный раствор (красного цвета) перемешивали при 40°С в течение 2 дней. Реакционный раствор охлаждали до комнатной температуры и разбавляли водой (10 мл). Затем раствор несколько раз экстрагировали EtOAc (3х20 мл). Объединяли органические экстракты и промывали 1М лимонной кислотой (водная, 20 мл), водой (20 мл) и солевым раствором (20 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали путем FCC (Biotage SNAP 50; элюировали с градиентом 40-50% смесями EtOAc в гексанах за 15 CV). Это приводило к получению титульного соединения (173 мг, 26,3%) в виде белого порошка. ЖХ-МС (ИЭР, m/z): 500 [М+Н]+.
Стадия 4. Гидрохлорид N-(4'-(пиперазин-1-карбонил)бифенил-3-ил)циклобутансульфонамида
В раствор трет-бутил-4-(3'-(циклобутансульфонамидо)бифенилкарбонил)пиперазин-1-карбоксилата (175,0 мг, 0,350 ммоль) в диоксане (2,0 мл) добавляли хлороводород (4,0н. в 1,4-диоксане) (0,438 мл, 1,751 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 3 часов, что приводило к образованию беловатого осадка. В реакционную смесь добавляли диэтиловый эфир (10 мл), а затем собирали осадок путем фильтрования. Осадок дополнительно промывали диэтиловым эфиром (10 мл), собирали и сушили в вакууме с получением титульного соединения (153 мг, 100%) в виде беловатого порошка. ЖХ-МС (ИЭР, m/z): 400 [М+Н]+.
Стадия 5. N-(4'-(4-(1-гидроксициклопропанкарбонил)пиперазин-1-карбонил)бифенил-3-ил)циклобутансульфонамид
В раствор гидрохлорида N-(4'-(пиперазин-1-карбонил)бифенил-3-ил)циклобутансульфонамида (164,5 мг, 0,377 ммоль), 1-гидроксициклопропанкарбоновой кислоты (38,5 мг, 0,377 ммоль) и HBTU (172 мг, 0,453 ммоль) в ДМФ (2,0 мл) добавляли DIEA (0,264 мл, 1,509 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный бикарбонат натрия (водный, 10 мл), затем воду (10 мл). Затем реакционный раствор несколько раз экстрагировали EtOAc (3х20 мл). Объединяли органические экстракты и промывали водой (2х20 мл) и солевым раствором (20 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением неочищенного продукта в виде оранжевой маслянистой жидкости. Неочищенный продукт очищали путем FCC (Biotage SNAP 25; элюент: 100% EtOAc за 10 CV), затем проводили дополнительную очистку путем FCC (Biotage SNAP 25; элюировали с градиентом 0-5% смесями МеОН в метиленхлориде). Это приводило к получению титульного соединения (55 мг, 30,3%) в виде стекловидного прозрачного твердого вещества. ЖХ-МС (ИЭР, m/z): 484 [М+Н]+.
Способ 34.
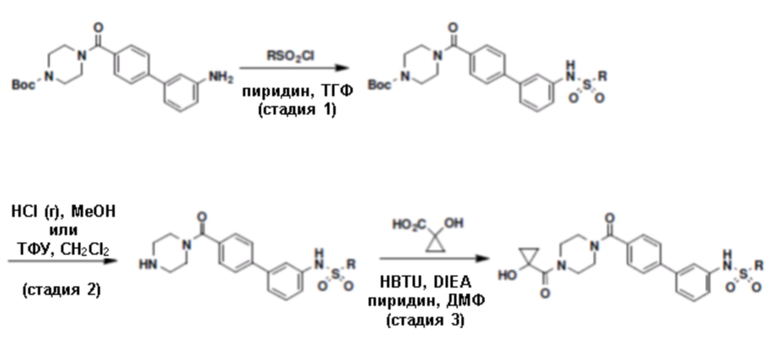
Стадия 1. Трет-бутил-4-(3'-(циклобутилметилсульфонамидо)-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилат
В ледяной раствор трет-бутил-4-(3'-амино-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилата (300 мг, 0,79 ммоль) в пиридине (4 мл) по каплям добавляли циклобутилметансульфонилхлорид (200 мг, 1,2 ммоль). Полученную смесь оставляли нагреваться до комнатной температуры на 4 часа, затем концентрировали и выдерживали в глубоком вакууме в течение ночи. Остаток растворяли в EtOAc, промывали водой, 5% уксусной кислотой, насыщенным NaHCO3, солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очищали путем хроматографии на колонке с силикагелем, элюируя с градиентом смесями EtOAc в CH2Cl2, с получением 295 мг (выход 73%) титульного соединения.
Стадия 2 (HCl). Гидрохлорид 1-циклобутил-N-(4'-(пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)метансульфонамида
Трет-бутил-4-(3'-(циклобутилметилсульфонамидо)-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилат (295 мг, 0,57 ммоль) растворяли в 18 мл безводного МеОН, насыщенного HCl (газ), и перемешивали при комнатной температуре в течение ~3 часов. Концентрировали полученный раствор досуха при пониженном давлении. Остаток растирали с 2% МеОН в Et2O, затем растирали с Et2O и сушили с получением 200 мг (выход 78%) титульного соединения в виде гидрохлоридной соли, которое использовали на следующей стадии без дополнительной очистки.
Стадия 2 (ТФУ). Трифторацетат N-(4'-(пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)тетрагидро-2Н-пиран-4-сульфонамида
В раствор трет-бутил-4-(3'-(тетрагидро-2Н-пиран-4-сульфонамидо)-[1,1'-бифенил]-4-карбонил)пиперазин-1-карбоксилата (70 мг, 0,13 ммоль) в ДХМ (3 мл) добавляли ТФУ (0,5 мл). Смесь перемешивали при комнатной температуре в течение 2 часов. После удаления растворителей при пониженном давлении неочищенное титульное соединение (69 мг, 100%) использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 3. 1-циклобутил-N-(4'-(4-(1-гидроксициклопропанкарбонил)пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)метансульфонамид
В раствор гидрохлорида 1-циклобутил-N-(4'-(пиперазин-1-карбонил)-[1,1'-бифенил]-3-ил)метансульфонамида (295 мг, 0,45 моль) и 1-гидроксициклопропан-1-карбоновой кислоты (77 мг, 0,76 ммоль) в 3 мл ДМФ добавляли HBTU (300 мг, 0,81 ммоль), затем добавляли DIEA (0,42 мл, 2,4 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой и нас. NaHCO3 и несколько раз экстрагировали CH2Cl2 и смесью растворителей MeOH/CHCl3. Объединяли органические слои, промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество дважды очищали на системе ISCO (40 г золотая колонка с силикагелем), элюируя с градиентом смесями МеОН в CH2Cl2, а затем растирали в 2-3% МеОН в Et2O с получением 129 мг (выход 58%) титульного соединения. ЖХ-МС (ИЭР, m/z): 498 [М+Н]+.
Способ 35.
