Область техники
[0001]
Настоящее изобретение относится к зонду для визуализации белка β-листовой структуры, который может использоваться для диагностики конформационных заболеваний, в частности, заболеваний (тауопатии), имеющих такой кардинальный симптом, как внутримозговое накопление белка tau (тау), например, болезни Альцгеймера.
Уровень техники
[0002]
Известно, что при болезни Альцгеймера накопление сенильных бляшек, содержащих амилоидный бета-белок (далее совместно именуемый Aβ), как основной компонент, и нейроволоконных сплетений, содержащих гиперфосфорилированный белок тау (далее совместно именуемый тау) в качестве основного компонента продолжается до такой степени, что ее невозможно излечить, когда люди, окружающие пациента, или врач замечают специфические клинические симптомы заболевания. Другими словами, если текущий диагноз болезни Альцгеймера сравнить с диагнозом рака, то она выявляется только при достижении ею конечной стадии.
[0003]
Недавно было выявлено, что даже в случаях крайне ранней стадии очень легкой болезни Альцгеймера, которая соответствует легкому когнитивному нарушению (MCI), которое считается частично предшествующим болезни Альцгеймера состоянием, полученные при аутопсии образцы показывают накопление множества Aβ и тау, и патологоанатомически состояние представляет собой почти развившуюся болезнь Альцгеймера. Поэтому при болезни Альцгеймера патогистология проявляется задолго до появления симптома потери памяти. Другими словами, существует достаточно большое различие между патогистологией и клинической картиной болезни Альцгеймера (так называемое различие между патоморфологической болезнью Альцгеймера и клинической болезнью Альцгеймера).
[0004]
Как показано на фиг. 1, считается, что накопление Aβ начинается на 10 и более лет раньше, чем накопление тау в мозге при болезни Альцгеймера. Как очевидно из фиг. 1, поскольку прослеживание Aβ считалось наиболее целесообразным для диагностики болезни Альцгеймера на самой ранней стадии или до ее развития, то почти все зонды PET (позитронно-эмиссионной томографии) для диагностики болезни Альцгеймера представляли собой так называемые зонды для визуализации амилоида для прослеживания Aβ с 20-го по 21-ый век. Сначала, главным образом использовали зонды, меченные [11C], но затем предпринимались попытки разработки зондов, меченных [18F], который имеет длительный период полувыведения и легко используется в клинических условиях. На фиг. 2 иллюстрируются примеры зондов для визуализации амилоида, которые были разработаны до настоящего времени.
[0005]
В начале 2002 г. были впервые в мире представлены изображения, показывающие введение PET зондов для визуализации амилоида у пациентов с болезнью Альцгеймера (см. непатентный документ 1). Этой чести была удостоена команда Barrio et al., UCLA (Калифорнийский Университет, Лос-Анджелес), и использованные зонды представляли собой [18F] FDDNP (2-(1-{6-[(2-[фтор-18]фторэтил)(метил)амино]-2-нафтил}-этилиден)малононитрил). Однако затем основным направлением зондов для визуализации амилоида стал [11C] PIB (полиизобутилен), разработанный компанией General Electric в Питтсбургском Университете, который возможно используется в настоящее время более чем для 1000 клинических случаев, стал основным направлением разработки зондов для визуализации амилоида (см. непатентный документ 2).
[0006]
Многие исследователи предполагали, что визуализация амилоида при диагностике болезни Альцгеймера станет так называемым методом широкого назначения, который обеспечивает возможность диагностики с высокой чувствительностью и специфичностью, а также ранней диагностики, дифференциальной диагностики, диагностики тяжести (или прогрессирования) и преклинической диагностики (так называемого выявления пациентов с пресимптоматическим высоким риском).
[0007]
Однако по мере продвижения клинических исследований, постепенно появлялись проблемы при визуализации амилоида, которую считали методом диагностики широкого назначения. Эти проблемы объясняются приведением [11C] PIB в качестве примера следующим образом:
[0008]
Во-первых, диагностика тяжести (или прогрессирования) невозможна. Другими словами, через 2 года после диагностики у пациента болезни Альцгеймера с использованием [11C] PIB, не было изменения накопления зонда, независимо от прогрессирования клинических симптомов (см. непатентный документ 3). Причиной считается то, что накопление Aβ, с которым связывается [11C] PIB, достигает плато задолго до выявления MCI перед развитием болезни Альцгеймера. Поэтому тяжесть или прогрессирование болезни Альцгеймера невозможно диагностировать с использованием [11C] PIB.
[0009]
Во-вторых, существует проблема в том, что наблюдается значительное количество ложноположительных результатов. К удивлению, на ADNI (Инициативе Визуализации при Болезни Альцгеймера), проведенной перед Международной Конференцией по Болезни Альцгеймера в Чикаго в июле 2008 г., сообщалось, что у 53% здоровых пожилых лиц отмечены положительные результаты при использовании [11C] PIB (см. непатентный документ 4). Хотя считается, что заболеваемость болезнью Альцгеймера составляет от 4 до 6% среди населения в возрасте 65 или более лет, по данным ADNI, у 53% пожилых людей, кроме пациентов с болезнью Альцгеймера, были положительные результаты при использовании [11C] PIB. Хотя заявители настоящего изобретения считают цифру 53% переоценкой, сами разработчики [11C] PIB признают возможность значительного количества ложноположительных результатов (см. непатентный документ 5).
[0010]
Считают, что причиной этих многих ложноположительных результатов является значительная дисперсия при накоплении Aβ у всех нормальных здоровых пациентов, пациентов с MCI и пациентов с болезнью Альцгеймера.
[0011]
Кроме того, с июня по июль 2008 г. последовательно сообщалось, что эффекты групп терапевтических лекарственных средств (вакцин и ингибиторов секретазы), которые, как ожидали, обеспечат основные лекарственные средства на основании гипотезы болезни Альцгеймера/амилоида (или Aβ), были далеки от ожидания. Самым шокирующим было сообщение Holmes et al. в журнале Lancet, что вакцины Aβ вообще не могут остановить прогрессирование клинических симптомов, хотя Aβ был удален из мозга пациентов с болезнью Альцгеймера (см. непатентный документ 6).
[0012]
Однако в сообщении в журнале Lancet была представлена другая важная информация; все накопление белка тау у пациентов в статье в Lancet прогрессировало до конечной стадии. На фиг. 3 иллюстрируется стадия Braak накопления Aβ и тау при болезни Альцгеймера. Для стадии Braak случая вскрытия 7 и 8 из сообщения в журнале Lancet, считалось, что Aβ не накапливался (или стадия A), хотя степень накопления тау составляла стадию VI. Это подразумевает, что в обоих случаях накопление Aβ было легким или меньшим по степени, хотя накопление тау имело самый высокий уровень стадии VI.
[0013]
Было несколько сообщений, что в начале 1990-х г.г. патогистология коррелировалась с клиническими симптомами болезни Альцгеймера, что было связано скорее с тау, чем с Aβ (непатентный документ 7). Это было неожиданно снова подтверждено сообщением Holmes et al.
[0014]
Эти данные убедительно свидетельствуют о том, что вакцины Aβ были менее эффективны в качестве терапевтических лекарственных средств после развития болезни Альцгеймера, и что степень накопления Aβ не всегда отражает тяжесть болезни Альцгеймера, а также, что более целесообразно прослеживать тау, чем Aβ, для диагностики тяжести болезни Альцгеймера.
[0015]
Заявители настоящего изобретения считают, что с учетом клинических исходов применения вакцин, других терапевтических лекарственных средств и зондов для визуализации амилоида, связь между амилоидом (или Aβ) и тау при болезни Альцгеймера следует пересмотреть в соответствии с фиг. 4. Как показано на фиг. 4, когда имеется низкое накопление амилоида, MCI и болезнь Альцгеймера развиваются, когда накопление тау достигает пороговой величины, и когда накопление амилоида очень высокое, MCI и болезнь Альцгеймера не развиваются, когда накопление тау не достигает порогового уровня. То есть, количественное накопление амилоида не связано с развитием MCI и болезни Альцгеймера, хотя накопление тау определяет это развитие. Предложено говорить, что «амилоид (или Aβ) не имеет порога, но тау имеет порог».
[0016]
Как описано выше, визуализация тау, вероятно, превосходит визуализацию амилоида для диагностики тяжести (или прогрессирования) болезни Альцгеймера, или для правильного выявления пресимптоматических пациентов с высоким риском болезни Альцгеймера.
[0017]
Заявители настоящего изобретения считают вероятным, что «визуализация тау будет играть ведущую роль в диагностике болезни Альцгеймера, в будущем с добавлением визуализации амилоида».
[0018]
Документ в релевантной области техники включает, например, (i) публикацию Okamura et al., J. Neurosci., 25 (4&), 10857-10862 (2005), (ii) европейский патент EP 1574500 A1, (iii) заявку на патент США от компании Siemens US 2010/0239496 A1, и (iv) патент Кореи KR 2010-0112423 A.
Документы уровня техники
Патентный документ
[0019]
Непатентный документ 1: Shoghi-Jadid K, Small GW, Agdeppa ED, Kepe V, Ercoli LM, Siddarth P, Read S, Satyamurthy N, Petric A, Huang SC, Barrio JR: Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am. J. Geriatr. Psychiatry 10, 24-35. 2002.
Непатентный документ 2: Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath МЛ, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA and Langstrom B.: Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann. Neurol. 55. 306-319 (2004).
Непатентный документ 3: Engler H, Forsberg A, Almkvist O, Blomquist G, Larsson E, Savitcheva I, Wall A, Ringheim A, Långström B, Nordberg A: Two-year follow-up of amyloid deposition in patients with Alzheimer's disease. Brain. 129. 2856-2866. 2006.
Непатентный документ 4: Weiner: International Conference on Alzheimer disease (ICAD) meeting, Chicago, 2008. Jul 19.
Непатентный документ 5: Aizenstein HJ, Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, Ziolko SK, James JA, Snitz BE, Houck PR, Bi W, Cohen AD, Lopresti BJ, DeKosky ST, Halligan EM, Klunk WE.: Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 65. 1509-1517. 2008.
Непатентный документ 6: Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA : Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 372. 2132-2142. 2008.
Непатентный документ 7: Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT: Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer''s disease. Neurology. 42. 631-639. 1992.
Описание изобретения
Проблемы, подлежащие решению изобретением
[0020]
Целью настоящего изобретения является получение соединения, которое является высокоспецифичным для тау и может визуализировать тау с удовлетворительной чувствительностью, а также имеет высокий мозговую транзицию, низкую или не выявленную остеотропность и низкую или не выявленную токсичность.
Средства для решения проблем
[0021]
В свете указанных выше проблем, заявители настоящего изобретения провели обширные исследования и обнаружили, что соединение формулы (I), его соль или сольват представляет собой соединение, которое является высокоспецифичным в отношении тау и может визуализировать тау с удовлетворительной чувствительностью, а также имеет высокую мозговую транзицию, низкую или не выявленную остеотропность и низкую или не выявленную токсичность. Заявители также обнаружили, что соединение формулы (I') может использоваться в качестве предшественника соединения формулы (I), его соли или сольвата. Таким образом, заявители завершили создание настоящего изобретения.
[0022]
То есть, настоящее изобретение относится к следующим аспектам.
(1) Соединение формулы (I):
 ,
,
где
A обозначает
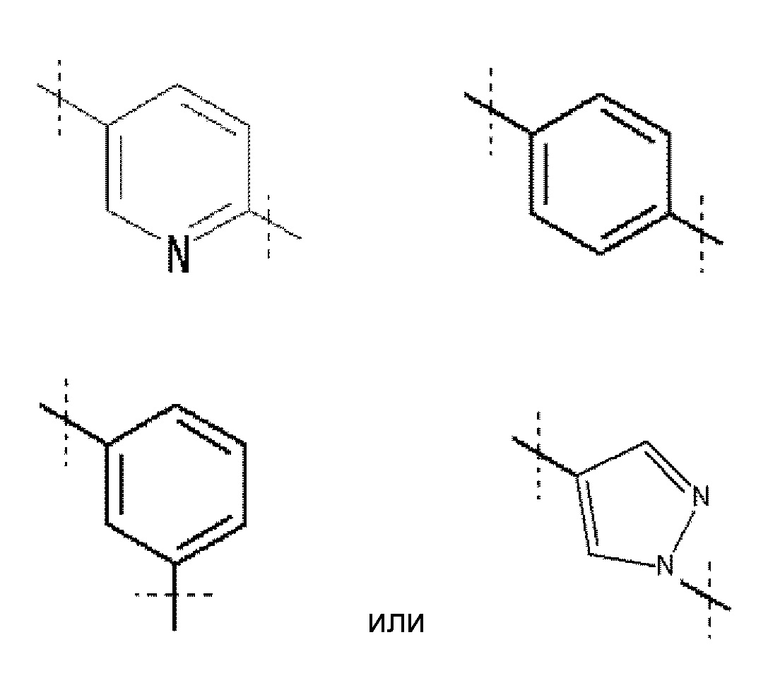 ,
,
R1 обозначает галоген, a -C(=O)-низшую алкильную группу (каждая алкильная группа может быть независимо замещена одним или более заместителями, выбранными из NRaRb, галогена и гидрокси группы), низшую алкильную группу (каждая алкильная группа может быть независимо замещена одним или более заместителями, выбранными из галогена и гидрокси группы), -O-низшую алкильную группу (каждая алкильная группа может быть независимо замещена одним или более заместителями, выбранными из галогена и гидрокси группы) или
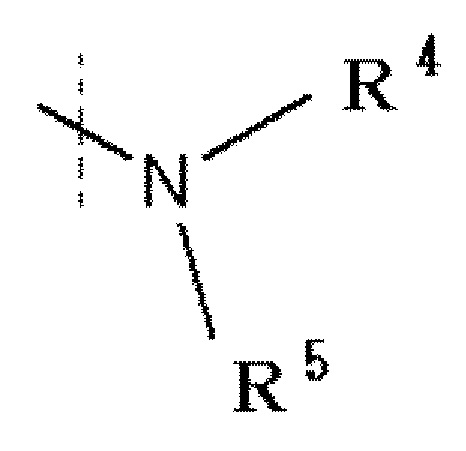 ,
,
в которой
R4 и R5 каждый независимо представляет водород, низшую алкильную группу или циклоалкильную группу, или R4, R5 и атом азота, к которому они присоединены, вместе образуют 3-8-членное азотсодержащее алифатическоое кольцо (один или более атомов углерода, составляющие азотсодержащее алифатическое кольцо могут быть замещены атомом азота, атомом серы или атомом кислорода, и когда атом углерода замещен атомом азота, то атом азота может быть замещен низшей алкильной группой),
или R4 и атом азота, к которому он присоединен, вместе с кольцом A образуют 8-16-членное азотсодержащее конденсированного бициклического кольца (один или более атомов углерода, составляющих азотсодержащее конденсированное бициклическое кольцо, могут быть замещены атомом азота, атомом серы или атомом кислорода, и когда атом углерода замещен атомом азота, то атом азота может быть замещен низшей алкильной группой), и R5 представляет водород, низшую алкильную группу или циклоалкильную группу,
в которой линия, которую пересекает пунктирная линия, означает связь указанной выше общей формулы с другой структурной составляющей,
R2 или R3 каждый независимо представляет галоген, OH, COOH, SO3H, NO2, SH, NRaRb, низшую алкильную группу (каждая алкильная группа может быть независимо замещена одним или более заместителями, выбранными из галогена и гидрокси группы) или -O-низшую алкильную группу (каждая алкильная группа может быть независимо замещена одним или более заместителями, выбранными из галогена, гидрокси группы и -O-низшей алкильной-O-низшей алкильной группы (каждая алкильная группа независимо может быть замещена галогеном)),
кольцо A является незамещенным или замещено R6 (в которой R6 обозначает один или более заместителей, независимо выбранных из галогена, OH, COOH, SO3H, NO2, SH, NRaRb, низшей алкильной группы (каждая алкильная группа может быть независимо замещена одним или более заместителями, выбранными из галогена и гидрокси группы) и -O-низшей алкильной группы (каждая алкильная группа может быть независимо замещена одним или более заместителями, выбранными из галогена и гидрокси группы),
Ra и Rb каждый независимо представляет водород или низшую алкильную группу (каждая алкильная группа может быть независимо замещена одним или более заместителями, выбранными из галогена и гидрокси группы),
m обозначает целое число от 0 до 4, и
n обозначает целое число от 0 до 4, или его фармацевтически приемлемая соль или сольват.
[0023]
(2) Соединение по п. (1), где R1 обозначает галоген, -C(=O)-низшую алкильную группу (каждая алкильная группа может независимо быть замещена NH2), низшую алкильную группу (каждая алкильная группа может быть независимо замещена гидрокси группой), -O-низшую алкильную группу или
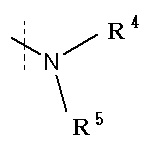
в которой
R4 и R5 каждый независимо представляет водород или низшую алкильную группу, или его фармацевтически приемлемая соль или сольват.
(3) Соединение по п. (1) или (2), где по меньшей мере один из R2, R3 и R6 обозначает -O-низшую алкильную группу, замещенную одной гидрокси группой и одним галогеном, или его фармацевтически приемлемая соль или сольват.
(4) Соединение по п. (3), где по меньшей мере один из R2, R3 и R6 представлен
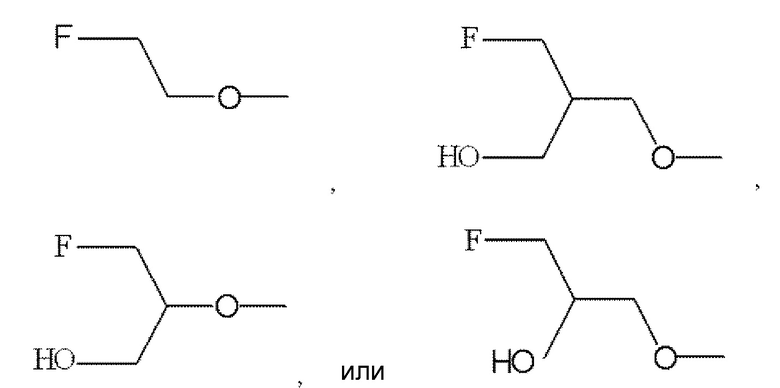 ,
,
или его фармацевтически приемлемая соль или сольват.
(5) Соединение по п. (1) или (2), где по меньшей мере один из R2, R3 и R6 обозначает NRaRb, и Ra и Rb каждый независимо представляет водород или незамещенную низшую алкильную группу, или его фармацевтически приемлемая соль или сольват.
[0024]
(6) Соединение по п. (1), где соединение формулы (I) представляет собой соединение, выбранное из группы, состоящей из:
2-(4-аминофенил)-8-(1―фторметил-2-гидроксиэтокси)хинолина,
2-(4-диэтиламинофенил)-6-(1-фторметил-2-гидрокси)хинолина,
2-(4-диэтиламинофенил)-7-(2-фторметил-2-гидроксиэтокси)хинолина,
2-(4-диэтиламинофенил)-8-(1-фторметил-2-гидроксиэтокси)хинолина,
2-(4-диэтиламинофенил)-7-(1-фторметил-2-гидроксиэтокси)хинолина,
2-(4-диэтиламинофенил)-4-(3-фтор-2-гидроксипропокси)хинолина,
2-(4-диэтиламинофенил)-5-(1-фторметил-2-гидроксиэтокси)хинолина,
2-(4-диэтиламинофенил)-3-(1-фторметил-2-гидроксиэтокси)хинолина,
2-(4-диэтиламинофенил)-8-[(3-фтор-2-гидрокси)пропокси]хинолина,
2-(4-фторметил-2-гидроксиэтокси)-2-(4-диметиламинофенил)хинолина,
7-(1-фторметил-2-гидроксиэтокси)-2-(4-метиламинофенил)хинолина,
2-(4-этилметиламинофенил)-7-(1-фторметил-2-гидроксиэтокси)хинолина,
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-диметиламинофенил)хинолина,
7-[(3-фтор-2-гидрокси)пропокси]-2-(4-метиламинофенил)хинолина,
7-[(3-фтор-2-гидрокси)пропокси]-2-(4-диметиламинофенил)хинолина,
2-(4-этилметиламинофенил)-7-[(3-фтор-2-гидрокси)пропокси]хинолина,
2-(4-аминофенил)-6-[(3-фтор-2-гидрокси)пропокси]хинолина,
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-метиламинофенил)хинолина,
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-диэтиламинофенил)хинолина,
7-амино-2-(4-фторфенил)хинолина,
2-(4-фторфенил)-7-диметиламинохинолина,
5-амино-2-(4-фторфенил)хинолина,
2-(4-фторфенил)-5-диметиламинохинолин оксалата,
8-амино-2-(4-фторфенил)хинолина,
2-(4-фторфенил)-8-диметиламинохинолина,
6-амино-2-(4-фторфенил)хинолина,
2-(4-фторфенил)-6-диметиламинохинолина,
2-(2-аминопирид-5-ил)-7-(1-фторметил-2-гидроксиэтокси)хинолина,
6-этилметиламино-2-(4-фторфенил)хинолина,
6-диэтиламино-2-(2-фторпирид-5-ил)хинолина,
8-этилметиламино-2-(2-фторпирид-5-ил)хинолина,
5-этиламино-2-(2-фторпирид-5-ил)хинолина,
5-диэтиламино-2-(2-фторпирид-5-ил)хинолина,
7-диэтиламино-2-(2-фторпирид-5-ил)хинолина,
7-этилметиламино-2-(2-фторпирид-5-ил)хинолина,
2-(4-этиламинофенил)-6-[(3-фтор-2-гидрокси)пропокси]хинолина,
2-(2-аминопирид-5-ил)-6-[(3-фтор-2-гидрокси)пропокси]хинолина,
2-(2-метиламинопирид-5-ил)-6-[(3-фтор-2-гидрокси)пропокси]хинолина,
6-[(3-фтор-2-гидрокси)пропокси]-2-(2-диметиламинопирид-5-ил)хинолина,
2-(2-диэтиламинопирид-5-ил)-6-[(3-фтор-2-гидрокси)пропокси]хинолина,
2-(2-этиламинопирид-5-ил)-6-[(3-фтор-2-гидрокси)пропокси]хинолина,
1-фтор-3-{2-[4-(4-метилпиперазин-1-ил)фенил]хинолин-6-илокси)пропан-2-ола,
1-фтор-3-{2-[6-(пиперазин-1-ил)пиридин-3-ил]хинолин-6-илокси)пропан-2-ола,
1-фтор-3-{2-[6-(4-метилпиперазин-1-ил)пиридин-3-ил]хинолин-6-илокси)пропан-2-ола,
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-метил-3,4-дигидро-2H-пиридо[3,2-b][1,4]оксазин-7-ил)хинолина,
6-[(3-фтор-2-гидрокси)пропокси]-2-(1-метил-1,2,3,4-тетрагидрохинолин-6-ил)хинолина,
6-[(3-фтор-2-гидрокси-1,1-диметил)пропокси]-2-(1-метил-1,2,3,4-тетрагидрохинолин-6-ил)хинолина,
2-(4-амино-3-фторфенил)-6-диметиламинохинолина,
2-[4-(амино)-3-[(3-фтор-2-гидрокси)пропокси]фенил]-6-метиламинохинолина,
2-[3-(3-фтор-2-гидрокси-1,1-диметил)пропокси]-4-(диметиламино)фенил]-6-диметиламинохинолина,
6-амино-2-[4-(амино)-3-[(3-фтор-2-гидрокси)пропокси]фенил]хинолина,
2-[3-[(3-фтор-2-гидрокси)пропокси]-4-(диметиламино)фенил]-6-диметиламинохинолина,
2-[3-[(3-фтор-2-гидрокси)пропокси]-4-(метиламино)фенил]-6-метиламинохинолина,
2-[4-(амино)-3-[(3-фтор-2-гидрокси)пропокси]фенил]-6-диметиламинохинолина,
6-амино-2-[3-[(3-фтор-2-гидрокси)пропокси]-4-(диметиламино)фенил]хинолина,
2-[3-[(3-фтор-2-гидрокси)пропокси]-4-(диметиламино)фенил]-6-метиламинохинолина,
2-[3-[2-[2-(2-фторэтокси)этокси]этокси-4-(метиламино)фенил]-6-диметиламинохинолина,
2-[3-[(3-фтор-2-гидрокси)пропокси]-4-(метиламино)фенил]-6-диметиламинохинолина,
6-амино-2-[3-[(3-фтор-2-гидрокси)пропокси]-4-(метиламино)фенил]хинолина,
2-[3-[(3-фтор-2-гидрокси)пропокси]-2-(диметиламино)пирид-5-ил]-6-диметиламинохинолина,
2-[3-[(3-фтор-2-гидрокси)пропокси]-2-(диметиламино)пирид-5-ил]хинолина,
6-[[2-(тетрагидро-2H-пиран-2-илокси)тозилокси]пропокси]-2-(4-метил-3,4-дигидро-2H-пиридо[3,2-b][1,4]оксазин-7-ил)хинолина,
6-[(3-фтор-2-гидрокси)пропокси]-2-(6-фторпиридин-3-ил)хинолина,
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-метоксифенил)хинолина,
6-[(3-фтор-2-гидрокси)пропокси]-2-[4-(гидроксиметил)фенил]хинолина,
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-этанонфенил)хинолина,
6-[(3-фтор-2-гидрокси)пропокси]-2-(6-метоксипиридин-3-ил)хинолина,
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-этоксифенил)хинолина,
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-амино-3-метоксифенил)хинолина,
6-[(3-фтор-2-гидрокси)пропокси]-2-(бензамидо-4-ил)хинолина,
6-[(3-фтор-2-гидрокси)пропокси]-2-(3-аминофенил)хинолина и
6-[(3-фтор-2-гидрокси)пропокси]-2-(1-метил-пиразол-4-ил)хинолина, или его фармацевтически приемлемая соль или сольват.
[0025]
(7) Соединение по любому из пп. (1)-(6), где соединение является меченым, или его фармацевтически приемлемая соль или сольват.
(8) Соединение по п. (7), где метка представляет собой радиоактивный нуклид, или его фармацевтически приемлемая соль или сольват.
(9) Соединение по п. (8), где радиоактивный нуклид представляет собой нуклид, испускающий рентгеновское излучение, или его фармацевтически приемлемая соль или сольват.
(10) Соединение по п. (7), где метка представляет собой нуклид, испускающий позитроны, или его фармацевтически приемлемая соль или сольват.
(11) Соединение по п. (10), где испускающий позитроны нуклид выбран из группы, состоящей из 11C, 13N, 15O, 18F, 35mCl, 76Br, 45Ti, 48V, 60Cu, 61Cu, 62Cu, 64Cu, 66Ga, 89Zr, 94mTc и 124I, или его фармацевтически приемлемая соль или сольват.
(12) Соединение по п. (11), где испускающий позитроны нуклид представляет собой 11C или 18F, или его фармацевтически приемлемая соль или сольват.
[0026]
(13) Фармацевтическая композиция, содержащая соединение по любому из пп. (1)-(12) или его фармацевтически приемлемую соль или сольват.
(14) Фармацевтическая композиция, содержащая соединение по любому из пп. (1)-(12) или его фармацевтически приемлемую соль или сольват и солюбилизирующий агент.
(15) Фармацевтическая композиция по п. (14), где солюбилизирующий агент выбран из группы, состоящей из Полисорбата 80, полиэтиленгликоля, этанола, пропиленгликоля и комбинации двух или более их видов.
(16) Фармацевтическая композиция по любому из пп. (13)-(15), которая представляет собой раствор для инъекций.
(17) Композиция для диагностики конформационного заболевания, содержащая соединение по любому из пп. (1)-(12) или его фармацевтически приемлемую соль или сольват.
(18) Фармацевтическая композиция для лечения/или профилактики конформационного заболевания, содержащая соединение по любому из пп. (1)-(12) или его фармацевтически приемлемую соль или сольват.
(19) Набор для диагностики конформационного заболевания, включающий соединение по любому из пп. (1)-(12) или его фармацевтически приемлемую соль или сольват в качестве активного ингредиента.
(20) Композиция или набор для выявления или окрашивания белка β-листовой структуры, содержащий соединение по любому из пп. (1)-(12) или его фармацевтически приемлемую соль или сольват в качестве активного ингредиента.
(21) Набор по п. (19) или (20) для визуализирующей диагностики.
[0027]
(22) Способ лечения и/или профилактики конформационного заболевания у пациента, который включает введение пациенту соединения по любому из пп. (1)-(12) или его фармацевтически приемлемой соли или сольвата.
(23) Способ диагностики конформационного заболевания у пациента, который включает введение пациенту соединения по любому из пп. (1)-(12) или его фармацевтически приемлемой соли или сольвата.
(24) Применение соединения по любому из пп. (1)-(12) или его фармацевтически приемлемой соли или сольвата для получения композиции или набора для диагностики конформационного заболевания у пациента.
(25) Применение соединения по любому из пп. (1)-(12) или его фармацевтически приемлемой соли или сольвата для получения фармацевтической композиции для лечения и/или профилактики конформационного заболевания у пациента.
(26) Способ выявления или окрашивания белка β-листовой структуры в образце, который включает окрашивание образца с использованием соединения по любому из пп. (1)-(12) или его фармацевтически приемлемой соли или сольвата.
(27) Применение соединения по любому из пп. (1)-(12) или его фармацевтически приемлемой соли или сольвата для получения композиции или набора для выявления или окрашивания белка β-листовой структуры.
(28) Композиция, набор, способ или применение по любому из пп. (17)-(27), где конформационное заболевание представляет собой тауопатию, в частности, болезнь Альцгеймера, и белок β-листовой структуры представляет собой белок тау.
[0028]
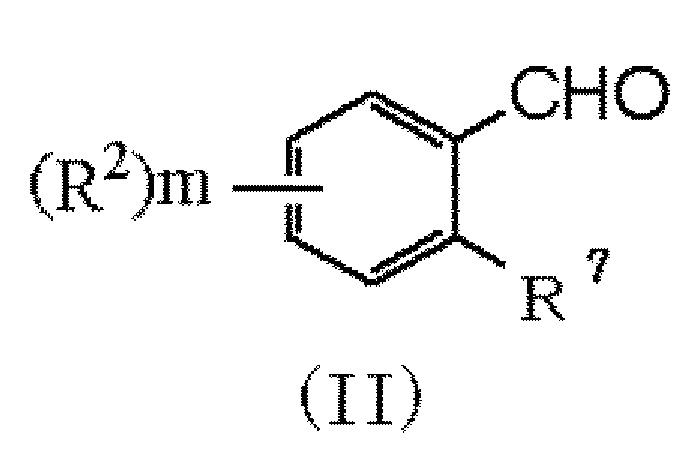
(29) Способ получения соединения формулы (I), который включает следующие стадии:
(i) взаимодействие соединения формулы (II):
 ,
,
в которой R2 и m обозначают, как определено в формуле (I), и R7 представляет NH2 или NO2, с соединением формулы (III):
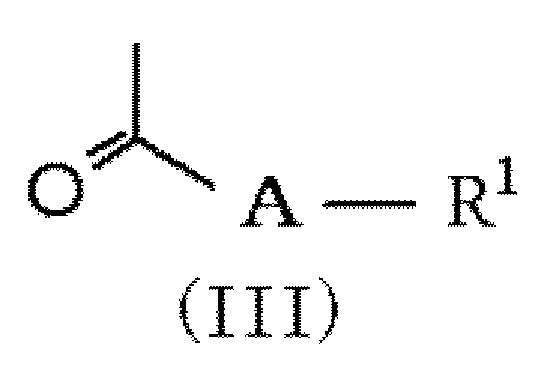 ,
,
в которой A и R1 обозначают, как определено в формуле (I), с получением соединения формулы (IV):
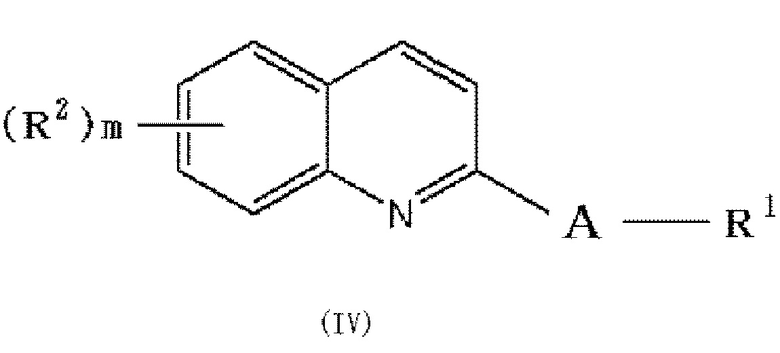 ,
,
и выделение этого соединения в виде соединения формулы (I), или
(ii) необязательно, превращение соединения формулы (IV) в другое соединение формулы (I) и выделение соединения.
[0029]
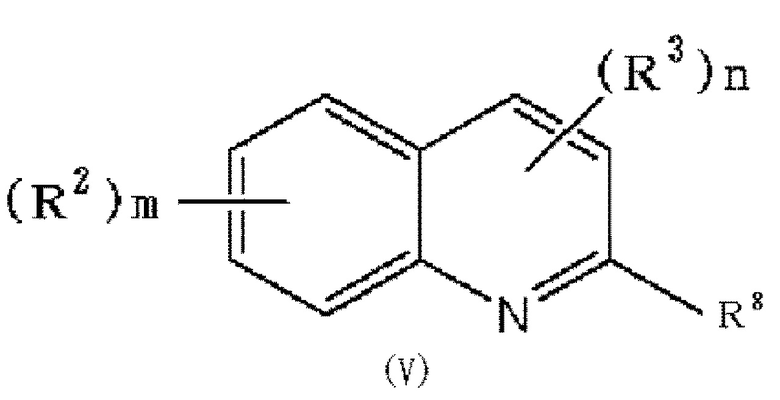
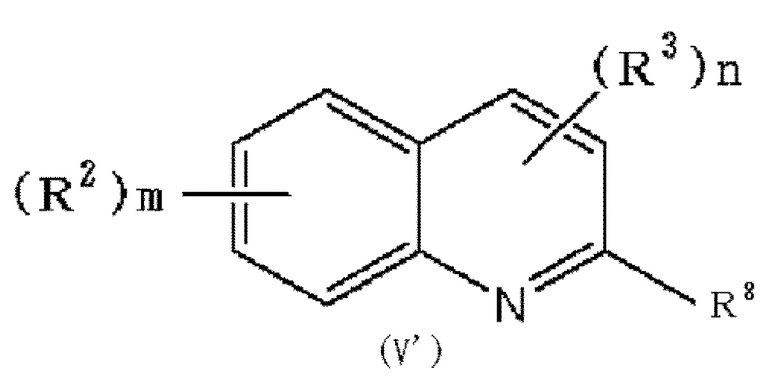
(30) Способ получения соединения формулы (I), который включает следующие стадии:
(i) взаимодействие соединения формулы (V):
 ,
,
в которой R2, R3, m и n обозначают, как определено в формуле (I), и R8 обозначает гидроксильную группу или галоген, при условии, что по меньшей мере один из R2 или R3 обозначает гидрокси группу, с соединением формулы: OH-Ark (Ark каждая независимо представляет низшую алкильную группу, которая может быть замещена одним или более заместителями, выбранными из группы, состоящей из галогена и гидрокси группы), с получением соединения формулы (V'):
 ,
,
в которой R2, R3, m и n обозначают, как определено в формуле (I), и R8 обозначает гидроксильную группу или галоген, при условии, что по меньшей мере один из R2 или R3 обозначает -O-Ark (Ark обозначает, как определено выше), и
(ii) взаимодействие соединения формулы (V') с соединением формулы (VI) или (VII):
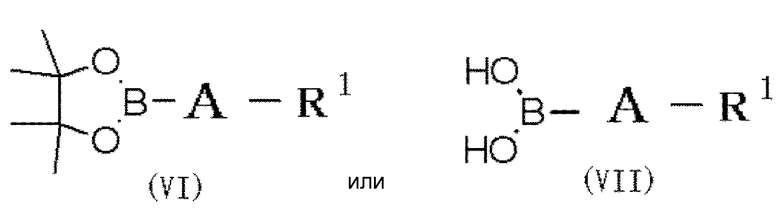 ,
,
в которой A и R1 обозначают, как определено в формуле (I), с получением соединения формулы (I), в которой по меньшей мере один из R2 и R3 обозначает -O-низшую алкильную группу (каждая алкильная группа может быть независимо замещена одним или более заместителями, выбранными из группы, состоящей из галогена и гидрокси группы), или выделение соединения, или
(iii) необязательно, превращение полученного соединения формулы (I) в другое соединение формулы (I), и выделение соединения.
[0030]
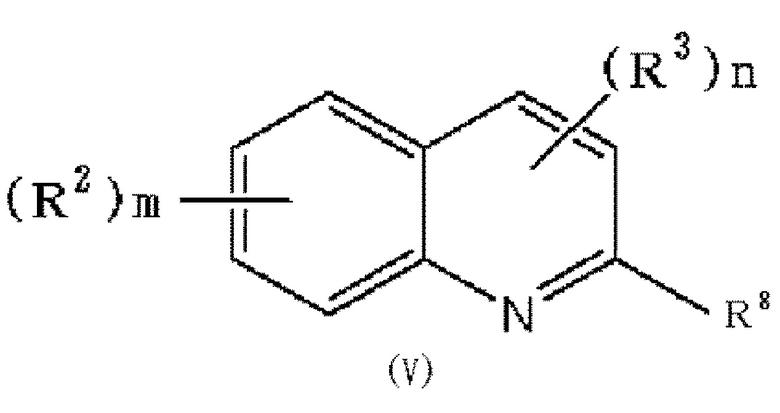
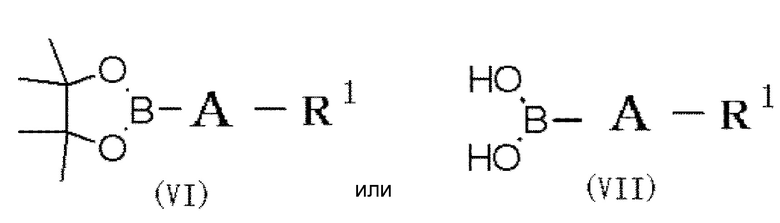
(31) Способ получения соединения формулы (I), в которой по меньшей мере один из R2 и R3 обозначает -O-низшую алкильную группу (каждая алкильная группа может быть независимо замещена одним или более заместителями, выбранными из группы, состоящей из галогена и гидрокси группы), который включает следующие стадии:
(i) взаимодействие соединения формулы (V):
 ,
,
в которой R2, R3, m и n обозначают, как определено в формуле (I), и R8 обозначает гидроксильную группу или галоген, при условии, что по меньшей мере один из R2 или R3 обозначает гидрокси группу, с соединением формулы (VI) или (VII):
 ,
,
в которой A и R1 обозначают, как определено в формуле (I), с получением соединения формулы (V”):
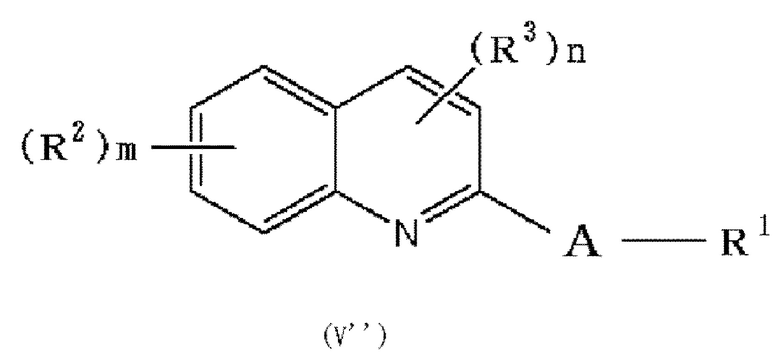 ,
,
в которой R1, R2, R3, A, m и n обозначают, как определено в формуле (I), при условии, что по меньшей мере один из R2 или R3 обозначает гидрокси группу, и
(ii) взаимодействие соединения формулы (V”) с соединением формулы: OH-Ark (Ark каждая независимо представляет низшую алкильную группу, которая может быть замещена одним или более заместителями, выбранными из группы, состоящей из галогена и гидрокси группы), с получением соединения формулы (I), в которой по меньшей мере один из R2 и R3 обозначает -O-низшую алкильную группу (каждая алкильная группа может быть независимо замещена одним или более заместителями, выбранными из группы, состоящей из галогена и гидрокси группы), и выделение соединения, или
(iii) необязательно, превращение полученного соединения формулы (I) в другое соединение формулы (I) и выделения соединения.
[0031]
(32) Способ по п. (31), где соединение формулы (I) выбрано из:
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-диметиламинофенил)хинолина,
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-метиламинофенил)хинолина и
2-(4-этиламинофенил)-6-[(3-фтор-2-гидрокси)пропокси]хинолина.
[0032]
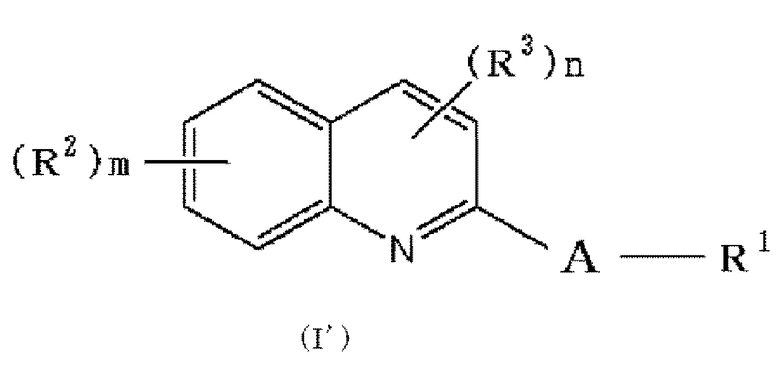
(33) Соединение формулы (I'):
 ,
,
в которой
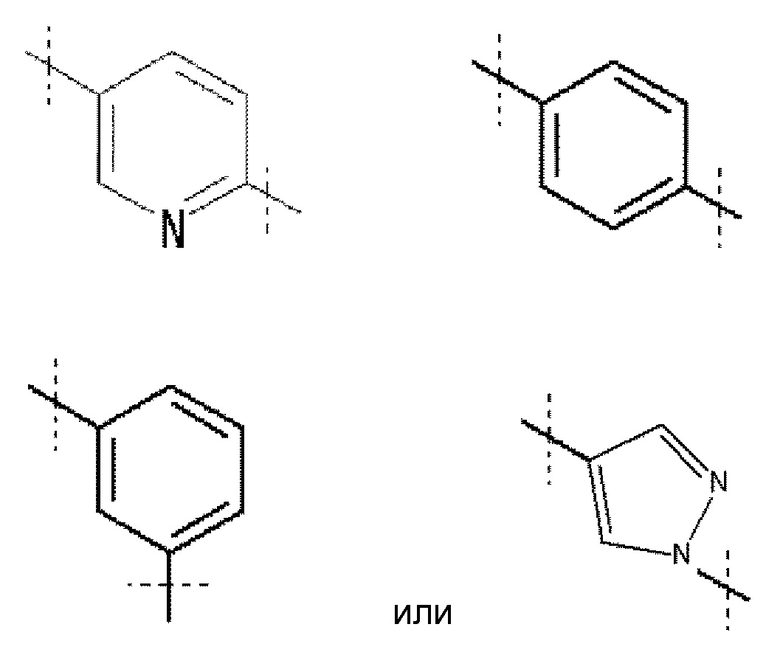
A обозначает
 ,
,
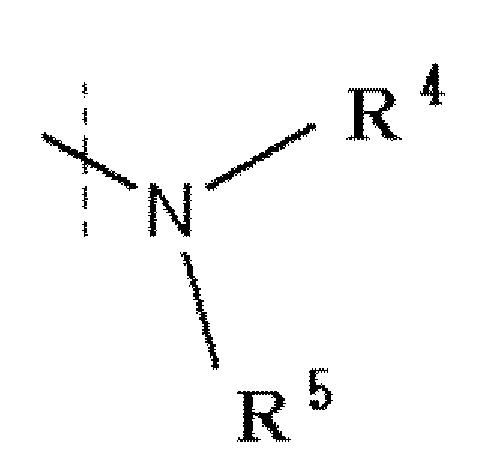
R1 обозначает галоген, -C(=O)-низшую алкильную группу (алкильная группа каждая независимо может быть замещена одним или более заместителями, выбранными из NRaRb, галогена и гидрокси группы), низшую алкильную группу (алкильная группа каждая независимо может быть замещена одним или более заместителями, выбранными из галогена и гидрокси группы), -O-низшую алкильную группу (алкильная группа каждая независимо может быть замещена одним или более заместителями, выбранными из галогена и гидрокси группы) или
 ,
,
в которой
R4 и R5 каждый независимо представляет водород, низшую алкильную группу или циклоалкильную группу, или R4, R5 и атом азота, к которому они присоединены, вместе образуют 3-8-членное азотсодержащее алифатическое кольцо (один или более атомов углерода, составляющие азотсодержащее алифатическое кольцо, могут быть замещены атомом азота, атомом серы или атомом кислорода, и когда атом углерода замещен атомом азота, то атом азота может быть замещен низшей алкильной группой), или R4 и атом азота, к которому он присоединен, вместе с кольцом A образуют 8-16-членное азотсодержащее конденсированное бициклическое кольцо (один или более атомов углерода, составляющие азотсодержащее конденсированное бициклическое кольцо, могут быть замещены атомом азота, атомом серы или атомом кислорода, и когда атом углерода замещен атомом азота, то атом азота может быть замещен низшей алкильной группой), R5 обозначает водород, низшую алкильную группу или циклоалкильную группу,
в которой линия, которую пересекает пунктирная линия, означает связь представленной выше общей формулы с другой структурной составляющей,
R2 или R3 каждый независимо может быть замещен галогеном, OH, COOH, SO3H, NO2, SH, NRaRb, низшей алкильной группой (низшая алкильная группа каждая независимо может быть замещена одним или более заместителями, выбранными из п-толуолсульфонилокси группы (тозилокси группы, TsO), метансульфонилокси группы, трифторметансульфонилокси группы или 2-тетрагидропиранилокси (OTHP), галогена и гидрокси группы) или -O-низшей алкильной группой (низшая алкильная группа замещена п-толуолсульфонилокси группой (тозилокси группой, TsO), метансульфонилокси группой, трифторметансульфонилокси группой или 2-тетрагидропиранилокси (OTHP), а также может быть замещена гидрокси группой),
кольцо A является незамещенным или замещено R6 (в которой R6 обозначает один или более заместителей, выбранных независимо из галогена, OH, COOH, SO3H, NO2, SH, NRaRb, низшей алкильной группы (низшая алкильная группа каждая независимо может быть замещена одним или более заместителями, выбранными из группы, состоящей из п-толуолсульфонилокси группы (тозилокси группы, TsO), метансульфонилокси группы, трифторметансульфонилокси группы или 2-тетрагидропиранилокси (OTHP), галогена и гидрокси группы) и -O-низшей алкильной группы (алкильная группа каждая независимо может быть замещена одним или более заместителями, выбранными из п-толуолсульфонилокси группы (тозилокси группы, TsO), метансульфонилокси группы, трифторметансульфонилокси группы или 2-тетрагидропиранилокси (OTHP), галогена, гидрокси группы, и -O-низшей алкильной группы-O-низшей алкильной группы (алкильная группа каждая независимо может быть замещена галогеном))),
Ra и Rb независимо представляют водород или низшую алкильную группу (алкильная группа и низшая алкильная группа каждая независимо может быть замещена одним или более заместителями, выбранными из п-толуолсульфонилокси группы (тозилокси группы, TsO), метансульфонилокси группы, трифторметансульфонилокси группы или 2-тетрагидропиранилокси (OTHP), галогена и гидрокси группы),
m обозначает целое число от 0 до 4, и
n обозначает целое число от 0 до 4,
при условии, что по меньшей мере один из R2, R3 и R6 представляет -O-низшую алкильную группу (низшая алкильная группа замещена п-толуолсульфонилокси группой (тозилокси группой, TsO), метансульфонилокси группой, трифторметансульфонилокси группой или 2-тетрагидропиранилокси (OTHP), и может быть также замещена одним или более заместителями, выбранными из галогена и гидрокси группы), или его фармацевтически приемлемая соль или сольват.
[0033]
(34) Соединение по п. (33), где по меньшей мере один из R2, R3 и R6 обозначает группу формулы:
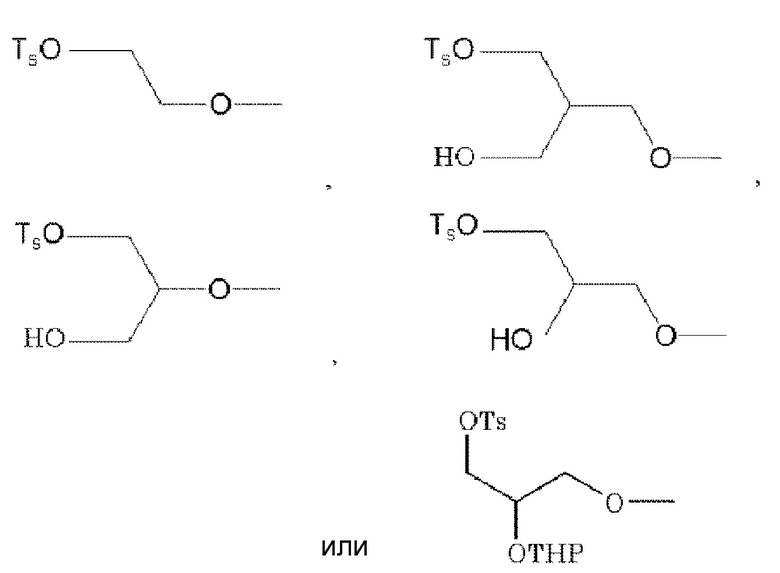 .
.
[0034]
(35) Соединение по п. (33), где соединение формулы (I') представляет собой соединение, выбранное из группы, состоящей из:
2-(4-диэтиламинофенил)-6-[(2-гидрокси-1-тозилоксиметил)этокси]хинолина,
2-(4-аминофенил)-8-(2-гидрокси-1-тозилоксиметилэтокси)хинолина,
2-(4-диэтиламинофенил)-8-(2-гидрокси-1-тозилоксиметилэтокси)хинолина,
2-(4-диэтиламинофенил)-8-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолина,
2-(4-диэтиламинофенил)-7-(2-гидрокси-1-тозилоксиметилэтокси)хинолина,
2-(4-диэтиламинофенил)-7-[[(2-тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолина,
7-(2-гидрокси-1-тозилоксиметилэтокси)-2-(4-диметиламинофенил)хинолина,
7-(2-гидрокси-1-тозилоксиметилэтокси)-2-(4-метиламинофенил)хинолина,
2-(4-этилметиламинофенил)-7-(2-гидрокси-1-тозилоксиметилэтокси)хинолина,
2-(4-диэтиламинофенил)-5-(2-гидрокси-1-тозилоксиметилэтокси)хинолина,
2-(4-этилметиламинофенил)-7-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолина,
2-(4-метиламинофенил)-7-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолина,
2-(4-диметиламинофенил)-7-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолина,
2-(4-метиламинофенил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолина,
2-(4-диэтиламинофенил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолина,
2-(4-диметиламинофенил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолина,
2-(4-аминофенил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолина,
2-(4-этиламинофенил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолина,
2-(2-аминопирид-5-ил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолина,
2-(2-метиламинопирид-5-ил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолина,
2-(2-диметиламинопирид-5-ил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолина,
2-(2-диэтиламинопирид-5-ил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолина,
2-(2-этиламинопирид-5-ил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолина,
2-[4-(метиламино)-3-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]фенил]-6-диметиламинохинолина и
2-[4-(диметиламино)-3-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]фенил]-6-диметиламинохинолина.
[0035]
(36) Набор для получения меченого соединения по любому из пп. (33)-(35) или его фармацевтически приемлемой соли или сольвата, включающий:
соединение по любому из пп. (33)-(35) или его фармацевтически приемлемую соль или сольват,
метящий агент и,
необязательно, инструкции по проведению мечения.
(37) Набор по п. (36), где метящий агент представляет собой радиоактивный нуклид.
(38) Набор по п. (37), где радиоактивный нуклид представляет собой нуклид, испускающий γ-лучи.
(39) Набор по п. (36), где метящий агент представляет собой нуклид, испускающий позитроны.
(40) Набор по п. (39), где испускающий позитроны нуклид выбран из группы, состоящей из 11C, 13N, 15O, 18F, 35mCl, 76Br, 45Ti, 48V, 60Cu, 61Cu, 62Cu, 64Cu, 66Ga, 89Zr, 94mTc и 124I.
(41) Набор по п. (40), где испускающий позитроны нуклид представляет собой 11C или 18F.
(42) Способ получения соединения по п. (7), который включает стадию взаимодействия соединения по п. (34) с метящим агентом.
(43) Способ по п. (42), где метящий агент представляет собой радиоактивный нуклид.
Эффекты изобретения
[0036]
Настоящее изобретение относится к соединению, обладающему очень высокой безопасностью, которое высокоспецифично к тау и может с удовлетворительной чувствительностью получать изображение тау, а также обладает высокой мозговой транзицией, низкой или не выявленной остеотропностью и низкой или не выявленной токсичностью, и к его предшественнику. Соответственно, диагностику, лечение и/или профилактику тауопатии можно проводить с использованием соединения по настоящему изобретению. Также, в соответствии с настоящим изобретением, становится возможным проведение визуализирующей диагностики тауопатии, в частности, визуализирующей диагностики с использованием PET (позитронно-эмиссионной томографии). Соответственно, в соответствии с настоящим изобретением, становится возможным проведение точной диагностики, эффективного лечения и профилактики на ранних стадиях тауопатии, в частности, болезни Альцгеймера.
Краткое описание чертежей
[0037]
Фиг. 1 представляет собой график, показывающий расхождение между клинической картиной и патогистологической картиной при болезни Альцгеймера. Ссылка из публикации Alzheimer's Disease (автор Yasuo IHARA, Hiroyuki ARAKI), Asahi Shimbun Company, 2007, Tokyo, частично отредактированной. В начале болезни Альцгеймера у пациентов в возрасте 80 лет накопление Aβ начинается в возрасте 50 лет и уже достигло плато в возрасте 60 лет. С другой стороны, накопление тау продолжается зависимо от возраста в возрасте 70 лет.
Фиг. 2 иллюстрирует зонды PET для визуализации амилоида, которые были разработаны до настоящего времени.
Фиг. 3 представляет собой диаграмму, показывающую стадии накопления Aβ и накопления тау при болезни Альцгеймера. Ссылка из публикации Braak & Braak: Neurobiol aging. 18. 351-357. 1997, частично отредактированной. Ссылаясь на стадии по Braak после смерти в случаях 7 и 8 в непатентном документе 6, накопление Aβ считалось как случаи, лишенные амилоида (или стадии A), в то время как накопление тау было на стадии VI. То есть, это значит, что хотя накопление Aβ является незначительным или глобально легким в обоих случаях, накопление тау было на стадии, на которой уровень накопления является самым высоким.
Фиг. 4 представляет собой график, показывающий связь между амилоидом (или Aβ) и тау при болезни Альцгеймера (предложенный заявителями настоящего изобретения). Как показано в верхней, средней и нижней колонках, когда накопление амилоида небольшое, MCI и болезнь Альцгеймера развиваются, накопление достигает порогового уровня, и когда имеется сильное накопление амилоида, MCI и болезнь Альцгеймера не развиваются, когда накопление тау не достигает порогового уровня. То есть, количественное накопление амилоида не связано с развитием MCI и болезни Альцгеймера, хотя накопление тау определяет это развитие. Другими словами, «амилоид (или Aβ) не имеет порогового уровня, но тау имеет».
Верхняя панель фиг. 5 представляет собой изображение при окраске THK-5035 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 5 представляет собой изображение при окраске THK-5038 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 6 представляет собой изображение при окраске THK-5058 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 6 представляет собой изображение при окраске THK-5064 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 7 представляет собой изображение при окраске THK-5065 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 7 представляет собой изображение при окраске THK-5066 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 8 представляет собой изображение при окраске THK-5071 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 8 представляет собой изображение при окраске THK-5077 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 9 представляет собой изображение при окраске THK-5078 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 9 представляет собой изображение при окраске THK-5079 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 10 представляет собой изображение при окраске THK-5080 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 10 представляет собой изображение при окраске THK-5081 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 11 представляет собой изображение при окраске THK-5082 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 11 представляет собой изображение при окраске THK-5087 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 12 представляет собой изображение при окраске THK-5088 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 12 представляет собой изображение при окраске THK-5089 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 13 представляет собой изображение при окраске THK-5091 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 13 представляет собой изображение при окраске THK-5092 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 14 представляет собой изображение при окраске THK-5097 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 14 представляет собой изображение при окраске THK-5098 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 15 представляет собой изображение при окраске THK-5059 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 15 представляет собой изображение при окраске THK-5075 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 16 представляет собой изображение при окраске THK-5076 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 16 представляет собой изображение при окраске THK-5086 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 17 представляет собой изображение при окраске THK-5100 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 17 представляет собой изображение при окраске THK-5105 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 18 представляет собой изображение при окраске THK-5106 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 18 представляет собой изображение при окраске THK-5107 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 19 представляет собой изображение при окраске THK-5112 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 19 представляет собой изображение при окраске THK-5116 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 20 представляет собой изображение при окраске THK-5117 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 20 представляет собой изображение при окраске THK-932 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
На фиг. 21 показано авторадиографическое изображение (соответственно верхнее левое и верхнее правое), полученное окрашиванием [18F] BF-227 и [18F] THK-5035, окрашиванием тиофлавином S (TF-S) (нижнее левое) в серийных срезах, и изображение с окрашиванием анти-Тау антителом (Тау) (нижнее правое).
На фиг. 22 показано изображение ауторадиографии [18F] THK-5105 (верхнее левое) и изображение с окрашиванием анти-фосфорилированным антителом к тау (pTau) срезов гиппокампа пациентки с болезнью Альцгеймера (женщины 83 лет и с массой мозга 900 г). На нижней панели показаны, слева направо, изображения с бόльшим увеличением авторадиографии [18F] THK-5105 окрашивания анти-фосфорилированным антителом к тау (pTau) и окрашивания немеченой THK-5105 с вкладками, представляющими гораздо большее увеличение при окрашивании анти-фосфорилированным антителом к тау (pTau) и окрашивании немеченой THK-5105.
На фиг. 23 показаны изображения авторадиографии [18F] THK-5105, [18F] THK-5117 и [18F] THK-5125 срезов латеральной височной коры и медиальной височной коры от пациентки с болезнью Альцгеймера (женщины 77 лет и с массой мозга 1100 г).
На фиг. 24 показаны величины аффинитета связывания (Kis) различных зондов к тау. Использованный тау представлял собой агрегаты мутированного тау (K18-ΔK280), и использованный радиоактивный лиганд представлял собой [18F] THK-5105.
Верхняя панель фиг. 25 представляет собой изображение при окраске THK-5136 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 25 представляет собой изображение при окраске THK-5153 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 26 представляет собой изображение при окраске THK-5157 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 26 представляет собой изображение при окраске THK-5128 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 27 представляет собой изображение при окраске THK-5147 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 28 представляет собой изображение при окраске THK-5155 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 28 представляет собой изображение при окраске THK-5156 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Верхняя панель фиг. 29 представляет собой изображение при окраске THK-5164 в срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения. Нижняя панель фиг. 28 представляет собой изображение при окраске THK-5154 на срезе мозга пациентов с болезнью Альцгеймера. Стрелками указаны нейроволоконные сплетения.
Способ осуществления изобретения
[0038]
Соединения по настоящему изобретению представляют собой соединения формул (I) и (I'), описанных ниже, или их соли или сольваты. Пока нет других уточнений, используемые в настоящем описании термины «соединение по настоящему изобретению» и «соединение в соответствии с настоящим изобретением» включают соединения формул (I) и (I'), описанных ниже, и их соли и сольваты.
[0039]
Используемый в настоящем описании термин «низшая алкильная группа» означает линейную или разветвленную алкильную группу, имеющую от 1 до 6 атомов углерода, и ее определенные примеры включают метильную группу, этильную группу, пропильную группу, изопропильную группу, бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, пентильную группу, изоамильную группу, неопентильную группу, изопентильную группу, 1,1-диметилпропильную группу, 1-метилбутильную группу, 2-метилбутильную группу, 1,2-диметилпропильную группу, гексильную группу, изогексильную группу, 1-метилпентильную группу, 2-метилпентильную группу, 3-метилпентильную группу, 1,1-диметилбутильную группу, 1,2-диметилбутильную группу, 2,2-диметилбутильную группу, 1,3-диметилбутильную группу, 2,3-диметилбутильную группу, 3,3-диметилбутильную группу, 1-этилбутильную группу, 2-этилбутильную группу, 1,2,2-триметилпропильную группу, 1-этил-2-метилпропильную группу и тому подобные. Термин «низший алкокси» означает -O-низший алкил.
[0040]
Используемый в настоящем описании термин «циклоалкильная группа» означает циклоалкильную группу, имеющую от 3 до 7 атомов углерода, и ее определенные примеры включают циклопропильную группу, циклобутильную группу, циклопентильную группу, циклогексильную группу и циклогептильную группу.
[0041]
Используемый в настоящем описании термин «галоген» означает фтор, хлор, бром или йод.
[0042]
Используемые в настоящем описании термины «белок тау» и «тау» имеют одинаковые значения. Используемые в настоящем описании термины «белок амилоидбета», «белок амилоида β», «белок Aβ», «амилоидбета», «амилоид β» и «Aβ» имеют одинаковые значения.
[0043]
В случае существования асимметричного атома углерода в соединении по настоящему изобретению, смесь изомеров и отдельный изомер также включены в соединение по настоящему изобретению.
[0044]
Например, в случае существования асимметричного углерода в соединении по настоящему изобретению, каждое оптически активное соединение может быть синтезировано отдельно, или отдельный оптический изомер может быть отделен колоночной хроматографией. Например, в случае отделения оптического изомера колоночной хроматографией, подлежащая использованию колонка включает, например, CHIRALPAK AD (выпускаемую DAICEL CHEMICAL INDUSTRIES, LTD.) или подобную. Также растворитель, используемый при колоночной хроматографии, может представлять собой растворитель, который обычно используется для отделения изомера. Например, хлороформ, ацетонитрил, этилацетат, метанол, этанол, ацетон, гексан, вода и тому подобные используются отдельно, или два или более видов этих растворителей могут также использоваться в комбинации.
[0045]
Для более определенного описания соединения формулы (I):
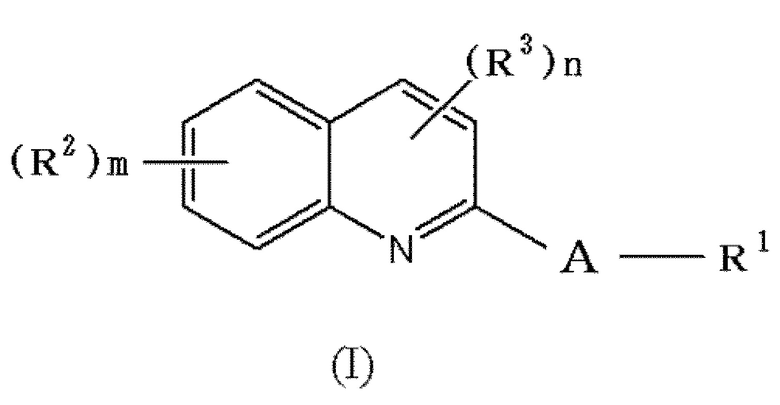 ,
,
[где соответствующие символы обозначают, как определено выше] в соответствии с настоящим изобретением, различные символы, используемые в формуле (I), будут описаны путем приведения определенных примеров.
[0046]
Кольцо A обозначает
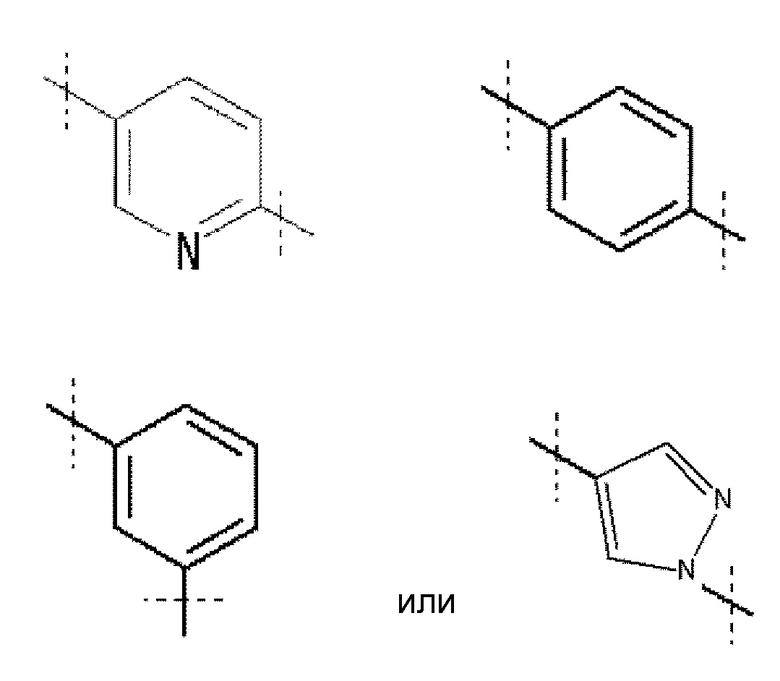 ,
,
где линия, которую пересекает пунктирная линия, означает связь представленной выше общей формулы с другой структурной составляющей. То есть, связи, существующие в положениях 2 и 5 пиридинового кольца соответственно присоединены к R1 и хинолиновому кольцу общей формулы (I). Кольцо A является незамещенным или замещено одним-четырьмя заместителями, и предпочтительно не замещено или замещено одним заместителем, выбранным из фтора, (3-фтор-2-гидрокси)пропокси, (3-фтор-2-гидрокси-1,1-диметил)пропокси, 2-[2-(2-фторэтокси)этокси]этокси и метокси.
[0047]
R1 обозначает галоген, -C(=O)-низшую алкильную группу (алкильная группа каждая независимо может быть замещена одним или более заместителями, выбранными из NRaRb, галогена и гидрокси группы), низшую алкильную группу (алкильная группа каждая независимо может быть замещена одним или более заместителями, выбранными из галогена и гидрокси группы), -O-низшую алкильную группу (алкильная группа каждая независимо может быть замещена одним или более заместителями, выбранными из галогена и гидрокси группы) или
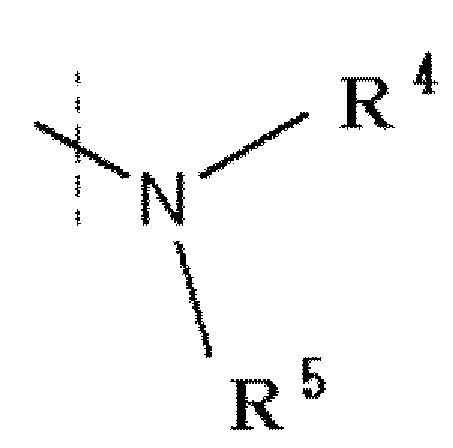 ,
,
в которой
R4 и R5 каждый независимо представляет водород, низшую алкильную группу или циклоалкильную группу, или R4, R5 и атом азота, к которому они присоединены, вместе образуют 3-8-членное азотсодержащее алифатическое кольцо (один или более атомов углерода, составляющие азотсодержащее алифатическое кольцо, могут быть замещены атомом азота, атомом серы или атомом кислорода, и когда атом углерода замещен атомом азота, то атом азота может быть замещен низшей алкильной группой, или R4 и атом азота, к которому он присоединен, вместе с кольцом A образуют 8-16-членное азотсодержащее конденсированное бициклическое кольцо (один или более атомов углерода, составляющих азотсодержащее конденсированное бициклическое кольцо, могут быть замещены атомом азота, атомом серы или атомом кислорода, и когда атом углерода представляет собой атом азота, то атом углерода замещен атомом азота, атом азота может быть замещен низшей алкильной группой), R5 обозначает водород, низшую алкильную группу или циклоалкильную группу.
[0048]
«Низшая алкильная группа», представленная R4 и R5, обозначает те же группы, как группы в низшей алкильной группе, определенные выше. Среди этих групп предпочтительными являются метильная группа, этильная группа и пропильная группа, и предпочтительнее метильная группа.
«Циклоалкильная группа», представленная R4 и R5, обозначает те же группы, что и группы в циклоалкильной группе, определенные выше.
[0049]
Определенные примеры 3-8-членного азот-содержащего алифатического кольца, образованного взятыми вместе R4, R5 и атомом азота, к которому они присоединены (атомы углерода, составляющие азотсодержащее алифатическое кольцо, могут быть замещены атомом азота, атомом серы или атомом кислорода, и в случае если атомы углерода замещены атомом азота, то атом азота может быть замещен низшей алкильной группой), включают группы формулы:
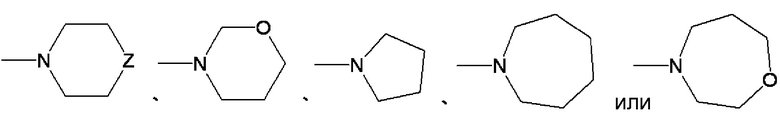 ,
,
где Z обозначает O, S, CH2 or NRe, и Re представляет водород или C1-4 алкильную группу. Среди этих групп предпочтительными являются морфолино группа, пиперазиновая группа и 4-метилпиперазиновая группа.
[0050]
Определенные примеры 8-16-членного азот-содержащего конденсированного бициклического кольца, образованного взятыми R4 и атомом азота, к которому он присоединен, вместе с кольцом A (один или более атомов углерода, составляющих азотсодержащее конденсированное бициклическое кольцо, могут быть замещены атомом азота, атомом серы или атомом кислорода, и в случае если атомы углерода замещены атомом азота, то атом азота может быть замещен низшей алкильной группой), включают группы формулы:
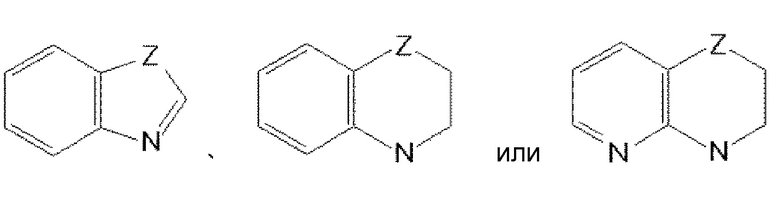 ,
,
где Z обозначает O, S, CH2 или NRe, и Re представляет водород или C1-4 алкильную группу.
Среди этих групп особенно предпочтительной является
 .
.
[0051]
R2, R3 и R6 каждый независимо представляет галоген, OH, COOH, SO3H, NO2, SH, NRaRb или низшую алкильную группу (алкильная группа замещена атомом галогена, а также может быть замещена гидрокси группой) или -O-низшую алкильную группу (алкильная группа каждая независимо может быть замещена одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси группы и -O-низшей алкильной группы-O-низшей алкильной группы (алкильная группа каждая независимо может быть замещена галогеном)).
