ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым соединениям, которые являются ингибиторами MALT1 (транслокационного белка 1 лимфомы лимфоидной ткани слизистых оболочек). Такие соединения могут быть полезными для лечения заболевания, синдрома, состояния или нарушения, в частности, связанного с MALT1 заболевания, синдрома, состояния или нарушения, в том числе без ограничения рака и иммунологических заболеваний. Настоящее изобретение также относится к фармацевтическим композициям, содержащим одно или несколько таких соединений, к способам получения таких соединений и композиций, и к применению таких соединений или фармацевтических композиций для лечения рака и аутоиммунологических заболеваний, синдромов, нарушений или состояний, ассоциированных с ингибиторами MALT1.
ПРЕДПОСЫЛКИ К СОЗДАНИЮ ИЗОБРЕТЕНИЯ
MALT1 (транслокационный белок 1 лимфомы лимфоидной ткани слизистых оболочек) является ключевым медиатором классического сигнального пути NFΚB. MALT1 является единственной паракаспазой человека и передает сигналы от B-клеточного рецептора (BCR) и T-клеточного рецептора (TCR). MALT1 является активной субъединицей комплекса CBM, который образуется в результате активации рецепторов. Комплекс CBM состоит из нескольких субъединиц из трех белков: CARD11 (члена 11 семейства белков, содержащих домен рекрутирования каспаз), BCL10 (белка B-клеточной CLL/лимфомы 10) и MALT1. MALT1 оказывает воздействие на пути передачи сигнала NFΚB посредством двух механизмов: во-первых, MALT1 выполняет функцию поддерживающего белка и рекрутирует сигнальные белки NFΚB, такие как TRAF6, TAB-TAK1 или NEMO-IKKα/β; а, во-вторых, MALT1, в качестве цистеиновой протеазы, расщепляет и за счет этого деактивирует отрицательные регуляторы передачи сигнала NFΚB, такие как RelB, A20 или CYLD. Основная конечная точка активности MALT1 заключается в транслокации комплекса транскрипционного фактора NFΚB в ядро и активации передачи сигнала NFΚB (Jaworski et al., Cell Mol Life Science 2016. 73, 459-473).
Конститутивная активация передачи сигнала NFΚB представляет собой отличительный признак ABC-DLBCL (подтипа диффузной В-крупноклеточной лимфомы из активированных В-клеток), более агрессивной формы DLBCL. DLBCL является наиболее распространенной формой неходжкинской лимфомы (NHL), на долю которой приходится примерно 25% случаев лимфомы, тогда как ABC-DLBCL составляет примерно 40% DLBCL. Активация пути NFΚB обуславливается мутациями компонентов передачи сигнала, таких как CD79A/B, CARD11, MYD88 или A20, у пациентов с ABC-DLBCL (Staudt, Cold Spring Harb. Perspect. Biol., 2010, 2; Lim et al, Immunol. Rev., 2012, 246, 359-378).
Применение ингибиторов BTK, например, ибрутиниба, обеспечивает клиническое подтверждение правильности концепции, заключающейся в том, что ингибирование передачи сигнала NFΚB при ABC-DLBCL является эффективным. MALT1 следует после BTK в пути передачи сигнала NFΚB, и ингибитор MALT1 может обеспечивать целенаправленное воздействие для пациентов с ABC-DLBCL, не восприимчивых к ибрутинибу, главным образом пациентов с мутациями CARD11, а также обеспечивать лечение пациентов, у которых наблюдается приобретенная резистентность к ибрутинибу.
Ингибиторы протеазы MALT1 на основе низкомолекулярных фармакологически активных соединений показали эффективность в доклинических моделях ABC-DLBCL (Fontan et al., Cancer Cell, 2012, 22, 812-824; Nagel et al., Cancer Cell, 2012, 22, 825-837). Примечательно, что были описаны ковалентный каталитический сайт и аллостерические ингибиторы функционирования протеазы MALT1, что указывает на то, что ингибиторы этой протеазы могут быть полезными в качестве фармацевтических средств (Demeyer et al., Trends Mol Med 2016, 22, 135-150).
Хромосомная транслокация, приводящая к возникновению слитого онкобелка API2-MALT1, является наиболее распространенной мутацией, определяемой при лимфоме MALT (лимфоидной ткани слизистых оболочек). API2-MALT1 является эффективным активатором пути NFΚB (Rosebeck et al., World J. Biol. Chem., 2016, 7, 128-137). API2-MALT1 имитирует лигандсвязанный рецептор TNF, способствует зависимому от TRAF2 убиквитинированию RIP1, что выступает в качестве поддерживающей структуры для активации канонической передачи сигнала NFΚB. Кроме того, как было показано, API2-MALT1 расщепляет и образует стабильный, конститутивно активный фрагмент индуцирующей NFΚB киназы (NIK) и тем самым активирует неканонический путь NFΚB (Rosebeck et al., Science, 2011, 331, 468-472).
Было показано что, помимо лимфом, MALT1 играет важную роль во врожденном и приобретенном иммунитете (Jaworski M, et al., Cell Mol. Life Sci., 2016). Ингибитор протеазы MALT1 может смягчать начало и прогрессирование заболевания при экспериментальном аллергическом энцефаломиелите у мышей, мышиной модели рассеянного склероза (Mc Guire et al., J. Neuroinflammation, 2014, 11, 124). Мыши, экспрессирующие каталитически неактивный мутантный вариант MALT1, продемонстрировали потерю B-клеток маргинальной зоны и B-клеток B1 и общий иммунодефицит, характеризующийся сниженной активацией и пролиферацией T- и B-клеток. Однако у этих мышей также развилось спонтанное аутоиммунное воспаление многих органов в возрасте от 9 до 10 недель. Все еще малопонятно, почему у мышей с нокин-мутацией по летальному аллелю протеазы MALT1 наблюдается утрата толерантности, тогда как у обычных мышей с нокаутом по MALT1 этого не происходит. В соответствии с одной из гипотез предполагается, что разбалансированность иммунного гомеостаза у мышей с нокин-мутацией по летальному аллелю протеазы MALT1 может являться результатом неполного дефицита T- и B-клеток при существенном дефиците иммунорегуляторных клеток (Jaworski et al., EMBO J., 2014; Gewies et al., Cell Reports, 2014; Bornancin et al., J. Immunology, 2015; Yu et al., PLOS One, 2015). Подобным образом, у людей дефицит MALT был связан с комбинированным иммунодефицитом (McKinnon et al., J. Allergy Clin. Immunol., 2014, 133, 1458-1462; Jabara et al., J. Allergy Clin. Immunol., 2013, 132, 151-158; Punwani et al., J. Clin. Immunol., 2015, 35, 135-146). С учетом разницы между генетической мутацией и фармакологическим ингибированием, фенотип, характерный для мышей с нокин-мутацией по летальному аллелю протеазы MALT1, может не иметь сходство с таковым у пациентов, которых лечат ингибиторами протеазы MALT1. Уменьшение количества иммуносупрессивных T-клеток путем подавления протеазы MALT1 может быть полезным для пациентов с раком за счет возможного повышения противоопухолевого иммунитета.
Таким образом, ингибиторы MALT1 по настоящему изобретению могут обеспечивать терапевтическую пользу для пациентов, страдающих раком и/или иммунологическими заболеваниями.
В WO 2018020474 описаны замещенные тиазолопиридиновые соединения в качестве ингибиторов MALT1.
В WO 2015181747 описаны производные пиразолопиримидина и их применение в качестве ингибиторов MALT1.
В WO 2017081641 описаны производные пиразолопиримидина.
В WO 2018226150 описаны пиразолопиримидиновые соединения в качестве ингибиторов MALT1.
В WO 2018119036 описаны производные пиразола в качестве ингибиторов MALT1.
В WO 2019243964 описаны производные пиразола в качестве ингибиторов MALT1.
В WO 2019243965 описаны производные пиразола в качестве ингибиторов MALT1.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение направлено на соединения формулы (I),
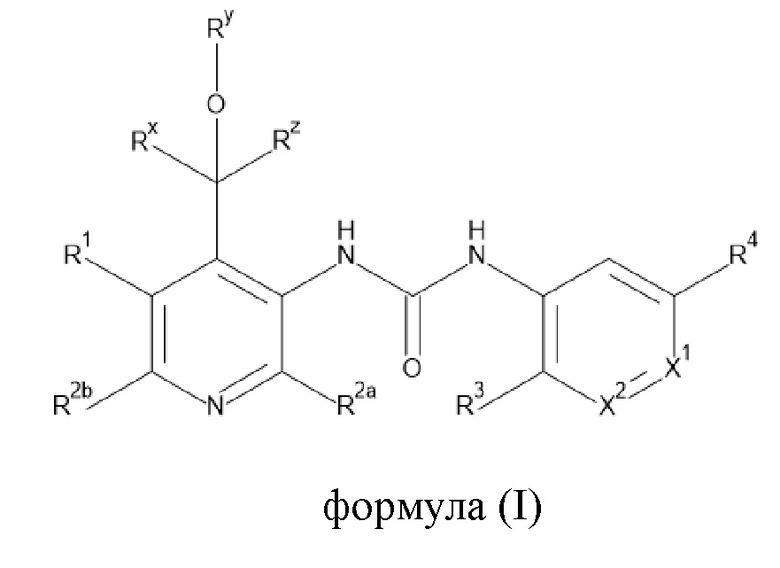
где
Rx представляет собой водород, C1-4алкил или C3-6циклоалкил;
Ry представляет собой водород, C1-4алкил или C3-6циклоалкил; и
Rz представляет собой водород;
или
Rx и Ry взяты вместе с образованием двухвалентного радикала -Rx-Ry-, где -Rx-Ry- представляет собой
-(CH2)n- или -CH2-O-(CH2)2-; где n представляет собой 2, 3, 4 или 5; и
Rz представляет собой водород;
или
Ry представляет собой водород, C1-4алкил или C3-6циклоалкил;
Rx и Rz взяты вместе с атомом углерода, к которому они присоединены, с образованием C3-6циклоалкила;
R1 выбран из группы, состоящей из водорода, -OR5, C1-4алкила, C2-4алкенила, галогена,
-CN, C3-6циклоалкила, Heta, -C(=O)-OH, -C(=O)-O-C1-4алкила, -NR6aR7a и
-C(=O)-NR6bR7b;
каждый из R2a и R2b независимо выбран из группы, состоящей из водорода,
-O-C1-4алкила, галогена, -NR6cR7c, C3-6циклоалкила, C1-4алкила и C1-4алкила, замещенного 1, 2 или 3 атомами галогена;
X1 представляет собой N или CRa;
X2 представляет собой N или CRb;
при условии, что только один из X1 и X2 представляет собой N в любом случае;
R3 представляет собой водород, C1-4алкил или -O-C1-4алкил;
R4 представляет собой галоген, циано или трифторметил;
R5 выбран из группы, состоящей из водорода, C1-4алкила, C3-6циклоалкила, Hetb и C1-4алкила, замещенного одним или двумя заместителями, каждый из которых независимо выбран из группы, состоящей из -OH, галогена, -C(=O)-NR8R9, -C(=O)-OH, -C(=O)-O-C1-4алкила, C3-6циклоалкила и фенила;
каждый из R6a, R6b, R6c, R7a, R7b, R7c, R8 и R9 независимо выбран из группы, состоящей из водорода и C1-4алкила;
Heta представляет собой моноциклический 4-7-членный неароматический гетероциклил, содержащий один или два гетероатома, выбранные из азота, кислорода и серы;
Hetb представляет собой моноциклический 4-7-членный неароматический гетероциклил, содержащий один или два гетероатома, выбранные из азота, кислорода и серы;
Ra представляет собой C1-4алкил или -O-C1-4алкил, каждый из которых необязательно замещен одним, двумя или тремя заместителями, представляющими собой атом галогена;
или
Ra представляет собой 2H-1,2,3-триазол-2-ил или C3-6циклоалкил; при этом каждый из них необязательно замещен по одному или двум атомам углерода посредством заместителя, который в каждом случае независимо выбран из группы, состоящей из C1-4алкила, и при этом C1-4алкил замещен одним -OH;
Rb представляет собой водород;
или их энантиомер, диастереоизомер, сольват или фармацевтически приемлемую солевую форму.
Специалисту в данной области техники будет понятно, что все ссылки ниже на формулу (I), в контексте данного изобретения могут также относиться к энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме данного соединения, даже при отсутствии ссылок на них в явной форме, и что они также включены в объем настоящего изобретения.
В настоящем изобретении также предусмотрена фармацевтическая композиция, содержащая фармацевтически приемлемый носитель, фармацевтически приемлемое вспомогательное вещество и/или фармацевтически приемлемый разбавитель и соединение формулы (I) или его фармацевтически приемлемую соль, состоящая и/или по сути состоящая из таковых.
Также предусмотрены способы получения фармацевтической композиции, предусматривающие смешивание соединения формулы (I) и фармацевтически приемлемого носителя, фармацевтически приемлемого вспомогательного вещества и/или фармацевтически приемлемого разбавителя, заключающиеся и/или по сути заключающиеся в нем.
В настоящем изобретении дополнительно предусмотрены способы лечения или уменьшения интенсивности заболевания, синдрома, состояния или нарушения у субъекта, в том числе млекопитающего и/или человека, у которых на заболевание, синдром или состояние воздействуют путем подавления MALT1, в том числе без ограничения рак и/или иммунологические заболевания, с применением соединения формулы (I).
Настоящее изобретение также направлено на применение любого из соединений, описанных в данном документе, в получении лекарственного препарата, где лекарственный препарат получен для лечения заболевания, синдрома, состояния или нарушения, на которые воздействуют путем подавления MALT1, таких как рак и/или иммунологические заболевания.
Настоящее изобретение также направлено на получение соединений формулы (I), которые действуют в качестве ингибиторов MALT1.
Примером настоящего изобретения являются способы лечения заболевания, синдрома, состояния или нарушения, опосредованных MALT1, выбранных из группы, состоящей из видов лимфомы, видов лейкоза, видов карциномы и видов саркомы, например, неходжкинской лимфомы (NHL), B-клеточной NHL, диффузной В-крупноклеточной лимфомы (DLBCL), лимфомы из клеток мантийной зоны (MCL), фолликулярной лимфомы (FL), лимфомы лимфоидной ткани слизистых оболочек (MALT), лимфомы из клеток маргинальной зоны, T-клеточной лимфомы, ходжкинской лимфомы, лимфомы Беркитта, множественной миеломы, хронического лимфоцитарного лейкоза (CLL), мелкоклеточной лимфоцитарной лимфомы (SLL), макроглобулинемии Вальденстрема, лимфобластного Т-клеточного лейкоза, хронического миелогенного лейкоза (CML), волосатоклеточного лейкоза, острого лимфобластного Т-клеточного лейкоза, плазмоцитомы, иммунобластного крупноклеточного лейкоза, мегакариобластного лейкоза, острого мегакариобластного лейкоза, промиелоцитарного лейкоза, эритролейкоза, (форм глиомы) головного мозга, видов глиобластомы, рака молочной железы, колоректального рака/рака толстой кишки, рака предстательной железы, рака легкого, в том числе немелкоклеточного, рака желудочно-кишечного тракта, рака эндометрия, меланомы, рака поджелудочной железы, рака печени, рака почки, плоскоклеточной карциномы, рака яичника, саркомы, остеосаркомы, рака щитовидной железы, рака мочевого пузыря, рака головы и шеи, рака яичка, саркомы Юинга, рабдомиосаркомы, медуллобластомы, нейробластомы, рака шейки матки, почечно-клеточного рака, уротелиального рака, рака вульвы, рака пищевода, рака слюнных желез, рака носоглотки, рака щеки, рака ротовой полости и GIST (гастроинтестинальной стромальной опухоли), предусматривающие введение субъекту, нуждающемуся в этом, терапевтически эффективного количества любого из соединений или фармацевтических композиций, описанных в настоящем изобретении, заключающиеся и/или по сути заключающиеся в нем.
В другом варианте осуществления настоящее изобретение направлено на соединение формулы (I) для применения в лечении заболевания, синдрома, состояния или нарушения, на которые воздействуют путем подавления MALT1, выбранных из группы, состоящей из видов лимфомы, видов лейкоза, видов карциномы и видов саркомы, например, неходжкинской лимфомы (NHL), B-клеточной NHL, диффузной В-крупноклеточной лимфомы (DLBCL), лимфомы из клеток мантийной зоны (MCL), фолликулярной лимфомы (FL), лимфомы лимфоидной ткани слизистых оболочек (MALT), лимфомы из клеток маргинальной зоны, T-клеточной лимфомы, ходжкинской лимфомы, лимфомы Беркитта, множественной миеломы, хронического лимфоцитарного лейкоза (CLL), мелкоклеточной лимфоцитарной лимфомы (SLL), макроглобулинемии Вальденстрема, лимфобластного Т-клеточного лейкоза, хронического миелогенного лейкоза (CML), волосатоклеточного лейкоза, острого лимфобластного Т-клеточного лейкоза, плазмоцитомы, иммунобластного крупноклеточного лейкоза, мегакариобластного лейкоза, острого мегакариобластного лейкоза, промиелоцитарного лейкоза, эритролейкоза, (форм глиомы) головного мозга, видов глиобластомы, рака молочной железы, колоректального рака/рака толстой кишки, рака предстательной железы, рака легкого, в том числе немелкоклеточного, рака желудочно-кишечного тракта, рака эндометрия, меланомы, рака поджелудочной железы, рака печени, рака почки, плоскоклеточной карциномы, рака яичника, саркомы, остеосаркомы, рака щитовидной железы, рака мочевого пузыря, рака головы и шеи, рака яичка, саркомы Юинга, рабдомиосаркомы, медуллобластомы, нейробластомы, рака шейки матки, почечно-клеточного рака, уротелиального рака, рака вульвы, рака пищевода, рака слюнных желез, рака носоглотки, рака щеки, рака ротовой полости и GIST (гастроинтестинальной стромальной опухоли).
В другом варианте осуществления настоящее изобретение направлено на композицию, содержащую соединение формулы (I), для лечения заболевания, синдрома, состояния или нарушения, на которые воздействуют путем подавления MALT1, выбранных из группы, состоящей из видов лимфомы, видов лейкоза, видов карциномы и видов саркомы, например, неходжкинской лимфомы (NHL), B-клеточной NHL, диффузной В-крупноклеточной лимфомы (DLBCL), лимфомы из клеток мантийной зоны (MCL), фолликулярной лимфомы (FL), лимфомы лимфоидной ткани слизистых оболочек (MALT), лимфомы из клеток маргинальной зоны, T-клеточной лимфомы, ходжкинской лимфомы, лимфомы Беркитта, множественной миеломы, хронического лимфоцитарного лейкоза (CLL), мелкоклеточной лимфоцитарной лимфомы (SLL), макроглобулинемии Вальденстрема, лимфобластного Т-клеточного лейкоза, хронического миелогенного лейкоза (CML), волосатоклеточного лейкоза, острого лимфобластного Т-клеточного лейкоза, плазмоцитомы, иммунобластного крупноклеточного лейкоза, мегакариобластного лейкоза, острого мегакариобластного лейкоза, промиелоцитарного лейкоза, эритролейкоза, (форм глиомы) головного мозга, видов глиобластомы, рака молочной железы, колоректального рака/рака толстой кишки, рака предстательной железы, рака легкого, в том числе немелкоклеточного, рака желудочно-кишечного тракта, рака эндометрия, меланомы, рака поджелудочной железы, рака печени, рака почки, плоскоклеточной карциномы, рака яичника, саркомы, остеосаркомы, рака щитовидной железы, рака мочевого пузыря, рака головы и шеи, рака яичка, саркомы Юинга, рабдомиосаркомы, медуллобластомы, нейробластомы, рака шейки матки, почечно-клеточного рака, уротелиального рака, рака вульвы, рака пищевода, рака слюнных желез, рака носоглотки, рака щеки, рака ротовой полости и GIST (гастроинтестинальной стромальной опухоли).
В другом варианте осуществления настоящее изобретение направлено на композицию, содержащую соединение формулы (I), для лечения заболевания, синдрома, состояния или нарушения, на которые воздействуют путем подавления MALT1, выбранных из группы, состоящей из видов лимфомы, видов лейкоза, видов карциномы и видов саркомы, например, неходжкинской лимфомы (NHL), B-клеточной NHL, диффузной В-крупноклеточной лимфомы (DLBCL), лимфомы из клеток мантийной зоны (MCL), фолликулярной лимфомы (FL), лимфомы лимфоидной ткани слизистых оболочек (MALT), лимфомы из клеток маргинальной зоны, T-клеточной лимфомы, ходжкинской лимфомы, лимфомы Беркитта, множественной миеломы, хронического лимфоцитарного лейкоза (CLL), мелкоклеточной лимфоцитарной лимфомы (SLL), макроглобулинемии Вальденстрема, лимфобластного Т-клеточного лейкоза, хронического миелогенного лейкоза (CML), волосатоклеточного лейкоза, острого лимфобластного Т-клеточного лейкоза, плазмоцитомы, иммунобластного крупноклеточного лейкоза, мегакариобластного лейкоза, острого мегакариобластного лейкоза, промиелоцитарного лейкоза, эритролейкоза, (форм глиомы) головного мозга, видов глиобластомы, рака молочной железы, колоректального рака/рака толстой кишки, рака предстательной железы, рака легкого, в том числе немелкоклеточного, рака желудочно-кишечного тракта, рака эндометрия, меланомы, рака поджелудочной железы, рака печени, рака почки, плоскоклеточной карциномы, рака яичника, саркомы, остеосаркомы, рака щитовидной железы, рака мочевого пузыря, рака головы и шеи, рака яичка, саркомы Юинга, рабдомиосаркомы, медуллобластомы, нейробластомы, рака шейки матки, почечно-клеточного рака, уротелиального рака, рака вульвы, рака пищевода, рака слюнных желез, рака носоглотки, рака щеки, рака ротовой полости и GIST (гастроинтестинальной стромальной опухоли).
В другом варианте осуществления настоящее изобретение направлено на композицию, содержащую соединение формулы (I), для лечения заболевания, синдрома, состояния или нарушения, на которые воздействуют путем подавления MALT1, выбранных из группы, состоящей из диффузной В-крупноклеточной лимфомы (DLBCL), лимфомы из клеток мантийной зоны (MCL), фолликулярной лимфомы (FL) и лимфомы лимфоидной ткани слизистых оболочек (MALT).
Один вариант осуществления настоящего изобретения направлен на композицию, содержащую соединение формулы (I), для лечения иммунологических заболеваний, на которые воздействуют путем подавления MALT1, в том числе без ограничения аутоиммунных и воспалительных нарушений, например, артрита, воспалительного заболевания кишечника, гастрита, анкилозирующего спондилита, язвенного колита, панкреатита, болезни Крона, целиакии, рассеянного склероза, системной красной волчанки, волчаночного нефрита, ревматической атаки, подагры, отторжения органов или трансплантатов, хронического отторжения аллотрансплантата, острой или хронической реакции "трансплантат против хозяина", дерматита, в том числе атопического, дерматомиозита, псориаза, болезни Бехчета, увеита, тяжелой миастении, болезни Грейвса, тиреоидита Хашимото, синдрома Шегрена, нарушений при пузырчатке, синдромов васкулита, опосредованного антителами, иммунокомплексного васкулита, аллергических нарушений, астмы, бронхита, хронического обструктивного заболевания легкого (COPD), муковисцидоза, пневмонии, заболеваний легкого, в том числе отека, эмболии, фиброза, саркоидоза, гипертонии и эмфиземы, силикоза, дыхательной недостаточности, острого респираторного дистресс-синдрома, заболевания BENTA, бериллиоза и полимиозита.
В другом варианте осуществления настоящее изобретение направлено на композицию, содержащую соединение формулы (I), для лечения заболевания, синдрома, состояния или нарушения, на которые воздействуют путем подавления MALT1, выбранных из группы, состоящей из ревматоидного артрита (RA), псориатического артрита (PsA), псориаза (Pso), язвенного колита (UC), болезни Крона, системной красной волчанки (SLE), астмы и хронического обструктивного заболевания легкого (COPD).
Другой вариант осуществления настоящего изобретения направлен на фармацевтическую композицию, содержащую соединение формулы (I).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
При ссылке на заместители термин "независимо" означает ситуацию, где имеется несколько заместителей, выбранных независимо друг от друга, и они могут быть одинаковыми или отличными друг от друга.
Приставка "Cx-y" (где x и y представляют собой целые числа), используемая в данном документе, означает число атомов углерода в данной группе. Таким образом, C1-4алкильная группа содержит от 1 до 4 атомов углерода и т. д.
Термин "C1-4алкил", используемый в данном документе в качестве названия группы или части группы, означает насыщенный углеводородный радикал с неразветвленной или разветвленной цепью, содержащий от 1 до 4 атомов углерода, такой как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил и т. п.
Термин "C2-4алкенил", используемый в данном документе в качестве названия группы или части группы, представляет собой неразветвленную или разветвленную углеводородную группу, содержащую от 2 до 4 атомов углерода и содержащую двойную связь углерод-углерод, такую как без ограничения этенил, пропенил, бутенил и т.п.
"Неароматический гетероциклил" охватывает ненасыщенные гетероциклические кольцевые системы неароматического характера, частично насыщенные и полностью насыщенные гетероциклические кольцевые системы. Термин "частично насыщенный" означает кольца, где кольцевая(кольцевые) структура(структуры) содержит(содержат) по меньшей мере одну кратную связь, например, связь C=C, N=C. Термин "полностью насыщенный" означает кольца, где кратные связи между атомами кольца отсутствуют. Специалисту в данной области техники будет понятно, что "неароматический гетероциклил" содержит по меньшей мере один гетероатом, такой как N, O или S, если иное не указано или ясно не следует из контекста.
Неограничивающие примеры моноциклических 4-7-членных, неароматических гетероциклилов, содержащих один или два гетероатома, выбранные из азота, кислорода и серы, включают без ограничения азетидинил, оксетанил, пирролидинил, тетрагидрофуранил, пиперидинил, пиперазинил, пиранил, дигидропиранил, тетрагидропиранил, морфолинил и тиоморфолинил.
Термин "C3-6циклоалкил", используемый в данном документе в качестве названия группы или части группы, означает насыщенный циклический углеводородный радикал, содержащий от 3 до 6 атомов углерода, такой как циклопропил, циклобутил, циклопентил и циклогексил.
Термин "галоген" или "галогено" означает атомы фтора, хлора, брома и йода.
Обозначение "R" в стереоцентре означает, что стереоцентр имеет абсолютную R-конфигурацию, в соответствии с определением, принятым в данной области техники; аналогично, обозначение "S" означает, что стереоцентр имеет абсолютную S-конфигурацию. Используемые в данном документе обозначения "*R" или "*S" в стереоцентре используются для указания на то, что стереоцентр имеет абсолютную, но неизвестную конфигурацию. Используемое в данном документе обозначение "RS" означает стереоцентр, который существует в виде смеси R- и S-конфигураций.
Соединение, содержащее один стереоцентр, изображенный без указания стереохимического положения связи, представляет собой смесь двух энантиомеров. Соединение, содержащее два стереоцентра, оба из которых изображены без указания стереохимического положения связи, представляет собой смесь четырех диастереоизомеров.
Необозначенные стереоцентры, изображенные без указания стереохимического положения связи, являются смесями R- и S-конфигураций. Для необозначенных стереоцентров, изображенных с указаниями стереохимического положения связи, относительная и абсолютная стереохимия является такой, как показано.
Если не указано иное, предполагается, что определение любого заместителя или переменной в конкретном месте в молекуле будет независимым от его определений где-нибудь в другом месте в этой молекуле. Понятно, что заместители и схемы замещений в соединениях по настоящему изобретению могут быть выбраны специалистом средней квалификации в данной области техники для получения соединений, которые являются химически стабильными, и которые можно легко синтезировать с помощью методик, известных в данной области техники, а также тех способов, которые изложены в данном документе.
Термин "субъект" означает животное, предпочтительно млекопитающее, наиболее предпочтительно человека, которое является объектом лечения, наблюдения или эксперимента. Термин "терапевтически эффективное количество" означает количество активного соединения или фармацевтического средства, в том числе соединения по настоящему изобретению, которое вызывает биологический или медицинский ответ в системе тканей, животном или человеке, который стремится получить исследователь, ветеринар, врач или другой клиницист, в том числе снижение или подавление активности фермента или белка, или уменьшение интенсивности симптомов, облегчение состояний, замедление или задержку развития заболевания, или предупреждение заболевания.
В одном варианте осуществления термин "терапевтически эффективное количество" означает количество соединения по настоящему изобретению, которое при введении субъекту является эффективным для (1) по меньшей мере частичного уменьшения интенсивности, подавления, предупреждения и/или облегчения состояния, или нарушения, или заболевания, (i) опосредованного MALT1; или (ii) ассоциированного с активностью MALT1; или (iii) характеризующегося активностью (нормальной или атипичной) MALT1; или (2) снижения или подавления активности MALT1; или (3) снижения или подавления экспрессии MALT1; или (4) модификации уровней белка MALT1.
Термин "композиция" означает продукт, который содержит указанные ингредиенты в терапевтически эффективных количествах, а также любой продукт, который получают, непосредственно или опосредованно, из комбинаций указанных ингредиентов в указанных количествах.
Термин "опосредованное MALT1" означает любое заболевание, синдром, состояние или нарушение, которые теоретически способны возникать при отсутствии MALT1, но практически могут возникать при наличии MALT1. Подходящие примеры заболевания, синдрома, состояния или нарушения, опосредованных MALT1, включают без ограничения виды лимфомы, виды лейкоза, виды карциномы и виды саркомы, например, неходжкинской лимфомы (NHL), B-клеточной NHL, диффузной В-крупноклеточной лимфомы (DLBCL), лимфомы из клеток мантийной зоны (MCL), фолликулярной лимфомы (FL), лимфомы лимфоидной ткани слизистых оболочек (MALT), лимфомы из клеток маргинальной зоны, T-клеточной лимфомы, ходжкинской лимфомы, лимфомы Беркитта, множественной миеломы, хронического лимфоцитарного лейкоза (CLL), мелкоклеточной лимфоцитарной лимфомы (SLL), макроглобулинемии Вальденстрема, лимфобластного Т-клеточного лейкоза, хронического миелогенного лейкоза (CML), волосатоклеточного лейкоза, острого лимфобластного Т-клеточного лейкоза, плазмоцитомы, иммунобластного крупноклеточного лейкоза, мегакариобластного лейкоза, острого мегакариобластного лейкоза, промиелоцитарного лейкоза, эритролейкоза, (форм глиомы) головного мозга, видов глиобластомы, рака молочной железы, колоректального рака/рака толстой кишки, рака предстательной железы, рака легкого, в том числе немелкоклеточного, рака желудочно-кишечного тракта, рака эндометрия, меланомы, рака поджелудочной железы, рака печени, рака почки, плоскоклеточной карциномы, рака яичника, саркомы, остеосаркомы, рака щитовидной железы, рака мочевого пузыря, рака головы и шеи, рака яичка, саркомы Юинга, рабдомиосаркомы, медуллобластомы, нейробластомы, рака шейки матки, почечно-клеточного рака, уротелиального рака, рака вульвы, рака пищевода, рака слюнных желез, рака носоглотки, рака щеки, рака ротовой полости и GIST (гастроинтестинальной стромальной опухоли).
При использовании в данном документе термин "ингибитор MALT1" означает средство, которое подавляет или снижает тяжесть по меньшей мере одного состояния, симптома, синдрома, нарушения и/или заболевания, связанного с MALT1.
При использовании в данном документе, если не отмечено иное, термин "влияние" или "под воздействием" включает (при ссылках на заболевание, синдром, состояние или нарушение, на которые воздействуют путем подавления MALT1) снижение частоты и/или тяжести одного или нескольких симптомов или проявлений указанного заболевания, синдрома, состояния или нарушения; и/или включает предупреждение развития одного или нескольких симптомов или проявлений указанного заболевания, синдрома, состояния или нарушения, либо развитие заболевания, синдрома, состояния или нарушения.
При использовании в данном документе термин "лечить", "осуществление лечения" или "лечение" любого заболевания, состояния, синдрома или нарушения означает в одном варианте осуществления облегчение заболевания, состояния, синдрома или нарушения (т.е. замедление, или приостановку, или снижение развития заболевания или по меньшей мере одного из его клинических симптомов). В другом варианте осуществления "лечить", "осуществление лечения" или "лечение" означает облегчение или уменьшение интенсивности по меньшей мере одного физического параметра, в том числе таких, которые могут быть незаметны для пациента. В дополнительном варианте осуществления "лечить", "осуществление лечения" или "лечение" означает модуляцию заболевания, состояния, синдрома или нарушения либо физически (например, путем стабилизации заметного симптома), либо физиологически (например, путем стабилизации физического параметра), либо и так, и так. В еще одном варианте осуществления "лечить", "осуществление лечения" или "лечение" означает профилактику или задержку начала, или развития, или прогрессирования заболевания, состояния, синдрома или нарушения.
Соединения по настоящему изобретению являются полезными в способах лечения заболевания, синдрома, состояния или нарушения или улучшения состояния при таковых, на которые воздействуют путем подавления MALT1. Такие способы предусматривают введение субъекту, в том числе животному, млекопитающему и человеку, нуждающемуся в таком лечении, улучшении состояния и/или профилактике, терапевтически эффективного количества соединения формулы (I) или его энантиомера, диастереомера, сольвата или фармацевтически приемлемой соли, заключаются и/или по сути заключаются в нем.
Один вариант осуществления настоящего изобретения направлен на способ лечения зависящего от MALT1 или опосредованного MALT1 заболевания или состояния у субъекта, нуждающегося в этом, в том числе животного, млекопитающего и человека, нуждающихся в таком лечении, предусматривающий введение субъекту терапевтически эффективного количества соединения формулы (I).
В другом варианте осуществления зависящее от MALT1 или опосредованное MALT1 заболевание или состояние выбрано из видов рака гемопоэтического происхождения или солидных опухолей, таких как хронический миелогенный лейкоз, миелоидный лейкоз, неходжкинская лимфома и другие виды B-клеточной лимфомы.
В частности, соединения формулы (I) или их энантиомеры, диастереоизомеры, сольваты или фармацевтически приемлемые солевые формы являются полезными для лечения или уменьшения интенсивности заболеваний, синдромов, состояний или нарушений, таких как диффузная В-крупноклеточная лимфома (DLBCL), лимфома из клеток мантийной зоны (MCL), фолликулярная лимфома (FL) и лимфома лимфоидной ткани слизистых оболочек (MALT).
Более конкретно, соединения формулы (I) или их энантиомеры, диастереомеры, сольваты или фармацевтически приемлемые солевые формы являются полезными для лечения или уменьшения интенсивности диффузной В-крупноклеточной лимфомы (DLBCL), лимфомы из клеток мантийной зоны (MCL), фолликулярной лимфомы (FL) и лимфомы лимфоидной ткани слизистых оболочек (MALT), предусматривающих введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его энантиомера, диастереомера, сольвата или фармацевтически приемлемой солевой формы, определенных в данном документе.
Кроме того, соединения формулы (I) или их энантиомер, диастереомер, сольват или фармацевтически приемлемая солевая форма являются полезными для лечения или уменьшения интенсивности иммунологического заболевания, синдрома, нарушения или состояния, выбранных из группы, состоящей из ревматоидного артрита (RA), псориатического артрита (PsA), псориаза (Pso), язвенного колита (UC), болезни Крона, системной красной волчанки (SLE), астмы и хронического обструктивного заболевания легкого (COPD).
Варианты осуществления настоящего изобретения включают соединение формулы (I), где
Rx представляет собой C1-4алкил или C3-6циклоалкил;
Ry представляет собой C1-4алкил; и
Rz представляет собой водород;
R1 выбран из группы, состоящей из водорода, -OR5, C1-4алкила, C2-4алкенила, галогена,
-CN, C3-6циклоалкила, Heta, -C(=O)-OH, -C(=O)-O-C1-4алкила, -NR6aR7a и
-C(=O)-NR6bR7b;
каждый из R2a и R2b независимо выбран из группы, состоящей из водорода,
-NR6cR7c, C3-6циклоалкила, C1-4алкила и C1-4алкила, замещенного 1, 2 или 3 атомами галогена;
X1 представляет собой N или CRa;
X2 представляет собой N или CRb;
при условии, что только один из X1 и X2 представляет собой N в любом случае;
R3 представляет собой водород, C1-4алкил или -O-C1-4алкил;
R4 представляет собой галоген, циано или трифторметил;
R5 выбран из группы, состоящей из водорода, C1-4алкила, C3-6циклоалкила, Hetb и C1-4алкила, замещенного одним или двумя заместителями, каждый из которых независимо выбран из группы, состоящей из -C(=O)-NR8R9, -C(=O)-OH, -C(=O)-O-C1-4алкила, C3-6циклоалкила и фенила;
каждый из R6a, R6b, R6c, R7a, R7b, R7c, R8 и R9 независимо выбран из группы, состоящей из водорода и C1-4алкила;
Heta представляет собой моноциклический 4-7-членный неароматический гетероциклил, содержащий один или два гетероатома, выбранные из азота, кислорода и серы;
Hetb представляет собой моноциклический 4-7-членный неароматический гетероциклил, содержащий один или два гетероатома, выбранные из азота, кислорода и серы;
Ra представляет собой -O-C1-4алкил, каждый вариант которого необязательно замещен одним, двумя или тремя заместителями, представляющими собой атом галогена;
или
Ra представляет собой 2H-1,2,3-триазол-2-ил или C3-6циклоалкил; при этом каждый из них необязательно замещен по одному атому углерода посредством заместителя, который в каждом случае независимо выбран из группы, состоящей из C1-4алкила, и при этом C1-4алкил замещен одним -OH;
Rb представляет собой водород;
или его энантиомер, диастереоизомер, сольват или фармацевтически приемлемую солевую форму.
Варианты осуществления настоящего изобретения включают соединение формулы (I), где
Rx представляет собой C1-4алкил;
Ry представляет собой C1-4алкил; и
Rz представляет собой водород;
R1 выбран из группы, состоящей из -OR5, галогена и -CN;
R2a представляет собой водород;
R2b выбран из группы, состоящей из водорода, -NR6cR7c и C1-4алкила;
X1 представляет собой CRa;
X2 представляет собой N;
R3 представляет собой водород;
R4 представляет собой трифторметил;
R5 представляет собой C1-4алкил;
R6c и R7c представляют собой водород;
Ra представляет собой 2H-1,2,3-триазол-2-ил;
или его энантиомер, диастереомер, сольват или фармацевтически приемлемую солевую форму.
Варианты осуществления настоящего изобретения включают соединение формулы (I), где
Rx представляет собой C1-4алкил;
Ry представляет собой C1-4алкил; и
Rz представляет собой водород;
R1 выбран из группы, состоящей из галогена и -CN;
R2a представляет собой водород;
R2b представляет собой водород, -NR6cR7c, C1-4алкил и C1-4алкил, замещенный 1, 2 или 3 атомами галогена;
X1 представляет собой CRa;
X2 представляет собой N;
R3 представляет собой водород или -O-C1-4алкил;
R4 представляет собой галоген или трифторметил;
R6c и R7c представляют собой водород;
Ra представляет собой 2H-1,2,3-триазол-2-ил;
или его энантиомер, диастереомер, сольват или фармацевтически приемлемую солевую форму.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
Rx представляет собой водород, C1-4алкил или C3-6циклоалкил;
Ry представляет собой водород, C1-4алкил или C3-6циклоалкил; и
Rz представляет собой водород.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
Rx и Ry взяты вместе с образованием двухвалентного радикала -Rx-Ry-, где -Rx-Ry- представляет собой
-(CH2)n- или -CH2-O-(CH2)2-; где n представляет собой 2, 3, 4 или 5; и
Rz представляет собой водород.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
Ry представляет собой водород, C1-4алкил или C3-6циклоалкил;
Rx и Rz взяты вместе с атомом углерода, к которому они присоединены, с образованием C3-6циклоалкила.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
R1 выбран из группы, состоящей из -OR5, C1-4алкила, C2-4алкенила, галогена, -CN,
C3-6циклоалкила, Heta, -C(=O)-OH, -C(=O)-O-C1-4алкила, -NR6aR7a и -C(=O)-NR6bR7b.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
R2a представляет собой водород.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
R2a представляет собой водород; и
R2b представляет собой водород, -NR6cR7c, C1-4алкил и C1-4алкил, замещенный 1, 2 или 3 атомами галогена.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
Ra представляет собой C1-4алкил или -O-C1-4алкил, каждый из которых необязательно замещен одним, двумя или тремя заместителями, представляющими собой атом галогена.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
Ra представляет собой 2H-1,2,3-триазол-2-ил или C3-6циклоалкил; при этом каждый из них необязательно замещен по одному или двум атомам углерода посредством заместителя, который в каждом случае независимо выбран из группы, состоящей из C1-4алкила, и при этом C1-4алкил замещен одним -OH.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
Ra представляет собой 2H-1,2,3-триазол-2-ил или C3-6циклоалкил; при этом каждый из них необязательно замещен по одному или двум атомам углерода посредством заместителя, который в каждом случае независимо выбран из группы, состоящей из C1-4алкила и -CH(OH)-C0-3алкила.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
Ra представляет собой C1-4алкил или -O-C1-4алкил, каждый из которых необязательно замещен одним, двумя или тремя заместителями, представляющими собой атом галогена;
или
Ra представляет собой 2H-1,2,3-триазол-2-ил или C3-6циклоалкил; при этом каждый из них необязательно замещен по одному или двум атомам углерода посредством заместителя, который в каждом случае независимо выбран из группы, состоящей из C1-4алкила и -CH(OH)-C0-3алкила.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
Ra представляет собой 2H-1,2,3-триазол-2-ил.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
X1 представляет собой N; и
X2 представляет собой CRb.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
X1 представляет собой CRa; и
X2 представляет собой N.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
X1 представляет собой CRa; и
X2 представляет собой CRb.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
R6c и R7c представляют собой водород.
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
Heta представляет собой моноциклический 4-7-членный полностью насыщенный гетероциклил, содержащий один или два гетероатома, выбранные из азота, кислорода и серы;
Hetb представляет собой моноциклический 4-7-членный полностью насыщенный гетероциклил, содержащий один или два гетероатома, выбранные из азота, кислорода и серы;
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где
Heta представляет собой моноциклический 4-7-членный полностью насыщенный гетероциклил, содержащий один атом кислорода;
Hetb представляет собой моноциклический 4-7-членный полностью насыщенный гетероциклил, содержащий один атом кислорода;
В одном варианте осуществления настоящее изобретение относится к таким соединениям формулы (I) и к их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, или любой их подгруппе, которые указаны в любом из других вариантов осуществления, где Heta и Hetb представляют собой оксетанил, в частности, 3-оксетанил.
Что касается применения в медицине, соли соединений формулы (I) означают нетоксичные "фармацевтически приемлемые соли". Однако в получении соединений формулы (I) или их фармацевтически приемлемых солевых форм могут быть полезными другие соли. Подходящие фармацевтически приемлемые соли соединения формулы (I) включают соли присоединения кислоты, которые могут быть образованы, например, путем смешивания раствора соединения с раствором фармацевтически приемлемой кислоты, такой как хлористоводородная кислота, серная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, уксусная кислота, бензойная кислота, лимонная кислота, винная кислота, угольная кислота или фосфорная кислота. Кроме того, если соединения формулы (I) имеют кислотный фрагмент, то их подходящие фармацевтически приемлемые соли могут включать соли щелочных металлов, такие как соли натрия или калия; соли щелочноземельных металлов, такие как соли кальция или магния; и соли, образованные с помощью пригодных органических лигандов, такие как соли четвертичного аммония. Таким образом, иллюстративные фармацевтически приемлемые соли включают ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, кислую соль винной кислоты, борат, бромид, эдетат кальция, камзилат, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, эзилат, фумарат, глюцептат, глюконат, глутамат, гликоллиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, йодид, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат, напсилат, нитрат, аммониевую соль N-метилглюкамина, олеат, памоат (эмбонат), пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, сульфат, основную соль уксусной кислоты, сукцинат, таннат, тартрат, теоклат, тозилат, триэтйодид и валерат. Иллюстративные кислоты и основания, которые можно применять в получении фармацевтически приемлемых солей, включают кислоты, в том числе уксусную кислоту, 2,2-дихлоруксусную кислоту, ацилированные аминокислоты, адипиновую кислоту, альгиновую кислоту, аскорбиновую кислоту, L-аспарагиновую кислоту, бензолсульфоновую кислоту, бензойную кислоту, 4-ацетамидобензойную кислоту, (+)-камфорную кислоту, камфорсульфоновую кислоту, (+)-(1S)-камфор-10-сульфоновую кислоту, каприновую кислоту, капроновую кислоту, каприловую кислоту, коричную кислоту, лимонную кислоту, цикламовую кислоту, додецилсерную кислоту, этан-1,2-дисульфоновую кислоту, этансульфоновую кислоту, 2-гидроксиэтансульфоновую кислоту, муравьиную кислоту, фумаровую кислоту, галактаровую кислоту, гентизиновую кислоту, глюкогептоновую кислоту, D-глюконовую кислоту, D-глюкуроновую кислоту, L-глутаминовую кислоту, α-оксоглутаровую кислоту, гликолевую кислоту, гиппуровую кислоту, бромистоводородную кислоту, хлористоводородную кислоту, (+)-L-молочную кислоту, (±)-DL-молочную кислоту, лактобионовую кислоту, малеиновую кислоту, (−)-L-яблочную кислоту, малоновую кислоту, (±)-DL-миндальную кислоту, метансульфоновую кислоту, нафталин-2-сульфоновую кислоту, нафталин-1,5-дисульфоновую кислоту, 1-гидрокси-2-нафтойную кислоту, никотиновую кислоту, азотную кислоту, олеиновую кислоту, оротовую кислоту, щавелевую кислоту, пальмитиновую кислоту, памоевую кислоту, фосфорную кислоту, L-пироглутаминовую кислоту, салициловую кислоту, 4-аминосалициловую кислоту, себациновую кислоту, стеариновую кислоту, янтарную кислоту, серную кислоту, дубильную кислоту, (+)-L-винную кислоту, тиоциановую кислоту, п-толуолсульфоновую кислоту и ундециленовую кислоту; и основания, в том числе аммиак, L-аргинин, бенетамин, бензатин, гидроксид кальция, холин, деанол, диэтаноламин, диэтиламин, 2-(диэтиламино)этанол, этаноламин, этилендиамин, N-метилглюкамин, гидрабамин, 1H-имидазол, L-лизин, гидроксид магния, 4-(2-гидроксиэтил)морфолин, пиперазин, гидроксид калия, 1-(2-гидроксиэтил)пирролидин, гидроксид натрия, триэтаноламин, трометамин и гидроксид цинка. Варианты осуществления настоящего изобретения включают пролекарства на основе соединений формулы (I). Как правило, такие пролекарства будут представлять собой функциональные производные соединений, которые легко превращаются in vivo в требуемое соединение. Таким образом, в способах лечения или предупреждения в соответствии с вариантами осуществления настоящего изобретения термин "введение" охватывает лечение или предупреждение различных описанных заболеваний, состояний, синдромов и нарушений с помощью конкретно раскрытого соединения или с помощью соединения, которое может не быть конкретно раскрытым, но которое in vivo превращается в указанное соединение после введения пациенту. Обычные процедуры для выбора и получения подходящих производных пролекарств описаны, например, в "Design of Prodrugs", ed. H. Bundgaard, Elsevier, 1985.