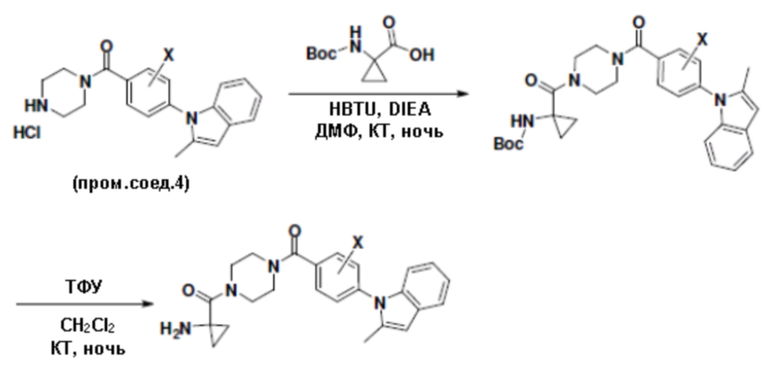
Стадия 1. Трет-бутил-(1-(4-(2-фтор-4-(2-метил-1Н-индол-1-ил)бензоил)пиперазин-1-карбонил)циклопропил)карбамат
В 100 мл круглодонную колбу помещали гидрохлорид 1-[3-фтор-4-[(пиперазин-1-ил)карбонил]фенил]-2-метил-1Н-индола (промежуточное соединение 4, 600 мг, 1,60 ммоль, 1,00 экв.), 1-[[(трет-бутокси)карбонил]амино]циклопропан-1-карбоновую кислоту (322 мг, 1,60 ммоль, 1,00 экв.), N,N-диметилформамид (40 мл), HBTU (729 мг, 1,92 ммоль, 1,20 экв.), DIEA (828 мг, 6,41 ммоль, 3,99 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли 50 мл ЭА, промывали 3х50 мл воды и 50 мл солевого раствора. Концентрировали органическую фазу в вакууме. Остаток помещали в колонку с силикагелем, элюировали смесью этилацетат/петролейный эфир (70:30). Это приводило к получению 300 мг (36%) титульного соединения в виде желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 521 [М+Н]+.
Стадия 2. (4-(1-аминоциклопропан-1-карбонил)пиперазин-1-ил)(2-фтор-4-(2-метил-1Н-индол-1-ил)фенил)метанон
В 100 мл круглодонную колбу помещали трет-бутил-N-[1-[(4-[[2-фтор-4-(2-метил-1Н-индол-1-ил)фенил]карбонил]пиперазин-1-ил)карбонил]циклопропил]карбамат (300 мг, 0,58 ммоль, 1,00 экв.), дихлорметан (20 мл), трифторуксусную кислоту (5 мл). Полученный раствор перемешивали в течение ночи при комнатной температуре. Концентрировали полученную смесь в вакууме. Остаток очищали путем препаративной ВЭЖХ в следующих условиях (Waters I): колонка SunFire Prep C18, 5 мкм, 19*100 мм; мобильная фаза, вода (0,05% NH4HCO3) и CH3CN (от 65% CH3CN от 85% за 7 минут); детектор, УФ, 220 и 254 нм. Это приводило к получению 73,3 мг (30%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 421 [М+Н]+.
Способ 36.
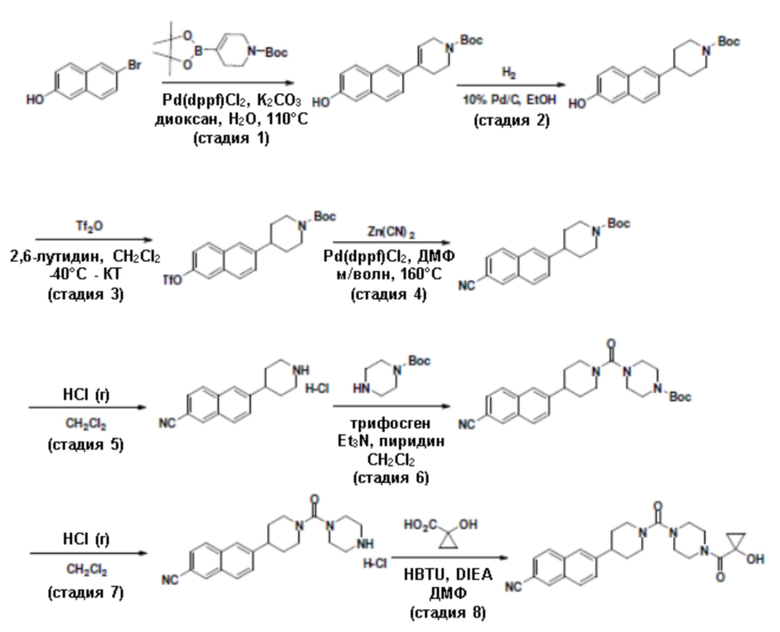
Стадия 1. Трет-бутил-4-(6-гидроксинафталин-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат
В 250 мл круглодонную колбу помещали 6-бромнафталин-2-ол (2 г, 8,97 ммоль, 1,00 экв.), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат (2,7 г, 8,70 ммоль, 1,00 экв.), карбонат калия (3,6 г, 26,05 ммоль, 3,00 экв.) и Pd(dppf)Cl2 (100 мг, 0,14 ммоль, 0,01 экв.) в 1,4-диоксане (30 мл) и воде (10 мл). Полученный раствор перемешивали в течение ночи при 110°С на масляной бане. Охлаждали реакционную смесь, разбавляли EtOAc (120 мл) и промывали солевым раствором (2х100 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали путем хроматографии на силикагеле, элюируя смесью дихлорметан/метанол (50:1). Это приводило к получению 1,9 г (65%) титульного соединения в виде желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 326 [М+Н]+.
Стадия 2. Трет-бутил-4-(6-гидроксинафталин-2-ил)пиперидин-1-карбоксилат
В 250 мл круглодонную колбу помещали трет-бутил-4-(6-гидроксинафталин-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат (1,9 г, 5,84 ммоль, 1,00 экв.), этанол (50 мл) и 10% палладий на подложке углерода (0,5 г). Полученную суспензию заполняли газообразным водородом. Полученный раствор перемешивали в течение 2 часов при КТ. Отфильтровывали твердые вещества и концентрировали раствор в вакууме с получением титульного соединения (1,9 г, 99%) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 328 [М+Н]+.
Стадия 3. Трет-бутил-4-(6-(((трифторметил)сульфонил)окси)нафталин-2-ил)пиперидин-1-карбоксилат
В 100 мл круглодонную колбу помещали трет-бутил-4-(6-гидроксинафталин-2-ил)пиперидин-1-карбоксилат (1,9 г, 5,79 ммоль, 1,00 экв.), дихлорметан (20 мл), 2,6-лутидин (3,4 мл, 3,00 экв.) и ангидрид трифторметансульфокислоты (1,5 мл, 1,50 экв.). Полученный раствор перемешивали при -40°С в течение 10 минут, а затем медленно нагревали до КТ в течение 2 часов. Реакцию гасили путем добавления воды (100 мл). Затем органический слой промывали солевым раствором (2х100 мл) и 5% хлороводородной кислотой (2х50 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали путем хроматографии на силикагеле, элюируя смесями EtOAc/петролейный эфир (1:5). Это приводило к получению 2,1 г (79%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 461 [М+Н]+.
Стадия 4. Трет-бутил-4-(6-цианонафталин-2-ил)пиперидин-1-карбоксилат
В 30 мл пробирку помещали трет-бутил-4-(6-(((трифторметил)сульфонил)окси)нафталин-2-ил)пиперидин-1-карбоксилат (1,5 г, 3,26 ммоль, 1,00 экв.), цианид цинка (840 мг, 7,15 ммоль, 2,20 экв.) и Pd(dppf)Cl2 (100 мг, 0,14 ммоль, 0,04 экв.) в ДМФ (10 мл). Полученный раствор облучали в микроволновом реакторе при 160°С в течение 15 минут. Охлаждали реакционную смесь, разбавляли 100 мл EtOAc и промывали водой (2х100 мл) и солевым раствором (2х50 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали путем хроматографии на силикагеле, элюируя смесью EtOAc/петролейный эфир (1:10). Это приводило к получению 1,0 г (91%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 337 [М+Н]+.
