Область техники
Настоящее изобретение относится к производным бензодиазепинов и их применению для лечения или предотвращения респираторно-синцитиальной вирусной (РСВ) инфекции.
Уровень техники
РСВ представляет собой вирус семейства парамиксовирусов (Paramyxoviridae) с отрицательно-полярной однонитевой РНК. РСВ легко передается через выделения от инфицированного человека через поверхности или при рукопожатии. В отличие от гриппа, он не передается воздушно-капельным путем. После успешной инокуляции инкубационный период продолжается от четырех до шести дней, и за это время вирус распространяется от носоглотки до нижних дыхательных путей посредством слияния инфицированных клеток с неинфицированными и отслоения некротизированного эпителия. В сочетании с повышенной секрецией слизи и отеком у младенцев это может привести к закупорке слизью, что вызывает чрезмерное раздувание и коллапс дистальной легочной ткани, характерные для бронхиолита. При этом обычно наблюдается гипоксия и из-за нарушенного дыхания часто затрудняется возможность кормления. При пневмонии, вызванной РСВ, воспалительная инфильтрация дыхательных путей состоит из одноядерных клеток и является более генерализованной с вовлечением бронхиол, бронхов и альвеол. Было обнаружено, что продолжительность и степень выделения вируса в среду коррелируют с клиническими признаками и тяжестью заболевания.
РСВ является основной причиной тяжелых инфекций дыхательных путей у младенцев и детей младшего возраста во всем мире. Самая высокая заболеваемость и смертность наблюдаются у детей, родившихся недоношенным, а также у детей с хроническими заболеваниями легких или сердца, хотя многие младенцы, госпитализированные с РСВ-инфекцией, не имеют других заболеваний. Тяжелая РСВ-инфекция в младенческом возрасте может привести к возникновению рецидивирующей бронхиальной обструкции в течение нескольких лет и связана с развитием астмы впоследствии.
РСВ также является основной причиной заболеваемости и смертности у пожилых людей и у детей и взрослых с ослабленным иммунитетом, а также тех, кто страдает от хронической обструктивной болезни легких (ХОБЛ) и застойной сердечной недостаточности (ЗСН). Заболеваемость РСВ имеет сезонный характер; ее можно очень точно прогнозировать, и заболевания РСВ наблюдаются в зимний период в обоих полушариях, с сентября по май в Европе и Северной Америке, при этом пик заболеваемости приходится на декабрь и январь, а в тропических странах заболевания РСВ могут наблюдаться в течение всего года. Инфекция РСВ поражает >90% младенцев и детей младшего возраста до двух лет, и, поскольку естественный иммунитет является нестойким, многие будут повторно инфицироваться каждый год. Как и в случае гриппа, у пожилых людей заболевания РСВ являются причиной примерно 10% госпитализаций в зимний период, при этом связанная с ними смертность составляет 10%.
Существующее лечение заболеваний РСВ, включает применение моноклонального антитела к РСВ под названием паливизумаб. Такое применение паливизумаба является профилактическим, а не терапевтическим лечением РСВ заболеваний. Хотя данное антитело часто является эффективным, его применение ограничивается применением у недоношенных детей и младенцев групп высокого риска. Действительно, ограниченная применимость паливизумаба означает, что он недоступен для многих людей, нуждающихся в лечении РСВ заболеваний. Соответственно, существует острая необходимость в разработке эффективных альтернатив существующему лечению РСВ заболеваний.
В качестве ингибиторов РСВ были также предложены низкомолекулярные синтетические соединения. К ним относятся бензимидазолы и бензодиазепины. Например, открытие и первоначальная разработка соединения RSV604, бензодиазепинового соединения, обладающего субмикромолярной активностью против РСВ, описаны в работе Antimicrobial Agents and Chemotherapy, September. 2007, 3346-3353 (Chapman et al). Бензодиазепиновые ингибиторы РСВ также раскрыты в публикациях, включая WO2004/026843 и WO2005/089770 (Arrow Therapeutics Limited); WO2016/166546 и WO2018/033714 (Durham University); и WO2017/015449, WO2018/129287 и WO2018/226801 (Enanta Pharmaceuticals, Inc.).
Существует потребность в выявлении и других соединений, которые бы обладали анти-РСВ активностью, в частности соединений, сочетающих в себе мощную противовирусную активность и благоприятные фармакокинетические свойства.
Краткое описание изобретения
К настоящему моменту было установлено, что новая серия производных бензодиазепина обладает мощной анти-РСВ активностью с благоприятными фармакокинетическими и физико-химическими свойствами. Соответственно, в настоящем изобретении предложено соединение, представляющее собой производное бензодиазепина и имеющее формулу (I):
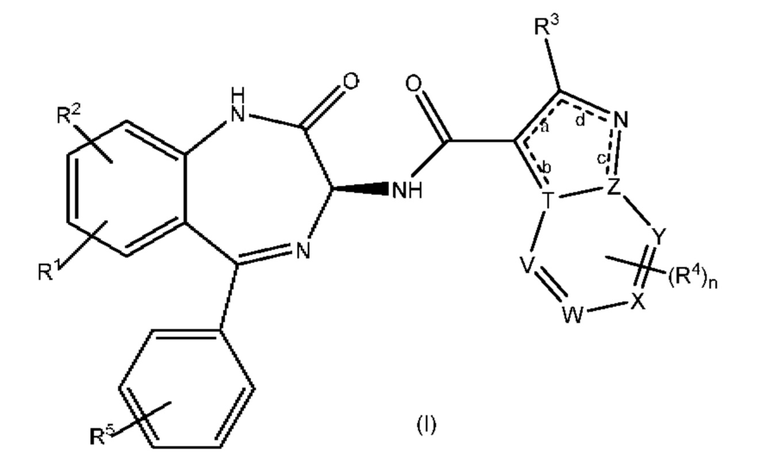
где:
каждый из R1 и R2 независимо представляет собой Н или галоген;
(i) Т представляет собой N, Z представляет собой С, 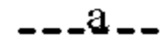 и
и 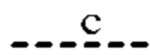 представляют собой связи, а
представляют собой связи, а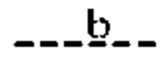 и
и 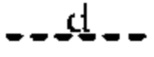 отсутствуют; или
отсутствуют; или
(ii) Т представляет собой С, Z представляет собой N, 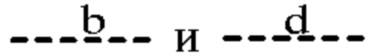 представляют собой связи, а
представляют собой связи, а 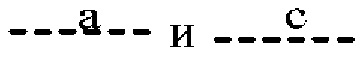 отсутствуют;
отсутствуют;
каждый из R3 и R4 независимо представляет собой галоген,-OR6,-NR6R7,-COR8,-C(О)OR8,-CON(R8)2 или-R6;
R5 представляет собой H или галоген;
каждый из R6 и R7 независимо представляет собой Н или группу, выбранную из C1-C6 алкила, С3-С10 циклоалкила, С6-С10 арила, 4-10-членного гетероциклила и 4-10-членного гетероарила, причем указанная группа является незамещенной или замещенной;
R8 представляет собой Н или C1-С6 алкил, при этом, если в формуле имеются два радикала R8, они могут быть одинаковыми или разными; n равно 0 или 1;
и
один из V, W, X и Y представляет собой N или СН, а остальные три представляют собой СН; или его фармацевтически приемлемая соль.
Соединения согласно настоящему изобретению имеют два атома N в пятичленном кольце бициклического гетероарильного кольца, которое связано через амидную группу с бензодиазепинильной кольцевой системой. Считается, что эта структурная особенность важна для свойств обсуждаемых ниже соединений.
Подробное описание изобретения
Когда любая группа, кольцо, заместитель или фрагмент, определенные в данном описании, замещены, они обычно замещены группой Q согласно приведенным выше определениям.
C1-6 алкильная группа или фрагмент являются неразветвлеиными или разветвленными. C1-6 алкильная группа обычно представляет собой С1-4 алкильную группу или С4-6 алкильную группу. Примеры C1-6 алкильных групп и фрагментов включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-пентил, изопентил (т.е., 3-метилбут-1-ил), трет-пентил (т.е., 2-метилбут-2-ил), неопентил (т.е., 2,2-диметилпропан-1-ил), н-гексил, изогексил (т.е., 4-метилпентан-1-ил), трет-гексил (т.е., 3-метилпентан-3-ил) и неопентил (т.е., 3,3-диметилбутан-1-ил). Во избежание сомнений, если в группе присутствуют два алкильных фрагмента, алкильные фрагменты могут быть одинаковыми или разными. C1-6 алкильная группа является незамещенной или замещенной обычно одной или более группами Q согласно приведенным выше определениям. Например, С1-6 алкильная группа является незамещенной или замещена 1, 2 или 3 группами Q согласно приведенным выше определениям.
Q представляет собой галоген, нитрогруппу,-CN, ОН, C1-6 алкоксигруппу, C1-6 гидроксиалкил, C1-6 алкоксиалкил, незамещенный С1-6 алкил, С1-6 алкилтиогруппу, С1-6 галогеналкил, С1-4 галогеналкоксигруппу,-CO2R''',-NR'2,-SR',-S(=О)R',-S(=О)2R', С3-С10 циклоалкил, 4-10-членный гетероциклил, С6-С10 арил или 4-10-членный гетероарил, где каждый R' независимо выбран из Н, C1-6 алкила, С3-10 циклоалкила, 4-10-членного гетероциклила, С6-С10 арила и 4-10-членного гетероарила.
C1-6 алкоксигруппа является линейной или разветвленной. Обычно это С1-4 алкоксигруппа, например, метоксигруппа, этоксигруппа, пропоксигруппа, изопропоксигруппа, н-пропоксигруппа, н-бутоксигруппа, втор-бутоксигруппа или трет-бутоксигруппа. C1-6 алкоксигруппа является незамещенной или замещена, как правило, одной или более группами Q согласно приведенным выше определениям.
C1-6 алкилтиогруппа является неразветвленной или разветвленной. Это обычно С1-4 ал килтио группа, например, метилтио-, этилтио-, пропилтио-, изопропилтио-, н-пропилтио-, н-бутилтио-, втор-бутилтио-или трет-бутилтиогруппа. С1-6 алкилтиогруппа является незамещенной или замещена, как правило, одной или более группами Q согласно приведенным выше определениям.
Галоген или галогеновая группа представляет собой F, Cl, Br или I. Обычно это F или Cl. C1-6 алкильная группа, замещенная галогеном, может быть обозначена как ''С1-6 галогеналкил'', что означает С1-6 алкильную группу согласно приведенным выше определениям, в которой один или более атомов водорода замещено галогеном. Аналогично, C1-6 алкоксигруппа, замещенная галогеном, может быть обозначена как ''С1-6 галогеналкоксигруппа'', что означает С1-6 алкоксигруппу согласно приведенным выше определениям, в которой один или более атомов водорода замещено галогеном. Обычно С1-6 галогеналкил или С1-6 галогеналкоксигруппа замещены 1, 2 или 3 вышеупомянутыми атомами галогена. Галогеналкильные группы и галогеналкоксигруппы включают пергалогеналкильные и пергалогеналкоксигруппы, такие как-СХ3 и-ОСХ3, где X представляет собой галоген, например,-CF3-CCl3-OCF3 и-OCCl3.
С1-6 гидроксиалкильная группа представляет собой Сьв алкильную группу согласно приведенным выше определениям, замещенную одной или более ОН группами. Обычно она замещена одной, двумя или тремя группами ОН. Предпочтительно она замещена одной группой ОН.
С1-6 алкоксиалкильная группа представляет собой С1-6 алкильную группу согласно приведенным выше определениям, замещенную C1-6 алкоксигруппой согласно приведенным выше определениям. Это может быть, например, метоксиалкил или этоксиалкил, в котором алкильный фрагмент представляет собой C1-6 алкильную группу согласно приведенным выше определениям.
С6-С10 арильная группа представляет собой ароматическую карбоциклическую группу, содержащую от 6 до 10 атомов углерода. Она представляет собой моноциклическую или конденсированную бициклическую кольцевую систему, в которой ароматическое кольцо конденсировано с другим ароматическим карбоциклическим кольцом. Примеры С6-С10 арильной группы включают фенил и нафтил. В случае замещения арильная группа обычно содержит группу Q согласно приведенным выше определениям, например, 1, 2 или 3 группы, выбранные из вариантов группы Q согласно приведенным выше определениям. Более конкретно, замещенная арильная группа, такая как замещенная фенильная группа, замещена 1 или 2 группами, выбранными из C1-С6 алкила, галогена,-OR8 и-N(R8)2, где R8 представляет собой Н или C1-C6 алкил, при этом, если в формуле имеются два радикала R8, они могут быть одинаковыми или разными.
С3-10 циклоалкильная группа представляет собой насыщенное углеводородное кольцо, содержащее от 3 до 10 атомов углерода. С3-10 циклоалкильная группа может представлять собой, например, С3-С7 циклоалкил, такой как циклопропил, циклобутил, циклопентил, циклогексил, или циклогептил. Обычно она представляет собой С3-С6 циклоалкил или С4-С6 циклоалкил, например, циклобутил, циклопентил или циклогексил. В одном варианте реализации она представляет собой циклобутил. С3-10 циклоалкильная группа является незамещенной или замещена, как правило, одной или более группами Q согласно приведенным выше определениям.
4-10-членная гетероарильная группа или фрагмент представляет собой 4-10-членную ароматическую гетероциклическую группу, содержащую 1, 2, 3 или 4 гетероатома, выбранных из О, N и S. Она является моноциклической или бициклической. Обычно она содержит один атом N и 0, 1, 2 или 3 дополнительных гетероатома, выбранных из О, S и N. Она может представлять собой, например, моноциклическую 5-7-членную гетероарильную группу, например, 5-или 6-членную N-содержащую гетероарильную группу. Примеры включают такие группы, как пиридил, пиразинил, пиримидинил, пиридазинил, фуранил, тиенил, пиразолидинил, пирролил, оксадиазолил, оксазолил, изоксазолил, тиазолил, тиадиазолил, имидазолил и пиразолил. Предпочтительными являются такие группы, как фуранил, тиенил, имидазолил, пиридил и пиримидил. Как вариант, это может быть бициклическая гетероарильная группа, например, 8-10-членная бициклическая гетероарильная группа. Примеры включают хинолил, изохинолил, хиназолил, хиноксалинил, индолил, изоиндолил, индазолил, имидазопиридазинил, пирролопиридинил, пиразолопиримидинил и пирролопиримидинил. В случае замещения гетероарильная группа (моноциклическая или бициклическая) обычно замещена одной или более, например, 1, 2 или 3, группами, выбранными из С1-4 алкила и группы Q согласно приведенным выше определениям.
4-10-членная гетероциклильная группа представляет собой моноциклическую или бициклическую неароматическую насыщенную или ненасыщенную кольцевую систему, содержащую от 5 до 10 атомов углерода и по меньшей мере один атом или группу, выбранные из N, О, S, SO, SO2 и СО, чаще всего N или О. Когда кольцевая система является бициклической, одно кольцо может быть насыщенным, а другое ненасыщенным. Обычно это С4-10 кольцевая система, в которой 1, 2 или 3 атома углерода в кольце замещены на атом или группу, выбранные из О, S, SO2, СО и NH. Чаще всего это моноциклическое кольцо, предпочтительно С4-С6 моноциклическое кольцо. Примеры 4-10-членной гетероциклильной группы включают такие фрагменты, как азетидинил, пиперидил, пиперазинил, морфолинил, тиоморфолинил, S,S-диоксотиоморфолинил, 1,3-диоксоланил, пирролидинил, имидазол-2-онил, пирролидин-2-онил, тетрагидрофуранил и тетрагидропиранил, пиперидин-2,6-дионил и пиперидин-2-онил. В частности, 4-10-членная гетероциклильная группа может представлять собой азетидинил, пиперидил, пиперазинил, морфолинил или тиоморфолинил.
В случае замещения гетероциклильная группа (моноциклическая или бициклическая) обычно замещена одной или более, например, 1, 2 или 3, группами, выбранными из ненасыщенного С1-4 алкила и группы Q согласно приведенным выше определениям. Она также может быть замещена узловым атомом мостиковой структуры, которая связывает два атома в кольце, как правило, два атома углерода в кольце. Например, пиперазиновая группа или морфолиновая группа может быть замещена углеродным мостиком. Полученная бициклическая структура может представлять собой, соответственно, 2,5-диазабицикло[2.2.1]гептановую или 2-окса-5-азабицикло[2.2.1]гептановую группу.
Во избежание неопределенности, хотя приведенные выше определения гетероарильных и гетероциклильных групп связаны с атомом ''N'', который может присутствовать в кольце, специалисту-химику будет понятно, что любой такой атом N будет нести протон (атом водорода) или другой заместитель, согласно приведенным выше определениям, если он связан с каждым из своих соседних атомов в кольце посредством одинарных связей. Такие протонированные формы охватываются определениями гетероарильных групп и гетероциклильных групп, приведенными в настоящем описании.
В одном из вариантов реализации формулы (I), согласно приведенным выше определениям, R2 представляет собой галогенный заместитель, в частности F, в положении 9 кольцевой системы бензодиазепинила. Примерами таких соединений являются соединения следующей формулы (I'):
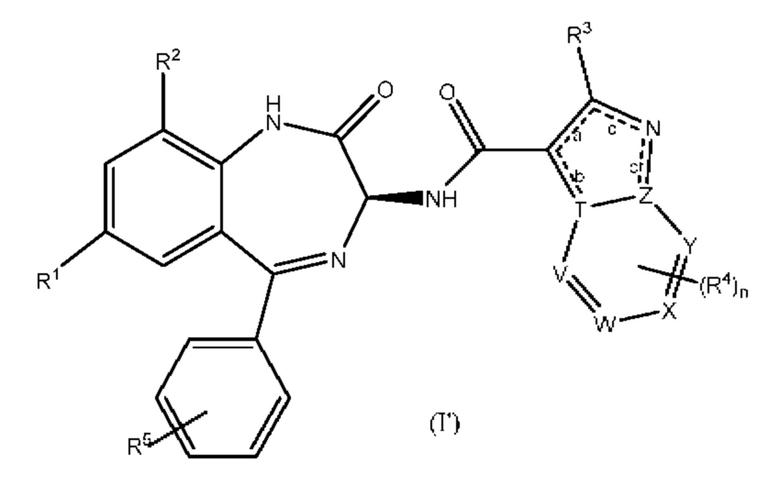
где R1 представляет собой Н или галоген, R2 представляет собой Н или галоген, а остальные группы и переменные являются такими, как это определено выше для формулы (I). Как правило, R1 представляет собой Н или F, и R2 представляет собой Н или F. Например, R1 представляет собой Н или F, a R2 представляет собой F.
Согласно одному из вариантов реализации формулы (I) Т представляет собой N, a Z представляет собой С. В таких соединениях 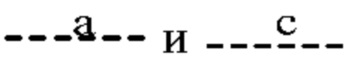 представляют собой связи, тогда как
представляют собой связи, тогда как 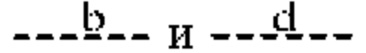 отсутствуют. Такие соединения имеют следующую формулу (Ia):
отсутствуют. Такие соединения имеют следующую формулу (Ia):
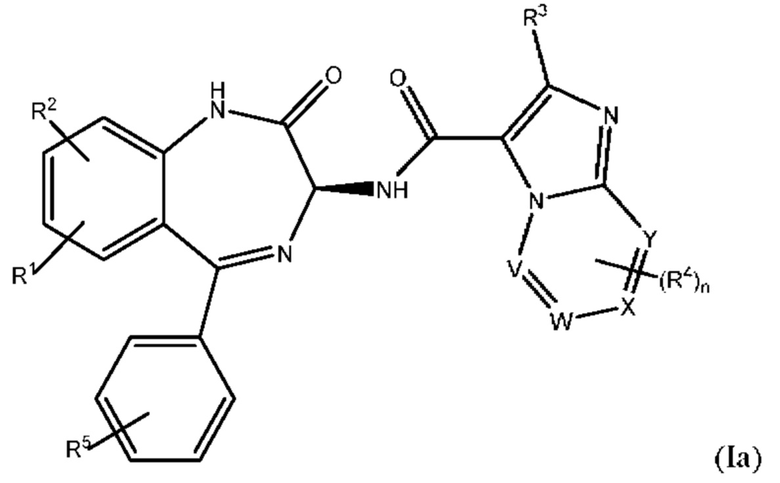
в которой все группы и переменные являются такими, как это определено выше для формулы (I) или (I').
Согласно другому варианту реализации формулы (I) Т представляет собой С, a Z представляет собой N. В таких соединениях 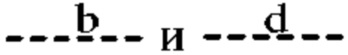 представляют собой связи, тогда как
представляют собой связи, тогда как 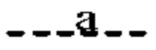 и
и 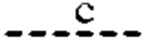 отсутствуют. Такие соединения имеют следующую формулу (Ib):
отсутствуют. Такие соединения имеют следующую формулу (Ib):
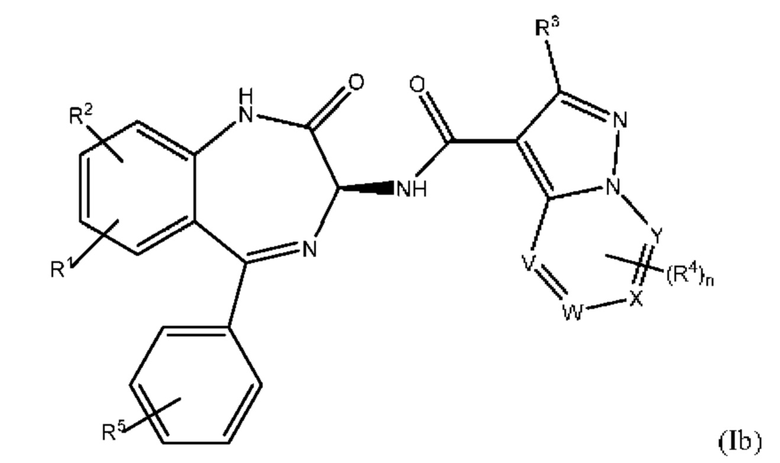
в которой все группы и переменные являются такими, как это определено выше для формулы (I) или (I').
В приведенных выше формулах (I), (I'), (Ia) и (Ib) V, как правило, представляет собой N, а каждый из W, X и Y представляет собой СН. Примеры таких структур включают бензодиазепинилимидазопиридазины приведенной ниже формулы (Ia') и бензодиазепинилпиразолопиримидины приведенной ниже формулы (Ib'):
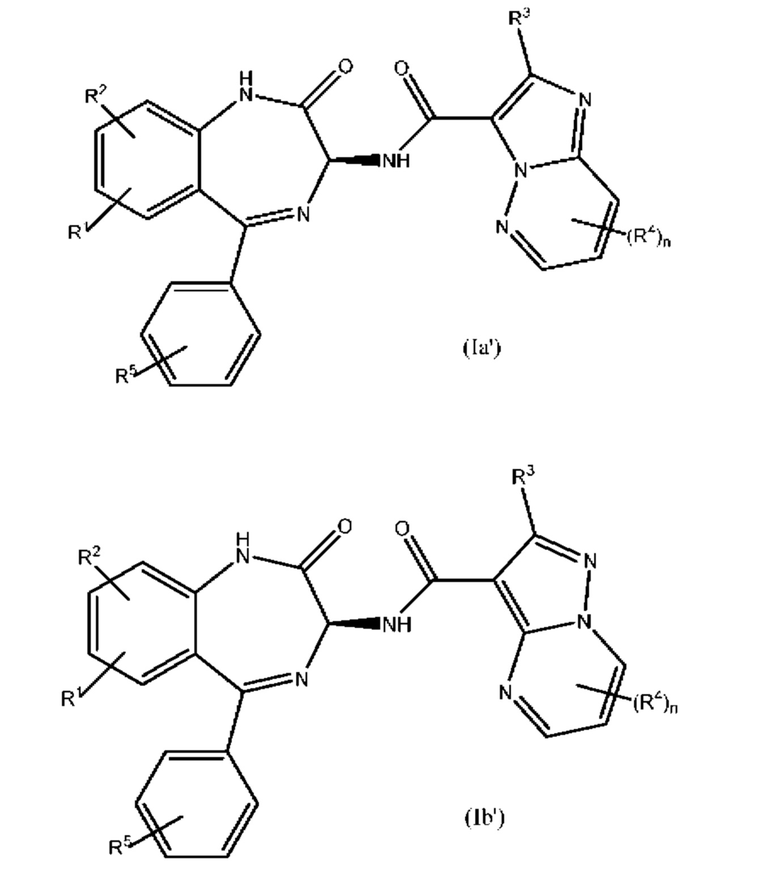
В формулах (Ia') и (Ib') каждый из R1-R5 и n является таким, как это определено выше для формулы (I) или (I').
В одном из вариантов реализации соединений согласно настоящему изобретению, имеющих любую из структурных формул (I), (I'), (Ia), (Ib), (Ia') и (Ib') согласно приведенным выше определениям, R1 представляет собой Н или F, a R2 представляет собой F в кольцевом положении 9 бензодиазепинильной кольцевой системы.
В одном из вариантов реализации соединений формул (Ia') и (Ib'), R1 и R2 соответствуют определениям и кольцевым положениям, указанным для формулы (I') выше. Такие соединения представляют собой бензодиазепинилимидазопиридазины с приведенной ниже формулой (Ia''):
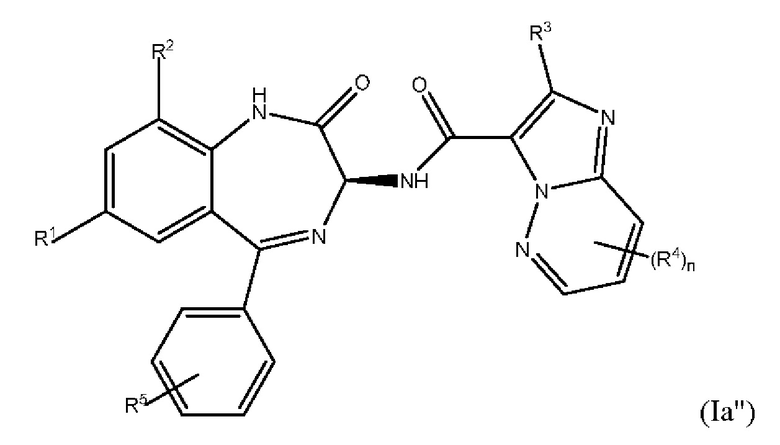
где каждый из R1-R5 и n является таким, как это определено выше для формулы (I'); и бензодиазепинилпиразолопиримидины с приведенной ниже формулой (Ib''):
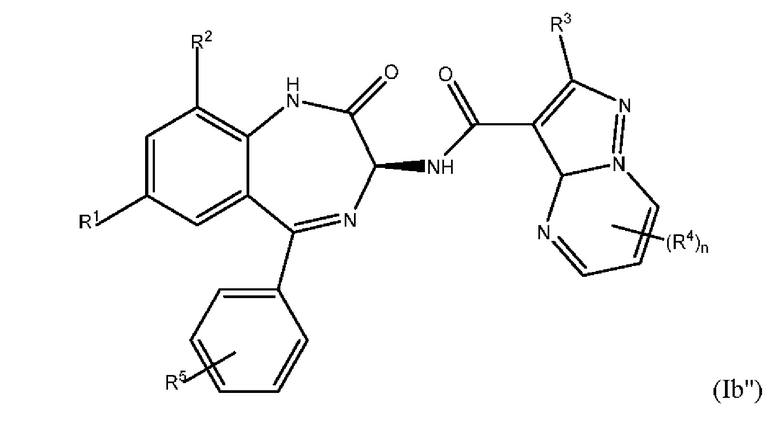
где каждый из R1-R5 и n является таким, как это определено выше для формулы (I').
В соединениях согласно настоящему изобретению, имеющих любую из указанных выше структурных формул, R3 обычно представляет собой группу, выбранную из C1-C6 алкила, С3-С10 циклоалкила, С6-С10 арила, 4-10-членного гетероциклила и 4-10-членного гетероарила, причем эта группа является незамещенной или замещена одной или двумя группами Q согласно приведенным выше определениям. Например, группа R3 согласно приведенным выше определениям может быть незамещенной или замещенной, выбранной из C1-С6 алкила, С3-С6 циклоалкила, C1-C6 алкоксигруппы, C1-С6 алкоксиалкила, C1-C6 трифторалкила, галогена,-OR8,-N(R8)2, где R8 представляет собой Н, C1-С6 алкил или С3-С6 циклоалкил, при этом, если в формуле имеются две группы R8, они могут быть одинаковыми или разными, и-N(R9)2, где группы R9 совместно образуют кольцо, выбранное из морфолина, пиперидина, пиперазина и пирролидина, при этом указанное кольцо является незамещенным или замещенным C1-С6 алкилом.
В соединениях согласно настоящему изобретению, имеющих любую из указанных выше структурных формул, R3 обычно представляет собой группу, выбранную из C1-C6 алкила, C1-С6 галогеналкила (такого как C1-С6 дифторалкил или C1-С6 трифторалкил), дигидроиндола, феноксигруппы и фенила, при этом каждая группа является незамещенной или замещена 1, 2 или 3 группами, выбранными из галогена,-OR8 и-N(R8)2, где R8 представляет собой Н, C1-С6 алкил или С3-С6 циклоалкил, при этом, если в формуле имеются две группы R8, они могут быть одинаковыми или разными.
В одном из вариантов реализации указанных выше структурных формул R3 представляет собой группу с приведенной ниже формулой (II):
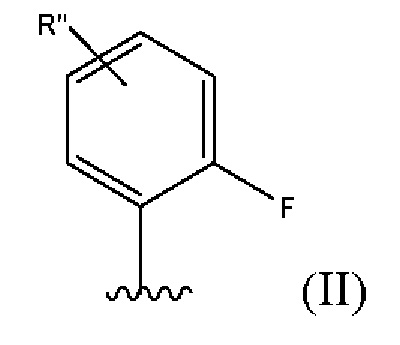
где R'' представляет собой Н, галоген,-OR8 или-N(R8)2, согласно приведенным выше определениям.
В другом варианте реализации указанных выше структурных формул R3 представляет собой 4-10-членную гетероарильную группу, например, выбранную из пиридила, пирролопиридила и индазолила. 4--10-членная гетероарильная группа является незамещенной или замещена группой Q согласно приведенным выше определениям, например, группой, выбранной из C1-С6 алкила, С3-С6 циклоалкила, C1-C6 алкоксигруппы, C1-C6 алкоксиалкила, C1-С6 трифторалкила, галогена,-OR8,-N(R8)2, где R8 представляет собой Н, C1-C6 алкил или С3-С6 циклоалкил, при этом, если в формуле имеются две группы R8, они могут быть одинаковыми или разными, и-N(R9)2, где группы R9 совместно образуют кольцо, выбранное из морфолина, пиперидина, пиперазина и пирролидина, при этом указанное кольцо является незамещенным или замещено C1-С6 алкилом или узловым атомом углерода мостиковой системы, связывающей два кольцевых атома.
Примеры групп R3 включают следующее:
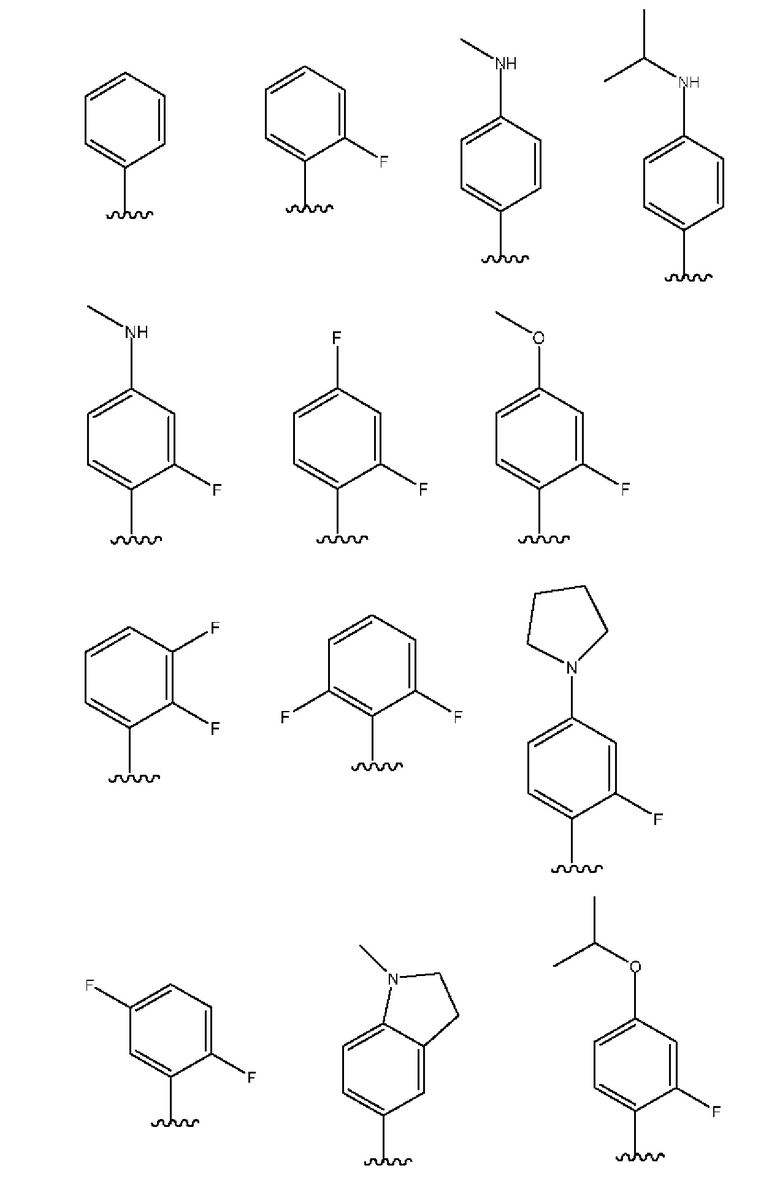
В соединениях согласно настоящему изобретению, имеющих любую из указанных выше структурных формул, R4 связано с любым доступным атомом углерода в шестичленном кольце. Таким образом, в указанных выше структурных формулах (I), (I'), (Ia), (Ib), (Ia') и (Ib') группа R4, при ее наличии, замещает Н в одной из кольцевых СН-групп, представленных любым из V, W, X и Y. Следовательно, один из V, W, X и Y представляет собой N, СН или CR4, а другие три представляют собой СН или CR4 при том условии, что присутствует только одна группа CR4.
Согласно одному из вариантов реализации соединений согласно настоящему изобретению, имеющих любую из указанных выше структурных формул, V представляет собой N, один из W, X и Y представляет собой CR4, а два остальных представляют собой СН. В другом варианте реализации V представляет собой N, Y представляет собой СН, один из W и X представляет собой CR4, а другой представляет собой СН.
В соединениях согласно настоящему изобретению R4 выбран из галогена,-OR6,-NR6R7,-COR8,-C(O)OR8,-CON(R8)2 и-R6, где каждый из R6 и R7 независимо представляет собой Н или группу, выбранную из C1-C6 алкила, С3-С10 циклоалкила, С6-С10 ар ила, 4-10-членного гетероциклила и 4-10-членного гетероарила, при этом указанная группа является незамещенной или замещенной, и где R8 представляет собой Н или C1-С6 алкил, при этом, если в формуле имеются две группы R8, они могут быть одинаковыми или разными.
В одном из вариантов реализации R4 представляет собой галоген,-OR6,-NR6R7 или группу, выбранную из C1-C6 алкила, С3-С10 циклоалкила, С6-С10 арила, 4-10-членного гетероциклила и 4-10-членного гетероарила, при этом указанная группа является незамещенной или замещенной. Если указанная группа является замещенной, она обычно имеет замещение группой Q согласно приведенным выше определениям. Если R4 представляет собой-OR6, где R6 представляет собой замещенный C1-C6 алкил, заместитель может представлять собой C1-С6 алкоксигруппу, так что R6 представляет собой C1-C6 алкоксиалкил. Если R4 представляет собой-OR6, где R6 представляет собой С4-С10 гетероциклильную группу, указанная гетероциклильная группа может представлять собой, например, азетидинил, незамещенный или замещенный, например, замещенный C1-С6 алкилом, таким как метил. Если R4 представляет собой-OR6, где R6 представляет собой C1-C6 алкил, при этом один или более атомов Н в этой алкильной группе могут быть замещены на D. Например, R4 может представлять собой группу-OCD3.
Как правило, R4 представляет собой галоген (например, C1),-OR6,-NR6R7, C1-C6 алкил, С3-С6 циклоалкил, фенил, фуранил, тиенил, имидазолил, пиридил, пиримидил, азетидинил, пирролиндинил, пиперидинил, пиперазинил или морфолинил, каждый из которых является незамещенным или замещен группой Q согласно приведенным выше определениям. Если R4 является замещенным, предпочтительным заместителем является галоген или C1-C6 алкил. Например, когда R4 представляет собой замещенный C1-С6 алкил, он может представлять собой моно-, ди-или тригалогензамещенную C1-С6 алкильную группу, такую как дифторалкильная или трифторалкильная группа. Если R4 является замещенным азетидинилом, пирролидинилом, пиперидинилом, пиперазинилом или морфолинилом, заместитель, как правило, представляет собой C1-C6 алкил. Другой предпочтительный заместитель пиперидинила, пиперазинила и морфолинила представляет собой узловой атом углерода мостиковой системы, связывающей два кольцевых атома.
Конкретные соединения согласно настоящему изобретению включают следующее:
2-[4-(Метиламино)фенил]-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-[4-(Метиламино)фенил]-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенилпиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-[4-(пропан-2-иламино)фенил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(1-Метил-2,3-дигидроиндол-6-ил)-Н-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2-Фтор-4-метоксифенил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2-Фтор-4-пропан-2-илоксифенил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-[2-Фтор-4-(метиламино)фенил]-Н-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенилпиразоло[1,5-а]пиримидин-3-карбоксамид;
6-Хлор-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-метилимидазо[1,2-b]пиридазин-3-карбоксамид;
6-Хлор-2-метил-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-(фуран-3-ил)-2-метилпиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2-Фтор-5-метилфенил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2-Фтор-5-метилфенил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(5-Хлорпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(5-Хлорпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(6-Циклопропилпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(6-Циклопропилпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(5-Циклопропилпиридин-3-ил)-Х-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2-Фтор-5-метилпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2-Фторфенил)-6-метил-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-метилпиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2,4-Дифторфенил)-5-метил-Х-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2,4-Дифторфенил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-5-метилпиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2-Фторфенил)-7-метил-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-7-метилпиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2-Фторфенил)-5-(морфолин-4-ил)-N-[(3S)-2-оксо-5-фенил-2,3-Дигидро-1Н-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-5-пирролидин-1-илпиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2-Фторфенил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-5-пирролидин-1-илпиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2,3-Дифторфенил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2,3-Дифторфенил)-N-[(3S)-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2,6-Дифторфенил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2-Фтор-4-пирролидин-1-илфенил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
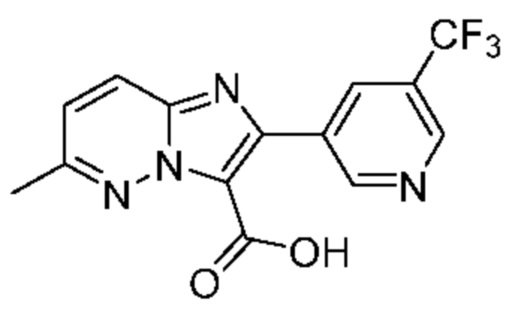
6-Метил-Н-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-[5-(трифторметил)пиридин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метил-2-[5-(трифторметил)пиридин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
2-[6-(Циклопропиламино)-2-фторпиридин-3-ил]-Н-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(6-Метилпиридин-3-ил)-Н-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(6-метилпиридин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-[6-(2-Метоксиэтил)пиридин-3-ил]-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-метилпиридин-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(6-Этоксипиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(6-Этилпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(6-Этилпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(6-Этил-2-метилпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-2,3-Дигидро-1Н-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(6-Этил-2-метилпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-2,3-Дигидро-1Н-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид
2-(6-Пропан-2-илпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(6-Пропан-2-илпиридин-3-ил)-Н-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-[2-Метил-6-(пропан-2-иламино)пиридин-3-ил]-Н-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-[2-Метил-6-(пропан-2-иламино)пиридин-3-ил]-Н-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-[4-Метил-6-(пропан-2-иламино)пиридин-3-ил]-Н-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(1-метилиндазол-5-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(5-метилпиридин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-[6-(Этиламино)-2-фторпиридин-3-ил]-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-[6-(Этиламино)-2-фторпиридин-3-ил]-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-[6-(3-Метилморфолин-4-ил)пиридин-3-ил]-N-[(3S)-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-[6-(3-Метилморфолин-4-ил)пиридин-3-ил]-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-[2-Фтор-6-(пропан-2-иламино)пиридин-3-ил]-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-[2-фтор-6-(пропан-2-иламино)пиридин-3-ил]пиразоло[1,5-а]пиримидин-3-карбокс амид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(1-метилпирроло[2,3-b]пиридин-5-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(6-морфолин-4-илпиридин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-[6-(пропан-2-ил амино)пиридин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-[6-(пропан-2-ил амино)пиридин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(1Н-пирроло[2,3-b]пиридин-5-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(6-метоксипиридин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-[6-(2-Гидрокси-2-метилпропил)пиридин-3-ил]-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-З-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(5-фторпиридин-2-ил)-6-метилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-морфолин-4-илимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-[(1S,4S)-5-метил-2,5-диазабицикло[2.2.1]гептан-2-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-[(1R,4R)-5-метил-2,5-диазабицикло[2.2.1]гептан-2-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-[(1R,4R)-2-окса-5-азабицикло[2.2.1]гептан-5-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-морфолин-4-ил-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-5-[(1R,4R)-5-метил-2,5-диазабицикло[2.2.1]гептан-2-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2-Фтор-6-метилпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
2-(2-Метокси-6-метилпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
2-(2,4-Дифторфенил)-N-[(3S)-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
2-(2,4-Дифторфенил)-Н-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
2-(2,4-Дифторфенил)-6-метил-Н-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
2-(2,4-Дифторфенил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]-6-метилимидазо[1,2-b]пиридазин-3-карбоксамид;
6-Метил-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метил-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
6-(Азетидин-1-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-(4-метилпиперазин-1-ил)имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метил-2-пиридин-3-илимидазо[1,2-b]пиридазин-3-карбоксамид;
6-Метил-2-(2-метилпиридин-4-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метил-2-(2-метилпиридин-4-ил)имидазо[1,2-b]пиридазин-3-карбоксамид;
2-(3-Фторпиридин-4-ил)-6-метил-N-[(3S)-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(3-фторпиридин-4-ил)-6-метилимидазо[1,2-b]пиридазин-3-карбоксамид;
2-(5-Фторпиридин-3-ил)-6-метил-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(5-фторпиридин-3-ил)-6-метилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метил-2-(5-метилпиридин-3-ил)имидазо[1,2-b]пиридазин-3-карбоксамид;
6-Метил-2-(6-морфолин-4-илпиридин-3-ил)-Н-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метил-2-(6-морфолин-4-илпиридин-3-ил)имидазо[1,2-b]пиридазин-3-карбоксамид;
6-(Этиламино)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-(1-метилазетидин-3-ил)оксиимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-(1-метил азетидин-3-ил)окси-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-(2-метоксиэтокси)имидазо[1,2-b]пиридазин-3-карбоксамид;
6-Метокси-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-2-оксо-5-фенил-1,3-Дигидро-1,4-бензодиазепин-3-ил]-2-фенил-6-(тридеутериометокси)имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метокси-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенил-6-(тридеутериометокси)имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-метоксиимидазо[1,2-b]пиридазин-3-карбоксамид;
6-Хлор-Н-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)имидазо[1,2-b]пиридазин-3-карбоксамид;
6-Хлор-2-(6-этилпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метокси-2-(5-метилпиридин-3-ил)имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(3-фторпиридин-4-ил)-6-метоксиимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метокси-2-(6-метилпиридин-3-ил)имидазо[1,2-b]пиридазин-3-карбоксамид;
2-(6-Этилпиридин-3-ил)-6-метокси-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
6-Этокси-2-(6-этилпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
2-(6-Этилпиридин-3-ил)-N-[(3S)9-фтор-2-оксо-5-фенил-2,3-дигидро-1Н-1,4-бензодиазепин-3-ил]-6-(2Н3)метоксиимидазо[1,2-b]пиридазин-3-карбоксамид;
2-(6-[(3S*)-3-метилморфолин-4-ил]пиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-2,3-дигидро-1Н-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-2,3-дигидро-1Н-1,4-бензодиазепин-3-ил]-2-{6-[(3S*)-3-метилморфолин-4-ил]пиридин-3-ил}пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(1Н-индазол-5-ил)имидазо[1,2-b]пиридазин-3-карбоксамид;
или их фармацевтически приемлемые соли.
