ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к гетеробициклическим производным, в частности, но без ограничения, к хинолиноновым и хиназолиноновым производным, которые являются ингибиторами вируса гепатита C (HCV), к их синтезу и их применению, отдельно или в комбинации с другими ингибиторами HCV, при лечении или профилактике HCV.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
HCV является однониточным вирусом с положительно-полярной РНК, принадлежащий семейству вирусов Flaviviridae роду гепатовирусов. Вирусный геном транслируется в отдельную открытую рамку считывания, которая кодирует множество структурных и неструктурных белков.
После начальной острой инфекции у большинства инфицированных индивидуумов развивается хронический гепатит, поскольку HCV реплицируется преимущественно в гепатоцитах, но не являющийся непосредственно цитопатическим. В частности, отсутствие сильного T-лимфоцитного ответа и высокая склонность вируса к мутации, по-видимому, способствует высокой скорости развития хронической инфекции. Хронический гепатит может развиваться до фиброза печени, приводящего к циррозу, конечной стадии болезни печени, и HCC (гепатоцеллюлярному раку), что делает его основной причиной трансплантации печени.
Существует 6 основных генотипов и свыше 50 подтипов HCV, которые распространены в разных географических зонах. 1 генотип HCV является доминирующим генотипом в Европе и США. Обширная генетическая гетерогенность HCV имеет важные диагностические и клинические последствия, возможно объясняющие трудности в разработке вакцины и отсутствие ответной реакции на существующую терапию.
Перенос HCV может осуществляться через контакт с зараженной кровью или продуктами, полученными из зараженной крови, например, в результате переливания крови или внутривенного применения лекарственных средств. Внедрение диагностических тестов, используемых при отборе крови, привело к тенденции снижения посттрансфузионной заболеваемости HCV. Однако вследствие медленного развития заболевания печени до конечной стадии развития болезни, инфекции, уже имеющие место в настоящее время, будут продолжать представлять серьезную медицинскую и экономическую проблему на протяжении десятилетий.
Применяющиеся в настоящее время терапевтические методы лечения HCV основываются на (пэгилированном) интерфероне-альфа (IFN-α) в комбинации с рибофлавином. Такая комбинированная терапия приводит к стабильному вирусологическому ответу у более чем 40% пациентов, инфицированных HCV генотипа 1, и примерно у 80% пациентов, инфицированных генотипами 2 и 3. Помимо ограниченной эффективности в отношении HCV генотипа 1 такая комбинированная терапия сопровождается значительными побочными эффектами, в том числе подобными гриппу симптомами, гематологическими патологиями и нейропсихическими симптомами. Следовательно, существует потребность в более эффективных, удобных и легче переносимых способах лечения.
Практический опыт применения лекарственных средств против HIV, в частности ингибиторов протеазы HIV, показывает, что субоптимальная фармакокинетика и сложные схемы дозировки быстро приводят к случайным нарушениям режима и схемы приема лекарственных средств. Это, в свою очередь, означает, что 24-часовая остаточная концентрация (минимальная концентрация в плазме) для соответствующих лекарственных средств в режиме лечения HIV зачастую на длительные периоды времени в течение суток падает ниже IC90 или ED90. Считается, что при 24-часовом остаточном уровне, по меньшей мере, IC50, точнее IC90 или ED90, существенно замедляется развитие мутантов, «отключающих действие лекарственных средств». Достижение нужной фармакокинетики и скорости метаболизма лекарственного средства для получения таких остаточных уровней обуславливает жесткое требование к разработке лекарственного средства.
Белок NS5A HCV располагается в прямом направлении относительно белка NS4B и в обратном направлении относительно белка NS5B. При посттрансляционном расщеплении вирусной сериновой протеазой NS3/4A NS5A созревает до содержащего цинк трехдоменного фосфопротеина, который существует либо в виде гипофосфорилированных (56-кДа, p56), либо гиперфосфорилированных частиц (58-кДа, p58). NS5A HCV включается во многие аспекты жизненного цикла вируса, в том числе в вирусную репликацию и сборку инфекционных частиц, а также в модуляцию окружающей среды его клетки-хозяина. Хотя ни одна из ферментативных функций не была приписана белку, сообщается о взаимодействии с многочисленными вирусными и клеточными факторами.
В ряде патентов и патентных заявок раскрываются соединения с ингибиторной активностью против HCV, в частности, нацеливание на NS5A. В WO2006/133326 раскрываются стильбеновые производные, а в WO 2008/021927 и WO 2008/021928 раскрываются бифенильные производные, обладающие ингибиторной активностью по отношению к NS5A HCV. В WO 2008/048589 раскрываются 4-(фенилэтинил)-1H-пирозоловые производные и их антивирусное применение. В WO 2008/070447 раскрывается широкий диапазон ингибирующих HCV соединений, включающих бензимидазольный фрагмент. В обеих из WO-2010/017401 и WO-2010/065681 раскрываются бис-имидазольных ингибиторов HCV NS5A.
Существует потребность в ингибиторах HCV, которые смогут преодолеть недостатки существующей в настоящее время терапии HCV, такие как побочные эффекты, ограниченная эффективность, возникновение устойчивости и неудачи в соблюдении режима лечения, а также улучшить устойчивый ответ на вирусную нагрузку.
Настоящее изобретение относится к группе ингибирующих HCV гетеробициклических производных и, в частности, но без ограничения, хинолиноновых и хиназолиноновых производных с полезными свойствами, касающимися одного или нескольких из следующих параметров: противовирусная эффективность, подходящий профиль развития устойчивости, пониженная токсичность и генотоксичность или отсутствие таковых, подходящая фармакокинетика и фармакодинамика, удобство составления и введения, а также ограниченные межлекарственные взаимодействия с другими веществами-лекарственными средствами, в частности, с другими средствами против-HCV, или отсутствие таковых.
Соединения в соответствии с настоящим изобретением также могут быть привлекательными благодаря тому факта, что у них отсутствует активность против других вирусов, в частности против HIV. Инфицированные HIV пациенты часто страдают от сопутствующих инфекций, таких как HCV. Лечение таких пациентов ингибитором HCV, который также ингибирует HIV, может привести к возникновению устойчивых штаммов HIV.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном аспекте настоящее изобретение относится к новым соединениям формулы I:
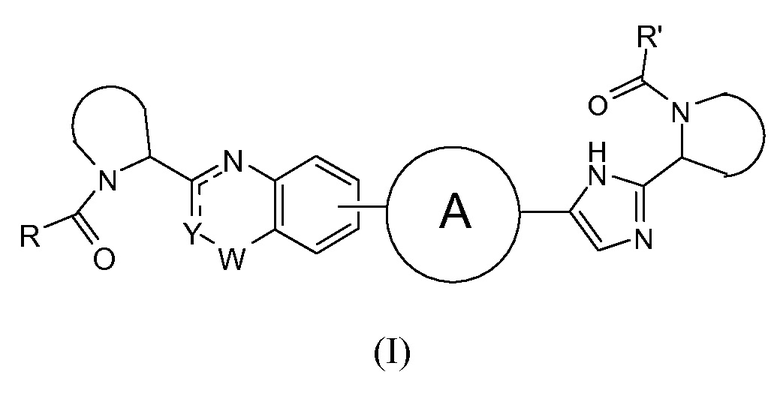
или его стереоизомер, где
Y представляет собой CH или N, CR4;
W представляет собой карбонил, сульфонил или CR5R6;
 представляет собой
представляет собой 
 независимо выбран из группы, содержащей
независимо выбран из группы, содержащей

R и R' независимо выбраны из -CR1R2R3, арила, необязательно замещенного 1 или 2 заместителями, выбранными из галогена и метила или гетероциклоалкила, где
R1 выбран из C1-4алкила, C2-4алкила, замещенного метокси или гидроксилом, и фенила, необязательно замещенного 1 или 2 заместителями, независимо выбранными из галогена и метила;
R2 представляет собой гидроксил, амино, моно- или ди-C1-4алкиламино, C1-4алкил-карбониламино, C1-4алкилоксикарбониламино;
R3 представляет собой водород или C1-4алкил;
R4 представляет собой водород, C1-4алкил или фтор;
каждый из R5 и R6 независимо представляет собой C1-4алкил; или
CR5R6 вместе образуют C3-7циклоалкил, оксетан, тетрагидрофуран;
или его фармацевтически приемлемые соли или сольваты.
Кроме того, настоящее изобретение относится к продукту, содержащему (a) соединение согласно настоящему изобретению и (b) другой ингибитор HCV, в качестве комбинированного препарата для одновременного, раздельного или последовательного применения в лечение инфекций HCV.
В следующем аспекте настоящее изобретение относится к применению соединений формулы Ia-c или их подгрупп, определенных в настоящем документе, для ингибирования HCV. В качестве альтернативы, предусматривается применение указанных соединений для производства лекарственного препарата для ингибирования HCV.
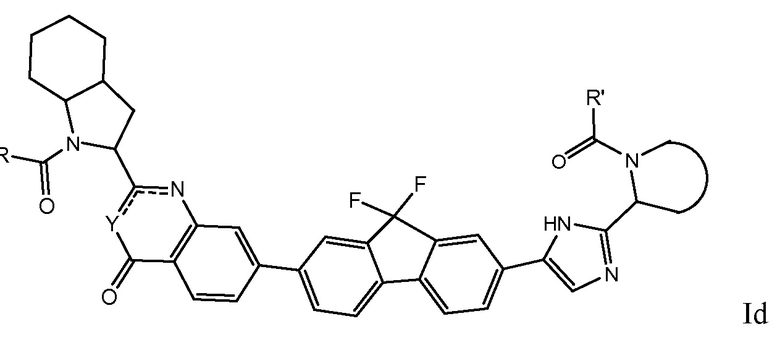
В первом варианте осуществления настоящее изобретение относится к подгруппе соединений формулы I, которые могут быть представлены формулой (Ia);
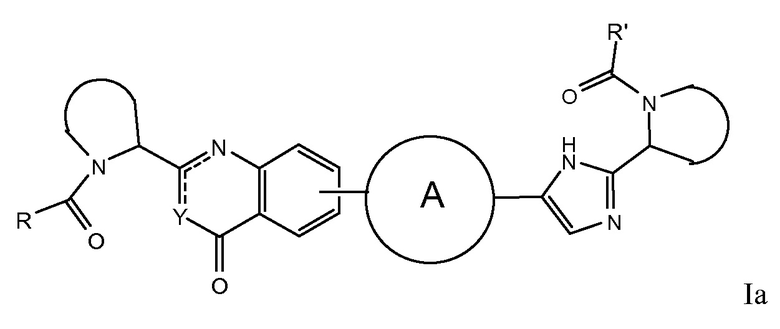
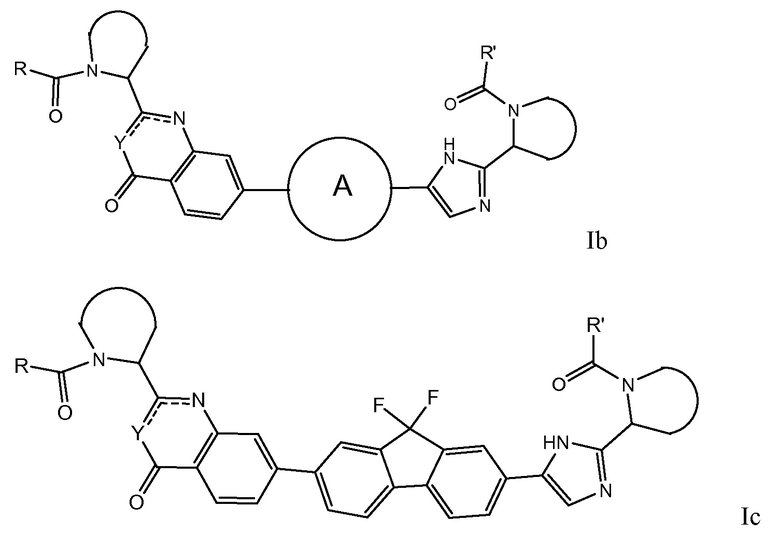
Особый интерес представляют соединения формулы I или их подгруппы, определенные в настоящем документе, которые соответствуют формуле Ib и Ic.
В предпочтительном варианте осуществления  независимо выбран из группы, включающей
независимо выбран из группы, включающей

Предпочтительно, по меньшей мере один  представляет собой
представляет собой  .
.
Более предпочтительно соединениями в соответствии с настоящим изобретением являются соединения, которые могут быть представлены формулой Id
В следующем варианте настоящего изобретения R2 выбран из группы, включающей C1-4алкилкарбониламино или C1-4алкилоксикарбониламино.
В еще одном варианте осуществления настоящего изобретения R1 выбран из разветвленного C3-4алкила; C2-3алкила, замещенного метокси; и фенила, необязательно замещенного 1 заместителем, выбранным из галогена и метила.
В другом варианте осуществления настоящего изобретения R3 представляет собой водород.
В следующем варианте осуществления R и R’ являются идентичными.
В еще одном варианте осуществления R2 представляет собой C1-4алкилкарбониламино или C1-4алкилоксикарбониламино, и R3 представляет собой водород.
В следующем аспекте настоящее изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, гидрату или сольвату для применения при лечении или профилактике (или производстве лекарственного препарата для лечения или профилактики) инфекции HCV. Типичные генотипы HCV применительно к лечению или профилактике в соответствии с настоящим изобретением включают без ограничения генотип 1b (преобладающий в Европе) и 1a (преобладающий в Северной Америке). Настоящее изобретение также относится к способу лечения или профилактики инфекции HCV, в частности, генотипа 1a или 1b, предусматривающему введение субъекту при необходимости терапевтически эффективное количество соединения, определенного выше.
Чистые стереоизомерные формы соединений и промежуточных соединений, как упомянуто в настоящем документе, определяются как изомеры, по сути не содержащие другие энантиомерные или диастереомерные формы одной и той же основной молекулярной структуры указанных соединений или промежуточных соединений. В частности, термин “стереоизомерно чистый” относится к соединениям или промежуточным соединениям, характеризующимся стереоизомерным избытком по меньшей мере 80% (т.е. минимум 90% одного изомера и максимум 10% других возможных изомеров) и до стереоизомерного избытка 100% (т.е. 100% одного изомера и отсутствие другого), более конкретно, к соединениям или промежуточным соединениям, характеризующимся стереоизомерным избытком от 90% до 100%, еще более конкретно, характеризующимся стереоизомерным избытком от 94% до 100%, и наиболее конкретно, характеризующимся стереоизомерным избытком от 97% до 100%. Термины “энантиомерно чистый” и “диастереомерно чистый” следует понимать подобным образом, но в отношении энантиомерного избытка и диастереомерного избытка, соответственно, представляющей интерес смеси.
Чистые стереоизомерные формы или стереоизомеры соединений и промежуточных соединений в соответствии с настоящим изобретением можно получать посредством применения процедур, известных из уровня техники. Например, энантиомеры можно отделять друг от друга при помощи селективной кристаллизации их диастереомерных солей с оптически активными кислотами или основаниями. Их примерами являются винная кислота, дибензоилвинная кислота, дитолуоилвинная кислота и камфорсульфоновая кислота. В альтернативном случае, энантиомеры можно разделять при помощи хроматографических методик с использованием хиральных неподвижных фаз. Указанные чистые стереохимические изомерные формы также можно получать из соответствующих чистых стереоизомерных форм подходящих исходных материалов, при условии, что реакция протекает стереоспецифично. Предпочтительно, если необходим конкретных стереоизомер, то указанное соединение можно синтезировать стереоспецифическими способами получения. В этих способах преимущественно используют энантиомерно чистые исходные материалы.
Диастереомерные рацематы соединений формулы I можно получать отдельно традиционными способами. Подходящими способами физического разделения, которые можно преимущественно использовать, являются, например, селективная кристаллизация и хроматография, например, колоночная хроматография или сверхкритическая флюидная хроматография.
Соединения формулы I и подгруппы соединений формулы I, определенные выше, имеют несколько центров хиральности. Интерес представляют стереогенные центры пирролидинового кольца на 2-ом атоме углерода. Конфигурация в этом положении может соответствовать L-пролину, т.е.
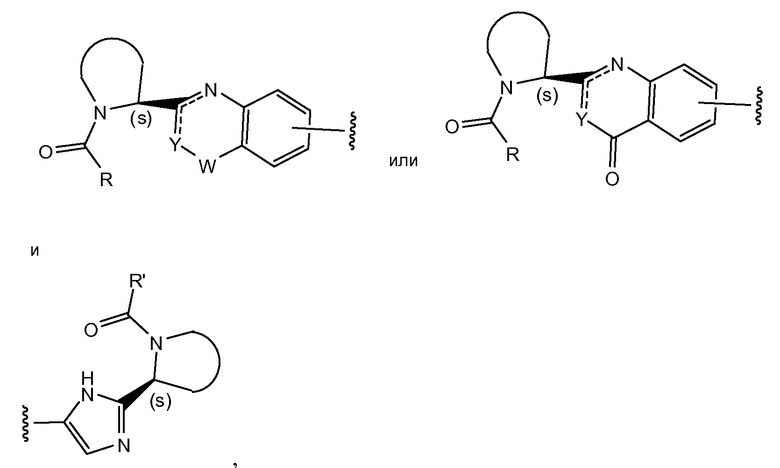
или соответствовать D-пролину, т.е.
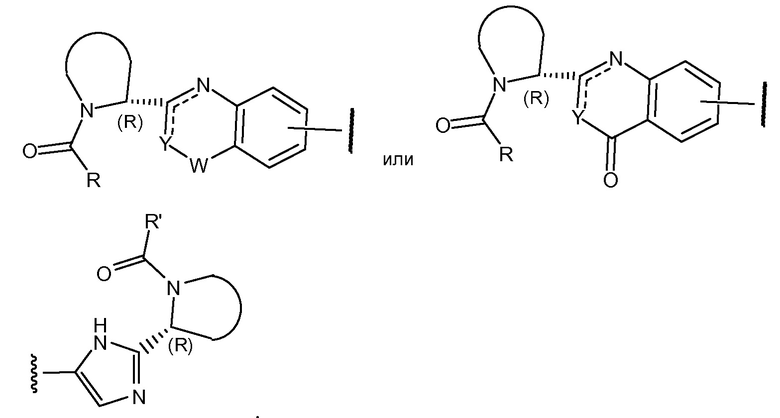
Также представляет интерес конфигурация группы –CR1R2R3, где R3 представляет собой H, если R1 выбран из разветвленного C3-4алкила; C2-3алкила, замещенного метокси, то предпочтительной является S-конфигурация; если R1 выбран из фенила, необязательно замещенного 1 или 2 заместителями, независимо выбранными из галогена и метила, то предпочтительной является R-конфигурация.
Фармацевтически приемлемые аддитивные соли включают в себя терапевтически активные нетоксические кислотно- и основно-аддитивные формы солей соединений формулы (I) или их подгрупп. Представляют интерес свободные, т.е. несолевые, формы соединений формулы I или какой-либо подгруппы соединений формулы I, определенных в настоящем документе.
Фармацевтически приемлемые кислотно-аддитивные соли традиционно можно получать путем обработки основной формы такой соответствующей кислотой. Соответствующие кислоты включают в себя, например, неорганические кислоты, такие как галогенводородные кислоты, например соляная или бромоводородная кислота, серная, азотная, фосфорная и подобные кислоты; или органические кислоты, такие, как, например, уксусная, пропионoвая, гликолевая, молочная, пировиноградная, щавелевая (т.е. этандиовая), малоновая, янтарная (т.е. бутандиовая), малеиновая, фумаровая, яблочная (т.е. гидроксилбутандиовая кислота), винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и подобные кислоты. И наоборот, указанные солевые формы можно превратить путем обработки соответствующим основанием в свободную основную форму.
Соединения формулы (I), содержащие кислотный протон, также можно превратить в их основно-аддитивные соли, в частности, в металло- или амино-аддитивные солевые формы, путем обработки соответствующими органическими и неорганическими основаниями. Приемлемые основные солевые формы включают в себя, например, аммониевые соли, соли щелочных и щелочно-земельных металлов, например, соли лития, натрия, калия, магния, кальция и т.п., соли органических оснований, например, бензатиновая, N-метил-D-глюкаминовая, гидрабаминовая соли, и соли с аминокислотами, такими как, например, аргинин, лизин и т.п.
Термин "сольваты" охватывает любые фармацевтически приемлемые сольваты, которые могут быть образованы соединениями формулы I, а также их солями. Такими сольватами являются, например, гидраты, алкоголяты, например, этаноляты, пропаноляты и т.п.
Некоторые из соединений формулы I могут также существовать в таутомерной форме. Например, таутомерными формами амидных (-C(=O)-NH-) групп являются иминоспирты (-C(OH)=N-). Таутомерные формы, хотя они явно и не указаны в представленных в настоящем документе формулах, предназначены быть включенными в объем настоящего изобретения.
Как используется в настоящем документе, "C1-4алкил" как группа или часть группы, обозначает насыщенные с прямой или разветвленной цепью углеводородные группы с 1-4 атомами углерода, такие как, например, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил, 2-метил-2-пропил. Для цели настоящего изобретения из C1-4алкила интерес представляет C3-4алкил, т.е. с прямой или разветвленной цепью углеводородные группы с 3 или 4 атомами углерода, такие как 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил, 2-метил-2-пропил. Особый интерес может представлять разветвленный C3-4алкил, такой как 2-пропил, 2-бутил, 2-метил-1-пропил, 2-метил-2-пропил.
Термин “C3-6циклоалкил”, как группа или ее часть, означает насыщенные циклические углеводородные группы с 3-6 атомами углерода, которые вместе образуют циклическую структуру. Примеры C3-6циклоалкила включают в себя циклопропил, циклобутил, циклопентил и циклогексил.
“C1-4алкокси”, как группа или часть группы, означает группу формулы -O-C1-4алкил, где C1-4алкил определен выше. Примерами C1-4алкокси являются метокси, этокси, н-пропокси, изопропокси.
Термин "галоген" является общим для атомов фтора, хлора, брома и йода.
Используемый в настоящем документе термин “(=O)” или “оксо” образует карбонильный фрагмент при присоединении к атому углерода. Следует отметить, что атом может быть только замещен оксогруппой, если позволяет валентность этого атома.
Используемый в настоящем документе с целью определения “арил”, как группа или ее часть, означает ароматическую кольцевую структуру, необязательно включающую один или два гетероатома, выбранные из N, O и S, в частности, из N и O. Указанная ароматическая кольцевая структура может содержать 5 или 6 кольцевых атомов.
Используемая в настоящем документе приставка “гетеро-” в определении группы означает, что группа содержит по меньшей мере 1 гетероатом, выбранный из N, O и S, в частности, N и O. Например, термин “гетероарил” означает ароматическую кольцевую структуру, определенную для термина “арил”, содержащую по меньшей мере 1 гетероатом, выбранный из N, O и S, в частности, из N и O, например, фуранил, оксазолил, пиридинил. В качестве альтернативы, термин “гетероC3-6циклоалкил” означает насыщенную циклическую углеводородную группу, определенную для “C3-6циклоалкила”, дополнительно содержащую по меньшей мере 1 гетероатом, выбранный из N, O и S, в частности, из N и O, например, тетрагидрофуранил, тетрагидропиранил, пиперидинил.
Если положение группы в молекулярном фрагменте не определено (например, заместитель в фениле) или представлено плавающей связью, такая группа может быть расположена на любом атоме такого фрагмента, при условии, что полученная структура химически стабильна. Если какая-либо переменная представлена более чем один раз в молекуле, каждое определение является.
Во всех случаях использования в настоящем документе термин “соединения формулы I” или “соединения в соответствии с настоящим изобретением” или подобные термины означают соединения формулы I, в том числе возможные стереоизомерные формы, и их фармацевтически приемлемые соли и сольваты.
Общие способы синтеза
Схема 1a

