Изобретение относится к способу получения этилена и необязательно других олефинов из водных растворов соответствующих спиртов в сжатой фазе с применением гетерогенного катализатора. Данный способ пригоден в частности для непрерывно эксплуатируемой системы реакторов.
До 1945 большая часть производства этилена основывалась на дегидратации этанола в газовой фазе на гетерогенных катализаторах. Реакцию проводили в реакторах с псевдоожиженным слоем при 350-400°C, время контакта между катализатором и исходными веществами составляло 1-10 секунд. В качестве катализатора применяли «кислые» твердые вещества, например, катализаторы на основе диоксида кремния и оксида алюминия, или различные цеолиты. Выход готового продукта составлял 99% (US 134926), селективность - 98%. Позднее выбирали способ парового крекинга для относительно более экономичных углеводородов.
С 1944 года существует предложенная методика каталитической дегидратации этанола при температуре выше 250°C и произвольном высоком давлении (GB 587378). В качестве возможных катализаторов упоминались фосфорная кислота, концентрированная серная кислота, γ-оксид алюминия и карбонат натрия; единственный вариант осуществления демонстрирует преобразование спирта в присутствии 5% фосфорной кислоты в периодическом процессе. После отделения образующейся в реакции воды полученный этилен был достаточно чистый для целей, которые не были упомянуты.
В настоящее время дегидратация этанола снова представляет интерес, так как этанол поступает в большом количестве из процесса ферментации биомассы. Однако при этом дегидратацию в газовой фазе не принимают в расчет, так как для этого требуется чистый этанол, для чего воду удаляют ректификацией из ферментного раствора, а затем ее нужно удалить из азеотропной смеси. Затраты энергии и расходы при таком способе настолько высоки, что данный способ не принимается во внимание в большинстве ведущих индустриальных государств в качестве реалистичной альтернативы.
В WO 2007/003899 A1, WO 2007/003901 и WO 2007/003910 A1 описан способ получения олефинов, в том числе преимущественно этилена, который позволяет присутствие до 50 масс. % воды. При более высоком содержании воды, например, в случае не подготовленного предварительно биоспирта, необходимо добавить процесс перегонки для того, чтобы понизить содержание воды. При повышенном давлении и повышенной температуре, которые не охарактеризованы подробно, исходные материалы преобразуются; образуется алкен и простой эфир, которые затем разделяют. Обсуждается конкуренция лежащих в основе реакций; слишком низкое содержание этилена можно скомпенсировать комбинацией реакции и перегонки. В качестве катализаторов равным образом предлагаются гетерогенные и гомогенные катализаторы, в том числе цеолиты, сульфонированные носители, серная и фосфорная кислоты. Гетерополикислоты упоминаются как предпочтительные. В единственном примере 80% водный раствор этанола без катализатора подвергают реакционной перегонке.
В WO 2011/115656 было установлено, что при дегидратации этанола замена применяемого катализатора с чистого оксида алюминия на смесь оксида алюминия с оксидом иттрия снижает образование продуктов окисления и что среди продуктов окисления преимущественно образуется легко отделяемый CO2, в то время как образование ацетальдегида находится ниже предела обнаружения.
Существует ряд исследований преобразования пропанола в сверхкритической воде. M. Watanabe и другие представляют в Green Chemistry, 2003(5), 539-544 результаты, которые они получили при применении катализаторов. В соответствии с этим серная кислота способствует образованию пропилена из 2-пропанола, в то время как в присутствии NaOH преимущественно происходит образование ацетона и почти не образуется пропилен. С различными оксидами металлов в качестве катализаторов всегда образуется пропилен, из-за чего авторы предполагают, что на данных материалах в сверхкритической воде образуются кислые центры. Однако выход готового продукта от оксида к оксиду очень различался, с ZrO2 и TiO2 также образовывался ацетон (измерения в большинстве случаев происходили после 15 минут протекания реакции). Найденная специфичность была сравнимой для серной кислоты и большинства применяемых оксидов и составляла примерно 70 моль.%. V.I. Anikeev и другие исследовали кинетические и термодинамические свойства 2-пропанола (The Journal of Supercritical Fluids 32, 123-135 (2004)), но только в отношении влияния плотности реакционной среды.
В то время как в сверхкритической воде пропанол можно превращать в пропен сравнительно без проблем, аналогичная реакция с этанолом проходит не так просто. В обзоре G. Herbert Vogel в Chemie Ingenieur Technik 2011, 83, s. 1-9 данный спирт описывается как «крепкий орешек». При этом проблема частично состоит в низкой степени превращения, частично в низкой специфичности. Xiadong Xu и другие исследовали дегидратацию этанола при различных концентрациях спирта в воде, различных концентрациях серной кислоты и различном времени выдержки (The Journal of Supercritial Fluids, 1990(3), 228-232; Ind. Eng. Chem. Res. 30(7), 1991, 1478-1485). Результаты показали, что хотя в присутствии серной кислоты дегидратация должна обладать высокой специфичностью к этанолу, однако выход готового продукта неудовлетворительный. К похожим результатам пришли S. Ramayya и другие в FUEL 66 (1987), 1364-1371; в представленных данной группой результатах следует обратить внимание на то, что углеродный баланс в эксперименте составлял только 88%, хотя не было обнаружено никаких побочных продуктов. При повышении концентрации этанола в потоке подходящем к реактору выход этилена повышался лишь ненамного, в то время как образование простого диэтилового эфира снижалось очень сильно. Поэтому в литературе для улучшения баланса предлагается возвращение образующегося простого эфира при более высоких концентрациях этанола в реактор (Halvorsen et al. в Process Optimization for the Supercritical Degidration of Ethanol to Ethylene in AIChENational Meeting, Chicago, 1990).