Специалист средней квалификации в данной области техники поймет, что соединения, описанные в данном документе, могут существовать в виде таутомеров, и что являются возможными другие таутомерные конфигурации структур, изображенных в данном документе. Таутомеры представляют собой структурные изомеры, которые легко взаимопревращаются друг в друга. Понятно, что все таутомерные формы охватываются структурой, в которой описана одна возможная таутомерная конфигурация групп соединения, даже если она конкретно не указана.
Если соединения в соответствии с вариантами осуществления данного изобретения имеют по меньшей мере один хиральный центр, они могут, следовательно, существовать в виде энантиомеров. Если соединения имеют два или более хиральных центров, они могут дополнительно существовать в виде диастереомеров. Следует понимать, что все такие изомеры и их смеси находятся в пределах объема настоящего изобретения. Кроме того, некоторые кристаллические формы соединений могут существовать в виде полиморфов, и предполагается их включение в настоящее изобретение как таковых. Кроме того, некоторые из соединения могут образовывать сольваты с водой (т.е. гидраты) или обычными органическими растворителями, при этом также предполагается, что такие сольваты охватываются объемом настоящего изобретения. Специалисту в данной области техники будет понятно, что термин "соединение" при использовании в данном документе подразумевает включение сольватированных соединений формулы (I).
Если способ получения соединений в соответствии с некоторыми вариантами осуществления настоящего изобретения обеспечивает получение смеси стереоизомеров, эти изомеры можно разделять с помощью обычных методик, таких как препаративная хроматография. Соединения можно получать в рацемической форме, или можно получать отдельные энантиомеры либо с помощью энантиоспецифического синтеза, либо путем разделения. Соединения можно, например, разделять на составляющие их энантиомеры с помощью стандартных методик, таких как образование диастереоизомерных пар путем образования соли с оптически активной кислотой, такой как (−)-ди-п-толуоил-d-винная кислота и/или (+)-ди-п-толуоил-d-винная кислота, с последующей фракционной кристаллизацией и восстановлением свободного основания. Соединения можно также разделять путем образования диастереомерных сложных эфиров или амидов с последующим хроматографическим разделением и удалением хирального вспомогательного вещества. В качестве альтернативы, соединения можно разделять с помощью колонки для хиральной HPLC.
Один вариант осуществления настоящего изобретения направлен на композицию, в том числе фармацевтическую композицию, содержащую (+)-энантиомер соединения формулы (I), состоящую и/или по сути состоящую из него, где указанная композиция по сути не содержит (−)-изомера указанного соединения. В данном контексте "по сути не содержит" означает менее чем приблизительно 25%, предпочтительно менее чем приблизительно 10%, более предпочтительно менее чем приблизительно 5%, еще более предпочтительно менее чем приблизительно 2% и еще более предпочтительно менее чем приблизительно 1% (−)-изомера, рассчитанного как
 .
.
Другой вариант осуществления настоящего изобретения представляет собой композицию, в том числе фармацевтическую композицию, содержащую (−)-энантиомер соединения формулы (I), состоящую и по сути состоящую из него, где указанная композиция по сути не содержит (+)-изомера указанного соединения. В данном контексте "по сути не содержит" означает менее чем приблизительно 25%, предпочтительно менее чем приблизительно 10%, более предпочтительно менее чем приблизительно 5%, еще более предпочтительно менее чем приблизительно 2% и еще более предпочтительно менее чем приблизительно 1% (+)-изомера, рассчитанного как
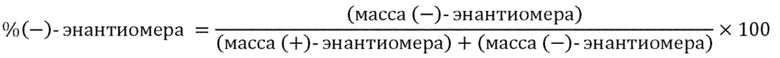 .
.
Предполагается, что в рамках настоящего изобретения любые один или несколько элементов, в частности, при упоминании в отношении соединения формулы (I), будут содержать все изотопы и изотопные смеси указанного(указанных) элемента(элементов), либо встречающиеся в природе, либо полученные синтетическим путем, либо с природным изотопным составом, либо в изотопно-обогащенной форме. Например, упоминание водорода включает в свой объем 1H, 2H (D) и 3H (T). Подобным образом, упоминания углерода и кислорода включают в свой объем 12C, 13C и 14C и 16O и 18O соответственно. Изотопы могут быть радиоактивными или нерадиоактивными. Меченные радиоактивным изотопом соединения формулы (I) могут содержать один или несколько радиоактивных изотопов, выбранных из группы 3H, 11C, 18F, 122I, 123I, 125I, 131I, 75Br, 76Br, 77Br и 82Br. Предпочтительно данный изотоп выбран из группы, состоящей из 2H, 3H, 11C и 18F. В частности, предполагается, что дейтерированные соединения включены в объем настоящего изобретения.
В ходе любого из способов получения соединений из различных вариантов осуществления настоящего изобретения может быть необходимой и/или желательной защита неустойчивых или реакционноспособных групп в какой-либо из рассматриваемых молекул. Это может достигаться с помощью традиционных защитных групп, таких как описанные в Protective Groups in Organic Chemistry, Second Edition, J.F.W. McOmie, Plenum Press, 1973; T.W. Greene & P.G.M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 1991; и T.W. Greene & P.G.M. Wuts, Protective Groups in Organic Synthesis, Third Edition, John Wiley & Sons, 1999. Защитные группы можно удалять на подходящей последующей стадии с применением способов, известных из уровня техники. Даже несмотря на то, что соединения из вариантов осуществления настоящего изобретения (в том числе их фармацевтически приемлемые соли и фармацевтически приемлемые сольваты) можно вводить в чистом виде, их обычно будут вводить в смеси с фармацевтически приемлемым носителем, фармацевтически приемлемым вспомогательным веществом и/или фармацевтически приемлемым разбавителем, выбранными в зависимости от предполагаемого пути введения и стандартной фармацевтической или ветеринарной практики. Таким образом, конкретные варианты осуществления настоящего изобретения направлены на фармацевтические и ветеринарные композиции, содержащие соединения формулы (I) и по меньшей мере один приемлемый носитель, фармацевтически приемлемое вспомогательное вещество и/или фармацевтически приемлемый разбавитель. В качестве примера, в фармацевтических композициях из вариантов осуществления настоящего изобретения соединения формулы (I) можно смешивать с любым(любыми) подходящим(подходящими) связывающим(связывающими) веществом(веществами), смазывающим(смазывающими) веществом(веществами), суспендирующим(суспендирующими) средством(средствами), средством(средствами) для нанесения покрытия, солюбилизирующим(солюбилизирующими) средством(средствами) и их комбинациями.
Твердые лекарственные формы для перорального введения, такие как таблетки или капсулы, содержащие соединения по настоящему изобретению, в случае необходимости можно вводить по меньшей мере в одной лекарственной форме за один прием. Также возможно введение соединений в составах с замедленным высвобождением.
Дополнительные формы для перорального введения, в которых можно вводить соединения в соответствии с настоящим изобретением, включают настойки, растворы, сиропы и суспензии; причем каждая необязательно содержит ароматизаторы и красители. В качестве альтернативы, соединения формулы (I) можно вводить путем ингаляции (внутритрахеальной или интраназальной) или в виде суппозитория, или пессария, или их можно наносить местно в виде лосьона, раствора, крема, мази или присыпки. Например, их можно вводить в состав крема, содержащего водную эмульсию полиэтиленгликолей или жидкого парафина, состоящего и/или по сути состоящего из таковых. Их также можно вводить в концентрации от приблизительно 1% до приблизительно 10% по весу крема в состав мази, содержащей основу из воска или мягкого парафина, состоящей и/или по сути состоящей из нее, вместе с любыми стабилизаторами и консервантами, которые могут потребоваться. Альтернативные пути введения включают трансдермальное введение с применением накожного или трансдермального пластыря. Фармацевтические композиции по настоящему изобретению (а также соединения по настоящему изобретению, взятые отдельно) можно также вводить посредством инъекции парентерально, например, внутрикавернозно, внутривенно, внутримышечно, подкожно, внутрикожно или интратекально. В этом случае композиции будут также включать по меньшей мере одно из подходящего носителя, подходящего вспомогательного вещества и подходящего разбавителя. Для парентерального введения фармацевтические композиции по настоящему изобретению лучше всего использовать в виде стерильного водного раствора, который может содержать другие вещества, например, достаточное количество солей и моносахаридов для получения раствора, изотоничного крови. Для буккального или подъязычного введения фармацевтические композиции по настоящему изобретению можно вводить в виде таблеток или пастилок, которые можно составлять обычным способом. В качестве дополнительного примера, фармацевтические композиции, содержащие по меньшей мере одно из соединений формулы (I) в качестве активного ингредиента, можно получать путем смешивания соединения(соединений) с фармацевтически приемлемым носителем, фармацевтически приемлемым разбавителем и/или фармацевтически приемлемым вспомогательным веществом в соответствии с обычными методиками составления фармацевтических препаратов. Носитель, вспомогательное вещество и разбавитель могут принимать широкое разнообразие форм в зависимости от необходимого пути введения (например, перорального, парентерального и т.д.). Таким образом, в случае жидких лекарственных препаратов для перорального введения, таких как суспензии, сиропы, настойки и растворы, подходящие носители, вспомогательные вещества и разбавители включают воду, гликоли, масла, спирты, ароматизаторы, консерванты, стабилизаторы, красители и т.п.; в случае твердых лекарственных средств для перорального введения, таких как порошки, капсулы и таблетки, подходящие носители, вспомогательные вещества и разбавители включают крахмалы, сахара, разбавители, средства, способствующие гранулированию, смазывающие вещества, связывающие вещества, разрыхлители и т.п. Твердые лекарственные препараты для перорального введения также могут быть необязательно покрыты веществами, такими как сахара, или иметь энтеросолюбильное покрытие для того, чтобы изменить преобладающую локализацию абсорбции и дезинтеграции. Для парентерального введения носитель, вспомогательное вещество и разбавитель будут обычно включать стерильную воду, а другие ингредиенты можно добавлять для повышения растворимости и сохранности композиции. Суспензии или растворы для инъекций также можно получать с помощью водных носителей вместе с подходящими добавками, такими как солюбилизаторы и консерванты.
Терапевтически эффективное количество соединения формулы (I) или его фармацевтической композиции включает диапазон доз от приблизительно 0,1 мг до приблизительно 3000 мг или любое конкретное количество или диапазон в пределах вышеуказанного; хотя специалисту в данной области техники очевидно, что терапевтически эффективное количество для соединения формулы (I) будет варьировать в зависимости от заболеваний, синдромов, состояний и нарушений, которые необходимо лечить.
Оптимальные дозировки соединения формулы (I), которые следует вводить, могут быть легко определены и будут изменяться в зависимости от конкретного применяемого соединения, пути введения, активности препарата и прогрессирования заболевания, синдрома, состояния или нарушения. Кроме того, факторы, связанные с конкретным субъектом, подвергаемым лечению, в том числе пол, возраст, вес, рацион субъекта и время введения, будут приводить в результате к необходимости регулировки доз для достижения соответствующего терапевтического уровня и желаемого терапевтического эффекта. Вышеуказанные дозировки, таким образом, являются иллюстративными для усредненного случая. Разумеется, могут существовать отдельные случаи, где являются необходимыми более высокие или более низкие диапазоны дозировок, и они находятся в пределах объема настоящего изобретения. Соединения формулы (I) можно вводить в любой из вышеуказанных композиций и схем дозирования или посредством таких композиций и схем дозирования, которые установлены в данной области техники, когда применение соединения формулы (I) требуется для субъекта, нуждающегося в этом.
Было обнаружено, что соединения по настоящему изобретению подавляют активность MALT1.
В некоторых вариантах осуществления подавление MALT1 с помощью предусмотренного соединения может быть полезным для лечения или предупреждения, в частности лечения, видов рака из неограничивающего перечня, описанного в данном документе.
Настоящее изобретение относится к соединениям формулы (I) или их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме для применения в качестве лекарственного препарата.
Настоящее изобретение относится к соединениям формулы (I) или их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме для применения в подавлении активности MALT1.
Настоящее изобретение относится к соединениям формулы (I) или их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме для применения в лечении заболеваний, упомянутых в данном документе.
Настоящее изобретение относится к соединениям формулы (I) или их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме для лечения или предупреждения, в частности для лечения, указанных заболеваний.
Настоящее изобретение относится к соединениям формулы (I) или их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме для лечения или предупреждения, в частности, при лечении, опосредованных MALT1 заболеваний или состояний.
Настоящее изобретение относится к соединениям формулы (I) или их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме для изготовления лекарственного препарата.
Настоящее изобретение относится к соединениям формулы (I) или их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме для изготовления лекарственного препарата для подавления MALT1.
Настоящее изобретение относится к соединениям формулы (I) или их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме для изготовления лекарственного препарата для лечения или предупреждения, в частности, для лечения, любого болезненного состояния из числа упомянутых в данном документе.
Настоящее изобретение относится к соединениям формулы (I) или их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме для изготовления лекарственного препарата для лечения любого болезненного состояния из числа упомянутых в данном документе.
Настоящее изобретение относится к соединениям формулы (I) или их энантиомеру, диастереомеру, сольвату или фармацевтически приемлемой солевой форме, которые можно вводить млекопитающим, предпочтительно людям, для лечения или предупреждения любого из заболеваний, упомянутых в данном документе.
Учитывая применимость соединений формулы (I) или их энантиомера, диастереомера, сольвата или фармацевтически приемлемой солевой формы, предусматривается способ лечения теплокровных животных, в том числе людей, страдающих любым из упомянутых в данном документе заболеваний, или способ предупреждения такового у теплокровных животных, в том числе у людей, подверженных любому из упомянутых в данном документе заболеваний.
В одном варианте осуществления виды рака, в отношении которых может быть достигнут положительный результат лечения с помощью ингибиторов MALT1 по настоящему изобретению, включают без ограничения виды лимфомы, виды лейкоза, виды карциномы и виды саркомы, например, неходжкинскую лимфому (NHL), B-клеточную NHL, диффузную В-крупноклеточную лимфому (DLBCL), лимфому из клеток мантийной зоны (MCL), фолликулярную лимфому (FL), лимфому лимфоидной ткани слизистых оболочек (MALT), лимфому из клеток маргинальной зоны, T-клеточную лимфому, ходжкинскую лимфому, лимфому Беркитта, множественную миелому, хронический лимфоцитарный лейкоз (CLL), мелкоклеточную лимфоцитарную лимфому (SLL), макроглобулинемию Вальденстрема, лимфобластный Т-клеточный лейкоз, хронический миелогенный лейкоз (CML), волосатоклеточный лейкоз, острый лимфобластный Т-клеточный лейкоз, плазмоцитому, иммунобластный крупноклеточный лейкоз, мегакариобластный лейкоз, острый мегакариобластный лейкоз, промиелоцитарный лейкоз, эритролейкоз, (формы глиомы) головного мозга, виды глиобластомы, рак молочной железы, колоректальный рак/рак толстой кишки, рак предстательной железы, рак легкого, в том числе немелкоклеточный, рак желудочно-кишечного тракта, рак эндометрия, меланому, рак поджелудочной железы, рак печени, рак почки, плоскоклеточную карциному, рак яичника, саркому, остеосаркому, рак щитовидной железы, рак мочевого пузыря, рак головы и шеи, рак яичка, саркому Юинга, рабдомиосаркому, медуллобластому, нейробластому, рак шейки матки, почечно-клеточный рак, уротелиальный рак, рак вульвы, рак пищевода, рак слюнных желез, рак носоглотки, рак щеки, рак ротовой полости и GIST (гастроинтестинальную стромальную опухоль).
В другом варианте осуществления ингибиторы MALT1 по настоящему изобретению можно использовать для лечения иммунологических заболеваний, в том числе без ограничения аутоиммунных и воспалительных нарушений, например, артрита, воспалительного заболевания кишечника, гастрита, анкилозирующего спондилита, язвенного колита, панкреатита, болезнь Крона, целиакию, рассеянный склероз, системную красную волчанку, волчаночный нефрит, ревматическую атаку, подагру, отторжение органов или трансплантатов, хроническое отторжение аллотрансплантата, острую или хроническую реакцию "трансплантат против хозяина", дерматит, в том числе атопический, дерматомиозит, псориаз, болезнь Бехчета, увеит, тяжелую миастению, болезнь Грейвса, тиреоидит Хашимото, синдром Шегрена, нарушения при пузырчатке, синдромы васкулита, опосредованного антителами, иммунокомплексный васкулит, аллергические нарушения, астму, бронхит, хроническое обструктивное заболевание легкого (COPD), муковисцидоз, пневмонию, заболевания легкого, включая отек, эмболию, фиброз, саркоидоз, гипертонию и эмфизему, силикоз, дыхательную недостаточность, острый респираторный дистресс-синдром, заболевание BENTA, бериллиоз и полимиозит.
В другом варианте осуществления настоящего изобретения соединения по настоящему изобретению можно использовать в комбинации с одним или несколькими другими медицинскими средствами, более конкретно с другими противораковыми средствами, например, химиотерапевтическими, антипролиферативными или иммуномодулирующими средствами, или со вспомогательными средствами в терапии рака, например, иммуносупрессорами или противовоспалительными средствами.
ОБЩИЕ СПОСОБЫ СИНТЕЗА
В этом разделе, как и во всех других разделах, если в контексте не указано иное, ссылки на формулу (I) также включают все другие подгруппы и их примеры, определенные в данном документе.
Общие способы получения некоторых типичных примеров соединений формулы (I) описаны в данном документе и в конкретных примерах, и, как правило, их получают из исходных веществ, являющихся либо коммерчески доступными, либо получаемыми с помощью стандартных способов синтеза, обычно применяемых специалистами в области органической химии. Подразумевается, что следующие схемы только демонстрируют примеры настоящего изобретения и ни в коей мере не ограничивают настоящее изобретение.
В качестве альтернативы, промежуточные соединения или соединения по настоящему изобретению также можно получать с помощью протоколов реакций, аналогичных нижеописанным на общих схемах и в конкретных примерах, в комбинации со стандартными способами синтеза, традиционно применяемыми специалистами в данной области техники, в том числе также аналогичных протоколам реакций, описанным в WO 2018020474, WO 2015181747 и WO 2017081641.
Специалисту в данной области техники будет понятно, что в реакциях, описанных на схемах, хотя это и не всегда явно показано, может потребоваться защита реакционноспособных функциональных групп (например гидрокси-, амино- или карбоксигрупп), если они являются необходимыми в конечном продукте, для того, чтобы избежать их нежелательного участия в реакциях. В целом, можно применять традиционные защитные группы в соответствии со стандартной практикой. Защитные группы можно удалять на подходящей последующей стадии с применением способов, известных из уровня техники.
Специалисту в данной области техники будет понятно, что в реакциях, описанных на схемах, может быть целесообразным или необходимым проводить реакцию в инертной атмосфере, например, в такой как атмосфера газообразного N2, например, при использовании в реакции NaH, LDA или MeMgBr.
Специалисту в данной области техники будет очевидно, что может быть необходимым охлаждать реакционную смесь перед выделением продукта реакции (касается ряда манипуляций, необходимых для выделения и очистки продукта(продуктов) химической реакции, таких как, например, гашение, колоночная хроматография, экстракция).
Специалисту в данной области техники будет понятно, что нагревание реакционной смеси при перемешивании может увеличить выход реакции. В некоторых реакциях можно применять нагревание с помощью микроволнового излучения вместо традиционного нагревания для сокращения общего времени реакции.
Специалисту в данной области техники будет понятно, что другая последовательность химических реакций, показанная на схемах ниже, также может приводить к образованию требуемого соединения формулы (I).
Специалисту в данной области техники будет понятно, что промежуточные соединения и конечные соединения, показанные на схемах ниже, могут быть дополнительно функционализированы в соответствии со способами, общеизвестными специалисту в данной области техники. Промежуточные соединения и соединения, описанные в данном документе, можно выделять в свободной форме или в виде их соли или сольвата. Промежуточные соединения и соединения, описанные в данном документе, могут быть синтезированы в виде смеси таутомеров и стереоизомерных форм, которые можно отделить друг от друга в соответствии с процедурами разделения, известными в данной области техники.
В отношении сокращений, применяемых на нижеизложенных схемах, см. таблицу с сокращениями из раздела "Примеры".
Общая схема 1
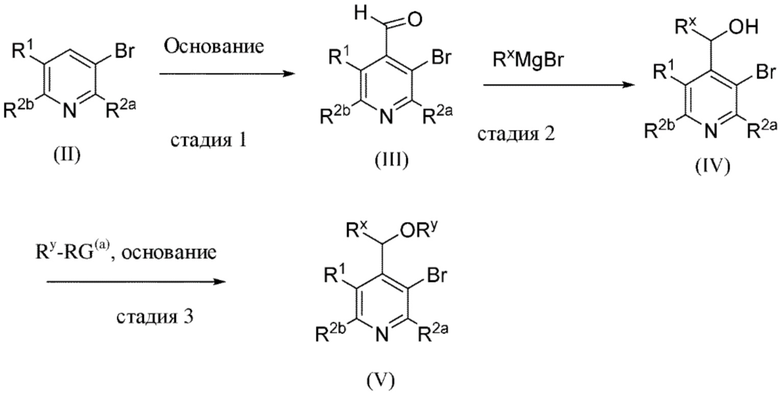
На схеме 1 "RG(a)" определена как подходящая реакционноспособная группа, такая как, например, йод, бром или тозил. В частности, схема 1 может использоваться для получения промежуточных соединений, где Rx и Ry не взяты вместе. Все другие переменные на схеме 1 определены в пределах объема настоящего изобретения.
На схеме 1, как правило, применяются следующие условия реакции.
1. Промежуточное соединение формулы (II) вводят в реакцию с основанием, таким как диизопропиламид лития (LDA), как правило, в апротонном растворителе, таком как, например, безводный THF, в пределах подходящего температурного диапазона, такого как, например, от −70°C до комнатной температуры, и в присутствии донора формильной группы, такого как DMF.
2. Промежуточное соединение формулы (III) вводят в реакцию с реактивом Гриньяра RxMgBr, как правило, в апротонном растворителе, таком как, например, безводный THF, в пределах подходящего температурного диапазона, такого как, например, от 0°C до комнатной температуры.
3. Промежуточное соединение формулы (IV) вводят в реакцию с алкилирующим средством Ry-RG(a), как правило в апротонном растворителе, таком как, например, безводный THF, и в присутствии подходящего основания, такого как гидрид натрия (NaH) или трет-бутоксид калия (KOtBu), или аналогичного им, в пределах подходящего температурного диапазона, такого как, например, от 0°C до комнатной температуры.
Общая схема 1a
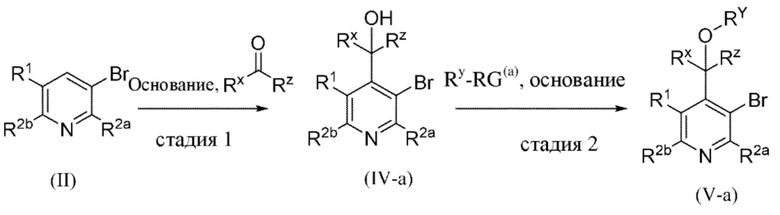
На схеме 1a "RG(a)" определена как подходящая реакционноспособная группа, такая как, например, йод, бром, тозил. В частности, схема 1a может использоваться для получения промежуточных соединений, где Rx и Ry не взяты вместе. Все другие переменные на схеме 1a определены в пределах объема настоящего изобретения.
На схеме 1a, как правило, применяются следующие условия реакции.
1. Промежуточное соединение формулы (II) вводят в реакцию с основанием, таким как LDA, как правило в апротонном растворителе, таком как, например, безводный THF, в пределах подходящего температурного диапазона, такого как, например, от −70°C до комнатной температуры, и в присутствии источника карбонила, Rx-C(O)-Ry.
2. Промежуточное соединение формулы (IV-a) вводят в реакцию с алкилирующим средством Ry-RG(a), как правило, в апротонном растворителе, таком как, например, безводный THF, и в присутствии подходящего основания, такого как (NaH) или аналогичного ему, в пределах подходящего температурного диапазона, такого как, например, от 0°C до комнатной температуры.
Общая схема 2
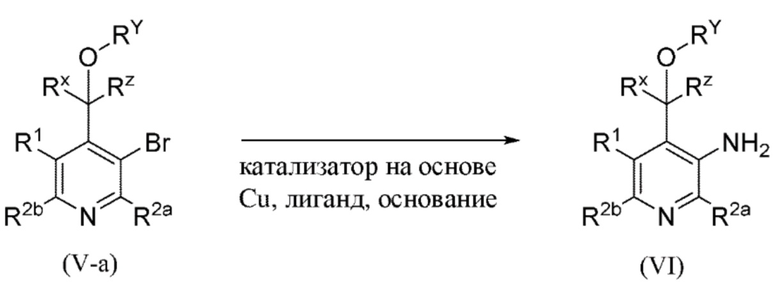
Все переменные на схеме 2 определены в пределах объема настоящего изобретения.
На схеме 2, как правило, применяются следующие условия реакции.
Промежуточное соединение формулы (V-a) вводят в реакцию с источником аминогруппы, таким как водный раствор аммиака, как правило, в растворителе, таком как, например, DMSO, в присутствии медного катализатора, такого как, например, йодид меди(I) (CuI), добавки, такой как L-пролин, и основания, такого как карбонат калия, в пределах подходящего температурного диапазона, такого как, например, от 60°C до 120°C.
Общая схема 2a
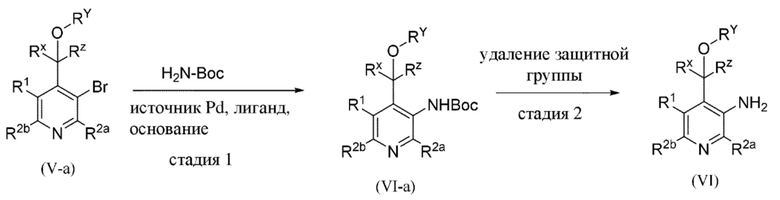
Все переменные на схеме 2a определены в пределах объема настоящего изобретения.
На схеме 2a, как правило, применяются следующие условия реакции.
1. Промежуточное соединение формулы (Va) вводят в реакцию с источником аминогруппы, таким как H2N-Boc ("Boc" означает "трет-бутилоксикарбонил"), как правило, в растворителе, таком как, например, толуол или 1,4-диоксан, в присутствии палладиевого катализатора, такого как ацетат палладия (Pd(OAc)2) или трис(дибензилиденацетон)дипалладий(0) (Pd2(dba)3), лиганда, такого как 4,5-бис(дифенилфосфино)-9,9-диметилксантен (xantphos) и основания, такого как карбонат цезия, в пределах подходящего температурного диапазона, такого как, например, от 100°C до 125°C;
2. В присутствии подходящей кислоты, такой как, например, трифторуксусная кислота (TFA) в дихлорметане (DCM), при подходящем температурном диапазоне, таком как, например, от 0°C до комнатной температуры.
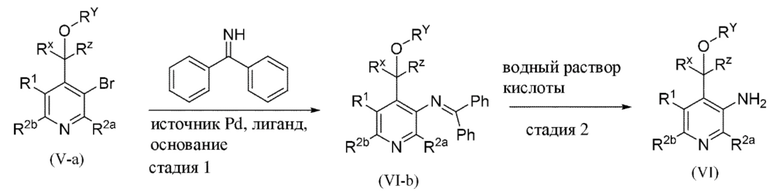
Общая схема 2b
Все переменные на схеме 2b определены в пределах объема настоящего изобретения.
На схеме 2b, как правило, применяются следующие условия реакции.
1. Промежуточное соединение формулы (Va) вводят в реакцию с источником аминогруппы, таким как дифенилметанимин, как правило, в растворителе, таком как, например, 1,4-диоксан, в присутствии палладиевого катализатора, такого как Pd2(dba)3, лиганда, такого как xantphos или BINAP, и основания, такого как трет-бутоксид натрия, в пределах подходящего температурного диапазона, такого как, например, от 80°C до 125°C.
2. В присутствии подходящей кислоты, такой как, например, водный раствор HCl, при концентрации от 1 М до 4 М в дихлорметане (DCM), при подходящем температурном диапазоне, таком как, например, от 20°C до 40°C.
Общая схема 3
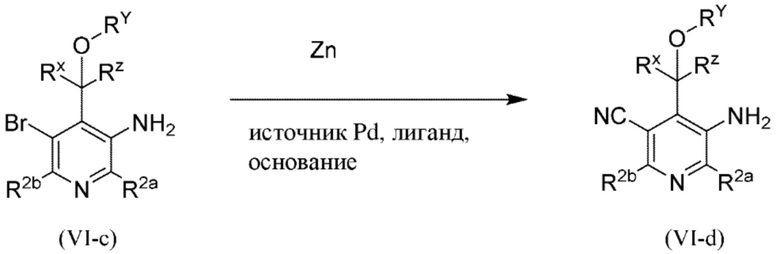
Все переменные на схеме 3 определены в пределах объема настоящего изобретения.
На схеме 3, как правило, применяются следующие условия реакции.
Промежуточное соединение формулы (VI-c) вводят в реакцию с источником цианогруппы, таким как цианид цинка, в присутствии цинка, как правило, в растворителе, таком как, например, DMF, в присутствии палладиевого катализатора, такого как Pd2(dba)3 или Pd(dppf)Cl2, в присутствии лиганда, такого как dppf, в пределах подходящего температурного диапазона, такого как, например, от 100°C до 120°C.
Общая схема 4
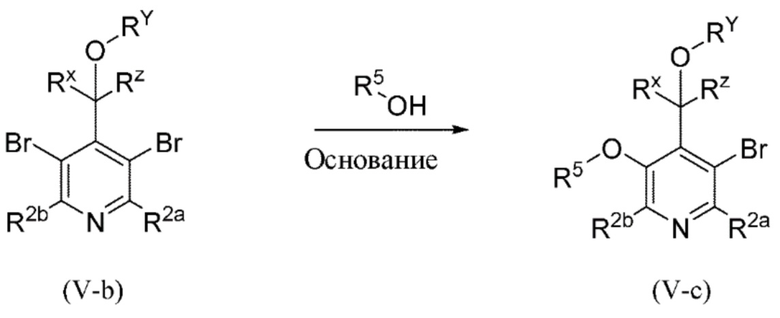
Все переменные на схеме 4 определены в пределах объема настоящего изобретения. Тем не менее специалисту в данной области техники будет понятно, что R5 является отличным от водорода.
На схеме 4, как правило, применяются следующие условия реакции.
Промежуточное соединение формулы (V-b) вводят в реакцию со спиртом R5-OH и основанием, таким как гидрид натрия, в присутствии катализатора, такого как порошок меди, как правило, в растворителе, таком как, например, DMF, в пределах подходящего температурного диапазона, такого как, например, от 20°C до 80°C.
Общая схема 4a
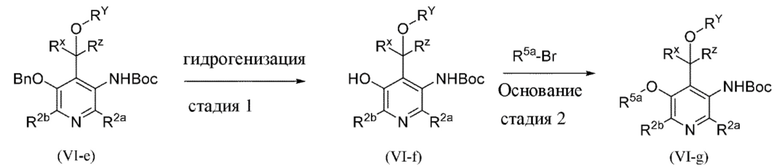
На схеме 4a "R5a" определен как C1-4алкил (необязательно замещенный) или C3-6циклоалкил; все другие переменные на схеме 4a определены в пределах объема настоящего изобретения.
На схеме 4a, как правило, применяются следующие условия реакции.
1. Промежуточное соединение формулы (VI-e) вводят в реакцию с газообразным водородом, как правило, при давлении 15 фунтов/кв. дюйм, в присутствии палладиевого катализатора, такого как палладий на угле, необязательно в присутствии кислоты, такой как хлористоводородная кислота, в подходящем растворителе, таком как метанол или THF, при подходящей температуре, составляющей 25°C.
2. В присутствии подходящего алкилирующего средства, такого как R5a-Br, в присутствии добавки, такой как йодид натрия, и подходящего основания, такого как карбонат цезия, в подходящем растворителе, таком как DMF или DMA, в пределах подходящего температурного диапазона, такого как, например, от 20°C до 140°C.
Общая схема 5
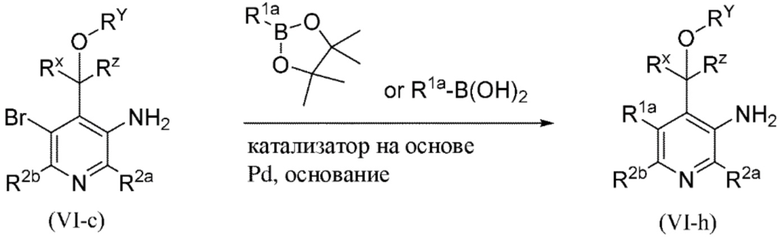
На схеме 5 "R1a" определен как C1-4алкил, C2-4алкенил или C3-6циклоалкил. Все другие переменные на схеме 5 определены в пределах объема настоящего изобретения.
На схеме 5, как правило, применяются следующие условия реакции.
Промежуточное соединение формулы (VI-c) вводят в реакцию со сложным эфиром бороновой кислоты, как правило, в растворителе, таком как, например, 1,4-диоксан или толуол, необязательно в присутствии воды, и в присутствии палладиевого катализатора, такого как [1,1′-бис(ди-трет-бутилфосфино)ферроцен]дихлорпалладий(II) (Pd(dtbpf)Cl2 (CAS 95408-45-0)), и подходящего основания, такого как фосфат калия, в пределах подходящего температурного диапазона, такого как, например, от 90°C до 120°C. В качестве альтернативы, реакцию можно осуществлять с применением подходящей бороновой кислоты R1a-B(OH)2, в присутствии палладиевого катализатора, такого как Pd(OAc)2, и подходящего лиганда, такого как трициклогексилфосфин, в присутствии основания, такого как фосфат калия, в подходящем растворителе, таком как, например, 1,4-диоксан или толуол, необязательно в присутствии воды, в пределах подходящего температурного диапазона, такого как, например, от 100°C до 140°C.
Общая схема 5a
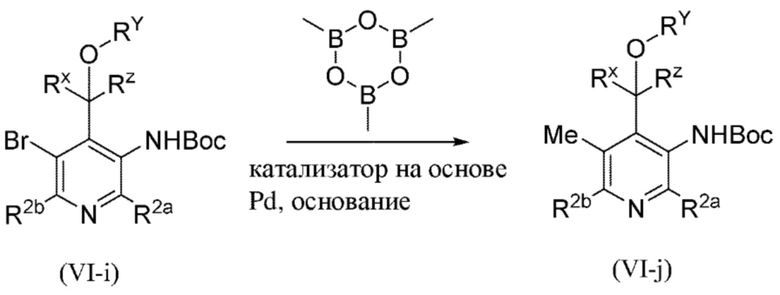
На схеме 5a все переменные определены в пределах объема настоящего изобретения.
На схеме 5a, как правило, применяются следующие условия реакции.
Промежуточное соединение формулы (VI-i) вводят в реакцию с 2,4,6-триметил-1,3,5,2,4,6-триоксатриборинаном (CAS 823-96-1), как правило, в растворителе, таком как, например, 1,4-диоксан или толуол, необязательно в присутствии воды, и в присутствии палладиевого катализатора, такого как тетракис(трифенилфосфин)палладий(0) (Pd(PPh3)4), и подходящего основания, такого как карбонат калия, в пределах подходящего температурного диапазона, такого как, например, от 90°C до 120°C.
Общая схема 5b

На схеме 5b "R1b" определен как C1-4алкил, C3-6циклоалкил или Het (например, оксетан). Все другие переменные на схеме 5b определены в пределах объема настоящего изобретения.
На схеме 5b, как правило, применяются следующие условия реакции.
Промежуточное соединение формулы (VI-i) вводят в реакцию с соединением R1b-Br в присутствии каталитической системы, состоящей из иридиевого катализатора, такого как гексафторфосфат [4,4'-бис(трет-бутил)-2,2'-бипиридин]-бис[3,5-дифтор-2-[5-(трифторметил)-2-пиридинил]фенил]иридия(III) ((Ir[dF(CF3)ppy]2(dtbpy))PF6 (CAS 870987-63-6)), и никелевого каталитического комплекса, такого как NiCl2·глим, в присутствии лиганда, такого как 4,4′-ди-трет-бутил-2,2′-бипиридин (CAS 72914-19-3). Для данной реакции также требуется присутствие трис(триметилсилил)силана, и она протекает под действием облучения, например, при использовании света от синего LED, в растворителе, подобном DME, при подходящей температуре, такой как 25°C.
Общая схема 6
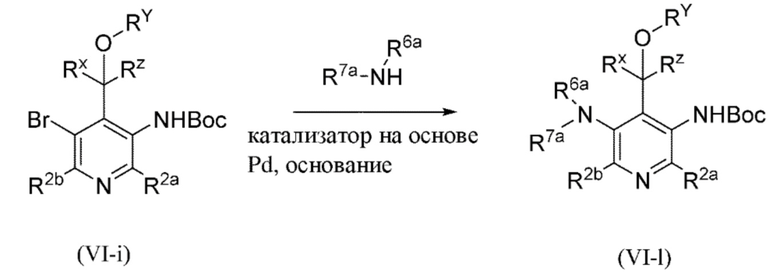
На схеме 6 все переменные определены в пределах объема настоящего изобретения.
На схеме 6, как правило, применяются следующие условия реакции.
Промежуточное соединение формулы (VI-i) вводят в реакцию с амином R7a-NH-R6a, как правило, в растворителе, таком как, например, толуол, в присутствии палладиевого катализатора, такого как Pd2(dba)3, и подходящего основания, такого как трет-бутоксид натрия, в пределах подходящего температурного диапазона, такого как, например, от 100°C до 140°C.
Общая схема 7
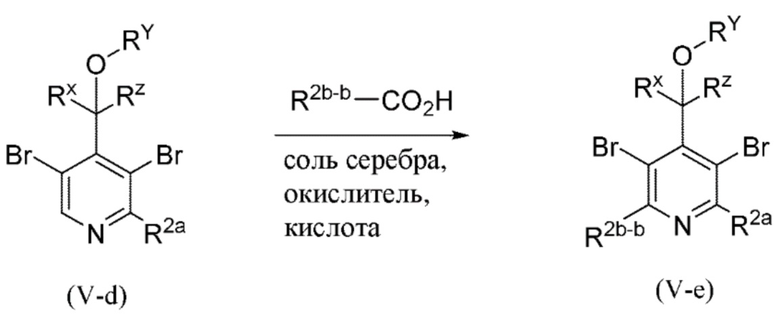
На схеме 7 "R2b-b" определен как C1-4алкил, необязательно замещенный 1, 2 или 3 атомами галогена, или
C3-6циклоалкил. Все другие переменные на схеме 7 определены в пределах объема настоящего изобретения.
На схеме 7, как правило, применяются следующие условия реакции.
Промежуточное соединение формулы (V-d) вводят в реакцию с карбоновой кислотой R2b-b-CO2H в присутствии окислителя, такого как персульфат аммония, соли серебра, такой как нитрат серебра, необязательно в присутствии сильной кислоты, такой как серная кислота. Реакция протекает в растворителе, таком как ацетонитрил или DMSO, в пределах подходящего температурного диапазона, такого как от 60°C до 100°C.
Специалисту в данной области техники будет понятно, что в случае, если R2a представляет собой водород, то возможно, что реакция, описанная на схеме 7, будет протекать повторно.
Общая схема 8
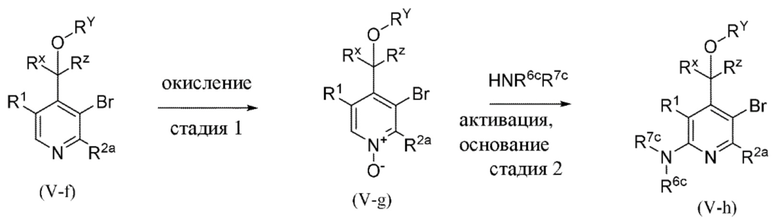
На схеме 8 все переменные определены в пределах объема настоящего изобретения.
На схеме 8, как правило, применяются следующие условия реакции.
1. Промежуточное соединение формулы (V-f) вводят в реакцию с окислителем, таким как м-CPBA, в подходящем растворителе, таком как дихлорметан, при подходящей температуре, составляющей от 0°C до 25°C.
2. В присутствии амина HNR6cR7c, в присутствии активирующего средства, такого как PyBrOP, и подходящего основания, такого как DIPEA, в подходящем растворителе, таком как THF, в пределах подходящего температурного диапазона, такого как, например, от 60°C до 80°C.
Общая схема 9
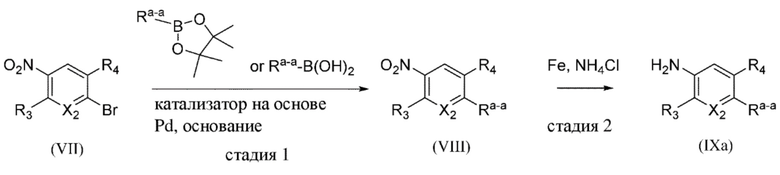
На схеме 9 X1 ограничен CRa, в котором Ra ограничен Ra-a, который определяется как C1-4алкил или C3-6циклоалкил. Все другие переменные на схеме 9 определены в пределах объема настоящего изобретения.
На схеме 9, как правило, применяются следующие условия реакции.
1. Промежуточное соединение формулы (VII) вводят в реакцию с бороновой кислотой или сложным эфиром бороновой кислоты, содержащим амин Ra-a в качестве заместителя, как правило, в растворителе, таком как, например, толуол и вода, в присутствии палладиевого катализатора, такого как Pd(OAc)2, и подходящего основания, такого как фосфат калия, необязательно в присутствии лиганда, такого как трициклогексилфосфин (PCy3), в пределах подходящего температурного диапазона, такого как, например, от 100°C до 140°C.
2. В присутствии восстановителя, такого как железо, в присутствии хлорида аммония в подходящей смеси растворителей, такой как метанол, THF и вода, в пределах подходящего температурного диапазона, такого как, например, от 25°C до 65°C.
Общая схема 10
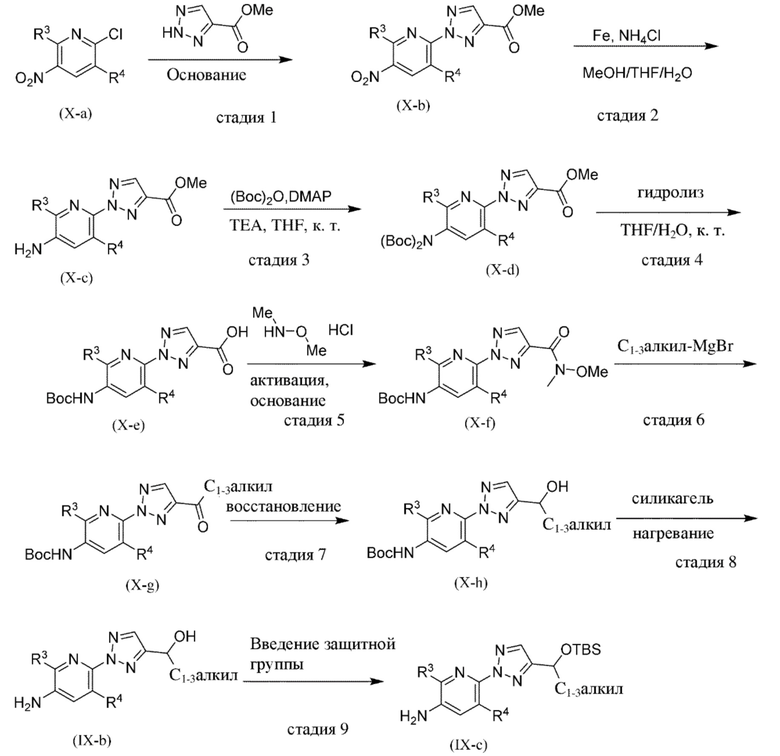
На схеме 10 все переменные определены в пределах объема настоящего изобретения.
На схеме 10, как правило, применяются следующие условия реакции.
1. Промежуточное соединение формулы (X-a) вводят в реакцию с метил-2H-1,2,3-триазол-4-карбоксилатом, как правило, в растворителе, таком как, например, ацетонитрил, в присутствии подходящего основания, такого как карбонат калия, в пределах температурного диапазона, такого как, например, от 25°C до 50°C.
2. В присутствии восстановителя, такого как железо, в присутствии хлорида аммония в подходящей смеси растворителей, такой как метанол, THF и вода, в пределах подходящего температурного диапазона, такого как, например, от 25°C до 65°C.
3. С применением защитной группы в виде Boc2O, с использованием подходящего основания, такого как DMAP, необязательно в присутствии триэтиламина, в подходящем растворителе, таком как THF, при подходящей температуре, такой как, например, комнатная температура.
4. Гидролиз с применением гидроксида лития, в подходящей смеси растворителей, такой как THF и вода, при подходящей температуре, такой как, например, комнатная температура.
5. Образование амида Вайнреба с применением гидрохлорида N, O-диметилгидроксиламина, в присутствии активирующего средства, такого как HATU, и подходящего основания, такого как DIPEA, в подходящем растворителе, таком как DMF, при подходящей температуре, такой как, например, комнатная температура.
6. Промежуточное соединение формулы (X-f) вводят в реакцию с реактивом Гриньяра C1-3алкил-MgBr, как правило, в апротонном растворителе, таком как, например, безводный THF, в пределах подходящего температурного диапазона, такого как, например, от 0°C до комнатной температуры.
7. Восстановление с применением, например, борогидрида натрия, в подходящем растворителе, таком как метанол, при подходящей температуре, такой как, например, комнатная температура.
8. Удаление защитной группы с применением слабой кислоты, такой как, например, силикагель, в подходящем растворителе, таком как толуол, при подходящей температуре, такой как от 100°C до 120°C.