Стадия 5. Гидрохлорид 6-(пиперидин-4-ил)-2-нафтонитрила
В 100 мл круглодонную колбу помещали трет-бутил-4-(6-цианонафталин-2-ил)пиперидин-1-карбоксилат (1,0 г, 2,97 ммоль, 1,00 экв.) в дихлорметане (20 мл). Реакционный раствор барботировали HCl (газ) и перемешивали в течение 30 минут при КТ, что приводило к образованию осадка. Собирали твердое вещество и сушили в вакууме с получением титульного соединения (0,7 г, 100%) в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 237 [М+Н]+.
Стадия 6. Трет-бутил-4-(4-(6-цианонафталин-2-ил)пиперидин-1-карбонил)пиперазин-1-карбоксилат
В 100 мл круглодонную колбу помещали гидрохлорид 6-(пиперидин-4-ил)-2-нафтонитрила, трифосген (1,0 г, 3,37 ммоль, 1,10 экв.), Et3N (1,8 мл, 4,80 экв.) в дихлорметане (20 мл). Реакционную смесь перемешивали в течение 2 часов, а затем добавляли пиридин (0,35 мл, 1,50 экв.) и трет-бутил-пиперазин-1-карбоксилат (700 мг, 3,76 ммоль, 1,20 экв.). Полученный раствор перемешивали в течение ночи при КТ. Затем реакционную смесь промывали водой (2х100 мл) и солевым раствором (2х50 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением титульного соединения (0,8 г, 60%) в виде желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 449 [М+Н]+.
Стадия 7. Гидрохлорид 6-(1-(пиперазин-1-карбонил)пиперидин-4-ил)-2-нафтонитрила
В 100 мл круглодонную колбу помещали трет-бутил-4-(4-(6-цианонафталин-2-ил)пиперидин-1-карбонил)пиперазин-1-карбоксилат (800 мг, 1,78 ммоль, 1,00 экв.) в дихлорметане (15 мл). Реакционный раствор барботировали HCl (газ) и перемешивали в течение 2 часов при КТ. Концентрировали реакционный раствор в вакууме с получением титульного соединения (0,6 г, 97%) в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 349 [М+Н]+.
Стадия 8. 6-(1-(4-(1-гидроксициклопропан-1-карбонил)пиперазин-1-карбонил)-пиперидин-4-ил)-2-нафтонитрил
В 100 мл круглодонную колбу помещали 1-гидроксициклопропан-1-карбоновую кислоту (180 мг, 1,76 ммоль, 1,00 экв.), HBTU (900 мг, 2,37 ммоль, 2,30 экв.) и гидрохлорид 6-(1-(пиперазин-1-карбонил)пиперидин-4-ил)-2-нафтонитрила (600 мг, 1,72 ммоль, 1,00 экв.) в ДМФ (15 мл). Добавляли DIEA (0,8 мл, 5,00 экв.) и полученный раствор перемешивали в течение ночи при КТ. Затем реакционный раствор разбавляли 100 мл EtOAc и промывали водой (2х100 мл) и солевым раствором (2х50 мл). Затем органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали путем хроматографии на силикагеле, элюируя смесью дихлорметан/метанол (20:1), а затем путем препаративной ВЭЖХ в следующих условиях (Waters I): колонка, Xbridge Prep C18 OBD, 5 мкм, 19*150 мм; мобильная фаза, вода (0,05% NH4HCO3) и CH3CN (от 33% CH3CN до 38% за 12 минут). Это приводило к получению 106,9 мг (14%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 433 [М+Н]+.
Промежуточные соединения
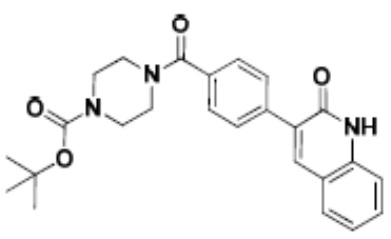
Трет-бутил-4-(4-(2-оксо-1,2-дигидрохинолин-3-ил)бензоил)пиперазин-1-карбоксилат
В смесь трет-бутил-4-(4-бромбензоил)пиперазин-1-карбоксилата (0,250 г, 0,677 ммоль), 2-фторхинолин-3-илбороновой кислоты (0,129 г, 0,677 ммоль) и карбоната натрия (0,287 г, 2,71 ммоль) в диоксане (5,0 мл) и воде (1,0 мл) добавляли тетракис(трифенилфосфин)-палладий (0) (0,078 г, 0,068 ммоль). Смесь перемешивали в микроволновом реакторе при 60°С в течение 1,5 часа. Фильтровали реакционную смесь и концентрировали с получением желтой маслянистой жидкости. Полученное вещество очищали путем колоночной хроматографии на силикагеле (25 г колонка Biotage, элюировали с градиентом 50-100% смесями этилацетат-гексан) с получением трет-бутил-4-(4-(2-оксо-1,2-дигидрохинолин-3-ил)бензоил)пиперазин-1-карбоксилата (0,228 г, 0,526 ммоль, выход 78%) в виде белого твердого вещества. МС (ИЭР, пол.ионы) m/z: 434 (М+1).

5-бромпиколиноилхлорид
К 5-бромпиколиновой кислоте (5,00 г, 24,75 ммоль) добавляли тионилхлорид (10 мл, 137 ммоль). Добавляли каплю ДМФ и смесь грели при 80°С в течение 2 часов. Удаляли избыток тионилхлорида с получением 5-бромпиколиноилхлорида (5,413 г, 24,55 ммоль, выход 99%) в виде бледно-желтого твердого вещества. Аликвоту гасили метанолом с получением метил-5-бромпиколината. МС (ИЭР, пол.ионы) m/z: 216, 218 (М+1).
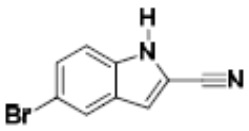
5-бром-1Н-индол-2-карбонитрил
Стадия 1. 5-бром-1Н-индол-2-карбоксамид
В дихлорметан (10 мл) добавляли 5-бром-1Н-индол-2-карбоновую кислоту (0,75 г, 3,12 ммоль) с получением суспензии. Постепенно при помощи шприца добавляли тионилхлорид (0,616 мл, 8,44 ммоль). Добавляли каплю ДМФ и суспензию грели при температуре обратной конденсации в течение 2,5 часа. Охлаждали смесь до комнатной температуры, а затем выливали при перемешивании в 5н. гидроксид аммония (20 мл, 100 ммоль) в 18 мл льда. Осадок перемешивали в течение 2 часов. Разбавляли водную эмульсию, а затем дважды экстрагировали этилацетатом. Сушка над MgSO4 и концентрирование приводили к получению желтого неочищенного твердого вещества, которое растирали с гексанами, фильтровали, промывали гексанами и сушили с получением 5-бром-1Н-индол-2-карбоксамида (0,81 г, 3,15 ммоль, выход 93%) в виде коричневатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 239, 241 (М+1).
Стадия 2. 5-бром-1Н-индол-2-карбонитрил
В толуол (20 мл) добавляли 5-бром-1Н-индол-2-карбоксамид (0,68 мг, 2,84 ммоль) с получением суспензии. Постепенно при помощи шприца добавляли оксихлорид фосфора (1,326 мл, 14,22 ммоль). Суспензию грели при температуре обратной конденсации в течение 1,5 часа. Реакционную смесь выливали в насыщенный NaHCO3 и оставляли для гашения. Слои дважды экстрагировали 50 мл дихлорметана. Объединенные слои промывали 20 мл солевого раствора, сушили над MgSO4 и концентрировали с получением неочищенного продукта. Полученное вещество очищали путем колоночной хроматографии на силикагеле (25 г колонка Biotage, элюировали с градиентом 0-5% смесями этилацетат/дихлорметан) с получением 5-бром-1Н-индол-2-карбонитрила (0,408 г, 1,85 ммоль, 65%) в виде коричневатого твердого вещества. МС (ИЭР, пол.ионы): m/z 219/221.