Соединения по настоящему изобретению могут содержать асимметричные или хиральные центры и, следовательно, могут существовать в различных стереоизомерных формах. Предполагается, что все стереоизомерные формы соединений по настоящему изобретению, включая, но не ограничиваясь ими, диастереомеры, энантиомеры и атропоизомеры, а также их смеси, такие как рацемические смеси, составляют часть настоящего изобретения. Соединения формулы (I), содержащие один или более хиральных центров, можно применять в энантиомерной или диастереоизомерно чистой форме или в форме смеси изомеров.
Настоящее изобретение охватывает все геометрические и позиционные изомеры соединений по настоящему изобретению, как определено выше. Например, если соединение по настоящему изобретению содержит двойную связь или конденсированное кольцо, в объем изобретения включены цис- и трансформы, а также их смеси. Как отдельные позиционные изомеры, так и смесь позиционных изомеров также входят в объем настоящего изобретения.
Соединения по настоящему изобретению могут существовать в несольватированных, а также в сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и тому подобное, и настоящее изобретение охватывает как сольватированные, так и несольватированные формы.
Соединения по настоящему изобретению могут существовать в разных таутомерных формах, и все такие формы включены в объем настоящего изобретения. Термин «таутомер» или «таутомерная форма» относится к структурным изомерам с различными энергиями, которые являются взаимопревращаемыми через низкоэнергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимопревращения посредством миграции протона, такие как кето-енольные таутомеризации. Валентные таутомеры включают взаимопревращения путем реорганизации некоторых связующих электронов.
Соединения согласно настоящему изобретению могут быть получены методами синтеза, описанными в приведенных ниже Примерах, или по аналогии с такими методами путем использования подходящих исходных материалов и методик, известных опытным химикам.
Производное бензодиазепина формулы (I) может быть преобразовано в его фармацевтически приемлемую соль, а соль может быть преобразована в свободное соединение традиционными способами. Например, производное бензодиазепина формулы (I) можно привести в контакт с фармацевтически приемлемой кислотой, в результате чего и будет получена его фармацевтически приемлемая соль. Фармацевтически приемлемая соль представляет собой соль с фармацевтически приемлемой кислотой или основанием.
Фармацевтически приемлемые кислоты включают как неорганические кислоты, такие как хлористоводородная, серная, фосфорная, дифосфорная, бромистоводородная или азотная кислота, так и органические кислоты, такие как лимонная, фумаровая, малеиновая, яблочная, аскорбиновая, янтарная, винная, бензойная, уксусная, метансульфоновая, этансульфоновая, бензолсульфоновая или n-толуолсульфоновая кислота. Фармацевтически приемлемые основания включают гидроксиды щелочных металлов (например, натрия или калия) и щелочноземельных металлов (например, кальция или магния) и органические основания, например, алкиламины, аралкиламины и гетероциклические амины.
В ходе биологических испытаний было установлено, что соединения согласно настоящему изобретению являются ингибиторами респираторно-синцитиального вируса (РСВ). Они сочетают в себе мощную анти-РСВ активность с благоприятной биодоступностью и физико-химическими характеристиками. Это сочетание свойств делает указанные соединения терапевтически полезными и в качестве потенциальных лекарственных препаратов превосходящими многие соединения, раскрытые в ранее обсуждавшейся научной литературе предшествующего уровня техники.
Соответственно, согласно настоящему изобретению дополнительно предложено соединение, которое представляет собой производное бензодиазепина формулы (I) согласно приведенному выше определению или его фармацевтически приемлемую соль, для применения в способе лечения организма человека или животного терапевтическими методами.
В данном изобретении также предложено соединение согласно приведенному выше определению для применения в способе лечения или профилактики РСВ-инфекции. Кроме того, настоящее изобретение относится к применению соединения по настоящему изобретению согласно приведенному выше определению при получении лекарственного средства, предназначенного для лечения или профилактики РСВ-инфекции. Таким образом, субъекта, страдающего от РСВ-инфекции или подверженного ей, можно лечить способом, включающим введение ему соединения по настоящему изобретению согласно приведенному выше определению. Тем самым состояние субъекта можно улучшить или облегчить.
РСВ-инфекция обычно представляет собой инфекцию дыхательных путей. РСВ-инфекция может представлять собой инфекцию у ребенка, например, ребенка в возрасте до десяти лет или ребенка в возрасте до двух лет. В одном из вариантов реализации настоящего изобретения предложено соединение согласно приведенному выше определению для применения при лечении или профилактике РСВ-инфекции у педиатрических пациентов. Как вариант, инфекция может представлять собой инфекцию у взрослого или пожилого человека, например, взрослого человека старше 60 лет, взрослого человека старше 70 лет или взрослого человека старше 80 лет. В настоящем изобретении дополнительно предложено соединение для применения при лечении или профилактике РСВ-инфекции у гериатрических пациентов.
РСВ-инфекция может представлять собой инфекцию у индивидуума с ослабленным иммунитетом или у индивидуума, страдающего ХОБЛ или ЗСН. В другом варианте реализации настоящего изобретения РСВ-инфекция представляет собой инфекцию у индивидуума с неослабленным иммунитетом, например, индивидуума, который в остальном здоров.
Соединение согласно настоящему изобретению можно вводить в различных лекарственных формах, например, перорально в виде таблеток, капсул, таблеток, покрытых сахарной или пленочной оболочкой, жидких растворов или суспензий, или парентерально, например, внутримышечно, внутривенно или подкожно. Следовательно, соединение может быть введено путем инъекции, вливания или посредством ингалятора или небулайзера. Соединение предпочтительно вводят перорально.
Дозировка зависит от множества факторов, включая возраст, вес и состояние пациента, а также путь введения. Суточные дозы могут варьироваться в широких пределах и будут подбираться в соответствии с индивидуальными требованиями в каждом конкретном случае. Обычно дозировка, принятая для каждого пути введения, когда соединение вводят отдельно взрослым людям, составляет от 0,0001 до 650 мг/кг, чаще всего в диапазоне от 0,001 до 10 мг/кг массы тела, например, от 0,01 до 1 мг/кг. Такую дозировку можно вводить, например, от 1 до 5 раз в сутки. Для внутривенной инъекции подходящая суточная доза составляет от 0,0001 до 1 мг/кг массы тела, предпочтительно от 0,0001 до 0,1 мг/кг массы тела. Суточную дозу можно вводить в виде разовой дозы или в соответствии с графиком разделенных доз.
Единичная дозированная форма, такая как таблетка или капсула, обычно содержит 1-250 мг активного ингредиента. Например, соединение формулы (I) можно вводить пациенту в дозе 100-250 мг один раз в сутки, два или три раза в сутки. Например, соединение формулы (I) можно вводить пациенту в дозе 100-250 мг один раз в сутки, два или три раза в сутки.
Соединения формулы (I) и их фармацевтически приемлемые соли можно применять сами по себе. Альтернативно их можно вводить в форме фармацевтической композиции. Следовательно, настоящее изобретение также относится к фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, как определено выше, в сочетании с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем. Обычные процедуры выбора и приготовления подходящих фармацевтических составов описаны, например, в ''Pharmaceuticals-The SCience of Dosage Form Designs'', M. E. Aulton, Churchill Livingstone, 1988.
В зависимости от способа введения фармацевтическая композиция предпочтительно будет содержать от 0,05 до 99 масс. % (массовых процентов), более предпочтительно от 0,05 до 80 масс. %, еще более предпочтительно от 0,10 до 70 масс. %, и еще более предпочтительно от 0,10 до 50 масс. % активного ингредиента, при этом все массовые проценты приведены по общей композиции.
Настоящее изобретение, кроме того, относится к способу получения фармацевтической композиции согласно настоящему изобретению, включающему смешивание соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем.
Соединения по настоящему изобретению можно вводить в различных дозированных формах. Таким образом, их можно вводить перорально, например, в виде таблеток, формованных пастилок, пастилок для рассасывания, водных или масляных суспензий, растворов, диспергируемых порошков или гранул. Соединения согласно настоящему изобретению также можно вводить парентерально, будь то подкожно, внутривенно, внутримышечно, внутрикожно, трансдермально, с помощью инфузионных методов или путем ингаляции или распыления с помощью небулайзера. Соединения также можно вводить в виде суппозиториев.
Твердые пероральные формы фармацевтической композиции согласно настоящему изобретению могут содержать наряду с активным соединением разбавители, например, лактозу, декстрозу, сахарозу, целлюлозу, кукуру3Ный крахмал или картофельный крахмал; смазывающие вещества, например, диоксид кремния, тальк, стеариновую кислоту, стеарат магния или кальция, и/или полиэтиленгликоли; связующие вещества, например, крахмалы, аравийскую камедь, желатин, метилцеллюлозу, карбоксиметилцеллюлозу или поливинилпирролидон; дезагрегирующие агенты, например, крахмал, альгиновую кислоту, альгинаты или крахмалгликолят натрия; вспенивающиеся смеси; красители; подсластители; смачивающие агенты, такие как лецитин, полисорбаты, лаурилсульфаты; и, в целом, нетоксичные и фармакологически неактивные вещества, используемые в фармацевтических составах. Такие фармацевтические препараты можно производить известными способами, например, с помощью процессов смешивания, гранулирования, таблетирования, нанесения сахарного или пленочного покрытия.
Жидкие дисперсии для перорального введения могут представлять собой сиропы, эмульсии и суспензии. Сиропы могут содержать в качестве носителей, например, сахарозу или сахарозу с глицерином и/или маннитом и/или сорбитом.
Суспензии и эмульсии могут содержать в качестве носителя, например, природную камедь, агар, альгинат натрия, пектин, метилцеллюлозу, карбоксиметилцеллюлозу или поливиниловый спирт. Суспензия или растворы для внутримышечных инъекций могут содержать совместно с активным соединением фармацевтически приемлемый носитель, например, стерильную воду, оливковое масло, этилолеат, гликоли, например, пропиленгликоль, и, если требуется, подходящее количество гидрохлорида лидокаина. Другие подходящие носители для суспензий включают стерильную воду, гидрокеипропилметилцеллюлозу (ГПМЦ), полисорбат 80, поливинилпирролидон (ПВП), аэрозоль АОТ (т.е. 1,2-бис(2-этилгексоксикарбонил)этансульфонат натрия), плюроник F127 и/или каптизол (т.е. сульфобутиловый эфир бета-циклодекстрина).
Соединения согласно настоящему изобретению могут, например, быть приготовлены в виде водных суспензий в носителе, выбранном из:
(i) 0,5% масс./об. гидроксипропилметилцеллюлозы (ГПМЦ) / 0,1% масс/об. полисорбата 80;
(ii) 0,67% масс/об. поливинилпирролидона (ПВП) / 0,33% масс./об. аэрозоля АОТ (1,2-бис(2-этилгексоксикарбонил)этансульфоната натрия);
(iii) 1% масс./об. плуроника F 127; и
(iv) 0,5% масс/об. полисорбата 80.
Носители можно получать посредством стандартных процедур, известных специалистам в данной области. Например, каждый из носителей (i)-(iv) можно получать следующим образом: берут навеску необходимого количества вспомогательного вещества в подходящем сосуде, добавляют примерно 80% конечного объема воды и перемешивают на магнитной мешалке до образования раствора. Затем носитель доводят до конечного объема водой. Водные суспензии соединений формулы I можно приготовить следующим образом: берут навеску необходимого количества соединения формулы I в подходящем сосуде, добавляют 100% требуемого объема носителя и перемешивают на магнитной мешалке.
Растворы для инъекций или вливаний могут содержать в качестве носителя, например, стерильную воду или, предпочтительно они могут быть в форме стерильных водных изотонических солевых растворов.
Соединения согласно настоящему изобретению также можно вводить в комбинации с другими соединениями, применяемыми для лечения вирусных инфекций. Таким образом, настоящее изобретение относится к комбинированной терапии, в которой соединение согласно настоящему изобретению, или его фармацевтически приемлемая соль, или фармацевтическая композиция или состав, содержащие соединение согласно настоящему изобретению, вводят одновременно или последовательно или в виде комбинированного препарата с другим терапевтическим агентом или агентами, для лечения или профилактики вирусной инфекции, в частности, РСВ-инфекции.
Следует понимать, что в настоящем документе термин ''комбинация'' относится к одновременному, раздельному или последовательному введению. В одном аспекте настоящего изобретения ''комбинация'' относится к одновременному введению. В одном аспекте настоящего изобретения ''комбинация'' относится к раздельному введению. В одном аспекте настоящего изобретения ''комбинация'' относится к последовательному введению. В тех случаях, когда введение является последовательным или раздельным, задержка введения второго компонента должна быть такой, чтобы не потерять полезный эффект комбинации.
Подходящие терапевтические агенты для применения в комбинированной терапии включают
(i) ингибиторы слияния РСВ
(ii) другие ингибиторы (М)-белка нуклеокапсида РСВ
(iii) другие ингибиторы белков РСВ, такие как ингибиторы белка фосфопротеина (Р) и большого (L) белка;
(iv) нуклеозидные или полимеразные ингибиторы, которые ингибируют L-белок;
(v) моноклональные антитела к РСВ, такие как антитела к F-белку;
(vi) иммуномодулирующие соединения toll-подобных рецепторов;
(vii) прочие противореспираторновирусные средства, такие как противогриппозные и противориновирусные соединения; и/или
(viii) противовоспалительные соединения.
Нуклеокапсидный (N)-белок РСВ играет ключевую роль в транскрипции и репликации вируса, опосредуя взаимодействие между геномной РНК и кодируемой вирусом PHN-зависимой PHN-полимеразой. Р- и L-белки РСВ являются компонентами кодируемой вирусом PHN-зависимой PHN-полимеразы РСВ.
Согласно другому аспекту настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль согласно приведенному выше определению в комбинации с одним или более терапевтическими агентами, перечисленными выше в пп. (i)-(vi), для применения при лечении РСВ-заболеваний.
Приведенные ниже Примеры даны с целью дополнительной иллюстрации изобретения. Подготовительные образцы касаются подготовки исходных материалов и промежуточных продуктов, используемых для получения соединений, указанных в Примерах. Ни Примеры, ни Подготовительные образцы никоим образом не ограничивают изобретение.
Примеры
Реактивы получали из коммерческих источников и использовались без дополнительной очистки. Безводные растворители закупали у коммерческих поставщиков, использовали в поставленном виде и хранили в атмосфере N2. Реакции проводили с безводными растворителями в атмосфере N2, если не было указано иное. Все значения температуры приведены в °С. Тонкослойную хроматографию (ТСХ) проводили на пластинах с силикагелем на алюминиевой основе с индикатором флуоресценции при 254 нМ (средний размер пор 60Å). Колоночную флэш-хроматографию проводили посредством системы Biotage Isolera One с использованием колонок KP-Sil или Ultra silica gel или посредством системы Isco CombiFlash Rf с использованием колонок FlashPure или RediSep Rf/RediSep Rf Gold silica gel. Обращенно-фазовую колоночную хроматографию проводили посредством системы Isco CombiFlash Rf с использованием обращенно-фазовых колонок Teledyne Isco RediSep Rf С18. Ионообменную хроматографию проводили на картриджах для твердофазной экстракции Isolute SCX-2 с кремнеземом и пропилсульфоновой кислотой, промываемых соответствующими растворителями, выбранными из воды, MeCN и МеОН. Элюирование основных соединений производили раствором NH3 в МеОН (0,7 н. или 7 н. в зависимости от субстрата).
Препаративную ВЭЖХ проводили на колонке при комнатной температуре следующими методами. ВЭЖХ-Метод 1: Колонка Gemini NX (30 мм × 150 мм, 5 мкм) со скоростью пропускания 42 мл/мин и УФ-детекцией при 210 нм. ВЭЖХ-Метод 2: Колонка Waters X-Select CSH C18 (30×100 мм, 5 мкм) со скоростью пропускания 50 мл/мин и УФ-детекцией при 215 нм. ВЭЖХ-Метод 3: Колонка Waters X-select CDH С18 (19×50 мм, 5 мкМ) со скоростью пропускания 42 мл/мин и УФ-детекцией на всех длинах волн с фотодиодной матрицей. ВЭЖХ-Метод 4: Колонка Waters XSelect CSH С18, (30×100 мм, 5 мкм) со скоростью пропускания 42 мл/мин и УФ-детекции на всех длинах волн с фотодиодной матрицей. Препаративную хиральную ВЭЖХ проводили при комнатной температуре колонки на системе Gilson HPLC (УФ-детекция при 230 нм) с колонкой ChiralPAK IC (20×250 мм; 5 мкм) при скорости пропускания 15 мл/мин. Аналитическую хиральную ВЭЖХ проводили при комнатной температуре колонки на системе Agilent 1100 HPLC (УФ-детекция при 260 нМ) с колонкой ChiralPAK 1С (2,1×150 мм; размер частиц 3 мкм) при скорости пропускания 0,4 мл/мин и временем хроматографирования 10 мин. Препаративную хиральную СФХ проводили на системе Waters SFC Prep 15 (УФ-детекция с диодно-матричным детектором (DAD) при 210-400 нм; скорость пропускания 15 мл/мин; температура колонки 40°С; противодавление 120 бар) и колонке Phenomenex Lux® Cellulose-4 (1x25 см; 5 мкм). Аналитическую хиральную СФХ проводили на системе Waters SFC ACQUITY UPC2 (УФ-детекция с диодно-матричным детектором (DAD) при 220 400 нм; скорость пропускания 1,5 мл/мин; температура колонки 40°С; противодавление 1750 psi) с временем хроматографирования 3 мин на колонках Phenomenex Lux® Cellulose-4 (1×25 см; 5 мкм).
Спектры ЯМР регистрировали на спектрометре 400 или 500 МГц при комнатной температуре зонда (номинально 298К). значения химических сдвигов (δ) приведены в ppm и откалиброваны по остаточному пику растворителя, взятому в качестве внутреннего стандарта (CDCl3, δ = 7,26 ppm; ДМСО-d6, δ = 2,50 ppm). Константы взаимодействия приведены в герцах (Гц). Спектры МСНР регистрировали с помощью компактного масс-спектрометра Advion Plate Express expressionL, оснащенного источником ионов APCI.
Анализ методом ЖХ-МС проводили на установке Waters Acquity UPLC с колонкой CSH С18 или ВЕН С18 (2,1×30 мм) при 40°С со скоростью пропускания 0,77 мл/мин с линейным градиентом ацетонитрила 5-95%, подходящим для липофильности соединения в течение 3 или 10 минут. Водная часть подвижной фазы представляла собой 0,1% раствор муравьиной кислоты (колонка CSH С18) или 10 мМ раствор бикарбоната аммония (колонка ВЕН С18).
Хроматограммы ЖХ-УФ регистрировали с помощью диодно-матричного детектора (DAD)
Waters Acquity в диапазоне от 210 до 400 нм. Масс-спектры регистрировали с помощью детектора Waters Acquity QDa с переключением ИЭР между режимами положительных и отрицательных ионов.
Метод А: 3 мин - Кислотный
Метод В: 3 мин - Основной
Метод С: 10 мин - Кислотный
Метод D: 10 мин - Основной
Метод Е: 1 мин - Основной
Метод F: 1 мин - Кислотный
Подготовительные образцы соединений (3S)-3-амино-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-она и (3S)-3-амино-9-фтор-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-она получали способами, описанными в заявках WO/2004/026843, WO/2005/090319 и WO/2017/015449.


Подготовительные образцы
1А Этил-5-амино-3-бром-1Н-пиразол-4-карбоксилат
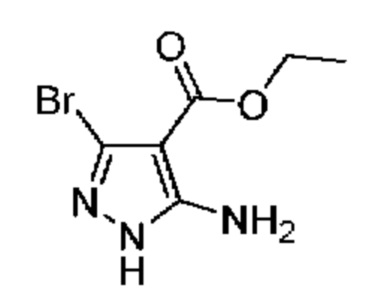
К охлажденному (до 0°С) раствору этилового эфира 5-амино-1Н-пиразол-4-карбоновой кислоты (10,00 г, 64,45 ммоль) в ТГФ (250 мл) в течение 25 минут по каплям добавляли раствор N-бромсукцинимида (13,77 г, 77,34 ммоль) в MeCN (270 мл). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь адсорбировали на силикагеле, летучие вещества удаляли при пониженном давлении, и остаток очищали посредством колоночной хроматографии[10-50% (EtOH:CH2Cl2:NH4OH; 50:8:1) в CH2Cl2], в результате чего получали твердое вещество бежевого цвета, которое тритурировали с CH2Cl2 (~20 мл) и получали твердое вещество белого цвета (5,93 г, выход 39%). МСНР (APCI+) m/z 234, 1/236,1 [М+Н]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 12,16 (s, 1H), 6,25 (s, 2Н), 4,18 (q, J=7,1 Гц, 2Н), 1,25 (t, J=7,1 Гц, 3Н).
2А Этил-2-бромпиразоло[1,5-а]пиримидин-3-карбоксилат
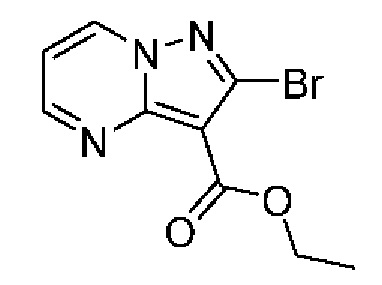
Раствор промежуточного соединения 1А (2,27 г, 9,70 ммоль) и 1,1,3,3-тетраметоксипропана (1,93 мл, 11,64 ммоль) в АсОН (33,9 мл) нагревали при 70°С в течение 68 часов. После охлаждения до комнатной температуры летучие вещества удаляли при пониженном давлении, остаток растворяли в EtOAc (50 мл) и нейтрализовали насыщенным водным раствором NaHCO3 до уровня рН ≈ 7. Смесь экстрагировали CH2Cl2 (3×50 мл), и объединенные органические слои сушили посредством Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии (0-3% МеОН в CH2Cl2), в результате чего получали твердое вещество бежевого цвета (2,31 г, выход 88%). ЖХ-МС (Метод A): m/z 292,0[M+Na]+ при 0,93 мин. 1H ЯМР (500 МГц, ДМСО-d6) δ 9,24 (dd, J=7,0, 1,8 Гц, 1Н), 8,86 (dd, J=4,3, 1,8 Гц, 1H), 7,33 (dd, J=6,9, 4,2 Гц, 1H), 4,33 (q, J=7,1 Гц, 2Н), 1,33 (t, J=7,1 Гц, 3Н).
3А Этил-2-бром-5-метилпиразоло[1,5-а]пиримидин-3-карбоксилат
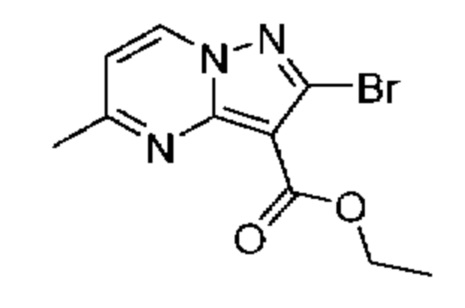
Раствор промежуточного соединения 1А (877 мг, 3,750 ммоль) и 4,4-диметоксибутан-2-она (0,99 мл, 7,490 ммоль) в толуоле (6,3 мл) нагревали при 100°С в течение 19 часов. Летучие вещества удаляли при пониженном давлении, а остаток очищали посредством колоночной хроматографии (30-57% EtOAc в гептане), в результате чего получали твердое вещество белого цвета (539 мг, выход 51%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,07 (d, J=7,1 Гц, 1H), 7,21 (d, J=1,1 Гц, 1H), 4,30 (d, J=1,1 Гц, 2Н), 2,62 (s, 3Н), 1,32 (t, J=1,1 Гц, 3Н). МСНР (APCI+) m/z 240,2[М-ОСН2СН3]+
4А Этил-6-бром-2-метилпиразоло[1,5-а]пиримидин-3-карбоксилат
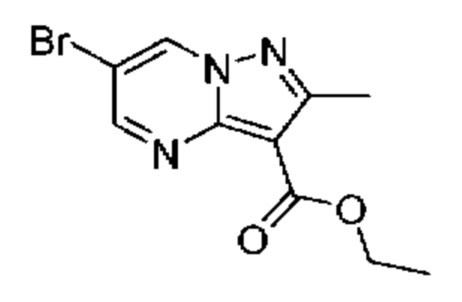
К раствору 2-броммалональдегида (1,50 г, 9,94 ммоль) в EtOH (19,4 мл) добавляли этиловый эфир 5-амино-3-метил-1Н-пиразол-4-карбоновой кислоты (1,64 г, 9,69 ммоль) и АсОН (8,32 мл, 145,41 ммоль), и полученную смесь нагревали при 75°С в течение ночи. Летучие вещества удаляли при пониженном давлении, CH2Cl2 (75 мл), после чего добавляли насыщенный водный раствор NaHCO3 (50 мл) и водный слой экстрагировали CH2Cl2 (2×50 мл). Объединенные органические экстракты промывали насыщенным водным раствором NaHCO3 и солевым раствором (по 25 мл каждого), сушили над Na2SO4, и удаляли растворитель при пониженном давлении. Очистка методом колоночной хроматографии (18-50% EtOAc в гептане) дала твердое вещество белого цвета (1,98 г, выход 72%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,65 (d, J=2,2 Гц, 1H), 8,85 (d, J=2,2 Гц, 1H), 4,30 (q, J=7,1 Гц, 2Н), 2,60 (s, 3Н), 1,31 (t, J=1,1 Гц, 3Н). МСНР (APCI+) m/z 238,1[М-ОСН2СН3]+
5А 3-Фтор-N-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин
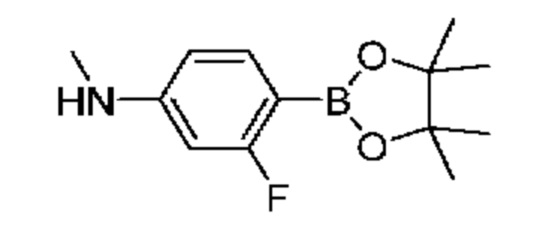
К раствору 3-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (300 мг, 1,27 ммоль), формальдегида (0,1 мл, 1,33 ммоль) и АсОН (0,11 мл, 1,90 ммоль) в CH2Cl2 (6 мл) добавляли триацетоксиборгидрид натрия (268 мг, 1,27 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Затем добавляли АсОН (0,11 мл, 1,90 ммоль) и триацетоксиборгидрид натрия (268 мг, 1,27 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разделяли в системе CH2Cl2 (10 мл) + вода (10 мл), отделяли органический слой, который затем пропускали через гидрофобный фильтр-сепаратор, и растворитель удаляли при пониженном давлении. Остаток очищали посредством колоночной хроматографии (0-100% EtOAc в изогексанах), в результате чего получали твердое вещество желтого цвета (100 мг, выход 28%). ЖХ-МС (Метод A) m/z 252,2 [М+Н]+ (ES+) при 1,48 мин.
6А Этил-2-фенилпиразоло[1,5-а]пиримидин-3-карбоксилат
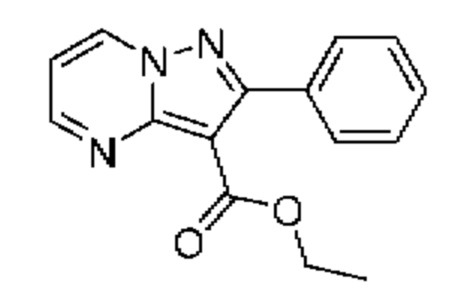
Смесь промежуточного соединения 2А (300 мг, 1,11 ммоль), фенилбороновой кислоты (167 мг, 1,37 ммоль) и K2CO3 (464 мг, 3,36 ммоль) в смеси 1,4-диоксана и воды (2:1, 3,0 мл) дегазировали посредством пропускания N2 в течение 5 мин. Затем добавляли Pd(PPh3)4 (192 мг, 0,170 ммоль), реакционную смесь дополнительно дегазировали путем пропускания N2 в течение 5 минут, после чего нагревали до 100°С в течение ночи. Реакционную смесь охлаждали до к.т., разбавляли CH2Cl2 (10 мл), и органический слой промывали солевым раствором (3×10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии (0-80% EtOAc в гексане), в результате чего получали твердое вещество желтого цвета (111 мг, выход 36%). ЖХ-МС (Метод A) m/z 222 [М+Н]+ (ES+) при 1,13 мин. 1Н ЯМР (500 МГц, CDCl3) δ 8,79 (dd, J=4,2, 1,8 Гц, 1H), 8,75 (dd, J=6,9, 1,9 Гц, 1H), 7,79-7,75 (m, 2H), 7,49-7,45 (m, 3Н), 7,03 (dd, J=6,9, 4,1 Гц, 1H), 4,38 (q, J=7,1 Гц, 2H), 1,29 (t, J=7,1 Гц, 3Н).
Приведенные далее промежуточные соединения получали по той же методике, что и описанная для промежуточного соединения 6А. Подготовительные образцы соединений 6М и 6N готовили, исходя из соответствующего пинаколового эфира бороновой кислоты. Подготовительный образец соединения 6О готовили с использованием 2 экв. (2,3-дифторфенил)бороновой кислоты.
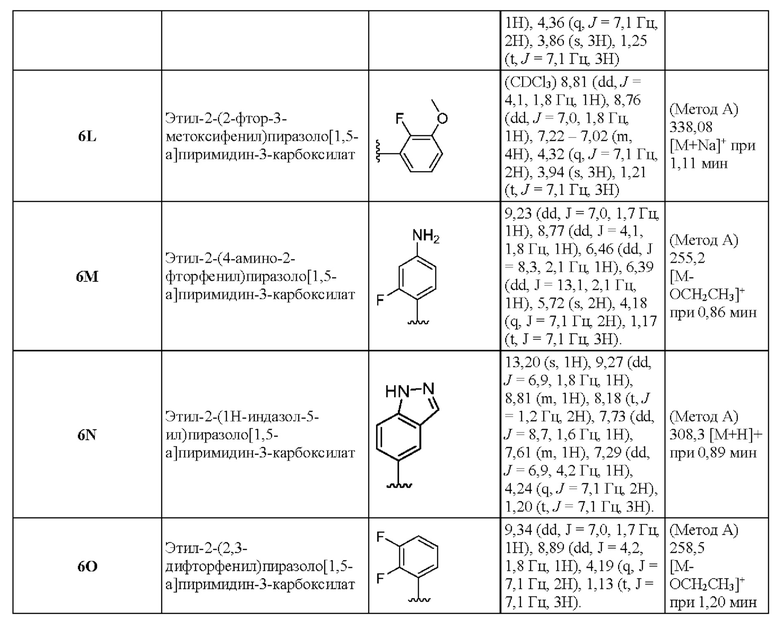
6Р Этил-2-(2-фторфенил)-5-[(1R,4R-5-метил-2,5-диазабицикло[2.2.1]гептан-2-ил]пиразоло[1,5-а]пиримидин-3-карбоксилат
Раствор промежуточного соединения 24С (149 мг, 0,386 ммоль), 2-фторфенилбороновой кислоты (141 мг, 1,00 ммоль) и K2CO3 (161 мг, 1,162 ммоль) в смеси 1,4-диоксана и воды (2:1, 1,9 мл) барботировали N2 в течение ~5 минут. Затем добавляли Pd(PPh3)4 (134 мг, 0,116 ммоль), и реакционную смесь нагревали при 100°С в течение ночи. Реакционную смесь разбавляли CH2Cl2 (25 мл) и подкисляли (до рН≈4) добавлением АсОН. Затем смесь очищали посредством ионообменной хроматографии (2 г SCX-2). Полученный раствор концентрировали при пониженном давлении, в результате чего получали твердое вещество коричневого цвета (124 мг, выход 73%). ЖХ-МС (Метод A) m/z 396,4 [М+Н]+ (ESI+) при 0,74 мин. 1Н ЯМР (500 МГц, CDCl3) 8,33 (d, J=7,6 Гц, 1H), 7,58-7,50 (m, 1H), 7,46-7,38 (m, 1H), 7,24 (t, J=7,6 Гц, 1H), 7,15 (t, J=9,1 Гц, 1H), 6,29-6,18 (m, 1H), 4,28-4,18 (m, 2H), 4,04-3,92 (m, 1H), 3,86-3,73 (m, 1H), 3,62-3,50 (m, 1H), 3,21-3,03 (m, 1H), 2,61 (s, 3H), 2,55-2,46 (m, 1H), 2,26-2,12 (m, 1H), 2,05-1,93 (m, 1H), 1,40-1,31 (m, 1H), 1,19 (t, J=7,1 Гц, 3H),
7A Этил-2-(2-фторфенил)-5-метилпиразоло[1,5-а]пиримидин-3-карбоксилат
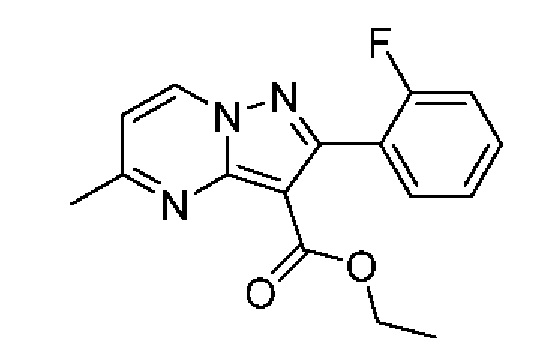
Получен по той же методике, что была описана для получения промежуточного соединения 6А из промежуточного соединения 3А. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,15 (d, J=7,1 Гц, 1H), 7.60-7,49 (m, 2Н), 7,37-7,28 (m, 2Н), 7,22 (d, J=7,1 Гц, 1H), 4,13 (q, J=7,1 Гц, 2Н), 2,64 (s, 3Н), 1,08 (t, J=7,1 Гц, 3Н). МСНР (APCI+) m/z 254,3 [М-OCH2CH3]+
8 А Этил-6-(фуран-3-ил)-2-метилпиразоло[1,5-а]пиримидин-3-карбоксилат
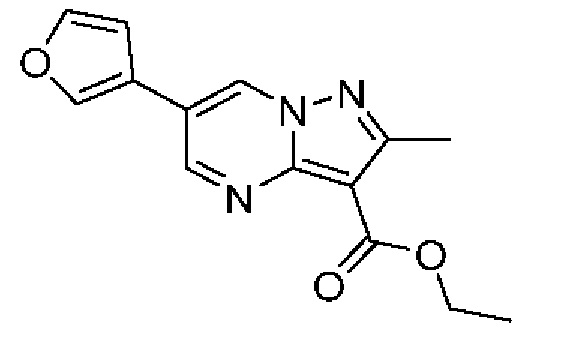
Раствор промежуточного соединения 4А (200 мг, 0,70 ммоль), фуран-3-бороновой кислоты (118 мг, 1,06 ммоль) и K2CO3 (292 мг, 2,11 ммоль) в смеси 1,4-диоксана и воды (3:1; 3,52 мл) во флаконе микроволнового реактора дегазировали путем пропускания N2 в течение-10 мин. Добавляли Pd(dppf)Cl2 (39 мг, 0,05 ммоль) и нагревали закрытый флакон при 80°С в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли EtOAc и фильтровали через слой Celite® на фильтровальной бумаге из стекловолокна, промывая EtOAc. Растворитель удаляли при пониженном давлении, а остаток очищали методом колоночной хроматографии (18-70% EtOAc в гептане). Полученное твердое вещество растирали с охлажденной (до 0°С) смесью Et2O и гептана (1:1), затем с гептаном (2×) и сушили при пониженном давлении, в результате чего получали твердое вещество розового цвета (85 мг, выход 45%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,50 (d, J=2,2 Гц, 1H), 9,14 (d, J=2,2 Гц, 1H), 8,45-8,43 (m, 1H), 7,85 (t, J=1,7 Гц, 1H), 7,19 (dd, J=1,9, 0,9 Гц, 1H), 4,30 (q, J=7,1 Гц, 2Н), 2.61 (s, 3Н), 1,33 (t, J=7,1 Гц, 3Н). МСНР (APCI+) m/z 226,2 [М-ОСН2СН3]+
9А 2-(2-Фторфенил)пиразоло[1,5-а]пиримидин-3-карбоновая кислота
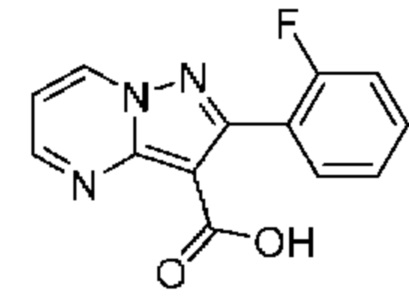
Раствор промежуточного соединения 6В (124 мг, 0,320 ммоль) и LiOH (1,5М водный раствор; 0,9 мл, 1,350 ммоль) в смеси ТГФ и МеОН (1:1, 4 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (20 мл) и промывали EtOAc (2×25 мл). Водный слой подкисляли путем добавления по каплям 1М водного раствора HCl до уровня рН ≈ 2, затем экстрагировали смесью CHCl3 и iPrOH (3:1; 3×25 мл). Объединенные органические экстракты сушили над Na2SO4, растворитель удаляли при пониженном давлении, в результате чего получали твердое вещество желтого цвета (48 мг, выход 56%). ЖХ-МС (Метод A) m/z 280,4 [M+Na]+ при 0,82 мин. 1Н ЯМР (500 МГц, ДМСО-d6) 12,19 (br s, 1H), 9,29 (dd, J=6,9, 1,8 Гц, 1H), 8,84 (dd, J=4,2, 1,8 Гц, 1H), 7,63-7,49 (m, 2Н), 7,36-7,26 (т, 2Н).
Приведенные ниже промежуточные соединения получали по той же методике, которая была описана для получения промежуточного соединения 9А.

9К 6-Метил-2-[5-(трифторметил)пиридин-3-ил]имидазо[1,2-b]пиридазин-3-карбоновая кислота
Получен по той же методике, что описана для получения промежуточного соединения 9А с нагреванием при 40°С. ЖХ-МС (Метод A) m/z 322,7 [М+Н]+ при 1,03 мин. 1H ЯМР (500 МГц, ДМСО-d6) 13,48 (s, 1H), 9,30 (d, J=2,0 Гц, 1H), 9,03 (d, J=2,2 Гц, 1H), 8,62-8,58 (m, 1H), 8,24 (d, J=9,3 Гц, 1H), 7,44 (d, J=9,3 Гц, 1H), 2,62 (s, 3Н),
10А 2-(2-Хлорфенил)пиразоло[1,5-а]пиримидин-3-карбоновая кислота
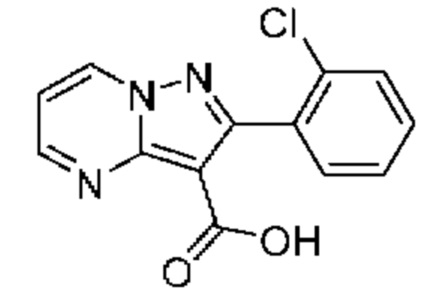
Смесь промежуточного соединения 9G (250 мг, 1,03 ммоль), 2-хлорбензолбороновой кислоты (170 мг, 1,08 ммоль) и K2CO3 (432 мг, 3,13 ммоль) в смеси 1,4-диоксана и воды (2:1, 3,0 мл) дегазировали пропусканием N2 в течение 5 мин. Добавляли Pd(PPh3)4 (119 мг, 0,100 ммоль), реакционную смесь дополнительно дегазировали пропусканием N2 в течение 5 мин, после чего нагревали до 100°С в течение ночи. Реакционную смесь охлаждали до к.т.и концентрировали при пониженном давлении. Добавляли NaOH (2М водный раствор; 30 мл), и полученный раствор промывали МТБЭ (3×50 мл). Затем водный раствор подкисляли 1М водным раствором HCl (до уровня рН≈2) и экстрагировали смесью CHCl3 и iPrOH (3:1; 3×50 мл). Объединенные органические экстракты сушили над MgSO4 и концентрировали под вакуумом, в результате чего получали твердое вещество белого цвета (105 мг, выход 30%). ЖХ-МС (Метод A) m/z 255,99 [М-ОСН2СН3]+ при 0,86 мин.