Общая схема 10a
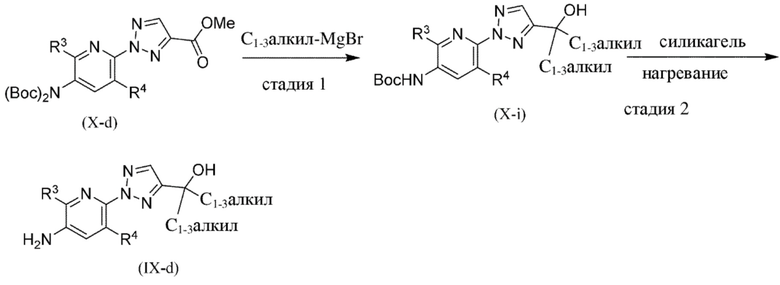
На схеме 10a все переменные определены в пределах объема настоящего изобретения.
На схеме 10a, как правило, применяются следующие условия реакции:
1. Промежуточное соединение формулы (X-d) вводят в реакцию с реактивом Гриньяра C1-3алкил-MgBr, как правило, в апротонном растворителе, таком как, например, безводный THF, в пределах подходящего температурного диапазона, такого как, например, от 0°C до комнатной температуры.
2. Удаление защитной группы с применением слабой кислоты, такой как, например, силикагель, в подходящем растворителе, таком как толуол, при подходящей температуре, такой как от 100°C до 120°C.
Общая схема 11
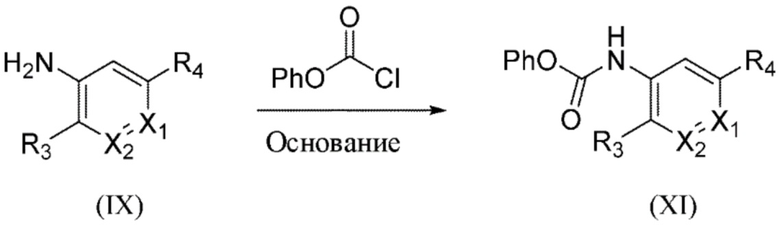
На схеме 11 все переменные определены в пределах объема настоящего изобретения.
На схеме 11, как правило, применяются следующие условия реакции.
Промежуточное соединение формулы (IX) вводят в реакцию с фенилхлорформиатом, как правило, в растворителе, таком как, например, THF, в присутствии подходящего основания, такого как пиридин, в пределах подходящего температурного диапазона, такого как, например, от 0°C до 20°C.
Специалисту в данной области техники будет понятно, что также, кроме фенилформиатных, могут применяться другие активирующие группы, например, изоцианатные.
Общая схема 12
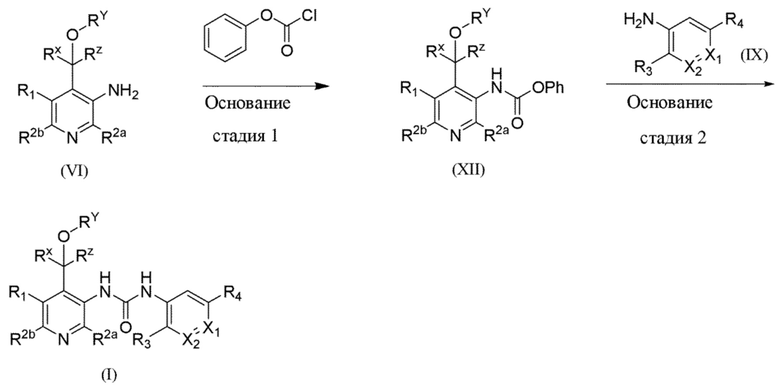
На схеме 12 все переменные определены в пределах объема настоящего изобретения.
На схеме 12, как правило, применяются следующие условия реакции.
1. Промежуточное соединение формулы (VI) вводят в реакцию с фенилхлорформиатом, как правило, в растворителе, таком как, например, THF, в присутствии подходящего основания, такого как пиридин, в пределах подходящего температурного диапазона, такого как, например, от 0°C до 20°C.
2. Промежуточное соединение формулы (XII) вводят в реакцию с промежуточным соединением формулы (IX) в подходящем растворителе, таком как THF, в присутствии подходящего основания, такого как триэтиламин или DMAP, или аналогичного им, в пределах подходящего температурного диапазона, такого как от 20°C до 80°C.
Специалисту в данной области техники будет понятно, что также, кроме фенилформиатных, могут применяться другие активирующие группы, например, изоцианатные.
Общая схема 13
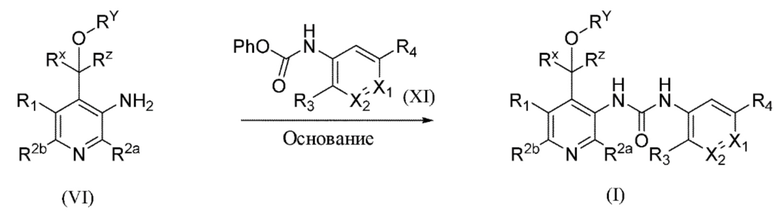
На схеме 13 все переменные определены в пределах объема настоящего изобретения.
На схеме 13, как правило, применяются следующие условия реакции.
Промежуточное соединение формулы (VI) вводят в реакцию с промежуточным соединением формулы (XI) в подходящем растворителе, таком как THF, в присутствии подходящего основания, такого как триэтиламин или DMAP, или аналогичного им, в пределах подходящего температурного диапазона, такого как от 20°C до 80°C.
Специалисту в данной области техники будет понятно, что также, кроме фенилформиатных, могут применяться другие активирующие группы, например, изоцианатные.
Общая схема 14
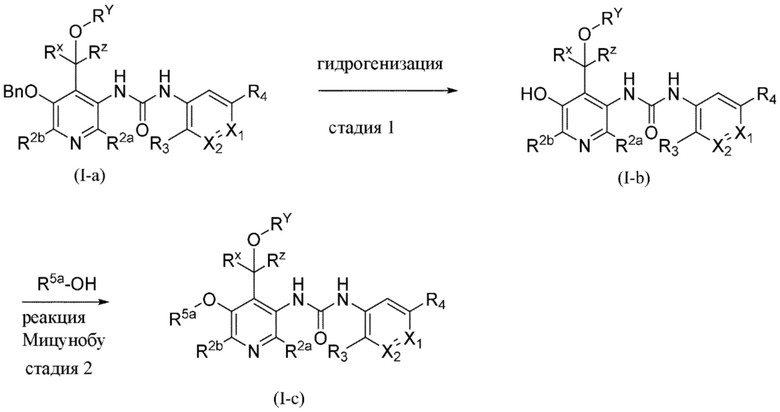
На схеме 14 "R5a" определен как C1-4алкил (необязательно замещенный) или C3-6циклоалкил. Все другие переменные на схеме 14 определены в пределах объема настоящего изобретения.
На схеме 14, как правило, применяются следующие условия реакции.
1. Соединение формулы (I-a) вводят в реакцию с газообразным водородом, как правило при давлении в 15 фунтов/кв. дюйм (фунтов на квадратный дюйм), в присутствии палладиевого катализатора, такого как палладий на угле, необязательно в присутствии кислоты, такой как хлористоводородная кислота, в подходящем растворителе, таком как метанол или THF, при подходящей температуре, составляющей 25°C.
2. В присутствии спирта R5a-OH, в присутствии (E)-диизопропилдиазен-1,2-дикарбоксилата (DIAD) и трифенилфосфина (PPh3), в подходящем растворителе, таком как THF или DMF, или аналогичном им, в пределах подходящего температурного диапазона, такого как, например, от 0°C до 40°C.
Общая схема 15
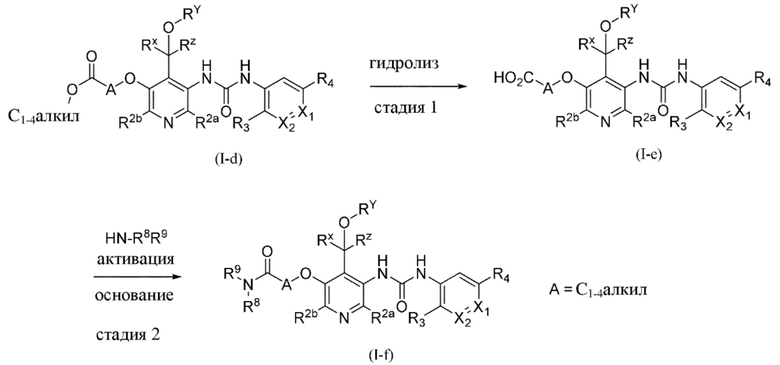
На схеме 15 "A" определен как C1-4алкил. Все другие переменные на схеме 15 определены в пределах объема настоящего изобретения.
На схеме 15, как правило, применяются следующие условия реакции.
1 . Гидролиз с применением гидроксида лития, в подходящей смеси растворителей, такой как THF и вода, и спирт, такой как этанол, при подходящей температуре, такой как, например, комнатная температура.
2. Образование амида из соединения (I-c) с применением амина с общей формулой HNR8R9, в присутствии активирующего средства, такого как HATU, и подходящего основания, такого как диизопропилэтиламин (DIPEA), в подходящем растворителе, таком как DMF, при подходящей температуре, такой как, например, комнатная температура.
Общая схема
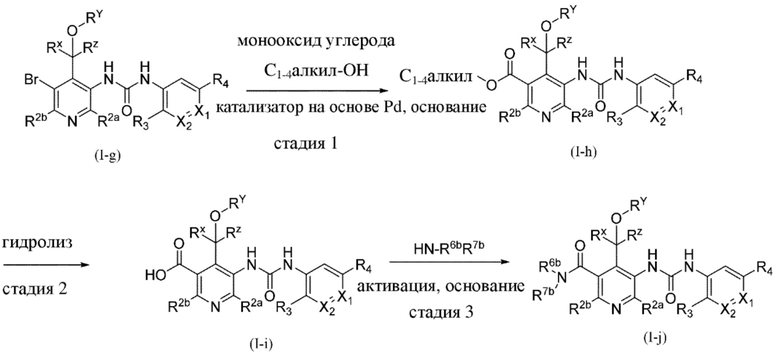
Все переменные на схеме 16 определены в пределах объема настоящего изобретения.
На схеме 16, как правило, применяются следующие условия реакции.
1 . Соединение формулы (I-g) обрабатывают в атмосфере монооксида углерода при подходящем давлении, например, 60 фунтов/кв. дюйм, в присутствии катализатора, такого как Pd(dppf)Cl2, в подходящем растворителе, таком как C1-4алкил-OH, необязательно в присутствии THF, в пределах подходящего температурного диапазона от 60°C до 100°C.
2. Гидролиз с применением гидроксида лития, в подходящей смеси растворителей, такой как вода и спирт, такой как метанол или этанол, необязательно в присутствии THF, в пределах подходящего температурного диапазона от 20°C до 40°C;
3. Образование амида из соединения (I-c) с использованием амина с общей формулой HNR6bR7b, в присутствии активирующего средства, такого как HATU, и подходящего основания, такого как диизопропилэтиламин (DIPEA), в подходящем растворителе, таком как DMF, при подходящей температуре, такой как, например, комнатная температура.
В ходе получения соединений по настоящему изобретению может оказаться необходимым обеспечение защиты труднодоступных функциональных групп (напр., первичного амина) у промежуточных соединений. Потребность в обеспечении такой защиты будет изменяться в зависимости от природы труднодоступных функциональных групп и от условий в способах их получения. Подходящие защитные группы для аминогрупп (NH-Pg) включают трет-бутоксикарбонил (Boc), ацетил и т.д. Необходимость в обеспечении такой защиты легко определяется специалистом в данной области техники.
Следует понимать, что если имеются соответствующие функциональные группы, соединения различных формул или какие-либо промежуточные соединения, используемые для их получения, можно подвергать дополнительной дериватизации с помощью одного или нескольких стандартных способов синтеза с применением реакций конденсации, замещения, окисления, восстановления или расщепления. Конкретные подходы относительно замещения включают традиционные процедуры алкилирования, арилирования, гетероарилирования, ацилирования, сульфонилирования, галогенирования, нитрования, формилирования и сочетания.
Соединения формулы (I) можно синтезировать в форме рацемических смесей энантиомеров, которые можно отделить друг от друга в соответствии с процедурами разделения, известными в данной области техники. Рацемические соединения формулы (I), содержащие основный атом азота, можно превращать в соответствующие диастереомерные солевые формы посредством реакции с подходящей хиральной кислотой. Затем указанные диастереомерные солевые формы разделяют, например, с помощью селективной или фракционной кристаллизации, а энантиомеры выделяют оттуда с помощью щелочи. Альтернативный способ разделения энантиомерных форм соединений формулы (I) включает жидкостную хроматографию с применением хиральной неподвижной фазы. Указанные чистые стереохимически изомерные формы также можно получать из соответствующих чистых стереохимически изомерных форм соответствующих исходных веществ при условии, что реакция протекает стереоспецифически.
В ходе получения соединений по настоящему изобретению может оказаться необходимым обеспечение защиты труднодоступных функциональных групп (например, первичного или вторичного амина) у промежуточных соединений. Потребность в обеспечении такой защиты будет изменяться в зависимости от природы труднодоступных функциональных групп и от условий в способах их получения. Подходящие защитные группы для аминогрупп (NH-Pg) включают ацетил, трифторацетил, трет-бутоксикарбонил (Boc), бензилоксикарбонил (CBz) и 9-флуоренилметиленоксикарбонил (Fmoc). Необходимость в обеспечении такой защиты легко определит специалист в данной области техники. Для общего описания защитных групп и их применения см. T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 4th ed., Wiley, Hoboken, Нью-Джерси, 2007.
Конкретные примеры
В следующих примерах некоторые продукты синтеза указаны как выделенные в виде остатка. Специалисту средней квалификации в данной области техники будет понятно, что термин "остаток" не ограничивает физическое состояние, в котором этот продукт был выделен, и может предусматривать, например, твердое вещество, масло, пену, смолу, сироп и т. п.
Специалисту в данной области техники будет понятно, что в примерах, описанных ниже, может быть целесообразным или необходимым (даже если не указано явно) проводить реакцию в инертной атмосфере, такой, например, как атмосфера газообразного N2, например, при использовании в реакции NaH, LDA или MeMgBr (например, синтез промежуточного соединения 55 или 56 проводили в инертной атмосфере).
Синтез соединений 1, 2 и 3
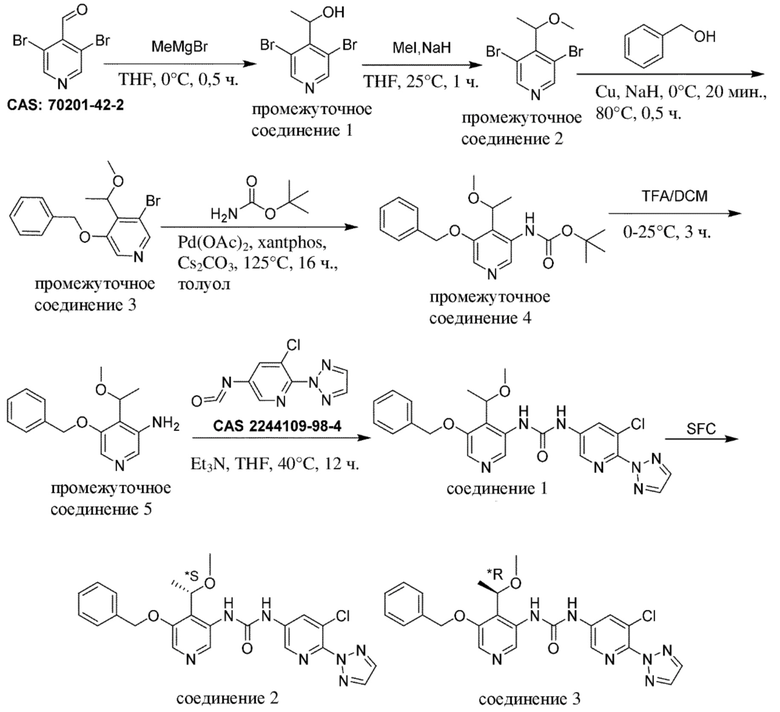
Получение промежуточного соединения 1
К раствору 3,5-дибромизоникотинальдегида (50 г, 189 ммоль) в THF (200 мл) добавляли бромид метилмагния (3 М в THF, 189 мл, 566 ммоль) при 0°C. Обеспечивали нагревание смеси до 20°C и перемешивали при 20°C в течение 1 часа. Смесь гасили насыщ. водн. раствором NH4Cl. Смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~15% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 1 (40 г, выход: 75%) в виде светло-желтого твердого вещества.
Получение промежуточного соединения 2
Промежуточное соединение 1 (40 г, 142 ммоль) растворяли в THF (150 мл) и добавляли гидрид натрия (60%, в минеральном масле, 8,5 г, 214 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 10 мин. Затем добавляли MeI (50,5 г, 356 ммоль) и смесь перемешивали при 25°C в течение 1 часа. Добавляли насыщ. водн. раствор NH4Cl и два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали под вакуумом с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (градиентное элюирование: 0~10% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 2 (39 г, выход: 93%) в виде белого твердого вещества.
Получение промежуточного соединения 3
К смеси бензилового спирта (7,3 г, 69 ммоль) в DMF (125 мл) добавляли гидрид натрия (60%, в минеральном масле, 2,7 г, 68 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 20 мин. По каплям добавляли раствор промежуточного соединения 2 (5,0 г, 17 ммоль) в DMF (25 мл). Затем к смеси добавляли порошок Cu (108 мг, 1,7 ммоль) и реакционную смесь перемешивали при 80°C в течение 0,5 часа. Обеспечивали нагревание смеси до 25°C и затем добавляли солевой раствор. Смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали над Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~5% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 3 (3,8 г, выход: 66%) в виде белого твердого вещества.
Получение промежуточного соединения 4
К смеси промежуточного соединения 3 (3,8 г, 11 ммоль) и трет-бутилкарбамата (2,6 г, 23 ммоль) в толуоле (150 мл) добавляли Cs2CO3 (14,7 г, 45,1 ммоль). Смесь дегазировали и затем на 10 мин заполняли N2. Затем добавляли Pd(OAc)2 (380 мг, 1,7 ммоль) и xantphos (652 мг, 1,1 ммоль) и смесь перемешивали при 125°C в течение 16 часов в атмосфере N2. Обеспечивали охлаждение смеси до 25°C и фильтровали. Фильтрат концентрировали с получением неочищенного продукта в виде желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~27% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 4 (2,8 г, выход: 68%) в виде белого твердого вещества.
Получение промежуточного соединения 5
К смеси промежуточного соединения 4 (2,8 г, 7,7 ммоль) в CH2Cl2 (45 мл) добавляли TFA (9 мл) при 0°C. Смесь нагревали до 25°C и перемешивали в течение 3 часов. Смесь подвергали нейтрализации насыщ. водн. раствором Na2CO3 и два раза экстрагировали с помощью CH2Cl2. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~70% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 5 (1,8 г, выход: 90%) в виде светло-желтого твердого вещества.
Получение соединения 1
К смеси 3-хлор-5-изоцианато-2-(2H-1,2,3-триазол-2-ил)-пиридина CAS 2244109-98-4 (1,1 г, 4,9 ммоль) и триметиламина (1,4 мл, 10 ммоль) в THF (20 мл) добавляли промежуточное соединение 5 (0,6 г, 2,3 ммоль) при 25°C. Смесь нагревали до 40°C и перемешивали в течение 12 часов. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта в виде светло-желтого твердого вещества. К неочищенному продукту добавляли MeOH и смесь перемешивали при 25°C в течение 15 мин. Затем смесь фильтровали, осадок на фильтре собирали и высушивали в вакууме с получением соединения 1 (1,0 г, выход: 89%) в виде белого твердого вещества.
LC/MS: масса/заряд 480,0 [M+H]+, способ: B, чистота: 99,5%, время удерживания: 0,727 мин.
Получение соединений 2 и 3
Соединение 1 (100 мг, 0,2 ммоль) разделяли посредством SFC. [Колонка: DAICEL CHIRALCEL OJ-H (250 мм*30 мм, 5 мкм), условия: A: сверхкритический CO2, B: 0,1% NH3 H2O EtOH; вначале: A (55%) и B (45%), в конце: A (55%) и B (45%), расход: 40 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 2 (42 мг, выход: 43%) и соединения 3 (46 мг, выход: 46%) в виде белого твердого вещества.
Соединение 2:
LC/MS: масса/заряд 480,2 [M+H]+, rt 3,47 мин. Чистота 100%, способ K
SFC: чистота 100%, rt 5,66 мин. Способ: SFC1
Соединение 3:
LC/MS: масса/заряд 480,2 [M+H]+, rt 3,47 мин. Чистота 100%, способ K
SFC: чистота 100%, rt 6,76 мин. Способ: SFC1
Синтез соединений 4, 5 и 6
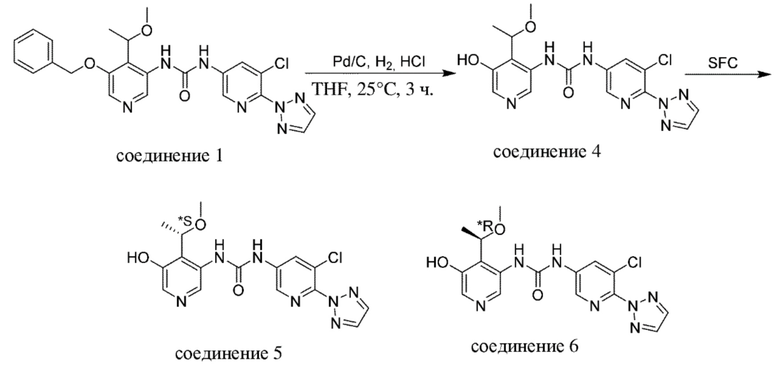
Получение соединения 4
Смесь соединения 1 (500 мг, 1,1 ммоль) в THF (100 мл) в присутствии концентрированной HCl (1 мл) гидрогенизировали при 25°C (15 фунтов на кв. дюйм) с Pd/C (500 мг, 10%, влажный) в качестве катализатора. Реакционную смесь перемешивали при 25°C в течение 3 часов. После поглощения H2 (1 экв.) катализатор отфильтровывали и фильтраты подвергали нейтрализации насыщ. водн. раствором NaHCO3 и два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и выпаривали в вакууме с получением соединения 4 (330 мг, выход: 75,9%) в виде белого твердого вещества.
LC/MS: масса/заряд 390,0 [M+H]+, rt: 0,77 мин, чистота: 93%, способ: A
Получение соединений 5 и 6
Соединение 4 (100 мг, 0,2 ммоль) отделяли посредством SFC. [Колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: растворитель, A: сверхкритический CO2. Растворитель, B: 0,1% водный раствор аммиака в EtOH. Вначале: A (65%) и B (35%), в конце: A (65%) и B (35%), расход: 70 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и смесь лиофилизировали до сухого состояния с получением соединения 5 (40 мг, выход: 42%) и соединения 6 (41 мг, выход: 43%) в виде белого твердого вещества.
Соединение 5:
HPLC-MS: масса/заряд 390,1 [M+H]+, rt: 2,84 мин, чистота: 97,2%, способ: M
SFC: чистота 100%, rt: 1,87 мин, способ: SFC9
Соединение 6:
HPLC-MS: масса/заряд 390,1 [M+H]+, rt: 2,84 мин, чистота: 95,9%, способ: M
SFC: чистота 99,6%, rt: 2,13 мин, способ: SFC9
Синтез соединения 7
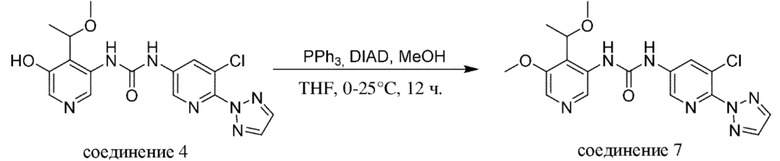
Получение конечного соединения 7
К смеси соединения 4 (250 мг, 0,6 ммоль), метанола (200 мг, 6,4 ммоль) и трифенилфосфина (336 мг, 1,3 ммоль) в THF (12 мл) добавляли (E)-диизопропилдиазен-1,2-дикарбоксилат (259 мг, 1,3 ммоль) при 0°C. Реакционную смесь перемешивали при 25°C в течение 16 часов. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии. [Колонка: Phenomenex Gemini 150×25 мм*10 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN. Вначале: A (82%) и B (18%), в конце: A (52%) и B (48%), время градиентного элюирования: 8 мин; 100% B, время удерживания: 2 мин; расход: 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 7 (146 мг, выход: 56,5%) в виде белого твердого вещества.
Соединение 7:
LC/MS: масса/заряд 404,1 [M+H]+, rt: 3,61 мин, чистота: 99,7%, способ: K.
SFC: чистота 48,6%/51,4%, rt: 6,30 мин/7,14 мин, способ: SFC6.
Синтез соединений 8, 9 и 10
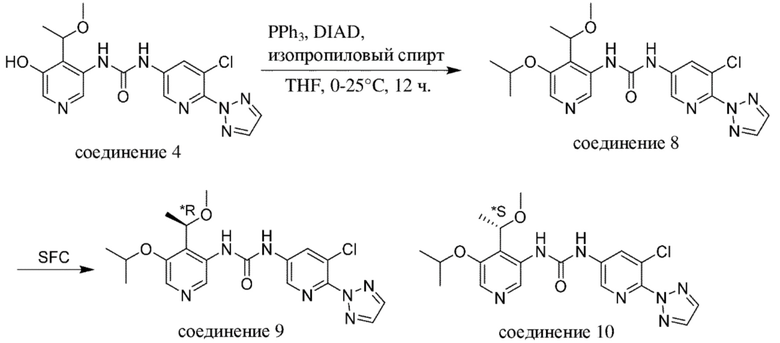
Получение соединения 8
Соединение 8 получали по аналогии с процедурой, описанной для соединения 7, с применением изопропилового спирта. Соединение очищали посредством препаративной высокоэффективной жидкостной хроматографии. [Колонка: Phenomenex Gemini 150 × 25 мм*10 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN. Вначале: A (60%) и B (40%), в конце: A (30%) и B (70%), время градиентного элюирования: 8 мин; 100% B, время удерживания: 2 мин; расход: 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 8 (100 мг, выход: 29,5%) в виде белого твердого вещества.
LC/MS: масса/заряд 432,0 [M+H]+, rt: 0,69 мин, чистота: 100%, способ: A.
SFC: чистота 49,6%/50,4%, rt: 1,64 мин/2,04 мин, способ: SFC9
Получение соединений 9 и 10
Соединение 8 (100 мг, 0,2 ммоль) отделяли посредством SFC. [Колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: растворитель, A: сверхкритический CO2. Растворитель, B: 0,1% водный раствор аммиака в EtOH. Вначале: A (55%) и B (45%), в конце: A (55%) и B (45%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и смесь подвергали лиофилизации до сухого состояния с получением соединения 9 (37 мг, выход: 37%) и соединения 10 (36 мг, выход: 35%) в виде белого твердого вещества.
Соединение 9:
LC/MS ESI-MS: масса/заряд 432,2 [M+H]+, rt: 3,87 мин, чистота: 99,8%, способ: K
SFC: чистота 100%, rt: 1,66 мин, способ: SFC9
Соединение 10:
LC/MS ESI-MS: масса/заряд 432,2 [M+H]+, rt: 3,88 мин, чистота: 98,4%, способ: K
SFC: чистота 99,8%, rt: 2,01 мин, способ: SFC9
Синтез соединения 11
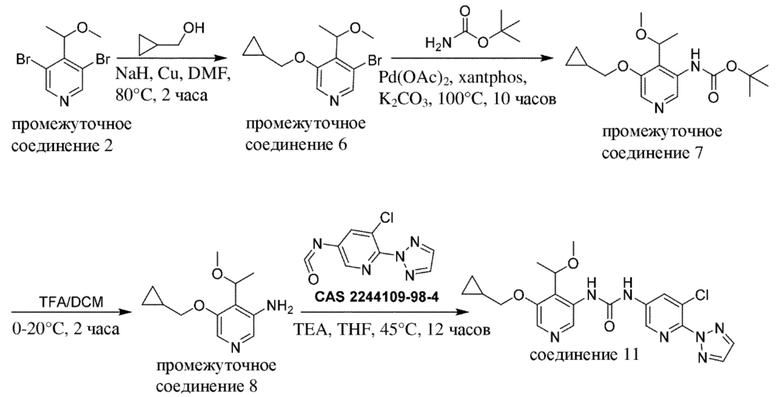
Получение промежуточного соединения 6
К смеси циклопропилметанола (978 мг, 13,6 ммоль) в DMF (20 мл) добавляли гидрид натрия (542 мг, 13,6 ммоль, 60%, в минеральном масле) и смесь перемешивали при 25°C в течение 1 часа. По каплям добавляли раствор промежуточного соединения 2 (1,0 г, 3,4 ммоль) в DMF (5 мл). Затем добавляли порошок меди (22 мг, 0,34 ммоль) и смесь перемешивали при 80°C в течение 0,5 часа. Обеспечивали охлаждение смеси до 25°C и гасили насыщ. водн. раствором NH4Cl. Смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~6% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 6 (600 мг, выход: 60%) в виде белого твердого вещества.
Получение промежуточного соединения 7
Смесь промежуточного соединения 6 (0,5 г, 1,7 ммоль), трет-бутилкарбамата (0,4 г, 3,4 ммоль) и Cs2CO3 (2,2 г, 6,8 ммоль) в толуоле (25 мл) дегазировали с помощью N2 в течение 10 мин. Затем добавляли Pd(OAc)2 (57 мг, 0,26 ммоль) и xantphos (98 мг, 0,17 ммоль) и смесь перемешивали при 120°C в течение 12 часов в атмосфере N2. Смесь охлаждали до 25°C и фильтровали. Фильтрат концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~27% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 7 (0,5 г, выход: 89,3%) в виде бесцветного масла.
Получение промежуточного соединения 8
К раствору промежуточного соединения 7 (0,3 г, 0,9 ммоль) в CH2Cl2 (5 мл) добавляли TFA (1 мл) при 25°C. Смесь перемешивали при 25°C в течение 2 часов. Добавляли насыщ. водн. раствор NaHCO3 и смесь два раза экстрагировали с помощью CH2Cl2. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~30% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 8 (0,3 г, выход: 96%) в виде светло-желтого твердого вещества.
Получение соединения 11
К раствору 3-хлор-5-изоцианато-2-(2H-1,2,3-триазол-2-ил)пиридина (CAS 2244109-98-4) (540 мг, 2,4 ммоль) и триэтиламина (0,7 мл, 4,8 ммоль) в THF (20 мл) добавляли раствор промежуточного соединения 8 (200 мг, 0,9 ммоль) в THF (5 мл) при 25°C. Реакционную смесь перемешивали при 40°C в течение 12 часов. Обеспечивали достижение температуры смеси, составляющей 25°C, и фильтровали. Фильтрат разбавляли с помощью H2O и полученную смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии. [Колонка: Phenomenex Gemini 150 × 25 мм*10 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN. Вначале: A (55%) и B (45%), в конце: A (25%) и B (75%), время градиентного элюирования: 8 мин; 100% B, время удерживания: 2 мин; расход: 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 11 (156 мг, выход: 40%) в виде белого твердого вещества.
Соединение 11:
LC/MS: масса/заряд 444,2 [M+H]+, rt: 3,97 мин, способ: K, чистота: 99,2%,
SFC: чистота 49,1%/50,9%, rt: 2,99 мин/3,29 мин, способ: SFC3
Синтез соединения 12
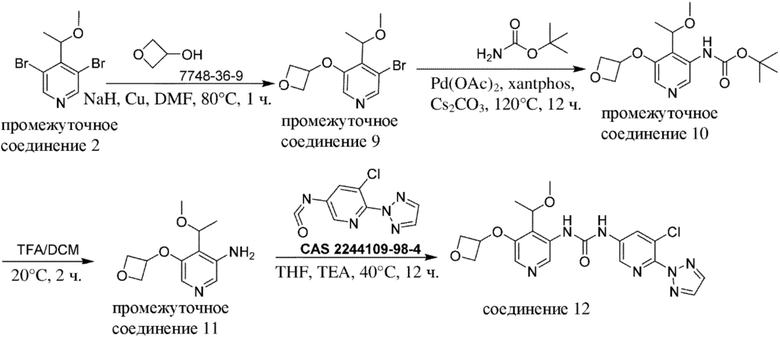
Получение промежуточного соединения 9
К смеси оксетан-3-ола (CAS 7748-36-9, 0,99 г, 13,3 ммоль) в DMF (20 мл) добавляли гидрид натрия (0,53 г, 13,3 ммоль, 60%, в минеральном масле) и смесь перемешивали при 20°C в течение 20 мин. По каплям добавляли раствор промежуточного соединения 2 (1,0 г, 3,3 ммоль) в DMF (5 мл). Затем добавляли порошок меди (22 мг, 0,34 ммоль) и смесь перемешивали при 80°C в течение 1 часа. Обеспечивали охлаждение реакционной смеси до 25°C и гасили насыщ. водн. раствором NH4Cl. Смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~22% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 9 (560 мг, выход: 56,7%) в виде белого твердого вещества.
Получение промежуточного соединения 10
Смесь промежуточного соединения 9 (560 мг, 1,9 ммоль), трет-бутилкарбамата (442 мг, 3,8 ммоль) и Cs2CO3 (2,5 г, 7,6 ммоль) в толуоле (35 мл) дегазировали и затем на 10 мин заполняли N2. Затем добавляли Pd(OAc)2 (64 мг, 0,28 ммоль) и xantphos (109 мг, 0,19 ммоль) и смесь перемешивали при 120°C в течение 16 часов в атмосфере N2. Обеспечивали достижение температуры смеси, составляющей 25°C, и фильтровали. Фильтрат концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~50% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 10 (600 мг, выход: 96%) в виде бесцветного масла.
Получение промежуточного соединения 11
К раствору промежуточного соединения 10 (300 мг, 0,9 ммоль) в CH2Cl2 (5 мл) добавляли TFA (1 мл) при 25°C. Смесь перемешивали при 25°C в течение 2 часов. Добавляли насыщ. водн. раствор NaHCO3 и смесь два раза экстрагировали с помощью CH2Cl2. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~100% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 11 (170 мг, выход: 83%) в виде белого твердого вещества.
Получение соединения 12
К раствору 3-хлор-5-изоцианато-2-(2H-1,2,3-триазол-2-ил)пиридина (CAS 2244109-98-4) (410 мг, 1,8 ммоль) в THF (15 мл) добавляли триэтиламин (0,5 мл, 3,6 ммоль) при 25°C. Добавляли раствор промежуточного соединения 11 (150 мг, 0,7 ммоль) в THF (5 мл), смесь нагревали до 40°C и перемешивали в течение 12 часов. Обеспечивали охлаждение реакционной смеси до 25°C и фильтровали. Фильтрат промывали с помощью H2O и два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии. [Колонка: Phenomenex Gemini 150 × 25 мм*10 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN. Вначале: A (77%) и B (23%), в конце: A (47%) и B (53%), время градиентного элюирования: 8 мин; 100% B, время удерживания: 2 мин; расход: 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 12 (113 мг, выход: 37%) в виде белого твердого вещества.
Соединение 12:
LC/MS: масса/заряд 446,1 [M+H]+, rt: 3,39 мин, чистота: 97,8%, способ: K.
SFC: чистота 49,3%/50,7%, rt: 3,69 мин/4,15 мин, способ: SFC 2
Синтез соединения 13
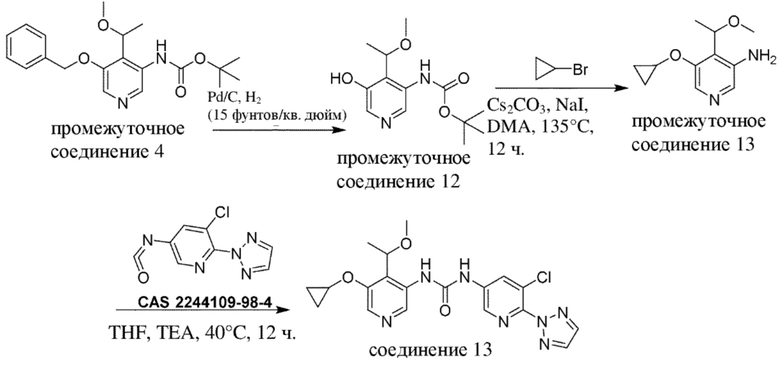
Получение промежуточного соединения 12
Смесь промежуточного соединения 4 (2 г, 5,6 ммоль) в MeOH (100 мл) гидрогенизировали при 25°C (15 фунтов на кв. дюйм) с Pd/C (1 г, 10%, влажный) в качестве катализатора. Реакционную смесь перемешивали при 25°C в течение 2 часов. После поглощения H2 (1 экв.) катализатор отфильтровывали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~30% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 12 (1,4 г, выход: 92%) в виде белого твердого вещества.
Получение промежуточного соединения 13
К смеси промежуточного соединения 12 (0,5 г, 1,8 ммоль), Cs2CO3 (1,8 г, 5,5 ммоль) и NaI (28 мг, 0,2 ммоль) в DMA (20 мл) добавляли бромциклопропан (0,45 г, 3,7 ммоль) при 25°C. Смесь перемешивали при 135°C в течение 12 часов. Обеспечивали охлаждение реакционной смеси до 25°C и фильтровали. К фильтрату добавляли H2O и смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~40% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 13 (100 мг, выход: 23,5%) в виде желтого твердого вещества.
Получение соединения 13
К раствору 3-хлор-5-изоцианато-2-(2H-1,2,3-триазол-2-ил)пиридина (CAS 2244109-98-4) (287 мг, 1,3 ммоль) в THF (15 мл) добавляли триэтиламин (0,9 мл, 6,5 ммоль). К реакционной смеси добавляли раствор промежуточного соединения 13 (100 мг, 0,4 ммоль) в THF (5 мл) при 25°C. Смесь нагревали до 40°C и перемешивали в течение 12 часов. Обеспечивали достижение температуры реакционной смеси, составляющей 25°C, и фильтровали. Фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии. [Колонка: Phenomenex Gemini 150 × 25 мм*10 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN. Вначале: A (65%) и B (35%), в конце: A (35%) и B (65%), время градиентного элюирования: 8 мин; 100% B, время удерживания: 2 мин; расход: 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 13 (32 мг, выход: 17%) в виде белого твердого вещества.
Соединение 13:
LC/MS: масса/заряд 430,1 [M+H]+, rt: 3,66 мин, чистота: 98,9%, способ: K.
SFC: чистота 49,9%/50,1%, rt: 3,72 мин/4,02 мин, способ: SFC 4
Синтез соединений 14, 15 и 16
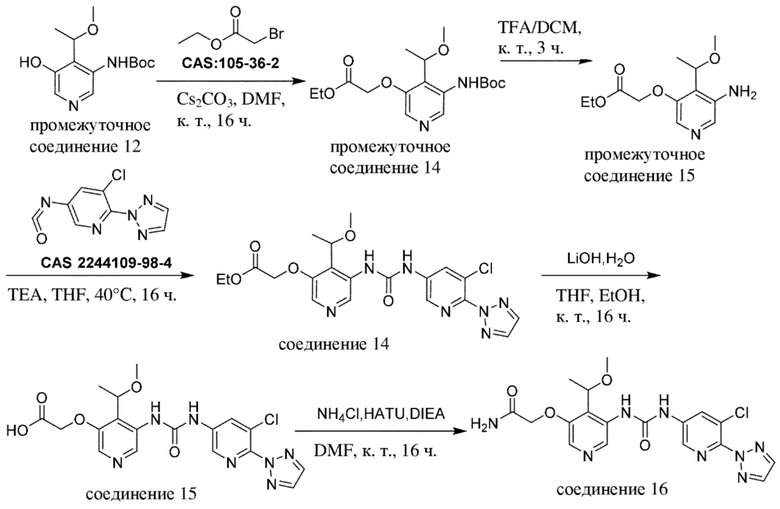
Получение промежуточного соединения 14
К смеси промежуточного соединения 12 (200 мг, 0,74 ммоль) и этилбромацетата (249 мг, 1,5 ммоль) в DMF (5 мл) добавляли Cs2CO3 (971 мг, 3 ммоль). Смесь перемешивали при 25°C в течение 16 часов. Смесь гасили солевым раствором и два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~50% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 14 (130 мг, выход: 42%) в виде желтого масла.
Получение промежуточного соединения 15
К раствору промежуточного соединения 14 (260 мг, 0,7 ммоль) в DCM (10 мл) добавляли TFA (2 мл) при 0°C. Смесь перемешивали при 25°C в течение 3 часов. Большую часть растворителя удаляли под вакуумом с получением желтой смолы. Желтую смолу растворяли в CH2Cl2. Добавляли насыщ. водн. раствор Na2CO3 к смеси и смесь два раза экстрагировали с помощью CH2Cl2. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~25% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 15 (160 мг, выход: 86%) в виде желтого твердого вещества.
Получение соединения 14
К раствору 3-хлор-5-изоцианато-2-(2H-1,2,3-триазол-2-ил)пиридина (CAS 2244109-98-4) (471 мг, 2,1 ммоль) в THF (10 мл) добавляли триэтиламин (1,6 мл, 11,8 ммоль). Затем добавляли раствор промежуточного соединения 15 (150 мг, 0,6 ммоль) в THF (5 мл) при 25°C. Смесь перемешивали при 40°C в течение 16 часов. Смесь концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт перемешивали в (смеси петролейного эфира/этилацетата=1:1) в течение 10 мин. Смесь фильтровали и фильтрат концентрировали под вакуумом с получением неочищенного продукта в виде желтого твердого вещества. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии. [Колонка: Xtimate C18 10μ, 250 мм*50 мм, условия: A: вода (0,04% NH3 H2O+10 мМ NH4HCO3), B: MeCN. Вначале: A (60%) и B (40%), в конце: A (30%) и B (70%), время градиентного элюирования: 8 мин; 100% B, время удерживания: 0 мин; расход: 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 14 (150 мг, выход: 33%) в виде белого твердого вещества.
LC/MS: масса/заряд 476,1 [M+H]+, rt 3,77 мин, чистота 95,1%, способ K.
SFC: чистота 49,9%/50,1%, rt: 4,26 мин/4,56 мин, способ: SFC4
Получение соединения 15
К раствору соединения 14 (200 мг, 0,2 ммоль) в THF (4 мл), H2O (1 мл), EtOH (0,2 мл) добавляли LiOH (50 мг, 1,2 ммоль). Смесь перемешивали при 25°C в течение 16 часов. К смеси добавляли воду и смесь экстрагировали с помощью EtOAc. Значение pH водного слоя доводили до 6 с помощью HCl (2 М в воде). Водный слой два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением соединения 15 (200 мг, неочищенное) в виде белого твердого вещества.
LC/MS: масса/заряд 448,1 [M+H]+, rt: 1,04 мин, чистота: 49,4%, способ: E
Получение соединения 16
К раствору соединения 15 (180 мг, 0,2 ммоль) и NH4Cl (32 мг, 0,59 ммоль) в DMF (20 мл) добавляли HATU (113 мг, 0,3 ммоль) и N, N-диизопропилэтиламин (0,1 мл, 0,6 ммоль) при 25°C. Смесь перемешивали при 25°C в течение 16 часов. Реакционную смесь фильтровали. Фильтраты концентрировали под вакуумом с получением неочищенного продукта в виде коричневого масла. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии. [Колонка: Xtimate C18 10μ, 250 мм*50 мм, условия: A: вода (0,04% NH3 H2O+10 мМ NH4HCO3), B: MeCN. Вначале: A (77%) и B (23%), в конце: A (47%) и B (53%). Время градиентного элюирования (мин): 8; 100% B, время удерживания 0 (мин); расход: 25 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом, лиофилизировали до сухого состояния с получением соединения 16 (10 мг, выход 11%) в виде белого твердого вещества.
HPLC-MS: масса/заряд 447,1 [M+H]+, rt: 3,63 мин, чистота: 98,5%, способ: M.
SFC: чистота: 50,6%/49,4%, rt: 4,98 мин/5,49 мин, способ: SFC8
Синтез соединений 17, 18 и 19
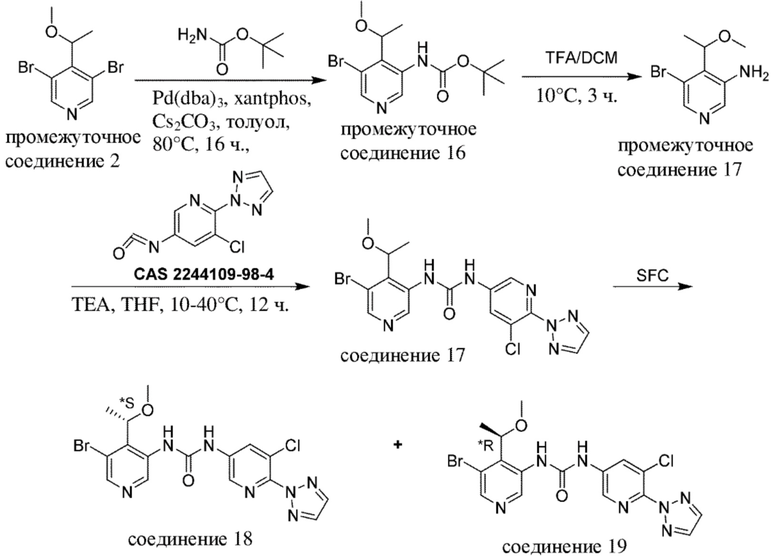
Получение промежуточного соединения 16
Смесь промежуточного соединения 2 (10 г, 33,9 ммоль), трет-бутилкарбамата (4 г, 33,9 ммоль) и Cs2CO3 (22 г, 67,8 ммоль) в толуоле (50 мл) дегазировали с помощью N2 в течение 10 мин. Затем добавляли Pd2(dba)3 (2,5 г, 2,9 ммоль), xantphos (2,59 г, 5 ммоль) и смесь перемешивали при 80°C в течение 16 часов в атмосфере N2. Смесь фильтровали и фильтрат концентрировали с получением неочищенного продукта в виде желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~20% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 16 (6,5 г, выход: 58%) в виде белого твердого вещества.
Получение промежуточного соединения 17
К раствору промежуточного соединения 16 (5,9 г, 17,8 ммоль) в CH2Cl2 (100 мл) добавляли 2,2,2-трифторуксусную кислоту (40 мл). Смесь перемешивали при 10°C в течение 3 часов. Большую часть растворителя удаляли под вакуумом с получением желтой смолы. Желтую смолу растворяли в CH2Cl2. К смеси добавляли NaHCO3 и смесь два раза экстрагировали с помощью CH2Cl2. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением желтого твердого вещества. Желтое твердое вещество очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~25% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 17 (3,3 г, выход: 81%) в виде белого твердого вещества.
