Настоящее изобретение относится к макроциклическим пиразинопирролопиридазиндионовым производным, обладающим свойствами ингибирования репликации ВИЧ (Вирус Иммунодефицита Человека), их получению и фармацевтическим композициям, включающим эти соединения.
Предпосылки изобретения
Изначально лечение ВИЧ инфекции состояло из монотерапии с использованием нуклеозидных производных, и, несмотря на то, что такая терапия была успешной в подавлении вирусной репликации, эти лекарственные средства быстро теряли свою эффективность из-за появления лекарственно-резистентных штаммов. Очевидно, что высокая скорость мутации в сочетании с быстрой репликацией сделали ВИЧ особенно интересной мишенью для противовирусной терапии. Введение комбинированной терапии, включающей несколько средств против ВИЧ, обеспечивало лучший терапевтический результат. Существующий стандарт лечения представляет собой так называемую HAART (высокоактивная антиретровирусная терапия), которая предлагает сильное и продолжительное подавление вируса. HAART обычно включает комбинацию нуклеозидных или нуклеотидных ингибиторов обратной транскриптазы (NRTI или NtRTI, соответственно) с не-нуклеозидным ингибитором обратной транскриптазы (NNRTI), ингибитором протеазы (PI) и ингибитором интегразы или ингибитором входных ворот инфекции. Существующие руководства для антиретровирусной терапии рекомендуют, по меньшей мере, режим тройной комбинированной терапии даже для начального лечения. Хотя HAART способна подавлять ВИЧ вплоть до неопределяемых уровней, резистентность может возникать из-за проблем совместимости. Также было показано, что резистентный вирус передается новым инфицированным субъектам, оставляя в результате строго ограниченные варианты терапии для этих не имеющих лекарственного привыкания пациентов.
Поэтому существует постоянная потребность в новых и эффективных соединениях, которые можно использовать в качестве анти-ВИЧ лекарственных средств. В частности, существует потребность в дополнительных ингибиторах ВИЧ интегразы, которые являются более эффективными в том, что касается активности против вируса дикого типа, но также против мутированных штаммов, в частности, в отношении мутированных штаммов, выбранных по резистентности к известным ингибиторам интегразы, таким как ралтегравир и элвитегравир. Первичные мутации, наиболее часто развивающиеся в ходе терапевтического лечения ралтегравиром, включают N155H и Q148K/R/H, и не очень часто Y143R/C. Как было обнаружено, приобретение N155 или Q148 мутаций приводит к перекрестной резистентности к структурно несхожим ингибиторам интегразы. Неудачное лечение ралтегравиром связывают с мутациями интегразы в по меньшей мере 3 разных генетических путях, определяемых 2 или более мутациями, включая сигнатурную (главную) мутацию, представляющую собой одну из первичных мутаций в Q148H/K/R, N155H или Y143R/H/C, и одну или несколько дополнительных малых мутаций. Малые мутации, описанные в Q148H/K/R пути, включают L74M плюс E138A, E138K или G140S. Наиболее распространенная картина мутаций в этом пути представляет собой Q148H плюс G140S, которая также сообщает наибольшую потерю восприимчивости к лекарственному средству. (V. A. Johnson et al. (2009) Topics in HIV Medicine 17(5), 138-145).
Существует потребность в ингибиторах интегразы, которые имеют преимущества, связанные с их фармакокинетическим и/или фармакодинамическим профилем. Другие аспекты, которые следует учитывать при разработке дополнительных ингибиторов интегразы, включают благоприятный профиль безопасности, дозирование и/или отсутствие необходимости в ревакцинации.
Другие ингибиторы ВИЧ интегразы известны из уровня техники. Например, WO0255079, WO0230931, WO0230930 и WO0230426 раскрывают аза- и полиазанафталинилкарбоксамиды, полезные в качестве ингибиторов ВИЧ интегразы. WO0236734 раскрывает дополнительно аза- и полиазанафталинилкетоны, полезные в качестве ингибиторов ВИЧ интегразы. Roggo et al, Journal of antibiotics (1996) раскрывает спиродигидробензофуранлактамы в качестве антагонистов эндотелина и в качестве ингибиторов ВИЧ-1 протеазы.
Полициклические карбамоилпиридоны также были раскрыты в качестве ингибиторов ВИЧ интегразы в EP1874117. WO2005118593 раскрывает ряд бициклических гетероциклов в качестве ингибиторов интегразы, и WO2004103278 раскрывает ряд ацилсульфонамидов в качестве ингибиторов ВИЧ интегразы. WO2005028478 раскрывает ряд азахинолинолфосфонатных соединений в качестве ингибиторов интегразы, и WO2004035577 раскрывает ряд имеющих предварительно определенное молекулярное расположение трициклических ингибиторов интегразы. Кроме того, ряд пиридопиразиновых и пиримидопиразиндионовых соединений раскрыт в WO2005087766. Кроме того, тетрагидро-4H-пиридо(1,2-a)пиримидины и родственные соединения были раскрыты в WO2004058757 Instituto di Ricerche di Biologia Moleculare p Angeletti Spa. Japan Tobacco Inc раскрывает 4-оксихинолиновые соединения в качестве ингибиторов ВИЧ интегразы в WO2004046115 и 6-(гетероцикл-замещенный бензил)-4-оксохинолиновое соединение в качестве ингибитора ВИЧ в US20080207618. WO2005110414 и WO2005110415 раскрывают гидрокси-замещенные пиразинопирролопиридазиндионовые соединения в качестве ингибиторов ВИЧ интегразы и ингибиторов ВИЧ репликации.
Настоящее изобретение направлено на обеспечение конкретного нового ряда пиразинопирролопиридазиндионовых производных, обладающих свойствами ингибирования ВИЧ репликации и ВИЧ интегразы.
Описание изобретения
Соединения по настоящему изобретению отличаются от соединений предшествующего уровня техники по своей структуре, противовирусной активности и/или фармакологической активности. Было обнаружено, что соединения по настоящему изобретению являются не только высокоактивными против вируса дикого типа, но также против мутантных штаммов, в частности, против штаммов, которые демонстрируют резистентность к одному или нескольким известным ингибиторам интегразы, такие штаммы называют лекарственно- или полилекарственно-резистентными ВИЧ штаммами. Также было обнаружено, что соединения по настоящему изобретению демонстрируют благоприятные фармакокинетические и/или фармакодинамические свойства.
Таким образом, в одном аспекте настоящее изобретение относится к соединениям формулы I, включая их стереохимически изомерные формы, которые могут быть представлены формулой I:
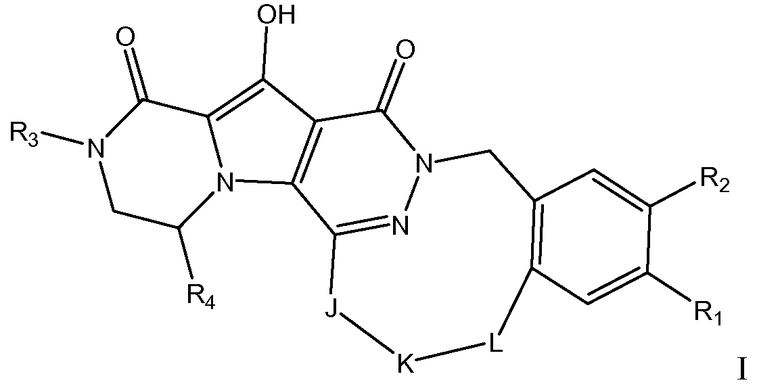
где
R1 представляет собой F или Cl;
R2 представляет собой H, F или Cl;
R3 представляет собой C1-4алкил, C1-4алкоксиC1-4алкил, циклопропил или тетрагидрофуранил;
R4 представляет собой водород или метил;
J представляет собой ---N(R5)-SO2-, ---C(=O)-N(R5)-, ---N(R5)-, 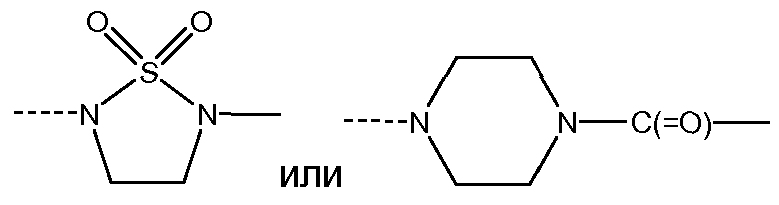 ,
,
где пунктирная линия означает точку присоединения к пиридазиноновому кольцу;
K представляет собой -(CHR6)р, *-(CH2)q-CH=CH-CH2- или *-(CH2)q-CH≡CH-CH2-, где * означает точку присоединения к группе J;
L представляет собой -O-, -O-CH2-* или -N(R5)-C(=O)-*, где * означает точку присоединения к фенильному кольцу; и
R5 представляет собой водород, C1-4алкил или C3-5циклоалкил;
каждый R6 независимо представляет собой водород или C1-3алкил;
p равно 3, 4, 5 или 6;
q равно 0, 1, 2 или 3;
или их фармацевтически приемлемую соль или сольват.
В следующем аспекте изобретение относится к применению соединений формулы I или подгрупп таких соединений, определенных в настоящей заявке, для ингибирования цикла репликации ВИЧ.
Альтернативно, предусматривается применение указанных соединений для получения лекарственного средства для ингибирования цикла репликации ВИЧ, или соединения формулы I для применения в качестве лекарственного средства для ингибирования репликации ВИЧ.
Как используется в настоящей заявке, "C1-3алкил", в качестве группы или части группы, означает насыщенные линейные или разветвленные углеводородные группы, содержащие от 1 до 3 атомов углерода, такие как, например, метил, этил, 1-пропил или 2-пропил.
Как используется в настоящей заявке, "C1-4алкил", в качестве группы или части группы, означает насыщенные линейные или разветвленные углеводородные группы, содержащие от 1 до 4 атомов углерода, такие как, например, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил, 2-метил-2-пропил.
Термин "C3-5циклоалкил" является общим для циклопропила, циклобутила и циклопентила.
Термин "C1-4алкокси", в качестве группы или части группы, означает группу формулы -O-C1-4алкил, где C1-4алкил определен выше. Примерами C1-4алкокси являются метокси, этокси, н-пропокси, изопропокси, 1-бутокси, 2-бутокси и трет-бутокси.
В любом случае, когда встречается радикал в определении соединений формулы I или в любой из подгрупп, определенных в настоящей заявке, указанный радикал независимо имеет значение, определенное выше в определении соединений формул I или в более ограниченных определениях далее в настоящей заявке.
Также следует отметить, что положения радикалов в любой молекулярной группе, используемой в определениях, могут представлять собой любое положение в такой группе, при условии, что группа является химически стабильной. Например, бутил включает 1-бутил и 2-бутил.
Некоторые соединения формулы I также могут существовать в их таутомерной форме. Такие формы, хотя они определенным образом не указаны в структурных формулах, раскрытых в настоящей заявке, предусматриваются для включения в объем настоящего изобретения.
Настоящее изобретение также включает любые изотопы атомов, присутствующих в соединениях по настоящему изобретению. Например, изотопы водорода включают тритий и дейтерий, а изотопы углерода включают C-13 и C-14.
В любом случае, когда выше или ниже в настоящей заявке используются термины "соединения формулы I", "представленные соединения", "соединения по настоящему изобретению" или любые эквивалентные термины, предполагается, что они включают соединения общей формулы I, а также их соли, сольваты и стереоизомеры. Подобным образом, термины "подгруппы соединений формулы I", "подгруппы представленных соединений", "подгруппы соединений по настоящему изобретению" или любые эквивалентные термины включают подгруппы соединений общей формулы I, а также их соли, сольваты и стереоизомеры.
Когда какая-либо переменная неоднократно встречается в какой-либо группе, каждое ее определение является независимым. Любые ограниченные определения радикалов, представленные в настоящей заявке, применимы к группе соединений формулы I, а также к любой подгруппе, определенной или указанной в настоящей заявке. Например, когда K представляет собой -(CHR6)n-, и p равно 5, тогда каждая из 5 присутствующих переменных R6 определена независимо, и это значит, что, в качестве примера, следующие группы охватываются определением K: -CH2-CH2-CH(CH3)-CH2-CH2- или -CH(CH3)-CH2-CH2-CH2-CH2- или подобные.
Представляющие интерес подгруппы соединений формулы I представляют собой такие соединения формулы I, к которым применимо одно или несколько из следующих ограничений:
- R1 представляет собой F;
- R2 представляет собой H или F;
- R2 представляет собой H;
- R3 представляет собой C1-4алкил или циклопропил;
- R3 представляет собой этил, изопропил или циклопропил;
- R3 представляет собой этил или изопропил;
- R4 представляет собой метил;
- R4 представляет собой метил, и стереоконфигурация, в которой углерод, к которому присоединена метильная группа, представляет собой (S);
J представляет собой ---N(R5)-SO2-, 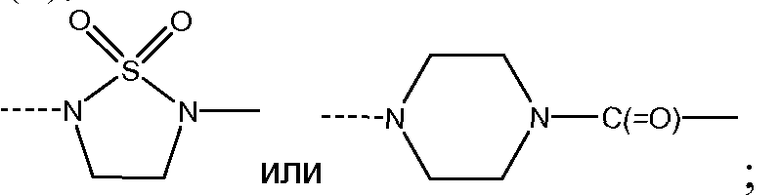
J представляет собой ---N(R5)-SO2- или 
K представляет собой -(CHR6)p-, где p равно 3, 4, 5 или 6 и каждый R6 независимо представляет собой H или CH3;
K представляет собой -(CHR6)p-, где p равно 3, 4, 5 или 6 и каждый R6 представляет собой H;
K представляет собой -(CHR6)p-, где p равно 4 или 5 и каждый R6 представляет собой H;
- K представляет собой *-(CH2)q-CH=CH-CH2-, где q равно 2 или 3;
- L представляет собой -O- или -O-CH2-,
- R5 представляет собой C1-4алкил или C3-5циклоалкил,
- R5 представляет собой метил, этил или циклопропил,
- R5 представляет собой метил,
- связывающая цепь -JKL-, т.е. атомы, образующие связь между фенильным кольцом и пиридазиноновым кольцом формулы I, представляет собой цепь длиной 8-11 атомов.
Фармацевтически приемлемые солевые формы, которые могут образовывать соединения по настоящему изобретению, легко можно получить с использованием подходящих кислот или оснований.
Соединения формулы I, содержащие основную функциональную группу, могут образовывать фармацевтически приемлемые кислотно-аддитивные соли с подходящими кислотами, такими как неорганические кислоты, например, галогенводородные кислоты (например, хлористоводородная или бромистоводородная кислота), серная, гемисерная, азотная, фосфорная и подобные; или органические кислоты, такие как метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая и подобные. И наоборот, указанные кислотно-аддитивные солевые формы можно преобразовать в форму свободного основания путем обработки подходящим основанием.
Соединения формулы I, содержащие кислотные протоны, можно преобразовать в их фармацевтически приемлемые формы солей присоединения металла или амина путем обработки подходящими органическими и неорганическими основаниями. Подходящие основные солевые формы включают, например, соли аммония, соли щелочных и щелочноземельных металлов, например, соли лития, натрия, калия, магния, кальция и подобные, соли с органическими основаниями, например, первичными, вторичными и третичными алифатическими и ароматическими аминами, такие как соли метиламина, этиламина, пропиламина, изопропиламина, четырех бутиламиновых изомеров, диметиламина, диэтиламина, диэтаноламина, дипропиламина, диизопропиламина, ди-н-бутиламина, пирролидина, пиперидина, морфолина, триметиламина, триэтиламина, трипропиламина, хинуклидина, пиридина, хинолина и изохинолина, бензатина, N-метил-D-глюкамина, 2-амино-2-(гидроксиметил)-1,3-пропандиола, гидрабамина, и соли с аминокислотами, такими как, например, аргинин, лизин и подобные. И наоборот, солевую форму можно преобразовать путем обработки кислотой в форму свободной кислоты.
Термин "сольват" охватывает любой фармацевтически приемлемый сольват, который могут образовывать соединения формулы I, а также любая фармацевтически приемлемая соль такого соединения. Такие сольваты представляют собой, например, гидраты, алкоголяты, например, этаноляты, пропаноляты и подобные.
Чистые стереоизомерные формы соединений и промежуточных соединений, указанных в настоящей заявке, определены как изомеры, по существу не содержащие другие энантиомерные или диастереомерные формы такой же основной молекулярной структуры указанных соединений или промежуточных соединений. В частности, термин "стереоизомерно чистый" относится к соединениям или промежуточным соединениям, имеющим стереоизомерный избыток по меньшей мере 80% (т.е. минимально 90% одного изомера и максимально 10% других возможных изомеров), вплоть до стереоизомерного избытка 100% (т.е. 100% одного изомера и отсутствие других), более конкретно, к соединениям или промежуточным соединениям, имеющим стереоизомерный избыток от 90% вплоть до 100%, еще более конкретно, имеющим стереоизомерный избыток от 94% вплоть до 100%, и наиболее точно, имеющим стереоизомерный избыток от 97% вплоть до 100%. Термины "энантиомерно чистый" и "диастереомерно чистый" следует рассматривать как аналогичные, обращая при этом внимание на энантиомерный избыток и диастереомерный избыток, соответственно, рассматриваемой смеси.
Чистые стереоизомерные формы или стереоизомеры соединений и промежуточных соединений по настоящему изобретению можно получить путем применения процедур, известных из уровня техники. Например, энантиомеры можно отделить друг от друга путем селективной кристаллизации их диастереомерных солей с использованием оптически активных кислот или оснований. Примеры включают винную кислоту, дибензоилвинную кислоту, дитолуоилвинную кислоту и камфорсульфоновую кислоту.
Альтернативно, энантиомеры можно разделить хроматографическими методами с использованием хиральных стационарных фаз. Указанные чистые стереохимически изомерные формы также можно получить из соответствующих чистых стереоизомерных форм подходящих исходных веществ, при условии, что реакция осуществляется стереоспецифически. Предпочтительно, когда желательно получение конкретного стереоизомера, указанное соединение синтезируют стереоспецифическими способами получения. В этих способах преимущественно используют энантиомерно чистые исходные вещества.
Диастереомерные рацематы соединений формулы I можно получить отдельно традиционными способами. Подходящие способы физического разделения, которые предпочтительны для использования, представляют собой, например, селективную кристаллизацию и хроматографию, например, колоночную хроматографию или сверхкритическую жидкостную хроматографию.
Соединения формулы I или подгруппы таких соединений могут иметь несколько центров хиральности. Особый интерес представляет стереогенный центр пиперазинонового кольца по R4-замещенному атому углерода. Конфигурация в этом положении может быть (R) или (S), в частности, конфигурация в этом положении представляет собой (S) конфигурацию, как проиллюстрировано формулой I(S).
В случае, когда K в линкере -JKL- содержит двойную связь, тогда Z-конфигурация такой двойной связи представляет интерес.
Соединение в соответствии с настоящим изобретением, как правило, можно получить путем последовательного осуществления стадий, каждая из которых известна специалистам в данной области. В частности, соединения, раскрытые в настоящей патентной заявке, можно получить в соответствии с одним или несколькими из следующих способов получения. На следующих схемах, если не указано иное, все используемые переменные такие, как определено для соединений формулы I.
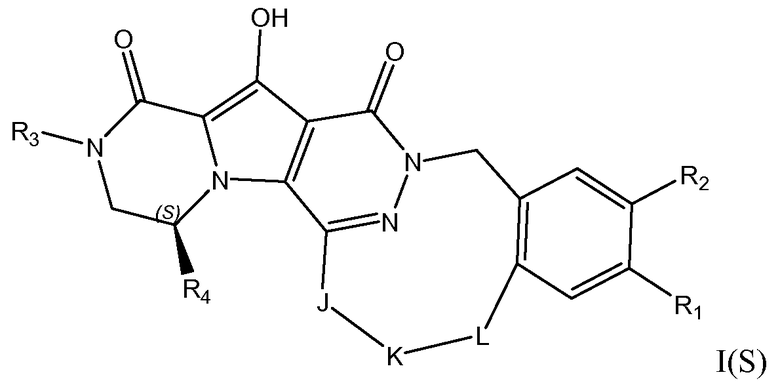
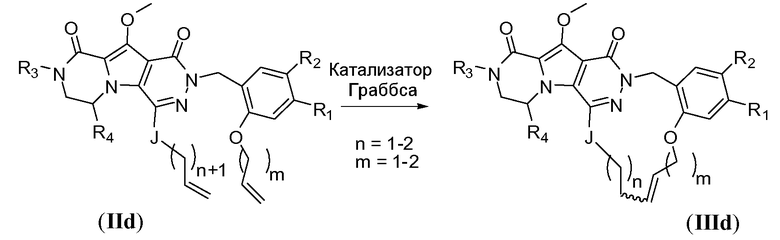
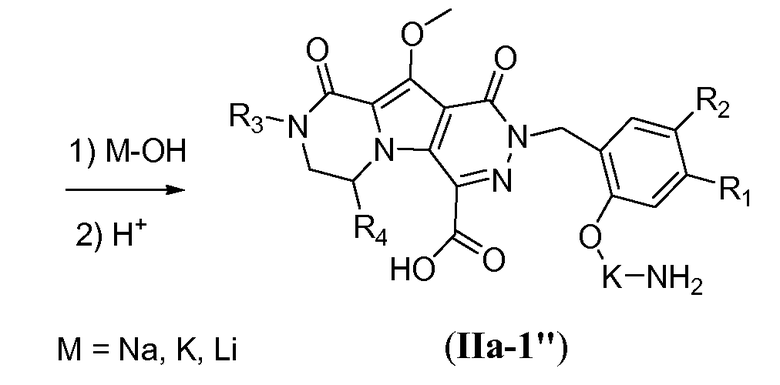
Макроциклы общей формулы I по настоящему изобретению можно получить посредством реакции циклизации, включающей «предшественник с разомкнутым циклом» общей формулы IIa, IIa' или IIa'', в котором гидроксильная функциональная группа пиррольного кольца в I защищена путем метилирования. Указанную макроциклизацию можно осуществить посредством образования амидной связи, эфирной связи или алкеновой связи, и ее, как правило, осуществляют в области линкера J-K-L, как проиллюстрировано на схеме 2a. Удаление метильной защитной группы в соединениях общей формулы III можно осуществить различными способами (схема 1). В первом варианте осуществления предшественник III обрабатывают хлоридом металла, таким как хлорид лития, в полярном апротонном растворителе, таком как диметилформамид (ДМФА). Это преобразование наиболее предпочтительно осуществляют при температуре в пределах от 90°C до 150°C. Во втором варианте осуществления макроцикл общей формулы III можно обработать йодидом натрия и тетрахлорсиланом в смеси растворителей, состоящей из полярного апротонного растворителя, такого как ацетонитрил или подобные, и ароматического неполярного растворителя, такого как толуол или подобные. Указанное преобразование предпочтительно осуществляют при температуре в пределах от 0°C до комнатной температуры. В третьем варианте осуществления предшественник III обрабатывают реагентом на основе бора, таким как трибромид бора (BBr3), в апротонном растворителе, таком как дихлорметан, при низкой температуре, такой как -78°C.
Схема 1
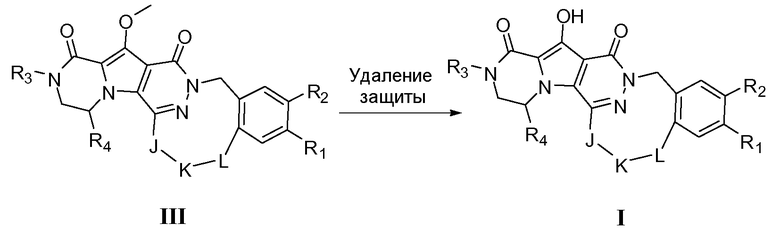
Макроциклы общей формулы IIIa, IIIa' и IIIa'' можно синтезировать по реакции макролактамизации, как показано на схеме 2a. Для этого преобразования требуется присутствие дегидратирующего реагента. Примерами таких традиционно используемых веществ являются HBTU (гексафторфосфат O-бензотриазол-N,N,N',N'-тетраметилурония), EDCI (1-этил-3-(3-диметиламинопропил)карбодиимид)(1-этил-3-(3-диметиламинопропил)карбодиимид), EDAC (гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида) или FDPP (пентафторфенил дифенилфосфинат). В конкретном варианте осуществления указанный дегидратирующий реагент представляет собой HBTU или FDPP. Реакцию обычно осуществляют путем медленного добавления предшественника с разомкнутым циклом общей формулы IIa, IIa' или IIa'' к смеси, содержащей указанный дегидратирующий агент и избыточное количество третичного амина, такого как диизопропилэтиламин. Полезный растворитель представляет собой апротонный растворитель или, более предпочтительно, полярный апротонный растворитель. Примеры апротонных растворителей включают CH2Cl2 (DCM), ДМФА, CH3CN, CHCl3 и т.п. Примеры полярных апротонных растворителей включают ДМФА, диметилацетамид (DMA), N-метилпирролидон (NMP) или диметилсульфоксид (ДМСО). В некоторых обстоятельствах предпочтительно использование гидроксибензотриазола (HOBT) или подобных соединений в качестве добавки в реакции сочетания. В предпочтительном варианте осуществления реакцию циклизации осуществляют при низкой концентрации предшественника с разомкнутым циклом, такой как в пределах 1-10 мМ.
Схема 2a
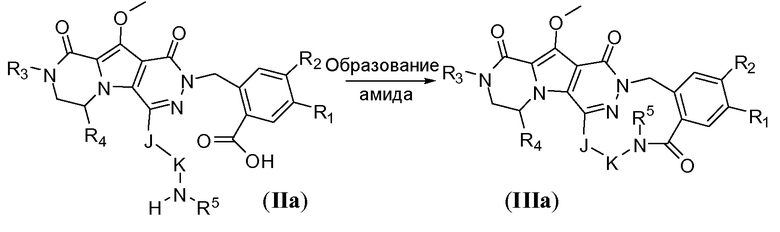
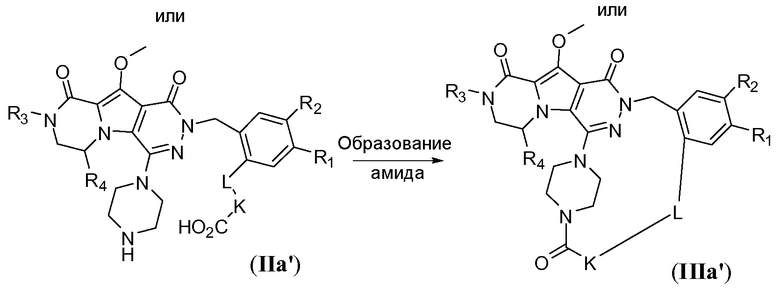
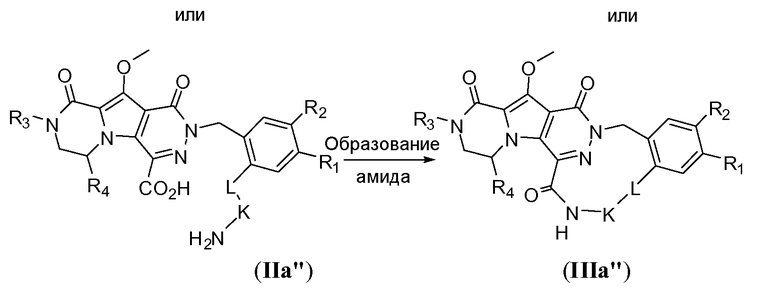
Макроциклы общей формулы IIIb можно синтезировать по реакции Мицунобу, как показано на схеме 2b. Это преобразование можно осуществить путем обработки дигидрокси-предшественника с разомкнутым циклом общей формулы IIb фосфином, таким как трифенилфосфин или трибутилфосфин, и диалкилазодикарбоксилатным реагентом, таким как диизопропилазодикарбоксилат (DIAD) или диэтилазодикарбоксилат (DEAD). Реакцию предпочтительно осуществляют в полярном апротонном растворителе, таком как тетрагидрофуран (ТГФ), или в неполярном растворителе, таком как толуол, и требуется температура реакции в пределах от -20°C до 50°C.
Схема 2b
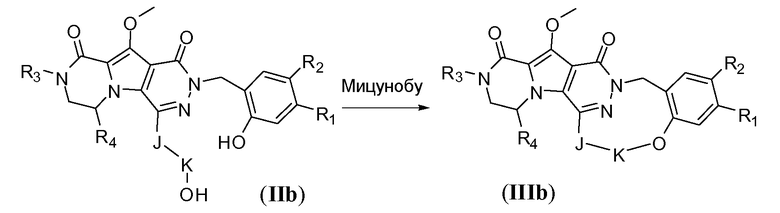
Макроциклы общей формулы IIIc можно синтезировать по реакции макро-этерификации, как показано на схеме 2c. Это преобразование можно осуществить путем обработки предшественника с разомкнутым циклом IIc, содержащего гидроксиалкильный заместитель и бензилгалогенид, такой как хлорид, сильным неорганическим основанием, таким как KOtBu, в полярном апротонном растворителе, таком как DMA. Температура реакции находится в пределах от -10°C до 20°C, в частности, составляет около 0°C.
Схема 2c
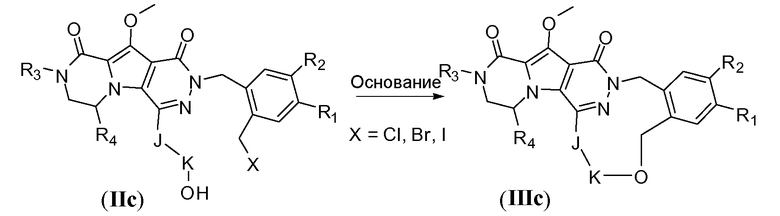
Макроциклы общей формулы IIId, т.е. где K представляет собой C3-6алкенилен, можно синтезировать по реакции макроциклизации с олефиновым метатезисом (обменом олефинов), как показано на схеме 2d. Указанное преобразование осуществляют с использованием рутениевого катализатора, такого как катализатор Граббса первого поколения (например, 1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)(дихлорфенилметилен)-(трициклогексилфосфин)рутений) или катализатор Ховейда-Граббса второго поколения (например, (1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(o-изопропоксифенилметилен)рутений). Предпочтительный растворитель представляет собой галогенированный растворитель, такой как дихлорметан, и температура реакции находится в пределах от 30°C до 90°C. Указанное преобразование может дать макроцикл общей формулы IIId в виде E-изомера или Z-изомера, или их смеси, которую можно разделить с использованием хроматографических методов, известных специалистам в данной области, таких как сверхкритическая CO2 хроматография. Исходное соединение IId получают, следуя реакционным путям, описанным для схемы 5b для введения аллилокси-замещенной фенильной группы, и реакционному пути, описанному для схем 7a-b и 8a для взаимодействия фторпиридазинона с подходящей группой A-(CH2)n-CH=CH2.
Схема 2d
Макроциклы, где K представляет собой C3-6алкинилен, можно получить способом, подобным процедуре получения макроциклов общей формулы IIId, но с использованием реакции макроциклизации с алкиновым метатезисом, известной из уровня техники.
Макроциклы можно подвергнуть дальнейшей дериватизации, как проиллюстрировано на схемах 3a и 3b. Например, на схеме 3a макроцикл общей формулы IIIa'' подвергают алкилированию путем обработки алкилгалогенидом R5-X, предпочтительно йодидом и/или первичным алкилом, таким как йодметан или йодэтан, и требуется присутствие сильного неорганического основания, такого как NaH. Преобразование предпочтительно осуществляют в полярном апротонном растворителе, таком как ТГФ, при температуре реакции в пределах от 0°C до 20°C.
Схема 3a
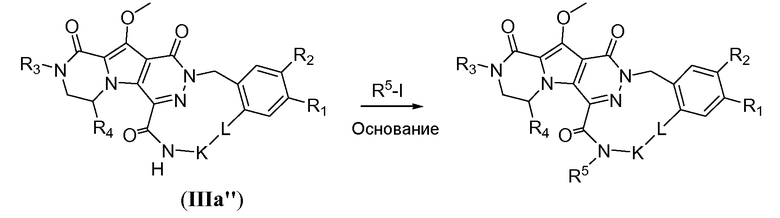
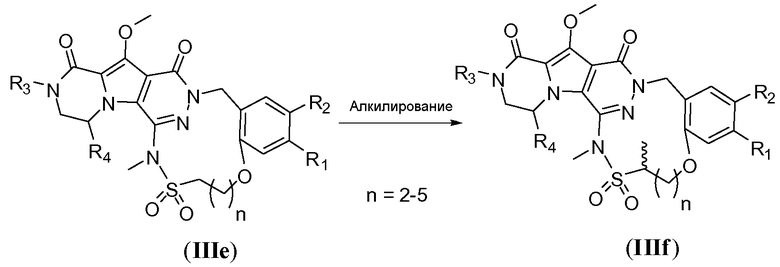
Альтернативно, макроцикл общей формулы IIIe может быть алкилирован с получением макроцикла IIIf путем обработки первичным алкилгалогенидом (схема 3b), предпочтительно йодидом, таким как йодметан, и требуется присутствие сильного литийамидного основания, такого как диизопропиламид лития (LDA) или гексаметилдисилазид лития (LiHMDS). Реакцию осуществляют в полярном апротонном растворителе, таком как ТГФ, при температуре в пределах от -78°C до 20°C.
Схема 3b
Макроциклические предшественники общей формулы IIa можно получить способами, проиллюстрированными на схемах 4a-4e.
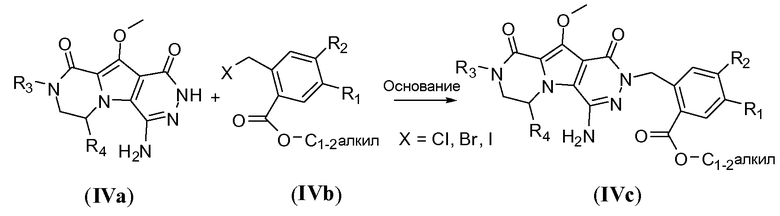
Бензилирование трициклического амина IVa для получения структуры общей формулы IVc можно осуществить путем обработки сильным литийамидным основанием, таким как LiHMDS, в апротонном полярном растворителе, таком как ДМФА, при температуре в пределах от 0°C до 20°C, в частности, примерно при 10°C. Бензильная группа представлена формулой IVb, где X представляет собой галоген, такой как бром или хлор.
Схема 4a
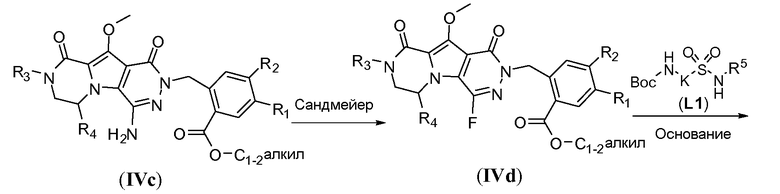
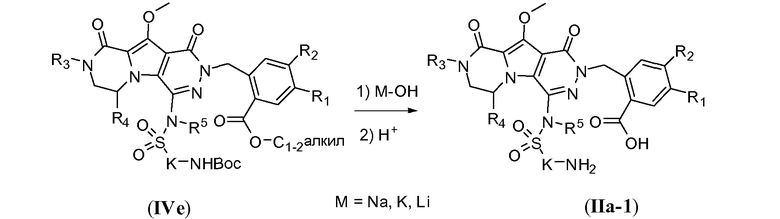
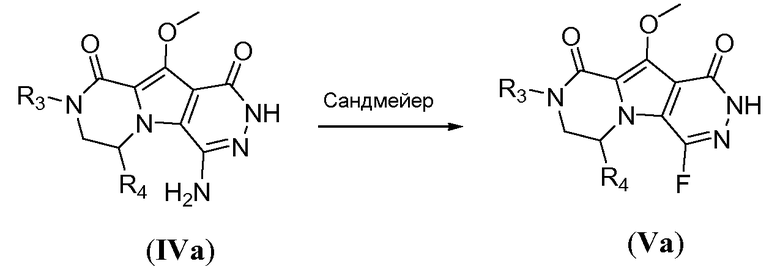
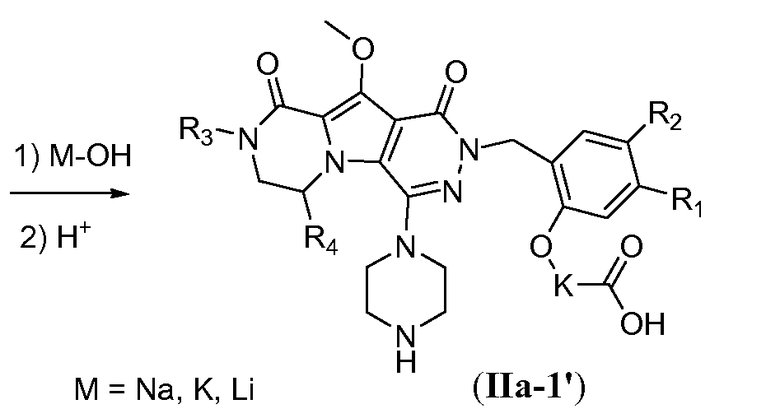
Введение сульфонамидосодержащего линкера (J представляет собой -N(R5)-SO2-) можно осуществить, исходя из IVc, как проиллюстрировано на схеме 4b. Сначала аминогруппу преобразуют во фтор по реакции Сандмейера, которую осуществляют путем обработки соединения IVc нуклеофильным фторидным реагентом, таким как фтористый водород, в пиридине в присутствии агента диазотирования, такого как нитрит натрия, при температуре в пределах от 0°C до 20°C, с получением фторсодержащего соединения IVd. Затем фторсодержащий предшественник IVd обрабатывают сульфонамидосодержащим линкером, имеющим формулу LI, в присутствии неорганического основания, такого как карбонат цезия, в полярном органическом растворителе, таком как ДМСО, при температуре в пределах от 50°C до 100°C, с получением защищенного макроциклического предшественника общей формулы IVe. Затем осуществляют удаление защитных групп линкерного предшественника. Группу сложного эфира карбоновой кислоты в IVe гидролизуют. Это можно осуществить с использованием гидроксида металла (M-OH), такого как гидроксид калия, гидроксид натрия или гидроксид лития. Реакцию осуществляют в водной среде, и наиболее предпочтительно ее осуществляют в присутствии по меньшей мере одного смешиваемого с водой органического сорастворителя, такого как метанол, этанол или ТГФ. Удаление Boc-защитной группы амина можно осуществить путем обработки полученной Boc-карбоновой кислоты раствором, содержащим трифторуксусную кислоту, необязательно в присутствии триизопропилсилана, в апротонном растворителе, таком как дихлорметан, с получением макроциклического предшественника общей формулы IIa-1. В предпочтительном варианте осуществления удаление защитной Boc-группы осуществляют при температуре в пределах от 0°C до комнатной температуры. Альтернативно, указанное удаление защиты можно осуществить путем обработки Boc-карбоновой кислоты раствором хлористоводородной кислоты в полярном апротонном растворителе, таком как диоксан, в частности, 4 н. раствором HCl в диоксане.
Схема 4b
Введение аминосодержащего линкера (J представляет собой -N(R5)-) можно осуществить путем восстановительного аминирования исходя из IVc, как проиллюстрировано на схеме 4c. Сначала получают промежуточный имин путем обработки амина IVc альдегидом L2 в протонном органическом растворителе, таком как 2-пропанол, в присутствии кислоты, такой как карбоновая кислота, например, уксусная кислота. Эта реакция требует повышенной температуры, такой как в пределах от примерно 60°C до примерно 90°C. Вторую стадию восстановительного аминирования проводят при более низкой температуре, такой как 0°C, и требуется восстановитель, такой как боргидрид натрия, и протонный органический растворитель, такой как метанол. Защитная группа в L2 может представлять собой кислотно-лабильную бензильную функциональную группу, такую как 2,4-диметоксибензил.
Схема 4c

Амин в IVf-1 может быть алкилирован с получением третичного амина общей формулы IVf-2 (R5 представляет собой C1-4алкил или циклопентил), как показано на схеме 4d. Амин IVf-1 обрабатывают сильным основанием, таким как NaH, в полярном апротонном растворителе, таком как ТГФ, при температуре в пределах от -10°C до 5°C. Алкилйодид формулы R5-I затем используют для взаимодействия с анионом.
Схема 4d
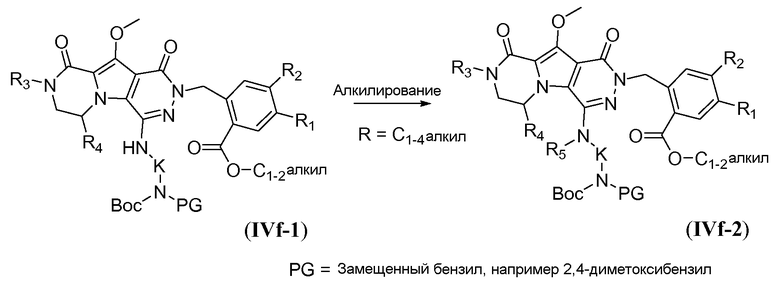
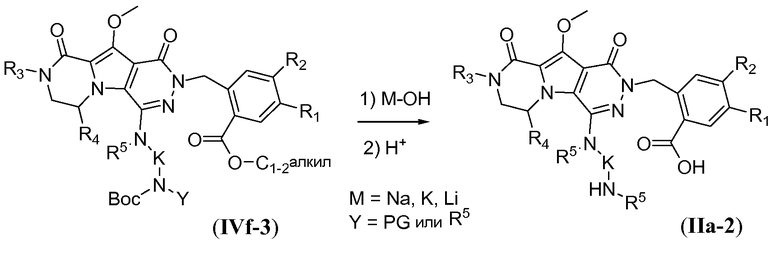
Процедура удаления защиты, подобная той, которая описана для схемы 4b выше, но исходя из соединения общей формулы IVf-3, обеспечивает получение аминокислотного макроциклического предшественника общей формулы IIa-2, и это проиллюстрировано на схеме 4e.
Схема 4е
Макроциклические предшественники общей формулы IIa' можно получить способами, проиллюстрированными на схемах 5a-5d, как показано для получения соединения IIa-1' (соединения формулы IIa', где L представляет собой -O-). Трициклический амин IVa преобразуют в соответствующее фторсодержащее соединение Va (схема 5a) в соответствии с процедурами, подобными описанным выше (схема 4a).
Схема 5a
Пиридазиноновую группу в Va можно бензилировать, следуя 2 альтернативным протоколам (схема 5b). В первом варианте осуществления это бензилирование осуществляют по реакции Мицунобу, подобной той, которая описана в настоящей заявке выше (схема 2b), с использованием защищенного бензилового спирта A1. Во втором варианте осуществления бензилирование осуществляют аналогично тому, как описано в настоящей заявке выше для схемы 4a, используя защищенный бензилгалогенид A2.
Схема 5b
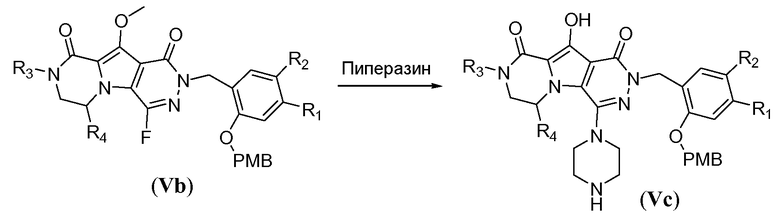
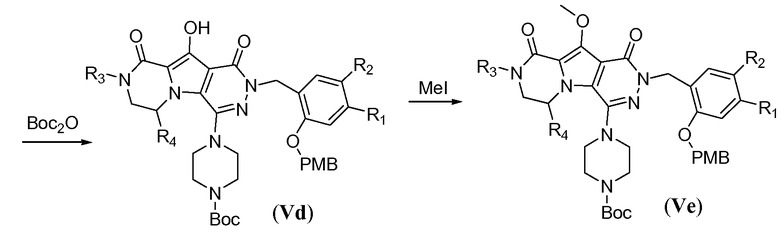
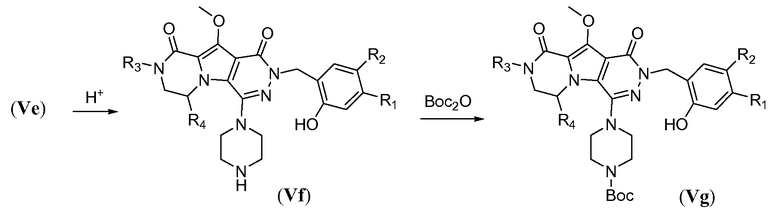
Пиперазинильную группу (для соединений формулы I, где J представляет собой  ) вводят путем обработки фторпиридазинона Vb пиперазином в полярном апротонном растворителе, таком как NMP, при температуре в пределах от 110°C до 130°C. В этих реакционных условиях осуществляют деметилирование метоксигруппы в пиррольном кольце с получением соответствующего гидроксила в полученном соединении общей формулы Vc. Пиперазинил защищают Boc-группой путем обработки с помощью Boc2O в протонном растворителе, таком как метанол, при температуре в пределах от 0°C до 20°C, с получением соединения формулы Vd. Гидроксил снова защищают путем обработки йодметаном в присутствии неорганического основания, такого как карбонат калия, в полярном апротонном растворителе, таком как ДМФА, при температуре в пределах от 0°C до 20°C, с получением соединения формулы Ve.
