Настоящее изобретение касается открытия новых селективных и обратимых ингибиторов убихитин-специфических протеаз, процесса их получения и их терапевтического применения.
Убихитин-специфические протеазы (USP) представляют собой цистеиновые протеазы, которые принадлежат семейству дезубихитинирующих (DUBs) ферментов.
Дезрегуляция убихитинпротеасомной системы вовлечена в патогенез многих заболеваний человека, включая рак (Hoeller et al. Nat Rev Cancer 2006, 6(10), 776-788), нейродегенеративные расстройства (Rubinsztein, Nature 2006, 443(7113), 780-786) и вирусные заболевания (Gao & Luo Can J Physiol Pharmacol 2006, 84(1), 5-14). Успех на рынке протеасомного ингибитора Velcade® (bortezomib) для лечения множественной или рассеянной миеломы и лимфомы клеток мантии упрочил данную систему как вескую мишень для лечения рака (Adams, Nat Rev Cancer 2004, 4(5), 349-360). Обещающей альтернативой нацеливанию протеасомы самой по себе было бы помешать с помощью механизма предшествующего конъюгирования/деконъюгирования убихитина, генерировать более специфические, менее токсичные противораковые агенты.
Моно- и полиубихитинирование может реверсироваться дезубихитинирующими ферментами, которые специфично расщепляют изопептидную связь на С-окончании убихитина. Убихитин-специфические протеазные и убихитин С-концевые гидролазные (UCH) ферменты являются наиболее хорошо охарактеризованными членами семейства DUB (Komander et al. Nat. Rev. Mol. Biol. 2009, 10(8), 550-63; Nijman et al. Cell 2005, 123(5), 773-786). Полагают, что UCHs расщепляют предпочтительно небольшие протеиновые субстраты и вовлекаются главным образом в обработку и рециркуляцию убихитина, но их конкретные функции остаются плохо понятными. USPs составляют наиболее крупное подсемейство DUBs, с более чем 60 членами. Они удаляют убихитин из специфических белковых субстратов, предотвращая таким образом их нацеливание на протеасому или регулирование их субклеточной локализации и активирование (Daviet & Colland, Biochimie 2008, 90(2), 270-83). USPa возникают как потенциальные мишени для фармакологического вмешательства в механизм регулирования убихитина, основанный на их протеазной активности и вовлечении в отдельные заболевания человека (Colland, Biochem Soc Trans 2010, 38, 137-43).
USP7 (Убихитин-специфическая протеаза 7) HAUSP (ассоциированная с герпесом убихитин-специфическая протеаза) является 135 kDa белком семейства USP. Было показано, что USP7 взаимодействует с вирусными белками, такими как ICP0 (Vmw 110), немедленно-ранним геном вируса простого герпеса, стимулирующим инициирование вирусного литического цикла (Everett et al., J Virol 73, 1999, 417-426), и EBNA1 (Epstein-Barr Nuclear Antigen-1 (ядерный антиген-1)) (Holowaty et al., J Biol Chem 2003, 278, 29987-29994 и 47753-47761). Человеческие белки, такие как р53, и важная Е3 лигаза белка р53, Mdm2, были также идентифицированы как соучастники и субстраты USP7 (Cummins et al. Nature 2004, 486, Cummins & Vogelstein, Cell Cycle, 2004, 3, 689-692; Li et al. Mol Cell 2004, 13, 879-886; Li et al. Nature 2002, 416, 648-653). Более обычно USP7 может дезубихитинировать различные мишени, включая Mdm2 и p53, и чистое дезубизихитинирование данных последних мишеней в конечном счете определяет функциональные уровни р53. Согласуясь с недавними сообщениями, было показано также, что USP7 глушение увеличивает уровни р53 в устойчивом состоянии путем промотирования деградации Mdm2. Недавно было показано, что связывание USP7 с р53 регулируется с помощью TSPYL5, белком, возможно вовлеченным в онкогенез груди вследствие конкуренции с р53 за связывание с той же областью USP7 (Epping et al., Nat Cell Biol. 2011, 13(1):102-8). Опять же недавно было показано, что регулирование и вверх и вниз ингибирует пролиферацию раковых клеток ободочной кишки in vitro и рост опухоли in vivo, приводя в результате к существенно высоким уровням р53 (Beker et al. Cell Cycle 2008, 7(9), 1205-13).
USP7 также изменяет уровень подавителя р16iNK4a опухоли вследствие Bmi1/Mel18 стабилизации (Maertens et al., Embo J. 2010, 29, 2553-2565). С помощью USP7 также стабилизируются дополнительные белки, вовлеченные в геномную целостность/регулирование, такие как DNMT1 DNA метилаза и Claspin адаптер (Du et al., Science Signaling 2010, 3(146):(146):ra80; Faustrup et al., J Cell Biol 2009, 184(1):13-9). Важно отметить, избыток USP7 и DNMT 1, белка, вовлеченного в поддержание эпигенетического метилирования, требуемого для того, чтобы заглушить гены, вовлекаемые в развитие рака, находится в определенном соотношении при раке ободочной кишки человека (Du et al., Science Signaling, 2010, 3(146):ra80). Было также показано, что USP7 в человеческих клетках дезубихитинируют хорошо известный ген подавителя опухоли PTEN, который вызывает его ядерный экспорт и отсюда его инактивирование (Song et al., Nature 2008, 455(7214), 813-7). Более важно, сообщалось о сверхэкспрессии USP7 в первое время при раке простаты, и данная сверхэкспрессия непосредственно связана с агрессивностью опухоли (Song et al., Nature 2008, 455(7214), 813-7).
Сообщалось также, что USP7 в человеческих клетках дезубихитинирует FOXO4, который вызывает его ядерный экспорт и отсюда его инактивирование; в результате онкогенный PI3K/PKB сигнальный путь активировался (van der Horst et al., Nat Cell Biol. 2006, 8, 1064-1073). Наконец, USP7 играет важную роль в р53-опосредуемых клеточных ответных реакциях на различные типы стресса, такие как ДНК повреждение и оксидативный стресс (Marchenko et al., Embo J. 2007 26, 923-934, Meulmeester et al., Mol Cell 2005, 18, 565-576., van der Horst et al., Nat Cell Biol. 2006, 8, 1064-1073).
Сообщалось о синтетических ингибиторах USP7 белка, связывающих содержащий полипептидный фрагмент P1-Gly-P3-Ser, в котором P1 представляет собой остаток глютаминовой кислоты или аминокислоты с неполярной боковой цепью, а P3 представляет собой остаток глицина или аминокислоты с неполярной боковой цепью (WO 2006072048).
Фенотипы, ассоциируемые с USP7 глушением и известными связями между USP7 и существенными вирусными белками и онкогенными путями, такими как p53/Mdm2 и PI3K/PKB пути, определенно подсказывают, что нацеливание на USP7 ингибиторами с малыми молекулами может быть благоприятным при лечении раковых и вирусных заболеваний (Sippl et al., Future Oncology 2011, 7, 619-32). Недавно сообщалось об ингибиторах USP7 (Colland et al. Molecular Cancer Therapeutics 2009, 8, 2286-95 и EP 1749822 и PCT/EP 2011/050523.2).
Однако до настоящего времени, кажется, не сообщалось ни о каких конкретных и реверсируемых USP7 ингибиторах с малыми молекулами.
В соответствии с первым объектом настоящее изобретение касается соединений формулы (I):
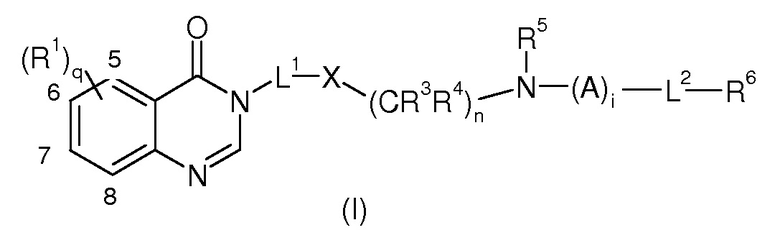
в которой
R1, каждый идентичный или различный, выбран из группы, состоящей из галогена, R, OR, NRR’, CN, CF3, C(O)R, C(O)OR, C(O)NRR’, NO2, (C1-C6)алкилен-OR, (C1-C6)алкилен-NRR’, (C1-C6)алкилен-CO2R, (C1-C6)алкилен-C(O)NRR’, -O-(C1-C6)алкилен-CO2R, -O-(C1-C6)алкилен-C(O)NRR’, CO2-(C1-C6)алкилен-OR, CO2-(C1-C6)алкилен-NRR’, C(O)NH-(C1-C6)алкилен-OR, C(O)NH-(C1-C6)алкилен-NRR’, OCF3, SO2R, SO3H, SO2NRR’, NHSO2R, R10C≡CR11, (R10)(R11)C=C(R11)2, (C1-C6)алкилен-C(O)R, NHC(O)R или (C1-C6)алкил, прерываемый, по крайней мере, одним гетероатомом, выбранным среди O, N или S, предпочтительно O;
L1 представляет собой линейный или разветвленный (C1-C6) алкилен, необязательно замещенный одним или более =O, CN, C(O)R, C(O)OR, или C(O)NRR’, или линейный или разветвленный CH2(C1-C6) алкилен, в котором последний (C1-C6)алкилен необязательно замещен одним или более галогенами, OR, NRR’ или CF3;
X представляет собой CR2R7, NR2, арил, гетероарил, циклоалкил или гетероцикл, где арил, гетероарил циклоалкил или гетероцикл необязательно замещен одним или более из линейного или разветвленного C1-C6(алкила), галогена, OR, NRR’, CN, CF3, C(O)R, C(O)OR или C(O)NRR’;
R2 представляет собой линейный или разветвленный (C1-C6)алкилен и связан с R5=линейным или разветвленным (C1-C6)алкиленом, образуя с -X-(CR3R4)n-N-, к которому они присоединены, гетероцикл, предпочтительно гетероцикл, имеющий 5-7 членов, необязательно замещенных одним или более из OR, прямого или разветвленного (C1-C6)алкила, галогена, NRR’, CN, CF3, C(O)R, C(O)OR, C(O)NRR’ или =O;
R5 выбран среди Н и линейного или разветвленного (C1-C6)алкила, (C1-C6)алкилена;
R3, R4, каждый идентичный или различный, выбраны из группы, состоящей из H, линейного или разветвленного (C1-C6)алкила, галогена, OR, NRR’, CN, CF3, C(O)R, C(O)OR, C(O)NRR’ или =O;
q представляет собой 0, 1, 2, 3 или 4
n представляет собой 0, 1, 2 или 3;
R7 представляет собой OR, H, галоген, линейный или разветвленный (C1-C6)алкил-OR, C(O)OR, C(O)NRR’, CN, OR9, NRR’ или SR;
i представляет собой или 0 или 1;
A выбран из группы, состоящей из:
линейного или разветвленного -[C1-C6(алкил)]0-1-C(O)-;
линейного или разветвленного -[C1-C6(алкил)]0-1-C(O)NH-;
линейного или разветвленного -[C1-C6(алкил)]0-1SO2-; или
линейного или разветвленного -[C1-C6(алкил)]0-1SO2N-;
L2 представляет собой линейный или разветвленный (C1-C6)алкилен-O или линейный или разветвленный (C1-C6)алкилен необязательно прерываемый, по крайней мере одним гетероатомом, выбранным из O, NR или S, и/или необязательно замещенный заместителем: R, OR, NRR’, (C1-C6)алкил-OR, (C1-C6)алкил-NRR’, OC(O)R, NHC(O)R, NHC(O)NRR’, CN, C(=NH)NHOR;
R6 выбран из группы, состоящей из арила, гетероарила циклоалкила, гетероцикла, H, где арил, гетероарил, циклоалкил или гетероцикл является моно или полициклическим и необязательно замещен одним или более линейным или разветвленным (C1-C6)алкилом, галогеном, NRR’, CN, CF3, OR, =O, C(O)R, C(O)OR, NHC(O)R, OC(O)R, линейным или разветвленным (C2-C6)алкениленом или C(O)NRR’;
R9 выбран из группы, состоящей из -C(O)R, -C(O)NHR, -C(O)OR, -C(O)CH2-NRR’, -C(O)-CH2-CH2-CO2R, -C(O)-CH2-SO3H, -C(O)-(C5H4N), -PO3H2 или их ионизированных форм;
R10 независимо идентичный или различный выбран из связи, линейного или разветвленного (C1-C6)aлкила;
R11 независимо идентичный или различный выбран из атома водорода, линейного или разветвленного (C1-C6)aлкила или арила, алкил или арил необязательно замещен OH, NH2, C(O)OH или C(O)NH2;
каждый R и R’, идентичные или различные, независимо выбраны из H, линейного или разветвленного (C1-C6)aлкила, циклоалкила, арила, ароматического или неароматического гетероцикла, линейного или разветвленного -(C1-C6)алкил-арила или линейного или разветвленного -(C1-C6)алкил-гетероцикла, где гетероцикл является ароматическим или неароматическим; необязательно замещенных или нет OH, CO2H, C(O)NH2, NH2;
или их фармацевтически приемлемых солей или их оптических изомеров, рацематов, диастереоизомеров, энантиомеров или таутомеров.
Формула (I) изобретения относится к любому из следующих воплощений или к любому из их сочетаний.
Предпочтительно, в соединении формулы (I), R1, каждый идентичный или различный выбран из группы, состоящей из линейного или разветвленного (C1-C6)алкила, галогена, OR, NRR’, CN, CF3, C(O)R, C(O)OR, C(O)NRR’; NO2; (C1-C6)aлкилен-OR, (C1-C6)aлкилен-NRR’, (C1-C6)aлкилен-CO2R, (C1-C6)aлкилен-C(O)NRR’, -O-(C1-C6)aлкилен-CO2R, -O-(C1-C6)aлкилен-C(O)NRR’, CO2-(C1-C6)aлкилен-OR, CO2-(C1-C6)aлкилен-NRR’, C(O)NH-(C1-C6)aлкилен-OR, C(O)NH-(C1-C6)aлкилен-NRR’ или NHC(O)R.
Предпочтительно, в соединении формулы (I), L1 представляет собой линейный или разветвленный (C1-C6)aлкилен, необязательно замещенный одним или более из =O, CN, C(O)R, C(O)OR или C(O)NRR’; или линейный или разветвленный CH2(C1-C6)aлкилен, где последний (C1-C6)aлкилен необязательно замещен одним или более из заместителей галогена, OR, NRR’ или CF3.
Предпочтительно, в соединении формулы (I), R2 представляет собой линейный или разветвленный (C1-C6)aлкилен и связан вместе с R5 = линейный или разветвленный (C1-C6)aлкилен, образуя с -X-(CR3R4)n-N-, к которому они присоединены, гетероцикл из 5 или 6 членов, необязательно замещенный одним или более из OR, линейного или разветвленного (C1-C6)aлкила, галогена, NRR’, CN, CF3, C(O)R, C(O)OR, C(O)NRR’ или =O.
Предпочтительно, в соединении формулы (I), R7 представляет собой OR, OR9, галоген, линейный или разветвленный (C1-C6)aлкил-OR, C(O)OR, C(O)NRR’ или CN. Более предпочтительно R7 представляет OR, OR9. Более предпочтительно R7 представляет OH или OR9, предпочтительно OH.
R6 выбран из группы, состоящей из арила, гетероарила, циклоалкила, гетероцикла, Н, где арил, гетероарил, циклоалкил или гетероцикл является моно или полициклическим и необязательно замещен одним или более из линейного или разветвленного (C1-C6)aлкила, галогена, NRR’, CN, CF3, OR, C(O)R, C(O)OR, NHC(O)R, OC(O)R или C(O)NRR’.
Предпочтительно, в соединении формулы (I), A выбран из группы, состоящей из:
-C(O)-;
-C(O)NH-;
-SO2-; или
-SO2N-.
Предпочтительно, в соединении формулы (I), L2 представляет собой линейный или разветвленный (C1-C6)aлкилен, необязательно прерываемый, по крайней мере одним гетороатомом, выбранным из O, NR или S, и/или необязательно замещен заместителем: R, OR, NRR’, (C1-C6)алкил-OR, (C1-C6)алкил-NRR’, OC(O)R, NHC(O)R, NHC(O)NRR’, CN, C(=NH)NHOR.
Предпочтительно, следует понимать, что L2 не представляет собой O-(C1-C6)aлкилен.
Предпочтительно, в соединении формулы (I):
- NR5 непосредственно связан с, по крайней мере, одной из C(O), C(O)N, SO2 или SO2N групп; и/или
- i=0, n представляет 1, 2 или 3 и CR3R4, связанный с NR5, представляет собой C(O); или i=1, A представляет собой -C(O)-, C(O)NH, SO2 или SO2N; и/или
- i=0, n представляет 1, 2 или 3 и CR3R4, связанный с NR5, представляет собой C(O); или i=1, A представляет -C(O)-, C(O)NH, SO2 или SO2N, X представляет собой CR2R7 или NR2 и R2 и R5, идентичные или различные представляют собой линейный или разветвленный (C1-C6)aлкилен и образуют вместе с -X-(CR3R4)n-N-, к которому они присоединены, гетероцикл из 5-7 членов, необязательно замещенный одним или более из OR, линейного или разветвленного (C1-C6)aлкила, галогена, NRR’, CN, CF3, C(O)R, C(O)OR, C(O)NRR’ и R3, R4, каждый из которых идентичный или различный, выбраны из группы, состоящей из H, линейного или разветвленного (C1-C6)aлкила, галогена, OR, NRR’, CN, CF3, C(O)R, C(O)OR, C(O)NRR’; и/или
- i=1 и A представляет собой -C(O)-, X CR2R7 или NR2 и R2 и R5, идентичные или различные, представляют собой линейный или разветвленный (C1-C6)aлкилен и образуют вместе с -X-(CR3R4)n-N-, к которому они присоединены, гетероцикл из 5-7 членов, необязательно замещенный одним или более из OR, линейного или разветвленного (C1-C6)aлкила, галогена, NRR’, CN, CF3, C(O)R, C(O)OR, C(O)NRR’ и R3, R4, каждый идентичный или различный, выбраны из группы, состоящей из Н, линейного или разветвленного (C1-C6)aлкила, галогена, OR, NRR’, CN, CF3, C(O)R, C(O)OR, C(O)NRR’; и/или
- R1, каждый идентичный или различный, выбран из группы, состоящей из линейного или разветвленного (C1-C6)aлкила, галогена, OR, NRR’, CN, CF3, C(O)R, C(O)OR, C(O)NRR’ или NHC(O)R; и/или
- R1, каждый идентичный или различный, выбран из группы, состоящей из линейного или разветвленного C1-C6(алкила), галогена, OH или линейного или разветвленного -O-(C1-C6)aлкила; и/или
- R1, каждый идентичный или различный, выбран из группы, состоящей из галогена или линейного или разветвленного -O-(C1-C6)aлкила; и/или
- q представляет собой 0, 1 или 2; и/или
- X представляет собой CR2R7 или NR2 и R2 и R5, идентичные или различные, представляют собой линейный или разветвленный (C1-C6)aлкилен и образуют вместе с -X-(CR3R4)n-N-, к которому они присоединены, гетероцикл из 5-7 членов, необязательно замещенный одним или более из OR, линейного или разветвленного (C1-C6)aлкила, галогена, NRR’, CN, CF3, C(O)R, C(O)OR или C(O)NRR’. Предпочтительно, гетероцикл, образуемый группой -XR2-(CR3R4)n-NR5-, является неароматическим гетероциклом; и/или
- R3, R4, каждый идентичный или различный, выбраны из группы, состоящей из Н, линейного или разветвленного (C1-C6)aлкила, галогена, =O, OR, NRR’, CN, CF3, C(O)R, C(O)OR или C(O)NRR’; и/или
- R3, R4, каждый идентичный или различный, выбраны из группы, состоящей из Н, -O-(C1-C6)aлкила, OH и =O. Предпочтительно, R3, R4, каждый идентичный или различный, выбраны из группы, состоящей из H и OH; и/или
- X представляет собой CR2R7, или арил и R7 представляет собой OR, OR9, линейный или разветвленный (C1-C6)aлкил-OR, галоген, C(O)OH, NRR’, C(O)NH2 или SR. Предпочтительно, X представляет собой CR2R7, или арил и R7 представляет собой OR, OR9, NRR’ или SR. Более предпочтительно, R7 представляет собой OH или OR9, предпочтительно OH. R9, является таким, как определен выше; и/или
- X представляет собой арил, предпочтительно фенил, и/или
- L1 представляет собой линейный или разветвленный (C1-C6)aлкилен, необязательно замещенный одним или более =O, или представляет собой CH2-C1-C6(aлкилен), где последний алкилен необязательно замещен одним или более -OH; и/или
- L2 представляет собой линейный или разветвленный C1-C6(aлкилен)-O или линейный или разветвленный C1-C6(aлкилен), необязательно прерываемый, по крайней мере, одним гетероатомом, выбранным из O или S, и/или необязательно замещенный одним или более из: R, OR, NRR’, (C1-C6)aлкил-OR, (C1-C6)aлкил-NRR’, OC(O)R, NHC(O)R, NHC(O)NRR’, CN, C(=NH)NHOR. Более предпочтительно L2 представляет собой линейный или разветвленный C1-C6(aлкилен) или линейный или -[C1-C6(aлкилен)]-O-; и/или
- R6 выбран из группы, состоящей их арила, гетероарила, циклоалкила или Н, где арил, гетероарил или циклоалкил необязательно замещен галогеном, линейным или разветвленным О-(C1-C6)aлкилом; и/или
- R6 выбран из группы, состоящей из фенила, тиофенила, циклопентила и Н, где фенил необязательно замещен галогеном, линейным или разветвленным O-(C1-C6)aлкилом.