Получение соединения 17
К раствору 3-хлор-5-изоцианато-2-(2H-1,2,3-триазол-2-ил)пиридина (CAS 2244109-98-4) (3,5 г, 15,8 ммоль) в THF (15 мл) добавляли триметиламин (4 мл, 30 ммоль) при 10°C. Затем добавляли раствор промежуточного соединения 17 (0,5 г, 2,1 ммоль) в THF (15 мл). Смесь перемешивали при 40°C в течение 12 часов. Смесь охлаждали до 25°C. Смесь выпаривали под вакуумом с получением желтого твердого вещества. Смесь растворяли в метаноле и перемешивали при 10°C в течение 0,5 часа. Смесь фильтровали и фильтрат выпаривали под вакуумом с получением желтого твердого вещества. Желтое твердое вещество растворяли в метаноле. Смесь перемешивали при 10°C в течение 0,5 часа с получением осадка. Смесь фильтровали и получали соединение 17 (420 мг, 0,9 ммоль) в виде желтого твердого вещества.
LC/MS: масса/заряд 452 [M+H]+, rt 0,92 мин, чистота 96,9%, способ A.
Получение соединений 18 и 19
Соединение 17 (124 мг, 0,26 ммоль) отделяли посредством SFC [колонка: YMC CHIRAL Amylose-C (250 мм*30 мм, 5 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в EtOH, вначале: A (55%) и B (45%), в конце: A (55%) и B (45%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 18 (44 мг, выход: 36,3%) в виде белого твердого вещества и соединения 19 (43 мг, выход: 35,9%) в виде белого твердого вещества.
Соединение 18:
LC/MS: масса/заряд 452 [M+H]+, rt 4,43 мин. Чистота 99,5%, способ K
SFC: чистота 100%, rt 1,73 мин. Способ: SFC15
Соединение 19:
LC/MS: масса/заряд 452 [M+H]+, rt 4,43 мин. Чистота 99,8%, способ K
SFC: чистота 99,2%, rt 2,36 мин. Способ: SFC15
Синтез соединения 20
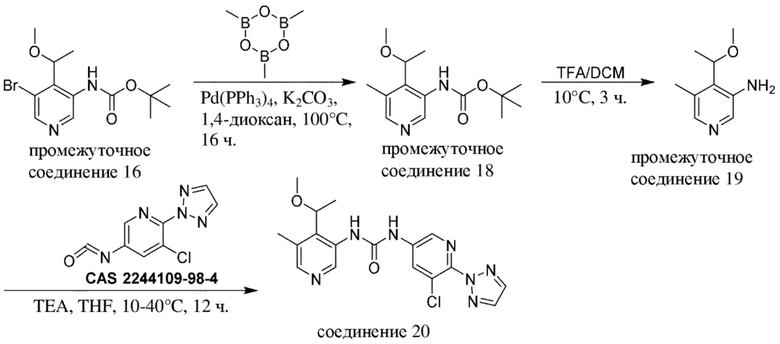
Получение промежуточного соединения 18
Смесь промежуточного соединения 16 (500 мг, 1,5 ммоль), 2,4,6-триметил-1,3,5,2,4,6-триоксатриборинана CAS 823-96-1 (417 мг, 1,7 ммоль), Pd(PPh3)4 (174 мг, 0,1 ммоль), карбоната калия (417 мг, 3,0 ммоль) в 1,4-диоксане (5 мл) дегазировали и снова заполняли с помощью N2 три раза. Смесь перемешивали при 100°C в течение 16 часов в атмосфере N2. Обеспечивали достижение температуры смеси, составляющей 25°C. Смесь гасили насыщ. водн. раствором NH4Cl и смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением желтого твердого вещества. Желтое твердое вещество очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~35% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 18 (330 мг, выход: 80,6%) в виде желтого твердого вещества.
Получение промежуточного соединения 19
Промежуточное соединение 19 получали по аналогии с процедурой, описанной для промежуточного соединения 17. Соединение очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~45% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 19 (180 мг, выход: 89,7%) в виде желтого твердого вещества.
Получение соединения 20
К раствору 3-хлор-5-изоцианато-2-(2H-1,2,3-триазол-2-ил)пиридина (CAS 2244109-98-4) (1,2 г, 5,3 ммоль) и триэтиламина (1,5 мл, 10,6 ммоль) в THF (8 мл) добавляли промежуточное соединение 19 (130 мг, 0,76 ммоль) при 25°C. Смесь перемешивали при 40°C в течение 12 часов. Смесь охлаждали до 25°C. Смесь выпаривали под вакуумом, затем растворяли в метаноле и перемешивали при 25°C в течение 0,5 часа. Смесь фильтровали и фильтрат выпаривали под вакуумом с получением желтого твердого вещества. Желтое твердое вещество очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Boston Prime C18 150 × 30 мм, 5 мкм, условия: A: вода (0,05% гидроксида аммония), B: MeCN, вначале: A (72%) и B (28%), в конце: A (42%) и B (58%), время градиентного элюирования 8 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 20 (214 мг, выход: 71,1%) в виде белого твердого вещества.
LC/MS: масса/заряд 388,1 [M+H]+, rt 3,28 мин, чистота 98,5%, способ: K.
SFC: чистота 50,1%/49,9%, rt 4,50 мин/5,16 мин. Способ: SFC5
Синтез соединения 21
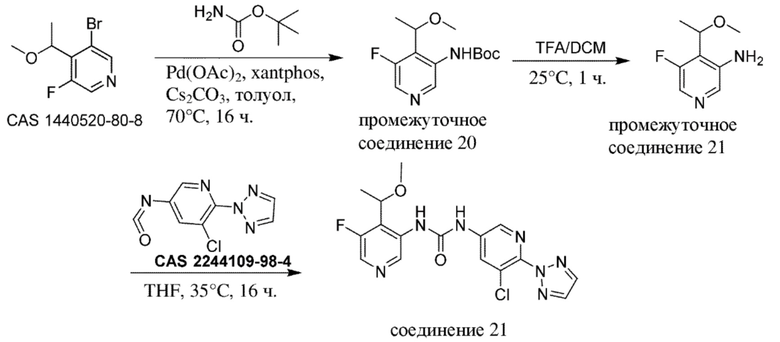
Получение промежуточного соединения 20
Смесь, состоящую из CAS 1440520-80-8 (657 мг, 2,8 ммоль), трет-бутилкарбамата (395 мг, 3,4 ммоль) и Cs2CO3 (1,8 г, 5,6 ммоль) в диоксане (20 мл), дегазировали с помощью N2 в течение 10 мин. Затем добавляли Pd(OAc)2 (32 мг, 0,14 ммоль) и xantphos (162 мг, 0,28 ммоль) и смесь перемешивали при 100°C в течение 16 часов в атмосфере N2. Смесь фильтровали и фильтрат концентрировали с получением неочищенного продукта в виде желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~30% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 20 (663 мг, выход: 87%) в виде желтого твердого вещества.
Получение промежуточного соединения 21
К раствору промежуточного соединения 20 (663 мг, 2,5 ммоль) в CH2Cl2 (10 мл) добавляли TFA (1 мл) при 25°C. Смесь перемешивали при 25°C в течение 2 часов. К смеси добавляли насыщ. водн. раствор NaHCO3 и смесь два раза экстрагировали с помощью CH2Cl2. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~70% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 21 (350 мг, выход: 68%) в виде желтого твердого вещества.
Получение соединения 21
Соединение 21 получали по аналогии с процедурой, описанной для соединения 20. Соединение очищали посредством препаративной высокоэффективной жидкостной хроматографии. [Колонка: Phenomenex Gemini 150 × 25 мм*10 мкм, условия: A: вода (0,05% гидроксида аммония), B: MeCN. Вначале: A (65%) и B (35%), в конце: A (35%) и B (65%), время градиентного элюирования: 10 мин; 100% B, время удерживания: 3 мин; расход: 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 21 (28 мг, выход: 23,2%) в виде белого твердого вещества.
LC/MS: масса/заряд 392,1 [M+H]+, rt 4,06 мин, чистота 95,5%, способ K
Синтез соединения 22
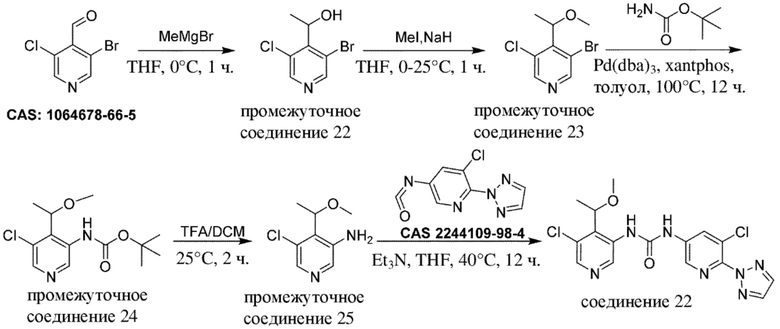
Получение промежуточного соединения 22
К раствору 3-бром-5-хлоризоникотинальдегида (1 г, 4,5 ммоль) в THF (20 мл) добавляли бромид метилмагния (3 М в THF, 2,3 мл, 6,8 ммоль) при 0°C. Смесь перемешивали при 25°C в течение 1 часа. Смесь гасили насыщ. водн. раствором NH4Cl и два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~15% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 22 (0,9 г, выход: 84%) в виде белого твердого вещества.
Получение промежуточного соединения 23
К смеси промежуточного соединения 22 (0,9 г, 3,8 ммоль) в THF (15 мл) добавляли гидрид натрия (230 мг, 5,7 ммоль, 60%, в минеральном масле) при 0°C и смесь перемешивали в течение 10 мин. Добавляли йодметан (3,7 г, 25,7 ммоль) и смесь перемешивали при 25°C в течение 2 часов. Смесь гасили насыщ. водн. раствором NH4Cl и два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~5% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 23 (0,8 г, выход: 84%) в виде белого твердого вещества.
Получение промежуточного соединения 24
Смесь промежуточного соединения 23 (0,7 г, 2,8 ммоль), трет-бутилкарбамата (393 мг, 3,4 ммоль) и Cs2CO3 (3,6 г, 11,2 ммоль) в толуоле (40 мл) дегазировали с помощью N2 в течение 10 мин. Затем добавляли Pd(OAc)2 (94 мг, 0,4 ммоль) и xantphos (162 мг, 0,3 ммоль) и смесь перемешивали при 100°C в течение 12 часов в атмосфере N2. Смесь охлаждали до 25°C и затем фильтровали. Фильтрат концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~4% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 24 (0,60 г, выход: 71%) в виде белого твердого вещества.
Получение промежуточного соединения 25
К раствору промежуточного соединения 24 (0,6 г, 2,0 ммоль) в CH2Cl2 (15 мл) добавляли TFA (3 мл) при 25°C. Смесь перемешивали при 25°C в течение 2 часов. Добавляли насыщ. водн. раствор NaHCO3 к смеси и смесь два раза экстрагировали с помощью CH2Cl2. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~26% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 25 (360 мг, выход: 96%) в виде белого твердого вещества.
Получение соединения 22
К раствору 3-хлор-5-изоцианато-2-(2H-1,2,3-триазол-2-ил)пиридина (CAS 2244109-98-4) (328 мг, 1,5 ммоль) и триэтиламина (0,4 мл, 3 ммоль) в THF (15 мл) добавляли раствор промежуточного соединения 25 (100 мг, 0,5 ммоль) в THF (5 мл) при 25°C. Реакционную смесь перемешивали при 40°C в течение 12 часов. Обеспечивали охлаждение смеси до 25°C и фильтровали. Фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии. [Колонка: Phenomenex Gemini 150 × 25 мм*10 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN. Вначале: A (65%) и B (35%), в конце: A (35%) и B (65%), время градиентного элюирования: 8 мин; 100% B, время удерживания: 2 мин; расход: 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 22 (55 мг, выход: 25%) в виде белого твердого вещества.
Соединение 22:
LC/MS: масса/заряд 408,0 [M+H]+, rt: 4,29 мин, чистота: 96,4%, способ: K
SFC: чистота 49,9%/50,1%, rt: 5,27 мин/5,93 мин, способ: SFC1
Синтез соединений 23 и 24
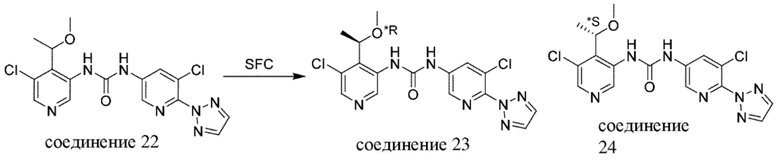
Соединение 22 (300 мг, 0,7 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: растворитель, A: сверхкритический CO2. Растворитель, B: 0,1% водный раствор аммиака в EtOH. Вначале: A (60%) и B (40%), в конце: A (60%) и B (40%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и смесь лиофилизировали до сухого состояния с получением соединения 23 (145 мг, выход: 48,3%) и соединения 24 (144 мг, выход: 47,8%) в виде белого твердого вещества.
Соединение 23:
LC/MS: масса/заряд 408,1 [M+H]+, rt: 4,37 мин, чистота: 99,8%, способ: K.
SFC: чистота 100%, rt: 5,24 мин, способ: SFC1
Соединение 24:
LC/MS: масса/заряд 408,1 [M+H]+, rt: 4,38 мин, чистота: 100%, способ: K
SFC: чистота 100%, rt: 5,89 мин, способ: SFC1
Синтез соединения 25
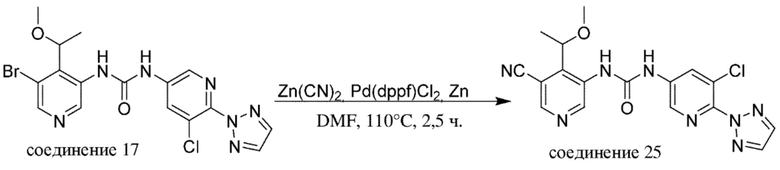
Получение конечного соединения 25
К смеси соединения 17 (50 мг, 0,11 ммоль), Zn(CN)2 (16 мг, 0,14 ммоль) и Zn (2 мг, 0,02 ммоль) в DMF (5 мл) добавляли Pd(dppf)Cl2 (12 мг, 0,02 ммоль) при 25°C. Реакционную смесь перемешивали при 110°C в течение 2,5 часов. Обеспечивали охлаждение смеси до 25°C и фильтровали. Фильтрат концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии. [Колонка: Phenomenex Gemini 150 × 25 мм*10 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN. Вначале: A (70%) и B (30%), в конце: A (40%) и B (60%), время градиентного элюирования: 8 мин; 100% B, время удерживания: 2 мин; расход: 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 25 (8 мг, выход: 18%) в виде белого твердого вещества.
LC/MS: масса/заряд 399,1 [M+H]+, rt: 4,06 мин, чистота: 99,8%, способ: K.
Синтез соединений 25, 26 и 27
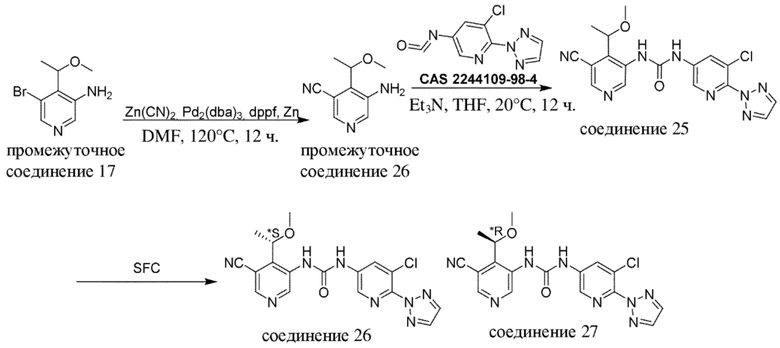
Получение промежуточного соединения 26
Смесь промежуточного соединения 17 (0,8 г, 3,5 ммоль), Zn(CN)2 (0,25 г, 2,1 ммоль) и Zn (68 мг, 1,1 ммоль) в DMF (20 мл) дегазировали с помощью N2 в течение 10 мин. Затем добавляли Pd2(dba)3 (159 мг, 0,17 ммоль) и dppf (192 мг, 0,35 ммоль) и смесь перемешивали при 120°C в течение 12 часов в атмосфере N2. Обеспечивали охлаждение смеси до 25°C и фильтровали. Фильтрат концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~30% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 26 (0,55 г, выход: 85%) в виде белого твердого вещества.
Получение соединения 25 (альтернативная процедура)
К раствору 3-хлор-5-изоцианато-2-(2H-1,2,3-триазол-2-ил)пиридина (CAS 2244109-98-4) (664 мг, 3,0 ммоль) и триэтиламина (0,8 мл, 6,0 ммоль) в THF (30 мл) добавляли раствор промежуточного соединения 26 (200 мг, 1,1 ммоль) в THF (10 мл) при 25°C. Реакционную смесь перемешивали при 40°C в течение 12 часов. Обеспечивали достижение температуры смеси, составляющей 25°C, и фильтровали. Фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии. [Колонка: Phenomenex Gemini 150 × 25 мм*10 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN. Вначале: A (65%) и B (35%), в конце: A (35%) и B (65%), время градиентного элюирования: 8 мин; 100% B, время удерживания: 2 мин; расход: 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 25 (120 мг, выход: 27,7%) в виде белого твердого вещества.
Получение соединений 26 и 27
Соединение 25 (120 мг, 0,3 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: растворитель, A: сверхкритический CO2, растворитель, B: 0,1% водный раствор аммиака в EtOH. Вначале: A (60%) и B (40%), в конце: A (60%) и B (40%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и смесь лиофилизировали до сухого состояния с получением соединения 26 (41 мг, выход: 34%) и соединения 27 (43 мг, выход: 36%) в виде белого твердого вещества.
Соединение 26:
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,52 (d, J=6,8 Гц, 3 H), 3,27 (s, 3 H), 4,82 (q, J=6,8 Гц, 1 H), 8,15 (s, 2 H), 8,47 (d, J=2,4 Гц, 1 H), 8,55 (d, J=2,4 Гц, 1 H), 8,77 (s, 1 H), 9,24 (s, 1 H);
HPLC/MS: масса/заряд 399,1 [M+H]+, rt: 4,20 мин, чистота: 98%, способ: M.
SFC: чистота 99,8%, rt: 4,74 мин, способ: SFC7
Соединение 27:
LC/MS: масса/заряд 399,1 [M+H]+, rt: 4,12 мин, чистота: 100%, способ: K
SFC: чистота 99,5%, rt: 5,30 мин, способ: SFC7
Синтез соединения 28
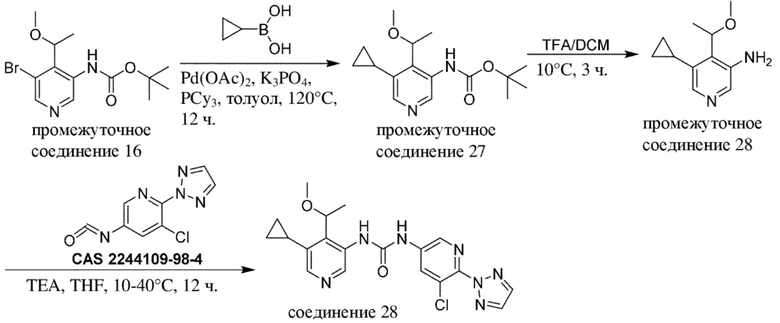
Получение промежуточного соединения 27
К раствору промежуточного соединения 16 (300 мг, 0,91 ммоль), циклопропилбороновой кислоты (156 мг, 1,8 ммоль) и фосфата калия (385 мг, 1,8 ммоль) в толуоле (2 мл) и H2O (0,5 мл) добавляли Pd(OAc)2 (10 мг, 0,04 ммоль) и трициклогексилфосфин (25 мг, 0,09 ммоль) в атмосфере N2. Реакционную смесь перемешивали при 120°C в течение 12 часов в атмосфере N2. Смесь охлаждали до 25°C. Смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали под вакуумом с получением смолы. Желтую смолу очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~30% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 27 (200 мг, выход: 71,9%) в виде светло-желтого масла.
Получение промежуточного соединения 28
Промежуточное соединение 28 получали по аналогии с процедурой, описанной для промежуточного соединения 17. Соединение очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~40% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 28 (111 мг, выход: 88,5%) в виде желтого твердого вещества.
Получение соединения 28
Соединение 28 получали по аналогии с процедурой, описанной для соединения 20. Соединение очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Boston Prime C18 150 × 30 мм, 5 мкм, условия: A: вода (0,05% гидроксида аммония), B: MeCN, вначале: A (70%) и B (30%), в конце: A (40%) и B (60%), время градиентного элюирования 9 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 28 (75 мг, выход: 27%) в виде белого твердого вещества.
LC/MS: масса/заряд 414,2 [M+H]+, rt 3,54 мин, чистота 99,3%, способ K.
SFC: чистота 50,3%/49,7%, rt 4,78 мин/5,40 мин. Способ: SFC7
Синтез соединения 29
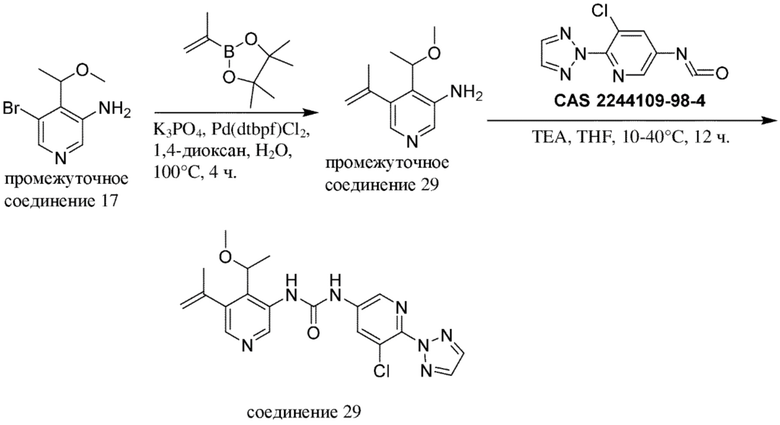
Получение промежуточного соединения 29
Смесь промежуточного соединения 17 (300 мг, 1,3 ммоль), 4,4,5,5-тетраметил-2-(проп-1-ен-2-ил)-1,3,2-диоксаборолана CAS 126726-62-3 (250 мг, 1,5 ммоль), фосфата калия (547 мг, 2,6 ммоль) в 1,4-диоксане (20 мл) и H2O (4 мл) барботировали с помощью N2 в течение 5 минут и затем обрабатывали с помощью Pd(dtbpf)Cl2 CAS 95408-45-0 (84 мг, 0,1 ммоль). Смесь барботировали с помощью N2 в течение еще 5 минут и затем перемешивали при 100°C в течение 4 часов. Обеспечивали достижение смеси комнатной температуры, гасили с помощью H2O и смесь два раза экстрагировали с помощью EtOAc. Органические слои разделяли, промывали солевым раствором, высушивали над Na2SO4 и выпаривали под вакуумом с получением желтой смолы. Желтую смолу очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~41% EtOAc в петролейном эфире) с получением промежуточного соединения 29 (205 мг, выход: 80,9%) в виде желтого твердого вещества.
Получение соединения 29
Соединение 29 получали по аналогии с процедурой, описанной для соединения 20. Соединение очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Boston Prime C18 150 × 30 мм, 5 мкм, условия: A: вода (0,05% гидроксида аммония), B: MeCN, вначале: A (70%) и B (30%), в конце: A (40%) и B (60%), время градиентного элюирования 8 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 29 (83 мг, выход: 19%) в виде белого твердого вещества.
LC/MS: масса/заряд 414,2 [M+H]+, rt 3,74 мин. Чистота 99,8%, способ K.
SFC: чистота 48,8%; 51,2%, rt 1,84 мин, 2,12 мин. Способ: SFC9.
Синтез соединения 30
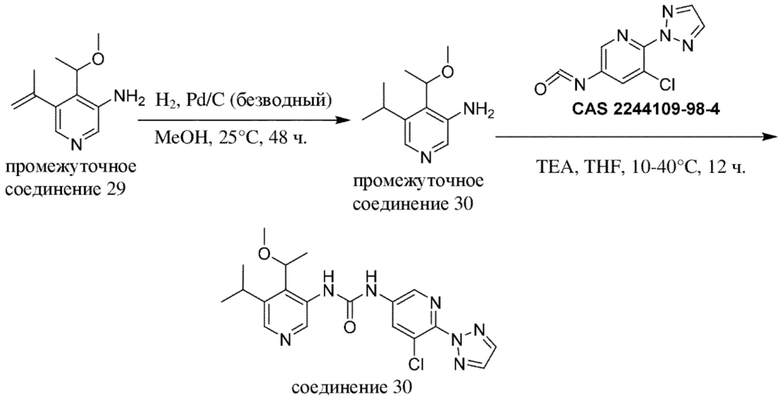
Получение промежуточного соединения 30
Смесь промежуточного соединения 29 (270 мг, 1,38 ммоль) в метаноле (50 мл) гидрогенизировали при 25°C (40 фунтов на кв. дюйм) с Pd/C (100 мг) в качестве катализатора. Реакционную смесь перемешивали в течение 48 часов. После поглощения H2 (1 экв.) катализатор отфильтровывали и фильтрат выпаривали. Фильтраты концентрировали под вакуумом с получением желтой смолы. Желтую смолу очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~60% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 30 (140 мг, выход: 51,9%) в виде светло-желтого твердого вещества.
Синтез соединения 30
Соединение 30 получали по аналогии с процедурой, описанной для соединения 20. Соединение очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Boston Prime C18 150 × 30 мм, 5 мкм, условия: A: вода (0,05% гидроксида аммония), B: MeCN, вначале: A (68%) и B (32%), в конце: A (38%) и B (62%), время градиентного элюирования 9 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 30 (73,5 мг, выход: 24,0%) в виде белого твердого вещества.
LC/MS: масса/заряд 416,2 [M+H]+, rt 3,63 мин. Чистота 97,2%, способ K
SFC: чистота 49,3%/50,7%, rt 4,42 мин/4,71 мин. Способ: SFC7.
Синтез соединения 31
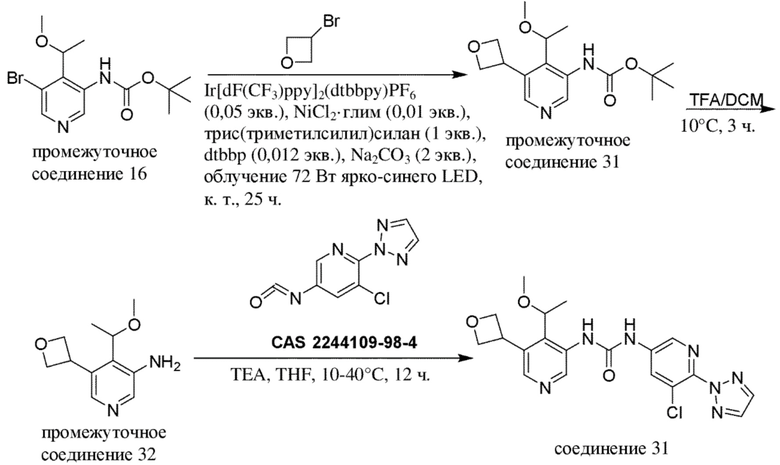
Получение промежуточного соединения 31
К смеси желтого цвета промежуточного соединения 16 (1100 мг, 3,3 ммоль), 3-бромоксетана (478 мг, 3,5 ммоль), трис(триметилсилил)силана (826 мг, 3,3 ммоль), 4,4′-ди-трет-бутил-2,2′-дипиридила (CAS 72914-19-3) (10,7 мг, 0,04 ммоль) и Na2CO3 (704 мг, 6,6 ммоль) в DME (3 мл) добавляли NiCl2•глим (7,3 мг, 0,03 ммоль) и (Ir[dF(CF3)ppy]2(dtbbpy))PF6 (CAS 870987-63-6) (74 мг, 0,07 ммоль). Смесь барботировали с помощью N2 и перемешивали при к. т. в атмосфере N2 в течение 25 часов при LED-освещении ярко-синим светом при 72 Вт. Смесь фильтровали и фильтрат выпаривали под вакуумом с получением желтого масла. Желтое масло очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~50% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 31 (380 мг, выход: 32,2%) в виде желтого твердого вещества.
Получение промежуточного соединения 32
Промежуточное соединение 32 получали по аналогии с процедурой, описанной для промежуточного соединения 17. Соединение очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~40% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 32 (140 мг, выход: 57,6%) в виде желтого твердого вещества.
Получение соединения 31
Соединение 31 получали по аналогии с процедурой, описанной для соединения 20. Соединение очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Boston Prime C18 150 × 30 мм, 5 мкм, условия: A: вода (0,05% гидроксида аммония), B: MeCN, вначале: A (77%) и B (23%), в конце: A (62%) и B (38%), время градиентного элюирования 9 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 31 (44 мг, выход: 38%) в виде белого твердого вещества.
HPLC/MS: масса/заряд 430,1 [M+H]+, rt 3,81 мин, чистота 96,6%, способ M.
SFC: чистота 49,6%/50,4%, rt 3,03 мин/3,42 мин. Способ: SFC16
Синтез соединения 32
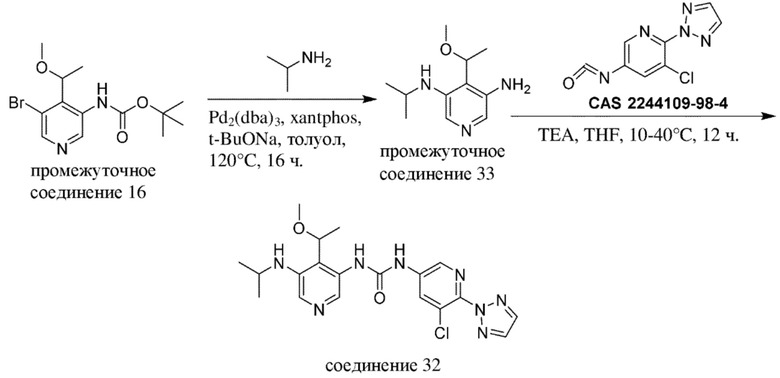
Получение промежуточного соединения 33
Смесь промежуточного соединения 16 (100 мг, 0,3 ммоль), xantphos (35 мг, 0,06 ммоль), Pd2(dba)3 (28 мг, 0,03 ммоль) и трет-бутоксида натрия (87 мг, 0,9 ммоль) в толуоле (4 мл) барботировали с помощью N2 в течение 1 мин. Затем к вышеупомянутой смеси добавляли пропан-2-амин (125 мг, 2,1 ммоль) в толуоле (1 мл). Полученную смесь перемешивали при 120°C в течение 16 часов в атмосфере N2. Смесь охлаждали до 25°C. Смесь фильтровали и фильтрат выпаривали под вакуумом с получением желтой смолы. Желтую смолу очищали посредством преп. TLC (CH2Cl2/MeOH=10/1) с получением промежуточного соединения 33 (25 мг, выход: 40%) в виде желтого твердого вещества.
Синтез соединения 32
Соединение 32 получали по аналогии с процедурой, описанной для соединения 20. Соединение очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Waters Xbridge 150 × 25, 5 мкм, условия: A: вода (10 мМ NH4HCO3), B: MeCN, вначале: A (70%) и B (30%), в конце: A (52%) и B (48%), время градиентного элюирования 8 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 32 (13,5 мг, выход: 14,2%) в виде белого твердого вещества.
LC/MS: масса/заряд 431,2 [M+H]+, rt 3,78 мин, чистота 95,2%, способ: K.
Синтез соединения 33
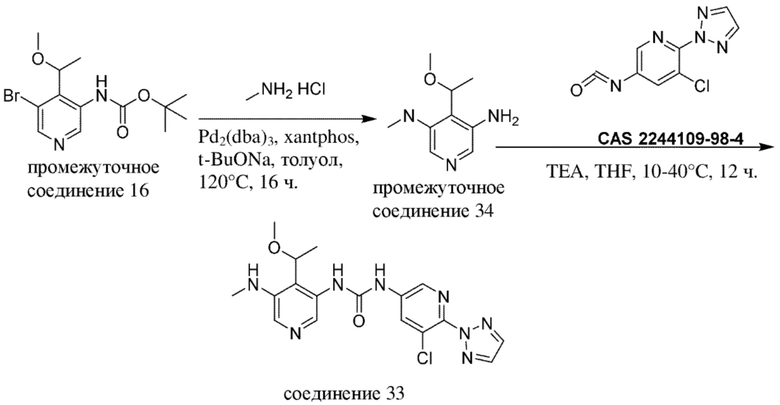
Получение промежуточного соединения 34
Промежуточное соединение 34 получали по аналогии с процедурой, описанной для промежуточного соединения 33. Соединение очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~10% MeOH в DCM). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 34 (270 мг, выход: 86,1%) в виде желтого твердого вещества.
Синтез соединения 33
Соединение 33 получали по аналогии с процедурой, описанной для соединения 20. После осуществления реакции смесь выпаривали под вакуумом с получением желтого твердого вещества. Желтое твердое вещество растворяли в MeOH. Смесь перемешивали при 20°C в течение 1 часа. Смесь фильтровали и фильтрат выпаривали под вакуумом с получением желтого твердого вещества. Желтое твердое вещество растворяли в DMSO и EtOAc. Смесь перемешивали при 20°C в течение 1 часа. Смесь фильтровали и осадок на фильтре промывали с помощью EtOAc и MeOH. Осадок на фильтре растворяли в H2O и MeOH. Водный слой лиофилизировали до сухого состояния с получением соединения 33 (192 мг, выход: 36,3%) в виде белого твердого вещества.
HPLC/MS: масса/заряд 403,2 [M+H]+, rt 3,89 мин, чистота 98,9%, способ: M.
Синтез соединений 34, 35, 36 и 37
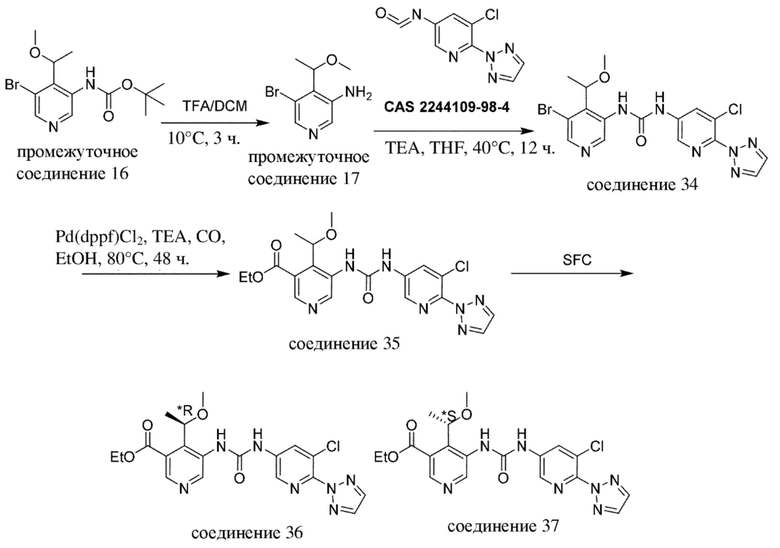
Получение промежуточного соединения 17
К раствору промежуточного соединения 16 (5,9 г, 17,8 ммоль) в CH2Cl2 (100 мл) добавляли TFA (40 мл) при 10°C. Смесь перемешивали при 10°C в течение 3 часов. Смесь обрабатывали насыщ. водн. раствором NaHCO3 и два раза экстрагировали с помощью CH2Cl2. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~25% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 17 (3,4 г, выход: 81%) в виде желтого твердого вещества.
Получение соединения 34
К раствору 3-хлор-5-изоцианато-2-(2H-1,2,3-триазол-2-ил)-пиридина CAS 2244109-98-4 (2,3 г, 10,3 ммоль) и триэтиламина (2,8 мл, 20,6 ммоль) в THF (80 мл) добавляли промежуточное соединение 17 (1 г, 4,3 ммоль). Смесь перемешивали при 40°C в течение 12 часов. Смесь концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт промывали с помощью MeOH/EtOAc и осадок на фильтре высушивали с получением соединения 34 (1,2 г, выход: 62%) в виде белого твердого вещества.
Получение соединения 35
Раствор соединения 34 (100 мг, 0,2 ммоль) и TEA (154 мкл, 1,1 ммоль) в EtOH (20 мл) обрабатывали в атмосфере CO при 80°C и 50 фунтах на кв. дюйм с Pd(dppf)Cl2 в качестве катализатора в течение 48 часов. Смесь фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~50% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением соединения 35 (85 мг, выход: 86%) в виде желтого твердого вещества.
LC/MS: масса/заряд 446,0 [M+H]+, rt 1,68 мин, чистота 94,2%, способ C
Получение соединений 36 и 37
Соединение 35 (85 мг, 0,19 ммоль) отделяли посредством SFC. [Колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в EtOH, вначале: A (60%) и B (40%), в конце: A (60%) и B (40%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 36 (8,1 мг, выход: 7,9%) в виде белого твердого вещества и соединения 37 (7,6 мг, выход: 7,5%) в виде белого твердого вещества.
Соединение 36:
LC/MS: масса/заряд 446,2 [M+H]+, rt 4,315 мин. Чистота 96,5%, способ K
SFC: чистота 97,7%, rt 4,019 мин. Способ: SFC17
Соединение 37:
HPLC/MS: масса/заряд 446,1 [M+H]+, rt 4,404 мин. Чистота 97,7%, способ M;
SFC: чистота 95,8%, rt 4,471 мин. Способ: SFC17
Синтез соединений 38 и 39
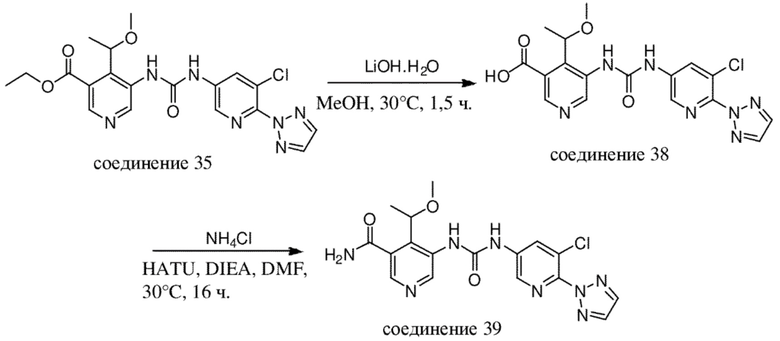
Получение соединения 38
К раствору соединения 35 (160 мг, 0,4 ммоль) в метаноле (5 мл) добавляли LiOH.H2O (1,8 мл, 3,7 ммоль, 2 М). Смесь перемешивали при 30°C в течение 1,5 часов. Смесь охлаждали до 25°C. Значение pH смеси доводили до 6 с помощью HCl (1 н.). Смесь экстрагировали с помощью MeOH/DCM (объем/объем=1/3) (20 мл x 5). Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали под вакуумом с получением соединения 38 в виде желтого твердого вещества. (160 мг, выход: 99,6%) в виде желтого твердого вещества.
LC/MS: масса/заряд 418,0 [M+H]+, rt 1,41 мин, чистота 96,4%, способ C.
Получение соединения 39
Смесь соединения 38 (160 мг, 0,4 ммоль), HATU (210 мг, 0,5 ммоль), N-этил-N-изопропилпропан-2-амина (191 мг, 1,5 ммоль) в DMF (5 мл) перемешивали при 30°C в течение 10 минут. К смеси добавляли NH4Cl (30 мг, 0,5 ммоль) и перемешивали при 30°C в течение 16 часов. Смесь фильтровали и фильтрат выпаривали под вакуумом с получением желтой смолы. Желтую смолу очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Xtimate C18 10μ, 250 мм*50 мм, условия: A: вода (0,05% гидроксида аммония), B: MeCN, вначале: A (80%) и B (20%), в конце: A (60%) и B (40%), время градиентного элюирования 9 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 39 (56 мг, выход: 345%) в виде белого твердого вещества.
HPLC/MS: масса/заряд 417,1 [M+H]+, rt 3,47 мин, чистота 95,2%, способ M.
SFC: чистота 50,1%/49,9%, rt 4,82 мин/5,03, способ: SFC1
Синтез соединения 40
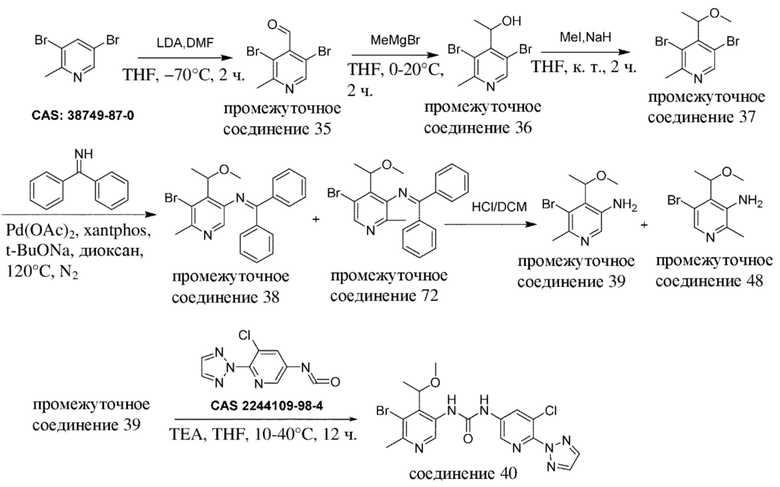
Получение промежуточного соединения 35
3,5-Дибром-2-метилпиридин (15 г, 60 ммоль) растворяли в THF (300 мл) и смесь охлаждали до -70°C, добавляли LDA (2 М в THF и гептанах, 35,9 мл, 71,8 ммоль). Реакционную смесь перемешивали при -70°C в течение 1 часа. К смеси добавляли DMF (6,9 мл, 90 ммоль) и смесь перемешивали при -70°C в течение 1 часа. Реакционную смесь гасили насыщ. водн. раствором NH4Cl при температуре от -20°C до -70°C и затем добавляли H2O и нагревали до комнатной температуры. Смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали под вакуумом с получением неочищенного продукта в виде желтого твердого вещества. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~7% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 35 (9,5 г, выход: 57%) в виде светло-желтого твердого вещества.
Получение промежуточного соединения 36
К раствору промежуточного соединения 35 (16 г, 57 ммоль) в THF (400 мл) добавляли бромид метилмагния (3 М в THF, 28,7 мл, 86 ммоль) при 0°C. Обеспечивали нагревание смеси до 20°C и перемешивали при 20°C в течение 1 часа. Смесь гасили насыщ. водн. раствором NH4Cl. Смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~15% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 36 (15 г, выход: 89%) в виде светло-желтого твердого вещества.
Получение промежуточного соединения 37
К раствору промежуточного соединения 36 (15 г, 50 ммоль) в THF (200 мл) добавляли NaH (60%, в минеральном масле, 3 г, 75 ммоль) при 0°C в течение 10 мин. Добавляли CH3I (26 г, 184 ммоль) при 0°C. Обеспечивали нагревание смеси до к. т. и перемешивали при к. т. в течение 2 часов. Смесь гасили насыщ. водн. раствором NH4Cl и смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~4% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 37 (14 г, выход: 90%) в виде белого твердого вещества.
Получение промежуточного соединения 38 и промежуточного соединения 72
Смесь промежуточного соединения 37 (6,0 г, 19 ммоль), дифенилметанимина (5,3 г, 29 ммоль) и t-BuONa (2,8 г, 29 ммоль) в диоксане (120 мл) продували с помощью N2 в течение 10 мин. Добавляли Pd(OAc)2 (0,44 г, 1,9 ммоль) и xantphos (2,2 г, 3,9 ммоль). Реакционную смесь перемешивали при 120°C в течение 16 часов. Обеспечивали охлаждение реакционной смеси до 25°C и фильтровали. Остаток промывали с помощью EtOAc (400 мл). Фильтраты концентрировали под вакуумом с получением неочищенного продукта в виде желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~10% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением смеси промежуточного соединения 38 и промежуточного соединения 72 (5,2 г, чистота: 73%) в виде желтого масла.
Получение промежуточного соединения 39 и промежуточного соединения 48
Смесь промежуточных соединений 38 и 72 (5,2 г, чистота 73%) растворяли в DCM (50 мл). Добавляли водный раствор HCl (4 мл, 12 М) и смесь перемешивали при 40°C в течение 18 часов. Значение pH реакционной смеси доводили до 8 с применением насыщ. раствора NaHCO3 и экстрагировали с помощью EtOAc (100 мл*3). Объединенные органические слои разделяли, промывали солевым раствором, высушивали над Na2SO4, фильтровали и фильтраты выпаривали под вакуумом с получением желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~30% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 39 (1,1 г) в виде желтого твердого вещества и промежуточного соединения 48 (800 мг).
Альтернативная процедура для получения промежуточного соединения 38
Смесь промежуточного соединения 37 (10 г, 32,4 ммоль), дифенилметанимина (6,5 г, 35,6 ммоль) и t-BuONa (3,1 г, 32,4 ммоль) в толуоле (200 мл) продували с помощью N2 в течение 10 мин. Добавляли Pd2(dba)3 (1,5 г, 1,6 ммоль) и BINAP (3,0 г, 4,8 ммоль). Реакционную смесь перемешивали при 120°C в течение 16 часов. Обеспечивали охлаждение реакционной смеси до 25°C и фильтровали. Остаток промывали с помощью EtOAc (500 мл). Фильтраты концентрировали под вакуумом с получением неочищенного продукта в виде желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~12% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением неочищенного промежуточного соединения 38 (20 г, чистота: 39%) в виде желтого масла.
Альтернативная процедура для получения промежуточного соединения 39
Неочищенное промежуточное соединение 38 (полученное посредством альтернативной процедуры для получения промежуточного соединения 38) (20 г, чистота 39%) растворяли в DCM (60 мл). Добавляли водный раствор HCl (10 мл, 2 М) и смесь перемешивали при 40°C в течение 5 часов. Значение pH реакционной смеси доводили до 8 с применением насыщ. раствора NaHCO3 и экстрагировали с помощью EtOAc (100 мл*3). Объединенные органические слои разделяли, промывали солевым раствором, высушивали над Na2SO4, фильтровали и фильтраты выпаривали под вакуумом с получением желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~30% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 39 (4 г, выход: 50,4%, выход с двух стадий) в виде желтого твердого вещества.