11А 2-(2-Фторфвнил)-5-мвтилпиразоло[1,5-а]пиримидин-3-карбоновая кислота
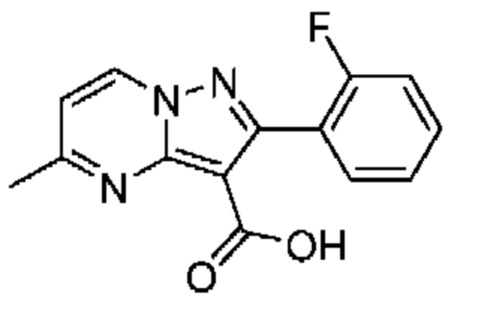
Раствор промежуточного соединения 7А (222 мг, 0,740 ммоль) и LiOH (1М водный раствор, 2,23 мл, 2,230 ммоль) в смеси ТГФ и МеОН (3:1; 3,7 мл) нагревали при 50°С в течение 16 часов. Летучие вещества удаляли при пониженном давлении, а остаток растирали с Et20, подкисляли раствором HCl (1М; 2,2 мл) и насыщенным водным раствором NH4Cl (5 мл), после чего экстрагировали смесью МеОН и CH2Cl2 (1:4; 4×15 мл). Объединенные органические экстракты промывали солевым раствором (5 мл), сушили над MgSO4, растворитель удаляли при пониженном давлении, в результате чего получали твердое вещество бледно-желтого цвета (151 мг, выход 75%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,16 (br s, 1H), 9,12 (d, J=7,1 Гц, 1H), 7,57-7,48 (m, 2Н), 7,33-7,27 (т, 2Н), 7,19 (d, J=7,1 Гц, 1H), 2,64 (s, 3Н). МСНР (APCI+) m/z 272,2 [М+Н]+.
Приведенные далее промежуточные соединения получали по той же методике, что была описана для получения промежуточного соединения 11А.
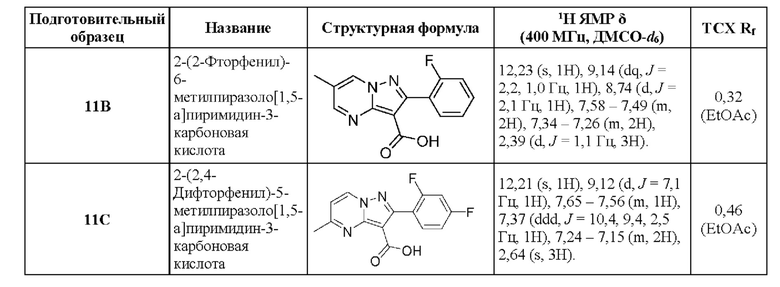
11D 2-(2-Фторфвнил)-5-морфолин-4-илпиразоло[1,5-а]пиримидин-3-карбоновая кислота
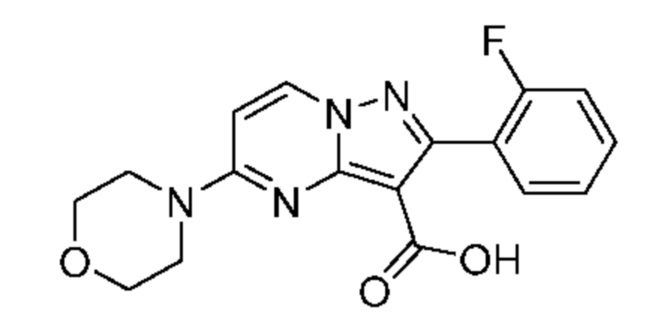
Раствор промежуточного соединения 26D (192 мг, 0,518 ммоль) и LiOH (1М водный раствор, 2,205 мл, 2,205 ммоль) в смеси THF и МеОН (3:1; 2,58 мл) нагревали при 50°С в течение 19 часов. Затем добавляли LiOH (1М водный раствор, 1,036 мл, 1,036 ммоль), и реакционную смесь нагревали в течение 23 часов. Процедура выделения, аналогичная описанной для промежуточного соединения 11А, сопровождающаяся растиранием в Et2O, давала твердое вещество беловатого цвета (158 мг, выход 58%). 1Н ЯМР (400 Мгц, ДМСО-d6) δ 11,57 (s, 1H), 8,79 (d, J=7,9 Гц, 1H), 7,52-7,44 (m. 2Н), 7,29-7,21 (т, 2Н), 6,91 (d, J=7,9 Гц, 1H), 3,82-3,66 (m, 8Н). МСНР (APCI+) m/z 343,1 [М+Н]+.
НЕ 2-(2-Фторфенил)-5-пирролидин-1-илпиразоло[1,5-а]пиримидин-3-карбоновая кислота
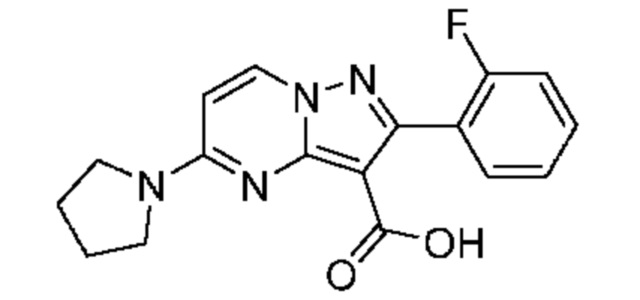
Получали по той же методике, которая была описана для получения промежуточного соединения 11D. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,42 (s, 1H), 8,75 (d, J=7,7 Гц, 1H), 7,54-7,44 (m, 2H), 7,32-7,22 (m, 2H), 6,59 (d, J=7,8 Гц, 1H), 3,68-3,49 (m, 4H), 2,08-1,89 (m, 4H). MCHP (APCI+) m/z 327,2 [М+Н]+.
12А 6-(Фуран-3-ил)-2-метилпиразоло[1,5-а]пиримидин-3-карбоновая кислота
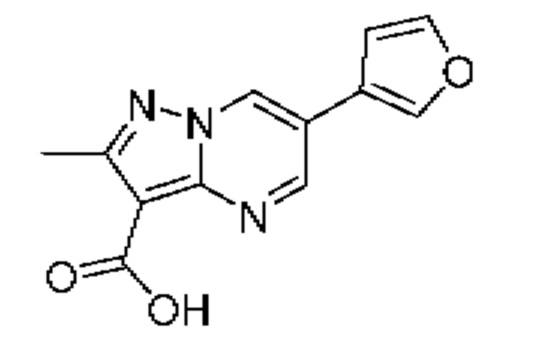
Раствор этил-6-(фуран-3-ил)-2-метилпиразоло[1,5-а]пиримидин-3-карбоксилата, промежуточного соединение 6А и LiOH (1М водный раствор; 0,94 мл, 0,94 ммоль) в смеси ТГФ и МеОН (3:1, 4 мл) нагревали при 50°С в течение 2 часов, затем при 40°С в течение 48 часов. Летучие вещества удаляли при пониженном давлении и полученный сырой продукт растирали с Et2O, затем подкисляли HCl (1М водный раствор, 0,8 мл) и насыщенным водным раствором NH4Cl (5 мл) до уровня рН≈2, после чего экстрагировали смесью EtOAc и EtOH (-3:1; 3x25 мл). Объединенные органические экстракты промывали солевым раствором (5 мл), сушили над Na2SO4, и удаляли растворитель при пониженном давлении. Остаток суспендировали в воде, фильтровали, промывали водой, и полученный осадок сушили при пониженном давлении, в результате чего получали сырой продукт в виде твердого вещества бледно-коричневого цвета (56 мг, выход 74%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,30 (br s, 1Н), 9,48 (d, J=2,3 Гц, 1H), 9,08 (d, J=2,3 Гц, 1H), 8,45 (s, 1H), 7,85 (s, 1H), 7,19 (s, 1H), 2,60 (s, 3Н). MCHP (APCI+) m/z 244,2 [М+Н]+.
13А Этил-2-хлор-3-(2-фторфен ил)-3-оксопропаноат
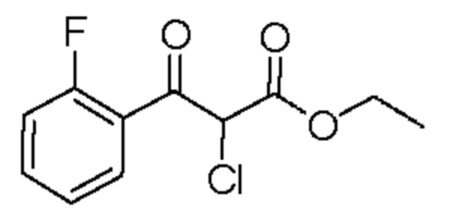
Раствор этил(2-фторбензоил)ацетата (0,43 мл, 2,38 ммоль) в МТБЭ (1 мл) по каплям добавляли к перемешиваемому раствору сульфурилдихлорида (0,21 мл, 2,62 ммоль) в МТБЭ (5 мл) при 0°С, и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Дополнительно добавляли сульфурилдихлорид (0,08 мл, 0,95 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили насыщенным водным раствором NaHCO3 (60 мл) и EtOAc (60 мл) и смесь перемешивали в течение 30 мин. Органический слой отделяли, промывали солевым раствором (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении, в результате чего получали маслянистую жидкость желтого цвета (346 мг, выход 55%). 1H ЯМР (500 МГц, CDCl3) δ 7,95 (td, J=7,6, 1,9 Гц, 1H), 7,65-7,57 (m, 1H), 7,33-7,26 (т, 1H), 7,24-7,14 (т, 1H), 5,63 (s, 1H), 4,30 (q, J=7,1 2Н), 1,27 (t, J=7,1 Гц, 3Н).
14А Этил-2-(2-фторфенил)имидазо[1,2-b]пиридазин-3-карбоксилат
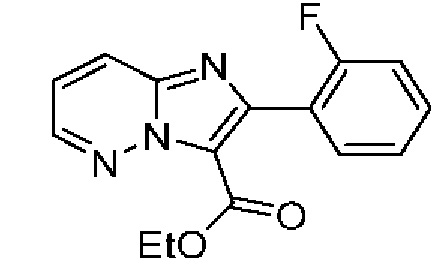
Раствор пиридазин-3-амина (201 мг, 2,110 ммоль) и промежуточного соединения 13А (0,27 мл, 2,640 ммоль) в EtOH (4 мл) нагревали до 100°С в течение 2 часов с помощью МВИ. Реакционную смесь охлаждали до к.т., упаривали под вакуумом, и полученный остаток очищали посредством колоночной хроматографии (0-100% EtOAc в изогексанах), в результате чего получали твердое вещество желтого цвета (70 мг, выход 9%). ЖХ-МС (Метод A) m/z 286,01 [М+Н]+ при 1,13 мин. 1Н ЯМР (500 МГц, ДМСО-d6) δ 8,77 (dd, J=4,5, 1,7 Гц, 1H), 8,33 (dd, J=9,3, 1,7 Гц, 1H), 7,69 (td, J=7,5, 1,8 Гц, 1Н), 7,58-7,48 (m, 2Н), 7,38-7,29 (т, 2Н), 4,23 (q, J=7,1 Гц, 2Н), 1,11 (t, J=7,1 Гц, 3Н).
15А Этил-2-(2-фторфенил)-6-метилимидазо[1,2-b]пиридазин-3-карбоксилат
Получали по той же методике, которая была описана для получения промежуточного соединения 14А. ЖХ-МС (Метод A) m/z 300,14 [М+Н]+ при 1,21 мин. 1Н ЯМР (500 МГц, ДМСО-d6) δ 8,20 (d, J=9,3 Гц, 1Н), 7,68 (td, J=7,5, 1,8 Гц, 1Н), 7,56-7,48 (m, 1H), 7,42 (d, J=9,3 Гц, 1H), 7,37-7,27 (т, 2Н), 4,21 (q, J=7,1 Гц, 2Н), 2,60 (s, 3Н), 1,10 (t, J=7,1 Гц, 3Н).
16А 2-(2-Фторфенил)-6-метилимидазо[1,2-b]пиридазин-3-карбоновая кислота
Получали по той же методике, которая была описана для получения промежуточного соединения 9А из промежуточного соединения 15А с нагреванием при 40°С в течение-18 ч. ЖХ-МС (Метод A) m/z 272,04 [М+Н]+ при 0,86 мин. 1H ЯМР (500 МГц, ДМСО-d6) δ 13,01 (s, 1H), 8,18 (d, J=9,3 Гц, 1H), 7,65 (td, J=7,5, 1,8 Гц, 1H), 7,53-7,45 (m, 1H), 7,39 (d, J=9,3 Гц, 1H), 7,34-7,25 (т, 2Н), 2,60 (s, 3Н).
17А 3-Бром-6-этил-2-метилпиридин
К охлажденному (до 0°С) раствору 3,6-дибром-2-метилпиридина (2,5 г, 9,96 ммоль) в ТГФ (40 мл) в атмосфере азота добавляли Pd(PPh3)4 (576 мг, 0,50 ммоль), после чего по каплям добавляли раствор диэтилцинка (1М в гексане; 11,96 мл, 11,96 ммоль). Реакционную смесь перемешивали в течение 1 часа при 0°С, затем в течение 16 часов при комнатной температуре. Реакцию гасили насыщенным водным раствором NH4Cl (25 мл), разбавляли EtOAc (10 мл), и отделенный водный слой экстрагировали EtOAc (3×20 мл). Объединенные органические экстракты сушили над Na2SO4 и удаляли растворитель при пониженном давлении. Очистка посредством колоночной хроматографии (0-10% ЕЮ Ас в изогексанах) давала бесцветную маслянистую жидкость (1,3 г, выход 62%). ЖХ-МС (Метод В) m/z 200,1/202,1 [М+Н]+ при 1,35 мин. 1Н ЯМР (500 МГц, CDCl3) 7,71 (d, J=8,1 Гц, 1H), 6,89 (d, J=8,1 Гц, 1Н), 2,77 (q, J=7,6 Гц, 2Н), 2,66 (s, 3Н), 1,30 (t, J=7,6 Гц, 3Н).
18А 5-Бром-6-метил-п-пропан-2-илпиридин-2-амин
Раствор изопропиламина (5,09 мл, 59,2 ммоль) и 3-бром-6-фтор-2-метилпиридина (2,5 г, 13,2 ммоль) в ДМСО (6 мл) нагревали с помощью МВИ при 120°С в течение 12 часов. Полученную реакционную смесь разбавляли водой (100 мл), экстрагировали CH2Cl2 (3×15 мл), объединенные органические экстракты сушили над Na2SO4, и растворитель удаляли при пониженном давлении. Очистка методом колоночной хроматографии (5-50% EtOAc в изогексанах) давала бесцветную маслянистую жидкость (2,30 г, 76%). ЖХ-МС (Метод В): m/z 229,1/231,1 [М+Н]+ при 1,54 мин. 1Н ЯМР (500 МГц, CDCl3) 7,48 (d, J=8,7 Гц, 1H), 6,11 (d, J=8,7 Гц, 1Н), 4,38 (d, J=7,6 Гц, 1Н), 3,80-3,74 (m, 1H), 2,47 (s, 3Н), 1,23 (d, J=6,4 Гц, 6Н).
18В 5-Бром-4-метил-п-пропан-2-илпиридин-2-амин
Получали по той же методике, которая была описана для получения промежуточного соединения 18А из 5-бром-2-фтор-4-метилпиридина (2 г, 10,5 ммоль) и изопропиламина (4,52 мл, 52,6 ммоль). ЖХ-МС (Метод В): m/z 229, 1/231,2 [М+Н]+ при 0,45 мин. 1Н ЯМР (500 МГц, CDCl3) 8,09 (s, 1H), 6,27 (s, 1H), 4,40-4,22 (m, 1H), 3,84 (m, 1H), 2,30 (s, 3Н), 1,23 (d, J=6,4 Гц, 6Н).
19А 5-Бром-2-(2-метоксиэтил)пиридин
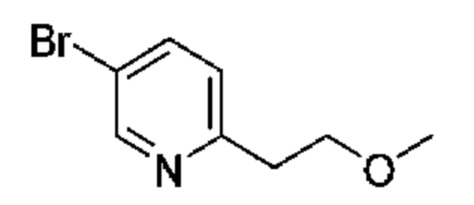
К охлажденному (до -78°С) раствору 2-(5-бромпиридин-2-ил)этанола (500 мг, 2,48 ммоль) в ТГФ (10 мл) добавляли NaH (60%-ная дисперсия в минеральном масле, 109 мг, 2,73 ммоль), смесь перемешивали при-78°С в течение 30 минут, затем добавляли MeI (0,17 мл, 2,73 ммоль). Реакционную смесь оставляли до достижения к.т. и перемешивали в течение ночи, после чего реакцию гасили насыщенным водным раствором NH4Cl (5 мл). Отделенный водный слой экстрагировали CH2Cl2 (3×10 мл), объединенные органические экстракты сушили над Na2SO4, и удаляли растворитель при пониженном давлении. Очистка методом колоночной хроматографии (0-30% EtOAc в гептане) давала бесцветную маслянистую жидкость (401 мг, выход 68%). ЖХ-МС (Метод В): m/z 216,5/218,5 [М+Н]+ при 1,00 мин. 1H ЯМР (500 МГц, ДМСО-d6) 8,59 (dd, J=2,5, 0,7 Гц, 1H), 7,94 (dd, J=8,3, 2,5 Гц, 1Н), 7,29 (dd, J=8,3, 0,7 Гц, 1H), 3,66 (t, J=6,6 Гц, 2Н), 3,22 (s, 3Н), 2,93 (t, J=6,5 Гц, 2Н).
20А 1-(5-Бромпиридин-2-ил)-2-метилпропан-2-ол
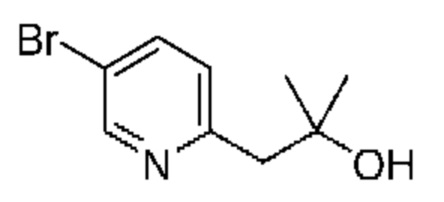
К охлажденному (до -78°С) раствору 5-бром-2-метилпиридина (1 г, 5,81 ммоль) в ТГФ (10 мл) по каплям добавляли диизопропиламид лития (раствор 2М в смеси ТГФ-гептан-этилбензол, 4,1 мл, 8,20 ммоль). Раствор перемешивали при -78°С в течение 20 минут, затем к нему по каплям добавляли ацетон (5,8 мл, 17,4 ммоль). Реакционную смесь оставляли до достижения комнатной температуры и перемешивали в течение 3 ч, затем гасили насыщенным водным раствором NH4Cl (40 мл) и экстрагировали CH2Cl2 (2×50 мл). Объединенные органические экстракты сушили над Na2SO4, концентрировали при пониженном давлении и очищали методом колоночной хроматографии (0-20% EtOAc в CH2Cl2), в результате чего получали бесцветную маслянистую жидкость (435 мг, выход 32%). ЖХ-МС (Метод В): m/z 214,1 [М-ОН]+ при 0,95 мин. 1H ЯМР (500 МГц, CDCl3) δ 8,59 (dd, J=2,5, 0,8 Гц, 1Н), 7,78 (dd, J=8,3, 2,5 Гц, 1H), 7,07 (d, J=8,3 Гц, 1H), 2,90 (s, 2Н), 1,24 (s, 6Н).
21А Этил-2-бром-6-метилпиразоло[1,5-а]пиримидин-3-карбоксилат
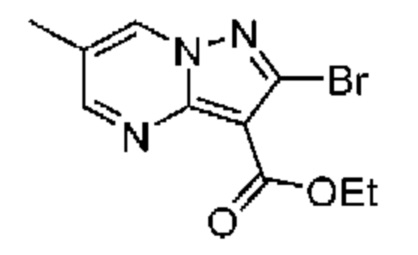
Раствор промежуточного соединения 1А (963 мг, 4,11 ммоль) и 3-этоксиметакролеина (0,5 мл, 4,22 ммоль) в АсОН (11,8 мл) нагревали при 75°С в течение 24 часов. После охлаждения до к.т.летучие вещества удаляли при пониженном давлении. Остаток разделяли в системе из EtOAc (50 мл) и насыщенного водного раствора NaHCO3 (50 мл), и отделенную водную фазу экстрагировали EtOAc (3×50 мл). Объединенные органические слои последовательно промывали насыщенным водным раствором NaHCO3, водой и солевым раствором (по 50 мл каждого), сушили над Na2SO4, и удаляли растворитель при пониженном давлении. Очистка методом колоночной хроматографии (30-79% EtOAc в гептане) давала твердое вещество белого цвета (936 мг, выход 80%). MCHP (APCI+) m/z 238,0, 240,0 [М-ОСН2СН3]+ 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,11 (d,J=1,1 Гц, 1Н), 8,76 (d, J=2,2 Гц, 1Н), 4,31 (d, J=7,1 Гц, 2Н), 2,36 (d, J=1,1 Гц, 3Н), 1,32 (t,J=7,l Гц, 3Н).
22А Этил-2-бром-5-оксо-4Н-пиразоло[1,5-а]пиримидин-3-карбоксилат
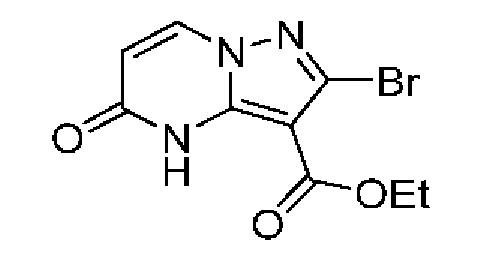
К суспензии этил-3-этоксиакрилата (6,58 мл, 45,6 ммоль) и Cs2CO3 (15,1 г, 46,0 ммоль) в ДМ ФА (120 мл) при комнатной температуре добавляли этил-5-амино-3-бром-1Н-пиразол-4-карбоксилат (7,88 г, 30,4 ммоль), и полученную смесь нагревали при 110°С в течение 1,5 часа. Реакционную смесь охлаждали до комнатной температуры, выливали в ледяную воду (~500 мл) с уксусной кислотой (3,48 мл, 60,8 ммоль), дополнительно смывая реакционный сосуд водой (~100 мл). Полученный осадок отделяли фильтрованием, промывали водой и затем сушили при пониженном давлении, в результате чего получали твердое вещество беловатого цвета (8,09 г, выход 87%). ЖХ-МС (Метод A) m/z 240,0/242,5 [М-ОСН2СН3]+ при 0,93 мин. 1H ЯМР (500 МГц, ДМСО-d6) δ 11,77 (s, 1H), 8,49 (d, J=7,9 Гц, 1H), 6,16 (d, J=7,9 Гц, 1H), 4,30 (q, J=7,0 Гц, 2Н), 1,28 (t, J=7,0 Гц, 3Н).
23А Этил-2-бром-5-хлорпиразоло[1,5-а]пиримидин-3-карбоксилат
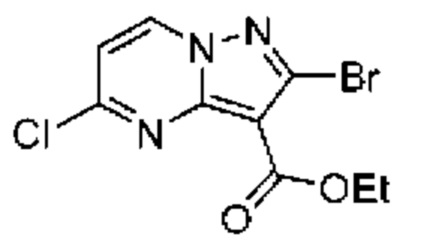
Раствор промежуточного соединения 22А (8,09 г, 26,1 ммоль) в POCl3 (25 мл, 268 ммоль) кипятили с обратным холодильником в течение 3 часов. Реакционную смесь охлаждали до к.т., и летучие вещества удаляли при пониженном давлении. Остаток разбавляли CH2Cl2 (~200 мл) и медленно (осторожно, процесс идет с выделением тепла) выливали в насыщенный водный раствор NaHCO3 (300 мл). Выделение образующегося тепла контролировали путем добавления в смесь льда. Фазы разделяли, и водный слой экстрагировали CH2Cl2 (2×200 мл). Объединенные органические экстракты сушили над Na2SO4, и удаляли растворитель при пониженном давлении, в результате чего получали твердое вещество серого цвета (7,94 г, выход 94%). ЖХ-МС (Метод A): m/z 326,6/328,5 [M+Na]+ при 1,18 мин. 1H ЯМР (500 МГц, ДМСО-d6) δ 9,27 (d, J=7,2 Гц, 1H), 7,44 (d, J=7,2 Гц, 1H), 4,33 (q, J=7,1 Гц, 2Н), 1,32 (t, J=7,1 Гц,3Н).
24А Этил-2-бром-5-морфолин-4-илпиразоло[1,5-а]пиримидин-3-карбоксилат
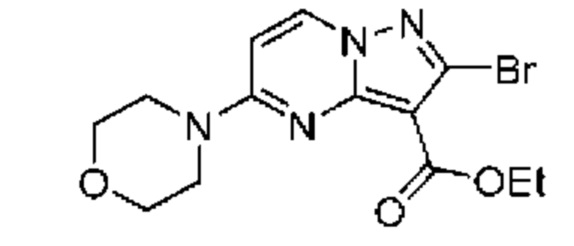
Раствор промежуточного соединения 23А (200 мг, 0,657 ммоль), морфолина (75 мкл, 0,857 ммоль) и ДИПЭА(232 мкл, 1,306 ммоль) в ДМФА(3,28 мл) нагревали в герметичной пробирке при 60°С в течение 7 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли ЕЮ Ас (30 мл) и промывали насыщенным водным раствором NH4Cl (3×10 мл). Водные смывы нейтрализовали насыщенным водным раствором NaHCO3 (~20 мл) и экстрагировали EtOAc (3×25 мл). Объединенные органические слои последовательно промывали водой (3×30 мл) и солевым раствором (30 мл), сушили над Na2SO4, и удаляли растворитель при пониженном давлении. Очистка методом колоночной хроматографии (35-100% EtOAc в гептане) давала твердое вещество белого цвета (194 мг, выход 83%). МСНР (APCI+) m/z 354,9/356,9 [М+Н]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,70 (d, J=7,9 Гц, 1H), 6,88 (A, J=1,9 Гц, 1H), 4,19 (t, J=7,1 Гц, 2Н), 3,80-3,67 (m, 8Н), 1,29 (t, J=7,1 Гц, 3Н).
24В Этил-2-бром-5-пирролидин-1-илпиразоло[1,5-а]пиримидин-3-карбоксилат
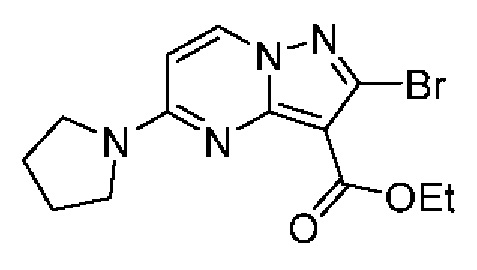
Раствор промежуточного соединения 23А (300 мг, 0,985 ммоль), пирролидина (107 мкл, 1,286 ммоль) и ДИПЭА (232 мкл, 1,306 ммоль) в ДМФА (4,23 мл) нагревали в закрытом флаконе при 60°С в течение 65 часов. Реакцию охлаждали до к.т. и гасили водой. Полученный осадок отделяли фильтрованием, промывали водой, затем растворяли в EtOAc, промывали водой (2×15 мл) и солевым раствором (15 мл), сушили над Na2SO4, и удаляли растворитель при пониженном давлении. Очистка методом колоночной хроматографии (33-70% EtOAc в гептане) давала твердое вещество белого цвета (280 мг, выход 84%). МСНР (APCI+) m/z 339,0/341,0 [М+Н]+. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,62 (d, J=7,8 Гц, 1H), 6,52 (d, J=7,7 Гц, 1H), 4,19 (q, J=7,1 Гц, 2Н), 3,59 (t, J=6,7 Гц, 2Н), 3,49 (t, J=6,7 Гц, 2Н), 2,07-1,87 (m, 4Н), 1,30 (t, J=7,1 Гц, 3Н).
24С Этил-2-бром-5-[(1R,4R-5-метил-2,5-диазабицикло[2.2.1]гептан-2-ил]пиразоло[1,5-а]пиримидин-3-карбоксилат
Раствор промежуточного соединения 23А (519 мг, 1,551 ммоль), (2R,5R)-1,2,5-триметилпиперазин-1,4-диий дихлорида (390 мг, 1,939 ммоль) и ДИПЭА (1 мл, 5,741 ммоль) в ДМСО (6,5 мл) нагревали при 70°С в течение 66 часов. Реакционную смесь охлаждали до к.т., разбавляли солевым раствором (25 мл), экстрагировали EtOAc (3×25 мл), и удаляли растворитель при пониженном давлении. Остаток растворяли в МеОН (5 мл) и очищали методом ионообменной хроматографии (2 г SCX-2). Водную фракцию также очищали методом ионообменной хроматографии (2 г SCX-2). Объединенные растворы концентрировали при пониженном давлении, в результате чего получали твердое вещество коричневого цвета (444 мг, выход 59%). ЖХ-МС (Метод A) m/z 380,3/382,6 [M+Na]+ при 0,59 мин. 1H ЯМР (500 МГц, CDCl3) δ 8,15 (d, J=7,7 Гц, 1Н), 6,33-6,00 (m, 1H), 5,27-5,10 (m, 1H), 4,37 (q, J=7,1 Гц, 2H), 3,65 (s, 1H), 3,50-3,34 (m, 1H), 3,21-2,76 (m, 2H), 2,61 (s, 1H), 2,49 (s, 3H), 2,15-2,00 (m, 1H), 1,94-1,78 (m, 1H), 1,41 (t, J=7,1 Гц, 3H).
25A Этил-2-бром- 7-метилпиразоло[1,5-а]пиримидин-3-карбоксилат
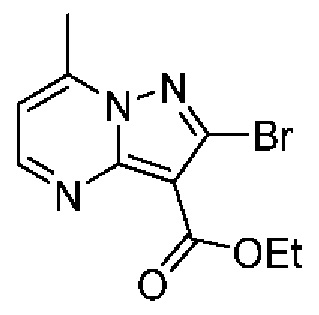
В реакционную емкость загружали этил-5-амино-3-бром-1Н-пиразол-4-карбоксилат (700 мг, 2,990 ммоль), 4,4-диметоксибутан-2-он (0,48 мл, 3,590 ммоль), 70%-ный этанол (9,00 мл) и концентрированную HCl (0,25 мл, 2,99 ммоль), и полученную смесь нагревали посредством МВИ при 140°С в течение 30 мин. Реакционную смесь выливали в насыщенный водный раствор NaHCO3 (100 мл), перемешивали в течение 15 мин, и полученный осадок отделяли фильтрованием, промывая водой. Полученное твердое вещество очищали методом колоночной хроматографии (0-20% CH2Cl:EtOH:NH3 [50:8:1] в CH2Cl2), в результате чего получали твердое вещество белого цвета (568 мг, выход 69%). МСНР m/z [М+Н]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,74 (d, J=4,4 Гц, 1Н), 7,28 (dd, J=4,4, 1,0 Гц, 1Н), 4,33 (q, J=7,1 Гц, 2Н), 2,75 (d, J=0,8 Гз, 3Н), 1,33 (t, J=7,1 Гц, 3Н).
26А Этил-2-(2-фторфенил)-6-метилпиразоло[1,5-а]пиримидин-3-карбоксилат
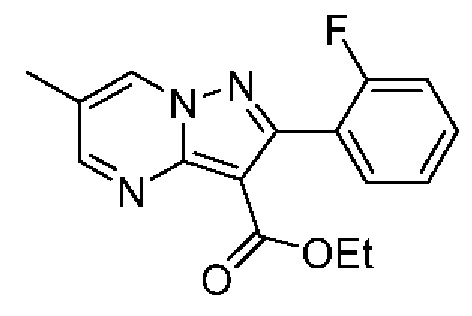
Раствор промежуточного соединения 21А (264 мг, 0,929 ммоль), 2-фторфенилбороновой кислоты (195 мг, 1,394 ммоль) и K2CO3 (389 мг, 2,813 ммоль) в смеси 1,4-диоксанаи воды (2:1, 2,79 мл) барботировали N2 в течение ~10 минут. Затем добавляли Pd(PPh3)4 (149 мг, 0,139 ммоль), и реакционную смесь нагревали при 100°С в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли CH2Cl2 и фильтровали через слой Celite® на фильтровальной бумаге из стекловолокна, промывая CH2Cl2. Растворитель удаляли при пониженном давлении, а полученный остаток очищали методом колоночной хроматографии (30-70% EtOAc в гептане), в результате чего получали твердое вещество бледно-желтого цвета (222 мг, выход 80%). МСНР (APCI+) m/z 254,1 [М-ОСН2СН3]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,17 (dd, J=2,1, 1,1 Гц, 1Н), 8,77 (d, J=2,2 Гц, 1H), 7,61-7,51 (m, 2Н), 7,36-7,29 (m, 2Н), 4,15 (q, J=7,1 Гц, 2Н), 2,40 (d, J=1,1 Гц, 3Н), 1,11 (t, J=7,1 Гц, 3Н).
Приведенные далее промежуточные соединения получали тем же способом, что и промежуточное соединение 26А.
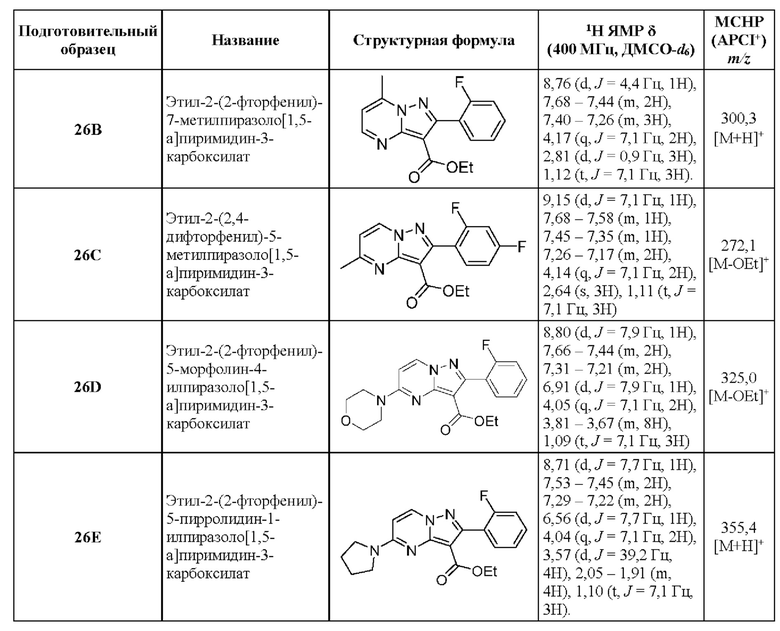
27А Этил-2-(5-хлорпиридин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксилат
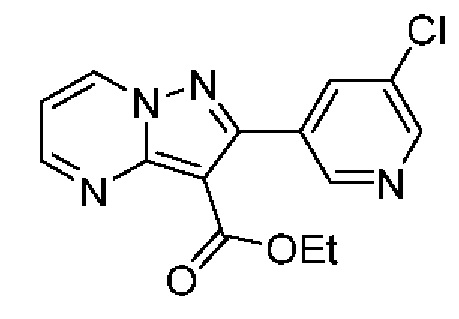
В реакционный сосуд загружали промежуточное соединение 2А (502 мг, 1,86 ммоль), (5-хлорпиридин-3-ил)бороновую кислоту (583 мг, 3,70 ммоль), K3PO4 (621 мг, 2,92 ммоль), XPhos (63 мг, 0,133 ммоль) и (кротил)Pd(XPhos)Cl (Pd-170; 122 мг, 0,180 ммоль), после чего смесь барботировали N2 в течение 5 минут. Затем добавляли ТГФ (9 мл) и воду (3 мл), реакционную смесь снова барботировали N2 в течение 5 минут и затем нагревали при 65°С в течение 3 часов. После этого реакционную смесь охлаждали до комнатной температуры, разбавляли CH2Cl2 (50 мл) и промывали солевым раствором (50 мл). Водный слой экстрагировали CH2Cl2 (2×50 мл), и объединенные органические слои сушили над Na2SO4, и удаляли растворитель при пониженном давлении. Очистка посредством колоночной хроматографии (0-3,5% МеОН в CH2Cl2) давала твердое вещество желтого цвета (409 мг, выход 70%). ЖХ-МС (Метод A) m/z 303,2 [М+Н]+ (ESI+) при 1,06 мин. 1H ЯМР (500 МГц, ДМСО-d6) 9,35 (dd, J=7,0, 1,8 Гц, 1H), 8,90 (dd, J=4,2, 1,8 Гц, 1H), 8,86 (d, J=1,8 Гц, 1H), 8,76 (d, J=2,4 Гц, 1H), 8,32 (t, J=2,1 Гц, 1H), 7,38 (dd, J=7,0, 4,2 Гц, 1H), 4,25 (q, J=7,1 Гц, 2Н), 1,22 (t, J=7,1 Гц, 3Н).
Приведенные далее промежуточные соединения получали тем же способом, что и промежуточное соединение 27А. Подготовительный образец соединения 27G получали из соответствующего пинаколового эфира бороновой кислоты.

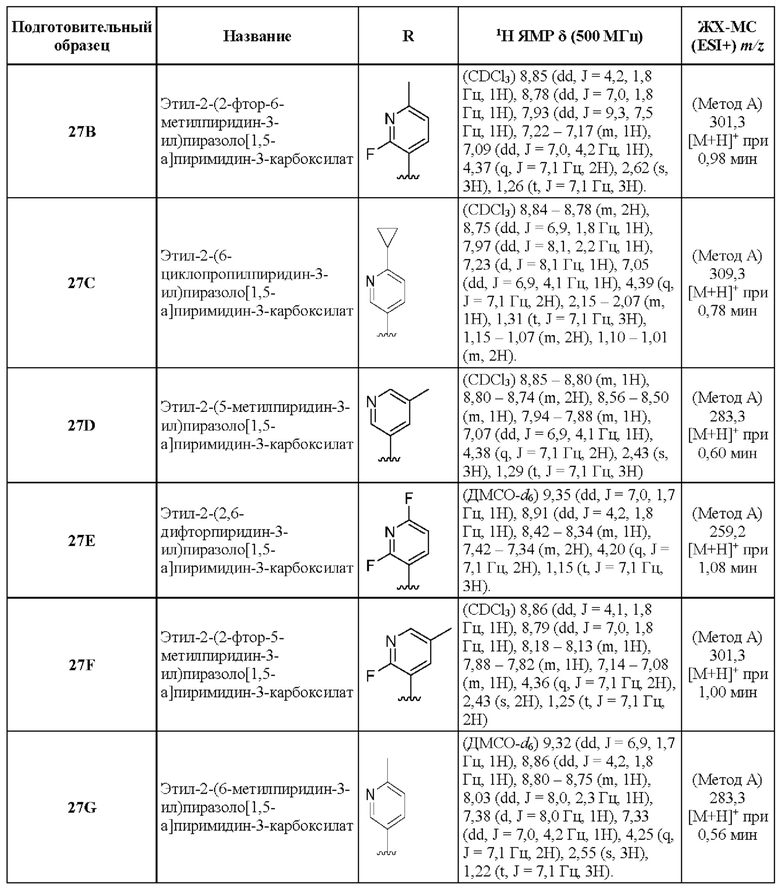
28А Этил-2-(6-метоксипиридин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксилат
К суспензии промежуточного соединения 2А (170 мг, 0,504 ммоль), (6-метоксипиридин-3-ил)бороновой кислоты (154 мг, 1,008 ммоль) и CsF (348 мг, 2,266 ммоль) в смеси 1,4-диоксана и воды (4:1; 5 мл) добавляли XPhos Pd G2 (59 мг, 0,076 ммоль), полученную реакционную смесь барботировали N2 и затем нагревали при 100°С в течение ночи. Реакционную смесь охлаждали до к.т., разбавляли водой и EtOAc (по 10 мл каждого), и отделенный водный слой экстрагировали EtOAc (3×10 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4, и удаляли растворитель при пониженном давлении. Очистка методом колоночной хроматографии (0-100% EtOAc в гексане) давала твердое вещество желтого цвета (149 мг, выход 99%). ЖХ-МС (Метод A) m/z 299,3 [М+Н]+ (ESI+) при 1,00 мин. 1H ЯМР (500 МГц, ДМСО-d6) 9,30 (dd, J=6,9, 1,8 Гц, 1H), 8,85 (dd, J=4,2, 1,8 Гц, 1H), 8,54 (dd, J=2,4, 0,8 Гц, 1H), 8,07 (dd, J=8,6, 2,5 Гц, 1H), 7,32 (dd, J=6,9, 4,2 Гц, 1H), 6,94 (dd, J=8,6, 0,8 Гц, 1H), 4,26 (q, J=7,1 Гц, 2Н), 3,93 (s, 3Н), 1,24 (t, J=7,1 Гц, 3Н).
Приведенные далее промежуточные соединения получали тем же способом, что и промежуточное соединение 28А. Подготовительные образцы 29С и 29D получали из соответствующего пинаколового эфира бороновой кислоты.