) вводят путем обработки фторпиридазинона Vb пиперазином в полярном апротонном растворителе, таком как NMP, при температуре в пределах от 110°C до 130°C. В этих реакционных условиях осуществляют деметилирование метоксигруппы в пиррольном кольце с получением соответствующего гидроксила в полученном соединении общей формулы Vc. Пиперазинил защищают Boc-группой путем обработки с помощью Boc2O в протонном растворителе, таком как метанол, при температуре в пределах от 0°C до 20°C, с получением соединения формулы Vd. Гидроксил снова защищают путем обработки йодметаном в присутствии неорганического основания, такого как карбонат калия, в полярном апротонном растворителе, таком как ДМФА, при температуре в пределах от 0°C до 20°C, с получением соединения формулы Ve.
Схема 5c
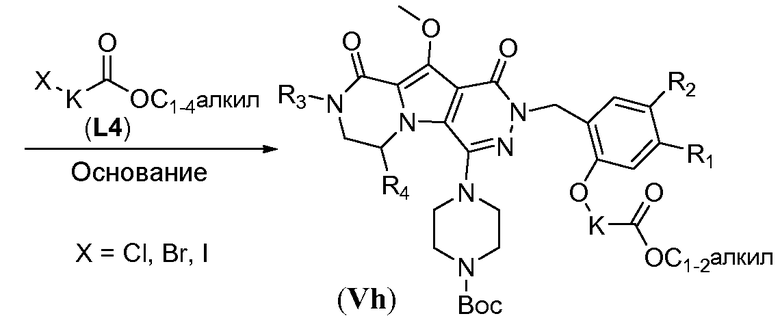
Удаление защитной пара-метоксибензильной (PMB) группы осуществляют путем обработки сильной кислотой, такой как HCl, в апротонном растворителе, таком как 1,4-диоксан, или трифторуксусной кислотой (ТФУК), необязательно в присутствии галогенированного сорастворителя, такого как DCM, при температуре в пределах от 0°C до 20°C, с получением соединения формулы Vf. Как следствие, происходит одновременное удаление защитной группы Boc, и необходима повторная защита пиперазина с использованием BoC2O аналогично тому, как описано в настоящей заявке выше, для получения соединения формулы Vg. Введение углеродного линкера (-K-) осуществляют путем обработки галогенированным алканоатом L4 в присутствии неорганического основания, такого как карбонат калия, в полярном апротонном растворителе, таком как ДМФА, при температуре в пределах от 0°C до 20°C, с получением соединения формулы Vh. Процедура удаления защиты, аналогичная той, которая описана для схемы 4b, исходя из соединения общей формулы Vh, приводит к получению аминокислотного макроциклического предшественника общей формулы IIa-1', как показано на схеме 5d.
Схема 5d
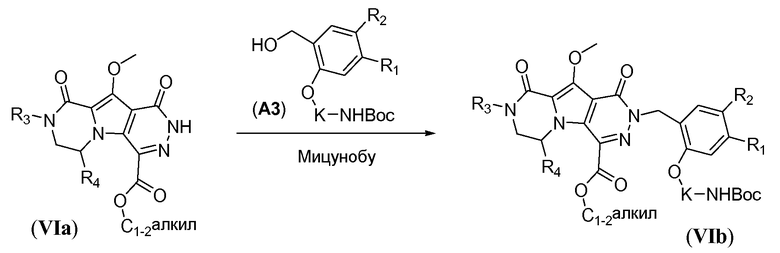
Макроциклические предшественники общей формулы IIa'' можно получить способами, проиллюстрированными на схеме 6, как показано для получения соединения IIa-1'' (соединение IIa'', где L представляет собой -O-). Первая стадия включает реакцию Мицунобу, как показано на схеме 6, которую осуществляют аналогично реакции, описанной для схемы 2b, с использованием соединения общей формулы VIa и бензилового спирта A3 с фосфином, таким как трифенилфосфин или трибутилфосфин, и диалкилазодикарбоксилатного реагента, такого как диизопропилазодикарбоксилат (DIAD) или диэтилазодикарбоксилат (DEAD). Реакцию предпочтительно проводят в полярном апротонном растворителе, таком как ТГФ, или неполярном растворителе, так как толуол, и требуется температура реакции в пределах от -5°C до 20°C, с получением соединения формулы VIb. Последовательное осуществление процедур удаления защиты, аналогично тому, как описано для схемы 4b, исходя из соединения общей формулы VIb, приводит к получению аминокислотного макроциклического предшественника общей формулы IIa-1''.
Схема 6
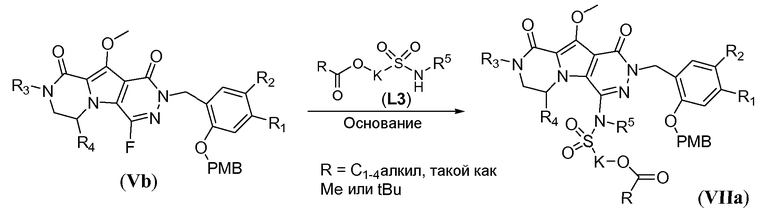
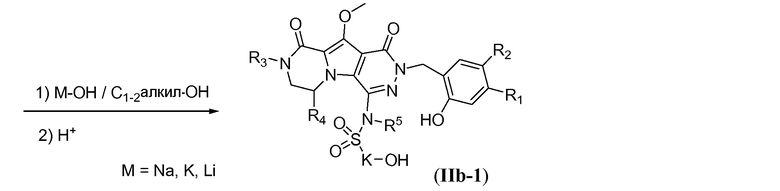
Макроциклические предшественники общей формулы IIb можно получить способами, проиллюстрированными на схемах 7a-7b, на примере синтеза соединений IIb-1 (соединение IIb, где J представляет собой -N(R5)-SO2-) и IIb-2 (соединение IIb, где J представляет собой  ) соответственно. Как проиллюстрировано на схеме 7a, сульфонамидосодержащий линкер L3 вводят путем реакции нуклеофильного замещения фтортрициклического соединения Vb с L3, в которой спирт защищен в виде алканоата -C(=O)-R. Указанную реакцию проводят в полярном растворителе, таком как ДМСО, и требуется присутствие неорганического основания, такого как карбонат цезия. Реакцию наиболее предпочтительно проводят при температуре в пределах от 50°C до 80°C с получением соединения формулы VIIa. Удаление алканоатной защитной группы можно осуществить путем обработки основанием, таким как NaOH, LiOH, в протонном растворителе, таком как метанол или этанол, при комнатной температуре. Удаление PMB защитной группы можно осуществить путем обработки кислотой, такой как ТФУК, в галогенированном растворителе, таком как DCM, или HCl в полярном растворителе, таком как 1,4-диоксан, с получением макроциклического предшественника IIb-1.
) соответственно. Как проиллюстрировано на схеме 7a, сульфонамидосодержащий линкер L3 вводят путем реакции нуклеофильного замещения фтортрициклического соединения Vb с L3, в которой спирт защищен в виде алканоата -C(=O)-R. Указанную реакцию проводят в полярном растворителе, таком как ДМСО, и требуется присутствие неорганического основания, такого как карбонат цезия. Реакцию наиболее предпочтительно проводят при температуре в пределах от 50°C до 80°C с получением соединения формулы VIIa. Удаление алканоатной защитной группы можно осуществить путем обработки основанием, таким как NaOH, LiOH, в протонном растворителе, таком как метанол или этанол, при комнатной температуре. Удаление PMB защитной группы можно осуществить путем обработки кислотой, такой как ТФУК, в галогенированном растворителе, таком как DCM, или HCl в полярном растворителе, таком как 1,4-диоксан, с получением макроциклического предшественника IIb-1.
Схема 7a
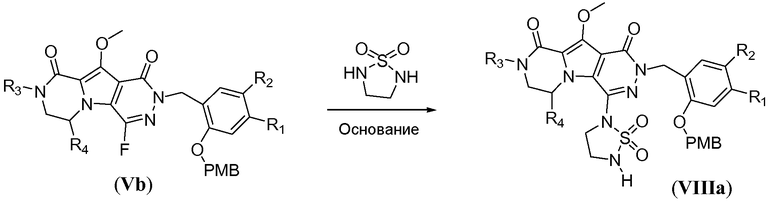
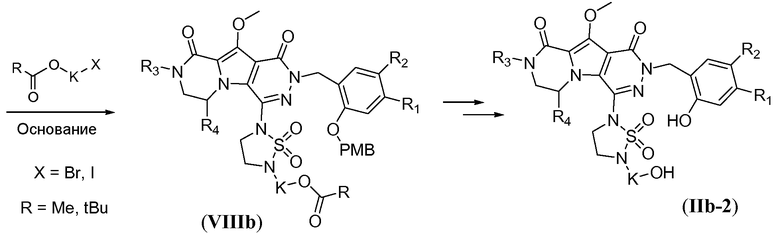
Синтез макроциклических предшественников общей формулы (IIb-2, схема 7b) начинается с нуклеофильного замещения атома фтора в соединении формулы Vb с использованием 1,1-диоксида 1,2,5-тиадиазолидина, с получением соединения формулы VIIIa, аналогично тому, как описано в настоящей заявке выше для схемы 7a. Последующее конструирование линкера включает алкилирование ацилзащищенным галогеналканолом L4. Указанное алкилирование осуществляют путем обработки с помощью L4 в присутствии сильного неорганического основания, такого NaH, в полярном растворителе, таком как ДМФА, при температуре в пределах от 80°C до 120°C, в частности, примерно при 100°C, с получением соединения формулы VIIIb. Удаление защиты с получением соединения формулы IIb-2 осуществляют аналогично тому, как описано в настоящей заявке выше для схемы 7a.
Схема 7b
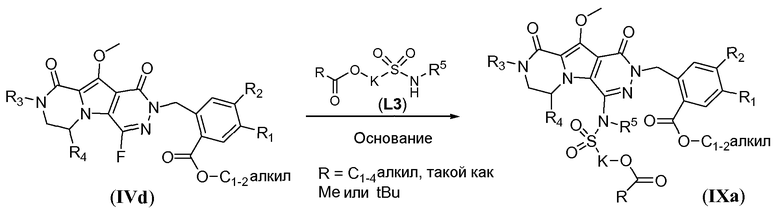
Макроциклические предшественники общей формулы IIc можно получить способом, проиллюстрированным на схеме 8a, как показано для соединений формулы IIc-1 (где J представляет собой -N(R5)-SO2-)-. Сульфонамидосодержащий линкер L3 вводят путем реакции нуклеофильного замещения фтортрициклического соединения IVd с использованием L3, в котором спирт защищен в виде алканоата, аналогично тому, как описано в настоящей заявке выше (схема 7a), с получением соединения формулы IXa.
Схема 8a
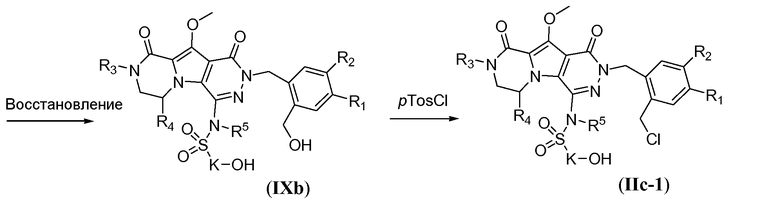
Восстановление двух сложноэфирных функциональных групп осуществляют с использованием металлгидридного реагента, такого как NaBH4, в системе растворителей, включающей полярный, апротонный растворитель, такой как ТГФ, и протонный растворитель, такой как спиртовой растворитель, такой как метанол. Реакцию осуществляют при температуре в пределах от 50°C до 80°C, такой как 65°C, с получением бис-спирта формулы IXb. Макроциклический предшественник формулы IIc-1 можно получить путем взаимодействия с pTosCl в присутствии третичного аминового основания, такого как триэтиламин, в галогенированном растворителе, таком как DCM, при температуре в пределах от 0°C до 20°C (схема 8a).
Аминотрициклическое соединение формулы IVa можно получить из обычного предшественника Xa, как проиллюстрировано на схемах 9a-9b. Первая стадия включает обработку соединения Xa первичным амином R3-NH2 в качестве растворителя, при температуре в пределах от 20°C до 90°C. Ацилирование бромацетилбромидом осуществляют в основной бифазной системе растворителей, состоящей из этилацетата и насыщенного водного раствора NaHCO3, при 0°C с получением бромида формулы Xc. Циклизацию осуществляют путем обработки сильным неорганическим основанием, таким как NaH, в полярном растворителе, таком как ТГФ, при температуре в пределах от 0°C до комнатной температуры, с получением карбобензилокси(Cbz)-защищенного пиперазинона Xd. Восстановительное удаление Cbz защитной группы осуществляют в атмосфере водорода в присутствии палладиевого катализатора, такого как палладий на углероде, в протонном растворителе, таком как метанол, с получением пиперазинона Xe. Конструирование бициклической системы формулы Xg включает две стадии. Сначала проводят реакцию добавления-удаления с использованием диэтилэтоксиметиленмалоната в ароматическом растворителе, таком как толуол, при температуре в пределах от 20°C до 120°C, с получением промежуточного соединения общей формулы Xf. Последующую конденсацию путем реакции Дикмана осуществляют в присутствии сильного основания, такого как LiHMDS, в полярном растворителе, таком как ТГФ, при температуре в пределах от -70°C до комнатной температуры, с получением бицикла общей формулы Xg.
Схема 9a
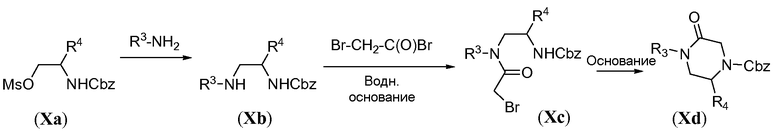
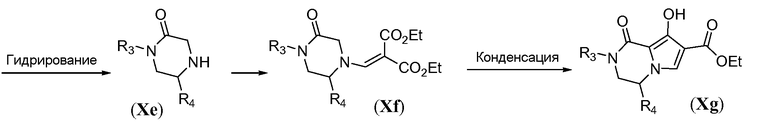
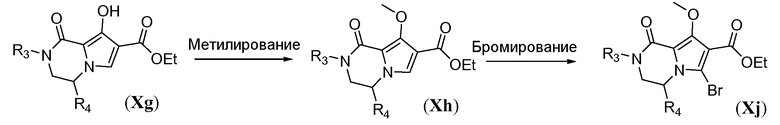
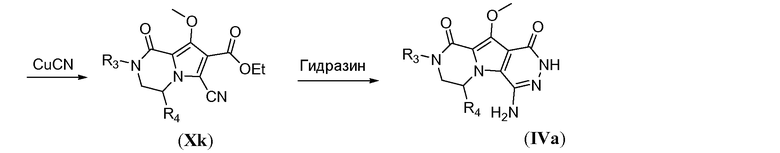
Схема 9b иллюстрирует синтез амино-функционализованного трицикла формулы IVa. Гидроксильную функциональную группу в Xg защищают путем метилирования с использованием йодметана в полярном растворителе, таком как ДМФА, в присутствии неорганического основания, такого как карбонат калия, с получением бициклического метоксипиррола общей формулы Xh. Бромирование в свободном положении в пирроле общей формулы Xh осуществляют путем обработки N-бромсукцинимидом в галогенированном растворителе, таком как дихлорэтан, при температуре в пределах от 0°C до 25°C. Нуклеофильное замещение бромида в Xj осуществляют с помощью CuCN в полярном апротонном растворителе, таком как ДМФА, при температуре в пределах от 90°C до 130°C, с получением цианопиррола общей формулы Xk. В заключение, введение третьего кольца осуществляют путем взаимодействия Xk с избыточным количеством гидразингидрата в протонном растворителе, таком как этанол или tBuOH, при температуре в пределах от 70°C до 90°C, с получением аминотрициклического соединения общей формулы IVa.
Схема 9b
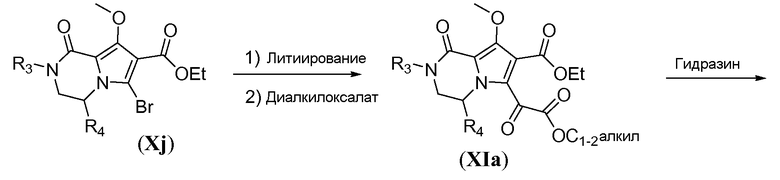
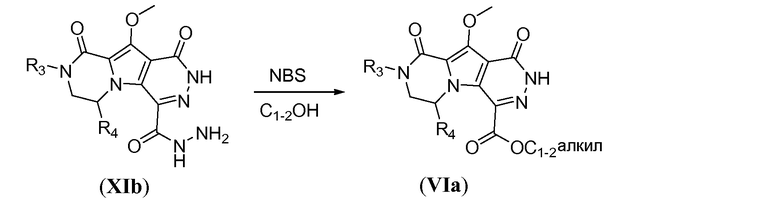
Алкоксикарбонил-функционализованный трицикл формулы VIa можно получить из бромбициклического соединения Xj, как показано на схеме 10. Литиирование Xj осуществляют путем обработки алкиллитиевым реагентом, таким как н-бутиллитий, в полярном растворителе, таком как ТГФ, при температуре в пределах от -70°C до -80°C. Гашение литиевого аниона диалкилоксалатом, таким как диэтилоксалат, при температуре в пределах от -60°C до -70°C дает соединение общей формулы XIa. Взаимодействие с гидразином в комбинации растворителей, включающей протонный растворитель, такой как метанол, и полярный апротонный растворитель, такой как ТГФ, дает трициклическое соединение общей формулы XIb, которое образуется в виде гидразида. Гидразид XIb можно преобразовать в соответствующее алкоксикарбонильное производное путем реакции окисления в присутствии N-бромсукцинимида (NBS) в алканольном растворителе, при температуре в пределах от 10 до 25°C, с получением соответствующего сложного эфира VIa.
Схема 10
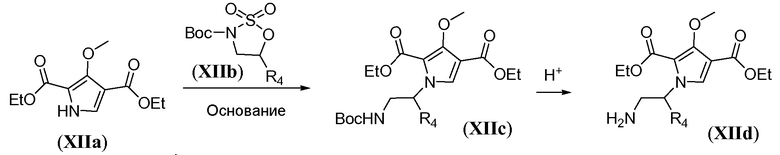
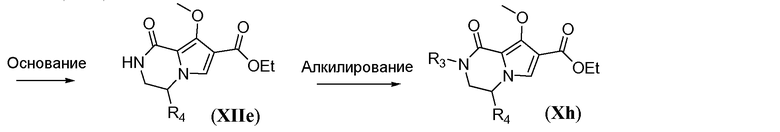
Бициклическое соединение общей формулы Xh также можно получить из пиррола XIIa, как показано на схеме 11, за исключением случая, когда R3 представляет собой циклопропил. Сначала пиррол подвергают алкилированию путем обработки сильным неорганическим основанием, таким как гидрид натрия, и необязательно замещенным 2,2-диоксо-[l,2,3]оксатиазолидином XIIb в полярном растворителе, таком как ДМФА, при температуре в пределах от 0°C до 25°C, с получением Boc-защищенного амина общей формулы XIIc. Эта реакция протекает с инверсией стереохимии, что является подходящим, когда R4 не является атомом водорода. Затем защитную группу Boc удаляют в условиях, известных из уровня техники, таких как использование HCl в 1,4-диоксане, как описано в настоящей заявке выше. Полученный амин общей формулы XIId подвергают циклизации путем обработки неорганическим основанием, таким как карбонат калия, в протонном растворителе, таком как этанол, при температуре реакции в пределах от 60°C до 90°C, с получением бициклического соединения общей формулы XIIe. Введение группы R3 можно осуществить путем обработки соединения XIIe сильным неорганическим основанием, таким как гидрид натрия, в полярном растворителе, таком как ДМФА, при температуре в пределах от -10°C до 5°C, с последующим гашением подходящим R3-X, где X предпочтительно представляет собой бром или йод, с получением бициклического соединения общей формулы Xh. Это преобразование можно осуществить, только когда R3 не является циклопропилом.
Схема 11
Синтез бензильных предшественников формул IVb-1 и A1-A2 проиллюстрирован на схемах 11a и 11b. Бензилбромид формулы IVb-1 получают путем взаимодействия сложного эфира общей формулы IVb-2 с N-бромсукцинимидом в галогенированном растворителе, таком как тетрахлорид углерода, в присутствии радикального инициатора, такого как бензойный пероксиангидрид, при повышенной температуре, в частности, при температуре кипения с обратным холодильником (схема 11a).
Схема 11a
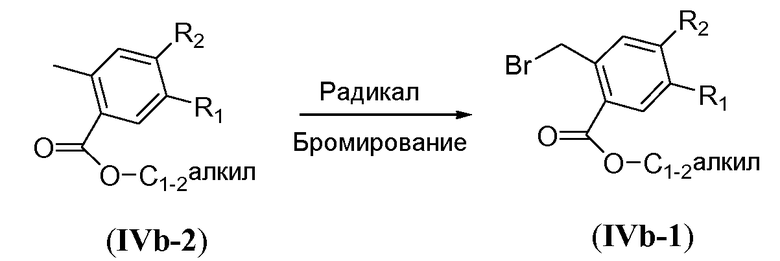
Синтез PMB-защищенных бензиловых спиртов общей формулы A1 и бензилбромидов A2-1 проиллюстрирован на схеме 11b. 2-Гидроксибензоат формулы XIIIa обрабатывают неорганическим основанием, таким как карбонат калия, и п-метоксибензилхлоридом в полярном растворителе, таком как ацетонитрил, при повышенной температуре, такой как температура кипения с обратным холодильником, с получением PMB-защищенного фенола формулы XIIIb. Сложноэфирную функциональную группу восстанавливают металлгидридным реагентом, таким как боргидрид натрия, в системе растворителей, состоящей из полярного растворителя, такого как ТГФ, и протонного растворителя, такого как метанол, при температуре в пределах от 50°C до 75°C, например, при 65°C, с получением соединения формулы A1. Преобразование функциональной группы с получением бензилбромида формулы A2-1 осуществляют путем обработки тетрабромидом углерода и трифенилфосфином в галогенированном растворителе, таком как DCM, предпочтительно при комнатной температуре.
Схема 11b
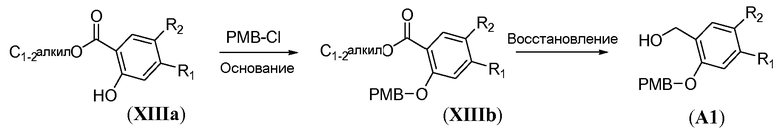

Бензиловые спирты формулы A3 можно получить способами, показанными на схемах 12a-b. Реакцию Мицунобу алкил 2-гидроксибензоата общей формулы XIIIa и N-Boc-защищенного аминоспирта формулы L5 осуществляют, как описано в настоящей заявке выше (схема 2b), с получением предшественника формулы XIVa. Восстановление сложного эфира карбоновой кислоты можно осуществить двумя путями. Сложный эфир карбоновой кислоты можно гидролизовать в присутствии водного основания, аналогично тому, как описано в настоящей заявке выше для схемы 4b, с получением карбоновой кислоты формулы XIVb, с последующим восстановлением в присутствии борана, необязательно в виде диметилсульфидного комплекса, в полярном растворителе, таком как ТГФ, при температуре в пределах от 0°C до 20°C, с получением бензилового спирта формулы A3. Альтернативно, сложный эфир карбоновой кислоты может быть восстановлен металлгидридным реагентом, таким как NaBH4, аналогично тому, как описано в настоящей заявке выше для схемы 8a, с получением бензилового спирта формулы A3.
Схема 12a
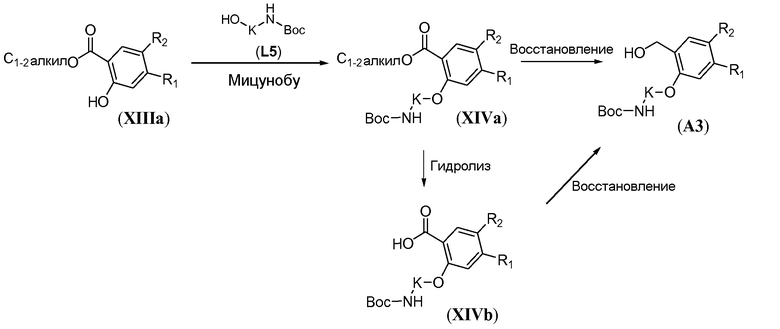
В некоторых случаях, когда R1=R2=F, предпочтительно введение линкерной группы по реакции алкилирования (схема 12b). В частности, алкил 2-гидроксибензоат общей формулы XIIIa обрабатывают неорганическим основанием, таким как карбонат цезия, и дважды защищенным бромзамещенным линкером L5-1 в полярном растворителе, таком как ДМФА, при комнатной температуре, с получением промежуточного соединения формулы XIVa-1. Восстановление сложного эфира карбоновой кислоты можно осуществить путем обработки металлгидридным реагентом, таким как литийалюминийгидрид, в полярном растворителе, таком как ТГФ, при комнатной температуре, с получением бензилового спирта формулы A3-1.
Схема 12b
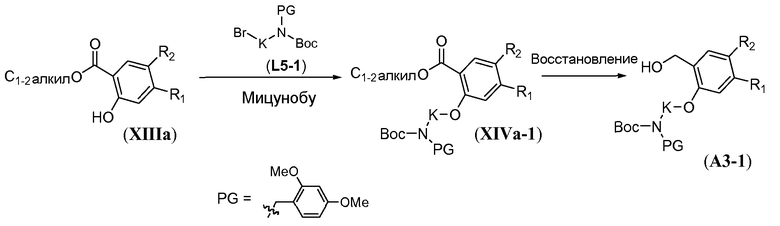
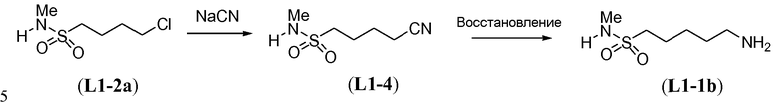
Линкер формулы L5-1 можно получить в три стадии из аминоспирта L5-2 (схема 13). Восстановительное аминирование L5-2 в присутствии 2,4-диметоксибензальдегида можно осуществить с использованием гидрида металла, такого как боргидрид натрия, и карбоновой кислоты, такой как уксусная кислота, в протонном органическом растворителе, таком как метанол. Это преобразование осуществляют при температуре в пределах от -10°C до 20°C с получением монозащищенного аминоспирта формулы L5-3. Boc-защиту осуществляют в условиях, известных из уровня техники, как описано в настоящей заявке выше для схемы 5d, с получением дважды амин-защищенного аминоспирта формулы L5-4. Преобразование спирта в бром осуществляют с использованием тетрабромида углерода в присутствии трифенилфосфина, как описано в настоящей заявке выше для схемы 11b, с получением дважды защищенного аминобромалкана формулы L5-1.
Схема 13
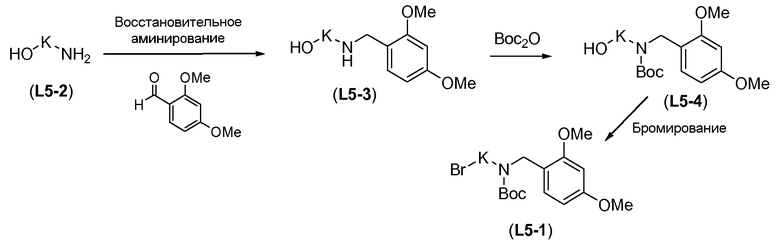
Boc-амино-функционализованный линкер общей формулы L1 можно получить из аминосульфонамида общей формулы L1-1 (схема 14). Обычно, раствор амина L1-1 в хлорированном углеводородном растворителе, таком как DCM, обрабатывают Boc2O при комнатной температуре.
Схема 14
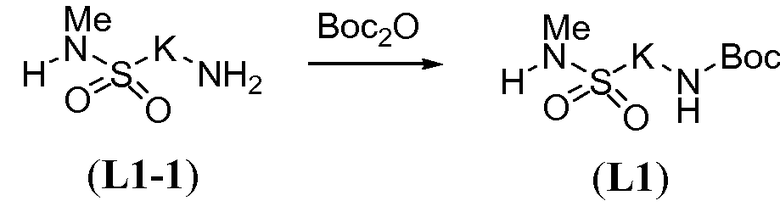
Амино-функционализованный линкер общей формулы L1-1 можно получить разными путями, в зависимости от природы K. Например, когда K представляет собой C3-4алкил, приводя к линкеру формулы L1-1a, можно использовать последовательность реакций, показанную на схеме 15a. Хлорсульфонамид L1-2 сначала обрабатывают йодидом натрия в полярном апротонном растворителе, таком как ДМФА, при комнатной температуре. Затем добавляют азид натрия и смеси дают прореагировать при температуре в пределах от 20 до 70°C, в частности, при 60°C, с получением азида L1-3. На следующей стадии азид восстанавливают с получением амина L1-1a. Это преобразование можно осуществить, помещая азид L1-3 в атмосферу водорода, обычно при 1 атм, в протонном органическом растворителе, таком как метанол. Использование катализатора, такого как палладий на углероде, является важным для осуществления указанной реакции гидрирования.
Схема 15a
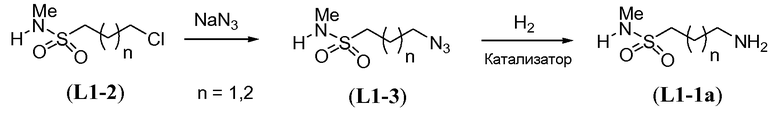
В другом примере, когда K представляет собой пентилен, приводя к линкеру формулы L1-1b, может быть использован способ, проиллюстрированный на схеме 15b. Хлорсульфонамид L1-2a сначала обрабатывают йодидом натрия в полярном апротонном растворителе, таком как ДМФА, при комнатной температуре. Затем добавляют цианид натрия и смеси дают прореагировать при температуре в пределах от примерно 20 до примерно 70°C, в частности, примерно при 60°C, с получением нитрила L1-4. На следующей стадии нитрил восстанавливают с получением амина L1-1b. Это преобразование можно, например, осуществить путем обработки L1-4 комплексом боран-диметилсульфид при комнатной температуре, в полярном апротонном растворителе, таком как ТГФ, с получением первичного амина L1-1b.
Схема 15b
Линкерные предшественники общей формулы L2 можно получить разными способами. Можно использовать реакцию окисления первичной гидроксильной функциональной группы в L5-4 (схема 16a). В частности, указанное окисление осуществляют в условиях реакции Сверна, которая включает взаимодействие между оксалилхлоридом и ДМСО в галогенированном растворителе, таком как DCM, при температуре в пределах от примерно -60°C до примерно -70°C, с последующим добавлением спирта общей формулы L5-4 и третичного амина, такого как триэтиламин, в тех же температурных пределах, с получением альдегида формулы L2-1.
Схема 16a
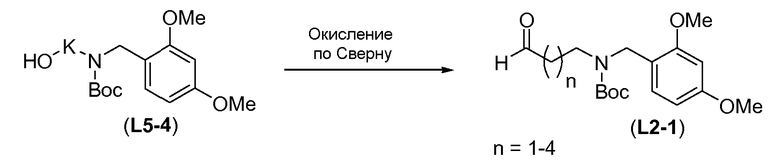
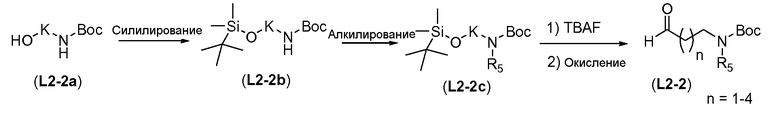
Альтернативно, можно следовать процедуре, проиллюстрированной на схеме 16b. На первой стадии Boc-защищенный ω-аминоспирт формулы L2-2a защищают силильной группой, такой как трет-бутилдиметилсилил, путем взаимодействия спирта L2-2a с трет-бутилдиметилхлорсиланом в присутствии аминового основания, такого как имидазол, в галогенированном растворителе, таком как DCM, при температуре в пределах от 0°C до 25°C. Затем Boc-защищенный амин алкилируют для введения R5 заместителя путем обработки сильным неорганическим основанием, таким как гидрид натрия, и R5-X (где X=бром или йод), в полярном апротонном растворителе, таком как ТГФ, при температуре в пределах от 0°C до 20°C, с получением силил-защищенного спирта формулы L2-2c. Удаление защитной силильной группы путем обработки фторидным реагентом, таким как TBAF, в полярном растворителе, таком как ТГФ, при температуре в пределах от 0°C до 20°C дает спирт формулы L2-2d. Окисление по Сверну аналогично тому, как описано в настоящей заявке выше для схемы 16a, дает альдегид общей формулы L2-2.
Схема 16b
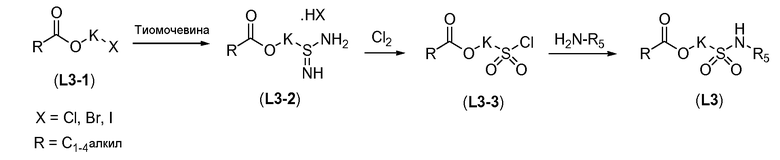
Алканоил-защищенный гидрокси-функционализованный линкер общей формулы L3 можно получить из алканоил-защищенного галогеналканола общей формулы L3-1, как показано на схеме 17. На первой стадии сложный эфир галогенкарбоновой кислоты общей формулы L3-1 преобразуют в соответствующий (аминоиминометил)тиоэфир формулы L3-2. Это преобразование осуществляют путем нагревания смеси тиомочевины и сложного эфира галогенкарбоновой кислоты L3-2 в органическом протонном растворителе, таком как этанол или тому подобное, при температуре в пределах от примерно 70 до 100°C. На второй стадии сульфонилхлорид общей формулы L3-3 получают путем обработки (аминоиминометил)тиоэфира формулы L3-2 хлором в воде в качестве растворителя при температуре около 0°C. На третьей стадии сульфонилхлорид общей формулы L3-3 преобразуют в соответствующий алкил или циклоалкилсульфонамид общей формулы L3, путем обработки смеси сульфонилхлорида L3-3 первичным амином R5-NH2 в бифазной системе растворителей, состоящей из воды и галогенированного углеводорода, такого как DCM. Указанное преобразование осуществляют необязательно в присутствии неорганического основания, такого как карбонат калия, при температуре в пределах от примерно 0 до примерно 20°C.
Схема 17
Соединения формулы I демонстрируют антиретровирусные свойства (свойства ингибирования интегразы), в частности, против ВИЧ, этиологического агента Синдрома Приобретенного Иммунодефицита (СПИД) человека. ВИЧ вирус преимущественно инфицирует человеческие T-4 клетки и разрушает их или изменяет их нормальную функцию, в частности, координацию иммунной системы. Как результат, инфицированный пациент имеет постоянно уменьшающееся количество клеток T-4, которые, кроме того, аномально функционируют. Следовательно, система иммунозащиты не может справиться с инфекциями и новообразованиями, и ВИЧ-инфицированный субъект обычно умирает от инфекций, вызываемых условно-патогенными организмами, таких как пневмония, или от рака.
Соединения по настоящему изобретению также демонстрируют активность против лекарственно - и полилекарственно-резистентных штаммов ВИЧ, в частности, против штаммов ВИЧ, которые приобрели резистентность к одному или нескольким клинически используемым ингибиторам ВИЧ интегразы, в частности, к ралтегравиру и/или элвитегравиру. Главные первичные мутации резистентности, связанные с лечением ралтегравиром, включают N155H и Q148K/R/H. Неблагоприятный результат лечения ралтегравиром также связан с мутациями интегразы по меньшей мере в 3 разных генетических путях, определяемых 2 или более мутациями, включая сигнатурную (или главную) мутацию, являющуюся одной из первичных мутаций в Q148H/K/R, N155H или Y143R/H/C, и одну или несколько дополнительных малых мутаций. Малые мутации, описанные в Q148H/K/R пути, включают L74M плюс E138A, E138K или G140S. Наиболее обычная картина мутаций в этом пути представляет собой Q148H плюс G140S, которая также сообщает наибольшую потерю восприимчивости к лекарственному средству (V.A. Johnson et al. (2009) Topics in HIV Medicine 17(5), 138-145). Как проиллюстрировано примерами ниже, соединения формулы I или подгруппы таких соединений обладают предпочтительными терапевтическими или профилактическими свойствами благодаря их противовирусным свойствам в отношении ВИЧ дикого типа, а также штаммов ВИЧ с одной и двойными мутациями, связанными с резистентностью против ралтегравира и/или элвитегравира.
Благодаря их антиретровирусным свойствам, в частности, их анти-ВИЧ свойствам, соединения формулы I или любая их подгруппа, их фармацевтически приемлемые аддитивные соли и их стереоизомерные формы являются полезными при лечении субъектов, инфицированных ВИЧ, и для профилактики этих инфекций. Соединения по настоящему изобретению также могут найти применение в лечении теплокровных животных, инфицированных вирусами, существование которых опосредовано ферментом протеазы или зависит от него. Состояния, которые можно предотвратить или лечить с помощью соединений по настоящему изобретению, особенно состояния, связанные с ВИЧ, включают СПИД, СПИД-связанный комплекс (ARC), прогрессирующую генерализованную лимфаденопатию (PGL), а также хронические заболевания центральной нервной системы, вызванные ретровирусами, такие как, например, ВИЧ-опосредованная деменция и рассеянный склероз.
Соединения по настоящему изобретению или любую подгруппу таких соединений можно поэтому использовать в качестве лекарственных средств против вышеуказанных состояний. Указанное применение в качестве лекарственного средства или способ лечения включает введение ВИЧ-инфицированным субъектам количества, эффективного для борьбы с состояниями, связанными с ВИЧ и другими патогенными ретровирусами, особенно ВИЧ-1. В частности, соединения формулы I можно использовать для получения лекарственного средства для лечения или профилактики ВИЧ-инфекций.
В следующем аспекте настоящее изобретение предусматривает способ лечения людей, страдающих от вирусных инфекций, или способ профилактики таких инфекций, особенно ВИЧ-инфекций. Указанный способ включает введение человеку эффективного количества соединения формулы I, его фармацевтически приемлемой аддитивной соли, фармацевтически приемлемого сольвата или их возможной стереоизомерной формы.
Настоящее изобретение также обеспечивает композиции для лечения вирусных инфекций, содержащие терапевтически эффективное количество соединения формулы I и фармацевтически приемлемый носитель или разбавитель.
Соединения по настоящему изобретению или любая подгруппа таких соединений могут быть составлены в различные фармацевтические формы для введения. В качестве подходящих композиций можно указать все композиции, обычно используемые для системного введения лекарственных средств. Для получения фармацевтических композиций по настоящему изобретению эффективное количество конкретного соединения, необязательно в форме аддитивной соли, в качестве активного ингредиента объединяют в однородной смеси с фармацевтически приемлемым носителем, при этом такой носитель может принимать разнообразные формы в зависимости от формы препарата, желательной для введения. Такие фармацевтические композиции желательны в виде стандартной лекарственной формы, подходящей, в частности, для введения пероральным, ректальным, чрескожным путем или путем парентеральной инъекции. Например, для получения композиций в пероральной лекарственной форме можно использовать любую обычную фармацевтическую среду, такую как, например, вода, гликоли, масла, спирты и подобные, в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры, эмульсии и растворы; или твердые носители, такие как крахмалы, сахара, каолин, разбавители, смазывающие вещества, связующие, разрыхлители и подобные, в случае порошков, пилюль, капсул и таблеток. С учетом простоты их введения, таблетки и капсулы представляют собой наиболее предпочтительные пероральные стандартные лекарственные формы, причем очевидно, что в этом случае используют твердые фармацевтические носители. Для парентеральных композиций носитель обычно включает стерильную воду, по меньшей мере, в значительной степени, хотя могут быть включены и другие ингредиенты, например, для улучшения растворимости. Можно получить, например, растворы для инъекций, где носитель включает физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Также можно получить суспензии для инъекций, причем в этом случае можно использовать подходящие жидкие носители, суспендирующие агенты и подобные. Также включены твердые формы препаратов, которые можно преобразовать, непосредственно перед использованием, в жидкую форму. В композициях, подходящих для чрескожного введения, носитель необязательно включает агент, усиливающий пенетрацию, и/или подходящее смачивающее вещество, необязательно в сочетании с подходящими добавками любой природы в малых пропорциях, при этом такие добавки не должны оказывать какого-либо серьезного вредного воздействия на кожу. Указанные добавки могут облегчать введение на кожу и/или могут способствовать получению желательных композиций. Такие композиции можно вводить различными путями, например, в виде трансдермального пластыря, путем нанесения в виде пятна, в виде мази.
Соединения по настоящему изобретению также можно вводить путем ингаляции или инсуффляции с использованием способов и композиций, известных из уровня техники для введения таким путем. Таким образом, соединения по настоящему изобретению, как правило, можно вводить в легкие в форме раствора, суспензии или сухого порошка. Любая система, разработанная для доставки растворов, суспензий или сухих порошков посредством пероральной или назальной ингаляции или инсуффляции, является подходящей для введения соединений по настоящему изобретению.
Особенно предпочтительно, когда указанные выше фармацевтические композиции составлены в виде стандартной лекарственной формы для простоты введения и равномерного дозирования. Стандартная лекарственная форма, как это используется в настоящей заявке, относится к физически дискретным единицам, подходящим в качестве стандартных доз, где каждая единица содержит предварительно определенное количество активного ингредиента, рассчитанное для обеспечения желаемого терапевтического эффекта, в ассоциации с необходимым фармацевтическим носителем. Примерами таких стандартных лекарственных форм являются таблетки (включая таблетки с насечкой или с покрытием), капсулы, пилюли, пакеты с порошком, пластинки, суппозитории, растворы или суспензии для инъекций и т.п. и их раздельные кратные количества.
Специалисты в области лечения ВИЧ-инфекции смогут определить эффективное суточное количество на основании результатов испытаний, представленных в настоящей заявке. Обычно предполагается, что эффективное суточное количество должно составлять от 0,01 мг/кг до 50 мг/кг массы тела, более предпочтительно от 0,1 мг/кг до 10 мг/кг массы тела. Может быть подходящим введение необходимой дозы в виде разделенной на две, три, четыре или более доз через подходящие интервалы в течение дня. Указанные дробные дозы могут быть составлены в виде стандартной лекарственной формы, например, содержащей от 1 до 1000 мг и, в частности, от 5 до 200 мг активного ингредиента на стандартную единицу лекарственной формы.
Точная доза и частота введения зависят от конкретного используемого соединения формулы I, конкретного состояния, подлежащего лечению, тяжести состояния, подлежащего лечению, возраста, массы тела и общего физического состояния конкретного пациента, а также другого лечения, которое может получать субъект, как хорошо известно специалистам в данной области. Кроме того, очевидно, что указанное эффективное суточное количество может быть уменьшено или увеличено в зависимости от ответной реакции субъекта, которого лечат, и/или в зависимости от оценки лечащего врача, прописывающего соединения по настоящему изобретение. Поэтому указанные выше диапазоны эффективного суточного количества являются только общим указанием и не предназначены для ограничения объема или применения настоящего изобретения в какой-либо степени.
Как проиллюстрировано примерами, соединения формулы I или подгруппы таких соединений обладают предпочтительными терапевтическими или профилактическими свойствами, благодаря их фармакокинетическим свойствам, таким как высокая метаболическая стабильность, соотносящаяся с низкой скоростью выведения из плазмы, как продемонстрировано на крысах. В частности, соединения формулы I или подгруппы таких соединений могут обладать благоприятными терапевтическими или профилактическими свойствами, благодаря сочетанию их противовирусных и фармакокинетических свойств, по сравнению с соединениями, известными из уровня техники.