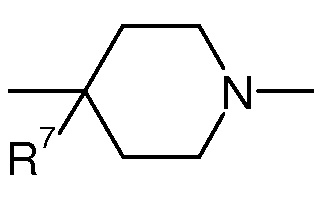
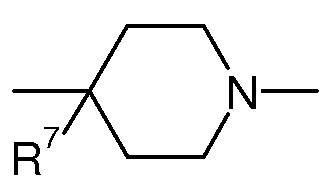
В одном воплощении в соединении формулы (I), X представляет собой CR2R7 или NR2 и R2, и R5 образуют вместе с -X-(CR3R4)n-N-, к которому присоединены, гетероцикл из 5-7 членов, необязательно замещенный одним или более OH. Предпочтительно в данном конкретном воплощении n представляет 0, 1 или 2 и/или X представляет собой CR2R7, где R7 представляет собой OR, OR9, линейный или разветвленный (C1-C6)aлкил-OR, галоген, C(O)OH, C(O)NH2, NRR’ или SR, и/или L1 представляет собой (CH2)k, где k представляет 1 или 2, предпочтительно k представляет 1, -C(O)-, -CH2-CH(OH)- или -CH2-C(O)-. Предпочтительно R7 представляет собой OR, OR9, NRR’ или SR. Более предпочтительно, R7 представляет собой OH или OR9, предпочтительно OH. R9 имеет значения, определенные выше.
В еще одном воплощении в соединений формулы (I), X представляет собой арил, гетероарил, циклоалкил или гетероцикл, где арил, гетероарил, циклоалкил или гетероцикл, необязательно замещен одним или более заместителем из линейного или разветвленного C1-C6(алкила), галогена, OR, NRR’, CN, CF3, C(O)R, C(O)OR или C(O)NRR’, предпочтительно X представляет собой арил и R5 представляет H или линейный или разветвленный C1-C6(алкил), предпочтительно H. Предпочтительно в данном конкретном воплощении, n представляет 0 и/или X представляет собой арил и/или L1 представляет собой -CH2-CH(OH)-.
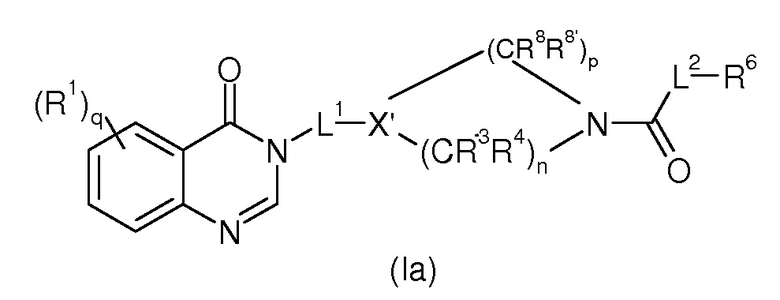
Согласно конкретному воплощению соединения изобретения могут быть соединениями следующей формулы (Ia)
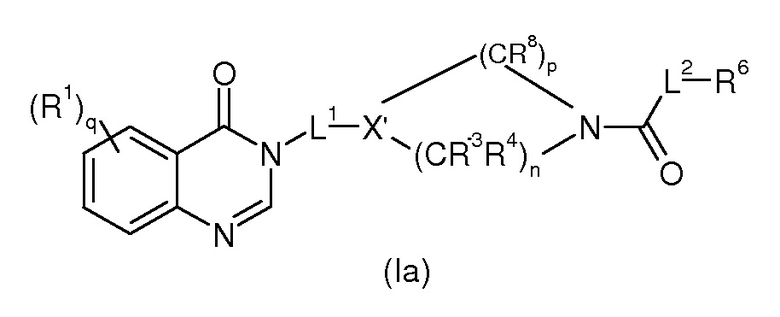
в которой
R1, q, L1, L2, R6 и R7 имеют значения, определенные для формулы (I);
X’ представляет собой CR7 или N;
n представляет 0, 1 или 2;
p представляет 1, 2 или 3;
R3, R4 и R8, каждый идентичный или различный, выбраны из группы, состоящей из Н, линейного или разветвленного (C1-C6)алкила, галогена, OH, -O-(C1-C6)алкила, NRR’, CN, CF3, OR, C(O)R, C(O)OR или C(O)NRR’.
Предпочтительно, в соединении формулы (Ia), R3, R4 и R8, каждый идентичный или различный, выбраны из группы, состоящей из H или OH; и/или p представляет 1 или 2.
Предпочтительно, в соединении формулы (Ia)
представляет собой
t представляет собой 0, 1 или 2, предпочтительно
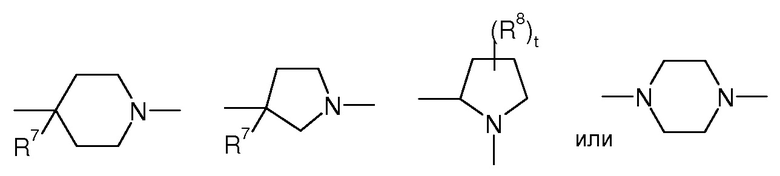
где R7 представляет собой OR, галоген, линейный или разветвленный (C1-C6)алкил-OR, C(O)OR, C(O)NRR’, CN, OR9, NRR’ или SR, более предпочтительно OR, OR9, NRR’ или SR, предпочтительно OH или OR9, p представляет 1 или 2 и R8 выбран из группы, состоящей из H или OH.
Согласно конкретному воплощению соединения изобретения могут быть соединениями следующей формулы (Ia)
в которой
R1, q, L1, L2, R6 и R7 имеют значения, определенные для формулы (I);
X’ представляет собой CR7 или N;
n представляет 0, 1 или 2;
p представляет 1, 2 или 3;
R3, R4, R8 и R8’, каждый идентичный или различный, выбраны из группы, состоящей из Н, линейного или разветвленного (C1-C6)алкила, галогена, OH, -O-(C1-C6)алкила, NRR’, CN, CF3, OR, C(O)R, C(O)OR или C(O)NRR’.
Предпочтительно, в соединении формулы (Ia), R3, R4, R8 и R8’, каждый идентичный или различный, выбраны из группы, состоящей из H или OH; и/или p представляет 1 или 2.
Предпочтительно, в соединении формулы (Ia)
представляет собой
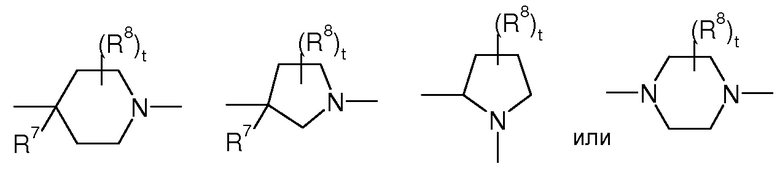 ,
,
t представляет 0, 1 или 2, предпочтительно
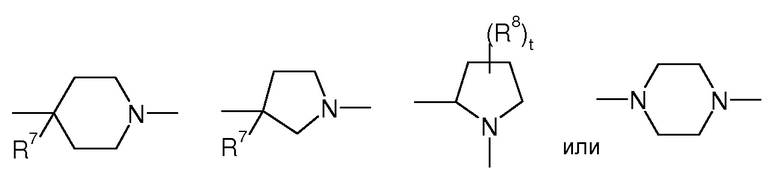
где R7 представляет собой OR, галоген, линейный или разветвленный (C1-C6)алкил-OR, C(O)OR, C(O)NRR’, CN, OR9, NRR’ или SR, более предпочтительно OR, OR9, NRR’ или SR, предпочтительно OH или OR9, p представляет 1 или 2 и R8 выбран из группы, состоящей из H или OH.
Согласно конкретному воплощению соединения изобретения могут быть соединениями следующей формулы (Ib)
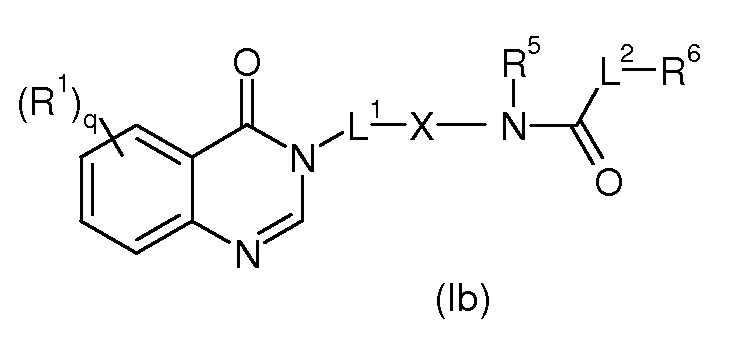
в которой
R1, q, L2 и R6 имеют значения, определенные для формулы (I);
X представляет собой арил, гетероарил, циклоалкил или гетероцикл, где арил, гетероарил, циклоалкил или гетероцикл, необязательно замещен одним или более из линейного или разветвленного C1-C6(алкила), галогена, OH, линейного или разветвленного -O-(C1-C6)алкила, NRR’, CN, CF3, OR, C(O)R, C(O)OR или C(O)NRR’;
R5 представляет собой Н или линейный или разветвленный (C1-C6)алкил;
L1 представляет собой линейный или разветвленный (C1-C6)aлкил, замещенный одним или более OH.
Предпочтительно, в соединении формулы (Ib), X представляет собой фенил.
Предпочтительно, в соединении формулы (Ib), R5 представляет собой H.
Согласно конкретному воплощению соединения изобретения могут быть соединениями следующей формулы (I’)
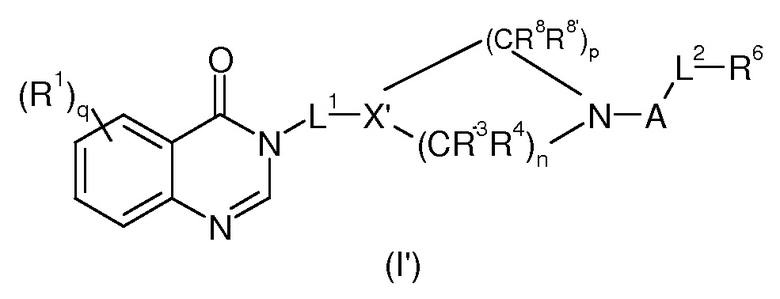
в которой
R1, q, L1, A, L2, R, R’ и R6 имеют значения, определенные для формулы (I);
X’ представляет собой CR7;
R7 представляет собой OR, галоген, линейный или разветвленный (C1-C6)алкил-OR, C(O)OR, C(O)NRR’, CN, OR9, NRR’ или SR, более предпочтительно OR, OR9, NRR’ или SR;
R9 имеет значения, определенные для формулы (I);
n представляет собой 0,1 или 2;
p представляет собой 1, 2 или 3;
R3, R4, R8 и R8’, каждый, идентичный или различный, выбран из группы, состоящей из Н, линейного или разветвленного -O-(C1-C6)алкила, галогена, ОН, -О-(С1-С6)алкила, NRR’, CN, CF3, OR, C(O)R, C(O)OR или C(O)NRR’.
Предпочтительно, в соединении формулы (I), A выбран из группы, состоящей из:
-C(O)-;
-C(O)NH-;
-SO2-; или
-SO2N-.
Предпочтительно, в соединениях формулы (I’), R3, R4, R8 и R8’, каждый идентичный или различный, выбраны из группы, состоящей из H или OH, и/или p представляет 1 или 2.
Предпочтительно, в соединениях формулы (I’) R7 представляет собой OR, галоген, линейный или разветвленный (C1-C6)алкил-OR, C(O)OR, C(O)NRR’, CN, более предпочтительно OH.
Предпочтительно, в соединениях формулы (I’) R7 представляет собой OR, OR9, NRR’ или SR, более предпочтительно R7 представляет собой OR, OR9, предпочтительно OH или OR9, например, OH.
Предпочтительно, в соединениях формулы (I’) p+n=4; более предпочтительно p представляет 2 и n представляет 2.
Предпочтительно, в соединении формулы (I’)
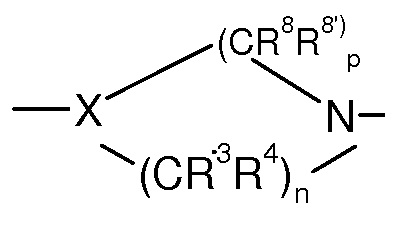
представляет собой
 .
.
Предпочтительно, в соединениях формулы (I’) L1 представляет собой CH2.
Предпочтительно, в соединениях формулы (I’) p представляет 2 и n представляет 2, R7 представляет собой OR, OR9, NRR’ или SR, более предпочтительно R7 представляет собой OR, OR9, предпочтительно OH или OR9, например, OH.
Предпочтительно в формуле (I’) p представляет 2 и n представляет 2, и L1 представляет собой CH2.
Предпочтительно в соединениях формулы (I’) L1 представляет собой CH2 и R7 представляет собой OR, OR9, NRR’ или SR, более предпочтительно R7 представляет собой OR, OR9, предпочтительно OH или OR9, например, OH.
Предпочтительно, в соединении формулы (I’) p представляет 2 и n представляет 2, L1 представляет собой CH2, R7 представляет собой OR, OR9, NRR’ или SR, более предпочтительно R7 представляет собой OR, OR9, предпочтительно OH или OR9, например, OH.
Согласно конкретному воплощению в соединениях формулы (I’) A представляет собой C=O, соединение таким образом является соединением формулы (Ia’)
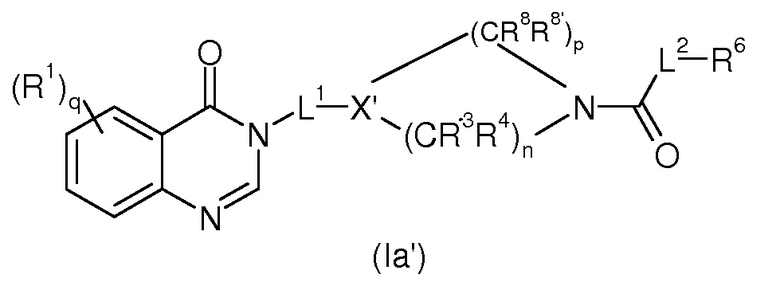
в которой R1, q, L1, n, p, X’, R3, R4, R8, R8’, L2, R6 и R7 имеют значения, определенные для формулы (I’).
Предпочтительно, в соединении формулы (Ia’)
представляет собой
 .
.
Согласно конкретному воплощению изобретение относится к соединениям формулы (I), определенной выше, за исключением следующего соединения:
- q представляет 0, L1 представляет собой CH2, X-(CR3R4)n-NR5 образует пиперидин, X представляет собой CR2R7 и R7 представляет собой OH, i представляет 1, A представляет собой C=O, L2 представляет собой C2H4 и R6 представляет собой
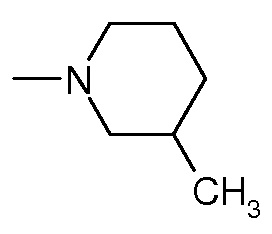 ;
;
- q представляет собой 1, R1 представляет собой Cl в 7 положении, L1 представляет собой СН2, X-(CR3R4)n-NR5 образует пиперидин, X представляет собой CR2R7 и R7 представляет собой OH, i представляет собой 1, A представляет собой C=O, L2R6 представляет собой CH(CH2CH3)2.
Согласно особому воплощению, соединения формулы (I) выбраны из:
3-({4-гидрокси-1-[3-(2-метоксифенил)пропаноил]пиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-она
7-хлор-3-{[1-(2-этилбутаноил)-4-гидроксипиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она
3-({1-[2-(3-фторфенокси)-ацетил]-4-гидроксипиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-она
3-{[4-гидрокси-1-(2-метилпропаноил)пиперидин-4-ил]метил}-6,7-диметокси-3,4-дигидрохиназолин-4-она
3-{[4-гидрокси-1-(2-метилпропаноил)пиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она
4-гидрокси-1-[2-метил-3-(тиофен-2-ил)пропаноил]пиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-она
7-хлор-3-{[1-(3-циклопентилпропаноил)-4-гидроксипиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она
3-{[1-(3-циклопентилпропаноил)4-гидроксипиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она
7-хлор-3-{[4-гидрокси-1-(3-фенилпропаноил)пиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она
3-{[4-гидрокси-1-(3-фенилпропаноил)пиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она
7-хлор-3-({4-гидрокси-1-[2-метил-3-(тиофен-2-ил)пропаноил]пиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-она
3-({4-гидрокси-1-[3-(тиофен-2-ил)пропаноил]пиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-она
3-{[1-(2-бензилпропаноил)-4-гидроксипиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она
3-{[1-(2-бензилпропаноил)-4-гидроксипиперидин-4-ил]метил}-7-хлор-3,4-дигидрохиназолин-4-она,
или их фармацевтически приемлемых солей или их оптических изомеров, рацематов, диастереомеров, энантиомеров или таутомеров.
Применяемый в описании выше или далее:
“Алкил” означает алифатическую углеводородную группу, которая может быть прямой или разветвленной, имеющую 1-6 атомов углерода в цепи. “Разветвленная” означает, что одна или более низших алкильных групп, таких как метил, этил или пропил, присоединены к линейной алкильной цепи. Примеры алкильных групп включают метил, этил, н-пропил, изо-пропил, н-бутил, трет-бутил, н-пентил, 3-пентил.
Применяемый в описании термин “циклоалкил” относится к не ароматическому моноциклическому или мультициклическому углеводородному кольцу из 3-10 атомов углерода образованному путем удаления одного углеводородного атома. Обозначение, такое как “С5-С7 циклоалкил” относится к циклоалкильному радикалу, содержащему от 5 до 7 атомов углерода. Примеры включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, адамантил и т.д., так же как и системы, образованные конденсацией их или конденсацией с фенильной группой.
“Алкен” или алкенил означает алифатическую углеводородную группу, содержащую углерод-углеродную двойную связь и которая может быть прямой или разветвленной, имеющую 2-6 атомов углерода в цепи. Предпочтительно алкенильные группы имеют 2-6 атомов углерода в цепи; и более предпочтительно примерно 2-4 углеродных атома в цепи. Примеры алкенильных групп включают этенил, пропенил, н-бутенил, изо-бутенил, 3-метилбут-2-енил, н-пентенил.
“Атом галогена” относится к атому фтора, хлора, брома или йода; предпочтительно атому фтора и хлора.
“Арил” означает ароматическую моноциклическую или мультициклическую углеводородную кольцевую систему из 6-14, атомов углерода, предпочтительно из 6-10 атомов углерода, замещенных или нет. Примеры алрильных групп включают фенил или нафтил.
Применяемые в описании термины “гетероцикл” или “гетероциклический” относятся к насыщенным, частично ненасыщенным или ненасыщенным, неароматическим стабильным 3-14-, предпочтительно 5-10-членным моно, би или мультициклическим кольцам, где, по крайней мере, один член кольца представляет собой гетероатом, замещенный или нет. Обычно, гетероатомы включают, но не ограничиваются ими, атомы кислорода, азота, серы, селена и фосфора. Предпочтительно гетероатомами являются кислород, азот и сера.
Подходящие гетероциклы также описаны в The Handbook of Chemistry and Physics, 76е Edition, CRC Press, Inc., 1995-1996, p.2-25 - 2-26, описание которых включено путем ссылки на них. Примером ароматического гетероцикла является тиофенил.
Предпочтительные не ароматические гетероциклы включают, но не ограничиваются ими, пирролидинил, пиразолидинил, имидазолидинил, оксиранил, тетрагидрофуранил, диоксоланил, тетрагидро-пиранил, диоксанил, диоксоланил, пиперидил, пиперазинил, морфолинил, пиранил, имидазолинил, пирролинил, пиразолинил, тиазолидинил, тетрагидротиопиранил, дитианил, тиоморфолинил, дигидро-пиранил, тетрагидропиранил, дигидропиранил, тетрагидро-пиридил, дигидропиридил, тетрагидропиринидил, дигидротиопиранил, азепанил, так же как и сконденсированные системы, получающиеся от конденсирования с фенильной группой, каждая из которых замещена или нет.
Применяемый в описании термин “гетероарил” или ароматические гетероциклы относится к 5-14-, предпочтительно 5-10-членным ароматическому гетеро-, моно-, би- или мультициклическому кольцу. Примеры включают пирролил, пиридил, пиразолил, тиенил, пиримидинил, пиразинил, тетразолил, индолил, хинолинил, пуринил, имидазолил, тиенил, тиазолил, бензотиазолил, фуранил, бензофуранил, 1,2,4-тиадиазолил, изотиазолил, триазолил, тетразолил, изохинолил, бензотиенил, изобензофурил, пиразолил, карбазолил, бензимидазолил, изоксазолил, пиридил-N-оксид, так же как и сконденсированные системы, получающиеся от конденсирования с фенильной группой.
Термин “алкил”, “циклоалкил”, “арил”, “гетероарил”, “гетероцикл” и аналогичные относится также к соответствующему “алкилену”, “циклоалкилену”, “арилену”, “гетероарилену”, “гетероциклену” и аналогичным, которые образуются путем удаления двух атомов водорода. Алкил и алкилен применяются в описании взаимозаменяемо.