[0052]
По меньшей мере один из R2, R3 и R6 обозначает предпочтительно -O-низшую алкильную группу (алкильная группа замещена атомом галогена, а также может быть замещена гидрокси группой). Среди этих групп предпочтительной является -O-низшая алкильная группа, замещенная атомом галогена, или -O-низшая алкильная группа, замещенная атомом галогена и гидрокси группой, и предпочтительнее группа формулы:
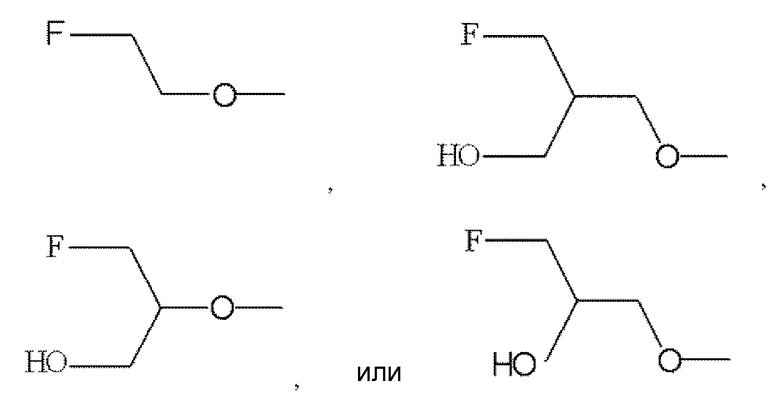 .
.
[0053]
Ra и Rb независимо представляют водород или низшую алкильную группу (алкильная группа замещена атомом галогена, а также может быть замещена гидрокси группой). Предпочтительно, Ra и Rb обозначают атомы водорода.
[0054]
m обозначает целое число от 0 до 4, и предпочтительно 1.
n обозначает целое число от 0 до 4. Предпочтительно, все R4(s) обозначают атомы водорода.
[0055]
Примеры предпочтительных соединений формулы (I) включают:
2-(4-аминофенил)-8-(1―фторметил-2-гидроксиэтокси)хинолин (THK-5004),
2-(4-диэтиламинофенил)-6-(1-фторметил-2-гидрокси)хинолин (THK-5035),
2-(4-диэтиламинофенил)-7-(2-фторметил-2-гидроксиэтокси)хинолин (THK-5038),
2-(4-диэтиламинофенил)-8-(1-фторметил-2-гидроксиэтокси)хинолин (THK-5051),
2-(4-диэтиламинофенил)-7-(1-фторметил-2-гидроксиэтокси)хинолин (THK-5058),
2-(4-диэтиламинофенил)-4-(3-фтор-2-гидроксипропокси)хинолин (THK-5059),
2-(4-диэтиламинофенил)-5-(1-фторметил-2-гидроксиэтокси)хинолин (THK-5064),
2-(4-диэтиламинофенил)-3-(1-фторметил-2-гидроксиэтокси)хинолин (THK-5065),
2-(4-диэтиламинофенил)-8-[(3-фтор-2-гидрокси)пропокси]хинолин (THK-5066),
2-(4-фторметил-2-гидроксиэтокси)-2-(4-диметиламинофенил)хинолин (THK-5071),
7-(1-фторметил-2-гидроксиэтокси)-2-(4-метиламинофенил)хинолин (THK-5077),
2-(4-этилметиламинофенил)-7-(1-фторметил-2-гидроксиэтокси)хинолин (THK-5078),
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-диметиламинофенил)хинолин (THK-5105),
7-[(3-фтор-2-гидрокси)пропокси]-2-(4-метиламинофенил)хинолин (THK-5106),
7-[(3-фтор-2-гидрокси)пропокси]-2-(4-диметиламинофенил)хинолин (THK5107),
2-(4-этилметиламинофенил)-7-[(3-фтор-2-гидрокси)пропокси]хинолин (THK-5112),
2-(4-аминофенил)-6-[(3-фтор-2-гидрокси)пропокси]хинолин (THK-5116),
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-метиламинофенил)хинолин (THK-5117),
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-диэтиламинофенил)хинолин (THK-5122),
7-амино-2-(4-фторфенил)хинолин (THK-5075),
2-(4-фторфенил)-7-диметиламинохинолин (THK-5076),
5-амино-2-(4-фторфенил)хинолин (THK-5079),
2-(4-фторфенил)-5-диметиламинохинолин оксалат (THK-5080),
8-амино-2-(4-фторфенил)хинолин (THK-5081),
2-(4-фторфенил)-8-диметиламинохинолин (THK-5082),
6-амино-2-(4-фторфенил)хинолин (THK-5086),
2-(4-фторфенил)-6-диметиламинохинолин (THK-5087),
2-(2-аминопирид-5-ил)-7-(1-фторметил-2-гидроксиэтокси)хинолин (THK-932),
6-этилметиламино-2-(4-фторфенил)хинолин (THK-5100),
6-диэтиламино-2-(2-фторпирид-5-ил)хинолин (THK-5088),
8-этилметиламино-2-(2-фторпирид-5-ил)хинолин (THK-5089),
5-этиламино-2-(2-фторпирид-5-ил)хинолин (THK-5091),
5-диэтиламино-2-(2-фторпирид-5-ил)хинолин (THK-5092),
7-диэтиламино-2-(2-фторпирид-5-ил)хинолин (THK-5097),
7-этилметиламино-2-(2-фторпирид-5-ил)хинолин (THK-5098),
2-(4-этиламинофенил)-6-[(3-фтор-2-гидрокси)пропокси]хинолин (THK-5125),
2-(2-аминопирид-5-ил)-6-[(3-фтор-2-гидрокси)пропокси]хинолин (THK-5127),
6-[(3-фтор-2-гидрокси)пропокси]-2-(2-диметиламинопирид-5-ил)хинолин (THK-5129),
2-(2-диэтиламинопирид-5-ил)-6-[(3-фтор-2-гидрокси)пропокси]хинолин (THK-5130),
2-(2-этиламинопирид-5-ил)-6-[(3-фтор-2-гидрокси)пропокси]хинолин (THK-5142),
2-(2-метиламинопирид-5-ил)-6-[(3-фтор-2-гидрокси)пропокси]хинолин (THK-5151),
1-фтор-3-{2-[4-(4-метилпиперазин-1-ил)фенил]хинолин-6-илокси)пропан-2-ол (THK-5177),
1-фтор-3-{2-[6-(пиперазин-1-ил)пиридин-3-ил]хинолин-6-илокси)пропан-2-ол (THK-5178),
1-фтор-3-{2-[6-(4-метилпиперазин-1-ил)пиридин-3-ил]хинолин-6-илокси)пропан-2-ол (THK-5180),
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-метил-3,4-дигидро-2H-пиридо[3,2-b][1,4]оксазин-7-ил)хинолин (THK-5136),
6-[(3-фтор-2-гидрокси)пропокси]-2-(1-метил-1,2,3,4-тетрагидрохинолин-6-ил)хинолин (THK-5153),
6-[(3-фтор-2-гидрокси-1,1-диметил)пропокси]-2-(1-метил-1,2,3,4-тетрагидрохинолин-6-ил)хинолин (THK-5157),
2-(4-амино-3-фторфенил)-6-диметиламинохинолин (THK-5128),
2-[4-(амино)-3-[(3-фтор-2-гидрокси)пропокси]фенил]-6-метиламинохинолин (THK-5147),
2-[3-(3-фтор-2-гидрокси-1,1-диметил)пропокси]-4-(диметиламино)фенил]-6-диметиламинохинолин (THK-5148),
6-амино-2-[4-(амино)-3-[(3-фтор-2-гидрокси)пропокси]фенил]хинолин (THK-5155),
2-[3-[(3-фтор-2-гидрокси)пропокси]-4-(диметиламино)фенил]-6-диметиламинохинолин (THK-5156),
2-[3-[(3-фтор-2-гидрокси)пропокси]-4-(метиламино)фенил]-6-метиламинохинолин (THK-5158),
2-[4-(амино)-3-[(3-фтор-2-гидрокси)пропокси]фенил]-6-диметиламинохинолин (THK-5159),
6-амино-2-[3-[(3-фтор-2-гидрокси)пропокси]-4-(диметиламино)фенил]хинолин (THK-5160),
2-[3-[(3-фтор-2-гидрокси)пропокси]-4-(диметиламино)фенил]-6-метиламинохинолин (THK-5161),
2-[3-[2-[2-(2-фторэтокси)этокси]этокси-4-(метиламино)фенил]-6-диметиламинохинолин (THK-5162),
2-[3-[(3-фтор-2-гидрокси)пропокси]-4-(метиламино)фенил]-6-диметиламинохинолин (THK-5164),
6-амино-2-[3-[(3-фтор-2-гидрокси)пропокси]-4-(метиламино)фенил]хинолин (THK-5165),
2-[3-[(3-фтор-2-гидрокси)пропокси]-2-(диметиламино)пирид-5-ил]-6-диметиламинохинолин (THK-5154),
2-[3-[(3-фтор-2-гидрокси)пропокси]-2-(диметиламино)пирид-5-ил]хинолин (THK-5166),
6-[(3-фтор-2-гидрокси)пропокси]-2-(6-фторпиридин-3-ил)хинолин (THK-5170),
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-метоксифенил)хинолин (THK-5171),
6-[(3-фтор-2-гидрокси)пропокси]-2-[4-(гидроксиметил)фенил]хинолин (THK-5172),
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-этанонфенил)хинолин (THK-5173),
6-[(3-фтор-2-гидрокси)пропокси]-2-(6-метоксипиридин-3-ил)хинолин (THK-5174),
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-этоксифенил)хинолин (THK-5175),
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-амино-3-метоксифенил)хинолин (THK-5176),
6-[(3-фтор-2-гидрокси)пропокси]-2-(бензамидо-4-ил)хинолин (THK-5179),
6-[(3-фтор-2-гидрокси)пропокси]-2-(3-аминофенил)хинолин (THK-5181 и
6-[(3-фтор-2-гидрокси)пропокси]-2-(1-метил-пиразол-4-ил)хинолин (THK-5182).
[0056]
Примеры более предпочтительного соединения формулы (I) включают:
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-диметиламинофенил)хинолин (THK-5105),
6-[(3-фтор-2-гидрокси)пропокси]-2-(4-метиламинофенил)хинолин (THK-5117) и
2-(4-этиламинофенил)-6-[(3-фтор-2-гидрокси)пропокси]хинолин (THK-5125).
[0057]
Как показано в Примерах, соединение формулы (I) высокоспецифично в отношении тау, а также имеет высокий захват мозгом. Также соединение формулы (I) представляет собой соединение, имеющее очень высокую безопасность, которое имеет низкую или не установленную остеотропность и низкую или не установленную токсичность. Соответственно, диагностика тауопатии может проводиться с использованием соединения формулы (I) в качестве зонда против тау, а также лечение и/или профилактика тауопатии может проводиться с использованием соединения формулы (I). В частности, соединение формулы (I) подходит для визуализационной диагностики тауопатии, в частности, визуализационной диагностики с использованием PET. Соответственно, становится возможным проведение точной диагностики, эффективного лечения и профилактики на ранних стадиях тауопатии, в частности, болезни Альцгеймера, с использованием соединения формулы (I).
[0058]
Конформационное заболевание представляет собой заболевание, при котором накапливается белок, имеющий специфическую β-листовую структуру, и существуют разнообразные заболевания, характеризуемые отложением нерастворимого волокнистого белка в различных внутренних органах и тканях. Эти заболевания включают болезнь Альцгеймера, прионовую болезнь, деменцию с тельцами Леви, болезнь Паркинсона, болезнь Хантингтона, спинальную и бульбарную атрофию, дентато-рубро-паллидо-луизиановую атрофию, спиномозжечковую дегенерацию, болезнь Мачадо-Джозефа, боковой амиотрофический склероз (ALS), синдром Дауна, болезнь Пика, FTDP-17 (лобно-височную деменцию и паркинсонизм, сцепленные с хромосомой 17), LNTD (деменция, связанная с лимбическими нейроволоконными сплетениями), суданофильную лейкодистрофию, амилоидоз и тому подобные.
[0059]
В настоящем изобретении конформационное заболевание предпочтительно означает заболевание (таутопатию), имеющее такой кардинальный симптом, как внутримозговое накопление белка тау. Тауопатия включает болезнь Альцгеймера, болезнь Пика, прогрессирующий супрануклеарный паралич (PSP) и тому подобные.
[0060]
Для описания соединения формулы (I'):
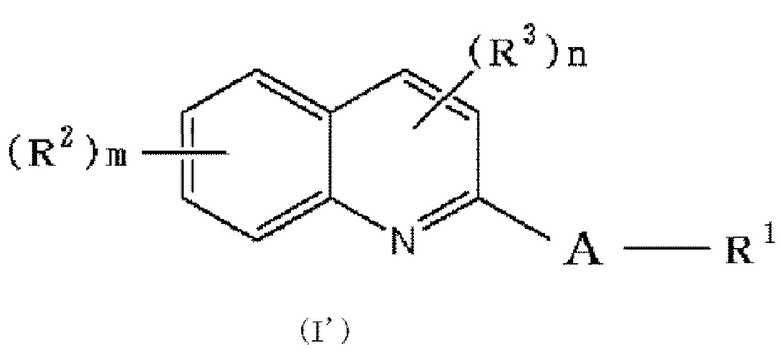 ,
,
[где соответствующие символы обозначают, как определено выше], которое представляет собой предшественник соединения формулы (I) в соответствии с настоящим изобретением, конкретнее, различные символы, используемые в формуле (I'), будут описаны путем приведения определенных примеров.
[0061]
A и R1 обозначают то же, что определено в формуле (I), как описано выше. Ra и Rb также обозначают то же, что определено в формуле (I), как описано выше.
[0062]
R2, R3 и R6 каждый независимо представляет галоген, -OH, -COOH, -SO3H, -NO2, -SH, -NRaRb (Ra и Rb независимо представляют водород или низшую алкильную группу (алкильная группа каждая независимо может быть замещена одним или более заместителями, выбранными из галогена и гидрокси группы)), низшую алкильную группу (низшая алкильная группа каждая независимо может быть замещена одним или более заместителями, выбранными из п-толуолсульфонилокси группы (тозилокси группы, TsO), метансульфонилокси группы, трифторметансульфонилокси группы или 2-тетрагидропиранилокси (OTHP), галогена и гидрокси группы), -O-низшую алкильную группу (низшая алкильная группа каждая независимо может быть замещена одним или более заместителями, выбранными из п-толуолсульфонилокси группы (тозилокси группы, TsO), метансульфонилокси группы, трифторметансульфонилокси группы или 2-тетрагидропиранилокси (OTHP), галогена, гидрокси группы и -O-низшей алкильной группы-O-низшей алкильной группы (алкильная группа каждая независимо может быть замещена галогеном)), в которой по меньшей мере один из R2, R3 и R6 обозначает -O-низшую алкильную группу (низшая алкильная группа замещена п-толуолсульфонилокси группой (тозилокси группой, TsO), метансульфонилокси группой, трифторметансульфонилокси группой или 2-тетрагидропиранилокси (OTHP), и также может быть замещена одним или более заместителями, выбранными из галогена, гидрокси группы и -O-низшей алкильной группы-O-низшей алкильной группы (алкильная группа каждая независимо может быть замещена галогеном)).
[0063]
По меньшей мере один из R2, R3 и R6 обозначает предпочтительно группу формулы:
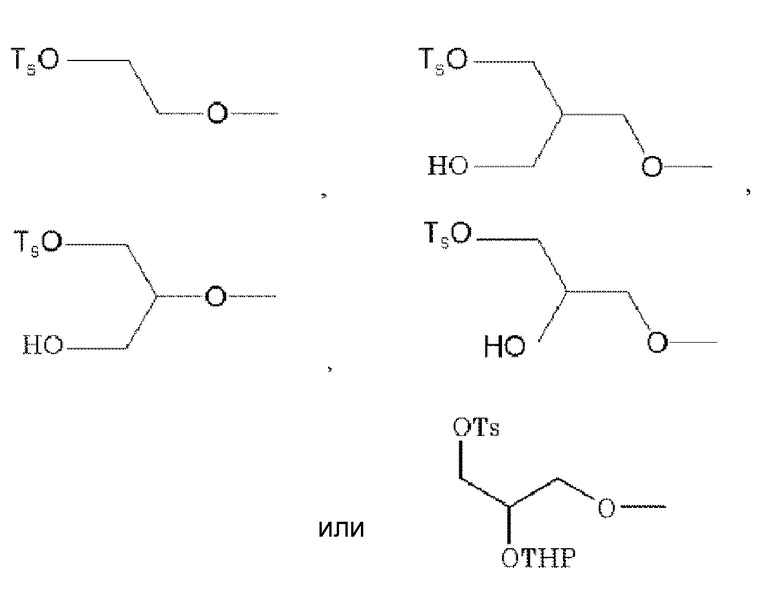 ,
,
[0064]
m обозначает целое число от 0 до 4. Предпочтительно, m=0.
[0065]
n обозначает целое число от 0 до 4. Предпочтительно, n=0.
[0066]
Примеры предпочтительного соединения формулы (I') включают:
2-(4-диэтиламинофенил)-6-[(2-гидрокси-1-тозилоксиметил)этокси]хинолин (THK-5039),
2-(4-аминофенил)-8-(2-гидрокси-1-тозилоксиметилэтокси)хинолин (THK-5041),
2-(4-диэтиламинофенил)-8-(2-гидрокси-1-тозилоксиметилэтокси)хинолин (THK-5050),
2-(4-диэтиламинофенил)-8-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолин (THK-5070),
2-(4-диэтиламинофенил)-7-(2-гидрокси-1-тозилоксиметилэтокси)хинолин (THK-5072),
2-(4-диэтиламинофенил)-7-[[(2-тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолин (THK-5073),
7-(2-гидрокси-1-тозилоксиметилэтокси)-2-(4-диметиламинофенил)хинолин (THK-5090),
7-(2-гидрокси-1-тозилоксиметилэтокси)-2-(4-метиламинофенил)хинолин (THK-5095),
2-(4-этилметиламинофенил)-7-(2-гидрокси-1-тозилоксиметилэтокси)хинолин (THK-5096),
2-(4-диэтиламинофенил)-5-(2-гидрокси-1-тозилоксиметилэтокси)хинолин (THK-5099),
2-(4-этилметиламинофенил)-7-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолин (THK-5111),
2-(4-метиламинофенил)-7-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолин (THK-5113),
2-(4-диметиламинофенил)-7-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолин (THK-5115),
2-(4-метиламинофенил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолин (THK-5119),
2-(4-диэтиламинофенил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолин (THK-5120),
2-(4-диметиламинофенил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолин (THK-5121),
2-(4-аминофенил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолин (THK-5123),
2-(4-этиламинофенил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолин (THK-5131),
2-(2-аминопирид-5-ил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолин (THK-5150),
2-(2-метиламинопирид-5-ил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолин (THK-5152),
2-(2-диметиламинопирид-5-ил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолин (THK-5135),
2-(2-диэтиламинопирид-5-ил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолин (THK-5138),
2-(2-этиламинопирид-5-ил)-6-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]хинолин (THK-5143),
2-[4-(метиламино)-3-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]фенил]-6-диметиламинохинолин (THK-5163),
6-[[2-(тетрагидро-2H-пиран-2-илокси)тозилокси]пропокси]-2-(4-метил-3,4-дигидро-2H-пиридо[3,2-b][1,4]оксазин-7-ил)хинолин (THK-5167) и
2-[4-(диметиламино)-3-[[2-(тетрагидро-2H-пиран-2-илокси)-3-тозилокси]пропокси]фенил]-6-диметиламинохинолин (THK-5168).
[0067]
Соединение формулы (I') может использоваться в качестве синтетического предшественника соединения формулы (I). Способ превращения в соединение формулы (I) из соединения формулы (I') хорошо известен среднему специалисту в данной области, и соединение формулы (I) может быть легко получено.
[0068]
Соли соединения по настоящему изобретению также включены в настоящее изобретение. Соль может быть получена в соответствии с обычным способом с использованием соединения формулы (I) или (I'), обеспечиваемого настоящим изобретением.
[0069]
В частности, когда соединение формулы (I) или (I') имеет, например, основную группу, происходящую из аминогруппы, пиридильной группы и тому подобных в молекуле, то соединение может быть превращено в соответствующую соль обработкой кислотой.
[0070]
Примеры кислотно-аддитивных солей включают галогеноводороды, такие как гидрохлорид, гидрофторид, гидробромид и гидройодид; соли неорганических кислот, такие как нитрат, перхлорат, сульфат, фосфат и карбонат; соли серной кислоты с низшими алкилами, такие как метансульфонат, трифторметансульфонат и этансульфонат; соли арилсульфоновой кислоты, такие как бензолсульфонат и п-толуолсульфонат; соли органических кислот, такие как фумарат, сукцинат, цитрат, тартрат, оксалат и малеат; и кислотно-аддитивные соли аминокислот, такие как глутамат и аспартат.
[0071]
Также, когда соединение по настоящему изобретению имеет кислотную группу, такую как карбоксильная группа в молекуле, то соединение может быть также превращено в соответствующую фармацевтически приемлемую соль обработкой основанием. Примеры основно-аддитивной соли включают соли щелочных металлов, таких как натрий и калий; соли щелочноземельных металлов, таких как кальций и магний; соли органических оснований, такие как соли аммония, гуанидин, триэтиламин и дициклогексиламин.
[0072]
Кроме того, соединение по настоящему изобретению может присутствовать в виде свободного соединения или произвольно гидрата или сольвата его соли.
[0073]
В зависимости от выбора исходных материалов и способов, соединения по настоящему изобретению могут существовать в виде одной формы в возможных изомерах или их смесях, например, по существу чистых геометрических (цис- или транс-) изомеров, оптических изомеров (энантиомеров, антиподов), рацемических форм и их смесей. Указанные выше возможные изомеры или их смеси включены в объем настоящего изобретения.
[0074]
Все получаемые смеси изомеров могут быть разделены на чистые геометрические или оптические изомеры, диастереомеры или рацемические формы на основании физико-химического различия компонента, например, хроматографией или фракционной кристаллизацией.
[0075]
Все из полученных рацемических форм конечного продукта или промежуточного соединения могут быть оптически разделены на оптические антиподы известным способом, например, отделяют соль диастереоизомера, полученную из оптически активного кислотного или основного соединения, и отделяется каждое оптически активное кислотное или основное соединение. Продукты из рацемических форм могут быть также разделены хиральной хроматографией, например, высокоэффективной жидкостной хроматографией с использованием хирального адсорбента.
[0076]
В исходных соединениях и предшественниках, которые превращаются в соединения по настоящему изобретению способом по настоящему описанию, существующие функциональные группы, такие как амино, тиольные, карбоксильные и гидрокси группы, могут быть необязательно защищены общей обычной защитной группой в препаративной органической химии. Замещенные таким образом амино, тиольные, карбоксильные и гидрокси группы могут быть превращены в свободные амино, тиольные, карбоксильные и гидрокси группы в мягких условиях, не вызывающих разрушения молекулярной сети или другой нежелательной небольшой реакции.
[0077]
Защитная группа вставляется так, чтобы защитить функциональную группу от нежелательной реакции с реакционным компонентом в условиях, используемых для выполнения желательного химического превращения. Необходимость и отбор защитной группы для определенной реакции известны специалистам в данной области и зависят от свойств функциональной группы, подлежащей защите (гидрокси группы, аминогруппы и т.д.), структуры и устойчивости молекулы с заместителем, составляющим его часть, и условий реакции. Примеры защитной группы включают OT, OTHP, метоксиметил и OAc. Защитная группа представляет собой предпочтительно защитную группу, которая удаляется в кислотных условиях.
[0078]
При диагностике тауопатии, соединение по настоящему изобретению может использоваться в качестве зонда без мечения. Например, присутствие или отсутствие подлежащей окрашиванию части может исследоваться контактом соединения по настоящему изобретению с образцом биопсии ткани. Однако обычно используется меченое соединение по настоящему изобретению в качестве зонда для диагностики тауопатии. Примеры метки включают флуоресцентное вещество, аффинное вещество, ферментный субстрат, радиоактивный нуклид и тому подобные. Зонд, меченный радиоактивным нуклидом, обычно используется при визуализирующей диагностике тауопатии. Возможно мечение соединения по настоящему изобретению различными радиоактивными нуклидами способами, которые хорошо известны в данной области. Например, 3H, 14C, 35S, 131I и тому подобные представляют собой радиоактивные нуклиды, которые использовались в течение длительного времени, и часто используются in vivo. Общими требованиями к визуализирующим диагностическим зондам и средствам для их выявления являются обеспечение возможности постановки диагноза in vivo, вызов меньшего вреда для пациентов (в частности, быть не инвазивными), наличие высокой чувствительности выявления, наличие соответствующего периода полувыведения (для наличия соответствующего периода времени для получения меченых зондов и для диагностики) и тому подобные. Соответственно, недавно была тенденция использования позитронно-эмиссионной томографии (PET) с использованием рентгеновского излучения, проявляющей высокую чувствительность и проницаемость через материалы, или компьютерной томографии (SPECT) с испускающими рентгеновское излучение нуклидами. Среди них PET, которая выявляет два рентгеновских луча, испускаемых в противоположных направлениях из испускающего позитроны нуклеотида, посредством одновременного подсчета парой детекторов, обеспечивает информацию, которая очень значима при разделении и количественной оценке, и, таким образом, предпочтительна. Для SPECT соединение по настоящему изобретению может быть мечено испускающим рентгеновское излучение нуклидом, таким как 99mTc, 111In, 67Ga, 201Tl, 123I, 133Xe и тому подобные. 99mTc и 123I часто используются для SPECT. Для PET соединение по настоящему изобретению может быть мечено испускающим позитроны нуклидом, таким как 11C, 13N, 15O, 18F, 62Cu, 64Cu, 68Ga, 76Br и тому подобные. Среди испускающих позитроны нуклидов предпочтительны 11C, 13N, 15O и 18F, более предпочтительны 18F и 11C, особенно предпочтителен 18F, с точки зрения наличия соответствующего периода полувыведения, легкости мечения и тому подобного. Хотя положение метящего соединения по настоящему изобретению с испускающим излучением нуклидом, таким как нуклиды, испускающие позитроны, или нуклид, испускающий рентгеновское излучение, может представлять собой любое положение, мечение может предпочтительно проводиться в алкильной группе и на фенильном кольце в соединении. Такие меченые соединения по настоящему изобретению также включены в настоящее изобретение. Например, когда соединение по настоящему изобретению мечено 18F, любое положение боковой цепи может быть мечено 18F, или водород на кольце может быть замещен 18F. Например, водород, содержащийся в любом из алкильных заместителей, может быть замещен 18F. Также, когда соединение по настоящему изобретению мечено 11C, углерод, содержащийся в любом из алкильных заместителей в боковой цепи, может быть замещен 11C. Хотя для среднего специалиста в данной области очевидно, что m 99mTc обозначает ядерный изомер в квази-стабильном состоянии.
[0079]
Радионуклиды, используемые в соединениях в соответствии с настоящим изобретением, генерируются на приборе, именуемом циклотроном или генератором. Средний специалист в данной области может выбрать способы и приборы для получения, в зависимости от подлежащих получению нуклидов. Полученные таким образом нуклиды могут использоваться для мечения соединений по настоящему изобретению.
[0080]
Способы получения меченых соединений, которые были мечены этими радионуклидами, хорошо известны в данной области. Типичные способы включают способы химического синтеза, обмена изотопов и биосинтеза. Способы химического синтеза традиционно и широко использовались и являются по существу такими же, как обычные способы химического синтеза, за исключением того, что используются радиоактивные исходные материалы. Различные нуклиды вводятся в соединения этими химическими способами. Способы обмена изотопов представляют собой способы, которыми 3H, 35S, 125I или тому подобные, содержащиеся в соединении простой структуры, переносятся в соединение, имеющее сложную структуру, посредством этого получая соединение, имеющее сложную структуру, которое было мечено этими нуклидами. Способы биосинтеза представляют собой способы, которыми соединение, меченное 14C, 35S или тому подобными изотопами, вводится в клетки, такие как микроорганизмы, для получения его метаболитов, имеющих эти нуклиды, введенные в них.
[0081]
В отношении положения мечения, аналогично обычному синтезу, могут быть спроектированы синтетические схемы, в зависимости от цели, так что метка может быть введена в желательное положение. Такое проектирование хорошо известно среднему специалисту в данной области.
[0082]
При использовании испускающих позитроны нуклидов, таких как 11C, 13N, 15O и 18F, которые имеют относительно короткие периоды полувыведения, например, возможно также генерирование желательного нуклида из циклотрона очень маленького размера, помещенного в такое учреждение, как больница, который в свою очередь используется для мечения желательного соединения в его желательном положении любым из описанных выше способов, с последующим проведением немедленной диагностики, обследования, лечения или тому подобного.
[0083]
Эти способы хорошо известны среднему специалисту в данной области и обеспечивают возможность проведения мечения введением желательного нуклида в соединение по настоящему изобретению в его желательное положение.
[0084]
Соединение по настоящему изобретению, которое было мечено, может вводиться пациентам местно или системно. Пути введения включают внутридермальные, внутрибрюшинные, внутривенные, внутриартериальные инъекции или инфузии в спинальную жидкость и тому подобные, и могут быть выбраны в зависимости от таких факторов, как тип заболевания, используемый нуклид, используемое соединение, состояние пациента, подлежащий исследованию участок. Подлежащий исследованию участок может обследоваться такими средствами, как PET, SPECT, введением зонда по настоящему изобретению, с последующим прохождением достаточного периода времени для обеспечения возможности его связывания с белком тау и распада. Эти процедуры могут быть соответствующим образом выбраны в зависимости от таких факторов, как тип заболевания, используемый нуклид, используемое соединение, состояние пациента, подлежащий обследованию участок.
[0085]
Доза соединения по настоящему изобретению, которое было мечено радионуклидом, варьируется в зависимости от типа заболевания, используемого нуклида, используемого соединения, возраста, физического состояния и пола пациента, степени заболевания, подлежащего обследованию участка и тому подобного. В частности, должно уделяться достаточное внимание дозам, воздействующим на пациента. Например, количество радиоактивности соединения, меченного испускающим позитроны нуклидом, таким как 11C, 13N, 15O и 18F, по настоящему изобретению, обычно находится в пределах диапазона от 3,7 мегабеккерелей до 3,7 гигабеккерелей, и предпочтительно от 18 мегабеккерелей до 740 мегабеккерелей.
[0086]
Соединение по настоящему изобретению или его соль или сольват подходит для использования в способе лечения тауопатии, способе диагностики, композиции для лечения, композиции для диагностики, наборе для диагностики, применения для получения этих композиций и наборов и других видов применения, которые будут описаны ниже. Предпочтительны соединения или их соли или сольваты, приведенные в качестве примеров в приведенном выше описании о соединениях формул (I)-(VI), и особенно предпочтительны те, которые включены в соединение формулы (I) или его соль или сольват. Среди соединений по настоящему изобретению, соединения, имеющие в качестве R2, R3 или R6 в формуле (I)
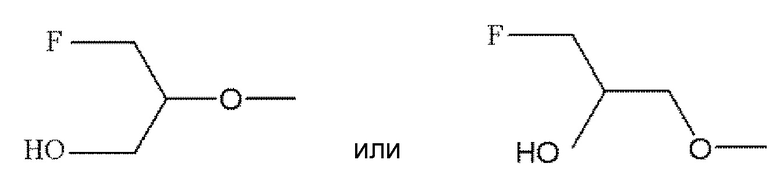 ,
,
подходят для введения в организм человека ввиду значительно меньшего или очень слабого накопления в костной ткани.
[0087]
Настоящее изобретение относится к композиции для визуализирующей диагностики тауопатии, содержащей соединение по настоящему изобретению. Композиция по настоящему изобретению содержит соединение по настоящему изобретению и фармацевтически приемлемый носитель. Предпочтительно, чтобы соединение по настоящему изобретению в композиции было меченым. Хотя возможны различные способы мечения, как описано выше, мечение радионуклидами (в частности, испускающими позитроны нуклидами, такими как 11C, 13N, 15O и 18F для PET) желательно для видов применения при визуализирующей диагностике in vivo. По назначению, предпочтительно, чтобы форма композиции по настоящему изобретению представляла собой ту, которая обеспечивает возможность инъекции или инфузии. Соответственно, фармацевтически приемлемый носитель представляет предпочтительно жидкость, и ее примеры включают без ограничения водные растворители, такие как буфер фосфата калия, физиологический солевой раствор, раствор Рингера и дистиллированная вода; и неводные растворители, такие как полиэтиленгликоль, растительное масло, этанол, глицерин, диметилсульфоксид и пропиленгликоль. Соотношение смешивания носителя к соединению по настоящему изобретению может быть соответствующим образом выбрано в зависимости от участка применения, средства выявления и тому подобного, и обычно составляет от 100000:1 до 2:1, и предпочтительно от 10000:1 до 10:1. Композиция по настоящему изобретению может дополнительно содержать известные антимикробные средства (например, антимикробное лекарственное средство и т.д.), местные анестетики (например, прокаина гидрохлорид и т.д.), буферы (например, Трис-гидрохлоридный буфер и буфер HEPES и т.д.), осмолиты (например, глюкозу, сорбит, хлорид натрия и т.д.) и тому подобные.
[0088]
Кроме того, настоящее изобретение относится к набору для визуализирующей диагностики тауопатии, содержащему соединение по настоящему изобретению в качестве активного ингредиента. Обычно набор представляет собой упаковку, в которой каждый из компонентов, такой как соединение по настоящему изобретению, растворитель для растворения соединения, буфер, осморегуляторный агент, антимикробное средство, местный анестетик, упакован отдельно в соответствующие контейнеры, или некоторые из компонентов упакованы вместе в соответствующие контейнеры. Соединение по настоящему изобретению может быть немеченым или меченым. Соединение по настоящему изобретению, если оно не меченое, может метиться перед использованием обычными способами, как описано выше. Кроме того, соединение по настоящему изобретению может быть представлено в виде твердого вещества, такого как лиофилизированный порошок, или в растворе в соответствующих растворителях. Растворители могут быть аналогичны носителям, используемым в указанной выше композиции по настоящему изобретению. Каждый из компонентов, таких как буфер, осморегулирующий агент, антимикробное средство, местный анестетик, также может быть аналогичен тем, которые используются в описанной выше композиции по настоящему изобретению. Хотя в соответствии с целесообразностью могут быть выбраны различные контейнеры, они могут иметь формы, подходящие для проведения введения метки в соединение по настоящему изобретению, или изготовлены из экранирующих свет материалов, в зависимости от природы соединений, или принимать такую форму, как флаконы или шприцы, с тем, чтобы быть удобными для введения пациенту. Набор также содержит при целесообразности инструменты, необходимые для диагностики, например, шприцы, инфузионный комплект, или устройство для использования в аппарате PET или SPECT. Набор обычно имеет приложенные к нему инструкции.
[0089]
Кроме того, соединения по настоящему изобретению специфически связаны с белком тау, и, таким образом, соединения по настоящему изобретению могут также использоваться, например, для выявления и количественного определения белка тау с мечением или без него путем обеспечения контакта с образцами пробы in vitro. Например, соединение по настоящему изобретению может использоваться для окрашивания белка тау в микроскопических образцах, для калориметрического определения белка тау в образцах, или для количественного определения белка тау с использованием сцинтилляционного счетчика. Получение микроскопического образца и окрашивание с использованием соединения по настоящему изобретению может проводиться обычным способом, известным среднему специалисту в данной области.
[0090]
Как описано выше, соединения по настоящему изобретению высокоспецифичны в отношении белка тау. Поэтому соединения по настоящему изобретению полезны, например, для исследований заболеваний с накоплением белка тау или при их диагностике до и после смерти, и могут использоваться, например, в качестве агентов для окрашивания нейроволоконных сплетений в срезах мозга пациентов с болезнью Альцгеймера. Окрашивание образцов, например, срезов мозга с использованием соединения по настоящему изобретению, может проводиться обычным способом, известным среднему специалисту в данной области.
[0091]
Как описано выше, среди соединений по настоящему изобретению, соединения, имеющие в качестве R2, R3 или R6 в формуле (I):
 ,
,
могут вызвать значительно меньшее или очень небольшое накопление в костной ткани. Соответственно, такие соединения по настоящему изобретению представляют собой не только достаточно безопасные зонды для диагностики тауопатии, но также проявляют высокую безопасность даже при использовании в качестве лекарственных средств или профилактических средств, описанных ниже.
[0092]
Соответственно, настоящее изобретение направлено на композицию для окрашивания белка амилоида β, в частности, тау в образце, содержащую соединение по настоящему изобретению или его фармацевтически приемлемую соль или сольват, и набор для окрашивания белка амилоида β, в частности, тау в образце, содержащий соединение по настоящему изобретению или его фармацевтически приемлемую соль или сольват в качестве активных ингредиентов. Кроме того, настоящее изобретение направлено на способ окрашивания белка амилоида β, в частности, тау в образце, причем способ включает использование соединения по настоящему изобретению или его фармацевтически приемлемой соли или сольвата. Образцы, подходящие для указанного выше окрашивания, представляют собой срезы мозга.
[0093]
Как описано выше, было обнаружено, что нейротоксичность присутствует в белке амилоида β или тау β-листовой структуры. Считается, что соединение по настоящему изобретению специфически связывается с белком амилоида β β-листовой структуры, в частности, тау, и, таким образом, нейротоксичность ингибируется. Соответственно, считается, что соединение по настоящему изобретению служит в качестве лекарственного средства или профилактического средства в отношении причин заболевания, в частности тауопатии, например, болезни Альцгеймера, поскольку сам белок имеет β-листовую структуру.
[0094]
Соответственно, настоящее изобретение относится к:
способу лечения и/или профилактики заболеваний с накоплением белка амилоида β, в частности, тауопатии, причем способ включает введение соединения формулы (I) или его соли или сольвата;
способу диагностики заболеваний с накоплением белка амилоида β, в частности, тауопатии, причем способ включает использование соединения формулы (I) или его соли или сольвата; и
к применению соединения формулы (I) или его соли или сольвата для получения композиции или набора для лечения, профилактики или диагностики заболеваний с накоплением белка амилоида β, в частности, тауопатии.
[0095]
Формы таких фармацевтических композиций конкретно не ограничиваются, но предпочтительны жидкие препаративные формы, в частности, препаративные формы для инъекций. Такие препаративные формы для инъекций могут инфузироваться непосредственно в мозг, или, альтернативно, указанные выше фармацевтические композиции могут составляться для внутривенной инъекции или капельной инфузии и вводиться, поскольку соединения по настоящему изобретению имеют высокую проницаемость, через гематоэнцефалический барьер, как показано в Примерах. Такие жидкие препаративные формы могут быть получены способами, хорошо известными в данной области. Растворы могут быть получены, например, растворением соединения по настоящему изобретению в соответствующем носителе, воде для инъекций, физиологическом солевом растворе, растворе Рингера или тому подобном, стерилизацией раствора через фильтр или тому подобное, и затем заполнением стерилизованного раствора в соответствующие контейнеры, например, флаконы или ампулы. Растворы могут быть также лиофилизироваться, и при использовании их влагосодержание может восстанавливаться соответствующим носителем. Суспензии могут быть получены, например, стерилизацией соединения по настоящему изобретению, например, воздействием этиленоксидом, и затем суспендированием его в стерилизованном жидком носителе.
[0096]
Когда такая фармацевтическая композиция используется в жидкой препаративной форме, в частности, препаративной форме для инъекций, то раствор для инъекций может быть получен добавлением солюбилизирующего агента к производному хинолина в соответствии с настоящим изобретением.
[0097]
Возможно использование в качестве солюбилизирующего агента неионных поверхностно-активных веществ, катионных поверхностно-активных веществ, амфотерических поверхностно-активных веществ и тому подобных, используемых в данной области. Среди них предпочтительным является Полисорбат 80, полиэтиленгликоль, этанол или пропиленгликоль, и более предпочтительным является Полисорбат 80.
[0098]
Количество соединения по настоящему изобретению, подлежащее введению человеку при указанном выше способе лечения, способе профилактики и применении варьируется в зависимости от состояния, пола, возраста, массы тела пациента и тому подобного, и в целом находится в диапазоне от 0,1 мг до 1 г, предпочтительно от 1 мг до 100 мг, предпочтительнее от 5 мг до 50 мг, в день для взрослых людей с массой тела 70 кг. Возможно проведение лечения такой дозой в течение определенного периода времени с последующим увеличением или уменьшением дозы, в соответствии с исходом.
[0099]
Кроме того, соединение по настоящему изобретению или его фармацевтически приемлемая соль или сольват может также применяться в качестве зонда для диагностики конформационного заболевания, в частности тауопатии, предпочтительно визуализирующего диагностического зонда, меченного радиоактивным нуклидом. Кроме того, соединения по настоящему изобретению эффективны для лечения и/или профилактики конформационного заболевания, в частности, тауопатии.
[0100]
Соответственно, настоящее изобретение также направлено на:
соединение по настоящему изобретению, применяемое в качестве визуализирующего диагностического зонда конформационного заболевания, в частности, тауопатии, или его соль или сольват;
композицию или набор для визуализирующей диагностики конформационного заболевания, в частности, тауопатии, включающие соединение по настоящему изобретению или его соль или сольват;
фармацевтическую композицию для профилактики и/или лечения конформационного заболевания, в частности, тауопатии, включающую соединение по настоящему изобретению или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель;
способ диагностики конформационного заболевания, в частности, тауопатии, причем способ включает применение соединения по настоящему изобретению или его фармацевтически приемлемой соли или сольвата;
применение соединения по настоящему изобретению или его фармацевтически приемлемой соли или сольвата для диагностики конформационного заболевания, в частности, тауопатии;
способ профилактики и/или лечения конформационного заболевания, в частности, тауопатии, причем способ включает введение пациенту соединения по настоящему изобретению или его фармацевтически приемлемой соли или сольвата;
применение соединения по настоящему изобретению или его фармацевтически приемлемой соли или сольвата для профилактики и/или лечения конформационного заболевания, в частности, тауопатии; и
применение соединения по настоящему изобретению при получении фармацевтической композиции для профилактики и/или лечения конформационного заболевания, в частности, тауопатии.
[0101]
Доза соединения по настоящему изобретению, подлежащая введению человеку-пациенту при указанных выше способах лечения и способах профилактики, составляет, как описано выше.
[0102]
Соединения по настоящему изобретению распознают нейроволоконные сплетения, содержащие избыточно фосфорилированный белок тау в качестве составляющего ингредиента, и, таким образом, соединения могут использоваться в качестве зонда для выявления нейроволоконных сплетений, или в качестве агента для окрашивания нейроволоконных сплетений. Соответственно, настоящее изобретение также относится к применению соединения по настоящему изобретению или его соли или сольвата в качестве зонда для выявления нейроволоконных сплетений, в частности, зонда для диагностики изображений. Примеры соединений по настоящему изобретению, которые предпочтительны для окрашивания нейроволоконных сплетений, включают THK-5004, THK-5035, THK-5038, THK-5051, THK-5058, THK-5064, THK-5065, THK-5066, THK-5071, THK-5077, THK-5078, THK-5105, THK-5106, THK-5107, THK-5112, THK-5116, THK-5117, THK-5122 и тому подобные.
[0103]
Соответственно, настоящее изобретение относится к композиции для выявления или окрашивания нейроволоконных сплетений, содержащей соединение по настоящему изобретению или его соль или сольват;
к набору для выявления или окрашивания нейроволоконных сплетений, содержащему соединение по настоящему изобретению или его соль или сольват;
способу выявления или окрашивания нейроволоконных сплетений, который включает применение соединения по настоящему изобретению или его соли или сольвата; и
применению соединения по настоящему изобретению или его соли или сольвата для получения композиции для выявления или окрашивания нейроволоконных сплетений.
[0104]
Способы, известные среднему специалисту в данной области, могут применяться в способах получения и окрашивания образца пробы при выявлении или окрашивании указанных выше нейроволоконных сплетений.
[0105]
Кроме того, настоящее изобретение относится к набору с получением соединения по настоящему изобретению или его фармацевтически приемлемой соли или сольвата, причем набор включает соединение по настоящему изобретению или его фармацевтически приемлемую соль или сольват, метящий агент и, необязательно, инструкции по мечению соединения. Метящий агент представляет собой, например, радиоактивный нуклид или испускающий позитроны нуклид. Радиоактивный нуклид представляет собой, например, нуклид, испускающий γ-лучи. Испускающий позитроны нуклид выбран, например, из группы, состоящей из 11C, 13N, 15O, 18F, 35mCl, 76Br, 45Ti, 48V, 60Cu, 61Cu, 62Cu, 64Cu, 66Ga, 89Zr, 94mTc и 124I. Предпочтительно, испускающий позитроны нуклид представляет собой 11C или 18F. Метящий агент представляет собой агент, который подходит для мечения соединения, и он известен специалистам в данной области.
[0106]
Типичные примеры соединения формулы (I), предпочтительно используемые в настоящем изобретении, показаны ниже.
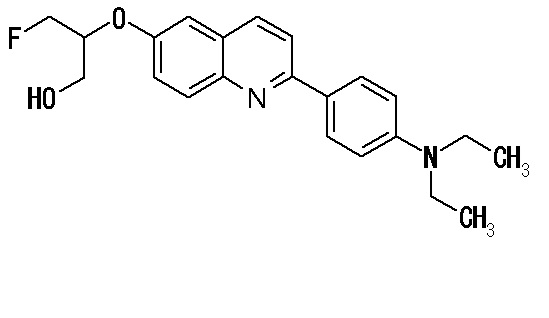
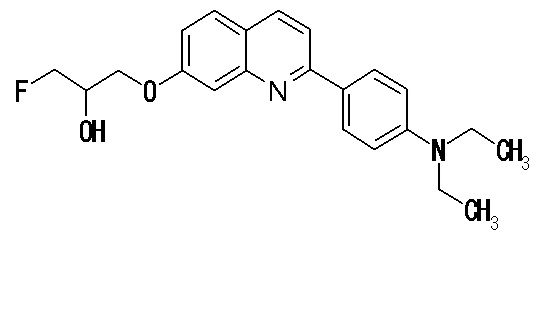
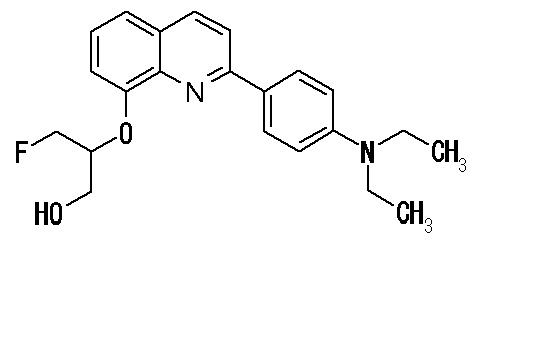
[0107]
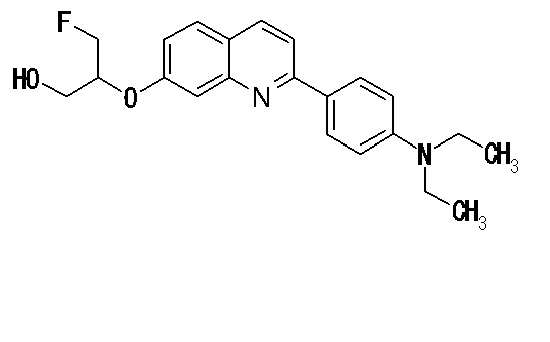
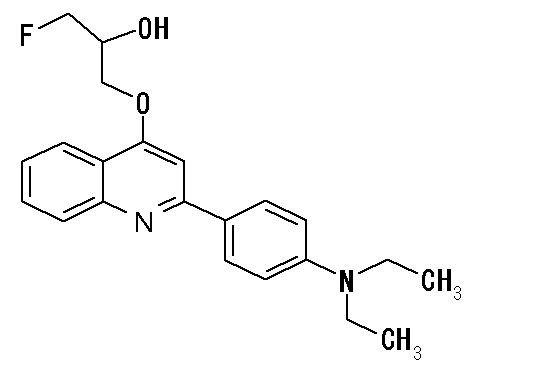
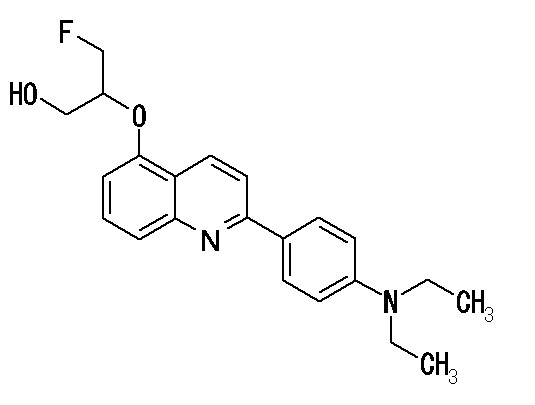
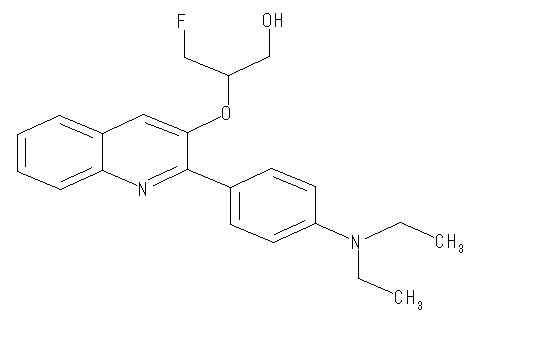
[0108]
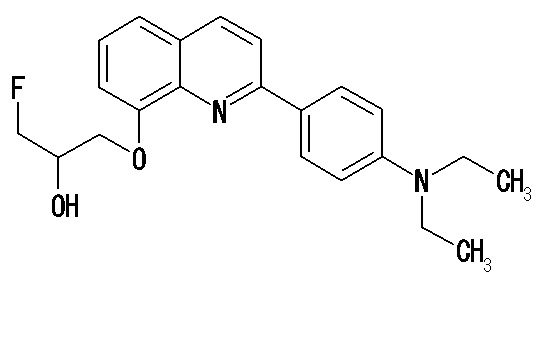
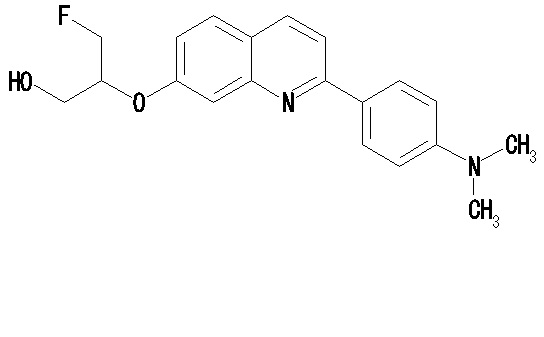
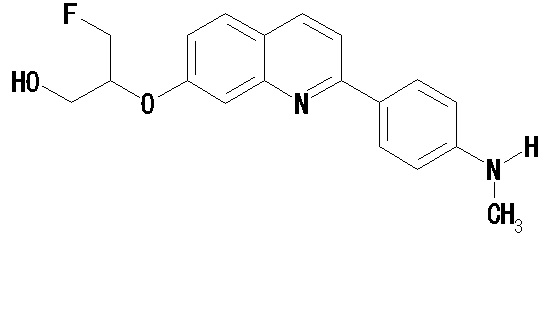
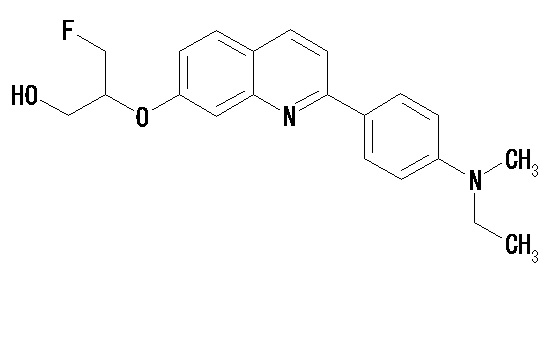
[0109]
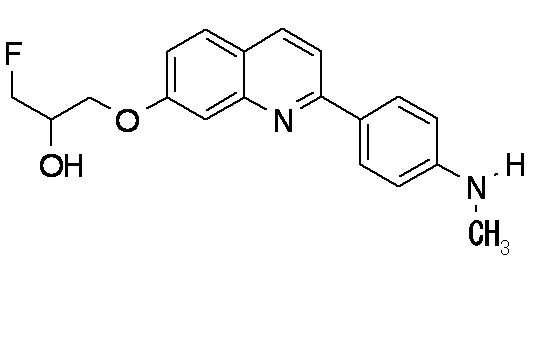
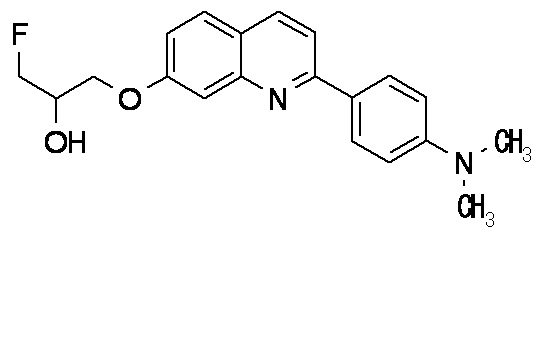
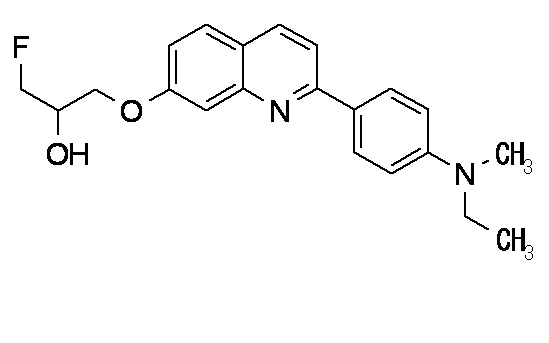
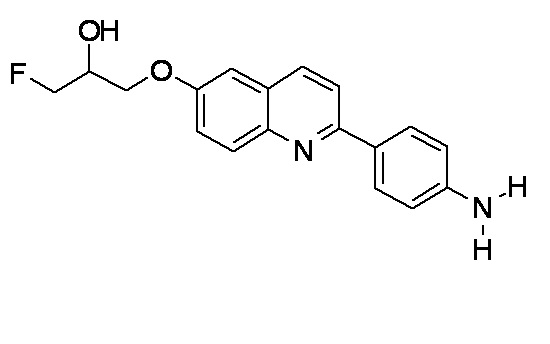
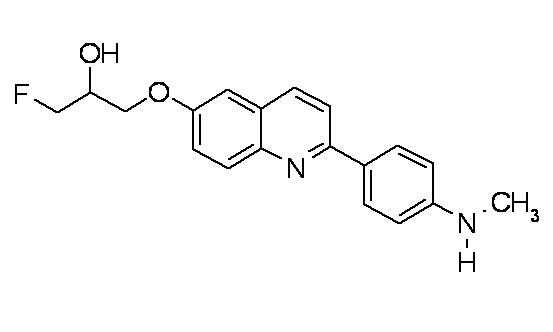
[0110]
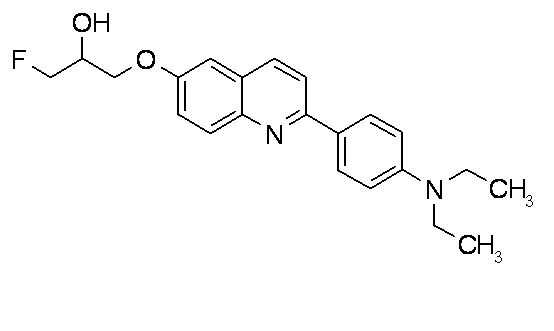
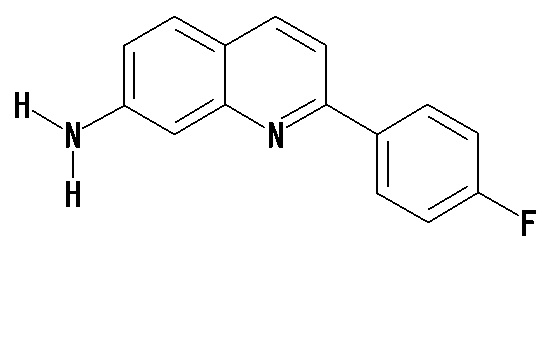
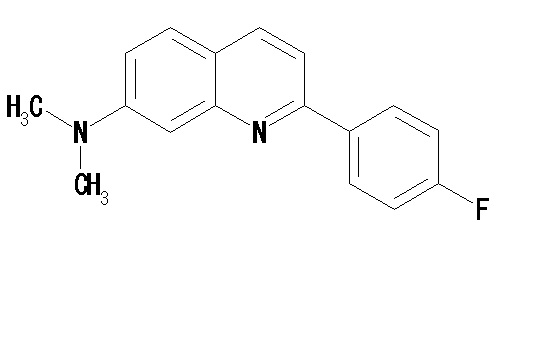
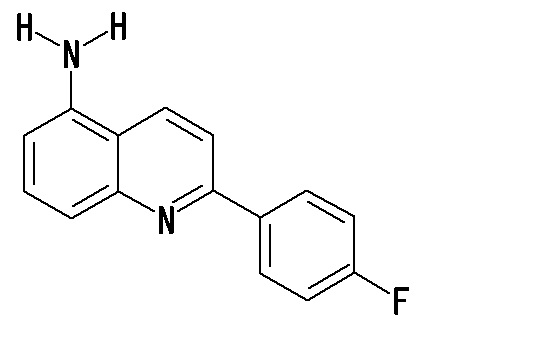
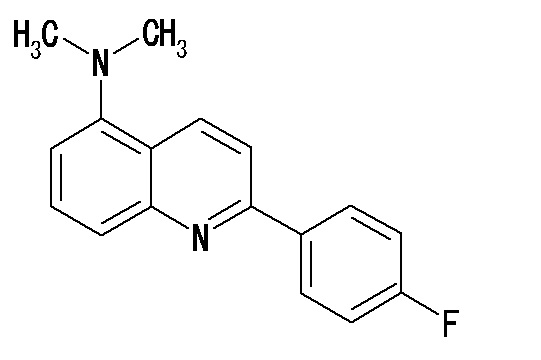
[0111]
[0112]
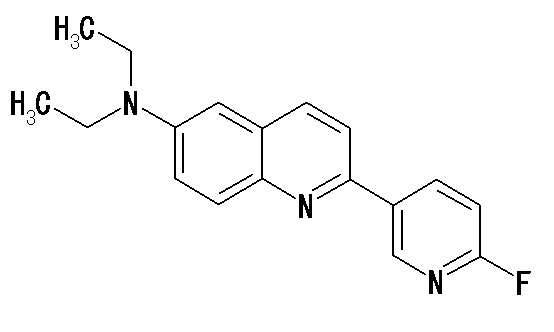
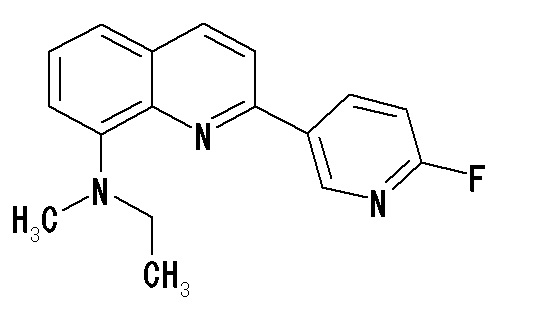
[0113]
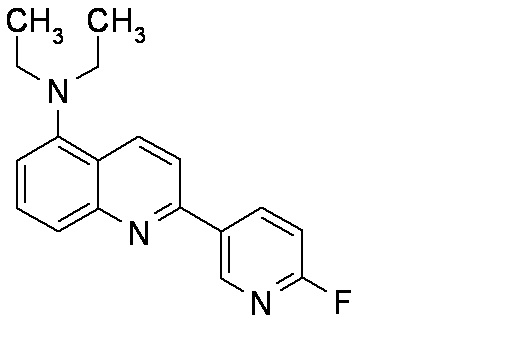
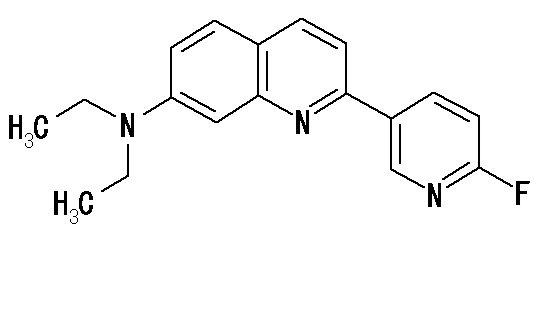
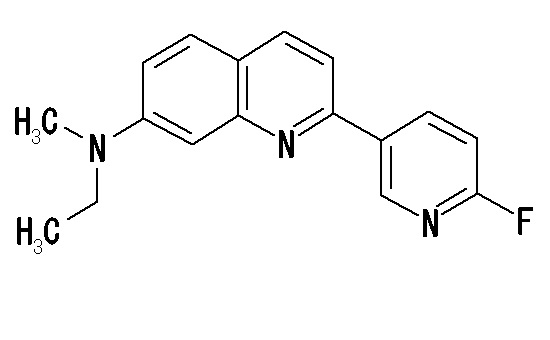
[0114]
хинолин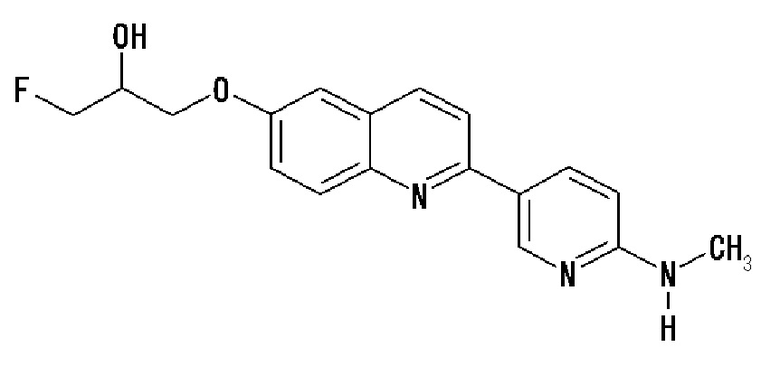
хинолин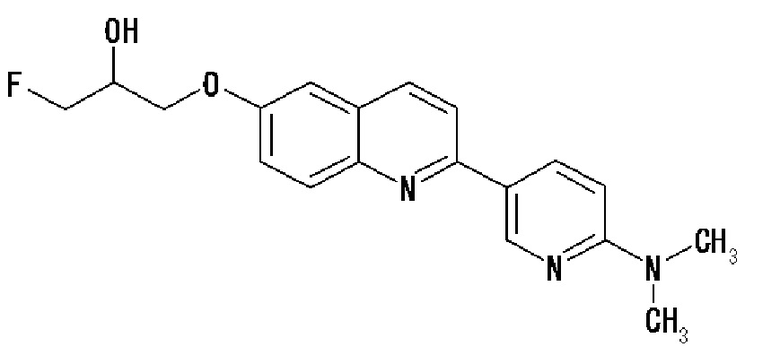
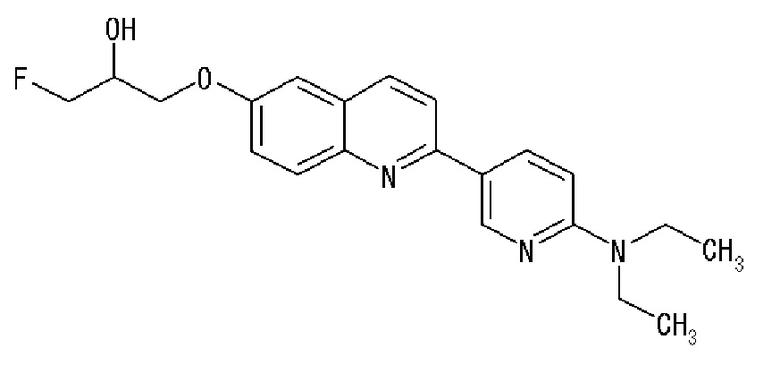
хинолин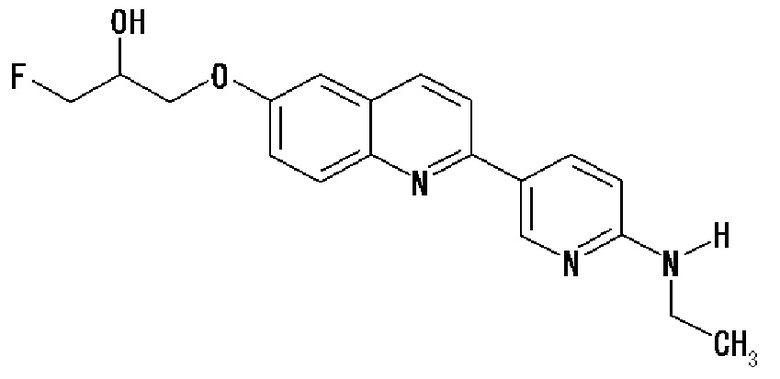
хинолин
[0115]
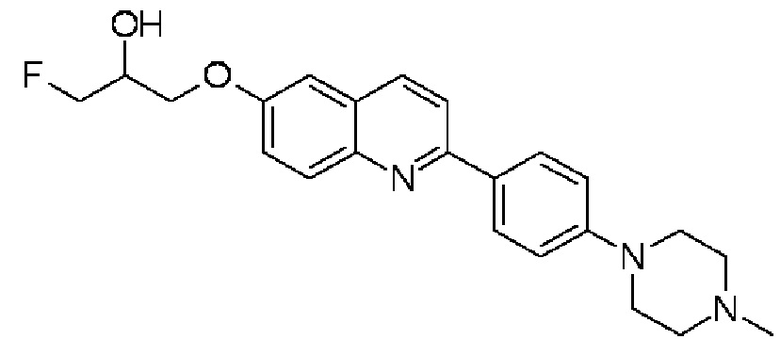
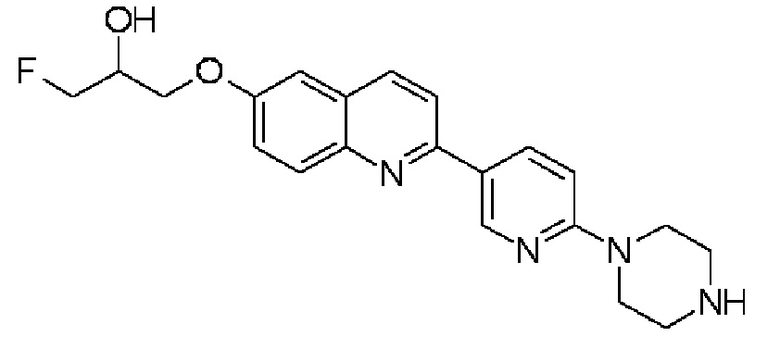
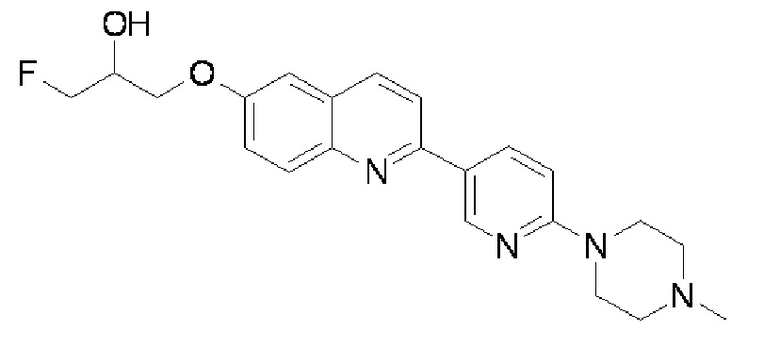
[0116]
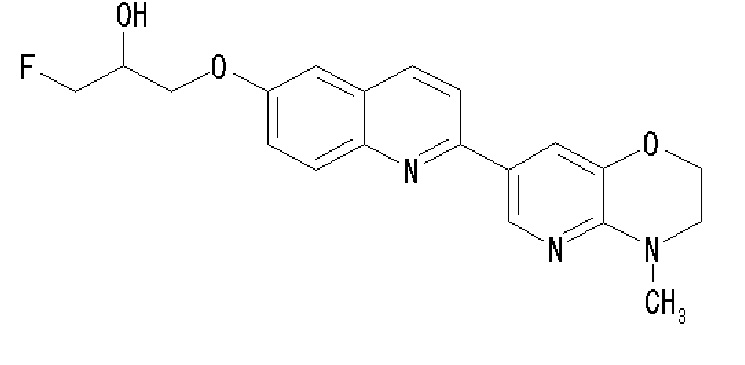
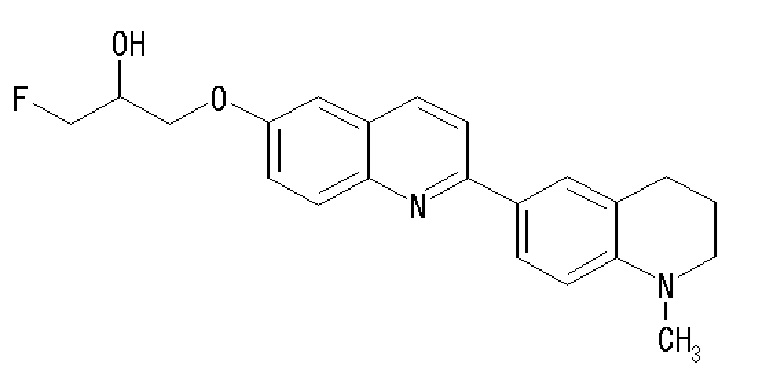
[0117]
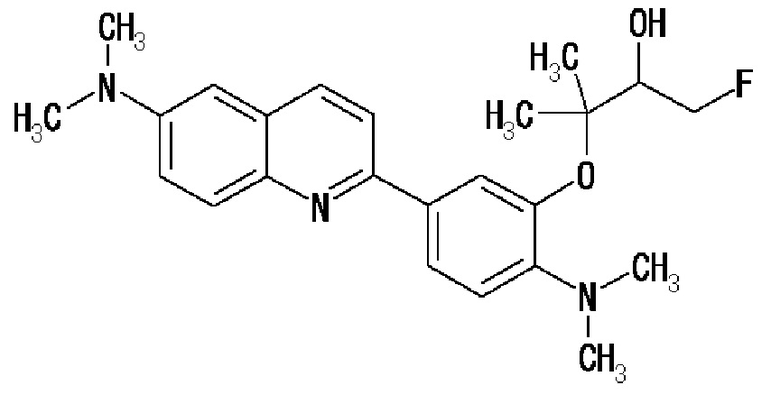
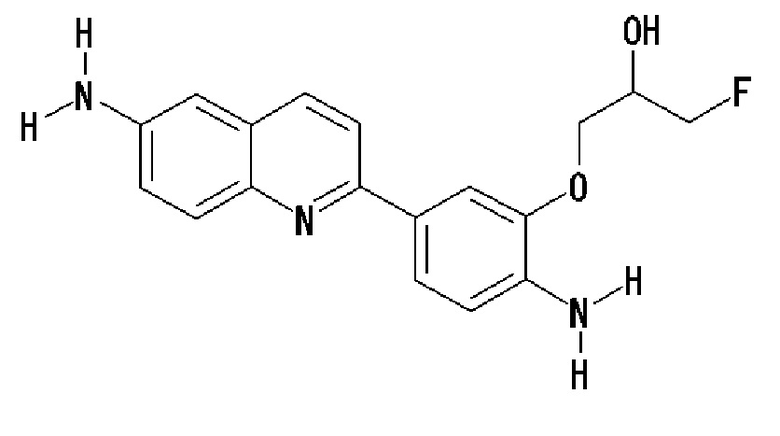
хинолин
[0118]
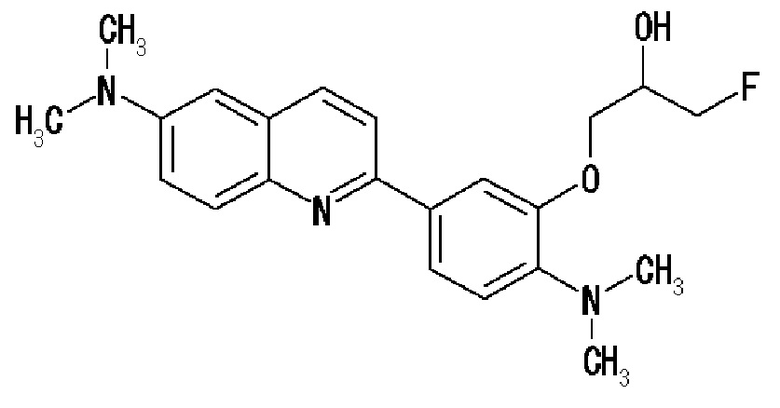

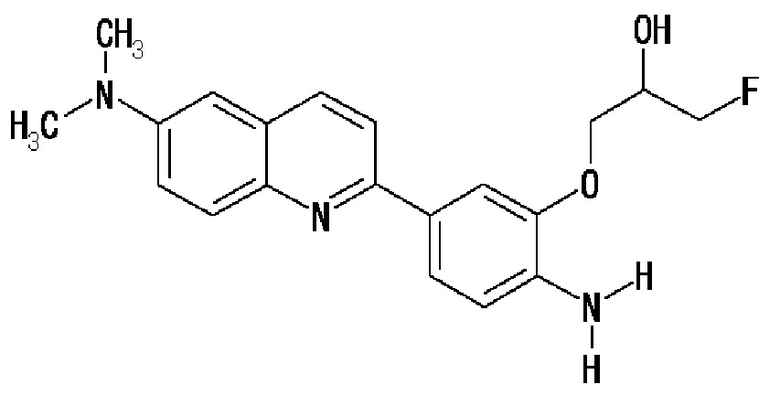
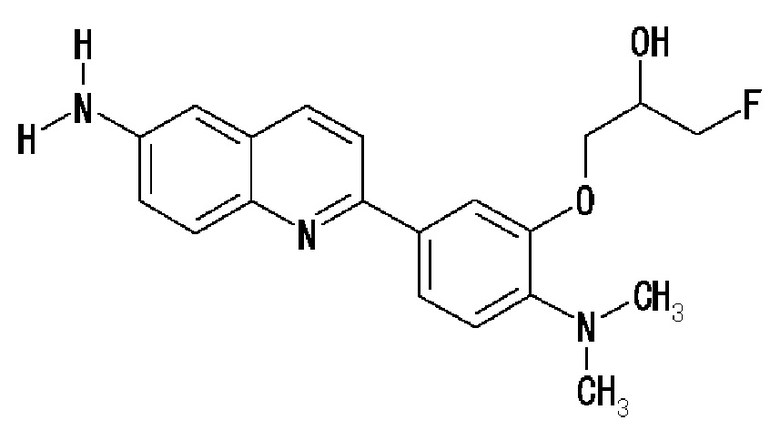
фенил]хинолин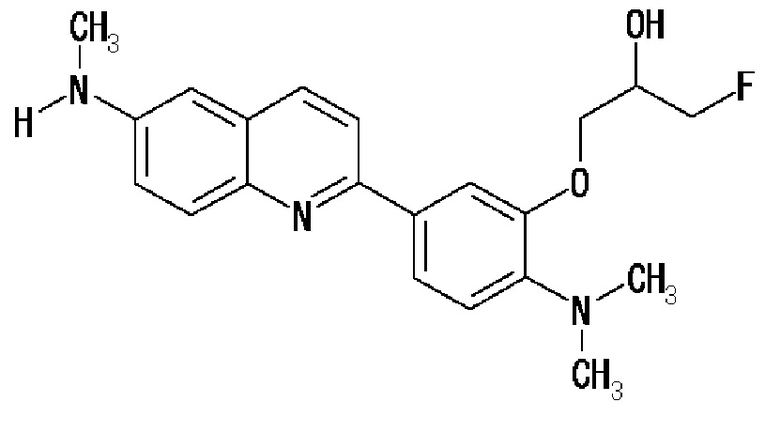
[0119]
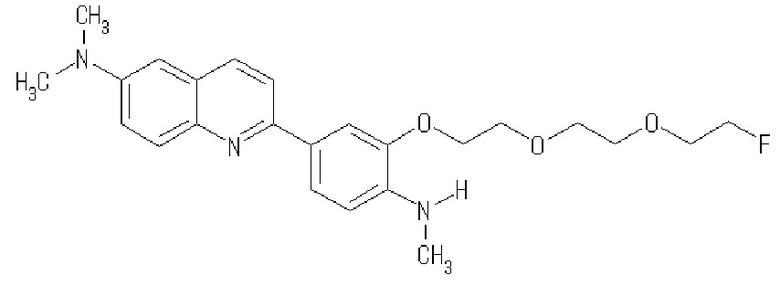
этокси-4-(метиламино)фенил]-6-диметиламинохинолин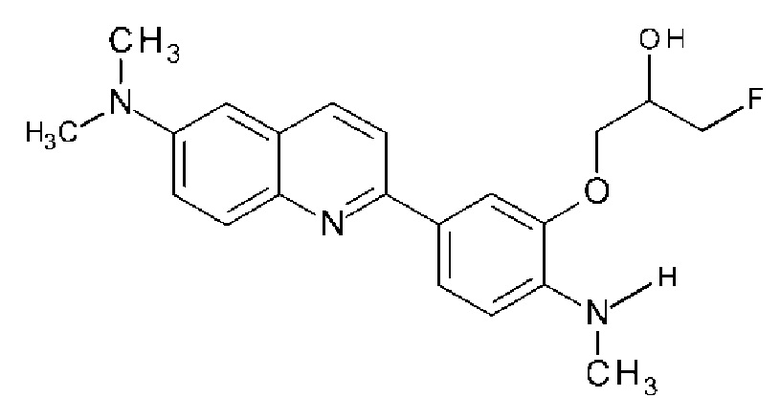
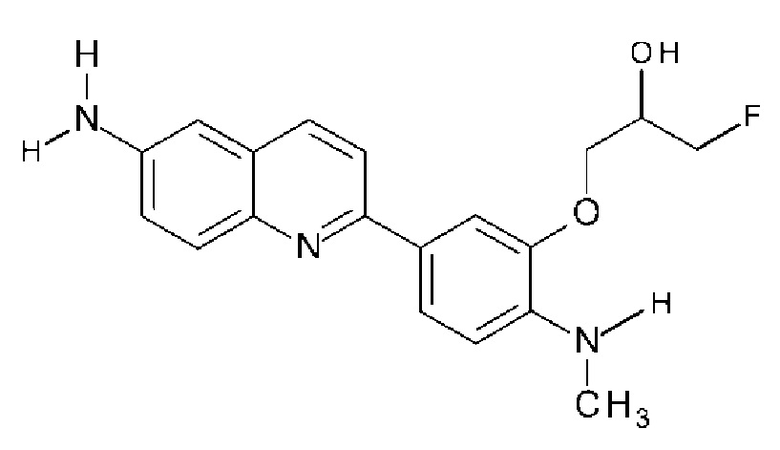
[0120]
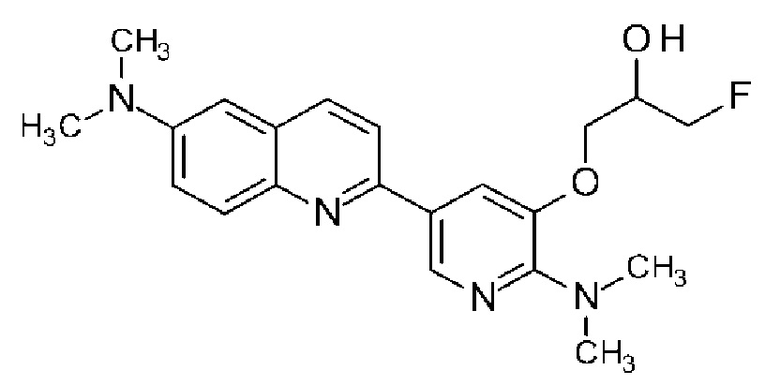
[0121]
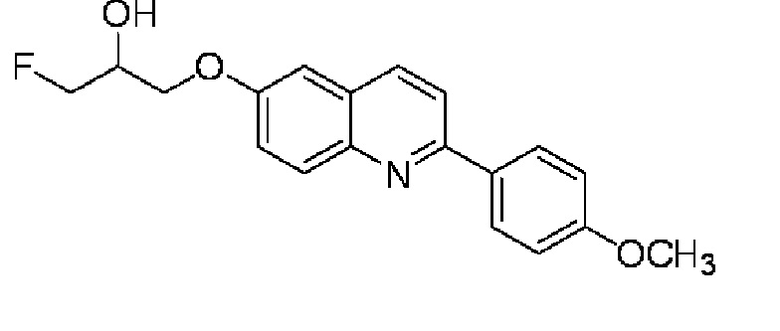
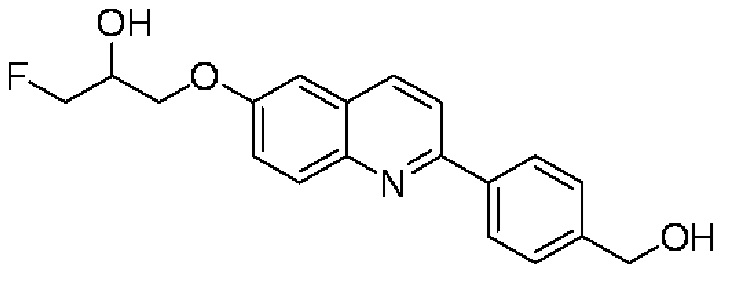
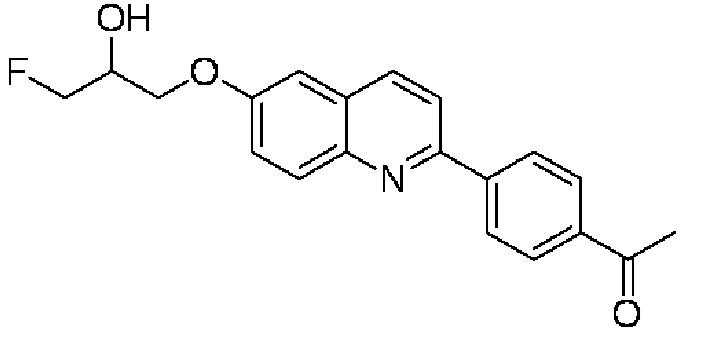
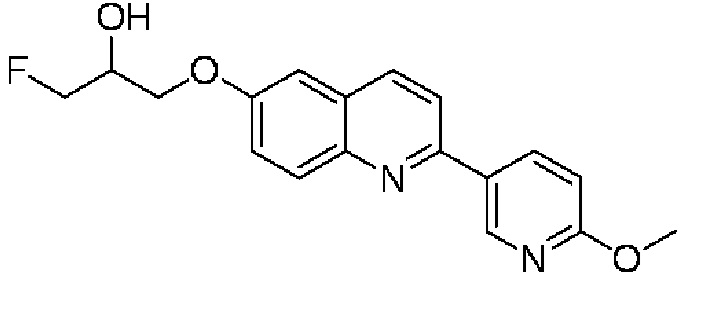
[0122]
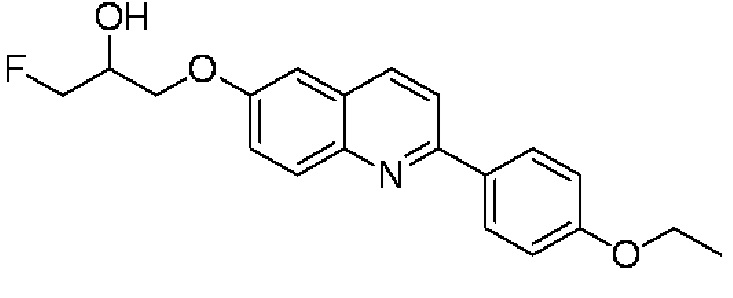
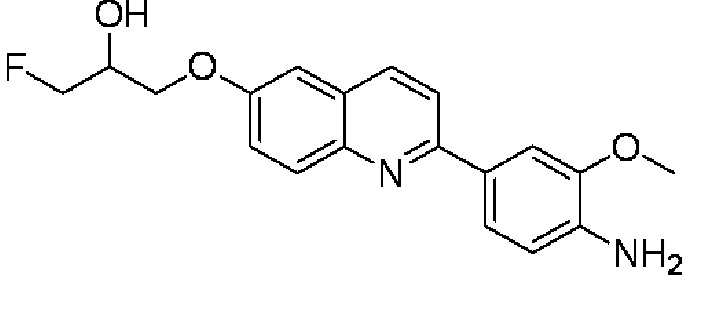
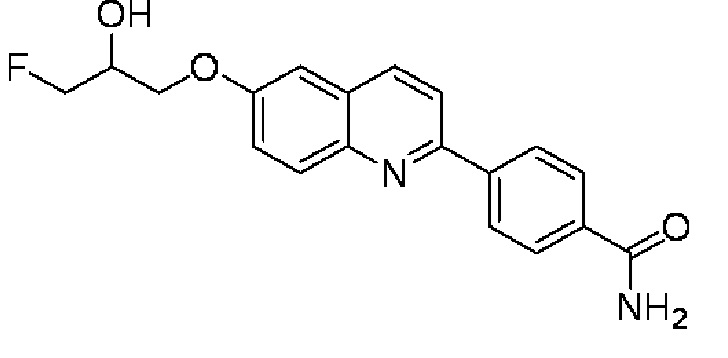
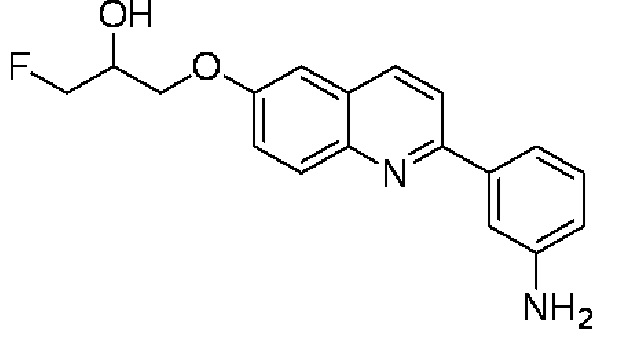
[0123]
Типичные примеры соединения формулы (I'), предпочтительно используемого в настоящем изобретении, показаны ниже. Среди этих соединений THK-5039 может применяться в качестве синтетического предшественника THK-5035, THK-5041 может применяться в качестве синтетического предшественника THK-5004, THK-5050 может применяться в качестве синтетического предшественника THK-5051, THK-5070 может применяться в качестве синтетического предшественника THK-5066, THK-5072 может применяться в качестве синтетического предшественника THK-5058, THK-5073 может применяться в качестве синтетического предшественника THK-5038, THK-5090 может применяться в качестве синтетического предшественника THK-5071, THK-5095 может применяться в качестве синтетического предшественника THK-5077, THK-5096 может применяться в качестве синтетического предшественника THK-5078, THK-5099 может применяться в качестве синтетического предшественника THK-5064, THK-5111 может применяться в качестве синтетического предшественника THK-5112, THK-5113 может применяться в качестве синтетического предшественника THK-5106, THK-5115 может применяться в качестве синтетического предшественника THK-5107, THK-5119 может применяться в качестве синтетического предшественника THK-5117, THK-5120 может применяться в качестве синтетического предшественника THK-5122, THK-5121 может применяться в качестве синтетического предшественника THK-5105, THK-5123 может применяться в качестве синтетического предшественника THK-5116, THK-5131 может применяться в качестве синтетического предшественника THK-5125, THK-5150 может применяться в качестве синтетического предшественника THK-5127, THK-5152 может применяться в качестве синтетического предшественника THK-5151, THK-5135 может применяться в качестве синтетического предшественника THK-5129, THK-5138 может применяться в качестве синтетического предшественника THK-5130, THK-5143 может применяться в качестве синтетического предшественника THK-5142, THK-5163 может применяться в качестве синтетического предшественника THK-5164, THK-5167 может применяться в качестве синтетического предшественника THK-5136, и THK-5168 может применяться в качестве синтетического предшественника THK-5156.