Также, комбинацию одного или нескольких дополнительных антиретровирусных соединений и соединения формулы I можно использовать в качестве лекарственного средства. Таким образом, в следующем аспекте настоящее изобретение также относится к продукту, содержащему (a) соединение формулы I и (b) одно или несколько дополнительных антиретровирусных соединений, в виде комбинированного препарата для одновременного, раздельного или последовательного применения в анти-ВИЧ лечении. Различные лекарственные средства могут быть объединены в раздельные препараты или в один препарат, вместе с фармацевтически приемлемыми носителями. Указанные другие антиретровирусные соединения могут представлять собой любые известные антиретровирусные соединения, такие как нуклеозидные ингибиторы обратной транскриптазы (NRTI), например, зидовудин (AZT), диданозин (ddl), залцитабин (ddC), ламивудин (3TC), ставудин (d4T), эмтрицитабин (FTC), абакавир (ABC), амдоксовир (DAPD), элвуцитабин (ACH-126,443), априцитабин (AVX 754, (-)-dOTC), фозалвудин тидоксил (FZT, HDP-990003), фосфазид, KP-1461, рацивир (PSI-5004), MIV-210 и GS-9131; не-нуклеозидные ингибиторы обратной транскриптазы (NNRTI), такие как делавирдин (DLV), эфавиренц (EFV), невирапин (NVP), дапивирин (TMC120), этравирин (ETR, TMC125), рилпивирин (TMC278), IDX899, RDEA-806, UK-453601, RDEA-427 и UC-781; нуклеотидные ингибиторы обратной транскриптазы (NtRTI), например, тенофовир и его пролекарство тенофовир дисопроксил фумарат (TDF); ингибиторы протеазы, например, ритонавир (RTV), саквинавир (SQV), лопинавир (ABT-378, LPV), индинавир (IDV), апренавир (VX-478), нелфинавир (AG-1343), атазанавир (BMS 232,632), дарунавир (TMC114), фосампренавир (GW433908 или VX-175), бреканавир (GW-640385, VX-385), типранавир (PNU-140690), DG-17, SPI256, PPL-100 (MK 8122) и TMC310911; ингибиторы входных ворот инфекции, которые включают ингибиторы слияния (например, энфувиртид (T-20) сифувиртид, HRG-214, албувиртид, SUC-HAS и maC46/M87o), ингибиторы присоединения, модуляторы внутриклеточного холестерина и биосинтеза кортикостероидов (например, SP-01A) и ингибиторы ко-рецепторов, причем последние включают антагонисты CCR5 (например, CCR5mAb004, маравирок (UK-427,857), PRO-140, TAK-220, TAK-652, PF232798, викривирок (SCH-D, SCH-417,690), GSK-706769, нифевирок и SCH-532706) и антагонисты CXR4 (например, AMD-070), дополнительные примеры ингибиторов входных ворот инфекции включают TNX-355, INCB 9471, BMS-488043, нонакин и VGV-1; ингибиторы созревания, например, бевиримат (PA-457) и вивекон; и ингибиторы вирусной интегразы, например, ралтегравир (MK-0518), элвитегравир (JTK-303, GS-9137), BMS-538158, S-349572, JTK-656 S-247303, GS-265744 и S/GSK-1349572.
Следующие примеры предназначены для иллюстрации настоящего изобретения, а не для ограничения его объема.
ПРИМЕРЫ
A. ХИМИЧЕСКИЙ СИНТЕЗ СОЕДИНЕНИЙ ФОРМУЛЫ I
Пример 1: промежуточное соединение I-2
Подготавливали три параллельные реакции и неочищенные реакционные смеси объединяли для обработки. Каждую реакцию проводили с использованием одной трети количеств, описанных ниже.
Трициклический амин I-1 (0,62 моль) растворяли в смеси HF/пиридин (450 мл) при 25°C. Добавляли порциями NaNO2 (0,93 моль) при 0°C. Смесь перемешивали в течение ночи при 25°C. Смесь добавляли по каплям к насыщенному раствору NaHCO3 и затем экстрагировали с помощью CH2Cl2 (3×1000 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Полученное твердое вещество промывали CH2Cl2. Выход: 90 г промежуточного соединения I-2 (выход 50%).
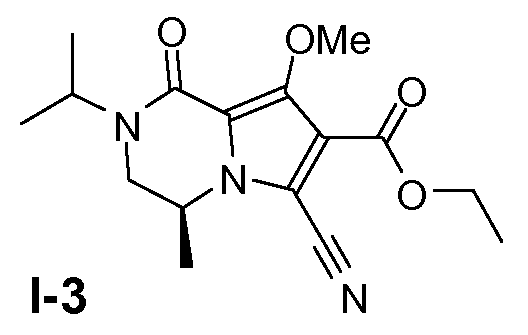
Пример 2: (S)-этил 6-циано-2-изопропил-8-метокси-4-метил-1-оксо-1,2,3,4-тетрагидропирроло[1,2-a]пиразин-7-карбоксилат (I-3)
Подготавливали три параллельные реакции и неочищенную реакционную смесь объединяли для обработки. Каждую реакцию проводили с использованием одной трети количеств, описанных ниже.
(S)-этил 6-бром-2-изопропил-8-метокси-4-метил-1-оксо-1,2,3,4-тетрагидропирроло[l,2-a]пиразин-7-карбоксилат (1,2 моль) и Cu(I)CN (2,4 моль) растворяли в диметилформамиде (ДМФА; 2,2 л) при 20°C в атмосфере N2. Смесь перемешивали в течение 2 часов при 110°C. Добавляли концентрированный NH3.H2O (650 мл) при 60°C. Смесь перемешивали в течение 30 минут при 60°C и затем охлаждали до 20°C. Смесь отфильтровывали. Добавляли этилацетат (3000 мл) и H2O (2000 мл) и органический слой отделяли. Водный слой экстрагировали этилацетатом (3×1000 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили и концентрировали в вакууме. Неочищенный продукт и небольшую партию из предыдущего получения объединяли и промывали метил-трет-бутиловым эфиром с получением твердого вещества. Выход: 350 г промежуточного соединения I-3 (чистота 95%, общий выход 80%).
Пример 3: промежуточное соединение I-4
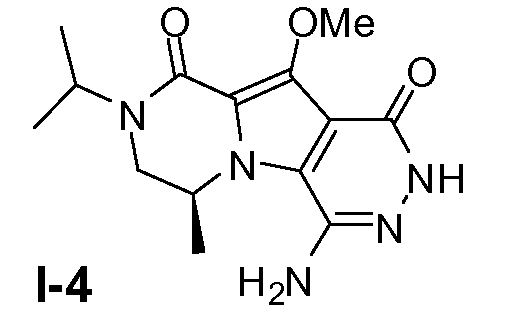
Промежуточное соединение I-3 (1,1 моль) и гидразингидрат (11 моль) в трет-бутиловом спирте (2,7 л) нагревали при температуре кипения с обратным холодильником в течение 18 часов. Смесь концентрировали в вакууме. Полученный остаток очищали колоночной хроматографией (градиентное элюирование: CH2Cl2, затем CH2Cl2/метанол от 50:1 до 30:1 до 20:1) с получением желаемого соединения. Выход: 220 г (выход 66%)
Пример 4: промежуточное соединение I-5
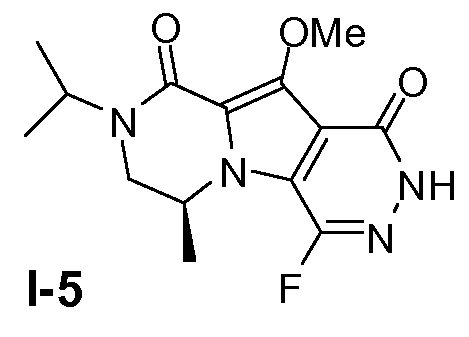
Промежуточное соединение I-5 получали из промежуточного соединения I-4 аналогично тому, как описано в примере 1.
Пример 5: (S)-бензил 1-(циклопропиламино)пропан-2-илкарбамат (I-6)
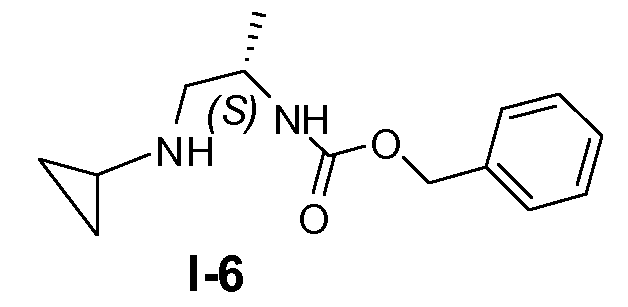
(S)-2-(бензилоксикарбониламино)пропилметансульфонат (48 ммоль) и циклопропиламин (480 ммоль) смешивали вместе и перемешивали при комнатной температуре в течение 48 часов. Оставшийся циклопропиламин удаляли на роторном испарителе и оставшуюся смесь добавляли к 500 мл этилацетата и промывали 5% водным раствором Na2CO3 и насыщенным солевым раствором. После сушки (K2CO3) и удаления растворителя оставшееся полутвердое вещество перекристаллизовывали из CH2Cl2 и петролейного эфира с получением целевого соединения с выходом 60%.
Пример 6: (S)-бензил 1-(2-бром-N-циклопропилацетамидо)пропан-2-илкарбамат (I-7)
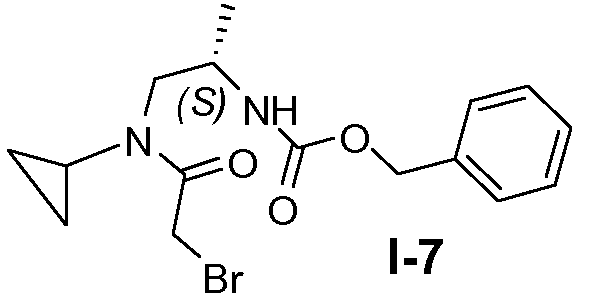
Промежуточное соединение I-6 (0,26 моль) добавляли к смеси этилацетата (530 мл) и насыщенного водного раствора NaHCO3 (322 мл), которую затем охлаждали до 0°C. Затем добавляли по каплям бромацетилбромид (0,29 моль) в атмосфере N2. После перемешивания при комнатной температуре в течение 1 часа слои разделяли и водную фазу экстрагировали этилацетатом. Объединенные органические фазы промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. После фильтрования и концентрирования получали целевое соединение с выходом 90%.
Пример 7: (S)-бензил 4-циклопропил-2-метил-5-оксопиперазин-1-карбоксилат (I-8)
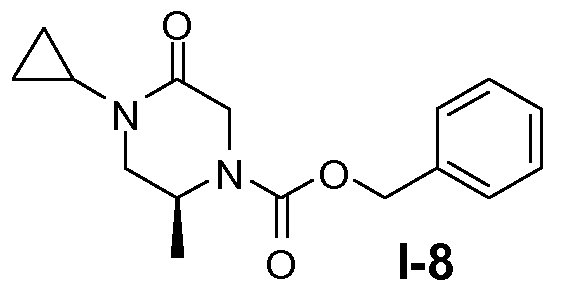
Промежуточное соединение I-7 (40 ммоль) растворяли в 90 мл безводного тетрагидрофурана (ТГФ; 690 мл), к смеси добавляли порциями 60% NaH (44 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов и гасили лимонной кислотой. Смесь концентрировали досуха и остаток распределяли между CH2Cl2 и насыщенным раствором NaHCO3. После сушки (Na2SO4) и концентрирования остаток очищали колоночной хроматографией на силикагеле, используя для элюирования от 1% до 6% метанола в хлороформе, с получением целевого соединения в виде твердого вещества белого цвета с 70% выходом.
Пример 8: (S)-1-циклопропил-5-метилпиперазин-2-он, гидрохлорид (I-9)
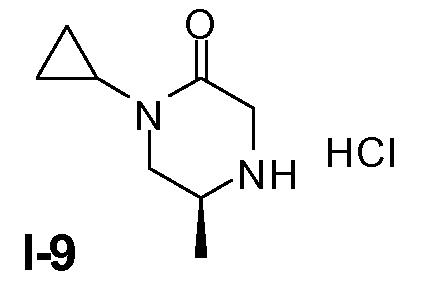
Промежуточное соединение I-8 (17,4 ммоль) растворяли в метаноле (50 мл) и добавляли 0,5 г (10%, 0,5 ммоль) Pd/C. Реакционную смесь перемешивали в атмосфере H2, предоставленного в баллоне для H2. Через 18 часов смесь фильтровали через слой целита и промывали метанолом. После концентрирования неочищенный продукт растворяли в диэтиловом эфире и добавляли 1,5 мл 4 н. раствора HCl в диэтиловом эфире. Затем получали белую суспензию. После перемешивания в течение ночи суспензию фильтровали и твердое вещество промывали диэтиловым эфиром с получением целевого соединения в виде твердого вещества белого цвета. (Выход 70%).
Пример 9: (S)-диэтил 2-((4-циклопропил-2-метил-5-оксопиперазин-1-ил)метилен)малонат (I-10)
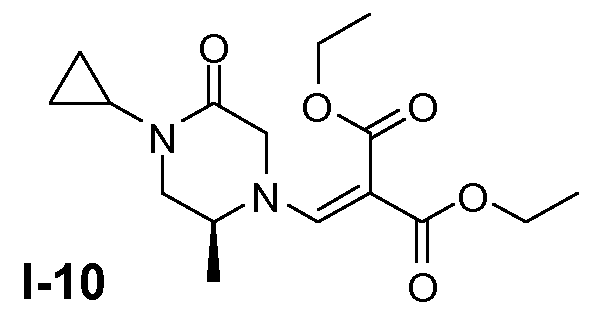
Промежуточное соединение I-9 (169 ммоль) суспендировали в этилацетате (300 мл). Добавляли насыщенный раствор K2CO3 (30 мл). Органический слой промывали насыщенным солевым раствором и сушили (Na2SO4). Растворитель удаляли в вакууме. Полученный остаток и диэтилэтоксиметиленмалонат (169 ммоль) растворяли в толуоле (260 мл). Смесь перемешивали при 25°C в течение 0,5 часа и затем при 120°C в течение 0,5 часа. Смесь упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2, затем этилацетат). Этилацетатные фракции собирали и упаривали. Выход: 60 г (чистота 62%; выход 80%).
Пример 10: (S)-этил 2-циклопропил-8-гидрокси-4-метил-1-оксо-1,2,3,4-тетрагидропирроло[1,2-a]пиразин-7-карбоксилат (I-11)
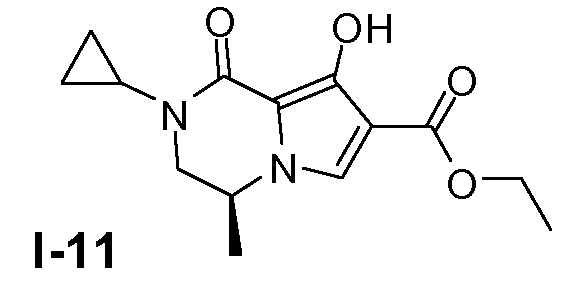
Промежуточное соединение I-10 (135 ммоль, чистота 80%) растворяли в безводном ТГФ (500 мл) при 20°C. Добавляли по каплям раствор гексаметилдисилазида лития (1M в ТГФ, 148 ммоль), поддерживая температуру ниже -70°C. Смеси давали нагреться до 20°C и затем гасили водным раствором NH4Cl. Смесь упаривали в вакууме. Добавляли CH2Cl2 и 1 н. раствор HCl. Органический слой промывали насыщенным солевым раствором и сушили (MgSO4). Растворитель удаляли в вакууме. Остаток очищали колоночной хроматографией с использованием SiO2 (элюент: петролейный эфир/этилацетат 1:1). Выход: 17,7 г (чистота 90%, выход 47%).
Пример 11: (S)-этил 2-циклопропил-8 -метокси-4-метил-1-оксо-1,2,3,4-тетрагидропирроло[1,2-a]пиразин-7-карбоксилат (I-12)
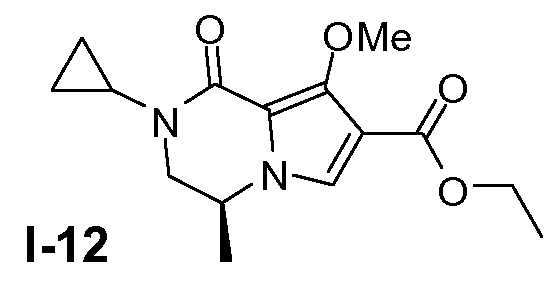
Промежуточное соединение I-11 (64 ммоль) растворяли в ДМФА (180 мл) при 20°C. Добавляли K2CO3 (254 ммоль). Добавляли по каплям метилйодид (76 ммоль) при 0°C. Смесь перемешивали в течение ночи при 20°C. Затем смесь отфильтровывали и фильтрат разбавляли насыщенным солевым раствором и CH2Cl2. Органический слой собирали и промывали насыщенным солевым раствором. Растворитель сушили (MgSO4) и затем выпаривали. Выход: 16 г (чистота 90%; выход 86%).
Пример 12: (S)-этил 6-бром-2-циклопропил-8-метокси-4-метил-1-оксо-1,2,3,4-тетрагидропирроло[1,2-a]пиразин-7-карбоксилат (I-13)
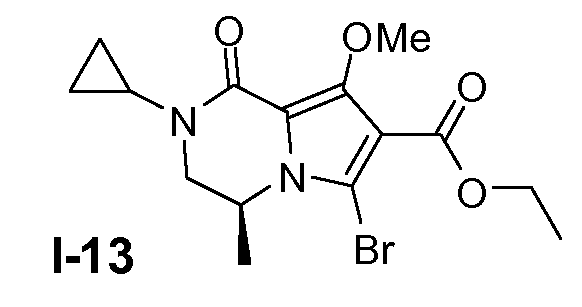
Промежуточное соединение I-12 (55 ммоль) растворяли в дихлорэтане (200 мл). К смеси добавляли N-бромсукцинимид (NBS; 60 ммоль) при 0°C и смесь перемешивали при 25°C в течение 3 часов. Добавляли насыщенный раствор Na2S2O3 (100 мл) и смесь перемешивали в течение 5 минут. Добавляли CH2Cl2 (200 мл) и 0,5 н. раствор NaOH (100 мл). Органический слой промывали насыщенным солевым раствором и сушили (Na2SO4). Растворитель удаляли в вакууме. Остаток очищали колоночной хроматографией с использованием SiO2 (элюент: петролейный эфир/этилацетат 1:3). Выход: 11 г (чистота 90%, выход 54%).
Пример 13: (S)-этил 6-(2-этокси-2-оксоацетил)-2-этил-8-метокси-4-метил-1-оксо-1,2,3,4-тетрагидропирроло[1,2-a]пиразин-7-карбоксилат (I-14)
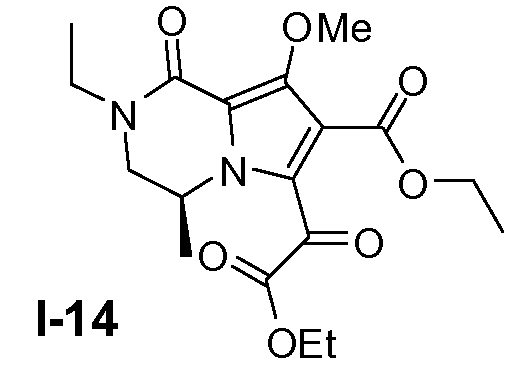
(S)-этил 6-бром-2-этил-8-метокси-4-метил-1-оксо-1,2,3,4-тетрагидропирроло[1,2-a]пиразин-7-карбоксилат (0,70 моль) растворяли в ТГФ (4500 мл) при 20°C. Добавляли молекулярные сита (4Å; 250 г). Смесь перемешивали в течение 30 минут при 20°C и затем охлаждали до -78°C. Добавляли по каплям раствор н-бутиллития (2,5M; 0,77 моль) ниже -65°C. Смесь перемешивали в течение 1 часа при -78°C. Добавляли по каплям диэтилоксалат (1,6 моль) в ТГФ (600 мл) ниже -65°C и затем смесь перемешивали в течение 3 часов. Смесь выливали в раствор водной H2SO4 (4200 мл, 2M) и ТГФ (3500 мл). Органический слой отделяли и водный слой снова экстрагировали этилацетатом (3×2000 мл). Объединенные органические слои собирали и упаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: петролейный эфир/этилацетат от 100:0 до 1:1). Чистые фракции собирали и органический растворитель выпаривали. Выход: 170 г (выход 67%).
Пример 14: промежуточное соединение I-15
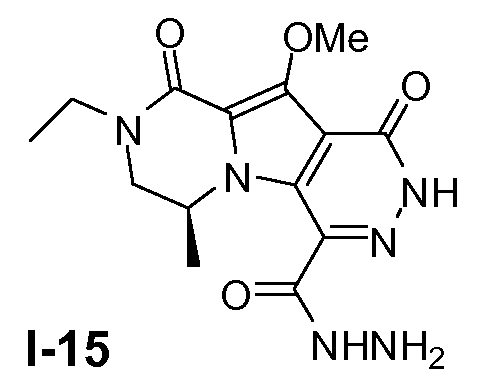
Промежуточное соединение I-14 (0,46 моль) растворяли в метаноле (2 л) при 20°C. Добавляли раствор гидразина в ТГФ (1M; 2,8 моль) при 10°C. Смесь перемешивали в течение 2 часов при 20°C. Растворитель удаляли в вакууме и полученный остаток использовали для следующей стадии без очистки. Выход: 180г неочищенного промежуточного соединения I-15 (чистота 86%).
Пример 15: промежуточное соединение I-16
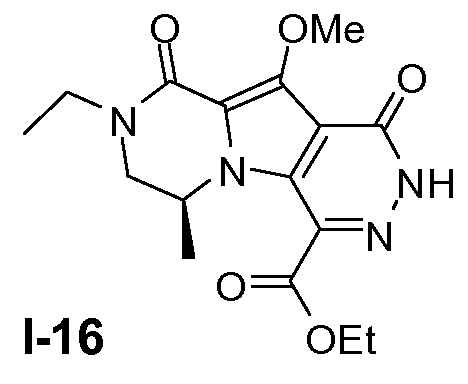
Неочищенное промежуточное соединение I-15 (0,46 моль) растворяли в безводном этаноле (3,5 л) при 20°C. Добавляли порциями NBS (1,2 моль), поддерживая температуру ниже 30°C. Смесь перемешивали в течение ночи при 25°C. Растворитель удаляли в вакууме и добавляли CH2Cl2. Смесь гасили 10% водным раствором Na2SO3 и затем промывали 10% раствором K2CO3 для удаления сукцинимида. Органический слой концентрировали досуха. Остаток очищали хроматографией на силикагеле (CH2Cl2/метанол от 100:0 до 10:1). Чистые фракции собирали и летучие вещества удаляли в вакууме. Неочищенное соединение промывали трет-бутилметиловым эфиром. Выход: 100 г (выход 62% для двух стадий).
Пример 16: промежуточное соединение I-17
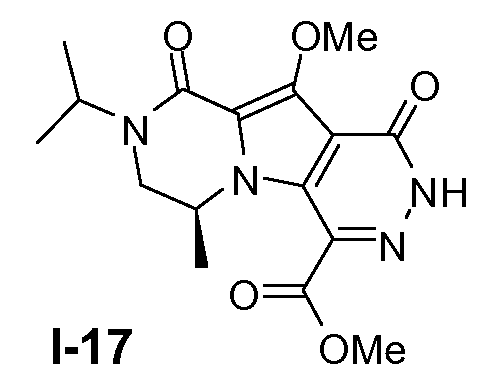
Промежуточное соединение I-17 получали из (S)-этил 6-бром-2-изопропил-8-метокси-4-метил-1-оксо-1,2,3,4-тетрагидропирроло[1,2-a]пиразин-7-карбоксилата аналогично тому, как описано в примерах 13-15.
Пример 17: промежуточное соединение I-18
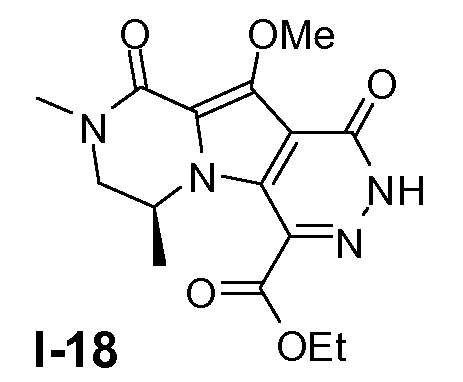
Промежуточное соединение I-18 получали из (S)-этил 6-бром-8-метокси-2,4-диметил-1-оксо-1,2,3,4-тетрагидропирроло[l,2-a]пиразин-7-карбоксилата аналогично тому, как описано в примерах 13-15.
Пример 18: диэтил 1-(2-(трет-бутоксикарбониламино)этил)-3-метокси-1Н-пиррол-2,4-дикарбоксилат (I-19)
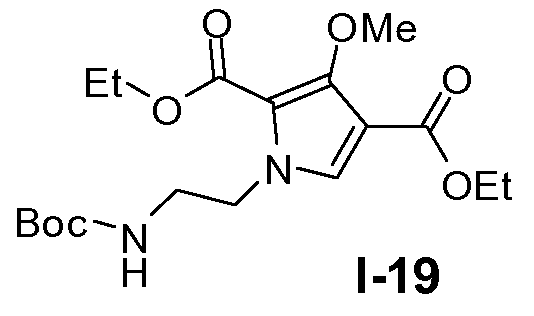
К раствору диэтил 3-метокси-1Н-пиррол-2,4-дикарбоксилата (93 ммоль) в ДМФА (340 мл) добавляли порциями 60% NaH (140 ммоль) при 0°C и реакционную смесь перемешивали при 25°C в течение 0,5 часа. Добавляли трет-бутиловый эфир 2,2-диоксо-[1,2,3]оксатиазолидин-3-карбоновой кислоты (93 ммоль) при 0°C и смеси давали нагреться до 25°C и перемешивали в течение 21 часа. Реакционную смесь гасили этанолом при 0°C. Растворитель выпаривали в вакууме. Выход: 35 г (выход 98%)
Пример 19: диэтил 1-(2-аминоэтил)-3-метокси-1Н-пиррол-2,4-дикарбоксилат, гидрохлорид (I-20)
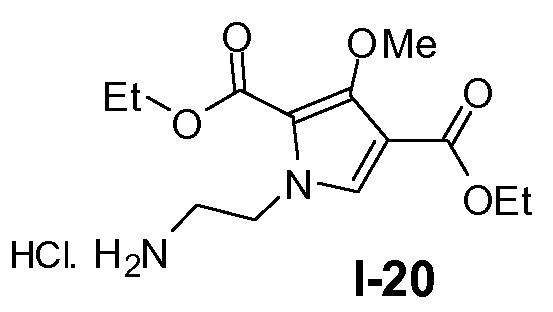
Раствор промежуточного соединения I-19 (91 ммоль) в 4 н. HCl в диоксане (350 мл) перемешивали при 60°C-70°C в течение 1 часа. Растворитель выпаривали в вакууме и продукт использовали как таковой на следующей стадии. Выход: 28,8 г (выход 99%)
Пример 20: этил 8-метокси-1-оксо-1,2,3,4-тетрагидропирроло[l,2-a]пиразин-7-карбоксилат (I-21)
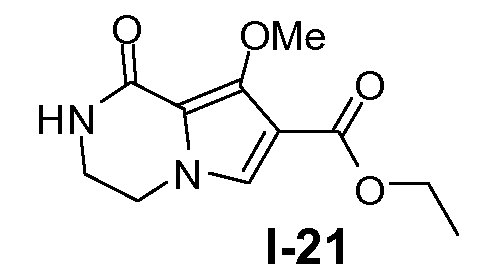
Смесь промежуточного соединения I-20 (90 ммоль), K2CO3 (463 ммоль) в этаноле (450 мл) нагревали при температуре кипения с обратным холодильником в течение 19 часов. Растворитель выпаривали в вакууме и к полученному остатку добавляли этилацетат. Смесь отфильтровывали. Фильтрат упаривали в вакууме и к остатку добавляли CH2Cl2. Полученный органический слой промывали насыщенным солевым раствором и упаривали в вакууме. Остаток очищали колоночной флэш-хроматографией (элюент: CH2Cl2, затем этилацетат). Чистую фракцию собирали и упаривали в вакууме. Остаток промывали трет-бутилметиловым эфиром с получением желаемого продукта. Выход: 12,7 г (выход 59%).
Пример 21: этил 2-этил-8-метокси-1-оксо-1,2,3,4-тетрагидропирроло[l,2-a]пиразин-7-карбоксилат (I-22)
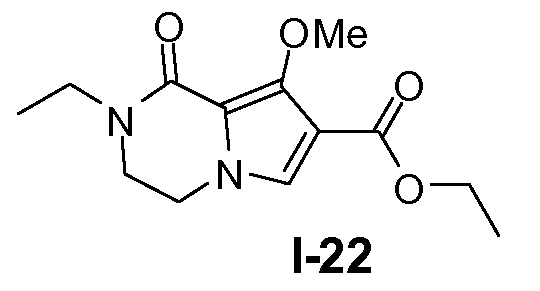
К раствору промежуточного соединения I-21 (53 ммоль) в ДМФА (400 мл) добавляли порциями 60% NaH (80 ммоль) при 0°C и реакционную смесь перемешивали при 25°C в течение 40 минут. Добавляли йодэтан (80 ммоль) при 0°C и реакционную смесь перемешивали при 25°C в течение 20 часов. Реакционную смесь гасили этанолом и затем водой при 0°C. Добавляли CH2Cl2. Органический слой отделяли, промывали насыщенным солевым раствором и сушили над Na2SO4. Выход: 14 г (выход 98%)
Пример 22: промежуточное соединение I-23
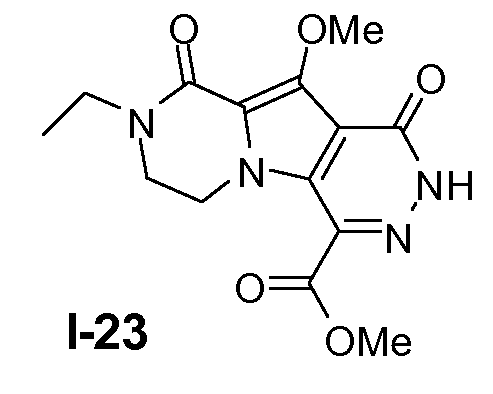
Промежуточное соединение I-23 получали из промежуточного соединения I-22 аналогично тому, как описано в примерах 12-15.
Пример 23: промежуточное соединение I-24
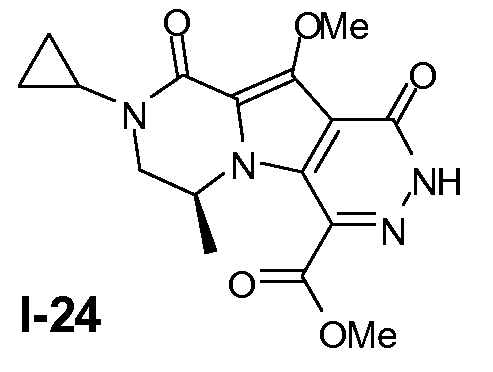
Промежуточное соединение I-24 получали из промежуточного соединения I-13 аналогично тому, как описано в примерах 13-15.
Пример 24: метил 2-(5-(трет-бутоксикарбониламино)пентилокси)-4-фторбензоат (I-25)

Реакцию проводили в атмосфере N2. Трифенилфосфин (0,52 моль) добавляли к раствору метил 4-фтор-2-гидроксибензоата (0,47 моль), трет-бутил 5-гидроксипентилкарбамата (0,47 моль) и диизопропилазодикарбоксилата (DIAD; 0,94 моль) в ТГФ (1 л) по каплям при -20°C. Реакционной смеси давали нагреться до комнатной температуры и перемешивали при комнатной температуре в течение 2 часов. Полученную смесь использовали непосредственно на следующей стадии без дополнительной очистки.
Пример 25: 2-(5-(трет-бутоксикарбониламино)пентилокси)-4-фторбензойная кислота (I-26)
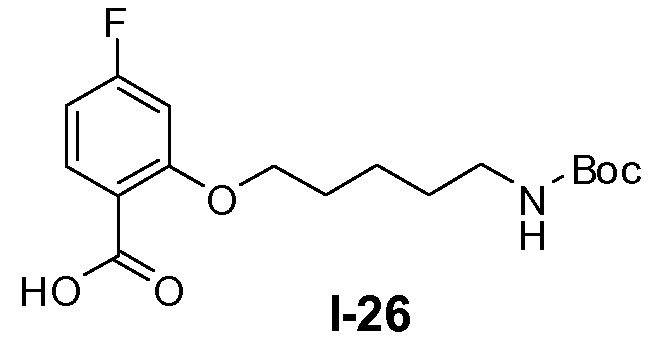
Метанол (500 мл) и воду (500 мл) добавляли к неочищенной реакционной смеси, полученной в примере 24. Добавляли гидрат LiOH (1,4 моль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем добавляли воду (500 мл) и органический растворитель выпаривали в вакууме. Смесь экстрагировали метил-трет-бутиловым эфиром для удаления примесей. Водный слой доводили до pH 8-9 и снова экстрагировали этилацетатом (3×500 мл). Водный слой доводили до pH 4-5 и добавляли этилацетат. Органический слой отделяли, промывали насыщенным солевым раствором, сушили и концентрировали в вакууме. Выход: 120 г (чистота 80%, выход от 2 стадий 75%).
Пример 26: трет-бутил 5-(5-фтор-2-(гидроксиметил)фенокси)пентилкарбамат (I-27)
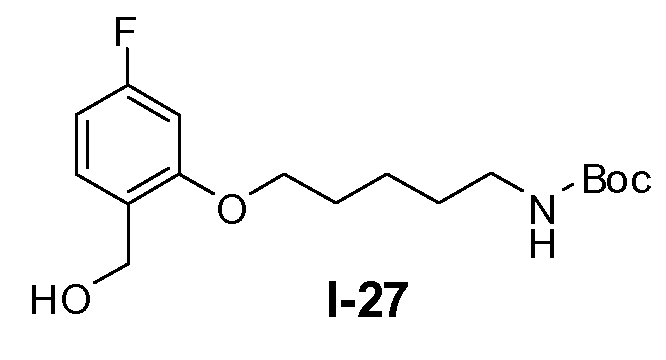
Промежуточное соединение I-26 (0,43 моль) растворяли в ТГФ (1 л). К раствору добавляли по каплям комплекс боран-Me2S (0,55 моль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 6 часов. К смеси добавляли по каплям метанол (500 мл) при 0°C. Органический растворитель выпаривали и остаток очищали колоночной флэш-хроматографией с использованием SiO2 (петролейный эфир/этилацетат от 100:0 до 2:1). Выход: 80 г (чистота 80%, выход 57%).
Пример 27: метил 2-(4-(трет-бутоксикарбониламино)бутокси)-4-фторбензоат (I-28)
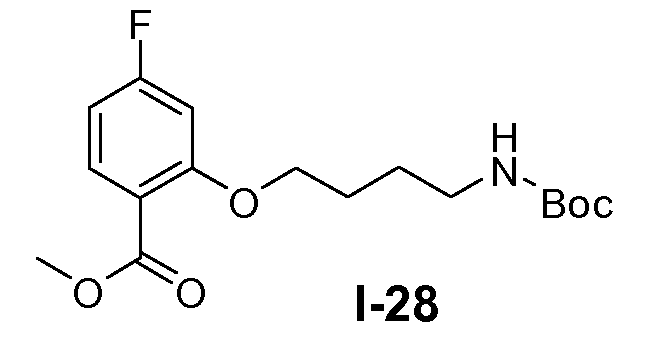
Метил 4-фтор-2-гидроксибензоат (21 ммоль), трет-бутил 4-гидроксибутилкарбамат (21 ммоль) и трифенилфосфин (24 ммоль) в ТГФ (70 мл) охлаждали до -10°C. Затем добавляли DIAD (43 ммоль). Смесь перемешивали в течение 30 минут при -10°C в атмосфере N2. Ледяную баню удаляли и смесь перемешивали в течение ночи при 20°C. Растворитель удаляли в вакууме. Остаток промывали петролейным и диэтиловым эфиром. Твердое вещество белого цвета фильтровали, и было подтверждено, что оно представляет собой оксид трифенилфосфина (ΡΡh3=O). Фильтрат упаривали досуха. Остаток очищали колоночной флэш-хроматографией (петролейный эфир/этилацетат от 100:0 до 5:1). Выход: 8,18 г (чистота 90%, выход 98%).
Пример 28: трет-бутил 4-(5-фтор-2-(гидроксиметил)фенокси)бутилкарбамат (I-29)
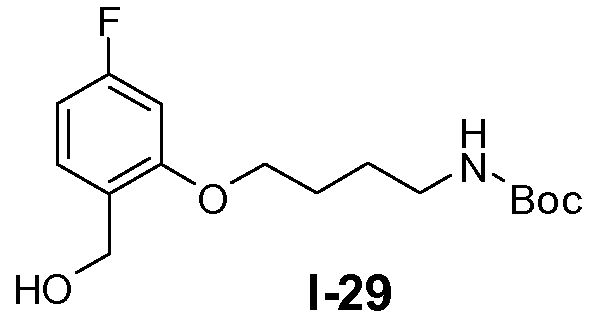
Промежуточное соединение I-28 (17 ммоль) и NaBH4 (188 ммоль) суспендировали в ТГФ. Полученную смесь перемешивали в течение 15 минут при 65°C. Добавляли по каплям метанол (20 мл) в течение 0,5 часа. Перемешивание при 65°C продолжали в течение ночи. Затем реакционную смесь охлаждали до 15°C и гасили насыщенным водным раствором NH4C1 (120 мл). Перемешивание продолжали в течение 1 часа. Добавляли этилацетат (200 мл) и органический слой отделяли. Водный слой экстрагировали этилацетатом (2×200 мл). Органические слои объединяли и сушили над Na2SO4. Растворитель удаляли в вакууме с получением бесцветного масла. Остаток очищали колоночной флэш-хроматографией с использованием SiO2, с элюентом (петролейный эфир/этилацетат от 100:0 до 80:20). Выход: 3,1 г (чистота 98%, выход 58%).
Пример 29: трет-бутил 5-(4-хлор-5-фтор-2-(гидроксиметил)фенокси)пентилкарбамат (I-30)
Промежуточное соединение I-30 получали из метил 5-хлор-4-фтор-2-гидроксибензоата и трет-бутил 5-гидроксипентилкарбамата аналогично тому, как описано в примерах 27-28.
Пример 30: трет-бутил 5-(4-хлор-2-(гидроксиметил)фенокси)пентилкарбамат (I-31)
Промежуточное соединение I-31 получали из метил 5-хлор-2-гидроксибензоата и трет-бутил 5-гидроксипентилкарбамата аналогично тому, как описано в примерах 24-26.
Пример 31: 5-(2,4-диметоксибензиламино)пентан-1-ол (I-32)
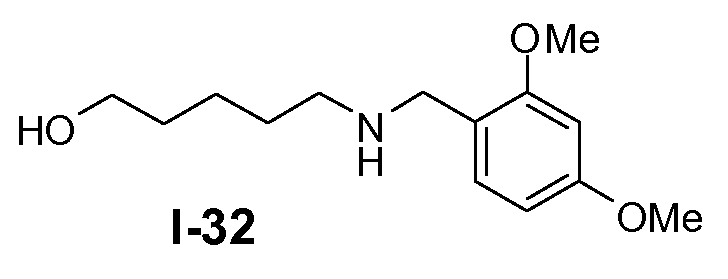
Раствор 5-аминопентан-1-ола (194 ммоль), 2,4-диметоксибензальдегида (213 ммоль) и уксусной кислоты (213 ммоль) в метаноле (500 мл) перемешивали при 10°C в течение 0,5 часа. Добавляли порциями NaBH4 (22 г) при -10°C и смесь перемешивали в течение 10 минут. Затем реакционной смеси давали нагреться до 10°C и перемешивали в течение 1,5 часов. Добавляли насыщенный раствор NaHCO3 и смесь перемешивали в течение 0,5 часа. Смесь экстрагировали с использованием CH2Cl2 (3×100 мл). Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением желтого масла. Выход: 46 г (чистота 69%, выход 65%).
Пример 32: трет-бутил 2,4-диметоксибензил(5-гидроксипентил)карбамат (I-33)
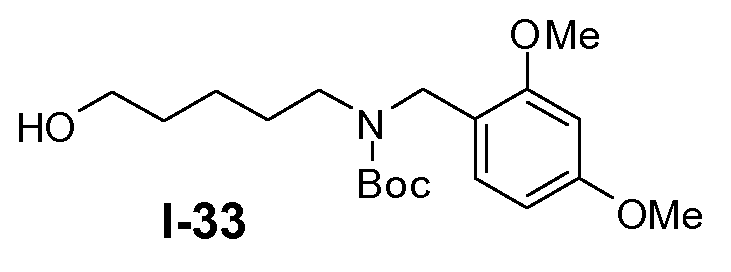
Раствор промежуточного соединения I-32 (182 ммоль) и Boc2O (182 ммоль) в метаноле (1 л) перемешивали при 10°C в течение ночи. Реакционную смесь упаривали в вакууме, остаток очищали SiO2 колоночной флэш-хроматографией с использованием SiO2 (элюент: петролейный эфир/этилацетат от 8:1 до 2:1), получая чистое промежуточное соединение I-33. Выход: 43 г (чистота 95%, выход 67%).
Пример 33: трет-бутил 5-бромпентил(2,4-диметоксибензил)карбамат (I-34)
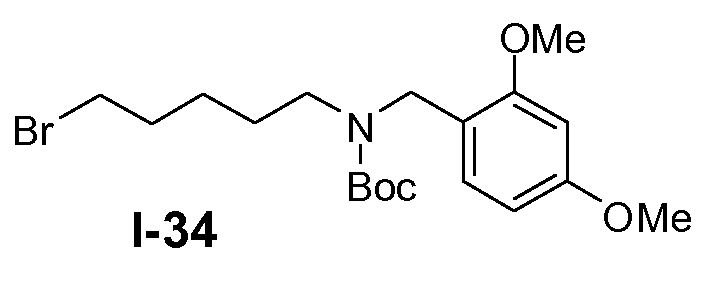
Трифенилфосфин (37 ммоль) добавляли к смеси промежуточного соединения I-33 (28 ммоль) в CH2Cl2 (200 мл) при 0°C в атмосфере N2. Затем к смеси добавляли по каплям CBr4 (37 ммоль) при 0°C. Смеси давали нагреться до 20°C и перемешивали в течение 2 часов. Смесь упаривали досуха и очищали колоночной флэш-хроматографией с использованием SiO2 (элюент: петролейный эфир/этилацетат от 20:1 до 10:1), получая чистое промежуточное соединение I-34. Выход: 9,0 г (чистота 95%, выход 76%).
Пример 34: метил 4,5-дифтор-2-гидроксибензоат (I-35)
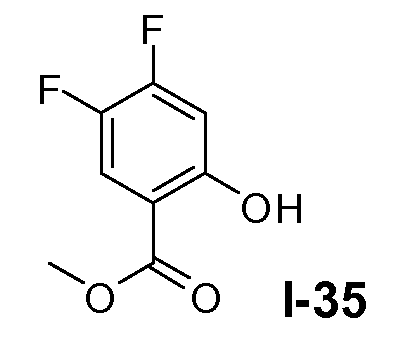
Смесь 4,5-дифтор-2-гидроксибензойной кислоты (20 ммоль), метанола (100 мл) и концентрированной H2SO4 (5 мл) нагревали при температуре кипения с обратным холодильником в течение 48 часов. Затем реакционную смесь концентрировали. Остаток растворяли в этилацетате, промывали насыщенным солевым раствором и насыщенным раствором NaHCO3 и нейтрализовали лимонной кислотой. Органический слой сушили над MgSO4 и концентрировали в вакууме с получением продукта. Выход: 2,8 г (чистота 97%, выход 74%).
Пример 35: метил 2-(5-(трет-бутоксикарбонил(2,4-диметоксибензил)амино)пентилокси)-4,5-дифторбензоат (I-36)
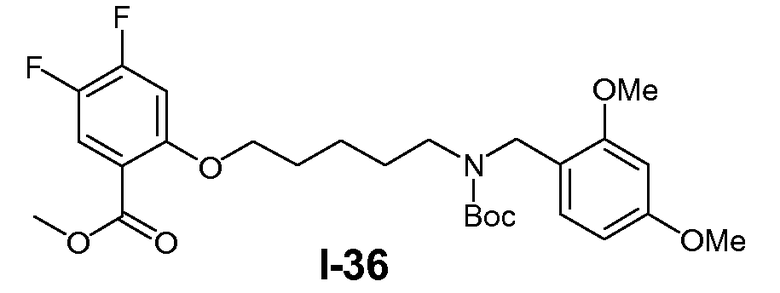
Смесь промежуточного соединения I-34 (11 ммоль), промежуточного соединения I-35 (11 ммоль) и Cs2CO3 (34 ммоль) в ДМФА (50 мл) перемешивали при комнатной температуре в течение ночи. Затем смесь разбавляли CH2Cl2 и фильтровали. Фильтрат промывали насыщенным солевым раствором, сушили над Na2SO4, концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (элюент: петролейный эфир/этилацетат от 16:1 до 8:1) с получением чистого промежуточного соединения I-36. Выход: 5,0 г (чистота 95%, выход 86%).