Применяемый в описании термин “пациент” относится или к животному, такому как животное, полезное для целей разведения, сохранения или общественных целей или предпочтительно к человеку или ребенку, которые страдают от заболевания или имеют возможность быть подверженными одному или более заболеваниям и состояниям описанным выше.
Применяемый в описании термин “терапевтически эффективное количество” относится к количеству соединения данного изобретения, которое является эффективным для предотвращения, снижения, устранения, лечения или контролирования симптомов вышеописанных заболеваний и состояний. Термин “контролирование” предназначен для ссылки на все процессы, в которых может быть замедление, прерывание, задержка или остановка развития заболеваний и состояний, раскрытых в описании, но нет необходимости отмечать полное устранение всех симптомов заболевания или состояния, и предназначен для включения пролиферативного лечения.
Применяемое в описании выражение “фармацевтически приемлемое” относится к тем соединениям, материалам, наполнителям, композициям или дозировочным формам, которые в области тщательного медицинского обследования являются подходящими для контакта с тканями человека или животных без излишней токсичности, болезненной чувствительности, аллергической ответной реакции или других проблемных осложнений соразмерных с благоразумным соотношением польза/риск.
Применяемые в описании термины “фармацевтически приемлемые соли” относятся к производным раскрытых в описании соединений, в которых исходное соединение модифицируют путем изготовления их кислотных или основных солей. Фармацевтически приемлемые соли включают общепринятые нетоксичные соли или четвертичные аммониевые соли исходных соединений, образованные, например, из нетоксичных неорганических или органических кислот. Например, такие общепринятые нетоксичные соли включают такие, которые происходят от неорганических кислот, таких как соляная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная и аналогичные, включая их моно, ди или три-соли; и соли полученные из органических кислот, таких как уксусная, пропионовая, янтарная, винная, лимонная, метансульфоновая, бензолсульфоновая, глюкуроновая, глютаминовая, бензойная, салициловая, толуолсульфоновая, щавелевая фумаровая, малеиновая молочная и аналогичные. Далее дополнительные соли включают аммониевые соли, такие как трометамин, меглумин, эполамин, и т.д., соли металлов, таких как натрий, калий, кальций, цинк или магний.
Фармацевтические приемлемые соли настоящего изобретения могут синтезироваться из основного соединения, которое содержит основный или кислотный фрагмент, с помощью общепринятых химических методов. Обычно, такие соли могут быть получены с помощью реакции форм свободной кислоты или основания данных соединений со стехиометрическим количеством соответствующего основания или кислоты в воде или в органическом растворителе, или в смеси двух. Обычно предпочитаются неводные среды, как простой эфир, этилацетат, этанол, изопропанол или ацетонитрил. Перечни подходящих солей находятся в работе Remington’s Pharmaceutical Sciences, 20th ed., Mack Publishing Company, Easton, PA, 2000, содержание которой включено путем ссылки на нее.
Соединения общей формулы (I), имеющие геометрические и стереоизомеры, также являются частью изобретения.
Согласно дальнейшему объекту настоящее изобретение также касается процесса получения соединений формулы (I).
Соединения в процессе настоящего изобретения могут быть получены с помощью ряда путей хорошо известных специалистам в данной области. Соединения могут синтезироваться, например, путем применения или адаптации методов, описанных ниже, или их видоизменений, понятных специалистам в данной области. Соответствующие модификации и замены, по-видимому, легко очевидны и хорошо известны специалистам в данной области или легко доступны из научной литературы.
В частности, такие способы можно найти в работе R.C. Larock, Comprehensive Organic Transformations, Wiley-VCH Publishers, 1999.
Понятно, что соединения настоящего изобретения могут содержать один или более асимметрически замещенных атомов углерода, и могут выделяться в оптически активных или рацемических формах. Таким образом, подразумеваются все хиральные, диастереомерные, рацемические формы, изомерные формы структур, если не указана конкретная стереохимия или изомерная форма. В технике хорошо известно как получать и выделять такие оптически активные формы. Например, смеси стереоизомеров могут разделяться с помощью стандартных приемов, включая, но не ограничиваясь ими, повторное растворение рацемических форм, нормальную, с обращенной фазой и хиральную хроматографию, образование предпочтительной соли, перекристаллизацию и аналогичные, или с помощью хирального синтеза или из хиральных исходных материалов, или с помощью преднамеренного синтеза целевых хиральных центров.
Соединения настоящего изобретения могут быть получены с помощью множества синтетических методов. Реагенты и исходные материалы являются промышленно доступными или свободно синтезируются с помощью хорошо известных обычным специалистам в данной области приемов. Все заместители, если не указано иное, имеют значения, определенные ранее.
В реакциях, описываемых здесь ниже, может быть необходимо защищать реакционно-способные функциональные группы, например, гидроксил, амино, имино, тио или карбокси группы, когда они желательны в конечном продукте, чтобы избежать их нежелательное участие в реакциях. Могут использоваться общепринятые защитные группы в соответствии со стандартной практикой, см., например, T.W. Greene и P.G.M. Wuts в работе Protective Groups in Organic Chemistry, 4th ed.(2007), John Wiley & Sons Inc., 1999; J.F.W. McOmie в работе Protective Groups in Organic Chemistry, Plenum Press, 1973.
Некоторые реакции могут осуществляться в присутствии основания. В отношении характера основания, используемого в данной реакции, нет никаких особых ограничений, здесь в равной степени может использоваться любое основание, обычно используемое в реакциях данного типа, при условии, что оно не оказывает никакого пагубного воздействия на другие части молекулы. Примеры подходящих оснований включают в себя гидроксид натрия, карбонат калия, триэтиламин, гидриды щелочных металлов, такие как гидрид натрия и гидрид калия; алкиллитиевые соединения, такие как метиллитий и бутиллитий; и алкоксиды щелочных металлов, такие как метоксид натрия и этоксид натрия.
Обычно реакции осуществляются в подходящем растворителе. Может использоваться множество растворителей при условии, что оно не оказывает пагубного никакого влияния на реакцию или на вовлекаемые реагенты. Примеры подходящих растворителей включает: углеводороды, которыми могут быть ароматические, алифатические или циклоалифатические углеводороды, такие как гексан, циклогексан, бензол, толуол и ксилол; амиды, такие как диметилформамид; спирты, такие как этанол и метанол, и простые эфиры, такие как диэтиловый эфир и тетрагидрофуран.
Реакции могут происходить в широком интервале температур. Обычно найдено удобным осуществлять реакцию при температуре от 0°C до 150°C (более предпочтительно примерно от комнатной температуры до 100°C). Время, требуемое для реакции, также может широко варьировать в зависимости от многих факторов, а именно температуры и характера реагентов. Однако, при условии, что реакция проводится в предпочтительных условиях, указанных выше, обычно будет достаточным период от 3 часов до 20 часов.
Соединение, получаемое таким образом, может выделяться из реакционной смеси с помощью обычных средств. Например, соединения могут выделяться с помощью отгонки растворителя из реакционной смеси или, если необходимо, после отгонки растворителя из реакционной смеси, выливания растворителя в воду с последующей экстракцией несмешиваемым с водой растворителем и отгонкой растворителя из экстракта. Дополнительно, продукт может при желании далее очищаться различными хорошо известными приемами, такими как перекристаллизация, переосаждение или различные приемы хроматографии, а именно колоночная хроматография или препаративная тонкослойная хроматография.
Процесс получения соединений формулы (I) изобретения является дополнительным объектом настоящего изобретения.
Согласно первому аспекту, соединение изобретения формулы (I) может быть получено с помощью реакции соединения формулы (II) с соединением формулы (III) для того, чтобы образовать вторичные или третичные амины, карбоксамиды, мочевину, сульфонамиды или тиомочевины,
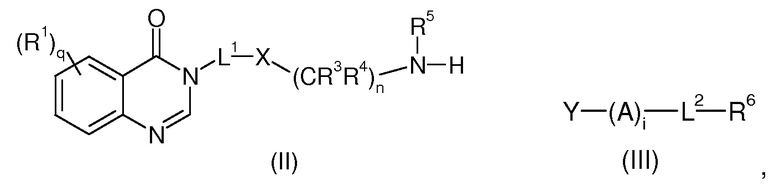
где R1, R3, R4, R5, R6, L1, L2, X, A, q, n и i имеют значения, определенные выше для формулы (I), Y представляет собой уходящую группу.
Уходящей или удаляемой группой является такая, чтобы реактивные функции соединений (II) и (III) вели к -NR5-(A)i- группе, как в формуле (I).
Предпочтительно, уходящая группа Y выбирается из галогена, OH, активированной OH, такой как группа R-S(O)2O-, где R представляет собой арил или линейный или разветвленный C1-C6(алкил). Предпочтительно, R-S(O)2O- представляет собой Ts- или Ms- группу, Ts представляет собой
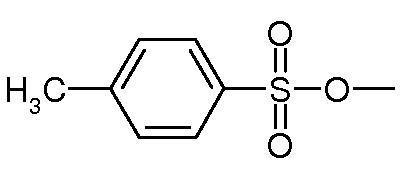
и Ms представляет собой
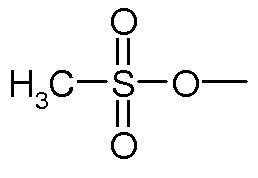 .
.
Более конкретно, когда группа, состоящая из -NR5-(A)i- в соединениях (I) представляет собой:
Вторичные или третичные амины: Y является уходящей группой, выбранной из галогена, OH, активированной OH, такой как группа R-S(O)2O-, в которой R представляет собой арил или линейный или разветвленный C1-C6(алкил). Предпочтительно, R-S(O)2O- представляет собой a Ts- или Ms- group, Ts является группой
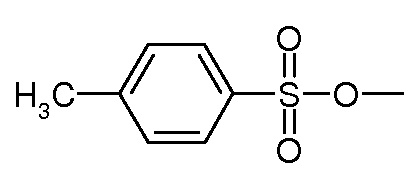
и Ms является группой
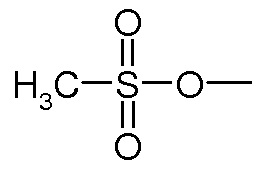
и i=0. Обычно данными реакциями являются реакции алкилирования, реакции Мицунобу, и проводятся они согласно методам, хорошо известным в данной области техники; или
Карбоксамиды: Y и A образуют хлорангидрид кислоты или карбоновую кислоту. Обычно, когда Y представляет OH, i представляет 1 и A представляет собой C(O), используются условия реакции пептидного сочетания.
Обычно, когда Y представляет собой OH, i представляет 1 и A представляет собой C(O), данная реакция осуществляется в присутствии реагентов сочетания, таких как EDCl (гидрохлорид (1-этил-3-[3-(диметиламино)карбодиимида] карбодиимида) и HOBt (N-гидроксибензотриазол), с или без основания (например, Et3N) в апротонном растворителе, таком как дихлорметан или диметилформамид;
или
Мочевины: Y и A образуют изоцианат; или
Сульфонамиды: Y и A образуют сульфонилхлорид; или
Тиомочевины: Y и A образуют тиоизоцианат.
Предпочтительно, соединением формулы (II) является соединение формулы (IIa)
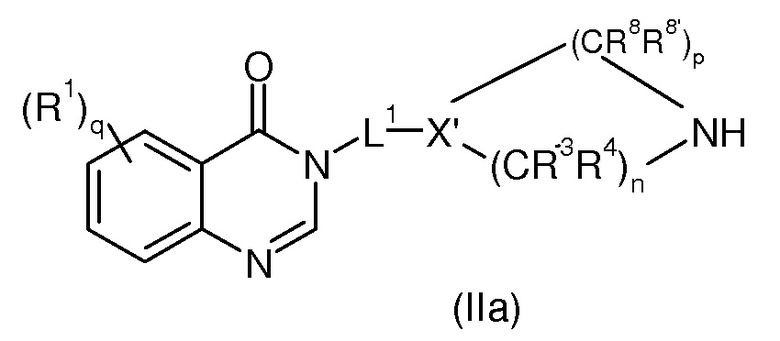
где R1, q, L1, X’, R8, R8’, p, R3, R4 и n имеют значения, определенные для формулы (I’) и (Ia’).
Согласно второму аспекту, соединение изобретения формулы (I) может быть получено с помощью реакции соединения формулы (IV) с соединением формулы (V)
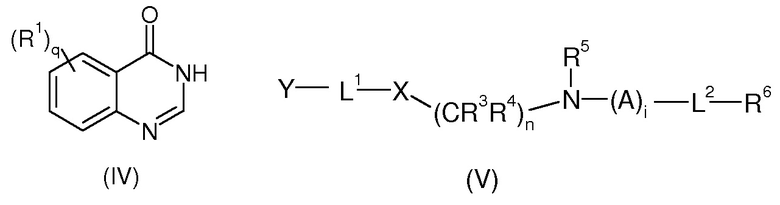
где R1, R3, R4, R5, R6, L1, L2, X, q, n и i имеют значения, определенные выше, и Y представляет собой уходящую группу.
Предпочтительно, Y выбран из эпокси, галогена, активированного ОН, определенного выше.
Более предпочтительно соединением формулы (V) является соединение формулы (Va)
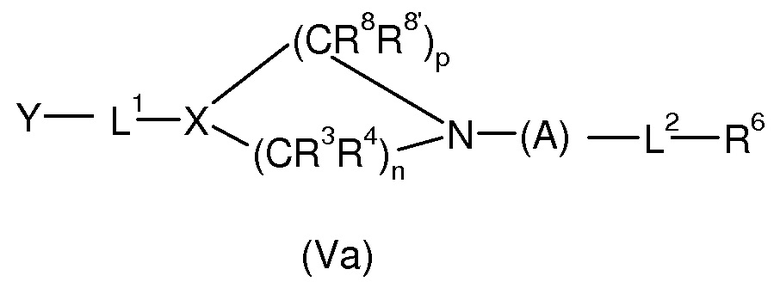
где, X, R8, R8’, p, R3, R4, n, A, L2 и R6 имеют значения, определенные для формулы (I’) и (Ia’).
Данная реакция обычно осуществляется в присутствии основания, предпочтительно неорганического основания, и в растворителе, предпочтительно полярном апротонном растворителе.
Соединения формулы (III) и (IV) являются промышленно доступными или могут быть получены специалистом в данной области техники на основе общих знаний в органической химии.
Соединения формулы (II) и (V) получаются, как описано ниже в общей процедуре.
Согласно третьему аспекту соединение изобретения формулы (I) может быть получено по реакции соответствующего соединения формулы (Ic)
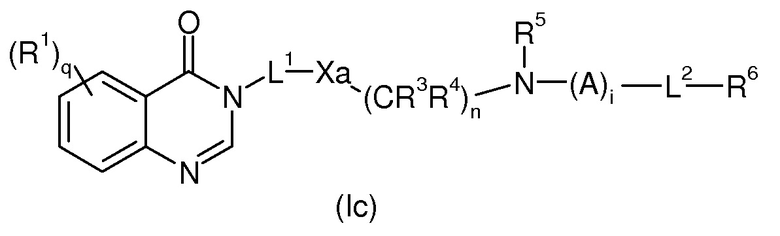
в которой R1, R2, R3, R4, R5, R6, L1, L2, q, n и i имеют значения, определенные для формулы (I), и Xa представляет собой предшествующую группу группы Х. Соединение формулы (Ic) может быть получено из соответствующих соединений формулы (IIc) и (IIIc) или (IVc) и (Vc), соответственно, по аналогии с соединениями формулы (I), как описано выше.
Термин “предшественник (предшествующий)” используется здесь для упоминания соединений, которые отличаются от указанных или желаемых соединений присутствием и/или отсутствием групп или функций. Такие группы или функции могут вводиться, трансформироваться и/или удаляться с помощью обычных реакций функционализации известных специалистам в данной области.
Реакция функционализации может осуществляться с помощью применения или адаптации известных способов.
Предпочтительно, предшествующей группой является такая, которая дает возможность с помощью одной или более стадий получить Х, исходя из Ха, таких как, например, восстановление, амидирование, окисление, гидролиз, сложная этерификация.
Указанные выше реакции могут осуществляться специалистами с помощью применения или адаптации способов, проиллюстрированных здесь ниже в примерах.
Далее, процесс изобретения может также включать дополнительную стадию выделения соединения формулы (I), (I’), (Ia), (Ib) или (I’a). Это может быть проделано специалистом с помощью любых известных общепринятых средств, таких как способы выделения, описанные выше.
Обычно исходные продукты промышленно доступны главным образом от Aldrich или Acros или других типичных химических поставщиков или могут быть получены с помощью применения или адаптации любых известных методов или методов, описанных в примерах.
Согласно дополнительному объекту настоящее изобретение касается также фармацевтической композиции, включающей в свой состав соединение формулы (I), (I’), (Ia), (Ib) или (I’a), определенное выше, с фармацевтически приемлемым наполнителем или эксцепиентом.
Предпочтительными воплощениями соединений формулы (I), (I’), (Ia), (Ib) или (I’a) являются воплощения, описанные выше в отношении соединений изобретения, и согласно любому из предпочтительных признаков или воплощений.
Согласно дальнейшему еще объекту настоящее изобретение касается соединения формулы (I) изобретения для ингибирования цистеиновой протеазы.
Соединения формул (I), (I’), (Ia), (Ib) или (I’a) преимущественно обеспечивают возможность селективного и обратимого ингибирования цистеиновой протеазы.
Соединения и фармацевтические композиции изобретения полезны для ингибирования цистеиновых протеаз, в частности, специфических ферментов дез-убихитинирования, таких как USPs, и более конкретно USP-7 у пациентов, нуждающихся в этом.
Соединения и фармацевтическая композиция изобретения особенно полезны для лечения и/или предотвращения рака и метастаз, более конкретно раков простаты и/или ободочной кишки, нейродегенеративных заболеваний, таких как болезнь Альцгеймера и болезнь Паркинсона, иммунологических расстройств, болезней костей и суставов, остеопороза, артритных воспалительных расстройств, сердечно-сосудистых заболеваний, вирусных инфекций и заболеваний, и/или вирусной инфекционности и/или латентного состояния, бактериальных инфекций и заболеваний.
Соединение и фармацевтическая композиция изобретения могут использоваться на пациентах, которые не имеют бета-амилоидных бляшек, которые действуют на старческое слабоумие, особенно болезнь Альцгеймера.
В частности, указанные вирусные инфекции и заболевания выбраны из вирусных инфекций Нerpes simplex-1 или -2 гепатита А, гепатита С, SARS коронавирусной инфекции и болезни, вируса Epstein-Barr, риновирусных инфекций и заболеваний, аденовирусных инфекций и заболеваний, полиомиелита.
Согласно одному аспекту указанные соединения ингибируют одну или более вирусных цистеиновых протеаз.
Бактериальные цистеиновые протеазы могут быть выбраны из стрептопаина, клострипаина, стафилококковых цистеиновых протеаз, гингипаина.
Настоящее изобретение касается также комбинационных препаратов, включающих соединение формулы (I), определенное выше, с одним или более активными агентами, выбранными из противораковых агентов, неврологических агентов, тромболитических агентов, антиоксидантов, противо-инфекционных, противо-гипертензивных агентов, диуретиков, тромболитических агентов, иммуноподавляющих агентов, сердечнососудистых агентов, иммуномодуляторных, противовоспалительных, антивирусных, анти-бактериальных агентов.
Настоящее изобретение касается также соответствующих способов лечения, предусматривающих введение пациенту, нуждающемуся в этом, соединения изобретения вместе с фармацевтически приемлемым носителем или наполнителем.
Согласно изобретению термины “пациент” или “пациент, нуждающийся в этом” предназначаются для обозначения животного или человека, пораженного или вероятно склонного быть подверженным поражению патологическим состоянием, вовлекающим в его патогенез активную цистеиновую протеазу. Пациентом, предпочтительно, является человек.
Идентификация данных субъектов, которые нуждаются в лечении описываемых здесь заболеваний и состояний, находится вполне в пределах способности и знаний специалиста в данной области. Ветеринар или врач, осведомленный в данной области, может свободно путем использования клинических испытаний, физического осмотра, медицинской истории семьи или биологических и диагностических испытаний определить тех субъектов, которые нуждаются в таком лечении.
“Терапевтически эффективное количество” обозначает количество соединения/медикамента согласно настоящему изобретению эффективное в профилактике или лечении патологического состояния, требующего ингибирование активной цистеиновой протеазы, вовлекаемой в его патогенез.
Терапевтически эффективное количество может быть легко определено обслуживающим диагностом, осведомленным в данной области, путем использования общепринятых приемов и наблюдения результатов, получаемых в аналогичных обстоятельствах. При определении терапевтически эффективного количества обслуживающим диагностом учитывается ряд факторов, включая, но не ограничиваясь ими; вид или особенности субъекта, его рост, возраст и общее состояние здоровья; конкретно затрагиваемое заболевание, степень вовлечения или тяжести заболевания; ответная реакция отдельного субъекта; конкретно вводимое соединение; способ введения; характеристики биологической доступности вводимого препарата; выбранный режим дозирования; и другие существенные обстоятельства.