Серная кислота, а также сульфат алюминия в качестве катализатора во многих случаях вызывает коррозию; подвергшиеся коррозии стенки емкости со своей стороны могут проявлять каталитическое действие, что описывали G. Aras и другие в заключительном отчете для исследовательского учреждения Max-Buchner в январе 2011 в работе с названием «Reaktionstechnische Untersuchungen zur Ethylenherstellung aus wässrigen Ethanollösungen (Fermenterausträgen) - Eine nachhaltige Alternative zum Steam-Cracker» (технический университет Дармштатт). Определенные этой группой степени превращения с различными другими гомогенными катализаторами никогда не превышали 10%; в частности сульфат цинка, который очень эффективно катализирует дегидратацию пропанола в пропен, оказался совершенно непригоден. Это может подтвердить предположение от том, что этанол по причине менее стабилизированного карбо-катиона наиболее тяжело подвергается дегидратации. Исследование повышения температуры или продолжительности взаимодействия в работе не описано, так как тогда начала бы доминировать реакция с образованием синтез-газа.
В общем, существующий уровень техники относительно дегидратации спиртов в сверхкритической воде позволяет сделать вывод от том, что (a) жидкую кислоту в качестве катализатора очень сложно контролировать из-за конкурирующей реакции на стенках емкости и (b) дегидратацию этанола нельзя сравнить с дегидратацией более высших спиртов, так как выход готового продукта и в частности специфичность по причине различной ситуации с электронами в переходном состоянии у более высших кислот существенно лучше. Однако чисто спекулятивные утверждения старого издания GB 587378 и противоречивые результаты более современных исследований оставляют специалистов без ясного представления.
В основе данного изобретения лежит задача найти непрерывный способ, которым из водного спиртового раствора даже с низкой концентрацией и в частности содержащего этанол, с помощью дегидратации экономично и с небольшими затратами можно получить соответствующие алкены, при этом способ в частности должен обеспечить высокую специфичность дегидратации в соотношении с конкурирующими реакциями. Также необходимо добиться образования только устраняемых без ущерба для окружающей среды остатков.
Для решения данной задачи автор предлагает способ, который осуществляют при условиях реакции выше критической точки реакционной смеси, а именно с применением неподвижного слоя катализатора с особыми свойствами. Выбор неподвижного катализатора делает возможным экономичное проведение способа независимо от вида спирта, и вместе с этим также использование способа для дегидратации этанола. Неподвижный слой катализатора располагают в пригодном реакторе, и через него может протекать большое количество исходных материалов. В качестве исходного материала применяют содержащий воду или, соответственно водный спиртовой раствор, например, полученный из биомассы, которую подвергали ферментации. Так как применяют только твердый катализатор и поэтому в растворе до и после преобразования не содержится жидкой или растворенной кислоты, то разделение или другая очистка потока продукта не является необходимой, а остаток обладает значением pH, которое делает возможным удаление отходов не нанося ущерба для окружающей среды без дополнительных мер.
Авторы совершенно неожиданно установили, что предусмотренные для данного способа неподвижные катализаторы при выборе подходящей относительно короткой продолжительности взаимодействия позволяют получить очень высокую степень превращения почти без побочных реакций. В сверхкритической области можно достичь степени превращения почти 95% с почти полной специфичностью. Причину можно видеть в том, что по причине низкой вязкости реакционной смеси при одновременно преобладающей очень высокой плотности в сверхкритическом состоянии можно достичь очень высокой скорости диффузии. Это позволяет достигать высокого объемного выхода и выхода готового продукта в единицу времени, что ведет к более высокой продуктивности (или, соответственно к возможному уменьшению объема реактора). Также вода при сверхкритических условиях обладает хорошими растворяющими свойствами для неполярных веществ, таких как олефины, так как относительная диэлектрическая проницаемость сильно уменьшается. Поэтому авторы предполагают, без того, чтобы ограничиваться этим, что исходные вещества, промежуточные продукты преобразования и получающиеся олефины всегда находятся в гомогенной, сжатой фазе и не имеется никаких препятствий для диффузии. Только это состояние позволяет применять сильно разбавленные водой исходные продукты, и это то, чего добиваются в данном изобретении.
В качестве катализаторов для данного способа пригодны оксиды металлов, которые ведут себя как кислоты Бренстеда, то есть являются донорами протонов, и в том числе в частности такие, у которых поверхность модифицирована кислотными группами. Далее, пригодными являются нерастворимые фосфаты металлов или полуметаллов и материалы с высокой удельной поверхностью, которая покрыта молекулами неорганических кислот, если только данные материалы не разрушаются при преобладающих во время преобразования условиях среды. Такие материалы известны специалистам; пригодными являются, например, модифицированная пемза и модифицированный активированный уголь, но не цеолитные материалы.