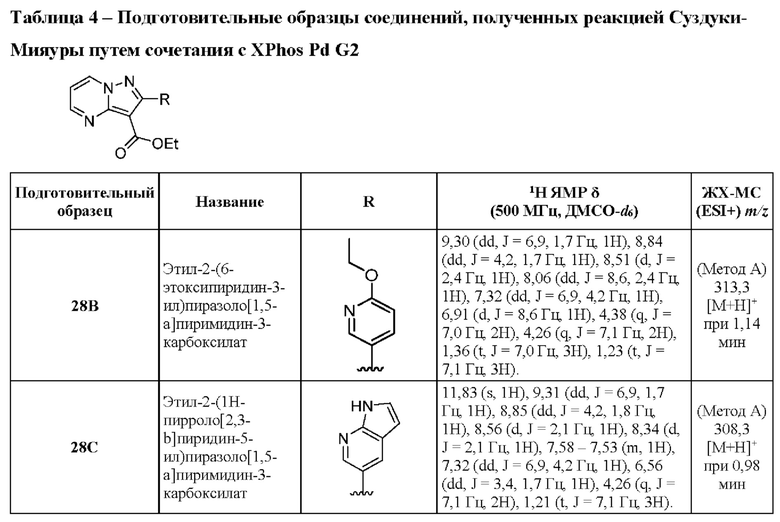
28Е Этил-2-(2,6-дифторфенил)пиразоло[1,5-а]пиримидин-3-карбоксилат
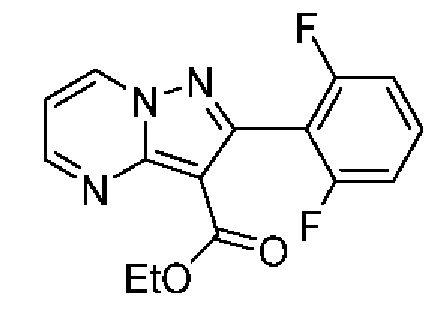
Получен по той же методике, которая была описана для получения промежуточного соединения 28А из промежуточного соединения 2А (76 мг, 0,276 ммоль) и 2,6-дифторфенилбороновой кислоты (440 мг, 2,786 ммоль) с использованием Xphos Pd G2 (57 мг, 0,072 ммоль) и CsF (169 мг, 1,272 ммоль) в смеси 1,4-диоксана и воды (3:1; 4 мл). Представляет собой твердое вещество белого цвета (74 мг, 0,244 ммоль). ЖХ-МС (Метод A) m/z 283,0 [М+Н]+ (ESI+) при 0,49 мин. 1Н ЯМР (500 МГц, ДМСО-d6) 9,35 (dd, J=7,0, 1,7 Гц, 1H), 8,91 (dd, J=4,2, 1,8 Гц, 1H), 7,68-7,58 (m, 1H), 7,38 (dd, J=7,0, 4,2 Гц, 1H), 7,30-7,22 (m, 2Н), 4,15 (q, J=7,1 Гц, 2Н), 1,09 (t, J=7,1 Гц, 3Н).
29А Этил-2-(2-метилпиридин-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксилат
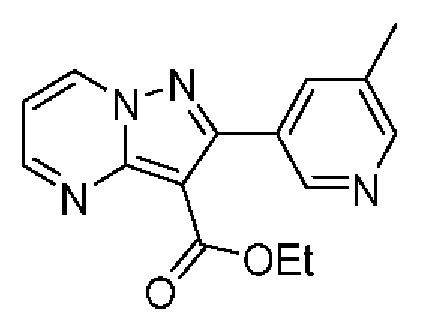
К суспензии промежуточного соединения 2А (170 мг, 0,504 ммоль), 2-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (221 мг, 1,00 ммоль) и CsF (347 мг, 2,266 ммоль) в смеси 1,4-диоксана и воды (4:1; 5 мл) добавляли XPhos Pd G2 (59 мг, 0,076 ммоль), полученную реакционную смесь барботировали N2 и нагревали при 70°С в течение ночи. Реакционную смесь охлаждали до комнатной температуры, повторно обрабатывали XPhos Pd G2 (59 мг, 0,076 ммоль), 2-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридином (221 мг, 1,01 ммоль) и CsF (347 мг, 2,266 ммоль), и реакционную смесь нагревали до 70°С в течение ночи. Процедура выделения и очистки, аналогичная описанной для промежуточного соединения 28А, давала твердое вещество желтого цвета (75 мг, выход 53%). ЖХ- МС (Метод A) m/z 283,0 [М+Н]+ (ESI+) при 0,49 мин. 1Н ЯМР (500 МГц, ДМСО-d6) 9,32 (dd, J=7,0, 1,7 Гц, 1Н), 8,87 (dd, J=4,2, 1,7 Гц, 1H), 8,56 (d, J=5,1 Гц, 1Н), 7,61-7,57 (m, 1H), 7,52 (dd, J=5,1, 1,7 Гц, 1H), 7,36 (dd, J=7,0, 4,2 Гц, 1H), 4,25 (q, J=7,1 Гц, 2Н), 2,55 (s, 3Н), 1,21 (t, J=7,1 Гц, 3Н).
30А Этил-2-(6-фторпиридин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксилат
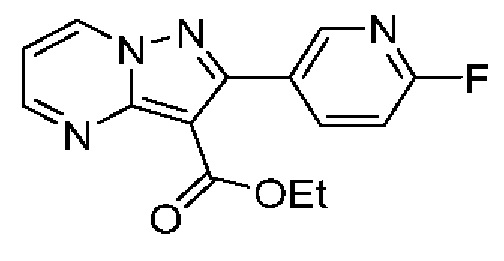
К суспензии промежуточного соединения 2А (1 г, 3,59 ммоль), 2-фторпиридин-5-бороновой кислоты (607 мг, 4,31 ммоль) и K3PO4 (1,53 г, 7,21 ммоль) в смеси 1,4-диоксана и воды (4:1; 10 мл) добавляли XPhos Pd G2 (198 мг, 0,25 ммоль), реакционную смесь барботировали N2 и нагревали при 100°С в течение 4 часов. Процедура выделения и очистки, аналогичная описанной для промежуточного соединения 28А, давала твердое вещество желтого цвета (850 мг, выход 83%). ЖХ-МС (Метод A) m/z 287,4 [М+Н]+ (ESI+) при 0,96 мин. 1Н ЯМР (500 МГц, ДМСО-d6) 9,36-9,31 (m, 1H), 8,89 (dd, J=4,2, 1,8 Гц, 1H), 8,58 (d, J=2,5 Гц, 1H), 8,35 (td, J=8,2, 2,5 Гц, 1H), 7,39-7,31 (m, 2Н), 4,25 (q, J=7,1 Гц, 2Н), 1,22 (t, J=7,1 Гц, 3Н).
31А Этил-2-(6-этилпиридин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксилат
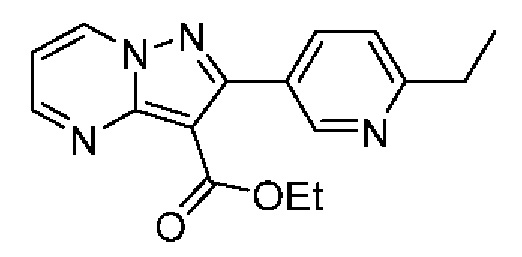
К суспензии бис(пинаколато)дибора (600 мг, 2,36 ммоль) и 5-бром-2-этилпиридина (400 мг, 2,15 ммоль) в диоксане (3,3 мл) добавляли KOAc (633 мг, 6,45 ммоль). Смесь барботировали пропусканием N2, добавляли Pd(dppf)Cl2 (118 мг, 0,161 ммоль) и нагревали при 100°С в течение 1 часа. Полученную реакционную смесь охлаждали до комнатной температуры, разбавляли EtOAc (30 мл) и фильтровали через целит, промывая EtOAc (40 мл). Фильтрат концентрировали при пониженном давлении, в результате чего получали остаток в виде маслянистой жидкости красного цвета (501 мг), который непосредственно направляли на следующую стадию синтеза. Часть неочищенного остатка (251 мг, 1,08 ммоль) суспендировали в смеси 1,4-диоксана и воды (4:1; 5 мл), к ней добавляли промежуточное соединение 2А (170 мг, 0,504 ммоль) и CsF (347 мг, 2,266 ммоль) и затем XPhos Pd G2 (59 мг, 0,076 ммоль). Реакционную смесь барботировали пропусканием N2 в течение 5 минут, затем нагревали при 100°С в течение 2 часов. Процедура выделения и очистки, аналогичная описанной для промежуточного соединения 28А, давала твердое вещество оранжевого цвета (112 мг, выход 73%). ЖХ-МС (Метод A) m/z 291,4 [М+Н]+ (ESI+) при 0,66 мин. 1H ЯМР (500 МГц, ДМСО-d6) 9,32 (dd, J=6,9, 1,8 Гц, 1H), 8,86 (dd, J=4,2, 1,8 Гц, 1H), 8,79 (dd, J=2,3, 0,9 Гц, 1H), 8,05 (dd, J=8,0, 2,3 Гц, 1H), 7,39 (d, J=8,0 Гц, 1H), 7,33 (dd, J=6,9, 4,2 Гц, 1H), 4,25 (q, J=7,1 Гц, 2Н), 2,83 (q, J=7,6 Гц, 2Н), 1,28 (t, J=7,6 Гц, 3Н), 1,22 (t, J=7,1 Гц, 3Н).
Следующие промежуточные соединения получали аналогично промежуточному соединению 31А. Подготовительные образцы соединений 31F и 31G подвергали дополнительной очистке методом ионообменной хроматографии (SCX-2). Реакционную смесь при получении соединения 31G нагревали при 70°С.
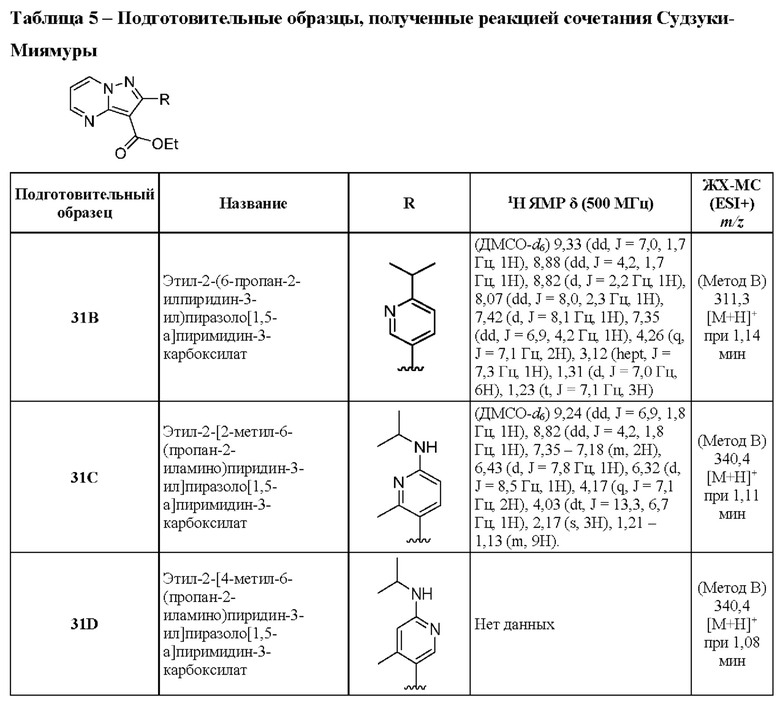
32А Этил-2-(5-циклопропилпиридин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксилат
К раствору промежуточного соединения 27А (95 мг, 0,313 ммоль) в толуоле (1,7 мл) и воде (0,2 мл) в атмосфере N2 добавляли циклопропилтрифторборат калия (143 мг, 0,964 ммоль), K2CO3 (187 мг, 1,353 ммоль), 2-дициклогексилфосфино-2',6'-диизопропоксибифенил (RuPhos; 29 мг, 0,063 ммоль) и Pd(OAc)2 (7,1 мг, 0,031 ммоль), после чего реакционную смесь нагревали посредством МВИ при 130°С в течение 2 часов. Затем реакционную смесь объединяли со второй серией, полученной с использованием 49 мг 27А (т.е. всего было взято 144 мг, 0,476 ммоль). Объединенные реакционные смеси разбавляли CH2Cl2 (25 мл) и промывали солевым раствором (25 мл). Водный слой экстрагировали CH2Cl2 (2×25 мл), объединенные органические экстракты сушили над Na2SO4, и удаляли растворитель при пониженном давлении. Очистка методом колоночной хроматографии (0-3% МеОН в CH2Cl2) давала маслянистую жидкость янтарно-желтого цвета (104 мг, совокупный выход 71%). ЖХ-МС (Метод A) m/z 309,3 [М+Н]+ (ESI+) при 0,80 мин. 1Н ЯМР (500 МГц, CDCb) 8,88-8,83 (m, 1H), 8,83-8,76 (m, 2Н), 8,55-8,50 (m, 1H), 7,86-7,81 (m, 1H), 7,10 (dd, J=7,0, 4,1 Гц, 1H), 4,41 (q, J=7,1 Гц, 2H), 2,07-2,01 (m, 1H), 1,33 (t, J=7,1 Гц, 3H), 1,16-1,07 (m, 2H), 0,91-0,80 (m, 2H).
33А Этил-2-[6-(пропан-2-иламино)пиридин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксилат
К раствору промежуточного соединения 30А (240 мг, 0,84 ммоль) в ДМСО (2 мл) добавляли ДИПЭА (0,18 мл, 1,04 ммоль) и изопропиламин (0,36 мл, 4,22 ммоль), и полученную смесь нагревали при 100°С в течение ночи. Реакционную смесь разделяли в системе EtOAc (10 мл) и насыщенного водного раствора NH4Cl (10 мл). Отделенный водный слой экстрагировали EtOAc (3×10 мл), и объединенные органические экстракты промывали солевым раствором (20 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Очистка методом колоночной хроматографии (0-100% EtOAc в гексане) давала твердое вещество желтого цвета (127 мг, выход 42%). ЖХ-МС (Метод В): m/z 326,4 [М+Н]+ при 1,06 мин. 1Н ЯМР (500 МГц, ДМСО-d6) 9,23 (dd, J=6,9, 1,8 Гц, 1H), 8,78 (dd, J=4,2, 1,8 Гц, 1Н), 8,38 (d, J=2,5 Гц, 1H), 7,75 (dd, J=8,7, 2,4 Гц, 1H), 7,25 (dd, J=6,9, 4,2 Гц, 1H), 6,73 (d, J=7,6 Гц, 1Н), 6,51 (dd, J=8,8, 0,8 Гц, 1H), 4,27 (q, J=7,1 Гц, 2Н), 4,14-4,02 (m, 1H), 1,26 (t, J=7,1 Гц, 3Н), 1,18 (d, J=6,4 Гц, 6Н).
Приведенные далее промежуточные соединения получали аналогично промежуточному соединению 33А. Реакционную смесь для получения промежуточного соединения 33С нагревали при 50°С в течение 3 часов.

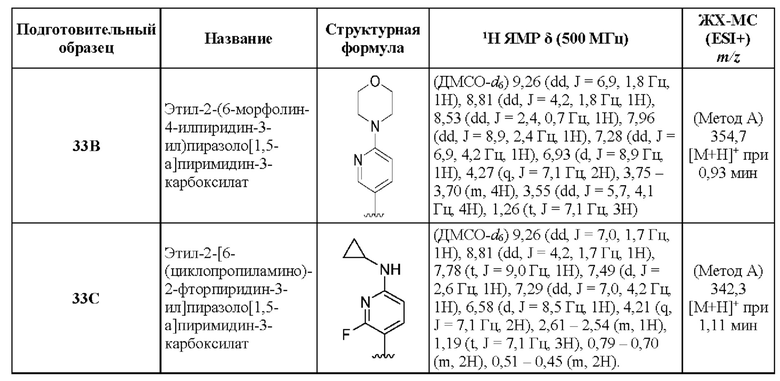
34А Этил-2-[6-(этиламино)-2-фторпиридин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксилат
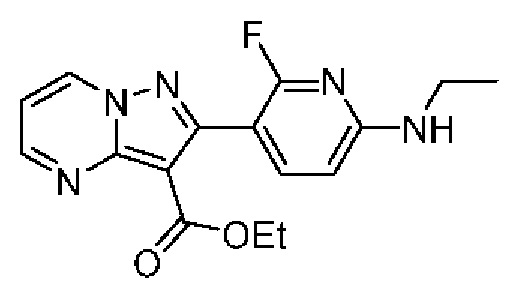
К раствору промежуточного соединения 27Е (144 мг, 0,47 ммоль) в ДМСО (2 мл) добавляли ДИПЭА (99 мкл, 0,57 ммоль) и этиламин (2М раствор в ТГФ; 583 мкл, 1,12 ммоль), и смесь нагревали посредством МВИ при 100°С в течение 30 минут. Процедура выделения, аналогичная описанной для промежуточного соединения 33А, с последующей очисткой методом колоночной хроматографии (0-2% МеОН в CH2Cl2), давала твердое вещество желтого цвета (84 мг, выход 52%). ЖХ-МС (Метод A) m/z 352,3 [M+Na]+ при 1,09 мин. 1H ЯМР (500 МГц, CDCl3) 8,80 (dd, J=4,2, 1,8 Гц, 1H), 8,75 (dd, J=7,0, 1,8 Гц, 1H), 7,84-7,78 (m, 1H), 7,04 (dd, J=7,0, 4,2 Гц, 1Н), 6,37-6,33 (m, 1H), 4,40 (q, J=7,1 Гц, 2Н), 3,40 (q, J=7,2 Гц, 2Н), 1,35-1,29 (m, 6Н).
35А Этил-2-[2-фтор-6-(пропан-2-иламино)пиридин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксилат
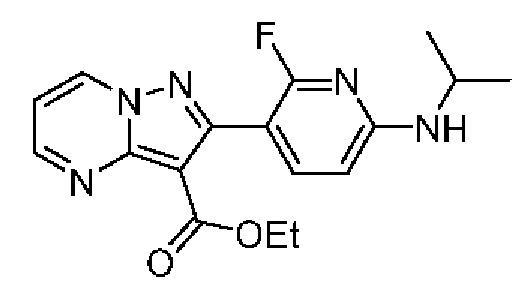
К раствору промежуточного соединения 27Е (168 мг, 0,55 ммоль) в ДМСО (2 мл) добавляли ДИПЭА (116 мкл, 0,67 ммоль) и изопропиламин (117 мкл, 1,36 ммоль), и смесь перемешивали при к.т. в течение 4 часов, после чего нагревали при 40°С в течение 1 часа. Процедура выделения, аналогичная описанной для промежуточного соединения 33А, с последующей очисткой методом колоночной хроматографии (0-3% МеОН в CH2Cl2), давала твердое вещество белого цвета (89 мг, выход 46%). ЖХ-МС (Метод A) m/z 344,2 [М+Н]+ при 1,19 мин. 1H ЯМР (500 МГц, ДМСО-d6) 9,24 (dd, J=7,0, 1,7 Гц, 1H), 8,80 (dd, J=4,2, 1,8 Гц, 1H), 7,67 (dd, J=9,9, 8,3 Гц, 1H), 7,27 (dd, J=6,9, 4,2 Гц, 1H), 7,14 (d, J=7,6 Гц, 1H), 6,43 (dd, J=8,2, 1,8 Гц, 1H), 4,20 (q, J=7,1 Гц, 2Н), 4,01-3,90 (m, 1H), 1,22-1,15 (m, 9Н).
36А Этил-2-[6-(3-метилморфолин-4-ил)пиридин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксилат
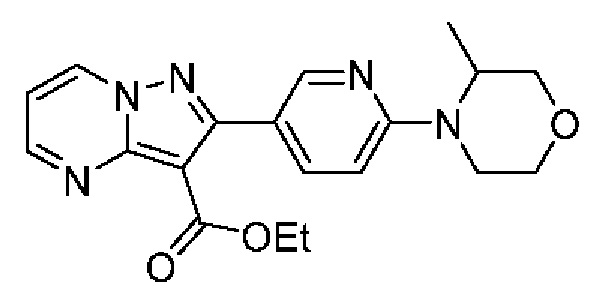
Раствор промежуточного соединения 30А (150 мг, 0,49 ммоль), 3-метил мор фол ина (39 мкл, 0,50 ммоль) и ДИПЭА (107 мкл, 0,62 ммоль) в ДМСО (2 мл) нагревали при 100°С в течение ночи. Затем добавляли 3-метилморфолин (330 мкл, 2,42 ммоль) и DIPEA (600 мкл, 3,45 ммоль), и полученную смесь нагревали при 145°С в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли EtOAc (25 мл) и промывали насыщенным водным раствором NaHCO3 (3×25 мл). Органический слой сушили над Na2SO4, растворитель удаляли при пониженном давлении, и остаток очищали методом колоночной хроматографии (0-4% МеОН в CH2Cl2), в результате чего получали коричневую маслянистую жидкость (140 мг, выход 65%). ЖХ-МС (Метод A) m/z 368,4 [М+Н]+ при 0,75 мин. 1H ЯМР (500 МГц, CDCl3) 8,81-8,77 (м, 1H), 8,77-8,74 (м, 1H), 8,70-8,66 (м, 1H), 8,04-8,00 (м, 1H), 7,07-7,01 (м, 1H), 6,71-6,67 (м, 1H), 4,49-4,41 (м, 3Н), 4,10-4,04 (м, 1H), 4,00-3,96 (м, 1H), 3,88-3,79 (м, 2Н), 3,72-3,63 (м, 1H), 3,34-3,30 (м, 1H), 1,39 (m, J=7,1 Гц, 3Н), 1,34-1,30 (м, 3Н).
37А Этил-2-(1-метилиндазол-5-ил)пиразоло[1,5-а]пиримидин-3-карбоксилат
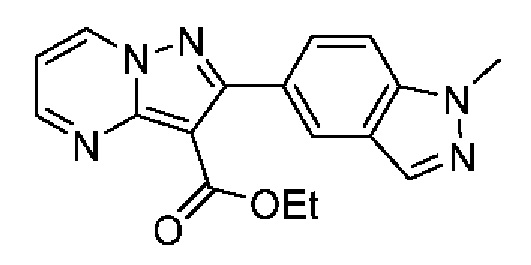
К охлажденному (до 0°С) раствору промежуточного соединения 6N (139 мг, 0,45 ммоль) в ДМФА (2 мл) добавляли NaH (60%-ную дисперсию в минеральном масле, 55 мг, 1,38 ммоль), и реакционную смесь перемешивали 30 мин при 0°С. Затем добавляли йодистый метил (34 мкл, 0,54 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь гасили водой (5 мл), разбавляли EtOAc (25 мл), и органический слой промывали солевым раствором (3×25 мл), сушили над Na2SO4, и растворитель удаляли при пониженном давлении. Очистка методом колоночной хроматографии (0-2% МеОН в CH2Cl2) давала твердое вещество белого цвета (47 мг, выход 32%). ЖХ-МС (Метод A) m/z 322,7 [М+Н]+ при 1,00 мин. 1Н ЯМР (500 МГц, CDCl3) 8,81-8,73 (m, 2Н), 8,20 (dd, J=1,6, 1,0 Гц, 1H), 8,07 (d, J=1,0 Гц, 1H), 7,83 (dd, J=8,7, 1,6 Гц, 1H), 7,48 (dt, J=8,8, 1,0 Гц, 1H), 7,03 (dd, J=6,9, 4,1 Гц, 1H), 4,39 (q, J=7,1 Гц, 2H), 4,13 (s, 3H), 1,29 (t, J=7,1 Гц, 3Н).
38A Этил-2-(2-фтор-4-пирролидин-1-илфенил)пиразоло[1,5-а]пиримидин-3-карбоксилат
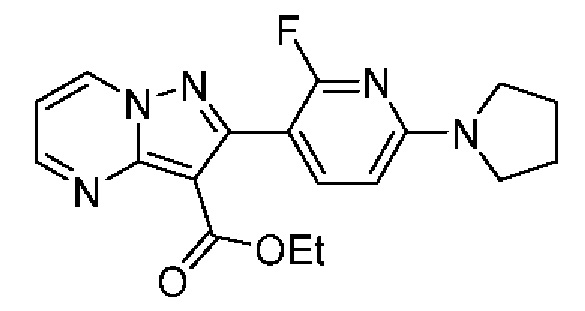
К раствору промежуточного соединения 6М (170 мг, 0,57 ммоль), K2CO3 (156 мг, 1,13 ммоль) и KI (45 мг, 1,15 ммоль) в MeCN (10 мл) добавляли 1,4-дибромбутан (67 мкл, 0,57 ммоль), и реакционную смесь нагревали до 90°С в течение ~3 суток. Летучие вещества удаляли при пониженном давлении, а остаток разделяли в двухфазной системе EtOAc (10 мл) и воды (10 мл). Отделенный водный слой экстрагировали EtOAc (3×10 мл), и объединенные органические экстракты промывали солевым раствором (20 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Очистка методом колоночной хроматографии (0-100% EtOAc в гексане) давала твердое вещество оранжевого цвета (120 мг, выход 49%). ЖХ-МС (Метод А) m/z 355,1 [М+Н]+ при 1,43 мин. 1Н ЯМР (500 МГц, ДМСО-d6) 9,24 (dd, J=6,9, 1,8 Гц, 1Н), 8,78 (dd, J=4,1, 1,8 Гц, 1H), 7,25 (dd, J=6,9, 4,2 Гц, 1Н), 6,45 (dd, J=8,6, 2,3 Гц, 1H), 6,39 (dd, J=13,8, 2,3 Гц, 1H), 4,19 (q, J=7,1 Гц, 2Н), 3,31-3,25 (m, 4Н), 2,02-1,94 (m, 4Н), 1,18 (t, J=7,1 Гц, 3Н).
39А Этил-6-этилпиридин-3-карбоксилат
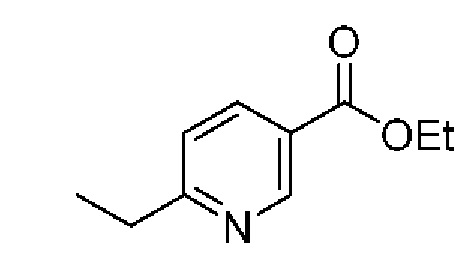
К охлажденному (до -75°С) раствору этил-6-хлорпиридин-3-карбоксилата (15,11 г, 81,41 ммоль), ацетилацетоната железа(III) (2,99 г, 8,30 ммоль) и 1-метилпирролидин-2-она (11 мл, 114,3 ммоль) в ТГФ (400 мл) по каплям добавляли EtMgBr (3,0 М раствор в Et2O; 41 мл, 123 ммоль). Смесь перемешивали при -75°С в течение 1 часа, затем давали нагреться до 0°С и гасили, добавляя по каплям воду (250 мл). Полученную смесь экстрагировали EtOAc (3×250 мл), и объединенные органические слои концентрировали при пониженном давлении. Остаток разбавляли EtOAc (250 мл), промывали солевым раствором (3×250 мл), сушили над Na2SO4, и растворитель удаляли при пониженном давлении. Очистка методом колоночной хроматографии (0-15% EtOAc в изогексанах) давала маслянистую жидкость желтого цвета (13,45 г, выход 90%). ЖХ-МС (Метод A) m/z 180,3 [М+Н]+ (ESI+) при 0,98 мин. 1Н ЯМР (500 МГц, ДМСО-d6) 8,99 (d, J=2,3 Гц, 1Н), 8,19 (dd, J=8,1, 2,3 Гц, 1H), 7,43 (d, J=8,1 Гц, 1H), 4,33 (q, J=7,1 Гц, 2Н), 2,83 (q, J=7,6 Гц, 2Н), 1,32 (t, J=7,1 Гц, 3Н), 1,24 (t, J=7,6 Гц, 3Н).
40А 6-Этилпиридин-3-карбоновая кислота
Раствор промежуточного соединения 39А (13,45 г, 73,30 ммоль) и LiOH (9,22 г, 219,67 ммоль) в системе МеОН-ТГФ-вода (1:1:1; 23 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь подкисляли (до уровня рН≈4-5) добавлением по каплям 1М водного раствора HCl и экстрагировали смесью CHCl3 и iPrOH (3:1; 3×250 мл). Объединенные органические экстракты сушили над Na2SO4, и растворитель удаляли при пониженном давлении. Водную фазу подкисляли (до уровня рН≈3-4) добавлением по каплям 1М водного раствора HCl и экстрагировали смесью CHCl3 и iPrOH (3:1; 3×250 мл). Объединенные органические слои сушили над Na2SO4, растворитель удаляли при пониженном давлении, полученный остаток объединяли с остатком от предыдущих органических экстрактов, в результате чего получали твердое вещество беловатого цвета (8,05 г, выход 71%). ЖХ-МС (Метод A) m/z 152,4 [М+Н]+ (ESI+) при 0,26 мин. 1Н ЯМР (500 МГц, ДМСО-d6) 13,23 (s, 1H), 8,98 (d, J=2,2 Гц, 1H), 8,17 (dd, J=8,1, 2,2 Гц, 1H), 7,40 (d, J=8,1 Гц, 1H), 2,83 (q, J=7,6 Гц, 2H), 1,25 (t, J=7,6 Гц, 3Н).
41А Этил-3-(5-фторпиридин-2-ил)-3-оксопропаноат
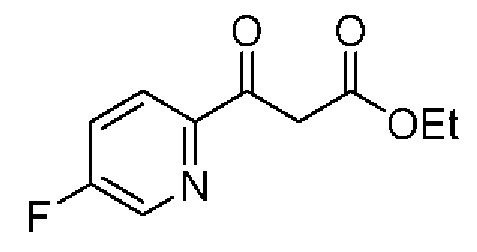
К раствору 5-фторпиридин-2-карбоновой кислоты (1 г, 7,09 ммоль) в ТГФ (10 мл) добавляли КДИ (1,21 г, 7,44 ммоль), и полученную смесь кипятили с обратным холодильником в течение 1 часа. Реакционную смесь охлаждали до к.т., добавляли калий 3-этокси-3-оксопропановую кислоту (1,33 г, 7,80 ммоль), затем MgCl2 (0,81 г, 8,50 ммоль), и полученную смесь нагревали до 60°С в течение ночи. Летучие вещества удаляли при пониженном давлении и остаток разделяли в двухфазной системе насыщенного водного раствора NH4Cl (50 мл) и EtOAc (50 мл). Отделенный водный слой экстрагировали EtOAc (50 мл), и объединенные органические экстракты сушили над Na2SO4 и концентрировали при пониженном давлении. Очистка методом колоночной хроматографии (0-50% EtOAc в изогексанах) давала бесцветную маслянистую жидкость (450 мг, выход 29%). ЖХ-МС (Метод A) m/z 166,3 [М-OCH2CH3]+ (ESI+) при 0,88 мин. 1Н ЯМР (500 МГц, ДМСО-d6) 8,73 (d, J=2,7 Гц, 1H), 8,11 (dd, J=8,7, 4,7 Гц, 1H), 7,99-7,91 (m, 1H), 4,16-4,00 (m, 4Н), 1,14 (t, J=7,1 Гц, 3Н).
42А Этил-3-(2-метилпиридин-4-ил)-3-оксопропаноат
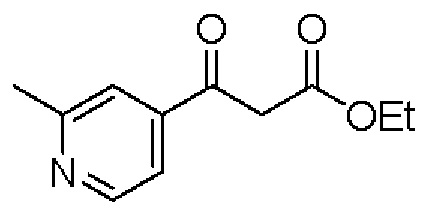
К охлажденной (до 0°С) суспензии MgCl2 (1,25 г, 13,13 ммоль) в MeCN (25 мл) добавляли калий 3-этокси-3-оксопропановую кислоту (2,05 г, 12,03 ммоль) и NEt3 (4,57 мл, 32,81 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 3 часов (флакон А). В отдельном флаконе к суспензии 2-метилпиридин-4-карбоновой кислоты (1,5 г, 10,94 ммоль) в смеси ТГФ и MeCN (1:1; 20 мл) добавляли КДИ (1,86 г, 11,48 ммоль), и полученную смесь перемешивали при 40°С в течение 3 часов (флакон В). По истечении этого времени содержимое флакона А добавляли с помощью пипетки во флакон В, и объединенную реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь выливали в ледяную воду (~10 мл), подкисляли концентрированной HCl до уровня рН≈6 и экстрагировали EtOAc (2×15 мл). Органические вещества промывали солевым раствором (50 мл), сушили над MgSO4 и концентрировали при пониженном давлении, в результате чего с получали маслянистую жидкость желтого цвета (2,05 г, выход 90%), которую использовали далее без дополнительной очистки. ЖХ-МС (Метод A) m/z 208,3 [М+Н]+ (ESI+) при 0,88 мин.
Приведенные далее промежуточные соединения получали аналогично промежуточному соединению 42А.
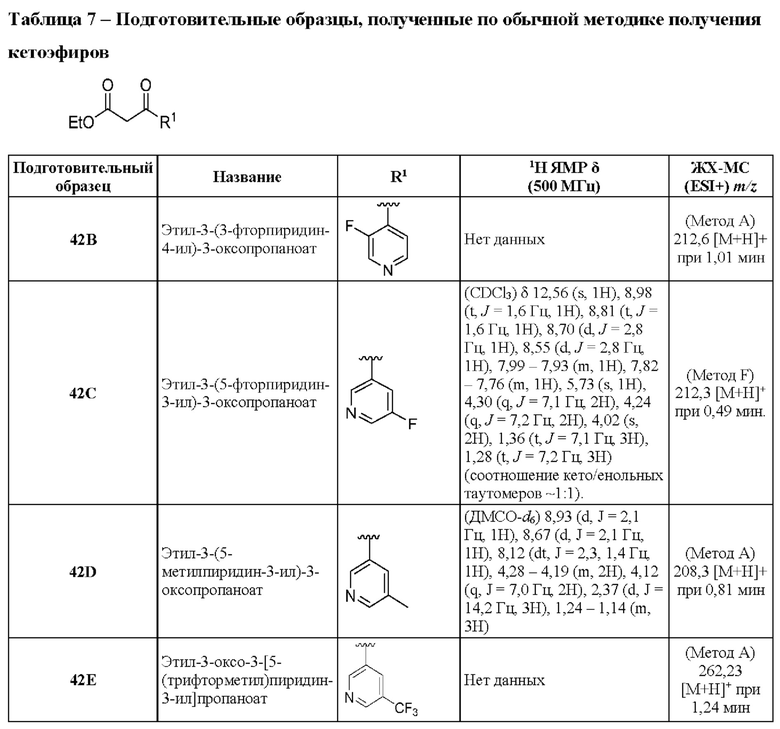
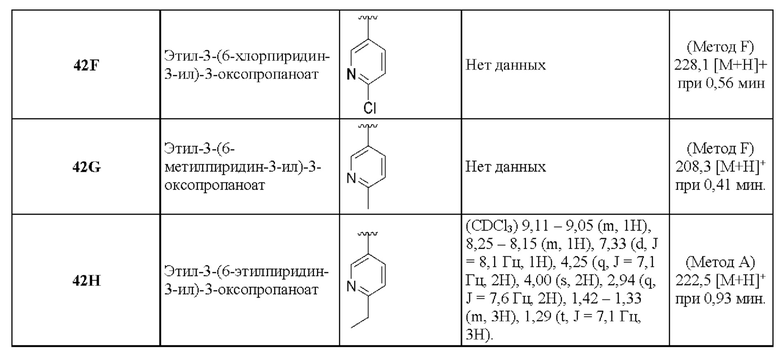
43А Этил-2-хлор-3-оксо-3-фенилпропаноат
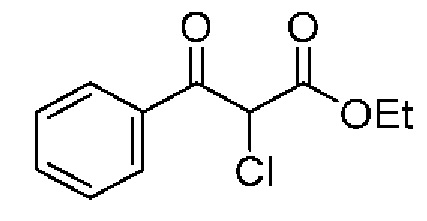
К охлажденному (до 0°С) раствору этил-3-оксо-3-фенилпропаноата (3 мл, 17,32 ммоль) в CH2Cl2 (65 мл) по каплям добавляли хлористый сульфурил (1,47 мл, 18,19 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь разделяли в двухфазной системе CH2Cl2 (30 мл) и насыщенного водного раствора NaHCO3 (50 мл). Отделенный водный слой экстрагировали CH2Cl2 (2×20 мл), а объединенные органические слои промывали солевым раствором (50 мл), пропускали через фазоразделительный картридж и концентрировали при пониженном давлении, в результате чего получали маслянистую жидкость желтого цвета (3,93 г, выход 80%). 1H ЯМР (500 МГц, CDCl3) 8,04-8,00 (m, 2Н), 7,68-7,63 (m, 1H), 7,55-7,51 (m, 2Н), 5,63 (s, 1H), 4,34-4,29 (m, 2Н), 1,27 (t, J=7,1 Гц, 3Н).
43В Этил-2-хлор-3-(2,4-дифторфенил)-3-оксопропаноат
Получали по той же методике, которая была описана для получения промежуточного соединения 43А. 1Н ЯМР (500 МГц, CDCl3) 8,07-8,00 (m, 1H), 7,08-7,03 (m, 1H), 6,96-6,91 (m, 1H), 5,60 (s, 1H), 4,32 (q, J=7,2, 0,7 Гц, 2Н), 1,30 (t, J=7,1 Гц, 3Н).
44А Этил-2-бром-3-оксо-3-пиридин-3-илпропаноат
К раствору этил-3-оксо-3- (3-пиридил)пропионата (400 мг, 2,07 ммоль) в CHCl3 (10 мл) по каплям добавляли бром (111 мкл, 2,17 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь разбавляли CH2Cl2 (20 мл), промывали водным раствором Na2S2O3 (10% масс./об.; 50 мл), насыщенным водным раствором NaHCO3 (50 мл) и солевым раствором (50 мл), пропускали через фазоразделительный картридж и концентрировали при пониженном давлении, в результате чего получали маслянистую жидкость оранжевого цвета (501 мг, выход 62%), которое использовали далее без дополнительной очистки. ЖХ-МС (Метод A) m/z 272,2/274,2 [М+Н]+ (ESI+) при 1,02 мин. 1H ЯМР (500 МГц, CDCl3) 9,23-9,19 (m, 1H), 8,86-8,80 (m, 1H), 8,32-8,28 (m, 1H), 7,47 (ddt, J=7,3, 4,7, 1,6 Гц, 2Н), 5,59 (s, 1H), 4,31 (q, J=7,2 Гц, 2Н), 1,28 (t, J=7,2 Гц, 3Н).
Приведенные далее промежуточные соединения получали аналогично промежуточному соединению 44А.
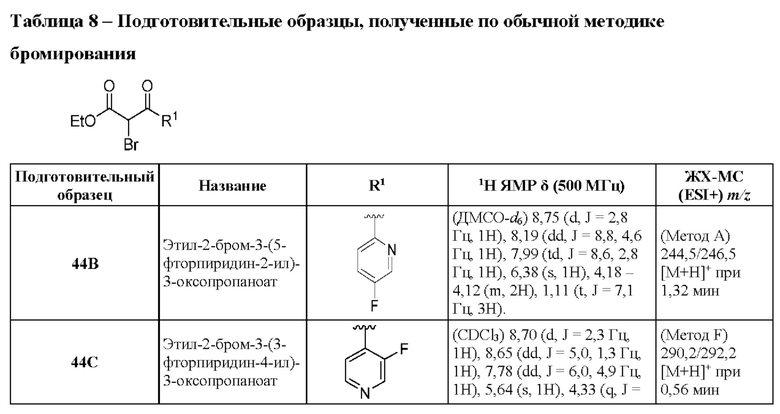
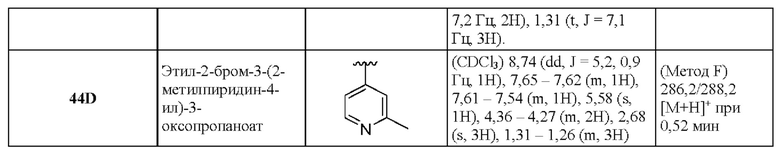
45А Этил-2-фенилимидазо[1,2-b]пиридазин-3-карбоксилат
Суспензию неочищенного промежуточного соединения 43А (0,6 г, 1,85 ммоль) и пиридазин-3-амина (0,18 г, 1,85 ммоль) в EtOH (4 мл) нагревали при 150°С в течение 12 часов с помощью МВИ. Реакционную смесь охлаждали до к.т., упаривали при пониженном давлении, и полученный остаток очищали методом колоночной хроматографии (0-100% EtOAc в изогексанах), в результате чего получали твердое вещество коричневого цвета (147 мг, выход 29%). ЖХ-МС (Метод Е) m/z 268,3 [М+Н]+ (ESI+) при 0,55 мин. 1Н ЯМР (500 МГц, CDCl3) 8,60 (dd, J=4,5, 1,7 Гц, 1H), 8,07 (dd, J=9,1, 1,7 Гц, 1H), 7,86-7,79 (m, 2H), 7,52-7,42 (m, 3Н), 7,26 (dd, J=9,1, 4,5 Гц, 1H), 4,42 (q, J=7,1 Гц, 2H), 1,29 (t, J=7,2 Гц, 3Н).
Приведенные далее промежуточные соединения получали аналогично промежуточному соединению 45А. Подготовительные образцы соединений 45G-45K получали из соответствующего неочищенного 2-бромкетоэфира с 1,3 экв. соответствующего пиридазин-3-амина.
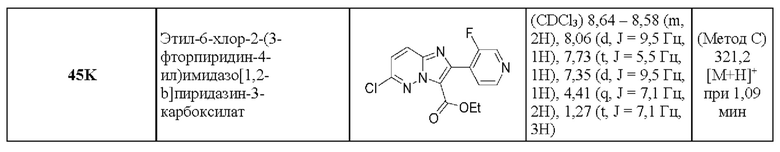
46А Этил-6-метил-2-[5-(трифторметил)пиридин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксилат
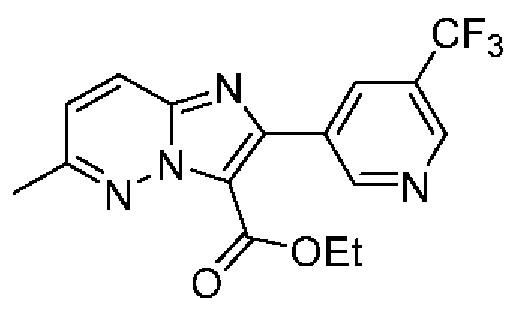
К охлажденному (до 0°С) раствору промежуточного соединения 42Е (2,7 мл, 3,46 ммоль) в CH2Cl2 (20 мл) по каплям добавляли раствор брома (186 мкл, 3,63 ммоль) в CH2Cl2 (5 мл), и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли CH2Cl2 (50 мл), промывали насыщенным водным раствором NaHCO3 (100 мл), сушили над Na2SO4 и концентрировали при пониженном давлении, в результате чего получали маслянистую жидкость оранжевого цвета (918 мг), которую непосредственно использовали в следующей реакции. Остаток растворяли в EtOH (3 мл), добавляли 6-метилпиридазин-3-амин (383 мг, 3,51 ммоль), и смесь нагревали при 150°С в течение 3 часов посредством МВИ. Реакционную смесь охлаждали до к.т., упаривали при пониженном давлении, и полученный остаток очищали методом колоночной хроматографии (0-100% EtOAc в изогексанах), в результате чего получали твердое вещество беловатого цвета (185 мг, выход 20%). ЖХ-МС (Метод A) m/z 351,3 [М+Н]+ (ESI+) при 1,28 мин. 1Н ЯМР (500 МГц, ДМСО-d6) 9,25 (d, J=2,0 Гц, 1H), 9,08-9,03 (m, 1H), 8,60-8,55 (m, 1H), 8,25 (d, J=9,3 Гц, 1H), 7,47 (d, J=9,4 Гц, 1H), 4,30 (q, J=7,1 Гц, 2Н), 2,62 (s, 3Н), 1,19 (t, J=7,1 Гц, 3Н).
Приведенные далее промежуточные соединения получали таким же способом, что и промежуточное соединение 46А, при нагревании с помощью МВИ до 140°С.
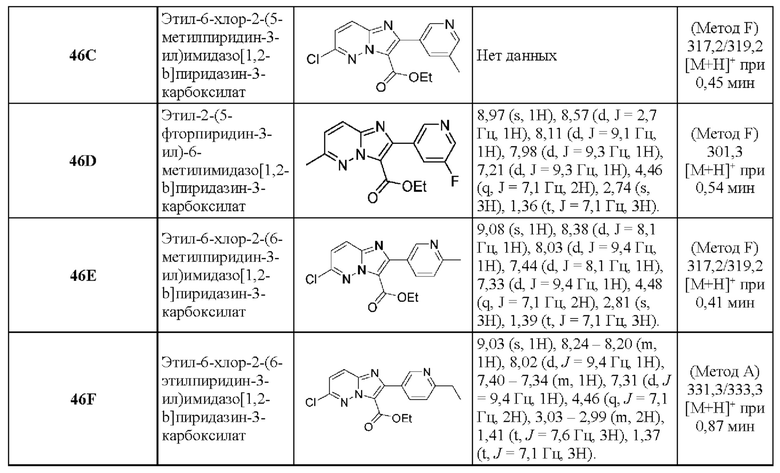
47А Этил-6-метил-2-(6-морфолин-4-илпиридин-3-ил)имидазо[1,2-b]пиридазин-3-карбоксилат
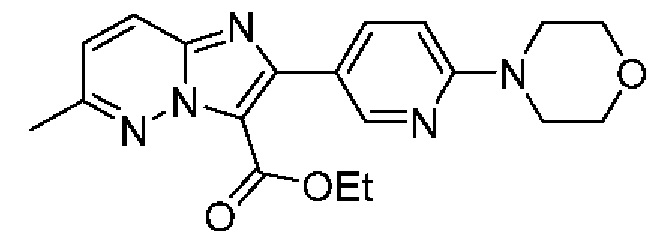
К охлажденному (до 0°С) раствору промежуточного соединения 42F (3,04 мл, 13,35 ммоль) в CH2Cl2 (40 мл) по каплям добавляли раствор брома (720 мкл, 14,02 ммоль) в CH2Cl2 (10 мл), и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли CH2Cl2 (50 мл), промывали насыщенным водным раствором NaHCO3 (100 мл) и солевым раствором (100 мл), пропускали через фазоразделительный картридж и концентрировали при пониженном давлении, в результате чего получали маслянистую жидкость желтого цвета (4,15 г), которую использовали в следующей реакции непосредственно. Неочищенный остаток растворяли в EtOH (15 мл), добавляли 6-метилпиридазин-3-амин (1,11 г, 10,15 ммоль), и полученную смесь нагревали посредством МВИ при 140°С в течение 2,5 ч. Реакционную смесь охлаждали до к.т., упаривали при пониженном давлении, и полученный остаток очищали методом колоночной хроматографии (0-10% МеОН в CH2Cl2), в результате чего получали твердое вещество желтого цвета (2,24 г), которое использовали в следующей реакции непосредственно. Неочищенный (сырой) остаток (200 мг) растворяли в ДМСО (6 мл) и обрабатывали морфолином (0,07 мл, 0,85 ммоль) и затем ДИПЭА (0,3 мл, 1,70 ммоль), после чего перемешивали в течение 18 часов при комнатной температуре. Затем добавляли еще морфолин (0,07 мл, 0,85 ммоль) и ДИПЭА (0,3 мл, 1,70 ммоль), и полученную смесь перемешивали при комнатной температуре еще в течение 4 суток. Реакционную смесь разбавляли водой (20 мл) и экстрагировали EtOAc (3×20 мл). Объединенные органические экстракты промывали солевым раствором (3×20 мл), сушили над MgSO4, и растворитель удаляли при пониженном давлении, в результате чего получали твердое вещество светло-желтого цвета (230 мг). ЖХ-МС (Метод F) m/z 368,3 [М+Н]+ (ESI+) при 0,42 мин. 1Н ЯМР (500 МГц, CDCl3) δ 8,69 (d, J=2,4 Гц, 1H), 8,02 (dd, J=8,8, 2,4 Гц, 1H), 7,91 (d, J=9,2 Гц, 1H), 7,12 (d, J=9,2 Гц, 1H), 6,72 (d, J=8,8 Гц, 1H), 4,44 (q, J=7,2 Гц, 2H), 3,87 (t, J=4,9 Гц, 4H), 3,65-3,60 (m, 4H), 2,70 (s, 3H), 1,37 (t, J=7,1 Гц, 3Н).
48A Этил-6-(азетидин-1-ил)-2-(2-фторфенил)имидазо[1,2-b]пиридазин-3-карбоксилат
Раствор промежуточного соединения 45D (72 мг, 0,230 ммоль), азетидина (50 мкл, 0,680 ммоль) и ДИПЭА (240 мкл, 1,350 ммоль) в ДМСО (3 мл) нагревали при 100°С в течение 18 часов. Реакционную смесь охлаждали до к.т., разбавляли водой (20 мл) и экстрагировали EtOAc (3×20 мл). Объединенные органические экстракты промывали солевым раствором (50 мл), пропускали через фазоразделительный картридж и концентрировали при пониженном давлении, в результате чего получали твердое вещество оранжевого цвета (68 мг, выход 85%), которое использовали далее без дополнительной очистки. ЖХ-МС (Метод Е) m/z 341,4 [М+Н]+ (ESI+) при 0,62 мин. 1H ЯМР (500 МГц, CDCl3) 7,77 (d, J=9,6 Гц, 1Н), 7,66 (td, J=7,4, 1,8 Гц, 1H), 7,44-7,36 (m, 1H), 7,25 (td, J=7,5, 1,2 Гц, 1H), 7,16-7,10 (m, 1H), 6,57 (d, J=9,6 Гц, 1H), 4,32 (q, J=7,2 Гц, 2H), 4,21 (t, J=7,5 Гц, 4H), 2,55-2,45 (m, 2H), 1,21 (t, J=7,1 Гц, 3Н).
Приведенные далее промежуточные соединения получали аналогично промежуточному соединению 48А.
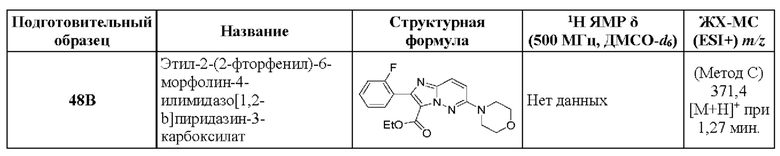
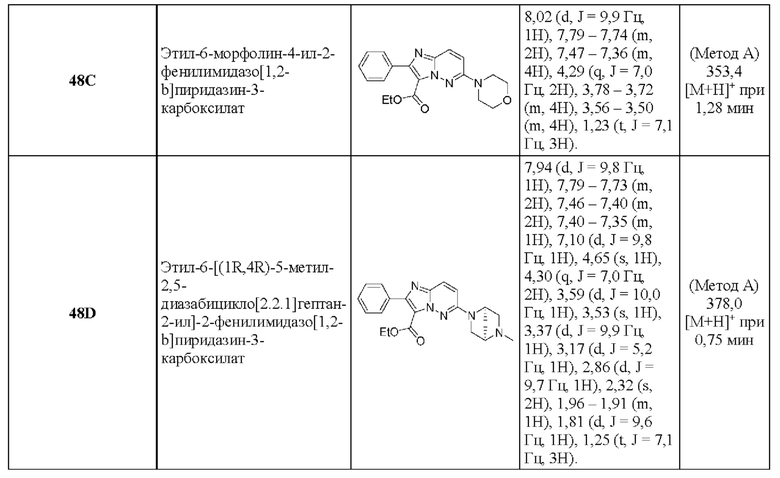
48E Этил-2-(2-фторфенил)-6-(4-метилпиперазин-1-ил)имидазо[1,2-b]пиридазин-3-карбоксилат
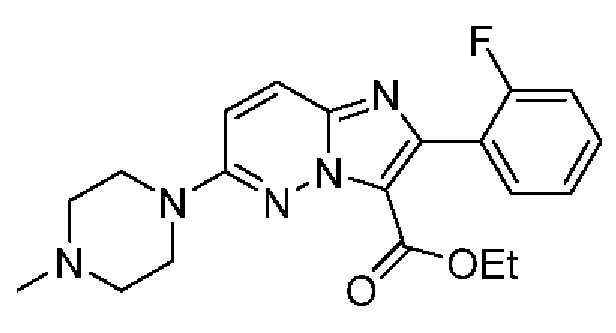
Получали по той же методике, которая была описана для получения промежуточного соединения 48А с промежуточным соединением 45D (162 мг, 0,42 ммоль), 1-метилпиперазином (240 мкл, 2,15 ммоль) и ДИПЭА (240 мкл, 1,35 ммоль) в ДМСО (5 мл). ЖХ-МС (Метод В): m/z 384,4 [М+Н]+ (ESI+) при 1,21 мин. 1Н ЯМР (500 МГц, CDCl3) 7,81 (d, J=9,9 Гц, 1H), 7,65 (td, J=7,4, 1,8 Гц, 1H), 7,43-7,38 (m, 1H), 7,25 (td, J=7,5, 1,1 Гц, 1H), 7,13 (ddd, J=9,7, 8,3, 1,2 Гц, 1H), 7,01 (d, J=9,9 Гц, 1H), 4,31 (q, J=7,2 Гц, 2H), 3,67 (t, J=5,1 Гц, 4H), 2,61-2,55 (m, 4H), 2,39 (s, 3Н), 1,20 (t, J=7,1 Гц, 3Н).
49A Этил-6-(этиламино)-2-(2-фторфенил)имидазо[1,2-b]пиридазин-3-карбоксилат
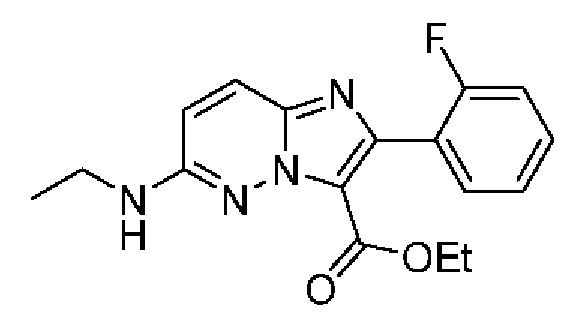
Раствор промежуточного соединения 45D (80 мг, 0,21 ммоль), этиламина (70% масс./масс. в воде; 68 мкл, 0,85 ммоль), ДИПЭА (296 мкл, 1,70 ммоль) в ДМСО (6 мл) нагревали при 100°С в течение 18 часов. Затем еще добавляли этиламин (70% масс./масс. в воде; 1 мл, 17,97 ммоль), и полученную реакционную смесь нагревали при 100°С еще в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (10 мл) и экстрагировали EtOAc (2×10 мл). Объединенные органические вещества промывали солевым раствором (20 мл), сушили над MgSO4, и растворитель удаляли при пониженном давлении, в результате чего получали твердое вещество оранжевого цвета (74 мг, выход 85%), которое далее использовали без дополнительной очистки. ЖХ-МС (Метод В): m/z 329,4 [М+Н]+ (ESI+) при 1,30 мин.
50А Этил-6-[(1R,4R-2-окса-5-азабицикло[2.2.1]гептан-5-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксилат
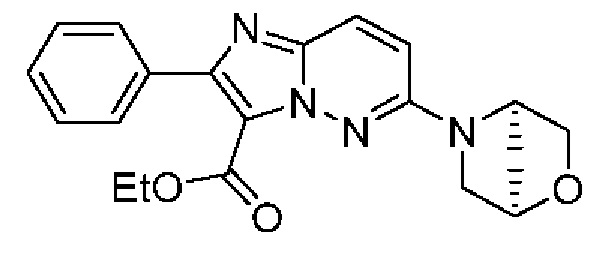
Раствор промежуточного соединения 45С (150 мг, 0,5 ммоль), (1R,4R)-2-окса-5-азониабицикло[2.2.1]гептанхлорида(81 мг, 0,6 ммоль) и ДИПЭА (296 мкл, 1,7 ммоль) в ДМСО (2 мл), нагревали при 100°С в течение ночи. Реакционную смесь охлаждали до кт, еще раз обрабатывали (1R,4R)-2-окса-5-азониабицикло[2.2.1]гептанхлоридом (40 мг, 0,3 ммоль) и ДИПЭА (113 мкл, 0,646 ммоль) и нагревали до 100°С в течение ночи. Затем реакционную смесь охлаждали до комнатной температуры, разбавляли водой (20 мл) и экстрагировали EtOAc (2×20 мл). Объединенные органические экстракты промывали солевым раствором (3×40 мл), пропускали через фазоразделительный картридж и концентрировали при пониженном давлении. Очистка методом колоночной хроматографии (0-100% EtOAc в изогексанах) давала твердое вещество беловатого цвета (86 мг, выход 46%). ЖХ-МС (Метод A) m/z 365,3 [М+Н]+ (ESI+) при 1,22 мин. 1Н ЯМР (500 МГц, ДМСО-d6) δ 7,98 (d, J=9,8 Гц, 1H), 7,76 (d, J=7,3 Гц, 2Н), 7,47-7,40 (m, 2Н), 7,40-7,35 (m, 1Н), 7,14 (d, J=9,8 Гц, 1H), 4,89 (s, 1H), 4,71 (s, 1H), 4,30 (q, J=7,1 Гц, 2Н), 3,81 (d, J=7,5 Гц, 1H), 3,75 (d, J=7,5 Гц, 1H), 3,54 (d, J=10,2 Гц, 1H), 3,40 (d, J=10,2 Гц, 1H), 1,98 (d, J=9,9 Гц, 1H), 1,92 (d, J=9,9 Гц, 1H), 1,25 (t, J=7,1 Гц, 3Н).
51А Этил-6-[(1S,4S)-5-метил-2,5-диазабицикло[2.2.1]гептан-2-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксилат
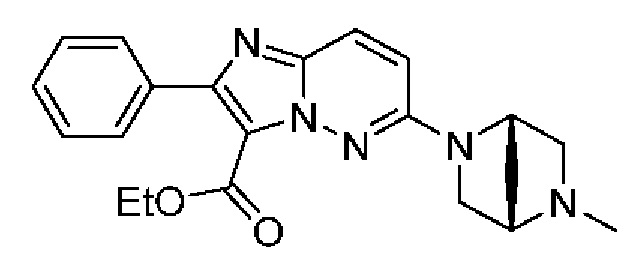
Раствор промежуточного соединения 45С (175 мг, 0,58 ммоль), (2S,5S)-1,2,5-триметилпиперазин-1,4-диий дибромида (204 мг, 0,70 ммоль) и ДИПЭА (0,61 мл, 3,48 ммоль) в ДМСО (2 мл) нагревали при 70°С в течение ночи. Реакционную смесь охлаждали до кт, дополнительно обрабатывали (1R,4R)-2-окса-5-азониабицикло[2.2.1]гептанхлоридом (200 мг, 0,690 ммоль) и ДИПЭА (0,61 мл, 3,480 ммоль) и нагревали до 70°С в течение выходных дней. Реакционную смесь охлаждали до к.т., разбавляли солевым раствором (10 мл) и экстрагировали EtOAc (4×10 мл). Объединенные органические экстракты промывали солевым раствором (10 мл), пропускали через фазоразделительный картридж и концентрировали при пониженном давлении, в результате чего получали твердое вещество почти белого цвета (143 мг, выход 65%). ЖХ-МС (Метод A) m/z 378,4 [М+Н]+ (ESI+) при 0,74 мин. 1Н ЯМР (500 МГц, ДМСО-d6) 7,95 (d, J=9,8 Гц, 1H), 7,79-7,73 (m, 2Н), 7,46-7,41 (m, 2Н), 7,41-7,35 (m, 1H), 7,10 (d, J=9,8 Гц, 1H), 4,65 (s, 1H), 4,30 (q, J=7,1 Гц, 2Н), 4,09 (q, J=5,2 Гц, 1H), 3,62-3,57 (m, 1H), 3,38 (d, J=10,1 Гц, 1H), 3,17 (d, J=5,2 Гц, 2Н), 2,87 (d, J=9,6 Гц, 1H), 2,33 (s, 2Н), 1,95-1,92 (m, 1H), 1,85-1,79 (m, 1Н), 1,25 (t, J=7,1 Гц, 3Н).
52А 2-(2-Фторфенил)-7-метилпиразоло[1,5-а]пиримидин-3-карбоновая кислота
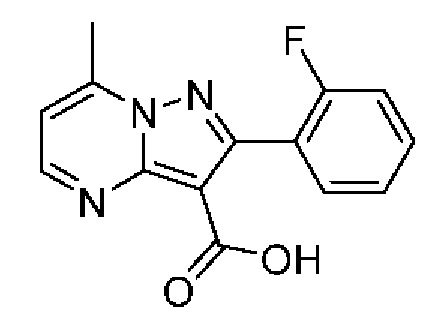
Раствор промежуточного соединения 26В (190 мг, 0,635 ммоль) и 5М водного раствора NaOH (0,635 мл, 3,174 ммоль) в смеси EtOH и воды (3:1, 4 мл) нагревали при 75°С в течение 1 часа. Реакционную смесь охлаждали до к.т., и EtOH удаляли при пониженном давлении. Остаточный водный раствор разбавляли водой (5 мл) и подкисляли (до рН≈1) 1М водным раствором НС1. Полученный осадок отделяли фильтрованием и очищали методом колоночной хроматографии (0-10% EtOH в EtOAc), в результате чего получали твердое вещество бледно-желтого цвета (128 мг, выход 74%). МСНР (APCI+) m/z 272,3 [М+Н]+
53А 2-(2,3-Дифторфенил)пиразоло[1,5-а]пиримидин-3-карбоновая кислота
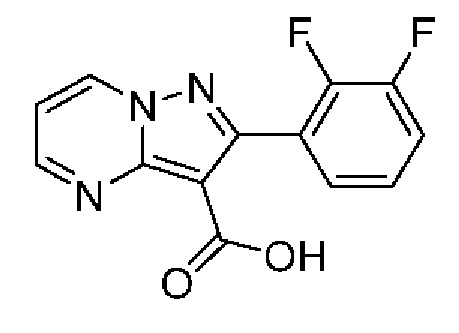
Раствор промежуточного соединения 6O (129 мг, 0,425 ммоль) и LiOH (1,5 М водный раствор; 2,83 мл, 4,25 ммоль) в смеси ТГФ и МеОН (1:1; 4 мл) перемешивали при 40°С в течение ночи. Реакционную смесь охлаждали до комнатной температуры и промывали МТБЭ (3×10 мл). Водный слой подкисляли (до рН≈1) 1М водным раствором HCl, и затем экстрагировали смесью CHCl3 и iPrOH (3:1; 3×10 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали при пониженном давлении, в результате чего получали твердое вещество оранжевого цвета (135 мг, выход 97%), которое далее использовали без дополнительной очистки. ЖХ-МС (Метод A) m/z 258,7 [М-ОСН2СН3]+ при 0,89 мин. 1H ЯМР (500 МГц, ДМСО-d6) 12,39 (s, 1H), 9,31 (dd, J=7,0, 1,8 Гц, 1H), 8,85 (dd, J=4,2, 1,8 Гц, 1H), 7,63-7,53 (m, 1H), 7,43-7,36 (m, 1H), 7,37-7,29 (m, 2Н).
Приведенные ниже промежуточные соединения получали по той же методике, которая была описана для получения промежуточного соединения 53А.
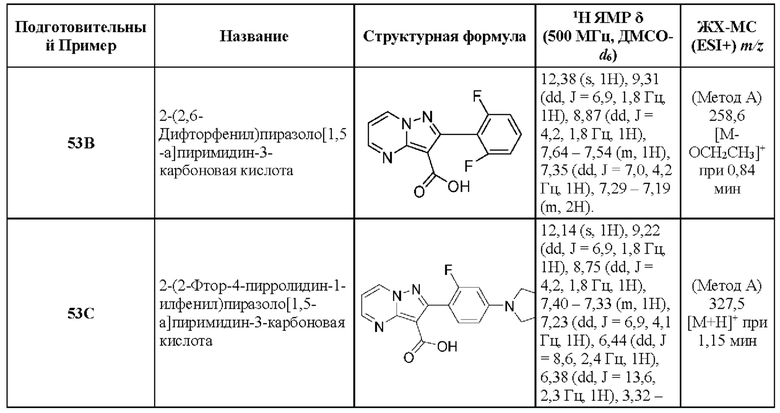

54А 2-(2-Фтор-5-метилпиридин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоновая кислота
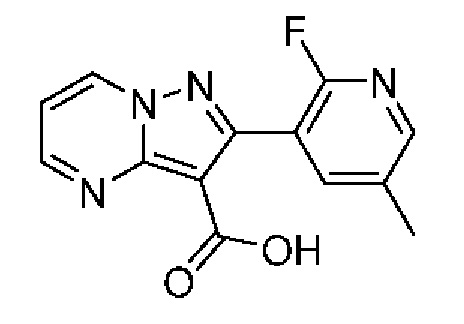
Раствор промежуточного соединения 27F (334 мг, 0,855 ммоль) и LiOH (1,5 М водный раствор; 2 мл, 3 ммоль) в ТГФ (9 мл) нагревали при 40°С в течение ночи. Затем добавляли еще LiOH (1,5 М водный раствор; 2 мл, 3 ммоль) и МеОН (2 мл), и полученную реакционную смесь нагревали при 60°С в течение 2 часов. Затем реакционную смесь охлаждали до комнатной температуры, разбавляли водой (20 мл) и промывали МТБЭ (2×25 мл). Водный слой подкисляли (до рН≈4), добавляя по каплям 1М водный раствор HCl, и экстрагировали смесью CHCl3 и iPrOH (3:1; 3×25 мл). Объединенные органические экстракты сушили над Na2SO4, растворитель удаляли при пониженном давлении, в результате чего получали твердое вещество оранжевого цвета (200 мг, выход 70%). ЖХ-МС (Метод A) m/z 273,6 [М+Н]+ при 0,74 мин. 1Н ЯМР (500 МГц, CDCl3) 12,27 (s, 1H), 9,30 (dd, J=7,0, 1,7 Гц, 1H), 8,85 (dd, J=4,1, 1,8 Гц, 1H), 8,18-8,14 (m, 1H), 7,98-7,92 (m, 1H), 7,33 (dd, J=7,0, 4,2 Гц, 1H), 2,37 (s, 3H).
55A 2-(2-Фтор-5-метилфенил)пиразоло[1,5-а]пиримидин-3-карбоновая кислота
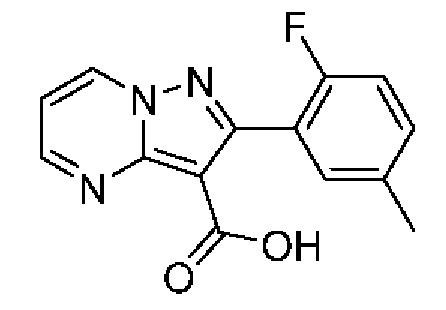
Смесь промежуточного соединения 2А (250 мг, 0,907 ммоль), (2-фтор-5-метилфенил)бороновой кислоты (172 мг, 1,117 ммоль) и K2CO3 (432 мг, 2,721 ммоль) в смеси 1,4-диоксана и воды (2:1, 3,0 мл) барботировали N2 в течение 5 мин. Затем добавляли Pd(PPh3)4 (157 мг, 0,136 ммоль), реакционную смесь дегазировали пропусканием N2 в течение 5 минут, после чего нагревали до 100°С в течение ночи. Реакционную смесь охлаждали до к.т., добавляли 2М водный раствор NaOH (3,0 мл), после чего перемешивали при комнатной температуре в течение 30 часов. Смесь концентрировали при пониженном давлении с удалением 1,4-диоксана, затем разбавляли водой (30 мл) и промывали МТБЭ (3×20 мл). Водный раствор подкисляли (до рН≈2) 1М водным раствором HCl и экстрагировали смесью CHCl3 и iPrOH (3:1; 3×50 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали при пониженном давлении, в результате чего получали твердое вещество коричневого цвета (238 мг, выход 74%). ЖХ-МС (Метод A) m/z 294,0 [M+Na]+ при 0,94 мин. 1Н ЯМР (500 МГц, ДМСО-d6) 12,26 (s, 1H), 9,28 (dd, J=7,0, 1,8 Гц, 1H), 8,83 (dd, J=4,2, 1,8 Гц, 1H), 7,35 (m, 1H), 7,31 (m, 2Н), 7,19 (m, 1H), 2,36 (s, 3Н).
56А 2-(2-Фторфенил)-6-(1-метилазетидин-3-ил)оксиимидазо[1,2-b]пиридазин-3-карбоновая кислота
К охлажденному (до 0°С) раствору гидрохлорида 1-метилазетидин-3-ола (47 мг, 0,378 ммоль) в ДМФ А (4 мл) добавляли NaH (60%-ная дисперсия в минеральном масле, 31 мг, 0,774 ммоль), и полученную смесь перемешивали в течение 15 минут. Затем добавляли раствор промежуточного соединения 45D (55 мг, 0,172 ммоль) в ДМФА (2 мл), и полученную реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли LiOH (103 мг, 4,301 ммоль), и раствор перемешивали в течение еще 1 часа. Реакционную смесь подкисляли до уровня рН~4 с помощью 1М водного раствора HCl, летучие вещества удаляли при пониженном давлении, и полученный остаток очищали методом колоночной обращенно-фазовой хроматографии (5-65% MeCN в 10 мМ водном растворе (NH4)2CO3), в результате чего получали твердое вещество беловатого цвета (55 мг, выход 93%). ЖХ-МС (Метод В): m/z 343,4 [М+Н]+ (ESI+) при 0,47 мин.
56В 2-(2-Фторфенил)-6-(2-метоксиэтокси)имидазо[1,2-b]пиридазин-3-карбоновая кислота
Получали по той же методике, которая была описана для получения промежуточного соединения 56А. ЖХ-МС (Метод В): m/z 332,3 [М+Н]+ (ESI+) при 0,49 мин.
57А 6-(1-Метилазетидин-3-ил)окси-2-фенилимидазо[1,2-b]пиридазин-3-карбоновая кислота
К охлажденному (до 0°С) раствору гидрохлорида 1-метилазетидин-3-ола (135 мг, 1,094 ммоль) в ДМФА (10 мл) добавляли NaH (60% дисперсия в минеральном масле, 89 мг, 2,237 ммоль), и полученную смесь перемешивали в течение 15 минут.Затем добавляли раствор промежуточного соединения 45С (150 мг, 0,497 ммоль) в ДМФА (2 мл), полученную реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 2 часов. Затем еще раз добавляли суспензию NaH (89 мг, 2,237 ммоль) и гидрохлорида 1-метилазетидин-3-ола (135 мг, 1,094 ммоль) в ДМФА (3 мл), которую предварительно перемешивали в отдельной колбе при комнатной температуре в течение 45 минут, и полученную реакционную смесь перемешивали в течение еще 1 часа. Затем добавляли LiOH (298 мг, 12,43 ммоль), и полученный раствор перемешивали в течение 72 часов при к.т. Реакционную смесь подкисляли до уровня рН~4 с помощью 1М водного раствора HCl, летучие вещества удаляли при пониженном давлении, и полученный остаток очищали методом колоночной обращенно-фазовой хроматографии (5-65% MeCN в 10 мМ водном растворе (NH4)2СО3), в результате чего получали твердое вещество желтого цвета (87 мг, выход 53%). ЖХ-МС (Метод В): m/z 324,6 [М+Н]+ (ESI+) при 0,51 мин.
58А 2-(6-Этилпиридин-3-ил)-6-(тридеутериометокси)имидазо[1,2-b]пиридазин-3-карбоновая кислота
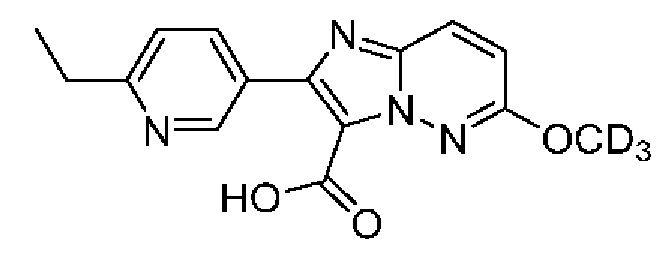
К раствору промежуточного соединения 46F (52 мг, 0,147 ммоль) в метаноле-d4 (1,2 мл, 29,5 ммоль) и воде (2 мл) добавляли LiOH (1,5 М водный раствор; 0,49 мл, 0,736 ммоль), и полученную смесь нагревали до 100°С в течение 1 часа посредством МВИ. Реакционную смесь охлаждали до комнатной температуры, подкисляли 1М водным раствором HCl до уровня рН~4, затем концентрировали при пониженном давлении, в результате чего получали твердое вещество бежево-белого цвета (40 мг, выход 86%), которое далее использовали без дополнительной очистки. ЖХ-МС (Метод A) m/z 301,7 [М+Н]+ (ESI+) при 0,20 мин.
Примеры
1. 2-[4-(Метиламино)фенил]-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид (Процедура А)
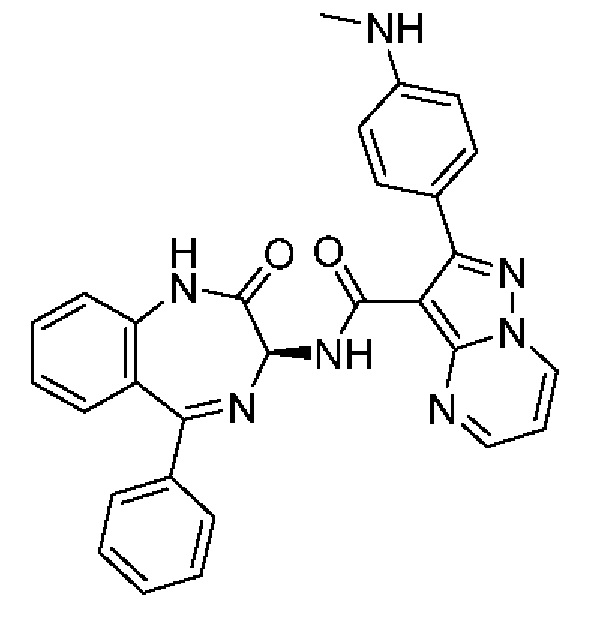
К смеси (3S)-3-амино-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-она (130 мг, 0,520 ммоль), HATU (214 мг, 0,560 ммоль) и NEt3 (0,200 мл, 1,430 ммоль) в ДМФА (3 мл) добавляли промежуточное соединение 9В (122 мг, 0,450 ммоль), и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой (15 мл), и полученный осадок отфильтровывали, промывая водой (2×15 мл). Осадок сушили под вакуумом, растворяли в CH2Cl2 и очищали методом колоночной хроматографии (0-90% EtOAc в изогексанах), в результате чего получали твердое вещество желтого цвета (95 мг, выход 40%). ЖХ-МС (Метод D) 502,29 [М+Н]+ при 3,80 мин. 1Н ЯМР (500 МГц, ДМСО-d6) 10,98 (s, 1H), 9,91 (d, J=7,8 Гц, 1H), 9,31 (dd, J=6,9, 1,7 Гц, 1H), 8,87 (dd, J=4,3, 1,7 Гц, 1H), 7,81-7,74 (m, 2Н), 7,68-7,42 (m, 6Н), 7,37-7,32 (m, 2Н), 7,31-7,24 (m, 2Н), 6,58-6,51 (m, 2Н), 5,98 (q, J=5,0 Гц, 1H), 5,52 (d, J=7,8 Гц, 1H), 2,71 (d, J=5,0 Гц, 3Н).
Приведенные далее соединения согласно настоящему изобретению были получены с (3S)-3-амино-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-оном и (3S)-3-амино-9-фтор-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-оном согласно процедуре амидного связывания (Процедуре А), описанной для получения соединения согласно Примеру 1.
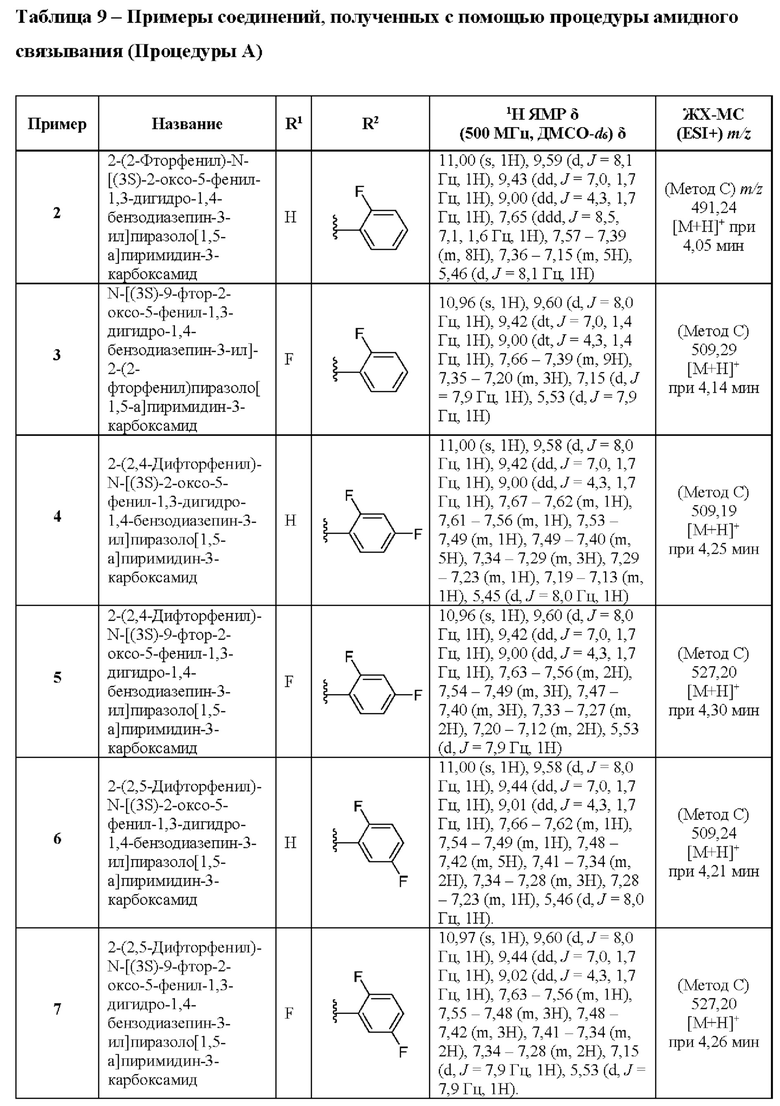
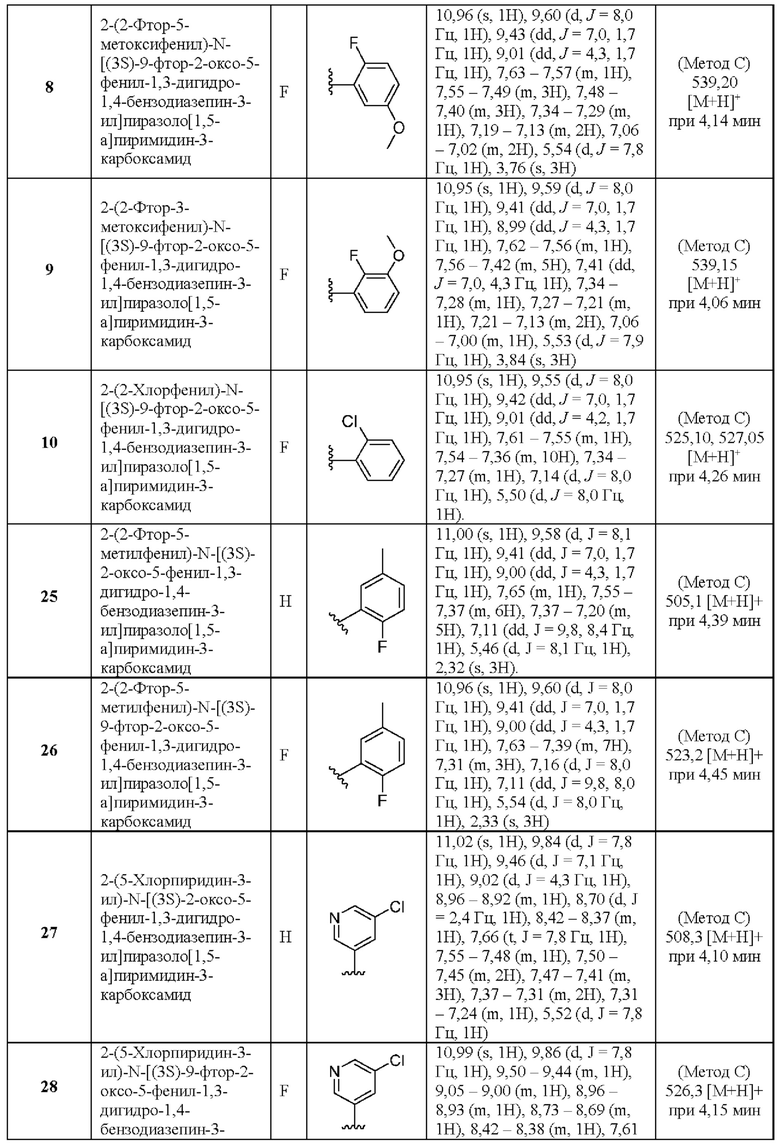
11. 2-(1-Метил-2,3-дигидроиндол-6-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид (Процедура В)
К раствору промежуточного соединения 6Е (12 мг, 0,038 ммоль) в смеси ТГФ и МеОН (1:1, 4 мл) добавляли LiOH (1,5М водный раствор, 0,447 мл, 0,671 ммоль), и полученную реакционную смесь перемешивали при 40°С в течение ночи. Затем смесь охлаждали до к.т., нейтрализовали добавлением HCl (1М водный раствор, 0,671 мл, 0,671 ммоль) и концентрировали под вакуумом. Полученный сырой продукт растворяли в ДМФА (1,5 мл), добавляли NEt3 (11 мкл, 0,076 ммоль), HATU (22 мг, 0,057 ммоль) и (3S)-3-амино-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-он (13 мг, 0,052 ммоль), и полученную реакционную смесь перемешивали при 40°С в течение 2 часов. Реакцию гасили водой (10 мл), и полученный осадок отфильтровывали, промывая водой. Осадок очищали методом колоночной хроматографии (0-5% МеОН в CH2Cl2), в результате чего получали твердое вещество желтого цвета (6 мг, выход 29%). ЖХ-МС (Метод С) 528,30 [М+Н]+ при 4,13 мин 1Н ЯМР (500 МГц, ДМСО-d6) δ 10,99 (s, 1H), 9,91 (d, J=7,8 Гц, 1H), 9,33 (dd, J=6,9, 1,7 Гц, 1Н), 8,89 (dd, J=4,3, 1,7 Гц, 1H), 7,72-7,63 (m, 3Н), 7,56-7,42 (m, 5Н), 7,38-7,23 (m, 4Н), 6,52 (d, J=8,2 Гц, 1H), 5,53 (d, J=7,8 Гц, 1Н), 3,36-3,26 (m, 2Н), 2,92 (t, J=8,3 Гц, 2Н), 2,76 (s, 3Н).
Приведенные далее соединения согласно настоящему изобретению были получены согласно процедуре амидного связывания (Процедура В), описанной для получения соединения согласно Примеру 11.
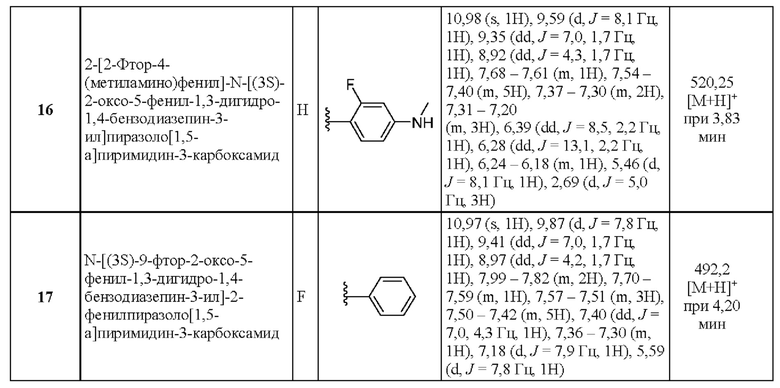
18. N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)имидазо[1,2-b]пиридазин-3-карбоксамид
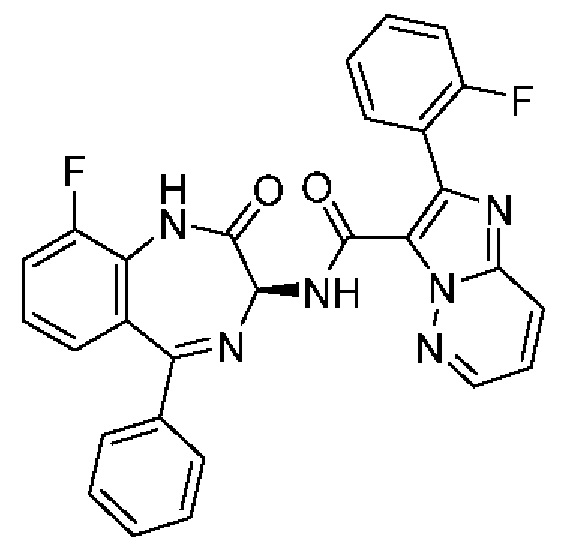
Получали с помощью процедуры амидного связывания (Процедуры В), описанной для получения соединения согласно Примеру 11 из промежуточного соединения 14А. ЖХ-МС (Метод С) m/z 509,1 [М+Н]+ при 3,88 мин. 1Н ЯМР (500 МГц, ДМСО-d6) δ 11,01 (s, 1H), 10,18 (d, J=7,6 Гц, 1Н), 8,98 (dd, J=4,6, 1,6 Гц, 1H), 8,45 (dd, J=9,2, 1,6 Гц, 1Н), 7,63-7,49 (m, 6Н), 7,49-7,42 (m, 3Н), 7,36-7,18 (m, 3Н), 7,16 (d, J=7,9 Гц, 1H), 5,57 (d, J=7,5 Гц, 1Н).