Количество соединения формулы (I), (I’), (Ia), (Ib) или (I’a), которое требуется для достижения желаемого биологического эффекта, будет меняться в зависимости от ряда факторов, включая химические характеристики (например, гидрофобность) применяемых соединений, силу действия соединений, тип заболевания, виды, к которым относится пациент, болезненное состояние пациента, способ введения, биологическая доступность соединения при выбранном способе, все факторы, которые диктуют требуемые количества дозы, доставка и режим введения.
“Фармацевтически” или “фармацевтически приемлемый” относится к молекулярным сущностям и композициям, которые не вызывают пагубные, аллергические или другие неблагоприятные реакции, когда они вводятся животному или человеку, кому предназначены.
Используемое здесь выражение “фармацевтически приемлемый эксципиент” включает в себя любые носители, разбавители, адъюванты или наполнители, такие как консервирующие агенты или антиоксиданты, заполнители, дезинтегрирующие, смачивающие, эмульгирующие, суспендирующие агенты, растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые агенты, изотонические и задерживающие абсорбцию агенты и аналогичные. Использование таких сред и агентов для фармацевтически активных веществ хорошо известно хорошо известно в технике. Предполагается их использование в терапевтических композициях, за исключением любых традиционных сред или агентов, которые, насколько известно, являются несовместимыми с активным ингредиентом. При подходящих терапевтических комбинациях в композиции могут также включаться дополнительные активные ингредиенты.
В контексте изобретения термин “лечение”, используемый здесь, обозначает реверсирование, смягчение, ингибирование развития или предотвращения расстройства или состояния, к которому такой термин применяется, или одного или более симптомов такого расстройства или состояния.
Обычно соединения изобретения могут предоставляться в водном физиологическом буферном растворе, содержащем 0,1-10% вес/объем соединения для парентерального введения. Типичные интервалы доз составляют от 1 мкг/кг до 0,1 г/кг веса тела в день; предпочтительный интервал доз составляет от 0,01 мг/кг до 100 мг/кг веса тела в день или эквивалентную дозу ребенку человека. Предпочтительная дозировка вводимого лекарства вероятно зависит от таких переменных, как тип и степень прогрессирования болезни или расстройства, общее состояние здоровья конкретного пациента, относительная биологическая эффективность выбранного соединения, рецептурной формы соединения, способа введения (внутривенный, внутримышечный или иной), фармакокинетические свойства соединения при выбранном способе, и скорость (болюс или непрерывная инфузия) и график введения (число повторений на протяжении заданного периода времени).
Соединения настоящего изобретения могут также вводиться в форме единичных доз, при этом “единичная доза” обозначает одиночную дозу, которая способна вводиться пациенту и которая может быть свободной в обращении и упаковываться, оставаясь в виде физически и химически стабильной единичной дозы, включающей или само активное соединение, или в виде фармацевтически приемлемой композиции, как описано ниже. Как таковые, типичные интервалы общей дневной дозы составляют от 0,01 до 100 мг/кг веса тела. В виде общего руководства, дозы для людей колеблются от 1 мг до 3000 мг в день. Предпочтительно интервал единичной дозы составляет от 1 до 500 мг, вводимых от одного до шести раз в день, и даже более предпочтительно от 10 мг до 500 мг один раз в день. Предлагаемые здесь соединения могут формулироваться в виде фармацевтических композиций смешением с одним или более фармацевтически приемлемыми наполнителями. Такие композиции единичных доз могут получаться для использования путем орального введения, особенно в форме таблеток, простых капсул или мягких коллоидных капсул; или внутриназально, в частности в форме порошков, носовых капель, или аэрозолей; или кожным путем, например, местно в виде мазей, кремов, лосьонов, гелей или аэрозолей, или трансдермальных бляшек.
Композиции могут удобно вводиться в форме единичных дозировок и могут получаться любым из способов, хорошо известных в области фармацевтики, например, как описано в публикации Remington: The Science and Practice of Pharmacy, 20th ed.; Gennaro, A. R., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, 2000.
Предпочтительные рецептурные формы включают фармацевтические композиции, в которых соединение настоящего изобретения формулируется для орального или парентерального введения.
Для орального введения таблетки, пилюли, порошки, капсулы, пастилки и аналогичные могут содержать один или более из любых следующих ингредиентов или соединений похожего характера: связующее, такое как микрокристаллическая целлюлоза или камедь трагаканта; разбавитель, такой как крахмал или лактоза; дезинтегрирующий агент, такой как крахмал и производные целлюлозы; смазочный агент, такой как стеарат магния; агент скольжения, такой как коллоидная двуокись кремния; подслащивающий агент, такой как сахароза или сахарин; или вкусовой агент, такой как перечная мята или метилсалицилат. Капсулы могут быть в форме твердых или мягких капсул, которые обычно приготавливаться из желатиновых смесей, необязательно смешанных с пластификаторами, также как крахмальные капсулы. В дополнение, формы дозированных единиц могут содержать различные другие материалы, которые видоизменяют физическую форму дозированной единицы, например, покрытия из сахара, шеллак или тонкокишечные (энтерические) агенты. Другие оральные дозировочные формы, сироп или эликсир, могут содержать подслащивающие агенты, консерванты, красители, оттеночные и вкусовые агенты. В дополнение, активные соединения могут включаться в состав быстро растворимых препаратов и рецептурных форм, препаратов модифицированного высвобождения или длительного высвобождения, и такие рецептурные формы длительного высвобождения являются предпочтительно би-модальными. Предпочтительные таблетки содержат лактозу, кукурузный крахмал, силикат магния, натриевую кроскармелозу, повидон, стеарат магния или тальк в любом сочетании.
Жидкие препараты для парентерального введения включают в их число стерильные водные или неводные растворы, суспензии и эмульсии. Жидкие композиции могут также включать в свой состав связующие, буферные агенты, консерванты, хелатирующие агенты, подсластители, вкусовые и подкрашивающие или оттеночные агенты, и аналогичные. Неводные растворители включают в их число спирты, пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, и органические сложные эфиры, такие как этилолеат. Водные носители включают смеси спиртов и воды, буферные среды и солевые растворы. В частности, биосовместимый биоразлагаемый лактидный полимер, лактид/полигликолидный сополимер или полиоксиэтилен-полиоксипропиленовые сополимеры могут быть полезными наполнителями для регулирования высвобождения активных соединений. Внутривенные наполнители могут включать в себя жидкие питательные наполнители, электролитные наполнители, такие как наполнители на основе декстрозы Рингера, и аналогичные. Другие потенциально полезные системы парентеральной доставки для данных активных соединений включают в себя этилен-винилацетатные сополимерные частицы, осмотические нагнетающие агенты, имплантируемые системы инфузии и липосомы.
Альтернативные формы введения включают в себя рецептурные формы для ингаляции, которые включают такие средства, как сухой порошок, аэрозоль или капли. Они могут представлять собой водные растворы, содержащие, например, полиоксиэтилен-9-лауриловый эфир, гликохолат и дезоксихолат, или масляные растворы для введения в виде носовых капель или в виде геля, применяемого в нос. Рецептурные формы для щечного введения включают, например, пастилки или лепешки, и могут также включать ароматизирующую или придающую вкус основу, такую как сахароза или камедь акации, и другие наполнители, такие как гликохолат. Рецептурные формы для ректального введения предпочтительно представлены в виде единично дозированных суппозиториев с носителем на основе твердого вещества, и могут включать салицилат. Рецептурные формы для местного или топического применения к коже предпочтительно принимают форму мази, крема, лосьона, пасты, геля, спрея, аэрозоля или масла. Носители, которые могут быть использованы, включают в себя вазелин, ланолин, полиэтиленгликоли, спирты или их сочетания. Рецептурные формы, подходящие для введения через кожу, могут быть представлены в виде дискретных частиц и могут представлять собой липофильные эмульсии или забуференные водные растворы, растворенные и/или диспергированные в полимере или адгезиве.
Изобретение далее иллюстрируется, но не ограничивается, описанием в следующих ниже примерах и фигурах в качестве неограничивающей иллюстрации селективного ингибирования USP7 дезубихитинирующей активности на ряде активных DUBs в физиологических условиях.
Фиг. 1 показывает конкурентный HAUbVS гель для соединения примера 2 и примера 5 (12,5-25-50-100-200 мкM) с использованием HEK293 протеома.
Фиг. 2 показывает конкурентные HAUbVS гели сравнения активности соединения примера 2 и примера 5 против USP7 и дополнительных дезубихитинирующих ферментов (USP8, USP5, USP10, CYLD, UCH-L3) с использованием HEK293 протеома.
Фиг. 3 показывает эксперименты в зависимости от времени с USP7 и соединением примера 2.
Фиг. 4 показывает результаты обратимости, полученные следуя экспериментам гель-фильтрации с USP7 в присутствии соединений примера 2 и примера 3.
Фиг. 5 показывает результаты обратимости, полученные следуя быстрому и обильному разбавлению USP7 в присутствии соединений примера 2 и примера 3.
Фиг. 6 представляет характеристику комплексов, включающих в себя USP7 и соединения примеров 1 и 2 по оценке с помощью ESI-MS в естественных условиях.
ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ
Характерные соединения изобретения приводятся в таблице ниже:
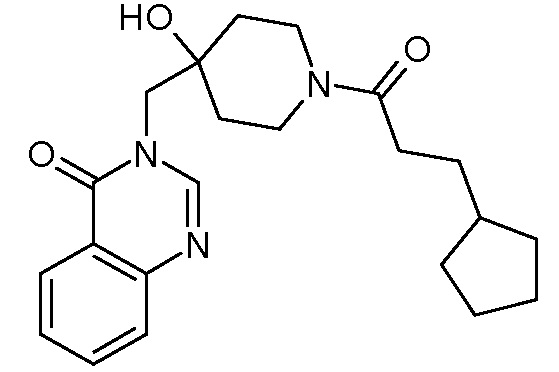
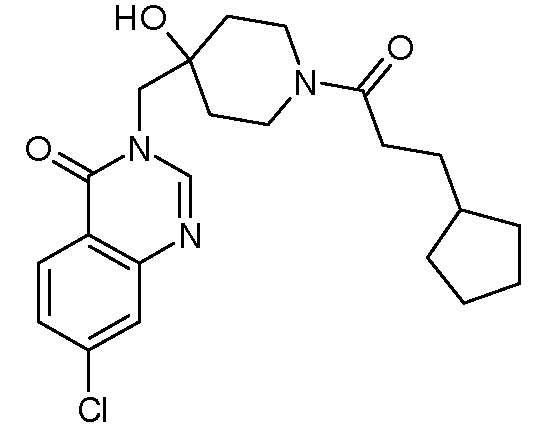

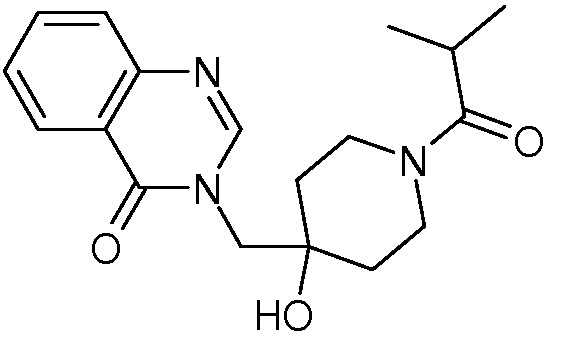
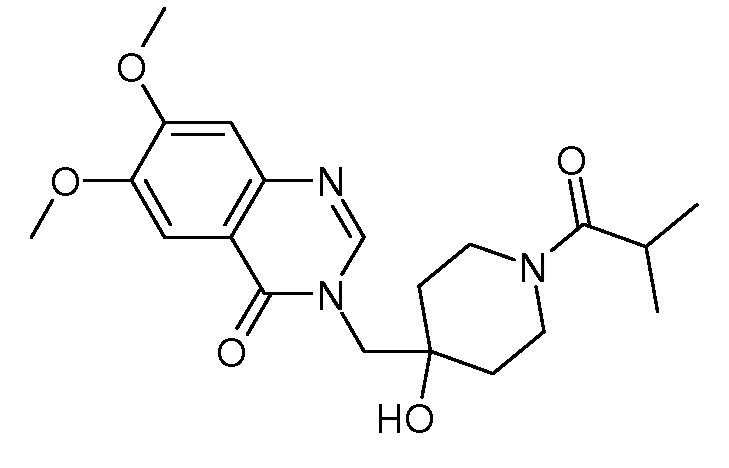
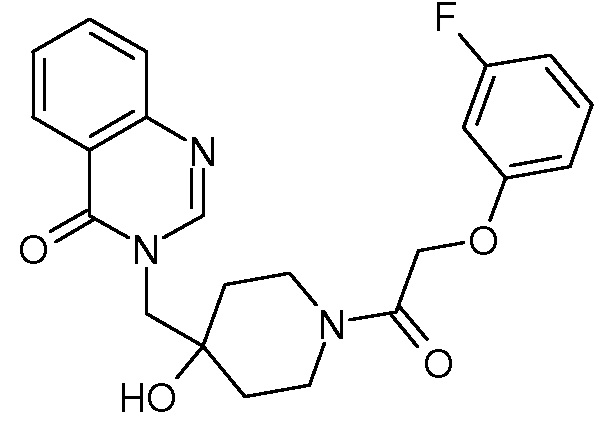
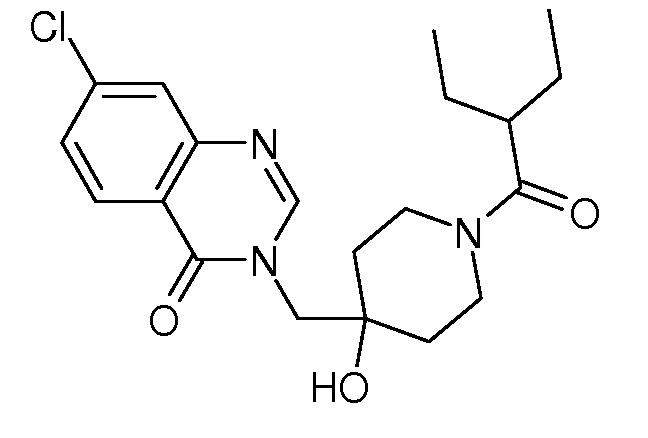
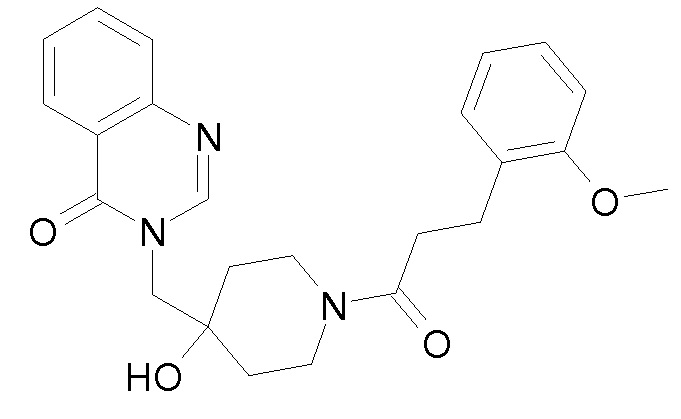
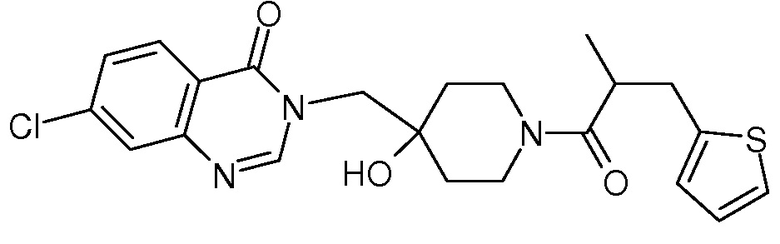
Характерные соединения изобретения могут быть синтезированы в соответствии со следующими процедурами.
Процедуры общего анализа
Данные спектра ЯМР записывались при 300 или 400 МГц для 1Н и при 75 или 100 МГц для 13С на спектрометре Bruker или Varian с использованием CDCl3 или DMSO-d6 (диметилсульфоксида) в качестве растворителя. Химические сдвиги даются в м.д., в отношении к внутреннему сигналу TMS (Триметилсилил) или дейтерированного растворителя.
Анализ LC-MS применяли для анализа и очистки целевых соединений. Анализы LC-MS осуществляли с применением масс спектрометров Waters Micromass, Druker Esquire 3000 (ESI-IT) или Agilent Iontrap XCT-Plus и Agilemt 1100 Series LS систем c UV или DAD детекцией. Колонки: Waters XTerra MS C18, 30×2,1 мм (3,5 мкм), Atlantis T3 C18, 3 мкм, 50 мм × 2,1 мм или Intersil C8, 250 мм, 4,6 мм, 5 мкм. Скорость потока: 0,8-1,2 мл/мин, Градиенты: а) вода 10% МеОН (метанол), формиат аммония 10 мМ, до 100% МеОН или b) 95% Вода-ацетонитрил, 0,1% НСООН до 95% ацетонитрил. UV детекция: 190-400 нм. Чистота всех соединений была >95%.
Типичная процедура 1:
Примеры получения 1-14
Реакционная схема 1
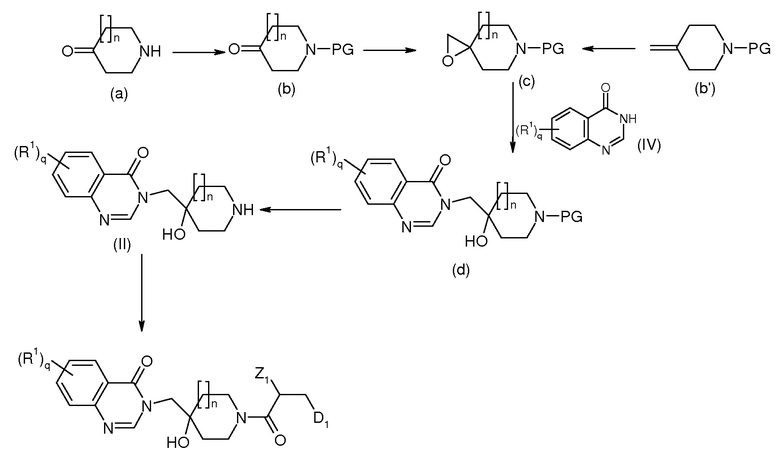
Соединения формулы (с) получают или из соединения (b) с помощью реакции Корей-Чайковского, как описано в J.Amer. Chem. Soc. 1965, 87, 1353-1364 и WO2005054249 или любым эпоксидированием двойной связи производных (b’). Соединение (b) является промышленно доступным или его получают из соединения (а) после реакции защиты, хорошо известной в данной области техники. Защищающей группой (PG) является, например, бензил (Bn), бензоил (Bz), трет-бутилоксикарбонил (BOC) или Карбобензилокси (Cbz).
Раскрытие кольца оксиран (с) с помощью хиназолинона (IV) осуществляют в присутствии основания, такого как NaH, KF, K2CO3, Cs2CO3 при нагревании между 50 и 100°С в диметилформамиде, ацетоне, и т.д.
Снятие защиты азота пиперидина, осуществляемое согласно известным способам, дает соединение (II).
Реакцию присоединения пептида осуществляют согласно хорошо известным способам в данной области между соединением (II) и производными кислоты HO-C(O)-CH2(Z1)-CH2-D (соединение III). Для некоторых примеров предпочтительными были условия идентичные EDCI (гидрохлорид 1-этил-3-[3-(диметиламино)пропил]карбодиимида), HOBt (N-гидроксибензотриазол) и Et3N в Дихлорметане (CH2Cl2). Образование амидной связи также возможно, когда реакция происходит с хлоридным производным кислоты.
Соединения (IV) являются промышленно доступными или их получают согласно опубликованным процедурам.
Следующие соединения получают путем применения типичной процедуры 1:
- q представляет собой 0, n представляет собой 1, Z1 представляет собой H и D1 представляет собой фенил (пример 1, описанный в экспериментальной части)
- q представляет собой 1, n представляет собой 1, R1 представляет собой Cl, Z1 представляет собой H и D1 представляет собой фенил (пример 2)
- q представляет собой 0, n представляет собой 1, Z1 представляет собой H и D1 представляет собой циклопентил (пример 3)
- q представляет собой 1, n представляет собой 1, R1 представляет собой Cl, Z1 представляет собой H и D1 представляет собой циклопентил (пример 4)
- q представляет собой 0, n представляет собой 1, Z1 представляет собой СH3 и D1 представляет собой тиофенил (пример 5)
- q представляет собой 0, n представляет собой 1, Z1 представляет собой СH3 и D1 представляет собой Н2 (пример 6)
- q представляет собой 2, R1 представляет собой ОМе, n представляет собой 1, Z1 представляет собой СH3 и D1 представляет собой Н2 (пример 7)
- q представляет собой 1, R1 представляет собой Cl, n представляет собой 1, Z1 представляет собой СН2СH3 и D1 представляет собой СН3 (пример 9)
- q представляет собой 0, n представляет собой 1, Z1 представляет собой H, D1 представляет собой 2-Оме-фенил (пример 10)
- q представляет собой 1, R1 представляет собой Cl, n представляет собой 1, Z1 представляет собой СH3, D1 представляет собой тиофенил (пример 11)
- q представляет собой 0, n представляет собой 1, Z1 представляет собой H, D1 представляет собой тиофенил (пример 12)
- q представляет собой 0, n представляет собой 1, Z1 представляет собой СH3, D1 представляет собой фенил (пример 13)
- q представляет собой 1, R1 представляет собой Cl, n представляет собой 1, Z1 представляет собой СH3, D1 представляет собой фенил (пример 14).