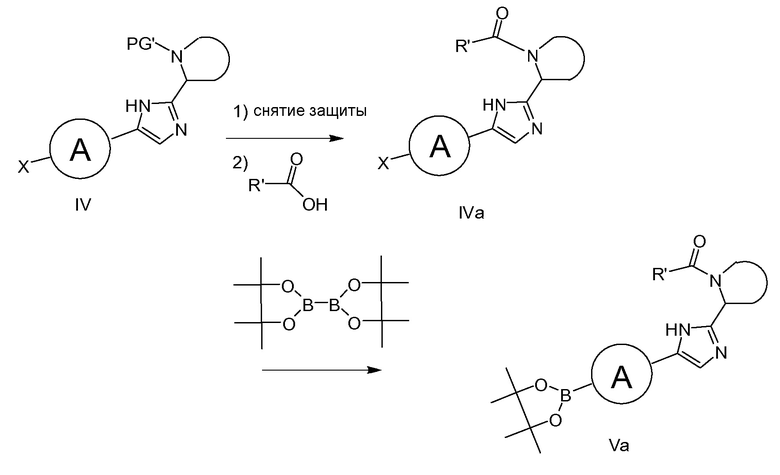
Структурные блоки, используемые в синтезе соединений формулы I, описываются в схеме 1. α-аминокетон IIa (схема 1, A=NH2), с X, являющимся галогеном, в частности, бромом или йодом, соединяют с приемлемым образом защищенным производным III, где PG’ представляет собой защитную группу на азоте, предпочтительно трет-бутоксикарбонил, в присутствие реагента присоединения для ацилирования аминогруппы, предпочтительно HATU, в присутствие основания, такого как DIPEA. Образованное таким образом промежуточное соединение циклизируют с имидазольным соединением общей формулы IV путем обработки ацетатом аммония, предпочтительно при температуре, варьирующей от 0°C до 150°C.
В качестве альтернативы, промежуточное соединение имидазол IV можно получать путем соединения α-галогенкетона IIb, где каждый Х и A независимо представляет атом галогена, Х предпочтительно выбран из йода или брома, а A предпочтительно выбран из хлора, брома или йода, с приемлемым образом защищенным соединением III, где PG' является защитной группой на азоте, предпочтительно трет-бутоксикарбонил, в присутствие приемлемого основания, например, DIPEA, с последующей циклизацией до имидазольного промежуточного соединения IV, как описано выше. Это промежуточное соединение IV может быть трансформировано до боронового сложного эфира формулы V при условиях Pd катализа, например, в присутствие Pd(dppf)Cl2, бис(пинаколато)диборона и основания, например, ацетата калия.
Подобным образом, соединения формулы IVa могут быть превращены в соединения формулы Va, как показано на схеме 1b. IVa можно получать путем удаления защитной группы PG' (например, при использовании HCl в диоксане или TMSOTf/лютидине в CH2Cl2 в случае, если PG’ равняется равняется трет-бутилоксикарбонилу) с последующим соединением полученного в результате амина с кислотой формулы R'(CO)OH при типичных условиях образования амидной связи (например, путем обработки с помощью HATU или HBTU и основания. такого как DIPEA, или при использовании EDCI/HOBt/DIPEA).
Схема 1b
Другие структурные блоки описаны на схемах 2a, 2b, 2c и 3a, 3b, 3c, 3d, 3e.
Схема 2a
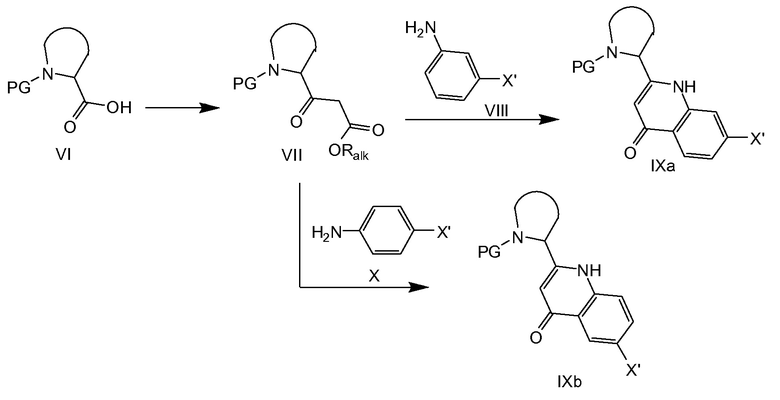
На схеме 2a кислотное производное VI превращают в сложный β-кетоэфир VII с помощью способов, известных в литературе, например, путем активации карбоновой кислоты с DCC или CDI, за которой следует, например, реакция с кислотой Мельдрума и последующее декарбоксилирование в присутствии спирта, или, в качестве альтернативы, путем конденсации с магниевой солью моноалкилмалоната с последующим декарбоксилированием. Сложный β-кетоэфир VII (Ralk относится к C1-4алкилу) затем конденсируют с VIII или Х с последующей циклизацией в IXa и IXb соответственно (X’ представляет собой галоген, выбранный из йода или брома, предпочтительно брома). Конденсацию можно осуществлять в толуоле в присутствии уксусной кислоты. Циклизация в соединения формулы IXa и IXb можно осуществлять термически путем нагревания с обратным холодильником в Dowtherm™ A (смесь дифенилоксида и бифенила). Предпочтительным примером защитной группы PG является бензилоксикарбонил (CBz).
Схема 2b
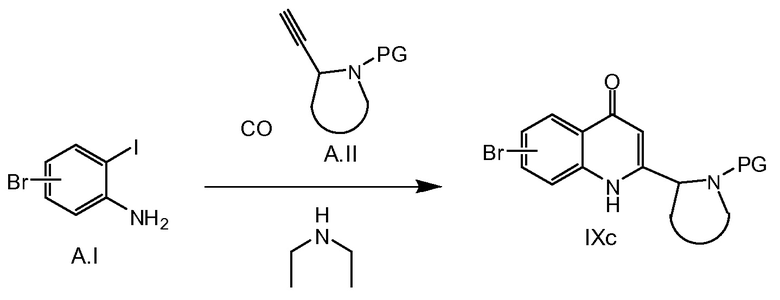
В качестве альтернативы, соединения общей формулы IXc можно получать путем последовательности катализируемых Pd реакций карбонилирования по Соногашире/циклизации, как описано на схеме 2b. Исходя из йоданилинового соединения A.I при процедурах, подобных описанным в Applied Catalysis, A: General 2009, 369, 1-2, 125-132, и ссылок, упомянутых там.
Схема 2c

Соединения общей формулы IXd, в которых R4 равняется H или C1-4алкилу, можно получать, как показано на схеме 2c. Соединение VIII (X’ представляет собой галоген, выбранный из йода или брома, предпочтительно брома) можно превратить в соединение A.IV, например, путем обработки VIII BCl3 в растворителе, таком как бензол, при температуре ниже комнатной температуры, например, при охлаждении на льду, с последующей обработкой AlCl3 и нитрилом A.III (R4 равняется H или C1-4алкилу), например, при нагревании с обратным холодильником в бензоле. После гидролиза можно получить соединение A.IV. Образование амидной связи, исходя из A.IV и VI, приводит в результате к образованию соединения A.V. Эта реакция может быть осуществлена путем превращения соединения VI в галогенангидрид кислоты, например, фторангидрид кислоты или хлорангидрид кислоты, с последующей реакцией с A.IV в присутствии основания. Другим примером является образование A.V из VI и A.IV путем применения реагента присоединения 4-(4, 6-диметокси[1.3.5]триазин-2-ил)-4-метилморфолиния хлорида или соли BF4 (DMTMM). Циклизация A.V при основных условиях, например, KOH в EtOH, или NaOH в диоксане, дает в результате соединение IXd.
Схема 3a
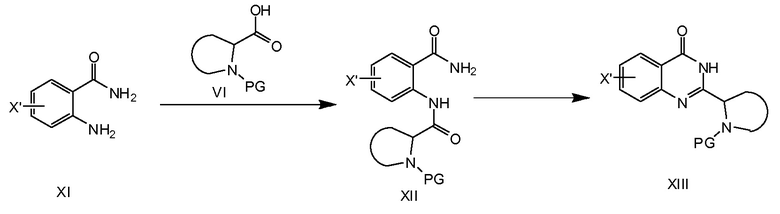
На схеме 3a описан синтез соединений формулы XIII. Образование амидной связи, исходя из XI (X' представляет собой галоген, выбранный из йода или брома, предпочтительно брома) и VI, приводит к образованию соединения XII. Эта реакция может быть осуществлена путем превращения соединения VI в галогенангидрид кислоты, например, фторангидрид кислоты или хлорангидрид кислоты, с последующей реакцией с XI в присутствии основания. Другим примером является образование XII из VI и XI путем применения реагента присоединения 4-(4, 6-диметокси[1.3.5]триазин-2-ил)-4-метилморфолиния хлорида (DMTMM). Затем соединения XII превращают в соединения общей формулы XIII при основных условиях, например, KOH или Na2CO3 в этаноле.
Схема 3b
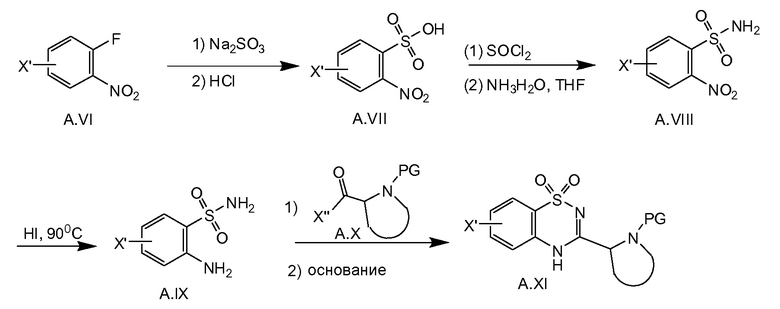
Соединение общей формулы A.XI (X' представляет собой галоген, выбранный из йода или брома, предпочтительно брома) можно получать, как показано на схеме 3b. С использованием способов, описанных в литературе (WO 2007039578; Tet. Lett. 2001, 42, 33, 5601-5603), фторид A.VI можно превратить в A.IX. Последнее соединение соединяют с галогенангидридом кислоты A.X (где X'' равняется хлор или фтор) в присутствии основания, например триэтиламина, с последующей циклизацией в соединение A.XI при основных условиях, таких как, например, 2N водный K2CO3 при нагревании с обратным холодильником.
Схема 3c
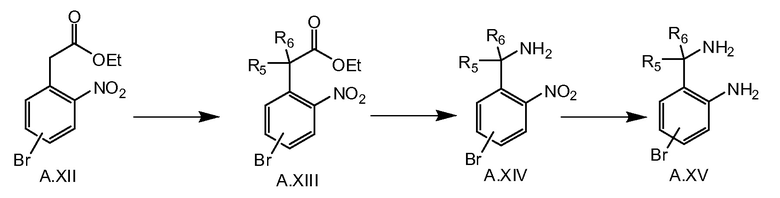
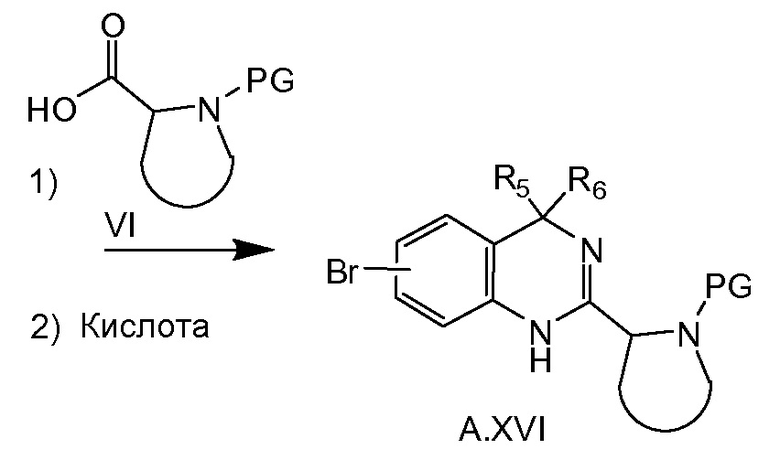
Соединение общей формулы A.XVI можно получать, как показано на схеме 3c. Диалкилирование сложного эфира A.XII соответствующим алкилгалогенидом, например, MeI в случае, если R5=R6=метил, в присутствии основания, например NaH, дает в результате соединение A.XIII. Этот сложный эфир можно превратить в соединение A.XIV путем последующего гидролиза, образования ацилазида (например, при обработке соответствующей кислоты A.XIII дифенилфосфорилазидом) и реакции Курциуса. После восстановления соединения A.XIV до A.XV, последнее соединение превращают в соединение A.XVI путем соединения с кислотой VI, например, при обработке HATU и основанием, таким как триэтиламин, и последующей циклизации в соединение A.XVI при кислотных условиях, например, в уксусной кислоте при 50°C.
Схема 3d
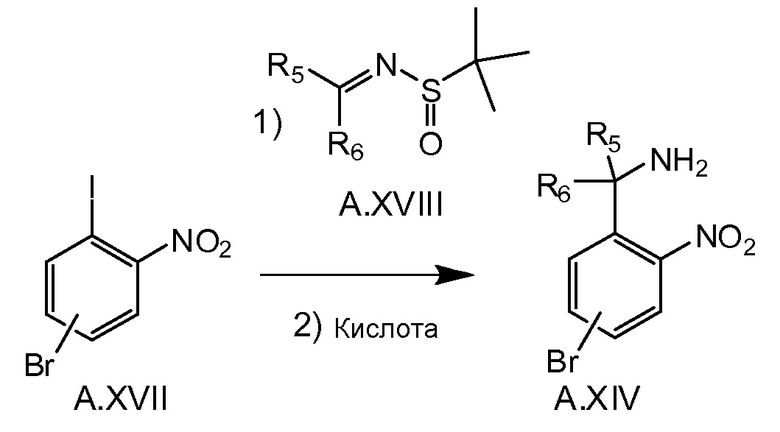
Альтернативная процедура синтеза соединения A.XIV (например, в случае, если R5 и R6 вместе с атомом углерода, который соединяет их, образуют оксетан) изображен на схеме 3d. Анион, образованный в реакции переметаллирования, например бутиллитий, и соединение A.XVII при низкой температуре, например -78°C, можно подвергнуть реагированию с сульфинамидом A.XVIII. После снятия защиты с образованного сульфинамида при кислотных условиях получают соединение A.XIV, которое далее можно превратить в A.XVI, как описано на схеме 3c.
Схема 4

Структурные блоки A.XIX, полученные с помощью способов, подобных описанным на схеме 2
(a, b, c) и схеме 3 (a, b, c и d), и V (схема 1) можно превратить в структуру XIV с использованием условий Сузуки-Мияуры (схема 4). Подобную реакцию Сузуки-Мияуры можно осуществлять, если V заменен на Va и/или A.XIX - на A.XIXa, что дает в результат соединения с общей формулой XXI, XXIII или I. A.XIXa можно получать из A.XIX путем избирательного удаления защитной группы PG (например, при использовании HCl в диоксане, TMSOTf/лютидина в CH2Cl2 или TFA в CH2Cl2 в случае, если PG равняется трет-бутилоксикарбонилу, или HBr в HOAc/H2O в случае, если PG равняется бензилоксикарбонилу) с последующим соединением полученного в результате амина с кислотой формулы R(CO)OH при типичных условиях образования амидной связи (например, путем обработки с помощью HATU или HBTU и основания. такого как DIPEA, или при использовании EDCI/HOBt/DIPEA) (схема 3e).
Схема 3e
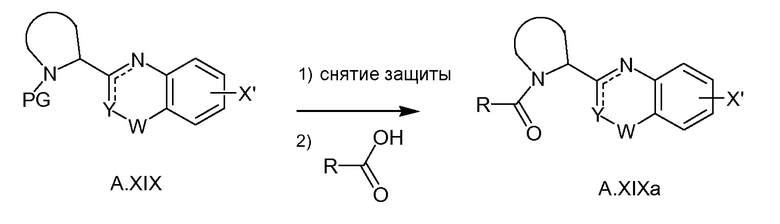
Схема 5

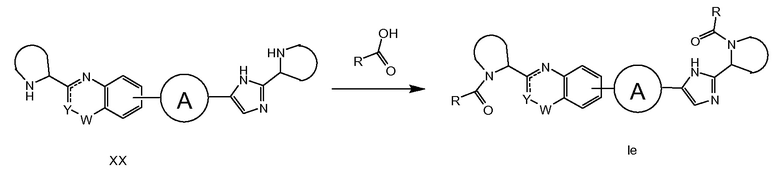
Если PG' и PG на схемах 1-4 представляет R'(C=O)- и R(C=O)-, соответственно, соединения общей структуры XIV попадают под определение соединений формулы I. В таком случае на схеме 4 описан синтез соединений формулы I. В качестве альтернативы, с XIV может быть снята защита, как описано на схеме 5. Например, путем обработки кислотой (например, HCl в iPrOH), если PG или PG’ представляют трет-бутилоксикарбонил (Boc). Соединение XX может быть превращено в соединение формулы Ie, где R и R' являются идентичными, путем классического образования амида между кислотой R-(C=O)OH и бисамином XX, как описано на схеме 6. Предпочтительными способами являются применение HATU в присутствие основания, такого как DIPEA, или HOBt в присутствии EDCI и NEt3.
Схема 6
Если PG' отличается от PG, возможно селективное снятие защитной группы, описанное на схеме 5, что дает соединения XVIII или XIX, исходя из XIV. Например, в случае, если PG’ равняется трет-бутилоксикарбонилу (Boc), а PG равняется бензилоксикарбонилу (Cbz), селективное снятие защитной группы может быть осуществлено путем удаления Boc-защитной группы в кислотных условиях, таких как HCl в iPrOH при комнатной температуре, или путем удаления CBz-защитной группы в восстановительных условиях, например, водород в присутствие катализатора, например, Pd(OH)2.
Если PG' представляет R'(C=O)-, или PG представляет R(C=O)-, синтез соединений XIV, описанных на схемах 1-4, дает соединения формулы XXI (схема 7) или XXIII (схема 8), соответственно. Соединения XXI и XXIII можно получать из соединения XIX и R’(C=O)OH или XVIII и R(C=O)OH, соответственно, при типичных условиях образования амида. Затем эти соединения могут быть превращены в соединения формулы I. Селективное снятие защитной группы XXI с получением XXII с последующим образованием амидной связи между XXII и R(C=O)-OH дает соединения формулы I. Затем аналогичная последовательность реакций может быть применена для превращения XXIII в XXIV, а затем соединения формулы I.
Схема 7