В качестве оксида металла принципиально пригодны все оксиды, если только они имеют определенные выше свойства кислот Бренстеда, и среди них прежде всего оксиды металлов 3-й главной подгруппы и 4-6-й побочных групп (переходные элементы) периодической системы, а также оксиды элементов из группы лантаноидов, например, оксид церия. Предпочтительными среди них являются оксиды 3-й главной группы, а также модифицированные кислотами оксиды металлов, и среди последних в свою очередь оксиды элементов 3-й главной группы и 4-6-й побочных групп (переходные элементы) периодической системы. В качестве кислотных групп пригодны в частности сульфатные или фосфатные группы. Пригодными являются, например, оксиды алюминия, циркония или титана, у которых поверхность покрыта кислотными группами, или пористые материалы, такие как пемза или уголь, в частности активированный уголь, у которых поверхность покрыта кислотными группами. Данные материалы особенно предпочтительно покрыты сульфатными или фосфатными группами.
Следует указать на то, что выражение «оксид металла» включает как немодифицированные, так и модифицированные (например, кислотными группами) материалы.
Данный способ пригоден в частности для преобразования и вместе с этим переработки этанола, но не ограничивается этим, так как он подходит также, конечно, для преобразования более высших спиртов, таких как пропанол, бутанол или пентанол (во всех изомерных формах). Часто применяют исходные материалы, которые содержат смесь спиртов. Данные смеси могут, например, содержать определенное количество (например, по меньшей мере 10 масс. %, предпочтительно по меньшей мере 20 масс. %, более предпочтительно по меньшей мере 50 масс. %) этанола, по отношению к всей массе спиртов, однако данные смеси могут не содержать этанола. В качестве спирта предпочтительно принимают во внимание C2-C4-спирты.
Преобразование происходит при по меньшей мере 300°C, предпочтительно при по меньшей мере 350°C, в частности в случае преобразования этанола. Также предпочтительно не превышать верхнюю границу 600°C. Минимальное давление должно составлять примерно 220 бар (2,2×107 Па). Давление и температуру необходимо выбирать таким образом, чтобы исходный спиртовой раствор достигал сверхкритического состояния и в данной смеси это состояние сохраняется также в ходе изменения соотношения воды и спирта при преобразовании спирта в олефин. Предпочтительной в зависимости от концентрации спирта является температура 350°C или выше, предпочтительно 400°C или выше, при давлении 250 бар или выше.
Исходная концентрация спирта в водном растворе может варьировать в широкой области, которая в частности включает от 5 до 95 об.%. Авторы установили, что также хорошо преобразуются исходные материалы с относительно низкой концентрацией спирта, например, в области от 10 до 500 г/л, особенно предпочтительно в области от 15 до 300 г/л.
Содержание воды в растворе не является критическим и может колебаться в широкой области. Однако его необходимо удерживать на таком уровне, чтобы оно было относительно небольшим, чтобы количество катализатора было достаточным. Как правило, количество катализатора составляет по меньшей мере 1 или лучше 2 масс. % от раствора. Водный раствор спирта может содержать другие жидкие (растворенные) содержащие углерод компоненты, такие как органические кислоты, при этом речь идет, например, о побочных продуктах ферментативной или нефтехимической предварительной реакции. При этом вид данных компонентов, а также общее содержание углерода в растворе не ограничено.
Время контакта спирта с катализатором и вместе с этим продолжительность взаимодействия при заданных условиях можно выбирать по потребности в зависимости от исходной концентрации или выбранного катализатора. Неожиданно было установлено, что совершенно достаточно промежутка времени в области секунд или минут, например, от 5 до 300 сек. Такая небольшая продолжительность пребывания в частности делает возможным выполнять данный способ как непрерывный способ с высокой производительностью. Более низкие концентрации еще укорачивают продолжительность пребывания так, что как правило, способ проводят при продолжительности контакта/пребывания не больше 100 сек, при этом также наблюдаются неожиданные результата относительно общей степени преобразования, а также специфичности. Особенно предпочтительно продолжительность пребывания составляет от 10 до 80 сек, более предпочтительно от 20 до 35 сек, и это в комбинации с упомянутыми давлением и температурой. Данная низкая продолжительность пребывания в частности делает возможным проводить данный способ как непрерывный с высокой производительностью.
Разделение газовой и жидкой фазы предпочтительно происходит под давлением. Вместе с этим олефин после реакции еще находится под давлением, и его можно дальше преобразовывать или расфасовывать.
Если в жидкой фазе остались остатки спирта, а также необязательно образовавшийся диалкиловый простой эфир, то поток жидкой фазы для достижения лучшей степени преобразования можно еще раз вернуть в реактор, например в виде подмешивания к новому исходному материалу.
Способ по изобретению пригоден в частности для переработки обработанных ферментацией биоматериалов или содержащих спирт растворов, которые являются побочными продуктами в химическом производстве (например, в нефтехимии). Необязательно имеющиеся твердые вещества, такие как целлюлозные материалы необходимо предварительно отделить для того, чтобы предотвратить нежелательное коксование и дезактивацию катализатора. Как средство для этого часто выбирают одноступенчатое выпаривание (мгновенная дистилляция). Растворенные соли как правило не мешают, пока при условиях реакции не устанавливается кислое значение pH, однако они по существу не остаются после одноступенчатого выпаривания. В этом отношении способ по изобретению имеет существенное преимущество по сравнению с применяемой ранее дегидратацией в газовой фазе, так как для нее необходимо проводить очистку исходной смеси путем дорогостоящей ректификации.