Синтез соединения 40
Соединение 40 получали по аналогии с процедурой, описанной для соединения 20. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Xtimate C18 10μ, 250 мм*50 мм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN, вначале: A (50%) и B (50%), в конце: A (20%) и B (80%), время градиентного элюирования 8 мин; 100% B, время удерживания 0 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 40 (6 мг, выход: 8,4%) в виде белого твердого вещества.
HPLC/MS: масса/заряд 466 [M+H]+, rt 4,716 мин. Чистота 98,7%, способ M.
SFC: чистота 53,1%; 46,9%, rt 5,535 мин, 6,995 мин. Способ: SFC1
Синтез соединений 41, 42 и 43
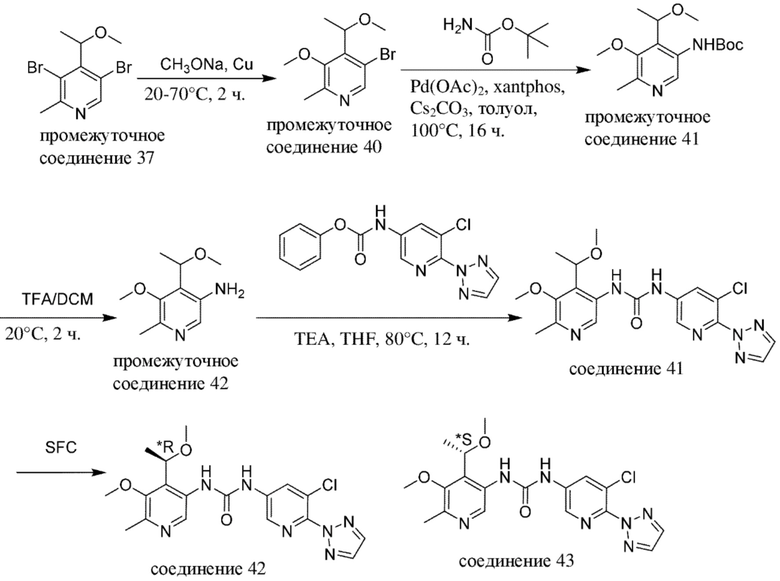
Получение промежуточного соединения 40
К смеси промежуточного соединения 37 (1 г, 3 ммоль) в DMF (10 мл) добавляли CH3ONa (810 мг, 15 ммоль) и порошок Cu (20 мг, 0,3 ммоль) при 20°C. Смесь перемешивали при 70°C в течение 2 часов. К смеси добавляли солевой раствор и смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~5% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 40 (520 мг, выход: 53,3%) в виде бесцветного масла.
Получение промежуточного соединения 41
Смесь, состоящую из промежуточного соединения 40 (240 мг, 0,9 ммоль), трет-бутилкарбамата (162 мг, 1,4 ммоль) и Cs2CO3 (1,2 г, 3,7 ммоль) в толуоле (6 мл), дегазировали с помощью N2 в течение 10 мин. Затем добавляли Pd(OAc)2 (31 мг, 0,14 ммоль), xantphos (53 мг, 0,1 ммоль) и смесь перемешивали при 100°C в течение 16 часов в атмосфере N2. Смесь фильтровали и фильтрат концентрировали с получением неочищенного продукта в виде желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~10% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 41 (140 мг, выход: 48%) в виде белого твердого вещества.
Получение промежуточного соединения 42
К раствору промежуточного соединения 41 (280 мг, 0,9 ммоль) в CH2Cl2 (10 мл) добавляли TFA (2 мл) при 20°C. Смесь перемешивали при 20°C в течение 2 часов. Смесь обрабатывали насыщ. водн. раствором NaHCO3 и два раза экстрагировали с помощью CH2Cl2. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~30% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 42 (130 мг, выход: 70,1%) в виде белого твердого вещества.
Получение соединения 41
К раствору промежуточного соединения 42 (110 мг, 0,6 ммоль) и сложного фенилового эфира N-[5-хлор-6-(2H-1,2,3-триазол-2-ил)-3-пиридинил]-карбаминовой кислоты (CAS 2178988-79-7) (226 мг, 0,7 ммоль) в THF (8 мл) добавляли TEA (233 мкл, 1,68 ммоль) при 20°C. Реакционную смесь перемешивали при 80°C в течение 12 часов. Обеспечивали охлаждение смеси до комнатной температуры и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Phenomenex Gemini 150 × 25 мм*10 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN, вначале: A (65%) и B (35%), в конце: A (35%) и B (65%), время градиентного элюирования 8 мин; 100% B, время удерживания 8 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 41 (150 мг, выход: 64%) в виде белого твердого вещества.
Получение соединений 42 и 43
Соединение 41 (150 мг, 0,36 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в EtOH, вначале: A (55%) и B (45%), в конце: A (55%) и B (45%), расход: 70 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 42 (71 мг, выход: 48%) в виде белого твердого вещества и соединения 43 (71 мг, выход: 48%) в виде белого твердого вещества.
Соединение 42:
LC/MS: масса/заряд 418,1 [M+H]+, rt 3,470 мин. Чистота 100%, способ K
SFC: чистота 100%, rt 5,66 мин. Способ: SFC1
Соединение 43:
LC/MS: масса/заряд 418,1 [M+H]+, rt 3,47 мин. Чистота 100%, способ K
SFC: чистота 100%, rt 6,76 мин. Способ: SFC1
Синтез соединений 44, 45 и 46
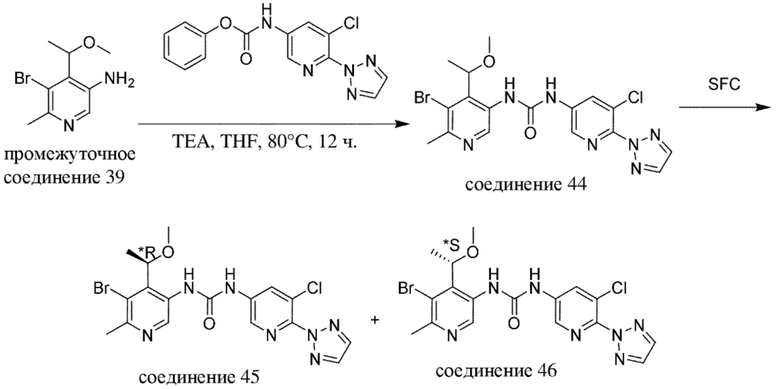
Получение соединения 44
Соединение 44 получали по аналогии с процедурой, описанной для соединения 41, начиная с промежуточного соединения 39 и сложного фенилового эфира N-[5-хлор-6-(2H-1,2,3-триазол-2-ил)-3-пиридинил]-карбаминовой кислоты (CAS 2178988-79-7). Реакционную смесь концентрировали под вакуумом с получением неочищенного продукта в виде белого твердого вещества. К смеси добавляли MeOH (100 мл) и перемешивали при 70°C в течение 1 ч. Фильтровали и фильтраты концентрировали под вакуумом с получением неочищенного соединения 44 в виде желтого масла. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Boston Prime C18 150 × 30 мм, 5 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN, вначале: A (55%) и B (45%), в конце: A (25%) и B (75%), время градиентного элюирования 8 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 44 (460 мг, выход: 48%) в виде белого твердого вещества.
LC/MS: масса/заряд 466,1/468,1 [M+H]+, rt 1,015 мин, чистота 99,4%, способ G.
Получение соединений 45 и 46
Соединение 44 (500 мг, 1,07 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AD (250 мм*30 мм, 10 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в EtOH, вначале: A (55%) и B (45%), в конце: A (55%) и B (45%), расход: 70 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 45 (235 мг, выход: 47,5%) в виде белого твердого вещества и соединения 46 (235,8 мг, выход: 47,7%) в виде белого твердого вещества.
Соединение 45:
LC/MS: масса/заряд 466,1/468,1 [M+H]+, rt 4,338 мин. Чистота 99,8%, способ K;
SFC: чистота 100%, rt 1,880 мин. Способ: SFC14.
Соединение 46:
LC/MS: масса/заряд 466,1/468,1 [M+H]+, rt 4,326 мин. Чистота 100%, способ K;
SFC: чистота 100%, rt 2,347 мин. Способ: SFC14.
Синтез соединений 47 и 48
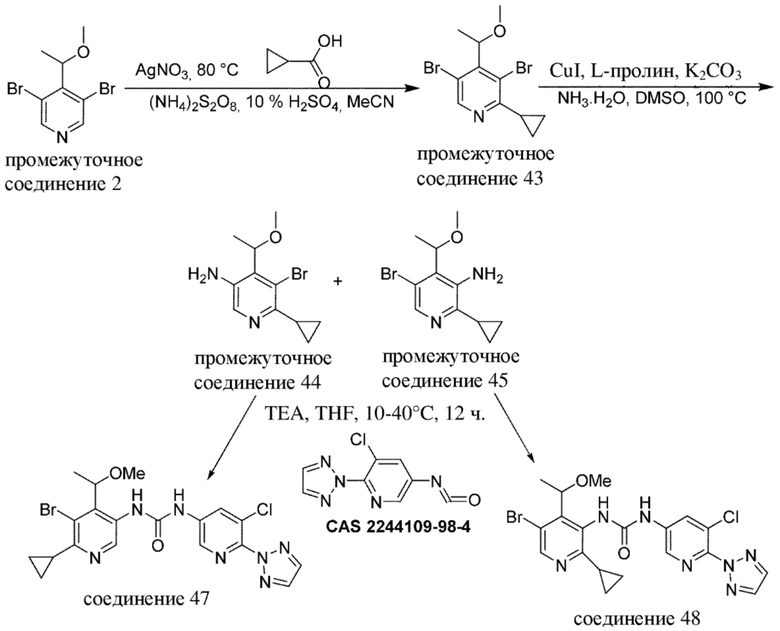
Получение промежуточного соединения 43
AgNO3 (3 г, 18 ммоль) и циклопропанкарбоновую кислоту (4,6 г, 54 ммоль) добавляли к раствору промежуточного соединения 2 (5,4 г, 18 ммоль) в смеси MeCN (90 мл) и 10% H2SO4 (90 мл). Реакционную смесь нагревали до температуры 70-80°C. К смеси медленно добавляли свежеприготовленный раствор (NH4)2S2O8 (12,3 г, 54 ммоль) в H2O (150 мл). Реакционную смесь перемешивали при 80°C в течение 4 часов. Обеспечивали достижение температуры смеси, составляющей 25°C, и значение pH доводили до 10 с применением NH3.H2O. Смесь три раза экстрагировали с помощью EtOAc. Объединенные органические слои разделяли, промывали солевым раствором, высушивали над Na2SO4, фильтровали и фильтраты выпаривали под вакуумом с получением желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~5% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением неочищенного продукта (2 г, чистота 60%), который очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Phenomenex Synergi Max-RP 250 × 50 мм*10 мкм; условия: A: вода (0,225% FA), B: MeCN, вначале: A (70%) и B (30%), в конце: A (15%) и B (85%), время градиентного элюирования 24 мин; 100% B, время удерживания 8 мин; расход 100 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением промежуточного соединения 43 (600 мг, выход: 7,5%) в виде бесцветного масла.
Получение промежуточных соединений 44 и 45
Смесь промежуточного соединения 43 (500 мг, 1,5 ммоль), CuI (57 мг, 0,3 моль), L-пролина (69 мг, 0,6 ммоль), K2CO3 (311 мг, 2,25 ммоль), NH3.H2O (8 мл) в DMSO (5 мл) продували с помощью N2. Смесь перемешивали при 100°C в течение 4 часов. Добавляли H2O и смесь три раза экстрагировали с помощью EtOAc. Объединенные органические слои разделяли, промывали солевым раствором, высушивали над Na2SO4, фильтровали и фильтраты выпаривали под вакуумом с получением желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~15% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 44 (30 мг, выход: 6%) в виде белого твердого вещества и промежуточного соединения 45 (60 мг, выход: 12%) в виде желтого масла.
Синтез соединения 47
Соединение 47 получали по аналогии с процедурой, описанной для соединения 20, с применением промежуточного соединения 44 и 3-хлор-5-изоцианато-2-(2H-1,2,3-триазол-2-ил)-пиридина (CAS 2244109-98-4). Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Xtimate C18 10μ, 250 мм*50 мм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN, вначале: A (50%) и B (50%), в конце: A (20%) и B (80%), время градиентного элюирования 8 мин; 100% B, время удерживания 0 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 47 (6 мг, выход: 13%) в виде белого твердого вещества.
LC/MS: масса/заряд 492 [M+H]+, rt 5,138 мин. Чистота 99,8%, способ K
SFC: чистота 50,7%; 49,3%, rt 4,758 мин, 5,371 мин. Способ: SFC1
Синтез соединения 48
Соединение 48 получали по аналогии с процедурой, описанной для соединения 20, с применением промежуточного соединения 45 и 3-хлор-5-изоцианато-2-(2H-1,2,3-триазол-2-ил)-пиридина (CAS 2244109-98-4). Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Boston Prime C18 150 × 30 мм, 5 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN, вначале: A (50%) и B (50%), в конце: A (20%) и B (80%), время градиентного элюирования 8 мин; 100% B, время удерживания 0 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 48 (3 мг, выход: 5%) в виде белого твердого вещества.
LC/MS: масса/заряд 492 [M+H]+, rt 4,772 мин. Чистота 98,9%, способ K
SFC: чистота 47,9%; 52,1%, rt 3,945 мин, 4,493 мин. Способ: SFC1.
Синтез соединений 49 и 50
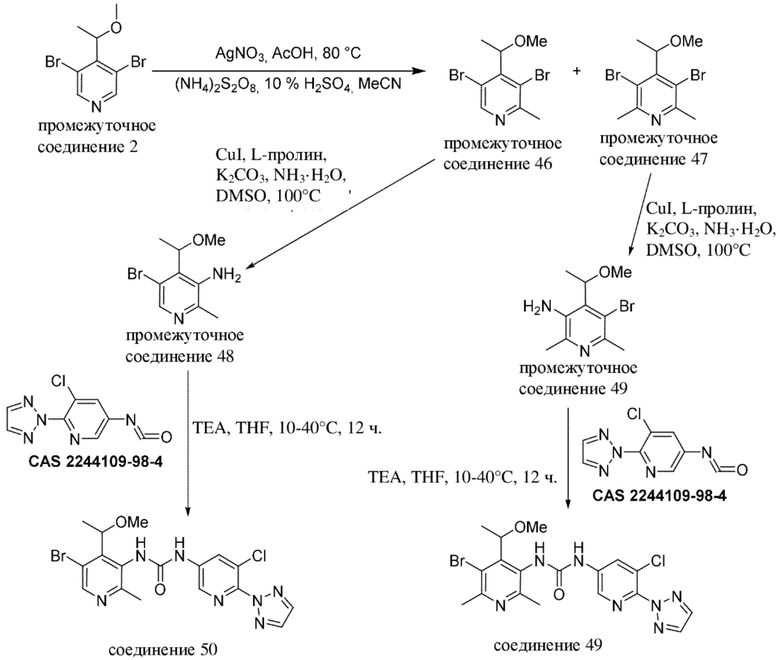
Получение промежуточных соединений 46 и 47
Промежуточные соединения 46 и 47 получали по аналогии с процедурой, описанной для промежуточного соединения 43. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~5% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением неочищенного продукта, который очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Phenomenex Synergi Max-RP 250 × 50 мм*10 мкм; условия: A: вода (0,225% FA), B: MeCN, вначале: A (80%) и B (20%), в конце: A (25%) и B (75%), время градиентного элюирования 24 мин; 100% B, время удерживания 3 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением промежуточного соединения 47 (150 мг, выход: 2%) и промежуточного соединения 46 (650 мг, выход: 8%) в виде желтых твердых веществ.
Получение промежуточного соединения 49
Промежуточное соединение 49 получали по аналогии с процедурой, описанной для промежуточного соединения 45. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~15% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 49 (20 мг, 25%) в виде белого твердого вещества.
Синтез соединения 49
Соединение 49 получали по аналогии с процедурой, описанной для соединения 20. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Boston Prime C18 150 × 30 мм, 5 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN, вначале: A (55%) и B (45%), в конце: A (25%) и B (75%), время градиентного элюирования 8 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 49 (4 мг, выход: 7%) в виде белого твердого вещества.
LC/MS: масса/заряд 480,0/482,0 [M+H]+, rt 3,911 мин. Чистота 99,8%, способ K
SFC: чистота 51,7%, 48,3%, rt 4,483 мин, 4,944 мин. Способ: SFC1
Получение промежуточного соединения 48
Промежуточное соединение 48 получали по аналогии с процедурой, описанной для промежуточного соединения 45, или как описано в экспериментальной процедуре для получения соединения 40. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~15% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 48 (40 мг, выход: 8%) в виде желтого твердого вещества.
Получение соединения 50
Соединение 50 получали по аналогии с процедурой, описанной для соединения 20, начиная с промежуточного соединения 48. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Xtimate C18 10μ, 250 мм*50 мм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN, вначале: A (60%) и B (40%), в конце: A (30%) и B (70%), время градиентного элюирования 8 мин; 100% B, время удерживания 0 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 50 (4 мг, выход: 5%) в виде белого твердого вещества.
LC/MS: масса/заряд 466 [M+H]+, rt 4,255 мин. Чистота 97,3%, способ K
SFC: чистота 52,9%; 47,1%, rt 6,058 мин, 7,033 мин. Способ: SFC13
Синтез соединений 51, 52 и 53

Получение промежуточного соединения 50
2-Бром-5-нитро-3-(трифторметил)пиридин (20 г, 74 ммоль) и циклопропилбороновую кислоту (13 г, 148 ммоль) растворяли в смеси растворителей из толуола (160 мл) и H2O (40 мл) и затем добавляли K3PO4 (31 г, 148 ммоль), PCy3 (3 г, 11 ммоль), Pd(OAc)2 (1 г, 5 ммоль) в атмосфере N2. Реакционную смесь перемешивали при 120°C в течение 12 часов в атмосфере N2. После охлаждения раствора до комнатной температуры смесь фильтровали и остаток два раза промывали с помощью 200 мл этилацетата. Органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~10% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 50 (13 г, выход: 76%) в виде желтого твердого вещества.
Получение промежуточного соединения 51
При комнатной температуре добавляли NH4Cl (15 г, 280 ммоль) к раствору промежуточного соединения 50 (13 г, 56 ммоль) в MeOH (40 мл), THF (80 мл) и H2O (20 мл). Медленно добавляли порошок железа (16 г, 280 ммоль). Реакционную смесь перемешивали при 65°C в течение 2 часов. Обеспечивали охлаждение смеси до 25°C и затем фильтровали. Остаток два раза промывали с помощью 300 мл этилацетата. Органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~30% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 51 (10,5 г, выход: 89%) в виде желтого твердого вещества.
Получение промежуточного соединения 52
К раствору промежуточного соединения 51 (2 г, 10 ммоль) в THF (50 мл) добавляли пиридин (1,2 мл, 14,8 ммоль) при комнатной температуре. Медленно добавляли фенилхлорформиат (2 г, 13 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Смесь гасили насыщ. водн. раствором NH4Cl. Смесь два раза экстрагировали с помощью EtOAc (100 мл). Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~30% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 52 (3 г, выход: 75%) в виде белого твердого вещества.
Получение соединения 51
К раствору промежуточного соединения 52 (657 мг, 1,6 ммоль) и промежуточного соединения 39 (200 мг, 0,8 ммоль) в THF (5 мл) добавляли TEA (340 мкл, 2,5 ммоль) при 25°C. Реакционную смесь перемешивали при 80°C в течение 16 часов. Обеспечивали достижение смеси комнатной температуры и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Xtimate C18 10μ, 250 мм*50 мм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN, вначале: A (40%) и B (60%), в конце: A (10%) и B (90%), время градиентного элюирования 8 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 51 (240 мг, выход: 57,3%) в виде белого твердого вещества.
LC/MS: масса/заряд 473,1 [M+H]+, rt 2,37 мин, чистота 92,2%, способ: D
Получение соединений 52 и 53
Соединение 51 (240 мг, 0,43 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALCEL OD-H (250 мм*30 мм, 5 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в MeOH, вначале: A (70%) и B (30%), в конце: A (70%) и B (30%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 52 (105 мг, выход: 46,8%) в виде белого твердого вещества и соединения 53 (110 мг, выход: 49,7%) в виде белого твердого вещества.
Соединение 52:
LC/MS: масса/заряд 473,1 [M+H]+, rt 5,15 мин. Чистота 98,7%, способ: K
SFC: чистота 100%, rt 3,98 мин. Способ: SFC11
Соединение 53:
LC/MS: масса/заряд 473,1 [M+H]+, rt 5,15 мин. Чистота 99,9%, способ: K
SFC: чистота 99,7%, rt 4,56 мин. Способ: SFC11
Синтез соединений 54, 55 и 56
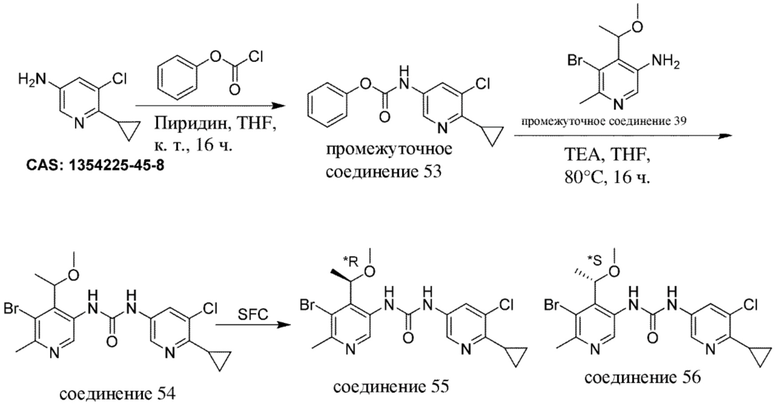
Получение промежуточного соединения 53
К раствору 5-хлор-6-циклопропил-3-пиридинамина (500 мг, 2,8 ммоль) в THF (10 мл) добавляли пиридин (0,4 мл, 4,3 ммоль) при комнатной температуре. Медленно добавляли фенилхлорформиат (0,5 мл, 3,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Смесь гасили насыщ. водн. раствором NH4Cl. Смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~33% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 53 (755 мг, выход: 93%) в виде белого твердого вещества.
Получение соединения 54
К раствору промежуточного соединения 53 (353 мг, 1,2 ммоль) и промежуточного соединения 39 (200 мг, 0,8 ммоль) в THF (10 мл) добавляли триэтиламин (340 мкл, 2,5 ммоль) при 25°C. Реакционную смесь перемешивали при 80°C в течение 16 часов. Обеспечивали охлаждение смеси до комнатной температуры и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Xtimate C18 10μ, 250 мм*50 мм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN, вначале: A (40%) и B (60%), в конце: A (10%) и B (90%), время градиентного элюирования 8 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 54 (186 мг, выход: 51,8%) в виде белого твердого вещества.
LC/MS: масса/заряд 439,1 [M+H]+ , rt: 2,30 мин, чистота: 100%, способ: B
Получение соединений 55 и 56
Соединение 54 (186 мг, 0,42 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALCEL OD-H (250 мм*30 мм, 5 мкм). Условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в MeOH. Вначале: A (60%) и B (40%), в конце: A (60%) и B (40%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 55 (79 мг, выход: 42%) в виде белого твердого вещества и соединения 56 (90 мг, выход: 48%) в виде белого твердого вещества.
Соединение 55:
LC/MS: масса/заряд 439,1 [M+H]+, rt: 4,85 мин. Чистота: 99,3%, способ: K
SFC: чистота 100%, rt: 5,01 мин. Способ: SFC12
Соединение 56:
LC/MS: масса/заряд 439,1 [M+H]+, rt 4,84 мин. Чистота 99%, способ: K
SFC: чистота 98,9%, rt: 5,57 мин. Способ: SFC12
Синтез соединений 57, 58 и 59
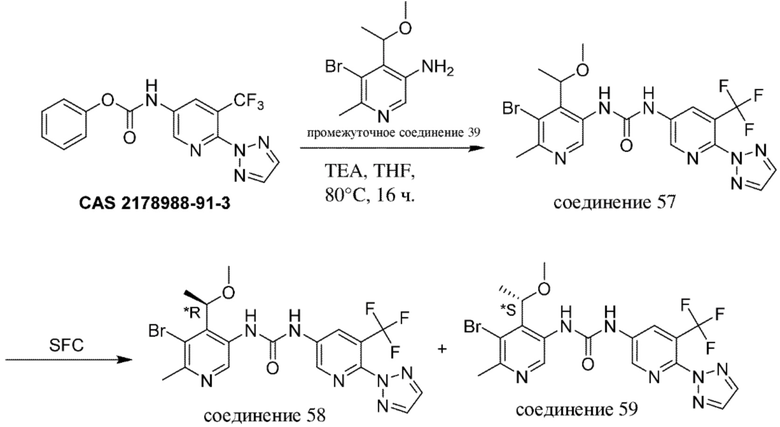
Получение соединения 57
К раствору сложного фенилового эфира N-[6-(2H-1,2,3-триазол-2-ил)-5-(трифторметил)-3-пиридинил]-карбаминовой кислоты (CAS 2178988-91-3) (427 мг, 1,2 ммоль) и промежуточного соединения 39 (200 мг, 0,8 ммоль) в THF (5 мл) добавляли TEA (0,34 мл, 2,5 ммоль) при 25°C. Реакционную смесь перемешивали при 80°C в течение 16 часов. Обеспечивали достижение смеси комнатной температуры и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Xtimate C18 10μ, 250 мм*50 мм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN, вначале: A (48%) и B (52%), в конце: A (18%) и B (82%), время градиентного элюирования 8 мин; 100% B, время удерживания 1 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 57 (240 мг, выход: 58%) в виде белого твердого вещества.
LC/MS: масса/заряд 500 [M+H]+, rt 2,08 мин, чистота 99,1%, способ: D.
Получение соединений 58 и 59
Соединение 57 (240 мг, 0,48 ммоль) отделяли посредством SFC [колонка: Phenomenex-Amylose-1 (250 мм*30 мм, 5 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в EtOH, вначале: A (70%) и B (30%), в конце: A (70%) и B (30%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 58 (95 мг, выход: 39%) в виде белого твердого вещества и соединения 59 (80 мг, выход: 33%) в виде белого твердого вещества.
Соединение 58:
LC/MS: масса/заряд 500,1 [M+H]+, rt 4,62 мин. Чистота 96,7%, способ: K
SFC: чистота 98,5%, rt 4,21 мин. Способ: SFC1
Соединение 59:
LC/MS: масса/заряд 500,1 [M+H]+, rt 4,62 мин. Чистота 99,3%, способ: K
SFC: чистота 99,6%, rt 3,77 мин. Способ: SFC1
Синтез соединений 60, 61 и 62
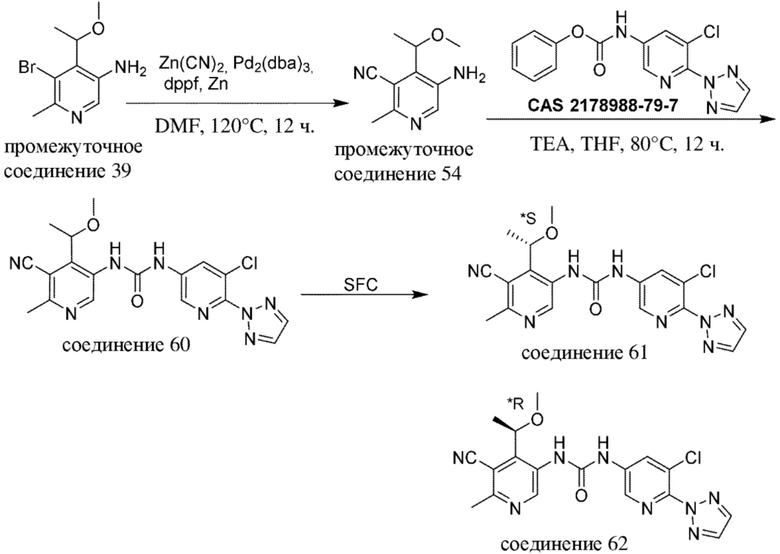
Получение промежуточного соединения 54
Смесь промежуточного соединения 39 (7,0 г, 28 ммоль), Zn(CN)2 (2,1 г, 18 ммоль) и Zn (0,55 г, 8,4 ммоль) в DMF (150 мл) дегазировали с помощью N2 в течение 5 мин. Добавляли Pd2(dba)3 (1,3 г, 1,4 ммоль) и dppf (1,6 г, 2,8 ммоль). Смесь перемешивали при 120°C в течение 12 часов в атмосфере N2. Смесь фильтровали и фильтрат концентрировали с получением неочищенного продукта в виде желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~30% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 54 (5,0 г, чистота: 90%) в виде желтого твердого вещества.
Получение соединения 60
К смеси промежуточного соединения 54 (150 мг, 0,78 ммоль) и сложного фенилового эфира N-[5-хлор-6-(2H-1,2,3-триазол-2-ил)-3-пиридинил]-карбаминовой кислоты (CAS 2178988-79-7) (316 мг, 0,94 ммоль) в THF (6 мл) добавляли триэтиламин (0,32 мл, 2,4 ммоль) при 25°C. Реакционную смесь перемешивали при 80°C в течение 12 часов. Обеспечивали охлаждение смеси до 25°C и фильтровали. Фильтрат концентрировали в вакууме с получением неочищенного продукта в виде желтого твердого вещества. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии. [Колонка: Phenomenex Gemini 150 × 25 мм*10 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN. Вначале: A (65%) и B (35%), в конце: A (35%) и B (65%), время градиентного элюирования: 8 мин; 100% B, время удерживания: 2 мин; расход: 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 60 (100 мг, выход: 30%) в виде белого твердого вещества.
Получение соединений 61 и 62
Соединение 60 (100 мг, 0,24 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: растворитель, A: сверхкритический CO2, растворитель, B: 0,1% водный раствор аммиака в EtOH. Вначале: A (65%) и B (35%), в конце: A (65%) и B (35%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и смесь подвергали лиофилизации до сухого состояния с получением соединения 61 (27 мг, выход: 28%) и соединения 62 (27 мг, выход: 28%) в виде белого твердого вещества.
Соединение 61:
LC/MS: масса/заряд 413,1 [M+H]+, rt: 4,27 мин, чистота: 100%, способ: K.
SFC: чистота 99,8%, rt: 4,38 мин, способ: SFC10.
Соединение 62:
LC/MS: масса/заряд 413,2 [M+H]+, rt: 4,26 мин, чистота: 98,4%, способ: K
SFC: чистота 99,2%, rt: 4,87 мин, способ: SFC10.
Альтернативное получение соединения 60
К раствору промежуточного соединения 54 (500 мг, 2,5 ммоль) и сложного фенилового эфира N-[5-хлор-6-(2H-1,2,3-триазол-2-ил)-3-пиридинил]-карбаминовой кислоты (CAS 2178988-79-7) (1,3 г, 3,8 ммоль) в THF (20 мл) добавляли DMAP (619 мг, 5,1 ммоль) при 20°C. Реакционную смесь перемешивали при 60°C в течение 2 часов. Смесь концентрировали в вакууме с получением неочищенного продукта. Смесь петролейного эфира:этилацетата=1:1 (50 мл) добавляли к неочищенному продукту и смесь перемешивали при 25°C в течение 10 мин. Полученное твердое вещество собирали посредством фильтрации и промывали смесью петролейного эфира:этилацетата=1:1 (20 мл). Твердый остаток собирали, обрабатывали с помощью MeCN (200 мл) и суспензию перемешивали при 25°C в течение 10 мин. Смесь фильтровали и фильтрат, содержащий продукт, концентрировали in vacuo с получением соединения 60 (450 мг, выход: 43%) в виде белого твердого вещества.
LC/MS: масса/заряд 413,0 [M+H]+, rt 0,75 мин, чистота 100%, способ A
Альтернативное получение соединений 61 и 62
Соединение 60 (450 мг, 1,09 ммоль) отделяли посредством SFC [колонка: Phenomenex-Amylose-1 (250 мм*30 мм, 5 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в EtOH, вначале: A (70%) и B (30%), в конце: A (70%) и B (30%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остаткам добавляли MeCN и H2O и их лиофилизировали до сухого состояния с получением соединения 61 (166 мг, выход: 37%) в виде белого твердого вещества и соединения 62 (173,3 мг, выход: 38,3%) в виде белого твердого вещества.
Соединение 61:
HPLC-MS: масса/заряд 413,1 [M+H]+, rt 4,25 мин. Чистота 100%, способ K;
SFC: чистота 100%, rt 4,40 мин. Способ: SFC10.
Соединение 62:
HPLC-MS: масса/заряд 413,1 [M+H]+, rt 4,25 мин. Чистота 99,59%, способ K;
SFC: чистота 99,38%, rt 4,88 мин. Способ: SFC10.
Синтез соединений 63, 64 и 65
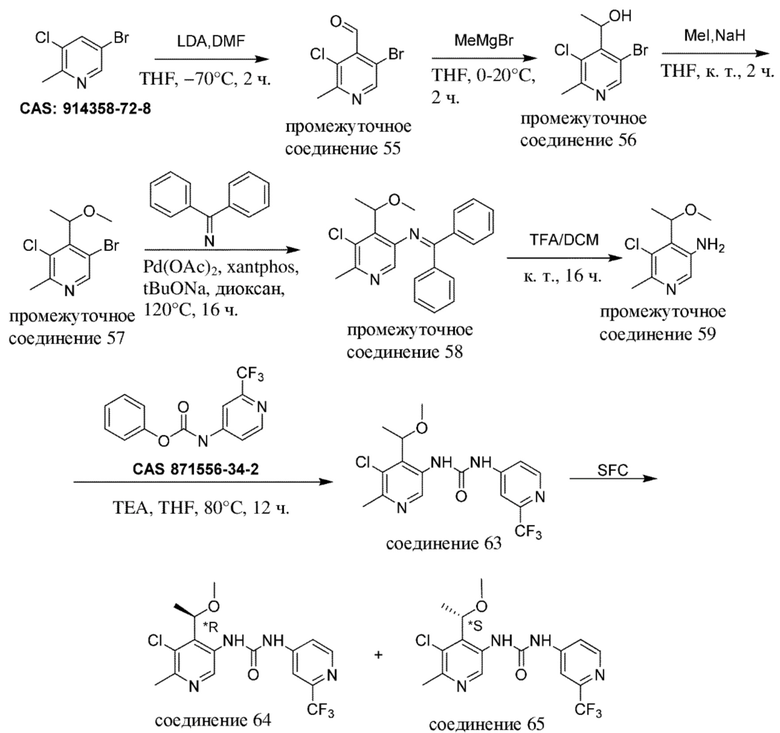
Получение промежуточного соединения 55
5-Бром-3-хлор-2-метилпиридин (6,5 г, 31,5 ммоль) растворяли в THF (130 мл) и охлаждали до -70°C. По каплям добавляли LDA (2 М в THF и гептанах, 19 мл, 38 ммоль). Реакционную смесь перемешивали при -70°C в течение 1 ч. К смеси добавляли DMF (4,9 мл, 63 ммоль) и перемешивали при -70°C в течение 1 часа. Реакционную смесь гасили насыщ. водн. раствором NH4Cl при температуре от -20°C до -70°C и затем добавляли H2O и смесь нагревали до комнатной температуры. Смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали под вакуумом с получением неочищенного продукта в виде желтого твердого вещества. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~3% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 55 (6 г, выход: 81%) в виде светло-желтого твердого вещества.
Получение промежуточного соединения 56
Промежуточное соединение 55 (6 г, 26 ммоль) растворяли в THF (150 мл) и перемешивали при 0°C. Добавляли раствор бромида метилмагния (3 М в THF, 17,1 мл, 51 ммоль) при 0°C. Обеспечивали нагревание смеси до 20°C и перемешивали при 20°C в течение 1 часа. Смесь гасили насыщ. водн. раствором NH4Cl. Смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного промежуточного соединения 56 (5,9 г, выход: 89%) в виде желтого твердого вещества.
Получение промежуточного соединения 57
Промежуточное соединение 56 (6,3 г, 25 ммоль) растворяли в THF (65 мл) и перемешивали при 0°C. Добавляли NaH (60%, в минеральном масле, 1,5 г, 38 ммоль) и смесь перемешивали при 0°C в течение 0,5 ч. Добавляли MeI (13 г, 93 ммоль) и перемешивали при к.т. в течение еще 16 часов. Смесь гасили насыщ. водн. раствором NH4Cl и смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~9% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 57 (6,1 г, выход: 91%) в виде желтого масла.
Получение промежуточного соединения 58
Промежуточное соединение 57 (3,9 г, 19 ммоль) и дифенилметанимин (4,0 г, 22 ммоль) растворяли в диоксане (60 мл). Затем добавляли Pd(OAc)2 (329 мг, 1,5 ммоль), xantphos (1,7 г, 2,9 ммоль) и tBuONa (2 г, 22 ммоль) и смесь продували с помощью N2. Реакционную смесь перемешивали при 120°C в течение 16 часов. К смеси добавляли насыщ. водн. раствор NH4Cl и смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~8% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 58 (6 г, выход 68,6%) в виде желтого масла.
Получение промежуточного соединения 59
Промежуточное соединение 58 (2 г, 5,5 ммоль) растворяли в DCM (40 мл) и добавляли TFA (20 мл). Реакционную смесь перемешивали при к. т. в течение 16 часов. Смесь концентрировали под вакуумом с удалением TFA. Неочищенный продукт разбавляли с помощью EtOAc и добавляли насыщ. раствор NaHCO3 с получением pH=7. Водную фазу два раза экстрагировали с помощью EtOAc. Органические слои промывали солевым раствором и высушивали с помощью MgSO4, фильтровали и выпаривали с получением желтого твердого вещества. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~40% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 59 (0,8 г, выход 73%) в виде желтого твердого вещества.
Получение соединения 63
К раствору промежуточного соединения 59 (200 мг, 1,0 ммоль) и сложного фенилового эфира N-[2-(трифторметил)-4-пиридинил]-карбаминовой кислоты (CAS 871556-34-2) (445 мг, 1,5 ммоль) в THF (10 мл) добавляли TEA (303 мг, 3,0 ммоль) при 25°C. Реакционную смесь перемешивали при 80°C в течение 12 часов. Смесь концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~60% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением соединения 63 (265 мг, выход: 68%) в виде желтого твердого вещества.
LC/MS: масса/заряд 389,1 [M+H]+, rt 0,79 мин, чистота 100%, способ A
Получение соединений 64 и 65
Соединение 63 (265 мг, 0,68 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в EtOH, вначале: A (85%) и B (15%), в конце: A (85%) и B (15%), расход: 60 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 64 (130 мг, выход: 49%) в виде белого твердого вещества и соединения 65 (135 мг, выход: 51%) в виде белого твердого вещества.
Соединение 64:
LC/MS: масса/заряд 389,1 [M+H]+, rt 5,54 мин. Чистота 100%, способ K
SFC: чистота 100%, rt 2,82 мин. Способ: SFC10
Соединение 65:
LC/MS: масса/заряд 389,1 [M+H]+, rt 5,54 мин. Чистота 100%, способ K
SFC: чистота 100%, rt 3,02 мин. Способ: SFC10
Синтез соединений 66, 67 и 68
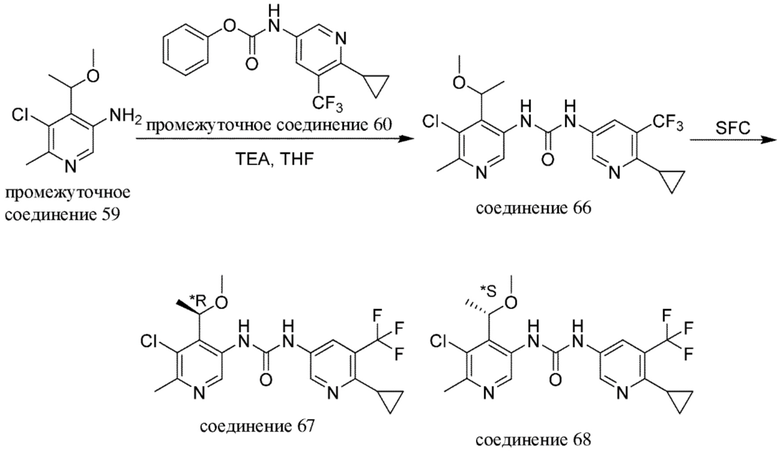
Получение соединения 66
К смеси промежуточного соединения 59 (200 мг, 1 ммоль) и промежуточного соединения 60 (полученного по аналогии с протоколами в WO2018020474) (385 мг, 1,2 ммоль) в THF (5 мл) добавляли триэтиламин (0,4 мл, 3 ммоль) при 25°C. Реакционную смесь перемешивали при 80°C в течение 12 часов. Затем добавляли дополнительное количество промежуточного соединения 60 (160 мг, 0,5 ммоль). Реакционную смесь перемешивали при 80°C в течение 12 часов. Обеспечивали достижение температуры смеси 25°C и концентрировали под вакуумом с получением неочищенного продукта в виде желтого твердого вещества. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~50% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали с получением продукта в виде желтого твердого вещества. Желтое твердое вещество промывали смесью петролейного эфира/этилацетата (5:1) с получением белого твердого вещества, представляющего собой соединение 66 (250 мг, выход: 58%).
LC/MS: масса/заряд 429,1 [M+H]+, rt: 2,33 мин, чистота: 100%, способ: C
SFC: чистота 49,9/50,1%, rt: 4,95/5,59, способ: SFC6
Получение соединений 67 и 68
Соединение 66 (250 мг, 0,6 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: растворитель, A: сверхкритический CO2, растворитель, B: 0,1% водный раствор аммиака в EtOH. Вначале: A (60%) и B (40%), в конце: A (60%) и B (40%), расход: 60 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и смесь лиофилизировали до сухого состояния с получением соединения 67 (100 мг, выход: 40%) и соединения 68 (103 мг, выход: 41%) в виде белых твердых веществ.
Соединение 67:
LC/MS: масса/заряд 429,2 [M+H]+, rt: 5,09 мин, чистота: 100%, способ: K.
SFC: чистота 100%, rt: 4,94 мин, способ: SFC6
Соединение 68:
LC/MS: масса/заряд 429,2 [M+H]+, rt: 5,10 мин, чистота: 99,8%, способ: K
SFC: чистота 100%, rt: 5,57 мин, способ: SFC6
Синтез соединений 69, 70 и 71
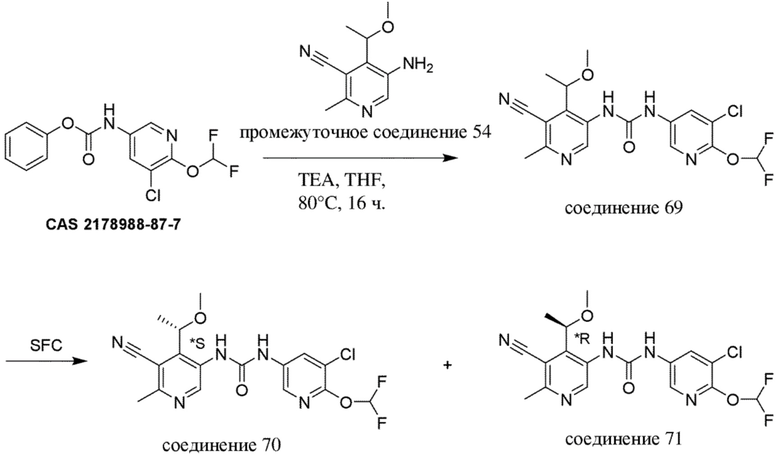
Получение соединения 69
Соединение 69 получали по аналогии с процедурой, описанной для соединения 57, с применением сложного фенильного эфира N-[5-хлор-6-(дифторметокси)-3-пиридинил]-карбаминовой кислоты (CAS 2178988-87-7) и промежуточного соединения 54. Обеспечивали достижение смеси комнатной температуры и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка Boston Prime C18, 150 × 30 мм, 5 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN, вначале: A (50%) и B (50%), в конце: A (20%) и B (80%), время градиентного элюирования 8 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 69 (125 мг, выход: 39%) в виде белого твердого вещества.
LC/MS: масса/заряд 412,2 [M+H]+, rt: 1,85 мин, чистота 99,9%, способ: C.
Получение соединений 70 и 71
Соединение 69 (125 мг, 0,3 ммоль) отделяли посредством SFC [колонка: Phenomenex-Amylose-1 (250 мм*30 мм, 5 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в EtOH, вначале: A (85%) и B (15%), в конце: A (85%) и B (15%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 70 (42 мг, выход: 33%) в виде белого твердого вещества и соединения 71 (48 мг, выход: 38,3%) в виде белого твердого вещества.
Соединение 70:
HPLC/MS: масса/заряд 412,1 [M+H]+, rt: 4,96 мин, чистота 98,5%, способ: K;
SFC: чистота 99,8%, rt: 2,87 мин, способ: SFC1.
Соединение 71:
HPLC/MS: масса/заряд 412,1 [M+H]+, rt: 4,96 мин, чистота 99,7%, способ: K;
SFC: чистота 100%, rt: 3,12 мин, способ: SFC1.
Синтез соединений 72, 73 и 74
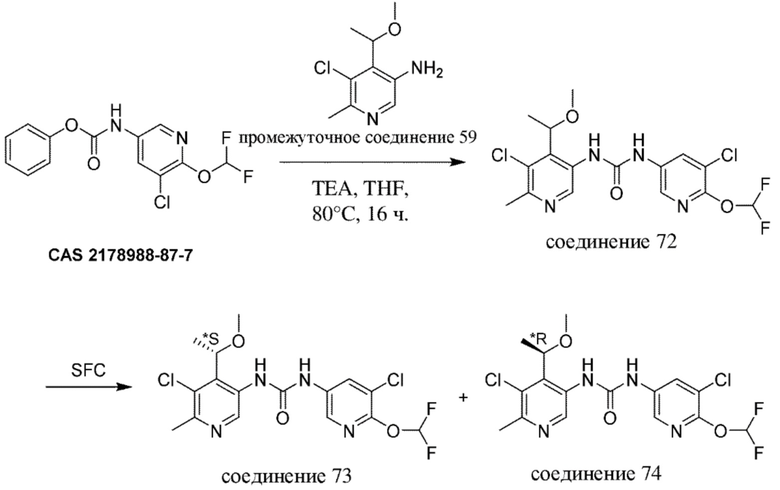
Получение соединения 72
Соединение 72 получали по аналогии с процедурой, описанной для соединения 57, с применением карбамата CAS 2178988-87-7 и промежуточного соединения 59. Обеспечивали достижение смеси комнатной температуры и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Xtimate C18 10μ, 250 мм*50 мм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN, вначале: A (45%) и B (55%), в конце: A (15%) и B (85%), время градиентного элюирования 15 мин; 100% B, время удерживания 0 мин; расход 60 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 72 (170 мг, выход: 53,4%) в виде белого твердого вещества.