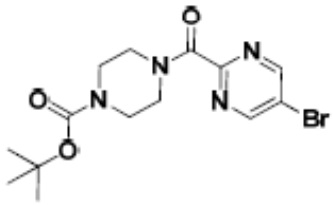
Трет-бутил-4-(5-бромпиримидин-2-карбонил)пиперазин-1-карбоксилат
Стадия 1. 5-бромпиридин-2-карбонилхлорид
К 5-бромпиридин-2-карбоновой кислоте (2,0 г, 9,85 ммоль) добавляли тионилхлорид (3,96 мл, 54,2 ммоль). Добавляли каплю ДМФ и смесь грели при 80°С в течение 2 часов. Удаляли избыток тионилхлорида в вакууме с получением 5-бромпиридин-2-карбонилхлорида в виде бледно-желтого твердого вещества. Вещество использовали без очистки на следующей стадии.
Стадия 2. Трет-бутил-4-(5-бромпиримидин-2-карбонил)пиперазин-1-карбоксилат
5-бромпиридин-2-карбонилхлорид (2,181 г, 9,85 ммоль) и N,N-диизопропилэтиламин (5,16 мл, 29,6 ммоль) растворяли в ДМФ (30 мл). Добавляли трет-бутил-пиперазин-1-карбоксилат и реакционную смесь перемешивали в течение 1,25 часа. Добавляли воду (100 мл) и перемешивали смесь. Затем водную смесь дважды экстрагировали 100 мл этилацетата. Во второй раз получали эмульсию, но насыщение NaCl приводило к разделению фаз. Объединенные органические фазы промывали один раз солевым раствором, сушили над MgSO4 и концентрировали. Неочищенный остаток очищали путем колоночной хроматографии на силикагеле (Biotage, элюировали с градиентом смесями 0-10% метанол/дихлорметан) с получением трет-бутил-4-(5-бромпиримидин-2-карбонил)-пиперазин-1-карбоксилата (3,1 г, 7,93 ммоль, 81%) в виде светло-коричневого твердого вещества. МС (ИЭР, пол.ионы) m/z: 371/373 (М+1).
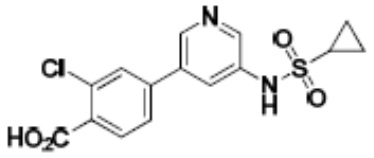
2-хлор-4-(5-(циклопропансульфонамидо)пиридин-3-ил)бензойная кислота
Стадия 1. N-(5-бромпиридин-3-ил)циклопропансульфонамид
5-бромпиридин-3-амин (0,304 мл, 3,0 ммоль) и пиридин (0,534 мл, 1,1 ммоль) объединяли в дихлорметане (15 мл) с получением коричневого раствора. Постепенно при помощи шприца добавляли циклопропансульфонилхлорид (0,973 мл, 9,00 ммоль). Полученную смесь перемешивали при КТ в течение 1 дня. Добавляли дополнительное количество пиридина (3,0 ммоль) и 1 эквивалент циклопропансульфонилхлорида (3,00 ммоль). Взаимодействие завершалось через 1 день. Реакционную смесь разбавляли 75 мл дихлорметана и промывали 40 мл насыщенного NaHCO3. Водный слой экстрагировали 10 мл дихлорметана и объединенные органические фазы промывали 20 мл солевого раствора. Затем сушили раствор MgSO4 и концентрировали. Неочищенный продукт очищали путем колоночной хроматографии на силикагеле (100 г колонка Biotage, элюировали с градиентом 0-50% смесями этилацетат/гексаны) с получением N-(5-бромпиридин-3-ил)циклопропансульфонамида (0,81 мг, 2,92 ммоль, 73%) в виде персикового твердого вещества. МС (ИЭР, пол.ионы) m/z: 277, 279 (М+1).
Стадия 2. 2-хлор-4-(5-(циклопропансульфонамидо)пиридин-3-ил)бензойная кислота
N-(5-бромпиридин-3-ил)циклопропансульфонамид (0,810 г, 2,92 ммоль), карбонат натрия (1,24 г, 11,69 ммоль) и 2-хлор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензойную кислоту (0,991 г, 3,51 ммоль) объединяли в 1,4-диоксане (32 мл) и воде (6,4 мл), затем добавляли тетракис(трифенилфосфин)палладий (0) (0,34 г, 0,292 ммоль) с получением светло-желтой суспензии. Суспензию помещали в атмосферу азота и грели в течение 16 часов при 80°С. Реакционную смесь фильтровали через Celite и промывали твердые вещества 1,4-диоксаном, затем этилацетатом. Концентрирование растворителей приводило к получению бледно-желтого липкого твердого вещества, которое суспендировали в 50 мл воды и подщелачивали 1н. раствором NaOH до рН 12. Основный слой промывали один раз 25 мл этилацетата. После повторно экстракции органической фазы 20 мл воды при рН=12 объединенные основные фазы подкисляли 1н. HCl до достижения рН=5. После отстаивания в течение ночи собирали осадок и сушили с получением 2-хлор-4-(5-(циклопропансульфонамидо)пиридин-3-ил)бензойной кислоты (0,438 г, 1,24 ммоль, 35%) в виде беловатого твердого вещества. МС (ИЭР, пол.ионы) m/z: 353, 355 (М+1).
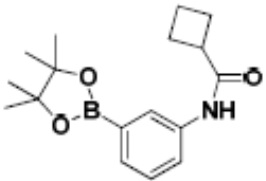
N-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутанкарбоксамид
3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (0,50 г, 2,28 ммоль) и N,N-диизопропилэтиламин (1,20 мл, 6,85 ммоль) объединяли в дихлорметане (5 мл) с получением светло-желтого раствора. Постепенно при помощи шприца добавляли циклобутанкарбонилхлорид (0,260 мл, 2,282 ммоль). Смесь перемешивали в течение 2,5 часа. Реакционную смесь разбавляли 70 мл дихлорметана, а затем дважды промывали 20 мл воды. Затем органический слой промывали 20 мл солевого раствора и сушили над MgSO4. Концентрирование приводило к получению белого твердого вещества, которое очищали путем колоночной хроматографии на силикагеле (50 г колонка Biotage, элюировали с градиентом 0-40% смесями этилацетат/гексаны) с получением N-(4'-(4-(1-гидроксициклопропанкарбонил)пиперазин-1-карбонил)бифенил-3-ил)оксетан-3-карбоксамида в виде восковидного белого твердого вещества. МС (ИЭР, пол.ионы) m/z 302 (М+1).
