Область изобретения
[001] В данном раскрытии представлены замещенные пиразолопиримидины и замещенные пурины в качестве ингибиторов убиквитин-специфической процессирующей протеазы 1 (USP1) и терапевтические способы лечения состояний и заболеваний, при которых ингибирование USP1 дает пользу. В частности, в данном раскрытии представлены способы лечения рака путем введения ингибитора USP1.
Уровень техники
[002] Убиквитин представляет собой небольшой (76 аминокислот) белок, который посттранс крипционно прикрепляется к целевым белкам. Последствия убиквитинирования определяются количеством и топологией связи молекул убиквитина, конъюгированных с целевым белком. Например, белки, демонстрирующие цепи полиубиквитина, связанные с лизином 48, обычно нацелены на протеасому для деградации, в то время как цепи моноубиквитина или полиубиквитина, связанные через другие лизины, регулируют непротеолитические функции, такие как регуляция клеточного цикла, восстановление повреждений ДНК, транскрипция и эндоцитоз. Убиквитинирование представляет собой обратимый процесс, и ферменты, называемые деубиквитиназами, удаляют убиквитин из целевых белков.
[003] USP1 представляет собой деубиквитиназу, которая участвует в восстановлении повреждений ДНК. USP1 взаимодействует с UAF1 (ассоциированный с USP1 фактор 1) с образованием комплекса, необходимого для активности деубиквитиназы. Комплекс USP1/UAF1 деубиквитинирует моноубиквитинированный PCNA (ядерный антиген пролиферирующих клеток) и моноубиквитинированный FANCD2 (группа комплементации группы анемии Фанкони D2), которые являются белками, которые играют важные функции в синтезе трансфузии (TLS) и пути анемии Фанкони (FA), соответственно. Комплекс USP1/UAF1 также деубиквитинирует группу комплементации анемии Фанкони I (FANCI). Эти два пути важны для восстановления повреждений ДНК, вызванных сшивающими ДНК агентами, такими как цисплатин и митомицин С (ММС).
[004] Безопасные и эффективные методы лечения, нацеленные на деубиквитиназы, неизвестны, еще не доступны в продаже или еще не разработаны в клинических условиях.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ РАСКРЫТИЯ
[005] В одном аспекте данное раскрытие относится к соединениям или их фармацевтически приемлемой соли или сольвату, имеющим Формулу I (также называемым в данном документе Соединениями раскрытия):
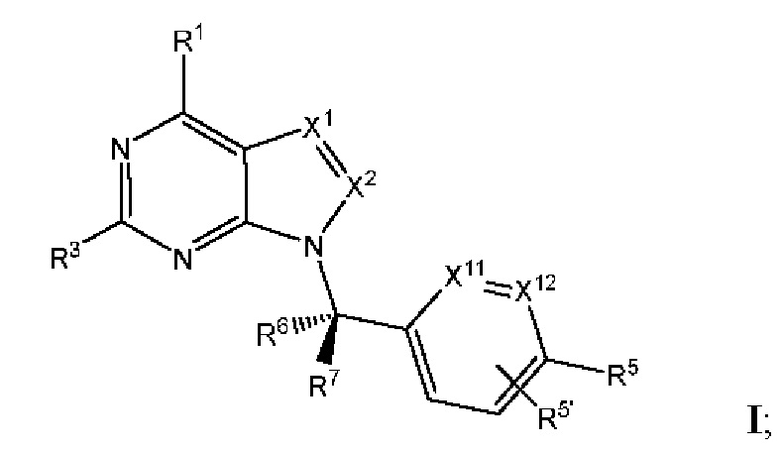
где:
[006] каждый из X1 и X2 независимо выбран из N и CR2;
[007] каждый из R1 и R2 независимо выбран из водорода, галогена, циано, необязательно
замещенного алкила, необязательно замещенного алкенила и необязательно замещенного алкинила;
[008] R3 представляет собой необязательно замещенный фенил, необязательно замещенный пиридил, необязательно замещенный пиримидинил, необязательно замещенный пиразинил, необязательно замещенный пиридазинил или необязательно замещенный пиразолил;
[009] каждый из X11 и Х12 независимо выбран из N и СН;
[010] R5' независимо выбран из водорода, необязательно замещенного (C1-С6) алкила, необязательно замещенного (С2-C6) алкенила, необязательно замещенного (C2-С6) алкинила, необязательно замещенного (C1-C6) алкокси, (C1-C6) галогеналкила, (C1-C6) галогеналкокси, (C1-С6) гидроксиалкила, циано, галогена, сульфонамидо,-C(=О)R23,-C(=О)OR24,-NR32aR32b,-NR31aC(=О)R25,-NR31aC(=О)NR31aR31b,-C(=О)NR31a R31b,-S(О)2R27,-NR31aSO2R27, необязательно замещенного (C6-C14) арила, необязательно замещенного (С6-С14) ар-(С1-С2) алкила, необязательно замещенного гетероарила, необязательно замещенного гетероар-(C1-C2) алкила, необязательно замещенного (С3-C8) циклоалкила, необязательно замещенного ((С3-C8) циклоалкил)-(С1-С2) алкила, необязательно замещенного гетероцикло, необязательно замещенного гетероцикло-(С1-С2) алкила, необязательно замещенного-О-(С6-С14) арила, необязательно замещенного-О-(C6-C14) ap-(C1-С2) алкила, необязательно замещенного-О-гетероарила, необязательно замещенного-О-гетероар-(C1-C2) алкила, необязательно замещенного-О-(C3-C8) циклоалкила, необязательно замещенного-О-((С3-С8) циклоалкил)-(С1-С2) алкила, необязательно замещенного-О-гетероцикло и необязательно замещенного-O-гетероцикло-(С1-С2) алкила;
[011] R5 независимо выбран из необязательно замещенного (C1-C6) алкила, необязательно замещенного (С2-С6) алкенила, необязательно замещенного (С2-С6) алкинила, необязательно замещенного (C1-С6) алкокси, (C1-С6) галогеналкила, (C1-С6) галогеналкокси, (C1-С6) гидроксиалкила, циано, галогена, сульфонамидо,-C(=О)R23,-C(=О)OR24,-NR32aR32b,-NR31aC(=О)R25,-NR31aC(=О)NR31aR31b,-C(=О)NR31aR31b,-S(O)2R27,-NR31aSO2R27, необязательно замещенного (C6-C14) арила, необязательно замещенного (С6-С14) ар-(С1-С2) алкила, необязательно замещенного гетероарила, необязательно замещенного гетероар-(С1-С2) алкила, необязательно замещенного (С3-C8) циклоалкила, необязательно замещенного ((С3-C8) циклоалкил)-(С1-С2) алкила, необязательно замещенного гетероцикло, необязательно замещенного гетероцикло-(С1-С2) алкила, необязательно замещенного-О-(С6-С14) арила, необязательно замещенного-О-(С6-С14) ар-(С1-С2) алкила, необязательно замещенного-О-гетероарила, необязательно замещенного-О-гетероар-(С1-С2) алкила, необязательно замещенного-О-(С3-С8) циклоалкила, необязательно замещенного-О-((С3-С8) циклоалкил)-(С1-С2) алкила, необязательно замещенного-О-гетероцикло и необязательно замещенного-О-гетероцикло-(С1-С2) алкила; или
[012] один из R5 и один из R5' на соседних атомах взяты вместе с атомами, к которым они присоединены, с образованием необязательно замещенного (С6-С14) арильного кольца; или один из R5 и один из R5' на соседних атомах взяты вместе с атомами, к которым они присоединены, с образованием необязательно замещенного гетероарильного кольца; или один из R5 и один из R5' на соседних атомах взяты вместе с атомами, к которым они присоединены, с образованием необязательно замещенного (С3-C8) циклоалкильного кольца; или один из R5 и один из R5' на соседних атомах взяты вместе с атомами, к которым они присоединены, с образованием необязательно замещенного гетероциклоалкильного кольца; или один из R5 и один из R5 на соседних атомах того же атома, к которому они присоединены, взятые вместе, образуя необязательно замещенное спироциклоалкильное кольцо; или один из R5 и один из R5' на соседних атомах того же атома, к которому они присоединены, взятые вместе, образуя необязательно замещенное спирогетероциклоалкильное кольцо;
[013] каждый из R6 и R7 независимо выбран из водорода, галогена, циано, необязательно замещенного алкила, необязательно замещенного алкенила и необязательно замещенного алкинила;
[014] R23 выбран из группы, состоящей из водорода, необязательно замещенного алкила, необязательно замещенного алкенила, необязательно замещенного алкинила, амино, алкиламино, диалкиламино, циклоалкиламино, гидроксиалкила, (амино)алкила, (алкиламино)алкила, (диалкиламино)алкила, (циклоалкиламино)алкила, (циклоалкил)алкила, аралкила, (гетер оцикло)алкила, (гетероарил)алкила, (амино)(гидрокси)алкила, (аралкиламино)алкила, необязательно замещенного гетероцикло, необязательно замещенного гетероарила, необязательно замещенного арила и необязательно замещенного циклоалкила;
[015] R31a и R31b каждый независимо выбран из группы, состоящей из водорода, необязательно замещенного алкила, необязательно замещенного алкенила, необязательно замещенного алкинила, гидроксиалкила, (амино)алкила, (алкиламино)алкила, (диалкиламино)алкила, алкоксиалкила, циклоалкила, (циклоалкил)алкила, (гетероцикло)алкила, аралкила и (гетероарил)алкила; и
[016] каждый из R24, R25, R27, R32a, и R32b независимо выбран из группы, состоящей из водорода, необязательно замещенного алкила, необязательно замещенного алкенила, необязательно замещенного алкинила, амино, алкиламино, диалкиламино, циклоалкиламино, гидроксиалкила, (амино)алкила, (алкиламино)алкила, (диалкиламино)алкила, (циклоалкиламино)алкила, (циклоалкил)алкила, аралкила, (гетероцикло)алкила, (гетероарил)алкила, (амино)(гидрокси) алкила, (аралкиламино)алкила, алкоксиалкила, необязательно замещенного гетероцикло, необязательно замещенного гетероарила, необязательно замещенного арила и необязательно замещенного циклоалкила.
[017] В некоторых вариантах реализации Соединения раскрытия демонстрируют улучшенную растворимость, например, как измерено с помощью анализа растворимости ADME, как раскрыто в данном документе.
[018] В некоторых вариантах реализации Соединения раскрытия демонстрируют улучшенную метаболическую стабильность, например, как измерено с помощью анализов метаболической стабильности микросом печени и гепатоцитов, как описано в данном документе.
[019] В других вариантах реализации Соединения раскрытия демонстрируют улучшенную продолжительность действия и пероральное воздействие in vivo.
[020] В некоторых вариантах реализации Соединение раскрытия представляет собой соединение Формулы I, в которой R5' выбран из водорода, галогена и необязательно замещенного (C1-C6) алкила.
[021] В некоторых вариантах реализации Соединение раскрытия представляет собой соединение, имеющее формулу I, где по меньшей мере один из X11 и X12 представляет собой N. В некоторых вариантах реализации X11 представляет собой N. В некоторых вариантах реализации X12 представляет собой N.
[022] В другом варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где по меньшей мере один из X11 и X12 представляет собой СН. В некоторых вариантах реализации X11 представляет собой СН. В некоторых вариантах реализации X12 представляет собой СН.
[023] В одном варианте реализации Соединение раскрытия представляет собой соединение, имеющее формулу I, где каждый из X11 и Х12 представляет собой N.
[024] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где каждый из X11 и Х12 представляет собой СН.
[025] В некоторых вариантах реализации один из X11 и X12 представляет собой N, а другой из X11 и X12 представляет собой СН. В одном варианте реализации X11 представляет собой N, а X12 представляет собой СН. В другом варианте реализации X11 представляет собой СН и X12 представляет собой N.
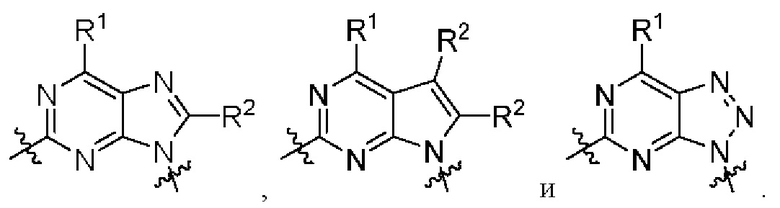
[026] В некоторых вариантах реализации 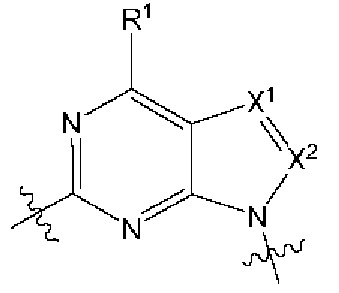 представляет собой
представляет собой 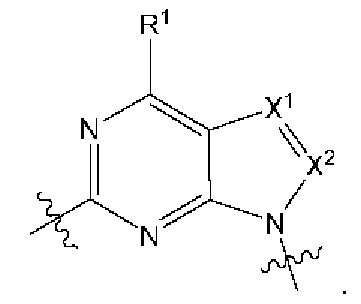
[027] В некоторых вариантах реализации 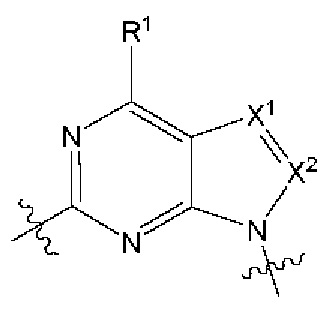 выбран из:
выбран из:
[028] В некоторых вариантах реализации необязательные заместители на R3 независимо выбраны из водорода, галогена, нитро, циано, гидрокси, амино, алкиламино, диалкиламино, галогеналкила, гидроксиалкила, алкокси, галогеналкокси, арилокси, гетероарилокси, аралкиларалкилокси, алкилтио, карбоксамидо, сульфонамидо, сульфонамидо. алкилкарбонила, арилкарбонила алкилсульфонила, арилсульфонила, карбокси, карбоксиалкила, алкила, необязательно замещенного циклоалкила, алкенила, алкинила, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного гетероцикло, алкоксиалкила, (амино)алкила, гидроксиалкиламино, (алкиламино) алкила, (диалкиламино) алкила, (циано)алкила, (карбоксамидо)алкила, меркаптоалкила, (гетероцикло) алкила, (циклоалкиламино)алкила, (С1-4 галогеналкокси)алкила и (гетероарил)алкила; или
[029] два из необязательных заместителей в R3 взяты вместе с атомами углерода или азота, к которым они присоединены, с образованием необязательно замещенной циклоалкильной, необязательно замещенной гетероцикло, необязательно замещенной арильной или необязательно замещенной гетероарильной группы.
[030] В некоторых вариантах реализации R3 представляет собой необязательно замещенный фенил, где фенил необязательно замещен в положении 2, необязательно замещен в положении 6, необязательно дизамещен в положениях 2 и 6 или необязательно дизамещен в положениях 2 и 3.
[031] В некоторых вариантах реализации R3 представляет собой необязательно замещенный
пирид-3-ил или необязательно замещенный пирид-4-ил, где пирид-3-ил необязательно замещен во положении 2, необязательно замещен в положении 4 или необязательно дизамещен во 2-м и 4-м положениях; и где пирид-4-ил необязательно замещен в положении 3, необязательно замещен в положении 5 или необязательно дизамещен в положениях 3 и 5.
[032] В некоторых вариантах реализации R3 представляет собой необязательно замещенный пиримидин-5-ил, где пиримидин-5-ил необязательно замещен в положении 4, необязательно замещен в положении 6, необязательно дизамещен в положениях 4 и 6, или необязательно тризамещен в 2-, 4- и 6-положениях.
[033] В некоторых вариантах реализации R3 представляет собой необязательно замещенный пиразол-5-ил, где пиразол-5-ил необязательно замещен в положении 1, необязательно замещен в положении 4 или необязательно дизамещен в положениях 1 и 4.
[034] В некоторых вариантах реализации R3 замещен, и заместители независимо выбраны из метокси, дейтерометокси, этокси, изопропокси, трет-бутокси, дифторметокси, 2-фторэтокси, 2-метоксиэтокси, циклопропокси, циклобутокси, (тетрагидрофуран-3-ил)окси, бензилокси, метила, этила, изопропила, 2-фторизопропила, трет-бутила, циклопропила, циклобутила, метилциклопропила, пирролидин-1-ила, азетидин-1-ила, метиламино, диметиламино, циано, галогена, метилтио, метилсульфонила и этилсульфонила.
[035] В некоторых вариантах реализации R3 выбран из группы, состоящей из:
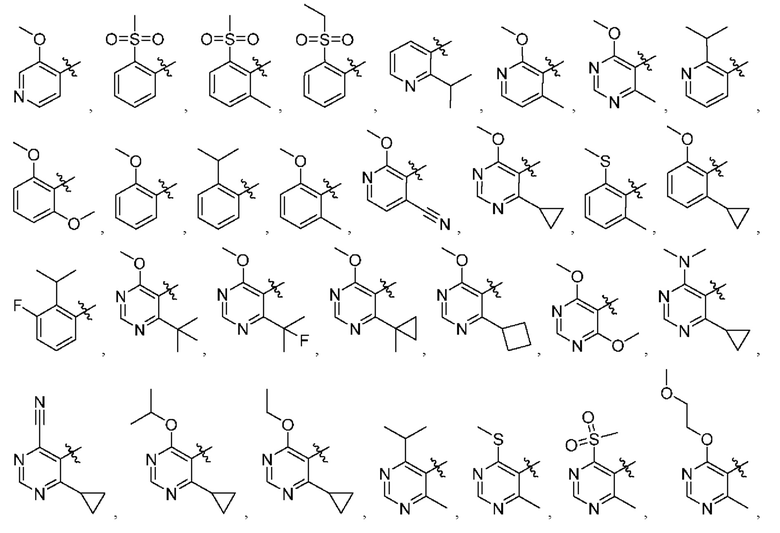
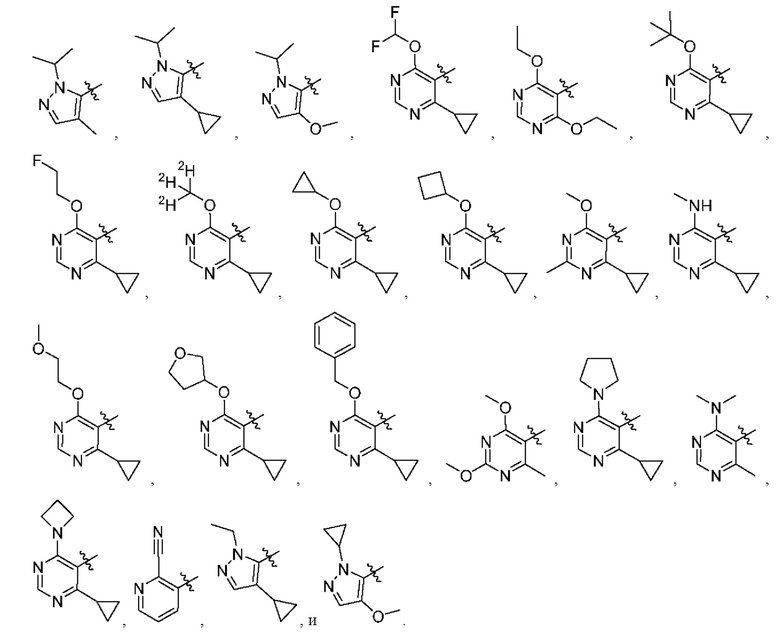
[036] В некоторых вариантах реализации необязательные заместители R5 независимо выбраны из водорода, галогена, нитро, циано, необязательно замещенного алкила, необязательно замещенного алкенила, необязательно замещенного алкинила, алкокси, гидрокси, карбокси, карбоксиалкила, амино, алкиламино, диалкиламино, циклоалкиламино, гетероциклоалкиламино, аралкиламино, гетероаралкиламино, алкилтио, галогеналкила, галогеналкокси, гидроксиалкила, гидроксиалкиламино, алкоксиалкила, (алкоксиалкил)амино, (амино)алкила, (алкиламино)алкила, (диалкиламино)алкила, (циклоалкиламино)алкила, (карбоксамидо)алкила, меркаптоалкила, (циано)алкила, (циклоалкил)алкила, аралкила, аралкилокси, алкилкарбонила, арилкарбонила, (гетероцикло)алкила, (гетероарил)алкила, (амино)(гидрокси)алкила, (аралкиламино)алкила, (С1-4 галогеналкокси)алкила, необязательно замещенного гетероцикло, необязательно замещенного гетероарила, необязательно замещенного арила, необязательно замещенного циклоалкила, карбоксамидо, сульфонила, сульфонамидо, сульфамидо, алкилсульфонила, алкилсульфонамидо, алкил сульфамид о, ар ил сульфонила, арилокси, гетероарилокси,-C(=О)R23,-C(=О)OR24,-C(=О)NR31a R31b,-NR31aC(=О)R25,-NR31aC(=О)OR26,-NR31aC(=О)NR31aR31b,-NR31aSQ2R27,-OC(=О)R28,-OC(=О)OR29,-OC(=О)NR31a R31b,-OSO2R30, и-NR32aR32b; или
[037] два из необязательных заместителей R5 взяты вместе с атомами углерода или азота, к которым они присоединены, с образованием циклоалкильной, гетероцикло-, арильной или гетеро ар ильной группы; а также
[038] каждый из R24, R25, R26, R27, R28, R29, R30, R32a, и R32b выбран из группы, состоящей из водорода, необязательно замещенного алкила, необязательно замещенного алкенила, необязательно замещенного алкинила, амино, алкиламино, диалкиламино, циклоалкиламино, гидроксиалкила, (амино)алкила, (алкиламино)алкила, (диалкиламино)алкила, (циклоалкиламино)алкила, (циклоалкил)алкила, аралкила, (гетероцикло)алкила, (гетероарил)алкила, (амино)(гидрокси) алкила, (аралкиламино)алкила, алкоксиалкила, необязательно замещенный гетероцикло, необязательно замещенного гетероарипа, необязательно замещенного ар ила и необязательно замещенного циклоалкила.
[039] В некоторых вариантах реализации R5 выбран из необязательно замещенного (C1-C6) алкила, необязательно замещенного (С2-С6) алкенила, необязательно замещенного (C1-C6) алкинила, необязательно замещенного (C1-C6) алкокси, (C1-C6) галогеналкила, (C1-C6) галогеналкокси, (C1-C6) гидроксиалкила, циано, галогена, сульфонамидо,-C(=О)R23,-C(=О)OR24,-NR32aR32b, NR31aC(=О)R25,-NR31aC(=О)NR31aR31b,-C(=О)NR31a R31b,-S(O)2R27, и-NR31aSO2R27, необязательно замещенного-O-(C6-C14) арила, необязательно замещенного-О-(С6-С14) ар-(С1-С2) алкила, необязательно замещенного-О-гетероарила, необязательно замещенного-О-гетероар-(C1-C2) алкила, необязательно замещенного-О-(C3-C8) циклоалкила, необязательно замещенного-O-((С3-С8) циклоалкил)-(С1-С2) алкила, необязательно замещенного-О-гетероцикло, необязательно замещенного-O-гетероцикло-(С1-С2) алкила.
[040] В некоторых вариантах реализации R5 выбран из необязательно замещенного (С6-С14) арила, необязательно замещенного (С6-С14) ap-(C1-C2) алкила, необязательно замещенного гетероарипа, необязательно замещенного гетероар-(С1-С2) алкила, необязательно замещенного (С3-C8) циклоалкила, необязательно замещенного ((С3-C8) циклоалкил)-(С1-С2) алкила, необязательно замещенного гетероцикло, необязательно замещенного гетероцикло-(С1-С2) алкила.
[041] В некоторых вариантах реализации R5 представляет собой необязательно замещенный пирролил, необязательно замещенный имидазолил, необязательно замещенный пиразолил, необязательно замещенный триазолил или необязательно замещенный тетразолил.
[042] В некоторых вариантах реализации R5 представляет собой необязательно замещенный имидазолил.
[043] В некоторых вариантах реализации R5 представляет собой необязательно замещенный пиразолил.
[044] В некоторых вариантах реализации R5 представляет собой необязательно замещенный триазолил.
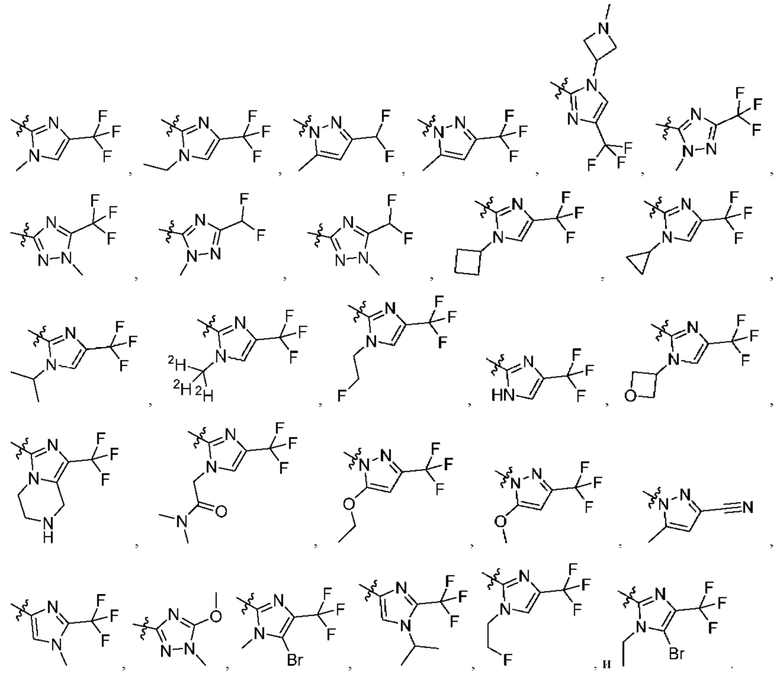
[045] В некоторых вариантах реализации R5 представляет собой необязательно замещенный гетероарил, такой как имидазолил, пиразолил или триазолил, где заместители независимо выбраны из галогена, метила, этила, изо пропила, циклопропила, циклобутила, метокси, этокси, триазолила, циано, необязательно замещенного алкила, амино, алкиламино, диалкиламино, дифторметила, трифторметила, метилсульфонила, оксетан-3-ила и метилазетидинила.
[046] В некоторых вариантах реализации R5 выбран из группы, состоящей из:
[047] В некоторых вариантах реализации соединение имеет Формулу II:
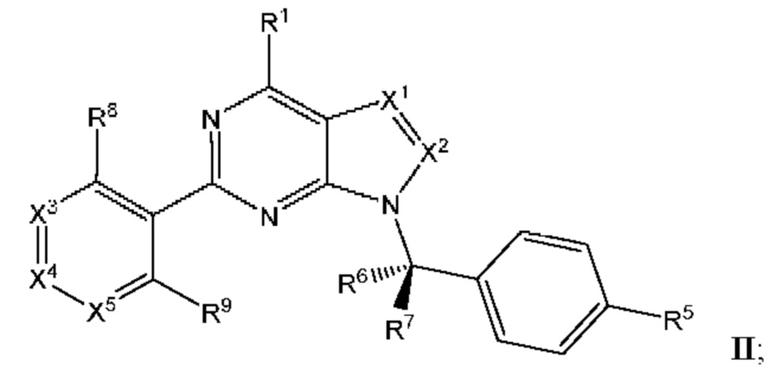
где X3 выбран из N и CR10; X4 выбран из N и CR11; Xs выбран из N и CR12; и
[048] каждый из R8, R9, R10, R11, и R12 независимо выбран из группы, состоящей из водорода, галогена, нитро, циано, гидрокси, амино, алкиламино, диалкиламино, галогеналкила, гидроксиалкила, алкокси, галогеналкокси, арилокси, гетероарилокси, аралкиларалкилокси, алкилтио, карбоксамидо, сульфонамидо, алкилкарбонила, арилкарбонила, алкилсульфонила, арилсульфонила, карбокси, карбоксиалкила, алкила, необязательно замещенного циклоалкила, алкенила, алкинила, необязательно замещенного ар ила, необязательно замещенного гетероарила, необязательно замещенного гетероцикло, алкоксиалкила, (амино)алкила, гидроксиалкиламино, (алкиламино)алкила, (диалкиламино) алкила, (циано)алкила, (карбоксамидо)алкила, меркаптоалкила, (гетероцикло)алкила, (циклоалкиламино)алкила, (C1-4 галогеналкокси)алкила, или (гетероарил)алкила.
[049] В некоторых вариантах реализации Соединения раскрытия имеют Формулу III, формулу IV, формулу V, формулу VT или формулу VIa:
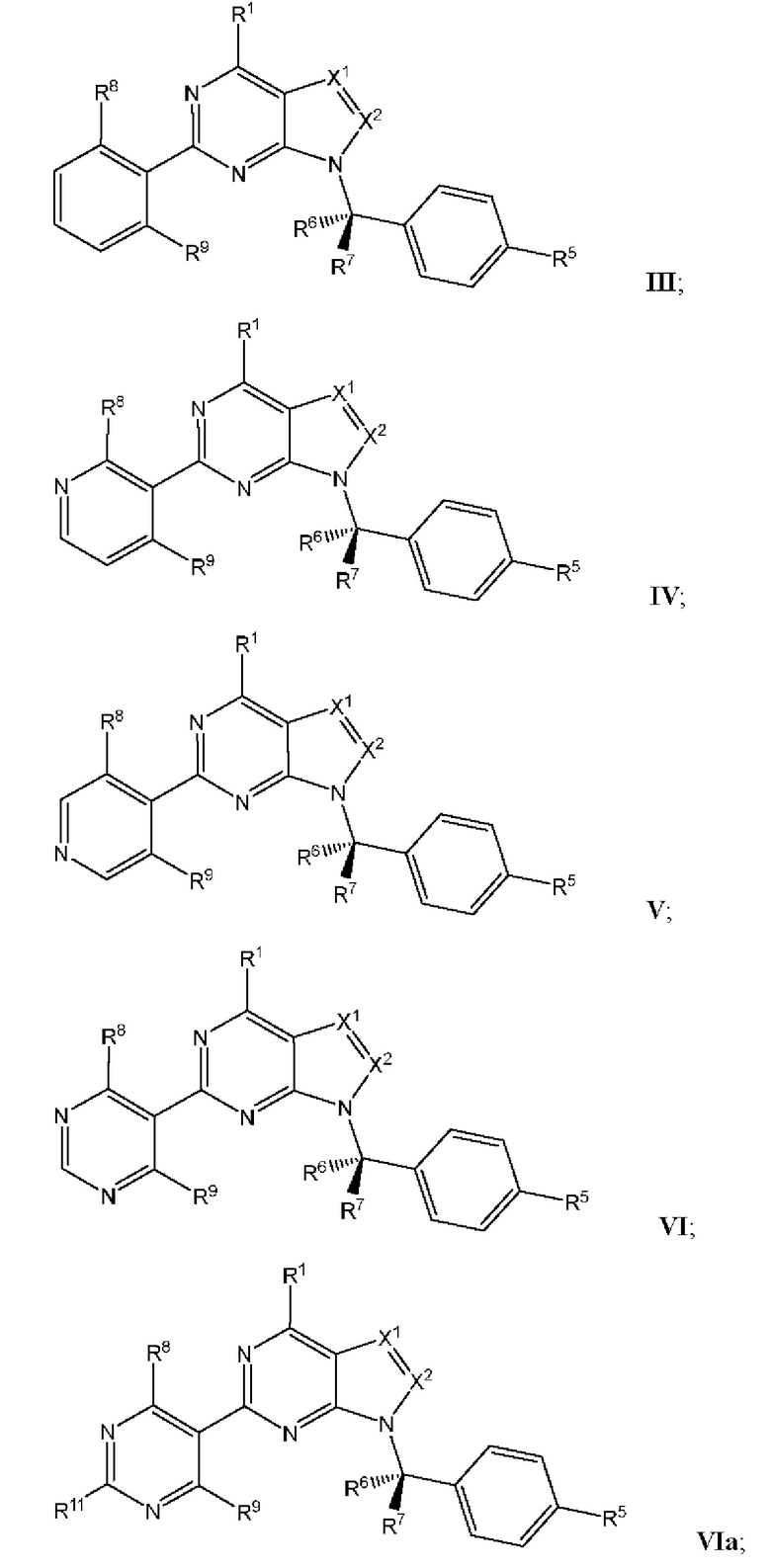
где каждый из X1, X2, R1, R5, R6, R7, R8, R9, и R11 имеет значения, указанные выше для Формулы II.
[050] В некоторых вариантах реализации R5 представляет собой:
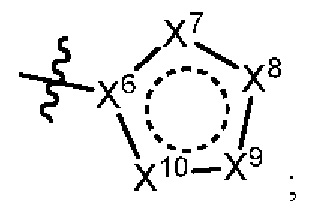
[051] где X6 выбран из NR13 и CR18; X7 выбран из NR14 и CR19; X8 выбран из NR15 и CR20; X9 выбран из NR16 и CR21; X10 выбран из NR17 и CR22; и
[052] каждый из R13, R14, R15, R16, и R17 отсутствует или независимо выбран из водорода, галогена, метила, этила, изопропила, циклопропила, метокси, триазолила, циано, необязательно замещенного алкила, амино, алкиламино, диалкиламино, дифторметила, трифторметила, метилсульфонила и метил азетидинила.
[053] каждый из R18, R19, R20, R21 и R22 независимо выбран из водорода, галогена, метила, этила, изопропила, циклопропила, метокси, триазолила, циано, необязательно замещенного алкила, амино, алкиламино, диалкиламино, дифторметила, трифторметила, метилсульфонила и метилазетидинила.
[054] В некоторых вариантах реализации Соединения раскрытия имеют Формулу VII, формулу VIII, формулу IX, формулу X, формулу XI или формулу XII.
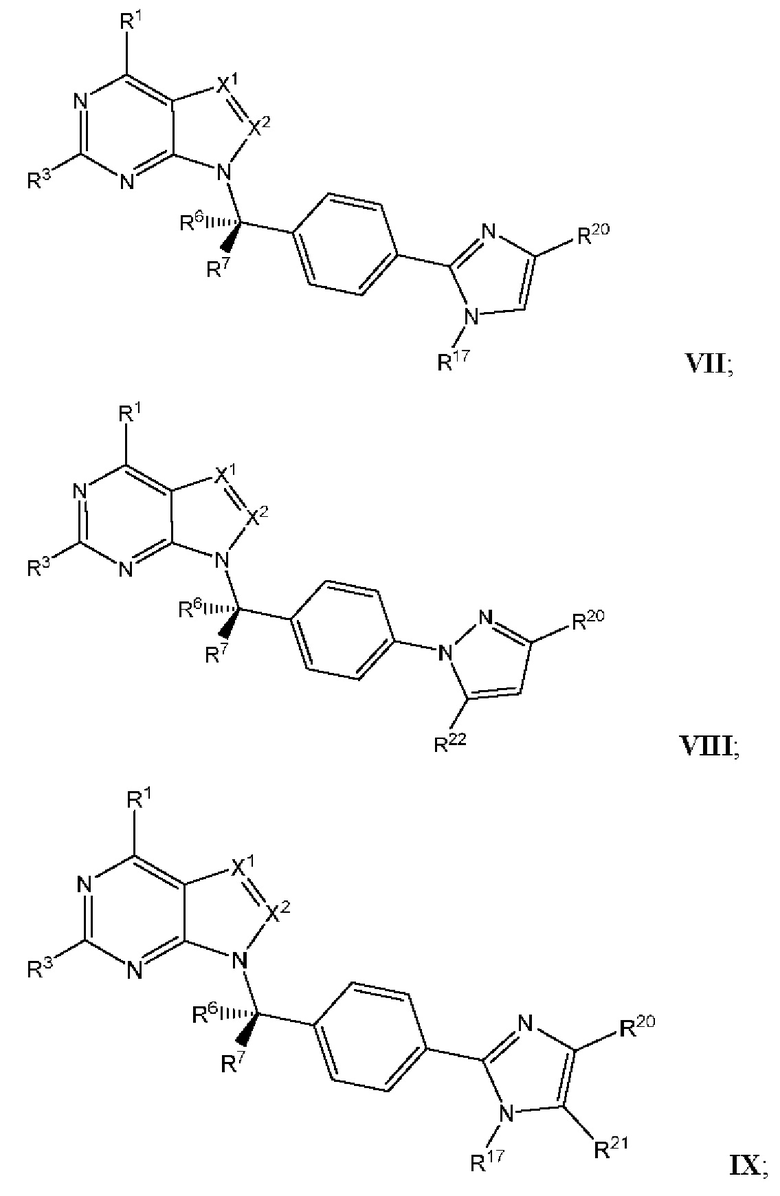
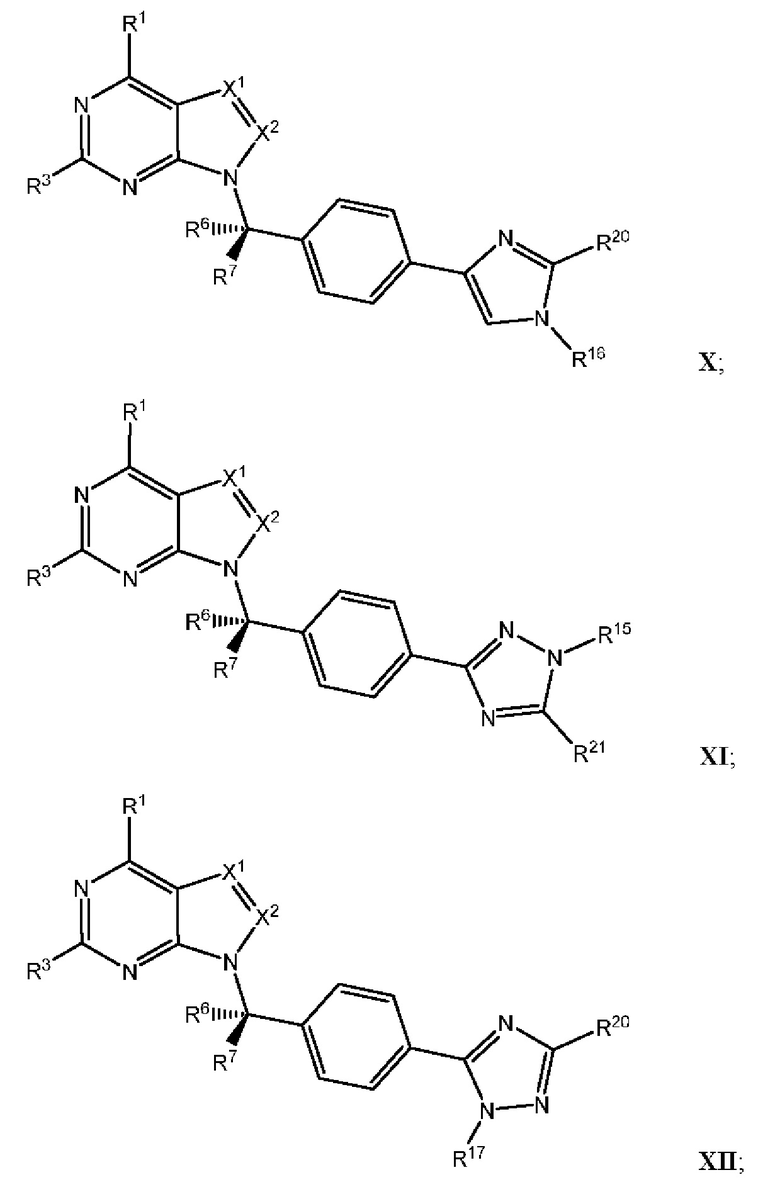
где каждый из X1, X2, R1, R3, R6, R7, R15, R16, R17, R20, R21, и R22 имеют значения, указанны выше для Формулы II.
[055] В другом аспекте данное раскрытие относится к Соединениям раскрытия или их фармацевтически приемлемой соли или сольвату, имеющим Формулу XIII:
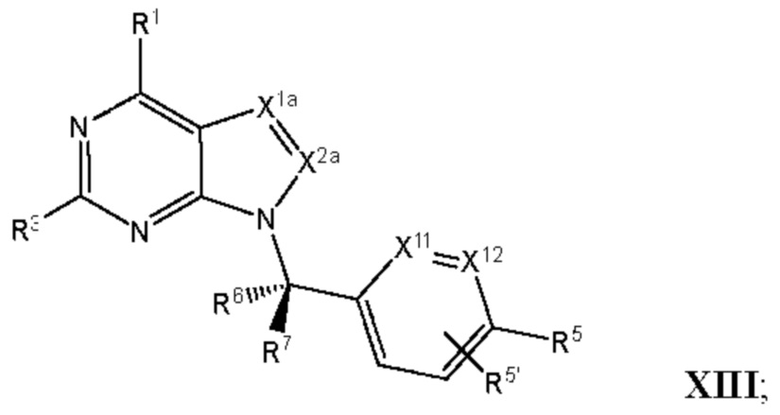
где:
[056] каждый из Х1а и Х2а независимо выбран из N и CR2a;
[057] R2a независимо выбран из водорода, галогена, пиано, необязательно замещенного алкила, необязательно замещенного алкенила, необязательно замещенного алкинила, алкокси, алкоксиалкила, алкилсульфонила, алкилтио, арила, гетероарила и гетероцикло; и
[058] каждый из оставшихся заместителей определен, как раскрыто в данном документе.
[059] В некоторых вариантах реализации Соединение раскрытия представляет собой одно из конкретных соединений, перечисленных в подробном описании, или его фармацевтически приемлемую соль или сольват.
[060] В некоторых вариантах реализации Соединение раскрытия ингибирует белок USP1.
[061] В некоторых вариантах реализации Соединение раскрытия ингибирует белок USP1 со значением IC50 менее около 1 мкМ в анализе деубиквитинирования Ub-Rho.
[062] В некоторых вариантах реализации изобретения анализ деубиквитинирования Ub-Rho представляет собой анализ, описанный в Примере 26.
[063] В одном аспекте данное раскрытие относится к способу лечения рака у пациента, включающему введение пациенту терапевтически эффективного количества соединения согласно данному описанию или его фармацевтически приемлемой соли или сольвата.
[064] В одном аспекте данное раскрытие относится к фармацевтической композиции, содержащей Соединение по раскрытию, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель.
[065] В некоторых вариантах реализации данное раскрытие относится к фармацевтической композиции для лечения рака.
[066] В некоторых вариантах реализации данное раскрытие относится к Соединению раскрытия для использования при лечении рака.
[067] В некоторых вариантах реализации данное раскрытие относится к применению Соединения раскрытия для производства лекарственного средства для лечения рака.
[068] В одном аспекте данное раскрытие относится к набору, содержащему Соединение по раскрытию, или его фармацевтически приемлемую соль или сольват, или к фармацевтической композиции, содержащей Соединение по раскрытию, и инструкциям по введению соединения или фармацевтически приемлемой соли, или его сольват, или фармацевтическая композиция для пациента, страдающего раком.
[069] В одном аспекте данное раскрытие относится к способу лечения рака у пациента, включающему введение пациенту Соединения по раскрытию, или его фармацевтически приемлемой соли или сольвата, или фармацевтической композиции, содержащей Соединение по раскрытию.
[070] В некоторых вариантах реализации рак выбран из группы, состоящей из гематологического рака, лимфатического рака и рака, лишенного пути репарации повреждений ДНК.
[071] В некоторых вариантах реализации рак включает раковые клетки с мутацией в гене, кодирующем р53. В некоторых вариантах реализации мутация в гене, кодирующем р53, представляет собой мутацию зародышевой линии. В некоторых вариантах реализации мутация в гене, кодирующем р53, является соматической мутацией. В некоторых вариантах реализации рак включает раковые клетки с мутацией потери функции в гене, кодирующем р53.
[072] В некоторых вариантах реализации рак выбран из группы, состоящей из рака легких, немелкоклеточного рака легких (NSCLC), рака толстой кишки, рака мочевого пузыря, остеосаркомы, рака яичников и рака груди.
[073] В некоторых вариантах реализации рак представляет собой немелкоклеточный рак легкого (NSCLC).
[074] В некоторых вариантах реализации рак представляет собой рак толстой кишки.
[075] В некоторых вариантах реализации рак представляет собой рак мочевого пузыря.
[076] В некоторых вариантах реализации рак представляет собой рак яичников или рак груди.
[077] В некоторых вариантах реализации рак представляет собой рак яичников.
[078] В некоторых вариантах реализации рак представляет собой рак груди.
[079] В некоторых вариантах реализации рак представляет собой тройной отрицательный рак молочной железы.
[080] В некоторых вариантах реализации рак включает раковые клетки с повышенным уровнем RAD18.
[081] В некоторых вариантах реализации повышенные уровни RAD18 представляют собой повышенные уровни белка RAD18.
[082] В некоторых вариантах реализации повышенные уровни RAD18 представляют собой повышенные уровни мРНК RAD18.
[083] В некоторых вариантах реализации повышенные уровни RAD18 были обнаружены до введения.
[084] В другом аспекте данное раскрытие относится к способу, который дополнительно включает определение уровней RAD18 в образце рака, полученном от субъекта.
[085] В некоторых вариантах реализации рак выбран из группы, состоящей из рака кости, включая остеосаркому и хондросаркому; рака мозга, включая глиому, глиобластому, астроцитому, медуллобластому и менингиому; рака мягких тканей, включая рабдоид и саркому; рака почки; рака мочевого пузыря; рака кожи, включая меланому; и рака легкого, включая немелкоклеточный рак легкого; рака толстой кишки, рака матки; рака нервной системы; рака головы и шеи; панкреатического рака; и рака шейки матки.
[086] В некоторых вариантах реализации рак представляет собой рак с недостаточностью пути репарации повреждений ДНК.
[087] В некоторых вариантах реализации рак включает раковые клетки с мутацией в гене, кодирующем р53. В некоторых вариантах реализации мутация в гене, кодирующем р53, представляет собой мутацию зародышевой линии. В некоторых вариантах реализации мутация в гене, кодирующем р53, является соматической мутацией. В некоторых вариантах реализации рак включает раковые клетки с мутацией потери функции в гене, кодирующем р53.
[088] В некоторых вариантах реализации рак представляет собой мугантный рак BRCA1. В некоторых вариантах реализации мутация BRCA1 представляет собой мутацию зародышевой линии. В некоторых вариантах реализации мутация BRCA1 является соматической мутацией. В некоторых вариантах реализации мутация BRCA1 приводит к дефициту BRCA1.
[089] В некоторых вариантах реализации рак представляет собой мугантный рак BRCA2. В некоторых вариантах реализации мутация BRCA2 представляет собой мутацию зародышевой линии. В некоторых вариантах реализации мутация BRCA2 является соматической мутацией. В некоторых вариантах реализации мутация BRCA2 приводит к дефициту BRCA2.
[090] В некоторых вариантах реализации рак представляет собой мугантный рак BRCA1 и мутантный рак BRCA2.
[091] В некоторых вариантах реализации рак представляет собой рак с дефицитом BRCA1.
[092] В некоторых вариантах реализации рак представляет собой рак с дефицитом BRCA2.
[093] В некоторых вариантах реализации рак представляет собой рак с дефицитом BRCA1 и рак с дефицитом BRCA2.
[094] В некоторых вариантах реализации рак представляет собой рефрактерный или резистентный рак к ингибитору поли (АДФ-рибозы) полимеразы («PARP»). В некоторых вариантах реализации рак представляет собой резистентный к ингибитору PARP или рефрактерный рак BRCA1, BRCA2 или BRCA1 и мутантный BRCA2. В некоторых вариантах реализации рак представляет собой рак, резистентный к ингибиторам PARP или рефрактерный BRCA1, BRCA2 или BRCA1 и BRCA2-дефицитный рак.
[095] В некоторых вариантах реализации рак имеет мутацию в гене, кодирующем протеинкиназу с мутацией атаксии и телеангиэктазии (ATM). В некоторых вариантах реализации мутация ATM представляет собой мутацию зародышевой линии. В некоторых вариантах реализации мутация ATM является соматической мутацией. В некоторых вариантах реализации рак представляет собой рак с дефицитом ATM.
[096] В некоторых вариантах реализации рак имеет мутацию в гене, кодирующем по меньшей мере два из р53, BRCA1, BRCA2 и ATM.
[097] В другом аспекте данное раскрытие относится к способу лечения расстройства,
опосредованного белком USP1, включающему введение пациенту, который в этом нуждается, Соединения Раскрытия, или его фармацевтически приемлемой соли или сольвата, или фармацевтической композиции, содержащей Соединение Раскрытие, в эффективном количестве для лечения расстройства, опосредованного белком USP1.
[098] В некоторых вариантах реализации белок USP1 содержит аминокислотную последовательность SEQ ID NO: 1.
[099] В другом аспекте данное раскрытие относится к способу ингибирования белка USP1, включающему контактирование белка USP1 с соединением по раскрытию, или его фармацевтически приемлемой солью или сольватом, или с фармацевтической композицией, содержащей соединение по раскрытию.
[0100] В некоторых вариантах реализации контактирование происходит in vitro.
[0101] В некоторых вариантах реализации контактирование происходит in vivo.
[0102] В другом аспекте данное раскрытие относится к способу лечения рака у пациента, включающему введение субъекту ингибитора USP1, где рак включает раковые клетки с мутацией в гене, кодирующем р53.
[0103] В некоторых вариантах реализации мутация в гене, кодирующем р53, была обнаружена до введения.
[0104] В некоторых вариантах реализации способ дополнительно включает обнаружение мутации в гене, кодирующем р53, в образце рака, полученном от пациента.
[0105] В другом аспекте данное раскрытие относится к способу выбора пациента с раком для лечения ингибитором USP1, включающему определение того, содержит ли рак раковые клетки с мутацией в гене, кодирующем р53, причем если рак включает раковые клетки с мутации в гене, кодирующем р53, пациента отбирают для лечения ингибитором USP1.
[0106] В другом аспекте данное раскрытие относится к ингибитору USP1 для использования при лечении рака у пациента, при этом рак включает раковые клетки с мутацией в гене, кодирующем р53.
[0107] В некоторых вариантах реализации изобретения установлено, что пациент реагирует на лечение ингибитором USP1 путем обнаружения раковых клеток с мутацией в гене, кодирующем р53, в образце рака, полученном от пациента, при этом раковые клетки с мутацией в гене, кодирующем р53 в образце рака идентифицируют пациента как чувствительного к лечению ингибитором USP1.
[0108] В другом аспекте данное раскрытие относится к ингибитору USP1 для использования при лечении рака у субъекта, который, как было установлено, реагирует на лечение ингибитором USP1 раковыми клетками с мутацией в гене, кодирующем р53 в образце рака, полученном у субъекта, у которого раковые клетки с мутацией в гене, кодирующем р53 в образце рака, идентифицируют пациента, который реагирует на лечение ингибитором USP1.
[0109] В некоторых вариантах реализации ингибитор USP1 не вводят субъекту в случае, если мутация в гене, кодирующем р53, не обнаруживается в образце рака, полученном от субъекта.
[0110] В другом аспекте данное раскрытие относится к способу in vitro идентификации субъекта с раком, чувствительного к лечению ингибитором USP1, включающему обнаружение мутации в гене, кодирующем р53, в образце рака, полученном от субъекта, при этом мутация в гене, кодирующем р53 в образце рака, свидетельствует о том, что пациент реагирует на лечение ингибитором USP1.
[0111] В другом аспекте данное раскрытие относится к применению in vitro по меньшей мере одного агента, способного специфически обнаруживать мутацию в гене, кодирующем р53, для идентификации субъекта с раком, чувствительного к лечению ингибитором USP1.
[0112] В некоторых вариантах реализации мутация в гене, кодирующем р53, представляет собой мутацию потери функции.
[0113] В другом аспекте данное раскрытие относится к способу лечения рака у пациента,
включающему введение субъекту ингибитора USP1, где рак включает раковые клетки с мутацией в гене, кодирующем BRCA1.
[0114] В некоторых вариантах реализации мутация в гене, кодирующем BRCA1, была обнаружена до введения.
[0115] В некоторых вариантах реализации способ дополнительно включает обнаружение мутации в гене, кодирующем BRCA1, в образце (например, образце рака или образце крови), полученном от пациента.
[0116] В другом аспекте данное раскрытие относится к способу выбора пациента с раком для лечения ингибитором USP1, включающему определение того, содержит ли рак раковые клетки с мутацией в гене, кодирующем BRCA1, причем, если рак включает раковые клетки с мутации в гене, кодирующем BRCA1, пациента отбирают для лечения ингибитором USP1.
[0117] В другом аспекте данное раскрытие относится к ингибитору USP1 для использования при лечении рака у пациента, где рак включает раковые клетки с мутацией в гене, кодирующем BRCA1.
[0118] В некоторых вариантах реализации идентифицировано, что пациент реагирует на лечение ингибитором USP1 путем обнаружения мутации в гене, кодирующем BRCA1, в образце (например, образце рака или образце крови), полученном от пациента, при этом мутация в ген, кодирующий BRCA1 в образце, идентифицирует пациента, чувствительного к лечению ингибитором USP1.
[0119] В другом аспекте данное раскрытие относится к ингибитору USP1 для использования при лечении рака у субъекта, у которого выявлена чувствительность к лечению ингибитором USP1 раковыми клетками с мутацией в гене, кодирующем BRCA1 в образце (например, образец рака или образец крови), полученном от субъекта, при этом раковые клетки с мутацией в гене, кодирующем BRCA1 в образце, идентифицируют пациента, который реагирует на лечение ингибитором USP1.
[0120] В некоторых вариантах реализации ингибитор USP1 не вводят субъекту в случае, если мутация в гене, кодирующем BRCA1, не обнаруживается в образце (например, образце рака или образце крови), полученном от субъекта.
[0121] В другом аспекте данное раскрытие относится к способу in vitro идентификации субъекта с раком, чувствительного к лечению ингибитором USP1, включающему обнаружение мутации в гене, кодирующем BRCA1, в образце (например, образце рака или образце крови), полученном от субъекта, причем мутация в гене, кодирующем BRCA1 в образце, свидетельствует о том, что пациент реагирует на лечение ингибитором USP1.
[0122] В другом аспекте данное раскрытие относится к применению in vitro по меньшей мере одного агента, способного специфически обнаруживать мутацию в гене, кодирующем BRCA1, для идентификации субъекта с раком, чувствительного к лечению ингибитором USP1.
[0123] В некоторых вариантах реализации мутация в гене, кодирующем BRCA1, представляет собой мутацию потери функции
[0124] В другом аспекте данное раскрытие относится к способу лечения рака у пациента,
включающему введение субъекту ингибитора USP1, при этом рак включает раковые клетки с мутацией в гене, кодирующем BRCA2.
[0125] В некоторых вариантах реализации мутация в гене, кодирующем BRCA2, была обнаружена до введения.
[0126] В некоторых вариантах реализации способ дополнительно включает обнаружение мутации в гене, кодирующем BRCA2, в образце (например, образце рака или образце крови), полученном от пациента.
[0127] В другом аспекте данное раскрытие относится к способу выбора пациента с раком для лечения ингибитором USP1, включающему определение того, содержит ли рак раковые клетки с мутацией в гене, кодирующем BRCA2, причем если рак включает раковые клетки с мутации в гене, кодирующем BRCA2, пациента отбирают для лечения ингибитором USP1.
[0128] В другом аспекте данное раскрытие относится к ингибитору USP1 для использования при лечении рака у пациента, при этом рак включает раковые клетки с мутацией в гене, кодирующем BRCA2.
[0129] В некоторых вариантах реализации установлено, что пациент реагирует на лечение ингибитором USP1 путем обнаружения мутации в гене, кодирующем BRCA2, в образце (например, образце рака или образце крови), полученном от пациента, при этом мутация в ген, кодирующий BRCA2 в образце, идентифицирует пациента, чувствительного к лечению ингибитором USP1.
[0130] В другом аспекте данное раскрытие относится к ингибитору USP1 для использования при лечении рака у субъекта, у которого выявлена чувствительность к лечению ингибитором USP1 раковыми клетками с мутацией в гене, кодирующем BRCA2 в образце (например, образец рака или образец крови), полученном от субъекта, при этом раковые клетки с мутацией в гене, кодирующем BRCA2, в образце идентифицируют пациента, который реагирует на лечение ингибитором USP1.
[0131] В некоторых вариантах реализации ингибитор USP1 не вводят субъекту в случае, если мутация в гене, кодирующем BRCA2, не обнаруживается в образце (например, образце рака или образце крови), полученном от субъекта.
[0132] В другом аспекте данное раскрытие относится к способу in vitro идентификации субъекта с раком, чувствительного к лечению ингибитором USP1, включающему обнаружение мутации в гене, кодирующем BRCA2, в образце (например, образце рака или образце крови), полученном от субъекта, причем мутация в гене, кодирующем BRCA2 в образце, свидетельствует о том, что пациент реагирует на лечение ингибитором USP1.
[0133] В другом аспекте данное раскрытие относится к применению in vitro по меньшей мере одного агента, способного специфически обнаруживать мутацию в гене, кодирующем BRCA2, для идентификации субъекта с раком, чувствительного к лечению ингибитором USP1.
[0134] В некоторых вариантах реализации мутация в гене, кодирующем BRCA2, представляет собой мутацию потери функции
[0135] В другом аспекте данное раскрытие относится к способу лечения рака у пациента, включающему введение субъекту ингибитора USP1, где рак включает раковые клетки с мутацией в гене, кодирующем ATM.
[0136] В некоторых вариантах реализации мутация в гене, кодирующем ATM была обнаружена до введения.
[0137] В некоторых вариантах реализации способ дополнительно включает обнаружение мутации в гене, кодирующем ATM, в образце, полученном от пациента.
[0138] В другом аспекте данное раскрытие относится к способу выбора пациента с раком для лечения ингибитором USP1, включающему определение того, содержит ли рак раковые клетки с мутацией в гене, кодирующем ATM, причем если рак включает раковые клетки с мутации в гене, кодирующем ATM, пациента отбирают для лечения ингибитором USP1.
[0139] В другом аспекте данное раскрытие относится к ингибитору USP1 для использования при лечении рака у пациента, где рак включает раковые клетки с мутацией в гене, кодирующем ATM.
[0140] В некоторых вариантах реализации установлено, что пациент реагирует на лечение ингибитором USP1 путем обнаружения раковых клеток с мутацией в гене, кодирующем ATM в образце рака, полученном от пациента, при этом раковые клетки с мутацией в гене, кодирующем ATM в образце рака идентифицируют пациента как чувствительного к лечению ингибитором USP1.
[0141] В другом аспекте данное раскрытие относится к ингибитору USP1 для использования при лечении рака у субъекта, который, как было установлено, реагирует на лечение ингибитором USP1 раковыми клетками с мутацией в гене, кодирующем ATM в образце рака, полученном от субъекта, у которого раковые клетки с мутацией в гене, кодирующем ATM в образце рака идентифицируют пациента, как чувствительного к лечению ингибитором USP1.
[0142] В некоторых вариантах реализации ингибитор USP1 не вводят субъекту в случае, если мутация в гене, кодирующем ATM не обнаруживается в образце рака, полученном от субъекта.
[0143] В другом аспекте данное раскрытие относится к способу in vitro идентификации субъекта с раком, чувствительного к лечению ингибитором USP1, включающему обнаружение мутации в гене, кодирующем ATM в образце рака, полученном от субъекта, при этом мутация в гене, кодирующем ATM в образце рака, свидетельствует о том, что пациент реагирует на лечение ингибитором USP1.
[0144] В другом аспекте данное раскрытие относится к применению in vitro по меньшей мере одного агента, способного специфически обнаруживать мутацию в гене, кодирующем ATM Для идентификации субъекта, больного раком, отвечающего на лечение ингибитором USP1.
[0145] В некоторых вариантах реализации мутация в гене, кодирующем ATM представляет собой мутацию потери функции.
[0146] В некоторых вариантах реализации ингибитор USP1 представляет собой Соединение раскрытия.
[0147] В одном аспекте (А1) данное раскрытие обеспечивает способ лечения рака у субъекта, включающий введение субъекту ингибитора USP1, при этом рак включает раковые клетки с повышенными уровнями RAD 18.
[0148] В одном аспекте (А2) данное раскрытие обеспечивает способ лечения тройного отрицательного рака груди у субъекта, включающий введение субъекту ингибитора USP1. В одном из аспектов А1 (A3) рак включает раковые клетки с повышенным уровнем RAD18.
[0149] В одном аспекте (А4) А1 или A3 повышенные уровни RAD18 были обнаружены до введения. В одном из аспектов (А5) А4 способ дополнительно включает определение уровней RAD18 в образце рака, полученном от субъекта.
[0150] В одном аспекте (А5) данное раскрытие обеспечивает способ выбора субъекта с раком для лечения ингибитором USP1, включающий определение того, содержит ли рак клетки с повышенными уровнями RAD18, причем, если рак включает клетки с повышенными уровнями RAD18, субъект выбран для лечения ингибитором USP1.
[0151] В одном аспекте (А6) данное раскрытие обеспечивает ингибитор USP1 для применения при лечении рака у субъекта, при этом рак включает раковые клетки с повышенными уровнями RAD18.
[0152] В одном аспекте (А7) данное раскрытие обеспечивает ингибитор USP1 для использования при лечении тройного отрицательного рака груди у субъекта.
[0153] В одном из аспектов (А9) А7 или А8 субъект идентифицируется как отвечающий на лечение ингибитором USP1 путем обнаружения уровней RAD18 в образце рака, полученном от субъекта, при этом повышенные уровни RAD18 в образце рака идентифицируют пациента, как чувствительного к лечению ингибитором USP1. как реагирующего
[0154] В одном аспекте (А10) данное раскрытие обеспечивает ингибитор USP1 для использования при лечении рака у субъекта, который, как было установлено, реагирует на лечение ингибитором USP1, путем обнаружения повышенных уровней RAD18 в образце рака, полученном от субъекта, при этом повышенные уровни RAD18 в образце рака указывают на то, что пациент реагирует на лечение ингибитором USP1.
[0155] В одном аспекте (АН) данное раскрытие обеспечивает ингибитор USP1 для использования любого из А7-А10, где ингибитор USP1 не вводится субъекту в случае, если в образце рака, полученном от субъекта, обнаружены не повышенные уровни RAD18..
[0156] В одном аспекте (А12) данное раскрытие обеспечивает способ in vitro идентификации субъекта с раком, чувствительного к лечению ингибитором USP1, включающий определение уровней RAD18 в образце рака, полученном от субъекта, при этом повышенные уровни RAD18 в образце рака указывают на то, что пациент реагирует на лечение ингибитором USP1.
[0157] В одном аспекте (А13) данное раскрытие обеспечивает использование in vitro по меньшей мере одного агента, способного специфически детектировать RAD18, для идентификации субъекта с раком, чувствительного к лечению ингибитором USP1.
[0158] В одном аспекте (А 14) данное раскрытие предоставляет набор или набор частей, включающий: (а) фармацевтическую композицию, содержащую ингибитор USP1 и один или несколько фармацевтически приемлемых наполнителей, и (b) диагностический набор, содержащий по меньшей мере один агент, способный специфически обнаруживать RAD18.
[0159] В одном аспекте (А15) А13 или А14 по меньшей мере один агент, способный специфически обнаруживать RAD18, способен специфически гибридизоваться с MPHKRAD18. В одном аспекте (А16) А13 или А14 по меньшей мере один агент, способный специфически обнаруживать RAD18, способен специфически связываться с белком RAD18.
[0160] В одном аспекте (А17) А10, А12 или А13 рак представляет собой тройной отрицательный рак молочной железы.
[0161] В одном аспекте (А18) А10, А12, А13 или А17 рак включает раковые клетки с повышенными уровнями RAD18.
[0162] В одном аспекте (А19) данное раскрытие обеспечивает способ классификации рака у субъекта, включающий обнаружение повышенных уровней RAD18 в образце рака, полученном от субъекта.
[0163] В одном аспекте (А20) любого из А1-А19 рак не является мутантным раком BRCA1. В одном аспекте (А21) любого из А1-А19 рак не является мутантным раком BRCA2. В одном аспекте (А22) любого из А1-А19 рак не является мутантным раком BRCA1 или мутантным раком BRCA2.
[0164] В одном аспекте (А23) любого из А1-А19 рак не является раком с дефицитом гомологичной рекомбинации. В одном аспекте (А24) любого из А1-А19 рак представляет собой рак с дефицитом гомологичной рекомбинации.
[0165] В одном аспекте (А25) любого из А1-А23 рак включает раковые клетки с мутацией в гене, кодирующем р53, необязательно, где мутация в гене, кодирующем р53, представляет собой мутацию с потерей функции.
[0166] В одном аспекте (А26) любого из A1, А3-А7, А9-А12 и А18-А25 повышенные уровни RAD18 представляют собой повышенные уровни белка RAD18. В одном аспекте (А27) А26 обнаружение повышенных уровней белка RAD18 осуществляется вестерн-блоттингом. В одном аспекте (А28) А26 обнаружение повышенных уровней белка RAD18 осуществляется с помощью сортировки клеток, активируемых флуоресценцией (FACS). В одном аспекте (А29) А26 обнаружение повышенных уровней белка RAD18 осуществляется с помощью иммуногистохимии.
[0167] В одном аспекте (А30) любого из A1, А3-А7, А9-А12 и А18-А25 повышенные уровни RAD18 представляют собой повышенные уровни мРНК RAD18. В одном аспекте (А31) А30 обнаружение повышенных уровней мРНК RAD18 осуществляется с помощью количественной полимеразной цепной реакции (PCR) с обратной транскриптазой (RT), RNA-Seq или микроматрицы.
[0168] В одном аспекте (А32) любого из A1, А3-А7, А9-А12 и А18-А31 повышенные уровни RAD18 по меньшей мере такие же высокие, как уровни RAD18 в клетках ES2. В одном аспекте (А33) любого из A1, А3-А7, А9-А12 и А18-А31 повышенные уровни RAD18 выше, чем уровни RAD18 в клетках НЕР3В217.
[0169] В одном аспекте (А34) любого из A1, А4-А7, А9-А16 и А18-А33 рак представляет собой рак яичников. В одном аспекте (А35) любого из A1, А4-А7, А9-А16 и А18-А33 рак представляет собой рак груди. В одном аспекте (А36) А35 рак груди представляет собой рак груди с тройным отрицательным результатом.
[0170] В одном аспекте (А37) любого из А1-А19 и А24-А36 рак представляет собой мутантный рак BRCA1. В одном аспекте (А38) любого из А1-А19 и А24-А36 рак представляет собой мутантный рак BRCA2. В одном аспекте (А39) любого из А1-А19 и А24-А36 рак представляет собой мутантный рак BRCA1 и мутантный рак BRCA2.
[0171] В одном аспекте (А40) любого из А1-А18 и А20-А39 ингибитор USP1 выбран из небольшой молекулы, миРНК, антисмыслового олигонуклеотида, пептида или аптамера. В одном аспекте (А41) любого из А1-А18 и А20-А40 ингибитор USP1 специфически связывает белок USP1. В одном аспекте (А42) А41 белок USP1 содержит аминокислотную последовательность 
[0172] В одном аспекте (A43) любого из А1-А18 и А20-А42 ингибитор USP1 ингибирует образование и/или активность комплекса USP1/UAF1.
[0173] В одном аспекте (А44) любого из А1-А18 и А20-А40 ингибитор USP1 специфически связывается с комплексом USP1/UAF1.
[0174] В одном аспекте (А45) любого из А1-А18 и А20-А40 ингибитор USP1 специфически связывает мРНК USP1.
[0175] В одном аспекте (А46) любого из А1-А18 и А20-А45 ингибитор USP1 увеличивает моноубиквитинированную PCNA.
[0176] В одном аспекте (А47) любого из А1-А18 и А20-А46 ингибитор USP1 увеличивает моноубиквитинированный FANCD2.
[0177] В одном аспекте (А48) любого из А1-А18 и А20-А39 ингибитор USP1 представляет собой пуринон. В одном аспекте (А49) любого из А1-А18 и А20-А39 ингибитор USP1 представляет собой GW7647. В одном аспекте (А50) любого из А1-А18 и А20-А39 ингибитор USP1 представляет собой пимозид. В одном аспекте (А51) любого из А1-А18 и А20-А39 ингибитор USP1 представляет собой ML323. [0178] В одном аспекте (А52) любого из А1-А18 и А20-А51 субъект является человеком.
[0179] Дополнительные варианты реализации и преимущества раскрытия будут изложены частично в нижеследующем описании и будут вытекать из описания или могут быть изучены при практическом использовании раскрытия. Варианты реализации и преимущества раскрытия будут реализованы и достигнуты посредством элементов и комбинаций, конкретно указанных в прилагаемой формуле изобретения.
[0180] Следует понимать, что как приведенное выше краткое изложение, так и последующее подробное описание являются только иллюстративными и пояснительными и не ограничивают заявленное изобретение.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0181] На ФИГ. 1 представлен профиль отсева USP1 для около 500 линий раковых клеток. Более низкий показатель отсева указывает на большую чувствительность к потере USP1. Линии клеток рака груди показаны белым цветом, а линии клеток рака яичников показаны черным. Символы указывают на мутации BRCA1/2 или статус тройного отрицательного рака груди (TNBC).
[0182] На ФИГ. 2 представлена схема убиквитинирования PCNA от Jacquemont С. и Taniguchi Т., ВМС Biochemistry 8 (Suppl 1): S10 (2007). Е3-лигаза Rad18 моноубиквитинатирует PCNA. USP1 деубиквитнитирует моноубиквитинированную PCNA.
[0183] На ФИГ. 3 показано распределение экспрессии мРНК Rad18 в около 500 линиях раковых клеток. Ось абсцисс показывает нормированные значения экспрессии мРНК Rad18, а ось ординат показывает количество клеточных линий, для которых значение экспрессии указано на оси абсцисс.20 клеточных линий с самыми низкими показателями отсева по USP1 показаны темными полосами, доходящими до верхней части графика.
[0184] На ФИГ. 4 представлены графики, показывающие показатели отсева USP1 по уровням экспрессии мРНК Rad 18 в клеточных линиях рака молочной железы (вверху слева), в линиях клеток рака яичников (вверху справа) и в линиях клеток тройного отрицательного рака молочной железы (внизу).
[0185] На ФИГ. 5 показаны уровни мРНК Rad18 в линиях клеток, чувствительных к USP1 (59М и ES2), и линиях нечувствительных клеток (OVISE, JHH7 и НЕР3В217), измеренные с помощью qRT-PCT, нормализованные по экспрессии GAPDH и нормализованные по OVISE.
[0186] На ФИГ. 6 показаны уровни белка RAD18 в линиях клеток, чувствительных к USP1 (59М и ES2), и в линиях нечувствительных клеток (OVISE, JHH7 и НЕР3В217). Верхняя панель показывает уровни белка в вестерн-блоте, а нижняя панель обеспечивает количественное определение уровней белка вестерн-блоттинга. Было определено, что «НСС1395» в Вестерн-анализе (верхняя панель) загрязнен другой линией клеток и, следовательно, не использовался в количественном анализе (нижняя панель).
[0187] На ФИГ. 7 представлены графики, демонстрирующие, что удаление Rad18 спасает удаление USP1. Графики показывают относительную жизнеспособность клеток в нокаутных клетках OR1A1 (отрицательный контроль) (левая панель) или в нокаутных клетках Rad 18 (правая панель) с добавлением руководств против отрицательного контроля OR1A1 («отрицательный контроль»), пан-летального положительного контроля EEF2 («летальный контроль») или 4 разных руководства USP1 («USP1_1», «USP1_2», «USP1_3» и «USP1_4»).
ПОЛНОЕ ОПИСАНИЕ РАСКРЫТИЯ
[0188] Один аспект данного раскрытия основан на использовании Соединений раскрытия в качестве ингибиторов белка убиквитин-специфической процессинговой протеазы 1 (USP1). Ввиду этого свойства Соединения раскрытия полезны для ингибирования белка USP1 и для лечения заболеваний, расстройств или состояний, например рака, которые реагируют на ингибирование белка USP1.
[0189] В некоторых вариантах реализации Соединения раскрытия демонстрируют улучшенную растворимость, например, как измерено с помощью анализа растворимости ADME, как раскрыто в данном документе.
[0190] В некоторых вариантах реализации Соединения раскрытия демонстрируют улучшенную метаболическую стабильность, например, как измерено с помощью анализов метаболической стабильности микросом печени, как раскрыто в данном документе.
[0191] В других вариантах реализации Соединения раскрытия демонстрируют улучшенную продолжительность действия и пероральное воздействие in vivo.
[0192] В одном варианте реализации Соединения раскрытия представляют собой соединения, имеющие Формулу I:
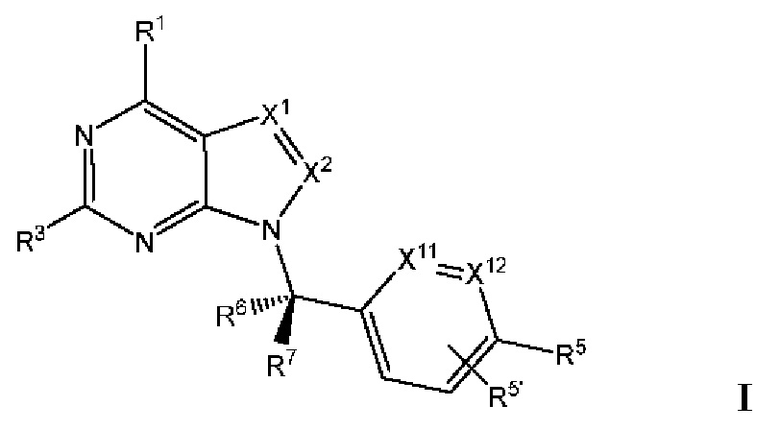
и их фармацевтически приемлемые соли или сольваты, например гидраты, где:
[0193] каждый из X1 и X2 независимо выбран из N и CR2;
[0194] каждый из R1 и R2 независимо выбран из водорода, галогена, циано, необязательно замещенного алкила, необязательно замещенного алкенила и необязательно замещенного алкинила;
[0195] R3 представляет собой необязательно замещенный фенил, необязательно замещенный пиридил, необязательно замещенный пиримидинил, необязательно замещенный пиразинил, необязательно замещенный пиридазинил или необязательно замещенный пиразолил;
[0196] R5' выбран из водорода, необязательно замещенного (C1-C6) алкила, необязательно замещенного (С2-С6) алкенила, необязательно замещенного (С2-С6) алкинила, необязательно замещенного (C1-C6) алкокси, (C1-C6) галогеналкила, (C1-C6) галогеналкокси, (C1-C6) гидроксиалкила, циано, галогена, сульфонамидо,-C(=О)R23,-C(=О)OR24,-NR32aR32b,-NR31aC(=О)R25,-NR31aC(=О)NR31aR31b,-C(=О)NR31a R31b,-S(O)2R27,-NR31aSO2R27, необязательно замещенного (С6-С14) арила, необязательно замещенного (С6-С14) ар-(С1-С2) алкила, необязательно замещенного гетероарила, необязательно замещенного гетероар-(C1-C2) алкила, необязательно замещенного (С3-C8) циклоалкила, необязательно замещенного ((С3-C8) циклоалкил)-(С1-С2) алкила, необязательно замещенного гетероцикло, необязательно замещенного гетероцикло-(С1-С2) алкила, необязательно замещенного-О-(C6-C14) арила, необязательно замещенного-О-(С6-С14) ap-(C1-C2) алкила, необязательно замещенного-О-гетероарила, необязательно замещенного-О-гетероар-(C1-C2) алкила, необязательно замещенного-О-(С3-C8) циклоалкила, необязательно замещенного-О- ((С3-C8) циклоалкил)-(С1-С2) алкила, необязательно замещенного-О-гетероцикло, необязательно замещенного-О-гетероцикло-(С1-С2) алкила;
[0197] R5 выбран из необязательно замещенного (C1-С6) алкила, необязательно замещенного (С2-C6) алкенила, необязательно замещенного (С2-С6) алкинила, необязательно замещенного (C1-C6) алкокси, (C1-C6) галогеналкила, (C1-C6) галогеналкокси, (C1-C6) гидроксиалкила, циано, галогена, сульфонамидо,-C(=О)R23,-C(=О)OR24,-NR32aR32b,-NR31aC(=О)R25,-NR31aC(=О)NR31aR31b,-C(=О)NR31a R31b,-S(O)2R27,-NR31aSO2R27, необязательно замещенного (С6-С14) арила, необязательно замещенного (С6-С14) ар-(С1-С2) алкила, необязательно замещенного гетероарила, необязательно замещенного гетероар-(С1-С2) алкила, необязательно замещенного (С3-C8) циклоалкила, необязательно замещенного ((С3-C8) циклоалкил)-(С1-С2) алкила, необязательно замещенного гетероцикло, необязательно замещенного гетероцикло-(С1-С2) алкила, необязательно замещенного-O-(C6-C14) арила, необязательно замещенного-O-(C6-C14) ар-(С1-С2) алкила, необязательно замещенного-О-гетероарила, необязательно замещенного-O-гетероар-(C1-C2) алкила, необязательно замещенного-О-(C3-C8) циклоалкила, необязательно замещенного-О-((С3-C8) циклоалкил)-(С1-С2) алкила, необязательно замещенного-О-гетероцикло, необязательно замещенного-О-гетероцикло-(С1-С2) алкила; или
[0198] один из R5 и один из R5' на соседних атомах взяты вместе с атомами, к которым они присоединены, с образованием необязательно замещенного (С6-C14) арильного кольца; или один из R5 и один из R5' на соседних атомах взяты вместе с атомами, к которым они присоединены, с образованием необязательно замещенного гетероарильного кольца; или один из R5 и один из R5' на соседних атомах взяты вместе с атомами, к которым они присоединены, с образованием необязательно замещенного (С3-C8) циклоалкильного кольца; или один из R5 и один из R5' на соседних атомахвзяты вместе с атомами, к которым они присоединены, с образованием необязательно замещенного гетероциклоалкильного кольца; или один из R5 и один из R5' на соседних атомах того же атома, к которому они присоединены, взяты вместе с образованием необязательно замещенного спироциклоалкильного кольца; или один из R5 и один из R5' на соседних атомах того же атома, к которому они присоединены, взяты вместе с образованием необязательно замещенного спирогетероциклоалкильного кольца;
[0199] каждый из R6 и R7 независимо выбран из водорода, галогена, циано, необязательно замещенного алкила, необязательно замещенного алкенила и необязательно замещенного алкинила;
[0200] каждый из X11 и Х12 независимо выбран из N и СН;
[0201] R23 выбран из группы, состоящей из водорода, необязательно замещенного алкила, необязательно замещенного алкенила, необязательно замещенного алкинила, амино, алкиламино, диалкиламино, циклоалкиламино, гидроксиалкила, (амино)алкила, (алкиламино)алкила, (диалкиламино)алкила, (циклоалкиламино)алкила, (циклоалкил) алкила, аралкила, (гетероцикло)алкила, (гетероарил)алкила, (амино)(гидрокси)алкила, (аралкиламино)алкила, необязательно замещенного гетероцикло, необязательно замещенного гетероарила, необязательно замещенного арила и необязательно замещенного циклоалкила;
[0202] R31a и R31b каждый независимо выбран из группы, состоящей из водорода, необязательно замещенного алкила, необязательно замещенного алкенила, необязательно замещенного алкинила, гидроксиалкила, (амино)алкила, (алкиламино)алкила, (диалкиламино)алкила, алкоксиалкила, циклоалкила, (циклоалкил)алкила, (гетероцикло)алкила, аралкила и (гетероарил)алкила; и
[0203] каждый из R24, R25, R27, R32a, и R32b независимо выбран из группы, состоящей из водорода, необязательно замещенного алкила, необязательно замещенного алкенила, необязательно замещенного алкинила, амино, алкиламино, диалкиламино, циклоалкиламино, гидроксиалкила, (амино)алкила, (алкиламино)алкила, (диалкиламино)алкила, (циклоалкиламино)алкила, (циклоалкил)алкила, аралкила, (гетероцикло)алкила, (гетероарил)алкила, (амино)(гидрокси) алкила, (аралкиламино)алкила, алкоксиалкила, необязательно замещенного гетероцикло, необязательно замещенного гетероарила, необязательно замещенного арила и необязательно замещенного циклоалкила.
[0204] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где каждый из X1 и Х2 представляет собой CR2.
[0205] В другом варианте реализации Соединение раскрытия представляет собой соединение, имеющее формулу I, где по меньшей мере один из X1 и X2 представляет собой N. В некоторых вариантах реализации X1 представляет собой N. В некоторых вариантах реализации X2 представляет собой N.
[0206] В другом варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где по меньшей мере один из X1 и X2 представляет собой CR2. В некоторых вариантах реализации X1 представляет собой CR2. В некоторых вариантах реализации X2 представляет собой CR2
[0207] В другом варианте реализации Соединение раскрытия представляет собой соединение, имеющее формулу I, где каждый из Х1 и Х2 представляет собой N.
[0208] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, в которой R5' выбран из водорода, галогена и необязательно замещенного (C1-C6) алкила.
[0209] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где  независимо выбрано из:
независимо выбрано из:
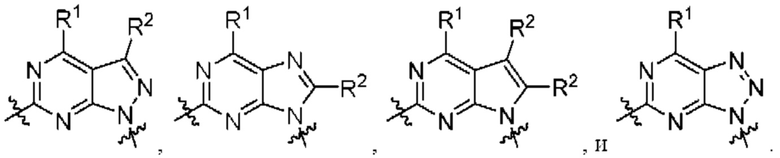
[0210] В другом варианте реализации 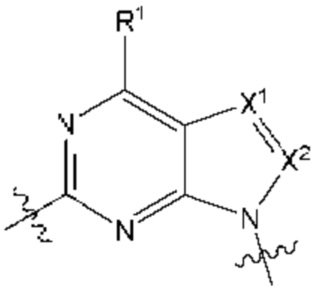 представляет собой
представляет собой 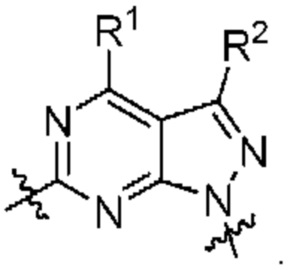
[0211] В другом варианте реализации 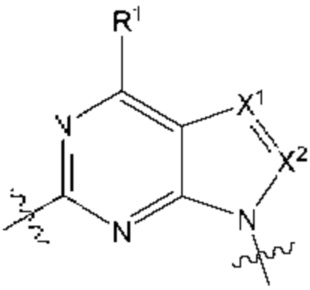 представляет собой
представляет собой 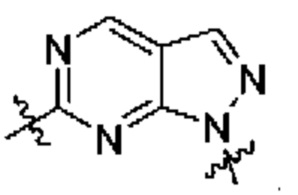
[0212] В другом варианте реализации 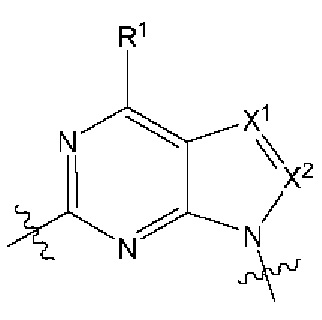 представляет собой
представляет собой 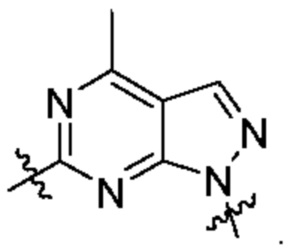
[0213] В другом варианте реализации 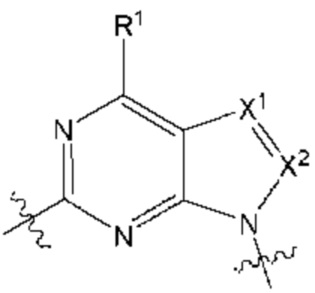 представляет собой
представляет собой 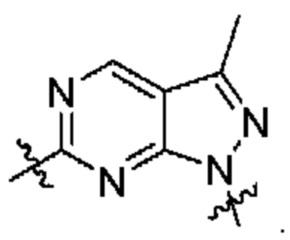
[0214] В другом варианте реализации 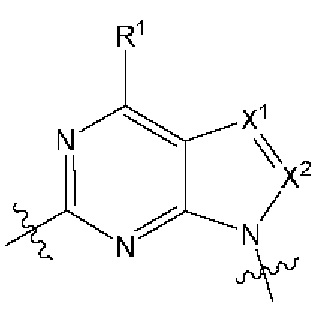 независимо выбран из:
независимо выбран из:
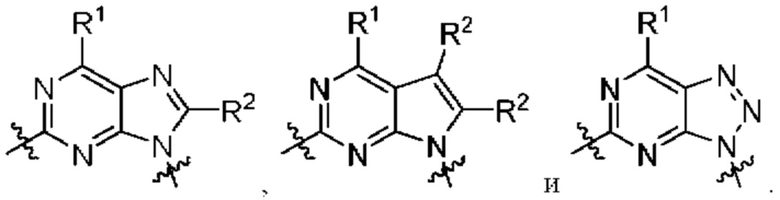
[0215] В другом варианте реализации 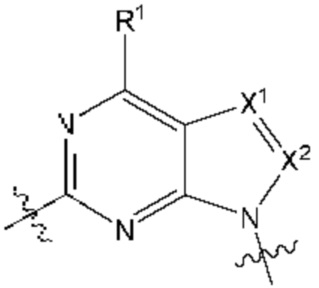 представляет собой
представляет собой 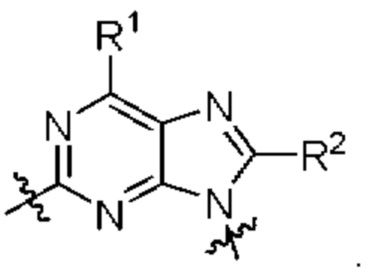
[0216] В другом варианте реализации 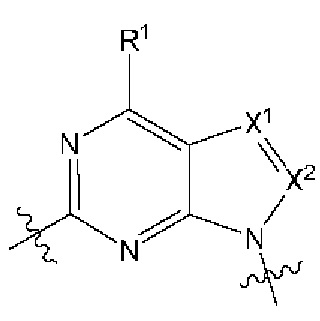 представляет собой
представляет собой 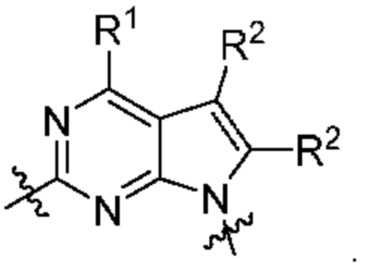
[0217] В другом варианте реализации 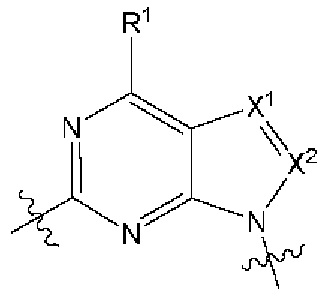 представляет собой
представляет собой 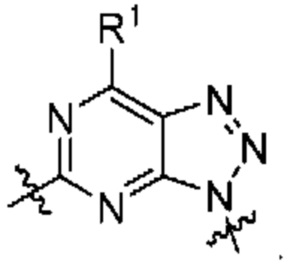
[0218] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, в которой каждый из R1 и R2 независимо выбран из водорода, галогена и циано. [0219] В другом варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где каждый из R1 и R2 независимо выбран из необязательно замещенного (С1-4) алкила, необязательно замещенного (С2-4) алкенила и необязательно замещенного (С2-4) алкинила. В некоторых вариантах реализации каждый из R1 и R2 независимо выбран из необязательно замещенного (С1-4) алкила. В некоторых вариантах реализации каждый из R1 и R2 независимо выбран из необязательно замещенного (С2-4) алкенила. В некоторых вариантах реализации каждый из R1 и R2 независимо выбран из необязательно замещенного (С2-4) алкинила.
[0220] В другом варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R3 представляет собой необязательно замещенный фенил.
[0221] В другом варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R3 представляет собой необязательно замещенный пиридил.
[0222] В другом варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R3 представляет собой необязательно замещенный пиримидил.
[0223] В другом варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R3 представляет собой необязательно замещенный пиразинил.
[0224] В другом варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R3 представляет собой необязательно замещенный пиридазинил.
[0225] В другом варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R3 представляет собой необязательно замещенный пиразолил.
[0226] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где необязательные заместители R3 независимо выбраны из водорода, галогена, нитро, циано, гидрокси, амино, алкиламино, диалкиламино, галогеналкила, гидроксиалкила, алкокси, галогеналкокси, арилокси, гетероарилокси, аралкиларалкилокси, алкилтио, карбоксамидо, сульфонамидо, алкилкарбонила, арилкарбонила, алкилсульфонила, арилсульфонила, карбокси, карбоксиалкила, алкила, необязательно замещенного циклоалкила, алкенила, алкинила, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного гетероцикло, алкоксиалкила, (амино)алкила, гидроксиалкиламино, (алкиламино)алкила, (диалкиламино) алкила, (циано)алкила, (карбоксамидо)алкила, меркаптоалкила, (гетероцикло)алкила, (циклоалкиламино)алкила, (С1-4 галогеналкокси)алкила и (гетероарил)алкила.
[0227] В другом варианте реализации необязательные заместители в R3 независимо выбраны из водорода, галогена, нитро, циано, гидрокси, амино, (С1-С4) алкиламино, ди-(С1-С4) алкиламино, галоген-(С1-С4) алкила, гидрокси-(С1-С4) алкила, (С1-С4) алкокси, галоген-(С1-С4) алкокси, (С6-С10) арилокси, (C3-C6) гетероарилокси, ap-(C1-C4) алкила, ap-(C1-C4) алкилокси, (С1-С4) алкилтио, карбоксамидо, сульфонамидо, (С1-С4) алкилкарбонила, (С6-С10) арилкарбонила, (С1-С4) алкилсульфонила, (С6-С10) арилсульфонила, карбокси, карбокси-(С1-С4) алкила, (С1-С4) алкила, (С2-С4) алкенила, (С2-С4) алкинила, алкокси-(С1-С4) алкила, (амино)-(С1-С4) алкила, гидрокси-(С1-С4) алкиламино, (алкиламино)-(С1-С4) алкила, (диалкиламино)-(С1-С4) алкила, (циано)-(С1-С4) алкила, (карбоксамидо)-(С1-С4) алкила, меркапто-(С1-С4) алкила, (гетероцикло)-(С1-С4) алкила, (циклоалкиламино)-(С1-С4) алкила, (С14 галогеналкокси)-(С1-С4) алкила и (гетероарил)-(С1-С4) алкила.
[0228] В другом варианте реализации необязательные заместители на R3 независимо выбраны из (С3-C6) циклоалкила, (С6-С10) арила, (С3-С6) гетероарила и (С3-C8) гетероцикло.
[0229] В одном варианте реализации Соединение раскрытия представляет собой соединение, имеющее формулу I, где два из необязательных заместителей в R3 взяты вместе с атомами углерода или азота, к которым они присоединены, с образованием необязательно замещенного циклоалкила, необязательно замещенного гетероцикло, необязательно замещенный арила или необязательно замещенной гетероарильной группы.
[0230] В другом варианте реализации два из необязательных заместителей в R3 взяты вместе с атомами углерода или азота, к которым они присоединены, с образованием необязательно замещенного (С3-C8) циклоалкила, необязательно замещенного (С3-С8) гетероцикло, необязательно замещенного (С6-С10) арила или необязательно замещенной (С3-С6) гетероарильной группы.
[0231] В одном варианте реализации Соединение Раскрытия представляет собой соединение, имеющее формулу I, где R3 представляет собой необязательно замещенный фенил, где фенил необязательно замещен в 2-положении. В другом варианте реализации фенил необязательно замещен в 6-положении. В другом варианте реализации фенил необязательно дизамещен в 2- и 6-положениях. В другом варианте реализации фенил необязательно дизамещен в 2- и 3-положениях.
[0232] В одном варианте реализации Соединение Раскрытия представляет собой соединение, имеющее формулу I, где R3 представляет собой необязательно замещенный пирид-3-ип или необязательно замещенный пирид-4-ил.
[0233] В другом варианте реализации R3 представляет собой необязательно замещенный пирид-3-ил. В другом варианте реализации пирид-3-ил необязательно замещен в 2-положении. В другом варианте реализации пирид-3-ил необязательно замещен в 4-положении. В другом варианте реализации пирид-3-ил необязательно дизамещен в 2- и 4-положениях.
[0234] В другом варианте реализации R3 представляет собой необязательно замещенный пирид-4-ил. В другом варианте реализации пирид-4-ил необязательно замещен в 3-положении. В другом варианте реализации пирид-4-ил необязательно замещен в 5-положении. В другом варианте реализации пирид-4-ил необязательно дизамещен в 3- и 5-положениях.
[0235] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R3 представляет собой необязательно замещенный пиримидин-5-ил. В другом варианте реализации пиримидин-5-ил необязательно замещен в 2-м положении. В другом варианте реализации пиримидин-5-ил необязательно замещен в 4-м положении. В другом варианте реализации пиримидин-5-ил необязательно замещен в 6-м положении. В другом варианте реализации пиримидин-5-ил необязательно дизамещен в 4- и 6-положениях. В другом варианте реализации пиримидин-5-ил необязательно дизамещен в 2 и 6-положениях. В другом варианте реализации пиримидин-5-ил необязательно дизамещен в 2 и 4-положениях. В другом варианте реализации пиримидин-5-ил необязательно тризамещен в 2-, 4- и 6-положениях.
[0236] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R3 представляет собой необязательно замещенный пиразол-3-ил или необязательно замещенный пиразол-5-ил.
[0237] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R3 представляет собой необязательно замещенный пиразол-5-ил. В другом варианте реализации пиразол-5-ил необязательно замещен в 1-положении. В другом варианте реализации пиразол-5-ил необязательно замещен в 3-положении. В другом варианте реализации пиразол-5-ил необязательно замещен в 4-положении. В другом варианте реализации пиразол-5-ил необязательно дизамещен в 1- и 4-положениях. В другом варианте реализации пиразол-5-ил необязательно дизамещен в 1- и 3-положениях. В другом варианте реализации пиразол-5-ил необязательно дизамещен в 3- и 4-положениях.
[0238] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R3 замещен, а заместители независимо выбраны из метокси, дейтерометокси, этокси, изопропокси, трет-бутокси, дифторметокси, 2-фторэтокси, 2-метоксиэтокси, циклопропокси, циклобутокси, (тетрагидрофуран-3-ил)окси, бензил оке и, метила, этила, изопропила, 2-фторизопропила, трет-бутила, циклопропила, циклобутила, метилциклопропила, пирролидин-1-ила, азетидин-1-ила, метиламино, диметиламино, циано, галогена, метилтио, метилсульфонила и этилсульфонила.
[0239] В одном варианте реализации R3 выбран из группы, состоящей из:
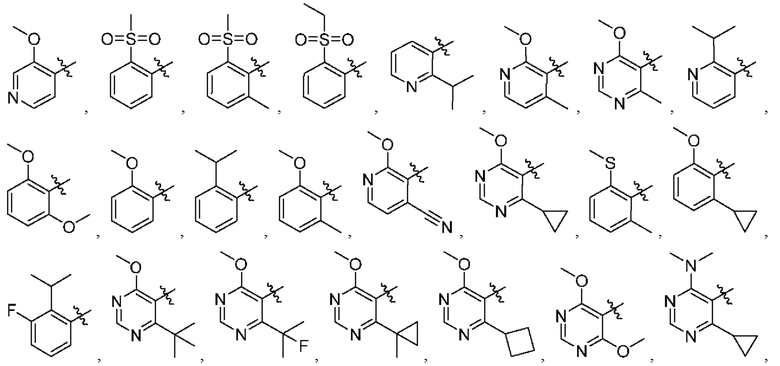
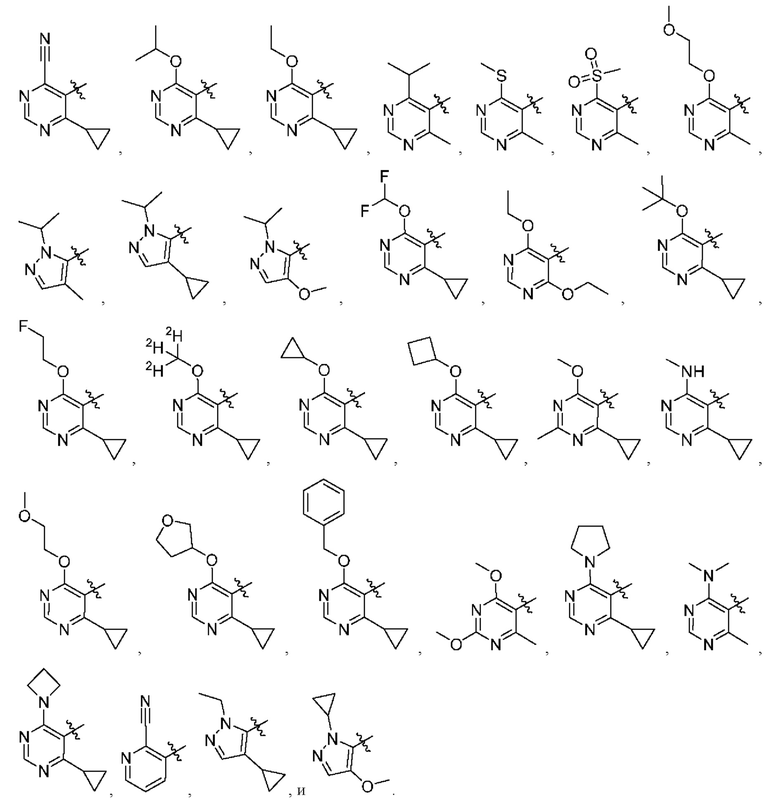
[0240] В другом варианте реализации Соединение раскрытия представляет собой соединение, имеющее формулу I, где по меньшей мере один из X11 и X12 представляет собой N. В некоторых вариантах реализации X11 представляет собой N. В некоторых вариантах реализации X12 представляет собой N.
[0241] В другом варианте реализации Соединение раскрытия представляет собой соединение формулы I, где по меньшей мере один из X11 и X12 представляет собой СН. В некоторых вариантах реализации X11 представляет собой СН. В некоторых вариантах реализации X12 представляет собой СН.
[0242] В одном варианте реализации Соединение раскрытия представляет собой соединение, имеющее формулу I, где каждый из X11 и Х12 представляет собой N.
[0243] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где каждый из X11 и Х12 представляет собой СН.
[0244] В некоторых вариантах реализации один из X11 и X12 представляет собой N, а другой из X11 и X12 представляет собой СН. В одном варианте реализации X11 представляет собой N, а X12 представляет собой СН. В другом варианте реализации X11 представляет собой СН и X12 представляет собой N.
[0245] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R5 выбран из необязательно замещенного (С1-С4) алкила, необязательно замещенного (С2-С4) алкенила, необязательно замещенного (С2-С4) алкинила, необязательно замещенного (С1-С4) алкокси, (С1-С4) галогеналкила, (С1-С4) галогеналкокси, (С1-С4) гидроксиалкила.
[0246] В другом варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R5 выбран из циано, галогена, сульфонамидо,-C(=О)R23,-C(=О)OR24,-NR32aR32b,-NR31aC(=О)R25,-NR31aC(=О)NR31aR31b,-C(=О)NR31a R31b,-S(О)2R27,-NR31aSO2R27.
[0247] В другом варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R5 выбран из необязательно замещенного (С6-С10) арила, необязательно замещенного (С6-С10) ap-(C1-C2) алкила, необязательно замещенного (С3-С6) гетероарипа, необязательно замещенного (С3-С6) гетероар-(C1-C2) алкила, необязательно замещенного (С3-C8) циклоалкила, необязательно замещенного ((С3-С6) циклоалкил)-(С1-С2) алкила, необязательно замещенного (С3-Св) гетероцикло, необязательно замещенного (С3-C8) гетероцикло-(С1-С2) алкила, необязательно замещенного-О-(С6-С10) арила, необязательно замещенного-О-(С6-С10) ap-(C1-C2) алкила, необязательно замещенного-О-(С3-С8) гетероарипа, необязательно замещенного-О-(С3-С8) гетероар-(С1-С2) алкила, необязательно замещенного-О-(С3-С8) циклоалкила, необязательно замещенного-О-((С3-С8) циклоалкил)-(С1-С2) алкила, необязательно замещенного-О-(С3-С8) гетероцикло, необязательно замещенного-О-(С3-С8) гетероцикло-(С1-С2) алкила.
[0248] В другом варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R5 представляет собой необязательно замещенный гетероарил. В другом варианте реализации R5 представляет собой необязательно замещенный 5- или 6-членный гетероарил, содержащий один или более атомов азота. В другом варианте реализации R5 представляет собой необязательно замещенный 5- или 6-членный гетероарил, содержащий только азот в качестве гетероатома или гетероатомов.
[0249] В другом варианте реализации Соединение раскрытия представляет собой соединение, имеющее формулу I, где один из R5 и один из R5' на соседних атомах взяты вместе с атомами, к которым они присоединены, с образованием необязательно замещенного (С6-С10) арильного кольца.
[0250] В другом варианте реализации Соединение раскрытия представляет собой соединение, имеющее формулу I, где один из R5 и один из R5 на соседних атомах взяты вместе с атомами, к которым они присоединены, с образованием необязательно замещенного (С3-С6) гетероарильного кольца.
[0251] В другом варианте реализации Соединение раскрытия представляет собой соединение, имеющее формулу I, где один из R5 и один из R5' на соседних атомах взяты вместе с атомами, к которым они присоединены, с образованием необязательно замещенного (С3-C8) циклоалкильного кольца.
[0252] В другом варианте реализации Соединение раскрытия представляет собой соединение,
имеющее формулу I, где один из R5 и один из R5'm соседних атомах взяты вместе с атомами, к которым они присоединены, с образованием необязательно замещенного (С3-C6) гетероциклоалкильного кольца.
[0253] В другом варианте реализации Соединение раскрытия представляет собой соединение, имеющее формулу I, где один из R5 и один из R5' на соседних атомах взяты вместе с атомами, к которым они присоединены, с образованием необязательно замещенного спироциклоалкильного кольца.
[0254] В другом варианте реализации Соединение раскрытия представляет собой соединение, имеющее формулу I, где один из R5 и один из R5' на соседних атомах взяты вместе с атомами, к которым они присоединены, с образованием необязательно замещенного спирогетероциклоалкильного кольца.
[0255] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где необязательные заместители в R5 независимо выбраны из водорода, галогена, нигро, циано, необязательно замещенного алкила, необязательно замещенного алкенила, необязательно замещенного алкинила, алкокси, гидрокси, карбокси, карбоксиалкила, амино, алкиламино, диалкиламино, циклоалкиламино, гетероциклоалкиламино, аралкиламино, гетероаралкиламино, алкилтио, галогеналкила, галогеналкокси, гидроксиалкила, гидроксиалкиламино, алкоксиалкила, (алкоксиалкил)амино, (амино)алкила, (алкиламино)алкила, (диалкиламино)алкила, (циклоалкиламино)алкила, (карбоксамидо)алкила, меркаптоалкила, (циано)алкила, (циклоалкил)алкила, аралкила, аралкилокси, алкилкарбонила, арилкарбонила, (гетероцикло)алкила, (гетероарил)алкила, (амино)(гидрокси)алкила, (аралкиламино)алкила, (С1-4 галогеналкокси)алкила, необязательно замещенного гетероцикло, необязательно замещенного гетероарила, необязательно замещенного арила, необязательно замещенного циклоалкила, карбоксамидо, сульфонила, сульфонамидо, сульфамидо, алкилсульфонила, алкилсульфонамидо, алкилсульфамидо, арилсульфонила, арилокси, гетероарилокси-C(=О)R23,-C(=О)OR24,-C(=О)NR31a R31b,-NR31aC(=О)R25,-NR31aC(=О)OR26,-NR31aC(=О)NR31aR31b,-NR31aSO2R27,-OC(=О)R28,-OC(=О)OR29,-OC(=О)NR31a R31b,-OSO2R30, и-NR32aR32b.
[0256] В другом варианте реализации необязательные заместители в R5 независимо выбраны из водорода, галогена, нитро, циано, необязательно замещенного (С1-С4) алкила, необязательно замещенного (С2-С4) алкенила, необязательно замещенного (С2-С4) алкинила, (С1-С4) алкокси, гидрокси, карбокси, карбокси-(С1-С4) алкила, амино, (С1-С4) алкиламино, ди-(C1-С4) алкиламино, (С3-C8) циклоалкиламино, гетероцикло-(С1-С4) алкиламино, ap-(C1-G4) алкиламино, гетероар-(C1-C4) алкиламино, (С1-С4) алкилтио, галоген-(С1-С4) алкила, галоген-(С1-С4) алкокси, гидрокси-(С1-С4) алкила, гидрокси-(С1-С4) алкиламино, алкокси-(С1-С4) алкила, (алкоксиалкил)амино, (амино)-(С1-С4) алкила, (алкиламино)-(С1-С4) алкила, (диалкиламино)-(С1-С4) алкила, (циклоалкиламино)-(С1-С4) алкила, (карбоксамидо)-(С1-С4) алкила, меркапто-(С1-С4) алкила, (циано)-(С1-С4) алкила, (циклоалкил)-(С1-С4) алкила, ap-(C1-C4) алкила, ap-(C1-C4) алкилокси, (С1-С4) алкилкарбонила, арилкарбонила, (гетероцикло)-(С1-С4) алкила, (гетероарил)-(С1-С4) алкила, (амино)(гидрокси)-(С1-С4) алкила, (аралкиламино)-(С1-С4) алкила и (С14 галогеналкокси)-(С1-С4) алкила.
[0257] В другом варианте реализации необязательные заместители в R5 независимо выбраны из необязательно замещенного (С3-C8) гетероцикло, необязательно замещенного (С3-С6) гетероарила, необязательно замещенного (С6-С10) арила, необязательно замещенного (С3-C8) циклоалкила.
[0258] В другом варианте реализации необязательные заместители R5 независимо выбраны из карбоксамидо, сульфонила, сульфонамидо, сульфамидо, (С1-С4) алкилсульфонила, (С1-С4) алкилсульфонамидо, (С1-С4) алкилсульфамидо, (С6-С10) арилсульфонила, (С6-С10) арилокси, (С3-С6) гетероарилокси,-C(=О)R23,-C(=О)OR24,-C(=О)NR31a R31b,-NR31aC(=О)R25,-NR31aC(=О)OR26,-NR31aC(=О)NR31aR31b,-NR31aSO2R27,-OC(=О)R28,-OC(=О)OR29,-OC(=О)NR31aR31b,-OSO2R30, и-NR32aR32b.
[0259] В одном варианте реализации Соединение раскрытия представляет собой соединение, имеющее формулу I, где два из необязательных заместителей в R5 взяты вместе с атомами углерода или азота, к которым они присоединены, с образованием необязательно замещенного циклоалкила, необязательно замещенного гетероцикло, необязательно замещенный арила или необязательно замещенной гетероарильной группы.
[0260] В другом варианте реализации два из необязательных заместителей в R5 взяты вместе с атомами углерода, к которым они присоединены, с образованием необязательно замещенного (С3-C8) циклоалкила, необязательно замещенного (С3-C8) гетероцикло, необязательно замещенного (С3-С10) арила или необязательно замещенной (С3-С6) гетероарильной группы.
[0261] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R5 выбран из необязательно замещенного (C1-C6) алкила, необязательно замещенного (С2-С6) алкенила, необязательно замещенного (C2-С6) алкинила, необязательно замещенного (C1-C6) алкокси, (C1-С6) галогеналкила, (C1-C6) галогеналкокси и (C1-С6) гидроксиалкила.
[0262] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R5 выбран из циано, галогена, сульфонамидо,-C(=О)R23,-C(=О)OR24,-NR32aR32b,-NR31aC(=О)R25,-NR31aC(=О)NR31aR31b,-C(=О)NR31a R31b,-S(О)2R27 и-NR31aSO2R27
[0263] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R5 выбран из необязательно замещенного-О-(C6-C14) арила, необязательно замещенного-О-(С6-C14) ap-(C1-C2) алкила, необязательно замещенного-О-гетероарила, необязательно замещенного-О-гетероар-(C1-C2) алкила, необязательно замещенного-О-(C3-C8) циклоалкила, необязательно замещенного-О-((C3-C8) циклоалкил)-(С1-С2) алкила, необязательно замещенного-О-гетероцикло, необязательно замещенного-О-гетероцикло-(С1-С2) алкила. В другом варианте реализации R5 выбран из необязательно замещенного-О-(С6-С10) арила, необязательно замещенного-О-(С6-С10) ар-(С1-С2) алкила, необязательно замещенного-О-(С3-С6) гетероарила, необязательно замещенного-О-(С3-С6) гетероар-(С1-С2) алкила, необязательно замещенного-О-(С3-С6) циклоалкила, необязательно замещенного-О-((С3-С6) циклоалкил)-(С1-С2) алкила, необязательно замещенного-О-(С3-С6) гетероцикло, необязательно замещенного-O-(C3-C8) гетероцикло-(С1-С2) алкила.
[0264] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R5 выбран из необязательно замещенного (С6-С14) арила, необязательно замещенного (С6-С14) ap-(C1-C2) алкила, необязательно замещенного гетероарила, необязательно замещенного гетероар-(С1-С2) алкила, необязательно замещенного (С3-C8) циклоалкила, необязательно замещенного ((С3-C8) циклоалкил)-(С1-С2) алкила, необязательно замещенного гетероцикло, необязательно замещенного гетероцикло-(С1-С2) алкила. В другом варианте реализации R5 выбран из необязательно замещенного (С6-С10) арила, необязательно замещенного (С6-С10) ap-(C1-C2) алкила, необязательно замещенного (С3-С6) гетероарила, необязательно замещенного (С3-С6) гетероар-(С1-С2) алкила, необязательно замещенного (С3-C8) циклоалкила, необязательно замещенного ((С3-C8) циклоалкил)-(С1-С2) алкила, необязательно замещенного (С3-C8) гетероцикло, необязательно замещенного (С3-C8)) гетероцикло-(С1-С2) алкила.
[0265] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R5 представляет собой необязательно замещенный пирролил, необязательно замещенный имидазолил, необязательно замещенный пиразолил, необязательно замещенный триазолил или необязательно замещенный тетразолил. В другом варианте реализации R5 представляет собой необязательно замещенный пирролил. В другом варианте реализации R5 представляет собой необязательно замещенный имидазолил. В другом варианте реализации R5 представляет собой необязательно замещенный пиразолил. В другом варианте реализации R5 представляет собой необязательно замещенный триазолил. В другом варианте реализации R5 представляет собой необязательно замещенный тетразолил.
[0266] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R5 замещен, а заместители независимо выбраны из галогена, метила, этила, изопропила, циклопроиила, циклобутила, метокси, этокси, триазолила, циано, необязательно замещенного алкила, амино, алкиламино, диалкиламино, дифторметила, трифторметила, метилсульфонила, оксетан-3-ила и метил азетидинила.
[0267] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R5 выбран из группы, состоящей из:
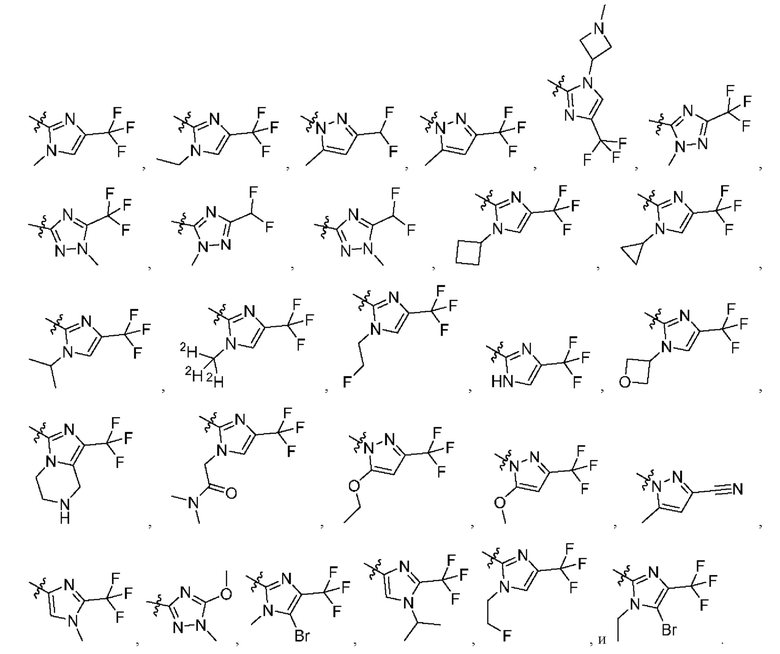
[0268] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где каждый из R6 и R7 независимо выбран из водорода, галогена и циано.
[0269] В другом варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где каждый из R6 и R7 независимо выбран из необязательно замещенного (С1-С4) алкила, необязательно замещенного (С2-С4) алкенила и необязательно замещенного (С2- С4) алкинил.
[0270] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где R23 выбран из группы, состоящей из водорода, необязательно замещенного (С1-С4) алкила, необязательно замещенного (С2-С4) алкенила, необязательно замещенного (С2-С4) алкинила, амино, (С1-С4) алкиламино, ди-(С1-С4) алкиламино, цикло-(С1-С4) алкиламино, гидрокси-(С1-С4) алкила, (амино)-(С1-С4) алкила, (алкиламино)-(С1-С4) алкила, (диалкиламино)-(С1-С4) алкила, (циклоалкиламино)-(С1-С4) алкила, (циклоалкил)-(С1-С4) алкила, ар-(С6-С10) алкилла, (гетероцикло)-(С1-С4) алкила, (гетероарил)-(С1-С4) алкила, (амино)(гидрокси)-(С1-С4) алкила, (аралкиламино-(С1-С4) алкила, необязательно замещенного (С3-C8) гетероцикло, необязательно замещенного (С3-С6) гетероарила, необязательно замещенного (С6-С10) арила и необязательно замещенного (С3-C8) циклоалкила.
[0271] В одном варианте реализации Соединение раскрытия представляет собой соединение, имеющее формулу I, где каждый из R31a и R31b независимо выбран из группы, состоящей из водорода, необязательно замещенного (С1-С4) алкила, необязательно замещенного (С2-С4) алкенила, необязательно замещенного (С2-С4) алкинила, гидрокси-(С1-С4) алкила, (амино)-(С1-С4) алкила, (алкиламино)-(С1-С4) алкила, (диалкиламино)-(С1-С4) алкила, алкокси-(С1-С4) алкила, (С3-C8) циклоалкила, (циклоалкил)-(С1-С4) алкила, (гетероцикло)-(С1-С4) алкила, ap-(C1-C4) алкила и (гетероарил)-(С1-С4) алкила.
[0272] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где каждый из R24, R25, R27, R32a, и R32b независимо выбран из группы, состоящей из водорода, необязательно замещенного (С1-С4) алкила, необязательно замещенного (С2-С4) алкенила, необязательно замещенного (С2-С4) алкинила, амино, (С1-С4) алкиламино, ди-(С1-С4) алкиламино, (С3-C8) циклоалкиламино, гидрокси-(С1-С4) алкила, (амино)-(С1-С4) алкила, (алкипамино)-(С1-С4)алкила, (диалкиламино)-(С1-С4) алкила, (циклоалкиламино)-(C1-C4) алкила, (циклоалкил)-(С1-С4) алкила, ap-(C1-C4) алкила, (гетероцикло)-(С1-С4) алкила, (гетероарил)-(С1-С4) алкила, (амино)(гидрокси)-(С1-С4) алкила, (аралкиламино)-(С1-С4) алкила, алкокси-(С1-С4) алкила, необязательно замещенного (С3-C8) гетероцикло, необязательно замещенного (С3-С6) гетероарипа, необязательно замещенного (C6-С10) арила и необязательно замещенного (С3-C8) циклоалкила.
[0273] В одном варианте реализации Соединение раскрытия представляет собой соединение Формулы I, где каждый из R24, R25, R26, R27, R28, R29, R30, R32a, и R32b выбран из группы, состоящей из водорода, необязательно замещенного алкила, необязательно замещенного алкенила, необязательно замещенного алкинила, амино, алкиламино, диалкиламино, циклоалкиламино, гидроксиалкила, (амино)алкила, (алкиламино)алкила, (диалкиламино)алкилла, (циклоалкиламино)алкила, (циклоалкил)алкила, аралкила, (гетероцикло)алкила, (гетероарил)алкила, (амино)(гидрокси) алкила, (аралкиламино)алкила, алкоксиалкипа, необязательно замещенного гетероцикло, необязательно замещенного гетероарила, необязательно замещенного арила и необязательно замещенного циклоалкила.
[0274] В другом варианте реализации каждый из R24, R25, R26, R27, R28, R29, R30, R32a, и R32b выбран из группы, состоящей из водорода, необязательно замещенного (С1-С4) алкила, необязательно замещенного (C2-С4) алкенила, необязательно замещенного (С2-С4) алкинила, амино, (С1-С4) алкиламино, ди-(С1-С4)алкиламино, (С3-C8) циклоалкиламино, гидрокси-(С1-С4) алкила, (амино)-(С1-С4) алкила, (алкиламино)-(С1-С4) алкила, (диалкиламино)-(С1-С4) алкила, (циклоалкиламино)-(С1-С4) алкила, (циклоалкил)-(С1-С4) алкила, ap-(C1-C4) алкила, (гетероцикло)-(С1-С4) алкила, (гетероарил)-(С1-С4) алкила, (амино)(гидрокси)-(С1-С4) алкила, (аралкиламино)-(С1-С4) алкила, алкокси-(С1-С4) алкила, необязательно замещенного (С3-C8) гетероцикло, необязательно замещенного (С3-С6) гетероарила, необязательно замещенного (C6-С10) арила и необязательно замещенного (С3-C8) циклоалкила.
[0275] В другом варианте реализации Соединения раскрытия представляют собой соединения, имеющие Формулу II:
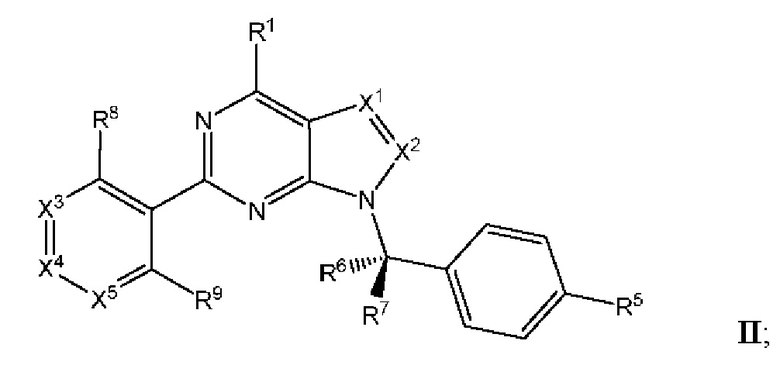
и их фармацевтически приемлемые соли или сольваты, например гидраты, где:
[0276] каждый из X1, X2, R1, R5, R6, и R7 имеет значения, указанные выше для Формулы I;
[0277] X3 выбран из N и CR10; X4 выбран из N и CR11; и X5 выбран из N и CR12; и
[0278] каждый из R8, R9, R10, R11, и R12 независимо выбран из группы, состоящей из водорода, галогена, нитро, циано, гидрокси, амино, алкиламино, диалкиламино, галогеналкила, гидроксиалкила, алкокси, галогеналкокси, арилокси, гетероарилокси, аралкиларалкилокси, алкилтио, карбоксамидо, сульфонамидо, алкилкарбонила, арилкарбонила, алкилсульфонила, арилсульфонила, карбокси, карбоксиалкила, алкила, необязательно замещенного циклоалкила, алкенила, алкинила, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного гетероцикло, алкоксиалкила, (амино)алкила, гидроксиалкиламино, (алкипамино)алкила, (диалкиламино) алкила, (циано)алкила, (карбоксамидо)алкила, меркаптоалкила, (гетероцикло)алкила, (циклоалкиламино)алкила, (C1-4 галогеналкокси)алкила, или (гетероарил)алкила.
[0279] В другом варианте реализации каждый из R8, R9, R10, R11, и R12 независимо выбран из водорода, галогена, нитро, циано, гидрокси, амино, (С1-С4) алкиламино, ди-(С1-С4) алкиламино, галоген-(С1-С4) алкила, гидрокси-(С1-С4) алкила, (С1-С4) алкокси, галоген-(С1-С4) алкокси, (С6-С10) арилокси, (С3-С6) гетероарилокси, ap-(C1-C4) алкил ap-(C1-C4) алкилокси, (С1-С4) алкилтио, карбоксамидо, сульфонамидо, (C1-С4) алкилкарбонила, (С6-С10) арилкарбонила, (С1-С4) алкилсульфонила, (С6-С10) арилсульфонила, карбокси, карбокси-(С1-С4) алкила, (С1-С4) алкила, (С2-С4) алкенила, (С2-С4) алкинила, алкокси-(С1-С4) алкила, (амино)-(С1-С4) алкила, гидрокси-(С1-С4) алкиламино, (алкиламино)-(С1-С4) алкила, (диалкиламино)-(С1-С4) алкила, (циано)-(С1-С4) алкила, (карбоксамидо)-(С1-С4) алкила, меркапто-(С1-С4) алкила, (гетероцикло)-(С1-С4) алкила, (циклоалкиламино)-(С1-С4) алкила, (С1-4 галогеналкокси)-(С1-С4) алкила или (гетероарил)-(С1-С4) алкила.
[0280] В другом варианте реализации каждый из R8, R9, R10, R11, и R12 независимо выбран из необязательно замещенного (С3-C8) циклоалкила, необязательно замещенного (С6-С10) арила, необязательно замещенного (С3-С6) гетероарила и необязательно замещенного (С3-Cs) гетероцикло.
[0281] В одном варианте реализации Соединения раскрытия представляют собой соединения, имеющие Формулу III, формулу IV, формулу V, формулу VI или формулу VIa:
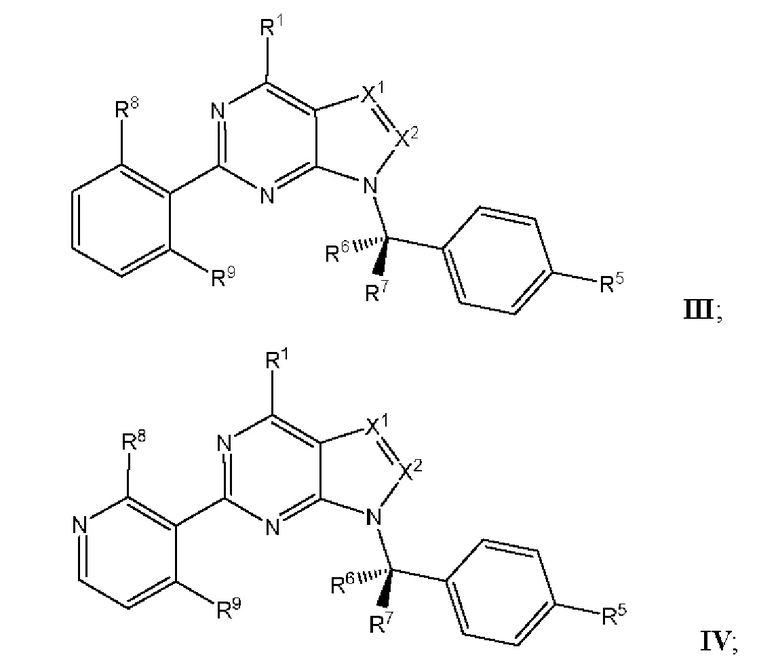
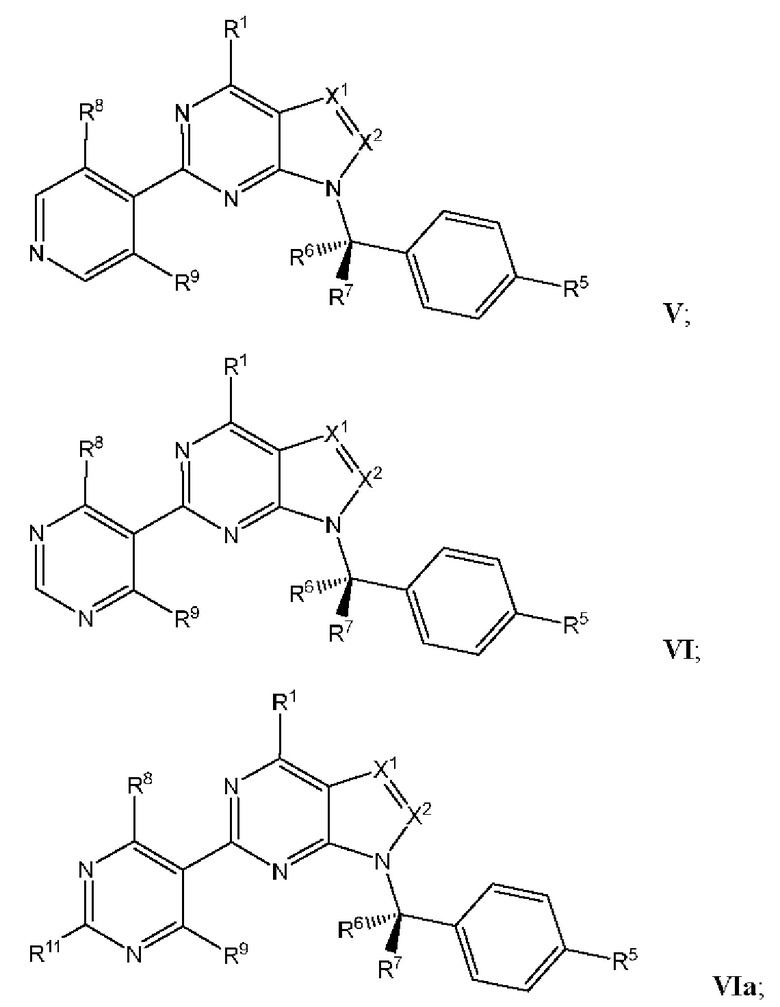
где каждый из X1, X2, R1, R5, R6, R7, R8, R9, и R11 имеет значения, указанные выше для Формулы II.
[0282] В другом варианте реализации R5 представляет собой:
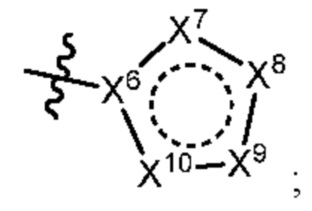
где:
[0283] X6 выбран из NR13 и CR18; X7 выбран из NR14 и CR19; X8 выбран из NR15 и CR20; X9 выбран из NR16 и CR21; X10 выбран из NR17 и CR22; и
[0284] каждый из R13, R14, R15, R16, и R17 отсутствует или независимо выбран из водорода, галогена, метила, этила, изопропила, циклопропила, циклобутила, метокси, этокси, триазолила, циано, необязательно замещенного алкила, амино, алкиламино, диалкиламино, дифторметила, трифторметила, метилсульфонила, оксетан-3-ила и метилазетидинила.
[0285] каждый из R18, R19, R20, R21 и R22 независимо выбран из водорода, галогена, метила, этила, изопропила, циклопропила, метокси, триазолила, циано, необязательно замещенного алкила, амино, алкиламино, диалкиламино, дифторметила, трифторметила, метилсульфонила и метилазетидинила.
[0286] В одном варианте реализации Соединения раскрытия представляют собой соединения, имеющие Формулу VII, формулу VIII, формулу IX, формулу X, формулу XI или формулу XII:
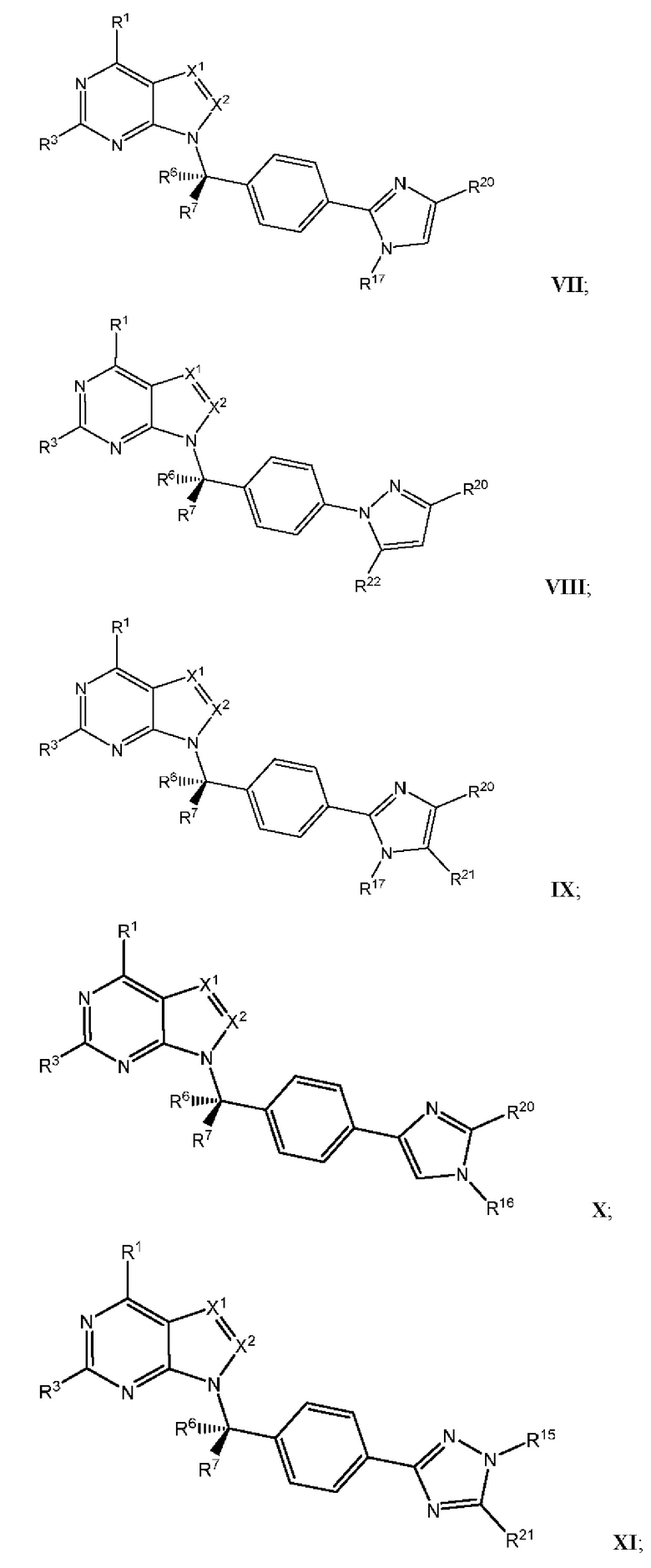
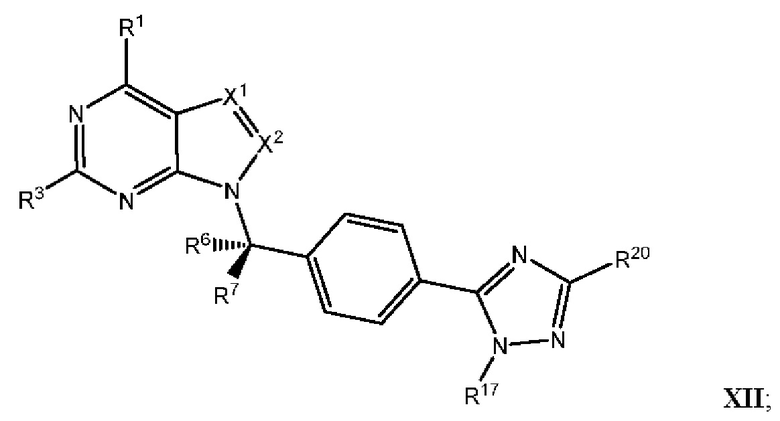
где каждый из X1, X2, R1, R3, R6, R7, R15, R16, R17, R20, R21, и R22 имеют значения, указанны выше для Формулы II.
[0287] В одном варианте реализации Соединения раскрытия представляют собой соединения, имеющие Формулу XIII:
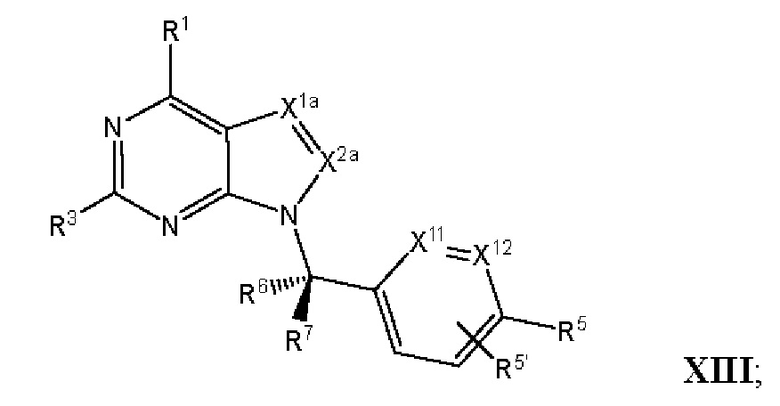
где:
[0288] каждый из Х1а и Х2а независимо выбран из N и CR2a;
[0289] R2a независимо выбран из водорода, галогена, циано, необязательно замещенного алкила, необязательно замещенного алкенила, необязательно замещенного алкинила, алкокси, алкоксиалкила, алкилсульфонила, алкилтио, арила, гетероарила и гетероцикло; и
[0290] каждый из оставшихся заместителей определен, как раскрыто в данном документе.
[0291] В одном варианте реализации Соединения Раскрытия представляют собой соединения, выбранные из группы, состоящей из:
6-(3-метоксипирцдин-4-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 8);
1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-6-(2-(метилсульфонил)фенил)-1Н-пиразоло[3,4-d]пиримидина (Пример 3);
1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-6-(2-метил-6-(метилсульфонил)фенил)-1Н-пиразоло[3,4-d]пиримидина (Пример 23);
6-(2-(этилсульфонил)фенил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 7);
1-(4-(3-(дифторметил)-5-метил-1Н-пиразол-1-ил)бензил)-6-(2-изопропилпиридин-3-ил)-1Н-пиразоло[3,4-d]пиримидина (Пример 20);
6-(2-метокси-4-метилпиридин-3-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 6);
6-(4-метокси-6-метилпиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 21);
6-(2-изопропилпиридин-3-ил)-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 18);
6-(2,6-диметоксифенил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 4);
6-(2-метоксифенил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидина (Пример 2);
6-(2-изопропилфенил)-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 15);
6-(2-метокси-6-метилфенил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 5);
2-метокси-3-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил)изоникотинонитрила (Пример 22);
2-(2-изопропилфенил)-9-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-9Н-пурина (Пример 24);
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 12);
6-(2-изопропилпиридин-3-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 9);
1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-6-(2-метил-6-(метилтио)фенил)-1Н-пиразоло[3,4-d]пиримидина (Пример 13);
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 19);
6-(2-циклопропил-6-метоксифенил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 11);
6-(2-изопропилфенил)-1-(4-(1-(1-метилазетидин-3-ил)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 25);
6-(2-изопропилфенил)-1-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этил)-1Н-пиразоло[3,4-d]пиримидина (Пример 16);
6-(2-изопропилфенил)-4-метил-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 14);
6-(3-фтор-2-изопропилфенил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 10);
6-(2-изопропилфенил)-3-метил-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 17);
6-(2-изопропилфенил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 1);
6-(4-(трет-бутил)-6-метоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-(2-фторпропан-2-ил)-6-метоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-метокси-6-(1-метилциклопропил)пиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклобутил-6-метоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
(S)-6-(4-циклопропил-6-метоксипиримвдин-5-ил)-1-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этил)-1Н-пиразоло[3,4-d]пиримидина;
(R)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-этоксипиримвдин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-изопропоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-циклопропил-5-(1-(4-(1-метил-4-(трифторметил)-1-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидин-6-ил)пиримидин-4-карбонитрила;
6-циклопропил-N,N-диметил-5-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил)пиримидин-4-амина;
6-(4,6-диметоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-изопропил-6-метилпир имидин-5-ил)-1-(4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-6-(4-метил-6-(метилтио)пиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина;
2-(4-циклопропил-6-метокс ипиримидин-5-ил)-8-метил-9-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-9Н-пурина;
1-(4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензил)-6-(4-метил-6-(метилсульфонил)пиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-(2-метоксиэтокси)-6-метилпиримидин-5-ил)-1-(4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-(1-изопропил-4-метил-1H-пиразол-5-ил)-1-(4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензил)-1H-пиразол[3,4-d]пиримидина;
6-(1-изопропил-4-метокси-1Н-пиразол-5-ил)-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-1-изопропил-1Н-пиразол-5-ил)-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразол[3,4-d]пиримидина;
2-(4-циклопропил-6-метоксипиримидин-5-ил)-7-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-7Н-пирроло[2,3-d]пиримидина; и
5-(4-циклопропил-6-метоксипиримидин-5-ил)-3-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-3Н-[1,2,3]триазоло[4,5-d]пиримидина,
или фармацевтически приемлемая соль или сольват, например гидрат, любого из вышеперечисленных.
[0292] В другом варианте реализации Соединения раскрытия представляют собой соединения, выбранные из группы, состоящей из:
6-(4,6-диэтоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-a]пиразин-3-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-метил-5-(трифторметил)-1Н-1,24-триазол-3-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(1-циклопропил-4-метокси-1H-пиразол-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имвдазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-((6-(5-метокси-3-(трифторметил)-1H-пиразол-1-ил)пирид-3-ил)метил)-1Н-пиразоло[3,4-d]пиримидина;
(R)-6-(4-циклобутил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)фенил)этил)-1Н-пиразоло[3,4-d]пиримидина;
(S)-6-(4-циклобутил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)фенил)этил)-1Н-пиразоло[3,4-d]пиримидина;
(R)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)фенил)этил)-1Н-пиразоло[3,4-d]пиримидина;
(S)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)фенил)этил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-метил-2-(трифторметил)-1H-имидазол-4-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-(4-(2-фторпропан-2-ил)-6-метоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(5-этокси-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-((6-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)пиридин-3-ил)метил)-1H-пиразоло[3,4-d]пиримидина;
1-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-5-метил-1Н-пиразол-3-карбонитрила;
6-(4-циклопропил-1-этил-1Н-пиразол-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразол[3,4-d]пиримидина;
6-(4-циклопропил-6-(метокси-d3)пиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-циклопропил-5-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидин-6-ил) пиримидин-4-карбонитрила;
6-(4-циклопропокси-6-циклопропилпиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклобутокси-6-циклопропилпиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-(дифторметокси)пиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метокси-2-метилпир имидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-(оксетан-3-ил)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-(трет-бутил)-6-метоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-(4-(трет-бутокси)-6-циклопропилпиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-(2-фторэтокси)пиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-(метил-d3)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
1-(4-(1-циклобутил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1H-пиразоло[3,4-d]пиримидина;
6-(4,6-диметоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-изопропил-4-(трифторметип)-1H-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
1-(4-(1-циклопропил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-(2-фторэтил)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(3-фтор-4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(2-фтор-4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(2,4-диметокси-6-метилпиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
б-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(5-метокси-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-(2-метоксиэтокси)пиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-(2-метоксиэтокси)пиримидин-5-ил)-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-метокси-6-(1-метилциклопропил)пиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-(1-изопропил-4-метил-1H-пиразол-5-ил)-1-(4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
(S)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)фенил)этил)-1Н-пиразоло[3,4-d]пиримидина;
(R)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этил)-1Н-пиразоло[3,4-d]пиримидина;
6-циклопропил-N,N-диметил-5-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил)пиримидин-4-амина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(5-(дифторметил)-1-метил-1Н-1,2,4-триазол-3-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-(пирролидин-1-ил)пиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-(азетидин-1-ил)-6-циклопропилпиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина; и
5-(4-циклопропил-6-метоксипиримидин-5-ил)-3-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-3Н-[1,2,3]триазоло[4,5-d]пиримидина,
или фармацевтически приемлемая соль или сольват, например гидрат, любого из вышеперечисленных.
[0293] В другом варианте реализации Соединения раскрытия представляют собой соединения, выбранные из группы, состоящей из:
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-3-(метилсульфонил)-1H-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-3-(метилтио)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-3-((4-метоксибензил)тио)-1Н-пиразоло[3,4-d]пиримидина;
(S)-1-(1-(4-(5-бром-1-метил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этил)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-3-метокси-1H-пиразоло[3,4-d]пиримидина;
2-(4-циклопропил-6-метоксипиримидин-5-ил)-7-(4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-7Н-пирроло[2,3-d]пиримидина;
2-(4-циклопропил-6-метоксипиримидин-5-ил)-9-(4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-8-(метоксиметил)-9Н-пурина;
2-(4-циклопропил-6-метоксипиримидин-5-ил)-8-изопропил-9-(4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-9Н-пурина;
3-циклобутокси-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-изопропил-2-(трифторметил)-1Н-имидазол-4-ил)бензил)-3-метокси-1H-пиразоло[3,4-d]пиримидина;
2-(4-циклопропил-6-метоксипиримидин-5-ил)-8-этил-9-(4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-9Н-пурина;
(S)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-(2-фторэтил)-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этил)-3-метокси-1H-пиразоло[3,4-d]пиримидина;
3-(азетидин-1-ил)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
1-(4-(5-бром-1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-3-метокси-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-(2-фторэтил)-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этил)-3-метокси-1H-пиразоло[3,4-d]пиримидина;
(S)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)фенил)этил)-3-метокси-1Н-пиразоло[3,4-d]пиримидина;
(S)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этил[-3-метокси-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-этоксипиримидин-5-ил)-3-этокси-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-3-этокси-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)-3-фторбензил)-3-метокси-1H-пиразоло[3,4-d]пиримидина;
2-(4-циклопропил-6-метоксипиримидин-5-ил)-9-(4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-9Н-пурина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-3-метокси-1Н-пиразоло[3,4-d]пиримидина;
2-(4-циклопропил-6-метоксипиримидин-5-ил)-8-метил-9-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-9Н-пурина;
8-циклопропил-2-(4-циклопропил-6-метоксипиримидин-5-ил)-9-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-9Н-пурина;
8-циклобутил-2-(4-циклопропил-6-метоксипиримидин-5-ил)-9-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-9Н-пурина;
2-(4-циклопропил-6-метоксипиримидин-5-ил)-8-изопропил-9-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-9Н-пурина;
2-(4-циклопропил-6-метоксипиримидин-5-ил)-8-этил-9-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-9Н-пурина;
2-(4-циклопропил-6-метоксипиримидин-5-ил)-7-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-7Н-пирроло[2,3-d]пиримидина;
2-(4-циклопропил-6-метоксипиримидин-5-ил)-8-метил-9-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-9Н-пурина;
5-(4-циклопропил-6-метоксипиримидин-5-ил)-3-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-3Н-[1,2,3]триазоло[4,5-d]пиримидина;
2-(4-метокси-6-метилпиримидин-5-ил)-9-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-9Н-пурина;
2-(4-циклопропил-6-метоксипиримидин-5-ил)-9-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-9Н-пурина;
2-(4-циклопропил-6-метоксипиримидин-5-ил)-9-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-9Н-пурина; и
2-(2-изопропилфенил)-9-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-9Н-пурина
Определения
[0294] Для целей данного раскрытия термин «алкил», используемый сам по себе или как часть другой группы, относится к алифатическому углеводороду с прямой или разветвленной цепью, содержащему от одного до двенадцати атомов углерода (т.е. C1-12 алкил) или числу обозначенных атомов углерода (т.е. C1-алкил, такой как метил, С2-алкил, такой как этил, С3-алкил, такой как пропил или изопропил, и т.д.). Алкильная группа может быть подходящим образом выбрана из С1-10алкильной группы с прямой цепью, С3-10алкильной группы с разветвленной цепью, С1-6алкильной группы с прямой цепью, С3-6алкильной группы с разветвленной цепью, С1-4алкильной группы с прямой цепью, С3-4алкильной группы с разветвленной цепью, С3-4алкильной группы с прямой или разветвленной цепью. Алкильная группа может быть частично или полностью дейтерированной, т.е. один или несколько атомов водорода алкильной группы заменены атомами дейтерия. He-ограничивающие примеры C1-10 алкильных групп включают метил (включая -CD3), этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, изо-бутил, 3-пентил, гексил, гептил, октил, нонил и децил. Неограничивающие примеры C1-4 алкильных групп включают метил, этил, пропил, изопропил, бутил, втор -бутил, трет-бутил и изо-бутил.
[0295] Для целей данного раскрытия термин «необязательно замещенный алкил», используемый сам по себе или как часть другой группы, означает, что алкил, как определено выше, либо незамещен, либо замещен одним, двумя или тремя заместителями, независимо выбранными из галогена, нитро, циано, гидрокси, амино, алкиламино, диалкиламино, галогеналкила, гидроксиалкила, алкокси, галогеналкокси, арилокси, аралкила, аралкилокси, алкилтио, карбоксамидо, сульфонамидо, алкилкарбонила, арилкарбонила, алкилсульфонила, арилсульфонила, карбокси, карбоксиалкила, алкоксиалкила, (амино)алкила, гидроксиалкиламино, (алкиламино) алкила, (диалкиламино)алкила, (циано)алкила, (карбоксамидо)алкила, меркаптоалкила, (гетероцикло)алкила или (гетероарил)алкила. Алкил может быть необязательно замещенным C1-4 алкилом. Необязательно замещенный алкил может быть замещен двумя заместителями или одним заместителем. He-ограничивающие примеры необязательно замещенных алкильных групп включают -CH2CH2NO2, -CH2CH2CO2H, -CH2CH2SO2CH3, -CH2CH2COPh и -CH2C6H11.
[0296] Для целей данного раскрытия термин «алкилен» или «алкиленил» относится к двухвалентному алкильному радикалу. Любая из вышеупомянутых одновалентных алкильных групп может быть алкиленом за счет отщепления второго атома водорода от алкила. Алкиленовая группа также может быть C1-C6 алкиленом или C1-C4 алкиленом. He-ограничивающие иллюстративные алкиленовые группы включают, -СН2-, -CH(СН3)-, -С(СН3)2-, -СН2СН2-, -СН2СН(СН3)-, -СН2С(СН3)2-, -CH2CH2CH2-, и -СН2СН2СН2СН2-.
[0297] Для целей данного раскрытия термин «циклоалкил», используемый сам по себе или как часть другой группы, относится к насыщенным и частично ненасыщенным (содержащим одну или две двойные связи) циклическим алифатическим углеводородам, содержащим от одного до трех колец, имеющих от трех до двенадцати атомы углерода (т.е. C3-12 циклоалкил) или количество обозначенных атомов углерода. Циклоалкильная группа может иметь два кольца или одно кольцо. Циклоалкильная группа может быть выбрана из С3-8 циклоалкильной группы и С3-6 циклоалкильной группы. Циклоалкильная группа может содержать одну или более двойных связей углерод-углерод или одну двойную связь углерод-углерод. Неограничивающие примеры циклоалкильных групп включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, норборнил, декалин, адамантил, циклогексенил и спиро[3.3]гептан.
[0298] Для целей данного раскрытия термин «необязательно замещенный циклоалкил», используемый сам по себе или как часть другой группы, означает, что циклоалкил, как определено выше, либо незамещен, либо замещен одним, двумя или тремя заместителями, независимо выбранными из галогена, нитро, циано, гидрокси, амино, алкиламино, диалкиламино, галогеналкила, гидроксиалкила, алкокси, галогеналкокси, арилокси, аралкила, аралкилокси, алкилтио, карбоксамидо, сульфонамидо, алкилкарбонила, арилкарбонила, алкилсульфонила, арилсульфонила, карбокси, карбоксиалкила, алкила, необязательно замещенного циклоалкила, алкенила, алкинила, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного гетероцикло, алкоксиалкила, (амино)алкила, гидроксиалкиламино, (алкиламино)алкила, (диалкиламино) алкила, (циано)алкила, (карбоксамидо)алкила, меркаптоалкила, (гетероцикло)алкила или (гетероарил)алкила. Необязательно замещенный циклоалкил может быть замещен двумя заместителями или одним заместителем.
[0299] Для целей данного раскрытия термин «алкенил», используемый сам по себе или как часть другой группы, относится к алкильной группе, как определено выше, содержащей одну, две или три двойные связи углерод-углерод. Алкенильная группа может быть выбрана из C2-6 алкенильной группы и С2-4 алкенильной группы. Неограничивающие примеры алкенильных групп включают этенил, пропенил, изопропенил, бутенил, втор-бутенил, пентенил и гексенил.
[0300] Для целей данного раскрытия термин «необязательно замещенный алкенил», используемый в данном документе сам по себе или как часть другой группы, означает, что алкенил, как определено выше, либо незамещен, либо замещен одним, двумя или тремя заместителями, независимо выбранными из галогена, нитро, циано, гидрокси, амино, алкиламино, диалкиламино, галогеналкила, гидроксиалкила, алкокси, галогеналкокси, арилокси, аралкилокси, алкилтио, карбоксамидо, сульфонамидо, алкилкарбонила, арилкарбонила, алкилсульфонила, арилсульфонила, карбокси, карбоксиалкила, необязательно замещенного циклоалкила, необязательно замещенного арила, необязательно замещенного гетероарила или необязательно замещенного гетероцикло.
[0301] Для целей данного раскрытия термин «алкинил», используемый сам по себе или как часть другой группы, относится к алкильной группе, как определено выше, содержащей от одной до трех тройных связей углерод-углерод. Алкинил может иметь одну тройную связь углерод-углерод. Алкинильная группа может быть выбрана из C2-6 алкинильной группы и C2-4 алкинильной группы. Неограничивающие примеры алкинильных групп включают группы этинил, пропилил, бутинил, 2-бутинил, пентинил и гексинил.
[0302] Для целей данного раскрытия термин «необязательно замещенный алкинил», используемый в данном документе сам по себе или как часть другой группы, означает, что алкинил, как определено выше, либо незамещен, либо замещен одним, двумя или тремя заместителями, независимо выбранными из галогена, нитро, циано, гидрокси, амино, алкиламино, диалкиламино, галогеналкила, гидроксиалкила, алкокси, галогеналкокси, арилокси, аралкилокси, алкилтио, карбоксамидо, сульфонамидо, алкилкарбонила, арилкарбонила, алкилсульфонила, арилсульфонила, карбокси, карбоксиалкила, циклоалкила, арила, гетероарила или гетероцикло.
[0303] Для целей данного раскрытия термин «галогеналкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной одним или несколькими атомами фтора, хлора, брома и/или йода. Алкильная группа может быть замещена одним, двумя или тремя атомами фтора и или хлора. Галогеналкильная группа может быть выбрана из C1-4 галогеналкильной группы. Неограничивающие примеры галогеналкильных групп включают фторметильную, дифторметильную, трифторметильную, пентафторэтильную, 1,1-дифторэтильную, 2,2-дифторэтильную, 2,2,2-трифторэтильную, 3,3,3-трифторпропильную, 4,4,4-трифторбутильную и трихлорметильную группы.
[0304] Для целей данного раскрытия термин «гидроксиалкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной одной или несколькими, например, одной, двумя или тремя гидроксигруппами. Гидроксиалкильная группа может быть выбрана из моногидроксиалкильной группы, т.е. замещенной одной гидроксигруппой, дигидроксиалкильной группой, т.е. замещенной двумя гидроксигруппами и С1-4 гидроксиалкильной группой. Неограничивающие примеры гидроксиалкильных групп включают гидроксиметил, гидроксиэтил, гидроксипропил и гидроксибутил группы, такие как 1-гидроксиэтил, 2-гидроксиэтил, 1,2-дигидроксиэтил, 2-гидроксипропил, 3-гидроксипропил, 3-гидроксибутил, 4-гидроксибутил, 2-гидрокси-1-метилпропил, и 1,3-дигидроксипроп-2-ил.
[0305] Для целей данного раскрытия термин «алкокси», используемый сам по себе или как часть другой группы, относится к необязательно замещенному алкилу, необязательно замещенному циклоалкилу, необязательно замещенному гетероцикло, необязательно замещенному алкенилу или необязательно замещенному алкинила, присоединенному к концевому атому кислорода. Алкоксигруппа может быть выбрана из C1-4 алкоксигруппы и C1-4 алкила, присоединенного к концевому атому кислорода, например, метокси, этокси и трет-бутокси.
[0306] Для целей данного раскрытия термин «алкилтио», используемый сам по себе или как часть другой группы, относится к атому серы, замещенному необязательно замещенной алкильной группой. Алкилтиогруппа может быть выбрана из C1-4 алкилтиогруппы. Неограничивающие примеры алкилтиогрупп включают -SCH3 (т.е. метилтио) и -SCH2CH3.
[0307] Для целей данного раскрытия термин «алкоксиалкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной алкоксигруппой. Неограничивающие иллюстративные алкоксиалкильные группы включают метоксиметил, метоксиэтил, метоксипропил, метоксибутил, этоксиметил, этоксиэтил, этоксипропил, этоксибутил, пропоксиметил, изопропоксиметил, пропоксиэтил, пропоксипропил, бутоксиметил, трет-бутоксиметил, изобутоксиметил, втор-бутоксиметил и пентилоксиметил.
[0308] Для целей данного раскрытия термин «галоген», используемый сам по себе или как часть другой группы, относится к атому галогена. Неограничивающие примеры галогеновых групп включают фтор, хлор, бром и йод.
[0309] Для целей данного раскрытия термин «галогеналкокси», используемый сам по себе или как часть другой группы, относится к галогеналкилу, присоединенному к концевому атому кислорода. He-ограничивающие примеры галогеналкоксигрупп включают фторметокси, дифторметокси, трифторметокси и 2,2,2-трифторэтокси.
[0310] Для целей данного раскрытия термин «гетероалкил», используемый сам по себе или как часть другой группы, относится к стабильному углеводородному радикалу с прямой или разветвленной цепью, содержащему от 1 до 10 атомов углерода и по меньшей мере два гетероатома, которые могут быть одинаковыми или разными, выбранными из О, N или S, при этом: 1) атом(ы) азота и атом(ы) серы необязательно могут быть окислены; и/или 2) атом(ы) азота необязательно может быть кватернизован. Гетероатомы могут быть размещены в любом внутреннем положении гетероалкильной группы или в положении, в котором гетероалкильная группа присоединена к остальной части молекулы. Гетероалкильная группа может содержать два атома кислорода, один атом кислорода и один атом азота или два атома азота. He-ограничивающие иллюстративные гетероалкильные группы включают -CH2OCH2CH2OCH3, -OCH2CH2OCH2CH2OCH3, -CH2NHCH2CH2OCH2, -OCH2CH2NH2, -NHCH2CH2N(H)CH3, -NHCH2CH2OCH3 и -OCH2CH2OCH3.
[0311] Для целей данного раскрытия термин «арил», используемый сам по себе или как часть другой группы, относится к моноциклической или бициклической ароматической кольцевой системе, содержащей от шести до четырнадцати атомов углерода (т.е. С6-14 арил). Арильная группа может быть выбрана из С6-14 арильной группы и С6-10 арильной группы. Неограничивающие примеры арильных групп включают фенильную (сокращенно Ph), нафтильную, фенантрильную, антрацильную, инденильную, азуленильную, бифенильную, бифениленильную и флуоренильную группы. Арильная группа может быть выбрана из фенила или нафтила. Арильная группа может быть фенилом.
[0312] Для целей данного раскрытия термин «необязательно замещенный арил», используемый здесь сам по себе или как часть другой группы, означает, что арил, как определено выше, либо незамещен, либо замещен от одного до пяти заместителями, независимо выбранными из группы, состоящей из галогена, нитро, циано, гидрокси, амино, алкиламино, диалкиламино, галогеналкила, гидроксиалкила, алкокси, галогеналкокси, арилокси, гетероарилокси, аралкиларалкилокси, алкилтио, карбоксамидо, сульфонамидо, алкилкарбонила, арилкарбонила, алкилсульфонила, арилсульфонила, карбокси, карбоксиалкила, алкила, необязательно замещенного циклоалкила, алкенила, алкинила, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного гетероцикло, алкоксиалкила, (амино)алкила, гидроксиалкиламино, (алкиламино)алкила, (диалкиламино)алкила, (циано)алкила, (карбоксамидо)алкила, меркаптоалкила, (гетероцикло)алкила, (циклоалкиламино)алкила, (C1-4 галогеналкокси)алкила, (гетероарил)алкила. Необязательно замещенный арил может быть необязательно замещенным фенилом. Необязательно замещенный фенил может иметь четыре заместителя, три заместителя, два заместителя или один заместитель. Необязательно замещенный фенил может иметь один амино, алкиламино, диалкиламино, (амино)алкильный, (алкиламино) алкильный или (диалкиламино)алкильный заместитель. Неограничивающие примеры замещенных арильных групп включают 2-метилфенил, 2-метоксифенил, 2-фторфенил, 2-хлорфенил, 2-бромфенил, 3-метилфенил, 3-метоксифенил, 3-фторфенил, 3-хлорфенил, 4-метилфенил, 4-этилфенил, 4-метоксифенил, 4-фторфенил, 4-хлорфенил, 2,6-дифторфенил, 2,6-дихлорфенил, 2-метил, 3-метоксифенил, 2-этил, 3-метоксифенил, 3,4-диметоксифенил, 3,5-дифторфенил, 3,5-диметилфенил, 3,5-диметокси, 4-метилфенил, 2-фтор-3-хлорфенил, 3-хлор-4-фторфенил и 2-фенилпропан-2-амин. Термин «необязательно замещенный арил» включает группы, содержащие конденсированные необязательно замещенные циклоалкильные и конденсированные необязательно замещенные гетероциклические кольца. Примеры включают:
[0313] Для целей данного раскрытия термин «арилокси», используемый сам по себе или как часть другой группы, относится к необязательно замещенному арилу, присоединенному к концевому атому кислорода. Неограничивающей иллюстративной арилокси-группой является PhO-.
[0314] Для целей данного раскрытия термин «гетероарилокси», используемый сам по себе или как часть другой группы, относится к необязательно замещенному гетероарилу, присоединенному к концевому атому кислорода.
[0315] Для целей данного раскрытия термин «аралкилокси» или «арилалкилокси», используемый сам по себе или как часть другой группы, относится к аралкильной группе, присоединенной к концевом атому кислорода. Неограничивающей иллюстративной аралкилоксигруппой является PhCH2O-.
[0316] Для целей данного раскрытия термин «гетероарил» или «гетеро ароматический» относится к моноциклическим и бициклическим ароматическим кольцевым системам, имеющим от 5 до 14 кольцевых атомов (т.е. C5-14 гетероарил) и 1, 2, 3 или 4 гетероатома независимо, выбираются из кислорода, азота или серы. Гетероарильная группа может быть выбрана из C5-14 гетероарильной группы и С3-6 гетероарильной группы. Гетероарил может иметь три гетероатома, два гетероатома или один гетероатом. Гетероарил может быть С5 гетероарилом или С6 гетероарилом. Неограничивающие примеры гетероарильных групп включают тиенил, бензо[b]тиенил, нафто[2,3-b]тиенил, тиантренил, фурил, бензофурил, пиранил, изобензофуранил, бензооксазонил, хроменил, ксантенил, 2H-пирролил, пирролил, имидазолил, пиразолил, пиридил, пиразинил, пиримидинил, пиридазинил, изоиндолил, 3H-индолил, индолил, индазолил, пуринил, изохинолил, хинолил, фталазинил, нафтиридинил, циннолинил, хиназолинил, птеридинил, 4aH-карбазолил, карбазолил, β-карболинил, фенантридинил, акридинил, фенантролинил, феназинил, тиазолил, изотиазолил, фенотиазолил, изоксазолил, фуразанил, триазолил, тетразолил и феноксазинил. Гетероарил может быть выбран из тиенила (напр., тиен-2-ил и тиен-3-ил), фурила (напр., 2-фурил и 3-фурил), пирролила (напр., 1Н-пиррол-2-ил и 1H-пиррол-3-ил), имидазолила (напр., 2Н-имидазол-2-ил и 2Н-имидазол-4-ил), пиразолила (напр., 1H-пиразол-3-ил, 1Н-пиразол-4-ил и 1Н-пиразол-5-ил), пиридила (напр., пиридин-2-ил, пиридин-3-ил и пиридин-4-ил), пиримидинила (напр., пиримидин-2-ил, пиримидин-4-ил и пиримидин -5-ил), тиазолила (напр., тиазол-2-ил, тиазол-4-ил и тиазол-5-ил), изотиазолила (напр., изотиазол-3-ил, изотиазол-4-ил и изотиазол-5-ил), оксазолила (напр., оксазол-2-ил, оксазол-4-ил и оксазол-5-ил) изоксазолил (напр., изоксазол-3-ил, изоксазол-4-ил и изоксазол-5-ил), триазолила (напр., 1,2,4-триазолил и 1,2,3-триазолил). Термин «гетероарил» также включает возможные N-оксиды. Примеры N-оксидов включают пиридил N-оксид.
[0317] Для целей данного раскрытия термин «необязательно замещенный гетероарил», используемый сам по себе или как часть другой группы, означает, что гетероарил, как определено выше, либо незамещен, либо замещен одним-четырьмя заместителями, например одним или двумя заместителями, независимо выбранными из галогена, нитро, пиано, гидрокси, амино, алкиламино, диалкиламино, галогеналкила, гидроксиалкила, алкокси, галогеналкокси, аралкиларилокси, аралкилокси, алкилтио, карбоксамидо, сульфонамидо, алкилкарбонила, арилкарбонила, алкилсульфонила, арилсульфонила, карбокси, карбоксиалкила, алкила, необязательно замещенного циклоалкила, алкенила, алкинила, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного гетероцикло, алкоксиалкила, (амино)алкила, гидроксиалкиламино, (алкиламино)алкила, (диалкиламино)алкила, (пиано)алкила, (карбоксамидо)алкила, меркаптоалкила, (гетероцикло)алкила, (гетероарил)алкила, -N(R33)(R34), или -N(H)C(=O)-R35, где R33 представляет собой водород или C1-4 алкил; R34 представляет собой алкоксиалкил, (гетероцикло)алкил, (амино)алкил, (алкиламино)алкил или (диалкиламино)алкил; и R35 представляет собой алкил, необязательно замещенный арил или необязательно замещенный гетероарил. Необязательно замещенный гетероарил может иметь один заместитель. Заместитель может быть амино, алкиламино, диалкиламино, (амино)алкилом, гидроксиалкиламино, (алкиламино)алкилом, (диалкиламино)алкилом, (гетероцикло)алкилом, -N(R33)(R34) или -N(H)C(=O)-R35. Необязательно замещенный гетероарил может быть необязательно замещенным пиридилом, напр., 2-, 3- или 4-пиридилом. Любой доступный атом углерода или азота может быть замещен.
[0318] Для целей данного раскрытия термин «гетероцикл» или «гетероцикло», используемый сам по себе или как часть другой группы, относится к насыщенным и частично ненасыщенным (напр., содержащим одну или две двойные связи) циклическим группам, содержащим одно, две, или три кольца, содержащие от трех до четырнадцати членов в кольце (напр., от 3 до 14-членного гетероцикло) и по меньшей мере один гетероатом. Гетероцикло группа может быть выбрана из С3-14 гетероцикло группы и С3-8 гетероцикло группы Каждый гетероатом независимо выбран из группы, состоящей из кислорода, серы, включая сульфоксид и сульфон, и/или атомов азота, которые могут быть кватернизованы. Термин «гетероцикло» предназначен для включения циклических уреидогрупп, таких как имидазолидинил-2-он, циклических амидных групп, таких как β-лактам, γ-лактам, δ-лактам и ε-лактам и циклических карбаматных групп, таких как оксазолидинил-2-он.. Термин «гетероцикло» также предназначен для включения групп, имеющих конденсированные необязательно замещенные ар ильные группы, напр., индолинил, индолинил-2-он, бензо[d]оксазолил-2(3Н)-он. Термин «гетероцикло» также предназначен для включения групп, содержащих конденсированные необязательно замещенные гетероарильные группы, напр., 5,6,7,8-тетрагидроимидазо[1,5-а]пиразин. Гетероцикло группа может быть выбрана из 4-, 5-, 6-, 7- или 8-членной циклической группы, содержащей одно кольцо и один или два атома кислорода и/или азота, 5- или 6-членной циклической группы, содержащей одно кольцо и один или два атома азота, 8-, 9-, 10-, 11- или 12-членной циклической группы, содержащей два кольца и один или два атома азота. Гетероцикло необязательно может быть связан с остальной частью молекулы через атом углерода или азота. He-ограничивающие примеры гетероцикло-групп включают 2-оксопирролидин-3-ил, 2-имидазолидинон, пиперидинил, морфолинил, пиперазинил, пирролидинил, азетидинил, 8-азабицикло[3.2.1]октан (нортропан), 6-азаспиро[2.5]октан, 6-азаспиро[3.4]октан, индолинил, индолинил-2-он, 1,3-дигидро-2Н-бензо[d]имидазол-2-он.
[0319] Для целей данного раскрытия термин «необязательно замещенный гетероцикло», используемый в данном документе сам по себе или как часть другой группы, означает, что гетероцикло, как определено выше, является либо незамещенным, либо замещенным от одного до четырех заместителями, независимо выбранными из галогена, нитро, пиано, гидрокси, амино, алкиламино, диалкиламино, галогеналкила, гидроксиалкила, алкокси, галогеналкокси, арилокси, аралкиларалкилокси, алкилтио, карбоксамидо, сульфонамидо, алкилкарбонила, арилкарбонила, алкилсульфонила, арилсульфонила, карбокси, карбоксиалкила, алкила, циклоалкила, алкенила, алкинила, арила, гетероарила, гетероцикло, алкоксиалкила, (амино)алкила, гидроксиалкиламино, (алкиламино)алкила, (диалкиламино)алкила, (циано)алкила, (карбоксамидо)алкила, меркаптоалкила, (гетероцикло)алкила и (гетероарил)алкила. Замещение может происходить у любого доступного атома углерода или азота и может образовываться спироцикл.
[0320] Для целей данного раскрытия термин «амино», используемый сам по себе или как часть другой группы, относится к -NH2.
[0321] Для целей данного раскрытия термин «алкиламино», используемый сам по себе или как часть другой группы, относится к -NHR36, где R36 представляет собой C1-6 алкил. R36 может быть C1-4 алкилом. Неограничивающие примеры алкиламиногрупп включают -N(Н)СН3 и -N(Н)СН2СН3.
[0322] Для целей данного раскрытия термин «диалкиламино», используемый сам по себе или как часть другой группы, относится к -NR37aR37b, где R37a и R37b каждый независимо представляет собой C1-6 алкил. R37a и R37b каждый независимо может означать C1-4 алкил. Неограничивающие примеры диалкиламиногрупп включают -N(CH3)2 и -N(СН3)СН2СН(СН3)2.
[0323] Для целей данного раскрытия термин «гидроксиалкиламино», используемый сам по себе или как часть другой группы, относится к -NHR38, где R38 представляет собой гидроксиалкил.
[0324] Для целей данного раскрытия термин «циклоалкиламино», используемый сам по себе или как часть другой группы, относится к -NR39aR39b, где R39a представляет собой необязательно замещенный циклоалкил, a R39b представляет собой водород или C1-4 алкил.
[0325] Для целей данного раскрытия термин «аралкиламино», используемый сам по себе или как часть другой группы, относится к -NR40aR40b, где R40a представляет собой аралкил, a R40b представляет собой водород или C1-4 алкил. Неограничивающие иллюстративные аралкиламиногруппы включают -N(H)CH2Ph и -N(СН3)CH2Ph.
[0326] Для целей данного раскрытия термин «(амино)алкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной аминогруппой. Алкил может быть C1-4 алкилом. Неограничивающие иллюстративные (амино)алкильные группы включают -CH2NH2, -С(NH2)(Н)СН3, -CH2CH2NH2, -СН2С(NH2)(Н)СН3, -CH2CH2CH2NH2, -CH2CH2CH2CH2NH2, и -СН2С(СН3)2CH2NH2
[0327] Для целей данного раскрытия термин «(алкиламино)алкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной алкиламиногруппой Алкил может быть C1-4 алкилом. Неограничивающей иллюстративной (алкиламино)алкильной группой является -CH2CH2N(Н)СН3.
[0328] Для целей данного раскрытия термин «(диалкиламино)алкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной диалкиламино группой. Алкил может быть C1-4 алкилом. He-ограничивающими иллюстративными (диалкиламино) алкильными группами являются -CH2CH2N(СН3)2.
[0329] Для целей данного раскрытия термин «(циклоалкиламино) алкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной циклоалкиламиногруппой. Алкил может быть C1-4 алкилом. Неограничивающие иллюстративные (циклоалкиламино) алкильные группы включают -CH2N(Н)циклопропил, -CH2N(Н)циклобутил и -CH2N(Н)циклогексил.
[0330] Для целей данного раскрытия термин «(аралкиламино) алкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной аралкиламиногруппой. Алкил может быть C1-4 алкилом. He-ограничивающей иллюстративной (аралкиламино)алкильной группой является -CH2CH2CH2N(H)CH2Ph.
[0331] Для целей данного раскрытия термин «(циано)алкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной одной или несколькими циано, напр., -CN, группами. Алкил может быть C1-4 алкилом. Неограничивающие иллюстративные (циано)алкильные группы включают -CH2CH2CN, -CH2CH2CH2CN, и -CH2CH2CH2CH2CN..
[0332] Для целей данного раскрытия термин «(амино)(гидрокси) алкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной одной амино, алкиламино или диалкиламиногруппой и одной гидроксигруппой. Алкил представляет собой C1-6 алкил или C1-4 алкил.
[0333] Для целей данного раскрытия термин «(амино)(арил)алкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной одной амино, алкиламино или диалкиламиногруппой и одной необязательно замещенной арильной группой.. Алкил может быть C1-6 алкилом. Необязательно замещенная арильная группа может быть необязательно замещенным фенилом.
[0334] Для целей данного раскрытия термин «(циклоалкил)алкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной одной необязательно замещенной циклоалкильной группой. Алкил может быть C1-4 алкилом или С3-6 циклоалкилом. Необязательно замещенная циклоалкильная группа может быть замещена амино или (амино)алкильной группой.
[0335] Для целей данного раскрытия термин «(гидрокси)(арил) алкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной одной гидроксигруппой и одной необязательно замещенной арильной группой. Алкил может быть C1-6 алкилом. Необязательно замещенная арильная группа может быть необязательно замещенным фенилом. He-ограничивающие иллюстративные (гидрокси)(арил)алкильные группы включают:
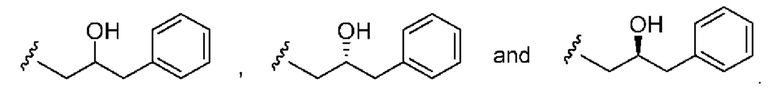
[0336] Для целей данного раскрытия термин «карбоксамидо», используемый сам по себе или как часть другой группы, относится к радикалу формулы -C(=O)NR41aR41b, где R41a и R41b каждый независимо представляют собой водород, необязательно замещенный алкил, необязательно замещенный арил или необязательно замещенный гетероарил, или R41a и R41b, взятые вместе с атомом азота, к которому они присоединены, из 3-8-членной гетероцикло группы. R41a и R41b каждый независимо может представлять собой водород или необязательно замещенный алкил. Неограничивающие иллюстративные карбоксамидные группы включают -CONH2, -CON(H)CH3, -CON(СН3)2, и -CON(H)Ph.
[0337] Для целей данного раскрытия термин «(карбоксамидо)алкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной карбоксамидной группой.
Неограничивающие иллюстративные (карбоксамидо) алкильные группы включают -CH2CONH2, -С(Н)СН3-CONH2, и -CH2CON(Н)СН3.
[0338] Для целей данного раскрытия термин «сульфонамидо», используемый сам по себе или как часть другой группы, относится к радикалу формулы -SO2NR42aR42b, где каждый и R42a и R42b независимо представляет собой водород, необязательно замещенный алкил или необязательно замещенный арил, или R42a и R42b, взятые вместе с азотом, к которому они присоединены из 3-8-членной гетероцикло группы. Неограничивающие примеры сульфонамидогрупп включают -SO2NH2, -SO2N(H)CH3, и -SO2N(H)Ph.
[0339] Для целей данного раскрытия термин «алкилкарбонил», используемый сам по себе или как часть другой группы, относится к карбонильной группе, напр., -С(=О)-, замещенной алкильной группой. He-ограничивающей иллюстративной алкилкарбонильной группой является -СОСН3.
[0340] Для целей данного раскрытия термин «арилкарбонил», используемый сам по себе или как часть другой группы, относится к карбонильной группе, напр., -С(=O)-, замещенной необязательно замещенной арильной группой. He-ограничивающей иллюстративной арилкарбонильной группой является -COPh.
[0341] Для целей данного раскрытия термин «алкилсульфонил», используемый сам по себе или как часть другой группы, относится к сульфонильной группе, напр., -SO2-, замещенной любой из выше-упомянутых необязательно замещенных алкильных групп. Неограничивающими примерами алкилсульфонильных групп являются -SO2CH3 (напр., метилсульфонил) и -SO2CH2CH3 (напр., этилсульфонил).
[0342] Для целей данного раскрытия термин «арилсульфонил», используемый сам по себе или как часть другой группы, относится к сульфонильной группе, напр., -SO2-, замещенной любой из вышеупомянутых необязательно замещенных арильных групп. He-ограничивающей иллюстративной арил сульфонильной группой является -SO2Ph.
[0343] Для целей данного раскрытия термин «меркаптоалкил», используемый сам по себе или как часть другой группы, относится к любой из вышеупомянутых алкильных групп, замещенных группой -SH.
[0344] Для целей данного раскрытия термин «карбокси», используемый сам по себе или как часть другой группы, относится к радикалу формулы -СООН.
[0345] Для целей данного раскрытия термин «карбоксиалкил», используемый сам по себе или как часть другой группы, относится к любой из вышеупомянутых алкильных групп, замещенных -СООН. He-ограничивающей иллюстративной карбоксиалкильной группой является -CH2CO2H.
[0346] Для целей данного раскрытия термин «алкоксикарбонил», используемый сам по себе или как часть другой группы, относится к карбонильной группе, напр., -С(=O)-, замещенной алкоксигруппой. He-ограничивающими примерами алкоксикарбонильных групп являются -CO2Me и -CO2Et.
[0347] Для целей данного раскрытия термин «аралкил» или «арилалкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной одной, двумя или тремя необязательно замещенными арильными группами. Аралкильная группа может быть C1-4 алкилом, замещенным одной необязательно замещенной арильной группой. Неограничивающие примеры аралкильных групп включают бензин, фенэтил, -CHPh2, -CH2(4-OH-Ph) и -CH(4-F-Ph)2.
[0348] Для целей данного раскрытия термин «(гетероцикло)алкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной одной, двумя или тремя необязательно замещенными гетероцикло группами. (Гетероцикло)алкил может быть C1-4 алкилом, замещенным одной необязательно замещенной гетероцикло группой. Гетероцикло может быть связан с алкильной группой через атом углерода или азота. He-ограничивающие иллюстративные (гетероцикло) алкильные группы включают:
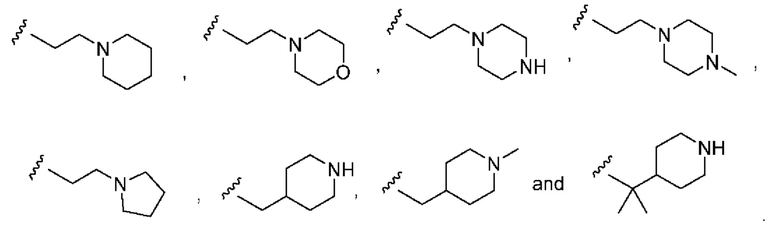
[0349] Для целей данного раскрытия термин «гетероаралкил» или «(гетероарил)алкил», используемый сам по себе или как часть другой группы, относится к алкильной группе, замещенной одной, двумя или тремя необязательно замещенными гетероарильными группами. (Гетероарил)алкильная группа может быть C1-4 алкилом, замещенным одной необязательно замещенной гетероарильной группой. He-ограничивающие иллюстративные (гетероарил) алкильные группы включают:
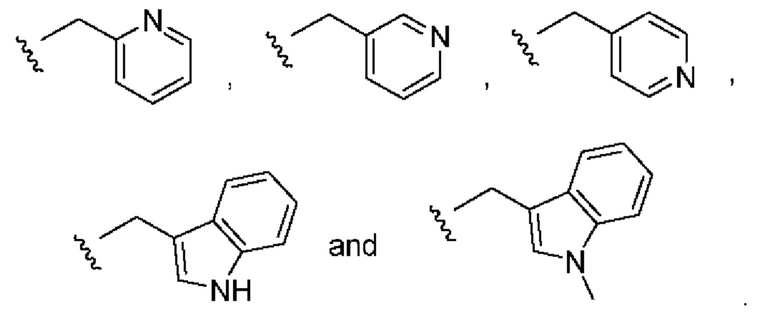
[0350] Для целей данного раскрытия термин «алкилкарбониламино», используемый сам по себе или как часть другой группы, относится к алкилкарбонильной группе, присоединенной к амино. Неограничивающей иллюстративной алкилкарбониламино группой является -NHCOCH3.
[0351] Данное раскрытие охватывает любое из Соединений раскрытия, меченых изотопами (напр., радиоактивно меченых), посредством замены одного или нескольких атомов атомом, имеющим другую атомную массу или массовое число. Примеры изотопов, которые могут быть включены в описанные соединения, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2H (или дейтерий (D)), 3H, 11С, 13С, 14С, 15N, 18O, 17О, 31Р, 32Р, 35S, 18F, и 36Cl, соответственно, напр., 3H, 11С и 14С. Данное раскрытие также обеспечивает композицию, в которой практически все атомы в положении в Соединении раскрытия заменены атомом, имеющим другую атомную массу или массовое число. Данное раскрытие также обеспечивает композицию, в которой часть атомов в положении в пределах Соединения раскрытия заменена, напр.. Соединение Раскрытия обогащено в положении атомом, имеющим другую атомную массу или массовое число. В одном варианте реализации данное раскрытие обеспечивает композицию, в которой Соединение раскрытия имеет от 1 до 8 атомов водорода, замененных дейтерием. Меченые-изотопами Соединения раскрытия могут быть получены способами, известными в данной области.
[0352] Соединения раскрытия могут содержать один или несколько асимметричных центров и, таким образом, могут давать энантиомеры, диастереомеры и другие стереоизомерные формы. Данное раскрытие охватывает использование всех таких возможных форм, а также их рацемических и разделенных форм и их смесей. Индивидуальные энантиомеры могут быть разделены способами, известными в данной области техники с учетом данного раскрытия. Когда описанные в данном документе соединения содержат олефиновые двойные связи или другие центры геометрической асимметрии, и если не указано иное, предполагается, что они включают оба Е и Z геометрических изомера. Все таутомеры также охватываются данным раскрытием.
[0353] Используемый в данном документе термин «стерео изомеры» является общим термином для всех изомеров отдельных молекул, которые различаются только ориентацией своих атомов в пространстве Он включает энантиомеры и изомеры соединений с более чем одним хиральным центром, которые не являются зеркальным отображением друг друга (диастереомеры).
[0354] Термин «хиральный центр» или «асимметричный атом углерода» относится к атому углерода, к которому присоединены четыре разные группы.
[0355] Термины «энантиомер» и «энантиомерный» относятся к молекуле, которая не может быть наложена на ее зеркальное изображение и, следовательно, является оптически активной, когда энантиомер вращает плоскость поляризованного света в одном направлении, а его соединение зеркального отображения вращает плоскость поляризованного света в противоположном направлении.
[0356] Термин «рацемический» относится к смеси равных частей энантиомеров, и эта смесь оптически неактивна.
[0357] Термин «абсолютная конфигурация» относится к пространственному расположению атомов хирального молекулярного объекта (или группы) и его стереохимическому описанию, напр., R или S.
[0358] Стереохимические термины и условные обозначения, используемые в описании, должны соответствовать тем, которые описаны в Pure & Appl. Chem 68: 2193 (1996), если не указано иное.
[0359] Термин «энантиомерный избыток» или «ее» относится к мере того, сколько присутствует одного энантиомера по сравнению с другим. Для смеси энантиомеров R и S процент энантиомерного избытка определяется как |R-S|*100, где R и S - соответствующие мольные или массовые доли энантиомеров в смеси, так чтобы R+S=1. Зная оптическое вращение хирального вещества, процент энантиомерного избытка определяется как ([α]obs/[α]max)*100, где [α]obs представляет собой оптическое вращение смеси энантиомеров, а [α]max представляет собой оптическое вращение чистого энантиомера. Определение энантиомерного избытка возможно с использованием различных аналитических методов, включая ЯМР-спектроскопию, хиральную колоночную хроматографию или оптическую поляриметрию.
[0360] Термины «энантиомерно чистый» или «энантиочистый» относятся к образцу хирального вещества, все молекулы которого (в пределах обнаружения) имеют один и тот же смысл хиральности.
[0361] Термины «энантиомерно обогащенный» или «энантиобогащенный» относятся к образцу хирального вещества, энантиомерное соотношение которого превышает 50:50. Энантиомерно обогащенные соединения могут быть энантиомерно чистыми.
[0362] Понятно, что варианты реализацииния изобретения, описанные в данном документе, включают варианты «состоящие» и/или «состоящие по существу из». Используемые в данном документе формы единственного числа включают множественные ссылки, если не указано иное. Использование термина «или» в данном документе не означает, что альтернативы являются взаимоисключающими.
[0363] В этой заявке использование «или» означает «и или», если явно не указано или не понятно специалисту в данной области. В контексте множественного зависимого пункта формулы использование «или» относится к более чем одному предшествующему независимому или зависимому пункту формулы.
[0364] Используемый в данном документе термин «около» включает указанное число ± 10%. Таким образом, «около 10» означает от 9 до 11. Как понятно специалисту в данной области техники, ссылка на «около» значения или параметра в данном документе включает (и описывает) примеры, которые направлены на это значение или параметр как таковые. Например, описание, относящееся к «около X», включает описание «X».
[0365] Данное раскрытие охватывает получение и использование солей Соединений раскрытия, включая нетоксичные фармацевтически приемлемые соли. Примеры фармацевтически приемлемых аддитивных солей включают аддитивные соли неорганических и органических кислот и основные соли. Фармацевтически приемлемые соли включают, но не ограничиваются ими, соли металлов, такие как натриевая соль, калиевая соль, цезиевая соль и тому подобное; соли щелочноземельных металлов, такие как соль кальция, соль магния и тому подобное; соли органических аминов, такие как соль триэтиламина, соль пиридина, соль пиколина, соль этаноламина, соль триэтаноламина, соль дициклогексиламина, соль N,N-дибензилэтилендиамина и т.п.; соли неорганических кислот, такие как гидрохлорид, гидробромид, фосфат, сульфат и т.п.; соли органических кислот, такие как цитрат, лактат, тартрат, малеат, фумарат, манделат, ацетат, дихлорацетат, трифторацетат, оксалат, формиат и т.п.; сульфонаты, такие как метансульфонат, бензолсульфонат, п-толуолсульфонат и т.п.; и соли аминокислот, такие как аргинат, аспаргинат, глутамат и т.п. Термин «фармацевтически приемлемая соль», используемый в данном документе, относится к любой соли, например, полученной реакцией с кислотой или основанием Соединения раскрытия, которая физиологически переносится целевым пациентом (например, млекопитающим, например, человеком).
[0366] Кислотно-аддитивные соли могут быть образованы путем смешивания раствора конкретного Соединения раскрытия с раствором фармацевтически приемлемой нетоксичной кислоты, такой как соляная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, уксусная кислота, лимонная кислота, винная кислота, угольная кислота, фосфорная кислота, щавелевая кислота, дихлоруксусная кислота и т.п. Основные соли могут быть образованы путем смешивания раствора соединения данного раскрытия с раствором фармацевтически приемлемого нетоксичного основания, такого как гидроксид натрия, гидроксид калия, гидроксид холина, карбонат натрия и т.п.
[0367] Данное раскрытие охватывает получение и использование сольватов Соединений раскрытия. Сольваты обычно существенно не изменяют физиологическую активность или токсичность соединений и, как таковые, могут действовать как фармакологические эквиваленты. Термин «сольват», используемый в данном документе, представляет собой комбинацию, физическую ассоциацию и/или сольватацию соединения данного раскрытия с молекулой растворителя, такой как, например, дисольват, моносольват или гемисольват, где соотношение молекулы растворителя к соединению данного раскрытия составляет около 2:1, около 1:1 или около 1:2 соответственно. Эта физическая ассоциация включает различную степень ионной и ковалентной связи, включая водородную связь. В некоторых случаях сольват можно выделить, например, когда одна или несколько молекул растворителя включены в кристаллическую решетку кристаллического твердого вещества. Таким образом, «сольват» включает как сольваты, находящиеся в фазе раствора, так и выделяемые сольваты. Соединения раскрытия могут присутствовать в сольватированных формах с фармацевтически приемлемым растворителем, таким как вода, метанол, этанол и т.п., и предполагается, что раскрытие включает как сольватированные, так и несольватированные формы Соединений раскрытия. Одним из типов сольватов является гидрат. «Гидрат» относится к конкретной подгруппе сольватов, в которых молекулой растворителя является вода. Сольваты обычно могут действовать как фармакологические эквиваленты. Приготовление сольватов известно в данной области. См., например, М. Caira et al, J. Pharm. Sci, 93 (3): 601-611 (2004), в котором описано получение сольватов флуконазола с этилацетатом и водой. Аналогичное получение сольватов, гемисольватов, гидратов и т.п. описано Е.С. van Tonder et al., AAPS Pharm. Sci. Tech, 5(1):Article 12 (2004), и A.L. Bmgham et al, Chem. Commun. 603-604 (2001). Типичный, неограничивающий процесс получения сольвата включает растворение Соединения раскрытия в желаемом растворителе (органическом, воде или их смеси) при температурах от более 20°С до около 25°С с последующим охлаждением раствора, со скоростью, достаточной для образования кристаллов, и выделение кристаллов известными методами, например фильтрацией. Аналитические методы, такие как инфракрасная спектроскопия, могут использоваться для подтверждения присутствия растворителя в кристалле сольвата.
[0368] Поскольку Соединения раскрытия являются ингибиторами белков USP1, данное раскрытие обеспечивает способ ингибирования белка USP1, включающий приведение в контакт белка USP1 или композиции, содержащей белок USP1, с одним или несколькими Соединениями раскрытия.
[0369] Поскольку Соединения раскрытия являются ингибиторами белков USP1, ряд заболеваний, состояний или нарушений, опосредованных белками USP1, можно лечить с помощью этих соединений. Данное раскрытие, таким образом, в целом направлено на способ лечения заболевания, состояния или расстройства, реагирующего на ингибирование белков USP1, у животного, страдающего этим расстройством или имеющего риск заболеть этим расстройством, причем способ включает введение животному эффективного количества одного или нескольких Соединений раскрытия.
[0370] Данное раскрытие дополнительно направлено на способ ингибирования белков USP1 у животного, нуждающегося в этом, причем способ включает введение животному терапевтически эффективного количества по меньшей мере одного Соединения раскрытия.
[0371] Используемый в данном документе термин «лечение» представляет собой подход к достижению благоприятных или желаемых клинических результатов. Используемый в данном документе термин «лечение» охватывает любое введение или применение терапевтического средства для лечения заболевания у млекопитающего, включая человека. Для целей данного раскрытия полезные или желаемые клинические результаты включают, но не ограничиваются ими, любой один или несколько из следующего: облегчение одного или нескольких симптомов, уменьшение степени заболевания, предотвращение или замедление распространения (например, метастазирования) заболевания, предотвращение или отсрочка рецидива заболевания, задержка или замедление прогрессирования заболевания, улучшение болезненного состояния, подавление заболевания или прогрессирования заболевания, подавление или замедление заболевания или его прогрессирования, остановка его развития и ремиссия (частичная или полная). «Лечение» также включает уменьшение патологических последствий пролиферативного заболевания. Способы, представленные в данном документе, предполагают любой один или несколько из этих аспектов лечения. В соответствии с вышеизложенным, термин «лечение» не требует полного устранения всех аспектов расстройства.
[0372] В контексте рака термин «лечение» включает, но не ограничивается этим, подавление роста раковых клеток, подавление репликации раковых клеток, уменьшение общей опухолевой нагрузки и задержку, остановку или замедление роста опухоли, прогрессирования или метастазирования.
[0373] Используемый в данном документе термин «задержка» означает отсрочку, затруднение, замедление, торможение, стабилизацию, подавление и/или отсрочку развития или прогрессирования заболевания (такого как рак). Эта задержка может быть различной продолжительности в зависимости от истории болезни и/или человека, которого лечат.
[0374] «Терапевтически эффективное количество» вещества может варьироваться в зависимости от таких факторов, как болезненное состояние, возраст, пол и масса индивидуума, а также способность вещества вызывать желаемый ответ у индивидуума. Терапевтически эффективным количеством также является такое количество, при котором любые токсические или вредные эффекты вещества перевешиваются терапевтически полезными эффектами. Терапевтически эффективное количество может быть доставлено за одно или несколько введений. Терапевтически эффективное количество относится к количеству, эффективному в дозировках и в течение периодов времени, необходимых для достижения желаемого терапевтического эффекта.
[0375] Термины «вводить», «введение», «применение» и т.п. относятся к способам, которые можно использовать для обеспечения доставки терапевтического агента к желаемому месту биологического действия. Методы введения, которые можно использовать с описанными здесь агентами и способами, можно найти, например, в Goodman and Gilman, The Pharmacological Basis of Therapeutics, текущее издание.; Пергамон; и Remington's, Pharmaceutical Sciences (текущее издание). Mack Publishing Co., Истон, Пенсильвания.
[0376] Термины «фармацевтический состав» и «фармацевтическая композиция» относятся к препарату, который находится в такой форме, чтобы обеспечить эффективность биологической активности активного ингредиента (ов), и который не содержит дополнительных компонентов, которые являются неприемлемо токсичными для субъекта, которому будет вводиться состав. Такие составы могут быть стерильными.
[0377] «Фармацевтически приемлемый носитель» относится к нетоксичному твердому, полутвердому или жидкому наполнителю, разбавителю, инкапсулирующему материалу, вспомогательному составу или носителю, обычному в данной области для использования с терапевтическим агентом, которые вместе составляют «фармацевтическую композицию» для введения субъекту. Фармацевтически приемлемый носитель нетоксичен для реципиентов в используемых дозировках и концентрациях и совместим с другими ингредиентами состава. Фармацевтически приемлемый носитель подходит для используемого состава.
[0378] «Стерильный» состав является асептическим или практически не содержит живых микроорганизмов и их спор.
[0379] Термин «контейнер» означает любую емкость и крышку, поэтому подходящие для хранения, транспортировки, выдачи и/или обращения с фармацевтическим продуктом.
[0380] Термин «вкладыш» или «вкладыш в упаковке» означает информацию, прилагаемую к фармацевтическому продукту, которая содержит описание того, как вводить продукт, а также данные о безопасности и эффективности, необходимые для того, чтобы врач, фармацевт и пациент могли принять обоснованное решение относительно использование продукта. Вкладыш в упаковку обычно рассматривается как «этикетка» для фармацевтического продукта.
[0381] Термин «заболевание», или «состояние», или «расстройство» в контексте данного описания относится к состоянию, при котором лечение необходимо и/или желательно, и обозначает нарушения и/или аномалии, которые, как правило, рассматриваются как патологические состояния или функции, и которые могут проявляться в виде определенных признаков, симптомов и/или дисфункций. Как показано ниже, Соединения согласно раскрытию ингибируют белки USP1 и могут быть использованы для лечения заболеваний и состояний, таких как пролиферативные заболевания, при которых ингибирование белков USP1 дает преимущество.
[0382] Термины «полипептид» и «белок» используются взаимозаменяемо для обозначения полимера из аминокислотных остатков и не ограничиваются минимальной длиной. Такие полимеры аминокислотных остатков могут содержать природные или неприродные аминокислотные остатки и включают, но не ограничиваются ими, пептиды, олигопептиды, димеры, тримеры и мультимеры аминокислотных остатков. Это определение охватывает как полноразмерные белки, так и их фрагменты. Термины также включают постэкспрессионные модификации полипептида, например гликозилирование, сиалирование, ацетилирование, фосфорилирование и тому подобное. Кроме того, для целей данного раскрытия «полипептид» относится к белку, который включает модификации, такие как делении, добавления и замены (обычно консервативные по природе), в нативной последовательности, пока белок сохраняет желаемую активность.. Эти модификации могут быть преднамеренными, например, посредством сайт-направленного мутагенеза, или могут быть случайными, например, посредством мутаций хозяев, которые продуцируют белки, или ошибок из-за амплификации ПЦР.
[0383] Используемые в данном документе термины «USP1» и «убиквитин-специфическая процессирующая протеаза 1» относятся к любому нативному полипептиду или полинуклеотиду, кодирующему USP1. Термин «USP1» охватывает «полноразмерный» необработанный полипептид USP1, а также любые формы USP1, которые возникают в результате процессинга внутри клетки (например, удаления сигнального пептида). Термин также включает встречающиеся в природе варианты USP1, например, те, которые кодируются вариантами сплайсинга и аллельными вариантами. Описанные в данном документе полипептиды USP1 могут быть выделены из множества источников, таких как типы тканей человека или из другого источника, или получены рекомбинантными или синтетическими методами. Последовательности USP1 человека известны и включают, например, последовательности, общедоступные как UniProt No. 094782 (включая изоформы). Используемый в данном документе термин «белок USP1 человека» относится к белку USP1, содержащему аминокислотную последовательность, указанную в SEQ ID NO: 1 в предварительной заявке на патент США №62/857 986, поданной 6 июня 2019 г.
[0384] USP1 представляет собой деубиквитинирующий фермент, который действует как часть комплекса с UAF1. «Деубиквитиназная активность» USP1 включает его способность деубиквитинировать как часть комплекса USP1-UAF1.
[0385] Термин «специфически связывается» с белком или доменом белка - это термин, который хорошо понятен в данной области, и способы определения такого специфического связывания также хорошо известны в данной области. Говорят, что молекула проявляет «специфическое связывание» или «предпочтительное связывание», если она реагирует или связывается чаще, быстрее, с большей продолжительностью и/или с большим сродством с конкретным белком или доменом белка, чем с альтернативными белками или доменами. Следует понимать, что молекула, которая специфически или предпочтительно связывается с первым белком или доменом, может или не может специфически или предпочтительно связываться со вторым белком или доменом. По существу, «специфическое связывание» или «предпочтительное связывание» не обязательно требует (хотя оно может включать) исключительного связывания. Обычно, но не обязательно, ссылка на привязку означает предпочтительную привязку. Например, ингибитор USP1, который специфически связывается с USP1, UAF1 и/или комплексом USP1-UAF 1, может не связываться с другими деубиквитиназами, другими белками USP или другими комплексами UAF1 (например, USP46-UAF1) или может связываться с другими деубиквитиназами, другими белками USP или другими комплексами UAF1 (например, USP46-UAF1) с пониженным сродством по сравнению со связыванием с USP1.
[0386] Термины «уменьшение» или «уменьшать», или «ингибирование», или «ингибировать» относятся к уменьшению или прекращению какой-либо фенотипической характеристики или к уменьшению или прекращению встречаемости, степени или вероятности этой характеристики. «Уменьшать» или «ингибировать» означает понижать, уменьшать или останавливать активность, функцию и/или количество по сравнению с эталоном. В некоторых вариантах реализации под «снижением» или «ингибированием» подразумевается способность вызывать общее снижение на 20% или более. В некоторых вариантах реализации под «снижением» или «ингибированием» подразумевается способность вызывать общее снижение на 50% или более. В некоторых вариантах реализации под «снижением» или «ингибированием» подразумевается способность вызывать общее снижение на 75%, 85%, 90%, 95% или более. В некоторых вариантах реализации указанное выше количество подавляется или уменьшается в течение периода времени по сравнению с контролем за тот же период времени.
[0387] В некоторых вариантах реализации ингибирование белков USP1 представляет собой ингибирование одной или нескольких активностей или функций белков USP1. Следует понимать, что активность или функция одного или нескольких белков USP1 может ингибироваться in vitro или in vivo. Неограничивающие примеры активностей и функций USP1 включают активность деубиквитиназы и образование комплекса с UAF1 и описаны в данном документе. Примеры уровней ингибирования активности одного или нескольких белков USP1 включают по меньшей мере 10% ингибирование, по меньшей мере 20% ингибирование, по меньшей мере 30% ингибирование, по меньшей мере 40% ингибирование, по меньшей мере 50% ингибирование, по меньшей мере 60% ингибирование, по меньшей мере 70% ингибирование, по меньшей мере 80% ингибирование, по меньшей мере 90% ингибирование и ингибирование до 100%.
[0388] Термины «индивидуум» или «субъект» используются в данном документе взаимозаменяемо для обозначения животного; например, млекопитающего, такого как человек. В некоторых случаях предоставляются способы лечения млекопитающих, включая, но не ограничиваясь ими, людей, грызунов, обезьян, кошачьих, псовых, лошадей, крупного рогатого скота, свиней, овечьих, козьих, лабораторных животных млекопитающих, сельскохозяйственных животных млекопитающих, спортивных животных млекопитающих и млекопитающих домашних животных. В некоторых примерах «индивидуум» или «субъект» относится к индивиду или субъекту, нуждающимся в лечении заболевания или расстройства. В некоторых случаях субъект, получающий лечение, может быть пациентом, что указывает на тот факт, что субъект был идентифицирован как страдающий нарушением, имеющим отношение к лечению, или подверженным особому риску подцепить расстройство.
[0389] Используемые в данном документе термины «рак» и «опухоль» относятся или описывают физиологическое состояние у млекопитающих, в котором популяция клеток характеризуется нерегулируемым ростом клеток. Эти термины охватывают солидный и гематологический/лимфатический рак. Примеры рака включают, но не ограничиваются ими, раковые образования с недостаточностью пути репарации повреждений ДНК. Дополнительные примеры рака включают, но не ограничиваются ими, рак яичников, рак груди (включая тройной негативный рак груди), немелкоклеточный рак легкого (NSCLC) и остеосаркому. Рак может быть BRCA1 или BRCA2 дикого типа. Рак также может быть мутантным по BRCA1 или BRCA2. Рак, кроме того, может представлять собой рак, устойчивый к ингибитору PARP или рефракторный рак, или рак, устойчивый к ингибитору PARP или рефракторный к BRCA1 или BRCA2-мутантный.
[0390] Используемый в данном документе термин «мутация с потерей функции» относится к мутации, которая приводит к отсутствию гена, снижению экспрессии гена или продукции генного продукта (например, белка), имеющего пониженную активность или отсутствие активности. Мутации с потерей функции включают, например, миссенс-мутации, вставки нуклеотидов, делеции нуклеотидов и делеции генов. Мутации с потерей функции также включают доминантно-отрицательные мутации. Таким образом, раковые клетки с мутацией потери функции в гене, кодирующем р53, включают раковые клетки, которые содержат миссенс-мутации в гене, кодирующем р53, а также раковые клетки, в которых отсутствует ген, кодирующий р53.
Ингибиторы USP1
[0391] В различных вариантах реализации Соединения раскрытия представляют собой ингибиторы USP1, которые снижают уровень белка USP1 и/или ингибируют или снижают по меньшей мере одну биологическую активность белка USP1
[0392] В некоторых вариантах реализации Соединения раскрытия специфически связываются с белком USP1. В некоторых вариантах реализации Соединения раскрытия специфически связываются с белком USP1 в комплексе USP1-UAF1. В некоторых вариантах реализации Соединения раскрытия специфически связываются с мРНК USP1. В некоторых вариантах реализации Соединения раскрытия специфически связываются с белком USP1 (отдельно или в комплексе USP1-UAF1) или мРНК USP1. В некоторых вариантах реализации Соединения раскрытия специфически связываются с UAF1 (отдельно или в комплексе USP1-UAF 1) и ингибируют или снижают образование или активность комплекса USP1-UAF1.
[0393] В некоторых вариантах реализации Соединения раскрытия уменьшают образование комплекса USP1-UAF1. В некоторых вариантах реализации Соединения Раскрытия снижают активность комплекса USP1-UAF1. В некоторых вариантах реализации Соединения раскрытия снижают активность деубиквитиназы USP1. В некоторых вариантах реализации Соединения раскрытия увеличивают моноубиквигинированный PCNA. В некоторых вариантах реализации Соединения раскрытия увеличивают моноубиквигинированный FANCD2. В некоторых вариантах реализации Соединения раскрытия увеличивают моноубиквитинированный FANCI.
[0394] В некоторых вариантах реализации Соединения раскрытия не связываются с другими деубиквитиназами, другими белками USP или другими комплексами UAF1 (например, USP46-UAF1) или связывают деубиквитиназы, другие белки USP или другие комплексы UAF1 (например, USP46-UAF1). с по меньшей мере 5-кратным, по меньшей мере 10-кратным, по меньшей мере 20-кратным или по меньшей мере 100-кратным снижением аффинности по сравнению со сродством к USP1 (т.е. kd ингибитора USP1 для других деубиквитиназ, других белков USP, или других комплексов UAF1 (например, USP46-UAF1) по меньшей мере в 5 раз, по меньшей мере в 10, по меньшей мере в 20 раз или по меньшей мере в 100 раз выше, чем KD для USP1).
[0395] В некоторых вариантах реализации Соединения раскрытия ингибируют активность деубиквитиназы USP1 с IC50 менее около 50 нМ, от около 50 нМ до около 200 нМ, от около 200 нМ до около 2 пМ или более 2 пМ, например, как измеряется с использованием анализа, описанного в публикации патентной заявки США №2017/0145012, или IC50 от 50 до 1000 нМ, например, при измерении с использованием анализа, описанного в Liang et al., Nat Chem Biol 10: 289-304(2014). В некоторых вариантах реализации Соединения раскрытия ингибируют активность деубиквитиназы USP1 с IC50, измеренным с использованием анализа, описанного в Chen, et al, Chem Biol., 18 (II): 1390-1400 (2011). В некоторых вариантах реализации Соединения раскрытия не ингибируют активность других деубиквитиназ, других белков USP или других комплексов UAF1 (например, USP46-UAF1) или ингибируют активность других деубиквитиназ, других белков USP или других комплексов UAF1 (например, USP46-UAF1) с по меньшей мере 5-кратным, по меньшей мере 10-кратным, по меньшей мере 20-кратным или по меньшей мере 100-кратным более высоким IC50 по сравнению с IC50 для ингибирования активности деубиквитиназы USP1.
[0396] В некоторых вариантах реализации Соединения раскрытия связываются с белком USP1 со сродством в диапазоне от 1 пМ до 100 мкМ, или от 1 пМ до 1 мкМ, или от 1 пМ до 500 нМ, или от 1 пМ до 100 нМ. В некоторых вариантах реализации Соединения раскрытия связываются с белком USP1 со сродством от около 1 пМ до около 100 мкМ, от около 1 нМ до около 100 мкМ, от около 1 мкМ до около 100 мкМ, от около 1 мкМ до около 50 мкМ, от около 1 мкМ до около 40 мкМ, от около 1 мкМ до около 30 мкМ, от около 1 мкМ до около 20 мкМ или от около 1 мкМ до около 10 мкМ, около 1 мкМ, около 5 мкМ, около 10 мкМ, около 15 мкМ, около 20 мкМ, около 25 мкМ, около 30 мкМ, около 35 мкМ, около 40 мкМ, около 45 мкМ, около 50 мкМ, около 60 мкМ, около 70 мкМ, около 80 мкМ, около 90 мкМ или около 100 мкМ. В некоторых вариантах реализации Соединения раскрытия связываются с белком USP1 со сродством от около 100 нМ до около 1 мкМ, от около 100 нМ до около 900 нМ, от около 100 нМ до около 800 нМ, от около 100 нМ до около 700 нМ, от около 100 нМ до около 600 нМ, от около 100 нМ до около 500 нМ, от около 100 нМ до около 400 нМ, от около 100 нМ до около 300 нМ, от около 100 нМ до около 200 нМ, от около 200 нМ до около 1 мкМ, около 300 нМ до около 1 мкМ, от около 400 нМ до около 1 мкМ, от около 500 нМ до около 1 мкМ, от около 600 нМ до около 1 мкМ, от около 700 нМ до около 1 мкМ, от около 800 нМ до около 1 мкМ, от около 900 нМ до около 1 мкМ, около 100 нМ, около 200 нМ, около 300 нМ, около 400 нМ, около 500 нМ, около 600 нМ, около 700 нМ, около 800 нМ или около 900 нМ. В некоторых вариантах реализации Соединения раскрытия связываются с белком USP1 со сродством от около 1 нМ до около 100 нМ, от 1 нМ до около 90 нМ, от 1 нМ до около 80 нМ, от 1 нМ до около 70 нМ, от 1 нМ до около 60 нМ, от 1 нМ до около 50 нМ, от 1 нМ до около 40 нМ, от 1 нМ до около 30 нМ, от 1 нМ до около 20 нМ, от 1 нМ до около 10 нМ, от около 10 нМ до около 100 нМ, от около 20 нМ до около 100 нМ, от около 30 нМ до около 100 нМ, от около 40 нМ до около 100 нМ, от около 50 нМ до около 100 нМ, от около 60 нМ до около 100 нМ, от около 70 нМ до около 100 нМ, от около 80 нМ до около 100 нМ, от около 90 нМ до около 100 нМ, около 1 нМ, около 2 нМ, около 3 нМ, около 4 нМ, около 5 нМ, около 6 нМ, около 7 нМ, около 8 нМ, около 9 нМ, около 10 нМ, около 20 нМ, около 30 нМ, около 40 нМ, около 50 нМ, около 60 нМ, около 70 нМ, около 80 нМ, около 90 нМ или около 100 нМ. В некоторых вариантах реализации Соединения раскрытия связываются с белком USP1 со сродством менее 1 мкМ, менее 500 нМ, менее 100 нМ, менее 10 нМ или менее 1 нМ. В некоторых вариантах реализации Соединения раскрытия связываются с белком USP1 со сродством менее 1 нМ.
[0397] В некоторых вариантах реализации Соединения раскрытия ингибируют активность USP1 с IC50 от 1 пМ до 100 мкМ, или от 1 пМ до 1 мкМ, или от 1 пМ до 500 нМ, или от 1 пМ до 100 нМ. В некоторых вариантах реализации Соединения раскрытия ингибируют активность USP1 с IC50 от около 1 пМ до около 100 мкМ, от около 1 нМ до около 100 мкМ, от около 1 мкМ до около 100 мкМ, от около 1 мкМ до около 50 мкМ, от около 1 мкМ до около 40 мкМ, от около 1 мкМ до около 30 мкМ, от около 1 мкМ до около 20 мкМ или от около 1 мкМ до около 10 мкМ, около 1 мкМ, около 5 мкМ, около 10 мкМ, около 15 мкМ, около 20 мкМ, около 25 мкМ, около 30 мкМ, около 35 мкМ, около 40 мкМ, около 45 мкМ, около 50 мкМ, около 60 мкМ, около 70 мкМ, около 80 мкМ, около 90 мкМ или около 100 мкМ. В некоторых вариантах реализации Соединения раскрытия ингибируют активность USP1 с IC50 от около 100 нМ до около 1 мкМ, от около 100 нМ до около 900 нМ, от около 100 нМ до около 800 нМ, от около 100 нМ до около 700 нМ, около 100 нМ. до около 600 нМ, от около 100 нМ до около 500 нМ, от около 100 нМ до около 400 нМ, от около 100 нМ до около 300 нМ, от около 100 нМ до около 200 нМ, от около 200 нМ до около 1 мкМ, от около 300 нМ до около 1 мкМ, от около 400 нМ до около 1 мкМ, от около 500 нМ до около 1 мкМ, от около 600 нМ до около 1 мкМ, от около 700 нМ до около 1 мкМ, от около 800 нМ до около 1 мкМ, от около 900 нМ до около 1 мкМ, около 100 нМ, около 200 нМ, около 300 нМ, около 400 нМ, около 500 нМ, около 600 нМ, около 700 нМ, около 800 нМ или около 900 нМ. В некоторых вариантах реализации Соединения раскрытия ингибируют активность USP1 с IC50 от около 1 нМ до около 100 нМ, от 1 нМ до около 90 нМ, от 1 нМ до около 80 нМ, от 1 нМ до около 70 нМ, от 1 нМ до около 60 нМ, от 1 нМ до около 50 нМ, от 1 нМ до около 40 нМ, от 1 нМ до около 30 нМ, от 1 нМ до около 20 нМ, от 1 нМ до около 10 нМ, от около 10 нМ до около 100 нМ, от около 20 нМ до около 100 нМ, от около 30 нМ до около 100 нМ, от около 40 нМ до около 100 нМ, от около 50 нМ до около 100 нМ, от около 60 нМ до около 100 нМ, от около 70 нМ до около 100 нМ, от около 80 нМ до около 100 нМ, от около 90 нМ до около 100 нМ, около 1 нМ, около 2 нМ, около 3 нМ, около 4 нМ, около 5 нМ, около 6 нМ, около 7 нМ, около 8 нМ, около 9 нМ, около 10 нМ, около 20 нМ, около 30 нМ, около 40 нМ, около 50 нМ, около 60 нМ, около 70 нМ, около 80 нМ, около 90 нМ или около 100 нМ. В некоторых вариантах реализации Соединения раскрытия ингибируют активность USP1 с IC50 менее 1 мкМ, менее 500 нМ, менее 100 нМ, менее 10 нМ или менее 1 нМ. В некоторых вариантах реализации Соединения раскрытия ингибируют активность USP1 с IC50 менее 1 нМ.
Иллюстративные анализы
[0398] Любой подходящий анализ в данной области может быть использован для определения активности, обнаружения результата или эффекта, определения эффективности и т.д. Описаны некоторые неограничивающие примерные анализы, которые можно использовать в способах, представленных в данном документе.
[0399] В некоторых случаях способ определения того, ингибирует ли Соединение раскрытия активность деубиквитиназы USP1, измеряет изменение массы при расщеплении диубиквитином связывания деубиквитиназы. Например, альдегид убиквитина и винилсульфон убиквитина образуют ковалентные необратимые связи с деубиквитиназами, что приводит к наблюдаемым изменениям массы деубиквитиназ. Точно так же расщепление диубиквигинов приводит к наблюдаемому изменению массы.
[0400] В некоторых случаях способ определения того, ингибирует ли Соединение раскрытия активность деубиквитиназы USP1, включает увеличение люминесценции или флуоресценции при расщеплении, например, это можно контролировать с помощью планшет-ридера. В таких анализах можно использовать убиквитин, связанный с флурофором через линкерную связь, такой как убиквитин-7-амино-4-метилкумарин (Ub-AMC) или убиквитин-родамин 110. В таких анализах также можно использовать диубиквитин, содержащий изопептидную связь. Типичные диубиквитины могут включать флуофор на одном убиквитине и гаситель на другом убиквитине, так что флуоресценция увеличивается с последующим расщеплением диубиквитина. В таких анализах можно также использовать связанные с ферментом системы, в которых убиквитин связан с ферментом, который активен в производстве флуоресцентного ферментного продукта только при высвобождении из убиквитина.
[0401] Иллюстративный анализ деубиквитинирования активности USP1/UAF1 и тестирование ингибиторов. Активность деубиквитиназы можно измерить с использованием убиквитин-родамина 110 в качестве субстрата. Разрыв амидной связи между родамином и с-концевым глицином убиквитина приводит к усилению сигнала флуоресценции. Анализ можно проводить в 20 мкл общего объема буфера для анализа (50 мМ трис-HCl, рН 7,8, 0,5 мМ EDTA 0,01% бычий сывороточный альбумин, 1 мМ DTT, 0,01% Tween-20) и 0,05 нМ фермента USP1/UAF1. Реакцию можно инициировать добавлением 150 нМ субстрата убиквитин-родамин (Boston Biochem).
[0402] Соединения раскрытия могут быть растворены в ДМСО и протестированы в формате доза-ответ, начиная с 10 мкМ.
[0403] Соединения раскрытия можно добавить к смеси фермент/аналитический буфер и инкубировать 10 мин. Смесь субстрата может быть добавлена, и реакционная смесь может быть считана в кинетическом режиме в течение 30 минут при Ех480/Em540, и могут быть построены кривые отклика IC50. См., Например, Chen, et al., Chem Biol., 18 (II): 1390-1400 (2011).
Способы использования
[0404] В некоторых вариантах реализации Соединения раскрытия можно использовать для ингибирования активности белка USP1. Например, в некоторых вариантах реализации способ ингибирования белка USP1 включает контактирование белка USP1 с Соединением раскрытия.
Контактирование может происходить in vitro или in vivo.
[0405] В некоторых вариантах реализации Соединения раскрытия можно использовать для лечения «нарушения, опосредованного белком USP1». Нарушение, опосредованное белком USP1, представляет собой любое патологическое состояние, в котором, как известно, играет роль белок USP1. В некоторых вариантах реализации нарушение, опосредованное белком USP1, представляет собой пролиферативное заболевание, такое как рак.
[0406] В данном документе представлены различные способы лечения заболеваний и нарушений с помощью Соединений раскрытия. Типичные заболевания и нарушения, которые можно лечить с помощью Соединений раскрытия, включают, но не ограничиваются ими, рак.
[0407] В некоторых вариантах реализации представлены способы лечения рака с помощью Соединений раскрытия. Такие способы включают введение субъекту, страдающему раком, терапевтически эффективного количества Соединения раскрытия.
[0408] В некоторых вариантах реализации рак, подлежащий лечению Соединением раскрытия, выбран из гематологического рака, лимфатического рака и рака, лишенного пути репарации повреждений ДНК. В некоторых вариантах реализации рак, подлежащий лечению Соединением раскрытия, представляет собой рак, который включает раковые клетки с мутацией в гене, кодирующем р53. В некоторых вариантах реализации рак, подлежащий лечению Соединением раскрытия, представляет собой рак, который включает раковые клетки с мутацией потери функции в гене, кодирующем р53. В некоторых вариантах реализации рак, подлежащий лечению с помощью Соединения раскрытия, представляет собой рак, который включает раковые клетки с мутацией в гене, кодирующем BRCA1. В некоторых вариантах реализации рак, подлежащий лечению с помощью Соединения раскрытия, представляет собой рак, который включает раковые клетки с мутацией в гене, кодирующем BRCA2. В некоторых вариантах реализации рак, подлежащий лечению с помощью Соединения раскрытия, представляет собой рак, который включает раковые клетки с мутацией потери функции в гене, кодирующем ATM.
[0409] В некоторых вариантах реализации рак, подлежащий лечению с помощью соединения раскрытия, выбран из немелкоклеточного рака легкого (NSCLC), остеосаркомы, рака яичников и рака груди. В некоторых вариантах реализации рак представляет собой рак яичников или рак груди. В некоторых вариантах реализации рак представляет собой рак яичников. В некоторых вариантах реализации рак представляет собой рак груди. В некоторых вариантах реализации рак представляет собой рак груди с тройным отрицательным результатом.
[0410] В некоторых вариантах реализации рак, подлежащий лечению соединением раскрытия, выбран из группы, состоящей из рака кости, включая остеосаркому и хондросаркому; рака мозга, включая глиому, глиобластому, астроцитому, медуллобластому и менингиому; рака мягких тканей, включая рабдоид и саркому; рака почки; рака мочевого пузыря; рака кожи, включая меланому; и рака легкого, включая немелкоклеточный рак легкого; рака толстой кишки, рака матки; рака нервной системы; рака головы и шеи; рака поджелудочной железы; и рака шейки матки.
[0411] В данном документе представлены различные способы лечения рака с помощью Соединения раскрытия. В некоторых вариантах реализации терапевтически эффективное количество Соединение раскрытия вводится субъекту с раком, где рак включает раковые клетки с повышенными уровнями RAD18. В некоторых вариантах реализации повышенные уровни RAD18 представляют собой повышенные уровни белка RAD18. В некоторых вариантах реализации повышенные уровни RAD18 представляют собой повышенные уровни мРНК RAD18. В некоторых вариантах реализации повышенные уровни RAD18 (например, белка RAD18 и/или мРНК RAD18) были обнаружены (например, в образце рака, полученном от субъекта) до введения. То есть в некоторых вариантах реализации рак субъекта был протестирован на белок RAD18 или мРНК до начала лечения ингибитором USP1.
[0412] В некоторых вариантах реализации такие способы включают (а) идентификацию рака у субъекта как рака, чувствительного к ингибитору USP1, и затем (b) введение субъекту терапевтически эффективного количества Соединения раскрытия.
[0413] В некоторых вариантах реализации такие способы включают (а) определение уровней RAD18 (например, белка RAD18 и/или мРНК RAD18) в раковых клетках (например, в образце рака, полученном от субъекта), а затем (b) введение терапевтически эффективного количества соединения раскрытия субъекту, страдающему раком, содержащим клетки с повышенными уровнями RAD18.
[0414] В некоторых вариантах реализации такие способы включают введение субъекту с тройным отрицательным раком молочной железы терапевтически эффективного количества Соединения раскрытия.
[0415] В некоторых вариантах реализации Соединение раскрытия используется для лечения рака, при этом рак представляет собой рак, дефицитный по гомологичной рекомбинации. В некоторых вариантах реализации Соединение раскрытия используется для лечения рака, при этом рак включает раковые клетки с мутацией в гене, кодирующем р53. В некоторых вариантах реализации Соединение раскрытия используется для лечения рака, при этом рак включает раковые клетки с мутацией потери функции в гене, кодирующем р53. В некоторых вариантах реализации Соединение раскрытия используется для лечения рака, который не имеет дефекта в пути гомологичной рекомбинации.
[0416] В некоторых вариантах реализации Соединение раскрытия используется для лечения рака, при этом рак представляет собой мутантный рак BRCA1. В некоторых вариантах реализации Соединение раскрытия используется для лечения рака, при этом рак представляет собой мутантный рак BRCA2. В некоторых вариантах реализации Соединение раскрытия используется для лечения рака, при этом рак представляет собой мутантный рак BRCA1 и мутантный рак BRCA2. В некоторых вариантах реализации рак не является BRCA1 мутантным раком или BRCA2 мутантным раком. В некоторых вариантах реализации рак представляет собой рак с дефицитом BRCA1. В некоторых вариантах реализации рак представляет собой рак с дефицитом BRCA2. В некоторых вариантах реализации рак представляет собой рак с дефицитом BRCA1 и мутантный рак BRCA2.
[0417] В некоторых вариантах реализации Соединение Раскрытия используется для лечения рака, при этом рак представляет собой мутантный рак ATM. В некоторых вариантах реализации рак не является мутантным раком ATM. В некоторых вариантах реализации рак представляет собой рак с дефицитом ATM.
[0418] В некоторых вариантах реализации Соединение раскрытия используется для лечения рака, при этом рак представляет собой устойчивый к ингибитору PARP или рефрактерный рак В некоторых вариантах реализации Соединение раскрытия используется для лечения рака, при этом рак представляет собой устойчивый к ингибитору PARP или рефрактерный рак с дефицитом BRCA1.
[0419] В некоторых вариантах реализации рак представляет собой мутантный рак BRCA1 и/или BRCA2, при этом рак включает клетки с повышенными уровнями RAD18, например, в которых повышенные уровни RAD18 по меньшей мере такие же высокие, как уровни белка RAD18 и/или мРНК в клетках ES2 или в которых повышенные уровни RAD18 выше, чем уровни белка RAD18 и/или мРНК в клетках НЕРЗВ217. В некоторых вариантах реализации тройной отрицательный рак молочной железы представляет собой мутантный рак BRCA1 и/или BRCA2.
[0420] В некоторых случаях рак представляет собой солидный рак. В некоторых случаях рак представляет собой гематологический/лимфатический рак. В некоторых случаях рак представляет собой рак с недостаточностью пути восстановления повреждений ДНК. В некоторых случаях рак представляет собой рак с дефицитом гомологичной рекомбинации. В некоторых случаях рак включает раковые клетки с мутацией в гене, кодирующем р53. В некоторых случаях рак включает раковые клетки с мутацией потери функции в гене, кодирующем р53. В некоторых случаях рак выбирают из группы, состоящей из немелкоклеточного рака легкого (NSCLC), остеосаркомы, рака яичников и рака груди (включая тройной отрицательный рак груди). В некоторых случаях рак представляет собой рак яичников или рак груди (включая рак груди с тройным отрицательным результатом). В некоторых случаях рак представляет собой рак яичников. В некоторых случаях рак представляет собой рак груди (включая рак груди с тройным отрицательным результатом).
[0421] В некоторых вариантах реализации Соединение раскрытия используется в комбинации с одним или несколькими дополнительными терапевтическими агентами для лечения рака. Сообщалось, что статус р53 определяет сенсибилизацию ингибитора PARP (Sa et al. Genome Biology, (2019) 20:253) и что статус BRCA1/2 позволяет прогнозировать эффективность ингибиторов PARP в клинике (Audeh et al. Lancet (2010) 376 (9737), 245-51).. Как показано ниже, мутантный рак р53 и мутантный рак BRCA обладают повышенной чувствительностью к ингибиторам USP1. Соответственно, в некоторых вариантах реализации Соединение раскрытия используется в комбинации с ингибитором PARP для лечения рака.
[0422] В некоторых вариантах реализации в данном документе представлены Соединения раскрытия для использования в качестве лекарственного средства или для использования при приготовлении лекарственного средства, например, для лечения рака. В некоторых вариантах реализации в данном документе представлены Соединения раскрытия для использования в способе лечения рака.
Фармацевтические композиции
[0423] Соединения раскрытия можно вводить млекопитающему в форме необработанного химического вещества без каких-либо других компонентов, или Соединения раскрытия также могут вводиться млекопитающему как часть фармацевтической композиции, содержащей соединение в сочетании с подходящим фармацевтически приемлемым носителем (см., например, Gennaro, Remington: The Science and Practice of Pharmacy with Facts and Comparisons: Drugfacts Plus, 20th ed. (2003); Ansel et al.. Pharmaceutical Dosage Forms and Drug Delivery Systems, 7th ed., Lippencott Williams and Wilkins (2004); Kibbe et al., Handbook of Pharmaceutical Excipients, 3rd ed., Pharmaceutical Press (2000)). Такой носитель может быть выбран из фармацевтически приемлемых эксципиентов и вспомогательных веществ. Термин «фармацевтически приемлемый носитель» или «фармацевтически приемлемая несущая среда» охватывает любой из стандартных фармацевтических носителей, растворителей, поверхностно-активных веществ или несущих сред. Стандартные фармацевтические носители и их составы описаны в Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton. PA, 19th ed. 1995.
[0424] Фармацевтическая композиция по данному раскрытию может быть приготовлена в виде жидких суспензий или растворов с использованием жидкости, такой как масло, вода, спирт и их комбинаций.
[0425] Фармацевтическая композиция по данному раскрытию может быть приготовлена в виде стерильной инъекции, которая может быть водной или масляной суспензией. Эти суспензии могут быть составлены согласно методикам, известным в данной области.
[0426] Фармацевтическая композиция по данному раскрытию может вводиться перорально в любой перорально приемлемой дозированной форме, включая капсулы, таблетки, водные суспензии или растворы.
[0427] Фармацевтическая композиция по данному раскрытию может вводиться в форме суппозиториев для ректального введения.
[0428] Фармацевтическую композицию по данному раскрытию можно также вводить местно, особенно когда цель лечения включает области или органы, легко доступные для местного применения, включая заболевания глаз, кожи или нижних отделов кишечного тракта. Местное применение для нижних отделов кишечного тракта может осуществляться в виде ректальных суппозиториев (см. выше) или подходящих составов для клизмы. Также можно использовать местно трансдермальные пластыри. Для местного применения фармацевтические композиции могут быть составлены в виде подходящей мази, лосьона или крема, содержащих активный компонент, суспендированный или растворенный в одном или более носителях.
[0429] Фармацевтическую композицию по данному раскрытию можно также вводить офтальмологически и составлять в виде микронизированных суспензий в изотоническом стерильном физиологическом растворе с отрегулированным рН или, предпочтительно, в виде растворов в изотоническом стерильном физиологическом растворе с отрегулированным рН, либо с консервантом, таким как хлорид бензилалкония, либо без него. В альтернативном варианте, для офтальмологического применения фармацевтические композиции могут быть приготовлены в виде мази, такой как вазелин.
[0430] Фармацевтическая композиция по данному раскрытию также может вводиться с помощью назального аэрозоля или ингаляции. Такие композиции готовят согласно методикам, хорошо известным в области фармацевтических препаратов, и могут быть приготовлены в виде растворов в физиологическом растворе с использованием бензилового спирта или других подходящих консервантов, промоторов абсорбции для повышения биодоступности, фгоруглеродов и/или других обычных солюбилизирующих или диспергирующих агентов.
[0431] Фармацевтические композиции, используемые для введения in vivo, могут быть стерильными. Это легко достигается путем фильтрации, например, через стерильные фильтрующие мембраны.
[0432] Фармацевтические композиции в рамках данного раскрытия включают все композиции, в которых Соединение раскрытия объединено с одним или несколькими фармацевтически приемлемыми носителями. В одном варианте реализации Соединение раскрытия присутствует в композиции в количестве, которое эффективно для достижения его предполагаемой терапевтической цели.
[0433] Фармацевтическая композиция по данному раскрытию может быть введена любому пациенту, который может испытывать положительные эффекты Соединения раскрытия. В первую очередь среди таких пациентов - млекопитающие, например люди и домашние животные, хотя данное раскрытие не предназначено для такого ограничения. В одном варианте реализации пациентом является человек. В другом варианте реализации фармацевтические композиции по данному раскрытию можно вводить пациенту, страдающему устойчивым к ингибитору PARP или рефракторным раком. В другом варианте реализации фармацевтические композиции по данному раскрытию можно вводить пациенту, имеющему рак, устойчивый к ингибиторам PARP или рефракторный с дефицитом BRCA1. В другом варианте реализации фармацевтические композиции по данному раскрытию можно вводить пациенту в комбинации с ингибитором PARP.
[0434] В другом варианте реализации данное раскрытие предоставляет наборы, которые содержат Соединение раскрытия (или композицию, содержащую Соединение раскрытия), упакованные таким образом, чтобы облегчить их использование на практике способов данного раскрытия. В одном варианте реализации набор включает Соединение раскрытия (или композицию, содержащую Соединение раскрытия), упакованное в контейнер, например герметичную бутылку или сосуд, с этикеткой, прикрепленной к контейнеру или включенной в набор, которая описывает использование соединения или композиции для практического применения способа согласно раскрытию. В одном варианте реализации соединение или композиция упакованы в виде стандартной лекарственной формы. Набор дополнительно может включать устройство, подходящее для введения композиции в соответствии с предполагаемым путем введения. В некоторых вариантах реализации данное раскрытие предоставляет набор, который включает Соединение раскрытия, или его фармацевтически приемлемую соль или сольват, и инструкции по введению соединения или его фармацевтически приемлемой соли или сольвата пациенту, страдающему раком.
[0435] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую Соединение раскрытия или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель.
[0436] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую соединение, имеющее формулу I, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель.
[0437] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую соединение, имеющее формулу II, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель.
[0438] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую соединение, имеющее формулу III, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель.
[0439] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую соединение, имеющее формулу IV, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель.
[0440] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую соединение, имеющее формулу V, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель.
[0441] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую соединение, имеющее формулу VI, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель.
[0442] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую соединение, имеющее формулу VIa или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель.
[0443] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую соединение, имеющее формулу VII, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель.
[0444] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую соединение, имеющее формулу VIII, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель.
[0445] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую соединение, имеющее формулу IX, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель.
[0446] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую соединение, имеющее формулу X, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель.
[0447] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую соединение, имеющее формулу XI, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель.
[0448] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую соединение, имеющее формулу XII, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель.
[0449] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую Соединение раскрытия или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, причем соединение связывается с белком, кодируемым геном USP1.
[0450] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую Соединение раскрытия или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, причем фармацевтическая композиция предназначена для использования при лечении рака.
[0451] В некоторых вариантах реализации данное раскрытие обеспечивает фармацевтическую композицию, содержащую Соединение раскрытия или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, причем фармацевтическая композиция предназначена для производства лекарственного средства для лечения рака.
ПРИМЕРЫ Общие синтетические методы
[0452] Соединения раскрытия получают с использованием способов, известных специалистам в данной области с учетом этого раскрытия, или иллюстративных способов, показанных на общих схемах ниже. В любой из общих схем подходящие защитные группы могут быть использованы в синтезе. (См. Wuts, P.G.M.; Greene, Т.W., "Greene's Protective Groups in Organic Synthesis", 4th Ed., J. Wiley & Sons, NY, 2007).
[0453] Если не указано иное, все реагенты использовали без дополнительной очистки. Спектры 1H-ЯМР записывали в ДМСО-d6 или CD3OD при комнатной температуре на приборе Bruker 300 МГц. Когда был обнаружен более чем один конформер, сообщается о химических сдвигах для наиболее распространенного из них. Химические сдвиги спектров 1H ЯМР регистрировали в частях на миллион (м.д.) по шкале δ от внутреннего стандарта остаточного растворителя. Картины расщепления имеют вид с, синглет; д, дублет; т, триплет; к, квартет; м, мультиплет; шир, широкий. Условия ЖХ-МС и препаративной ВЭЖХ описаны ниже:
ЖХМС Метод А
ЖХМС, Метод В
ЖХМС, Метод С
Колонка А для ВЭЖХ: Agilent SB-C18 4,6×150 мм, 3,5 мкм
Препаративная колонка A: Phenomenex Luna 5u 100А, 21,2×250 мм, 5 мкм
Препаративная колонка В: X-Select C18, 30×250 мм, 5 мкм
Препаративная колонка С: Nucleodur C18, 30×250 мм, 5 мкм
Препаративная колонка D: X-Select C18, 30×250 мм, 5 мкм
Препаративная колонка Е: Nucleodur C18, 30×250 мм, 5 мкм
Метод SFC: Green SepESDiol, 250×4.6 мм, 5 мкм
Хиральный метод A: DIACEL CHIRAL PAK-IC (250×4,6 мм, 5 мкм); Длина волны: 260 нм
Хиральный метод В: DIACEL CHIRAL PAK-IG (250×4,6 мм, 5 мкм); Длина волны: 280 нм
[0454] В тексте используются следующие сокращения:
РЕ = петролейный эфир
ЕА или EtOAc = этилацетат
DMSO = диметилсульфоксид
DMF = N,N-диметилформамид
DMA = N,N-диметилацетамид
МеОН = метанол
МТВЕ = метил-трет-бутиловый эфир
DCM: дихлорметан
TEA = триметиламин
DIPEA = диизопропилэтиламин
TFA = трифторуксусная кислота
MeCN или ACN = ацетонитрил
TLC = тонкослойная хроматография
(BPin)2 = бис(пинаколато)диборон
HFIP = 1,1,1,3,3,3-гексафторпропан-2-ол
DIBAL-H = гидрид диизобутилалюминия
MeI = йодметан
hex или n-hex = н-гексан
DCE = 1,2-дихлорэтан
TBSCl = трет-бутилдиметилсилилхлорид
Tf2O = ангидрид трифторметансульфоновой кислоты
n-BuLi = н-бутиллитий
DMAP = 4-диметиламинопиридин
KOAc = ацетат калия
NaOAc = ацетатнатрия
TFAA = трифторуксусный ангидрид
m-СРВА = метахлорпероксибензойная кислота
DME = 1,2-диметоксиэтан
ТРР = трифенилфосфин
DTAD = ди-трет-бутилазодикарбоксилат
DEAD = диэтилазодикарбоксилат
DIAD = диизопропилазодикарбоксилат
PS-ТРР = трифенилфосфиннаполистирольной основе
NBS = N-бромсукцинимид.
Получение общего промежуточного продукта I-1:
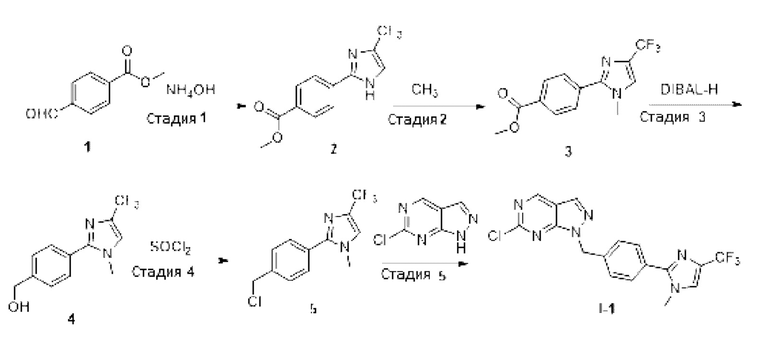
Стадия 1: Синтез метил 4-(4-(трифторметил)-1Н-имидазол-2-ил)бензоата:
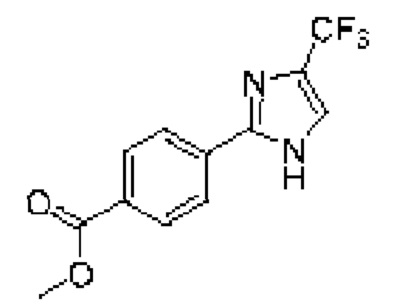
[0455] К смеси 3,3-дибром-1,1,1-трифторпропан-2-она (3,1 г, 11,49 ммоль) в воде (6 мл) добавляли NaOAc (951,8 мг, 11,60 ммоль). Смесь нагревали при 100°С в течение 1 часа, затем охлаждали до комнатной температуры. Затем к реакционной смеси при комнатной температуре добавляли раствор метил-4-формилбензоата (1,7 г, 10,34 ммоль) в МеОН (47 мл) и NH4OH (11 мл). Через 40 мин реакционную смесь нагревали до 100°С и перемешивали 2 часа. Смесь гасили водой (50 мл), а затем экстрагировали ЕА (100 мл × 2). Органический слой сушили над Na2SO4 (30 г), фильтровали и упаривали. Концентрированный остаток очищали хроматографией на силикагеле (РЕ:ЭА = от 20:1 до 5:1) с получением 1,9 г указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 271 (М+Н)+; 1H-ЯМР (300 МГц, CD3OD) δ 8,12 (д, J=8,7 Гц, 2Н), 8,01 (д, J=8,7 Гц, 2Н), 7,72 (с, 1Н), 3,93 (с, 3Н).
Стадия 2: Синтез Метил 4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензоата:
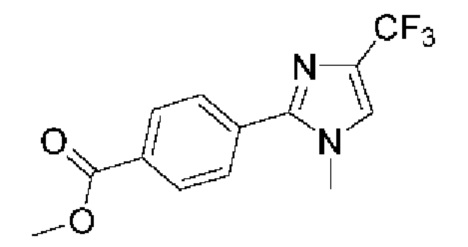
[0456] К раствору метил 4 (4-(трифторметил)-1H-имидазол-2-ил) бензоата (1,9 г, 7,03 ммоль) в ТГФ при 0°С (50 мл) добавляли NaH (60% дисперсия) (338 мг, 8,44 ммоль) порциями в течение 5 мин. После перемешивания смеси при 0°С в течение 30 минут к реакционной смеси по каплям в течение 2 минут добавляли MeI (1,2 г, 8,44 ммоль). Реакционную смесь нагревали до комнатной температуры в течение 3 часов, затем гасили ледяной водой (30 г). Полученный раствор экстрагировали ЕА (50 мл × 3), и объединенный органический слой сушили над Na2SO4 (30 г), фильтровали и упаривали. Полученный остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 20:1 до 5:1) с получением 1,4 г указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 285 (М+Н)+; 1H-ЯМР (300 МГц, CD3OD) δ 8,17 (д, J=8,4 Гц, 2Н), 7,80 (д, J=8,4 Гц, 2Н), 7,75 (с, 1Н), 3,94 (с, 3Н), 3,83 (с, 3Н).
Стадия 3: Синтез (4-(1-Метил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)метанола:
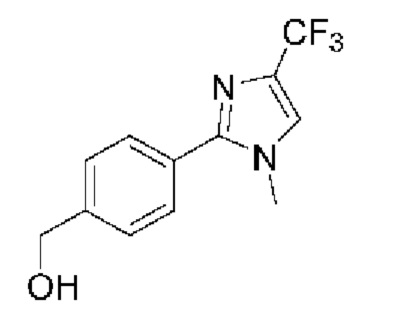
[0457] К раствору метил 4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензоата (1,4 г, 4,93 ммоль) в ТГФ (40 мл) при 0°С, был DIBAL-H (24 мл, 24,6 ммоль, 1 М в толуоле) через шприц. После добавления реакционную смесь перемешивали при 0°С в течение 20 минут, а затем нагревали до комнатной температуры в течение 30 минут. После завершения реакции, как показывает анализ ТСХ, смесь гасили ледяной водой (10 мл) и рН доводили до 5 с помощью 1 н. раствора HCl. Полученную смесь экстрагировали ЕА (50 мл × 2), и объединенный органический слой промывали солевым раствором (30 мл), сушили над Na2SO4 (30 г), фильтровали и упаривали в вакууме. Полученный остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 20:1 до 5:1) с получением 1,2 г указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 257 (М+Н)+; 1H-ЯМР (300 МГц CD3OD) δ 7,69 (с, 1Н), 7,63 (д, J=8,1 Гц, 2Н), 7,52 (д, J=8,1 Гц, 2Н), 4,69 (с, 2Н), 3,78 (с, 3Н).
Стадия 4: Синтез 2-(4-(хлорметил)фенил)-1-метил-4-(трифторметил)-1Н-имидазола:
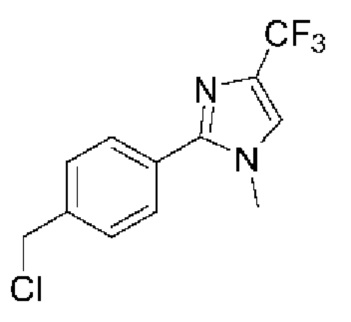
[0458] К смеси (4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил) фенил)метанола (1,2 г, 4,68 ммоль) в DCE (40 мл) добавляли SOCl2 (1,7 г, 14,1 ммоль) одной порцией. После добавления реакционную смесь перемешивали при 50°С в течение 20 минут, затем упаривали досуха с получением 1,28 г сырого указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 275, 277 (М+Н)+; 1H-ЯМР (300 МГц, CD3OD) δ 8,04 (с, 1Н), 7,74 (д, J=8,4 Гц, 2Н), 7,68 (д, J=8,4 Гц, 2Н), 4,75 (с, 2Н), 3,87 (с, 3Н).
Стадия 5: Синтез 6-Хлор-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (I-1):
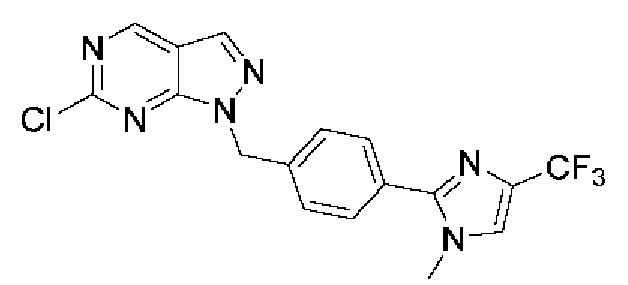
[0459] К раствору 2-(4-(хлорметил)фенил)-1-метил-4-(трифторметил)-1Н-имидазола (1 г, 3,64 ммоль) в ДМФА (25 мл) добавляли 6-хлор-1H-пиразоло[3,4-d]пиримидин (675 мг, 4,37 ммоль) и K2CO3 (1,26 г, 9,10 ммоль). Полученную смесь перемешивали при 35°С в течение 20 часов, затем гасили водой (50 мл) и экстрагировали ЕА (100 мл × 2). Объединенную органическую фазу промывали водой и солевым раствором, сушили над Na2SO4 (50 г), фильтровали и упаривали в вакууме. Полученный остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 20:1 до 10:1) с получением 580 мг указанного в заголовке соединения с небольшим количеством его региоизомера. ЖХ-МС (Способ А) (ИЭР+): m/z 393, 395 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 9,06 (с, 1Н), 8,18 (с, 1Н), 7,61 (д, J=8,1 Гц, 2Н), 7,47 (д, J=8,1 Гц, 2Н), 7,30 (с, 1Н), 5,69 (с, 2Н), 3,74 (с, 3Н).
Следующие промежуточные продукты были получены из соответствующих гетероциклов и алкилирующих реагентов в соответствии со способом получения для I-1.
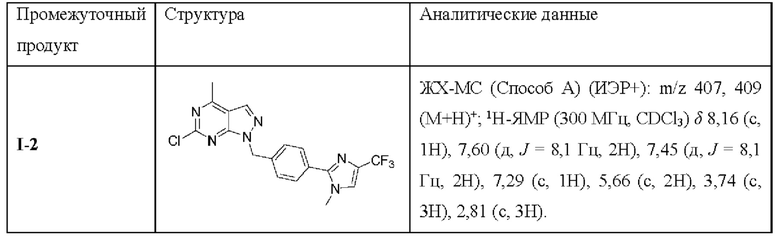
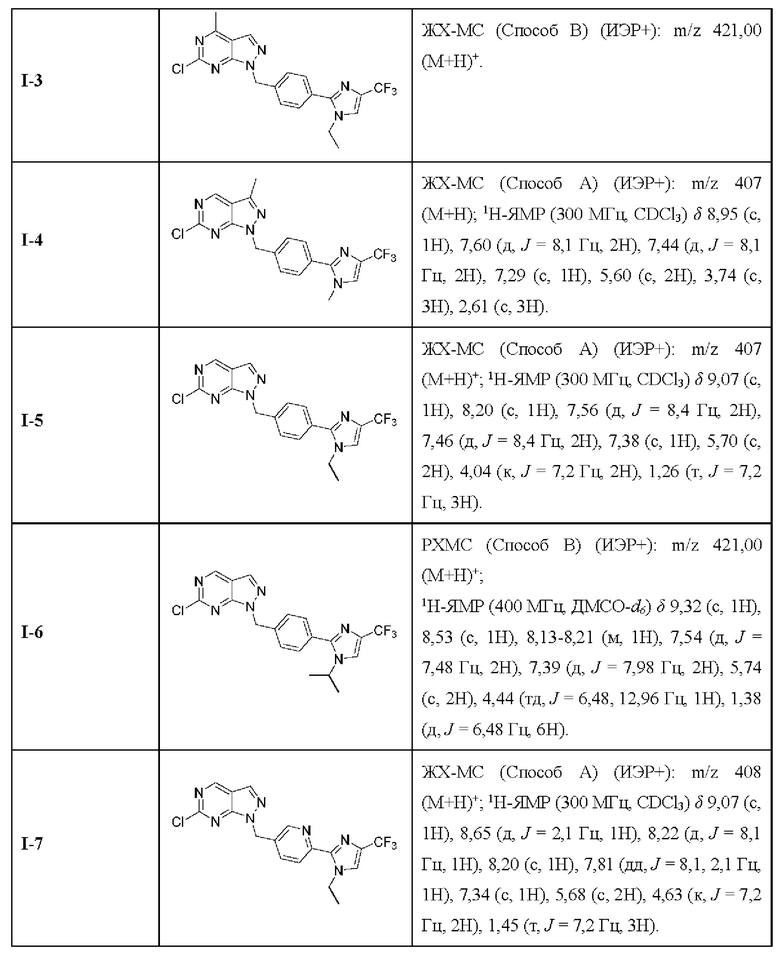
Получение общего промежуточного продукта I-8:
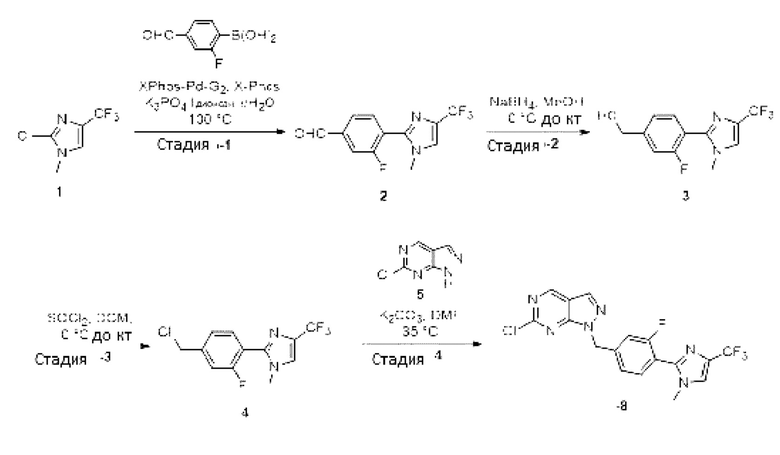
Стадия 1: Синтез 3-Фтор-4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензальдегида:
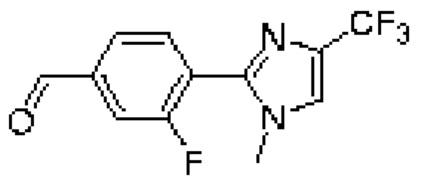
[0460] Раствор 2-хлор-1-метил-4-(трифторметил)-1H-имидазола (1,00 г, 5,40 ммоль), (2-фтор-4-формилфенил)бороновой кислоты (1,00 г, 6,50 ммоль) и K3PO4 (3,40 г, 16.30 ммоль) в диоксане (20 мл) и Н2О (5 мл), дегазировали аргоном 6 течение 5 мин. К полученной реакционной смеси последовательно добавляли Xphos-Pd-G2 (0,42 г, 0,54 ммоль) и X-Phos (1,03 г, 2,10 ммоль), и реакционную смесь нагревали в запаянной пробирке при 100°С в течение 16 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли водой и экстрагировали EA. Органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Полученное таким образом неочищенное соединение очищали эфоматографией на силикагеле, используя 0-30% ЕА в гексане, с получением 1,40 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 272,90 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 10,09 (с, 1Н), 8,08 (с, 1Н), 7,83-7,94 (м, 3Н), 3,66 (с, 3Н).
Стадия 2: Синтез (3-фтор-4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)фенил)метанола:
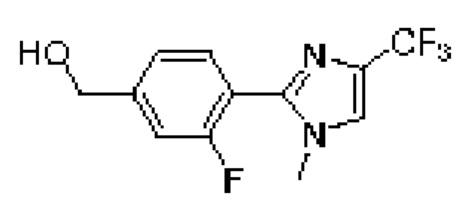
[0461] К охлажденному льдом раствору 3-фтор-4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензальдегида (1,40 г, 5,14 ммоль) 6 МеОН (30 мл) добавляли NaBH4 (0,293 г 7,70 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Неочищенную реакционную смесь растворяли в воде и экстрагировали ЕА. Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением 1,40 г указанного в заголовке соединения, которое использовали без дополнительной очистки. ЖХ-МС (Способ В) (ИЭР+): m/z 274,85 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 7,99 (с, 1Н), 7,54 (т, J=7,8 Гц, 1Н), 7,28-7,34 (м, 2Н), 5,46 (т, J=5,6 Гц, 1Н), 4,60 (д, J=5,4 Гц, 2Н), 3,59 (с, 3Н).
Стадия 3: Синтез 2-(4-(Хлорметил)-2-фторфенил)-1-метил-4-(трифторметил)-1H-имидазола:
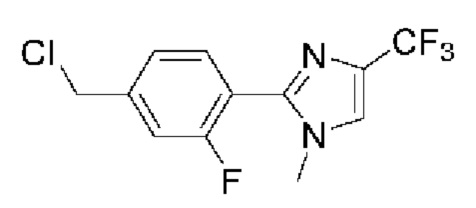
[0462] К охлаждаемому льдом раствору (3-фтор-4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)фенил)метанола (1,40 г, 5,10 ммоль) в DCM (15 мл), добавляли SOCl2 (1,80 г, 15,0 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили насыщенным раствором NaHCO3 и экстрагировали DCM. Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением 1,42 г указанного в заголовке соединения, которое использовали без дополнительной очистки. ЖХ-МС (Метод В) (ИЭР+): m/z 292.85 (М+Н)+.
Стадия 4: Синтез 6-Хлор-1-(3-фтор-4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидина (I-8):
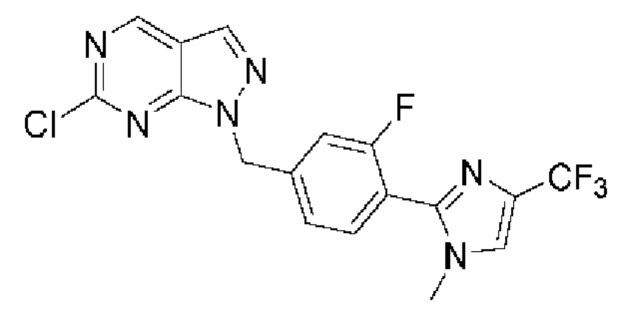
[0463] Раствор 2-(4-(хлорметил)-2-фторфенил)-1-метил-4-(трифторметил)-1H-имидазола (1,41 г, 4,80 ммоль), 6-хлор-1H-пиразоло[3,4-d]пиримидина 5 (0,65 г, 4,20 ммоль) и K2CO3 (1,45 г, 10,50 ммоль) в ДМФА (12 мл) перемешивали при 35°С в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой и экстрагировали ЕА. Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-15% ЕА в гексане, с получением 0,70 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 410,85 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,33 (с, 1Н), 8,54 (с, 1Н), 7,99 (с, 1Н), 7,57 (т, J=7,6 Гц, 1Н), 7,31 (д, J=10,8 Гц, 1Н), 7,19 (д, J=7,8 Гц, 1Н), 5,76 (с, 2Н), 3,58 (с, 3Н).
Следующий промежуточный продукт был получен из соответствующих альдегида и алкилирующих реагентов в соответствии с процедурой для промежуточного продукта I-8.
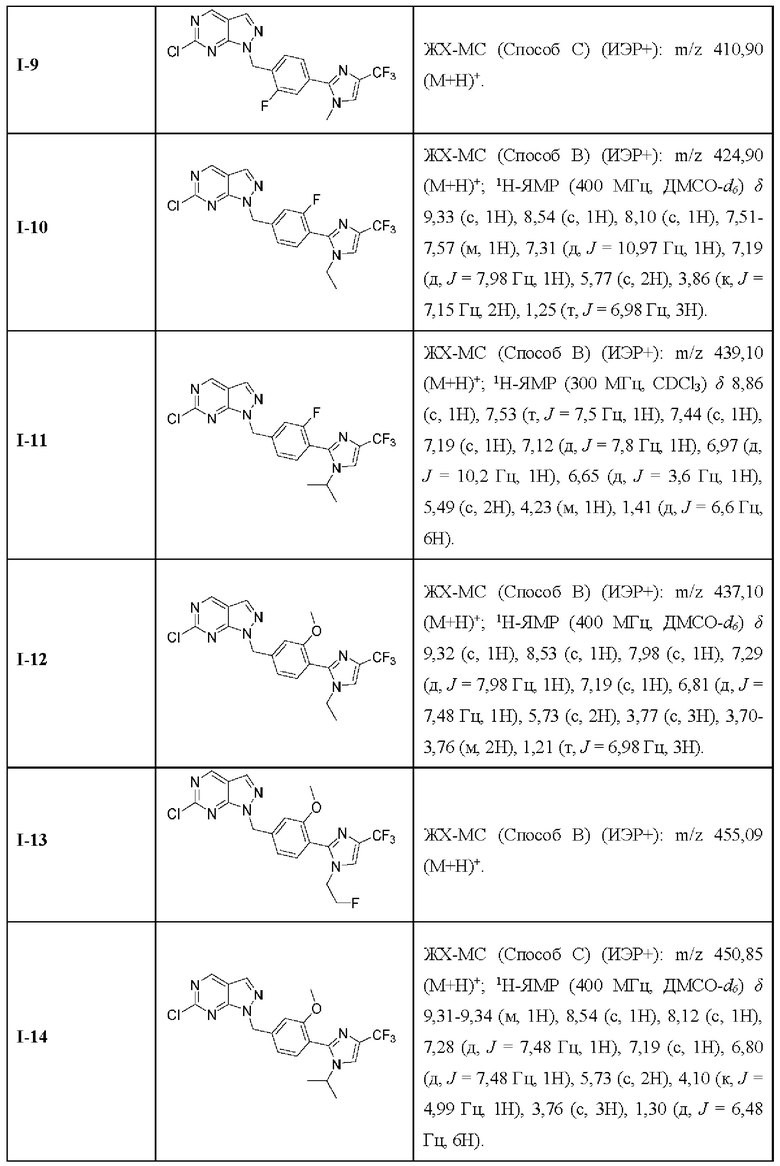
Получений общего промежуточного продукта I-15:
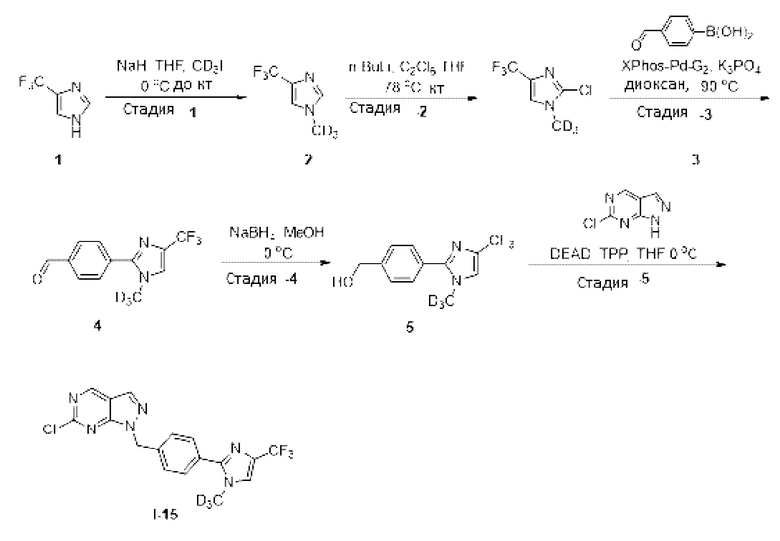
Стадия 1: Синтез 1-(Метил-d3)-4-(трифторметил)-1H-имидазола:
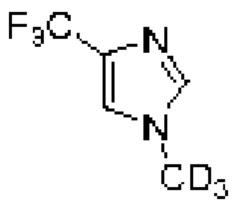
[0464] К перемешиваемому раствору 4-(трифторметил)-1H-имидазола (4,0 г, 29,4 ммоль) в суком ТГФ (100 мл) при 0°С добавляли гидрид натрия (60% дисперсия, 1,294 г, 32,35 ммоль). Полученную смесь перемешивали в течение 15 минут, затем добавляли йодметан-d3 (4,26 г, 29,4 ммоль) при 0°С, и реакционную смесь перемешивали в течение 30 минут. Затем реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 4 часов. После завершения реакции (под контролем ТСХ) реакционную смесь гасили насыщенным раствором хлорида аммония (50 мл) и экстрагировали ЕА (3×50 мл). Объединенный органический слой сушили над безводным Na2SO4, фильтровали и сушили под вакуумом. Неочищенный продукт очищали хроматографией на силикагеле (50-100% ЕА в н-генсане) с получением 3,0 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 154,45 (М+Н)+; 1Н-ЯМР (400 МГц, CDCl3) δ 7,4S (с, 1Н), 7,27 (с, 1Н)
Стадия 2: Синтез 2-Хлор-1-(метил-d3)-4-(трифторметил)-1H-имидазола:
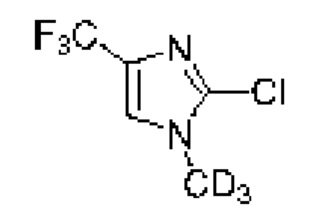
[0465] К перемешиваемому раствору 1-(метил-d3)-4-(трифторметил)-1H-имидазола (2,70 г, 17,6 ммоль) в ТГФ (50 мл) при -78°С добавляли 2,5 М раствор н-бутиллития в н-гексане (7 мл, 17,64 ммоль). Полученную смесь перемешивали при -78°С в течение 30 минут, а затем обрабатывали гексахлорэтаном (2,08 г, 8,82 ммоль). Через 1 час реакционную смесь нагревали до комнатной температуры, после чего перемешивание продолжали еще 3 часа. После завершения реакции (под контролем ТСХ) реакционную смесь гасили насыщенным раствором хлорида аммония (25 мл) и экстрагировали ЕА (3×50 мл). Объединенный органический слой сушили над безводным Na2SO4, фильтровали и упаривали под вакуумом. Неочищенное соединение очищали хроматографией на силикагеле (30-100% ЕА в н-гексане) с получением 1,30 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 188,30 (М+Н)+.
Стадия 3: Синтез 4-(1-(Метил-d3)-4-(трифторметил)-1H-имидазол-2-ил)бензальдегида:
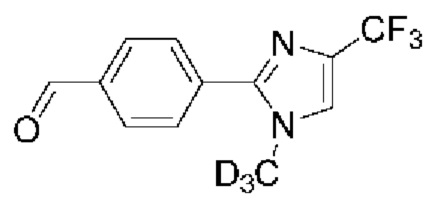
[0466] К раствору 2-хлор-1-(метил-d3)-4-(трифторметил)-1H-имидазола (1,40 г, 7,49 ммоль) в 1,4-диоксане (60 мл) добавляли X-Phos-Pd-G2 катализатор (0,294 г, 0,374 ммоль). Полученную смесь продували газообразным аргоном в течение 10 мин, после чего добавляли твердый K3PO4 (4,76 г, 22,5 ммоль) и (4-формилфенил)бороновую кислоту (2,24 г, 14,97 ммоль) с последующей дополнительной продувкой газообразным аргоном еще 5 мин. Затем реакционную смесь нагревали при 90°С в течение 16 часов. После завершения реакции (под контролем ТСХ) смесь разбавляли водой (60 мл) и ЕА (60 мл). Органический слой отделяли, а водный слой экстрагировали ЕА (2×50 мл). Объединенный органический слой сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле (0-30% ЕА в н-гексане) с получением 1,62 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 258,00 (М+Н)+; 1H-ЯМР (400 МГц, CDCl3) δ 10,09 (с, 1Н), 8,00 (д, J=7,88 Гц, 2Н), 7,86 (д, J=7,88 Гц, 2Н), 7,37 (с, 1Н).
Стадия 4: Синтез (4-(1-(Метил-d3)-4-(трифторметил)-1H-имидазол-2-ил)фенил)метанола:
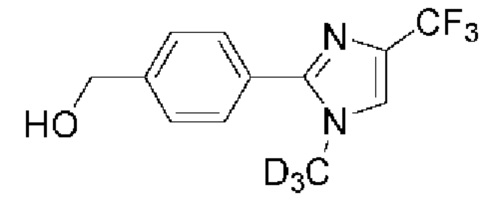
[0467] К перемешиваемому раствору 4-(1-(метил-d3)-4-(трифторметил)-1H-имидазол-2-ил)бензальдегида (1,60 г, 6,23 ммоль) в метаноле (50 мл) при 0°С, добавляли NaBH4 (0,118 г, 3,11 ммоль). После завершения реакции (под контролем ТСХ) смесь гасили ледяной водой (30 мл) и экстрагировали 10% метанолом в DCM (4×50 мл). Объединенный органический слой сушили над безводным Na2SO4, фильтровали и упаривали под вакуумом. Неочищенное соединение очищали хроматографией на силикагеле (0-60% ЕА в н-гексане) с получением 1,30 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 259,80 (М+Н)+; 1H-ЯМР (400 МГц, CDCl3) δ 7,60 (д, J=8,31 Гц, 2Н), 7,45 (д, J=7,83 Гц, 2Н), 7,31 (с, 1Н), 4,76 (с, 2Н).
Стадия 5: Синтез 6-Хлор-1-(4-(1-(метил-d3)-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидина (I-15):
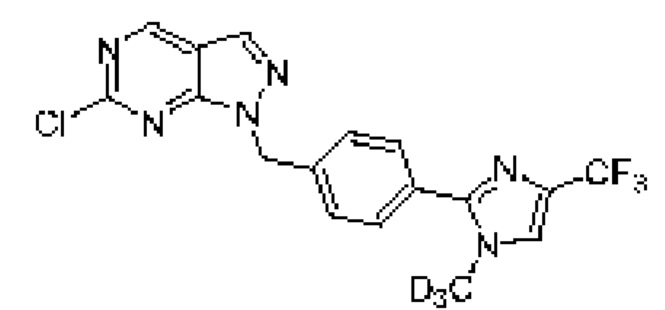
[046S] К смеси (4-(1-(метил-d3)-4-(трифторметил)-1H-имидазол-2-ил)фенил)метанола (0,500 г, 1,930 ммоль) и 6-хлор-1H-пиразоло[3,4-d]пиримидина 7 (0,335 г, 3,181 ммоль) в сухом ТГФ (5 мл) при 0°С, добавляли трифенилфосфин (0,657 г, 2,509 ммоль). Полученную смесь перемешивали в течение 5 минут, а затем по каплям обрабатывали диэтилазодикарбоксилатом (0,434 г, 2,509 ммоль). Смесь дополнительно перемешивали при 0°С в течение 2 часов. После завершения реакции (под контролем ТСХ) смесь разбавляли водой (30 мл) и экстрагировали ЕА (3×30 мл). Объединенный органический спой промывали солевым раствором (3×30 мл), сушили над безводным Na2SO4, фильтровали и упаривали под вакуумом Неочищенное соединение очищали хроматографией на силикагеле (0-3% метанол в DCM) с получением 0,400 г указанного в заголовке соединения. ЖХ-МС (Способ В) (НЭР+): m/z 396,00 (М+Н)+; 1H-ЯМР (400 МГц, CDCl3) δ 9,05 (с, 1H), 8,18 (c, 1Н), 7,61 (д J=8,31 Гц 2Н), 7,47 (д J=7,82 Гц, 2Н), 7,30 (с, 1Н), 5,69 (с, 2Н).
Получение общего промежуточного продукта I-16:
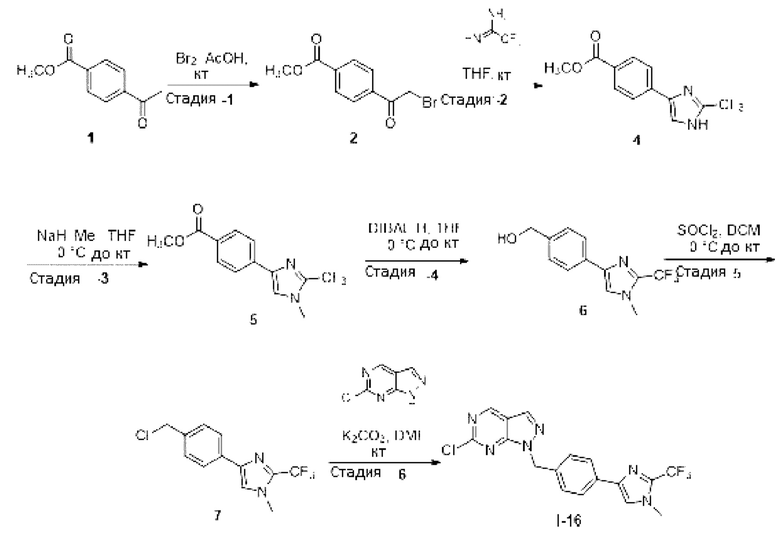
Стадия 1: Синтез Метил 4-(2-бромацетил)бензоата:
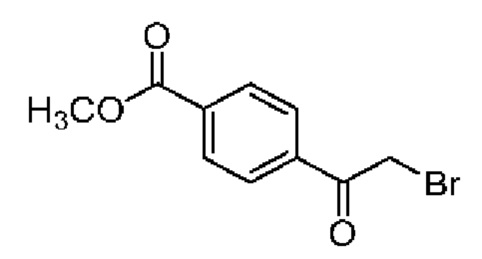
[0469] К перемешиваемому раствору метил 4-ацетилбензоата (5,00 г, 28,0 ммоль) в АсОН (40 мл) по каплям добавляли Br2 (1,00 мл, 19,6 ммоль) в АсОН (10 мл) и реакционную смесь перемешивали при комнатной температуре в течение 6 ч. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь выливали в ледяную воду (100 мл), и полученное твердое вещество фильтровали и сушили. Неочищенное соединение очищали хроматографией на силикагеле, используя 5-7% ЕА в гексане, с получением 3,50 г указанного в заголовке соединения. ЖХ-МС (Способ С) (ИЭР+): m/z 278,95 (М+Na)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,04-8,15 (м, 4Н), 5,00 (с, 2Н), 3,89 (с, 3Н).
Стадия 2: Синтез Метил 4-(2-(трифторметил)-1H-имидазол-4-ил)бензоата:
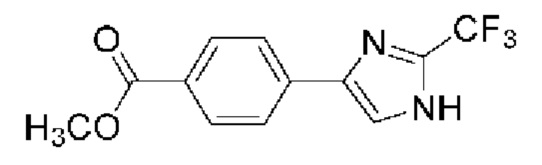
[0470] К перемешиваемому раствору 2,2,2-трифторацетимидамида (4,37 г, 39,0 ммоль) в ТГФ (30 мл) добавляли метил 4-(2-бромацетил)бензоат (2,00 г, 7,80 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли ЕА (20 мл) и промывали солевым раствором (2×20 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-15% ЕА в гексане, с получением 0,81 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 270,90 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 13,89 (с, 1Н), 8,14 (с, 1Н), 7,98 (с, 4Н), 3,86 (с, 3Н).
Стадия 3: Синтез Метил 4-(1-метил-2-(трифторметил)-1H-имидазол-4-ил)бензоата:
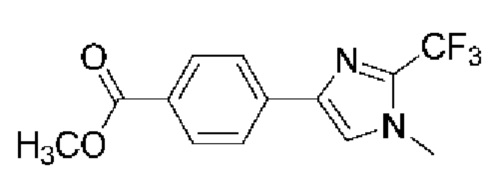
[0471] К перемешиваемому раствору метил 4-(2-(трифторметил)-1H-имидазол-4-ил)бензоата (0,80 г, 2,9 ммоль) в ТГФ (15 мл) при 0°С добавляли NaH (60% дисперсия в минеральном масле, 0,178 г, 4,40 ммоль) порциями, и реакционную смесь перемешивали при той же температуре в течение 15 мин. К полученной реакционной смеси добавляли метилиодид (0,624 г, 4,40 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили ледяной водой (50 мл) и экстрагировали ЕА (2×30 мл). Объединенный органический слой промывали солевым раствором (2×30 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением 0,80 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 284,95 (M+H)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,12-8,17 (м, 1H), 7,96-8,02 (м, 2Н), 7,88-7,94 (м, 2Н), 3,86 (с, 6Н).
Стадия 4: Синтез (4-(1-Метил-2-(трифторметил)-1H-имидазол-4-ил)фенил)метанола:
[0472] К перемешиваемому раствору метил 4-(1-метил-2-(трифторметил)-1H-имидазол-4-ил)бензоата (0,80 г, 2,8 ммоль) в ТГФ (10 мл) при 0°С добавляли DIBAL-H (1M в толуоле, 5,63 мл, 5,63 ммоль) по каплям, и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили 2 н. HCl (20 мл) и экстрагировали ЕА (2×30 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением 0,65 г указанного в заголовке соединения, которое использовали в последующих реакциях без дополнительной очистки. ЖХ-МС (Способ В) (ИЭР+): m/z 256,85 (M+H)+.
Стадия 5: Синтез 4-(4-(Хлорметил)фенил)-1-метил-2-(трифторметил)-1H-имидазола:
[0473] К перемешиваемому раствору (4-(1-метил-2-(трифторметил)-1H-имидазол-4-ил)фенил)метанола (0,65 г, 2,5 ммоль) в DCM (10 мл) при 0°С добавляли SOCl2 (0,27 мл, 3,80 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли DCM (50 мл) и промывали насыщенным раствором NaHCO3 (3×30 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением 0,65 г указанного в заголовке соединения, которое использовали в последующих реакциях без дополнительной очистки. ЖХ-МС (Способ В) (ИЭР+): m/z 274,90 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,00 (с, 1H), 7,77 (д, J=7,8 Гц, 2Н), 7,46 (д, J=7,8 Гц, 2Н), 4,78 (с, 2Н), 3,84 (с, 3Н).
Стадия 6: Синтез 6-Хлор-1-(4-(1-метил-2-(трифторметил)-1H-имидазол-4-ил)бензил)-1H-пиразоло[3,4-d]пиримидина (I-16):
[0474] К перемешиваемому раствору 6-хлор-1H-пиразоло[3,4-d]пиримидина (0,40 г, 2,5 ммоль) в ДМФА (10 мл) добавляли K2CO3 (0,517 г, 3,70 ммоль) одной порцией. После перемешивания в течение 10 минут при комнатной температуре добавляли 4-(4-(хлорметил)фенил)-1-метил-2-(трифторметил)-1H-имидазол (0,638 г, 2,30 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь выливали в ледяную воду (50 мл) и экстрагировали ЕА (2 × 30 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-50% ЕА в гексане, с получением 0,15 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 392,95 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,30 (с, 1H), 8,49 (с, 1H), 7,94 (с, 1H), 7,72 (д, J=8,3 Гц, 2Н), 7,29 (д, J=8,3 Гц, 2Н), 5,63 (с, 2Н), 3,82 (с, 3H).
Следующий промежуточный продукт был получен из соответствующих реагентов в соответствии с процедурой для промежуточного продукта I-16.
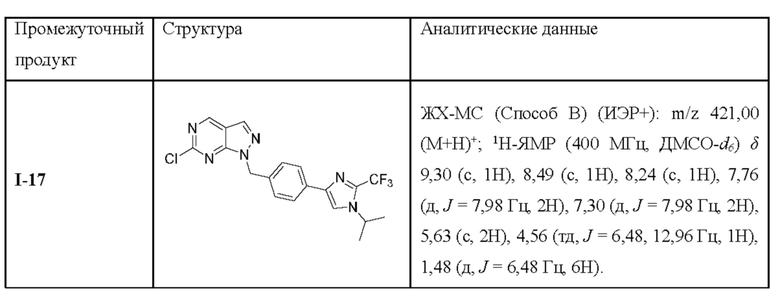
Получение общего промежуточного продукта I-18:
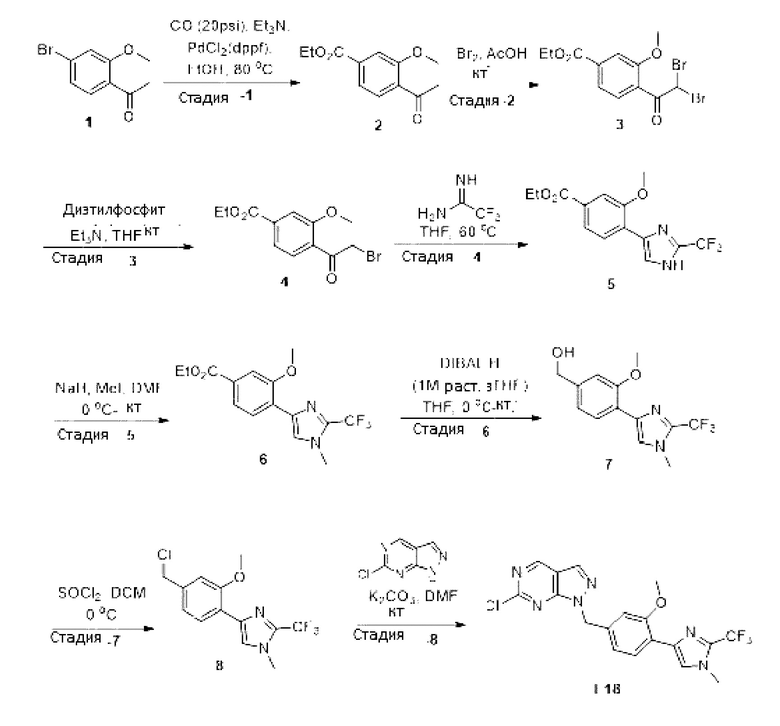
Стадия I: Синтез этил 4-ацетил-3-метоксибензоата:
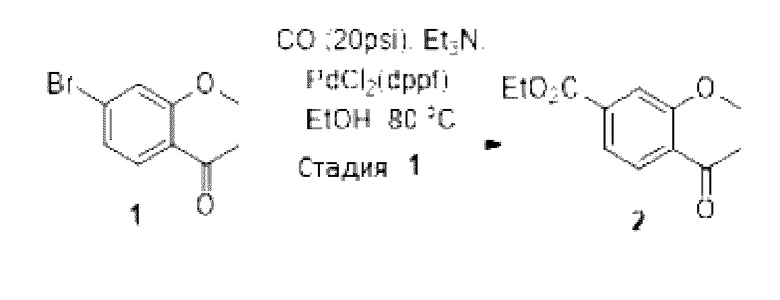
[0475] К перемешиваемому раствору 1-(4-бром-2-метоксифенил) этан-1-она 1 (3,00 г, 13,1 ммоль) в этаноле (60 мл) добавляли триэтиламин (3,67 ми, 26,2 ммоль) и смесь дегазировали аргоном в течение 10 мин. К реакционной смеси добавляли PdCl2(dppf) (0,9:57 г, 0,131 ммоль) при комнатной температуре, и полученную смесь нагревали при 80°С в атмосфере монооксида углерода (давление 20 фунтов на квадратный дюйм) в течение 3 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Неочищенное соединение очищали хроматографий на силикагеле, используя 0-5% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (2,50 г). ЖХ-МС (Способ В) (ИЭР+): m/z 223,00 (М+Н)-; 1Н-ЯМР (400 МГц, CD Cl3) δ 7,72-7,76 (м, 1H), 7,64-7,70 (м, 2H), 4,42 (дк, J=3,99, 7,15 Гц, 2H), 4,00 (уш. с, 1H), 3,99 (c, 2H), 2,65 (с, 1H), 2,64 (с, 2H), 1,43 (дг, J=3,99, 6,98 Гц, 3H).
Стадия 2: Синтез этил 4-(2,2-дибромацетил)-3-метоксибензоата:
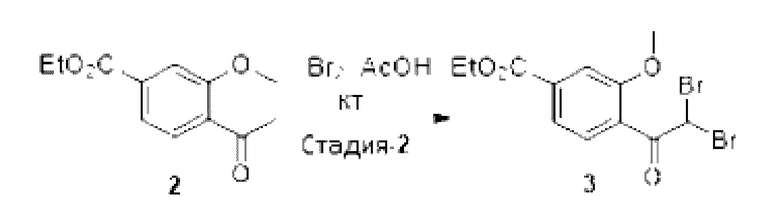
[0476] К перемешиваемому раствору этил 4-ацетил-3-метоксибензоата 2 (1,00 г, 4,50 ммоль) в уксусной кислоте (10 мл) добавляли бром (0,16 мл, 3,153 ммоль) при комнатной температуре. Полученную смесь перемешивали е течение 3 ч, и за ходом реакции следили с помощью ТСХ После завершения реакционную смесь упаривали при пониженном давлении. Неочищенный остаток разбавляли водой (30 мл) и ЕА (50 мл), а затем нейтрализовали насыщенным раствором Na2CO3. Органический слой промывали солевым раствором (30 мл), сушили над безводным сульфатам натрия, фильтровали и упаривали при пониженном давлении с получением указанного в заголовке соединения (1,20 г). 1Н-ЯМР (400 МГц, CD Cl3) δ 7,83 (д, J=7,98 Гц, 1H), 7,69 (д, J=8,48 Гц, 1Н), 7,67 (с, 1Н), 4,59 (с, 1Н), 4,41 (к, J=7,15 Гц, 2H), 4,01 (с, 3H), 1,42 (т, J=6,98 Гц, 3H).
Стадия 3: Синтез этил 4-(2-бромацетил)-3-метоксибензоата:
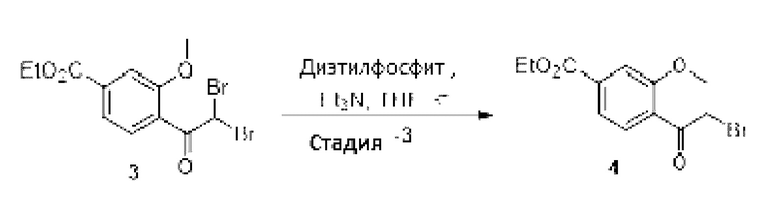
[0477] К перемешиваемому раствору этил 4-(2,2-дибромацетил)-3-метоксибензоата 3 (1,20 г, 3,17 ммоль) в ТГФ (15 мл) добавляли триэтиламин (0,4 4 мл, 3 Д 7 ммоль) и диэтилфосфит (0,433 г, 3,166 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение 3 ч, и за ходом реакции следили с помощью TCS. После завершения реакционную смесь разбавляли ЕА (30 мл) и промывали солевым раствором (3 × 10 мл). Органический спой сушили над безводный сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-15% ЕА в гексане в качестве злюента, с получением указанного в заголовке соединения (0,900 г). ЖХ-МС (Способ В) (ИЭР+): m/z 300,80 (М)+; 1H-ЯМР (400 МГц. CDCl3) δ 7,84 (д, J=7,98 Гц, 1Н), 7,69 (д, J=7,92 Гц, 1Н), 7,67 (с, 1Н), 4,60 (с, 2Н), 4,41 (к, J=7,15 Гц 2Н), 4,02 (с, 3H), 1,42 (т, J=6,98 Гц, 3H).
Стадии 4: Синтез этил 3-метокси-4-(2-(трифторметил)-1H-имидазол-4-ил)бензоата:
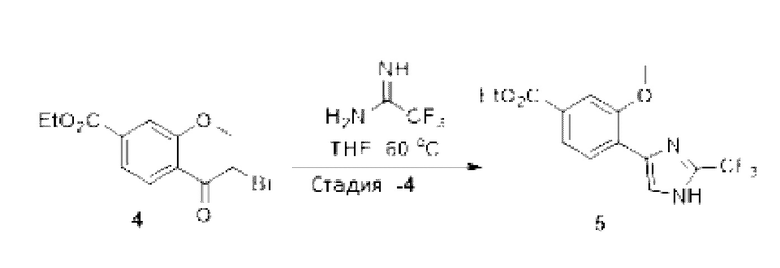
[0478] К перемешиваемому раствору этил 4-(2-бромацетил)-3-метоксибензоата 4 (0,900 г, 3,00 ммоль) в ТГФ (20 мл) добавляли 2,2,2-трифторацетимидамид (1,68 г, 15,0 ммоль) при комнатной температуре. Затем реакционную смесь нагревали при 60°С в течение 16 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли ЕА (50 мл) и промывали насьпценньпл раствором NaHCO3 (2 30 мл). Органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный остаток очищали хроматографией на силикагеле, используя 20-30% ЕА в гексане в качестве злюента, с получением указанного в заголовке соединения (0,300 г). ЖХ-МС (Способ В) (ИЭР+): m/z 315,10 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 13,32 (уш. с, 1Н), 8,17 (д, J=7,48 Гц. 1Н), 7,92 (уш. с, 1Н), 7,64 (д, J=7,98 Гц, 1Н), 7,59 (с, 1Н), 4,33 (к, J=7,31 Гц, 2Н), 4,00 (с, 3H), 1,34 (т, J=6,93 Гц 3H).
Стадии 5: Синтез этил 3-метокси-4-(1-метил-2-(трифторметил)-1Н-имидазол-4-ил)бензоата:
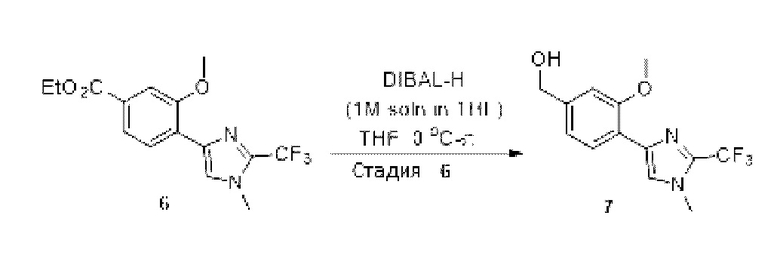
[0479] К перемешиваемому раствору этил 3-метокси-4-(2-(трифторметил)-1H-имидазол-4-ид)бензоата 5 (0,300 г, 0,955 ммоль) в ДМФА (10 мл) при 0°С добавляли 60% дисперсию гидрида натрия в масле (0,057 г, 1,43 ммоль) и метилиодид (0,089 мл, 1,43 ммоль). Реакционной смеси давали нагреться до комнатной температурь; а затем перемешивали в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь выливали в ледяную воду (50 мл) и экстрагиовали ЕА (2 × 30 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-15% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,240 г). ЖХ-МС (Способ В) (ИЭР+), m/z 328,90 (М+Н)+, 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,14 (д, J=8,48 Гц, 1Н), 8,04 (с, 1Н), 7,64 (дд, J=1,50, 7,98 Гц, 1Н), 7,59 (с, 1Н), 4,33 (к, J=7,15 Гц, 2Н), 4,00 (с, 3H), 3,87 (с, 3H), 1,33 (т, J=7,23 Гц, 3H).
Стадия 6: Синтез (3-метокси-4-(1-метил-2-(трифторметил)-1H-имидазол-4-ил)фенил)метанола:
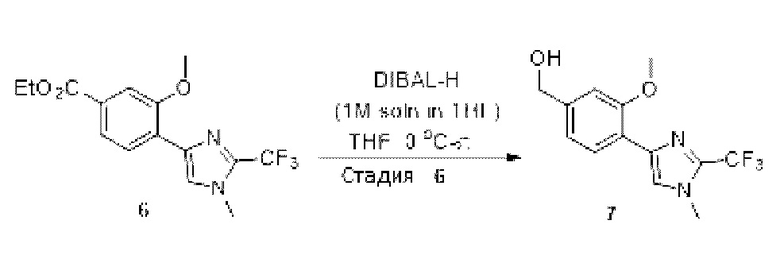
[0480] К перемешиваемому раствору этил 3-метокси-4-(1-метил-2-(трифторметил)-1Н-имидазол-4-ил)бензоата 6 (0,320 г, 0,975 ммоль) в сухом ТГФ (10 мл) при 0°С добавляли DIBAL-H (1 М раствор в толуоле, 1,46 мл, 1,46 ммоль). Реакционной смеси давали нагреться до комнатной температуры, а затем перемешивали в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили 1 н. HCl (20 мл) и экстрагировали ЕА (3 × 20 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением неочищенного указанного в заголовке соединения (0,260 г). ЖХ-МС (Способ В) (ИЭР+): rn/z 287,30 (М+Н)+, 1H-ЯМР (400 МГц, ДМСО-d6) δ 7,94 (д, J=7,83 Гц, 1Н), 7,84 (с, 1Н), 7,05 (с, 1Н), 6,95 (д, J=7,82 Гц, 1Н), 5,21 (т, J=4,89 Гц, 1Н), 4,51 (д, J=4,89 Гц, 2Н), 3,92 (с, 3H), 3,84 (с, 3H).
Стадия 7: Синтез 4-(4-(хлорметил)-2-метоксифенил)-1-метил-2-(трифторметил)-1H-имидазола:
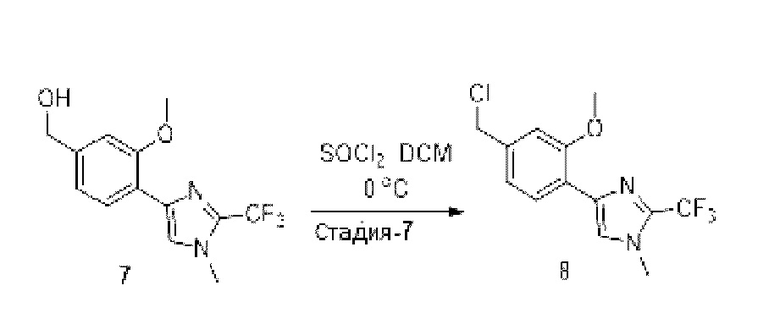
[0481] К перемешиваемому раствору (3-метокси4-(1-метил-2-(трифторметил)-1H-имидазол-4-ил)фенил)метанола 7 (0,260 г, 0,909 ммоль) в DCM (10 мл) при 0°С, добавляли тионилхлорид (0,065 мл, 0,91 ммоль). Смесь перемешивали в течение 1 ч, и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли DCM (20 мл) и промывали насыщенным раствором Na2CO3 (3 × 20 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением неочищенного указанного в заголовке соединения (0,260 г). ЖХ-МС (Способ В) (ИЭР+): m/z 304,80 (М+Н)+, 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,00 (д, J=1,98 Гц, 1Н), 7,90 (с, 1Н), 7,18 (с, 1Н),7,08 (д, J=7,98 Гц, 1Н), 4,78 (с, 2Н), 3,94 (с, 3H), 3,85 (с, 3H).
Стадия 8: Синтез 6-хлор-1-(3-метокси-4-(1-метил-2-(трифторметил)-1Н-имидазол-4-ил)безил)-1Н-пиразоло[3,4-d]пиримидина (I-18):
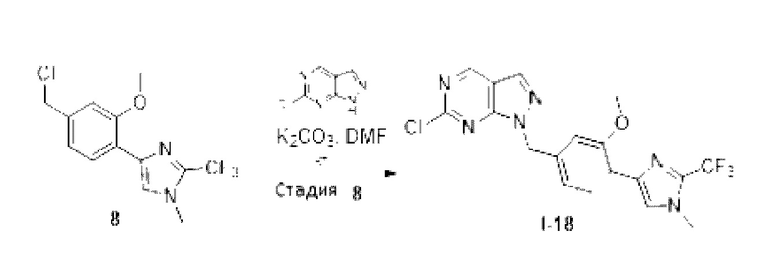
[0482] К перемешиваемому раствору 4-(4-(хлорметил)-2-метоксифенил)-1-метил-2-(трифторметил)-1Н-имидазола S (0,250 г, 0,822 ммоль) в ДМФА (5 мл) добавляли калий карбонат (0,170 г, 1,23 ммоль) и 6-хлор-1Н-пиразоло[3,4-d]пиримидин (0,126 г, 0,822 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение 16 ч, и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь выливали в ледяную воду (30 мл) и экстрагировали ЕА (2 × 30 мл). Объединенный органический слой промывали солевым раствором (2 × 30 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 30-50% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,150 г). ЖХ-МС (Способ В) (ИЭР+): m/z 423,00 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,31 (с, 1Н), 8,50 (с, 1Н), 7,92 (д, J=7,98 Гц, 1Н), 7,86 (с, 1Н), 7,11 (с, 1Н), 6,82 (д, J=7,98 Гц, 1Н), 5,64 (с, 2Н), 3,89 (с, 3H), 3,83 (с, 3H).
Получение общею промежуточного продукта I-19:
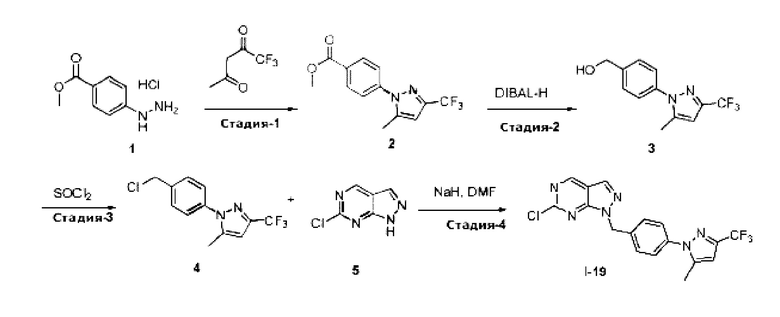
Стадия 1: Синтез Метил 4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензоата:
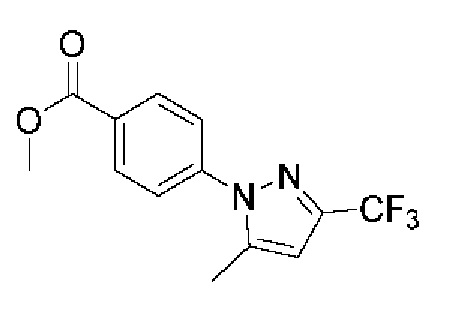
[0483] К раствору гидрохлорида метил 4-гидразинилбензоата (1,0 г, 4,92 ммоль) и 1,1,1-трифторпентан-2,4-диона (758 мг, 4,92 ммоль) в HFIP (5 мл), охлажденному до 0°С, добавляли раствор TEA (994 мг, 9,84 ммоль) в HFIP (3 мл) по каплям в течение 5 мин. После добавления полученную смесь перемешивали при комнатной температуре в течение 1 часа, затем гасили водой (20 мл) и экстрагировали DCM (100 мл × 3). Объединенные органические слои сушили безводным Na2SO4 (30 г), фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (ЕА: n-Нех=1:20) с получением 1,1 г указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 285 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 8,18 (д, J=8,7 Гц, 2Н), 7,57 (д, J=8,7 Гц, 2Н), 6,50 (с, 1H), 3,96 (с, 3H), 2,41 (с, 3H).
Стадия 2: Синтез (4-(5-Метил-3-(трифторметил)-1Н-пиразол-1-ил) фенил)метанола:
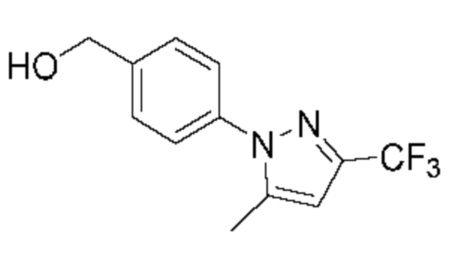
[0484] К раствору метил 4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензоата (1,0 г, 3,52 ммоль) в сухом ТГФ (20 мл) при 0°С добавляли DIBAL-H (10,5 мл, 10,56 ммоль, 1 M в толуоле) в течение 10 мин. После добавления реакционную смесь нагревали до комнатной температуры. После завершения реакции, как показывает анализ ТСХ, реакцию гасили насыщенным раствором NH4Cl (20 мл), экстрагировали ЕА (100 мл × 3). Объединенные органические слои сушили безводным Na2SO4 (50 г), фильтровали и упаривали. Неочищенный продукт очищали хроматографией на силикагеле (РЕ:ЕА = от 20:1 до 10:1) с получением 1,1 г указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CDCl3) δ 7,40-7,49 (м, 4Н), 6,46 (с, 1H), 4,76 (с, 2Н), 2,34 (с, 3H), 2,17 (уш. с, 1H).
Стадия 3: Синтез 1-(4-(Хлорметил)фенил)-5-метил-3-(трифторметил)-1Н-пиразола:
[0485] К раствору (4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил) фенил)метанола (0,82 г, 3,20 ммоль) в DCE (20 мл) добавляли SOCl2 (1,14 г, 9,61 ммоль) за одну порцию. После перемешивания реакционной смеси при 50°С в течение 30 мин смесь охлаждали до 0°С и гасили водой (10 мл). Полученный раствор нейтрализовали насыщенным раствором NaHCO3 (водн.) и экстрагировали DCM (3 × 100 мл). Объединенный органический слой сушили над безводным Na2SO4 (50 г), фильтровали и упаривали досуха с получением 480 мг сырого продукта, который использовали непосредственно в следующей стадии. ЖХ-МС (Способ А) (ИЭР+): m/z 275 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 7,53 (д, J=8,7 Гц, 2Н), 7,46 (д, J=8,7 Гц, 2Н), 6,47 (с, 1Н), 4,64 (с, 2Н), 2,37 (с, 3H).
Стадия 4: Синтез 6-Хлор-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (I-19):
[0486] К раствору 1-(4-(хлорметил)фенил)-5-метил-3-(трифторметил)-1Н-пиразола (0,48 г, 1,75 ммоль) в ДМФА (8 мл) добавляли 6-хлор-1Н-пиразоло[3,4-с!]пиримидин (0,27 г, 1,75 ммоль) и K2CO3 (0,60 г, 4,38 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем гасили водой (20 мл) и экстрагировали ЕА (50 мл × 3). Объединенный органический слой сушили над безводным Na2SO4 (30 г), фильтровали и упаривали в вакууме. Полученный остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 20:1 до 10:1) с получением 370 мг указанного в заголовке продукта с небольшим количеством региоизомера. ЖХ-МС (Способ А) (ИЭР+): m/z 393,395 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 9,06 (с, 1H), 8,19 (с, 1H), 7,51 (д, J=8,7 Гц, 2Н), 7,42 (д, J=8,7 Гц, 2Н), 6,44 (с, 1H), 5,70 (с, 2Н), 2,33 (с, 3H).
Следующие общие промежуточные продукты были получены в соответствии с процедурой I-19 из соответствующих реагентов:
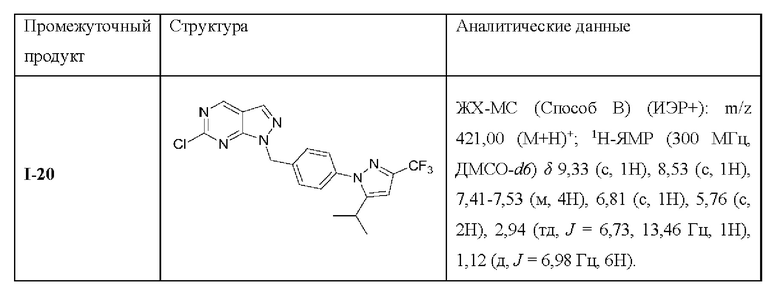
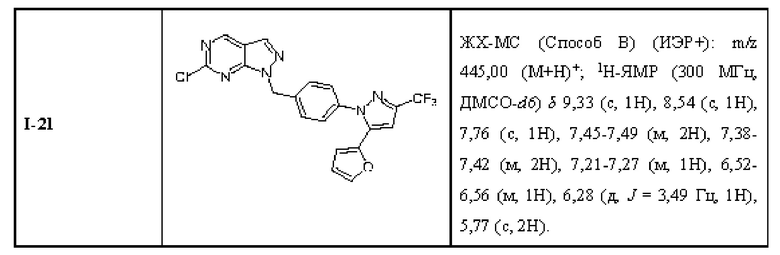
Получение общего промежуточного продукта I-22:
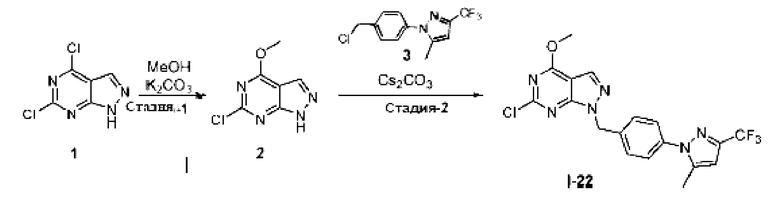
Стадия 1: Синтез 6-хлор-4-метокси-1Н-пиразоло[3,4-d]пиримидина:
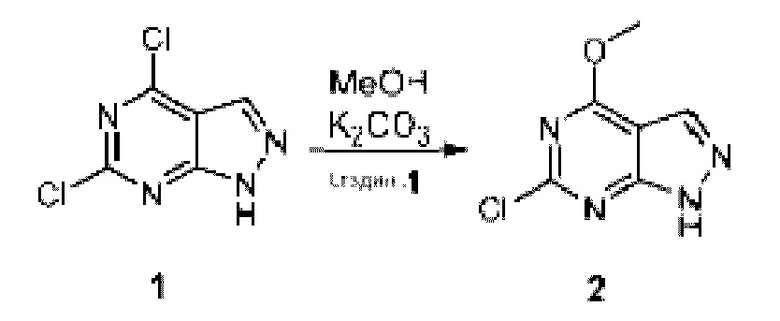
[0487] К раствору 4,6-дихлор-1 Н-пиразол о[3,4-сЦпиримидина 1 (330 мг, 1,74 ммоль) в метаноле (10 мл) добавляли карбонат калия (240 мг, 1,74 ммоль). После перемешивания смеси при комнатной температуре 6 течение 24 ч реакцию гасили насыщенным раствором хлорида аммония (30 мл) и экстрагировали ЕА (20 мл × 3). Объединенный органический слой сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (элюент: ЕА/nHex = 1/10) с получением 120 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 185 (М+Н)+; 1Н-ЯМР(300 МГц, CDCl3) δ 10,69 (уш. с, 1Н), 8,09 (с, 1Н), 4,19 (с, 3H).
Стадия 2: Синтез 6-хлор-4-метокси-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (I-22):
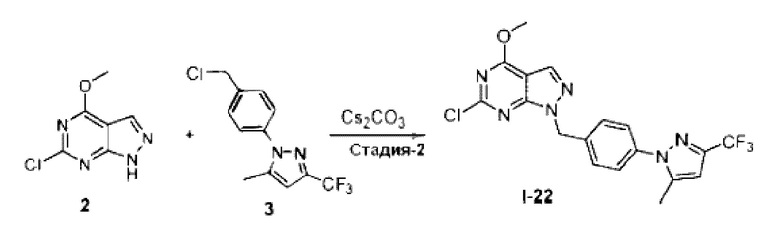
[0488] Соединение синтезировали в соответствии с процедурой получения общего промежуточного продукта I-19. ЖХ-МС (Способ А) (ИЭР+): m/z 423 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 8,05 (с, 1Н), 7,45 (м, 4Н), 6,44 (с, 1Н), 5,63 (с, 2Н), 4,15 (с, 3H), 2,31 (с, 3H).
Следующий общий промежуточный продукт был получен в соответствии с процедурой I-22 из соответствующих реагентов:
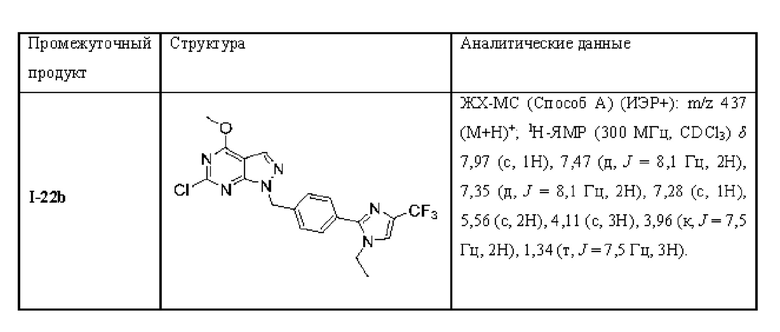
Получение общею промежуточного продукта I-23:
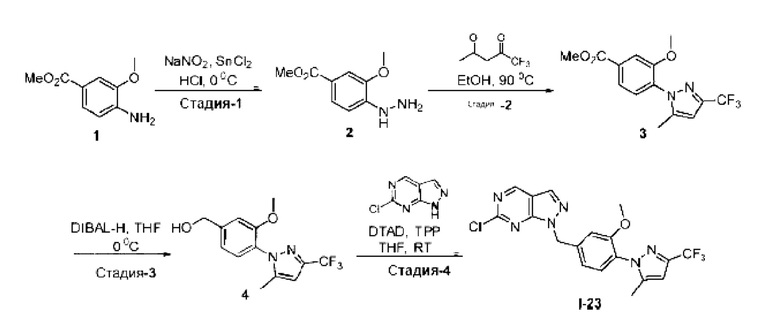
Стадия 1: Синтез метил 4-гидразинеил-3-метоксибензоата:
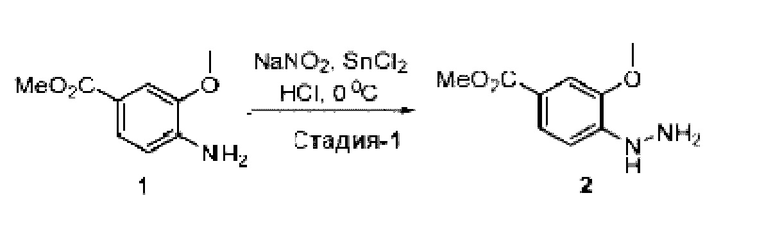
[0489] К перемешиваемому раствору метил 4-амино-3-метоксибензоата 1 (5,0 г, 27,6 ммоль) в конц. HCl (55 мл) при -10°С по каплям добавляли водный раствор NaNO2 (1,99 г, 29,0 ммоль) в воде (5 мл). К полученной реакционной смеси по каплям добавляли раствор SnCl2 (26,0 г, 138 ммоль) в конц. HCl (35 мл), и реакционную смесь перемешивали в течение 2 часов. За кодом реакции следили с помощью ТСХ. После завершения полученное твердое вещество фильтровали и сушили при пониженном давлении. Неочищенное соединение промывали диэтиловымэ фиром (50 мл), фильтровали и сушили при пониженном давлении с получением неочищенного указанного в заголовке соединения (8,0 г), которое использовали в следующей стадии без очистки. 1H-ЯМР (400 МГц, ДМСО-d6) δ 10,43 (уш. с, 2Н), 7,95-8,40 (м, 1Н), 7,57 (д, J=8,31 Гц 1Н), 7,44 (с, 1Н), 7,09 (д, J=8,31 Гц 1Н), 3,88 (с, 3H).
Стадия 2: Синтез метил 3-метокси-4-(5-метил-3-(трифторметил)-1H-лиразол-1-ил)бензоата:
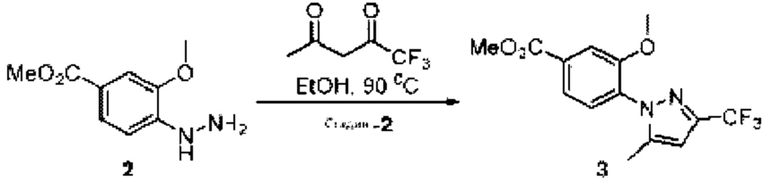
[0490] Перемешиваемый раствор метил 4-гидразинил-3-метоксибензоата 2 (2,0 г, 12,98 ммоль) и 1,1,1-трифторпентан-2,4-диона (6,09 г, 25,97 ммоль) в этаноле (20 мл) нагревали при 90°С в течение 16 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры и упаривали при пониженном давлении Неочищенное соединение очищали хроматографией на силикагеле, используя 0-30% ЕА в гексане, с получением указанного в заголовке соединения(2,5 г). ЖХ-МС (Способ В) (ИЭР+): m/z 315,05 (М+Н)+.
Стадия 3: Синтез (3-метокси-4-(5-метнл-3-(трифторметил)-1Н-пиразол-1-ил)фенил)метанола:
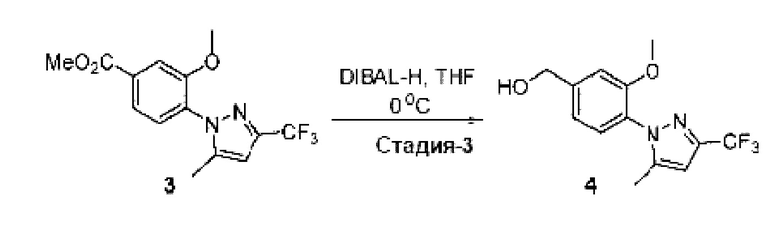
[0491] К перемешиваемому раствору метил 3-метокси-4(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензоата 3 (2,5 г, 7,96 ммоль) в ТГФ (40 мл) при 0°С, по каплям добавляли DIBAL-H (16 мл, 15,9 ммоль, 1,0 М раствор в ТГФ) Полученную смесь перемешивали в течение 2 ч при 0°С, и за кодом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили 1 н. HCl (4 мл) при 0°С и экстрагировали ЕА (100 мл). Объединенный органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении, получая неочищенный 4 (1,5 г). Неочищенный продукт использовали без очистки. ЖХ-МС (Способ В) (ИЭР+): m/z 287,45 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) 7,31 (д, J=7,9% Гц, 1Н), 7,21 (с, 1Н), 7,05 (д, J=7,98 Гц, 1Н), 6,67 (с, 1Н), 5,40 (уш. с, 1Н), 4,59 (уш. с, 2Н), 3,79 (с, 3H), 2,09 (с, 3H).
Стадия 4: Синтез 6-хлор-1-(3-метокси-4-(5-метил-3-(трифторметил)-1H-пираазол-1-ил)бензил)-1H-пиразоло [3,4-d]пиримидина (I-23):
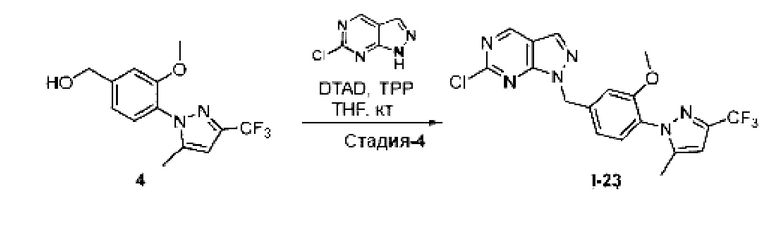
[0492] К охлажденному льдом раствору (3-метокси-4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)фенил)метанола 4 (1,0 г, 3,494 ммоль) в ТГФ (10 мл), добавляли 6-хлор-1H-пиразол о [3,4-d]пиримидин (0,486 г, 3,144 ммоль), DTAD (1,20 г, 5,240 ммоль) и ТРР (2,747 г, 10,48 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (30 мл) и экстрагировали ЕА (3 × 50 мл). Объединенный органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-40% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (1,0 г). ЖХ-МС (Способ В) (Т1ЭР+): m/z 423 (М+Н)+.
Получение общего промежуточного продукта I-24:
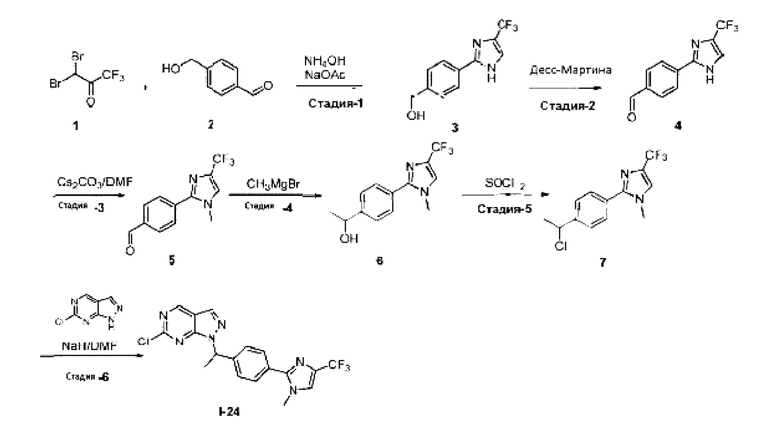
Стадия 1: Синтез (4-(4-(Трифторметил)-1Н-имидазол-2-ил)фенил)метанола:
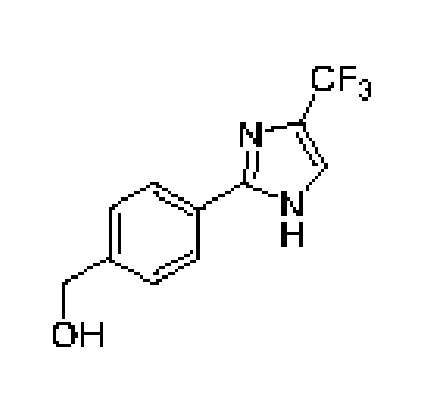
[0493] Раствор 3,3-дибром-1,1,1-трифторпропан-2-она (1,0 г, 7,3 ммоль) и NaOAc (533,5 мг, 8,1 ммоль) в Н2О (7,3 мл) перемешивали при 100°С в течение 1 час. Затем реакционную смесь охлаждали до 0°С ив течение 10 мин добавляли раствор 4-(гидроксиметил)бензальдегида (2,18 г, 8,08 ммоль) и NH4OH (9,3 мл) в МеОН (37 мл). После добавления реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Полученную смесь упаривали при пониженном давлении для удаления большей части растворителя. К концентрированному остатку добавляли воду (20 мл) и смесь экстрагировали ЕА(100 мл × 3). Объединенный органический слой промывали солевым раствором, сушили над безводный Na2SO4 (50 г), фильтровали и упаривали до сук а в вакууме. Полученный неочищенный продукт суспендировали в DCM (30 мл), собирали фильтрованием и сушили в вакууме с получением 1,1 г указанного в заголовке соединения 1Н-ЯМР (300 МГц, CD3OD) δ 7,88 (д, J=8,4 Гц, 2Н), 7,64(с, 1Н), 7,48 (д, J=8,4 Гц 2Н), 4,66 (с, 2Н).
Стадия 2: Синтез 4-(4-(Трифторметил)-1Н-нмндазол-2-ил)бензальдегида:
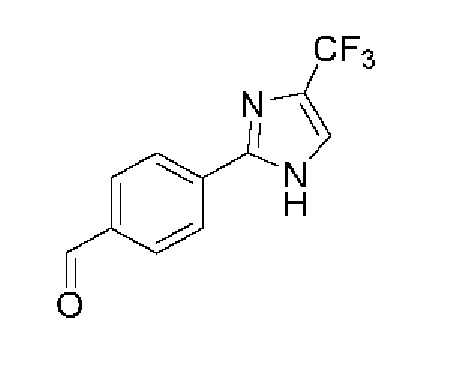
[0494] К раствору (4-(4-(трифторметил)-1Н-имидазол-2-ил)фенил) метанола (1 г, 4,1 ммоль) в ЕА (80 мл) добавляли реагент Десс-Мартина (2,6 г, 6,2 ммоль) порциями при 0 "С в течение 5 мин. После добавления реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Полученную суспензию фильтровали, и осадок на фильтре промывали ЕА (20 мл). Фильтрат промывали насыщенным водным раствором NH4Cl (20 мл), сушили безводным Na2SO4 (20 г), фильтровали и упаривали в вакууме. Концентрированный остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 20:1 до 5:1) с получением 0,9 г указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CD3OD) δ 10,04 (с, 1H), 8,12 (д, J=8,4 Гц, 2Н), 8,03 (д, J=8,4 Гц, 2Н), 7,74 (с, 1H).
Стадия 3: Синтез 4-(1-Метил-4-(трифторметил)-1Н-имидазол-2-ил)бензальдегида:
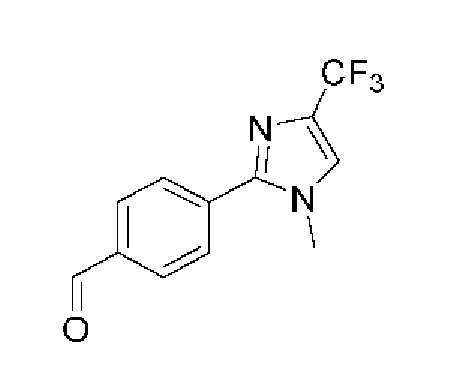
[0495] К раствору 4-(4-(трифторметил)-1Н-имидазол-2-ил)бензальдегида (0,9 г, 3,8 ммоль) в ДМФА (40 мл) при комнатной температуре добавляли Cs2CO3 (3,7 г, 11,2 ммоль) и MeI (1,1 г, 7,5 ммоль) за одну порцию. После перемешивания реакционной смеси при температуре окружающей среды в течение 1 часа реакцию гасили водой (100 мл) и экстрагировали ЕА (100 мл × 2). Объединенный органический слой промывали водой (50 мл × 2), сушили безводным Na2SO4 (50 г) и упаривали в вакууме. Концентрированный остаток очищали хроматографией на силикагеле (РЕ: ЕА = от 20:1 до 5:1) с получением 0,8 г указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CDCl3) δ 10,09 (с, 1H), 8,00 (д, J=8,4 Гц, 2Н), 7,86 (д, J=8,4 Гц, 2Н), 7,38 (с, 1H), 3,85 (с, 3H).
Стадия 4: Синтез 1-(4-(1-Метил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этан-1-ола:
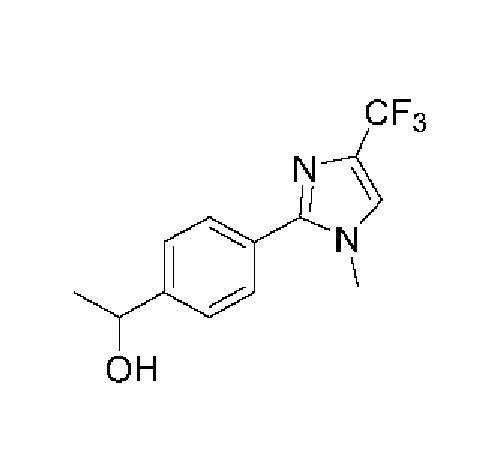
[0496] К раствору 4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензальдегида (0,8 г, 3,2 ммоль) в ТГФ (30 мл) добавляли CH3MgBr (4,7 мл, 4,7 ммоль, 1 M в THF) по каплям в течение 10 минут при 0°С. После добавления реакционную смесь перемешивали при 0°С в течение 1,5 часов, а затем гасили добавлением насыщенного водного раствора NH4Cl (20 мл). Полученную смесь экстрагировали ЕА (100 мл × 2), и объединенный органический слой промывали солевым раствором (20 мл), сушили над безводным Na2SO4 (30 г) и упаривали в вакууме с получением 0,9 г неочищенного указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CDCl3) δ 7,60 (д, J=8,1 Гц, 2Н), 7,47 (д, J=8,1 Гц, 2Н), 7,31 (с, 1Н), 4,97 (к, J=6,3, 1H), 3,81 (с, 3H), 1,52 (a, J=6,3 Гц, 3H).
Стадия 5: Синтез 2-(4-(1-хлорэтил)фенил)-1-метил-4-(трифторметил)-1Н-имидазола:
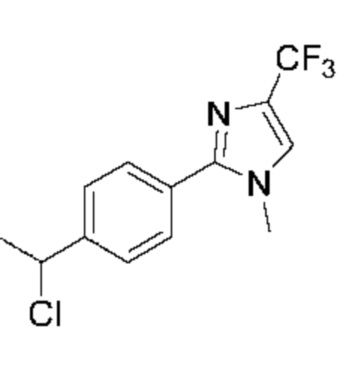
[0497] К раствору 1-(4-(1-метил-4(трифторметил)-1Н-имидазол-2-ил) фенил)этан-1-ола (570 мг, 2,1 ммоль) в DCE (20 мл) добавляли SOCl2 (753,8 мг, 6,3 ммоль) одной порцией. После добавления реакционную смесь перемешивали при 60°С в течение 1 часа, а затем упаривали досуха в вакууме. Остаток растворяли в ЕА (100 мл), а затем обрабатывали насыщенным раствором NHCO3. После разделения органический слой сушили безводным Na2SO4 (30 г) и упаривали в вакууме. Концентрированный неочищенный продукт очищали хроматографией на силикагеле (РЕ:ЕА = 20:1) с получением 330 мг указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CDCl3) δ 7,64 (д, J=8,4 Гц, 2Н), 7,53 (д, J=8,4 Гц, 2Н), 7,32 (с, 1H), 5,13 (к, J=6,6 Гц, 1H), 3,79 (с, 3H), 1,87 (д, J=6,6 Гц, 3H).
Стадия 6: Синтез 6-Хлор-1-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этил)-1Н-пиразоло[3,4-(1]пиримидина (I-24):
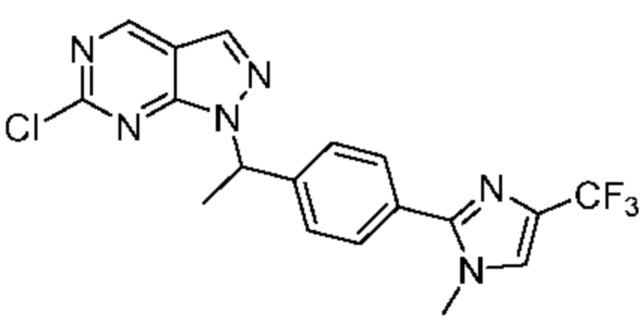
[0498] К раствору 6-хлор-1Н-пиразоло[3,4-d]пиримидина (321,2 мг, 2,1 ммоль) в ДМФА (14 мл) добавляли 2-(4-(1-хлорэтил)фенил)-1-метил-4-(трифторметил)-1Н-имидазол (400 мг, 1,4 ммоль) и K2CO3 (574,5 мг, 4,2 ммоль). После добавления реакционную смесь перемешивали при 65°С в течение ночи, затем гасили водой (30 мл) и экстрагировали ЕА (50 мл × 2). Объединенный органический слой промывали водой (20 мл × 2), сушили над безводным Na2SO4 (30 г), фильтровали и упаривали досуха в вакууме. Концентрированный остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 5:1 до 3:1) с получением 40 мг указанного в заголовке соединения. Его региоизомер был получен как основной продукт после очистки. 1Н-ЯМР (300 МГц, CDCl3) δ 9,03 (с, 1Н), 8,19 (с, 1Н), 7,50-7,64 (м,4Н), 7,29 (с, 1Н), 6,26 (к, J=7,2, 1Н), 3,75 (с, 3H), 2,04 (д, J=7,2 Гц, 3H).
Следующие общие промежуточные продукты были получены в соответствии с процедурой I-24 из соответствующих реагентов:
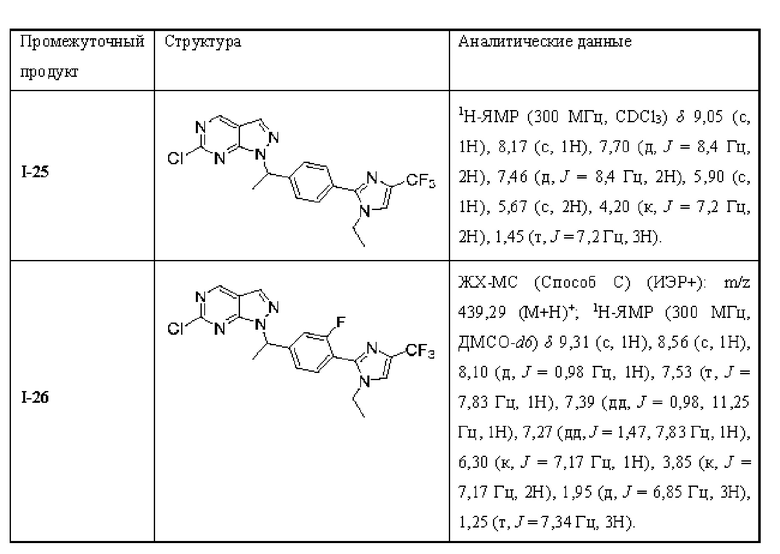
Получение общего промежуточного продукта I-27:
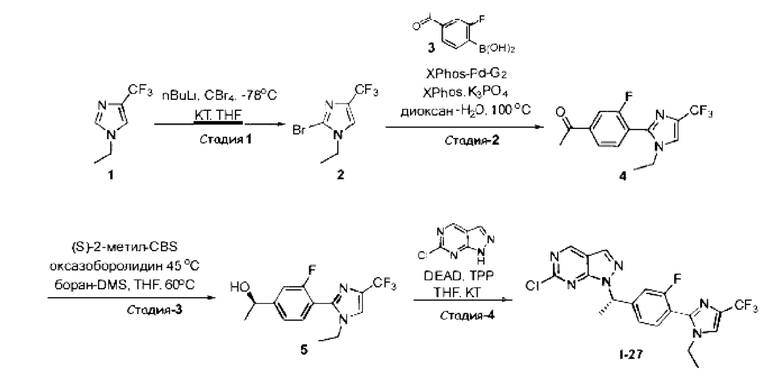
Стадия 1: Синтез 2-бром-1-этил-4-(трифторметил)-1Н-имидазола:
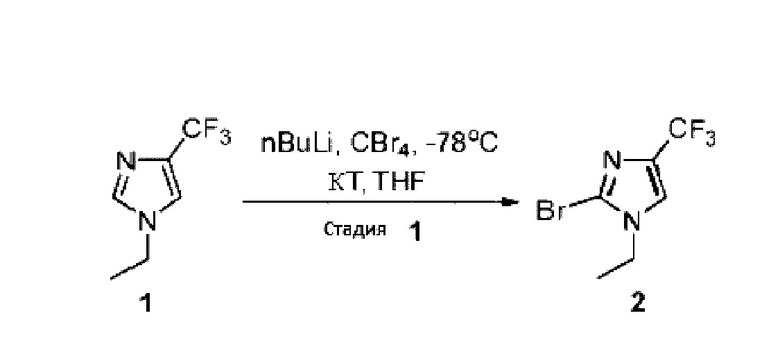
[0499] К перемешиваемому раствору 1-этил-4 (трифторметил)-1Н-имидазола 1 (12,0 г, 73,2 ммоль) в ТГФ (75 мл) при -78°С добавляли н-BuLi (1,6 М в гексане, 68,5 мл, 110 ммоль) по каплям. Полученную смесь перемешивали в течение 30 минут при -78°С, а затем обрабатывали раствором CBr4 (36,3 г, 109,75 ммоль) в сухом ТГФ (75 мл). Реакционной смеси давали нагреться до комнатной температуры и затем перемешивали в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили насыщенным раствором NH4Cl и экстрагировали ЕА (3 × 50 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на колонке с силикагелем, элюируя 0-10% ЕА в гексане, с получением указанного в заголовке соединения (8,00 г). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,09 (с, 1Н),4,01 (к, J=7,34 Гц, 2Н), 1,33 (т, J=7,34 Гц, 3H).
Стадия 2: Синтез 1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)-3-фторфенил)этан-1-она:
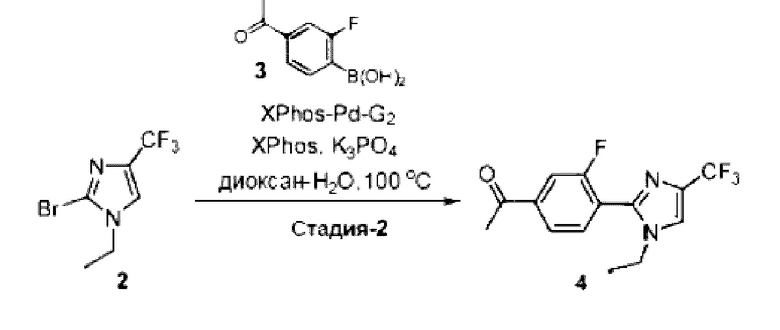
[0500] К перемешиваемому раствору 2-бром-1-этил-4-(трифторметил)-1H-имидазола-2 ммоль) в диоксане Н2О (8:2 мл) добавляли К3РО4 (3,94 г, 18,6 ммоль). и (4-ацетил-2-фторфенил)бороновую кислоту 3 (1,48 г, 8,18 ммоль). Полученную смесь дегазировали аргоном в течение 10 мин, а затем обрабатывали X-Phos (0,708 г, 1,49 ммоль) и X-Phos-Pd-G2 (0,292 г, 0,372 ммоль) в запаянной пробирке при комнатной температуре. Реакционную смесь дополнительно дегазировали аргоном в течение 10 мин, а затем нагревали при 100°С в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, элюируя 0-20% ЕА в гексане, с получением указанного в заголовке соединения (1,85 г). ЖХ-МС (Способ В) (ИЭР+): m/z 301,15 (М+Н)+; 1Н-ЯМР(400 МГц, ДМСО-d6) δ 8,14-8,20 (м. 1 Н), 7,93 (д, J=9,29 Гц, 2Н), 7,76 (т, J=7,34 Гц, 1Н), 3,93 (к, J=7,01 Гц, 2Н), 2,66 (с, 3H), 1,29 (т, J=7,34 Гц, 3H).
Стадия 3: Синтез (R)-1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ола:
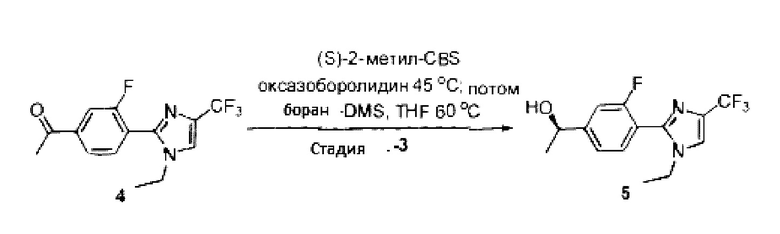
[0501] К перемешиваемому раствору 1-(4-(1 -этил-4-(трифторметил)-1Н-имидззол-2-ил)-3-фторфенил)этан-1-она 4 (2,30 г, 7,67 ммоль) в ТГФ (18 мл), добавляли (S)-2-метил-CBS- оксазоборолидин (0,42 мл, 1,53 ммоль) при комнатной температуре. Затем полученную смесь нагревали при 45°С в течение 1 часа К полученной реакционной смеси добавляли боран-DMS (1,09 мл, 11,5 ммоль), и смесь нагревали при 60°С в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, гасили метанолом (10 мл) и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на колонке с силикагелем, элюируя 0-30% ЕА в гексане, с получением указанного в заголовке соединения (2,10 г). ЖХ-МС (Способ В) (ИЭР+): m/z 303,00 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,10 (с, 1Н), 7,50 (т, J=7,82 Гц, 1Н), 7,30-7,37 (м, 2Н), 5,43 (д, J=4,40 Гц, 1Н), 4,81 (м, J=5,87 Гц, 1Н), 3,88 (к, J=7,17 Гц, 2Н), 1,37 (д, J=6,36 Гц, 3H), 1,27 (т, J=7,09 Гц, 3H).
Стадия 3: Синтез (S)-6-хлор-1-(1-(4-(1-этил-4-(трифторме1ил)-1Н-имидазол-2-ил)-1Н-пиразоло[3,4-d]пиримидина (I-27):
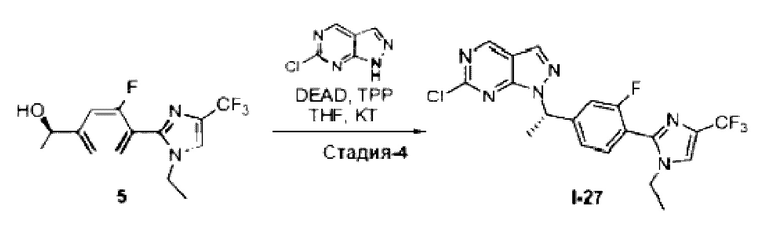
[0502] К перемешиваемому раствору (R)-1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)-3-фторфенил)этан-1-ола 5 (2,10 г, 6,95 ммоль) в ТГФ (12 мл) при 0°С, добавляли 6-хлор-1H-пиразоло[3,4-d6]пиримидин (1,07 г, 6,95 ммоль), DEAD (2,41 г, 13,9 ммоль) и ТРР (2,25 г, 13,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (10 мл) и экстрагировали ЕА (3 × 10 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный продукт очищали хроматографией на колонке с силикателем, используя 0-30% ЕА в гексане, с получением указанного в заголовке соединения (2,20 г). ЖХ-МС (Способ В) (ИЭР+): m/z 439,22 (М+Н)+.
Следующие общие промежуточные продукты были получены в соответствии с процедурой I-27 из соответствующих кетонов и восстанавливающих реагентов CBS:
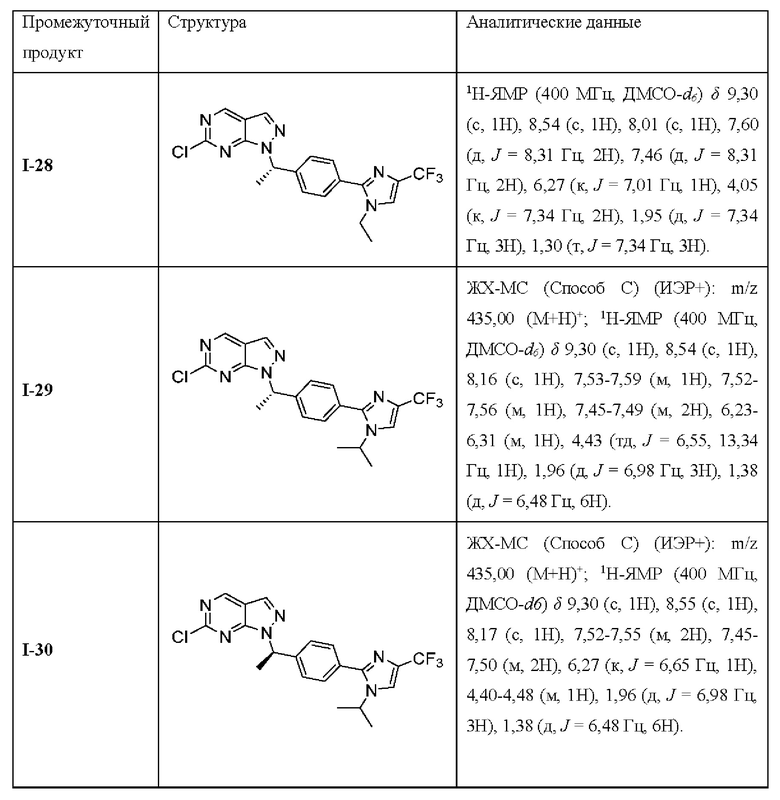
Получение общего промежуточного продукта I-31:
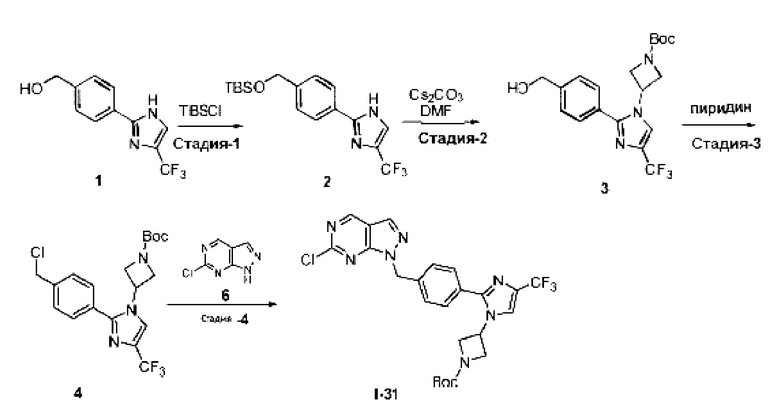
Стадия 1: Синтез 2-(4-(((Трет-бутилдиметилсилил)окси)метил)фенил)-4-(трифторметил)-1Н-имидазола:
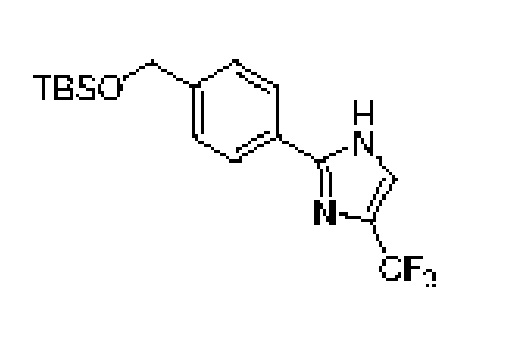
[0503] К раствору (4-(4-(трифторметил)-1Н-имидазол-1-ил)фенил) метанола (1,90 г, 7,85 ммоль), DMAP (95,8 мг, 0,79 ммоль) и TEA (1,19 г, 11,78 ммоль) в ТГФ (40 мл) при комнатной температуре добавляли TBSC1 (1,42 г, 9,42 ммоль) одной порцией. После добавления реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Затем реакционную смесь гасили водой (20 мл) и экстрагировали ЕА (150 мл × 3). Объединенный органический слой сушили над безводным Na2SO4 (50 г), фильтровали и упаривали в вакууме. Концентрированный остаток очищали хроматографией на силикагеле (ЕА:n-Нех = от 1:10 до 1:1) с получением 2,69 г указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 357 (М+Н)+; 1H-ЯМР (300 МГц CDCl2) δ 9,82 (уш. с, 1Н), 7,68 (д, J=8,7 Гц 2Н), 7,26-7,31 (м, 3H), 4,66 (с, 2Н), 0,84 (с, 9Н), 0,00 (с, 6Н).
Стадия 2: Синтез 3-(2-(4-(гидроксиметил)фенил)-4-(трифторметил)-1Н-нмндазол-1-ил)азетидин-1-карбоксилата:
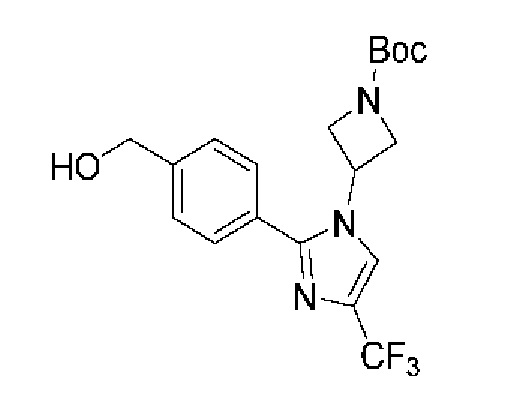
[0504] К раствору 2-(4-(((трет-бутилдиметилсилил)окси)метил)фенил)-4-(трифторметил)-1Н-имидазола (2,50 г, 7,02 ммоль) в ДМФА (40 мл) добавляли трет-бутил 3-йодоазетидин-1-карбоксилат (2,58 г, 9,13 ммоль) и Cs2CO5 (6,87 г, 21,06 ммоль). Реакционную смесь перемешивали при 120°С в течение 4 часов. Затем реакцию гасили водой (80 мл) и экстрагировали ЕА (150 мл × 3). Объединенный органический слой промывали водой (50 мл × 2), сушили безводным Na2SO4 (100 г), фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (DCM: МеОН = от 100:1 до 20:1) с получением 917 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 398 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 7,73 (с, 1Н), 7,37-7,44 (м, 4Н), 5,06 (м, 1H), 4,74 (с, 2Н), 4,37-4,43 (м, 2Н), 4,07-4,16 (м, 2Н), 2,86 (уш. с, 1H), 1,46 (с, 9Н).
Стадия 3: Синтез трет-Бутил 3-(2-(4-(хлорметил)фенил)-4-(трифторметил)-1Н-имидазол-1-ил)азетидин-1-карбоксилата:
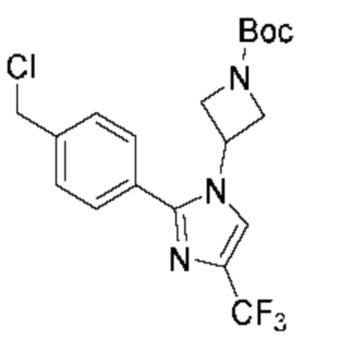
[0505] К раствору трет-бутил 3-(2-(4-(гидроксиметил)фенил)-4-(трифторметил)-1Н-имидазол-1-ил)азетидин-1-карбоксилата (0,92 г, 2,32 ммоль) в DCE (22 мл), добавляли пиридин (1,10 г, 13,92 ммоль) и SOCl2 (0,83 г, 6,95 ммоль). Затем реакционную смесь перемешивали при комнатной температуре в течение ночи перед упариванием досуха в вакууме. Полученный остаток очищали колоночной хроматографией (ЕА:n-Нех = от 1:10 до 1:4) с получением 480 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 416 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 7,75 (с, 1H), 7,52 (д, J=8,1 Гц, 2Н), 7,46 (д, J=8,1 Гц, 2Н), 5,07 (м, 1H), 4,64 (с, 2Н), 4,41-4,47 (т, J=9,0 Гц, 2Н), 4,09-4,14 (м, 2Н), 4,47 (с, 9Н).
Стадия 4: Синтез трет-Бутил 3-(2-(4-((6-хлор-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-4-(трифторметил)-1Н-имидазол-1-ил)азетидин-1-карбоксилата (I-31):
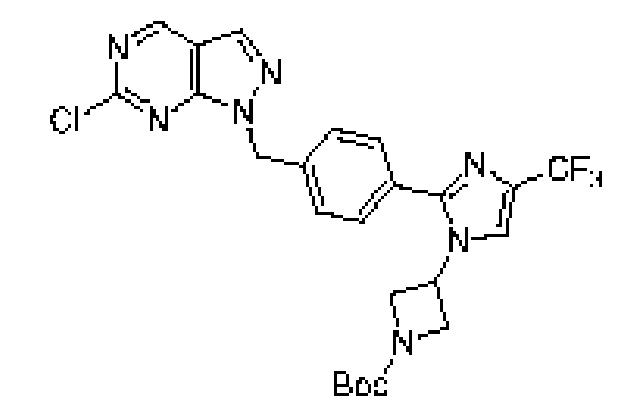
[0506] Раствор трет-бутил 3-(2-(4-(хлорметил)фенил)-4-(трифторметил)-1Н-имидазол-1-ил)азетидин-1-карбоксилата (0,45 г, 1,07 ммоль), 6-хлор-1Н-пиразоло[3,4-d]пиримидина (0,18 г, 1,18 ммоль) и K2CO3 (0,37 г, 2,68 ммоль) в ДМФА (13 мл) перемешивали при комнатной температуре в течение 3 часов. Затем смесь гасили водой (30 мл) и экстрагировали ЕА (100 мл × 3). Объединенный органический слой промывали водой (20 мл) и сушили над безводным Na2SO4 (50 г), фильтровали и упаривали е вакууме. Остаток очищали хроматографией на силикагеле (ЕА:Нех = от 1:10 до 1:0) с получением 280 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 534(М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 9,07 (с, 1Н), 8,19 (с, 1Н), 7,72 (с, 1Н), 7,41-7,49 (м. 4Н), 5,70 (с, 2Н), 5,01 (м. 1Н), 4,36-4,42 (т, J=8,7 Гц, 2Н), 4,06-4,13 (м, 2Н), 1,42 (с, 9Н).
Получение общего промежуточного продукта I-32:
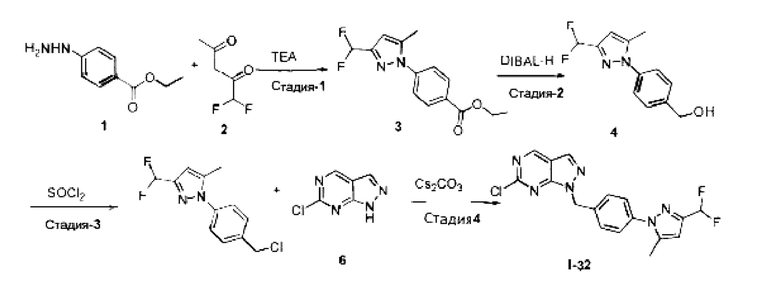
Стадия 1: Синтез Этил4-(3-(дифторметил)-5-метил-1Н-пиразол-1-ил)бензоата:
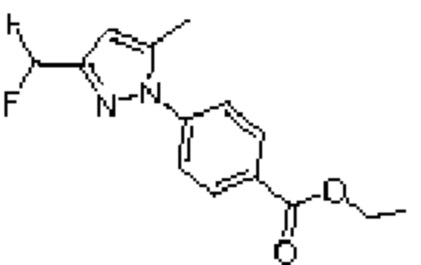
[0507] К раствору гидрохлоридной соли этил 4-гидразинилбензоата (300 мг, 1,5 ммоль) и 1,1-дифторпентан-2,4-диона (201,5 мг, 1,5 ммоль) в HFIP (2 мл) при 0°С добавляли раствор TEA (299,6 мг, 3,0 ммоль) в HFIP (1 мл) по каплям в течение 2 мин. После добавления реакционную смесь перемешивали при комнатной температуре в течение 2,5 часов. Затем ре акцию гасили водой (10 мл) и экстрагировали DCM (20 мл × 2). Объединенный органический слой промывали водой (10 мл), сушили над 6езводным Na2SO4 (20 г) и упаривали е вакууме. Остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 15:1 до 3:1) с получением 300 мг указанного в заголовке соединения. 1H-ЯМР (300 МГц, CDCl3) δ 8,17 (дд, J=6,9, 1,8 Гц, 2Н), 7,56 (дд, J=6,9, 1,8 Гц 2Н), 6,71 (т, J=54,9 Гц 1H), 6,48 (с, 1Н), 3,96 (с, 3H), 2,42 (с, 3H).
Стадия 2: Синтез (4-(3-(Дифторметил)-5-метил-1Н-пиразол-1-ил)фенил)метанола:
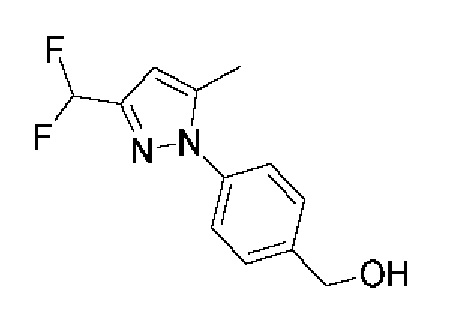
[0508] Указанное в заголовке соединение синтезировали согласно стадии 3 общего промежуточного продукта I-1. 1H-ЯМР (300 МГц, CDCl2) δ 7,49 (дд, J=6,3, 2,1 Гц, 2Н), 7,42 (дд, J=6,3, 2,1 Гц, 2Н), 6,70 (т, J=55,2 Гц, 1H), 6,44 (с, 1H), 4,78 (д, J=5,7 Гц 2Н), 2,35 (с, 3H), 1,90 (т, J=5,7 Гц 1H).
Стадия 3: Синтез 1-(4-(хлорметил)фенил)-3-(дифторметил)-5-метил-1Н-пиразола:
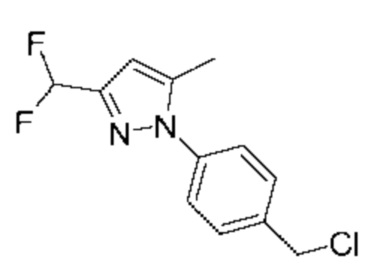
[0509] К раствору (4-(3-(дифторметил)-5-метил-1Н-пиразол-1-ил) фенил)метанола (290 мг, 1,2 ммоль) в DCE (10 мл) добавляли SOCl3 (426,4 мг, 3,6 ммоль) одной порцией. После добавления реакционную смесь перемешивали при 60°С в течение 1 часа, затем упаривали в вакууме с получением 300 мг неочищенного указанного в заголовке соединения. 1H-ЯМР (300 МГц, CDCl2) δ 7,52 (дд, J=6,3, 2,1 Гц, 2Н), 7,44 (дд, J=6,3, 2,1 Гц 2Н), 6,70 (т, J=54,9 Гц 1H), 6,44 (с, 1Н), 4,65 (с, 2Н), 2,37 (с, 3H).
Стадия 4: Синтез 6-Хлор-1-(4-(3-(дифторметил)-5-метил-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (I-32):
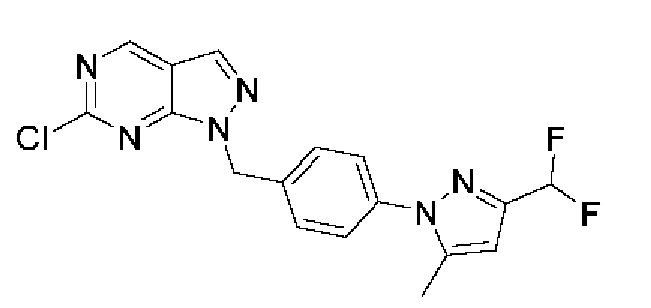
[0510] Раствор 1-(4-(хлорметил)фенил)-3-(дифторметил)-5-метил-1Н-пиразола (300 мг, 1,2 ммоль), 6-хлор-1Н-пиразоло[3,4-d]пиримидина (270,9 мг, 1,8 ммоль) и K2CO3 (484,6 мг, 351 ммоль) в ДМФА (12 мл) перемешивали при 90°С в течение 1,5 часов. Затем реакцию гасили водой (25 мл) и экстрагировали ЕА (50 мл × 2). Объединенный органический слой промывали водой (20 мл × 2), сушили над безводным Na2SO4 (30 г) и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 5:1 до 3:1) с получением 150 мг указанного в заголовке соединения с небольшим количеством региоизомера. 1H-ЯМР (300 МГц, CDCl3) δ 9,06 (с, 1Н),8,19 (с, 1Н),7,50 (да J=6,6, 1,8 Гц, 2Н), 7,41 (дд, J=6,6, 1,8 Гц, 2Н), 6,67 (т, J=54,9 Гц, 1Н), 6,42 (с, 1Н), 5,69 (с, 2Н), 2,33 (с, 3H).
Получение общего промежуточного продукта I-33 и I-34:
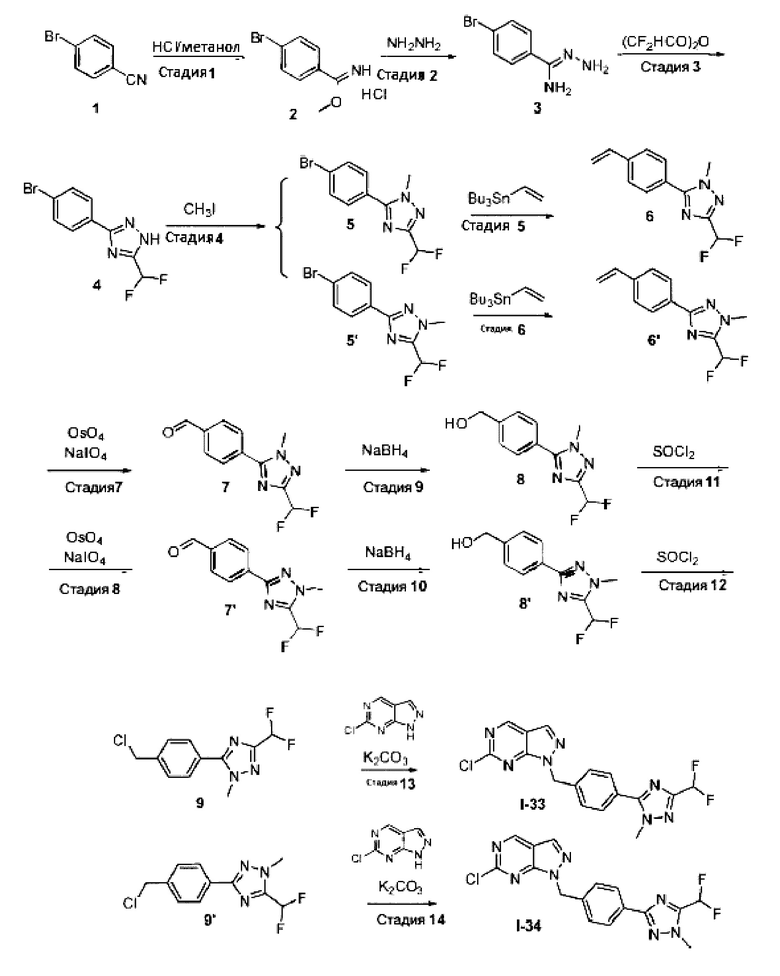
Стадия 1: Синтез гидрохлоридной соли этил-4-бромбензимидата:
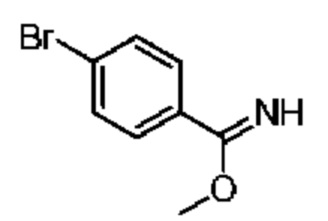
[0511] Газообразный HCl барботировали через интенсивно перемешиваемый раствор 4-бромбензонитрила (4,2 г, 23,07 ммоль) в безводном МеОН (40 мл) при 0°С в течение 3 часов. Затем смесь перемешивали при комнатной температуре в течение ночи. Полученную реакционную смесь выливали в Et2O (40 мл), и полученный твердый осадок собирали фильтрованием. Твердое вещество суспендировали в холодном DCM (80 мл) и нейтрализовали насыщенным водным раствором NaHCO3 (80 мл). После отделения органической фазы водную фазу экстрагировали DCM (40 мл × 2). Объединенную органическую фазу сушили сульфатом натрия (40 г), фильтровали и упаривали, получая 5,0 г указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 214 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 7,81 (уш. с, 1H), 7,51-7,68 (м, 4Н), 3,92 (с, 3H).
Стадия 2: Синтез 4-бромбензогидразонамида:
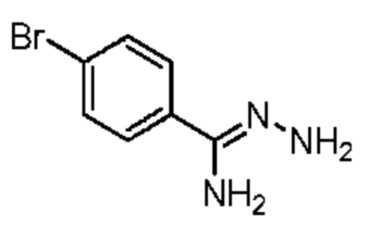
[0512] К раствору метил 4-бромбензимидата (4,6 г, 21,6 ммоль) в IPA (80 мл) при 0°С по каплям в течение 30 минут добавляли гидразингидрат (1,6 г, 25,9 ммоль) в IPA (20 мл). Реакционную смесь перемешивали на бане с ледяной водой в течение 1 ч, а затем нагревали до комнатной температуры в течение ночи. После завершения реакции, как показывает анализ ТСХ, реакционную смесь упаривали досуха. Остаток разбавляли Et2O (35 мл), и выпадало в осадок большое количество твердого вещества. Суспензию перемешивали при комнатной температуре в течение 1 ч, затем твердое вещество собирали фильтрованием, получая 3,8 г неочищенного указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 214 (М+Н)+; 1Н-ЯМР (300 МГц, ДМСО) δ 7,63 (д, J=8,7 Гц, 2Н), 7,52 (д, J=8,7 Гц, 2Н), 5,66 (уш. с, 2Н), 5,08 (уш. с, 2Н).
Стадия 3: Синтез 3-(4-Бромфенил)-5-(дифторметил)-1Н-1,2,4-триазола:
[0513] Раствор 2,2-дифторуксусного ангидрида (1,63 г, 9,38) в DCM (20 мл) добавляли по каплям в течение 5 минут к смеси 4-бромбензогидразонамида (2,0 г, 9,38 ммоль) и TEA (2,84 г, 28,14 ммоль) в DCM (70 мл) при 0°С. После добавления реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. После завершения реакции, как показывает анализ ТСХ, смесь гасили солевым раствором (80 мл) и экстрагировали DCM (80 мл × 2). Объединенный органический слой сушили сульфатом натрия (30 г), фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 8:1 до 4:1) с получением 1,2 г указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 274 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 7,85 (д, J=8,4 Гц, 2Н), 7,66 (д, J=8,4 Гц, 2Н), 6,81 (т, J=53,4 Гц, 1Н).
Стадия 4: Синтез 5-(4-Бромфенил)-3-(дифторметил)-1-метил-1Н-1,2,4-триазола и 3-(4-бромфенил)-5-(дифторметил)-1-метил-1Н-1,2,4-триазола:
[0514] К раствору 3-(4-бромфенил)-5-(дифторметил)-1Н-1,2,4-триазола (1,05 г, 3,85 ммоль) в ДМФА (25 мл) при 0°С добавляли гидрид натрия (200 мг, 5,0 ммоль) порциями в течение 5 мин. После перемешивания полученной смеси при 0°С в течение 20 минут по каплям в течение 1 минуты добавляли MeI (1,09 г, 7,69 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. После завершения реакции, как показывает анализ ТСХ, реакцию гасили ледяной водой (50 мл) и экстрагировали ЕА (70 мл × 2). Объединенный органический слой промывали водой (60 мл × 2), сушили сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 20:1 до 15:1) с получением 260 мг 5-(4-бромфенил)-3-(дифторметил)-1-метил-1Н-1,2,4-триазола и 750 мг 3-(4-бромфенил)-5-(дифторметил)-1-метил-1Н-1,2,4-триазола.
5-(4-бромфенил)-3-(дифторметил)-1-метил-1Н-1,274-триазол: ЖХ-МС (Способ А) (ИЭР+): m/z 288 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 7,70 (д, J=6,6, 1,8 Гц, 2Н), 7,59 (д, J=6,6, 1,8 Гц, 2Н), 6,73 (т, J=53,7 Гц, 1Н), 4,03 (с, 3H).
3-(4-бромфгпил)-5-(дифторметил)-1-метил-1Н-1,2,4-триазол: ЖХ-МС (Способ А) (ИЭР+): m/z 288 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 7,93 (д, J=8,7 Гц, 2Н), 7,58 (д, J=8,7 Гц, 2Н), 6,87 (т, J=53,7 Гц, 1H), 4,09 (с, 3H).
Стадия 5: Синтез 3-(Дифторметил)-1-метил-5-(4-винилфенил)-1Н-1,2,4-триазола:
[0515] В атмосфере азота раствор 5-(4-бромфенил)-3-(дифторметил)-1-метил-1Н-1,2,4-триазола (1,6 г, 5,57 ммоль) в толуоле (50 мл) обрабатывали трибутилвинилстаннаном (2,65 г, 8,36 ммоль) и Pd(PPh3)4 (644 мг, 0,56 ммоль) в одной порции. Реакционную смесь перемешивали при 90°С в течение 5 часов, затем гасили водой (50 мл) и экстрагировали ЕА (50 мл × 2). Объединенный органический слой промывали солевым раствором (60 мл), сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали флэш-хроматографией на силикагеле (РЕ:ЕА = 10:1) с получением 1,2 г указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 236 (М+Н)+; 1Н-ЯМР (300 МГц CDCl3) δ 7,68 (д, J=8,1 Гц 2Н), 7,56 (д, J=8,1 Гц, 2Н), 6,74 (т, J=53,7 Гц, 1H), 6,73 (м, 1H), 5,88 (д, J=17,4 Гц, 1H), 5,40 (д, J=11,1 Гц, 1H), 4,05 (с, 3H).
Стадия 6: Синтез 5-(Дифторметил)-1-метил-3-(4-винилфенил)-1Н-1,2,4-триазола:
[0516] Соединение синтезировали в соответствии с процедурой стадии 5 общего промежуточного продукта I-33. ЖХ-МС (Способ А) (ИЭР+): m/z 236 (М+Н)+; 1Н-ЯМР (300 МГц CDCl3) δ 8,02 (д, J=8,1 Гц 2Н), 7,51 (д,.J=8,1 Гц, 2Н), 6,90 (т, J=53,7 Гц, 1H), 6,78 (м, 1H), 5,83 (д, J=17,2 Гц, 1H), 5,31 (д, J=10,8 Гц, 1H), 4,10 (с, 3H).
Стадия 7: Синтез 4-(3-(Дифторметил)-1-метил-1Н-1,2,4-триазол-5-ил)бензальдегида:
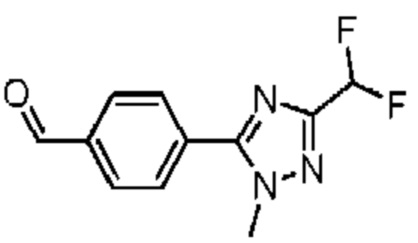
[0517] К раствору 3-(дифторметил) 1-метил-5-(4-винилфенил)-1Н-1,2,4-триазола (450 мг, 1,91 ммоль) в ТГФ (15 мл) и воде (8 мл), при комнатной температуре добавляли NaIO4 (1,23 г, 5,75 ммоль) и OsO4 (4,2 мг, 1 моль %) одной порцией. После завершения реакции, как показывает анализ ТСХ, реакционную смесь гасили насыщенным раствором хлорида аммония (20 мл) и экстрагировали ЕА (30 мл × 2). Объединенный органический слой промывали солевым раствором (40 мл), сушили над сульфатом натрия (20 г), фильтровали и упаривали с получением 450 мг неочищенного указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 238 (М+Н)+.
Стадия 8: Синтез 4-(5-(Дифторметил)-1-метил-1Н-1,2,4-триазол-3-ил)бензальдегида:
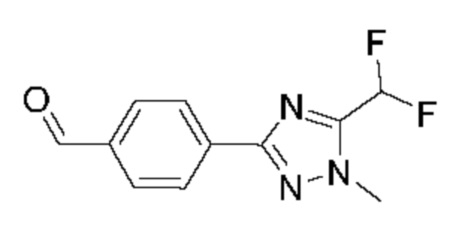
[0518] Соединение синтезировали в соответствии с процедурой стадии 7 общего промежуточного соединения I-33. ЖХ-МС (Способ А) (ИЭР+): m/z 238 (М+РР/. 1Н-ЯМР (300 МГц, CDCl3) δ 9,99 (с, 1H), 8,17 (д, J=8,1 Гц, 2Н), 7,90 (д, J=8,1 Гц, 2Н), 6,84 (т, J=52,5 Гц, 1H), 4,06 (с, 3H).
Стадия 9: Синтез (4-(3-(Дифторметил)-1-метил-1Н-1,2,4-триазол-5-ил)фенил)метанола:
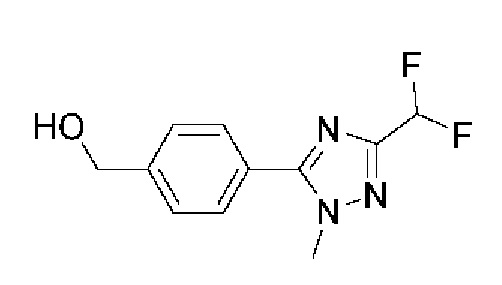
[0519] К раствору 4-(3-(дифторметил)-1-метил-1Н-1,2,4-триазол-5-ил)бензальдегида (450 мг, 1,90 ммоль) в ТГФ (15 мл) добавляли NaBH4 (94 мг, 2,47 ммоль) за одну порцию. После завершения реакции, как показывает анализ ТСХ, реакцию гасили ледяной водой (20 мл) и экстрагировали ЕА (40 мл × 2). Объединенный органический слой промывали солевым раствором (40 мл), сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ:ЕА = 3:2) с получением 340 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 240 (М+Н)+; 1Н-ЯМР (300МГц, CDCl3) δ 7,71 (д, J=8,1 Гц, 2Н), 7,54 (д, J=8,1 Гц, 2Н), 6,74 (т, J=53,7 Гц, 1Н), 4,81 (д, J=5,7 Гц, 2Н) 4,04 (с, 3H).
Стадия 10: Синтез (4-(5-(Дифторметил)-1-метил-1Н-1,2,4-триазол-3-ил)фенил)метанола:
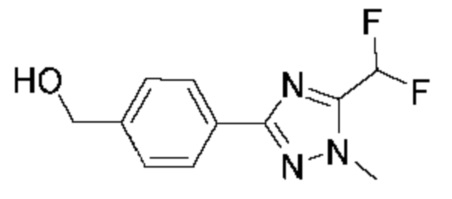
[0520] Соединение синтезировали в соответствии с процедурой стадии 9 общего промежуточного продукта I-14. ЖХ-МС (Способ А) (ИЭР+): m/z 240 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 8,06 (д, J=8,4 Гц, 2Н), 7,45 (д, J=8,4 Гц, 2Н), 7,89 (т, J=52,5 Гц, 1H), 4,75(д, J=6,0 Гц, 2Н) 4,13 (с, 3H).
Стадия 11: Синтез 5-(4-(хлорметил)фенил)-3-(дифторметил)-1-метил-1Н-1,2,4-триазола:
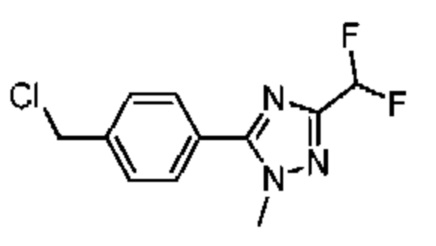
[0521] К раствору (4-(3-(дифторметил)-1-метил-1Н-1,2,4-триазол-5-ил)фенил)метанола (320 мг, 1,34 ммоль) в DCE (15 мл) добавляли SOCl2 (318 мг, 2,68 ммоль) одной порцией. Реакционную смесь перемешивали при 60°С в течение 1 часа. После завершения реакции, как показывает анализ ТСХ, реакционную смесь упаривали досуха с получением 340 мг неочищенного указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 258 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 7,72 (д, J=8,1 Гц 2Н), 7,57 (д, J=8,1 Гц, 2Н), 6,74 (т, J=53,7 Гц, 1H), 4,66 (с, 2Н), 4,05 (с, 3H).
Стадия 12: Синтез 3-(4-(хлорметил)фенил)-5-(дифторметил)-1-метил-1Н-1,2,4-триазола:
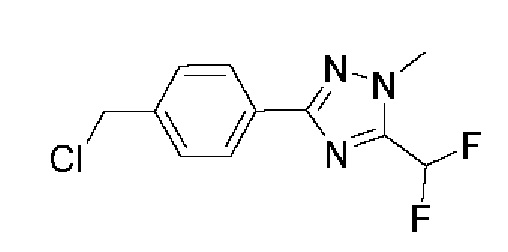
[0522] Соединение синтезировали в соответствии с процедурой стадии 11 общего промежуточного продукта I-33. ЖХ-МС (Способ А) (ИЭР+): m/z 258 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 8,06 (д, J=8,1 Гц, 2Н), 7,45 (д, J=8,1 Гц, 2Н), 6,89 (т, J=52,5 Гц, 1H), 4,63 (с, 2Н), 4,10 (с, 3H).
Стадия 13: Синтез 6-Хлор-1-(4-(3-(дифторметил)-1-метил-1Н-1,2,4-триазол-5-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина. (I-33):
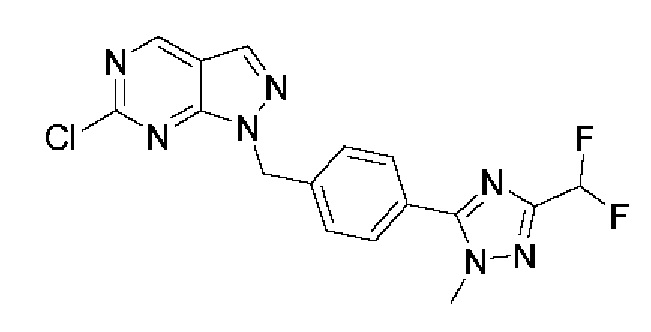
[0523] К раствору 5-(4-(хлорметил)фенил)-3-(дифторметил)-1-метил-1Н-1,2,4-триазола (340 мг, 1,32 ммоль) и 6-хлор-1Н-пиразоло[3,4-d]пиримидина (244 мг, 1,58 ммоль) в ДМФА (15 мл), добавляли Cs2CO3 (474 мг, 1,45 ммоль) одной порцией. Реакционную смесь перемешивали при 80 "С в течение 1 ч, затем охлаждали до комнатной температуры.м Затем реакцию гасили ледяной водой (40 мл) и затем экстрагировали ЕА (50 мл × 2). Объединенный органический слой промывали водой (40 мл × 3), сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 1:1 до 1:3) с получением 180 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 376 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 9,07 (с, 1H), 8,20 (с, 1H), 7,69 (д, J=8,4 Гц, 2Н), 7,52 (д, J=8,4 Гц, 2Н), 6,71 (т, J=53,7 Гц, 1Н), 5,72 (с, 2Н), 4,00 (с, 3H).
Стадия 14: Синтез 6-Хлор-1-(4-(5-(дифторметил)-1-метил-1Н-1,2,4-триазол-3-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина. (I-34):
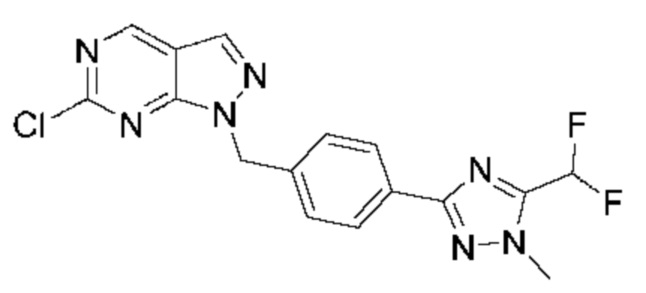
[0524] Соединение синтезировали в соответствии с процедурой стадии 13 общего промежуточного соединения I-33. ЖХ-МС (Способ А) (ИЭР+): m/z 376 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 9,05 (с, 1H), 8,18 (с, 1H), 8,03 (д, J=8,4 Гц, 2Н), 7,43 (д, J=8,4 Гц, 2Н), 6,86 (т, J=58,2 Гц, 1H), 5,67 (с, 2Н),4,10(с, 3H).
Получение общего промежуточного продукта I-35:
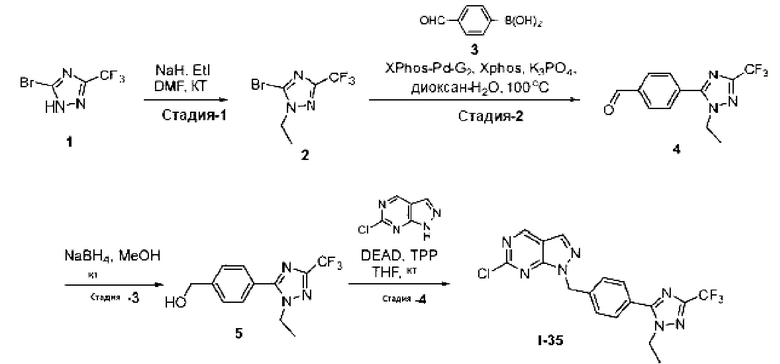
Стадия 1: Синтез 5-6ром-1-этил-3-(трифторметил)-1Н-1,2,4-триазола:
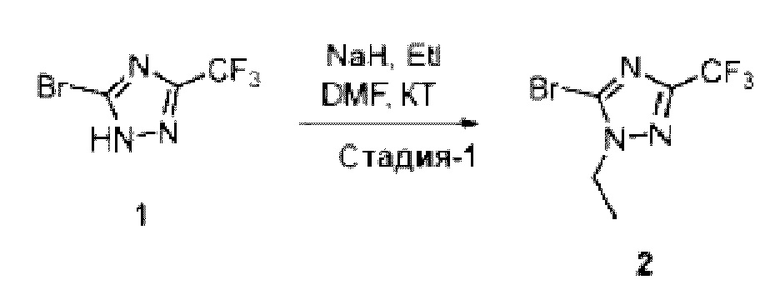
[0525] К охлажденному льдом раствору 5-6ром-3-(трифторметил)-1Н-1,2,4-триазола 1 (0,500 г, 2,31 ммоль) в ДМФА (10 мл) добавляли порционно гидрид натрия (0,138 г, 3,47 ммоль). Полученную смесь перемешивали в течение 15 минут, а затем добавляли этилиодид (0,360 мл, 0,708 г, 0,462 ммоль). Реакционной смеси давали нагреться до комнатной температуры и затем перемешивали в течение 16 часов. Заходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (20 мл) и экстрагировали ЕА (2 × 30 мл). Объединенный органический слой промывали водой (20 мл) и солевым раствором (20 мл), затем сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-5% ЕА в н-гексане, с получением указ энного в заголовке соединения (0,280 г). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 4,28 (к, J=7,01 Гц, 2Н), 1,40 (т, J=7,09 Гц, 3H).
Стадия 2: Синтез 4-(1-этил-3-(трифторметил)-1H-1,2,4-триазол-5-ил)бензальдегида:
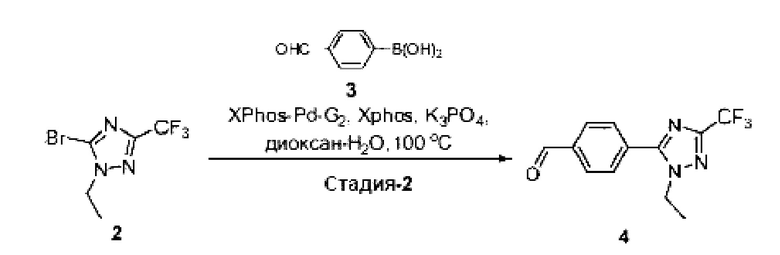
[0526] К перемешиваемому раствору 5-бром-1-этил-3-(трифторметил)-1Н-1,2,4-триазола 2 (0,250 г, 1,03 ммоль) в диоксане: Н2О (10:4 мл) 6 запаянной пробирке добавляли K3PO4 (0,436 г, 2,07 ммоль) и (4-формилфенил)бороновую кислоту 3 (0,200 г, 1,34 ммоль). Полученную смесь дегазиравали аргоном в течение 10 минут, а затем добавляли X-Phos (0,049 г, 0,102 ммоль) и X-Phos-Pd-G2 (0,040 г, 0,051 ммоль) при комнатной температуре. Реакционную смесь дополнительно дегазировали аргоном в течение 10 мин, а затем нагревали при 100°С в течение 2 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь оклаждапи до комнатной температурь; гасили водой (20 мл) и экстрагиров апи ЕА (3 × 30 мл). Органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, элюируя 0-25% ЕА в гексане, с получением указанного в заголовке соединения (0,220 г). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 10,13 (с, 1Н), 3,11 (д, J=6,35 Гц 2Н), 3,00 (д, J=7,34 Гц. 2Н), 4,33 (к, J=6,35 Гц 2Н), 1,44 (т, J=6,35 Гц, 3H).
Стадия 3: Синтез (4-(1-этил-3 (трифторметил)-1Н-1,2,4-триазол-5-ил) фенил)метанола:
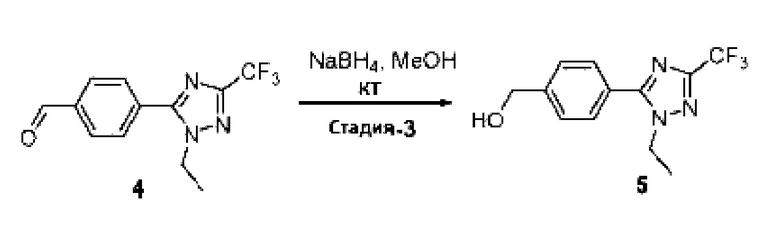
[0527] К перемешиваемому раствору 4-(1-этил-3-(трифторметил)-1Н-1,2,4-триазол-5-ил)бензальдегида 4 (0,200 г, 0,743 ммоль) в метаноле (5 мл) добавляли натрий боргидрид (0,056 г, 1,49 ммоль) порциями Реакционной смеси давали нагреться до комнатной температуры и дополнительно перемешивали в течение 2 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, гасили водой (20 мл) и экстрагировали ЕА (3 × 30 мл). Органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, элюируя 0-40% ЕА в гексане, с получением указанного в заголовке соединения (0,150 г). ЖХ-МС (Способ В) (ИЭР+): m/z 271,40 (М+Н)+; 1Н-ЯМР (400 МГц ДМСО-d6) δ 7,72 (д, J=3,31 Гц 2Н), 7,53 (д. J=7,82 Гц 2Н), 5,33 (т, J=5,62 Гц 1Н), 4,61 (д, J=5,37 Гц 2Н), 4,34 (к, J=7,34 Гц 2Н), 1,42 (т, J=7,34 Гц 3H).
Стадия 4: Синтез 6-хлор-1-(4-(1-этнл-3-(трифторметил)-1Н,-1,2,4-триазол-5-ил)бензил)-1Н-пиразоло [3,4-d]пиримндина (I-35):
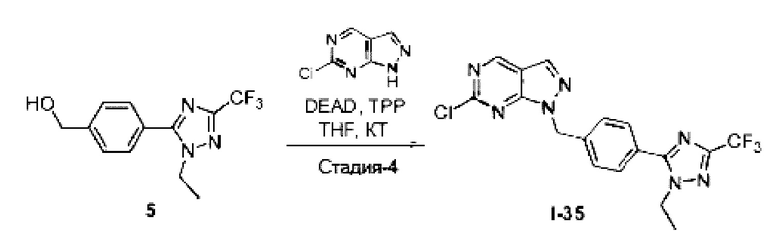
[0528] К перемешиваемому раствору (4-(1-этил-3-(трифторметил)-1Н-1,2,4-триазол-5-ил)фенил)метанола 5 (0,130 г, 0,479 ммоль) в ТГФ (5 мл) при 0°С добавляли 6-хлор-1Н-пиразоло[3,4-d]пиримидин (0,066 г, 0,43 ммоль), DEAD (0,163 г, 0,959 ммоль) и ТРР (0,246 г, 0,959 ммоль). Реакционной смеси давали нагреться до комнатной температуры и затем перемешивали в течение 6 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (20 мл) и экстрагировали ЕА (3 × 30 мл). Объединенный органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-30% ЕА в гексане в качестве злюента, с получением указанного в заголовке соединения (0,100 г). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,33 (с, 1Н), 3,53 (с, 1 Н), 7,73 (д, J=8,31 Гц, 2Н), 7,45 (д, J=7,83 Гц, 2Н), 5,77 (с, 2Н), 4,30 (к, J=7,17 Гц, 2Н), 1,39 (т, J=7,34 Гц 3H).
Следующие промежуточные продукты были получены из соответствующих гетероциклов и алкилирующих реагентов в соответствии со способом получения для I-35:
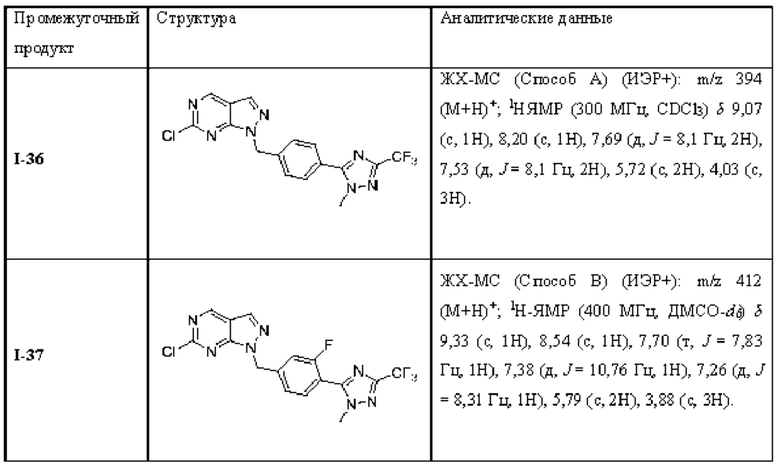
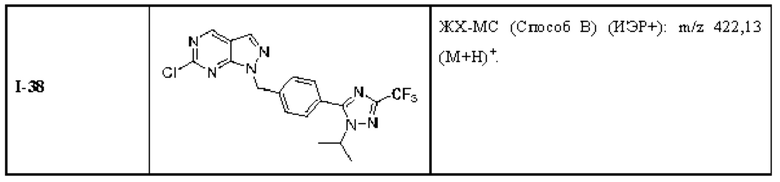
Получение общего промежуточного продукта I-39:
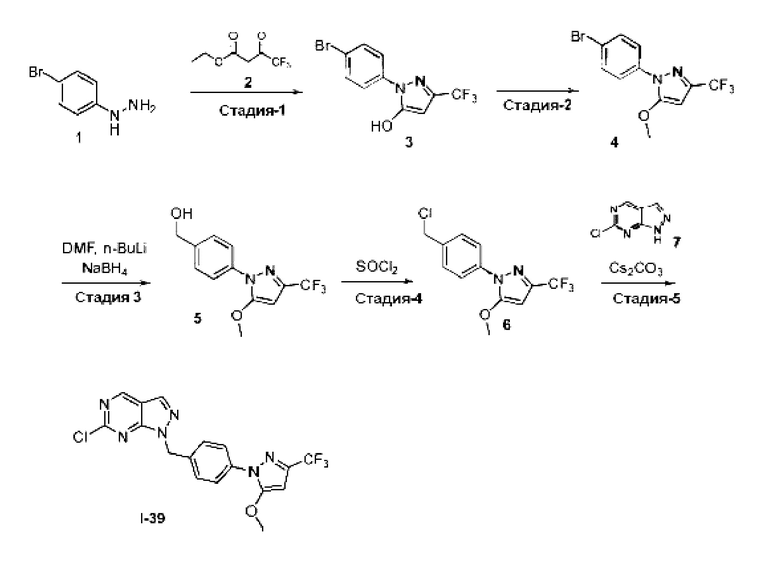
Стадия 1: Синтез 1-(4-бромфенил)-3-(трифторметил)-1Н-пиразол-5-ола:
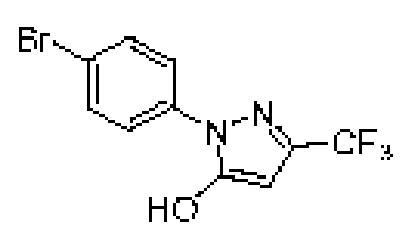
[0529] К раствору гидрохлорида (4-бромфенил)гидразина (4,9 г, 22 ммоль) в этаноле (60 мл) при комнатной температуре добавляли гидроксид натрия (898 мг, 22,5 ммоль) одной порцией. Полученную смесь перемешивали при комнатной температуре в течение 30 мин, затем к реакционной смеси одной порцией добавляли раствор этил 4,4,4-трифтор-3-оксобутаноата (4,9 г, 27 ммоль) в этаноле (10 мл).. Реакционную смесь кипятили с обратным холодильником и перемешивали в течение ночи. После завершения реакции, как показывает анализ ТСХ, реакцию охлаждали до комнатной температуры и гасили водой (150 мл). Полученную смесь подкисляли до рН 5 разбавленным раствором HCl экстрагировали ЕА (100 мл × 3). Объединенный органический слой сушили над сульфатом натрия (50 г), фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (РЕ:ЭА = от 30:1 до 6:1) с получением 6,2 г указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 307 (М+Н)+; 1H-ЯМР (300 МГц, CD3OD) δ 7,62-7,74 (м, 4Н), 5,84 (с, 1H).
Стадия 2: Синтез 1-(4-бромфенил)-5-метокси-3-(трифторметил)-1Н-пиразола:
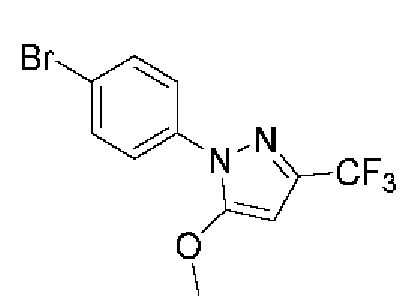
[0530] К раствору метил 1-(4-бромфенил)-3-(трифторметил)-1Н-пиразол-5-ола (6,2 г, 20 ммоль) в ДМФА (65 мл) при 0°С был добавлен гидрид натрия (60% дисперсия, 973 мг, 0,024 ммоль) порциями в течение 5 мин. После перемешивания смеси при 0°С в течение 30 минут к реакционной смеси в течение 2 минут по каплям добавляли йодметан (4,26 г, 0,03 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем гасили смесью ледяной воды (200 мл) и экстрагировали ЕА (100 мл × 3). Органическую фазу промывали водой (100 мл × 2), сушили над сульфатом натрия (40 г), фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (от РЕ к РЕ:ЕА = 100:1) с получением 4,4 г указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 321 (М+Н)+; 1Н-ЯМР (300 МГц, CD3OD) δ 7,55-7,63 (м, 4Н), 5,95 (с, 1Н), 4,00 (с, 3H).
Стадия 3: Синтез 4-(5-Метокси-3-(трифторметил)-1Н-пиразол-1-ил)бензальдегида:
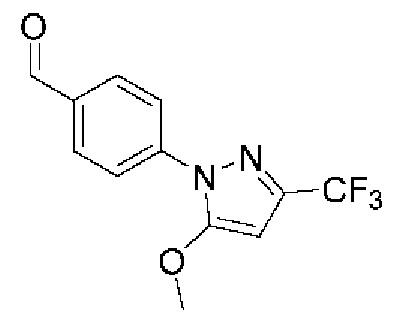
[0531] К раствору 1-(4-бромфенил)-5-метокси-3-(трифторметил)-1Н-пиразола (1 г, 3,13 ммоль) в ТГФ (10 мл) при -78°С добавляли H-BuLi (1,5 мл, 3,76 ммоль) по каплям в течение 15 мин. После перемешивания реакционной смеси при -78°С в течение 1 часа к реакционной смеси в течение 5 минут по каплям добавляли безводный ДМФА (0,48 мл, 6,26 ммоль). После добавления реакционную смесь перемешивали при -78°С в течение дополнительных 30 мин, а затем нагревали до комнатной температуры. После завершения реакции, как показывает анализ ТСХ, реакцию гасят ледяной водой (20 мл). Полученную смесь экстрагировали ЕА (30 мл × 2), сушили над сульфатом натрия (30 г), фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 30:1 до 20:1) с получением 570 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 271 (М+Н)+; 1H-ЯМР (300 МГц, CD3OD) δ 10,04 (с, 1H), 7,97 (д, J=6,6 Гц, 4Н), 3,99 (с, 1H), 4,05 (с, 3H).
Стадия 4: Синтез (4-(5-Метокси-3-(трифторметил)-1Н-пиразол-1-ил)фенил)метанола:
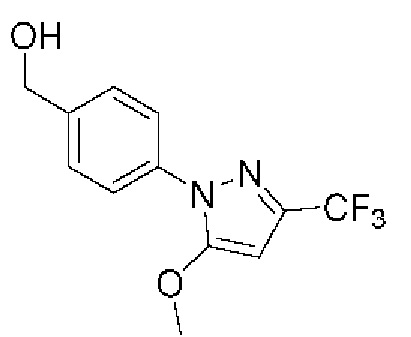
[0532] К раствору 4-(5-метокси-3-(трифторметил)-1Н-пиразол-1-ил)бензальдегида (570 мг, 2,11 ммоль) в ТГФ (10 мл) при комнатной температуре добавляли NaBH4 (80 мг, 2,11 ммоль). ммоль) в одной порции. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После завершения реакции, как показывает анализ ТСХ, реакцию гасили водой (20 мл) и экстрагировали ЕА (20 мл × 2). Объединенный органический слой сушили над сульфатом натрия (20 г), фильтровали и упаривали в вакууме с получением 620 мг указанного в заголовке соединения, которое использовали без очистки в последующих стадиях. ЖХ-МС (Способ А) (ИЭР+): m/z 273 (М+Н)+; 1Н-ЯМР (300 МГц, CD3OD) δ 7,68 (д, J=8,4 Гц, 2Н), 7,45 (д, J=8,4 Гц, 2Н), 5,96 (с, 1H), 4,75 (д, J=5,1 Гц, 2Н), 3,99 (с, 3H), 1,58 (с, 1H).
Стадия 5: Синтез 1-(4-(хлорметил)фенил)-5-метокси-3-(трифторметил)-1Н-пиразола:
[0533] К раствору (4-(5-метокси-3-(трифторметил)-1Н-пиразол-1-ил)фенил)метанола (570 мг, 2,10 ммоль) в DCE (20 мл) добавляли SOCl2 (748 мг, 6,29 ммоль) одной порцией. После добавления реакционную смесь перемешивали при 50°С в течение 20 мин. После завершения реакции, как показывает анализ ТСХ, реакционную смесь упаривали досуха в вакууме с получением 500 мг неочищенного указанного в заголовке соединения, которое использовали без очистки в последующих стадиях. ЖХ-МС (Способ А) (ИЭР+): m/z 291 (М+Н)+.
Стадия 6: Синтез 6-Хлор-1-(4-(5-метокси-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (I-39):
[0534] К раствору 1-(4-(хлорметил)фенил)-5-метокси-3-(трифторметил)-1Н-пиразола (520 мг, 1,79 ммоль) в ДМФА (25 мл) добавляли 6-хлор-1Н-пиразоло[3,4-d]пиримидин (276 мг, 1,79 ммоль) и карбонат калия (741 мг, 5,37 ммоль). После добавления реакционную смесь перемешивали при 70°С в течение 1 часа. После завершения реакции, как показывает анализ ТСХ, реакцию гасили водой (30 мл) и экстрагировали ЕА (30 мл × 2). Объединенную органическую фазу промывали водой и солевым раствором, сушили над сульфатом натрия (20 г), фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 10:1 до 5:1) с получением 330 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 409 (М+Н)+; 1Н-ЯМР (300 МГц, CD3OD) δ 9,05 (с, 1H), 8,17 (с, 1H), 7,67 (д, J=8,4 Гц, 2Н), 7,47 (д, J=8,4 Гц, 2Н), 5,93 (с, 1H), 5,66 (с, 2Н), 3,99 (с, 3H).
Следующие промежуточные продукты были получены из соответствующих гетероциклов и алкилирующих реагентов в соответствии со способом получения для I-39:
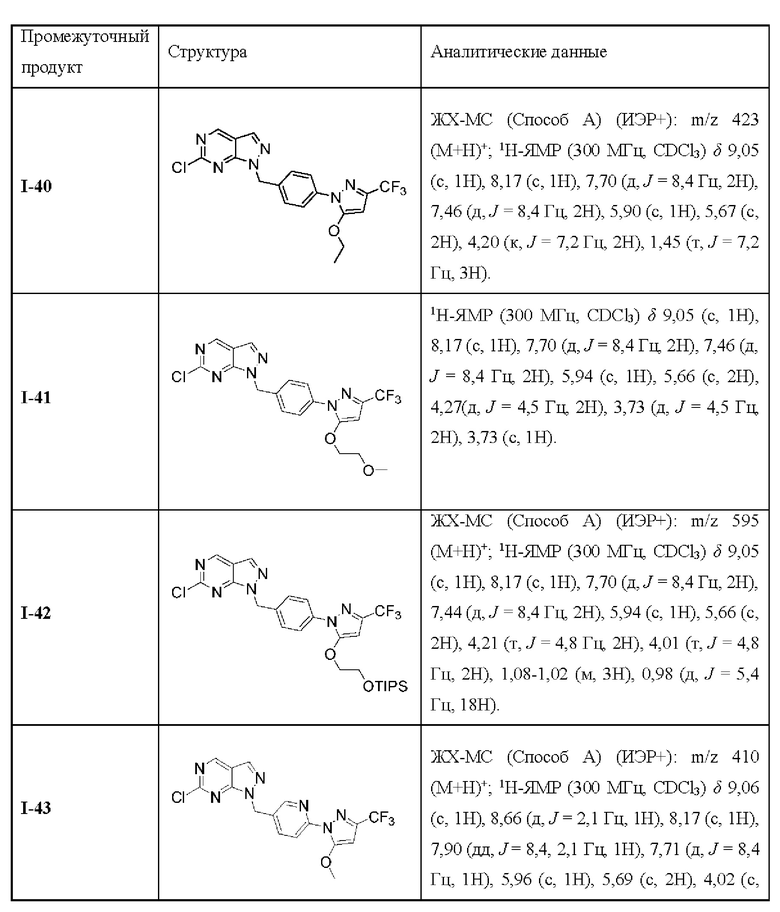
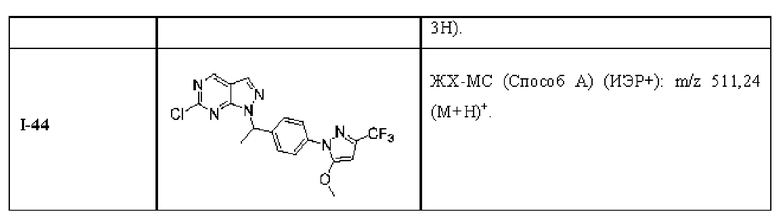
Получение общего промежуточного продукта I-45:
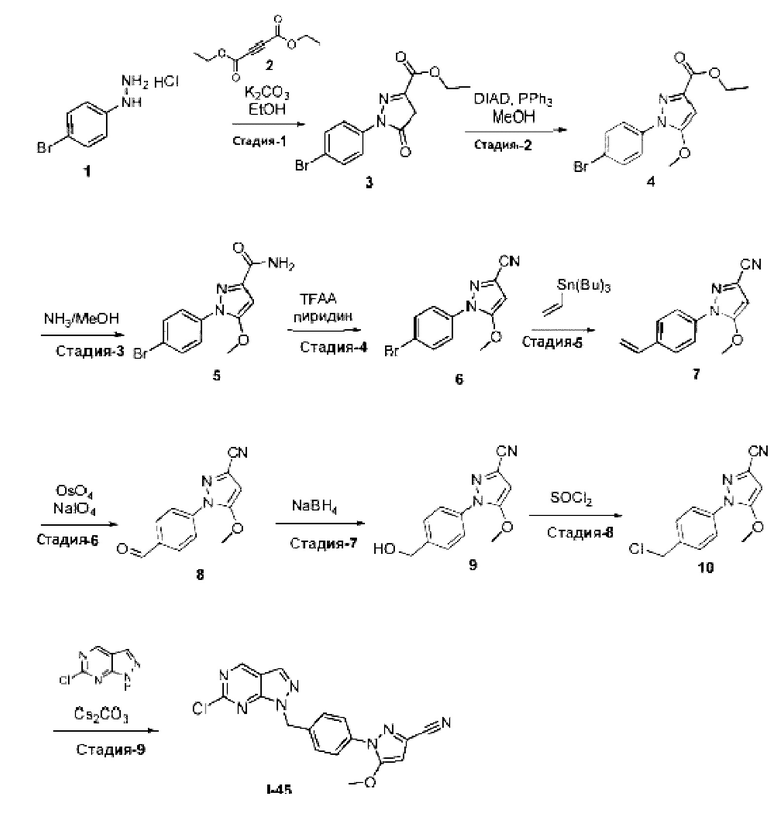
Стадня 1: Синтез этил 1-(4-бромфенил)-5-окео-4,5-дигидро-1Н-пиразол-3-карбоксилата:
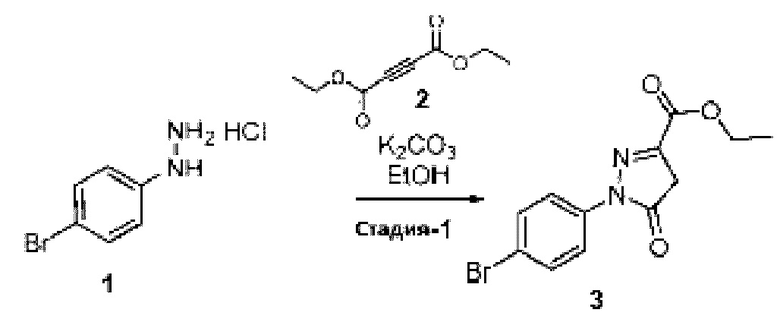
[0535] К раствору гидрохлорида (4-бромфенил)гидразина (1,0 г, 4,5 ммоль) в этаноле (40 мл) добавляли карбонат калия (1,23 г, 8,94 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 30 минут к реакционной смеси по каплям в течение 5 минут добавляли диэтил бут-2-индиоат (761 мг, 4,47 ммоль) в этаноле (10 мл). После перемешивания реакционной смеси с обратным холодильником в течение 4 ч реакцию гасили водой (50 мл) и доводили до рН 5 разбавленным раствором соляной кислоты. Полученную смесь экстрагировали ЕА (40 мл × 3). Объединенный органический слой сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА = от 10/1 до 5:1) с получением 320 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 311,313 (М+Н)+, 1Н-ЯМР (300 МГц, CD3OD) δ 7,74 (д, J=9,0 Гц, 2Н), 7,71 (д, J=9,0 Гц,2Н), 6,01 (с, 1Н), 4,30-4,33 (к, J=6,9 Гц 2Н), 1,32-1,33 (т, J=6,9 Гц 3H).
Стадия 2: Синтез этил 1-(4-бромфенил)-5-метокси-1Н-пиразол-3-карбоксилата:
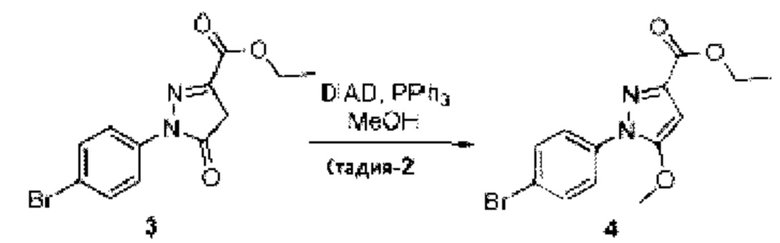
[0536] К смеси этил 1-(4-бромфенил)-5-оксо-4,5-дигидро-1Н-пиразол-3-карбоксилата (720 мг, 2,32 ммоль), трифенилфосфина (913 мг, 3,43 ммоль) и метанола (96 мг, 3,02 ммоль) в толуоле (40 мл) при 0°С, добавляли DIAD (704 мг, 3,43 ммоль) по каплям в течение 5 минут. После перемешивания смеси при 0°С в течение 30 минут реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение ночи. После завершения реакции, как показывает анализ ТСХ, смесь гасили водой (20 мл) и экстрагировали ЕА (30 мл × 3). Объединенный органический слой сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА = от 20/1 до 10/1) с получением 650 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 325, 327 (М+Н)+; 1Н-ЯМР (300 МГц CDCl3) δ 7,65 (д, J=3,7 Гц 2Н), 7,56 (д, J=8,7 Гц 2Н), 6,22 (с, 1Н), 4,39-4,46 (к, J=7,2 Гц 2Н), 3,93 (с, 3H), 1,39-1,44 (т, J=7,2 Гц 3H).
Стадия 3: Синтез метил 1-(4-6ромфенил)-5-метокси-1Н-пиразол-3-карбоксамида:
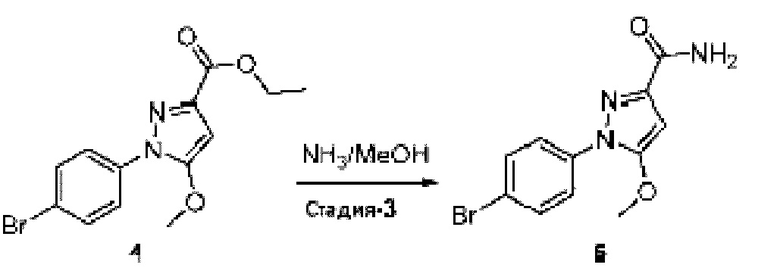
[0537] В герметичной пробирке раствор этил 1-(4-бромфенил)-5-метокси-1Н-пиразол-3-карбоксилата(650 мг, 2,01 ммоль) в насыщенном растворе аммиака в метаноле (30 мл) перемешивали при 60°С в течение 7 ч. После завершения реакции, как показывает анализ ТСХ, смесь гасили водой (30 мл) и экстрагировали ЕА (20 мл × 3). Объединенный органический слой сушили над сульфатом натрия (30 г), фильтровали и упаривали Остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА = от 5/1 до 0/1) с получением 405 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 296, 292 (М+Н)+; 1Н-ЯМР (300 МГц. CDCl3) δ 7,55-7,65 (м, 4Н), 6,32 (с, 1Н), 6,26 (с, 1Н), 5,40 (с, 1Н),3,99 (с, 3H).
Стадия 4: Синтез 1-(4-бромфенил)-5-метокси-1Н-пиразол-3-карбонитрила:
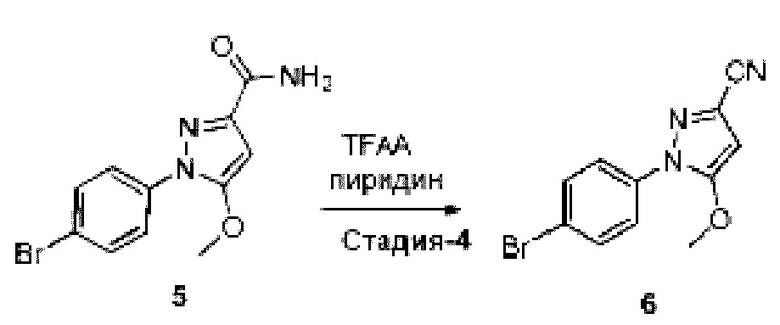
[0538] К раствору метил 1-(4-бромфенил)-5-метокси-1Н-пиразол-3-карбоксамида (400 мг, 1,36 ммоль) в DCM (40 мл) и пиридине (2 мл) при 0°С добавляли ангидрид трифторуксусной кислоты (1,6 мл) по каплям в течение 5 мин. Реакционную смесь перемешивали при 0°С в течение 1 часа После завершения реакции, как показывает анализ ТСХ, смесь гасили насыщенным водным раствором NH4Cl (40 мл) и экстрагировали DCM (40 мл × 3). Органический слой промывали в од ой (20 мл) и солевым раствором(20 мл) и сушили над сульфатом натрия (40 г), фильтровали и упаривали. Полученный остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА = от 20/1 до 10/1) с получением 295 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 278, 280 (М+Н)+; 1Н-ЯМР (300 МГц CDCl3) δ 7,59 (с,4Н), 6,06(с,1Н),4,00(с, 3H).
Стадия 5: Синтез 5-метокси-1-(4-винилфенил)-1Н-пиразол-3-карбонитрила:
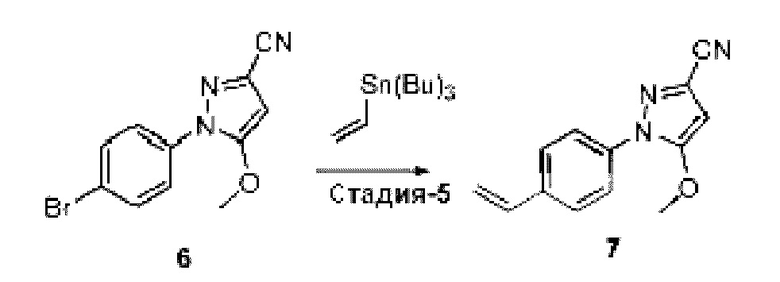
[0539] К раствору 1-(4-бромфенил)-5-метокси-1Н-пиразол-3-карбонитрила (290 мг, 1,05 ммоль) в толуоле (8 мл) добавляли три6утил(винил)станнан (498 мг, 1,57 ммоль) и Pd(Ph3)4 (121 мг, 0,11 ммоль). Реакционную смесь перемешивали при 90°С в течение 6 часов. После завершения реакции, как показывает анализ ЖХ-МС, смесь гасили в одой (10 мл) и экстрагировали ЕА (10 мл × 2). Объединенный органический спой сушили над сульфатом натрия (20 г), фильтровали и упаривали. Полученный остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА = от 20/1 до 10/1) с получением 200 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 226 (М+Н)+; 1Н-ЯМР (300 МГц CDCl3) δ 7,64 (д, J=8,7 Гц2Н), 7,49 (д, J=8,7 Гц2Н), 6,74 (дд, J=17,7 Гц J=10,8 Гц 1Н),6,0б (с, 1Н), 5,80 (д J=17,7 Гц 1Н), 5,33 (д, J=10,8 Гц 1Н), 3,99 (с, 3H).
Стадия 6: Синтез (4-формилфенил)-5-метокси-1Н-пиразол-3-карбонитрила:
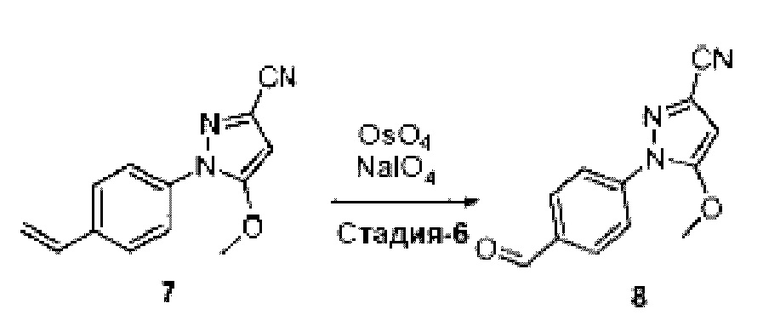
[0540] К раствору 5-метокси-1-(4-винилфенил)-1Н-пиразол-3-карбонитрила (20 мг, 0,89 ммоль) в ТГФ/Н2О (10 мл/5 мл) добавляли NaIO4 (570 мг, 2,67 ммоль). ммоль) и OsO4 (22 мг, 0,09 ммоль) за одну порцию. Реакционную смесь перемешивали при комнатной температуре 30 мин. После завершения реакции, как показывает анализ ТСХ смесь гасили водным раствором NH4Cl (10 мл) и экстрагировал и ЕА (20 мл × 2). Объединенные органические соединения сушили над сульфатом натрия (20 г), фильтровали и упаривали с получением 210 мг неочищенного указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 228 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 10,05 (с, 1Н), 7,84-8,05 (м. 4Н), 6,01 (с, 1Н), 3,99 (с, 3H).
Стадия 7: Синтез 1-(4-(гидроксиметил)фенил)-5-метокси-1Н-пиразол-3-карбонитрила:
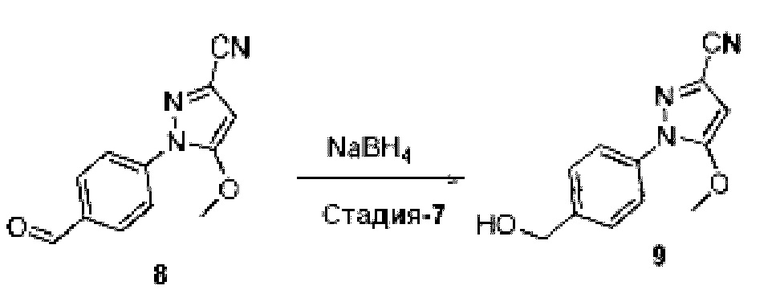
[0541] К раствору 1-(4-формилфенил)-5-метокси-1Н-пиразол-3-карбонитрила (210 мг, 0,93 ммоль) в ТГФ (30 мл) добавляли NaBH4 (46 мг, 1,2 ммоль) порциями через 5 мин. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа После завершения реакции, как показывает анализ ТСХ смесь гасили водой (20 мл) и экстрагировали ЕА (30 мл × 2). Объединенный органический слой сушили над сульфатом натрия (20 г) и упаривали Остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА = от 5/1 до 2/1) с получением 170 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 230 (М+Н)+; 1H-HMP (300 МГц, CDCl3) δ 7,67 (д, J=8,7 Гц 2Н), 7,47 (д, J=8,7 Гц 2Н), 6,07 (с, 1Н), 4,76 (д. J=6 Гц 2Н), 3,99 (с, 3H), 1,75 (т, J=6 Гц 1Н).
Стадия 8: Синтез 1-(4-(хлорметил)фенил)-5-метокси-1Н-пиразол-3-карбоннтрила:
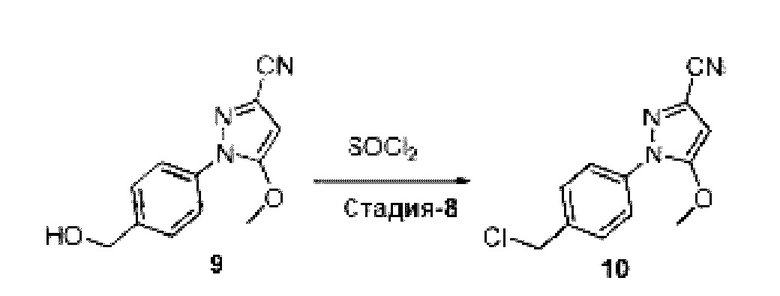
[0542] К раствору 1-(4-(гидроксиметил)фенил)-5-метокси-1Н-пиразол-3-карбонитрила (170 мг, 0,74 ммоль) е DCE (10 мл) добавляли SOCl2 (176 мг, 1,43 ммоль) одной порцией, и реакционную смесь перемешивали при 50°С в течение 1 часа. После завершения реакции, как показывает анализ ТСХ, реакционную смесь упаривали досуха с получением 205 мг неочищенного указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 248 (М+Н)+-; 1Н-ЯМР (300 МГц. CDCl3) δ 7,70 (д, J=8,4 Гц, 2Н), 7,49 (Д, J=8,4 Гц, 2Н), 6,07 (с, 1Н), 4,62 (с, 2Н), 4,00 (с, 3H).
Стадия 9: Синтез 1-(4-((б-хлор-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-5-метокси-1Н-пиразол-3-карбонитрила (I-45):
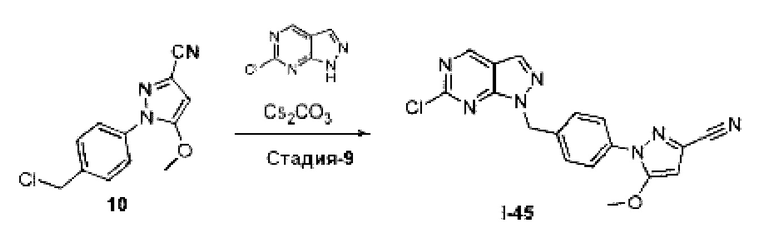
[0543] К раствору 1-(4-(хлорметил)фенил)-5-метокси-1Н-пиразол-3-карбонитрила (205 мг, 0,83 ммоль) в ДМФА (5 мл) добавляли 6-хлор-1Н-пиразоло[3,4-d]пиримидин (153 мг, 1,0 ммоль) и карбонат калия (812 мг, 2,49 ммоль) в одной порции. После добавления реакционную смесь перемешивали при 70°С в течение 1 часа. После того, как реакция была завершена, как показал анализ ТСХ, смесь гасили водой (10 мл) и экстрагировали ЕА (30 мл × 2). Объединенный органический слой промывали водой (20 мл) и солевым раствором (10 мл), сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА = от 10/1 до 5/1) с получением 57 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 366 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 9,05 (с, 1Н), 8,18 (с, 1Н), 7,66 (д. J=8,4 Гц 2Н), 7,48 (д, 1=8,4 Гц 2Н), 6,04 (с, 1Н), 5,67 (с, 2Н), 3,97 (с, 3H).
Получение общего промежуточного продукта I-46:
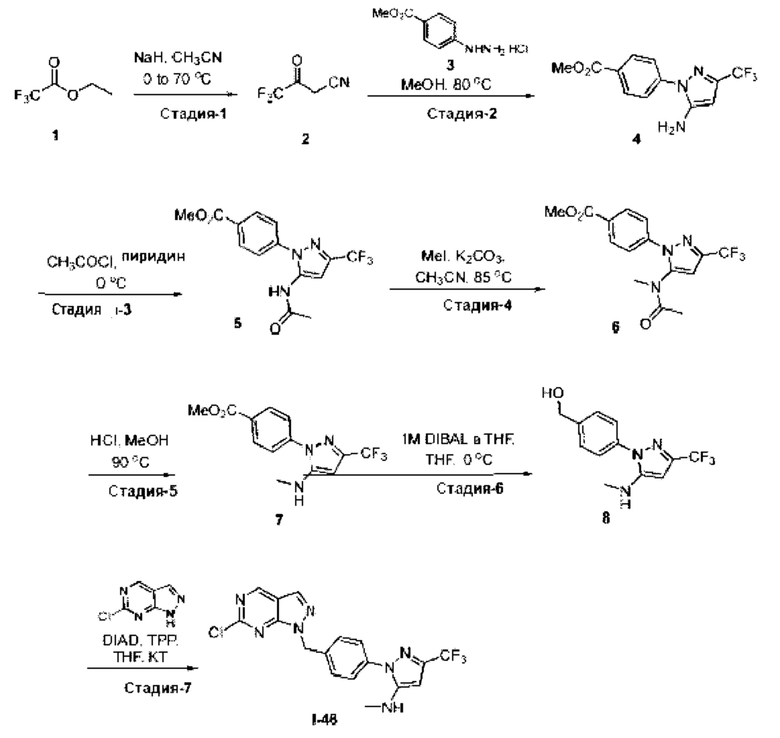
Стадия 1: Синтез 4,4,4-трифтор-3-оксобутаннитрила:
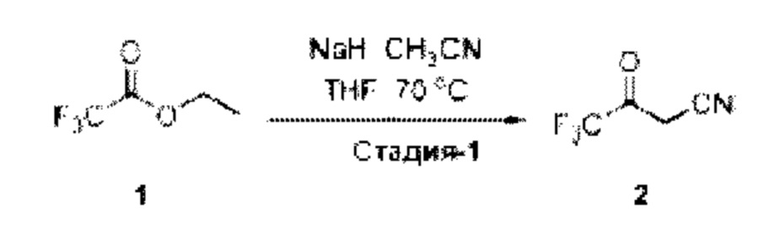
[0544] К перемешиваемой суспензии NaH (10,5 г, 264 ммоль) в ТГФ (150 мл) при 0°С одновременно добавляли этил 2,2,2-трифторацетат 1 (15,0 г, 106 ммоль) и MeCN (12,4 г, 159 ммоль) в атмосфере аргона. Полученную смесь перемешивали при комнатной температуре в течение 10 мин, а затем нагревали до 70°С в течение 16ч. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили ледяной водой (50 мл) и упаривали при пониженном давлении для удаления органической фазы. Оставшийся водный слой экстрагировали диэтиловым эфиром (2 × 30 мл). Затем водный слой доводили до рН 2, используя конц. РСд, затем экстрагировали диэтиловым эфиром (3 × 50 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением неочищенного указанного в заголовке соединения 2 (18,0 г), которое использовали в последующих стадиях без дополнительной очистки.
Стадия 2: Синтез этил 4-(5-амино-3-(трифторметил)-1Н-пиразол-1-ил)бензоата:
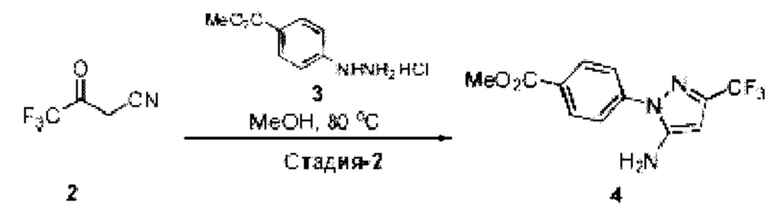
[0545] К перемешиваемому раствору 4,4,4-трифтор-3-оксобутаннитрила 2 (3,39 г, 24,8 ммоль) в метаноле (30 мл) при комнатной температуре добавляли гидрохлорид метил-4-гидразинилбензоата 3 (2,50 г, 12,47 ммоль). Реакционную смесь нагревали до 80°С в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 20-25% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (2,00 г). 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,09 (д, J=8,48 Гц. 2Н), 7,79 (д, J=8,48 Гц, 2Н), 5,98 (уш. с, 2Н), 5,84 (с, 1Н),3,89 (с, 3Н).
Стадия 3: Синтез метил-4-(5-ацетамидо-3-(трифторметил)-1H-пиразол-1-ил)бензоата:
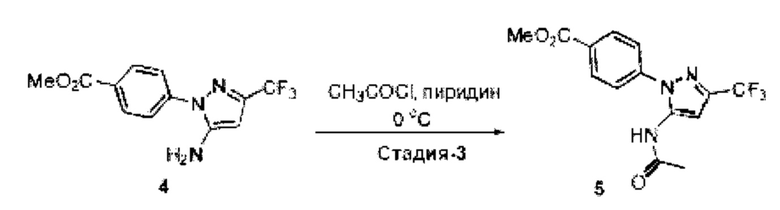
[0546] К охлажденному льдом раствору этил 4-(5-амино-3-(трифторметил)-1Н-пиразол-1-ил)бензоата 4 (1,00 г, 3,51 ммоль) в пиридине (10 мл) добавляли по каплям CH3COCl (0,37 мл, 5,3 ммоль). Полученную смесь перемешивали в течение 30 мин, и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили насыщенным раствором CuSO4 (30 мл) и экстрагировали ЕА (2 × 50 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-40% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,800 г). ЖХ-МС (Способ В) (ИЭР+): Опред.: 323,05 (М+Н)+; 1H-ЯМР (400 МГц, CDCl3) δ 8,21 (д, J=8,48 Гц, 2Н), 7,59 (д, J=8,48 Гц, 2Н), 6,95-7,00 (м, 1Н), 3,96 (с, 3Н), 2,17 (с, 3Н).
Стадия 3: Синтез метил 4-(5-(N-метилацетамидо)-3-(трифторметил)-1H-пиразол-1-ил)бензоата:
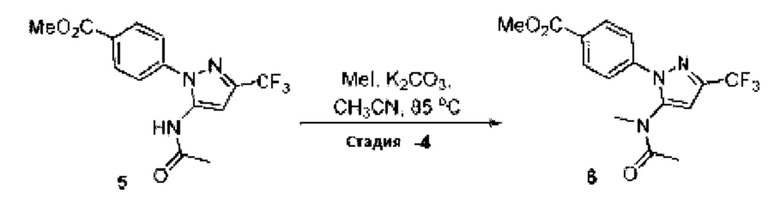
[0547] К перемешиваемому раствору метил 4-(5-ацетамидо-3-(трифторметил)-1Н-пиразол-1-ил)бензоата 5 (0,800 г, 2,45 ммоль) в CH3CN (15 мл) добавляли карбонат калия (0,506 г, 3,67 ммоль) и метилиодид (0,45 мл, 7,34 ммоль) при комнатной температуре. Затем реакционную смесь нагревали в запаянной пробирке при 85°С в течение 16 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли ЕА (100 мл) и промывали водой (2 × 25 мл), а затем солевым раствором (20 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-30% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,650 г). ЖХ-МС (Способ В) (ИЭР+): Опред.: 342,10 (М+Н)+; 1H-ЯМР (400 МГц, CDCl3) δ 8,19 (д, J=8,48 Гц, 2Н), 7,57 (д, J=8,48 Гц, 2Н), 6,62 (с, 1Н), 3,96 (с, 3Н), 3,15 (с, 3Н), 1,85 (с, 3Н).
Стадия 5: Синтез метил 4-(5-(метиламино)-3-(трифторметил)-1H-пиразол-1-ил)бензоата:
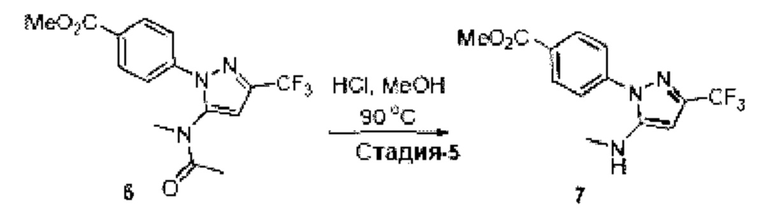
[0548] К перемешиваемому раствору метил 4-(5-(N-метилацетамидо)-3-(трифторметил)-1H-пиразол-1-ил)бензоата 6 (0,650 г, 1,91 ммоль) в метаноле (10 мл) добавляли конц. HCl (7 мл) при комнатной температуре. Затем реакционную смесь нагревали при 90°С в течение 4 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, разбавляли водой (20 мл) и экстрагировали ЕА (3×25 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-30% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,50 0 г). ЖХ-МС (Способ С) (ИЭР+): m/z 299,92 (М+Н)+; 1H-ЯМР (400 МГц, CDCl3) δ 8,16 (д, J=8,48 Гц, 2Н), 7,68 (д, J=8,48 Гц, 2Н), 5,80 (с, 1Н), 3,95 (с, 3Н), 2,89 (с, 3Н).
Стадия 6: Синтез (4-(5-(метиламино)-3-(трифторметил)-1H-пиразол-1-ил)фенил)метанола:
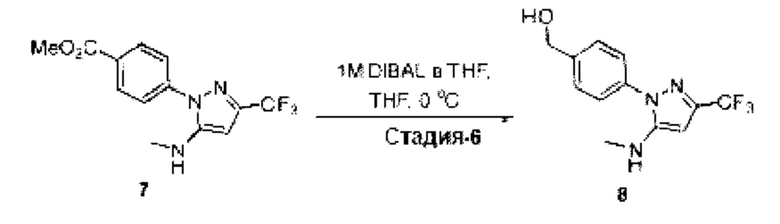
[0549] К перемешиваемому раствору метил 4-(5-(метиламино)-3-(трифторметил)-1Н-пиразол-1-ил)бензоата 7 (0,500 г, 1,67 ммоль) в ТГФ (15 мл) при 0°С добавляли DIBAL-H (1M в толуоле, 5,00 мл, 5,02 ммоль). Полученную смесь перемешивали в течение 30 мин, и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водным NH4Cl (10 мл). Полученный раствор разбавляли ЕА (20 мл) и фильтровали через подушку из целита. Водный слой отделяли и повторно экстрагировали ЕА (3 × 15 мл). Объединенный органический слой промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 50-80% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,400 г) 1H-ЯМР (400 МГц, ДМСО-d6) δ 7,42-7,51 (м, 4Н), 5,89 (с, 1Н), 5,83 (д, J=4,40 Гц, 1Н), 5,33 (т, J=5,62 Гц, 1Н), 4,56 (д, J=5,87 Гц, 2Н), 2,69 (д, J=4,89 Гц, 3Н).
Стадия 7: Синтез 1-(4-((6-хлор-1H-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-N-метил-3-(трифторметил)-1H-пиразол-5-амина (I-46):
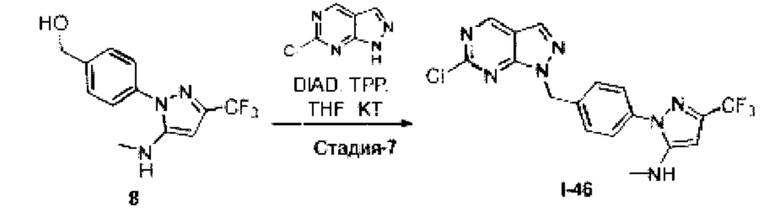
[0550] К перемешиваемому раствору (4-(5-(метиламино)-3-(трифторметил)-1Н-пиразол-1-ил)фенил)метанола 8 (0,200 г, 0,738 ммоль) в ТГФ (10 мл) добавляли 6-хлор-1Н-пиразоло [3,4-d]пиримидин (0,102 г, 0,664 ммоль), DIAD (0,336 г, 1,48 ммоль) и ТРР (0,382 г, 1,48 ммоль). Реакционную смесь перемешивали при комнатной температуре 6 течение 1 ч, и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (20 мл) и экстрагировали ЕА (3 × 30 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-40% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,160 г). ЖХ-МС (Способ С) (ИЭР+): m/z 407,86 (М+Н)+.
Получение общего промежуточного продукта I-47:
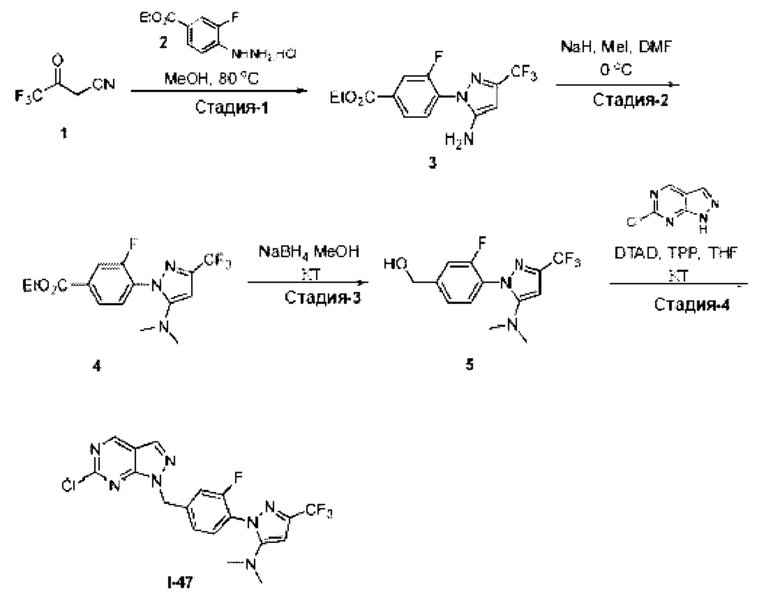
Стадия 1: Синтез этил 4-(5-амино-3-(трифторметил)-1H-пиразол-1-ил)-3-фторбензоата:
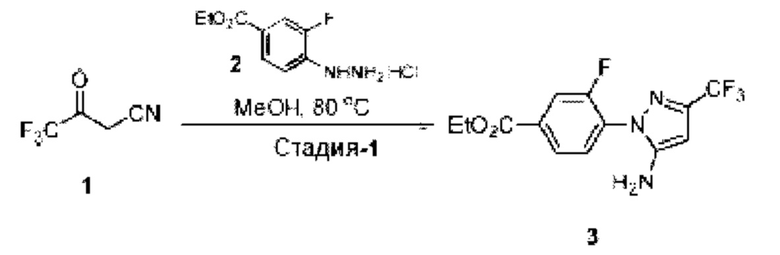
[0551] К перемешиваемому раствору 4,4,4-трифтор-3-оксобутаннитрила 1 (0,500 г, 2,136 ммоль) в метаноле (20 мл) добавляли гидрохлорид этил 3-фтор-4-гидразинеилбензоата 2 (0,731 г, 5,341 ммоль), при комнатной температуре. Затем реакционную смесь нагревали до 80°С в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 15-25% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,070 г). 1H-ЯМР (400 МГц, ДМСО-d6) δ 7,92 (д, J=9,29 Гц, 2Н), 7,72 (т, J=7,83 Гц 1Н), 5,91 (с, 2Н), 5,75 (с, 1Н), 4,37 (к, J=6,85 Гц, 2Н), 1,35 (т, J=7,09 Гц, 3Н).
Стадия 2: Синтез этил 4-(5-(диметиламино)-3-(трифторметил)-1H-пиразол-1-ил)-3-фторбензоата:
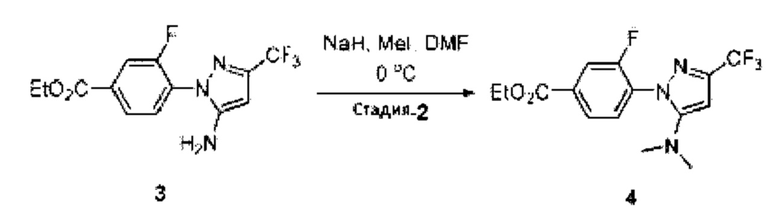
[0552] К перемешиваемому раствору этил 4-(5-амино-3-(трифторметил)-1Н-пиразол-1-ил)-3-фторбензоата 3 (0,250 г, 0,788 ммоль) в ДМФА(10 мл) при 0°С, добавляли 60% дисперсию гидрида натрия в масле (0,063 г, 1,577 ммоль) и метилиодид (0,145 мл, 2,365 ммоль). Полученную смесь перемешивали в течение 16 ч, и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили ледяной водой (10 мл) и экстрагировали ЕА (2 × 20 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-10% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,070 г). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 7,94-8,00 (м, 2Н), 7,79-7,85 (м, 1Н),6,40 (с, 1Н), 4,37 (к, J=6,85 Гц, 2Н), 2,56 (с, 6Н), 1,35 (т, J=7,09 Гц, 3Н).
Стадия 3: Синтез (4-(5-(диметиламино)-3-(трифторметил)-1Н-пиразол-1-ил)-3-фторфенил)метанола:
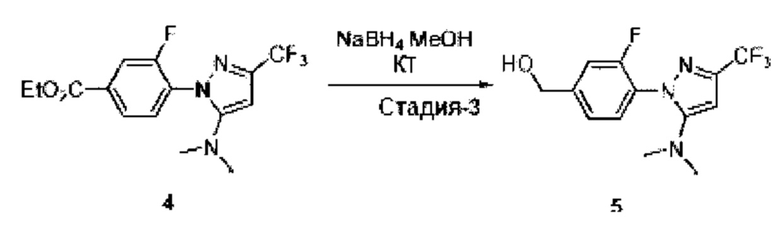
[0553] К охлажденному льдом раствору этил 4-(5-(диметиламино)-3-(трифторметил)-1Н-пиразол-1-ил)-3-фторбензоата 4 (0,150 г, 0,434 ммоль) в метаноле (10 мл) добавляли порциями боргидрид натрия (0,066 г, 1,739 ммоль). После завершения добавления реакционную смесь перемешивали в течение 5 ч и за кодом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водным раствором NH4Cl (10 мл) и экстрагировали ЕА (3 × 20 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, элюируя 40-50% ЕА в гексане, с получением указанного в заголовке соединения (0,090 г). ЖХ-МС (Способ В) (ИЭР+): m/z 304,00 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 7,56 (т, J=7,92 Гц, 1Н), 7,38 (д, J=11,47 Гц 1Н), 7,32 (д J=7,98 Гц, 1Н), 6,35 (с, 1Н), 5,49 (т, J=5,73 Гц 1Н), 4,59 (д, 3=5,49 Гц, 2Н), 2,54 (с, 6Н).
Стадия 4: Синтез 1-(4-((6-хлор-1H-пиразоло[3,4-d]пиримидни-1-ил)метил)-2-фторфенил)-N,N-диметил-3-(трифторметил)-1-пиразол-5-амина (I-47):
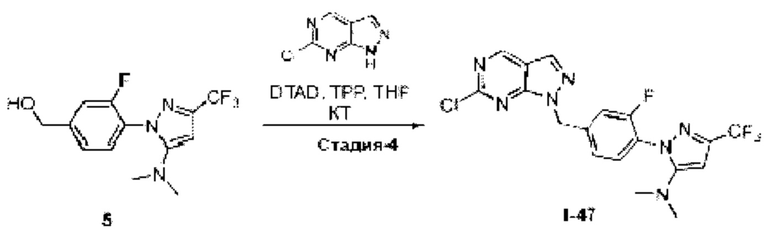
[0554] К перемешиваемому раствору (4-(5-(диметиламино)-3-(трифторметил)-1Н-пиразол-1-ил)-3-фторфенил)метанола 5 (0,080 г, 0,264 ммоль) в ТГФ (5 мл), добавляли 6-хлор-1H-пиразоло[3,4-d]пиримидин (0,036 г, 0,237 ммоль), DTAD (0,119 г, 0,528 ммоль) и ТРР (0,136 г, 0,528 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение 1 ч, и за кодом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, элюируя 0-60% ЕА в гексане, с получением указ энного в заголовке соединения (0,060 г). ЖХ-МС (Способ В) (ИЭР+): m/z 440,00 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,33 (с, 1Н), 8,54 (с, 1Н), 7,60 (т, J=8,07 Гц 1Н), 7,38 (д, 3=10,76 Гц 1Н), 7,21 (д, J=8,31 Гц 1Н), 6,35 (с, 1Н), 5,77 (с, 2Н), 2,52 (с, 6Н).
Следующие промежуточные продукты были получены из соответствующих гетероциклов и алкилирующих реагентов в соответствии со способом получения I-47:
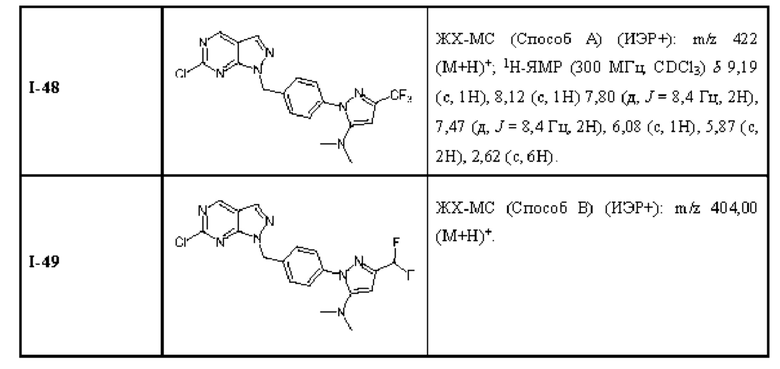
Получение общего промежуточного продукта I-50:
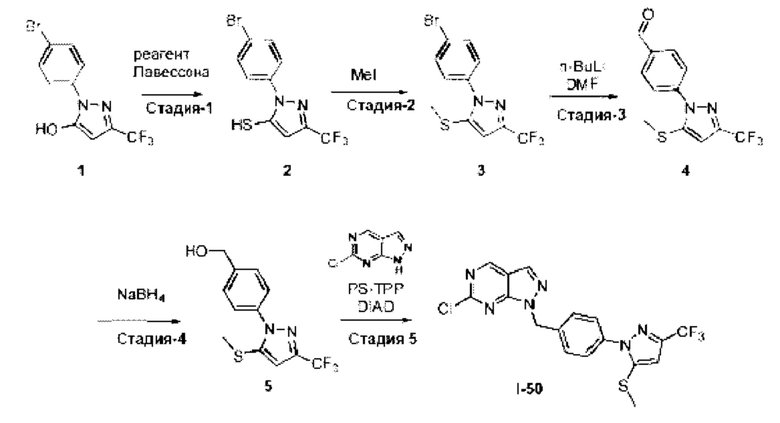
Стадия 1: Синтез 1-(4-бромфенил)-3-(трифторметил)-1Н-пиразол-5-тиола:
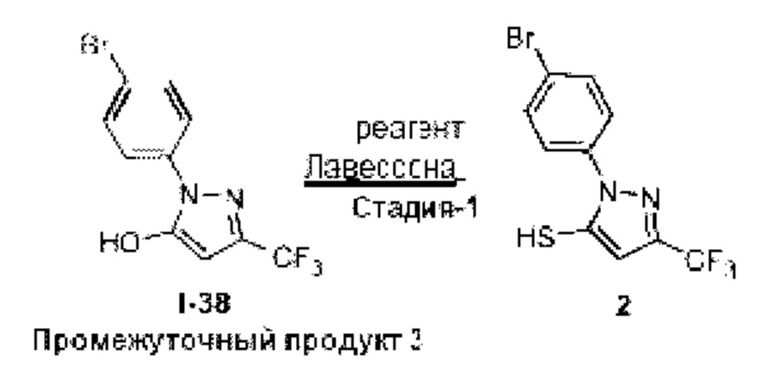
[0555] К раствору 1-(4-бромфенил)-3-(трифторметил)-1Н-пиразол-5-ола (900 мг, 2,94 ммоль) в толуоле (40 мл) добавляли реагент Лавессона (2,38 г, 5,88 ммоль). Затем реакционную смесь перемешивали при кипячении с обратным холодильником в течение 4 часов. После завершения реакции, как показывает анализ ТСХ, суспензию фильтровали и фильтрат упаривали досуха с получением 2,6 г неочищенного указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 323, 325 (М+Н)+.
Стадия 2: Синтез 1-(4-бромфенил)-5-(метилтио)-3-(трифторметил)-1Н-пиразола:
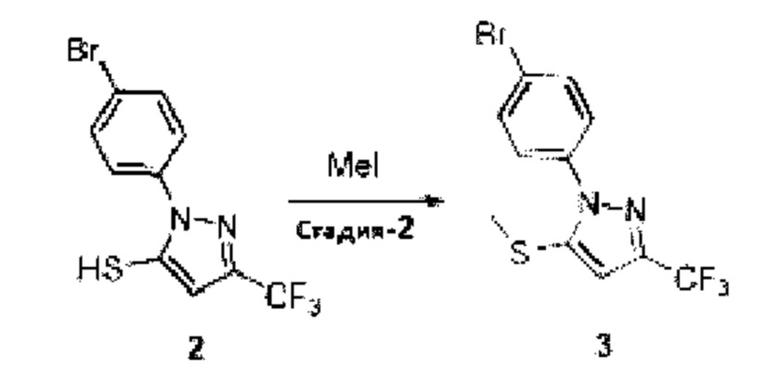
[0556] К раствору неочищенного метил 1-(4-бромфенил)-3-(трифторметил)-1Н-пиразол-5-тиола (2,6 г, 3,1 моль) в ДМФА (20 мл) при 0°С добавляли NaH (388 мг, 9.69 ммоль) порциями в течение 5 мин. После перемешивания смеси при 0°С в течение 30 минут по каплям в течение 2 минут добавляли MeI (1,72 г, 12,1 ммоль). Реакционную смесь сначала перемешивали при 0°С, а затем нагревали до комнатной температуры в течение 3 часов. После завершения реакции, как показывает анализ ТСХ, смесь гасили ледяной водой (20 мл) и экстрагировали ЕА (20 мл × 3). Органический слой промывали водой (20 мл × 2) и солевым раствором (20 мл), сушили над сульфатом натрия (20 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА = от 50/0 до 50/1) с получением 650 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 337, 339 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 7,63 (д, J=6,9 Гц 2Н), 7,48 (д, J=6,9 Гц, 2Н), 6,57 (с, 1Н), 2,44 (с, 3Н).
Стадия 3: Синтез 4-(5-(метилтио)-3-(трифторметил)-1Н-пиразол-1-ил)бензальдегида:
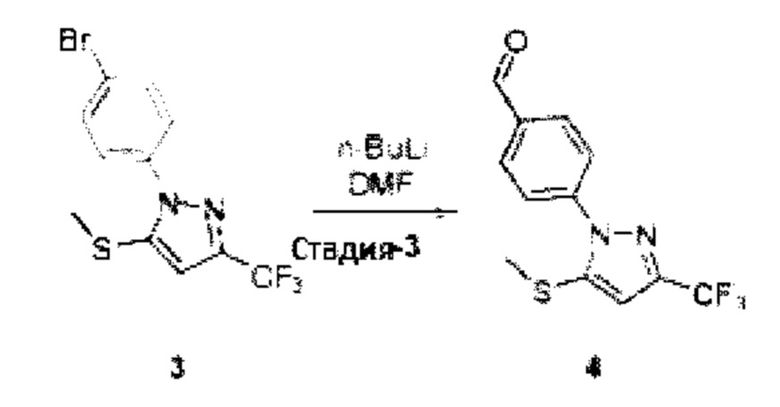
[0557] К раствору 1-(4-бромфенил[)-5-(метилтио)-3-(трифторметил)-1Н-пиразола 3 (550 мг, 1,64 ммоль) в безводном ТГФ (10 мл) при -78°С добавляли H-BuLi (0,98 мл, 2,46 ммоль) по каплям в течение 5 мин. После перемешивания в течение 1 часа при-78°С добавляли ДМФА (240 мг, 3,28 ммоль) по каплям в течение 5 минут и смесь медленно нагревали до комнатной температуры в течение 2 часов. После завершения реакции, как показывает анализ ТСХ, смесь гасили ледяной водой (20 мл). Полученную смесь экстрагировали ЕА (30 мл × 2). Органический слой сушили над сульфатом натрия (30 г), фильтровали и упаривали Полученный остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА = от 100/1 до 50/1) с получением 100 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 287 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 10,08 (с, 1Н), 8,02 (д, J=8,7 Гц, 2Н), 7,84 (д, J=8,7 Гц, 2Н), 6,61 (с, 1Н), 2,48 (с, 3Н).
Стадия 4: Синтез (4-(5-(метилтио)-3-(трифторметил)-1Н-пиразол-1-ил)фенил)метанола:
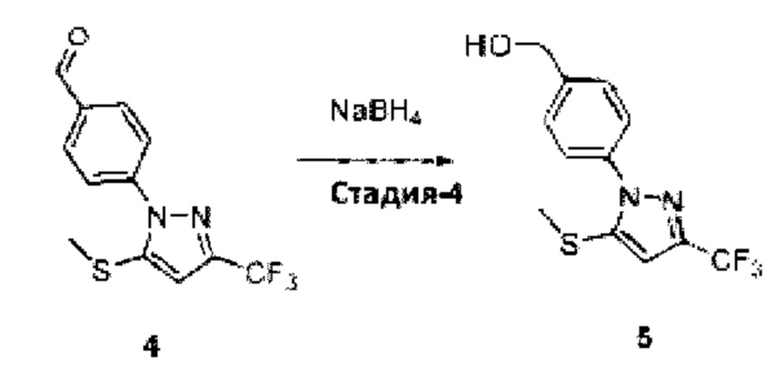
[0558] Получен в соответствии с процедурой восстановления 1-8. ЖХ-МС (Способ А) (ИЭР+): m/z 289 (М+Н)+; 1Н-ЯМР (300 МГц. CDCl3) δ 7,57 (д, J=8,4 Гц, 2Н),7,49 (д, J=8,4 Гц, 2Н), 6,57 (с, 1Н), 4,78 (с, 2Н),2,43(с, 3Н), 1,83(уш. с, 1Н).
Стадия 5: Синтез 6-хлор-1-(4-(5-(метилтио)-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (I-50):
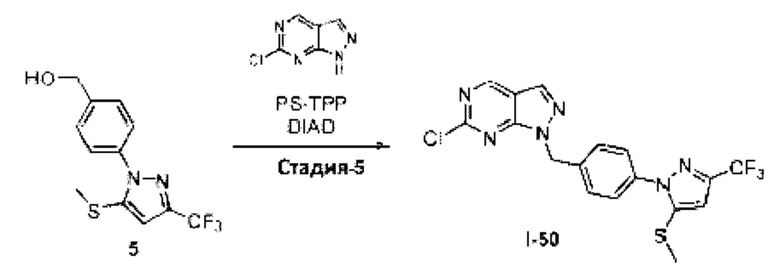
[0559] К суспензии 5 (130 мг, 0,45 моль), 6-хлор-1Н-пиразопо[3,4-d]пиримидина (90 мг, 0,59 ммоль) и PS-ТРР (300 мг, 0,9 ммоль) в безводном ТГФ (10 мл) при 0°С, добавляли DIAD (136 мг, 0,68 ммоль) по каплям в течение 5 минут. Реакционную смесь перемешивали на бане с ледяной в одой в течение 30 минут, а затем медленно нагревали до комнатной температуры в течение ночи. После завершения реакции, как показывает анализ ТСХ, реакцию гасили водой (20 мл) и экстрагировали ЕА (20 мл × 2). Объединенный органический слой сушили и упаривали. Остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА = от 10/1 до 1/0) с получением 160 мг неочищенного продукта. Неочищенный продукт дополнительно очищали препаративной ВЭЖХ (метод А) с получением 30 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (НЭР+): m/z 425 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 9,06 (с, 1Н), 8,19 (с, 1Н), 7,59-7,45 (м, 4Н), 6,55 (с, 1Н), 5,70 (с, 2Н), 2,42 (с, 3Н).
Получение общего промежуточного продукта I-51:
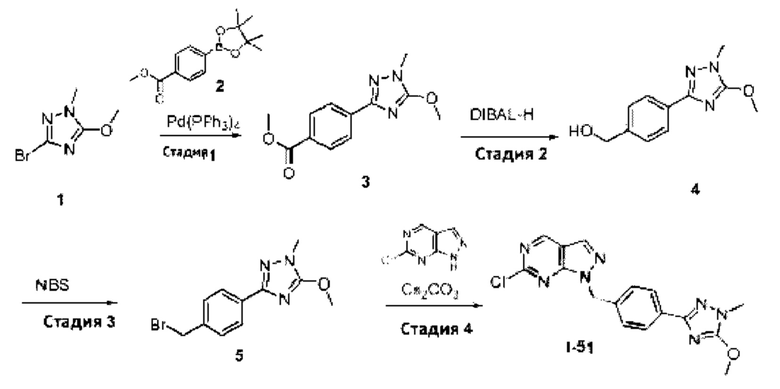
Стадия 1: Синтез метил 4-(5-метокси-1-метил-1Н-1,2,4-триазол-3-ил)бензоата:
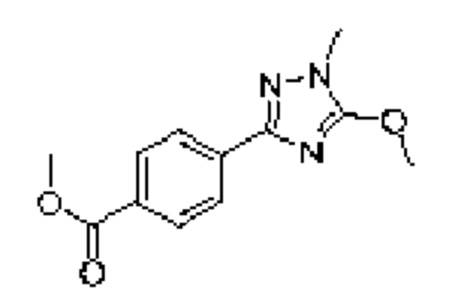
[0560] К смеси 3-бром-5-метокси-1-метил-1Н-1,2,4-триазола (500 мг, 2,60 ммоль), метил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоата (1,02 г, 3,91 ммоль), трициклогексилфосфина (219,07 мг, 0,78 ммоль), K3PO4-3H2O (2,08 г, 7,81 ммоль) в диоксане (26 мл) и Н2О (1,3 мл), добавляли Pd2(dba)3 (238,45 мг, 0,26 ммоль) в атмосфере азота. Полученную смесь перемешивали при 90°С в течение 1 часа. После завершения реакции, как показывает анализ ТСХ, реакцию гасили водой (30 мл) и экстрагировали ЕА (25 мл × 3). Объединенный органический слой сушили над безводным Na2SO4 (30 г), фильтровали и упаривали, получая неочищенный продукт. Неочищенный продукт очищали хроматографией на силикагеле (РЕ:ЕА = 10:1) с получением 650 мг указанного в заголовке соединения. 1H-ЯМР (300 МГц CDCl3) δ 8,08 (с,, 4Н), 4,17 (с, 3Н),3,93(с, 3Н), 3,68 (с, 3Н).
Стадия2: Синтез (4-(5-Метокси-1-метил-1Н-1,2,4-триазол-3-ил)фенил)метанола:
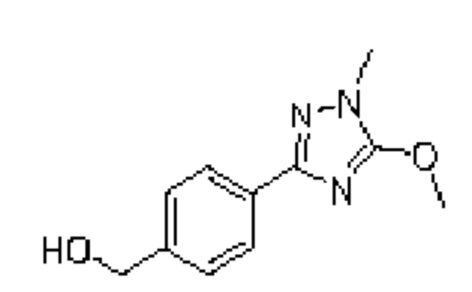
[0561] К раствору метил 4-(5-метокси-1-метил-1Н-1,2,4-триазол-3-ил)бензоата (650 мг, 2,63 ммоль) в ТГФ (26 мл) при 0°С добавляли DIBAL-H (1,5 М, 7,9 мл, 11,9 ммоль) по каплям в течение 15 мин. Полученную смесь перемешивали при 0°С в течение 1 ч, затем медленно разбавляли насыщенным водным раствором NH4Cl (10 мл). Полученную суспензию перемешивали на бане с ледяной водой в течение дополнительных 30 мин, а затем фильтровали для удаления твердых веществ. Осадок на фильтре промывали ЕА (30 мл × 4), и полученный фильтрат промывали солевым раствором (50 мл). После разделения органический слой сушили над безводным Na2SO4 (30 г), фильтровали и упаривали с получением неочищенного остатка, который очищали хроматографией на силикагеле (РЕ:ЕА = от 3:1 до 2:1), получая 500 мг указанного в заголовке соединения. 1H-ЯМР (300 МГц, CDCl3) δ 8,00 (д, J=8,4 Гц, 2Н), 7,41 (д, J=8,4 Гц, 2Н), 4,73 (д, J=6,0 Гц, 2Н), 4,16 (с, 3Н), 3,67 (с, 3Н), 1,70 (т, J=6,0 Гц 1H).
Стадия 3: Синтез 3-(4-(хлорметил)фенил)-5-метокси-1-метил-1Н-1,2,4-триазола:
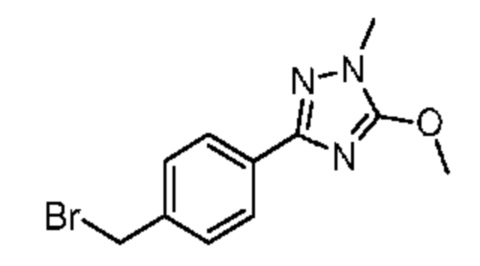
[0562] К раствору (4-(5-метокси-1-метил-1H-1,2,4-триазол-3-ил)фенил)метанола (450 мг, 2,05 ммоль) в DCM (25 мл) при к.т. добавляли PPh3 (592 мг, 2,26 ммоль) и NBS (438 мг, 2,05 ммоль) одной порцией. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. После завершения реакции, как показывает анализ ТСХ, реакционную смесь упаривали в вакууме и неочищенный остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 5:1 до 3:1) с получением 480 мг указанного в заголовке соединения.. 1H-ЯМР (300 МГц, CDCl3) δ 7,98 (д, J=8,4 Гц, 2Н), 7,43 (д, J=8,4 Гц 2Н), 4,53 (с, 2Н), 4,16 (с, 3Н), 3,66 (с, 3Н).
Стадия 4: Синтез 6-Хлор-1-(4-(5-метокси-1-метил-1H-1,2,4-триазол-3-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (I-51):
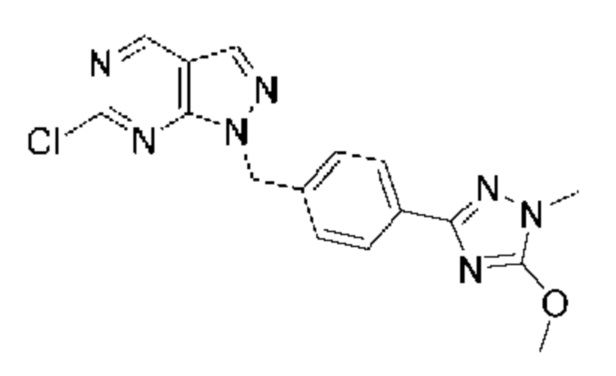
[0563] К раствору 3-(4-(хлорметил)фенил)-5-метокси-1-метил-1Н-1,2,4-триазола (480 мг, 1,70 ммоль) и 6-хлор-1Н-пиразоло[3,4-d]пиримидин (316 мг, 2,04 ммоль) в ДМФА (17 мл), добавляли Cs2CO3 (1,66 г, 5,10 ммоль) одной порцией и реакционную смесь перемешивали при 80°С в течение 1 часа. После завершения реакции (на что указывает анализ ТСХ) смесь охлаждали до комнатной температуры. Затем реакцию гасили ледяной водой (30 мл) и экстрагировали ЕА (50 мл × 2). Объединенный органический слой промывали водой (40 мл × 3), сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 1:1 до 1:3) с получением 220 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 356 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 9,03 (с, 1Н), 8,16 (с, 1H), 7,95 (д, J=81 Гц 2Н), 7,40 (д, J=8,1 Гц 2Н), 5,65 (с, 2Н), 4,13 (с, 3Н), 3,64 (с, 3Н).
Следующее промежуточный продукт был получен из соответствующего гетероцикла и алкилирующего реагента в соответствии со способом получения I-51:
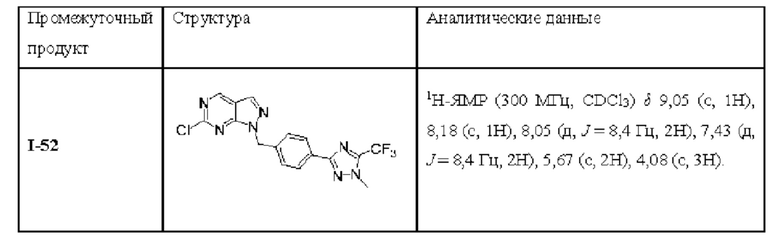
Получение общего промежуточного продукта I-53
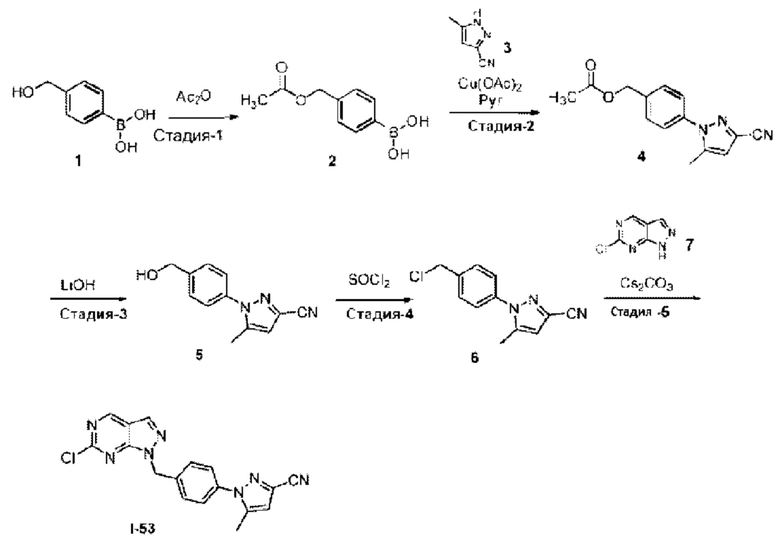
Стадия 1: Синтез (4-(Ацетоксиметил)фенил)бороновой кислоты:
[0564] Смесь (4-(гидроксиметал)фенил)бороновой кислоты (3 г, 0,02 моль) и уксусного ангидрида (4,08 г, 0,04 моль) в пиридине (5 мл, 0,062 моль) перемешивали при комнатной температуре в течение 4 часов. После завершения реакции, как показывает анализ ТСХ, смесь выливали в разбавленный водный раствор HCl (2 н, 20 мл) и экстрагировали ЕА (30 мл × 2). Объединенную органическую фазу промывали насыщенным раствором бикарбоната натрия (50 мл), сушили над сульфатом натрия (30 г), фильтровали и упаривали в вакууме с получением 4,5 г неочищенного указанного в заголовке соединения. 1H-ЯМР (300 МГц, CD3OD) δ 8,04 (д, J=7,8 Гц, 2Н), 7,38 (д, J=7,8 Гц, 2Н), 5,13 (с, 2Н), 2,11 (с, 3Н).
Стадия 2: Синтез 4-(3-циано-5-метил-1Н-пиразол-1-ил)бензилацетата:
[0565] К смеси (4-(ацетоксиметил)фенил)бороновой кислоты (1,087 г, 5,60 ммоль), 5-метил-1H-пиразол-3-карбонитрила (500 мг, 11,60 ммоль), триэтиламина (709 мг, 4,67 ммоль), пиридина (1,11 г, 14,07 ммоль) в DCM (20 мл), добавляли моногидрат ацетата меди (1,14 г, 6,26 ммоль), и полученную смесь перемешивали при 40°С в течение ночи. После завершения реакции, как показывает анализ ТСХ, реакцию гасили добавлением воды (20 мл) и экстрагировали DCM (20 мл × 2). Объединенную органическую фазу промывали водой (20 мл), сушили над сульфатом натрия (30 г), фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле на диоксиде кремния (РЕ:ЕА = от 30:1 до 6:1) с получением 390 мг указанного в заголовке соединения.
ЖХ-МС (Способ А) (ИЭР+): m/z 256 (М+Н)+; 1Н-ЯМР (300 МГц, CD3OD) δ 7,51 (д, J=8,7 Гц, 2Н), 7,44 (д, J=8,7 Гц, 2Н), 6,60 (с, 1Н), 5,18 (с, 2Н), 2,36 (с, 3Н), 2,15 (с, 3Н).
Стадия 3 Синтез 1-(4-(гидроксиметил)фенил)-5-метил-1Н-пиразол-3-карбонитрила:
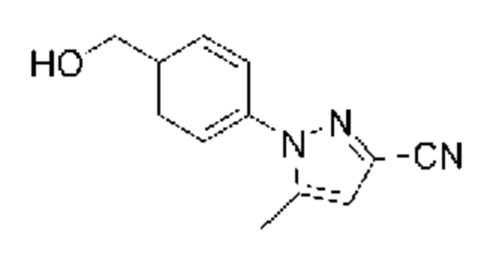
[0566] К раствору 4-(3-циано-5-метил-1H-пиразол-1-ил)бензилацетата (390 мг, 1,53 ммоль) в ТГФ (40 мл) и воде (5 мл) добавляли гидроксид лития (128 мг, 3,06 ммоль) одной порцией. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. После завершения реакции, как показывает анализ ТСХ, реакцию гасят ледяной водой (10 мл). Полученную смесь экстрагировали ЕА (10 мл × 2) и слои разделяли. Объединенный органический слой сушили над сульфатом натрия (10 г), фильтровали и упаривали с получением 300 мг неочищенного указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 214 (М+Н)+; 1H-ЯМР (300 МГц, CD3OD) δ 7,56 (д, J=8,4 Гц 2Н), 7,47 (д, J=8,4 Гц, 2Н), 6,75 (с, 1H), 4,70 (с, 2Н), 2,37 (с, 3Н).
Стадия 4: Синтез 1-(4-(хлорметил)фенил)-5-метил-1Н-пиразол-3-карбонитрила:
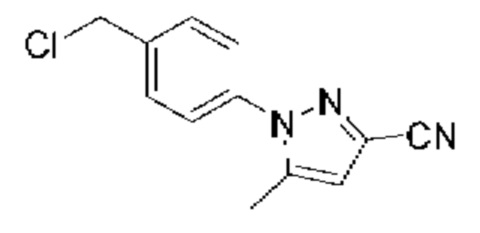
[0567] К раствору 1-(4-(гидроксиметил)фенил)-5-метил-1H-пиразол-3-карбонитрила (300 мг, 0,96 ммоль) в DCE (6 мл) добавляли SOCl2 (341 мг, 2,87 ммоль) одной порцией. После добавления реакционную смесь перемешивали при 60°С в течение 1 часа. После того, как реакция завершилась, как показал анализ ТСХ, реакционную смесь упаривали досуха в вакууме с получением 330 мг неочищенного указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 232 (М+Н)+; 1Н-ЯМР (300 МГц CD3OD) δ 7,55 (д, J=8,4 Гц, 2Н), 7,44 (д, J=8,4 Гц 2Н), 6,10 (с, 1H), 4,65 (с, 2Н), 2,38 (с, 3Н).
Стадия 5: Синтез 1-(4-((6-Хлор-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-5-метил-1Н-пиразол-3-карбонитрила (I-53).:
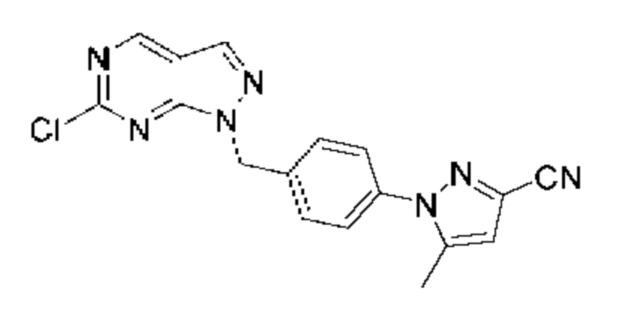
[0568] К раствору 1-(4-(хлорметил)фенил)-5-метил-1H-пиразол-3-карбонитрила (330 мг, 1,43 ммоль) в ДМФА (5 мл) добавляли 6-хлор-1H-пиразоло[3,4-d]пиримидин (220 мг, 1,43 ммоль) и карбонат калия (590 мг, 4,29 ммоль). После добавления реакционную смесь перемешивали при 70°С в течение 1 часа. После завершения реакции, как показывает анализ ТСХ, реакцию гасили водой (10 мл) и экстрагировали ЕА (10 мл × 2). Объединенную органическую фазу промывали водой, сушили над сульфатом натрия (10 г), фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (РЕ:ЕА = от 10:1 до 2:1) с получением 188 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 350 (М+Н)+; 1H-ЯМР (300 МГц, CD3OD) δ 9,07 (с, 1H), 8,20 (с, 1H), 7,52 (д, J=8,4 Гц 2Н), 7,41 (д, J=8,4 Гц 2Н), 6,58 (с, 1H), 5,71 (с, 2Н), 2,36 (с, 3Н).
Получение общего промежуточного продукта I-54:
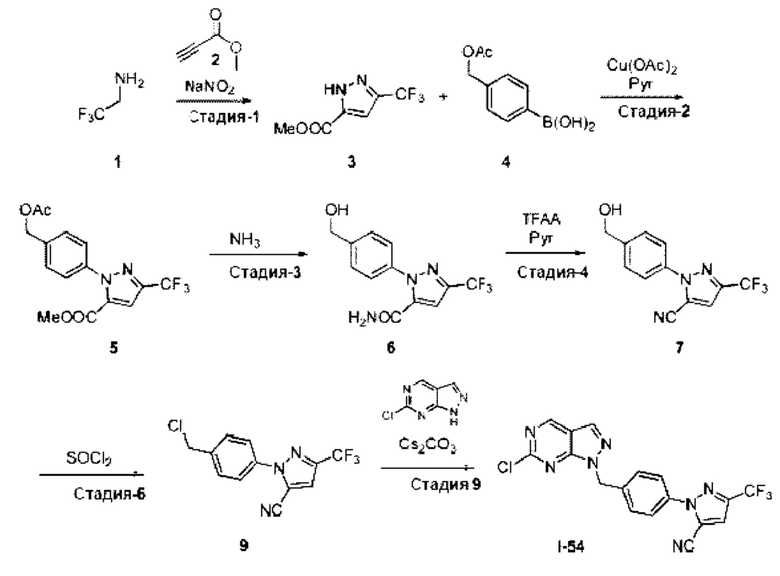
Стадия 1: Синтез метил-3-(трифторметил)-1Н-пиразол-5-карбоксилата:
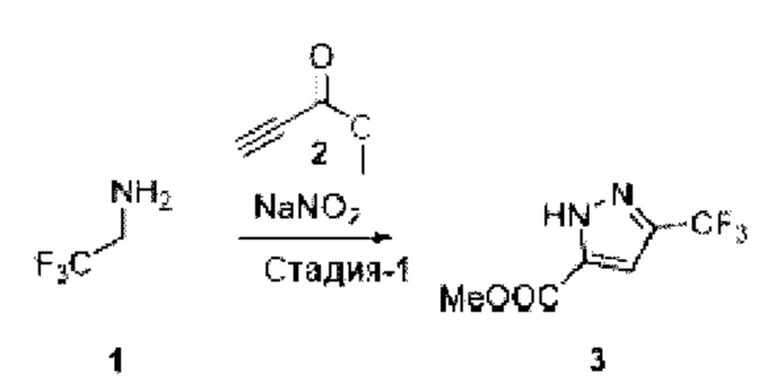
[0569] К раствору метилпропиолата 2 (500 мг, 5,94 ммоль) в DCM (4,0 мл) при комнатной температуре добавляли водный раствор NaNO2 (613 мг NaNO2 в 2,0 мл воды) частями. После добавления реакционную смесь охлаждали на бане с ледяной водой и к реакционной смеси порциями в течение 5 минут добавляли гидрохлорид 2,2,2-трифторэтанамина 1 (1,2 г, 8,9 ммоль). Затем реакционную смесь перемешивали при 0 в течение 1 ч, а затем при комнатной температуре в течение ночи. Смесь гасили водой (30 мл) и экстрагировали DCM (30 мл × 2). Органический слой сушили над сульфатом натрия (20 г), фильтровали и упаривали, получая 1,1 г неочищенного указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 195 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 11,41 (уш. с, 1Н), 7,10 (с, 1Н), 3,98 (с, 3Н).
Стадия 2: Синтез метил 1-(4-(ацетоксиметил)фенил)-3-(трифторметил)-1Н-пиразол-5-карбоксилата:
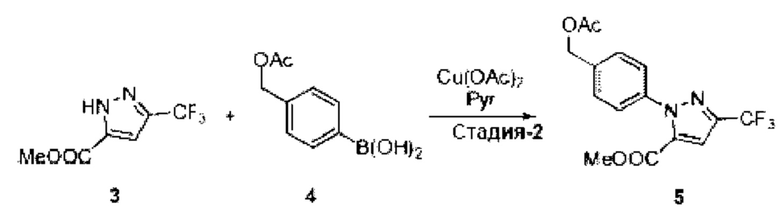
[0570] К смеси метил 3-(трифторметил)-1Н-пиразол-5-карбоксилата 3 (870 мг, 4,48 ммоль) и (4-(ацетоксиметил)фенил)бороновой кислоты (1,1 г, 5,8 ммоль) в DCM (20 мл) и DCE (20 мл), добавляли Cu(ОАс)2 (1,2 г, 6,7 ммоль) и пиридин (709 мг, 8,96 ммоль). Затем реакционную смесь перемешивали при 40°С в течение ночи. После завершения реакции, как показывает анализ ТСХ, смесь фильтровали и осадок на фильтре промывали DCM (30 мл). Объединенный фильтрат промывали водой (30 мл × 2), сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА=9/1) с получением 1,1 г указ энного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 343 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 7,43-7,51 (м, 4Н), 7,08 (с, 1Н), 5,18 (с, 2Н), 3,84 (с, 3Н), 2,14 (с, 3Н).
Стадия 3: Синтез 1(4-(гидроксиметил)фенил)-3-(трифторметил)-1Н-пиразол-5-карбоксамида:
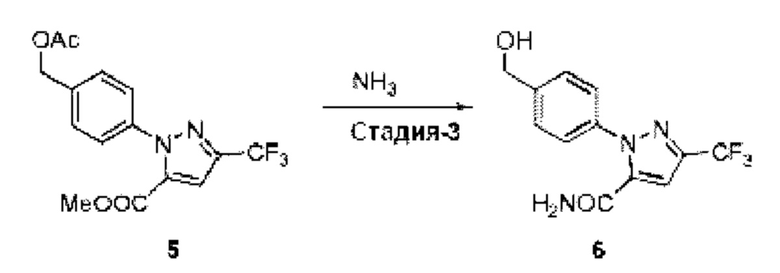
[0571] К насыщенному метанольному раствору аммиака (10 мл, 7 н.) добавляли метил 1-(4-(ацетоксиметил)фенил)-3-(трифторметил)-1Н-пиразол-5-карбоксилат 5 (1,1 г, 3,2 ммоль) за один прием. Реакционную смесь перемешивали в закрытой пробирке при 50°С в течение 6 часов. После завершения реакции, как показывает анализ ТСХ, смесь гасили насыщенным раствором хлорида аммония (40 мл) и экстрагировали ЕА (50 мл × 2). Объединенный органический слой промывали водой (30 мл), солевым раствором (30 мл), сушили над сульфатом натрия (30 г), фильтровали и упаривали остаток очищали хроматографией на силикагеле (элюент: DCM/MeOH = от 50/1 до 20/1) с получением 530 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 286 (М+Н)+; 1H-ЯМР (300 МГц, CD3OD) δ 7,43-7,51(м. 4Н), 7,19 (с, 1Н), 4,68 (с, 2Н).
Стадия 4: Синтез 1-(4(гидроксиметил)фенил)-3-(трифторметил)-1Н-пиразол-5-карбонитрила:
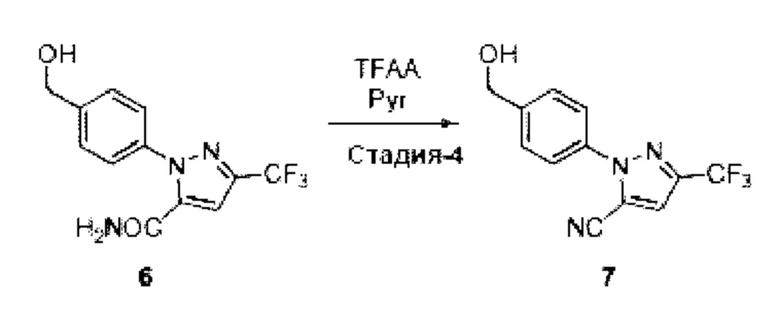
[0572] Соединение синтезировали в соответствии с процедурой из I-44, стадия 4. ЖХ-МС (Способ А) (ИЭР+): m/z 267 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 7,72 (д, J=8,7 Гц, 2Н), 7,58 (д, J=8,4 Гц, 2Н), 7,27 (с, 1Н), 4,83 (с, 2Н).
Стадия 5: Синтез 1-(4-(хлорметил)фенил)-3-(трифторметил)-1Н-пиразол-5-карбонитрила:
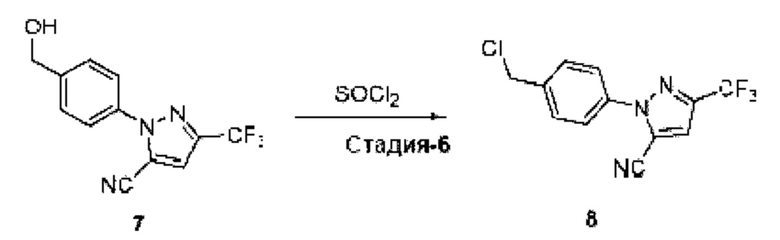
[0573] Соединение синтезировали в соответствии с процедурой получения общего промежуточного продукта I-8. ЖХ-МС (Способ А) (ИЭР+): m/z 286 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 7,74 (д, J=8,1 Гц, 2Н), 7,61 (д, J=8,1 Гц, 2Н), 7,29 (с, 1Н), 4,66 (с, 2Н).
Стадия 6: Синтез 1-(4-((6-хлор-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-3-(трифторметил)-1Н-пиразол-5-карбонитрила (I-54):
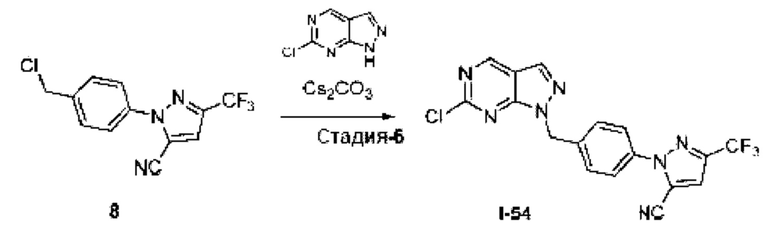
[0574] Соединение синтезировали в соответствии с процедурой получения общего промежуточного продукта I-8. ЖХ-МС (Способ А) (ИЭР+): m/z 404 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 9,08 (с, 1Н), 8,20 (с, 1Н) 7,71 (д, J=8,4 Гц, 2Н), 7,58 (д, J=8,4 Гц, 2Н), 7,27 (с, 1 Н), 5,72 (с, 2Н).
[0575]
Получение общего промежуточного продукта I-56:
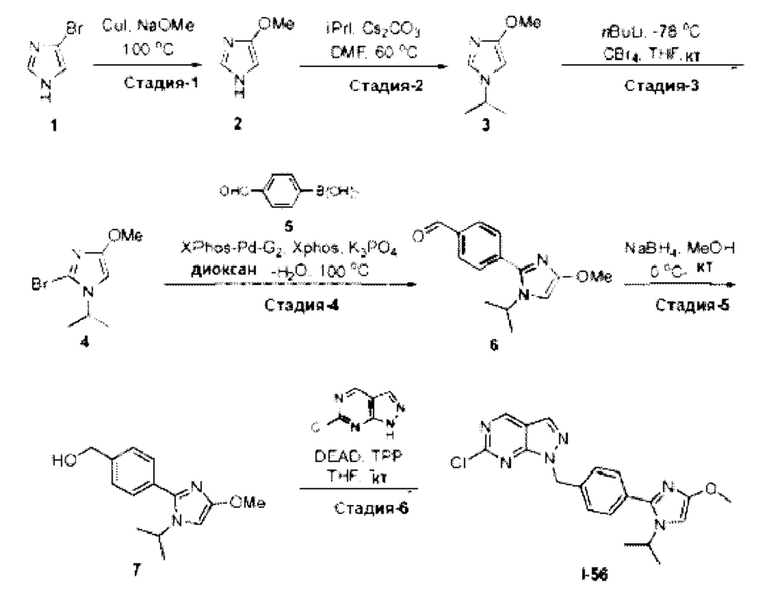
Стадия 1: Синтез 4-метокси-1H-имидазола:
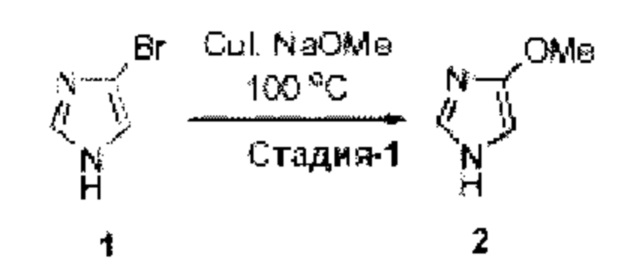
[0576] К перемешиваемому раствору 4-бром-1Н-имидазола 1 (15,0 г, 102 ммоль) в растворе NaOMe (150 мл) добавляли иодид меди (3,87 г, 20,4 ммоль) при комнатной температуре. Реакционную смесь нагревали до 100°С в течение 16 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, элюируя 0-80% ЕА в гексане, с получением указанного в заголовке соединения (1,50 г). 1H-ЯМР (400 МГц, ДМСО-d6) δ 11,61 (уш. с, 1Н), 7,25 (уш. с, 1Н), 6,39 (уш. с, 1Н), 3,64 (с, 3Н).
Стадия 2: Синтез 1-изопропил-4-метокси-1H-имидазола:
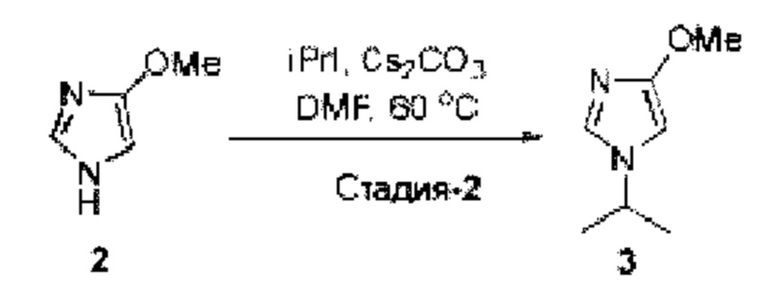
[0577] К перемешиваемому раствору 4-метокси-1Н-имидазола 2 (1,5 0 г, 15,3 ммоль) в ТГФ (15 мл) добавляли Cs2CO3 (9,90 г, 30,6 ммоль) и изопропилиодид (3,88 г, 23,0 ммоль) при комнатной температуре.. Затем реакционную смесь нагревали до 60°С в течение 16 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (50 мл) и экстрагировали ЕА (3 × 30 мл). Органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле по размеру, используя 0-50% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (1,00 г). ЖХ-МС (Способ В) (ИЭР+): m/z 140,98 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 7,29 (с, 1Н), 6,55 (с, 1Н), 4,26 (тд, J=6,79, 13,33 Гц, 1Н), 3,63 (д, J=0,98 Гц, 3Н), 1,35 (дд, J=0,98, 6,85 Гц, 6Н).
Стадия 3: Синтез 2-бром-1-изопропил-4-метокси-1Н-имидазола:
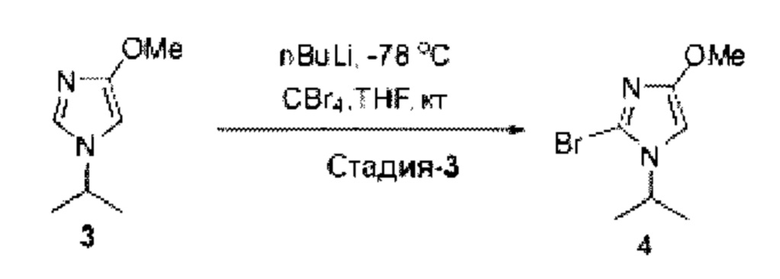
[0578] К перемешиваемому раствору 1-изопропил-4-метокси-1Н-имидазола 3 (1,00 г, 7,14 ммоль) в ТГФ (10 мл) при -78°С добавляли h-BuLi (1,6 М в гексане, 6,69 мл, 10,7 ммоль) по каплям. Полученный раствор перемешивали в течение 15 минут при -78°С, а затем добавляли CBr4 (3,50 г, 10,7 ммоль). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили насыщенным раствором NH4Cl (30 мл) и экстрагировали ЕА (3 × 50 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-30% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,70 0 г). ЖХ-МС (Способ В) (ИЭР+): m/z 221,19 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 6,81 (с, 1Н), 4,34(тд, J=6,79, 13,33 Гц, 1Н), 3,65 (с, 3Н), 1,34 (д, J=6,85 Гц, 6Н).
Стадия4: Синтез 4-(1-изопропил-4-метокси-1Н-имидазол-2-ил)бензальдегида:
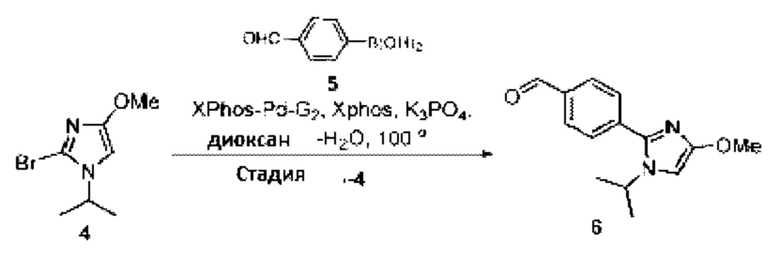
[0579] К перемешиваемому раствору 2-бром-1-изопропил-4-метокси-1Н-имидазола 4 (0,700 г, 3,21 ммоль) в диоксане Н2О (15:3 мл) добавляли K3PO4 (1,36 г, 6,42 ммоль) и (4-формилфенил)бороновую кислоту 5 (0,621 г, 4,17 ммоль). Полученную смесь дегазировали аргоном в течение 10 мин, а затем обрабатывали XPhos (0,152 г, 0,321 ммоль) и X-Phos-Pd-G2 (0,126 г, 0,160 ммоль) в герметичной пробирке. Реакционную смесь дополнительно дегазировали аргоном в течение 10 мин, а затем нагревали при 100°С в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, гасили водой (20 мл) и экстрагировали ЕА (3 × 30 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-20% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,400 г).
Стадия 5: Синтез (4-(1-изопропил-4-метокси-1Н-имидазол-2-ил)фенил)метанола:
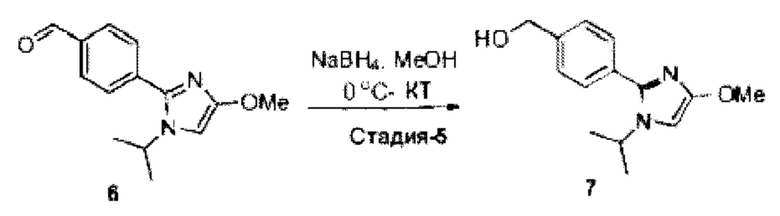
[0580] К перемешиваемому раствору 4-(1-изопропил-4-метокси-1Н-имидазол-2-ил)бензальдегида 6 (0,400 г, 1,64 ммоль) в метаноле (4 мл) при 0°С добавляли боргидрид натрия (0,063 г, 1,64 ммоль). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (10 мл) и экстрагировали ЕА (3 × 30 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-70% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,250 г). ЖХ-МС (Способ В) (ИЭР+): m/z 247,04 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 7,37-7,46 (м, 4Н), 6,75 (с, 1Н), 4,54 (д, J=5,87 Гц, 2Н), 4,43 (тт, J=6,79, 13,27 Гц, 2Н), 3,70 (с, 3Н), 1,35 (д, J=6,85 Гц, 6Н).
Стадия 6: Синтез 6-хлор-1-(4-(1-изопропил-4-метокси-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (I-56):
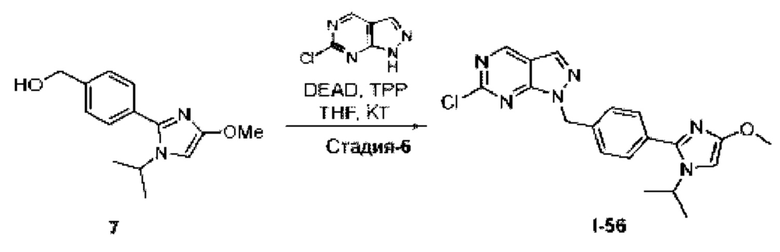
[0581] К перемешиваемому раствору (4-(1-изопропил-4-метокси-1Н-имидазоп-2-ил)фенил)метанола 7 (0,250 г, 1,02 ммоль) в ТГФ (4 мл) при 0°С добавляли 6-хлор-1Н-пиразоло[3,4-d]пиримидин (0,156 г, 1,02 ммоль), DEAD (0,357 г, 2,03 ммоль) и ТРР (0,532 г, 2,03 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (20 мл) и экстрагировали ЕА (3 × 30 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-50% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,120 г). ЖХ-МС (Способ В) (Т1ЭР+): m/z 383,04 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,32 (с, 1Н), 8,51 (с, 1Н), 7,45 (д, J=8,31 Гц,2Н), 7,34 (д, J=8,31 Гц, 2Н), 6,76 (с, 1Н),5,70 (с, 2Н), 4,36-4,42 (м. 1Н), 3,68 (с, 3Н), 1,33 (д, J=6,85 Гц, 6Н).
Получение общего промежуточного продукта I-57:
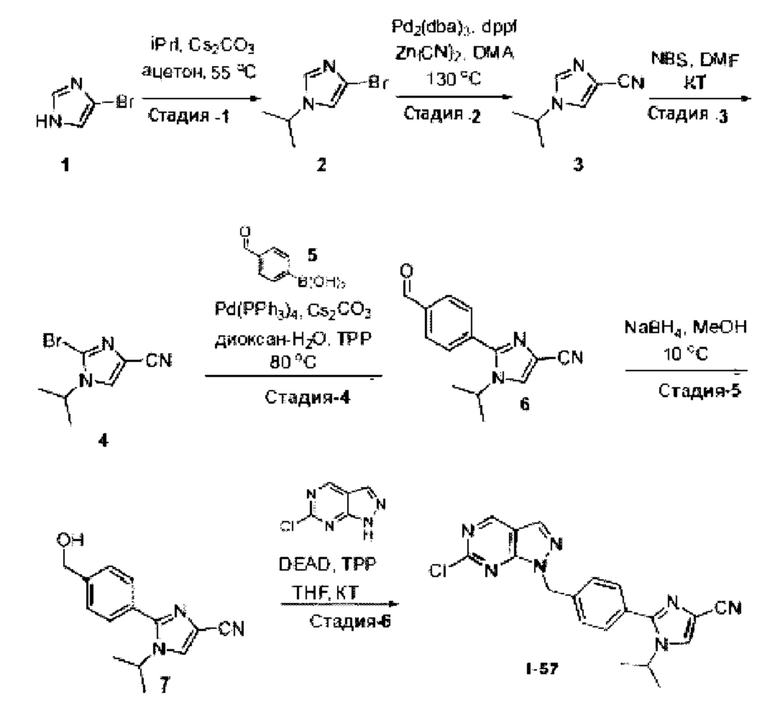
Стадия 1: Синтез 4-бром-1-изопропил-1Н-имидазола:
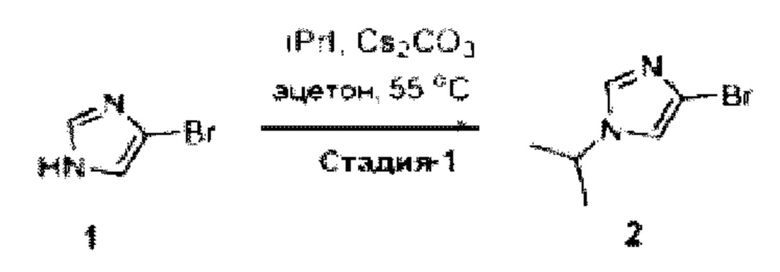
[D582] К перемешиваемому раствору 4-бром-1H-имидазола 1 (10,0 г, 68,0 ммоль) в ацетоне (200 мл) добавляли Cs2CO2 (22,1 г, 74,8 ммоль) и изопропилиодид (12,7 г, 74,8 ммоль) при комнатной температуре.. Затем реакционную смесь нагревали при 55°С в течение 15 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры и фильтровали через слой целита. Фильтрат упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-30% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (9,00 г). ЖХ-МС (Способ С) (ИЭР+): m/z 191 (M+H)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 7,69 (д, J=1,00 Гц, 1Н), 7,43 (д, J=1,50 Гц, 1Н), 4,39 (тд, J=6,73, 13,46 Гц, 1Н), 1,38 (д, J=6,98 Гц, 6Н).
Стадии 2: Синтез 1-изопропил-1Н-имидазол-4-карбонитрила:
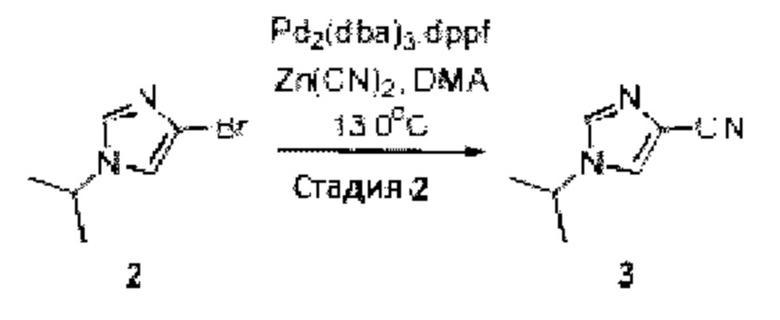
[0583] К перемешиваемому раствору 4-бром-1-изопропил-1Н-имидазола 2 (5,00 г, 26,4 ммоль) в DMA (50 мл) при комнатной температуре добавляли Zn(CN)2 (6,17 г, 52,9 ммоль). Затем полученную смесь дегазировали аргоном в течение 10 мин. К полученной реакционной смеси добавляли Pd(dba)3 (2,40 г, 2,64 ммоль) и 1,1'-бис(дифенилфосфино)ферроцен (2,92 г, 5,29 ммоль) при комнатной температуре. Затем реакционную смесь нагревали в запаянной пробирке при 130°С в течение 6 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температурь; гасили водным аммиаком (30 мл) и экстрагировали ЕА (100 мл). Органический спой промывали водой (2 × 200 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-40% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (2,10 г). ЖХ-МС (Способ В) (ИЭР+): m/z 135,9 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,27 (с, 1Н), 7,98 (с, 1Н), 4,49 (квин, J=6,60, 13,21 Гц, 1Н), 1,41 (д, J=6,85 Гц, 6Н).
Стадии 3: Синтез 2-бром-1-изопропил-1Н-имидазол-4-карбонитрила:
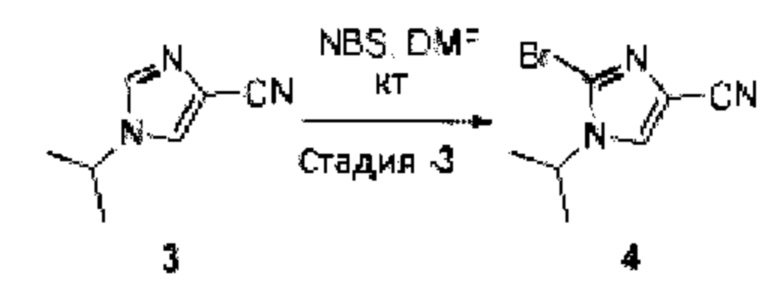
[0584] К перемешиваемому раствору метил 1-изопропип-1Н-имидазол-4-карбонитрила 3 (2,00 г, 14,79 ммоль) в ДМФА (20 мл) при комнатной температуре добавляли NBS (5,26 г, 29,6 ммоль). Полученную смесь перемешивали в течение 4 ч, и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (50 мл) и экстрагировали ЕА (3 × 50 мл). Объединенный органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-60% ЕА в гексане, с получением указанного в заголовке соединения (1,50 г). ЖХ-МС (Способ В) (ИЭР-), m/z 213,8 (М-Н); 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,25 (с, 1Н), 4,40-4,49 (м, 1Н), 1,45 (д, J=6,48 Гц, 6Н).
Стадия 4: Синтез 2-(4-формилфенил)-1-изопропил-1Н-имидазол-4-карбонитрила:
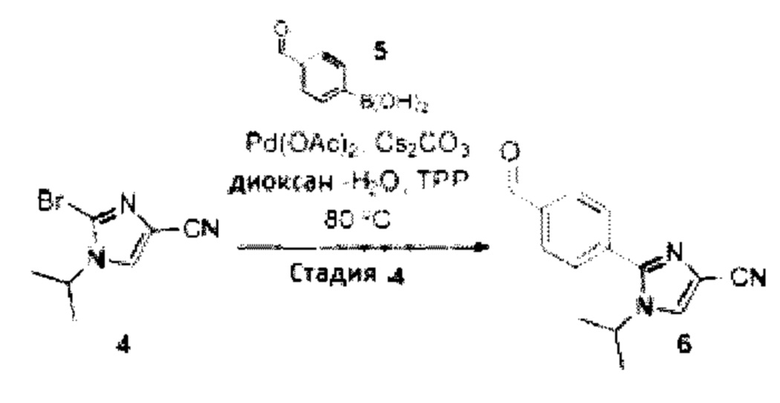
[0585] К перемешиваемому раствору 2-бром-1-изопропил-1Н-имидазол-4-карбонитрила 4 (0,8 40 г, 5,60 ммоль) в диоксане: H2O (2:1) (75 мл) добавляли Cs2CO3 (3,64 г, 11,2 ммоль) и (4-формилфенил) бороновую кислоту 5 (1,2 г, 5,60 ммоль). Полученную смесь дегазировали аргоном в течение 10 мин. К реакционной смеси добавляли трифенилфосфин (0,586 г, 2,24 ммоль) и Pd(OAc)2 (0,251 г, 1,12 ммоль) при комнатной температуре. Реакционную смесь нагревали в запаянной пробирке при 80°С в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры; разбавляли водой (10 мл) и экстрагировали ЕА (60 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-40% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,800 г). ЖХ-МС (Способ В) (ИЭР+): m/z 240,10 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 10,11 (с, 1Н), 8,32 (с, 1Н), 8,11 (д, J=7,83 Гц, 2Н),7,78 (д, J=7,83 Гц,2Н), 4,33 (тд, J=6,42, 13,08 Гц, 1Н), 1,42 (д, J=6,85 Гц, 6Н).
Стадия 5: Синтез 2-(4-(гидроксиметил)фенил)-1-изопропил-1H-имидазол-4-карбонитрила:
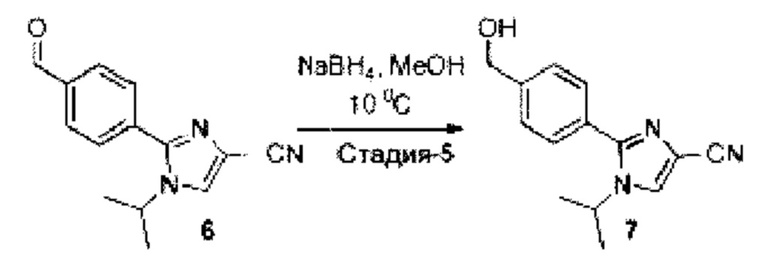
[0586] К перемешиваемому раствору 2-(4-формилфенил)-1-изопропил-1Н-имидазол-4-карбонитрила 6 (0,750 г, 3,134 ммоль) в метаноле (15 мл) при 10°С добавляли боргидрид натрия (0,059 г, 1,567 ммоль). Полученную смесь перемешивали при той же температуре в течение 30 мин. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (50 мл) и экстрагировали DCM (100 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-30% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,700 г). ЖХ-МС (Способ В) (ИЭР+): m/z 242 (М+Н)+; 1Н-ЯМР (400 МГЦ, ДМСО-d6) δ 8,23 (с, 1Н), 7,51-7,55 (м, 2Н), 7,44-7,49 (м, 2Н), 5,34 (т, J=5,87 Гц, 1Н), 4,59 (д, J=5,87 Гц, 2Н), 4,29 (тд, J=6,60, 13,21 Гц, 1Н), 1,40 (д, J=6,85 Гц, 6Н).
Стадия 6: Синтез 2-(4-((6-хлор-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-изопропил-1Н-имидазол-4-карбонитрила (I-57).
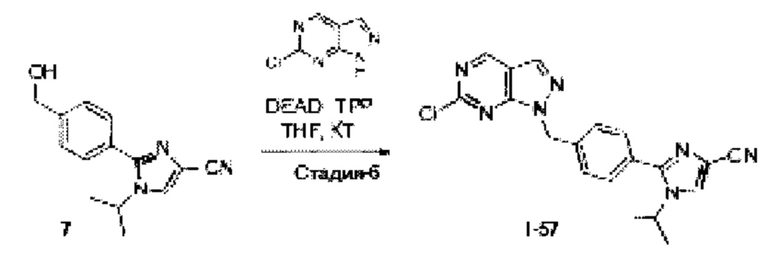
[0587] К перемешиваемому раствору 2-(4-(гидроксиметил)фенил)-1-изопропил-1Н-имидазол-4-карбонитрила 7 (0,319 г, 2р7 ммоль)и ТРР (0,812 г, 3,11 ммоль) в ТГФ (5 мл) добавляли DEAD (0,540 г,3,11 ммоль) и 6-хлор-1H-пиразоло[3,4-пиримидин (0,500 г, 2,07 ммоль) при комнатной температуре. Полученную смесь перемешивали е течение 1 ч при комнатной температуре, и за ходом реакции следили с помощью ТСХ После завершения реакционную смесь упаривали при пониженном давлении, а оставшийся остаток растворяли е воде (50 мл) и экстрагировали ЕА (50 мл). Органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-50% ЕА в гексане, с получением указанного в заголовке соединения(0,250 г). ЖХ-МС (Способ В)(ИЭР+): m/x 377,9 (М+Н)+.
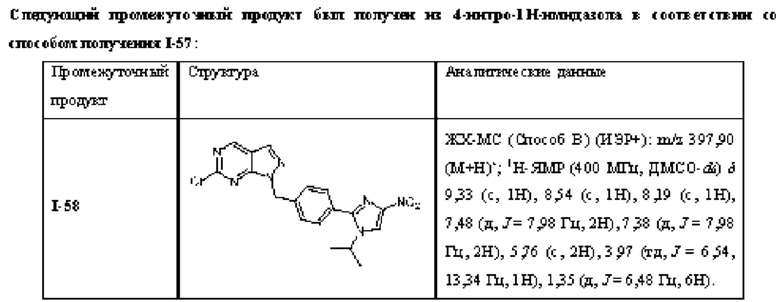
Получение общего промежуточного продукта I-59:
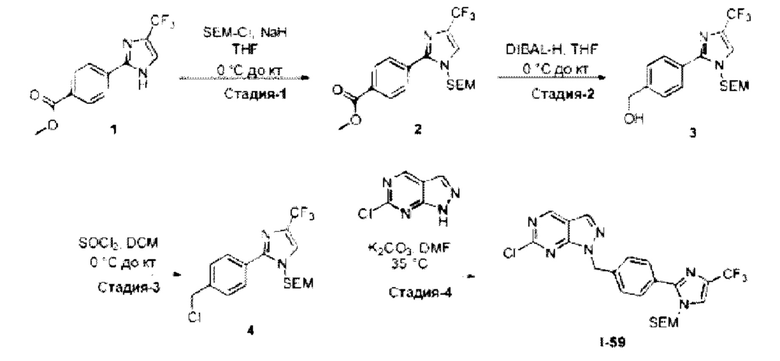
Стадия 1: Синтез Метил-4-(4-(трифторметил)-1-((2-(триметилсилил)этокси)метил)-1Н-имидазал-2-ил)бензоата:
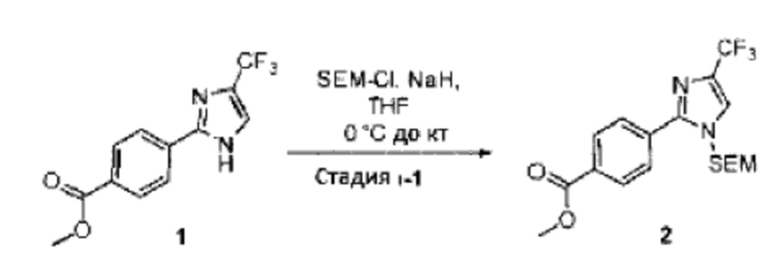
[0588] К охлажденному льдом раствору метил 4-(4-(трифторметил)-1Н-имидазол-2-ил)бензоата (14,5 г, 53,0 ммоль) в ТГФ (300 мл) добавляли NaH (60% дисперсия в минеральном масле) (3,23 г, 80,0 ммоль) порциями в течение 5 мин. Полученную смесь перемешивали при той же температуре в течение 30 минут, затем добавляли по каплям SEM-Cl (14,2 мл, 80,0 ммоль) в течение 30 минут. Реакционную смесь перемешивали при комнатной температуре еще 3 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили ледяной водой и экстрагировали ЕА. Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-3,5% ЕА в гексане, с получением 15,00 г указанного в заголовке соединения и 1,20 г нежелательного региоизомера. ЖХ-МС (Способ В) (ИЭР+): m/z 400,98 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,23 (с, 1Н), 8,06 (ц, J=7,8 Гц, 2Н), 7,96 (д, J=8,3 Гц, 2Н), 5,44 (с, 2Н), 3,87 (с, 3Н), 3,57 (т, J=7,8 Гц, 2Н), 0,83 (т, J=8,1 Гц, 2Н), -0,06 (с, 9Н).
Стадия 3: Синтез (4-(4-(Трифторметил)-1-((2-(триметилсилил)этокси)метил)-1H-имидазал-2-ил)фенил)метанола:
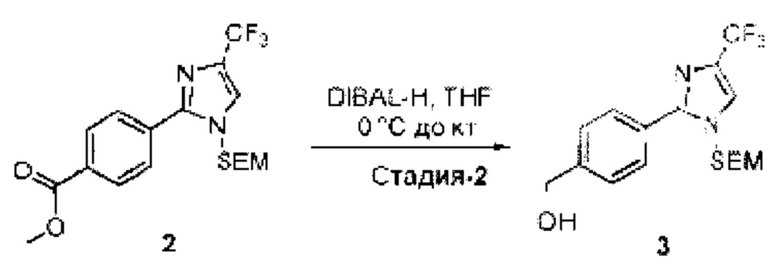
[0589] К перемешиваемому раствору метил 4-(4-(трифторметил)-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)бензоата (10,0 г, 25,0 ммоль) в ТГФ (200 мл) при 0°С добавляли DIBAL-H (1М в толуоле, 125 мл, 125 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. За кодом реакции следили с помощью ТСХ. После завершения к реакционной смеси добавляли воду и экстрагировали ЕА. Органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением 8,10 г указанного в заголовке соединения. ЖХ-МС (Способ В) (НЭР+): m/z 372,97 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,13 (с, 1Н), 7,73 (д, J=8,3 Гц, 2Н), 7,43 (д, J=8,3 Гц, 2Н), 5,36 (с, 2Н), 5,29 (т, J=5,6 Гц, 1Н), 4,54(д, J=5,9 Гц, 2Н), 3,56 (т, J=8,1 Гц, 2Н), 0,79-0,86 (м, 2Н), -0,04 (с, 9Н).
Стадия 4: Синтез 2-(4-(хлорметил)фенил)-4-(трифторметил)-1-((2-(триметилсилил)этокси)метил)-1Н-имидазола:
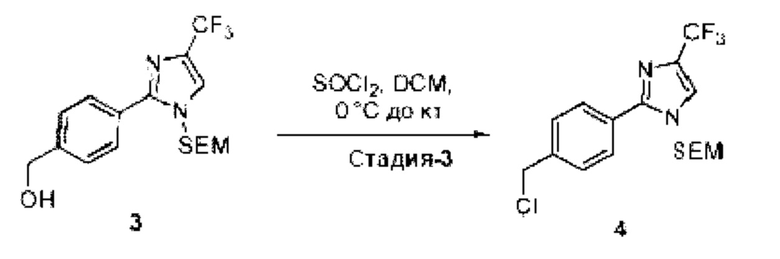
[0590] К перемешиваемому раствору (4-(4-(трифторметил)-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)фенил)метанола (2,50 г, 6,72 ммоль) в DCM (50 мл) при 0°С, добавляли SOCl2 (1,46 мл, 20,1 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили насыщенным раствором NaHCO3 (~ 50 мл) при 0°С, разбавляли водой (200 мл) и экстрагировали DCM (2 × 200 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением 2,40 г указанного в заголовке соединения, которое использовали как таковое в следующей реакции без дополнительной очистки. ЖХ-МС (Способ В) (ИЭР+): m/z 390,85 (М+Н)+; 1Н-ЯМР (400 МГц, CDCl3) δ 7,81 (д, J=7,9 Гц, 2Н), 7,51 (д, J=8,4 Гц, 2Н), 7,47 (с, 1Н), 5,30 (с, 2Н), 4,64 (с, 2Н), 3,62 (т, J=8,1 Гц, 2Н), 0,91-1,01 (м, 2Н), -0,06 (м, 9Н).
Стадия 5: Синтез 6-Хлор-1-(4-(4-(трифторметил)-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин (I-59):
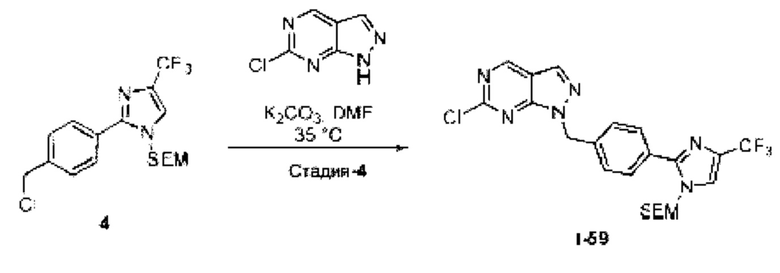
[0591] Раствор2-(4-(хлорметил)фенил)-4-(трифторметил)-1-((2-(триметилсилио)этокси)метил)-1Н-имидазола (2,52 г, 6,46 ммоль), 6-хлор-1Н-пиразоло[3,4-пиримидин (1,00 г, 6,46 ммоль) и K4CO3 (2,32 г, 16,10 ммоль) е ДМФА (15 мл) перемешивали при 35°С в течение 16 часов. За ходом реакции следили с помощью ТСХ и ЖХМС. После завершения реакционную смесь разбавляли холодной водой (100 мл) и экстрагировали ЕА (3 × 200 мл). Объединенный органический слой промывали ледяной водой(3 × 200 мл), а затем солевым раствором (200 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 50-100% ЕА в гексане, с получением 1,25 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 509,01 (М+Н)+; 1Н-ЯМР (400 МГц CDCl3) δ 9,06 (с, 1Н), 8,19 (с, 1Н), 7,78 (д, J=7,4 Гц, 2Н), 7,41-7,49 (м, 3Н), 5,70 (с, 2Н), 5,26 (с, 2Н), 3,59 (т, J=1,9 Гц, 2Н), 0,94 (т, J=8,1 Гц, 2Н), -0,01 (с, 9Н).
Получение общего промежуточного продукта I-60:
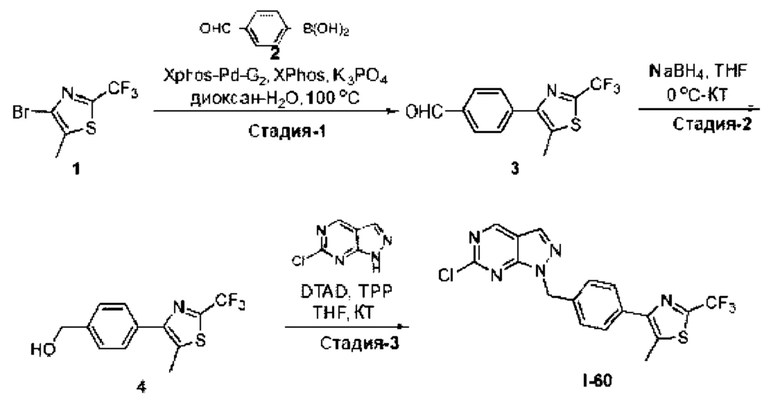
Стадия 1: Синтез 4-(5-метил-2-(трифторметил)тиазол-4-ил)бензальдегида:
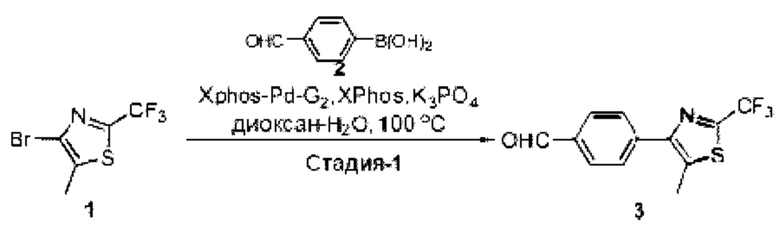
[0592] К перемешиваемому раствору 4-бром-метил-2-(трифторметил)тиазола 1 (1,00 г, 4,10 ммоль) в диоксане: Н2О (4:1, 12,5 мл) добавляли K3PO4 (1,73 г, 8,20 ммоль), (4-формилфенил)бороновую кислоту 2 (0,730 г, 4,92 ммоль) и смесь дегазировали аргоном в течение 15 минут. К полученной реакционной смеси добавляли X-Phos (0,195 г, 0,410 ммоль) и Pd-XPhos-G2 (0,161 г, 0,205 ммоль) при комнатной температуре. Реакционную смесь дополнительно нагревали в герметичной трубке при 100°С в течение 4 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, разбавляли водой (50 мл) и экстрагировали ЕА (3 × 50 мл). Объединенный органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-50% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (1,00 г). ЖХ-МС (Способ В) (ИЭР+): m/z 271,99 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 10,08 (с, 1Н), 8,05 (д, J=7,98 Гц, 2Н), 7,94-7,98 (м, 2Н), 2,73 (с, 3Н).
Стадия 2: Синтез (4-(5-метил-2-(трифторметил)тиазол-4-ил)фенил)метанола:
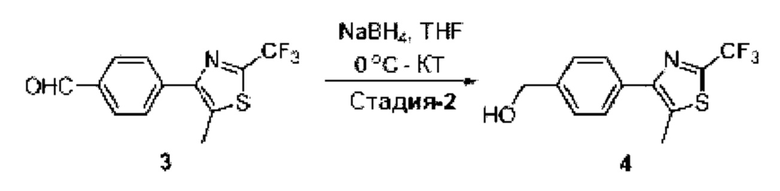
[0593] К охлажденному льдом раствору 4-(5-метил-2-(трифторметил)тиазол-4-ил)бензальдегида 3 (1,00 г, 3,690 ммоль) в метаноле (10 мл) добавляли боргидрид натрия (0,140 г, 3,690 ммоль). Реакционной смеси давали нагреться до комнатной температуры, а затем перемешивали в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (10 мл) и упаривали при пониженном давлении досуха. Полученный неочищенный остаток растворяли в воде (20 мл) и экстрагировали ЕА (2 × 30 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-50% ЕА в гексане, с получением указанного в заголовке соединения (0,800 г). ЖХ-МС (Способ В) (ИЭР+): m/z 273,95 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 7,66 (д, J=7,82 Гц, 2Н), 7,45 (д, J=7,83 Гц, 2Н), 5,27 (т, J=5,62 Гц, 1Н), 4,56 (д, J=5,87 Гц, 2Н), 2,66 (с, 3Н).
Стадия 3: Синтез 4-(4-((6-хлор-1H-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-5-метил-2-(трифторметил)тиазола (I-60):
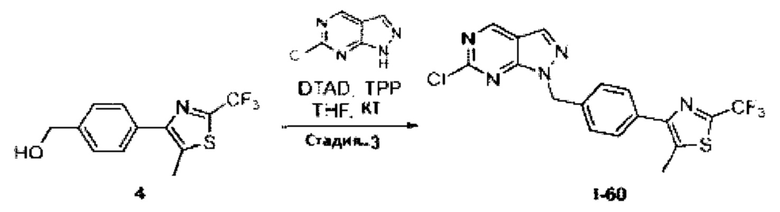
[0594] К перемешиваемому раствору (4-(5-метил-2-(трифторметил)тиазол-4-ил)фенил)метанола 4 (0,500 г, 1,831 ммоль) в ТГФ (10 мл) добавляли 6-хлор-1H-пиразоло[3,4-d]пиримидин (0,282 г, 1,831 ммоль), DTAD (0,842 г, 3,662 ммоль) и ТРР (0,959 г, 3,662 ммоль) при комнатной температуре и смесь перемешивали в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (20 мл) и экстрагировали ЕА (2 × 30 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-50% ЕА в гексане, с получением указанного в заголовке соединения (0,450 г). ЖХ-МС (Способ В): (ИЭР+): m/z 410,02 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,32 (с, 1Н), 8,52 (с, 1Н), 7,67 (д, J=7,98 Гц, 2Н), 7,39 (д, J=7,98 Гц, 2Н), 5,71 (с, 2Н), 2,63 (с, 3Н).
Получение общего промежуточного продукта I-61:
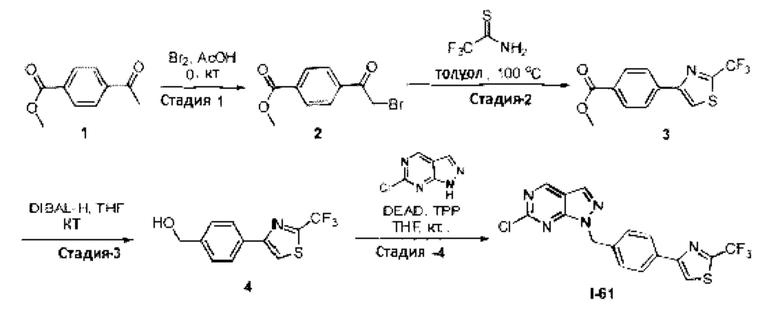
Стадия 1: Синтез 4-(2-бромацетил)фенилацетата:
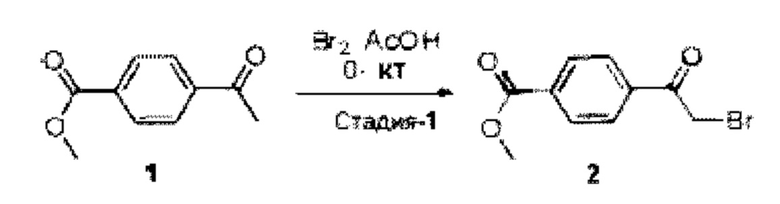
[0595] К охлажденному льдом раствору 4-ацетилфенилацетата 1 (5,00 г, 28,08 ммоль) в уксусной кислоте (50 мл) добавляли бром (1,15 мл, 22,47 ммоль), и реакционную смесь перемешивали в течение 4 часов при комнатной температуре. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь выливали в ледяную воду (10 мл) и полученное твердое вещество собирали на воронке Бюхнера. Твердое вещество промывали водой (20 мл) и сушили при пониженном давлении с получением неочищенного указанного в заголовке соединения (5,10 г). ЖХ-МС (Способ В) (ИЭР+): m/z 258,95 (М+Н)+; 1Н-ЯМР (400 МГц, CDCl3) δ 8,16 (д, J=7,98 Гц, 2Н), 8,05 (д, J=8,48 Гц, 2Н), 4,47 (с, 2Н), 3,96 (с, 3Н).
Стадия 2: Синтез метил 4-(2-(трифторметил)тиазол-4-ил)бензоата:
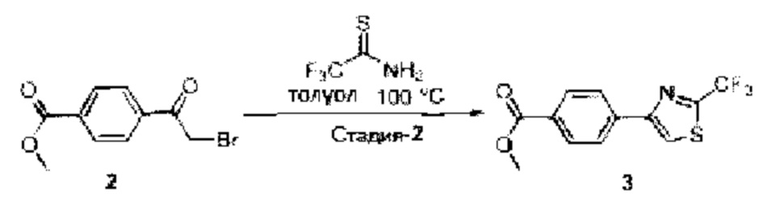
[0596] К перемешиваемому раствору 4-(2-бромацетил)фенилацетата 2 (0,700 г, 2,73 ммоль) в толуоле (15 мл) при комнатной температуре добавляли 2,2,2-трифторэтантиоамид (1,05 г, 8,20 ммоль). Реакционную смесь нагревали в закрытой пробирке при 100°С в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 5-20% ЕА в гексане, с получением указанного в заголовке соединения (0,700 г) 1Н-ЯМР (400 МГц, CDCl3) δ 8,13 (д, J=7,98 Гц, 2Н), 8,01 (д, J=8,48 Гц, 2Н),7,82 (с, 1Н), 3,95 (с, 3Н).
Стадия 3: Синтез (4-(2-(трифторметил)тиазол-4-ил)фенил)метанола:

[0597] К охлажденному льдом раствору метил 4-(2-(трифторметил)тиазол-4-ил)бензоата 3 (0,500 г, 1,74 ммоль) в ТГФ (5 мл) добавляли DIBAL-H (5,22 мл, 5,22 ммоль, 1,0 М раствор в ТГФ) по каплям. После добавления смесь перемешивали в течение 1 часа при комнатной температуре. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили насыщенным раствором NH4Cl (5 мл) и экстрагировали ЕА (3×10 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 1-20% ЕА в гексане в качестве элюента, с получением (0,400 г). ЖХ-МС (Способ В) (ИЭР+): m/z 260,00 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,54 (с, 1Н), 7,96 (д, J=7,83 Гц, 2Н), 7,44 (д, J=8,31 Гц, 2Н), 5,27 (т, J=5,87 Гц, 1Н), 4,55 (д, J=5,38 Гц, 2Н).
Стадия 4: Синтез 4(4-((6-хлор-1H-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-2-(трифторметил)тиазола (I-61):
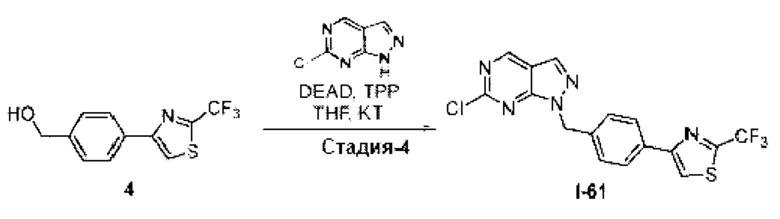
[0598] К перемешиваемому раствору (4-(2-(трифторметил)тиазол-4-ил)фенил)метанол 4 (0,400 г, 1,544 ммоль) и ТРР (0,606 г, 2,316 ммоль) в ТГФ (5 мл) добавляли DEAD (0,363 г, 2,316 ммоль) ил-хлор-1Н-пиразоло[3,4-d]пиримидин (0,214 г, 1,389 ммоль) при комнатной температуре и перемешивали в течение 30 мин при комнатной температуре. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-30% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,350 г). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,31 (с, 1Н), 8,55 (с, 1Н), 8,51 (с, 1Н), 7,96 (д, J=8,31 Гц, 2Н), 7,38 (д, J=7,83 Гц, 2Н), 5,69 (с, 2Н).
Получение общего промежуточного продукта I-62:
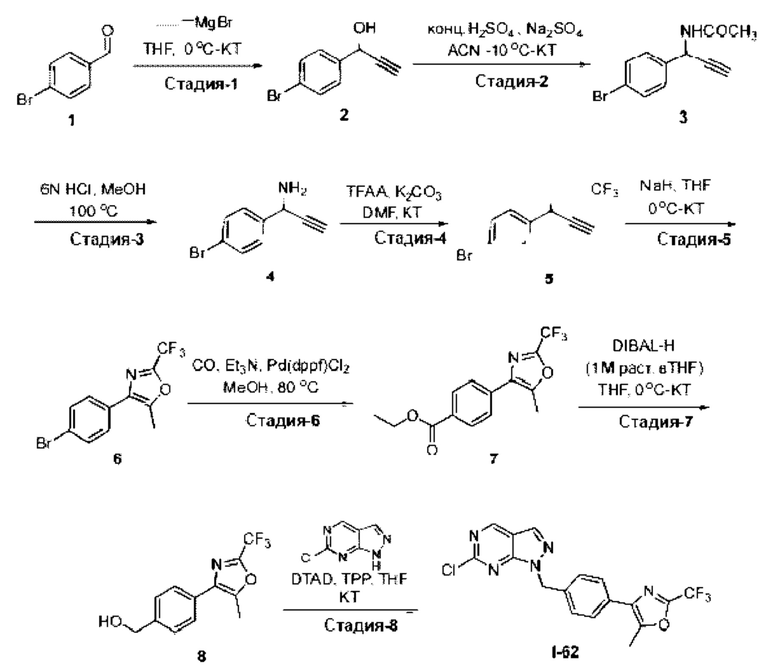
Стадия 1: Синтез 1-(4-бромфенил)проп-2-ин-1-ола:
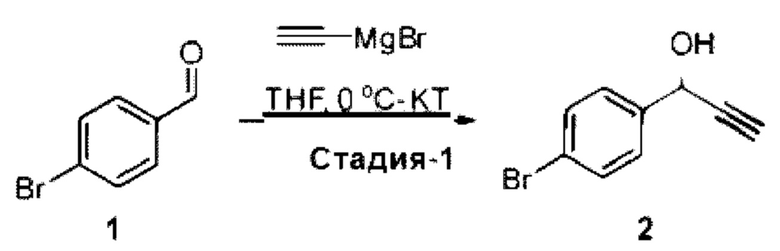
[0599] К охлажденному льдом раствору 4-бромбензальдегида 1 (5,00 г, 27,0 ммоль) в сухом ТГФ (75 мл) по каплям в атмосфере азота добавляли этинилмагнийбромид (0,5 М раствор в ТГФ, 119 мл, 67,6 ммоль). Реакционной смеси давали нагреться до комнатной температуры, а затем перемешивали в течение 3 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили разбавленной HCl (150 мл) и экстрагировали ЕА(3 × 50 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-20% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (3,00 г). ЖХ-МС (Способ В) (ИЭР+): m/z 212,00 (М+Н)+; 1Н-5МР (400 МГц, ДМСО-d6) δ 7,56 (д, J=7,92 Гц, 2Н), 7,41 (д, J=7,92 Гц, 2Н), 6,15 (д, J=5,98 Гц, 1Н), 5,33-5,37 (м, 1Н), 3,54 (д, J=2,49 Гц, 1Н).
Стадия 2: Синтез N-(1-(4-бромфенил)проп-2-ин-1-ил)ацетамида:
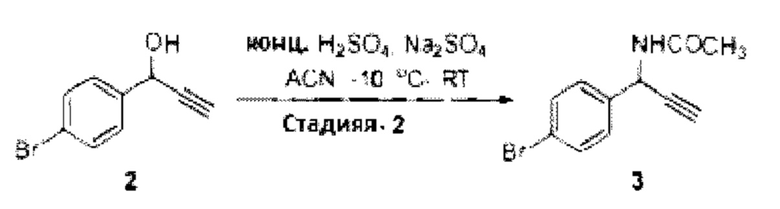
[0600] К перемешиваемому раствору 1-(4-бромфенил)проп-2-ин-1-ола 2 (1,00 г, 4,76 ммоль) в ацетонитриле (20 мл) при -10°С добавляли сульфат натрия (0,676 г, 4,76 ммоль) и перемешивали 5 мин. К реакционной смеси добавляли конц. H2SO4 (2,33 г, 23,8 ммоль) по каплям. Полученную реакционную смесь перемешивали 16 ч при комнатной температуре. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь выпивали в ледяную воду (50 мл), подщелачивали карбонатом натрия и экстрагировали ЕА (3 × 30 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 30-40% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,710 г). ЖХ-МС (Способ С) (ИЭР+): m/z 252,00 (М)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,87 (д, J=8,31 Гц, 1Н), 7,58 (д, J=8,31 Гц,2Н), 7,37 (д J=8,31 Гц, 2Н), 5,79 (д, J=8,31 Гц, 1Н), 3,50 (д, J=2,45 Гц, 1Н), 1,86 (с, 3Н).
Стадия 3: Синтез 1-(4-бромфенил)проп-2-ин-1-амниа:
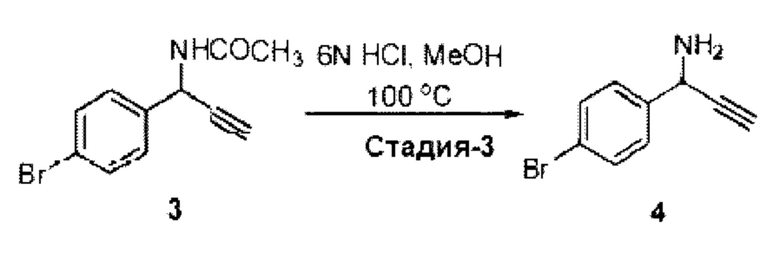
[0601] К перемешиваемому раствору N-(1-(4-бромфенил)проп-2-ин-1-ил)ацетамида 3 (0,700 г, 2,79 ммоль) в метаноле (15 мл) добавляли 6 н. HCl (10 мл) при комнатной температуре. Реакционную смесь нагревали при 100°С в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Полученный неочищенный остаток растворяли в смеси воды (20 мл) и ЕА (30 мл), а затем нейтрализовали твердым Na2CO3. Водный спой экстрагировали ЕА (2 × 10 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на сидикагеле, используя 25-30% ЕА в гексане, с получением указанного в заголовке соединения (0,400 г). 1H-ЯМР (400 МГц, ДМСО-d6) δ 7,51-7,56 (м, 2Н), 7,45 (д, J=7,98 Гц, 2Н), 4,67 (с, 1Н), 3,33 (шир. С, 1Н, слияние в H2O пика растворителя ДМСО), 2,28 (шир. с, 2Н).
Стадия 4: Синтез N-(1-(4-бромфенил)проп-2-ин-1-ил-2,2,2-трифторацетамида:

[0602] К перемешиваемому раствору 1-(4-бромфенил)проп-2-ин-1-амина 4 (0,350 г, 1,67 ммоль) в ДМФА (5 мл) добавляли карбонат калия (0,277 г, 2,01 ммоль) с последующим добавлением трифторуксусного ангидрида (0,28 мл, 0,42 г, 2,01 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь выливали в ледяную воду (30 мл) и экстрагировали ЕА (2 × 20 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 4-5% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,260 г). 1Н-ЯМР (400 МГц, CDCl3) δ 7,54 (д, J=8,48 Гц, 2Н), 7,40 (д, J=8,48 Гц, 2Н), 6,70 (уш. с, 1Н), 5,95 (да, J=1,75, 8,23 Гц, 1Н), 2,63 (д, J=2,49 Гц, 1Н).
Стадия 5: Синтез 4-(4-бромфенил)-5-метил-2-(трифторметил)оксазола:
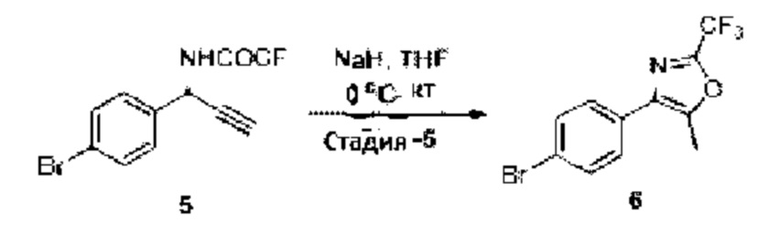
[0603] К перемешиваемому раствору N-(1-(4-бромфенил)проп-2-ин-1-ил)-2,2,2-трифторацетамида 5 (0,250 г, 0,819 ммоль) в сухом ТГФ (5 мл) при 0°С, добавляли 60% дисперсию гидрида натрия в масле (0,033 г, 0,82 ммоль). Реакционной смеси давали нагреться до комнатной температуры и затем перемешивали в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь выливали в ледяную воду (30 мл), хорошо перемешивали и экстрагировали ЕА (2 × 20 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 3-5% ЕА в гексане в качестве элюента, с получением указ энного в заголовке соединения (0,200 г). ЖХ-МС (Способ В) (ИЭР+): m/z 308,08 (М+Н)+; 1H-ЯМР (400 МГц, CDCl3) δ 7,52-7,61 (м, 4Н), 2,61 (с, 3Н).
Стадия 6: Синтез этил 4-(5-метил-2-(трифторметил)оксазол-4-ил) бензоата:
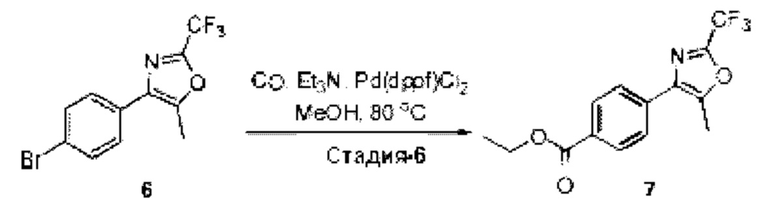
[0604] К перемешиваемому раствору 4-(4-бромфенил)-5-метил-2-(трифторметил)оксазола 6 (0,200 г, 0,655 ммоль) и этанола (10 мл) во флаконе под давлением добавляли триэтиламин (0,180 мл, 1,31 ммоль). Полученную смесь дегазировали аргоном в течение 10 мин, а затем обрабатывали Pd(dppf)Cl2 (0,048 г, 0,065 ммоль) при комнатной температуре. Затем реакционную смесь нагревали при 80°С в атмосфере монооксида углерода (давление 30 фунтов на квадратный дюйм в течение 16 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 5-10% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,170 г). ЖХ-МС (Способ В) (ИЭР+): m/z 299,80 (М+Н)+; 1Н-ЯМР (400 МГц, CDCl3) δ 8,13 (д, J=8,31 Гц, 2Н), 7,76 (д, J=8,31 Гц, 2Н), 4,40 (к, J=7,17 Гц, 2Н), 2,66 (с, 3Н), 1,42 (т, J=7,09 Гц, 3Н).
Стадия 7: Синтез (4-(5-метил-2-(трифторметил)оксазол-4-ил)фенил)метанола:
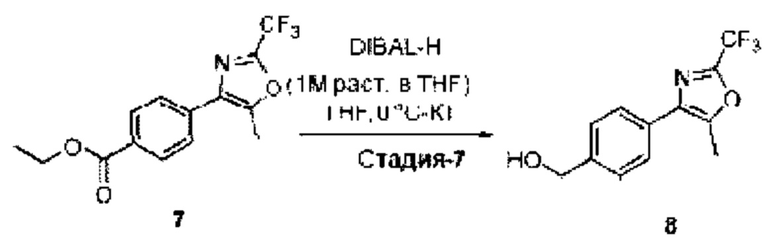
[0605] К перемешиваемому раствору этил 4-(5-метил-2-(трифторметил)оксазол-4-ил)бензоата 7 (0,170 г, 0,568 ммоль) в сухом ТГФ (5 мл) при 0°С добавляли DIBAL-H (1М раствор в толуоле, 0,850 мл, 0,850 ммоль). Реакционной смеси давали нагреться до комнатной температуры и затем перемешивали в течение 1 часа. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили разбавленной HCl (5 мл) и экстрагировали ЕА (2 × 20 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением неочищенного указанного в заголовке соединения (0,130 г). 1Н-ЯМР(400 МГц, CDCl3) δ 7,67 (д, J=7,83 Гц, 2Н), 7,46 (д, J=7,83 Гц, 2Н), 4,75 (с, 2Н), 2,62 (с, 3Н).
Стадия 8: Синтез 4-(4-((6-хлор-1H-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-5-метил-2-(трифторметил)оксазола (I-62):
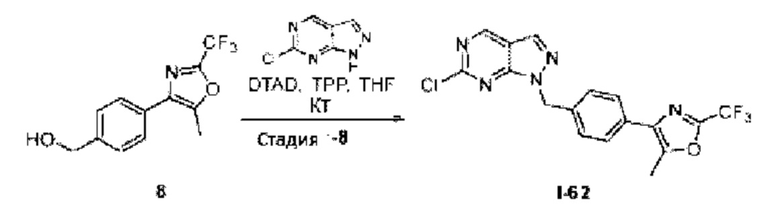
[0606] К перемешиваемому раствору (4-(5-метил-2-(трифторметил)оксазол-4-ил)фенил)метанола 8 (0,120 г, 0,466 ммоль) в ТГФ (5 мл) добавляли 6-хлор-1Н-пиразоло[3,4-d]пиримидин (0,072 г, 0,47 ммоль), DTAD (0,161 г, 0,699 ммоль) и ТРР (0,183 г, 0,699 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение 3 ч, и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (10 мл) и экстрагировали ЕА (3×10 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле, используя 15-20% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,100 г). ЖХ-МС (Способ В) (ИЭР+): m/z 393,95 (М+Н)+, 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,31 (с, 1Н), 8,51 (с, 1Н),7,67 (д, J=8,31 Гц, 2Н), 7,38 (ц, J=8,31 Гц, 2Н), 5,69 (с, 2Н), 2,61 (с, 3Н).
Получение общего промежуточного продукта I-63:
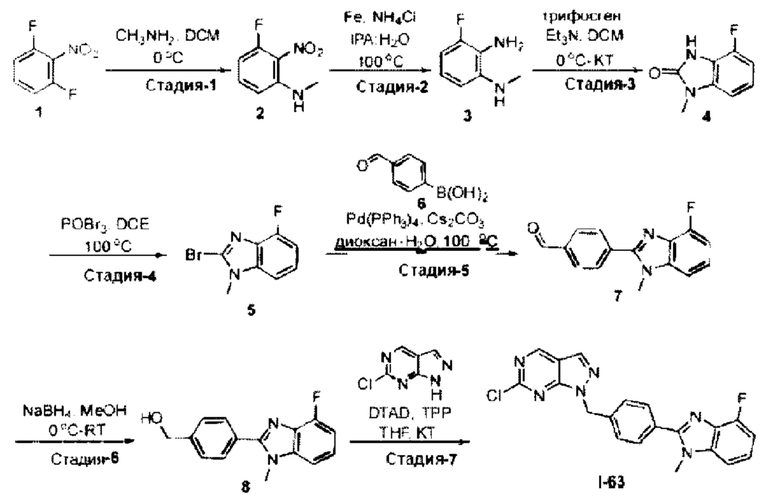 Стадия 1: Синтез 3-фтор-N-метил-2-нитроанилина:
Стадия 1: Синтез 3-фтор-N-метил-2-нитроанилина:
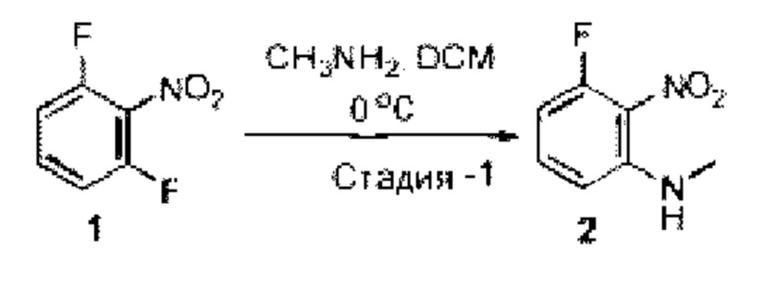
[0607] К перемешиваемому раствору 1,3-дифтор-2-нитробензола 1 (14,0 г, 88,1 ммоль) в DCM(150 мл) при 0°С добавляли метиламин (7,00 г, 228 ммоль) и смесь перемешивали в течение 3 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили ледяной водой (50 мл). Органический спой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением неочищенного указанного в заголовке соединения (9,00 г). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,29 (с, 1Н), 7,41 (дд, J=4,24, 8,73 Гц, 1Н), 7,01 (дд, J=8,73, 10,22 Гц, 1Н), 4,09 (с, 3Н).
Стадия2: Синтез 3-фтор-N1-метилбензол-1,2-диамина:
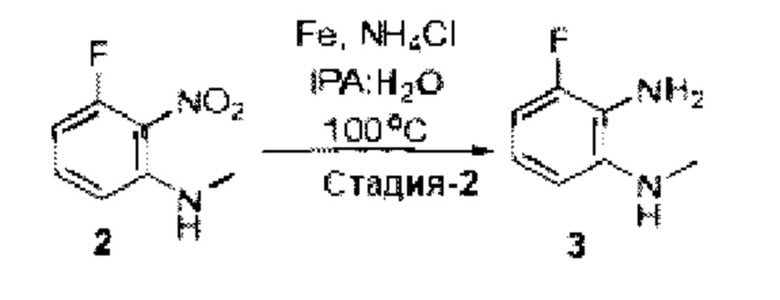
[0608] К перемешиваемому раствору 3-фтор-N-метил-2-нитроанилина 2 (9,0 г, 53 ммоль) в IPA:Н2О (4:1, 100 мл) добавляли хлорид аммония (28,2 г, 529 ммоль) и железа порошок (29,5 г, 529 ммоль) при комнатной температуре. Затем реакционную смесь нагревали в запаянной пробирке при 100°С в течение 3 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры и фильтровали через целит. Фильтрат упаривали при пониженном давлении, остаток разбавляли водой (25 мл) и экстрагировали DCM (3 × 100 мл). Объединенный органический слой промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении Полученное таким образом неочищенное соединение очищали хроматографией на силикагеле, используя 0-15% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения(4,67 г). ЖХ-МС (Способ В) (ИЭР+): m/z 141,0 (М+Н)+.
Стадия 3: Синтез 4-фтор-1-метил-1,3-дигидро-2Н-бензо[d]имидазол-2-она
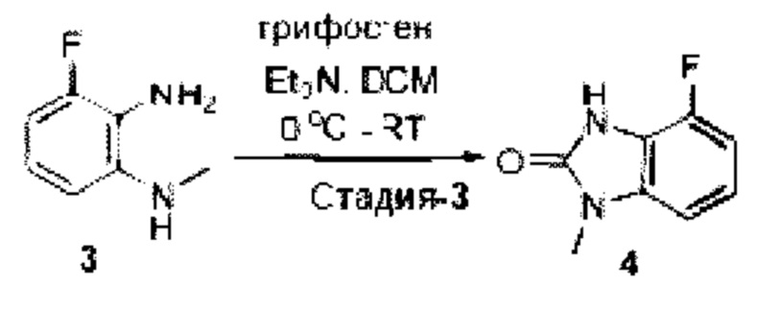
[0609] К перемешиваемому раствору 3-фтор-N1-метилбензол-1,2-диамина 3 (1,5 г, 10,70 ммоль) в DCM добавляли триэтиламин (1,4 г, 14 ммоль) и трифосген (0,950 г, 3,21 ммоль) при комнатной температуре. Смесь перемешивали в течение 12 ч, и за кодом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли DCM (10 мл) и промывали насыщенным раствором NaHCO3 (10 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, элюируя 0-40% ЕА в гексане, с получением указанного в заголовке соединения (0,770 г). ЖХ-МС (Способ В) (НЭР+): m/z 166 (М+Н)+.
Стадия4: Синтез 2-бром-4-фтор-1-метал-1H-бензо[d]имидазола:
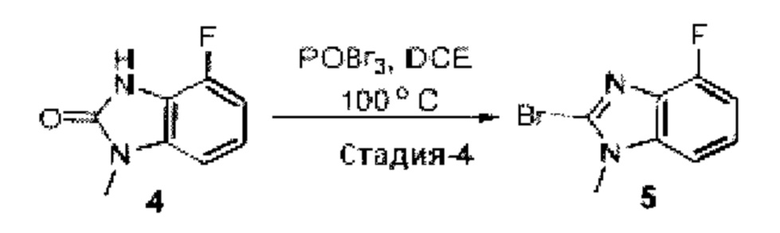
[0610] К перемешиваемому раствору 4-фтор-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-она 4 (0,770 г, 4,63 ммоль) в дихлорэтане (15 мл) при 0°С добавляли POBr3 (0,650 г, 2,27 ммоль). Реакционную смесь дополнительно нагревали при 90°С в течение 12 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры и упаривали при пониженном давлении. Полученный неочищенный остаток разбавляли ледяной водой (20 мл) и доводили рН до 8, используя 0,500 г NaHCO3). Затем смесь экстрагировали DCM (3 × 10 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-5% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,806 г). ЖХ-МС (Способ В) (ИЭР+): m/z 230,95 (М+Н)+; 1H-HMP (400 МГц, ДМСО-d6) δ 7,46 (д, J=7,9% Гц, 1Н), 7,27 (дт, J=4,74, 8,10 Гц, 1Н), 7,05 (дд, J=7,98, 10,97 Гц, 1Н), 3,81 (с, 3Н).
Стадия 5: Синтез 4-(4-фтор-1-метил-1Н-бензо[d]имидазол-2-ил)бензальдегида:
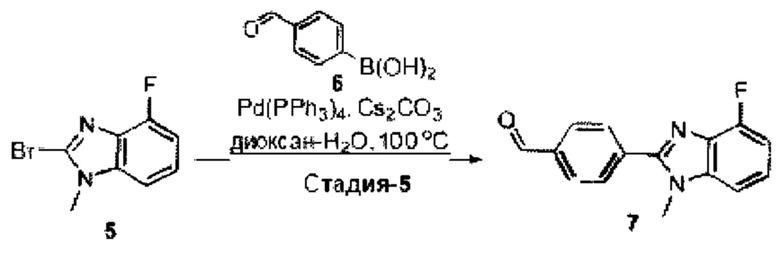
[0611] К перемешиваемому раствору 2-бром-4-фтор-1-метил-1Н-бензо[d]имидазола 5 (0,600 г, 2,62 ммоль) в диоксане:H2O (7:3 мл) добавляли K3PO4 (1,2 г, 7,9 ммоль) и (4-формилфенил) бороновую кислоту 6 (0,471 г, 3,14 ммоль) при комнатной температуре. Затем смесь дегазировали аргоном в течение 15 мин с последующим добавлением Х-фос (0,099 г, 0,21 ммоль) и Х-фос-Pd-G2 (0,082 г, 0,10 ммоль) при комнатной температуре. Реакционную смесь дополнительно дегазировали аргоном в течение 5 мин, а затем нагревали в запаянной пробирке при 100°С в течение 1 часа. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Полученный остаток растворяли в DCM (10 мл) и промывали Н2О (10 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-20% ЕА в гексане в качестве элюента, с получением указанного в заголовке с о единения (0,433 г). ЖХ-МС (Способ С) (ИЭР+): m/z 255,0 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 10,14 (с, 1Н), 8,09-8,15 (м, 4Н), 7,53 (д, J=8,48 Гц, 1Н),7,32 (дт, J=4,74, 8,10 Гц, 1Н), 7,10 (дд, J=7,98, 10,97 Гц, 1Н), 3,95 (с, 3Н).
Стадия 6: Синтез (4-(4-фтор-1-метил-1H-бензо[d]имидазол-2-ил) фенил)метанола:
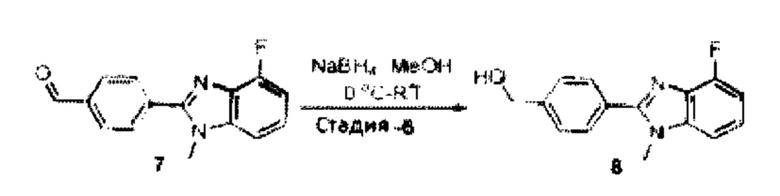
[0612] К перемешиваемому раствору 4-(4-фтор-1-метил-1Н-бензо[d]имидазол-2-ил)бензальдегида 5 (0,432 г, 1,70 ммоль) в метаноле (15 мл) при 0°С добавляли натрий борогидрид (0,128 г, 3,40 ммоль). Реакционной смеси давали нагреться до комнатной температуры, а затем перемешивали в течение 3 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили насыщенным раствором NH4Cl (5 мл) и упаривали при пониженном давлении Полученный неочищенный остаток растворяли в DCM (20 мл) и промывали солевым раствором (5 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-50% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,300 г). ЖХ-МС (Способ В) (ИЭР+): m/z. 256,90 (М+Н)+.
Стадия 7: Синтез 6-хлор-1-(4-(4-фтор-1-метнл-1Н-бензо[d]имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (1-63):
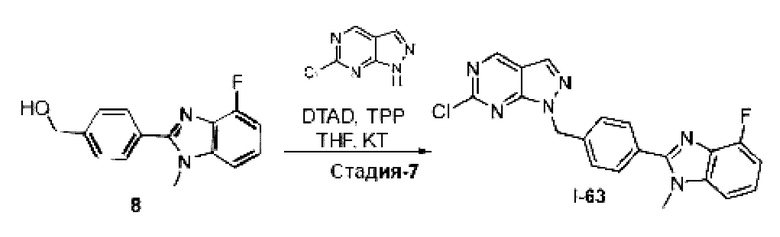
[0613] К охлажденного льдом раствору (4-(4-фтор-1-метил-1Н-бензо[d]имидазол-2-ил)фенил)метанола 8 (0,300 г, 1,17 ммоль) в ТГФ (10 мл) добавляли 6-хлор-1Н-пиразоло[3,4-d]пиримидин (0,162 г, 1,05 ммоль), DTAD (0,403 г, 1,76 ммоль) и ТРР (0,460 г, 1,76 ммоль). Реакционной смеси давали нагреться до комнатной температуры и затем перемешивали в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (10 мл) и экстрагировали DCM (2×20 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-60% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,160 г). ЖХ-МС (Способ С) (ИЭР+): m/z 393,00 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,33 (с, 1Н), 8,54 (с, 1Н), 7,85 (д, J=7,98 Гц, 2Н), 7,43-7,49 (м, 3Н), 7,27 (дт, J=4,49, 7,98 Гц, 1Н), 7,05 (дд, J=8,23, 10,72 Гц, 1Н), 5,77 (с, 2Н),3,87 (с, 3Н).
Получение общего промежуточного продукта 1-64:
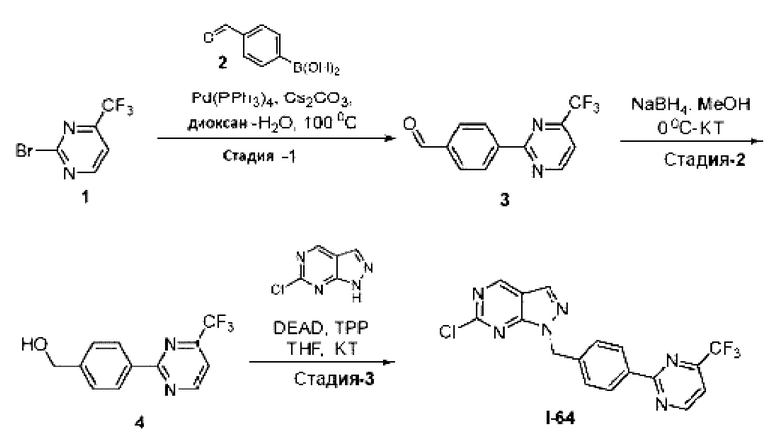
Стадия 1: Синтез 4-(4-(трифторметил)пиримидин-2-ил)бензальдегида:
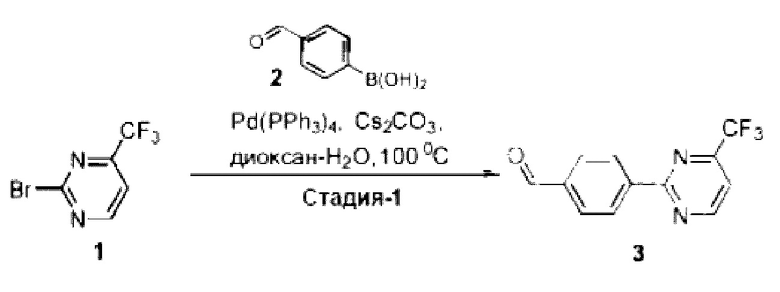
[0614] Раствор 2-бром-4-(трифторметил)пиримидина 1 (2,00 г, 8,81 ммоль), (4-формилфенил)бороновой кислоты 2 (1,50 г, 10,6 ммоль) и Cs2CO3 (7,15 г, 22,0 ммоль) в диоксане-Н2О (5:1, 25 мл), продували аргоном в течение 30 мин. К полученной реакционной смеси добавляли Pd(PPh3)4 (1,01 г, 0,881 ммоль) при комнатной температуре. Реакционную смесь нагревали в закрытой пробирке при 100°С в течение 12 ч. За ходом реакции следили с помощью ТСХ и ЖХМС. После завершения реакционную смесь охлаждали до комнатной температуры, разбавляли водой (250 мл) и экстрагировали ЕА (2×250 мл). Объединенный органический слой сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 20-50% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (1,50 г). ЖХ-МС (Способ В) (ИЭР+): m/z 253,2 (М+Н)+, 1Н-ЯМР (400 МГц, CDCl3) δ 10,14 (с, 1Н), 9,11 (д, J=4,99 Гц, 1Н), 8,70 (д, J=8,48 Гц, 2Н), 8,04 (д, J=7,98 Гц, 2Н), 7,60 (д, J=4,99 Гц, 1Н).
Стадия 2: Синтез (4-(4-(трифторметил)пиримидин-2-ил)фенил)метанола:
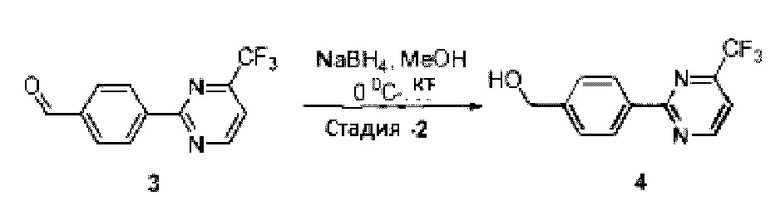
[0615] К перемешиваемому раствору 4-(4-(трифторметил)пиримидин-2-ил) бензальдегида 3 (1,50 г, 5,95 ммоль) в метаноле (30 мл) при 0°С добавляли порционно NaBH4 (0,449 г, 11,9 ммоль). Реакционную смесь дополнительно перемешивали при комнатной температуре в течение 2 ч, и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении, гасили ледяной водой (50 мл) и экстрагировали ЕА (100 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 20-40% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,400 г).
Стадия 3: Синтез 6-хлор-1-(4-(4-(трифторметил)пиримидин-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (1-64):
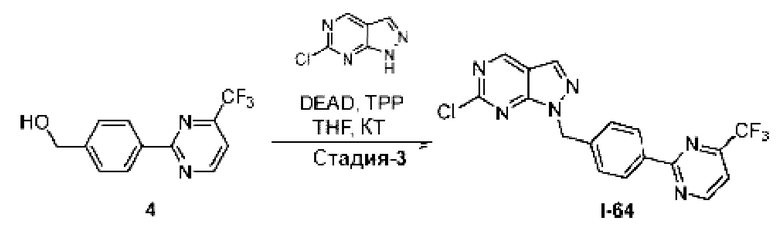
[0616] К перемешиваемому раствору (4-(4-(трифторметил) пиримидин-2-ил)фенил)метанола 4
(0,700 г, 2,75 ммоль) в ТГФ (7 мл) добавляли 6-хлор-1Н-пиразоло[3,4-d]пиримидин (0,388 г, 2,48 ммоль) и трифенилфосфин (1,083 г, 4,130 ммоль) при комнатной температуре. Полученную смесь перемешивали 5 мин. Затем смесь охлаждали до 0°С и обрабатывали DEAD (0,719 г, 4,13 ммоль). Реакционной смеси давали нагреться до комнатной температуры, а затем перемешивали в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (100 мл) и экстрагировали ЕА (2×250 мл). Объединенный органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-30% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,550 г). ЖХ-МС (Способ В) (ИЭР+): m/z 390,90 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,32 (с, 1Н), 9,26 (д, J=4,49 Гц, 1Н), 8,53 (с, 1Н), 8,37 (д, J=7,98 Гц, 2Н), 7,95 (д, J=4,49 Гц, 1Н), 7,44 (д, J=7,98 Гц, 2Н), 5,73-5,77 (м, 2Н).
Получение общего строительного блока (ВВ-1):
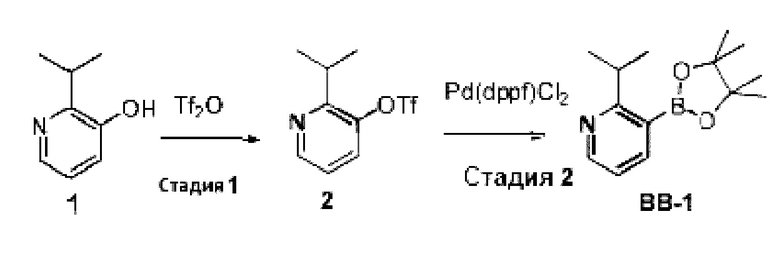
Стадия 1: Синтез 2-Изопропилпиридин-3-илтрифторметансульфоната:
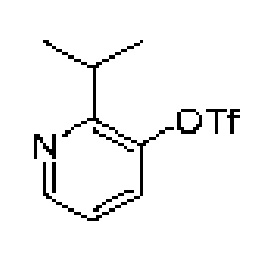
[0617] К раствору 2-изопропилпиридин-3-ола (0,40 г, 2,92 ммоль) в пиридине (4 мл) по каплям при 0°С в течение 2 минут добавляли Tf2O (0,82 г, 2,92 ммоль). После добавления смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. После завершения реакции, как показывает анализ ТСХ, смесь гасили холодной водой (10 мл) и нейтрализовали насыщенным водным раствором NaHCO3. Полученную смесь экстрагировали DCM (50 мл ×3), сушили безводным Na2SO4 (30 г), фильтровали и упаривали в вакууме. Остаток очищали колоночной хроматографией (ЕА:Нех=от 0:1 до 1:100) с получением 420 мг указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CDCl3) δ 8,60 (д, J=4,2 Гц 1Н), 7,57 (м, 1Н),7,24(м, 1Н), 3,42 (м, 1Н), 1,32-1,37 (д, J=7,2 Гц, 6Н).
Стадия 2: Синтез 2-Изопропил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (ВВ-1):
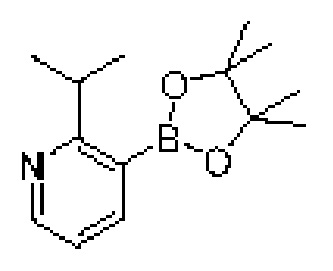
[0618] В атмосфере N2 смесь 2-изопрорилпиридин-3-илтрифторметансульфоната (0,28 г, 1,04 ммоль), (Bpin)2 (0,53 г, 2,08 ммоль), KOAc (0,20 г, 2,08 ммоль) и Pd(dppf)Cl2 (85 мг, 10 мол.%) в диоксане (5 мл) перемешивали при 95°С в течение 5 часов. Затем смесь гасили водой (15 мл) и экстрагировали ЕА (50 мл ×3). Объединенный органический слой сушили над безводным Na2SO4 (30 г), фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (ЕА: n-Нех=от 1:20 до 1:5) с получением 300 мг указанного в заголовке соединения. 1Н-ЯМР (300 МГц. CDCl3) δ 8,60 (дд, J=4,8, 2,1 Гц, 1Н), 8,00 (м, 1Н), 7,08 (м,1Н), 3,75 (м, 1Н), 1,32-1,37 (д, J=1,2 Гц, 6Н), 1,24-1,29 (с, 12Н).
Получение общего строительного блока (ВВ-2):
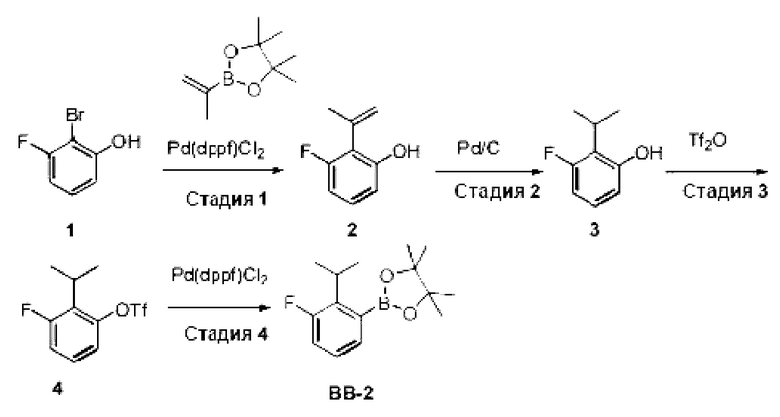
Стадня1: Синтез 3-фтор-2-(проп-1-ен-2-ил)фенола:
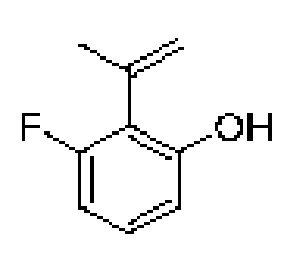
[0619] К смеси 2-бром-3-фторфенола(2 г, 10,5 ммоль), 4,4,5,5-тетраметил-2-проп-1-ен-2-ил)-1,3,2-диоксаборолана (2,37 г, 14,1 ммоль) и K2CO3 (2,9 г, 21 ммоль) в диоксане и воде (30 мл:6 мл) добавляли Pd(dppf)Cl2 (800 мг, 1 ммоль) в атмосфере азота. Полученную смесь перемешивали при 90°С в течение ночи, а затем гасили водой (50 мл) и экстрагировали ЕА (100 мл ×2). Объединенный органический слой сушили над Na2SO4 (30 г) и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (РЕ:ЕА=от 20:1 до 5:1) с получением 1,5 г указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CDCl3) δ 7,12 (м, 1Н), 6,74 (д, J=8,1 Гц, 1Н), 6,63 (т, J=8,4 Гц, 1Н), 5,72 (с, 1Н), 5,54 (с, 1Н), 5,16 (с, 1Н), 2,08 (с, 3Н).
Стадия 2: Синтез 3-фтор-2-изопропилфенола:
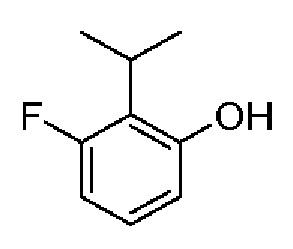
[0620] К раствору 3-фтор-2-(проп-1-ен-2-ил)фенола (1,5 г, 0,98 ммоль) в МеОН (20 мл) добавляли Pd/C (100 мг, 10% масс.) и перемешивали в атмосфере водорода. После завершения реакции, как показывает анализ ТСХ, суспензию фильтровали через слой целита. Осадок на фильтре промывали МеОН (10 мл). Фильтрат упаривали досуха с получением 1,3 г неочищенного указанного в заголовке соединения, которое использовали непосредственно в следующей стадии. 1H-ЯМР (300 МГц, CDCl3) δ 6,96 (м, 1Н), 6,59 (м, 1Н), 6,51 (д, J=8,1 Гц, 1Н),4,96(уш. с, 1Н), 3,40 (м, 1Н), 1,32-1,35 (д, J=6,6 Гц, 6Н).
Стадия 3: Синтез 3-фтор-2-изопропилфенилтрифторметансульфоната:
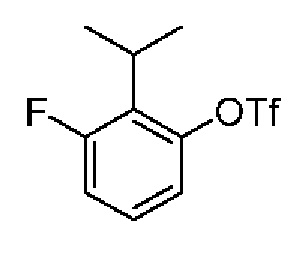
[0621] К раствору 3-фтор-2-изопропилфенола (1,2 г, 7,8 ммоль) и пиридина (3 мл) в DCM (20 мл) при 0°С добавляли избыточное количество Tf2O (2 мл). Реакционную смесь перемешивали при 0-5°С в течение 2 часов, затем гасили, выливая в холодную воду (20 мл). Полученную смесь обрабатывали насыщенным раствором NH4Cl (20 мл), а затем экстрагировали DCM (100 мл ×2). Объединенный органический слой сушили над Na2SO4 (30 г) и упаривали досуха, получая неочищенный продукт. Неочищенный продукт очищали хроматографией на силикагеле (РЕ:ЕА=100:1) с получением 1,8 г указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CDCl3) δ 7,19 (м, 1Н), 7,04-7,10 (м, 2Н), 3,32 (м, 1Н), 1,33-1,37 (д, J=6,6 Гц, 6Н)
Стадия 4: Синтез 2-(3-фтор-2-изопропилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (ВВ-2):
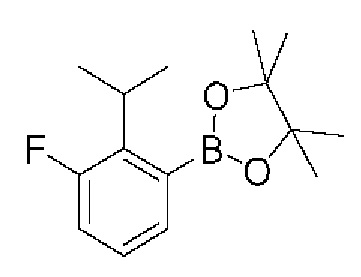
[0622] К раствору 3-фтор-2-изопропилфенилтрифторметансульфоната (10 0 мг, 0,35 ммоль), (Bpin)2 (178 мг, 0,7 ммоль) и KOAc (69 мг, 0,7 ммоль) в диоксане (3 мл) добавляли Pd(dppf)Cl2 (29 мг, 0,035 ммоль). После добавления полученную смесь нагревали до 90°С и перемешивали в течение 16 часов. Реакционную смесь фильтровали, и осадок на фильтре промывали ЕА (5 мл). Фильтрат упаривали досуха и остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА=100:1 до 30:1) с получением47 мг указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CDCl3) δ 7,43 (д, J=6,3 Гц, 1Н), 7,14 (м, 1Н), 7,02 (м, 1Н), 3.62 (м, 1Н), 1,35 (с, 12Н), 1,24-1,26 (д, J=6,0 Гц, 6Н) Получение общего строительного блока (ВВ-3):
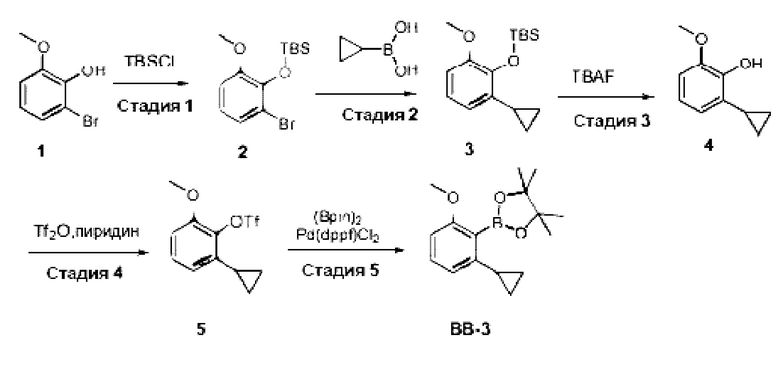
Стадия 1: Синтез (2-бром-6-метоксифенокси)(трет-бутил)диметилсилана:
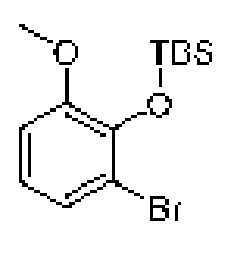
[0623] К раствору 2-бром-6-метоксифенола (500 мг, 2,46 ммоль) в DCM (10 мл) добавляли TBSC1 (446 мг, 2,96 ммоль), DMAP (30 мг, 0,246 ммоль) и TEA (373 мг, 3,69 ммоль). После завершения реакции, как показывает анализ ТСХ, реакцию гасили водой (10 мл) и экстрагировали ЕА (20 мл ×3). Объединенный органический слой сушили над безводным Na2SO4 (20 г), фильтровали и упаривали досуха в вакууме с получением 800 мг неочищенного указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CDCl3) δ 7,10 (м, 1Н), 6,74-6,78 (м, 2Н), 3,78 (с, 3Н), 1,05 (с, 9Н), 0,25 (с, 6Н).
Стадия 2: Синтез трет-бутил(2-циклопропил-6-метоксифенокси)диметилсилана:
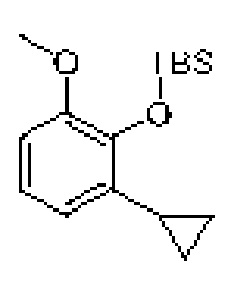
[0624] Смесь (2-бром-6-метоксифенокси)(трет-бутил)диметилсилана (700 мг, 2,21 ммоль), циклопропилбороновой кислоты (379 мг, 4,42 ммоль), K3PO4 (2,81 мг, 13,3 ммоль) и Pd(PPh3)4 (255 мг, 0,220 ммоль) в толуоле (35 мл) и H2O (1,75 мл) перемешивали при 95°С в течение ночи. Полученную смесь экстрагировали ЕА (50 мл ×3). Объединенный органический слой сушили над безводным Na2SO4 (30 г), фильтровали и упаривали досуха в вакууме. Концентрированный остаток очищали хроматографией на силикагеле (ЕА: н-гексагон=от 0:1 до 1:200) с получением 500 мг указанного в заголовке соединения 1Н-ЯМР (300 МГц, CDCl3) δ 6,78 (т, J=7,8 Гц, 1Н), 6,67 (д, J=8,1 Гц, 1Н), 6,38 (д, J=8,1 Гц, 1Н), 3,77 (с, 3Н), 2,21 (м, 1Н), 1,05 (с, 9Н), 0,89-0,95 (м, 2Н), 0,63-0,68 (м, 2Н), 0,19 (с, 6Н).
Стадия 3: Синтез 2-циклопропил-6-метоксифенола:
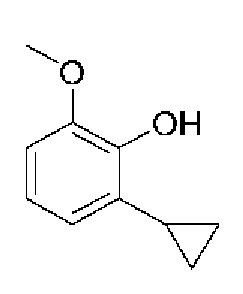
[0625] К раствору трет-бутил-(2-циклопропил-6-метоксифенокси)диметилсилана (500 мг, 1,8 ммоль) в ТГФ (5 мл) при комнатной температуре добавляли раствор TBAF (3,6 мл, 1 М) одной порцией. После добавления реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем гасили водой (15 мл) и экстрагировали ЕА (10 мл ×3). Объединенный органический слой сушили над безводным Na2SO4 (30 г), фильтровали и упаривали досуха в вакууме. Концентрированный остаток очищали хроматографией на силикагеле (ЕА:н-гексан=от 0:1 до 1:100) с получением 210 мг указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CDCl3) δ 6,69-6,79 (м, 2Н), 6,47 (д, J=7,8 Гц 1Н), 5,77 (с, 1Н), 3,88 (с, 3Н), 2,13 (м, 1Н), 0,91-0,98 (м, 2Н), 0,65-0,71 (м, 2Н).
Стадия 4: Синтез 2-Циклопропил-6-метоксифенилтрифторметансульфоната:
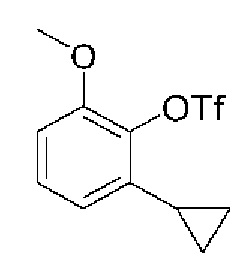
[0626] Раствор 2-циклопропил-6-метоксифенола (90 мг, 0,55 ммоль), пиридина (0,5 мл) и Tf2O (0,5 мл) в DCM (5 мл) перемешивали при кипячении с обратным холодильником в течение 30 минут. Затем реакционную смесь охлаждали до комнатной температуры и гасили насыщенным раствором NH4Cl (15 мл). Затем смесь экстрагировали DCM (10 мл ×3), и объединенный органический слой сушили над безводным Na2SO4 (20 г), фильтровали и упаривали досуха в вакууме. Остаток очищали хроматографией на силикагеле (ЕА:n-Нех=1:200 до 1:100) с получением 160 мг указанного в заголовке соединения. 1Н-ЯМР (300 МГц CDCl3) δ 7,19 (т, J=8,1 Гц, 1Н), 6,82 (дд, J=8,1, 0,9 Гц, 1Н), 6,54 (дд, J=8,1, 0,9 Гц, 1Н), 3,88 (с, 3Н), 2,08 (м, 1Н), 1,01-1,09 (м, 2Н), 0,71-0,79 (м, 2Н).
Стадия 5: Синтез 2-(2-Циклопропил-6-метоксифенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (ВВ-3):
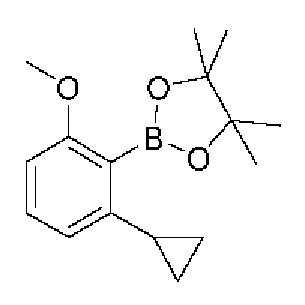
[0627] В атмосфере N2 смесь 2-циклопропил-6-метоксифенилтрифторметансульфоната (96 мг, 0,33 ммоль), (BPin)2 (166 мг, 0,650 ммоль), KOAc (96 мг, 0,98 ммоль) и Pd(PPh3)4 (35 мг, 0,03 ммоль) в диоксане (2 мл) перемешивали при 80°С в течение ночи. Затем реакционную смесь разбавляли водой (10 мл) и экстрагировали ЕА (10 мл ×3). Объединенные органические фазы сушили над безводным Na2SO4 (20 г), фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (ЕА:n-Нех=от 1:200 до 1:100) с получением 30 мг указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CDCl3) δ 7,16-7,21 (т, J=8,1 Гц, 1Н), 6,62 (д, J=8,1 Гц, 1Н), 6,50 (д, J=8,1 Гц 1Н), 3,76 (с, 3Н), 1,96 (м, 1Н), 1,37 (с, 12Н), 0,85-0,91 (м, 2Н), 0,71-0,73 (м, 2Н).
Получение общего строительного блока (ВВ-4):
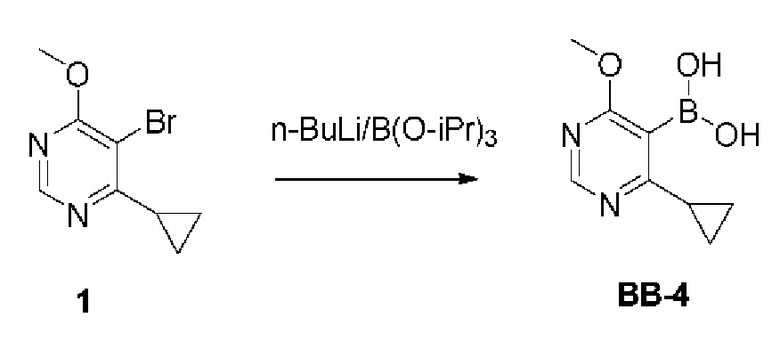
Стадия 1: Синтез (4-Циклопропил-6-метоксипиримидин-5-ил)бороновой кислоты:
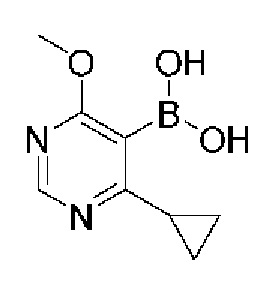
[0628] К раствору 5-бром-4-циклопропил-6-метоксипиримидина (500 мг, 2,2 ммоль) и триизопропилбората (533,7 мг, 2,8 ммоль) в толуоле (5 мл) и ТГФ (1,5 мл) при -78°С по каплям в течение 30 мин добавляли h-BuLi (1,1 мл, 2,8 ммоль). После перемешивания реакционной смеси при -78°С в течение 30 минут реакционную смесь нагревали до -20°С в течение 1 часа. Затем реакцию гасили водным 1 н. раствором HCl (3,4 мл). После перемешивания смеси при комнатной температуре в течение 30 минут смесь обрабатывали насыщенным водным раствором Na2CO3 для доведения рН до 8. Смесь экстрагировали ЕА (100 мл ×2). Объединенный органический слой сушили над безводным Na2SO4 (30 г) и упаривали в вакууме с получением неочищенного указанного в заголовке соединения (0,450 г). 1Н-ЯМР (300 МГц, CDCl3) δ 8,61 (с, 1Н), 6,56 (уш. с, 2Н), 4,01 (с, 3Н), 3,02 (м, 1Н), 1,38-1,35 (м, 1Н), 1,18-1,23 (м, 2Н), 1,00-1,06 (м, 2Н).
Следующее соединение было получено из коммерческого бромида в соответствии с процедурой ВВ-4:


Получение общего строительного блока (ВВ-6):
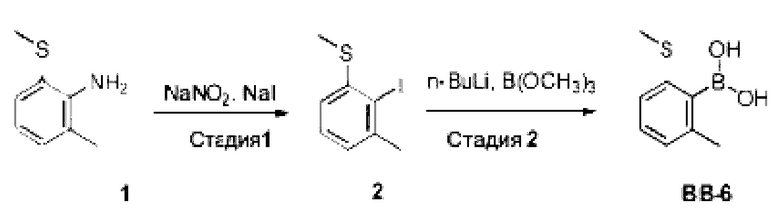
Стадия 1: Синтез (2-йод-3-метилфенил)(метил)сульфана:
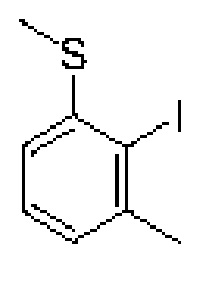
[0629] Раствор 2-метил-6-(метилтио)анилина (1,0 г, 6,53 ммоль) в ацетоне (20 мл) и конц. HCl (1,36 мл) перемешивали при 0°С в течение 10 минут. Затем к полученной смеси добавляли раствор NaNO2 (540 мг, 7,84 ммоль) в Н2О (8 мл) и раствор NaI (2,06 г, 13,7 ммоль) в Н2О (8 мл) в течение 10 минут. После добавления реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь гасили насыщенным раствором NH4Cl (20 мл) и экстрагировали ЕА (10 мл ×3). Объединенный органический слой сушили над безводным Na2SO4 (20 г), фильтровали и упаривали в вакууме. Остаток очищали колоночной хроматографией (ЕА:n-Нех=от 0:100 до 1:100) с получением 970 мг указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CDCl3) δ 7,22 (м, 1Н), 7,02 (дд, J=8,4, 0,6 Гц, 1Н), 6,88 (д, J=8,4,0,6 Гц, 1Н), 2,47 (с, 3Н), 2,45 (с,3Н).
Стадия 2: Синтез (2-Метил-6-(метилтио)фенил)бороновой кислоты (ВВ-6):
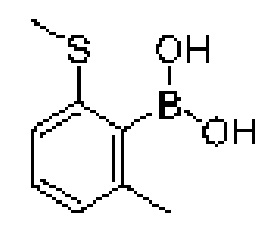
[0630] К раствору (2-иод-3-метилфенил)(метил)сульфана (200 мг, 0,76 ммоль) в безводном ТГФ (4 мл) добавляли h-BuLi (0,37 мл, 2,5 М) при -78°С в течение 10 мин. После перемешивания реакционной смеси при -78°С в течение 45 минут по каплям в течение 10 минут добавляли В(ОСН3)3 (237 мг, 2,28 ммоль). После добавления полученную смесь перемешивали при -78°С в течение дополнительных 30 минут и давали нагреться до комнатной температуры в течение 40 минут. После того как реакцию гасили водным 1 н. раствором HCl (2 мл), реакционную смесь перемешивали при комнатной температуре в течение около 30 минут. Попущенную смесь экстрагировали ЕА (5 мл ×3). Объединенные органнческие слои сушили над безводным Na2SO4 (10 г), фильтровали и упаривали в вакууме. Остаток суспендировали в н-гексане (5 мл), затем фильтровали для сбора твердых веществ, которые затем сушили в вакууме с получением 75 мг указанного в заголовке соединения. 1Н-ЯМР (300 МГц ДМСО-d6) δ 7,11-7,21 (м, 2Н), 6,95 (д, J=7,2 Гц, 1Н), 2,40 (с, 3Н), 2,23 (с, 3Н).
Общая процедура получения строительных блоков на основе алкилированной пиримидинбороновой кислоты с помощью  Синтез (4-Циклопропил-6-((терагидрофуран-3-ил)окси) пиримидин-5-ил)бороновой кислоты (BB-7):
Синтез (4-Циклопропил-6-((терагидрофуран-3-ил)окси) пиримидин-5-ил)бороновой кислоты (BB-7):
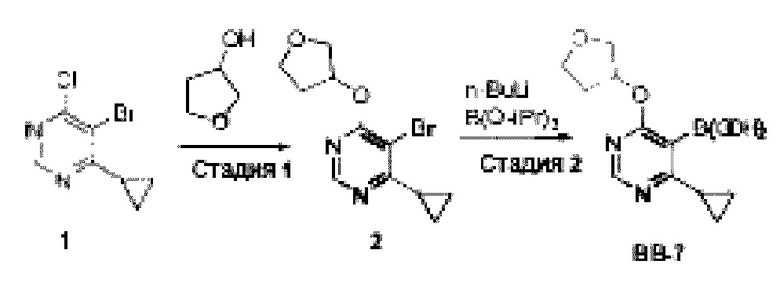
Стадия 1: Синтез 5-бром-4-циклопропил-6-((тетрагидрофуран-3-ил)окси)пиримидина:
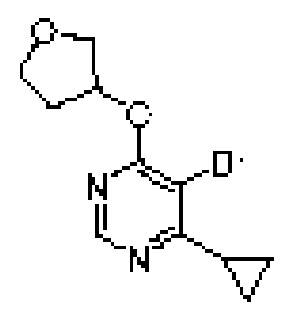
[0631] К раствору тетрагидрофуран-3-ола (107 мг, 1.95 ммоль) в сухом ТГФ (5 мл) при 0°С добавляли 60% дисперсию гидрида натрия (76 мг, 1,95 ммоль) порциями в течение 5 минут. После перемешивания смеси при 0°С в течение 1 часа к реакционной смеси по каплям в течение 5 минут добавляли раствор 5-бром-4-хлор-6-циклопропилпиримидина (300 мг, 1,30 ммоль) в сухом ТГФ (5 мл). После перемешивания реакционной смеси при комнатной температуре в течение 1 ч реакцию гасили водой (10 мл) и экстрагировали ЕА (5 мл ×3). Объединенный органический слой сушили над сульфатом натрия (20 г), фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (РЕ:ЕА=20:1) с получением 358 мг указанного в заголовке соединения ЖХ-МС (Способ А) (ИЭР+): m/z 285 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 8,33 (с, 1Н), 5,53 (м, 1Н), 3,93-4,06 (м, 2Н), 3,84-3,97 (м, 2Н), 2,45 (м, 1Н), 2,12-2,23 (м, 2Н), 1,06-1,13 (м, 2Н), 1,00-1,06 (м, 2Н).
Стадия 2: Синтез (4-Циклопропил-6-((тетрагидрофуран-3-ил)окси)пиримидин-5-ил)бороновой кислоты (ВВ-7):
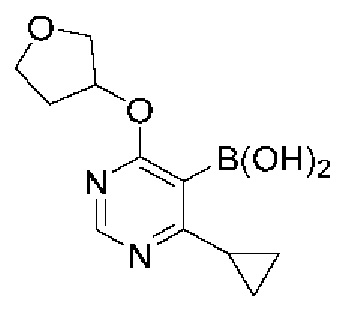
[0632] К смеси 5-бром-4-циклопропил-6-((тетрагидрофуран-3-ил)окси)пиримидина (200 мг, 0,7 ммоль) и триизопропилбората (171 мг, 0,9 ммоль) в толуоле (4 мл) и ТГФ (1 мл) при -78°С, по каплям в течение 10 минут добавляли н-бутиллитий (0,36 мл, 0,9 ммоль). Смесь перемешивали при -78°С в течение 30 минут, затем нагревали до -20°С и перемешивали в течение 1 часа. Реакцию гасили водным 1 н. раствором HCl (3 мл), а затем перемешивали при комнатной температуре в течение 30 минут. Добавляли насыщенный водный раствор Na2CO3, чтобы довести рН до 8, а затем смесь экстрагировали ЕА (25 мл ×3) Объединенный органический слой сушили над безводным Na2SO4 (30 г), фильтровали и упаривали в вакууме с получением 146 мг неочищенного указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 251 (М+Н)+; 1Н-ЯМР (300 МГц, ДМСО-d6) δ 8,51 (с, 1Н), 8,44 (с, 2Н), 5,50 (м, 1Н), 3,90 (м, 1Н), 3,70-3,88 (м, 3Н), 2,18 (м, 1Н), 1,86-1,98 (м, 2Н), 0,90-1,05 (м, 4Н).
Следующие соединения были получены в соответствии с методом получения ВВ-7:
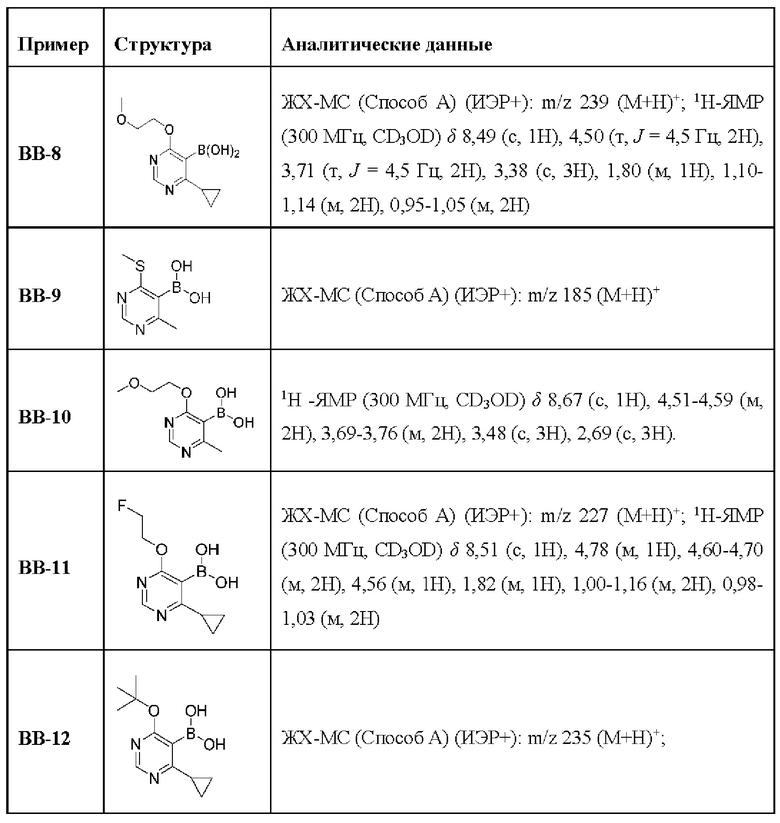
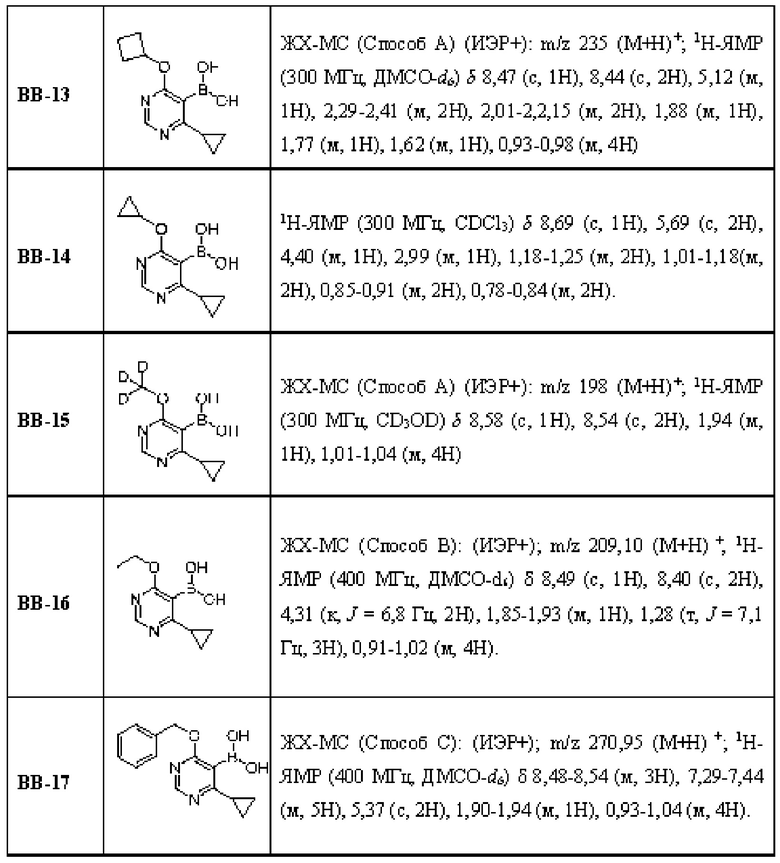
Получение общего строительного блока (ВВ-18):
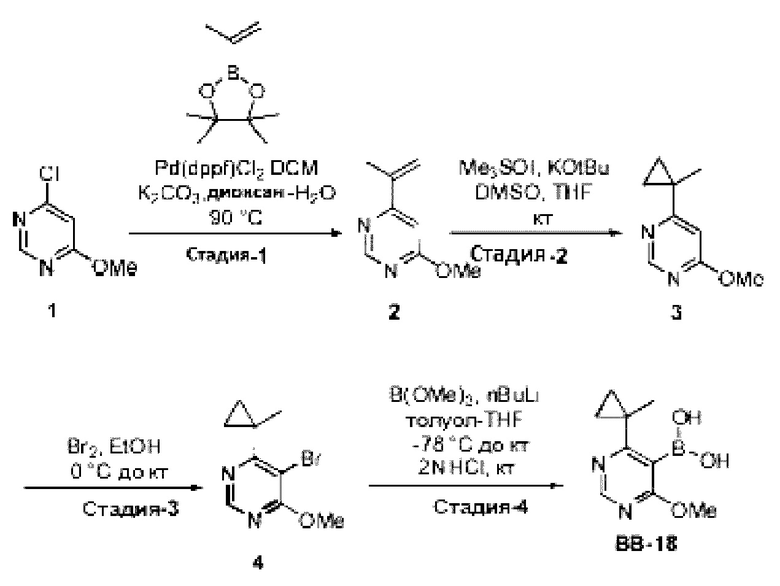
Стадия 1: Синтез 4-метокси-6-(проп-1-ен-2-ил)пиримидина:
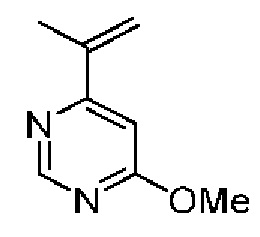
[0633] К перемешиваемому раствору 4-хлор-6-метоксипиримидина (10,0 г, 69,2 ммоль) в диоксане (100 мл) и воде (15 мл) добавляли K2CO3 (19,1 г, 138 ммоль). Полученную смесь дегазировали аргоном в течение 15 мин с последующим добавлением 4,4,5,5-тетраметил-2-(проп-1-ен-2-ил)-1,3,2-диоксаборолана (13,90 г, 83,00 ммоль) и Pd(dppf)Cl2-DCM (2,80 г, 3,40 ммоль). Реакционную смесь дополнительно дегазировали аргоном в течение 10 мин и нагревали до 90°С в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли ЕА и фильтровали через слой целита. Органический слой собирали и сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-15% ЕА в гексане, с получением 7,80 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 150,85 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,74 (с, 1Н), 7,00 (с, 1Н), 6,18 (с, 1Н), 5,44 (с, 1Н), 3,93 (с, 3Н), 2,09 (с, 3Н)
Стадия 2: Синтез 4-метокси-6-(1-метилциклопропил)пиримидина:
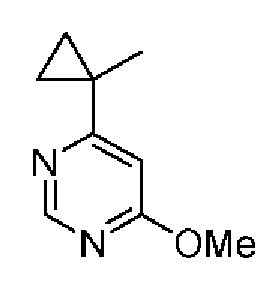
[0634] К перемешиваемому раствору иодида триметилсульфоксония (16,5 г, 74,9 ммоль) в ДМСО
(100 мл) добавляли KOtBu (8,42 г, 74,9 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. К полученной реакционной смеси добавляли раствор 4-метокси-6-(проп-1-ен-2-ил)пиримидина 2 (7,50 г, 49,9 ммоль) в ТГФ (10 мл), и реакционную смесь дополнительно перемешивали при комнатной температуре в течение 3 ч. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли насыщенным раствором NaHCO3 и экстрагировали ЕА (2×20 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали колоночной хроматографией, используя 5-10% ЕА в гексане, с получением 3,80 г указанного в заголовке соединения. ЖХМС (Способ В) (ИЭР+): m/z 164,85 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,59 (с, 1Н), 6,79 (с, 1Н), 3,89 (с, 3Н), 1,40 (с, 3Н), 1,19-1,25 (м, 2Н), 0,85 (к, J=3,4 Гц, 2Н).
Стадия 3: Синтез 5-бром-4-метокси-6-(1-метилциклопропил)пиримидина:
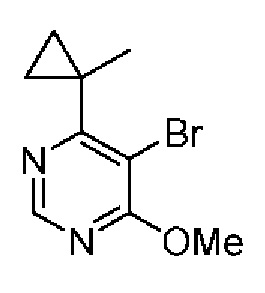
[0635] К перемешиваемому раствору 4-метокси-6-(1-метилциклопропил)пиримидина (3,00 г, 18,2 ммоль) в EtOH (50 мл) при 0°С добавляли Br2 (2,91 мл, 54,0 ммоль), и полученную реакционную смесь перемешивали при комнатной температуре 72 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили насыщенным раствором бисульфита натрия и экстрагировали ЕА (2×50 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 2-5% ЕА в гексане, с получением 3,0 г указанного в заголовке соединения. ЖХМС (Способ В) (ИЭР+): m/z 244,80 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,63 (с, 1Н), 3,98 (с, 3Н), 1,35 (с, 3Н), 0,88-0,93 (м, 2Н), 0,78-0,84 (м, 2Н).
Стадия 4: Синтез (4-Метокси-6-(1-метилциклопропил)пиримидин-5-ил)бороновой кислоты (ВВ-18):
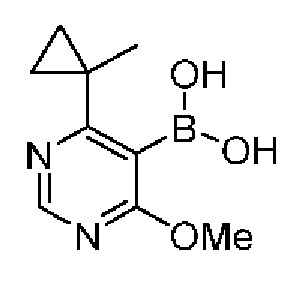
[0636] К перемешиваемому раствору 5-бром-4-метокси-6-(1-метилциклопропил)пиримидина (0,50 г, 2,1 ммоль) в толуоле:ТГФ (3:1,8 мл) добавляли триметилборат (0,32 г, 3,1 ммоль). К полученной реакционной смеси при -78°С добавляли н-BuLi (1,6 М, 1,93 мл, 3,1 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили 2 н. HCl (10 мл) и перемешивали при комнатной температуре в течение 2 часов. Затем рН доводили до 7 добавлением водн. раствора NaHCO3. Затем смесь экстрагировали ЕА (2×10 мл), и объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение растирали с Et2O, фильтровали и сушили, получая 0,11 г указанного в заголовке соединения ЖХМС (Способ С) (ИЭР+): m/z 209,00 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,56 (с, 1Н), 8,29 (с, 2Н), 3,84 (с, 3Н), 1,41 (с, 3Н), 1,10 (д, J=2,0 Гц, 2Н), 0,65-0,69 (м, 2Н)
Получение общего строительного блока (ВВ-19):
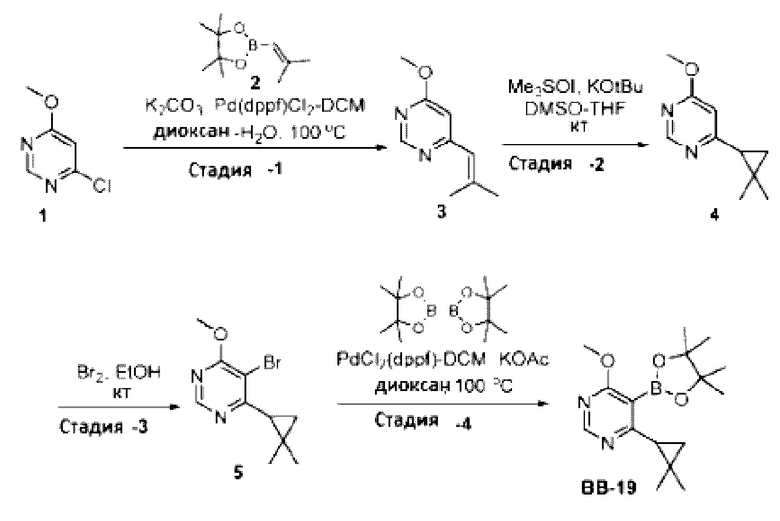
Стадия 1.: Синтез 4-метокси-6-(2-метилпроп-1-ен-1-ил)пиримидина:
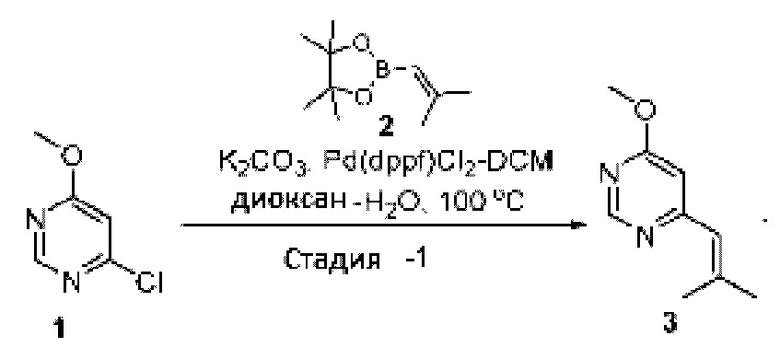
[0637] К перемешиваемому раствору 4-хлор-6-метоксипиримидина 1 (5,00 г, 34,7 ммоль), 4,4,5,5- тетраметил-2-(2-метилпроп-1-ен-1-ил)-1,3,2-диокса6оролана 2 (7,50 г, 41,7 ммоль) в диоксане (50 мл) и воде (8 мл), добавляли K2CO3 (14,30 г, 104,2 ммоль) при комнатной температуре. Смесь продували аргоном в течение 30 минут, а затем обрабатывали Pd(dppf)Cl2-DCM (2,83 г, 3,47 ммоль). Затем смесь нагревали в запаянной пробирке при 100°С в течение 16 ч, и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, разбавляли водой (50 мл) и экстрагировали ЕА (2×30 мл). Объединенный органический слой сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-2% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (5,20 г). ЖХ-МС (Способ В) (ИЭР+):m/z 164,8 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6)δ 8,70 (с, 1Н), 6,70 (с, 1Н), 6,20 (с, 1Н), 3,90(с, 3Н), 2,18 (с, 3Н), 1,93 (с, 3Н).
Стадия 2: Синтез 4-(2,2-диметилциклопропил)-6-метоксипиримидина:
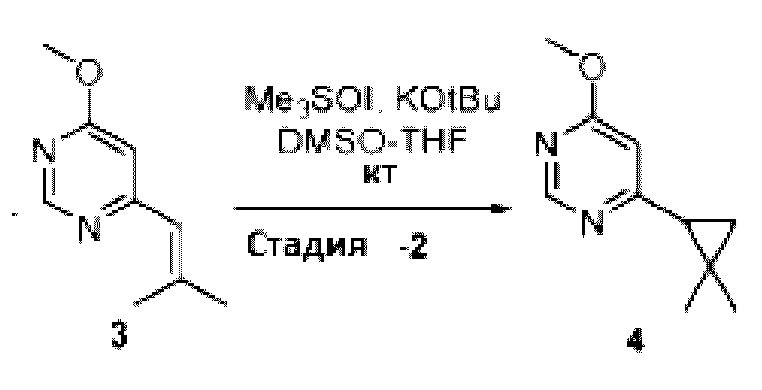
[0638] К перемешиваемому раствору йодида триметилсульфоксония (9,09 г, 41,1 ммоль) и трет-бутоксида калия (4,5 г, 41,1 ммоль) в ДМСО (50 мл) и ТГФ (5 мл) добавляли 4-метокси-6-(2-метилпроп-1-ен-1-ил)пиримидин 3 (4,5 г, 27,4 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение 16 ч, и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (50 мл) и экстрагировали ЕА (2×40 мл). Объединенный органический слой сушили над безводным сульфатом натрия и упаривали при пониженном давлении с получением указанного в заголовке соединения (4,5 г), которое использовали как таковое в следующей стадии без дополнительной очистки. ЖХ-МС (Способ В) (ИЭР+): m/z 178,9(М+Н)+.
Стадия 3: Синтез 4-(2,2-диметилциклопропил)-6-метоксипиримидина:
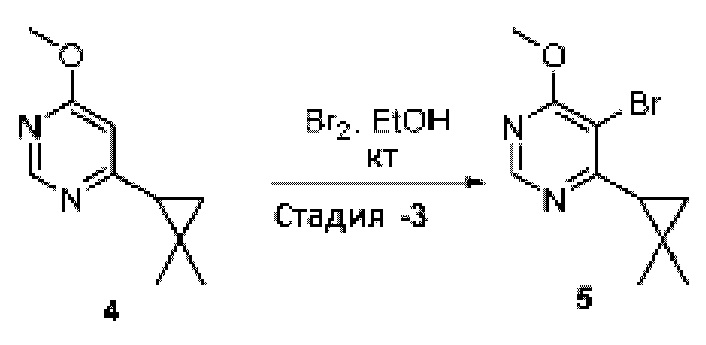
[0639] К перемешиваемому раствору 4-(2,2-диметилциклопропил)-6-метоксипиримидина 4 (4,50 г,2,53 ммоль) в этаноле (50 мл) при 0°С по каплям добавляли бром (6,02 г, 3,79 ммоль). После завершения добавления реакционную смесь перемешивали при комнатной температуре в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (50 мл) и экстрагировали гексаном (3×20 мл) Объединенный органический слой сушили над безводным сульфатом натрия и упаривали при пониженном давлении с получением неочищенного указанного в заголовке соединения (1,50 г), которое использовали без очистки в следующей стадии. ЖХ-МС (Способ В) (ИЭР+): m/z 256,8 (М+); 1Н-ЯМР(400 МГц, ДМСО-d6) δ 3,60 (с, 1Н), 3,99 (с, 3Н), 2,21 (дд, J=5,98,7,48 Гц, 1Н), 1,47 (т, J=4,74 Гц, 1Н), 1,30 (с, 3Н), 0,97 (дд, J=3,99, 7,48 Гц, 1Н), 0,84 (с, 3Н).
Стадия 4: Синтез 4-(2,2-диметилциклопропил)-6-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидина (ВВ-19):
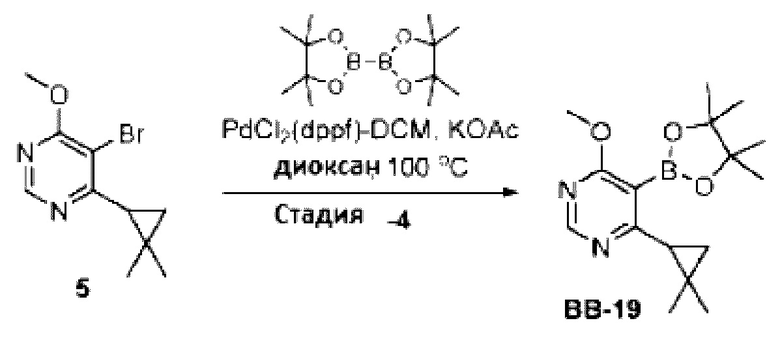
[0640] К раствору 5-бром-4-(2,2-диметилциклопропил)-6-метоксипиримидина 5 (0,500 г, 1,95 ммоль) и 4,4,4',4',5,5,5'5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (2,40 г, 9,77 ммоль) в диоксане (10 мл) добавляли KOAc (0,382 г, 3,91 ммоль) при комнатной температуре. Полученную смесь продували аргоном в течение 30 минут, а затем обрабатывали Pd(dppf)Cl2-DCM (0,159 г, 0,195 ммоль). Затем реакционную смесь нагревали в закрытой пробирке при 100°С в течение 2 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-20% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,330 г). ЖХ-МС (Способ В) (ИЭР+): m/z 304,00 (М+).
Получение общего строительного блока (ВВ-20):
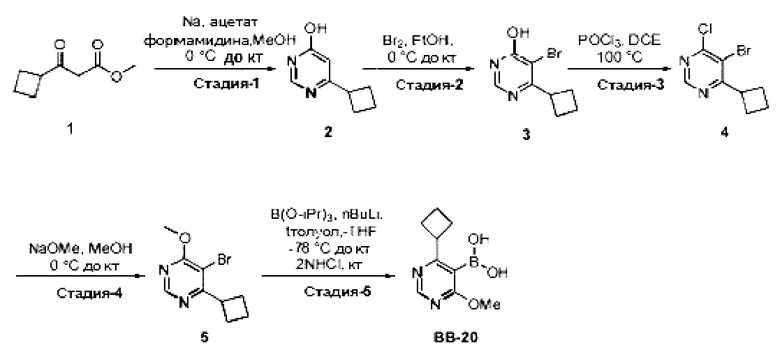
Стадия 1: Синтез 6-циклобутилпиримидин-4-ола:
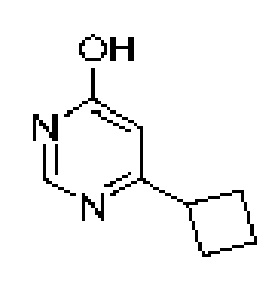
[0641] Металлический натрий (1,47 г, 64,0 ммоль) добавляли к МеОН (40 мл) и перемешивали при комнатной температуре в течение 1 часа. К полученной реакционной смеси добавляли ацетат формамидина (4,00 г, 38,4 ммоль) и метил 3-циклобутил-3-оксопропаноата (4,00 г, 25,6 ммоль) при 0°С. Полученную смесь перемешивали при комнатной температуре в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (20 мл) и доводили до рН=4 с помощью АсОН. Затем реакционную смесь упаривали при пониженном давлении. Неочищенную реакционную смесь экстрагировали 10% МеОН в DCM (3×50 мл), и объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле на диоксиде кремния, используя 5-10% МеОН в DCM, с получением 1,70 г указанного в заголовке соединения. ЖХ-МС (Способ С) (ИЭР+): m/z 150,93 (М+Н)+; 1Н-ЯМР (400 МГц ДМСО-d6) δ 11,66-12,56 (м, 1Н), 8,12 (с, 1Н), 6,09 (с, 1Н), 3,27-3,40 (м, 1Н), 2,08-2,19 (м, 4Н), 1,87-2,01 (м, 1Н), 1,74-1,86 (м, 1Н).
Стадия 2: Синтез 5-бром-6-циклобутилпиримидин-4-ола:
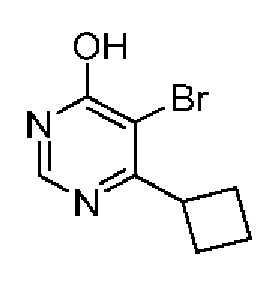
[0642] К охлажденному льдом раствору 6-циклобутилпиримидин-4-ола (1,70 г, 11,3 ммоль) в EtOH (30 мл) медленно добавляли Br2 (1,80 г, 11,3 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Неочищенную реакционную смесь гасили насыщенным водн. раствором Na2S2O3 (50 мл) и полученное твердое вещество фильтровали, промывали избытком раствора Na2S2O3 и сушили с получением 2,0 г указанного в заголовке соединения. ЖХ-МС (Способ С) (ИЭР+): m/z 230,86 (М+Н)+ 81Br.
Стадия 3: Синтез 5-бром-4-хлор-6-циклобутилпиримидина:
[0643] К перемешиваемому раствору 5-бром-6-циклобутилпиримидин-4-ола (2,00 г, 8,70 ммоль) в 1,2-DCE (30 мл) добавляли POCl3 (5 мл) и реакционную смесь нагревали до 100°С в течение 3 ч. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Неочищенную реакционную смесь разбавляли Н2О (15 мл), подщелачивали до рН=8, используя насыщенный раствор NaHCO3, и экстрагировали ЕА (3×30 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали флэш-хроматографией на диоксиде кремния, используя 0-10% ЕА в гексане, с получением 1,30 г указанного в заголовке соединения. ЖХ-МС (Способ С) (ИЭР+): m/z 248,80 (М+Н)+ Br; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,96 (с, 1Н), 3,92-4,02 (м, 1Н), 2,29-2,39 (м, 4Н), 1,96-2,10 (м, 1Н), 1,76-1,88 (м, 1Н).
Стадия 4: Синтез 5-Бром-4-циклобутил-6-метоксипиримидина:
[0644] К охлажденному льдом раствору 5-бром-4-хлор-6-циклобутилпиримидина (1,30 г, 5,20 ммоль) в МеОН (30 мл) добавляли метоксид натрия (1,17 г, 21,00 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Остаток растворяли в воде (10 мл) и экстрагировали ЕА (3×20 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением 1,0 г указанного в заголовке соединения. ЖХ-МС (Способ С) (ИЭР+): m/z 244,90 (М+Н)+ 81Br; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,71 (с, 1Н), 3,98 (с, 3Н), 3,85-3,94 (м, 1Н), 2,23-2,39 (м, 4Н), 1,95-2,08 (м, 1Н), 1,77-1,87 (м, 1Н).
Стадия 5: Синтез (4-Циклобутил-6-метоксипиримидин-5-ил)бороновой кислоты (ВВ-20):
[0645] К перемешиваемому раствору 5-бром-4-циклобутил-6-метоксипиримидина (1,00 г, 4,11 ммоль) в толуоле: ТГФ (5:1, 30 мл) в атмосфере азота добавляли триизопропилборат (2,86 мл, 12,30 ммоль) и реакционную смесь охлаждали до -78°С. Затем по каплям добавляли н-BuLi (1,6 М, 3,85 мл, 6,17 ммоль) в течение 45 минут. Перемешивание продолжали при той же температуре в течение 30 минут, а затем реакционную смесь дополнительно перемешивали при комнатной температуре в течение 3 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили 2 н. HCl (10 мл) и перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь подщелачивали до рН 8, используя K2CO3 (твердый), и экстрагировали ЕА (3×50 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение растирали с Et2O и н-пентаном (10 мл), фильтровали для сбора твердого вещества и сушили с получением 0,42 г указанного в заголовке соединения. ЖХ-МС (Способ С) (ИЭР+): m/z 209,03 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,70 (с, 1Н), 8,33 (с, 2Н), 3,85 (с, 3Н), 3,46-3,58 (м, 1Н), 2,26-2,38 (м, 2Н), 2,10-2,20 (м, 2Н), 1,89-2,03 (м, 1Н), 1,74-1,86 (м, 1Н).
Следующие строительные блоки были приготовлены в соответствии с процедурой ВВ-20 из соответствующего β-кетоэфира или 6-алкилпиримидин-4-ола:
Получение общего строительного блока (ВВ-25):
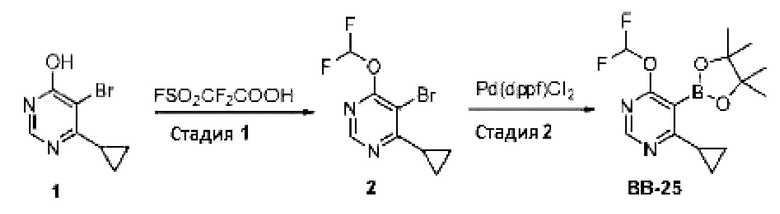
Стадия 1: Синтез 5-Бром-4-циклопропил-6-(дифторметокси)пиримидина:
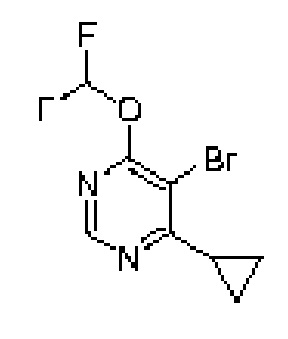
[0646] К суспензии 5-бром-6-циклопропилпиримидин-4-ола (1,0 г, 4,7 ммоль) в ACN (100 мл) при комнатной температуре добавляли NaH (60% дисперсия, 560 мг, 14,0 ммоль) порциями в течение 10 мин. После перемешивания смеси при комнатной температуре в течение 20 минут по каплям в течение 10 минут добавляли 2,2-дифтор-2-(фторсульфонил)уксусную кислоту (1,4 г, 7,9 ммоль). После добавления смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции, как показывает анализ ТСХ, реакцию гасили насыщенным раствором NH4Cl (20 мл) и экстрагировали ЕА (50 мл ×2) Объединенный органический слой сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ:ЭА=от 20:1 до 10:1) с получением 600 мг указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CDCl3) δ 8,46 (с, 1Н), 7,48 (т, J=71,4 Гц), 2,58 (м, 1Н), 1,10-1,30 (м, 4Н).
Стадия 2: Синтез 4-Циклопропил-6-(дифторметокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидина (ВВ-25):
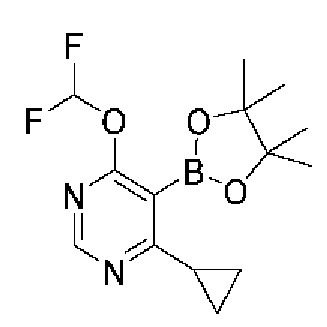
[0647] К смеси 5-бром-4-циклопропил-6-(дифторметокси)пиримидина (100 мг, 0,38 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (192 мг, 0,75 ммоль) и KOAc (112 мг, 1,14 ммоль) в диоксане (3 мл) в атмосфере азота добавляли Pd(dppf)Cl2 (33,00 мг, 10 мол.%). Полученную смесь перемешивали при 95°С в течение ночи. После завершения реакции на основании анализа ТСХ реакцию гасили водой (15 мл) и экстрагировали ЕА (20 мл ×3). Объединенную органическую фазу сушили над безводным Na2SO4 (20 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (ЕА: n-Нех=1:20) с получением 93 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 313 (М+Н)+.
Получение общего строительного блока (ВВ-26):
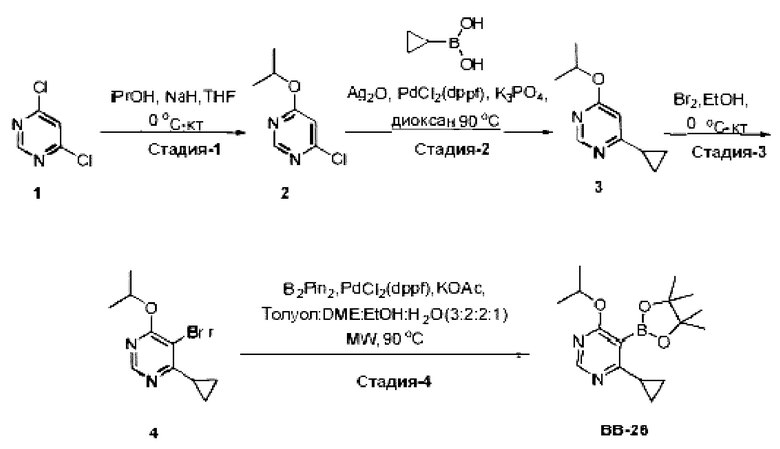
Стадия 1: Синтез 4-хлор-6-нзопропоксипиримидина:
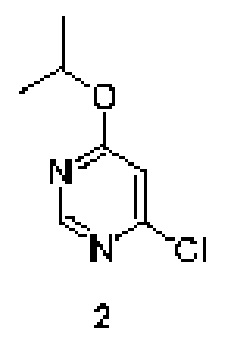
[0648] Смесь гидрида натрия (60% дисперсия, 1,07 г, 26,8 ммоль) в ТГФ (40 мл) охлаждали до 0°С и по каплям добавляли раствор изопропилового спирта (2,04 мл, 26,8 ммоль) в ТГФ (15 мл). После добавления реакционную смесь перемешивали при комнатной температуре в течение 30 минут, а затем по каплям добавляли 4,6-дихлорпиримидин (4,0 г, 26,84 ммоль) в ТГФ (15 мл) при комнатной температуре. Полученную смесь перемешивали еще 1,5 ч, а затем гасили холодной водой (100 мл) и экстрагировали ЕА (3×50 мл). Объединенный органический спой сушили над безводным Na2SO4, фильтровали и сушили под вакуумом. Неочищенное соединение очищали хроматографией на силикагеле (0-10% ЕА в н-гексане) с получением 1,75 г указанного в заголовке соединения. 1Н-ЯМР (400 МГц, CDCl3) δ 8,55 (с, 1Н), 6,70 (с, 1Н), 5,34-5,42 (м, 1Н), 1,36 (дд, J=1,22, 6,11 Гц, 6Н).
Стадия 2: Синтез 4-Циклопропил-6-изопропоксипиримидина:
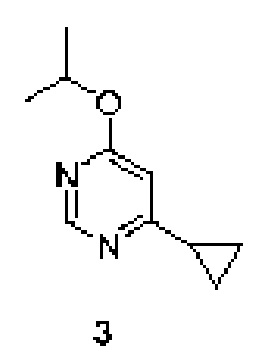
[0649] Смесь 4-хлор-6-изопропоксипиримидина (1,50 г, 8,720 ммоль), циклопропилбороновой кислоты (1,49 г, 17,4 ммоль), трикалия фосфата (5,54 г, 26,2 ммоль) и Ag2O (1,00 г, 4,36 ммоль) в 1,4 диоксане (15 мл) продували газообразным аргоном в течение 20 мин. К полученному раствору добавляли PdCl2(dppf) (0,637 г, 0,872 ммоль) при комнатной температуре. Затем реакционную смесь нагревали при 90°С в течение 8 часов. После завершения реакции (под контролем ТСХ) смесь фильтровали через слой целита и промывали DCM (50 мл). Фильтрат упаривали при пониженном давлении. Полученное неочищенное соединение очищали хроматографией на силикагеле (0-5% ЕА в н-гексане) с получением 0,700 г указанного в заголовке соединения. 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,53 (с, 1Н), 6,76 (с, 1Н), 5,27 (тд, J=6,11, 12,23 Гц, 1Н), 1,95-2,05 (м, 1Н), 1,35 (д, J=6,36 Гц, 2Н), 1,28 (д, J=6,36 Гц, 6Н), 0,98 (д, J=3,91 Гц, 2Н).
Стадия 3: Синтез 5-Бром-4-циклопропил-6-изопропоксипиримидина:
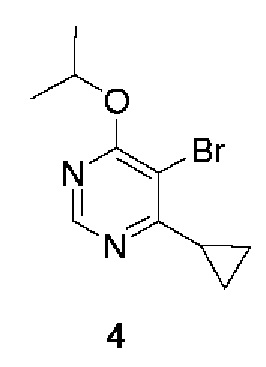
[0650] К перемешиваемому раствору 4-циклопропил-6-изопропоксипиримидина (0,600 г, 3,370 ммоль) в этаноле (7 мл) при 0°С по каплям добавляли бром (0,20 мл, 4,04 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 4 часов. После завершения реакции (под контролем ТСХ) смесь гасили насыщенным раствором NaHCO3 (30 мл). Водный слой экстрагировали ЕА (3×30 мл), и объединенный органический слой сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Полученное неочищенное соединение очищали хроматографией на силикагеле (0-10% ЕА в н-гексане) с получением 0,610 г указанного в заголовке соединения 1Н-ЯМР (400 МГц, CDCl3) δ 8,40 (с, 1Н), 5,38 (тд, J=6,11, 12,23 Гц, 1Н), 2,47-2,57 (м, 1Н), 1,40 (д, J=5,87 Гц, 6Н), 1,16 (д, J=1,96 Гц, 2Н), 1,08 (дд, J=3,18, 4,65 Гц, 2Н).
Стадия 4: Синтез 4-Цикпопропил-6-изопропокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидина (ВВ-26):
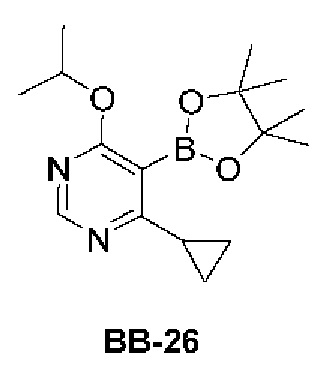
[0651] Смесь 5-бром-4-циклопропил-6-изопропоксипиримидина 4 (0,250 г, 0,972 ммоль), биспинаколатодиборана (0,246 г, 0,972 ммоль) и KOAc (0,095 г, 0,972 ммоль) в толуоле:DME:EtOH:H2O (3:2:2:1, 3 мл) объединяли при комнатной температуре и продували газообразным аргоном в течение 20 мин. К полученному раствору добавляли PdCl2(dppf) (0,071 г, 0,097 ммоль). Реакционную смесь нагревали в микроволновой печи при 90°С в течение 20 мин. После завершения реакции (под контролем ТСХ) смесь без обработки использовали в последующих реакциях сочетания.
Получение общего строительного блока (ВВ-27):
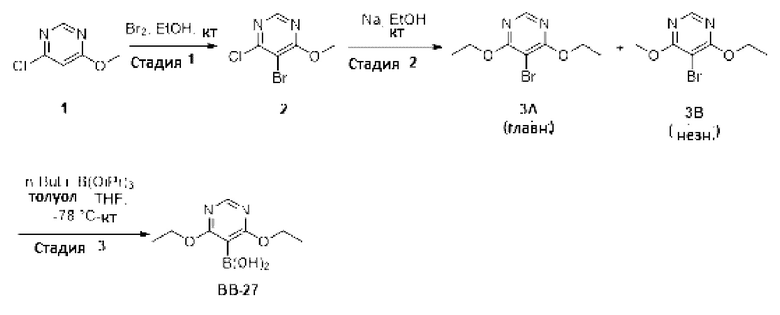
Стадии 1: Синтез 5-Бром-4-хлор-6-метоксипиримидина:
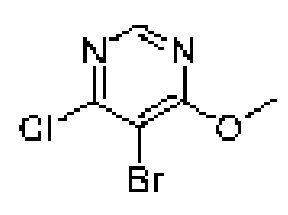
[0652] К перемешиваемому раствору 4-хлор-6-метоксипиримидина (5,00 г, 34,70 ммоль) в EtOH (50 мл) добавляли Br2 (3,20 г, 41,60 ммоль) при комнатной температуре, и реакционную смесь перемешивали при комнатной температуре в течение 16 час. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили раствором бисульфата натрия и экстрагировали н-гексаном (3×100 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле на диоксиде кремния, используя 0-3% ЕА в гексане, с получением 3,90 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 224,70 (М+Н)+ 81Br; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,64 (с, 1Н), 4,04 (с, 3Н).
Стадия 2: Синтез 5-Бром-4,6-диэтоксипиримидина:
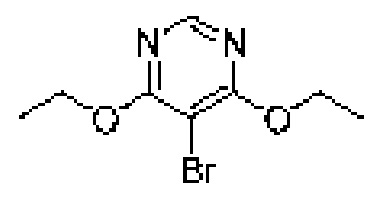
[0653] К перемешиваемому раствору EtOH (20 мл) добавляли порциями металлический натрий (0,092 г) при комнатной температуре в атмосфере аргона. К полученной реакционной смеси добавляли 5-бром-4-хлор-6-метоксипиримидин (0,90 г, 4,00 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении, и полученный таким образом остаток гасили водой (50 мл) и экстрагировали ЕА (2×25 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении Та же самая последовательность была повторена с другой партией исходного материала в масштабе 500 мг, и объединенное неочищенное соединение двух партий очищали вместе с помощью препаративной ВЭЖХ с получением 0,38 г указанного в заголовке соединения. ЖХ-МС (Способ С) (ИЭР+): m/z 246,85 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,40 (с, 1Н), 4,44 (к, J=7,2 Гц, 4Н), 1,33 (т, J=7,1 Гц, 6Н).
Стадия 3: Синтез (4,6-Диэтоксипиримидин-5-ил)бороновой кислоты (ВВ-27):
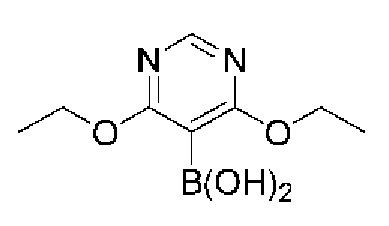
[0654] К перемешиваемому раствору 5-бром-4,6-диэтоксипиримидина (0,38 г, 1,54 ммоль) в ТГФ (2,5 мл) и толуоле (5 мл) добавляли триизопропилборат (0,435 г, 2,31 ммоль) в атмосфере аргона. К полученной реакционной смеси по каплям добавляли н-BuLi (1,6 М в гексане, 1,35 мл, 2,16 ммоль) при -76°С, перемешивали в течение 30 минут при той же температуре и дополнительно перемешивали при комнатной температуре в течение 30 минут. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили 2 н. HCl (5 мл), перемешивали в течение 2 часов и доводили до рН 7,5-8, используя Na2CO3 (2 г). Водный слой экстрагировали ЕА (3×20 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение растирали с н-пентаном (10 мл), полученное твердое вещество собирали и сушили с получением 0,065 г неочищенного указанного в заголовке соединения, которое использовали как есть в следующей стадии без дополнительной очистки. ЖХ-МС (Способ С) (ИЭР+): m/z 212,90 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,35-8,43 (м, 1Н), 8,11 (с, 1Н), 4,27-4,37 (м, 4Н), 1,21-1,35 (м, 6Н).
Следующий промежуточный продукт был получен из коммерчески доступного арилбромида с помощью процедуры, описанной для получения ВВ-27:
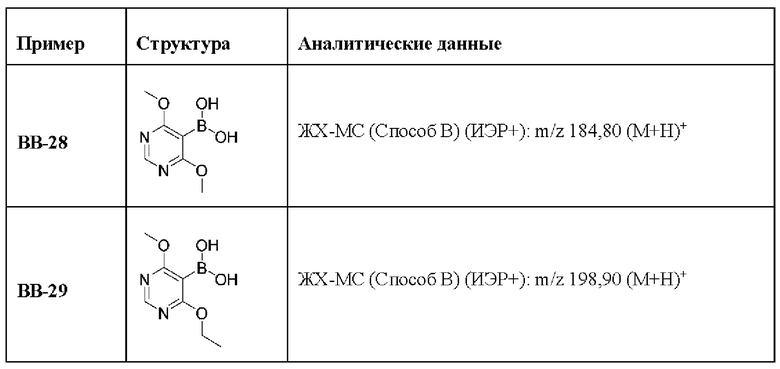