Схема 8

В следующем аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы I, определенного в настоящем документе, и фармацевтически приемлемый носитель. Терапевтически эффективным количеством в этом контексте является количество, достаточное для стабилизации или снижения инфекции HCV у инфицированных субъектов, или количество, достаточное для предупреждения инфекции HCV у субъектов с риском инфицирования. В следующем аспекте настоящее изобретение относится к способу получения фармацевтической композиции, определенной в настоящем документе, которая предусматривает тщательное смешивание фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения формулы I, определенного в настоящем документе.
Таким образом, соединения в соответствии с настоящим изобретением или любая их подгруппа могут быть составлены в различные фармацевтические формы для целей введения. В качестве подходящих композиций могут приводиться все композиции, обычно используемые для систематического введения лекарственных средств. Для получения фармацевтических композиций в соответствии с настоящим изобретением эффективное количество конкретного соединения, необязательно в форме аддитивной соли или комплекса с металлом, в качестве активного ингредиента объединяют в однородную смесь с фармацевтически приемлемым носителем, при этом носитель может принимать широкое разнообразие форм в зависимости от формы препарата, необходимой для введения. Данные фармацевтические композиции являются желательными в единичной лекарственной форме, приемлемой, в частности, для введения перорально, ректально, чрескожно или путем парентеральной инъекции. Например, при получении композиций в виде пероральной лекарственной формы можно использовать любую общепринятую фармацевтическую среду, такую как, например, вода, гликоли, масла, спирты и т.п., в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие вещества, средства для улучшения распадаемости и т.п., в случае порошков, пилюль, капсул и таблеток. Благодаря своей простоте введения таблетки и капсулы представляют собой наиболее предпочтительные пероральные формы единицы дозирования, в случае которых очевидно используются твердые фармацевтические носители. Для парентеральных композиций носитель будет, как правило, по меньшей мере в значительной степени включать стерильную воду, хотя может включать и другие ингредиенты, например, для улучшения растворимости. Например, могут быть получены растворы для инъекций, в которых носитель включает физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Также могут быть получены суспензии для инъекций, в случае которых могут использоваться подходящие жидкие носители, суспендирующие средства и т.п. Также включены препараты в твердой форме, которые предназначены для преобразования, непосредственно перед применением, в препараты в жидкой форме. В композициях, подходящих для чрескожного введения, носитель необязательно включает средство усиления проникновения и/или подходящее смачивающее средство, необязательно комбинированное с подходящими добавками любой природы в минимальных пропорциях, при этом добавки не оказывают никакого значительного вредного воздействия на кожу. Соединения в соответствии с настоящим изобретением также могут быть введены путем пероральной ингаляции или инсуффляции в форме раствора, суспензии или сухого порошка с использованием любой известной в уровне техники системы доставки.
Особенно предпочтительно составление вышеуказанных фармацевтических композиций в единичной лекарственной форме для простоты введения и равномерности дозирования. Единичная лекарственная форма, применяемая по данному документу, относится к физически отдельным единицам, подходящим в качестве единичных дозировок, при этом каждая единица содержит предварительно установленное количество активного ингредиента, рассчитанное для получения желаемого терапевтического эффекта в сочетании с необходимым фармацевтическим носителем. Примерами таких единичных лекарственных форм являются таблетки (включая делимые таблетки или покрытые таблетки), капсулы, пилюли, суппозитории, пакетики с порошком, пластинки, растворы для инъекций или суспензии и т.п., а также их отдельные множества.
Соединения формулы I показывают активность против HCV и могут быть использованы в лечение и профилактике инфекции HCV или заболеваний, ассоциированных с HCV. Последние включают в себя прогрессирующий фиброз печени, воспаление и некроз, ведущие к циррозу, конечной стадии болезни печени, и печеночноклеточной карциноме. Ряд соединений в соответствии с настоящим изобретением, более того, известен как активный против мутантных штаммов HCV. Кроме того, соединения в соответствии с настоящим изобретением могут обладать привлекательными свойствами, касающимися биодоступности, показывать благоприятный фармакокинетический профиль, в том числе приемлемые период полувыведения, AUC (площадь под кривой), пиковые и минимальные значения, и отсутствие неблагоприятного явления, такого как недостаточно быстрое наступление действия или удерживание в тканях.
In vitro противовирусная активность против HCV соединений формулы I может быть тестирована в клеточной системе репликона HCV, основанной на Lohmann et al. (1999) Science 285:110-113, с дополнительными модификациями, описанными в Krieger et al. (2001) Journal of Virology 75: 4614-4624, и Lohmann et al. (2003) Journal of Virology 77: 3007-3019 для генотипа 1b, и в Yi et al. (2004) Journal of Virology 78: 7904-7915 для генотипа 1a (включенных в настоящий документ посредством ссылки), которые далее проиллюстрированы в разделе примеров. Эта модель, хотя и не полная модель инфекции HCV, в настоящее время получила широкое признание в качестве наиболее надежной и эффективной модели автономной репликации РНК HCV. Следует иметь в виду, что очень важно различать соединения, которые специфично препятствуют функциям HCV, и те, которые проявляют цитотоксические или цитостатические эффекты в модели репликона HCV и, как следствие, вызывают снижение концентрации РНК HCV или связанного репортерного фермента. В данной области известны анализы для оценки клеточной цитотоксичности, например, на основе активности митохондриальных ферментов с использованием флуорогенных окислительно-восстановительных красителей, таких как резазурин. Кроме того, существуют обратные скрининги клетки для оценки неселективного ингибирования активности связанного репортерного гена, такого как ген люциферазы светлячка. Приемлемые типы клеток могут быть обеспечены стабильной трансфекцией с репортерным геном люциферазы, экспрессия которого зависит от конститутивно активного промотора гена, и такие клетки могут быть использованы как обратный скрининг для устранения неселективных ингибиторов.
Благодаря своим свойствам против HCV соединения формулы I или их подгруппы, определенные в настоящем документе, применимы в ингибировании репликации HCV, в частности, при лечении теплокровных животных, в частности, людей, инфицированных HCV, и для профилактики инфекций HCV у теплокровных животных, в частности, людей. Кроме того, настоящее изобретение относится к способу лечения теплокровного животного, в частности, человека, инфицированного HCV или с риском инфекции HCV, при этом указанный способ предусматривает введение терапевтически или профилактически эффективного количества соединения формулы I, определенного выше.
Поэтому соединения формулы I, определенные в настоящем документе, могут быть использованы в качестве медицинского препарата, в частности, в качестве медицинского препарата против HCV. Указанные применение в качестве медицинского препарата или способ лечения предусматривают систематическое введение инфицированным HCV субъектам или субъектам, восприимчивым к инфекции HCV, количества, эффективного для облегчения или предупреждения симптомов и состояний, ассоциированных с инфекцией HCV.
Настоящее изобретение также относится к применению соединений в соответствии с настоящим изобретением при изготовлении лекарственного препарата для лечения или предупреждения инфекции HCV.
В общем предполагается, что эффективное противовирусное ежесуточное количество должно составлять от приблизительно 0,01 до приблизительно 50 мг/кг или от приблизительно 0,02 до приблизительно 30 мг/кг массы тела. Может быть целесообразным введение необходимой дозы в виде двух, трех, четырех или более частей дозы при подходящих интервалах в течение дня. Указанные субдозы могут быть составлены в виде единичных лекарственных форм, например, содержащих от приблизительно 1 до приблизительно 500 мг, или от приблизительно 1 до приблизительно 300 мг, или от приблизительно 1 до приблизительно 100 мг, или от приблизительно 2 до приблизительно 50 мг активного ингредиента на единичную лекарственную форму.
Комбинированная терапия
Настоящее изобретение также относится к комбинации соединения формулы I, его фармацевтически приемлемой соли или сольвата и другого противовирусного соединения, в частности, другого соединения против HCV. Термин “комбинация” относится к продукту, содержащему (a) соединение формулы I, определенное выше, и (b) другой ингибитор против HCV, в качестве комбинированного препарата для одновременного, раздельного или последовательного применения в лечение инфекций HCV.
Комбинации в соответствии с настоящим изобретением могут быть использованы в качестве лекарственных препаратов. Следовательно, настоящее изобретение относится к применению соединения формулы (I) или любой его подгруппы, как определено выше, для изготовления лекарственного препарата, применимого для ингибирования активности HCV у млекопитающего, инфицированного вирусами HCV, при этом указанный лекарственный препарат используют в комбинированной терапии, указанная комбинированная терапия, в частности, предусматривает соединение формулы (I) и по меньшей мере одно другое средство против HCV, например, IFN-α, пэгилированный IFN-α, рибавирин, альбуферон, тарибавирин, нитазоксанид Debio025 или их комбинация.
Другие средства, которые могут быть скомбинированы с соединениями в соответствии с настоящим изобретением, включают, например, нуклеозидные и ненуклеозидные ингибиторы полимеразы HCV, ингибиторы протеазы, ингибиторы геликазы, ингибиторы NS4B и средства, которые функционально ингибируют внутренний сайт связывания рибосомы (IRES) и другие средства, которые ингибируют присоединение HCV к клетке или вхождение вируса, трансляцию РНК HCV, транскрипцию РНК HCV, репликацию или созревание HCV, сборку или высвобождение вируса. Конкретные соединения в этих классах включают в себя ингибиторы протеазы HCV, такие как телапревир (VX-950), боцепревир (SCH-503034), нарлапревир (SCH-900518), ITMN-191 (R-7227), TMC-435350 (TMC-435), MK-7009, BI-201335, BI-2061 (цилупревир), BMS-650032, ACH-1625, ACH-1095, GS 9256, VX-985, IDX-375, VX-500, VX-813, PHX-1766, PHX2054, IDX-136, IDX-316, ABT-450, EP-013420 (и конгнеры) и VBY-376; нуклеозидные ингибиторы полимеразы HCV, применимые в настоящем изобретении, включают в себя TMC649128, R7128, PSI-7851, PSI 7977, INX-189,IDX-184, IDX-102, R1479, UNX-08189, PSI-6130, PSI-938 и PSI-879 и различные другие нуклеозидные и нуклеотидные аналоги, а ингибиторы HCV включают в себя полученные как 2'-C-метил-модифицированные нуклеозиды, 4'-аза-модифицированные нуклеозиды, и 7'-деаза-модифицированные нуклеозиды. Ненуклеозидные ингибиторы полимеразы HCV, применимые в настоящее изобретение, включают в себя HCV-796, HCV-371, VCH-759, VCH-916, VCH- 222, ANA-598, MK-3281, ABT-333, ABT-072, PF-00868554, BI-207127, GS-9190, A-837093, JKT-109, GL-59728, GL-60667, ABT-072, AZD-2795 и TMC647055.
Следующие примеры предназначены для иллюстрации настоящего изобретения и не должны истолковываться как ограничивающие его объем.
Экспериментальная часть
Способы LCMS
Способ A: Общие условия: подвижная фаза A: H2O (0,1% TFA; B: CH3CN (0,05% TFA). Время остановки: 2 минуты; градиент времени (минуты) [%A/%B] 0,01 [90/10] - 0,9 [20/80] - 1,5 [20/80] - 1,51 [90/10]; поток: 1,2 мл/минута; температура колонки: 50°С
Способ A1: Shimadzu LCMS 2010, Shim-pack XR-ODS, 3*30 мм.
Способ A2: Xtimate C18 2,1*30 мм, 3 мкм.
Способ A3: SHIMADZU Shim pack 2*30
Способ B: Agilent 1100, YMC-PACK ODS-AQ, 50×2,0 мм, 5 мкм, подвижная фаза A: H2O (0,1% TFA); B: CH3CN (0,05% TFA). Время остановки: 0 минут; градиент времени (минуты) [%A/%B] 0 [100/0] - 1 [100/0] - 5[40/60] - 7,5 [40/60] - 8 [100/10]; поток: 0,8 мл/минута; температура колонки: 50°С
Способ C: Agilent 1100, YMC-PACK ODS-AQ, 50×2,0 мм, 5 мкм, подвижная фаза A: H2O (0,1%TFA; B: CH3CN (0,05% TFA); Время остановки: 0 минут; градиент времени (минуты) [%A/%B] 10 [90/10] - 0,8 [90/10] - 4,5[20/80] - 7,5 [20/80] - 8 [90/10]; поток: 0,8 мл/минута; температура колонки: 50°С
Способ D: Shimadzu LCMS 2010, Shim-pack XR-ODS, 3*30 мм, подвижная фаза A: H2O (0,1% TFA; B: CH3CN (0,05% TFA). Время остановки: 2 минуты; градиент времени (минуты) [%A/%B] 0,01 [100/0] - 0,9 [70/30] - 1,5 [70/30] - 1,51 [100/0]; поток: 1,2 мл/минута; температура колонки: 50°С
Способ E: Жидкостная хроматография: Waters Alliance 2695, UV детектор: Waters 996 PDA, диапазон: 210-400 нм; масс-детектор: Waters ZQ, источник ионов: используемая ES+, ES-Column: SunFire C18 3,5 μ 4,6×100 мм, подвижная фаза A: 10 мM NH4OOCH + 0,1% HCOOH в H2O; подвижная фаза B: CH3OH; температура колонки: 50°C; поток: 1,5 мл/минута, градиент времени (минуты) [%A/%B] 0 [65/35] - 7 [5/95] - 9,6 [5/95] - 9,8 [65/35] - 12 [65/35].
Способ F: Xtimate C18 2,1*30 мм, 3 мкм, подвижная фаза A: H2O (1,5 мл TFA/4 л); B: CH3CN (0,75 мл TFA/4 л) Время остановки: 3 минуты; градиент времени (минуты) [%A/%B] 0,0 [90/10] - 1,35 [20/80] - 2,25 [20/80] - 2,26 [90/10]; 3,0 [90/10], поток: 0,8 мл/минута; температура колонки: 50°С
Способ G: Общие условия: подвижная фаза A: H2O (1,5 мл TFA/4 л); B: CH3CN (0,75 мл TFA/4 л) Время остановки: 2 минуты; градиент времени (минуты) [%A/%B] 0,0 [100/0] - 0,9 [40/60] - 1,5 [40/60] - 1,51 [100/0]; 2,0 [100/0], поток: 1,2 мл/минута; температура колонки: 50°С
Способ G1: Xtimate C18, 2,1*30 мм, 3 мкм.
Способ H: Общие условия: подвижная фаза A: H2O (0,1 % TFA); B: CH3CN (0,05% TFA). Время остановки: 10 минут; градиент времени (минуты) [%A/%B] 0,0 [90/10] - 0,8 [90/10] - 4,5 [20/80] - 7,5 [20/80]; 9,5 [90/10], поток: 0,8 мл/минута; температура колонки: 50°С
Способ H1: Agilent TC-C18, 2,1*50 мм, 5 мкм.
Способ I: Shimadzu LCMS 2010, Shim-pack XR-ODS, 3*30 мм, подвижная фаза A: H2O (0,1% TFA; B: CH3CN (0,05% TFA). Время остановки: 7 минут; градиент времени (минуты) [%A/%B] 0,01 [90/10] - 6,0 [20/80] - 6,5 [20/80] - 6,51 [90/10]; поток: 0,8 мл/минута; температура колонки: 50°С
Способ J: Agilent TC-C18, 50×2,1 мм, 5 мкм, подвижная фаза A: H2O (0,1% TFA; B: CH3CN (0,05% TFA). Время остановки: 10 минут; время после начала: 0,5 минуты; градиент времени (минуты) [%A/%B] 0 [100/0] - 1 [100/0] - 5[40/60] - 7,5 [15/85] - 9,5 [100/10]; поток: 0,8 мл/минута; температура колонки: 50°С
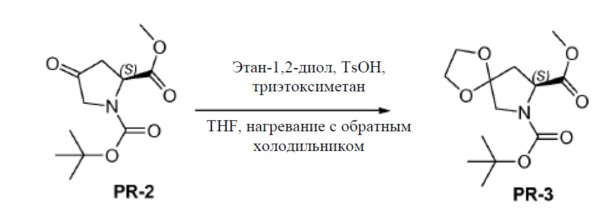
Соединение PR-2 (30 г, 123 ммоля) в THF (120 мл), этан-1,2-диол (53,6 г, 864 ммоля), триэтоксиметан (54,6 г, 369 ммоль) и TsOH (3 г, 3,69 ммоля) добавляли при 25°C. Смесь перемешивали при нагревании с обратным холодильником в течение 5 часов. Смесь вливали в NH4Cl (400 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали солевым раствором и сушили над Na2SO4. Органическую фазу концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан:эфир ацетат = 10:1) с получением соединения PR-3 (8,4 г).

К перемешиваемому раствору соединения PR-3 (8,4 г, 29,3 ммоля) в THF/H2O (100 мл, 1:1) добавляли NaOH (5,85 г, 146 ммоль). Реакционную смесь перемешивали при 20°C в течение 1 часа и обрабатывали этилацетатом (20 мл). Неорганический слой отделяли, доводили до pH=4 2 н. HCl и экстрагировали с помощью CH2Cl2 (3×50 мл). Объединенный органический слой промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме с получением соединения PR-4 (5,9 г).
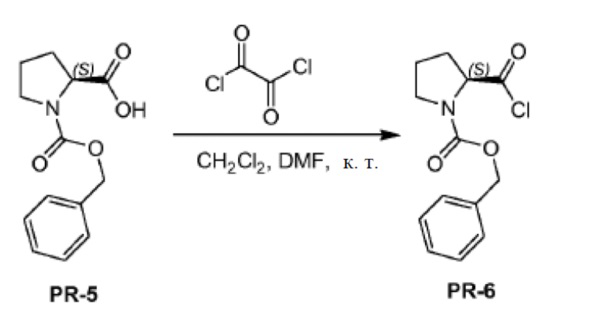
Соединение PR-5 (15,7 г, 63,1 ммоля) растворяли в сухом CH2Cl2 (250 мл) и к раствору добавляли DMF (1,5 мл). Оксалилхлорид (13,5 мл, 157,5 ммоля) каплями добавляли при комнатной температуре. Реакционную смесь перемешивали в течение 0,5 часа при комнатной температуре. Реакционную смесь концентрировали в вакууме и остаток (PR-6, 22 г) использовали непосредственно без дополнительной очистки.
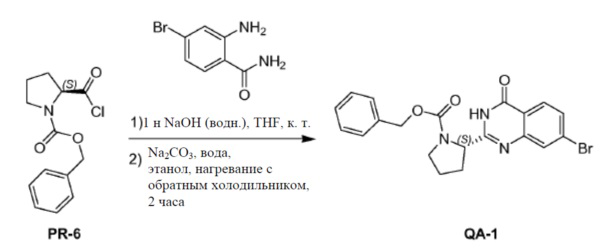
К раствору соединения PR-6 (неочищенного, 22 г) в сухом THF (250 мл) добавляли 2-амино-4-бромбензамид (7,6 г, 35,3 ммоля) и 1 н. NaOH (водн., 85 мл, 85 ммоля). Смесь перемешивали в течение 1 часа при комнатной температуре. Реакционную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали 1 н. NaOH в воде (15 мл), солевым раствором, сушили над Na2SO4 и концентрировали в вакууме с получением неочищенного остатка (17 г). Неочищенный остаток, полученный, как описано выше (25 г), и Na2CO3 (17,8 г, 168 ммоль) в этаноле (250 мл) и H2O (250 мл) нагревали с обратным холодильником в течение 2 часов. Органический растворитель удаляли в вакууме. Смесь экстрагировали дихлорметаном (2×200 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и очищали с помощью колоночной хроматографии на силикагеле (элюент:этилацетат). Желаемые фракции выпаривали досуха. Полученный остаток перемешивали в этилацетате (50 мл), осадок отфильтровывали и промывали этилацетатом с получением соединения QA-1 (17 г).

Соединение QA-1 (8 г, 18,6 ммоля) растворяли в HOAc (80 мл) и добавляли 40% HBr (40 мл). Смесь перемешивали при 80°С в течение ночи. Большую часть растворителя удаляли в вакууме. Осадок отфильтровывали и промывали метил-трет-бутиловым эфиром. Твердое вещество совместно выпаривали с толуолом (2×20 мл) с получением неочищенного остатка (6,5 г). Затем часть этого остатка (6,4 г), (S)-2-(метоксикарбониламино)-3-метилбутановую кислоту (4,5 г, 25,6 ммоля), EDCI (4,9 г, 25,6 ммоля) и HOBt (1,15 г, 8,5 ммоля) в CH2Cl2 (120 мл) охлаждали до 0°С. Добавляли DIPEA (14,8 мл, 85,0 ммоля). Смесь перемешивали в течение 1,5 часа при 20°С. Органический слой промывали насыщенным водным NaHCO3 (100 мл) и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (градиент элюента: петролейный эфир:этилацетат: от 100:0 до 0:100) с получением соединения QA-2 (3,3 г).
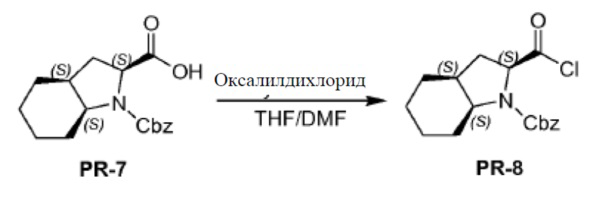
Соединение PR-7 (7,0 г, 23,21 ммоля) в THF (70 мл) перемешивали при 0°С. Каплями добавляли оксалилдихлорид (7 мл, 46,2 ммоля) и DMF (2 капли) добавляли и смесь перемешивали в течение 10 минут при 0°С. Смесь перемешивали и нагревали с обратным холодильником в течение 1 часа. Смесь охлаждали и выпаривали в вакууме с получением соединения PR-8 (7 г).
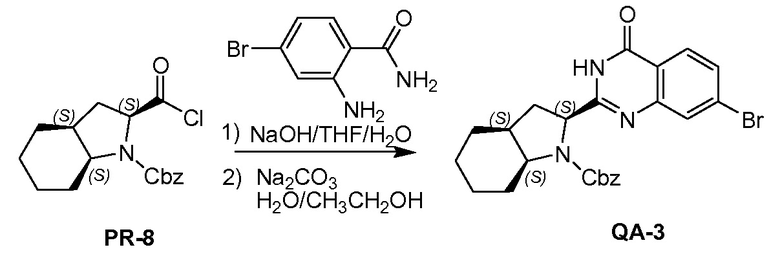
К раствору соединения PR-8 (7 г, 21 ммоль) в THF (70 мл) добавляли 2-амино-4-бромбензамид (4,5 г, 21 ммоля) и 1 н NaOH (42 мл, 42 ммоля). Смесь перемешивали в течение 1 часа при 25°С. Смесь экстрагировали этилацетатом. Органические слои собирали, промывали 0,5 н. NaOH, солевым раствором, сушили и концентрировали в вакууме с получением неочищенного остатка (9 г). Этот остаток (9 г) и Na2CO3 (5,7 г, 54 ммоля) в H2O (200 мл) и THF (200 мл) перемешивали и нагревали с обратным холодильником в течение 2 часов. Смесь концентрировали в вакууме и экстрагировали с помощью CH2Cl2 (2x), промывали солевым раствором, сушили и выпаривали в вакууме. Остаток растворяли в CH2Cl2 и промывали 1 н. HCl (3x), солевым раствором, сушили и выпаривали в вакууме с получением QA-3 (4,4 г). Способ A2; Rt: 1,27 минуты, масса/заряд = 484,0 (M+H)+. Точная масса: 483,1.
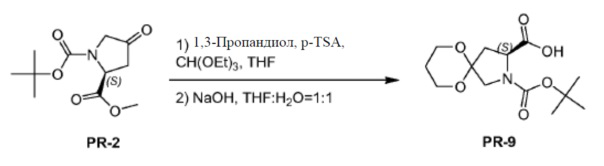
К соединению PR-2 (10 г, 41,2 ммоля) в THF (100 мл) добавляли 1,3-пропандиол (22 г, 288 ммоль), триэтилортоформиат (18,3 г, 123,6 ммоля) и толуол-4-сульфоновую кислоту (1 г, 0,2 ммоля) при 25°C. Смесь перемешивали при нагревании с обратным холодильником в течение 2 часов. Смесь вливали в водный NH4Cl (400 мл), экстрагировали этилацетатом (3×50 мл) и отделяли. Объединенные органические слои промывали солевым раствором и сушили над Na2SO4. Органическую фазу концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан:эфир ацетат = 5:1) и полученное соединение (3,8 г) растворяли в THF/H2O (40 мл, 1:1). Добавляли NaOH (2,52 г, 63 ммоля), реакционную смесь перемешивали при комнатной температуре в течение 1 часа и обрабатывали этилацетатом (20 мл). Объединенный неорганический слой отделяли, регулировали pH до 4 с помощью 2 н. HCl и экстрагировали с помощью CH2Cl2 (3×20 мл). Объединенный органический слой промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме с получением соединения PR-9 (5,9 г).

Оксалилдихлорид (2,5 мл, 13,11 ммоля) каплями добавляли к смеси соединения PR-9 (2,5 г, 8,74 ммоля), 2-амино-4-бромбензамида (2,5 г, 10,49 ммоля) в дихлорметане (20 мл) и пиридине (20 мл) при комнатной температуре. Смесь перемешивали в течение 1 часа при комнатной температуре. Растворитель удаляли в вакууме. Остаток очищали колоночной хроматографией (петролейный эфир: эфир ацетат = 1:1). Полученное промежуточное амидное соединение (0,98 г), Na2CO3 (1,08 г, 10,15 ммоля), H2O (5 мл) и CH3CH2OH (5 мл) перемешивали в течение 2 часов при нагревании с обратным холодильником. Большую часть CH3CH2OH удаляли в вакууме и полученный остаток экстрагировали этилацетатом. Органический слой сушили над Na2SO4 и концентрировали в вакууме. Остаток промывали трет-бутилметиловым эфиром с получением соединения QA-4 (0,89 г).
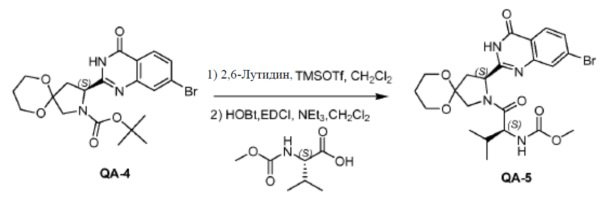
К перемешиваемому раствору соединения QA-4 (0,89 г, 1,92 ммоля) и лютидина (0,41 г, 3,84 ммоля) в сухом CH2Cl2(10 мл) при 0°С каплями добавляли TMSOTf (1,7 г, 7,68 ммоля). Реакционную смесь перемешивали при 0°C в течение 30 минут, гасили насыщенным водным NH4Cl и экстрагировали этилацетатом, объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Полученный остаток использовали как таковой в следующей реакции (0,3 г). Способ A2; Rt: 0,68 минуты, масса/заряд = 368,0 (M+H)+. Точная масса: 367,0. NEt3 (0,5 мл, 2,46 ммоля) добавляли к раствору полученного выше остатка (0,3 г), (S)-2-(метоксикарбониламино)-3-метилбутановой кислоты (0,22 г, 1,23 ммоля), HOBt (0,17 г, 1,23 ммоля) и EDCI (0,24 г, 1,23 ммоля) в дихлорметане (15 мл) в водяной бане со льдом. Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Затем смесь растворяли дихлорметаном (20 мл) и промывали насыщенным NaHCO3, солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали колоночной хроматографией (гексан:этилацетат = 1:1) с получением в результате соединения QA-5 (0,2 г).
Способ A2; Rt: 1,14 минуты, масса/заряд = 547,1 (M+Na)+. Точная масса: 524,1.
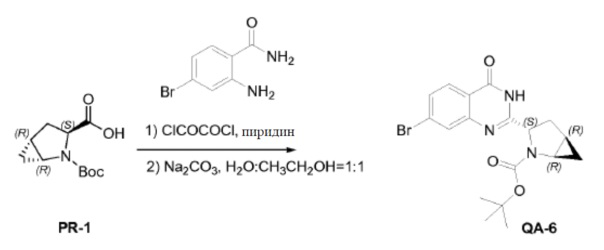
Оксалилхлорид (2,9 мл, 33 ммоля) каплями добавляли к смеси соединения PR-1 (5 г, 22 ммоля), 2-амино-4-бромбензамида (4,7 г, 22 ммоля) и пиридина (50 мл). Смесь перемешивали в течение 1 часа при комнатной температуре. Растворитель удаляли в вакууме. Полученный остаток очищали хроматографией (петролейный эфир:эфир ацетат = 5:1) с получением в результате промежуточного соединения (3,6 г). Способ A2; Rt: 1,15 минуты, масса/заряд = 447,7 (M+Na)+. Точная масса: 425,1. Полученное выше промежуточное соединение (3,6 г), Na2CO3 (2,7 г, 25,4 ммоля), H2O (20 мл) и CH3CH2OH (20 мл) перемешивали в течение 2 часов при нагревании с обратным холодильником. Большую часть CH3CH2OH удаляли в вакууме. Остаток экстрагировали этилацетатом (3×20 мл). Органический слой сушили над Na2SO4 и концентрировали в вакууме. Остаток промывали трет-бутилметиловым эфиром с получением соединения QA-6 (3,4 г).
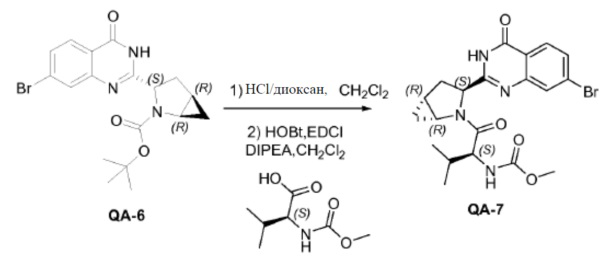
Соединение QA-6 (3,4 г, 8,4 ммоля) растворяли в дихлорметане (30 мл) и каплями добавляли HCl/диоксане (3 мл) к смеси при 0°C. Реакционную смесь перемешивали в течение 5 часов при комнатной температуре. Растворитель удаляли в вакууме. Остаток промывали трет-бутилметиловым эфиром и полученный неочищенный остаток использовали как таковой (2,7 г). К раствору неочищенного продукта (2,7 г), (S)-2-(метоксикарбониламино)-3-метилбутановой кислоты (2,75 г, 15,76 ммоля), HOBt (2,42 г, 17,33 ммоля) и EDCI (3,32 г, 17,33 ммоля) в дихлорметане (20 мл), охлажденному в водяной бане со льдом, добавляли DIPEA (14 мл, 78,8 ммоля). Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Смесь растворяли дихлорметаном (20 мл) и промывали насыщенным NaHCO3, солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан:эфир ацетат = 1:1) с получением соединения QA-7 (2,5 г). SFC: Колонка: AD-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2. B: EtOH (0,05% диэтиламин); 5-40% B в A: Rt: 9,99 минуты.
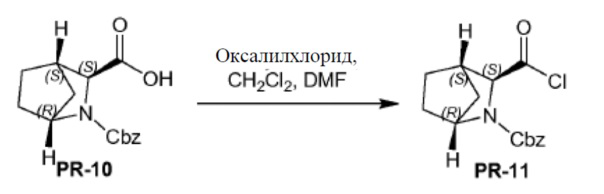
Соединение PR-10 (2,0 г, 7,3 ммоля) в CH2Cl2 (20 мл) перемешивали при 0°C. Каплями добавляли оксалилдихлорид (2,3 г, 18,2 ммоля) и DMF (2 капли) и смесь перемешивали в течение 10 минут при 0°C. Смесь перемешивали в течение 1 часа при 20°C. Смесь охлаждали и выпаривали в вакууме. Остаток дважды растворяли толуолом (2×10 мл) и выпаривали с получением остатка (PR-11, 2,5 г).