Далее представлены выбранные данные некоторых из соединений, которые были получены путем применения или адаптации способов описанных выше:
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
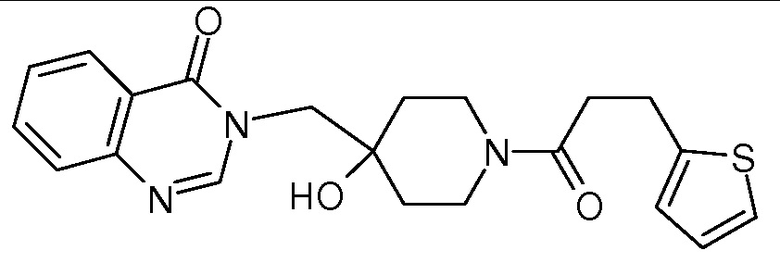
Пример 1: 3-{[4-гидрокси-1-(3-фенилпропаноил)пиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-он
Стадия 1: Получение трет-бутил 1-окса-6-азаспиро[2.5]октан-6-карбоксилата
К перемешиваемому раствору 1-Вос-4-пиперидина (6,1 г, 30,6 ммоль, 1 экв.) в тетрагидрофуране (200 мл) добавляли йодид триметилсульфоксония (6,8 г, 30,6 ммоль) и трет-бутоксид калия (4,0 г, 30,6 ммоль, 1 экв.). Смесь нагревали с обратным холодильником в течение 18 часов и концентрировали в вакууме. Сырой неочищенный продукт растворяли в АсОЕt (100 мл) и промывали водой (100 мл). Слои разделяли, и водный слой экстрагировали АсОЕt. Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Твердое вещество очищали с помощью колоночной флеш хроматографии на силикагеле (элюент: циклогексан/этилацетат 9/1), получая соединение трет-бутил 1-окса-6-азаспиро[2.5]октан-6-карбоксилат (3,3 г, 52%) в виде белого порошка.
Стадия 2: Получение трет-бутил 4-гидрокси-4-[(4-оксо-3,4-дигидрохиназолин-3-ил)метил]пиперидин-1-карбоксилата
К раствору 4-гидроксихиназолина (1,65 г, 11,3 ммоль, 1,1 экв.) в ДМФ (20 мл) добавляли соединение из стадии 1 (2,35 г, 1 экв.) и карбонат цезия (10,34 г, 3 экв.). Смесь нагревали при 80°С в течение ночи. Смесь промывали насыщенным раствором NH4Cl, и водный слой экстрагировали АсОЕt. Органический экстракт сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой неочищенный продукт очищали с помощью флеш хроматографии на силикагеле (элюент: циклогексан/этилацетат 3/7), получая трет-бутил 4-гидрокси-4-[(4-оксо-3,4-дигидрохиназолин-3-ил)метил]пиперидин-1-карбоксилат (1,6 г, 40%) в виде бесцветного масла.
MS(ES+, m/z): 360,2[M+H]+, 719,6[2M+H]+
Стадия 3: Получение трифторацетатной соли 3-[(4-гидроксипиперидин-4-ил)метил]-3,4-дигидрохиназолин-4-она
Соединение из стадии 2 (1,0 г, 2,8 ммоль) растворяли в TFA (трифторуксусная кислота)(80 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Полученную смесь концентрировали в вакууме. Сырую неочищенную трифторацетатную соль 3-[(4-гидроксипиперидин-4-ил)метил]-3,4-дигидрохиназолин-4-она использовали в следующей стадии без очистки.
MS(ES+, m/z): 260,1[M+H]+
1Н ЯМР (DMSO-d6) δ: 8,53 (шир.м, 1H), 8,25 (с, 1H), 8,27 (шир.м, 1H), 8,17 (дд, 1H), 7,85 (дд, 1H), 7,70 (дд, 1H), 7,57 (дд, 1H), 4,07 (с, 2H), 3,18 (м, 2H), 3,02 (м, 2H), 1,60 (м, 2H), 1,50 (м, 2H).
Стадия 4: 3-{[4-гидрокси-1-(3-фенилпропаноил)пиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-он
К раствору соли TFA из стадии 3 (0,8 ммоль, 1 экв.) в CH2Cl2 (10 мл) последовательно добавляли DIEA (N,N диизопропилэтиламин) (0,4 мл, 2,4 ммоль, 3 экв.), оксикоричную кислоту (150 мг, 0,96 ммоль, 1,2 экв.), EDCI (306 мг, 1,6 ммоль, 2 экв.) и HoBt (216 мг, 1,6 ммоль, 2 экв.). Реакционную смесь нагревали при комнатной температуре в течение ночи и затем концентрировали в вакууме. Сырой неочищенный продукт очищали с помощью колоночной флеш хроматографии на силикагеле (элюент: циклогексан/этилацетат от 2/8 до 0/10 затем этилацетат/МеОН 9/1), получая соединение Примера 1 (256 мг, 82%) в виде белого твердого вещества.
MS(ES+, m/z): 392,2[M+H]+
1Н ЯМР (DMSO-d6) δ: 8,25 (с, 1H), 8,17 (дд, 1H), 7,84 (дд, 1H), 7,69 (дд, 1H), 7,55 (дд, 1H), 7,21 (м, 5H), 4,96 (с, 1H), 4,04 (м, 3H), 3,64 (м, 1H), 3,21 (м, 1H), 2,93 (м, 1H), 2,80 (м, 2H), 2,62 (м, 2H), 1,41 (м, 4H).
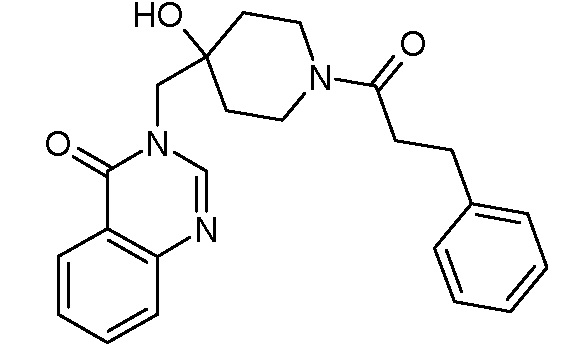
Пример 5: 3-({4-гидрокси-1-[2-метил-3-(тиофен-2-ил) пропаноил]пиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-он
К раствору соли TFA из стадии 3 примера 1 (0,42 ммоль, 1 экв.) в CH2Cl2 (10 мл) последовательно добавляли DIEA (0,37 мл, 2,12 ммоль, 5 экв.), 2-метил-3-(2-тиенил)пропановую кислоту (71 мг, 0,42 ммоль, 1 экв., Organometallics, 2002, 21, 2842), EDCI (161 мг, 0,84 ммоль, 2 экв.) и HoBt (114 мг, 0,84 ммоль, 2 экв.). Реакционную смесь нагревали при комнатной температуре в течение ночи и затем концентрировали в вакууме. Сырой неочищенный продукт очищали с помощью колоночной флеш хроматографии на силикагеле (элюент: циклогексан/этилацетат от 2/8 до 0/10 затем этилацетат/МеОН 9/1). Вторую очистку с помощью колоночной хроматографии осуществляли с применением тех же условий, получая после выпаривания растворителя и сушки в высоком вакууме, соединение Примера 5 (117 мг, 68%) в виде белого твердого вещества.
MS(ES+, m/z): 412,2[M+H]+
1Н ЯМР (DMSO-d6) δ: 8,24 (с, 1H), 8,17 (м, 1H), 7,84 (м, 1H), 7,70 (м, 1H), 7,56 (м, 1H), 7,27 (м, 1H), 6,88 (м, 1H), 6,82 (с, 1H), 4,96 (с, 1H), 4,06 (м, 1H), 3,90 (м, 2H), 3,68 (м, 1H), 3,20 (м, 5H), 1,34 (м, 4H), 1,02 (м, 3H).
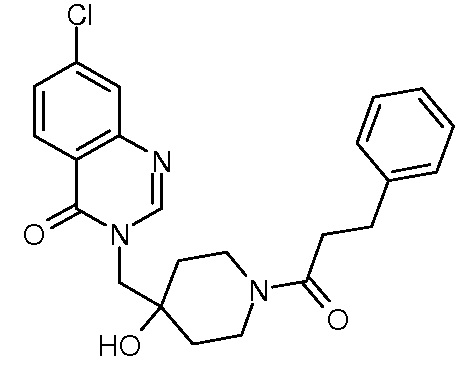
Пример 11: 7-хлор-3-({4-гидрокси-1-[2-метил-3-(тиофен-2-ил) пропаноил]пиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-он
Стадия 1: Получение трет-бутил 4-[(7-хлор-4-оксо-3,4-дигидрохиназолин-3-ил)метил]-4-гидроксипиперидин-1-карбоксилата
К раствору 7-хлор-3,4-дигидроксихиназолин-4-она (0,40 г, 2,2 ммоль, 1 экв.) в ДМФ (5 мл) добавляли соединение из стадии 1 примера 1 (0,47 г, 1 экв.) и карбонат цезия (2,17 г, 3 экв.). Реакционную смесь нагревали при 80°С в течение ночи, а затем давали достичь комнатной температуры. Смесь промывали насыщенным раствором NH4Cl, и затем экстрагировали АсОЕt. Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой неочищенный продукт очищали с помощью флеш хроматографии на силикагеле (элюент: циклогексан/этилацетат 3/7). Проводили вторую очистку с помощью флеш хроматографии на силикагеле (элюент: циклогексан/этилацетат от 6/4 до 0/10), получая трет-бутил 4-[(7-хлор-4-оксо-3,4-дигидрохиназолин-3-ил)метил]-4-гидроксипиперидин-1-карбоксилат (0,6 г, 68%) в виде бесцветного масла.
Стадия 2: Получение трифторацетатной соли 7-хлор-3-[(4-гидроксипиперидин-4-ил)метил]-3,4-дигидрохиназолин-4-она
Соединение из стадии 1 выше (0,6 г, 1,5 ммоль) растворяли в TFA (5 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Полученную смесь концентрировали в вакууме. Сырую неочищенную трифторацетатную соль 7-хлор-3-[(4-гидроксипиперидин-4-ил)метил]-3,4-дигидрохиназолин-4-она использовали в следующей стадии без очистки.
Стадия 3: 7-хлор-3-({4-гидрокси-1-[2-метил-3-(тиофен-2-ил)пропаноил]пиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-он
К раствору соли TFA из стадии 2 (0,51 ммоль, 1 экв.) в CH2Cl2 (18 мл) последовательно добавляли DIEA (0,45 мл, 2,5 ммоль, 5 экв.), 2-метил-3-(2-тиенил)пропановую кислоту (86 мг, 0,51 ммоль, 1 экв., Organometallics, 2002, 21, 2842), EDCI (196 мг, 1,02 ммоль, 2 экв.) и HoBt (138 мг, 1,02 ммоль, 2 экв.). Реакционную смесь нагревали при комнатной температуре в течение ночи и затем концентрировали в вакууме. Сырой неочищенный продукт очищали с помощью колоночной флеш хроматографии на силикагеле (элюент: циклогексан/этилацетат от 2/8 до 0/10 затем этилацетат/МеОН 9/1). Вторую очистку с помощью колоночной флеш хроматографии на силикагеле осуществляли для удаления остаточных реагентов. Продукт солюбилизировали в EtOH/H2O 1/1 и раствор сушили сублимацией, получая соединение Примера 11 (85 мг, 40% за две стадии) в виде белого безе-подобного вещества.
MS(ES+, m/z): 446,2[M+H]+
1Н ЯМР (DMSO-d6) δ: 8,27 (м, 1H), 8,17 (м, 1H), 7,77 (м , 1H), 7,59 (м, 1H), 7,25 (м, 1H), 6,90 (м, 1H), 6,82 (м, 1H), 4,96 (с, 1H), 4,09 (м, 3H), 3,68 (м, 1H), 3,20 (м, 5H), 1,50 (м, 4H), 1,03 (м, 3H).
Пример 12: 3-({4-гидрокси-1-[3-(тиофен-2-ил)пропаноил]пиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-он
К раствору соли TFA из стадии 3 примера 1 (0,42 ммоль, 1 экв.) в CH2Cl2 (10 мл) последовательно добавляли DIEA (0,37 мл, 2,1 ммоль, 5 экв.), 3-(2-тиофен-2-ил)пропановую кислоту (80 мг, 0,51 ммоль, 1,2 экв.), EDCI (161 мг, 0,84 ммоль, 2 экв.) и HoBt (114 мг, 0,84 ммоль, 2 экв.). Реакционную смесь нагревали при комнатной температуре в течение 4 дней и затем концентрировали в вакууме. Сырой неочищенный продукт очищали с помощью колоночной флеш хроматографии на силикагеле (элюент: циклогексан/этилацетат от 2/8 до 0/10 затем этилацетат/МеОН 9/1). Продукт солюбилизировали в EtOH/H2O и раствор сушили сублимацией, получая соединение Примера 12 (125 мг, 75%) в виде белого безе-подобного вещества.
MS(ES+, m/z): 398,2[M+H]+
1Н ЯМР (DMSO-d6) δ: 8,24 (с, 1H), 8,16 (дд, J=8 Гц, 1H), 7,84 (дд, J=8 Гц, J=8 Гц, 1H), (д, J=8 Гц, 1H), 7,55 (дд, J=8 Гц, J=8 Гц, 1H), 7,27 (м, 1H), 6,90 (м, 3H), 4,98 (с, 1H), 4,09 (м, 1H), 3,99 (м, 2H), 3,64 (м, 1H), 3,23 (м, 1H), 3,00 (м, 2H), 2,92 (м, 1H), 2,66 (м, 2H), 1,45 (м, 4H).
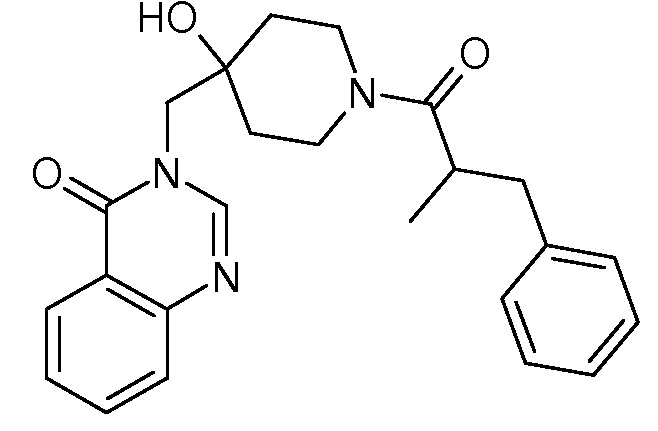
Пример 13: 3-{[1-(2-бензилпропаноил)-4-гидроксипиперидин-4-ил] метил}-3,4-дигидрохиназолин-4-он
К раствору соли TFA из стадии 3 примера 1 (2,37 ммоль, 1 экв.) в CH2Cl2 (30 мл) последовательно добавляли DIEA (1,24 мл, 7,11 ммоль, 7 экв.), 2-метил-3-фенилпропановую кислоту (466 мг, 2,84 ммоль, 1,2 экв.), EDCI (909 мг, 4,74 ммоль, 2 экв.) и HoBt (640 мг, 4,74 ммоль, 2 экв.). Реакционную смесь нагревали при комнатной температуре в течение ночи и затем концентрировали в вакууме. Сырой неочищенный продукт очищали с помощью колоночной флеш хроматографии на силикагеле (элюент: циклогексан/этилацетат от 2/8 до 0/10 затем этилацетат/МеОН 9/1), получая соединение Примера 13 (640 мг, 66% за две стадии) в виде белого твердого вещества.
MS(ES+, m/z): 406,3[M+H]+
1Н ЯМР (DMSO-d6) δ: 8,20 (м, 2H), 7,84 (м, 1H), 7,68 (м, 1H), 7,56 (м, 1H), 7,20 (м, 5H), 4,90 (м, 1H), 3,85 (м, 4H), 3,11 (м, 2H), 2,82 (м, 2H), 2,52 (м, 1H), 1,48 (м, 4H), 1,00 (м, 3H).
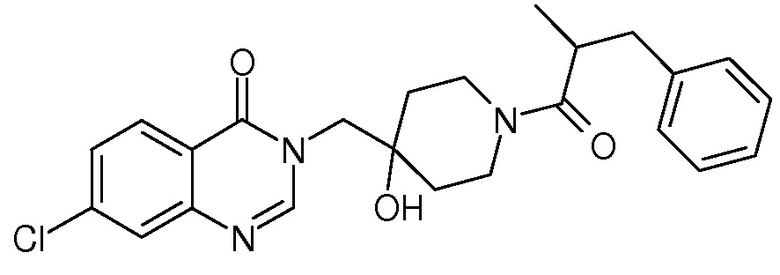
Пример 14: 3-{[1-(2-бензилпропаноил)-4-гидроксипиперидин-4-ил] метил}-7-хлор-3,4-дигидрохиназолин-4-он
К раствору соли TFA из стадии 2 примера 11 (0,34 ммоль, 1 экв.) в CH2Cl2 (10 мл) последовательно добавляли DIEA (0,18 мл, 1,02 ммоль, 3 экв.), 2-метил-3-фенилпропановую кислоту (67 мг, 0,41 ммоль, 1,2 экв.), EDCI (130 мг, 0,68 ммоль, 2 экв.) и HoBt (104 мг, 0,68 ммоль, 2 экв.). Реакционную смесь нагревали при комнатной температуре в течение ночи и затем концентрировали в вакууме. Сырой неочищенный продукт очищали с помощью колоночной флеш хроматографии на силикагеле (элюент: циклогексан/этилацетат от 2/8 до 0/10 затем этилацетат/МеОН 9/1), получая соединение Примера 14 (56 мг, 37% за две стадии) в виде белого твердого вещества.
MS(ES+, m/z): 440,3[M+H]+
1Н ЯМР (DMSO-d6) δ: 8,29 (д, J=8 Гц, 1H), 7,20 (т, J=8 Гц, 1H), 7,79 (шир.с, 1H), 7,64-7,61 (м, 1H), 7,32-7,26 (м, 2H), 7,22-7,16 (м, 3H), 4,94 (д, J=6 Гц, 1H), 4,15-3,79 (м, 3H), 3,68 (т, J=14 Гц, 1H), 3,24-3,09 (м, 2H), 2,89-2,82 (м, 2H), 1,59-1,21 (м, 4H), 1,03 (д, J=6 Гц, 3H), 0,78-0,75 (м, 1H).
Характерные цистиновые протеазы
Получение и очистка USP7 белка
cDNA Кодирующий USP7 получали с помощью PCR амплификации из mRNA плаценты. USP7 cDNA субклонировали PCR в экспрессионный вектор бациловируса (pFastBac-HT; Invitrogen). Человеческая USP7 полной длины дикого типа и ее каталитический мутант (цистеин 223, замененный аланином, С223А) получали в виде N-концевых His-меченых вставок слияния в клетках Spodoptera frugiperda (Sf9, Invitrogen), с использованием Bac-to-Bac Baculovirus системы от Invitrogen согласно инструкциям производителя. pFastBac-HT-B-USP7 использовали для трансформирования DH10bac клеток (Invitrogen), и осуществляли селекцию голубой/белый на X-gal/IPTG агаровых пластинах. Бакмидную ДНК получали с помощью щелочной лизисной процедуры. Целостность наборов бакмид и их ориентацию проверяли с помощью PCR, с использованием характерных и специфических праймеров. Клетки насекомых Sf9 культивировали в среде InsectXpress (Cambrex) при 27°C и трансфицировали соответствующей бакмидой (челночный вектор на основе генома AcMNPV, способный существовать в клетках E.coli и клетках насекомых), используя GeneShuttle 40 (Q-BIOgen). Через 72 часа после инфицирования вирусы выделяли в надосадочную жидкость. Вирусы амплифицировали заражением клеток насекомых (Sf9 или High Five cells; invitrogen) в 50 мл InsectXpress среды в 150 cм2 сосуде для клеточной культуры с 500 мкл надосадочной жидкости от инфицированных Sf9 клеток. После второго цикла амплификации зараженные клетки выделяли с помощью быстрого SDS лизиса, кипятили в течение 5 мин при 100°C, подвергали краткой обработке звуком и центрифугировали в течение 20 мин при 14,000 g. Экспрессионные уровни в зараженных Sf9 клетках сравнивали с таковыми в незараженных клетках. Белкам слияния затем давали возможность связываться с TALON шариками (BD Biosciences, TALON металло афинная смола) в течение 30 мин при 4°C с мягким покачиванием. Шарики обширно промывали (500 мM натрий-фосфатный буфер рН 7,0, 500 мМ NaCl, 10 мM имидазол, 0,5% Triton X-100 и 10% глицерин) и связанные белки элюировали в промывном буфере с добавлением 250 мМ имидазола (Sigma). Элюированные фракции разлагали на 4-12% NuPAGE гелях (Novex, Invitrogen). Фракции, содержащие высокие концентрации очищенных белков (чистота>95%) диализировали (20 мM Tris HCl pH 7,6, 200 мM NaCl, 1 мM DTT, 1 мM EDTA и 10% глицерин) аликвотировали и мгновенно замораживали в жидком азоте перед хранением при -80°C.