[0124]
хинолин
хинолин
[0125]
хинолин
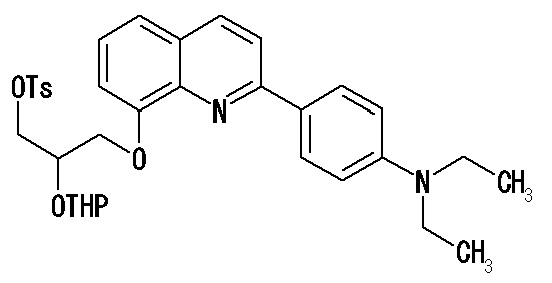
хинолин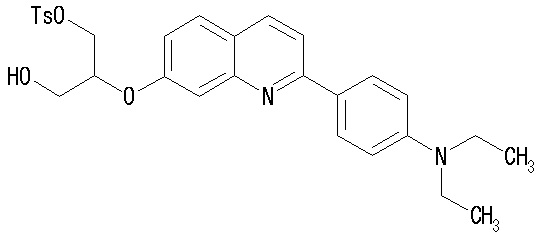
хинолин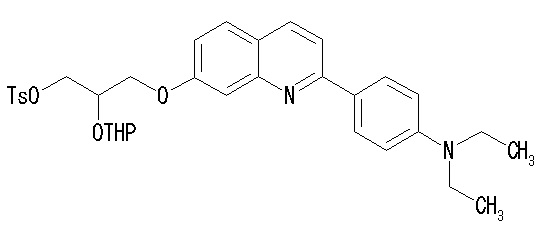
хинолин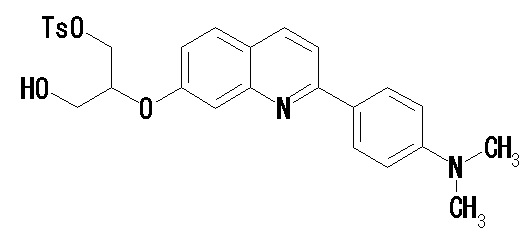
хинолин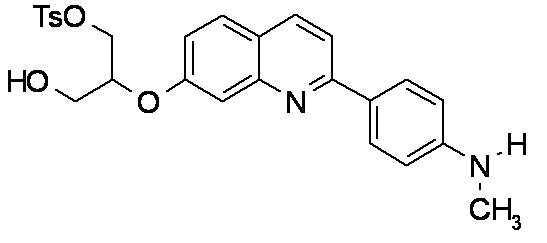
хинолин
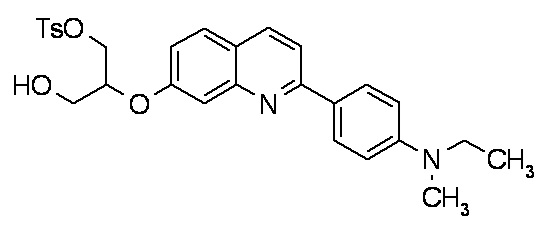
хинолин
[0126]
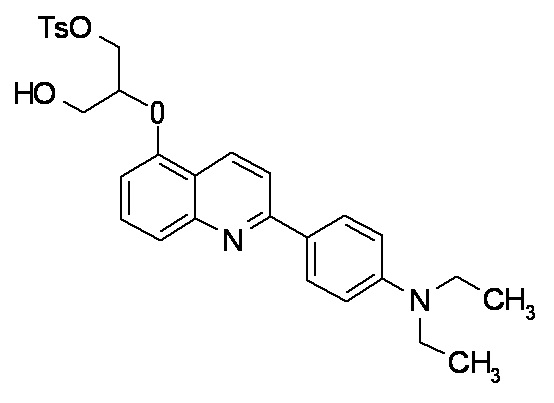
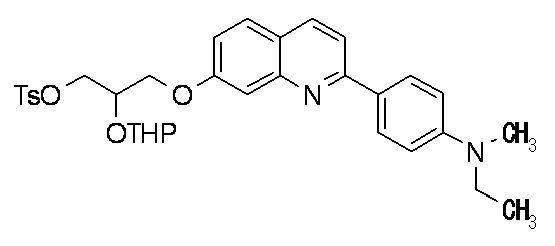
хинолин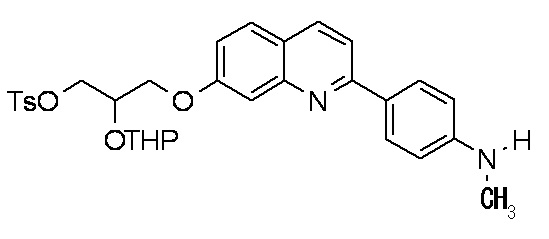
хинолин
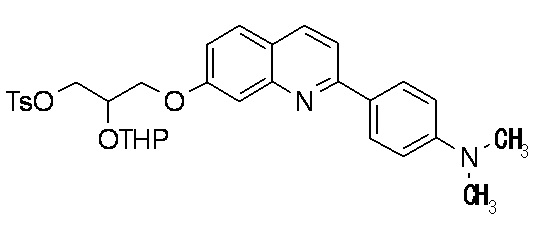
хинолин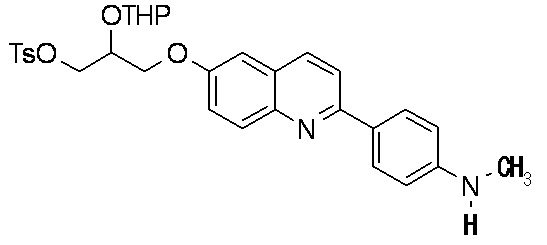
хинолин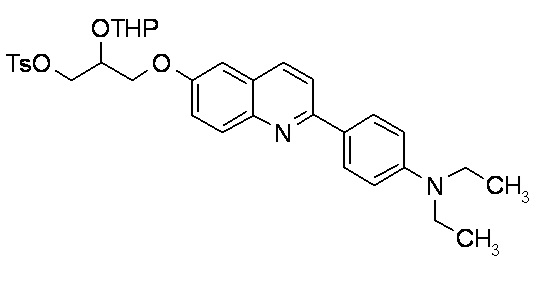
[0127]
хинолин

хинолин
[0128]
хинолин
хинолин
хинолин
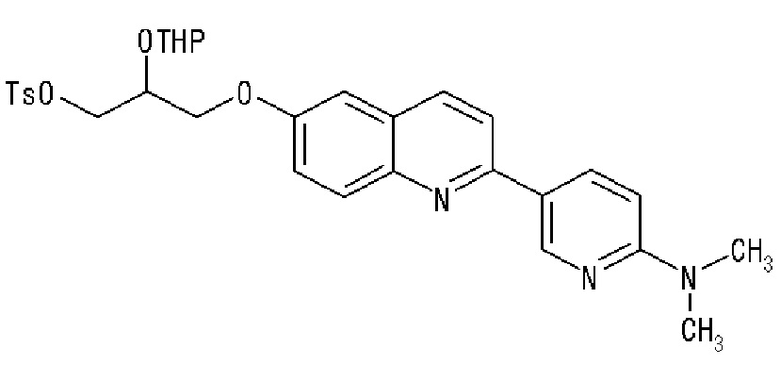
хинолин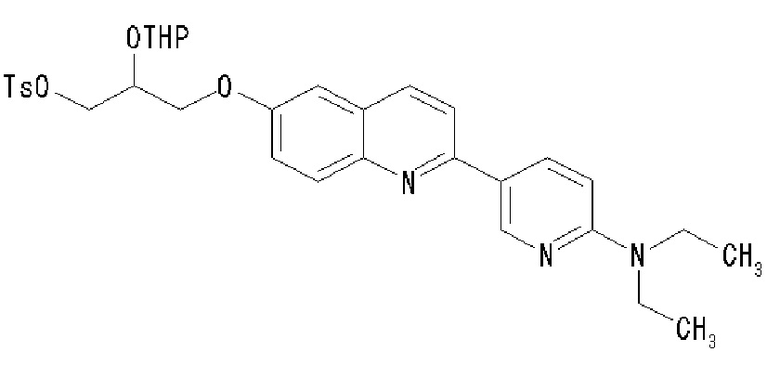
хинолин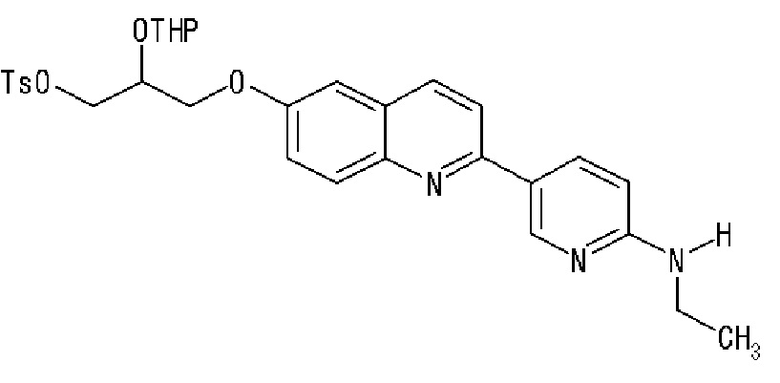
[0129]
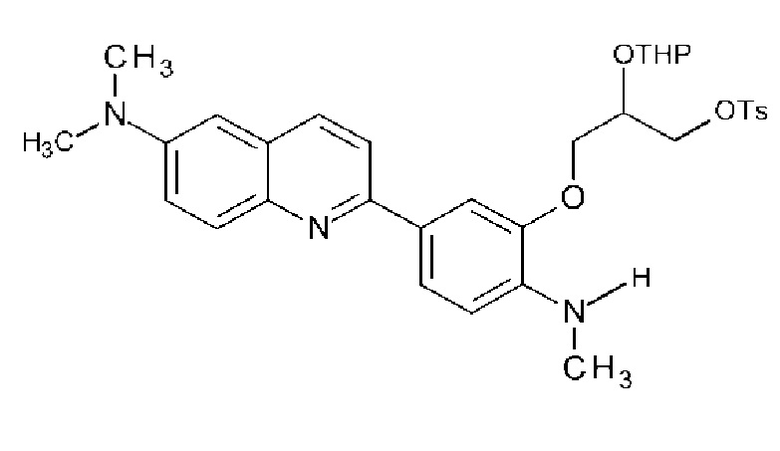
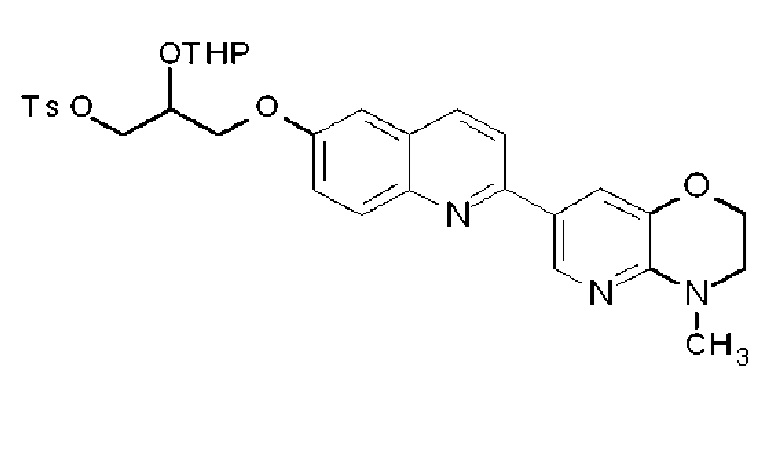
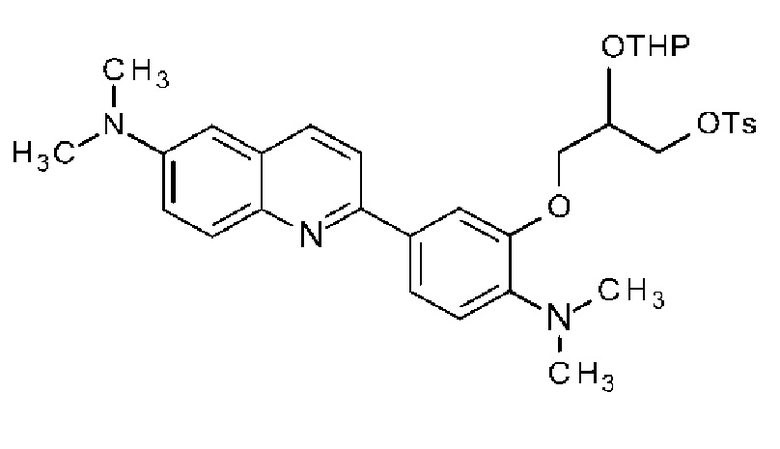
фенил]-6-диметиламинохинолин
[0130]
Настоящее изобретение будет более подробно и определенно описано ниже в виде Примеров, но настоящее изобретение не ограничивается Примерами.
[Пример 1]
[0131]
Синтез соединений по настоящему изобретению
Силикагель BW300, выпускаемый компанией Fuji Silysia Chemical Ltd., использовали при колоночной хроматографии на силикагеле Примеров. Chromatorex NH-DM1020, выпускаемый компанией Fuji Silysia Chemical Ltd., использовали при основной колоночной хроматографии на силикагеле с использованием типа силикагеля, связанного с аминогруппой.
1H-ЯМР измеряли, используя устройство UNITY INOVA500 (500 МГц), выпускаемое компанией VARIAN, JNM-LA400 (400 МГц), выпускаемое компанией JEOL, Ltd., и Gemini 2000 (300 МГц) тетраметилсилан, выпускаемое компанией VARIAN в качестве стандартных веществ, и все величины δ измеряли в ppm (м.д.).
Масс-спектр измеряли химической ионизацией при атмосферном давлении (APCI) с использованием устройства LCQ-Advantage, выпускаемого компанией ThermoQuest, или SSQ-7000C, выпускаемого компанией FinniganMAT.
Инфракрасные спектры измеряли с использованием устройства Paragon1000 FT-IR, выпускаемого компанией Perkin-Elmer, и B-545, выпускаемого компанией BUCHI, использовали для измерения точки плавления.
[0132]
Значения аббревиатур в измерении ЯМР показаны ниже.
с: синглет
д: дублет
дд: двойной дублет
ддд: тройной дублет
т: триплет
дт: двойной триплет
кв.: квартет
м: мультиплет
уш.: широкий
J: константа соединения
Гц: Герц
CDCl3: дейтерированный хлороформ
ДМСО-d6: дейтерированный диметилсульфоксид
[0133]
Примеры Синтеза и Пример Метки соединений по настоящему изобретению показаны ниже.
[0134]
Способ синтеза соединения THK-5004
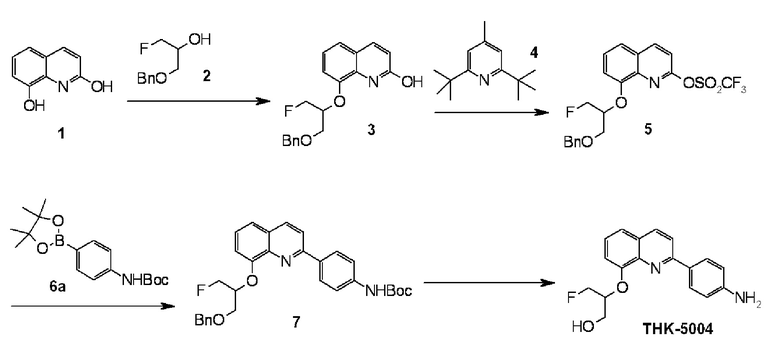
[0135]
Синтез соединения 3
К смеси соединения 1 (1,29 г, 8,0 ммоль), 2 (1,66 г, 9,0 ммоль), трифенилфосфина (2,36 г, 9,0 ммоль) и тетрагидрофурана (20 мл) добавляли по каплям при охлаждении льдом и перемешивании раствор в тетрагидрофуране (10 мл) диизопропилазодикарбоксилата (1,82 г, 9,0 ммоль), и смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=2/3) с получением 3 (2,62 г, 100%) в виде бледно-желтого маслянистого вещества.
[0136]
Синтез соединения 5
К раствору в метиленхлориде (20 мл) соединения 3 (2,62 г, 8,0 ммоль) и соединения 4 (2,2 г, 10,4 ммоль) добавляли по каплям при охлаждении льдом и перемешивании раствор в метиленхлориде (10 мл) трифторметансульфонового ангидрида (2,7 г, 9,6 ммоль), и смешанный раствор перемешивали при комнатной температуре в течение 16 часов. Добавляли соединение 4 (2,2 г, 10,4 ммоль) и трифторметансульфоновый ангидрид (2,7 г, 9,6 ммоль), с последующим дальнейшим перемешиванием при комнатной температуре в течение 16 часов. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=20/1) с получением 5 (1,47 г, 40%) в виде бесцветного маслянистого вещества.
APCI-MS m/z 460[M+H]+
[0137]
Синтез соединения 7
К смеси соединения 5 (1,45 г, 3,15 ммоль), соединения 6a (1,00 г, 3,15 ммоль) и 1,2-диметоксиэтана (20 мл) добавляли водный 2M раствор карбоната натрия (3,5 мл, 7,0 ммоль) и тетракистрифенилфосфин палладия (146 мг, 0,126 ммоль), и смесь нагревали в сосуде с обратным холодильником в течение 16 часов в атмосфере аргона. Реакционному раствору давали возможность вернуться к комнатной температуре, и растворитель отгоняли при пониженном давлении, и затем к остатку добавляли воду, и раствор экстрагировали хлороформом. Экстракицонную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=9/1) с получением соединения 7 (1,55 г, 98%) в виде бледно-желтого пеноподобного вещества.
APCI-MS m/z 503[M+H]+
[0138]
Синтез соединения THK-5004
К смеси соединения 7 (1,54 г, 3,0 ммоль) и анизола (0,98 мл) добавляли метансульфоновую кислоту (4,9 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли ледяную воду, и раствор делали основным водным раствором карбоната калия и затем экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (элюирующий раствор: н-гексан/этилацетат=2/1→3/2→1/4) и затем промывали диизопропиловым эфиром с получением соединения THK-5004 (593 мг, 62%) в виде желтого твердого вещества.
точка плавления 97-100°C, 1H ЯМR (400 МГц, CDCl3) δ 3,70-3,80 (2H, м), 3,80-3,90 (2H, уш. с), 4,40-4,50 (1Н, м), 4,70-5,00 (2H, м), 6,29 (1H, т, J=6,4 Гц), 6,75-6,85 (2H, м), 7,49 (1Н, т, J=7,6 Гц), 7,59 (1H, д, J=7,6 Гц), 7,82 (1H, д, J=8,8 Гц), 7,92 (2H, д, J=8,8 Гц), 8,19 (1H, д, J=8,8 Гц)
ИК (Nujol) 1600 см-1
APCI-MS m/z 313[M+H]+
[0139]
Способ синтеза соединения THK-5035
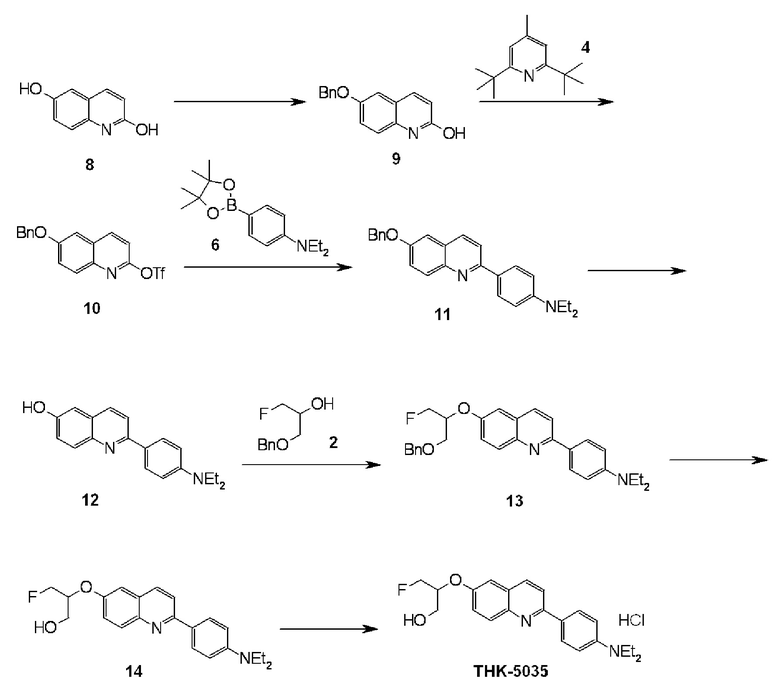
[0140]
Синтез соединения 9
К суспензии в N,N-диметилформамиде (50 мл) соединения 8 (3,00 г, 18,6 ммоль) добавляли карбонат калия (2,83 г, 20,5 ммоль) и бензилхлорид (2,25 мл, 19,6 ммоль) и смесь перемешивали при 105°C в течение 1 часа. Реакционному раствору давали возможность вернуться к комнатной температуре и добавляли воду, и затем раствор экстрагировали теплым этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем раствор концентрировали примерно до 100 мл при пониженном давлении. После обеспечения возможности охладиться, осажденный кристалл собирали фильтрацией и затем сушили с получением соединения 9 (2,66 г, 57%) в виде бесцветных кристаллов.
точка плавления 220-221°C, ИК (Nujol) 1661, 1623 см-1
APCI-MS m/z 252[M+H]+
[0141]
Синтез соединения 10
К суспензии в метиленхлориде (70 мл) соединения 9 (2,66 г, 10,6 ммоль) и соединения 4 (3,04 г, 14,8 ммоль) добавляли по каплям трифторметансульфоновый ангидрид (2,14 мл, 12,7 ммоль) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли соединение 4 (1,52 г, 7,41 ммоль) и трифторметансульфоновый ангидрид (1,07 мл, 6,35 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 3 дней. К реакционному раствору добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=20/1, 9/1) и затем перекристаллизовывали из этилацетат-н-гесана с получением соединения 10 (3,75 г, 92%) в виде бесцветных кристаллов.
точка плавления 109,5-110°C, APCI-MS m/z 384[M+H]+
[0142]
Синтез соединения 11
К смеси соединения 10 (2,19 г, 5,7 ммоль), соединения 6 (1,65 г, 6,0 ммоль) и 1,2-диметоксиэтана (20 мл) добавляли водный 2M раствор карбоната натрия (5,5 мл, 11 ммоль) и тетракистрифенилфосфин палладия (277 мг, 0,240 ммоль), и смесь перемешивали в атмосфере аргона при 90°C в течение 16 часов. Реакционному раствору давали возможность возвратиться к комнатной температуре, и нерастворимые вещества удаляли фильтрацией и затем промывали хлороформом. Фильтрат и промывную жидкость объединяли, и смесь экстрагировали хлороформом. Экстракционную жидкость промывали насыщенным солевым раствором и затем сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: хлороформ) с получением соединения 11 (1,86 г, 85%) в виде оранжевого твердого вещества.
APCI-MS m/z 383[M+H]+
[0143]
Синтез соединения 12
К смеси соединения 11 (1,85 г, 4,84 ммоль) и анизола (1 мл) добавляли метансульфоновую кислоту (5 мл), и смесь перемешивали при комнатной температуре в течение 30 минут. К реакционному раствору добавляли воду, и раствор промывали этилацетатом. Водный слой делали основным гидрокарбонатом натрия и экстрагировали хлороформом-тетрагидрофураном. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток промывали н-гексаном с получением соединения 12 (1,25 г, 88%) в виде оранжевого твердого вещества.
APCI-MS m/z 293[M+H]+
[0144]
Синтез соединения 13
К смеси соединения 12 (468 мг, 1,6 ммоль), соединения 2 (354 мг, 1,9 ммоль), трифенилфосфина (504 мг, 1,9 ммоль) и тетрагидрофурана (20 мл) добавляли по каплям при охлаждении льдом и перемешивании раствор диизопропилазодикарбоксилата (388 мг, 1,9 ммоль) в тетрагидрофуране (10 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. Добавляли трифенилфосфин (150 мг, 0,57 ммоль) и диизопропилазодикарбоксилат (120 мг, 0,59 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 2 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: хлороформ) с получением соединения 13 (733 мг, 100%) в виде оранжевого твердого вещества.
[0145]
Синтез соединения 14
К смеси соединения 13 (733 мг, 1,6 ммоль) и анизола (1 мл) добавляли метансульфоновую кислоту (5 мл) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 50 минут. К реакционному раствору добавляли ледяную воду (40 мл), и раствор промывали этилацетатом. Водный слой делали основным карбонатом калия и экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: хлороформ/метанол=99/1) и затем промывали диизопропиловым эфиром с получением 14 (400 мг, 68%) в виде желтого твердого вещества.
APCI-MS m/z 369[M+H]+
[0146]
Синтез соединения THK-5035
К раствору в этаноле (10 мл) соединения 14 (400 мг, 1,09 ммоль) добавляли 4М HCl/этилацетат (1 мл, 4 ммоль), и растворитель отгоняли при пониженном давлении. Остаток промывали диэтилэфиром и затем перекристаллизовывали из изопрапонола-ацетона с получением соединения THK-5035 (368 мг, 84%) в виде оранжевых кристаллов.
точка плавления 207-209°C, 1H ЯМR (400 МГц, ДМСО-d6+D20) δ 2,53 (6H, т, J=7,0 Гц), 3,52 (4H, кв., J=7,0 Гц), 4,6-4,9 (3H, м), 6,99 (2H, д, J=8,5 Гц), 7,7-7,8 (2H, м), 8,13 (2H, д, J=9,1 Гц), 8, 27 (2H, д, J=9,1 Гц), 8,71 (1H, д, J=9,1 Гц)ИК (Nujol) 1595, 1460, 1216 см-1
APCI-MS m/z 369[M+H]+
[0147]
Способ синтеза соединения THK-5038
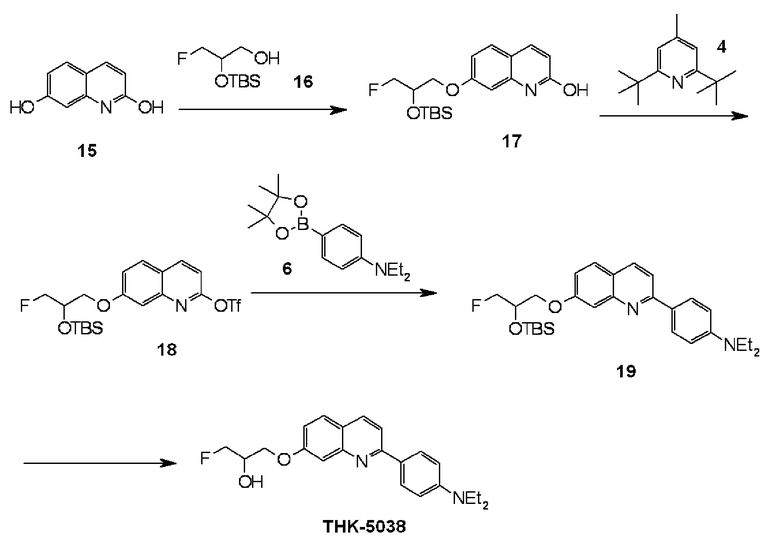
[0148]
Синтез соединения 17
К смеси соединения 15 (1,00 г, 6,21 ммоль), соединения 16 (1,55 г, 7,45 ммоль), трифенилфосфина (1,95 г, 7,45 ммоль) и тетрагидрофурана (80 мл) добавляли по каплям при охлаждении льдом и перемешивании диизопропилазодикарбоксилат (1,48 мл, 7,45 ммоль), и смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2) с получением соединения 17 (1,80 г, 82%) в виде бледно-розового твердого вещества.
точка плавления 132-135°C, APCI-MS m/z 352[M+H]+
[0149]
Синтез соединения 18
К раствору в метиленхлориде (25 мл) соединения 17 (1,80 г, 5,12 ммоль) и соединения 4 (1,47 г, 7,17 ммоль) добавляли по каплям трифторметансульфоновый ангидрид (1,03 мл, 6,14 ммоль) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 70 минут. К реакционному раствору добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан→н-гексан/этилацетат=50/1→30/1) с получением соединения 18 (2,09 г, 84%) в виде бледно-розового маслянистого вещества.
APCI-MS m/z 484[M+H]+
[0150]
Синтез соединения 19
К смеси соединения 18 (1,20 г, 2,48 ммоль), соединения 6 (680 мг, 2,48 ммоль) и 1,2-диметоксиэтана (30 мл) добавляли карбонат калия (1,03 г, 7,44 ммоль) и воду (0,62 мл) и добавляли тетракистрифенилфосфин палладия (124 мг, 0,124 ммоль) в атмосфере аргона с последующим перемешиванием при 80°C в течение 2,5 часов. Реакционному раствору давали возможность возвратиться к комнатной температуре, добавляли воду и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=20/1) и затем перекристаллизовывали из н-гексана с получением соединения 19 (700 мг, 59%) в виде бледно-желтых кристаллов.
точка плавления 102-102,5°C, APCI-MS m/z 483[M+H]+
[0151]
Синтез соединения THK-5038
К раствору в тетрагидрофуране (10 мл) соединения 19 (700 мг, 1,45 ммоль) добавляли 1M тетра-н-бутиламмонийфторид/тетрагидрофуран (1,45 мл, 1,45 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=2/1) и затем перекристаллизовывали из этилацетата-н-гесана с получением соединения THK-5038 (482 мг, 90%).
точка плавления 110-110,5°C, 1Н ЯМR (500 МГц, ДМСО-d6) δ 1,14 (6H, т, J=7,0 Гц), 3,42 (4H, кв., J=7,0 Гц), 4,10-4,19 (3H, м), 4,45-4,64 (2H, м), 5,55 (1H, д, J=4,8 Гц), 6,78 (2H, д, J=9,0 Гц), 7,12 (1H, дд, J=8,7, 2,6 Гц), 7,35 (1H, д, J=2,6 Гц), 7,81 (1H, д, J=9,0 Гц), 7,85 (1Н, д, J=9,0 Гц), 8,10 (2H, д, J=9,0 Гц), 8,21 (1H, д, J=9,0 Гц)
ИК (Nujol) 1622, 1596 см-1
APCI-MS m/z 369[M+H]+
[0152]
Способ синтеза соединения THK-5039
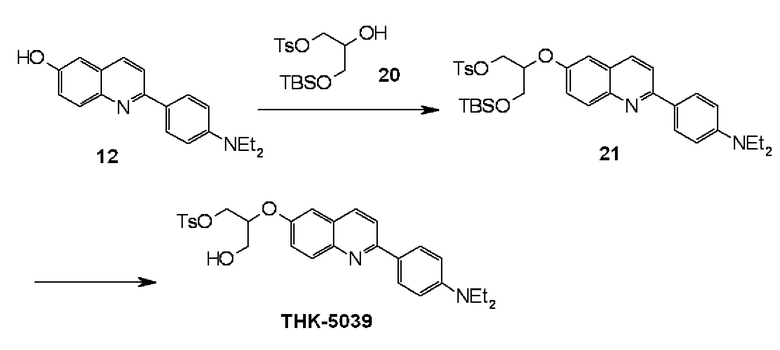
[0153]
Синтез соединения 21
К смеси соединения 12 (400 мг, 1,37 ммоль), соединения 20 (592 мг, 1,64 ммоль), трифенилфосфина (431 мг, 1,64 ммоль) и тетрагидрофурана (20 мл) добавляли по каплям диизопропилазодикарбоксилат (0,325 мл, 1,64 ммоль) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 16 часов. Добавляли соединение 20 (592 мг, 1,64 ммоль), трифенилфосфин (431 мг, 1,64 ммоль) и тетрагидрофуран (10 мл) с последующим дополнительным перемешиванием при комнатной температуре в течение 24 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9) с получением соединения 21 (870 мг, 100%) в виде желтого маслянистого вещества.
APCI-MS m/z 635[M+H]+
[0154]
Синтез соединения THK-5039
К раствору в хлороформе (12 мл) соединения 21 (870 мг, 1,37 ммоль) по каплям добавляли трифторуксусную кислоту (8 мл) и затем воду (2 мл) в условиях охлаждения льдом и перемешивания и смесь перемешивали при комнатной температуре в течение 1,5 часов. К реакционному раствору добавляли ледяную воду и этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 9 с использованием водного раствора карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=4/1, 2/1) и затем перекристаллизовывали из этилацетата-н-гесана с получением соединения THK-5039 (219 мг, 31%).
точка плавления 119-120°C, 1H ЯМR (500 МГц, ДМСО-d6) δ 1,14 (6H, т, J=7,0 Гц), 2,33 (3H, с), 3,42 (4H, кв., J=7,0 Гц), 4,07 (5H, м), 5,62 (1Н, д, J=4,8 Гц), 6,78 (2H, д, J=9,0 Гц), 7,21 (2H, м), 7,40 (2H, д, J=8,0 Гц), 7,78 (2H, д, J=8,0 Гц), 7,86 (1H, д, J=8,0 Гц), 7,97 (1H, д, J=8,7 Гц), 8,07 (2H, д, J=8,7 Гц), 8,20 (1H, д, J=8,7 Гц)
APCI-MS m/z 521[M+H]+
[0155]
Способ синтеза соединения THK-5050
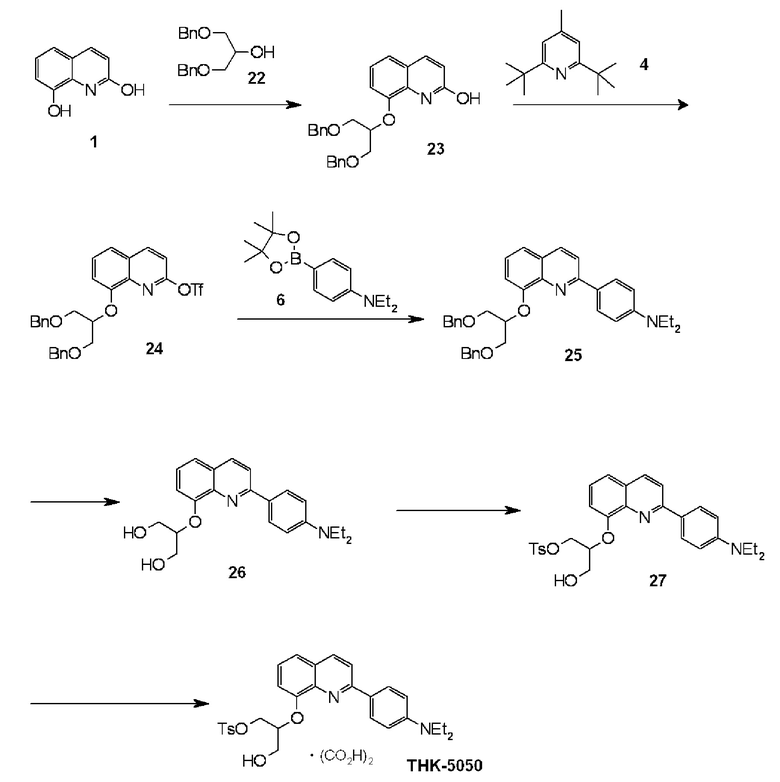
[0156]
Синтез соединения 23
К смеси соединения 1 (2,00 г, 12,4 ммоль), соединения 22 (4,06 г, 14,9 ммоль), трифенилфосфина (3,91 г, 14,9 ммоль) и тетрагидрофурана (40 мл) добавляли по каплям диизопропилазодикарбоксилат (2,95 мл, 14,9 ммоль) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 3 дней. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4, 2/3) с получением соединения 23 (3.45 г, 67%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 416[M+H]+
[0157]
Синтез соединения 24
К раствору в метиленхлориде (40 мл) соединения 23 (3,44 г, 8,28 ммоль) и соединения 4 (2,38 г, 11,6 ммоль) добавляли по каплям трифторметансульфоновый ангидрид (1,67 мл, 9,94 ммоль) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан→н-гексан/этилацетат=9/1) с получением соединения 24 (4,16 г, 92%) в виде бесцветного маслянистого вещества.
APCI-MS m/z 548[M+H]+
[0158]
Синтез соединения 25
К смеси соединения 24 (2,00 г, 3,65 ммоль), соединения 6 (1,01 г, 3,65 ммоль) и 1,2-диметоксиэтана (30 мл) добавляли карбонат калия (1,51 г, 11 ммоль) и воду (0,63 мл), и тетракистрифенилфосфин палладия (210 мг, 0,183 ммоль) добавляли в атмосфере аргона, и смесь перемешивали при 80°C в течение 2 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и затем раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=9/1) с получением соединения 25 (1,95 г, 98%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 547[M+H]+
[0159]
Синтез соединения 26
К смеси соединения 25 (1,94 г, 3,55 ммоль) и анизола (3 мл) добавляли по каплям метансульфоновую кислоту (9 мл) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 20 минут. К реакционному раствору добавляли ледяную воду, и затем этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 9 с использованием водного раствора карбоната калия. Экстракционную жидкость промывали насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=4/1→1/2→1/3) с получением соединения 26 (1,22 г, 94%) в виде желтого порошка.
APCI-MS m/z 367[M+H]+
[0160]
Синтез соединения 27
К раствору в 1,2-диметоксиэтане (120 мл) соединения 26 (1,36 г, 3,71 ммоль) добавляли паратолуолсульфоновый ангидрид (1,21 г, 3,71 ммоль) и триэтиламин (0,78 мл, 5,57 ммоль) и смесь перемешивали при комнатной температуре в течение 16 часов. Реакционный раствор промывали насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=3/1) с получением соединения 27 (500 мг, 26%) в виде оранжевого пеноподобного вещества.
[0161]
Синтез соединения THK-5050
Соединение 27 (638 мг, 1,23 ммоль) растворяли в этилацетате, и раствор подвергали короткой хроматографии на силикагеле (элюирующий растворитель: этилацетат) для удаления исходного вещества. После растворения добавлением щавелевой кислоты (221 мг, 2,45 ммоль) к элюату, растворитель концентрировали до примерно 100 мл при пониженном давлении. После растворения осадка добавлением этанола (100 мл), растворитель концентрировали примерно до 100 мл при пониженном давлении. Нерастворимые вещества удаляли фильтрацией, и фильтрат концентрировали примерно до 50 мл при пониженном давлении. Добавляли изопропанол (50 мл), и растворитель концентрировали при пониженном давлении для осаждения кристаллов. После отстоя при 5°С в течение 16 часов, осажденные кристаллы собирали фильтрацией и сушили с получением соединения THK-5050 (520 мг, 69%) в виде оранжевых кристаллов.
точка плавления 124-125°C, 1H ЯМR (400 МГц, ДМСО-d6) δ 1,15 (6H, т, J=7,0 Гц), 2,30 (3H, с), 3,50 (4H, кв., J=7,0 Гц), 3,82-3,88 (2H, м), 4,42-4,49 (1Н, м), 4,85 (1Н, дд, J=14, 3,0 Гц), 4,92 (1Н, дд, J=14, 9,4 Гц), 6,93 (2H, д, J=9,1 Гц), 7,15 (2H, д, J=7,9 Гц), 7,46 (2H, д, J=7,9 Гц), 7,66-7,70 (3H, м), 7,85 (1H, т, J=8,2 Гц), 7,93 (1H, дд, J=8,2, 1,2 Гц), 8,06 (1H, д, J=9,0 Гц), 8,95 (1H, д, J=9,0 Гц)
ИК (Nujol) 1633, 1590 см-1
APCI-MS m/z 521[M+H]+
[0162]
Способ синтеза соединения THK-5051
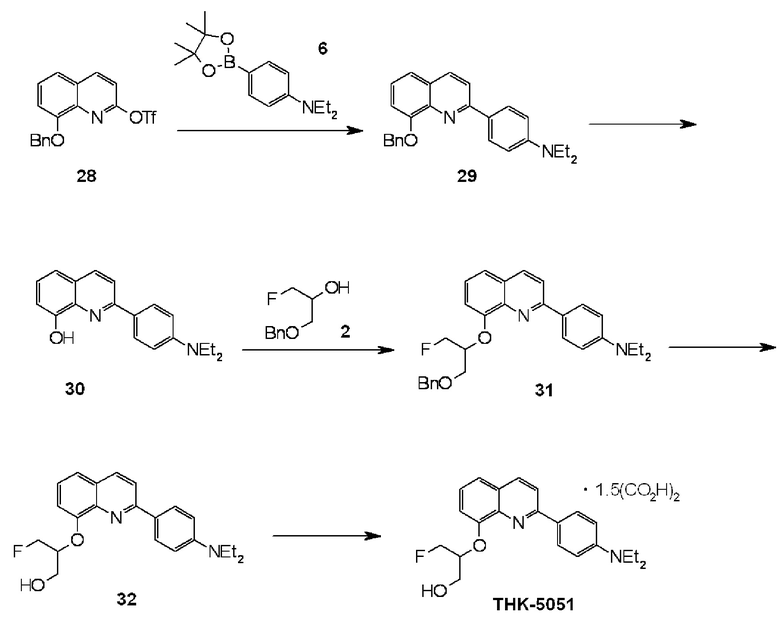
[0163]
Синтез соединения 29
К смеси соединения 28 (894 мг, 2,30 ммоль), соединения 6 (706 мг, 2,57 ммоль) и 1,2-диметоксиэтана (10 мл) добавляли водный 2M раствор карбоната натрия (2,5 мл, 5,0 ммоль) и тетракистрифенилфосфин палладия (119 мг, 0,10 ммоль) в атмосфере аргона, и смесь перемешивали при 90°C в течение 2 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, и нерастворимые вещества удаляли фильтрацией, и затем раствор промывали хлороформом. Фильтрат и промывную жидкость объединяли, и смесь экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=9/1→4/1) с получением соединения 29 (880 мг, 100%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 383[M+H]+
[0164]
Синтез соединения 30
К смеси соединения 29 (880 мг, 2,30 ммоль) и анизола (1 мл) добавляли по каплям метансульфоновую кислоту (5 мл) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли воду, и раствор промывали этилацетатом. Водный слой экстрагировали хлороформом после доведения pH до 9 с использованием водного насыщенного раствора гидрокарбоната натрия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении с получением соединения 30 (670 мг, 100%) в виде желтого маслянистого вещества.
APCI-MS m/z 293[M+H]+
[0165]
Синтез соединения 31
К смеси соединения 30 (670 мг, 2,3 ммоль), соединения 2 (516 мг, 2,8 ммоль), трифенилфосфина (918 мг, 3,5 ммоль) и тетрагидрофурана (10 мл) добавляли по каплям раствор в тетрагидрофуране (10 мл) диизопропилазодикарбоксилата (708 мг, 3,5 ммоль) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/19→1/9) с получением соединения 31 (780 мг, 73%) в виде бледно-зеленого маслянистого вещества.
APCI-MS m/z 459[M+H]+
[0166]
Синтез соединения 32
К смеси соединения 31 (775 мг, 1,69 ммоль) и анизола (1 мл) добавляли метансульфоновую кислоту (5 мл) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 1,5 часов. К реакционному раствору добавляли воду, и раствор промывали этилацетатом. Водный слой делали основным водным насыщенным раствором гидрокарбоната натрия и экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением соединения 32 (608 мг, 97%) в виде оранжевого маслянистого вещества.
APCI-MS m/z 369[M+H]+
[0167]
Синтез соединения THK-5051
К раствору в ацетоне соединения 32 (576 мг, 1,56 ммоль) добавляли щавелевую кислоту (281 мг, 3,12 ммоль) для образования оксалата, который перекристаллизовывали из метанола-диэтилового эфира с получением соединения THK-5051 (414 мг, 53%) в виде оранжевых кристаллов.
точка плавления 126-128°C, 1H ЯМR (400 МГц, ДМСО-d6+D2O) δ 1,15 (6H, т, J=7,0 Гц), 3,44 (4H, кв., J=7,0 Гц), 3,6-3,8 (1H, м), 4,7-4,9 (3H, м), 6,84 (2H, д, J=9,0 Гц), 7,39 (1Н, д, J=7,4 Гц), 7,44 (1Н, т, J=7,7 Гц), 7,60 (1H, д, J=7,7 Гц), 8,05 (1H, д, J=9,0 Гц), 8,12 (2H, д, J=9,0 Гц), 8,35 (1H, д, J=9,0 Гц)
APCI-MS m/z 369[M+H]+
[0168]
Способ синтеза соединения THK-5058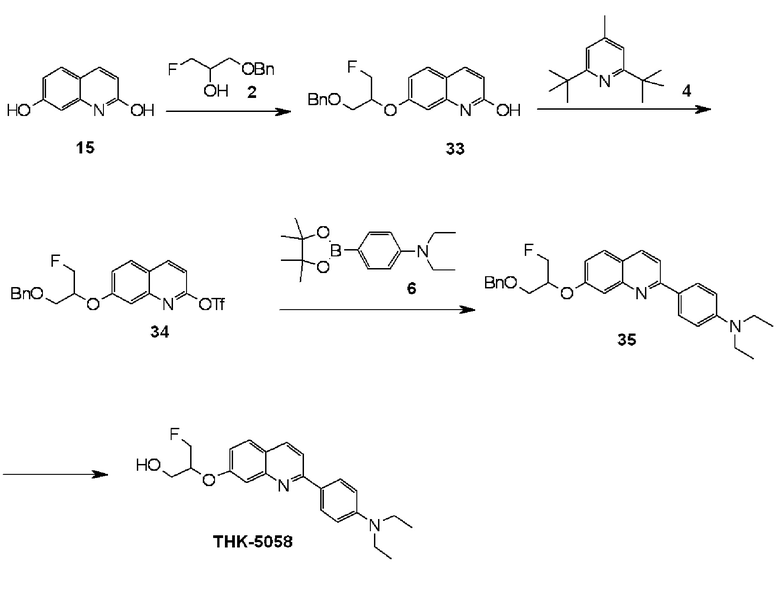
[0169]
Синтез соединения 33
К смеси соединения 15 (1,00 г, 6,2 ммоль), соединения 2 (1,37 г, 7,45 ммоль), трифенилфосфина (1,95 г, 7,45 ммоль) и тетрагидрофурана (50 мл) добавляли по каплям диизопропилазодикарбоксилат (1,48 мл, 7,45 ммоль) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли соединение 2 (0,67 г, 3,73 ммоль), трифенилфосфин (0,98 г, 3,73 ммоль), тетрагидрофуран (10 мл) и диизопропилазодикарбоксилат (0,74 мл, 3,73 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 6 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: хлороформ, хлороформ/метанол/концентрированная аммиачная вода=50/1/0,1) с получением соединения 33 (1,66 г, 82%) в виде бледно-коричневого вещества.
APCI-MS m/z 328[M+H]+
[0170]
Синтез соединения 34
К раствору в метиленхлориде (25 мл) соединения 33 (1,65 г, 5,0 ммоль) и соединения 4 (1,45 г, 7,06 ммоль) добавляли по каплям трифторметансульфоновый ангидрид (1,02 мл, 6,05 ммоль) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли соединение 4 (0,21 г, 1,01 ммоль) и трифторметансульфоновый ангидрид (0,17 мл, 1,01 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 30 минут. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан, н-гексан/этилацетат=4/1) с получением соединения 34 (1,94 г, 84%) в виде бесцветного твердого вещества.
точка плавления 81-82°C, APCI-MS m/z 460[M+H]+
[0171]
Синтез соединения 35
К смеси соединения 34 (1,00 г, 2,18 ммоль), соединения 6 (0,6 г, 2,18 ммоль) и 1,2-диметоксиэтана (18 мл) добавляли карбонат калия (0,90 г, 6,54 ммоль), воду (0,38 мл) и тетракистрифенилфосфин палладия (130 мг, 0,109 ммоль) в атмосфере аргона, и смесь перемешивали при 80°C в течение 1,5 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=9/1, 6/1) с получением соединения 35 (1,00 г, 100%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 459[M+H]+
[0172]
Синтез соединения THK-5058
К смеси соединения 35 (0,99 г, 2,16 ммоль) и анизола (3 мл) добавляли метансульфоновую кислоту (9 мл) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли ледяную воду и затем этилацетат. Раствор делали основным водным раствором карбоната калия и экстрагировали этилацетатом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=4/1, 2/1) и затем промывали н-гексаном/этилацетатом (4/1) с получением соединения THK-5058 (636 мг, 80%) в виде бледно-желтых кристаллов.
точка плавления 89-90°C, 1H ЯМR (500 МГц, ДМСО-d6) δ 1,14 (6H, т, J=7,0 Гц), 3,42 (4H, кв., J=7,0 Гц), 3,65-3,77 (2H, м), 4,65-4,87 (3H, м), 5,14 (1H, т, J=5,6 Гц), 6,78 (2H, д, J=8,7 Гц), 7, 17 (1Н, дд, J=8,8, 2,4 Гц), 7,47 (1H, д, J=2,4 Гц), 7,82 (1Н, д, J=8,8 Гц), 7,85 (1Н, д, J=8,8 Гц), 8,10 (2H, д, J=8,8 Гц), 8,21 (1H, д, J=8,8 Гц)
ИК (Nujol) 1598 см-1
APCI-MS m/z 369[M+H]+
[0173]
Способ синтеза соединения THK-5059
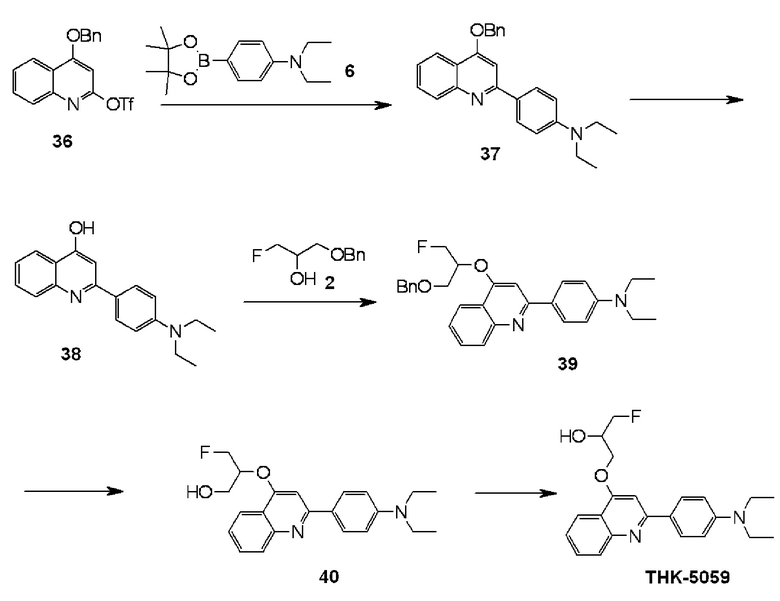
[01734]
Синтез соединения 37
К смеси соединения 36 (1,68 г, 4,38 ммоль), соединения 6 (1,21 г, 4,38 ммоль) и 1,2-диметоксиэтана (36 мл) добавляли карбонат калия (1,82 г 13,2 ммоль), воду (0,76 мл) и тетракистрифенилфосфин палладия (250 мг, 0,22 ммоль) в атмосфере аргона, и смесь перемешивали при 80°C в течение 1,5 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=20/1, 9/1) с получением соединения 37 (1,73 г) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 383[M+H]+
[0175]
Синтез соединения 38
К смеси соединения 37 (1,73 г) и анизола (4 мл) добавляли метансульфоновую кислоту (12 мл) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 3 дней. К реакционному раствору добавляли ледяную воду и затем этилацетат. Раствор делали основным водным раствором карбоната калия и экстрагировали этилацетатом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток промывали н-гексаном/этилацетатом (1/1) и затем сушили с получением соединения 38 (1,20 г, 94% из соединения 36) в виде бледно-желтых кристаллов.
точка плавления 290-291°C, APCI-MS m/z 293[M+H]+
[0176]
Синтез соединения 39
К смеси соединения 38 (600 мг, 2,05 ммоль), соединения 2 (454 мг, 2,46 ммоль), трифенилфосфина (646 мг, 2,46 ммоль) и тетрагидрофурана (20 мл) добавляли по каплям диизопропилазодикарбоксилат (0,49 мл, 2,46 ммоль) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=9/1) с получением соединения 39 (940 мг, 100%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 459[M+H]+
[0177]
Синтез соединения 40
К смеси соединения 39 (940 мг, 2,05 ммоль) и анизола (2 мл) добавляли метансульфоновую кислоту (6 мл) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 10 минут. К реакционному раствору добавляли ледяную воду и затем этилацетат. Раствор делали основным водным раствором карбоната калия и экстрагировали этилацетатом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=4/1, 2/1) и затем перекристаллизовывали из этилацетата с получением соединения 40 (627 мг, 83%) в виде бледно-желтых кристаллов.
точка плавления 146-147°C, APCI-MS m/z 369[M+H]+
[0178]
Синтез соединения THK-5059
К раствору этилацетата (100 мл) соединения 40 (700 мг, 1,90 ммоль) добавляли суспензию этилацетата (10 г/20 мл) силикагеля и смесь перемешивали при комнатной температуре в течение 3 дней. Силикагель удаляли фильтрацией, и раствор промывали этилацетатом. Фильтрат и промывной раствор объединяли, и растворитель отгоняли при пониженном давлении. Остаток перекристаллизовывали из н-гексана/этилацетата (1/1) с получением соединения THK-5059 (484 мг, 69%) в виде бледно-желтых кристаллов.
точка плавления 155-155,5°C, 1H ЯМR (400 МГц, ДМСО-d6) δ 1,15 (6H, т, J=7,0 Гц), 3,42 (4H, кв., J=7,0 Гц), 4,19-4,41 (3H, м), 4,61 (1H, ддд, J=48, 9,7, 5,0 Гц), 4,65 (1H, ддд, J=48, 10, 4,5 Гц), 5,64 (1H, д, J=5,4 Гц), 6,77 (2H, д, J=9,1 Гц), 7,42 (1H, с), 7,42-7,47 (1Н, м), 7,66-7,71 (1Н, м), 7,89 (1H, д, J=8,2 Гц), 8,10-8,13 (3H, м)
ИК (Nujol) 3150, 1610 см-1
APCI-MS m/z 369[M+H]+
[0179]
Способ синтеза соединения THK-5064
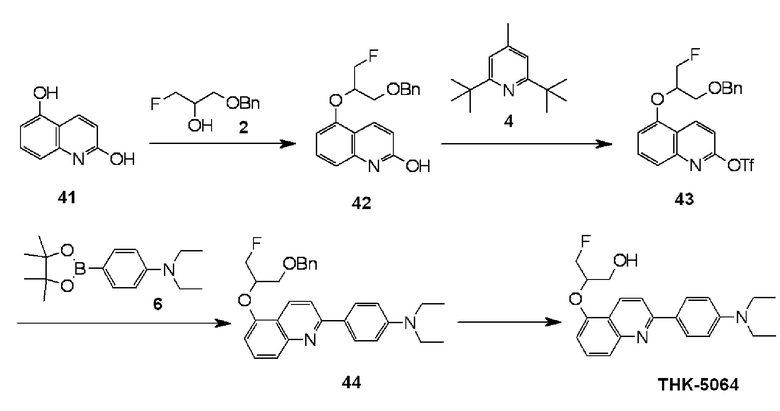
[0180]
Синтез соединения 42
К смеси соединения 41 (520 мг, 3,23 ммоль), соединения 2 (710 мг, 3,87 ммоль), трифенилфосфина (1,02 г, 3,87 ммоль) и тетрагидрофурана (30 мл) добавляли по каплям диизопропилазодикарбоксилат (0,77 мл, 3,87 ммоль) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли соединение 2 (360 мг, 1,94 ммоль), трифенилфосфин (0,51 г, 1,94 ммоль), диизопропилазодикарбоксилат (0,38 мл, 1,94 ммоль) и тетрагидрофуран (10 мл) с последующим дополнительным перемешиванием при комнатной температуре в течение 2 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=2/1, 1/1) с получением соединения 42 (600 мг, 57%) в виде бледно-коричневого твердого вещества.