LC/MS: масса/заряд 421,1 [M+H]+, rt: 1,96 мин, чистота 98,1%, способ: C.
Получение соединений 73 и 74
Соединение 72 (170 мг, 0,4 ммоль) отделяли посредством SFC [колонка: Phenomenex-Amylose-1 (250 мм*30 мм, 5 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в EtOH, вначале: A (75%) и B (25%), в конце: A (75%) и B (25%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 73 (70 мг, выход: 41,9%) в виде белого твердого вещества и соединения 74 (65 мг, выход: 38,5%) в виде белого твердого вещества.
Соединение 73:
HPLC/MS: масса/заряд 421,1 [M+H]+, rt: 4,88 мин, чистота 99,9%, способ: K;
SFC: чистота 99,7%, rt: 3,53 мин, способ: SFC10.
Соединение 74:
HPLC/MS: масса/заряд 421,1 [M+H]+, rt: 4,88 мин, чистота 98,8%, способ: K;
SFC: чистота 98,1%, rt: 4,87 мин, способ: SFC10.
Синтез соединений 75, 76 и 77
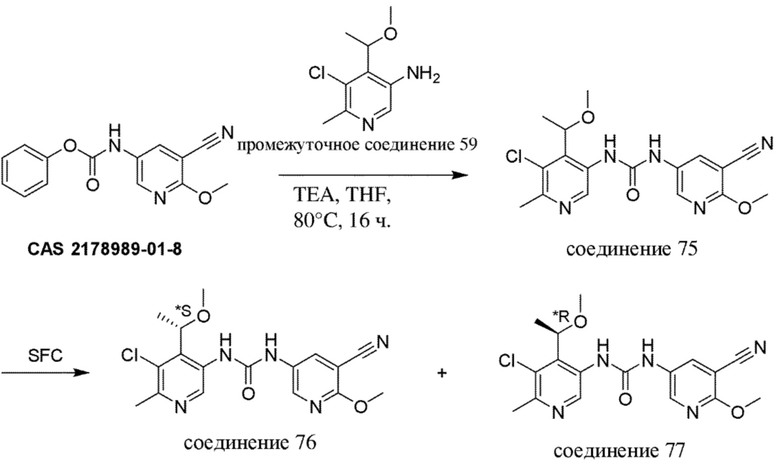
Получение соединения 75
Соединение 75 получали по аналогии с процедурой, описанной для соединения 57, с применением карбамата CAS 2178989-01-8 и промежуточного соединения 59. Обеспечивали достижение смеси комнатной температуры и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Boston Prime C18 150 × 30 мм, 5 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN, вначале: A (60%) и B (40%), в конце: A (30%) и B (70%), время градиентного элюирования 8 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 75 (160 мг, выход: 42,1%) в виде белого твердого вещества.
LC/MS: масса/заряд 376,2 [M+H]+, rt: 1,71 мин, чистота 97,7%, способ: C.
Получение соединений 76 и 77
Соединение 75 (160 мг, 0,42 ммоль) отделяли посредством SFC [колонка: Phenomenex-Amylose-1 (250 мм*30 мм, 5 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в EtOH, вначале: A (70%) и B (30%), в конце: A (70%) и B (30%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 76 (52 мг, выход: 33,2%) в виде белого твердого вещества и соединения 77 (55 мг, выход: 35,2%) в виде белого твердого вещества.
Соединение 76:
HPLC/MS: масса/заряд 376,1 [M+H]+, rt: 4,17 мин. Чистота 99,7%, способ: K.
SFC: чистота 99,9%, rt: 4,39 мин, способ: SFC10.
Соединение 77:
HPLC/MS: масса/заряд 376,2 [M+H]+, rt: 4,17 мин. Чистота 100%, способ: K.
SFC: чистота 99,1%, rt: 4,94 мин, способ: SFC10.
Синтез соединений 78, 79 и 80
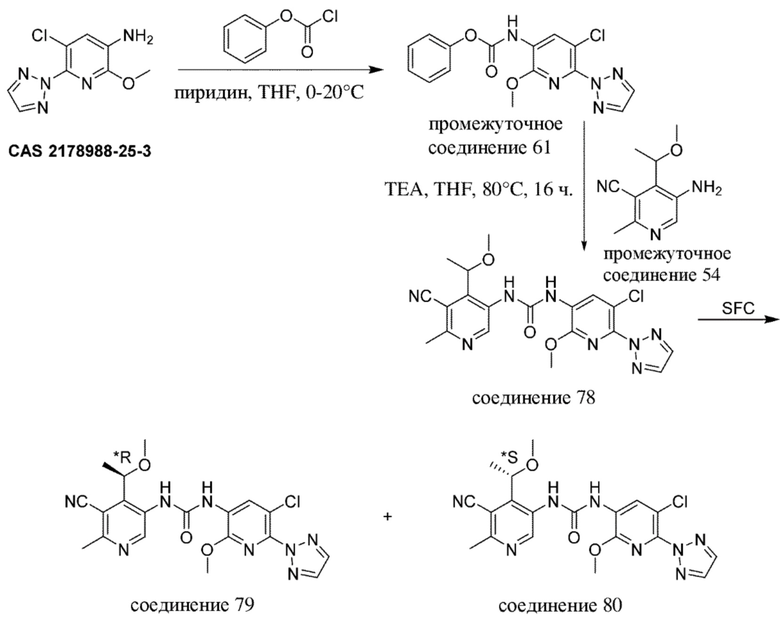
Получение промежуточного соединения 61
К смеси аминопиридина CAS 2178988-25-3 (1,6 г, 7 ммоль) в THF (30 мл) добавляли пиридин (1,1 мл, 14 ммоль) при 20°C и фенилхлорформиат (1,6 г, 10,5 ммоль) при 0°C. Смесь перемешивали при 20°C в течение 16 часов. Смесь гасили насыщ. водн. раствором NH4Cl. Смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~30% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 61 (2 г, выход: 82%) в виде белого твердого вещества.
Получение соединения 78
Соединение 78 получали по аналогии с процедурой, описанной для соединения 57, с применением промежуточного соединения 61 и промежуточного соединения 54. Обеспечивали достижение смеси комнатной температуры и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~50% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением соединения 78 (150 мг, выход: 32%) в виде белого твердого вещества.
LC/MS: масса/заряд 443,2 [M+H]+, rt: 0,83 мин, чистота 98,4%, способ: B.
Получение соединений 79 и 80
Соединение 78 (150 мг, 0,3 ммоль) отделяли посредством SFC. [Колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: растворитель, A: сверхкритический CO2, растворитель, B: 0,1% водный раствор аммиака в MeOH. Вначале: A (45%) и B (55%), в конце: A (55%) и B (45%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и смесь лиофилизировали до сухого состояния с получением соединения 79 (65 мг, выход: 44%) в виде белого твердого вещества и соединения 80 (65 мг, выход: 42%) в виде белого твердого вещества.
Соединение 79:
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,49 (d, J=6,8 Гц, 3 H), 2,67 (s, 3 H), 3,20 (s, 3 H), 3,99 (s, 3 H), 4,77 (q, J=6,8 Гц, 1 H), 8,13 (s, 2 H), 8,70 (s,1 H), 8,93 (s, 1 H), 8,98 (br s, 1 H), 9,51 (br s, 1 H)
HPLC/MS: масса/заряд 443,2 [M+H]+, rt: 4,62 мин, чистота 99,3%, способ: K;
SFC: чистота 100%, rt: 4,43 мин, способ: SFC19.
Соединение 80:
HPLC/MS: масса/заряд 443,2 [M+H]+, rt: 4,64 мин, чистота 95,1%, способ: K;
SFC: чистота 100%, rt: 3,53 мин, способ: SFC19.
Синтез соединений 81, 82 и 83
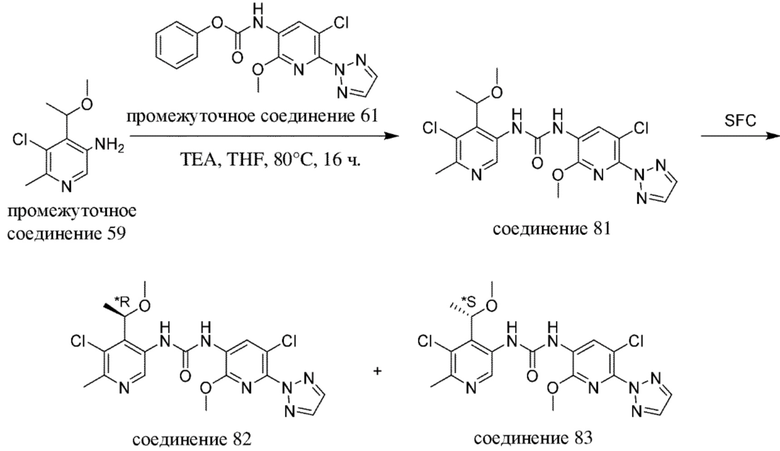
Получение соединения 81
Соединение 81 получали по аналогии с процедурой, описанной для соединения 57, с применением промежуточного соединения 61 и промежуточного соединения 59. Обеспечивали достижение смеси комнатной температуры и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~50% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением соединения 81 (200 мг, выход: 45%) в виде белого твердого вещества.
LC/MS: масса/заряд 452,1 [M+H]+, rt: 0,83 мин, чистота 100%, способ: B.
Получение соединений 82 и 83
Соединение 81 (200 мг, 0,4 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: растворитель, A: сверхкритический CO2, растворитель, B: 0,1% водный раствор аммиака в MeOH. Вначале: A (50%) и B (50%), в конце: A (50%) и B (50%), расход: 70 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и смесь лиофилизировали до сухого состояния с получением соединения 82 (80 мг, выход: 40%) в виде белого твердого вещества и соединения 83 (80 мг, выход: 40%) в виде белого твердого вещества.
Соединение 82:
HPLC/MS: масса/заряд 452,0 [M+H]+, rt: 4,7 мин, чистота 99,5%, способ: K;
SFC: чистота 98,8%, rt: 2,72 мин, способ: SFC19.
Соединение 83:
HPLC/MS: масса/заряд 452,0 [M+H]+, rt: 4,7 мин, чистота 99,8%, способ: K;
SFC: чистота 100%, rt: 1,38 мин, способ: SFC19.
Синтез соединений 84, 85 и 86

Получение соединения 84
Соединение 84 получали по аналогии с процедурой, описанной для соединения 57, с применением промежуточного соединения 61 и промежуточного соединения 39. Обеспечивали достижение смеси комнатной температуры и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~50% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением соединения 84 (150 мг, выход: 36%) в виде белого твердого вещества.
LC/MS: масса/заряд 496 [M+H]+, rt: 0,84 мин, чистота 98%, способ: B.
Получение соединений 85 и 86
Соединение 84 (150 мг, 0,3 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: растворитель, A: сверхкритический CO2, растворитель, B: 0,1% водный раствор аммиака в MeOH. Вначале: A (50%) и B (50%), в конце: A (50%) и B (50%), расход: 70 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и смесь лиофилизировали до сухого состояния с получением соединения 85 (60 мг, выход: 39%) в виде белого твердого вещества и соединения 86 (60 мг, выход: 40,9%) в виде белого твердого вещества.
Соединение 85:
HPLC/MS: масса/заряд 496 [M+H]+, rt: 4,79 мин, чистота 96,3%, способ: K;
SFC: чистота 100%, rt: 3,30 мин, способ: SFC19.
Соединение 86:
HPLC/MS: масса/заряд 496 [M+H]+, rt: 4,79 мин, чистота 100%, способ: K;
SFC: чистота 100%, rt: 1,55 мин, способ: SFC19.
Синтез соединений 87, 88 и 89
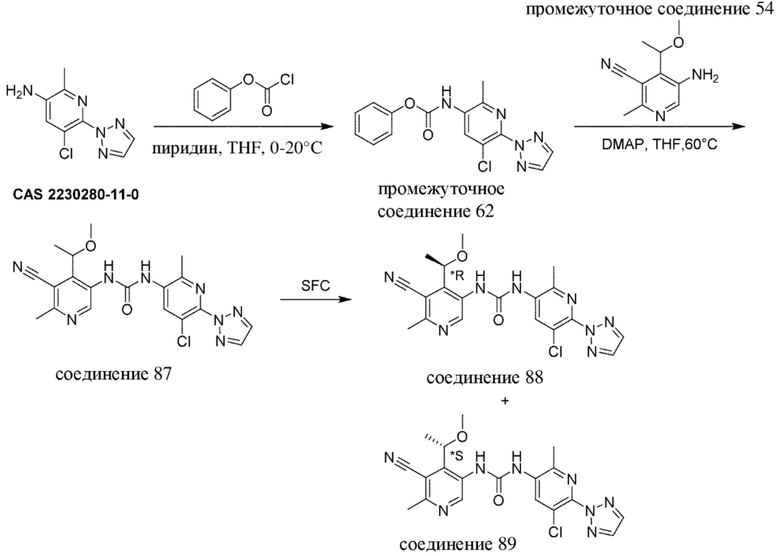
Получение промежуточного соединения 62
К смеси CAS 2230280-11-0 (10 г, 47 ммоль) в THF (200 мл) добавляли пиридин (11,5 мл, 143 ммоль) при 20°C и фенилхлорформиат (9 мл, 71 ммоль) при 0°C. Смесь перемешивали при 20°C в течение 16 часов. Смесь гасили насыщ. водн. раствором NH4Cl. Смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~50% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 62 (9 г, выход: 51%) в виде желтого твердого вещества.
Получение соединения 87
К смеси промежуточного соединения 54 (200 мг, 1 ммоль) в THF (10 мл) добавляли промежуточное соединение 62 (561 мг, 1,5 ммоль) и DMAP (247 мг, 2 ммоль) при 25°C. Реакционную смесь перемешивали при 60°C в течение 2 часов. Обеспечивали достижение температуры смеси, составляющей 25°C, и фильтровали. Фильтрат концентрировали в вакууме с получением неочищенного продукта в виде желтого твердого вещества. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Phenomenex Gemini 150 × 25 мм*10 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN. Вначале: A (70%) и B (30%), в конце: A (40%) и B (60%), время градиентного элюирования: 8 мин; 100% B, время удерживания: 2 мин; расход: 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 87 (120 мг, выход: 27,4%) в виде желтого твердого вещества.
LC/MS: масса/заряд 427,0 [M+H]+, rt: 0,75 мин, чистота: 98,9%, способ: A.
Получение соединений 88 и 89
Соединение 87 (120 мг, 0,28 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK IC (250 мм*30 мм, 10 мкм), условия: MeOH, A: сверхкритический CO2, растворитель, B: MeOH. Вначале: A (45%) и B (55%), в конце: A (45%) и B (55%), расход: 80 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и смесь лиофилизировали до сухого состояния с получением соединения 88 (41,3 мг, выход: 35%) в виде белого твердого вещества и соединения 89 (44,1 мг, выход: 37%) в виде белого твердого вещества.
Соединение 88:
HPLC/MS: масса/заряд 427,2, [M+H]+, rt: 4,25 мин, чистота: 100%, способ: K.
SFC: чистота 100%, rt: 1,93 мин, способ: SFC19.
Соединение 89:
HPLC/MS: масса/заряд 427,2, [M+H]+, rt: 4,25 мин, чистота: 99,9%, способ: K.
SFC: чистота 99,99%, rt: 4,40 мин, способ: SFC19.
Синтез соединений 90, 91 и 92
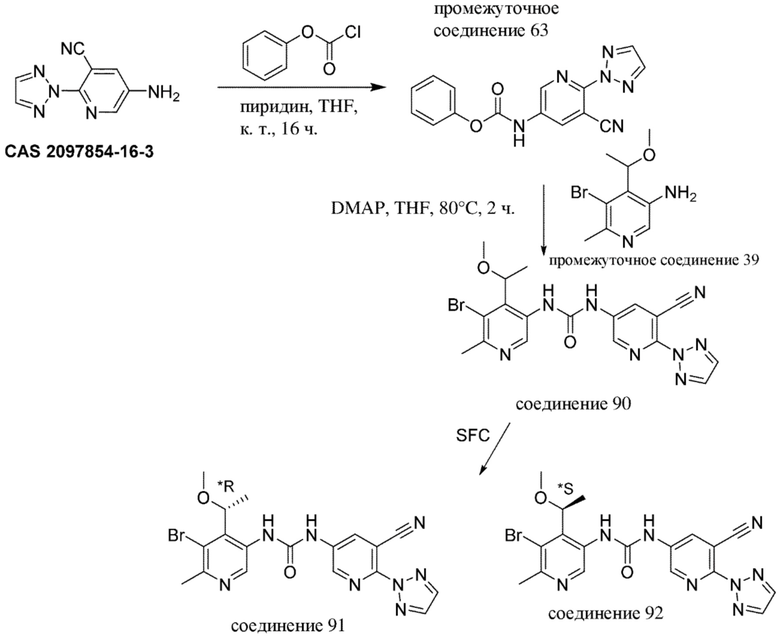
Получение промежуточного соединения 63
К раствору CAS 2097854-16-3 (10 г, 54 ммоль) и пиридина (8,7 мл, 108 ммоль) в THF (10 мл) добавляли фенилхлорформиат (11 г, 70 ммоль) при 20°C. Реакционную смесь перемешивали при 20°C в течение 16 часов. Смесь концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~40% EtOAc в петролейном эфире). Чистые фракции собирали и растворитель выпаривали под вакуумом с получением промежуточного соединения 63 (13 г, выход: 70%) в виде белого твердого вещества.
Получение соединения 90
К раствору промежуточного соединения 39 (200 мг, 0,7 ммоль) и промежуточного соединения 63 (300 мг, 1,0 ммоль) в THF (10 мл) добавляли DMAP (159 мг, 1,3 ммоль) при 20°C. Реакционную смесь перемешивали при 80°C в течение 2 часов. Смесь концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: PhenomenexGemini 150 × 25 мм*10 мкм, условия: A: вода (0,04% NH3 H2O+10 мМ NH4HCO3), B: MeCN, вначале: A (64%) и B (36%), в конце: A (34%) и B (66%), время градиентного элюирования 8,5 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 90 (180 мг, выход: 60%) в виде белого твердого вещества.
LC/MS: масса/заряд 457,1 [M+H]+, rt 1,0 мин, чистота 100%, способ G
Получение соединений 91 и 92
Соединение 90 (180 мг, 0,39 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 10 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в EtOH, вначале: A (50%) и B (50%), в конце: A (50%) и B (50%), расход: 70 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 91 (86 мг, выход: 47,5%) в виде белого твердого вещества и соединения 92 (86 мг, выход: 47,3%) в виде белого твердого вещества.
Соединение 91:
HPLC-MS: масса/заряд 457,1 [M+H]+, rt 4,10 мин. Чистота 99,5%, способ K
SFC: чистота 100%, rt 0,59 мин. Способ: SFC18
Соединение 92:
HPLC-MS: масса/заряд 457 [M+H]+, rt 4,10 мин. Чистота 99,1%, способ K
SFC: чистота 100%, rt 1,51 мин, способ: SFC18
Синтез соединений 96, 97 и 98
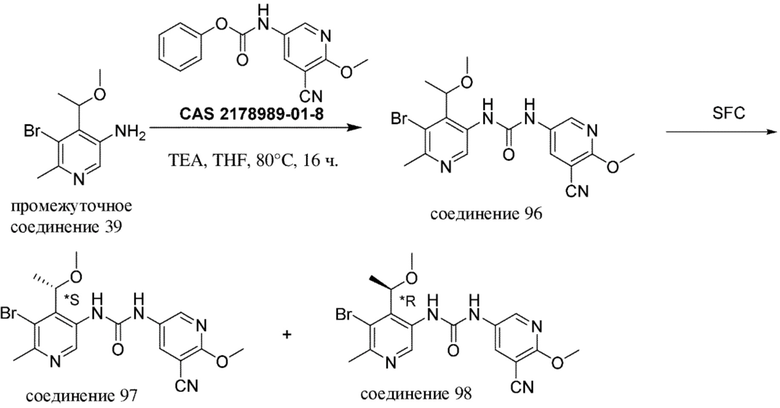
Получение соединения 96
Соединение 96 получали по аналогии с процедурой, описанной для соединения 57, с применением CAS 2178989-01-8 и промежуточного соединения 39 в качестве исходных материалов. Обеспечивали достижение смеси комнатной температуры и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Boston Prime C18 150 × 30 мм, 5 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN, вначале: A (60%) и B (40%), в конце: A (30%) и B (70%), время градиентного элюирования 8 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 96 (200 мг, выход: 57%) в виде белого твердого вещества.
Получение соединений 97 и 98
Соединение 96 (200 мг, 0,47 ммоль) отделяли посредством SFC [колонка: Phenomenex-Amylose-1 (250 мм*30 мм, 5 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в EtOH, вначале: A (70%) и B (30%), в конце: A (70%) и B (30%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 97 (66 мг, выход: 33,2%) в виде белого твердого вещества и соединения 98 (70 мг, выход: 35,6%).
Соединение 97:
LC/MS: масса/заряд 420,1 [M+H]+, rt 4,24 мин. Чистота 99,2%, способ K;
SFC: чистота 99,7%, rt 4,68 мин. Способ: SFC1
Соединение 98:
LC/MS: масса/заряд 420,1 [M+H]+, rt 4,24 мин. Чистота 100%, способ K;
SFC: чистота 98,3%, rt 5,25 мин. Способ: SFC1
Синтез соединений 99, 100 и 101
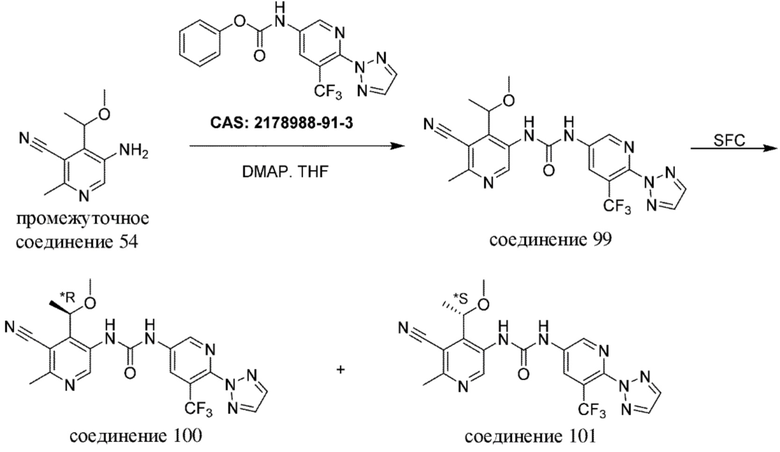
Получение соединения 99
К раствору промежуточного соединения 54 (1 г, 5,1 ммоль) и CAS 2178988-91-3 (2,6 г, 7,6 ммоль) в THF (30 мл) добавляли DMAP (1,2 г, 10 ммоль) при 20°C. Реакционную смесь перемешивали при 80°C в течение 3 часов. Смесь концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~55% EtOAc в петролейном эфире). Чистые фракции собирали и растворитель выпаривали под вакуумом с получением продукта в виде белого твердого вещества. Соединение очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: PhenomenexGemini 150 × 25 мм*10 мкм, условия: A: вода (0,225% FA)-ACN, B: MeCN, вначале: A (70%) и B (30%), в конце: A (40%) и B (60%), время градиентного элюирования 8 (мин); 100% B, время удерживания 2 (мин); расход: 60 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 99 (1,1 г, выход: 47%) в виде белого твердого вещества.
LC/MS: масса/заряд 447,0 [M+H]+, rt 0,783 мин. Чистота 98,6%, способ A
Получение соединений 100 и 101
Соединение 99 (1,1 г, 2,4 ммоль) отделяли посредством SFC. [Колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: A: CO2, B: 0,1% NH3H2O EtOH, вначале: A (75%) и B (25%), в конце: A (75%) и B (25%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водные слои лиофилизировали до сухого состояния с получением соединения 100 (502 мг, выход: 47%) и соединения 101 (505 мг, выход: 47%) в виде белого твердого вещества.
Соединение 100:
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,52 (d, J=6,8 Гц, 3 H), 2,67 (s, 3 H) 3,27 (s, 3 H), 4,82 (q, J=6,8 Гц, 1 H), 8,17 (s, 2 H), 8,69 (d, J=2,4 Гц, 1 H), 8,77 (br s, 1 H), 8,84 (d, J=2,4 Гц, 1 H), 9,10 (s, 1 H), 10,46 (br s, 1 H)
HPLC/MS: масса/заряд 447,1 [M+H]+, rt: 4,59 мин. Чистота: 99,9%, способ: K;
SFC: чистота 99,9%, rt: 4,85 мин, способ: SFC13.
Соединение 101:
HPLC/MS: масса/заряд 447,2 [M+H]+, rt: 4,55 мин. Чистота: 100%, способ: K;
SFC: чистота 99,7%, rt: 5,36 мин, способ: SFC13.
Синтез соединений 102, 103 и 104
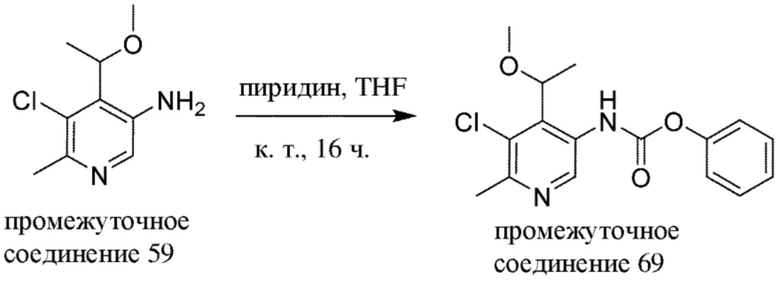
Получение промежуточного соединения 69
Промежуточное соединение 59 (350 мг, 1,74 ммоль) и пиридин (0,21 мл, 2,8 ммоль) растворяли в THF (4 мл) и перемешивали при 0°C, к смеси по каплям добавляли фенилхлорформиат (0,4 мл, 3,5 ммоль) и обеспечивали нагревание до к. т. в течение 16 ч. Добавляли насыщ. раствор NH4Cl и два раза экстрагировали с помощью EtOAc. Объединенные органические слои высушивали с помощью Na2SO4, фильтровали и концентрировали под вакуумом с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (градиентное элюирование: 10~30% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 69 (450 мг, выход: 80,4%).
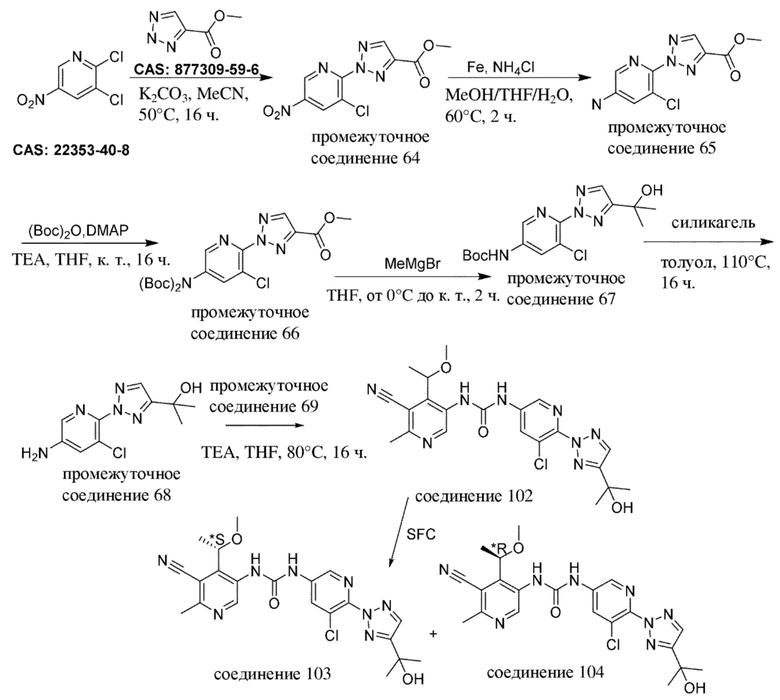
Получение промежуточного соединения 64
К смеси 2,3-дихлор-5-нитропиридина (16,7 г, 86,5 ммоль) и метил-2H-1,2,3-триазол-4-карбоксилата (10,0 г, 78,7 ммоль) в MeCN (200 мл) добавляли K2CO3 (32,6 г, 236,0 ммоль) и смесь перемешивали при 50°C в течение 16 часов. Смесь охлаждали до 25°C и фильтровали и фильтрат концентрировали с получением промежуточного соединения 64 (22 г, выход: 98,6%) в виде желтого твердого вещества.
Получение промежуточного соединения 65
Порошок Fe (4,9 г, 88,1 ммоль) и NH4Cl (4,7 г, 88,1 ммоль) добавляли к смеси промежуточного соединения 64 (10 г, 17,6 ммоль) в MeOH (40 мл), THF (80 мл) и H2O (20 мл) и смесь перемешивали при 60°C в течение 2 часов. Смесь охлаждали до 25°C и фильтровали. Фильтрат концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~60% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 65 (3,2 г, выход: 35,8%) в виде желтого твердого вещества.
Получение промежуточного соединения 66
К раствору промежуточного соединения 65 (6 г, 23,7 ммоль), DMAP (289 мг, 2,4 ммоль) и TEA (7,2 г, 70,9 ммоль) в THF (100 мл) медленно добавляли (Boc)2O (25,8 г, 118,3 ммоль) при 25°C. Реакционную смесь перемешивали при 25°C в течение 16 часов. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~30% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 66 (7,5 г, выход: 69,8%) в виде белого твердого вещества.
Получение промежуточного соединения 67
Промежуточное соединение 66 (2,9 г, 6,4 ммоль) растворяли в THF (40 мл) и бромид метилмагния (3 М в THF, 8,9 мл, 26,8 ммоль) добавляли при 0°C. Смесь нагревали до 25°C и перемешивали при 25°C в течение 2 часов. Смесь гасили насыщ. водн. раствором NH4Cl. Смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~60% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 67 (2,2 г, выход: 96%) в виде желтого твердого вещества.
Получение промежуточного соединения 68
Силикагель (15 г) добавляли к смеси промежуточного соединения 67 (2,2 г, 6,1 ммоль) в толуоле (50 мл), перемешивали при 110°C в течение 16 часов. Смесь охлаждали до 25°C и фильтровали. Фильтрат концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~100% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 68 (1,5 г, выход: 97%) в виде желтого твердого вещества.
Получение соединения 102
Соединение 102 получали по аналогии с процедурой, описанной для соединения 57, с применением промежуточных соединений 68 и 69. Обеспечивали достижение смеси комнатной температуры и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Boston Prime C18 150 × 30 мм, 5 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN. Вначале: A (65%) и B (35%), в конце: A (35%) и B (65%), время градиентного элюирования: 8 мин; 100% B, время удерживания: 2 мин; расход: 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 102 (114,7 мг, выход: 30,3%).
LC/MS: масса/заряд 480,1 [M+H]+, rt: 1,87 мин, чистота: 100%, способ: C.
Получение соединений 103 и 104
Соединение 102 (114,7 мг, 0,24 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AD (250 мм*30 мм, 10 мкм), условия: A: сверхкритический CO2, B: 0,1% NH3H2O EtOH, вначале: A (55%) и B (45%), в конце: A (55%) и B (45%), расход: 70 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 103 (44 мг, выход: 38,3%) в виде белого твердого вещества и соединения 104 (44 мг, выход: 40%) в виде белого твердого вещества.
Соединение 103:
HPLC/MS: масса/заряд 480,1 [M+H]+, rt 4,21 мин, чистота 99,8%, способ K.
SFC: чистота 100%, rt 5,57 мин. Способ: SFC1.
Соединение 104:
HPLC/MS: масса/заряд 480,2 [M+H]+, rt 4,21 мин, чистота 100%, способ K.
SFC: чистота 100%, rt 7,02 мин. Способ: SFC1.
Синтез соединения 105
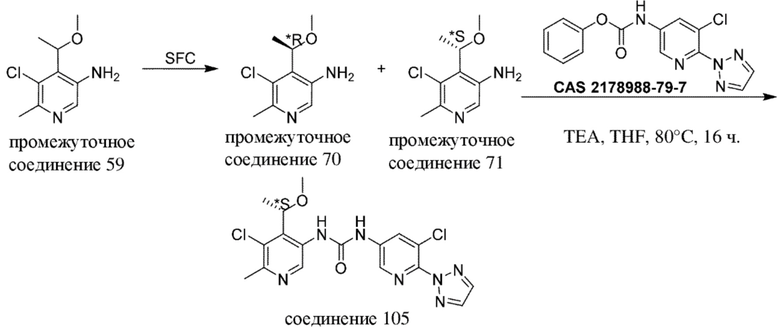
Получение промежуточных соединений 70 и 71
Промежуточное соединение 59 (500 мг, 2,46 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в MeOH, вначале: A (85%) и B (15%), в конце: A (85%) и B (15%), расход: 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением промежуточного соединения 70 (220 мг, выход: 44%) в виде белого твердого вещества и промежуточного соединения 71 (210 мг, выход: 42%) в виде белого твердого вещества.
Промежуточное соединение 70 SFC: чистота 100%, rt 2,594 мин. Способ: SFC10.
Промежуточное соединение 71 SFC: чистота 99,87%, rt 2,848 мин. Способ: SFC10.
Получение соединения 105
Соединение 105 получали по аналогии с процедурой, описанной для соединения 57, с применением CAS 2178988-79-7 и промежуточного соединения 71. Смесь концентрировали в вакууме с получением неочищенного продукта в виде светло-желтого твердого вещества. Смесь петролейного эфира: этилацетата=1:1 (50 мл) добавляли к неочищенному веществу; смесь перемешивали при 25°C в течение 10 мин и фильтровали. Осадок на фильтре промывали с помощью еще 20 мл смеси растворителей. Осадок на фильтре собирали, добавляли THF (20 мл) и смесь перемешивали при 25°C в течение 10 мин. Фильтрат концентрировали в вакууме с получением соединения 105 (164,4 мг, выход: 37%) в виде белого твердого вещества.
HPLC/MS: масса/заряд 422,2 [M+H]+, rt 4,225 мин. Чистота 99,47%, способ K;
SFC: чистота 99,93%, rt 1,653 мин. Способ: SFC18.
Синтез соединения 106
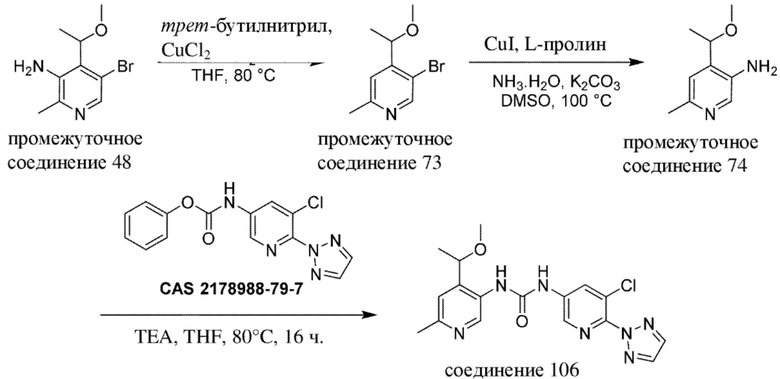
Получение промежуточного соединения 73
Смесь промежуточного соединения 48 (500 мг, 2 ммоль; получено по аналогии с промежуточным соединением 39), трет-бутилнитрита (630 мг, 6,1 ммоль) и CuCl2 (55 мг, 0,4 ммоль) в THF (15 мл) перемешивали при 80°C в течение 16 часов. Обеспечивали охлаждение смеси до 25°C. Добавляли H2O (30 мл) и экстрагировали с помощью EtOAc (20 мл*2). Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и фильтраты концентрировали под вакуумом с получением неочищенного вещества в виде желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~10% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали in vacuo с получением промежуточного соединения 73 (270 мг, чистота 83%; желтое масло).
Получение промежуточного соединения 74
Смесь промежуточного соединения 73 (220 мг, чистота 83%), CuI (15 мг, 0,08 ммоль), L-пролина (18 мг, 0,16 ммоль), K2CO3 (165 мг, 1,2 ммоль), NH3.H2O (5 мл) растворяли в DMSO (5 мл). Смесь перемешивали при 100°C в течение 16 часов. Реакционную смесь гасили нас. раствором NH4Cl (20 мл), экстрагировали с помощью EtOAc (20 мл*2). Объединенные органические слои разделяли, промывали солевым раствором, высушивали над Na2SO4, фильтровали и фильтраты выпаривали под вакуумом с получением желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~100% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 74 (40 мг, выход: 29,5%) в виде желтого масла.
Получение соединения 106
Соединение 106 получали по аналогии с процедурой, описанной для соединения 57, с применением сложного фенилового эфира N-[5-хлор-6-(2H-1,2,3-триазол-2-ил)-3-пиридинил]-карбаминовой кислоты и промежуточного соединения 74. Реакционную смесь концентрировали под вакуумом с получением неочищенного продукта в виде белого твердого вещества. К смеси добавляли MeOH (20 мл) и перемешивали при 80°C в течение 15 мин. Фильтровали и фильтраты концентрировали под вакуумом с получением неочищенного вещества в виде желтого масла. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Boston Prime C18 150 × 30 мм, 5 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN, вначале: A (75%) и B (25%), в конце: A (45%) и B (55%), время градиентного элюирования 8 мин; 100% B, время удерживания 2 мин; расход 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 106 в виде смеси 1:1 2 энантиомеров (21 мг, выход: 23%) в виде белого твердого вещества.
HPLC/MS: масса/заряд 388,1 [M+H]+, rt 3,903 мин. Чистота 100%, способ M;
SFC: чистота 49,79%; 50,21%, rt 5,813 мин, 8,012 мин. Способ: SFC1
Синтез соединений 110, 111 и 112
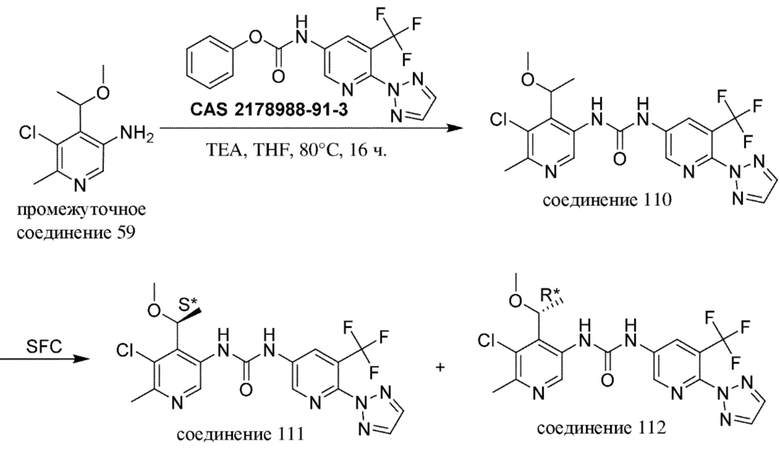
Получение соединения 110
Соединение 110 получали по аналогии с процедурой, описанной для соединения 57, с применением CAS 2178988-91-3 и промежуточного соединения 59. Обеспечивали достижение смеси комнатной температуры и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Boston Prime C18 150 × 30 мм, 5 мкм, условия: A: вода (0,04% водный раствор аммиака+10 мМ NH4HCO3), B: MeCN. Вначале: A (55%) и B (45%), в конце: A (25%) и B (75%), время градиентного элюирования: 8 мин; 100% B, время удерживания: 2 мин; расход: 25 мл/мин]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 110 (200 мг, выход: 44%) в виде белого твердого вещества.
LC/MS: масса/заряд 456,1 [M+H]+, rt: 1,85 мин, чистота: 100%, способ: C.
Получение соединений 111 и 112
Соединение 110 (200 мг, 0,44 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: A: сверхкритический CO2, B: 0,1% NH3H2O EtOH, вначале: A (75%) и B (25%), в конце: A (75%) и B (25%), расход: 60 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 111 (89,5 мг, выход: 44,7%) в виде белого твердого вещества и соединения 112 (98,2 мг, выход 47,5%).
Соединение 111:
HPLC/MS: масса/заряд 456,2 [M+H]+, rt 4,55 мин. Чистота 100%, способ K
SFC: чистота 100%, rt 3,54 мин. Способ: SFC1.
Соединение 112:
HPLC/MS: масса/заряд 456,0 [M+H]+, rt 4,55 мин. Чистота 96,6%, способ K
SFC: чистота 100%, rt 3,92 мин. Способ: SFC1.
Синтез соединений 113, 114 и 115

Получение промежуточного соединения 75
К смеси 2,3-дихлор-5-нитропиридина (16,7 г, 86,5 ммоль) и метил-2H-1,2,3-триазол-4-карбоксилата (10,0 г, 78,7 ммоль) в MeCN (200 мл) добавляли K2CO3 (32,6 г, 236,0 ммоль) и смесь перемешивали при 50°C в течение 16 часов. Смесь охлаждали до 25°C и фильтровали и фильтрат концентрировали с получением промежуточного соединения 75 (22 г, выход: 98,6%) в виде желтого твердого вещества.
Получение промежуточного соединения 76
Порошок Fe (4,9 г, 88,1 ммоль) и NH4Cl (4,7 г, 88,1 ммоль) добавляли к смеси промежуточного соединения 75 (10 г, 17,6 ммоль) в MeOH (40 мл), THF (80 мл) и H2O (20 мл). Смесь перемешивали при 60°C в течение 2 часов. Смесь охлаждали до 25°C и фильтровали. Фильтрат концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~60% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 76 (3,2 г, выход: 35,8%) в виде желтого твердого вещества.
Получение промежуточного соединения 77
К раствору промежуточного соединения 76 (6 г, 23,7 ммоль), DMAP (289 мг, 2,4 ммоль) и TEA (7,2 г, 70,9 ммоль) в THF (100 мл) медленно добавляли (Boc)2O (25,8 г, 118,3 ммоль) при 25°C. Реакционную смесь перемешивали при 25°C в течение 16 часов. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~30% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 77 (7,5 г, выход: 69,8%) в виде белого твердого вещества.
Получение промежуточного соединения 78
К раствору промежуточного соединения 77 (3 г, 6,6 ммоль) в THF (24 мл) и H2O (6 мл) добавляли LiOH (2,8 г, 66,0 ммоль) при 25°C. Реакционную смесь перемешивали при 25°C в течение 16 часов. Значение pH реакционной смеси доводили до 3~4 с применением водного раствора HCl (5 М) и экстрагировали с помощью EtOAc (50 мл × 3). Объединенные органические слои разделяли, промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного промежуточного соединения 78 (2,2 г, выход: 97,3%) в виде белого твердого вещества.
Получение промежуточного соединения 79
К раствору промежуточного соединения 78 (2,2 г, 6,4 ммоль), гидрохлорида N, O-диметилгидроксиламина (0,94 г, 9,6 ммоль) и DIEA (4,8 мл, 28,9 ммоль) в DMF (30 мл) медленно добавляли HATU (3,7 г, 9,6 ммоль) при 25°C. Реакционную смесь перемешивали при 25°C в течение 16 часов. Смесь разбавляли с помощью H2O и два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~60% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 79 (2,4 г, выход: 96%) в виде белого твердого вещества.
Получение промежуточного соединения 80
Промежуточное соединение 79 (2,4 г, 6,2 ммоль) растворяли в THF (60 мл) и добавляли бромид метилмагния (3 М в THF, 8,3 мл, 24,8 ммоль) при 0°C. Смесь нагревали до 25°C и перемешивали при 25°C в течение 2 часов. Смесь гасили насыщ. водн. раствором NH4Cl. Смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~50% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 80 (2 г, выход: 95%) в виде желтого твердого вещества.
Получение промежуточного соединения 81
Промежуточное соединение 80 (2 г, 5,9 ммоль) растворяли в MeOH (30 мл) и медленно добавляли NaBH4 (1,1 г, 29,6 ммоль) при 25°C. Смесь перемешивали при 25°C в течение 16 часов. Смесь гасили насыщ. водн. раствором NH4Cl. Смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~100% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 81 (1,7 г, выход: 84%) в виде желтого твердого вещества.
Получение промежуточного соединения 82
Силикагель (8 г) добавляли к смеси промежуточного соединения 81 (1,1 г, 3,2 ммоль) в толуоле (30 мл). Смесь перемешивали при 110°C в течение 18 часов. Смесь охлаждали до 25°C и фильтровали. Фильтрат концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~100% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 82 (650 мг, выход: 82,3%) в виде желтого твердого вещества.
Получение промежуточного соединения 83
Трет-бутилхлордиметилсилан добавляли к смеси промежуточного соединения 82 (650 мг, 2,7 ммоль) и имидазола (910 мг, 13,3 ммоль) в DMF (10 мл) при 0°C в атмосфере N2. Реакционную смесь перемешивали при к.т. в течение 16 часов. Обеспечивали нагревание смеси до 25°C. Добавляли H2O (30 мл) и экстрагировали с помощью EtOAc (30 мл*2). Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и фильтраты концентрировали под вакуумом с получением неочищенного вещества в виде желтого твердого вещества. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~60% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 83 (700 мг, чистота: 74%) в виде белого твердого вещества.
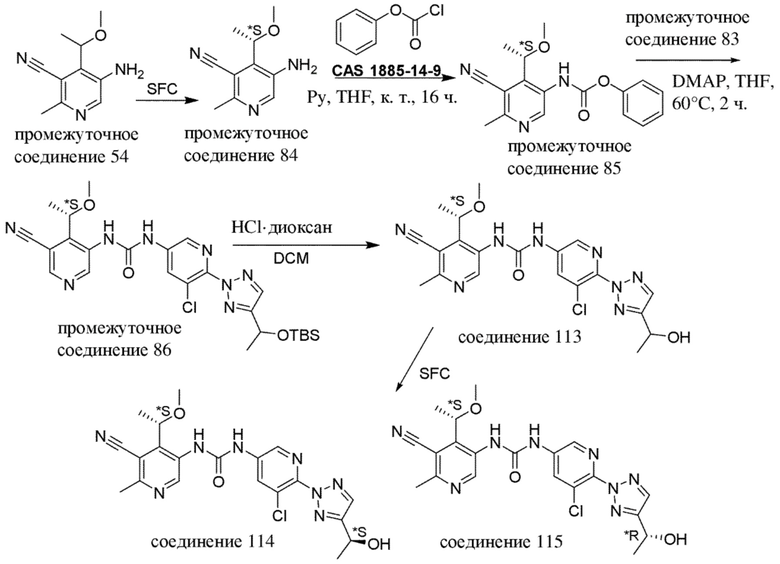
Получение промежуточного соединения 84
Промежуточное соединение 54 (2,6 г, 13,6 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AY (250 мм*50 мм, 10 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в EtOH, вначале: A (85%) и B (15%), в конце: A (85%) и B (15%), расход: 180 (мл/мин)]. Чистые фракции собирали и концентрировали в вакууме с получением промежуточного соединения 84 (1,1 г, чистота: 100%) в виде желтого твердого вещества.