К раствору соединения PR-11 (2,5 г) в THF (30 мл) добавляли 2-амино-4-бромбензамидe (1,57 г, 7,3 ммоля) и 1 н. NaOH (14,6 мл, 14,6 ммоля). Смесь перемешивали в течение 1 часа при 25°С. Смесь экстрагировали этилацетатом (2 x). Органические слои объединяли, промывали 0,5н NaOH, солевым раствором, сушили и концентрировали в вакууме с получением остатка (3,5 г), который перемешивали с Na2CO3 (2,32 г, 21,9 ммоля) в H2O (50 мл) и THF (50 мл) и нагревали с обратным холодильником в течение 2 часов. Летучие вещества удаляли в вакууме. Смесь экстрагировали с помощью CH2Cl2 (2x), промывали солевым раствором, сушили и летучие вещества удаляли в вакууме. Остаток растворяли в CH2Cl2 и промывали с помощью 1 н. HCl (3x), солевым раствором, сушили и летучие вещества удаляли в вакууме с получением соединения QA-8 (1,5 г). Способ A2; Rt: 1,15 минуты, масса/заряд = 453,9 (M+H)+. Точная масса: 453,1.

ClCOCOCl (44,4 мл, 510,2 ммоля) каплями добавляли к смеси PR-13 (100,6 г, 374 ммоля), 2-амино-4-бромбензамида (73,2 г, 340 ммоля) и пиридина (760 мл) в азоте при 0°С. Смесь перемешивали в течение 2 часа при комнатной температуре. Растворитель удаляли в вакууме. К остатку добавляли насыщенный NaHCO3 и полученную смесь экстрагировали этилацетатом три раза. Объединенные органические слои промывали насыщенным NaHCO3, солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Полученный остаток очищали хроматографией (CH2Cl2:MeOH = 50:1) с получением промежуточного амидного соединения (50,6 г). Способ A2; Rt: 1,15 минуты, масса/заряд = 490,1 (M+Na)+. Точная масса: 467,1. Раствор полученного выше промежуточного соединения (50,61 г), Na2CO3 (34,51 г, 325,6 ммоля), H2O (300 мл) и CH3CH2OH (300 мл) перемешивали в течение 3 часов при нагревании с обратным холодильником. EtOH удаляли в вакууме и смесь экстрагировали этилацетатом (3×300 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме. Полученный остаток промывали трет-бутилметиловым эфиром с получением соединения QA-9 (39,2 г). Способ A2; Rt: 1,37 минуты, масса/заряд = 448,1 (M+H)+. Точная масса: 447,1.
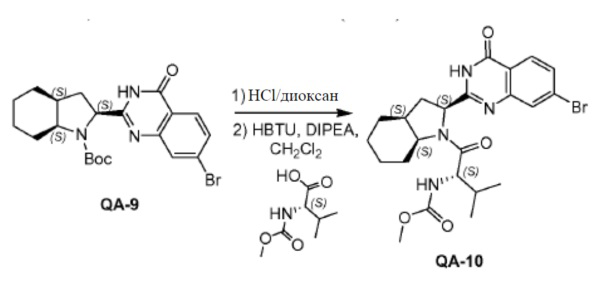
QA-9 (39,2 г, 87,5 ммоля) растворяли в дихлорметане (400 мл). HCl/диоксан (470 мл) каплями добавляли к смеси при 0°C. Реакционную смесь перемешивали в течение 3,5 часов при комнатной температуре. Растворитель осторожно удаляли в вакууме. Полученный остаток промывали трет-бутилметиловым эфиром с получением остатка (30,8 г). Способ A2; Rt: 0,92 минуты, масса/заряд = 348,1 (M+H)+. Точная масса: 347,1.
DIPEA (54,2 мл, 308 ммоля) добавляли при 0°C к раствору вышеупомянутого остатка (30,84 г, 61,6 ммоля), (S)-2-(метоксикарбониламино)-3-метилбутановой кислоты (11,9 г, 67,8 ммоля) и HBTU (35,0 г, 92,4 ммоля) в дихлорметане (265 мл) в атмосфере азота. Затем реакционную смесь перемешивали в течение 3 часов в атмосфере азота при комнатной температуре. Реакционную смесь растворяли дихлорметаном и промывали насыщенным NaHCO3, солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Полученный остаток очищали колоночной хроматографией (петролейный эфир:этилацетат = 1:1) с получением соединения QA-10 (31,1 г). Способ A2; Rt: 1,28 минуты, масса/заряд = 507,2 (M+H)+. Точная масса: 506,1.
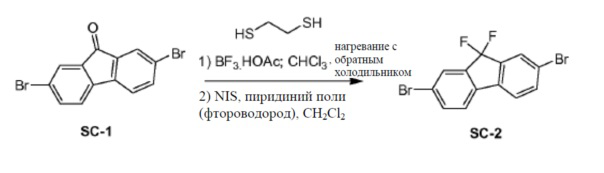
Соединение SC-1 (100 г, 296 ммоль) растворяли в CHCl3 (3720 мл). 1,2-Этандитиол (53 мл, 592 ммоль) и комплекс трифторид бора-уксусная кислота (44 мл, 296 ммоль) добавляли под защитой N2. Смесь нагревали с обратным холодильником в течение 16 часов. Твердое вещество отфильтровывали и сушили при условиях сильного разрежения с получением в результате промежуточного соединения-тиокеталя (98 г). В сосуде из фторполимера NIS (183 г, 811 ммоль, 4,8 экв.) растворяли в сухом CH2Cl2 (1600 мл). Фтороводород-пиридин добавляли при -75°C. Смесь перемешивали при -75°C в течение 10 минут. Часть полученного ранее тиокеталя (70 г, 169 ммоль) в сухом CH2Cl2 (1000 мл) добавляли каплями. Смесь перемешивали при -75°C в течение 15 минут. Смесь разводили CH2Cl2 (800 мл) и пропускали через прокладку из геля основного оксида алюминия. Растворитель концентрировали до 600 мл и промывали насыщенным раствором Na2SO3 (500 мл) и насыщенным раствором K2CO3 (500 мл). Органический слой сушили над Na2SO4 и выпаривали в вакууме с получением соединения SC-2 (48 г).
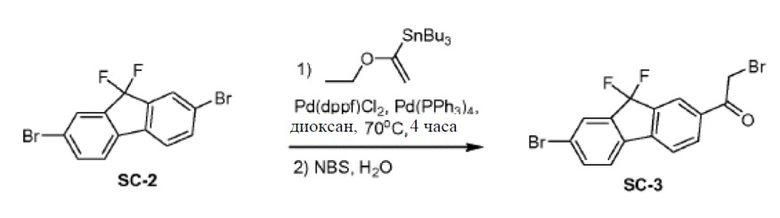
Pd(PPh3)4 (6,5 г, 5,6 ммоля, 0,2 экв.) и Pd(dppf)2Cl2 (4 г, 5,6 ммоля, 0,2 экв.) добавляли к смеси соединения SC-2 (10 г, 28 ммоль, 1 экв.), трибутил(1-этоксивинил)олова (10 г, 28 ммоль, 1 экв.) и сухого диоксана (200 мл). Смесь перемешивали при 70°С в атмосфере N2 в течение 4 часов. Смесь охлаждали до 20°С. Добавляли H2O (50 мл) и NBS (20 г, 112 ммоль) и смесь перемешивали при 20°С в атмосфере N2 в течение 12 часов. Добавляли CH2Cl2 (200 мл) и H2O (100 мл). Органический слой сушили над Na2SO4 и выпаривали. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир:этилацетат = 3:1) с получением соединения SC-3 (2 г).

Соединение SC-3 (2 г, 5 ммоль) растворяли в CH3CN (20 мл). Добавляли Boc-L-пролин (4,3 г, 20 ммоль) и триэтиламин (2,4 мл, 17,5 ммоль). Смесь перемешивали при 25°C в течение 3 часов. Растворитель удаляли в вакууме с получением в результате неочищенного остатка (4 г). Этот остаток (4 г) растворяли в толуол (40 мл). Добавляли CH3COONH4 (7,7 г, 100 ммоль). Смесь перемешивали при 100°С в течение 2 часов. Раствор разводили этилацетатом (20 мл) и промывали H2O (2×10 мл). Органический слой сушили над Na2SO4 и выпаривали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир:этилацетат = 3:1) с получением соединения SC-4 (2,6 г).

Pd(dppf)Cl2 (0,54 г, 0,74 ммоль) добавляли к смеси соединения SC-4 (4,8 г, 7,4 ммоль), KOAc (1,45 г, 14,8 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (3,76 г, 14,8 ммоль) и толуола (48 мл). Смесь перемешивали при 85°C в течение 12 часов. После охлаждения растворитель выпаривали в вакууме, добавляли CH2Cl2 и смесь промывали H2O (200 мл) и насыщенным раствором Na2CO3 (200 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/этилацетат =1:3) с получением соединения SC-5 (2,8 г).
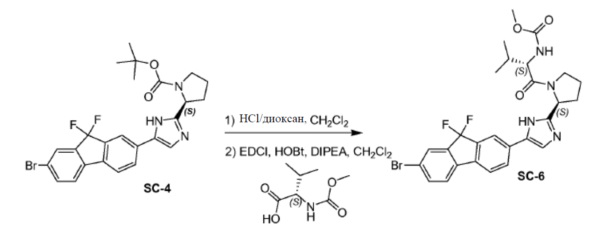
Соединение SC-4 (2,6 г, 5 ммоль) растворяли в CH2Cl2 (26 мл) и добавляли 4 н HCl/диоксан (2 мл, 8 ммоль) при 0°C. Смесь перемешивали при 25°С в течение 20 минут. Растворитель удаляли в вакууме с получением в результате остатка (2,5 г). Способ A2; Rt: 0,95 минуты, масса/заряд: 415,9 (M+H)+. Точная масса: 415,1. Данный остаток (2,5 г), (S)-2-(метоксикарбониламино)-3-метилбутановую кислоту (1,9 г, 11 ммоль), EDCI (2,1 г, 11 ммоль) и HOBt (1,5 г, 11 ммоль) в CH2Cl2 (25 мл) охлаждали до 0°C и добавляли DIPEA (8,7 мл, 50 ммоль). Смесь перемешивали при 20°C в течение 12 часов. Смесь разводили CH2Cl2 (20 мл) и H2O (5 мл). Органический слой отделяли и промывали насыщенным водным NaHCO3 (5 мл), солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир:этилацетат = 1:8) с получением соединения SC-6 (2,2 г). Способ A2; Rt: 0,97 минуты, масса/заряд: 575,0 (M+H)+. Точная масса: 574,1.
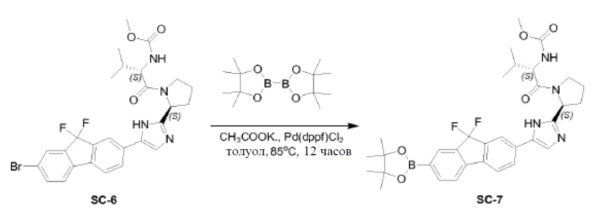
Pd(dppf)Cl2 (0,14 г, 0,19 ммоль) добавляли к смеси соединения SC-6 (2,2 г, 3,8 ммоля), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (1,95 г, 7,6 ммоля), CH3COOK (0,75 г, 7,6 ммоля) и сухого толуола (45 мл). Смесь перемешивали при 85°C в течение 12 часов. После охлаждения растворитель выпаривали в вакууме, добавляли CH2Cl2 и смесь промывали H2O (200 мл) и насыщенным раствором Na2CO3 (200 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир:этилацетат = 1:3) с получением соединения SC-7 (2,05 г). Способ A2; Rt: 0,98 минуты, масса/заряд: 621,1 (M+H)+. Точная масса: 620,3.
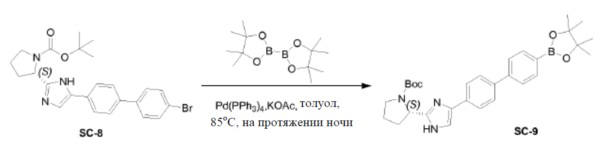
Pd(PPh3)4 (0,4 г, 0,35 ммоль) добавляли к смеси соединения SC-8 (3,3 г, 7 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (3,6 г, 14 ммоль), KOAc (1,4 г, 14 ммоль) и толуол (75 мл). Смесь перемешивали при 85°C в течение 12 часов. После охлаждения добавляли CH2Cl2 и смесь промывали насыщенным раствором Na2CO3 (200 мл) и солевым раствором (200 мл). Воду экстрагировали CH2Cl2 (3×200 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/метанол=10:1). Растворитель удаляли в вакууме с получением в результате соединения SC-9 (1,6 г). Способ A2; Rt: 1,11 минуты, масса/заряд: 516,1 (M+H)+. Точная масса: 515,3.
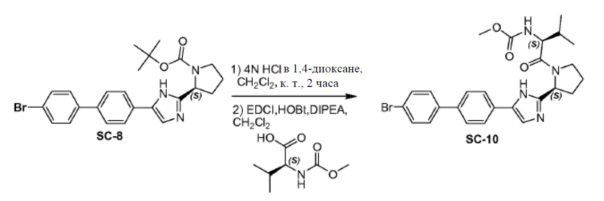
Соединение SC-8 (10 г, 21 ммоль) растворяли в CH2Cl2 (100 мл) и добавляли каплями 4н HCl/диоксан (50 мл). Смесь перемешивали в течение 30 минут при 25°C. Растворитель удаляли в вакууме и полученный остаток совместно выпаривали с толуолом (2×20 мл) и использовали в следующем этапе без дальнейшей очистки.
Способ A2; Rt: 0,96 минуты, масса/заряд: 368,1 (M+H)+. Точная масса: 367,1
Полученный ранее остаток (S)-2-(метоксикарбониламино)-3-метилбутановой кислоты (5,6 г, 32 ммоль), EDCI (6,1 г, 32 ммоль) и HOBt (1,4 г, 10 ммоль) в CH2Cl2 (180 мл) охлаждали до 0°C. DIPEA (18,6 г, 106 ммоль) добавляли каплями. Смесь перемешивали в течение 1 часа при 25°C. Органический слой промывали насыщенным раствором водный слой NaHCO3 (20 мл) и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (градиент элюента: этилацетат:метанол: от 100:0 до 20:1) с получением в результате соединения SC-10 (9,9 г) в виде белого порошка. Способ A2; Rt: 1,02 минуты, масса/заряд: 527,1 (M+H)+. Точная масса: 526,1.

Смесь соединения SC-10 (2 г, 3,80 ммоля), 4,4,4',4',5,5,5',5'-октаметил-2,2’-би(1,3,2-диоксаборолана) (1,9 г, 7,6 ммоля), Pd(dppf)Cl2 (0,28 г, 0,38 ммоля), KOAc (0,75 г, 7,6 ммоля) в сухом диоксане (20 мл) перемешивали в течение 2 часов при 100°С в атмосфере N2. Твердое вещество отфильтровывали и фильтрат выпаривали досуха. Остаток очищали с помощью колоночной хроматографии на силикагеле (градиент элюента: петролейный эфир: этилацетат: от 100:0 до 0:100) с получением в результате соединения SC-11 (1,88 г) в виде белого порошка. Способ A2; Rt: 1,08 минуты, масса/заряд: 573,1 (M+H)+. Точная масса: 572,3.
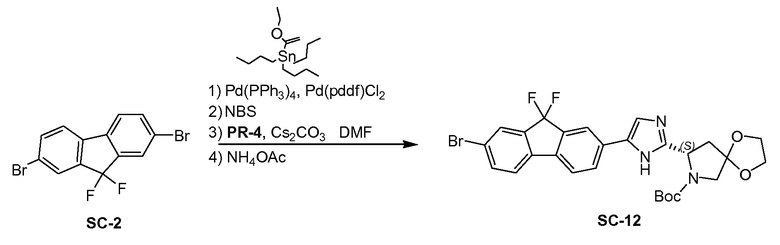
SC-2 (20 г, 55,5 ммоля), трибутил(1-этоксивинил)олово (20 г, 55,5 ммоля), Pd(PPh3)4 (13 г, 12 ммоль) и Pd(ddpf)Cl2 (8 г, 12 ммоль) суспендировали в 1,4-диоксане (100 мл) при 20°С. Смесь перемешивали при нагревании с обратным холодильником в течение 4 часов. Смесь вливали в H2O (30 мл) при 20°C. Добавляли NBS (40 г, 110,0 ммоль) и полученную в результате смесь перемешивали при 20°C в течение 12 часов. Смесь вливали в H2O (50 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали солевым раствором и сушили над Na2SO4. Органическую фазу концентрировали в вакууме с получением в результате неочищенного SC-3 (21 г). Полученный остаток в следующей реакции без очистки. Cs2CO3 (20,0 г, 61,38 ммоля) добавляли к перемешиваемому раствору PR-4 (7,6 г, 27,81 ммоля) в DMF (40 мл). Реакционную смесь перемешивали при 20°C в течение 0,5 часа. Неочищенное SC-3 (21,0 г, 52,23 ммоля) добавляли к смеси. Реакционную смесь перемешивали при 20°C в течение 2 часов. Смесь промывали водой (20 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: гексан: эфир ацетат=5:1) с получением в результате (S)-8-(2-(7-бром-9,9-дифтор-9H-флуорен-2-ил)-2-оксоэтил)-7-трет-бутил-1,4-диокса-7-азаспиро[4.4]нонан-7,8-дикарбоксилата (8 г).
К перемешиваемому раствору (S)-8-(2-(7-бром-9,9-дифтор-9H-флуорен-2-ил)-2-оксоэтил) 7-трет-бутил-1,4-диокса-7-азаспиро[4.4]нонан-7,8-дикарбоксилата (8 г) в ксилоле (80 мл) в автоклаве добавляли NH4OAc (20 г, 260 ммоль). Реакционную смесь перемешивали при 140°C в течение 1 часа. Смесь промывали водой (90 мл) и экстрагировали этилацетатом (3×50 мл); объединенный органический слой промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Полученный остаток очищали с помощью препаративной высокоэффективной жидкостной хроматографии на RP-18 (элюент: CH3CN в H2O (0,5% NH4HCO3) от 40% до 80%, объем/объем). Чистые фракции собирали и летучие вещества удаляли в вакууме. Водный слой лиофилизировали досуха с получением в результате SC-12 (4,12 г). Способ A2; Rt: 1,12 минуты, масса/заряд: 576,1 (M+H)+. Точная масса: 575,1.
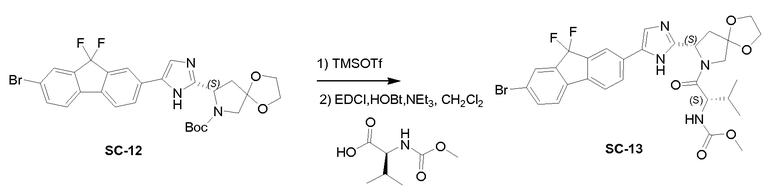
К перемешиваемому раствору соединения SC-12 (4,5 г, 7,85 ммоль) и 2,6-лютидина (1,68 г, 15,7 ммоль) в сухом CH2Cl2 (50 мл) при 0°C добавляли каплями TMSOTf (7 г, 31,4 ммоль). Реакционную смесь перемешивали при 0°C в течение 30 минут, гасили насыщенным водным NH4Cl и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме, с получением в результате (S)-8-(5-(7-бром-9,9-дифтор-9H-флуорен-2-ил)-1H-имидазол-2-ил)-1,4-диокса-7-азаспиро[4.4]нонана (2 г). NEt3 (0,5 г, 45 ммоль) добавляли к перемешиваемому раствору (S)-8-(5-(7-бром-9,9-дифтор-9H-флуорен-2-ил)-1H-имидазол-2-ил)-1,4-диокса-7-азаспиро[4.4]нонана (2 г, 4,2 ммоль), (S)-2-(метоксикарбониламино)-3-метилбутановой кислоты (0,89 г, 5 ммоль), EDCI (0,96 г, 5 ммоль) и HOBt (0,67 г, 5 ммоль) в сухом CH2Cl2 (50 мл). Реакционную смесь перемешивали при 20°C в течение 2 часов, гасили насыщенным водным Na2CO3 и экстрагировали CH2Cl2 (3×10 мл). Объединенный органический слой промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Полученный остаток очищали с помощью препаративной высокоэффективной жидкостной хроматографии на RP-18 (элюент: CH3CN в H2O (0,5% NH4HCO3) от 30% до 70%, объем/объем). Чистые фракции собирали и органические летучие вещества удаляли в вакууме. Водный слой лиофилизировали досуха с получением в результате SC-13 (1,2 г) в виде белого твердого вещества. Способ A2; Rt: 1,09 минуты, масса/заряд: 633,3 (M+H)+. Точная масса: 632,1.