APCI-MS m/z 328[M+H]+
[0181]
Синтез соединения 43
К раствору в метиленхлориде (15 мл) соединения 42 (590 мг, 1,80 ммоль) и соединения 4 (520 мг, 2,52 ммоль),добавляли по каплям трифторметансульфоновый ангидрид (0,36 мл, 2,16 ммоль) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли соединение 4 (74 мг, 0,36 ммоль) и трифторметансульфоновый ангидрид (0,061 мл, 0,37 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 30 минут. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан, н-гексан/этилацетат=9/1) с получением соединения 43 (560 мг, 68%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 460[M+H]+
[0182]
Синтез соединения 44
К смеси соединения 43 (550 мг, 1,20 ммоль), соединения 6 (330 мг, 1,20 ммоль) и 1,2-диметоксиэтана (20 мл) добавляли карбонат калия (500 мг, 3,59 ммоль), воду (0,21 мл) и тетракис трифенилфосфин палладия (69 мг, 0,06 ммоль) в атмосфере аргона, и смесь перемешивали при 80°C в течение 1,5 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, разбавляли этилацетатом и промывали водой. Органический слой сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=4/1) с получением соединения 44 (550 мг, 100%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 459[M+H]+
[0183]
Синтез соединения THK-5064
К смеси соединения 44 (540 мг, 1,18 ммоль) и анизола (1,0 мл) добавляли метансульфоновую кислоту (3,0 мл) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 20 минут. К реакционному раствору добавляли ледяную воду и затем этилацетат. Раствор делали основным водным раствором карбоната калия и экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=4/1) с получением соединения THK-5064 (403 мг, 93%) в виде оранжевого порошка.
1H ЯМR (500 МГц, ДМСО-d6) δ 1,14 (6H, т, J=7,0 Гц), 3,42 (4H, кв., J=7,0 Гц), 3,73-3,78 (2H, м), 4,70-4,88 (3H, м), 5,14 (1H, т, J=5,3 Гц), 6,79 (2H, д, J=9,0 Гц), 7,10 (1Н, д, J=7,4 Гц), 7,56 (1Н, д, J=8,7 Гц), 7,61 (1H, т, J=8,4 Гц), 7,98 (1H, д, J=8,7 Гц), 8,11 (2H, д, J=9,0 Гц), 8,51 (1H, д, J=9,0 Гц)
ИК (Nujol) 3400, 1610 см-1
APCI-MS m/z 369[M+H]+
[0184]
Способ синтеза соединения THK-5065
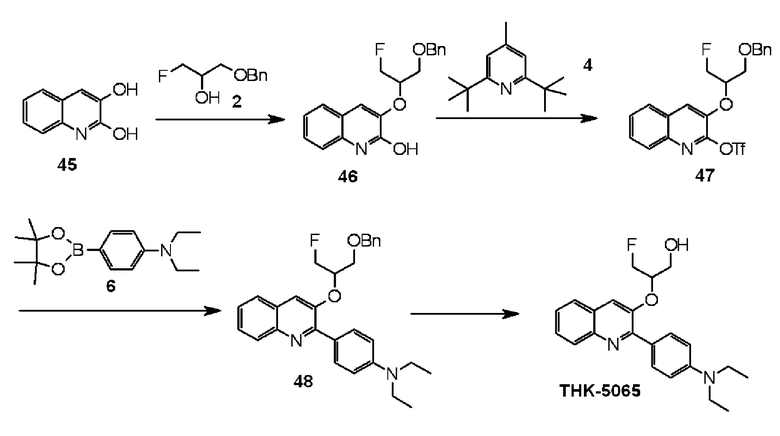
[0185]
Синтез соединения 46
К смеси соединения 45 (3,0 г, 18,6 ммоль), соединения 2 (3,5 г, 19,0 ммоль), трифенилфосфина (5,77 г, 22,0 ммоль) и тетрагидрофурана (60 мл) добавляли по каплям диизопропилазодикарбоксилат (4,45 г, 22,0 ммоль) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=2/1, 1/1) с получением соединения 46 (3,0 г, 49%) в виде бесцветного твердого вещества.
точка плавления 124-125°C
[0186]
Синтез соединения 47
К раствору в метиленхлориде (20 мл) соединения 46 (2,62 г, 8,0 ммоль) и соединения 4 (2,2 г, 10,4 ммоль) добавляли по каплям раствор в метиленхлориде (10 мл) трифторметансульфонового ангидрида (2,7 г, 9,6 ммоль) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 16 часов. Добавляли соединение 4 (2,2 г, 10,4 ммоль) и трифторметансульфоновый ангидрид (2,7 г, 9,6 ммоль) с последующим дальнейшим перемешиванием при комнатной температуре в течение 16 часов. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=20/1) с получением соединения 47 (1,47 г, 40%) в виде бесцветного маслянистого вещества.
APCI-MS m/z 460[M+H]+
[0187]
Синтез соединения 48
К смеси соединения 47 (1,20 г, 2,61 ммоль), соединения 6 (0,72 г, 2,61 ммоль) и 1,2-диметоксиэтана (30 мл) добавляли карбонат натрия (560 мг, 5,28 ммоль), воду (3 мл) и тетракистрифенилфосфин палладия (150 мг, 0,13 ммоль) в атмосфере аргона, и смесь нагревали в сосуде с обратным холодильником в течение 3 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, разбавляли этилацетатом, промывали по очереди водой и насыщенным солевым раствором и затем сушили. Растворитель отгоняли при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=8/1, 6/1) с получением соединения 48 (1,06 г, 88%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 459[M+H]+
[0188]
Синтез соединения THK-5065
К смеси соединения 48 (1,27 г, 2,77 ммоль) и анизола (1,0 мл) добавляли метансульфоновую кислоту (10 мл), и смесь перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор разбавляли этилацетатом и выливали в ледяную воду с последующим отделением жидкости. Водный слой делали основным концентрированной аммиачной водой и экстрагировали этилацетатом. Экстракционную жидкость промывали по очереди водой и насыщенным солевым раствором и затем сушили. Растворитель отгоняли при пониженном давлении, и остаток перекристаллизовывали из простого диизопропилового эфира с получением соединения THK-5065 (758 мг, 74%) в виде бледно-желтых кристаллов.
точка плавления 135-136°C, 1H ЯМR (400 МГц, CDCl3) δ 1,20 (6H, т, J=7,1 Гц), 1,95 (1Н, уш.), 3,42 (4H, кв., J=7,1 Гц), 3,77-3,88 (2H, м), 4,56-4,65 (1Н, м), 4,65-4,69 (1Н, м), 4,77-4,81 (1Н, м), 6,75 (2H, д, J=9,0 Гц), 7,47 (1Н, м), 7,57 (1Н, м), 7,61 (1Н, с), 7,68 (1H, дд, J=8,2, 1,5 Гц), 7,93 (2H, д, J=9,0 Гц), 8,07 (1H, д, J=8,5 Гц)
ИК (Nujol) 3200, 1617, 1606 см-1
APCI-MS m/z 369[M+H]+
[0189]
Способ синтеза соединения THK-5066
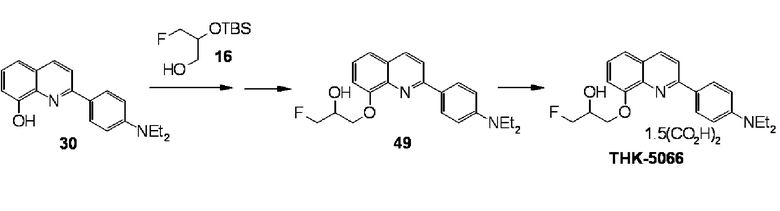
[0190]
Синтез соединения 49
К смеси соединения 30 (648 мг, 2,2 ммоль), соединения 16 (559 мг, 2,7 ммоль), трифенилфосфина (880 мг, 3,36 ммоль) и тетрагидрофурана (10 мл) добавляли по каплям раствор в тетрагидрофуране (5 мл) диизопропилазодикарбоксилата (678 мг, 3,36 ммоль), и смесь перемешивали при комнатной температуре в течение 16 часов. Добавляли соединение 16 (447 мг, 2,15 ммоль), трифенилфосфин (700 мг, 2,67 ммоль) и диизопропилазодикарбоксилат (542 мг, 2,68 ммоль) с последующим дальнейшим перемешиванием при комнатной температуре в течение 16 часов. Реакционный раствор отгоняли при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9) с получением маслянистого продукта (1,24 г). К полученному маслянистому продукту добавляли тетрагидрофуран (8 мл) и тетрабутиламмонийфторид (2,2 мл/1M раствора тетрагидрофурана) с последующим перемешиванием при комнатной температуре в течение 1 часа. Реакционный раствор отгоняли при пониженном давлении, и остаток растворяли в хлороформе. Раствор промывали водным насыщенным раствором гидрокарбоната натрия и сушили. Растворитель органического слоя отгоняли при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/19→1/9→1/4→1/2) с получением соединения 49 (497 мг, 61%) в виде бледно-желтого пеноподобного вещества.
APCI-MS m/z 369[M+H]+
[0191]
Синтез соединения THK-5066
К раствору в ацетоне (10 мл) соединения 49 (494 мг, 1,30 ммоль) добавляли щавелевую кислоту (181 мг, 2,01 ммоль) для образования оксалата. Ацетон отгоняли при пониженном давлении, и остаток кристаллизовали из диэтилового эфира с получением соединения THK-5066 (584 мг, 87%) в виде темно-оранжевого твердого вещества.
1H ЯМR (500 МГц, ДМСО-d6) δ 1,15 (6H, кв., J=7,0 Гц), 3,43 (4H, кв., J=7,0 Гц), 4,15-4,30 (3H, м), 4,6-4,8 (2H, м), 6,81 (2H, д, J=9,0 Гц), 7,24 (1H, д, J=7,7 Гц), 7,40 (1Н, т, J=7,7 Гц), 7,49 (1Н, д, J=7,7 Гц), 8,03 (1H, д, J=8,6 Гц), 8,14 (2H, д, J=9,0 Гц), 8,27 (1Н, д, J=8,6 Гц)
ИК (Nujol) 1592, 1461 см-1
APCI-MS m/z 369[M+H]+
[0192]
Способ синтеза соединения THK-5070
[0193]
Синтез соединения 50
К суспензии в N,N-диметилформамиде (20 мл) соединения 1 (2,00 г, 12,4 ммоль) добавляли по каплям тионилхлорид (3,56 мл, 49,6 ммоль) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 2 часов, и затем добавляли тионилхлорид (3,56 мл, 49,6 ммоль) с последующим дополнительным перемешиванием при 50°C в течение 3,5 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, выливали в ледяную воду и экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/20) с получением соединения 50 (1,72 г, 77%) в виде бледно-желтого твердого вещества.
точка плавления 79-80°C, APCI-MS m/z 180/182[M+H]+
[0194]
Синтез соединения 52
К смеси соединения 50 (1,71 г, 9,52 ммоль), соединения 51 (1,51 г, 11,4 ммоль), трифенилфосфина (3,00 г, 11,4 ммоль) и тетрагидрофурана (50 мл) добавляли по каплям диизопропилазодикарбоксилат (2,27 мл, 11,4 ммоль) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/19, 1/9) с получением соединения 52 (2,05 г, 73%) в виде бесцветного твердого вещества.
точка плавления 94-95°C, APCI-MS m/z 294/296[M+H]+
[0195]
Синтез соединения 53
К смеси соединения 52 (600 мг, 2,04 ммоль), соединения 6 (560 мг, 2,04 ммоль) и 1,2-диметоксиэтана (20 мл) добавляли карбонат калия (850 мг, 6,12 ммоль), воду (0,36 мл), тетракистрифенилфосфин палладия (120 мг, 0,102 ммоль) в атмосфере аргона, и смесь перемешивали при 80°C в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду и этилацетат, и нерастворимые вещества удаляли фильтрацией целитом с последующим отделением жидкости фильтрата. Органический слой промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9) с получением соединения 53 (820 мг, 98%) в виде бледно-желтого твердого вещества.
точка плавления 87-89°C
[0196]
Синтез соединения 54
Смесь соединения 53 (820 мг, 2,02 ммоль), метанола (50 мл) и 1н хлористоводородной кислоты (2,02 мл) перемешивали при 70°C в течение 1 часа. Реакционному раствору давали возможность охладиться до комнатной температуры и нейтрализовали гидрокарбонатом натрия, и затем добавляли этилацетат и сульфат натрия с последующим перемешиванием при комнатной температуре. Нерастворимые вещества удаляли фильтрацией, и растворитель фильтрата отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1, этилацетат) с получением соединения 54 (720 мг, 97%) в виде бледно-желтого пеноподобного вещества.
[0197]
Синтез соединения 55
К раствору в пиридине (5 мл)-тетрагидрофуране (5 мл) соединения 54 (480 мг, 1,31 ммоль) добавляли по каплям в течение 20 минут раствор в тетрагидрофуране (5 мл) паратолуолсульфонового ангидрида (641 мг, 1,97 ммоль) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при той же температуре в течение 10 минут. К реакционному раствору добавляли, метанол, и растворитель отгоняли при пониженном давлении, и затем добавляли содержащий толуол триэтиламин, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 1/1) с получением соединения 55 (340 мг, 50%) в виде желтого пеноподобного вещества.
[0198]
Синтез соединения THK-5070
К раствору в тетрагидрофуране (20 мл) соединения 55 (390 мг, 0,749 ммоль) добавляли 3,4-дигидро-2H-пиран (0,204 мл, 2,25 ммоль) и моногидрат паратолуолсульфоновой кислоты (168 мг, 0,976 ммоль), и смесь перемешивали при комнатной температуре в течение 18 часов, и затем добавляли 3,4-дигидро-2H-пиран (0,475 мл, 5,24 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 24 часов. Реакционный раствор нейтрализовали триэтиламином, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2) и затем очищали колоночной хроматографией на NH силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1) и кристаллизовали из метанола (15 мл) с получением соединения THK-5070 (96 мг, 21%) в виде бледно-желтых кристаллов.
точка плавления 80-81°C
ИК (Nujol) 1597 см-1, APCI-MS m/z 605[M+H]+
[0199]
Способ синтеза соединения THK-5071
[0200]
Синтез соединения 57
К смеси соединения 34 (735 мг, 1,6 ммоль), соединения 56 (435 мг, 1,76 ммоль) и 1,2-диметоксиэтана (8 мл) добавляли водный 2M раствор карбоната натрия (1,6 мл, 3,2 ммоль) и тетракистрифенилфосфин палладия (92 мг, 0,08 ммоль) в атмосфере аргона, и смесь перемешивали при 90°C в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, и нерастворимые вещества удаляли фильтрацией, и раствор промывали хлороформом. Фильтрат и промывной раствор объединяли, и смесь сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/5, 1/2) с получением соединения 57 (678 мг, 98%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 431[M+H]+
[0201]
Синтез соединения THK-5071
К смеси соединения 57 (638 мг, 1,48 ммоль) и анизола (5 мл) добавляли метансульфоновую кислоту (1 мл) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 30 минут. К реакционному раствору добавляли воду, и раствор промывали этилацетатом. Водный слой делали основным карбонатом калия и экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) и затем промывали диизопропиловым эфиром-хлороформом с получением соединения THK-5071 (394 мг, 78%) в виде желтого твердого вещества.
точка плавления 134-135,5°C, 1H ЯМR (500 МГц, ДМСО-d6) δ 3,01 (6H, с), 3,7-3,8 (2H, м), 4,7-4,9 (3H, м), 6,84 (2H, д, J=9 Гц), 7,18 (1H, дд, J=8,7, 2,3 Гц), 7,48 (1H, д, J=2,3 Гц), 7,83 (1Н, д, J=9 Гц), 7,88 (1Н, д, J=8,7 Гц), 8,13 (2H, д, J=9 Гц), 8,23 (1H, д, J=8,4 Гц)
ИК (Nujol) 1597, 1505, 1202 см-1,
APCI-MS m/z 341[M+H]+
[0202]
Способ синтеза соединения THK-5072
[0203]
Синтез соединения 58
К суспензии в N,N-диметилформамиде (30 мл) соединения 15 (2,87 г, 17,81 ммоль) добавляли по каплям тионилхлорид (5,11 мл, 71,23 ммоль) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 30 минут, с последующим перемешиванием при 70°C в течение 30 минут. Реакционному раствору давали возможность охладиться до комнатной температуры, выливали в ледяную воду и экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1, этилацетат) с получением соединения 58 (3,04 г, 95%) в виде коричневатого твердого вещества.
APCI-MS m/z 180/182[M+H]+
[0204]
Синтез соединения 59
К раствору в тетрагидрофуране (30 мл) соединения 58 (1,50 г, 8,35 ммоль) и простого этилвинилового эфира (1,21 г, 16,7 ммоль) добавляли соль пиридина паратолуолсульфоновой кислоты (210 мг, 0,84 ммоль) и смесь перемешивали при комнатной температуре в течение 16 часов. После того как pH реакционного раствора доводили до 8, используя триэтиламин, растворитель отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/20, 1/3) с получением соединения 59 (1,91 г, 91%) в виде розового маслянистого вещества.
APCI-MS m/z 252[M+H]+
[0205]
Синтез соединения 60
К раствору в 1,2-диметоксиэтане (62 мл) соединения 59 (1,90 г, 7,55 ммоль) и соединения 6 (2,08 г, 7,55 ммоль) добавляли карбонат калия (3,13 г, 22,65 ммоль), воду (1,31 мл) и тетракистрифенилфосфин палладия (436 мг, 0,38 ммоль) в атмосфере аргона, и смесь перемешивали при 80°C в течение 27 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/19) и затем перекристаллизовывали из этилацетата-н-гесана (1/9) с получением соединения 60 (1,50 г, 54%) в виде желтых кристаллов.
точка плавления 86-87°C
APCI-MS m/z 365[M+H]+
[0206]
Синтез соединения 61
К раствору в метиленхлориде (12 мл) соединения 60 (1,50 г, 4,12 ммоль) добавляли по каплям трифторуксусную кислоту (8 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 15 минут. К реакционному раствору добавляли ледяную воду и этилацетат и экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток перекристаллизовывали из этилацетата-н-гексана с получением соединения 61 (1,13 г, 94%) в виде желтых кристаллов.
точка плавления 223-224°C
APCI-MS m/z 293[M+H]+
[0207]
Синтез соединения THK-5072
К смеси соединения 61 (500 мг, 1,71 ммоль), соединения 62 (690 мг, 2,05 ммоль), трифенилфосфина (540 мг, 2,05 ммоль) и тетрагидрофурана (25 мл) добавляли по каплям диизопропилазодикарбоксилат (0,41 мл, 2,05 ммоль) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли соединение 62 (690 мг, 2,05 ммоль), трифенилфосфин (540 мг, 2,05 ммоль) и тетрагидрофуран (5 мл) в условиях охлаждения льдом и перемешивания, и по каплям добавляли диизопропилазодикарбоксилат (0,41 мл, 2,05 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 6 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением бледно-желтого маслянистого вещества (1,47 г). К раствору в анизоле (4 мл) настоящего продукта добавляли по каплям метансульфоновую кислоту (12 мл) при охлаждении льдом и перемешивании с последующим перемешиванием при комнатной температуре в течение 15 минут. Реакционный раствор выливали в ледяную воду и добавляли этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9, 1/1) с получением соединения THK-5072 (620 мг, 69%) в виде желтого пеноподобного вещества.
1H ЯМR (400 МГц, ДМСО-d6) δ 1,15 (6H, т, J=7,3 Гц), 2,33 (3H, с), 3,43 (4H, кв., J=7,3 Гц), 3,57-3,71 (2H, м), 4,30 (1H, дд, J=11, 5,7 Гц), 4,38 (1H, дд, J=11, 3,0 Гц), 4,71-4,77 (1Н, м), 5,11 (1H, т, J=5,6 Гц), 6,79 (2H, д, J=9,1 Гц), 7,01 (1H, дд, J=8,8, 2,4 Гц), 7,32 (1H, д, J=2,1 Гц), 7,39 (2H, д, J=8,2 Гц), 7,75 (2H, д, J=8,5 Гц), 7,78 (1H, д, J=8,8 Гц), 7,85 (1H, д, J=8,5 Гц), 8,10 (2H, д, J=9,1 Гц), 8,21 (1H, д, J=8,8 Гц)
ИК (Nujol) 1619, 1593 см-1,
APCI-MS m/z 521[M+H]+
[0208]
Способ синтеза соединения THK-5073
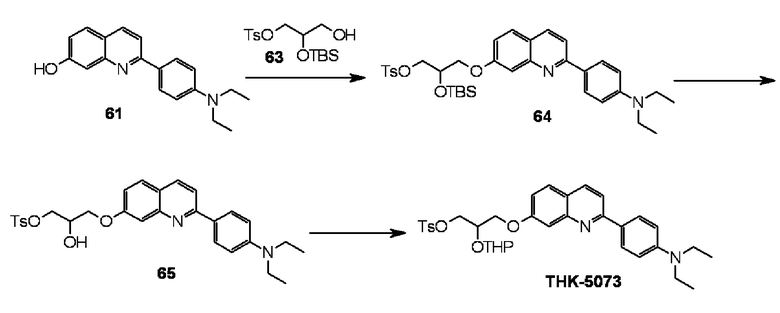
[0209]
Синтез соединения 64
К раствору в тетрагидрофуране (45 мл) соединения 61 (630 мг, 2,16 ммоль), соединения 63 (932 мг, 2,59 ммоль) и трифенилфосфина (678 мг, 2,59 ммоль) добавляли по каплям диизопропилазодикарбоксилат (0,513 мл, 2,59 ммоль) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли соединение 63 (788 мг, 2,18 ммоль), трифенилфосфин (573 мг, 2,18 ммоль) и тетрагидрофуран (5 мл) в условиях охлаждения льдом и перемешивания, и по каплям добавляли диизопропилазодикарбоксилат (0,433 мл, 2,18 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 3 дней. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9) с получением соединения 64 (720 мг, 53%) в виде бледно-желтого пеноподобного вещества.
APCI-MS m/z 635[M+H]+
[0210]
Синтез соединения 65
К раствору в метиленхлориде (12 мл) соединения 64 (710 мг, 1,12 ммоль) добавляли по каплям трифторуксусную кислоту (8 мл) при охлаждении льдом и перемешивании добавляли воду (2 мл) и смесь перемешивали при комнатной температуре в течение 5,5 часов. К реакционному раствору добавляли ледяную воду и добавляли этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 8, используя водный насыщенный раствор гидрокарбоната натрия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4, 1/2) с получением соединения 65 (520 мг, 85%) в виде желтого пеноподобного вещества.
APCI-MS m/z 221[M+H]+
[0211]
Синтез соединения THK-5073
К раствору в метиленхлориде (60 мл) соединения 65 (290 мг, 0,557 ммоль) добавляли 3,4-дигидро-2H-пиран (0,51 мл, 5,6 ммоль) и пиридиновую соль паратолуолсульфоновой кислоты (182 мг, 0,724 ммоль) и смесь перемешивали при комнатной температуре в течение 3 дней. К реакционному раствору добавляли моногидрат паратолуолсульфоновой кислоты (125 мг, 0,724 ммоль) и 3,4-дигидро-2H-пиран (0,51 мл, 5,6 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 1 часа. Реакционный раствор нейтрализовали триэтиламином, и растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/3, 1/2) с получением соединения THK-5073 (336 мг, 100%) в виде желтого маслянистого вещества.
1H ЯМR (500 МГц, ДМСО-d6) δ 1,15 (6H, т, J=7,2 Гц), 1,36-1,74 (7H, м), 2,34 (3H, с), 3,43 (4H, кв., J=7,2 Гц), 3,69-3,75, 3,84-3,90 (1Н, м), 4,13-4,37 (5H, м), 4,71-4,76, 4,86-4,88 (1Н, м), 6,79 (2H, д, J=9,0 Гц), 7,04 (1H, дд, J=9,0, 1,9 Гц), 7,25-7,29 (1Н, м), 7,39-7,44 (2H, м), 7,77-7,82 (3H, м), 7,86 (1H, д, J=8,7 Гц), 8,11 (2Н, д, J=8,7 Гц), 8,19-8,25 (1H, м)
APCI-MS m/z 605[M+H]+
[0212]
Способ синтеза соединения THK-5077
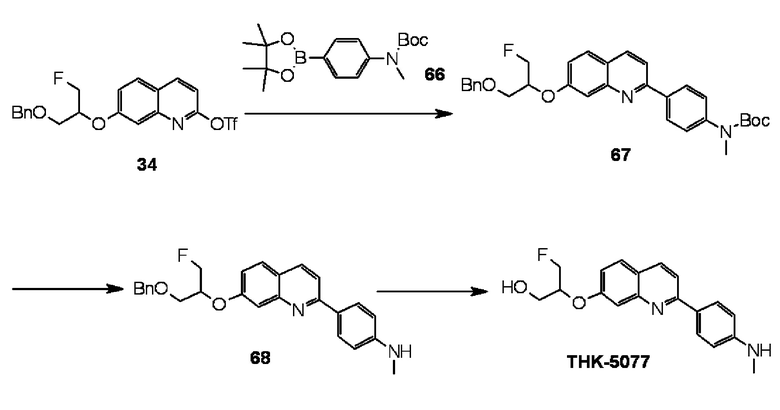
[0213]
Синтез соединения 67
К смеси соединения 34 (1,50 г, 3,3 ммоль), соединения 66 (1,2 г, 3,6 ммоль) и 1,2-диметоксиэтана (15 мл) добавляли водный 2M раствор карбоната натрия (3,3 мл, 6,6 ммоль) и тетракистрифенилфосфин палладия (189 мг, 0,16 ммоль) в атмосфере аргона, и смесь перемешивали при 90°C в течение 1,5 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, и нерастворимые вещества удаляли фильтрацией, и раствор промывали хлороформом. Фильтрат и промывной раствор объединяли, и смесь промывали водным насыщенным раствором гидрокарбоната натрия и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9, 1/4, 1/2) с получением соединения 67 (1,70 г, 100%) в виде бесцветного маслянистого вещества.
APCI-MS m/z 517[M+H]+
[0214]
Синтез соединения 68
К раствору в метиленхлориде (8 мл) соединения 67 (1,70 г, 3,29 ммоль) добавляли по каплям трифторуксусную кислоту (2 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель реакционного раствора отгоняли при пониженном давлении и добавляли хлороформ. Раствор делали основным водным насыщенным раствором гидрокарбоната натрия и карбонатом калия и экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=3/1) с получением соединения 68 (1,34 г, 97%) в виде желтого маслянистого вещества.
APCI-MS m/z 417[M+H]+
[0215]
Синтез соединения THK-5077
К смеси соединения 68 (101 мг, 0,243 ммоль) и тиоанизола (2 мл) добавляли метансульфоновую кислоту (0,4 мл) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 2 часов. К реакционному раствору добавляли ледяную воду, и раствор промывали этилацетатом. Этилацетатный слой экстрагировали водой, и слой экстракта и предыдущий водный слой объединяли. Смесь делали основной карбонатом калия и экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=2/1, 1/1, 1/2) и затем перекристаллизовывали из метанола-диизопропилового эфира с получением соединения THK-5077 (54 мг, 68%) в виде желтого твердого вещества.
точка плавления 143,5-145,5°C, 1H ЯМR (400 МГц, ДМСО-d6) δ 2,75 (3H, д, J=4,9 Гц), 3,6-3,8 (2Н, м), 4,6-4,9 (3H, м), 5,10-5,13 (1Н, м), 5,12 (1H, кв., J=4,9 Гц), 6,10 (1Н, уш. с), 6,66 (2H, д, J=9,0 Гц), 7,16 (1H, дд, J=8,8, 2,4 Гц), 7,45 (1H, д, J=2,4 Гц), 7,81 (1Н, д, J=8,9 Гц), 7,84 (1H, д, J=8,8 Гц), 8,07 (2H, д, J=9,0 Гц), 8,20 (1Н, д, J=7,5 Гц)
ИК (Nujol) 2854, 1612, 1461 см-1
APCI-MS m/z 327[M+H]+
[0216]
Способ синтеза соединения THK-5078
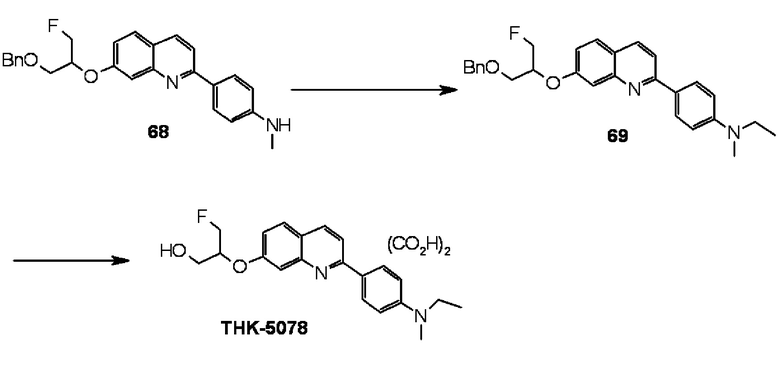
[0217]
Синтез соединения 69
Смесь соединения 68 (637 мг, 1,50 ммоль), ацетоальдегида (115 мг, 2,60 ммоль), пиколин-боранового комплекса (279 мг, 2,60 ммоль) и метанола (10 мл) в уксусной кислоте (1 мл) перемешивали при комнатной температуре в течение 16 часов. Добавляли ацетоальдегид (1,15 г, 26 ммоль) и пиколин-борановый комплекс (279 мг, 2,60 ммоль) с последующим дальнейшим перемешиванием при комнатной температуре в течение 16 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и к остатку добавляли водную 10% хлористоводородную кислоту (10 мл). После перемешивания при комнатной температуре в течение 30 минут раствор делали основным добавлением карбоната калия. Реакционный раствор экстрагировали хлороформом, и экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=4/1) для получения соединения 69 (640 мг, 94%) в виде желтого маслянистого вещества.
APCI-MS m/z 445[M+H]+
[0218]
Синтез соединения THK-5078
К смеси соединения 69 (639 мг, 1,44 ммоль) и тиоанизола (5 мл) добавляли метансульфоновую кислоту (1 мл) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли ледяную воду, и раствор промывали этилацетатом. Этилацетатный слой экстрагировали водой, и экстракционный слой и предыдущий водный слой объединяли. Смесь делали основной карбонатом калия и экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=2/1, 1/1) и затем оксалат образовывался в ацетоне с получением соединения THK-5078 (460 мг, 71%) в виде оранжевых кристаллов.
точка плавления 134-136°C, 1H ЯМR (400 МГц, ДМСО-d6) δ 1,10 (3H, т, J=7,0 Hz), 3,49 (2H, кв., J=7,0 Гц), 3,7-3,8 (2H, м), 4,6-4,9 (3H, м), 6,83 (2H, д, J=9,0 Гц), 7,20 (1H, дд, J=8,8, 2,4 Гц), 7,49 (1H, д, J=2,4 Гц), 7,85 (1H, д, J=9,0 Гц), 7,89 (1Н, д, J=7,3 Гц), 8,11 (2H, д, J=9,0 Гц), 8,26 (1H, д, J=9,0 Гц)
ИК (Nujol) 1603, 1458, 1214 см-1
APCI-MS m/z 355[M+H]+
[0219]
Способ синтеза соединения THK-5090
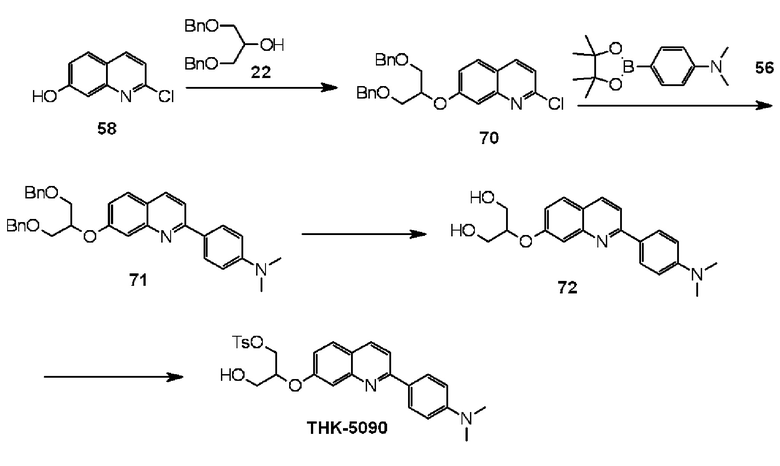
[0220]
Синтез соединения 70
К раствору в тетрагидрофуране (60 мл) соединения 58 (1,36 г, 7,57 ммоль), соединения 22 (2,48 г, 9,09 ммоль) и трифенилфосфина (2,38 г, 9,09 ммоль) добавляли по каплям диизопропилазодикарбоксилат (1,8 мл, 9,09 ммоль) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли соединение 22 (1,24 г, 4,55 ммоль), трифенилфосфин (1,19 г, 4,55 ммоль) и тетрагидрофуран (10 мл) в условиях охлаждения льдом и перемешивания, и добавляли по каплям диизопропилазодикарбоксилат (0,9 мл, 4,55 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 4 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9) с получением соединения 70 (3,04 г, 93%) в виде бледно-розового маслянистого вещества.
APCI-MS m/z 434[M+H]+
[0221]
Синтез соединения 71
К раствору в 1,2-диметоксиэтане (40 мл) соединения 70 (2,00 г, 4,61 ммоль) и соединения 56 (1,14 г, 4,61 ммоль) добавляли карбонат калия (1,91 г, 13,83 ммоль), воду (0,8 мл) и тетракистрифенилфосфин палладия (270 мг, 0,23 ммоль) в атмосфере аргона, и смесь перемешивали при 80°C в течение 2 дней. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9) с получением соединения 71 (1,22 г, 51%) в виде желтого маслянистого вещества.
APCI-MS m/z 519[M+H]+
[0222]
Синтез соединения 72
К смеси соединения 71 (1,21 г, 2,33 ммоль) и анизола (3 мл) добавляли по каплям метансульфоновую кислоту (9 мл) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор выливали в ледяную воду, и раствор экстрагировали этилацетатом-тетрагидрофураном. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток промывали этилацетатом-н-гексаном (1/2) с получением соединения 72 (680 мг, 86%) в виде желтых кристаллов.
точка плавления 193-194°C
APCI-MS m/z 339[M+H]+
[0223]
Синтез соединения THK-5090
К раствору в пиридине (15 мл) соединения 72 (690 мг, 2,04 ммоль) по каплям добавляли раствор в тетрагидрофуране (15 мл) паратолуолсульфонового ангидрида (1,00 г, 3,06 ммоль) в течение 35 минут в условиях охлаждения льдом и перемешивания, и смесь перемешивали при той же температуре в течение 30 минут. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении (толуол азеотроп). Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 1/1) и затем перекристаллизовывали из этилацетата-н-гесана с получением соединения THK-5090 (459 мг, 46%) в виде бледно-желтых кристаллов.
точка плавления 100-102°C, 1Н ЯМR (400 МГц, ДМСО-d6) δ 2,33 (3H, с), 3,03 (6H, с), 3,56-3,71 (2H, м), 4,30 (1Н, дд, J=11, 6,0 Гц), 4,39 (1H, дд, J=11, 2,7 Гц), 4,72-4,78 (1Н, м), 5,12 (1Н, т, J=5,4 Гц), 6,84 (2H, д, J=9,1 Гц), 7,02 (1H, дд, J=9,1, 2,4 Гц), 7,33 (1H, д, J=2,4 Гц), 7,39 (2H, д, J=7,9 Гц), 7,75 (2H, д, J=8,2 Гц), 7,79 (1H, д, J=9,1 Гц), 7,88 (1Н, д, J=8,5 Гц), 8,13 (2H, д, J=9,1 Гц), 8,23 (1Н, д, J=7,9 Гц)
ИК (Nujol) 1731, 1609, 1597 см-1
APCI-MS m/z 493[M+H]+
[0224]
Способ синтеза соединения THK-5075
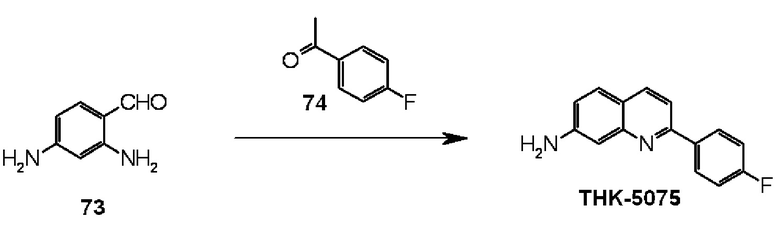
[0225]
Синтез соединения THK-5075
Смесь соединения 73 (1,19 г, 8,74 ммоль), соединения 74 (1,06 мл, 8,74 ммоль), гидроксида калия (590 мг, 10,5 ммоль) и толуола (40 мл) нагревали кипячением в сосуде с обратным холодильником в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и добавляли водный насыщенный раствор аммония хлорида и раствор экстрагировали этилацетатом. Нерастворимые вещества удаляли фильтрацией целитом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=9/1, 2/1) и затем перекристаллизовывали из этилацетата-н-гексана с получением соединения THK-5075 (737 мг, 35%) в виде бледно-желтых кристаллов.
точка плавления 160-161°C, 1H ЯМR (500 МГц, ДМСО-d6) δ 5,80 (2H, с), 6,96-7,00 (2H, м), 7,33 (2H, т, J=8,8 Гц), 7,63 (1H, д, J=9,6 Гц), 7,68 (1H, д, J=8,4 Гц), 8,11 (1H, д, J=8,4 Гц), 8,24 (2H, дд, J=9,0,5,5 Гц)
ИК (Nujol) 3388, 1619, 1600 см-1
APCI-MS m/z 239[M+H]+
[0226]
Способ синтеза соединения THK-5076
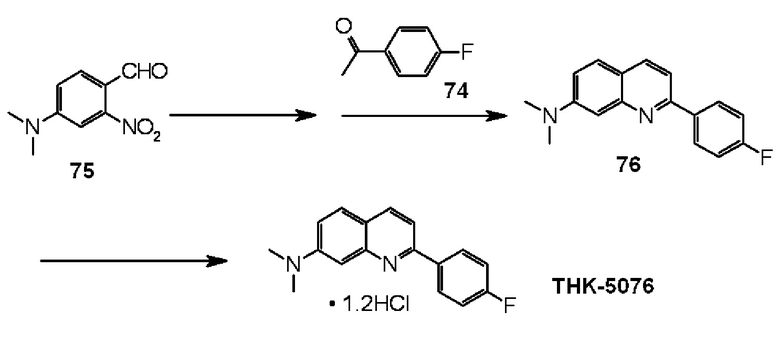
[0227]
Синтез соединения 76
К суспензии в этаноле (20 мл) соединения 75 (1,00 г, 5,15 ммоль) добавляли порошок железа (1,15 г) и 0,1н хлористоводородную кислоту (2,58 мл) и смесь нагревали кипячением в сосуде с обратным холодильником в течение 16 часов, и затем добавляли 0,1н хлористоводородную кислоту (7,72 мл) с последующим нагреванием в сосуде с обратным холодильником в течение 6 часов. К реакционному раствору добавляли соединение 74 (0,625 мл, 5,15 ммоль) и гидроксид калия (0,347 г, 6,18 ммоль) и смесь нагревали кипячением в сосуде с обратным холодильником в течение 3 дней. Реакционному раствору давали возможность охладиться до комнатной температуры, и нерастворимые вещества удаляли фильтрацией, и раствор промывали метанолом. Фильтрат и промывной раствор объединяли, и растворитель отгоняли при пониженном давлении. К остатку добавляли воду и раствор экстрагировали этилацетатом. Этилацетатный слой экстрагировали водной 10% хлористоводородной кислотой, и экстракционную жидкость хлористоводородной кислоты экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=9/1, 4/1) для получения соединения 76 (225 мг, 16%) в виде желтого маслянистого вещества.
APCI-MS m/z 267[M+H]+
[0228]
Синтез соединения THK-5076
К раствору в метаноле (20 мл) соединения 76 (540 мг, 2,03 ммоль) добавляли по каплям 4М хлористоводородную кислоту/этилацетат (1,02 мл), и растворитель отгоняли при пониженном давлении. Остаток перекристаллизовывали из этилацетата-изопропанола с получением соединения THK-5076 (478 мг) в виде оранжевых кристаллов.
точка плавления 250-251°C, 1H ЯМR (400 МГц, ДМСО-d6) δ 3,26 (6H, с), 7,46-7,57 (4H, м), 7,85 (1Н, д, J=8,2 Гц), 8,06 (1H, д, J=9,4 Гц), 8,23-8,29 (2H, м), 8,77 (1H, д, J=8,2 Гц), 15,0 (1H, уш.)
ИК (Nujol) 1634, 1599 см-1
APCI-MS m/z 267[M+H]+
[0229]
Способ синтеза соединения THK-5079
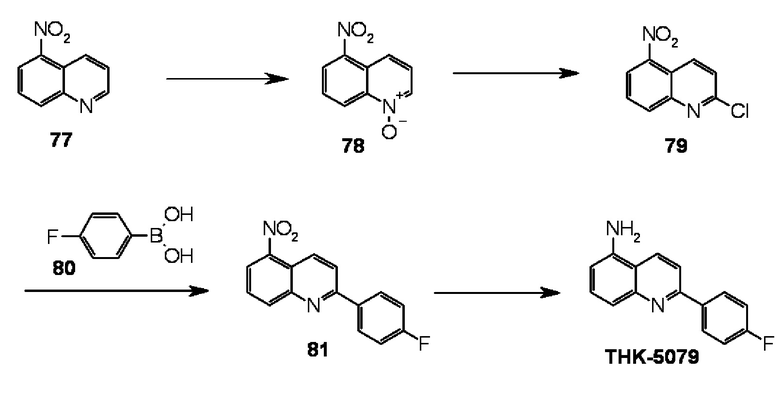
[0230]
Синтез соединения 78
К раствору в хлороформе (65 мл) соединения 77 (5,00 г, 28,71 ммоль) по каплям добавляли раствор в хлороформе (27 мл)-метаноле (7 мл) метанхлорбензойной кислоты (10,9 г, 63,16 ммоль) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли водный раствор тиосульфата натрия, и смешанный раствор перемешивали при комнатной температуре в течение 10 минут и экстрагировали хлороформом. Экстракционную жидкость промывали водным раствором карбоната калия, очищали в том виде, как он есть, колоночной хроматографией на силикагеле (элюирующий растворитель: хлороформ) и затем промывали н-гексаном-этилацетом (2/1) с получением соединения 78 (4,96 г, 91%) в виде желтых кристаллов.
точка плавления 164-165°C
APCI-MS m/z 191[M+H]+
[0231]
Синтез соединения 79
К соединению 78 (4,94 г, 26 ммоль) добавляли по каплям оксихлорид фосфора (22 мл) при охлаждении льдом и перемешивании и смесь нагревали в сосуде с обратным холодильником в течение 3 часов. Реакционный раствор охлаждали льдом и добавляли ледяную воду, и затем раствор делали основным аммиачной водой и экстрагировали хлороформом. Экстракционную жидкость промывали насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан/метиленхлорид=2/1, 1/1, 1/2) и затем промывали н-гексаном с получением соединения 79 (2,25 г, 42%) в виде бледно-желтого твердого вещества.
APCI-MS m/z 209/211[M+H]+
[0232]
Синтез соединения 81
К смеси соединения 79 (1,04 г, 5 ммоль), соединения 80 (770 мг, 5,5 ммоль) и 1,2-диметоксиэтана (25 мл) добавляли водный 2M раствор карбоната натрия (5 мл, 10 ммоль) и тетракистрифенилфосфин палладия (289 мг, 0,25 ммоль) в атмосфере аргона, и смесь перемешивали при 90°C в течение 3 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, и растворитель отгоняли при пониженном давлении. К остатку добавляли хлороформ, и нерастворимые вещества удаляли фильтрацией, и раствор промывали хлороформом. Фильтрат и промывной раствор объединяли, и смесь промывали насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: метиленхлорид/н-гексан=1/1) и затем промывали н-гексаном с получением соединения 81 (1,30 г, 97%) в виде бледно-желтого твердого вещества.
APCI-MS m/z 269[M+H]+
[0233]
Синтез соединения THK-5079
Смесь соединения 81 (1,29 г, 4,81 ммоль), 10% Pd-C (130 мг) и этанола (30 мл) перемешивали в атмосфере водорода при комнатной температуре в течение 2 часов. Катализатор удаляли фильтрацией, и растворитель фильтрата отгоняли при пониженном давлении, и затем остаток промывали н-гексаном с получением соединения THK-5079 (1,087 г, 95%) в виде желтовато-оранжевого твердого вещества.
точка плавления 105-108°C, 1H ЯМR (400 МГц, ДМСО-d6) δ 5,9-6,3 (2H, уш. с), 6,70 (1H, дд, J=8,1 Гц), 7,23 (1H, д, J=8 Гц), 7,36 (2H, т, J=9 Гц), 7,44 (1H, т, J=8 Гц), 7,98 (1H, д, J=9 Гц), 8,31 (2H, дд, J=9,5 Гц), 8,63 (1Н, д, J=9 Гц)
ИК (Nujol) 1614, 1592 см-1
APCI-MS m/z 239[M+H]+
[0234]
Способ синтеза соединения THK-5080
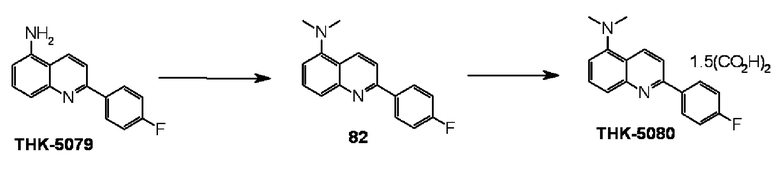
[0235]
Синтез соединения 82
Смесь соединения THK-5079 (450 мг, 1,9 ммоль), водного 35% раствора формалина (0,81 г, 9.4 ммоль), пиколин-боранового комплекса (303 мг, 2,8 ммоль) и метанола (10 мл)-уксусной кислоты (1 мл) перемешивали при комнатной температуре в течение 30 минут и добавляли пиколин-борановый комплекс (100 мг, 0,93 ммоль) с последующим перемешиванием при комнатной температуре в течение 30 минут. Растворитель реакционного раствора отгоняли при пониженном давлении, и к остатку добавляли водную 10% хлористоводородную кислоту (5 мл). После перемешивания при комнатной температуре в течение 30 минут раствор делали основным добавлением карбоната калия. Реакционный раствор экстрагировали хлороформом, и экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=49/1, 19/1) для получения соединения 82 (465 мг, 92%) в виде зеленовато-желтого маслянистого вещества.
APCI-MS m/z 267[M+H]+
[0236]
Синтез соединения THK-5080
После растворения добавлением щавелевой кислоты (312 мг, 3,47 ммоль) к раствору в ацетоне (10 мл) соединения 82 (462 мг, 1,73 ммоль) растворитель отгоняли при пониженном давлении. Остаток промывали диизопропиловым эфиром с получением соединения THK-5080 (620 мг, 89%) в виде оранжевого твердого вещества.
точка плавления 118-120°C, 1H ЯМR (500 МГц, ДМСО-d6) δ 2,87 (2H, с), 7,15 (1H, дд, J=6, 1,5 Гц), 7,39 (2H, т, J=9 Гц), 7,6-7,7 (2H, м), 8,11 (1H, д, J=9 Гц), 8,33 (2H, дд, J=9,5 Гц), 8,59 (1H, д, J=9 Гц)
ИК (Nujol) 1638, 1603 см-1
APCI-MS m/z 267[M+H]+
[0237]
Способ синтеза соединения THK-5081
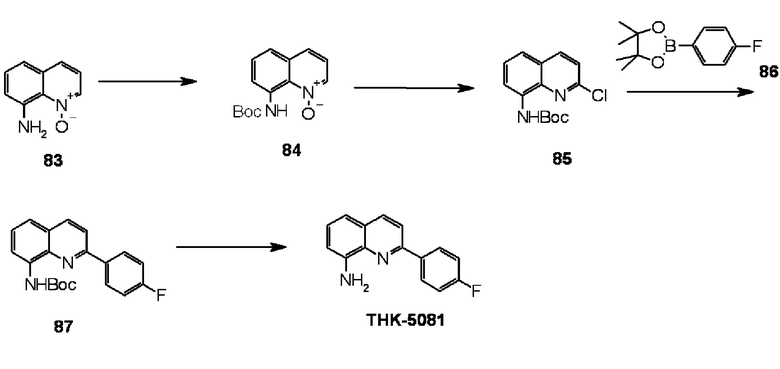
[0238]
Синтез соединения 84
Смесь соединения 83 (4,78 г, 29,84 ммоль) и Boc2O (10,28 мл, 44,8 ммоль) перемешивали при 80°C в течение 2 часов, при 100°C в течение 1 часа, с последующим перемешиванием при 120°C в течение 30 минут. Реакционный раствор очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=4/1, 1/1) с получением соединения 84 (6,54 г, 84%) в виде желтого твердого вещества.
точка плавления 126-128°C
APCI-MS m/z 261[M+H]+
[0239]
Синтез соединения 85
К раствору в хлороформе (120 мл) соединения 84 (6,62 г, 25,43 ммоль) добавляли палатолуолсульфонилхлорид (5,82 г, 30,5 ммоль) и карбонат калия (4,22 г, 30,5 ммоль) при комнатной температуре при перемешивании и смесь нагревали кипячением в сосуде с обратным холодильником в течение 5 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали хлороформом. Экстракционную жидкость очищали в том виде, как она есть, колоночной хроматографией на NH силикагеле (элюирующий растворитель: хлороформ) и затем проводили колоночную хроматографию на силикагеле (элюирующий растворитель: н-гексан/этилацетат=100/1 до этилацетата) с получением соединения 85 (3,70 г, 52%) в виде бледно-желтого твердого вещества.
точка плавления 99-100°C
APCI-MS m/z 279[M+H]+
[0240]
Синтез соединения 87
К раствору в 1,2-диметоксиэтане (44 мл) соединения 85 (1,50 г, 5,38 ммоль) и соединения 86 (1,20 г, 5,38 ммоль) добавляли карбонат калия (2,23 г, 16,13 ммоль), воду (0,94 мл) и тетракистрифенилфосфин палладия (310 мг, 0,27 ммоль) в атмосфере аргона и смесь перемешивали при 80°C в течение 3 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/500, 1/100) с получением соединения 87 (1.68 г, 92%) в виде бесцветного твердого вещества.
APCI-MS m/z 339[M+H]+
[0241]
Синтез соединения THK-5081
К раствору в хлороформе (12 мл) соединения 87 (1,50 г, 4,43 ммоль) добавляли по каплям трифторуксусную кислоту (8 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 3 часов. К реакционному раствору добавляли воду, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9) и затем перекристаллизовывали из н-гексана с получением соединения THK-5081 (404 мг, 38%) в виде бледно-желтых кристаллов.
точка плавления 87,5-88°C, 1H ЯМR (500 МГц, ДМСО-d6) δ 6,25 (2H, уш.), 6,91 (1H, д, J=7,7 Гц), 7,09 (1H, д, J=8,0 Гц), 7,29 (1H, т, J=8,0 Гц), 7,35 (2H, т, J=9,0 Гц), 8,08 (1H, д, J=8,7 Гц), 8,26 (1H, д, J=8,7 Гц), 8,43 (2H, дд, J=8,7, 5,5 Гц)
ИК (Nujol) 3433, 1616, 1600 см-1
APCI-MS m/z 239[M+H]+
[0242]
Способ синтеза соединения THK-5082
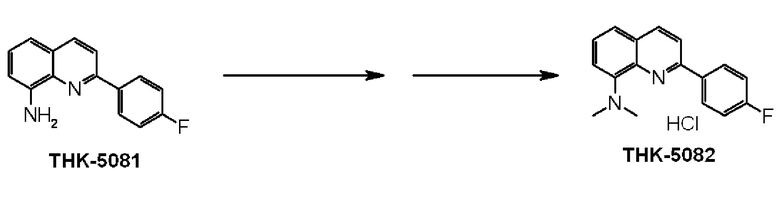
[0243]
Синтез соединения THK-5082
К раствору в метаноле (21 мл)-уксусной кислоте (2,1 мл) соединения THK-5081 (450 мг, 1,9 ммоль) и водного 35% раствора формалина (1,51 мл, 18,9 ммоль) добавляли пиколин-борановый комплекс (605 мг, 5,67 ммоль) в условиях охлаждения льдом и перемешивания и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли водный раствор карбоната калия, посредством этого доводя pH до 8, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=50/1), и затем проводили колоночную хроматографию на NH силикагеле (элюирующий растворитель: н-гексан/этилацетат=100/1) с получением свободной формы (430 мг) соединения THK-5082 в виде желтого маслянистого вещества. Настоящий продукт растворяли в этилацетате (10 мл) и по каплям добавляли 4M хлористоводородную кислоту/этилацетат (0,4 мл) с последующим перемешиванием при комнатной температуре в течение 30 минут. Преципитат собирали фильтрацией и сушили с получением соединения THK-5082 (430 мг, 75%) в виде бесцветных кристаллов.
точка плавления 216-216,5°C, 1H ЯМR (400 МГц, ДМСО-d6) δ 3,57 (6H, с), 7,41-7,48 (2H, м), 7,76 (1H, т, J=7,9 Гц), 8,15 (1Н, д, J=7,9 Гц), 8,24 (1H, д, J=7,6 Гц), 8,38 (1Н, д, J=8,8 Гц), 8,56-8,62 (2H, м), 8,66 (1H, д, J=8,8 Гц), 12,2 (1Н, уш.)
ИК (Nujol) 2252, 1602 см-1
APCI-MS m/z 267[M+H]+
[0244]
Способ синтеза соединения THK-5086
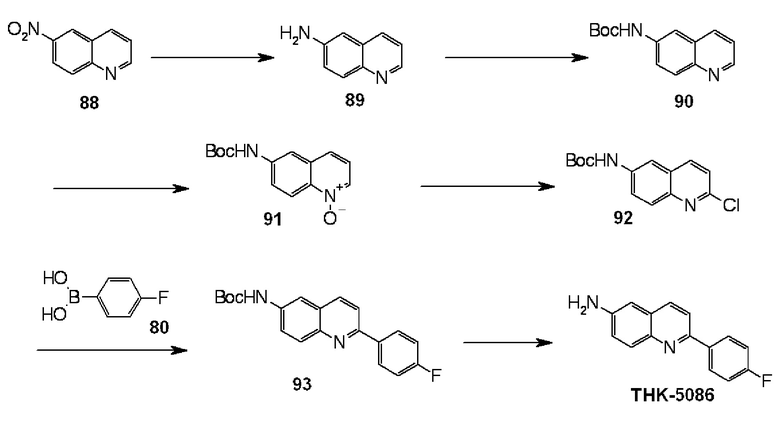
[0245]
Синтез соединения 89
К суспензии в метаноле (50 мл)-тетрагидрофуране (50 мл) соединения 88 (5,00 г, 28,71 ммоль) добавляли 10% Pd-C (содержание влаги примерно 50%; 1,00 г) в атмосфере водорода и смесь перемешивали при комнатной температуре в течение 2 часов. Катализатор удаляли фильтрацией, и растворитель фильтрата отгоняли при пониженном давлении с получением соединения 89 (4,14 г, 100%) в виде бледно-желтого твердого вещества.
точка плавления 116-117°C
APCI-MS m/z 145[M+H]+
[0246]
Синтез соединения 90
Смесь соединения 89 (4,14 г, 28,71 ммоль) и Boc2O (9,9 мл, 43,1 ммоль) перемешивали при 120°C в течение 30 минут. К реакционному раствору добавляли силикагель (30 мл) и толуол (100 мл) с последующим перемешиванием при 80°C в течение 1 часа. Реакционному раствору давали возможность охладиться до комнатной температуры и очищали в том виде, как он есть, колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=2/1, этилацетат) с получением соединения 90 (5,92 г, 84%) в виде бледно-желтого твердого вещества.
точка плавления 132-133°C
APCI-MS m/z 245[M+H]+
[0247]
Синтез соединения 91
К раствору в хлороформе (55 мл) соединения 90 (5,91 г, 24,19 ммоль) добавляли по каплям раствор в хлороформе (22 мл)-метаноле (6 мл) метанхлорбензойной кислоты (9,19 г, 53,2 ммоль) при комнатной температуре, и смешанный раствор перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли водный раствор тиосульфата натрия с последующим перемешиванием при комнатной температуре в течение 10 минут и дальнейшей экстракцией хлороформом. Экстракционную жидкость промывали водным раствором карбоната калия, очищали в том виде, как он есть, колоночной хроматографией на силикагеле (элюирующий растворитель: хлороформ) и затем промывали н-гексаном-этилацетом (2/1) с получением соединения 91 (5,25 г, 83%) в виде бесцветных кристаллов.
точка плавления 212-213°C
APCI-MS m/z 261[M+H]+
[0248]
Синтез соединения 92
К раствору в хлороформе (200 мл) соединения 91 (5,25 г, 20,17 ммоль) добавляли палатолуолсульфонилхлорид (4,61 г, 24,2 ммоль) и карбонат калия (3,35 г, 24,2 ммоль) при комнатной температуре при перемешивании и смесь нагревали кипячением в сосуде с обратным холодильником в течение 3,5 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали хлороформом. Экстракционую жидкость очищали в том виде, как она есть, колоночной хроматографией на NH силикагеле (элюирующий растворитель: хлороформ), и затем проводили колоночную хроматографию на силикагеле (элюирующий растворитель: н-гексан/этилацетат=9/1, 4/1) с получением соединения 92 (1,87 г, 33%) в виде бесцветного твердого вещества.
точка плавления 152-153°C
APCI-MS m/z 279/281[M+H]+
[0249]
Синтез соединения 93
К раствору в 1,2-диметоксиэтане (30 мл) соединения 92 (1,00 г, 3,59 ммоль) и соединения 80 (0,60 г, 4,31 ммоль) добавляли карбонат калия (1,50 г, 10,8 ммоль), воду (0,62 мл) и тетракистрифенилфосфин палладия (210 мг, 0,18 ммоль) в атмосфере аргона и смесь перемешивали при 80°C в течение 5,5 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) и затем перекристаллизовывали из этилацетата-н-гесана с получением соединения 93 (990 мг, 82%) в виде бесцветных кристаллов.
точка плавления 194-195°C
APCI-MS m/z 339[M+H]+
[0250]
Синтез соединения THK-5086
К суспензии в хлороформе (12 мл) соединения 93 (980 мг, 2,90 ммоль) добавляли по каплям трифторуксусную кислоту (8 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 1,5 часов. К реакционному раствору добавляли воду, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2) и затем перекристаллизовывали из этилацетата-н-гесана с получением соединения THK-5086 (635 мг, 92%) в виде бледно-желтых кристаллов.
точка плавления 145,5-146°C, 1H ЯМR (500 МГц, ДМСО-d6) δ 5,71 (2H, с), 6,82 ( 1H, д, J=2,6 Гц), 7,19 (1Н, дд, J=9,0, 2,6 Гц), 7,29-7,35 (2H, м), 7,75 (1H, д, J=9,0 Гц), 7,89 (1H, д, J=8,7 Гц), 8,03 (1H, д, J=8,7 Гц), 8,19-8,24 (2H, м)
ИК (Nujol) 3342, 1628 см-1
APCI-MS m/z 239[M+H]+
[0251]
Способ синтеза соединения THK-5087
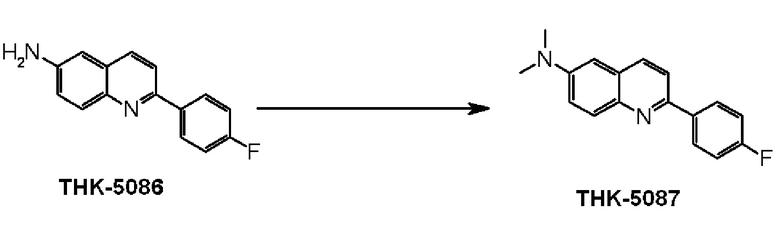
[0252]
Синтез соединения THK-5087
К раствору в метаноле (21 мл)-уксусной кислоте (2,1 мл) соединения THK-5086 (440 мг, 1,85 ммоль) и водному 37% раствору формалина (1,48 мл, 18,5 ммоль) добавляли пиколин-борановый комплекс (590 мг, 5,55 ммоль) в условиях охлаждения льдом и перемешивания и смесь перемешивали при комнатной температуре в течение 3,5 часов. После доведения pH реакционного раствора до 9 добавлением водного раствора карбоната калия, раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=9/1, 4/1) и затем перекристаллизовывали из н-гексана-этилацетата с получением соединения THK-5087 (415 мг, 84%) в виде бледно-желтых кристаллов.
точка плавления 172-173°C, 1H ЯМR (400 МГц, ДМСО-d6) δ 3,06 (6H, с), 6,96 (1H, д, J=2,7 Гц), 7,30-7,37 (2H, м), 7,47 (1H, дд, J=9,4, 3,0 Гц), 7,88 (1H, д, J=9,4 Гц), 7,96 (1Н, д, J=8,8 Гц), 8,17 (1H, д, J=8,8 Гц), 8,22-8,28 (2H, м)
ИК (Nujol) 1618 см-1
APCI-MS m/z 267[M+H]+
[0253]
Способ синтеза соединения THK-932
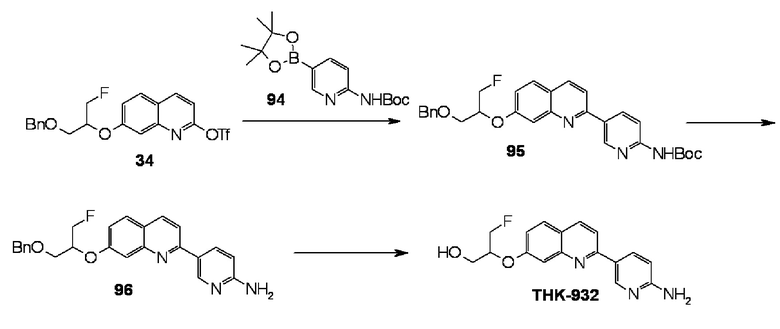
[0254]
Синтез соединения 95
К раствору в 1,2-диметоксиэтане (27 мл) соединения 34 (1,05 г, 2,28 ммоль) и соединения 94 (720 мг, 2,28 ммоль) добавляли карбонат калия (940 мг, 6,83 ммоль), воду (0,57 мл) и тетракистрифенилфосфин палладия (131 мг, 0,114 ммоль) в атмосфере аргона и смесь перемешивали при 80°C в течение 1,5 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, разбавляли этилацетатом, промывали водой и затем сушили. Растворитель отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=4/1→3/1) с получением соединения 95 (900 мг, 78%) в виде бледно-желтого твердого вещества.
точка плавления 150-152°C, APCI-MS m/z 504[M+H]+
[0255]
Синтез соединения 96
К раствору в хлороформе (12 мл) соединения 95 (900 мг, 1,79 ммоль) добавляли по каплям трифторуксусную кислоту (8 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор охлаждали льдом, нейтрализовали водным раствором карбоната калия и экстрагировали этилацетатом. Органический слой промывали водой и сушили, и затем растворитель концентрировали при пониженном давлении с получением соединения 96 (720 мг, 99%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 404[M+H]+
[0256]
Синтез соединения THK-932
К раствору в анизоле (5 мл) соединения 96 (720 мг, 1,78 ммоль) добавляли по каплям метансульфоновую кислоту (20 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор охлаждали льдом и по каплям добавляли ледяную воду, и раствор промывали этилацетатом. Водный слой экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Органический слой промывали водой и сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на NH силикагеле (элюирующий растворитель: н-гексан/этилацетат=1/1→этилацетат) и затем перекристаллизовывали из этилацетата с получением соединения THK-932 (390 мг, 69%) в виде бесцветных кристаллов.
точка плавления 160-161°C, 1H ЯМR (500 МГц, ДМСО-d6) δ 3,66-3,77 (2H, м), 4,65-4,86 (3H, м), 5,14 (1H, т, J=5,8 Гц), 6,41 (2H, с), 6,57 (1H, д, J=9,0 Гц), 7,20 (1H, дд, J=8,8, 2,4 Гц), 7,48 (1H, д, J=2,4 Гц), 7,84 (1H, д, J=8,8 Гц), 7,87 (1H, д, J=8,8 Гц), 8,18-8,38 (2H, м), 8,83 (1H, д, J=2,4 Гц)
ИК (Nujol) 3335, 1638, 1613 см-1
APCI-MS m/z 314[M+H]+
[0257]
Способ синтеза соединения THK-5088
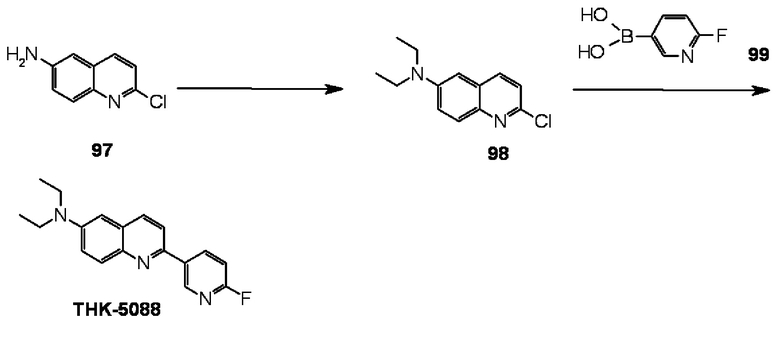
[0258]
Синтез соединения 98
К смеси соединения 97 (387 мг, 2,17 ммоль), ацетальдегида (2,39 г, 54,3 ммоль) и метанола (10 мл)-уксусной кислоты (1 мл) добавляли пиколин-борановый комплекс (1,16 г, 10,8 ммоль) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 3,5 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и к остатку добавляли водную 10% хлористоводородную кислоту (5 мл). После перемешивания при комнатной температуре в течение 30 минут раствор делали оснόвным добавлением карбоната калия. Реакционный раствор экстрагировали хлороформом, и экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=19/1) с получением соединения 98 (353 мг, 69%) в виде желтого маслянистого вещества.
APCI-MS m/z 235/237[M+H]+
[0259]
Синтез соединения THK-5088
К смеси соединения 98 (348 мг, 1,48 ммоль), соединения 99 (230 мг, 1,6 ммоль) и 1,2-диметоксиэтана (7,5 мл) добавляли водный 2M раствор карбоната натрия (1,5 мл, 3,0 ммоль) и тетракистрифенилфосфин палладия (86 мг, 0,074 ммоль) в атмосфере аргона, и смесь перемешивали при 90°C в течение 6 часов. К реакционному раствору добавляли соединение 99 (115 мг, 0,816 ммоль), водный 2M раствор карбоната натрия (0,75 мл, 1,5 ммоль) и тетракистрифенилфосфин палладия (43 мг, 0,037 ммоль) в атмосфере аргона с последующим дальнейшим перемешиванием при 90°C в течение 6 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и раствор экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=9/1) и затем проводили колоночную флэш-хроматографию на NH силикагеле (элюирующий растворитель: н-гексан, н-гексан/этилацетат=49/1, 19/1), и затем промывали холодным н-гексаном с получением соединения THK-5088 (380 мг, 87%) в виде желтовато-коричневого твердого вещества.
точка плавления 118-120°C, 1H ЯМR (500 МГц, ДМСО-d6) δ 1,18 (6H, т, J=7,0 Гц), 3,50 (4H, кв., J=7,0 Гц), 6,94 (1Н, с), 7,32 (1H, дд, J=6,1, 1,5 Гц), 7,43 (1H, д, J=8,4 Гц), 7,89 (1H, д, J=9 Гц), 8,01 (1H, д, J=9 Гц), 8,20 (1H, д, J=8,3 Гц), 8,74 (1Н, дт, J=8,3, 2,6 Гц), 9,01 (1H, д, J=2,6 Гц)
ИК (Nujol) 1465, 1376 см-1
APCI-MS m/z 296[M+H]+
[0260]
Способ синтеза соединения THK-5089
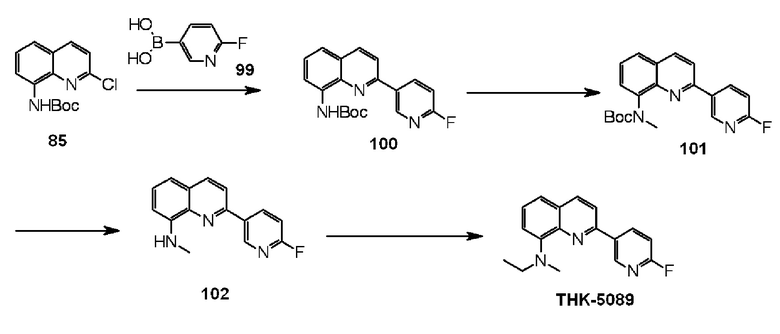
[0261]
Синтез соединения 100
К смеси соединения 85 (1,00 г, 3,59 ммоль), соединения 99 (0,61 г, 4,31 ммоль) и 1,2-диметоксиэтана (30 мл) добавляли карбонат калия (1,49 г, 10,76 ммоль), воду (0,62 мл) и тетракистрифенилфосфин палладий (210 мг, 0,18 ммоль) в атмосфере аргона, и смесь перемешивали при 80°C в течение 2,5 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9, 1/4) и затем перекристаллизовывали из этилацетата-н-гесана с получением соединения 100 (1,01 г, 83%) в виде бесцветных кристаллов.
точка плавления 189,5-190°C
APCI-MS m/z 340[M+H]+
[0262]
Синтез соединения 101
К раствору в N,N-диметилформамиде (33 мл) соединения 100 (1,00 г, 2,95 ммоль) добавляли 60% гидрид натрия (131 мг, 3,28 ммоль) в условиях охлаждения льдом и перемешивания в атмосфере аргона, и смесь перемешивали при той же температуре в течение 10 минут, и добавляли по каплям метилйодид (0,22 мл, 3,54 ммоль) при той же температуре с последующим перемешиванием при комнатной температуре в течение 30 минут. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9, 1/4) с получением соединения 101 (1,03 г, 98%) в виде бесцветного твердого вещества.