Пример 36: трет-бутил 5-((4,5-дифтор-2-гидроксиметил)фенокси)пентил-(2,4-диметоксибензил)карбамат (I-37)
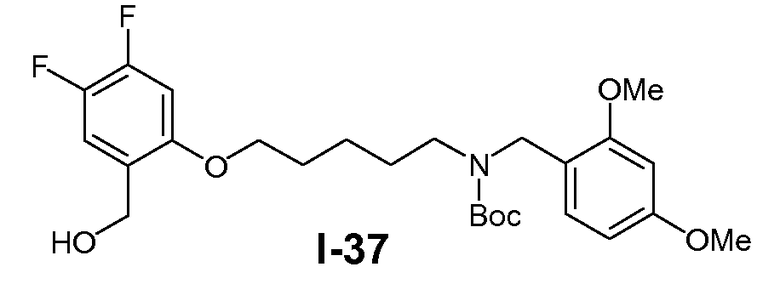
Смесь промежуточного соединения I-36 (9,5 ммоль) и LiAlH4 (38 ммоль) в безводном ТГФ (100 мл) перемешивали при комнатной температуре в течение 4 часов. Смесь гасили 0,8 мл 10% раствора NaOH в воде. Смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4. Раствор концентрировали с получением неочищенного промежуточного соединения I-37. Остаток очищали колоночной флэш-хроматографией (элюент: петролейный эфир/этилацетат 2:1) с получением чистого промежуточного соединения I-37. Выход: 3,6 г (чистота 85%, выход 75%).
Пример 37: метил 4-фтор-2-(4-метоксибензилокси)бензоат (I-38)
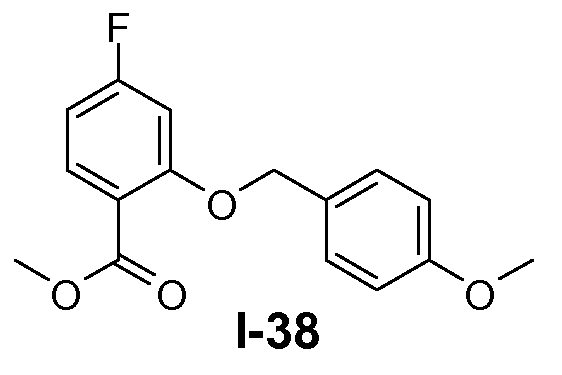
Метил 4-фтор-2-гидроксибензоат (0,88 моль) растворяли в CH3CN (1,5 л). Добавляли пара-метоксибензилхлорид (1,3 моль) и K2CO3 (2,2 моль). Смесь нагревали при температуре кипения с обратным холодильником в течение 12 часов. Смесь фильтровали для удаления твердого вещества. Растворитель удаляли в вакууме. Полученный остаток очищали колоночной флэш-хроматографией (элюент: CH2Cl2). Выход 270 г (чистота 90%, выход 95%).
Пример 38: (4-фтор-2-(4-метоксибензилокси)фенил)метанол (I-39)
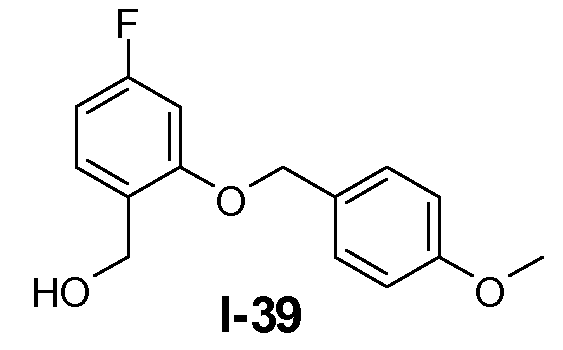
Промежуточное соединение I-38 (0,93 моль) и NaBH4 (9,3 моль) суспендировали в ТГФ (4,1 л). Полученную смесь перемешивали в течение 15 минут при 65°C. Добавляли по каплям метанол (1,3 л) в течение 1,5 часов и смесь перемешивали в течение 3 часов при 65°C. Реакционную смесь охлаждали до 15°C и гасили насыщенным водным раствором NH4C1 (3500 мл). Затем смесь перемешивали в течение 20 минут. Органический слой отделяли и водный слой экстрагировали этилацетатом (3×1500 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Выход 150 г (выход 61%).
Пример 39: метил 2-(аллилокси)-4-фторбензоат (I-40)
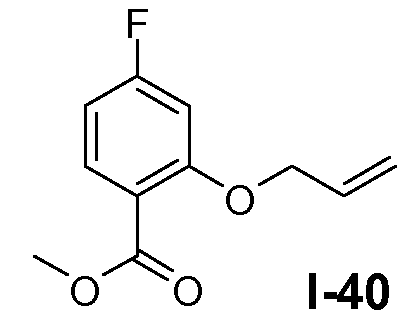
Метил 4-фтор-2-гидроксибензоат (0,27 моль), K2CO3 (0,41 моль) и KI (27 ммоль) загружали в реактор на 1 л в атмосфере N2. Добавляли ацетон (500 мл) с последующим добавлением 3-бром-пропена (0,33 моль). Смесь нагревали при температуре кипения с обратным холодильником в течение 6 часов в атмосфере N2. Смесь охлаждали до комнатной температуры и фильтровали для удаления твердого вещества. Растворитель удаляли в вакууме и остаток растворяли в воде (200 мл) и CH2Cl2 (300 мл). Органический и водный слои разделяли и водную фазу экстрагировали с использованием CH2Cl2. Органические слои объединяли и сушили с помощью Na2SO4. Растворитель удаляли в вакууме с получением промежуточного соединения I-40 в виде белого твердого вещества.
Пример 40: 1-(бромметил)-4-фтор-2-(4-метоксибензилокси)бензол (I-41)
Трифенилфосфин (30 ммоль) и CBr4 (30 ммоль) добавляли к промежуточному соединению I-39 (23 ммоль) в CH2Cl2 (120 мл) при 0°C и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь очищали колоночной флэш-хроматографией (элюент: CH2Cl2) с получением чистого продукта. Выход: 8,2 г (выход 99%)
Пример 41: (2-(аллилокси)-4-фторфенил)метанол (I-42)
Промежуточное соединение I-40 (0,24 моль) и NaBH4 (1,2 моль) суспендировали в диметоксиэтане (DME, 350 мл) в атмосфере N2. Полученную смесь перемешивали в течение 15 минут при 65°C-75°C. Добавляли по каплям метанол (175 мл) и смесь перемешивали в течение 14 часов при 65°C. Реакционную смесь охлаждали до 15°C и гасили насыщенным водным раствором NH4Cl (150 мл). Смесь экстрагировали этилацетатом (400 мл). Органический слой отделяли и водный слой экстрагировали этилацетатом (2×200 мл). Объединенные органические слои сушили над Na2SO4. Растворитель удаляли в вакууме с получением промежуточного соединения I-42 в виде твердого вещества белого цвета.
Пример 42: 4-(карбамимидоилтио)бутилпивалат, гидробромид (I-43)
4-Бромбутилпивалат (0,71 моль) и тиомочевину (0,78 моль) растворяли в этаноле (330 мл) и полученную смесь нагревали при температуре кипения с обратным холодильником в течение 1 часа. Растворитель удаляли в вакууме. Полученный остаток использовали на следующей стадии без дополнительной очистки. Выход: 210 г (выход 95%).
Пример 43: 4-(хлорсульфонил)бутилпивалат (I-44)
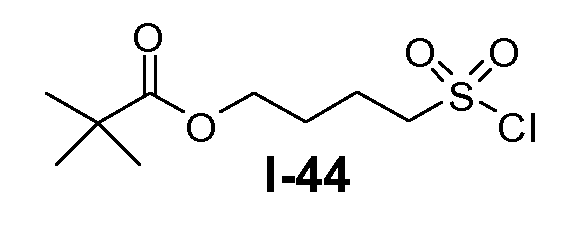
Неочищенное промежуточное соединение I-43 (0,38 моль) растворяли в воде (1,2 л). Добавляли этилацетат (200 мл) и в полученную смесь барботировали газообразный Cl2 при 0°C в течение 1 часа, затем барботировали N2 в течение 2 часов. Смесь экстрагировали этилацетатом (2×800 мл). Органический слой промывали 5% раствором NaHSO3 и насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Выход: 95 г (чистота 90%, выход 97%).
Пример 44: 4-(N-метилсульфамоил)бутилпивалат (I-45)
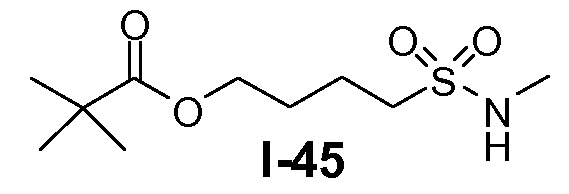
Промежуточное соединение I-44 (0,37 моль) и метиламин.HCl (1,5 моль) суспендировали в CH2Cl2 (1,5 л) и охлаждали до 0°C. Добавляли по каплям K2CO3 (1,5 моль) в воде (750 мл). Смесь перемешивали при 20°C в течение 2 часов. Органический слой промывали насыщенным солевым раствором и сушили (Na2SO4). Растворитель удаляли в вакууме. Остаток очищали колоночной хроматографией с использованием SiO2 (элюент: петролейный эфир/этилацетат 3:1). Выход: 65 г (чистота 90%, выход 70%).
Пример 45: 4-(N-этилсульфамоил)бутилацетат (I-46)
Промежуточное соединение I-46 получали из 4-бромбутилацетата аналогично тому, как описано в примерах 42-44.
Пример 46: 4-(N-циклопропилсульфамоил)бутил пивалат (I-47)
Промежуточное соединение I-47 получали из промежуточного соединения I-44 аналогично тому, как описано в примере 44.
Пример 47: 4-(N-метилсульфамоил)бутилацетат (I-48)
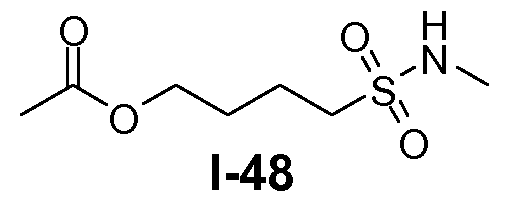
Промежуточное соединение I-48 получали из 4-бромбутилацетата аналогично тому, как описано в примерах 42-44.
Пример 48: 5-(N-метилсульфамоил)пентилпивалат (I-49)
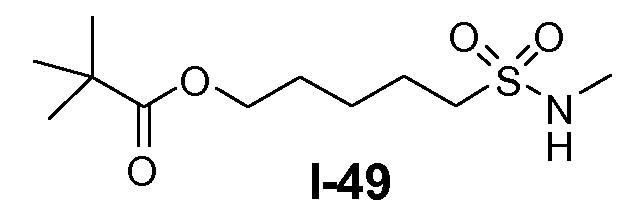
Промежуточное соединение I-49 получали из 4-бромпентилпивалата аналогично тому, как описано в примерах 42-44.
Пример 49: 5-(N-циклопропилсульфамоил)пентилпивалат (I-50)
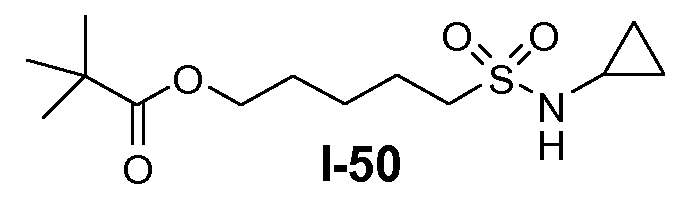
Промежуточное соединение I-50 получали из 4-бромпентилпивалата аналогично тому, как описано в примерах 42-44.
Пример 50: 5-(N-этилсульфамоил)пентилацетат (I-51)
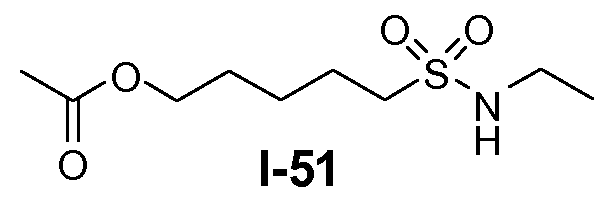
Промежуточное соединение I-51 получали из 5-бромпентилацетата, аналогично тому, как описано в примерах 42-44.
Пример 51: 5-(N-метилсульфамоил)пентилацетат (I-52)
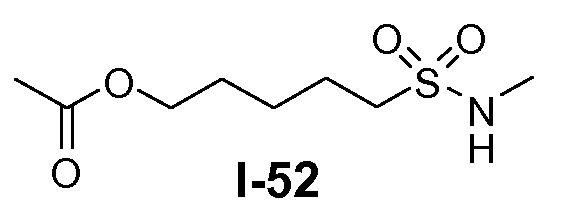
Промежуточное соединение I-52 получали из 5-бромпентилацетата, аналогично тому, как описано в примерах 42-44.
Пример 52: н-метилбут-3-ен-1-сульфонамид (I-53)
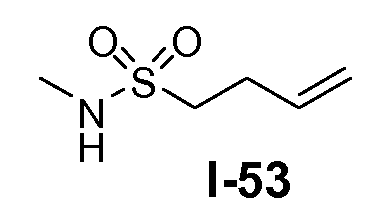
Промежуточное соединение I-53 получали из бут-3-ен-1-сульфонилхлорида аналогично тому, как описано в примере 44.
Пример 53: н-метилпент-4-ен-1-сульфонамид (I-54)
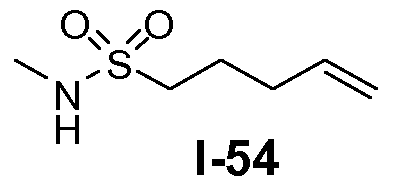
Промежуточное соединение I-54 получали из пент-4-ен-1-сульфонилхлорида аналогично тому, как описано в примере 44.
Пример 54: 3-(N-метилсульфамоил)пропилацетат (I-55)
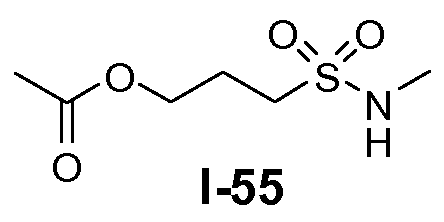
Промежуточное соединение I-55 получали из 3-(хлорсульфонил)пропилацетата аналогично тому, как описано в примере 44.
Пример 55: 3-азидо-N-метилпропан-1-сульфонамид (I-56)
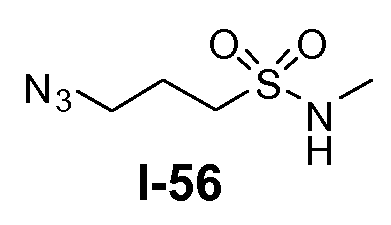
Смесь 3-хлор-N-метилпропан-1-сульфонамида (29 ммоль), NaN3 (29 ммоль) в ДМФА (50 мл) перемешивали при 100°C в течение 2 часов. Смесь выливали в 40 мл насыщенного солевого раствора. Смесь экстрагировали диэтиловым эфиром (3×30 мл). Органические слои объединяли и промывали 20 мл насыщенного солевого раствора. Органический слой сушили с помощью Na2SO4, фильтровали и фильтрат упаривали. Выход: 4,2 г (чистота 90%, выход 81%).
Пример 56: трет-бутил 3-(N-метилсульфамоил)пропилкарбамат (I-57)
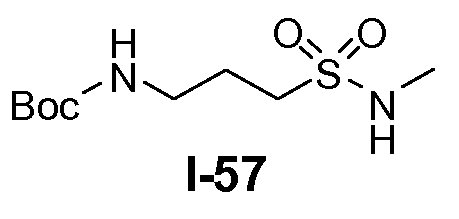
Смесь промежуточного соединения I-56 (14 ммоль), 10% Pd/C (1 г), ди-трет-бутилдикарбоната (Boc2O; 14 ммоль) в метаноле (25 мл) гидрировали при 25°C в течение 2 часов. После поглощения водорода (3 экв.) катализатор отфильтровывали. Фильтрат упаривали. Остаток очищали колоночной хроматографией с использованием SiO2 (элюент: петролейный эфир/этилацетат 3:1). Выход: 2 г (чистота 90%, выход 56%).
Пример 57: трет-бутил 4-(N-метилсульфамоил)бутилкарбамат (I-58)
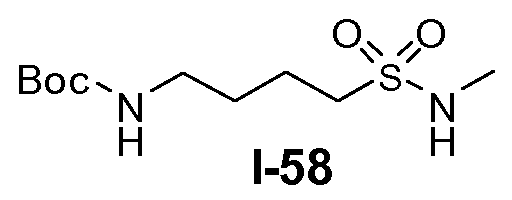
Промежуточное соединение I-58 получали из 4-хлор-N-метилбутан-1-сульфонамида аналогично тому, как описано в примерах 55-56.
Пример 58: 4-циано-N-метилбутан-1-сульфонамид (I-59)
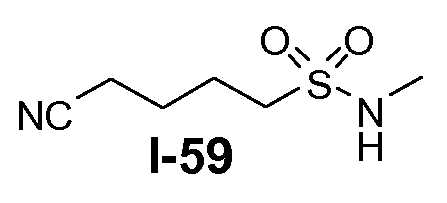
4-Хлор-N-метилбутан-1-сульфонамид (54 ммоль), KCN (108 ммоль) и 18-краунэфир-6 (11 ммоль) растворяли в CH3CN (100 мл). Смесь перемешивали при температуре кипения с обратным холодильником в течение ночи. Смесь охлаждали и фильтровали. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюент: CH2Cl2, затем этилацетат). Выход: 3,2 г (чистота 70%).
Пример 59: 5-амино-N-метилпентан-1-сульфонамид (I-60)
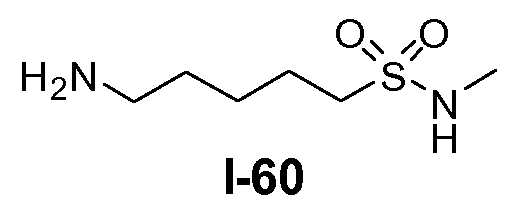
Промежуточное соединение I-59 (8,5 ммоль) растворяли в безводном ТГФ (30 мл) и смесь перемешивали при 0°C. Добавляли по каплям комплекс боран-Me2S (34 ммоль). Реакционную смесь перемешивали при комнатной температуре. Смесь охлаждали до 0°C и добавляли по каплям метанол вплоть до прекращения выделения газа. Растворитель выпаривали. Неочищенный продукт использовали для следующей стадии без дополнительной очистки. Выход: 1,5 г (выход 97%).
Пример 60: трет-бутил 5-(N-метилсульфамоил)пентилкарбамат (I-61)
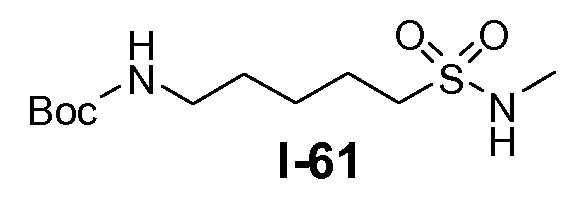
Промежуточное соединение I-60 (8,3 ммоль) растворяли в метаноле (50 мл). Смесь перемешивали при 0°C. Добавляли порциями Boc2O (8,3 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока завершение реакции не подтвердится методом тонкослойной хроматографии (ТСХ; петролейный эфир/этилацетат 5:1). Растворитель выпаривали. Неочищенный продукт промывали петролейным эфиром. Добавляли CH2Cl2 и органический слой промывали насыщенным раствором NaHCO3, насыщенным солевым раствором. Органический слой сушили и упаривали. Выход: 1,7 г (выход 73%).
Пример 61: трет-бутил 2,4-диметоксибензил(5-оксопентил)карбамат (I-62)
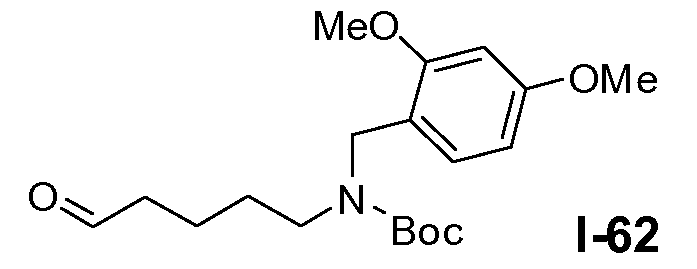
К перемешиваемому раствору оксалилхлорида (7,8 ммоль) в CH2Cl2 (13,5 мл) при температуре от -65°C до -70°C в атмосфере N2 добавляли диметилсульфоксид (ДМСО; 15,6 ммоль) в CH2Cl2 (4 мл). Через 2 минуты добавляли раствор промежуточного соединения I-33 (7,1 ммоль) в CH2Cl2 (7 мл). Через 20 минут добавляли триэтиламин (35 ммоль). Через 10 минут смеси давали нагреться до 10°C и добавляли воду (32,5 мл). После разделения водный слой экстрагировали с помощью CH2Cl2 (2×) и объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель выпаривали при пониженном давлении. Остаток сушили и использовали на следующей стадии без дополнительной очистки. Продукт получали в виде масла коричневого цвета. Выход: 2,4 г (выход 97%).
Пример 62: трет-бутил 2,4-диметоксибензил(6-оксогексил)карбамат (I-63)
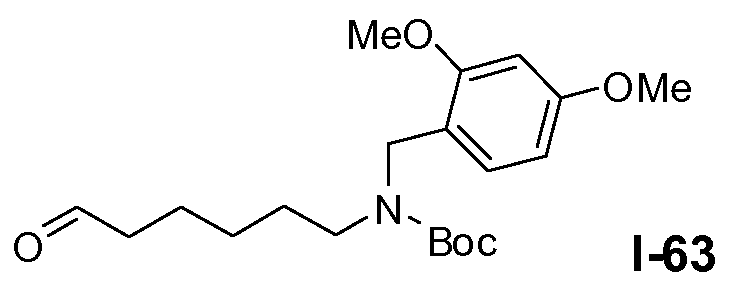
Промежуточное соединение I-63 получали из 6-аминогексан-1-ола аналогично тому, как описано в примерах 31-32 и 61.
Пример 63: трет-бутил 6-(трет-бутилдиметилсилилокси)гексилкарбамат (I-64)
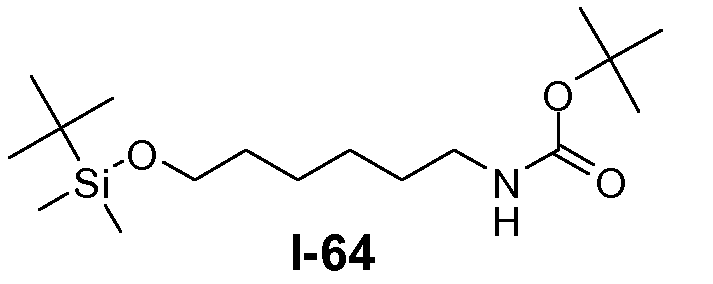
К раствору трет-бутил 6-гидроксигексилкарбамата (21 ммоль) в CH2Cl2 (30 мл) добавляли имидазол (27 ммоль) при 0°C. Реакционную смесь поддерживали при 25°C и перемешивали в течение 20 минут, затем охлаждали до 0°C. К полученной реакционной смеси добавляли по каплям трет-бутилдиметилхлорсилан (23 ммоль) в течение 30 минут при 0°C. Реакционной смеси давали нагреться до 25°C и перемешивали в течение 16 часов и затем фильтровали. Фильтрат промывали 1 н. раствором HCl (2×25 мл) и насыщенным солевым раствором (30 мл), сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией с использованием SiO2 (элюент: петролейный эфир и затем CH2Cl2). Выход 6,53 г (выход 87%).
Пример 64: трет-бутил 6-(трет-бутилдиметилсилилокси)гексил(метил)карбамат (I-65)
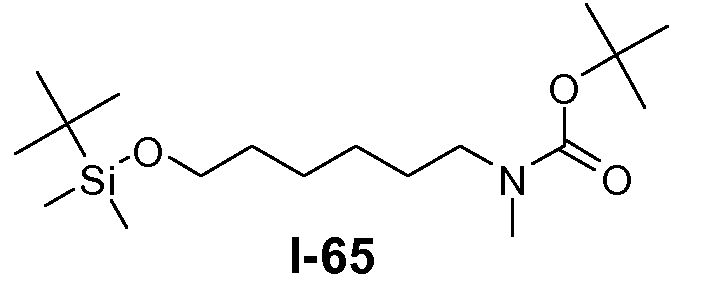
К раствору промежуточного соединения I-64 (18 ммоль) в безводном ТГФ (100 мл) добавляли порциями 60% раствор NaH (91 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 0,5 часа. К полученной реакционной смеси добавляли по каплям йодметан (54 ммоль) при 0°C. Реакционной смеси давали нагреться до 25°C и перемешивали в течение 16 часов. Добавляли по каплям насыщенный раствор NH4Cl (100 мл) при 0°C и смесь перемешивали в течение 0,5 часа и фильтровали. Фильтрат экстрагировали трет-бутилметиловым эфиром (3×50 мл). Объединенные органические части промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией с использованием SiO2 (элюент: петролейный эфир и затем петролейный эфир/этилацетат 10:1). Выход: 5,67 г (выход 85%).
Пример 65: трет-бутил 6-гидроксигексил(метил)карбамат (I-66)
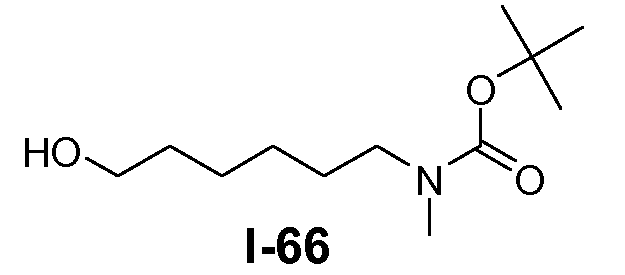
К раствору промежуточного соединения I-65 (15 ммоль) в ТГФ (85 мл) добавляли по каплям тетра-н-бутиламмонийфторид (TBAF) в виде 1M раствора в ТГФ (30 ммоль) при 0°C и реакционную смесь перемешивали в течение 2 часов. Добавляли дополнительное количество TBAF (15 ммоль) и реакционную смесь перемешивали в течение 16 часов при 25°C. Органический растворитель выпаривали в вакууме и добавляли ледяную воду (70 мл). Полученную смесь перемешивали в течение 1 часа. Смесь разделяли и органический слой промывали петролейным эфиром при -20°C. Остаток очищали колоночной флэш-хроматографией (элюент: петролейный эфир и затем петролейный эфир/этилацетат 10:1). Выход: 2,1 г (выход 60%).
Пример 66: трет-бутил метил(6-оксогексил)карбамат (I-67)
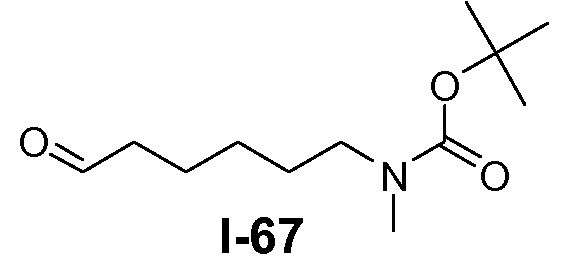
Промежуточное соединение I-67 получали, следуя процедуре, аналогичной той, которая описана в примере 61, исходя из промежуточного соединения I-66.
Пример 67: метил 5-хлор-4-фтор-2-(4-метоксибензилокси)бензоат (I-68)
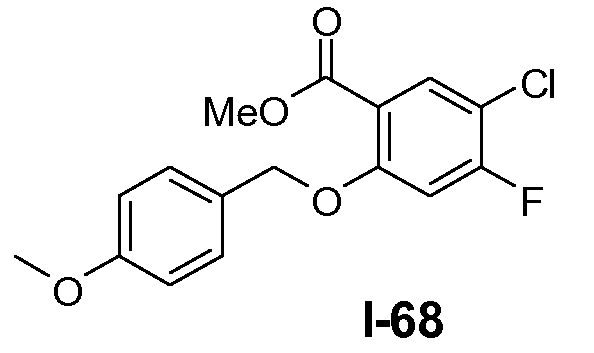
Промежуточное соединение I-68 получали из метил 5-хлор-4-фтор-2-гидроксибензоата аналогично тому, как описано в примере 37.
Пример 68: (5-хлор-4-фтор-2-(4-метоксибензилокси)фенил)метанол (I-69)
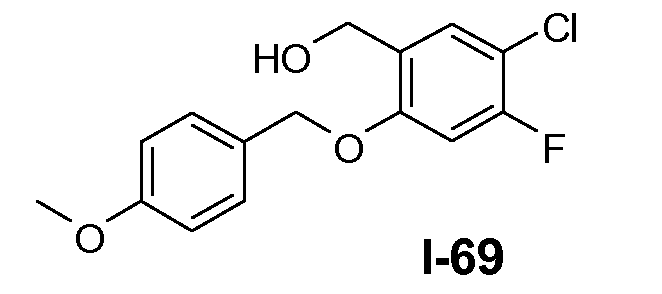
Промежуточное соединение I-68 (20,3 ммоль) и LiBH4 (203 ммоль) суспендировали в ТГФ. Полученную смесь перемешивали в течение 15 минут при 65°C. Добавляли по каплям метанол (100 мл) в течение 0,5 часа. Перемешивание при 65°C поддерживали в течение ночи. Реакционную смесь охлаждали до 15°C и гасили насыщенным водным раствором NH4C1 (100 мл). Перемешивание продолжали еще в течение 1 часа. Органический слой отделяли и водный слой экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали колоночной флэш-хроматографией с использованием SiO2 (элюент: петролейный эфир/этилацетат от 100:0 до 0:100), получая целевой продукт. Выход: 5,7 г (выход 95%).
Пример 69: метил 4,5-дифтор-2-(4-метоксибензилокси)бензоат (I-70)
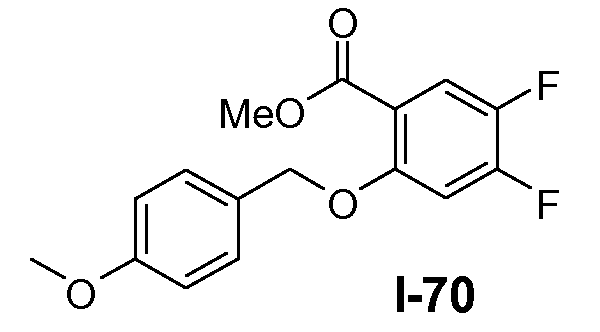
Промежуточное соединение I-70 получали из промежуточного соединения I-35 аналогично тому, как описано в примере 37.
Пример 70: (4,5-дифтор-2-(4-метоксибензилокси)фенил)метанол (I-71)
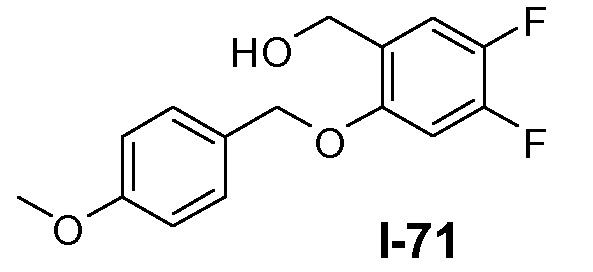
LiAlH4 (20 ммоль) добавляли к раствору промежуточного соединения I-70 (4,9 ммоль) в безводном ТГФ (30 мл). Смесь перемешивали при температуре окружающей среды в течение 4 часов. Реакционную смесь гасили 10% водным раствором NaOH (0,5 мл) и водой (2 мл). Смесь отфильтровывали и фильтрат экстрагировали этилацетатом. Органический слой сушили над Na2SO4 и растворитель выпаривали. Остаток очищали колоночной флэш-хроматографией с использованием SiO2 (петролейный эфир/этилацетат от 100:0 до 50:50). Желаемую фракцию собирали и чистый продукт сушили в вакууме. Выход: 1,1 г (выход 80%).
Пример 71: промежуточное соединение I-72
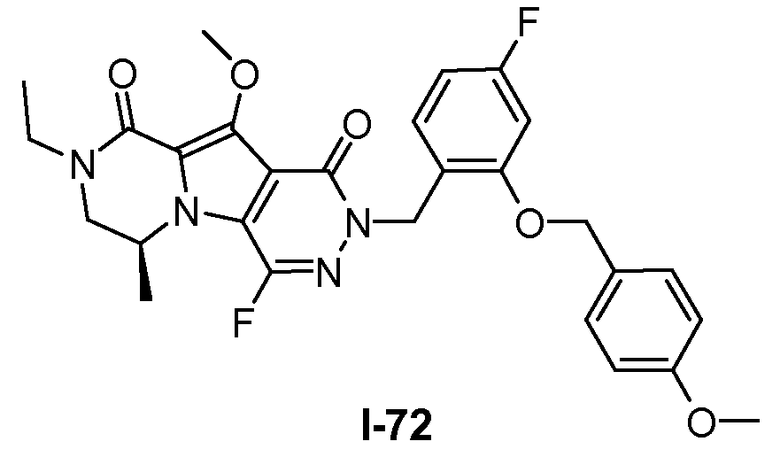
Промежуточное соединение I-2 (0,27 моль), промежуточное соединение I-39 (0,33 моль) и трифенилфосфин (0,41 моль) растворяли в безводном ТГФ (1,6 л) при 20°C. Добавляли DIAD (0,54 моль), поддерживая температуру реакции ниже -5°C. Смесь перемешивали в течение 5 часов при 0-10°C. Смесь упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (элюент: петролейный эфир/этилацетат от 100:1 до 0:100). Чистые фракции собирали и органический растворитель выпаривали. Выход: неочищенное промежуточное соединение I-72 200 г (чистота 50%), содержащее PPh3=O.
Альтернативно, можно следовать описанной ниже процедуре:
К раствору промежуточного соединения I-2 (32 ммоль) в ДМФА (30 мл) добавляли бис(триметилсилил)амид лития (LiHMDS; 1M раствор в ТГФ; 39 ммоль) при 0°C. Смесь перемешивали в течение 0,5 часа и затем обрабатывали свежеполученной партией промежуточного соединения I-41 (39 ммоль) в виде одной порции. Смеси давали нагреться до 20°C и перемешивали в течение ночи. Растворитель удаляли в вакууме. Остаток растворяли в CH2Cl2 (60 мл), раствор промывали 1 н. раствором HCl (60 мл) и насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали колоночной флэш-хроматографией (градиентное элюирование: CH2Cl2/метанол от 100:0 до 9:1). Выход: 10 г (чистота 90%, выход 58%).
Пример 72: промежуточное соединение I-73
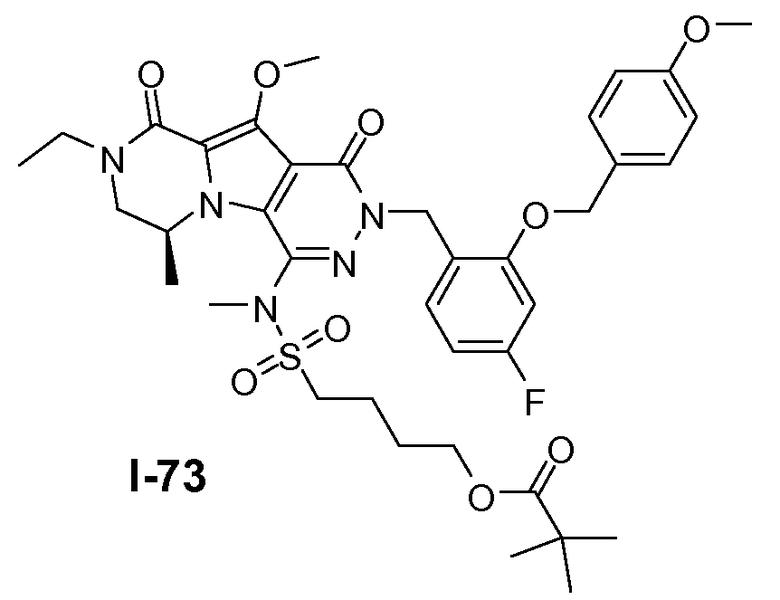
Промежуточное соединение I-72 (0,15 моль; чистота 50%), промежуточное соединение I-45 (0,18 моль) и CsCO3 (0,25 моль) в ДМСО (800 мл) перемешивали в течение ночи при 60°C. Добавляли насыщенный солевой раствор (800 мл). Смесь экстрагировали этилацетатом (3×400 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×200 мл) и сушили над Na2SO4. Растворитель удаляли в вакууме. Полученный остаток промывали трет-бутилметиловым эфиром. Выход: 70 г (чистота 80%).
Пример 73: промежуточное соединение I-74
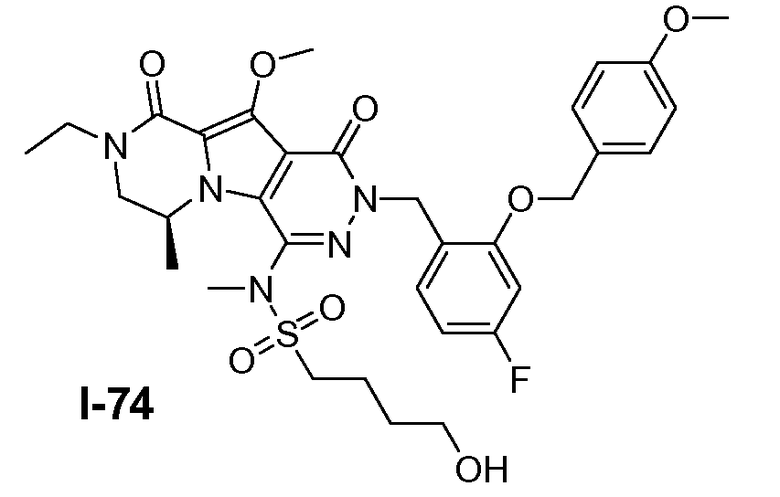
Смесь промежуточного соединения I-73 (91 ммоль) и NaOH (455 ммоль) в метаноле (700 мл) перемешивали в течение ночи при 20°C. Смесь упаривали. Добавляли 10% раствор лимонной кислоты до достижения pH 3. Смесь экстрагировали с помощью CH2Cl2 (2×800 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюент: петролейный эфир/этилацетат 100:1, затем 1:100 об./об.). Полученное твердое вещество дополнительно промывали трет-бутилметиловым эфиром. Выход: 42 г (общий выход от трех стадий 36%).
Пример 74: промежуточное соединение I-75
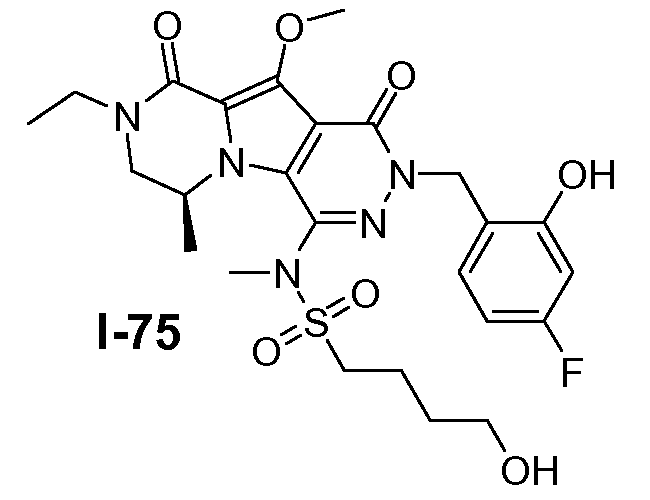
Смесь промежуточного соединения I-74 (61 ммоль) растворяли в CH2Cl2 (420 мл). Смесь охлаждали до 0°C. Добавляли по каплям раствор HCl в диоксане (4 н; 336 ммоль). Смесь перемешивали при 25°C в течение 1 часа. Растворитель удаляли в вакууме. Полученное твердое вещество промывали метанолом. Выход: 30 г (выход 87%).
Пример 75: макроциклизация (I-76)
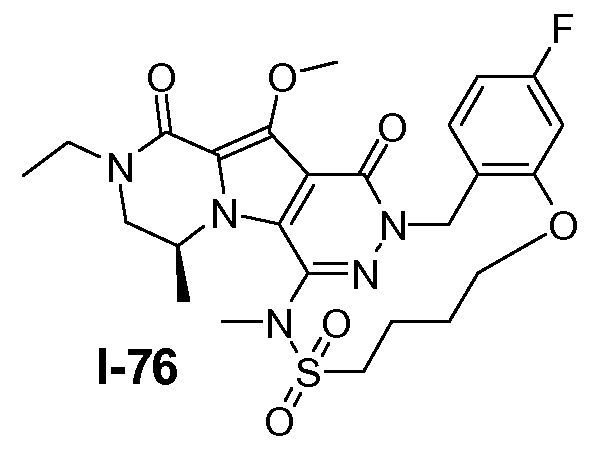
Раствор трифенилфосфина (265 ммоль) в безводном ТГФ (1500 мл) охлаждали до 0°C. Добавляли по каплям DIAD (265 ммоль) при 0°C. Смесь перемешивали в течение 5 минут при 0°C. Добавляли по каплям раствор промежуточного соединения I-75 (53 ммоль) в безводном ТГФ (1500 мл) при 0°C. Смесь перемешивали в течение 3 часов при 20°C. Растворитель удаляли в вакууме. Остаток очищали колоночной флэш-хроматографией с использованием SiO2 (градиентное элюирование: петролейный эфир/этилацетат от 3:2 до 1:1). Чистые фракции собирали и органический растворитель выпаривали. Полученный продукт дополнительно промывали трет-бутилметиловым эфиром. Выход: 25 г (выход 86%).
Пример 76: промежуточное соединение I-77
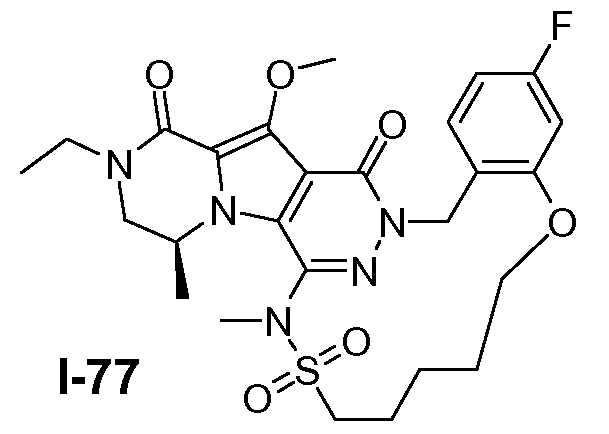
Промежуточное соединение I-77 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 72-75, с использованием промежуточного соединения I-72 и промежуточного соединения I-49.
Пример 77: промежуточное соединение I-78
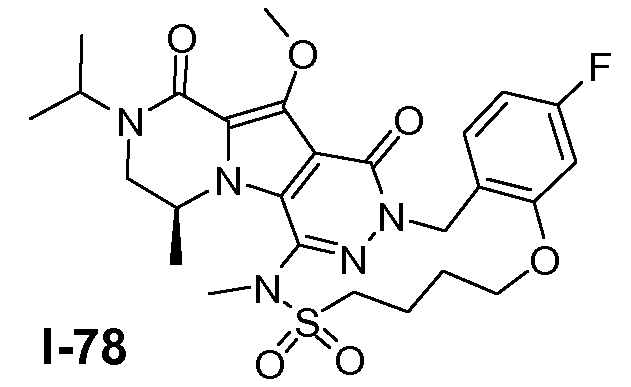
Промежуточное соединение I-78 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 71-75, с использованием промежуточных соединений I-5, I-39 и I-45.
Пример 78: промежуточное соединение I-79
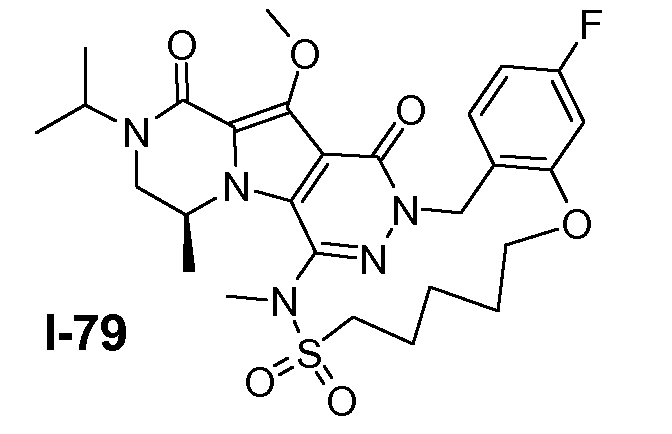
Промежуточное соединение I-79 получали, следуя процедуре, аналогичной той, которая описана в примерах 71-75, с использованием промежуточных соединений I-5, I-39 и I-49.