К перемешиваемому раствору соединения SC-13 (1,2 г, 1,9 ммоль) и Pd (dppf) Cl2 (0,1 г, 0,137 ммоль) в сухом диоксане (25 мл) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2’-би(1,3,2-диоксаборолан) (0,72 г, 2,8 ммоль) и KOAc (0,37 г, 3,76 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 30 минут, гасили водой и экстрагировали этилацетатом (3×20 мл). Объединенный органический слой промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан:эфир ацетат = 1:1) с получением соединения SC-14 (0,756 г) в виде желтого твердого вещества.
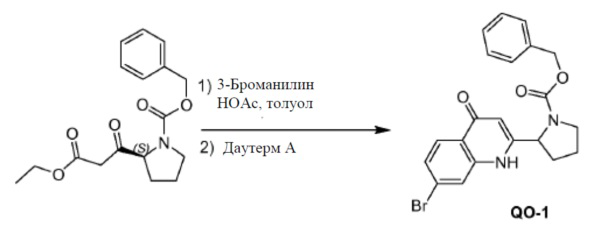
3-Броманилин (186 г,1080 ммоль) добавляли к смеси (S)-бензил-2-(3-этокси-3-оксопропаноил)пирролидин-1-карбоксилата (460 г, 1440 ммоль) в толуоле, содержащей уксусную кислоту (86,4 г, 1440 ммоль), и нагревали с обратным холодильником в течение 8 часов с использованием аппарата Дина-Старка для удаления воды из реакционной смеси. Смесь концентрировали при пониженном давлении и сушили в вакууме. Неочищенный продукт использовали в следующем этапе без дополнительной очистки (662 г). Колбу, оборудованную мешалкой, дефлегматором и капельной воронкой, продували азотом. Добавляли Dowtherm™ A (90 мл) и затем нагревали до 240°C. Раствор полученного ранее остатка (662 г) в Dowtherm™ A (900 мл) добавляли за 10 мин. при поддержании температуры в диапазоне 230-245°C. Смесь нагревали в течение еще 1 часа при 240°C и затем охлаждали до комнатной температуры. Добавляли петролейный эфир (2000 мл) и гептан (2400 мл). Образовавшийся маслянистый остаток и растворитель декантировали. Собранный масляный остаток очищали с помощью колоночной флэш-хроматографии (элюент: CH2Cl2: EtOAc=10: 1 до 1: 3) с получением в результате соединения 4 (38 г). Способ В; Rt: 5,20 минуты, масса/заряд: 429,0 (M+H)+. Точная масса: 428,1 Колонки: AD-H 50 мм * 4,6 мм, 3 мкм; поток: 4 мл/минута; подвижная фаза: A: CO2. B: EtOH (0,05% диэтиламина), от 5% до 40% B в A; температура: 40°C, изомер 4a: Rt: 1,53 минуты; 4b Rt: 1,73 минуты.

Соединение QO-1 (1,85 г, 4,3 ммоль) растворяли в CHCl3 (10 мл). Добавляли конц. HCl (10 мл) и смесь перемешивали в запечатанной пробирке при 60°С в течение 1 часа. Растворитель удаляли в вакууме. Полученный остаток (1,6 г) растворяли в метаноле (30 мл) и добавляли NEt3 (1,8 мл, 13,0 ммоль). Далее добавляли каплями Boc2O (1,1 г, 5,2 ммоль) при 0°С. После добавления смесь перемешивали в течение 0,5 часа при 20°С. Растворитель удаляли в вакууме и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (градиент элюента: петролейный эфир:этилацетат: от 100:0 до 0:100). Это давало в результате соединение QO-2 (1,08 г). Способ A2; Rt: 0,97 минуты, масса/заряд = 392,9 (M+H)+. Точная масса: 392,1.
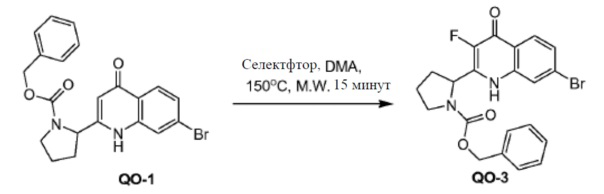
Соединение QO-1 (1 г, 2,3 ммоль) и Selectfluor (0,81 г, 2,3 ммоль) в DMA (10 мл) перемешивали при 150°С в течение 15 минут. Cмесь охлаждали до комнатной температуры и вливали в предварительно охлажденный насыщенный NaHCO3 (100 мл). Осадок фильтровали, промывали H2O и очищали хроматографией на силикагеле (элюент: CH2Cl2/EtOAc, 1/1). Собранные фракции объединяли и концентрировали в вакууме. Полученный остаток отверждали THF (3 мл) с получением в результате соединения QO-3 (0,13 г).
Способ A2; Rt: 1,55 минуты, масса/заряд = 447,0 (M+H)+. Точная масса: 446,1.
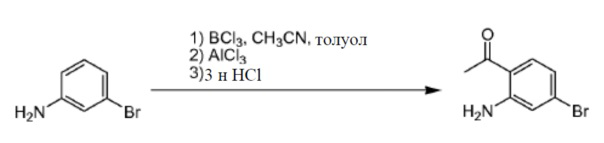
Ацетонитрил (23,7 г, 580 ммоль) добавляли к 3-броманилину (10 г, 58 ммоль) в толуоле (70 мл). Смесь охлаждали до 0°С и добавляли каплями BCl3 (1 M в CH2Cl2, 64 мл, 64 ммоль) при сохранении температуры ниже 10°С. Далее добавляли небольшими порциями AlCl3 (11,6 г, 87 ммоль) при 0°С. Реакционную смесь нагревали до 90°С в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и гасили водной HCl (2н, 100 мл). Смесь нагревали до 50°С в течение 1 часа, охлаждали до комнатной температуры и разделяли. Органический слой отделяли и промывали водой и солевым раствором. Органический слой собирали, сушили и концентрировали с получением в результате 1-(2-амино-4-бромфенил)этанона (4 г). Способ A2; Rt: 0,98 минуты, масса/заряд = 215,7 (M+H)+. Точная масса: 215,0.
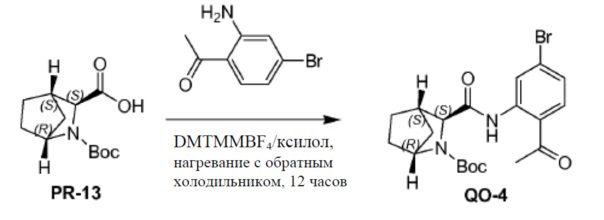
Соединение PR-13 (10,6 г, 43,9 ммоль), молекулярное сито 4 A (1,0 г) и 1-(2-амино-4-бромфенил)этанон (9,4 г, 43,9 ммоль) в ксилоле (100 мл) перемешивали и нагревали с обратным холодильником в течение 1 часа. Добавляли 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния BF4 (DMTMM.BF4, 15,8 г, 48,3 ммоль) и смесь перемешивали и нагревали с обратным холодильником в течение 12 часов. Смесь фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/CH2Cl2 = 5:1, затем петролейный эфир/этилацетат = 1/1 объем/объем) с получением в результате соединения QO-4 (10,9 г).
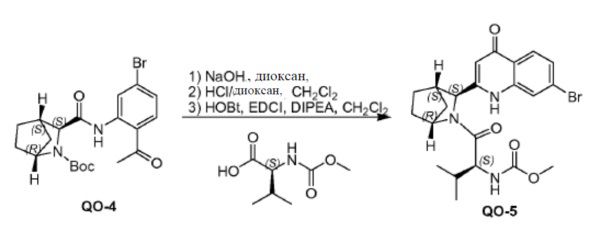
Соединение QO-4 (10,0 г, 22,9 ммоль) и NaOH (совместно выпариваемые с толуолом, 3,2 г, 80 ммоль) в диоксане (100 мл) перемешивали в течение 1 часа при 100°С в атмосфере N2. Смесь вливали в 10% NH4Cl (200 мл). Смесь экстрагировали с помощью CH2Cl2 (2×100 мл). Органические слои промывали солевым раствором, сушили и концентрировали в вакууме.
Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: CH2Cl2, затем этилацетат). Чистые фракции собирали и растворитель удаляли в вакууме. Полученный хинолинон (3,0 г) в CH2Cl2 (30 мл), 4 н HCl/диоксане (30 мл) добавляли каплями. Смесь перемешивали в течение 2 часов при 20°С, а затем летучие соединения удаляли в вакууме. Полученный остаток (3,0 г), (S)-2-(метоксикарбониламино)-3-метилбутановую кислоту (1,9 г, 10,8 ммоль), EDCI (2,1 г, 10,8 ммоль) и HOBt (0,49 г, 3,6 ммоль) в CH2Cl2 (30 мл) перемешивали при 0°С. Добавляли DIPEA (4,7 г, 36 ммоль). Смесь перемешивали в течение 2 часов при 20°С. Добавляли H2O (30 мл) и смесь отфильтровывали. Твердое вещество собирали и сушили с получением в результате соединения QO-5. Фильтрат разделяли и органический слой промывали H2O (2×30 мл), солевым раствором, сушили и выпаривали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: этилацетат, затем этилацетат: CH3OH = 10:1). Чистые фракции собирали и растворитель концентрировали в вакууме с получением в результате дополнительного соединения QO-5 (в сумме 3 г).

К перемешиваемому раствору соединения PR-14 (6 г, 22,3 ммоль) в CH2Cl2/пиридине (100 мл, 1/1) при 0°С добавляли каплями (COCl) 2 (5,6 г, 44,6 ммоль). Смесь перемешивали при 25°С в течение 0,5 часа. Затем смесь добавляли к раствору 1-(2-амино-4-бромфенил)этанона (4,7 г, 22,3 ммоль) в CH2Cl2 (30 мл). Смесь перемешивали при 25°С в течение 1 часа. Смесь вливали в H2O (100 мл), экстрагировали CH2Cl2 (3×50 мл) и разделяли. Объединенные органические слои промывали солевым раствором и сушили над Na2SO4. Органическую фазу концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии (гексан: эфир ацетат=5:1) с получением в результате соединения QO-6 (9 г) в виде твердого вещества.

К соединению QO-6 (9 г, 19,3 ммоля) в толуоле (100 мл) добавляли NaOH (3 г, 77,2 ммоля) при 25°С. Смесь перемешивали при нагревании с обратным холодильником в течение 1 часа. Смесь вливали в водный NH4Cl (50 мл), экстрагировали этилацетатом (3×100 мл) и разделяли. Объединенные органические слои промывали солевым раствором и сушили над Na2SO4. Органическую фазу концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии (гексан: эфир ацетат=10:1) с получением в результате соединения QO-7 (3,5 г). Способ A2; Rt: 1,25 минуты, масса/заряд= 449,1 (M+H)+. Точная масса: 448,1.

К раствору соединения QO-7 (3,5 г, 7,83 ммоль) в CH2Cl2 (100 мл) добавляли каплями TFA (10 мл) при 25°С. Смесь перемешивали при 25°С в течение 0,5 часа. Растворитель удаляли в вакууме. Остаток промывали трет-бутил-метиловым эфиром и сушили в вакууме, полученное в результате твердое вещество (2,4 г) перемешивали в CH2Cl2 (100 мл) с (S)-2-(метоксикарбониламино)-3-метилбутановой кислотой (1,45 г, 8,3 ммоль), HATU (3,15 г, 8,3 ммоль) и NEt3 (0,84 г, 8,3 ммоль) при 25°С. Смесь перемешивали при 25°С в течение 1 часа. Смесь вливали в H2O (50 мл), экстрагировали CH2Cl2 (3×50 мл) и разделяли. Объединенные органические слои промывали солевым раствором и сушили над Na2SO4. Органическую фазу концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографией (гексан: эфир ацетат=5:1) с получением в результате соединения QO-8 (1,5 г) в виде твердого вещества. Способ A2; Rt: 1,11 минуты, масса/заряд = 506,2 (M+H)+. Точная масса: 505.1; 1H ЯМР (400 МГц, DMSO-d6) δ м.д. 0,85 (д, J=6,7 Гц, 3 H), 0,90 (д, J=6,7 Гц, 3 H), 1,15-1,37 (м, 2 H), 1,37-1,56 (м, 2 H), 1,56-1,79 (м, 3 H), 1,82-2,11 (м, 3 H), 2,23-2,43 (м, 2 H), 3,55 (с, 3 H), 3,93 (т, J=8,7 Гц, 1 H), 4,45 (дт, J=11,7, 5,9 Гц, 1 H), 4,73 (дд, J=10,3, 7,4 Гц, 1 H), 5,89 (ушир.с., 1 H), 7,44 (дд, J=8,6, 1,9 Гц, 1 H), 7,54 (д, J=8,0 Гц, 1 H), 7,73 (д, J=1,9 Гц, 1 H), 7,95 (д, J=8,5 Гц, 1 H), 11,63 (ушир.с., 1 H).

1-(2-Амино-4-бром-5-фторфенил)этанон (0,36 г, 1,57 ммоль) и соединение PR-12 (0,34 г, 1,57 ммоль) в ксилоле (8 мл) нагревали с обратным холодильником в течение 1 часа. Добавляли DMTMM.BF4 (0,57 г, 1,73 ммоль) и смесь перемешивали и нагревали с обратным холодильником в течение 8 часов. Растворитель удаляли в вакууме и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (градиент элюента: петролейный эфир/этилацетат от 1 до 1/2) с получением в результате соединения QO-9 (0,5 г).
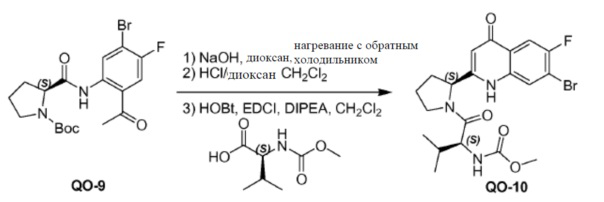
Соединение QO-9 (0,5 г, 1,16 ммоль) и NaOH (0,16 г, 4,0 ммоль) в сухом диоксане (5 мл) перемешивали при 100°С в течение 1 часа в атмосфере N2. Смесь вливали в 10% раствор NH4Cl (20 мл). Остаток экстрагировали CH2Cl2 (2×10 мл), сушили над Na2SO4 и выпаривали в вакууме. Полученный остаток (0,5 г) растворяли в CH2Cl2 (5 мл). 4н HCl/диоксан (2 мл) добавляли при 0°С и смесь затем перемешивали при 25°С в течение 20 минут. Растворитель удаляли в вакууме. Полученный остаток (0,5 г), (S)-2-(метоксикарбониламино)-3-метилбутановая кислота (0,45 г, 2,55 ммоль), EDCI (0,49 г, 2,55 ммоль) и HOBt(0,34 г, 2,55 ммоль) в CH2Cl2 (10 мл) охлаждали до 0°С и DIPEA (2,3 мл, 11,6 ммоль) добавляли. Смесь перемешивали при 20°С в течение 12 часов. Смесь разводили CH2Cl2 (20 мл) и H2O (5 мл). Органический слой отделяли и промывали насыщенным водным NaHCO3 (5 мл), солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: метанол/CH2Cl2=15%) с получением в результате соединения QO-10 (150 мг) в виде белого порошка.
Способ A2; Rt: 0,92 минуты, масса/заряд = 470,1 (M+H)+. Точная масса: 469,0
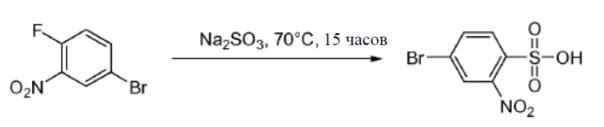
4-Бром-1-фтор-2-нитробензол (50 г, 227 ммоль) растворяли в этаноле (600 мл). Затем добавляли суспензию Na2SO3 (71,6 г, 568 ммоль) в этаноле (1000 мл) и воде (1250 мл). Суспензию перемешивали при 70°С в течение 15 часов. Затем при комнатной температуре реакционную смесь подкисляли HCl (2 н.) до pH=2 и концентрировали в вакууме. Остальной остаток растворяли при нагревании с обратным холодильником в солевом растворе (1000 мл). Затем добавляли воду (100 мл) и раствор охлаждали в ледяной бане. Осадок собирали фильтрованием с получением в результате 4-бром-2-нитробензолсульфоновой кислоты (57,3 г, 89 %).

К раствору тионилхлорида (50 мл) добавляли 4-бром-2-нитробензолсульфоновую кислоту (30 г, 106 ммоль) и DMF (1 капля) и реакционную смесь нагревали до возврата флегмы в течение 4 часов. После охлаждения реакционную смесь подвергали азеотропной перегонке с толуолом три раза. Остаток растворяли в минимальном количестве толуола, а затем полученную в результате смесь добавляли к смеси концентрированного водного раствор гидроксида аммония (1 мл) и THF (10 мл) при -10°С. После перемешивания в течение 2 часов эту реакцию гасили добавлением раствора 6 M водной соляной кислоты до pH=4. Органический слой отделяли, а затем сушили и концентрировали в вакууме. Петролейный эфир добавляли к полученной в результате взвеси и продукт собирали с помощью вакуум-фильтрации с получением в результате 4-бром-2-нитробензолсульфонамида.
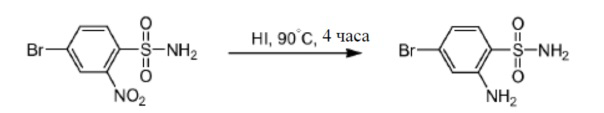
Суспензию 4-бром-2-нитробензолсульфонамида (21,2 г, 75 ммоль) в 57% HI (250 мл) нагревали до 90°C в течение 4 часов. После охлаждения до комнатной температуры темно-багряную смесь разбавляли этилацетатом (500 мл) и затем последовательно промывали насыщенным водн. Na2S2O3, насыщенным водн. NaHCO3 и солевым раствором. Бесцветный органический органический слой сушили над безводным MgSO4, фильтровали и концентрировали досуха. Неочищенный продукт очищали с помощью высокоэффективной жидкостной хроматографии (элюент: CH3CN/H2O от 22/78 до 52/48 с 0,01% NH3H2O в качестве буфера) с получением в результате 2-амино-4-бромбензолсульфонамида (18,6 г). Способ В; Rt: 3,36 минуты, масса/заряд = 250,9 (M+H)+. Точная масса: 249,9.

Триэтиламин (40,5 мл, 296 ммоль) добавляли к раствору 2-амино-4-бромбензолсульфонамида (18,6 г, 74 ммоль) в ацетоне (200 мл). Соединение PR-6 (12,8 г, 48 ммоль) добавляли к реакционной смеси при охлаждении. После перемешивания в течение 5 часов реакционную смесь разбавляли водой и подкисляли 2н HCl до pH 4. Полученный в результате осадок собирали с помощью фильтрования и затем переносили в другую колбу. Добавляли раствор K2CO3 (15 г) в воде (100 мл) и реакционную смесь нагревали с обратным холодильником в течение 2 часов до тех пор, пока реакционная смесь не становилась однородной. Реакционную смесь подкисляли 2 н. HCl до pH=4. Осадок отфильтровывали и промывали водой. Неочищенный продукт очищали с помощью высокоэффективной жидкостной хроматографии (элюент: CH3CN/H2O от 35/65 до 65/35 с 0,75% CF3COOH в качестве буфера) с получением в результате соединения TD-1 (8,3 г, 45 %). Способ A2; Rt: 1,05 минуты, масса/заряд: 487,8 (M+Na)+. Точная масса: 465,0.

Соединение TD-1 (2 г, 4,3 ммоль) растворяли в CH3COOH (20 мл). Добавляли 40% HBr (30 мл) и перемешивали при 50°С в течение 3 часов. Растворитель выпаривали в вакууме. Полученный остаток промывали трет-бутилметиловым эфиром. Твердое вещество фильтровали и сушили при условиях сильного разрежения. Раствор полученного в результате желтого порошка (1,7 г) и Boc2O (1,8 г, 8,2 ммоля) в метаноле (15 мл) охлаждали до 0°С. Добавляли триэтиламин (2,3 мл, 16,4 ммоль). Смесь перемешивали при 20°С в течение 2 часов и растворитель удаляли в вакууме. Добавляли CH2Cl2 (10 мл) и смесь промывали H2O (10 мл) и сушили над Na2SO4. Растворитель удаляли в вакууме и полученный остаток отверждали петролейным эфиром (5 мл) и фильтровали. После высушивания при условиях сильного разрежения получали соединение TD-2 (1,7 г). Способ G; Rt: 1,26 минуты, масса/заряд = 453,9 (M+Na)+. Точная масса: 431,0.

К оксетан-3-ону (5 г, 69 ммоль) в THF (50 мл) последовательно добавляли 2-метилпропан-2-сульфинамид (8,34 г, 69 ммоль) и Ti(OEt)4 (20 мл). Реакционную смесь нагревали до 50°С в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и гасили водой (200 мл). Осадок фильтровали и фильтрат экстрагировали CH2Cl2 (2×50 мл). Объединенный органический слой отделяли и промывали водой (50 мл) и солевым раствором (50 мл). Органический слой сушили и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (CH2Cl2) с получением в результате соединения OA-1 (5,5 г, выход 46%).
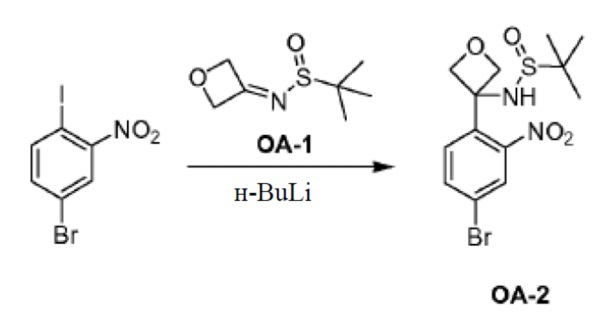
4-Бром-1-йод-2-нитробензол (3 г, 9,18 ммоль) растворяли в безводном THF (20 мл) в атмосфере N2 и колбу охлаждали до -78°С. Смесь перемешивали в течение 5 минут и медленно добавляли n-BuLi (4,4 мл, 2,5 моль/л). Реакционная смесь темнела, и перемешивание продолжали при -78°C в течение 15 минут. Затем соединение OA-1 (1,92 г, 11 ммоль) медленно добавляли к смеси. Реакционную смесь перемешивали в течение 30 минут при -78°С, а затем подогревали до комнатной температуры. Смесь вливали в воду (50 мл) и экстрагировали CH2Cl2 (2×20 мл). Органические фазы разделяли и промывали солевым раствором, сушили над Na2SO4 и концентрировали досуха. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 3/1) с получением в результате соединения OA-2 (1,3 г, выход 38%).
Способ A2; Rt: 1,04 минуты, масса/заряд = 378,7 (M+H)+. Точная масса: 378,0.