точка плавления 147-148°C
APCI-MS m/z 354[M+H]+
[0263]
Синтез соединения 102
К раствору в хлороформе (12 мл) соединения 101 (1,02 г, 2,89 ммоль) добавляли по каплям трифторуксусную кислоту (8 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 1,5 часов. К реакционному раствору добавляли воду, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9) с получением соединения 102 (732 мг, 100%) в виде желтого маслянистого вещества.
APCI-MS m/z 254[M+H]+
[0264]
Синтез соединения THK-5089
К раствору метанола (32 мл)-уксусной кислоты (3,2 мл), соединения 102 (730 мг, 2,88 ммоль) и ацетальдегида (1,63 мл, 28,8 ммоль) понемногу добавляли пиколин-борановый комплекс (930 мг, 8,64 ммоль) в условиях охлаждения льдом и перемешивания и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли воду и водный раствор карбоната калия, посредством этого доводя pH до 9, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=9/1, 4/1) и затем перекристаллизовывали из н-гексана с получением соединения THK-5089 (680 мг, 84%) в виде бледно-желтых кристаллов.
точка плавления 71-72°C, 1H ЯМR (400 МГц, ДМСО-d6) δ 1,20 (3H, т, J=7,1 Гц), 3,04 (3H, с), 3,72 (2H, кв., J=7,1 Гц), 7,07-7,12 (1Н, уш.), 7,39 (1Н, дд, J=8,5, 2,7 Гц), 7,42-7,49 (2H, м), 8,16 (1Н, д, J=8,5 Гц), 8,40 (1H, д, J=8,5 Гц), 8,79 (1H, тд, J=8,3, 2,7 Гц), 9,09 (1H, д, J=2,7 Гц)
ИК (Nujol) 1598 см-1
APCI-MS m/z 282[M+H]+
[0265]
Способ синтеза соединения THK-5095
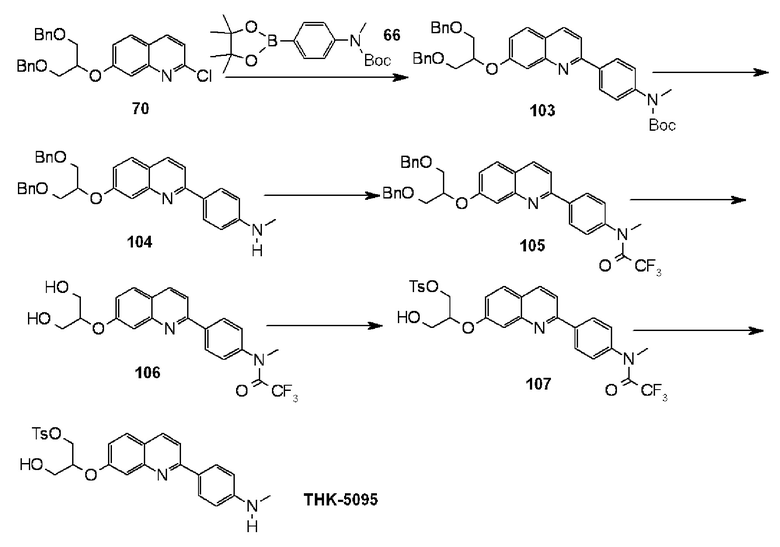
[0266]
Синтез соединения 103
К раствору в 1,2-диметоксиэтане (38 мл) соединения 70 (2,02 г, 4,66 ммоль) и соединения 66 (1,55 г, 4,66 ммоль) добавляли карбонат калия (1,93 г, 13,97 ммоль), воду (0,81 мл) и тетракистрифенилфосфин палладия (540 мг, 0,47 ммоль) в атмосфере аргона и смесь перемешивали при 80°C в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением соединения 103 (2,82 г, 100%) в виде бледно-розового маслянистого вещества.
APCI-MS m/z 605[M+H]+
[0267]
Синтез соединения 104
К раствору в хлороформе (24 мл) соединения 103 (2,81 г, 4,65 ммоль) добавляли по каплям трифторуксусную кислоту (16 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли воду и этилацетат, и раствор делали основным водным раствором карбоната калия и экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=3/1, 2/1) с получением соединения 104 (2,28 г, 97%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 505[M+H]+
[0268]
Синтез соединения 105
К раствору в хлороформе (20 мл) соединения 104 (1,14 г, 2,26 ммоль) добавляли по каплям триэтиламин (0,473 мл, 3,39 ммоль) и затем трифторуксусный ангидрид (0,383 мл, 2,71 ммоль) в условиях охлаждения льдом и перемешивания и смесь перемешивали при той же температуре в течение 2 часов. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением соединения 105 (1,36 г, 100%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 601[M+H]+
[0269]
Синтез соединения 106
К смеси соединения 105 (1,36 г, 2,26 ммоль) и анизола (2 мл) добавляли по каплям метансульфоновую кислоту (6 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 20 минут. Реакционный раствор охлаждали льдом и добавляли ледяную воду, и раствор делали основным водным раствором карбоната калия и экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1, этилацетат) с получением соединения 106 (890 мг, 94%) в виде бледно-желтого аморфного вещества.
APCI-MS m/z 421[M+H]+
[0270]
Синтез соединения 107
К раствору в пиридине (10 мл) соединения 106 (880 мг, 2,09 ммоль) добавляли по каплям раствор в тетрагидрофуране (15 мл) паратолуолсульфонового ангидрида (1,03 г, 3,14 ммоль) в течение 20 минут в условиях охлаждения льдом и перемешивания и смесь перемешивали при той же температуре в течение 1 часа. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, растворитель отгоняли при пониженном давлении (толуол азеотроп). Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 1/1, этилацетат) с получением соединения 107 (750 мг, 63%) в виде бесцветного аморфного вещества.
APCI-MS m/z 575[M+H]+
[0271]
Синтез соединения THK-5095
К раствору в тетрагидрофуране (10 мл)-воде (1 мл) соединения 107 (750 мг, 1,31 ммоль) добавляли гидроксид моногидрат лития (82 мг, 1,97 ммоль) в условиях охлаждения льдом и перемешивания и смесь перемешивали при той же температуре в течение 1 часа с последующим перемешиванием при комнатной температуре в течение 1 часа. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1, 2/1) и затем перекристаллизовывали из этилацетата-н-гексана с получением соединения THK-5095 (563 мг, 90%) в виде бесцветных кристаллов.
точка плавления 109-111°C, 1H ЯМR (400 МГц, ДМСО-d6) δ 2,33 (3H, с), 2,76 (3H, д, J=4,5 Гц), 3,56-3,71 (2H, м), 4,30 (1H, дд, J=11, 5,7 Гц), 4,38 (1H, дд, J=11, 3,0 Гц), 4,71-4,76 (1H, м), 5,11 (1H, т, J=5,6 Гц) 6,14 (1Н, уш.) 6,66 (2H, д, J=8,8 Гц), 7,01 (1Н, дд, J=8,8, 2,4 Гц), 7,31 (1H, д, J=2,1 Гц), 7,39 (2H, д, J=8,5 Гц), 7,75 (2H, д, J=8,8 Гц), 7,78 (1H, д, J=9,1 Гц), 7,84 (1H, д, J=8,8 Гц), 8,06 (2H, д, J=8,8 Гц), 8,20 (1H, д, J=8,5 Гц)
ИК (Nujol) 3359, 1741, 1608 см-1
APCI-MS m/z 479[M+H]+
[0272]
Способ синтеза соединения THK-5096
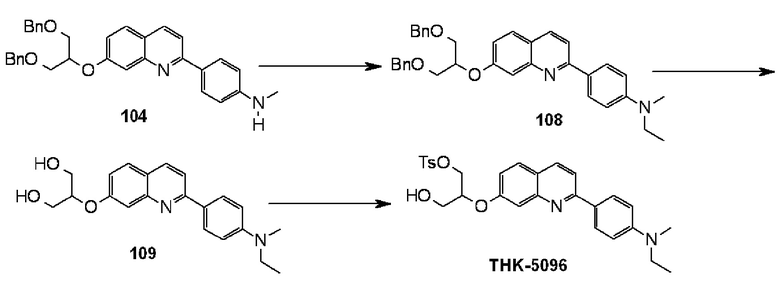
[0273]
Синтез соединения 108
К раствору в метаноле(25 мл)-уксусной кислоте (2,5 мл) соединения 104 (1,13 г, 2,24 ммоль) и ацетальдегида (1,26 мл, 22,4 ммоль) добавляли пиколин-борановый комплекс (720 мг, 6,72 ммоль) в условиях охлаждения льдом и перемешивания и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом доведением pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=9/1, 4/1) с получением соединения 108 (1,08 г, 91%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 533[M+H]+
[0274]
Синтез соединения 109
К смеси соединения 108 (1,15 г, 2,16 ммоль) и анизола (3 мл) добавляли по каплям метансульфоновую кислоту (9 мл) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 20 минут. Реакционный раствор охлаждали льдом и добавляли ледяную воду, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток промывали этилацетатом-н-гексаном (1/1) с получением соединения 109 (640 мг, 84%) в виде желтых кристаллов.
точка плавления 136-137°C
APCI-MS m/z 353[M+H]+
[0275]
Синтез соединения THK-5096
К раствору в пиридине (10 мл) соединения 109 (630 мг, 1,79 ммоль) добавляли по каплям раствор в тетрагидрофуране (10 мл) паратолуолсульфонового ангидрида (875 мг, 2,68 ммоль) в течение 30 минут в условиях охлаждения льдом и перемешивания и смесь перемешивали при той же температуре в течение 20 минут. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении (толуол азеотроп). Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, этилацетат) и затем перекристаллизовывали из этилацетата-н-гексана с получением соединения THK-5096 (450 мг, 50%) в виде желтых кристаллов.
точка плавления 89-90°C, 1H ЯМR (400 МГц, ДМСО-d6) δ 1,10 (3H, т, J=7,0 Гц), 2,33 (3H, с), 2,96 (3H, с), 3,48 (2H, кв., J=7,0 Гц), 3,37-3,71 (2H, м), 4,30 (1H, дд, J=11, 5,7 Гц), 4,38 (1H, дд, J=11, 2,7 Гц), 4,71-4,78 (1H, м), 5,11 (1H, т, J=5,4 Гц), 6,83 (2H, д, J=9,1 Гц), 7,02 (1H, дд, J=8,9, 2,3 Гц), 7,33 (1H, д, J=1,8 Гц), 7,39 (2Н, д, J=7,9 Гц), 7,75 (2H, д, J=8,2 Гц), 7,79 (1H, д, J=8,8 Гц), 7,87 (1H, д, J=8,8 Гц), 8,11 (2H, д, J=9,1 Гц), 8,21 (1H, д, J=9,1 Гц)
ИК (Nujol) 1620, 1597 см-1
APCI-MS m/z 507[M+H]+
[0276]
Способ синтеза соединения THK-5099
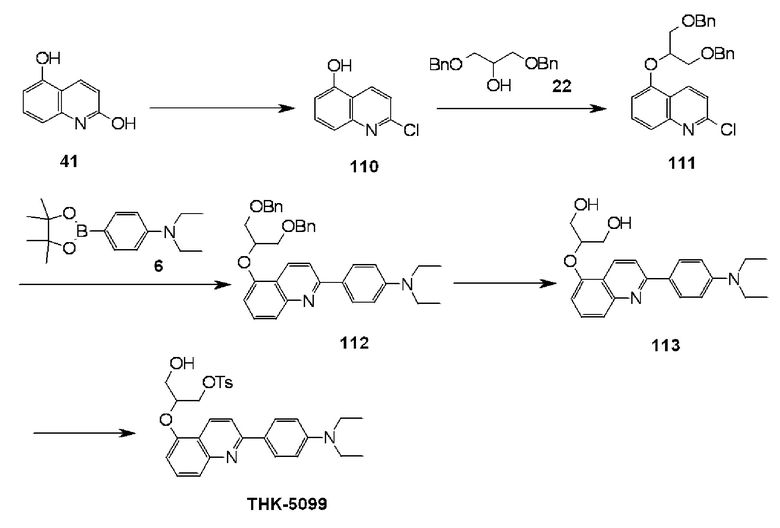
[0277]
Синтез соединения 110
К суспензии в N,N-диметилформамиде (30 мл) соединения 41 (5,84 г, 36,24 ммоль) добавляли по каплям тионилхлорид (10,39 мл, 145 ммоль) при охлаждении льдом и перемешивании и смесь перемешивали при той же температуре в течение 30 минут, с последующим перемешиванием при комнатной температуре в течение 16 часов. Реакционный раствор охлаждали льдом и добавляли ледяную воду, и раствор экстрагировали этилацетатом после доведения pH до 7 с использованием водного раствора карбоната калия. Экстракционную жидкость промывали водой, сушили, очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9, 1/4) и затем перекристаллизовывали из этилацетата―н-гексана с получением соединения 110 (3,88 г, 60%) в виде бледно-желтых кристаллов.
точка плавления 176-177°C
APCI-MS m/z 180[M+H]+
[0278]
Синтез соединения 111
К раствору в тетрагидрофуране (30 мл) соединения 110 (700 мг, 3,90 ммоль), соединения 22 (1,27 г, 4,68 ммоль) и трифенилфосфина (1,23 г, 4,68 ммоль) добавляли по каплям раствор в тетрагидрофуране (5 мл) диизопропилазодикарбоксилата (0,93 мл, 4,68 ммоль) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 3 дней. К реакционному раствору добавляли раствор в тетрагидрофуране (5 мл) соединения 22 (1,27 г, 4,68 ммоль) и диизопропилазодикарбоксилат (0,93 мл, 4,68 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 2 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/20, 1/15) с получением соединения 111 (1,18 г, 70%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 434[M+H]+
[0279]
Синтез соединения 112
К раствору в 1,2-диметоксиэтане (25 мл) соединения 111 (1,17 г, 2,70 ммоль) и соединения 6 (740 мг, 2,70 ммоль) добавляли карбонат калия (1,12 г, 8,09 ммоль), воду (0,5 мл) и тетракистрифенилфосфин палладия (310 мг, 0,27 ммоль) в атмосфере аргона, и смесь перемешивали при 80°C в течение 7 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9, 1/4) с получением соединения 112 (1,48 г, 100%) в виде желтого маслянистого вещества.
APCI-MS m/z 547[M+H]+
[0280]
Синтез соединения 113
К смеси соединения 112 (1,47 г, 2,69 ммоль) и анизола (4 мл) добавляли по каплям метансульфоновую кислоту (12 мл) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор выливали в ледяную воду, и раствор экстрагировали этилацетатом после доведения pH до 8 с использованием водного раствора карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток промывали гексаном с получением соединения 113 (900 мг, 91%) в виде бледно-желтых кристаллов.
точка плавления 165-166°C
APCI-MS m/z 367[M+H]+
[0281]
Синтез соединения THK-5099
К раствору в пиридине (15 мл) соединения 113 (890 мг, 2,43 ммоль) по каплям добавляли раствор в тетрагидрофуране (15 мл) паратолуолсульфонового ангидрида (1,19 г, 3,64 ммоль) в течение 30 минут в условиях охлаждения льдом и перемешивания и смесь перемешивали при той же температуре в течение 30 минут. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении (толуол азеотроп). Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) и затем перекристаллизовывали из этилацетата с получением соединения THK-5099 (658 мг, 52%) в виде желтых кристаллов.
точка плавления 143-143,5°C, 1H ЯМR (400 МГц, ДМСО-d6) δ 1,15 (6H, т, J=7,0 Гц), 2,31 (3H, с), 3,43 (4H, кв., J=7,0 Гц), 3,67-3,72 (2H, м), 4,37 (1H, дд, J=11, 5,7 Гц), 4,42 (1H, дд, J=11, 3,0 Гц), 4,71-4,77 (1H, м), 5,13 (1H, т, J=5,6 Гц), 6,80 (2H, д, J=9,1 Гц), 6,94 (1H, дд, J=5,6,3,2 Гц), 7,28 (2H, д, J=8,2 Гц), 7,51-7,54 (2H, м), 7,68 (2H, д, J=8,2 Гц), 7,93 (1H, д, J=9,1 Гц), 8,12 (2H, д, J=9,1 Гц), 8,25(1Н, д, J=8,8 Гц)
ИК (Nujol) 1593 см-1
APCI-MS m/z 521[M+H]+
[0282]
Способ синтеза соединения THK-5105
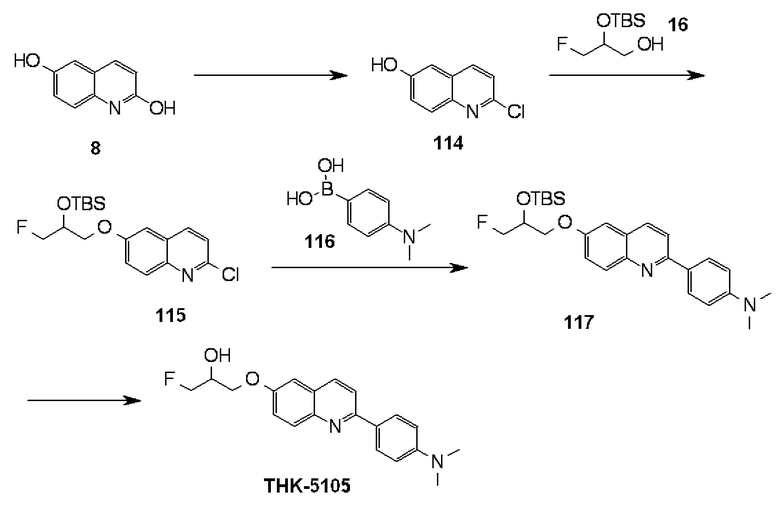
[0283]
Синтез соединения 114
К суспензии в N,N-диметилформамиде (20 мл) соединения 8 (1,16 г, 10 ммоль) добавляли по каплям тионилхлорид (5 г, 42 ммоль) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли водный насыщенный раствор гидрокарбоната натрия, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением соединения 114 (1,12 г, 62%) в виде бесцветного твердого вещества.
точка плавления 190-191°C
[0284]
Синтез соединения 115
К раствору в тетрагидрофуране (20 мл) соединения 114 (1,0 г, 5,57 ммоль), соединения 16 (1,2 г, 5,76 ммоль) и трифенилфосфина (1,9 г, 7,24 ммоль) добавляли по каплям диизопропилазодикарбоксилат (1,46 г, 7,22 ммоль) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 16 часов. Реакционный раствор отгоняли при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/7) с получением соединения 115 (1,78 г, 86%) в виде бледно-желтого твердого вещества.
точка плавления от 50 до 52°C
[0285]
Синтез соединения 117
К раствору в 1,2-диметоксиэтане (14 мл) соединения 115 (600 мг, 1,62 ммоль) и соединения 116 (294 мг, 1,78 ммоль) добавляли карбонат калия (670 мг, 4,87 ммоль), воду (0,28 мл) и тетракис трифенилфосфин палладий (190 мг, 0,16 ммоль) в атмосфере аргона и смесь перемешивали при 80°C в течение 3 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/19, 1/9) с получением соединения 117 (690 мг, 94%) в виде бледно-желтого твердого вещества.
точка плавления 131-133°C
APCI-MS m/z 455[M+H]+
[0286]
Синтез соединения THK-5105
К раствору в тетрагидрофуране (20 мл) соединения 117 (690 мг, 1,52 ммоль) добавляли тетрабутиламмония фторид (1,52 мл/1M раствор тетрагидрофурана) и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4, 1/1) и затем перекристаллизовывали из этилацетата с получением соединения THK-5105 (437 мг, 85%) в виде бледно-желтых кристаллов.
точка плавления 178-179°C, 1H ЯМR (400 МГц, ДМСО-d6) δ 4,07-4,18 (3H, м), 4,44-4,65 (2H, м), 5,52 (1H, уш.), 6,84 (2H, д, J=9,1 Гц), 7,36-7,41 (2H, м), 7,90 (1H, д, J=10 Гц), 7,99 (1H, д, J=8,8 Гц), 8,10 (2H, д, J=9,1 Гц), 8,23 (1H, д, J=8,8 Гц)
ИК (Nujol) 3406, 1616 см-1
APCI-MS m/z 341[M+H]+
[0287]
Способ синтеза соединения THK-5106
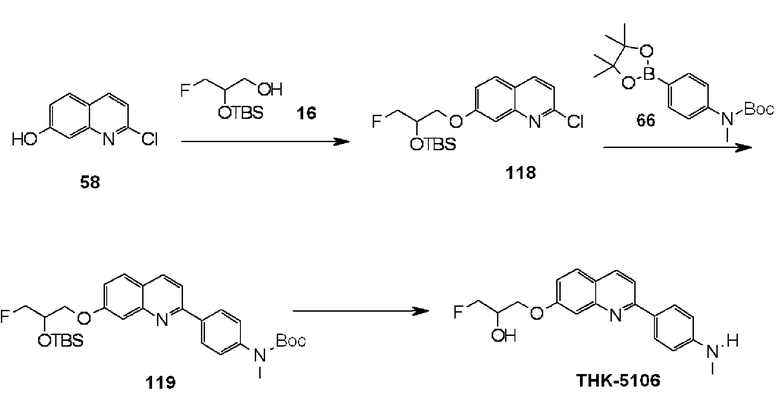
[0288]
Синтез соединения 118
К раствору в тетрагидрофуране (40 мл) соединения 58 (2,00 г, 10,6 ммоль), соединения 16 (2,64 г, 12,7 ммоль) и трифенилфосфина (3,32 г, 12,7 ммоль) добавляли по каплям раствор в тетрагидрофуране (10 мл) диизопропилазодикарбоксилата (2,51 мл, 12,7 ммоль) при охлаждении льдом и перемешивании, и смесь перемешивали при той же температуре в течение 30 минут, с последующим перемешиванием при комнатной температуре в течение 16 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/20) с получением соединения 118 (3,90 г, 100%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 370/372[M+H]+
[0289]
Синтез соединения 119
Смесь соединения 118 (1,83 г, 4,9 ммоль), соединения 66 (1,73 г, 5,2 ммоль), карбоната калия (1,37 г, 9,9 ммоль), тетракистрифенилфосфина палладия (300 мг, 0,26 ммоль) и 1,2-диметоксиэтана (45 мл)―воды (5 мл) перемешивали в атмосфере аргона при 85°C в течение 2 часов и добавляли тетракистрифенилфосфин палладия (283 мг, 0,245 ммоль) и карбонат калия (678 мг, 4,9 ммоль) с последующим перемешиванием при той же температуре в течение 4 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан, этилацетат/н-гексан=1/9) с получением соединения 119 (2,28 г, 85%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 541[M+H]+
[0290]
Синтез соединения THK-5106
К раствору в хлороформе (21 мл) соединения 119 (2,25 г, 3,33 ммоль) добавляли по каплям трифторуксусную кислоту (14 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 1 часа, и по каплям добавляли воду (7 мл) с последующим дополнительным перемешиванием при комнатной температуре в течение 1 часа. Растворитель реакционного раствора отгоняли при пониженном давлении и добавляли хлороформ, и раствор делали основным водным насыщенным раствором гидрокарбоната натрия и экстрагировали хлороформом/метанолом (19/1). Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: хлороформ, хлороформ/метанол=19/1) и затем промывали н-гексаном и диизопропиловым эфиром с получением соединения THK-5106 (1,0 г, 92%) в виде желтого твердого вещества.
точка плавления 137-139°C, 1H ЯМR (500 МГц, ДМСО-d6) δ 2,76 (3H, д, J=4,4 Гц), 4,1-4,2 (3H, м), 4,4-4,7 (2H, м), 5,53 (1H, д, J=4,8 Гц), 6,13 (1H, уш. с), 6,66 (2H, д, J=8,7 Гц), 7,15 (1Н, дд, J=9,0, 2,6 Гц), 7,35 (1H, д, J=2,6 Гц), 7,80 (1H, д, J=9,0 Гц), 7,83 (1H, д, J=8,7 Гц), 8,06 (2H, д, J=9,0 Гц), 8,20 (1H, д, J=8,3 Гц)
ИК (Nujol)1612, 1598 см-1
APCI-MS m/z 327[M+H]+
[0291]
Способ синтеза соединения THK-5107
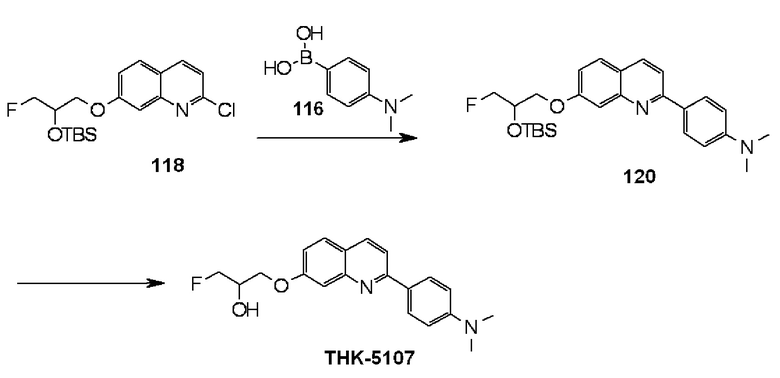
[0292]
Синтез соединения 120
К раствору в 1,2-диметоксиэтане (20 мл) соединения 118 (800 мг, 2,16 ммоль) и соединения 116 (390 мг, 2,38 ммоль) добавляли карбонат калия (900 мг, 6,49 ммоль), воду (0,38 мл) и тетракистрифенилфосфин палладия (380 мг, 0,22 ммоль) в атмосфере аргона и смесь перемешивали при 80°C в течение 3,5 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/19) с получением соединения 120 (860 мг, 88%) в виде бледно-желтого твердого вещества.
точка плавления 102-106°C
APCI-MS m/z 455[M+H]+
[0293]
Синтез соединения THK-5107
К раствору в тетрагидрофуране (10 мл) соединения 120 (850 мг, 1,87 ммоль) добавляли тетрабутиламмония фторид (1,87 мл/1M раствор тетрагидрофурана) и смесь перемешивали при комнатной температуре в течение 30 минут. К реакционному раствору добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4, 1/2) с получением соединения THK-5107 (352 мг, 55%) в виде желтых кристаллов.
точка плавления 163,5-164°C, 1Н ЯМR (400 МГц, ДМСО-d6) δ 3,01 (6H, с), 4,09-4,22 (3H, м), 4,44-4,65 (2H, м), 5,53 (1Н, уш.), 6,84 (2H, д, J=9,1 Гц), 7,18 (1H, дд, J=8,8, 2,4 Гц), 7,38 (1Н, д, J=2,4 Гц), 7,83 (1Н, д, J=9,1 Гц), 7,89 (1H, д, J=8,5 Гц), 8,13 (2H, д, J=9,1 Гц), 8,25 (1Н, д, J=8,8 Гц)
ИК (Nujol) 1619, 1608, 1595 см-1
APCI-MS m/z 341[M+H]+
[0294]
Способ синтеза соединения THK-5111
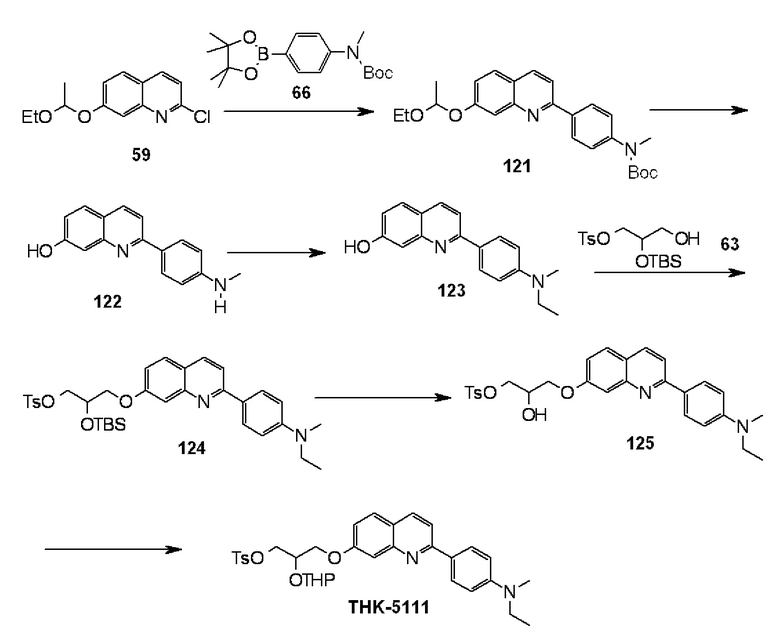
[0295]
Синтез соединения 121
К раствору в 1,2-диметоксиэтане (65 мл) соединения 59 (2,01 г, 7,99 ммоль) и соединения 66 (2,66 г, 7,99 ммоль) добавляли карбонат калия (3,31 г, 24,0 ммоль), воду (1,39 мл) и тетракистрифенилфосфин палладия (920 мг, 0,80 ммоль) в атмосфере аргона, и смесь перемешивали при 80°C в течение 8 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/19, 1/9, 1/4) с получением соединения 121 (3,38 г, 100%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 423[M+H]+
[0296]
Синтез соединения 122
К раствору в хлороформе (12 мл) соединения 121 (3,37 г, 7,98 ммоль) добавляли по каплям трифторуксусную кислоту (8 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 2 часов. К реакционному раствору добавляли воду, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток перекристаллизовывали из этилацетата с получением соединения 122 (1,71 г, 86%) в виде желтых кристаллов.
точка плавления 262-265°C
APCI-MS m/z 251[M+H]+
[0297]
Синтез соединения 123
К раствору в метаноле (35 мл)-уксусной кислоте (3,5 мл) соединения 122 (800 мг, 3,20 ммоль) и ацетальдегида (1,80 мл, 32,0 ммоль) добавляли пиколинборановый комплекс (1,03 г, 9,59 ммоль) в условиях охлаждения льдом и перемешивания и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток перекристаллизовывали из этилацетата с получением соединения 123 (840 мг, 94%) в виде желтых кристаллов.
точка плавления 212-213°C
APCI-MS m/z 279[M+H]+
[0298]
Синтез соединения 124
К раствору в тетрагидрофуране (30 мл) соединения 123 (830 мг, 2,98 ммоль), соединения 63 (1,29 г, 3,58 ммоль) и трифенилфосфина (940 мг, 3,58 ммоль) по каплям добавляли раствор в тетрагидрофуране (10 мл) диизопропилазодикарбоксилата (0,71 мл, 3,58 ммоль) в течение 1 часа в условиях охлаждения льдом и перемешивания, и смесь перемешивали при той же температуре 1 час с последующим перемешиванием при комнатной температуре в течение 16 часов. К реакционному раствору добавляли соединение 63 (540 мг, 1,49 ммоль) и трифенилфосфин (390 мг, 1,49 ммоль) в условиях охлаждения льдом и перемешивания, и добавляли диизопропилазодикарбоксилат (0,3 мл, 1,49 ммоль) и тетрагидрофуран (10 мл) с последующим дополнительным перемешиванием при комнатной температуре в течение 3 дней. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9) с получением соединения 124 (2,21 г) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 621[M+H]+
[0299]
Синтез соединения 125
К раствору в хлороформе (12 мл) соединения 124 (2,21 г) добавляли по каплям трифторуксусную кислоту (8 мл) при охлаждении льдом и перемешивании, добавляли воду (2 мл) и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли ледяную воду и этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 9 с использованием водного раствора карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 1/1) с получением соединения 125 (1,27 г, 84% из соединения 123) в виде желтого пеноподобного вещества.
APCI-MS m/z 507[M+H]+
[0300]
Синтез соединения THK-5111
К раствору в метиленхлориде (30 мл) соединения 125 (940 мг, 1,86 ммоль) добавляли 3,4-дигидро-2H-пиран (3,36 мл, 37,2 ммоль) и моногидрат паратолуолсульфоновой кислоты (415 мг, 2,42 ммоль) и смесь перемешивали при комнатной температуре в течение 40 минут. Реакционный раствор нейтрализовали триэтиламином, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением соединения THK-5111 (1,05 г, 96%) в виде желтого аморфного вещества.
1Н ЯМR (500 МГц, ДМСО-d6) δ 1,10 (3H, т, J=7,2 Гц), 1,36-1,70 (6H, м), 2,34 (3H, с), 2,97 (3H, с), 3,37-3,45 (1Н, м), 3,49 (2H, кв., J=7,2 Гц), 3,68-3,74, 3,84-3,90 (1H, м), 4,13-4,37 (5H, м), 4,71-4,74, 4,86-4,88 (1H, м), 6,84 (2H, д, J=8,7 Гц), 7,06 (1H, д, J=8,7 Гц), 7,31 (1H, уш.), 7,39-7,44 (2H, м), 7,78-7,84 (3H, м), 7,90 (1H, д, J=9,0 Гц), 8,12 (2H, д, J=9,0 Гц), 8,27 (1H, уш.)
APCI-MS m/z 591[M+H]+
[0301]
Способ синтеза соединения THK-5112
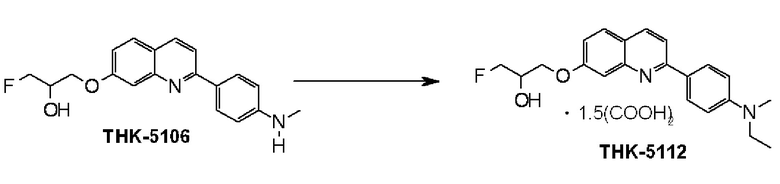
[0302]
Синтез соединения THK-5112
К раствору в метаноле (10 мл)-уксусной кислоте (1 мл) соединения THK-5106 (478 мг, 1,46 ммоль) и ацетальдегида (323 мг, 7,3 ммоль) добавляли пиколин-борановый комплекс (204 мг, 1,9 ммоль) в условиях охлаждения льдом и перемешивания и смесь перемешивали при комнатной температуре в течение 1 часа, и добавляли ацетальдегид (323 мг, 7,3 ммоль) и пиколин-борановый комплекс (204 мг, 1,9 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 5 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и к остатку добавляли водную 10% хлористоводородную кислоту (5 мл). После перемешивания при комнатной температуре в течение 30 минут, раствор делали основным добавлением карбоната калия. Реакционный раствор экстрагировали хлороформом, и экстракционную жидкость сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на NH силикагеле (элюирующий растворитель: н-гексан, н-гексан/этилацетат=1/1) с получением желтого маслянистого вещества. После растворения добавлением щавелевой кислоты (292 мг, 2,9 ммоль) к раствору в ацетоне (10 мл) настоящего продукта, растворитель отгоняли при пониженном давлении. Остаток промывали диизопропиловым эфиром и диэтиловым эфиром с получением соединения THK-5112 (660 мг, 91%) в виде оранжевого твердого вещества.
точка плавления 193-194,5°C (десятичн.), 1H ЯМR (400 МГц, ДМСО-d6) δ 1,10 (3H, т, J=7,0 Гц), 2,97 (3H, с), 3,49 (2H, кв., J=7,0 Гц), 4,0-4,2 (3H, м), 4,4-4,7 (2H, м), 6,83 (2H, д, J=9,1 Гц), 7,18 (1H, дд, J=8,7, 2,4 Гц), 7,38 (1H, д, J=2,4 Гц), 7,84 (1Н, д, J=9,1 Гц), 7,89 (1H, д, J=8,7 Гц), 8,12 (2H, д, J=9,0 Гц), 8,27 (1H, д, J=8,8 Гц)
ИК (Nujol) 2922, 2852 см-1
APCI-MS m/z 355[M+H]+0
[0303]
Способ синтеза соединения THK-5113
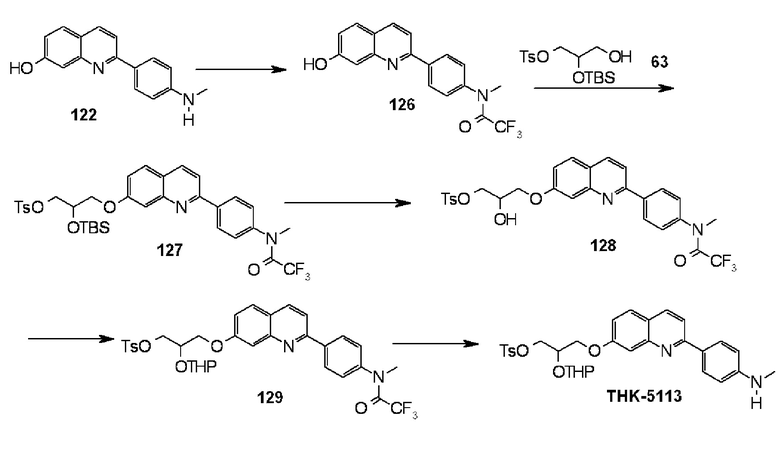
[0304]
Синтез соединения 126
К суспензии в метиленхлориде (20 мл) соединения 122 (900 мг, 3,60 ммоль) и триэтиламина (1,51 мл, 10,79 ммоль) добавляли по каплям трифторуксусный ангидрид (1,22 мл, 8,63 ммоль) при охлаждении льдом и перемешивании и смесь перемешивали при той же температуре в течение 30 минут. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали хлороформом/метанолом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем добавляли Остаток перекристаллизовывали из этилацетата-н-гексана с получением соединения 126 (1,24 г, 99%) в виде оранжевых кристаллов.
точка плавления 176-178°C
APCI-MS m/z 347[M+H]+
[0305]
Синтез соединения 127
К суспензии в тетрагидрофуране (30 мл) соединения 126 (1,23 г, 3,55 ммоль), соединения 63 (1,54 г, 4,26 ммоль) и трифенилфосфина (1,12 г, 4,26 ммоль) добавляли по каплям раствор в тетрагидрофуране (10 мл) диизопропилазодикарбоксилата (0,84 мл, 4,26 ммоль) в течение 20 минут в условиях охлаждения льдом и перемешивания, и смесь перемешивали при той же температуре в течение 2 часов с последующим перемешиванием при комнатной температуре в течение 16 часов. К реакционному раствору добавляли соединение 63 (640 мг, 1,78 ммоль), трифенилфосфин (470 мг, 1,78 ммоль) и диизопропилазодикарбоксилат (0,35 мл, 1,78 ммоль) с последующим перемешиванием при комнатной температуре в течение 4 часов. Кроме того, к реакционному раствору добавляли соединение 63 (640 мг, 1,78 ммоль), трифенилфосфин (470 мг, 1,78 ммоль) и диизопропилазодикарбоксилат (0,35 мл, 1,78 ммоль) с последующим перемешиванием при комнатной температуре в течение 16 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением соединения 127 (3,28 г) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 347[M+H]+
[0306]
Синтез соединения 128
К раствору в хлороформе (12 мл) соединения 127 (3,27 г) добавляли по каплям трифторуксусную кислоту (8 мл) и воду (2 мл) в условиях охлаждения льдом и перемешивания и смесь перемешивали при комнатной температуре в течение 8 часов. К реакционному раствору добавлял ледяную воду и этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 7 с использованием водного раствора карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 1/1) с получением соединения 128 (1,85 г, 91% из соединения 126) в виде бледно-желтого пеноподобного вещества.
APCI-MS m/z 575[M+H]+
[0307]
Синтез соединения 129
К раствору в тетрагидрофуране (60 мл) соединения 128 (1,84 г, 3,20 ммоль) и 3,4-дигидро-2H-пирана (5,81 мл, 64,1 ммоль) добавляли моногидрат паратолуолсульфоновой кислоты (720 мг, 4,16 ммоль) и смесь перемешивали при комнатной температуре в течение 16 часов. Реакционный раствор нейтрализовали триэтиламином, и растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9, 1/4, 1/2) с получением 129 (2,10 г, 99%) в виде бледно-желтого пеноподобного вещества.
APCI-MS m/z 659[M+H]+
[0308]
Синтез соединения THK-5113
К раствору в тетрагидрофуране (25 мл)-воде (10 мл) соединения 129 (2,09 г, 3,17 ммоль) добавляли гидроксид моногидрат лития (200 мг, 4,76 ммоль) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при той же температуре в течение 30 минут. Реакционный раствор экстрагировали этилацетатом, и экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2) с получением соединения THK-5113 (1,70 г, 95%) в виде бледно-желтого аморфного вещества.
1H ЯМR (400 МГц, ДМСО-d6) δ 1,36-1,72 (6H, м), 2,34 (3H, с), 2,76 (3H, д, J=4,2 Гц), 3,35-3,48, 3,67-3,76, 3,83-3,91 (2H, м), 4,11-4,37 (5H, м), 4,70-4,74, 14,85-4,88 (1Н, м), 6,14 (1Н, уш.), 6,66 (2H, д, J=8,8 Гц), 7,03 (1H, дд, J=8,8, 2,4 Гц), 7,24-7,28 (1Н, м), 7,39-7,44 (2H, м), 7,76-7,87 (4H, м), 8,07 (2H, д, J=8,8 Гц), 8,21 (1H, д, J=8,8 Гц)
ИК (Nujol) 3422, 1609, 1596 см-1
APCI-MS m/z 563[M+H]+
[0309]
Способ синтеза соединения THK-5115
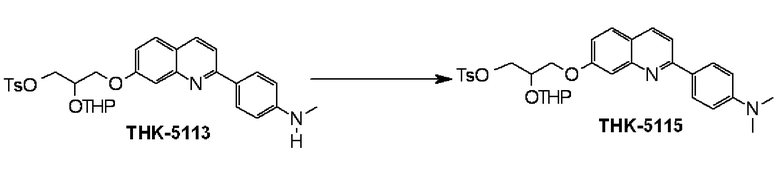
[0310]
Синтез соединения THK-5115
К раствору в метаноле (20 мл)-уксусной кислоте (2 мл) соединения THK-5113 (1,02 г, 1,81 ммоль) и водного 37% раствора формалина (1,45 мл, 18,1 ммоль) добавляли пиколин-борановый комплекс (580 мг, 5,44 ммоль) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при той же температуре в течение 5 минут с последующим перемешиванием при комнатной температуре в течение 1 часа. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=4/1) с получением соединения THK-5115 (980 мг, 93%) в виде желтого аморфного вещества.
1H ЯМR (500 МГц, ДМСО-d6) δ 1,36-1,71 (6H, м), 2,34 (3H, с), 3,01 (6H, с), 3,37-3,47 (1Н, м), 3,69-3,74, 3,84-3,90 (1Н, м), 4,13-4,37 (5H, м), 4,71-4,74, 4,85-4,88 (1Н, м), 6,85(2H, д, J=9,0 Гц), 7,05 (1H, д, J=8,7 Гц), 7,30 (1Н, уш.), 7,40-7,44 (2H, м), 7,78-7,84 (3H, м), 7,90 (1H, д, J=9,3 Гц), 8,14 (2H, д, J=8,7 Гц), 8,25 (1H, уш.)
ИК (Nujol) 1731, 1620, 1595 см-1
APCI-MS m/z 577[M+H]+
[0311]
Способ синтеза соединения THK-5116
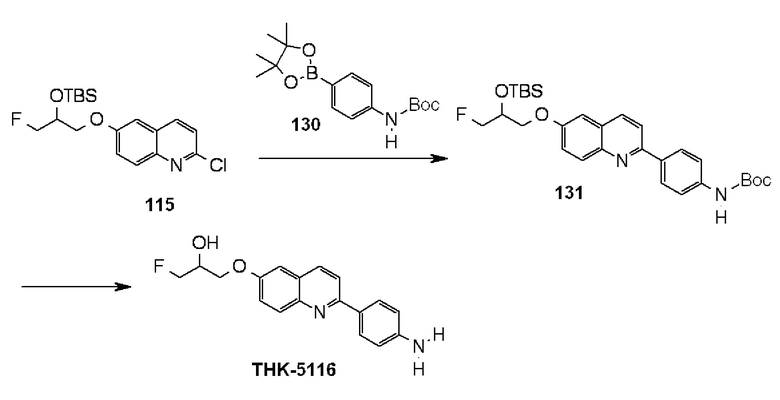
[0312]
Синтез соединения 131
Смесь соединения 115 (625 мг, 1,69 ммоль), 130 (593 мг, 1,86 ммоль), карбоната калия (467 мг, 3,40 ммоль), тетракистрифенилфосфина палладия (195 мг, 0,169 ммоль) и 1,2-диметоксиэтана (4,5 мл)―воды (0,5 мл) перемешивали в атмосфере аргона при 85°C в течение 16 часов и добавляли тетракистрифенилфосфин палладия (98 мг, 0,085 ммоль) с последующим дополнительным перемешивании при той же температуре в течение 8 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли хлороформ, и затем нерастворимые вещества удаляли фильтрацией, и раствор промывали хлороформом. Хлороформные слои объединяли и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/19, 1/9, 1/6) с получением соединения 131 (799 мг, 90%) в виде бледно-коричневого твердого вещества.
APCI-MS m/z 527[M+H]+
[0313]
Синтез соединения THK-5116
К раствору в хлороформе (10 мл) соединения 131 (790 мг, 1,50 ммоль) добавляли по каплям трифторуксусную кислоту (5 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 1 часа, и добавляли по каплям воду (5 мл) с последующим дополнительным перемешиванием при комнатной температуре в течение 1 часа. После отгонки органического растворителя при пониженном давлении реакционный раствор делали основным в условиях охлаждения льдом с использованием водного насыщенного раствора карбоната калия. Осадок собирали фильтрацией, промывали водой и затем сушили. Полученное твердое вещество очищали колоночной флэш-хроматографией на NH силикагеле (элюирующий растворитель: хлороформ, хлороформ/метанол=9/1, 1/1) и затем промывали метанолом с получением соединения THK-5116 (396 мг, 84%) в виде желтого твердого вещества.
точка плавления 219-221°C, 1H ЯМR (500 МГц, ДМСО-d6) δ 4,0-4,2 (3H, м), 4,4-4,7 (2H, м), 5,53 (3H, уш. с), 6,67 (2H, д, J=8,6 Гц), 7,3-7,4 (2H, м), 7,87 (1H, д, J=7,3 Гц), 7,92 (1Н, д, J=8,6 Гц), 7,95 (2H, д, J=8,7 Гц), 8,19 (1H, д, J=8,6 Гц)
ИК (Nujol) 1623, 1600 см-1
APCI-MS m/z 313[M+H]+
[0314]
Способ синтеза соединения THK-5117
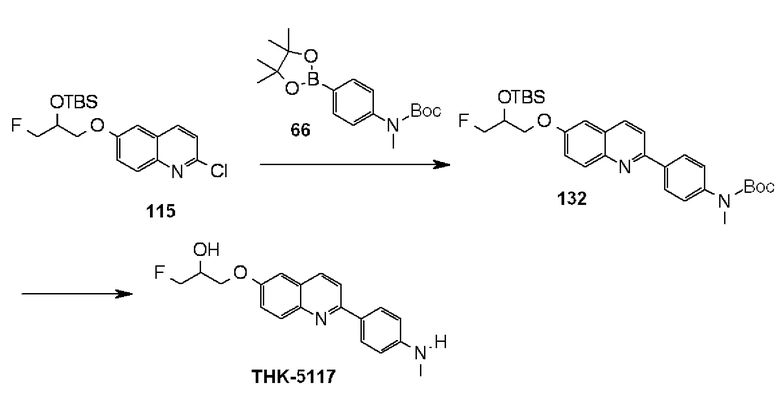
[0315]
Синтез соединения 132
Смесь соединения 115 (625 мг, 1,69 ммоль), соединения 66 (619 мг, 1,86 ммоль), карбоната калия (467 мг, 3,4 ммоль), тетракистрифенилфосфина палладия (195 мг, 0,169 ммоль) и 1,2-диметоксиэтана (4,5 мл)-воды (0,5 мл) перемешивали в атмосфере аргона при 85°C в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан, этилацетат/н-гексан=1/19, 1/9, 1/6) с получением соединения 132 (920 мг, 100%) в виде бесцветного маслянистого вещества.
APCI-MS m/z 541[M+H]+
[0316]
Синтез соединения THK-5117
К раствору в хлороформе (10 мл) соединения 132 (912 мг, 1,69 ммоль) добавляли по каплям трифторуксусную кислоту (5 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 1 часа, и добавляли по каплям воду (5 мл) с последующим дополнительным перемешиванием при комнатной температуре в течение 1 часа. После отгонки органического растворителя при пониженном давлении реакционный раствор делали основным в условиях охлаждения льдом с использованием водного насыщенного раствора карбоната калия, и раствор экстрагировали хлороформом-метанолом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток промывали диизопропиловым эфиром с получением соединения THK-5117 (468 мг, 85%) в виде желтого твердого вещества.
точка плавления 158-159°C, lH ЯМR (500 МГц, ДМСО-d6) δ 2,75 (3H, д, J=1,8 Гц), 4,0-4,2 (3H, м), 4,4-4,7 (2H, м), 5,53 (1H, д, J=4,8 Гц), 6,09 (1Н, уш. с), 6,65 (2H, д, J=8,9 Гц), 7,3-7,4 (2H, м), 7,87 (1H, д, J=8,7 Гц), 7,94 (1H, д, J=8,7 Гц), 8,03 (2H, д, J=8,6 Гц), 8,20 (1H, д, J=8,7 Гц)
ИК (Nujol) 1600 см-1
APCI-MS m/z 327[M+H]+
[0317]
Способ синтеза соединения THK-5100
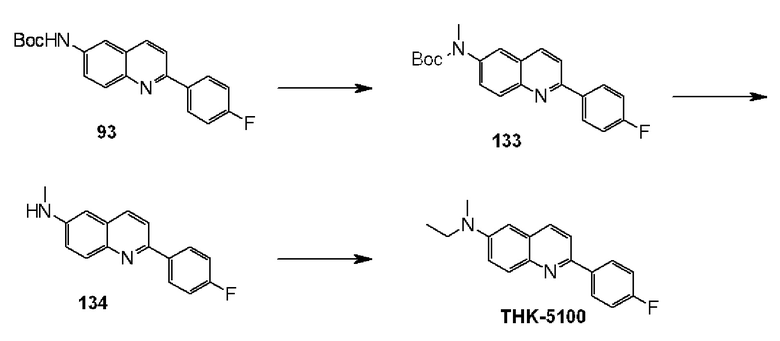
[0318]
Синтез соединения 133
К раствору в N,N-диметилформамиде (10 мл) соединения 93 (790 мг, 2,34 ммоль) добавляли 60% гидрид натрия (112 мг, 2,81 ммоль) в условиях охлаждения льдом и перемешивания в атмосфере аргона и смесь перемешивали при той же температуре в течение 5 минут, и добавляли по каплям метилйодид (0,22 мл, 3,5 ммоль) с последующим перемешиванием при той же температуре в течение 10 минут. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9, 1/4) с получением соединения 133 (800 мг, 97%) в виде бледно-желтого твердого вещества.
точка плавления 144-145°C
APCI-MS m/z 353[M+H]+
[0319]
Синтез соединения 134
К раствору в хлороформе (12 мл) соединения 133 (790 мг, 2,24 ммоль) добавляли по каплям трифторуксусную кислоту (8 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением соединения 134 (568 мг, 100%) в виде бледно-желтого твердого вещества.
точка плавления 134-134,5°C
APCI-MS m/z 253[M+H]+
[0320]
Синтез соединения THK-5100
К раствору в метаноле (24 мл)-уксусной кислоте (2,4 мл) соединения 134 (560 мг, 2,22 ммоль) и ацетальдегида (1,25 мл, 22,2 ммоль) небольшими порциям добавляли пиколин-борановый комплекс (710 мг, 6,66 ммоль) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=9/1) и затем промывали н-гексаном с получением соединения THK-5100 (475 мг, 76%) в виде бледно-желтых кристаллов.
точка плавления 137-138°C, 1Н ЯМR (400 МГц, ДМСО-d6) δ 1,11 (3H, т, J=7,1 Гц), 3,01 (3H, с), 3,55 (2H, кв., J=7,1 Гц), 6,93 (1H, д, J=2,7 Гц), 7,33 (2H, т, J=8,9 Гц), 7,46 (1H, дд, J=9,4, 3,0 Гц), 7,87 (1H, д, J=9,4 Гц), 7,95 (1H, д, J=8,8 Гц), 8,15 (1H, д, J=8,8 Гц), 8,24 (2H, дд, J=9,1, 5,7 Гц)
ИК (Nujol) 1618 см-1
APCI-MS m/z 281[M+H]+
[0321]
Способ синтеза соединения THK-5091 и соединения THK-5092
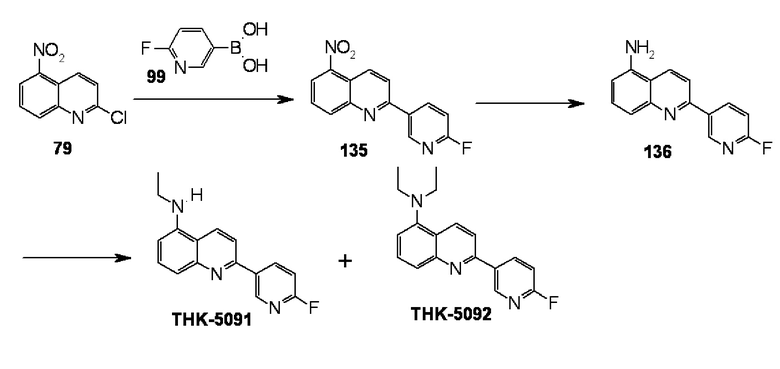
[0322]
Синтез соединения 135
К смеси соединения 79 (627 мг, 3,0 ммоль), соединения 99 (465 мг, 3,3 ммоль) и 1,2-диметоксиэтана (20 мл) добавляли водный 2M раствор карбоната натрия (3 мл, 6,0 ммоль) и тетракистрифенилфосфин палладия (173 мг, 0,15 ммоль) в атмосфере аргона и смесь нагревали в сосуде с обратным холодильником в течение 3 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, разбавляли этилацетатом и затем промывали по очереди водой и насыщенным солевым раствором. Органический слой сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=4/1, 3/1) с получением соединения 135 (700 мг, 87%) в виде бледно-желтого твердого вещества.
точка плавления 159-161°C
APCI-MS m/z 270[M+H]+
[0323]
Синтез соединения 136
Смесь соединения 135 (260 мг, 0,96 ммоль), 10% Pd-C (100 мг), этанола (30 мл) и этилацетата (10 мл) перемешивали в атмосфере водорода при комнатной температуре в течение 16 часов. Катализатор удаляли фильтрацией, и растворитель фильтрата отгоняли при пониженном давлении, и затем остаток промывали диизопропиловым эфиром с получением соединения 136 (178 мг, 77%) в виде твердого вещества.
точка плавления 156-157°C
APCI-MS m/z 240[M+H]+
[0324]
Синтез соединений THK-5091, THK-5092
К раствору в метаноле (20 мл)-уксусной кислоте (1 мл) соединения 136 (170 мг, 0,71 ммоль) и ацетальдегида (300 мг, 7 ммоль) небольшими порциями добавляли пиколин-борановый комплекс (230 мг, 2,14 ммоль) в условиях охлаждения льдом и перемешивания и смесь перемешивали при комнатной температуре в течение 8 часов. К реакционному раствору добавляли насыщенный солевой раствор, и раствор экстрагировали этилацетатом. Реакционный раствор промывали насыщенным солевым раствором и сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=10/1) с получением соединения THK-5092 (120 мг, 57%) и соединения THK-5091 (70 мг, 41%) в порядке элюирования.
[0325]
THK-5092
Желтое маслянистое вещество, 1Н ЯМR (400 МГц, CDCl3) δ 1,07 (6H, т, J=7,2 Гц), 3,22 (4H, кв., J=7,2 Гц), 7,09 (1H, дд, J=8,2, 2,4 Гц), 7,20 (1H, дд, J=7,3, 0,9 Гц), 7,67 (1H, дд, J=9,6, 8,6 Гц), 7,80 (1H, д, J=9,1 Гц), 7,85 (1H, д, J=8,5 Гц), 8,66 (1Н, м), 8,72 (1H, дд, J=8,8, 0,9 Гц), 8,94 (1H, д, J=2,7 Гц)
ИК (Nujol) 1586 см-1
APCI-MS m/z 296[M+H]+
[0326]
THK-5091
точка плавления 175-176°C, 1H ЯМR (400 МГц, CDCl3) δ 1,48 (3H, т, J=7,3 Гц), 3,35 (2H, кв., J=7,3 Гц), 4,27 (1Н, уш. с), 6,66 (1H, д, J=7,0 Гц), 7,08 (1H, дд, J=7,9, 2,1 Гц), 7,52 (1H, д, J=8,5 Гц), 7,61 (1Н, т), 7,75 (1H, д, J=8,8 Гц), 8,27 (1H, д, J=8,8 Гц), 8,66 (1H, м), 8,93 (1H, д, J=2,7 Гц)
ИК (Nujol) 3332, 1610 см-1
APCI-MS m/z 268[M+H]+
[0327]
Способ синтеза соединения THK-5097
[0328]
Синтез соединения 137
Смесь соединения 15 (3,73 г, 23 ммоль), триизопропилсилана хлорида (5,0 г, 26 ммоль), имидазола (2,07 г, 26 ммоль) и N,N-диметилформамида (50 мл) перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан, н-гексан/этилацетат=3/1, 1/1) и затем промывали н-гексаном с получением соединения 137 (6,52 г, 89%) в виде бледно-коричневого твердого вещества.
[0329]
Синтез соединения 138
К раствору в метиленхлориде (40 мл) соединения 137 (6,36 г, 20 ммоль) и соединения 4 (5,76 г, 28 ммоль) добавляли по каплям раствор в метиленхлориде (10 мл) трифторметансульфонового ангидрида (4,3 мл, 26 ммоль) при охлаждении льдом и перемешивании, и смесь перемешивали при той же температуре в течение 1 часа. К реакционному раствору добавляли ледяную воду, и нерастворимые вещества удаляли фильтрацией с последующим промыванием хлороформом. Органические слои объединяли и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан, н-гексан/этилацетат=19/1, 9/1) с получением соединения 138 (8,60 г, 95%) в виде коричневого маслянистого вещества.
[0330]
Синтез соединения 139
К смеси соединения 138 (4,24 г, 9,4 ммоль), соединения 99 (1,54 г, 10,9 ммоль) и 1,2-диметоксиэтана (80 мл) добавляли трикалий фосфат (3,98 г) и тетракистрифенилфосфин палладия (575 мг, 0,498 ммоль) в атмосфере аргона, и смесь нагревали кипячением в сосуде с обратным холодильником в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, и нерастворимые вещества удаляли фильтрацией с последующим промыванием хлороформом. Органические слои объединяли, и растворитель отгоняли при пониженном давлении. К остатку добавляли хлороформ-воду, и раствор экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан, н-гексан/этилацетат=50/1, 20/1) с получением соединения 139 (3,31 г, 88%) в виде бледно-коричневого вещества.
[0331]
Синтез соединения 140
К соединению 139 (3,31 г, 8,35 ммоль) добавляли тетрагидрофуран (4 мл), и затем добавляли тетрабутиламмоний фторид (8,5 мл/1M раствор тетрагидрофурана), и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток растворяли в хлороформе и промывали водой. Органический слой сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: хлороформ, хлороформ/метанол=50/1, 20/1, 10/1) и затем промывали н-гексаном и диизопропиловым эфиром с получением соединения 140 (1,87 г, 93%) в виде бледно-коричневого твердого вещества.
[0332]
Синтез соединения 141
К раствору в метиленхлориде (20 мл) соединения 140 (1,87 г, 7,8 ммоль) и этилдиизопропиламина (2,6 мл, 15,3 ммоль) добавляли по каплям раствор в метиленхлориде (10 мл) трифторметансульфонового ангидрида (2,35 мл, 14,3 ммоль) при охлаждении льдом и перемешивании, и смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционный раствор выливали в ледяную воду, и раствор экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан, н-гексан/этилацетат=50/1, 20/1, 10/1) с получением соединения 141 (1,81 г, 62%) в виде бледно-желтого твердого вещества.
[0333]
Синтез соединения 142
К смеси соединения 141 (2,03 г, 5,3 ммоль), ацетамида (393 мг, 6,65 ммоль) и 1,4-диоксана (50 мл) добавляли карбонат цезия (2,49 г), Pd2(dba)3 (251 мг) и Xantphos (316 мг) в атмосфере аргона, и смесь нагревали кипячением в сосуде с обратным холодильником в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, и нерастворимые вещества удаляли фильтрацией с последующим промыванием хлороформом. Органические слои объединяли, и растворитель отгоняли при пониженном давлении. К остатку добавляли хлороформ-воду, и раствор экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: хлороформ, хлороформ/метанол=50/1, 20/1) с получением соединения 142 (1,35 г, 88%) в виде бледно-коричневого твердого вещества.
[0334]
Синтез соединения 143
К раствору в N,N-диметилформамиде (8 мл) соединения 142 (650 мг, 2,3 ммоль) добавляли 60% гидрид натрия (111 мг, 2,7 ммоль) в условиях охлаждения льдом и перемешивания в атмосфере аргона и смесь перемешивали при той же температуре в течение 20 минут, и добавляли по каплям раствор в N,N-диметилформамиде (4 мл) этилйодида (396 мг, 2.5 ммоль) при той же температуре, с последующим перемешиванием при комнатной температуре в течение 1 часа. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 1/1, 2/1) и затем промывали н-гексаном с получением соединения 143 (575 мг, 80%) в виде бледно-коричневого твердого вещества.
APCI-MS m/z 310[M+H]+
[0335]
Синтез соединения 144
К раствору в тетрагидрофуране (2 мл) соединения 143 (531 мг, 1,7 ммоль) добавляли по каплям Zr(BH4)2 (14 мл/0,24M раствор тетрагидрофурана) при охлаждении льдом и перемешивании и смесь перемешивали при той же температуре в течение 30 минут. К реакционному раствору добавляли метанол, и растворитель отгоняли при пониженном давлении. К остатку добавляли водный насыщенный раствор гидрокарбоната натрия, и нерастворимые вещества удаляли фильтрацией, и затем водный слой экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/20, 1/10) с получением соединения 144 (249 мг, 49%) в виде желтого маслянистого вещества.
APCI-MS m/z 296[M+H]+
[0336]
Синтез соединения THK-5097
После растворения добавлением щавелевой кислоты (150 мг, 1,7 ммоль) к раствору в ацетоне (10 мл) соединения 144 (246 мг, 0,83 ммоль) растворитель отгоняли при пониженном давлении. Остаток промывали диизопропиловым эфиром с получением соединения THK-5097 (305 мг, 84%) в виде оранжевого твердого вещества.
точка плавления 127-129°C, 1Н ЯМR (400 МГц, ДМСО-d6) δ 1,19 (6H, т, J=7,0 Гц), 3,52 (4H, кв., J=7,0 Гц), 7,01 (1Н, д, J=2,5 Гц), 7,27 (1H, д, J=2,5 Гц), 7,34 (1Н, дд, J=8,6, 2,5 Гц), 7,7-7,8 (2H, м), 8,24 (1H, д, J=8,5 Гц), 8,7-8,8 (1Н, м), 9,02 (1H, д, J=9,7 Гц)
ИК (Nujol) 1659 см-1
APCI-MS m/z 296[M+H]+
[0337]
Синтез соединения THK-5098
[0338]
Синтез соединения 145
К раствору в N,N-диметилформамиде (6 мл) соединения 142 (650 мг, 2,3 ммоль) добавляли 60% гидрид натрия (111 мг, 2,7 ммоль) в условиях охлаждения льдом и перемешивания в атмосфере аргона и смесь перемешивали при той же температуре в течение 20 минут, и добавляли по каплям раствор в N,N-диметилформамиде (4 мл) этилйодида (360 мг, 2,5 ммоль) при той же температуре, с последующим перемешиванием при комнатной температуре в течение 1 часа. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 1/1, 2/1) и затем промывали н-гексаном с получением соединения 145 (518 мг, 76%) в виде бледно-коричневого твердого вещества.
[0339]
Синтез соединения 146
К раствору в тетрагидрофуране (4 мл) соединения 145 (517 мг, 1,75 ммоль) добавляли по каплям Zr(BH4)2 (14 мл/0,24M раствор тетрагидрофурана) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор охлаждали льдом и добавляли метанол, и растворитель отгоняли при пониженном давлении. К остатку добавляли метанол, и метанол отгоняли при пониженном давлении. К остатку добавляли водный насыщенный раствор гидрокарбоната натрия-этилацетата, и нерастворимые вещества удаляли фильтрацией, и затем водный слой экстрагировали этилацетатом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/20, 1/10) с получением соединения 146 (258 мг, 52%) в виде желтого маслянистого вещества.
[0340]
Синтез соединения THK-5098
После растворения добавлением щавелевой кислоты (164 мг, 1,8 ммоль) к раствору в ацетоне (10 мл) соединения 146 (257 мг, 0,91 ммоль) растворитель отгоняли при пониженном давлении. Остаток промывали диизопропиловым эфиром с получением соединения THK-5098 (333 мг, 88%) в виде оранжевого твердого вещества.
1H ЯМR (400 МГц, ДМСО-d6) δ 1,13 (3H, т, J=7,0 Гц), 3,05 (3H, с), 3,58 (2H, кв., J=7,0 Гц), 7,03 (1Н, д, J=2,5 Гц), 7,3-7,4 (2H, м), 7,8-7,9 (2H, м), 8,25 (1H, д, J=8,1 Гц), 8,7-8,8 (1Н, м), 9,03 (1H, д, J=2,5 Гц)
ИК (Nujol) 1646 см-1
APCI-MS m/z 282[M+H]+
[0341]
Способ синтеза соединения THK-5119
[0342]
Синтез соединения 147
К раствору в тетрагидрофуране (15 мл) соединения 114 (840 мг, 4,68 ммоль) и простого этилвинилового эфира (1,35 мл, 10,0 ммоль) добавляли соль паратолуолсульфоновой кислоты в виде соли пиридина (120 мг, 0,47 ммоль) и смесь перемешивали при комнатной температуре в течение 2,5 часов. После доведения pH реакционного раствора до 9 добавлением триэтиламина, растворитель отгоняли при пониженном давлении, и остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9) с получением соединения 147 (970 мг, 82%) в виде бесцветного маслянистого вещества.
APCI-MS m/z 252/254[M+H]+
[0343]
Синтез соединения 148
К раствору в 1,2-диметоксиэтане (70 мл) соединения 147 (2,08 г, 8,26 ммоль) и соединения 6a (2,90 г, 9,09 ммоль) добавляли карбонат калия (3,43 г, 24,8 ммоль), воду (1,44 мл) и тетракистрифенилфосфин палладия (960 мг, 0,83 ммоль) в атмосфере аргона, и смесь перемешивали при 80°C в течение 9 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9) с получением соединения 148 (3,00 г, 89%) в виде бесцветного твердого вещества.
точка плавления 115-117°C
APCI-MS m/z 409[M+H]+
[0344]
Синтез соединения 149
К раствору в N,N-диметилформамиде (25 мл) соединения 148 (1,79 г, 4,38 ммоль) добавляли 60% гидрид натрия (210 мг, 5,25 ммоль) в условиях охлаждения льдом и перемешивания и атмосфере аргона при той же температуре. После перемешивания при той же температуре в течение 10 минут добавляли по каплям метилйодид (0,41 мл, 6,57 ммоль) и смесь перемешивали при той же температуре в течение 10 минут. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением соединения 149 (1,85 г, 100%) в виде бесцветного смолянистого вещества.
APCI-MS m/z 423[M+H]+
[0345]
Синтез соединения 150
К раствору в хлороформе (12 мл) соединения 149 (1,85 г, 4,38 ммоль) добавляли по каплям трифторуксусную кислоту (8 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток измельчали и затем промывали этилацетатом/н-гексаном (1/4) с получением соединения 150 (1,04 г, 95%) в виде оранжевых кристаллов.
точка плавления 273-275°C
APCI-MS m/z 251[M+H]+
[0346]
Синтез соединения 151
К суспензии в метиленхлориде (40 мл) соединения 150 (1,03 г, 4,12 ммоль) и триэтиламина (1,72 мл, 12,36 ммоль) добавляли по каплям трифторуксусный ангидрид (1,40 мл, 9,89 ммоль) при охлаждении льдом и перемешивании и смесь перемешивали при той же температуре в течение 30 минут. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2) и затем очищали колоночной хроматографией на NH силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1, этилацетат) с получением соединения 151 (1,40 г, 98%) в виде бледно-желтого твердого вещества.
точка плавления 214-216°C
APCI-MS m/z 347[M+H]+
[0347]
Синтез соединения 152
К суспензии в тетрагидрофуране (40 мл) соединения 151 (1,39 г, 4,01 ммоль), соединения 63 (1,74 г, 4,82 ммоль) и трифенилфосфина (1,26 г, 4.82 ммоль) по каплям добавляли раствор в тетрагидрофуране (10 мл) диизопропилазодикарбоксилата (0,95 мл, 4,82 ммоль) в течение 30 минут в условиях охлаждения льдом и перемешивания, и смесь перемешивали при той же температуре в течение 1 часа, с последующим перемешиванием при комнатной температуре в течение 16 часов. К реакционному раствору добавляли соединение 63 (1,45 г, 4,01 ммоль), трифенилфосфин (1,05 г, 4,01 ммоль) и диизопропилазодикарбоксилат (0,80 мл, 4,01 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 1 часа. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением бесцветного маслянистого вещества (5,65 г). К раствору в хлороформе (24 мл) настоящего продукта (5,65 г) добавляли по каплям трифторуксусную кислоту (16 мл) при охлаждении льдом и перемешивании, и добавляли воду (16 мл) с последующим перемешиванием при комнатной температуре в течение 3 дней. Реакционный раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4, 2/3) с получением соединения 152 (2,15 г, 93%) в виде желтого аморфного вещества.
APCI-MS m/z 575[M+H]+
[0348]
Синтез соединения 153
К раствору в метиленхлориде (60 мл) соединения 152 (2,14 г, 3,72 ммоль) и 3,4-дигидро-2H-пирана (6,75 мл, 74 ммоль) добавляли моногидрат паратолуолсульфоновой кислоты (830 мг, 4,84 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. После доведения pH реакционного раствора до 9 с использованием триэтиламина, растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9, 1/2) с получением соединения 153 (2,36 г, 96%) в виде бледно-желтого аморфного вещества.
APCI-MS m/z 659[M+H]+
[0349]
Синтез соединения THK-5119
К раствору в тетрагидрофуране (28 мл)-воде (11 мл) соединения 153 (2,35 г, 3,57 ммоль) добавляли гидроксид моногидрат лития (225 мг, 5,35 ммоль) в условиях охлаждения льдом и перемешивания и смесь перемешивали при той же температуре в течение 30 минут. К реакционному раствору добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=2/3) с получением соединения THK-5119 (1,78 г, 89%) в виде бледно-желтых кристаллов.