По сравнению с классической дегидратацией в газовой фазе данное изобретение имеет следующие преимущества:
- экономия на многоступенчатой ректификации для концентрирования полученного ферментацией спирта,
- улучшение объемного выхода и выхода готового продукта за единицу времени по причине реакции в сжатой фазе,
- низкая потребность в энергии, так как нет необходимости в испарении, требуется только нагревание,
- олефин после реакции находится под высоким давлением - экономия на сжатии газа,
- отмена транспортных и складских расходов для, например, этилена, так как его можно производить непосредственно у потребителя (например, вблизи от производственного оборудования, которое не оснащено соединением с трубопроводом и поэтому не имеет непосредственного доступа, например, этилена или пропилена),
- разработанный способ может быть реализован в устройстве небольшого размера, так что нет необходимости в присоединении к трубопроводам.
Собственные эксперименты авторов данной заявки с жидкими кислотами привели к результатам, которые отчасти диаметрально противоположны результатам из литературы, по меньшей мере неожиданным, в частности относительно выхода готового продукта и специфичности (см., например, (не представляющий данное изобретение) пример 2, согласно которому серная кислота в качестве катализатора обладает существенно меньшей специфичностью для преобразования этанола, чем опубликованная специфичность для преобразования пропанола). Возможно в данном случае большую роль играет также описанная в уровне техники проблема агрессивности серной кислоты и ее воздействие на стенки емкости. Так как Watanabe и другие исследовали дегидратацию пропанола как с жидкой кислотой, так и с твердым оксидом и результат при применении твердого катализатора был незначительно хуже, чем с кислотой, нельзя было ожидать, что дегидратация «крепкого орешка» этанола на кислом твердом катализаторе пройдет успешно. Авторы данной заявки неожиданно установили, что степени преобразования этанола при применении серной кислоты и твердого катализатора примерно одинаковы и в каждом случае очень высокие, а специфичность и вместе с этим селективность выхода готового продукта этилена при применении оксида алюминия, однако, возрастает более чем в 10 раз. Кроме того они установили, что условия реакции давление/температура в сверхкритической области, например, температура ≥380°C и/или давление ≥240 бар значительно повышают степень преобразования, в то время как селективность несколько снижается. Дополнительного повышения степени преобразования удается достичь, если вместо оксида алюминия применять диоксид циркония или диоксид титана. Применение модифицированных кислотами катализаторов вызывает повышение селективности этилена.
Эти результаты оказались совершенно неожиданными. Тем не менее, только эти данные делают возможной успешную дегидратацию в виде непрерывного процесса на неподвижном слое катализатора.
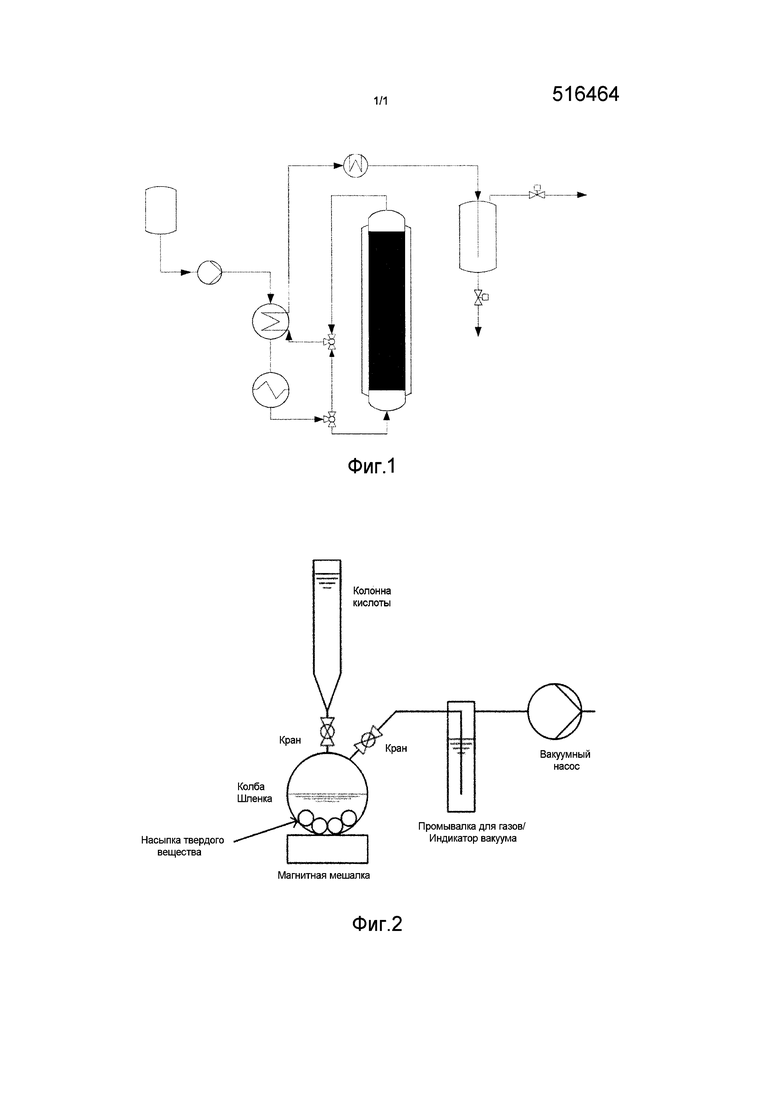
Предпочтительно преобразование проходит как указано ниже, при этом схема пригодного для этого устройства представлена на фиг. 1: водный спиртовой раствор, который, например, получен в ходе ферментативного или нефтехимического процесса, сначала из конденсатора, в частности емкости конденсатора 1, с помощью насоса (в данном случае насоса высокого давления 2) сжимают до по меньшей мере 220 бар или до давления из описанной выше области давления. Раствор нагревают до температуры предпочтительно от 350 до 600°C с помощью теплообменника с противотоком 3 и/или пароперегревателя 4. Для запуска устройства можно, например, использовать байпас 5. Сверхкритический реакционный раствор преобразуется в реакторе 6, который эксплуатируется в адиабатическом или изотермическом режиме, на неподвижном слое катализатора, например, засыпке. Конечно, возможно также применение монолитного материала катализатора. Неподвижный слой катализатора, который, как правило, технически обычно насыпан, предпочтительно заполняет весь объем реактора. Это имеет несколько преимуществ. Во первых таким образом достигают равномерного протекания через реактор. Во вторых реакционный раствор в течение всего пребывания в реакторе находится в контакте с катализатором при преобладающих в реакторе давлении и температуре.
Альтернативно также можно применять, например, каскад реакторов для повышения селективности. Продолжительность пребывания в реакторе составляет предпочтительно от 5 до 300 сек. В качестве катализатора можно применять упомянутые катализаторы, необязательно также в виде смеси или в виде модифицированного кислотой носителя. После выхода из реактора поток продукта попадает предпочтительно в охладитель 7, предпочтительно через упомянутый теплообменник с противотоком. В охладителе происходит охлаждение, например, до температуры ниже 50°C, однако при этом давление сохраняется. Из охладителя двухфазная смесь поступает в разделитель 8, в котором происходит разделение смеси на газовую фазу неочищенного олефина 9 и жидкую водную фазу 10. В заключении газовую фазу можно подвергать очистке, способом, известным из уровня техники.
Упомянутое преобразование в частности проходит на следующих катализаторах, которые получают способом пропитывания:
Кат 01 Активированный уголь + H2SO4 (кальцинированный в атмосфере азота).
Кат 02 Активированный уголь + H3PO4 (кальцинированный в атмосфере азота).
Кат 03 Al2O3 + H2SO4.
Кат 04 Al2O3 + H3PO4.
Кат 05 TiO2 + H2SO4.
Кат 06 TiO2 + H3PO4.
Кат 07 ZrO2 + H2SO4.
Кат 08 ZrO2 + H3PO4.
Данные катализаторы можно получать, например, описанным ниже способом и/или с помощью представленного на фиг. 2 устройства. При этом в колбе, в данном случае колбе Шленка, с помощью вакуумного насоса создают разряжение для того, чтобы обеспечить удаление всего капиллярного кислорода из материала катализатора перед пропитыванием твердого вещества кислотой. Индикатором вакуума служит наполненная водой промывалка для газов. Если ослабевает образование пузырьков газа в приемной трубке промывалки для газов, можно быть уверенным, что достигнут максимально возможный вакуум. Далее, водяная баня препятствует утечке мельчайших частиц. После достижения вакуума можно открывать колонну для кислоты в реакционный сосуд; вакуум должен оставаться. Благодаря диспергированию твердого катализатора в серной кислоте (например, 1Н) или фосфорной кислоте (например, 1Н) образуются активные центры серной или фосфорной кислот на поверхности частиц. После воздействия в течение, например, от 0,5 до 2 часов, предпочтительно, примерно, 1 часа, вакуум можно убирать и извлекать содержимое колбы через фильтр (например, синий ленточный фильтр. Фильтрационный осадок сушат, например, 24 часа при примерно 105°C в сушильном шкафу, и затем кальцинируют, например, 24 часа при 500°C.
Для получения фосфатов металлов и полуметаллов соответствующие соединения металлов или полуметаллов, например, оксиды, кислоты, хлориды или оксихлориды подвергают реакции с фосфорной кислотой. Примерами пригодных катионов является титан, цирконий и алюминий. При этом для реакции в лабораторных условиях отвешивают предпочтительно примерно от 5 до 50 г соответствующего соединения металла или полуметалла и помещают в реакционный сосуд. Туда же добавляют превышающую стехиометрическую массу 85% ортофосфорной кислоты (или другой фосфорной кислоты), и реакционную смесь для лучшей однородности и смешиваемости дополнительно разбавляют деионизированной водой (предпочтительно в количестве максимально 50% фосфорной кислоты). Реакционные сосуды, а также проведение реакции различаются для экзотермичных и эндотермичных реакций. Эндотермичные реакции проводят в нагреваемой круглодонной колбе в состоянии кипения с обратным холодильником, экзотермические реакции проводят в охлаждаемом химическом стакане. Температура кипения составляет от 90°C до 110°C. При этом продолжительность реакции для химического преобразования в фосфаты составляет предпочтительно примерно 4 часа при температуре в выбранной области.