Соединение OA-2 (1,3 г, 3,45 ммоль) растворяли в MeOH (10 мл) и медленно добавляли HCl/диоксан (4н, 10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут и смесь концентрировали с получением в результате остатка (0,89 г). Способ A2; Rt: 0,60 минуты, масса/заряд = 272,7 (M+H)+ К полученному остатку (0,89 г) в 50 мл колбе добавляли HATU (1,49 г, 3,94 ммоль), триэтиламин (0,66 г, 6,56 ммоль) и (S)-1-(трет-бутоксикарбонил)-пирролидин-2-карбоновую кислоту (0,84 г, 3,94 ммоль). Остаток растворяли в CH2Cl2 (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 40 минут. Смесь гасили водой (20 мл) и экстрагировали CH2Cl2 (2×10 мл). Фазы разделяли и органическую фазу промывали солевым раствором, сушили над Na2SO4, а затем концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 2/1) с получением в результате промежуточного нитро-соединения (1,27 г). Это промежуточное нитро-соединение (1 г, 2,1 ммоля) растворяли в MeOH/воде (20 мл, 1:1), порошок Fe (0,35 г, 6,3 ммоля) и добавляли NH4Cl (0,55 г, 10,5 ммоля) и смесь перемешивали при нагревании с обратным холодильником в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали досуха. Полученный остаток промывали водой (10 мл), и экстрагировали CH2Cl2 (2×10 мл). Органический слой отделяли и концентрировали в вакууме с получением в результате промежуточного соединения (0,77 г). Способ A2; Rt: 1,07 минуты, масса/заряд = 464,0 (M+Na)+. Точная масса: 441,1. Данное промежуточное соединение (0,77 г, 1,75 ммоль) растворяли в AcOH (20 мл). Полученный в результате раствор перемешивали при 80°C в течение 30 минут. Реакционную смесь концентрировали в вакууме и полученный в результате остаток очищали с помощью колоночной хроматографии (петролейный эфир: этилацетат=1:1) с получением в результате соединения OA-3 (0,49 г, 66%). Способ В; Rt: 4,06 минуты, масса/заряд = 422,0 (M+H)+. Точная масса: 421,1.
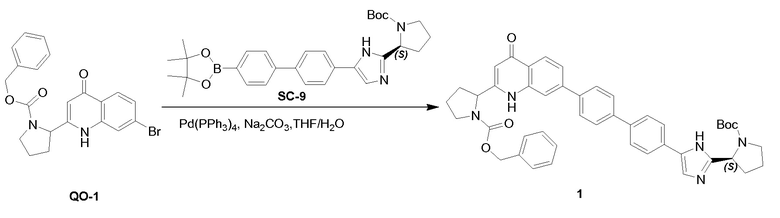
Pd (PPh3)4 (0,14 г, 0,12 ммоль) добавляли к смеси соединения SC-9 (0,5 г, 1,2 ммоль), соединения QO-1 (0,41 г, 1,2 ммоля), Na2CO3 (0,51 г, 4,8 ммоля), THF (20 мл) и H2O (10 мл) в атмосфере N2. Смесь перемешивали при микроволновом излучении при 80°С в течение 15 минут. CH2Cl2 (20 мл) и H2O (15 мл) добавляли к реакционной смеси. Органический слой отделяли, промывали солевым раствором и сушили над Na2SO4. Растворитель выпаривали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: этилацетат:петролейный эфир =3:1) с получением в результате соединения 1 (0,43 г). Способ A2; Rt: 0,89 минуты, масса/заряд = 736,3 (M+H)+. Точная масса: 735,3.
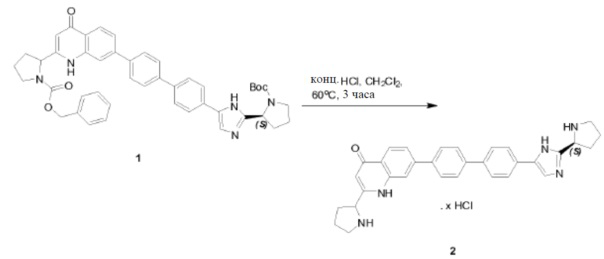
Соединение 1 (0,43 г, 0,58 ммоль) растворяли в CHCl3 (4,3 мл). Добавляли концентрированную HCl (4,3 мл). Смесь перемешивали при 60°С в запаянной пробирке в течение 1 часа. Растворитель выпаривали в вакууме с получением в результате соединения 2 (0,6 г). Способ A2; Rt: 0,92 минуты, масса/заряд= 502,3 (M+H)+. Точная масса: 501,3.
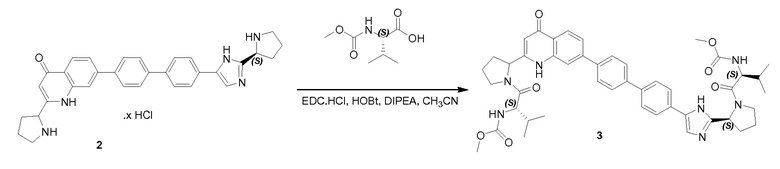
К раствору (S)-2-(метоксикарбониламино)-3-метилбутановой кислоты (0,49 г, 2,78 ммоль) в ацетонитриле (20 мл) добавляли EDCI (0,53 г, 2,78 ммоль) и HOBt (0,38 г, 2,78 ммоль). После перемешивания в течение 1 часа при 10°С добавляли соединение 2 (0,6 г). Смесь затем охлаждали до 0°С и добавляли DIPEA (1,5 г, 11,6 ммоль). Смесь перемешивали при 10°С в течение 12 часов. Твердое вещество фильтровали, полученный фильтрат концентрировали и разбавляли CH2Cl2 (20 мл) и 1 н. HCl (5 мл). Органический слой отделяли и промывали насыщенным водным NaHCO3 (5 мл) и солевым раствором, а затем сушили над Na2SO4. Растворитель удаляли в вакууме. Полученное соединение 3, смесь двух диастереоизомеров 3a и 3b, очищали с помощью высокоэффективной жидкостной хроматографии (колонка: Grace Vydac 250*20 мм*5 мкм, подвижная фаза A: вода (содержащая 0,075% TFA, объем/объем %; подвижная фаза B: ацетонитрил (содержащий 0,025% TFA, объем/объем %; скорость потока: 30 мл/минута; градиент: 35-50% B (объем/объем) от 0 до 11 минут). Две чистые фракции собирали и подщелачивали NaHCO3 до pH=8. Летучие вещества удаляли в вакууме. Остаток экстрагировали CH2Cl2 (2×10 мл). Органический слой промывали солевым раствором (10 мл) и сушили над Na2SO4. Растворитель удаляли в вакууме. Полученный остаток промывали ацетонитрилом (1 мл) и трет-бутилметиловым эфиром (1 мл). Твердое вещество сушили при условиях сильного разрежения с получением в результате двух отдельных диастереоизомеров, соединения 3a (14 мг) и соединения 3b (26 мг).
3a: Способ В; Rt: 4,71 минуты, масса/заряд: 816,3 (M+H)+. Точная масса: 815,4.
SFC: Колонка: OJ-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2. B: EtOH (0,05% диэтиламин); 5-40% B в A: Rt: 8,39 минуты
SFC: Колонка: OD-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2. B: MeOH (0,05% диэтиламин); 40% B в A: Rt: 7,67 минуты
3b: Способ В; Rt: 4,79 минуты, масса/заряд: 816,3 (M+H)+. Точная масса: 815,4.
SFC: Колонка: OJ-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2. B: MeOH (0,05% диэтиламин); 5-40% B в A: Rt: 7,41 минуты
SFC: Колонка: OD-H 150 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2. B: MeOH (0,05% диэтиламин); 5-40% B в A: Rt: 9,60 минуты
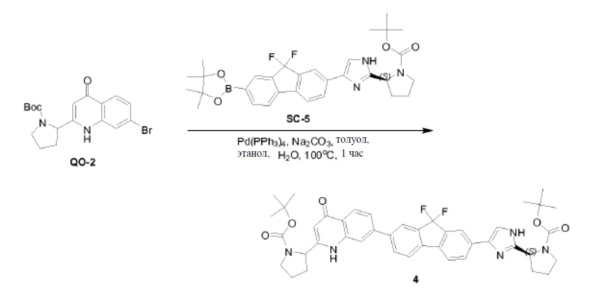
Смесь соединения QO-2 (1,0 г, 2,5 ммоль), соединения SC-5 (1,4 г, 2,5 ммоль), Na2CO3 (2,1 г, 20 ммоль), Pd(PPh3)4 (0,29 г, 0,25 ммоль) в H2O (10 мл), этаноле (10 мл) и толуоле (10 мл) перемешивали в течение 1 часа при 100°С в атмосфере N2. Добавляли воду (10 мл) и смесь экстрагировали дихлорметаном (2×30 мл). Объединенные органические слои сушили над Na2SO4. После фильтрования растворитель удаляли в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (градиент элюента: вначале этилацетат:метанол: от 100: 0 до 10:1; затем дихлорметан: метанол: от 10:1 до 1:1) с получением в результате соединения 4 (1,04 г).
Способ A2; Rt: 0,99 минуты, масса/заряд: 750,3 (M+H)+. Точная масса: 749,3.
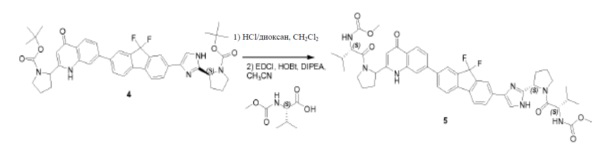
Соединение 4 (1 г, 1,3 ммоль) растворяли в CH2Cl2 (10 мл). Добавляли 4 н HCl/диоксан (10 мл). Смесь перемешивали в течение 20 минут при 25°С. Растворитель удаляли в вакууме. Остаток совместно выпаривали с толуолом (10 мл) с получением в результате 0,85 г остатка. Способ A2; Rt: 0,84 минуты, масса/заряд: 550,1 (M+H)+. Точная масса: 549,2. К этому остатку (0,85 г, 1,3 ммоля) в CH2Cl2 (10 мл) добавляли (S)-2-(метоксикарбониламино)-3-метилбутановую кислоту (0,51 г, 2,9 ммоль), EDCI (0,59 г, 2,9 ммоль) и HOBt (0,09 г, 0,67 ммоль) и смесь охлаждали до 0°С. Затем добавляли DIPEA (2,3 мл, 13,3 ммоль) и смесь перемешивали в течение 1,5 часа при 25°С. Добавляли воду (20 мл) и дихлорметан (20 мл) и органический слой отделяли и сушили над Na2SO4. После фильтрования растворитель удаляли в вакууме. Соединение 5 (смесь диастереоизомеров 5a и 5b) очищали колоночной хроматографией на силикагеле (градиент элюента: этилацетат:метанол: от 100:0 до 6:1) с получением в результате светло-желтого твердого вещества. Полученное твердое вещество промывали ацетонитрилом и дополнительно очищали с помощью сверхкритической жидкостной хроматографии (колонка: OJ 250 мм * 30 мм, 5 мкм; подвижная фаза: A: Сверхкритический CO2, B: изопропанол; 0,05% диэтиламина), A:B =65:35 при 55 мл/минута, температура колонки: 38°С, давление в сопле: 100 бар; температура сопла: 60°С, температура испарителя: 20°С, температура триммера: 25°С, длина волны: 220 нм). Полученную фракцию соединения 5a и 5b промывали ацетонитрилом и дополнительно очищали с помощью сверхкритической жидкостной хроматографии (колонка: OJ 250 мм*30 мм. 5 мкм; подвижная фаза: A: Сверхкритический CO2, B: изопропанол; 0,05% диэтиламина), A:B =65:35 при 55 мл/минута, температура колонки: 38°С; давление в сопле: 100 бар, температура сопла: 60°С, температура испарителя: 20°С, температура триммера: 25°С, длина волны: 220 нм). Она давала в результате соединение 5a (148 мг) и 5b (200 мг).
5a: Способ C; Rt: 3,66 минуты, масса/заряд: 864,4 (M+H)+. Точная масса: 863,4.
SFC: Колонка: OJ-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2 B: iPrOH (0,05% диэтиламина); 5-40% B в A: Rt: 8,18 минуты.
5b: Способ C; Rt: 3,72 минуты, масса/заряд: 864,4 (M+H)+. Точная масса: 863,4.
SFC: Колонка: OJ-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2 B: iPrOH (0,05% диэтиламина); 5-40 % B в A: Rt: 8,77 минуты.
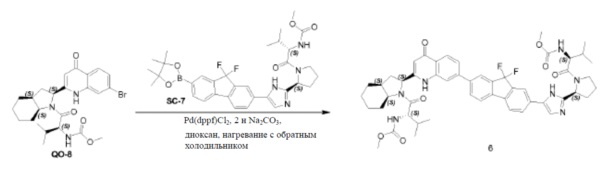
К перемешиваемому раствору соединения QO-8 (900 мг, 1,78 ммоля), соединения SC-7 (922 мг, 1,49 ммоля) и Pd (dppf) Cl2 (100 мг, 1,9 ммоля) в сухом THF (20 мл) добавляли Na2CO3 (10 мл, 2 н.). Реакционную смесь перемешивали при нагревании с обратным холодильником в течение 20 минут, гасили водой (20 мл) и экстрагировали этилацетатом (3×10 мл). Объединенный органический слой промывали солевым раствором, сушили над Na2SO4 и после фильтрования полученный фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью высокоэффективной жидкостной хроматографии (колонка: Phenomenex Synergi C18 150 * 20 мм * 5 мкм. Способ: A: H2O + 0,1% TFA B: MeCN. Скорость потока (мл/минута): 40). Чистые фракции собирали и нейтрализовали насыщенным NaHCO3. Органический растворитель концентрировали в вакууме. Осадок фильтровали, промывали с H2O (10 мл) и сушили в условиях сильного разрежения с получением соединения 6 (450 мг).
Способ H; Rt: 3,68 минуты, масса/заряд: 818,5 (M+H)+. Точная масса: 817,4.
SFC: Колонка: OJ-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2. B: MeOH (0,05% диэтиламин); 5-40% B в A: Rt: 8,24 минуты.
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 0,88 (д, J=6,5 Гц, 3 H), 0,88 (д, J=6,7 Гц, 3 H), 0,92 (д, J=6,6 Гц, 3 H), 0,95 (д, J=6,7 Гц, 3 H), 1,19-1,36 (м, 3 H), 1,45 (д, J=11,0 Гц, 1 H), 1,54 (г, J=12,0 Гц, 1 H), 1,60-1,69 (м, 1 H), 1,70-1,80 (м, 2 H), 1,89-2,09 (м, 5 H), 2,10-2,21 (м, 2 H), 2,31-2,44 (м, 2 H), 3,54 (с, 3 H), 3,56 (с, 3 H), 3,83 (т, J=6,2 Гц, 2 H), 3,95 (т, J=8,8 Гц, 1 H), 4,08 (т, J=8,4 Гц, 1 H), 4,48 (дт, J=11,0, 6,3 Гц, 1 H), 4,79 (т, J=8,9 Гц, 1 H), 5,09 (дд, J=7,0, 3,4 Гц, 1 H), 5,88 (с, 1 H), 7,34 (д, J=8,5 Гц, 1 H), 7,57 (д, J=7,9 Гц, 1 H), 7,71 (д, J=8,8 Гц, 1 H), 7,72 (с, 1 H), 7,84 (ушир.с., 1 H), 7,86 (д, J=7,9 Гц, 1 H), 7,93-8,00 (м, 3 H), 8,05 (s, 1 H), 8,08 (с, 1 H), 8,12 (д, J=8,4 Гц, 1 H), 11,76 (ушир.с., 1 H), 11,96 (ушир.с, 1 H).
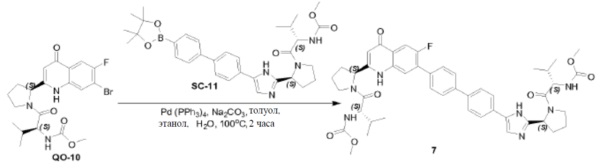
Смесь соединения QO-10 (0,12 г, 0,26 ммоль), соединения SC-11 (0,15 г, 0,26 ммоль), Pd(PPh3)4 (0,030 г, 0,026 ммоль) и Na2CO3 (0,22 г, 2,05 ммоль) в смеси толуола, этанола и H2O (1:1:1, 4,5 мл) перемешивали в течение 2 часов при 100°С в атмосфере N2. Летучие вещества удаляли в вакууме. Добавляли дихлорметан (15 мл) и воду (10 мл). Органический слой отделяли и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (градиент элюента: вначале: петролейный эфир: EtOAc: от 100:0 до 0:100, затем EtOAc: метанол: от 100:0 до 10:1). Полученное твердое вещество промывали ацетонитрилом и совместно выпаривали с метанолом. Полученное твердое вещество дополнительно очищали с помощью сверхкритической жидкостной хроматографии (колонки: OD-3 150×4,6 мм внутренний диаметр 3 мкм, поток: 2,5 мл/минута, подвижная фаза: 40% метанол (0,05% диэтиламина) в CO2) с получением в результате соединения 7 (0,1 г) в виде белого порошка. Способ H; Rt: 3,39 минуты, масса/заряд: 834,5 (M+H)+. Точная масса: 833,4; SFC: Колонка: OD-H 150 мм × 4,6 мм; 3 мкм. Поток: 2,5 мл/минута, подвижная фаза: A: CO2. B: MeOH (0,05% диэтиламин); 40% B в A: Rt: 6,56 минуты.
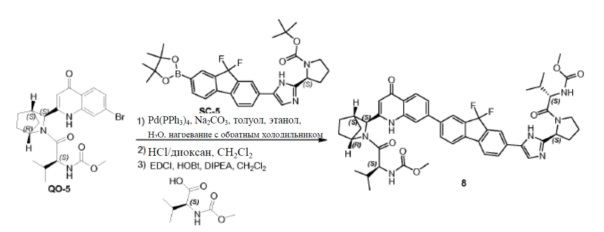
Соединение QO-15 (0,30 г, 0,63 ммоль), соединение SC-5 (0,35 г, 0,63 ммоль), Pd(PPh3)4 (0,22 г, 0,19 ммоль) и Na2CO3 (0,27 г, 2,5 ммоль) в толуоле (3 мл), этаноле (3 мл) и H2O (3 мл) нагревали с обратным холодильником в атмосфере N2 в течение 12 часов. Летучие вещества удаляли в вакууме. Смесь экстрагировали CH2Cl2 (2×10 мл). Органические слои промывали солевым раствором, сушили и выпаривали в вакууме с получением в результате остатка (0,5 г). Этот остаток (0,50 г) в CH2Cl2 (5 мл) перемешивали при 0°С. Добавляли 4 н. HCl/диоксан (5 мл). Смесь перемешивали в течение 1 часа при 20°С и летучие соединения удаляли в вакууме с получением в результате остатка (0,50 г). К этому остатку (0,5 г) в CH2Cl2 (5 мл) добавляли (S)-2-(метоксикарбониламино)-3-метилбутановую кислоту (0,13 г, 0,76 ммоль), EDCI (0,22 г, 1,14 ммоль) и HOBt (0,043 г, 0,32 ммоль) и смесь перемешивали при 0°С. Затем добавляли DIPEA (0,4 г, 3,2 ммоль). Смесь перемешивали в течение 2 часа при 20°С, а затем промывали H2O (2x) и солевым раствором, сушили над Na2SO4 и растворитель удаляли в вакууме. Полученный остаток очищали с помощью высокоэффективной жидкостной хроматографии (C18, элюент: CH3CN/H2O от 15/85 до 35/65 с 0,1% CF3COOH в качестве буфера). Чистые фракции собирали и смесь подщелачивали с помощью NaHCO3 до pH=9. Органический растворитель выпаривали и смесь отфильтровывали. Твердое вещество сушили и выпаривали в вакууме с получением в результате соединения 8 (140 мг). Способ H; Rt: 3,52 минуты, масса/заряд: 890,3 (M+H)+. Точная масса: 889,4; SFC: Колонка: AS-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,5 мл/минута, подвижная фаза: A: CO2. B: MeOH (0,05% диэтиламин); 40% B в A: Rt: 2,9 минуты; SFC: Колонка: OD-3 150 мм × 4,6 мм; 3 мкм. Поток: 2,5 мл/минута, подвижная фаза: A: CO2. B: EtOH (0,05% диэтиламин); 40% B в A: Rt: 5,2 минуты.

Раствор Na2CO3 (0,24 г, 2,3 ммоль) в H2O (6 мл) добавляли к смеси соединения SC-9 (0,6 г, 1,16 ммоль), соединения QA-1 (0,5 г, 1,16 ммоль), этанола (6 мл) и толуола (12 мл). Pd(PPh3)4 (55 мг, 0,058 ммоль) добавляли к смеси одной порцией в атмосфере азота. Смесь перемешивали в течение 10 часов при 90°С. Затем раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток растворяли в CH2Cl2 (20 мл) и промывали водой (3×10 мл). Раствор сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (градиент элюента: EtOAc: дихлорметан= от 1:3 до 2:1 и EtOAc: MeOH= 100:1 to 100:5). Необходимую фракцию собирали, растворитель удаляли в вакууме и полученный остаток сушили в вакууме с получением в результате соединения 9 (0,52 г). Способ A2; Rt: 1,03 минуты, масса/заряд: 737,3 (M+H)+. Точная масса: 736,3.
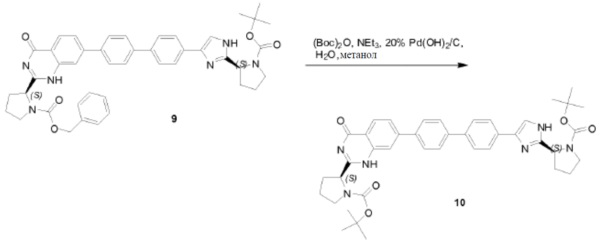
Смесь соединения 9 (0,52 г, 0,71 ммоль), (Boc)2O (0,307 г, 1,41 ммоль), NEt3 (0,212 г, 2,1 ммоль) и 20% Pd(OH)2/C (0,5 г) в метаноле (5 мл) гидрировали (1 атм.) при 10°С в течение 1,5 часа. Смесь фильтровали и летучие соединения удаляли в вакууме. Остаток растворяли в CH2Cl2 (10 мл) и промывали H2O (5 мл). Органический слой сушили над Na2SO4 и выпаривали в вакууме. Остаток промывали трет-бутилметиловым эфиром (3 мл). Твердое вещество фильтровали и сушили при условиях сильного разрежения с получением в результате соединения 10 (0,47 г). Способ A2; Rt: 1,03 минуты, масса/заряд: 703,3 (M+H)+. Точная масса: 702,4.
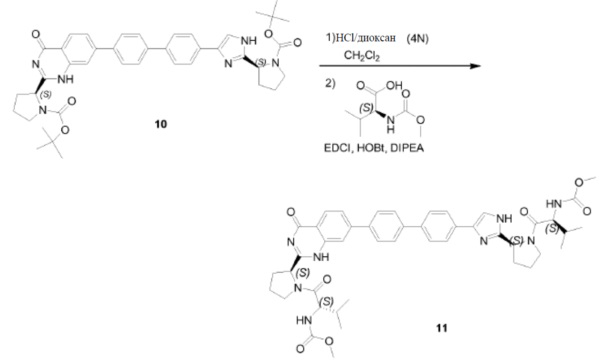
Соединение 10 (0,47 г, 0,67 ммоль) растворяли в CH2Cl2 (5 мл) и каплями добавляли HCl/диоксан (4 н) (0,5 мл, 2 ммоль) при 0°С. Смесь перемешивали при 10°С в течение 1 часов. Растворитель удаляли в вакууме, а полученный остаток отверждали с помощью трет-бутилметилового эфира (2 мл). Твердое вещество фильтровали и сушили при условиях высокого вакуума с получением в результате желтого порошка. Способ A2; Rt: 0,79 минуты, масса/заряд: 503,1 (M+H)+. Точная масса: 502,3. Этот порошок добавляли к раствору, который получали путем обработки (S)-2-(метоксикарбониламино)-3-метилбутановой кислоты (0,28 г, 1,61 ммоль) в ацетонитриле (5 мл) с помощью EDCI (0,31 г, 1,61 ммоль) и HOBt (0,217 г, 1,61 ммоль) при 200C в течение 1 часа. Взвесь охлаждали до 0°C и добавляли DIPEA (0,35 г, 2,7 ммоль). Смесь перемешивали при комнатной температуре в течение 15 часов. Смесь концентрировали и разбавляли с помощью CH2Cl2 (20 мл) и 1 н. водного раствора HCl (5 мл). Органический слой отделяли, промывали насыщенным водным NaHCO3 и солевым раствором и концентрировали в вакууме с получением неочищенного соединения. Неочищенную смесь очищали с помощью препаративной высокоэффективной жидкостной хроматографии (элюент: CH3CN/H2O = 30/70 - 60/40, 0,1% CF3COOH). Желаемую фракцию собирали и регулировали значение pH раствора до приблизительно 8 путем добавления твердого NaHCO3. Избыток ацетонитрила удаляли при пониженном давлении. Водный слой экстрагировали CH2Cl2 (3×50 мл), органические слои объединяли и сушили над Na2SO4. Полученный раствор концентрировали, а остаток далее сушили в вакууме с получением в результате соединения 11 (0,1 г). Способ В; Rt: 5,06 минуты, масса/заряд: 817,3 (M+H)+. Точная масса: 816,4, SFC: колонка: OD-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2. B: MeOH (0,05% диэтиламин); 40% B в A: Rt: 7,5 минуты; SFC: колонка: OD-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2. B: EtOH (0,05% диэтиламин); 40% B в A: Rt: 5,25 минуты.