19. N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-метилимидазо[1,2-b]пиридазин-3-карбоксамид
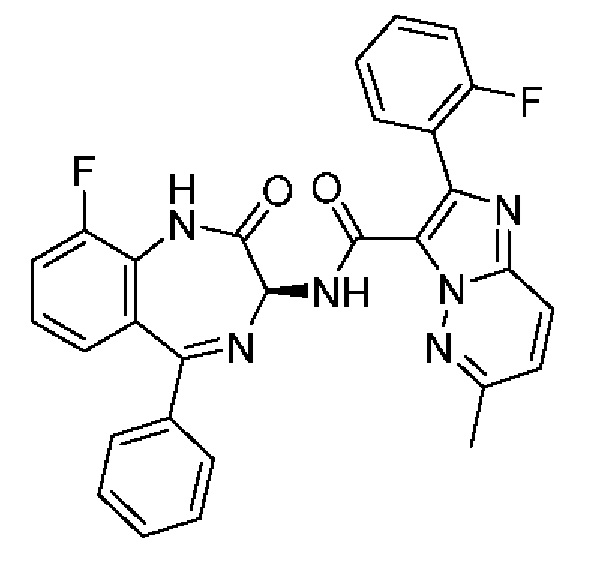
Получали с помощью процедуры амидного сочетания (Процедуры А), описанной для получения соединения согласно Примеру 1 из промежуточного соединения 16А с нагреванием при 40°С в течение ночи. ЖХ-МС (Метод С) m/z 523,15 [М+Н]+ при 4,12 мин. 1Н ЯМР (500 МГц, ДМСО-d6) δ 11,01 (s, 1H), 10,59 (d, J=7,2 Гц, 1H), 8,33 (d, J=9,3 Гц, 1H), 7,63-7,41 (m, 9Н), 7,36-7,28 (m, 1H), 7,28-7,20 (m, 2Н), 7,20-7,14 (m, 1H), 5,53 (d, J=7,2 Гц, 1H), 2,75 (s, 3Н).
20. 6-Хлор-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-метилимидазо[1,2-b]пиридазин-3-карбоксамид (Процедура С)
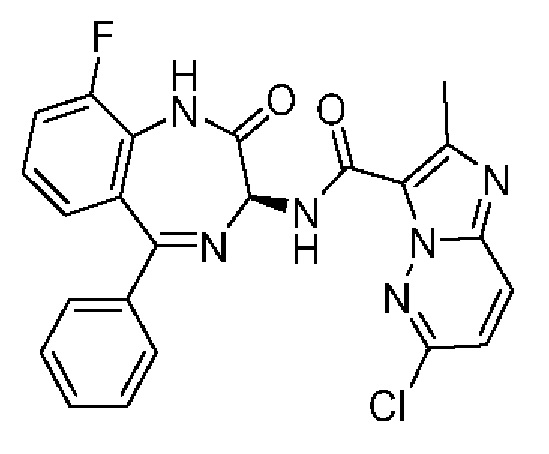
К раствору (3S)-3-амино-9-фтор-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-она (121 мг, 0,45 ммоль), 6-хлор-2-метилимидазо[1,2-b]пиридазин-3-карбоновой кислоты (99,8 мг, 0,47 ммоль) и ДИПЭА (172 мкл, 0,99 ммоль) в безводном ДМФА (2,88 мл) в атмосфере N2 добавляли HATU (205 мг, 0,540 ммоль), и полученную реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Реакцию гасили водой, что приводило к образованию осадка почти белого цвета, который отфильтровывали и промывали водой. Осадок растворяли в EtOAc, и растворитель удаляли при пониженном давлении. Очистка методом колоночной хроматографии [10-37% (EtOH:CH2Cl2:NH4OH; 50:8:1) в CH2Cl2] давала твердое вещество белого цвета (153 мг, выход 74%). 1H ЯМР (400 МГц, ДМСО-d6) δ 11,03 (s, 1H), 9,91 (d, J=7,1 Гц, 1H), 8,36 (d, J=9,5 Гц, 1H), 7,69-7,57 (m, 2Н), 7,57-7,42 (m, 5Н), 7,39-7,30 (m, 1H), 7,21 (d, J=7,9 Гц, 1H), 5,60 (d, J=7,2 Гц, 1H), 2,68 (s, 3Н). МСНР (APCI+) m/z 463,3 [М+Н]+
Приведенные далее соединения были получены согласно процедуре амидного связывания (Процедуре С), описанной для получения соединения согласно Примеру 20. Примеры соединений 40 и 41 получали с дополнительной очисткой методом ВЭЖХ 1 (система MeCN-вода с 0,2% об./об. муравьиной кислоты; 25-100% в течение 18 мин).
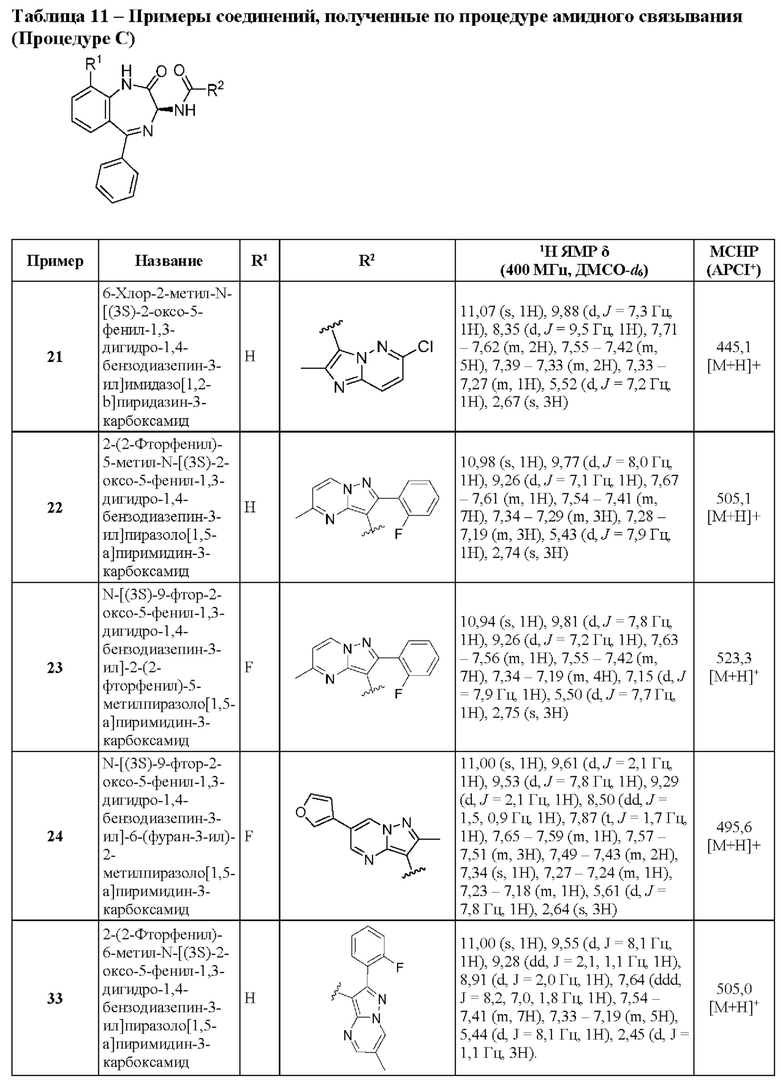
42. 2-(2,3-Дифторфенил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид (Процедура D)
К раствору промежуточного соединения 53А (57 мг, 0,206 ммоль) в ДМФА (2 мл) добавляли NEt3 (59 мкл, 0,423 ммоль), HATU (79 мг, 0,206 ммоль) и (3S)-3-амино-9-фтор-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-он (56 мг, 0,206 ммоль), и полученную реакционную смесь перемешивали при 40°С в течение 1 часа. Реакцию гасили водой, и полученный осадок фильтровали, промывая водой (100 мл). Осадок растворяли в CH2Cl2 (30 мл), концентрировали при пониженном давлении и очищали методом колоночной хроматографии (0-5% МеОН в CH2Cl2), в результате чего получали твердого вещества почти белого цвета (44 мг, выход 40%). ЖХ-МС (Метод С) m/z 527,3 [М+Н]+ при 4,39 мин. 1Н ЯМР (500 МГц, ДМСО-d6) 10,97 (s, 1H), 9,61 (d, J=8,0 Гц, 1H), 9,44 (dd, J=7,0, 1,7 Гц, 1H), 9,02 (dd, J=4,3, 1,7 Гц, 1H), 7,61-7,57 (m, 1H), 7,57-7,48 (m, 4Н), 7,47-7,42 (m, 3Н), 7,39-7,33 (m, 1H), 7,33-7,26 (m, 2Н), 7,16-7,13 (m, 1H), 5,54 (d, J=7,9 Гц, 1H).
Приведенные далее соединения согласно настоящему изобретению были получены согласно процедуре амидного связывания (Процедура D), описанной для получения соединения согласно Примеру 42.
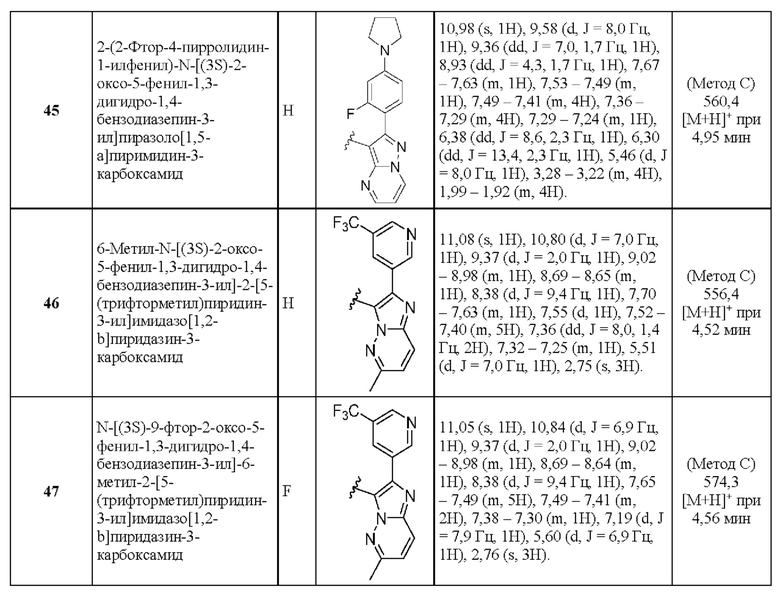
48. 2-[6-(Циклопропиламино)-2-фторпиридин-3-ил]-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид (Процедура Е)
Раствор промежуточного соединения 33С (70 мг, 0,205 ммоль) и LiOH (1,5 М водный раствор; 0,6 мл, 0,9 ммоль) в смеси ТГФ и МеОН (1:1; 1 мл) перемешивали при 40°С в течение ночи. Смесь охлаждали до к.т., нейтрализовали 1М водным раствором HCl (0,92 мл, 0,920 ммоль) и концентрировали при пониженном давлении. Полученный сырой остаток растворяли в ДМФА (1,26 мл), добавляли HATU (78 мг, 0. 206 ммоль), NEt3 (57 мкл, 0,41 ммоль) и затем (3S)-3-амино-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-он (41 мг, 0,205 ммоль), и полученную реакционную смесь перемешивали при 40°С в течение ночи. Процедура выделения и очистки, аналогичная описанной для соединения согласно Примеру 42, давала твердое вещество желтого цвета (39 мг, выход 35%). ЖХ-МС (Метод С) m/z 547,3 [М+Н]+ при 4,13 мин. 1Н ЯМР (500 МГц, ДМСО-d6) 10,99 (s, 1H), 9,61 (d, J=8,0 Гц, 1H), 9,38 (dd, J=7,0, 1,7 Гц, 1H), 8,95 (dd, J=4,3, 1,7 Гц, 1H), 7,75-7,68 (m, 1H), 7,68-7,61 (m, 1Н), 7,55-7,50 (m, 1Н), 7,50-7,40 (m, 4Н), 7,40-7,35 (m, 2Н), 7,35-7,30 (m, 2Н), 7,30-7,23 (m, 1H), 6,51 (d, J=8,1 Гц, 1H), 5,47 (d, J=8,0 Гц, 1H), 2,56-2,51 (m, 1H), 0,75-0,66 (m, 2Н), 0,48-0,41 (m, 2Н).
Приведенные далее соединения согласно настоящему изобретению были получены согласно процедуре амидного связывания (Процедура Е), описанной для получения соединения согласно Примеру 48. Процедура амидного связывания может быть выполнена при к.т. или при 40°С, а стехиометрию используемого NEt3 можно варьировать от 2 до 4 экв. Для получения соединений согласно Примерам 69-76 на стадии гидролиза сложного эфира использовали 10 экв. LiOH (1,5М водный раствор). Соединение согласно Примеру 75 подвергали дополнительной очистке методом обращенно-фазовой колоночной хроматографии (15-65% MeCN в воде с 1% муравьиной кислоты).
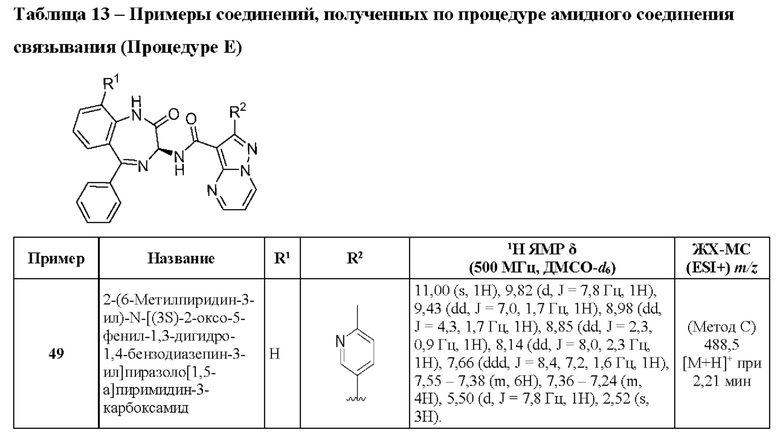
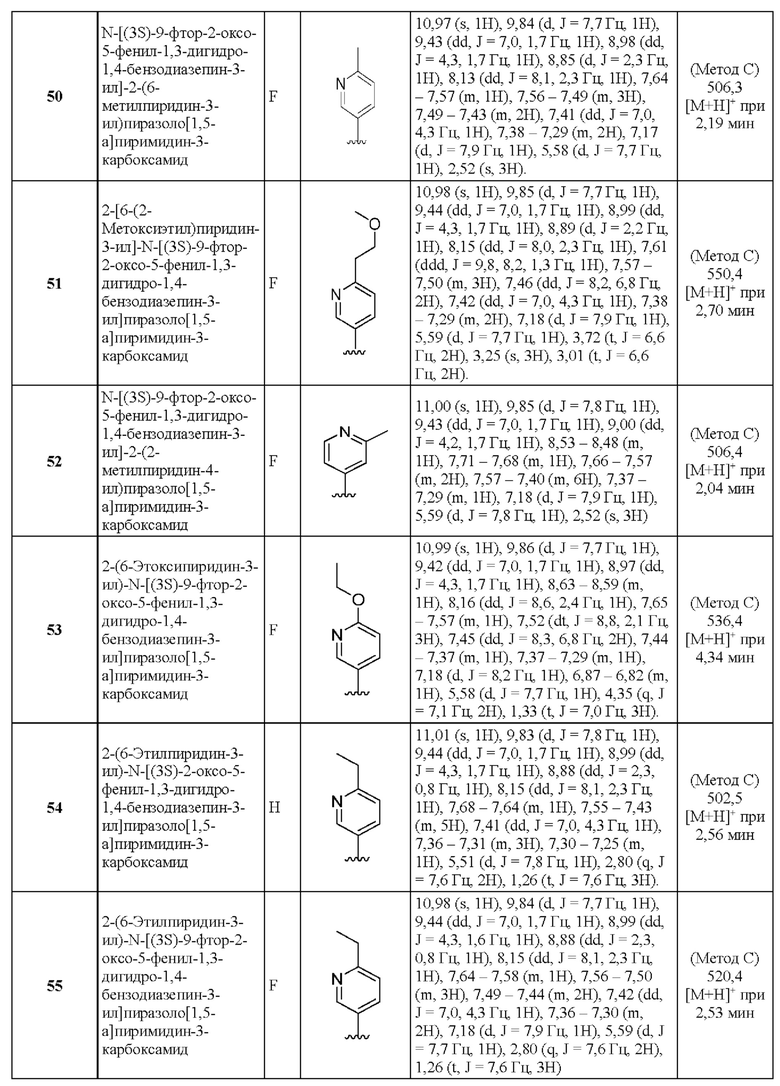
77. 2-[6-(2-Гидрокси-2-метилпропил)пиридин-3-ил]-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид
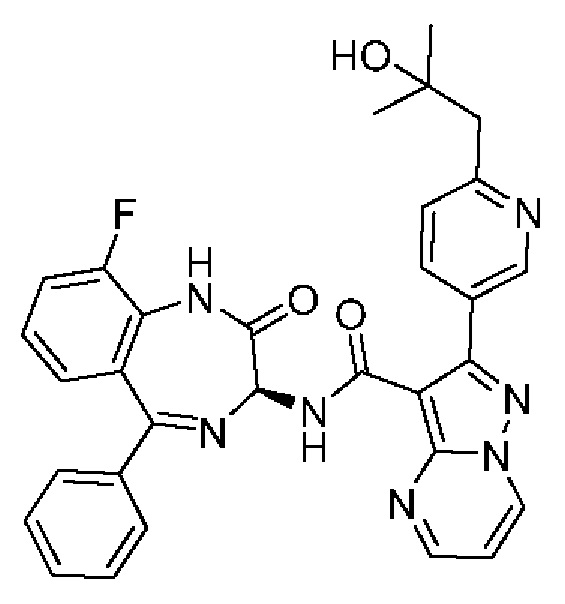
Получали способом амидного связывания (Процедура Е), описанным для получения соединения согласно Примеру 48, с дополнительной очисткой методом обращенно-фазовой колоночной хроматографии [5-40% MeCN в воде (0,1% муравьиной кислоте)], затем методом препаративной ВЭЖХ [Метод 3: 15-100% MeCN в воде (0,1% муравьиной кислоте)], в результате чего получали твердое вещество белого цвета (9 мг, выход 5%). ЖХ-МС (Метод С) m/z 564,4 [М+Н]+ при 2,40 мин. 1Н ЯМР (500 МГц, ДМСО-d6) δ 10,97 (s, 1Н), 9,86 (d, J=7,8 Гц, 1Н), 9,43 (dd, J=7,0, 1,7 Гц, 1Н), 8,98 (dd, J=4,3, 1,7 Гц, 1Н), 8,89 (d, J=2,3 Гц, 1Н), 8,16 (dd, J=8,1, 2,3 Гц, 1Н), 7,64-7,56 (m, 1Н), 7,56-7,49 (m, 3Н), 7,49-7,28 (m, 5Н), 7,18 (d, J=7,9 Гц, 1Н), 5,59 (d, J=7,8 Гц, 1Н), 4,72 (s, 1Н), 2,88 (s, 2Н), 1,12 (s, 6Н).
Приведенные далее соединения согласно настоящему изобретению были получены согласно процедуре амидного связывания (Процедура Е), описанной для получения соединения согласно Примеру 48. Для получения соединения согласно Примеру 79 на стадии гидролиза сложнофирной связи использовали 10 экв. LiOH (1,5М водный раствор). Соединение согласно Примеру 81 подвергали дополнительной очистке методом колоночной обращенно-фазовой хроматографии [15-70% MeCN в воде с 0,1% (NH4)2CO3].
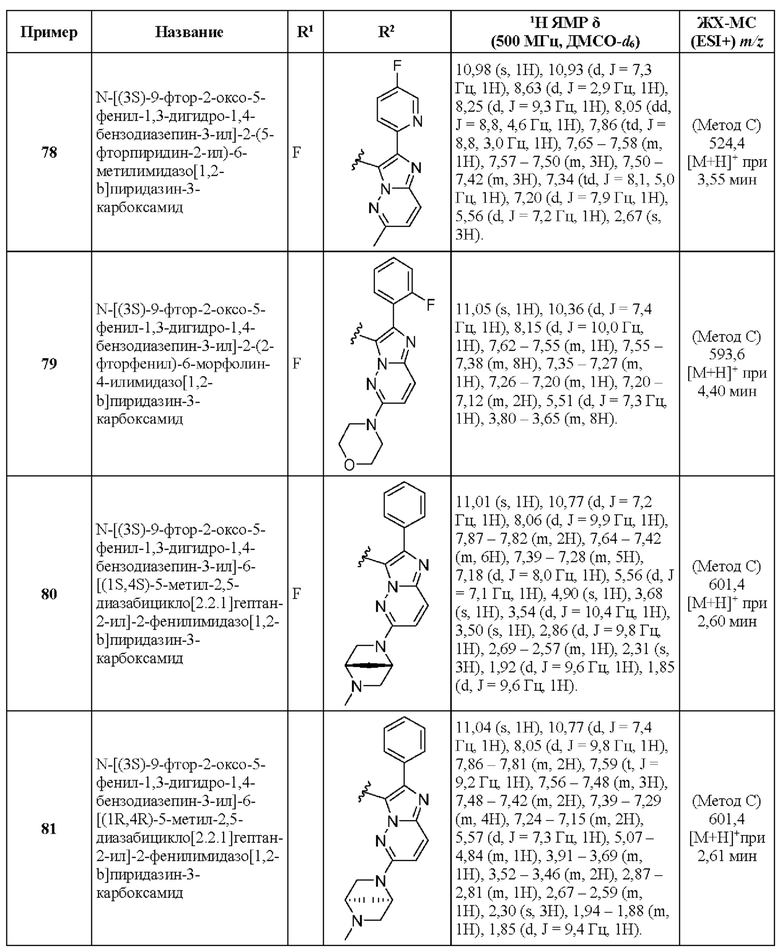
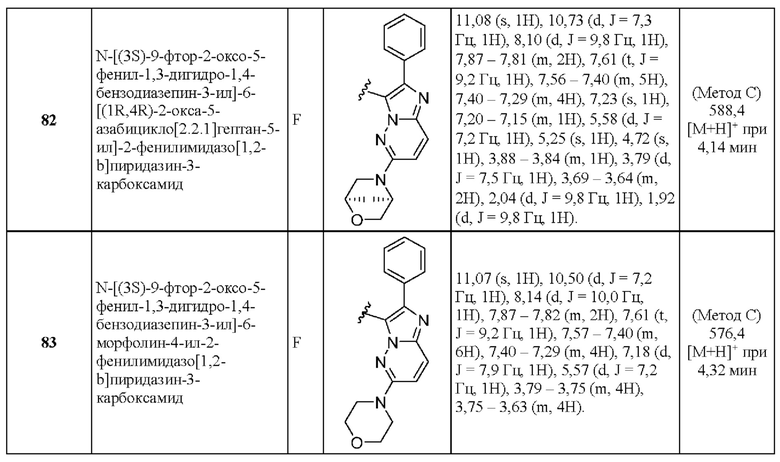
84. N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-5-[(1R,4R)-5-метил-2,5-диазабицикло[2.2.1]гептан-2-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид
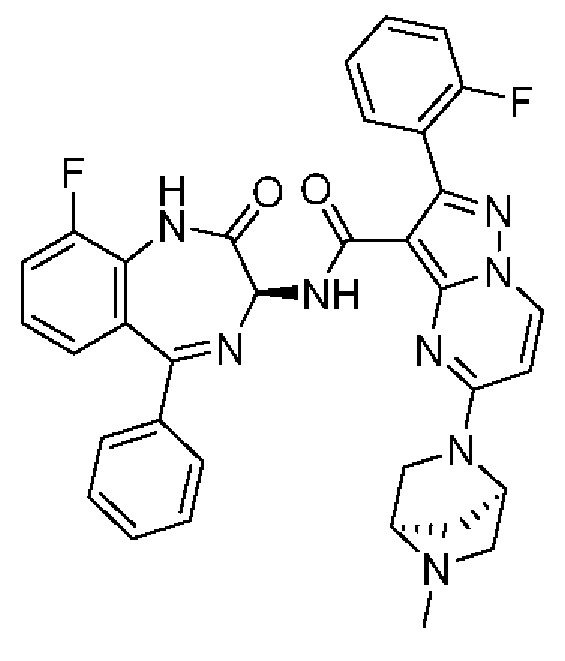
Раствор промежуточного соединения 6Р (124 мг, 0,282 ммоль) и LiOH (1,5М водный раствор; 0,55 мл, 0,825 ммоль) в смеси ТГФ и МеОН (1:1; 2,18 мл) перемешивали при комнатной температуре в течение ночи, затем при 40°С в течение 86 часов. Смесь охлаждали до к.т., подкисляли (до рН≈1) 1М водным раствором HCl (0,92 мл, 0,920 ммоль) и концентрировали при пониженном давлении. Часть полученной неочищенной кислоты (52 мг, 0,135 ммоль) использовали непосредственно в следующей реакции и растворяли в ДМФА (1,5 мл). Затем добавляли HATU (51 мг, 0. 135 ммоль), NEt3 (75 мкл, 0,541 ммоль), после чего добавляли (3S)-3-амино-9-фтор-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-он (36 мг, 0,135 ммоль), и полученную реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакцию гасили водой (5 мл), подкисляли (до рН≈2) посредством АсОН и очищали методом ионообменной хроматографии (2 г SCX-2). Полученный остаток дополнительно очищали методом колоночной хроматографии [0-7% (0,7 н. раствора NH3 в МеОН) в CH2Cl2], в результате чего получали твердое вещество кремового цвета (13 мг, выход 15%) в виде смеси ротамеров. 0,5Н соответствует 1Н ротамерного пика в системе отнесении сигналов 1Н ЯМР. ЖХ-МС (Метод С) m/z 619,4 [М+Н]+при 2,42 мин. 1H ЯМР (500 МГц, ДМСО-d6) δ 11,08 -10,89 (m, 1Н), 10,02-9,92 (m, 1Н), 8,85-8,75 (m, 1Н), 7,61-7,54 (m, 1Н), 7,54-7,39 (m, 6Н), 7,34-7,25 (m, 1Н), 7,24-7,10 (m, 3Н), 6,85 (d, J=7,8 Гц, 0,5Н), 6,54 (d, J=7,8 Гц, 0,5Н), 5,49-5,43 (m, 1Н), 5,40 (d, J=7,3 Гц, 0,5Н), 4,82 (s, 0,5Н), 4,16-4,09 (m, 0,5Н), 3,68-3,60 (m, 0,5Н), 3,58 (s, 0,5Н), 3,52-3,43 (m, 1,5Н), 2,95-2,87 (m, 0,5Н), 2,84-2,77 (m, 0,5Н), 2,70-2,63 (m, 0,5Н), 2,37-2,27 (m, 3Н), 2,01-1,94 (m, 0,5Н), 1,93-1,79 (m, 2Н), 1,75 (s, 1Н).
85. 2-(2-Фтор-6-метилпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид
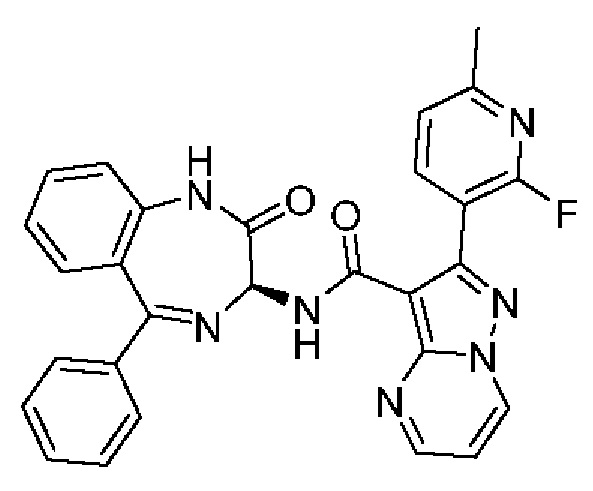
86. 2-(2-Метокси-6-метилпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид
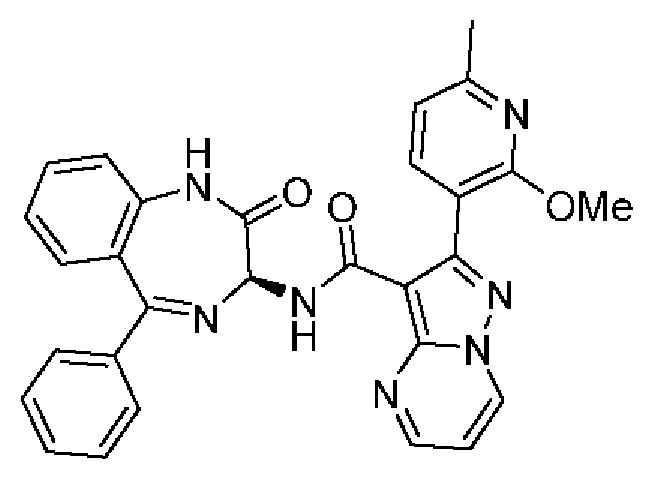
Раствор промежуточного соединения 27В (227 мг, 0,717 ммоль) и LiOH (1,5М водный раствор; 4,78 мл, 7,17 ммоль) в смеси NUA и МеОН (1:1; 6 мл) нагревали при 40°С в течение 48 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (20 мл) и промывали МТБЭ (3×10 мл). Водный слой подкисляли (до рН≈1) 1М водным раствором HCl и экстрагировали смесью CHCl3 и iPrOH (3:1; 3×10 мл). Объединенные органические экстракты сушили над Na2SO4, и удаляли растворитель при пониженном давлении, в результате чего получали твердое вещество белого цвета (151 мг), которое использовали далее без дополнительной очистки. Полученный остаток растворяли в ДМ ФА (1,5 мл), добавляли HATU (208 мг, 0,547 ммоль), NEt3 (123 мкл, 0,882 ммоль) и затем (3S)-3-амино-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-он (137 мг, 0,546 ммоль), и полученную реакционную смесь перемешивали при 40°С в течение 1 часа. Процедуру выделения и очистки, аналогичную описанной для соединения согласно Примеру 42, сопровождали дополнительной стадией очистки методом препаративной ВЭЖХ [Метод 2: 20-100% MeCN в воде (0,1% муравьиной кислоте)].
Пример 85: Твердое вещество белого цвета (28 мг, выход 8%). ЖХ-МС (Метод С) 506,4 [М+Н]+ при 3,73 мин. 1Н ЯМР (500 МГц, ДМСО-d6) δ 11,01 (s, 1Н), 9,56 (d, J=8,0 Гц, 1Н), 9,43 (dd, J=7,0, 1,7 Гц, 1Н), 9,01 (dd, J=4,3, 1,7 Гц, 1Н), 7,96 (dd, J=9,6, 7,5 Гц, 1Н), 7,68-7,61 (m, 1Н), 7,54-7,48 (m, 1Н), 7,48-7,40 (m, 5Н), 7,34-7,23 (m, 4Н), 5,45 (d, J=8,0 Гц, 1Н), 2,47 (s, 3Н).
Пример 86: Твердое вещество белого цвета (14 мг, выход 3%). ЖХ-МС (Метод С) m/z 518,5 [М+Н]+ при 3,96 мин. 1Н ЯМР (500 МГц, ДМСО-d6) δ 10,99 (s, 1Н), 9,50 (d, J=8,0 Гц, 1Н), 9,37 (dd, J=7,0, 1,7 Гц, 1Н), 8,95 (dd, J=4,3, 1,7 Гц, 1Н), 7,67-7,61 (m, 1Н), 7,59 (d, J=7,4 Гц, 1Н), 7,53-7,48 (m, 1Н), 7,48-7,41 (m, 4Н), 7,37 (dd, J=7,0, 4,3 Гц, 1Н), 7,34-7,28 (m, 2Н), 7,28-7,22 (m, 1Н), 6,89 (d, J=7,4 Гц, 1Н), 5,41 (d, J=7,9 Гц, 1Н), 3,74 (s, 3Н), 2,43 (s, 3Н).
87. N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид (Процедура F)
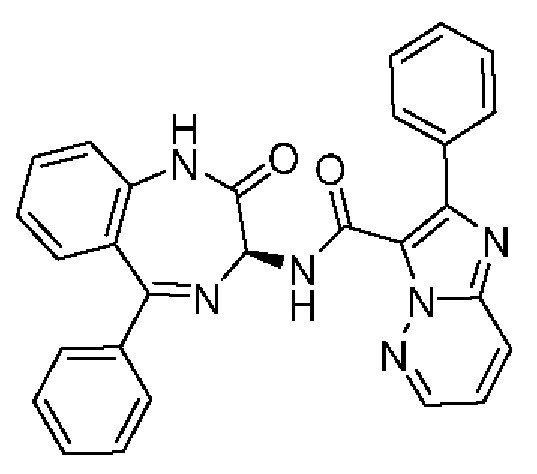
Раствор промежуточного соединения 45А (58 мг, 0,217 ммоль) и LiOH⋅H2O (91 мг, 2,174 ммоль) в смеси МеОН-ТГФ-вода (2:2:1; 5 мл) нагревали до 50°С в течение 2 часов.
Реакционную смесь охлаждали до к.т., нейтрализовали 1М водным раствором HCl до рН≈7 и затем концентрировали при пониженном давлении. Полученный сырой продукт растворяли в ДМФА (1,5 мл), и к раствору добавляли (3S)-3-амино-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-он (55 мг, 0,217 ммоль), NEt3 (91 мкл, 0,651 ммоль) и затем HATU (83 мг, 0,217 ммоль). Реакционную смесь перемешивали при кт в течение 18 ч, гасили водой (10 мл), и полученный осадок фильтровали, промывая водой. Затем осадок растворяли в 10% смеси МеОН и CH2Cl2, пропускали через фазоразделительный картридж, концентрировали при пониженном давлении и очищали методом колоночной хроматографии (0-10% МеОН в CH2Cl2), в результате чего получали твердое вещество беловатого цвета (30 мг, выход 29%). ЖХ-МС (Метод D) m/z 473,4 [М+Н]+ при 3,89 мин. 1H ЯМР (500 МГц, ДМСО-d6) δ 11,05 (s, 1Н), 10,34 (d, J=7,4 Гц, 1Н), 8,86 (dd, J=4,5, 1,6 Гц, 1Н), 8,38 (dd, J=9,2, 1,6 Гц, 1Н), 8,01-7,95 (m, 2Н), 7,68 (ddd, J=8,6, 7,2, 1,6 Гц, 1Н), 7,57-7,39 (m, 9Н), 7,36 (dd, J=8,1, 1,4 Гц, 2Н), 7,29 (ddd, J=8,3, 7,3, 1,2 Гц, 1Н), 5,54 (d, J=7,4 Гц, 1Н).
Приведенные далее соединения согласно настоящему изобретению были получены согласно процедуре амидного связывания (Процедура F), описанной для получения соединения согласно Примеру 87. Процедуру проводили с использованием 5 экв. NEt3 в случае Примеров 95-105 и с 10 экв. NEt3 в случае Примера 106. Соединение согласно Примеру 107 подвергали дополнительной очистке методом обращенно-фазовой колоночной хроматографии (5-60% MeCN в 10 мМ водном растворе (NH4)2СО3).