точка плавления 126-126,5°C
1H ЯМR (500 МГц, ДМСО-d6) δ 1,35-1,71 (6H, м), 2,34, 2,34 (3H, с), 2,75 (3H, уш.), 3,38-3,46 (1Н, м), 3,66-3,72, 3,81-3,88 (1Н, м), 4,08-4,37 (5H, м), 4,69-4,72, 4,85-4,87 (1Н, м), 6,10 (1Н, уш.), 6,66 (2H, д, J=9,0 Гц), 7,22-7,25 (1Н, м), 7,27-7,29 (1Н, м), 7,39-7,43 (2H, м), 7,77-7,81 (2H, м), 7,85 (1H, д, J=9,0 Гц), 7,96 (1H, д, J=8,7 Гц), 8,03 (2H, д, J=8,7 Гц), 8,18 (1H, д, J=8,7 Гц)
ИК (Nujol) 3377, 1621, 1610 см-1
APCI-MS m/z 563[M+H]+
[0350]
Способ синтеза соединения THK-5120
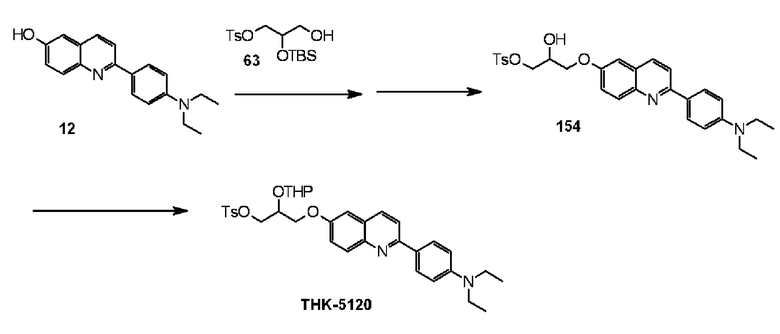
[0351]
Синтез соединения 154
К суспензии в тетрагидрофуране (25 мл) соединения 12 (340 мг, 1,16 ммоль), соединения 63 (500 мг, 1,39 ммоль) и трифенилфосфина (370 мг, 1,39 ммоль) добавляли по каплям раствор в тетрагидрофуране (5 мл) диизопропилазодикарбоксилата (0,28 мл, 1,39 ммоль) в течение 20 минут в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли соединение 63 (500 мг, 1,39 ммоль), трифенилфосфин (370 мг, 1,39 ммоль) и диизопропилазодикарбоксилат (0,28 мл, 1,39 ммоль) в условиях охлаждения льдом и перемешивания, с последующим дополнительным перемешиванием при комнатной температуре в течение 3 дней. К реакционному раствору добавляли соединение 63 (170 мг, 0,46 ммоль), трифенилфосфин (120 мг, 0,46 ммоль), и диизопропилазодикарбоксилат (0,09 мл, 0,46 ммоль) добавляли в условиях охлаждения льдом и перемешивания, с последующим дополнительным перемешиванием при комнатной температуре в течение 1 дня. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9). К раствору в хлороформе (12 мл) полученной основной фракции добавляли по каплям трифторуксусную кислоту (8 мл) при охлаждении льдом и перемешивании и воду (2 мл), с последующим дальнейшим перемешиванием при комнатной температуре в течение 16 часов. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом после доведения pH до 8 с использованием водного раствора карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/3, 1/1) и затем промывали этилацетатом/н-гексаном (1/2) с получением соединения 154 (410 мг, 68%) в виде бледно-желтых кристаллов.
точка плавления 167-168°C
APCI-MS m/z 521[M+H]+
[0352]
Синтез соединения THK-5120
К раствору в метиленхлориде (10 мл) соединения 154 (400 мг, 0,77 ммоль) добавляли 3,4-дигидро-2H-пиран (1,39 мл, 15,4 ммоль) и моногидрат паратолуолсульфоновой кислоты (172 мг, 1,00 ммоль) и смесь перемешивали при комнатной температуре в течение 10 минут. После доведения pH реакционного раствора до 9 с использованием триэтиламина растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением соединения THK-5120 (466 мг, 100%) в виде бледно-желтого аморфного вещества.
1Н ЯМR (500 МГц, ДМСО-d6) δ 1,14 (6H, т, J=6,9 Гц), 1,34-1,69 (6H, м), 2,33, 2,34 (3H, с), 3,22-3,32 (1Н, м), 3,42 (4H, кв., J=7,3 Гц), 3,66-3,72, 3,82-3,87 (1Н, м), 4,09-4,37 (5H, м), 4,69-4,72, 4,85-4,87 (1Н, м), 6,78 (2H, д, J=9,0 Гц), 7,22-7,26 (1Н, м), 7,28-7,31 (1Н, м), 7,38-7,43 (2H, м), 7,77-7,82 (2H, м), 7,86 (1H, д, J=9,0 Гц), 7,97 (1Н, д, J=8,7 Гц), 8,07 (2H, д, J=9,0 Гц), 8,20 (1H, д, J=8,3 Гц)
APCI-MS m/z 605[M+H]+
[0353]
Способ синтеза соединения THK-5121
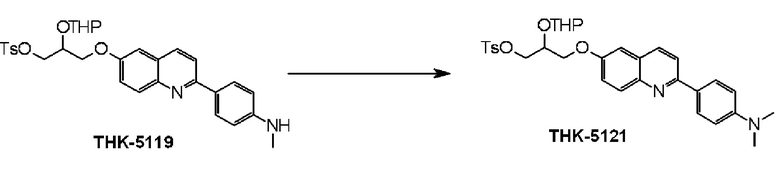
[0354]
Синтез соединения THK-5121
К раствору в этаноле (12 мл)-уксусной кислоте (1,2 мл)-хлороформе (10 мл) соединения THK-5119 (600 мг, 1,07 ммоль) и водному 20% раствору формальдегида (1,60 мл, 10,7 ммоль) небольшими порциями добавляли комплекс пиколина с бораном (342 мг, 3,21 ммоль) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор разбавляли этилацетатом и экстрагировали этилацетатом после доведения pH до 8 с использованием водного раствора карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=4/1) и затем перекристаллизовывали из н-гексана/этилацетата с получением соединения THK-5121 (566 мг, 92%) в виде бледно-желтых кристаллов.
точка плавления 152-154°C
1H ЯМR (500 МГц, ДМСО-d6) δ 1,34-1,68 (6H, м), 2,34 (3H, с), 3,01 (6H, с), 3,41-3,47 (1Н, м), 3,65-3,72, 3,81-3,87 (1Н, м), 4,10-4,37 (5H, м), 4,68-4,72, 4,85-4,87 (1Н, м), 6,85 (2H, д, J=7,4 Гц), 7,25-7,37 (2H, м), 7,39-7,43 (2H, м), 7,77-7,81 (2H, м), 7,88-7,96 (1H, м), 8,01-8,07 (1Н, м), 8,11 (2H, д, J=8,7 Гц), 8,23-8,32 (1Н, м)
ИК (Nujol) 1622 см-1
APCI-MS m/z 577[M+H]+
[0355]
Способ синтеза соединения THK-5122
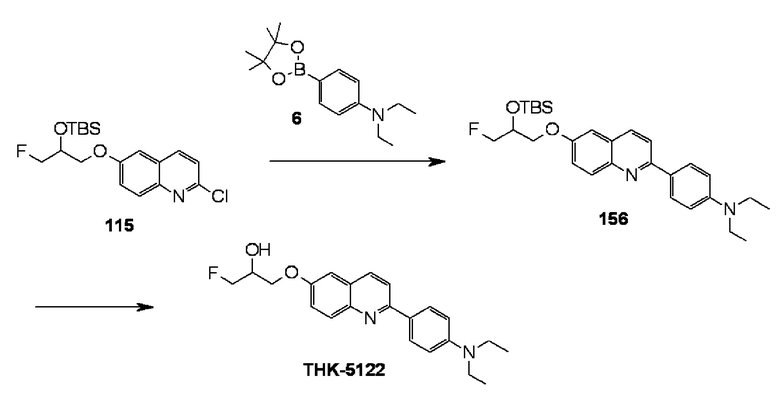
[0356]
Синтез соединения 156
К смеси соединения 115 (650 мг, 1,76 ммоль), соединения 6 (580 мг, 2,11 ммоль) и 1,2-диметоксиэтана (20 мл) добавляли карбонат натрия (372 мг, 3.50 ммоль) и воду (2 мл) и добавляли тетракистрифенилфосфин палладия (102 мг, 0,088 ммоль) в атмосфере аргона, и смесь нагревали кипячением в сосуде с обратным холодильником в течение 48 часов. К реакционному раствору добавляли тетракистрифенилфосфин палладия (102 мг, 0,088 ммоль) и смесь нагревали кипячением в сосуде с обратным холодильником в течение 24 часов в атмосфере аргона. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли силикагель, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=10/1) с получением соединения 156 (750 мг, 89%) в виде бледно-желтого твердого вещества.
точка плавления 104-105°C
[0357]
Синтез соединения THK-5122
К смеси соединения 156 (740 мг, 1,55 ммоль) и тетрагидрофурана (20 мл) добавляли 1M тетра-н-бутиламмонийфторид/тетрагидрофуран (2,0 мл, 2,00 ммоль), и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор выливали в воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=3/1, 2/1) и затем перекристаллизовывали из этилацетата/н-гексана (1/1) с получением соединения THK-5122 (448 мг, 78%) в виде кристаллов.
точка плавления 126-127°C
1H ЯМR (400 МГц, CDCl3) δ 1,21 (6H, т, J=7,0 Гц), 2,60 (1Н, уш. с), 3,43 (4H, кв., J=7,0 Гц), 4,14-4,23 (2H, м), 4,26-4,37 (1Н, м), 4,53-4,62 (1Н, м), 4,66-4,74 (1Н, м), 6,78 (2H, д, J=8,8 Гц), 7,06 (1H, д, J=2,7 Гц), 7,32 (1H, дд, J=9,0, 2,7 Гц), 7,78 (1H, д, J=9,0 Гц), 7,99 (1Н, д, J=7,0 Гц), 8,01 (1Н, д, J=7,0 Гц), 8,03 (2H, д, J=8,8 Гц)
ИК (Nujol) 3378, 1620, 1596 см-1
APCI-MS m/z 369[M+H]+
[0358]
Способ синтеза соединения THK-5123
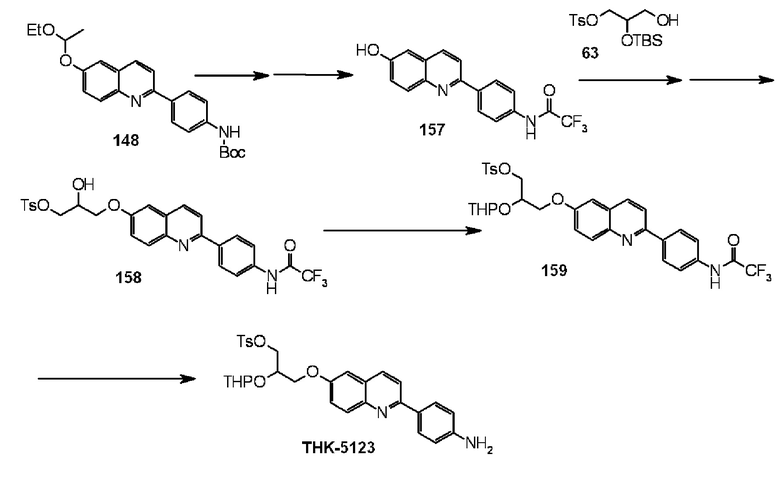
[0359]
Синтез соединения 157
К раствору в хлороформе (12 мл) соединения 148 (1,20 г, 2,94 ммоль) добавляли по каплям трифторуксусную кислоту (8 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 1,5 часов. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали хлороформом/метанолом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость сушили, растворитель отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1, этилацетат). К суспензии в метиленхлориде (20 мл) полученного оранжевого твердого вещества (650 мг) и триэтиламина (1,13 мл, 8,13 ммоль) добавляли по каплям трифторуксусный ангидрид (0,92 мл, 6,5 ммоль) при охлаждении льдом и перемешивании, с последующим перемешиванием при той же температуре в течение 30 минут. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали хлороформом/метанолом. Экстракционную жидкость сушили, растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1) и затем промывали этилацетатом/н-гексаном (1/4) с получением соединения 157 (360 мг, 37%) в виде бледно-коричневых кристаллов.
точка плавления 253-254°C
APCI-MS m/z 333[M+H]+
[0360]
Синтез соединения 158
К суспензии в тетрагидрофуране (25 мл) соединения 157 (350 мг, 1,05 ммоль), соединения 63 (911 мг, 2,53 ммоль) и трифенилфосфина (664 мг, 2,53 ммоль) добавляли по каплям раствор в тетрагидрофуране (8 мл) диизопропилазодикарбоксилата (0,5 мл, 2,52 ммоль) в течение 20 минут в условиях охлаждения льдом и перемешивания, и смесь перемешивали при той же температуре в течение 1 часа, с последующим перемешиванием при комнатной температуре в течение 3 дней. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4). К раствору в хлороформе (16 мл) полученной основной фракции добавляли по каплям трифторуксусную кислоту (11 мл) при охлаждении льдом и перемешивании и добавляли воду (2,5 мл) с последующим дальнейшим перемешиванием при комнатной температуре в течение 16 часов. Реакционный раствор разбавляли хлороформом и экстрагировали хлороформом после доведения pH до 8 с использованием водного раствора карбоната калия. Экстракционную жидкость сушили и очищали колоночной хроматографией на силикагеле (элюирующий растворитель: хлороформ, этилацетат/н-гексан=1/2, 1/1) с получением соединения 158 (490 мг, 83%) в виде бледно-желтого твердого вещества.
точка плавления 186-188°C
APCI-MS m/z 561[M+H]+
[0361]
Синтез соединения 159
К суспензии в метиленхлориде (20 мл) соединения 158 (480 мг, 0,86 ммоль) и 3,4-дигидро-2H-пирана (1,55 мл, 17,2 ммоль) добавляли моногидрат паратолуолсульфоновой кислоты (192 мг, 1,12 ммоль) и смесь перемешивали при комнатной температуре в течение 10 минут. После доведения pH реакционного раствора до 8 добавлением триэтиламина, растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4, 1/2) с получением соединения 159 (480 мг, 87%) в виде бледно-желтого твердого вещества.
точка плавления 112-115°C
APCI-MS m/z 645[M+H]+
[0362]
Синтез соединения THK-5123
К раствору в тетрагидрофуране (9 мл)-воде (3 мл) соединения 159 (470 мг, 0,73 ммоль) добавляли моногидрат гидроксида лития (46 мг, 1,09 ммоль) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при той же температуре в течение 1,5 часов, с последующим перемешиванием при комнатной температуре в течение 1 часа. Добавляли моногидрат гидроксида лития (46 мг, 1,09 ммоль) и метанол (2 мл) с последующим дальнейшим перемешиванием при комнатной температуре в течение 16 часов. Реакционный раствор разбавляли этилацетатом, промывали водой и сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=2/3, 1/1) с получением соединения THK-5123 (380 мг, 95%) в виде бледно-желтого аморфного вещества.
1H ЯМR (500 МГц, ДМСО-d6) δ 1,34-1,71 (6H, м), 2,33, 2,34 (3H, с), 3,37-3,46 (1Н, м), 3,66-3,72, 3,82-3,87 (1Н, м), 4,08-4,36 (5H, м), 4,69-4,72, 4,84-4,87 (1Н, м), 5,52 (2H, уш.), 6,68 (2H, д, J=8,7 Гц), 7,21-7,25 (1Н, м), 7,26-7,29 (1Н, м), 7,38-7,43 (2H, м), 7,77-7,81 (2H, м), 7,84 (1H, д, J=9,3 Гц), 7,93 (1H, д, J=8,7 Гц), 7,96 (1Н, д, J=8,7 Гц), 8,17 (1H, д, J=8,7 Гц)
ИК (Nujol) 3452, 1728, 1622 см-1
APCI-MS m/z 549[M+H]+
[0363]
Способ синтеза соединения THK-5125
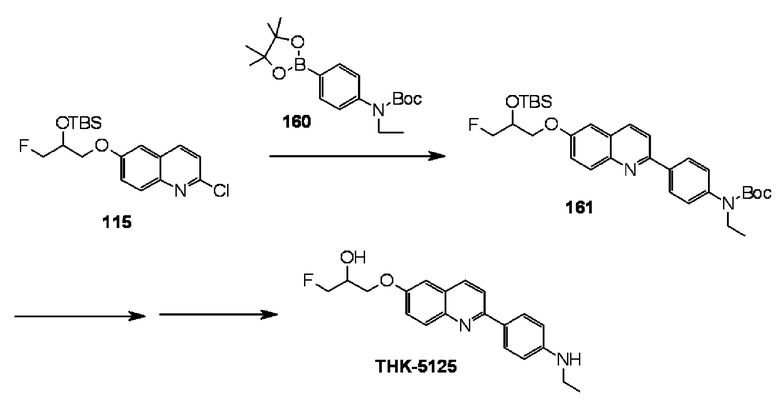
[0364]
Синтез соединения 161
К смеси соединения 115 (650 мг, 1,76 ммоль), соединения 160 (733 мг, 2,11 ммоль) и 1,2-диметоксиэтана (20 мл) добавляли карбонат натрия (372 мг, 3,50 ммоль) и воду (2 мл), и тетракистрифенилфосфин палладия (102 мг, 0,088 ммоль) добавляли в атмосфере аргона, и затем смесь нагревали кипячением в сосуде с обратным холодильником в течение 33 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, фильтровали целитом и затем очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=10/1) с получением соединения 161 (900 мг, 93%) в виде вязкого маслянистого вещества.
[0365]
Синтез соединения THK-5125
К смеси соединения 161 (900 мг, 1,62 ммоль) и тетрагидрофурана (20 мл) добавляли 1M тетра-н-бутиламмонийфторид/тетрагидрофуран (2,0 мл, 2,00 ммоль), и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор выливали в воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. К раствору в метиленхлориде (20 мл) остатка добавляли трифторуксусную кислоту (4 мл) с последующим перемешиванием при комнатной температуре в течение 1,5 часов. Реакционный раствор разбавляли этилацетатом, и раствор делали основным водным насыщенным раствором гидрокарбоната натрия. Органический слой промывали насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток перекристаллизовывали из этилацетата с получением соединения THK-5125 (350 мг, 63%) в виде бледно-коричневых кристаллов.
точка плавления 146-147°C
1H ЯМR (400 МГц, CDCl3) δ 1,29 (3H, т, J=7,1 Гц), 2,61 (1Н, уш.), 3,24 (2H, кв., J=7,1 Гц), 3,80 (1H, уш.), 4,14-4,23 (2H, м), 4,32 (1Н, уш. д, J=18 Гц), 4,54-4,62 (1Н, м), 4,65-4,74 (1Н, м), 6,71 (2H, д, J=8,8 Гц), 7,07 (1H, д, J=2,7 Гц), 7,33 (1H, дд, J=9,0, 2,7 Гц), 7,76 (1H, д, J=8,8 Гц), 8,01 (2H, д, J=8,8 Гц), 8,02 (1H, д, J=9,0 Гц), 7,90-8,20 (1H, уш.)
ИК (Nujol) 3432, 3356, 1621, 1598 см-1
APCI-MS m/z 341[M+H]+
[0366]
Способ синтеза соединения THK-5131
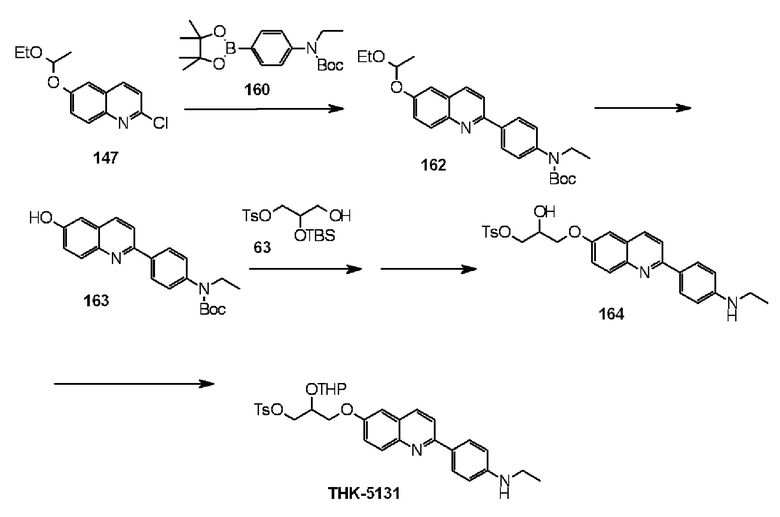
[0367]
Синтез соединения 162
К раствору в 1,2-диметоксиэтане (20 мл) соединения 147 (600 мг, 2,38 ммоль) и соединения 160 (830 мг, 2,38 ммоль) добавляли карбонат калия (990 мг, 7,14 ммоль), воду (0,41 мл) и тетракистрифенилфосфин палладия (275 мг, 0,24 ммоль) в атмосфере аргона и смесь перемешивали при 80°C в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9) с получением соединения 162 (1,02 г, 98%) в виде бледно-желтого вязкого маслянистого вещества.
APCI-MS m/z 437[M+H]+
[0368]
Синтез соединения 163
К раствору в хлороформе (10 мл) соединения 162 (1,01 г, 2,31 ммоль) добавляли по каплям трифторуксусную кислоту (1 мл) при охлаждении льдом и перемешивании, и смесь перемешивали при той же температуре в течение 40 минут, и добавляли трифторуксусную кислоту (0,5 мл) с последующим перемешиванием при той же температуре в течение 10 минут. К реакционному раствору добавляли ледяную воду―этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=4/1, 2/1) с получением соединения 163 (740 мг, 88%) в виде бледно-желтого твердого вещества.
точка плавления 205-206°C
APCI-MS m/z 365[M+H]+
[0369]
Синтез соединения 164
К раствору в тетрагидрофуране (30 мл) соединения 163 (730 мг, 2,00 ммоль), соединения 63 (1,74 г, 4,81 ммоль) и трифенилфосфина (1,26 г, 4,81 ммоль) добавляли по каплям раствор в тетрагидрофуране (10 мл) диизопропилазодикарбоксилата (0,95 мл, 4,81 ммоль) в течение 20 минут в условиях охлаждения льдом и перемешивания, и смесь перемешивали при той же температуре в течение 1 часа с последующим перемешиванием при комнатной температуре в течение 4 дней. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9). К раствору в хлороформе (24 мл) полученного бесцветного маслянистого вещества (2,53 г) добавляли по каплям трифторуксусную кислоту (16 мл) при охлаждении льдом и перемешивании, и добавляли воду (4 мл) с последующим перемешиванием при комнатной температуре в течение 16 часов. К реакционному раствору добавляли ледяную воду и этилацетат, и раствор экстрагировали этилацетатом доведением pH до 8 с использованием раствора карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, этилацетат) и затем промывали этилацетатом/н-гексаном (1/2) с получением соединения 164 (880 мг, 89%) в виде желтых кристаллов.
точка плавления 162-164°C
APCI-MS m/z 493[M+H]+
[0370]
Синтез соединения THK-5131
К суспензии в метиленхлориде (20 мл) соединения 164 (440 мг, 0,893 ммоль) добавляли 3,4-дигидро-2H-пиран (1,62 мл, 17,9 ммоль) и моногидрат паратолуолсульфоновой кислоты (200 мг, 1,16 ммоль) и смесь перемешивали при комнатной температуре в течение 15 минут. После доведения pH реакционного раствора до 8 с использованием триэтиламина растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2) и затем перекристаллизовывали из этилацетата/н-гексана с получением соединения THK-5131 (389 мг, 76%) в виде бледно-желтых кристаллов.
точка плавления 103-105°C
1H ЯМR (500 МГц, ДМСО-d6) δ 1,20 (3H, т, J=7,1 Гц), 1,34-1,69 (6H, м), 2,33, 2,34 (3H, с), 3,09-3,15 (2H, м), 3,37-3,46 (1Н, м), 3,66-3,72, 3,80-3,88 (1Н, м), 4,09-4,37 (5H, м), 4,69-4,72, 4,84-4,87 (1Н, м), 6,01 (1Н, уш.), 6,67 (2H, д, J=8,3 Гц), 7,23 (1H, д, J=9,0 Гц), 7,26-7,29 (1Н, м), 7,38-7,43 (2H, м), 7,77-7,81 (2H, м), 7,85 (1H, д, J=9,0 Гц), 7,95 (1H, д, J=8,7 Гц), 8,01 (2H, д, J=8,7 Гц), 8,18 (1H, д, J=8,0 Гц)
ИК (Nujol) 3360, 1619, 1608 см-1
APCI-MS m/z 577[M+H]+
[0371]
Способ синтеза соединения THK-5127
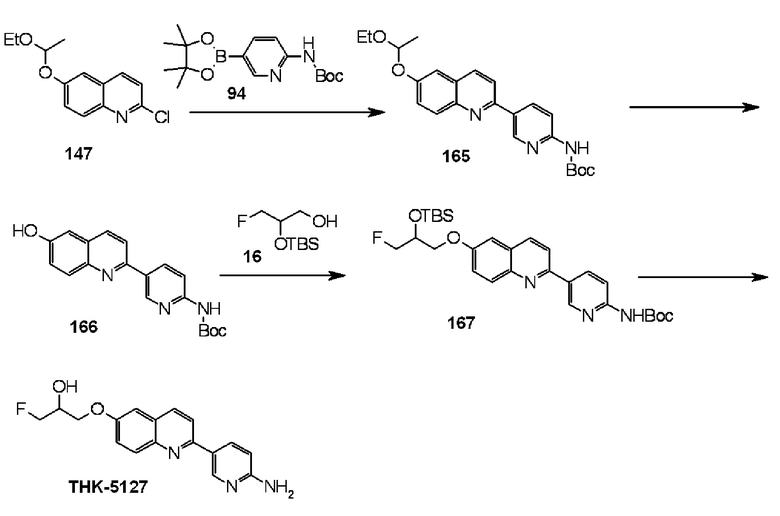
[0372]
Синтез соединения 165
К суспензии в 1,2-диметоксиэтане (63 мл) соединения 147 (1,94 г, 7,71 ммоль) и соединения 94 (2,72 г, 8,48 ммоль) добавляли карбонат калия (3,20 г, 23,1 ммоль), воду (1,34 мл) и тетракистрифенилфосфин палладия (890 мг, 0,77 ммоль) в атмосфере аргона и смесь перемешивали при 80°C в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) и затем перекристаллизовывали из этилацетата/н-гексана с получением соединения 165 (2,78 г, 88%) в виде бледно-желтых кристаллов.
точка плавления 182-183°C
APCI-MS m/z 410[M+H]+
[0373]
Синтез соединения 166
К раствору в хлороформе (40 мл) соединения 165 (2,77 г, 6,76 ммоль) добавляли по каплям трифторуксусную кислоту (4 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 20 минут. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом-тетрагидрофураном после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток перекристаллизовывали из этилацетата с получением соединения 166 (2,00 г, 88%) в виде бледно-желтых кристаллов.
точка плавления 325-326°C
APCI-MS m/z 338[M+H]+
[0374]
Синтез соединения 167
К раствору в тетрагидрофуране (40 мл) соединения 166 (1,50 г, 4,45 ммоль), соединения 16 (1,11 г, 5,35 ммоль) и трифенилфосфина (1,40 г, 5,35 ммоль) добавляли по каплям раствор в тетрагидрофуране (10 мл) диизопропилазодикарбоксилата (1,06 мл, 5,35 ммоль) в течение 30 минут в условиях охлаждения льдом и перемешивания, и смесь перемешивали при той же температуре в течение 1 часа с последующим перемешиванием при комнатной температуре в течение 3 дней. К реакционному раствору дополнительно добавляли соединение 16 (830 мг, 4,00 ммоль) и трифенилфосфин (1.05 г, 4,00 ммоль), и диизопропилазодикарбоксилат (0,79 мл, 4,00 ммоль) добавляли по каплям при охлаждении льдом и перемешивании с последующим дополнительным перемешиванием при комнатной температуре в течение 5 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9, 1/4) и затем перекристаллизовывали из этилацетата/н-гексана с получением соединения 167 (1,51 г, 64%) в виде бесцветных кристаллов.
точка плавления 188-189°C
APCI-MS m/z 528[M+H]+
[0375]
Синтез соединения THK-5127
К раствору в хлороформе (24 мл) соединения 167 (1,50 г, 2,84 ммоль) добавляли по каплям трифторуксусную кислоту (16 мл) при охлаждении льдом и перемешивании, добавляли воду (4 мл) и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли ледяную воду и затем этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 9 с использованием водного раствора карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат) и затем перекристаллизовывали из этилацетата с получением соединения THK-5127 (717 мг, 81%) в виде бледно-желтых кристаллов.
точка плавления 196-197°C
1Н ЯМR (400 МГц, ДМСО-d6) δ 4,06-4,19 (3H, м), 4,44-4,65 (2H, м), 5,54 (1Н, д, J=5,7 Гц), 6,33 (2H, с), 6,57 (1H, д, J=8,8 Гц), 7,36-7,41 (2H, м), 7,87-7,91 (1Н, м), 7,97 (1H, д, J=8,8 Гц), 8,21-8,27 (2H, м), 8,80 (1H, д, J=2,4 Гц)
ИК (Nujol) 3440, 1651, 1619 см-1
APCI-MS m/z 314[M+H]+
[0376]
Способ синтеза соединения THK-5150
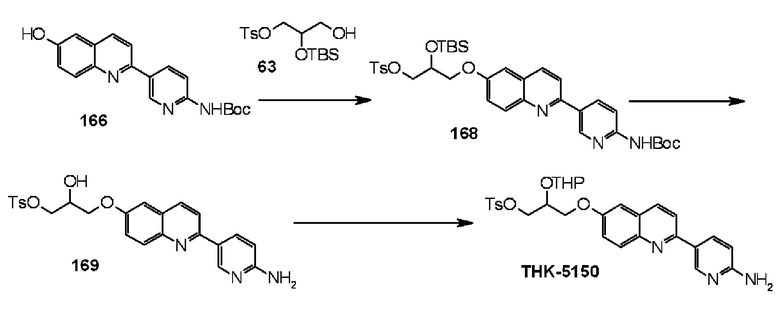
[0377]
Синтез соединения 168
К раствору в тетрагидрофуране (30 мл) соединения 166 (980 мг, 2,91 ммоль), соединения 63 (2,51 г, 6,98 ммоль) и трифенилфосфина (1,83 г, 6,98 ммоль) добавляли по каплям раствор в тетрагидрофуране (10 мл) диизопропилазодикарбоксилата (1,38 мл, 6,98 ммоль) в течение 10 минут в условиях охлаждения льдом и перемешивания, и смесь перемешивали при той же температуре в течение 1 часа, с последующим перемешиванием при комнатной температуре в течение 2 дней. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток промывали этилацетатом/н-гексаном=1/4) после измельчения с получением соединения 168 (1,38 г, 70%) в виде бесцветных кристаллов.
точка плавления 204-206°C
APCI-MS m/z 680[M+H]+
[0378]
Синтез соединения 169
К раствору в хлороформе (24 мл) соединения 168 (1,98 г, 2,91 ммоль) добавляли по каплям трифторуксусную кислоту (16 мл) при охлаждении льдом и перемешивании, и добавляли воду (4 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли ледяную воду и этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 8 с использованием водного раствора карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1, этилацетат) и затем промывали этилацетатом/н-гексаном (1/1) с получением соединения 169 (940 мг, 69%) в виде бесцветных кристаллов.
точка плавления 168-169°C
APCI-MS m/z 466[M+H]+
[0379]
Синтез соединения THK-5150
К раствору в этиленхлориде (20 мл) соединения 169 (930 мг, 2.00 ммоль) добавляли 3,4-дигидро-2H-пиран (3,62 мл, 40,0 ммоль) и моногидрат паратолуолсульфоновой кислоты (447 мг, 2,60 ммоль), и смесь перемешивали при комнатной температуре в течение 40 минут. Добавляли моногидрат паратолуолсульфоновой кислоты (344 мг, 2,0 ммоль) с последующим перемешиванием при комнатной температуре в течение 10 минут. После доведения pH реакционного раствора до 8 добавлением триэтиламина добавляли этилацетат и воду, с последующим перемешиванием при комнатной температуре в течение 10 минут. Органический слой отделяли и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1, 2/1, этилацетат) с получением соединения THK-5150 (510 мг, 46%) в виде бесцветного пеноподобного вещества.
1H ЯМR (500 МГц, ДМСО-d6) δ 1,35-1,68 (6H, м), 2,34, 2,34 (3H, с), 3,36-3,46 (1Н, м), 3,66-3,71, 3,82-3,87 (1Н, м), 4,09-4,37 (5H, м), 4,70, 4,84-4,87 (1Н, м), 6,37 (2H, с), 6,57 (1H, д, J=8,7 Гц), 7,23-7,27 (1Н, м), 7,29-7,31 (1Н, м), 7,41 (2H, дд, J=8,2, 1,8 Гц), 7,79 (2H, дд, J=8,7, 2,6 Гц), 7,87 (1H, д, J=9,0 Гц), 7,98(1Н, д, J=8,7 Гц), 8,21 (1Н, д, J=8,7 Гц), 8,25 (1H, дд, J=8,7, 2,6 Гц), 8,80 (1H, д, J=2,2 Гц)
ИК (Nujol) 1621 см-1
APCI-MS m/z 550[M+H]+
[0380]
Способ синтеза соединения THK-5152
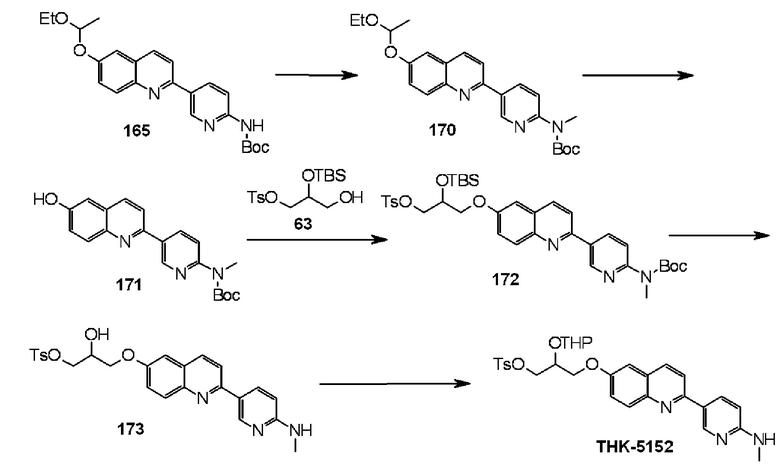
[0381]
Синтез соединения 170
К суспензии в N,N-диметилформамиде (30 мл) соединения 165 (1,87 г, 4,57 ммоль) небольшими порциями добавляли 60% гидрид натрия (220 мг, 5,5 ммоль) в условиях охлаждения льдом и перемешивания в атмосфере аргона, и смесь перемешивали при той же температуре в течение 15 минут, и добавляли по каплям метилйодид (0,43 мл, 6,9 ммоль) при той же температуре, с последующим перемешиванием при той же температуре в течение 5 минут и дополнительным перемешиванием при комнатной температуре в течение 30 минут. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9, 1/4) с получением соединения 170 (1,93 г, 100%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 424[M+H]+
[0382]
Синтез соединения 171
К раствору в хлороформе (20 мл) соединения 170 (1,93 г, 4,56 ммоль) добавляли по каплям трифторуксусную кислоту (4 мл) при охлаждении льдом и перемешивании и смесь перемешивали при той же температуре в течение 1 часа. К реакционному раствору добавляли воду и этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 8 с использованием водного раствора карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=2/1) и затем промывали н-гексаном/этилацетатом=2/1 с получением соединения 171 (1,57 г, 98%) в виде бледно-желтых кристаллов.
точка плавления 179-180°C
APCI-MS m/z 352[M+H]+
[0383]
Синтез соединения 172
К суспензии в тетрагидрофуране (20 мл) соединения 171 (500 мг, 1,42 ммоль), соединения 63 (1,23 г, 3,42 ммоль) и трифенилфосфина (900 мг, 3,42 ммоль) добавляли по каплям раствор в тетрагидрофуране (10 мл) диизопропилазодикарбоксилата (0,68 мл, 3,42 ммоль) в течение 5 минут в условиях охлаждения льдом и перемешивания, и смесь перемешивали при той же температуре в течение 1 часа, с последующим перемешиванием при комнатной температуре в течение 16 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9, 1/4) и затем перекристаллизовывали из этилацетата/н-гексана (1/9) с получением соединения 172 (950 мг, 96%) в виде бесцветных кристаллов.
точка плавления 112-113°C
APCI-MS m/z 694[M+H]+
[0384]
Синтез соединения 173
К раствору в хлороформе (18 мл) соединения 172 (940 мг, 1,35 ммоль) добавляли по каплям трифторуксусную кислоту (12 мл) при охлаждении льдом и перемешивании и добавляли воду (3 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли ледяную воду и этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 8 с использованием водного раствора карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1, этилацетат) с получением соединения 173 (400 мг, 62%) в виде бледно-желтого твердого вещества.
точка плавления 162-163°C
APCI-MS m/z 480[M+H]+
[0385]
Синтез соединения THK-5152
К суспензии в хлороформе (20 мл) соединения 173 (390 мг, 0,81 ммоль) добавляли 3,4-дигидро-2H-пиран (1,47 мл, 16,3 ммоль) и моногидрат паратолуолсульфоновой кислоты (322 мг, 1,87 ммоль) и смесь перемешивали при комнатной температуре в течение 20 минут. После доведения pH реакционного раствора до 8 добавлением триэтиламина растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=2/1, этилацетат) и затем перекристаллизовывали из этилацетата/н-гексана с получением соединения THK-5152 (442 мг, 97%) в виде бесцветных кристаллов.
точка плавления 129-130°C
1H ЯМR (400 МГц, ДМСО-d6) δ 1,33-1,71 (6H, м), 2,32 (3H, с), 2,85 (3H, д, J=4,8 Гц), 3,36-3,51 (1Н, м), 3,65-3,73, 3,81-3,89 (1Н, м), 4,09-4,38 (5H, м), 4,70, 4,86 (1Н, м), 6,58 (1H, д, J=8,8 Гц), 6,89 (1H, кв., J=4,8 Гц), 7,23-7,27 (1Н, м), 7,29-7,32 (1Н, м), 7,39-7,43 (2H, м), 7,79 (2H, дд, J=8,3, 2,0 Гц), 7,87 (1H, д, J=9,1 Гц), 7,99 (1H, д, J=8,8 Гц), 8,21 (1H, д, J=8,8 Гц), 8,26 (1H, дд, J=8,8, 2,4 Гц), 8,87 (1H, д, J=2,4 Гц)
ИК (Nujol) 3355, 1623, 1604 см-1
APCI-MS m/z 564[M+H]+
[0386]
Способ синтеза соединения THK-5129
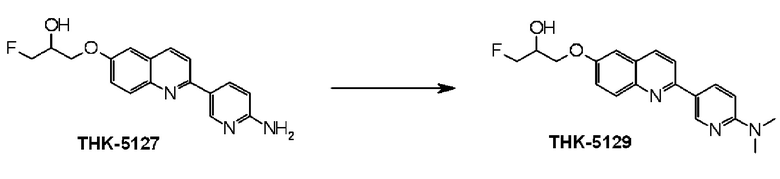
Синтез соединения THK-5129
К раствору в метаноле (8,6 мл)-уксусной кислоте (0,86 мл) соединения THK-5127 (123 мг, 0,39 ммоль) и водному 20% раствору формальдегида (1,18 мл, 7,8 ммоль) небольшими порциями добавляли пиколин-борановый комплекс (252 мг, 2,36 ммоль) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 3 часов. К реакционному раствору добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на NH силикагеле (элюирующий растворитель: н-гексан/этилацетат=1/2), очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=1/2) и затем перекристаллизовывали из этилацетата/н-гексана с получением соединения THK-5129 (100 мг, 75%) в виде бледно-желтых кристаллов.
точка плавления 172-173°C
1Н ЯМR (400 МГц, ДМСО-d6) δ 3,12 (6H, с), 4,06-4,19 (3H, м), 4,44-4,65 (2H, м), 5,54 (1H, д, J=4,8 Гц), 6,78 (1H, д, J=9,1 Гц), 7,37-7,41 (2H, м), 7,88-7,93 (1Н, м), 8,01 (1H, д, J=8,8 Гц), 8,25 (1H, д, J=8,8 Гц), 8,36 (1H, дд, J=9,1, 2,4 Гц), 8,95 (1Н, д, J=2,4 Гц)
ИК (Nujol) 3378, 1620, 1611 см-1
APCI-MS m/z 342[M+H]+
[0387]
Способ синтеза соединения THK-5135
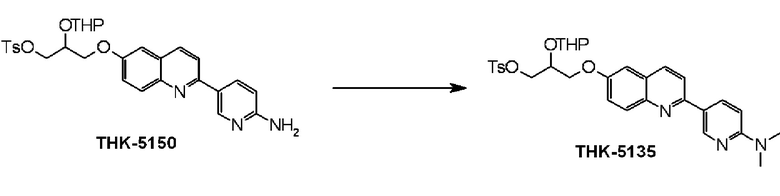
Синтез соединения THK-5135
К раствору в метаноле (20 мл)-уксусной кислоте (2 мл) соединения THK-5150 (500 мг, 0,91 ммоль) и водному 20% раствору формальдегида (2,73 мл, 18,2 ммоль) небольшими порциями добавляли пиколин-борановый комплекс (584 мг, 5,46 ммоль) при комнатной температуре в условиях перемешивания, и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли воду и этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=1/1) и затем перекристаллизовывали из этилацетата/н-гексана с получением соединения THK-5135 (331 мг, 63%) в виде бледно-желтых кристаллов.
точка плавления 135-138°C
1H ЯМR (400 МГц, ДМСО-d6) δ 1,34-1,70 (6H, м), 2,34 (3H, с), 3,12 (6H, с), 3,38-3,51 (1Н, м), 3,65-3,73, 3,81-3,89 (1Н, м), 4,08-4,38 (5H, м), 4,69-4,73, 4,84-4,88 (1Н, м), 6,79 (1Н, д, J=9,1 Гц), 7,24-7,28 (1Н, м), 7,30-7,33 (1Н, м), 7,39-7,43 (2H, м), 7,79 (2H, дд, J=8,5, 2,1 Гц), 7,88 (1H, д, J=9,1 Гц), 8,02 (1H, д, J=8,8 Гц), 8,23 (1H, д, J=8,8 Гц), 8,37 (1H, дд, J=9,1, 2,4 Гц), 8,96 (1H, д, J=2,4 Гц)
ИК (Nujol) 1624 см-1
APCI-MS m/z 578[M+H]+
[0388]
Способ синтеза соединения THK-5130
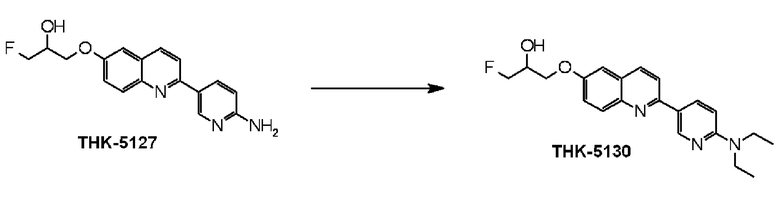
Синтез соединения THK-5130
К суспензии в метаноле (16 мл)-уксусной кислоте (1,6 мл) соединения THK-5127 (449 мг, 1,43 ммоль) и ацетальдегида (78 мг/мл раствора в метаноле: 3,24 мл, 5,73 ммоль) небольшими порциями добавляли пиколин-борановый комплекс (460 мг, 4,30 ммоль) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 4 часов. Добавляли ацетальдегид (78 мг/мл раствора в метаноле: 3,24 мл, 5,73 ммоль) и пиколин-борановый комплекс (460 мг, 4,30 ммоль) с последующим перемешиванием при комнатной температуре в течение 16 часов. К реакционному раствору добавляли воду и этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=2/1, 1/1) и затем перекристаллизовывали из этилацетата/н-гексана с получением соединения THK-5130 (479 мг, 91%) в виде бледно-желтых кристаллов.
точка плавления 131-132°C
1H ЯМR (400 МГц, ДМСО-d6) δ 1,15 (6H, т, J=7,1 Гц), 3,57 (4H, кв., J=7,1 Гц), 4,00-4,19 (3H, м), 4,44-4,65 (2H, м), 5,54 (1H, д, J=4,8 Гц), 6,74 (1H, д, J=9,1 Гц), 7,36-7,41 (2H, м), 7,87-7,92 (1Н, м), 8,00 (1H, д, J=8,8 Гц), 8,24 (1H, д, J=8,8 Гц), 8,33 (1Н, дд, J=9,1, 2,4 Гц), 8,93 (1H, д, J=2,4 Гц)
ИК (Nujol) 3275, 1622 см-1
APCI-MS m/z 370[M+H]+
[0389]
Способ синтеза соединения THK-5138
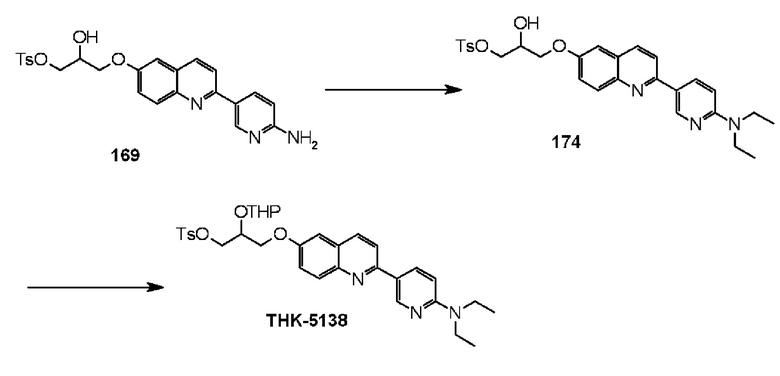
[0390]
Синтез соединения 174
К раствору в метаноле (13 мл)-уксусной кислоте (1,3 мл)-хлороформе (10 мл) соединения 169 (550 мг, 1,18 ммоль) и ацетальдегиде (0,66 мл, 11,8 ммоль) небольшими порциями добавляли пиколин-борановый комплекс (890 мг, 8,32 ммоль) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 4 дней. К реакционному раствору добавляли воду и этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 8 с использованием водного раствора карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=1/1, этилацетат) с получением соединения 174 (560 мг, 91%) в виде бледно-желтого пеноподобного вещества.
APCI-MS m/z 522[M+H]+
[0391]
Синтез соединения THK-5138
К раствору в хлороформе (20 мл) соединения 174 (550 мг, 1,05 ммоль) добавляли 3,4-дигидро-2H-пиран (1,19 мл, 21,1 ммоль), моногидрат паратолуолсульфоновой кислоты (420 мг, 2,43 ммоль), и смесь перемешивали при комнатной температуре в течение 10 минут. После доведения pH реакционного раствора до 8 добавлением триэтиламина растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4, 1/2) и затем промывали этилацетатом/н-гексаном с получением соединения THK-5138 (477 мг, 75%) в виде бледно-желтых кристаллов.
точка плавления 102-104°C
1H ЯМR (400 МГц, ДМСО-d6) δ 1,15 (6H, т, J=7,0 Гц), 1,30-1,50 (4H, м), 1,52-1,69 (2H, м), 2,34 (3H, с), 3,36-3,46 (1Н, м), 3,57 (4H, кв., J=6,7 Гц), 3,65-3,73, 3,81-3,88 (1Н, м), 4,08-4,38 (5H, м), 4,69-4,72, 4,84-4,88 (1Н, м), 6,73 (1Н, д, J=9,1 Гц), 7,23-7,27 (1Н, м), 7,29-7,32 (1Н, м), 7,41 (2H, д, J=7,3 Гц), 7,79 (2H, дд, J=8,2, 2,1 Гц), 7,87 (1H, д, J=9,1 Гц), 8,00 (1H, д, J=8,8 Гц), 8,22 (1H, д, J=8,8 Гц), 8,34 (1Н, дд, J=9,1, 2,4 Гц), 8,94 (1Н, д, J=2,4 Гц)
ИК (Nujol) 1620 см-1
APCI-MS m/z 606[M+H]+
[0392]
Способ синтеза соединения THK-5151
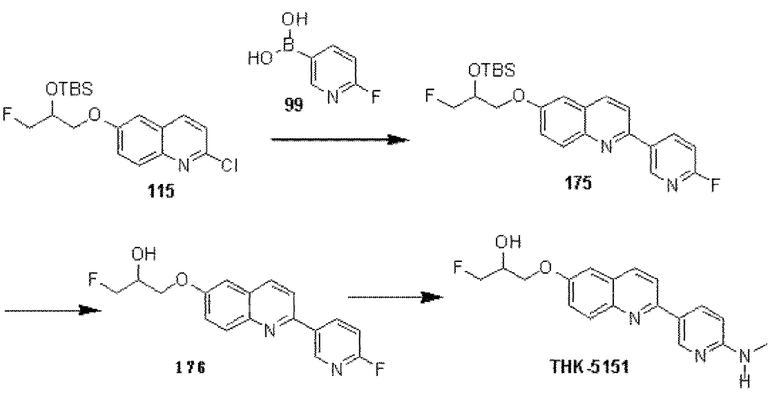
[0393]
Синтез соединения 175
К соединению 115 (175 мг, 0,473 ммоль), соединению 99 (71,1 мг, 0,505 ммоль), карбонату натрия (105 мг, 0,992 ммоль) и дихлорбис(трифенилфосфин)палладию (3,6 мг, 0,00513 ммоль) добавляли 2,5 мл смешанного раствора воды/этанола/1,2-диметоксиэтана (каждые в соотношении=0,55:0,4:1,25), и смесь нагревали кипячением в сосуде с обратным холодильником при 90°C в течение 90 минут. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду(40 мл), и раствор экстрагировали этилацетатом (2×40 мл) и затем сушили над сульфатом магния. Экстракционный растворитель отгоняли, и остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=85/15, 60/40) с получением соединения 175 (191 мг, 94%) в виде кристаллического вещества.
EI-MS m/z 430[M]+
[0394]
Синтез соединения 176
Соединение 175 (106 мг, 0,247 ммоль) растворяли в тетрагидрофуране (3,0 мл) и добавляли 1M раствор тетрабутиламмония фторида/ацетонитрила (0,37 мл, 0,37 ммоль) и смесь взаимодействовала при комнатной температуре в течение 90-минутной реакции. К реакционному раствору добавляли, вода (30 мл), и раствор экстрагировали этилацетатом (3× 30 мл) и затем сушили над сульфатом магния. Экстракционный растворитель отгоняли, и остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=45/55, 20/80) с получением соединения 176 (74,1 мг, 95%) в виде белых кристаллов.
EI-MS m/z 316[M]+.
[0395]
Синтез соединения THK-5151
Соединение 176 (52,4 мг, 0,166 ммоль) растворяли в метаноле (2,0 мл) и добавляли 40% раствор монометиламина/метанола (0,60 мл, 5,8 ммоль), и смесь нагревали при 40°C в течение 7 часов с последующим перемешиванием с нагреванием при 50°С в течение 17 часов. После охлаждения реакционного раствора до комнатной температуры растворитель отгоняли, и остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/метанол=100/0, 70/30) с получением соединения THK-5151 (46,0 мг, 85%) в виде желтых кристаллов. EI-MS m/z 327[M]+.
1Н ЯМR (600 МГц, CDCl3) δ 3,01 (3H, д, J=4,8 Гц), 4,20-4,25 (2H, м), 4,32-4,38 (1Н, м), 4,59-4,65 (1Н, м), 4,67-4,73 (1Н, м), 4,85 (1Н, уш.), 6,54 (1H, д, J=8,4 Гц), 7,11 (1H, д, J=3,0 Гц), 7,37 (1H, дд, J=3,0, 9,6 Гц), 7,76 (1H, д, J=8,4 Гц), 8,02 (1Н, д, J=9,6 Гц), 8,06 (1Н, д, J=8,4 Гц), 8,36 (1H, дд, J=1,8, 9,0 Гц), 8,82 (1Н, д, J=l, 8 Гц).
[0396]
Способ синтеза соединения THK-5177
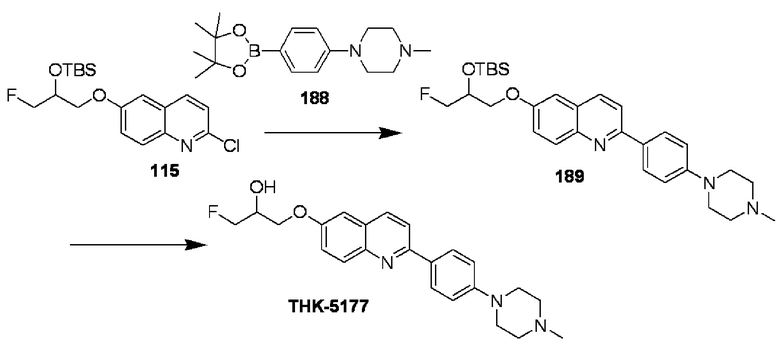
[0397]
Синтез соединения 189
Из соединения 115 (112 мг, 0,302 ммоль) и соединения 188 (91,0 мг, 0,301 ммоль) синтезировали соединение 189 (желтый кристалл, 94,6 мг, 62%) таким же образом, как соединение 175. EI-MS m/z 509[M]+.
[0398]
Синтез соединения THK-5177
Соединение 189 (75,0 мг, 0,147 ммоль) растворяли в тетрагидрофуране (3,0 мл) и добавляли 1M раствор тетрабутиламмония фторида/ацетонитрила (0,37 мл, 0,37 ммоль), и смесь взаимодействовала при комнатной температуре в течение 90-минутной реакции. К реакционному раствору добавляли воду (30 мл), и раствор экстрагировали этилацетатом (3×30 мл) и затем сушили над сульфатом магния. Экстракционный растворитель отгоняли, и остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=45/55, 20/80) для синтеза соединения THK-5177 (желтые кристаллы, 40,8 мг, 70%). EI-MS m/z 395[M]+.
1H ЯМR (600 МГц, CDCl3) δ 2,38 (3H, с), 2,54 (1Н, уш.), 2,61 (4H, т, J=4,8 Гц), 3,33 (4H, т, J=4,8 Гц), 4,20-4,24 (2H, м), 4,31-4,38 (1Н, м), 4,59-4,65 (1Н, м), 4,67-4,73 (1Н, м), 7,04 (1H, д, J=9,0 Гц), 7,10 (1H, д, J=3,0 Гц), 7,36 (1Н, дд, J=3,0, 9,0 Гц), 7,81 (1H, д, J=8,4 Гц), 8,04 (1H, д, J=9,6 Гц), 8,05 (1H, д, J=9,0 Гц), 8,08 (2Н, д, J=9,0 Гц)
[0399]
Способ синтеза соединения THK-5178
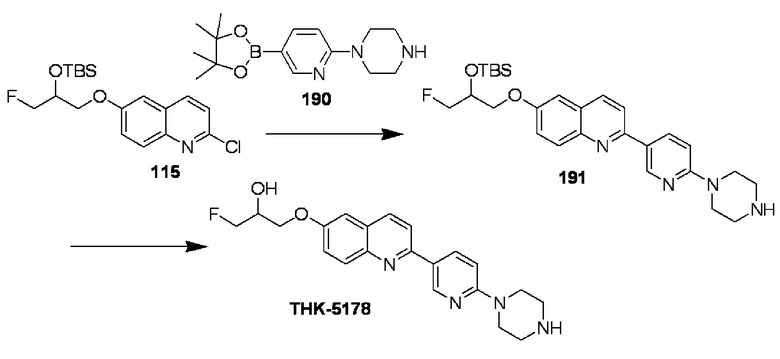
[0400]
Синтез соединения 191
Из соединения 115 (134 мг, 0,361 ммоль) и соединения 190 (146 мг, 0,505 ммоль) синтезировали соединение 191 (желтый кристалл, 144 мг, 80%) таким же образом, как соединение 175. EI-MS m/z 496[M]+.
[0401]
Синтез соединения THK-5178
Из соединения 191 (30,4 мг, 0,065 ммоль) синтезировали соединение THK-5178 (молочно-белый кристалл, 13,3 мг, 54%) таким же образом, как соединение THK-5177. EI-MS m/z 382[M]+.
1H ЯМR (600 МГц, CDCl3) δ 2,19 (1Н, с), 2,54 (1Н, уш.), 3,02 (4H, т, J=5,4 Гц), 3,63 (4H, т, J=5,4 Гц), 4,20-4,24 (2H, м), 4,32-4,38 (1Н, м), 4,59-4,65 (1Н, м), 4,67-4,73 (1Н, м), 6,78 (1H, д, J=9,0 Гц), 7,11 (1H, д, J=2,4 Гц), 7,37 (1H, дд, J=2,4, 9,0 Гц), 7,78 (1H, д, J=9,0 Гц), 8,02 (1H, д, J=9,6 Гц), 8,06 (1H, д, J=8,4 Гц), 8,38 (1H, дд, J=2,4, 9,0 Гц), 8,90 (1H, д, J=2,4 Гц).
[0402]
Способ синтеза соединения THK-5180
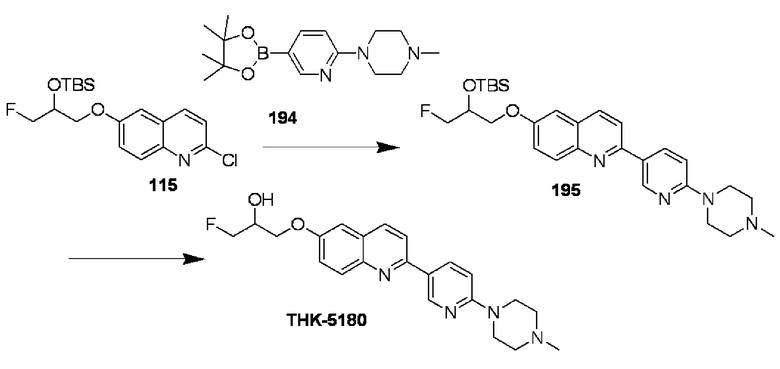
[0403]
Синтез соединения 195
Из соединения 115 (180 мг, 0,486 ммоль) и соединения 194 (157 мг, 0,517 ммоль) синтезировали соединение 195 (белый кристалл, 119 мг, 48%) таким же образом, как соединение 175. EI-MS m/z 510[M]+.
[0404]
Синтез соединения THK-5180
Из соединения 195 (86,0 мг, 0,168 ммоль) синтезировали соединение THK-5180 (белые кристаллы, 48,6 мг, 73%) таким же образом, как соединение THK-5177. EI-MS m/z 396[M]+.
1H ЯМR (600 МГц, CDCl3) δ 2,37 (3H, с), 2,46 (1H, д, J=5,4 Гц), 2,55 (4H, т, J=5,4 Гц), 3,69 (4H, т, J=5,4 Гц), 4,20-4,25 (2H, м), 4,31-4,38 (1Н, м), 4,59-4,65 (1Н, м), 4,67-4,73 (1Н, м), 6,79 (1H, д, J=9,0 Гц), 7,11 (1H, д, J=2,4 Гц), 7,37 (1H, дд, J=3,0, 9,6 Гц), 7,76 (1H, д, J=9,0 Гц), 8,02 (1H, д, J=9,0 Гц), 8,06 (1H, д, J=8,4 Гц), 8,38 (1Н, дд, J=2,4, 9,0 Гц), 8,90 (1H, д, J=2,4 Гц).
[0405]
Способ синтеза соединения THK-5142
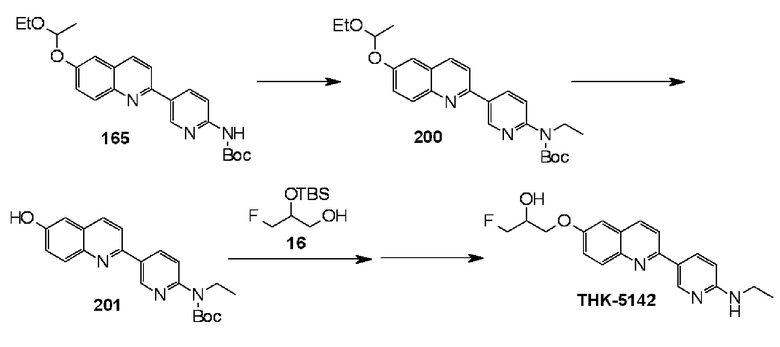
[0406]
Соединение 200
К суспензии в N,N-диметилформамиде (30 мл) соединения 165 (2,09 г, 5,10 ммоль) небольшими порциями добавляли 60% гидрид натрия (245 мг, 6,12 ммоль) в условиях охлаждения льдом и перемешивания и в атмосфере аргона. После перемешивания при той же температуре в течение 10 минут добавляли по каплям этилйодид (0,616 мл, 7,66 ммоль), и смесь перемешивали при той же температуре в течение 1 часа с последующим перемешиванием при комнатной температуре в течение 30 минут. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением соединения 200 (2,07 г, 92%) в виде бесцветного маслянистого вещества.
APCI-MS m/z 438[M+H]+
[0407]
Соединение 201
К раствору в хлороформе (30 мл) соединения 200 (2,06 г, 4,71 ммоль) добавляли трифторуксусную кислоту (5 мл) по каплям при охлаждении льдом и перемешивании, и смесь перемешивали при той же температуре в течение 20 минут. К реакционному раствору добавляли, воду, и раствор экстрагировали этилацетатом после доведения pH до 7 с использованием водного раствора карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и остаток очищали хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=2/1) и затем перекристаллизовывали из н-гексана/этилацетата с получением соединения 201 (1,26 г, 73%) в виде бледно-желтых кристаллов.
точка плавления 181-182°C
APCI-MS m/z 366[M+H]+
[0408]
Синтез соединения THK-5142
К раствору в тетрагидрофуране (20 мл) соединения 201 (750 мг, 2,05 ммоль), соединения 16 (1,03 г, 4,93 ммоль) и трифенилфосфина (1,29 г, 4,93 ммоль) добавляли по каплям раствор в тетрагидрофуране (10 мл) диизопропилазодикарбоксилата (0,98 мл, 4,93 ммоль) при охлаждении льдом и перемешивании, и смесь перемешивали при той же температуре в течение 1 часа с последующим перемешиванием при комнатной температуре в течение 16 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/20, 1/9) с получением бесцветного маслянистого вещества (1,69 г), содержащего целевой продукт. К раствору в хлороформе (18 мл) настоящего продукта (1,69 г) добавляли по каплям трифторуксусную кислоту (12 мл) при охлаждении льдом и перемешивании, и добавляли воду (3 мл) с последующим перемешиванием при комнатной температуре в течение 16 часов. К реакционному раствору добавляли ледяную воду и затем этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 9 с использованием водного раствора карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1, 2/1) и затем перекристаллизовывали из этилацетата с получением соединения THK-5142 (465 мг, 66%) в виде бесцветных кристаллов.
точка плавления 154-155°C
1H ЯМR (400 МГц, ДМСО-d6) δ 1,17 (3H, т, J=7,3 Гц), 3,29-3,38 (2H, м), 4,06-4,20 (3H, м), 4,44-4,65 (2H, м), 5,53 (1H, д, J=5,7 Гц), 6,58 (1Н, д, J=8,8 Гц), 6,90 (1Н, т, J=5,4 Гц), 7,39 (1H, д, J=2,7 Гц), 7,39 (1H, дд, J=8,8, 2,7 Гц), 7,90 (1H, д, J=8,8 Гц), 7,97 (1H, д, J=8,5 Гц), 8,23 (1H, д, J=8,5 Гц), 8,25 (1H, дд, J=8,8, 2,7 Гц), 8,85 (1Н, уш.)
ИК (Nujol) 3333, 1625 см-1
APCI-MS m/z 342[M+H]+
[0409]
Способ синтеза соединения THK-5143
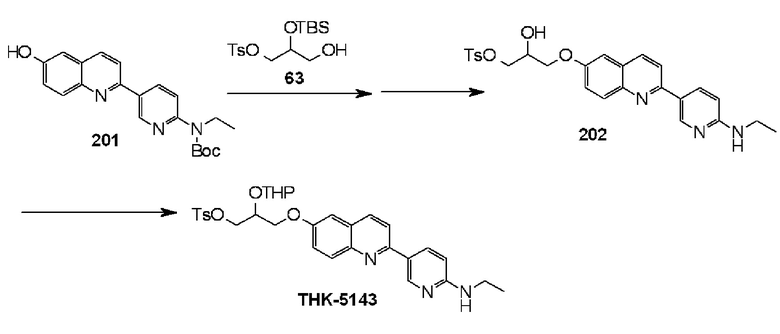
[0410]
Соединение 202
К раствору в тетрагидрофуране (20 мл) соединения 201 (500 мг, 1,37 ммоль), соединения 63 (1,18 г, 3,28 ммоль) и трифенилфосфина (860 мг, 3,28 ммоль) добавляли по каплям раствор в тетрагидрофуране (10 мл) диизопропилазодикарбоксилата (0,65 мл, 3,28 ммоль) при охлаждении льдом и перемешивании, с последующим перемешиванием при той же температуре в течение 1 часа и дополнительном перемешивании при комнатной температуре в течение 2 дней. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением бесцветного твердого вещества (1,51 г), содержащего целевой продукт. К раствору в хлороформе (18 мл) настоящего продукта (1,51 г) добавляли по каплям трифторуксусную кислоту (12 мл) при охлаждении льдом и перемешивании и добавляли воду (3 мл), с последующим перемешиванием при комнатной температуре в течение 3 дней. К реакционному раствору добавляли ледяную воду и этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 8 с использованием водного раствора карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=2/1, этилацетат) и затем промывали этилацетатом/н-гексаном=1 с получением соединения 202 (520 мг, 77%) в виде бесцветных кристаллов.
точка плавления 158-159°C
APCI-MS m/z 494[M+H]+
[0411]
Синтез соединения THK-5143
К суспензии в хлороформе (20 мл) соединения 202 (510 мг, 1,03 ммоль) добавляли 3,4-дигидро-2H-пиран (1,87 мл, 20,7 ммоль) и моногидрат паратолуолсульфоновой кислоты (409 мг, 2,38 ммоль) и смесь перемешивали при комнатной температуре в течение 20 минут. После доведения pH реакционного раствора до 8 добавлением триэтиламина растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1, 2/1) и затем перекристаллизовывали из этилацетата/н-гексана с получением соединения THK-5143 (508 мг, 85%) в виде бесцветных кристаллов.
точка плавления 93-94°C
1H ЯМR (400 МГц, ДМСО-d6) δ 1,17 (3H, т, J=7,3 Гц), 1,33-1,72 (6H, м), 2,34 (3H, с), 3,30-3,37 (2H, м), 3,37-3,47 (1Н, м), 3,69-3,73, 3,81-3,88 (1Н, м), 4,09-4,37 (5H, м), 4,71, 4,84-4,88 (1Н, м), 6,58 (1H, д, J=8,8 Гц), 6,91 (1H, т, J=5,3 Гц), 7,23-7,27 (1Н, м), 7,29-7,31 (1Н, м), 7,39-7,43 (2H, м), 7,77-7,82 (2H, м), 7,98 (1H, д, J=8,8 Гц), 7,97 (1Н, д, J=8,5 Гц), 8,21 (1H, д, J=8,5 Гц), 8,25 (1H, дд, J=9,1, 2,4 Гц), 8,86 (1Н, уш.)
ИК (Nujol) 3347, 1622, 1602 см-1
APCI-MS m/z 578[M+H]+
[0412]
Способ синтеза соединения THK-5136
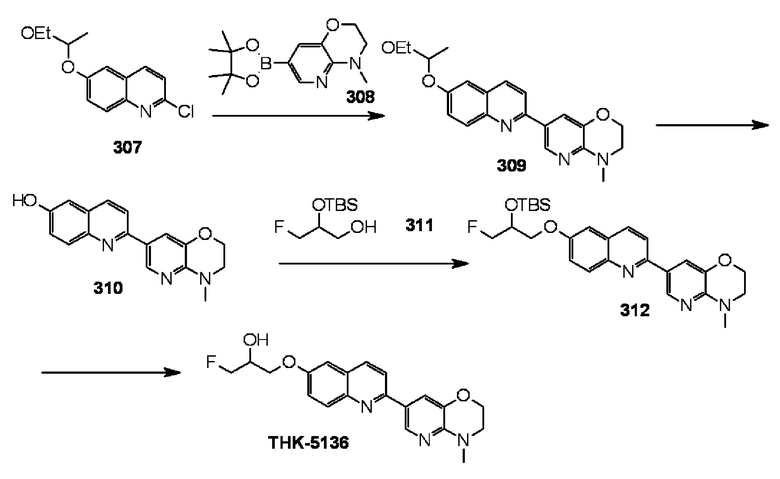
Синтез соединения 309
К раствору в 1,2-диметоксиэтане (30 мл) соединения 307 (900 мг, 3,58 ммоль) и соединения 308 (990 мг, 3,58 ммоль) добавляли карбонат калия (1,48 г, 10,7 ммоль), воду (0,62 мл) и тетракистрифенилфосфин палладия (410 мг, 0,36 ммоль) в атмосфере аргона и смесь перемешивали при 80°C в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2) с получением соединения 309 (1,31 г, 100%) в виде бледно-желтого твердого вещества.
точка плавления 112-113°C
APCI-MS m/z 366[M+H]+
[0413]
Синтез соединения 310
К раствору в хлороформе (20 мл) соединения 309 (1,31 г, 3,58 ммоль) добавляли по каплям трифторуксусную кислоту (2 мл) при охлаждении льдом и перемешивании и смесь перемешивали при той же температуре в течение 30 минут. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом после доведения pH до 8 с использованием водного раствора карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток промывали н-гексаном/этилацетатом=4/1 с получением соединения 310 (950 мг, 90%) в виде бледно-желтых кристаллов.
точка плавления 273-274°C
APCI-MS m/z 294[M+H]+
[0414]
Синтез соединения 312
К раствору в тетрагидрофуране (20 мл) соединения 310 (500 мг, 1,71 ммоль), соединения 311 (852 мг, 4,09 ммоль) и трифенилфосфина (1,07 г, 4,09 ммоль) добавляли по каплям раствор диизопропилазодикарбоксилата (0,81 мл, 4,09 ммоль) в тетрагидрофуране (10 мл) в течение 15 минут в условиях охлаждения льдом и перемешивания, и смесь перемешивали при той же температуре в течение 1 часа, с последующим перемешиванием при комнатной температуре в течение 4 дней. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4, 1/2) с получением соединения 312 (710 мг, 86%) в виде бледно-желтого твердого вещества.
точка плавления 139-140°C
APCI-MS m/z 484[M+H]+
[0415]
Синтез соединения THK-5136
К раствору в тетрагидрофуране (20 мл) соединения 312 (700 мг, 1,45 ммоль) добавляли по каплям 1M тетра-н-бутиламмонийфторид/тетрагидрофуран (1,45 мл, 1,45 ммоль) при комнатной температуре при перемешивании, и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 2/1) и затем перекристаллизовывали из этилацетата с получением соединения THK-5136 (466 мг, 87%) в виде бледно-желтых кристаллов.