Пример 79: промежуточное соединение I-80
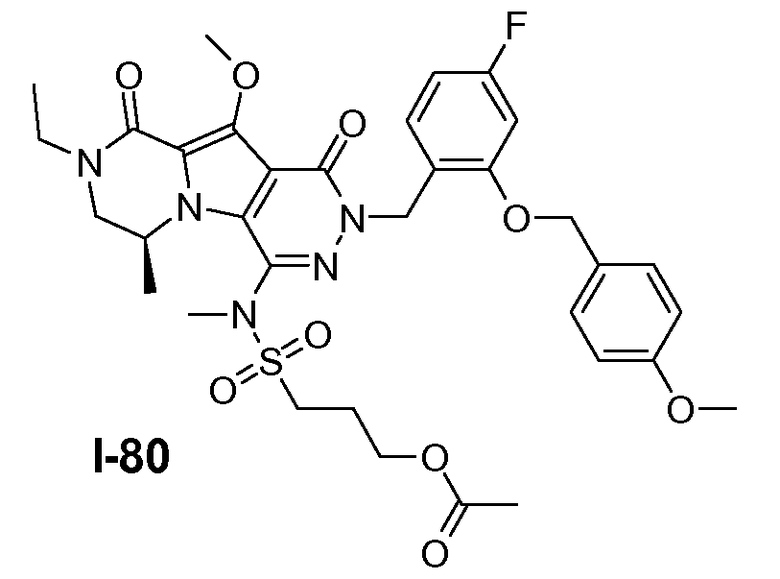
Промежуточное соединение I-80 получали, следуя процедуре, аналогичной той, которая описана в примере 72, с использованием промежуточных соединений I-72 и I-55.
Пример 80: промежуточное соединение I-81
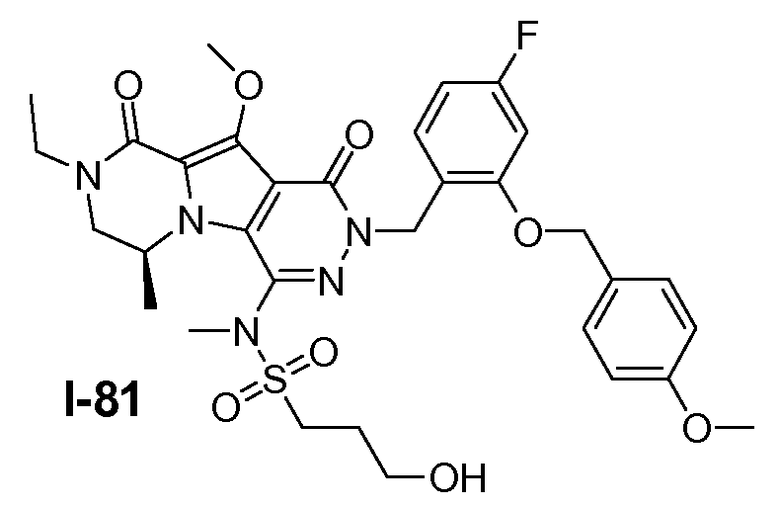
Раствор NaOH (25 ммоль) в воде (10 мл) добавляли к промежуточному соединению I-80 (2,8 ммоль) и перемешивали при 25°C в течение 1 часа. Смесь нейтрализовали 10% раствором лимонной кислоты и экстрагировали этилацетатом. Органический слой дважды промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией с использованием SiO2 (градиентное элюирование: от CH2Cl2 до смеси метанол/CH2Cl2 8:100). Выход: 1,3 г (чистота 80%).
Пример 81: промежуточное соединение I-82
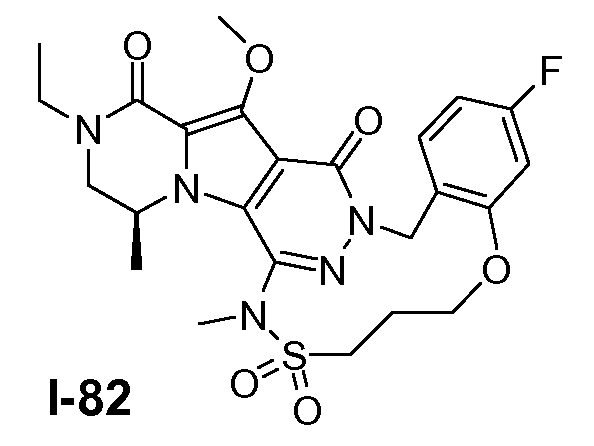
Промежуточное соединение I-82 получали, следуя процедуре, аналогичной той, которая описана в примерах 74 и 75, с использованием промежуточного соединения I-81.
Пример 82: промежуточное соединение I-83, 2 диастереомера
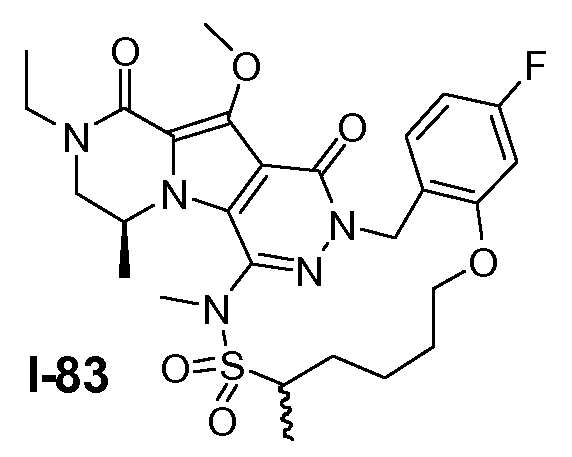
Макроцикл I-77 (0,45 ммоль) в безводном ТГФ (5 мл) охлаждали до -78°C. Добавляли по каплям гексаметилдисилазид лития (1M раствор в ТГФ; 0,49 ммоль) в атмосфере N2. Смесь перемешивали в течение 0,5 часа при -78°C. Добавляли по каплям йодметан (0,58 ммоль). Смеси давали нагреться до комнатной температуры и перемешивали в течение 3 часов при 25°C. Добавляли воду (5 мл), чтобы погасить реакцию. Смесь экстрагировали этилацетатом (3×10 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали высоко-эффективной жидкостной хроматографией (ВЭЖХ; C18, элюент: метанол/H2O от 15:85 до 45:55 с 0,1% трифторуксусной кислоты (ТФУК) в качестве буфера). Чистые фракции собирали, летучие вещества удаляли в вакууме и водный раствор подщелачивали насыщенным водным раствором NaHCO3 до pH 8 и экстрагировали с помощью CH2Cl2 (2×30 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток дополнительно очищали путем разделения методом сверхкритической жидкостной хроматографии (ChiralPak AS_H, 5 мкм; подвижная фаза: сверхкритический CO2: пропан-2-ол (0,05% диэтиламин), об./об.=70:30, 40 мл/мин, температура колонки: 38°C, давление в насадке: 100 бар, температура насадки: 60°C, температура испарителя: 20°C, температура триммера: 25°C, длина волны: 220 нм). Чистые фракции собирали и растворитель концентрировали в вакууме. Выход: 20 мг (чистота 95%, выход 8%) для первого элюирующего изомера; и 96 мг (чистота 95%, выход 38%) для второго элюирующего изомера.
Пример 83: промежуточное соединение I-84
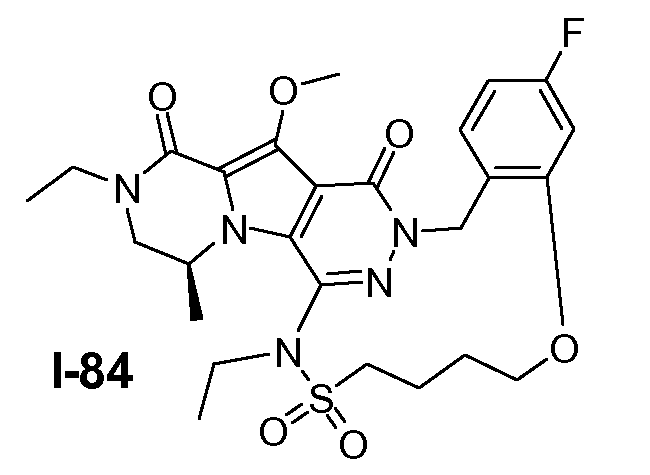
Промежуточное соединение I-84 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 72; 80; 74-75, с использованием промежуточных соединений I-72 и I-46.
Пример 84: промежуточное соединение I-85
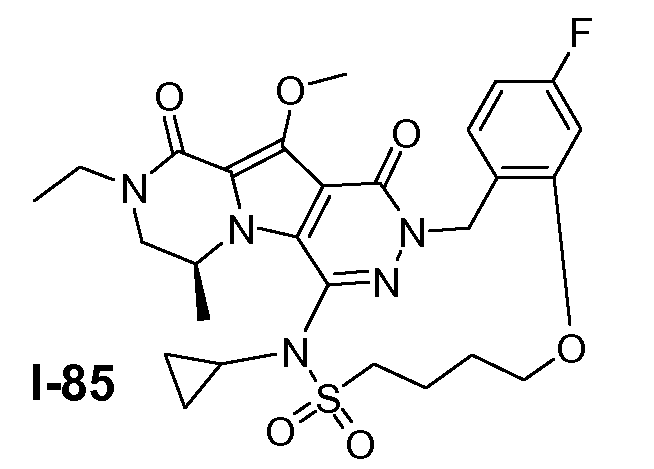
Промежуточное соединение I-85 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 72-75, с использованием промежуточных соединений I-72 и I-47.
Пример 85: промежуточное соединение I-86
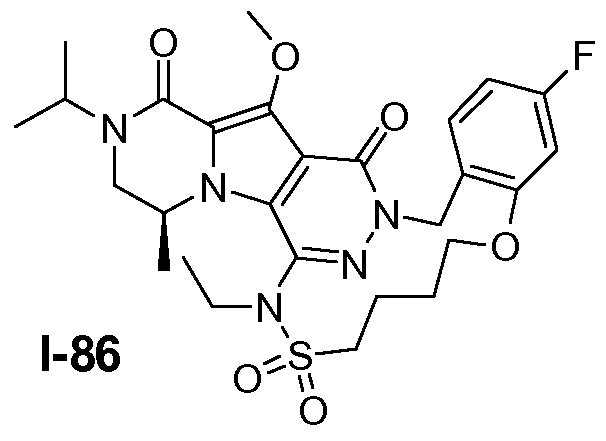
Промежуточное соединение I-86 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 71-72; 80; 74-75, с использованием промежуточных соединений I-5, I-41 и I-46.
Пример 86: промежуточное соединение I-87
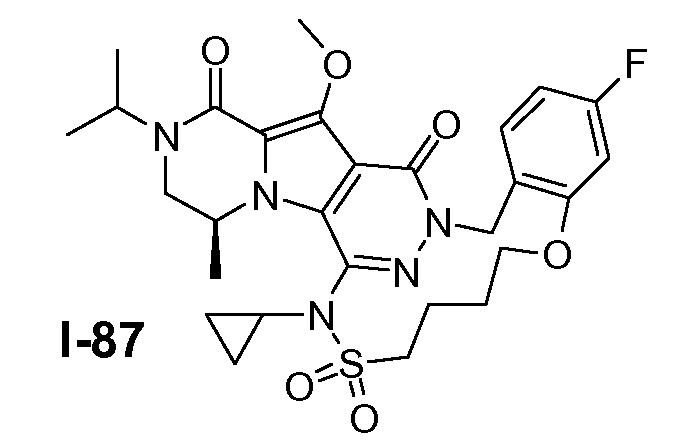
Промежуточное соединение I-87 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 71-75, с использованием промежуточных соединений I-5, I-41 и I-47.
Пример 87: промежуточное соединение I-88
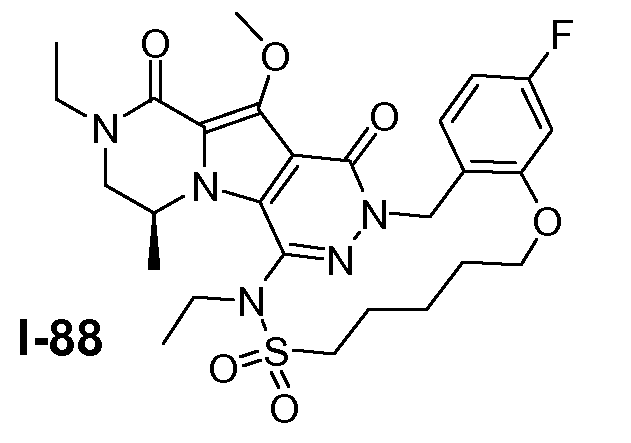
Промежуточное соединение I-88 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 71-72; 80; 74-75, с использованием промежуточных соединений I-2, I-41 и I-51.
Пример 88: промежуточное соединение I-89
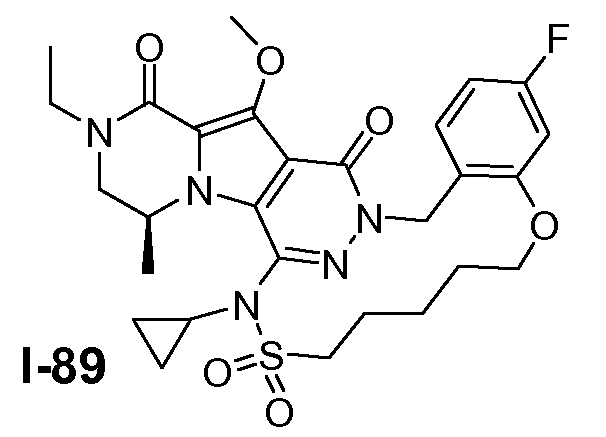
Промежуточное соединение I-89 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 71-72; с использованием промежуточных соединений I-2, I-41 и I-50.
Пример 89: промежуточное соединение I-90
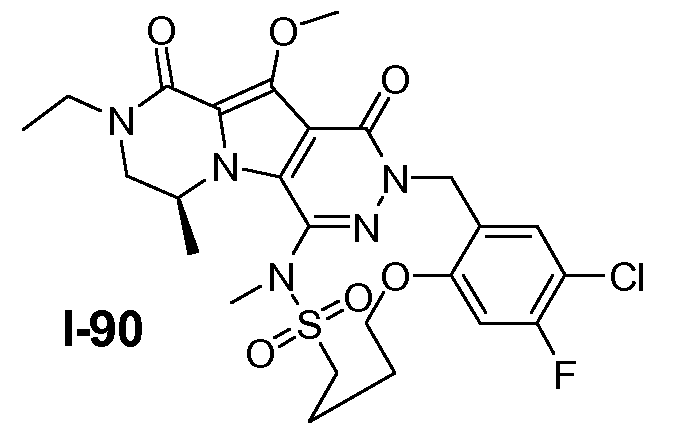
Промежуточное соединение I-90 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 71-75, с использованием промежуточных соединений I-2, I-69 и I-45.
Пример 90: промежуточное соединение I-91
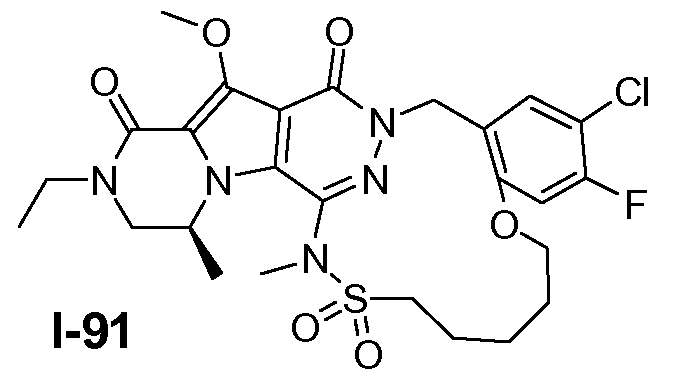
Промежуточное соединение I-91 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 71-75, с использованием промежуточных соединений I-2, I-69 и I-49.
Пример 91: промежуточное соединение I-92
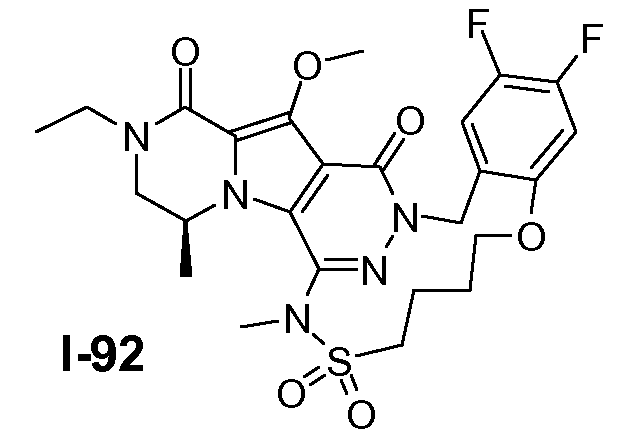
Промежуточное соединение I-92 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 71-72; 80; 74-75, с использованием промежуточных соединений I-2, I-71 и I-48.
Пример 92: промежуточное соединение I-93
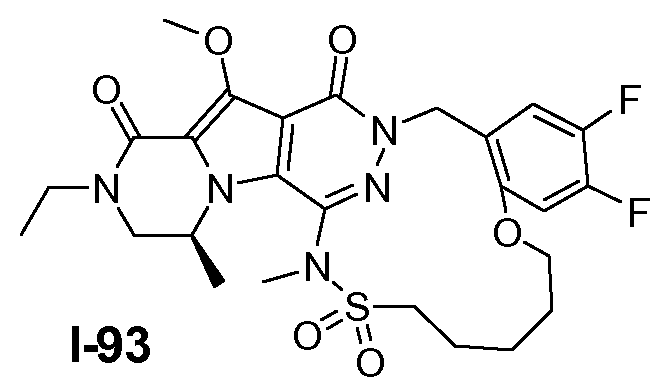
Промежуточное соединение I-93 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 71-75, с использованием промежуточных соединений I-2, I-71 и I-52.
Пример 93: промежуточное соединение I-95
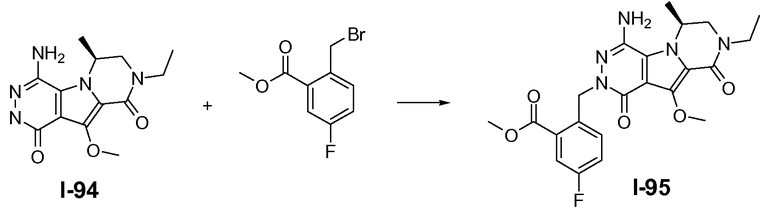
К раствору трициклического амина I-94 (33 ммоль) в ДМФА (100 мл) добавляли LiHMDS (1M в ТГФ, 40 ммоль) при 10°C. Смесь перемешивали в течение 30 минут и затем ее обрабатывали метил 2-(бромметил)-5-фторбензоатом (35 ммоль) в виде одной порции. Реакционную смесь перемешивали при 10°C в течение 3 часов и концентрировали в вакууме. Остаток распределяли между CH2Cl2 и 1 н. раствором HCl. Органический экстракт промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток промывали смесью диэтиловый эфир/CH2Cl2, фильтровали с получением неочищенного продукта, который растирали в порошок со смесью CH3CN/диэтиловый эфир 1:1, получая 3,5 г желтого порошка. Маточный раствор концентрировали и очищали колоночной хроматографией с использованием SiO2 (метанол/CH2Cl2 1:20), получая 4 г желтого порошка. Выход: 7,5 г (чистота 97%; выход 50%).
Пример 94: промежуточное соединение I-96
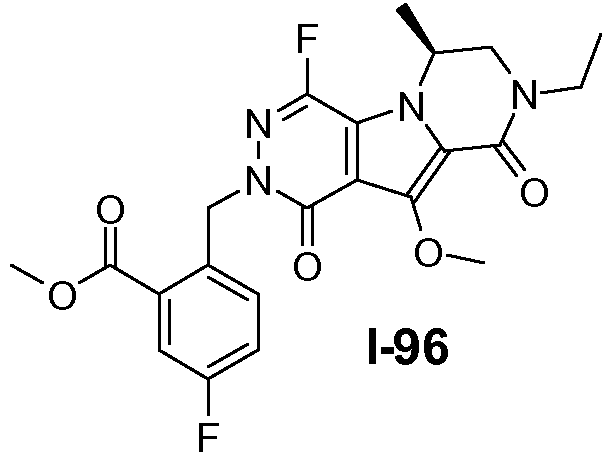
Промежуточное соединение I-95 (44 ммоль) растворяли в HF-пиридине (30 мл) в трехгорлой политетрафторэтиленовой (PTFE) колбе. Смесь перемешивали при 0°C. Добавляли порциями NaNO2 (48 ммоль) в течение 10 минут. Смесь перемешивали при комнатной температуре в течение 1 часа. Смесь добавляли по каплям к насыщенному водному раствору NaHCO3. Смесь экстрагировали этилацетатом. Органический слой отделяли, сушили и упаривали. Выход: 2 г (выход 98%).
Пример 95: промежуточное соединение I-97
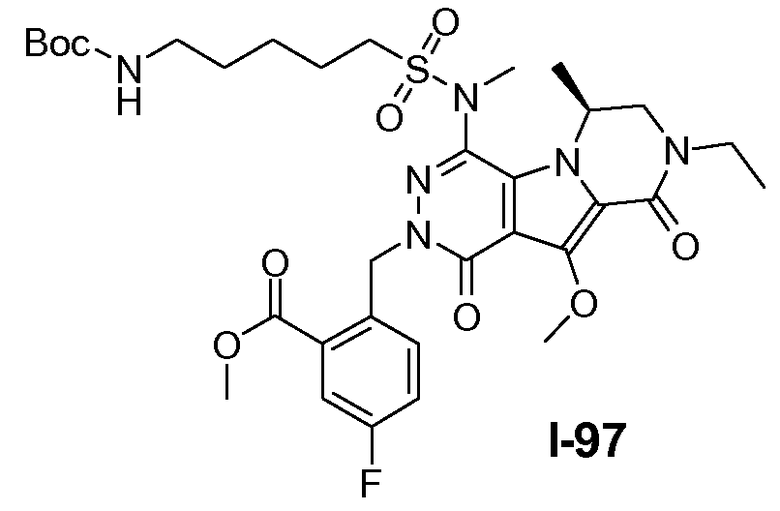
Промежуточные соединения I-96 (2,2 ммоль) и I-61 (4,3 ммоль) и CsCO3 (7,4 ммоль) растворяли в ДМСО (80 мл). Реакционную смесь перемешивали при 60°C-70°C в течение 17 часов. Реакционную смесь использовали на следующей стадии без дополнительной очистки.
Пример 96: промежуточное соединение I-98
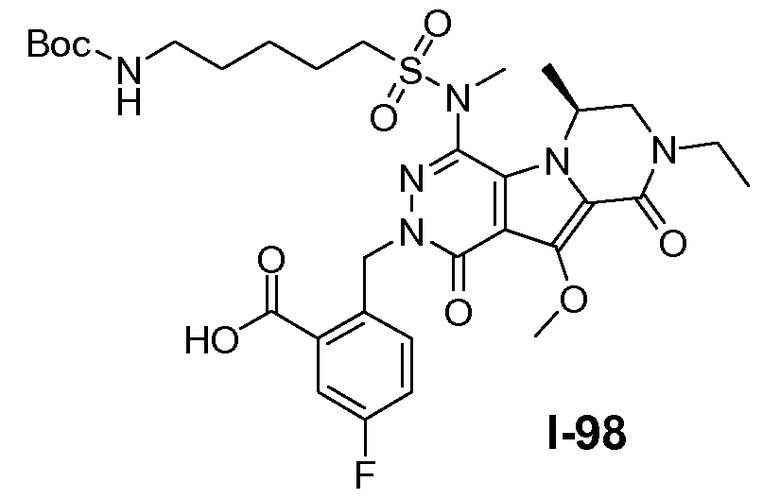
К неочищенному раствору промежуточного соединения I-97 добавляли воду (20 мл), с последующим добавлением NaOH (9,8 ммоль) при 17°C и реакционную смесь перемешивали в течение 40 минут. К реакционной смеси добавляли насыщенный солевой раствор (120 мл) и воду (50 мл). Водную часть подкисляли до pH 3 с использованием 1,0M HCl и экстрагировали с помощью CH2Cl2 (3×100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали препаративной ВЭЖХ (колонка: Luna 300×50 мм, 0,01 мм, элюент CH3CN/H2O от 35:65 до 65:35, 0,1% ТФУК). Чистые фракции собирали и органический растворитель выпаривали. Водную часть экстрагировали с помощью CH2Cl2. Объединенный органический слой промывали насыщенным солевым раствором, сушили над Na2SO4. Органический слой фильтровали и концентрировали в вакууме. Выход: 1,06 г (чистота 98%; выход 69%).
Пример 97: промежуточное соединение I-99
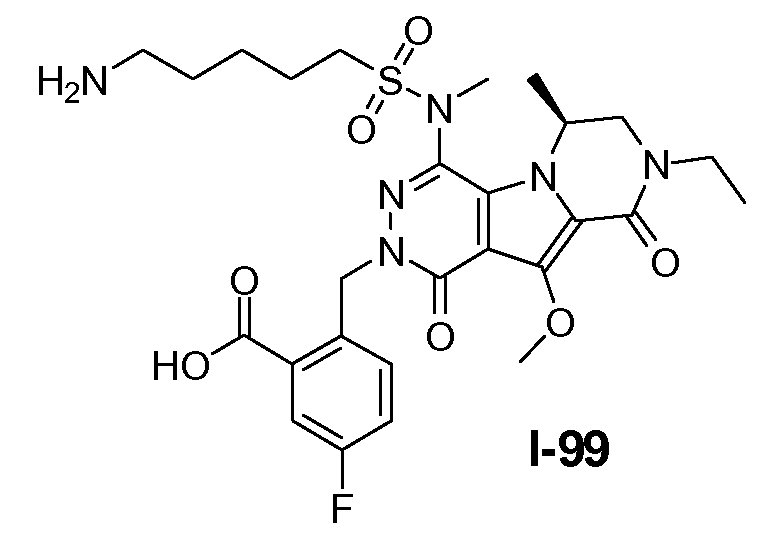
Промежуточное соединение I-98 (1,4 ммоль) растворяли в CH2Cl2 (10 мл) и смесь охлаждали до 0°C. К смеси добавляли по каплям ТФУК (10 мл) при 0°C. Реакционной смеси давали нагреться до 20°C и перемешивали в течение 0,5 часа. Растворитель выпаривали при пониженном давлении. Остаток совместно упаривали с толуолом (2×20 мл) и использовали на следующей стадии без дополнительной очистки. Выход: 1 г (соль ТФУК, чистота 99%, выход 98%).
Пример 98: промежуточное соединение I-100
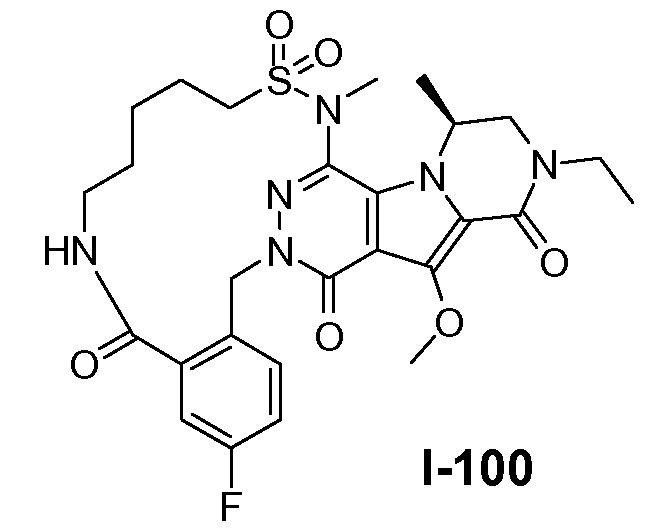
Пентафторфенилдифенилфосфинат (FDPP; 1,4 ммоль) и DIPEA (1,6 мл) растворяли в ДМФА (150 мл) при 18°C. Добавляли по каплям смесь промежуточного соединения I-99 (1,4 ммоль) и DIPEA (0,8 мл) в ДМФА (150 мл) при 18°C. Реакционную смесь перемешивали в течение ночи при 18°C. Смесь затем упаривали в вакууме. Остаток растворяли в CH2Cl2 (30 мл), промывали 1 н. водным раствором HCl (×16 мл), насыщенным раствором NaHCO3 (21 мл) и насыщенным солевым раствором (20 мл), сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток промывали диэтиловым эфиром (10 мл) и фильтровали с получением бледно-белого порошка. Выход: 0,65 г (чистота 93%; выход 80%).
Пример 99: промежуточное соединение I-101
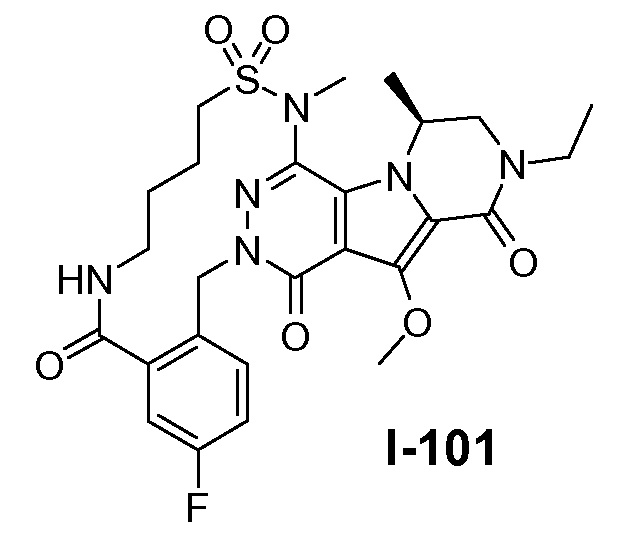
Промежуточное соединение I-101 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 95-98, с использованием промежуточных соединений I-96 и I-58.
Пример 100: промежуточное соединение I-102
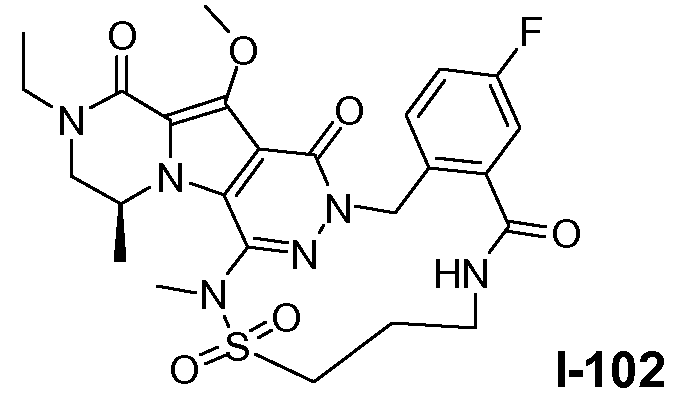
Промежуточное соединение I-102 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 95-98, с использованием промежуточных соединений I-96 и I-57.
Пример 101: промежуточное соединение I-103
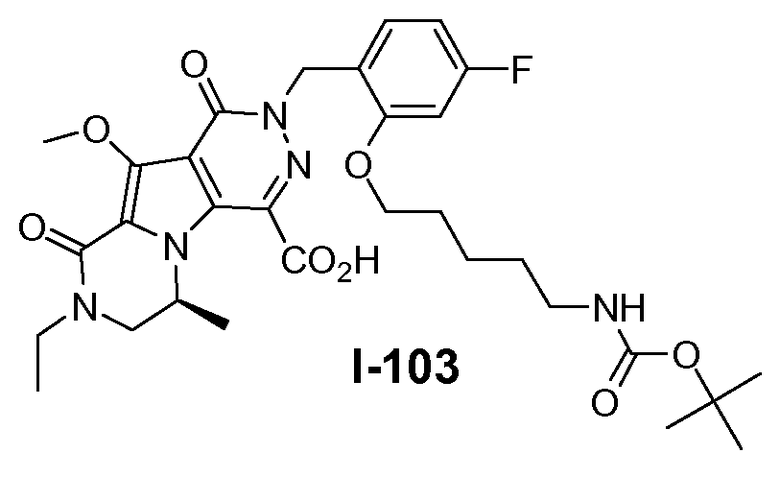
Промежуточные соединения I-16 (123 ммоль) и I-27 (185 ммоль) и трифенилфосфин (245 ммоль) растворяли в безводном ТГФ при 20°C. Добавляли DIAD (245 ммоль) ниже -5°C. Смесь перемешивали в течение 5 часов при 0-10°C. ЖХ-МС анализ показал, что исходное вещество израсходовано и что образовался желаемый продукт. Добавляли NaOH в смеси вода/метанол (1: 1; 0,17 н, 1,6 л). Смесь перемешивали в течение 0,5 часа при 30°C. Растворители выпаривали в вакууме. Смесь экстрагировали трет-бутилметиловым эфиром (3×100 мл) для удаления примесей. К водному слою добавляли 2 н. раствор HCl до достижения pH 2-3. Водную смесь затем экстрагировали этилацетатом (2×200 мл). Объединенный органический слой промывали насыщенным солевым раствором, сушили и упаривали. Неочищенный продукт использовали на следующей стадии без дополнительной очистки. Выход: 65 г (выход 84%).
Пример 102: промежуточное соединение I-104 (соль ТФУК)
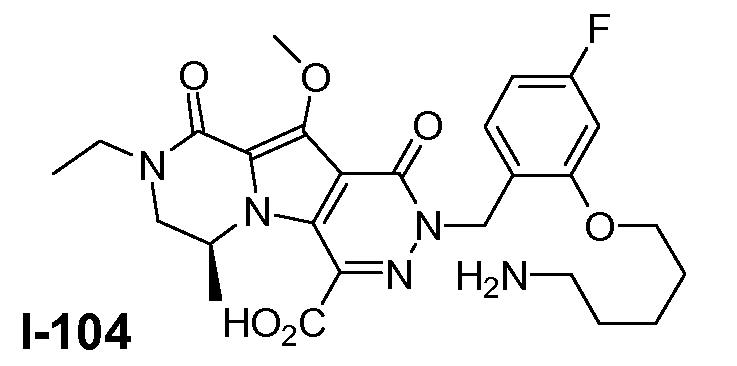
Промежуточное соединение I-103 (103 ммоль) растворяли в CH2Cl2 (650 мл) при 20°C. Добавляли по каплям ТФУК (260 мл) при 0°C. Смесь перемешивали в течение 0,5 часа при 20°C. Растворитель удаляли в вакууме. Остаток совместно упаривали с толуолом (2×50 мл) и использовали на следующей стадии без дополнительной очистки. Выход: 65 г (чистота 85%).
Пример 103: промежуточное соединение I-105
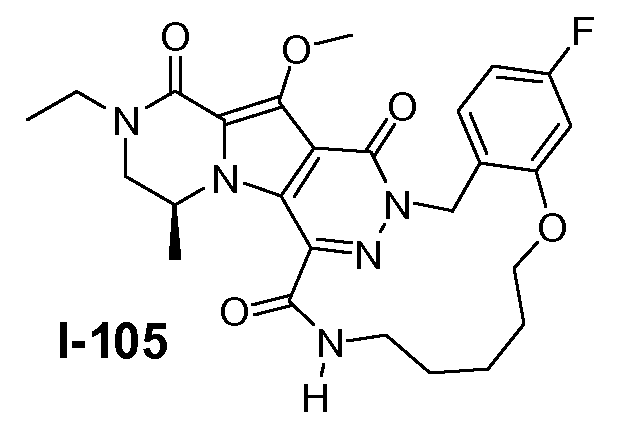
FDPP (113 ммоль) и DIPEA (1,24 моль) растворяли в ТГФ (7 л) при 30°C. Добавляли по каплям промежуточное соединение I-104 (103 ммоль) в ТГФ (7 л) при 30°C. Смесь перемешивали в течение ночи при 30°C. Растворитель удаляли в вакууме. Остаток растворяли в CH2Cl2 (2 л) и промывали 1 н. раствором HCl (2×500 мл), насыщенным водным раствором NaHCO3 и насыщенным солевым раствором. Органический слой сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюент: CH2Cl2/метанол 100:3, затем 100:7 об./об.). Чистые фракции собирали и концентрировали досуха. Полученный остаток промывали трет-бутилметиловым эфиром. Выход: 46 г (общий выход для двух стадий 79%).
Пример 104: промежуточное соединение I-106
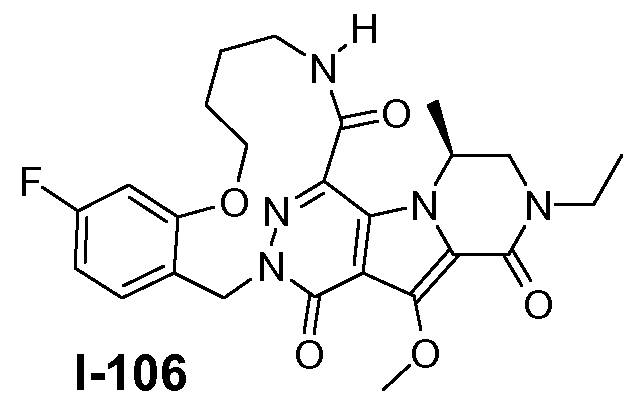
Промежуточное соединение I-106 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 101-103, с использованием промежуточных соединений I-16 и I-29.
Пример 105: промежуточное соединение I-107
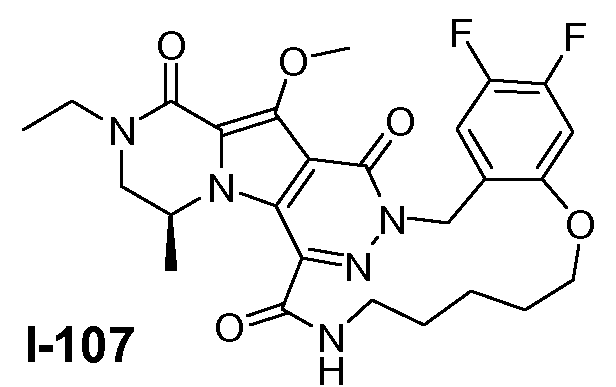
Промежуточное соединение I-107 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 101-103, с использованием промежуточных соединений I-16 и I-37.
Пример 106: промежуточное соединение I-108
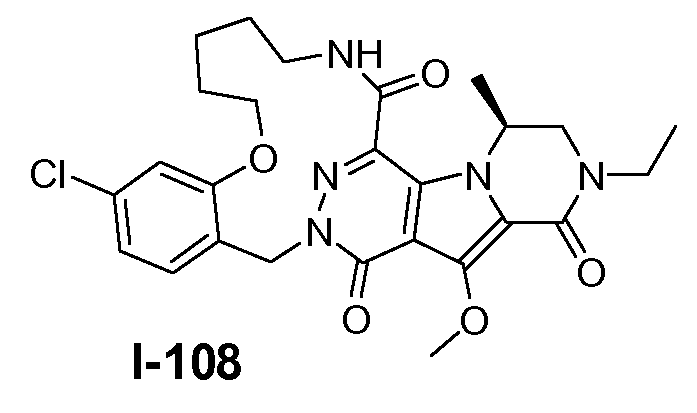
Промежуточное соединение I-108 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 101-103, с использованием промежуточных соединений I-16 и I-31.
Пример 107: промежуточное соединение I-109
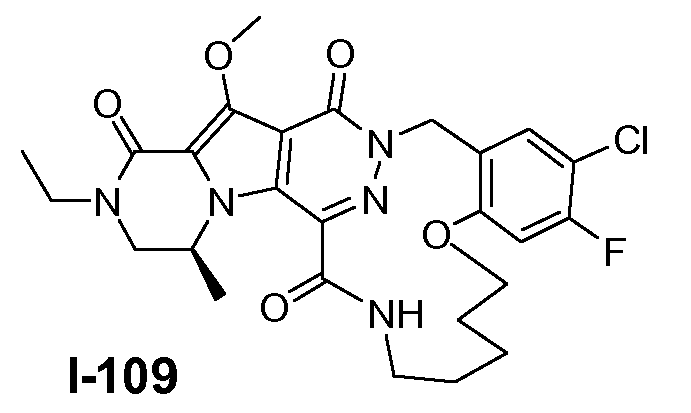
Промежуточное соединение I-109 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 101-103, с использованием промежуточных соединений I-16 и I-30.
Пример 108: промежуточное соединение I-110
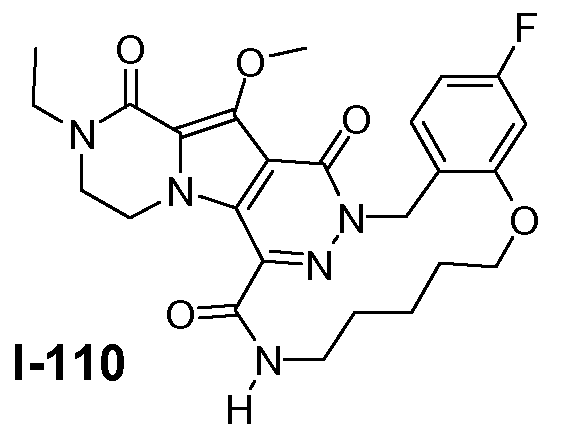
Промежуточное соединение I-110 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 101-103, с использованием промежуточных соединений I-23 и I-27.
Пример 109: промежуточное соединение I-111
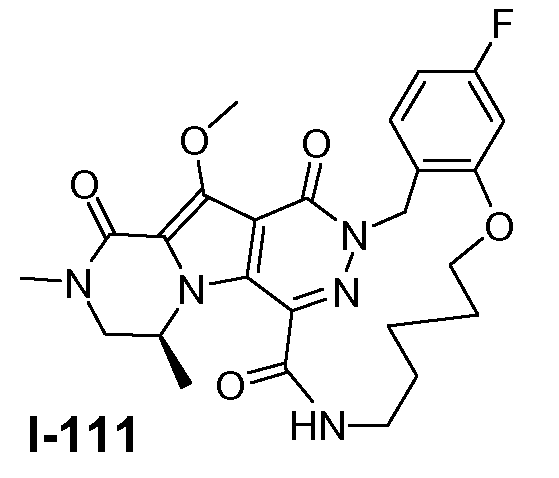
Промежуточное соединение I-111 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 101-103, с использованием промежуточных соединений I-18 и I-27.
Пример 110: промежуточное соединение I-112
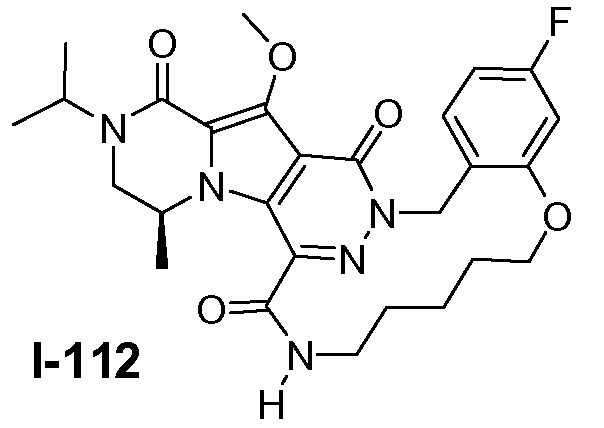
Промежуточное соединение I-112 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 101-103, с использованием промежуточных соединений I-17 и I-27.
Пример 111: промежуточное соединение I-113
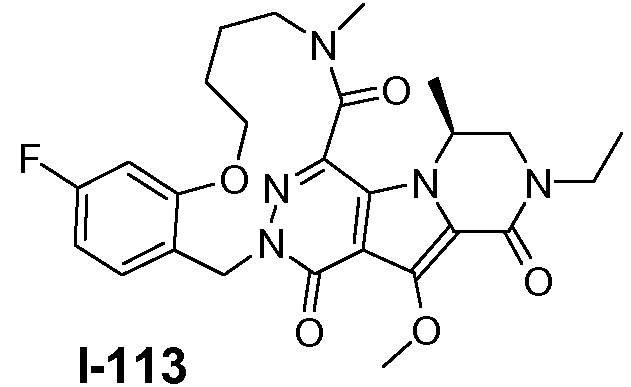
Промежуточное соединение I-106 (0,44 ммоль) растворяли в безводном ТГФ (7 мл). Добавляли NaH (60%; 0,66 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 15 минут. Затем добавляли по каплям йодметан (0,66 ммоль) и смесь перемешивали в течение 40 минут при 15°C. Смесь гасили насыщенным водным раствором NH4C1 и растворитель удаляли в вакууме. Смесь экстрагировали с помощью CH2Cl2 (2×10 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток промывали диэтиловым эфиром. Выход: 0,2 г (чистота 89%, выход 88%).