Na2CO3 (1,7 г, 16 ммоль, 10 экв.) в H2O (10 мл) добавляли к смеси соединения SC-7 (1 г, 1,6 ммоль, 1 экв.), соединения QA-2 (0,73 г, 1,6 ммоль, 1 экв.), толуола (10 мл) и этанола (10 мл). Добавляли Pd(PPh3)4 (0,18 г, 0,16 ммоль, 0,1 экв.). Смесь перемешивали при 100°С в течение 3 часов в атмосфере N2. Добавляли CH2Cl2 (10 мл) и H2O (5 мл). Органический слой отделяли, сушили над Na2SO4 и выпаривали с получением в результате неочищенного остатка (3 г). Часть этого неочищенного материала (0,9 г) очищали с помощью высокоэффективной жидкостной хроматографии (колонка: Grace Vydac 250 * 20 мм * 5 мкм, подвижная фаза A: вода; подвижная фаза B: ацетонитрил; скорость потока: 30 мл/минута; градиент: 35-50% B (объем/объем) от 0 до 11 минут). Чистую фракцию собирали и выпаривали в вакууме с получением в результате соединения 12 (0,1 г). Способ H; Rt: 3,56 минуты, масса/заряд: 865,4 (M+H)+. Точная масса: 864,4; SFC: колонка: OD-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2. B: EtOH (0,05% диэтиламин); 40% B в A; Rt: 4,80 минуты; SFC: колонка: AS-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2. B: MeOH (0,05% диэтиламин); 5-40% B в A: Rt: 9,0 минуты.

Na2CO3 (0,94 г, 8,9 ммоль) в H2O (5 мл) добавляли к смеси соединения SC-5 (0,5 г, 0,89 ммоль) и соединения TD-2 (0,38 г, 0,89 ммоль) в толуоле (5 мл) и этаноле (5 мл). N2 барботировали через раствор, а затем добавляли Pd(PPh3)4 (0,1 г, 0,089 ммоль). Смесь перемешивали при 100°С в течение 3 часов в атмосфере N2. Добавляли CH2Cl2 (20 мл) и H2O (10 мл) и после разделения органический слой сушили над Na2SO4, а растворитель удаляли в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: этилацетат:петролейный эфир от 75% до 100%) с получением в результате соединения 13 (0,5 г). Способ A2; Rt: 1,05 минуты, масса/заряд: 787,4 (M+H)+. Точная масса: 786,3.
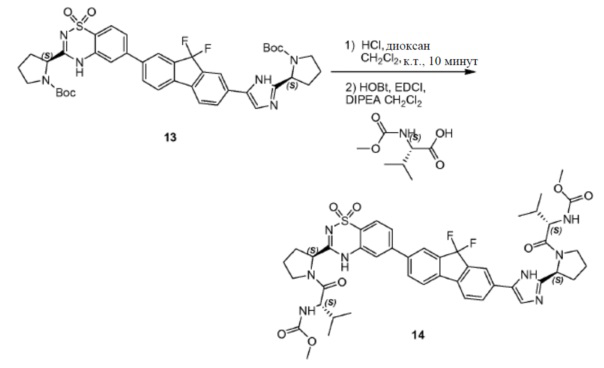
HCl/диоксан (2 мл) добавляли к смеси соединения 13 (0,5 г, 0,64 ммоль, 1 экв.) и CH2Cl2 (5 мл) при 0°С. Смесь перемешивали при 20°С в течение 10 минут. Растворитель удаляли в вакууме. Способ A2; Rt: 0,84 минуты, масса/заряд: 587,1 (M+H)+. Точная масса: 586,2. К полученному остатку в CH2Cl2 (5 мл) добавляли (S)-2-(метоксикарбониламино)-3-метилбутановую кислоту (0,24 г), EDCI (0,27 г, 1,4 ммоль) и HOBt (0,19 г, 1,4 ммоль), смесь охлаждали до 0°С и добавляли DIPEA (1,1 мл, 6,4 ммоль, 10 экв.). Смесь далее перемешивали при 20°С в течение 12 часов. Смесь разводили CH2Cl2 (20 мл) и H2O (5 мл). Органический слой отделяли и промывали насыщенным водным NaHCO3 (5 мл), солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали с помощью высокоэффективной жидкостной хроматографии (колонка: Grace Vydac 250 * 20 мм * 5 мкм; подвижная фаза A: вода (содержащая 0,075% TFA, % объем/объем), подвижная фаза B: ацетонитрил (содержащий 0,025% TFA, % объем/объем; скорость потока: 30 мл/минута; градиент: 35-50% B (объем/объем) от 0 до 11 минут). Собирали соответствующую фракцию и подщелачивали насыщенным раствором NaHCO3 до pH = 8. Летучие вещества удаляли в вакууме. Остаток экстрагировали CH2Cl2 (2×10 мл). Органический слой промывали солевым раствором (10 мл) и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток отделяли с помощью сверхкритической жидкостной хроматографии (Колонка: AS 250 мм × 30 мм, 5 мкм; подвижная фаза: A: сверхкритический CO2, B: MeOH (0,05% диэтиламин, A:B = 60:40 при 50 мл/минута; температура колонки: 38°С; давление в сопле: 100 бар; температура сопла: 60°C; температура выпарного аппарата: 20°С; температура триммера: 25°С; длина волны: 220 нм). Собирали соответствующую фракцию, а растворитель удаляли в вакууме. Остаток растворяли в CH2Cl2 (5 мл) и промывали насыщенным раствором NaHCO3 (2×5 мл). Органический слой сушили над Na2SO4 и выпаривали. Остаток промывали трет-бутилметиловым эфиром (2 мл) и фильтровали. Твердое вещество сушили при условиях высокого вакуума с получением в результате соединения 14 (0,153 г). Способ C; Rt: 3,98 минуты, масса/заряд: 901,3 (M+H)+. Точная масса: 900,3; SFC: колонка: OJ-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2. B: MeOH (0,05% диэтиламин); 5-40% B в A: Rt: 7,44 минуты.
SFC: Колонка: AS-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2. B: EtOH (0,05% диэтиламин); 5-40% B в A: Rt: 8,90 минуты.
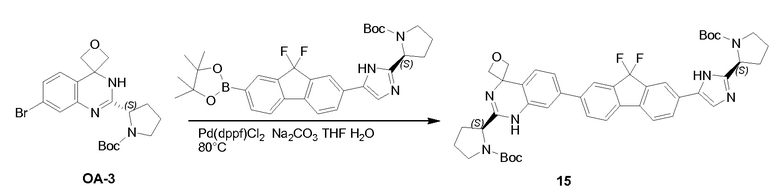
К соединению OA-3 (0,49 г, 1,16 ммоль) добавляли (S)-трет-бутил-2-(5-(9,9-дифтор-7-(4,4,5,5-тетрaметил-1,3,2-диоксаборолан-2-ил)-9H-флуорен-2-ил)-1H-имидазол-2-ил)пирролидин-1-карбоксилат (0,78 г, 1,4 ммоль), Pd(dppf)Cl2 (45 мг, 0,058 ммоль), THF (10 мл) и водный Na2CO3 (2 мл, 2 н). Смесь продували газообразным азотом (3x). Реакционную смесь перемешивали при 80 градусах в течение 15 минут, гасили водой (10 мл) и экстрагировали CH2Cl2 (2×5 мл). Фазы разделяли и органическую фазу промывали солевым раствором и сушили над Na2SO4. После удаления летучих веществ полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: CH2Cl2/метанол = 10/1) с получением в результате соединения 15 (0,49 г). Способ A; Rt: 0,99 минуты, масса/заряд: 779,4 (M+H)+. Точная масса: 778,4.
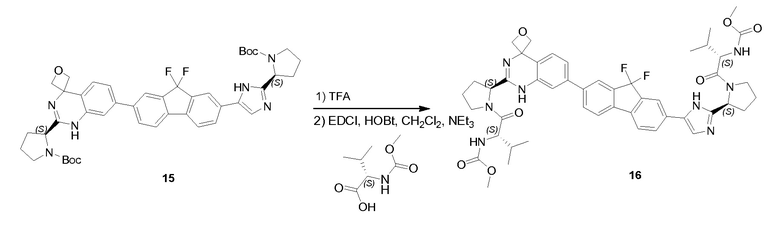
Соединение 15 (0,2 г, 0,28 ммоль) растворяли в CH2Cl2 (5 мл) и медленно добавляли TFA (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут и удаляли летучие вещества с получением в результате остатка (0,19 г). К раствору части полученного остатка (45 мг) в CH2Cl2 (5 мл) добавляли (S)-2-(метоксикарбониламино)-3-метилбутановую кислоту (26 мг, 0,15 ммоль), EDCI (29 мг, 0,15 ммоль), HOBt (8 мг, 0,058 ммоль) и NEt3 (23 мг, 0,23 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Смесь промывали водой (10 мл) и водный слой экстрагировали CH2Cl2 (2×10 мл). Объединенные органические слои сушили над Na2SO4, а фильтрат после фильтрации концентрировали с получением в результате остатка. Полученный остаток очищали с помощью высокоэффективной жидкостной хроматографии (колонка: Phenomenex Synergi C18 150 * 30 мм * 4 мкм; способ: от 30 до 50% B в A в течение 12 минут; A: H2O + 0,1% TFA B: MeCN. скорость потока (25 мл/минута)), к фракциям, содержащим продукт, добавляли Na2CO3 до значения pH 9. Органический растворитель удаляли в вакууме и экстрагировали водный слой с помощью CH2Cl2 (2×20 мл). Отделяли органический слой, сушили над Na2SO4, а после фильтрации растворитель удаляли с получением в результате соединения 16 (11 мг). Способ В; Rt: 4,58 минуты, масса/заряд: 892,4 (M+H)+. Точная масса: 893,3; SFC: колонка: OD-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2. B: EtOH (0,05% диэтиламин); 40% B в A: Rt: 6,17 минуты.

Соединение QO-3 (0,2 г, 0,44 ммоль), соединение SC-9 (0,25 г, 0,49 ммоль), Pd(PPh3)4 (0,10 г, 0,088 ммоль) и Na2CO3 (0,21 г, 1,98 ммоль) в THF (8 мл) и H2O (2,4 мл) перемешивали при 80°С при микроволновом излучении в течение 5 минут. Растворитель удаляли в вакууме, а полученный остаток растворяли в CHCl3 и фильтровали. Фильтрат концентрировали и очищали с помощью препаративной TLC (элюент: этилацетат/метанол, 10:1) с получением в результате соединения 17 (0,25 г).
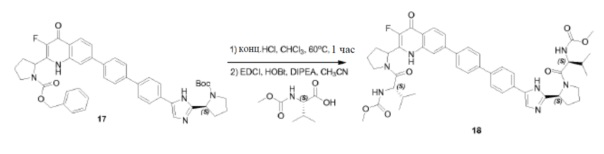
Соединение 17 (0,24 г, 0,32 ммоль) в CHCl3 (10 мл) и концентрированной HCl (10 мл) перемешивали при 60°С в закупоренной пробирке в течение 2 часов. Водный слой отделяли и концентрировали в вакууме. Остаток (0,2 г) совместно выпаривали с толуолом и THF и добавляли к раствору, который был образован путем добавления EDCI (0,25 г, 1,32 ммоль) и HOBt (0,18 г, 1,32 ммоль) к раствору (S)-2-(метоксикарбониламино)-3-метилбутановой кислоты (0,23 г, 1,32 ммоль) в CH3CN (5 мл) и перемешивания при 10°С в течение 1 часа. Затем смесь охлаждали до 0°С и добавляли DIPEA (1 мл, 5,6 ммоль). Смесь перемешивали при 10°С в течение ночи. Растворитель удаляли в вакууме и полученное соединение 18 (смесь диастереоизомеров 18a и 18b) очищали с помощью препаративной HPLC (C18, элюент: CH3CN, H2O, TFA, 40:60:0,05). Получали две фракции. Фракции нейтрализовали насыщенным NaHCO3. Органический растворитель удаляли в вакууме. Полученный в результате осадок фильтровали и сушили при условиях высокого вакуума с получением в результате 18a (33 мг; e.e. 99%) и 18b (33 мг; e.e. 90%). Соединение 18b очищали с помощью SFC. Колонка: OD 250 мм * 30 мм, 5 мкм; подвижная фаза: A: сверхкритический CO2, B: EtOH; 0,05% диэтиламин; A:B = 60:40 при 50 мл/минута; температура колонки: 38°С; давление в сопле: 100 бар; температура сопла: 60°С; температура выпарного аппарата: 20°С; температура триммера: 25°С; длина волны: 220 нм). Собранные фракции объединяли и концентрировали в вакууме. Остаток промывали насыщенным NaHCO3 и сушили при условиях высокого вакуума. В результате получали соединение 18b. (26 мг, e.e. 99%)
18a; способ B; Rt: 4,75 минуты, масса/заряд: 833,4 (M+H)+. Точная масса: 834,6; SFC: колонка: OD-3 150 мм × 4,6 мм; 3 мкм. Поток: 2,5 мл/минута, подвижная фаза: A: CO2. B: MeOH (0,05% диэтиламин); 40% B в A: Rt: 6,08 минуты.
18b; способ B; Rt: 4,87 минуты, масса/заряд: 833,4 (M+H)+. Точная масса: 834,5; SFC: колонка: OD-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2. B: EtOH (0,05% диэтиламин); 40% B в A; Rt: 4,25 минуты.
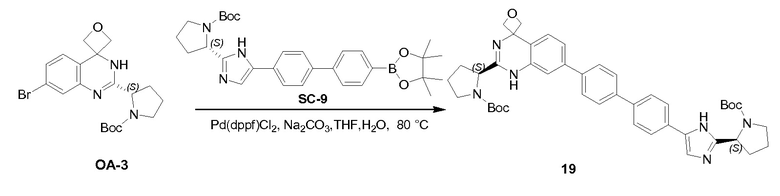
Смесь соединения OA-3 (0,46 г, 1,09 ммоль), SC-9 (0,67 г, 1,3 ммоль), Pd(dppf)Cl2 (45 мг, 0,058 ммоль), THF (10 мл) и водного Na2CO3 (2 мл, 2н) три раза продували газообразным азотом. Реакционную смесь перемешивали при 80 градусах в течение 15 минут. Смесь гасили водой (10 мл) и экстрагировали CH2Cl2 (2×5 мл). Фазы разделяли и органическую фазу промывали солевым раствором и сушили над Na2SO4. После удаления летучих веществ полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: CH2Cl2/метанол = 10/1) с получением в результате соединения 19 (0,42 г).
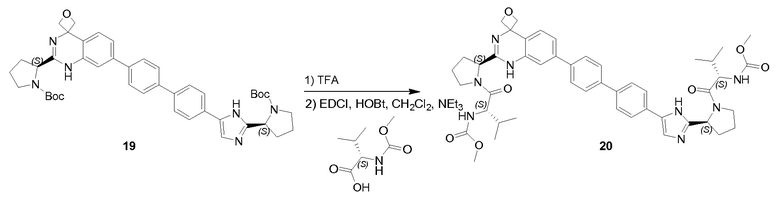
Соединение 19 (0,2 г, 0,28 ммоль) растворяли в CH2Cl2 (5 мл) и медленно добавляли TFA (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут и смесь концентрировали с получением в результате остатка (0,19 г). К части этого остатка (110 мг) в CH2CL2 (5 мл) добавляли (S)-2-(метоксикарбониламино)-3-метилбутановую кислоту (105 мг, 0,60 ммоль), EDCI (114 мг, 0,60 ммоль), HOBt (13,5 мг, 0,1 ммоль) и NEt3 (60 мг, 0,6 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Смесь промывали водой (10 мл) и экстрагировали CH2Cl2 (2×10 мл). Объединенные органические слои сушили и концентрировали с получением в результате остатка, который очищали с помощью высокоэффективной жидкостной хроматографии (MeCN/H2O (колонка: Diamonsil C18 150 * 20 мм * 5 мкм. Способ: от 20 до 40% B в A в течение 14 минут. A: H2O + 0,1% TFA, B: MeCN. скорость потока (40 мл/минута))). К фракциям, содержащим продукт, добавляли Na2CO3 до значения pH 9, органический растворитель удаляли, а водный слой промывали CH2Cl2 (2×20 мл). Органический слой отделяли и концентрировали досуха с получением в результате соединения 20 (40 мг). Способ C; Rt: 3,48 минуты, масса/заряд: 845,5 (M+H)+. Точная масса: 844,4; SFC: колонка: OD-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,35 мл/минута, подвижная фаза: A: CO2. B: MeOH (0,05% диэтиламин); 40% B в A: Rt: 8,48 минуты.

Na2CO3 (0,4 г, 3,8 ммоль, 2 экв.) в H2O (10 мл) добавляли к смеси соединения SC-9 (1 г, 1,9 ммоль), соединения TD-1 (0,9 г, 1,9 ммоль), этанола (10 мл) и толуола (20 мл). Добавляли Pd(PPh3)4 (0,11 г, 0,095 ммоль). Смесь перемешивали при 90°С в течение 10 часов при защите N2. Органический растворитель удаляли в вакууме. Остаток экстрагировали CH2Cl2 (10 мл). Органический слой промывали солевым раствором (5 мл) и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали с помощью колоночной флэш-хроматографии (элюент: CH2Cl2/метанол=10:1). Растворитель выпаривали с получением в результате соединения 21 (1,7 г).

Соединение 21 (1,7 г, 1,1 ммоль) растворяли в CH3COOH (20 мл). Добавляли 40% HBr в H2O (10 мл). Смесь перемешивали при 50°С в течение 3 часов. Растворитель выпаривали в вакууме. Остаток промывали смесью трет-бутилметилового эфира и метанола (1:1). Твердое вещество фильтровали и сушили при условиях высокого вакуума с получением в результате остатка (1,7 г). Часть этого остатка (0,7 г) добавляли в предварительно полученный раствор, образованный путем добавления EDCI (0,46 г, 2,4 ммоль) и HOBt (0,32 г, 2,4 ммоль) к (S)-2-(метоксикарбониламино)-3-метилбутановой кислоте (0,42 г, 2,4 ммоль) в ацетонитриле (14 мл) и перемешивания при 10°С в течение 1 часа. Взвесь охлаждали до 0°С и добавляли DIPEA (1 г, 8 ммоль). Смесь перемешивали при 10°С в течение 12 часов. Твердое вещество фильтровали. Фильтрат концентрировали и разбавляли CH2Cl2 (20 мл) и 1 н. HCl (5 мл). Органический слой отделяли и промывали насыщенным водным NaHCO3 (5 мл), солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали с помощью высокоэффективной жидкостной хроматографии; колонка: Grace Vydac 250 * 20 мм * 5 мкм; подвижная фаза A: вода, содержащая 0,075% TFA, % объем/объем, подвижная фаза B: ацетонитрил (содержащий 0,025% TFA, % объем/объем; скорость потока: 30 мл/минута; градиент: 35-50% B (объем/объем) от 0 до 11 минут). Чистые фракции собирали и подщелачивали с помощью NaHCO3 до pH=8. Летучие вещества удаляли в вакууме. Остаток экстрагировали CH2Cl2 (2×15 мл). Органический слой промывали солевым раствором (10 мл) и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток отделяли с помощью сверхкритической жидкостной хроматографии (колонка: AS 250 мм × 30 мм, 5 мкм; подвижная фаза: A: сверхкритический CO2, B: MeOH (0,05% диэтиламин), A:B = 60:40 при 50 мл/минута; температура колонки: 38°С; давление в сопле: 100 бар; температура сопла: 60°С;
температура выпарного аппарата: 20°С; температура триммера: 25°С; длина волны: 220 нм). Фракции собирали, а растворитель удаляли в вакууме. Остаток растворяли в CH2Cl2 (5 мл) и промывали насыщенным раствором NaHCO3 (5 мл) и солевым раствором (5 мл). Органический слой сушили над Na2SO4 и выпаривали с получением в результате соединения 22 (98 мг); способ B; Rt: 4,94 минуты, масса/заряд: 853,3 (M+H)+. Точная масса: 852,4; SFC: колонка: AS-H 250 мм × 4,6 мм; 5 мкм. Поток: 2,5 мл/минута, подвижная фаза: A: CO2. B: MeOH (0,05% диэтиламин); 40% B в A: Rt: 4,53 минуты.
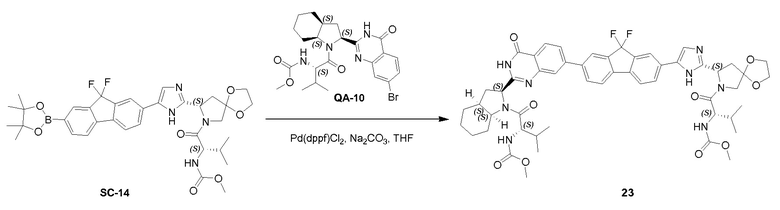
К перемешиваемому раствору SC-14 (800 мг, 1,18 ммоль), QA-10 (713 мг, 1,42 ммоль) и Pd(dppf)Cl2 (100 мг) в сухом THF (10 мл) добавляли Na2CO3 (5 мл, 2 н водн.). Реакционную смесь перемешивали при нагревании с обратным холодильником в предварительно нагретой масляной бане при 90°С в течение 20 минут. Смесь далее гасили водой (20 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Полученный остаток очищали с помощью высокоэффективной жидкостной хроматографии (колонка: Phenomenex Synergi C18 150 * 20 мм * 5 мкм. Способ: способ: 34-64% B в A: H2O + 0,1% TFA, B: MeCN. Скорость потока (мл/минута): 25). Чистые фракции собирали и нейтрализовали насыщенным NaHCO3. Смесь концентрировали в вакууме. Полученный продукт далее очищали с помощью сверхкритической жидкостной хроматографии (колонка: Chiralpak OD-3 50 * 4,6 мм, в.д. 3 мкм, подвижная фаза: A: метанол (0.05% диэтиламин), B: CO2, A/B=40/60, скорость потока: 2,5 мл/минута, длина волны: 220 нм). Чистые фракции собирали, а летучие вещества удаляли в вакууме. Полученный остаток растворяли в дихлорметане (20 мл). Органический слой промывали насыщенным водным NaHCO3 (10 мл) и сушили над Na2SO4. Растворитель удаляли в вакууме с получением в результате соединения 23 (247 г). Способ В; Rt: 5,84 минуты, масса/заряд: 977,7 (M+H)+. Точная масса: 976,4; 1H ЯМР (400 MГц, DMSO-d6) δ м.д. 12,29-12,54 (1 H, м), 12,03 (1 H, ушир.с.), 8,12-8,21 (1 H, м), 8,01-8,11 (2 H, м), 7,91-8,01 (3 H, м), 7,79-7,91 (2 H, м), 7,62-7,76 (2 H, м), 7,54 (1 H, d, J=7,3 Гц), 7,32 (1 H, d, J=8,3 Гц), 5,01-5,15 (1 H, м), 4,67-4,81 (1 H, м), 4,37-4,52 (1 H, м), 3,81-4,13 (7 H, м), 3,70-3,80 (1 H, м), 3,54 (6 H, ушир.с), 2,31-2,46 (3 H, м), 2,16-2,29 (1 H, м), 1,82-2,16 (5 H, м), 1,56-1,82 (3 H, м), 1,37-1,51 (1 H, м), 1,16-1,36 (2 H, м), 0,71-0,99 (12 H, м).
Биологические примеры – активность соединений формулы I против HCV
Анализ репликона
Соединения формулы (I) проверяли по ингибиторной активности по отношению к репликону HCV. Этот клеточный анализ основан на конструкте бицистронной экспрессии, как описано Lohmann et al. (Science (1999) 285: 110-113; Journal of Virology (2003) 77: 3007-3019), с модификациями, описанными Krieger et al. (Journal of Virology (2001) 75: 4614-4624), и Lohmann et al. (Journal of Virology (2003) 77: 3007-3019) для генотипа 1b, и Yi et al. (Journal of Virology (2004) 78: 7904-7915) для генотипа 1a в многоцелевой скрининговой стратегии.
Стабильная трансфекция
Способ заключался в следующем. Для анализа использовали стабильно трансфицированную клеточную линию Huh-7 luc/neo (далее называемую Huh-Luc). Эта клеточная линия содержит РНК, кодирующую бицистронный конструкт экспрессии, содержащий участки NS3-NS5B дикого типа HCV типа 1b, транслируемые на сайте внутренней посадки рибосомы (IRES) у вируса энцефаломиокардита (EMCV), с предшествующими репортерной частью (FfL-люцифераза) и селективной маркерной частью (neoR, неомицин-фосфотрансфераза). К конструкции примыкают 5' и 3' NTR (нетранслируемые участки) типа HCV 1b. Непрерывная культура клеток с репликонами в присутствии G418 (neoR) зависит от репликации РНК HCV. Для скрининга противовирусных соединений применяли стабильно трансфицированные клетки с репликонами, которые автономно и интенсивно реплицируют РНК HCV, кодирующую inter alia люциферазу.
Клетки с репликонами высевали в 384-луночные планшеты в присутствии тестируемых и контрольных соединений, которые добавляли при различных концентрациях. После инкубации в течение трех дней измеряли репликацию HCV путем анализирования люциферазной активности (с использованием стандартных люциферазных аналитических субстратов и реагентов, а также устройства для визуализации микропланшетов Perkin Elmer ViewLuxTM ultraHTS). Клетки с репликонами в контрольных культурах при отсутствии какого то ни было ингибитора отличались высокой экспрессией люциферазы. Осуществляли мониторинг ингибиторной активности соединения на клетках Huh-Luc, получая кривую зависимости доза-эффект для каждого тестируемого соединения. Затем рассчитывали значения EC50, которые соответствовали количеству соединения, необходимому для уменьшения уровня выявленной активности люциферазы на 50%, или, более конкретно, для снижения способности генетически связанной РНК репликона HCV к репликации.
Результаты
Для случаев, когда соединение формулы (I) при анализе репликонов тестировали белее чем один раз, среднее всех результатов тестирования приведено в таблице 1.