После окончания реакции раствор доводят до комнатной температуры. В зависимости от вида аниона исходного вещества может быть необходима вторая реакция для нейтрализации неорганической кислоты и одновременного осаждения фосфата. Например, при применении содержащего хлорид исходного материала образующуюся соляную кислоту нейтрализуют NaOH. Затем продукт фильтруют, например, через фильтр для тонкого осадка. При этом предпочтительно применяют синий ленточный фильтр.
Таким образом полученный фильтровальный осадок освобождают от поверхностной воды, например, сушат 24 часа при 105°C в сушильном шкафу. Затем предпочтительно фосфат кальцинируют, например, при повышенной температуре в муфельной печи. При этом удаляется капиллярная и кристаллизационная вода, вследствие чего катализатор приобретает активность. При этом по результатам измерений удельной поверхности и ее корреляции с каталитической активностью особенно предпочтительной оказалась область температур примерно от 300°C до 1100°C. Качество подготовки катализатора можно, например, определить гравиметрическим способом. Также процент покрытия поверхности кислотными группами, по отношению к максимально возможному покрытию, при необходимости можно определить. Часто образовавшиеся при кальцинировании куски материала затем измельчают в ступке.
Данное изобретение разъясняется подробнее с помощью приведенных ниже примеров.
Пример 1 (сравнительный пример)
В устройстве, таком как представлено на фиг. 1, реактор полностью наполняли 150 мл SiC в качестве несущего материала для того, чтобы добиться постоянного профиля потока. В качестве исходного материала применяли 25% раствор этанола в воде, который преобразовывался при температуре реакции 400°C и давлении 250 бар. Продолжительность пребывания в реакторе составляла 20 сек. При этом достигали степени преобразования 21,8% при селективности этилена 29,7%.
Пример 2 (сравнительный пример)
Пример 1 повторяли с тем изменением, что в раствор этанола перед входом в реактор по каплям подмешивали серную кислоту 98 масс. %. При этом достигали степени преобразования 89,7% при селективности этилена 6,8%.
Пример 3
Пример 1 повторяли с тем изменением, что реактор вместо SiC в качестве носителя содержал гранулы γ-Al2O3. При этом достигали степени преобразования 23,3% селективности этилена 91,5%.
Пример 4
Пример 3 повторяли с тем изменением, что продолжительность пребывания составила 30 сек. При этом достигали степени преобразования 92,5% при селективности этилена 97,6%.
Пример 5
В устройстве, таком как представлено на фиг. 1, реактор наполняли 44 мл ZrO2 (тетрагональный) в качестве материала катализатора. В качестве исходного материала применяли 39% раствор этанола в воде при температуре реакции 357°C и давлении 230 бар. Продолжительность пребывания в реакторе составляла 64 сек. При этом достигали степени преобразования 56,7% при селективности этилена 73,8%.
Пример 6
В устройстве, таком как представлено на фиг. 1, реактор наполняли 28 мл γ-Αl2O3 в качестве материала катализатора. В качестве исходного материала применяли 39% раствор этанола в воде при температуре реакции 357°C и давлении 230 бар. Продолжительность пребывания в реакторе составила 75 сек. При этом достигали степени преобразования 47,9 % при селективности этилена 70,4%.
Пример 7
В устройстве, таком как представлено на фиг. 1, реактор наполняли 33 мл TiO2 (анатаз) в качестве материала катализатора. В качестве исходного материала применяли 39% раствор этанола в воде при температуре реакции 357°C и давлении 230 бар. Продолжительность пребывания в реакторе составила 72 сек. При этом достигали степени преобразования 87,9 % при селективности этилена 11,5%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНА ПОСРЕДСТВОМ КАТАЛИТИЧЕСКОЙ КОНВЕРСИИ ПО МЕНЬШЕЙ МЕРЕ ОДНОГО СПИРТА | 2014 |
|
RU2660132C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОПИЛЕНА | 2006 |
|
RU2415832C2 |
| ДЕГИДРИРОВАНИЕ СМЕШАННЫХ СПИРТОВ | 2006 |
|
RU2412141C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВ | 2006 |
|
RU2419596C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛЕНА | 2006 |
|
RU2415121C2 |
| РЕАКЦИОННАЯ РЕКТИФИКАЦИЯ ДЛЯ ДЕГИДРАТАЦИИ СМЕШАННЫХ СПИРТОВ | 2006 |
|
RU2419595C2 |
| УДАЛЕНИЕ АЗОТСОДЕРЖАЩИХ ПРИМЕСЕЙ ИЗ КОМПОЗИЦИЙ СПИРТОВ | 2013 |
|
RU2641695C2 |
| РЕАКЦИОННАЯ РЕКТИФИКАЦИЯ С ВОЗВРАТОМ В ПРОЦЕСС ОЛЕФИНОВ | 2006 |
|
RU2419597C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКЕНОВ ИЗ ОКСИГЕНАТОВ С ИСПОЛЬЗОВАНИЕМ НАНЕСЕННЫХ НА НОСИТЕЛЬ ГЕТЕРОПОЛИКИСЛОТНЫХ КАТАЛИЗАТОРОВ | 2007 |
|
RU2446011C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКЕНА | 2011 |
|
RU2603636C2 |
Изобретение относится к способу непрерывного получения одного или нескольких олефинов из водного раствора одного или нескольких соответствующих спиртов, причем спирты выбирают из этанола, пропанола и бутанола. Способ включает стадии: подготовки не содержащего твердых веществ водного раствора спирта или спиртов, прохождения данного раствора через реактор, который наполнен неподвижным слоем катализатора таким образом, что данный раствор при прохождении через реактор контактирует с катализатором при температуре по меньшей мере 300°C и давлении по меньшей мере 220 бар в такой комбинации температуры и давления, что спирт или спирты подвергаются превращению при сверхкритических условиях, при этом продолжительность пребывания раствора в реакторе составляет от 5 до 300 с, и прохождения образовавшейся двухфазной смеси в разделитель, в котором происходит разделение смеси на газовую фазу неочищенного олефина и жидкую водную фазу. При этом катализатор выбирают из оксидов катионов 3 главной группы, а также побочных групп с 4-й по 6-ю Периодической системы, нерастворимых фосфатов металлов и фосфатов полуметаллов, а также пористых материалов, которые выбирают из пемзы и угля, у которых поверхность покрыта группами неорганических кислот. Данный способ при высокой степени превращения позволяет обеспечить низкое количество побочных реакций. 28 з.п. ф-лы, 7 пр., 2 ил.
1. Способ непрерывного получения одного или нескольких олефинов из водного раствора одного или нескольких соответствующих спиртов, причем спирты выбирают из этанола, пропанола и бутанола, включающий стадии:
подготовка не содержащего твердых веществ водного раствора спирта или спиртов,
прохождение данного раствора через реактор, который наполнен неподвижным слоем катализатора таким образом, что данный раствор при прохождении через реактор контактирует с катализатором, при температуре по меньшей мере 300°C и давлении по меньшей мере 220 бар в такой комбинации температуры и давления, что спирт или спирты подвергаются превращению при сверхкритических условиях, при этом продолжительность пребывания раствора в реакторе составляет от 5 до 300 с, и
прохождение образовавшейся двухфазной смеси в разделитель, в котором происходит разделение смеси на газовую фазу неочищенного олефина и жидкую водную фазу,
при этом катализатор выбирают из оксидов катионов 3 главной группы, а также побочных групп с 4-й по 6-ю Периодической системы, нерастворимых фосфатов металлов и фосфатов полуметаллов, а также пористых материалов, которые выбирают из пемзы и угля, у которых поверхность покрыта группами неорганических кислот.
2. Способ по п. 1, отличающийся тем, что продолжительность пребывания раствора в реакторе составляет от 5 до 100 с и предпочтительно от 10 до 80 с.
3. Способ по п. 1, отличающийся тем, что реактор полностью заполнен загрузкой катализатора.
4. Способ по п. 2, отличающийся тем, что реактор полностью заполнен загрузкой катализатора.
5. Способ по п. 1, отличающийся тем, что поверхность катализатора имеет группы неорганических кислот.
6. Способ по п. 2, отличающийся тем, что поверхность катализатора имеет группы неорганических кислот.
7. Способ по п. 3, отличающийся тем, что поверхность катализатора имеет группы неорганических кислот.
8. Способ по одному из пп. 1-7, отличающийся тем, что неподвижный слой катализатора выбирают из оксидов катионов 3 главной группы, а также побочных групп с 4-й по 6-ю Периодической системы, у которых поверхность покрыта группами неорганических кислот.
9. Способ по одному из пп. 1-7, отличающийся тем, что неподвижный катализатор выбирают из оксидов алюминия, циркония и титана, у которых поверхность предпочтительно покрыта группами неорганических кислот, пористых материалов, которые выбирают из пемзы или угля, у которых поверхность покрыта сульфатными или фосфатными группами, и фосфатов металлов.
10. Способ по одному из пп. 1-7, отличающийся тем, что неподвижный катализатор выбирают из фосфата алюминия, фосфата циркония, фосфатированного оксида алюминия, фосфатированного оксида титана, фосфатированного оксида циркония, фосфатированного угля, фосфатированной пемзы, сульфатированного оксида циркония, сульфатированного оксида титана, сульфатированного угля, сульфатированной пемзы и сульфатированного оксида алюминия.
11. Способ по одному из пп. 1-7, отличающийся тем, что водный раствор спирта или спиртов включает этанол, пропанол или бутанол в качестве единственного спирта или в качестве одного из спиртов в количестве по меньшей мере 10 мас.%, предпочтительно по меньшей мере 20 мас.%, более предпочтительно по меньшей мере 50 мас.%, по отношению ко всему весу имеющихся спиртов.
12. Способ по п. 11, отличающийся тем, что водный раствор спирта или спиртов включает этанол в качестве единственного спирта или в качестве одного из спиртов в количестве по меньшей мере 10 мас.%, предпочтительно по меньшей мере 20 мас.%, более предпочтительно по меньшей мере 50 мас.%, по отношению ко всей массе имеющихся спиртов.
13. Способ по одному из пп. 1-7, отличающийся тем, что температура составляет по меньшей мере 350°C и предпочтительно 400°C или более и/или давление составляет предпочтительно 250 бар или больше.