Пример 112: промежуточное соединение I-114
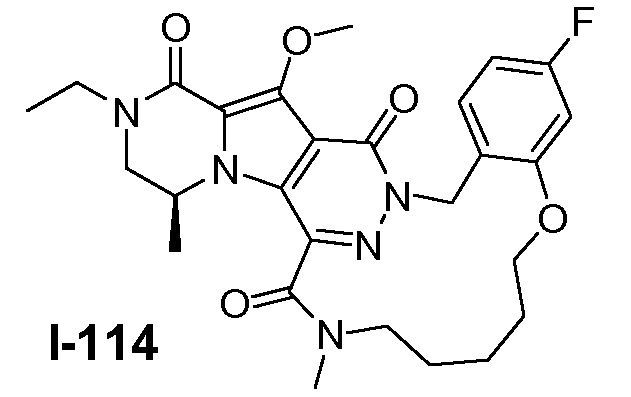
Промежуточное соединение I-114 получали, следуя процедуре, аналогичной той, которая описана в примере 111, с использованием промежуточного соединения I-105.
Пример 113: промежуточное соединение I-115
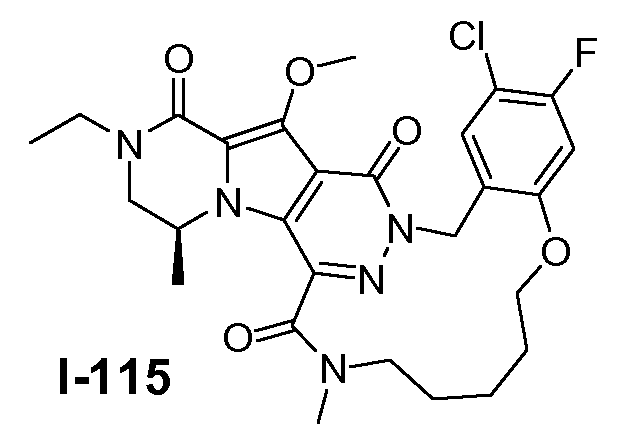
Промежуточное соединение I-115 получали, следуя процедуре, аналогичной той, которая описана в примере 111, с использованием промежуточного соединения I-109.
Пример 114: промежуточное соединение I-116
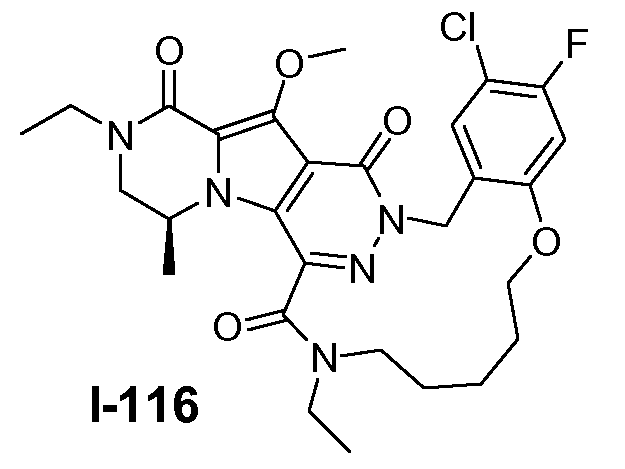
Промежуточное соединение I-116 получали, следуя процедуре, аналогичной той, которая описана в примере 111, с использованием промежуточного соединения I-109 и йодэтана.
Пример 115: промежуточное соединение I-117
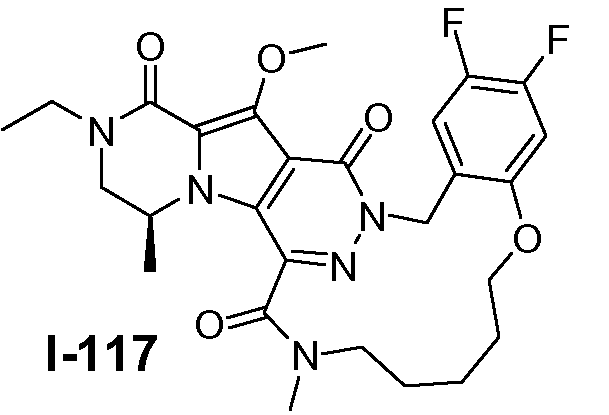
Промежуточное соединение I-117 получали, следуя процедуре, аналогичной той, которая описана в примере 111, с использованием промежуточного соединения I-107.
Пример 116: промежуточное соединение I-118
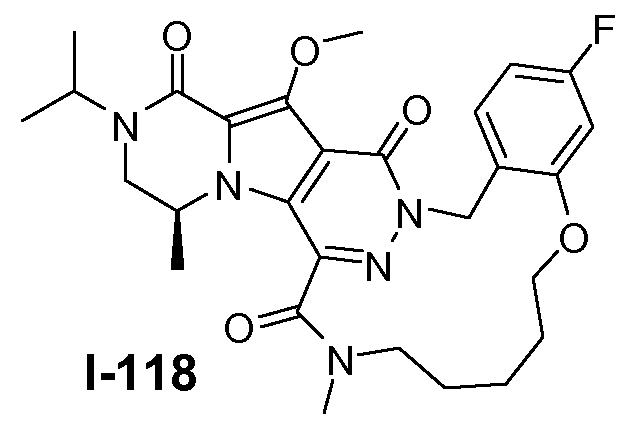
Промежуточное соединение I-118 получали, следуя процедуре, аналогичной той, которая описана в примере 111, с использованием промежуточного соединения I-112.
Пример 117: промежуточное соединение I-119
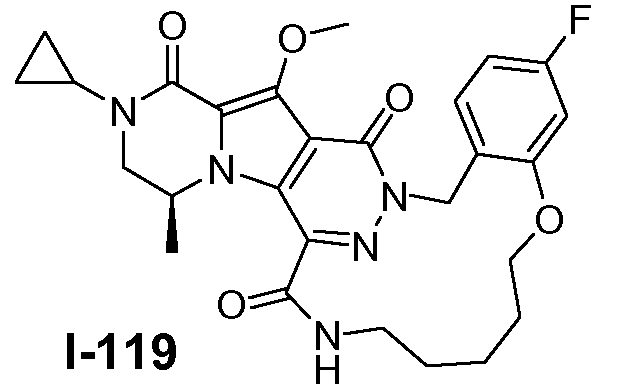
Промежуточное соединение I-119 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 101-103, с использованием промежуточных соединений I-24 и I-27.
Пример 118: промежуточное соединение I-120
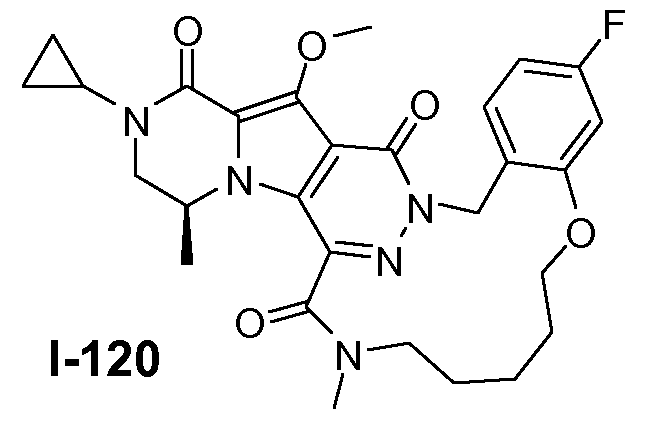
Промежуточное соединение I-120 получали, следуя процедуре, аналогичной той, которая описана в примере 111, с использованием промежуточного соединения I-119.
Пример 119: промежуточное соединение I-121
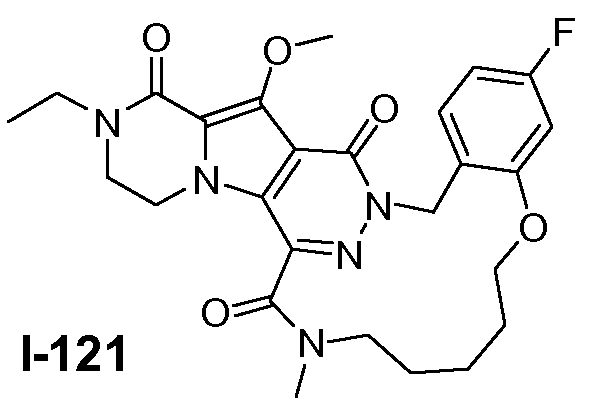
Промежуточное соединение I-121 получали, следуя процедуре, аналогичной той, которая описана в примере 111, с использованием промежуточного соединения I-110.
Пример 120: промежуточное соединение I-122
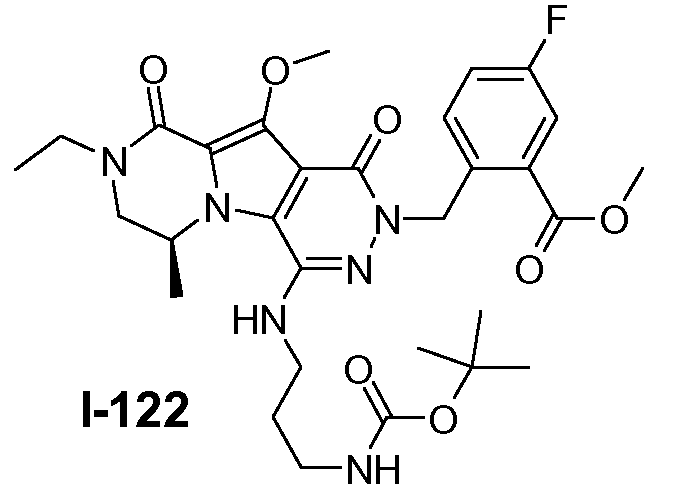
Промежуточное соединение I-95 (2,2 ммоль) растворяли в 2-пропаноле (30 мл). Добавляли трет-бутил 3-оксопропилкарбамат (11 ммоль) и уксусную кислоту (11 ммоль). Смесь нагревали при температуре кипения с обратным холодильником в течение 4 часов и охлаждали до 0°C. Добавляли по каплям метанол (30 мл), затем добавляли NaBH4 (22 ммоль) при 0°C и смесь перемешивали при 0°C в течение 1 часа. Добавляли NaHCO3 (насыщенный водный раствор, 10 мл) и CH2Cl2 (50 мл). Органический слой промывали насыщенным солевым раствором и сушили (Na2SO4). Растворитель удаляли в вакууме. Остаток очищали с помощью ВЭЖХ (C18, элюент: метанол/H2O от 15:85 до 45:55 с 0,1% ТФУК в качестве буфера). Чистые фракции собирали, летучие вещества удаляли в вакууме и водный раствор подщелачивали NaHCO3 до pH 8. Добавляли CH2Cl2 (200 мл) и органический слой промывали насыщенным солевым раствором и сушили (Na2SO4). Выход: 1,1 г (чистота 95%, выход 81%).
Пример 121: промежуточное соединение I-123
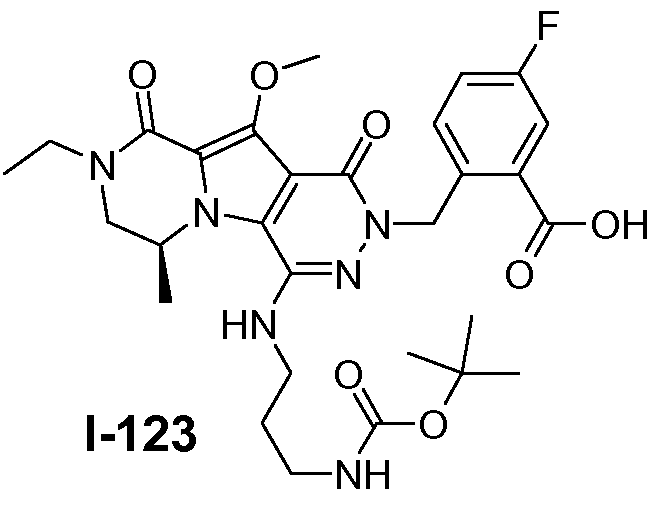
К раствору промежуточного соединения I-122 (1,8 ммоль) в метаноле (11 мл) и ТГФ (11 мл) добавляли гидрат LiOH (9,0 ммоль) в воде (11 мл) при 20°C. Смесь перемешивали при 20°C в течение 5 часов. Реакционную смесь концентрировали в вакууме. Затем добавляли воду (15 мл) и водный слой экстрагировали диэтиловым эфиром (2×20 мл). Водную часть подкисляли до pH 7 с использованием 1M HCl при 0°C и экстрагировали этилацетатом для удаления примесей. Водную часть затем подкисляли до pH 3 с использованием 1M HCl при 0°C и экстрагировали этилацетатом (2×20 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением кислоты в виде бледно-желтого порошка. Выход: 1 г (чистота 95%, выход 94%).
Пример 122: промежуточное соединение I-124
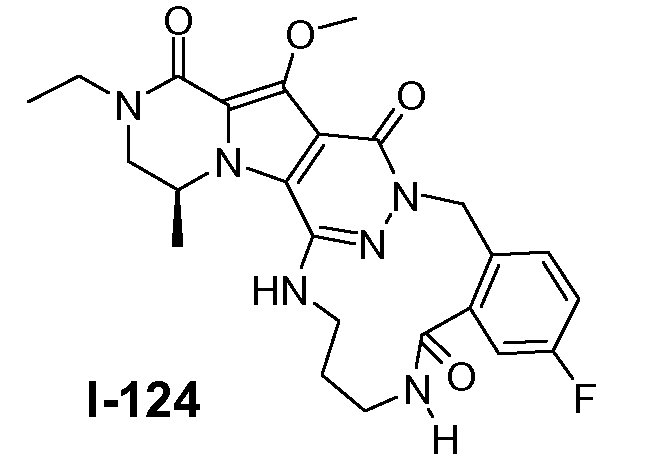
Промежуточное соединение I-124 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 97-98, с использованием промежуточного соединения I-123.
Пример 123: промежуточное соединение I-125
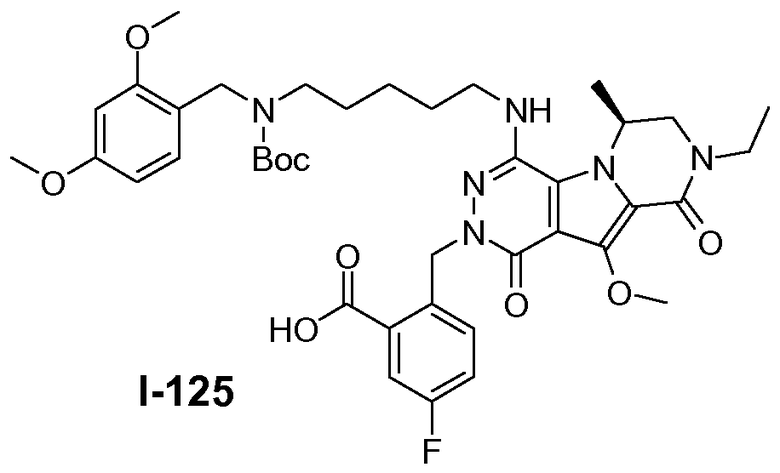
Промежуточное соединение I-125 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 120-121, с использованием промежуточных соединений I-95 и I-62.
Пример 124: промежуточное соединение I-126
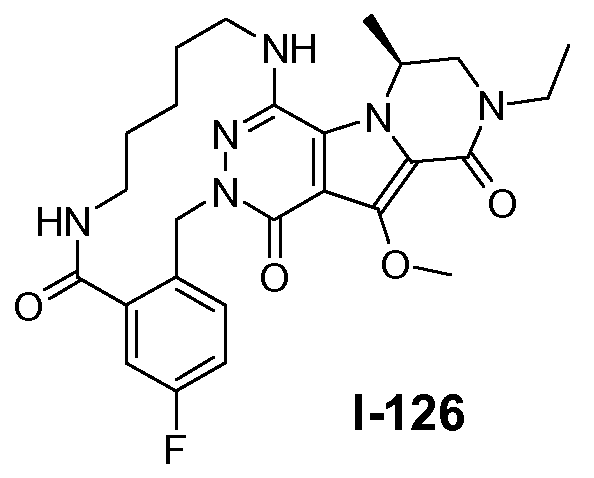
Промежуточное соединение I-126 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 97-98, с использованием промежуточного соединения I-125.
Пример 125: промежуточное соединение I-127
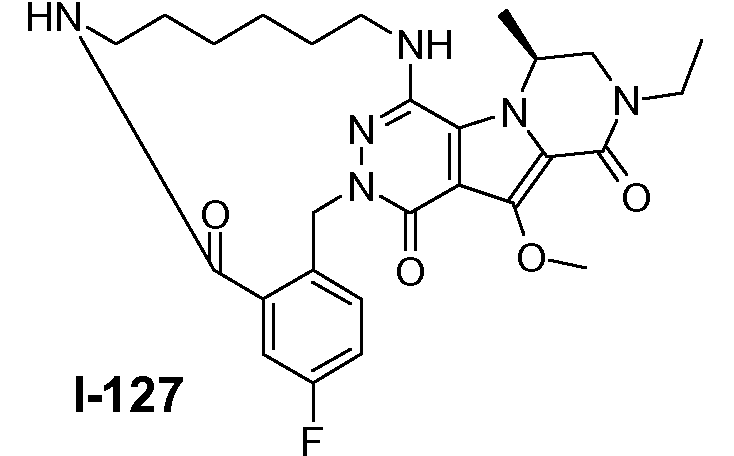
Промежуточное соединение I-127 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 120-121 и примерах 97-98, с использованием промежуточных соединений I-95 и трет-бутил 6-оксогексилкарбамата.
Пример 126: промежуточное соединение I-128
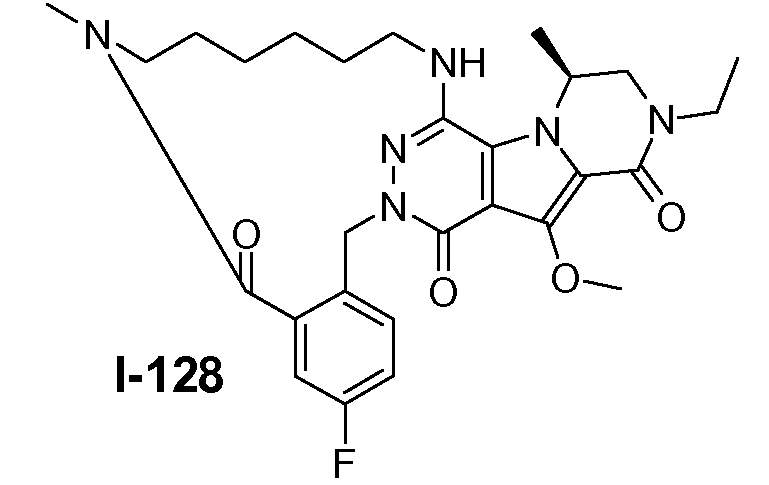
Промежуточное соединение I-128 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 120-121 и примерах 97-98, с использованием промежуточных соединений I-95 и I-67.
Пример 127: промежуточное соединение I-129
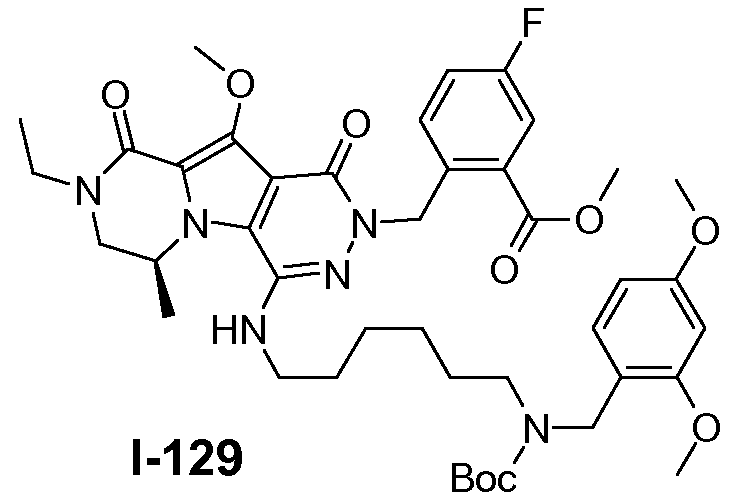
Промежуточное соединение I-129 получали, следуя процедуре, аналогичной той, которая описана в примере 120, с использованием промежуточных соединений I-95 и I-63.
Пример 128: промежуточное соединение I-130
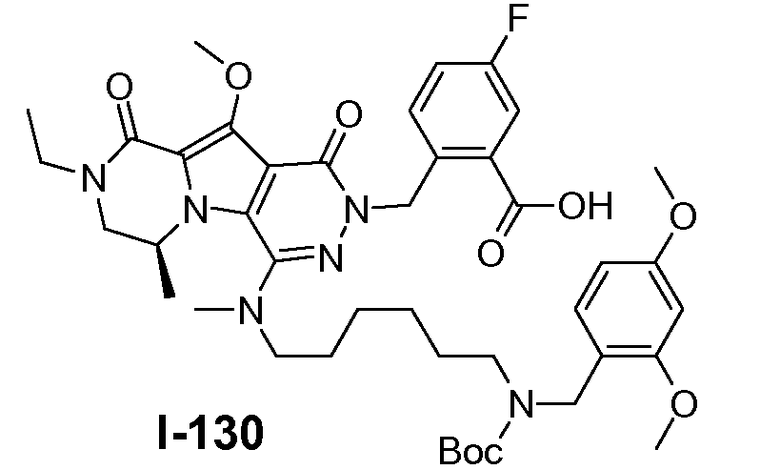
К раствору промежуточного соединения I-129 (0,69 ммоль) в безводном ТГФ (8 мл) добавляли NaH (60%; 0,69 ммоль) одной порцией при 0°C. Реакционную смесь перемешивали при 0°C в течение 15 минут. К реакционной смеси добавляли по каплям йодметан (1,6 ммоль) при 0°C. Реакционной смеси давали нагреться до 20°C и перемешивали в течение 1,5 часов. К реакционной смеси добавляли дополнительное количество NaH (60%, 0,30 ммоль) при 0°C и затем смесь перемешивали в течение 5 минут. Еще одну партию йодметана (1,5 ммоль) добавляли по каплям при 0°C и реакционную смесь перемешивали при 20°C еще в течение 0,5 часа. Методом жидкостной хроматографии-масс-спектрометрии (ЖХ/МС) был обнаружен гидролизованный продукт. К реакционной смеси добавляли воду (8 мл) и затем добавляли метил трет-бутиловый эфир (8 мл). Водный слой разделяли и подкисляли до pH 7 с использованием 1 н. раствора HCl и затем экстрагировали этилацетатом (8 мл) для удаления примесей. Водный слой затем разделяли, подкисляли до pH 2 с использованием 1 н. раствора HCl и экстрагировали этилацетатом (3×8 мл). Объединенный органический слой промывали насыщенным солевым раствором (15 мл), сушили над Na2SO4. Органический растворитель фильтровали и концентрировали в вакууме. Остаток очищали препаративной ВЭЖХ (колонка: Gemini 200×25 мм, 0,005 мм, элюент CH3CN/H2O от 68% до 88%, 0,05% ТФУК). Чистые фракции собирали и органический растворитель выпаривали. Водную часть экстрагировали с помощью CH2Cl2. Объединенный органический слой промывали насыщенным солевым раствором, сушили над Na2SO4. Органический слой фильтровали и концентрировали в вакууме. Выход: 300 мг (чистота 98%, выход 54%).
Пример 129: промежуточное соединение I-131
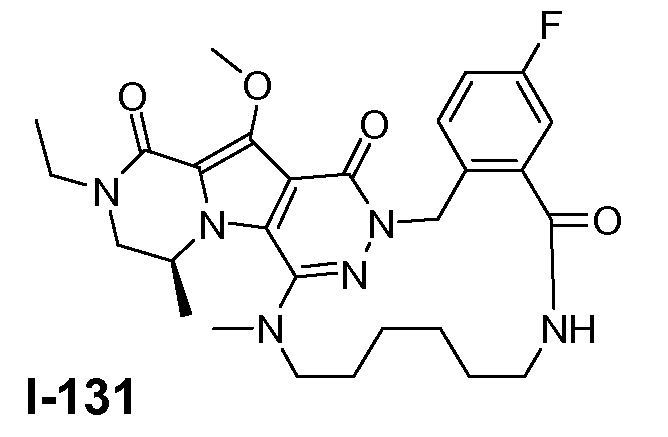
Промежуточное соединение I-131 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 97-98, с использованием промежуточного соединения I-130.
Пример 130: промежуточные соединения I-132 и I-133
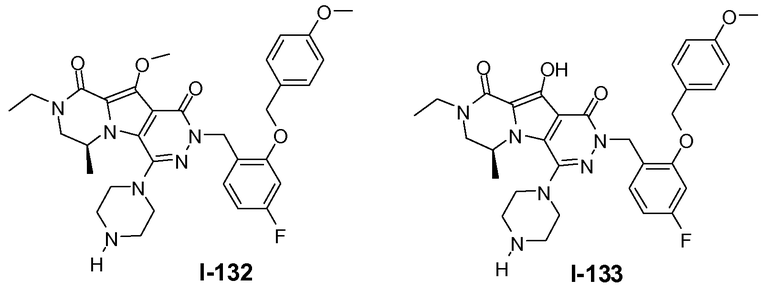
Промежуточное соединение I-72 (7,1 ммоль), пиперазин (141 ммоль) и 4 Å молекулярные сита (3,8 г) суспендировали в NMP (N-метилпирролидон). Смесь перемешивали в течение ночи при 120°C. Молекулярные сита отфильтровывали, затем реакционную смесь охлаждали до комнатной температуры. К полученному фильтрату добавляли воду (50 мл) и CH2Cl2 (100 мл). Происходило образование осадка, который собирали фильтрованием, и было обнаружено, что структура соответствует I-133; 1,1 г (чистота 90%).
Органический слой отделяли от фильтрата и полученный водный слой экстрагировали с помощью CH2Cl2 (100 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×100 мл) и сушили над Na2SO4. Растворитель удаляли в вакууме. ЖХ/МС анализ показывал смесь двух продуктов, промежуточных соединений I-132 и I-133 в соотношении 1:2, в сумме составляющих 3 г смеси.
Пример 131: промежуточное соединение I-134
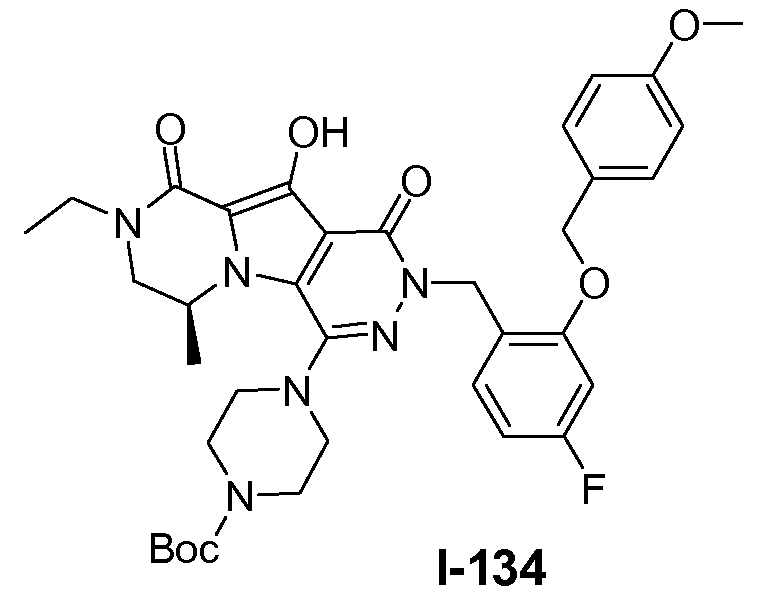
Смесь промежуточных соединений I-132 и I-133 (соотношение 1:2; 3 г) растворяли в метаноле (30 мл). Добавляли Boc2O (1,1 г) при 0°C. Смесь перемешивали в течение 1 часа при 15°C. Затем добавляли еще одну партию Boc2O (1,1 г). Смесь перемешивали в течение ночи при 10°C, после чего добавляли еще одну партию Boc2O (3,3 г). Смесь перемешивали еще в течение 3 часов при 15°C. Растворитель удаляли в вакууме. Остаток очищали колоночной флэш-хроматографией с использованием SiO2 (градиентное элюирование: CH2Cl2/метанол от 100:0 до 90:10). Выход: 2,1 г (чистота 80%; выход 60%).
Пример 132: промежуточное соединение I-135
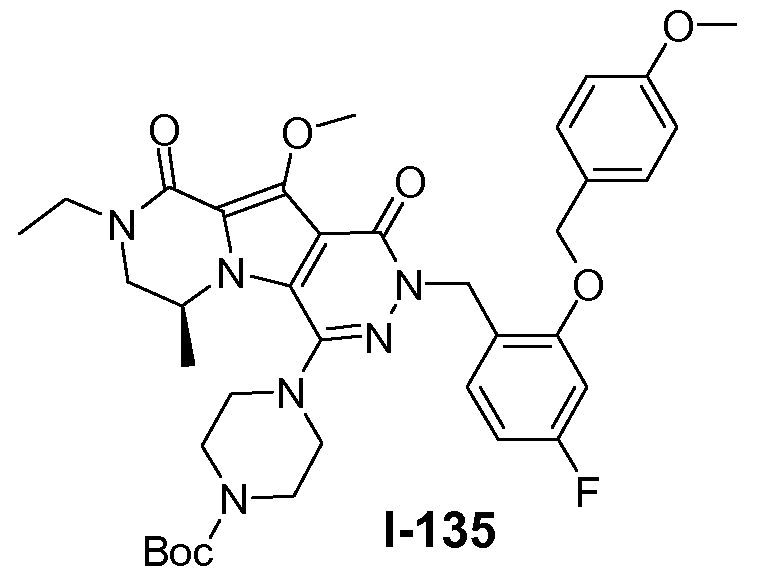
Промежуточное соединение I-134 (3,0 ммоль) растворяли в ДМФА (20 мл). Добавляли йодметан (15 ммоль) и K2CO3 (15 ммоль). Смесь перемешивали в течение ночи при 10°C. Добавляли воду (20 мл) и CH2Cl2 (40 мл). Органический слой отделяли, промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Выход: 1,7 г (чистота 85%).
Пример 133: промежуточное соединение I-136
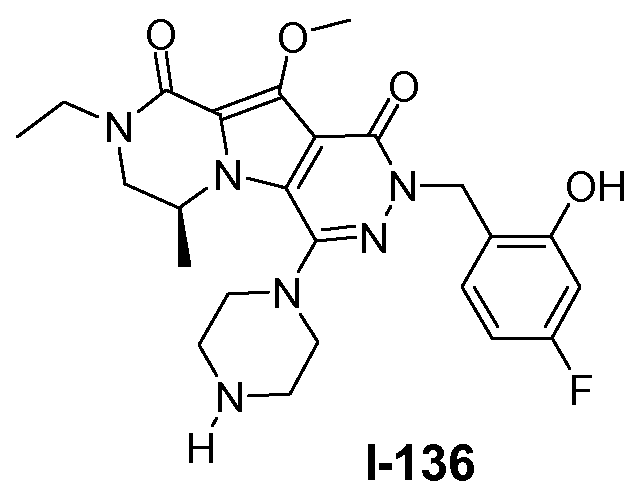
Промежуточное соединение I-136 получали (в виде соли HCl), следуя процедуре, аналогичной той, которая описана в примере 74, с использованием промежуточного соединения I-135.
Пример 134: промежуточное соединение I-137
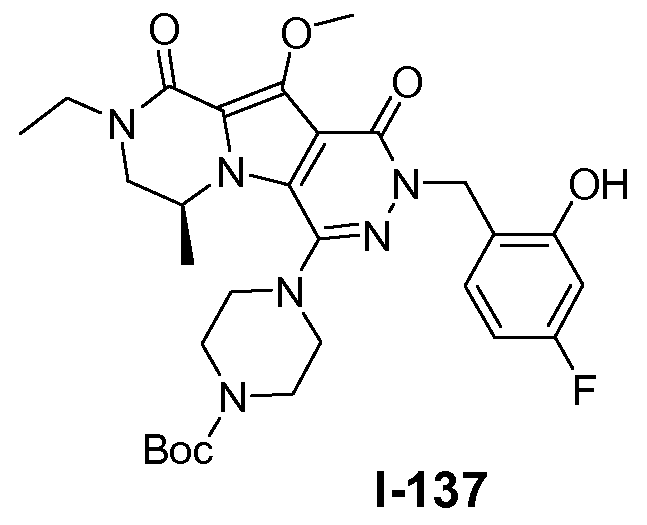
Промежуточное соединение I-137 получали, следуя процедуре, аналогичной той, которая описана в примере 131, с использованием промежуточного соединения I-136. Реакция требовала присутствия триэтиламина (3 экв.).
Пример 135: промежуточное соединение I-138
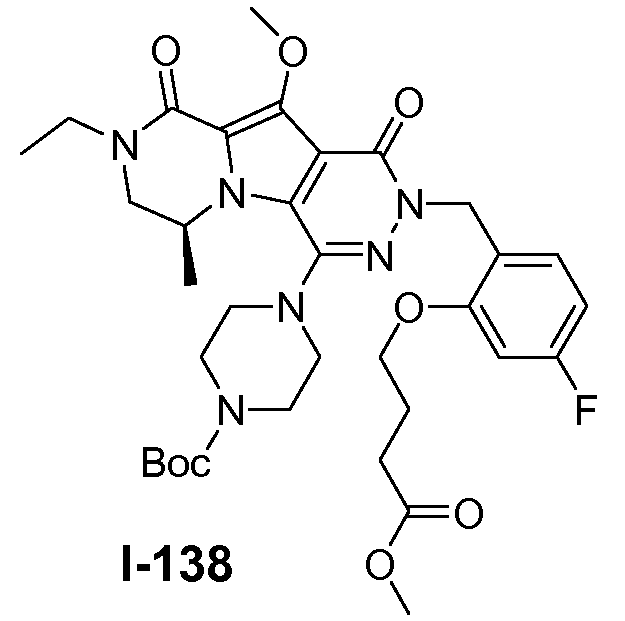
Промежуточное соединение I-137 (0,77 ммоль) растворяли в ДМФА (5 мл). Добавляли метил 4-бромбутаноат (0,92 ммоль) и K2CO3 (1,54 ммоль). Смесь перемешивали в течение ночи при 10°C. Добавляли воду (10 мл) и CH2Cl2 (20 мл). Органический слой отделяли и водный слой экстрагировали с помощью CH2Cl2 (20 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали колоночной флэш-хроматографией с использованием SiO2 (градиентное элюирование: CH2Cl2/метанол от 100:0 до 90:10). Выход: 0,45 г (чистота 90%, выход 85%).
Пример 136: промежуточное соединение I-139
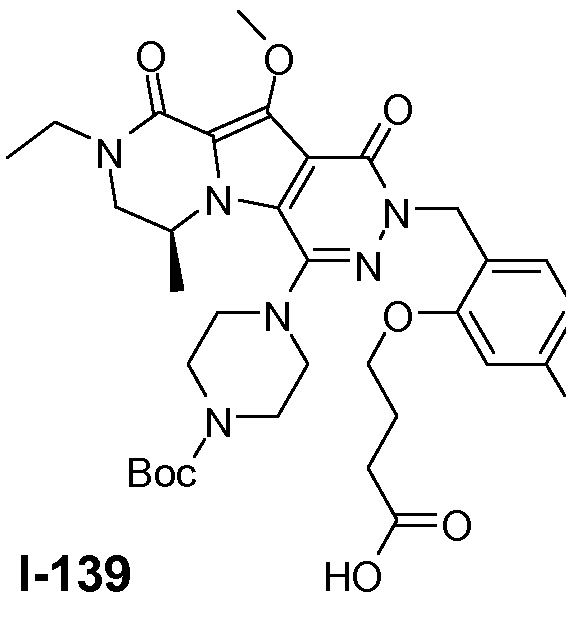
Промежуточное соединение I-138 (0,66 ммоль) растворяли в метаноле (5 мл) и ТГФ (5 мл). Добавляли водный раствор NaOH (1,3 ммоль; 5 мл). Смесь перемешивали в течение 1 часа при 15°C. Смесь экстрагировали метил-трет-бутиловым эфиром (20 мл). Водный слой доводили до pH 3-4 насыщенным водным раствором лимонной кислоты и полученную смесь экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Выход: 0,49 г (неочищенный продукт, чистота 88%).
Пример 137: промежуточное соединение I-140
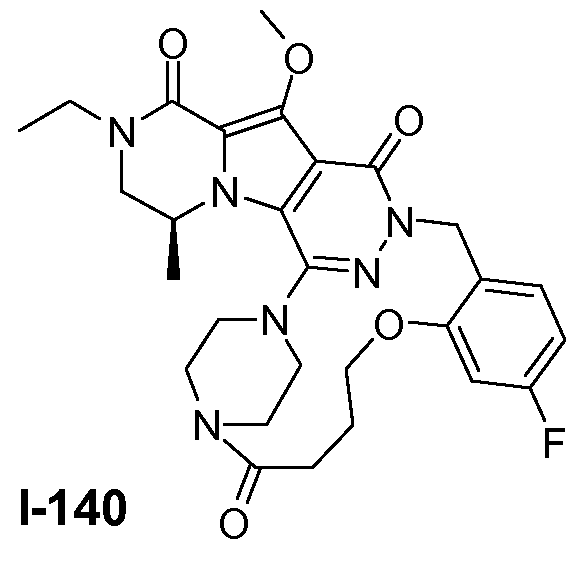
Промежуточное соединение I-140 получали, следуя процедурам, аналогичным тем, которые описаны в примере 74 и примере 103, с использованием промежуточного соединения I-139.
Пример 138: промежуточное соединение I-141
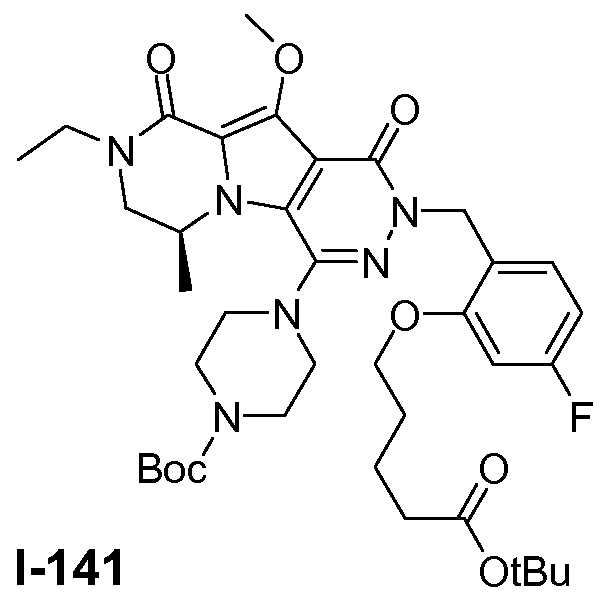
Промежуточное соединение I-141 получали, следуя процедуре, аналогичной той, которая описана в примере 135, с использованием промежуточного соединения I-137 и трет-бутил 5-бромпентаноата.
Пример 139: промежуточное соединение I-142
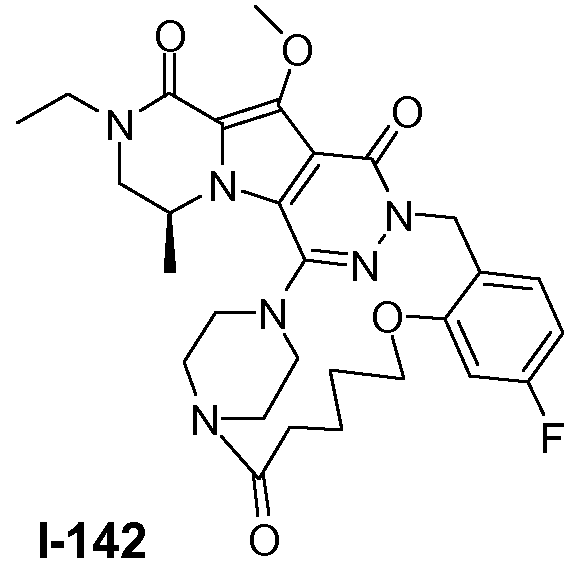
Промежуточное соединение I-142 получали, следуя процедурам, аналогичным тем, которые описаны в примерах 102-103, с использованием промежуточного соединения I-141.
Пример 140: промежуточное соединение I-144
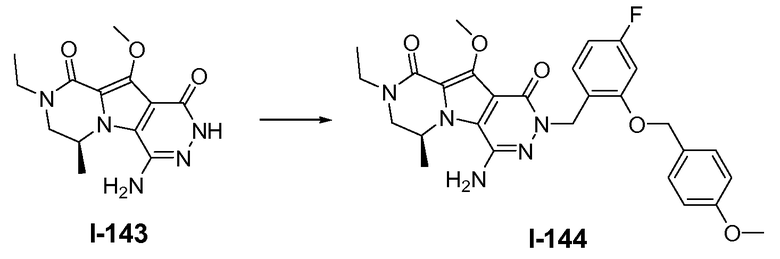
К раствору трициклического амина I-143 (12 ммоль) в ДМФА (100 мл) добавляли LiHMDS (1M в ТГФ; 16 ммоль) при 0°C. Смесь перемешивали в течение 0,5 часа и затем обрабатывали промежуточным соединением I-41 (15 ммоль) в виде одной порции. Смеси давали нагреться до 20°C и перемешивали в течение ночи. Растворитель удаляли в вакууме. Остаток растворяли в CH2Cl2 (50 мл), раствор промывали 1M раствором HCl (50 мл) и насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали с помощью ВЭЖХ (C18; элюент: метанол/H2O от 15:85 до 45:55 с 0,1% ТФУК в качестве буфера). Чистые фракции собирали, летучие вещества удаляли в вакууме и водный раствор подщелачивали насыщенным водным раствором NaHCO3 до pH 8 и экстрагировали с помощью CH2Cl2 (2×50 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Выход: 2,27 г (выход 35%).
Пример 141: промежуточное соединение I-145
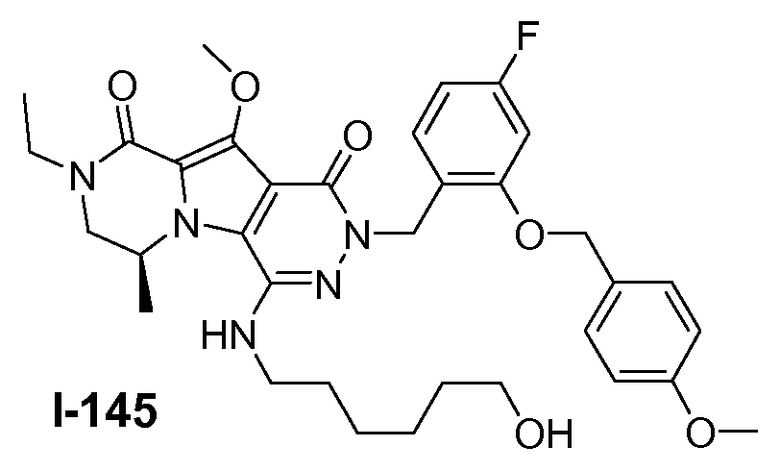
Промежуточное соединение I-145 получали, следуя процедуре, аналогичной той, которая описана в примере 120, с использованием промежуточных соединений I-144 и 6-гидроксигексаналя.
Пример 142: промежуточное соединение I-146
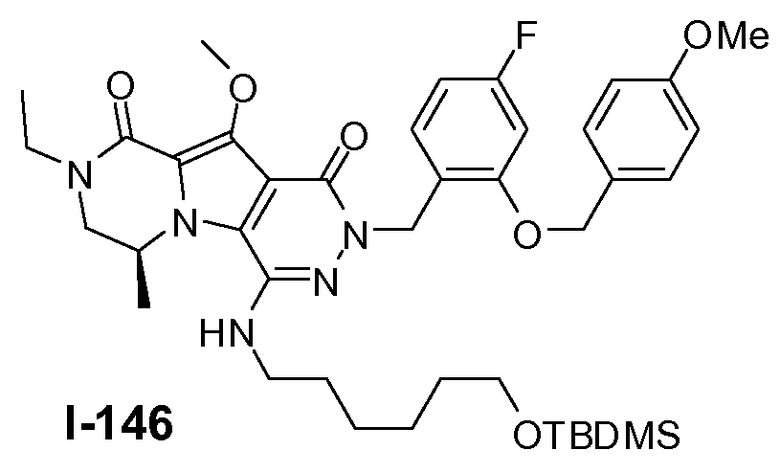
Смесь промежуточного соединения I-145 (1,4 ммоль), трет-бутилхлордиметилсилана (TBDMSCl; 14 ммоль) и имидазола (20 ммоль) в CH2Cl2 (20 мл) перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь промывали насыщенным раствором NaHCO3 и насыщенным солевым раствором. Органический слой концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией с использованием SiO2 (элюент: CH2Cl2/метанол от 100:0 до 90:10), получая чистое промежуточное соединение I-146. Выход: 1,1 г (выход 80%).