Анализ USP7 активности
USP7 разбавляли в USP буфере (50 мM Tris HCl; 0,5 мM EDTA (этилендиаминтетрауксусная кислота); 5 мM DTT; 0,01 % Triton X-100; бычий сывороточный альбумин 0,05 мг.мл-1 pH 7,6). Запасы соединений (10 мM) хранили при -20°C в ДМСО. Соединения испытывали в различных концентрациях: от 200 мкM до 91 нM.
Реакции проводили в виде дубликатов на Black 384 луночных планшетах (микропланшеты малого объема; Greiner; 10 мкл конечный реакционный объем). Концентрация субстрата для USP7 составляла 300 нM Ub-AMC (Chem. Biol., 2003, 10, стр. 837-846) (Boston Biochem). Концентрации фермента (USP7) в анализах на специфичность составляли 100 pM. Чтобы выполнить анализы на специфичность при начальных скоростях при фиксированной концентрации субстрата, определяли концентрации. Соединения предварительно инкубировали с ферментами в течение 30 минут при 25°C. Реакции инициировали добавлением субстрата на планшеты, содержащие ферменты (+/- соединения), разбавленные в аналитическом буфере. Реакционные смеси инкубировали в течение 60 минут при 37°C. Реакции останавливали добавлением уксусной кислоты (100 мM конечная). Считывания выполняли на Pherastar Fluorescent считывателе (BMG). λ Эмиссия 380 нм; λ Возбуждение = 460 нм. Данные (средние величины +/- стандартное отклонение) анализировали как % от контроля (никакого соединения) и наносили на график в виде процента против Log концентрации соединения с использованием GraphPad (Prism). Данные приспосабливали к сигмоидной модели (изменяемый наклон).
Анализ USP5 активности
USP5 разбавляли в USP буфере (50 мM Tris HCl; 0,5 мM EDTA; 5 мM DTT; 0,01% Triton X-100; бычий сывороточный альбумин 0,05 мг.мл-1 pH 7,6). Запасы соединений (100 мM) хранили при -20°C в ДМСО. Соединения испытывали при различных концентрациях: от 200 мкМ до 91 нМ.
Реакции проводили в виде дубликатов на Black 384 луночных планшетах (микропланшеты малого объема; Greiner; 10 мкл конечный реакционный объем). Концентрация субстрата для USP5 составляла 300 нM Ub-AMC (Boston Biochem). Концентрации фермента (USP5) в анализах на специфичность составляли 300 pM. Чтобы выполнить анализы на специфичность при начальных скоростях при фиксированной концентрации субстрата, определяли концентрации. Соединения предварительно инкубировали с ферментами в течение 30 минут при 25°C. Реакции инициировали добавлением субстрата на планшеты, содержащие ферменты (+/- соединения), разбавленные в аналитическом буфере. Реакционные смеси инкубировали в течение 60 минут при 37°C. Реакции останавливали добавлением уксусной кислоты (100 мM конечная). Считывания выполняли на Pherastar Fluorescent считывателе (BMG). λ Эмиссия 380 нм; λ Возбуждение = 460 нм. Данные (средние величины +/- стандартное отклонение) анализировали как % от контроля (никакого соединения) и наносили на график в виде процента против Log концентрации соединения с использованием GraphPad (Prism). Данные приспосабливали к сигмоидной модели (изменяемый наклон).
Клонирование и очистка USP8
cDNA кодирующую USP8 получали PCR амплификацией из плаценты mRNA. USP8 cDNA субклонировали PCR в бакуловирусный экспрессионный вектор (pFastBac-HT; Invitrogen). cDNA Кодирование мутированной USP8 производили мутагенным PCR. Соответствующий белок кодирует цистеин до аланинового замещения в остатке 786. Последовательности устанавливались секвенированием полной открытой рамки считывания. Бакмиды кодирующие USP8 производились с помощью DH10bac транспозиции. Соответствующие бакмиды вносили в клетки насекомых (Sf9). Вирусы выделялись из надосадочной жидкости культуры и амплифицировались дважды. Клетки насекомых (Sf9 или High Five; Invitrogen) заражали в течение 72 часов. Целые клеточные лизаты собирали и лизировали в буфере для лизиса (Tris HCl 50 мM pH 7,6; 0,75% NP40; 500 мM NaCl; 10% глицерин; 1 мM DTT; 10 мM имидазол; ингибитор протеазы Cocktail; AEBSF 20 мкг.мл-1; апротинин 10 мкг.мл-1). Белки афинно очищались на металло афинных смолах (Talon Metal affinity resin; BD Biosciences). Связанные материалы обширно промывали в промывном буфере (50 мМ фосфат натрия pH 7,0; 300 мM NaCl; 10 мM имидазол; 0,5% Triton X-100; 10% глицерин) и элюировали из смолы в 250 мМ содержащем имидазол промывном буфере. Белки подвергали диализу в буфере для диализа (Tris HCl pH 7,6 20 мM; NaCl 200 мM; DTT 1 мM; EDTA 1 мM; 10% глицерин). Очищенные белки анализировали на 4-12% NuPAGE (Invitrogen).
Анализ активности USP8
USP8 разбавляли в USP буфере (50 мM Tris HCl; 0,5 мM EDTA; 5 мM DTT; 0,01% Triton X-100; бычий сывороточный альбумин 0,05 мг.мл-1 pH 8,8). Запасы соединений (100 мM) хранили при -20°C в ДМСО. Соединения испытывали при различных концентрациях: от 200 мкМ до 91 нМ.
Реакции проводили в виде дубликатов на Black 384 луночных планшетах (микропланшеты малого объема; Greiner; 10 мкл конечный реакционный объем). Концентрация субстрата для USP8 составляла 300 нM Ub-AMC (Boston Biochem). Концентрация фермента (USP8) в анализах на специфичность составляла 1,36 nM. Чтобы выполнить анализы на специфичность при начальных скоростях при фиксированной концентрации субстрата, определяли концентрации. Соединения предварительно инкубировали с ферментами в течение 30 минут при 25°C. Реакции инициировали добавлением субстрата на планшеты, содержащие ферменты (+/- соединения), разбавленные в аналитическом буфере. Реакционные смеси инкубировали в течение 60 минут при 37°C. Реакции останавливали добавлением уксусной кислоты (100 мM конечная). Считывания выполняли на Pherastar Fluorescent считывателе (BMG). λ Эмиссия 380 нм; λ Возбуждение = 460 нм. Данные (средние величины +/- стандартное отклонение) анализировали как % от контроля (никакого соединения) и наносили на график в виде процента против Log концентрации соединения с использованием GraphPad (Prism). Данные применяли к сигмоидной модели (изменяемый наклон).
Анализ активности UCH-L1
UCH-L1 разбавляли в USP буфере (50 мM Tris HCl; 0,5 мM EDTA; 5 мM DTT (Дитиотреит); 0,01% Triton X-100; бычий сывороточный альбумин 0,05 мг.мл-1 pH7,6). Запасы соединений (100 мM) хранили при -20°C в ДМСО. Соединения испытывали при различных концентрациях: от 200 мкМ до 91 нМ.
Реакции проводили в виде дубликатов на Black 384 луночных планшетах (микропланшеты малого объема; Greiner; 10 мкл конечный реакционный объем). Концентрация субстрата для UCH-L1 составляла 300 нM Ub-AMC (Boston Biochem). Концентрация фермента (UCH-L1) в анализах на специфичность составляла 2,5 нМ. Чтобы выполнить анализы на специфичность при начальных скоростях при фиксированной концентрации субстрата, определяли концентрации. Соединения предварительно инкубировали с ферментами в течение 30 минут при 25°C. Реакции инициировали добавлением субстрата на планшеты, содержащие ферменты (+/- соединения), разбавленные в аналитическом буфере. Реакционные смеси инкубировали в течение 60 минут при 37°C. Реакции останавливали добавлением уксусной кислоты (100 мM конечная). Считывания выполняли на Pherastar Fluorescent считывателе (BMG). λ Эмиссия 380 нм; λ Возбуждение = 460 нм. Данные (средние величины +/- стандартное отклонение) анализировали как % от контроля (никакого соединения) и наносили на график в виде процента против Log концентрации соединения с использованием GraphPad (Prism). Данные приспосабливали к сигмоидной модели (изменяемый наклон).
Анализ активности UCH-L3
UCH-L3 разбавляли в USP буфере (50 мM Tris HCl; 0,5 мM EDTA; 5 мM DTT; 0,01% Triton X-100; бычий сывороточный альбумин 0,05 мг.мл-1 pH 7,6). Запасы соединений (100 мM) хранили при -20°C в ДМСО. Соединения испытывали при различных концентрациях: от 200 мкМ до 91 нМ.
Реакции проводили в виде дубликатов на Black 384 луночных планшетах (микропланшеты малого объема; Greiner; 10 мкл конечный реакционный объем). Концентрация субстрата для UCH-L3 составляла 300 нM Ub-AMC (Boston Biochem). Концентрация фермента (UCH-L3) в анализах на специфичность составляла 13 рМ. Чтобы выполнить анализы на специфичность при начальных скоростях при фиксированной концентрации субстрата, определяли концентрации. Соединения предварительно инкубировали с ферментами в течение 30 минут при 25°C. Реакции инициировали добавлением субстрата на планшеты, содержащие ферменты (+/- соединения), разбавленные в аналитическом буфере. Реакционные смеси инкубировали в течение 60 минут при 37°C. Реакции останавливали добавлением уксусной кислоты (100 мM конечная). Считывания выполняли на Pherastar Fluorescent считывателе (BMG). λ Эмиссия 380 нм; λ Возбуждение = 460 нм. Данные (средние величины +/- стандартное отклонение) анализировали как % от контроля (никакого соединения) и наносили на график в виде процента против Log концентрации соединения с использованием GraphPad (Prism). Данные приспосабливали к сигмоидной модели (изменяемый наклон).
Анализ активности каспазы 3
Каспазу 3 разбавляли в Каспазном 3 буфере (100 мM Hepes pH 7,5; 10% сахароза; 0,1% CHAPS). Запасы соединений (100 мM) хранили при -20°C в ДМСО. Соединения испытывали при различных концентрациях: от 200 мкМ до 91 нМ.
Реакции проводили в виде дубликатов на Black 384 луночных планшетах (микропланшеты малого объема; Greiner; 10 мкл конечный реакционный объем). Концентрация субстрата для анализа специфичности к каспазе 3 составляла 250 нM (Ac-DEVD-AMC; Promega). Концентрация фермента (каспаза 3) в анализах на специфичность составляла 1,6 нМ. Чтобы выполнить анализы на специфичность при начальных скоростях при фиксированной концентрации субстрата, определяли концентрации. Соединения предварительно инкубировали с ферментами в течение 30 минут при 25°C. Реакции инициировали добавлением субстрата на планшеты, содержащие ферменты (+/- соединения), разбавленные в аналитическом буфере. Реакционные смеси инкубировали в течение 60 минут при 37°C. Реакции останавливали добавлением уксусной кислоты (100 мM конечная). Считывания выполняли на Pherastar флуоресцентном считывателе (BMG). λ Эмиссия 380 нм; λ Возбуждение = 460 нм. Данные (средние величины +/- стандартное отклонение) анализировали как % от контроля (никакого соединения) и наносили на график в виде процента против Log концентрации соединения с использованием GraphPad (Prism). Данные приспосабливали к сигмоидной модели (изменяемый наклон).
Жизнеспособность клеток и способы пролиферации
Анализ жизнеспособности HCT116 клеток и пролиферации
HCT116 раковые клетки получали из ATCC (Американской коллекции типов культур), содержали в Mc Coy’s 5A среде, содержали 10% FBS, 3 мM глютамина и 1% пенициллин/стрептомицина. Клетки инкубировали при 37°C в увлажненной атмосфере, содержащей 5% CO2.
Жизнеспособность анализировали с использованием MTS приема на 96-луночных планшетах для сред (CellTiter 96® Aqueous Non-Radioactive Cell Proliferation Assay, Promega) согласно инструкциям производителя. MTS (3-(4,5-диметил-тиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2H-тетразолий) представляет собой MTT-производный тетразолий, который восстанавливается в метаболически активных клетках в растворимый, клетко-проницаемый формазан. Количество формазана, определенное по его спектральной поглощательной способности при 492 нм, пропорционально числу живых метаболически активных клеток.
На планшету высевали 103 HCT116 клеток. Спустя 24 часа, среду заменяли, и клетки трижды обрабатывали концентрациями каждого соединения от 100 мкМ до 50 нМ. Соединения разбавляли в 100% ДМСО, чью конечную концентрацию на клетках сохраняли при 0,5%.
Клетки инкубировали с соединениями в течение 72 часов, и их жизнеспособность анализировали затем добавлением MTS в течение 2 часов. Абсорбцию при 492 нм измеряли непосредственно с 96-луночных планшет для культуры. Концентрации GI50 (Ингибирование роста 50) для каждого соединения вычисляли с использованием подгонки уклона сигмоидной переменной (Prism 4.0, Graphpad Softwares). Величины представляют среднее от трех независимых экспериментов.
Способы оценки селективности соединений из панели дезубихитинирующих ферментов активных в клеточных лизатах
С-концевое модифицированное винилсульфоновое производное убихитина, UbVS, несомненно полезно для непосредственно визуализации активных DUBs в клетках. Данное средство, которое ковалентно связывается с активным сайтом дезубихитинирующих ферментов, успешно применялось для обнаружения и характеристики новых убихитин/убихитин-подобных протеаз и для профилирования активных дезубихитинирующих ферментов в нормальных, зараженных вирусом и злокачественных или болезнетворных клетках (Borodovsky et al., Chem Biol 2002, 9, 1149-1159, Hemelaar et al., Mol Cell Biol 2004, 24, 84-95, Ovaa et al., Proc Natl Acad Sci U S A 2004 101, 2253-2258).
В данном исследовании использовали пробу HA-Ub-VS (Гемаглютин tag-убихитин-винилсульфон) для непосредственно визуализации активности всех дезубихитинирующих ферментов из природных протеом. Данное средство использовали для оценки активности/селективности наших мелкомолекулярных соединений на USP7 в отношении всех дезубихитинирующих ферментов в физиологических условиях.
HEK293 Клетки собирали и лизировали на льду неденатурирующим буфером, содержащим Tris pH 7,4, 50 мM; NaCl, 150 мM; MgCl2, 5 мM; EDTA, 0,5 мM; DTT, 2 мM; ATP (аденозинтрифосфат), 2 мM; NP40 (нонил-феноксиполиэтоксилэтанол), 0,5% и глицерин, 10%. Образцы инкубировали при 4°C в течение 1 часа и делали прозрачными. Белки затем подвергали количественному определению по методу Bradford (Bio-Rad белковый анализ). 50 мкг белков из природных клеточных лизатов обрабатывали соединениями примеров 1 и 2 (от 12,5 мкМ до 200 мкМ) неспецифическим DUB ингибитором для контроля в течение 2 часов при комнатной температуре. Реакцию мечения убихитина инициировали добавлением HA-Ub-VS (8 мкг/мл) в буфере для мечения (Tris pH 7,6, 50 мM; MgCl2, 5 мM; EDTA, 0,5 мM; DTT, 2 мM; ATP, 2 мM; сахароза, 250 мM) и инкубировали при комнатной температуре в течение 15 мин. Образцы затем нагревали при 100°C в течение 10 минут и подвергали краткой звуковой обработке. Их растворяли с помощью SDS-полиакриламид-гелевого электрофореза (SDS-PAGE), переносили на нитроцеллюлозную мембрану и исследовали с антителами против USP7, HA, UCH-L3, CYLD, USP8, USP5, USP10 и Stat3. В качестве вторичных антител использовали (HRP)-сопряженные с пероксидазой хрена противо-мышиные (Jackson Laboratories, 115-035-003) или HRP-сопряженные противо-кроличьи (Cell Signaling, 7074) антитела. Сигналы детектировали с помощью усиленной хемилюминесценции (ECL; Amersham) согласно инструкциям производителя реагента.
РЕЗУЛЬТАТЫ
1. Использование Ub52 в качестве USP7 и USP8 субстрата для оценки USP модуляторов
Для композиций согласно изобретению in vitro анализы на USP7 и USP8 осуществляли согласно следующей процедуре
Получение слияний убихитин-рибосомного белка
cDNA Кодирующий белок слияния между убихитином и рибосомным белком L40 (ub52 или uba52 или убихитин-L40) амплифицировали из человеческой RNA с использованием частной библиотеки человеческой плаценты. cDNA субклонировали в вектор бактериальной экспрессии (pGEX-2T, GE Healthcare), включая дополнительный сигнальный ярлычок на карбоксильном конце кодированного белка. Использовали следующие праймеры для субклонирования в рамке с GST ярлычком убихитин-L40 в pGEX-2T: 5’-cgtggatccatgcagatctttgtgaagaccctc-3’ (SEQ ID NO:1) и 5’-gcgaattctttatcgtcatcgtctttgtagtctttgaccttcttcttgggacg-3’ (SEQ ID NO:2) в BamHI & EcoRI сайты рестрикции.
Для получения и очистки рекомбинантных белков плазмиду pGEX-2T-Ub52-flag трансформировали в E. coli BL21 (Stratagene), выращенный в LB среде с добавлением 100 мг/мл ампицилина (LB ampi) при 37°C на протяжении ночи, а затем разбавляли 1/100 в LB ampi. Клетки инкубировали при 37°C до тех пор, пока не достигали A600=0,6-0,8. После индуцирования 0,1 мM изопропил-β-D-тиогалактопиранозидом (IPTG), культуру инкубировали при 30°C в течение 180 мин.
Клетки собирали центрифугированием в течение 15 мин при 7000x g при 4°C. Бактериальные пеллеты лизировали в NETN (Tris HCl pH 8,0; EDTA 1 мM; NP40 0,5%; коктейль ингибитора протеазы, PMSF 1 м) и подвергали краткой звуковой обработке. Нерастворимый материал удаляли центрифугированием в течение 30 минут при 14000x g. GST-Ub52-flag белки очищали согласно Everett RD et al., EMBO J. (1997) 16, 1519-1530. Вкратце, растворимую фракцию инкубировали на глютатионовых шариках, предварительно уравновешенных в NETN буфере +0,5% молоко в течение 120 мин при 4°C. Фильтрат восстанавливали. Шарики экстенсивно или обширно промывали: последнюю промывку проводили в Tris HCl pH 7,6 20 мM; NaCl 100 мM; MgCl2 12 мM. Элюирования проводили с использованием 20 мM восстановленного глютатиона в 50 мM Tris HCl pH 8,0, NaCl 120 мM. Все фракции растворяли на 4-12% NuPAGE с последующей 0,1 M DTT обработкой и денатурацией и окрашивали Кумасси Бриллиантовым Голубым. Элюаты диализировали на протяжении ночи при 4°C в Tris HCl pH 7,6 20 мM; NaCl 50 мM; DTT 0,5 мM.
Анализ белка слияния (GST-Ub52-Flag) с использованием метода измерения гомогенной флуоресценции с временным разрешением (HTRF®)
Использование GST-Ub52-Flag основано на измерении временного разрешения флуоресценции, испускаемой при переносе в гомогенной среде.
Используемыми реагентами были следующие:
- Конъюгат Анти-flag антитело-криптат европия, называемый Анти-Flag-K (CIS bio international), раствор при 0,2 мкM в 0,8 M KF, 0,1 % бычий сывороточный альбумин, Tris HCl 25 мM pH 7,6.
- Конъюгат Анти-GST антитело-XL665 (CIS bio international), раствор при 2,6 мкM в 0,8 M KF, 0,1 % бычий сывороточный альбумин, Tris HCl 25 мM pH 7,6.
- GST-Ub52-Flag раствор при 14,75 мкM & MBP_Ub52 при 37,7 мкM, полученный из исходного раствора, описанного выше, в 50 мМ Tris HCl pH 7,6, EDTA 0,5 мM, бычий сывороточный альбумин 0,05%, DTT 5 мM.
Анализ осуществляют на многолуночных аналитических планшетах. Планшеты анализируют на PHERAstar флуориметре (BMG) после инкубирования на протяжении ночи при 4°C (возбуждение 337 нм, эмиссия 620 и 665 нм).
Анализ активности ферментов дезубихитинирующего типа с убихитин-рибосомным белком слияния
Используемыми реагентами были следующие:
- Раствор USP7 при 200 pM и USP8 при 400 pM в 50 мM Tris HCl pH 7,6, бычий сывороточный альбумин 0,05%, DTT 5 мM.
- Анти-Flag-K (CIS bio international), раствор при 0,2 мкM в 0,8 M KF, 0,1% бычий сывороточный альбумин, Tris HCl 25 мM pH 7,6.
- Конъюгат анти-GST антитело-XL665 (CIS bio international), раствор 2,6 мкM в 0,8 M KF, 0,1% бычий сывороточный альбумин, Tris HCl 25 мM pH 7,6.
- GST-Ub52-flag раствор при 14,75 мкM и MBP_Ub52 при 37,7 мкM получают 50 мM Tris HCl pH 7,6, EDTA 0,5 мM, бычий сывороточный альбумин 0,05%, DTT 5 мM.