точка плавления 169-170°C
lH ЯМR (400 МГц, ДМСО-d6) δ 3,12 (3H, с), 3,52 (2H, т, J=4,5 Гц), 4,06-4,19 (3H, м), 4,27 (2H, т, J=4,5 Гц), 4,44-4,65 (2H, м), 5,53 (1H, д, J=4,2 Гц), 7,37-7,41 (2H, м), 7,78 (1H, д, J=1,8 Гц), 7,88-7,92 (1Н, м), 8,00 (1H, д, J=8,5 Гц), 8,24 (1Н, д, J=8,8 Гц), 8,56 (1H, д, J=2,1 Гц)
ИК (Nujol) 1620 см-1
APCI-MS m/z 370[M+H]+
[0416]
Способ синтеза соединения THK-5153
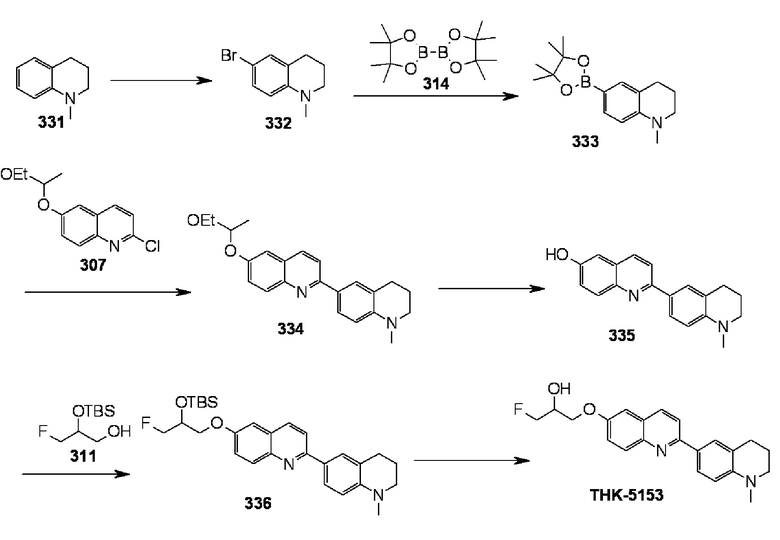
[0417]
Синтез соединения 332
К раствору в тетрагидрофуране (70 мл) соединения 331 (5,25 г, 35,66 ммоль) добавляли N-бромсукцинимид (6,35 г, 35,66 ммоль) при -78°C в атмосфере аргона и смесь перемешивали при той же температуре в течение 3 часов, с последующим перемешиванием при комнатной температуре в течение 16 часов. К реакционному раствору добавляли воду и этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 9, используя водный раствор карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/50) с получением соединения 332 (7,38 г, 91%) в виде бесцветного маслянистого вещества.
APCI-MS m/z 226/228[M+H]+
[0418]
Синтез соединения 333
К раствору в 1,4-диоксане (200 мл) соединения 332 (7,37 г, 32,6 ммоль) и соединения 314 (9,93 г, 39 ммоль) добавляли ацетат калия (9,6 г, 97,8 ммоль) и Pd(dppf)Cl2∙CH2Cl2 (1,19 г, 1,46 ммоль) в атмосфере аргона и смесь нагревали кипячением в сосуде с обратным холодильником при 100°C в течение 8 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, и нерастворимые вещества отфильтровывали целлитом, и растворитель фильтрата отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/50) с получением соединения 333 (4,71 г, 53%) в виде бледно-желтого твердого вещества.
точка плавления 129-130°C
APCI-MS m/z 274[M+H]+
[0419]
Синтез соединения 334
К раствору в 1,2-диметоксиэтане (107 мл) соединения 333 (4,46 г, 16,3 ммоль) и соединения 307 (3,28 г, 13,0 ммоль) добавляли карбонат калия (5,40 г, 39,0 ммоль), воду (2,27 мл) и тетракистрифенилфосфин палладия (1,51 г, 1,3 ммоль) в атмосфере аргона, и смесь перемешивали при 80°C в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли сульфат натрия с последующей сушкой и дополнительной фильтрацией целитом. Растворитель фильтрата отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9) с получением соединения 334 (4,52 г, 95%) в виде желтого маслянистого вещества.
APCI-MS m/z 363[M+H]+
[0420]
Синтез соединения 335
К раствору в хлороформе (24 мл) соединения 334 (4,51 г, 12,4 ммоль) добавляли по каплям трифторуксусную кислоту (16 мл) при охлаждении льдом и перемешивании и смесь перемешивали при той же температуре в течение 20 минут. К реакционному раствору добавляли воду и этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 8 с использованием водного раствора карбоната калия. Экстракционную жидкость промывали водой и сушили, и растворитель отгоняли при пониженном давлении, и затем остаток промывали этилацетатом/н-гексаном=1/1 с получением соединения 335 (3,17 г, 88%) в виде оранжевых кристаллов.
точка плавления 323-325°C
APCI-MS m/z 291[M+H]+
[0421]
Синтез соединения 336
К смеси соединения 335 (500 мг, 1,72 ммоль), соединения 311 (861 мг, 4,13 ммоль), трифенилфосфина (1,08 г, 4,13 ммоль) и тетрагидрофурана (20 мл) добавляли по каплям раствор в тетрагидрофуране (10 мл) диизопропилазодикарбоксилата (0,82 мл, 4,13 ммоль) в течение 10 минут в условиях охлаждения льдом и перемешивания, и смесь перемешивали при той же температуре в течение 1 часа, с последующим перемешиванием при комнатной температуре в течение 4 дней. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9) с получением соединения 336 (820 мг, 99%) в виде бледно-желтого твердого вещества.
точка плавления 84-85°C
APCI-MS m/z 481[M+H]+
[0422]
Синтез соединения THK-5153
К раствору в тетрагидрофуране (10 мл) соединения 336 (810 мг, 1,69 ммоль) добавляли по каплям 1M тетра-н-бутиламмонийфторид/тетрагидрофуран (1,69 мл, 1,69 ммоль) при комнатной температуре при перемешивании, и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2) и затем перекристаллизовывали из этилацетата с получением соединения THK-5153 (353 мг, 57%) в виде бледно-желтых кристаллов.
точка плавления 137-138°C
1H ЯМR (500 МГц, ДМСО-d6) δ 1,89-1,97 (2H, м), 2,81 (2H, т, J=6,4 Гц), 2,94 (3H, с), 3,30 (2H, т, J=5,8 Гц), 4,06-4,18 (3H, м), 4,46-4,63 (2H, м), 5,55 (1Н, с), 6,68 (1H, д, J=9,0 Гц), 7,38-7,43 (2H, м), 7,84 (1H, д, J=1,9 Гц), 7,91 (1H, дд, J=8,7, 2,2 Гц), 7,92-7,95 (1Н, м), 7,99 (1H, д, J=8,7 Гц), 8,26 (1H, д, J=8,7 Гц)
ИК (Nujol) 1598 см-1
APCI-MS m/z 367[M+H]+
[0423]
Способ синтеза соединения THK-5157
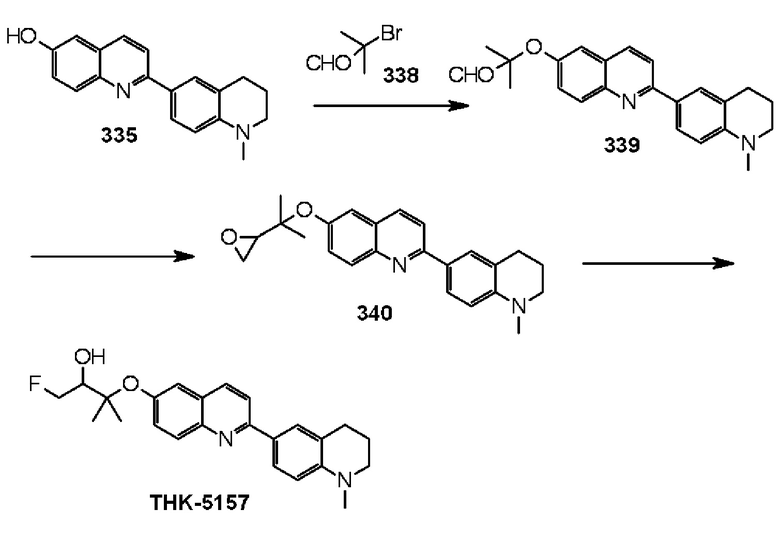
Синтез соединения 339
К суспензии в тетрагидрофуране (100 мл) соединения 335 (2,00 г, 6,89 ммоль) добавляли по каплям 1M трет-бутокси калий/тетрагидрофуран (7,58 мл, 7,58 ммоль) и растворяли при комнатной температуре при перемешивании и в атмосфере аргона. Растворитель реакционного раствора отгоняли при пониженном давлении, и к остатку добавляли 18-crown-6 (1,4,7,10,13,16-гексаоксициклооктадекан) (1,82 г, 6,89 ммоль) и ацетонитрил (40 мл). После охлаждения до -78°C добавляли по каплям раствор в ацетонитриле (10 мл) соединения 338 (1,87 г, 12,4 ммоль), смесь перемешивали в атмосфере аргона при комнатной температуре в течение 16 часов. К реакционному раствору добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4, этилацетат) и затем промывали этилацетатом/н-гексаном=1/100 с получением соединения 339 (1,81 г, 73%) в виде бледно-желтых кристаллов.
точка плавления 135-136°C
APCI-MS m/z 361[M+H]+
[0424]
Синтез соединения 340
К смеси соединения триметилсульфоксония йодида (1,65 г, 7,50 ммоль) и ДМСО (20 мл) добавляли 60% гидрид натрия (300 мг, 7,50 ммоль) в условиях охлаждения льдом и перемешивания, и смесь перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор охлаждали льдом, и добавляли по каплям раствор в тетрагидрофуране (10 мл) соединения 339 (1,80 г, 4,99 ммоль), с последующим перемешиванием при комнатной температуре в течение 1 часа. К реакционному раствору добавляли ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении, и остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением соединения 340 (1,74 г, 93%) в виде бледно-желтого маслянистого вещества.
APCI-MS m/z 375[M+H]+
[0425]
Синтез соединения THK-5157
Смесь соединения 340 (1,73 г, 4,62 ммоль), н-Bu4NH2F3 (232 мг, 0,46 ммоль) и KHF2 (722 мг, 9,24 ммоль) перемешивали при 120°C в течение 2 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, добавляли воду и этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 8 с использованием водного раствора карбоната калия. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4, 1/3) и затем перекристаллизовывали из этилацетата с получением соединения THK-5157 (474 мг, 26%) в виде бледно-желтых кристаллов.
точка плавления 163-164°C
1H ЯМR (400 МГц, ДМСО-d6) δ 1,21 (3H, с), 1,22 (3H, с), 1,90-1,96 (2H, м), 2,81 (2H, т, J=6,3 Гц), 2,93 (3H, с), 3,30 (2H, т, J=5,8 Гц), 4,54-4,61 (1Н, м), 4,67 (1Н, ддд, J=48, 10, 7,1 Гц), 4,93 (1H, ддд, J=48, 10, 2,9 Гц), 4,95 (1H, д, J=6,7 Гц), 6,68 (1Н, д, J=8,7 Гц), 7,44-7,49 (2H, м), 7,85 (1H, д, J=2,2 Гц), 7,89-7,94 (2H, м), 7,97 (1Н, д, J=8,7 Гц), 8,22 (1H, д, J=8,3 Гц)
ИК (Nujol) 3267, 1620, 1599 см-1
APCI-MS m/z 395[M+H]+
[0426]
Способ синтеза соединения THK-5128
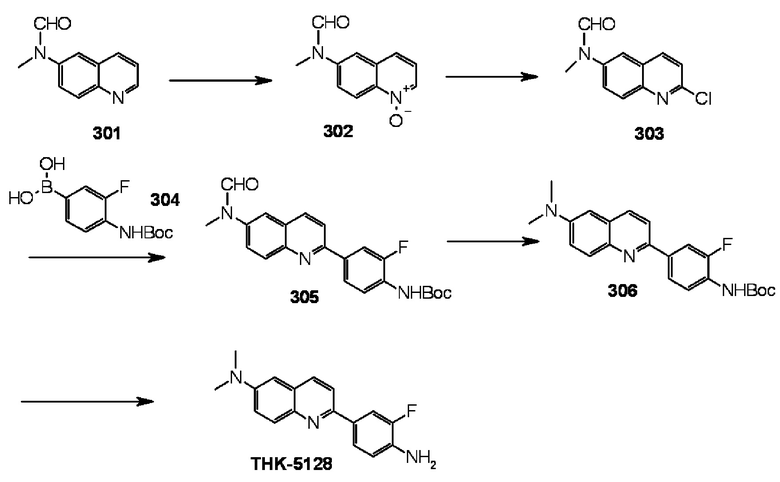
[0427]
Синтез соединения 302
Смесь соединения 301 (3,65 г, 19,6 ммоль), 75% метахлорбензойной кислоты (5,20 г, 22,6 ммоль) и хлороформа (40 мл) перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор промывали по очереди водным раствором тиосульфата натрия, водным насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: хлороформ/метанол=30/1, 10/1) и промывали диизопропиловым эфиром с получением соединения 302 (3,30 г, 83%) в виде твердого вещества.
точка плавления 145-146°C
[0428]
Синтез соединения 303
Смесь соединения 302 (3,20 г, 16,3 ммоль), паратолуолсульфонилхлорида (3,3 г, 17,3 ммоль), карбоната калия (2,5 г, 18,1 ммоль) и хлороформа (60 мл) нагревали кипячением в сосуде с обратным холодильником в течение 3 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали хлороформом. Экстракционную жидкость промывали по очереди водным насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором, и после сушки растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=2/1) с получением соединения 303 (2,30 г, 64%) в виде бесцветного твердого вещества.
точка плавления 110-111°C
APCI-MS m/z 221[M+H]+
[0429]
Синтез соединения 305
Смесь соединения 303 (883 мг, 4,0 ммоль), соединения 304 (1,224 г, 4,8 ммоль), 1,2-диметоксиэтана (20 мл), карбоната натрия (848 мг, 8,0 ммоль), воды (2 мл) и тетракистрифенилфосфина палладия (231 мг, 0,2 ммоль) нагревали кипячением в сосуде с обратным холодильником в атмосфере аргона в течение 18 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали по очереди водой и насыщенным солевым раствором, и после сушки растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/толуола=1/10, 1/4) с получением соединения 305 (1,38 г, 87%) в виде бесцветного твердого вещества.
точка плавления 160-161°C
[0430]
Синтез соединения 306
К суспензии в тетрагидрофуране (30 мл) соединения NaBH4 (380 мг, 10 ммоль) добавляли по каплям раствор в тетрагидрофуране (5 мл) BF3·Et2O (1,86 г, 13,1 ммоль) при 0°C и смесь перемешивали при той же температуре в течение 30 минут. К настоящему реакционному раствору добавляли по каплям раствор в тетрагидрофуране (10 мл) соединения 305 (1,30 г, 3,28 ммоль) при температуре от 0 до 5°C, и раствор перемешивали при той же температуре в течение 20 минут, с последующим перемешиванием при комнатной температуре в течение 1 часа. Реакционный раствор охлаждали льдом и добавляли по каплям водный насыщенный раствор гидрокарбоната натрия, и раствор перемешивали и затем экстрагировали этилацетатом. Экстракционную жидкость промывали по очереди водой и насыщенным солевым раствором, и после сушки растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4, 1/3) и затем промывали н-гексаном―диизопропиловым эфиром с получением соединения 306 (980 мг, 78%) в виде оранжевого твердого вещества.
точка плавления 175-176°C
APCI-MS m/z 382[M+H]+
[0431]
Синтез соединения THK-5128
К раствору в метиленхлориде (15 мл) соединения 306 (900 мг, 2,36 ммоль) добавляли по каплям трифторуксусную кислоту (3 мл) при охлаждении льдом и перемешивании и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор охлаждали льдом и разбавляли этилацетатом, и раствор делали основным 28% аммиачной водой с последующим разделением жидкости. Органический слой промывали по очереди водой и насыщенным солевым раствором, и после сушки растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: толуола, этилацетат/толуол=1/3) и затем перекристаллизовывали из этилацетата с получением соединения THK-5128 (545 мг, 82%) в виде желтых кристаллов.
точка плавления 204-205°C (dec.)
1H ЯМR (400 МГц, CDCl3) δ 3,08 (6H, с), 3,86 (2H, уш. с), 6,80 (1H, д, J=2,7 Гц), 6,87 (1Н, дд, J=9,2, 8,8 Гц), 7,36 (1H, дд, J=9,4, 2,7 Гц), 7,66 (1H, д, J=8,8 Гц), 7,72 (1Н, дд, J=8,8, 1,8 Гц), 7,86 (1Н, дд, J=13, 1,8 Гц), 7,96 (1H, д, J=8,8 Гц), 7,97 (1Н, д, J=9,4 Гц).
ИК (Nujol) 3460, 3304, 3181, 1645, 1619, 1589 см-1
APCI-MS m/z 282[M+H]+
[0432]
Синтез соединения THK-5147
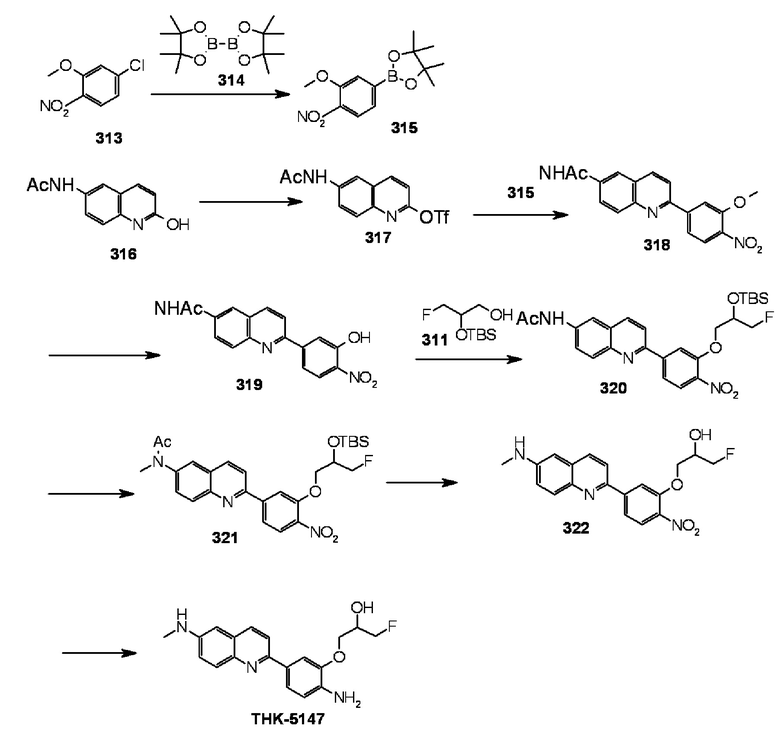
[0433]
Синтез соединения 315
Смесь соединения 313 (18,76 г, 0,1 моль), соединения 314 (27,93 г, 0,11 моль), ацетата калия (14,72 г, 0,15 моль), Pd2(dba)3 (2,75 г, 3 ммоль), трициклогексилфосфина (2,02 г, 7,2 ммоль) и 1,4-диоксана (200 мл) перемешивали в атмосфере аргона при 90°C в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и выливали в этилацетат-воду, и затем раствор экстрагировали этилацетатом. Экстракционную жидкость промывали насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан→н-гексан/этилацетат=19/1, 9/1, 6/1, 4/1) с получением соединения 315 (26,74 г, 96%) в виде бледно-желтого твердого вещества.
APCI-MS m/z 280[M+H]+
[0434]
Синтез соединения 317
К суспензии в пиридине (1 л) соединения 316 (29,51 г, 146 ммоль) добавляли по каплям трифторметансульфоновый ангидрид (48 мл, 292 ммоль) при температуре от -30 до -20°C и смесь перемешивали при 10°C в течение 1 часа. К реакционному раствору добавляли ледяную воду после отгонки летучего вещества при 40°C или ниже, раствор экстрагировали этилацетатом. Экстракционную жидкость промывали насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан→н-гексан/этилацетат=2/1, 1/1, 1/2, 1/5) и затем перекристаллизовывали из этилацетата-диизопропилового эфира с получением соединения 317 (30,86 г, 63%) в виде бледно-розовых кристаллов.
APCI-MS m/z 335[M+H]+
[0435]
Синтез соединения 318
Смесь соединения 317 (29,11 г, 87 ммоль), соединения 315 (26,74 г, 96 ммоль), водного 2M раствора карбоната натрия (100 мл), тетракистрифенилфосфина палладия (5 г, 4,3 ммоль) и 1,2-диметоксиэтана (300 мл) перемешивали в атмосфере аргона при 90°C в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, и 1,2-диметоксиэтан отгоняли при пониженном давлении и добавляли хлороформ/метанол (=1/1), и затем нерастворимые вещества удаляли фильтрацией. Фильтрат промывали насыщенным солевым раствором, и высушенный растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: хлороформ/тетрагидрофурана=4/1) и затем промывали хлороформом-диизопропиловым эфиром с получением соединения 318 (20,5 г, 76%) в виде желтого твердого вещества.
APCI-MS m/z 338[M+H]+
[0436]
Синтез соединения 319
Смесь соединения 318 (1,0 г, 29,6 ммоль), хлорида лития (1,26 г, 29,7 ммоль) и гексаметилфосфорного триамида (5 мл) перемешивали при 110°C в течение 2 дней. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду. Раствор подкисляли 10% водным раствором хлористоводородной кислоты и делали основным водным насыщенным раствором гидрокарбоната натрия, и затем экстрагировали этилацетатом-тетрагидрофураном. Экстракционную жидкость промывали насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: хлороформ/тетрагидрофурана=4/1, 3/2) и затем перекристаллизовывали из тетрагидрофурана-диизопропилового эфира с получением соединения 319 (745 мг, 78%) в виде желтого твердого вещества.
APCI-MS m/z 324[M+H]+
[0437]
Синтез соединения 320
К смеси соединения 319 (323 мг, 1,0 ммоль), соединения 311 (208 мг, 1,0 ммоль), трифенилфосфина (394 мг, 1,5 ммоль) и тетрагидрофурана (5 мл) добавляли по каплям раствор в тетрагидрофуране (2 мл) диизопропилазодикарбоксилата (300 мг, 1,5 ммоль) при охлаждении льдом и перемешивании, с последующим перемешиванием при той же температуре в течение 30 минут и дополнительном перемешивании при комнатной температуре в течение 20 часов. Реакционный раствор очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1) с получением соединения 320 (403 мг, 78%) в виде желтой карамели.
APCI-MS m/z 514[M+H]+
[0438]
Синтез соединения 321
К суспензии в N,N-диметилформамиде (20 мл) 60% гидрида натрия (345 мг, 8,6 ммоль) добавляли по каплям раствор в N,N-диметилформамиде (10 мл) соединения 320 (3,70 г, 7,2 ммоль) в атмосфере аргона и смесь перемешивали при 0-5°C с последующим перемешиванием при -15°C в течение 30 минут. Реакционному раствору давали возможность согреться до 0-5°C и добавляли по каплям раствор в N,N-диметилформамиде (2 мл) метилйодида (2,2 г, 15 ммоль), и раствор перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор выливали в ледяную воду, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1) с получением соединения 321 (3,30 г, 89%) в виде бледно-желтого твердого вещества.
точка плавления 97-99°C
APCI-MS m/z 528[M+H]+
[0439]
Синтез соединения 322
Смесь соединения 321 (2,18 г, 4,13 ммоль) и 48% HBr (25 мл) перемешивали при 90-95°C в течение 2 часов. Реакционный раствор охлаждали и делали основным 28% аммиачной водой, и затем экстрагировали этилацетатом. Экстракционную жидкость промывали насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток промывали диизопропиловым эфиром с получением соединения 322 (1,45 г, 95%) в виде оранжевого твердого вещества.
точка плавления 130-132°C
ИК (Nujol) 3379, 1624, 1605 см-1
APCI-MS m/z 372[M+H]+
[0440]
Синтез соединения THK-5147
Смесь соединения 322 (250 мг, 0,673 ммоль), SnCl2·2H2O (450 мг, 1,99 ммоль) и этанола (10 мл) нагревали кипячением в сосуде с обратным холодильником в течение 1 часа. Реакционному раствору давали возможность охладиться до комнатной температуры, разбавляли этилацетатом и затем делали основным 28% аммиачной водой. Нерастворимые вещества удаляли фильтрацией целитом, и органический слой фильтрата отделяли и промывали насыщенным солевым раствором. После сушки растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на NH силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением соединения THK-5147 (175 мг, 76%) в виде желтого твердого вещества.
точка плавления 122-124°C
1H ЯМR (400 МГц, CDCl3) δ 2,95 (3H, с), 3,96 (3H, уш.), 4,23-4,37 (3H, м), 4,49-4,61 (1Н, м), 4,62-4,73 (1Н, м), 6,69 (1H, д, J=2,7 Гц), 6,81 (1H, д, J=8,2 Гц), 7,08 (1Н, дд, J=9,0, 2,7 Гц), 7,53 (1H, дд, J=8,2, 1,8 Гц), 7,68 (1H, д, J=8,8 Гц), 7,74 (1H, д, J=1,8 Гц), 7,91 (1H, д, J=9,0 Гц), 7,94 (1Н, д, J=8,8 Гц)
ИК (Nujol) 3429, 3342, 1625, 1595 см-1
APCI-MS m/z 342[M+H]+
[0441]
Способ синтеза соединения THK-5148
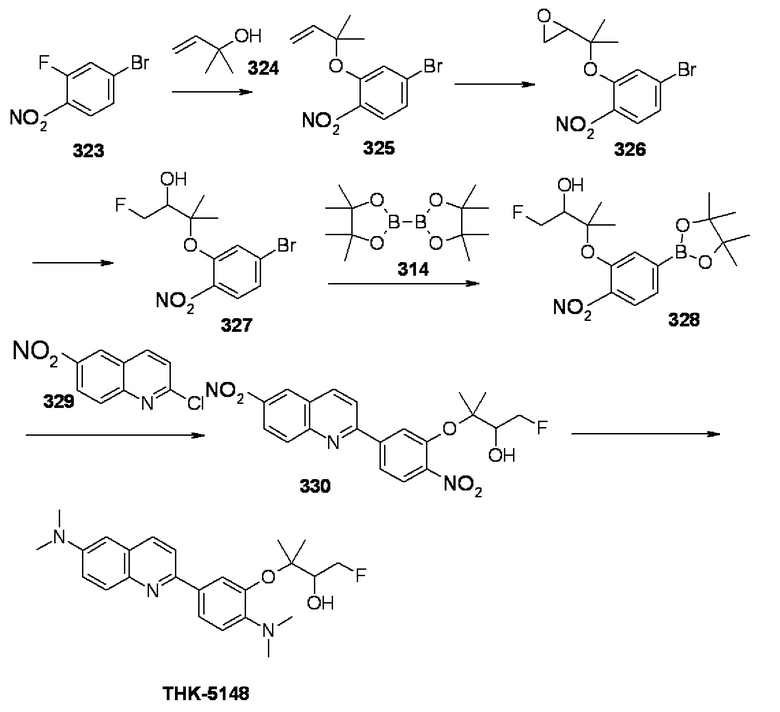
[0442]
Синтез соединения 325
В атмосфере аргона раствор в тетрагидрофуране (15 мл) соединения 324 (3,1 г, 36 ммоль) добавляли по каплям к суспензии в тетрагидрофуране (30 мл) 60% гидрида натрия (1,44 г, 36 ммоль) при комнатной температуре, и смесь нагревали кипячением в сосуде с обратным холодильником в течение 30 минут. Реакционный раствор охлаждали до 0-5°C, раствор в тетрагидрофуране (15 мл) соединения 323 (7,2 г, 33 ммоль) добавляли по каплям, и раствор перемешивали при комнатной температуре в течение 16 часов. Реакционный раствор выливали в холодный водный раствор хлорида аммония, и затем раствор экстрагировали этилацетатом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан→этилацетат/н-гексан=1/50, 1/20) с получением соединения 325 (8,46 г, 90%) в виде желтого маслянистого вещества.
APCI-MS m/z 303/305[M+NH4]+
[0443]
Синтез соединения 326
Смесь соединения 325 (7,0 г, 24,5 ммоль), 75% метахлорбензойной кислоты (20,26 г, 88,1 ммоль) и хлороформа (70 мл) перемешивали при комнатной температуре в течение 2 дней. Реакционный раствор охлаждали льдом и добавляли водный насыщенный раствор гидрокарбоната натрия, и раствор экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан→этилацетат/н-гексан=1/20, 1/10) с получением соединения 326 (4,197 г, 57%) в виде желтого маслянистого вещества.
APCI-MS m/z 319/321[M+NH4]+
[0444]
Синтез соединения 327
Смесь соединения 326 (4,119 г, 13,6 ммоль), н-Bu4NH2F3 (410 мг, 1,36 ммоль) и KHF2 (2,13 г, 27,27 ммоль) перемешивали при 120°C в течение 6 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли хлороформ, и затем нерастворимые вещества удаляли фильтрацией. Фильтрат сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан→этилацетат/н-гексан=1/20, 1/10, 1/6, 1/4) с получением соединения 327 (1,3 г, 30%) в виде желтого твердого вещества.
APCI-MS m/z 339/341[M+NH4]+
[0445]
Синтез соединения 328
Смесь соединения 327 (1,29 г, 4,0 ммоль), соединения 314 (1,06 г, 4,16 ммоль), ацетата калия (1,18 г, 12 ммоль), Pd(dppf)Cl2·CH2Cl2 (229 мг, 0,28 ммоль) и 1,4-диоксана (20 мл) нагревали кипячением в сосуде с обратным холодильником в течение 16 часов в атмосфере аргона. Реакционному раствору давали возможность охладиться до комнатной температуры, и растворитель отгоняли при пониженном давлении. К остатку добавляли этилацетат и водный насыщенный раствор гидрокарбоната натрия, и затем раствор экстрагировали этилацетатом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан→этилацетат/н-гексан=1/10, 1/4, 1/2) с получением соединения 328 (1,438 г, 97%) в виде коричневого маслянистого вещества.
APCI-MS m/z 387[M+NH4]+
[0446]
Синтез соединения 330
Смесь соединения 328 (1,4 г, 3,8 ммоль), соединения 329 (729 мг, 3,5 ммоль), водного 2M раствора карбоната натрия (3,5 мл), тетракистрифенилфосфина палладия (202 мг, 0,175 ммоль) и 1,2-диметоксиэтана (20 мл) перемешивали при 90°C в течение 5 часов в атмосфере аргона. Реакционному раствору давали возможность охладиться до комнатной температуры, 1,2-диметоксиэтан отгоняли при пониженном давлении. Добавляли хлороформ, и нерастворимые вещества удаляли фильтрацией с последующим промыванием хлороформом/метанолом (=9/1). Фильтрат и промывной раствор объединяли, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан→этилацетат/н-гексан=1/10, 1/4, 1/2) и затем промывали н-гексаном с получением соединения 330 (1,448 г, 99%) в виде желтого твердого вещества.
APCI-MS m/z 416[M+H]+
[0447]
Синтез соединения THK-5148
Соединение 330 (1,44 г, 3,47 ммоль), 10% Pd-C (300 мг) и этанол-метанол (60 мл-20 мл) перемешивали при давлении водорода (40 фунтов/дюйм2 (2068,6 мм рт.ст.) при комнатной температуре в течение 16 часов. Катализатор удаляли фильтрацией, и к фильтрату добавляли Pd-C (150 мг) с последующим дополнительным перемешиванием при давлении водорода (40 фунтов/дюйм2 (2068,6 мм рт.ст.) при комнатной температуре в течение 1,5 часов. Катализатор удаляли фильтрацией, и растворитель фильтрата отгоняли при пониженном давлении. К остатку добавляли метанол (50 мл)-уксусную кислоту (5 мл), водный 36% раствор формальдегида (3,1 г, 37,2 ммоль) и пиколин-борановый комплекс (1,5 г, 14 ммоль) с последующим перемешиванием при комнатной температуре в течение 16 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и водный насыщенный раствор гидрокарбоната натрия, остаток и раствор экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: хлороформ→хлороформ/метанол=50/1) и затем перекристаллизовывали из хлороформа-н-гексана с получением соединения THK-5148 (832 мг, 58%) в виде желтовато-оранжевого твердого вещества.
точка плавления 182-184°C
1H ЯМR (400 МГц, ДМСО-d6) δ 1,25, 1,23 (6H, каждый с), 2,05 (6H, с), 2,82 (6H, с), 4,45-4,51 (1Н, м), 4,81 (1H, ддд, J=48, 10, 6,5 Гц), 4,90 (1Н, ддд, J=46, 10, 2,4 Гц), 5,09 (1Н, уш. с), 6,94 (1H, д, J=2,7 Гц), 7,02 (1H, д, J=8,4 Гц), 7,44 (1H, дд, J=9,4, 2,7 Гц), 7,73 (1H, дд, J=8,5, 1,8 Гц), 7,85 (1H, д, J=9,4 Гц), 7,89 (1H, д, J=8,7 Гц), 7,91 (1H, д, J=2,1 Гц), 8,13 (1Н, д, J=8,8 Гц)
ИК (Nujol) 1819, 1375 см-1
APCI-MS m/z 412[M+H]+
[0448]
Способ синтеза соединения THK-5155
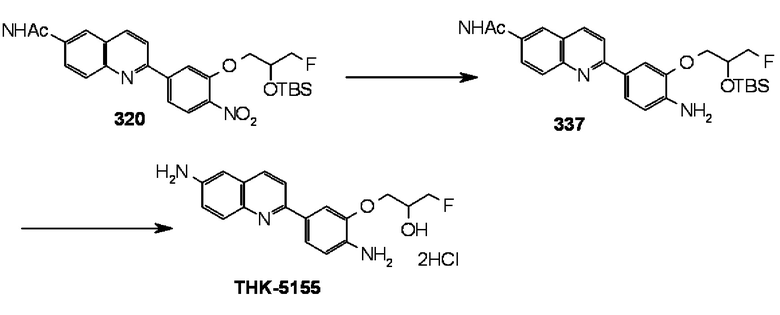
[0449]
Синтез соединения 337
Соединение 320 (1,18 г, 2,3 ммоль), 10% Pd-C (влажность 50%; 250 мг) и этанол (30 мл) перемешивали при давлении водорода 40 фунтов/дюйм2 (2068,6 мм рт.ст.) при комнатной температуре в течение 16 часов. Катализатор удаляли фильтрацией, и растворитель фильтрата отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: хлороформ) с получением соединения 337 (1,03 г, 93%) в виде желтого пеноподобного вещества.
[0450]
Синтез соединения THK-5155
Смесь соединения 337 (1,025 г, 2,12 ммоль) и 48% HBr (10 мл) перемешивали при 110°C в течение 5 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, и растворитель отгоняли при пониженном давлении. К остатку добавляли водный насыщенный раствор гидрокарбоната натрия, и раствор экстрагировали этилацетатом-тетрагидрофураном. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на NH силикагеле (элюирующий растворитель: хлороформ/метанол=50/1) с получением бледно-желтого аморфного вещества (684 мг). Настоящий продукт превращали в гидрохлорид обычным способом, используя 4M хлористоводородную кислоту/этилацетат, с получением соединения THK-5155 (670 мг, 79%) в виде оранжевого твердого вещества.
точка плавления 232-235°C
1H ЯМR (400 МГц, ДМСО-d6) δ 4,05-4,25 (3H, м), 4,50-4,70 (2H, м), 6,94 (1H, д, J=8,2 Гц), 7,18 (1Н, уш. с), 7,49 (1H, дд, J=9,0, 2,4 Гц), 7,72 (1H, дд, J=8,4, 1,8 Гц), 7,81 (1H, д, J=1,8 Гц), 8,19 (1H, д, J=9,0 Гц), 8,39 (1H, д, J=9,0 Гц), 8,62 (1Н, д, J=9,0 Гц)
ИК (Nujol) 1624, 1460 см-1
APCI-MS m/z 328[M+H]+
[0451]
Способ синтеза соединения THK-5156
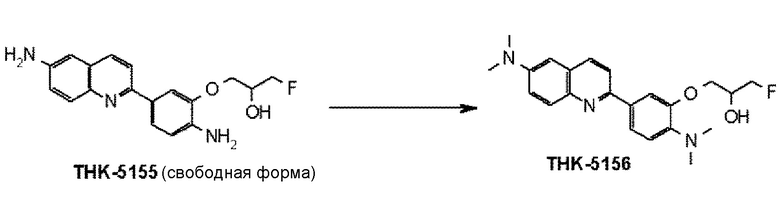
[0452]
Синтез соединения THK-5156
К смеси соединения THK-5155 (свободной формы) (450 мг, 1,4 ммоль), водного 36% раствора формальдегида (0,97 г, 11,6 ммоль) и метанола (10 мл)-уксусной кислоты (1 мл) небольшими порциями добавляли пиколин-борановый комплекс (441 мг, 4,1 ммоль), и смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и к остатку добавляли 10% водную хлористоводородную кислоту, и раствор перемешивали при комнатной температуре в течение 10 минут. Реакционный раствор экстрагировали хлороформом после придания основности водным раствором карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и затем остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: хлороформ→хлороформ/тетрагидрофуран=9/1, 6/1, 4/1) с получением соединения THK-5156 (328 мг, 62%) в виде оранжевого твердого вещества.
точка плавления 187-188,5°C
1H ЯМR (400 МГц, ДМСО-d6) δ 2,82 (6H, с), 3,05 (6H, с), 4,10-4,20 (3H, м), 4,40-4,70 (2H, м), 5,50 (1Н, уш. с), 6,95 (1H, д, J=2,4 Гц), 6,98 (1H, д, J=8,5 Гц), 7,45 (1Н, дд, J=9,0, 2,4 Гц), 7,72 (1H, дд, J=8,4, 1,8 Гц), 7,78 (1Н, д, J=1,8 Гц), 7,87 (1Н, д, J=9,4 Гц), 7,95 (1Н, д, J=8,8 Гц), 8,14 (1H, д, J=8,8 Гц)
ИК (Nujol) 1620, 1463, 1375 см-1
APCI-MS m/z 384[M+H]+
[0453]
Способ синтеза соединения THK-5158
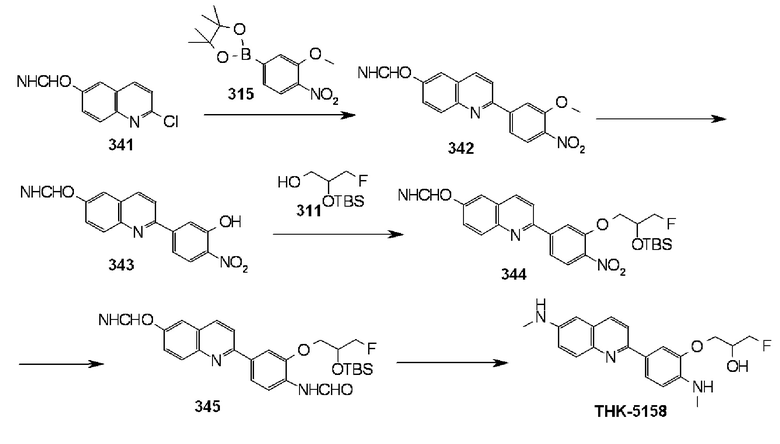
[0454]
Синтез соединения 342
К раствору в 1,2-диметоксиэтане (364 мл) соединения 341 (9,14 г, 44,23 ммоль) и соединения 315 (14,57 г, 52,20 ммоль) добавляли карбонат калия (18,34 г, 133 ммоль), воду (7,7 мл) и тетракистрифенилфосфин палладия (5,11 г, 4,42 ммоль) в атмосфере аргона, и смесь перемешивали при 80°C в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли этилацетат, и затем нерастворимые вещества удаляли фильтрацией целитом, и раствор промывали хлороформом/метанолом (=1/1). Фильтрат и промывной раствор объединяли, и растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1, 3/1) и затем промывали этилацетатом с получением соединения 342 (9,55 г, 67%) в виде оранжевых кристаллов.
точка плавления 216-217°C
APCI-MS m/z 324[M+H]+
[0455]
Синтез соединения 343
Смесь соединения 342 (6,5 г, 20,1 ммоль), хлорида лития (8,48 г, 0,2 моль) и гексаметилфосфорного триамида (65 мл) перемешивали при 110°C в течение 41 часа. Реакционному раствору давали возможность охладиться до комнатной температуры и выливали в водный раствор лимонной кислоты, и раствор перемешивали. Осадок собирали фильтрацией, промывали водой и затем растворяли в тетрагидрофуране. Раствор тетрагидрофурана сушили, и растворитель отгоняли при пониженном давлении, и затем остаток промывали этилацетатом с получением соединения 343 (5,74 г, 92%) в виде коричневого твердого вещества.
точка плавления 222-223°C
[0456]
Синтез соединения 344
К раствору в тетрагидрофуране (110 мл) соединения 343 (5,73 г, 18,5 ммоль), соединения 311 (5,65 г, 27,1 ммоль) и трифенилфосфина (8,26 г, 31,5 ммоль) добавляли по каплям раствор в тетрагидрофуране (20 мл) диизопропилазодикарбоксилата (6,37 г, 31,5 ммоль) при -5-0°C в течение 20 минут, и смесь перемешивали при той же температуре в течение 30 минут, с последующим перемешиванием при комнатной температуре в течение 66 часов. К реакционному раствору добавляли соединение 311 (565 мг, 2,7 ммоль), трифенилфосфин (826 мг, 3,15 ммоль) и диизопропилазодикарбоксилат (638 мг, 3,15 ммоль), и раствор дополнительно перемешивали при комнатной температуре в течение 6 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 1/1) с получением соединения 344 (7,67 г, 83%) в виде твердого вещества.
точка плавления 150-152°C
APCI-MS m/z 500[M+H]+
[0457]
Синтез соединения 345
Соединение 344 (1,0 г, 2 ммоль), 10% Pd-C (150 мг) и этанол (20 мл) перемешивали при давлении водорода 40 фунтов/дюйм2 (2068,6 мм рт.ст.) при комнатной температуре в течение 16 часов. Катализатор удаляли фильтрацией, и растворитель отгоняли при пониженном давлении. Бледно-желтое аморфное вещество в качестве остатка растворяли добавлением этилформиата (20 мл), и раствор нагревали кипячением в сосуде с обратным холодильником в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: тетрагидрофуран/хлороформ=1/19, 1/9, 1/4, 1/3, 1/1, метанол/хлороформ=1/9, 1/1) и затем промывали этилацетатом/н-гексаном с получением соединения 345 (860 мг, 86%) в виде оранжевого твердого вещества.
APCI-MS m/z 498[M+H]+
[0458]
Синтез соединения THK-5158
К суспензии в тетрагидрофуране (10 мл) NaBH4 (392 мг, 10,4 ммоль) добавляли по каплям раствор в тетрагидрофуране (10 мл) BF3·Et2O (1,96 г, 13,8 ммоль) при 0°C и смесь перемешивали при комнатной температуре в течение 1 часа. К настоящему реакционному раствору добавляли по каплям раствор в тетрагидрофуране (10 мл) соединения 345 (855 мг, 1,7 ммоль) при 0-5°C, и раствор перемешивали при комнатной температуре в течение 2 часов, с последующим перемешиванием при нагревании в сосуде с обратным холодильником в течение 3 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, и растворитель отгоняли при пониженном давлении. К остатку добавляли водный раствор 10% хлористоводородной кислоты с последующим перемешиванием при комнатной температуре в течение 1 часа. Реакционный раствор делали основным добавлением карбоната калия и затем экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: хлороформ→тетрагидрофуран/хлороформ=1/19, 1/9, 1/4), снова очищали флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан → этилацетат/н-гексан=1/1, 6/4) и затем промывали диизопропиловым эфиром и затем н-гексаном с получением соединения THK-5158 (331 мг, 54%) в виде оранжевого твердого вещества.
точка плавления 150-152°C
1H ЯМR (400 МГц, ДМСО-d6) δ 2,79 (3H, д, J=5,8 Гц), 2,80 (3H, д, J=5,1 Гц), 3,95-4,20 (3H, м), 4,50-4,70 (2H, м), 5,47 (1H, д, J=5,8 Гц), 5,53 (1H, д, J=5,1 Гц), 6,12 (1Н, д, J=5,1 Гц), 6,56 (1Н, д, J=8,8 Гц), 6,62 (1H, д, J=2,7 Гц), 7,13 (1H, дд, J=9,1, 2,4 Гц), 7,64-7,68 (2H, м), 7,70 (1H, д, J=9,1 Гц), 7,84 (1H, д, J=8,8 Гц), 7,99 (1H, д, J=8,7 Гц)
ИК (Nujol) 1621 см-1
APCI-MS m/z 356[M+H]+
[0459]
Способ синтеза соединения THK-5159
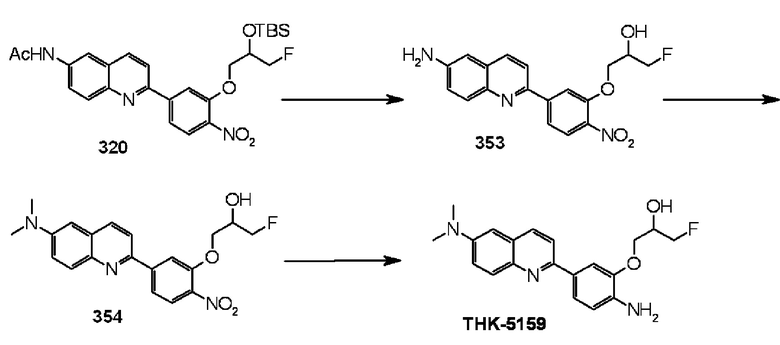
[0460]
Синтез соединения 353
Смесь соединения 320 (100 мг, 0,195 ммоль) и 48% HBr (1 мл) перемешивали при температуре от 90 до 95°C в течение 1 часа. Реакционному раствору давали возможность охладиться до комнатной температуры и делали основным концентрированной аммиачной водой с последующей экстракцией этилацетатом. Экстракционную жидкость промывали насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток промывали этилацетатом-диизопропиловым эфиром с получением соединения 353 (45 мг, 65%) в виде оранжевого твердого вещества.
точка плавления 153-154°C
APCI-MS m/z 358[M+H]+
[0461]
Синтез соединения 354
К смеси соединения 353 (167 мг, 0,467 ммоль), водного 36% раствора формальдегида (1 мл) и этанола (5 мл)-уксусной кислоты (0,5 мл) небольшими порциями добавляли пиколин-борановый комплекс (150 мг, 1,4 ммоль), с последующим перемешиванием при комнатной температуре в течение 3 часов. Реакционный раствор разбавляли этилацетатом, промывали по очереди водным насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2) и затем промывали этилацетатом/н-гексаном (=1/3) с получением соединения 354 (161 мг, 89%) в виде коричневого твердого вещества.
точка плавления 169-171°C
[0462]
Синтез соединения THK-5159
Смесь соединения 354 (270 мг, 0,7 ммоль), 10% Pd-C (влажность примерно 50%; 100 мг), формиата аммония (440 мг, 7 ммоль) и метанола (10 мл)-тетрагидрофурана (5 мл) перемешивали в атмосфере аргона при комнатной температуре в течение 3 часов. Реакционный раствор разбавляли этилацетатом, и нерастворимые вещества удаляли фильтрацией. Фильтрат промывали по очереди водой и насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на NH силикагеле (элюирующий растворитель: н-гексан/этилацетат=1/1, 1/2, 1/3, 1/4, 1/5) и затем промывали н-гексаном/диизопропиловым эфиром с получением соединения THK-5159 (216 мг, 87%) в виде бледно-коричневого твердого вещества.
точка плавления 169-170°C
1H ЯМR (400 МГц, CDCl3) δ 2,94 (1Н, уш.), 3,08 (6H, с), 3,96 (2H, уш.), 4,24-4,36 (3H, м), 4,52-4,61 (1Н, м), 4,64-4,73 (1Н, м), 6,81 (1H, д, J=3,0 Гц), 6,81 (1H, д, J=8,0 Гц), 7,36 (1H, дд, J=9,4, 3,0 Гц), 7,53 (1H, дд, J=8,0, 1,2 Гц), 7,68 (1H, д, J=8,8 Гц), 7,76 (1Н, уш.), 7,96 (1H, д, J=8,8 Гц), 7,99 (1H, д, J=9,4 Гц)
ИК (Nujol) 3345, 1621, 1590 см-1
APCI-MS m/z 356[M+H]+
[0463]
Способ синтеза соединения THK-5160
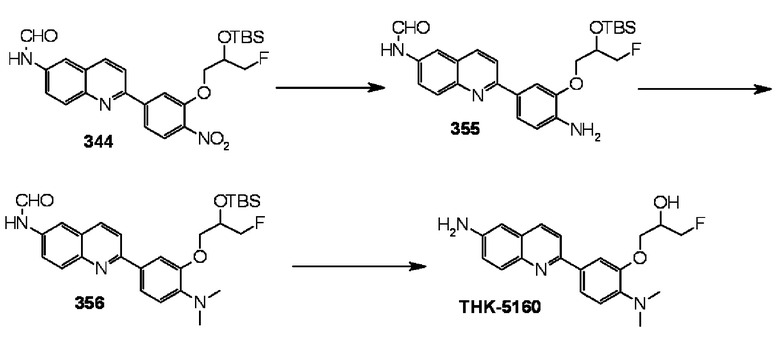
[0464]
Синтез соединения 355
Соединение 344 (2,65 г, 5,3 ммоль), 10% Pd-C (влажность примерно 50%; 350 мг) и этилацетат (40 мл)-тетрагидрофуран (20 мл) перемешивали в атмосфере водорода при комнатной температуре в течение 21 часа. Катализатор удаляли фильтрацией, и растворитель фильтрата отгоняли при пониженном давлении. Остаток растворяли в этилацетате (50 мл) и добавляли 10% Pd-C (влажность примерно 50%; 300 мг), и раствор перемешивали в атмосфере водорода при комнатной температуре в течение 2 дней. Катализатор удаляли фильтрацией, и растворитель фильтрата отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=1/1, 2/3, этилацетат) с получением соединения 355 (2,27 г, 91%).
APCI-MS m/z 470[M+H]+
[0465]
Синтез соединения 356
К смеси соединения 355 (1,20 г, 2,55 ммоль), водного 35% раствора формальдегида (2,2 г, 26 ммоль) и этанола (20 мл)-уксусной кислоты (2 мл) небольшими порциями добавляли пиколин-борановый комплекс (545 мг, 5,1 ммоль) с последующим перемешиванием при комнатной температуре в течение 6 часов. К реакционному раствору добавляли водный 35% раствор формальдегида (2,2 г, 26 ммоль), и добавляли пиколин-борановый комплекс (545 мг, 5,1 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 2 часов. Реакционный раствор разбавляли этилацетатом, промывали по очереди водой, разбавленной аммиачной водой и насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток промывали диизопропиловым эфиром с получением соединения 356 (922 мг, 73%) в виде бледно-коричневого твердого вещества.
точка плавления 164-166°C
APCI-MS m/z 498[M+H]+
[0466]
Синтез соединения THK-5160
Смесь соединения 356 (350 мг, 0,7 ммоль) и 48% HBr (3 мл) перемешивали при температуре от 90 до 95°C в течение 1 часа. Реакционному раствору давали возможность охладиться до комнатной температуры и разбавляли этилацетатом, и добавляли ледяную воду. Затем раствор делали основным концентрированной аммиачной водой и экстрагировали этилацетатом. Экстракционную жидкость промывали по очереди водой и насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=1/2, 1/4, этилацетат) с получением соединения THK-5160 (179 мг, 72%) в виде бежевого твердого вещества.
точка плавления 158-160°C
1H ЯМR (400 МГц, CDCl3) δ 2,85 (6H, с), 3,95 (2H, уш. с), 4,04-4,15 (1Н, м), 4,24-4,35 (2H, м), 4,43-4,52 (1Н, м), 4,55-4,63 (1Н, м), 5,20 (1Н, уш.), 6,92 (1H, д, J=2,4 Гц), 7,11 (1H, д, J=8,5 Гц), 7,17 (1H, дд, J=8,5, 2,7 Гц), 7,71 (1H, д, J=8,8 Гц), 7,72 (1H, дд, J=8,5, 2,1 Гц), 7,86 (1H, д, J=2,1 Гц), 7,94 (1H, д, J=8,5 Гц), 7,95 (1Н, д, J=8,8 Гц)
ИК (Nujol) 3462, 3345, 1633 см-1
APCI-MS m/z 356[M+H]+
[0467]
Способ синтеза соединения THK-5161
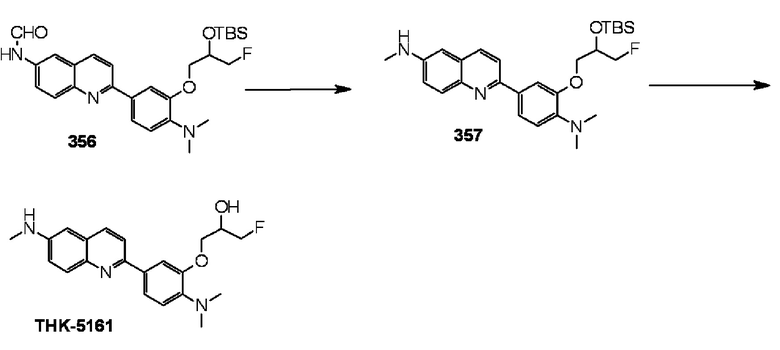
[0468]
Синтез соединения 357
В атмосфере аргона раствор в тетрагидрофуране (5 мл) BF3·Et2O (1,23 г, 8,7 ммоль) добавляли по каплям к суспензии в тетрагидрофуране (10 мл) NaBH4 (252 мг, 6,6 ммоль) при 0°C, и смесь перемешивали при той же температуре в течение 20 минут. К настоящему реакционному раствору добавляли по каплям раствор в тетрагидрофуране (3 мл) соединения 356 (630 мг, 1,27 ммоль) при 0-5°C с последующим перемешиванием при 5°C в течение 1 часа. К реакционному раствору добавляли ледяную воду и этилацетат, и раствор делали основным концентрированной аммиачной водой и затем экстрагировали этилацетатом. Экстракционную жидкость промывали по очереди водой и затем насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на NH силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/8, 1/6) с получением соединения 357 (440 мг, 72%) в виде бледно-желтого твердого вещества.
APCI-MS m/z 484[M+H]+
[0469]
Синтез соединения THK-5161
Смесь соединения 357 (440 мг, 0,91 ммоль) и 48% HBr (3 мл) перемешивали при 50°C в течение 10 минут. Реакционный раствор делали основным концентрированной аммиачной водой при охлаждении льдом и затем экстрагировали этилацетатом. Экстракционную жидкость промывали по очереди водой и затем насыщенным солевым раствором и сушили, растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на NH силикагеле (элюирующий растворитель: этилацетат/н-гексан=2/1, 3/1) с получением соединения THK-5161 (280 мг, 83%) в виде бледно-желтого твердого вещества.
точка плавления 140-141°C
1H ЯМR (400 МГц, CDCl3) δ 2,84 (6H, с), 2,96 (3H, уш. с), 4,00-4,15 (2H, м), 4,24-4,35 (2H, м), 4,43-4,51 (1Н, м), 4,55-4,63 (1Н, м), 5,26 (1Н, уш.), 6,71 (1H, д, J=2,7 Гц), 7,10 (1H, д, J=9,0 Гц), 7,10 (1Н, дд, J=9,0, 2,7 Гц), 7,71 (1H, д, J=8,5 Гц), 7,72 (1H, дд, J=8,5, 2,0 Гц), 7,86 (1H, д, J=2,0 Гц), 7,92 (1H, д, J=9,0 Гц), 7,98 (1H, д, J=8,8 Гц)
ИК (Nujol) 3462, 1627 см-1
APCI-MS m/z 370[M+H]+
[0470]
Способ синтеза соединения THK-5162
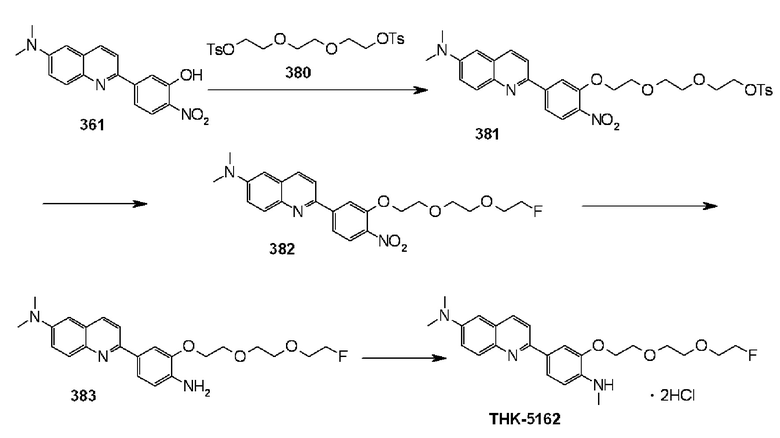
[0471]
Синтез соединения 381
К суспензии в N,N-диметилформамиде (30 мл) соединения 361 (900 мг, 2,91 ммоль) и соединения 380 (4,00 г, 8,7 ммоль) добавляли карбонат калия (1,21 г, 8,7 ммоль) при комнатной температуре при перемешивании и смесь перемешивали при комнатной температуре в течение 4 дней. Реакционный раствор экстрагировали этилацетатом после добавления воды и этилацетата. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 1/1) с получением соединения 381 (1,73 г, 100%) в виде красного маслянистого вещества.
APCI-MS m/z 596[M+H]+
[0472]
Синтез соединения 382
Смесь соединения 381 (1,73 г, 2,9 ммоль) и 1M тетра-н-бутиламмонийфторида/тетрагидрофурана (14,6 мл, 14,6 ммоль) перемешивали при комнатной температуре в течение 16 часов. Реакционный раствор разбавляли этилацетатом, промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 2/3) с получением соединения 382 (960 мг, 74%) в виде красного смолянистого вещества.
APCI-MS m/z 444[M+H]+
[0473]
Синтез соединения 383
Смесь соединения 382 (950 мг, 2,14 ммоль), 10% Pd-C (влажность примерно 50%; 190 мг), формиата аммония (1,35 г, 21,4 ммоль) и метанола (20 мл)-тетрагидрофурана (10 мл) перемешивали в атмосфере аргона при комнатной температуре в течение 1 часа. Реакционный раствор разбавляли этилацетатом, и нерастворимые вещества удаляли фильтрацией, и затем растворитель фильтрата отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 2/1) с получением соединения 383 (850 мг, 96%) в виде желтого смолянистого вещества.
APCI-MS m/z 414[M+H]+
[0474]
Синтез соединения THK-5162
Смесь соединения 383 (600 мг, 1,45 ммоль), водного 35% раствора формальдегида (373 мг, 4,35 ммоль), сульфата магния (7 г), тетрагидрофурана (40 мл) и изопропанола (20 мл) перемешивали при комнатной температуре в течение 3 часов. К реакционному раствору добавляли NaBH4 (274 мг, 7,2 ммоль), и раствор перемешивали при комнатной температуре в течение 3 дней. Нерастворимые вещества удаляли фильтрацией из реакционного раствора, и растворитель фильтрата отгоняли при пониженном давлении. Остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=2/1), и полученное оранжевое смолянистое вещество (580 мг; свободная форма) превращали в гидрохлорид обработкой 4M хлористоводородной кислотой/этилацетатом, и затем перекристаллизовывали из этанола с получением соединения THK-5162 (516 мг, 71%) в виде оранжевых кристаллов.
точка плавления 197-198°C
1H ЯМR (400 МГц, ДМСО-d6) δ 2,87 (3H, с), 3,10 (6H, с), 3,56-3,64 (3H, м), 3,64-3,68 (2H, м), 3,69-3,72 (1Н, м), 3,84-3,88 (2H, м), 4,38 (2H, дд, J=5,4, 3,9 Гц), 4,53 (2H, дт, J=48, 4,3 Гц), 6,72 (1H, д, J=8,8 Гц), 7,18 (1H, д, J=2,7 Гц), 7,68 (1H, дд, J=9,5, 2,9 Гц), 7,84 (1H, дд, J=8,5, 2,1 Гц), 7,90 (1H, д, J=2,1 Гц), 8,24 (1H, д, J=9,1 Гц), 8,58 (1Н, д, J=9,4 Гц), 8,63 (1H, д, J=9,1 Гц)
ИК (Nujol) 3376, 2582, 1643, 1603 см-1
APCI-MS m/z 428[M+H]+
[0475]
Способ синтеза соединения THK-5163
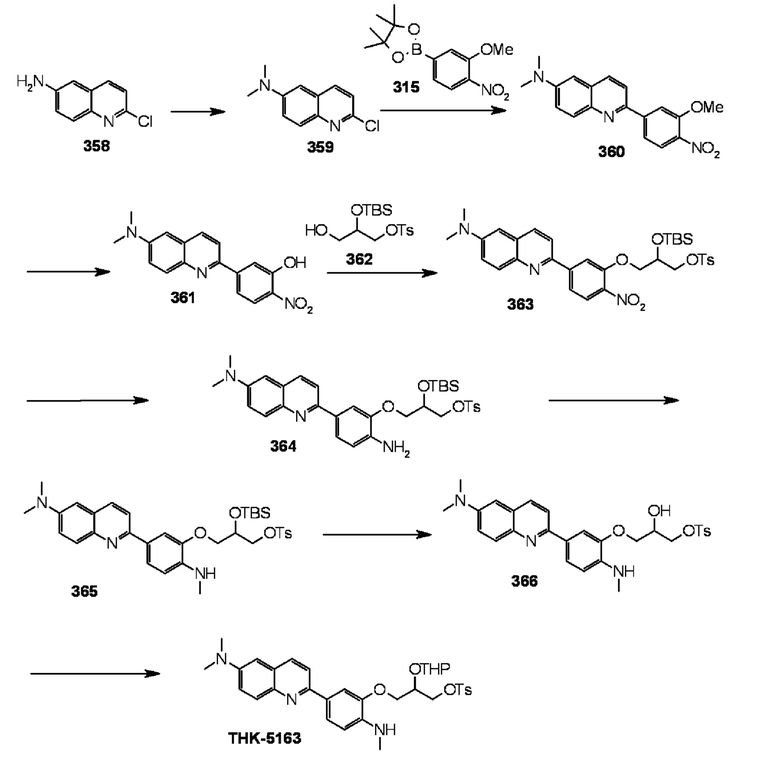
[0476]
Синтез соединения 359
К смеси соединения 358 (2,20 г, 12,3 ммоль), водного 36% раствора формальдегида (20,5 г, 246 ммоль) и метанола (250 мл)-уксусной кислоты (20 мл) небольшими порциями добавляли пиколин-борановый комплекс (7,91 г, 73,95 ммоль) с последующим перемешиванием при комнатной температуре в течение 16 часов. К реакционному раствору добавляли этилацетат-воду, и раствор экстрагировали этилацетатом после доведения pH до 8 с использованием водного раствора карбоната калия. Экстракционную жидкость промывали водой и сушили, растворитель отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением соединения 359 (2,54 г, 99%) в виде бледно-желтого твердого вещества.
точка плавления 74-75°C
APCI-MS m/z 207[M+H]+
[0477]
Синтез соединения 360
Смесь соединения 359 (2,54 г, 12,29 ммоль), соединения 315 (4,36 г, 15,6 ммоль), карбоната калия (5,10 г, 36,9 ммоль), тетракистрифенилфосфина палладия (1,42 г, 1,23 ммоль) и воды (2,2 мл)-1,2-диметоксиэтана (100 мл) перемешивали в атмосфере аргона при 80°C в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, разбавляли этилацетатом, сушили над сульфатом натрия и затем фильтровали целитом. Фильтрат концентрировали примерно до 100 мл при пониженном давлении, очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2) и затем промывали этилацетатом/н-гексаном (=1/2) с получением соединения 360 (3,66 г, 92%) в виде оранжевого твердого вещества.
точка плавления 175-175,5°C
APCI-MS m/z 324[M+H]+
[0478]
Синтез соединения 361
Смесь соединения 360 (3,11 г, 9,62 ммоль), хлорида лития (4,08 г, 96,2 ммоль) и гексаметилфосфорного триамида (31 мл) перемешивали в атмосфере аргона при 110°C в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли этилацетат и воу, и раствор экстрагировали этилацетатом. Экстракционную жидкость промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2) и затем перекристаллизовывали из этилацетата/н-гексана с получением соединения 361 (2,97 г, 100%) в виде коричневых кристаллов.
точка плавления 224-225°C
APCI-MS m/z 310[M+H]+
[0479]
Синтез соединения 363
К смеси соединения 361 (1,00 г, 3,23 ммоль), соединения 362 (1,40 г, 3,88 ммоль), трифенилфосфина (1,02 г, 3,88 ммоль) и тетрагидрофурана (40 мл) добавляли по каплям раствор диизопропилазодикарбоксилата (0,77 мл, 3,88 ммоль) в тетрагидрофуране (10 мл) в течение 20 минут при охлаждении льдом, с последующим перемешиванием при той же температуре в течение 1 часа и дополнительным перемешиванием при комнатной температуре в течение 16 часов. К реакционному раствору добавляли соединение 362 (700 мг, 1,94 ммоль), трифенилфосфин (510 мг, 1,94 ммоль), диизопропилазодикарбоксилат (0.38 мл, 1,92 ммоль) и тетрагидрофуран (10 мл), и раствор дополнительно перемешивали при комнатной температуре в течение 3 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9, 1/4) и затем перекристаллизовывали из этилацетата/н-гексана=1/4 с получением соединения 363 (1,94 г, 92%) в виде оранжевых кристаллов.
точка плавления 144-145°C
APCI-MS m/z 652[M+H]+
[0480]
Синтез соединения 364
Смесь соединения 363 (1,00 г, 1,53 ммоль), 10% Pd-C (влажность примерно 50%; 130 мг), формиата аммония (965 мг, 15,3 ммоль) и метанола (20 мл)-тетрагидрофурана (10 мл) перемешивали в атмосфере аргона, с последующим перемешиванием при комнатной температуре в течение 27 часов. Катализатор удаляли фильтрацией, и растворитель фильтрата отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=4/1, 2/1) с получением соединения 364 (900 мг, 94%) в виде оранжевого аморфного вещества.
APCI-MS m/z 622[M+H]+
[0481]
Синтез соединения 365
Соединение 364 (890 мг, 1,43 ммоль) и водный 35% раствор формальдегида (0,61 мл, 7,16 ммоль) почти растворяли в метаноле (50 мл)-тетрагидрофуране (10 мл) и добавляли сульфат магния (10 г), и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор охлаждали льдом и добавляли NaBH4 (271 мг, 7,16 ммоль) при той же температуре в течение 10 минут, с последующим перемешиванием при комнатной температуре в течение 16 часов. Нерастворимые вещества удаляли из реакционного раствора фильтрацией, и растворитель фильтрата отгоняли при пониженном давлении. Остаток почти растворяли в изопропиловом спирте (50 мл) и добавляли водный 35% раствор формальдегида (0,61 мл, 7,16 ммоль) и сульфат магния (10 г), с последующим перемешиванием при комнатной температуре в течение 30 минут. Реакционный раствор охлаждали льдом и добавляли NaBH4 (271 мг, 7,16 ммоль). После перемешивания при комнатной температуре в течение 2 дней добавляли NaBH4 (271 мг, 7,16 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 6 дней. Нерастворимые вещества удаляли из реакционного раствора фильтрацией, и растворитель фильтрата отгоняли при пониженном давлении. Остаток растворяли в этилацетате, и раствор промывали водой и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4) с получением соединения 365 (650 мг, 71%) в виде бледно-желтого аморфного вещества.
APCI-MS m/z 636[M+H]+
[0482]
Синтез соединения 366
К раствору в хлороформе (12 мл) соединения 365 (640 мг, 1,01 ммоль) добавляли по каплям трифторуксусную кислоту (8 мл) при охлаждении льдом и перемешивании, добавляли воду (2 мл) и затем смесь перемешивали при комнатной температуре в течение 2 дней. К реакционному раствору добавляли ледяную воду и затем этилацетат, и раствор экстрагировали этилацетатом после доведения pH до 9 с использованием водного раствора карбоната калия. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/1) с получением соединения 366 (520 мг, 99%) в виде оранжевого аморфного вещества.
APCI-MS m/z 522[M+H]+
[0483]
Синтез соединения THK-5163
К раствору в метиленхлориде (20 мл) соединения 366 (510 мг, 0,98 ммоль) и 3,4-дигидро-2H-пирана (1,77 мл, 19,6 ммоль) добавляли моногидрат паратолуолсульфоновой кислоты (387 мг, 2,25 ммоль) и смесь перемешивали при комнатной температуре в течение 20 минут. После доведения pH реакционного раствора до 8 добавлением триэтиламина, растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 2/3, 1/1) с получением соединения THK-5163 (569 мг, 96%) в виде оранжевого аморфного вещества.
1H ЯМR (400 МГц, ДМСО-d6) δ 1,32-1,51 (4H, м), 1,52-1,74 (2H, м), 2,34 (3H, с), 2,78 (3H, д, J=3,6 Гц), 3,34-3,46 (1Н, м), 3,64-3,72, 3,78-3,86 (1Н, м), 4,10 (2Н, д, J=5,4 Гц), 4,14-4,22 (1Н, м), 4,29-4,44 (2Н, м), 4,65-4,70, 4,85-4,88 (1Н, м), 5,31 (1Н, уш.), 6,57 (1H, дд, J=8,3, 1,4 Гц), 6,93 (1H, д, J=2,1 Гц), 7,39 (2H, д, J=7,9 Гц), 7,44 (1H, дд, J=9,1, 2,4 Гц), 7,61-7,64 (1Н, м), 7,70 (1H, д, J=8,2 Гц), 7,79 (2H, дд, J=8,3, 2,9 Гц), 7,83 (1H, д, J=9,4 Гц), 7,92 (1H, д, J=8,8 Гц), 8,08 (1H, д, J=8,5 Гц)
ИК (Nujol) 3433, 1733, 1619 см-1
APCI-MS m/z 606[M+H]+
[0484]
Способ синтеза соединения THK-5164
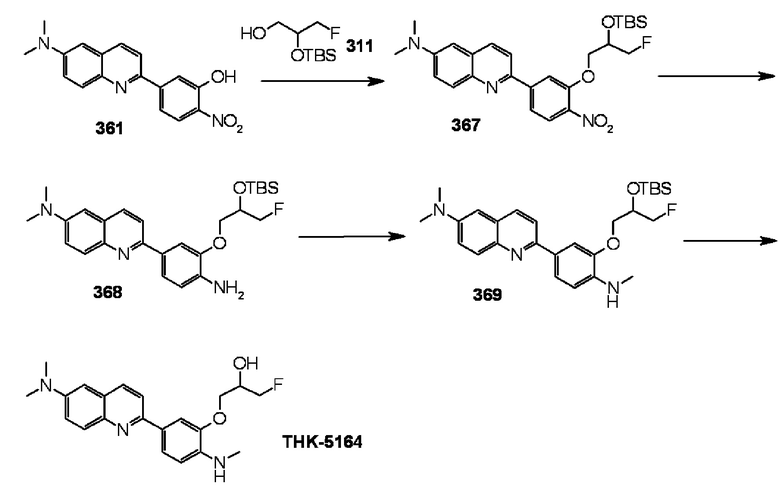
[0485]
Синтез соединения 367
К смеси соединения 361 (1,09 г, 3,52 ммоль), соединения 311 (1,04 г, 5,0 ммоль), трифенилфосфина (1,57 г, 5,98 ммоль) и тетрагидрофурана (25 мл) добавляли по каплям раствор в тетрагидрофуране (5 мл) диизопропилазодикарбоксилата (1,21 г, 5,98 ммоль) при охлаждении льдом и перемешивании, с последующим перемешиванием при той же температуре в течение 1 часа и дополнительном перемешивании при комнатной температуре в течение 16 часов. Реакционный раствор очищали колоночной флэш-хроматографией на NH силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/9) с получением соединения 367 (1,66 г, 94%) в виде оранжевого твердого вещества.