Пример 143: промежуточное соединение I-147
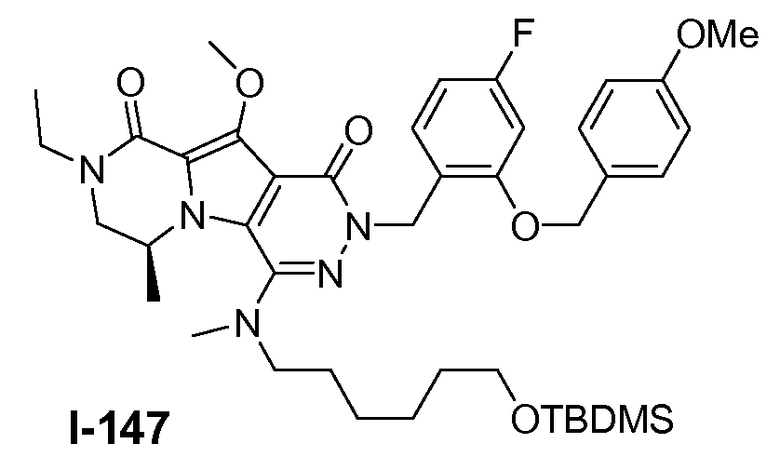
Промежуточное соединение I-146 (1,5 ммоль) растворяли в безводном ТГФ (10 мл). Добавляли NaH (60%; 2,3 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 15 минут. Добавляли по каплям йодметан (2,3 ммоль) и смесь перемешивали в течение 40 минут при 15°C. Смесь гасили насыщенным водным раствором NH4C1 и растворитель удаляли в вакууме. Смесь экстрагировали с помощью CH2Cl2 (2×20 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали колоночной флэш-хроматографией с использованием S1O2 (элюент: CH2Cl2/метанол от 100:0 до 90:10), получая чистое промежуточное соединение I-147. Выход: 0,6 г (выход 80%).
Пример 144: промежуточное соединение I-148
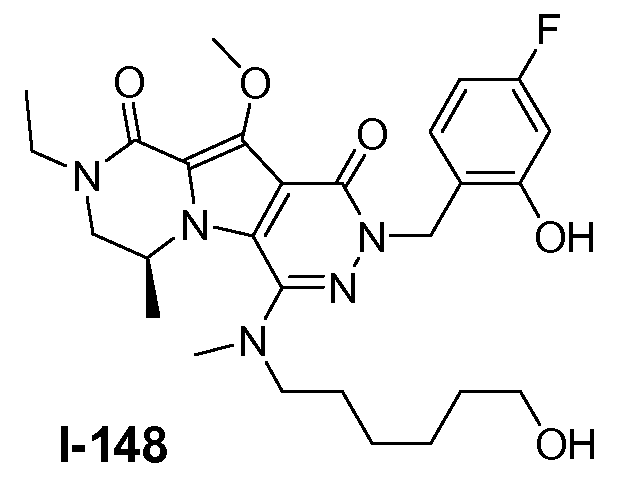
Промежуточное соединение I-148 получали, следуя процедуре, аналогичной той, которая описана в примере 74, с использованием промежуточного соединения I-147.
Пример 145: промежуточное соединение I-149
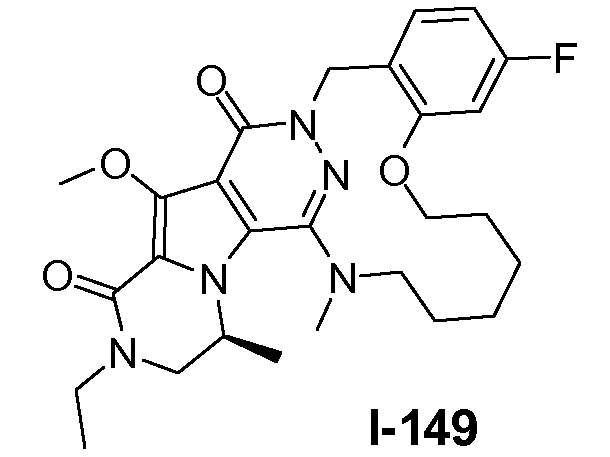
Промежуточное соединение I-149 получали, следуя процедуре, аналогичной той, которая описана в примере 75, с использованием промежуточного соединения I-148.
Пример 146: промежуточное соединение I-150
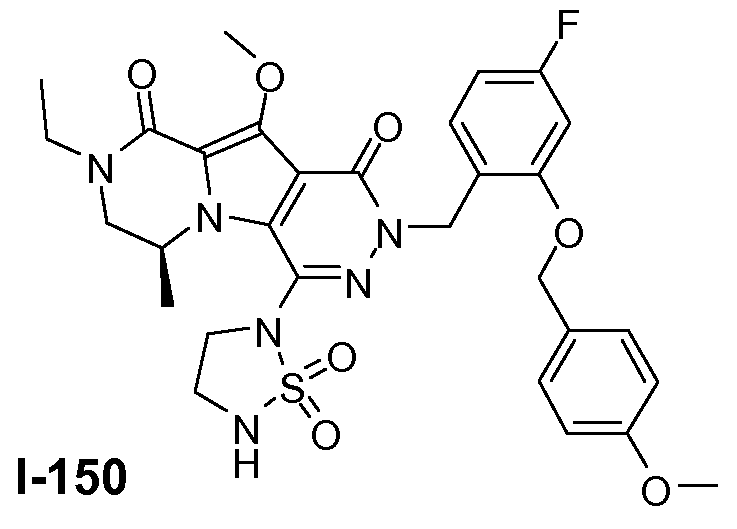
Промежуточное соединение I-150 получали, следуя процедуре, аналогичной той, которая описана в примере 72, с использованием промежуточного соединения I-72 и 1,1-диоксида 1,2,5-тиадиазолидина.
Пример 147: промежуточное соединение I-151
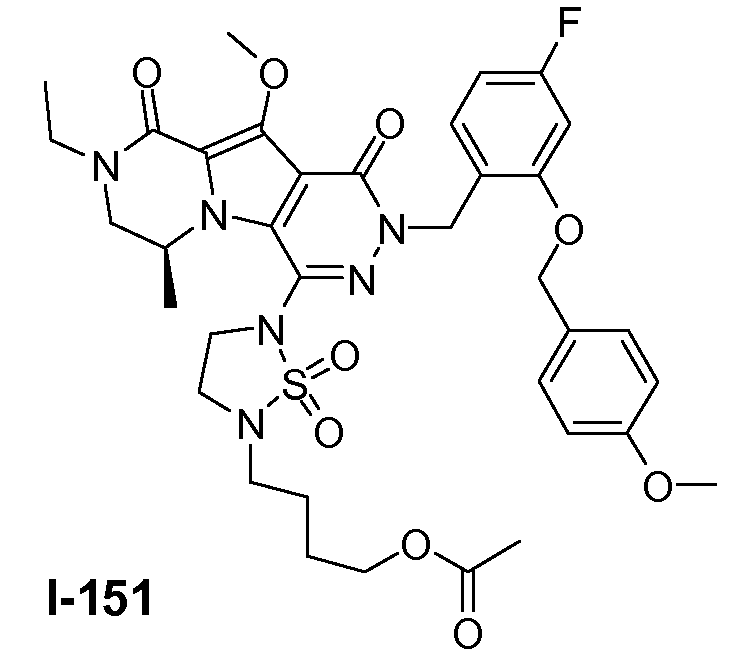
Промежуточное соединение I-150 (1,4 ммоль) растворяли в ДМФА (15 мл). Добавляли NaH (60%; 2,8 ммоль) и смесь перемешивали в течение 30 минут. Добавляли 4-бромбутилацетат (2,1 ммоль) и смесь перемешивали при 100°C в течение 5 часов. Добавляли 10% раствор NH4Cl и этилацетат. Органический слой промывали насыщенным солевым раствором три раза и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток промывали петролейным эфиром. Выход 1,1 г (чистота 70%).
Пример 148: промежуточное соединение I-152
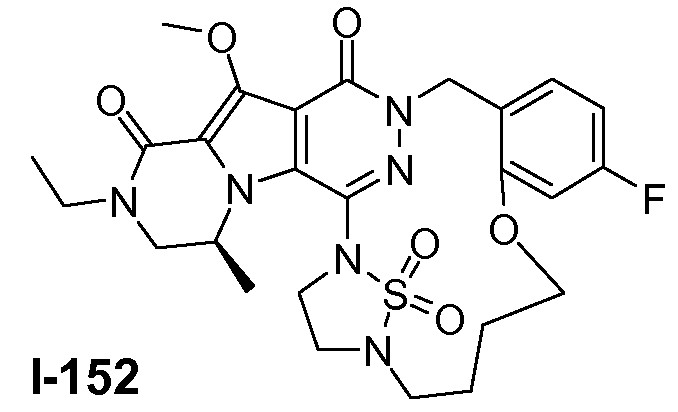
Промежуточное соединение I-152 получали, следуя процедурам, аналогичным тем, которые описаны в примере 80 и примерах 74-75, с использованием промежуточного соединения I-151.
Пример 149: промежуточное соединение I-153
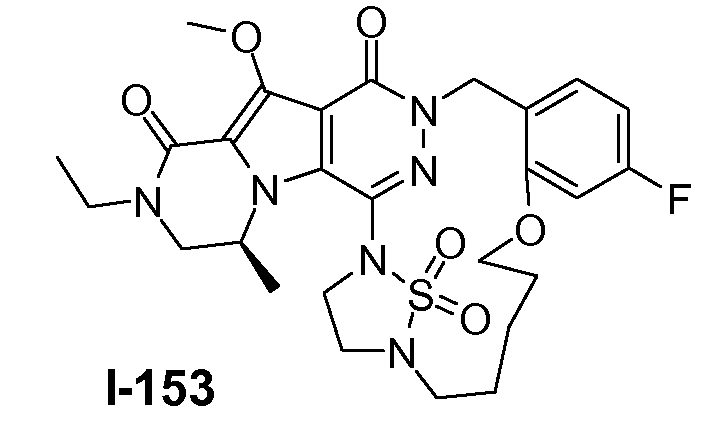
Промежуточное соединение I-153 получали, следуя процедурам, аналогичным тем, которые описаны в примере 147, примере 80 и примерах 74-75, с использованием 5-бромпентилацетата и промежуточного соединения I-150.
Пример 150: промежуточное соединение I-154
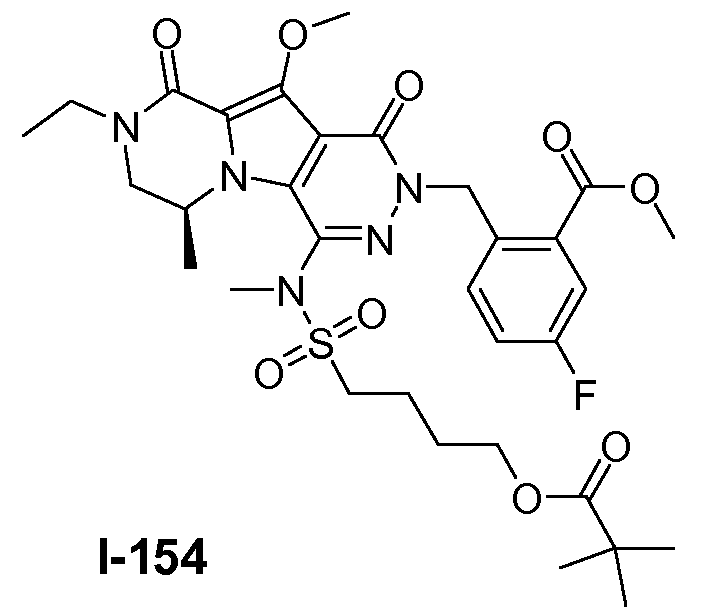
Промежуточное соединение I-154 получали, следуя процедуре, аналогичной той, которая описана в примере 95, с использованием промежуточных соединений I-96 и I-45.
Пример 151: промежуточное соединение I-155
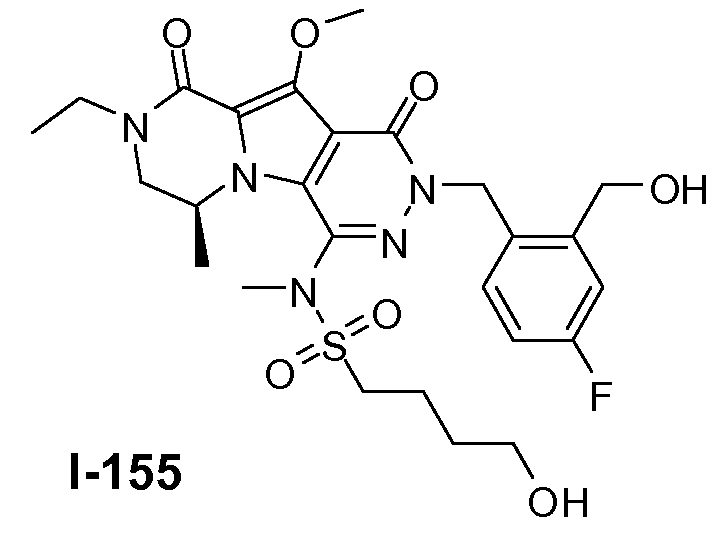
Промежуточное соединение I-154 (16,6 ммоль) и NaBH4 (166 ммоль) суспендировали в ТГФ (225 мл). Полученную смесь перемешивали в течение 15 минут при 65°C. Добавляли по каплям метанол (90 мл) в течение 0,5 часа. Перемешивание при 65°C поддерживали в течение ночи. Реакционную смесь охлаждали до 15°C и гасили насыщенным водным раствором NH4Cl (200 мл). Органический слой отделяли и водный слой экстрагировали этилацетатом (3×200 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали колоночной флэш-хроматографией с использованием SiO2 (градиентное элюирование: CH2Cl2/метанол от 100:0 до 90: 10). Выход: 5,5 г (чистота 90%; выход 57%).
Пример 152: промежуточное соединение I-156
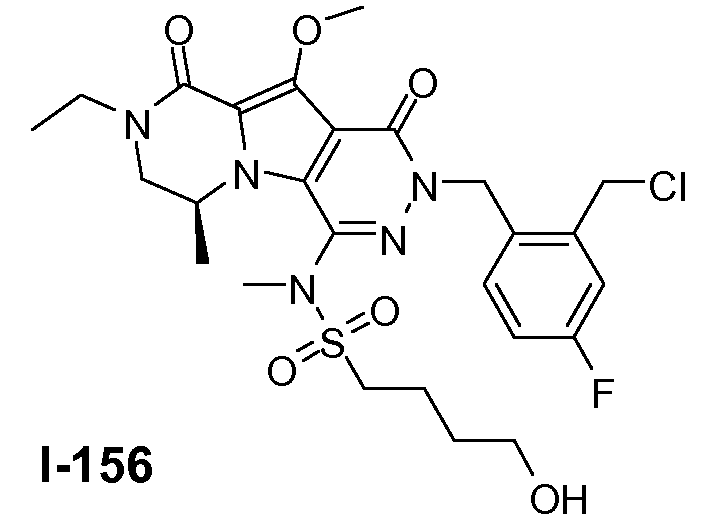
Промежуточное соединение I-155 (6,9 ммоль) и триэтиламин (21 ммоль) в CH2Cl2 (60 мл) охлаждали до 0°C. Добавляли пара-толуолсульфонилхлорид (7,6 ммоль) при 0°C. Смесь перемешивали в течение 2 часов при 0°C. Затем добавляли еще одну партию пара-толуолсульфонилхлорида (0,13 г) при 0°C. Смесь перемешивали в течение ночи при 20°C. Добавляли насыщенный водный раствор NaHCO3 (30 мл) и органический слой отделяли и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали с помощью ВЭЖХ (C18, элюент: метанол/H2O от 15:85 до 45:55 с 0,1% ТФУК в качестве буфера). Чистые фракции собирали и летучие вещества удаляли в вакууме. Затем водный раствор подщелачивали насыщенным водным раствором NaHCO3 до pH 7-8 и экстрагировали с помощью CH2Cl2 (120 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Выход: 1,36 г (чистота 98%; выход 33%).
Пример 153: промежуточное соединение I-157
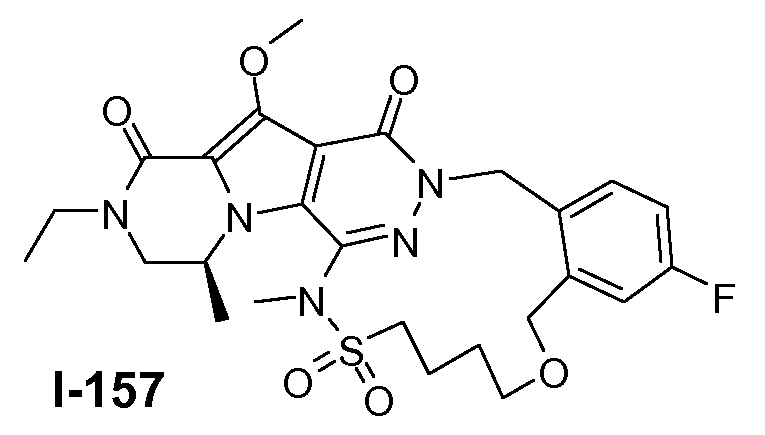
Промежуточное соединение I-156 (2,0 ммоль) растворяли в диметилацетамиде (DMA; 25 мл). Добавляли порциями трет-бутоксид калия (2,1 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 1 часа. Реакционную смесь выливали в насыщенный водный раствор лимонной кислоты (25 мл) и экстрагировали с помощью CH2Cl2 (2×50 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали колоночной флэш-хроматографией с использованием SiO2 (градиентное элюирование: CH2Cl2/метанол от 100:0 до 10:1). Полученное соединение дополнительно очищали с помощью ВЭЖХ (C18, элюент: метанол/H2O от 15:85 до 45:55 с 0,1% ТФУК в качестве буфера). Чистые фракции собирали и летучие вещества удаляли в вакууме. Затем водный раствор подщелачивали насыщенным водным раствором NaHCO3 до pH 7-8 и экстрагировали с помощью CH2Cl2 (2×20 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Выход: 0,17 г (чистота 95%; выход 15%).
Пример 154: промежуточное соединение I-158
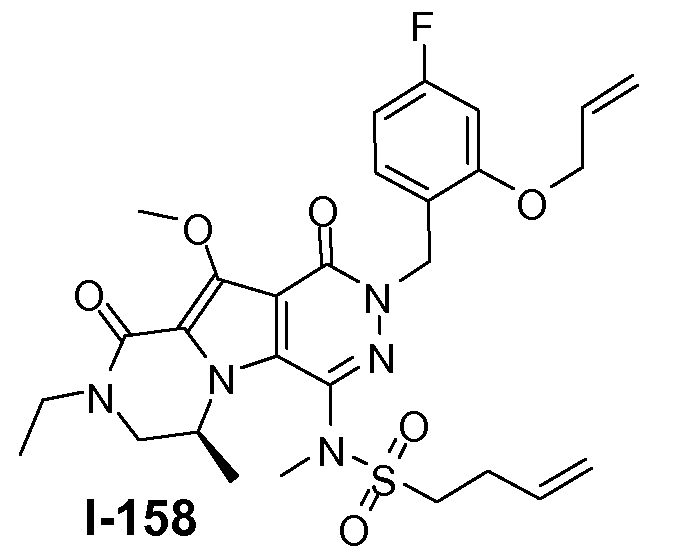
Промежуточное соединение I-158 получали, следуя процедуре, аналогичной той, которая описана в примерах 71 и 72, с использованием продуктов, полученных в примере 1, примере 41 и примере 52.
Пример 155: промежуточное соединение I-159
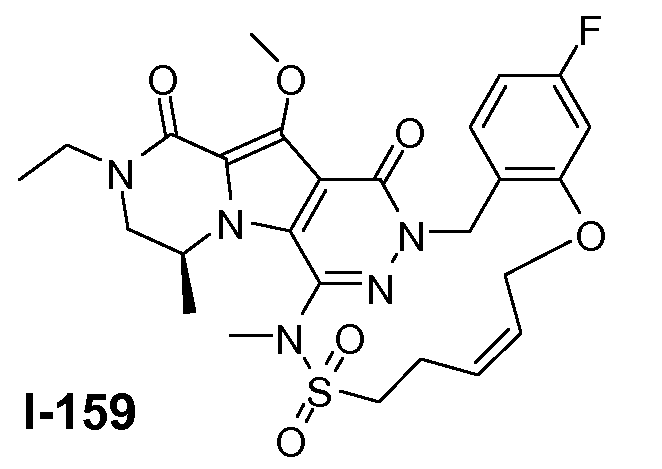
Промежуточное соединение I-158 (1,4 ммоль) и 1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)(дихлорфенилметилен)-(трициклогексилфосфин)рутений (0,14 ммоль) в безводном CH2Cl2 (200 мл) перемешивали и нагревали при температуре кипения с обратным холодильником в течение 2,5 часов. Смесь упаривали. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюент: петролейный эфир/этилацетат от 100:1 до 1:100). Чистые фракции собирали и органический растворитель выпаривали с получением 700 мг в виде смеси изомеров. Смесь очищали методом SFC разделения (колонка: ChiralPak AS, 5 мкм, Daicel Chemical Industries, Ltd 250×20 мм в.д.; подвижная фаза: A: сверхкритический CO2, B: изопропанол, A/B 75:25 при 50 мл/мин). Чистые фракции собирали и растворитель концентрировали в вакууме с получением 400 мг изомерной смеси (выход 57%), которая содержала два пика, присутствующих по данным SFC анализа. Эту смесь дополнительно очищали методом SFC разделения (колонка: ChiralPak AS, 5 мкм, Daicel Chemical Industries, Ltd 250×20 мм в.д.; подвижная фаза: A: сверхкритический CO2, B: изопропанол, A/B 70:30 при 40 мл/мин). Чистые фракции собирали и растворитель концентрировали в вакууме. Выход: 0,10 г.
Пример 156: промежуточное соединение I-160
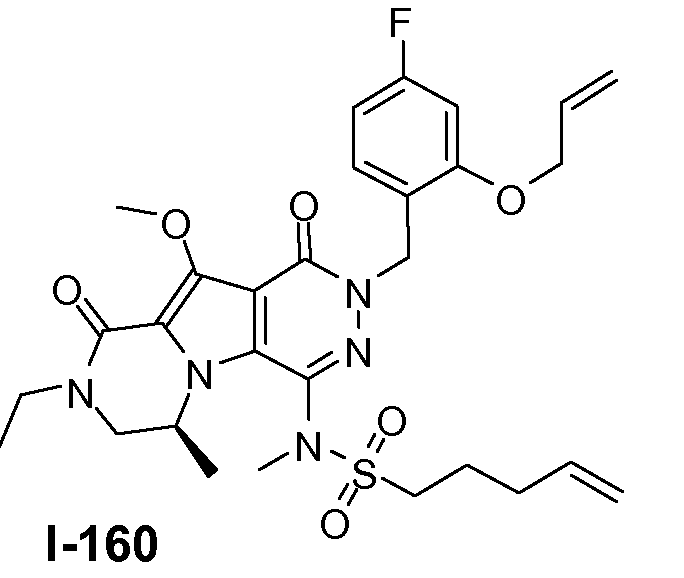
Промежуточное соединение I-160 получали, следуя процедуре, аналогичной той, которая описана в примерах 71 и 72, с использованием продуктов, полученных в примере 1, примере 41 и примере 53.
Пример 157: промежуточное соединение I-161 (Z-изомер) и промежуточное соединение I-162 (E-изомер)
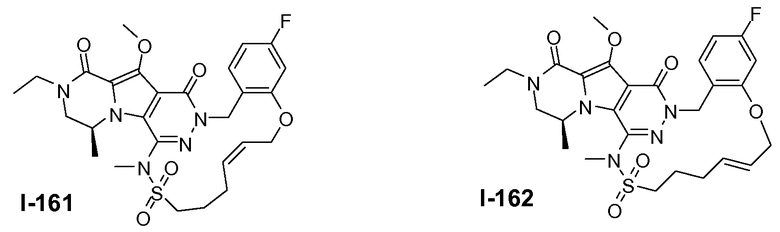
Промежуточное соединение I-160 (1,7 ммоль) и катализатор Ховейда-Граббса 2-го поколения 1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(o-изопропоксифенилметилен)рутений (0,17 ммоль) в безводном CH2Cl2 (200 мл) перемешивали и нагревали при температуре кипения с обратным холодильником в течение 2,5 часов в атмосфере N2. Смесь упаривали. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюент: петролейный эфир/этилацетат от 100:1 до 1:100). Чистые фракции собирали и органический растворитель выпаривали с получением 0,8 г в виде смеси изомеров. Эту смесь очищали методом SFC разделения (колонка: ChiralPak AD, 5 мкм, Daicel Chemical Industries, Ltd 250×30 мм в.д.; подвижная фаза: A: сверхкритический CO2, B: метанол, A/B 60:40 при 40 мл/мин). Чистые фракции собирали и растворитель концентрировали в вакууме с получением промежуточного соединения I-161 (0,4 г) и промежуточного соединения I-162 (0,1 г).
Пример 158: соединение 1 (удаление защиты, способ A)
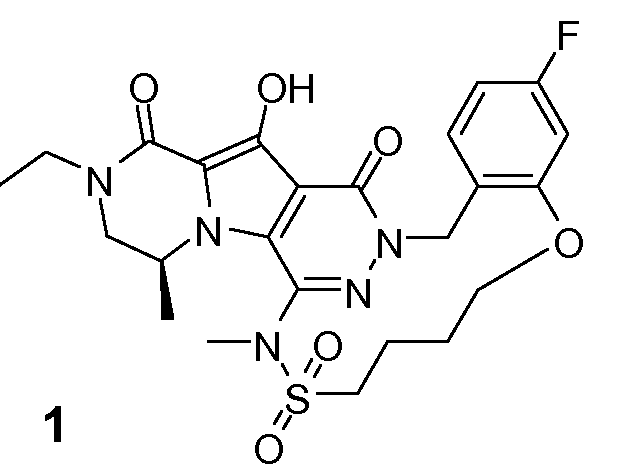
Промежуточное соединение I-76 (45,7 ммоль) и LiCl (457 ммоль) в ДМФА (250 мл) перемешивали в течение 3 часов при 140°C. Добавляли воду. Смесь экстрагировали с помощью CH2Cl2 (2×500 мл). Объединенные органические слои промывали водой (2×500 мл), 10% раствором лимонной кислоты (500 мл) и насыщенным солевым раствором (2×500 мл), сушили над Na2SO4 и упаривали в вакууме. Остаток растирали в порошок с трет-бутилметиловым эфиром (200 мл). Осадок собирали фильтрованием и затем растворяли в кипящем CH3CN (120 мл). Смеси давали медленно охладиться до 20°C. Твердое вещество отфильтровывали, сушили в условиях высокого вакуума при 45°C в течение 2 часов. Затем твердое вещество суспендировали в CH3CN (50 мл), нагревали при температуре кипения с обратным холодильником в течение 30 минут и затем перемешивали при 0°C в течение 15 минут. Смесь фильтровали. Осадок сушили в вакуумной печи при 45°C в течение ночи. Полученный продукт суспендировали в CH3CN (30 мл) и затем смесь перемешивали при 20°C в течение ночи. Смесь фильтровали. Собранный осадок сушили в условиях высокого вакуума при 20°C в течение 1 часа.
Выход: 10 г соединения 1 (выход 41%),
э.и.: 100%.
CHN моногидрат (% на основании теоретического количества): C, 52,26 (99,02%); H, 5,48 (97,23%); N, 12,70 (99,21%).
Оптическое вращение: α (589 нм, 20°C)=-64,93 (растворитель: CH2Cl2, c=5,06 мг/мл)
Пример 159: соединения 2a и 2b (стабильные ротамеры)
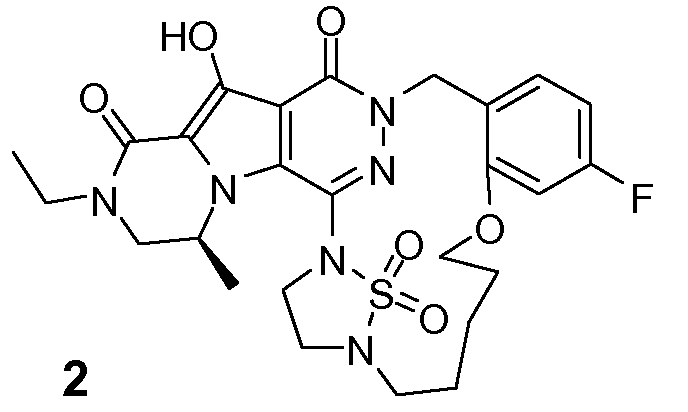
Соединения 2a и 2b получали, следуя процедуре, аналогичной той, которая описана для соединения 1, с использованием промежуточного соединения I-153. Аналитическая сверхкритическая жидкостная хроматография показала два отдельных пика на базовой линии, которые разделяли препаративно с использованием сверхкритической жидкостной хроматографии (SFC; колонка: AD 250×20 мм, 20 мкм, подвижная фаза: A: сверхкритический CO2, B: пропан-2-ол, A/B 45:55 при 80 мл/мин, длина волны: 220 нм).
Выход 20 мг соединения 2a в виде быстро движущегося изомера;
SFC: чистота 99%
Выход 30 мг соединения 2b в виде медленно движущегося изомера;
SFC: чистота 96,6%
Пример 160: соединение 3 (удаление защиты, способ B)
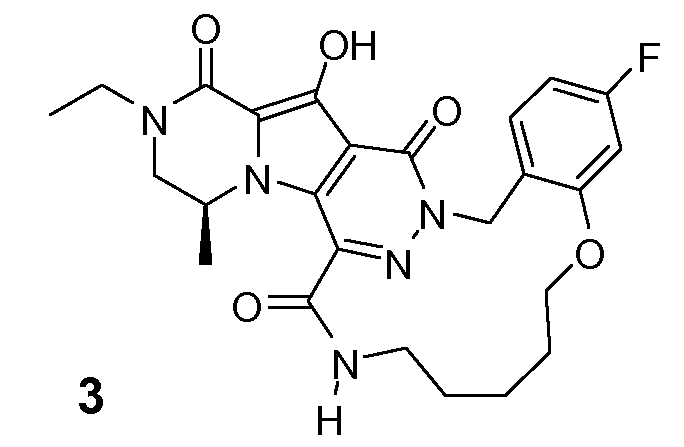
Промежуточное соединение I-105 (90 ммоль) и NaI (174 ммоль) растворяли в смеси CH3CN (760 мл) и толуола (760 мл) при 30°C. Добавляли по каплям тетрахлорсилан (174 ммоль) при 0°C. Смесь перемешивали в течение 2 часов при 30°C. Затем смесь выливали на лед. Добавляли CH2Cl2 (1500 мл) и воду (1500 мл). Органические слои собирали и промывали 10% раствором NaHCO3, 10% раствором лимонной кислоты, 10% раствором Na2SO3, 2 н. раствором HCl, затем насыщенным солевым раствором. Полученный органический слой сушили над Na2SO4 и фильтровали. Растворитель удаляли в вакууме. Полученный остаток промывали трет-бутилметиловым эфиром и метанолом. Полученный неочищенный продукт затем снова кристаллизовали из CH3CN (30 г продукта/300 мл CH3CN).
Выход: 18 г соединения 3 (выход 40%).
% э.и. по данным SFC-MS: 99,99%
CHN (% основан на теоретическом количестве: C, 60,49 (100,2%). H, 5,525 (97,4%). N, 14,44 (102,6%).
Оптическое вращение: α (589 нм, 20°C): 53,05 (c=9,61 мг/мл, CH2Cl2)
Пример 161: соединение 4 (удаление защиты, способ C)
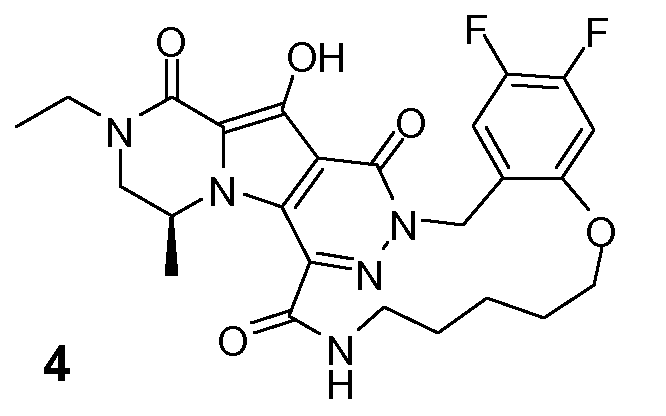
К раствору промежуточного соединения I-107 (0,56 ммоль) в CH2Cl2 (25 мл) добавляли по каплям BBr3 (5,6 ммоль, в виде 1M раствора в CH2Cl2) при -78°C. Смеси давали нагреться до 15°C и перемешивали в течение 0,5 часа. Добавляли воду (10 мл). Органический слой отделяли и водный слой экстрагировали с помощью CH2Cl2 (2×30 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали с помощью ВЭЖХ (C18, элюент: метанол/H2O от 15:85 до 45:55 с 0,1% ТФУК в качестве буфера). Чистые фракции собирали, летучие вещества удаляли в вакууме, водный раствор подщелачивали NaHCO3 до pH 8 и экстрагировали с помощью CH2Cl2 (2×80 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме.
Выход: 45 мг соединения 4 (чистота 99%; выход 20%)
э.и. по данным SFC-MS: 100%
Пример 162: ссылочные соединения 52r, 53r и 54r
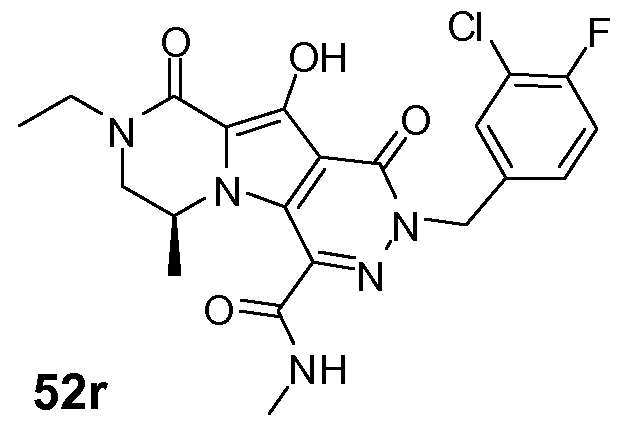
Соединение 52r, также известное как MK-2048, описано в публикации Vacca, J.P., J.S. Wai, et al. (2007; Discovery of MK-2048: subtle changes confer unique resistance properties to a series of tricyclic hydroxypyrrole integrase strand transfer inhibitors. 4th International AIDS Society (IAS) Conference, Sydney, Australia).
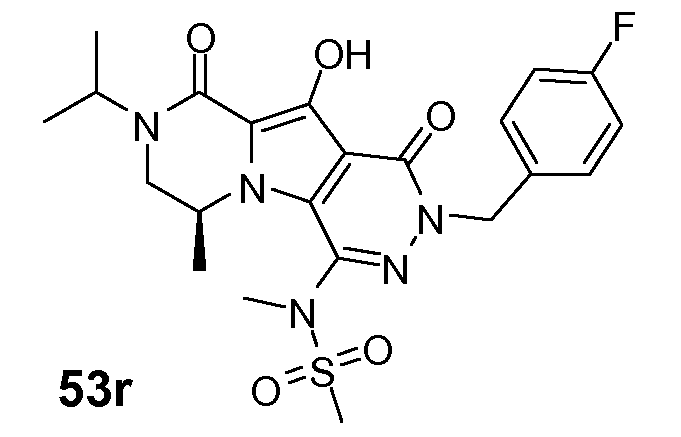
Соединение 53r описано в WO2005110414 (пример номер 159-A).
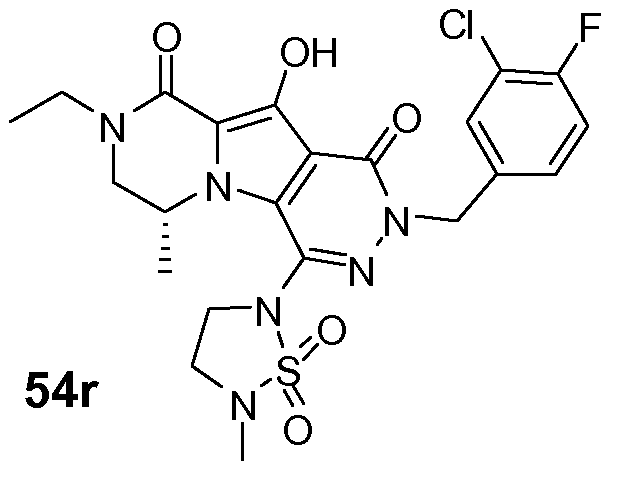
Соединение 54r описано в публикации Wai, J.S., T.E. Fisher, et al. (2007; Next Generation of Inhibitors of HIV-1 Integrase Strand Transfer Inhibitor: Structural Diversity and Resistance Profiles. CROI, Los Angeles, California).