14. Способ по п. 8, отличающийся тем, что температура составляет по меньшей мере 350°C и предпочтительно 400°C или более и/или давление составляет предпочтительно 250 бар или больше.
15. Способ по п. 9, отличающийся тем, что температура составляет по меньшей мере 350°C и предпочтительно 400°C или более и/или давление составляет предпочтительно 250 бар или больше.
16. Способ по п. 10, отличающийся тем, что температура составляет по меньшей мере 350°C и предпочтительно 400°C или более и/или давление составляет предпочтительно 250 бар или больше.
17. Способ по одному из пп. 1-7, отличающийся тем, что исходная концентрация спирта или спиртов в водном растворе составляет от 5 до 95% (мас./об.), предпочтительно от 15 до 30% (мас./об.).
18. Способ по одному из пп. 1-7, отличающийся тем, что водный раствор после прохождения через содержащий неподвижный слой катализатора реактор, еще находясь под давлением, охлаждается, вслед за чем происходит разделение газовой и жидкой фаз таким образом, что образовавшийся олефин и дальше остается под давлением.
19. Способ по п. 8, отличающийся тем, что водный раствор после прохождения через содержащий неподвижный слой катализатора реактор, еще находясь под давлением, охлаждается, вслед за чем происходит разделение газовой и жидкой фаз таким образом, что образовавшийся олефин и дальше остается под давлением.
20. Способ по п. 9, отличающийся тем, что водный раствор после прохождения через содержащий неподвижный слой катализатора реактор, еще находясь под давлением, охлаждается, вслед за чем происходит разделение газовой и жидкой фаз таким образом, что образовавшийся олефин и дальше остается под давлением.
21. Способ по п. 10, отличающийся тем, что водный раствор после прохождения через содержащий неподвижный слой катализатора реактор, еще находясь под давлением, охлаждается, вслед за чем происходит разделение газовой и жидкой фаз таким образом, что образовавшийся олефин и дальше остается под давлением.
22. Способ по п. 13, отличающийся тем, что водный раствор после прохождения через содержащий неподвижный слой катализатора реактор, еще находясь под давлением, охлаждается, вслед за чем происходит разделение газовой и жидкой фаз таким образом, что образовавшийся олефин и дальше остается под давлением.
23. Способ по одному из пп. 1-7, отличающийся тем, что образующуюся жидкую водную фазу после отделения от газовой фазы неочищенного олефина смешивают со свежим раствором спирта или спиртов и вместе с ним снова отправляют в реактор, причем продолжительность пребывания раствора в реакторе составляет от 5 до 100 с и предпочтительно от 10 до 80 с.
24. Способ по п. 8, отличающийся тем, что образующуюся жидкую водную фазу после отделения от газовой фазы неочищенного олефина смешивают со свежим раствором спирта или спиртов и вместе с ним снова отправляют в реактор, причем продолжительность пребывания раствора в реакторе составляет от 5 до 100 с и предпочтительно от 10 до 80 с.
25. Способ по п. 9, отличающийся тем, что образующуюся жидкую водную фазу после отделения от газовой фазы неочищенного олефина смешивают со свежим раствором спирта или спиртов и вместе с ним снова отправляют в реактор, причем продолжительность пребывания раствора в реакторе составляет от 5 до 100 с и предпочтительно от 10 до 80 с.
26. Способ по п. 10, отличающийся тем, что образующуюся жидкую водную фазу после отделения от газовой фазы неочищенного олефина смешивают со свежим раствором спирта или спиртов и вместе с ним снова отправляют в реактор, причем продолжительность пребывания раствора в реакторе составляет от 5 до 100 с и предпочтительно от 10 до 80 с.
27. Способ по п. 13, отличающийся тем, что образующуюся жидкую водную фазу после отделения от газовой фазы неочищенного олефина смешивают со свежим раствором спирта или спиртов и вместе с ним снова отправляют в реактор, причем продолжительность пребывания раствора в реакторе составляет от 5 до 100 с и предпочтительно от 10 до 80 с.
28. Способ по одному из пп. 1-7, отличающийся тем, что в качестве спиртового водного раствора применяют раствор, который получен из обработанного ферментацией биоматериала или содержащего спирт материала, которые образуются как побочный продукт в химической промышленности, предпочтительно в нефтехимии.
29. Способ по одному из пп. 1-7, отличающийся тем, что нагревание водного спиртового раствора происходит с помощью теплообменника с противотоком, в котором спиртовой раствор поступает противотоком к покидающему реактор раствору.
| NOURI S AND AMRAN T, "Supercritical water ethanol reforming for hydrogen production: (effect of different support on nickel based catalyst in supercritical water condition)", ASIAN JOURNAL OF CHEMISTRY, vol | |||
| Машина для добывания торфа и т.п. | 1922 |
|
SU22A1 |
| Masaru Watanabe et al, Conversions of some small organic compounds with metal oxides in supercritical water at 673 KPresented at The First International Conference on Green & Sustainable Chemistry, Green Chemistry, Vol:5, Nr:5, Page(s):539, 2003 | |||
| ДЕГИДРИРОВАНИЕ СМЕШАННЫХ СПИРТОВ | 2006 |
|
RU2412141C2 |