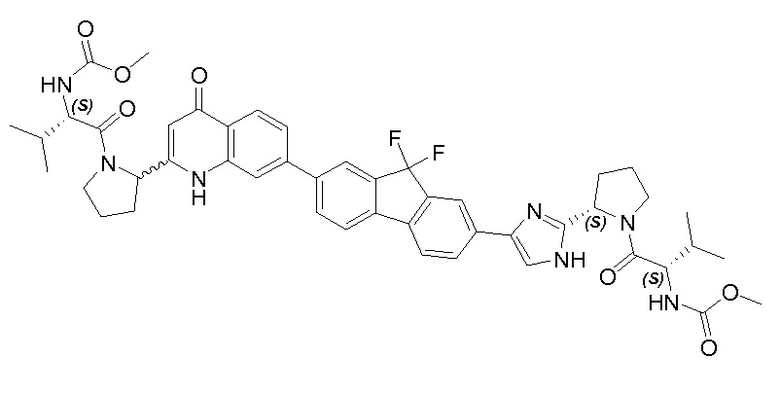
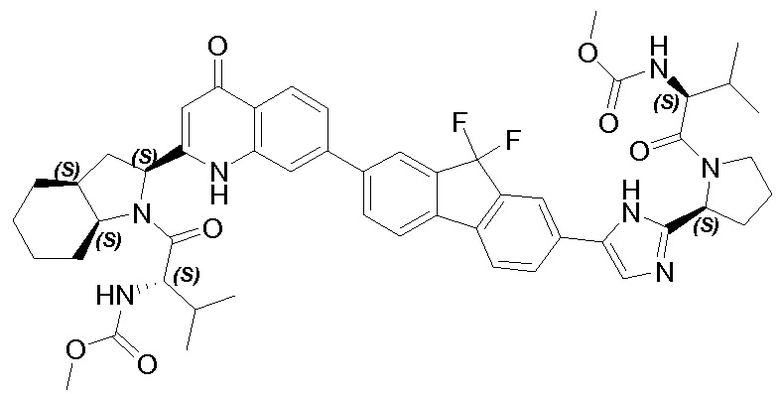
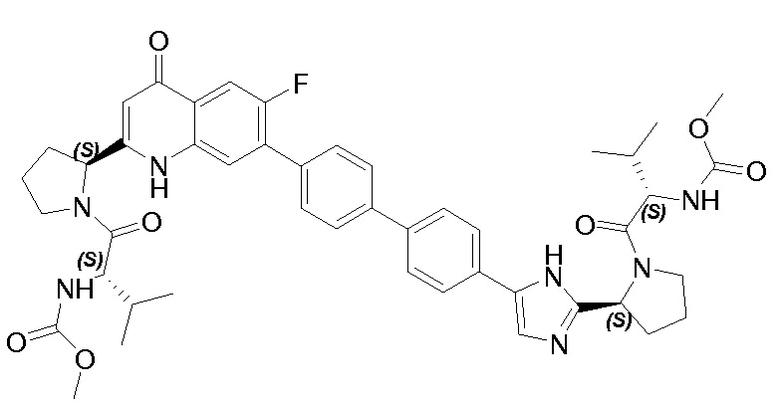
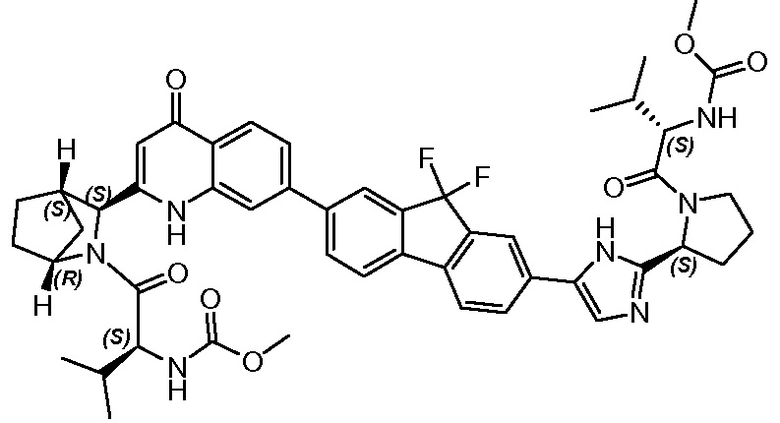

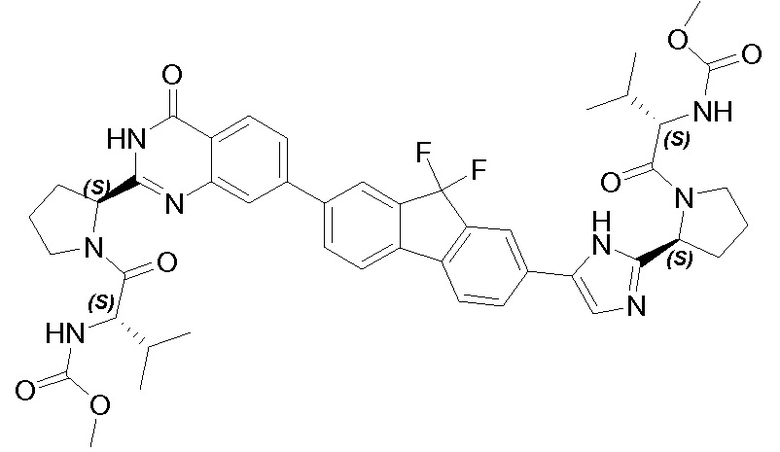
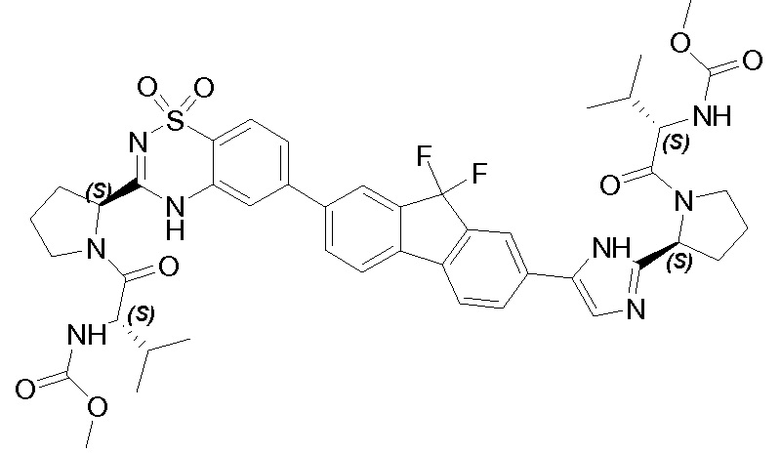
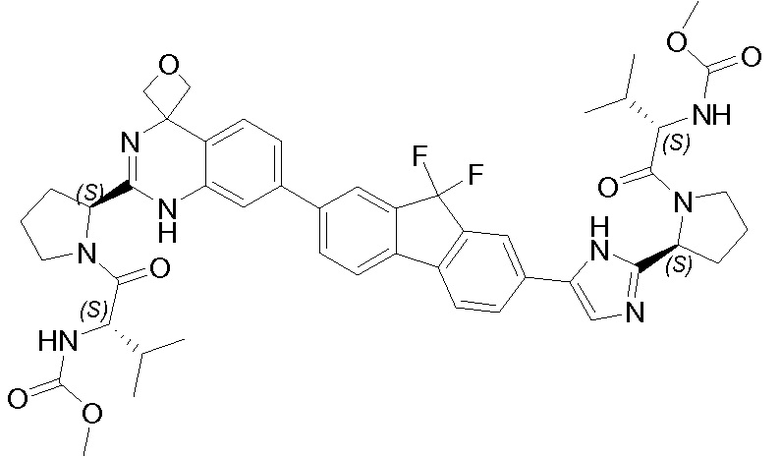
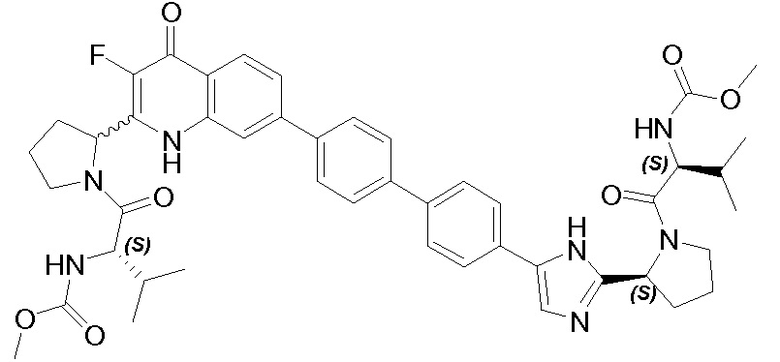

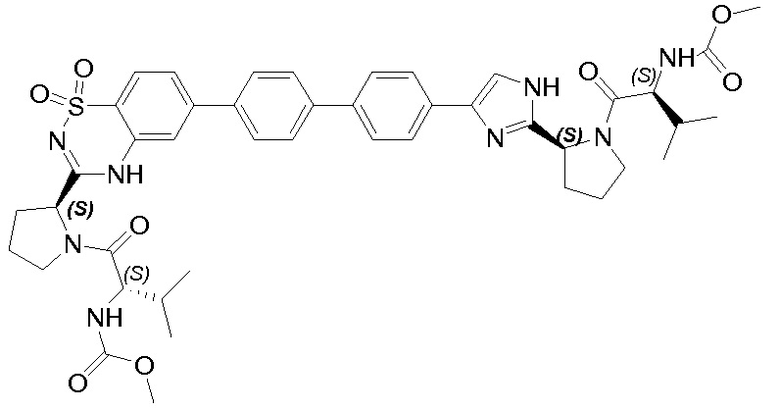
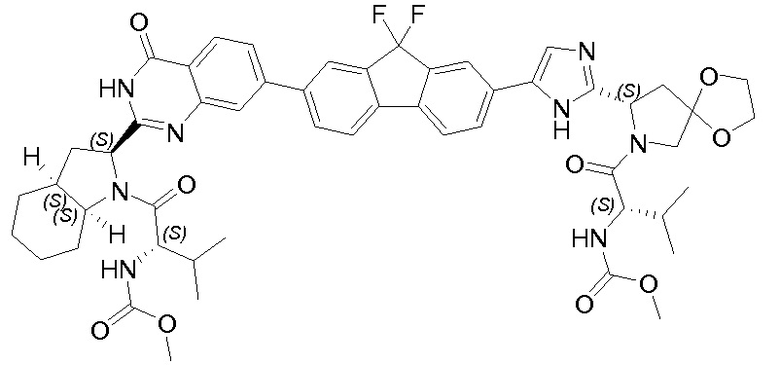
Транзиентная трансфекция
При транзиентной схеме линию гепатомных клеток Huh-7 Lunet транзиентно трансфицировали автономно реплицирующейся РНК, кодирующей бицистронный конструкт экспрессии. Этот конструкт содержит репортерный ген люциферазы светлячка, предшествующий субгеномному участку NS3-NS5B HCV (генотипа 1a H77 или 1b Con1). Трансляция субгеномного участка HCV опосредуется сайтом внутренней посадки рибосомы вируса энцефаломиокардита. Более того, конструкт фланкируется 5'- и 3'-нетранслируемыми участками HCV (генотипа 1a H77 или 1b Con 1, соответственно), которые обеспечивают репликацию РНК.
Клетки высевали в 384-луночные планшеты в присутствии тестируемых и контрольных соединений, которые добавляли при различных концентрациях. После инкубации в течение двух дней измеряли репликацию РНК субгеномного репликона HCV путем анализирования люциферазной активности (с использованием стандартных люциферазных аналитических субстратов и реагентов, а также устройства для визуализации микропланшетов Perkin Elmer ViewLuxTM ultraHTS). Клетки, содержащие субгеномный репликон HCV, в контрольных культурах при отсутствии какого то ни было ингибитора отличались высокой экспрессией люциферазы. Осуществляли мониторинг ингибиторной активности соединения, получая кривую зависимости доза-эффект для каждого тестируемого соединения. Затем рассчитывали значения EC50, которые соответствовали количеству соединения, необходимому для уменьшения уровня выявленной активности люциферазы на 50%, или, более конкретно, для снижения способности генетически связанной субгеномной РНК HCV к репликации.
Обратные скрининги
Обратный скрининг клеточных линий предусматривал линию гепатомных клеток Huh-7, содержащую конструкт с главным предранним промотором цитомегаловируса человека и Luc (Huh7-CMV-Luc), и линию Т-клеток MT4, содержащую Luc-репортер с длинным концевым повтором (MT4-LTR-Luc).
соединения
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА И ПИПЕРИДИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1997 |
|
RU2178414C2 |
| СОЕДИНЕНИЯ | 2016 |
|
RU2725616C2 |
| Ингибиторы ErbB/BTK | 2019 |
|
RU2764069C1 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ПЛАТИНЫ И ЛИПИДОВ И НАНОЧАСТИЦЫ | 2014 |
|
RU2737735C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ПЛАТИНЫ И ЛИПИДОВ И НАНОЧАСТИЦЫ | 2014 |
|
RU2679896C2 |
| СПИРОЦИКЛИЧЕСКИЕ АМИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ S1P | 2011 |
|
RU2602800C2 |
| PDE10 ИНГИБИТОРЫ И СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ | 2011 |
|
RU2545456C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ИНТЕГРАЗЫ | 2011 |
|
RU2567385C2 |
| ПРОИЗВОДНЫЕ СОЕДИНЕНИЙ, СОДЕРЖАЩИХ ПИРИДИНОВЫЕ КОЛЬЦА, В КАЧЕСТВЕ ИНГИБИТОРОВ MALT1 | 2020 |
|
RU2827961C2 |
Настоящее изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы (I) или к его фармацевтически приемлемой соли, где Y представляет собой СН, N или CR4; W представляет собой карбонил, сульфонил или CR5R6;  представляет собой
представляет собой 
или 
 независимо выбран из группы, содержащей
независимо выбран из группы, содержащей  R и R' независимо выбраны из -CR1R2R3, где R1 выбран из С1-4алкила; R2 представляет собой С1-4алкилоксикарбониламино; R3 представляет собой водород; R4 представляет собой водород или фтор; CR5R6 вместе образуют оксетан. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I), его применению и продукту на его основе. Технический результат: получены новые гетероциклические соединения, полезные при лечении инфекции HCV. 7 н. и 7 з.п. ф-лы, 2 табл.
R и R' независимо выбраны из -CR1R2R3, где R1 выбран из С1-4алкила; R2 представляет собой С1-4алкилоксикарбониламино; R3 представляет собой водород; R4 представляет собой водород или фтор; CR5R6 вместе образуют оксетан. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I), его применению и продукту на его основе. Технический результат: получены новые гетероциклические соединения, полезные при лечении инфекции HCV. 7 н. и 7 з.п. ф-лы, 2 табл.

1. Соединение формулы I, или его стереоизомер
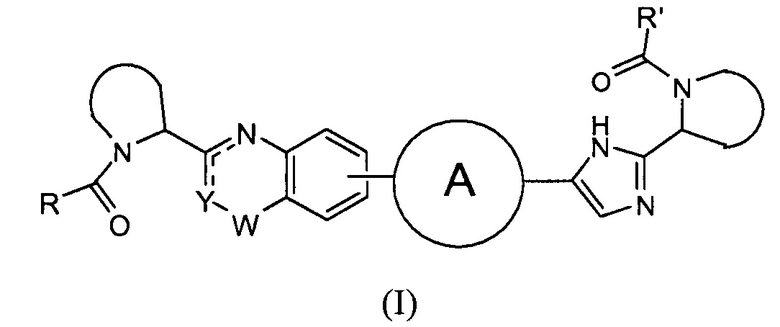
(I),
где
Y представляет собой СН, N или CR4;
W представляет собой карбонил, сульфонил или CR5R6;
 представляет собой
представляет собой  или
или 
 независимо выбран из группы, содержащей
независимо выбран из группы, содержащей

R и R' независимо выбраны из -CR1R2R3, где
R1 выбран из С1-4алкила;
R2 представляет собой С1-4алкилоксикарбониламино;
R3 представляет собой водород;
R4 представляет собой водород или фтор;
CR5R6 вместе образуют оксетан;
или его фармацевтически приемлемые соли.
2. Соединение по п.1, которое представлено формулой Ia,
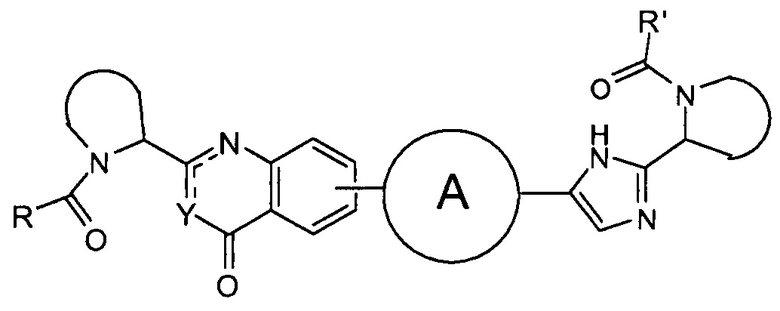
3. Соединение по п.1 или 2, которое представлено формулой Ib,
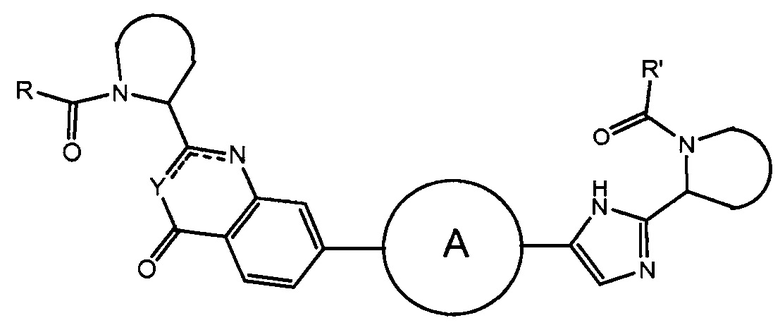
4. Соединение по п.1, которое представлено формулой Ic,

5. Соединение формулы I, Ia, Ib или Ic по п.1, где каждый
 независимо выбран из группы, содержащей
независимо выбран из группы, содержащей
6. Соединение по п.1, где по меньшей мере один
 представляет собой
представляет собой  .
.
7. Соединение по п.1, где R1 выбран из разветвленного С3-4алкила.
8. Соединение по любому из пп.4-7, которое представлено формулой Id,
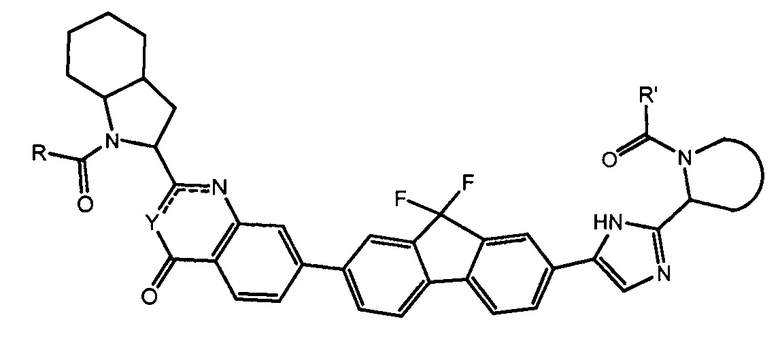
9. Фармацевтическая композиция для предупреждения или лечения у млекопитающего инфекции HCV, содержащая соединение по любому из пп.1-8 в эффективном количестве и фармацевтически приемлемый носитель.
10. Применение соединения по любому из пп.1-8 в качестве лекарственного препарата для предупреждения или лечения у млекопитающего инфекции HCV.
11. Применение фармацевтической композиции по п.9 в качестве лекарственного средства для предупреждения или лечения у млекопитающего инфекции HCV.
12. Применение соединения по любому из пп.1-8 при предупреждении или лечении у млекопитающего инфекции HCV.
13. Применение фармацевтической композиции по п.9 при предупреждении или лечении у млекопитающего инфекции HCV.
14. Продукт, содержащий (а) соединение по любому из пп.1-8 и (b) другой ингибитор HCV, в виде комбинированного препарата для одновременного, раздельного или последовательного применения при лечении инфекций HCV.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Прибор для проверки плотности постановки вставных втулок цилиндрических золотниковых коробок | 1928 |
|
SU15756A1 |