Получение общего строительного блока (ВВ-31):
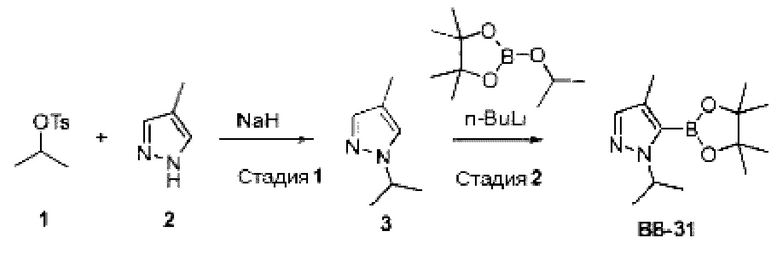
Стадия 1: Синтез 1-Изопропил-4-метил-1Н-пиразола:
[0655] К раствору 4-метил-1Н-пиразола (1,0 г, 12,2 ммоль) в ТГФ (30 мл) при 0°С добавляли NaH (60% дисперсия, 730 мг, 18,3 ммоль) порциями в течение 5 минут. Смесь перемешивали при 0°С в течение 30 минут, затем давали нагреться до комнатной температуры. К реакционной смеси одной порцией добавляли раствор изопропил 4-метилбензолсульфоната (2,6 г, 12,2 ммоль) в ТГФ (10 мл). После добавления реакционную смесь кипятили с обратным холодильником в течение ночи. Затем реакцию гасили водой (50 мл) и экстрагировали ЕА (50 мл ×2). Объединенный органический слой сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ:ЭА=от 50:1 до 10:1) с получением 0,44 г указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CD3OD) δ 7,30 (с, 1Н), 7,20 (с, 3Н),4,44(м, 1Н), 2,07 (с,3Н), 1,48 (д, J=6,6 Гц 6Н).
Стадия 2: Синтез 1-Изопропил-4-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пнразола (ВВ-31):
[0656] К раствору 1-изопропил-4-метил-1Н-пиразола (350 мг, 2,82 ммоль) в ТГФ (10 мл) при 0°С по каплям в течение 10 минут добавляли н-BuLi (2,3 мл, 5,7 ммоль). После перемешивания смеси при 0°С в течение 1 ч реакционную смесь охлаждали до -7S°С.К реакционной смеси по каплям в течение 5 минут добавляли раствор 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолана (1,05 г, 5,65 ммоль) в ТГФ (5 мл). После перемешивания реакционной смеси при-78°С в течение 30 минут реакционную смесь нагревали до комнатной температуры и перемешивали в течение 4 часов. Реакцию гасили водой (20 мл) и экстрагировали ЕА (20 мл ×2). Объединенный органический слой сушили над сульфатом натрия (10 г), фильтровали и упаривали. Остаток очищали колоночной хроматографией на диоксиде кремния (элюент: РЕ/ЕА=от 100/1 до 20/1) с получением 180 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 251 (М+Н)+; 1Н-ЯМР (300 МГц, CD3OD) δ 7,34 (с, 1Н), 5,05 (м, 1Н), 2,22 (с,3Н), 1,45 (д, J=6,6 Гц, 6Н), 1,33 (с, 12Н).
Следующие соединения были получены из соответствующего гетероцикла и алкилирующего агента в соответствии с процедурой, описанной для ВВ-31:
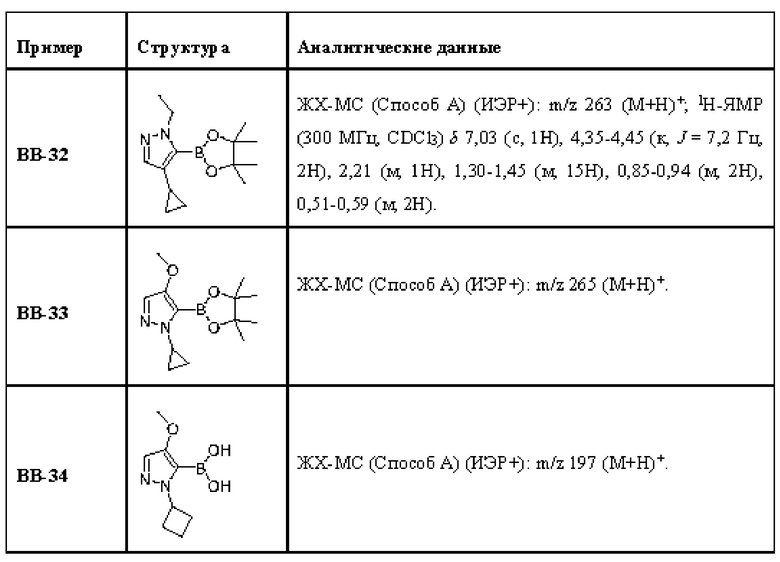
Получение общего строительного блока (ВВ-35):
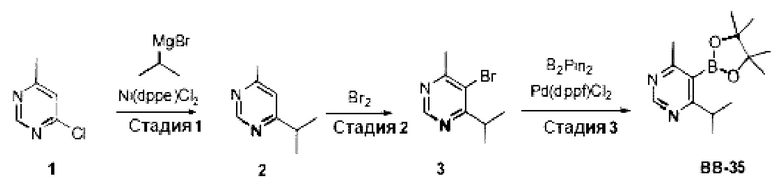
Стадия 1: Синтез 4-Изопропил-6-метилпиримидина:
[0657] К раствору 4-хлор-6-метилпиримидина (2 г, 15,63 ммоль) в Et2O (10 мл) при комнатной температуре добавляли Ni(dppe)Cl2 (165 мг, 0,3 ммоль) одной порцией. После охлаждения смеси до -10°С в течение 10 минут к реакционной смеси по каплям в течение 5 минут добавляли раствор изопропилмагнийбромида (1 М, 18,75 ммоль, 18,75 мл). Смесь перемешивали при -10°С в течение 1 ч, а затем гасили водой (30 мл) и экстрагировали ЕА (30 мл ×3). Объединенный органический слой сушили над сульфатом натрия (50 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле на диоксиде кремния (РЕ:ЕА=от 100:1 до 20:1) с получением 2,02 г указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 137 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 9,00 (с, 1Н), 7,06 (с, 1Н), 2,98 (м, 1Н), 2,51 (с, 3Н), 1,30 (д, J=6,9 Гц, 6Н).
Стадия 2: Синтез 5-Бром-4-изопропил-6-метилпиримидина:
[0658] К раствору 4-изопропил-6-метилпиримидина (500 мг, 3,6 ммоль) в EtOH (5 мл) при 0°С добавляли Br2 (588 мг, 3,6 ммоль) одной порцией. Полученную смесь перемешивали при комнатной температуре в течение ночи, затем гасили водой (20 мл) и экстрагировали ЕА (15 мл ×3). Объединенный органический слой сушили над сульфатом натрия (20 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле на диоксиде кремния (РЕ:ЕА=20:1) с получением 400 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 215 (М+Н)+; 1Н-ЯМР (300 МГц CDCl3) δ 8,88 (с, 1Н), 3,56 (м, 1Н), 2,66 (с, 3Н), 1,25-1,31 (м, 6Н).
Стадия 3: Синтез 4-Изопропил-6-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидина (ВВ-35):
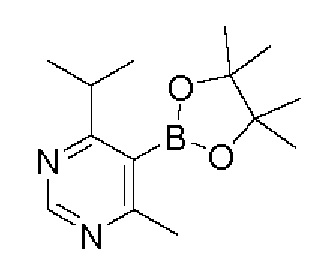
[0659] К раствору 5-бром-4-изопропил-6-метилпиримидина (400 мг, 1,86 ммоль) и триизопропилбората (457 мг, 2,43 ммоль) в толуоле (4 мл) и ТГФ (1 мл) при -78°С, добавляли н-бутиллитий (1 мл, 2,6 ммоль) по каплям в течение 10 минут. Через 30 минут при -78°С реакционную смесь нагревали до -20°С и перемешивали в течение 1 часа. Затем реакцию гасили водным раствором HCl (3 мл, 1 н.) и перемешивали при комнатной температуре в течение 30 минут. Затем рН доводили до 8 насыщенным водным раствором Na2CO3 и экстрагировали ЕА (25 мл ×3). Объединенный органический слой сушили над безводным Na2SO4 (30 г), фильтровали и упаривали в вакууме с получением 140 мг неочищенного указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 181 (М+Н)+; 1Н-ЯМР (300 МГц, CD3OD) δ 8,91 (с, 1Н),2,78(м, 1Н), 2,44 (с, 3Н), 1,26-1,32 (д, J=6,6 Гц, 6Н).
Получение общего строительного блока (ВВ-36):
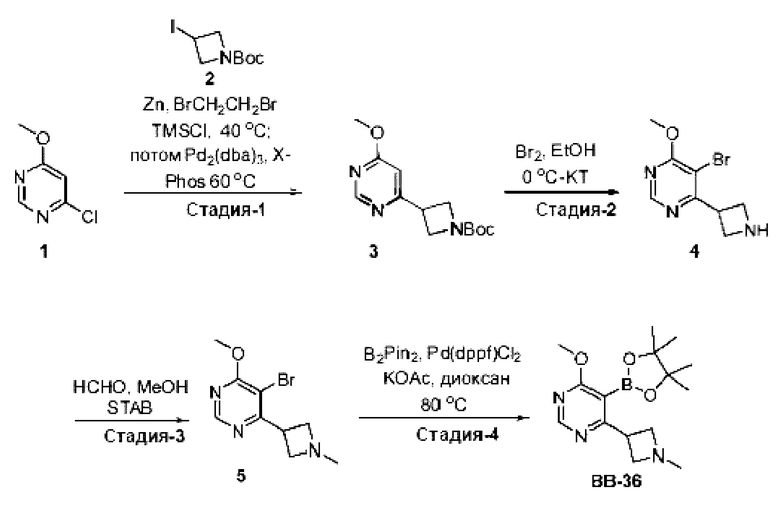
Стадия 1: Синтез трет-бутил-3-(6-метоксипиримидин-4-ил) азетидин-1-карбоксилата:
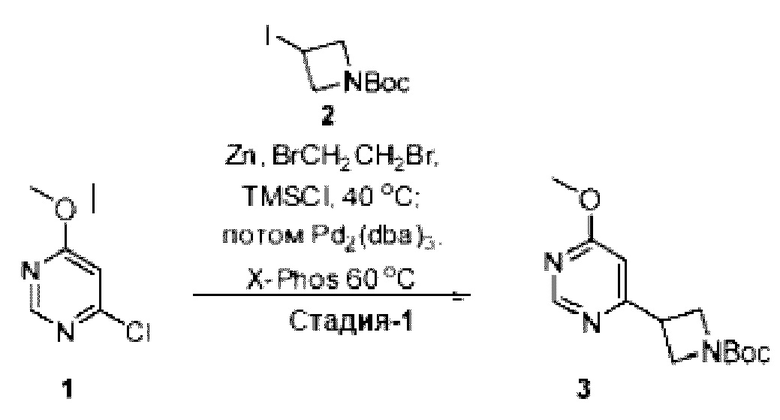
[0660] К перемешиваемому раствору цинка (3,78 г, 58,3 ммоль) в ТГФ (150 мл) добавляли TMSC1 (0,574 г, 5,30 ммоль) и 1,2-дибромэтан (0,962 г, 5,30 ммоль). Смесь перемешивали в течение 5 минут при комнатной температуре, а затем обрабатывали трет-бутил-3-йодоазетидин-1-карбоксилатом 2 (15 г, 53,0 ммоль) со скоростью, которая поддерживала внутреннюю температуру реакции ниже 40°С. Полученную смесь дополнительно перемешивали в течение 30 минут при комнатной температуре. К полученной реакционной смеси добавляли 4-хлор-6-метоксипиримидин 1 (7,64 г, 53,0 ммоль), X-Phos (5 г, 10,6 ммоль) и Pd2(dba)3 (4,84 г, 5,30 ммоль). Реакционную смесь нагревали до 60°С в герметичной пробирке в течение 4 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли водой (200 мл), экстрагировали ЕА (200 мл) и фильтровали через целит. Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-70% ЕА в гексане, с получением указанного в заголовке соединения (10,00 г). ЖХ-МС (Способ С) (ИЭР+): m/z 266,3 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,80 (с, 1Н),6,89 (с, 1Н), 4,09-4,20 (м, 2Н), 3,95-4,06 (м,2Н), 3,91 (с, 3Н), 3,79-3,86 (м, 1Н), 1,39 (с, 9Н).
Стадия 2: Синтез 4-(азетидин-3-ил)-5-бром-6-метоксипиримидина:
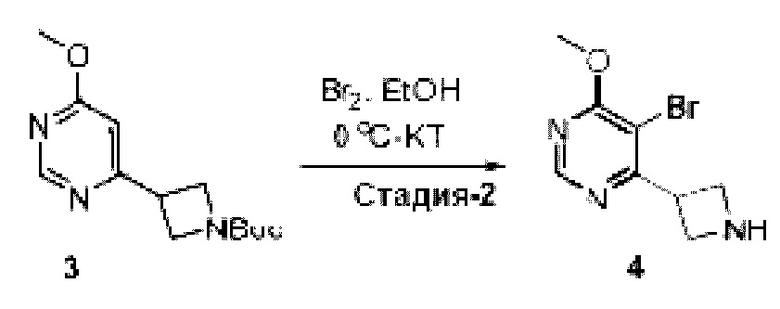
[0661] К перемешиваемому раствору трет- бутил-3-(6-метоксипиримидин- 4-ил)азетидин-1-карбоксилата 3 (3,0 г, 11,3 ммоль) в этаноле (100 мл) добавляли бром (5,42 г, 33,9 ммоль) при 0°С. Полученной смеси давали нагреться до комнатной температуры и перемешивали в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Остаток растворяли в водном растворе аммиака (30 мл) и экстрагировали DCM (100 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-10% метанол в DCM в качестве элюента, с получением указанного в заголовке соединения (1,7 г). ЖХ-МС (Способ С) (ИЭР+): m/z 245,90 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,77 (с, 1Н), 4,31 (тд, J=8,35, 16,21 Гц, 2Н), 4,01 (с, 3Н), 3,87-3,94 (м,3Н).
Стадия 3: Синтез 5-бром-4-метокси-6-(1-метилазетидин-3-ил)пиримидина:
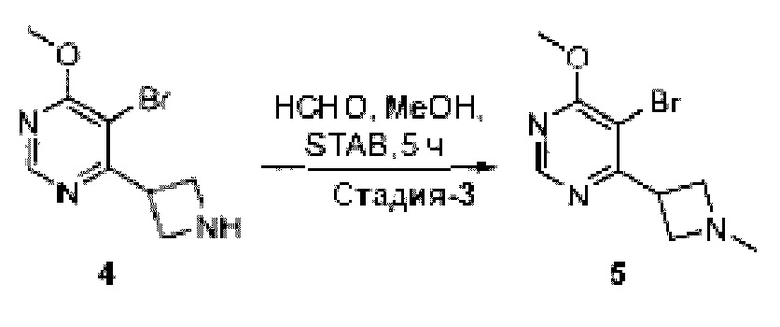
[0662] К перемешиваемому раствору 4-(азетидин-3-ил)-5-бром-6-метоксипиримидина 4 (1,0 г, 4,08 ммоль) в метаноле (20 мл) добавляли раствор формальдегида (10 мл, 37% в воде), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа К полученной реакционной смеси добавляли триацетоксиборгидрид натрия (4,3 г, 20,4 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение 5 ч. и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли DCM (50 мл), и раствор доводили до рН 8, используя насыщенный раствор бикарбоната натрия (10 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-10% метанол в DCM в качестве элюента, с получением указанного в заголовке соединения (0,719 г). ЖХ-МС (Способ С) (ИЭР): m/z 257,88 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 3,75 (с, 1Н), 4,11 (квин, J=7,83 Гц 1Н),4,01 (с, 3Н),3,90 (т, J=8,07 Гц,2Н), 3,62 (т, J=7,58 Гц, 2Н),2,45 (с, 3Н).
Стадия 4: Синтез 4-метокси-6-(1-метилазетидин-3-ил)-5-(4,4,5,5-тетраметил-1,3,2-диокса6оролан-2-ил)пиримидина (ВВ-36):
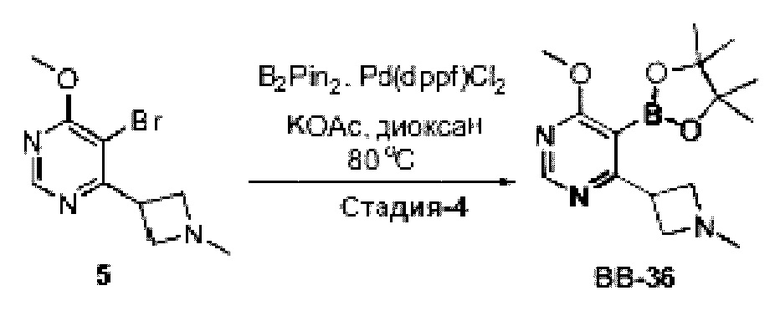
[0663] К перемешиваемому раствору 5-бром-4-метокси-6-(1-метилазетидин-3-ил)пиримидина 5 (0,600 г, 2,32 ммоль) и B2Pin2 (1,77 г, 6,97 ммоль) в диоксане (30 мл) добавляли KOAc (0,676 г, 6,97 ммоль), и реакционную смесь дегазировали газообразным аргоном в течение 15 мин. К полученной реакционной смеси добавляли Pd(dppf)Cl2-DCM (0,375 г, 0,465 ммоль) при комнатной температуре и смесь нагревали в запаянной пробирке при 80°С в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, разбавляли ЕА (100 мл) и фильтровали через слой целита. Полученный фильтрат упаривали при пониженном давлении с получением неочищенного указанного в заголовке соединения (0,709 г). Неочищенное соединение использовали как таковое в следующей стадии без дополнительной очистки.
Получение общего строительного блока (ВВ-37):
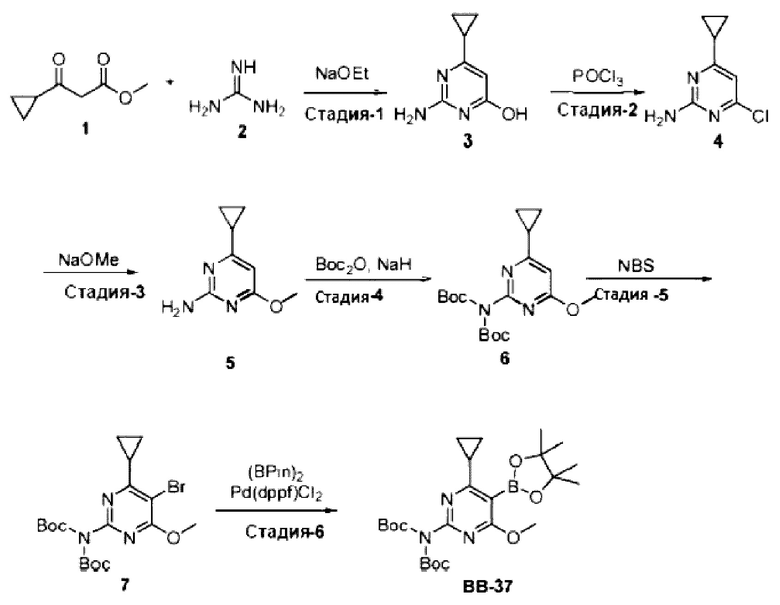
Стадия 1: 2-амино-6-циклопропилпиримидин-4-ол:
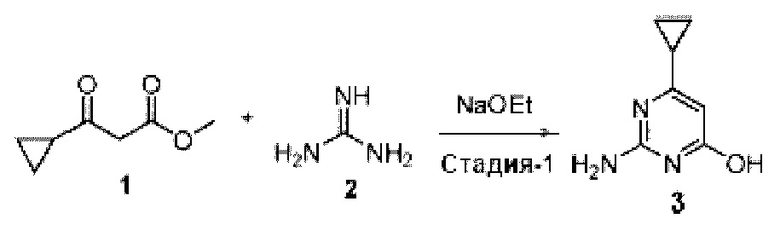
[0664] К суспензии метил-3-циклопропил-3-оксопропаноата (8,1 г, 57 ммоль) и гуанидинкарбоната (5,65 г, 63 ммоль) в этаноле (70 мл) при комнатной температуре добавляли порциями NaOEt (7,75 г, 114 ммоль) в течение 20 мин. После добавления полученную смесь кипятили с обратным холодильником и перемешивали в течение ночи. После завершения реакции, как показывает анализ ТСХ, полученную суспензию упаривали при пониженном давлении для удаления большей части этанола. Остаток разбавляли насыщенным раствором NH4Cl (100 мл) и экстрагировали ЕА (3×200 мл). Объединенный органический слой сушили над Na2SO4 (50 г), фильтровали и упаривали, получая 9 г неочищенного продукта. Неочищенный продукт суспендировали в смешанном растворителе (метанол/МТВЕ=1/10, 33 мл) при комнатной температуре в течение ночи. После фильтрации и сушки получали 7,2 г указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 152 (М+Н)+, 1Н-ЯМР (300 МГц, ДМСО-d6) δ 10,45 (с, 1Н), 6,37 (с, 2Н), 5,48 (с, 1Н), 1,65 (м, 1Н), 078-0,83 (м, 4Н).
Стадия 2: 4-хлор-6-циклопропилпиримидин-2-амин:
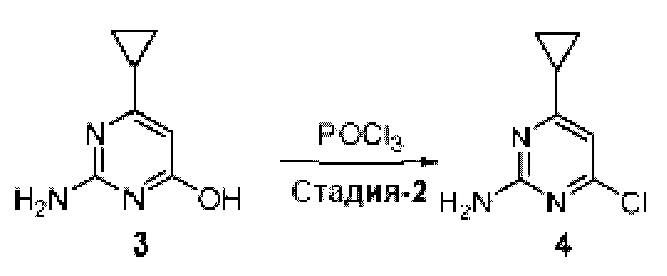
[0665] К суспензии 2-амино-6-циклопропилпиримидин-4-ола (1,5 г, 9,9 ммоль) в DCE (30 мл) добавляли POCl3 (20 мл). Реакционную смесь перемешивали при 75°С в течение ночи. После завершения реакции, как показывает ТСХ, смесь упаривали досуха. Остаток гасили холодным насыщенным раствором карбоната натрия (60 мл) и экстрагировали DCM (100 мл ×2). Объединенный органический слой сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ/ЕА=10:1), получая 1,0 г указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 170 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 6,52 (с, 1Н), 5,05 (уш. с, 2Н), 1,79 (м, 1Н), 0,94-1,10 (м, 4Н).
Стадия 3: 4-циклопропил-6-метоксипиримидин-2-амин:
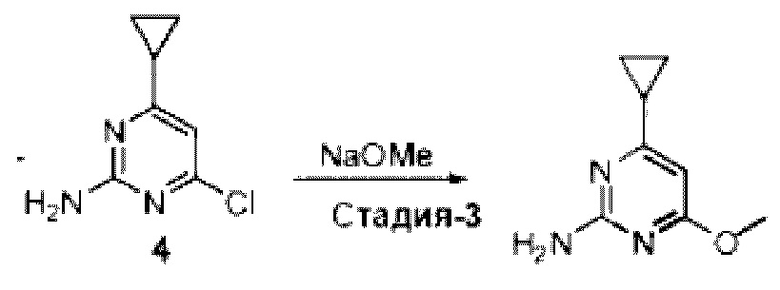
[0666] К раствору 4-хлор-6-циклопропилпиримидин-2-амина (850 мг, 5,03 ммоль) в МеОН (50 мл) добавляли раствор NaOMe (2,72 г, 15,1 ммоль, 30 мас. % в МеОН) одной порцией. Реакционную смесь перемешивали при 60°С в течение 3 часов. После завершения реакции, как показывает ТСХ, смесь гасили водой (60 мл) и экстрагировали ЕА (100 мл ×2). Объединенный органический слой промывали солевым раствором (120 мл), сушили над сульфатом натрия (30 г), фильтровали и упаривали, получая 800 мг неочищенного указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 166 (М+Н)+, 1Н-ЯМР (300 МГц, CDCl3) δ 5,91 (с, 1Н), 4,80 (уш. с, 2Н), 3,84 (с, 3Н), 1,79 (м, 1Н), 0,91-1,08 (м, 2Н), 0,85-0,91 (м, 2Н).
Стадия 4: Ди-Вос-защищенный 4-циклопропил-6-метоксипиримидин-2-амин:
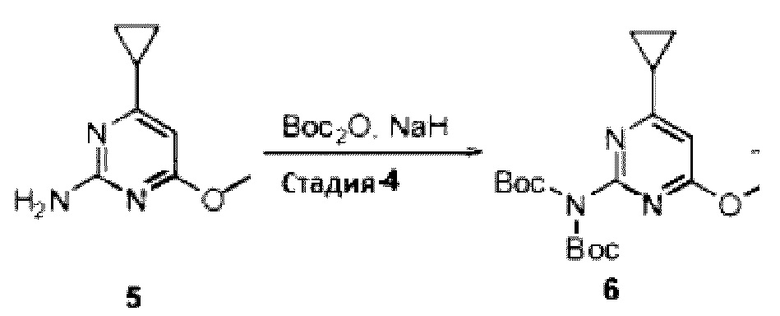
[0667] К раствору 4-циклопропил-6-метоксипиримидин-2-амина (900 мг, 5,45 ммоль) и Вос2О (5,96 г, 27,3 ммоль) в ТГФ (50 мл) добавляли NaH (654 мг, 16,4 ммоль, 60 мас. %) порциями в течение 5 мин. Реакционную смесь перемешивали при 60°С в течение ночи. После завершения реакции, как показывает анализ ТСХ, смесь гасили водным раствором NH4Cl (100 мл) и экстрагировали ЕА (100 мл ×2). Объединенный органический слой промывали солевым раствором (120 мл), сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ/ЕА=от 50:1 до 25:1) с получением 1,27 г указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 366 (М+Н)+, 1Н-ЯМР (300 МГц, CDCl3) δ 6,43 (с, 1Н), 3,92 (с, 3Н), 1,89 (м, 1Н), 1,44 (с, 18Н), 1,02-1,10 (м, 2Н), 0,91-1,02 (м, 2Н).
Стадия 5: Ди-Вос защищенный 5-бром-4циклопропил-6-метоксипиримидин-2-амин:
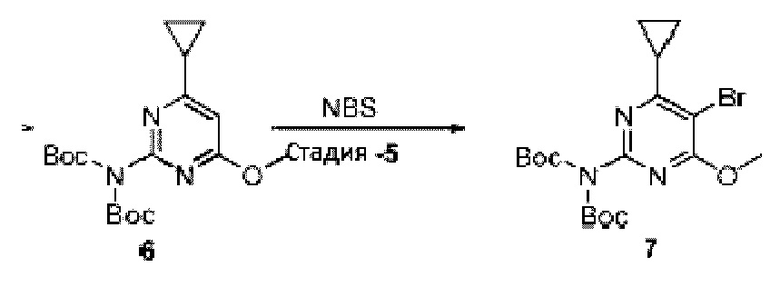
[0668] К раствору ди-Вос-защищенного 4-циклопропил-6-метоксипиримидин-2-амина (1,27 г, 3,47 ммоль) в DCM (35 мл) при 0°С добавляли NBS (1,24 г, 6,96 ммоль) порциями в течение 5 мин. Реакционную смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции, как показывает ТСХ, смесь гасили водой (30 мл) и экстрагировали DCM (40 мл ×2). Органический слой промывали солевым раствором (60 мл), сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ/ЕА=от 60:1 до 30:1) с получением 1,4 г указанного в заголовке соединения ЖХ-МС (Способ А) (ИЭР+): m/z 444 (М+Н)+, 1Н-ЯМР (300 МГц, CDCl3) δ 4,01 (с, 3Н), 2,52 (м, 1Н), 1,45 (с, 18Н), 1,09-1,17 (м, 2Н), 1,02-1,09 (м, 2Н).
Стадия 6: Ди-Вос-защищенный 4-циклопропил-6-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2-амин (ВВ-37):
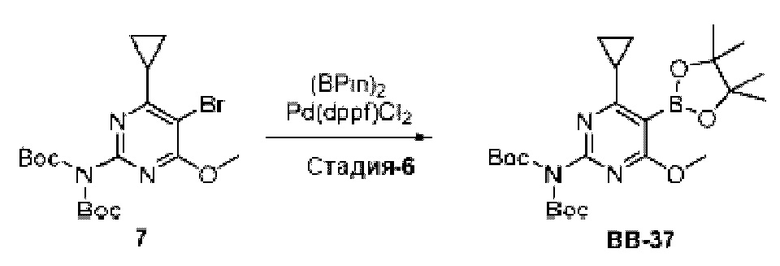
[0669] К раствору ди-Вос-защищенного 5-бром-4-циклопропил-6-метоксипиримидин-2-амина (1,5 г, 3,4 ммоль) в 1,4-диоксане (34 мл) добавляли бис(пинаколато)диборон (1,72 г, 6,77 ммоль), ацетат калия (997 мг, 10,2 ммоль) и Pd(dppf)Cl2 (249 мг, 0,34 ммоль). Затем реакционную смесь нагревали до 95°С в течение ночи. После того, как большая часть исходного материала с бромом была израсходована на основании анализа ТСХ, смесь гасили водой (40 мл) и нерастворенное твердое вещество фильтровали. Фильтрат экстрагировали ЕА (100 мл ×2). Объединенный органический слой промывали солевым раствором (60 мл), сушили над сульфатом натрия (30 г), фильтровали и упаривали. Полученный остаток очищали хроматографией на силикагеле (РЕ/ЕА=от 30:1 до 10:1) с получением 650 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 492 (М+Н)+, 1Н-ЯМР (300 МГц, CDCl3) δ 3,90 (с, 3Н), 2,18 (м, 1Н), 1,45 (с, 18Н), 1,33 (с, 12Н), 1,09-1,13 (м, 2Н), 0,89-0,96 (м, 2Н).
Общая экспериментальная процедура 1. Получение Примера 1:
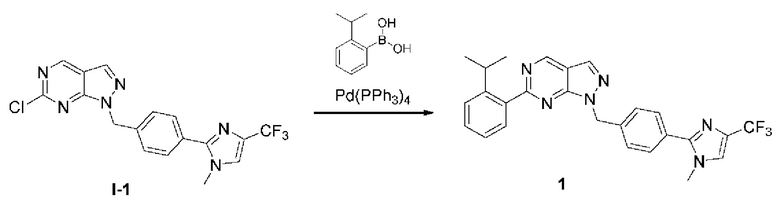
[0670] Смесь 6-хлор-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (100 мг, 0,25 ммоль), K3PO4.3Н2О (208 мг, 0,78 ммоль), (2-изопропилфенил)бороновой кислоты (84 мг, 0,51 ммоль) и Pd(PPh3)4 (30 мг, 0,025 ммоль) в 1,4-диоксане (4 мл) и воде (0,3 мл), перемешивали при 100°С в течение 2 часов. Полученную смесь разбавляли водой (10 мл) и экстрагировали ЕА (20 мл ×2). Объединенный органический слой сушили над Na2SO4 (10 г), фильтровали и упаривали досуха. Концентрированный остаток очищали хроматографией на силикагеле (РЕ: ЕА=от 30:1 до 5:1) с последующей препаративной ВЭЖХ с получением 62 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 477 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 9,30 (с, 1Н), 8,20 (с, 1Н), 7,72 (д, J=7,2 Гц, 1Н), 7,59 (д, J=8,1 Гц, 2Н), 7,44-7,51 (м, 4Н), 7,26-7,35 (м, 2Н), 5,75 (с, 2Н), 3,73 (с, 3Н), 3,56 (м, 1Н), 1,27 (д, J=6,9 Гц, 6Н).
Следующие соединения были получены в соответствии с Общей экспериментальной процедурой 1 с использованием Pd(PPh3)4/K3PO4 или XPhos-Pd-G2/XPhos/K3PO4 и соответствующих соединений бора (указаны структуры коммерческих бороновых кислот и боронатов):
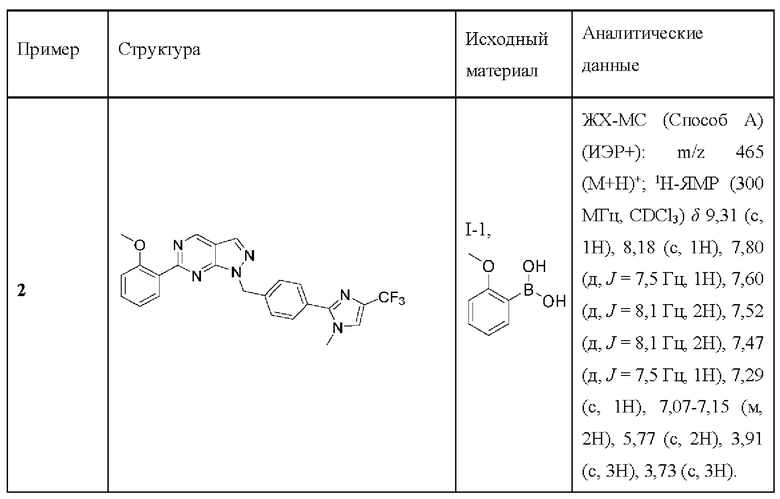
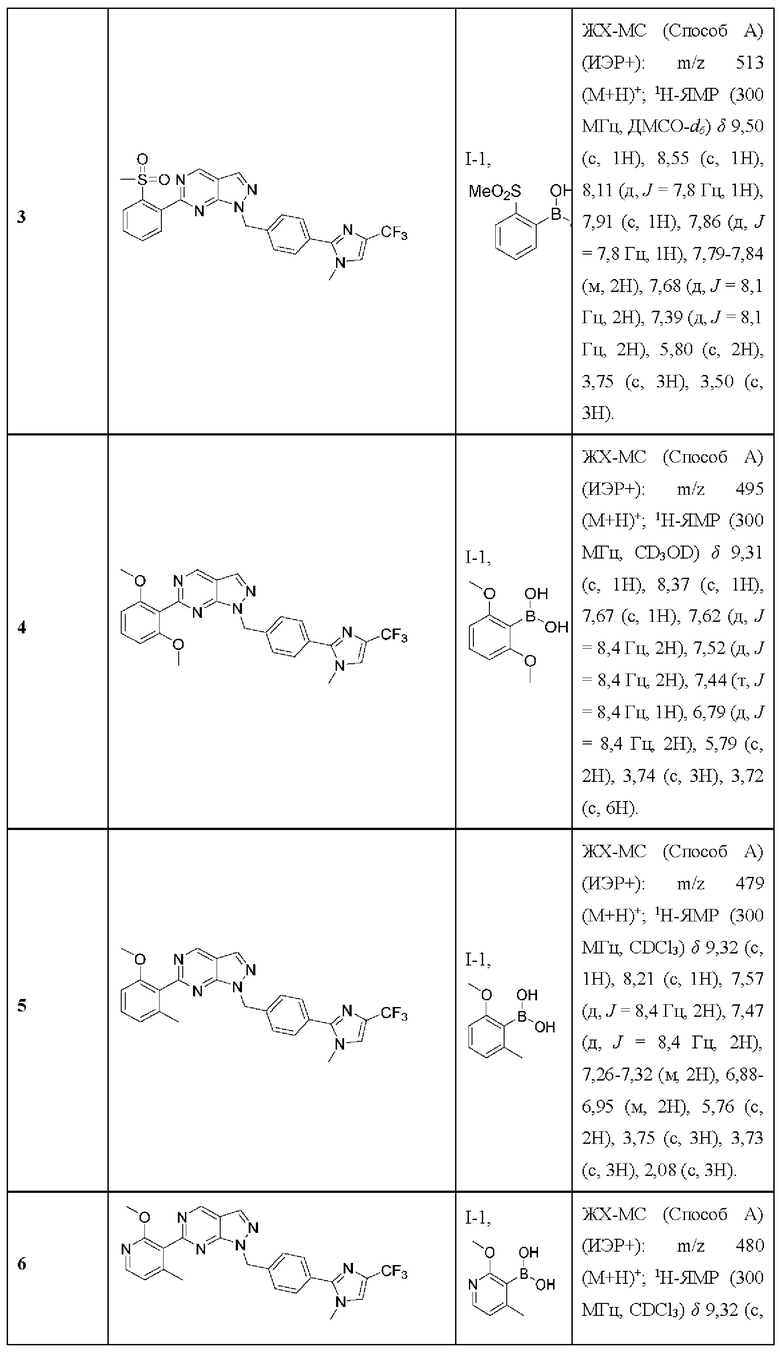
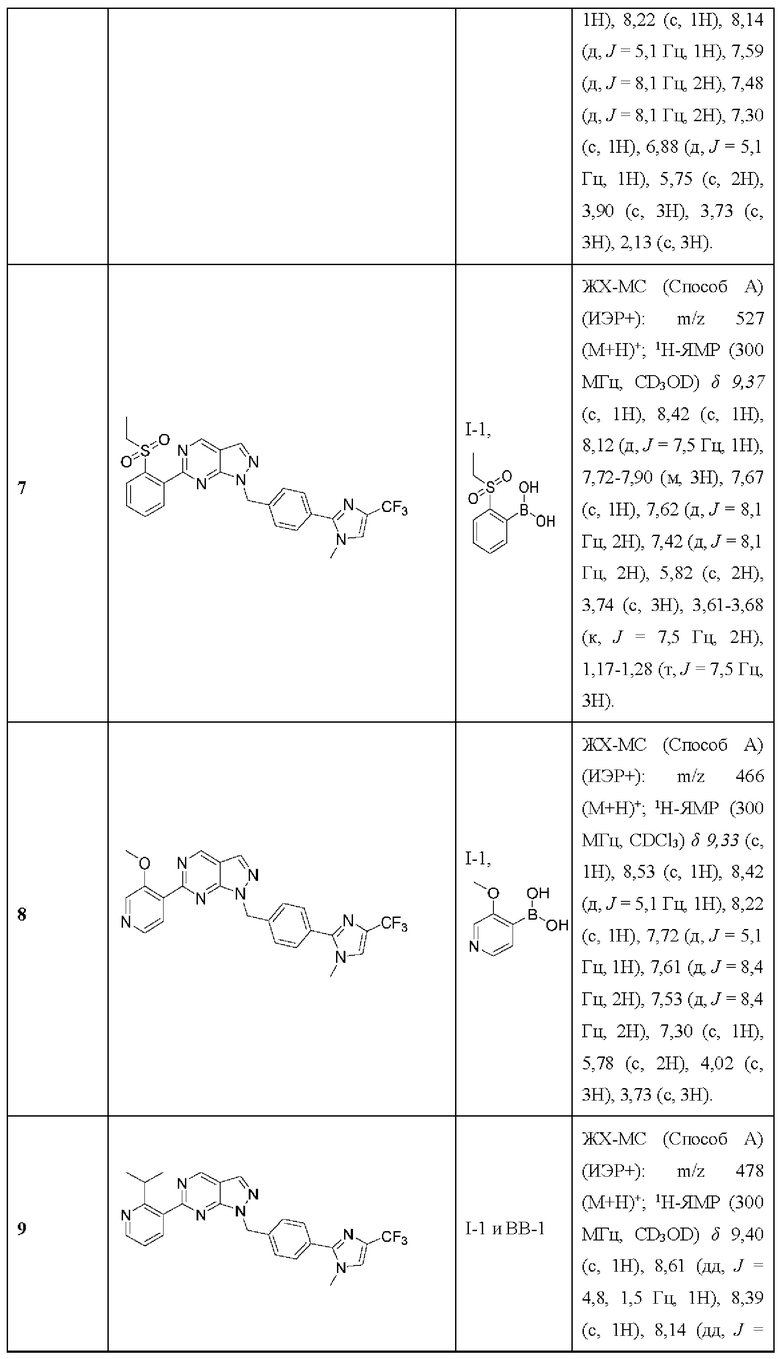
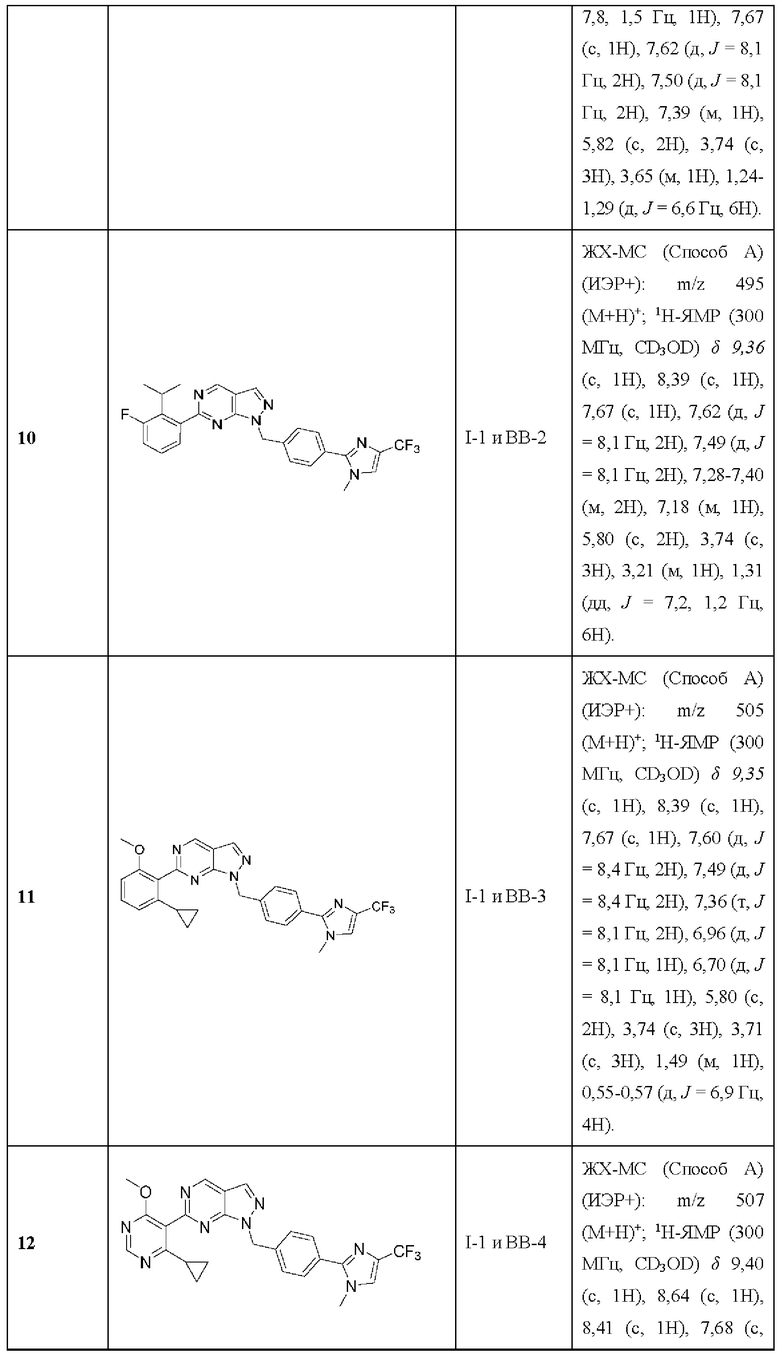
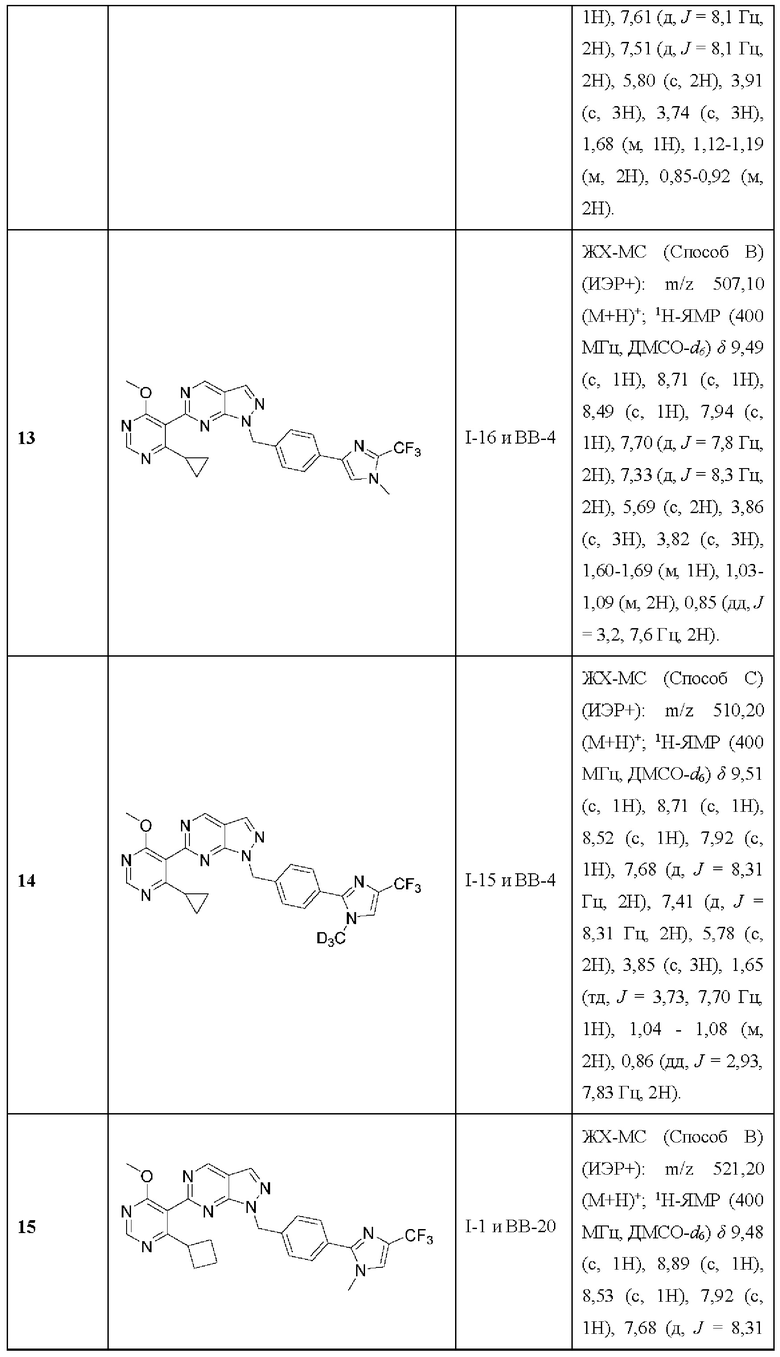
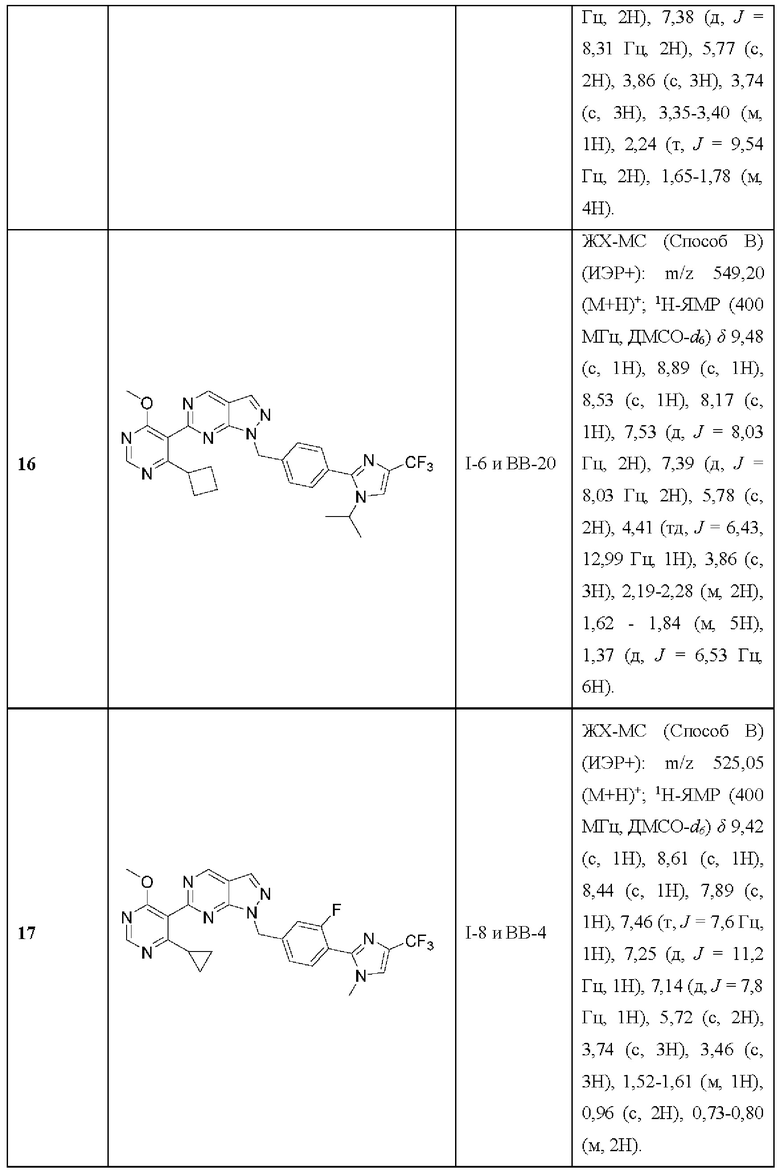
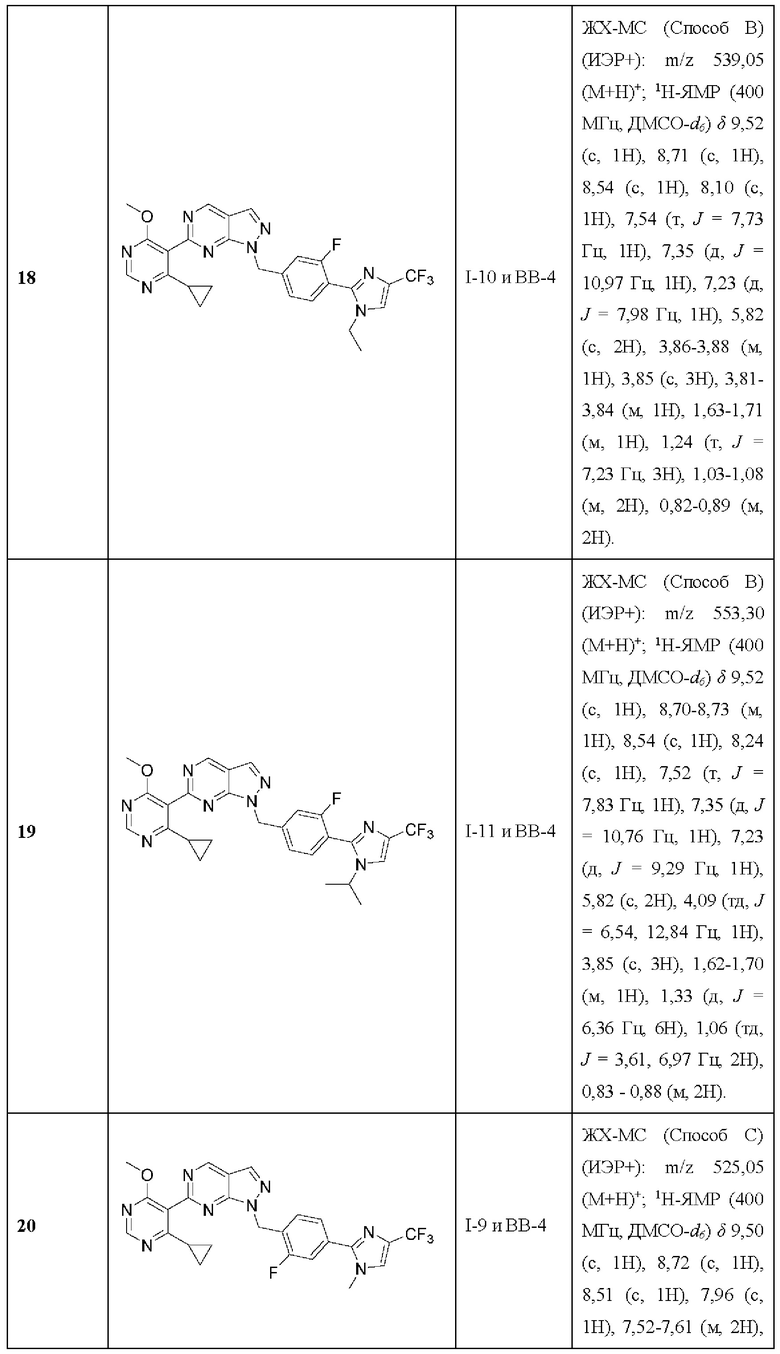
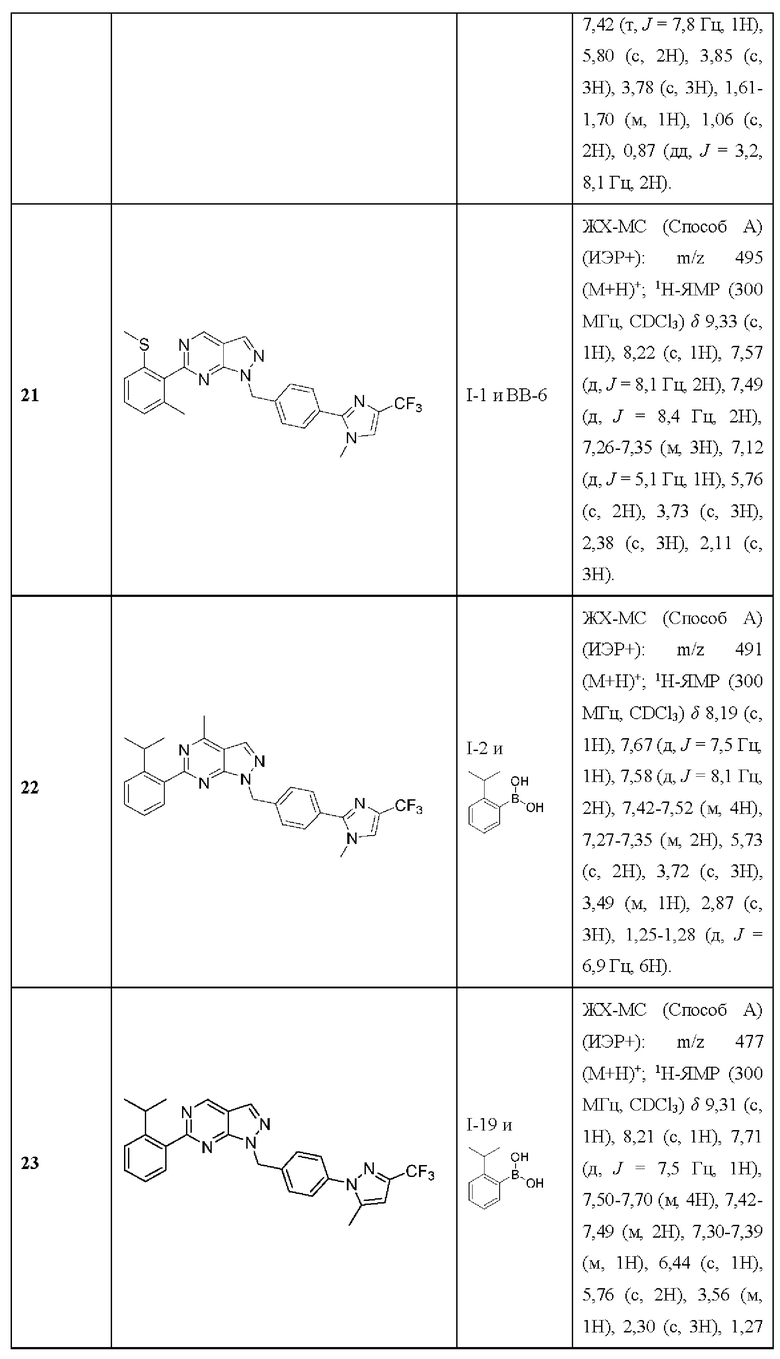
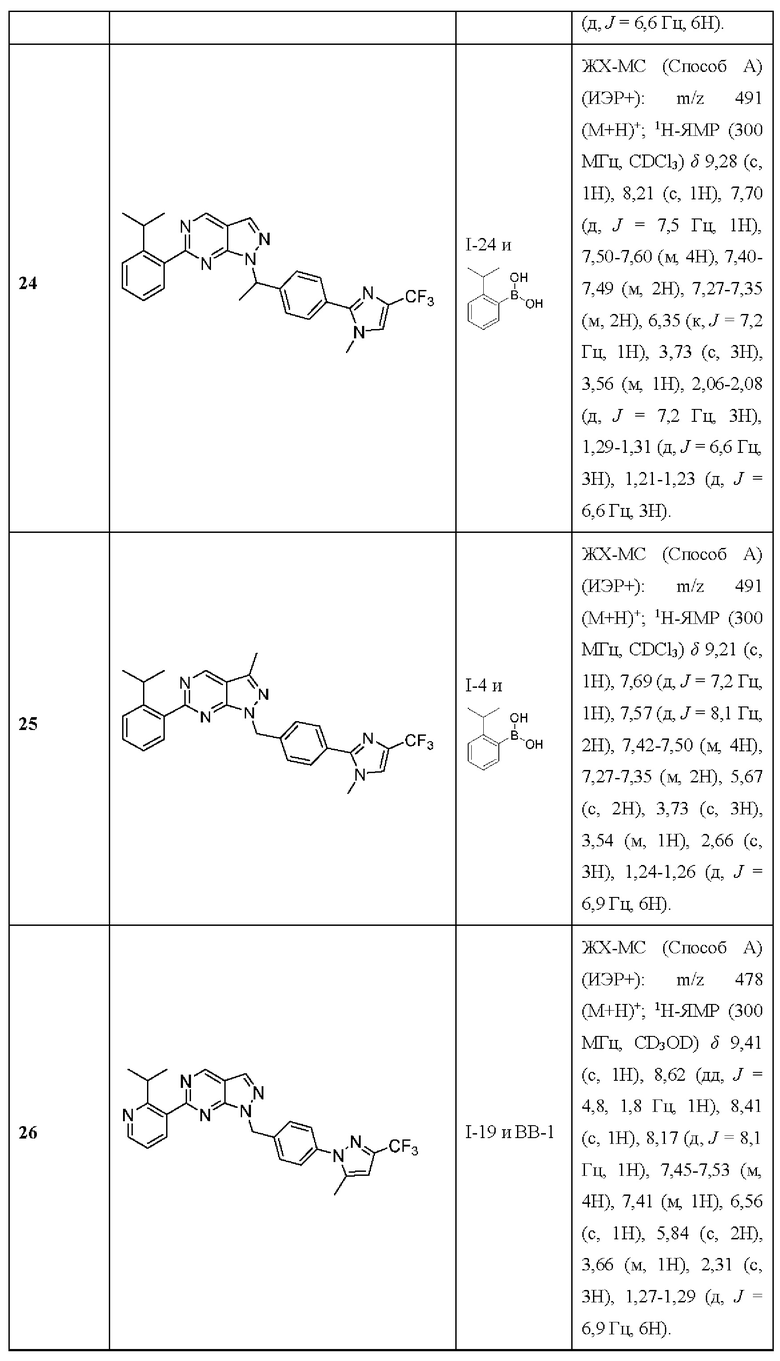
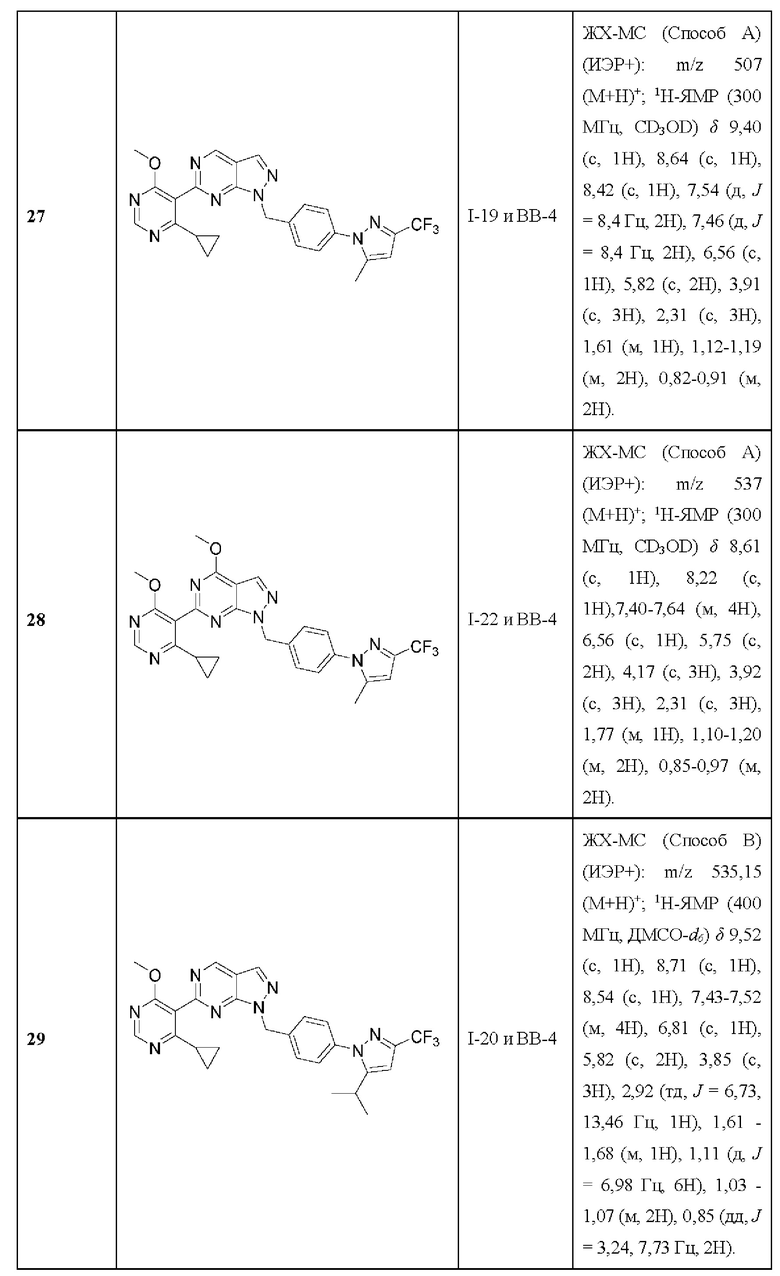
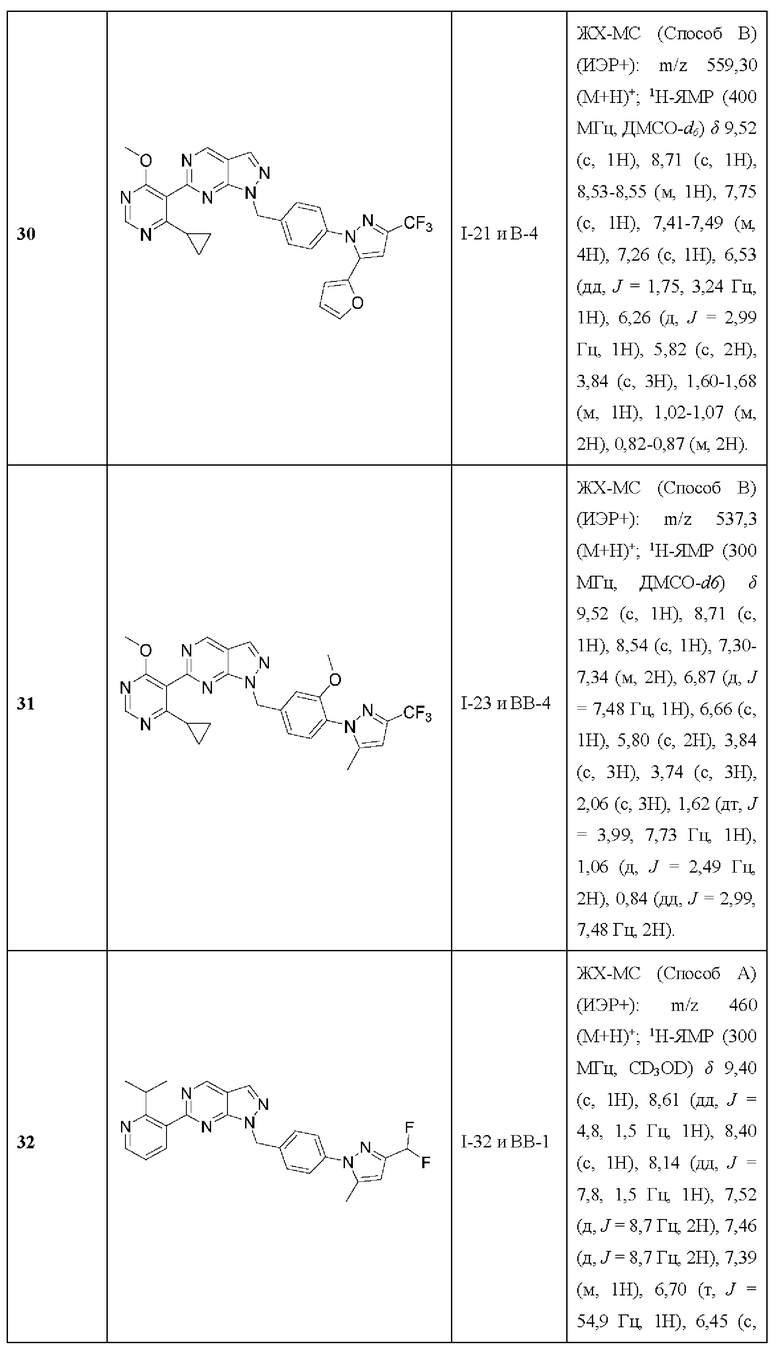
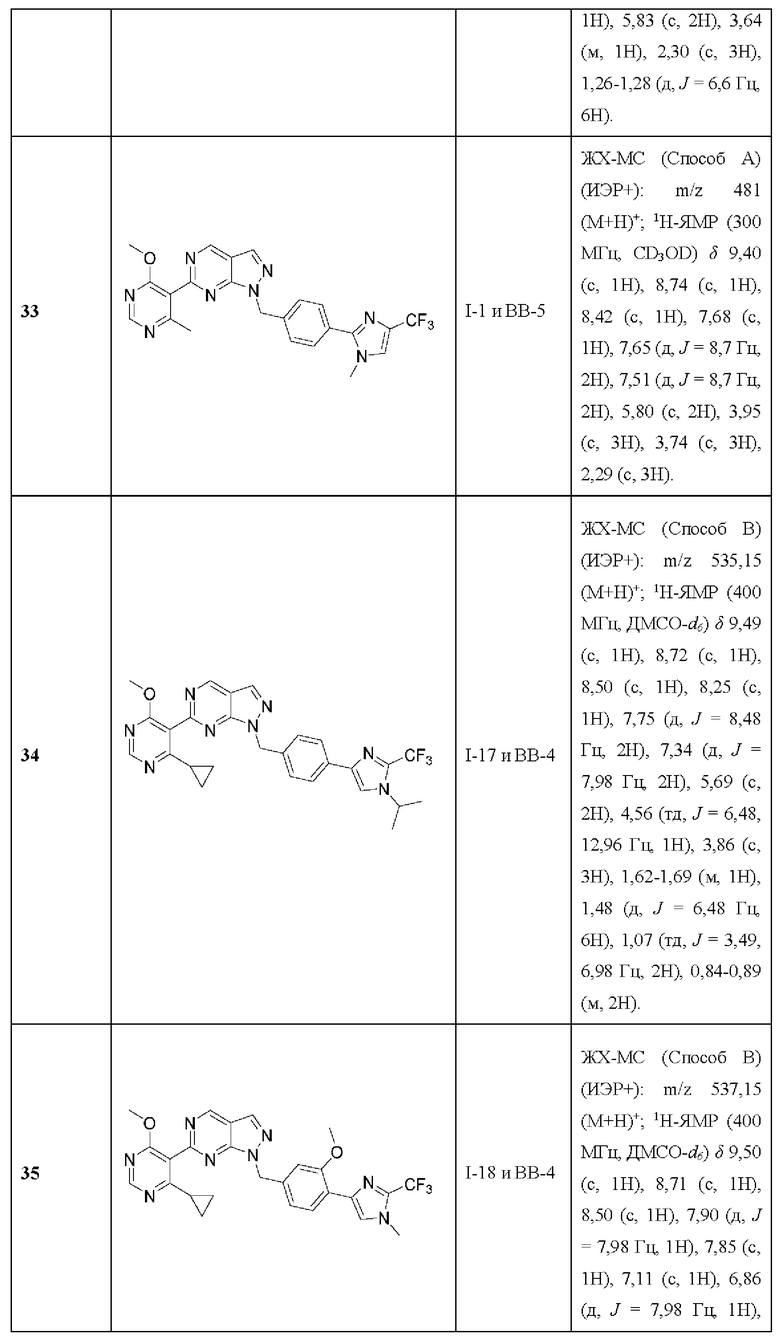
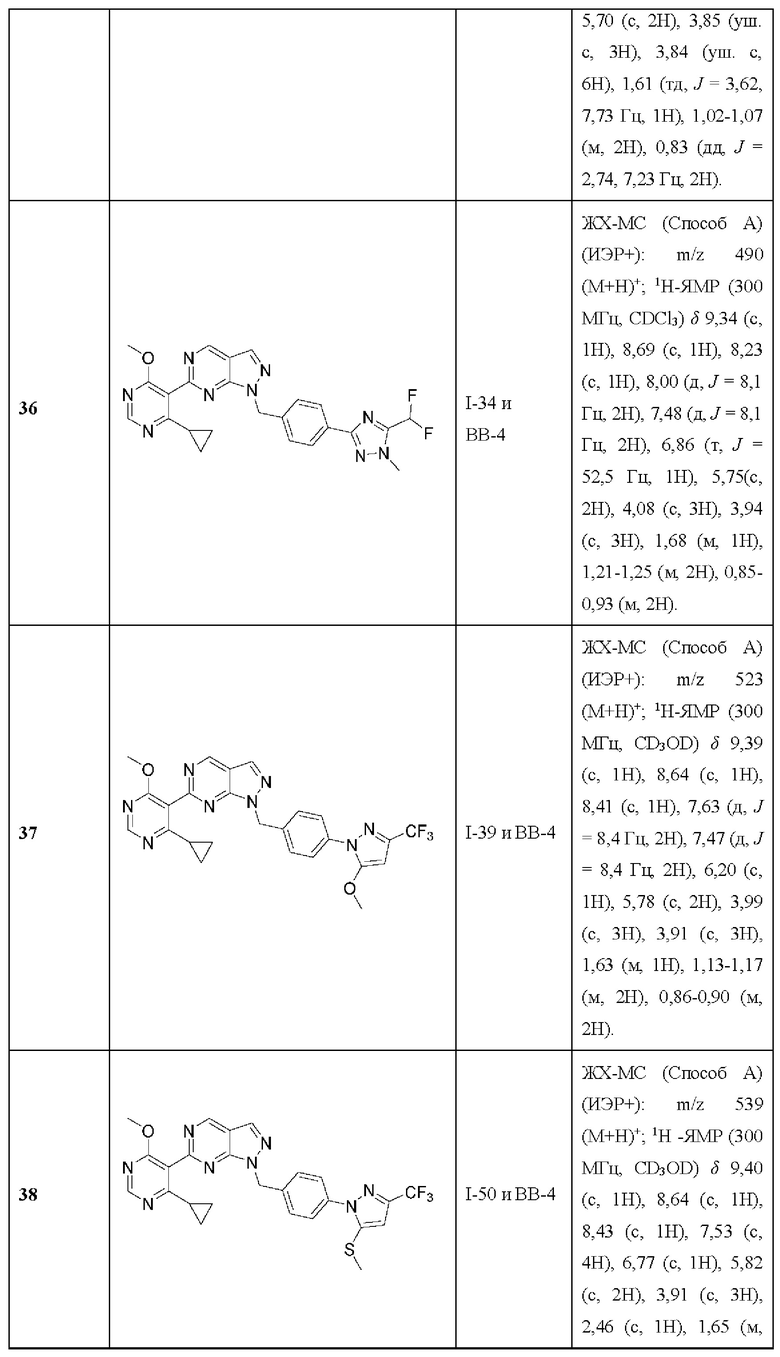
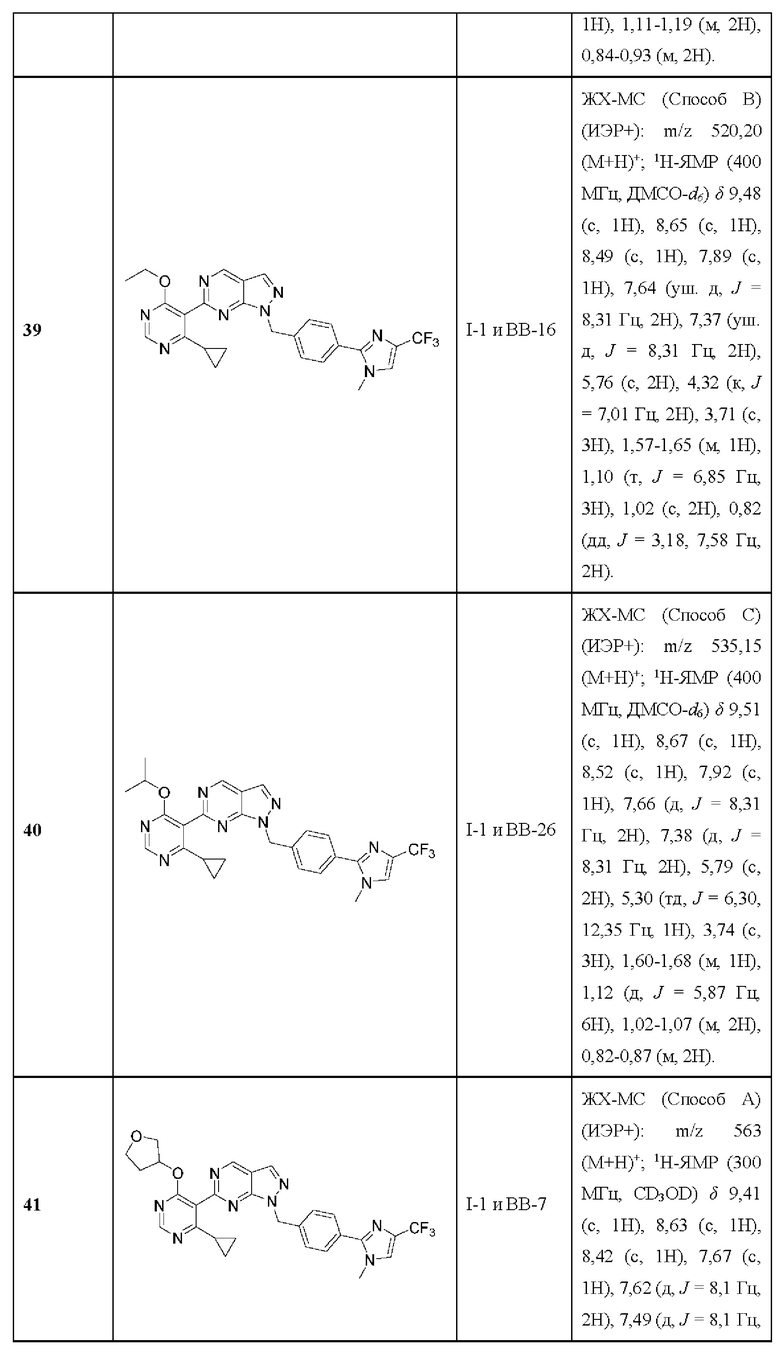

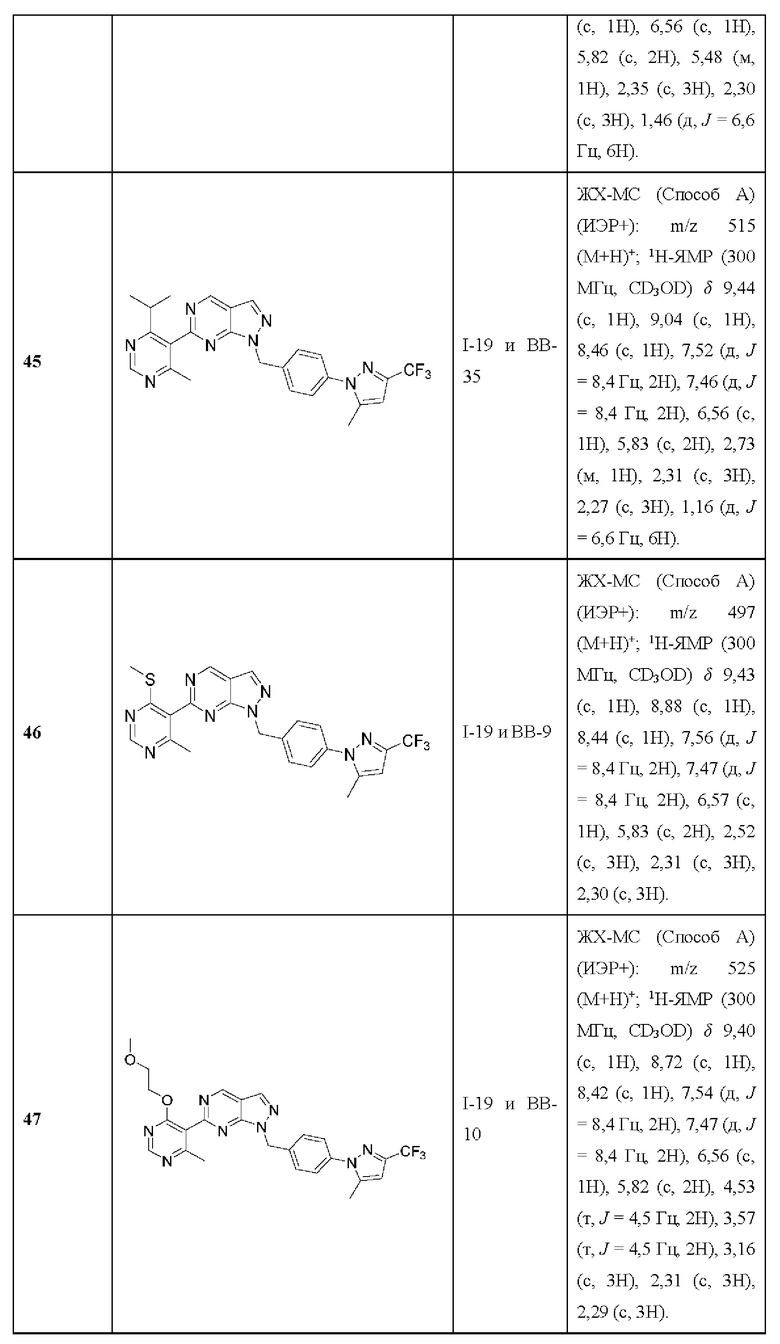
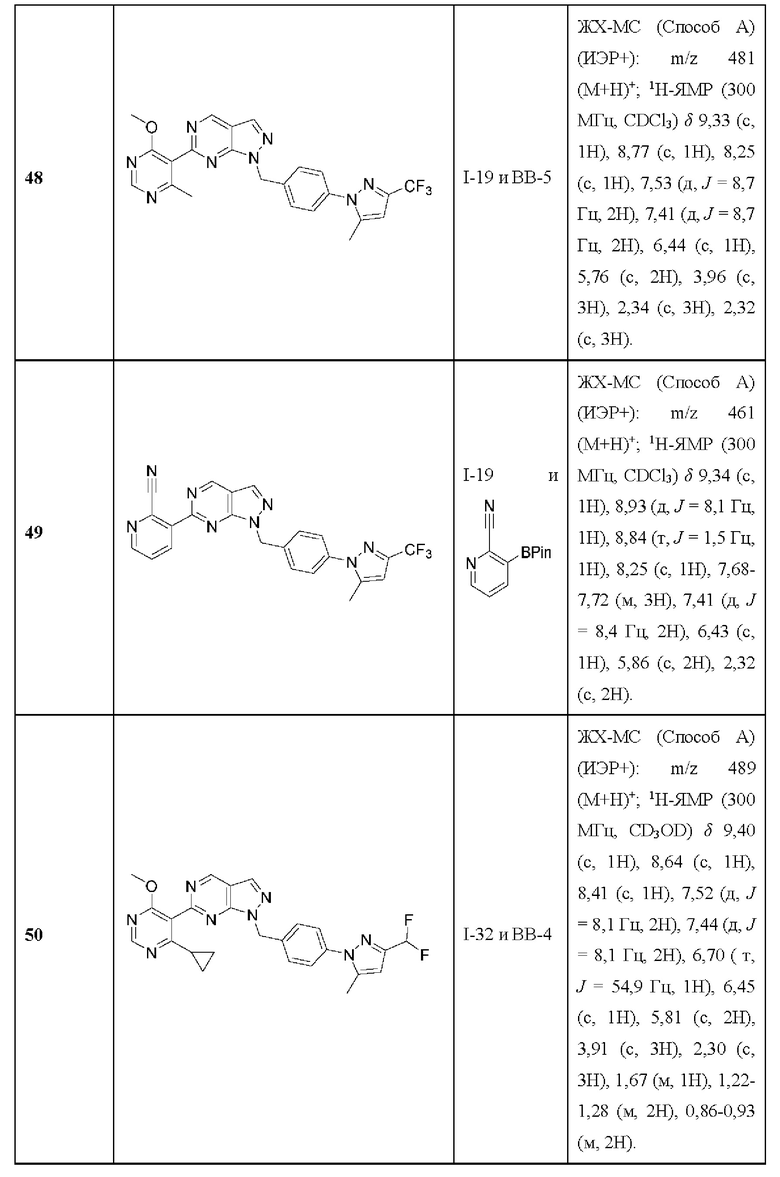
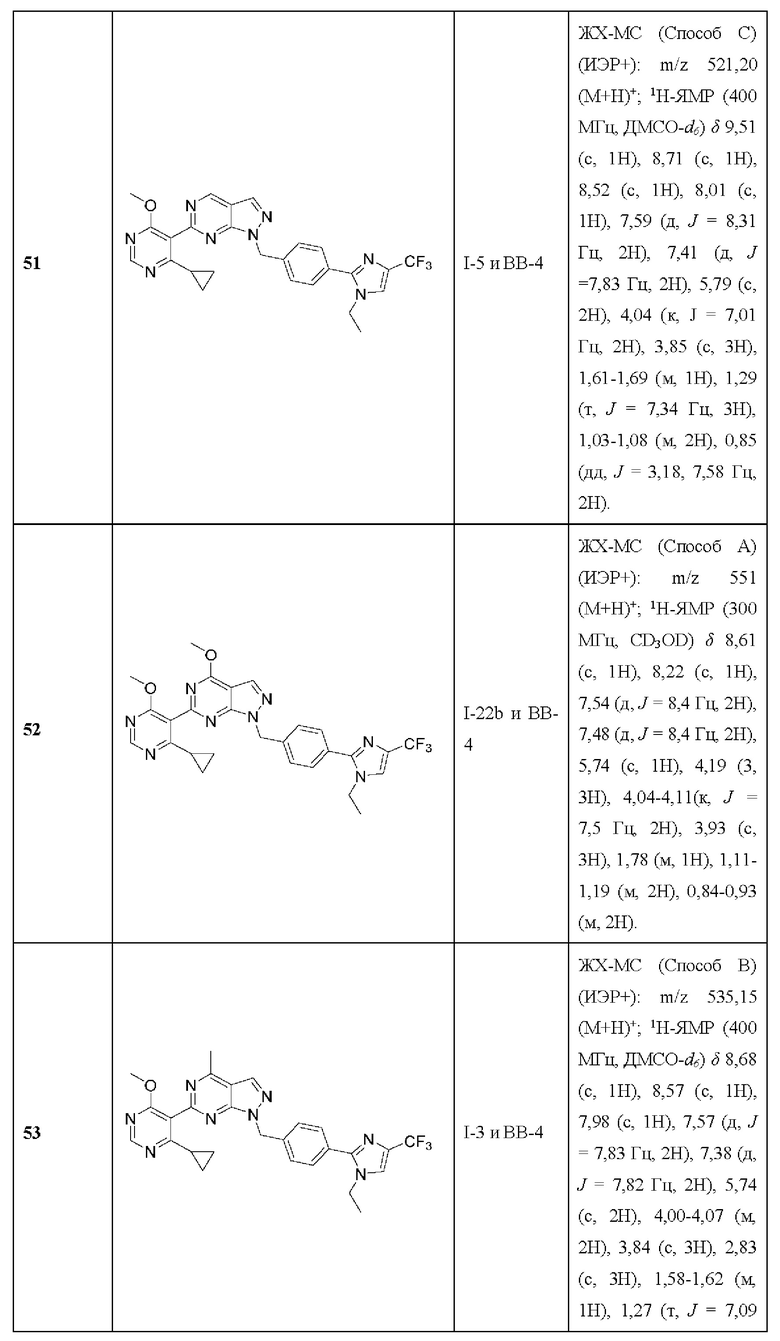
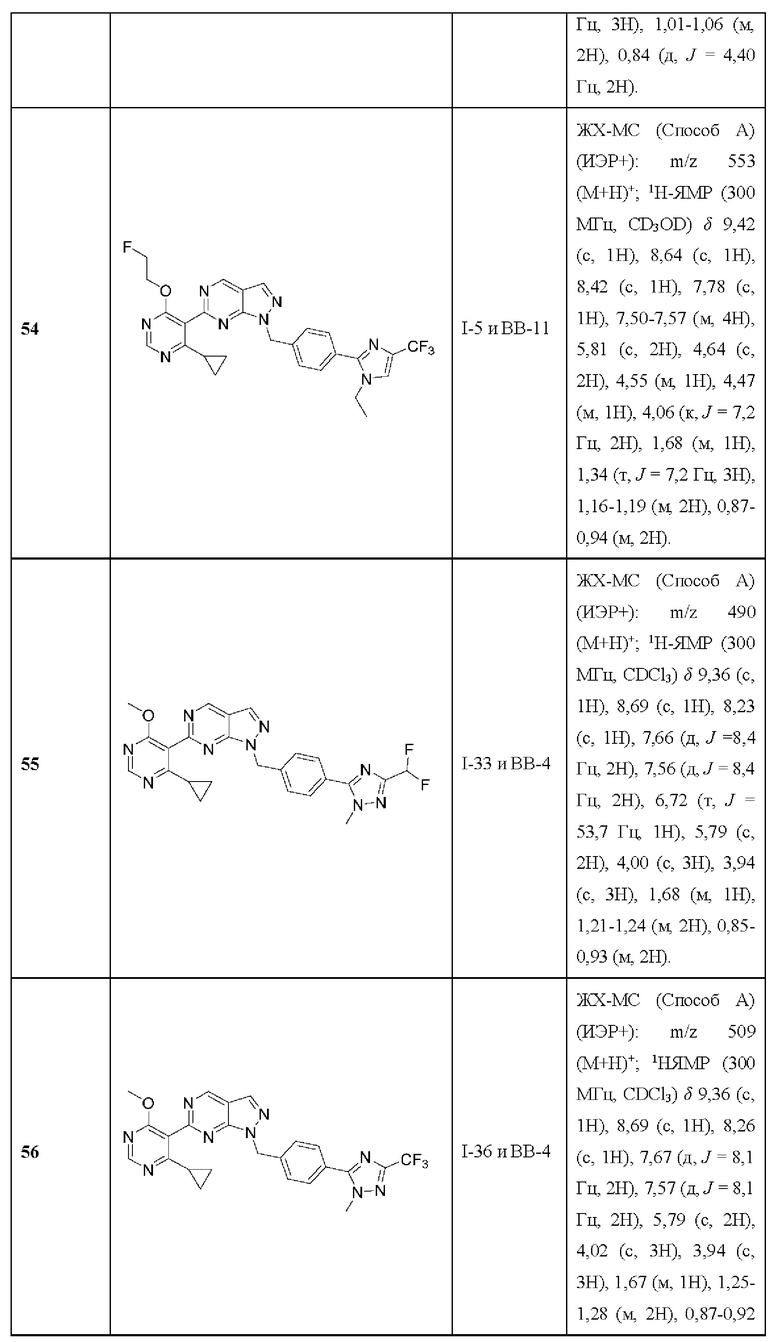
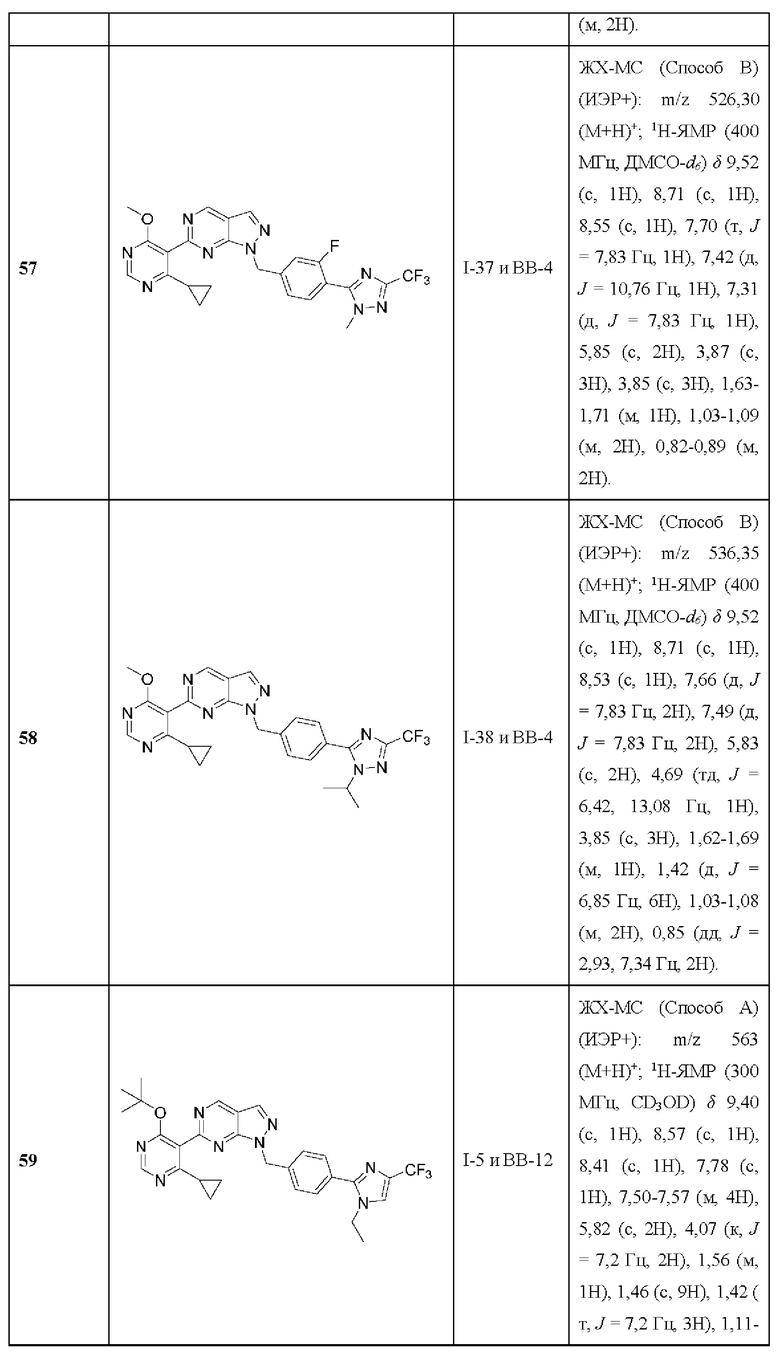
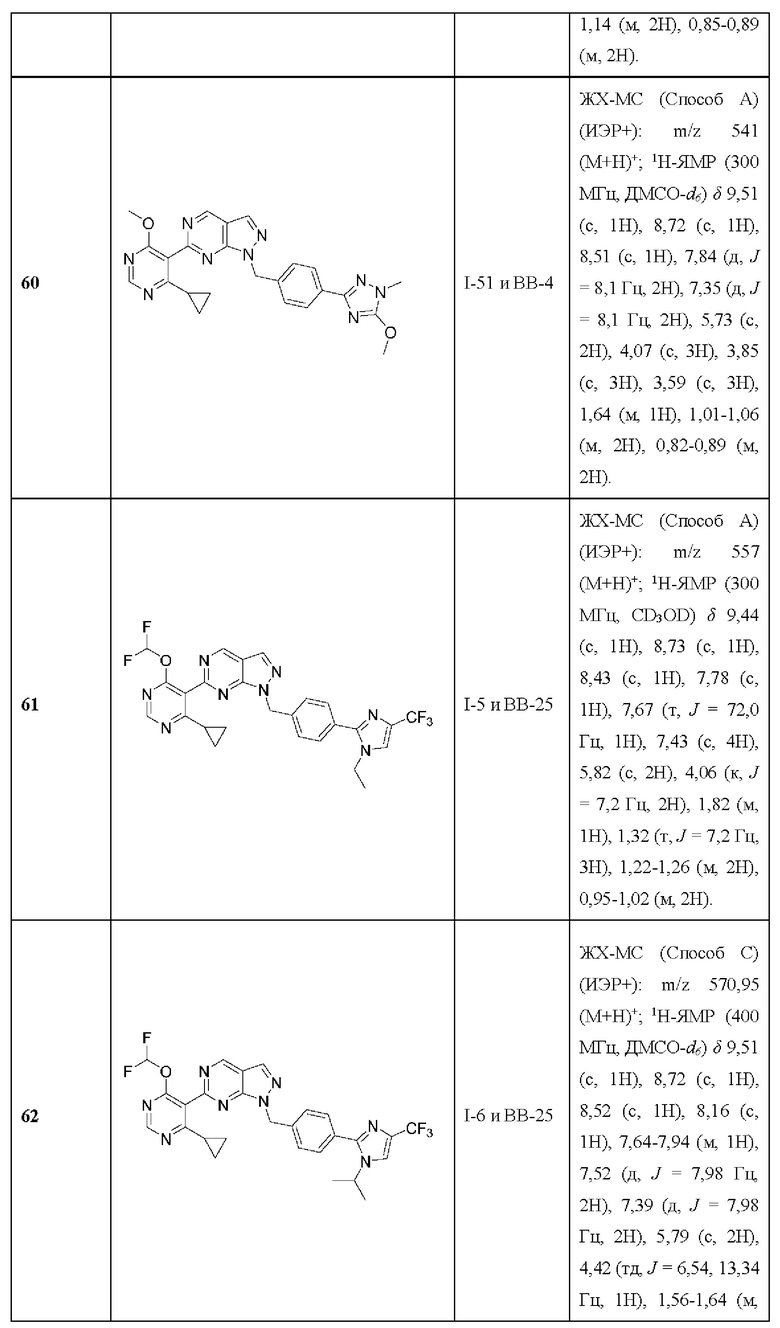
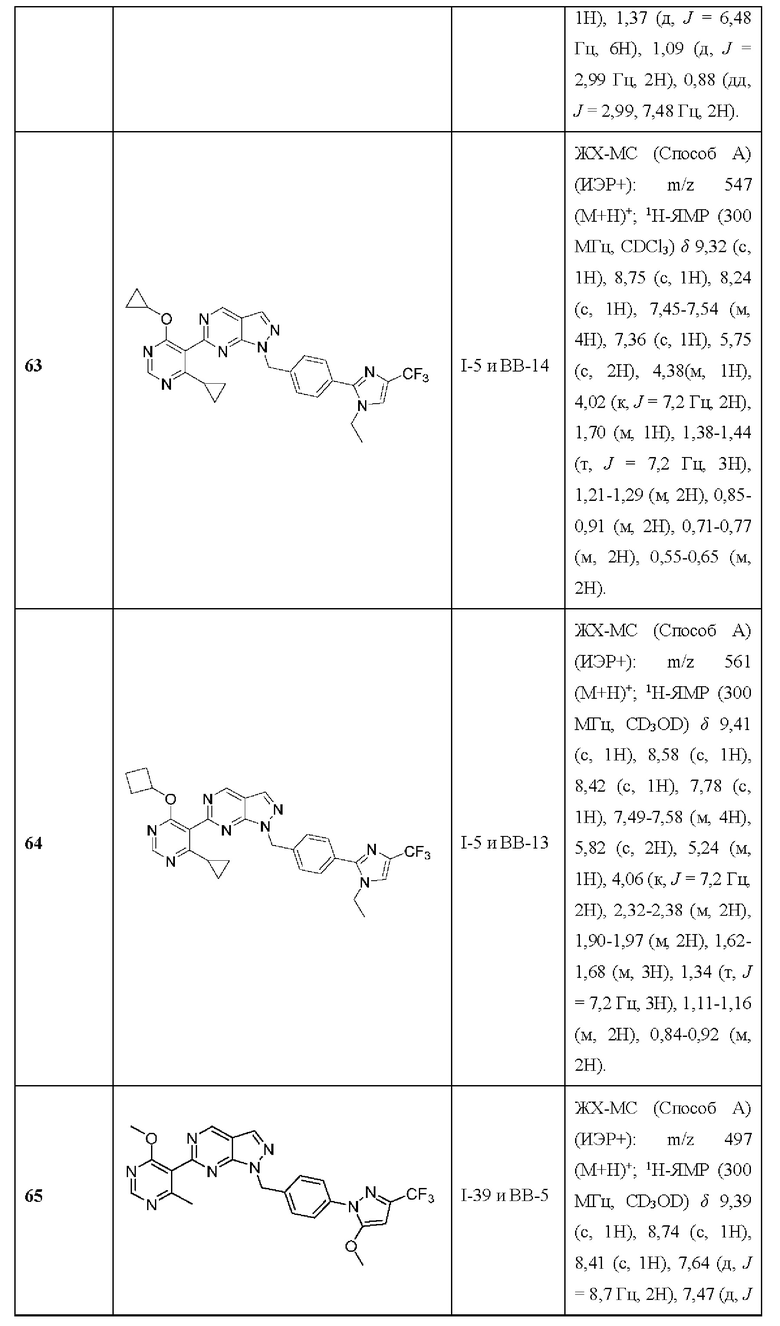
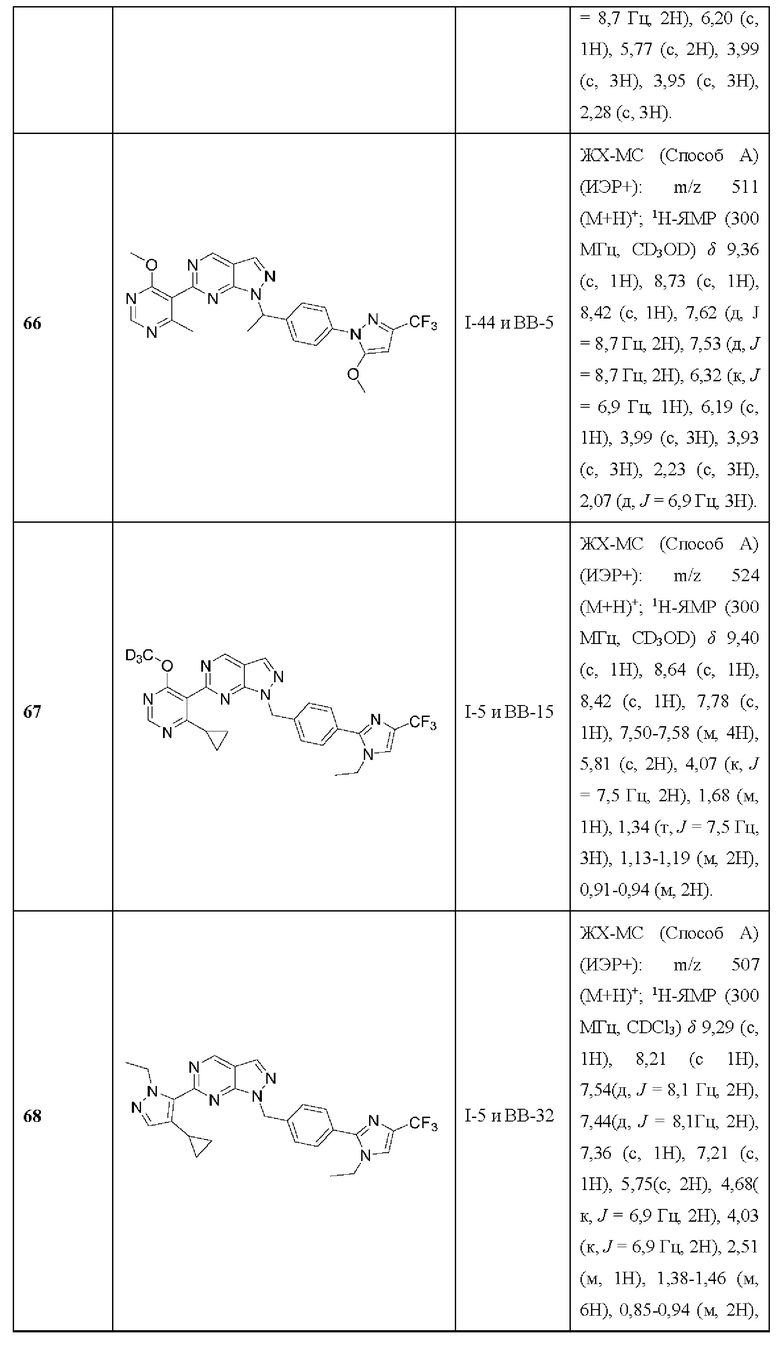
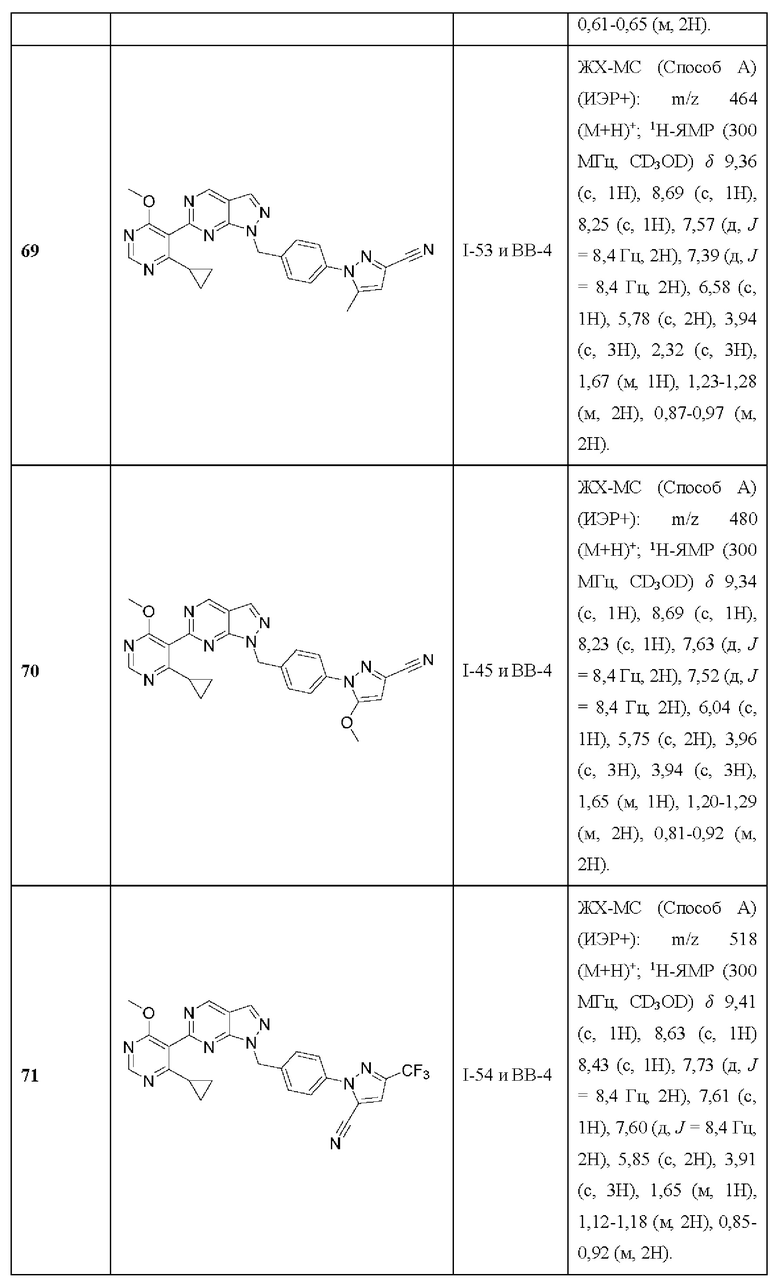
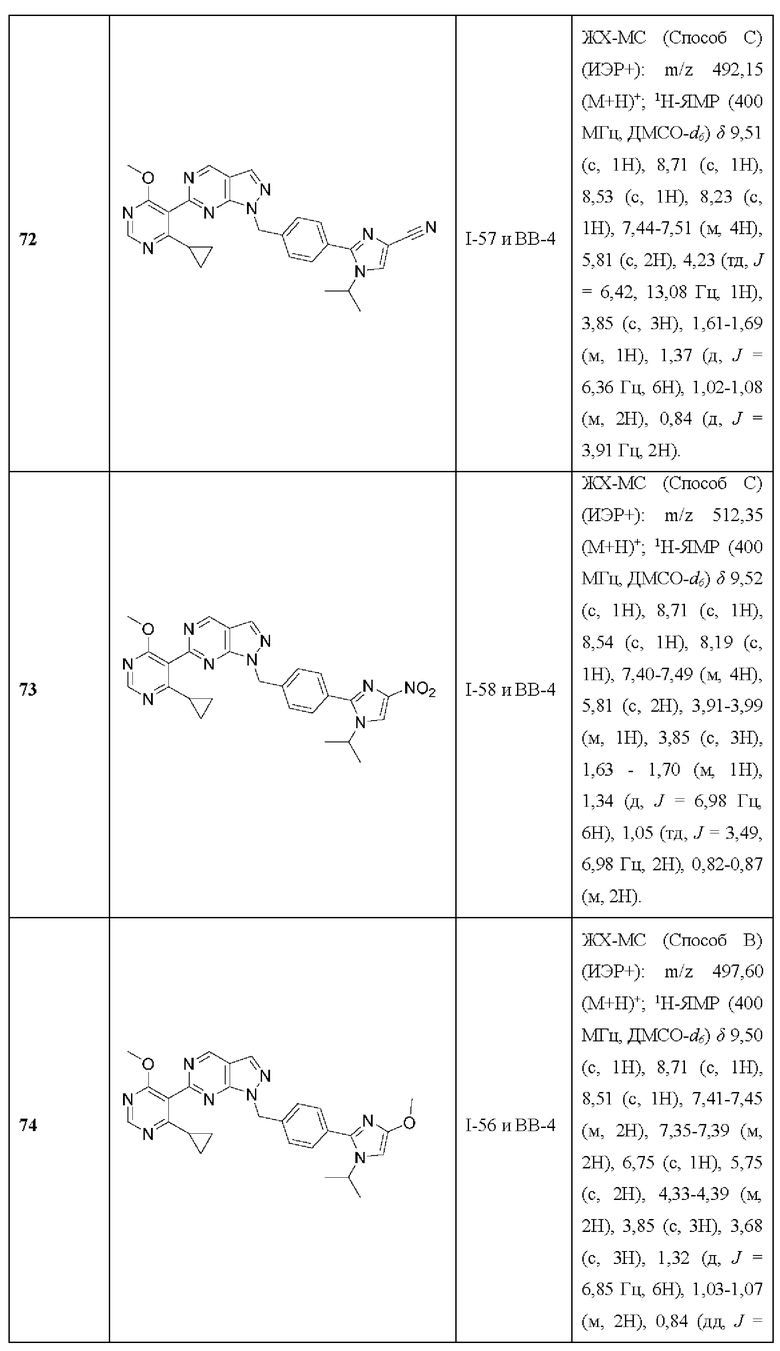
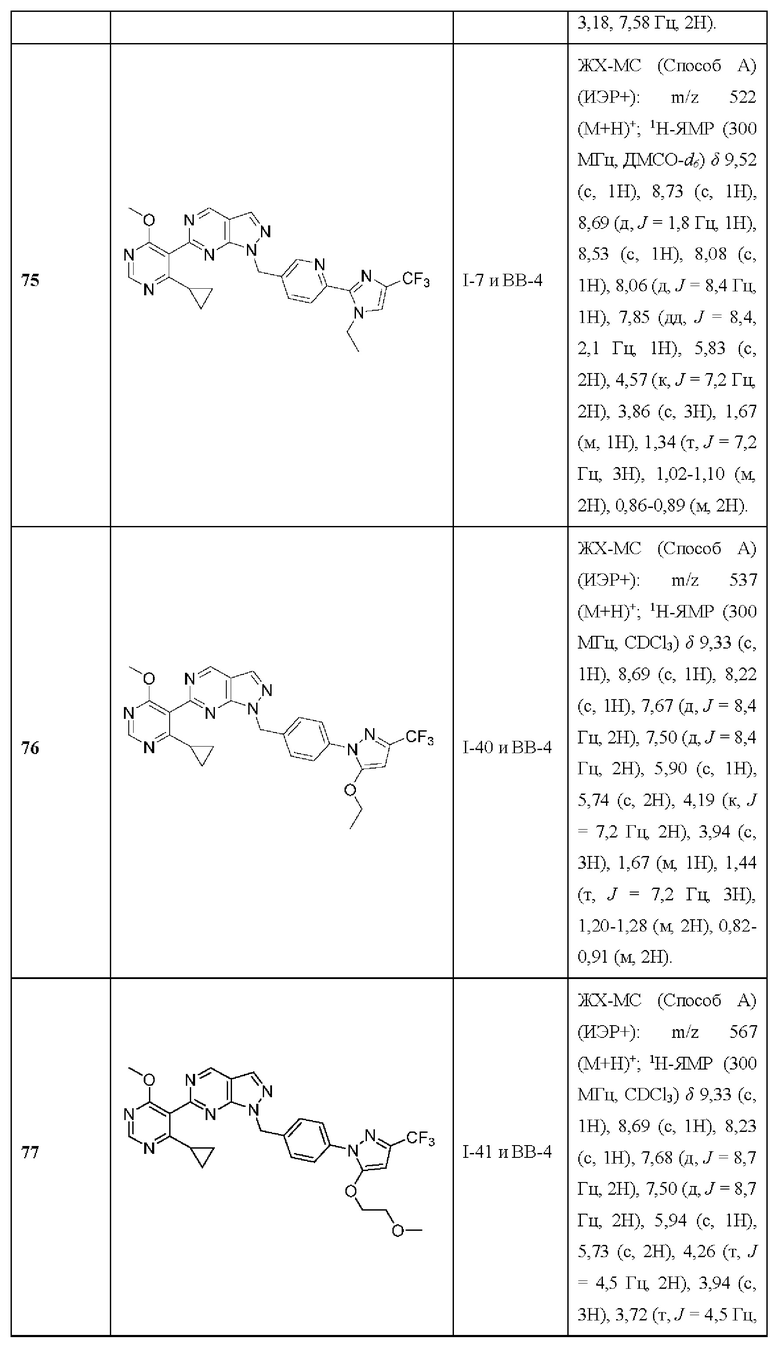
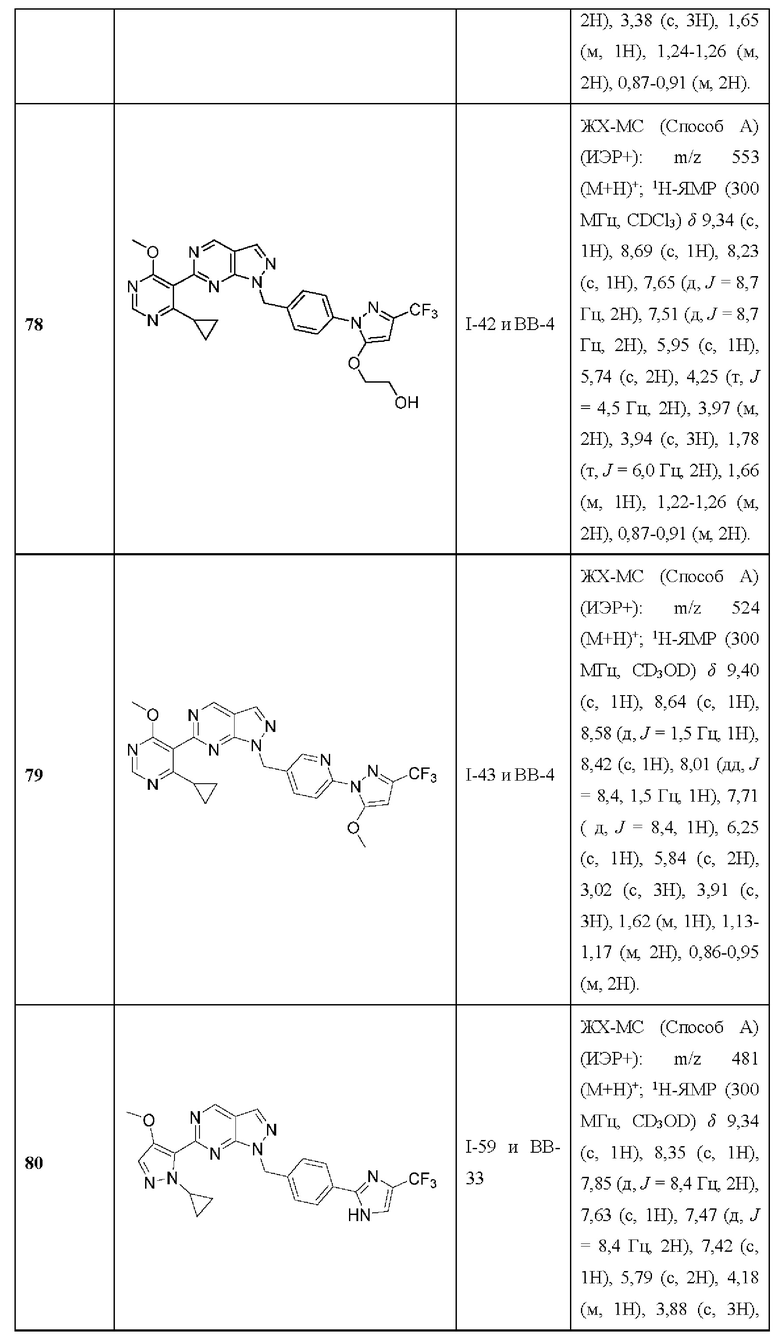
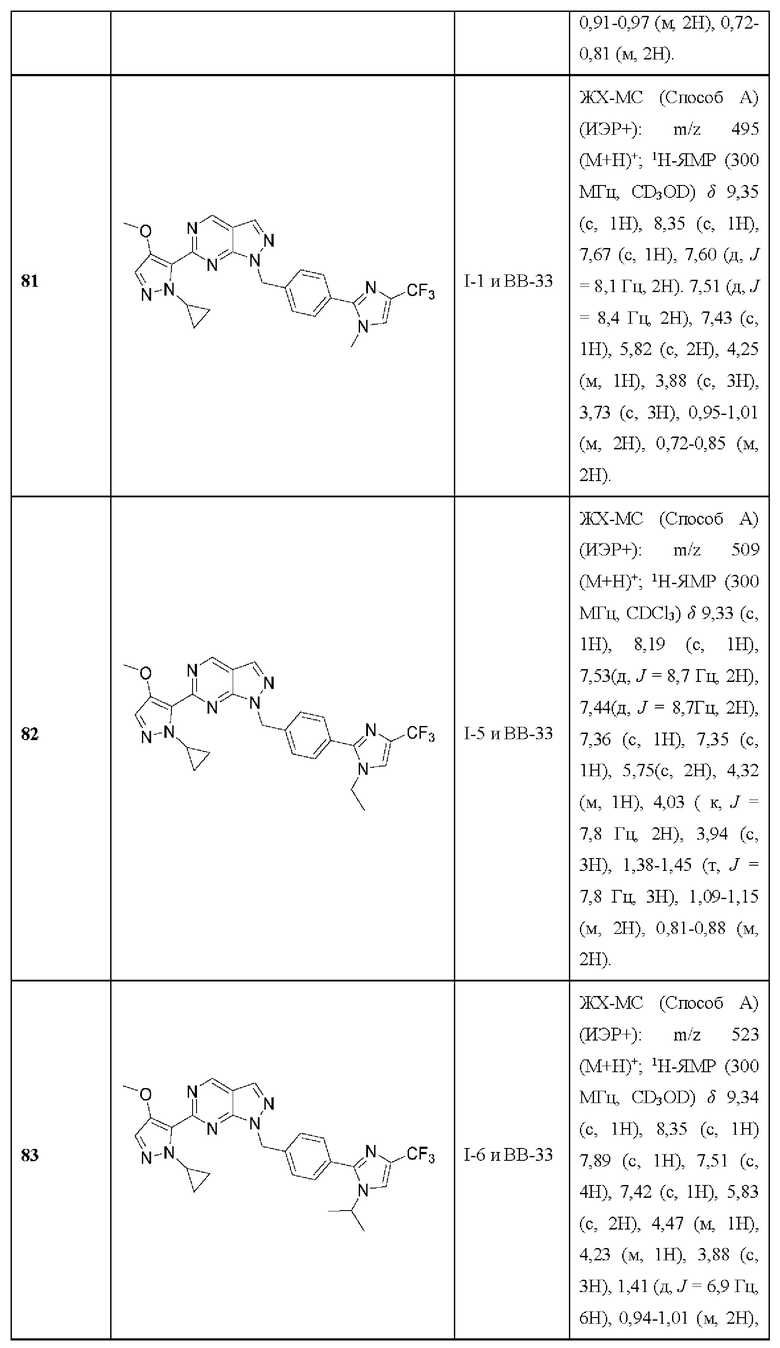
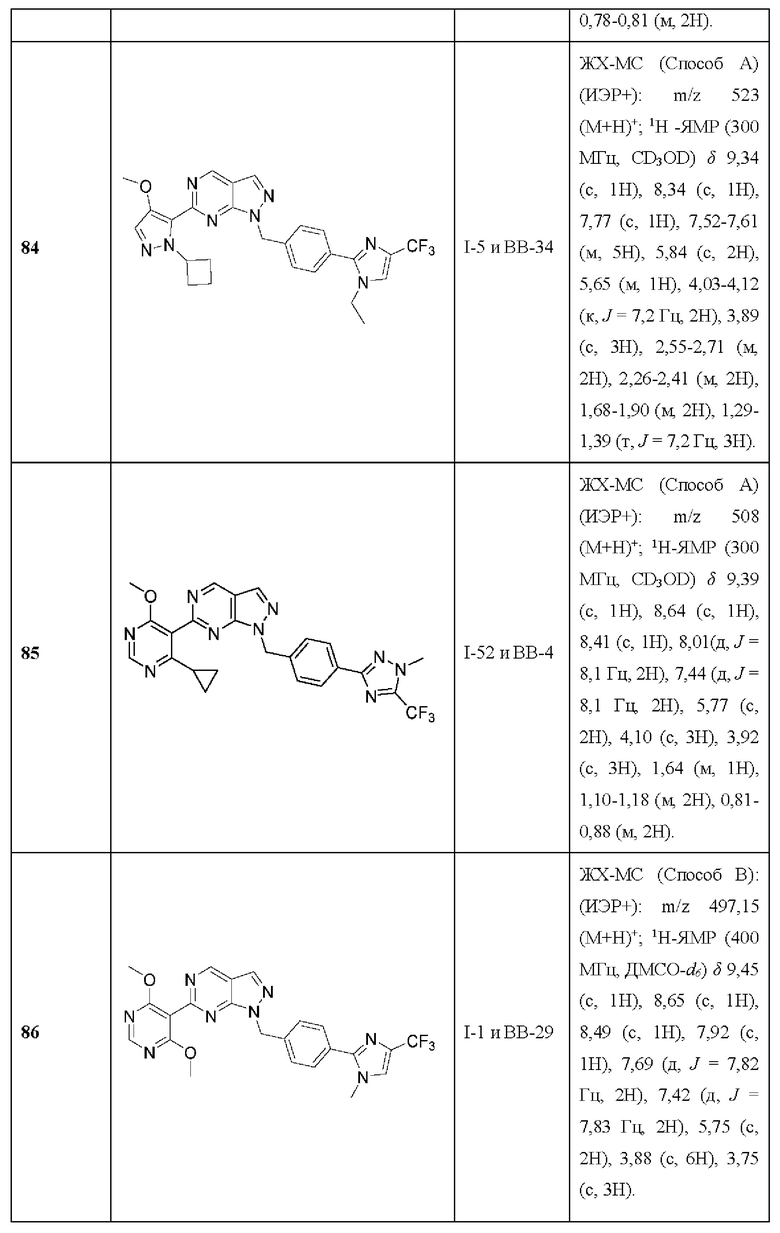
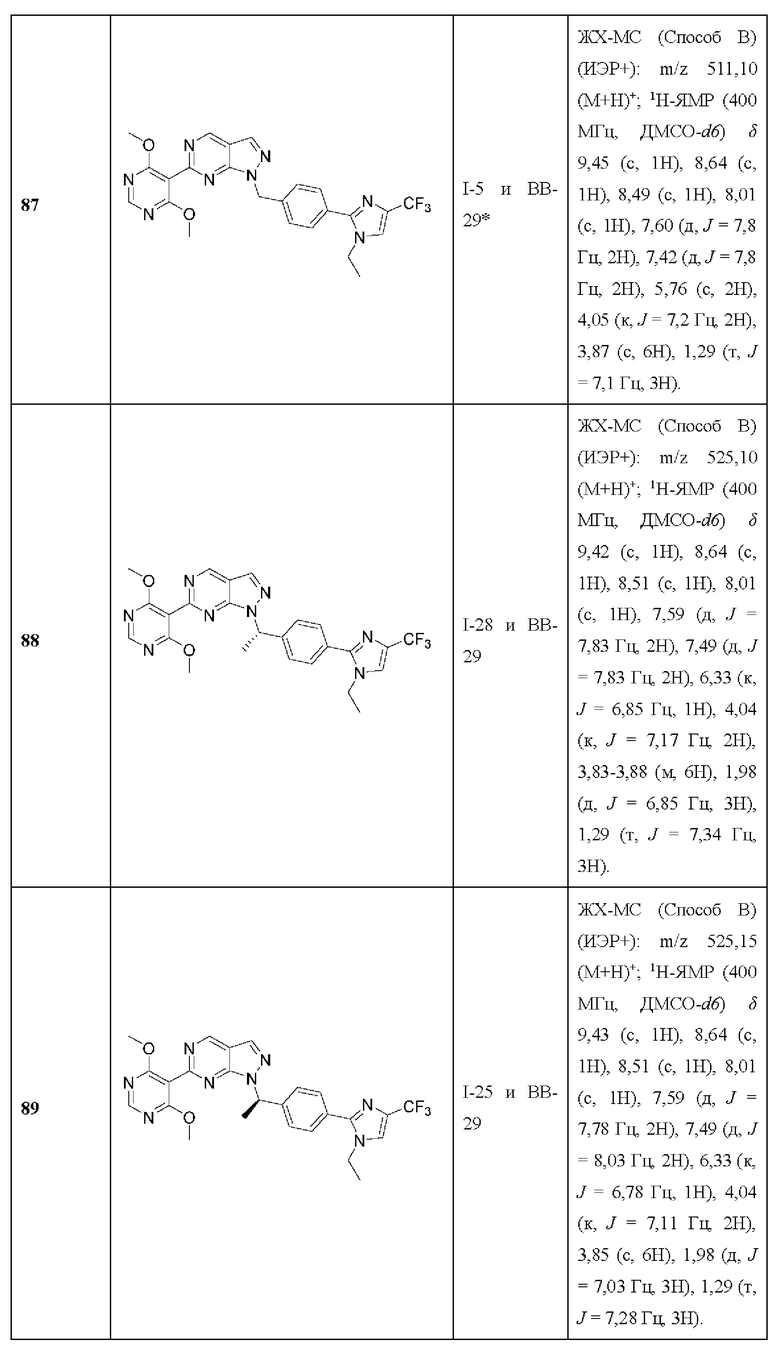
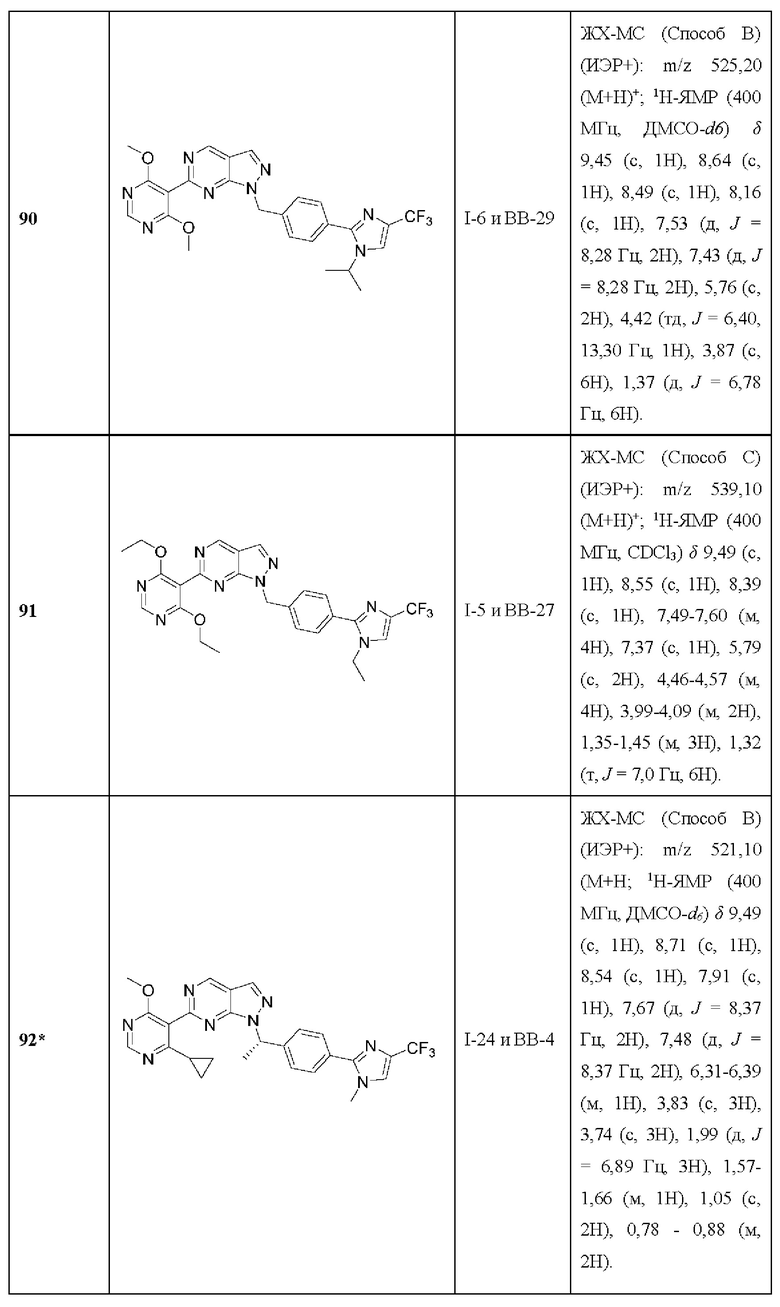
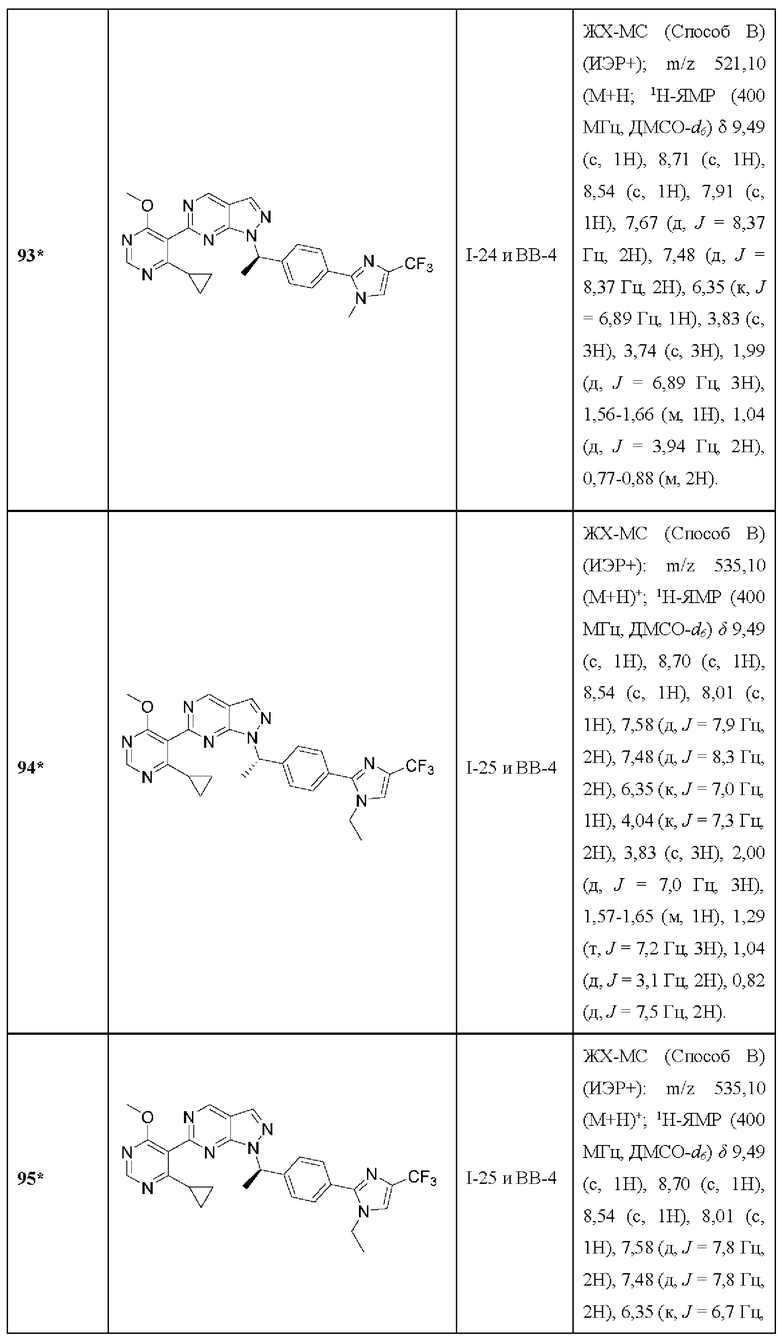
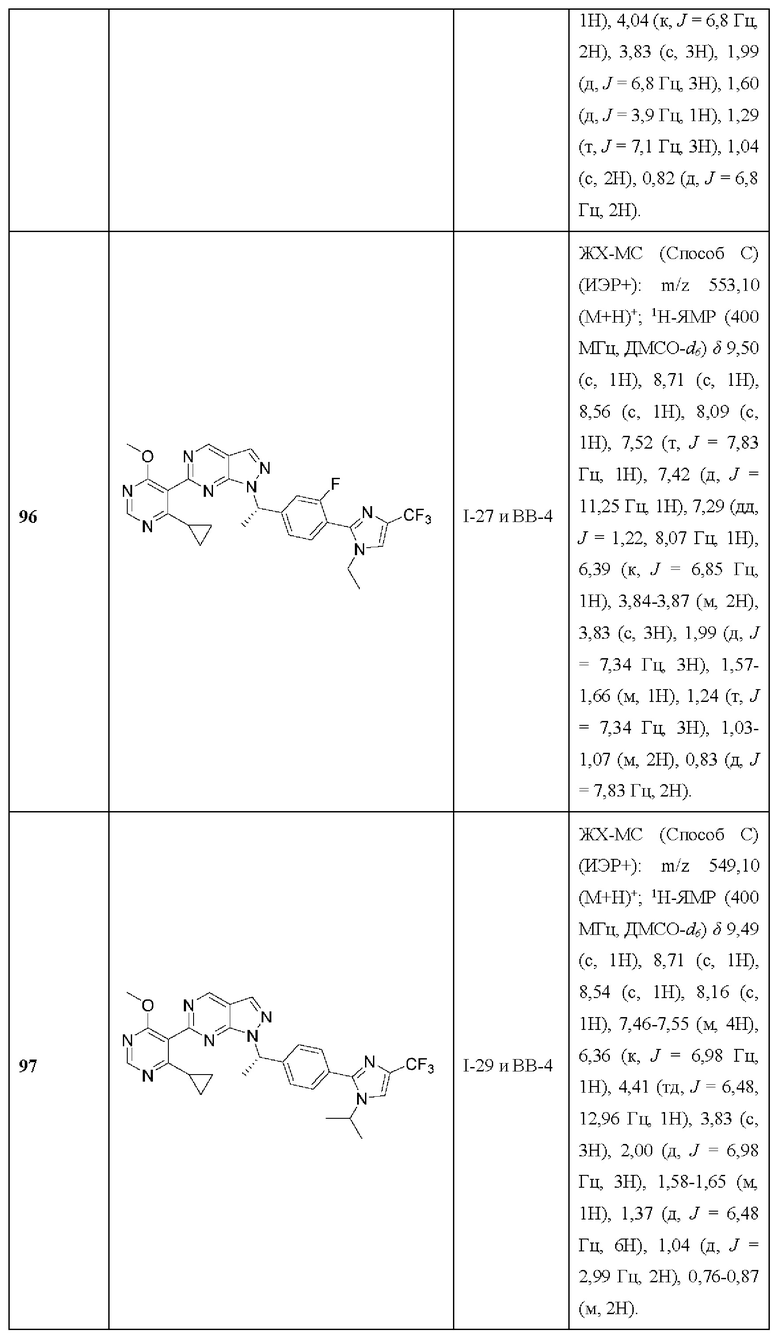

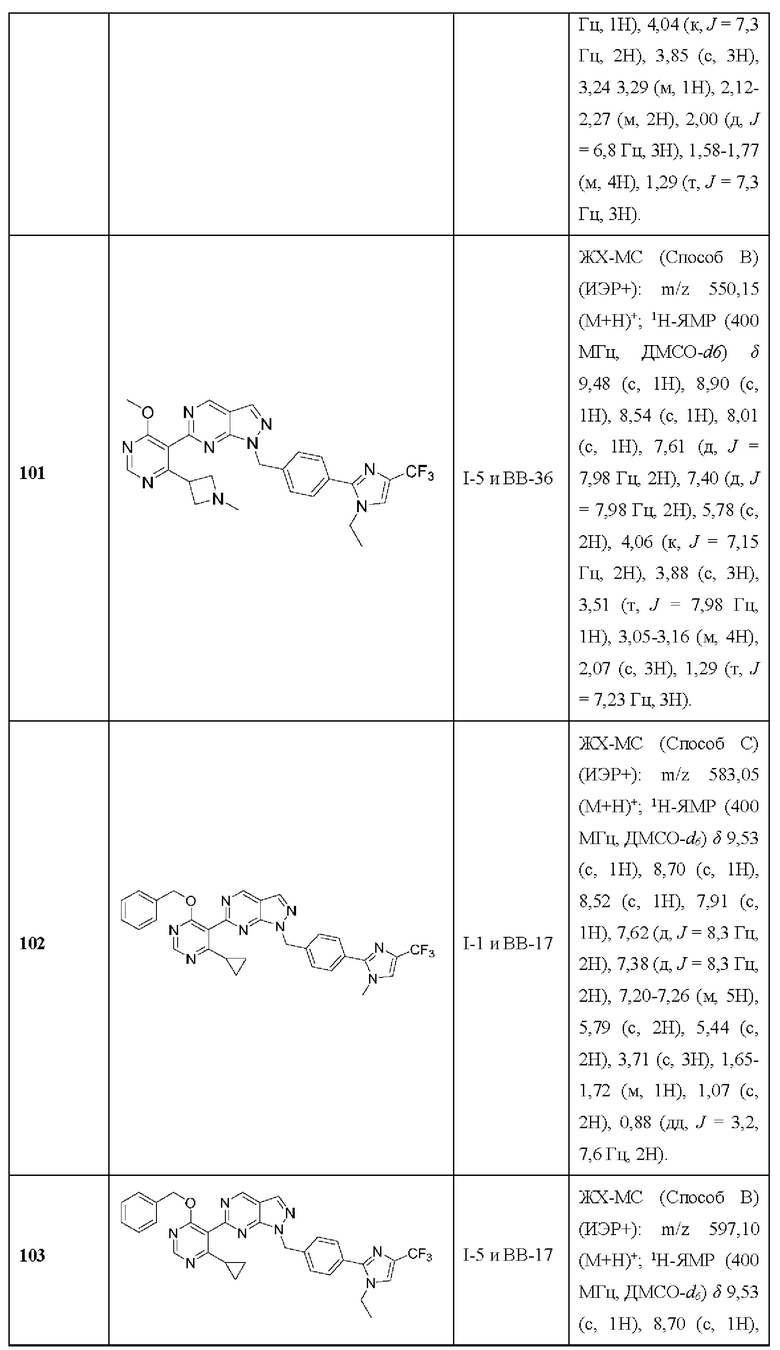
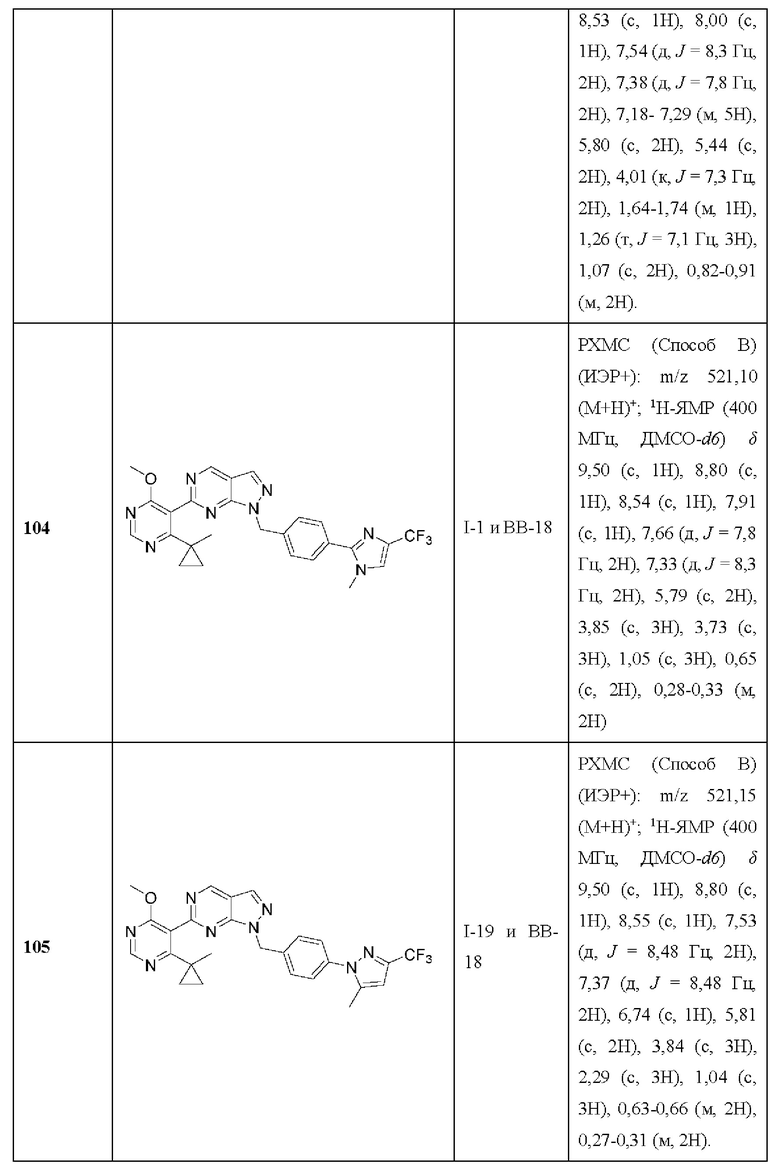
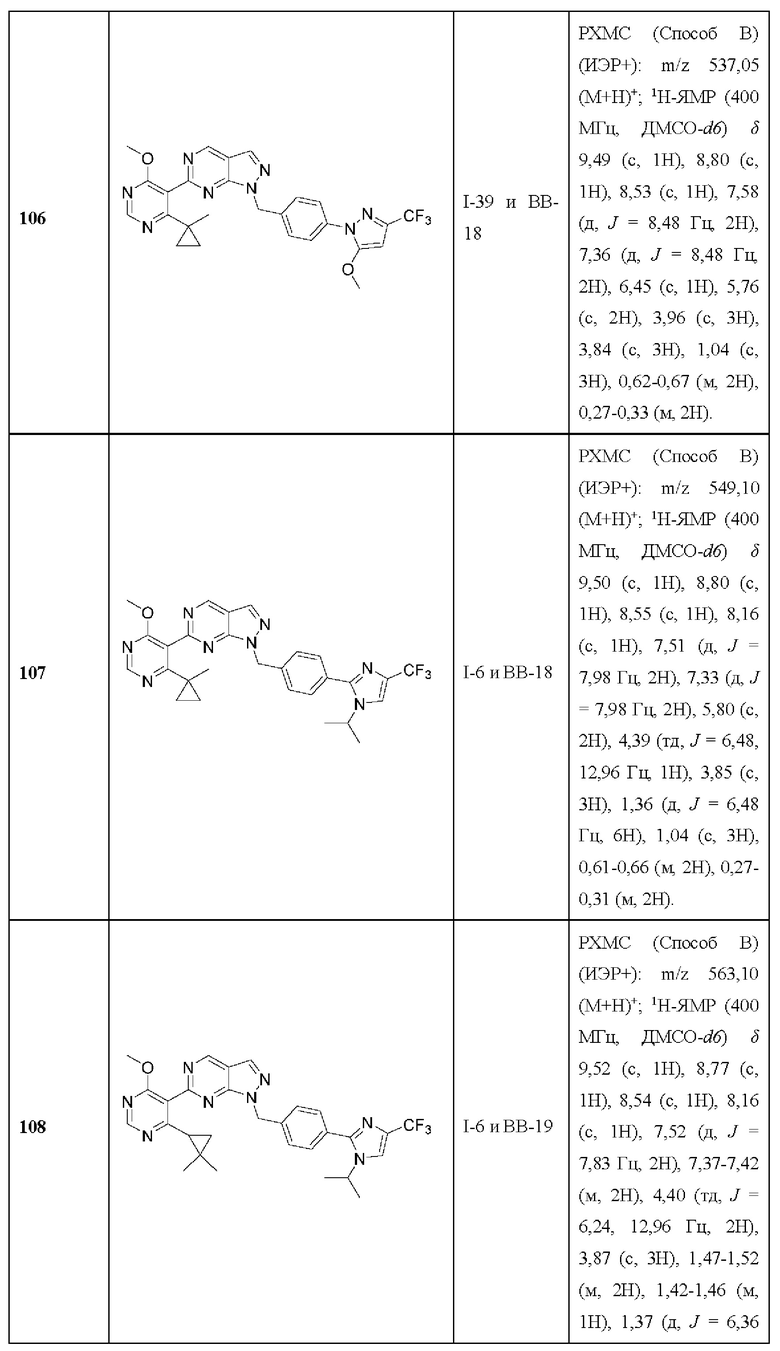
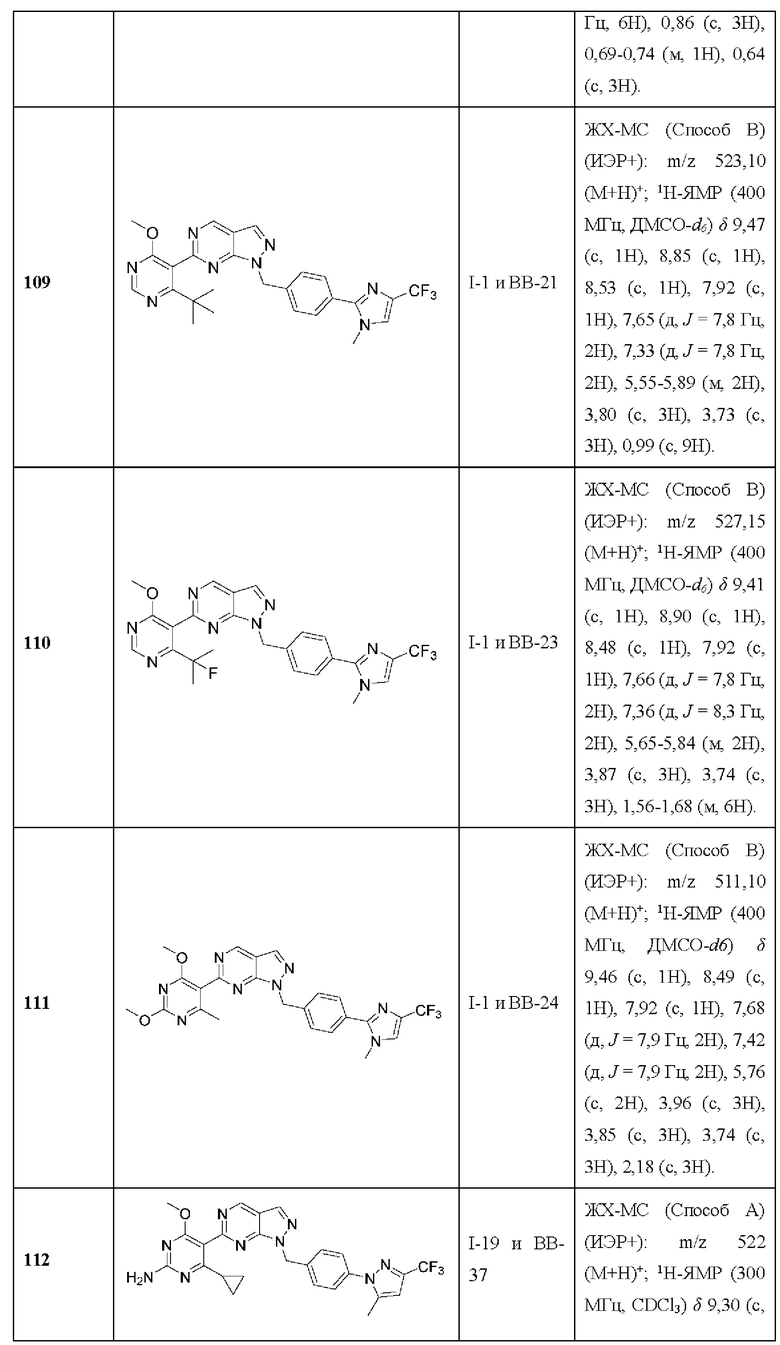
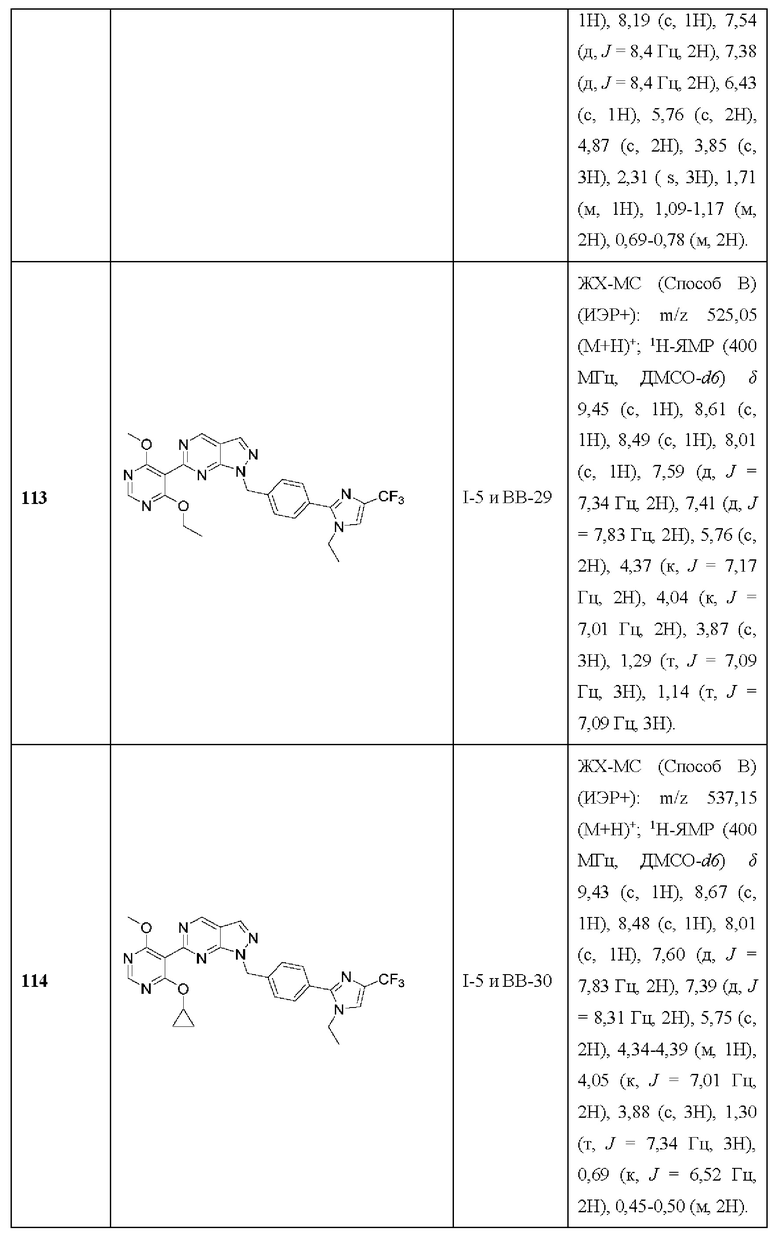
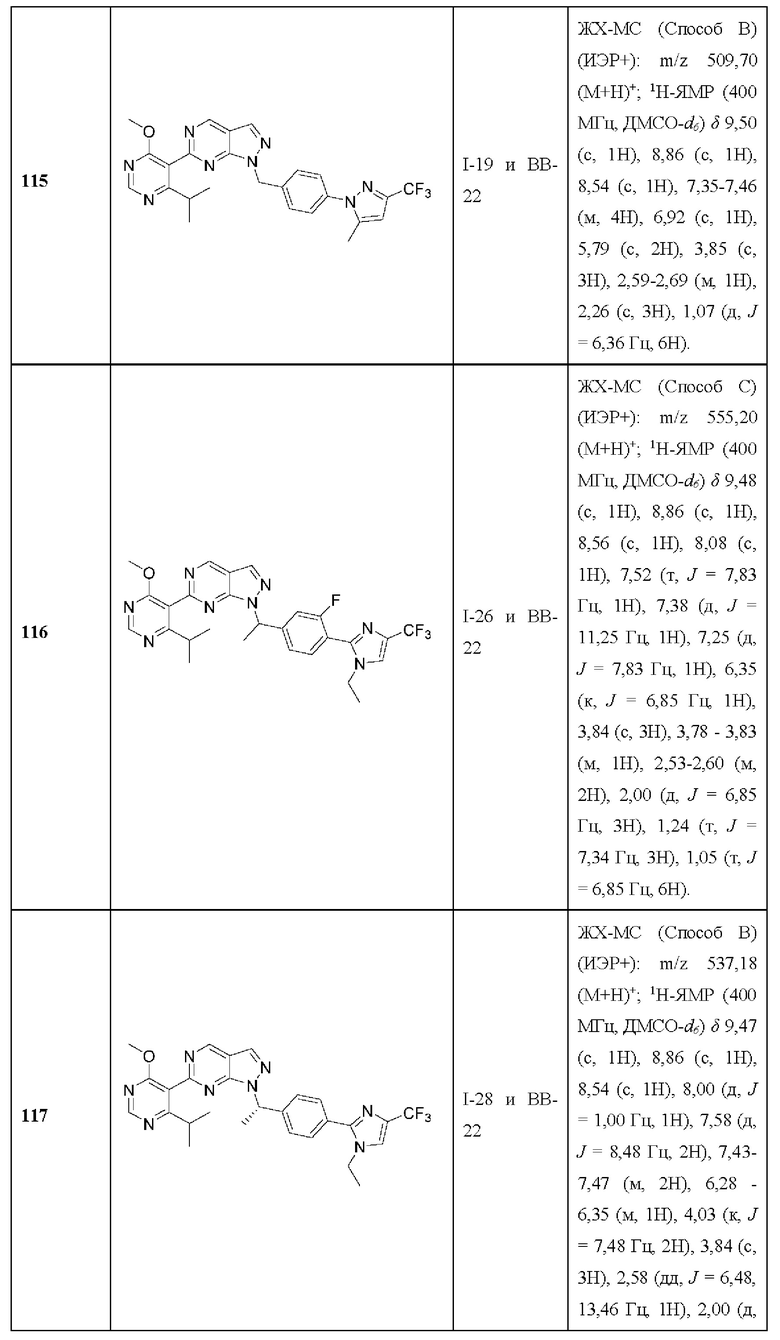
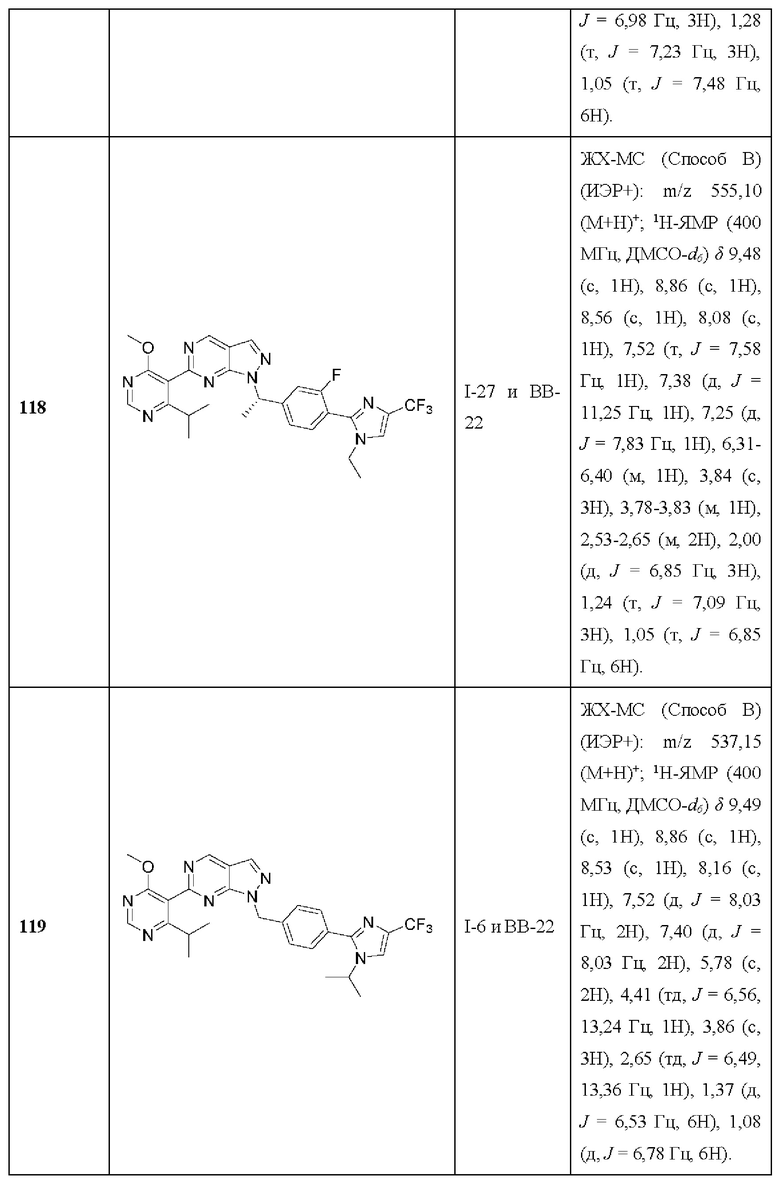
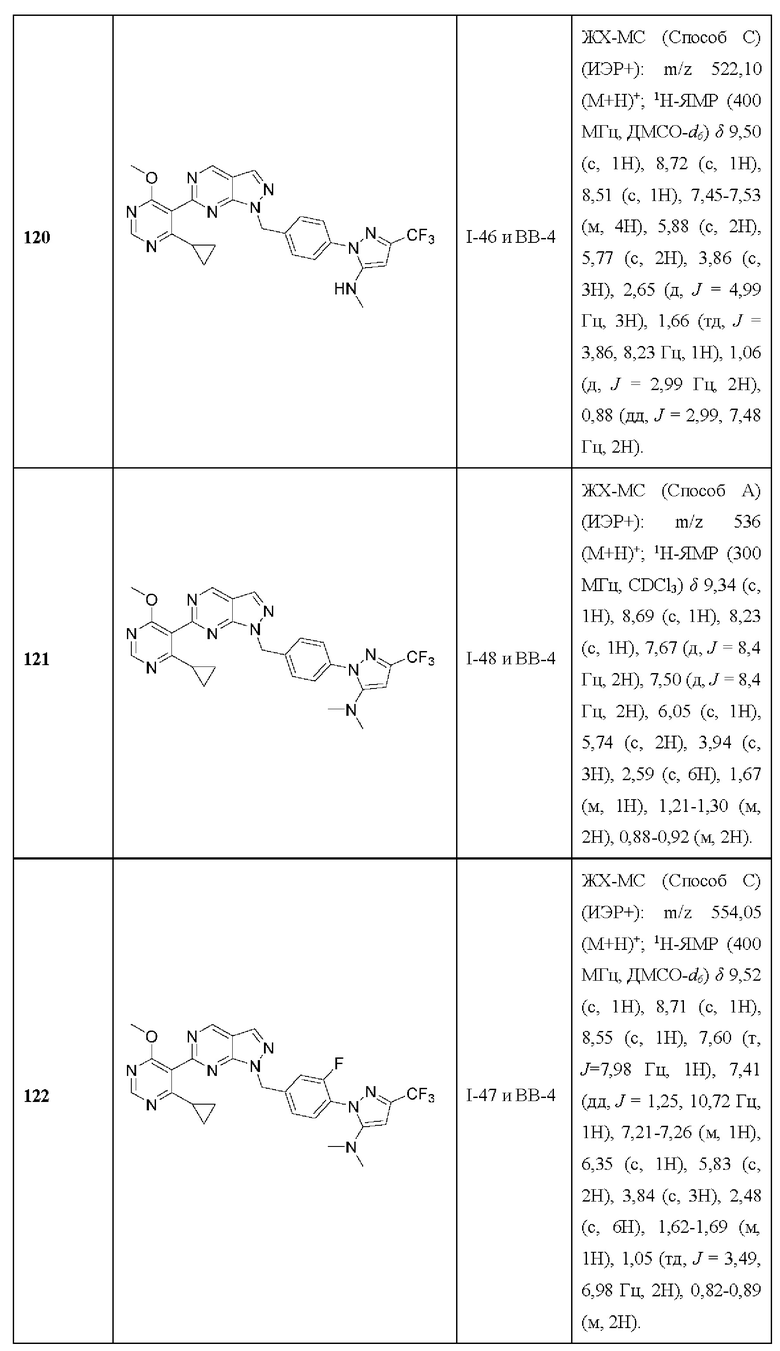
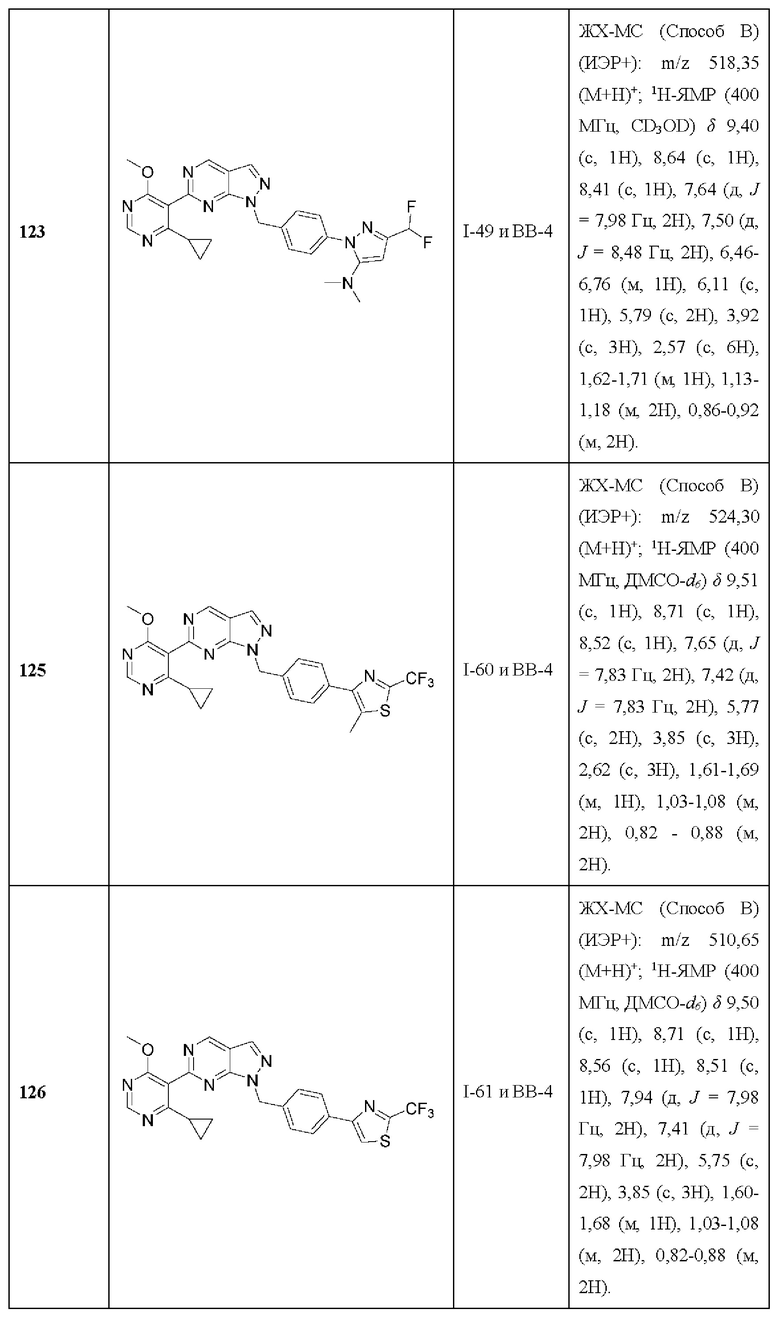
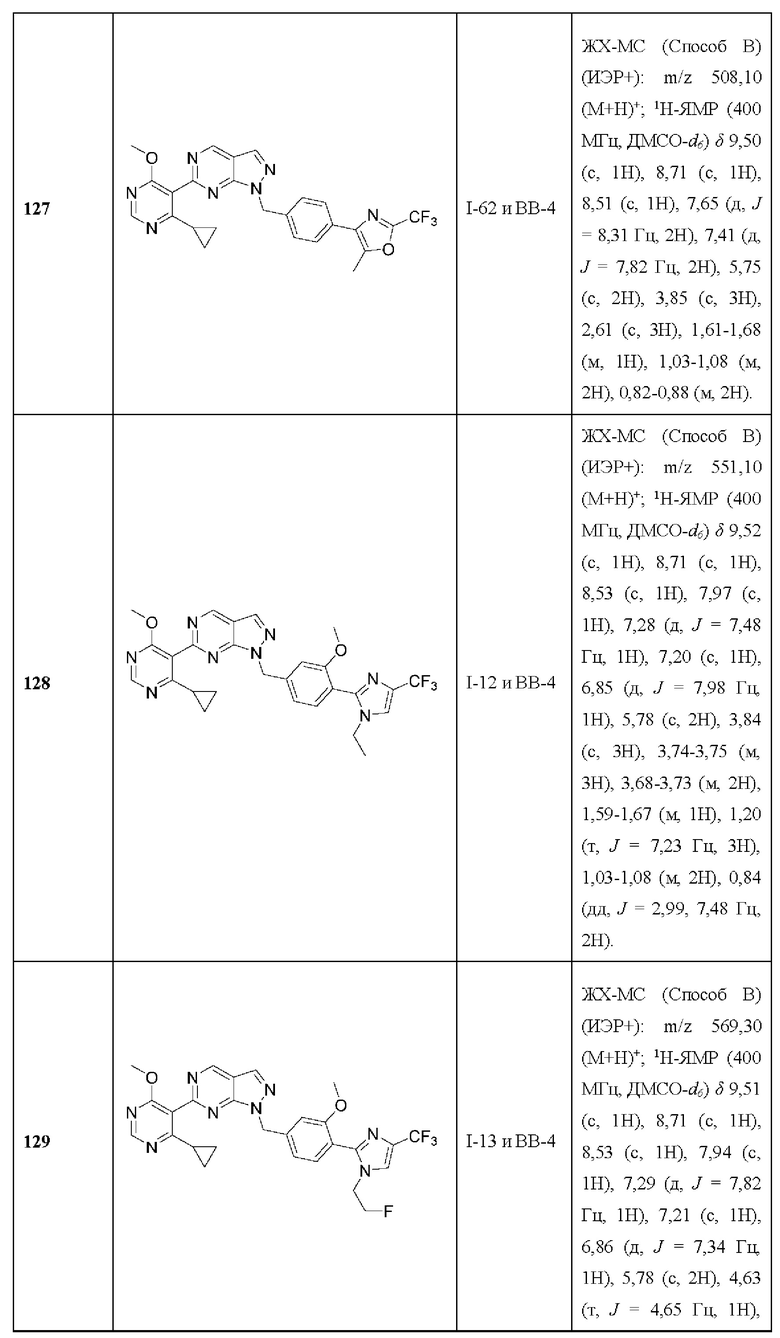
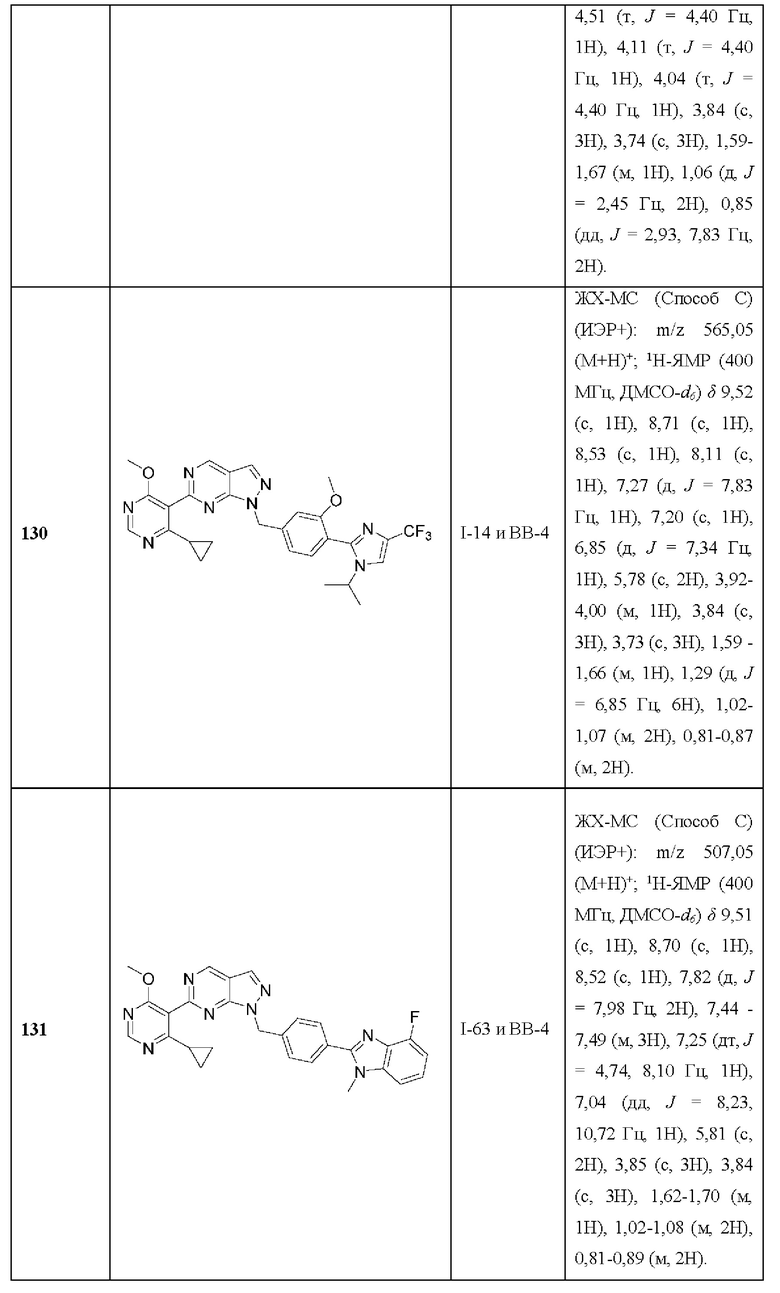
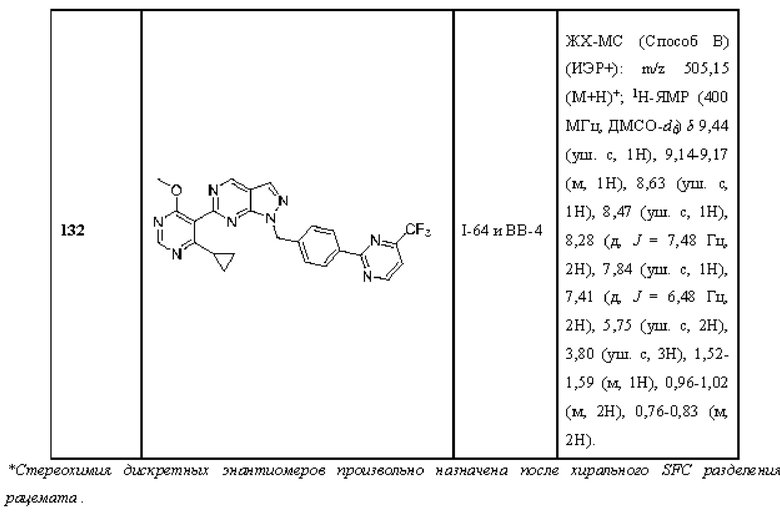
Пример 133 Синтез 6-(4-Циклопропил-6-метокси-2-метилепиримидин-5ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин (133):
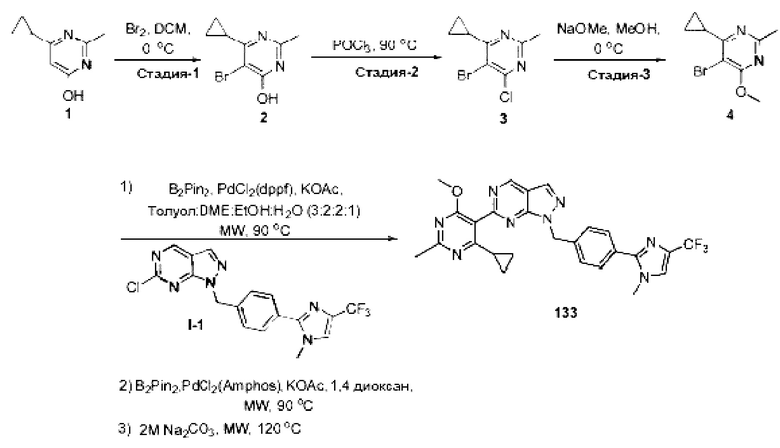
Стадия 1: Синтез 5-Бром-6-циклопропил-2-метилпиримидин-4-ола:
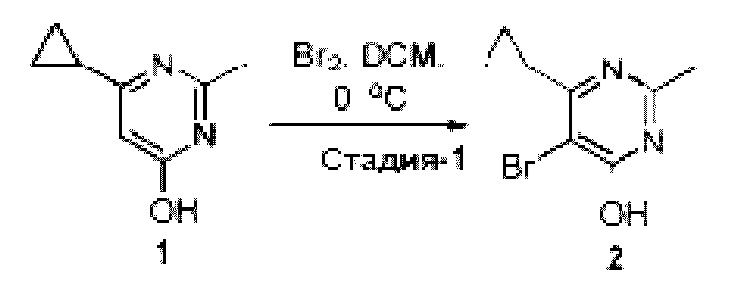
[0671] Раствор 6-циклопропил-2-метилпиримидин-4-ола (2,0 г, 13,33 ммоль) 6 DCM (50 мл), охлажденный до 0°С, обрабатывали по каплям бромом (0,75 мл, 14,66 ммоль). Полученную смесь перемешивали при 0°С в течение 4 часов. После завершения реакции (под контролем ТОХ) реакционную смесь гасили насыщенным раствором тиосульфата натрия (50 мл). Водный слой экстрагировали 10% метанолом в DCM (3×50 мл). Объединенный органический спой сушили над безводным Na2SO4 фильтровали и упаривали при пониженном давлении, получая неочищенное соединение. Неочищенное соединение очищали хроматографией на силикагеле (0 -10% метанол в DCM) с получением 1,50 г указанного в заголовке соединения ЖХ-МС (Способ В) (ИЭР+): m/z 230,90 (М+Н)+; 1H-HMP (400 МГц, ДМСО-d6) δ 12,62 (уш. с, 1Н), 2,31 (тд, J = 6,17, 12,59 Гц, 1Н), 2,20 (с, 3Н), 0,96-1,02 (м, 4Н).
Стадия 2: Синтез 5-Бром-4-хлор-6-циклопропил-2-метилпиримидина:
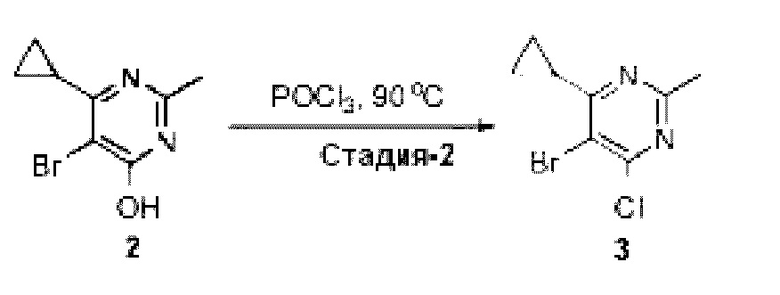
[0672] Раствор 5-бром-6-циклопропил-2-метилпиримидин-4-ола (1.5 г, 6.550 ммоль) в POCl3 (20 мл) нагревали при 90°С в течение 2 часов. После завершения реакции (под контролем ТСХ) реакционную смесь гасили льдом и экстрагировали ЕА (3×50 мл). Объединенный органический спой сушили над безводным Na2SO4, фильтровали и упаривали под вакуумом. Неочищенное соединение очищали колоночной хроматографией (силикагель; 0-20% ЕА в н-гексане) с получением 1,20 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 248,95 (М+Н)+.
Стадия 3: Синтез 5-Бром-4-циклопропил-6-метокси-2-метилпиримидина:
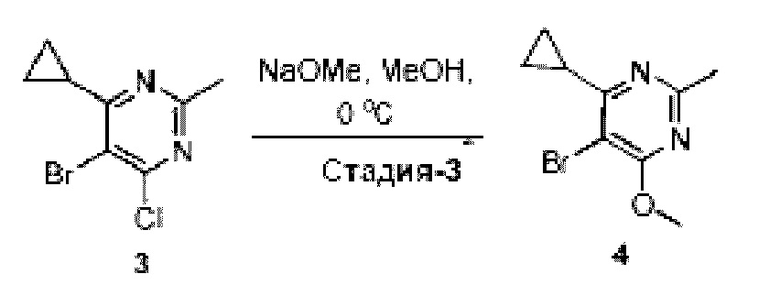
[0673] К ледяному раствору 5-бром-4-хлор-6-циклопропил-2-метилпиримидина (1,1 г, 4,453 ммоль) е метаноле (20 мл) добавляли 2 М раствор метоксида натрия (20 мл). Реакционную смесь перемешивали при 0°С в течение 2 часов. После завершения реакции (под контролем TCX) реакционную смесь гасили ледяной водой (50 мл) и ЕА (50 мл). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и упаривали под вакуумом. Неочищенное соединение очищали колоночной к хроматографией (силикагель; 0-10% ЕА в н-гексане) с получением 0,90 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 242,95 (М+Н)+; 1Н-ЯМР (400 МГц, CDCl3) δ 4,00 (с, 3Н), 2,47-2,51 (м, 1Н), 2,46 (с, 3Н), 1,12-1,17 (м, 2Н), 1,00-1,05 (м, 2Н).
Стадия 4: Синтез 6-(4-Циклопропил-6-метокси-2-метилпириминин-5-нл)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин (133):
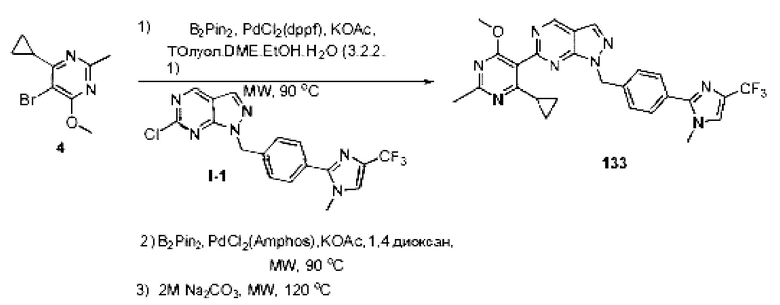
Стадия 1:
[0674] В 30 мл флакон для микроволновой печи добавляли 5-бром-4-циклопропил-6-метокси-2-метилпиримидин (0,100 г, 0,411 ммоль), биспинаколатодиборон (0,104 г, 0,411 ммоль), KOAc (0,040 г, 0,41 ммоль) и толуол: DME: EtOH: Н2О (3:2:2:1, 6 мл). Смесь продували аргоном в течение 20 мин, после чего добавляли PdCl2(dppf) (0,030 г, 0,041 ммоль). Затем реакционную смесь нагревали в микроволновой печи при 90°С в течение 20 минут. После завершения реакции (под контролем ТСХГ) смесь использовали без обработки в Стадии 3.
Стадия 2:
[0675] В другой 30 мл флакон для микроволновой печи был добавлен 6-клор-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил) бензил)-1Н-пиразоло[3,4-d]пиримидин (0,160 г, 0,408 ммоль), биспинаколатодиборан (0,103 г, 0,408 ммоль) и KOAc (0,119 г, 1,2 24 ммоль) в 1,4-диоксане (6 мл). Полученную смесь продували аргоном в течение 20 мин, после чего добавляли Pd(amphos) Cl2 (0,028 г, 0,040 ммоль). Реакционную смесь нагревали затем в микроволновой печи при 90°С в течение 20 мин. После завершения реакции (под контролем ТСХ) реакционную смесь без обработки использовали в Стадии 3.
Стадия 3:
[0676] Обе реакционные смеси со Стадии 1 и Стадии 2 объединяли и добавляли 2М водный Na2CO3 (4 мл) при комнатной температуре. Полученную реакционную смесь нагревали в микроволновой печи при 120°С в течение 30 мин. После завершения реакции (под контролем ТСХ) смесь разбавляли водой (50 мл) и экстрагировали ЕА (3×30 мл). Объединенный органический слой сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле (0-10% метанол в DCM) с получением 0,025 г указанного в заголовке соединения. ЖХ-МС (Метод В) (ИЭР+): m/z 521,10 (М+Н)+; 1H-ЯМР (400 МГц ДМСО-d6) δ 9,48 (с, 1Н), 8,50 (с, 1Н), 7,91 (с, 1Н), 7,67 (д, J=8,31 Гц, 2Н), 7,41 (д, J=8,31 Гц, 2Н), 5,76 (с, 2Н), 3,82 (с, 3Н), 3,74 (с, 3Н), 2,50 (шир. с, 3Н, протоны ОСН3 спились в пике остаточного растворителя), 1,59-1,67 (m, 1Н), 1,00-1,05 (м, 2Н), 0,81 (дд, J=3,18, 7,58 Гц, 2Н).
Пример 134: Синтез 6-(4-Циклопропи-6-метоксилиримидин-5-ил)-1-(4-(4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин (134):
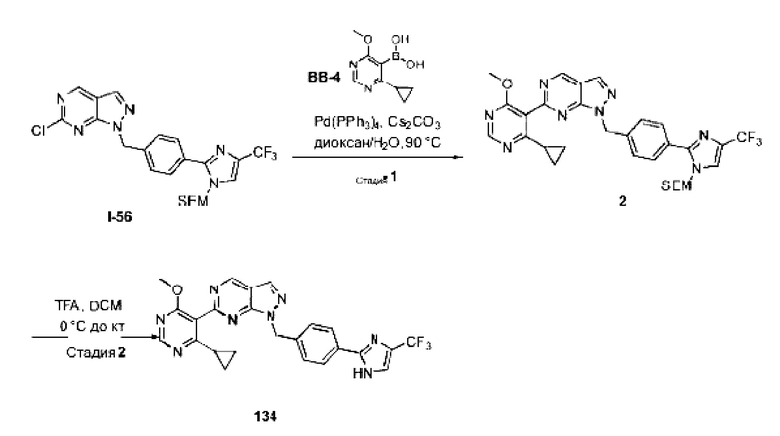
Стадия 1: Синтез 6-(4-Циклопропил-6-метоксипиримидин-5-ил)-1-(4-(4-(трифторметил)-1-((2-(триметилсилин)этокси)метил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин:
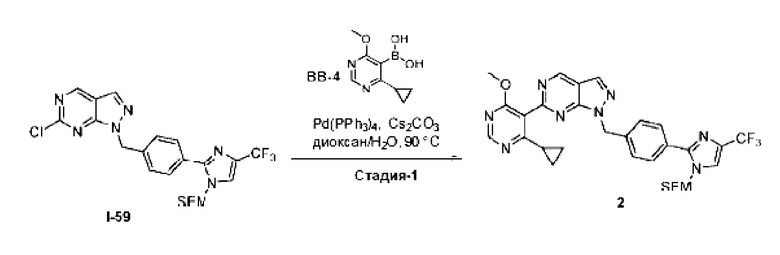
[0677] К перемешиваемому раствору 6-хлор-1-(4-(4-(трифторметил)-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина I-59 (0,50 г, 0,98 ммоль) и (4-циклопропил-6-метоксипиримидин-5-ил)бороновой кислоты (ВВ-4) (0,25 г, 1,27 ммоль) в диоксане : Н2О (5:1, 12 мл), добавляли CsCO3 (0,801 г, 2,46 ммоль). Реакционную смесь дегазировали аргоном в течение 10 мин с последующим добавлением Pd(PPh3)4 (0,113 г, 0,09 ммоль). Реакционную смесь дегазировали аргоном в течение 5 мин и нагревали до 90°С в течение 12 ч. За ходом реакции следили с помощью ТСХ и ЖХМС. После завершения реакционную смесь разбавляли водой (100 мл) и экстрагировали ЕА (2×200 мл). Объединенный органический слой промывали солевым раствором (200 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-30% ЕА в гексане, с получением 0,41 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 623,25 (М+Н)+.
Стадия 2: Синтез 6-(4-Циклопропил-6-метоксипиримидин-5-ил)-1-(4-(4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин (134):
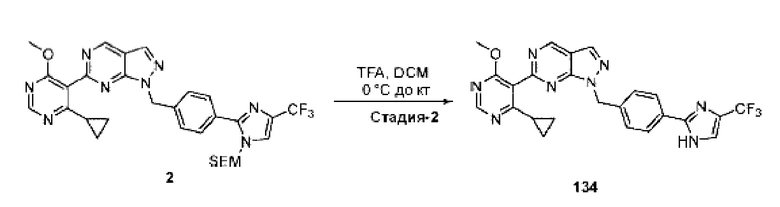
[0678] К перемешиваемому раствору 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4(4-(трифторметил)-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (0,07 г, 0,11 ммоль) в DCM (5 мл) при 0°С, медленно добавляли ТФУ (2 мл) и реакционную смесь перемешивают при комнатной температуре 4 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. К остатку добавляли воду, доводили рН до 7, используя раствор NaHCO3, и экстрагировали ЕА. Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-40% ЕА в гексане, с получением 0,025 г указанного в заголовке соединения. ЖХ-МС (Способ С) (ИЭР+): m/z 493,05 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 13,17 (с, 1Н), 9,50 (с, 1Н), 3,71 (с, 1Н), 2,51 (с, 1Н), 7,88-7,94 (м, 3Н), 7,40 (д, J=7,8 Гц, 2Н), 5,74 (с, 2Н), 3,86 (с, 3Н), 1,66 (с, 1Н), 1,06 (с, 2Н), 0,86 (д, J=3,9 Гц, 2Н).
Пример 135: Синтез 6-(4-(азетидин-3-нл)-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (135):
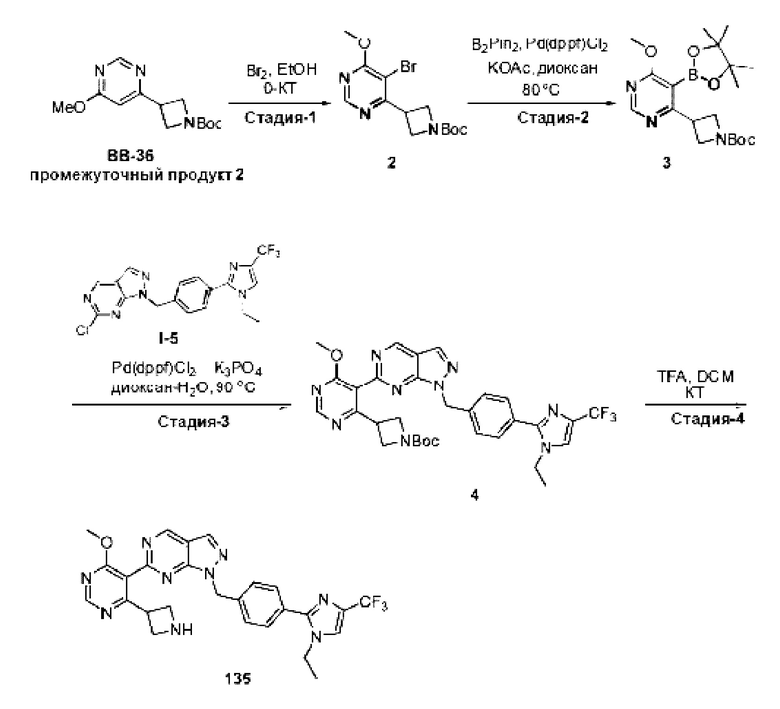
Стадия 1: Синтез трет-бутил 3-(5-бром-6-метоксипиримидин-4-ил)азетидин-1-карбоксилата:

[0679] К перемешиваемому раствору трет-бутил 3-(6-метоксипиримидин-4ил)азетидин-1-карбоксилата (10,0 г, 37,7 ммоль) в этаноле (200 мл) добавляли бром (9,0 г, 57 ммоль) при 0°С. Затем полученной смеси давали нагреться до комнатной температуры и перемешивали в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Полученный остаток растворяли в водном растворе аммиака (30 мл) и экстрагировали ЕА (200 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-50% ЕА в н-гексане, с получением указанного в заголовке соединения (10,0 г). ЖХ-МС (Способ С) (ИЭР+): m/z 289,90 (М+Н)+; 1Н-ЯМР (400 МГц. ДМСО-d6) δ 8,77 (с, 1Н), 4,15-4,21 (м, 3Н), 4,05-4,10 (м, 2Н), 4,01 (с, 3Н), 1,38 (с, 9Н).
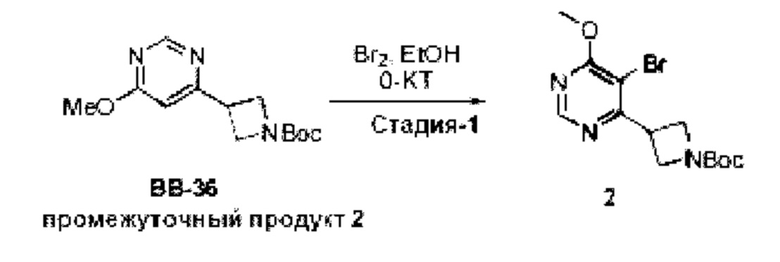
Стадия 2: Синтез трет-бутил 3-(6-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиринидин-4-ил)азетидин-1-карбоксилата:
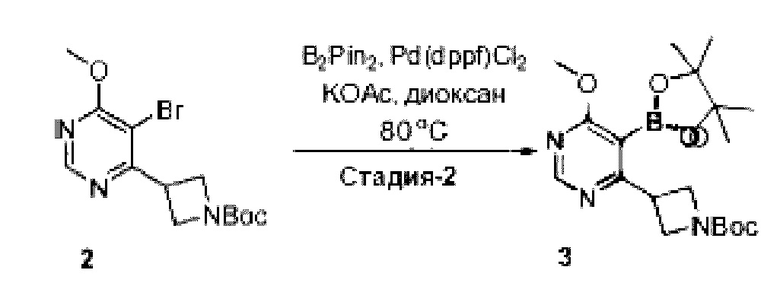
[0680] К перемешиваемому раствору трет-бутил 3-(5-бром-6-метоксипиримидин-4-ил)азетидин-1-карбоксилата 2 (5,0 г, 14,5 ммоль) и B2Pin2 (11,1 г, 43,6 ммоль) в диоксане (50 мл) и KOAc (4,26 г, 43,6 ммоль) и реакционную смесь дегазировали газообразным аргоном в течение 15 мин. К полученной реакционной смеси добавляли Pd(dppf)Cl2-DCM (2,36 г, 2,90 ммоль), а затем смесь нагревали в запаянной пробирке при 80°С в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, разбавляли ЕА (100 мл) и фильтровали через слой целита. Фильтрат упаривали при пониженном давлении и неочищенный остаток очищали хроматографией на силикагеле, используя 10-30% ЕА в н-гексане, с получением указанного в заголовке соединения (3,9 г). ЖХ-МС (Способ С) (ИЭР+): m/z 392,10 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 7,93 (с, 1Н), 4,05-4,15 (м, 3Н), 3,90-3,94 (м, 2Н), 3,89 (с, 3Н), 1,38 (с, 9Н), 1,16 (с, 12Н).
Стадия 3: Синтез трет-бутил 3-(5-(1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пириминдин-6-ил)-6-метоксипиримидин-4-ил)азетидин-1-карбоксилата:
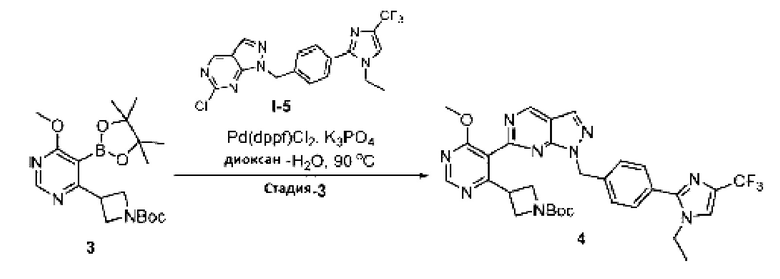
[0681] К перемешиваемому раствору трет-бутил 3-(6-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-4-ил)азетидин-1-карбоксилата 3 (1,0 г, 2,6 ммоль) в диоксане (20 мл) и воде (10 мл) добавляли I-5 (1,03 г, 2,6 ммоль) и фосфат калия (0,811 г, 3,83 ммоль)) при комнатной температуре. Полученную реакционную смесь дегазировали газообразным аргоном в течение 30 мин, а затем обрабатывали X-Phos-Pd-G2 (0,200 г, 0,255 ммоль) и X-Phos (0,243 г, 0,511 ммоль) при комнатной температуре. Затем реакционную смесь нагревали в запаянной пробирке при 90°С в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, разбавляли водой (20 мл) и экстрагировали ЕА (100 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-100% ЕА в н-гексане, с получением указанного в заголовке соединения (0,714 г). ЖХ-МС (условие 03) (ИЭР+): m/z 636,12 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,49 (с, 1Н), 8,97 (с, 1Н), 8,54 (с, 1Н), 8,01 (с, 1Н), 7,61 (д, J=7,83 Гц, 2Н), 7,41 (д, J=7,83 Гц, 2Н), 5,77 (с, 2Н), 4,04 (к, J=7,34 Гц, 3Н), 3,90 (с, 3Н), 3,74-3,87 (м, 3Н), 3,61-3,71 (м, 2Н), 1,31 (с, 9Н), 1,24-1,29 (м, 2Н).
Стадия 5: Синтез 6-(4-(азетидин-3-ил)-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (135):
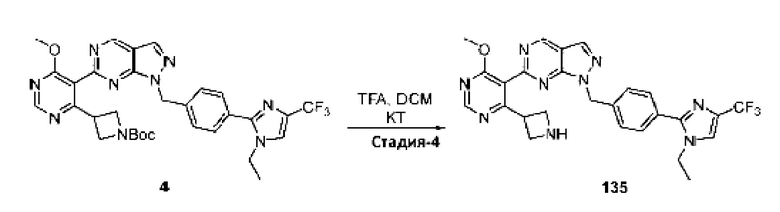
[0682] К перемешиваемому раствору трет-бутил 3-(5-(1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил)-6-метоксипиримидин-4-ил)азетидин-1-карбоксилата 4 (0,250 г, 0,393 ммоль) в DCM (20 мл) добавляли ТФУ (0,224 г, 1,97 ммоль) при 0°С. Полученный раствор дополнительно перемешивали при комнатной температуре в течение 4 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Оставшийся остаток растворяли в DCM (10 мл) и доводили рН до 9, используя водный раствор аммиака (10 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-10% метанола в DCM, с получением указанного в заголовке соединения (0,050 г). ЖХ-МС (Способ В) (ИЭР+): m/z 536,15 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,41 (уш. с, 1Н), 8,94 (уш. с, 1Н), 8,49 (уш. с, 1Н), 7,88 (уш с, 1Н), 7,54 (д, J=7,82 Гц, 2Н), 7,38 (д, J=7,34 Гц, 2Н), 5,71 (уш. с, 2Н), 4,16-4,24 (м, 2Н), 3,92-4,02 (м, 6Н), 3,86 (уш. с, 3Н), 1,23 (д, J=6,85 Гц, 3Н).
Пример 136: Синтез 6-(4-Циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-(трифторметил)-5,6,7,8-тетрагидронмидазо[1,5-а]пиразин-3-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (136):
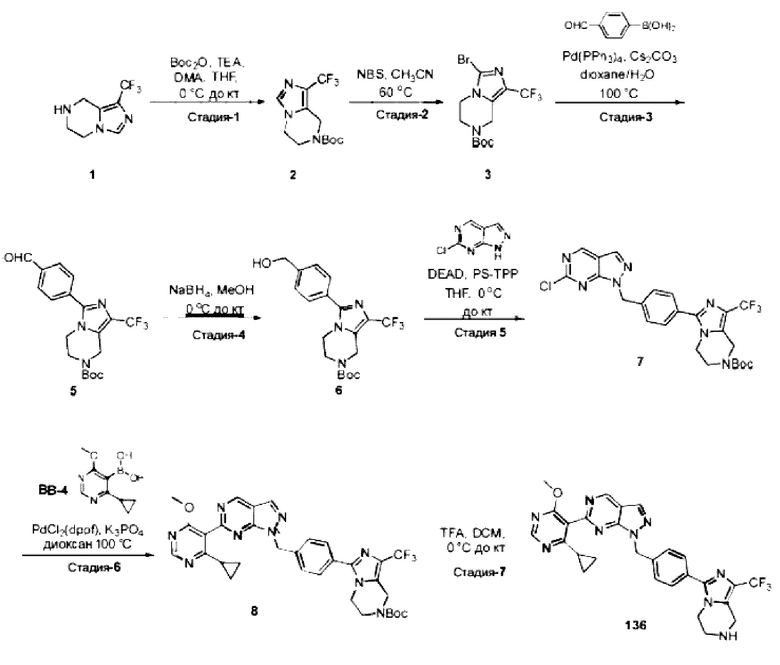
Стадия 1: Синтез трет-Бутил 1-(трифторметил)-5,6-дигидроимидазо[1,5-а]пиразин-7(8Н)-карбоксилата:
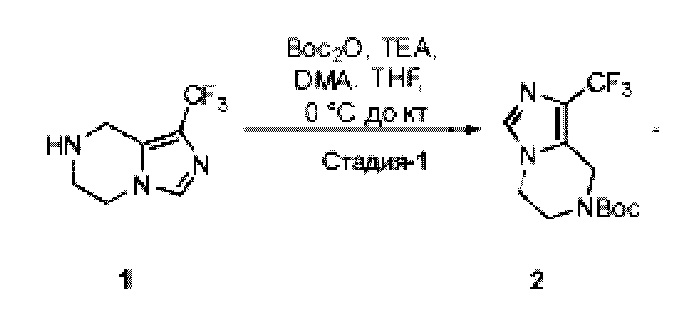
[0683] К перемешиваемому раствору 1-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразина (2,00 г, 10,40 ммоль) в ТГФ (20 мл) при 0°С добавляли триэталамин. (2,10 г, 20,80 ммоль), затем DMAP (0,12 г, 1,00 ммоль), и реакционную смесь перемешивали в течение 10 минут. К образовавшейся реакционной смеси добавляли Вос2О (3,42 г, 15,70 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой и экстрагировали ЕА (3×20 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-30% ЕА в гексане, с последующей промывкой н-пентаном с получением 3,00 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 292,05 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 7,82 (с, 1Н), 4,66 (с, 2Н), 4,09 (т, J=5,4 Гц, 2Н), 3,73 (т, J=5,4 Гц, 2Н), 1,43 (с, 9Н).
Стадия 2: Синтез трет-Бутил 3-бром-1-(трифторметил)-5,6-дигидроимидазо[1,5-а]пиразин-7(8Н)-карбоксилата:
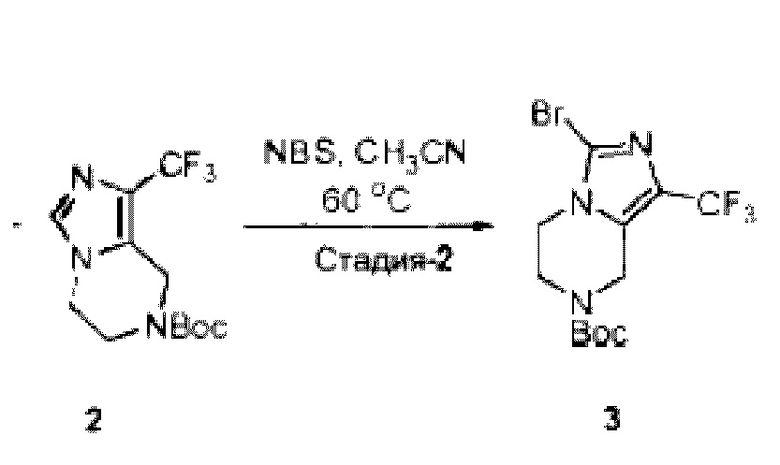
[0684] К перемешиваемому раствору трет-бутил 1-(трифторметил)-5,6-дигидроимидазо[1,5-а]пиразин-7(8Н)-карбоксилата (3,00 г, 10,00 ммоль) в MeCN (30 мл) добавляли NBS (2,10 г, 12,00 ммоль) при комнатной температуре, и реакционную смесь нагревали до 60°С в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой и экстрагировали ЕА (2×30 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением 3,00 г сырого указанного в заголовке соединения, которое использовали без очистки в последующих реакциям. 1Н-ЯМР (400 МГц, ДМСО-d6) δ 4,65 (с, 2Н), 3,91-3,96 (м, 2Н), 3,77 (т, J=5,4 Гц, 2Н), 2,56 (с, 1Н), 1,43 (с, 9Н).
Стадия 3: Синтез трет-Бутил 3-(4-формилфенил)-1-(трифторметил)-5,6-дигидроимидазо[1,5-а]пиразин-7(8Н)-карбоксилата:

[0685] Смесь трет-бутил 3-бром-1-(трифторметил)-5,6-дигидроимидазо[1,5-а]пиразин-7(8Н)-карбоксилата (3,00 г, 8,10 ммоль), (4-формилфенил)бороновой кислоты кислота (1,45 г, 9,70 ммоль) и водн. 2М Cr2CO3 (5,28 г, 16,20 ммоль) в диоксане (30 мл) дегазировали аргоном в течение 15 мин. К полученной реакционной смеси добавляли Pd(PPh3)4 (0,92 г, 0,81 ммоль), и реакционную смесь нагревали до 100°С в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли водой (50 мл) и экстрагировали ЕА (2×30 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-30% ЕА в гексане, с получением 1,40 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 396,00 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 10,09 (с, 1Н), 7,95-8,06 (м, 4Н), 4,78 (с, 2Н), 4,26 (д, J=4,4 Гц, 2Н), 3,73 (с, 2Н), 1,46 (с, РН).
Стадия 4: Синтез трет-Бутил 3-(4-(гидроксиметил)фенил)-1-(трифторметил)-5,6-дигидроимидазо[1,5-а]пиразин-7(8Н)-карбоксилата:
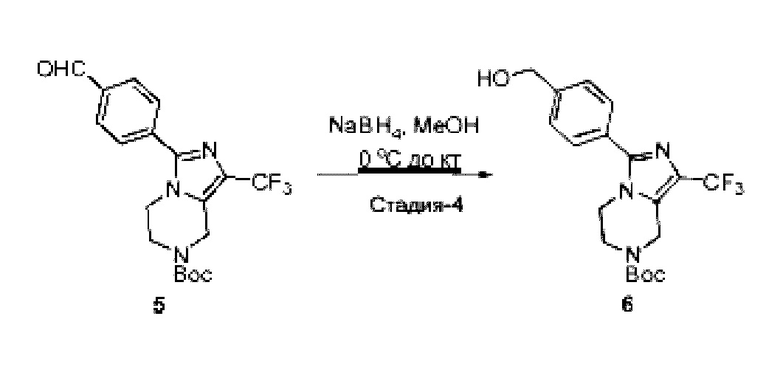
[0686] К перемешиваемому раствору трет-бутил 3-(4-формилфенил)-1-(трифторметил)-5,6-дигидроимидазо[1,5-а]пиразин-7(8Н)-карбоксилата (1,40 г, 3,50 ммоль) в МеОН (20 мл) при 0°С, добавляли NaBH4 (0,20 г, 5,30 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Полученный остаток гасили водой (20 мл) и экстрагировали DCM (2×20 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением 1,20 г указанного в заголовке соединения, которое использовали в последующих реакциях без дополнительной очистки. ЖХ-МС (Способ В) (ИЭР+): m/z 397,95 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 7,68 (д, J=8,0 Гц, 2Н), 7,44 (д, J=8,0 Гц, 2Н), 5,31 (т, J=5,5 Гц, 1Н), 4,75 (с, 2Н), 4,57 (д, J=5,5 Гц, 2Н), 4,17 (т, J=5,0 Гц, 2Н), 3,71 (д, J=4,5 Гц, 2Н), 1,45 (с, 9Н).
Стадия 5: Синтез трет-Бутил 3-(4-((6-хлор-1Н-пиразоло[3,4-d]пиримидин-1-ил)(метил)фенил)-1-(трифторметил)-5,6-дигидроимидазо[1,5-a]пиразин-7(8Н)-карбоксилата:

[0687] К перемешиваемому раствору трет-бутил 3-(4-(гидроксиметил)фенил)-1-(трифторметил)-5,6-дигидроимидазо[1,5-а]пиразин-7(8Н)-карбоксилата (1,20 г, 3,02 ммоль) и 6-хлор-1Н-пиразоло[3,4-d]пиримидина (0,41 г, 2,72 ммоль) в ТГФ (20 мл) при 0°С, добавляли связанный полимером трифенилфосфин (2,37 г, 9,06 ммоль), и реакционную смесь перемешивали в течение 25 минут. К полученной реакционной смеси добавляли диэтилазодикарбоксилат (DEAD) (1,05 г, 6,04 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь фильтровали через целит, и фильтрат упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-40% ЕА в гексане, с получением 0,50 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 534,10 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,32 (с, 1Н), 8,52 (с, 1Н), 7,69 (д, J=8,3 Гц, 2Н), 7,38 (д, J=7,8 Гц, 2Н), 5,72 (с, 2Н), 4,74 (с, 2Н), 4,15 (с, 2Н), 3,68 (с, 2Н), 1,44 (с, 9Н).
Стадия 6: Синтез трет-Бутил 3-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-(трифторметил)-5,6-дигидроимидазо[1,5-а]пиразин-7(8Н)-карбоксилата:
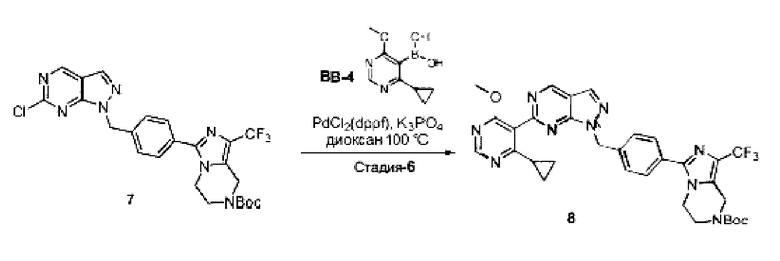
[0688] Смесь трет-бутил 3-(4-((6-хлор-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-(трифторметил)-5,6-дигидроимидазо[1,5-а]пиразин-7(8Н)-карбоксилата (0,30 г, 0,50 ммоль), (4-циклопропил-6-метоксипиримидин-5-ил) бороновой кислоты (0,11 г, 0,60 ммоль) и 2М водн. раствор 0.318 г (1.50 ммоль) K3PO4 в 5 мл диоксана дегазировали аргоном в течение 5 мин. К полученной реакционной смеси добавляли PdCl2(dppf) (0,04 г, 0,05 ммоль), и реакционную смесь нагревали до 100°С в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли водой и экстрагировали ЕА (2×30 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-60% ЕА в гексане, с получением 0,20 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 648,3 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,50 (с, 1Н), 8,71 (с, 1Н), 8,51 (с, 1Н), 7,67 (д, J=7,8 Гц, 2Н),7,42 (д, J=7,8 Гц, 2Н), 5,75-5,79 (м, 2Н), 4,74 (с, 2Н), 4,14 (с, 2Н), 3,86 (с, 3Н), 3,68 (с, 2Н), 1,44 (с, 9Н), 1,65 (с, 1Н), 1,06 (с, 2Н), 0,87 (д, J=4,4 Гц, 2Н).
Стадия 7: Синтез 6-(4-Циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-3-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (136):
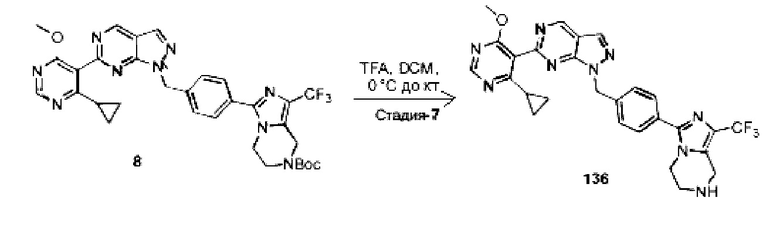
[0689] К перемешиваемому раствору трет-бутил 3-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-(трифторметил)-5,6-дигидроимидазо[1,5-а]пиразин-7(8Н)-карбоксилата (0,20 г, 0,40 ммоль) в DCM (5 мл) при 0°С, медленно добавляли ТФУ (0,097 г, 0,80 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Полученный таким образом остаток растворяли в воде, подщелачивали 2 н. раствором NaHCO3 и экстрагировали ЕА (2×10 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали препаративной ВЭЖХ с получением 0,052 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 548,10 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,50 (с, 1Н), 8,71 (с, 1Н), 8,51 (с, 1Н), 7,66 (д, J=8,0 Гц, 2Н), 7,40 (д, J=8,0 Гц, 2Н), 5,76 (с, 2Н), 4,03 (с, 2Н), 3,98 (с, 2Н), 3,85 (с, 3Н), 2,94-2,99 (м, 2Н), 1,61-1,69 (м, 1Н), 1,06 (с, 2Н), 0,85 (дд, J=3,0, 7,5 Гц, 2Н).
Общие процедуры синтеза N-алкилимидазолов путем функционализации Примера 134:
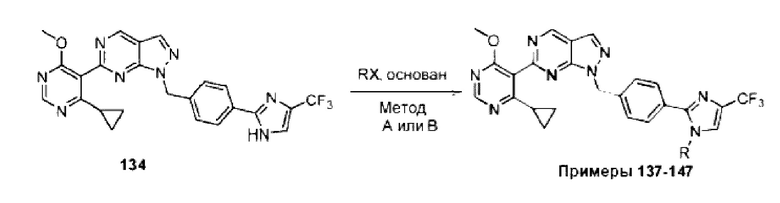
Пример 134 можно алкилировать по способу А или способу В, описанному ниже, с получением N-функционализированных имидазолов.
Общая процедура:
[0690] Метод-А: К охлажденному льдом раствору 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (134) (1 экв.) в ДМФА (5 мл) добавляли порциями NaH (60% дисперсия в минеральном масле) (1,2 экв.) и реакционную смесь перемешивали при той же температуре в течение 10 мин. К полученной реакционной смеси добавляли алкилгалогениды (1,20 экв.) и перемешивание продолжали при комнатной температуре в течение 16 часов. За ходом реакции следили с помощью ТСХ и ЖХМС. После завершения реакционную смесь разбавляли водой (50 мл) и экстрагировали ЕА (2×200 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенные соединения очищали препаративной ВЭЖХ с получением указанных в заголовке соединений.
Метод-В:
[0691] К перемешиваемому раствору 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (134) (1 экв.) в ДМФА (5 мл) последовательно добавляли Cs2CO3 (1 экв.) и алкилгалогенид 1 (1 экв.). Реакционную смесь нагревали до 90°С в течение 12 часов. За ходом реакции следили с помощью ТСХ и ЖХМС. После завершения реакционную смесь разбавляли водой (50 мл) и экстрагировали ЕА (2×100 мл). Объединенный органический слой промывали холодной водой (3×50 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Полученные таким образом неочищенные соединения очищали препаративной ВЭЖХ с получением указанных в заголовке соединений.
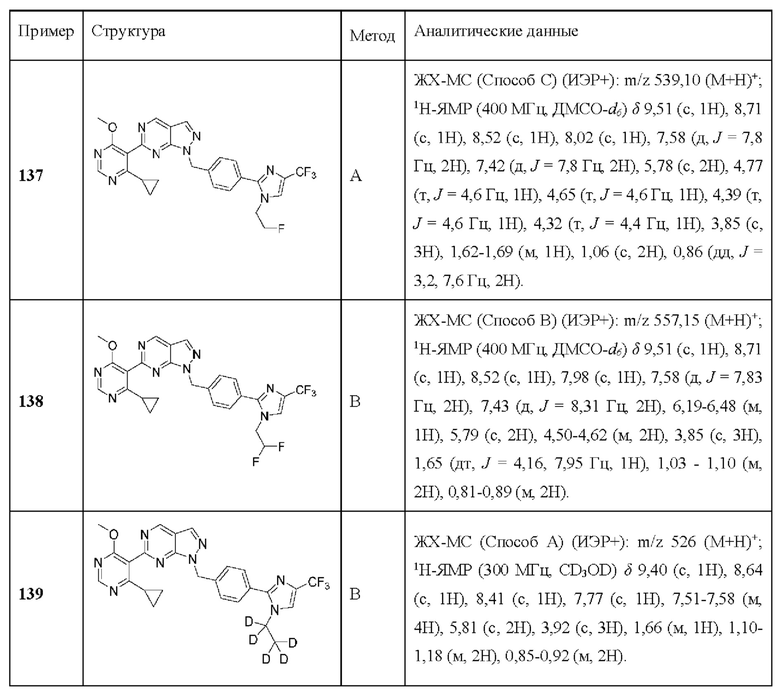
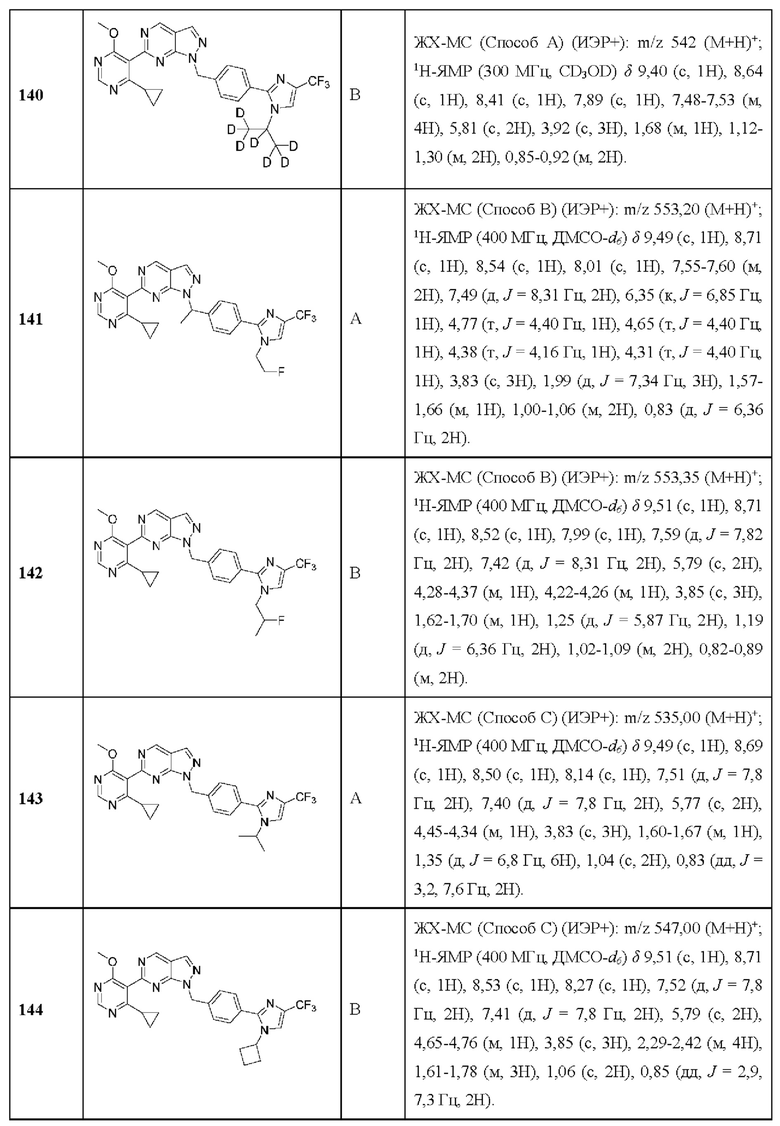
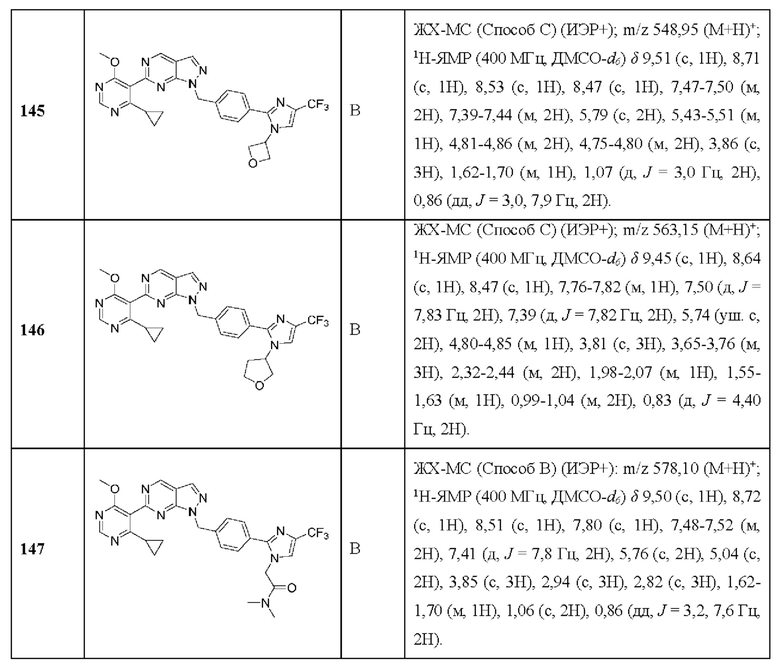
Пример 148: Синтез 2-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-этил-1Н-имидазол-4-карбонитрила (148):
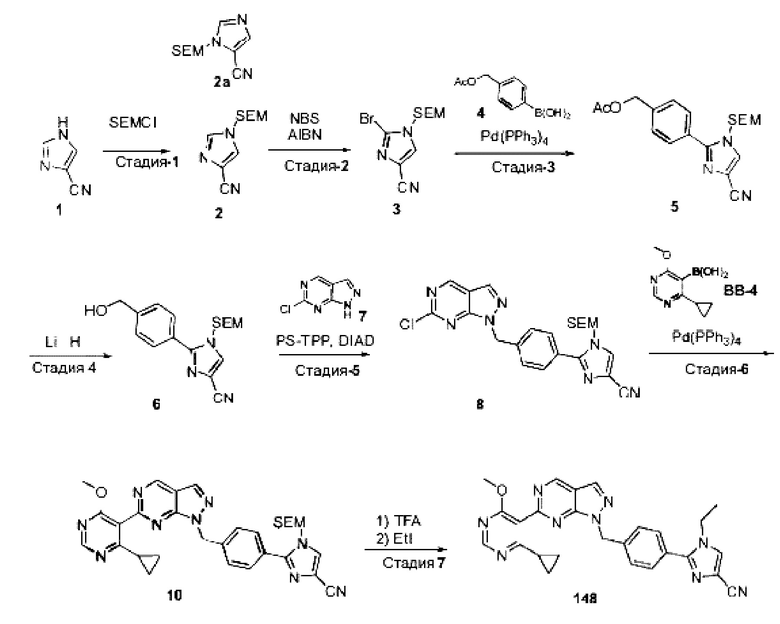
Стадия 1: Синтез 1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-4-карбонитрила и 1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-5-карбонитрила:
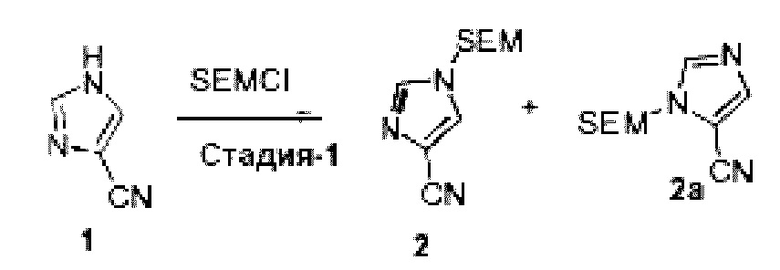
[0692] К суспензии 1Н-имидазол-4-карбонитрила (1 г, 10,76 ммоль) и карбоната калия (2,96 г, 21,5 ммоль) в ацетоне (20 мл) при 10°С по каплям добавляли SEMC1 (1,98 г, 11,8 ммоль) 30 мин, при этом внутренняя температура поддерживалась ниже 15°С. Затем реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. После завершения реакции реакцию гасили колодной водой (50 мл) и экстрагировали ЕА (50 мл ×3). Объединенный органический слой сушили над сульфатом натрия (30 г), фильтровали и упаривали. Концентрированный остаток очищали хроматографией на силикагеле (элюент: EA/n-Hex = 1/4) с получением 1,3 г смеси указанного в заголовке соединения и его региоизомера 2а. ЖХ-МС (Способ А) (ИЭР+): m/z 224 (М+Н)+.
Стадия 2: Синтез 2-бром-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-4-карбонитрила:
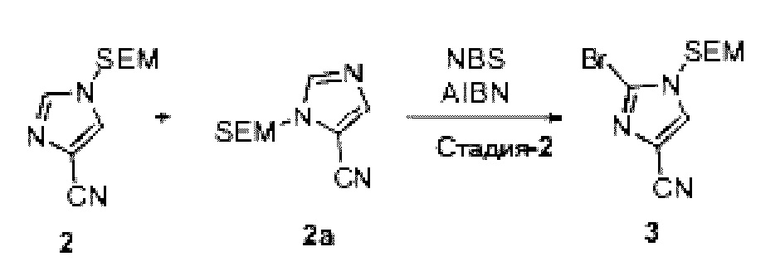
[0693] Раствор 1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-4-карбонитрила 2 и 1-((2(триметилсилил)этокси)метил)-1Н-имидазол-5-карбонитрила 2а (1,30 г, 3,83 ммоль) в CCl4 (30 мл) при 60°С добавляли порциями NBS (1,14 г, 6,41 ммоль) в течение 5 минут. Наблюдалась экзотермическая реакция, и внутренняя температура повысилась до 75°С. После добавления реакционную смесь перемешивали при 60°С в течение ночи. Реакционной смеси давали остыть до комнатной температуры и полученную суспензию фильтровали. Осадок на фильтре промывали CCl4 (20 мл). Объединенный фильтрат промывали насыщенным раствором NaHCO3 (60 мл). После разделения органический слой сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (элюент: ЕА/n-Нех = 1/10) с получением 800 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 324 (M+Na)+; 1Н-ЯМР (300 МГц CDCl3) δ 7,63 (с, 1Н), 5,30 (с, 2Н), 3,55 (т, J=8,1 Гц, 2Н), 0,93 (т, J=8,1 Гц, 2Н), 0,05 (с, 9Н).
Стадия 3: Синтез 4-(4-циано-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)бензилацетата:
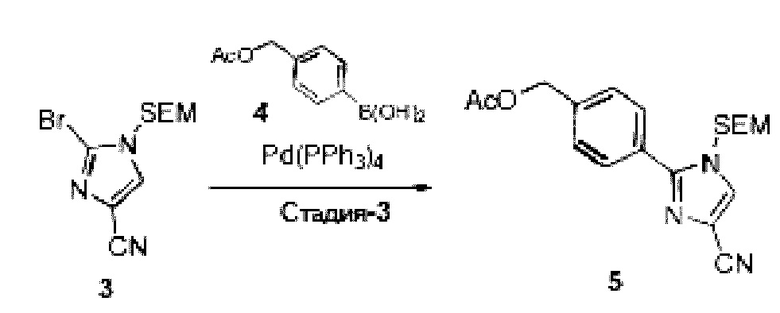
[0694] К раствору 2-бром-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-4-карбонитрила 3 (800 мг, 2,48 ммоль) в 1,4-диоксане/H2O (20 мл, 20:1) добавляли 4-(ацетоксиметил)фенил)бороновую кислоту (568 мг, 2,93 ммоль), K3PO4⋅3H2O (2,12 г, 7,98 ммоль) и Pd(PPh3)4 (307 мг, 0,27 ммоль) одной порцией. Затем реакционную смесь перемешивали при 110°С в течение ночи. Реакционную смесь охлаждали до комнатной температуры и гасили водой (50 мл). Полученную смесь экстрагировали ЕА (50 мл ×3). Органический слой сушили над сульфатом натрия (100 г), фильтровали и упаривали Остаток очищали хроматографией на силикагеле (элюент: EA/n-Hex = 1/3) с получением 650 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 372 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 7,76 (д, J=8,1 Гц, 2Н), 7,65 (с, 1Н), 7,46 (д, J=8,1 Гц 2Н), 5,28 (с, 2Н), 5,16 (с, 2Н), 3,58 (т, J=8,4 Гц 2Н), 2,12 (с, 3Н), 0,94 (т, J=8,4 Гц, 2Н), 0,05 (с, 9Н).
Стадия 4: Синтез 2-(4-(гидроксиметил)фенил)-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-4-карбокитрила:
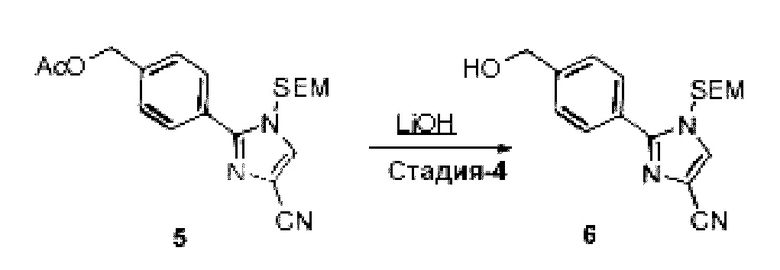
[0695] К раствору 4-(4-циано-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)бензилацетата 5 (650 мг, 1,75 ммоль) в ТГФ/воде, 1:1 (10 мл) добавляли LiOH.Н2О (150 мг, 3,50 ммоль) одной порцией. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После завершения реакции, как показывает анализ ТСХ, рН реакции доводили до 8 разбавленным раствором HCl и экстрагировали ЕА (50 мл ×2). Объединенный органический слой сушили сульфатом натрия (30 г), фильтровали и упаривали с получением 690 мг неочищенного указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 330 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 7,75 (д, J=7,8 Гц, 2Н), 7,65 (с, 1Н), 7,49 (д, J=7,8 Гц, 2Н), 5,28 (с, 2Н), 4,78 (д J=5,1 Гц, 2Н), 3,57 (т, J=8,1 Гц, 2Н), 1,80 (т, J=5,1 Гц, 1Н), 0,94 (т, J=8,1 Гц, 2Н), 0,05 (с, 9Н).
Стадия 5: Синтез 2-(4-((6-хлор-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-4-карбонитрила:
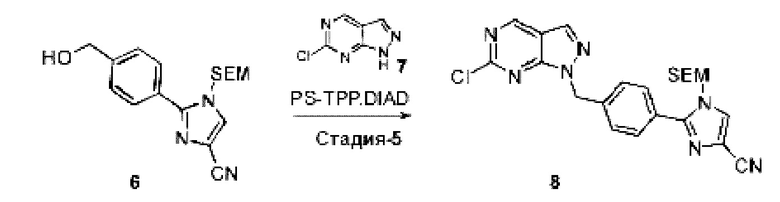
[0696] Соединение синтезировали в соответствии с процедурой получения общего промежуточного соединения I-47. ЖХ-МС (Способ А) (ИЭР+): m/z 466 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 9,06 (с, 1Н), 8,19 (с, 1Н), 7,76 (д, J=8,4 Гц, 2Н), 7,64 (с, 1Н), 7,47 (д, J=8,4 Гц, 2Н), 5,70 (с, 2Н), 5,26 (с, 2Н), 3,57 (т, J=8,1 Гц 2Н), 0,93 (т, J=8,1 Гц, 2Н), 0,05 (с, 9Н).
Стадия 6: Синтез 2-(4-((6-(4-циклопропил-6-метоксипиримидин-5-нл)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-4-карбонитрила:
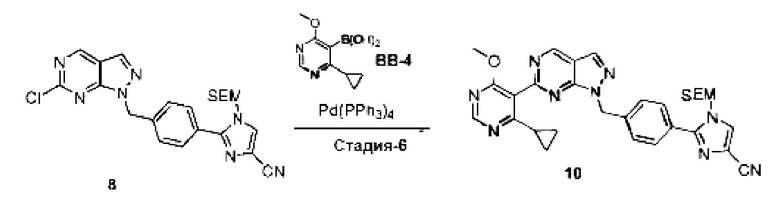
[0697] Соединение синтезировали согласно общей экспериментальной процедуре 1. ЖХ-МС (Способ А) (ИЭР+): m/z 580 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 9,35 (с, 1Н), 8,69 (с, 1Н), 8,24 (с, 1Н), 7,66-7,78 (м, 2H), 7,64 (с, 1Н), 7,42-7,58 (м, 2Н), 5,77 (с, 2Н), 5,25 (с, 2Н), 3,95 (с, 3Н), 3,58 (т, J=8,4 Гц, 2Н), 1,64-1,75 (м, 1Н), 1,20-1,31 (м, 2Н), 0,83-0,99 (м, 4Н), 0,05 (с, 9Н).
Стадия 7: Синтез 2-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-этил-1Н-имидазол-4-карбонитрила (148):
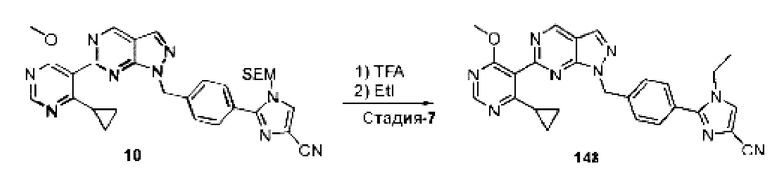
[0698] Раствор 2-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-4-карбонитрила 10 (400 мг, 0,69 ммоль) в ТФУ (10 мл) перемешивали при комнатной температуре в течение 3 часов. После завершения реакции на основании анализа ТСХ реакционную смесь упаривали досуха в вакууме. Остаток растворяли в ЕА (50 мл) и нейтрализовали насыщенным раствором бикарбоната натрия. После разделения водную фазу экстрагировали ЕА (50 мл ×2). Объединенный органический слой сушили, фильтровали и упаривали, получая 400 мг неочищенного промежуточного продукта со снятой защитой.
[0699] К раствору промежуточного соединения со снятой защитой (180 мг, 0,40 ммоль) в ДМФА (10 мл) добавляли карбонат цезия (156 мг, 0,48 ммоль) и EtI (75 мг, 0,48 ммоль). Реакционную смесь перемешивали при 40°С в течение 5 часов. После завершения реакции, как показывает анализ ТСХ, реакцию гасили водой (30 мл) и экстрагировали ЕА (50 мл ×3). Органический слой промывали водой (20 мл), сушили сульфатом натрия (50 г), фильтровали и упаривали. Остаток очищали препаративной ВЭЖХ (метод А), получая 53 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 478 (М+Н)+; 1Н-ЯМР (300 МГц, CD3OD) δ 9,40 (с, 1Н), 8,64 (с, 1Н), 8,42 (с, 1Н), 8,06 (с, 1Н), 7,51-7,62 (м, 4Н), 5,81 (с, 2Н), 4,09 (к, J=7,2 Гц, 2Н), 3,91 (с, 3Н), 1,60-1,75 (м, 1Н), 1,34 (т, J=7,2 Гц, 3Н), 1,10-1,20 (м. 2Н), 0,83-0,94 (м, 2Н).
Пример 149: Синтез 6-(2-изопропилфенил)-1-(4-(1-(1-метилазетидин-3-ил)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пириминдин (149):
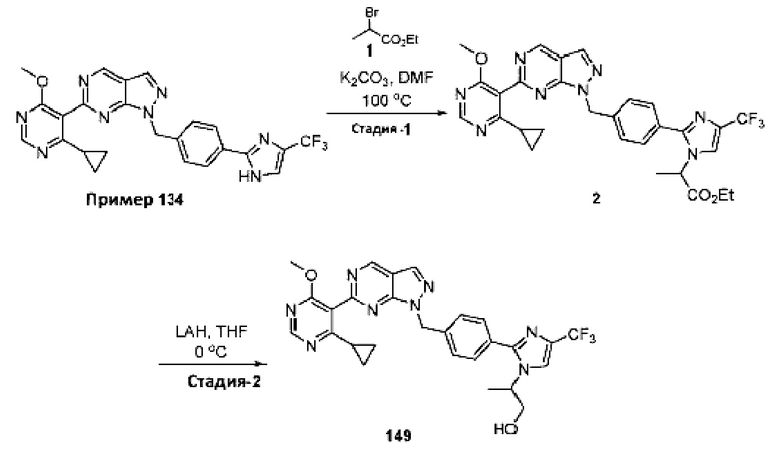
Стадия 1: Синтез этил 2-(2-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-4-(трифторметил)-1Н-имидазол-1-ил)пропаноата:
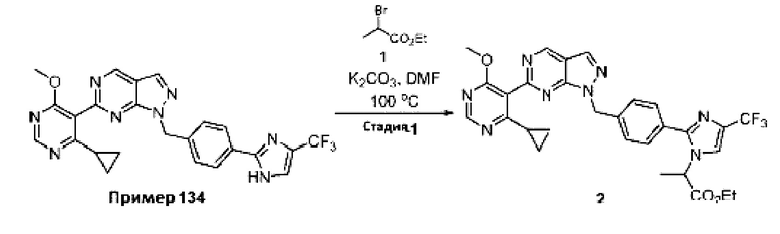
[0700] К перемешиваемому раствору 6-(4-циклопропил-6-метоксипиримидин-5-ил)4-(4-(4-(трифторметил)-1H-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидина (134) (0,320 г, 0,650 ммоль) в ДМФА(10 мл), добавляли K3CO3 (0,224 г, 1,63 ммоль) и этил 2-бромпропаноат 1 (0,141 г, 0,780 ммоль) при комнатной температуре. Реакционную смесь нагревали при 100°С в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, гасили водой (10 мл) и экстрагировали ЕА (3×5 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-10% МеОН в DCM, с получением указанного в заголовке соединения (0,240 г). ЖХ-МС (Способ С) (ИЭР+): m/z 593,1 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,51 (с, 1Н), 8,71 (с, 1Н), 8,52 (с, 1Н), 8,15 (уш. с, 1Н), 7,95 (с, 1Н), 7,47-7,51 2Н), 7,41-7,45 (м, 2Н), 5,78 (с, 2Н), 5,06-5,13 (м, 2Н), 4,01-4,08 (м, 2Н), 3,85 (с, 3Н), 1,68 (д, J=6,98 Гц, 3Н), 1,02-1,08 (м, 4Н), 0,82-0,88 (м, 2Н).
Стадия 2: Синтез 2-(2-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-4-(трифторметил)-1Н-имидазол-1-ил)пропан-1-ола (149):
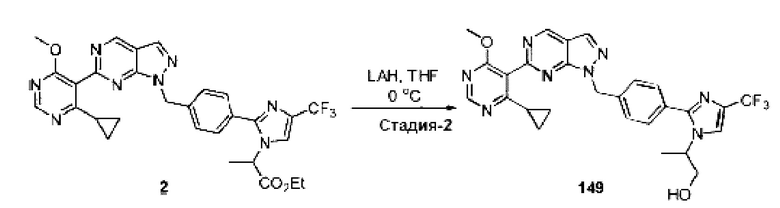
[0701] К охлажденному льдом раствору этил 2-(2-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-4-(трифторметил)-1Н-имидазол-1-ил)пропаноата 2 (0,175 г, 0,296 ммоль) в ТГФ (3 мл), добавляли LAH (0,022 г, 0,59 ммоль). Полученную смесь затем перемешивали в течение 5 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили ЕА (10 мл), фильтровали через слой целита и промывали ЕА (5 мл). Фильтрат упаривали при пониженном давлении, получая неочищенное соединение, которое очищали препаративной ВЭЖХ (метод С), получая указанное в заголовке соединение (0,042 г). ЖХ-МС (Способ С) (ИЭР+): m/z. 551,14 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,44 (с, 1Н), 8,64 (с, 1Н), 8,45-8,50 (м, 1Н), 8,00 (уш. с, 1Н), 7,53 (д, J=7,98 Гц, 2Н), 7,37 (д, J=7,48 Гц, 2Н), 5,73 (уш. с, 2Н), 4,28 (д, J=4,99 Гц, 1Н), 3,81 (с, 3Н), 3,51-3,62 (м, 3Н), 1,54-1,63 (м, 1Н), 1,25 (д, J=6,48 Гц, 3Н), 0,98-1,06 (м, 2Н), 0,81-0,86 (м, 2Н).
Пример 150: Синтез 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-(1-метоксипропан-2-ил)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (150):
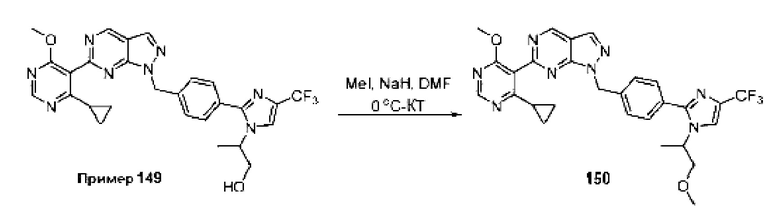
[0702] К перемешиваемому раствору 2-(2-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-4-(трифторметил)-1Н-имидазол-1-ип)пропан-1-ола (149) (0,050 г, 0,090 ммоль) в ДМФА (1,5 мл) при 0°С добавляли NaH (0,005 г, 0,14 ммоль) и смесь перемешивали 30 мин. К полученной реакционной смеси добавляли MeI (0,011 мл, 0,19 ммоль) и реакционную смесь дополнительно перемешивали при комнатной температуре в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили ледяной водой (5 мл) и экстрагировали ЕА (4×5 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали препаративной ВЭЖХ (Метод Е) с получением указанного в заголовке соединения (0,025 г). ЖХ-МС (Способ С) (ИЭР+): m/z 565 (М+Н)+; 1Н-ЯМР (400 МГц. ДМСО-d6) δ 9,51 (с, 1Н), 8,71 (с, 1Н), 8J2 (с, 1Н), 8,15 (с, 1Н), 7,55 (д, J=7,98 Гц, 2Н), 7,42 (д, J=7,98 Гц 2Н), 5,78 (с, 2Н), 4,45 (д, J=3,49 Гц, 1Н), 3,85 (с, 3Н), 3,57-3,64 (м, 1Н), 3,50 (дд, J=4,24, 10,22 Гц 1Н), 3,11 (c, 3Н), 1,66 (дт, J=3,99, 7,73 Гц, 1Н), 1,33 (д, J=6,98 Гц, 3Н), 1,03-1,08 (м, 2Н), 0,86 (дд, J=2,99,7,48 Гц, 2Н).
Пример 151: Синтез 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-(1-фторпропан-2-ил)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (151):
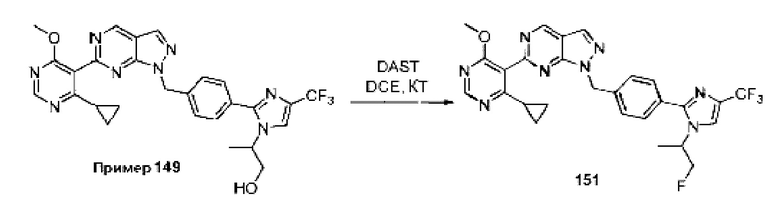
[0703] К перемешиваемому раствору 2-(2-(4-((6-(4-циклопропил-6-метоксигпиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-4-(трифторметил)-1Н-имидазол-1-ил)пропан-1-ола (149) (0,120 г, 0,2181 ммоль) в DCE (10 мл) добавляли DAST (0,140 г, 0,872 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение 4 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли водой (10 мл) и экстрагировали DCM (20 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-10% метанола в DCM, с получением указанного в заголовке соединения (0,090 г). ЖХ-МС (Способ В) (ИЭР+): m/z 553,20 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,51 (с, 1Н), 8,71 (с, 1Н), 8,52 (с, 1Н), 8,22 (с, 1Н), 7,50-7,54 (м, 2Н), 7,44 (д, J=8,31 Гц, 2Н), 5,79 (с, 2Н), 4,54-4,72 (м, 3Н), 3,85 (с, 3Н), 1,62-1,71 (м, 1Н), 1,38 (д, J=6,36 Гц, 3Н), 1,03-1,09 (м, 2Н), 0,83-0,89 (м, 2Н).
Пример 152: Синтез 6-(2-изопропилфенил)-1-(4-(1-(1-метилазетидин-3-ил)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин (152):
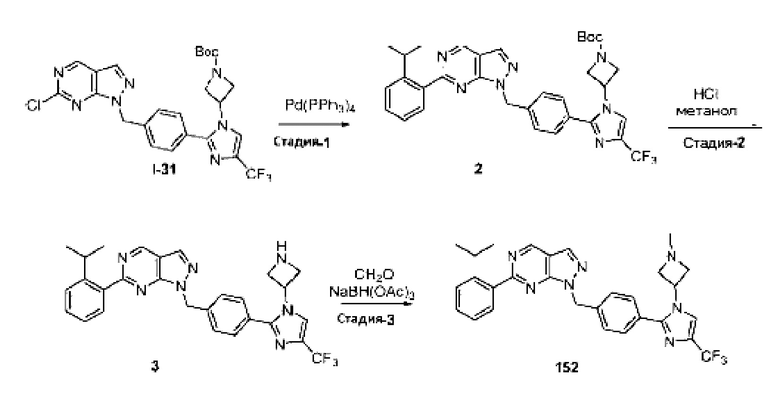
Стадия 1: Синтез трет-Бутил 3-(2-(4-((6-(2-изопропилфенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-4-(трифторметил)-1Н-имидазол-1-ил)азетидин-1-карбоксилата:
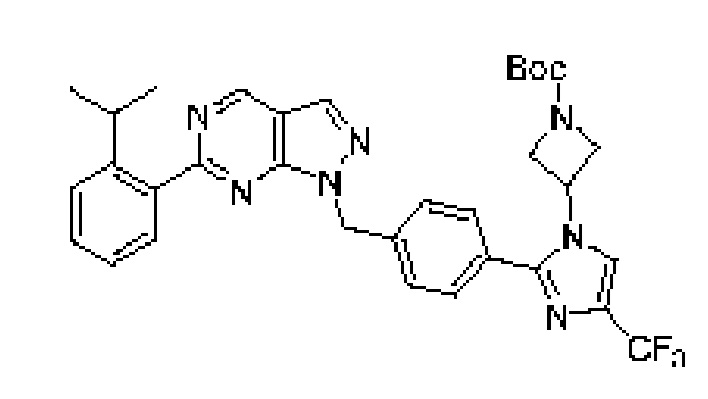
[0704] В атмосфере азота смесь трет-бутил 3-(2-(4-((6-хлор-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-4-(трифторметил)-1Н-имидазол-1-ип)азетидин-1-карбоксилата (I-31) (0,26 г, 0,49 ммоль), K3PO4 (0,39 г, 1,47 ммоль), (2-изопропипфенил)бороновой кислоты и Pd(PPh3)4 (57 мг, 10 мол. %) в диоксане (9 мл) и воде (0,9 мл) перемешивали при 90°С в течение 3 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (10 мл) и экстрагировали ЕА (100 мл ×3). Объединенный органический слой сушили над безводным Na2SO4 (20 г), фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (ЕА: n-Нех = от 1:10 до 1:4) с получением 280 мг указанного в заголовке соединения. 1Н-ЯМР (300 МГц, CDCl3) δ 9,31 (с, 1Н), 8,21 (с, 1Н), 7,73 (с, 1Н), 7,64-7,71 (м, 3Н), 7,52-7,56 (м, 2Н), 7,42-7,50 (м, 3Н), 5,76 (с, 2Н), 5,01 (м, 1Н), 4,35-4,40 (м, 2Н), 4,05-4,13 (м, 2Н), 3,56 (м, 1Н), 1,45 (с, 9Н), 1,26 (д, J=7,2 Гц, 6Н).
Стадия 2: Синтез 1-(4-(1-(Азетидин-3-ил)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-6-(2-изопропилфенил)-1Н-пиразоло[3,4-d]пиримидина:
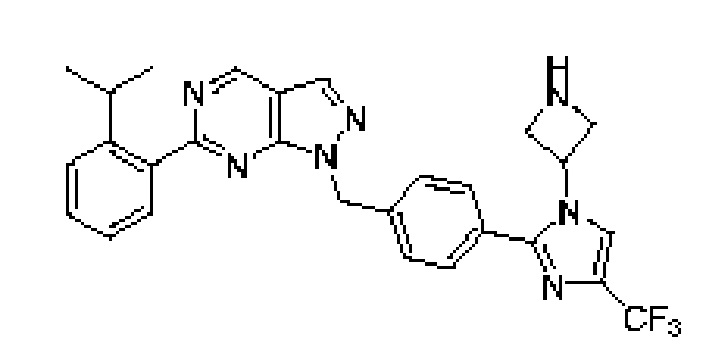
[0705] К раствору трет-бутил 3-(2-(4-((6-(2-изопропилфенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-4-(трифторметил)-1H-имидазол-1-ил)азетидин-1-карбоксилата (0,25 г, 0,41 ммоль) в DCM (15 мл), добавляли ТФУ (3 мл). Смесь перемешивали при комнатной температуре в течение 3 часов, а затем упаривали в вакууме. Неочищенный продукт напрямую использовали в следующей стадии. ЖХ-МС (ИЭР): m/z 518 (М+Н)+.
Стадия 3: Синтез 6-(2-изопропилфенил)-1-(4-(1-(1-метилазетидин-3-ил)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин (152):
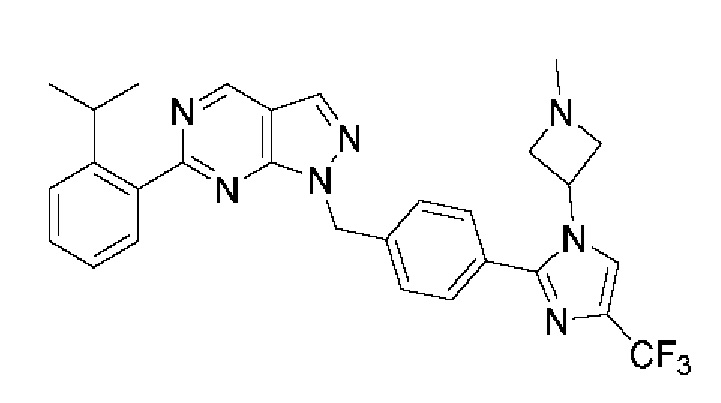
[0706] К раствору 3-(2-(4-((6-(2-изопропилфенил)-1H-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-4-(трифторметил)-1H-имидазол-1-ил)азетидин-1-иум-2,2,2-трифторацетата (соль ТФУ) (0,20 г, 0,41 ммоль) в DCM (10 мл) при комнатной температуре, добавляли 37% водный раствор формальдегида (2,04 мл) и NaBH(ОАс)3 (0,17 г, 0,82 ммоль) одной порцией. После перемешивания реакционной смеси при комнатной температуре в течение 30 минут добавляли дополнительно 70 мг NaBH(ОАс)3. Еще через 1 час реакцию гасили водой (20 мл) и экстрагировали DCM (50 мл ×3). Объединенный органический слой сушили над безводным Na2SO4 (20 г), фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (DCM:МеОН = от 100:1 до 20:1) с получением 38 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР): m/z 532 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 9,31 (с, 1H), 8,21 (с, 1H), 7,82 (с, 1H), 7,72 (д, J=7,5 Гц, 1H), 7,41-7,71 (м, 6Н), 7,33 (м, 1H), 5,75 (с, 2Н), 4,78-4,82 (м, 1H), 3,60-3,65 (м, 2Н), 3,55 (м, 1Н), 3,31-3,36 (м, 2Н), 2,37 (с, 3Н), 1,26-1,28 (д, J=6,9 Гц, 6Н).
Следующий пример был приготовлен из ВВ-4 в соответствии с Примером 152:
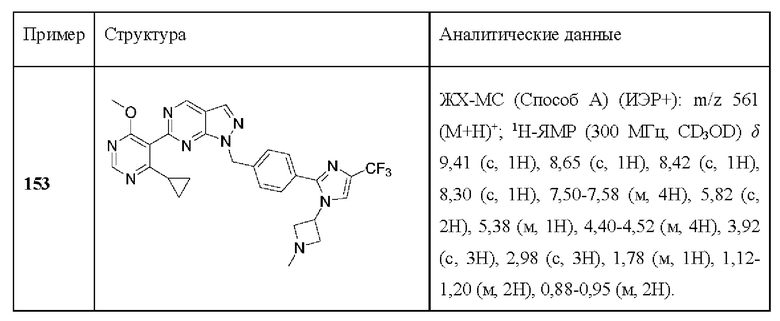
Пример 154: Синтез 1-(4-(1-Циклопропил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина (154):
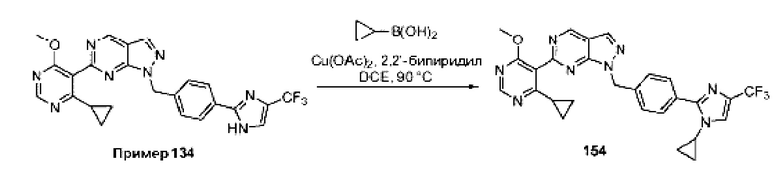
[0707] К перемешиваемому раствору 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (134) (0,20 г, 0,40 ммоль) и циклопропил бороновой кислоты (0,052 г, 0,60 ммоль) в DCE (20 мл), добавляли Cu(ОАс)2 (0,108 г, 0,60 ммоль) и 2,2'-бипиридил (0,074 г, 0,48 ммоль). Реакционную смесь дегазировали кислородом в течение 30 минут, а затем нагревали при 90°С под давлением баллона с кислородом в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь фильтровали через слой целита, промывали DCM и фильтрат упаривали при пониженном давлении. Неочищенное соединение очищали колоночной хроматографией с использованием 0-2% МеОН в DCM с получением неочищенного соединения, которое дополнительно очищали преп. ВЭЖХ с получением 0,042 г указанного в заголовке соединения. ЖХ-МС (Способ С) (ИЭР+): m/z 533,05 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,51 (с, 1Н), 8,71 (с, 1Н), 8,53 (с, 1Н), 7,92 (с, 1Н), 7,85 (д, J=7,8 Гц 2Н), 7,40 (д, J=8,3 Гц 2Н), 5,78 (с, 2Н), 3,85 (с, 3Н), 3,67-3,74 (м, 1Н), 1,62-1,69 (м, 1Н), 1,06 (с, 2Н), 0,92-1,00 (м, 2Н), 0,83-0,91 (м, 4Н).
Общая процедура получения амннопиримидинов: Синтез 6-Циклопропил-N,N-димел-5-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил)пиримидин-4-амнна (155):
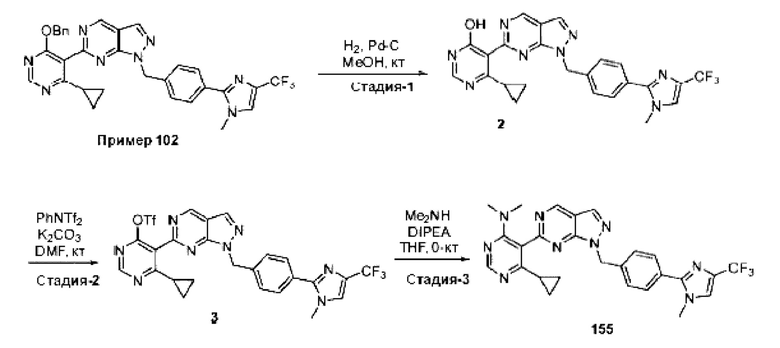
Стадия 1: Синтез 6-Циклопропил-5-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-нл)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил)пиримидин-4-ола:
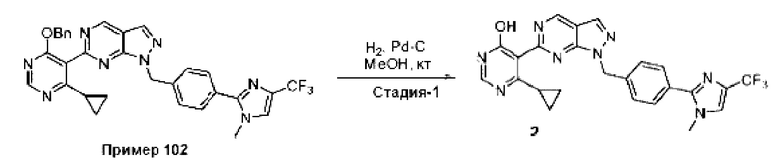
[0708] К перемешиваемому раствору 6-(4-(бензилокси)-6-цинклопропилпиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (102) (0,32 г, 0,55 ммоль) в МеОН (5 мл) добавляли PcDC (0,10 г) в атмосфере азота, и реакционную смесь перемешивали в атмосфере Н2 при комнатной температуре в течение 2 ч. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь фильтровали через целит, и фильтрат упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 7-10% МеОН в DCM, с получением 0,215 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 493,00 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 12,62 (с, 1Н), 9,47 (с, 1Н), 8,48 (с, 1Н), 8,17 (с, 1Н), 7,91 (с, 1Н), 7,67 (д, J=7,9 Гц, 2Н), 7,40 (д, J=8,4 Гц, 2Н), 5,77 (с, 2Н), 3,74 (с, 3Н), 1,46-1,54 (м, 1Н), 0,97-1,02 (м, 2Н), 0,74-0,80 (м, 2Н).
Стадии 2: Синтез 6-Циклопропил-5-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил)пиримидин-4-илтрифторметансульфоната:
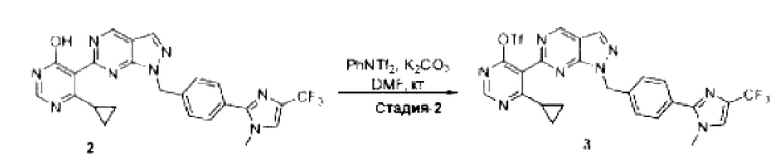
[0709] К перемешиваемому раствору 6-циклопропил-5-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина-6-ил)пиримидин-4-ола (0,20 г, 0,41 ммоль) в ДМФА (5 мл), добавляли K2CO3 (0,168 г, 1,22 ммоль). Затем добавляли N-фенил-бис(трифторметансульфонимид) (0,217 г, 0,61 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли Н2О (20 мл) и экстрагировали ЕА (2×20 мл). Объединенный органический слой промывали холодной Н2О (25 мл) и солевым раствором (25 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 1-2% МеОН в DCM, с получением 0,13 г указанного в заголовке соединения. ЖХ-МС (Способ В) (ИЭР+): m/z 625,00 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,62 (с, 1Н), 9,09 (с, 1Н), 8,61 (с, 1Н), 7,92 (с, 1Н), 7,67 (д, 3=7,9 Гц, 2Н), 7,40 (д, J=1,9 Гц, 2Н), 5,81 (с, 2Н), 3,74 (с, 3Н), 2,26-2,30 (м, 1Н), 1,25 (с, 2Н), 1,09-1,14 (м, 2Н).
Стадия 3: Синтез 6-Циклопропил-N,N-диметил-5-(1-(4-(1-метил-4-трифторметил)-1Н-имидазол-2ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил_пиримидин-4-амина (Пример 155):
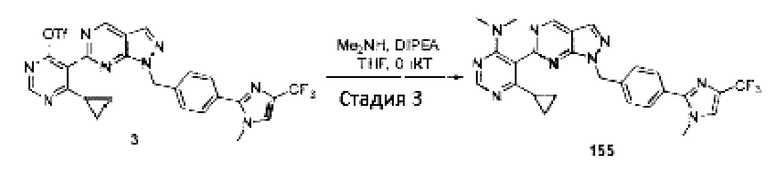
[0710] К охлажденному льдом раствору 6-циклопропил-5-(1-(4-(1-метил-4-(трифторетил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-а]пиримидин-6-ил)пиримидин-4-илтрифторметансульфоната (1 экв.) в сухом ТГФ (2 мл), последовательно добавляли DIPEA (2 экв.) и Me2NH (2 экв.). Полученную смесь перемешивали при комнатной температуре в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли водой (10 мл) и экстрагировали ЕА (2×15 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение растирали с н-пентаном (10 мл), фильтровали и полученное таким образом твердое вещество дополнительно очищали препаративной ВЭЖХ с получением мг указанного в заголовке соединения ЖХ-МС (Способ В) (ИЭР+): m/z 520,15 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,49 (с, 1Н), 8,51 (с, 1H), 8,41 (с, 1Н), 7,92 (с, 1H), 7,67 (д, J=7,8 Гц, 1H), 7,36 (д, J=8,3 Гц, 2Н), 5,78 (с, 2Н), 3,74 (с, 3Н), 2,61 (с, 6Н), 1,58-1,66 (м, 1Н), 0,93-0,97 (м, 2Н), 0,67-0,70 (м, 1Н).
Следующие соединения были получены из соответствующих аминов в соответствии со способом, описанным для Примера 155:
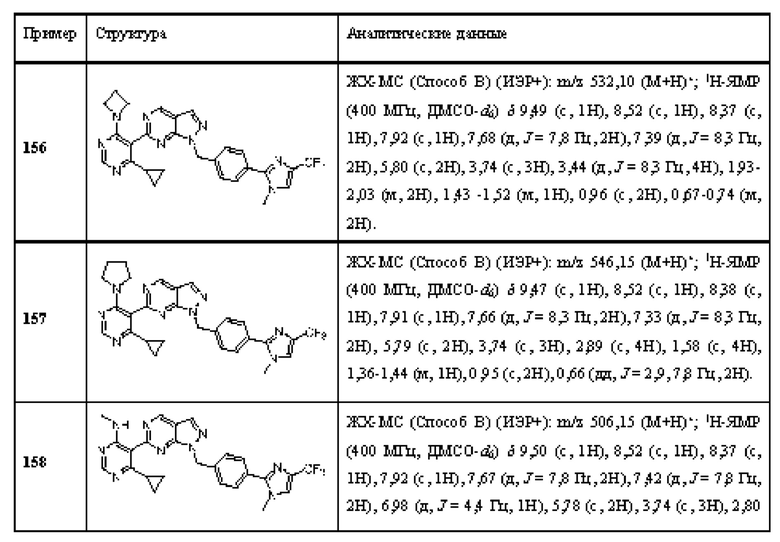
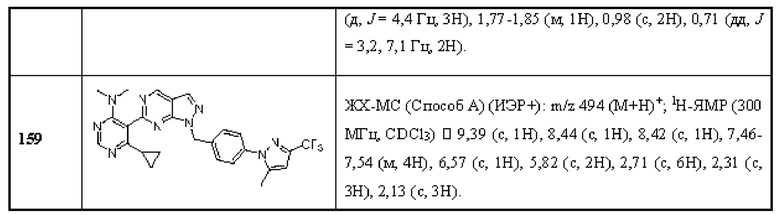
Пример 160: Синтез 6-Циклопропил-5-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил)пиримидин-4-илтрифторметансульфоната (160):
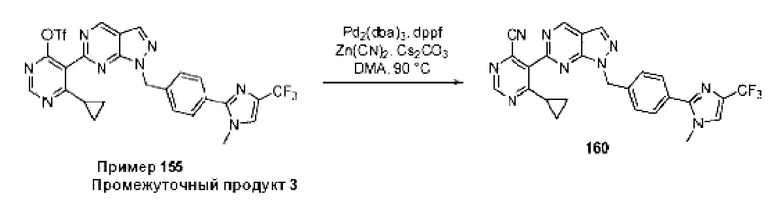
[0711] К перемешиваемому раствору 6-циклопропил-5-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил)пиримидин-4-илтрифторметансульфоната (0,15 г, 0,24 ммоль) в DMA (5 мл), добавляли Zn(CN)2 (0,141 г, 1,20 ммоль) и Cs2CO3 (0,156 г, 0,48 ммоль). Затем смесь дегазировали аргоном в течение 10 мин, после чего добавляли Pd2(dba)3 (0,022 г, 0,024 ммоль) и dppf (0,013 г, 0,024 ммоль). Реакционную смесь нагревали до 90°С в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Полученный остаток растворяли в Et2O (50 мл), фильтровали через целит и промывали Et2O (20 мл). Органический слой упаривали при пониженном давлении и неочищенное соединение очищали с помощью SFC, получая 0,037 г указанного в заголовке соединения. ЖХ-МС (Способ С) (ИЭР+): m/z 501,95 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,65 (с, 1H), 9,28 (с, 1Н), 8,63(с, 1Н), 7,92 (с, 1Н), 7,67 (д, J=7,8 Гц, 2Н), 7,46 (д, J=7,8 Гц, 2Н), 5,83 (с, 2Н),3,74(с, 3Н), 2,38-2,45 (м, 1Н), 1,23 (с, 2Н), 1,09-1,16 (м, 2Н).
Пример 161: Синтез 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-N,N-диметил-1Н-пиразоло[3,4-d]пиримиднн-4-амина (161):
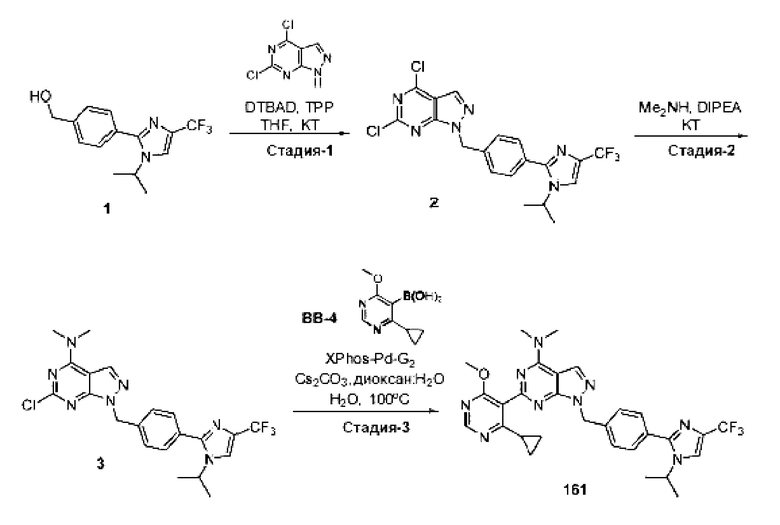
Стадия 1: Синтез 4,6-дихлор-1-(4-(1-изопропил-4-(трифторметил)-1Н-иминдазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина:
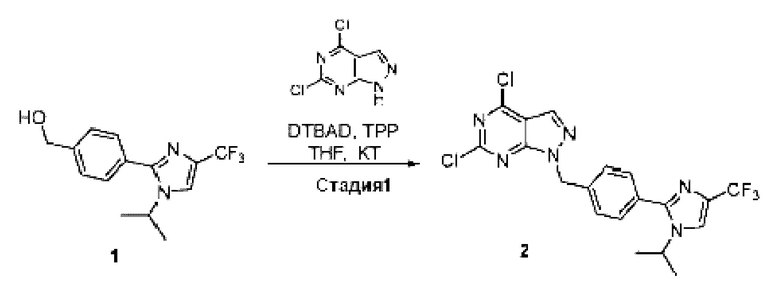
[0712] К перемешиваемому раствору (4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)метанола 1 (1,0 г, 3,52 ммоль), 4,6-диклор-1Н-пиразоло[3,4-d]пиримидина (0,665 г, 0,352 ммоль) и ТРР (1,84 г, 7,04 ммоль) в ТГФ (10 мл), добавляли DTBAD (1,61 г, 7,04 ммоль) порциями при комнатной температуре. Полученную смесь перемешивали в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении досуха, получая неочищенное соединение. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-40% ЕА в гексане, с получением указанного в заголовке соединения (0,230 г). ЖХ-МС (Способ В) (ИЭР+): m/z 455,10 (М)+; 1Н-ЯМР(400 МГц, ДМСО-d6) δ 8,63 (с, 1Н), 8,17 (с, 1Н), 7,54 (д, J=7,83 Гц, 2Н), 7,40 (д, J=7,83 Гц, 2Н), 5,73-5,77 (м, 2Н), 4,43 (тд, J=6,72, 12,96 Гц, 1Н), 1,38 (д, J=6,85 Гц, 6Н).
Стадия 2: Синтез 6-хлор -1-(4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-N,N-диметил-1Н-пиразоло[3,4-d]пиримидни-4-амина:
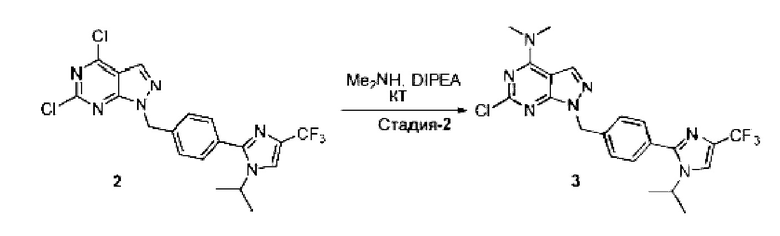
[0713] К перемешиваемому раствору 4,6-дихлор-1-(4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (0,230 г, 0,506 ммоль) в ТГФ (12 мл), добавляли DIPEA (0,263 мл, 1,519 ммоль) при комнатной температуре. К полученной смеси добавляли диметиламин (2М раствор в ТГФ, 0,506 мл, 1,01 ммоль) и реакционную смесь перемешивали в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли водой (20 мл) и экстрагировали ЕА (3×5 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-50% ЕА в гексане, с получением указанного в заголовке соединения (0,200 г). ЖХ-МС (Способ В) (ИЭР+): m/z 464,08 (М+Н)+, 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,31 (с, 1Н), 8,17 (с, 1H), 7,53 (д, J=7,83 Гц, 2Н), 7,32 (д, J=7,83 Гц, 2Н), 5,57 (с, 2Н), 4,10 (к, J=5,22 Гц, 1Н), 3,42 (уш. с, 3Н), 3,26 (уш. с, 3Н), 1,38 (д, J=6,36 Гц, 6Н).
Стадия 3: Синтез 6-(4-циклопропил-метоксипиримидин-5-ил)-1(4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-N,N-диметил-1H-пиразоло[3,4-d]пиримидин-4-амина (161):
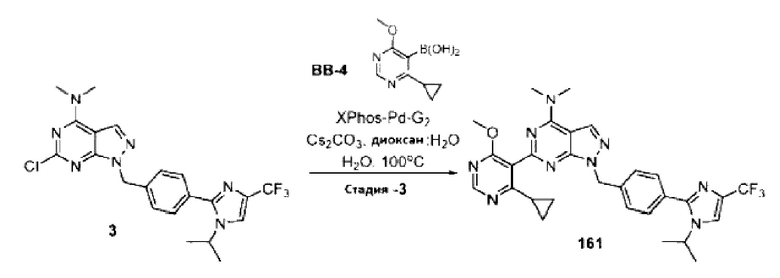
[0714] К перемешиваемому раствору 6-хлор-1-(4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-N,N-диметил-1H-пиразоло[3,4-d]пиримидин-4-амина 3 (0,090 г, 0,194 ммоль) и (4-циклопропил-6-метоксипиримидин-5-ил)бороновой кислоты ВВ-4 (0,041 г, 0,213 ммоль) в диоксане: Н2О (3:1 мл), добавляли Cs2CO3 (0,158 г, 0,485 ммоль) при комнатной температуре. Полученную смесь дегазировали аргоном в течение 5 минут, а затем обрабатывали X-Phos (0,018 г, 0,038 ммоль) и X-Phos-Pd-G2 (0,015 г, 0,019 ммоль). Смесь дополнительно дегазировали аргоном в течение 5 мин, а затем нагревали при 100°С в течение 8 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, разбавляли водой (100 мл) и экстрагировали ЕА (2×100 мл). Объединенный органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-50% ЕА в гексане, с получением (0,070 г) указанного в заголовке соединения. Этот материал растворяли в диоксане (7 мл) и перемешивали с диоксидом кремния PS-Thiol (0,007 г). Полученную суспензию нагревали до 100°С в течение 2 часов, затем охлаждали до комнатной температуры, фильтровали и упаривали при пониженном давлении с получением указанного в заголовке соединения (0,050 г).
[0715] Этот материал дополнительно очищали препаративной ВЭЖХ (Метод С) с получением указанного в заголовке соединения (0,020 г). ЖХ-МС (Способ В) (ИЭР+): m/z 578,30 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,61 (с, 1Н), 8,29 (с, 1Н), 8,11 (с, 1Н), 7,47 (д, J=7,83 Гц, 2Н), 7,34 (д, J=7,83 Гц, 2Н), 5,59 (с, 2Н), 4,38 (тд, J=6,24, 12,96 Гц, 1Н), 3,83 (с, 3Н), 3,19-3,47 (м, 6Н), 1,70-1,80 (м, 1Н), 1,35 (д, J=6,36 Гц, 6Н), 0,96-1,02 (м, 2Н), 0,80-0,86 (м, 2Н).
Получение I-65: Синтез 1-(4-(5-бром-1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина (I-65):
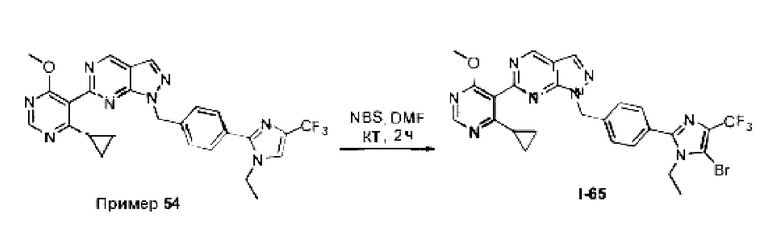
[0716] К перемешиваемому раствору 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (54) (1,80 г, 3,461 ммоль) в ДМФА (10 мл) добавляли NBS (1,23 г, 6,923 ммоль) одной порцией при комнатной температуре. Полученную смесь перемешивали в течение 2 ч и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили ледяной водой (15 мл) и экстрагировали ЕА (3×15 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Полученное таким образом неочищенное соединение очищали хроматографией на силикагеле, используя 0-50% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (1,00 г). ЖХ-МС (Способ В) (ИЭР+): m/z 600,40 (М+Н)+; 1Н-ЯМР(400 МГц, ДМСО-d6) δ 9,51 (с, 1Н), 8,71 (с, 1Н), 8,53 (с, 1Н), 7,54-7,64 (м, 3Н), 7,43 (д, J=7,98 Гц, 1Н), 5,80 (с, 2Н), 4,00-4,08 (м, 2Н), 3,85 (с, 2Н), 1,62-1,69 (м, 1Н), 1,24 (т, J=7,23 Гц, 3Н), 1,17 (т, J=7,23 Гц, 1Н), 1,02-1,08 (м, 2Н), 0,83-0,88 (м, 2Н).
Следующие Примеры соединений были получены из соответствующих строительных блоков в соответствии со способом, описанным для I-65:

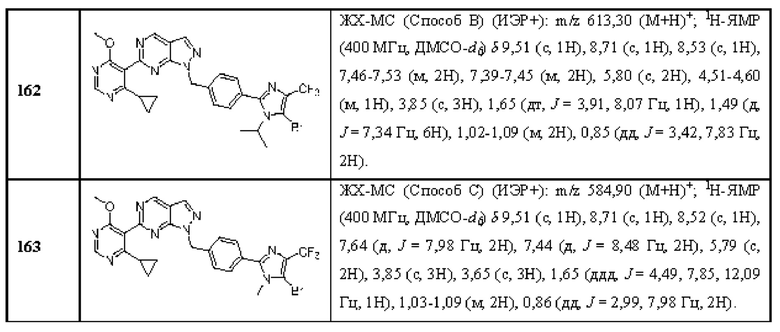
Пример 164: Синтез 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-5-метокси-4-(трифторметил)-1Н-имидазол-2-ил)бензнл)-1Н-пиразоло[3,4-d]пиримидина (164):
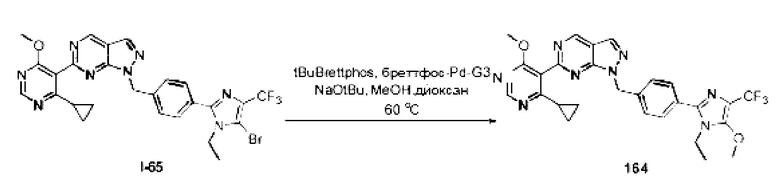
[0717] К перемешиваемому раствору 1-(4-(5-бром-1-этил-4-(трифторметил)-1Н-имидазол-2-ил)6ензил)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина I-65 (0,100 г, 0,167 ммоль) в диоксане: МеОН (1,2:0,3 мл) добавляли NaOtBu (0,032 г, 0,334 ммоль). Затем смесь дегазировали аргоном в течение 10 мин К реакционной смеси добавляли трет-бутил-Бреттфос (0,002 г, 0,003 ммоль) и трет-бутил-Бреттфос Pd-G3 (0,003 г, 0,003 ммоль) при комнатной температуре, и смесь дополнительно дегазировали аргоном в течение 5 минут. Затем реакционную смесь нагревали при 60°С в течение 48 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, элюируя 0-2% метанолом в DCM, с последующей повторной очисткой с использованием препаративной ВЭЖХ (Метод В) с получением указ энного в заголовке соединения (0,014 г). ЖХ-МС (Способ В) (ИЭР+): m/z 551,25 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,51 (с, 1Н), 8,71 (с, 1Н), 8,52 (с, 1Н), 7,58 (д, J=8,31 Гц, 2Н), 7,40 (д, J=8,31 Гц, 2Н), 5,78 (с, 2Н), 3,97 (с, 3Н), 3,90-3,96 (м, 2Н), 3,85 (с, 3Н), 1,61-1,69 (м, 1Н), 1,17 (т, J=7,09 Гц, 3Н), 1,03-1,08 (м, 2Н), 0,83-0,88 (м, 2Н).
Следующие соединения были получены из соответствующих строительных блоков в соответствии со способом, описанным для Примера 164:

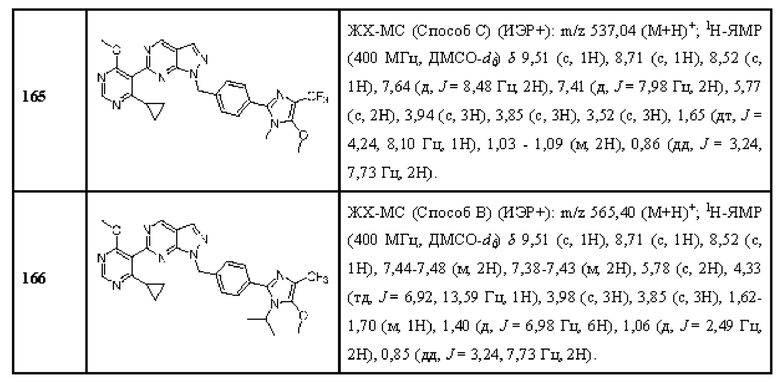
Пример 167: Синтез 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-5-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (167):
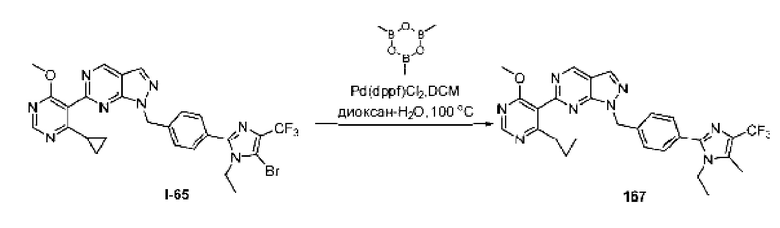
[0718] К перемешиваемому раствору 1-(4-(5-бром-1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина I-65 (0,200 г, 0,334 ммоль) и триметилбороксина (0,031 г, 0,502 ммоль) в диоксане (6 мл) и воде (2 мл) добавляли K2CO3 (0,090 г, 0,668 ммоль) при комнатной температуре, и реакционную смесь дегазировали газообразным аргоном в течение 15 мин. К полученной реакционной смеси добавляли Pd(dppf)Cl2-DCM (0,014 г, 0,016 ммоль) при комнатной температуре. Реакционную смесь нагревали в закрытой пробирке при 100°С в течение 6 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры и фильтровали через целит и промывали ЕА (5 мл). Полученный фильтрат упаривали при пониженном давлении. Полученное неочищенное соединение очищали преп. ВЭЖХ (Метод В), что дает указанное в заголовке соединение. К перемешиваемому раствору соединения 168 (0,70 г) в диоксане (7 мл) добавляли SP тиоловый диоксид кремния (0,007 г). Полученную реакционную смесь нагревали до 100°С в течение 1 ч, затем охлаждали до комнатной температуры, фильтровали и упаривали при пониженном давлении с получением указанного в заголовке соединения (0,027 г). ЖХ-МС (Способ В) (ИЭР+): m/z 535,15 (М+Н)+; 1Н-ЯМР (400 МГц ДМСО-d6) δ 9,51 (с, 1Н), 8,71 (с, 1Н), 8,52 (с, 1Н), 7,55 (д, J=7,82 Гц, 2Н), 7,41 (д, J=7,82 Гц, 2Н), 5,79 (с, 2Н), 3,92-4,01 (м, 2Н), 3,85 (с, 3Н), 2,37 (уш. с, 3Н), 1,66 (д, J=3,42 Гц, 1Н), 1,19 (т, J=7,09 Гц, 3Н), 1,02-1,08 (м, 2Н), 0,85 (д, J=3,91 Гц, 2Н).
Получение общего промежуточного продукта I-63: Синтез 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина (I-66):
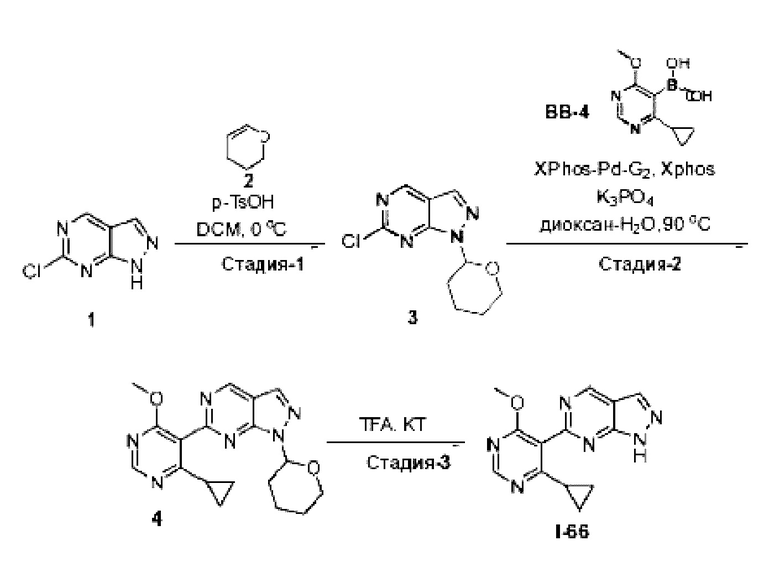
Стадия 1: Синтез 6-хлор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразоло[3,4-d]пиримидина:
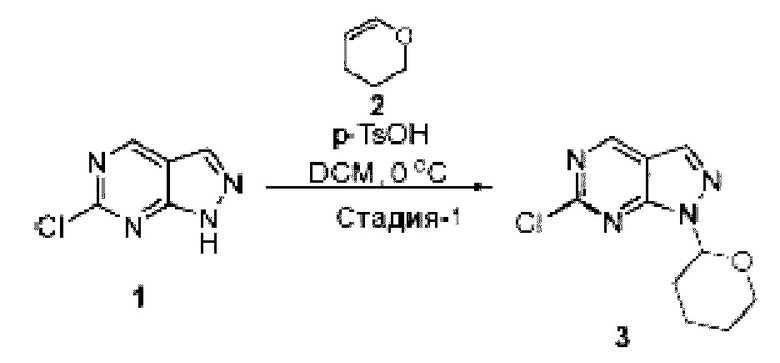
[0719] К перемешиваемому раствору 6-хлор-1Н-пиразоло[3,4-d]пиримидина 1 (2,5 г, 16,2 ммоль) в DCM (50 мл) добавляли n-TsOH (0,418 г, 2,43 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение 5 минут, а затем по каплям добавляли 3,4-дигидро-2Н-пиран 2 (2,29 г, 3,24 ммоль) при 0°С. После добавления смесь перемешивали в течение 1 ч при той же температуре. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили насыщенным раствором NaHCO3 (50 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-20% ЕА в гексане, с получением указанного в заголовке соединения (3,0 г). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,31-9,29 (м, 1Н), 8,50 (с, 1Н), 5,94 (дд, J=2,4, 10,3 Гц, 1Н), 3,98-3,92 (м, 1Н), 3,77-3,69 (м, 1Н), 2,48-2,36 (м, 1Н), 2,07-1,98 (м, 1Н), 1,97-1,89 (м, 1Н), 1,84-1,71 (м, 1Н), 1,62-1,54 (м, 2Н).
Стадия 2: Синтез 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразоло[3,4-d]пиримидина:
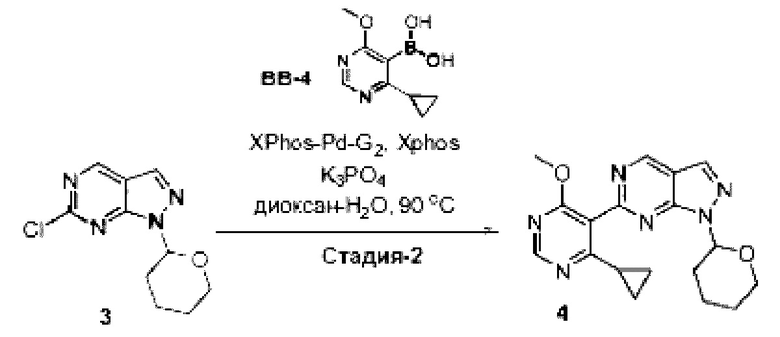
[0720] К перемешиваемому раствору 6-хлор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразоло[3,4-d]пиримидина 3 (2,5 г, 10,5 ммоль) и (4-циклопропил-6-метоксипиримидин-5-ил) бороновой кислоты ВВ-4 (2,03 г, 10,5 ммоль) в диоксане: Н2О (3:1, 40 мл) добавляли K3PO4 (2,43 г, 11,4 ммоль) при комнатной температуре. Реакционную смесь продували аргоном в течение 30 минут, а затем обрабатывали XPhos-Pd-G2 (0,818 г, 1,04 ммоль) и XPhos (0,991 г, 2,08 ммоль) при комнатной температуре. Полученную смесь нагревали до 90°С в герметичной пробирке в течение 1 ч. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, разбавляли водой (50 мл) и экстрагировали ЕА (50 мл). Органический слой отделяли, сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-40% ЕА в гексане, с получением указанного в заголовке соединения 4 (3,00 г). К перемешиваемому раствору соединения 4 (3,00 г) в диоксане (10 мл) добавляли SP тиоловый диоксид кремния (0,250 г). Полученную реакционную смесь нагревали до 100 С в течение 1 ч, а затем охлаждали до комнатной температуры, фильтровали и упаривали при пониженном давлении с получением указанного в заголовке соединения (2,92 г). ЖХ-МС (Способ С) (ИЭР+): m/z 353,05 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,49 (с, 1Н), 8,72 (с, 1Н), 8,51 (с, 1Н), 3,97-3,90 (м, 1Н), 3,84 (с, 3Н), 3,76-3,68 (м, 1Н), 2,05-1,98 (м, 3Н), 1,86-1,71 (м, 1Н), 1,63-1,52 (м, 3Н), 1,20-1,15 (м, 1Н), 1,11-1,02 (м, 2Н), 0,90-0,86 (м, 2Н).
Стадия 3: Синтез 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина (I-66):
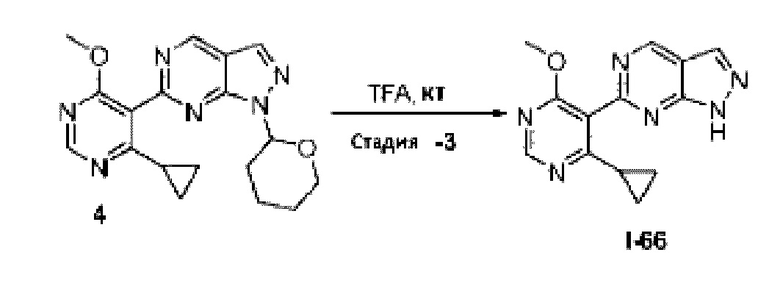
[0721] Твердый 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразоло[3,4-d]пиримидина 4 (2,9 г, 8,23 ммоль) растворяли при комнатной температуре в ТФУ (30 мл) и перемешивали в течение 15 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Остаток разбавляли DCM (50 мл) и подщелачивали до рН 8, используя насыщенный раствор NaHCO3 (10 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-20% ЕА в гексане, с получением указанного в заголовке соединения (1,87 г). ЖХ-МС (Способ С) (ИЭР+): m/z 268,95 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 14,20 (уш. с, 1Н), 9,46 (с, 1Н), 8,69 (с, 1Н), 8,43 (с, 1Н), 3,83 (с, 3Н), 1,67-1,59 (м, 1Н), 1,05 (тд, J=3,7, 7,1 Гц, 2Н), 0,91-0,84 (м, 2Н).
Пример 168: Синтез (S)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1(1-(4-(1-(2-фторэтил)-4-(трифторметил)-1Н-имидазол-2)-ил)-3-метоксифенил)этил)-1Н-пиразоло[3,4-d]пиримидина (16S):
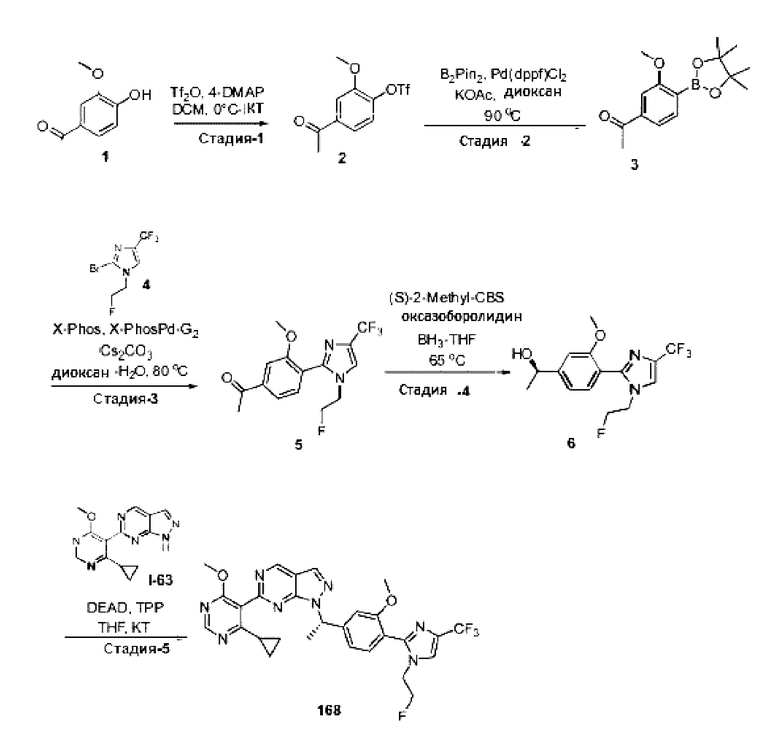
Стадия 1: Синтез 4-ацетил-2-метоксифенилтрифторметансульфоната:
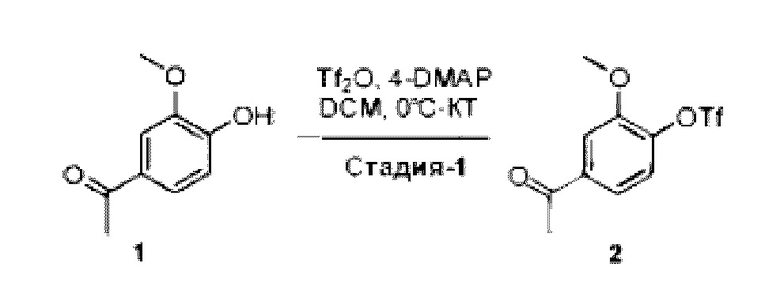
[0722] К перемешиваемому раствору 1-(4-гидрокси-3-метоксифенил)этан-1-она 1 (3 г, 18 ммоль) в DCM (60 мл) добавляли 4-диметиламинопиридин (3,3 г, 27 ммоль) при 0°С. К образовавшейся реакционной смеси по каплям добавляли трифликовый ангидрид (6,1 г, 22 ммоль). После завершения добавления смесь дополнительно перемешивали при 0°С в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили 1 н. HCl (50 мл) и экстрагировали DCM (50 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-20% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (4,57 г). 1Н-ЯМР (400 МГц, CDCl3) δ 7,66 (д, J=1,96 Гц, 1Н), 7,57 (дд, J=1,96, 8,31 Гц, 1Н), 7,32 (д, J=8,31 Гц, 1Н), 3,99 (с, 3Н), 2,61-2,64 (м, 3Н).
Стадия 2: Синтез 1-(3-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этан-1-она:
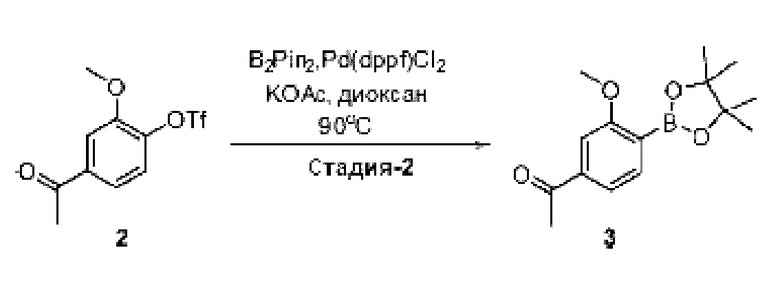
[0723] К перемешиваемому раствору 4-ацетил-2-метоксифенилтрифторметансульфоната 2 (4,5 г, 15,1 ммоль) и бис(пинаколато)диборана (19,15 г, 75,44 ммоль) в диоксане (100 мл) добавляли ацетат калия (4,44 г, 45,2 ммоль). ммоль). Полученную смесь дегазировали аргоном в течение 10 мин, а затем обрабатывали Pd(dppf)Cl2 (0,044 г, 1,508 ммоль) при комнатной температуре. Затем реакционную смесь нагревали в запаянной пробирке при 90°С в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры и фильтровали через слой целита. Фильтрат разбавляли водой (100 мл) и экстрагировали ЕА (2×50 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-15% ЕА в гексане в качестве элюента, с получением 3 (2,07 г). ЖХ-МС (Способ С) (ИЭР+): m/z 277,26 (М+Н)+; 1Н-ЯМР (400 МГц, CDCl3) δ 7,73 (д, J=7,34 Гц, 1Н), 7,49 (дд, J=1,22, 7,58 Гц, 1Н), 7,42-7,45 (м, 1Н), 3,89 (с, 3Н), 2,61 (с, 3Н), 1,37(с, 12Н).
Стадия 3: Синтез 1-(4-(1-(2-фторэтил)-4-(трифторметил)-1Н-имидазол-2-ил)-3-метоксифенил)этан-1-она:
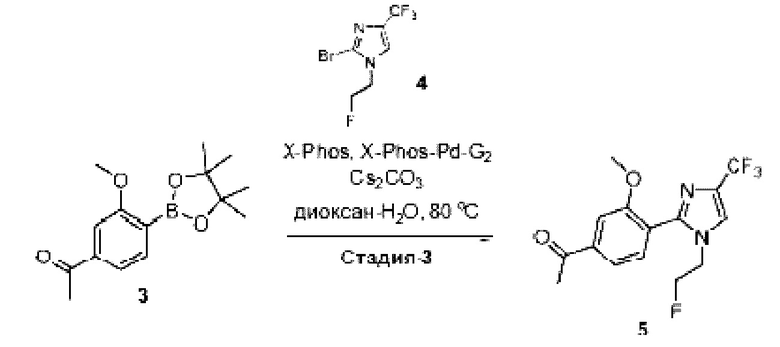
[0724] К перемешиваемому раствору 2-бром-1-(2-фторэтил)-4-(трифторметил)-1Н-имидазола 4 (2,0 г, 7,24 ммоль) и 1-(3-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборопан-2-ил)фенил)этан-1-она 3 (1,89 г, 7,24 ммоль) в диоксане: Н2О (4:1, 50 мл), добавляли Cs2CO3 (3,53 г, 10,96 ммоль). Полученную смесь продували аргоном в течение 10 мин, а затем обрабатывали XPhos-Pd-G2 (0,284 г, 0,362 ммоль) и XPhos (0,345 г, 0,724 ммоль) при комнатной температуре. Затем реакционную смесь нагревали до 80°С в герметичной пробирке в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, разбавляли водой (50 мл) и экстрагировали ЕА (2×25 мл). Объединенный органический слой сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-30% ЕА в гексане в качеств в элюента, с получением указанного в заголовке соединения (2,0 г). ЖХ-МС (Способ В) (ИЭР+): m/z 331,10 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,01 (с, 1Н), 7,67-7,72 (м, 1Н), 7,62 (с, 1Н), 7,53 (д, J=7,83 Гц, 1Н), 4,49-4,67 (м, 2Н), 4,08-4,20 (м, 2Н), 3,89 (с, 3Н), 2,63-2,70 (м, 4Н).
Стадия 4: Синтез (R)-1-(4-(1-(2-фторэтил)-4-(трифторметил)-1Н-имидазол-2-ил)-3-метоксифенил)этан-1-ола):
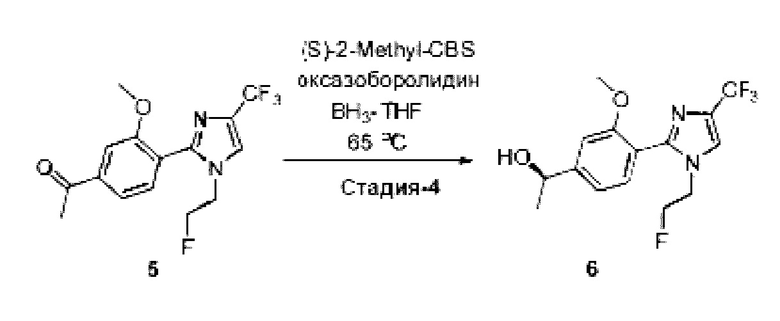
[0725] К перемешиваемому раствору 1-(4-(1-(2-фторэтил)-4-(трифторметил)-1Н-имидазол-2-ил)-3-метоксифенил)этан-1-она 5 (1,5 г, 4,5 ммоль) в ТГФ (30 мл) добавляли (S)-2-метип-CBS-оксазоборолидин (0,9 мл, 0,91 ммоль) при комнатной температуре. Затем смесь нагревали при 45°С в течение 1 часа. К полученной реакционной смеси добавляли боран-DMS (0,689 мг, 9,08 ммоль), и смесь нагревали при 65°С в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, медленно гасили метанолом (50 мл) и упаривали при пониженном давлении. Полученный неочищенный остаток разбавляли 1 н. HCl (20 мл) и экстрагировали ЕА (2×25 мл). Объединенный органический слой сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-60% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (1,04 г). ЖХ-МС (Способ В) (ИЭР+): m/z 333,4 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 7,94 (с, 1Н), 7,28 (д, J=7,83 Гц, 1Н), 7,15 (с, 1Н), 7,04 (д, J=7,34 Гц, 1Н), 5,31 (д, J=3,91 Гц, 1Н), 4,74-4,83 (м, 1Н), 4,67 (т, J=4,40 Гц 1Н), 4,55 (т, J=4,65 Гц, 1Н), 4,14(т, J=4,40 Гц, 1Н), 4,07 (т, J=4,65 Гц, 1Н), 3,80 (с, 3Н), 1,37 (д, J=6,36 Гц, 3Н).
Стадия 5: Синтез (S)-6-(4-циклолропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-(2-фторэтил)-4-(трифторметил)-1Н-имидазол-2)-ил)-3-метоксифенил)этил)-1Н-пиразоло[3,4-d]пиримидина (168):
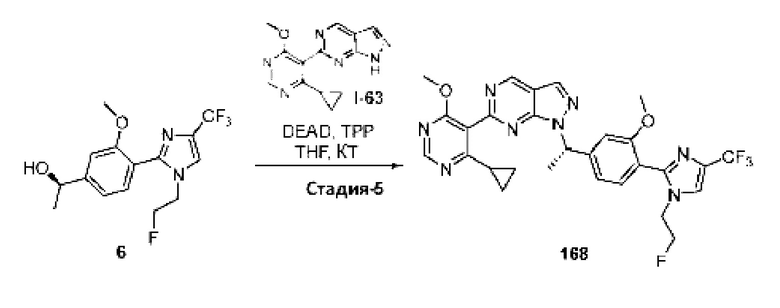
[0726] К перемешиваемому раствору (R)-1-(4-(1-(2-фторэтил)-4-(трифторметил)-1Н-имидазол-2-ил)-3-метоксифенил)этан-1-ола 6 (0,400 г, 1,20 ммоль), 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина I-66 (0,322 г, 1,20 ммоль) и трифенилфосфина (0,470 г, 1,80 ммоль) в ТГФ (4 мл), добавляли DEAD (0,133 г, 1,80 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение 30 мин, и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Полученный неочищенный остаток разбавляли водой (30 мл) и экстрагировали ЕА (2×25 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-70% ЕА в гексане в качестве элюента, с получением неочищенного соединения, которое дополнительно очищали препаративной ВЭЖХ (Метод D) и хиральной очисткой SFC (Метод В) с получением указанного в заголовке соединения (0,200 г). ЖХ-МС (Способ В) (ИЭР+): m/z 583,25 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,49 (с, 1Н), 8,71 (с, 1Н), 8,55 (с, 1Н), 7,93 (с, 1Н), 7,28 (д, J=7,83 Гц, 1Н), 6,97 (д, J=7,83 Гц, 1Н), 6,30-6,38 (м, 1Н), 4,63 (т, J=4,40 Гц, 1Н), 4,51 (т, J=4,40 Гц, 1Н), 4,11 (д, J=3,91 Гц, 1Н), 4,04 (д, J=4,40 Гц 1Н), 3,82 (с, 3Н), 3,74 (с, 3Н), 2,00 (д, 3=6,85 Гц, 3Н), 1,55-1,64 (м, 2Н), 1,01-1,07 (м, 2Н), 0,77-0,88 (м, 2Н).
Следующее соединение было получено из соответствующих строительных блоков в соответствии со способом, описанным дня Примера 168:
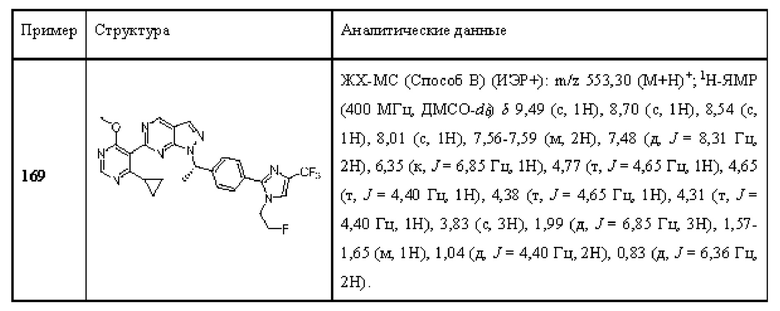
Пример 170: Синтез 3-бром-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (170):
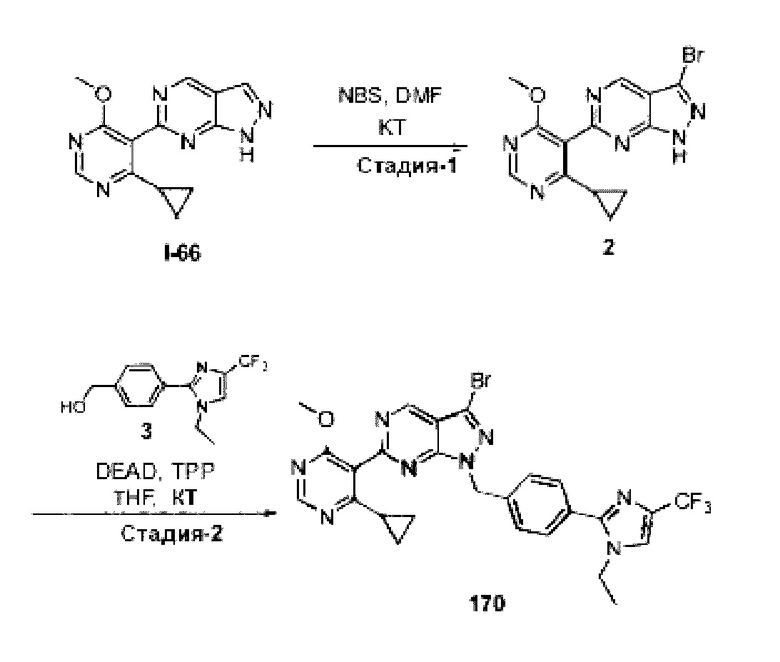
Стадия 1: Синтез 3-бром-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина:
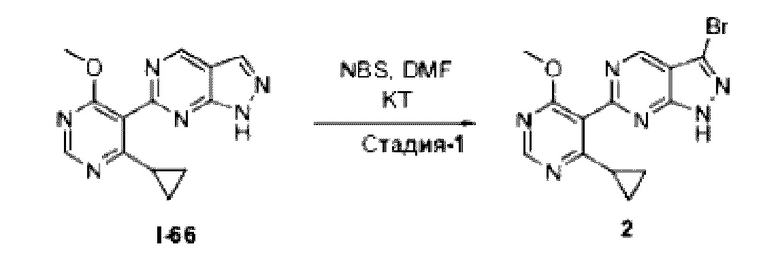
[0727] К перемешиваемому раствору 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1H-пиразоло[3,4-d]пиримидина I-66 (1,80 г, 6,71 ммоль) в ДМФА (18 мл) добавляли NBS (2,38 г, 13,4 ммоль) при комнатной температуре. Затем реакционную смесь перемешивали в течение 2 часов. После завершения реакционную смесь упаривали при пониженном давлении с получением неочищенного указанного в заголовке соединения 2. Неочищенное соединение растирали с DCM (20 мл), фильтровали и сушили в вакууме с получением указанного в заголовке соединения 2 (2,04 г). ЖХ-МС (Способ С) (ИЭР+): m/z 346,85 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 14,58 (уш. с, 1Н), 9,39 (с, 1Н), 8,71 (с, 1Н), 3,84 (с, 3Н), 1,71-1,62 (м, 1Н), 1,06 (уш. с, 2Н), 0,91-0,83 (м, 2Н).
Стадия 2: Синтез 3-бром-6-(4-циклопропил-6-метаксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (170):
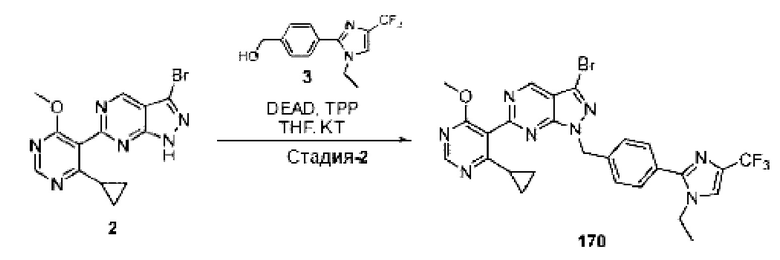
[0728] К перемешиваемой смеси (4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)метанола 3 (1,55 г, 5,74 ммоль), 3-бром-6-(4-циклопропил-6-метокстипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина 2 (2,00 г, 5,740 ммоль) и трифенилфосфина (2,26 г, 8,611 ммоль) в ТГФ (20 мл) добавляли DEAD (1,50 г, 8,611 ммоль) при комнатной температуре. Полученную смесь затем перемешивали в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли водой (50 мл) и экстрагировали ЕА (2×50 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 10-60% ЕА в н-гексане в качестве элюента, с получением указанного в заголовке соединения (2,41 г). ЖХ-МС (Способ В) (ИЭР+): m/z 599,15 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,44 (с, 1Н), 8,72 (с, 1Н), 8,02 (с, 1Н), 7,61 (д, J=7,83 Гц, 2Н), 7,45 (д, J=7,83 Гц, 2Н), 5,77 (с, 2Н), 4,05 (к, J=7,34 Гц, 2Н), 3,86 (с, 3Н), 1,70 (дд, J=3,67, 8,07 Гц, 1Н), 1,30 (т, J=7,34 Гц, 3Н), 1,04-1,09 (м, 2Н), 0,83-0,89 (м, 2Н).
Следующее соединение было получено из соответствующих строительных блоков в соответствии со способом, описанным дня Примера 170:
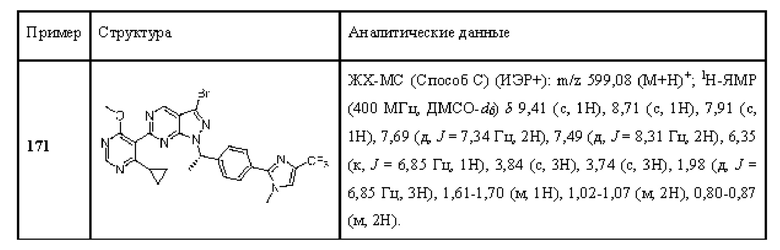
Пример 172: Синтез 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-3-карбонитрила (172):
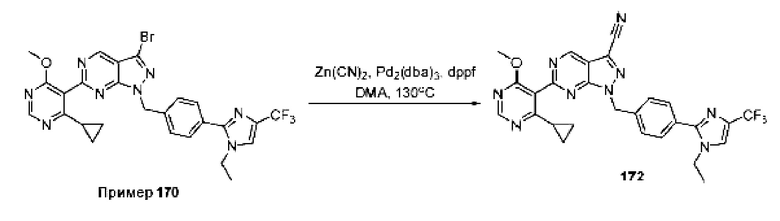
[0729] К перемешиваемому раствору 3-бром-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (170) (0,200 г, 0,334 ммоль) в DMA (10 мл), добавляли цианид цинка (0,078 г, 0,67 ммоль) при комнатной температуре. Полученную смесь дегазировали аргоном в течение 30 мин, а затем обрабатывали 1,1'-бис(дифенилфосфино)ферроценом (0,044 г, 0,080 ммоль) и Pd2(dba)3 (0,036 г, 0,040 ммоль) при комнатной температуре. Реакционную смесь нагревали в запаянной пробирке при 130°С в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и обрабатывали водным раствором аммиака (20 мл) в течение 30 минут. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь экстрагировали DCM (20 мл). Органический слой отделяли, промывали водой (10 мл) и солевым раствором (10 мл). Затем органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение обрабатывали смолой улавливающей металл PS-тиол, в ТГФ, фильтровали и упаривали в вакууме. Полученный остаток очищали хроматографией на силикагеле, используя 0-40% ЕА в гексане в качестве элюента, с получением неочищенного соединения, которое затем повторно очищали препаративной ВЭЖХ (Метод В) с получением указанного в заголовке соединения (0,070 г). ЖХ-МС (Способ С) (ИЭР+): m/z 546,10 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,80 (с, 1Н), 8,74 (с, 1Н), 8,03 (д, J=1,47 Гц, 1Н), 7,62 (д, J=8,31 Гц, 2Н), 7,49 (д, J=8,31 Гц, 2Н), 5,92 (с, 2Н), 4,05 (к, J=7,34 Гц, 2Н), 3,86 (с, 3Н), 1,70 (дdд, J=4,65, 7,95, 12,35 Гц, 1Н), 1,30 (т, J=7,09 Гц, 3Н), 1,06-1,11 (м, 2Н), 0,83-0,89 (м, 2Н).
Пример 173: Синтез (S)-6-(4-циклопропил-6-метоксимиримидин-5-ил)-1-(5-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)-2,3-дигидро-1Н-инден-1-ил)-1Н-пиразоло[3,4-d]пиримидина (173):
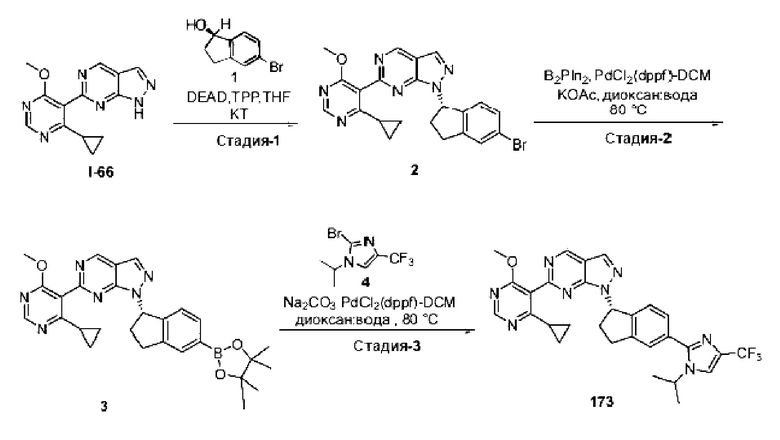
Стадия 1: Синтез (S)-1-(5-бром-2,3-дигидро-1Н-инден-1-ил)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина:
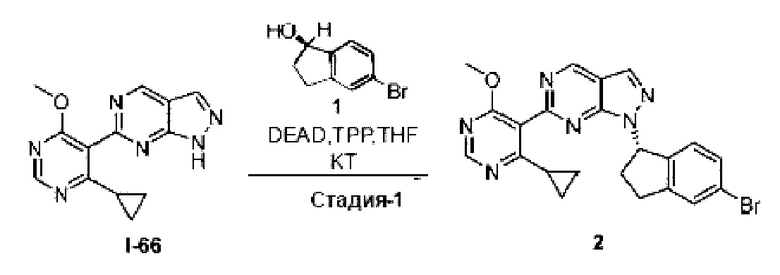
[0730] К перемешиваемому раствору 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина I-66 (0,800 г, 2,98 ммоль) в ТГФ (8 мл) при 0°С. добавляли (R)-5-бром-2,3-дигидро-1H-инден-1-ол 1 (0,635 г, 2,98 ммоль), DEAD (0,777 г, 4,47 ммоль) и ТРР (1,17 г, 4,47 ммоль). Реакционной смеси давали нагреться до комнатной температуры и затем перемешивали в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (20 мл) и экстрагировали ЕА (3×15 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, элюируя 10-90% ЕА в гексане, с получением указанного в заголовке соединения (0,897 г). ЖХ-МС (Способ В) (ИЭР+): m/z 463,00 (М+).
Стадия 2: Синтез (S)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-)-2,3-дигидро-1Н-инден-1-ил)-1Н-пиразало[3,4-d]пиримидина:
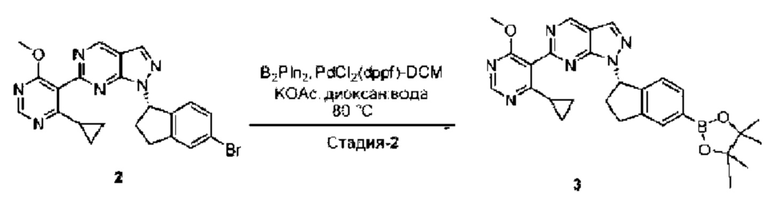
[0731] К перемешиваемому раствору (S)-1-(5-бром-2,3-дигидро-1H-инден-1-ил)-6-(4-циклопропил-6-метоксипиримидан-5-ил)-1H-пиразоло[3,4-d]пиримидина 2 (0,800 г, 1,72 ммоль) в диокеане (30 мл) добавляли КОАс (0,506 г, 5,16 ммоль) и B2Pin2 (2,19 г, 8,62 ммоль). Полученную смесь дегазировали аргоном в течение 10 минут, а затем обрабатывали PdCl2(dppf)-DCM (0,140 г, 0,171 ммоль) при комнатной температуре. Затем реакционную смесь нагревали при 80°С в течение 3 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь фильтровали через слой целита. Фильтрат разбавляли водой (50 мл) и ЕА (50 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, элюируя 0-40% ЕА в гексане, с получением указанного в заголовке соединения (0,550 г). ЖХ-МС (Способ С) (ИЭР+), m/z 511,25 (М+Н)+, 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,48 (с, 1Н), 8,71 (с, 1Н), 8,43 (с, 1Н), 7,65 (с, 1Н), 7,42 (д, J=7,48 Гц, 1Н), 6,91 (д, J=7,48 Гц, 1Н), 6,56 (т, J=7,23 Гц, 1Н), 3,87 (с, 3H), 3,21-3,29 (м, 2Н), 3,04 (тд, J=1,13, 15,46 Гц, 1Н), 2,60-2,76 (м, 3H), 1,64-1,72 (м, 1Н), 1,16 (с, 6Н), 1,07 (с, 6Н), 0,89 (т, J=6,13 Гц, 2Н).
Стадия 3: Синтез (S)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(5-(1-изопропил-4-(трифторметил)-1H-имидазал-2-ил)-2,3-дигидро-1H-инден-1 ил)-1H-пиразало[3,4-d]пиримидина (173):
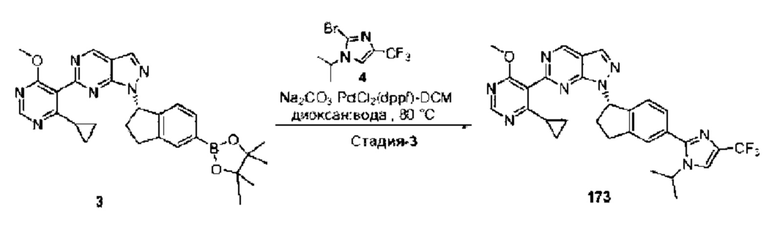
[0732] К перемешиваемому раствору (S)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2,3-дигидро-1H-инден-1-ил)-1H-пиразоло[3,4-d]пиримидина 3 (0,500 г, 0,979 ммоль) в диоксане:H2O (25:10 мл), добавляли Na2CO3 (0,155 г, 1,47 ммоль) и 2-бром-1-изопропил-4-(три фтор метил)-1H-имидазо л 4 (0,252 г, 0,979 ммоль) при комнатной температуре. Полученную смесь дегазировали аргоном в течение 10 мин, а затем обрабатывали PdCl2(dppf)-DCM (0,080 г, 0,097 ммоль) при комнатной температуре. Далее реакционную смесь дегазировали аргоном в течение 10 мин, а затем нагревали при 80°С в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли Н2О (50 мл) и ЕА (50 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикат еле, используя 10-90% ЕА в гексане в качестве элюента. Повторная очистка неочищенного соединения, полученного таким образом, с помощью препаративной ВЭЖХ (Метод D) дала 173. К перемешиваемому раствору 173 (0,090 г) в диоксане (9 мл) добавляли ЗР тиоловый диоксид кремния (0,009 г). Полученную реакционную смесь нагревали до 100°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали и упаривали при пониженном давлении с получением указанного в заголовке соединения (0,040 г). ЖХ-МС (Способ В) (ИЭР+): m/z 561,35 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,50 (с, 1Н), 8,72 (с, 1Н), 8,46 (с, 1Н), 8,16 (с, 1Н), 7,55 (с, 1Н),7,31 (д, J=7,48 Гц, 1Н), 7,07 (д, J=7,98 Гц, 1Н), 4,47 (тд J=6,67, 13,09 Гц, 1Н), 3,88 (с, 3H), 3,06 - 3,17 (м, 2Н), 2,64 - 2,82 (м, 3H), 1,68 - 1,76 (м, 1Н), 1,39 (д, J=6,48 Гц, 6Н), 1,06-1,10 (м, 2Н), 0,91 (тд, J=3,74,7,48 Гц, 2Н).
Следуюице соединения были приготовлены из соответствующих строительных блоков в соответствии со способом, описанным для Примера 173:
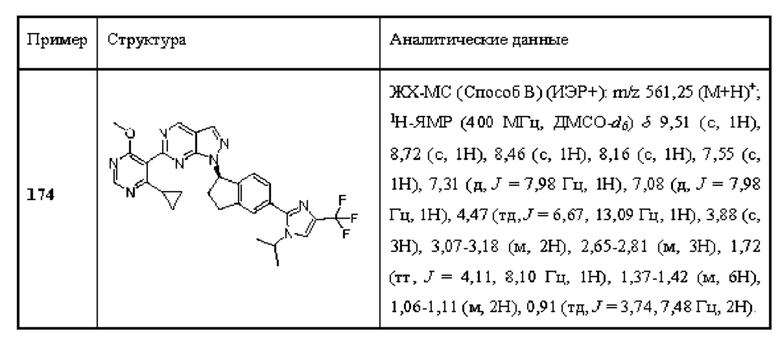
Пример 175: Синтез 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)-3-фторбензил)-3-метокси-1Н-пиразоло[3,4-d]пиримидина (175):
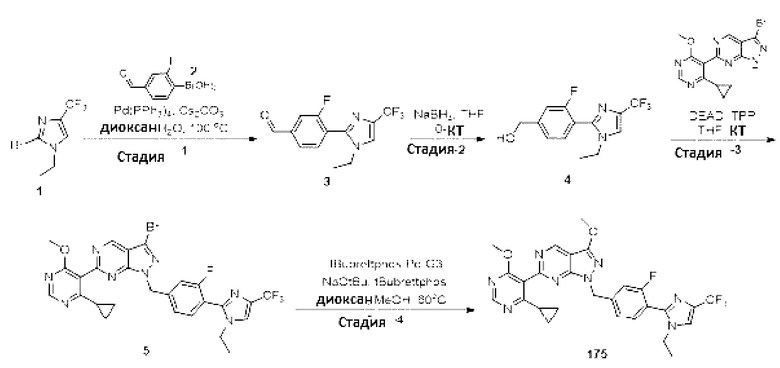
Стадия 1: Синтез 4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)-3-фторбензальдегида:
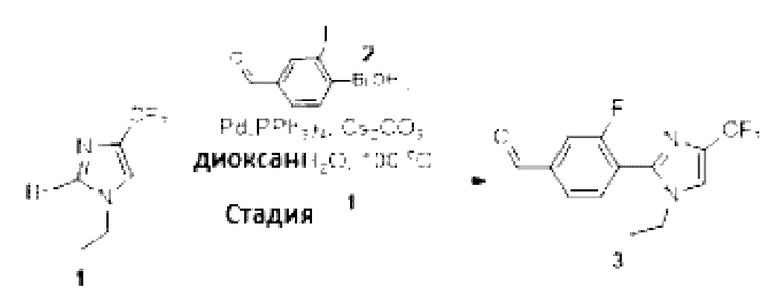
[0733] К перемешиваемому раствору 2-бром-1-этил-4-(трифторметил)-1H-имидазола 1(1,50 г, 6,17 ммоль) в диоксане: Н2О (5:1) (18 мл) добавляли Cs2CO3 (4,0 г, 12,3 ммоль) и (2-фтор-4-формилфенил)бороновую кислоту 2 (1,24 г, 7,41 итога). Полученную смесь дегазировали аргоном в течение 5 мин, а затеи обрабатывали Pd(PPh3)4 (0,709 г, 0,617 ммоль) при комнатной температуре. Реакционную смесь нагревали в запаянной пробирке при 100°С в течение 16 часов. За ходом реакции следили с помощью ТСХ. Посте завершения реакционную смесь охлаждали до комнатной температуры, разбавляли водой (50 мл) и экстрагировали ЕА (2 × 60 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженной давлении. Неочищенное соединение очищали с помощью силикагеля, используя 10-30% ЕА в гексане, с получением указанного в заголовке соединения (0,400 г). ЖХ-МС (Способ В) (ИЭР+): m/z 286,9 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 10,09 (с, 1Н), 8,19 (с, 1Н), 7,92 (д, J=7,83 Гц 2Н), 7,81-7,87 (м, 1Н), 3,94 (к, J=7,34 Гц, 2Н), 1,29 (т, J=7,34 Гц 3H).
Стадия 2: Синтез (4-(1-этил-4-(трифторметил)-1Н-эмидазол-2-ил)-3-фторфенил)метанола:
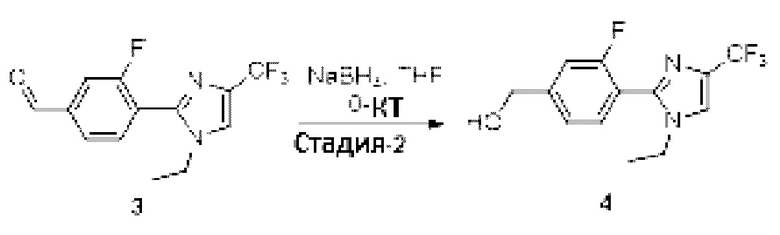
[0734] К перемешиваемому раствору 4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)-3-фторбензальдегида 3 (0,400 г, 1,398 ммоль) в метаноле (5 мл) при 0°С добавлял и боргидрид натрия (0,053 г, 1,398 ммоль). Реакционной смеси давали нагреться до комнатной температуры, а затем перемешивали в течение 2 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (10 мл) и упаривали при пониженном давлении. Оставшийся остаток растворяли в воде (20 мл) и экстрагировали ЕА (2 × 30 мл). Объединенный органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-50% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (0,250 г). ЖХ-МС (Способ В) (ИЭР+): m/z 289,0 (М+Н)+, 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,09 (с, 1Н), 7,51 (т, J=7,33 Гц, 1Н), 7,28-7,34 (м, 2H, 5,45 (уш. с, 1Н), 4,60 (уш. с, 2Н), 3,88 (к, J=7,01 Гц, 2Н), 1,26 (т, J=7,09 Гц, 3H).
Стадия 3: Синтез 3-бром-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)-3-фторбензил)-1H-пиразало[3,4-d]пиримидина:
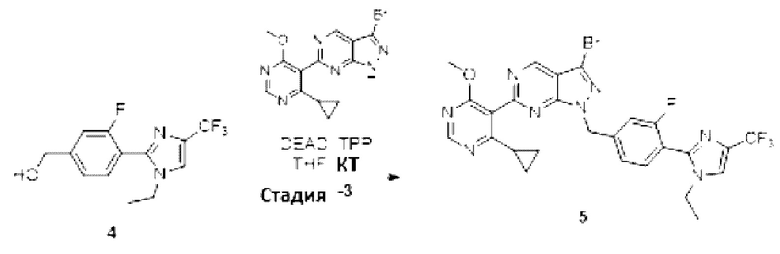
[0735] К перемешиваемому раствору (4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)-3-фторфенил)метанола 4 (0,250 г, 0,868 ммоль) в ТГФ (5 мл) добавляли 3-бром-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразапо [3,4-d]пиримидин (Промежуточный продукт 2, Пример 170) (0,301 г, 0,868 ммоль), DEAD (0,226 г, 1.302 ммоль) и ТРР (0.341 г, 1.302 ммоль). Реакционную смесь перемешивали при комната ой температуре в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили водой (20 мл) и экстрагировали ЕА (2 × 20 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, элюируя 0-40% ЕА в гексане, названном в заголовке соединением (0,200 г). ЖХ-МС (Способ В) (ИЭР+): m/z 619,05 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,45 (с, 1Н), 8,72 (с, 1Н), 8,09 (с, 1Н), 7,55 (т, J=7,83 Гц, 1H), 7,38 (д, J=10,76 Гц, 1Н), 7,28 (д, J=6,85 Гц, 1Н), 5,81 (с, 2Н), 3,86 (с, 3H), 3,84-3,85 (м, 2Н), 1,72 (тд, J=4,03, 7,58 Гц, 1Н), 1,25 (т, J=7,34 Гц, 3H), 1,04-1,09 (м, 2Н), 0,85 (дд, J=2,93, 7,83 Гц, 2Н).
Стадия 4: Синтез 6-(4-циклопропил-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)-3-фторбензил)-3-метокси-1H-пиразоло[3,4-d]пиримидина (175):
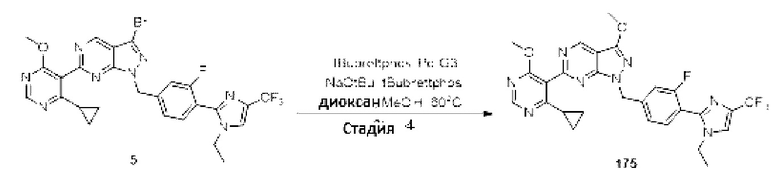
[0736] К перемешиваемому раствору 3-бром-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)-3-фторбензил)-1H-пиразоло [3,4-d]пиримидина 5 (0,180 г, 0,292 ммоль) в диоксане:H2O (4:1) (12,5 мл), добавляли трет-бутоксид натрия (0,042 г, 0,438 ммоль), а затем метанол (2,5 мл). Полученную смесь дегазировали аргоном в течение 15 минут, затем обрабатывали трет-бутил-бреттфос-Pd-G3 (0,024 г, 0,029 ммоль) и трет-бутил-бреттфос (0,028 г, 0,058 ммоль) при комнатной температуре. Затем реакционную смесь нагревали в запаянной пробирке при 60°С в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, разбавляли водой (20 мл) и экстрагировали ЕА (2 × 60 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 1-5% метанол в DCM в качестве элюента. Полученный таким образом продукт суспендировали с диоксидом кремния PS-Thiol в ТГФ при 100°С в течение 2 часов, затем фильтровали. Фильтрат упаривали в вакууме, получая указанное в заголовке соединение (0,025 г). ЖХ-МС (Способ С) (ИЭР+): m/z 569,15 (М+Н)+; 1H-ЯМР (400 МГц, CD3OD) δ 9,21 (с, 1H), 8,63 (с, 1H), 7,84 (с, 1H), 7,50 (т, J=7,48 Гц, 1H), 7,29-7,36 (м, 2Н), 5,63 (с, 2Н), 4,13-4,18 (м, 3H), 3,93-3,97 (м, 2Н), 3,92 (с, 3H), 1,70 (дdц, J=4,49, 8,10, 12,34 Гц, 1H), 1,32 (т, J=7,23 Гц, 3H), 1,14-1,19 (м, 2Н), 0,88-0,94 (м, 2Н).
Следующие соединения были получены из соответствующих строительных блоков и реагентов в соответствии со способом, описанным для Примера 175:
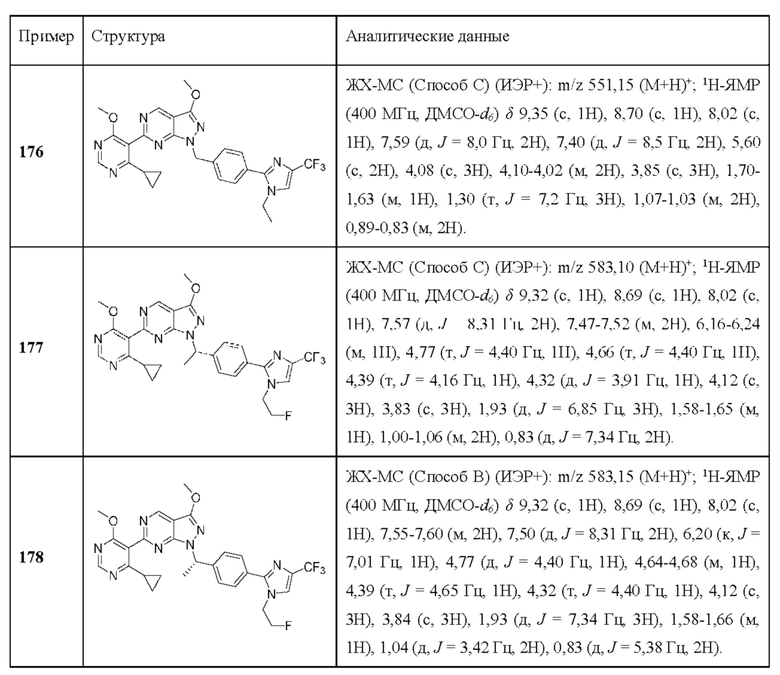
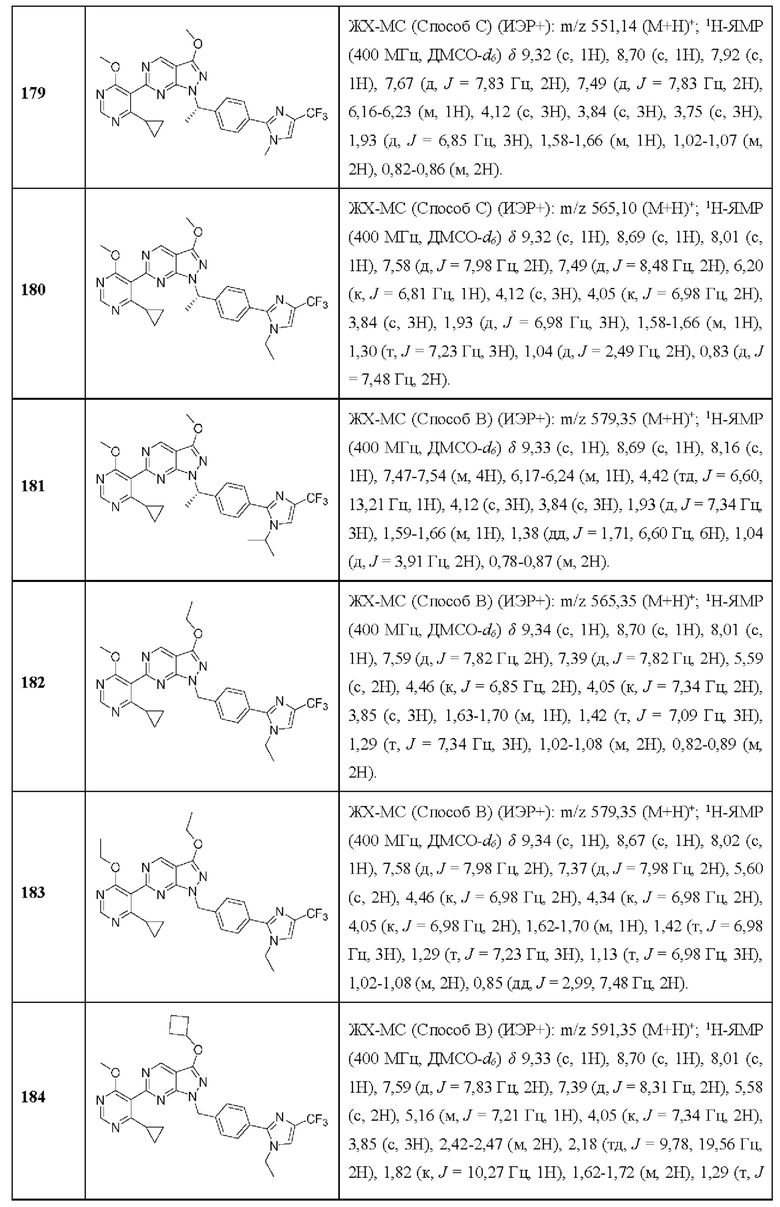
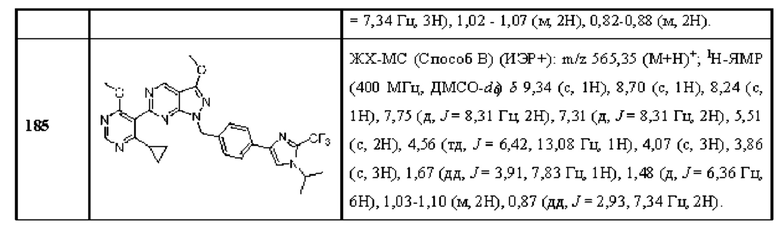
Пример 186: Синтез 3-(азетидин-1-ил)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)-1Н-пиразоло[3,4-d]пиримидина (186):
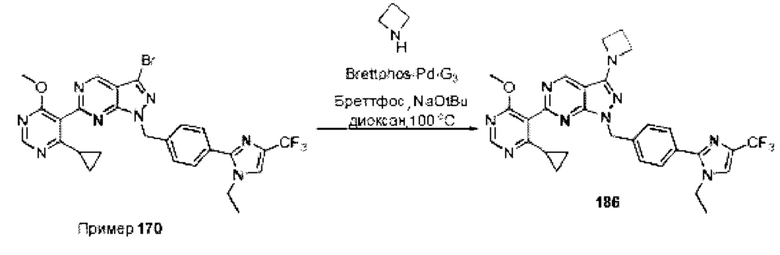
[0737] К перемешиваемому раствору 3-бром-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметип)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина 170 (0,150 г, 0,250 ммоль) и азетидина (0,1 43 г, 2,51 ммоль) в диоксане (6 мл), добавляли NaOtBu (0,036 г, 0,38 ммоль) и смесь дегазировали аргоном в течение 10 мин. К полученной реакционной смеси добавляли Brettphos (0,027 г, 0,050 ммоль) и Brettphos-Pd-G3 (0,023 г, 0,025 ммоль) при комнатной температуре. Полученную смесь дегазировали аргоном в течение 5 мин, а затем нагревали при 100°С в течение 2 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли водой (100 мл) и экстрагировали ЕА (2 × 100 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, элюируя 0-50% ЕА в н-гексане. Полученный продукт (0,080 г) растворяли в диоксане (8 мл) и обрабатывали диоксидом кремния PS-Thiol (0,008 г). Полученную смесь нагревали до 100°С в течение 2 часов, затем охлаждали до комнатной температуры, фильтровали и упаривали при пониженном давлении для удаления металлических примесей. Полученное таким образом твердое вещество затем очищали препаративной ВЭЖХ (Метод В) с получением указанного в заголовке соединения (0,035 г). ЖХ-МС (Способ В) (ИЭР+): m/z 576,30 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,21 (с, 1Н), 8,69 (с, 1Н), 8,01 (с, 1Н), 7,58 (д, J=8,31 Гц, 2Н), 7,37 (д, J=7,83 Гц, 2Н), 5,52 (с, 2Н), 4,17 (т, J=7,34 Гц, 4Н), 4,02-4,09 (м, 2Н), 3,85 (с, 3H), 2,42-2,47 (м, 2Н), 1,62-1,71 (м, 1Н), 1,30 (т, J=7,09 Гц, 3H), 1,02-1,07 (м, 2Н), 0,86 (дд, J=3,18, 7,58 Гц, 2Н).
Пример 187: Синтез 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-3-метил-1Н-пиразоло[3,4-d]пиримидина (187):
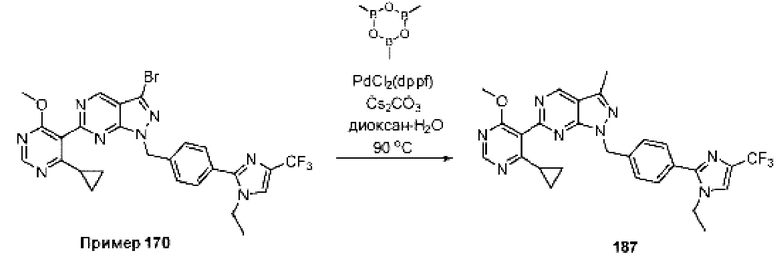
[0738] К перемешиваемому раствору 3-бром-6-(4-циклопропил-6-метоксипир (1-этил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1H-пиразоло-[3,4-d]пиримидина 170 (11,200 г, 0,334 ммоль) и триметилбороксина (0,031 г, 1,00 ммоль) в диоксане (10 мл) и воде (5 мл) добавляли Cs2CO3 (0,162 г, 0,5016 ммоль) при комнатной температуре. Смесь дегазировали аргоном в течение 15 мин, а затем обрабатывали Pd(dppf)Cl2.DCM (0,054 г, 0,066 ммоль) при комнатной температуре. Затем реакционную смесь нагревали в запаянной пробирке при 90°С в течение 1 часа. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, разбавляли ЕА (20 мл) и водой (10 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. К перемешиваемому раствору неочищенного соединения (0,70 г) в диоксане (7 мл) добавляли SP тиоповый диоксид кремния (0,007 г). Полученную реакционную смесь нагревали до 100°С в течение 1 часа, охлаждали до комнатной температуры, фильтровали и упаривали при пониженном давлении. Неочищенное соединение затем очищали препаративной ВЭЖХ (Метод D) с получением указанного в заголовке соединения (0,050 г). ЖХ-МС (Способ С) (ИЭР+): m/z 535,15 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,47 (с, 1Н), 8,71 (с, 1Н),8,02(с, 1Н),7,59(д J=8,48 Гц, 2Н), 7,40 (д, J=7,98 Гц, 2Н), 5,69 (с, 2Н), 4,05 (к, J=6,92 Гц, 2Н), 3,85 (с, 3H), 2,63 (с, 3H), 1,60 - 1,68 (м, 1Н), 1,29 (т, J=7,23 Гц, 3H), 1,03-1,07 (м, 2Н), 0,85 (дд, J=3,24, 7,23 Гц, 2Н).
Пример 188: Синтез (S)-(1-(4-(5-бром-1-метил-4-(трифторметил)-1H-имидазол-2-ил)фенил)этил)-6-(4-циклопропил-6-метоксипиридин-5-ил)-3-метокси-1H-пиразоло[3,4-d]пиримидина (188):
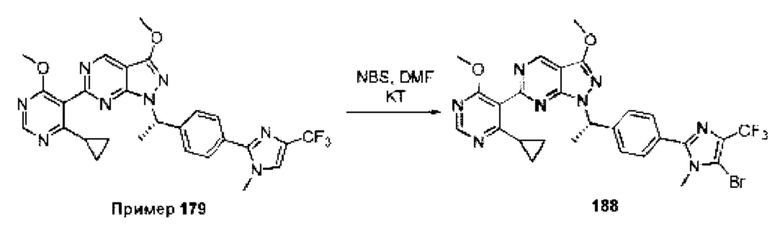
[0739] К перемешиваемому раствору (S)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-3-метокси-1-(1-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)фенил)этил)-1H-пиразоло[3,4-d]пиримидина 179 (0,220 г, 0,400 ммоль) в ДМФА(4 мл) добавляли NBS (0,142 г, 0,800 ммоль) при комнатной температуре и реакционную смесь перемешивали в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении, разбавляли водой (10 мл) и экстрагировали ЕА (2 × 20 мл). Объединенный органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикат еле, используя 0-40% ЕА в н-гексане в качестве элюента, с последующей дополнительной хиральной очисткой (Метод А) с получением указанного в заголовке соединения (0,035 г). ЖХ-МС (Способ В) (ИЭР+): m/z 629,15 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,32 (с, 1 Н), 8,69 (с, 1Н), 7,63 (д, J=8,31 Гц, 2Н), 7,51 (д, J=8,31 Гц, 2H), 6,20 (к, J=7,17 Гц, 1Н), 4,12 (с, 3H), 3,84 (с, 3H), 3,65 (с, 3H), 1,93 (д, J=7,34 Гц 3H), 1,58 - 1,67 (м, 1Н), 1,04 (д, J=3,42 Гц 2Н), 0,84 (дд, J=2,20, 7,58 Гц 2Н).
Следующие соединения были получены из соответствующих строительных блоков в соответствии со способом, описанным для: Примера 188:
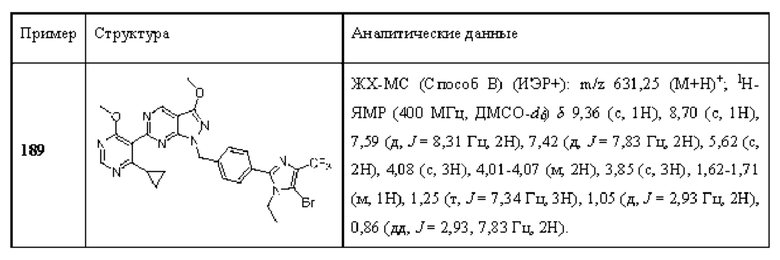
Пример 190: Синтез 6-(4-циклопротил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-3-(метилтио)-1H-пиразоло[3,4-d]пиримидина (190):
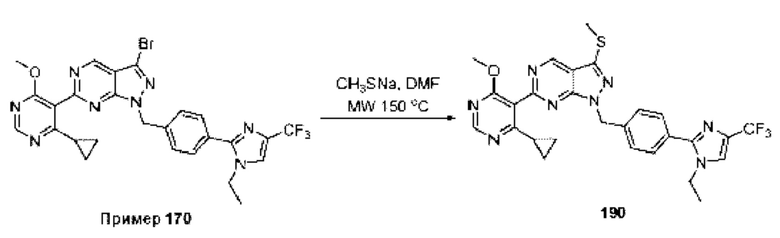
[0740] К перемешиваемому раствору 3-бром-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил) бензил)-1H-170 (0,200 г, 0,334 ммоль) в DMF (4 мл), добавляли метантиолат натрия (0,047 г, 0,67 мысль) при комнатной температуре. Реакционную смесь дополнительно нагревали в микроволновой печи при 150°С в течение 2 часов. Заходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли ледяной водой (50 мл) и экстрагировали ЕА (2 × 100 мл). Объединенный органический слой промывали ледяной водой (2 × 150 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, элюируя 0-40% ЕА в н-гексане, с последующей дополнительной очисткой с использованием препаративной ВЭЖХ (Метод В) с получением указанного в заголовке соединения (0,025 г ЖХ-МС (Способ В) (ИЭР+): m/z 567,30 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,47 (с, 1Н),8,71 (с, 1Н),8,02(с, 1Н), 7,60 (д, J=8,31 Гц, 2Н), 7,42 (д, J=8,31 Гц 2Н), 5,73 (с, 211), 4,05 (к, J=7,34 Гц 2Н), 3,85 (с, 3H), 2,72 (с, 3H), 1,64-1,71 (м, 1Н), 1,29 (т, J=7,34 Гц, 3H), 1,02-1,07 (м, 2Н), 0,85 (дд, J=3,18, 7,58 Гц, 2Н).
Пример 191: Синтез 6-(4-циклопропил-6-метоксипиридин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-3-(метилсульфонил)-1Н-пиразоло[3,4-d]пиримидина (191):
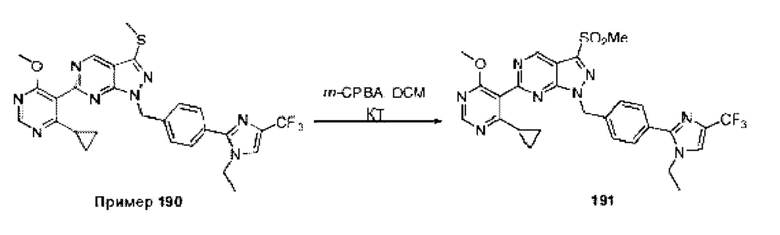
[0741] К охлажденному льдом раствору 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазоп-2-ип)бензил)-3-(метилтио)-1Н-пиразоло[3,4-d]пиримидина 190 (0,150 г, 0,265 ммоль) в DCM (15 мл) добавляли м-СРВА (0,182 г, 1,06 ммоль). Полученной смеси давали нагреться до комнатной температуры и дополнительно перемешивали в течение 2 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли в одой (100 мл) и экстрагировали DCM (2 × 100 мл). Объединенный органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-50% ЕА в н-гексане, с последующей повторной очисткой препаративной ВЭЖХ (Метод В) с получением указанного в заголовке соединения (0,030 г). ЖХ-МС (Способ В) (ИЭР+): m/z 599,20 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,67 (с, 1Н), 8,74 (с, 1Н), 8,02 (с, 1Н), 7,62 (д, J=8,31 Гц, 2Н), 7,50 (д, 3=8,31 Гц, 2Н), 5,93 (с, 2Н), 4,05 (к, J=7,17 Гц, 2Н), 3,86 (с, 3H), 3,53 (с, 3H), 1,69-1,76 (м, 1Н), 1,30 (т, J=7,09 Гц, 3H), 1,06-1,10 (м,2Н), 0,86 (дд, J=3,42, 7,83 Гц 2Н).
Пример 192: Синтез 6-(4-циклопропил-6-метоксипиридин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-3-((4-метоксибензил)тио)-1Н-пиразоло[3,4-(2]пиримидина (192):
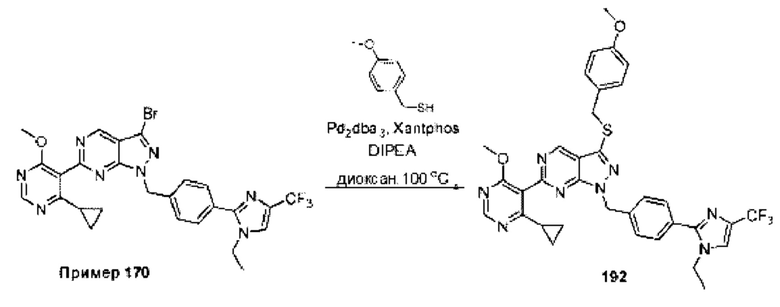
[0742] К перемешиваемому раствору 3-бром-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазоп-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина 170 (0,300 г, 0,501 ммоль) и (4-метоксифенип)метантиола (0,115 г, 0,752 ммоль) в диоксане (10 мл), добавляли DIPEA (0,620 мл, 3,51 ммоль). Полученный раствор дегазировали аргоном в течение 10 мин, затем обрабатывали ксантфосом (0,020 г, 0,035 ммоль) и Pd2(dba)3 (0,032 г, 0,035 ммоль) при комнатной температуре. Смесь дополнительно дегазировали аргоном в течение 5 мин, а затем нагревали при 100°С в течение 16 часов. За кодом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли водой (100 мл) и экстрагировали ЕА (2 × 100 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-30% ЕА в н-гексане в качестве элюента, с получением указанного в заголовке соединения (0,300 г). ЖХ-МС (Способ В) (ИЭР+): m/z 673,30 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,23 (д, J=1,96 Гц, 1Н), 8,70 (д, J=1,96 Гц, 1Н), 8,03 (с, 1Н), 7,62 (д J=6,85 Гц 2H),7,41 (д, J=7,34 Гц 2Н), 7,22 (д J=7,34 Гц 2Н), 6,74 (д J=6,85 Гц 2Н), 5,74 (с, 2Н), 4,40 (с, 2Н), 4,03-4,11 (м, 2Н),3,85 (с, 3H),3,65 (с, 3H), 1,59-1,67 (м, 1Н), 1,27-1,33 (м,3H), 1,02-1,07 (м, 2Н), 0,83-0,88 (м, 2Н).
Пример 193: Синтез 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-((метилсульфонил)метил)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (193):
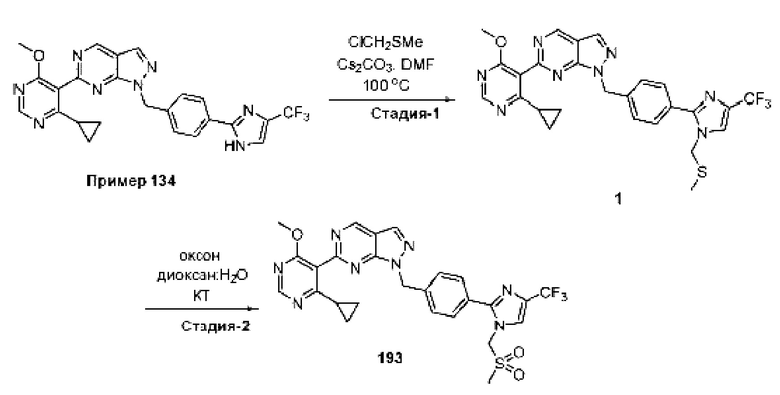
Стадия 1: Синтез 6-(4-циклопропил-6-метоксипиридин-5-ил)-1-(4-(1((метилтио)метил)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1H-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина:
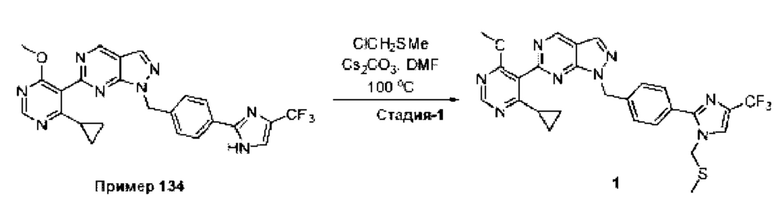
[0743] К перемешиваемому раствору 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина 134 (0,200 г, 0,406 ммоль) и (хлорметил(метил) сульфана (0,078 г, 0,813 ммоль) в ДМФА (2 мл) добавляли карбонат цезия (0,3 97 г, 1,219 ммоль) при комнатной температуре. Реакционную смесь нагревали при 100°С в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли ледяной водой (100 мл) и экстрагировали ЕА (2 × 100 мл). Объединенный органический слой промывали водой (3 × 100 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали препаративной ВЭЖХ (Метод В) с получением указанного в заголовке соединения (0,080 г). ЖХ-МС (Способ В) (ИЭР+): m/z 553,20 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,51 (с, 1Н), 8,71 (с, 1Н), 8,52 (с, 1Н), 8,06 (с, 1Н), 7,67 (д, J=8,31 Гц 2Н), 7,42 (д, J=7,83 Гц 2Н), 5,78 (с, 2Н), 5,22 (с, 2Н),3,85(с,3H), 2,01 (с,3H), 1,61-1,69 (м, 1Н), 1,03-1,07 (м. 2Н),0,86 (д, J=4,40 Гц 2Н).
Стадия 2: Синтез 6-(4-циклопропил-6-метоксипиридин-5-ил)-1-(4-(1-((метилсульфонил(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (193):
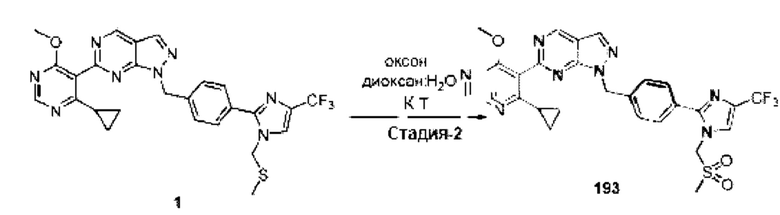
[0744] К перемешиваемому раствору 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-((метилтио)метил)-4-(трифторметил)-1H-имидазоп-2-ил)бензил)-1H-пиразопо[3,4-d]пиримидина 1 (0,070 г, 0,126 ммоль) в диоксане: Н2О (3:1 мл), добавляли оке он (0,233 г, 0,380 ммоль) при комнатной температуре. Смесь перемешивали в течение 1 ч, и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли в одой (50 мл) и экстрагировали ЕА (2 × 50 мл). Объединенный органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-5% метанола в DCM, а затем повторно очищали с помощью препаративной ВЭЖХ (Метод В) с получением указанного в заголовке соединения (0,018 г). ЖХ-МС (Способ В) (ИЭР+): m/z 585,10 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,51 (с, 1Н), 8,71 (с, 1Н), 8,52 (с, 1Н), 7,99 (с, 1Н), 7,69 (д, J=7,98 Гц, 2Н), 7,44 (д, J=7,98 Гц, 2Н), 5,79 (с, 2Н), 5,63 (с, 2Н), 3,85 (с, 3H), 3,04 (с, 3H), 1,63-1,70 (м, 1Н), 1,03-1,07 (м, 2Н), 0,84-0,92 (м, 2Н).
Пример 194: Синтез (2-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-этил-1H-имидазол-4-ил)метанола (194):
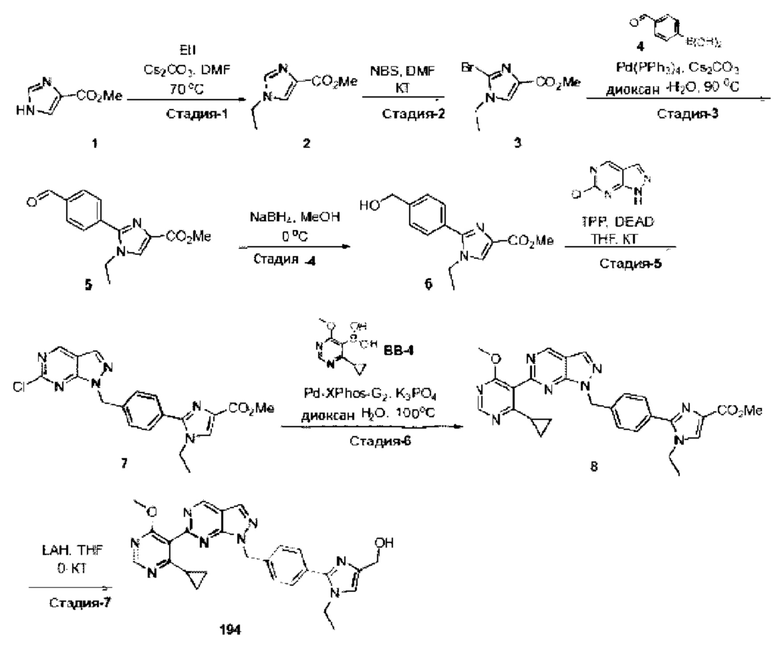
Стадия 1: Синтез метил 1-этил-1Н-имидазол-4-кар6оксилата:
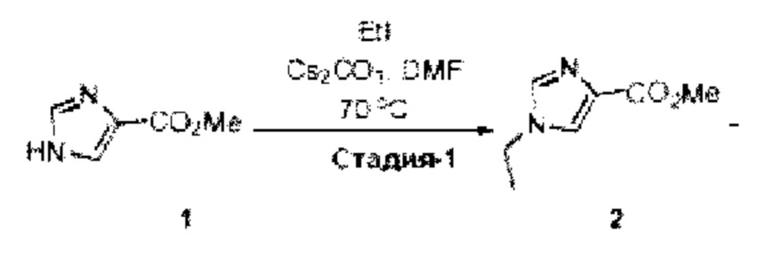
[0745] К перемешиваемому раствору метил 1Н-имидазол-4-карбоксилата 1 (15,0 г, 119,04 ммоль) в ДМФА (300 мл) добавляли Cs6CO3 (96,7 г, 297,61 ммоль) и этилиодид (27,8 г, 178,56 ммоль) при комнатной температуре. Реакционную смесь нагревали при 70°С в течение 12 часов, и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры и фильтровали через слой целита Фильтрат упаривали при пониженном давлении и неочищенное соединение очищали хроматографией на силикат еле, используя 20-90% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (6,0 г). 1H-ЯМР (400 МГц, ДМСО-d6) δ 7,96 (с, 1Н), 7,7 8 (с, 1Н), 4,03 (к, J=7,15 Гц, 2Н), 3,73 (с, 3H), 1,34 (т, J=7,23 Гц, 3H).
Стадия 2: Синтез метил-2-бром-1-этил-1H-имидазол-4-карбоксилата:
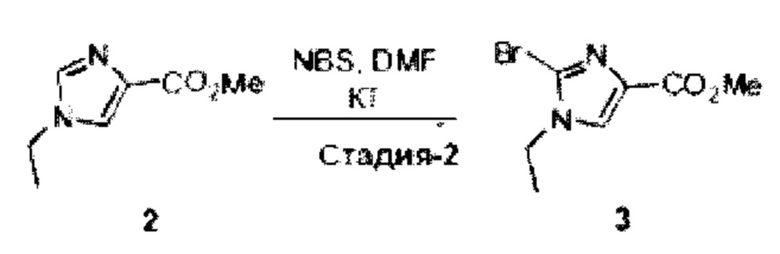
[0746] К перемешиваемому раствору метил 1-этил-1H-имидазол-4-карбоксилата 2 (1,50 г, 9,740 ммоль) в ДМ ФА (100 мл) при комнатной температуре добавляли NBS (1,73 г, 9,740 ммоль). Полученную смесь перемешивали в течение 16 ч, и за ходом реакции следили с помощью ТСХ. После завершения реакционную смесь разбавляли ледяной водой (100 мл) и экстрагировали ЕА (2 × 100 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-5% метанол в DCM в качестве элюента, с получением указанного в заголовке соединения (1,50 г). ЖХ-МС (Способ В) (ИЭР+): m/z 234,95 (М+Н)+.
Стадия 3: Синтез метил 1-этил-2 (4-формилфенил)-1H-имидазол-4-карбоксилата:
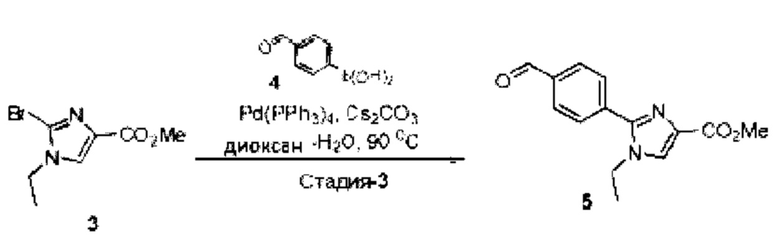
[0747] К перемешиваемому раствору метил 2-бром-1-этил-1Н-имидазол4-карбоксилата 3 (1,00 г, 4,291 ммоль) в диоксане:Н2О (90: 30 мл) добавляли Cs2CO3 (3,42 г, 10,72 ммоль) и (4-формилфенил)бороновую кислоту 4 (0,77 г, 5,149 ммоль). Полученную смесь дегазировали аргоном в течение 30 мин, а затем обрабатывали Pd(PPh3)4 (0,097 г, 0,085 ммоль) при комнатной температуре. Затем реакционную смесь нагревали в запаянной пробирке при 90°С в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры, разбавляли водой (50 мл) и экстрагировали ЕА (2 × 50 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 20-80% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (1,0 г). ЖХ-МС (Способ В) (ИЭР+): m/z 259 (М+Н)+.
Стадия 4: Синтез метил 1-этил-2-(4-(гидроксиметил)фенил)-1Н-имидазол-4-карбоксилата:
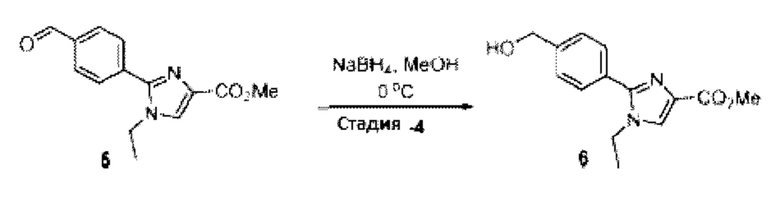
[0748] К перемешиваемому раствору метил 1-этил-2-(4-формилфенил)-1H-иминазол-4-карбоксилат а 5 (1,0 г, 3,375 ммоль) в метаноле (40 мл) при 0°С добавляли боргидрид натрия (0,073 г, 1.937 ммоль). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 30 минут. За ходом реакции следили с помощью ТСХ. По еле завершения реакционную смесь гасили водой (10 мл) и упаривали при пониженном давлении. Оставшийся остаток растворяли в воде (20 мл) и экстрагировали ЕА (2 × 100 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении с получением указанного в заголовке соединения (0,960 г). ЖХ-МС (Способ в) (ИЭР+): m/z 261 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 7,89 (с, 1Н), 7,39-7,44 (м, 2Н), 7,31-7,36 (м, 2Н), 5,29 (т, J=5,73 Гц, 1Н),4,57 (д, J=5,49 Гц, 2Н), 3,82 (к, J=6,98 Гц, 2Н), 3,57 (с, 3H), 1,12 (т, J=7,23 Гц, 3H).
Стадия 5: Синтез метил 2-(4-((6-хлор-1H-пиразоло [3,4-d] пиримидин-1-ил)метил) фенил)-1 -этил-1Н-имидазол-4-карбоксилата:
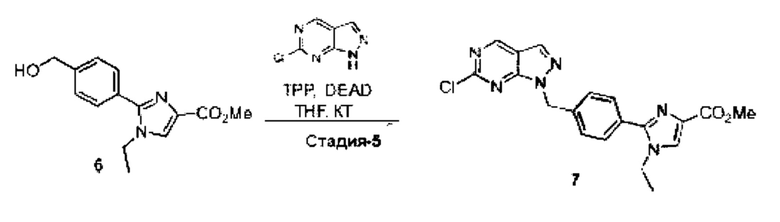
[0749] К перемешиваемому раствору метил 1-этил-2-(4-(гидроксиметил)фенил)-1H-имидазол-4-карбоксилата 6 (0,900 г, 3,461 ммоль) в ТГФ (20 мл) при 0°С добавляли 6- хлор-1Н-пиразоло[3,4-]пиримидин (0,533 г, 3,461 ммоль), DEAD (0,887 г, 5,192 ммоль) и ТРР (1,33 г, 5,192 ммоль). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 1 часа. За ходом реакции слепили с помощью ТСХ После завершения реакционную смесь гасили водой (20 мл) и экстрагировали ЕА (2 × 100 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикат еле, используя 20-70% ЕА в гексане в качестве элюента, с получением указанного в заголовке соединения (1,20 г). ЖХ-МС (Способ в) (ИЭР+): m/z 396,9 (М+Н)+.
Стадия б: Синтез метил 2-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1H-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-этил-1H-имидазол-4-карбоксилата:
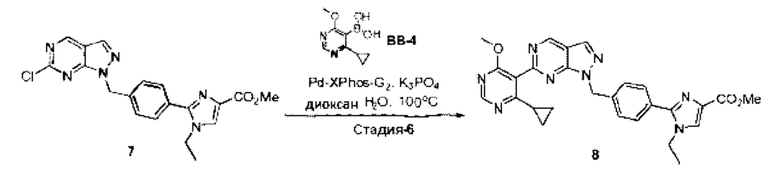
[0750] К перемешиваемому раствору метил 2-(4-((6-хлор-1H-пиразоло [3,4-d]пиримидин-1-ил)метил)фенил)-1-этил-1H-имидазол- -карбоксилата 7 (1,2 г, 3,03 ммоль) в диоксане: Н2О (20:4 мл) добавляли K3PO4 (0,320 г, 1,51 ммоль) и (4-циклопропил-6-метоксипиримидин-5-ил)бороновую кислоту (BB-4) (1,59 г, 7,58 ммоль) при комнатной температуре. Полученную смесь дегазировали аргоном в течение 30 мин, затем обрабатывали Х-фос (0,288 г, 0,606 ммоль) и Х-фос-Pd-G2 (0,118 г, 0,1515 ммоль) при комнатной температуре. Затем смесь дегазировали аргоном в течение 10 мин. Затем реакционную смесь нагревали в закрытой пробирке при 100°С в течение 16 часов. За ходом реакции слепили с помощью ТСХ. По еле завершения реакционную смесь гасили Н2О (20 мл) и экстрагировали ЕА (2×100 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-80% ЕА в гексане в качестве элюента, с последующим упариванием в вакууме с получением промежуточного соединения 8 (1,50 г). К перемешиваемому раствору 8 (1,50 г) в диоксане (15 мл) добавляли SP тиоловый диоксид кремния (0,150 г). Полученную реакционную смесь нагревали до 100°С в течение 2 часов. Затем реакционную смесь охлаждали до комнатной температуры, фильтровали и упаривали при пониженном давлении с получением указанного в заголовке соединения (1,20 г). ЖХ-МС (Способ В) (ИЭР+): m/z 511,30 (М+Н)+; 1Н-ЯМР(400 МГц, ДМСО-d6) δ 9,51 (с, 1Н), 8,71 (с, 1Н), 8,53 (с, 1Н), 7,88 (с, 1Н), 7,33-7,39 (м, 4Н), 5,78 (с, 2Н), 3,85 (с, 3H), 3,78 (к, J=7,01 Гц, 2Н), 3,54 (с, 3H), 1,65 (д, J=4,40 Гц, 1Н), 1,03-1,12 (м, 5Н), 0,85 (дд, J=2,93,4,40 Гц, 2Н).
Стадия 7: Синтез (2-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразолл[3,4-d]пиримидин-1-ил)метил)фенил)-1-этил-1Н-имидазол-4-ил)метанола (194):
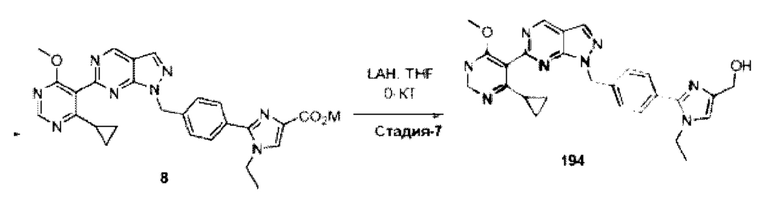
[0751] К перемешиваемому раствору метил 2-(4-((6-(4-циклопропил-6-метокспиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-этил-1Н-имидазол 8 (0,070 г, 0,1372 ммоль) в ТГФ (20 мл) при 0°С медленно добавляли 1,0 М раствор LAH в ТГФ (0,260 мл, 0,260 ммоль). Реакционной смеси давали нагреться до комнатной температуры, а затем перемешивали в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили насыщенным раствором Na2SO4 (10 мл) и фильтровали через целит. Фильтрат упаривали при пониженном давлении, и полученное неочищенное соединение очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (0,025 г). ЖХ-МС (Способ В) (ИЭР+): m/z 483,30 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,51 (с, 1Н), 8,71 (с, 1Н), 8,52 (с, 1Н), 7,69 (с, 1Н), 7,36-7,43 (м, 4Н), 5,76 (с, 2Н), 4,71 (т, J=5,24 Гц, 1Н), 4,14 (д, J=4,99 Гц, 2Н), 3,87-3,93 (м, 2Н), 3,85 (с, 3H), 1,66 (д, J=3,99 Гц, 1Н), 1,11 (т, J=6,98 Гц, 3H), 1,04-1,07 (м, 2Н), 0,82-0,88 (м, 2Н).
Следующее соединение было получено из соответствующих строительных блоков в соответствии со способом, описанным для Примера 194:
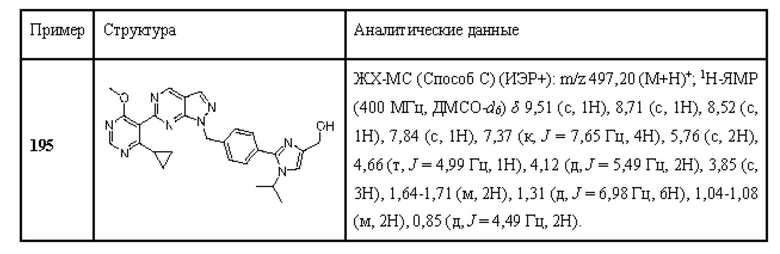
Пример 196: Синтез 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(4-(дифторметил)-1-этил-1H-имндазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидина (196):
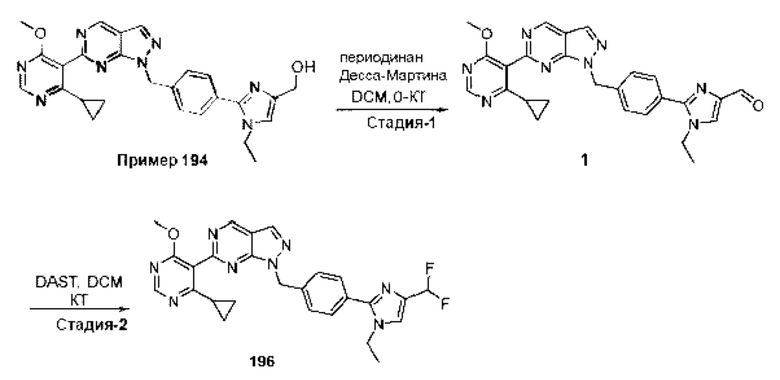
Стадия 1: Синтез 2-(4-((6-(4-циклопропил-6-метоксипиримидин-5ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-этил-1H-имидазол-4-карбальдегида:
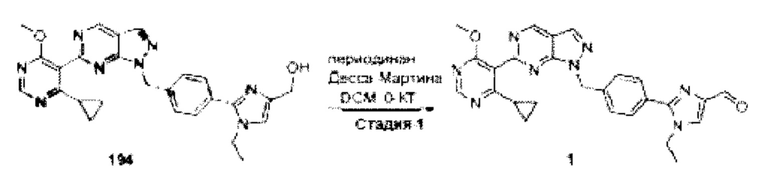
[0752] К охлажденному льдом раствору (2-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1H-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-этил-1H-имидахол-4-ил)метанола (194) (0,250 г, 0,518 ммоль) в DCM (40 мл), добавляли периодинан Десса-Мартина (0,330 г, 0,778 ммоль). Реакционной смеси давали нагреться до комнатной температуры, а затем перемешивали в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили насыщенным раствором NaHCO3 (10 мл) и насыщенным раствором тиосульфата натрия (10 мл) и перемешивали в течение 20 минут. Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле с использованием 0-5% метанола в DCM в качестве элюента с получением указанного в заголовке соединения (0,220 г). ЖХ-МС (Способ С) (ИЭР+): m/z 481,30 (М+Н)+; 1Н-ЯМР (400 МГц, CDCl3) δ 9,72 (с, 1Н), 9,36 (с, 1H), 8,69 (с, 1Н), 8,26 (с, 1Н), 7,67 (с, 1Н), 7,56 (д, J=7,98 Гц, 2Н), 7,37 (д, J=8,48 Гц, 2Н), 5,79 (с, 2Н), 3,94 (с, 3H), 3,86-3,93 (м, 2Н), 1,31 (т, J=7,23 Гц, 3H), 1,23-1,28 (м, 3H), 0,85-0,91 (м, 2Н).
Стадия 2: Синтез 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(4-(дифторметил)-1-этил-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4- d]пиримидина (196):
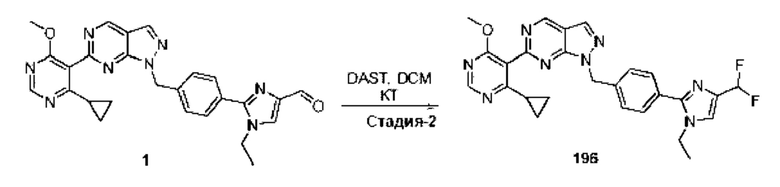
[0753] К перемешиваемому раствору 2-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1H-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-этил-1H-имидазол4-карбальдегида 1 (0,200 г, 0,416 ммоль) в DCM (20 мл) добавляли DAST (0,335 г, 2,08 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение 2 ч, и за ходом реакции следили с помощью ТСХ После завершения реакционную смесь разбавляли насыщенным раствором NaHCO3 (20 мл) и экстрагировали DCM (2 × 20 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение сначала очищали хроматографией на силикагеле, используя 0-25% ЕА в гексане в качестве элюента. Полученное таким образом нечистое соединение затем очищали препаративной ВЭЖХ (Метод В) с получением указанного в заголовке соединения (0,035 г). ЖХ-МС (Способ В) (ИЭР+): m/z 503,30 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,51 (с, 1Н), 8,71 (с, 1H), 8,53 (с, 1H), 7,90 (с, 1H), 7,39 (к, J=7,99 Гц, 4Н), 6,47-6,75 (м, 1H), 5,79 (с, 2Н), 3,86-3,90 (м, 2Н), 3,85 (с, 3H), 1,66 (тд, J=3,55, 7,58 Гц, 1H), 1,12 (т, J=7,09 Гц, 3H), 1,03-1,08 (м, 2Н), 0,84 (дд, J=2,93, 7,34 Гц, 2Н).
Следующие соединения были получены из соответствующих строительных блоков в соответствии со способом, описанным для Примера 196:
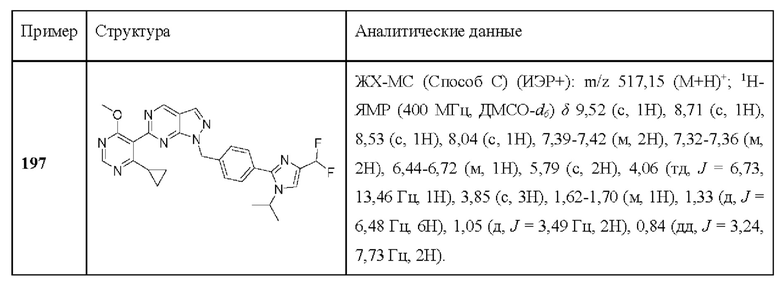
Пример 198: Синтез (1-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1H-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-3-(трифторметил)-1Н-пиразол-5-ил)метанола (198):
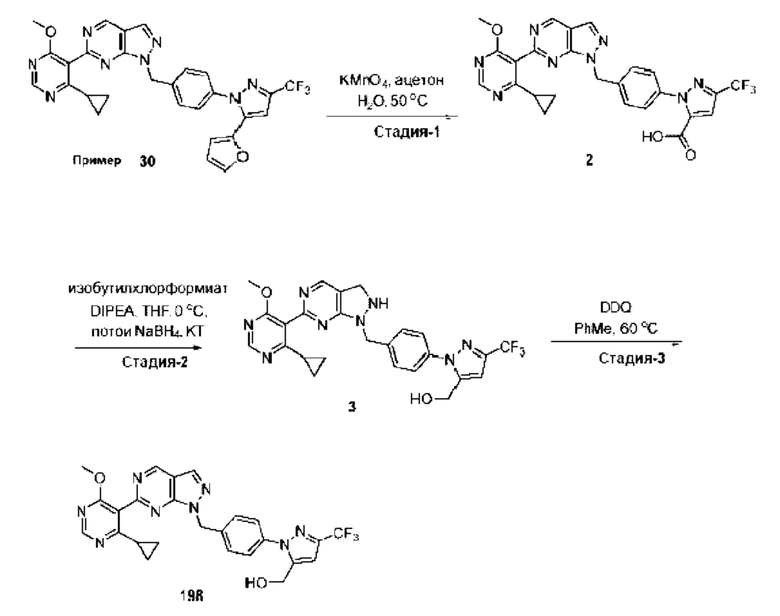
Стадия 1: Синтез 1-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-3-(трифторметил)-1H-пиразол-5-карбоновой кислоты:
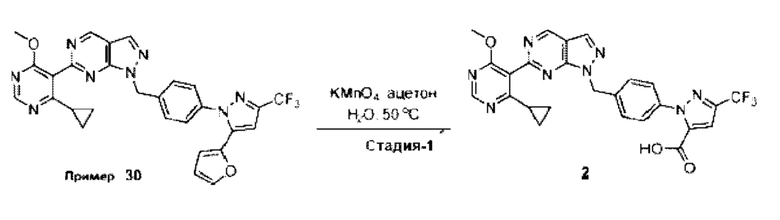
[0754] К перемешиваемому раствору 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(5-(фуран-2-ил)-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина (Пример 30) (1,00 г, 2,25 ммоль) в ацетоне:воде (2:1, 30 мл) и добавляли KMnO4 (0,707 г, 4,48 ммоль) при комнатной температуре. Затем реакционную смесь нагревали при 50°С в течение 4 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры и разбавляли Н2О (50 мл). Суспензию фильтровали, и полученный фильтрат упаривали при пониженном давлении. В одной слой промывали DCM (2 × 10 мл), и рН доводили до 5, используя 1 н. раствор HCl. Твердый осадок фильтровали, промывали водой и сушили в печи при 60°С в течение 2 часов с получением указанного в заголовке соединения (0,350 г). ЖХ-МС (Способ В) (ИЭР+): m/z 537,30 (М+Н)+; 1Н-ЯМР(400 МГц, ДМСО-d6) δ 9,51 (с, 1Н), 8,71 (с, 1Н), 8,53 (с, 1Н), 7,38-7,52 (м, 6Н), 5,80 (с, 2Н), 3,85 (с, 3H), 1,65 (д, J=3,99 Гц, 1Н), 1,02-1,07 (м, 2Н), 0,83-0,8 9 (м, 2Н).
Стадия 2: Синтез (1-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-2,3-дигидро-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-3-(трифторметил)-1H-пиразол-5-ил)метанола:
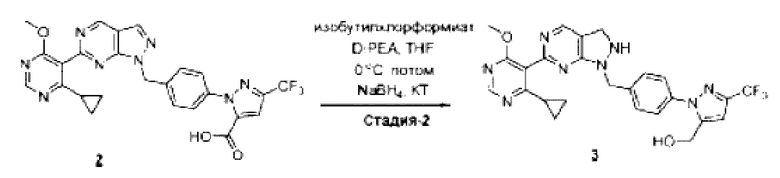
[0755] К охлажденному льдом раствору 1-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1H-пиразоло [3,4-d]пиримидин-1-ил)метил)фенил)-3-(трифторметил)-1H-пиразол-5-карбоновой кислоты 2 (0,300 г, 0,559 ммоль) в ТГФ (10 мл), добавляли DIPEA (0,145 мл, 0,838 ммоль) и изобутилхлорформиат (0,080 мл, 0,62 ммоль) по каплям.. Полученную смесь перемешивали 30 мин. К полученной реакционной смеси добавляли борогидрид натрия (0,021 г, 0,56 ммоль) одной порцией при той же температуре. Затем реакционной смеси давали нагреться до комнатной температуры и перемешивали еще 1 час. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении, затем разбавляли ЕА (20 мл) и водой (20 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикат еле, элюируя 0-10% метанолом в DCM, с получением указанного в заголовке соединения (0,050 г). ЖХ-МС (Способ В) (ИЭР+): m/z 525,30 (М+Н)+; 1Н-ЯМР(400 МГц, ДМСО-d6) δ 8,62 (с, 1H), 7,99 (с, 1Н), 7,58 (д, J=8,31 Гц, 2Н), 7,35 (д, J=8,31 Гц, 2Н), 7,15 (с, 1Н), 6,88 (с, 1Н), 5,58 (т, J=5,38 Гц, 1Н), 5,25 (с, 2Н), 4,74 (с, 2Н), 4,49 (д, J=5,38 Гц, 2Н), 3,93 (с, 3H), 2,10-2,18 (м, 1Н), 0,94-1,05 (м,4Н).
Стадия 3: Синтез (1-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1H-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-3-(трифторметил)-1H-пиразол-5-нл)метанола (198):
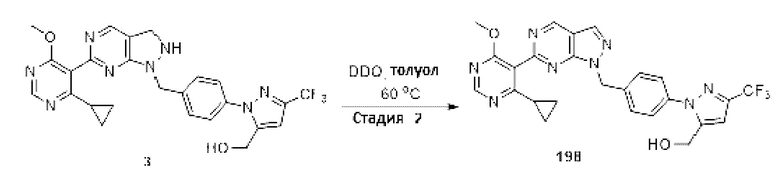
[0756] К перемешиваемому раствору (1-(4-((6-(4-циклопрогпта-6-метокс1 лшримидин-5-ип)-2,3-дигидро-1Н-пиразоло[3,4-d] пиримидин-1-ил)метил) фенил)-3-(трифторметил)-1Н-пиразол-5-ил)метанола 3 (0,040 г, 0,076 ммоль) в толуоле (4 мл), добавляли DDQ (0,026 г, 0,114 ммоль) и реакционную смесь нагревали при 60°С в течение 1 часа. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь упаривали при пониженном давлении. Очистка неочищенного соединения хроматографией на силикат еле с использованием 0-90% ЕА в гексане в качестве элюента давала нечистое соединение, которое дополнительно очищали препаративной ВЭЖХ (Метод D) с получением указанного в заголовке соединения (10,022 г). ЖХ-МС (Способ В) (ИЭР+): m/z 523,25 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,51 (с, 1Н), 3,71 (с, 1Н), 3,52 (с, 1Н), 7,61 (д, J=7,83 Гц, 2Н), 7,46 (д, J=8,31 Гц, 2Н), 6,88 (с, 1Н), 5,80 (с, 2Н), 5,57 (т, J=5,38 Гц, 1Н), 4,48 (д, J=5,38 Гц 2Н), 3,85 (с, 3H), 1,62 - 1,69 (м. 1Н), 1,02-1,07 (м, 2Н), 0,86 (дд, J=3,18,7,58 Гц 2Н).
Пример 199: Синтез 2-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-1-изопропил-1Н-имидазол-4-амина (199):
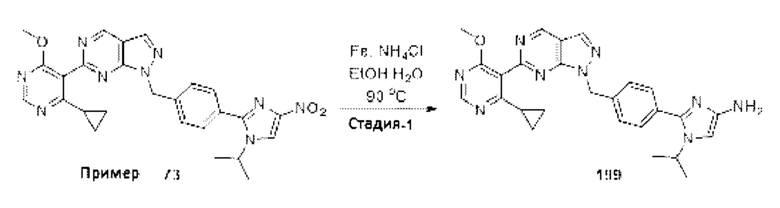
[0757] К перемешиваемому раствору 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-изопропил-4-нитро-1H-имидазоп-2-ил)бензил)-1H-пиразопо[3,4-d]пиримидина (Пример 73) (0,130 г, 0,254 ммоль) в EtOH:Н2О (4:2 мл) добавляли хлорид аммония (0,067 г, 1,27 ммоль) и порошок желез а (0,069 г, 1,27 ммоль) при комнатной температуре. Реакционную смесь нагревали в запаянной пробирке при 90°С в течение 2 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь охлаждали до комнатной температуры и упаривали при пониженном давлении. Полученный неочищенный остаток разбавляли водой (10 мл) и экстрагировали DCM (2 × 20 мл). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-5% МеОН в DCM, с получением указанного в заголовке соединения (0,550 г). ЖХ-МС (Способ В) (ИЭР+): m/z 482,05 (М+Н)+; 1Н-ЯМР (400 МГц ДМСО-d6) δ 9,49 (с, 1Н), 8,71 (с, 1Н), 8,50 (с, 1Н), 7,46 (с, 1Н), 7,36 (д, J=8,48 Гц 2Н), 7,25 (д, J=7,98 Гц 2Н),5,71 (с, 2Н), 4,11-4,18 (м, 1Н), 4,09 (с, 2Н), 3,85 (с, 3H), 1,27 (д, J=6,48 Гц 6Н), 1,03-1,09 (м, 3H), 0,83-0,89 (м, 2Н).
Пример 200: Синтез 1-(4-(5-(азетнднн-3-илокси)-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина (200):
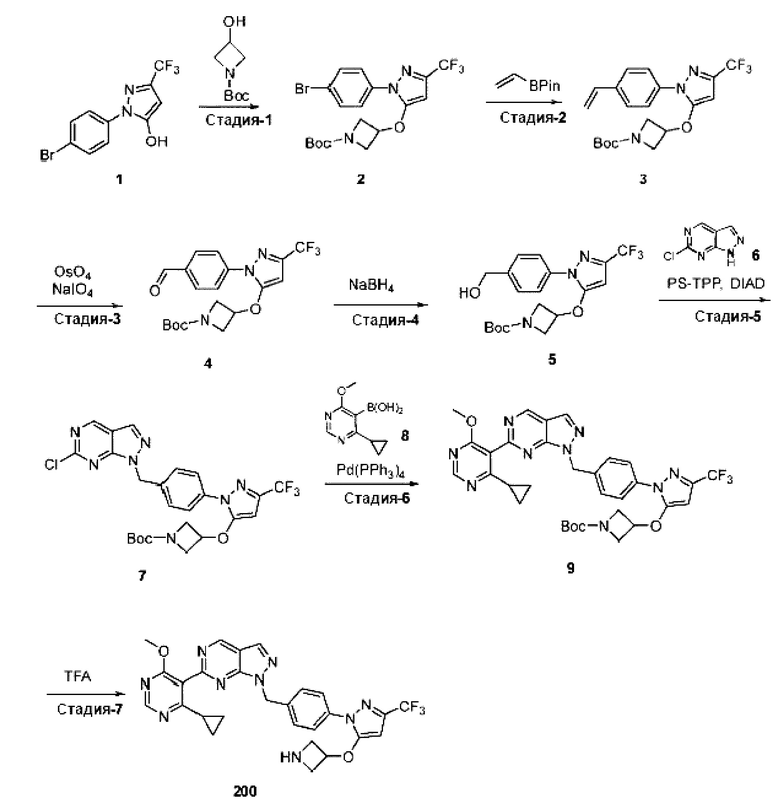
Стадия 1: Синтез трет-бутил 3-((1-(4-бромфенил)-3-(трифторметил)-1Н-пиразол-5-ил)окси)азетидин-1-карбоксилата:
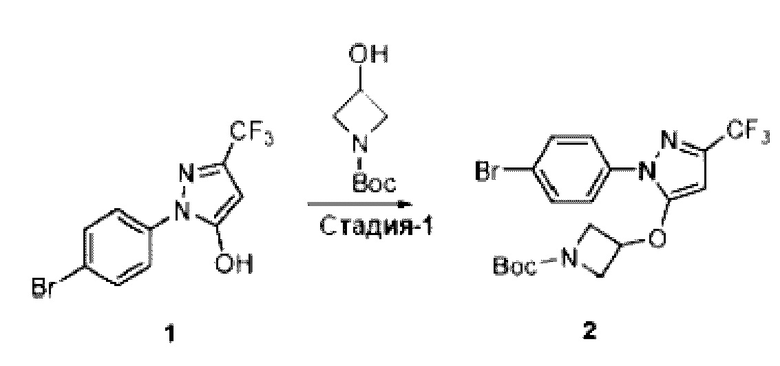
[0758] К раствору 1-(4-бромфенил)-3-(трифторметил)-1Н-пиразол-5-ола (1-39, промежуточный продукт 3) (550 мг, 1,63 ммоль) 6 ТГФ (10 мл) при 0°С добавляли трет-бутил 3-гидроксиазетидин-1-карбоксилат (339 мг, 1,96 ммоль) и PS-TPP (2,18 г, 4,52 ммоль) 6 одной порции. После перемешивания реакционной смеси при 0°С в течение 10 минут к реакционной смеси по каплям в течение 2 минут добавляли DIAD (495 мг, 2,45 ммоль). Реакционную смесь перемешивали при 0°С в течение 10 мин, затем нагревали до комнатной температуры и перемешивали в течение ночи. После завершения реакции, как показывает анализ ТСХ, смесь гасили в одой (20 мл) и экстрагировали ЕА (20 мл × 3). Органический слой сушили над сульфатом натрия (20 г), фильтровали и упаривали Полученный остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА = от 50/1 до 25/1) с получением 420 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 462, 464 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 7,60 (с,4Н), 5,74 (с, 1Н),4,94(м, 1Н), 4,31-4,39 (м. 2Н), 4,04-4,19 (м,2Н), 1,45 (с, 9Н).
Стадия 2: Синтез трет-бутил 3-((3-(трифторметил)-1-(4-винилфенил)-1Н-пиразол-5-ил)окси)азетидин-1-карбоксилата:
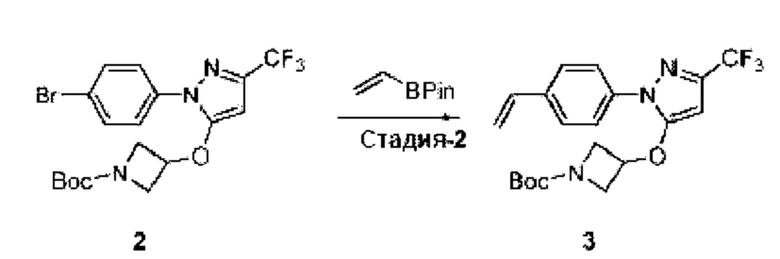
[0759] К раствору трет-бутил 3-((1-(4-бромфенил)-3-(трифторметил)-1Н-пиразоп-5-ил) окси) азетидин-1-карбоксилата 2 (370 мг, 0,80 ммоль) в толуоле (6 мл) добавляли трибутил(винил)станнан (382 мг, 1,20 ммоль) и Pd(PPh3)4 (92 мг, 0,08 ммоль). Затем реакционную смесь перемешивали при 90°С в течение ночи. После завершения реакции, как показывает анализ ЖХ-МС, смесь гасили водой (10 мл) и экстрагировали ЕА(15 мл × 2). Объединенные органические вещества сушили над сульфатом натрия (30 г), фильтровали и упаривали. Полученный остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА = от 30/1 до 20/1) с получением 300 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 410 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 7,65 (д, J=8,7 Гц, 2Н), 7,50 (д, J=8,7 Гц, 2Н), 6,78 (м. 1Н), 5,75-5,79 (м, 2Н), 5,32 (д, J=7,2 Гц 1Н), 4,92 (т,1Н), 4,30-4,39 (м. 2Н), 4,05-4,20 (м, 2Н), 1,45 (с, 9Н).
Стадия 3: Синтез трет-бутил 3-((1-(4-форишлфеннл)-3-(трифторметил)-1Н-пиразол-5-ил)окси)азетидин-1-карбоксилата:
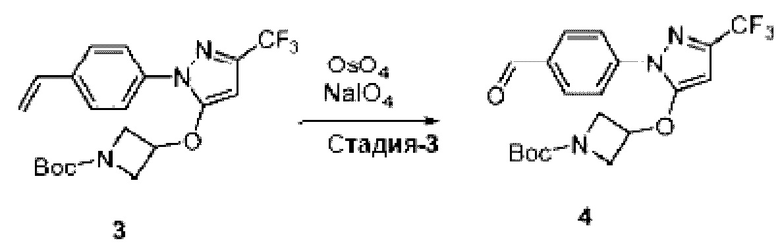
[0760] Соединение синтезировали в соответствии с процедурой получения общего промежуточного продукта 1-33. ЖХ-МС (Метод А) (ЭИР+): m/z 434,19 (M+Na)+.
Стадия 4: Синтез трет-бутил 3-((1-(4-(гидроксиметил)фенил)-3-(трифторметил)-1Н-пиразол-5-ил)окси)азетидин-1-карбоксилата:
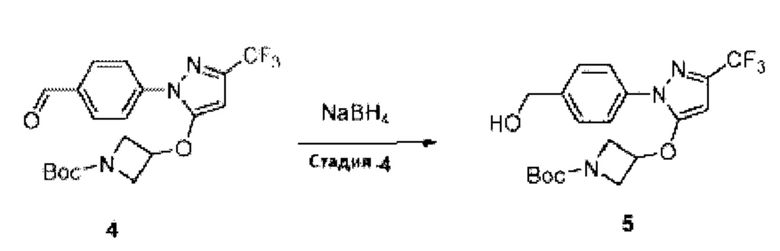
[0761] Соединение синтезировали в соответствии с процедурой получения общего промежуточного продукта I-33. ЖХ-МС (Способ А) (ИЭР+): m/z 414 (M+Na)+, 1H-ЯМР (300 МГц, CDCl3) δ 7,62 (д, J=8,1 Гц 2Н), 7,48 (д, 1=8,1 Гц 2Н), 5,75 (с, 1Н), 4,94 (м, 1Н), 4,76 (д, J=5,4 Гц 2Н), 4,30-4,35 (м, 2H), 4,04-4,13 (м, 2Н), 1,85 (м, 1Н), 1,45 (с, 9Н).
Стадия 5: Синтез трет-бутил 3-((1-(4-((6-хлор-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-3-(трифторметил)-1Н-пиразол-5-ил)окси)азетидин-1-карбоксилата:
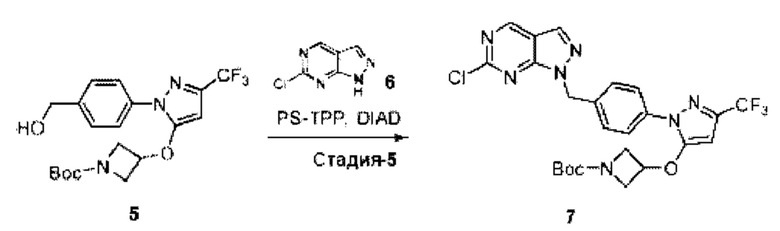
[0762] Соединение синтезировали в соответствии с процедурой получения, описанной в Примере 148. ЖХ-МС (Способ А) (ИЭР+): m/z 572,00 (M+Na)+.
Стадия 6: Синтез 3-((1-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-3-(трифторметил)-1Н-пиразол-5-ил)окси)азетидин-1-карбоксилата:
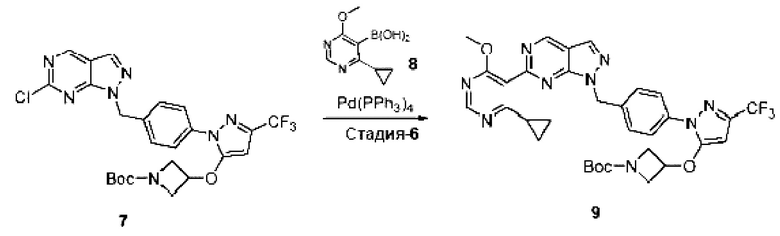
[0763] Соединение синтезировали согласно общей экспериментальной процедуры 1. ЖХ-МС (Способ А) (ИЭР+): m/z 664 (М+Н)+.
Стадия 7: Синтез 1-(4-(5-(азетидин-3-илокси)-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-6-(4-циклопропил-6-метоксипиримидин-5-нл)-1Н-пиразоло [3,4-d]пиримидина (200):
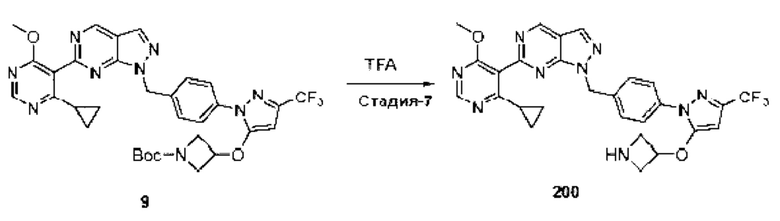
[0764] К раствору трет-бутил 3-((1-(4-((6-(4-циклопропип-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d] пиримидин-1 -ил) метил) фенил)-3-(трифторметил)-1Н-пиразол- 5-ил) окси) аз етидин-1 -карбоксилата 9 (90 мг, 0,14 ммоль) в DCM (10 мл) и ТФУ(1 мл) перемешивали при комнатной температуре в течение 1 часа. После завершения реакции, как показывает анализ ТСХ, смесь гасили водой (10 мл) и доводили рН до 8 с помощью раствора карбоната натрия. Смесь экстрагировали DCM (10 мл × 3), и органический слой сушили над сульфатом натрия (30 г), фильтровали и упаривали. Остаток очищали препаративной ВЭЖХ (Метод А), получая 24,6 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 564 (М+Н)+; 1H-ЯМР (300 МГц, CD3OD) δ 9,40 (с, 1Н), 8,64 (с, 1Н), 8,41 (с, 1Н), 7,68 (д, J=8,4 Гц, 2Н), 7,51 (д, J=8,4 Гц, 2Н), 6,06 (с, 1Н), 5,79 (с, 2Н), 5,12 (м. 1Н), 3,95-4,02 (м, 2Н), 3,92 (с, 3H), 3,65-3,71(м, 2Н), 1,65 (м,1Н), 1,09-1,19 (м. 2Н), 0,85-0,92 (м, 2Н).
Пример 201: Синтез 2-(4-циклопропил-6-метоксипиримидин-5-ил)-7-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-7Н-пирроло[2,3-d]пиримидина (201):
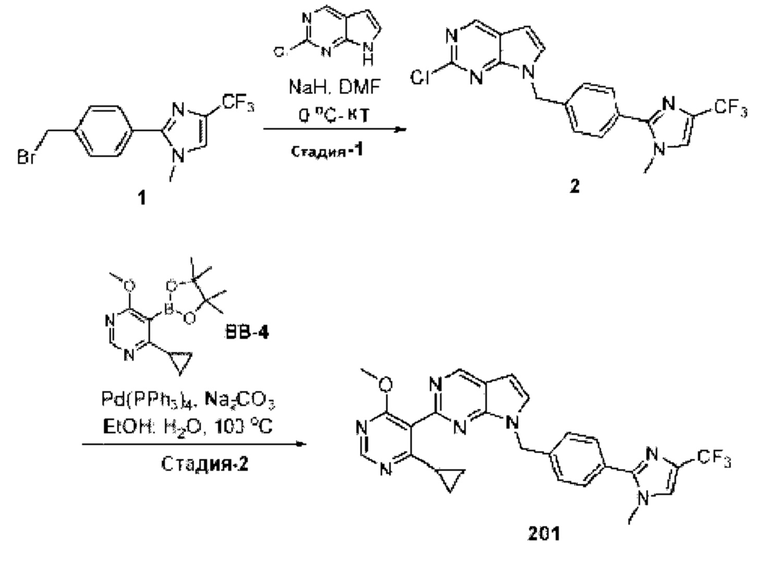
Стадия 1: Синтез 2-хлор-7-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-7Н-пирроло[2,3-d]пиримидина:
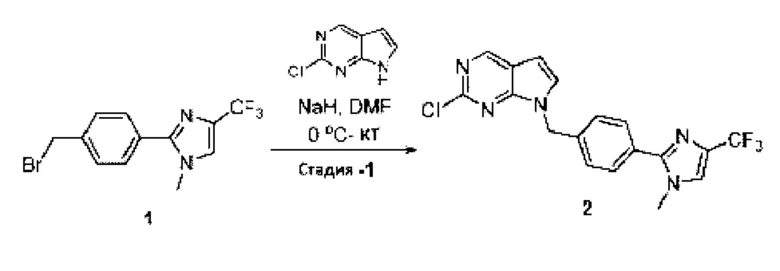
[0765] К охлажденному льдом раствору 2-хлор-7Н-пирроло[2,3-d]пиримидина (0,140 г, 0,914 ммоль) в ДМФА (5 мл) добавляли 60% дисперсию гидрида натрия в масле (0,073 г, 1,83 ммоль) по порциям Полученную смесь перемешивали в течение 30 минут и затем обрабатывали 2-(4-(бромметил)фенил)-1-метил[-4-(трифторметил)-1H-имидазолом 1 (0,292 г, 0,914 ммоль). Смесь дополнительно перемешивали в течение 2 часов при комнатной температуре. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили ледяной водой (10 мл) и экстрагировали ЕА (2 × 25 мл). Объединенный органический слой промывали холодной водой (10 мл), затем солевым раствором (10 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 30-50% ЕА в н-гексане, с получением указанного в заголовке соединения(0,260 г). ЖХ-МС (Способ В) (ИЭР+): m/z 391,90 (М+Н)+.
Стадия 2: Синтез 2-(4-циклопропил-6-метоксипиридин-5-ил)-7-(4-(1-метил-4-(трифторметил)-имидазол-2-ил)бензил)-7Н-пирроло[2,3-d]пиримидина (201):
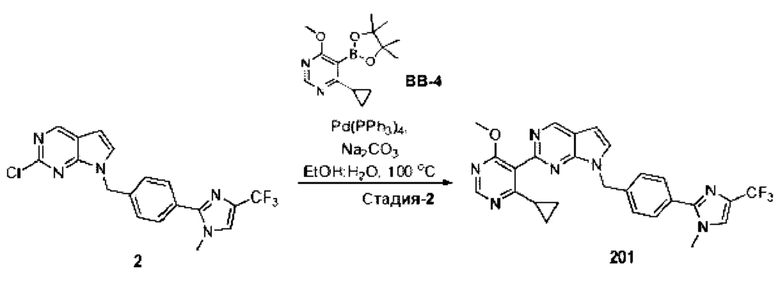
[0766] К перемешиваемому раствору 4-цикпопропил-6-метокси-5-(4,4,5,5-тетраметил-1,3,2 -диоксаборолан-2-ил)пиримидина ВВ-4 (0,243 г, 0,880 ммоль) и 2-хлор-7-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-7Н-пирропо[2,3-d]пиримидина 2 (0,230 г, 0,587 ммоль) в этаноле: Н2О (10:2 мл) добавляли Na2CO3 (0,186 г, 1,76 ммоль) при комнатной температуре. Полученную смесь дегазировали аргоном в течение 10 мин, а затем обрабатывали Pd(PPh3)4 (0,034 г, 0,029 ммоль) при комнатной температуре. Смесь дополнительно дегазировали аргоном в течение 5 мин, а затем нагревали в микроволновой печи при 100°С в течение 2 часов. За ходом реакции следили с помощью ТСХ После завершения реакционную смесь разбавляли ЕА (25 мл) и фильтровали через слой целита. Органический слой отделяли, промывали солевым раствором, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя 0-50% ЕА в н-гексане, с последующей повторной очисткой препаративной ВЭЖХ (Метод В) с получением указанного в заголовке соединения (0,032 г) ЖХ-МС (Способ В) (ИЭР+): m/z 506,15 (М+Н)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,16 (с, 1Н), 8,67 (с, 1H), 7,92 (с, 1Н), 7,85 (д, J=3,42 Гц, 1Н), 7,66 (д, J=7,83 Гц, 2Н), 7,40 (д,J=8,31 Гц, 2Н), 6,76 (д, J=3,42 Гц, 1Н),5,57(с, 2Н), 3,83 (с, 3H), 3,73 (с, 3H), 1,60-1,66 (м, 1Н), 1,00-1,04 (м, 2Н), 0,81 (дд, J=3,18, 7,58 Гц, 2Н).
Пример 202: Общая методика синтеза имидазолопиримидинов. Получение 2-(2-изопропилфенил)-9-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-9Н-пурина (202):
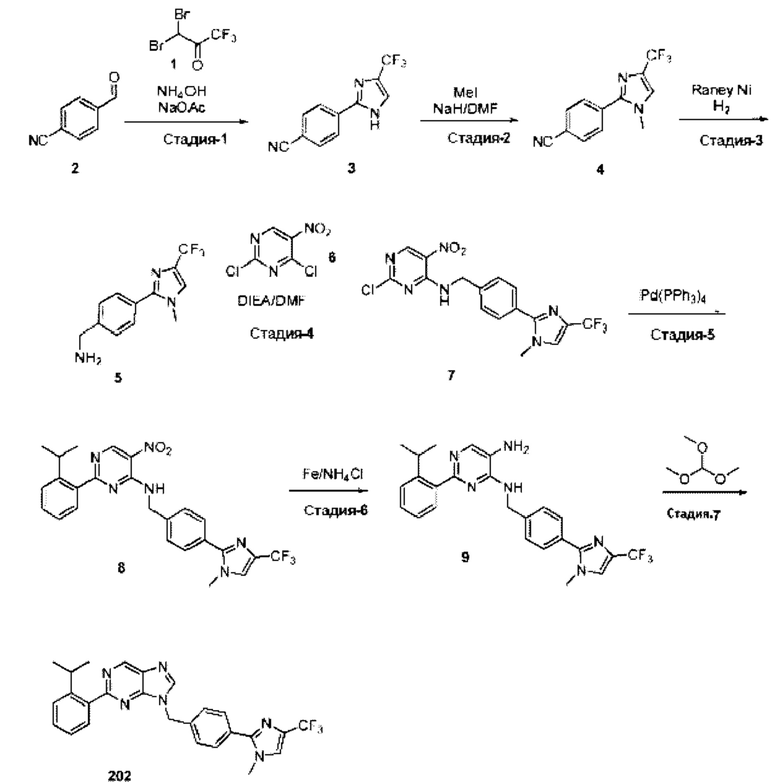
Стадия 1: Синтез 4- (4 - (Трифторметил)-1 Н-имидазол- 2 -ил) бензонитрила:
[0767] Смесь 3,3-дибром-1,1,1-трифторпропан-2-она 1 (3,06 г, 11,3 ммоль) и NaOAc (0,93 г, 11,3 ммоль) в воде (3 мл) перемешивали при 100°С в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и затем к реакционной смеси добавляли смесь 4-формилбензонитрила 2 (1,5 г, 11,3 ммоль) в метаноле (50 мл) и NH4OH (10 мл). Полученную реакционную смесь перемешивали при комнатной температуре в течение 40 минут, а затем при 100°С в течение 2 часов. Затем смесь охлаждали до комнатной температуры и гасили водой (100 мл), а затем экстрагировали ЕА (100 мл × 2). Объединенный органический слой сушили над Na2SO4 (30 г), фильтровали и упаривали досуха, получая неочищенный продукт, который очищали колоночной хроматографией (элюент: EA/n-Hex=1:10 к 1:5) с получением 1,5 g указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 238 (М+Н)+, 1Н-ЯМР (300 МГц, ДМСО-d6) δ 13,55 (уш. с, 1Н), 8,14(д, J=8,1 Гц, 1Н),8,07 (с, 1Н), 7,97 (д, J=8,1 Гц, 1Н).
Стадия 2: Синтез 4-(1-Метил-4-(трифторметил)-1Н-имидазол-2-ил) бензонитрила:
[0768] К раствору 4-(4-(трифторметил)-1Н-имидазол-2-ил)бензонитрила 3 (1,37 г, 5,8 ммоль) в ТГФ (15 мл) при 0°С добавляли NaH (290 мг, 7,25 ммоль) порционно в течение 2 мин. После добавления полученную суспензию перемешивали при 0°С в течение 1 часа. Затем по каплям в течение 2 минут добавляли MeI (1,03 г, 7,25 ммоль) и смесь перемешивали при 0°С в течение дополнительных 2,5 часов. Затем реакцию гасили насыщенным раствором NH4Cl (30 мл) и экстрагировали ЕА (50 мл × 2). Объединенный органический слой сушили над Na20O+(30 г), фильтровали и упаривали досуха. Остаток очищали колоночной хроматографией (элюент: EA/n-Hex=1: 10 к 1: 5) с получением 700 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 252 (М+Н); 1H-ЯМР (300 МГц, CDCl3) δ 7,76-7,83 (м, 4Н), 7,38 (с, 1Н), 3,83 (с, 3H).
Стадия 3: Синтез (4-(1-Метил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)метанамина:
[0769] Суспензию 4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил) бензонитрила 4 (2,5 г, 9,9 ммоль) и Ni Ренея (2 г, влажное твердое вещество) в ЕА (100 мл) и NH3.H3O (0,5 мл) гидрировали с использованием баллонного H2 (1 атм). После перемешивания реакционной смеси при комнатной температуре в течение 2,5 часов суспензию фильтровали через слой целит а. Осадок на фильтре промывали ЕА (10 мл) и фильтрат упаривали досуха, получая 2,5 г неочищенного указанного в заголовке соединения.
ЖХ-МС (Способ А) (ИЭР+): m/z 256 (М+Н)+, 1Н-ЯМР (300 МГц, CDCl3) δ 7,60 (д, J=8,1 Гц, 1Н), 7,46 (д, J=8,1 Гц 1Н),7,31 (с,1Н),3,95(с,2Н),3,77(с,3H).
Стадия 4: Синтез 2-Хлор-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-5-нитропиримидин-4-амина:
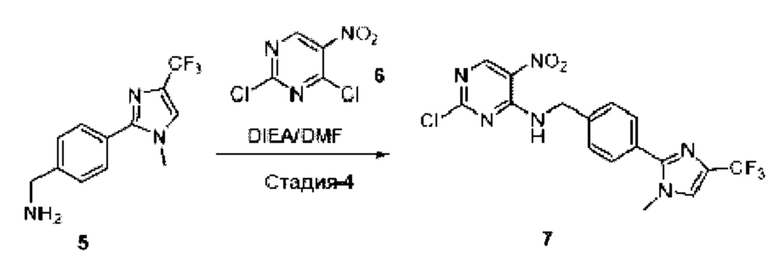
[0770] Смесь (4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ип)фенил) метанамина 5 (0,87 г, 3,4 ммоль), 2,4-дихлор-5-ншропиримидина 6 (0,66 г, 3,4 ммоль) и DIE А (0,88 г, 6,8 ммоль) в ДМФА (12 мл) перемешивали при комнатной температуре 6 течение 30 минут. Затем смесь гасили водой и экстрагировали ЕА (50 мл × 3). Объединенный органический слой промывали водой (30 мл), сушили над безводным Na2SO4 (30 г), фильтровали упаривали в вакууме. Остаток очищали хроматографией на силикагеле (элюент: ЕА: н-Нех = от 1:20 до 1:4) с получением 480 мг указанного в заголовке соединения. 1H-ЯМР (300 МГц CDCl3) δ 9,09 (с, 1Н), 8,74 (уш. с, 1Н), 7,66 (д, J=8,1 Гц, 2Н), 7,48 (д, J=8,1 Гц 2Н), 7,33 (с, 1Н), 4,91 (д, J=6,0 Гц 2Н),3,79 (с, 3H).
Стадия 5: Синтез 2-(2-изопропилфенил)-N-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-5-нитропиримидин-4-амина:
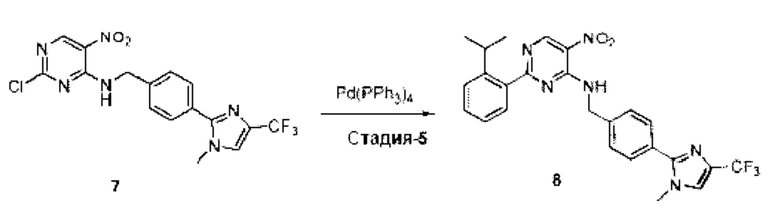
[0771] Смесь 2-хлор-N-(4-(1 -метип-4-(трифторметил)-1Н -имидазол-2 -ил) бензил)- 5 -нитропиримидин-4-амина 7 (0,30 г, 0,74 ммоль), К3РО4 (0,47 г, 2,2 ммоль), Pd(dppf)Cl2 (121 мг, 20 мол.%) и (2-изопропипфенил)бороновой кислоты (0,24 г, 1,5 ммоль) в DME (18 мл) перемешивали при 80°С в течение 3 часов. Затем смесь охлаждали до комнатной температуры и разбавляли водой (20 мл), а затем экстрагировали ЕА (50 мл × 3). Объединенный органический спой сушили над безводным Na2SO4 (20 г), фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (элюент: ЕА:n-Нех = от 1:10 до 1:5) с получением 300 мг указанного в заголовке соединения ЖХ-МС (Способ А) (НЭР): m/z 497 (М+Н)+.
Стадия 6: Синтез 2-(2-изопропилфенил)-N4-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)пиримидин-4,5-диамина:
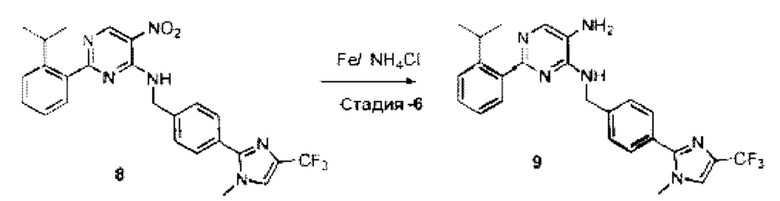
[0772] К раствору 2-(2-изопропилфенил)-N-(4-(1-митил-4-(трифторметил)-1Н-имидазоп-2-ил)бензил)-5-нитропиримидин-4-амина 8 (0,28 г 0,56 ммоль) в EtOH (30 мл), добавляли FeCl3 (18 мг, 0,11 ммоль), N2H4 (1,13 г, 17,9 ммоль) и древесный уголь (0,11 г) в атмосфере n2. Полученную смесь перемешивали при 70°С в течение 2 часов, а затем суспензию фильтровали через слой целита. Осадок на фильтре промывали ЕА (30 мл). Фильтрат разбавляли водой (50 мл) и экстрагировали ЕА (50 мл × 3). Объединенный органический спой промывали водой (20 мл) и сушили над безводным Na2SO4 (30 г), фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле (элюент: ЕА:n-Нех = от 1:10 до 2:1) с получением 330 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 467 (М+Н)+.
Стадия 7: Синтез 2-(2-изопропилфенил)-9-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-9Н-пурина (202):
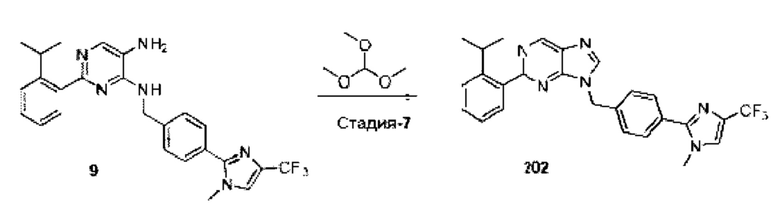
[0773] К раствору 2-(2-изопропилфенил)-N4-(4-(1-метил-4-(трифторметил)-1Н-имидазоп-2-ил) бензил) пиримидин-4,5-диамина 9 (60 мг, 0,13 ммоль) в диоксане (4,8 мл), добавляли метансульфоновую кислоту (0,30 мг, 2,5%) и триметоксиметан (1,2 мл) одной порцией. После добавления реакционную смесь перемешивали при 80°С в течение 1 часа. Затем реакционную смесь охлаждали до комнатной температуры, гасили водой (10 мл) и экстрагировали ЕА (20 мл × 3). Объединенный органический спой сушили над безводным Na2SO4 (20 г), фильтровали и упаривали в вакууме. Остаток очищали препаративной ВЭЖХ (Метод А) с получением 28 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 477 (М+Н)+; 1H-ЯМР (300 МГц, CD3OD) δ 9,18 (с, 1Н), 8,69 (с, 1Н), 7,64-7,68 (м, 3H), 7,53-7,56 (м, 3H), 7,40-7,48 (м. 2Н),7,27 (м. 1Н), 5,66 (с, 2Н), 3,74 (с, 3H), 3,37 (м, 1Н), 1,16 (д, J=6,9 Гц, 6Н).
Следующее соединение было получено в соответствии с общей экспериментальной процедурой 202:

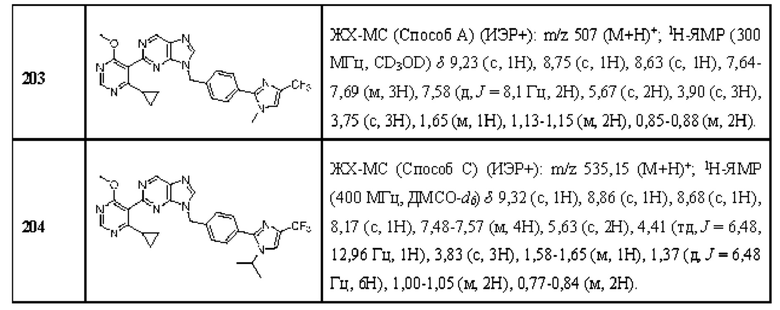
Пример 205: Получение 2-(4-циклопропил-6-метоксипиримидин-5-ил)-8-этил-9-(4-(1-изопропил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-9Н-пурина (205):
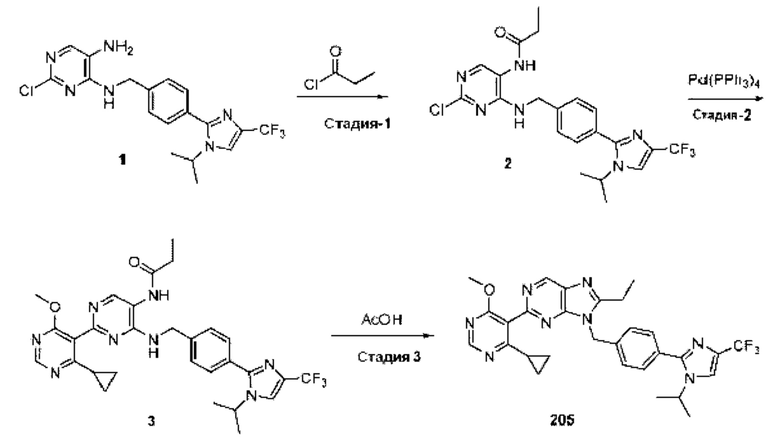
Синтез 2-клор-N4-(4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)пиримидин-4,5-диамина:
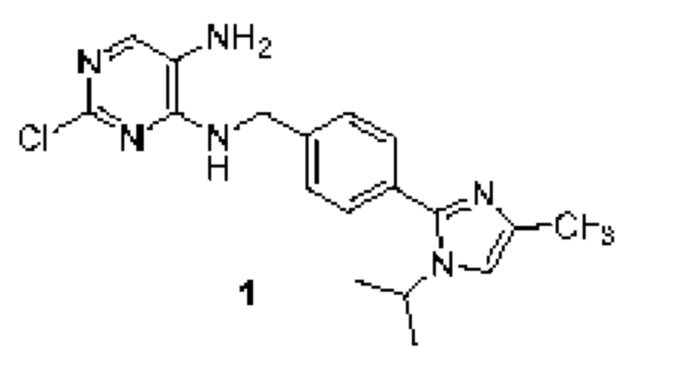
[0774] Соединение синтезировали в соответствии с общей экспериментальной процедурой 202. ЖХ-МС (Способ А) (ИЭР+): m/z 411 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 1,59 (с, 1Н), 7,47 (д, J=8,4 Гц, 2Н), 7,39-7,42 (м, 3H), 5,61 (уш. с, 1Н), 4,71 (д, J=5,1 Гц 2Н), 4,49-4,58 (м, 1Н), 3,22 (уш. с, 2Н), 1,45 (д, J=6,6 Гц, 6Н).
Стадня 1: Синтез N-(2- хлор -4-((4-(1-изопропил-4-(трифторметил)- 1Н-имидазол-2-нл)бензил)амино)пиримидин-5-ил) пропнонамида:
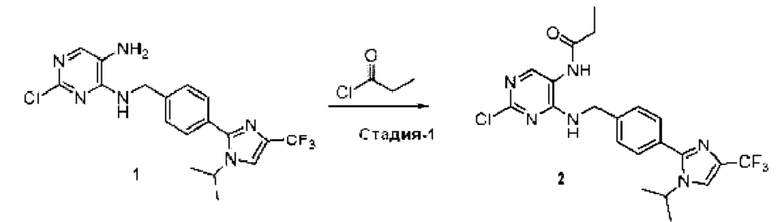
[0775] К раствору 2-клор-N4-(4-(1-изопропип-4-(трифторметил)-1Н-имндазол-2-ил) бензил)пиримидин-4,5-диамина 1 (250 мг, 0,61 ммоль) в к безводному ТГФ (10 мл) по каплям в течение 2 минут добавляли пропионилхлорид (65 мг, 0,70 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 часа После завершения реакции, как показывает анализ ТСХ реакцию гасили водным раствором NaHCO3 (10 мл) и экстрагировали ЕА (20 мл × 2). Объединенный органический слой сушили над Na2SO4, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ/ЕА = от 3/1 до 1/1), получая 320 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 467 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 8,27 (с, 1Н), 7,98 (с, 1Н), 7,44 (с, 1Н), 7,31-7,38 (м. 4Н), 6,13 (м. 1Н), 4,65 (д, J=5,1 Гц 2Н), 4,48 (м, 1Н), 2,40 (к, J=1,5 Гц 2Н), 1,41-1,46 (д, J=6,6 Гц 6Н), 1,25 (т, J=1,5 Гц 3H).
Стадия 2: Синтез N-(4'-циклопропил-4-((4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензиламино)-6'-метокси-[2,5'-бипиримидин]-5-ил)пропионамида:
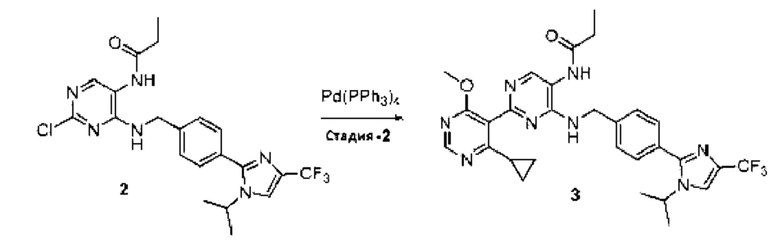
[0776] Соединение синтезировали в соответствии с общей экспериментальной процедурой 1.
ЖХ-МС (Способ А) (ИЭР+): m/z 581 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 8,60 (с, 1Н), 8,26 (с, 1Н), 7,40-7,42 (м, 6Н), 6,10 (м, 1H), 4,75 (д, J=5,4 Гц, 2Н), 4,50 (м, 1Н), 3,91 (с, 3H), 2,43 (к, J=7,2 Гц, 2Н), 2,10-2,13 (м, 1Н), 1,41-1,46 (м, 6Н), 1,26-1,28 (м, 4Н), 0,85-0,89 (м, 2Н).
Стадия 3: Синтез 2-(4-циклопропил-6-метоксипиримидин-5-ил)-8-этил-9-(4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-9Н-пурина (205):
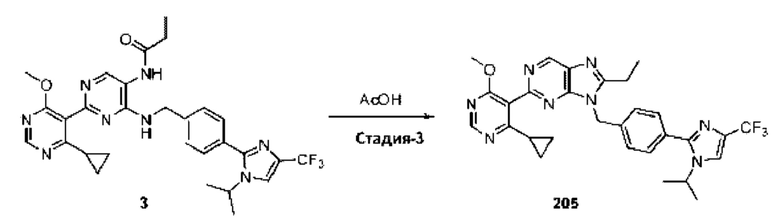
[0777] Раствор N-(4'-циклопропил-4-((4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил) бензил)амино)-6'-метокси- [2,5'-бипиримидин]-5-ил)пропионамида 3 (80 мг, 0,14 ммоль) в АсОН (5 мл) перемешивали при кипячении с обратным холодильником в течение 7 часов. После завершения реакции, как показывает анализ ТСХ, реакционную смесь упаривали до сук а. Остаток растворяли в ЕА (50 мл), промывали водным раствором NaHCO3 (20 мл). Органический слой сушили, фильтровали и упаривали, получая неочищенный продукт. Неочищенный продукт очищали препаративной ВЭЖХ (Метод А), получая 57 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 563 (М+Н)+; 1H-ЯМР (300 МГц. CD3OD) δ 9,11 (с, 1Н),8,61 (с, 1Н), 7,91 (с, 1Н), 7,54(д, J=8,1 Гц, 2Н), 7,44 (д, J=8,1 Гц, 2Н), 5,67 (с, 2Н), 4,52 (м. 1Н), 3,91 (с, 3H), 3,03 (к, J=1,5 Гц, 2Н), 1,68 (м, 1Н), 1,38-1,44 (м, 9Н), 1,10-1,18 (м, 2Н), 0,84-0,92 (м, 2Н).
Следующие соединения были получены в соответствии с общей экспериментальной процедурой 205:

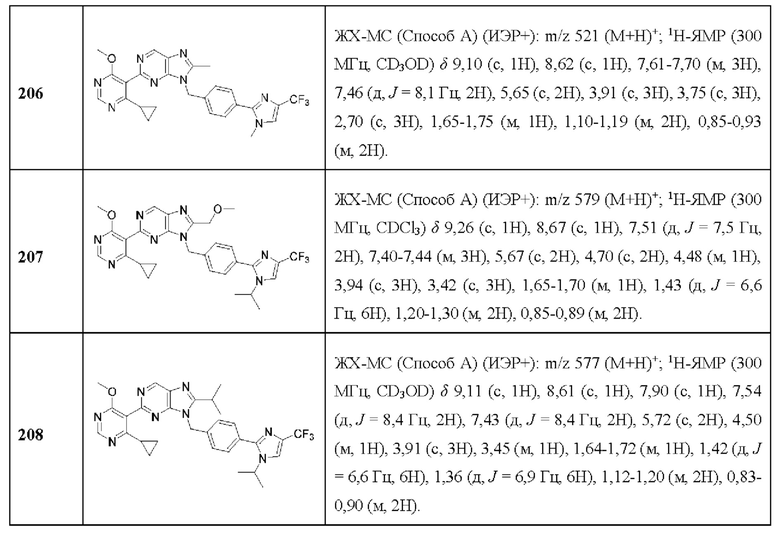
Пример 209: Синтез 2-(4-циклопропил-6-метоксипиримидин-5-ил)-8-этил-9-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил) бензил)-9Н-пурина (209):
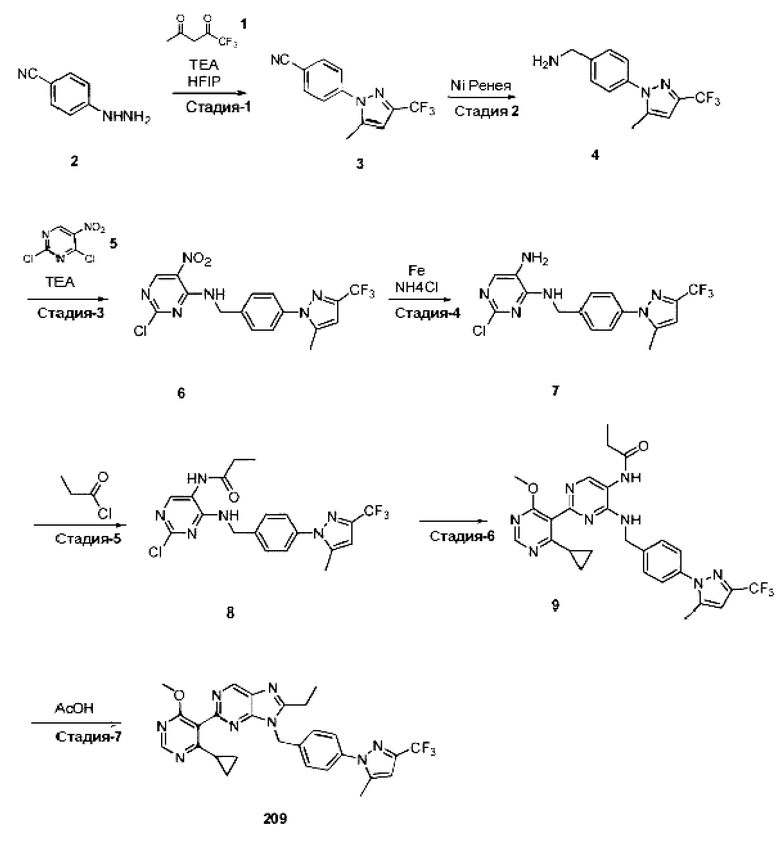
Стадия 1: Синтез 4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил) бензонитрила:
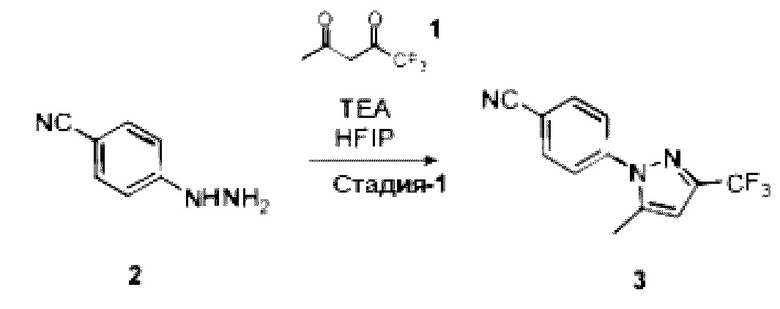
[0778] Соединение синтезировали в соответствии с процедурой получения общего промежуточного продукта I-19. ЖХ-МС (Способ А) (ИЭР+): m/z 252 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 7,79-7,85 (д, J=8,7 Гц, 2Н), 7,65 (д, J=8,7 Гц, 2Н), 6,53 (с, 1Н), 2,45 (с, 3H).
Стадия 2: Синтез (4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)фенил)метанамина:

[0779] Соединение синтезировали 6 соответствии с процедурой Примера 202. ЖХ-МС (Способ А) (ИЭР+): m/z 256 (М+Н)+; 1Н-ЯМР (300МГц, CDCl2) δ 7,39-7,47 (м, 4Н), 6,46 (с, 1Н), 3,96(с, 2Н), 2,34(с,3H).
Стадия 3: Синтез 2-хлор -N-(4-(5- метил-3-(трифторметил)-1Н -пиразол-1-ил)бензил)-5-нитропиримидин-4-амина:
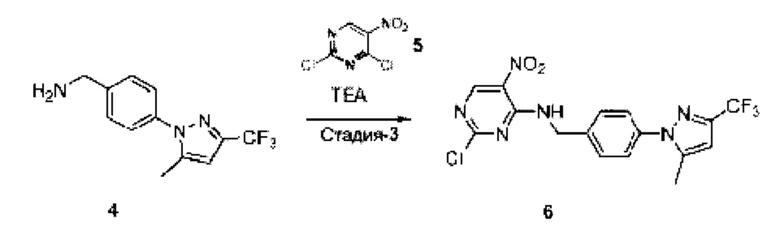
[0780] Соединение синтезировали в соответствии с процедурой Примера 202. ЖХ-МС (Способ А) (ИЭР+): m/z 413 (М+Н)+; 1Н-ЯМР(300 МГц, CDCl3) δ 9,10 (с, 1Н), 8,73 (с, 1Н), 7,45-7,53 (м, 4Н), 6,48 (с, 1Н), 4,91 (д J=6,0 Гц, 2Н), 2,37 (с, 3H).
Стадия 4: Синтез 2-хлор-N4-(4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензил)пиримидин-4,5-диамина:
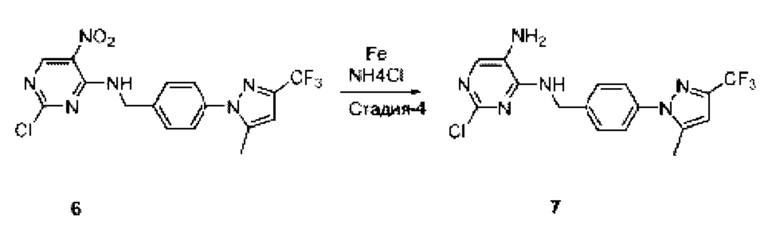
[0781] Соединение синтезировали в соответствии с процедурой Примера 202. ЖХ-МС (Способ А) (ИЭР+): m/z 383 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 7,65 (с, 1Н), 7,48 (д, J=8,4 Гц, 2Н), 7,41 (д, J=8,4 Гц, 2Н),6,47 (с, 1Н),5,43 (уш. с, 1Н),4,74(д, J=5,7 Гц, 2Н),3,07 (уш. с, 2Н), 2,36 (с, 3H).
Стадия 5: Синтез N-(2-хлор-4-((4-(5-метал-3-(трифторметил)-1Н-пиразол-1-ил)бензил)амино)пиримидин-5-ил)пропионамида:
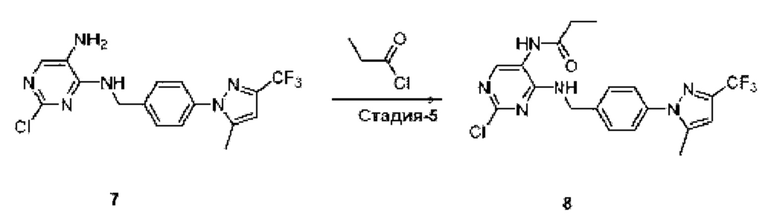
[0782] Соединение синтезировали е соответствии с процедурой Примера 205. ЖХ-МС (Способ А) (ИЭР+): m/z 439 (М+Н)+; 1H-ЯМР (300 МГц, CD3OD) δ 8,23 (с, 1Н), 7,58 (д, J=8,4 Гц, 2Н), 1,49 (д, J=8,4 Гц, 2Н), 6,59 (с, 1Н), 4,84 (с, 2Н), 2,49 (к, J=7,5 Гц 2Н), 2,34 (с, 3H), 1,20 (т, J=7,5 Гц 3H).
Стадия 6: Синтез N-(4'-циклопропил-6'-метокси-4-((4-(5-метил-3-(трифторметил)-1Н-пиразол-1-нл)бензил)амино)-[2,5'-бипиримидин]-5-ил)пропнонамида:
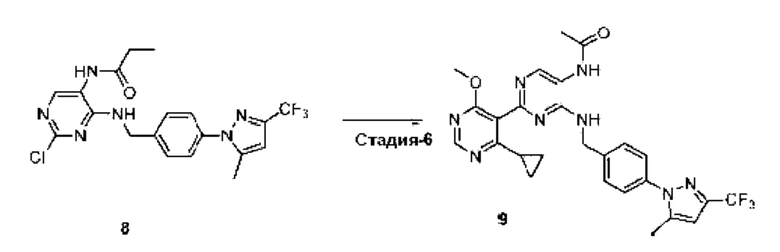
[0783] Соединение синтезировали в соответствии с процедурой Примера205. ЖХ-МС (Способ А) (ИЭР+): m/z 553 (М+Н)+; 1H-ЯМР (300 МГц CD3OD) δ 8,53 (с, 1Н), 8,18 (с, 1Н), 7,53 (д, 3=8,4 Гц 2Н), 7,42 (д, J=8,4 Гц, 2Н), 6,57 (с, 1Н), 4,78 (с, 2Н),3,88 (с,3H), 2,52 (к, J=7,5 Гц,2Н), 2,32 (с, 3H), 1,72 (м. 1Н), 1,24 (т, J=7,5 Гц, 3H), 1,03-1,10 (м,2Н), 0,78-0,92 (м,2Н).
Стадия 7: Синтез 2-(4-циклопропил-6-метоксипиримидин-5-ил)-8-этил-9-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-9Н-пурина (209):
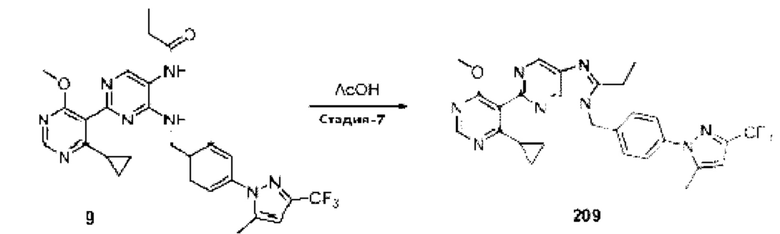
[0784] Соединение синтезировали 6 соответствии с процедурой Примера 205. ЖХ-МС (ИЭР+): m/z 535 (М+Н)+; 1H-ЯМР (300 МГц, CD3OD) δ 9,11 (с, 1Н), 8,61 (с, 1Н), 7,44-7,52 (м, 4Н), 6,57 (с, 1Н), 5,67 (с, 2Н), 3,91 (с, 3H), 3,04 (к, J=7,2 Гц, 2Н), 2,32 (с, 3H), 1,68 (м, 1Н), 1,41 (т, 1=1,2 Гц, 3H), 1,10-1,16 (м, 2Н), 0,84-0,89 (м, 2Н).
Следующие примеры были получены в соответствии с процедурой Примера 209 с использованием либо триметилортоформната, либо соответствующих хлорангидридов:
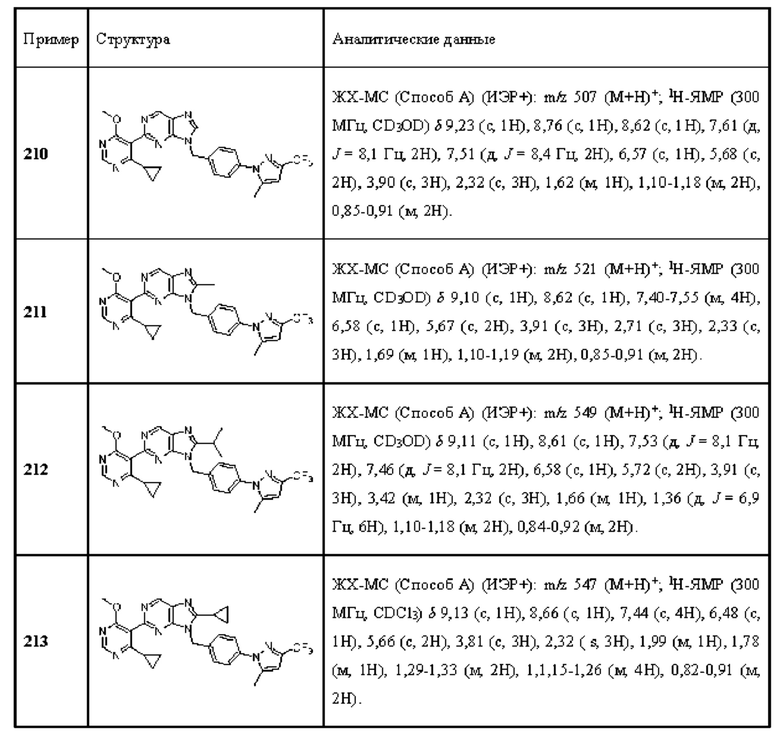
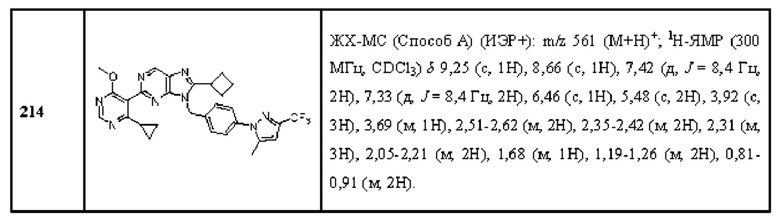
Пример 215: Синтез 5-(4-циклопропил-6-метоксипиримидин-5-ил)-3-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-3H-[1,2,3]триазоло[4,5-d]пиримидина (215):
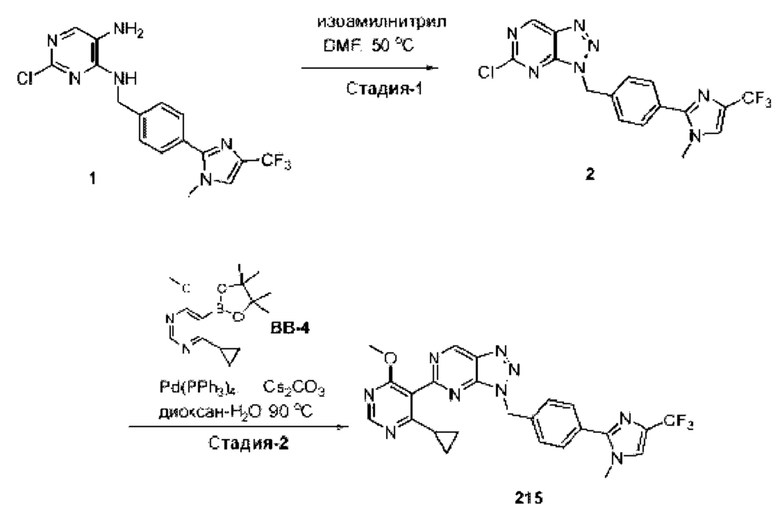
Стадия 1: Синтез 5-хлор-3-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-3Н-[1,2,3]триазоло[4,5-d]пиримидина:
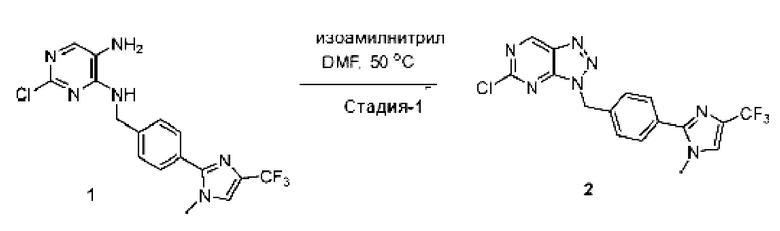
[0785] К перемешиваемому раствору 2-хлор-N4-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензил)пиримидин-4,5-диамина 1 (0,500 г, 1,31 ммоль) в ДМФА (5 мл) добавляли изоамилнитрил (0,229 г, 1,96 ммоль) при комнатной температуре. Реакционную смесь нагревали при 50°С в течение 2 часов. За кодом реакции следили с помощью ТСХ и ЖХМС. После завершения реакционную смесь гасили насыщенным раствором Na2CO3 (20 мл) медленно при перемешивании и экстрагировали ЕА (2×10 мл). Объединенный органический слой промывали ледяной водой (10 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали растиранием с диэтиловым эфиром (20 мл), фильтровали и сушили с получением указанного в заголовке соединения (0,380 г). ЖХ-МС (Способ В) (ИЭР+): m/z 394,25 (М+Н)+; 1Н-ЯМР (400 МГц. ДМСО-d6) δ 9,78 (с, 1Н), 7,94 (с, 1Н),7,73 (д, J=8,31 Гц, 2Н), 7,51 (д, J=8,31 Гц, 2Н), 6,03 (с, 2Н), 3,76 (с, 3Н).
Стадия 2: Синтез 5-(4-циклопропил-6-метоксипиримидин-5-ил)-3-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-3H-[1,2,3]триазоло[4,5-d]пиримидина (215):
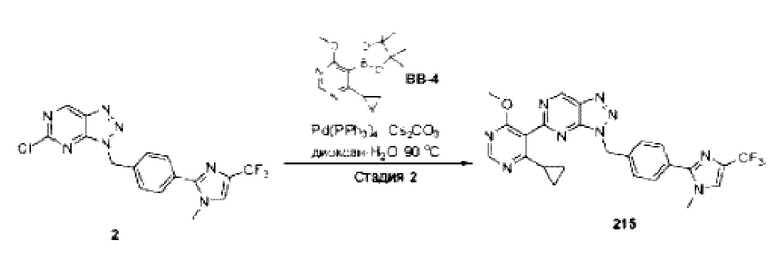
[0786] К перемешиваемому раствору 5-хлор-3-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-3H-[1,2,3]триазоло[4,5-d]пиримидина 2 (0,150 г, 0,380 ммопь) в диоксане:H2O (5:1 мл) добавляли карбонат цезия (0,308 г, 0,950 ммоль), 4-циклопропил-6-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин ВВ-4 (0,157 г, 0,571 ммоль) при комнатной температуре. Полученную смесь дегазировали аргоном в течение 10 мин, а затем обрабатывали Pd(PPh3)4 (0,065 г, 0,057 ммоль) при комнатной температуре. Смесь дополнительно дегазировали аргоном в течение 10 мин, а затем нагревали при 90°С в течение 16 часов. За ходом реакции следили с помощью ТСХ. После завершения реакционную смесь гасили H2O (20 мл) и экстрагировали ЕА (2×20 мл). Объединенный органический спой сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенное соединение очищали препаративной ВЭЖХ (Метод D) с получением указанного в заголовке соединения (0,050 г). ЖХ-МС (Способ В) (ИЭР+): m/z 508,20 (М+Н)+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,94 (с, 1Н), 8,73 (с, 1Н),7,93 (с, 1Н), 7,72 (д, J=8,31 Гц, 2Н), 7,54 (д, J=8,31 Гц, 2Н), 6,09 (с, 2Н), 3,85 (с, 3Н), 3,75 (с, 3Н), 1,69 (тд, J=3,79, 8,07 Гц, 1Н), 1,05-1,09 (м, 2Н), 0,85 (дд, J=2,93, 7,83 Гц, 2Н).
Пример 216 и 217: Синтез 5-бром-2-(4-циклопропил-6-метоксипиримидин-5-ил)-7-(3-фтор-4-(1-изопропил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-7Н-пирроло[2,3-d]пиримидина (216) и 2-(4-циклопропил-6-метоксипиримидин-5-ил)-7-(3-фтор-4-(1-изопропил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-5,7-дигидро-6Н-пирроло[2,3-d]пиримидин-6-она (217):
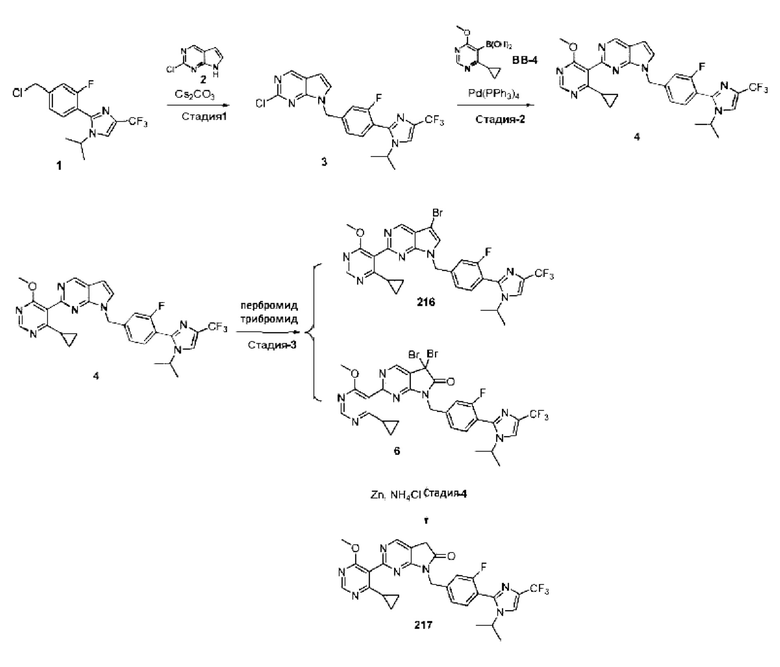
Стадия 1: Синтез 2-хлор-7-(3-фтор-4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-7Н-пирроло[2,3-d]пиримидина:
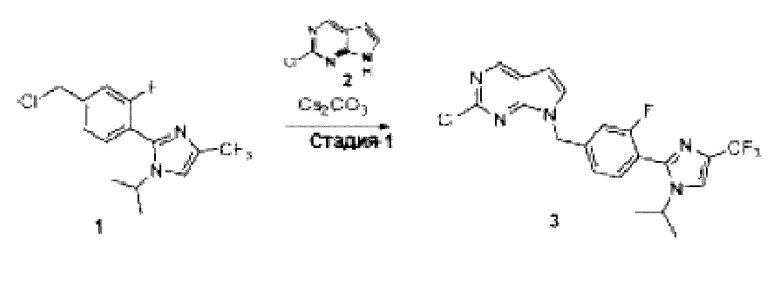
[0787] Соединение синтезировали в соответствии с общей процедурой для синтеза общего промежуточного продукта I-8. ЖХ-МС (Способ А) (ИЭР+): m/z 438 (М+Н)+; 1H-ЯМР (300 МГц, CDCl3) δ 8,86 (с, 1Н), 7,53 (т, J=7,5 Гц 1Н), 7,44 (с, 1Н), 7,19 (с, 1Н),7,12 (д, J=7,8 Гц, 1Н), 6,97 (д, J=10,2 Гц 1Н), 6,65 (д, J=3,6 Гц 1Н), 5,49 (с, 2Н), 4,23 (м, 1Н), 1,41 (д, J=6,6 Гц 6Н).
Стадия 2: Синтез 2-(4-цнклопропнл-6-метоксипиримидин-5-нл)-7-(3-фтор-4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-7Н-пирроло[2,3-d]пиримидина:
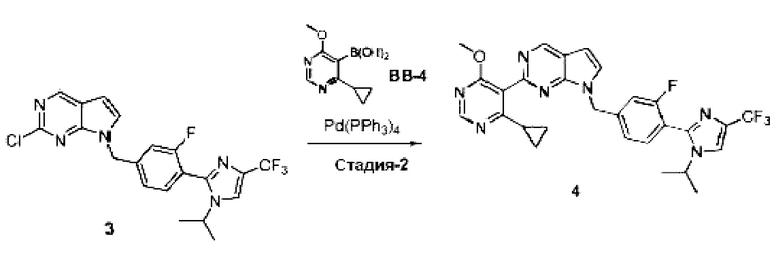
[0788] Соединение синтезировали в соответствии с общей экспериментальной процедурой 1. ЖХ-МС (Способ А) (ИЭР+): m/z 552 (М+Н)+; 1H-ЯМР (300 МГц CDCl3) δ 9,16 (с, 1Н), 8,66 (с, 1Н), 7,52 (т, J=7,2 Гц, 1Н), 7,43 (с, 1Н), 7,28 (с, 1Н), 7,20 (д, J=8,1 Гц 1Н),7,10 (д, J=9,6 Гц, 1Н), 6,69 (д, J=3,6 Гц, 1Н), 5,54 (с,2Н), 4,19(м. 1Н), 3,93 (с,3Н), 1,72 (м. 1Н), 1,40 (д, J=6,6 Гц, 6Н), 1,19-1,25 (м, 2Н), 0,82-0,89 (м, 2Н).
Стадия 2: Синтез 5,5-дибром-2-(4-циклопропил-6-метоксипиримидин-5-нл)-7-(3-фтор-4-(1-изопропил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-5,7-дигидро-6Н-пирроло[2,3-d]пиримидин-6-она (6) и 5-бром-2-(4-циклопропил-6-метоксипиримидин-5-ил)-7-(3-фтор-4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-7Ни-пирроло[2,3-d]пиримидина (216):
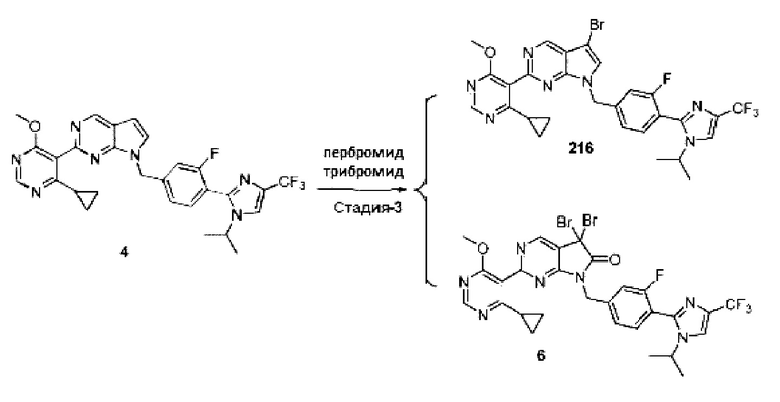
[0789] К раствору 2-(4-циклопропил-6-метоксипиримидин-5-ил)-7-(3-фтор-4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил) бензил)-7Н-пирроло[2,3-d]пиримидина (60 мг, 0,11 ммоль) в АсОН (1 мл) и t-BuOH (2 мл) добавляли пербромид бромид пиридиния (104 мг, 0,33 ммоль) одной порцией. Полученную смесь перемешивали при комнатной температуре в течение 5 часов. После завершения реакции, как показывает анализ ТСХ, реакционную смесь упаривали досуха. Остаток растворяли в ЕА (10 мл) и промывали водным раствором NaHCO3 (10 мл). Органический слой сушили, фильтровали и упаривали. Остаток очищали флэш-хроматографией на силикагеле (РЕ/ЕА=2/1), получая 14 мг (5-бром-2-(4-циклопропил-6-метоксипиримидин-5-ил)-7-(3-фтор-4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-7Н-пирроло[2,3-d]пиримидин) 216 и 47 мг 5,5-дибром-2-(4-циклопропил-6-метоксипиримидин-5-ил)-7-(3-фтор-4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-5,7-дигидро-6Н-пирроло[2,3-d]пиримидин-6-он 6, содержащий некоторые примеси.
Аналитические данные 5-бром-2-(4-циклопропил-6-метоксипиримидин-5-ил)-7-(3-фтор-4-(1-изопропил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-7Н-пирроло[2,3-d]пиримидина 216: ЖХ-МС (Способ А) (ИЭР+): m/z 630 (М+Н)+, 1H-ЯМР (300 МГц, CD3OD) δ 9,03 (с, 1Н), 8,62 (с, 1Н), 7,96 (с, 1Н), 7,87 (с, 1Н), 7,48 (т, J=7,5 Гц, 1Н), 7,31 (д, J=9,0 Гц, 1Н), 5,62 (с, 2Н), 4,21 (м, 1Н), 3,91 (с, 3Н), 1,66 (м, 1Н), 1,40 (д, J=6,6 Гц, 6Н), 1,19-1,25 (м, 2Н), 0,82-0,89 (м, 2Н).
Аналитические данные 5,5-дибром-2-(4-циклопропил-6-метоксипиримидин-5-ил)-7-(3-фтор-4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-5,7-дигидро-6Н-пирроло[2,3-d]пиримидин-6-она 6. ЖХ-МС (Способ А)(ИЭР+): m/z 724,726,728 (М+Н)+.
Стадия 4: Синтез 2-(4-циклопропил-6-метоксипиримидин-5-ил)-7-(3-фтор-4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-5,7-дигидро-6Н-пирроло[2,3-d]пиримидин-6-она (217):
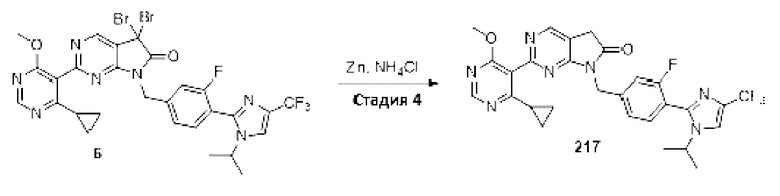
[0790] К раствору 5,5-дибром-2-(4-циклопропил-6-метоксипиримидин-5-ил)-7-(3-фтор-4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-5,7-дигидро-6Н-пирроло[2,3-d]пиримидин-6-она 6 (47 мг, 0,065 ммоль) в ТГФ (1,5 мл) и насыщенном водном растворе NH4Cl (1,5 мл) добавляли порошок Zn (65 мг, 0,26 ммоль) одной порцией. Полученную смесь перемешивали при комнатной температуре в течение 1,5 часов. После завершения реакции, как показывает анализ ТСХ, реакцию гасили H2O (1 мл) и экстрагировали ЕА (3 мл × 3).ов Объединенный органический спой сушили над Na2SO4, фильтровали и упаривали. Остаток очищали препаративной ВЭЖХ (Метод А), получая 7 мг указанного в заголовке соединения. ЖХ-МС (Способ А) (ИЭР+): m/z 568 (М+Н)+; 1Н-ЯМР (300 МГц, CDCl3) δ 8,66 (с, 1Н), 8,52 (с, 1Н), 7,35-7,52 (м; 4Н), 5,02 (с, 2Н), 4,23 (м, 1Н), 3,93 (с, 3Н), 3,68 (с, 2Н), 1,75 (м, 1Н), 1,40 (д, J=6,6 Гц, 6Н), 1,22-1,26 (м, 2Н), 0,87-0,95 (м, 2Н).
ПРИМЕР 218
Анализ на деубиквитинирование активности USP1 /UAF1 и тестирование ингибиторов
[0791] Некоторые Соединения раскрытия оценивали на активность USP1/UAF1 в анализе убиквитина родамина, модифицированном по сравнению с описанным ранее.
[0792] Активность деубиквитиназы измеряли с использованием убиквитин-родамина 110 в качестве субстрата. Разрыв амидной связи между род амином и с-концевым глицином убиквитина приводит к усилению сигнала флуоресценции. Анализ проводили в 20 мкл общего объема буфера для анализа (50 мМ Трис-HCl, рН 7,8, 0,5 мМ ЭДТА 0,01% бычьего сывороточного альбумина, 1 мМ DTT, 0,01% Твин-20) и 0,05 нМ фермента USP1/UAF1. Реакцию инициировали добавлением 150 нМ субстрата убиквитин-род амин (Boston Biochem).
[0793] Соединения, растворенные в ДМСО, тестировали в формате доза-ответ, начиная с 10 мкМ.
[0794] Соединения, изображенные ниже, добавляли к смеси фермент/буфер для анализа и инкубировали 10 мин. Добавляли субстратную смесь, реакционную смесь считывали в кинетическом режиме в течение 30 мин при Ех480/Em540 и строили кривые отклика IC50.
[0795] Данные для всех форматов анализа рассчитывали как процент ингибирования по сравнению с контрольными лунками. Процент ингибирования рассчитывали с использованием следующего уравнения % ингибирования = 100×[1-(X-min)/(max-min)], где X - считывание необработанных данных, min - среднее значение контрольных лунок без фермента (n=32), max - среднее значение лунки с ДМСО-контролем (n=32). Значения IC50 рассчитывали с использованием стандартного алгоритма подбора кривой с четырьмя параметрами либо в программном обеспечении Prism GraphPad (La Jolla, СА), либо в Collaborative Drag Discovery (Burlingame, CA) CDD Vault. См. Chem. Biol. 20(1): 55-62 (January 24, 2013); Bioorg. Med. Chem. Lett. 23(20): 5660-5666 (October 15, 2013).
[0796] Следующие Соединения раскрытия ингибируют активность USP1 со значениями IC50, показанными в Таблице 2, Таблице 3 и Таблице 4 ниже.
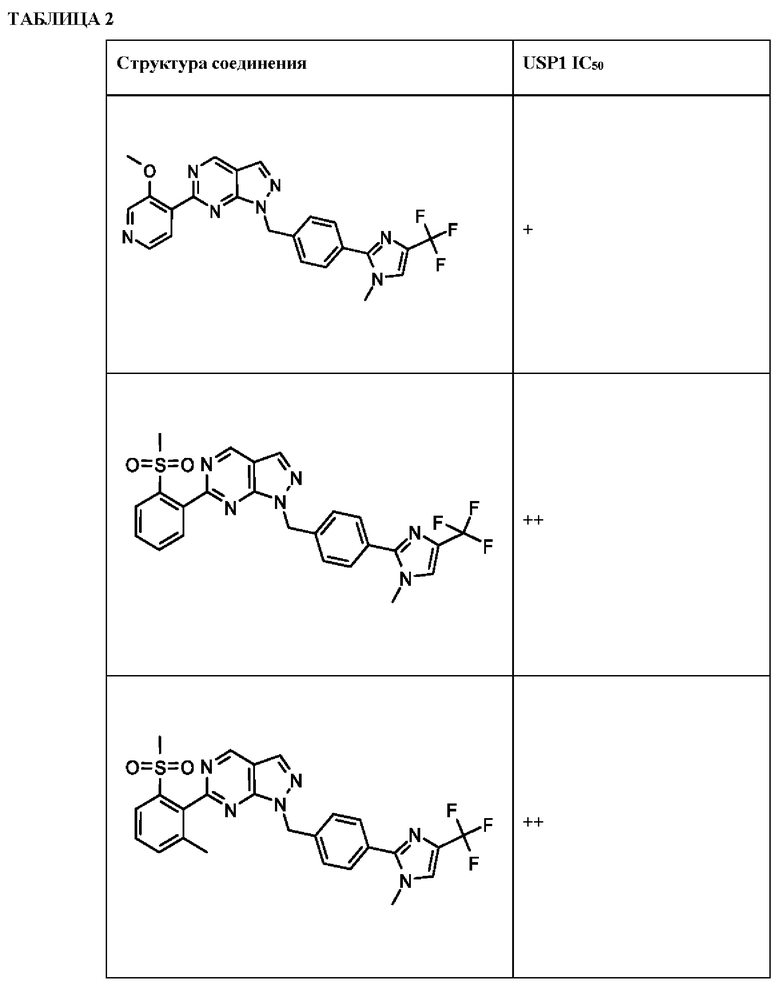