Ферментную реакцию осуществляют смешением GST-Ub52-flag раствора с 5 мкл USP7 раствора (200 pM конечная) или 5 мкл USP8 (400 pM конечная). Данную смесь инкубируют в течение одного часа при комнатной температуре на много-луночной планшете для анализа. В каждую лунку много-луночной планшеты для анализа добавляют 10 мкл смеси 5 мкл анти-Flag-K раствора (0,2 мкМ) плюс 5 мкл анти-GST-XL665 антител (2,6 мкM). После инкубации на протяжении ночи при 4°C производят считывание с планшеты на PHERAstar флуориметре (BMG).
Уменьшение сигнала находится в определенном соотношении с увеличением активности фермента, т.е. расщеплением GST-Ub52-Flag субстрата. Используемый формат является следовательно целиком подходящим для метода анализа на ферменте дезубихитинирующего типа, такого как убихитин специфическая протеаза, а также для определения модулятора активности данного фермента.
Определение модулятора активности фермента дезубихитинирующего типа
Осуществляют те же процедуры, упомянутые выше для анализа активности ферментов дезубихитинирующего типа, но инкубируются различные реакционные смеси с идентичной концентрацией фермента, в присутствии или отсутствие соединений 1-14. Данные (средние величины +/- стандартное отклонение) анализировали как % от контроля (никакого соединения) и наносили на график в виде процента против Log концентрации соединения с использованием GraphPad (Prism). Данные приспосабливали к сигмоидной модели (изменяемый наклон) и определяли IC50 (мкM) и они представлены в следующей таблице:
2. Селективное ингибирование USP7 дезубихитинирующей активности с использованием UbAMC субстрата
Результаты суммированы в следующей таблице (мкМ):
3. Ингибирование жизнеспособности/пролиферации клеток
Результаты суммированы в следующей таблице (мкМ):
4. Селективное ингибирование USP7 дезубихитинирующей активности относительно активных DUBs в физиологических условиях:
Чтобы подтвердить специфичность рекомбинантных ферментов, наблюдаемых in vitro мы провели конкурентные анализы с использованием основанной на активности HAUbVS пробы (ABP), которая ковалентно связывается активным сайтом цистеина дезубихитинирующих ферментов (Borodovsky et al., Chem Biol 2002, 9, 1149-1159, Hemelaar et al., Mol Cell Biol 2004, 24, 84-95, Ovaa et al., Proc Natl Acad Sci U S A 2004 101, 2253-2258). В сравнении с рекомбинантными ферментами данный анализ имеет преимущество в оценке действия ингибитора против многочисленных дезубихитинирующих ферментов параллельно непосредственно в природных протеомах в блот анализе Western с использованием анти-НА антител. В данном анализе ингибитор добавляют к целому клеточному лизату и ингибирование определяют мечением остаточно активных дезубихитинирующих ферментов с помощью HAUbVS. Данное мечение, сопровождаемое иммуноблотированием анти-HA антителами позволяло идентифицировать все активные дезубихитинирующие ферменты из HEK293 клеточных лизатов (Фигура 1, полоса 2). Данное мечение специфичное к активной форме DUBs, ингибируется неспецифичным DUB ингибитором неселективным образом (Фигура 1, полоса 14). Когда лизированные HEK293 клетки обрабатывали HAUbVS в отсутствие и присутствии различных доз соединений примеров 2 и 5, не замечалось никакого изменения в иммуноблотированном образце за исключением одной полосы, которая почти полностью элиминировалась при размере, соответствующем HA-Ub-VS-USP7 (Фигура 1). Данное действие на USP7 активность подтверждалось анти-USP7 антителами, о чем указывает изменчивость наблюдаемого сдвига между обработанными и необработанными образцами (Фигура 2).
Далее мы оценивали действие USP7 ингибиторов на эффективность HAUbVS мечения проверкой отдельно нескольких дезубихитинирующих ферментов. Для этого сначала проверяли эффективность HAUbVS мечения на USP7, USP8, USP5, USP10, CYLD и UCH-L3 из HEK293 клеточных лизатов. Данное мечение подтверждалось всеми испытанными DUBs, о чем указывает изменчивость наблюдаемого сдвига между HAUbVS-обработанными и необработанными образцами, хотя с различными степенями эффективности (Фигура 2, полосы 1 и 2). Потенциальное ингибирование данных DUBs оценивали в присутствии соединений примеров 2 и 5, также как и неспецифического DUB ингибитора в качестве контроля. Как и ожидалось, мы обнаружили, что неспецифический DUB ингибитор ингибировал все испытанные DUBs (Фигура 2, полоса 14). Интересно, мы нашли, что соединения примеров 2 и 5 эффективно блокировали мечение USP7, но не USP8, USP5, USP10, CYLD или UCH-L3, показывая его USP7 специфичность по сравнению с активными DUBs в физиологических условиях (Фигура 2). Эти данные, взятые вместе, указывают на то, что соединения примеров 2 и 5 попадают непосредственно в USP7 в природных протеомах в физиологических условиях и зависимым от дозы образом без какой-либо перекрестной реакции со всеми испытанными дезубихитинирующими ферментами.
5. USP7-специфические соединения являются обратимыми ингибиторами
Для того, чтобы лучше понять механизм ингибирования USP7 нашими USP7-специфическими соединениями, были проведены несколько экспериментов. Сначала мы охарактеризовали ингибиторный механизм соединения примера 2 предварительным инкубированием соединения примера 2 (100 мкМ) с USP7 (100 pM) в различные периоды времени (30 мин, 1 час, 2 часа, 4 часа, 6 часов). Затем оценивали дезубихитинирующую активность с Ub-AMC субстратом, как описано выше, и сравнивали с активностью образцов, обработанных ДМСО. Интересно, было найдено, что USP7 ингибирование соединением примера 2 (~75%) было зависимым от времени предварительного инкубирования (Фигура 3). Эти данные ясно показывают, что соединение примера 2 является зависимым от времени ингибитором USP7.
Затем определяли обратимость ингибирования с помощью измерения возвращения ферментной активности после гель-фильтрации. Мы инкубировали USP7 (100 pM) в течение 4 часов при комнатной температуре в присутствии соединений примеров 2 и 3 (100 мкМ) или контрольного соединения, используемого в качестве обратимого USP7 ингибитора (25 мкМ). Мы пропускали некоторое количество каждого образца через G50 гель-фильтрационную систему (Sephadex, G50 высшего сорта, Sigma Aldricht). Дезубихитинирующую активность G50 оценивали с Ub-AMC субстратом, как описано выше, и сравнивали с таковой образцов, неподверженных G50 фильтрации. Реакции контролировали с использованием PHERAstar (BMG Labtech), в отсутствие фильтрации, USP7 активность ингибировали соединениями примеров 2 и 3 и контрольным ингибитором, используемым в качестве необратимого соединения (Фигура 4A, -G50). Используя образцы, содержащие одни данные соединения, мы подтвердили, что все соединения, профильтрованные через G50 колонку, целиком оставались на колонке (Фигура 4В). Фильтрация образцов, содержащих USP7 и соединения примеров 2 и 3, через G50 колонку вела к полному восстановлению USP7 активности, тогда как USP7 ингибирование необратимым контрольным соединением не могла возвратиться фильтрацией (Фигура 4A, +G50). Поскольку ожидается, что обратимый комплекс остается ингибированным после промывки, наши открытия демонстрируют, что соединения примеров 2 и 3 являются обратимыми USP7 ингибиторами.
Чтобы подтвердить данные открытия, мы измерили возврат ферментной активности после быстрого и обширного разбавления комплексом фермент-соединение. Мы инкубировали USP7 при 100-кратной концентрации (10 нМ) по сравнению с концентрацией, требуемой для анализа активности с концентрацией ингибитора эквивалентной 10-кратной IC50 (Фигура 5A). После разумного времени уравновешивания (1 час) смесь разбавляют 100-кратно в реакционном буфере, содержащем UbAMC (300 нM) для инициирования реакции (Фигура 5А). Разбавление образцов, содержащих USP7 и соединения примеров 2 и 3, вело к почти полному возврату USP7 активности, тогда как USP7 ингибирование необратимым контрольным соединением не могло возвращаться разбавлением (Фигура 5В). Диссоциация соединений примеров 2 и 3 из USP7, ведущая к восстановлению более, чем приблизительно 90% ферментной активности после быстрого и обширного разбавления ясно демонстрирует, что соединения примеров 2 и 3 являются быстрыми обратимыми ингибиторами.
Чтобы охарактеризовать комплексы, включающие в себя USP7 и соединения примеров 1, 2, 3 и 5, мы оценивали непосредственное связывание с помощью ESI-MS в естественных условиях (7 мМ ацетат аммония рН 7,5), как описано ранее (Vivat Hannah et al., Future Med Chem. 2010 2(1):35-50). Сначала мы проверяли взаимодействие инкубацией белка в присутствии 3-5-10 молярных эквивалентов соединений примеров 1, 2, 3 и 5. Все данные соединения образовывали комплексы с USP7 в различных степенях и показали 1:1 стехиометрию связывания с USP7, когда испытывались при 3 или 5 молярном эквивалентном избытке, как иллюстрируется соединениями примеров 1 и 2 на Фигуре 6. Никакого различия массы соединений не наблюдалось в присутствии или отсутствие USP7. Для того, чтобы диссоциировать слабые не-ковалентные комплексы, комплексы USP7/ингибитор разбавляли в 50/50/1 H2O/CH3CN/HCOOH и анализировали в энергетических инструментальных условиях (Vc=120 V, Pi=4 мбар). В данных условиях не наблюдалось никакого связывания между USP7 и соединениями 1, 2, 3 и 5, что предполагает, что USP7 и соединения примеров 1, 2, 3 и 5 образуют не-ковалентные комплексы.
Все эти данные ясно указывают на то, что данные USP7-специфические ингибиторы связывают USP7 обратимым образом.
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИНОТИОЛЭФИРНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2833575C2 |
| НОВЫЕ ГИДРОКСИСЛОЖНОЭФИРНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2734418C2 |
| СПОСОБ СИНТЕЗА НУКЛЕОЗИДНЫХ АНАЛОГОВ | 2000 |
|
RU2200738C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОИЗОХИНОЛИНА, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ СИНАПТИЧЕСКОГО ЗАХВАТА ДОПАМИНА И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2293728C2 |
| СОЕДИНЕНИЯ 2,3-ДИГИДРОХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ NaV1.8 | 2020 |
|
RU2833870C2 |
| НОВЫЕ ТЕТРАЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ЦИСТЕИНОВЫХ ПРОТЕАЗ, ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ОБЛАСТИ ИХ ТЕРАПЕВТИЧЕСКОГО ПРИМЕНЕНИЯ | 2007 |
|
RU2481349C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ | 2018 |
|
RU2837261C2 |
| 6-, 7- ИЛИ 8-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ХИНАЗОЛИНОНА И КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ ИХ, И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2008 |
|
RU2476432C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ | 2010 |
|
RU2550352C2 |
| НОВЫЕ ФОСФАТНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2617682C2 |

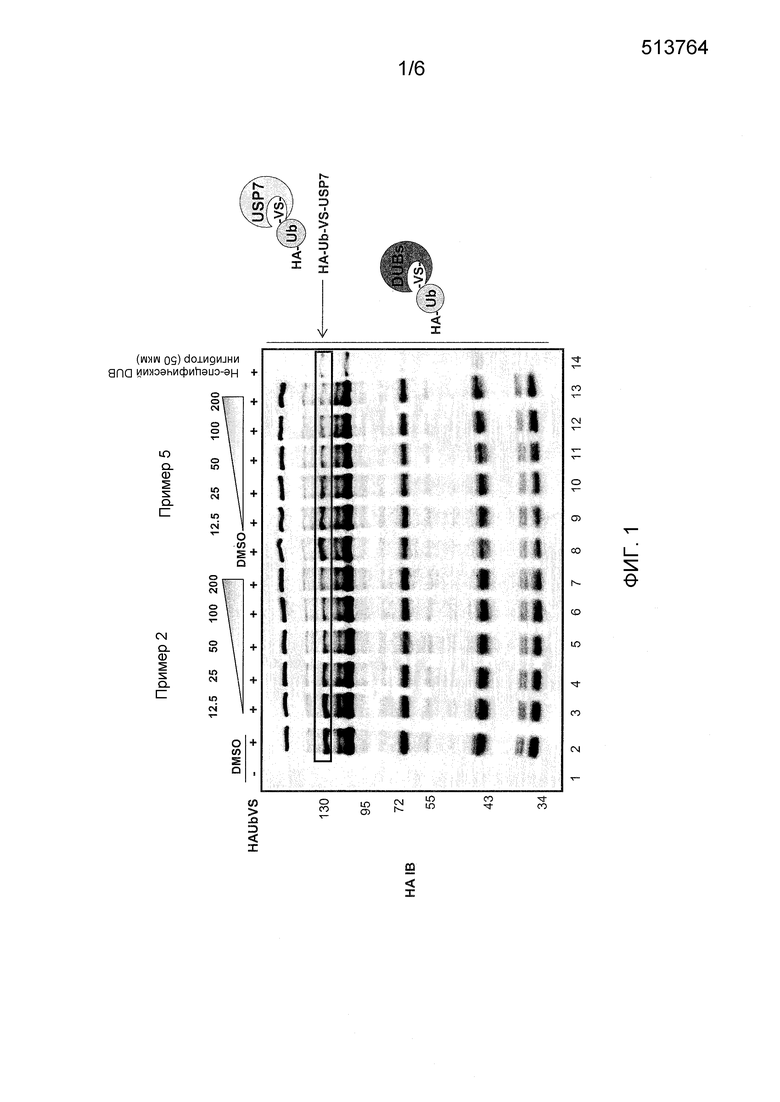
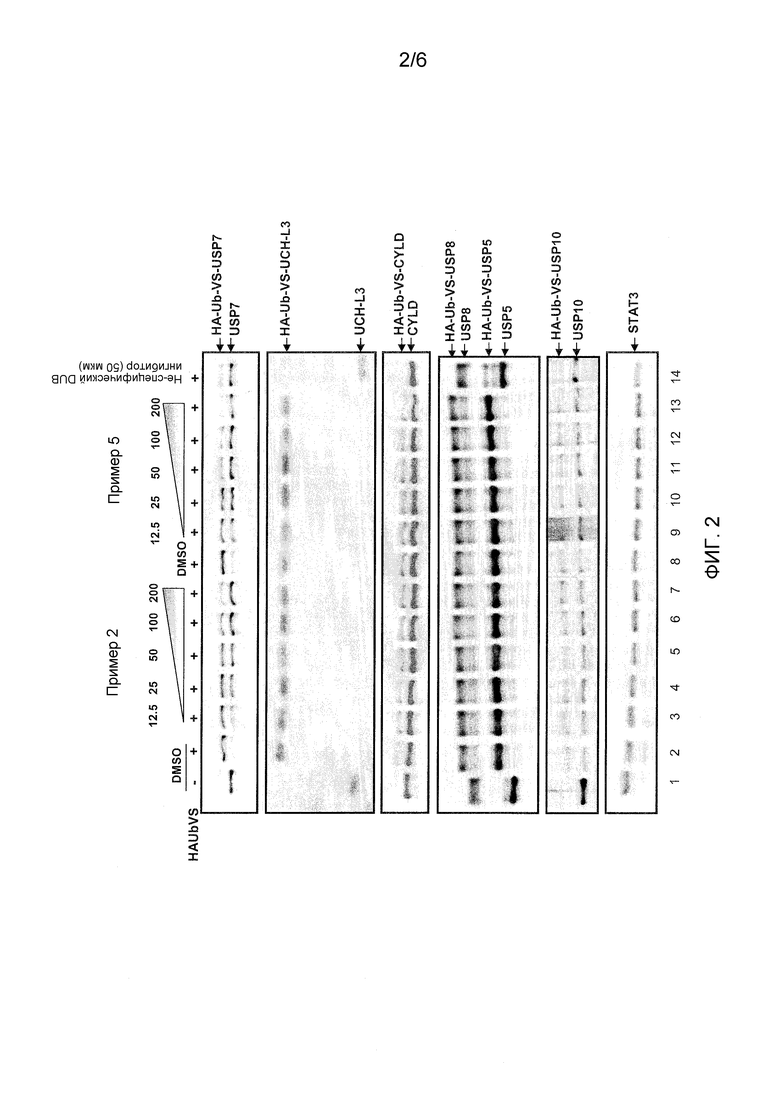
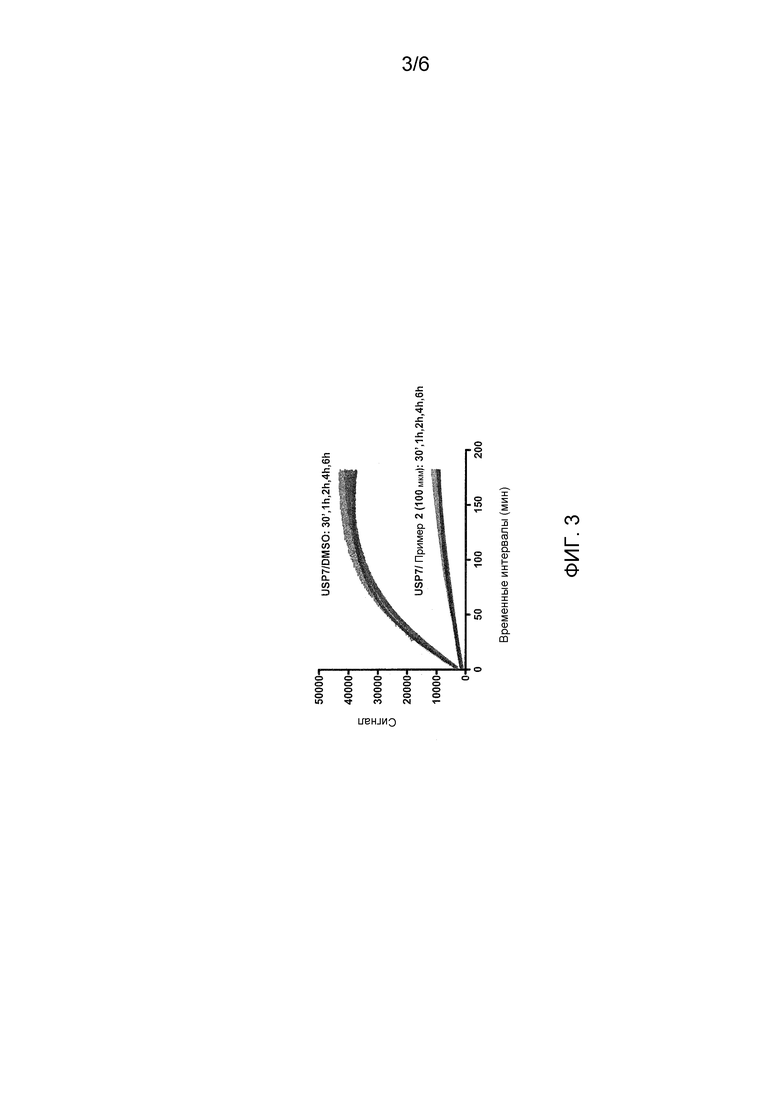
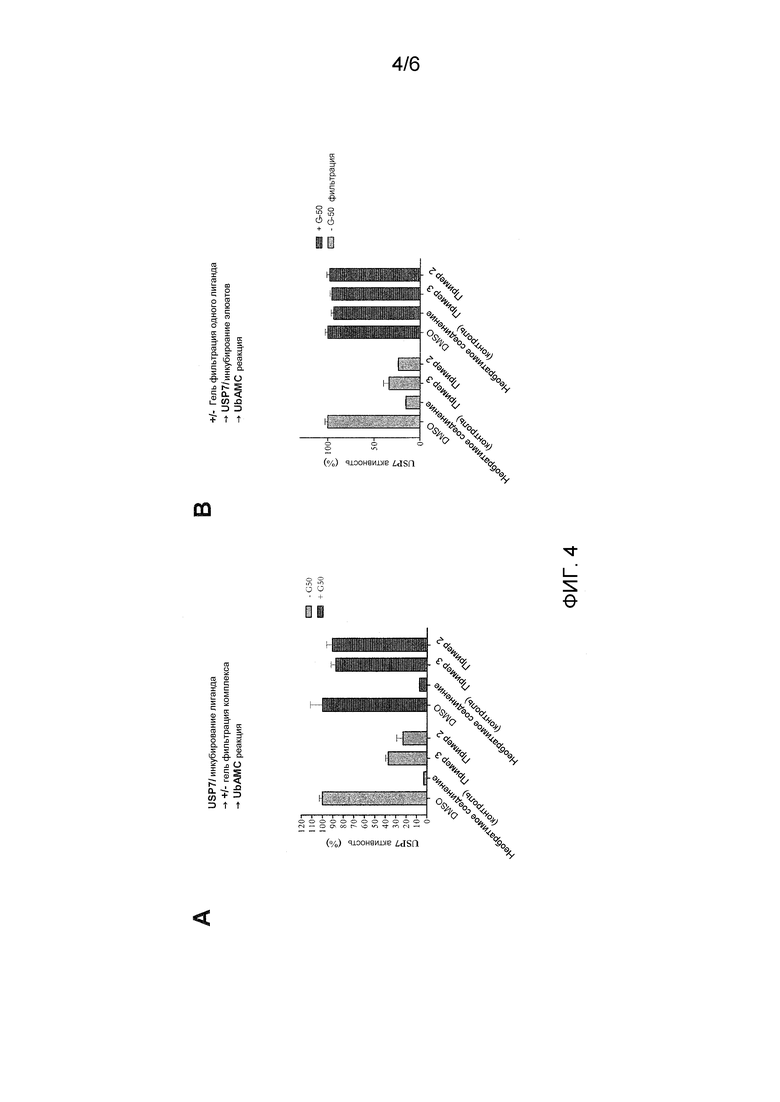

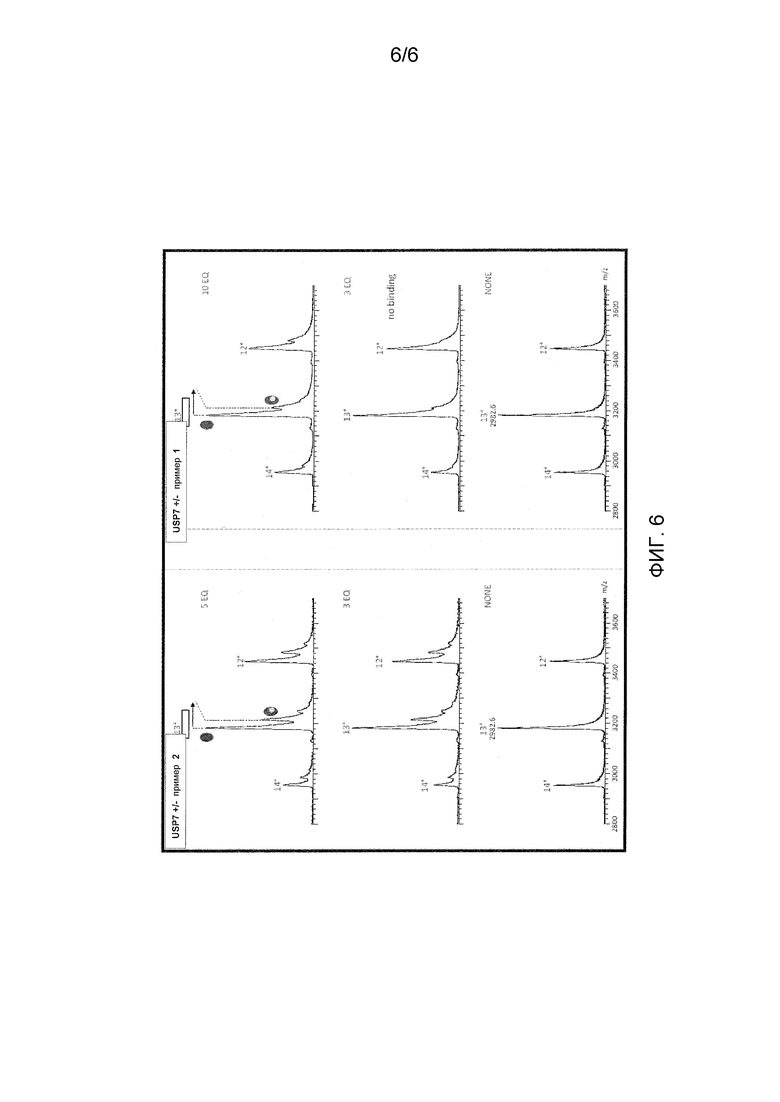
Изобретение относится к хиназолин-4-оновым соединениям формулы (I’) или их фармацевтически приемлемым солям, обладающим свойствами ингибирования дезубихитинирующего фермента. Соединения могут найти применение для получения препарата для лечения и/или профилактики рака и метастаз, вирусных инфекций и болезней, и/или вирусной инфекционности, и/или латентности, опосредованных активностью убихитин-специфических протеаз (USP). Вирусные инфекции и болезни выбраны из вирусных инфекций Herpes simplex-1 или -2, гепатита А, гепатита С, SARS коронавирусной инфекции и болезни, вируса Epstein-Barr, риновирусных инфекций и заболеваний, аденовирусных инфекций и заболеваний, полиомиелита. В формуле (I’)
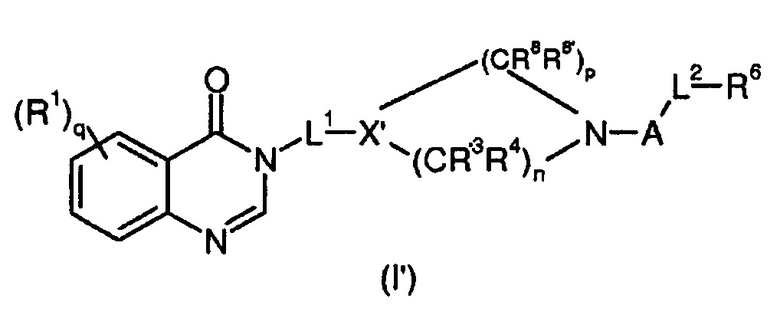
R1, каждый идентичный или различный, выбран из группы, состоящей из галогена, линейного или разветвленного (C1-С6)алкила, OR; L1 представляет собой линейный или разветвленный (C1-С6)алкилен; q представляет собой 0, 1, 2, 3 или 4; X' представляет собой CR7; R7 представляет собой OR; n представляет собой 0, 1 или 2; p представляет собой 1, 2 или 3; R3, R4, R8' и R8 каждый представляет собой Н; А представляет собой -С(О)-; L2 представляет собой линейный или разветвленный (C1-С6)алкилен, необязательно прерываемый по меньшей мере одним О; R6 выбран из группы, состоящей из арила, гетероарила, циклоалкила, Н, где арил, гетероарил, циклоалкил является моно- или полициклическим и необязательно замещен галогеном, OR; каждый R, идентичный или различный, независимо выбран из Н, линейного или разветвленного (C1-С6)алкила. При этом "арил" означает ароматическую моноциклическую углеводородную кольцевую систему из 6 атомов углерода, «циклоалкил» означает неароматическое, моноциклическое углеводородное кольцо из 3-10 атомов углерода; «гетероарил» означает 5-членное ароматическое моно-гетероциклическое кольцо. Изобретение также относится к вариантам способа получения соединений. 6 н. и 13 з.п. ф-лы, 6 ил., 4 табл., 14 пр.