SFC: чистота 100%, rt: 3,36 мин, способ: SFC20
Получение промежуточного соединения 85
К раствору промежуточного соединения 84 (300 мг, 1,6 ммоль) в THF (10 мл) добавляли пиридин (0,3 мл, 3,1 ммоль) при комнатной температуре. Медленно добавляли фенилхлорформиат (320 мг, 2,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Смесь гасили насыщ. водн. раствором NH4Cl и затем два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором и высушивали с помощью безводного Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~50% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 85 (450 мг, выход: 92%) в виде белого твердого вещества.
Получение промежуточного соединения 86
Промежуточное соединение 86 получали по аналогии с процедурой, описанной для соединения 87, начиная с промежуточного соединения 85 и промежуточного соединения 83. Обеспечивали достижение смеси комнатной температуры и концентрировали в вакууме с получением неочищенного вещества. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~60% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 86 (420 мг, выход: 88%) в виде белого твердого вещества.
Получение соединения 113
К раствору промежуточного соединения 86 (350 мг, 0,6 ммоль) в DCM (5 мл) добавляли HCl.диоксан (4 М) (5 мл) при 25°C. Реакционную смесь перемешивали при 25°C в течение 2 часов. Значение pH реакционной смеси доводили до 8~9 с применением насыщ. раствора NaHCO3 и экстрагировали с помощью EtOAc (30 мл*3). Объединенные органические слои разделяли, промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~100% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением соединения 113 (260 мг, выход: 91%) в виде белого твердого вещества.
LC/MS: масса/заряд 457,2 [M+H]+, rt: 0,82 мин, чистота 98,6%, способ: B
SFC: чистота 50,0%/50,0%, rt: 2,3 мин/6,0 мин, способ: SFC21
Получение соединений 114 и 115
Соединение 113 (260 мг, 0,56 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK IC (250 мм*30 мм, 10 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в EtOH, вначале: A (60%) и B (40%), в конце: A (60%) и B (40%), расход: 70 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 114 (89,0 мг, выход: 34%) в виде белого твердого вещества и соединения 115 (95,0 мг, выход: 36%)
Соединение 114:
HPLC/MS: масса/заряд 457,0 [M+H]+, rt 4,60 мин. Чистота 98,2%, способ M;
SFC: чистота 100%, rt: 5,57 мин, способ: SFC21
Соединение 115:
HPLC/MS: масса/заряд 457,0 [M+H]+, rt 4,67 мин. Чистота 97,4%, способ M.
SFC: чистота 99,2%, rt: 6,3 мин, способ: SFC21
Синтез соединений 116, 117, 118 и 119
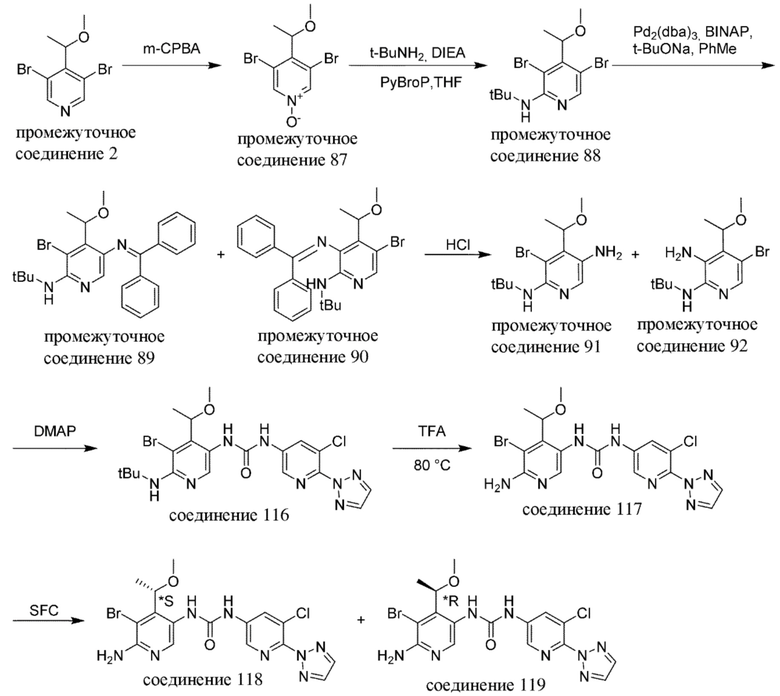
Получение промежуточного соединения 87
Промежуточное соединение 87 (10 г, 40 ммоль) растворяли в DCM (100 мл) и добавляли м-CPBA (85%, 13,7 г, 688 ммоль). Смесь перемешивали при 20°C в течение 16 часов. Значение pH доводили до 12 с применением NaOH (5 М), добавляли воду (100 мл) и смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и концентрировали под вакуумом с получением неочищенного продукта. Неочищенный продукт промывали с помощью 100 мл смеси растворителей (петролейный эфир/этилацетат=10:1). Твердое вещество собирали и концентрировали под вакуумом с получением промежуточного соединения 87 (9,8 г, выход: 90%) в виде белого твердого вещества.
Получение промежуточного соединения 88
К раствору промежуточного соединения 87 (30 г, 96,5 ммоль) в THF (200 мл) добавляли трет-бутиламин (8,82 г, 120,6 ммоль), PyBroP CAS 132705-51-2 (58,5 г, 125,4 ммоль) и DIPEA (46,7 г, 361,8 ммоль). Реакционную смесь перемешивали при 80°C в течение 16 часов. Смесь охлаждали до 25°C и затем фильтровали и остаток два раза промывали с помощью 150 мл этилацетата. Добавляли насыщ. раствор NH4Cl (400 мл), объединенные органические слои промывали солевым раствором и высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~10% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 88 (12,5 г, выход: 35,4%) в виде белого твердого вещества.
Получение смеси промежуточных соединений 89 и 90
Смесь промежуточных соединений 89 и 90 получали по аналогии с процедурой, описанной для промежуточного соединения 38. Остаток промывали с помощью EtOAc (200 мл). Фильтраты концентрировали под вакуумом с получением неочищенного продукта в виде желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~8% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением смеси промежуточных соединений 89 и 90 (6 г, неочищенные) в виде желтого масла.
Получение промежуточных соединений 91 и 92
Смесь промежуточных соединений 89 и 90 (6 г, неочищенные) растворяли в DCM (20 мл). Добавляли водный раствор HCl (80 мл, 1 М) и смесь перемешивали при 25°C в течение 16 часов. Значение pH реакционной смеси доводили до 8 с применением водн. раствора NaOH (5 М) и экстрагировали с помощью DCM (80 мл*2). Объединенные органические слои разделяли, промывали солевым раствором, высушивали над Na2SO4, фильтровали и фильтраты выпаривали под вакуумом с получением желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~10% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 91 (1,5 г, выход: 25% с двух стадий) в виде белого твердого вещества и непрореагировавшего промежуточного соединения 90 (4 г, неочищенное) в виде желтого масла. Затем промежуточное соединение 90 (4 г, неочищенное) растворяли в DCM (10 мл). Добавляли водный раствор HCl (30 мл, 3 М) и смесь перемешивали при 25°C в течение 16 часов. Значение pH реакционной смеси доводили до 8 с применением водн. раствора NaOH (5 М) и экстрагировали с помощью DCM (50 мл*2). Объединенные органические слои разделяли, промывали солевым раствором, высушивали над Na2SO4, фильтровали и фильтраты выпаривали под вакуумом с получением желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~12% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 92 (700 мг, выход: 12% с трех стадий) в виде желтого твердого вещества.
Получение соединения 116
Соединение 116 получали по аналогии с процедурой, описанной для соединения 99. Реакционную смесь охлаждали до 25°C, фильтровали и остаток промывали с помощью EtOAc (50 мл). Объединенные фильтраты концентрировали под вакуумом с получением неочищенного вещества в виде желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~60% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением соединения 116 (1,0 г, выход: 60%) в виде желтого твердого вещества.
LC/MS: масса/заряд 523,1 [M+H]+, rt 2,230 мин, чистота 99,3%, способ C.
Получение соединения 117
Соединение 116 (1,0 г, 1,9 ммоль) растворяли в DCM (10 мл), добавляли TFA (10 мл). Реакционную смесь перемешивали при 80°C в течение 16 часов. Реакционную смесь концентрировали под вакуумом с получением неочищенного продукта в виде желтого масла. Значение pH реакционной смеси доводили до 7 с применением насыщ. раствора NaHCO3, добавляли 50 мл воды и перемешивали при к.т. в течение 30 мин. Продукт собирали посредством фильтрации и остаток собирали и высушивали под вакуумом с получением соединения 117 (700 мг, выход: 72%) в виде белого твердого вещества.
LC/MS: масса/заряд 467,0 [M+H]+, rt 1,488 мин, чистота 90,8%, способ C.
Получение соединений 118 и 119
Соединение 117 (600 мг, 1,165 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK IC (250 мм*30 мм, 10 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водный раствор аммиака в EtOH, вначале: A (45%) и B (55%), в конце: A (45%) и B (55%), расход: 60 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 118 (252 мг, выход: 46,3%) в виде белого твердого вещества и соединения 119 (255 мг, выход: 46,8%) в виде белого твердого вещества.
Соединение 118:
HPLC/MS: масса/заряд 467 [M+H]+, rt: 3,526 мин, чистота 98,4%, способ: K;
SFC: чистота 100%, rt: 7,808 мин, способ: SFC22.
Соединение 119:
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,37 (d, J=6,8 Гц, 3 H) 3,21 (s, 3 H) 4,89 (q, J=6,8 Гц, 1 H) 6,14 (s, 2 H) 8,11 (s, 2 H) 8,20 (s, 1 H) 8,25 (br s, 1 H) 8,42-8,44 (m, 1 H) 8,45-8,47 (m, 1 H) 10,17 (br s, 1 H)
HPLC/MS: масса/заряд 467 [M+H]+, rt: 3,536 мин, чистота 98,8%, способ: K;
SFC: чистота 100%, rt: 9,907 мин, способ: SFC22.
Синтез соединений 120, 121 и 122
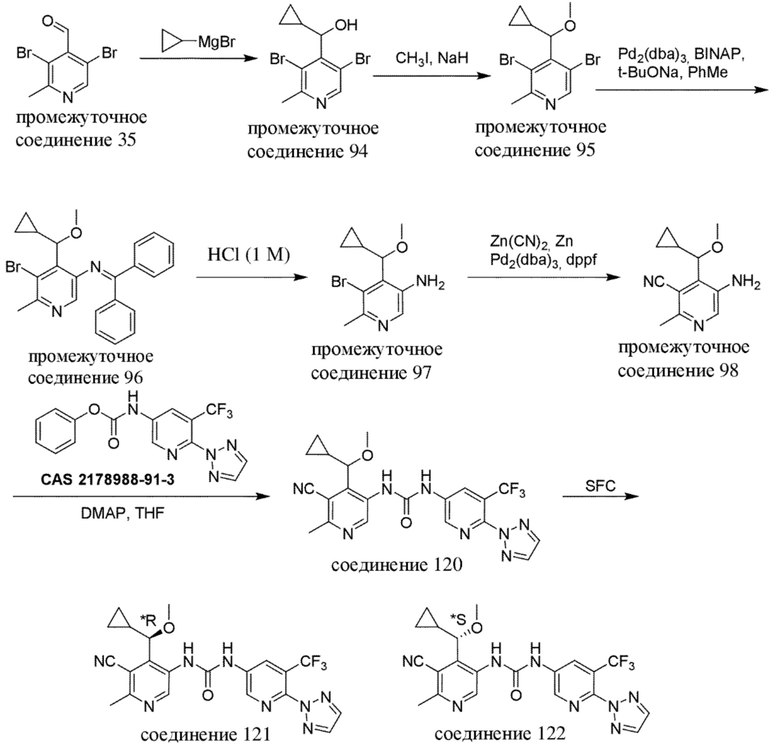
Получение промежуточного соединения 94
Промежуточное соединение 35 (2,0 г, 7,2 ммоль) растворяли в THF (20 мл) и добавляли бромид циклопропилмагния (28,7 мл, 14,3 ммоль, 0,5 М в THF) при −78°C. Реакционную смесь перемешивали при −78°C в течение 2 часов. TLC демонстрировала завершение реакции. Реакционную смесь гасили насыщ. водн. раствором NH4Cl, затем добавляли H2O и смесь два раза экстрагировали с помощью EtOAc. Объединенные органические слои высушивали с помощью Na2SO4, фильтровали и фильтрат концентрировали под вакуумом с получением неочищенного продукта в виде желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~20% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 94 (1,8 г, выход: 78%) в виде желтого масла.
Получение промежуточного соединения 95
К раствору промежуточного соединения 94 (1,8 г, 5,6 ммоль) в THF (20 мл) добавляли NaH (336 мг, 8,4 ммоль, 60%, в минеральном масле) при 0°C. Смесь перемешивали при 0°C в течение 0,5 часа. Затем добавляли MeI (3,2 г, 22,4 ммоль). Смесь перемешивали при 20°C в течение 2 часов. Смесь гасили насыщ. водн. раствором NH4Cl и два раза экстрагировали с помощью EtOAc. Объединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~20% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 95 (1,4 г, выход: 74%) в виде желтого масла.
Получение промежуточного соединения 96
К смеси промежуточного соединения 95 (1,4 г, 4,2 ммоль) в толуоле (16 мл) добавляли дифенилметанимин (0,8 г, 4,6 ммоль) и t-BuONa (0,4 г, 4,2 ммоль) и смесь продували с помощью N2 в течение 10 мин. Добавляли Pd2(dba)3 (0,2 г, 0,2 ммоль) и BINAP (0,4 г, 0,6 ммоль). Реакционную смесь перемешивали при 120°C в течение 12 часов. TLC демонстрировала завершение реакции. Смесь фильтровали и фильтрат концентрировали с получением неочищенного продукта в виде желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~20% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 96 (1,0 г, выход: 36%) в виде желтого масла.
Получение промежуточного соединения 97
К раствору промежуточного соединения 96 (1,0 г, 1,5 ммоль) в DCM (15 мл) добавляли HCl (3 мл, 1 М). Смесь перемешивали при 40°C в течение 2 часов. Значение pH реакционной смеси доводили до 8 с применением насыщ. водн. раствора NaHCO3 и два раза экстрагировали с помощью CH2Cl2. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и выпаривали под вакуумом с получением желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~55% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 97 (330 мг) в виде белого твердого вещества.
Получение промежуточного соединения 98
К смеси промежуточного соединения 97 (330 мг, 1,1 ммоль) добавляли Zn (CN)2 (82 мг, 0,7 ммоль) и Zn (23 мг, 0,3 ммоль) в DMF (10 мл). Смесь продували с помощью N2 в течение 5 мин. Добавляли Pd2(dba)3 (53 мг, 0,1 ммоль) и dppf CAS 12150-46-8 (65 мг, 0,1 ммоль). Реакционную смесь перемешивали при 120°C в течение 12 часов. Смесь фильтровали и фильтрат концентрировали с получением неочищенного продукта в виде желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~52% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 98 (250 мг, выход: 88%) в виде желтого твердого вещества.
Получение соединения 120
К раствору промежуточного соединения 98 (150 мг, 0,6 ммоль) и CAS 2178988-91-3 (322 мг, 0,9 ммоль) в THF (20 мл) добавляли DMAP (150 мг, 1,2 ммоль) при 20°C. Реакционную смесь перемешивали при 80°C в течение 3 часов. Смесь концентрировали в вакууме с получением неочищенного вещества. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~45% EtOAc в петролейном эфире). Чистые фракции собирали и растворитель выпаривали под вакуумом с получением продукта в виде белого твердого вещества. Соединение очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: PhenomenexGemini 150 × 25 мм*10 мкм, условия: A: вода (0,04%NH3H2O+10 мМ NH4HCO3)-ACN, B: MeCN, вначале: A (58%) и B (42%), в конце: A (28%) и B (72%), время градиентного элюирования 8 (мин); 100% B, время удерживания 2 (мин); расход: 25 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением соединения 120 (70 мг, выход: 24%) в виде белого твердого вещества.
LC/MS: масса/заряд 473,1 [M+H]+, rt: 0,96 мин, чистота 100%, способ: A.
SFC: чистота 50,1%/49,9%, rt: 5,2 мин/6,9 мин, способ: SFC13.
Получение соединений 121 и 122
Соединение 120 (70 мг, 0,1 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AD-H (250 мм*30 мм, 5 мкм), условия: A: CO2, B: 0,1% NH3H2O EtOH, вначале: A (60%) и B (40%), в конце: A (60%) и B (40%), расход: 70 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водные слои лиофилизировали до сухого состояния с получением соединения 121 (29,5 мг, выход: 42%) и соединения 122 (28,6 мг, выход: 40%) в виде белого твердого вещества.
Соединение 121:
HPLC/MS: масса/заряд 473,2 [M+H]+, rt: 4,87 мин. Чистота 100%, способ: K;
SFC: чистота 100%, rt: 5,19 мин, способ: SFC13.
Соединение 122:
HPLC/MS: масса/заряд 473,2 [M+H]+, rt: 4,87 мин. Чистота: 100%, способ: K;
SFC: чистота 100%, rt: 6,87 мин, способ: SFC13.
Синтез соединений 123, 124 и 125
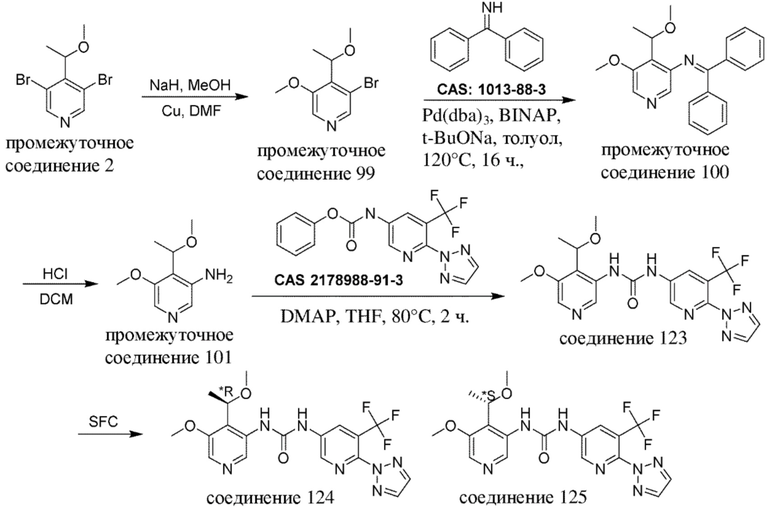
Получение промежуточного соединения 99
Метанол (0,83 г, 26 ммоль) растворяли в DMF (30 мл) и добавляли NaH (1,0 г, 26 ммоль, 60%, в минеральном масле) при 0°C. Смесь перемешивали при комнатной температуре в течение 1 часа. Затем к смеси медленно добавляли промежуточное соединение 2 (2,0 г, 6,5 ммоль) и порошок Cu (0,040 г, 0,65 ммоль). Смесь перемешивали при 80°C в течение 20 мин. Реакционную смесь охлаждали до 0°C и гасили посредством добавления по каплям воды (30 мл) и затем два раза экстрагировали с помощью EtOAc (50 мл × 2). Объединенные органические слои промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и концентрировали под вакуумом с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (градиентное элюирование: 0~60% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 99 (1,13 г, выход: 70,4%) в виде бесцветной жидкости.
Получение промежуточного соединения 100
Смесь промежуточного соединения 99 (1,13 г, 4,58 ммоль), дифенилметанимина CAS 1013-88-3 (0,9 г, 5 ммоль) и t-BuONa (0,44 г, 4,58 ммоль) в толуоле (20 мл) продували с помощью N2 в течение 10 мин. Затем добавляли Pd2(dba)3 (0,21 г, 0,23 ммоль) и BINAP (0,43 г, 0,69 ммоль). Реакционную смесь перемешивали при 120°C в течение 16 часов. Реакционную смесь охлаждали до 25°C и фильтровали. Остаток промывали с помощью EtOAc (50 мл). Фильтраты концентрировали под вакуумом с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~70% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением неочищенного промежуточного соединения 100 (1,78 г, выход: 65,9%) в виде желтого масла.
Получение промежуточного соединения 101
Неочищенное промежуточное соединение 100 (1,78 г, чистота 58,7%) растворяли в DCM (20 мл). Добавляли водн. раствор HCl (6 мл, 1 М) и смесь перемешивали при комнатной температуре в течение 24 часов. Значение pH реакционной смеси доводили до 8 с применением насыщ. раствора NaHCO3 и экстрагировали с помощью DCM (30 мл × 2). Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и фильтраты выпаривали под вакуумом с получением желтого масла. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~90% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения 101 (0,47 г, выход: 56% с двух стадий) в виде белого твердого вещества.
Получение соединения 123
К раствору промежуточного соединения 101 (350 мг, 1,92 ммоль) и CAS 2178988-91-3 (805 мг, 2,3 ммоль) в THF (10 мл) добавляли DMAP (469 мкл, 3,84 ммоль) при 20°C. Реакционную смесь перемешивали при 80°C в течение 2 часов. Обеспечивали охлаждение смеси до комнатной температуры и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~90% EtOAc в петролейном эфире). Необходимые фракции собирали и растворитель концентрировали в вакууме с получением соединения 123 (0,6 г, выход: 66%) в виде белого твердого вещества.
LC/MS: масса/заряд 438,1 [M+H]+, rt: 0,699 мин, чистота 92,8%, способ: B.
Получение соединений 124 и 125
Соединение 123 (600 мг, 1,27 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALPAK AD (250 мм*50 мм, 10 мкм), условия: растворитель A: сверхкритический CO2, растворитель B: 0,1% водн. раствор аммиака в EtOH, вначале: A (65%) и B (35%), в конце: A (65%) и B (35%), расход: 80 (мл/мин)]. Чистые фракции собирали и растворитель выпаривали в вакууме с получением неочищенного продукта (260 мг, чистота: 93,5%) в виде белого твердого вещества. Неочищенное вещество очищали посредством колоночной флеш-хроматографии на силикагеле (градиентное элюирование: 0~90% EtOAc в петролейном эфире). Чистые фракции собирали и органический растворитель выпаривали под вакуумом. К остатку добавляли MeCN и H2O и его лиофилизировали до сухого состояния с получением соединения 124 (207 мг, выход: 37%) и соединения 125 (253 мг, выход: 45%) в виде белых твердых веществ.
Соединение 124:
HPLC/MS: масса/заряд 438,2 [M+H]+, rt: 3,762 мин, чистота 99,9%, способ: K.
SFC: чистота 100%, rt: 0,928 мин, способ: SFC1.
Соединение 125:
LC/MS: масса/заряд 438,2 [M+H]+, rt: 3,765 мин, чистота 100%, способ: K.
SFC: чистота 100%, rt: 1,163 мин, способ: SFC1.
Синтез соединений 126, 127 и 128
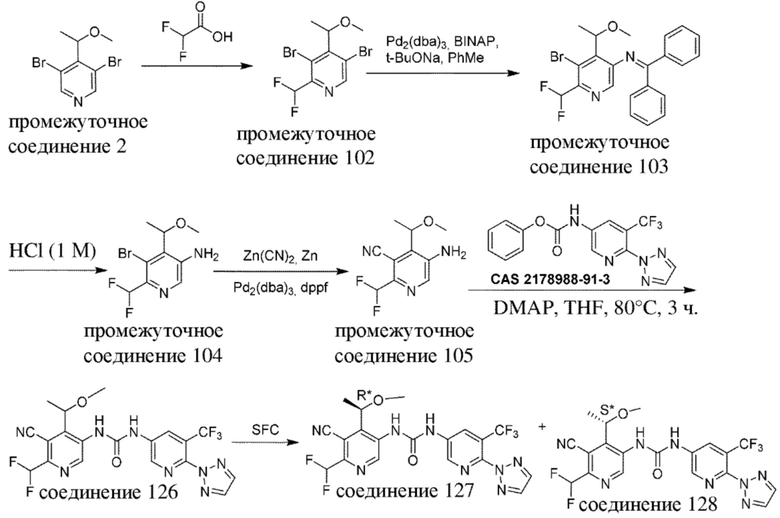
Получение промежуточного соединения 102
(NH4)2S2O3 (10 экв.) добавляли к смеси промежуточного соединения 2 (5,0 г; 17 ммоль)), 2,2-дифторуксусной кислоты (5 экв.) и AgNO3 (5 экв.) в смеси CH3CN (250 мл) и H2O (125 мл). Реакционную смесь нагревали до 60°C. Реакционную смесь перемешивали при 60°C в течение 36 часов. Значение pH реакционной смеси доводили до 10 с применением NH3.H2O и экстрагировали с помощью EtOAc (100 мл*3). Объединенные органические слои высушивали с помощью Na2SO4, фильтровали и фильтраты концентрировали под вакуумом с получением желтого масла. Неочищенное вещество очищали посредством колоночной флеш-хроматографии на силикагеле (петролейный эфир/этилацетат от 100/0 до 95/5). Необходимую фракцию собирали и растворитель концентрировали под вакуумом с получением неочищенного промежуточного соединения 102 (2,5 г) в виде желтого масла, которое очищали посредством пре-HPLC: колонка: Xtimate C18 150 × 25 мм*5 мкм, условия: A: вода (0,225% FA), B: MeCN, вначале: A (60%) и B (40%), в конце: A (30%) и B (70%); время градиентного элюирования 7 (мин); 100% B, время удерживания 2 (мин); расход 30 (мл/мин). Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Водный слой лиофилизировали до сухого состояния с получением промежуточного соединения 102 (800 мг, выход: 15,5%) в виде белого твердого вещества.
Получение промежуточного соединения 103
Промежуточное соединение 102 (800 мг, 2,32 ммоль) и дифенилметанимин (462 мг, 2,55 ммоль) растворяли в толуоле (20 мл), Pd2(dba)3 CAS:51364-51-3 (106 мг, 0,12 ммоль). К раствору добавляли BINAP (216 мг, 0,35 ммоль) и NaOtBu (223 мг, 2,32 ммоль) и раствор продували с помощью N2. Реакционную смесь перемешивали при 120°C в течение 16 часов. Реакционную смесь охлаждали до к. т., смесь фильтровали и осадок на фильтре промывали с помощью EtOAc (20 мл). Фильтраты концентрировали под вакуумом с получением неочищенного продукта. Неочищенное вещество очищали посредством колоночной флеш-хроматографии на силикагеле (петролейный эфир/этилацетат от 100/0 до 85/15). Необходимую фракцию собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 103 (750 мг, выход: 72,3%) в виде желтого масла.
Получение промежуточного соединения 104
Промежуточное соединение 103 (750 мг, 1,68 ммоль) растворяли в DCM (6 мл) и добавляли HCl (6 мл, 1 М водный раствор). Реакционную смесь перемешивали при к. т. в течение 16 часов. Значение pH реакционной смеси доводили до 8 с применением насыщ. раствора NaHCO3 и экстрагировали с помощью DCM (30 мл * 2). Объединенные органические слои высушивали с помощью Na2SO4, фильтровали и фильтраты концентрировали под вакуумом с получением неочищенного вещества в виде желтого масла. Неочищенное вещество очищали посредством колоночной флеш-хроматографии на силикагеле (элюент: петролейный эфир/этилацетат от 100/0 до 45/55). Необходимые фракции собирали и растворитель концентрировали под вакуумом с получением промежуточного соединения 104 (350 мг, выход: 73,9%) в виде желтого твердого вещества.
Получение промежуточного соединения 105
Раствор промежуточного соединения 104 (350 мг, 1,24 ммоль), Zn(CN)2 (150 мг, 1,28 ммоль) и пыль Zn (49 мг, 0,75 ммоль) в DMF (10 мл) дегазировали в течение 5 мин. Затем добавляли Pd2(dba)3 (57 мг, 0,06 ммоль), dppf (69 мг, 0,12 ммоль) и смесь перемешивали при 120°C в течение 16 часов в атмосфере N2. Смесь фильтровали и фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной флеш-хроматографии на силикагеле (элюент: петролейный эфир/EtOAc от 100/0 до 62/38). Необходимые фракции собирали и растворитель концентрировали до сухого состояния под вакуумом с получением промежуточного соединения 105 (210 мг, выход: 65,6%) в виде желтого твердого вещества.
Получение соединения 126
К раствору промежуточного соединения 105 (210 мг, 0,82 ммоль) и CAS 2178988-91-3 (428 мг, 1,22 ммоль) в THF (10 мл) добавляли DMAP (299 мг, 2,45 ммоль) при к. т. Реакционную смесь перемешивали при 80°C в течение 16 часов. К смеси добавляли MeCN (10 мл) и фильтровали. Фильтрат концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии [колонка: Waters Xbridge Prep OBD C18 150 × 40 мм*10 мкм, условия: A: вода (10 мМ NH4HCO3)B: MeCN, вначале: A (62%) и B (38%), в конце: A (32%) и B (68%); время градиентного элюирования 15 (мин); 100% B, время удерживания 1 (мин); расход: 25 (мл/мин)]. Чистые фракции собирали и растворитель выпаривали до сухого состояния с получением соединения 126 (80 мг, выход 20%) в виде белого твердого вещества.
LC/MS: масса/заряд 483,1 [M+H]+, rt: 0,98 мин, чистота 99,2%, способ: A.
Получение соединений 127 и 128
Соединение 126 (80 мг, 0,16 ммоль) отделяли посредством SFC [колонка: DAICEL CHIRALCEL OD (250 мм*30 мм, 10 мкм), условия: подвижная фаза: A: сверхкритический CO2, B: 0,1% NH3H2O IPA. Вначале: B (40%), в конце: B (40%), расход 50 (мл/мин)]. Чистые фракции собирали и органический растворитель выпаривали под вакуумом. Добавляли 2 мл CH3CN и 20 мл H2O и смесь лиофилизировали до сухого состояния с получением соединения 127 (35 мг, чистота: 99%, выход: 43,8%) и соединения 128 (35 мг, чистота: 100%, выход: 44,1%) в виде белых твердых веществ.
Соединение 127:
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,54 (d, J=6,8 Гц, 3 H), 3,30 (s, 3 H), 4,93 (q, J=6,8 Гц, 1 H), 6,94-7,31 (m, 1 H), 8,15 (s, 2 H), 8,67 (d, J=2,4 Гц, 1 H), 8,83 (d, J=2,4 Гц, 1 H), 9,07 (br s, 1 H), 9,44 (s, 1 H), 10,71 (br s, 1 H)
LC/MS: масса/заряд 483,1 [M+H]+, rt: 5,04 мин, чистота 99,3%, способ: K;
SFC: чистота 100%, rt: 5,43 мин, способ: SFC23
Соединение 128:
LC/MS: масса/заряд 483,2 [M+H]+, rt: 5,04 мин, чистота 100%, способ: K;
SFC: чистота 99,6%, rt: 5,90 мин, способ: SFC23.
Соединения в таблице ниже получали по аналогии с одним из ранее описанных соединений. В таблице ниже для столбца "Синтез" ссылка сделана на процедуры, описанные на общих схемах.
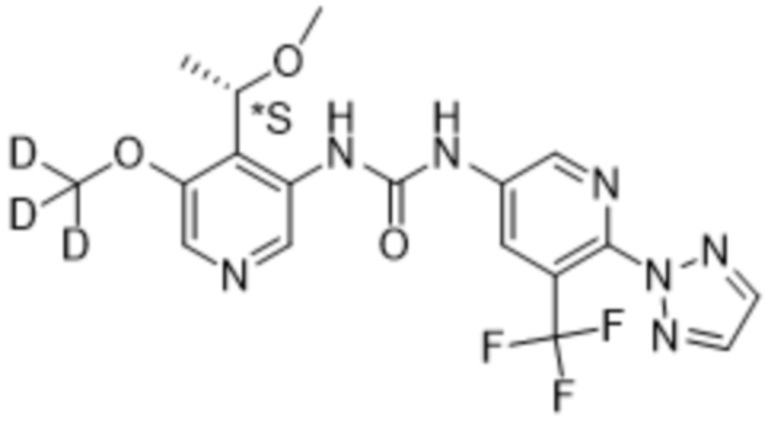
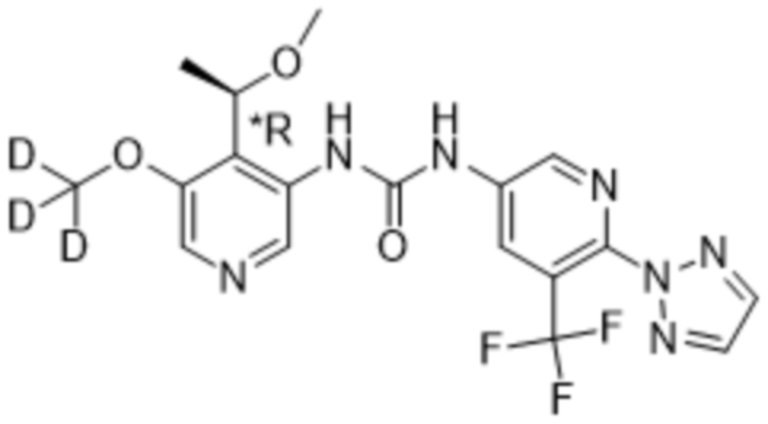
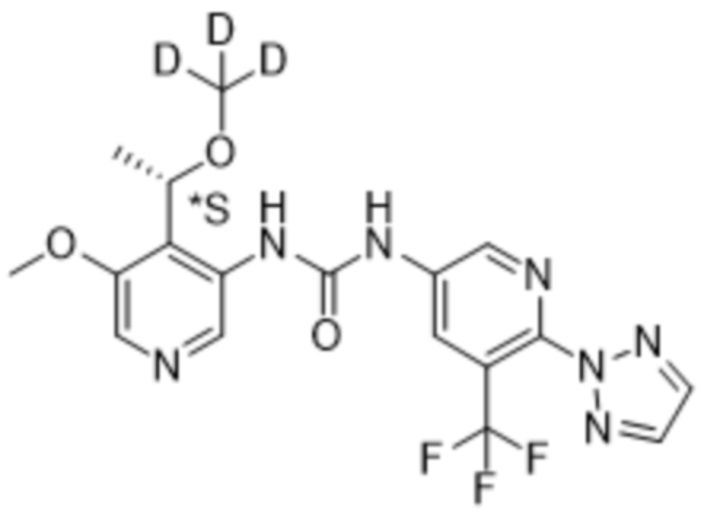
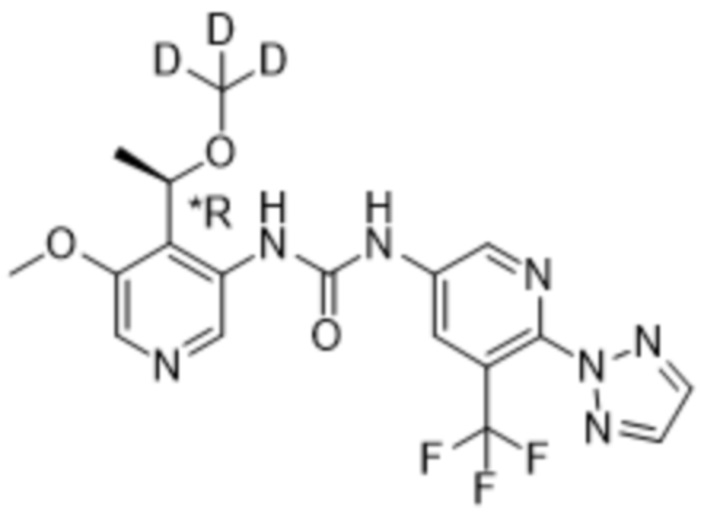
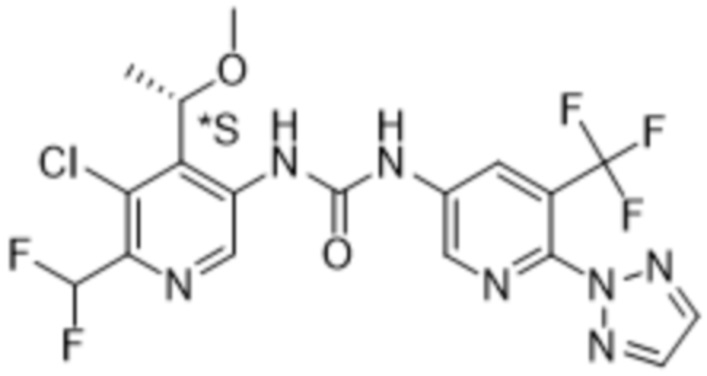
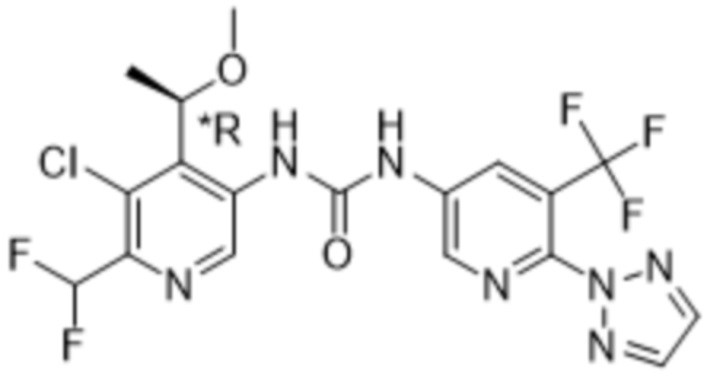
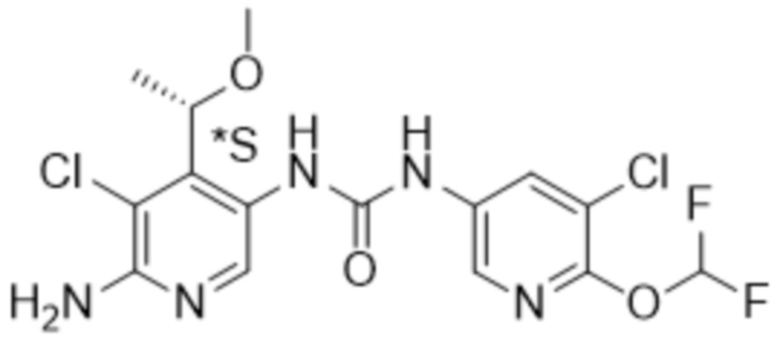
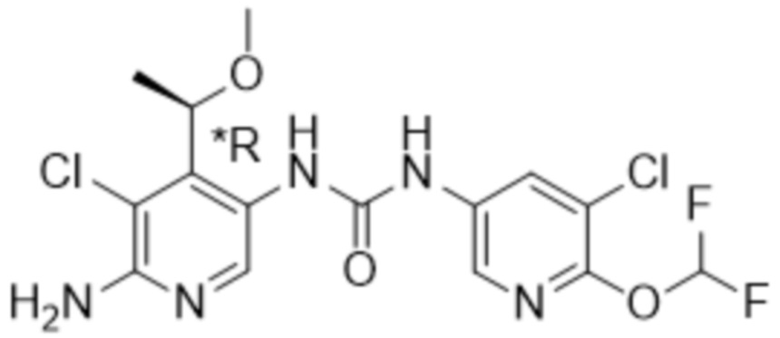
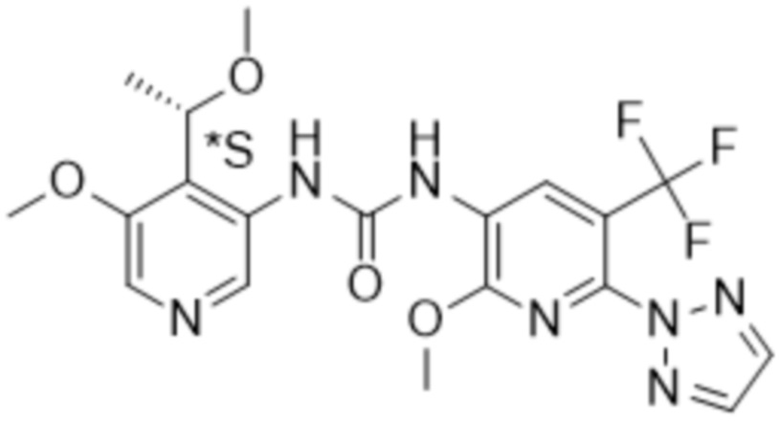
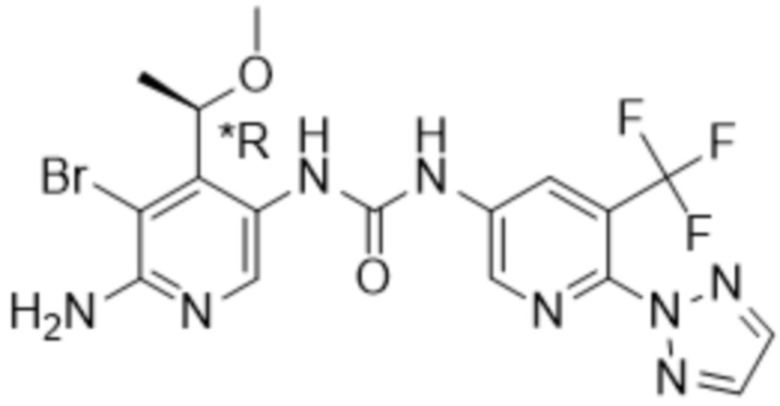
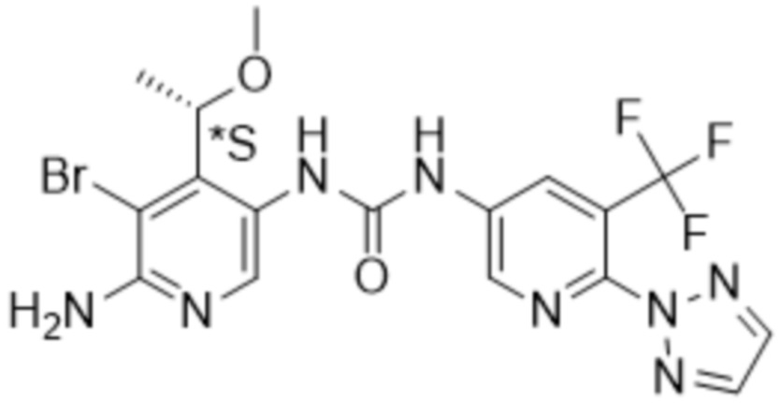
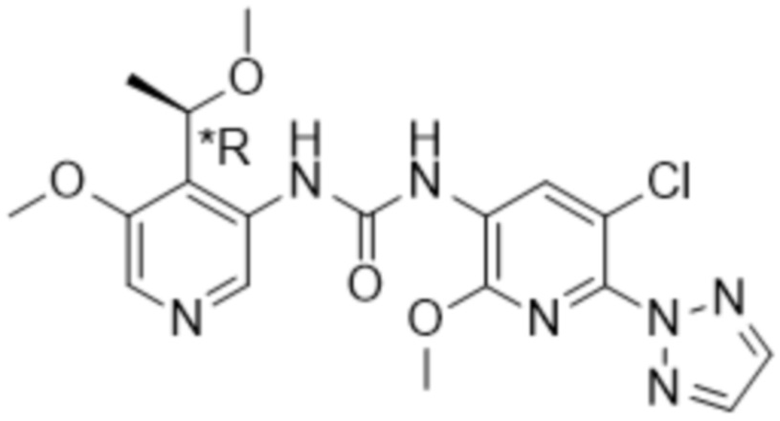
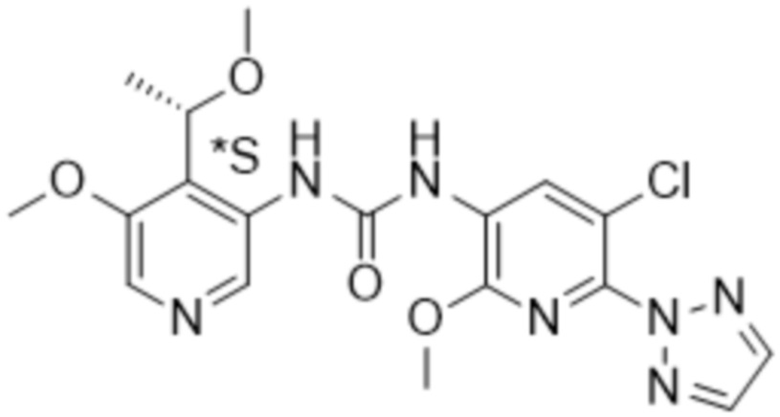
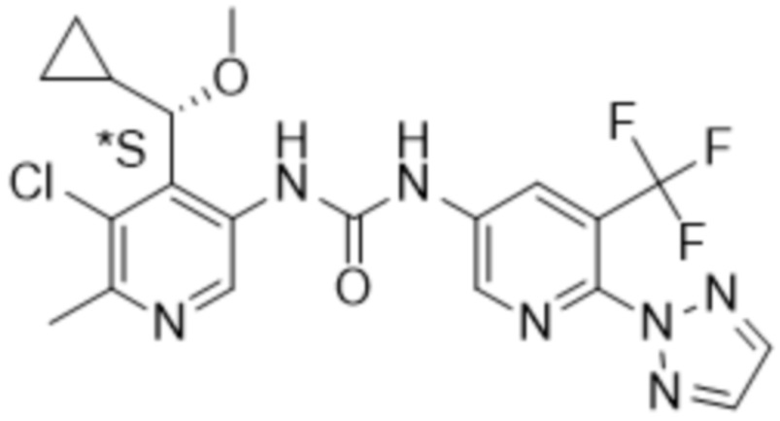
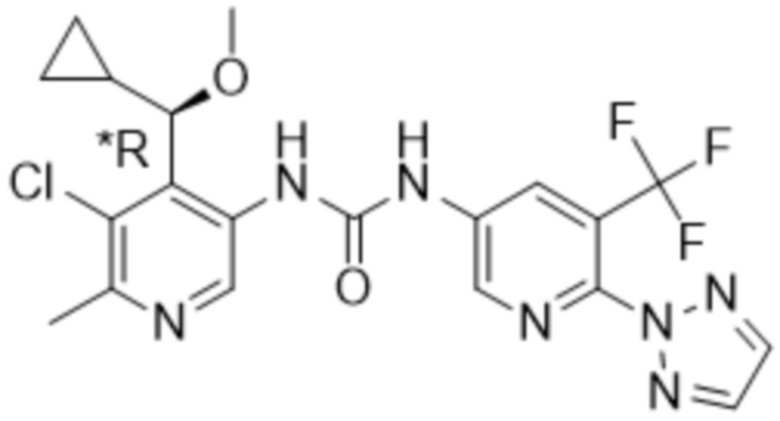
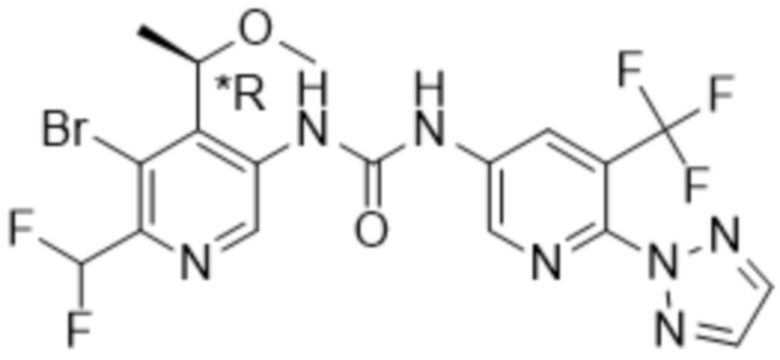
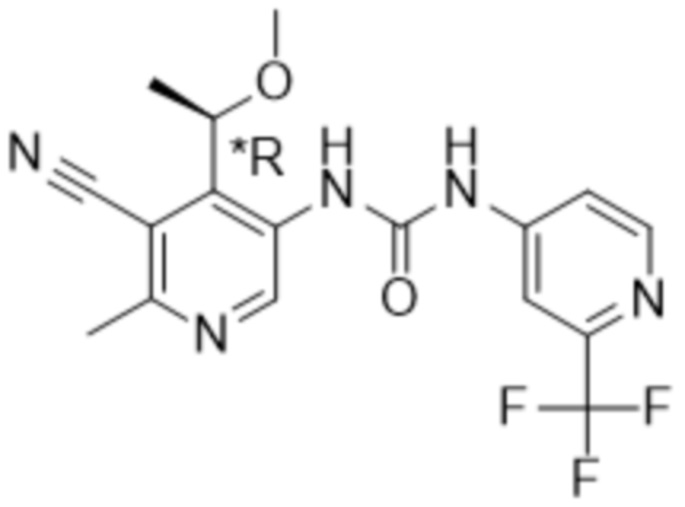
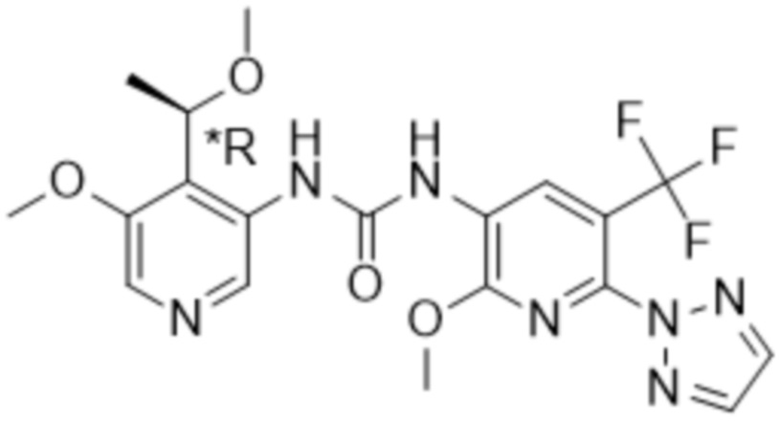
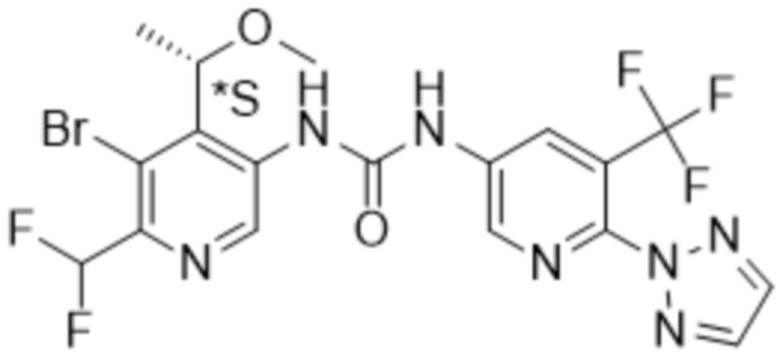
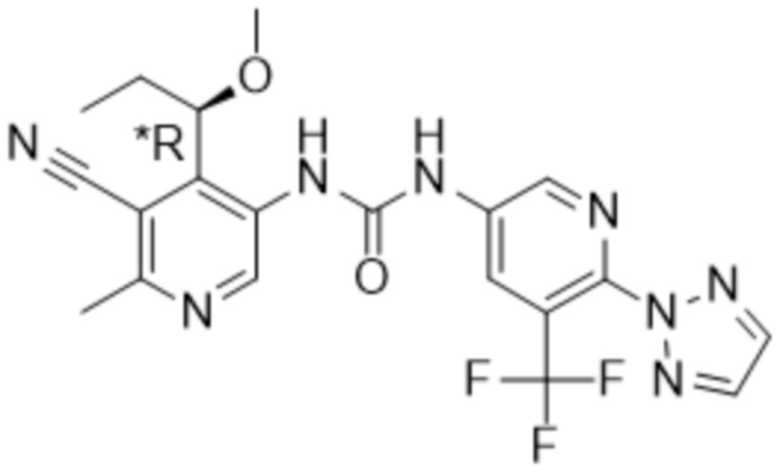
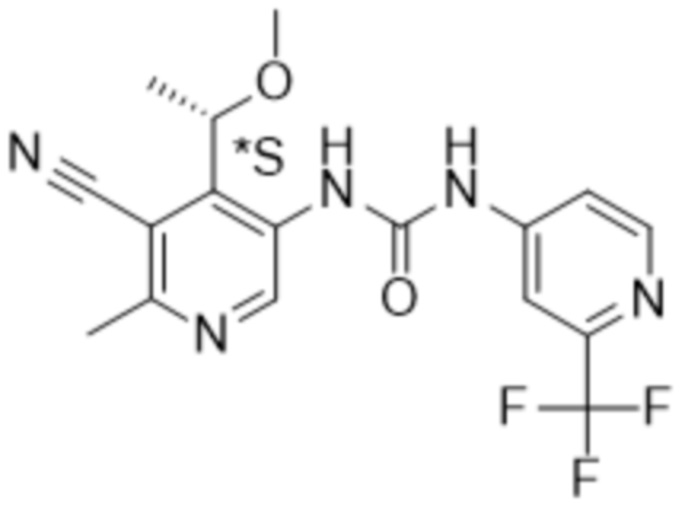
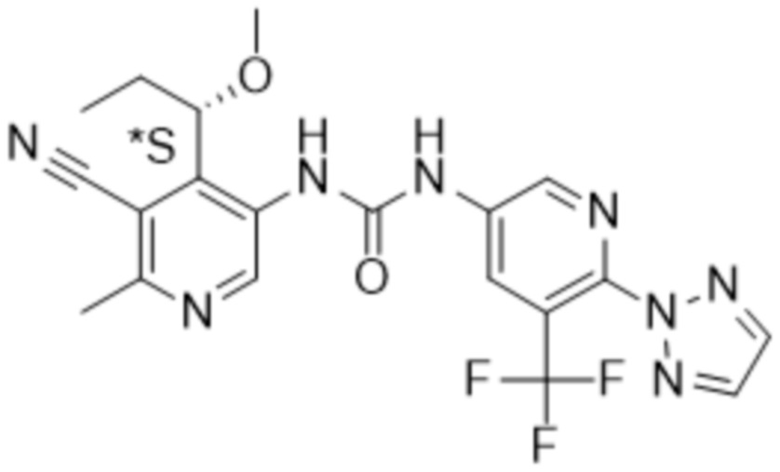
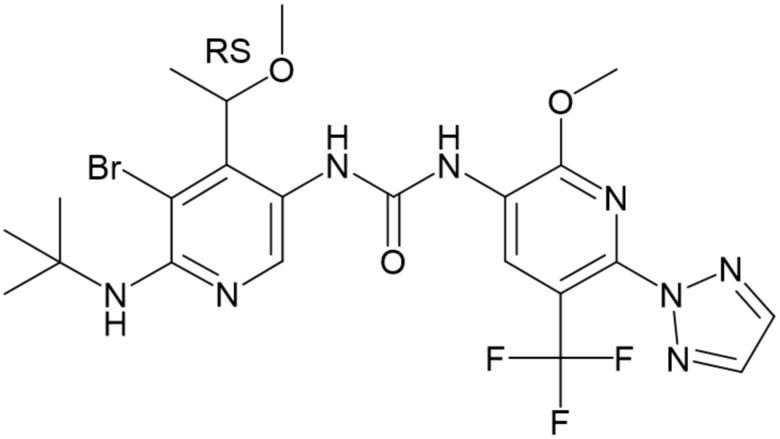
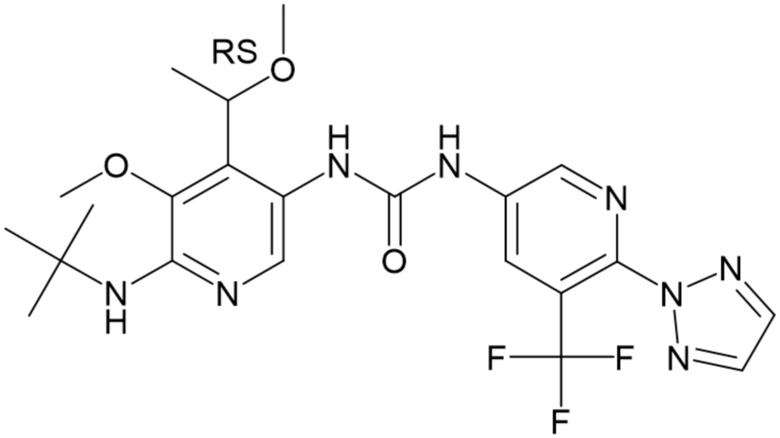
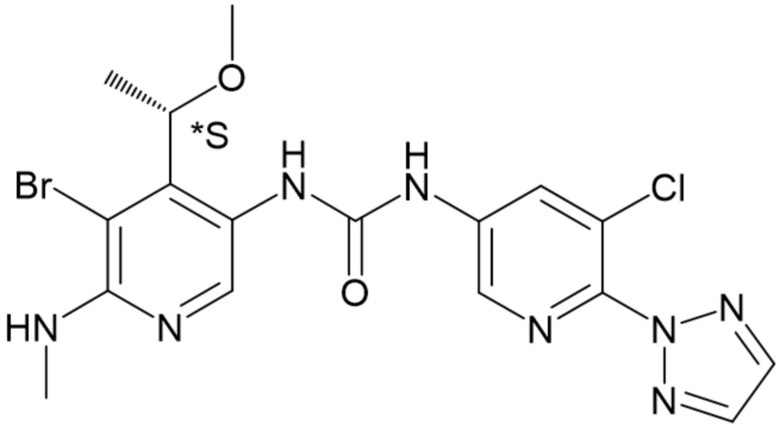
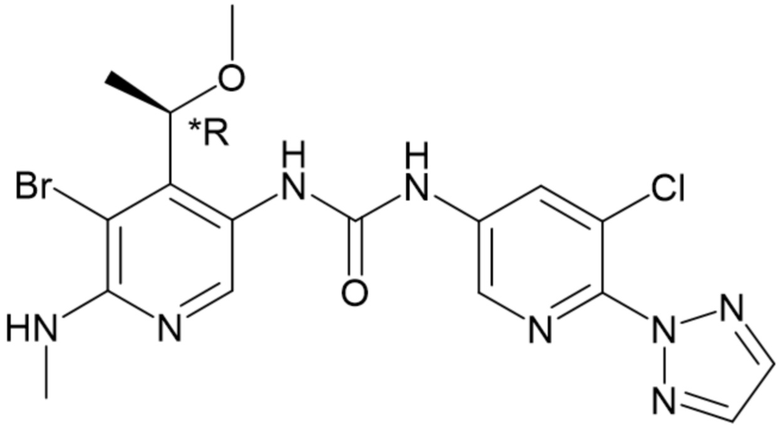
Соединение 130: 1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,39 (d, J=6,8 Гц, 3 H), 3,24 (s, 3 H), 4,98 (d, J=6,4 Гц, 1 H), 8,08 (s, 1 H), 8,13 (s, 2 H), 8,66 (d, J=2,4 Гц, 1 H), 8,76 (s, 1 H), 8,81 (d, J=2,4 Гц, 1 H), 8,85 (s, 1 H), 10,61 (br s, 1 H);
HPLC/MS: масса/заряд 441,1, [M+H]+, rt: 3,81 мин. Чистота: 100%, способ: K.
SFC: чистота: 99,48%, rt: 1,20 мин, способ: SFC30.
Соединение 129:
HPLC/MS: масса/заряд 441,1, [M+H]+, rt: 3,81 мин. Чистота: 100%, способ: K;
SFC: чистота: 100%, rt: 1,40 мин, способ: SFC30.
Соединение 132: 1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,39 (d, J=6,8 Гц, 3 H), 3,28 (s, 3 H), 3,88 (s, 3 H), 4,97 (q, J=6,8 Гц, 1 H), 8,08 (s, 1 H), 8,13 (s, 2 H), 8,66 (d, J=2,4 Гц, 1 H), 8,77 (s, 1 H), 8,81 (d, J=2,4 Гц, 1 H), 8,85 (s, 1 H), 10,61 (s, 1 H);
HPLC/MS: масса/заряд 441,1, [M+H]+, rt: 4,04 мин, чистота: 100%, способ: M;
SFC: чистота: 99,72%, rt: 1,21 мин, способ: SFC30.
Соединение 131:
HPLC/MS: масса/заряд 441,1 [M+H]+, rt: 4,15 мин, чистота: 100%, способ: M;
SFC: чистота: 99,69%, rt: 1,40 мин, способ: SFC30.
Соединение 134:
LC/MS: масса/заряд 492,1, 494,1 [M+H]+, rt 5,1 мин. Чистота 100%, способ: K;
SFC: чистота 99,9%, rt 5,3 мин. Способ: SFC 13.
Соединение 133:
LC/MS: масса/заряд 492,1, 494,1 [M+H]+, rt 5,1 мин. Чистота 100%, способ: K;
SFC: чистота 99,3%, rt 5,6 мин, способ: SFC 13.
Соединение 136:
HPLC/MS: масса/заряд 422,1, 423,1, 424,1 [M+H]+, rt 4,2 мин. Чистота 98,6%, способ: K;
SFC: чистота 100%, rt 5,9 мин. Способ: SFC1.
Соединение 135:
HPLC/MS: масса/заряд 422,1, 423,1, 424,1 [M+H]+, rt 4,2 мин. Чистота 98,6%, способ: K;
SFC: чистота 100%, rt 7,3 мин. Способ: SFC1.
Соединение 146:
LC/MS: масса/заряд 468,2 [M+H]+, rt 4,0 мин. Чистота 99,6%, способ: K;
SFC: чистота 100%, rt 4,5 мин. Способ: SFC25.
Соединение 137:
LC/MS: масса/заряд 468,2 [M+H]+, rt 4,0 мин. Чистота 99,8%, способ: K;
SFC: чистота 98%, rt 4,7 мин. Способ: SFC25.
Соединение 139: 1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,41 (d, J=6,8 Гц, 3 H), 3,25 (s, 3 H), 4,93 (q, J=6,8 Гц, 1 H), 6,21 (s, 2 H), 8,16 (s, 2 H), 8,25 (s, 1 H), 8,29 (s, 1 H), 8,70 (d, J=2,4 Гц, 1 H), 8,79 (d, J=2,4 Гц, 1 H), 10,34 (br s, 1 H);
HPLC/MS: масса/заряд 501,1, 503,1 [M+H]+, rt: 3,875 мин, чистота 99%, способ: K;
SFC: чистота 100%, rt: 0,666 мин, способ: SFC 41.
Соединение 138:
HPLC/MS: масса/заряд 501,1, 503,1 [M+H]+, rt: 3,877 мин, чистота 98,7%, способ: K;
SFC: чистота 99,89%, rt: 1,128 мин, способ: SFC 41.
Соединение 140:
HPLC/MS: масса/заряд 434,2, 436,2 [M+H]+, rt 3,8 мин. Чистота 99,9%, способ: K.
SFC: чистота 100%, rt 3,3 мин. Способ: SFC 25
Соединение 141:
HPLC/MS: масса/заряд 434,2, 436,2 [M+H]+, rt 3,8 мин. Чистота 100%, способ: K.
SFC: чистота 100%, rt 5,0 мин, способ: SFC 25
Соединение 143:
HPLC/MS: масса/заряд 482,2, 484,1 [M+H]+, rt: 3,61 мин. Чистота: 100%, способ: L;
SFC: чистота 99,9%, rt: 3,51 мин, способ: SFC1.
Соединение 142:
HPLC/MS: масса/заряд 482,1, 484,1 [M+H]+, rt: 3,60 мин. Чистота: 100%, способ: L;
SFC: чистота 100%, rt: 4,40 мин, способ: SFC1.
Соединение 144:
HPLC/MS: масса/заряд 536,0, 538,0 [M+H]+, rt: 5,29 мин. Чистота: 99%, способ: K;
SFC: чистота 100%, rt: 1,24 мин, способ: SFC33.
Соединение 147:
HPLC/MS: масса/заряд 535,9, 537,9 [M+H]+, rt: 5,28 мин. Чистота: 98%, способ: K;
SFC: чистота 99,8%, rt: 1,34 мин, способ: SFC33.
Соединение 145:
HPLC/MS: масса/заряд 380,1 [M+H]+, rt: 4,43 мин. Чистота 100%, способ: K.
SFC: чистота 100%, rt: 3,73 мин, способ: SFC39.
Соединение 149:
HPLC/MS: масса/заряд 380,2 [M+H]+, rt: 4,43 мин. Чистота: 100%, способ: K;
SFC: чистота 99%, rt: 3,55 мин, способ: способ: SFC39.
Соединение 148: 1H ЯМР (400 МГц, DMSO-d6) δ ppm 0,89 (t, J=7,2 Гц, 3 H), 1,69-1,81 (m, 1 H), 1,94 (dt, J=14, 7,2 Гц, 1 H), 2,64 (s, 3 H), 3,26 (s, 3 H), 4,56 (t, J=6,8 Гц, 1 H), 8,13 (s, 2 H), 8,65 (s, 2 H), 8,80 (s, 1 H), 9,09 (s, 1 H), 10,46 (br s, 1 H);
HPLC/MS: масса/заряд 461,2, [M+H]+, rt: 4,80 мин, чистота: 100%, способ: K;
SFC: чистота 100%, rt: 1,45, мин, способ: SFC35.
Соединение 150:
HPLC/MS: масса/заряд 461,2, [M+H]+, rt: 4,79 мин, чистота: 100%, способ: K;
SFC: чистота 100%, rt: 1,58 мин, способ: SFC35.
Соединение 151:
HPLC/MS: масса/заряд 587,1, 589,1 [M+H]+, rt: 4,649 мин, чистота 96,77%, способ: L
Соединение 152:
HPLC/MS: масса/заряд 509,3, [M+H]+, rt: 3,06 мин. Чистота: 100%, способ: L
Соединение 153:
LC/MS: масса/заряд 481,1, 482,0, 483,1, [M+H]+, rt 3,7 мин. Чистота 99,1%, способ: K;
SFC: чистота 100%, rt 4,0 мин, способ: SFC27
Соединение 154:
LC/MS: масса/заряд 481,1, 482,0, 483,1, [M+H]+, rt 3,7 мин. Чистота 98%, способ: K;
SFC: чистота 100%, rt 2,1 мин. Способ: SFC27
Описание ЯМР
Для некоторых соединений ЯМР-эксперименты проводили с применением спектрометра Bruker Avance III 400 при температуре окружающей среды (295 K) с применением внутренней дейтериевой стабилизации, оснащенного головкой датчика PABBO BB-1H/D на 5 мм с z-градиентами и работающего при 400 МГц для протонов и 100 для атомов углерода, или с применением спектрометра Varian VNMRS 400M при температуре окружающей среды (295 K) с применением внутренней дейтериевой стабилизации, оснащенного датчиком PFG 4Nuc на 5 мм и работающего при 400 МГц для протонов и 100 МГц для атомов углерода. Химические сдвиги (δ) приведены в частях на миллион (ppm). Значения J выражены в Гц.
В качестве альтернативы, некоторые ЯМР-эксперименты проводили с применением спектрометра Varian MR 400 МГц при температуре окружающей среды (295 K) с применением внутренней дейтериевой стабилизации, оснащенного датчиком PFG 4Nuc на 5 мм и работающего при 400 МГц для протонов и 100 МГц для атомов углерода. Химические сдвиги (δ) приведены в частях на миллион (ppm). Значения J выражены в Гц.
LCMS (жидкостная хроматография/масс-спектрометрия)
Общая процедура
Измерение с помощью высокоэффективной жидкостной хроматографии (HPLC) осуществляли с применением насоса для LC, детектора на диодной матрице (DAD) или УФ-детектора, и колонки, указанной в соответствующих способах. При необходимости включали дополнительные детекторы (см. приведенную ниже таблицу способов).
Поток из колонки направляли в масс-спектрометр (MS), который был оснащен источником ионизации при атмосферном давлении. В компетенции специалиста в данной области техники находится выставление настраиваемых параметров (например, диапазона сканирования, минимального времени измерения и т.п.) с целью получения ионов, позволяющих определить номинальную моноизотопную молекулярную массу (MW) соединения. Сбор данных проводили с помощью соответствующего программного обеспечения.
Соединения описаны по их значениям экспериментального времени удерживания (Rt) и ионам. Если не указано иное, то в таблице данных указанный молекулярный ион представляет собой [M+H]+ (протонированную молекулу) и/или [M−H]− (депротонированную молекулу). В случае, если соединение не было непосредственно способно к ионизации, указывали тип аддукта (т. е. [M+NH4]+, [M+HCOO]− и т. д.). Для молекул со сложными изотопными распределениями (Br, Cl и т.д.) описанное значение представляет собой значение, которое получали для наименьшей массы изотопа. Все результаты получали с экспериментальными погрешностями, которые обычно связаны с применяемым способом.
Далее в данном документе "SQD" означает одиночный квадрупольный детектор, "к.т." означает комнатную температуру, "BEH" означает мостиковый гибрид этилсилоксана/диоксида кремния, "HSS" означает диоксид кремния повышенной прочности, "DAD" означает детектор на диодной матрице.
Таблица 1a. Коды способов LCMS (расход выражен в мл/мин; температура колонки (T) в °C; время анализа в минутах)
(мл/мин)
-------
Т кол.
(°С)
(мин)
LC20-MS2010
УФ 220, 254 нм
(25 × 2 мм)
B: 0,02% TFA в CH3CN
5% A за 0,7 мин, удерживание 0,4 мин,
обратно до 95% A за 0,01 мин, удерживание 0,39 мин
-------
50
LC1200-MS6110
УФ 220, 254 нм
(25 × 2 мм)
B: 0,02% TFA в CH3CN
5% A за 0,7 мин, удерживание 0,4 мин,
обратно до 95% A за 0,01 мин, удерживание 0,39 мин
-------
50
УФ 220, 254 нм
(3,5 мкм, 2,1 × 30 мм)
B: ацетонитрил
20% A за 2,0 мин, удерживание 0,48 мин,
обратно до 90% A за 0,01 мин, удерживание 0,51 мин
-------
50
LC1100-MS1946D
УФ 220, 254 нм
(5 мкм, 2,1 × 50 мм)
B: ацетонитрил
20% A за 2,0 мин, удерживание 0,48 мин,
обратно до 90% A за 0,01 мин, удерживание 0,51 мин
-------
30
LC1200-MS6110
УФ 220, 254 нм
(3 мкм, 2,1 × 30 мм)
B: 0,02% TFA в CH3CN
40% A за 0,9 мин, удерживание 0,6 мин.,
обратно до 100% A за 0,01 мин, удерживание 0,49 мин
------
50
LC1200-MS6110,
УФ 220, 254
(3 мкм, 2,1 × 30 мм)
B: 0,02% TFA в CH3CN
20% A за 0,9 мин, удерживание 0,6 мин.,
обратно до 90% A за 0,01 мин, удерживание 0,49 мин
-------
50
LC1200-MSD6110
УФ 220 нм
(5 мкм, 2 × 50 мм)
B: ацетонитрил с 0,02% TFA
40% A за 4 мин., до 15% A за 2,5 мин, обратно
до 100% A за 2 мин, удерживание 0,5 мин.
-------
50
LC1200-MSD6110
УФ 220 нм
(5 мкм, 2 × 50 мм)
B: ацетонитрил с 0,02% TFA
20% A за 3,7 мин, удерживание 3 мин, обратно до 90% A за 2 мин, удерживание 0,5 мин
-------
50
LC1200-MSD6110
УФ 220 нм
B: ацетонитрил
40% A за 4 мин, до 5% A за 2,5 мин, обратно до 100% A за 2 мин, удерживание 0,5 мин
-------
40
Аналитическая SFC
Общая процедура для способов SFC
Измерения в ходе SFC проводили с применением аналитической системы для сверхкритической жидкостной хроматографии (SFC), укомплектованной насосом для двухкомпонентных смесей для доставки диоксида углерода (CO2) и модификатора, автоматическим дозатором, термостатом для колонок, детектором на диодной матрице, оснащенным проточной кюветой для работы под высоким давлением, выдерживающей до 400 бар. Сбор данных проводили с помощью соответствующего программного обеспечения.
Таблица 2a. Коды способов аналитической SFC (расход выражен в мл/мин; температура колонки (T) в °C; время анализа в минутах, противодавление (BPR) в барах. "ACN" означает ацетонитрил; "MeOH" означает метанол; "EtOH" означает этанол; "DEA" означает диэтиламин; "HEP" означает н-гептан. Все другие сокращения, используемые в таблице ниже, определены ранее.
-------
Т кол.
--------
BPR
B: EtOH+0,05% DEA
-------
40
-------
100
B: MeOH+0,05% DEA
-------
40
-------
100
B: EtOH+0,05% DEA
-------
40
-------
100
B: EtOH+0,05% DEA
-------
35
-------
100
B: EtOH+0,05% DEA
-------
40
-------
100
B: IPA+0,1% этаноламина
-------
40
-------
100
B: EtOH+0,05% DEA
-------
40
-------
100
B: EtOH+0,05% DEA
-------
35
-------
100
B: EtOH+0,05% DEA
-------
40
-------
100
B: EtOH+0,05% DEA
-------
40
-------
100
B: MeOH+0,05% DEA
-------
35
-------
100
B: EtOH+0,05% DEA
-------
40
-------
100
B: EtOH+0,05% DEA
-------
35
-------
100
B: MeOH+0,05% DEA
-------
35
-------
100
B: EtOH+0,05% DEA
-------
35
-------
100
B: MeOH+0,05% DEA
-------
40
-------
100
B: MeOH+0,05% DEA
-------
40
-------
100
B: EtOH+0,05% DEA
-------
35
-------
100
B: EtOH+0,05% DEA
-------
35
-------
100
B: EtOH+0,05% DEA
-------
40
-------
100
B: IPA+0,05% DEA
-------
40
-------
100
B: EtOH+0,05% DEA
-------
35
-------
100
B: IPA+0,05% DEA
-------
35
-------
100
B: EtOH+0,05% DEA
-------
35
-------
100
B: EtOH+0,05% DEA
-------
35
-------
100
B: EtOH+0,05% DEA
-------
35
-------
100
B: MeOH+0,05% DEA
-------
35
-------
100
B: EtOH+0,05% DEA
-------
35
-------
100
B: EtOH+ 0,05% DEA
-------
40
-------
100
B: EtOH+0,05% DEA
-------
35
-------
100
Биологические примеры
Анализы in vitro предусматривают анализы, в ходе которых определяют морфологию клеток, экспрессию белка и/или цитотоксичность, ингибирующую активность в отношении ферментов и/или функциональные последствия, являющиеся результатом обработки клеток соединениями по настоящему изобретению. Альтернативные или дополнительные анализы in vitro могут применяться для количественной оценки способности ингибитора связываться с молекулами белков или нуклеиновых кислот внутри клетки.
Уровень связывания ингибитора может быть измерен путем введения радиоизотопной метки в ингибитор перед связыванием, выделения комплекса ингибитор/целевая молекула и определения количества радиоизотопной метки по результатам связывания. В качестве альтернативы или дополнения, уровень связывания ингибитора может быть определен путем проведения экспериментального исследования в отношении конкурентного связывания, в котором новые ингибиторы инкубируют с очищенными белками или нуклеиновыми кислотами, связанными с известными радиоактивными лигандами. Подробные условия иллюстративных систем для проведения анализа соединения формулы (I) по настоящему изобретению в качестве ингибитора MALT1 изложены в биологических примерах далее.
Такие анализы являются иллюстративными и не предназначены для ограничения объема настоящего изобретения. Квалифицированный специалист-практик сможет понять, что в отношении традиционных видов анализа могут быть произведены модификации для разработки эквивалентных или иных видов анализа, которые могут быть использованы для того, чтобы аналогичным образом оценить активность или как-либо иначе охарактеризовать соединения и/или композиции, описанные в данном документе.
Представленные в таблицах ниже значения IC50 приведены в соответствии с допустимыми погрешностями, которые обусловлены применяемыми анализом и оборудованием.
Анализы in vitro
Биологический пример 1
Биохимический анализ в отношении протеазной активности MALT1
Протеазную активность MALT1 оценивали в ходе анализа in vitro с применением в нем тетрапептида в качестве субстрата и полноразмерного белка MALT1 (Strep-MALT1(1-824)-His), выделенного из инфицированных бакуловирусом клеток насекомых. Тетрапептид LRSR связан с AMC (7-амино-4-метилкумарином) и представляет собой субстрат для протеазы MALT1 (SM Biochemicals) для проведения флуоресцентного анализа и характеризуется угасающей флуоресценцией. Отщепление AMC от остатка аргинина приводит к усилению флуоресценции кумарина, измеряемой на 460 нм (при возбуждении на 355 нм). Итоговый буфер для анализа состоял из 10 нМ полноразмерного (FL) белка MALT1, 200 мкМ Ac-LRSR-AMC, 50 мМ Трис с pH 7,5, 0,6 М цитрата, 1 мМ DTT, 1 мМ EDTA, 0,05% BSA и 1,5% DMSO. Пробы исследуемых соединений наносили в объеме 50 нл в 100% DMSO в расчете на одну лунку в черном 384-луночном планшете Proxiplate (Perkin Elmer). Концентрации исследуемых соединений варьировались от 30 мкМ до 0,5 нМ, при использовании разбавления в 11 стадий (1:3). Фоновый сигнал измеряли в контрольных лунках, содержащих буфер для анализа без фермента, которые использовались в качестве контроля нижнего уровня (LC). Значения контроля верхнего уровня (HC) были получены с применением реакционной системы с ферментом, но без обработки соединениями. Соединения предварительно инкубировали с ферментом MALT1 в течение 50 минут при к.т. Затем добавляли субстрат и измеряли флуоресценцию на Labsystems Fluoroskan при возбуждении при 355 нм и излучении при 460 нм для определения 0 момента времени. После этого реакционную систему инкубировали в течение 4 ч. при к. т. и осуществляли измерение флуоресценции. Для вычисления IC50 значения для 0 момента времени вычитались из таковых для момента времени через 4 ч. для внесения поправки на любую возможную автофлуоресценцию соединений. На протяжении 4 ч. периода инкубации ферментативная реакция имела линейный характер. Определение характеристик субстрата Ac-LRSR-AMC обеспечило возможность установить значение константы Михаэлиса KM в 200 мкМ.
Значения IC50 рассчитывали с применением следующей формулы (Z-фактор должен быть >0,5):
LC =медиана значений контроля нижнего уровня
= контроль нижнего уровня: реакция без фермента
HC=медиана значений контроля верхнего уровня
= контроль верхнего уровня: реакция с ферментом
% эффекта=100 − [(значение для образца − LC) / (HC − LC) × 100]
% контроля=(значение для образца/HC) × 100
% мин контроля=(значение для образца − LC) / (HC − LC) × 100
Кривую наилучшего приближения подбирали с помощью метода наименьших квадратов для графика зависимости % мин контроля от концентрации соединения. На его основании можно получать значение IC50 (ингибирующей концентрации, которая вызывает 50% ингибирования). Также была получена оценка угла наклона для графика в отношении коэффициента Хилла.
Расчет IC50:
y=LB+UB − LB _
1+10(h(pConc−pIC50))
где y =расчетный ответ
UB=верхняя граница значения
LB=нижняя граница значения
h=коэффициент Хилла
Применяли "Lexis Dose Response Curve Fitting", версия 1.0. Полученные в результате данные приведены в таблице A.
(Ac-LRSR-amc)
IC50 (мкМ)
Биологический пример 2
Анализ в отношении IL6/IL10 человека от Meso Scale
Передача сигнала NFΚB контролирует секрецию многих цитокинов, в том числе IL6 и IL10. Секрецию цитокинов IL6 и IL10 клетками OCI-LY3 ABC-DLBCL измеряли с применением анализа от Meso Scale. Ингибирование передачи сигнала NFΚB с помощью ингибиторов MALT1 приводит к снижению секреции IL6/10.
Клетки OCI-LY3 размножали в RPMI-1640 (Sigma Aldrich), дополненной 10% фетальной бычьей сыворотки (HyClone), 1 мМ пирувата натрия (Invitrogen), 2 мМ L-глутамина (Sigma Aldrich) и 1% PenStrep (Sigma Aldrich). Число пассажей клеток не должно превышать 30. Во время культивирования количество клеток должно поддерживаться на уровне 0,5-2,5 миллиона клеток на мл, и для клеток каждые 2-3 дня следует проводить дополнительное внесение свежего бета-меркаптоэтанола в количестве 50 мкМ. При проведении анализа от Meso Scale бета-меркаптоэтанол не применяли.
Для проведения анализа от Meso Scale в 96-луночные планшеты черного цвета с прозрачным дном (Corning № 3904) высеивали по 100000 клеток OCI-LY3 на лунку и добавляли исследуемые соединения в 9 разбавлениях (1:2) в диапазоне от 15 мкМ до 58,6 нМ (конечная концентрация DMSO 0,3%). Контрольные лунки с DMSO применяли для определения максимального сигнала (контроля верхнего уровня (HC)). Обработка с помощью ингибитора BTK RN486 в диапазоне доз от 30 нМ до 131 пМ (9 разбавлений в соотношении 1:2) выполняла функцию положительного контроля для ингибирования пути NFΚB и использовалась для определения максимального ингибирования (контроль нижнего уровня (LC)). Соединения и клетки инкубировали в течение 24 ч. при 37°C и 5% CO2 (объем для анализа составлял 150 мкл). После 24 ч. инкубирования переносили 50 мкл надосадочной жидкости на планшет MSD (набор для проведения анализов у человека V-Plex Proinflammation Panel 1 от Meso Scale (MSD)) и инкубировали в течение 2 ч. при энергичном встряхивании (600 об/мин) при комнатной температуре. После окончания инкубирования планшеты 3 раза промывали с помощью PBS+0,05% Tween-20 и добавляли по 25 мкл раствора детекторных антител (антитела к IL6 и IL10 в разбавителе 3 (MSD)) на лунку с последующим инкубированием в течение 2 ч. при энергичном встряхивании (600 об/мин) при комнатной температуре. После 3-кратного промывания с помощью PBS+0,05% Tween-20 планшеты инкубировали в 150 мкл буфера для считывания Read Buffer T 2x и считывали на устройстве для визуализации SECTOR. Полученные в результате данные приведены в таблице B.
Таблица B
(OCI-LY3)
IC50 (мкМ)
(OCI-LY3)
IC50 (мкМ)
Несмотря на то, что в вышеуказанном описании изложены основные идеи настоящего изобретения с примерами, приведенными в иллюстративных целях, понятно, что практическое осуществление настоящего изобретения охватывает все обычные варианты, адаптации и/или модификации, которые подпадают под объем приведенной далее формулы изобретения, и их эквиваленты.
| название | год | авторы | номер документа |
|---|---|---|---|
| Имидазопирролопиразиновые производные, полезные для лечения заболеваний, вызванных аномальной активностью протеинкиназ Jak1, Jak3 или Syk | 2010 |
|
RU2711869C2 |
| ИМИДАЗОПИРРОЛОПИРАЗИНОВЫЕ ПРОИЗВОДНЫЕ, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ АНОМАЛЬНОЙ АКТИВНОСТЬЮ ПРОТЕИНКИНАЗ Jak1, Jak3 ИЛИ Syk | 2010 |
|
RU2570416C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИДИНОНА | 2018 |
|
RU2806751C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИДИНОНА | 2018 |
|
RU2806625C2 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2799820C2 |
| ПРОИЗВОДНЫЕ 1,2,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ГИСТОНДЕЗАЦЕТИЛАЗЫ 6 | 2018 |
|
RU2797613C2 |
| ОКСИСТЕРОЛЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2017 |
|
RU2754130C2 |
| ИНГИБИТОРЫ ФАКТОРА XIa | 2016 |
|
RU2728783C2 |
| НОВЫЕ СПИРОБИЦИКЛИЧЕСКИЕ АНАЛОГИ | 2018 |
|
RU2789377C2 |
| аЭКЗО-АЗАСПИРО-ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2018 |
|
RU2795096C2 |
Изобретение относится к соединению формулы (I) или его энантиомеру, диастереомеру или фармацевтически приемлемой солевой форме. В формуле (I): Rx представляет собой C1-4алкил или C3-6циклоалкил; Ry представляет собой C1-4алкил; и Rz представляет собой водород; R1 выбран из группы, состоящей из водорода, -OR5, C1-4алкила, C2-4алкенила, галогена, -CN, C3-6циклоалкила, Heta, -C(=O)-OH, -C(=O)-O-C1-4алкила, -NR6aR7a и -C(=O)-NR6bR7b; каждый из R2a и R2b независимо выбран из группы, состоящей из водорода, -NR6cR7c, C3-6циклоалкила, C1-4алкила и C1-4алкила, замещенного 1, 2 или 3 атомами галогена; X1 представляет собой N или CRa; X2 представляет собой N или CRb; при условии, что только один из X1 и X2 представляет собой N в любом случае; R3 представляет собой водород, C1-4алкил или -O-C1-4алкил; R4 представляет собой галоген, циано или трифторметил; R5 выбран из группы, состоящей из водорода, C1-4алкила, C3-6циклоалкила, Hetb и C1-4алкила, замещенного одним или двумя заместителями, каждый из которых независимо выбран из группы, состоящей из -C(=O)-NR8R9, -C(=O)-OH, -C(=O)-O-C1-4алкила, C3-6циклоалкила и фенила; каждый из R6a, R6b, R6c, R7a, R7b, R7c, R8 и R9 независимо выбран из группы, состоящей из водорода и C1-4алкила; Heta представляет собой моноциклический 4-7-членный неароматический гетероциклил, содержащий один или два гетероатома, выбранные из азота, кислорода и серы; Hetb представляет собой моноциклический 4-7-членный неароматический гетероциклил, содержащий один или два гетероатома, выбранные из азота, кислорода и серы; Ra представляет собой -O-C1-4алкил, каждый из которых необязательно замещен одним, двумя или тремя заместителями, представляющими собой атом галогена; или Ra представляет собой 2H-1,2,3-триазол-2-ил или C3-6циклоалкил; при этом каждый из них необязательно замещен по одному или двум атомам углерода посредством заместителя, который в каждом случае независимо выбран из группы, состоящей из C1-4алкила и C1-4алкил, замещенного одним -OH; Rb представляет собой водород. Также предложены фармацевтическая композиция, содержащая соединение формулы (I), и способ лечения с использованием соединения формулы (I). Предложенные соединения являются ингибиторами MALT1 (транслокационного белка 1 лимфомы лимфоидной ткани слизистых оболочек) и могут быть полезными для лечения заболевания, синдрома, состояния или нарушения, связанного с MALT1 заболевания, синдрома, состояния или нарушения, в том числе без ограничения рака и иммунологических заболеваний. 3 н. и 6 з.п. ф-лы, 5 табл., 2 пр.

1. Соединение формулы (I):
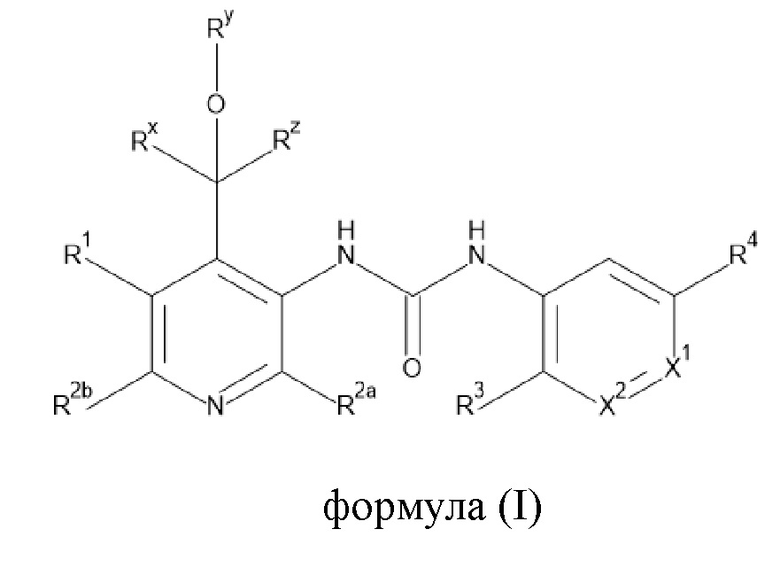
где
Rx представляет собой C1-4алкил или C3-6циклоалкил;
Ry представляет собой C1-4алкил; и
Rz представляет собой водород;
R1 выбран из группы, состоящей из водорода, -OR5, C1-4алкила, C2-4алкенила, галогена,
-CN, C3-6циклоалкила, Heta, -C(=O)-OH, -C(=O)-O-C1-4алкила, -NR6aR7a и
-C(=O)-NR6bR7b;
каждый из R2a и R2b независимо выбран из группы, состоящей из водорода,
-NR6cR7c, C3-6циклоалкила, C1-4алкила и C1-4алкила, замещенного 1, 2 или 3 атомами галогена;
X1 представляет собой N или CRa;
X2 представляет собой N или CRb;
при условии, что только один из X1 и X2 представляет собой N в любом случае;
R3 представляет собой водород, C1-4алкил или -O-C1-4алкил;
R4 представляет собой галоген, циано или трифторметил;
R5 выбран из группы, состоящей из водорода, C1-4алкила, C3-6циклоалкила, Hetb и C1-4алкила, замещенного одним или двумя заместителями, каждый из которых независимо выбран из группы, состоящей из -C(=O)-NR8R9, -C(=O)-OH, -C(=O)-O-C1-4алкила, C3-6циклоалкила и фенила;
каждый из R6a, R6b, R6c, R7a, R7b, R7c, R8 и R9 независимо выбран из группы, состоящей из водорода и C1-4алкила;
Heta представляет собой моноциклический 4-7-членный неароматический гетероциклил, содержащий один или два гетероатома, выбранные из азота, кислорода и серы;
Hetb представляет собой моноциклический 4-7-членный неароматический гетероциклил, содержащий один или два гетероатома, выбранные из азота, кислорода и серы;
Ra представляет собой -O-C1-4алкил, каждый из которых необязательно замещен одним, двумя или тремя заместителями, представляющими собой атом галогена;
или
Ra представляет собой 2H-1,2,3-триазол-2-ил или C3-6циклоалкил; при этом каждый из них необязательно замещен по одному или двум атомам углерода посредством заместителя, который в каждом случае независимо выбран из группы, состоящей из C1-4алкила и C1-4алкил, замещенного одним -OH;
Rb представляет собой водород;
или его энантиомер, диастереомер или фармацевтически приемлемая солевая форма.
2. Соединение по п. 1, где
Rx представляет собой C1-4алкил;
Ry представляет собой C1-4алкил; и
Rz представляет собой водород;
R1 выбран из группы, состоящей из -OR5, галогена и -CN;
R2a представляет собой водород;
R2b выбран из группы, состоящей из водорода, -NR6cR7c и C1-4алкила;
X1 представляет собой CRa;
X2 представляет собой N;
R3 представляет собой водород;
R4 представляет собой трифторметил;
R5 представляет собой C1-4алкил;
R6c и R7c представляют собой водород;
Ra представляет собой 2H-1,2,3-триазол-2-ил.
3. Соединение по п. 1, где
X1 представляет собой CRa;
X2 представляет собой N.
4. Соединение по п. 3, где Ra представляет собой 2H-1,2,3-триазол-2-ил.
5. Фармацевтическая композиция для лечения связанного с MALT1 заболевания, синдрома, состояния или нарушения, содержащая соединение по любому из пп. 1-4 и по меньшей мере один фармацевтически приемлемый носитель, фармацевтически приемлемое вспомогательное вещество или фармацевтически приемлемый разбавитель.
6. Соединение по п. 1 для применения в качестве лекарственного препарата для лечения заболевания, синдрома, состояния или нарушения, на которые воздействуют путем подавления MALT1.
7. Соединение по п. 1 для применения в лечении или предупреждении диффузной В-крупноклеточной лимфомы (DLBCL), лимфомы из клеток мантийной зоны (MCL), фолликулярной лимфомы (FL), лимфомы лимфоидной ткани слизистых оболочек (MALT), ревматоидного артрита (RA), псориатического артрита (PsA), псориаза (Pso), язвенного колита (UC), болезни Крона, системной красной волчанки (SLE), астмы и хронического обструктивного заболевания легкого (COPD).
8. Способ лечения заболевания, синдрома, состояния или нарушения, где на указанное заболевание, синдром, состояние или нарушение воздействуют путем подавления MALT1, предусматривающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по п. 1.
9. Способ по п. 8, где указанные заболевание, синдром, состояние или нарушение выбраны из группы, состоящей из диффузной В-крупноклеточной лимфомы (DLBCL), лимфомы из клеток мантийной зоны (MCL), фолликулярной лимфомы (FL), лимфомы лимфоидной ткани слизистых оболочек (MALT), ревматоидного артрита (RA), псориатического артрита (PsA), псориаза (Pso), язвенного колита (UC), болезни Крона, системной красной волчанки (SLE), астмы и хронического обструктивного заболевания легкого (COPD).
| WO 2018226150 A1, 13.12.2018 | |||
| US 20180030061 A1, 01.02.2018 | |||
| WO 2017081641 A1, 18.05.2017 | |||
| EA 201590916 A1, 29.02.2016. |