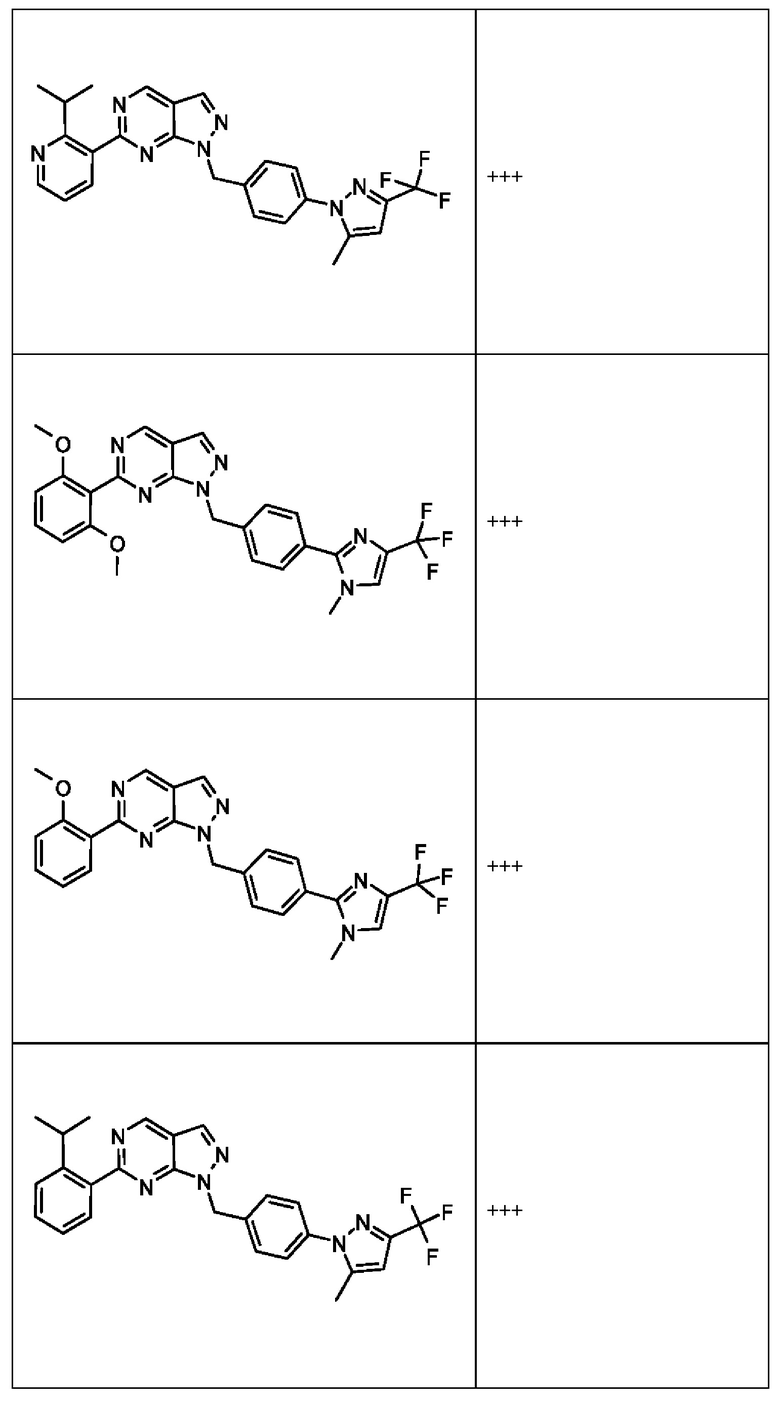
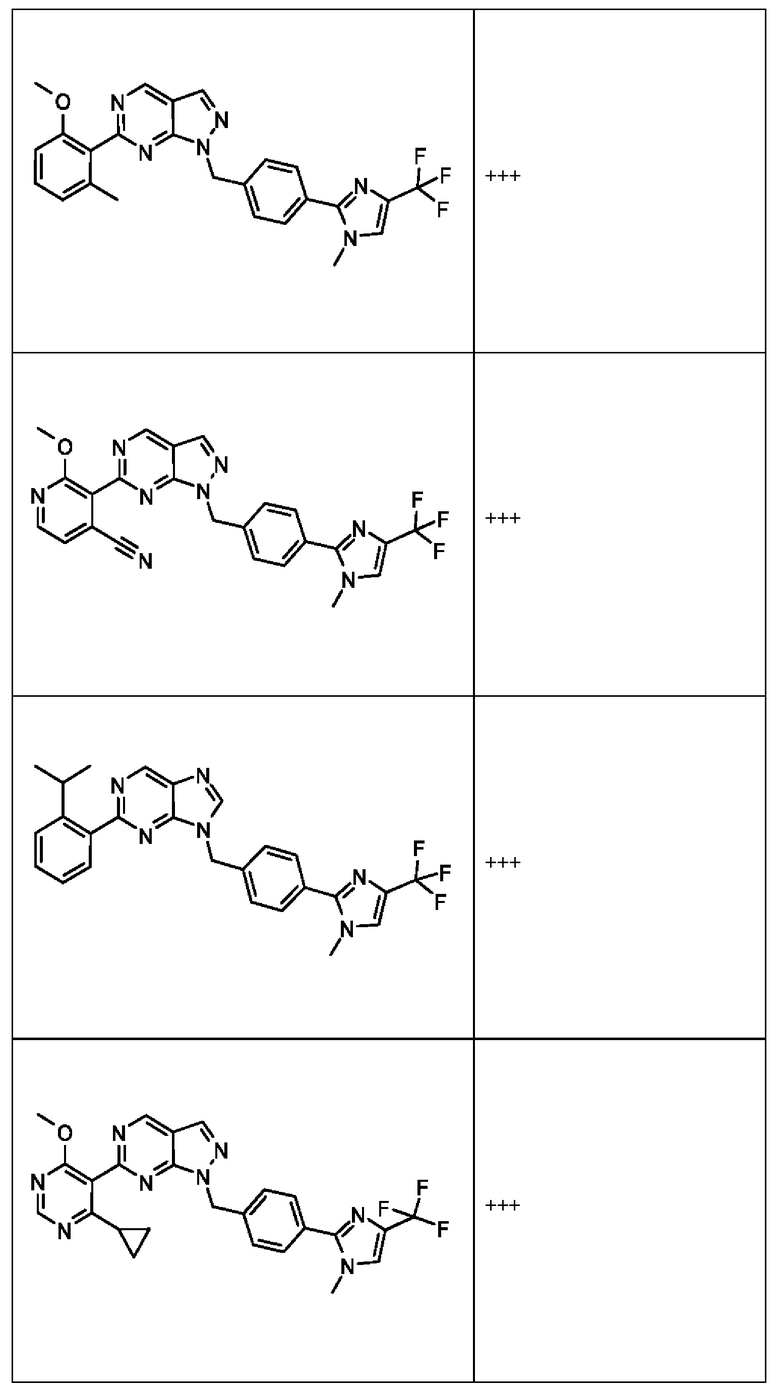
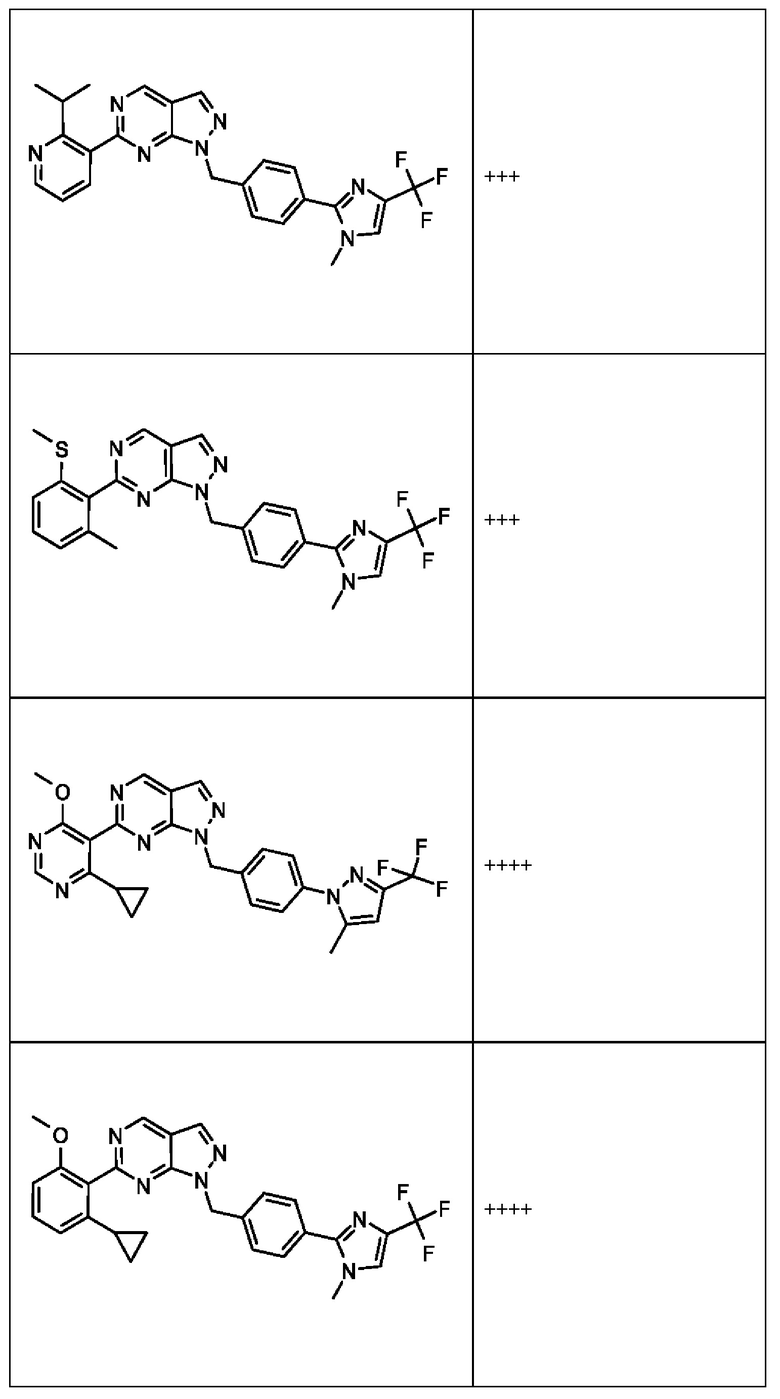
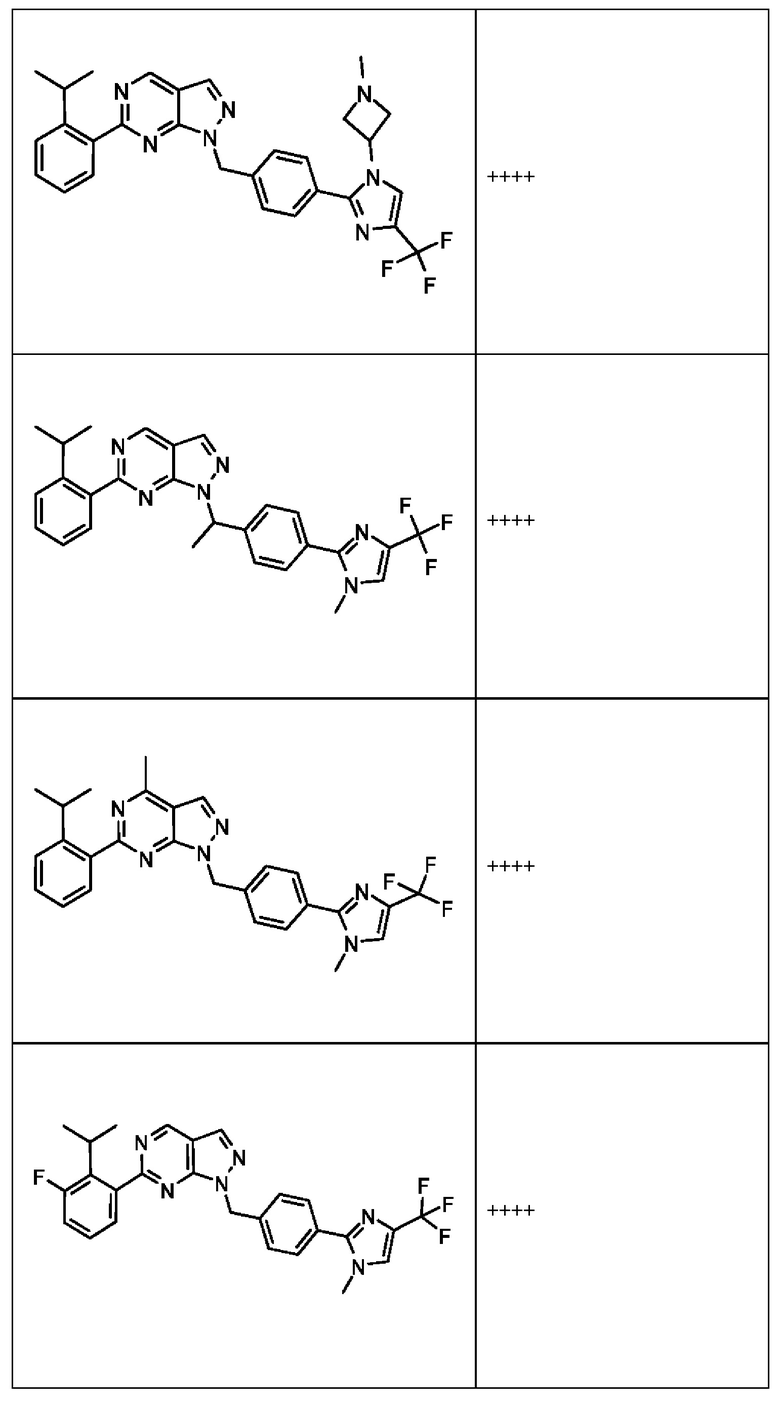
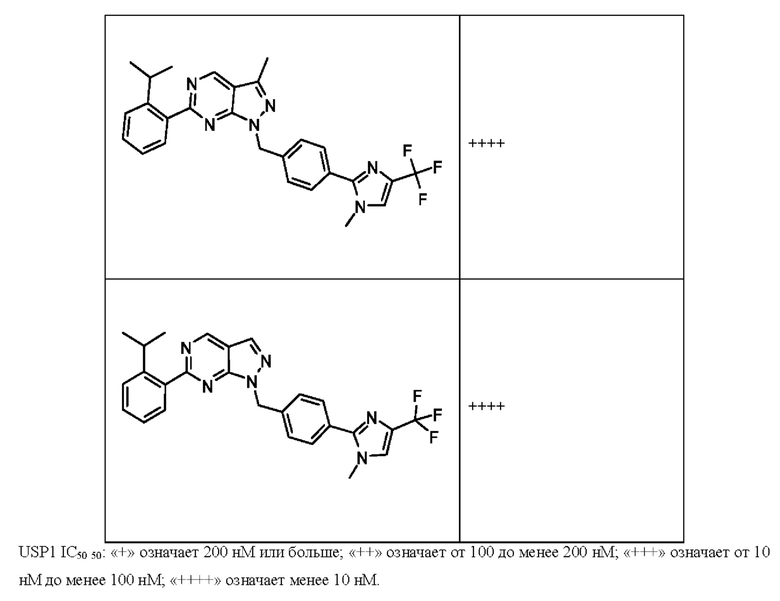
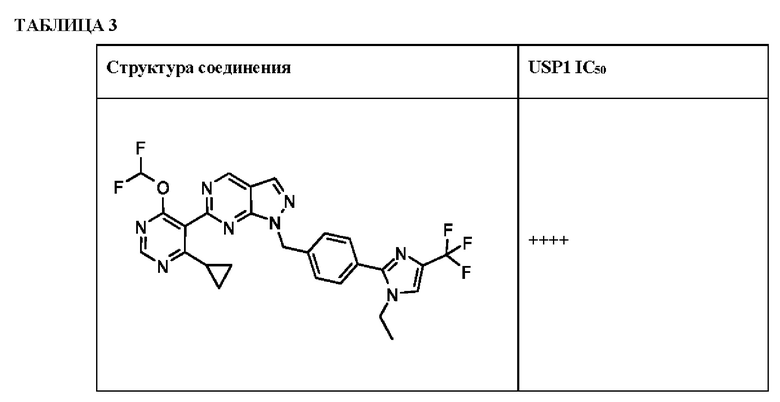
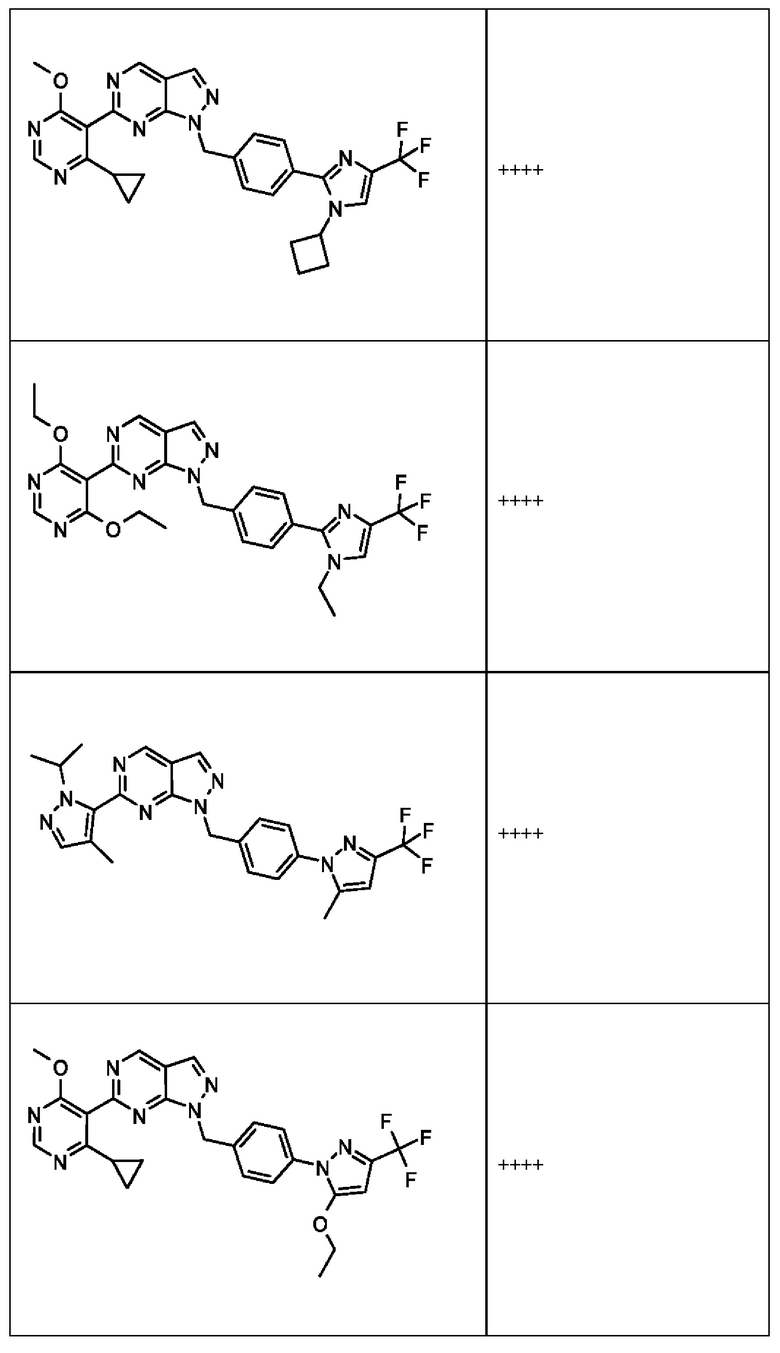
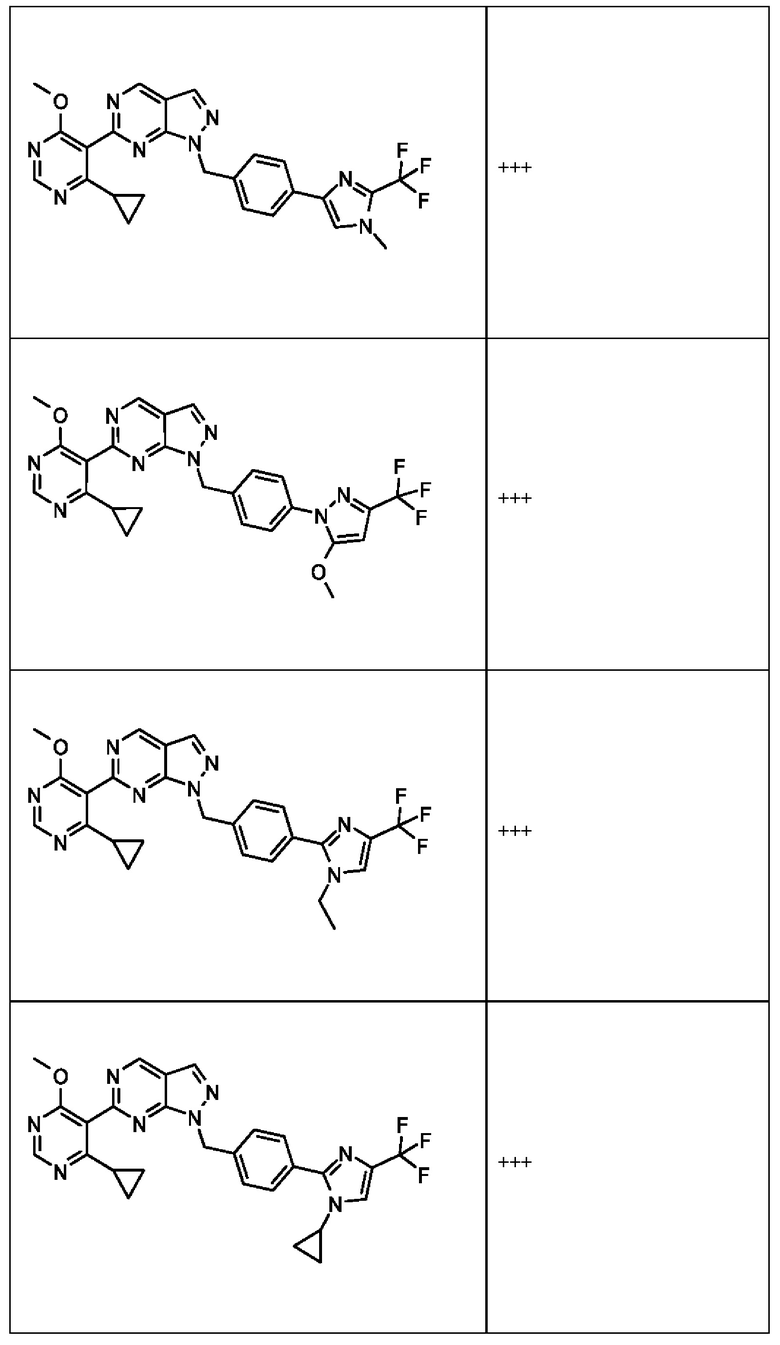
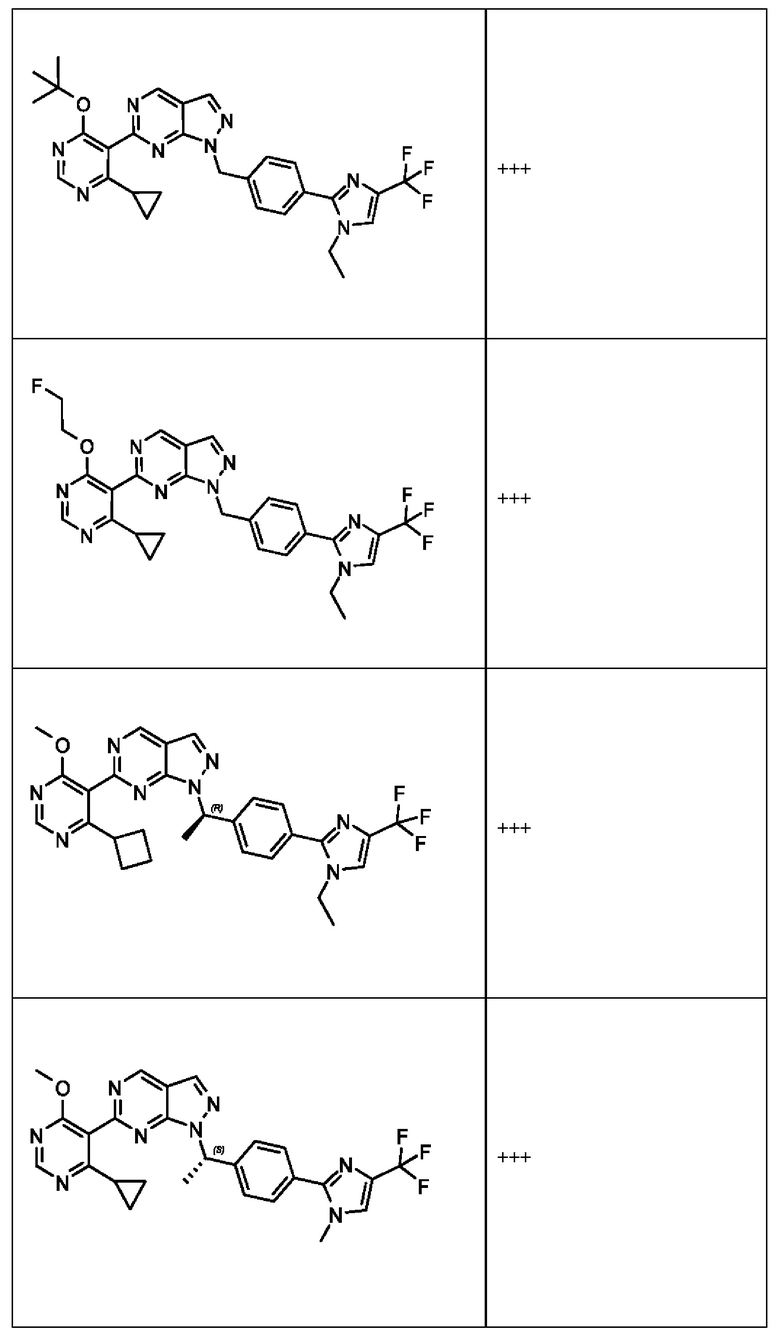
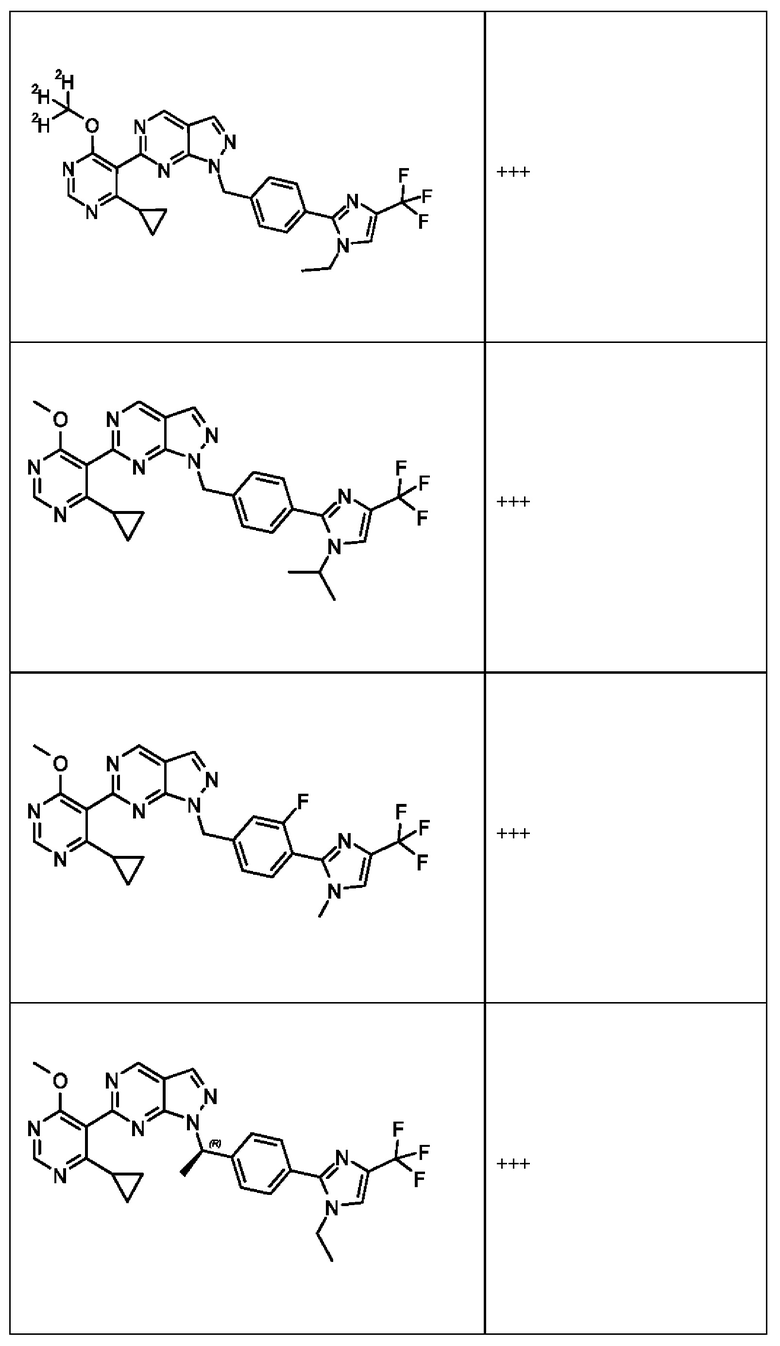
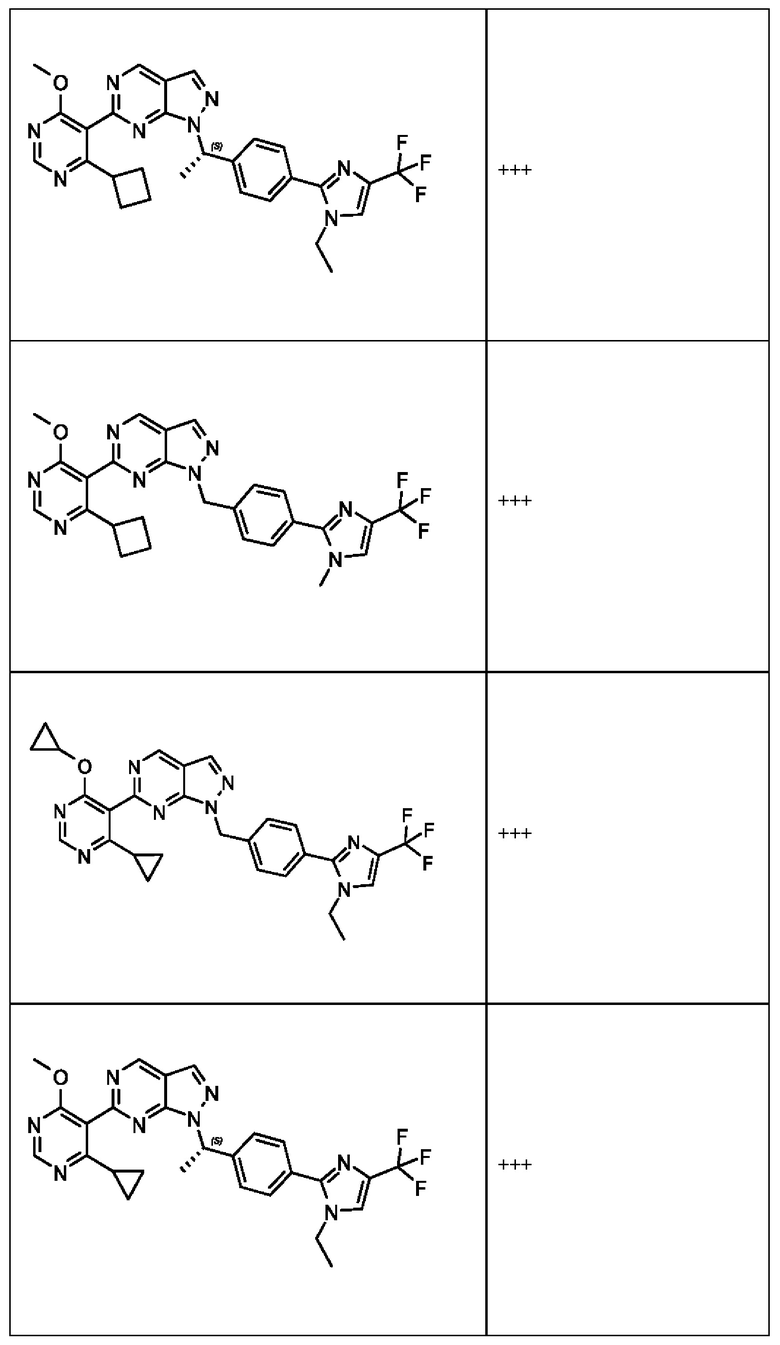
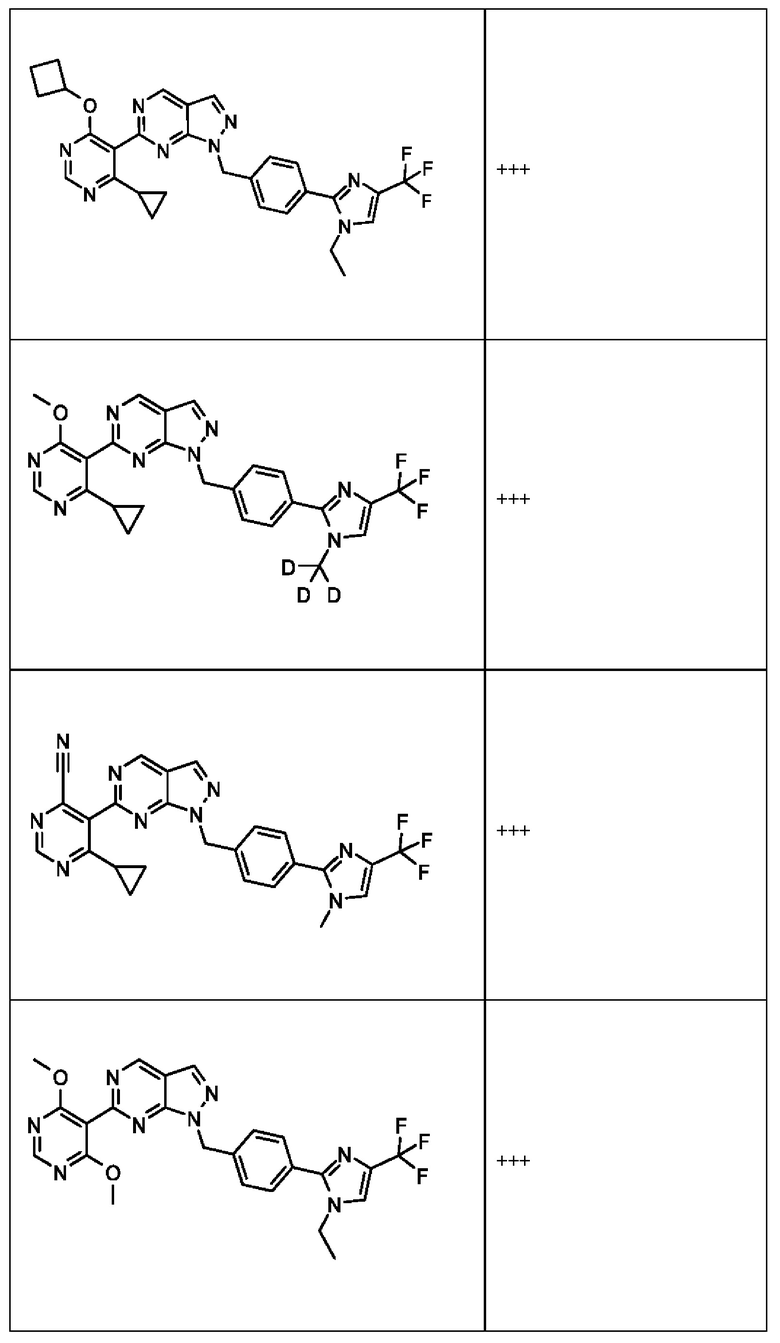
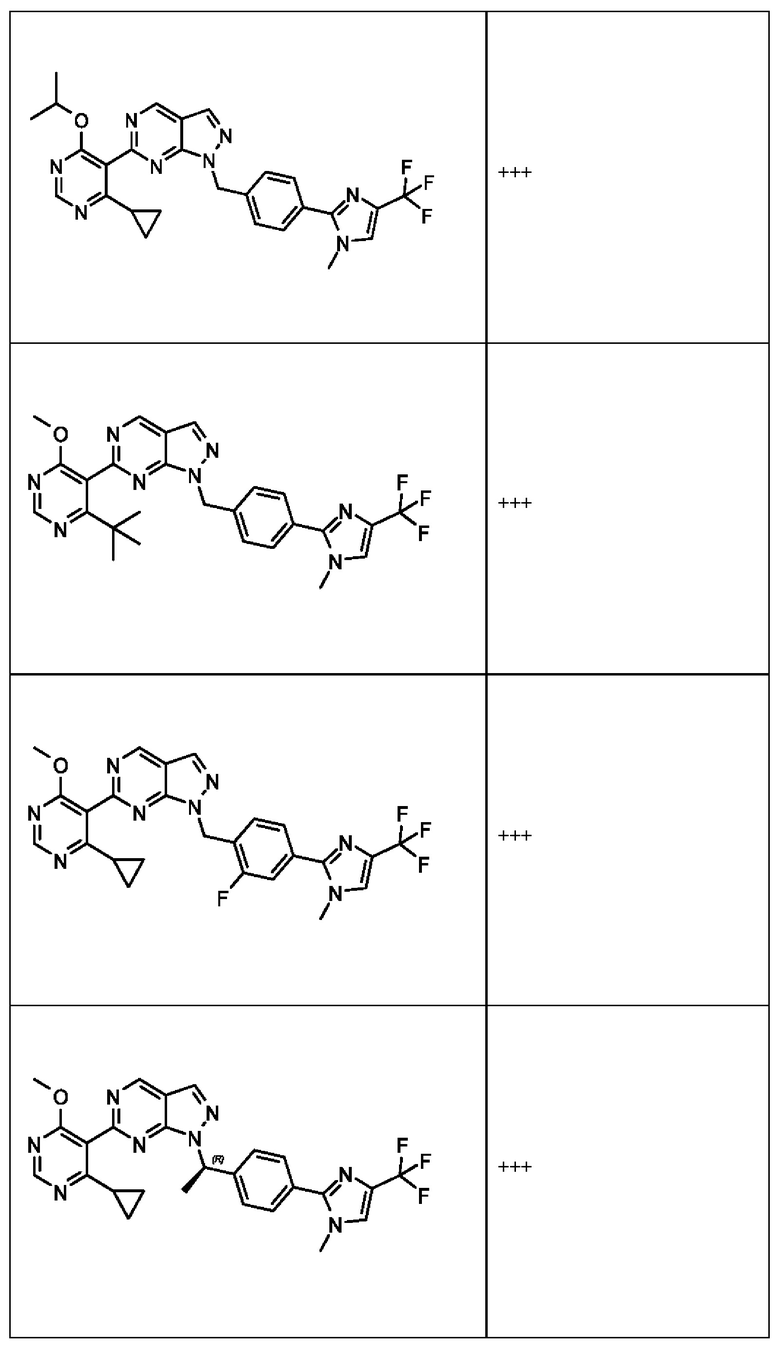
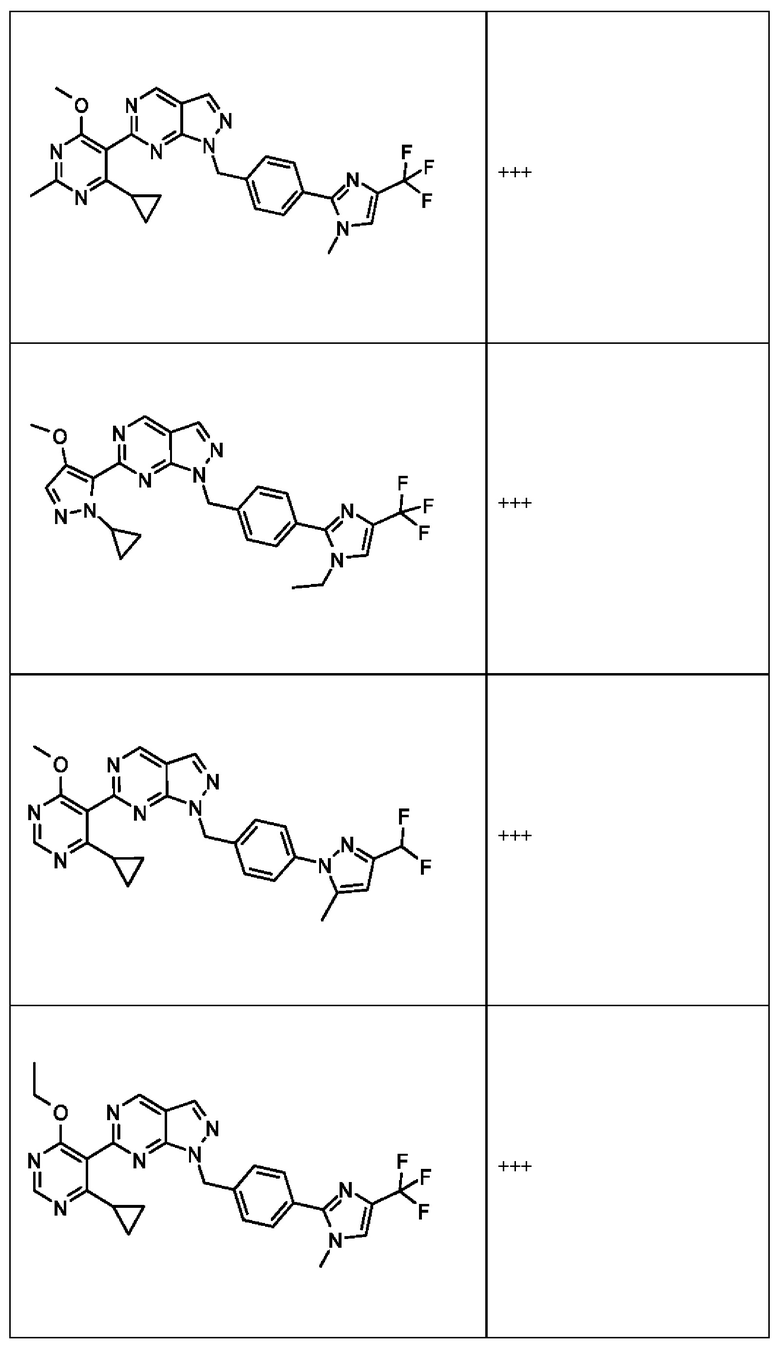
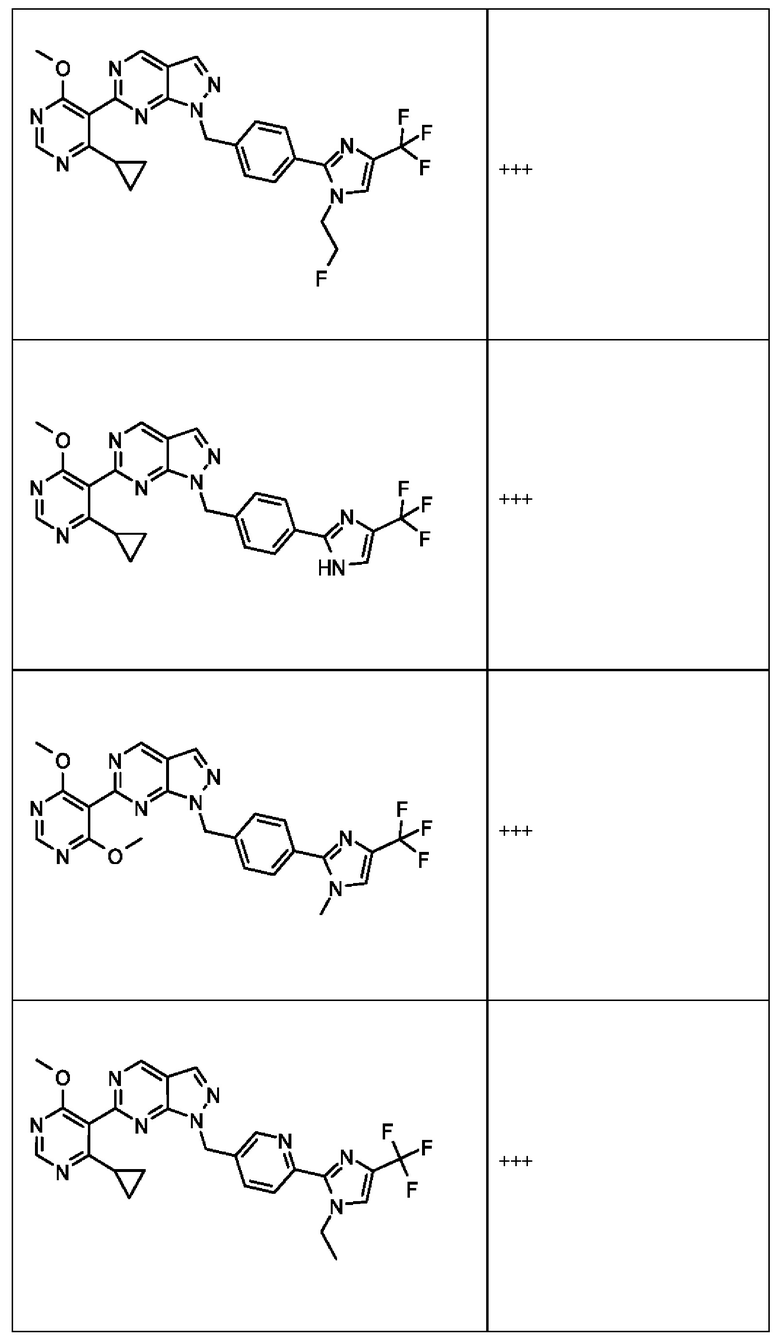
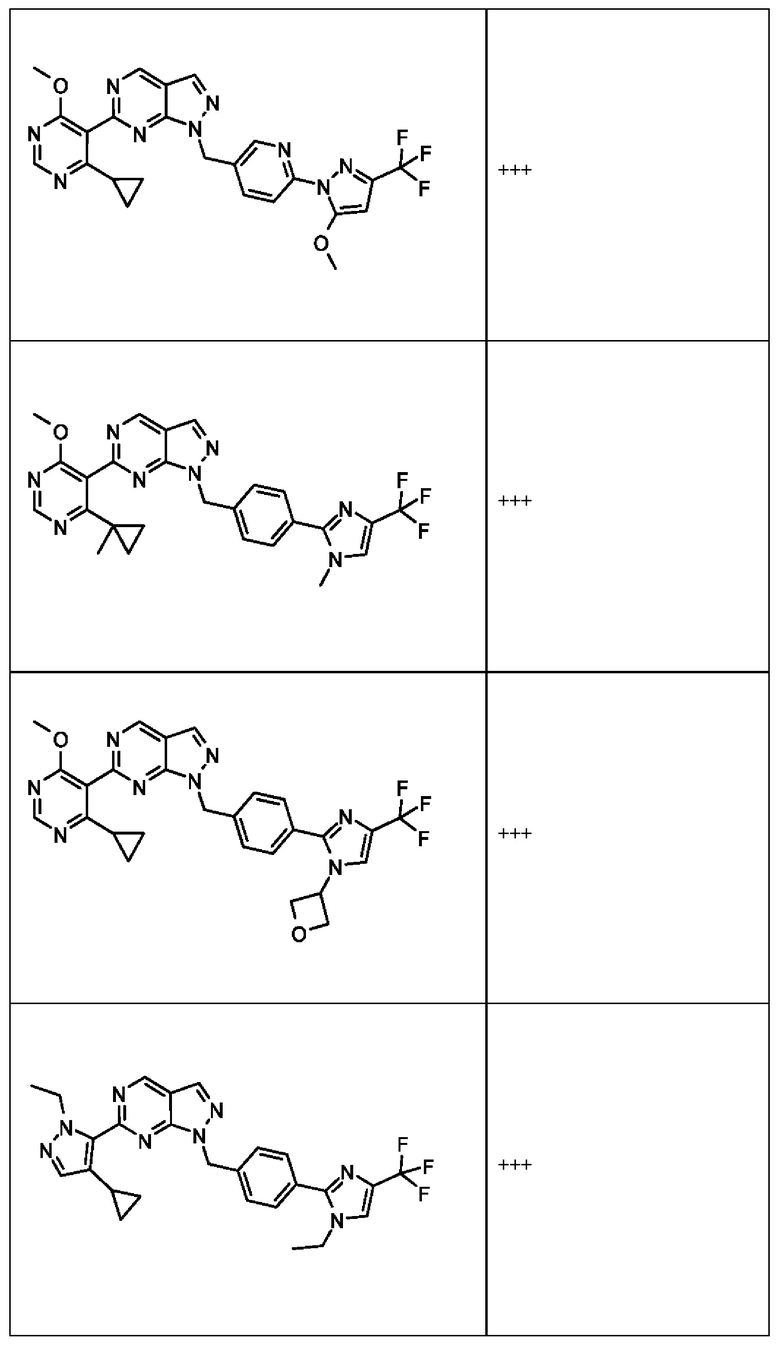
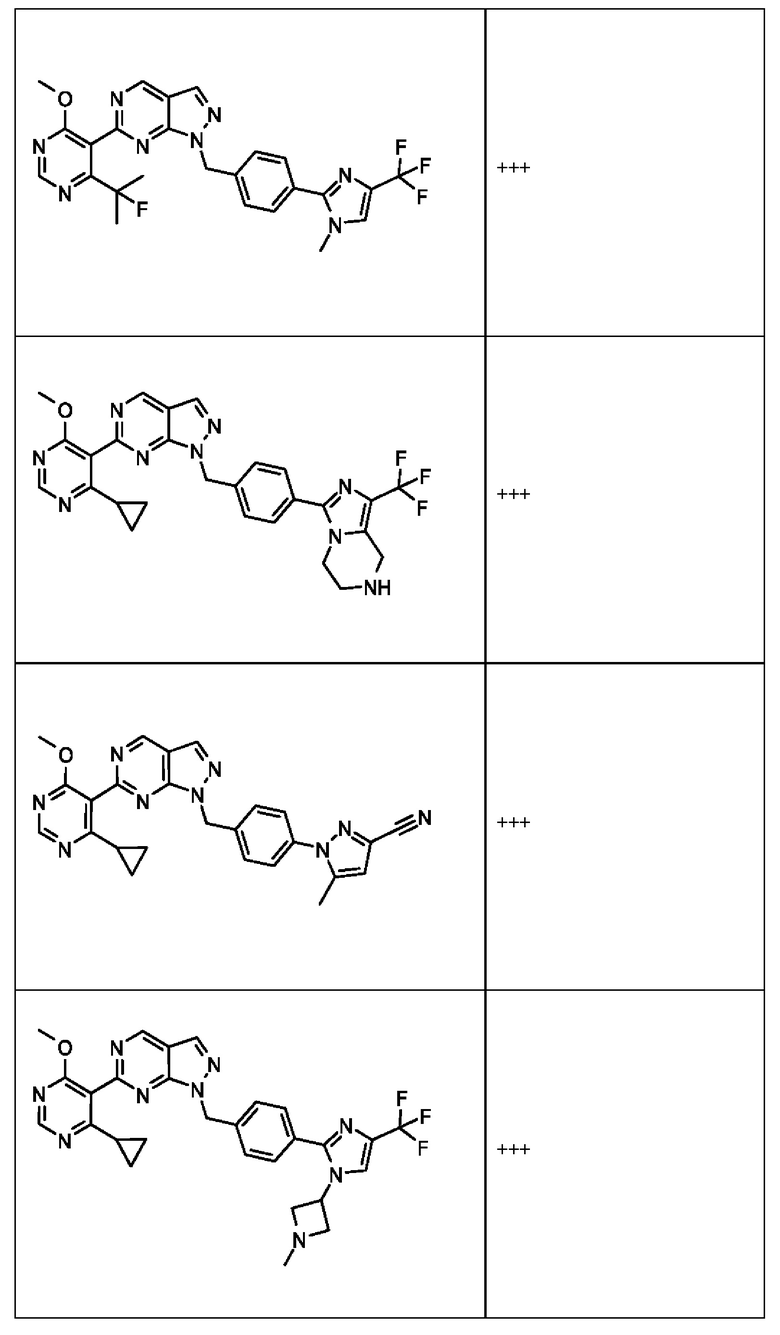
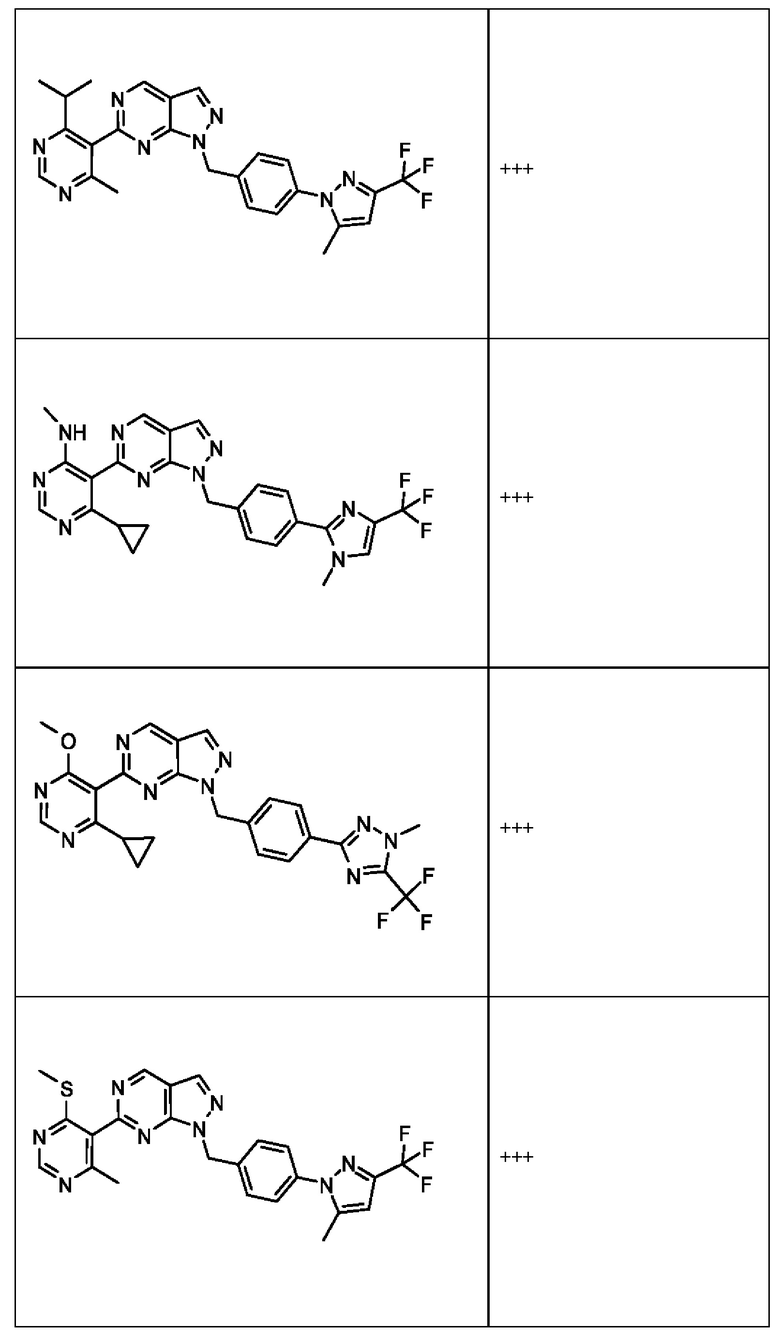
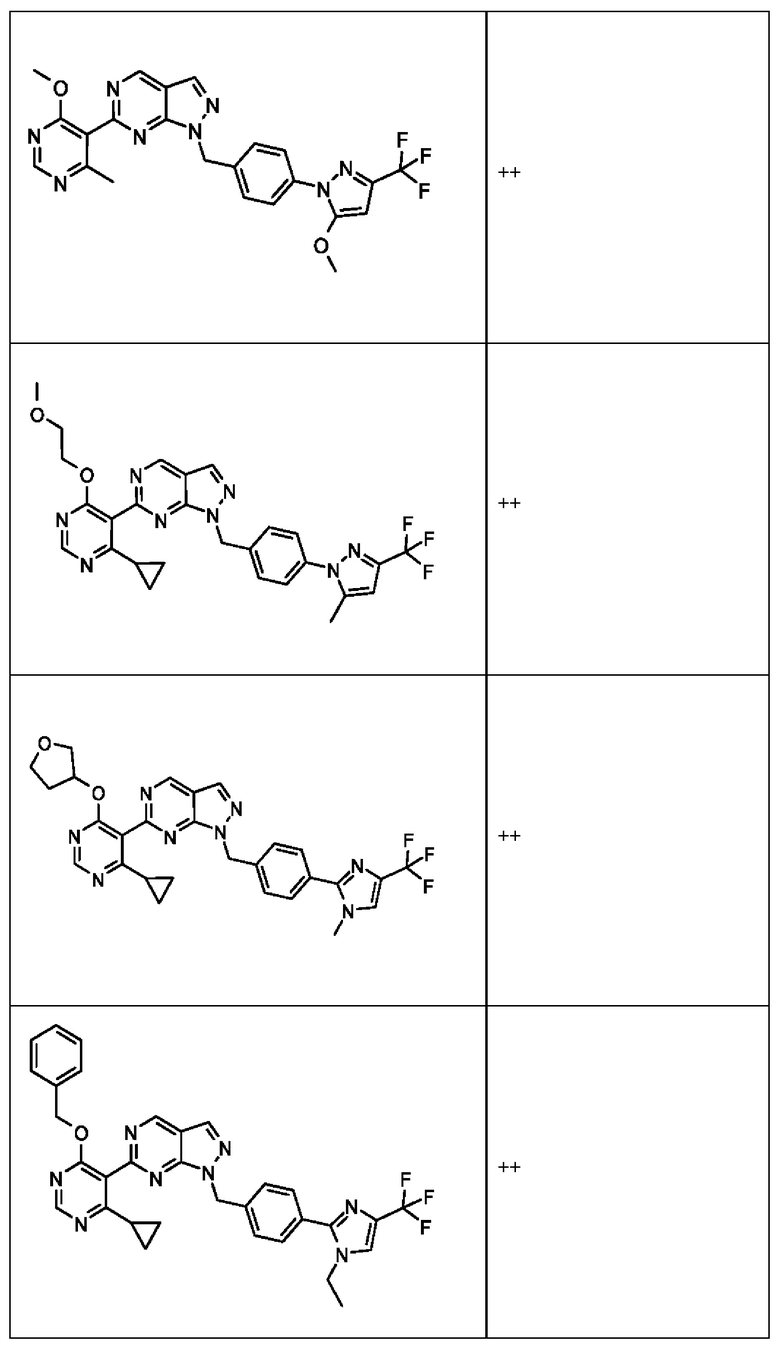
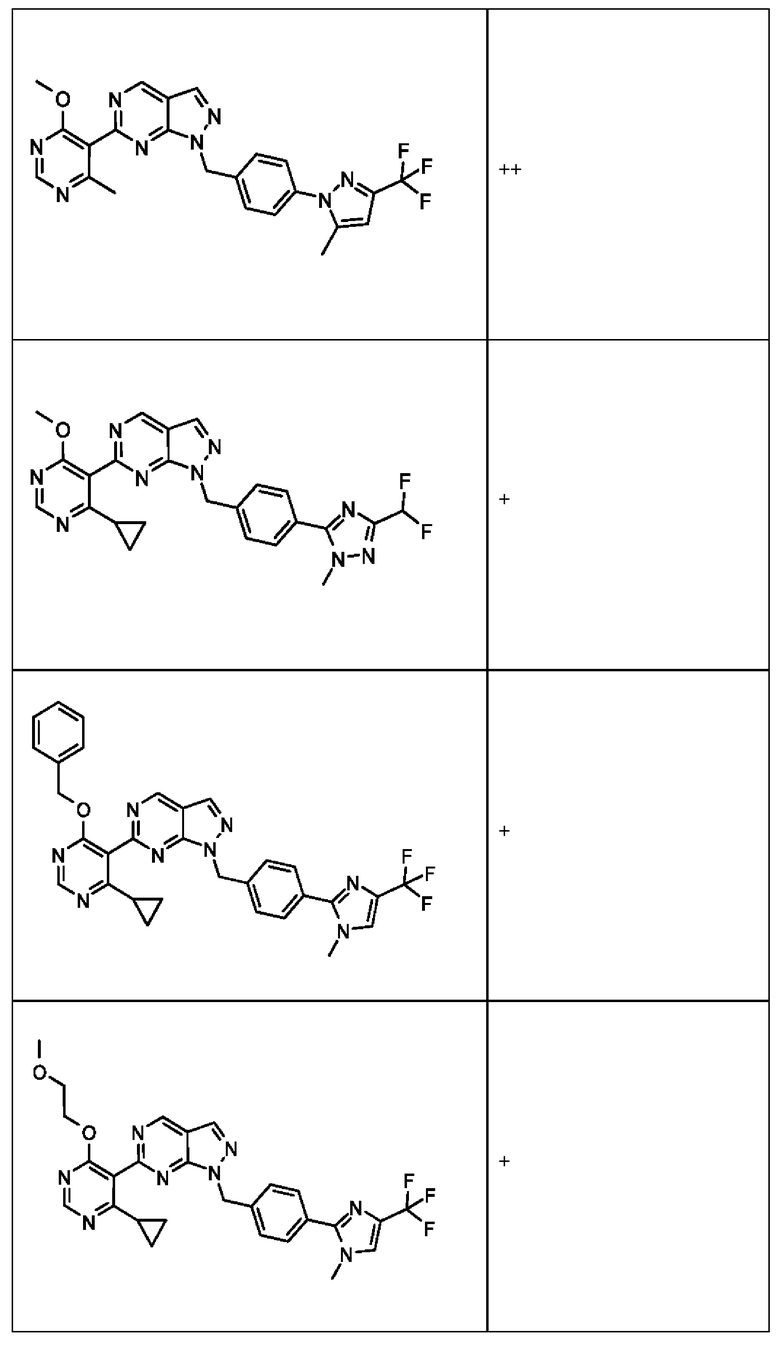
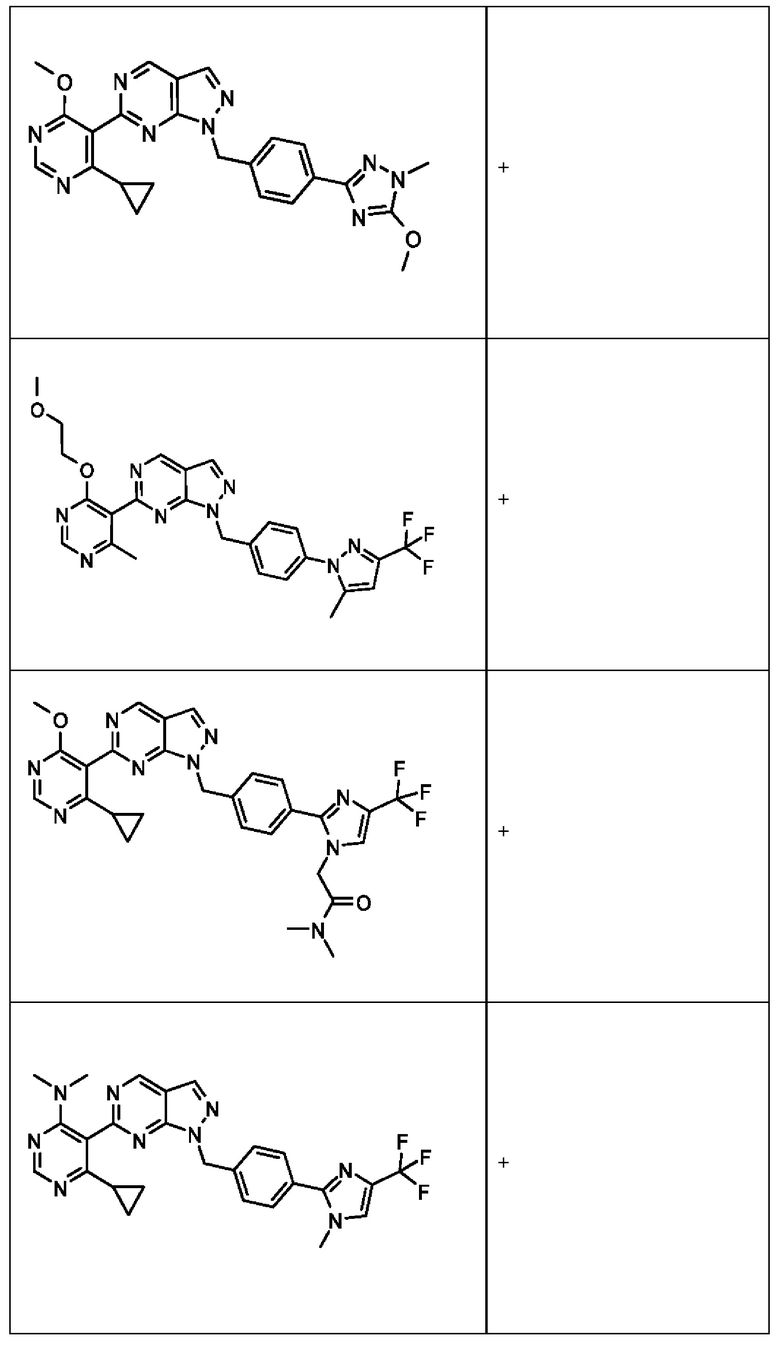
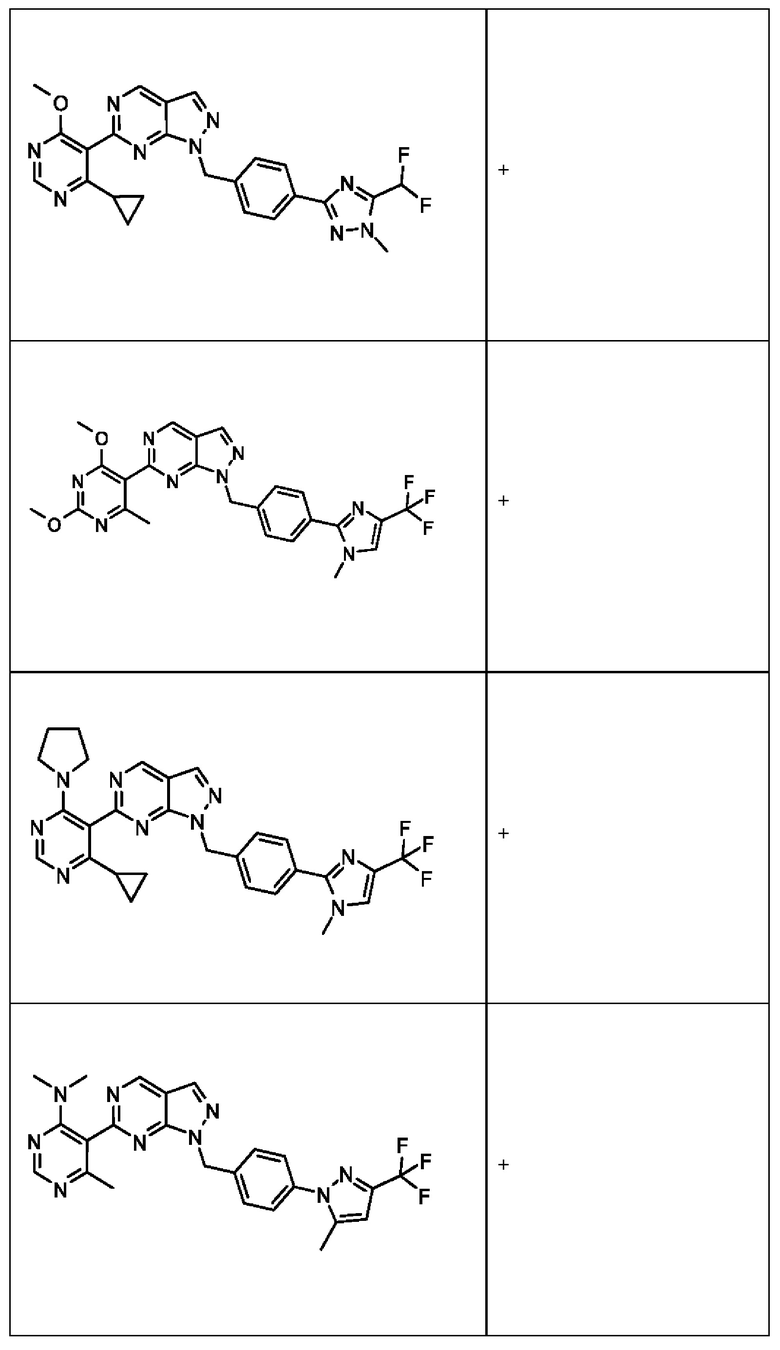
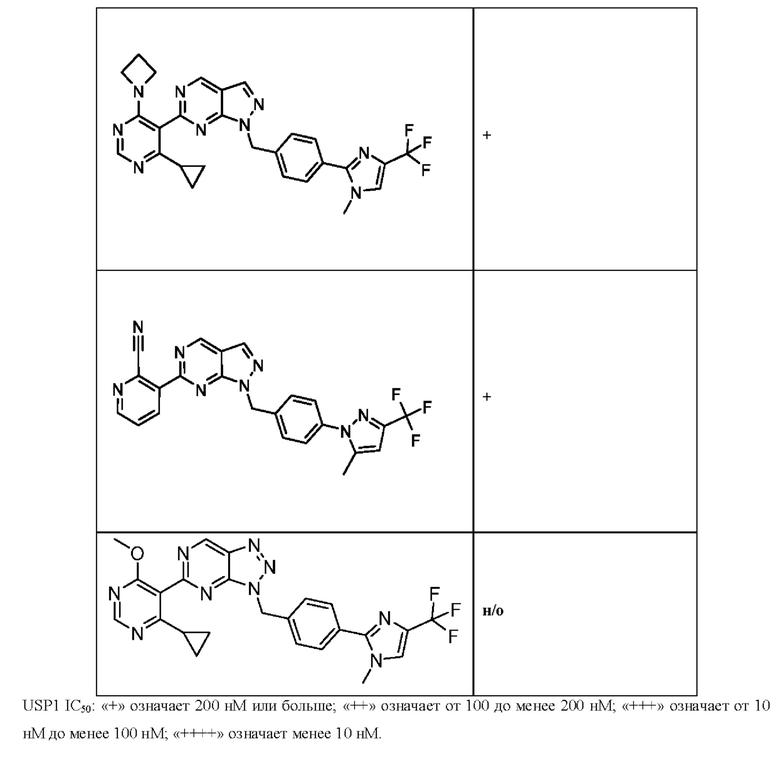
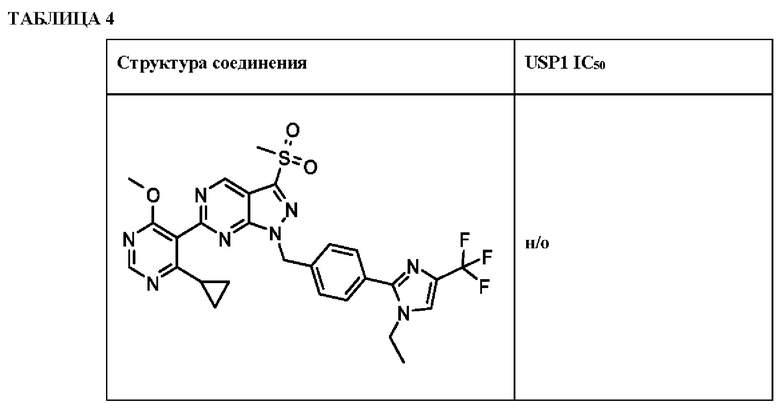

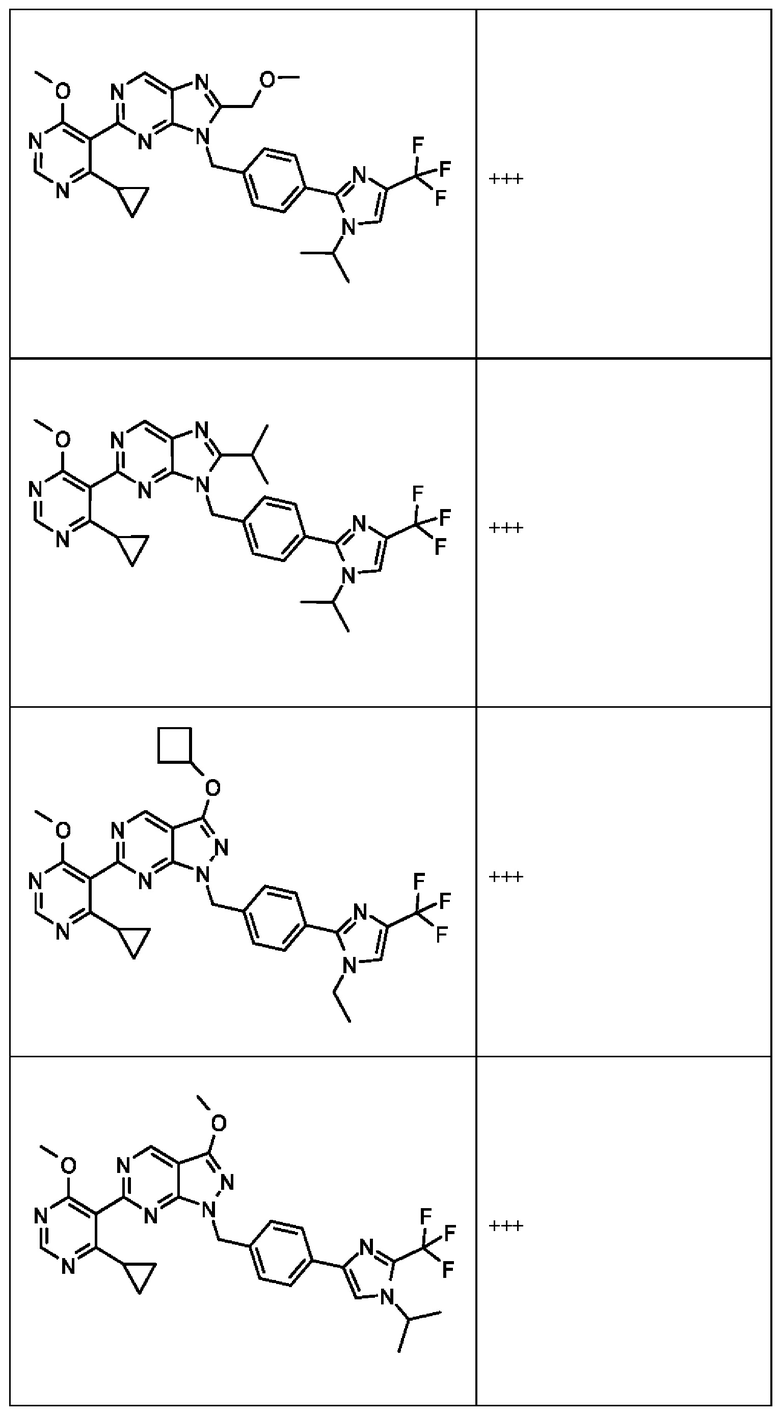
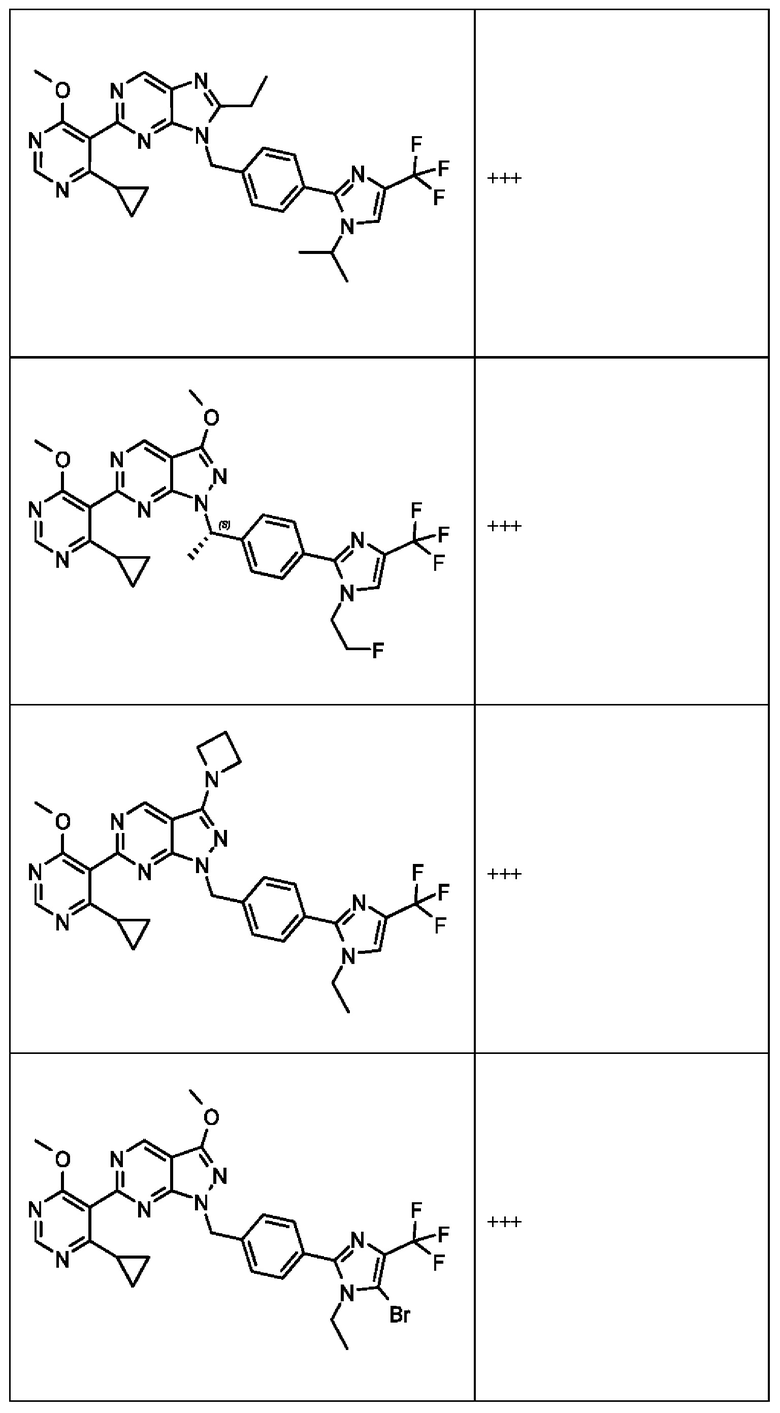
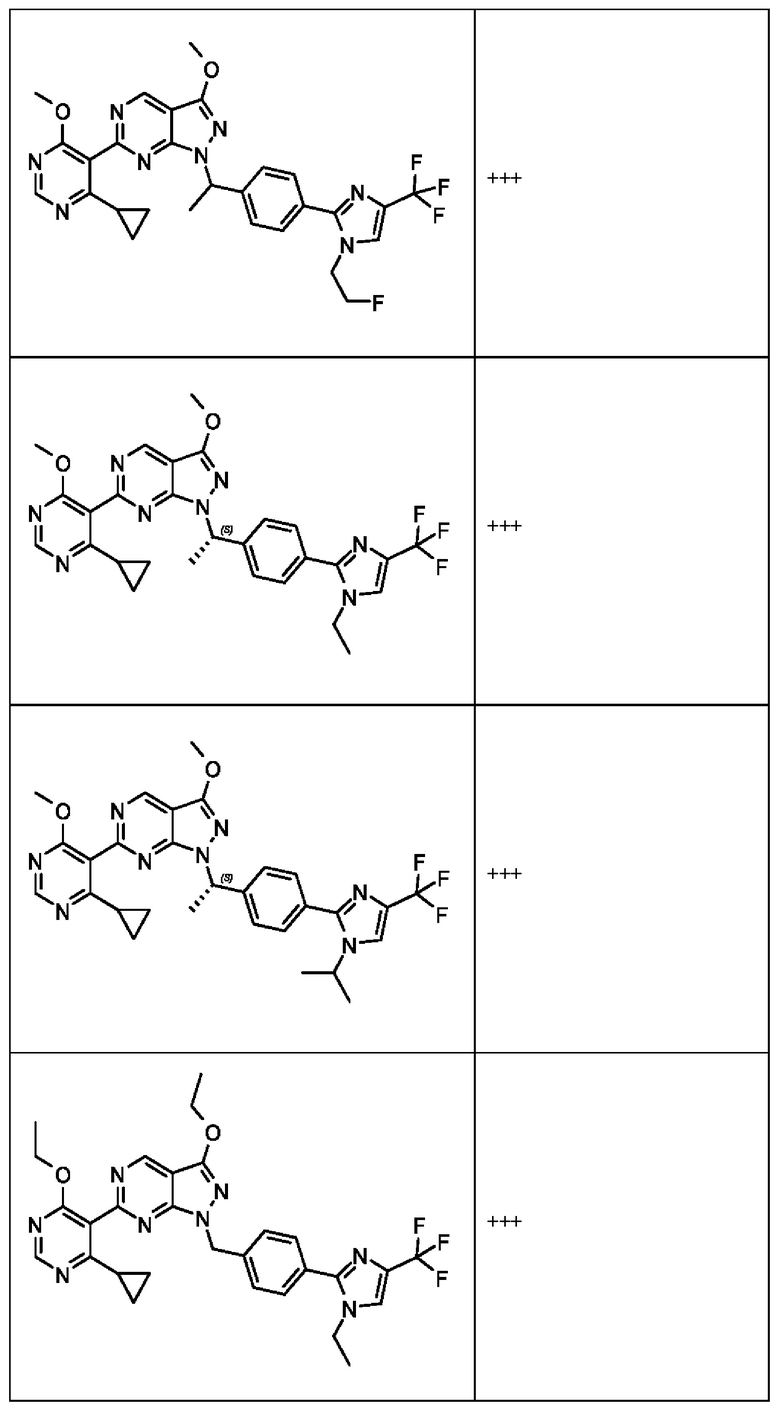
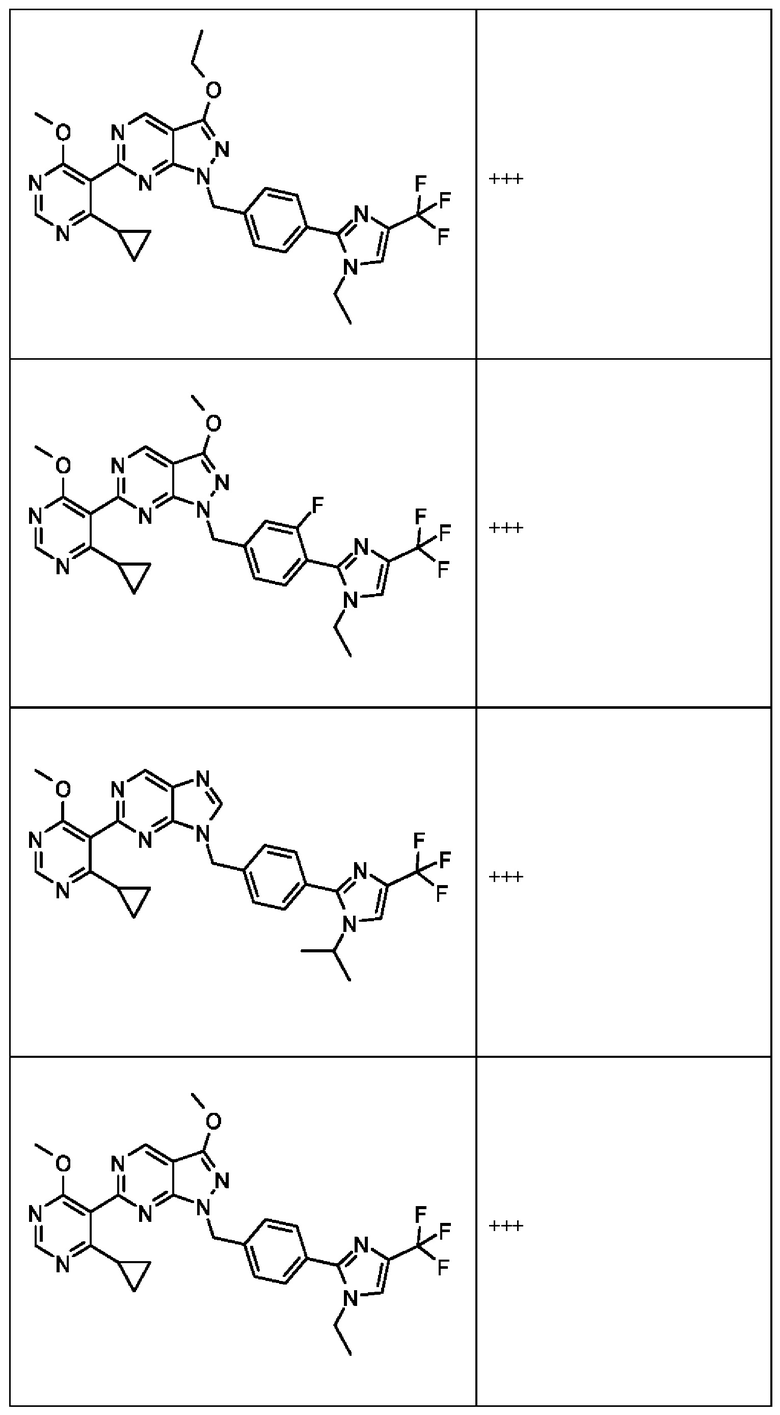
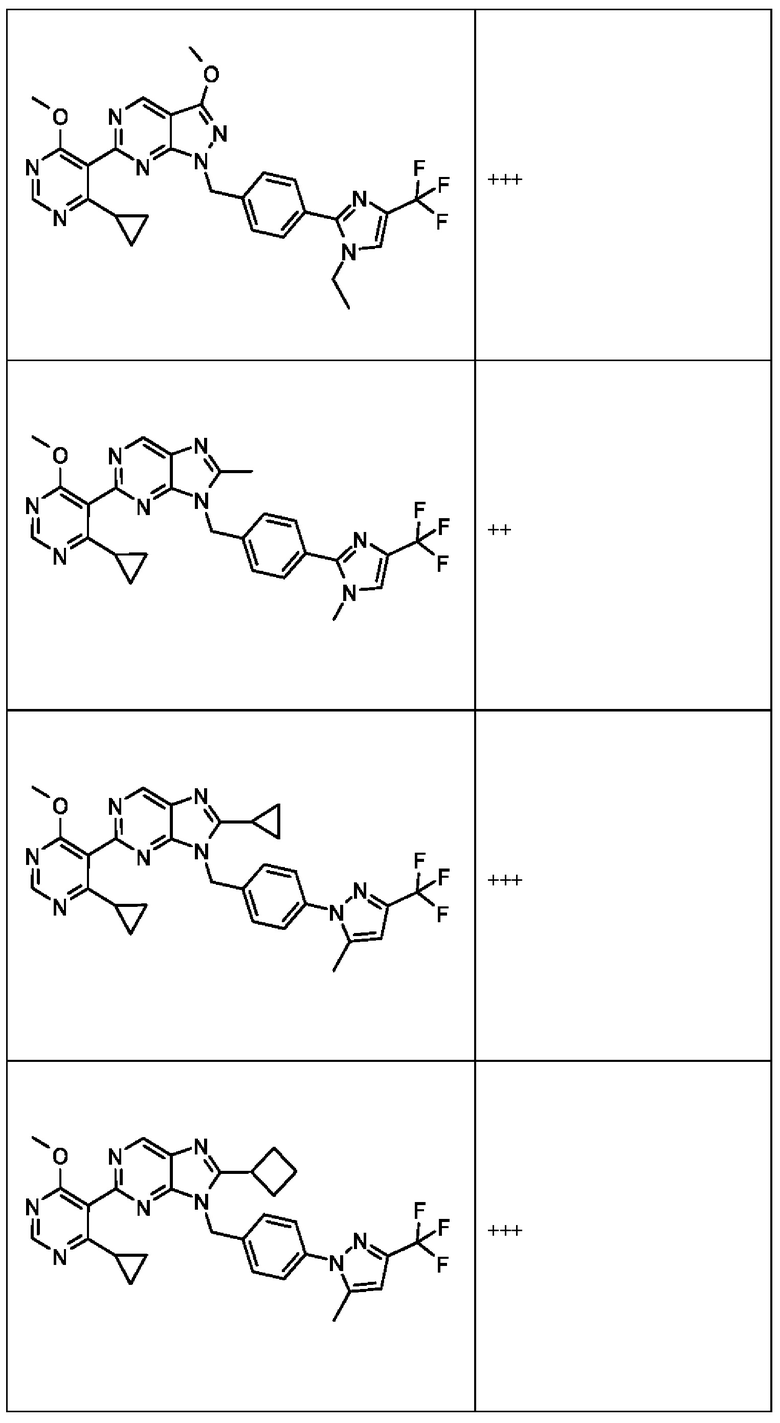
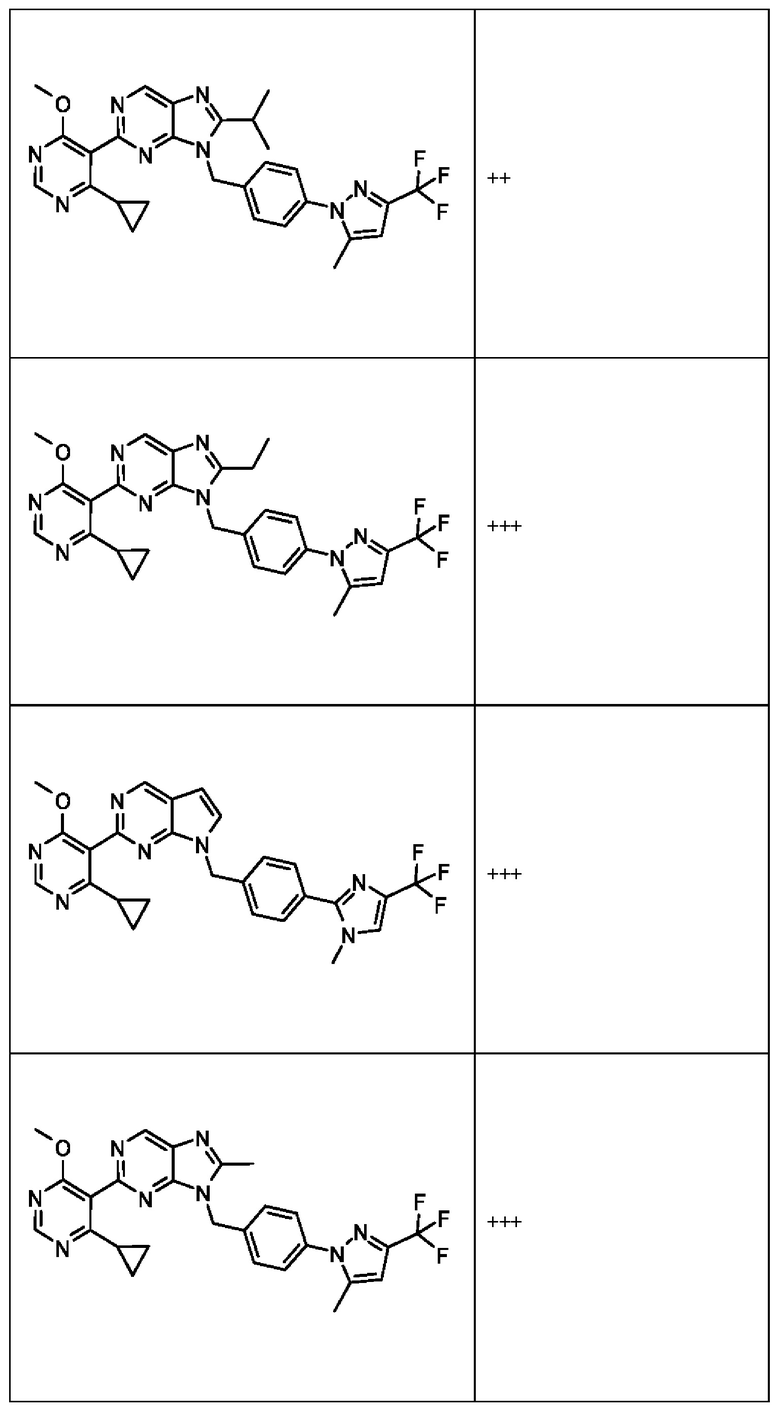
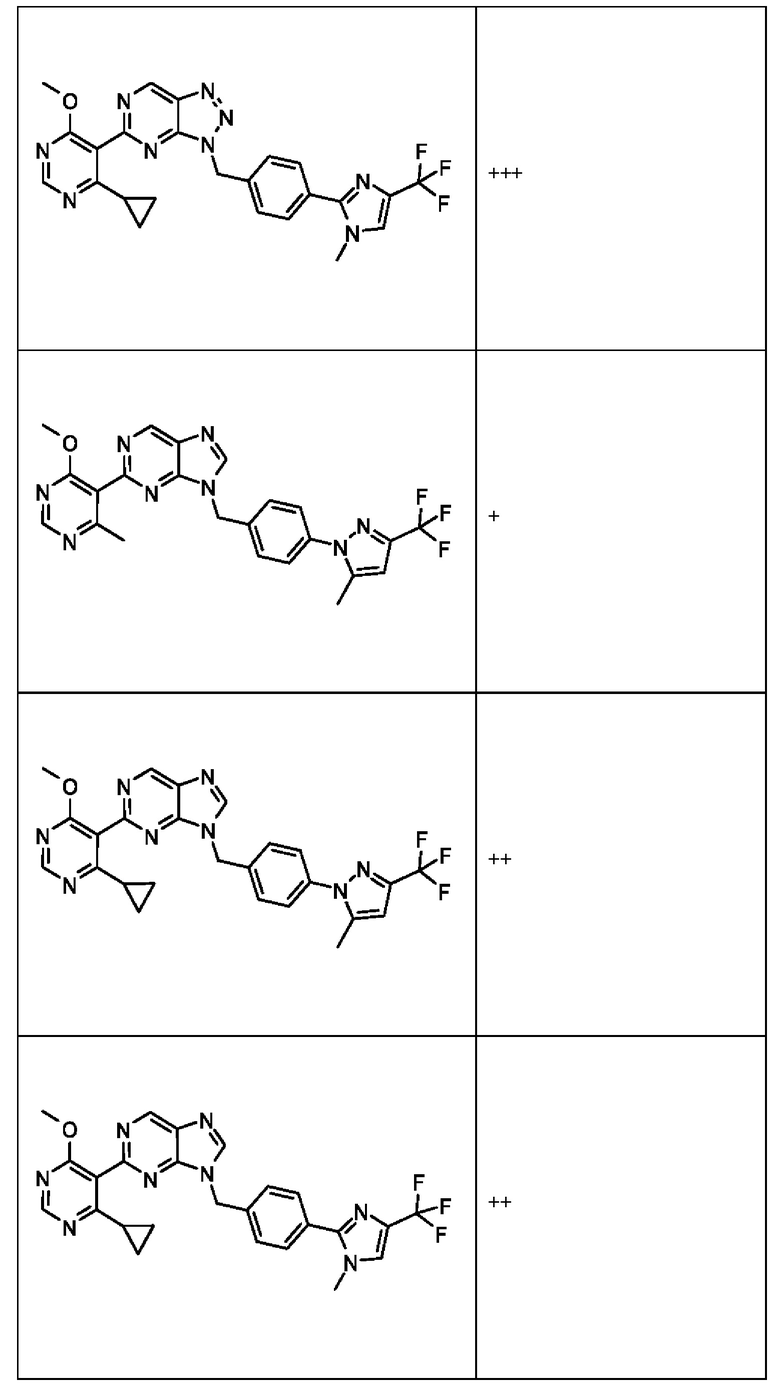
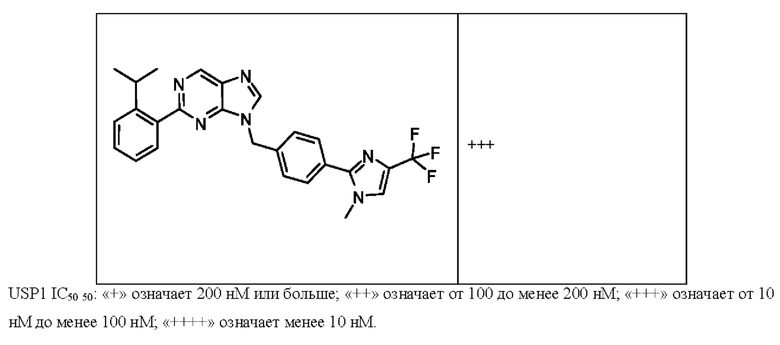
ПРИМЕР 219
Мутации Р53 и BRCA коррелируют с чувствительностью к ингибиторам USP1.
[0797] Чтобы идентифицировать рак, чувствительный к ингибиторам USP1, статус мутации р53 и статус мутации BRCA оценивали в линиях раковых клеток, которые были либо чувствительны, либо нечувствительны к ингибиторам USP1.
[0798] В этих экспериментах использовались два разных формата анализа. Первый формат анализа, называемый анализом долгосрочной пролиферации (LTP), включал посев линий раковых клеток в 6-луночные планшеты с очень низкой плотностью в объеме среды 3 мл с целью не расщепления в течение как минимум 10 дней (обычно 5-20 тыс.клеток/лунку). Клетки высевали в день -1, а в день 0 лунки обрабатывали ДМСО или увеличивающимися концентрациями ингибитора USP1. На протяжении всего анализа клетки, обработанные ДМСО, проверяли на конфлюэнтность, и если конфлюэнтность клеток достигала 80%, клетки снова разделяли на 20-40%. Затем соотношение, необходимое для достижения этого, применяли к другим лункам, обработанным ингибитором USP1. Каждые 3-4 дня меняли среду, содержащую соответствующие концентрации ДМСО или ингибитора USP1. В конце эксперимента рост клеток измеряли с использованием реагента CellTiter-Glo® (Promega), и результаты регистрировали с помощью планшет-ридера SynergyHTX. Второй формат анализа представлял собой анализ образования колоний (CFA). Этот анализ требовал сначала установить, какая плотность посева клеток позволила развить четко перемежающиеся колонии на шестилуночном планшете, когда они оставались для роста на около 14 дней. После определения этой плотности клетки высевали в день -1, а в день 0 лунки обрабатывали ДМСО или увеличивающимися концентрациями ингибитора USP1. На 8 день меняли среду, содержащую соответствующие концентрации ДМСО или ингибитора USP1. На 14 день или около того, когда в лунках, обработанных ДМСО, были видны четко перемежающиеся колонии, клетки фиксировали и окрашивали с использованием 0,1%. кристаллического фиолетового в 10% этаноле в течение 20 минут при комнатной температуре. Планшеты визуализировали, затем количество окрашивания кристаллического фиолетового в каждой лунке определяли количественно путем экстракции кристаллического фиолетового 10% уксусной кислоты и измерения оптической плотности при 565 нм. Результаты показаны в Таблице ниже.

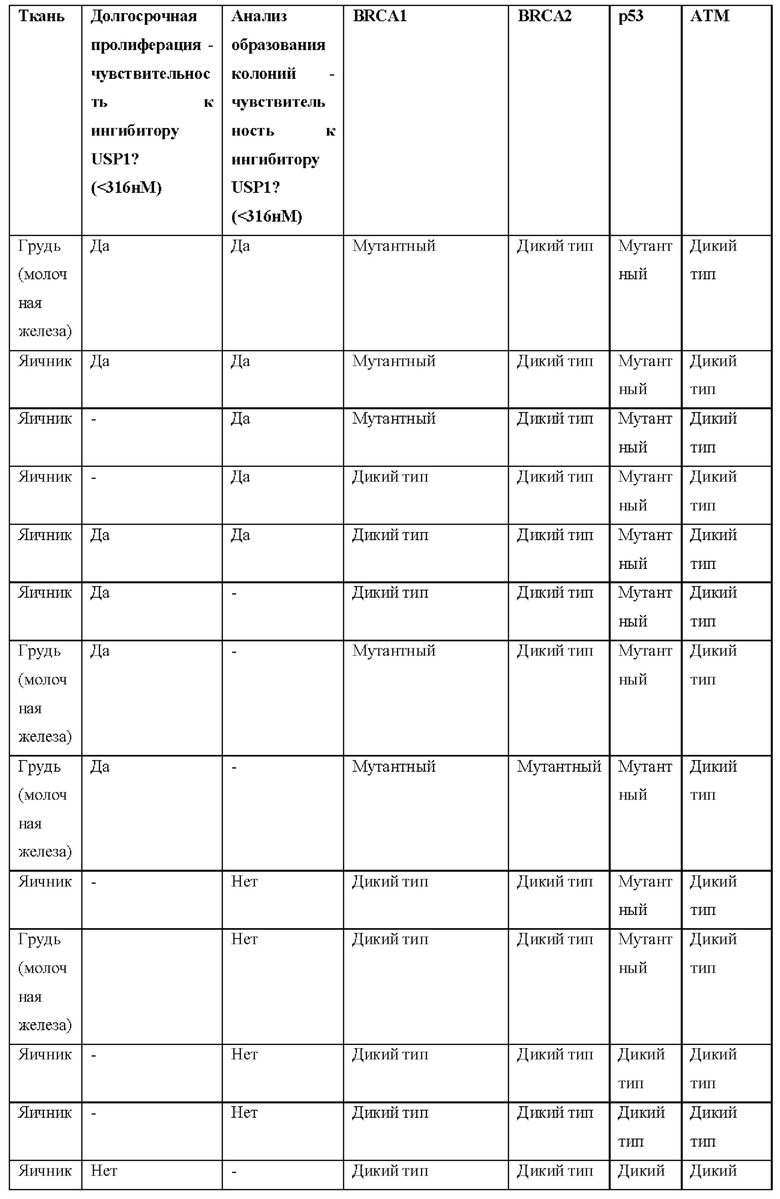
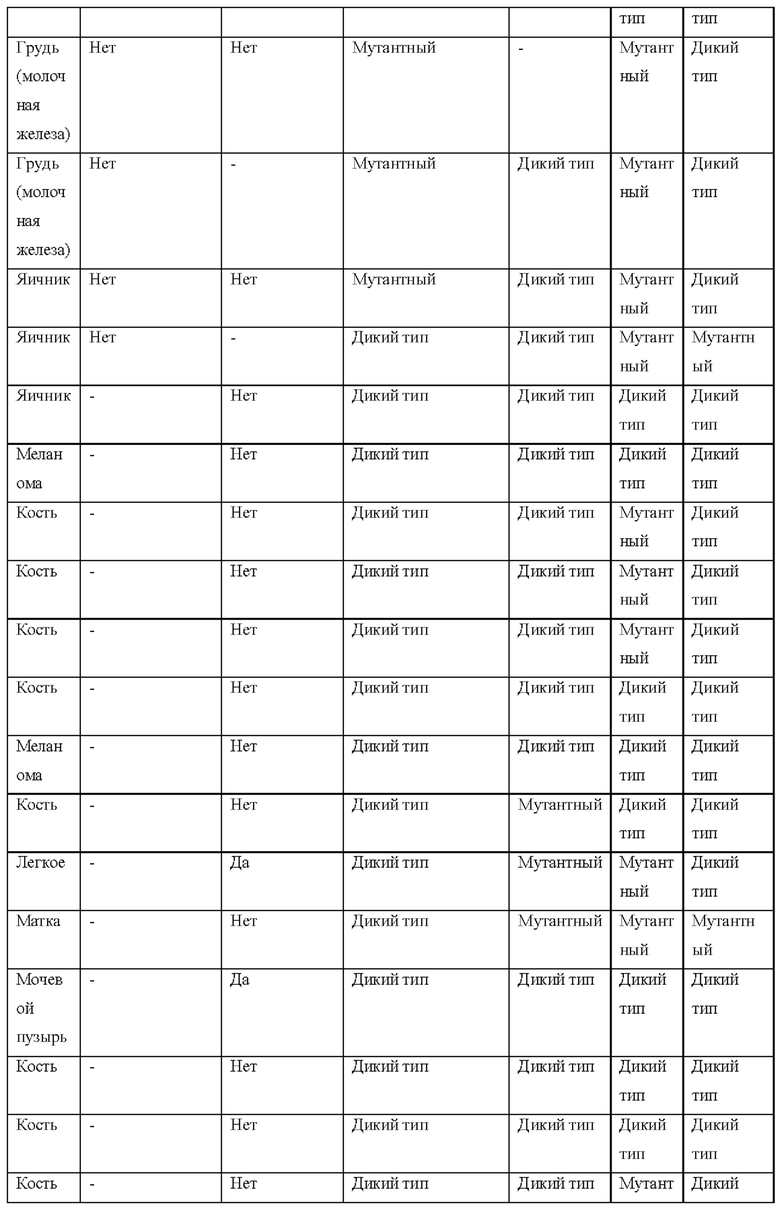
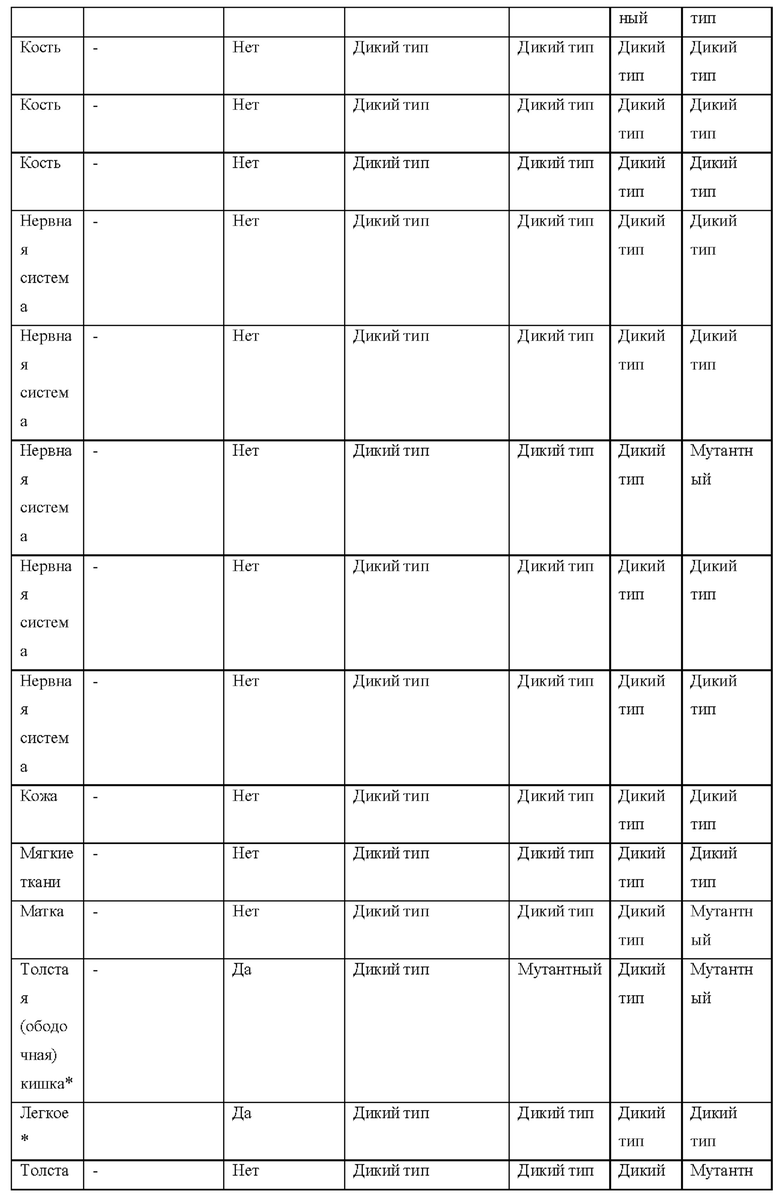
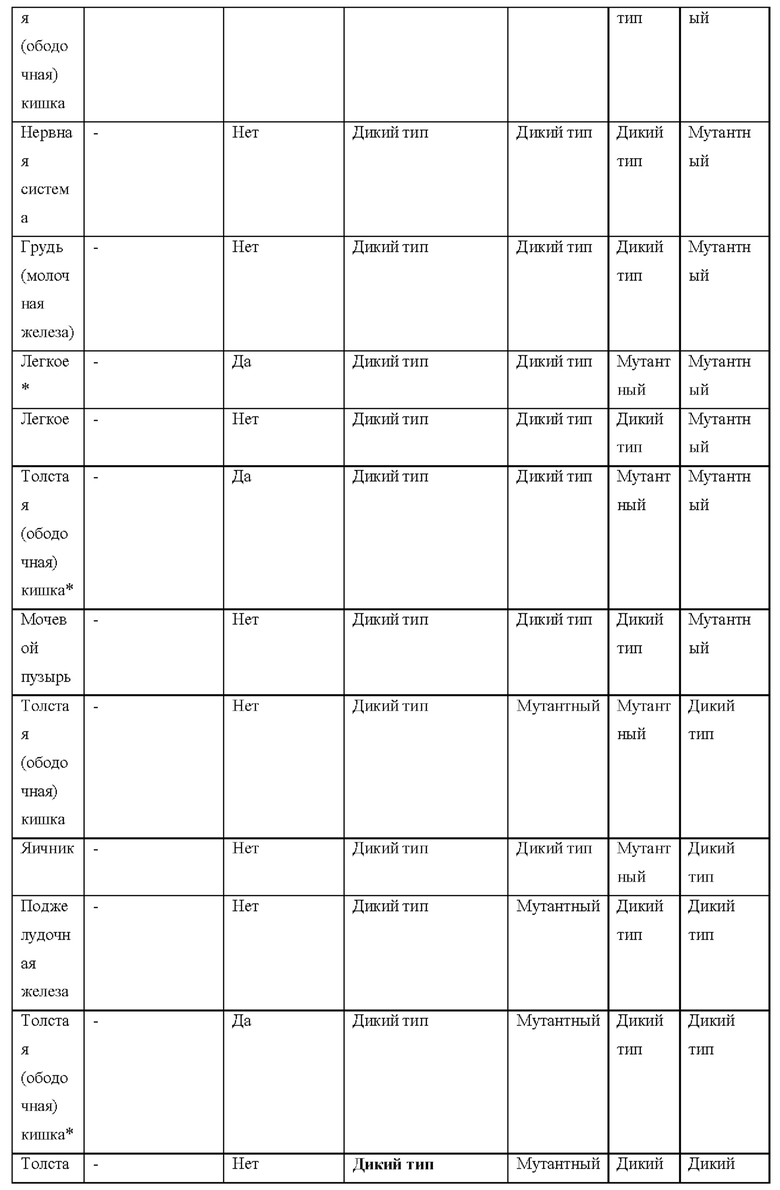
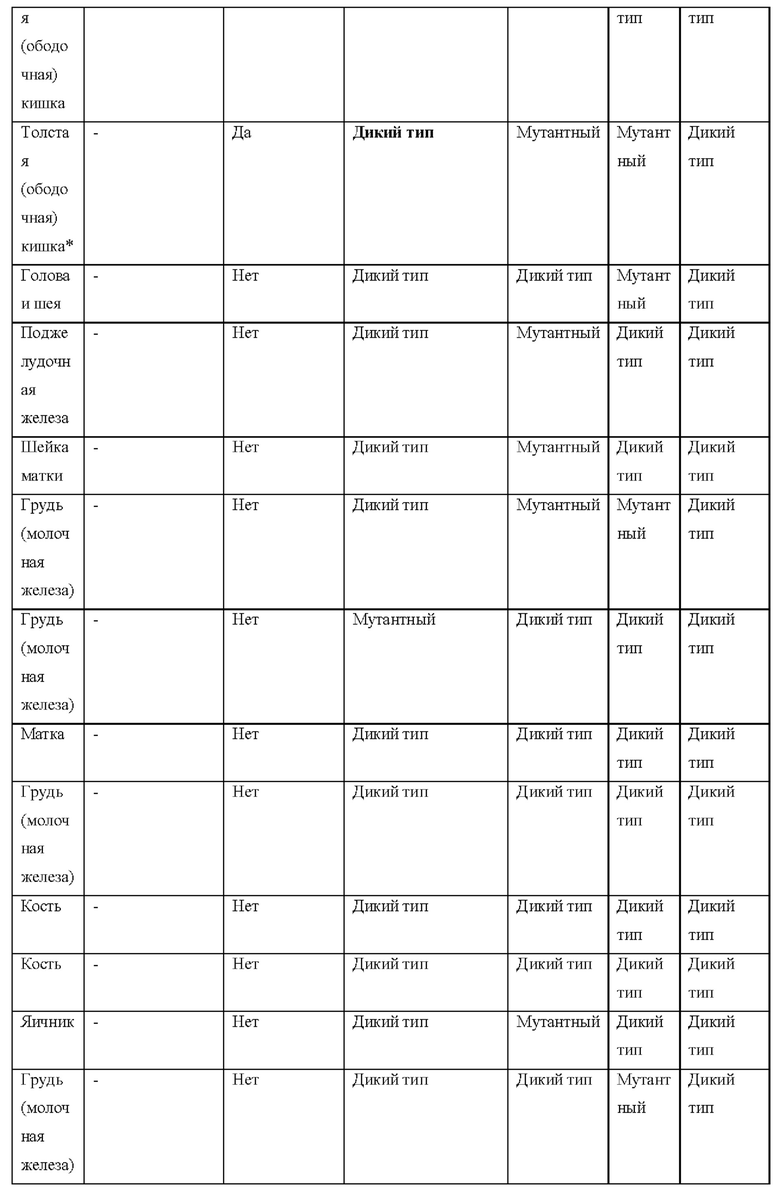
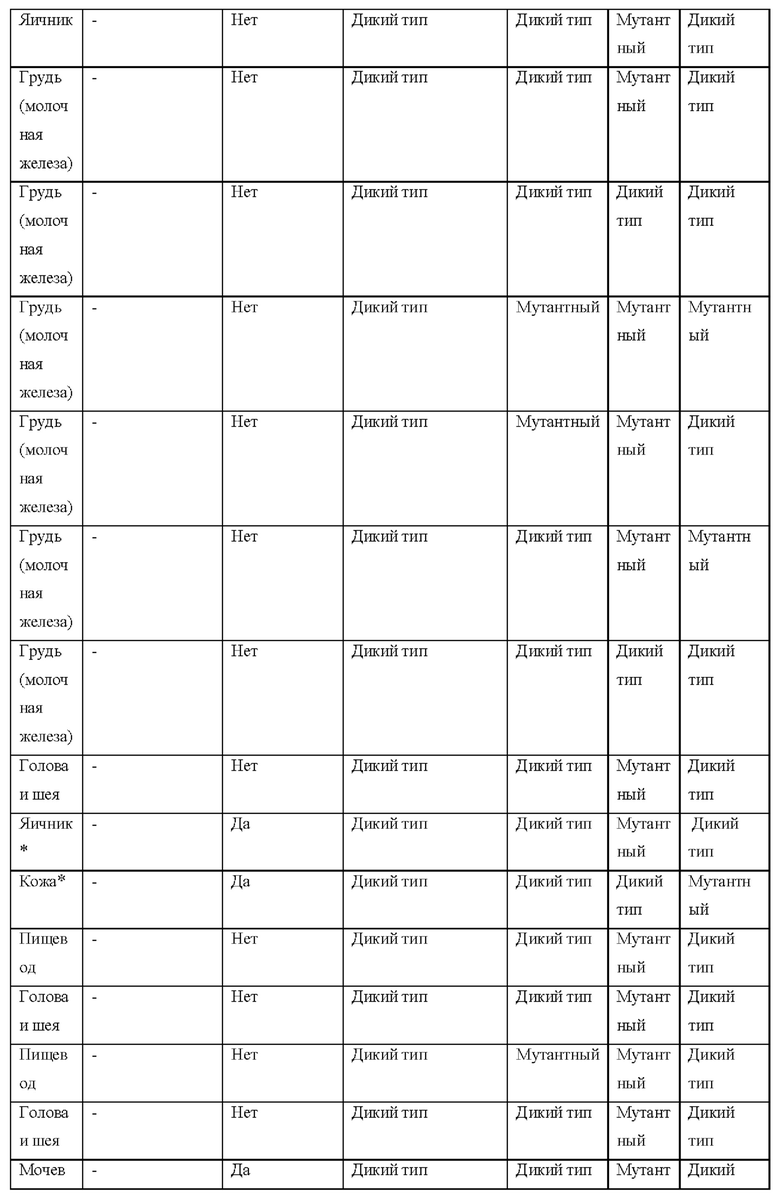
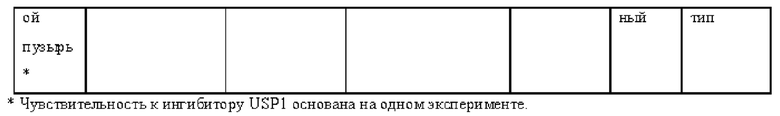
[0799] Эти результаты показывают, что злокачественные опухоли с мутантным р53 обладают повышенной чувствительностью к ингибиторам USP1, а злокачественные опухоли с мутантным BRCA имеют повышенную чувствительность к ингибиторам USP1. Ранее сообщалось, что статус р53 определяет сенсибилизацию ингибитора PARP (Sa et al. Genome Biology, (2019) 20:253) и что статус BRCA1/2 позволяет прогнозировать эффективность ингибиторов PARP в клинике (Audeh et al. Lancet (2010) 376 (9737), 245-51). Кроме того, эти результаты указывают на то, что раковые опухоли с мутантным ATM могут иметь повышенную чувствительность к ингибиторам USP1. Ранее сообщалось, что раковые клетки с мутациями в ATM чувствительны к ингибиторам PARP (Wang et al. Translational Oncology (2017) 10, 190-196). Таким образом, ингибиторы USP1 могут быть эффективными в комбинации с ингибиторами PARP.
ПРИМЕР 220
Определение растворимости
[0800] Определенные соединения раскрытия оценивали на растворимость ADME при рН 2,0 и рН 7,4.
[0801] Исходные растворы готовили путем добавления каждого соединения к ДМСО в концентрации 10 мМ. Образцы готовили, добавляя по 50 мкл каждого исходного раствора в отдельные флаконы. Флаконы загружали на 96-луночный штатив и сушили. 500 мкл забуференного фосфатом физиологического раствора (PBS) рН 7,4 или PBS рН 2,0 добавляли в каждый флакон. Затем флаконы встряхивали при 25°С и 1100 об/мин в течение 24 часов.
[0802] Через 24 часа флаконы центрифугировали при 3220 G и 25°С в течение 30 минут. Жидкий супернатант анализировали с помощью ЖХ-МС/МС в сравнении со стандартом известной концентрации. Затем растворимость каждого образца рассчитывалась с использованием следующего уравнения:
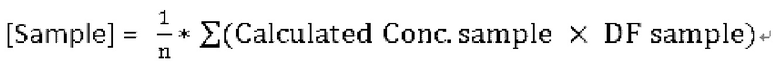
где DF - коэффициент разбавления.
[0803] Следующие Соединения раскрытия имеют значения растворимости ADME, показанные в Таблице 6 ниже.
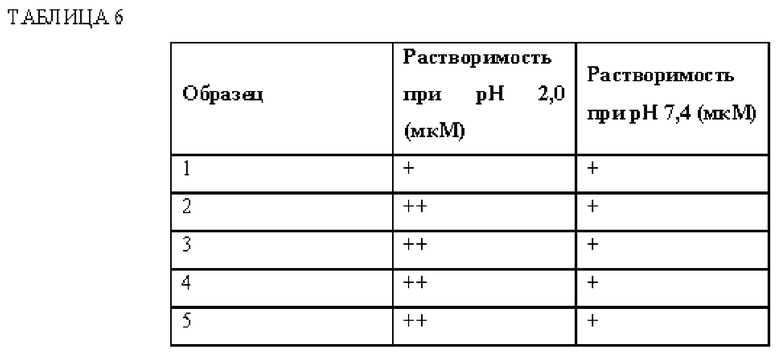
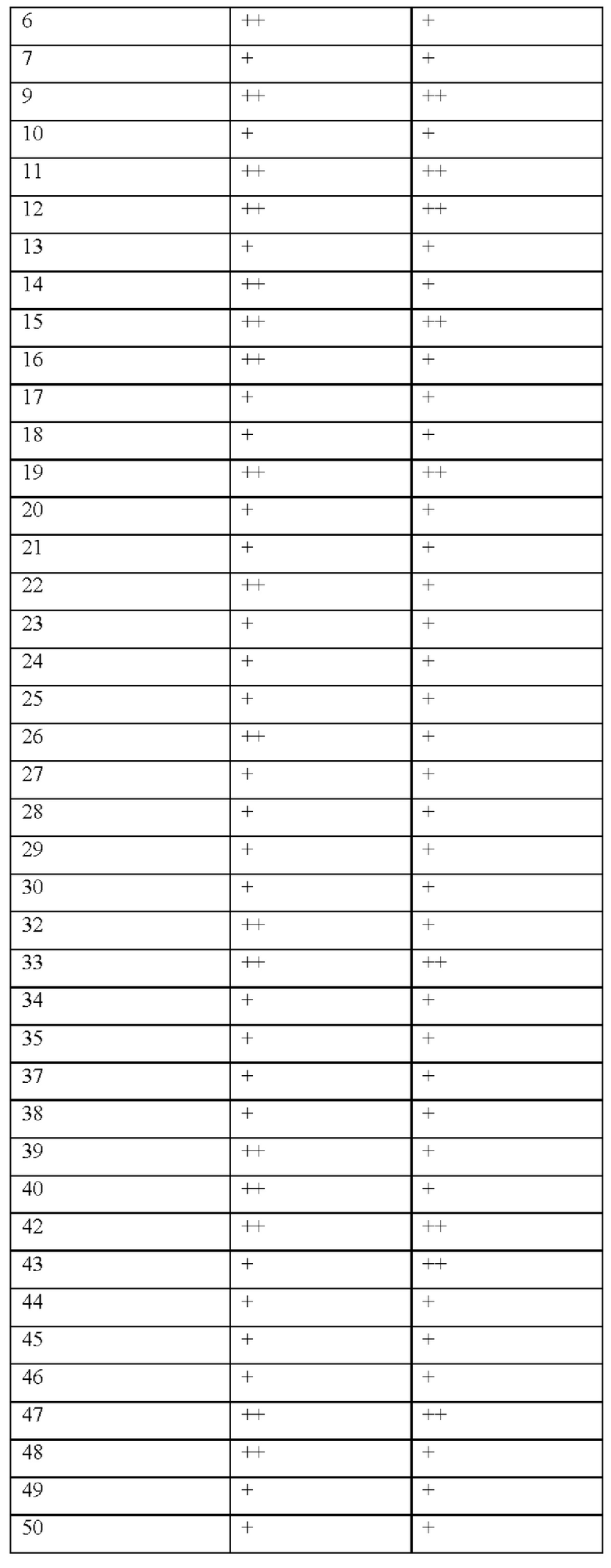
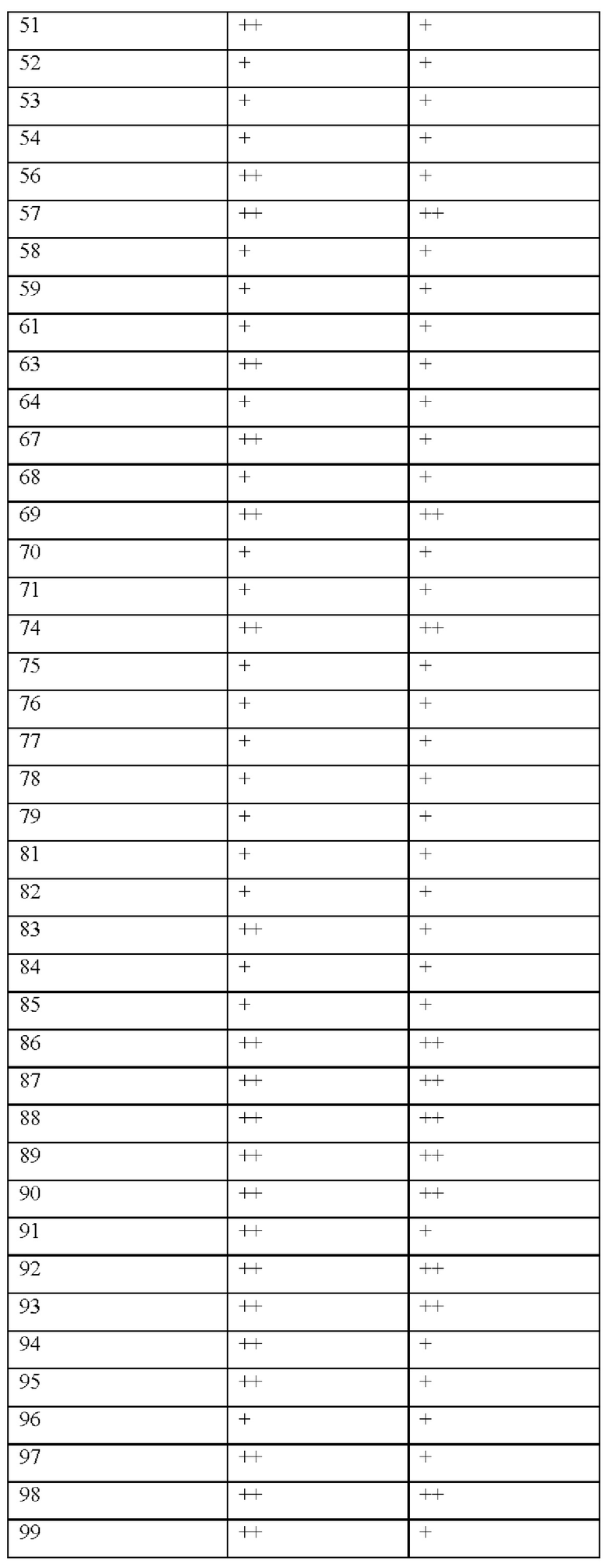
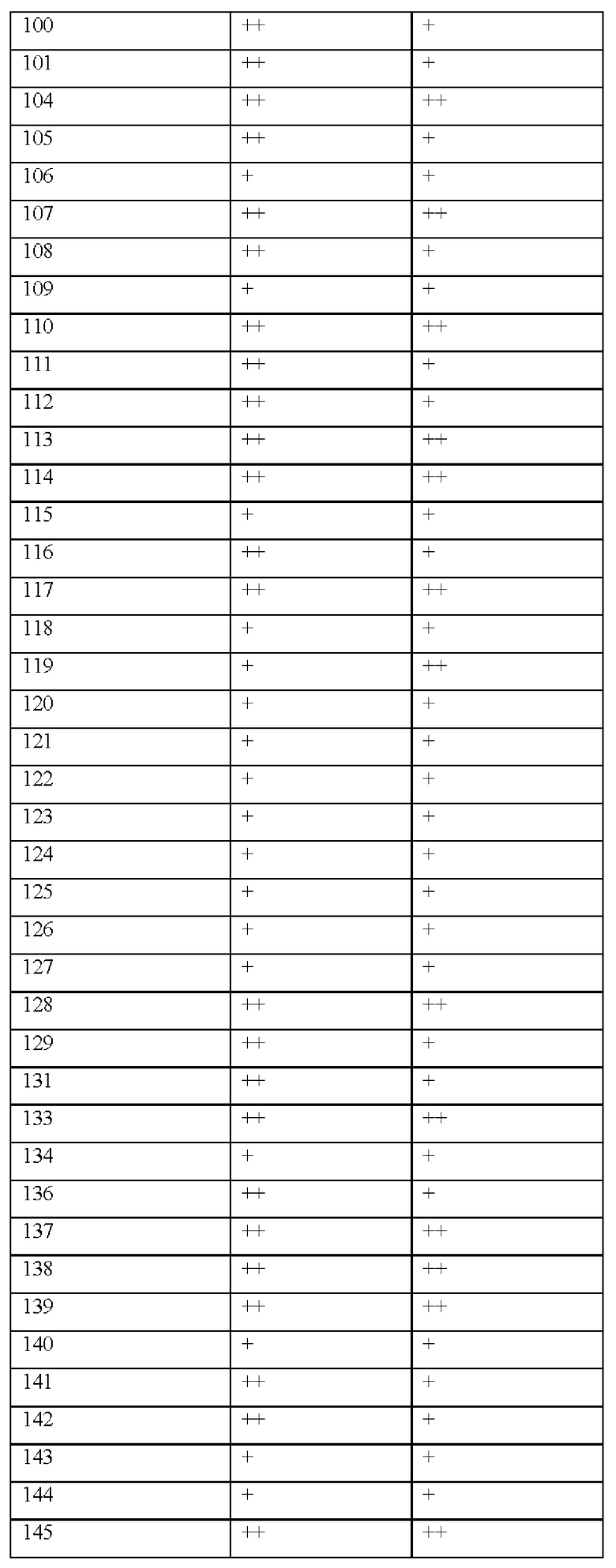
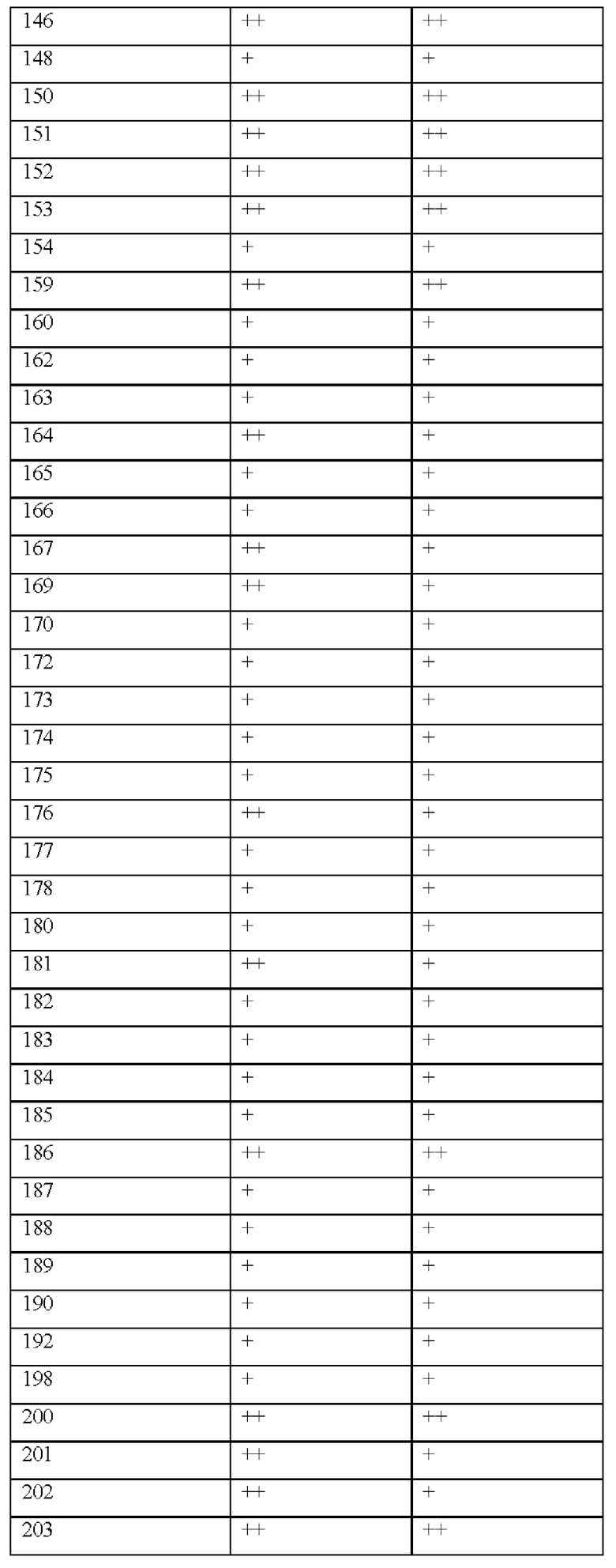
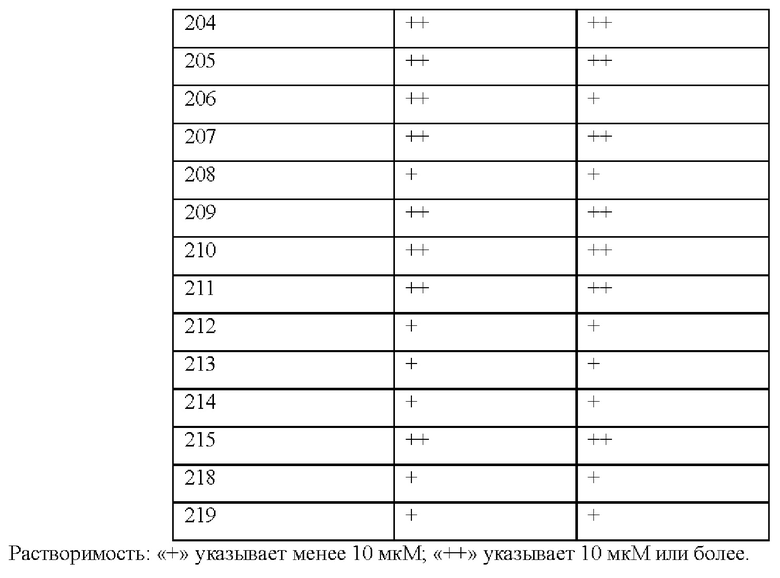
ПРИМЕР 221
Микросомальная стабильность печени
[0804] Определенные Соединения раскрытия оценивали на метаболическую стабильность ADME в микросомах печени человека (HLM) и микросомах печени крысы (RLM).
[0805] Образцы готовили путем добавления 222,5 мкл основного раствора (100 мМ фосфатного буфера и микросомы печени 1 мг/мл (HLM или RLM)) и 25 мкл 10 мМ раствора NADPH в планшеты для инкубации, которые затем нагревали в течение 10 мин. Каждое соединение отдельно растворяли в ДМСО с получением 10 мМ исходных растворов, которые затем разбавляли ацетонитрилом до 100 мкМ. Реакцию начинали добавлением 2,5 мкл 100 мкМ раствора каждого соединения в отдельные планшеты для инкубации таким образом, чтобы конечная концентрация каждого соединения в каждом планшете составляла 1 мкМ.
[0806] Отбирали аликвоты по 25 мкл каждого образца через 0,5, 5, 10, 15, 20 и 30 минут, и реакцию останавливали добавлением 5 объемов холодного ацетонитрила с IS (100 нМ алпразолама, 200 нМ кофеина и 100 нМ толбутамида). Затем образцы центрифугировали при 3220 G в течение 30 минут и 100 мкл над осадочной жидкости смешивали со 100 мкл сверхчистой Н2О.
[0807] Затем образцы анализировали с помощью ЖХ-МС/ МС. Площади пиков определяли по хроматограммам экстрагированных ионов. Значения наклона (к) определяли путем линейной регрессии натурального логарифма оставшегося процента соединения в зависимости от кривой времени инкубации. Период полувыведения in vitro (in vitro t1/2) определяли по величине наклона кривой с использованием следующего уравнения:
in vitro t1/2=-(0,693/k).
Период полувыведения in vitro (мин) был преобразован во внутренний клиренс in vitro (in vitro CLint, в мкл/мин/мг белка) с использованием следующего уравнения:
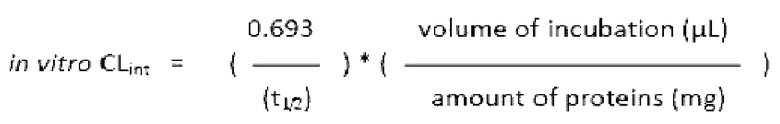
[0808] Следующие Соединения раскрытия имеют значения метаболической стабильности ADME, показанные в Таблице 7 ниже.
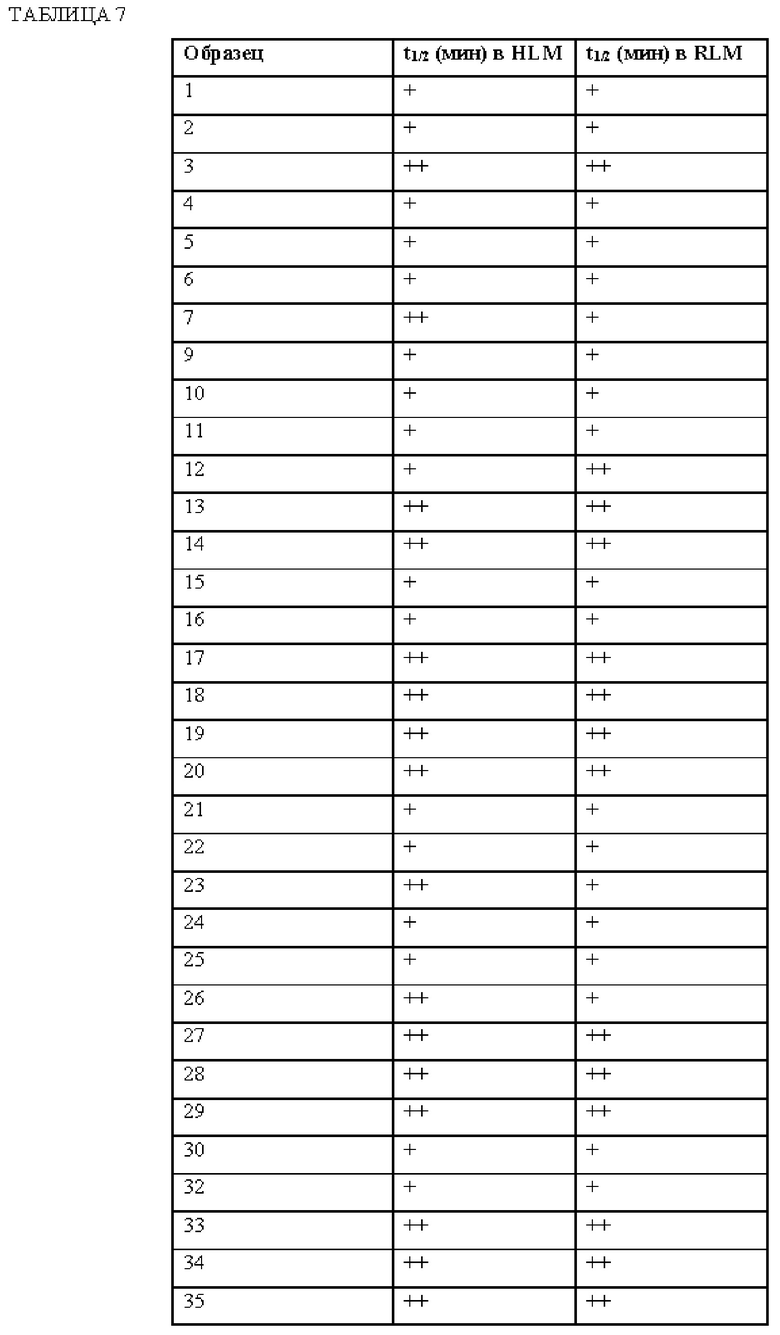
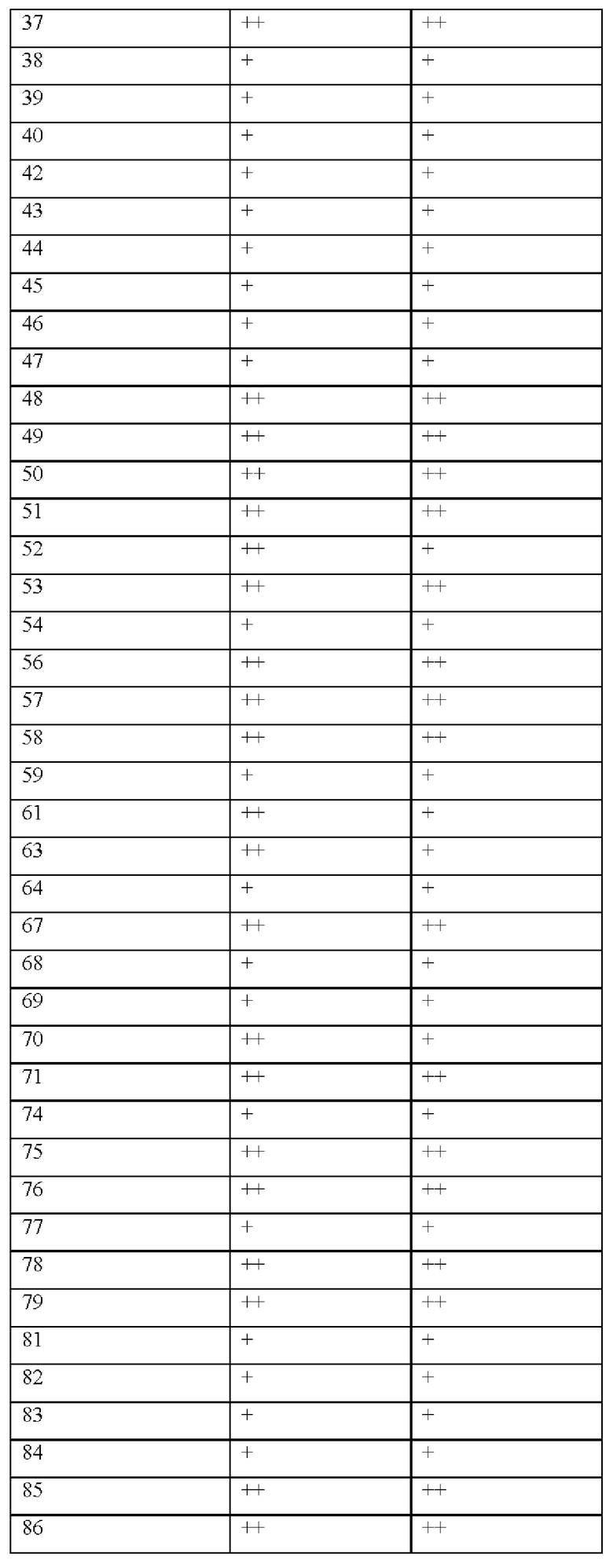
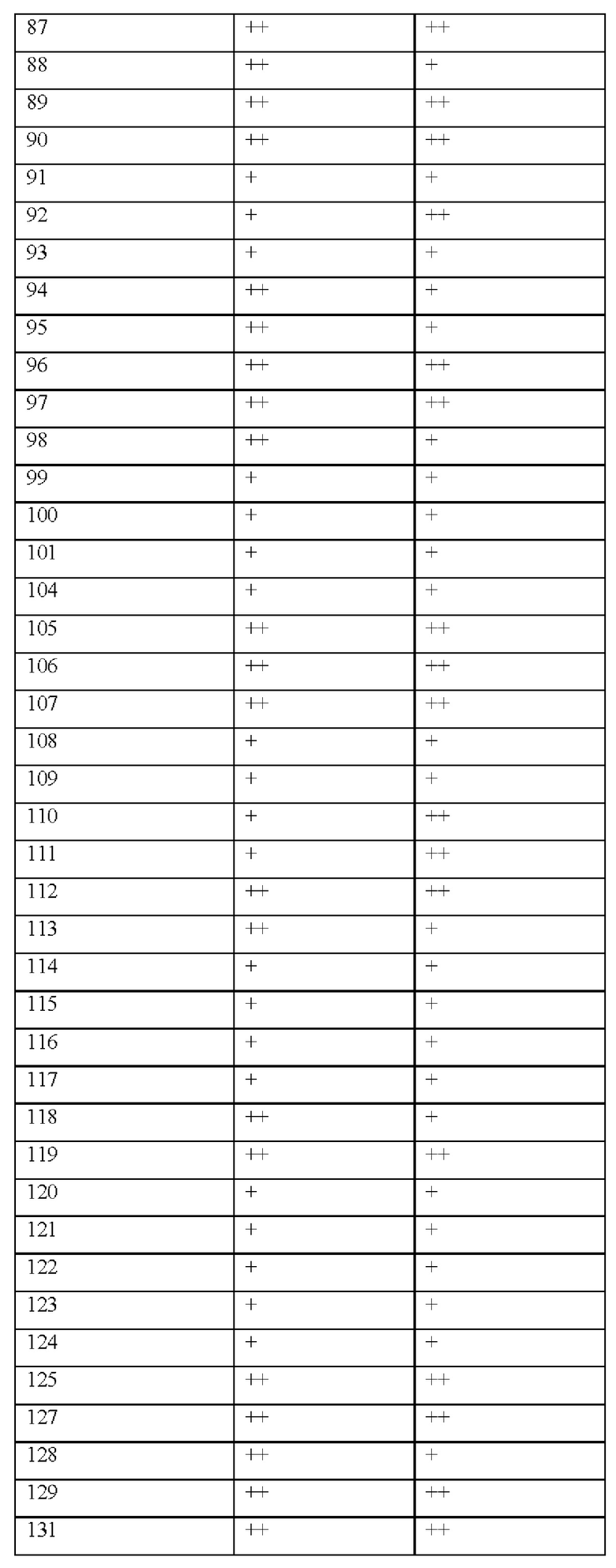
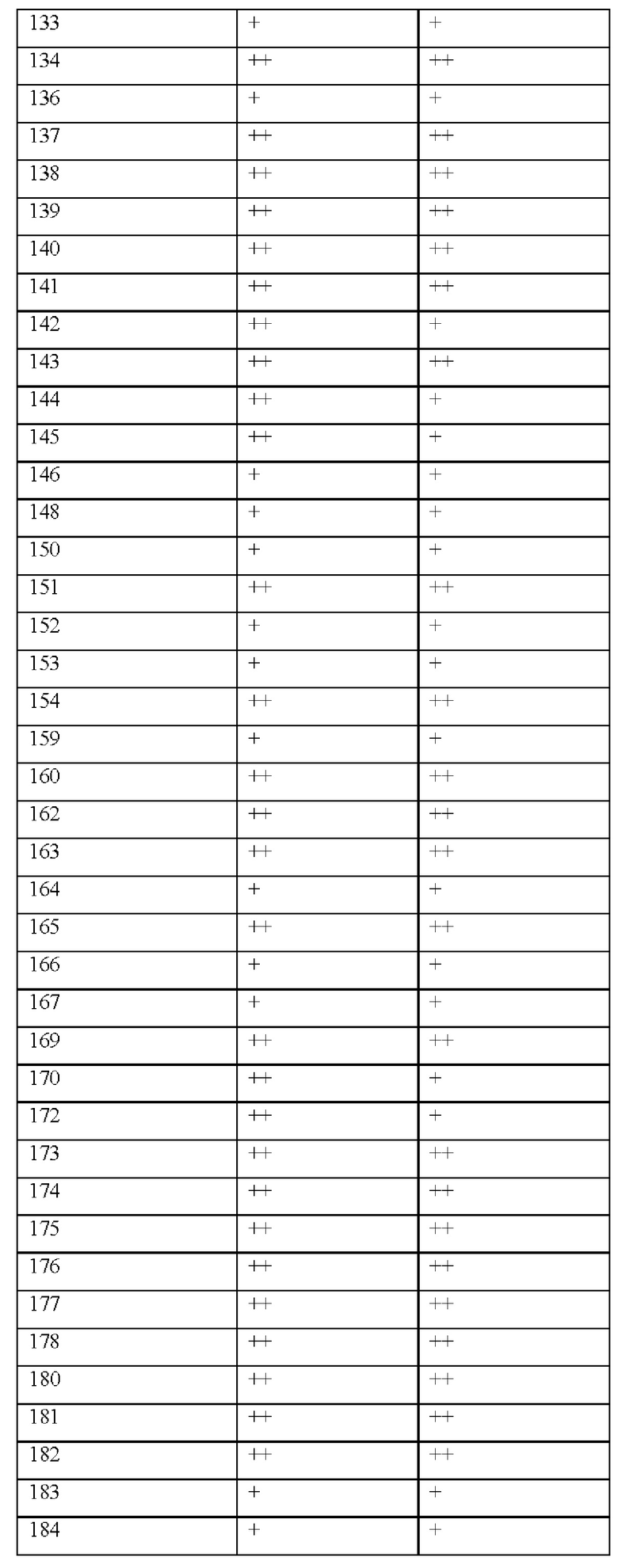
Стабильность HLM/RLM t1/2: «+» означает менее 25 минут; «++» означает 25 минут или более.
ПРИМЕР 222
USP1 необходим для жизнеспособности подмножества линий раковых клеток.
[0809] Для проведения скрининга истощения гена CRISPR-Cas9 было сконструировано около 500 линий раковых клеток для экспрессии Cas9, которые впоследствии были инфицированы лентивирусом, экспрессирующим направляющие РНК, нацеленные на каждый ген в геноме. Через 14 дней клетки собирали, геномную ДНК экстрагировали, и Illumina Sequencing использовали для определения репрезентативности направляющих, а показатель «отсева» использовали для измерения истощения направляющих, нацеленных на каждый ген в каждой клеточной линии. Более низкий показатель отсева указывает на большую чувствительность к потере гена. Показатели отсева для USP1 в различных клеточных линиях представлены на ФИГ. 1. Эти данные демонстрируют, что направляющие, нацеленные на USP1, были истощены в подмножестве клеточных линий рака яичников и рака молочной железы, что указывает на то, что USP1 необходим для жизнеспособности этих линий (ФИГ. 1). Линии клеток рака груди показаны белым цветом, а линии клеток рака яичников показаны черным. Семь из девяти наиболее чувствительных линий груди были тройным отрицательным раком груди (TNBC), но некоторые линии TNBC были нечувствительны к потере USP1.
[0810] USP1 представляет собой белок деубиквитиназы, который удаляет убиквитин из моноубиквитинированной PCNA (ФИГ. 2) и FANCD2. Генетическое истощение CRISPR-Cas9 или фармакологическое ингибирование USP1 приводит к усилению моноубиквитинирования как PCNA, так и FANCD2.
ПРИМЕР 223
Уровни Rad18 коррелируют с чувствительностью к потере USP1
[0811] Чтобы определить особенности, которые могут предсказать чувствительность к потере USP1, экспрессия генов была проанализирована во всех около 500 клеточных линиях. Было обнаружено, что уровни мРНК Rad18 коррелируют с чувствительностью к потере USP1. Rad18 представляет собой убиквитинлигазу Е3, которая убиквитинирует PCNA (ФИГ. 2). Ранее сообщалось, что нокдаун Rad18 приводил к потере моноубиквитинированной PCNA и что это спасало нестабильность репликационной вилки, вызванную ингибированием USP1 в опухолях, дефицитных по гомологичной рекомбинации с мутациями BRCA1 hBRCA2. (Lim K. et al., "USP1 is required for replication fork stability in BRCA1-deficient tumors," AACR 2018 Meeting, Abstract 333/14.) Поэтому было неожиданным, что линии клеток, чувствительных к USP1, включали линии клеток дикого типа BRCA1/2, а также линии мутантных клеток BRCA1/2 (ФИГ. 1).
[0812] RAD18 ранее не предлагался даже в качестве потенциального биомаркера чувствительности к ингибитору USP1, и, как показано в данном документе, Rad18 может быть обнаружен как в опухолях, чувствительных к USP1, так и нечувствительных к USP1 (см. ФИГ 3-6). Однако клеточные линии, которые были наиболее чувствительны к ингибиторам USP1, также имели тенденцию иметь высокие уровни Rad18. На ФИГ. 3 показана гистограмма экспрессии мРНК Rad18 в около 500 клеточных линиях, а 20 линий с самым низким показателем отсева USP1 указаны в темных столбцах, доходящих до верхней части графика. Подобным образом на ФИГ. 4 показано, что показатели отсева USP1 коррелировали с уровнями экспрессии мРНК Rad18 как в линиях рака молочной железы (включая TNBC), так и в линиях рака яичников.
[0813] Для дальнейшего анализа корреляции уровней Rad18 и чувствительности к ингибиторам USP1 мРНК Rad18 и уровни белка анализировали на панели чувствительных и нечувствительных к USP1 линий. Чувствительными клеточными линиями были 59М (линия рака яичников дикого типа BRCA1/2) и ES2 (линия рака яичников дикого типа BRCA1/2). Нечувствительными клеточными линиями были OVISE (линия рака яичников дикого типа BRCA1/2), Нер3В217 (линия рака печени дикого типа BRCA1/2) и JHH7 (линия рака печени дикого типа BRCA1/2). Уровни мРНК Rad18 определяли с помощью количественной ПЦР с обратной транскрипцией (qRT-PCR), и было обнаружено, что чувствительные линии имеют более высокую экспрессию мРНК Rad18 (ФИГ. 5). Уровни белка Rad18 исследовали с помощью вестерн-блоттинга в тех же клеточных линиях, и было обнаружено, что чувствительные линии имеют более высокую экспрессию уровней белка Rad18 (ФИГ. 6).
[0814] Эти данные демонстрируют, что, хотя Rad18 может быть обнаружен как в опухолях, чувствительных к USP1, так и нечувствительных к USP1, повышенные уровни мРНК Rad18 и белка коррелируют с чувствительностью к USP1.
ПРИМЕР 224
Удаление Rad18 спасает удаление USP1
[0815] Было обнаружено, что Rad18 играет функциональную роль в чувствительности к потере USP1. Клетки ES2 инфицировали лентивирусом, экспрессирующим Cas9, затем подвергали электропорации рибонуклеопротеинами, содержащими белок Cas9 и либо ориентиром для отрицательного контроля (OR1A1), либо ориентиром, нацеленным на Rad18. Затем клетки инфицировали лентивирусом, экспрессирующими направляющие, против отрицательного контроля (OR1A1), положительного летального контроля (EEF2) или одного из четырех различных направляющих нацеливания на USP1. В клетках отрицательного контроля обработка направляющими элементами USP1 снижает жизнеспособность. Однако генетическое истощение Rad18 полностью изменило эффект потери USP1 на жизнеспособность клеток. (ФИГ. 7.) Эти данные демонстрируют, что удаление Rad18 спасает удаление USP1.
[0816] После полного описания данного изобретения специалистам в данной области будет понятно, что то же самое может быть выполнено в широком и эквивалентном диапазоне условий, составов и других параметров, не влияя на объем изобретения или любой его вариант реализации.
[0817] Другие варианты реализации изобретения будут очевидны специалистам в данной области техники из рассмотрения описания и практического применения изобретения, раскрытого в данном документе. Предполагается, что описание и примеры следует рассматривать только как иллюстративные, с истинным объемом и смыслом изобретения, указанными в следующей формуле изобретения.
[0818] Все патенты и публикации, цитируемые в данном документе, полностью включены в данный документ посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| КОМПОЗИЦИИ ТЕТРАГИДРОХИНОЛИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ БРОМОДОМЕНА ВЕТ | 2014 |
|
RU2727169C2 |
| Соединения триазоло-пиримидина и их применение | 2019 |
|
RU2802866C2 |
| ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ 2Н-ПИРАЗОЛА | 2016 |
|
RU2722363C2 |
| ЗАМЕЩЕННЫЕ 1,2-ДИГИДРО-3Н-ПИРАЗОЛО[3,4-D]ПИРИМИДИН-3-ОНЫ | 2019 |
|
RU2812726C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2811612C2 |
| ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ И 7-ДЕАЗАПУРИНОВЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2606514C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЕСПЕЧИВАЮЩИЕ РАЗРУШЕНИЕ БЕЛКА IKAROS И БЕЛКА AIOLOS | 2020 |
|
RU2833608C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2011 |
|
RU2652638C2 |
Изобретение относится к соединениям формулы I или их фармацевтически приемлемым солям, где Х1, Х2, X11, Х12, R1, R3, R5, R5', R6 и R7 определены в формуле изобретения, в качестве ингибиторов USP1. Также предложены фармацевтические композиции, содержащие соединения формулы I, и способ ингибирования белка USP1. Соединения формулы I могут быть использованы для лечения расстройства, реагирующего на ингибирование белков USP1, в частности, для лечения рака. 6 н. и 7 з.п. ф-лы, 7 табл., 224 пр.
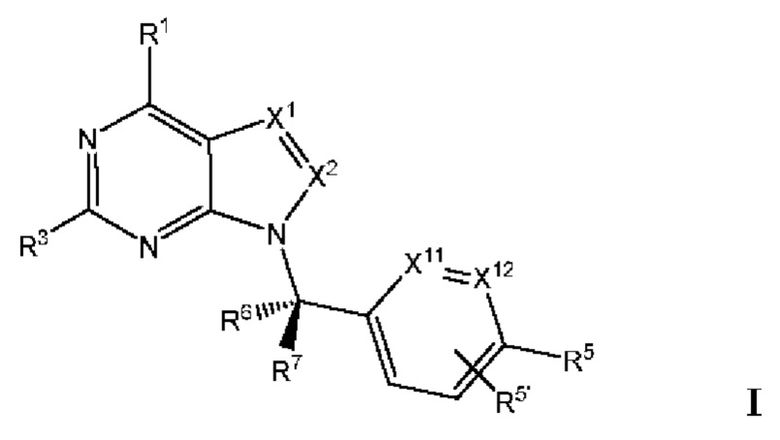
1. Соединение, представляющее собой 6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин.
2. Соединение формулы I:
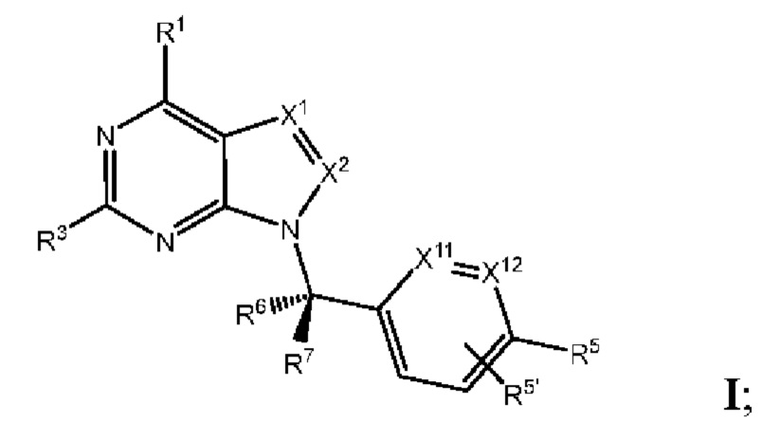
или его фармацевтически приемлемая соль, где:
 представляет собой
представляет собой 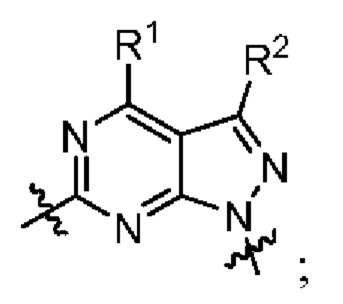
каждый из R1 и R2 независимо выбран из водорода, галогена, циано и (C1-C6)алкила;
R3 представляет собой необязательно замещенный фенил, необязательно замещенный пиридил, необязательно замещенный пиримидинил или необязательно замещенный пиразолил;
каждый из X11 и X12 независимо выбран из N и СН;
R5’ выбран из водорода, (C1-C6)алкокси и галогена;
R5 выбран из необязательно замещенного гетероарила и необязательно замещенного гетероцикло;
каждый из R6 и R7 независимо выбран из водорода и (C1-C6)алкила;
где указанные необязательные заместители для R3 независимо выбраны из галогена, циано, амино, (С1-6)алкиламино, ди(С1-6)алкиламино, (С1-6)галогеналкила, (C1-6)гидроксиалкила, (С1-6)алкокси, (С1-6)галогеналкокси, (С6-14)аралкилокси, (С1-6)алкилтио, (С1-6)алкилсульфонила, (С1-6)алкила, (С3-8)циклоалкила, необязательно замещенного одним или более (С1-6)алкилом, гетероцикло, необязательно замещенного одним или более (С1-6)алкилом, (С1-6)алкокси(С1-6)алкила, 2-метоксиэтокси, (тетрагидрофуран-3-ил)окси, циклопропокси, циклобутокси, бензилокси и дейтерометокси;
где указанные необязательные заместители для R5 независимо выбраны из галогена, нитро, циано, (С1-6)алкила, необязательно замещенного (С1-6)алкилсульфонилом, (C1-6)алкокси, амино, (С1-6)алкиламино, ди(С1-6)алкиламино, (С1-6)алкилтио, (С1-6)галогеналкила, (карбоксамидо)(С1-6)алкила, (С1-6)гидроксиалкила, (С1-6)алкокси(С1-6)алкила, гетероцикло, необязательно замещенного одним или более (С1-6)алкилом, гетероарила, (С3-8)циклоалкила и азетидин-3-илокси;
где каждый гетероарил или группа гетероар- содержит 5-14 атомов в кольце и 1-4 гетероатома, независимо выбранных из азота, кислорода и серы;
где каждая группа гетероцикло или гетероцикл содержит 3-14 атома в кольце и 1-4 гетероатома, независимо выбранных из азота, кислорода и серы.
3. Соединение по п. 2 или его фармацевтически приемлемая соль,
a) где R3 представляет собой необязательно замещенный фенил, причем фенил необязательно замещен в положении 2, необязательно замещен в положении 6, необязательно дизамещен во положениях 2 и 6, или необязательно дизамещен во положениях 2 и 3, или
b) где R3 представляет собой необязательно замещенный пирид-3-ил или необязательно замещенный пирид-4-ил,
причем пирид-3-ил необязательно замещен в положении 2, необязательно замещен в положении 4 или необязательно дизамещен в положениях 2 и 4; и
при этом пирид-4-ил необязательно замещен в положении 3, необязательно замещен в положении 5 или необязательно дизамещен в положениях 3 и 5, или
c) где R3 представляет собой необязательно замещенный пиримидин-5-ил, причем пиримидин-5-ил необязательно замещен в положении 4, необязательно замещен в положении 6, необязательно дизамещен в положениях 4 и 6 или необязательно тризамещен во положениях 2, 4 и 6, или
d) где R3 представляет собой необязательно замещенный пиразол-5-ил, причем пиразол-5-ил необязательно замещен в положении 1, необязательно замещен в положении 4 или необязательно дизамещен в положениях 1 и 4, или
e) где R3 выбран из группы, состоящей из:
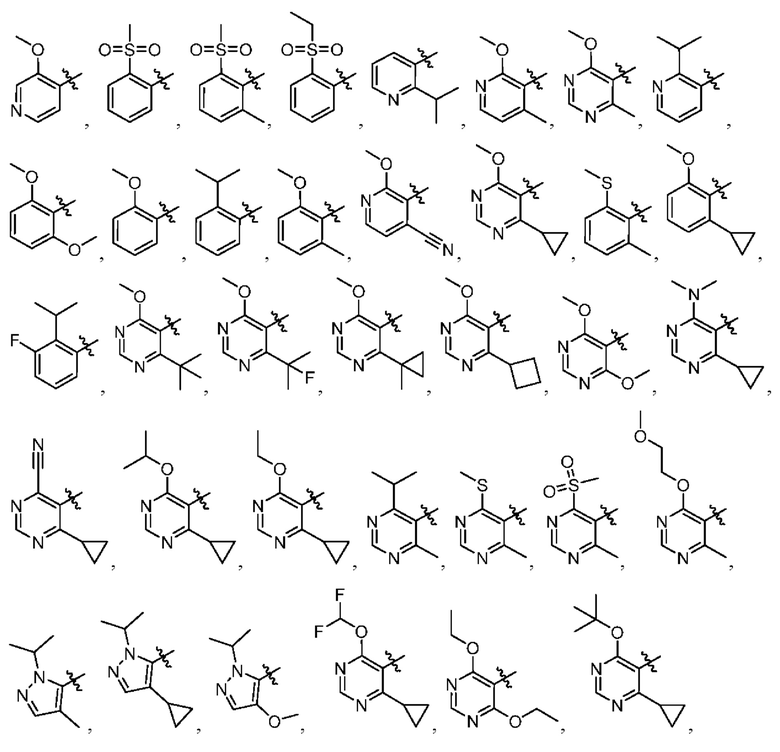
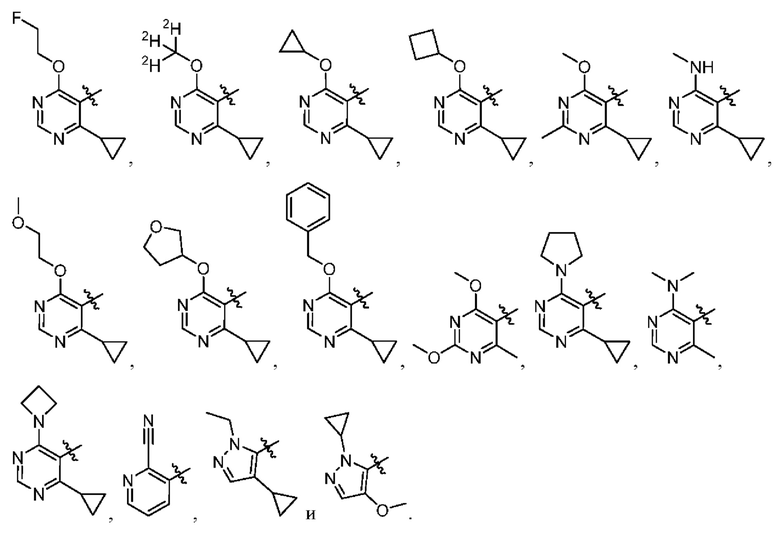
4. Соединение по п. 3 вариант а), вариант b), вариант с) или вариант d), где R3 замещен и заместители независимо выбраны из метокси, дейтерометокси, этокси, изопропокси, трет-бутокси, дифторметокси, 2-фторэтокси, 2-метоксиэтокси, циклопропокси, циклобутокси, (тетрагидрофуран-3-ил)окси, бензилокси, метила, этила, изопропила, 2-фторизопропила, трет-бутила, циклопропила, циклобутила, метилциклопропила, пирролидин-1-ила, азетидин-1-ила, метиламино, диметиламино, циано, галогена, метилтио, метилсульфонила и этилсульфонила.
5. Соединение по п. 2 или его фармацевтически приемлемая соль, где по меньшей мере один из X11 и X12 представляет собой N.
6. Соединение по п. 2 или его фармацевтически приемлемая соль, где R5 представляет собой необязательно замещенный гетероарил.
7. Соединение по п. 2 или его фармацевтически приемлемая соль,
а) где R5 представляет собой необязательно замещенный пирролил, необязательно замещенный имидазолил, необязательно замещенный пиразолил, необязательно замещенный триазолил или необязательно замещенный тетразолил, и/или
b) где R5 представляет собой необязательно замещенный имидазолил, или
c) где R5 представляет собой необязательно замещенный пиразолил, или
d) где R5 представляет собой необязательно замещенный триазолил, или
e) где R5 выбран из группы, состоящей из:
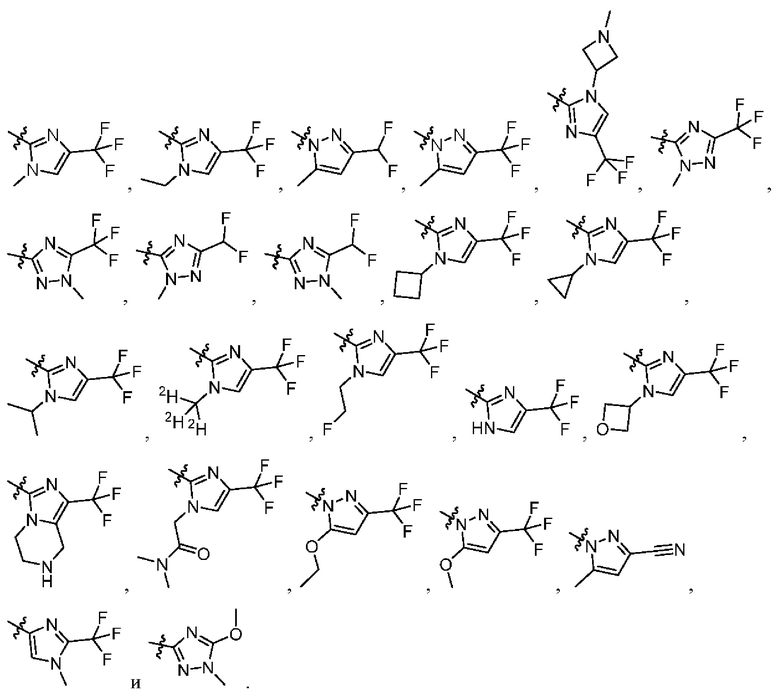
8. Соединение по п. 7 вариант а), вариант b) или вариант с), где R5 замещен, и заместители независимо выбраны из галогена, метила, этила, изопропила, циклопропила, циклобутила, метокси, этокси, триазолила, циано, (С1-C6)алкила, амино, (С1-С6)алкиламино, ди(С1-С6)алкиламино, дифторметила, трифторметила, метилсульфонила, оксетан-3-ила и метилазетидинила.
9. Соединение по п. 2 или его фармацевтически приемлемая соль, имеющее формулу II:
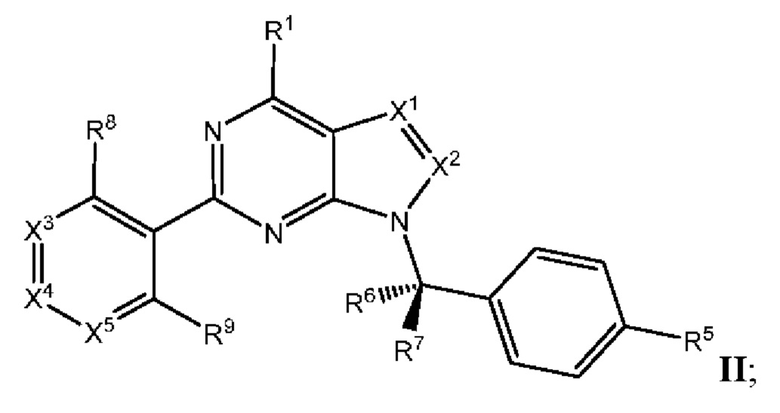
где:
X1 представляет собой CR2; X2 представляет собой N; X3 выбран из N и CR10; X4 выбран из N и CR11; X5 выбран из N и CR12; и
каждый из R8, R9, R10, R11, и R12 независимо выбран из группы, состоящей из водорода, галогена, циано, амино, (C1-С6)алкиламино, ди(С1-С6)алкиламино, (C1-С6)галогеналкила, (С1-С6)гидроксиалкила, (С1-С6)алкокси, (С1-С6)галогеналкокси, (С6-С14)аралкилокси, (C1-С6)алкилтио, (С1-С6)алкилсульфонила, (С1-С6)алкила, (С3-С8)циклоалкила, необязательно замещенного одним или более (С1-С6)алкилом, гетероцикло, необязательно замещенного одним или более (С1-С6)алкилом, и (С1-С6)алкокси(С1-С6)алкила, предпочтительно имеющее формулу III, формулу IV, формулу V, формулу VI или формулу VIa:
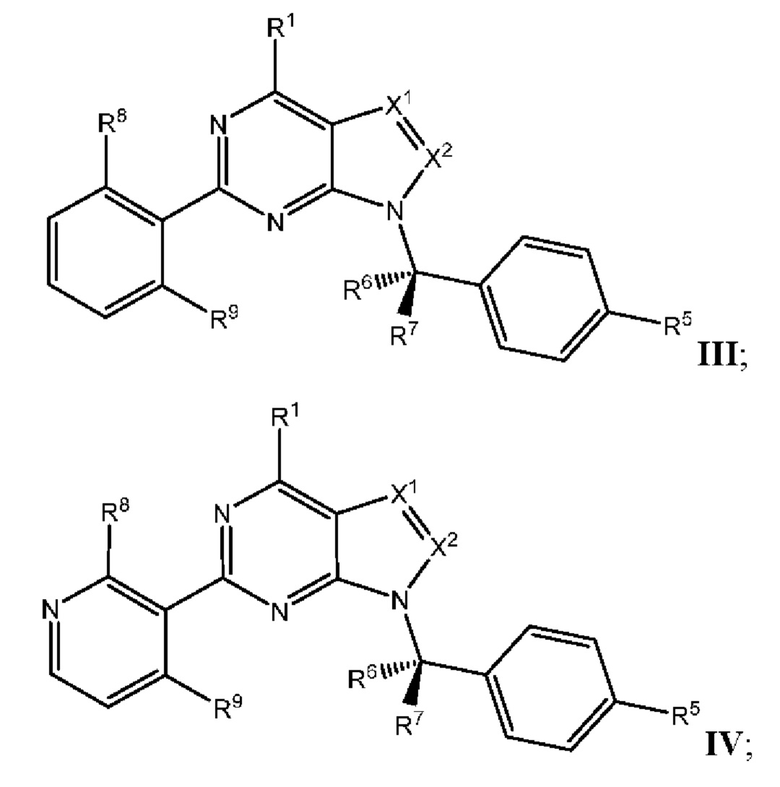
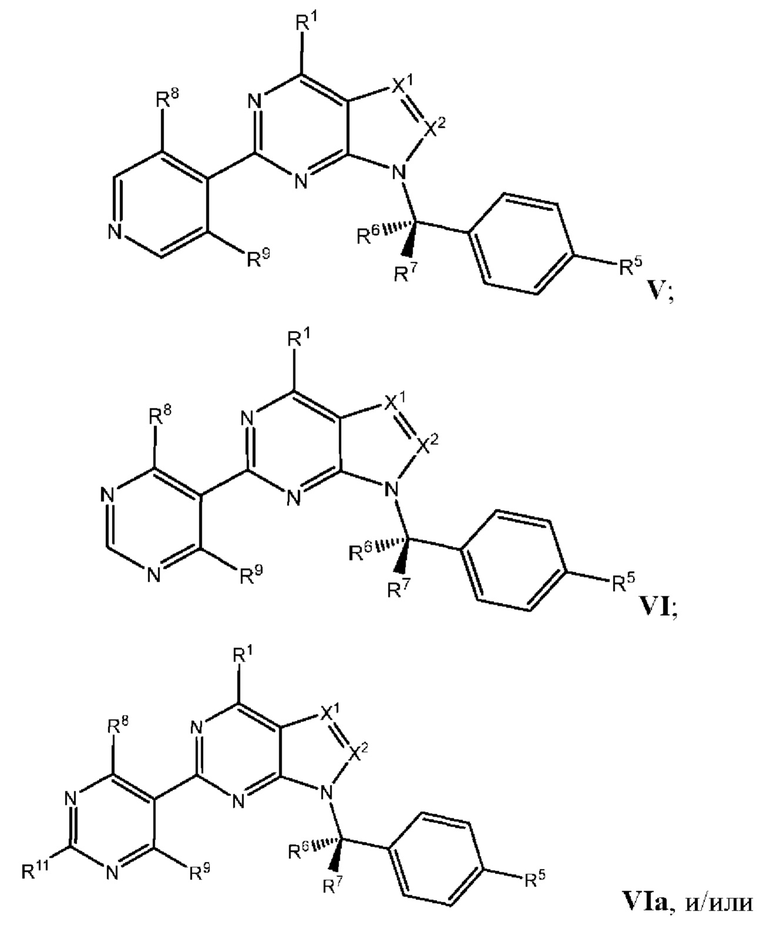
где R5 представляет собой:
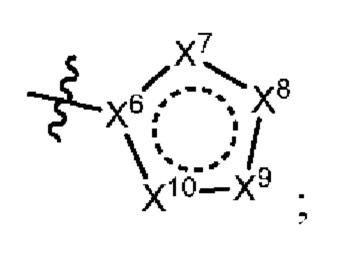
где:
X6 выбран из NR13 и CR18; X7 выбран из NR14 и CR19; X8 выбран из NR15 и CR20; X9 выбран из NR16 и CR21; X10 выбран из NR17 и CR22;
каждый из R13, R14, R15, R16 и R17 отсутствует или независимо выбран из водорода, галогена, метила, этила, изопропила, циклопропила, метокси, триазолила, циано, (C1-С6)алкила, амино, (C1-С6)алкиламино, ди(С1-С6)алкиламино, дифторметила, трифторметила, метилсульфонила и метилазетидинила; и
каждый из R18, R19, R20, R21 и R22 независимо выбран из водорода, галогена, метила, этила, изопропила, циклопропила, метокси, триазолила, циано, (С1-С6)алкила, амино, (C1-С6)алкиламино, ди(С1-С6)алкиламино, дифторметила, трифторметила, метилсульфонила и метилазетидинила, предпочтительно имеющее формулу VII, формулу VIII, формулу IX, формулу X, формулу XI или формулу XII:
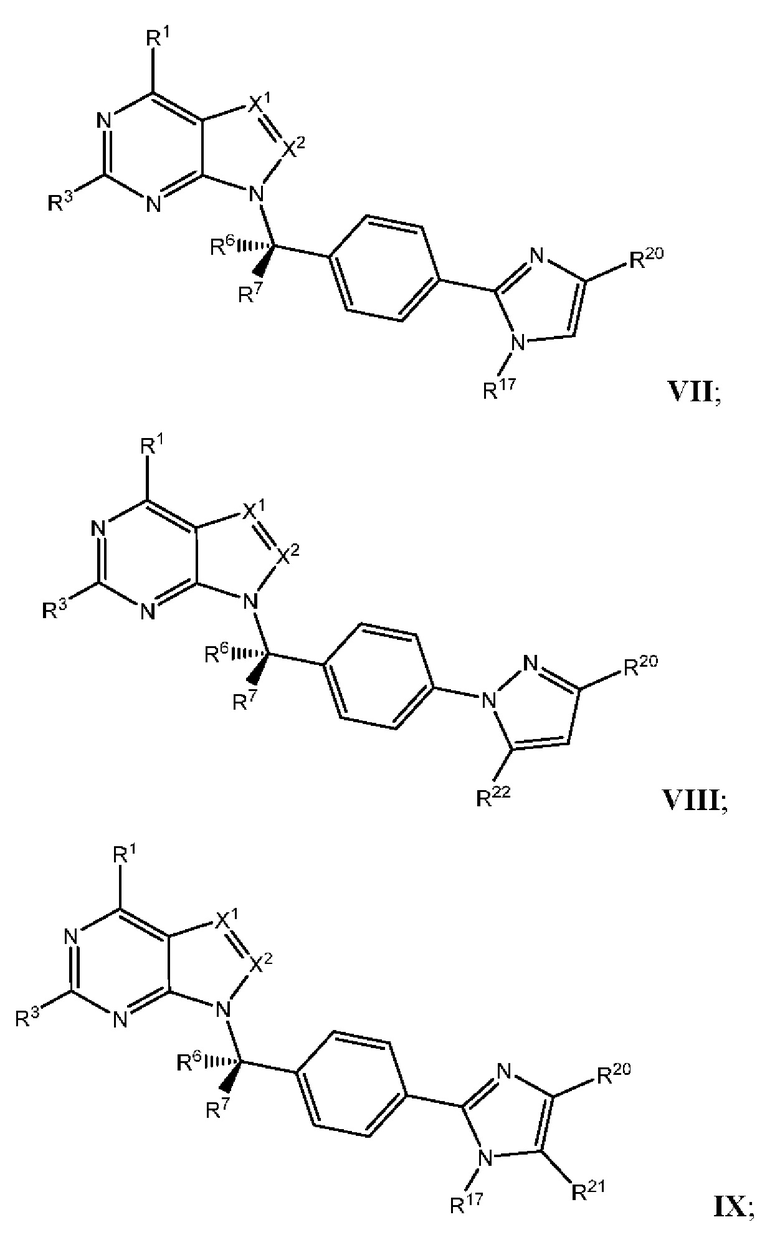
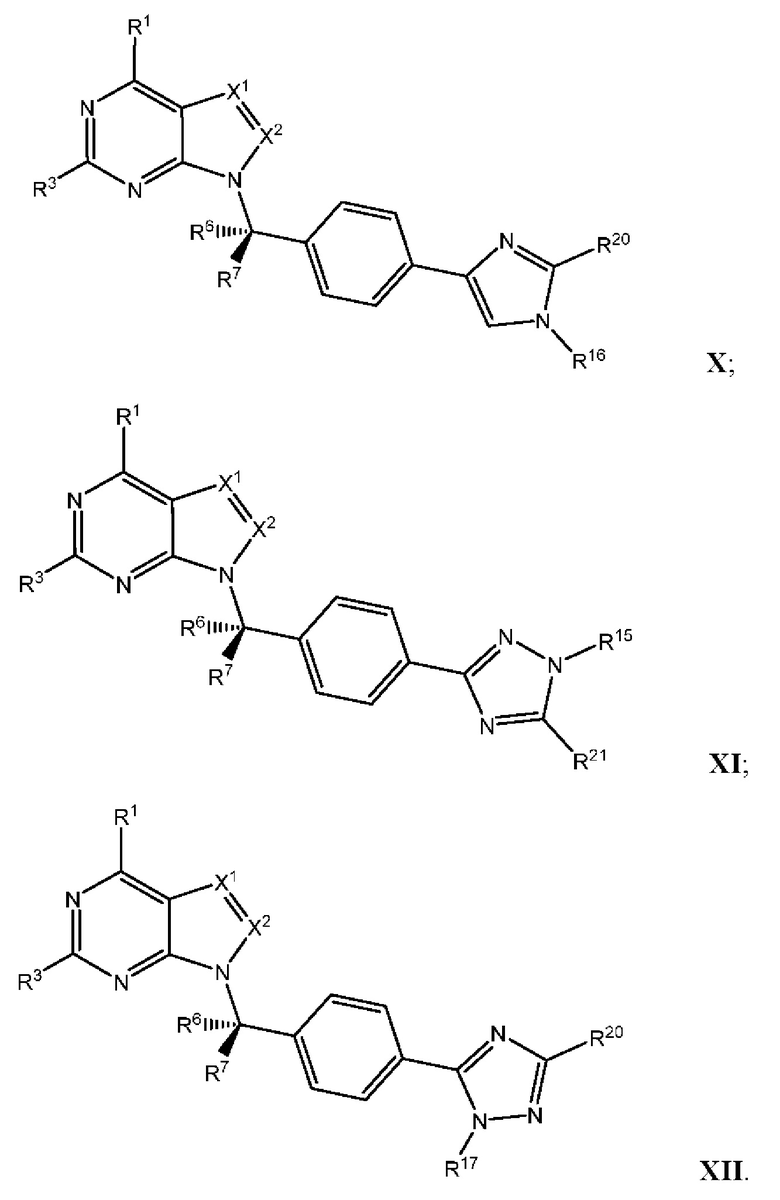
10. Соединение,
а) выбранное из группы, состоящей из:
6-(3-метоксипиридин-4-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-6-(2-(метилсульфонил)-фенил)-1Н-пиразоло[3,4-d]пиримидина;
1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-6-(2-метил-6-(метилсульфонил)фенил)-1Н-пиразоло[3,4-d]пиримидина;
6-(2-(этилсульфонил)фенил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
1-(4-(3-(дифторметил)-5-метил-1Н-пиразол-1-ил)бензил)-6-(2-изопропилпиридин-3-ил)-1Н-пиразоло[3,4-d]пиримидина;
6-(2-метокси-4-метилпиридин-3-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-метокси-6-метилпиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(2-изопропилпиридин-3-ил)-1-(4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(2,6-диметоксифенил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(2-метоксифенил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-(2-изопропилфенил)-1-(4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(2-метокси-6-метилфенил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
2-метокси-3-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил)изоникотинонитрила;
2-(2-изопропилфенил)-9-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-9Н-пурина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(2-изопропилпиридин-3-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-6-(2-метил-6-(метилтио)фенил)-1H-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-(2-циклопропил-6-метоксифенил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(2-изопропилфенил)-1-(4-(1-(1-метилазетидин-3-ил)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(2-изопропилфенил)-1-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этил)-1H-пиразоло[3,4-d]пиримидина;
6-(2-изопропилфенил)-4-метил-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(3-фтор-2-изопропилфенил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(2-изопропилфенил)-3-метил-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(2-изопропилфенил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-(4-(трет-бутил)-6-метоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-(2-фторпропан-2-ил)-6-метоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-метокси-6-(1-метилциклопропил)пиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклобутил-6-метоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
(S)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)фенил)этил)-1H-пиразоло[3,4-d]пиримидина;
(R)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-этоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-изопропоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-циклопропил-5-(1-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидин-6-ил)пиримидин-4-карбонитрила;
6-циклопропил-N,N-диметил-5-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил)пиримидин-4-амина;
6-(4,6-диметоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-изопропил-6-метилпиримидин-5-ил)-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
1-(4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензил)-6-(4-метил-6-(метилтио)пиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина;
2-(4-циклопропил-6-метоксипиримидин-5-ил)-8-метил-9-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-9Н-пурина;
1-(4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензил)-6-(4-метил-6-(метилсульфонил)пиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-(2-метоксиэтокси)-6-метилпиримидин-5-ил)-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(1-изопропил-4-метил-1Н-пиразол-5-ил)-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразол[3,4-d]пиримидина;
6-(1-изопропил-4-метокси-1Н-пиразол-5-ил)-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-1-изопропил-1Н-пиразол-5-ил)-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразол[3,4-d]пиримидина;
2-(4-циклопропил-6-метоксипиримидин-5-ил)-7-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-7Н-пирроло[2,3-d]пиримидина; и
5-(4-циклопропил-6-метоксипиримидин-5-ил)-3-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-3Н-[1,2,3]триазоло[4,5-d]пиримидина;
или его фармацевтически приемлемая соль, или
b) выбранное из группы, состоящей из:
6-(4,6-диэтоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-3-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-метил-5-(трифторметил)-1Н-1,2,4-триазол-3-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(1-циклопропил-4-метокси-1Н-пиразол-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-((6-(5-метокси-3-(трифторметил)-1Н-пиразол-1-ил)пиридин-3-ил)метил)-1Н-пиразоло[3,4-d]пиримидина;
(R)-6-(4-циклобутил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-этил-4-(трифторметил)-1H-имидазол-2-ил)фенил)этил)-1H-пиразоло[3,4-d]пиримидина;
(S)-6-(4-циклобутил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этил)-1Н-пиразоло[3,4-d]пиримидина;
(R)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этил)-1Н-пиразоло[3,4-d]пиримидина;
(S)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-метил-2-(трифторметил)-1Н-имидазол-4-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-(2-фторпропан-2-ил)-6-метоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(5-этокси-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-((6-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)пиридин-3-ил)метил)-1Н-пиразоло[3,4-d]пиримидина;
1-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-5-метил-1H-пиразол-3-карбонитрила;
6-(4-циклопропил-1-этил-1Н-пиразол-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1H-пиразол[3,4-d]пиримидина;
6-(4-циклопропил-6-(метокси-d3)пиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-циклопропил-5-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил) пиримидин-4-карбонитрила;
6-(4-циклопропокси-6-циклопропилпиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклобутокси-6-циклопропилпиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-(дифторметокси)пиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метокси-2-метилпиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-(оксетан-3-ил)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-(трет-бутил)-6-метоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-(трет-бутокси)-6-циклопропилпиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-(2-фторэтокси)пиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-(метил-d3)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
1-(4-(1-циклобутил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4,6-диметоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло [3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-изопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
1-(4-(1-циклопропил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-(2-фторэтил)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло [3,4-d] пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(3-фтор-4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(2-фтор-4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(2,4-диметокси-6-метилпиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(5-метокси-3-(трифторметил)-1H-пиразол-1-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-(2-метоксиэтокси)пиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-(2-метоксиэтокси)пиримидин-5-ил)-1-(4-(5-метил-3-(трифторметил)-1 Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-метокси-6-(1-метилциклопропил)пиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(1-изопропил-4-метил-1Н-пиразол-5-ил)-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1H-пиразол[3,4-d]пиримидина;
(S)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)фенил)этил)-1H-пиразоло[3,4-d]пиримидина;
(R)-6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)фенил)этил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклобутил-6-метоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-изопропоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-изопропил-6-метилпиримидин-5-ил)-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-этоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
N,N,6-триметил-5-(1-(4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил)пиримидин-4-амина;
1-(4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензил)-6-(4-метил-6-(метилтио)пиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4,6-диметоксипиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-(2-метоксиэтокси)-6-метилпиримидин-5-ил)-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(1-(1-метилазетидин-3-ил)-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-метокси-6-метилпиримидин-5-ил)-1-(4-(5-метил-3-(трифторметил)-1Н-пиразол-1-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
3-(1-(4-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил)пиколинонитрила;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(3-(дифторметил)-5-метил-1Н-пиразол-1-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-циклопропил-N-метил-5-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил)пиримидин-4-амина;
1-(4-(5-метокси-3-(трифторметил)-1H-пиразол-1-ил)бензил)-6-(4-метокси-6-метилпиримидин-5-ил)-1H-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-((тетрагидрофуран-3-ил)окси)пиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1H-имидазол-2-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-(4-(бензилокси)-6-циклопропилпиримидин-5-ил)-1-(4-(1-этил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(3-(дифторметил)-1-метил-1Н-1,2,4-триазол-5-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-(4-(бензилокси)-6-циклопропилпиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(5-метокси-1-метил-1Н-1,2,4-триазол-3-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
2-(2-(4-((6-(4-циклопропил-6-метоксипиримидин-5-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)фенил)-4-(трифторметил)-1Н-имидазол-1-ил)-N,N-диметилацетамида;
6-циклопропил-N,N-диметил-5-(1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидин-6-ил)пиримидин-4-амина;
6-(4-циклопропил-6-метоксипиримидин-5-ил)-1-(4-(5-(дифторметил)-1-метил-1Н-1,2,4-триазол-3-ил)бензил)-1H-пиразоло[3,4-d]пиримидина;
6-(4-циклопропил-6-(пирролидин-1-ил)пиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина;
6-(4-(азетидин-1-ил)-6-циклопропилпиримидин-5-ил)-1-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-1Н-пиразоло[3,4-d]пиримидина; и
5-(4-циклопропил-6-метоксипиримидин-5-ил)-3-(4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)бензил)-3H-[1,2,3]триазоло[4,5-d]пиримидина,
или его фармацевтически приемлемая соль.
11. Фармацевтическая композиция для лечения рака, поддающегося лечению ингибитором USP1, содержащая эффективное количество соединения по любому из пп. 1-10, или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.
12. Набор для лечения рака, поддающегося лечению ингибитором USP1, содержащий соединение по любому из пп. 1-10, или его фармацевтически приемлемую соль, или фармацевтическую композицию по п. 11, и инструкции по введению соединения.
13. Способ ингибирования белка USP1, включающий приведение белка USP1 в контакт с соединением по любому из пп. 1-10, или его фармацевтически приемлемой солью, или фармацевтической композицией по п. 11.
| СИСТЕМА ПОДАЧИ ТОПЛИВА | 2005 |
|
RU2298731C2 |
| WO 2009111354 A2, 11.09.2009 | |||
| WO 2017026718 A1, 16.02.2017 | |||
| US 20110144134 A1, 16.06.2011 | |||
| US 20170145012 A1, 25.05.2017 | |||
| RU 2010117737 A, 10.11.2011 | |||
| US 20130253005 A1, 26.09.2013 | |||
| YE LIANBAO et al | |||
| "Design, synthesis and molecular docking analysis of some novel 7-[(quinolin-6-yl)methyl] purines as potential c-Met | |||