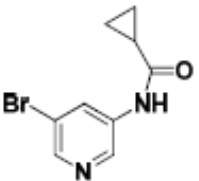
N-(5-бромпиридин-3-ил)циклопропанкарбоксамид
5-бромпиридин-3-амин (0,292 мл, 2,89 ммоль) и N,N-диизопропилэтиламин (0,757 мл, 4,33 ммоль) объединяли в дихлорметане (15 мл) с получением светло-желтого раствора. Постепенно при помощи шприца добавляли циклопропанкарбонилхлорид (0,263 мл, 2,89 ммоль). Реакционную смесь перемешивали при КТ в течение 1,5 часа. Реакционную смесь разбавляли 50 мл дихлорметана и промывали 25 мл насыщенного NaHCO3. После экстракции водного слоя 20 мл дихлорметана объединенные органические слои промывали 20 мл NaHCO3, 20 мл солевого раствора и сушили над MgSO4. Концентрировали растворитель с получением коричневого полутвердого вещества. Остаток очищали путем колоночной хроматографии на силикагеле (50 г колонка Biotage, элюировали с градиентом 0-4% смесями метанол/дихлорметан) с получением N-(5-бромпиридин-3-ил)циклопропанкарбоксамида (0,603 г, 2,50 ммоль, 87%) в виде светло-коричневого твердого вещества. МС (ИЭР, пол.ионы) m/z: 241, 243 (М+1).

N-(3-бромфенил)этансульфонамид
3-броманилин (0,158 мл, 1,453 ммоль) и пиридин (0,118 мл, 1,453 ммоль) объединяли в воде (10 мл) с получением коричневатого раствора и постепенно при помощи шприца добавляли этансульфонилхлорид (0,138 мл, 1,453 ммоль). Через 1 час добавляли дополнительное количество этансульфонилхлорида (0,138 мл, 1,453 ммоль) и продолжали перемешивать еще 1 час. Реакционную смесь дважды экстрагировали 15 мл этилацетата и объединенные органические слои промывали 10 мл солевого раствора и сушили над MgSO4. Концентрировали растворитель с получением бледно-красной маслянистой жидкости. Полученное вещество очищали путем колоночной хроматографии на силикагеле (25 г колонка Biotage, элюировали с градиентом 0-30% смесями этилацетат/гексаны) с получением N-(3-бромфенил)этансульфонамида (0,345 г, 1,24 ммоль, 85%) в виде бесцветной вязкой пленки, которая частично отверждалась при стоянии. МС (ИЭР, отр.ионы): m/z 262, 264 (М-1).

(3'-аминобифенил-4-ил)(4-(1-гидроксициклопропанкарбонил)пиперазин-1-ил)метанон
(4-(1-гидроксициклопропанкарбонил)пиперазин-1-ил)(3'-нитробифенил-4-ил)метанон (0,263 г, 0,665 ммоль, полученный согласно общему способу В) суспендировали в этаноле (6 мл) и воде (2 мл). Добавляли порошок железа (0,371 г, 6,65 ммоль) и хлорид аммония (8,89 мг, 0,166 ммоль) и суспензию грели при 80°С в течение 2 часов. Разбавляли суспензию метанолом и фильтровали через Celite. Концентрировали фильтрат и добавляли 50 мл хлороформа. Раствор промывали 30 мл насыщенного NaHCO3 и отделяли водную фазу и промывали 20 мл хлороформа. Объединенные органические слои промывали 20 мл солевого раствора и сушили над MgSO4. Концентрировали смесь и очищали остаток путем колоночной хроматографии на силикагеле (25 г колонка Biotage, элюировали с градиентом 0-6% смесями метанол/хлороформ) с получением янтарной пленки, которую помещали в дихлорметан и растирали с гексанами с получением (3'-аминобифенил-4-ил)(4-(1-гидроксициклопропанкарбонил)пиперазин-1-ил)метанона (0,152 г, 0,345 ммоль, 52%) в виде твердой пены. МС (ИЭР, пол.ионы) m/z 366 (М+1).
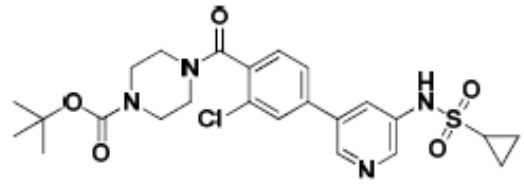
Трет-бутил-4-(2-хлор-4-(5-(циклопропансульфонамидо)пиридин-3-ил)бензоил)-пиперазин-1-карбоксилат
Трет-бутил-пиперазин-1-карбоксилат (0,139 г, 0,748 ммоль), 2-хлор-4-(5-(циклопропансульфонамидо)пиридин-3-ил)бензойную кислоту (0,240 г, 0,680 ммоль) и N,N-диизопропилэтиламин (0,131 мл, 0,748 ммоль) объединяли в ДМФ (5 мл) и добавляли гексафторфосфат О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (0,387 г, 1,020 ммоль) с получением коричневого раствора, который перемешивали в течение 1 дня. Реакционную смесь разбавляли 30 мл воды и перемешивали. Экстрагировали смесь 60 мл дихлорметана, а затем 15 мл дихлорметана. Объединенные органические слои промывали 20 мл воды и 20 мл солевого раствора и концентрировали. Остаток очищали путем колоночной хроматографии на силикагеле (50 г колонка Biotage, элюировали с градиентом 0-4% смесями метанол/дихлорметан) с получением трет-бутил-4-(2-хлор-4-(5-(циклопропансульфонамидо)-пиридин-3-ил)бензоил)пиперазин-1-карбоксилата (0,252 г, выход 71%) в виде янтарной стекловидной пленки. МС (ИЭР, пол.ионы) m/z: 521/523.

Трет-бутил-4-(2-хлор-4-(5-(циклопропанкарбоксамидо)пиридин-3-ил)бензоил)-пиперазин-1-карбоксилат
Трет-бутил-пиперазин-1-карбоксилат (0,137 г, 0,736 ммоль), 2-хлор-4-(5-(циклопропанкарбоксамидо)пиридин-3-ил)бензойную кислоту (0,212 г, 0,669 ммоль) и N,N-диизопропилэтиламин (0,129 мл, 0,736 ммоль) объединяли в ДМФ (5 мл), затем добавляли гексафторфосфат О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (0,381 мг, 1,004 ммоль) с получением коричневого раствора, который перемешивали в течение 1 дня. Добавляли воду (10 мл) и перемешивали реакционную смесь. Суспензию разбавляли 60 мл дихлорметана и 20 мл воды. Отделяли водную фазу и экстрагировали 15 мл дихлорметана. Объединенные органические слои промывали 20 мл воды и 20 мл солевого раствора, сушили над безводным сульфатом магния, фильтровали и концентрировали. Неочищенный продукт очищали путем колоночной хроматографии на силикагеле (50 г колонка Biotage, элюировали с градиентом 0-4% смесями метанол/дихлорметан) с получением трет-бутил-4-(2-хлор-4-(5-(циклопропанкарбоксамидо)пиридин-3-ил)бензоил)пиперазин-1-карбоксилата (0,454 г, 140%) в виде янтарного стекла. МС (ИЭР, пол.ионы) m/z: 485/487.
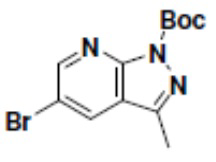
Трет-бутил-5-бром-3-метил-1Н-пиразоло[3,4-b]пиридин-1-карбоксилат
В 100 мл круглодонную колбу помещали 5-бром-3-метил-1Н-пиразоло[3,4-b]пиридин (800 мг, 3,77 ммоль, 1,00 экв.), Boc2O (1,22 г, 5,59 ммоль, 1,50 экв.), 4-диметиламинопиридин (55 мг, 0,45 ммоль, 0,12 экв.), ТЭА (756 мг, 7,47 ммоль, 2,00 экв.) и тетрагидрофуран (20 мл). Полученный раствор перемешивали в течение 18 часов при 20°С. Полученный раствор разбавляли 50 мл H2O, экстрагировали 3х20 мл этилацетата. Объединяли органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Это приводило к получению 1,18 г (95%) титульного соединения в виде белого твердого вещества. ЖХ-МС (ИЭР, m/z): 312, 314 [М+Н]+.