1. Соединение формулы (I')
в которой
R1, каждый идентичный или различный, выбран из группы, состоящей из галогена, линейного или разветвленного (C1-С6)алкила, OR;
L1 представляет собой линейный или разветвленный (C1-С6)алкилен;
q представляет собой 0, 1, 2, 3 или 4;
X' представляет собой CR7;
R7 представляет собой OR;
n представляет собой 0, 1 или 2;
p представляет собой 1, 2 или 3;
R3, R4, R8' и R8 каждый представляет собой Н;
А представляет собой -С(О)-;
L2 представляет собой линейный или разветвленный (C1-С6)алкилен, необязательно прерываемый по меньшей мере одним О;
R6 выбран из группы, состоящей из арила, гетероарила, циклоалкила, Н, где арил, гетероарил, циклоалкил является моно- или полициклическим и необязательно замещен галогеном, OR;
каждый R, идентичный или различный, независимо выбран из Н, линейного или разветвленного (C1-С6)алкила;
где "арил" означает ароматическую моноциклическую углеводородную кольцевую систему из 6 атомов углерода,
«циклоалкил» означает неароматическое, моноциклическое углеводородное кольцо из 3-10 атомов углерода;
«гетероарил» означает 5-членное ароматическое моно-гетероциклическое кольцо;
или его фармацевтически приемлемые соли,
за исключением
- q равен 0, L1 представляет собой СН2, X' представляет собой CR7 и R7 представляет собой ОН, А представляет собой С=O, L2 представляет собой С2Н4 и R6 представляет собой
- q равен 1, R1 представляет собой С1 в положении 7, L1 представляет собой СН2, X' представляет собой CR7 и R7 представляет собой ОН, А представляет собой С=O, L2R6 представляет собой СН(СН2СН3)2.
2. Соединение по п. 1, в котором R7 представляет собой ОН.
3. Соединение по п. 1, в котором R1 представляет собой ОН.
4. Соединение по п. 1, в котором L2 представляет собой линейный или разветвленный C1-С6(алкилен).
5. Соединение по п. 1, в котором R6 выбран из группы, состоящей из арила, гетероарила, циклоалкила или Н, где арил, гетероарил или циклоалкил необязательно замещен линейным или разветвленным O-(C1-С6) алкилом.
6. Соединение по п. 1, в котором p + n = 4.
7. Соединение по п. 1, в котором p равен 2 и n равен 2.
8. Соединение по п. 1, в котором L1 представляет собой СН2.
9. Соединение по п. 1, выбранное из группы, состоящей из
3-({4-гидрокси-1-[3-(2-метоксифенил)пропаноил]пиперидин-4-ил}метил)-3,4-дигидроксихиназолин-4-она,
3-({1-[2-(3-фторфенокси)ацетил]-4-гидроксипиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-она,
3-{[4-гидрокси-1-(2-метилпропаноил)пиперидин-4-ил]метил}-6,7-диметокси-3,4-дигидрохиназолин-4-она,
3-{[4-гидрокси-1-(2-метилпропаноил)пиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она,
4-гидрокси-1-([2-метил-3-(тиофен-2-ил)пропаноил]пиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-она,
7-хлор-3-{[1-(3-циклопентилпропаноил)-4-гидроксипиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она,
3-{[1-(3-циклопентилпропаноил)-4-гидроксипиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она,
7-хлор-3-{[4-гидрокси-1-(3-фенилпропаноил)пиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она,
3-{[4-гидрокси-1-(3-фенилпропаноил)пиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она,
7-хлор-3-({4-гидрокси-1-[2-метил-3-(тиофен-2-ил)пропаноил]пиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-она,
3-({4-гидрокси-1-[3-(тиофен-2-ил)пропаноил]пиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-она,
3-{[1-(2-бензилпропаноил)-4-гидроксипиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она,
3-{[1-(2-бензилпропаноил)-4-гидроксипиперидин-4-ил]метил}-7-хлор-3,4-дигидрохиназолин-4-она
или их фармацевтически приемлемых солей.
10. Способ получения соединения, заявленного в пп. 1-9, включающий реакцию соединения формулы (IIa) с соединением формулы (III)

в которой R1, R8, R3, R4, R5, R6, L1, L2, X', A, q, p, n имеют значения, определенные в любом из пп. 1-8, Y представляет собой уходящую группу.
11. Способ получения соединения, заявленного в пп. 1-9, включающий реакцию соединения формулы (IV) с соединением формулы (Va)
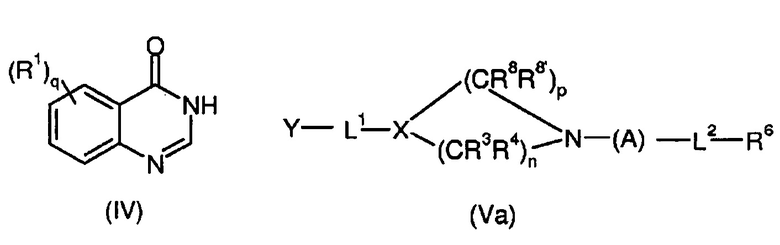
в которой R1, R8, R3, R4, R5, R6, L1, L2, X, q, p, n имеют значения, определенные в любом из пп. 1-8, и Y представляет собой уходящую группу, выбранную из эпокси, галогена, активированного ОН.
12. Фармацевтическая композиция, обладающая свойствами ингибирования дезубихитинирующего фермента, содержащая терапевтически эффективное количество соединения формулы (I')
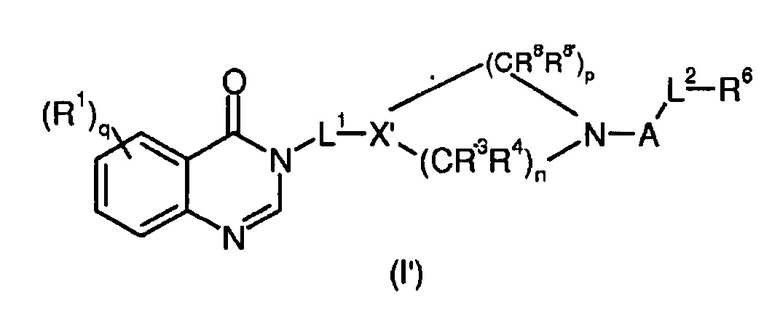
где
R1, каждый идентичный или различный, выбран из группы, состоящей из галогена, линейного или разветвленного (C1-С6)алкила, OR;
L1 представляет собой линейный или разветвленный (C1-С6)алкилен;
q представляет собой 0, 1, 2, 3 или 4;
X' представляет собой CR7;
R7 представляет собой OR;
n представляет собой 0, 1 или 2;
p представляет собой 1, 2 или 3;
R3, R4, R8' и R8 каждый представляет собой Н;
А представляет собой -С(O)-;
L2 представляет собой линейный или разветвленный (C1-С6)алкилен, необязательно прерываемый по крайней мере одним O;
R6 выбран из группы, состоящей из арила, гетероарила, циклоалкила, Н, где арил, гетероарил, циклоалкил является моно- или полициклическим и необязательно замещенным галогеном, OR;
каждый R, идентичный или различный, независимо выбран из Н, линейного или разветвленного (C1-С6)алкила;
где "арил" означает ароматическую моноциклическую углеводородную кольцевую систему из 6 атомов углерода,
«циклоалкил» означает неароматическое, моноциклическое углеводородное кольцо из 3-10 атомов углерода;
«гетероарил» означает 5-членное ароматическое гетеро-моноциклическое кольцо;
или его фармацевтически приемлемые соли с фармацевтически приемлемым наполнителем.
13. Фармацевтическая композиция по п. 12, в которой L2 представляет собой линейный или разветвленный (C1-С6)алкилен.
14. Фармацевтическая композиция по п. 13, в которой в соединении R1, R8, R3, R4, R5, R6, L1, X, q, n имеют значения, определенные в п. 12.
15. Фармацевтическая композиция по п. 12, в которой соединение формулы (I') выбрано из группы, состоящей из
3-({4-гидрокси-1-[3-(2-метоксифенил)пропаноил]пиперидин-4-ил}метил)-3,4-дигидроксихиназолин-4-она,
7-хлор-3-{[1-(2-этилбутаноил)-4-гидроксипиперидин-4-ил]метил}-3,4-дигидроксихиназолин-4-она,
3-({1-[2-(3-фторфенокси)ацетил]-4-гидроксипиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-она,
3-{[4-гидрокси-1-(2-метилпропаноил)пиперидин-4-ил]метил}-6,7-диметокси-3,4-дигидрохиназолин-4-она,
3-{[4-гидрокси-1-(2-метилпропаноил)пиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она,
4-гидрокси-1-([2-метил-3-(тиофен-2-ил)пропаноил]пиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-она,
7-хлор-3-{[1-(3-циклопентилпропаноил)-4-гидроксипиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она,
3-{[1-(3-циклопентилпропаноил)-4-гидроксипиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она,
7-хлор-3-{[4-гидрокси-1-(3-фенилпропаноил)пиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она,
3-{[4-гидрокси-1-(3-фенилпропаноил)пиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она,
7-хлор-3-({4-гидрокси-1-[2-метил-3-(тиофен-2-ил)пропаноил]-пиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-она,
3-({4-гидрокси-1-[3-(тиофен-2-ил)пропаноил]пиперидин-4-ил}метил)-3,4-дигидрохиназолин-4-она,
3-{[1-(2-бензилпропаноил)-4-гидроксипиперидин-4-ил]метил}-3,4-дигидрохиназолин-4-она,
3-{[1-(2-бензилпропаноил)-4-гидроксипиперидин-4-ил]метил}-7-хлор-3,4-дигидрохиназолин-4-она
или его фармацевтически приемлемых солей.
16. Применение соединения формулы (I')
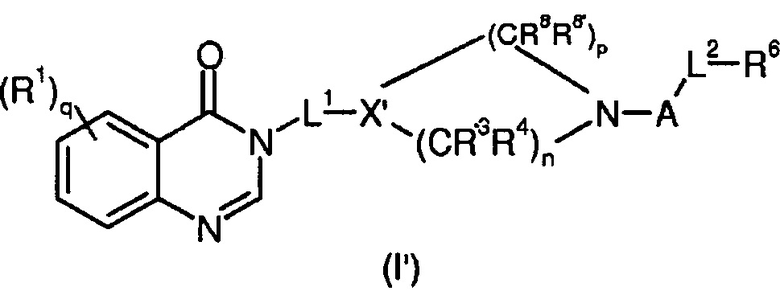
где
R1, каждый идентичный или различный, выбран из группы, состоящей из галогена, линейного или разветвленного (C1-С6)алкила, OR;
L1 представляет собой линейный или разветвленный (C1-С6)алкилен;
q представляет собой 0, 1, 2, 3 или 4;
X' представляет собой CR7;
R7 представляет собой OR;
n представляет собой 0, 1 или 2;
p представляет собой 1, 2 или 3;
R3, R4, R8' и R8 каждый представляет собой Н;
А представляет собой -C(O)-;
L2 представляет собой линейный или разветвленный (C1-С6)алкилен, необязательно прерываемый по крайней мере одним О;
R6 выбран из группы, состоящей из арила, гетероарила, циклоалкила, Н, где арил, гетероарил, циклоалкил является моно- или полициклическим и необязательно замещенным галогеном, OR;
каждый R, идентичный или различный, независимо выбран из Н, линейного или разветвленного (C1-С6)алкила; где "арил" означает ароматическую моноциклическую углеводородную кольцевую систему из 6 атомов углерода,
«циклоалкил» означает неароматическое, моноциклическое углеводородное кольцо из 3-10 атомов углерода;
«гетероарил» означает 5-членное ароматическое гетеро-моноциклическое кольцо;
или его фармацевтически приемлемые соли для ингибирования дезубихитинирующего фермента.
17. Применение по п. 16 для ингибирования USP7 (Убихитин Специфической Протеазы 7) HAUSP (Ассоциированная с Герпесом Убихитин Специфическая Протеаза).
18. Применение соединения формулы (I')
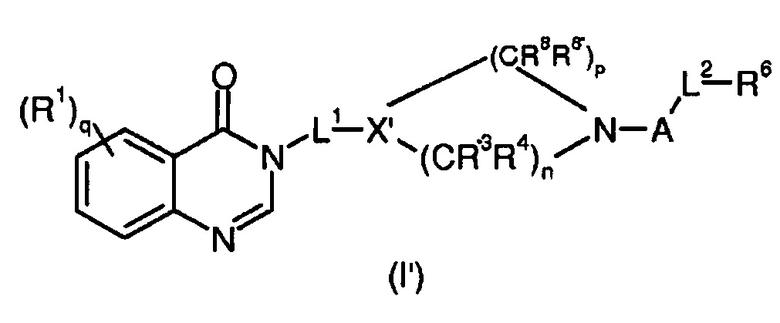
где
R1, каждый идентичный или различный, выбран из группы, состоящей из галогена, линейного или разветвленного (C1-С6)алкила, OR;
L1 представляет собой линейный или разветвленный (C1-С6)алкилен;
q представляет собой 0, 1, 2, 3 или 4;
X' представляет собой CR7;
R7 представляет собой OR;
n представляет собой 0, 1 или 2;
p представляет собой 1, 2 или 3;
R3, R4, R8' и R8 каждый представляет собой Н;
А представляет собой -С(О)-;
L2 представляет собой линейный или разветвленный (C1-С6)алкилен, необязательно прерываемый по крайней мере одним О;
R6 выбран из группы, состоящей из арила, гетероарила, циклоалкила, Н, где арил, гетероарил, циклоалкил является моно- или полициклическим и необязательно замещенным галогеном, OR;
каждый R, идентичный или различный, независимо выбран из Н, линейного или разветвленного (C1-С6)алкила; где "арил" означает ароматическую моноциклическую углеводородную кольцевую систему из 6 атомов углерода,
«циклоалкил» означает неароматическое, моноциклическое углеводородное кольцо из 3-10 атомов углерода;
«гетероарил» означает 5-членное ароматическое, гетеро-моноциклическое кольцо;
или его фармацевтически приемлемые соли,
для получения препарата для лечения и/или профилактики рака и метастаз, вирусных инфекций и болезней, и/или вирусной инфекционности, и/или латентности, опосредованных активностью убихитин-специфических протеаз (USP).
19. Применение по п. 18, при котором указанные вирусные инфекции и болезни выбраны из вирусных инфекций Herpes simplex-1 или -2, гепатита А, гепатита С, SARS коронавирусной инфекции и болезни, вируса Epstein-Barr, риновирусных инфекций и заболеваний, аденовирусных инфекций и заболеваний, полиомиелита.
| WO 2005095360 A1, 2005-10-13 | |||
| WO 2007147217 A1, 27.12.2007 | |||
| DATABAS REGISTRY, [Online]; CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US: retrieved from STN, 25.03.2010, no.1214606-92-4 (RN) | |||
| retrieved from STN, 05.07.2012, no.1381736-63-5 (RN) | |||
| retrieved from STN, 04.07.2012, no.1381217-56-6 (RN) | |||
| retrieved from STN, 25.03.2010, no.1214488-43-3 (RN) | |||
| FISHMAN; ET AL., Studie in Alkylation | |||
| I | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| WOLFGANG SIPPL et al., Ubiquitin-specific proteases as cancer drug targets FUTURE ONCOLOGY, 20010501 Vol:7, Nr:5, Page(s):619 - 632 | |||
| RU 2006124026 A, 27.01.2008. | |||