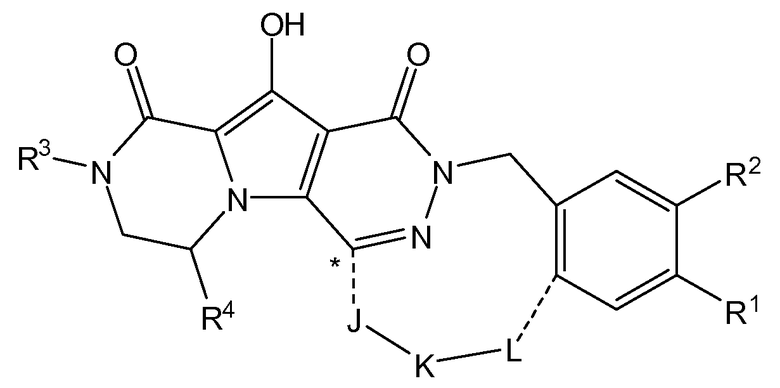
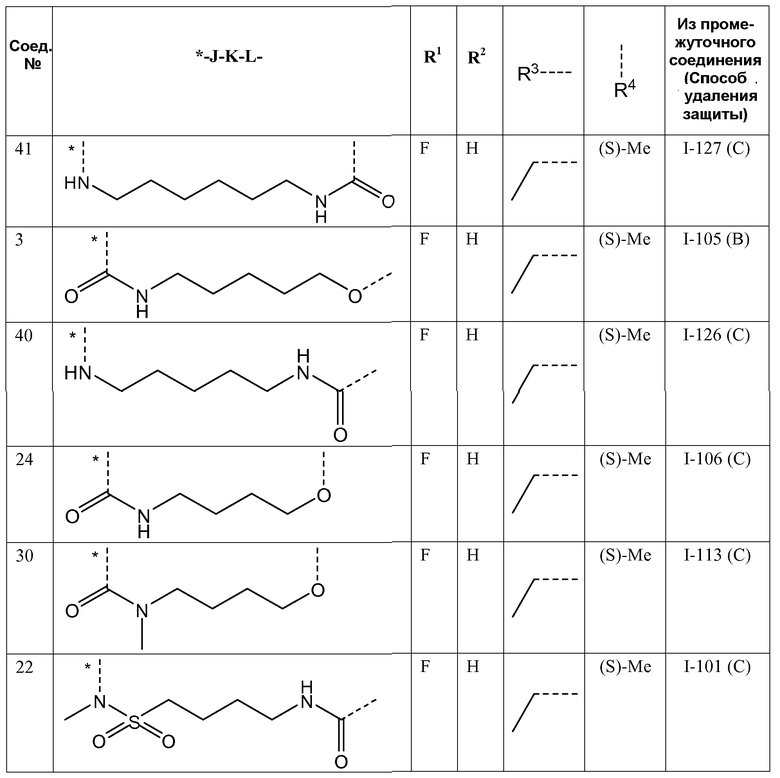
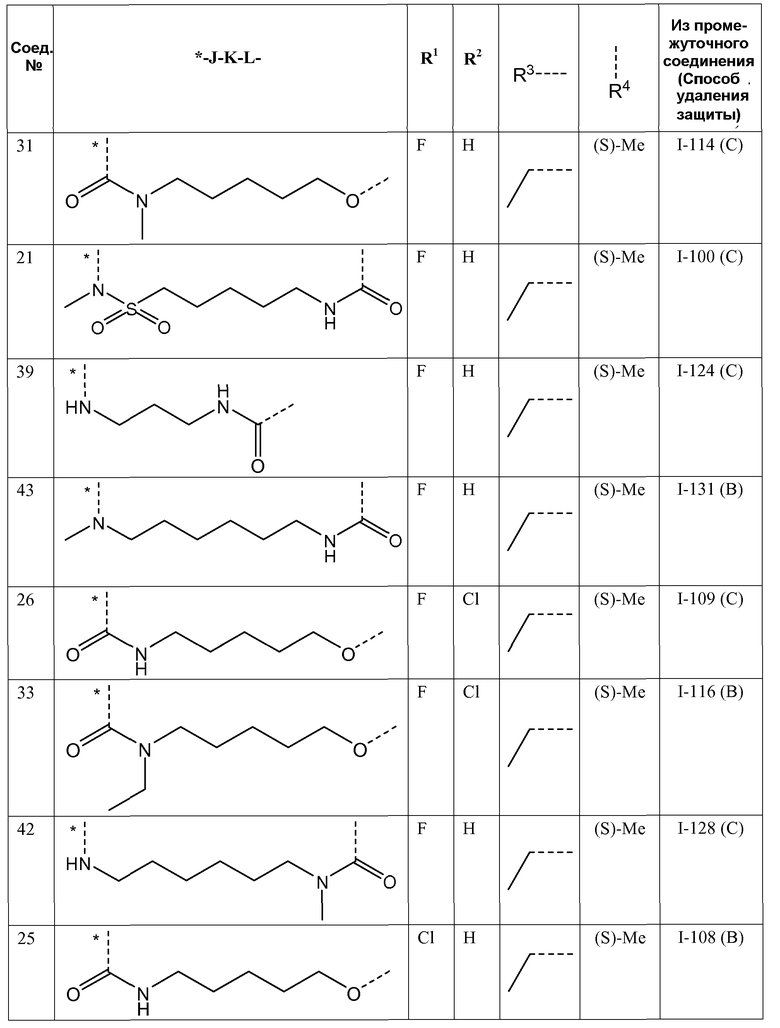
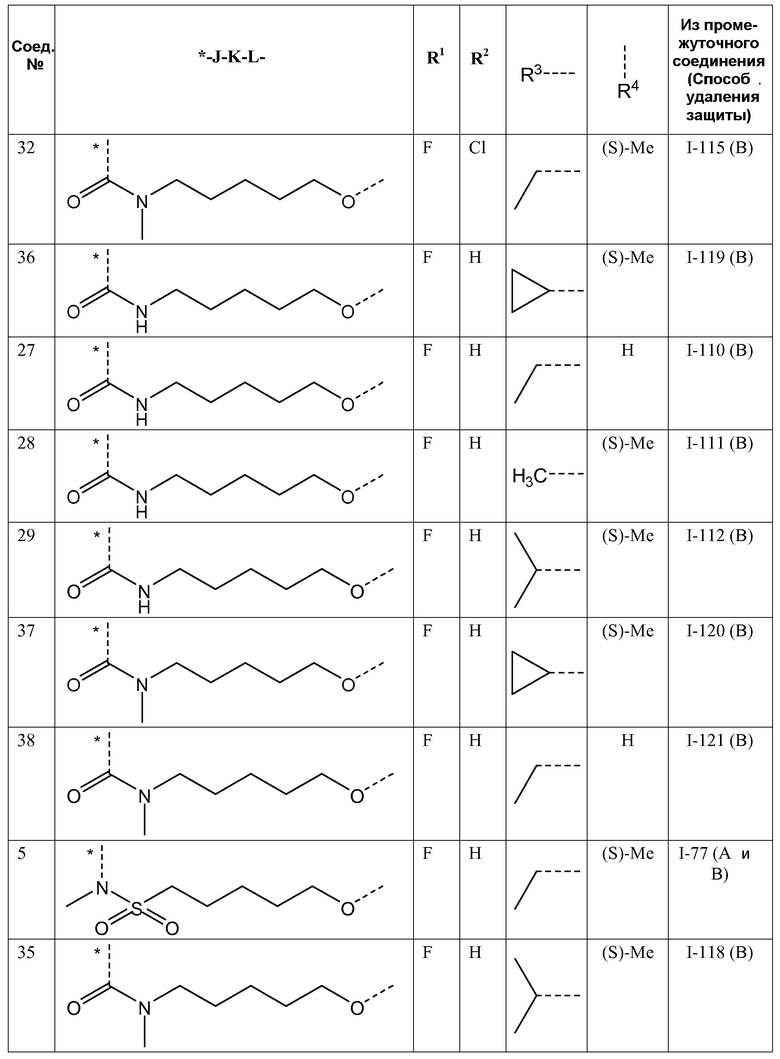
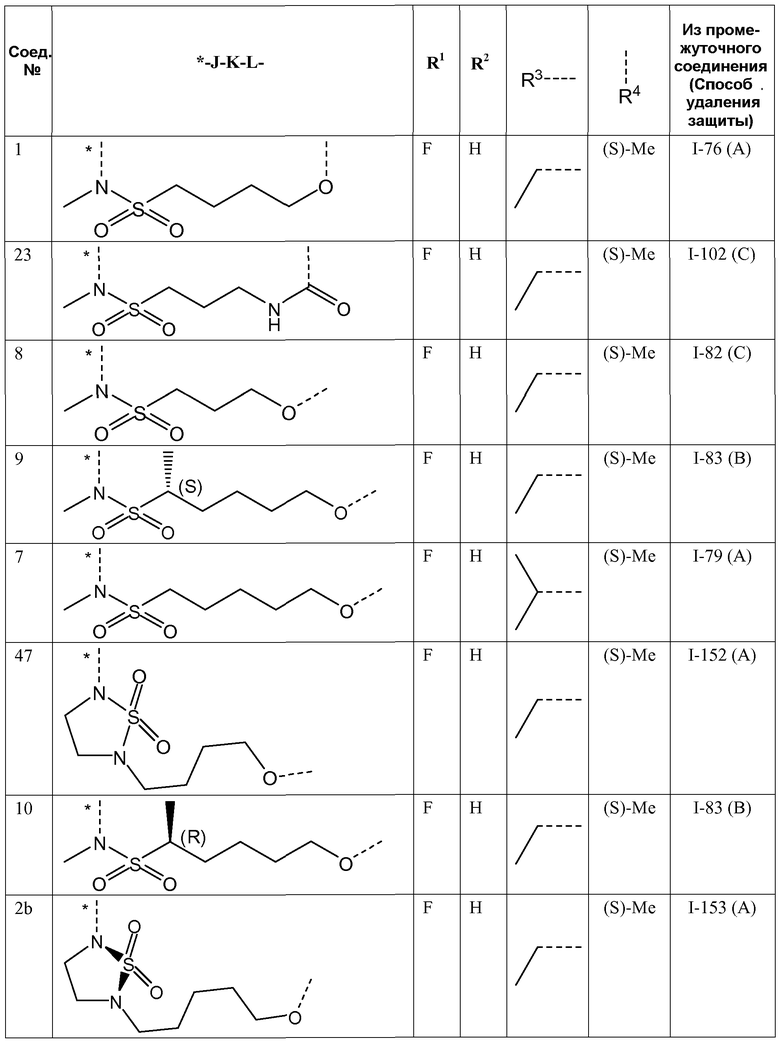
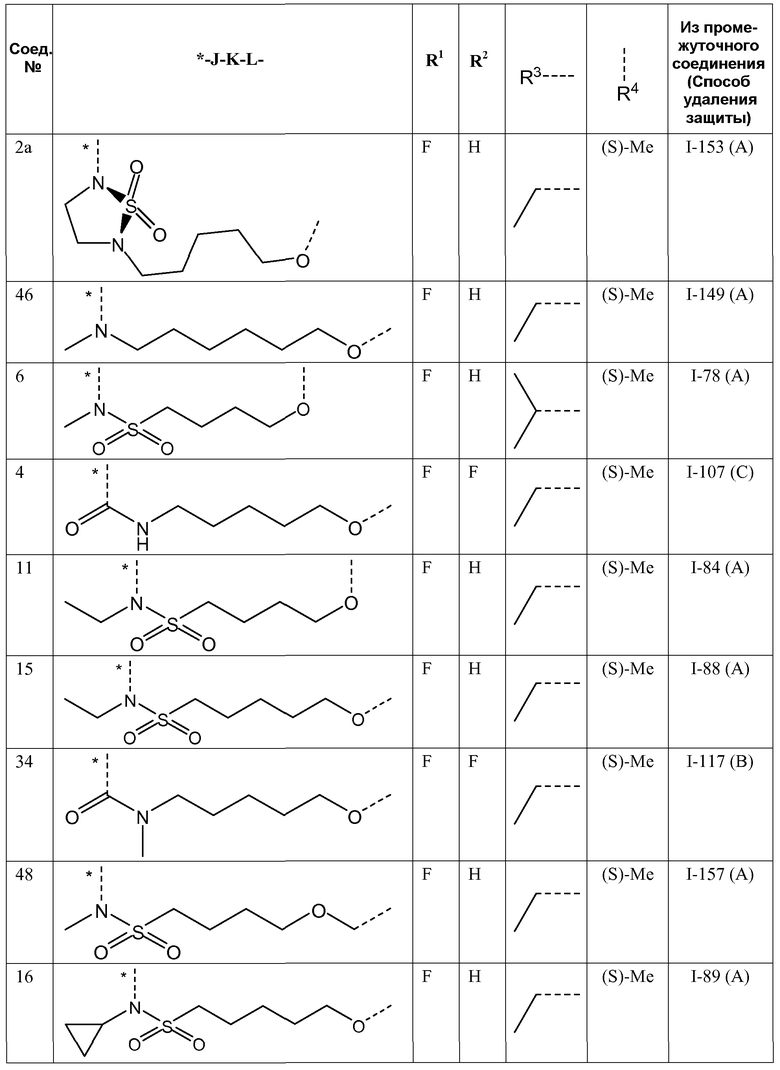
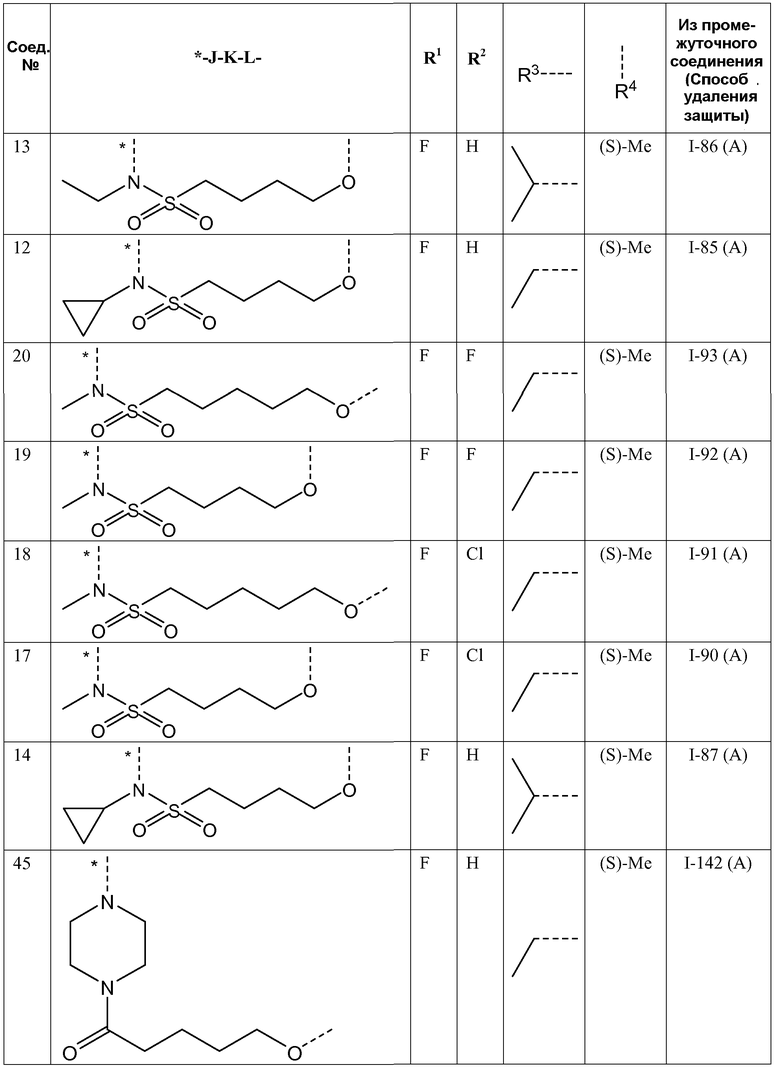
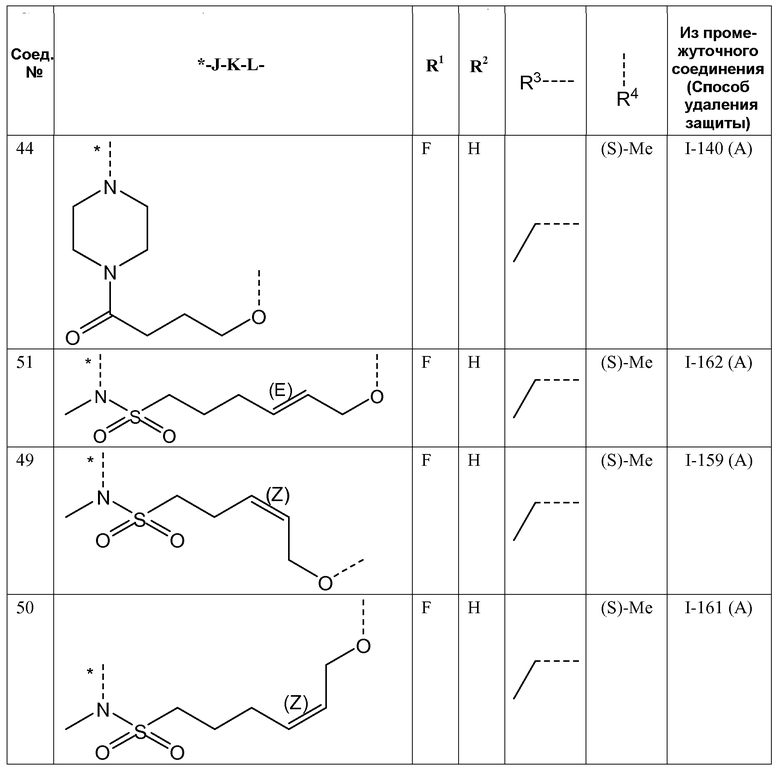
Пример 163: ЖХ/МС анализ
Общая процедура для М1 и M2
Определение характеристик методом ВЭЖХ осуществляли с использованием модуля Agilent 1100, включающего насос, детектор на диодной матрице (DAD) (используемая длина волны 220 нм), нагреватель для колонки и колонку, как определено в соответствующих способах ниже. Поток из колонки разделяли для отведения к Agilent MSD Series G1946C и G1956A. МС детектор был сконфигурирован с API-ES (ионизация электрораспылением при атмосферном давлении). Масс-спектры получали путем сканирования от 100 до 1000. Напряжение на игле капилляра было 2500В для режима положительной ионизации и 3000В для режима отрицательной ионизации. Напряжение фрагментации было 50В. Температуру сушильного газа поддерживали при 350°C при скорости потока 10 л/мин.
Способ (M1)
В дополнение к общей процедуре, осуществляли обращенно-фазную ВЭЖХ на колонке YMC-Pack ODS-AQ, 50×2,0 мм 5 мкм при скорости потока 0,8 мл/мин. Использовали две подвижные фазы (подвижная фаза A: вода с 0,1% ТФУК; подвижная фаза B: CH3CN с 0,05% ТФУК). Сначала 100% A удерживали в течение 1 минуты. Затем использовали градиент до 40% A и 60% B в течение 4 минут и удерживание в течение 2,5 минут. Использовали обычные объемы вводимых проб 2 мкл. Температура печи составляла 50°C. (МС полярность: положительная)
Способ (M2)
В дополнение к общей процедуре, обращенно-фазную ВЭЖХ осуществляли на колонке YMC-Pack ODS-AQ, 50×2,0 мм 5 мкм при скорости потока 0,8 мл/мин. Использовали две подвижные фазы (подвижная фаза A: вода с 0,1% ТФУК; подвижная фаза B: CH3CN с 0,05% ТФУК). Сначала 90% A и 10% B удерживали в течение 0,8 минут. Затем использовали градиент до 20% A и 80% B в течение 3,7 минут и удерживали в течение 3 минут. Использовали обычные объемы вводимых проб 2 мкл. Температура печи составляла 50°C. (МС полярность: положительная)
Способ (M3)
ЖХ анализ осуществляли с использованием системы Acquity UPLC (Ultra Performance Liquid Chromatography, Waters), включающей бинарный насос, пробоотборник, нагреватель для колонки (установленный на 55°C), детектор на диодной матрице (DAD) и колонку, как определено в соответствующих способах ниже. Поток из колонки разделяли для отведения к МС спектрометру. МС детектор был сконфигурирован с источником ионизации электрораспылением. Масс-спектры получали путем сканирования от 100 до 1000 в течение 0,18 секунд с использованием времени выдержки 0,02 секунд. Напряжение на игле капилляра было 3,5 кВ, и температурный источник поддерживали при 140°C. Азот использовали в качестве распылительного газа. Сбор данных осуществляли с использованием системы данных Waters-Micromass MassLynx-Openlynx. Обращенно-фазную UPLC осуществляли на параллельной колонке C18 с гибридом этилсилоксан/диоксид кремния (BEH) (1,7 мкм, 2,1×50 мм; Waters Acquity) при скорости потока 0,8 мл/мин. Использовали две подвижные фазы (25 мМ ацетата аммония в H2O/ацетонитрил 95:5; подвижная фаза B: ацетонитрил) с осуществлением условий градиента от 95% A и 5% B до 5% A и 95% B в течение 1,3 минут, и удерживание в течение 0,3 минут. Использовали объем вводимой пробы 0,5 мкл.
Напряжение на конусе было 10В для режима положительной ионизации и 20В для режима отрицательной ионизации.
Пример 164: определение температур плавления
DSC
Для ряда соединений температуры плавления определяли с использованием DSC823e (Mettler-Toledo). Температуры плавления измеряли с использованием температурного градиента 30°C/мин. Максимальная температура 400°C.
WRS-2A
Для ряда соединений температуры плавления определяли с использованием устройства для определения температуры плавления WRS-2A, которое закупали у компании Shanghai Precision and Scientific Instrument Co. Ltd. Температуры плавления измеряли с использованием линейного нагрева до 0,2-5,0°C/мин. Указанные значения представляют собой диапазоны температур плавления. Максимальная температура 300°C.
Rt
[M+H]+
°С
Пример 165: определение спектров ЯМР
1H спектры записывали на спектрометре Bruker AVANCE III (400МГц), Bruker AVANCE (600 МГц) или Varian 400-MR (400МГц).
1H ЯМР (400 МГц, CDCl3) δ м.д. 1,10 (д, J=6,9 Гц, 3H), 1,14 (д, J=6,9 Гц, 3H), 1,26 (д, J=6,7 Гц, 2,7H), 1,40 (д, J=6,3 Гц, 0,3H), 1,73-1,85 (м, 0,9H), 1,88 (шир.с, 0,1H), 2,00-2,19 (м, 2H), 2,61 (шир.с, 0,1H), 2,70-2,86 (м, 0,9H), 3,09-3,35 (м, 3,3H), 3,40 (c, 2,7H), 3,43-3,54 (м, 0,2H), 3,59-3,75 (м, 1,8H), 4,00-4,10 (м, 1H), 4,86 (квинтет, J=6,7 Гц, 1,1H), 5,02 (д, J=13,1 Гц, 0,9H), 5,09 (д, J=13,1 Гц, 0,1H), 5,30-5,40 (м, 1H), 5,33 (д, J=13,5 Гц, 0,9H), 6,44 (дд, J1=10,8 Гц, J2=2,1 Гц, 0,95H), 6,48 (м, 0,05H), 6,57 (дт, J1=8,2 Гц, J2=2,1 Гц, 1H), 7,78 (т, J=7,4 Гц, 1H), 8,48 (шир.с, 0,9 H), 8,52 (шир.c, 0,1H)
1H ЯМР (300 МГц, ДМСО) δ м.д. 1,29 (т, J=1,8 Гц, 3H), 1,33-1,41 (м, 6H), 1,50-1,99 (м, 5,5H), 2,70 (м, 0,5H), 3,21 (c, 1,5H), 3,25 (c, 1,5H), 3,42 (м, 1,5H), 3,54-3,79 (м, 3H), 3,93-4,28 (м, 2,5H), 4,70 (м, 0,5H), 5,11-5,20 (м, 2H), 5,35 (д, J=13,5 Гц, 0,5H), 6,73 (т, J=8,1 Гц, 1H), 6,85 (д, J=10,8 Гц, 0,5H), 6,95 (д, J=10,8 Гц, 0,5H), 7,62 (т, J=7,8 Гц, 0,5H), 7,70 (т, J=7,8 Гц, 0,5H), 9,09 (шир.с, 1H)
1H ЯМР (400 МГц, ДМСО-d) δ м.д. 0,86-1,41 (м, 6H), 1,21 (д, J=6,6 Гц, 2H), 1,27 (д, J=6,4 Гц, 1H), 1,75 (шир.с, 3H), 2,32 (шир.с, 1H), 3,35-3,59 (м, 4,33H), 3,59-3,91 (м, 4H), 4,01 (шир.с, 1,67H), 4,87-5,04 (м, 1,33H), 5,07-5,26 (м, 1,67H), 6,59-6,72 (м, 1H), 6,72-6,86 (м, 1H), 7,50-7,61 (м, 1H), 8,96 (шир.с, 0,67H), 9,01 (шир.с, 0,33H)
1H ЯМР (400 МГц, ДМСО-d) δ м.д. 0,33-0,41 (м, 0,5H), 0,46-0,61 (м, 1H), 0,69 (п, J=0,73 Гц, 0,5H), 0,82-1,00 (м, 2H), 1,08-1,19 (м, 3H), 1,26 (д, J=6,7 Гц, 1,5H), 1,39 (д, J=6,4 Гц, 1,5H), 1,83-2,02 (м, 2H), 2,02-2,24 (2 шир.с, 1H), 2,56-2,72 (м, 1H), 3,07-3,17 (м, 1H), 3,17-3,32 (м, 1,5H), 3,35-3,50 (м, 2H), 3,50-3,66 (м, 1H), 3,66-3,80 (м, 1H), 3,84 (тд, J1=12,9 Гц, J2=3,5 Гц, 1H), 3,88-3,96 (м, 0,5H), 3,96-4,06 (м, 1H), 4,81 (шир.с, 0,5H), 5,05 (д, J=13,4, 0,5H), 5,10 (д, J=13,3, 0,5H), 5,19 (шир.с, 0,5H), 5,30 (д, J=13,4 Гц, 0,5H), 5,37 (д, J=13,3 Гц, 0,5H), 6,41-6,51 (м, 1H), 6,57 (т, J=8,2 Гц, 1H), 7,68-7,79 (м, 1H), 8,38 (шир.с, 0,5H), 8,44 (шир.с, 0,5H)
1H ЯМР (400 МГц, ДМСО-d) δ м.д. 1,03 (т, J=7,3 Гц, 1,2H), 1,07-1,20 (м, 7,8H), 1,26 (д, J=6,5 Гц, 1,8H), 1,33 (д, J=6,2 Гц, 1,2H), 1,84 (шир.с, 2H), 1,92 (шир.с, 0,6H), 2,01 (шир.с, 0,4H), 2,43 (шир.с, 1H), 3,45-3,72 (м, 3,3H), 3,72-3,95 (м, 2,7H), 4,10 (шир.с, 1,60H), 4,73-4,83 (шир.с, 1,4H), 4,97-5,09 (м, 1,4H), 5,19-5,33 (м, 1,6H), 6,75 (шир.с, 1H), 6,85 (дд, J1=11,3 Гц, J2=2,6 Гц, 0,6H), 6,91 (дд, J1=11,3 Гц, J2=2,2 Гц, 0,4H), 7,60-7,67 (м, 1H), 9,07 (шир.с, 0,6H), 9,12 (шир.с, 0,4H)
1H ЯМР (400 МГц, ДМСО-d) δ м.д. 0,42 (шир.с, 1H), 0,53 (шир.с, 0,4H), 0,65 (шир.с, 0,6H), 0,72-0,94 (м, 2H), 0,95-1,09 (м, 6H), 1,12 (д, J=6,0, 1,8H), 1,21 (J=6,3 Гц, 1,2H), 1,81 (шир.с, 3H), 2,43 (шир.с, 1H), 3,07 (шир.с, 1H), 3,35-3,78 (м, 3,6H), 3,78-4,17 (м, 2,4H), 4,60-4,84 (м, 1,6H), 4,84-5,24 (м, 2,4H), 6,65 (т, J=8,3 Гц, 1H), 6,76 (д, J=11,4 Гц, 0,6H), 6,83 (д, J=11,0 Гц, 0,4H), 7,48-7,59 (м, 1H), 8,97 (2 шир.с, 1H)
1H ЯМР (300 МГц, ДМСО-d) δ м.д. 0,95 (т, J=7,2 Гц, 2H), 1,03 (т, J=6,9 Гц, 1H), 1,14 (т, J=6,9 Гц, 3H), 1,28-1,40 (м, 3H), 1,63 (шир.с, 2H), 1,86 (шир., 3H), 2,21 (шир. 0,4H), 2,41 (шир. 0,6H), 3,37-3,81 (м, 7H), 3,92-4,32 (м, 3H), 4,99 (м, 0,66H), 5,01-5,37 (м, 2,33H), 6,75 (дт, J1=8,4 Гц, J2=2,1 Гц, 1H), 6,92 (дд, J1=6,9 Гц, J2=2,4 Гц, 1H), 7,52 (т, J=7,23 Гц, 0,33H), 7,67 (т, J=7,8 Гц, 0,66H), 9,09 (шир.с, 0,33H), 9,14 (шир.с, 0,66H)
1H ЯМР (400 МГц, ДМСО-d) δ м.д. 0,34-0,44 (м, 0,6H), 0,44-0,52 (м, 0,4H), 0,52-0,67 (м, 1H), 0,67-0,86 (м, 2H),
1H ЯМР (400 МГц, ДМСО-d) δ м.д. 1,34 (т, J=7,2 Гц, 3H), 1,52 (д, J=6,8 Гц, 2,25H), 1,61 (д, J=6,5 Гц, 0,75H), 2,12 (шир. 3H), 2,43 (шир.с, 1H), 3,49 (c, 0,75H), 3,61 (шир.с, 3,25H), 3,66-3,74 (м, 0,75H), 3,75-3,88 (м, 2,25H), 4,00 (дд, J1=13,4 Гц, J2=4,25 Гц, 1H), 4,03-4,22 (м, 1,75H), 4,37 (шир.с, 1,25H), 5,07 (шир.с, 0,25H), 5,21 (д, J=13,6 Гц, 0,75H), 5,28 (д, J=13,9, 0,25H), 5,42-5,53 (м, 1,75H), 7,34 (д, J=11,8 Гц, 0,75H), 7,41 (д, J=11,8 Гц, 0,25H), 7,97 (д, J=8,3 Гц, 0,25H), 8,01 (д, J=8,7 Гц, 0,75H), 9,37 (шир.с, 1H)
1H ЯМР (400 МГц, CDCl3) δ м.д. 1,09-1,19 (м, 3H), 1,31 (д, J=6,7 Гц, 2H), 1,43 (д, J=6,4 Гц, 1H), 1,59-1,74 (м, 2H), 1,89 (2 шир.с, 2H), 2,05 (шир.с, 1H), 2,19 (шир.с, 0,67H), 2,35 (шир.с, 0,33H), 3,15 (шир.с, 1,67H), 3,18 (шир.с, 0,33H), 3,23 (c, 2H), 3,24-3,31 (м, 1,33H), 3,34-3,50 (м, 1,67H), 3,52-3,70 (м, 1H), 3,85-4,06 (м, 2,33H), 4,06-4,15 (м, 0,67H), 4,81 (шир.с, 0,33H), 5,07-5,17 (м, 1H), 5,24 (шир.с, 0,67H), 5,28-5,40 (м, 1H), 6,6 (д, J=11,0 Гц, 1H), 7,77 (д, J=8,1 Гц, 0,67H), 7,86 (д, J=8,1 Гц, 0,33H), 8,50 (шир.с, 0,67H), 8,55 (шир.с, 0,33H)
1H ЯМР (300 МГц, ДМСО-d) δ м.д. 1,13-1,18 (м, 3H), 1,34 (д, J=6,5 Гц, 1,5H), 1,39 (д, J=6,5 Гц, 1,5H), 1,59-1,77 (м, 2H), 1,91 (шир.с, 3H), 2,23 (шир.с, 1H), 2,47 (шир.с, 1H), 3,22 (c, 1,5H), 3,28 (c, 1,5H), 3,38-3,88 (м, 4H), 3,95-4,12 (м, 1,7H), 4,12-4,30 (м, 1,3H), 4,83 (шир.с, 0,5H), 5,11-5,32 (м, 2,5H), 7,14-7,28 (м, 1H), 7,60-7,22 (м, 1H), 9,15 (c, 0,5H), 9,18 (c, 0,5H)
1H ЯМР (400 МГц, ДМСО-d) δ м.д. 1,05-1,12 (м, 5H), 1,27 (д, J=6,7 Гц, 1,8H), 1,37 (д, J=6,7 Гц, 1,2H), 1,48-2,08 (м, 6H), 3,17 (c, 1,2H), 3,21 (c, 1,8H), 3,33-3,42 (м, 2H), 3,42-3,51 (м, 2H), 3,52-3,66 (м, 1H), 3,77 (дд, J1=13,1 Гц, J2=3,3 Гц, 0,6H), 3,95 (дд, J1=13,5 Гц, J2=3,6 Гц, 0,4H), 4,75-4,85 (м, 0,4H), 5,05 (д, J=14,6 Гц, 0,6H), 5,17 (м, 1H), 5,67 (д, J=13,5 Гц, 0,4H), 5,76 (д, J=13,9 Гц, 0,6H), 7,20-7,27 (м, 2H), 7,63 (дд, J1=8,17 Гц, J2=5,7 Гц, 0,6H), 7,69 (шир.с, 0,4H), 8,64 (т, J=5,3 Гц, 0,6H), 8,70 (т, J=5,3 Гц, 0,4H), 9,12 (2 шир.с, 1H)
1H ЯМР (300 МГц, ДМСО-d) δ м.д. 1,11 (т, J=7,5 Гц, 3H), 1,31 (д, J=6,7Гц, 1,2H), 1,36 (д, J=6,5Гц, 1,8H), 1,66-1,85 (м, 2H), 1,89-2,11 (м, 2H), 3,17 (c, 1,8H), 3,22 (c, 1,2H), 3,42-3,65 (м, 5H), 3,80 (дд, J1=13,5 Гц, J2=3,2 Гц, 0,4H), 3,96 (дд, J1=13,5 Гц, J2=3,2 Гц, 0,6H), 4,78 (шир.с, 1H), 5,20 (д, J=13,4 Гц, 0,6H), 5,20-5,30 (м,
1H ЯМР (300 МГц, ДМСО-d) δ м.д. 1,11 (т,J=7,1 Гц, 3H), 1,29-1,36 (дд, J=6,6 Гц, 6,3 Гц, 3H), 1,99-2,05 (м, 2H), 3,01-3,22 (м, 1H), 3,27-3,29 (д, J=7,5 Гц, 2H), 3,50-3,55 (м, 4H), 3,71-3,79 (м, 2H), 4,68-5,41 (шир.с, 1H), 5,36-5,44 (шир.с, 1H), 5,66-5,77 (м, 1H), 7,25-7,35 (м, 1H), 7,94 (м, 1H), 8,61-8,68 (2м, 1H), 9,14 (шир.с, 1H)
1H ЯМР (400 МГц, ДМСО-d) δ м.д. 1,11 (т, J=7,6 Гц, 3H), 1,20 (д, J=6,8 Гц, 2,6H), 1,36 (д, J=6,4 Гц, 0,4H), 1,40-1,57 (м, 2H), 1,57-1,93 (м, 4H), 3,04 (c, 3H), 3,12-3,20 (м, 1H), 3,29-3,50 (м, 2H), 3,54-3,68 (м, 1H), 3,89 (дд, J1=13,2 Гц, J2=3,6 Гц, 1H), 4,02-4,11 (м, 1H), 4,12-4,13 (м, 1H), 4,23-4,37 (м, 0,85H), 4,44 (шир.с, 0,15H), 4,56-4,67 (м, 0,85H), 4,83 (м, 0,15H), 5,13 (д, J=13,6 Гц, 1H), 5,28 (д, J=13,6 Гц, 1H), 6,68 (шир.с, 0,15H), 6,73 (дт,
1H ЯМР (300 МГц, ДМСО-d) δ м.д. 0,45 (м, 1H), 0,66-0,69 (м, 2H), 0,79-0,83 (м, 1H), 1,02-1,22 (д, J=9,6 Гц, 3H), 1,40 (м, 3H), 1,64 (м, 4H), 2,68-2,73 (м, 2H), 2,94 (c, 3H), 3,04-3,09 (м, 1H), 3,76-3,77 (дд, J=6,6 Гц, 2,2 Гц, 1H), 3,80-4,09 (м, 2H), 4,48 (шир.с, 1H), 5,01-5,21 (дд, J=28,6 Гц, 13,8 Гц, 2H), 6,64 (т, J=2,1 Гц, 1H), 6,66-6,81 (д, J=12,8 Гц, 1H), 7,42-7,47 (т, J=15,6 Гц, 1H), 9,03 (шир.с, 1H)
B. БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ СОЕДИНЕНИЙ ФОРМУЛЫ I
Пример 166: Определение ингибиторной активности в отношении репликации ВИЧ дикого типа, Q148R мутанта, N155H мутанта и G140S-Q148H мутанта
Клетки MT4-LTR-усиленный зеленый флуоресцентный белок (EGFP) получали путем трансфекции клеток MT4 с использованием селектируемой конструкции, включающей кодирующие последовательности для ВИЧ LTR, в качестве промотора для экспрессии EGFP и последующего отбора перманентно трансфицированных клеток. MT4-цитомегаловирус (CMV)-EGFP клетки получали путем отбора перманентно трансформированных клеток MT4 с CMV-EGFP репортерным геном. Клеточные линии поддерживали в среде RPMI 1640, дополненной 10% термоинактивированной фетальной телячьей сыворотки, 2 мМ L-глутамина, 0,1% NaHCO3 и антибиотиками (0,02% гентамицина и 0,8% G418), и инкубировали во влажном инкубаторе с атмосферой 5% CO2 при 37°C.
Кодирующие последовательности N155H и Q148R мутантной интегразы были сконструированы в pUC19-5'HXB2D векторе (XbaI-SalI фрагмент pHXB2D), содержащем ВИЧ-1 клон HXB2D IN кодирующую последовательность, с использованием набора QuikChange для сайт-направленного мутагенеза (Stratagene, La Jolla, CA) и ВЭЖХ-очищенных праймеров (Genset Oligos, La Jolla, CA). Измененные плазмидные последовательности подтверждали путем дидезоксирибоза-секвенирования. Для образования сайт-направленных мутантных (SDM) вирусных штаммов клетки MT4 субкультивировали при плотности 250000 клеток/мл за день до трансфекции. Клетки осаждали центрифугированием и ресуспендировали в фосфатно-буферном солевом растворе при концентрации 3,1×106 клеток/мл. Порцию объемом 0,8 мл (2,5×106 клеток/мл) использовали для каждой трансфекции. Трансфицирование осуществляли с использованием Bio-Rad Gene Pulser (Bio-Rad, Hercules, CA) с 0,4-см электродными кюветами (Bio-Rad). Клетки подвергали электропорации с использованием 10 мкг SalI-линеаризованного pUC19-3'HXB2D (SalI-XbaI фрагмент pHXB2D) и 5 мкг SalI-расщепленного SDM при 250 мкF и 300В, с последующей 30-минутной инкубацией при комнатной температуре. Десять мл свежей культуральной среды затем добавляли к суспензии трансфицированных клеток и инкубацию осуществляли при 37°C в увлажненной атмосфере с 5% CO2. Клеточные культуры отслеживали на появление цитопатического эффекта (CPE). Когда происходило проникновение вируса (полный CPE), культуральный супернатант собирали центрифугированием (обычно через 8-10 дней после трансфекции) и хранили при -80°C для последующего определения восприимчивости к лекарственному средству.
Ингибиторную противовирусную активность различных соединений определяли в клеточном анализе ВИЧ-1 репликации. Клетки MT4-LTR-EGFP (150000 клеток/мл) инфицировали ВИЧ-1 (IIIB или SDM штаммы Q148R, N155H или G140S-Q148H; множественность заражения [MOI] 0,0025) в присутствии или в отсутствие ингибитора. После 3 дней инкубации проводили количественное определение ингибирования ВИЧ репликации путем измерения EGFP флуоресценции и выражали как концентрацию соединения, необходимую для 50% ингибирования ВИЧ-1 репликации в клеточной культуре (EC50). Противовирусная активность различных соединений указана в таблице 4 и выражена как EC50 для ингибирования ВИЧ дикого типа (IIIB EC50) и как кратное изменение активности против мутантных штаммов (FC-Q148R, FC-N155H, FC-G140S-Q148H) по сравнению с активностью против ВИЧ дикого типа.
Пример 167: определение ингибиторной активности в отношении репликации ВИЧ дикого типа в присутствии человеческой сыворотки
Для противовирусного анализа в присутствии 50% человеческой сыворотки клетки MT-4-LTR-EGFP инфицировали ВИЧ-1 LAI (IIIB) при MOI 0,001-0,01 CCID50/клетка в среде RPMI1640. После 1 часа инкубации клетки промывали и высевали в 96-луночный планшет, содержащий серийные разведения испытуемого соединения в присутствии 10% фетальной телячьей сыворотки или 50% человеческой сыворотки. После 4 дней инкубации EC50 в присутствии 50% человеческой сыворотки (EC50/HS) определяли путем анализа жизнеспособности клеток, используя резазурин (как описано в публикации Fields, R.D. and M.V. Lancaster (1993) Am. Biotechnol. Lab. 11:48-50).
Пример 168: определение фармакокинетических характеристик у крыс
Для определения фармакокинетических свойств у крысы каждый ингибитор ВИЧ интегразы растворяли в PEG400/нормальном солевом растворе (70:30 (об./об.)), при номинальной концентрации 1 мг/мл, и вводили (2 мл/кг) самцам крыс Sprague-Dawley (n=3 на испытуемое соединение) в виде внутривенного болюса через подкожную вену конечности в дозе 2 мг/кг. Образцы крови собирали через хвостовую вену в последовательные моменты времени вплоть до 24 часов после введения дозы. Плазму получали путем центрифугирования и хранили при -20°C до анализа. Анализ осуществляли, используя жидкостную хроматографию (ЖХ) с тандемной масс-спектрометрической детекцией (МС/МС) в режиме положительной ионизации. Каждый ингибитор ВИЧ интегразы элюировали из колонки с обращенной фазой, используя градиент ацетонитрила и воды, содержащей 0,1% (об./об.) муравьиной кислоты. Непосредственно перед анализом образцы плазмы (50 мкл) размораживали и депротонировали 200 мкл ацетонитрила, содержащего 0,1% (об./об.) муравьиной кислоты, и центрифугировали. Аликвоты супернатанта (100 мкл) разбавляли 200 мкл воды, содержащей 25% (об./об.) метанола и 0,1% (об./об.) муравьиной кислоты, и вводили через электрораспылительное устройство непосредственно в масс-спектрометр. Стандарты калибровки и контроли качества получали в плазме крови крыс в то же время, что и образцы, и анализировали до и после образцов, содержащих ингибитор ВИЧ интегразы. Не-компартментный фармакокинетический анализ кривых концентрация в плазме-время осуществляли с использованием программы WinNonLin для оценки скорости выведения из плазмы (CLP), объема распределения в стабильном состоянии (Vss) и периода полувыведения конечной фазы элиминации (t1/2).
Пример 169: метаболическая стабильность в гепатоцитах крысы и человека
Для определения in vitro метаболической стабильности криоконсервированные гепатоциты крыс Sprague Dawley и человека (пул мужчин и женщин) получали от внешнего поставщика (Cellz Direct Inc., Paisly, UK). Испытуемые соединения растворяли в ДМСО и этот исходный раствор использовали для обработки клеточных культур гепатоцитов крысы и человека (106 клеток/мл) при конечной концентрации 1 мкМ (конечный процент ДМСО 0,02% (об./об.)). Инкубации проводили в течение 60 минут, приблизительно при 37°C, и прекращали путем добавления двух объемов ДМСО. Образцы затем центрифугировали и аликвоты супернатанта анализировали, используя жидкостную хроматографию (ЖХ) с масс-спектрометрической детекцией с использованием электрораспыления (МС/МС), для отслеживания исчезновения исходного соединения путем контроля отдельной реакции (SRM) и обращенно-фазной хроматографии с градиентом ацетонитрила и воды, содержащей 0,1% (об./об.) муравьиной кислоты. На основании площади пика рассчитывали процент исходного соединения, оставшегося после 15 и 60 минут инкубации, и результаты выражали в виде процента оборота (TO) относительно контролей на время ноль.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ N-ГИДРОКСИФОРМАМИДА И СОДЕРЖАЩЕЕ ЕГО ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2011 |
|
RU2569851C2 |
| ПИРРОЛИДИНИЛЬНЫЕ ПРОИЗВОДНЫЕ ГИДРОКСАМОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), СПОСОБ ЛЕЧЕНИЯ | 1996 |
|
RU2144917C1 |
| 2-АМИНОБЕНЗОКСАЗОЛКАРБОКСАМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-НТ3 | 2007 |
|
RU2448105C2 |
| ПОЛУСИНТЕТИЧЕСКИЙ СПОСОБ И НОВЫЕ СОЕДИНЕНИЯ | 2000 |
|
RU2237063C9 |
| ИНГИБИТОРЫ СЕТР | 2006 |
|
RU2513107C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИДНОГО СОЕДИНЕНИЯ | 2008 |
|
RU2459820C2 |
| 1,3-ОКСАТИОЛАН, ЕГО ГЕОМЕТРИЧЕСКИЕ И ОПТИЧЕСКИЕ ИЗОМЕРЫ, СМЕСИ ЭТИХ ИЗОМЕРОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АНТИВИРУСНУЮ АКТИВНОСТЬ | 1990 |
|
RU2092485C1 |
| СПИРОПИПЕРИДИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1993 |
|
RU2168512C2 |
| БИАРИЛ- ИЛИ ГЕТЕРОЦИКЛИЧЕСКИЕ БИАРИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСЕНА В КАЧЕСТВЕ ИНГИБИТОРОВ СЕТР | 2014 |
|
RU2627361C2 |
| 3'-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ НУКЛЕОЗИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ЛЕКАРСТВЕННОЕ СРЕДСТВО, СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ | 1995 |
|
RU2130029C1 |
Изобретение относится к макроциклическому соединению общей формулы I, к его стереохимически изомерной форме и к его фармацевтически приемлемой соли, где R1 представляет собой F; R2 представляет собой Н, F или Cl; R3 представляет собой C1-4алкил или циклопропил; R4 представляет собой метил; J представляет собой ---N(R5)-SO2-, ---С(=O)-N(R5)-, ---N(R5)-,  где пунктирная линия означает точку присоединения к пиридазиноновому кольцу; К представляет собой -(CHR6)P, или *-(СН2)q-CH=CH-CH2-, где * означает точку присоединения к группе J; L представляет собой -O-, -O-СН2-* или -N(R5)-С(=O)-*, где * означает точку присоединения к фенильному кольцу; и R5 представляет собой водород, C1-4алкил или C3-5циклоалкил; каждый R6 независимо представляет собой водород или C1-3алкил; p равно 3, 4, 5 или 6; q равно 2 или 3. Также изобретение относится к фармацевтической композиции на основе соединения формулы I и применению соединения формулы I. Технический результат: получены новые макроциклические соединения, полезные при лечении или профилактики ВИЧ-инфекции. 4 н. и 9 з.п. ф-лы. 6 табл., 169 пр.
где пунктирная линия означает точку присоединения к пиридазиноновому кольцу; К представляет собой -(CHR6)P, или *-(СН2)q-CH=CH-CH2-, где * означает точку присоединения к группе J; L представляет собой -O-, -O-СН2-* или -N(R5)-С(=O)-*, где * означает точку присоединения к фенильному кольцу; и R5 представляет собой водород, C1-4алкил или C3-5циклоалкил; каждый R6 независимо представляет собой водород или C1-3алкил; p равно 3, 4, 5 или 6; q равно 2 или 3. Также изобретение относится к фармацевтической композиции на основе соединения формулы I и применению соединения формулы I. Технический результат: получены новые макроциклические соединения, полезные при лечении или профилактики ВИЧ-инфекции. 4 н. и 9 з.п. ф-лы. 6 табл., 169 пр.
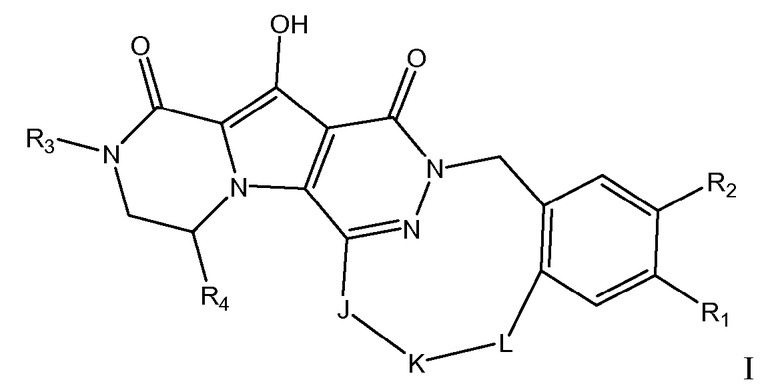
1. Соединение, имеющее формулу I, включая его стереохимически изомерную форму,
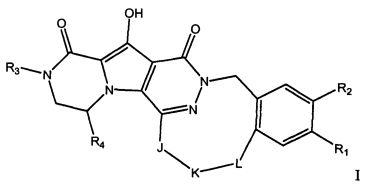
где
R1 представляет собой F;
R2 представляет собой Н, F или Cl;
R3 представляет собой C1-4алкил или циклопропил;
R4 представляет собой метил;
J представляет собой ---N(R5)-SO2-, ---С(=O)-N(R5)-, ---N(R5)-,
где пунктирная линия означает точку присоединения к пиридазиноновому кольцу;
К представляет собой -(CHR6)P, или *-(СН2)q-CH=CH-CH2-, где * означает точку присоединения к группе J;
L представляет собой -O-, -O-СН2-* или -N(R5)-С(=O)-*, где * означает точку присоединения к фенильному кольцу; и
R5 представляет собой водород, C1-4алкил или C3-5циклоалкил;
каждый R6 независимо представляет собой водород или C1-3алкил;
p равно 3, 4, 5 или 6;
q равно 2 или 3;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где R1 представляет собой F и R2 представляет собой Н.
3. Соединение по п.1, где R3 представляет собой этил, изопропил или циклопропил.
4. Соединение по п.2, где R3 представляет собой этил, изопропил или циклопропил.
5. Соединение по п.1, где J представляет собой -N(R5)-SO2- или 
6. Соединение по п.1, где К представляет собой -(CHR6)P, где p равно 3, 4, 5 или 6 и каждый R5 независимо представляет собой Н или СН3.
7. Соединение по п.1, где К представляет собой *-(CH2)q-СН=СН-СН2-, где q равно 2 или 3.
8. Соединение по п.1, где R5 представляет собой метил, этил или циклопропил.
9. Соединение по п.1, где L представляет собой -О- или -O-СН2-.
10. Соединение по п.1, где связывающая цепь -JKL-представляет собой цепь длиной 8-11 атомов.
11. Фармацевтическая композиция, включающая противовирусно эффективное количество соединения формулы I, определенного в любом из пп.1-10, и носитель.
12. Применение соединения по любому из пп.1-10 в качестве лекарственного средства для лечения или профилактики ВИЧ-инфекций.
13. Применение соединения по любому из пп.1-10 в качестве ингибитора репликации ВИЧ.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |