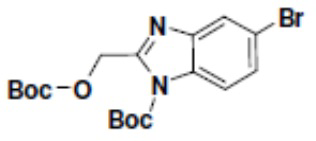
Трет-бутил-5-бром-2-(((трет-бутоксикарбонил)окси)метил)-1Н-бензо[d]имидазол-1-карбоксилат
В 50 мл круглодонную колбу помещали (5-бром-1Н-1,3-бензодиазол-2-ил)метанол (600 мг, 2,64 ммоль, 1,00 экв.), дихлорметан (20 мл), Boc2O (1 г, 4,58 ммоль, 1,73 экв.), ТЭА (800 мг, 7,91 ммоль, 2,99 экв.), 4-диметиламинопиридин (32 мг, 0,26 ммоль, 0,10 экв.). Полученный раствор перемешивали в течение 4 часов при 25°С. Смесь разбавляли 20 мл дихлорметана, промывали 3*30 мл воды и 30 мл солевого раствора. Органическую фазу сушили над безводным сульфатом натрия и концентрировали в вакууме. Это приводило к получению 600 мг (масса неочищенного вещества) титульного соединения в виде желтой маслянистой жидкости. ЖХ-МС (ИЭР, m/z): 429, 427 [М+Н]+.
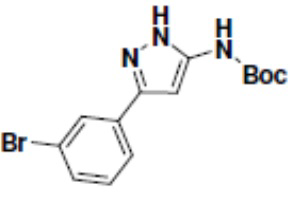
Трет-бутил-(3-(3-бромфенил)-1Н-пиразол-5-ил)карбамат
В 50 мл круглодонную колбу помещали раствор 3-(3-бромфенил)-1Н-пиразол-5-амина (500 мг, 2,10 ммоль, 1,00 экв.) в дихлорметане (20 мл), (Boc)2O (916 мг, 4,20 ммоль, 2,00 экв.), 4-диметиламинопиридин (25,6 мг, 0,21 ммоль, 0,10 экв.), ТЭА (636,5 мг, 6,29 ммоль, 3,00 экв.). Полученный раствор перемешивали в течение 1 часа при комнатной температуре. Затем реакционную смесь выливали в 100 мл воды, экстрагировали 2х100 мл дихлорметана. Объединяли органические слои, сушили над безводным сульфатом натрия и концентрировали в вакууме. Это приводило к получению 710 мг (100%) трет-бутил-N-[3-(3-бромфенил)-1Н-пиразол-5-ил]карбамата в виде коричневой маслянистой жидкости. ЖХ-МС (ИЭР, m/z): 338 [М+Н]+.
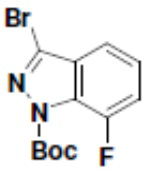
Трет-бутил-3-бром-7-фтор-1Н-индазол-1-карбоксилат
В 50 мл круглодонную колбу помещали 3-бром-7-фтор-1Н-индазол (700 мг, 3,26 ммоль, 1,00 экв.), дихлорметан (20 мл), Boc2O (1,5 г, 6,87 ммоль, 2,11 экв.), ТЭА (1 г, 9,88 ммоль, 3,04 экв.), 4-диметиламинопиридин (40 мг, 0,33 ммоль, 0,10 экв.). Полученный раствор перемешивали в течение 4 часов при 25°С. Полученную смесь выливали в 50 мл воды, экстрагировали и концентрировали в вакууме. Это приводило к получению 1 г (масса неочищенного вещества) титульного соединения в виде красной маслянистой жидкости. ЖХ-МС (ИЭР, m/z): 315, 317 [М+Н]+.

Трет-бутил-(5-бромбензо[d]изоксазол-3-ил)карбамат
В 50 мл круглодонную колбу помещали 5-бром-1,2-бензоксазол-3-амин (1 г, 4,69 ммоль, 1,00 экв.), дихлорметан (20 мл), Boc2O (2,1 г, 9,62 ммоль, 2,05 экв.), 4-диметиламинопиридин (57 мг, 0,47 ммоль, 0,10 экв.), ТЭА (1,4 г, 13,84 ммоль, 2,95 экв.). Реакционную смесь перемешивали в течение 3 часов при 25°С. Полученную смесь промывали 30 мл воды, 3х30 мл солевого раствора, сушили над безводным сульфатом натрия и концентрировали в вакууме. Это приводило к получению 650 мг (масса неочищенного вещества) титульного соединения в виде желтой маслянистой жидкости. ЖХ-МС (ИЭР, m/z): 313, 315 [М+Н]+.
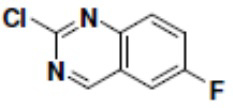
2-хлор-6-фторхиназолин
В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали 2,4-дихлор-6-фторхиназолин (1 г, 4,61 ммоль, 1,00 экв.), солевой раствор (23 мл), дихлорметан (25 мл), гидрат аммиака (2 мл), Zn (0,9 г, 13,8 ммоль, 3,0 экв.). Полученную смесь перемешивали в течение ночи при 50°С на масляной бане. Реакционную смесь охлаждали до комнатной температуры и разбавляли 50 мл H2O, экстрагировали 2х100 мл дихлорметана. Объединяли органические слои и концентрировали в вакууме. Остаток очищали на системе Combi-Flash в следующих условиях (IntelFlash-1): колонка с силикагелем; мобильная фаза, ЭА:ПЭ = 100%, повышали до ЭА:ПЭ = 60% за 40 минут; детектор, УФ 254 нм. Это приводило к получению 0,35 г (35%) титульного соединения в виде желтого твердого вещества. ЖХ-МС (ИЭР, m/z): 183 [М+Н]+.
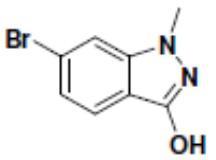
6-бром-1-метил-1Н-индазол-3-ол
В 5 мл герметичную пробирку помещали метил-4-бром-2-фторбензоат (300 мг, 1,29 ммоль, 1,00 экв.), метилгидразин (71,4 мг, 1,55 ммоль, 1,20 экв.), ДМА (2 мл). Полученный раствор перемешивали в течение ночи при 150°С на песчаной бане. После охлаждения до комнатной температуры реакционную смесь разбавляли 10 мл этилацетата, промывали 3х10 мл H2O и 1х20 мл солевого раствора. Концентрировали органическую фазу в вакууме. Это приводило к получению 225 мг (77%) 6-бром-1-метил-1Н-индазол-3-ола в виде беловатого твердого вещества. ЖХ-МС (ИЭР, m/z): 227, 229 [М+Н]+.

6-бром-1-(метилсульфонил)-1Н-индол
В раствор 6-броминдола (2,94 г, 15 ммоль, 1 экв.) в безводном ДМФ (45 мл), охлажденный на бане лед-вода, по частям добавляли NaH (0,72 г, 18 ммоль, 1,2 экв.) в атмосфере N2. Смесь перемешивали еще 20 минут, а затем по каплям добавляли MeSO2Cl (1,4 мл, 18 ммоль, 1,2 экв.). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 1 часа, после чего гасили во льду и разбавляли EtOAc. Органический слой промывали водой (2х) и солевым раствором (2х), сушили над Na2SO4 и концентрировали в вакууме. Неочищенный остаток очищали путем флэш-хроматографии (силикагель), элюируя смесями EtOAc/гексаны (0~20%), с получением титульного соединения в виде белого твердого вещества (0,94 г, 23%).
Ферментное исследование ингибиторов FASN
Исследования проводили в 384-луночном черном планшете для определения активности ингибирования FASN отдельными соединениями, предложенными в настоящем описании. 250 нл аликвоту соединения инкубировали с 10 мкл 40 нМ фермента FASN в буфере для исследования (50 мМ HEPES, pH=7,3, 0,5 мМ ЭДТА, 1 мМ аскорбат, 100 мМ NaCl и 0,04% Triton X-100) в каждой лунке при 25°С в течение 60 минут. После непродолжительного центрифугирования планшета в каждую лунку добавляли 10 мкл смеси субстратов (20 мкМ ацетил-СоА, 60 мкМ малонил-СоА и 100 мкМ НАДФ-Н в буфере для исследования). Реакционную смесь выдерживали при 25°С в течение 90 минут. Затем реакцию гасили путем добавления 10 мкл 90 мкМ 7-диэтиламино-3-(4'-малеимидилфенил)-4-метилкумарина в 50/50 растворе этанол/H2O. Планшет для исследования инкубировали при 25°С в течение 15 минут и исследовали на планшетном анализаторе при длинах волн возбуждения и испускания 360 нм и 530 нм, соответственно. Значение IC50 для данного соединения рассчитывали путем подстановки решений четырехпараметрового логистического уравнения в кривую зависимости доза-ответ.
Результаты
В таблице 2-1 приведены соединения, имеющие IC50 < 0,5 мкМ.
В таблице 2-2 приведены соединения, имеющие IC50 ≥ 0,5 мкМ и < 5,0 мкМ.
В таблице 2-3 приведены соединения, имеющие IC50 ≥ 5,0 мкМ.
Кроме того, также указаны молекулярная масса, результаты ионной масс-спектрометрии, время удерживания ВЭЖХ и способ, используемый для синтеза соединения.
Таблица 2-1
масса
Таблица 2-2
масса
(мин)
Таблица 2-3
масса
(мин)
Исследования действия ингибиторов FASN в отношении пролиферации клеток
Действие ингибиторов FASN в отношении пролиферации выращенных раковых клеток анализировали в исследованиях пролиферации клеток. Клетки PC3 выдерживали в стандартной питательной среде (среда F12K, дополненная 10% эмбриональной бычьей сывороткой, 1Х МЕМ заменимыми аминокислотами и 1Х пенициллин/стрептомицин). 2000-3000 клеток/100 мкл/лунка высевали в 96-луночный прозрачный планшет для клеточных культур. Клетки инкубировали в течение ночи при 5% СО2 и 37°С для слипания. Удаляли клеточную среду и заменяли на среду F12K, содержащую 10% сыворотку с пониженным содержанием липидов и соединение. Конечная концентрация ДМСО составляла 0,1%. Клетки выдерживали при 5% СО2 и 37°С в течение 4 дней. Определяли жизнеспособность клеток в исследованиях МТТ. Значение IC50 данного соединения рассчитывали путем подстановки решений четырехпараметрового логистического уравнения в кривую зависимости доза-ответ.
Результаты
В таблице 3-1 приведены соединения, имеющие IC50 < 0,5 мкМ.
В таблице 3-2 приведены соединения, имеющие IC50 ≥ 0,5 мкМ.
Кроме того, также указаны молекулярная масса, результаты ионной масс-спектрометрии, время удерживания ВЭЖХ и способ, используемый для синтеза соединения.
Таблица 3-1
масса
(мин)
Таблица 3-2
масса
(мин)
Несмотря на то, что настоящее изобретение описано конкретными вариантами реализации, приведенными выше, множество альтернатив, модификаций и других изменений должны быть очевидными специалистам в данной области техники. Предполагается, что все указанные альтернативы, модификации и изменения не выходят за рамки сущности и объема настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ FASN | 2014 |
|
RU2686323C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ БЕНЗОПИПЕРАЗИН, В КАЧЕСТВЕ ИНГИБИТОРОВ БРОМОДОМЕНОВ ВЕТ | 2014 |
|
RU2720237C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| ИНГИБИТОРЫ Р38 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2004 |
|
RU2357957C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ МОДУЛИРОВАНИЯ КИНАЗЫ И ПОКАЗАНИЯ К ИХ ПРИМЕНЕНИЮ | 2013 |
|
RU2666146C2 |
| ИНГИБИТОРЫ MAGL | 2017 |
|
RU2754536C1 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ IRAK И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2743360C2 |
| СОЕДИНЕНИЯ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ | 2019 |
|
RU2783414C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| ПРОИЗВОДНЫЕ ИНДОЛ-3-ИЛ-КАРБОНИЛ-ПИПЕРИДИН-БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРОВ V1А | 2006 |
|
RU2415139C2 |
Изобретение относится к конкретным соединениям для ингибирования FASN. Технический результат: получены новые соединения, обладающие способностью ингибировать FASN. 6 з.п. ф-лы, 6 табл.
1. Соединение, выбранное из группы, состоящей из:
1-[(2S)-4-[2-фтор-4-(1-метил-1H-1,3-бензодиазол-5-ил)бензоил]-2-метилпиперазин-1-карбонил]циклопропан-1-ола;
1-[(2S)-2-метил-4-[4-(1-метил-1H-индазол-6-ил)бензоил]пиперазин-1-карбонил]циклопропан-1-ола;
1-[(2S)-2-метил-4-[4-(1-метил-1H-1,3-бензодиазол-5-ил)бензоил]пиперазин-1-карбонил]циклопропан-1-ола;
1-[(2S)-2-метил-4-[4-(2-метил-2H-индазол-6-ил)бензоил]пиперазин-1-карбонил]циклопропан-1-ола;
1-[(2S)-4-[4-(6-хлор-1,3-бензоксазол-2-ил)бензоил]-2-метилпиперазин-1-карбонил]циклопропан-1-ола;
1-[(2R,6S)-2,6-диметил-4-[4-(1-метил-1H-1,3-бензодиазол-5-ил)бензоил]пиперазин-1-карбонил]циклопропан-1-ола;
1-[(2S)-4-[4-(4-хлор-2-фторфенил)бензоил]-2-метилпиперазин-1-карбонил]циклопропан-1-ола;
1-[(2S)-4-[4-(1,3-диметил-1H-индазол-5-ил)бензоил]-2-метилпиперазин-1-карбонил]циклопропан-1-ола;
1-[(3S)-3-метил-4-[4-(1-метил-1H-1,3-бензодиазол-5-ил)бензоил]пиперазин-1-карбонил]циклопропан-1-ола;
1-[(3R)-3-метил-4-[4-(1-метил-1H-1,3-бензодиазол-5-ил)бензоил]пиперазин-1-карбонил]циклопропан-1-ола;
1-[(3R,5S)-3,5-диметил-4-[4-(1-метил-1H-1,3-бензодиазол-5-ил)бензоил]пиперазин-1-карбонил]циклопропан-1-ола;
1-[(2R)-2-метил-4-[4-(1-метил-1H-1,3-бензодиазол-5-ил)бензоил]пиперазин-1-карбонил]циклопропан-1-ола;
1-[(2S,6R)-4-[4-(6-фтор-1,3-бензоксазол-2-ил)бензоил]-2,6-диметилпиперазин-1-карбонил]циклопропан-1-ола;
1-[(2R)-4-[4-(4-хлор-2-фторфенил)бензоил]-2-метилпиперазин-1-карбонил]циклопропан-1-ола;
1-[(2S)-2-метил-4-[4-(1-метил-1H-1,3-бензодиазол-5-ил)бензоил]пиперазин-1-карбонил]циклопропан-1-амина;
1-[(2S)-4-[4-(6-фтор-1,3-бензоксазол-2-ил)бензоил]-2-метилпиперазин-1-карбонил]циклопропан-1-ола;
1-{4-[4-(6-фтор-1,3-бензоксазол-2-ил)бензоил]-3,3-диметилпиперазин-1-карбонил}циклопропан-1-ола;
1-[(2S,6R)-4-[4-(4-хлор-2-фторфенил)бензоил]-2,6-диметилпиперазин-1-карбонил]циклопропан-1-ола;
1-[(2R)-4-[4-(6-фтор-1,3-бензоксазол-2-ил)бензоил]-2-метилпиперазин-1-карбонил]циклопропан-1-ола;
1-[(3R)-4-[4-(4-хлор-2-фторфенил)бензоил]-3-метилпиперазин-1-карбонил]циклопропан-1-ола;
1-[(3R)-4-[4-(6-фтор-1,3-бензоксазол-2-ил)бензоил]-3-метилпиперазин-1-карбонил]циклопропан-1-ола;
1-[(3S)-4-[4-(4-хлор-2-фторфенил)бензоил]-3-метилпиперазин-1-карбонил]циклопропан-1-ола;
1-[(2R)-2-(гидроксиметил)-4-[4-(1-метил-1H-1,3-бензодиазол-5-ил)бензоил]пиперазин-1-карбонил]циклопропан-1-ола;
1-[(2S,6R)-4-[4-(6-фтор-1,3-бензоксазол-2-ил)бензоил]-2,6-диметилпиперазин-1-карбонил]циклопропан-1-амина;
1-[(2S)-4-[4-(6-фтор-1,3-бензоксазол-2-ил)бензоил]-2-метилпиперазин-1-карбонил]циклопропан-1-амина;
1-[(2S)-4-{4-[3-(циклопропанeсульфонил)фенил]бензоил}-2-метилпиперазин-1-карбонил]циклопропан-1-ола;
1-[(3S)-4-[4-(6-фтор-1,3-бензоксазол-2-ил)бензоил]-3-метилпиперазин-1-карбонил]циклопропан-1-ола;
1-[(3S)-3-метил-4-[4-(1-метил-1H-1,3-бензодиазол-5-ил)бензоил]пиперазин-1-карбонил]циклопропан-1-амина;
1-{[(3S)-4-{[4-(4-хлор-2-фторфенил)фенил]карбонил}-3-(гидроксиметил)пиперазин-1-ил]карбонил}циклопропан-1-ола;
1-{[(2S,5R)-4-{[4-(изохинолин-6-ил)фенил]карбонил}-2,5-диметилпиперазин-1-ил]карбонил}циклопропан-1-ола;
1-{[(2S)-4-{[4-(4-хлор-2-фторфенил)фенил]карбонил}-2-(гидроксиметил)пиперазин-1-ил]карбонил}циклопропан-1-ола; and
(2S)-2-гидрокси-1-{4-[(4-фенилфенил)карбонил]пиперазин-1-ил}пропан-1-она
или его фармацевтически приемлемая соль.
2. Соединение по п.1, представляющее собой
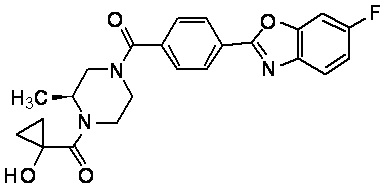
или его фармацевтически приемлемая соль.
3. Соединение по п.1, представляющее собой
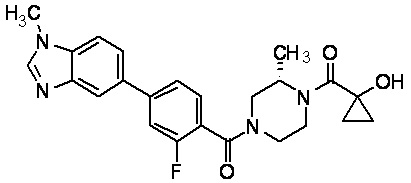
или его фармацевтически приемлемая соль.
4. Соединение по п.1, представляющее собой

или его фармацевтически приемлемая соль.
5. Соединение по п.1, представляющее собой
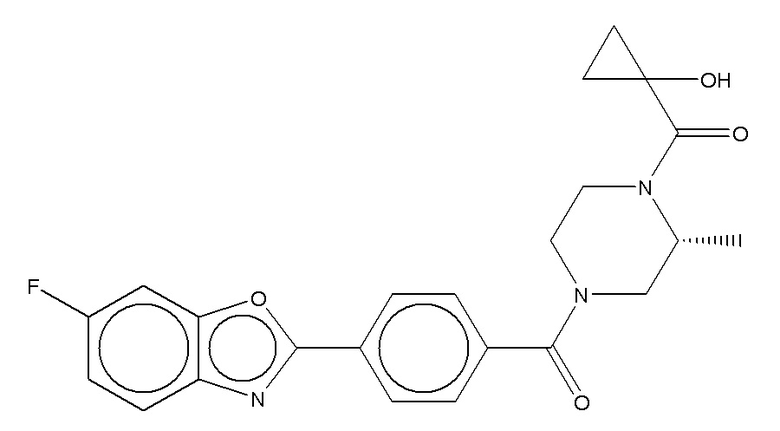
или его фармацевтически приемлемая соль.
6. Соединение по п.1, представляющее собой
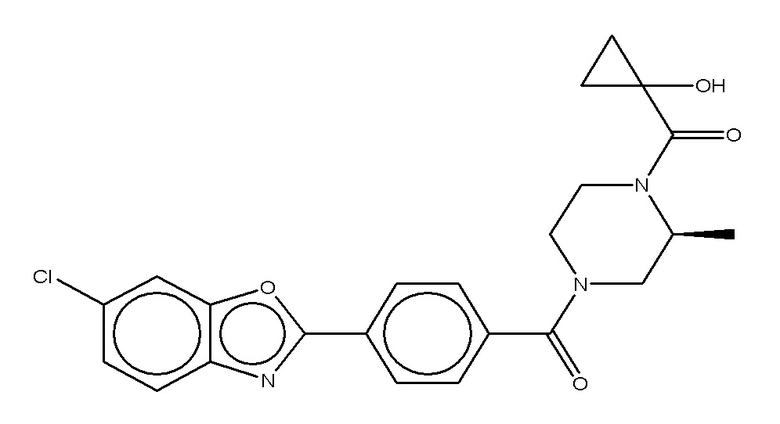
или его фармацевтически приемлемая соль.
7. Соединение по п.1, представляющее собой
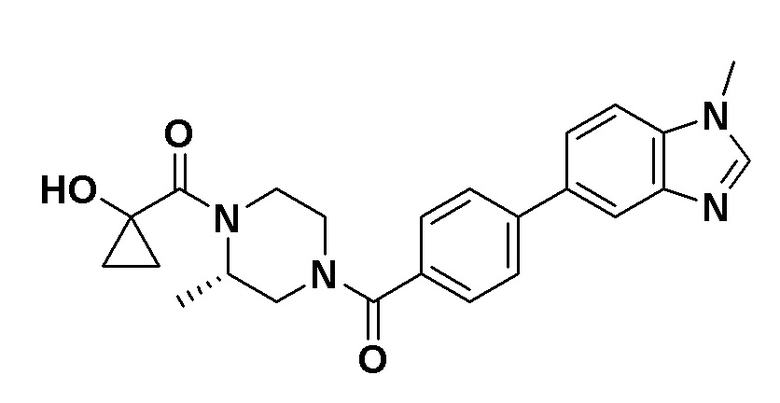
или его фармацевтически приемлемая соль.
| WO2013028495 A1, 28.02.2013 | |||
| CN102627610 A, 08.08.2012 | |||
| RU2011108493 A, 20.09.2012 | |||
| US2009197863 A1, 06.08.2009 | |||
| WO2005046685 A1, 26.05.2005 | |||
| ПРОИЗВОДНЫЕ БЕНЗАМИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2194044C2 |
| US2005043300 A1, 24.02.2005. | |||