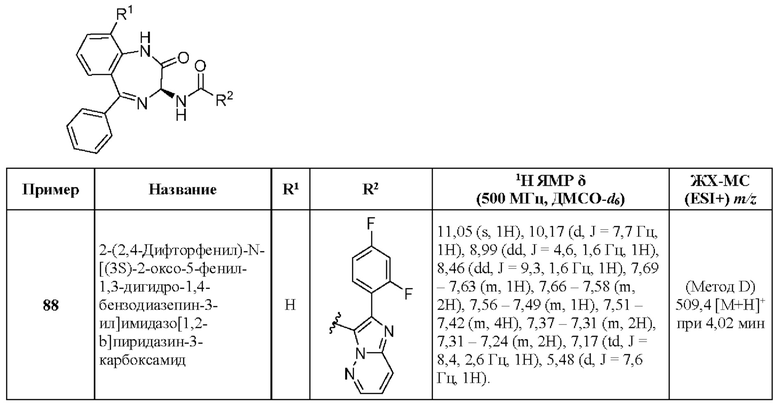
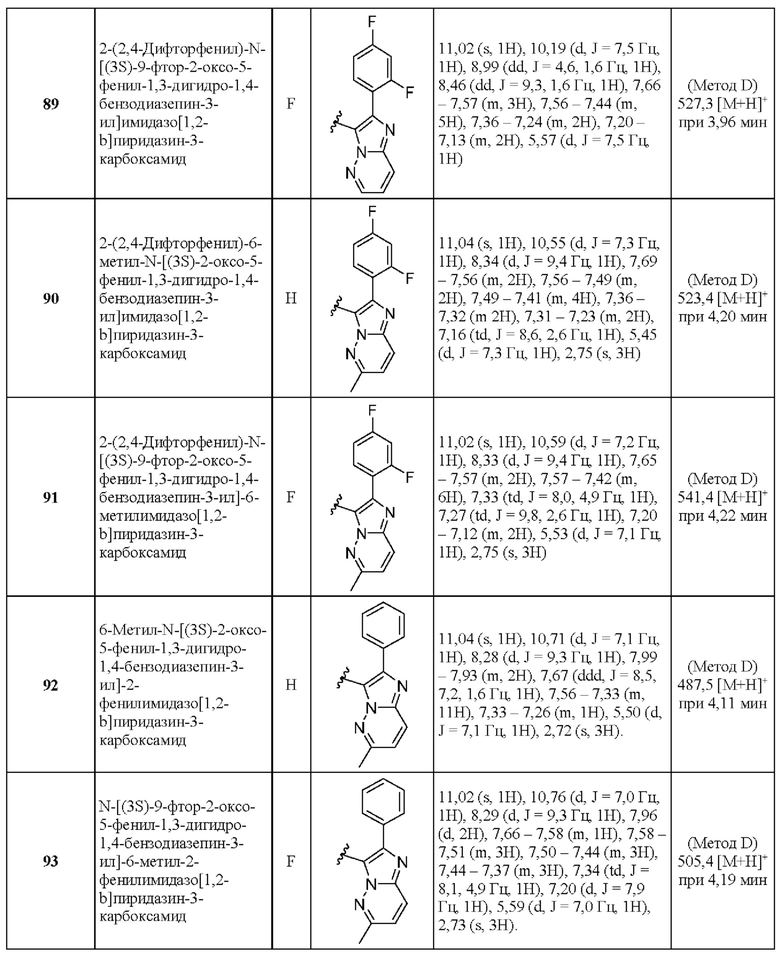
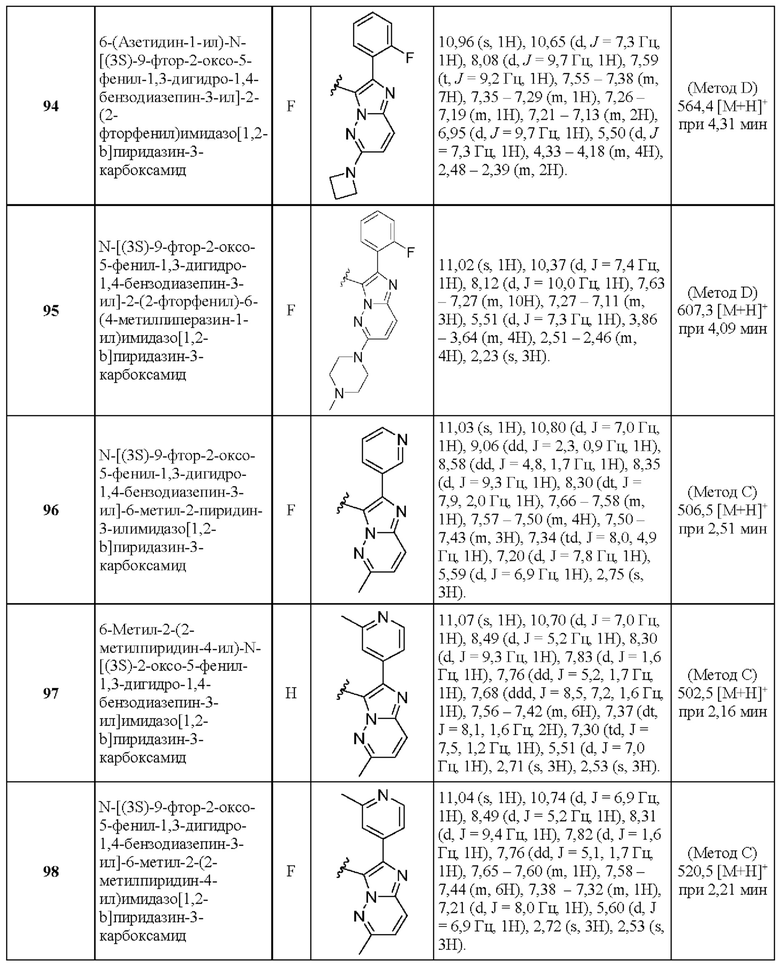
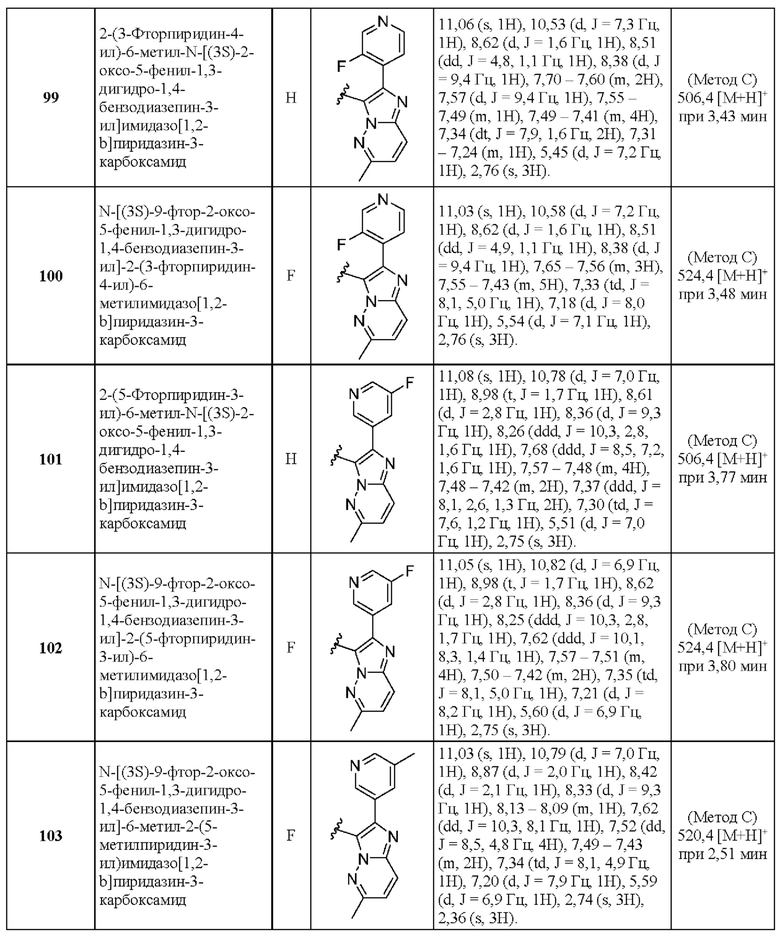
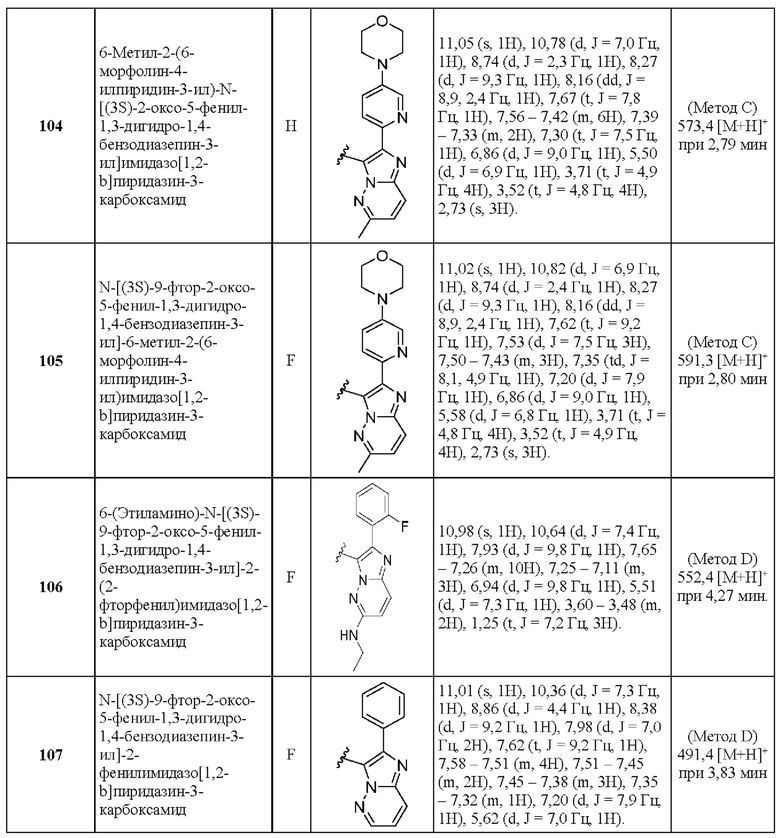
108. N-[(3S)-9-(фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-(1-метилазетидин-3-ил)оксиимидазо[1,2-b]пиридазин-3-карбоксамид
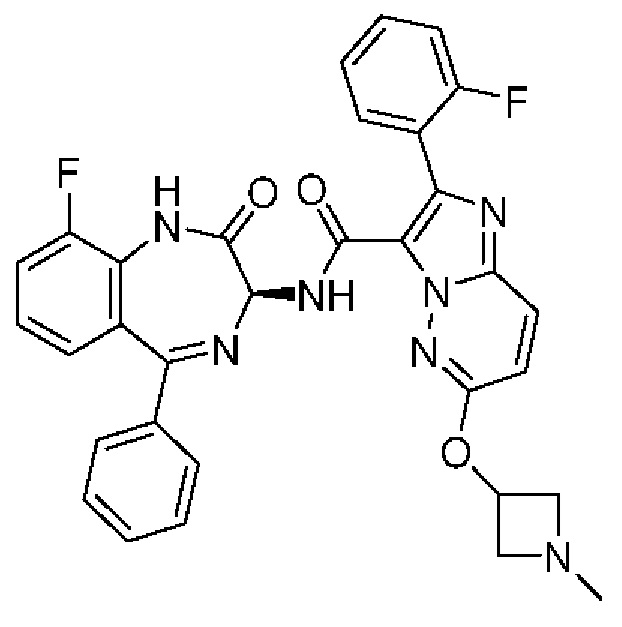
К раствору сырого промежуточного соединения 56А (55 мг, 0,161 ммоль), (3S)-3-амино-9-фтор-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-она (43 мг, 0,161 ммоль) и NEt3 (112 мкл, 0,803 ммоль) в ДМФА (3 мл) добавляли HATU (61 мг, 0,161 ммоль), и полученную реакционную смесь перемешивали при комнатной температуре в течение ночи. Процедура выделения и очистки, аналогичная описанной для соединения согласно Примеру 87, давала твердое вещество почти белого цвета (30 мг, выход 31%). ЖХ-МС (Метод D) m/z 594,4 [М+Н]+ при 3,85 мин. 1Н ЯМР (500 МГц, ДМСО-d6) δ 11,08 (s, 1Н), 9,89 (d, J=7,5 Гц, 1Н), 8,33 (d, J=9,7 Гц, 1Н), 7,65-7,41 (m, 8Н), 7,37-7,16 (M, 5Н), 5,55 (d, J=7,5 Гц, 1Н), 5,51 (р, J=5,4 Гц, 1Н), 3,92 (t, J=7,1 Гц, 1Н), 3,85 (t, J=7,2 Гц, 1Н), 3,27-3,19 (m, 2Н), 2,22 (s, 3Н).
109. N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-(1-метилазетидин-3-ил)окси-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид
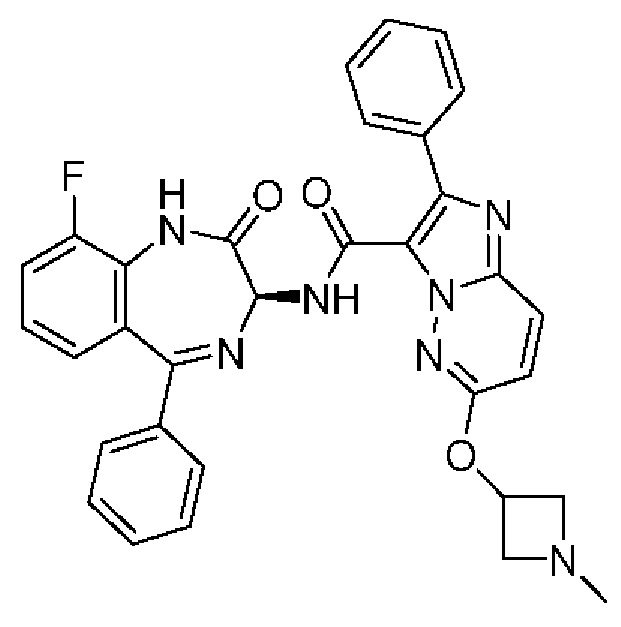
Получали по той же методике, которая была описана для получения соединения согласно Примеру 108. ЖХ-МС (Метод D) m/z 576,4 [М+Н]+ при 4,05 мин. 1Н ЯМР (500 МГц, ДМСО-d6) δ 11,07 (s, 1Н), 10,01 (d, J=7,3 Гц, 1Н), 8,29 (d, J=9,7 Гц, 1Н), 7,93-7,84 (m, 2H), 7,63 (ddd, J=9,9, 8,2, 1,4 Гц, 1Н), 7,60-7,52 (m, 3Н), 7,51-7,44 (m, 2H), 7,43-7,31 (m, 4H), 7,22 (dd, J=9,4, 7,8 Гц, 2Н), 5,62 (d, J=7,3 Гц, 1Н), 5,45 (р, J=5,4 Гц, 1Н), 3,91 (t, J=7,1 Гц, 1Н), 3,85 (t, J=7,4 Гц, 1H), 3,22 (d, J=15,4 Гц, 2H), 2,23 (s, 3H).
110. N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-(2-метоксиэтокси)имидазо[1,2-b]пиридазин-3-карбоксамид
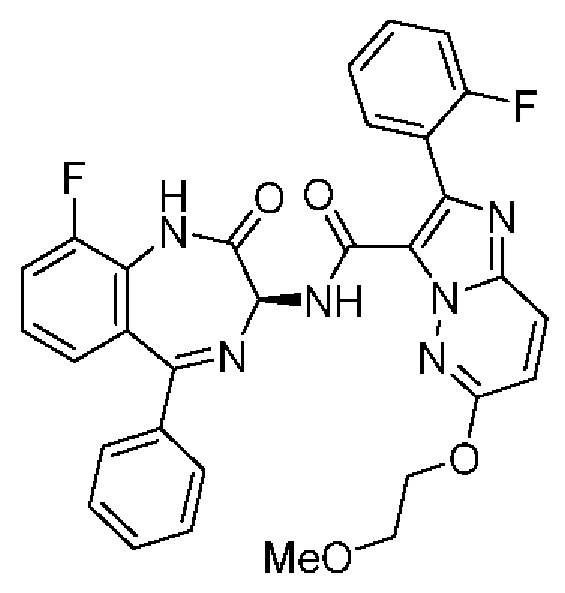
Получали по той же методике, которая была описана для получения соединения согласно Примеру 108, но с использованием 10 экв. NEt3 на стадии амидного сочетания и дополнительной очистки методом обращенно-фазовой колоночной хроматографии [10-60% MeCN в 10 мМ водном растворе (NH4)2CO3]. ЖХ-МС (Метод D) m/z 582,6 [М+Н]+ при 4,20 мин. 1H ЯМР (500 МГц, ДМСО-d6) δ 11,09 (s, 1Н), 10,23 (d, J=7,2 Гц, 1Н), 8,31 (d, J=9,7 Гц, 1Н), 7,59 (t, J=9,2 Гц, 1Н), 7,56-7,41 (m, 7Н), 7,33 (t, J=7,0 Гц, 1Н), 7,29 (d, J=9,8 Гц, 1Н), 7,25 (td, J=7,5, 1,1 Гц, 1Н), 7,23-7,18 (m, 1Н), 7,17 (d, J=7,9 Гц, 1Н), 5,52 (d, J=7,1 Гц, 1Н), 4,84 (ddd, J=12,0, 5,8, 3,1 Гц, 1Н), 4,71 (ddd, J=12,0, 5,4, 3,0 Гц, 1Н), 3,78 (ddd, J=7,1, 4,5, 3,1 Гц, 2Н), 3,33 (s, 3Н).
111. 6-Метокси-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид
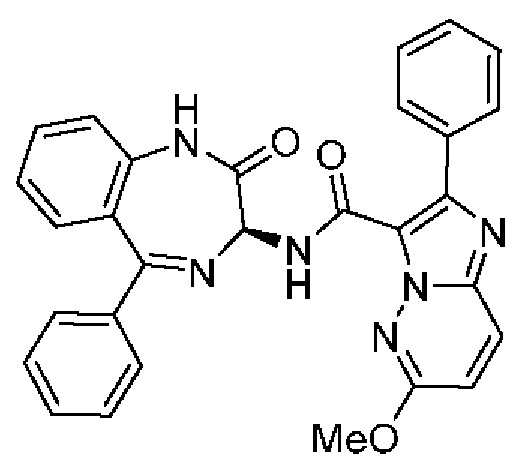
К раствору промежуточного соединения 45С (180 мг, 0,597 ммоль) в смеси МеОН и воды (1:1; 4 мл) добавляли LiOH (1,5М водный раствор; 1,75 мл, 2,625 ммоль), и полученную смесь нагревали до 100°С в течение 1 часа посредством МВИ. Реакционную смесь охлаждали до комнатной температуры, подкисляли 1М водным раствором HCl (3 мл) и затем концентрировали при пониженном давлении, в результате чего получали твердое вещество почти белого цвета (161 мг), которое затем непосредственно использовали в следующей реакции. Часть сырого остатка (80 мг, 0,297 ммоль) растворяли в ДМФА (1,8 мл), добавляли HATU (113 мг, 0,298 ммоль), NEt3 (124 мкл, 0,891 ммоль) и (3S)-3-амино-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-он (75 мг, 0,297 ммоль), и полученную реакционную смесь перемешивали при 40°С в течение ночи. Процедура выделения и очистки, аналогичная описанной для соединения согласно Примеру 42, давала твердое вещество белого цвета (32 мг, выход 22%). ЖХ-МС (Метод С) m/z 503,4 [М+Н]+ при 4,32 мин. 1Н ЯМР (500 МГц, ДМСО-d6) δ 11,06 (s, 1Н), 10,36 (d, J=7,2 Гц, 1Н), 8,26 (d, J=9,7 Гц, 1Н), 7,92-7,86 (m, 2H), 7,70-7,63 (m, 1H), 7,55-7,32 (m, 10H), 7,32-7,25 (m, 1H), 7,22 (d, J=9,7 Гц, 1H), 5,51 (d, J=7,1 Гц, 1H), 4,21 (s, 3H).
Приведенные далее соединения согласно настоящему изобретению были получены по процедуре, аналогичной описанной для соединения согласно Примеру 111, с дополнительной очисткой для соединений согласно Примерам 113 и 114 методом обращенно-фазовой колоночной хроматографии (10-65% MeCN в воде с 0,1% муравьиной кислоты).
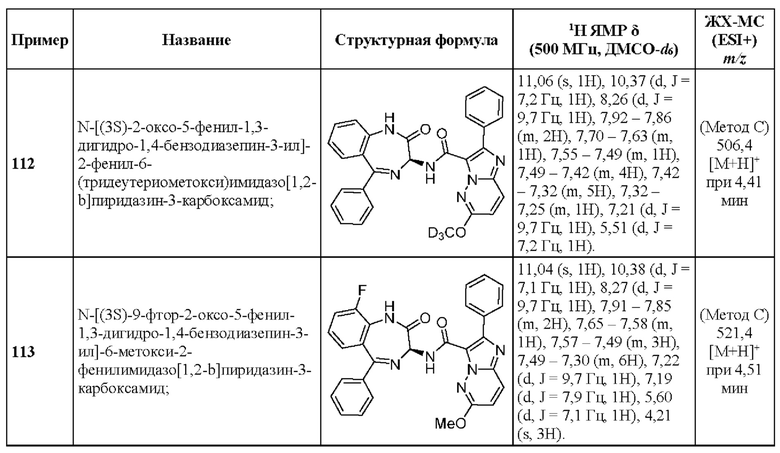
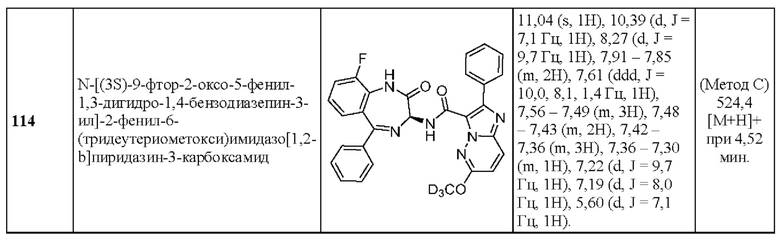
115. N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-метоксиимидазо[1,2-b]пиридазин-3-карбоксамид
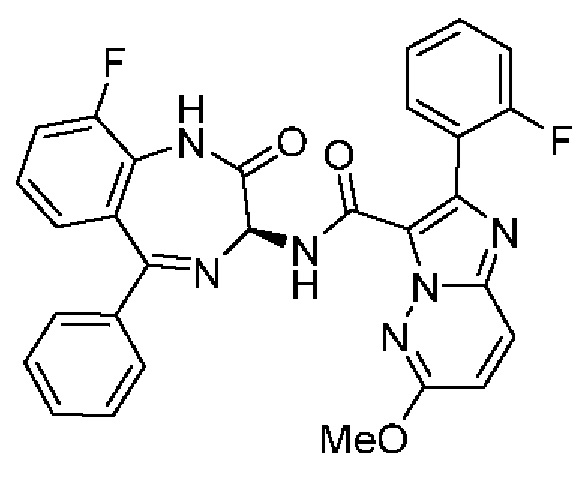
Раствор промежуточного соединения 45D (45 мг, 0,141 ммоль) и LiOH⋅H2O (59 мг, 1,41 ммоль) в смеси МеОН-ТГФ-вода (2:2:1; 5 мл) нагревали при 50°С в течение 18 часов. Реакционную смесь охлаждали до к.т., подкисляли 1М водным раствором HCl до рН≈4, и удаляли
растворитель при пониженном давлении. Полученный сырой остаток растворяли в ДМФА (1,5 мл), добавляли (3S)-3-амино-9-фтор-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-он (40 мг, 0,141 ммоль) и NEt3 (39 мкл, 0,281 ммоль) и затем HATU (54 мг, 0,141 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Процедура выделения и очистки, аналогичная описанной для соединения согласно Примеру 42, с последующей дополнительной очисткой методом колоночной обращенно-фазовой хроматографии [5-75% MeCN в 10 мМ водном растворе (NH4)2CO3], давала твердое вещество почти белого цвета (18 мг, выход 23%). ЖХ-МС (Метод С) m/z 539,4 [М+Н]+ при 4,13 мин. 1Н ЯМР (500 Мгц, ДМСО-d6) δ 11,04 (s, 1Н), 10,30 (d, J=7,2 Гц, 1Н), 8,31 (d, J=9,7 Гц, 1Н), 7,60 (t, 1Н), 7,55-7,42 (m, 7H), 7,32 (td, J=8,0, 4,9 Гц, 1Н), 7,29-7,15 (m, 4H), 5,54 (d, J=7,2 Гц, 1Н), 4,24 (s, 3Н).
116. 6-Хлор-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)имидазо[1,2-b]пиридазин-3-карбоксамид
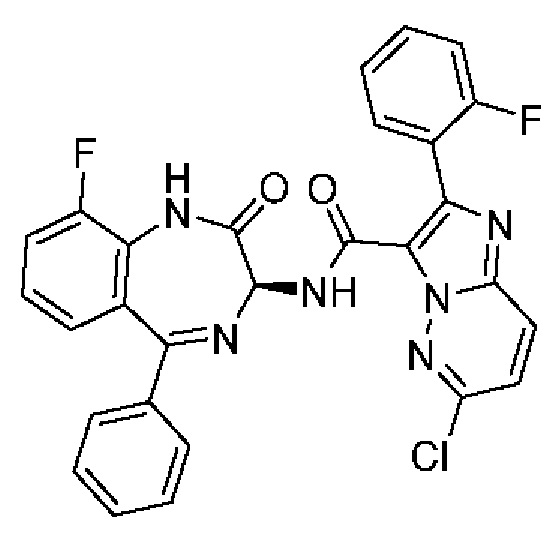
Раствор промежуточного соединения 45D (45 мг, 0,141 ммоль) и LiOH⋅H2O (59 мг, 1,41 ммоль) в смеси ТГФ и воды (2:1; 3 мл) нагревали до 50°С в течение 3 часов. Реакционную смесь охлаждали до к.т., нейтрализовали 1М водным раствором HCl до рН≈7 и затем концентрировали при пониженном давлении. Полученный сырой остаток растворяли в ДМФА (3 мл), добавляли (3S)-3-амино-9-фтор-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-он (38 мг, 0,141 ммоль), NEt3 (20 мкл, 0,141 ммоль) и затем HATU (54 мг, 0,141 ммоль), после чего полученную реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Процедура выделения и очистки, аналогичная описанной для соединения согласно Примеру 42, с последующей очисткой методом обращенно-фазовой колоночной хроматографии [15-75% MeCN в 10 мМ водном растворе (NH4)2СО3], давала твердое вещество почти белого цвета (13 мг, выход 17%). ЖХ-МС (Метод D) m/z 543,4 [М+Н]+ при 4,18 мин. 1Н ЯМР (500 МГц, ДМСО-d6) δ 11,01 (s, 1Н), 9,92 (d, J=7,4 Гц, 1Н), 8,50 (d, J=9,5 Гц, 1Н), 7,73 (d, J=9,5 Гц, 1Н), 7,65-7,58 (m, 2Н), 7,58-7,44 (m, 6Н), 7,37-7,22 (m, 3Н), 7,18 (d, J=7,7 Гц, 1Н), 5,54 (d, J=7,3 Гц, 1Н).
117. 6-Хлор-2-(6-этилпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид
Раствор промежуточного соединения 46F (63 мг, 0,179 ммоль) и 1,5М водный раствор LiOH (0,359 мл, 0,538 ммоль) в ТГФ (3 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь подкисляли 1М водным раствором HCl до рН≈4 и затем концентрировали при пониженном давлении. Полученный сырой остаток растворяли в ДМФА (1,8 мл), добавляли (3S)-3-амино-9-фтор-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-он (48 мг, 0,179 ммоль) и NEt3 (75 мкл, 0,538 ммоль) и затем HATU (68 мг, 0,179 ммоль), после чего полученную реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Процедура выделения и очистки, аналогичная описанной для соединения согласно Примеру 42, давала твердое вещество бежевого цвета (50 мг, выход 48%). ЖХ-МС (Метод С) m/z 554,4 [М+Н]+ при 2,77 мин. 1H ЯМР (500 МГц, ДМСО-d6) δ 11,04-11,00 (m, 1Н), 10,18 (d, J=7,2 Гц, 1Н), 9,05-9,00 (m, 1Н), 8,46 (d, J=9,5 Гц, 1Н), 8,26 (dd, J=8,1, 2,3 Гц, 1Н), 7,67 (d, J=9,5 Гц, 1Н), 7,66-7,60 (m, 1Н), 7,59-7,53 (m, 3Н), 7,48 (t, J=7,4 Гц, 2Н), 7,39-7,31 (m, 2Н), 7,21 (d, J=7,9 Гц, 1Н), 5,61 (d, J=7,2 Гц, 1Н), 2,82 (q, J=7,6 Гц, 2Н), 1,28 (t, J=7,6 Гц, 3Н).
118. N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метокси-2-(5-метилпиридин-3-ил)имидазо[1,2-b]пиридазин-3-карбоксамид
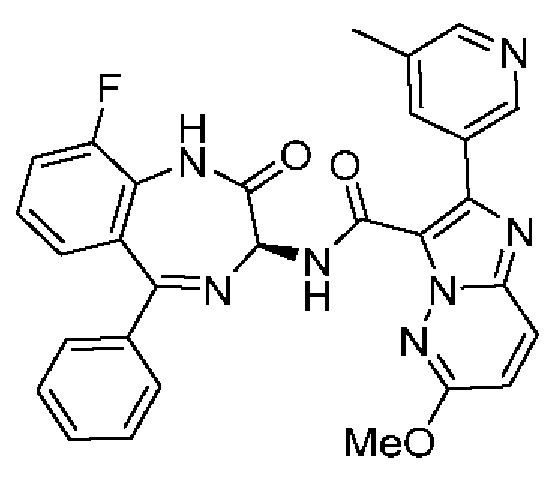
Раствор промежуточного соединения 46С (130 мг, 0,308 ммоль) и LiOH (74 мг, 3,083 ммоль) в смеси МеОН-ТГФ-вода (4:3:3; 10 мл) нагревали при 50°С в течение 1 часа. Реакционную смесь охлаждали до к.т., подкисляли 1М водным растовром HCl до уровня рН ≈4, и удаляли растворитель при пониженном давлении. Полученный сырой остаток растворяли в ДМФА (6 мл), добавляли (3S)-3-амино-9-фтор-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-он (83 мг, 0,308 ммоль), NEt3 (214 мкл, 1,539 ммоль) и затем HATU (117 мг, 0,308 ммоль), после чего полученную реакционную смесь перемешивали при комнатной температуре в течение 1 часов. Процедура выделения и очистки, аналогичная описанной для соединения согласно Примеру 87, давала твердое вещество желтого цвета (40 мг, выход 24%). ЖХ-МС (Метод С) m/z 536,4 [М+Н]+ при 2,63 мин. 1Н ЯМР (500 МГц, ДМСО-d6) δ 11,06 (s, 1Н), 10,42 (d, J=7,1 Гц, 1Н), 8,81 (d, J=2,0 Гц, 1Н), 8,41 (s, 1Н), 8,30 (d, J=9,7 Гц, 1Н), 8,05 (s, 1Н), 7,61 (t, J=9,3 Гц, 1Н), 7,54 - 7,49 (m, 3Н), 7,45 (t, J=7,5 Гц, 2H), 7,33 (d, J=5,9 Гц, 1Н), 7,26 (d, J=9,7 Гц, 1H), 7,19 (d, J=8,0 Гц, 1H), 5,60 (d, J=6,9 Гц, 1H), 4,23 (s, 3H), 2,35 (s, 3H).
Приведенные далее соединения согласно настоящему изобретению были получены согласно процедуре, описанной для получения соединения согласно Примеру 118.
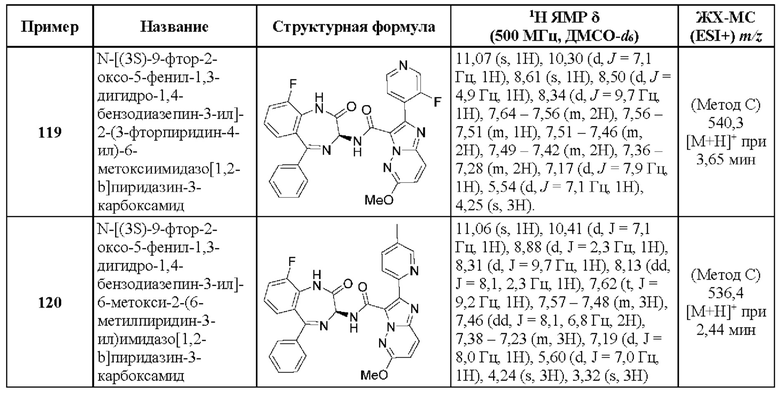
121. 2-(6-Этилпиридин-3-ил)-6-метокси-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид
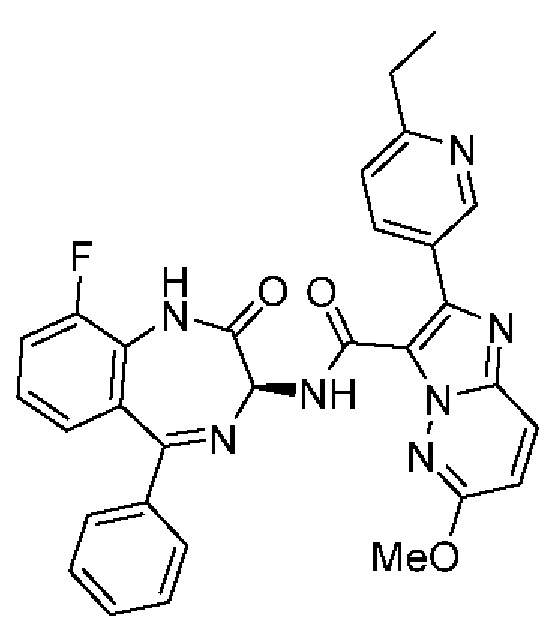
К раствору промежуточного соединения 46F (68 мг, 0,193 ммоль) в смеси ТГФ и МеОН (1:1; 4 мл) добавляли LiOH (1,5М водный раствор; 0,643 мл, 0,965 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 4 дней. Реакционную смесь подкисляли 1М водным раствором HCl до уровня рН ≈4 и затем концентрировали при пониженном давлении, в результате чего получали твердое вещество почти белого цвета (123 мг), которое затем непосредственно использовали в следующей реакции. Часть сырого остатка (58 мг, 0,193 ммоль) растворяли в ДМФА (1,5 мл), добавляли NEt3 (81 мг, 0,579 ммоль), (3S)-3-амино-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-он (53 мкл, 0,196 ммоль) и затем HATU (74 мг, 0,195 ммоль), после чего полученную реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакцию гасили водой (15 мл), и полученный осадок отфильтровывали, промывая водой (2×15 мл). Осадок растворяли в CH2Cl2, концентрировали при пониженном давлении и очищали методом колоночной хроматографии (0-5% МеОН в CH2Cl2). Дальнейшая очистка методом препаративной ВЭЖХ (Метод 4) [20-100% MeCN в воде (0,1% муравьиной кислоте)] давала твердое вещество белого цвета (9,7 мг, выход 9%). ЖХ-МС (Метод С) m/z 550,1 [М+Н]+ при 1,02 мин. 1Н ЯМР (500 МГц, ДМСО-d6) δ 11,05 (s, 1Н), 10,42 (d, J=7,0 Гц, 1Н), 8,93-8,88 (m, 1Н), 8,31 (d, J=9,7 Гц, 1Н), 8,14 (dd, J=8,1, 2,3 Гц, 1Н), 7,66-7,58 (m, 1Н), 7,56-7,49 (m, 3Н), 7,48-7,43 (m, 2Н), 7,37-7,32 (m, 1Н), 7,30 (d, J=8,1 Гц, 1Н), 7,26 (d, J=9,7 Гц, 1Н), 7,22-7,17 (m, 1Н), 5,60 (d, J=7,1 Гц, 1Н), 4,24 (s, 3Н), 2,79 (q, J=7,6 Гц, 2Н), 1,26 (t, J=7,6 Гц, 3Н).
122. 6-Этокси-2-(6-этилпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид
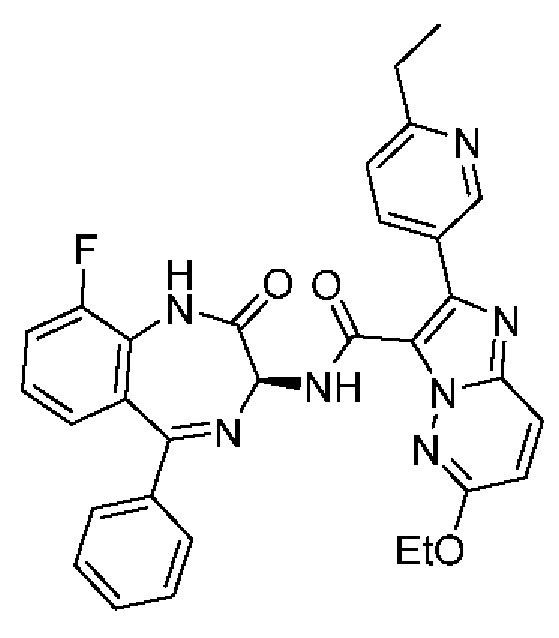
Раствор промежуточного соединения 46F (60 мг, 0,172 ммоль) и 1,5М водного раствора LiOH (0,572 мл, 0,858 ммоль) в смеси EtOH и ТГФ (1:1; 4 мл) нагревали при 50°С в течение 1 часа. Реакционную смесь охлаждали до к.т., подкисляли 1М водным растовром HCl до уровня рН≈4, и удаляли растворитель при пониженном давлении. Полученный сырой остаток растворяли в ДМФА (1,5 мл), добавляли (3S)-3-амино-9-фтор-5-фенил-1,3-дигидро-1,4-бензодиазепин-2-он (46 мг, 0,172 ммоль), NEt3 (72 мкл, 0,515 ммоль) и затем HATU (68 мг, 0,178 ммоль), после чего полученную реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь разбавляли EtOAc (50 мл), промывали солевым раствором (3×50 мл), сушили над Na2SO4, и удаляли растворитель при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии (0-3% МеОН в CH2Cl2) и обращенно-фазовой колоночной хроматографии (35-65% MeCN в 0,1% водном растворе муравьиной кислоты). Полученный материал растворяли в смеси CHCl3 и iPrOH (3:1; 25 мл), промывали насыщенным водным раствором NaHCO3 (2×20 мл) и сушили над Na2SO4, в результате чего получали твердого вещества белого цвета (17 мг, выход 18%). ЖХ-МС (Метод С) m/z 540,3 [М+Н]+ при 3,63 мин. 1H ЯМР (500 МГц, ДМСО-d6) δ 11,07 (s, 1Н), 10,30 (d, J=7,2 Гц, 1Н), 8,63-8,59 (m, 1Н), 8,53-8,48 (m, 1Н), 8,34 (d, J=9,8 Гц, 1Н), 7,64-7,57 (m, 2Н), 7,57-7,50 (m, 1Н), 7,52-7,42 (m, 4Н), 7,37-7,28 (m, 2Н), 7,17 (d, J=7,9 Гц, 1Н), 5,54 (d, J=1,1 Гц, 1Н), 4,25 (s, 3Н).
123. 2-(6-Этилпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-2,3-дигидро-1H-1,4-бензодиазепин-3-ил]-6-(*2Н3)метоксиимидазо[1,2-b]пиридазин-3-карбоксамид
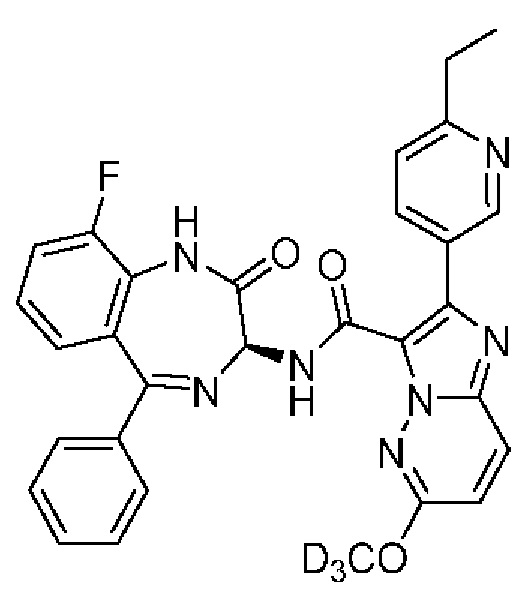
Получали с помощью процедуры амидного сочетания (Процедуры А), описанной для получения соединения согласно Примеру 1.
ЖХ-МС (Метод С) m/z 553,4 [М+Н]+ при 2,72 мин. 1Н ЯМР (500 МГц, ДМСО-d6) δ 11,05 (s, 1Н), 10,41 (d, J=7,1 Гц, 1Н), 8,92-8,87 (m, 1Н), 8,30 (d, J=9,7 Гц, 1Н), 8,14 (dd, J=8,1, 2,3 Гц, 1Н), 7,65-7,57 (m, 1Н), 7,55-7,48 (m, 3Н), 7,47-7,42 (m, 2Н), 7,36-7,31 (m, 1Н), 7,30 (d, J=8,1 Гц, 1Н), 7,25 (d, J=9,7 Гц, 1Н), 7,19 (d, J=7,9 Гц, 1Н), 5,59 (d, J=7,1 Гц, 1Н), 2,78 (q, J=7,6 Гц, 2Н), 1,25 (t, J=7,6 Гц, 3Н).
124. 2-(6-[(3S*)-3-метилморфолин-4-ил]пиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-2,3-дигидро-1Н-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид
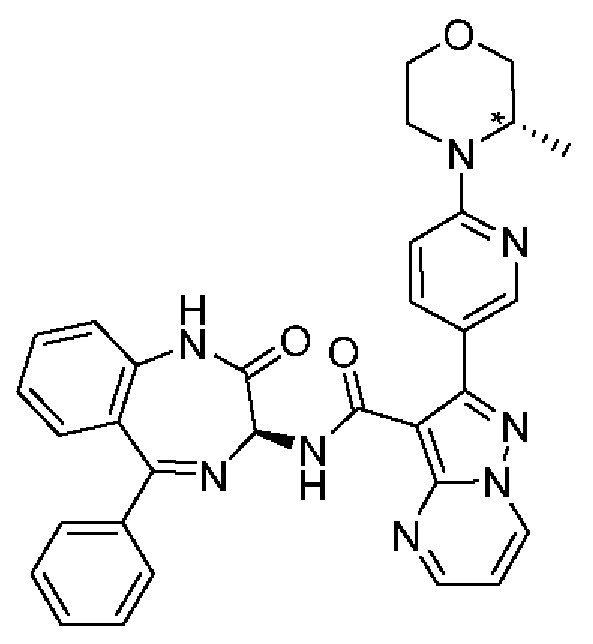
Соединение согласно Примеру 67 (40 мг) очищали методом препаративной хиральной ВЭЖХ [70% MeCN в воде (0,1% растворе аммиака)], в результате чего получали указанное в заголовке соединение в форме единого диастереомера с неизвестной абсолютной конфигурацией и произвольно определенным стереоцентром, помеченным звездочкой. Второй элюирующий энантиомер. Твердое вещество желтого цвета (12 мг, выход 30%). Аналитическая хиральная ВЭЖХ: 8,99 мин. ЖХ-МС (Метод D) 573,4 [М+Н]+ при 3,95 мин. 1H ЯМР (500 МГц, ДМСО-d6) δ 11,00 (s, 1Н), 9,87 (d, J=7,8 Гц, 1Н), 9,38 (dd, J=7,0, 1,7 Гц, 1Н), 8,93 (dd, J=4,3, 1,7 Гц, 1Н), 8,66 (d, J=2,3 Гц, 1Н), 8,09 (dd, J=8,9, 2,4 Гц, 1Н), 7,70-7,63 (m, 1Н), 7,56-7,41 (m, 5Н), 7,39-7,32 (m, 3Н), 7,32-7,25 (m, 1Н), 6,80 (d, J=9,0 Гц, 1Н), 5,52 (d, J=7,8 Гц, 1Н), 4,42-4,36 (m, 1Н), 3,98-3,91 (m, 2Н), 3,73 (d, J=11,3 Гц, 1Н), 3,63 (dd, J=11,3, 3,1 Гц, 1Н), 3,48 (td, J=11,3, 3,1 Гц, 1Н), 3,15-3,05 (m, 1Н), 1,15 (d, J=6,7 Гц, 3Н).
125. N-[(3S)-9-фтор-2-оксо-5-фенил-2,3-дигидро-1H-1,4-бензодиазепин-3-ил]-2-{6-[(3S*)-3-метилморфолин-4-ил]пиридин-3-ил}пиразоло[1,5-а]пиримидин-3-карбоксамид
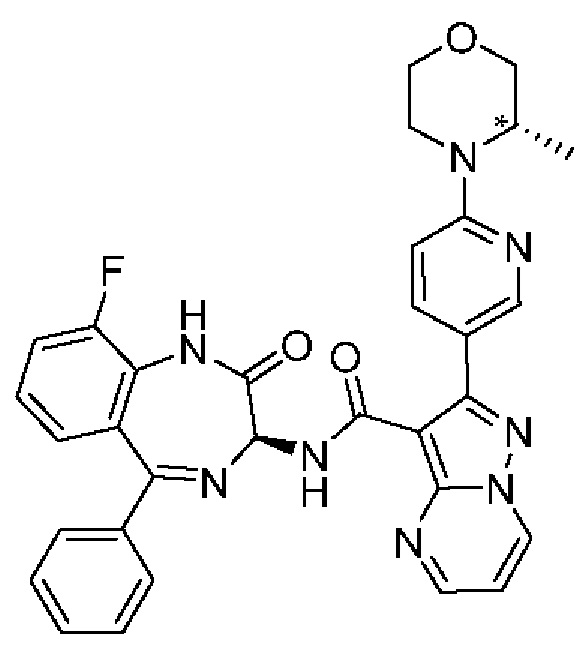
Соединение согласно Примеру 68 (40 мг) очищали методом препаративной хиральной ВЭЖХ [50% раствор смеси MeOH-MeCN (1:1) в 0,1% растворе аммиака], в результате чего получали указанное в заголовке соединение в форме единого диастереомера с неизвестной абсолютной конфигурацией и произвольно определенным стереоцентром, помеченным звездочкой. Второй элюирующий энантиомер. Твердое вещество желтого цвета (3,8 мг, выход 9%). Аналитическая СФХ: 7,86 мин. ЖХ-МС (Метод D) m/z 591,5 [М+Н]+ при 3,94 мин. 1Н ЯМР (500 МГц, ДМСО-в6) δ 9,88 (d, J=7,8 Гц, 1Н), 9,38 (dd, J=7,0, 1,7 Гц, 1Н), 8,93 (dd, J=4,3, 1,7 Гц, 1Н), 8,66 (d, J=2,3 Гц, 1Н), 8,09 (dd, J=8,9, 2,3 Гц, 1Н), 7,65-7,58 (m, 1Н), 7,57-7,50 (m, 3Н), 7,50-7,41 (m, 2Н), 7,39-7,30 (m, 2Н), 7,19 (d, J=7,9 Гц, 1Н), 6,80 (d, J=8,9 Гц, 1Н), 5,60 (d, J=7,7 Гц, 1Н), 4,42-4,36 (m, 1Н), 3,98-3,92 (m, 2Н), 3,76-3,70 (m, 1Н), 3,67-3,60 (m, 1Н), 3,52-3,43 (m, 1Н), 3,15-3,05 (m, 1Н), 1,15 (d, J=6,7 Гц, 3Н).
Пример 127: Эффективность в условиях in vitro
Соединения были подвергнуты анализу на подавление РСВ бляшкообразование согласно приведенному далее протоколу. Значения полуэффективной концентрации (ЕС50) по подавлению бляшкообразования и половинной цитотоксической концентрации (СС50) являются средними значениями по результатам по меньшей мере двух экспериментов, и полученные цифры округлены до целых единиц.
Анализ на подавление бляшкообразования
Клетки Hep-G2 (ЕСАСС, 85011430) пересевали в колбах и высевали в 24-луночные планшеты в среду DMEM, содержащую антибиотики, с добавлением 10% FBS. Во время инокуляции и последующего инкубирования клетки культивировали в среде DMEM, содержащей 2% FBS. В каждую лунку помещали по 100 бляшкообразующих единиц РСВ (РСВ А2 ЕСАСС, 0709161v) и смешивали их с восемью последовательными разбавлениями исследуемого соединения. Затем к конфлюэнтным монослоям клеток Hep-G2 добавляли по 100 мкл смесей вируса с исследуемым соединением. Клетки и смеси вирус/соединение инкубировали при температуре 37°С в увлажненном инкубаторе с 5% СО2 в течение 2 часов, после чего удаляли инокулят и добавляли 1 мл покровного слоя (среды DMEM, содержащей 2% FBS и 0,8% CMC) с соответствующими разбавлениями исследуемого соединения. Клетки и смеси инкубировали при 37°С в увлажненном инкубаторе с 5% СО2 в течение 2 суток.
Клетки промывали PBS в течение 3 минут, после чего добавляли смесь EtOH и МеОН (75/25% об./об.). Фиксатор удаляли, и планшеты промывали PBS. Предварительно оттитрованное количество первичного антитела добавляли к 200 мкл PBS с 2% сухого молока, и планшеты инкубировали в течение 90 минут при 37°С. Планшеты 3 раза промывали PBS с 0,05% Tween 20, после чего добавляли конъюгат кроличьих антител к Ig козы с пероксидазой хрена в 200 мкл PBS с 2% сухого молока и инкубировали в течение 1 часа при 37°С. После трех стадий промывки посредством PBS с 0,05% Tween 20 добавляли 200 мкл готового к применению TrueBlue, и планшеты инкубировали при комнатной температуре в течение 10-15 минут, после чего промывали водой. После удаления воды планшеты сушили на воздухе в темноте.
Затем планшеты сканировали и анализировали с помощью макроанализатора Immunospot S6, оснащенного аналитическим программным обеспечением BioSpot для подсчета иммуноокрашенных бляшек (вироспотов). Значения счета бляшек использовали для расчета % заражения относительно среднего счета бляшек в лунках вирусного контроля для РСВ. Величины ЕС50 рассчитывали по уменьшению сигнала на 50%, соответственно, путем интерполяции кривых ингибирования, аппроксимированных по 4-параметрической нелинейной регрессии с переменным наклоном в приложении Dotmatics. Величины полуэффективной концентрации (ЕС50) по подавлению бляшкообразования и половинной цитотоксической концентрации (СС50) брали как средние значения по результатам по меньшей мере двух экспериментов, и полученные цифры округляли до целых единиц.
Результаты
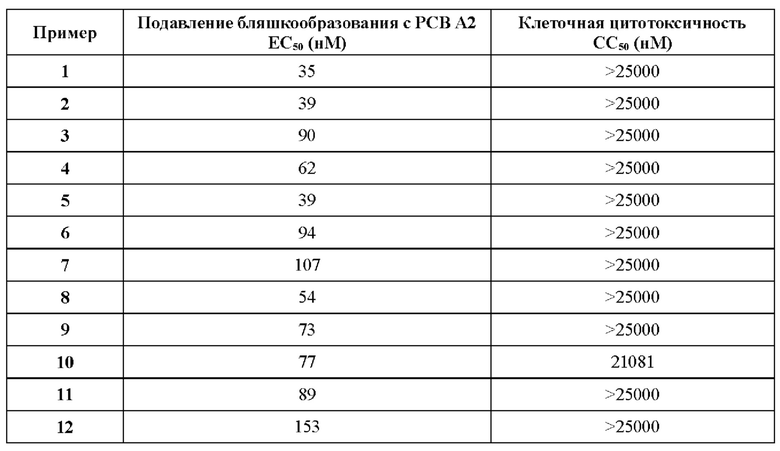
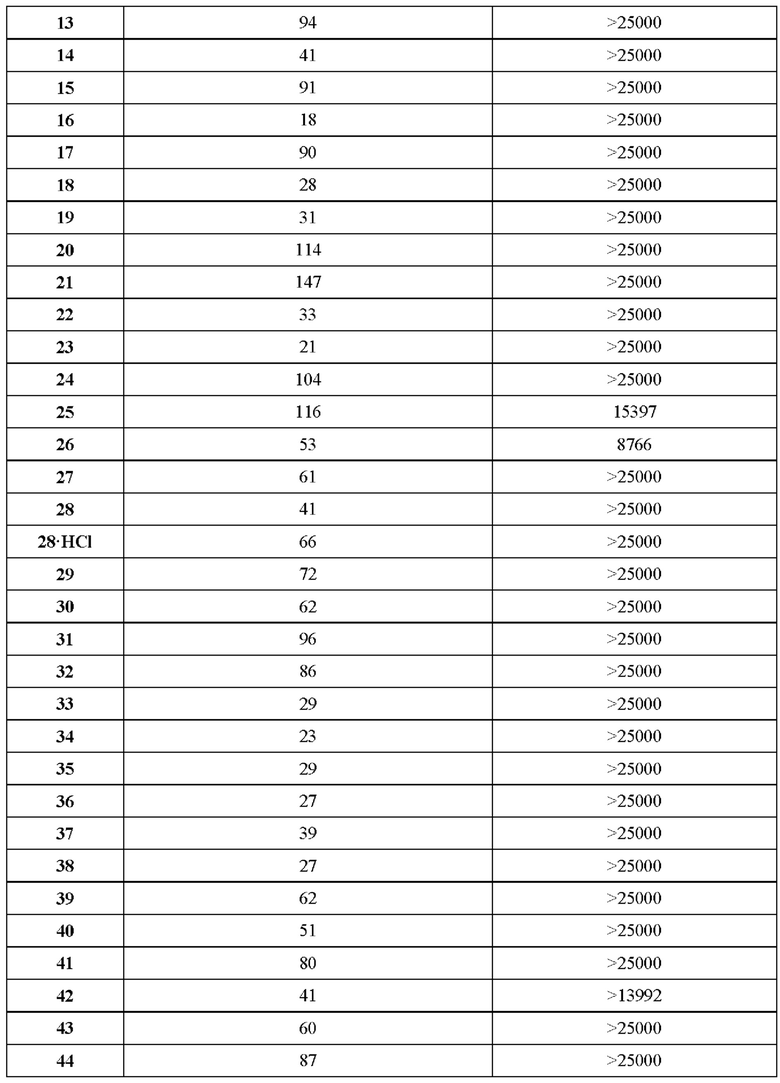
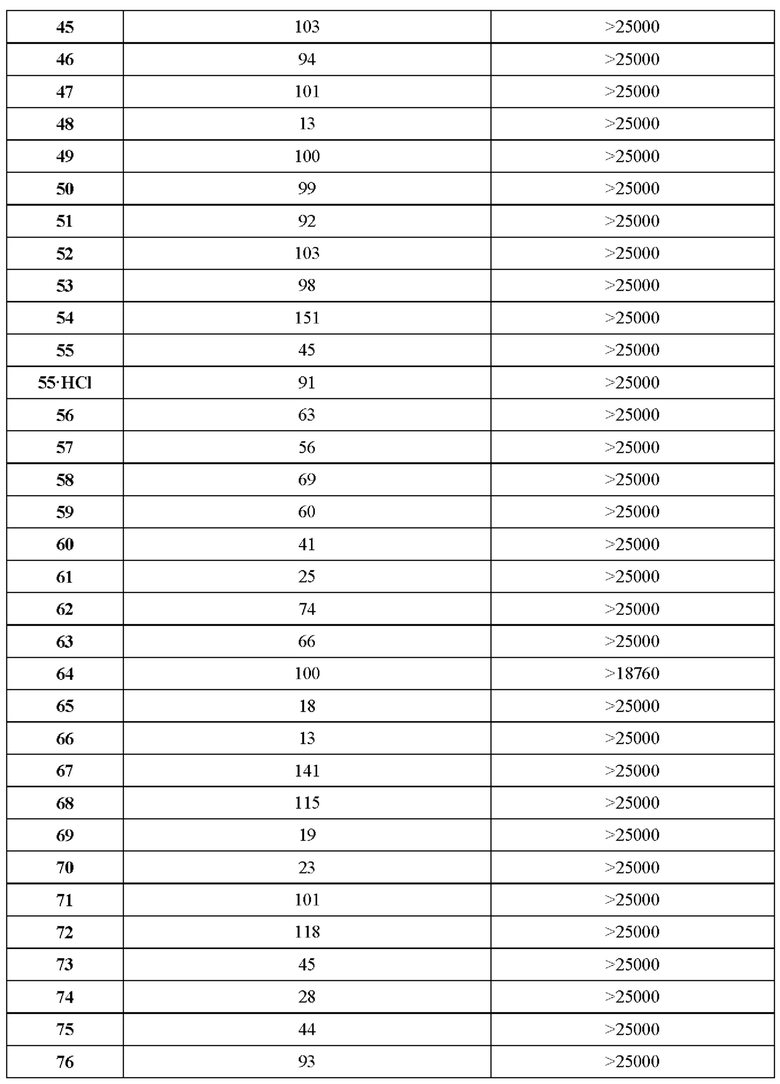
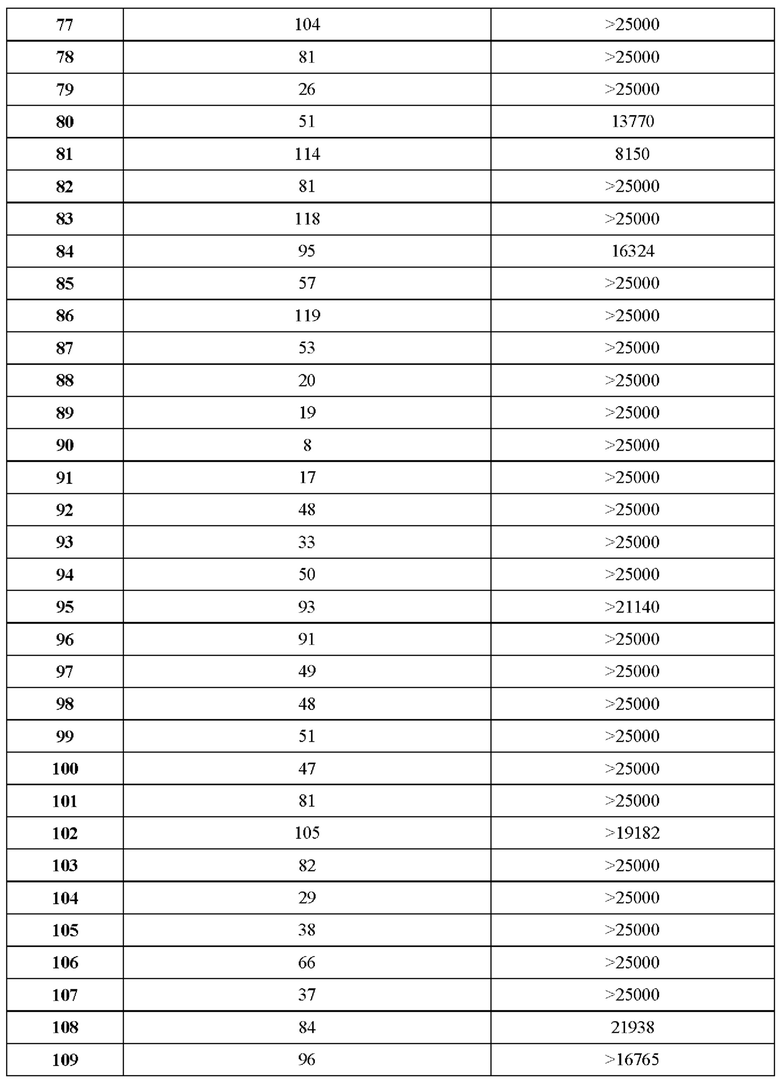
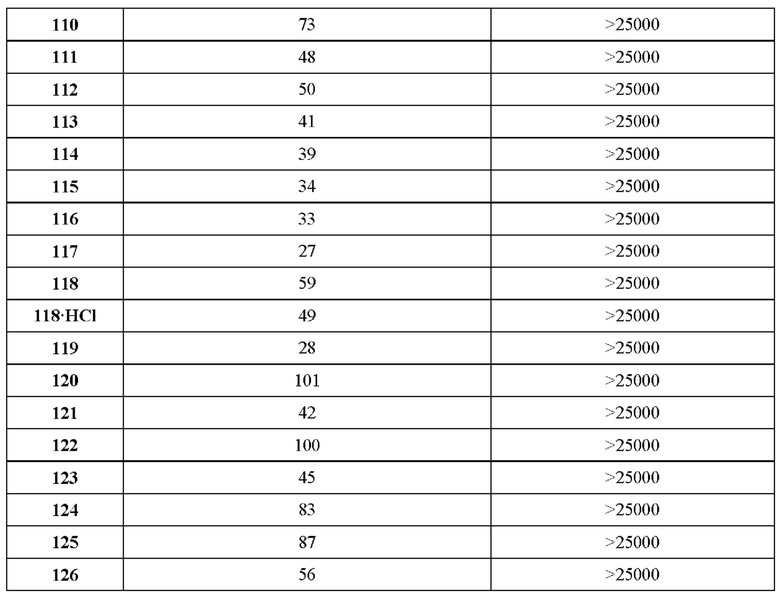
Пример 128: Термодинамическая растворимость
Во флаконе из янтарного стекла брали навеску испытуемого соединения (2,5 мг твердого вещества; n=1) и добавляли буфер (0,5 мл) (обычно фосфатно-солевой буфер, рН 7,4). Раствор перемешивали при температуре окружающей среды в течение ночи на роликовой мешалке для флаконов. Затем раствор фильтровали (через фильтр с размер пор 0,45 мкм без предварительного насыщения). Брали по две аликвоты (50 мкл) фильтрата и разбавляли их одним объемом смеси воды Milli-Q и метанола (1:1 об./об.), после чего подвергали анализу методом ВЭЖХ-УФ. Стандарт готовили в ДМСО в концентрации 10 мг/мл (n=1), который затем разбавляли в 10 раз смесью воды Milli-Q и метанола (1:1 об./об.), так чтобы получить раствор с концентрацией 1 мг/мл. Концентрацию тестируемого соединения в фильтрате количественно определяли относительно концентрации стандарта. Анализ проводили на градиентной системе ВЭЖХ-МС.
Результаты

Пример 129: Фармакокинетика в условиях in vitro
Соединения подвергали следующей процедуре анализа с целью исследования их стабильности в микросомах печени и гепатоцитах.
Микросомальная инкубация: Процедура эксперимента
Объединенные микросомы печени приобретали у заслуживающего доверия коммерческого поставщика и до использованием хранили при температуре -80°С. Микросомы (конечная концентрация белка 0,5 мг/мл), 0,1М фосфатный буфер с рН 7,4 и испытуемое соединение (конечная концентрация субстрата 1 мкМ; конечная концентрация ДМСО 0,25%) предварительно инкубировали при температуре 37°С, после чего добавляли NADPH (конечная концентрация 1 мМ) для инициирования реакции. Конечный инкубируемый объем составлял 50 мкл. По каждому исследуемому соединению проводили контрольную инкубацию, где вместо NADPH добавляли 0,1М фосфатный буфер с рН 7,4 (контроль «минус NADPH»). Для каждого вида микросомов также включали по два контрольных соединения. Все инкубации выполняли для каждого испытуемого соединения индивидуально. Каждое соединение инкубировали в течение 0, 5, 15, 30 и 45 мин. Контроль «минус NADPH» инкубировали в течение только 45 мин. Реакции останавливали путем переноса инкубата в ацетонитрил в соответствующие моменты времени в соотношении 1:3. Терминационные планшеты центрифугировали при 3000 об/мин в течение 20 мин при температуре 4°С для осаждения белка. После осаждения белка супернатанты образцов объединяли в кассеты, содержащие до 4 соединений, добавляли внутренний стандарт, и образцы анализировали методом ЖХ-МС/МС. Из графика временной зависимости ln соотношения площадей пиков (площадь пика соединения/площадь пика внутреннего стандарта) определяли наклон линии. Затем рассчитывали период полураспада (t1/2) и собственный клиренс (CLint). Соединения с низким клиренсом (>80%, остающиеся через 45 мин) в условиях анализа обозначали как t1/2 >140 мин.
Результаты
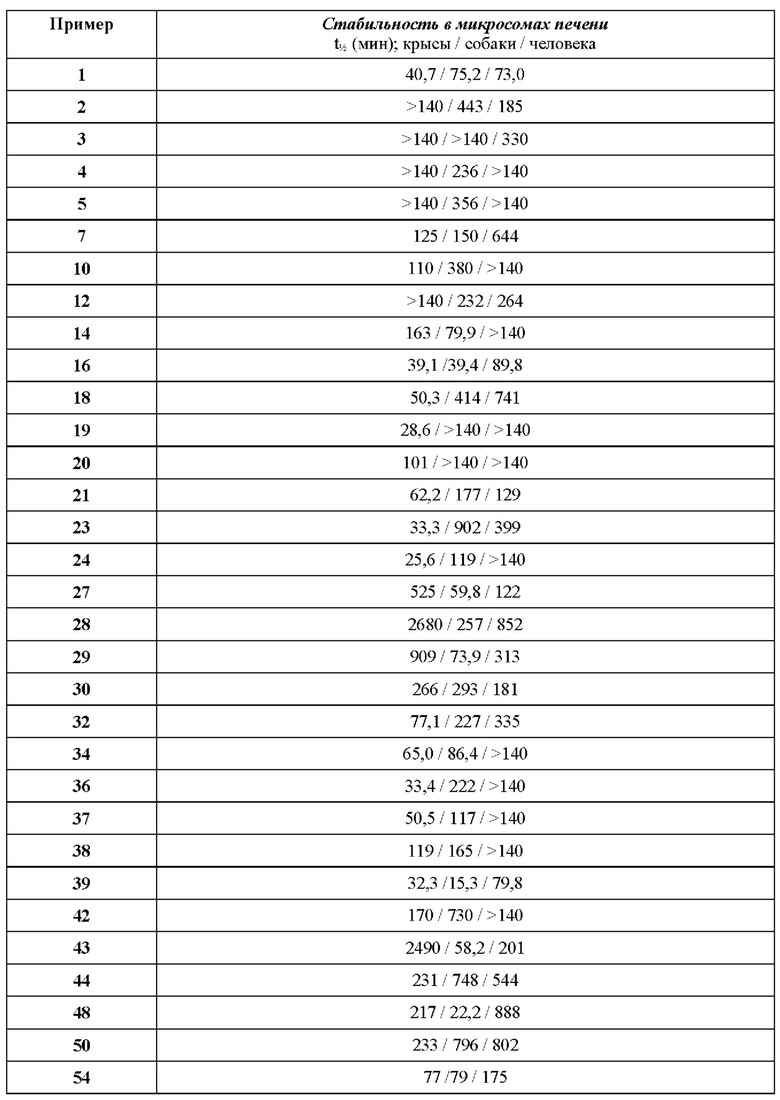
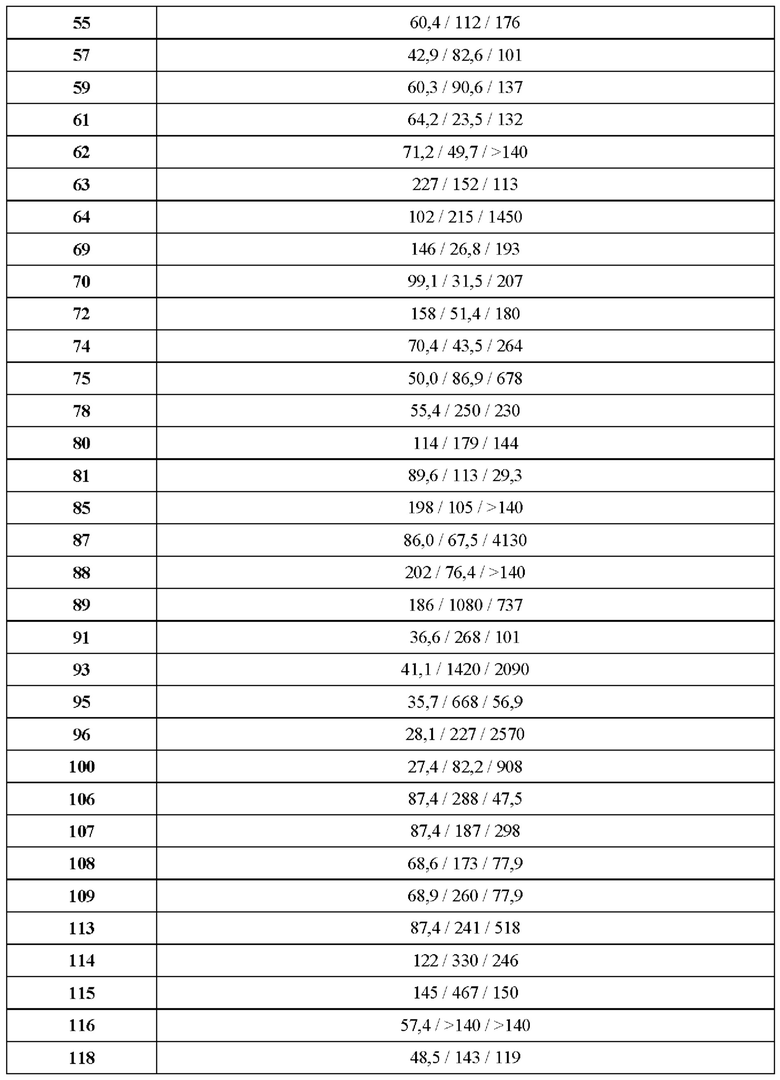

Гепатоцитарная инкубация: Процедура эксперимента
Криоконсервированные объединенные гепатоциты приобретали у заслуживающего доверия коммерческого поставщика и до использованием хранили в жидком азоте. Среду Williams Е с добавлением 2 мМ L-глутамина и 25 мМ HEPES и испытуемое соединение (конечная концентрация субстрата 3 мкМ; конечная концентрация ДМСО 0,25%) предварительно инкубировали при 37°С, после чего к ним добавляли суспензию криоконсервированных гепатоцитов (конечная плотность клеток 0,5×106 жизнеспособных клеток/мл в среде Williams Е, дополненной 2 мМ L-глутамина и 25 мМ HEPES) для инициирования реакции. Конечный объем инкубации составил 500 мкл. Для каждого вида гепатоцитов включали по два контрольных соединения, наряду с подходящим контрольным носителем. В подходящие моменты времени реакции останавливали путем переноса 50 мкл инкубата в 100 мкл ацетонитрила, содержащего внутренний стандарт. Образцы отбирали в 6 временных точках (0, 5, 15, 30, 45 и 60 мин) на протяжении 60 минут эксперимента. Терминационные планшеты центрифугировали при 2500 об/мин при 4°С в течение 30 мин для осаждения белка. После осаждения белка супернатанты образцов объединяли в кассеты, содержащие до 4 соединений и анализировали методом ЖХ-МС/МС в стандартных условиях. Из графика временной зависимости ln соотношения площадей пиков (площадь пика соединения/площадь пика внутреннего стандарта) определяли наклон линии. Затем рассчитывали период полураспада (t1/2) и собственный клиренс (CLint). Соединения с низким клиренсом (>80%, остающиеся через 60 мин) в условиях анализа обозначали как t1/2 > 186 мин.
Результаты
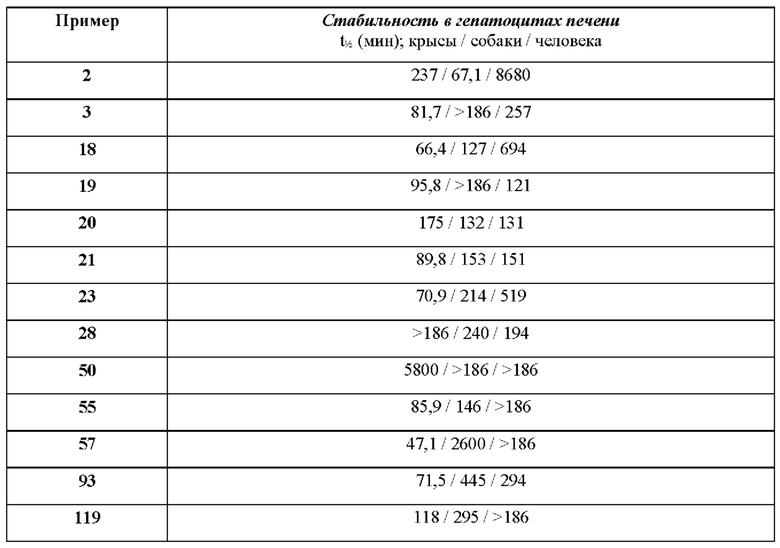
Пример 130: Фармакокинетика в условиях in vivo
Фармакокинетику исследуемых соединений изучали in vivo у крыс при дозах 1 мг/кг (внутривенно) и 10 мг/кг (перорально).
Фармакокинетика у крыс Процедура
Самцов крыс [Sprague Dawley (SD)], хирургически подготовленных установкой яремной венозной канюли, обрабатывали экспериментальными соединениями путем внутривенного введения (в/в; n=3; 1 мг/кг) или перорального введения (п/о; n=3; 10 мг/кг). Соединения были представлены в форме раствора в смеси диметилацетамида и солевого раствора 40:60 (при в/в введении) и раствора в смеси 10% ДМСО, 20% кремафора и воды (70%) (при п/о введении).
Животных наблюдали на предмет любых явных клинических признаков или симптомов. Серийные образцы крови отбирали из канюли через 0,02, 0,08, 0,25, 0,5, 1, 2, 4, 6, 8 и 24 ч после в/в дозирования соединения, и через 0,08, 0,25, 0,5, 1, 2, 4, 6, 8 и 24 ч после перорального дозирования соединения, плазму готовили центрифугированием и немедленно сохраняли при -80°С. Затем образцы размораживали, готовили для анализа осаждением белка ацетонитрилом и анализировали методом тандемной ЖХ-МС с использованием электрораспылительной ионизации и калибровочной кривой с согласованной матрицей. Из полученных данных рассчитывали фармакокинетические параметры.
Результаты
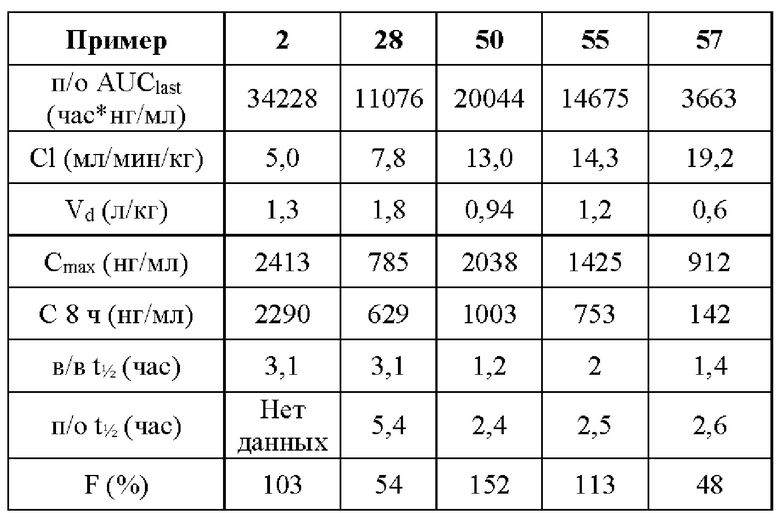
Фармакокинетика у собак
Фармакокинетику исследуемых соединений изучали in vivo у собак при дозах 0,5 мг/кг (внутривенно) и 4 мг/кг (перорально).
Процедура
Самцы собак породы бигль получали экспериментальные соединения путем внутривенного введения (n=2; 0,5 мг/кг) или перорального введения (n=2; 3 или 4 мг/кг). Состав соединений составляли в виде раствора (20% масс./об.) в смеси 20% диметилацетамида и 80% (2-гидроксипропил)-β-циклодекстрина (при в/в введении) или раствора (20% масс/об.) в смеси 10% диметилацетамиде и 90% (2-гидроксипропил)-β-циклодекстрина (при п/о введении). Животных наблюдали на предмет любых явных клинических признаков или симптомов.
Серийные образцы крови брали из яремной вены через 0,03, 0,08, 0,25, 0,5, 1, 2, 4, 6, 8 и 24 ч после в/в дозирования соединения, и через 0,08, 0,25, 0,5, 1, 2, 4, 6, 8 и 24 ч после перорального дозирования соединения, и плазму готовили центрифугированием и немедленно сохраняли при -80°С. Затем образцы размораживали, готовили для анализа осаждением белка ацетонитрилом и анализировали методом тандемной ЖХ-МС с использованием электрораспылительной ионизации и калибровочной кривой с согласованной матрицей. Из полученных данных рассчитывали фармакокинетические параметры.
Результаты
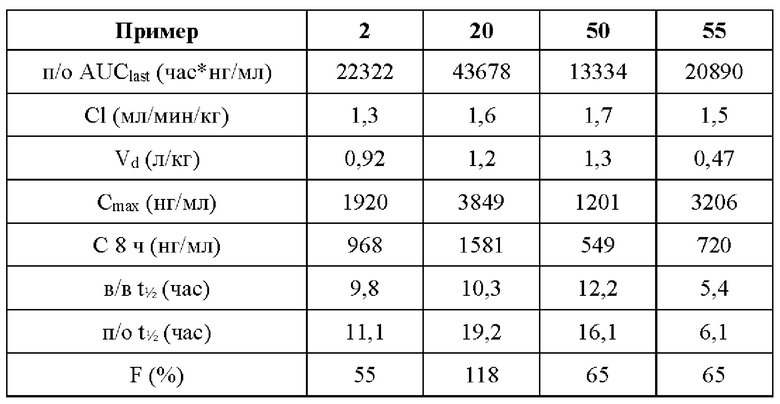
Пример 131: Форма водного раствора
Готовили состав соединения согласно Примеру 1 в форме раствора в 30% (масс./об.) растворе каптизола (т.е. сульфобутилового эфира бета-циклодекстрина) при рН=4 в соответствии со следующей процедурой.
Носитель, представляющий собой 30% (масс./об.) раствор каптизола (т.е. сульфобутилового эфира бета-циклодекстрина), получали путем взвешивания в подходящем сосуде необходимого количества каптизола, добавления примерно 80% от итогового объема воды и перемешивания на магнитной мешалке до образования раствора. Затем носитель доводили до конечного объема водой.
Водный раствор соединения согласно Примеру 1 получали путем взвешивания 175 мг указанного соединения в подходящем сосуде и добавления примерно 80% от требуемого объема носителя. Используя водный раствор хлористоводородной кислоты, рН получаемого раствора доводили до уровня рН=2, и полученную смесь перемешивали на магнитной мешалке до образования раствора. Затем объем водного раствора доводили до итогового уровня носителем, и его рН доводили до рН=4 водным раствором гидроксида натрия.
Пример 132: Таблеточная форма
Таблетки массой 0,15 г каждая, содержащие 25 мг соединения согласно настоящему изобретению, изготавливали следующим образом:
Состав для получения 10000 таблеток
Соединение согласно настоящему изобретению (250 г)
Лактоза (800 г)
Кукурузный крахмал (415 г)
Тальк, порошок (30 г)
Стеарат магния (5 г)
Смешивали соединение согласно настоящему изобретению, лактозу и половину кукурузного крахмала. Затем смесь продавливали через сито с ячейками 0,5 мм. Суспендировали кукурузный крахмал (10 г) в теплой воде (90 мл). Полученную пасту использовали для гранулирования порошка. Гранулят сушили и разбивали на мелкие фрагменты на сите с ячейками 1,4 мм. Добавляли оставшееся количество крахмала, тальк и магний, тщательно перемешивали, и формовали таблетки.
Пример 133: Инъекционная форма
Соединение согласно настоящему изобретению растворяли в большей части воды (при 35°С-40°С), и при необходимости рН полученного раствора доводили до уровня 4,0-7,0 хлористоводородной кислотой или гидроксидом натрия. Затем полученный раствор доводили до итогового объема водой и фильтровали через стерильный микропористый фильтр в стерильный флакон янтарного стекла (типа 1) объемом 10 мл, после чего его укупоривали стерильной пробкой с обжимным колпачком.
Пример 134: Форма для внутримышечных инъекций
Соединение согласно настоящему изобретению растворяли в гликофуроле. Затем добавляли и растворяли бензиловый спирт, и добавляли воду до итогового объема 3 мл. Затем смесь фильтровали через стерильный микропористый фильтр и герметизировали в стерильный стеклянный флакон (типа 1) объемом 3 мл.
Пример 135: Форма сиропа
Соединение согласно настоящему изобретению растворяли в смеси глицерина и большей части очищенной воды. Затем к раствору добавляли водный раствор бензоата натрия, после чего добавляли раствор сорбита и в последнюю очередь ароматизатор. Полученный раствор доводили до итогового объема очищенной водой и хорошо перемешивали.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2013 |
|
RU2646223C2 |
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| НОВЫЕ ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2607788C2 |
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2693629C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ПИРАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2686117C1 |
| НОВЫЕ ФОСФАТНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2617682C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
Группа изобретений относится к области органической химии и включает индивидуальные соединения, приведенные в формуле изобретения, их фармацевтически приемлемые соли и способ получения, а также фармацевтическую композицию, применение и способ лечения на их основе. Технический результат: производные бензодиазепина, представленные в формуле изобретения, которые являются ингибиторами РСВ (респираторно-синцитиальный вирус) и применяются для лечения или предотвращения РСВ-инфекции. 5 н. и 2 з.п. ф-лы, 14 табл., 135 пр.
1. Соединение, выбранное из:
2-[4-(Метиламино)фенил]-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2-Фторфенил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенилпиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-[4-(пропан-2-иламино)фенил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(1-Метил-2,3-дигидроиндол-6-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2-Фтор-4-метоксифенил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2-Фтор-4-пропан-2-илоксифенил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-[2-Фтор-4-(метиламино)фенил]-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенилпиразоло[1,5-a]пиримидин-3-карбоксамид;
6-Хлор-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-метилимидазо[1,2-b]пиридазин-3-карбоксамид;
6-Хлор-2-метил-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид; и
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-(фуран-3-ил)-2-метилпиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2-Фтор-5-метилфенил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид
2-(2-Фтор-5-метилфенил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид
2-(5-Хлорпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид
2-(5-Хлорпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(6-Циклопропилпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(6-Циклопропилпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(5-Циклопропилпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2-Фтор-5-метилпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2-Фторфенил)-6-метил-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-метилпиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2,4-Дифторфенил)-5-метил-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2,4-Дифторфенил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-5-метилпиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2-Фторфенил)-7-метил-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-7-метилпиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2-Фторфенил)-5-(морфолин-4-ил)-N-[(3S)-2-оксо-5-фенил-2,3-дигидро-1H-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-5-пирролидин-1-илпиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2-Фторфенил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-5-пирролидин-1-илпиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2,3-Дифторфенил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2,3-Дифторфенил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2,6-Дифторфенил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2-Фтор-4-пирролидин-1-илфенил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
6-Метил-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-[5-(трифторметил)пиридин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метил-2-[5-(трифторметил)пиридин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
2-[6-(Циклопропиламино)-2-фторпиридин-3-ил]-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(6-Метилпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(6-метилпиридин-3-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-[6-(2-Метоксиэтил)пиридин-3-ил]-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-метилпиридин-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(6-Этоксипиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(6-Этилпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(6-Этилпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(6-Этил-2-метилпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-2,3-дигидро-1H-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(6-Этил-2-метилпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-2,3-дигидро-1H-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(6-Пропан-2-илпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(6-Пропан-2-илпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-[2-Метил-6-(пропан-2-иламино)пиридин-3-ил]-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-[2-Метил-6-(пропан-2-иламино)пиридин-3-ил]-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-[4-Метил-6-(пропан-2-иламино)пиридин-3-ил]-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(1-метилиндазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(5-метилпиридин-3-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-[6-(Этиламино)-2-фторпиридин-3-ил]-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-[6-(Этиламино)-2-фторпиридин-3-ил]-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-[6-(3-Метилморфолин-4-ил)пиридин-3-ил]-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-[6-(3-Метилморфолин-4-ил)пиридин-3-ил]-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-[2-Фтор-6-(пропан-2-иламино)пиридин-3-ил]-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-[2-фтор-6-(пропан-2-иламино)пиридин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(1-метилпирроло[2,3-b]пиридин-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(6-морфолин-4-илпиридин-3-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-[6-(пропан-2-иламино)пиридин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-[6-(пропан-2-иламино)пиридин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(1H-пирроло[2,3-b]пиридин-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(6-метоксипиридин-3-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-[6-(2-Гидрокси-2-метилпропил)пиридин-3-ил]-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(5-фторпиридин-2-ил)-6-метилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-морфолин-4-илимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-[(1S,4S)-5-метил-2,5-диазабицикло[2.2.1]гептан-2-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-[(1R,4R)-5-метил-2,5-диазабицикло[2.2.1]гептан-2-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-[(1R,4R)-2-окса-5-азабицикло[2.2.1]гептан-5-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-морфолин-4-ил-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-5-[(1R,4R)-5-метил-2,5-диазабицикло[2.2.1]гептан-2-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2-Фтор-6-метилпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
2-(2-Метокси-6-метилпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
2-(2,4-Дифторфенил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
2-(2,4-Дифторфенил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
2-(2,4-Дифторфенил)-6-метил-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
2-(2,4-Дифторфенил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метилимидазо[1,2-b]пиридазин-3-карбоксамид;
6-Метил-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метил-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
6-(Азетидин-1-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-(4-метилпиперазин-1-ил)имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метил-2-пиридин-3-илимидазо[1,2-b]пиридазин-3-карбоксамид;
6-Метил-2-(2-метилпиридин-4-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метил-2-(2-метилпиридин-4-ил)имидазо[1,2-b]пиридазин-3-карбоксамид;
2-(3-Фторпиридин-4-ил)-6-метил-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(3-фторпиридин-4-ил)-6-метилимидазо[1,2-b]пиридазин-3-карбоксамид;
2-(5-Фторпиридин-3-ил)-6-метил-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(5-фторпиридин-3-ил)-6-метилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метил-2-(5-метилпиридин-3-ил)имидазо[1,2-b]пиридазин-3-карбоксамид;
6-Метил-2-(6-морфолин-4-илпиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метил-2-(6-морфолин-4-илпиридин-3-ил)имидазо[1,2-b]пиридазин-3-карбоксамид;
6-(Этиламино)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-(1-метилазетидин-3-ил)оксиимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-(1-метилазетидин-3-ил)окси-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-(2-метоксиэтокси)имидазо[1,2-b]пиридазин-3-карбоксамид;
6-Метокси-N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенил-6-(тридеутериометокси)имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метокси-2-фенилимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-фенил-6-(тридеутериометокси)имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)-6-метоксиимидазо[1,2-b]пиридазин-3-карбоксамид;
6-Хлор-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(2-фторфенил)имидазо[1,2-b]пиридазин-3-карбоксамид;
6-Хлор-2-(6-этилпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метокси-2-(5-метилпиридин-3-ил)имидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(3-фторпиридин-4-ил)-6-метоксиимидазо[1,2-b]пиридазин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-6-метокси-2-(6-метилпиридин-3-ил)имидазо[1,2-b]пиридазин-3-карбоксамид;
2-(6-Этилпиридин-3-ил)-6-метокси-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
6-Этокси-2-(6-этилпиридин-3-ил)-N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]имидазо[1,2-b]пиридазин-3-карбоксамид;
2-(6-Этилпиридин-3-ил)-N-[(3S)9-фтор-2-оксо-5-фенил-2,3-дигидро-1H-1,4-бензодиазепин-3-ил]-6-(2H3)метоксиимидазо[1,2-b]пиридазин-3-карбоксамид;
2-(6-[(3S*)-3-метилморфолин-4-ил]пиридин-3-ил)-N-[(3S)-2-оксо-5-фенил-2,3-дигидро-1H-1,4-бензодиазепин-3-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-2,3-дигидро-1H-1,4-бензодиазепин-3-ил]-2-{6-[(3S*)-3-метилморфолин-4-ил]пиридин-3-ил}пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-2,3-дигидро-1H-1,4-бензодиазепин-3-ил]-2-{6-[(3S*)-3-метилморфолин-4-ил]пиридин-3-ил}пиразоло[1,5-a]пиримидин-3-карбоксамид;
N-[(3S)-9-фтор-2-оксо-5-фенил-1,3-дигидро-1,4-бензодиазепин-3-ил]-2-(1H-индазол-5-ил)имидазо[1,2-b]пиридазин-3-карбоксамид;
или их фармацевтически приемлемые соли.
2. Фармацевтическая композиция, обладающая анти-РСВ активностью, содержащая эффективное количество соединения по п. 1 и фармацевтически приемлемый носитель или разбавитель.
3. Соединение по п. 1 для применения для лечения или предотвращения инфекции РСВ.
4. Применение соединения по п. 1 для получения лекарственного средства для лечения или предотвращения инфекции РСВ.
5. Способ лечения субъекта, страдающего от инфекции РСВ или восприимчивого к ней, включающий введение указанному субъекту эффективного количества соединения по п. 1.
6. Способ получения фармацевтически приемлемой соли по п. 1, включающий обработку соединения по п. 1 подходящей кислотой в подходящем растворителе.
7. Способ по п. 6, отличающийся тем, что указанная кислота выбрана из хлористоводородной кислоты, бромистоводородной кислоты, иодистоводородной кислоты, серной кислоты, азотной кислоты, фосфорной кислоты, метансульфоновой кислоты, бензолсульфоновой кислоты, муравьиной кислоты, уксусной кислоты, трифторуксусной кислоты, пропионовой кислоты, щавелевой кислоты, малоновой кислоты, янтарной кислоты, фумаровой кислоты, малеиновой кислоты, молочной кислоты, яблочной кислоты, винной кислоты, лимонной кислоты, этансульфоновой кислоты, аспарагиновой кислоты и глутаминовой кислоты.
| WO 2005089771 A1, 29.09.2005 | |||
| WO 2004026843 A1, 01.04.2004 | |||
| Ионный выпрямитель | 1929 |
|
SU23262A1 |
| WO 2006113140 A2, 26.10.2006 | |||
| Evans B.E | |||
| et al., Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists | |||
| Journal of medicinal chemistry, 1988, 31, p.2235-2246. | |||