точка плавления 119-120°C
APCI-MS m/z 500[M+H]+
[0486]
Синтез соединения 368
Смесь соединения 367 (1,65 г, 3,3 ммоль), 10% Pd-C (влажность примерно 50%; 350 мг), формиата аммония (2,08 г, 33 ммоль) и метанола (40 мл)-тетрагидрофурана (20 мл) перемешивали в атмосфере аргона при комнатной температуре в течение 2 часов. Катализатор удаляли фильтрацией, и растворитель фильтрата отгоняли при пониженном давлении. Остаток растворяли в этилацетате-тетрагидрофуране, и раствор промывали по очереди водой и насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=3/1) с получением соединения 368 (1,40 г, 90%) в виде бледно-желтого твердого вещества.
точка плавления 90-92°C
APCI-MS m/z 470[M+H]+
[0487]
Синтез соединения 369
К смеси соединения 368 (800 мг, 1,7 ммоль), водного 36% раствора формальдегида (708 мг, 8,5 ммоль) и изопропанола (20 мл)-тетрагидрофурана (5 мл) добавляли сульфат магния (5 г) с последующим перемешиванием при комнатной температуре в течение 1 часа. К реакционному раствору добавляли NaBH4 (323 мг, 8,5 ммоль), и раствор перемешивали при комнатной температуре в течение 24 часов. Добавляли изопропанол (5 мл) и NaBH4 (323 мг, 8,5 ммоль) с последующим перемешиванием при 80°C в течение 1 часа. Реакционному раствору давали возможность охладиться до комнатной температуры, и нерастворимые вещества удаляли фильтрацией, и фильтрат очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/5) с получением соединения 369 (720 мг, 88%) в виде бледно-желтого твердого вещества.
точка плавления 134-135°C
APCI-MS m/z 484[M+H]+
[0488]
Синтез соединения THK-5164
Смесь соединения 369 (700 мг, 1,45 ммоль) и 48% HBr (7 мл) перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор выливали в ледяную воду, и раствор делали основным концентрированной аммиачной водой и затем экстрагировали этилацетатом. Экстракционную жидкость промывали насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток промывали этилацетатом/н-гексаном=1/5 и затем перекристаллизовывали из этилацетата с получением соединения THK-5164 (490 мг, 91%) в виде бледно-желтого твердого вещества.
точка плавления 193-194°C
1H ЯМR (400 МГц, CDCl3) δ 2,75 (1Н, уш.), 2,92 (3H, с), 3,08 (6H, с), 4,25-4,38 (3H, м), 4,40 (1Н, уш.), 4,52-4,62 (1Н, м), 4,65-4,74 (1Н, м), 6,69 (1H, д, J=8,2 Гц), 6,81 (1H, д, J=2,7 Гц), 7,35 (1H, дд, J=9,4, 2,7 Гц), 7,63 (1H, дд, J=8,5, 1,8 Гц), 7,70 (1H, д, J=8,8 Гц), 7,79 (1H, д, J=1,8 Гц), 7,96 (1H, д, J=8,8 Гц), 8,00 (1Н, уш. д, J=9,4 Гц)
ИК (Nujol) 3417, 3250, 1620, 1592 см-1
APCI-MS m/z 370[M+H]+
[0489]
Способ синтеза соединения THK-5165
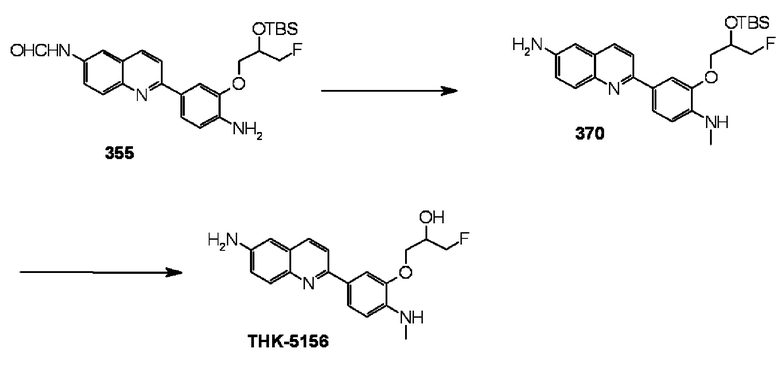
[0490]
Синтез соединения 370
К раствору в изопропаноле (50 мл) соединения 355 (836 мг, 1,78 ммоль) добавляли водный 35% раствор формальдегида (760 мг, 8,9 ммоль) и сульфат магния (10 г) и смесь перемешивали при комнатной температуре в течение 2 часов. К реакционному раствору добавляли NaBH4 (340 мг, 9 ммоль) три раза через каждые 24 часа, и раствор перемешивали при комнатной температуре в течение 2 дней и затем кипятили в сосуде с обратным холодильником в течение 1 часа. Реакционному раствору давали возможность охладиться до комнатной температуры, и нерастворимые вещества удаляли фильтрацией, и затем растворитель фильтрата отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 2/5) с получением соединения 370 (683 мг, 84%) в виде бледно-желтого аморфного вещества.
APCI-MS m/z 456[M+H]+
[0491]
Синтез соединения THK-5165
Смесь соединения 370 (676 мг, 1,48 ммоль) и 1M тетра-н-бутиламмонийфторида/тетрагидрофурана (5,0 мл, 5,0 ммоль) перемешивали при комнатной температуре в течение 1 часа. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: хлороформ→хлороформ/метанол=50/1, 24/1) и затем промывали н-гексаном-диизопропиловым эфиром с получением соединения THK-5165 (453 мг, 85%) в виде оранжевого твердого вещества.
точка плавления 79-82°C
1Н ЯМR (400 МГц, ДМСО-d6) δ 2,80 (3H, д, J=4,9 Гц), 2,80 (3H, с), 4,00-4,20 (3H, м), 4,50-4,70 (2H, м), 5,45-5,55 (4H, м), 6,56 (1H, д, J=8,2 Гц), 6,77 (1H, д, J=2,5 Гц), 7,11 (1Н, дд, J=8,8, 2,5 Гц), 7,60-7,70 (3H, м), 7,80 (1H, д, J=8,8 Гц), 7,90 (1H, д, J=8,8 Гц)
ИК (Nujol) 1503, 1462, 1377 см-1
APCI-MS m/z 342[M+H]+
[0492]
Способ синтеза соединения THK-5154
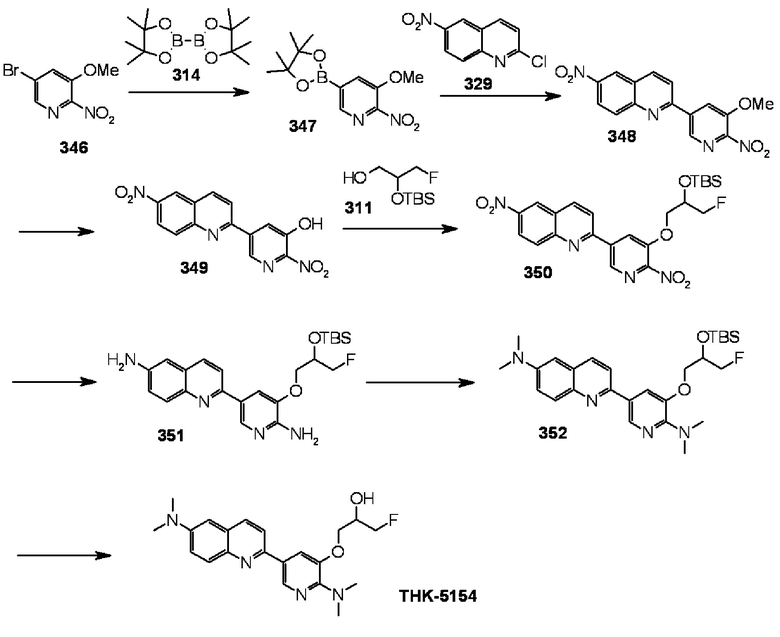
[0493]
Синтез соединения 347
Смесь соединения 346 (1,40 г, 6 моль), соединения 314 (1,53 г, 6 моль), ацетата калия (2,36 г, 24 моль), PdCl2(dppf)·CH2Cl2 (245 мг, 0,3 ммоль) и 1,4-диоксана (25 мл) перемешивали в атмосфере аргона при 100°C в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, и нерастворимые вещества удаляли фильтрацией, и растворитель фильтрата отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: н-гексан/этилацетат=2/1) и затем промывали н-гексаном с получением соединения 347 (1,17 г, 70%) в виде бесцветного твердого вещества.
точка плавления 130-131°C
APCI-MS m/z 281[M+H]+
[0494]
Синтез соединения 348
Смесь соединения 347 (1,10 г, 3,39 ммоль), соединения 329 (840 мг, 4 ммоль), карбоната натрия (850 мг, 8 ммоль), тетракистрифенилфосфина палладия (230 мг, 0,2 ммоль), воды (2 мл) и 1,2-диметоксиэтана (20 мл) перемешивали в атмосфере аргона при 90°C в течение 4 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, и нерастворимые вещества собирали фильтрацией и затем сушили с получением соединения 348 (836 мг, 66%).
точка плавления 253-255°C
APCI-MS m/z 327[M+H]+
[0495]
Синтез соединения 349
Смесь соединения 348 (326 мг, 1 ммоль), хлорида лития (424 мг, 10 ммоль) и гексаметилфосфорного триамида (7 мл) перемешивали в атмосфере аргона при 110°C в течение 19 часов. Реакционному раствору давали возможность охладиться до комнатной температуры и добавляли воду, и раствор подкисляли водным раствором лимонной кислоты и затем экстрагировали этилацетатом. Экстракционную жидкость промывали по очереди водой и насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток промывали диизопропиловым эфиром с получением соединения 349 (312 мг, 100%).
APCI-MS m/z 313[M+H]+
[0496]
Синтез соединения 350
К смеси соединения 349 (680 мг, 2,18 ммоль), соединения 311 (680 мг, 3,26 ммоль), трифенилфосфина (973 мг, 3,71 ммоль) и тетрагидрофурана (20 мл) добавляли по каплям раствор диизопропилазодикарбоксилата (750 мг, 3,71 ммоль) в тетрагидрофуране (2 мл) при охлаждении льдом и перемешивании, с последующим перемешиванием при комнатной температуре в течение 20 часов. Реакционный раствор очищали колоночной флэш-хроматографией (элюирующий растворитель: этилацетат/н-гексан=1/9-1/5) и затем промывали диизопропиловым эфиром с получением соединения 350 (820 мг, 75%) в виде серовато-белого твердого вещества.
точка плавления 167-168°C
APCI-MS m/z 503[M+H]+
[0497]
Синтез соединения 351
Смесь соединения 350 (510 мг, 1,01 ммоль), Fe (503 мг), NH4Cl (325 мг, 6,09 ммоль) и воды (3 мл)-этанола (15 мл) перемешивали при 70-75°C в течение 1,5 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, и затем разбавляли этилацетатом. Нерастворимые вещества удаляли фильтрацией целитом, и фильтрат промывали по очереди аммиачной водой и насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: хлороформ, хлороформ/метанол=40/1) с получением соединения 351 (385 мг, 86%) в виде оранжевого твердого вещества.
точка плавления 139-141°C
APCI-MS m/z 443[M+H]+
[0498]
Синтез соединения 352
К смеси соединения 351 (520 мг, 1,17 ммоль), водного 36% раствора формальдегида (1,95 г, 23,4 ммоль) и этанола (30 мл)-уксусной кислоты (3 мл) небольшими порциями добавляли пиколин-борановый комплекс (751 мг, 7,02 ммоль), с последующим перемешиванием при комнатной температуре в течение 4 часов. Реакцию завершали добавлением водного 36% раствора формальдегида (3 г, 27,8 ммоль), уксусной кислоты (1 мл) и пиколин-боранового комплекса (1,05 г, 9,82 ммоль). Реакционный раствор делали основным концентрированной аммиачной водой при охлаждении льдом и затем экстрагировали этилацетатом. Экстракционную жидкость промывали по очереди водой и насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/5) с получением соединения 352 (430 мг, 74%) в виде желтого твердого вещества.
APCI-MS m/z 499[M+H]+
точка плавления 136-137°C
[0499]
Синтез соединения THK-5154
Смесь соединения 352 (300 мг, 0,68 ммоль) и 48% HBr (3 мл) перемешивали при комнатной температуре в течение 10 минут. Реакционный раствор разбавляли водой, и раствор делали основным концентрированной аммиачной водой и затем экстрагировали этилацетатом. Экстракционную жидкость промывали по очереди водой и насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток промывали этилацетатом с получением соединения THK-5154 (220 мг, 84%) в виде желтого твердого вещества.
точка плавления 154-155°C
1H ЯМR (400 МГц, CDCl3) δ 3,06 (6H, с), 3,10 (6H, с), 3,26 (1Н, уш. с), 4,24-4,32 (3H, м), 4,51-4,60 (1Н, м), 4,64-4,72 (1Н, м), 6,81 (1H, д, J=2,7 Гц), 7,37 (1Н, дд, J=9,3, 2,7 Гц), 7,70 (1H, д, J=8,8 Гц), 7,98 (1H, д, J=8,8 Гц), 7,98 (1H, д, J=9,3 Гц), 8,04 (1H, д, J=1,9 Гц), 8,53 (1H, д, J=1,9 Гц).
ИК (Nujol) 1618, 1588 см-1
APCI-MS m/z 385[M+H]+
[0500]
Способ синтеза соединения THK-5166
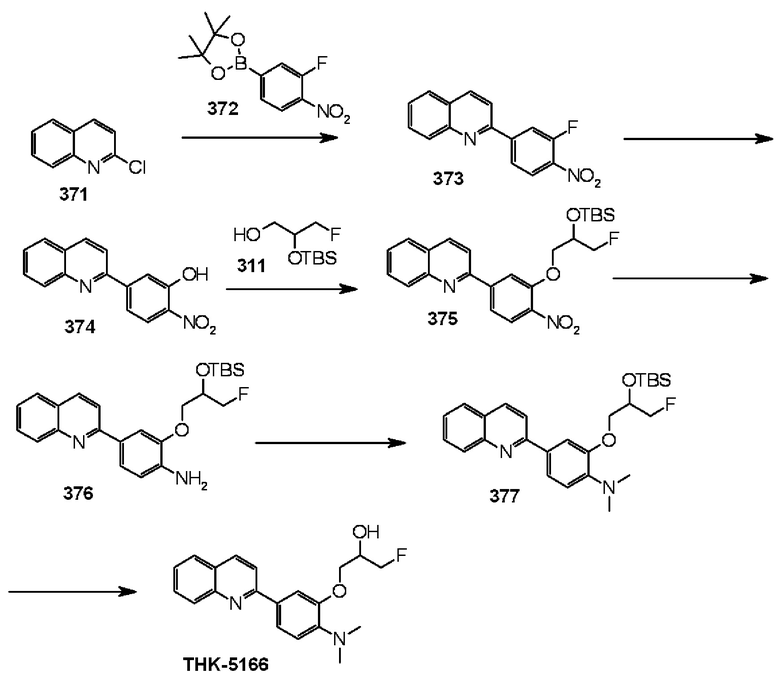
[0501]
Синтез соединения 373
Смесь соединения 371 (654 мг, 4 ммоль), соединения 372 (1,28 г, 4,8 ммоль), 2M карбоната натрия (4 мл, 8 ммоль), тетракистрифенилфосфина палладия (231 мг, 0,2 ммоль) и 1,2-диметоксиэтана (7 мл) перемешивали в атмосфере аргона при 90°C в течение 16 часов. Реакционному раствору давали возможность охладиться до комнатной температуры, и растворитель отгоняли. Добавляли хлороформ, и нерастворимые вещества удаляли фильтрацией. Хлороформный слой фильтрата отделяли и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан, хлороформ/н-гексан=1/4, 2/3, 3/2) и затем промывали н-гексаном с получением соединения 373 (986 мг, 92%) в виде бледно-желтого твердого вещества.
APCI-MS m/z 269[M+H]+
[0502]
Синтез соединения 374
К раствору в N,N-диметилформамиде (15 мл) соединения 373 (982 мг, 3,6 ммоль) и 2-метансульфонилэтанола (679 мг, 5,47 ммоль) добавляли 60% NaH (438 мг, 10,9 ммоль) при комнатной температуре при перемешивании и смесь перемешивали при комнатной температуре в течение 2 часов. После того как реакционный раствор подкисляли добавлением воды и 10% водного раствора хлористоводородной кислоты, раствор делали основным водным насыщенным раствором гидрокарбоната натрия и затем экстрагировали этилацетатом. Экстракционную жидкость промывали насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток перекристаллизовывали из этилацетата/н-гексана с получением соединения 374 (778 мг, 80%) в виде бледно-коричневых кристаллов.
APCI-MS m/z 267[M+H]+
[0503]
Синтез соединения 375
К смеси соединения 374 (773 мг, 2,9 ммоль), соединения 311 (844 мг, 4,0 ммоль), трифенилфосфина (1,21 г, 4,6 ммоль) и тетрагидрофурана (10 мл) добавляли по каплям раствор в тетрагидрофуране (5 мл) диизопропилазодикарбоксилата (936 мг, 4,6 ммоль) при охлаждении льдом и перемешивании, с последующим перемешиванием при комнатной температуре в течение 16 часов. Растворитель реакционного раствора отгоняли при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан, этилацетат/н-гексан=1/4) и затем промывали н-гексаном с получением соединения 375 (1,10 г, 83%) в виде бесцветного твердого вещества.
APCI-MS m/z 457[M+H]+
[0504]
Синтез соединения 376
Смесь соединения 375 (1,09 г, 2,4 ммоль), 10% Pd-C (влажность примерно 50%; 200 мг), формиата аммония (1,50 г, 24 ммоль) и метанола (20 мл)-тетрагидрофурана (10 мл) перемешивали в атмосфере аргона при комнатной температуре в течение 1 часа. Катализатор удаляли фильтрацией, и растворитель фильтрата отгоняли при пониженном давлении. К остатку добавляли воду, и раствор экстрагировали из хлороформа. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении с получением соединения 376 (1,02 г, 100%) в виде желтого маслянистого вещества.
APCI-MS m/z 427[M+H]+
[0505]
Синтез соединения 377
К смеси соединения 376 (1,02 г, 2,4 ммоль), водного 35% раствора формальдегида (1,0 г, 11,7 ммоль) и метанола (20 мл)-уксусной кислоты (2 мл) небольшими порциями добавляли пиколин-борановый комплекс (385 мг, 3,6 ммоль) в условиях охлаждения льдом и перемешивания, с последующим перемешиванием при комнатной температуре в течение 2,5 часов. К реакционному раствору добавляли водный 35% раствор формальдегида (0,5 г, 5,8 ммоль) и пиколин-борановый комплекс (260 мг, 2,4 ммоль), и раствор дополнительно перемешивали при комнатной температуре в течение 16 часов. Растворитель отгоняли из реакционного раствора при пониженном давлении, и к остатку добавляли водный насыщенный раствор гидрокарбоната натрия, и затем раствор экстрагировали хлороформом. Экстракционную жидкость сушили, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан, этилацетат/н-гексан=1/20, 1/12) и затем промывали холодным н-гексаном с получением соединения 377 (827 мг, 74%) в виде бесцветного твердого вещества.
APCI-MS m/z 455[M+H]+
[0506]
Синтез соединения THK-5166
Смесь соединения 377 (802 мг, 1,76 ммоль) и 1M тетра-н-бутиламмонийфторида/тетрагидрофурана (5,0 мл, 5,0 ммоль) перемешивали при комнатной температуре в течение 1 часа. Растворитель реакционного раствора отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: хлороформ→хлороформ/метанол=50/1), очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: н-гексан→этилацетат/н-гексан=1/1, 3/2) и затем промывали н-гексаном с получением соединения THK-5166 (453 мг, 75%) в виде бледно-желтого твердого вещества.
точка плавления 81-84°C
1H ЯМR (400 МГц, ДМСО-d6) δ 2,86 (6H, с), 4,10-4,20 (3H, м), 4,40-4,70 (2H, м), 5,54 (1Н, уш. с), 7,04 (1Н, уш. д, J=7,5 Гц), 7,54-7,58 (1Н, т подобный), 7,74-7,78 (1Н, т подобный), 7,83 (1H, дд, J=8,5, 1,8 Гц), 7,87 (1H, д, J=1,8 Гц), 7,97 (1H, д, J=7,6 Гц), 8,05 (1Н, д, J=8,5 Гц), 8,14 (1Н, д, J=8,8 Гц), 8,40 (1H, д, J=8,8 Гц)
ИК (Nujol) 1595, 1497, 1457, 1436 см-1
APCI-MS m/z 341[M+H]+
[0507]
Способ синтеза соединения THK-5167
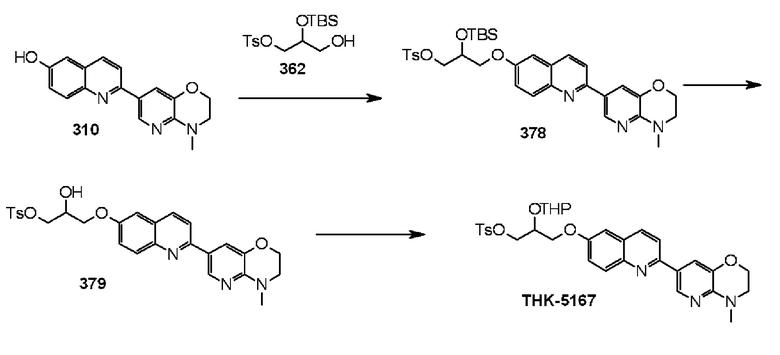
[0508]
Синтез соединения 378
К смеси соединения 310 (452 мг, 1,54 ммоль), соединения 362 (560 мг, 1,55 ммоль), трифенилфосфина (630 мг, 2,4 ммоль) и тетрагидрофурана (20 мл) добавляли по каплям раствор в тетрагидрофуране (2 мл) диизопропилазодикарбоксилата (485 мг, 2,4 ммоль) при охлаждении льдом и перемешивании, с последующим перемешиванием при той же температуре в течение 20 минут и дополнительном перемешивании при комнатной температуре в течение 16 часов. К реакционному раствору добавляли соединение 362 (250 мг, 0,69 ммоль), трифенилфосфин (320 мг, 1,27 ммоль) и диизопропилазодикарбоксилат (250 мг, 1,24 ммоль), и раствор дополнительно перемешивали при комнатной температуре в течение 20 часов. Реакционный раствор очищали колоночной флэш-хроматографией на NH силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/4, 1/3, 1/2, этилацетат, хлороформ/метанол=10/1) и затем промывали н-гексаном/этилацетатом с получением соединения 378 (683 мг, 70%).
точка плавления 135-138°C
[0509]
Синтез соединения 379
Смесь соединения 378 (700 мг, 1,1 ммоль), тетрагидрофурана (15 мл), воды (5 мл) и трифторуксусной кислоты (10 мл) перемешивали при комнатной температуре в течение 16 часов. Реакционный раствор разбавляли этилацетатом, и раствор промывали по очереди водным насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 1/1, 2/1) с получением соединения 379 (490 мг, 85%) в виде твердого вещества.
точка плавления 156-158°C
APCI-MS m/z 522[M+H]+
[0510]
Синтез соединения THK-5167
К раствору в метиленхлориде (30 мл)-тетрагидрофуране (20 мл) соединения 379 (480 мг, 0,92 ммоль) и 3,4-дигидро-2H-пирана (1,55 г, 18,4 ммоль) добавляли моногидрат паратолуолсульфоновой кислоты (200 мг, 1,16 ммоль) и смесь перемешивали при комнатной температуре в течение 78 часов. Реакционный раствор разбавляли этилацетатом (200 мл), и раствор промывали по очереди водным насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором и сушили, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (элюирующий растворитель: этилацетат/н-гексан=1/2, 2/3, 1/1) и затем перекристаллизовывали из этилацетата с получением соединения THK-5167 (380 мг, 68%) в виде бледно-желтого твердого вещества.
точка плавления 153-155°C
1H ЯМR (400 МГц, CDCl3) δ 1,45-1,62 (4H, м), 1,66-1,84 (2H, м), 2,34, 2,35 (3H, каждый с), 3,21 (3H, с), 3,44-3,55 (3H, м), 3,83, 3,92 (1Н, каждый м), 4,06-4,39 (7H, м), 4,74, 4,83 (1H, каждый уш. т), 6,98, 7,00 (1H, каждый д, J=2,7 Гц), 7,18, 7,19 (1Н, каждый дд, J=9,1, 2,5 Гц), 7,227 (1H, д, J=8,2 Гц), 7,234 (1Н, д, J=8,2 Гц), 7,73, 7,74 (1Н, каждый д, J=9,4 Гц), 7,75 (1Н, уш. д, J=8,3 Гц), 7,77 (1H, уш. д, J=8,3 Гц), 7,836, 7,838 (1Н, каждый д, J=1,8 Гц), 7,95 (1H, д, J=9,4 Гц), 8,00, 8,01 (1Н, каждый д, J=8,8 Гц), 8,486, 8,489 (1H, каждый д, J=1,8 Гц)
ИК (Nujol) 1621, 1598 см-1
APCI-MS m/z 606[M+H]+
[0511]
Способы синтеза соединений THK-5168, THK-5170, THK-5171, THK-5172, THK-5173, THK-5174, THK-5175, THK-5176, THK-5179, THK-5181 и THK-5182
Каждый способ синтеза такой же, как любой из указанных выше способов синтеза.
Пример 2
[0512]
Синтез с мечением [18F] THK-5035
18F- синтезировали облучением [18O] H2O, имеющей чистоту изотопа 95 или более, протонным пучком 12 МэВ, ускоренным циклотроном HM12 (выпускаемым компанией Sumitomo Heavy Industries, Ltd.). Затем его раствор пропускали через анионообменную смолу (AG1-X8), посредством этого, захватывая 18F- на смолу с последующим элюированием 33 мМ раствором K2CO3. После переноса этого водного содержащего 18F- раствора K2CO3 (200 мкл, 3,5 GBq (гигабеккерелей) в коричневый флакон добавляли криптофикс 222 (16 мг) и ацетонитрил (1,5 мл), и распыляли газообразный He при нагревании в масляной бане (110°C), и затем ацетонитрил полностью удаляли при азеотропной отгонке воды. Кроме того, повторяли операцию добавления ацетонитрила (1 мл) и удаления ацетонитрила таким же образом в условиях нагревания, посредством этого, превращая состояние внутри флакона в по существу лишенное влаги состояние. Добавляли раствор ДМСО (0,7 мл), содержащий соединение THK-5039 (4 мг), в качестве предшественника метки, с последующим нагреванием и перемешиванием в масляной бане (110°C) в течение 10 минут. Затем реакционный раствор разбавляли дистиллированной водой (7 мл) и загружали в кассету Sep-Pack tC18 (выпускаемую компанией Waters), и после промывания кассеты дистиллированной водой неочищенный продукт элюировали этанолом. Часть этанолового раствора разбавляли дистиллированной водой и подвергали полупрепаративной высокоэффективной жидкостной хроматографии (колонка: YMC-Pack Pro C18 RS (10×250 мм), подвижная фаза: MeCN/20 мМ NaH2PO4 =60/40, скорость потока: 5,0 мл/мин) и затем подавали происходящий из [18F] THK-5035 радиоактивный пик (121 MBq (мегабеккерелей), без коррекции затухания), который элюировали в пределах примерно от 14 до 15 минут. После разбавления фракции дистиллированной водой, [18F] THK-5035 подвергали твердофазной экстракции, используя кассету Sep-Pak tC18, и затем элюировали этанолом. В тесте авторадиографии этаноловый раствор использовали после соответствующего разбавления. В тесте биораспределения Полисорбат 80 добавляли к этаноловому раствору, и после отгонки этанола испарителем этанола радиоактивный остаток, содержащий [18F] THK-5035 во флаконе, растворяли в физиологическом солевом растворе и затем использовали в качестве инъекционного раствора в тесте оценки миграции в мозг.
[0513]
Тест окрашивания соединений по настоящему изобретению на срезах ткани мозга пациентов с болезнью Альцгеймера
Процедура теста окрашивания соединений по настоящему изобретению на срезе ткани мозга пациентов, страдающих болезнью Альцгеймера, описана ниже:
(1) Заявители использовали образцы ткани мозга височной доли или гиппокампа от пациентов, у которых определенно была диагностирована патология в виде болезни Альцгеймера. Образцы получали из медицинского института Чойу, Госпиталь Фукушимура, который является научно-исследовательским институтом, работающим в сотрудничестве с заявителями, и согласие на использование в исследовательских целях было получено от семей умерших пациентов (Одобрение Комитета по Этике № 20 госпиталя Фукушимура).
(2) Делали срезы залитой в парафин ткани мозга толщиной 6 или 8 мкм, раскладывали на предметное стекло и сушили. Парафинизированный срез ткани мозга депарафинировали промыванием ксиленом в течение 10 минут×2, 100% этанолом в течение 5 минут×2, 90% этанолом в течение 5 минут и проточной водой в течение 10 минут в указанном порядке.
(3) В качестве предварительной обработки для окрашивания соединений по настоящему изобретению, выполняли обработку липофусцином для удаления аутофлуоресценции. Во-первых, парафинизированный срез ткани мозга погружали в 0,25% раствор KMnO4 на 20 минут. Его промывали PBS (солевым раствором с фосфатным буфером) дважды в течение 2 минут, погружали в 0,1% раствор K2S2O5/щавелевой кислоты приблизительно на 5 минут и дополнительно промывали PBS 3 раза в течение 2 минут.
(4) Из 100 мкМ раствора соединений по настоящему изобретению, растворенного в 50% этаноле, приблизительно 150 мкл капали на срез для взаимодействия в течение 10 минут. После 5-кратного погружения в водопроводную воду, его обрабатывали реагентом Flour Save Reagent (Calbiochem) и исследовали флуоресцентным микроскопом (Nikon, ECLIPSE 80i). Изображения получали цифровой камерой (Nikon, Dxin1200F или Photometrics, Cool SNAP ES).
[0514]
Результаты указанного выше теста окрашивания соединений по настоящему изобретению иллюстрируются на фиг. 5-20. Было обнаружено, что все из соединений THK-5035, THK-5038, THK-5058, THK-5064, THK-5065, THK-5066, THK-5071, THK-5077, THK-5078, THK-5079, THK-5080, THK-5081, THK-5082, THK-5087, THK-5088, THK-5089, THK-5091, THK-5092, THK-5097, THK-5098, THK-5059, THK-5075, THK-5076, THK-5086, THK-5100, THK-5105, THK-5106, THK-5107, THK-5112, THK-5116, THK-5117 и THK-932 специфически и селективно связываются с нейроволоконными сплетениями в срезах мозга пациентов с болезнью Альцгеймера, и что соединение формулы (I) по настоящему изобретению обладает высокой специфичностью в отношении тау (фиг. 5-20).
[0515]
Результаты указанного выше теста окрашивания соединений по настоящему изобретению иллюстрируются на фиг. 25-29. Было обнаружено, что все из соединений THK-5136, THK-5153, THK-5157, THK-5128, THK-5147, THK-5155, THK-5156, THK-5164 и THK-5154 специфически и селективно связываются с нейроволоконными сплетениями в срезах мозга пациентов с болезнью Альцгеймера, и что соединение формулы (I) по настоящему изобретению обладает высокой специфичностью в отношении тау (фиг. 25-29).
[0516]
Тест авторадиографии
После депарафинирования залитого в парафин среза ткани мозга пациентов с болезнью Альцгеймера, его погружали в PBS на 10 минут. Приблизительно 400 мкКи/мл [18F] BF-227 и [18F] THK-5035 капали на срез для взаимодействия в течение 10 минут при комнатной температуре. Его погружали в дистиллированную воду на 2 минуты, слегка встряхивали 50% EtOH в течение 2 минут, снова погружали в дистиллированную воду на 2 минуты и сушили в устройстве для заливки парафина. Срез контактировали с визуализирующей пластиной и отверждали в течение ночи, и на следующий день изображения считывали прибором BAS5000 (Fujifilm Corporation). Кроме того, проводили иммуноокрашивание серийных срезов тиофлавином-S и антителом к тау (AT8).
[0517]
На фиг. 21 иллюстрируются авторадиографические изображения [18F] BF-227 и [18F] THK-5035, изображения при окрашивании тиофлавином-S (TF-S) на серийных срезах и окрашивании антителом к тау (Tau). На авторадиографических изображениях области, где накапливалось [18F] THK-5035 (область, окруженная прямоугольниками), не проявилось накопление [18F] BF-227, показывающее высокий аффинитет к амилоиду β. В этой области также наблюдались многочисленные структуры, положительные по иммуноокрашиванию антителом к тау. По морфологическим изображениям при визуализации с окрашиванием тиофлавином-S, который окрашивает нейроволоконные сплетения, имеются веские свидетельства того, что структуры, наблюдаемые в этой области, представляют собой нейроволоконные сплетения. Из представленных выше результатов подтверждается, что [18F] THK-5035 проявляет более высокий аффинитет к нейроволоконным сплетениям (белку тау), чем к амилоиду β.
[0518]
Оценка перехода меченых соединений в мышиный мозг
Физиологический солевой раствор, содержащий [18F] THK-5035, вводили в хвостовую вену самцов мышей ICR (в возрасте 6-7 недель). Путем учета накопления радиоактивности в ткани мозга и плазматической ткани через 2 минуты после введения, заявители оценивали захват мозговой тканью меченых соединений.
При оценке накопления радиоактивности, в качестве показателя использовали отношение радиоактивности на единицу массы ткани, подлежащей оценке всех видов введенной радиоактивности (% инъецированной дозы/г ткани; %ID/г). Для измерения радиоактивности использовали гамма-счетчик (1480 WIZARD, PerkinElmer, Inc.). В тесте меченые соединения вводили в хвостовую вену, и через 2 минуты у мышей выполняли смещение позвонков шейного отдела в условиях эфирного наркоза. Затем сразу брали кровь из сердца и иссекали весь мозг (включая мозжечок и ствол головного мозга). Затем измеряли радиоактивность и массу ткани каждого образца, и эти данные использовали для расчета %ID/г.
[0519]
Таблица 3 иллюстрирует результаты этого теста оценки.
[0520]
Величина 0,5% ID/г или более считается достаточной для указания на прохождение в мозг меченых соединений для нацеливания PET (позитронно-эмиссионной томографии) или SPECT (одиночнофотонной эмиссионной томографии) на центральную нервную систему. С этой точки зрения, было обнаружено, что соединения формулы (I) по настоящему изобретению, меченные [18F], представляют собой меченные [18F] соединения, имеющие высокий захват тканью мозга.
[0521]
Синтез метки, [18F] THK-5105
[18O]-H2O с чистотой изотопа 98% или выше облучали пучком протонов 12 МэВ, которые ускоряли циклотроном HM 12 (Sumitomo Heavy Industries, Ltd.), для получения 18F-. В последующем, полученный раствор пропускали через анионообменную смолу (AG 1-X8) для захвата 18F- смолой, которую элюировали 33 мМ раствором K2CO3. В янтарный флакон помещали 200 мкл содержащего 18F- водного раствора K2CO3 (3,13 GBq) и добавляли Криптофикс 222 (16 мг) и ацетонитрил (2,3 мл). Распыляли газообразный He при нагревании в масляной бане (110°C) с тем, чтобы полностью удалить ацетонитрил азеотропным выпариванием воды. Добавляли другой ацетонитрил (1,5 мл), и ацетонитрил удаляли при нагревании аналогичным образом. Эти процедуры повторяли дважды для обезвоживания внутренней среды флакона. Во флакон добавляли раствор ДМСО (0,70 мл) предшественника метки THK-5121 (3,0 мг), и смесь нагревали и перемешивали в течение 10 минут в масляной бане (110°C). После этого к реакционному раствору добавляли хлористоводородную кислоту (2M, 0,2 мл), и дополнительную реакцию проводили при 110°C в течение 3 минут. Затем реакционный раствор разбавляли раствором ацетата калия (4M, 0,1 мл) и дистиллированной водой (7,0 мл) и загружали на кассету Sep-Pak tC18 (Waters), которую затем промывали дистиллированной водой с последующим элюированием неочищенного продукта этанолом. Элюированную этанолом фракцию с самой высокой радиоактивностью разбавляли дистиллированной водой и подвергали полупрепаративной высокоэффективной жидкостной хроматографии, используя колонку Inertsil ODS-4 (10×250 мм), подвижная фаза MeCN/NaH2PO4 (20 мМ) (50/50), и скорость потока 5,0 мл/мин. Брали радиоактивный пик [18F] THK-5105, который элюировали примерно через 18,5 минут (988 MBq, без коррекции на затухание). Анализ взятого образца выявил радиохимическую чистоту 98% или более и удельную радиоактивность 137 GBq/мкмоль (при анализе).
[0522]
Фракцию препаративной ВЭЖХ [18F] THK-5105, которое было синтезировано в соответствии с процедурой синтеза, описанной выше, разбавляли дистиллированной водой и затем подвергали твердофазной экстракции с использованием кассеты Sep-Pak tC18 с последующим элюированием этанолом или ДМСО и соответствующим разбавлением, и использовали в тестах связывания и экспериментах авторадиографии. Для экспериментов по его распределению in vivo (оценке его доставки в мозг) Полисорбат 80 добавляли к элюированной этанолом фракции, из которой этанол удаляли, используя испаритель. [18F] THK-5105, содержащий радиоактивный остаток внутри колбы, растворяли в физиологическом солевом растворе, и полученный раствор использовали в качестве раствора для инъекции.
[0523]
Синтез метки, [18F] THK-5117
[18F] THK-5117 синтезировали в соответствии с описанным выше способом синтеза метки, [18F] THK-5105. При синтезе [18F] THK-5117 синтез проводили, используя 500 мкл содержащего 18F- водный раствор K2CO3 (3,10 GBq), 3,0 мг предшественника метки THK-5119 и MeCN/NaH2PO4 (20 мМ) (45/55), в виде подвижной фазы при полупрепаративной высокоэффективной жидкостной хроматографии, и был способен дать 770 MBq [18F] THK-5117 (без коррекции на затухание). Как при [18F] THK-5105, получали раствор для инъекций и использовали в экспериментах для оценки его доставки в мозг.
[0524]
Синтез метки, [18F] THK-5125
[18F] THK-5125 синтезировали в соответствии с описанным выше способом метки, [18F] THK-5105. При синтезе соединения [18F] THK-5125, синтез проводили, используя 200 мкл содержащего 18F- водного раствора K2CO3 (3,14 GBq), 3,3 мг предшественника метки THK-5131 и MeCN/NaH2PO4 (20 мМ) (45/55) в качестве подвижной фазы при полупрепаративной высокоэффективной хроматографии, и был способен дать 1,36 GBq [18F] THK-5125 (без коррекции на затухание). Как при [18F] THK-5105, получали раствор для инъекции и использовали в экспериментах для оценки его доставки в мозг.
[0525]
Синтез метки, [18F] FDDNP
В янтарный флакон помещали 300 мкл содержащего 18F- водного раствора K2CO3 (3,87 GBq), который получали таким же образом, как при [18F] THK-5105, и добавляли Криптофикс 222 (16 мг) и ацетонитрил (2,3 мл). Газообразный He распыляли при нагревании флакона и масляной бане (110°C) с тем, чтобы полностью удалить ацетонитрил азеотропным выпариванием воды. Добавляли еще ацетонитрил, и ацетонитрил удаляли при нагревании аналогичным образом. Эти процедуры повторяли три раза для обезвоживания внутренней среды флакона. Во флакон добавляли раствор в ацетонитриле (0,70 мл) предшественника метки (2,7 мг), который имеет группу тозила в качестве уходящей группы, присоединенной в положении фтора в FDDNP (Jie Liu et al., Molecular Imaging and Biology (2007), vol. 9, pp.6-16). Смесь нагревали и перемешивали в течение 10 минут в ледяной бане (110°C), в негерметизированном состоянии и с добавлением 0,1 мл ацетонитрила каждую вторую минуту. После этого реакционный раствор охлаждали воздухом, разбавляли дистиллированной водой (7,0 мл) и загружали на кассету Sep-Pak tC18 (Waters), которую затем промывали дистиллированной водой, с последующим элюированием неочищенного продукта этанолом. Элюированную этанолом фракцию с самой высокой радиоактивностью разбавляли дистиллированной водой и подвергали полупрепаративной высокоэффективной жидкостной хроматографии, используя колонку Inertsil ODS-4 (10×250 мм), подвижную фазу MeCN/NaH2PO4 (20 мМ) (60/40) и скорость потока 5,0 мл/мин. Брали радиоактивный пик [18F] FDDNP, который элюировали примерно через 19,5 минут (814 MBq, без коррекции на затухание). Как и при [18F] THK-5105, получали раствор для инъекции и использовали в экспериментах для оценки ее доставки в мозг.
[0526]
Оценка доставки в мозг [18F] THK-5105, [18F] THK-5117 и [18F] THK-5125 у мышей
Физиологический солевой раствор, содержащий [18F] THK-5105, [18F] THK-5117 или [18F] THK-5125, вводили в хвостовую вену самцов мышей ICR (в возрасте 6-7 недель), и доставку в мозг этих метящих соединений оценивали по накоплению радиоактивности в тканях мозга и плазме через 2 минуты после инъекции.
В качестве показателя при оценке радиоактивного накопления использовали процентную радиоактивность на единицу массы мозга относительно общей введенной радиоактивности (% Инъецированной дозы/г ткани; %ID/г). Для измерения радиоактивности использовали гамма-счетчик (1480 WIZARD, Perkin Elmer). Экспериментальные процедуры были следующие. Соединение метки вводили в хвостовую вену мышей. Через две минуты каждую мышь подвергали смещению позвонков шейного отдела под эфирным наркозом, и немедленно отделяли весь мозг, включая мозжечок и ствол мозга. Измеряли радиоактивность и массу ткани образца, и их данные использовали для расчета величин %ID/г для меченого соединения.
[0527]
В таблице 4 показаны величины %ID/г через 2 минуты и через 60 минут после введения, и величины %ID/г через 2 минуты после введения, деленные на величины через 60 минут после введения. Меченые зонды, которые нацелены на предназначенную патогистологию в мозге, должны быстро включаться в мозг и быстро вымываться из патогистологических областей, которые не являются их предназначенной патогистологией. [18F] THK-5105, [18F] THK-5117 и [18F] THK-5125 проявили достаточное внутримозговое содержание через 2 минуты после введения, и доли их внутримозгового содержания через 2 минуты после введения и 60 минут после введения были больше, чем величины для [18F] BAY94-9172 и [18F] AV-45, которые известны как зонды для визуализации амилоида, и величина для [18F] FDDNP. По этим результатам оказалось, что [18F] THK-5105, [18F] THK-5117 и [18F] THK-5125 превосходили по доставке в мозг и вымыванию из него. (Данные [18F] BAY94-9172 были взяты из публикации Zhang et al., Nuclear Medicine and Biology, 32, 799-809, 2005, и данные [18F] AV-45 - из публикации Choi et al., J Nucl Med, 50, 1887-1894, 2009.)
[0528]
Эксперименты авторадиографии с использованием [18F] THK-5105, [18F] THK-5117 и [18F] THK-5125
Залитые в парафин срезы мозга от пациентов с болезнью Альцгеймера депарафинировали и затем погружали в PBS на 10 минут. Приблизительно 400 мкКи/мл каждой из [18F] THK-5105, [18F] THK-5117 и [18F] THK-5125 добавляли по каплям в депарафинированные срезы и давали возможность взаимодействовать в течение 10 минут при комнатной температуре. После этого срезы погружали в дистиллированную воду на 2 минуты, слегка встряхивали в 50% EtOH в течение 2 минут, снова погружали в дистиллированную воду на 2 минуты и сушили в устройстве, распределяющем парафин по площади. Срез оставляли на ночь в контакте с пластиной для визуализации. На следующий день изображение считывали прибором BAS 5000 (Fujifilm Corporation). Кроме того, проводили иммуноокрашивание прилегающих срезов анти-фосфорилированным антителом к тау (AT8).
[0529]
На фиг. 22 показано изображение авторадиографии [18F] THK-5105 среза гиппокампа и изображения окрашивания немеченой THK-5105 и окрашивания анти-фосфорилированным антителом к тау (AT8) прилегающих к нему срезов. На авторадиографическом изображении зоны, где наблюдалось накопление [18F] THK-5105, были обнаружены в изобилии в структурах, которые были положительными (в отношении pTau), иммуноокрашиванием анти-фосфорилированным антителом к тау. Также, на их соответствующих изображениях с бόльшим увеличением изображение авторадиографии [18F] THK-5105 хорошо соответствовало изображению иммуноокрашивания анти-фосфорилированным антителом к тау, и, кроме того, изображение окрашивания немеченой THK-5105 хорошо соответствовало изображению иммуноокрашивания анти-фосфорилированным антителом к тау, включая их морфологические изображения.
[0530]
На фиг. 23 показано изображение авторадиографии [18F] THK-5105, [18F] THK-5117 и [18F] THK-5125 срезов латеральной височной коры и медиальной височной коры. Наблюдали накопление [18F] THK-5105, [18F] THK-5117 и [18F] THK-5125 в латеральной височной коре и медиальной височной коре, которые известны как зоны, где происходит продукция тау на поздних стадиях болезни Альцгеймера.
[0531]
По этим результатом было установлено, что [18F] THK-5105, [18F] THK-5117 и [18F] THK-5125 имеют высокий аффинитет к нейроволоконным сплетениям (белок тау).
[0532]
Оценка связывания с агрегатами белка тау
В отношении связывания визуализирующих тау зондов с агрегатами белка тау, проводили тест связывания и оценивали с использованием конструкта K18-ΔK280, который содержал структуру с 4 повторами, участвующую в образовании бета-листовых структур в белке htau40 protein. Для K18-ΔK280 (Martin von Bergen et al., THE JOURNAL OF BIOLOGICAL CHEMISTRY (2001), vol. 276, pp. 48165-48174; M. Goedert et al., NEURON (1989), vol. 3, pp.519-526), искусственный фрагмент гена, который был получен на основании его аминокислотной последовательности, клонировали в вектор pET-3a, полученную плазмиду использовали для трансформации компетентных клеток Escherichia coli BL21(DE3) для экспрессии белка, и рекомбинантную клетку Escherichia coli культивировали для экспрессии K18-ΔK280. Экспрессированную K18-ΔK280 очищали в соответствии со способом, описанным в литературе (Stefan Barghorn et al., Methods in Molecular Biology, vol. 299, pp.35-51). В тесте связывания были получены и использованы агрегаты очищенной K18-ΔK280.
[0533]
Агрегаты K18-ΔK280 получали приготовлением 20 мкМ PBS (pH 7,4) и инкубацией K18-ΔK280 в PBS при 37°C в течение 4 дней. Состояние, при котором образовывались бета-листовые структуры в агрегатах K18-ΔK280, подтверждали тестом связывания флуоресцентного тиофлавина S. Раствор агрегатов разбавляли до 400 нМ в аналитическом буфере (PBS, 0,1% BSA (бычий сывороточный альбумин)) и использовали в тесте связывания.
[0534]
Тест насыщения связывания THK-5105
В тесте связывания [18F] THK-5105 реакционные растворы получали так, чтобы агрегаты K18-ΔK280 находились в конечной концентрации 200 нМ в реакционной системе, а [18F] THK-5105 находилось в конечных концентрациях от 0,1 до 100 нМ, и инкубировали при комнатной температуре в течение одного часа. После инкубации [18F] THK-5105, связывающееся с агрегатами K18-ΔK280, и THK-5105, не связывающееся с агрегатами K18-ΔK280, отделяли, используя фильтровой планшет MultiScreen HTS (96 лунок, Millipore Corporation), и фильтр промывали аналитическим буфером (200 мкл, 3 раза). Общее связывание [18F] THK-5105 с агрегатами K18-ΔK280 рассчитывали по радиоактивности фильтра, которую измеряли на гамма-счетчике (AccuFLEX γ7000, Aloka). Неспецифическое связывание определяли добавлением немеченого THK-5105 (в конечной концентрации 2 мкМ) к реакционной системе и выполнением экспериментов аналогичным образом. Данные общего связывания и неспецифического связывания использовали и анализировали, используя аналитическое программное обеспечение GraphPad prism (версия 5), которое дало низкую величину константы диссоциации Kd 3,9 нМ, указывая на то, что THK-5105 проявляло высокий аффинитет связывания с агрегатами K18-ΔK280.
[0535]
Тест конкурентного связывания с использованием [18F] THK-5105 в качестве радиоактивного конкурента
Для визуализирующих тау зондов, отличных от THK-5105, связывание с агрегатами K18-ΔK280 оценивали тестом конкурентного связывания с использованием [18F] THK-5105 в качестве радиоактивного конкурента. В тесте связывания [18F] THK-5105 (в конечной концентрации 1,76 нМ) и тестируемое соединение (в конечной концентрации от 0,1 до 1000 нМ) сосуществовали в реакционной системе, к которой затем добавляли агрегаты K18-ΔK280 (в конечной концентрации 200 нМ), и смеси инкубировали при комнатной температуре в течение одного часа. Радиоактивность связывания [18F] THK-5105 с агрегатами определяли аналогичным образом в тесте насыщения связывания THK-5105. Процентное связывание [18F] THK-5105 в отсутствие каждого тестируемого соединения устанавливали на 100% и определяли процентные величины связывания [18F] THK-5105 при различных концентрациях каждого из тестируемых соединений. По этим данным рассчитывали константы ингибирования (Ki) соответствующих тестируемых соединений с использованием аналитического программного обеспечения GraphPad prism (версия 5). Результаты показаны в таблице 5 и на фиг. 24. Любое из тестируемых соединений проявляло высокий аффинитет связывания с агрегатами K18-ΔK280.
Промышленная применимость
[0536]
Соединения по настоящему изобретению очень полезны, например, при раннем выявлении, лечении и профилактике нейроволоконных сплетений, включая болезнь Альцгеймера, и могут использоваться в областях получения диагностических средств и диагностических наборов для этих заболеваний, областях получения лекарственных средств и профилактических средств для этих заболеваний, исследованиях этих заболеваний и тому подобных областях.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ЦИКЛОГЕКСАНОВОЕ ПРОИЗВОДНОЕ, ЕГО ПРОЛЕКАРСТВО И ЕГО СОЛЬ И СОДЕРЖАЩЕЕ ИХ ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ОТ ДИАБЕТА | 2005 |
|
RU2394015C2 |
| НОВОЕ ПРОИЗВОДНОЕ ФЕНИЛПИРРОЛА | 2009 |
|
RU2470917C2 |
| ХИНОЛИЛПИРРОЛПИРИМИДИЛЬНОЕ КОНДЕНСИРОВАННОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2013 |
|
RU2581039C1 |
| ПИРАЗОЛОХИНОЛИНОВОЕ ПРОИЗВОДНОЕ | 2012 |
|
RU2605096C2 |
| НОВОЕ ПРОИЗВОДНОЕ ФЕНОЛА | 2010 |
|
RU2536689C2 |
| НОВОЕ ПРОИЗВОДНОЕ ГЛЮЦИТОЛА, ЕГО ПРОЛЕКАРСТВО И ЕГО СОЛЬ И СОДЕРЖАЩИЙ ИХ ТЕРАПЕВТИЧЕСКИЙ АГЕНТ ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА | 2005 |
|
RU2386631C2 |
| ПРОИЗВОДНЫЕ СПИРОКЕТАЛЕЙ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА ПРОТИВ ДИАБЕТА | 2006 |
|
RU2416617C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОПИРАНА В КАЧЕСТВЕ ПРОТИВОАРИТМИЧЕСКИХ АГЕНТОВ | 2005 |
|
RU2380370C2 |
| ПРОЛЕКАРСТВЕННАЯ ФОРМА ЗАМЕЩЕННОГО ПОЛИЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО КАРБАМОИЛПИРИДОНА | 2011 |
|
RU2608519C2 |
| ЦИКЛОАЛКИЛ-ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ИМИДАЗОЛА, ОБЛАДАЮЩЕЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ TAFIa | 2011 |
|
RU2572814C2 |
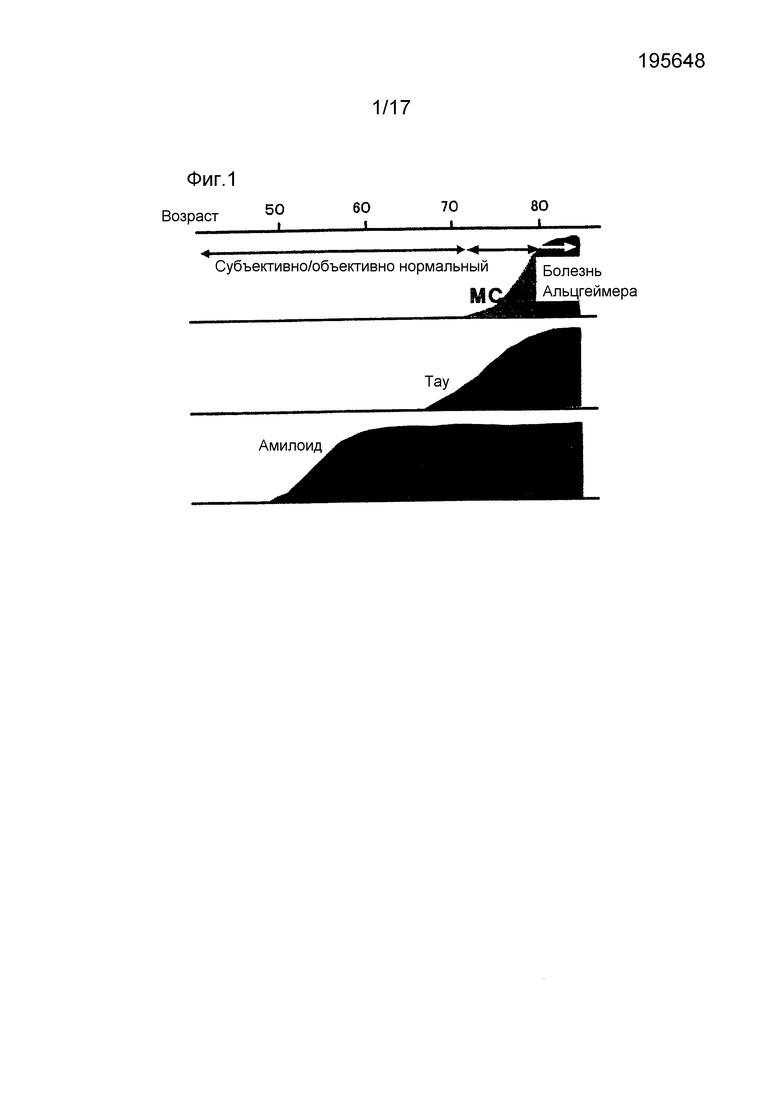
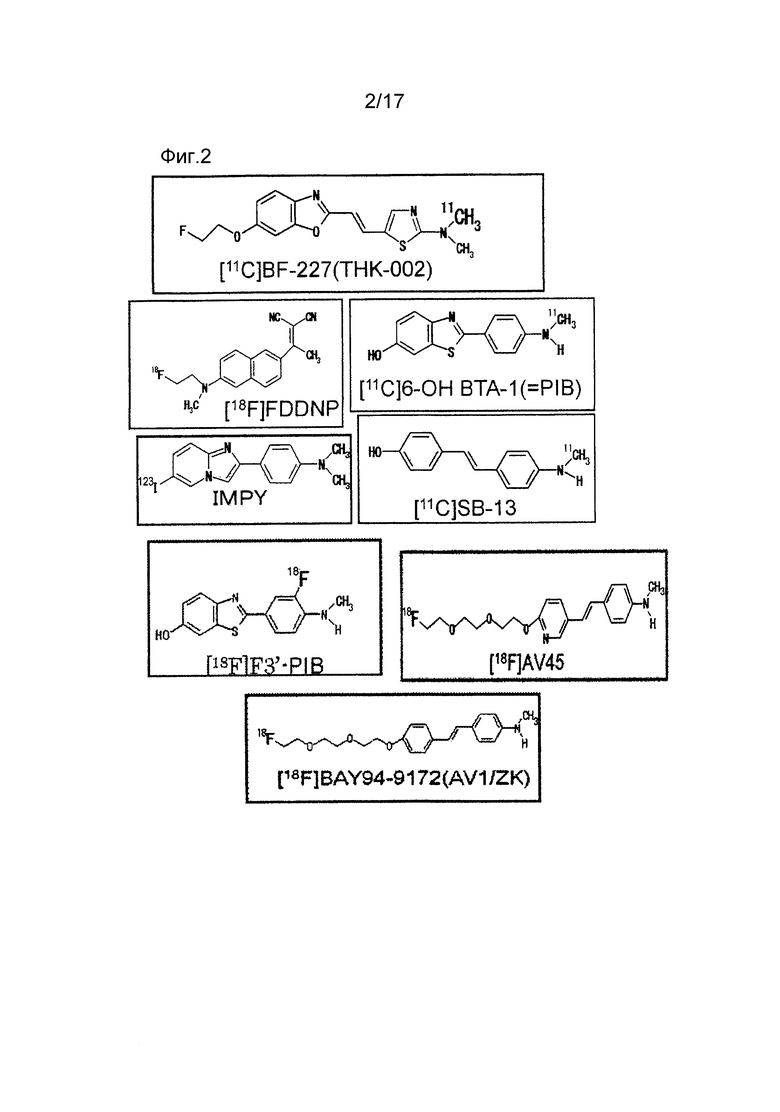

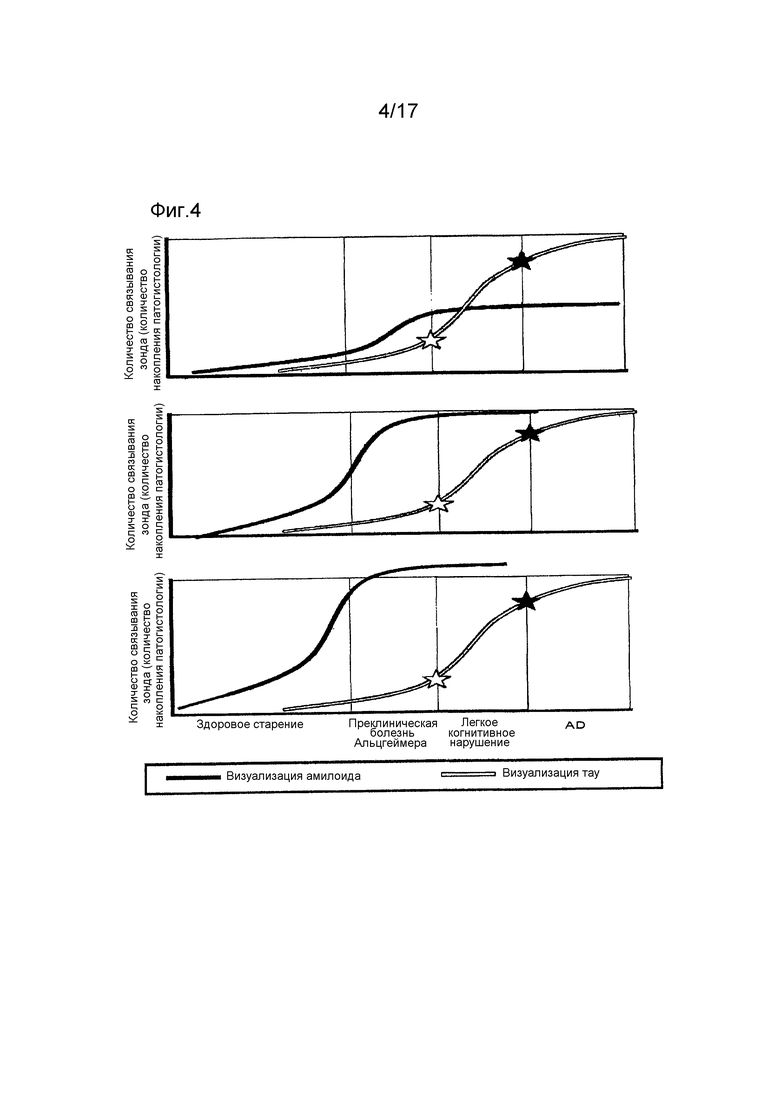







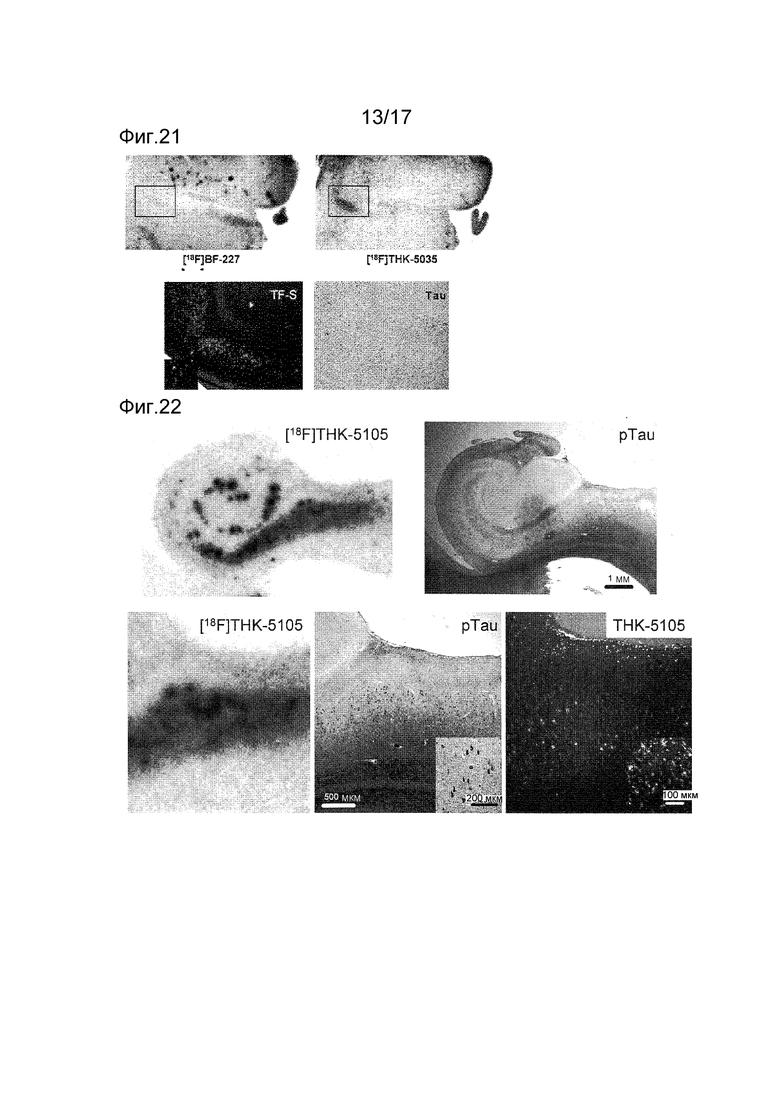
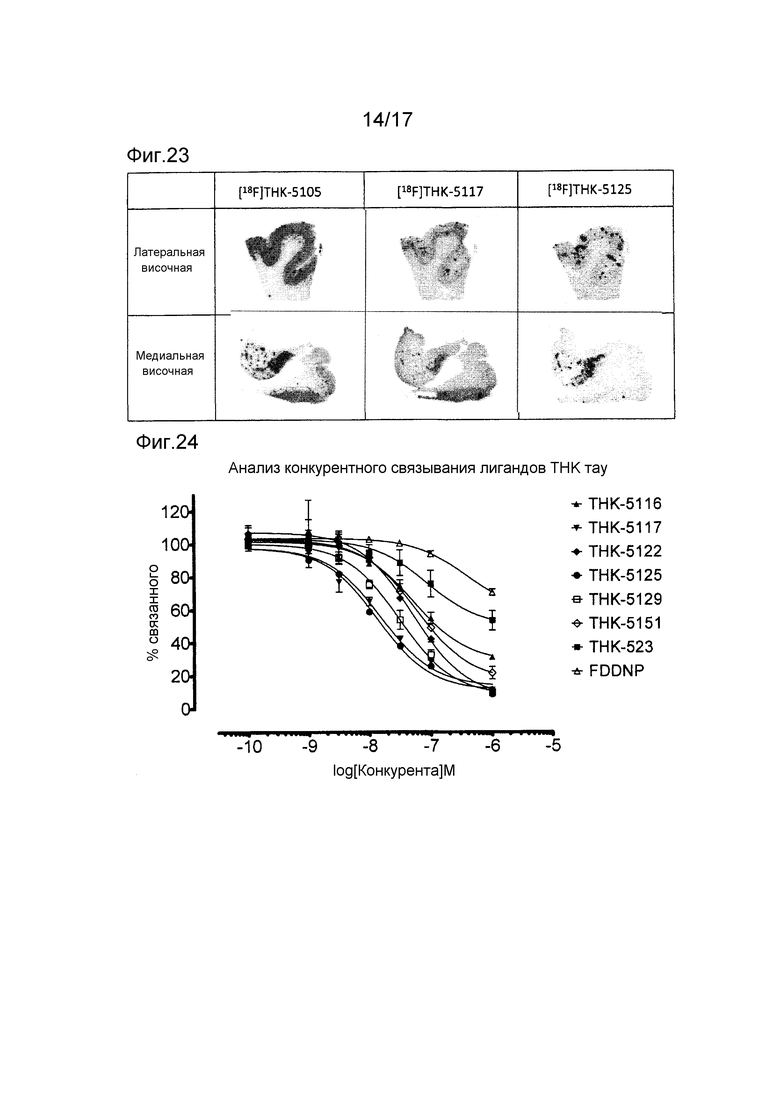



Изобретение относится к области органической химии, а именно к новому производному хинолина формулы (I) или к его фармацевтически приемлемой соли, где А обозначает 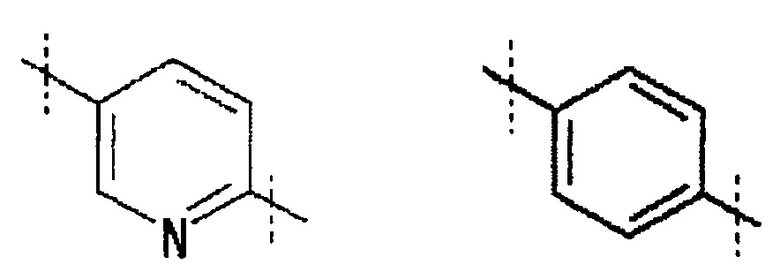 ,
,  или
или  , R1 обозначает галоген или
, R1 обозначает галоген или 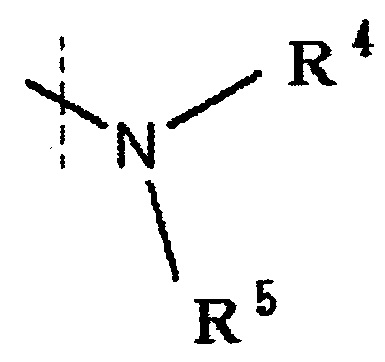 , в которой R4 и R5 каждый независимо представляет водород, низшую алкильную группу, или R4, R5 и атом азота, к которому они присоединены, вместе образуют 6-членное азотсодержащее алифатическое кольцо (при атом азота может быть замещен низшей алкильной группой), или R4 и атом азота, к которому он присоединен, вместе с кольцом А образуют 10-членное азотсодержащее конденсированное бициклическое кольцо (один атом углерода, составляющий азотсодержащее конденсированное бициклическое кольцо, может быть заменен атомом кислорода), и R5 представляет низшую алкильную группу, в которой линия, которую пересекает пунктирная линия, означает связь указанной выше общей формулы с другой структурной составляющей, R2 или R3 каждый независимо представляет NRaRb или -О-низшую алкильную группу (каждая алкильная группа может быть замещена 1-2 заместителями, выбранными из галогена, гидрокси группы), кольцо А является незамещенным или замещено R6 (где R6 обозначает один заместитель, выбранный из галогена, и -О-низшей алкильной группы (каждая алкильная группа может быть замещена 1-2 заместителями, выбранными из галогена, гидрокси группы, Ra и Rb каждый представляет водород или низшую алкильную группу, m и n обозначают целое число от 0 до 1, где по меньшей мере один из R2, R3 и R6 обозначает -O-низшую алкильную группу, замещенную одной гидрокси группой и одним галогеном. Также изобретение относится к фармацевтической композиции, композиции для диагностики, лечения или профилактики конформационного заболевания, набору для диагностики на основе соединения формулы (I), способу лечения, способу выявления или окрашивания белка β-листовой структуры, способу получения соединения формулы (I) и промежуточным соединениям. Технический результат: получены новые соединения, которые можно использовать для диагностики и лечения конформационных заболеваний, в частности, заболевания (тауопатии), имеющего такой кардинальный симптом, как внутримозговое накопление белка тау, например, болезни Альцгеймера. 10 н. и 7 з.п. ф-лы, 29 ил., 26 табл., 2 пр.
, в которой R4 и R5 каждый независимо представляет водород, низшую алкильную группу, или R4, R5 и атом азота, к которому они присоединены, вместе образуют 6-членное азотсодержащее алифатическое кольцо (при атом азота может быть замещен низшей алкильной группой), или R4 и атом азота, к которому он присоединен, вместе с кольцом А образуют 10-членное азотсодержащее конденсированное бициклическое кольцо (один атом углерода, составляющий азотсодержащее конденсированное бициклическое кольцо, может быть заменен атомом кислорода), и R5 представляет низшую алкильную группу, в которой линия, которую пересекает пунктирная линия, означает связь указанной выше общей формулы с другой структурной составляющей, R2 или R3 каждый независимо представляет NRaRb или -О-низшую алкильную группу (каждая алкильная группа может быть замещена 1-2 заместителями, выбранными из галогена, гидрокси группы), кольцо А является незамещенным или замещено R6 (где R6 обозначает один заместитель, выбранный из галогена, и -О-низшей алкильной группы (каждая алкильная группа может быть замещена 1-2 заместителями, выбранными из галогена, гидрокси группы, Ra и Rb каждый представляет водород или низшую алкильную группу, m и n обозначают целое число от 0 до 1, где по меньшей мере один из R2, R3 и R6 обозначает -O-низшую алкильную группу, замещенную одной гидрокси группой и одним галогеном. Также изобретение относится к фармацевтической композиции, композиции для диагностики, лечения или профилактики конформационного заболевания, набору для диагностики на основе соединения формулы (I), способу лечения, способу выявления или окрашивания белка β-листовой структуры, способу получения соединения формулы (I) и промежуточным соединениям. Технический результат: получены новые соединения, которые можно использовать для диагностики и лечения конформационных заболеваний, в частности, заболевания (тауопатии), имеющего такой кардинальный симптом, как внутримозговое накопление белка тау, например, болезни Альцгеймера. 10 н. и 7 з.п. ф-лы, 29 ил., 26 табл., 2 пр.
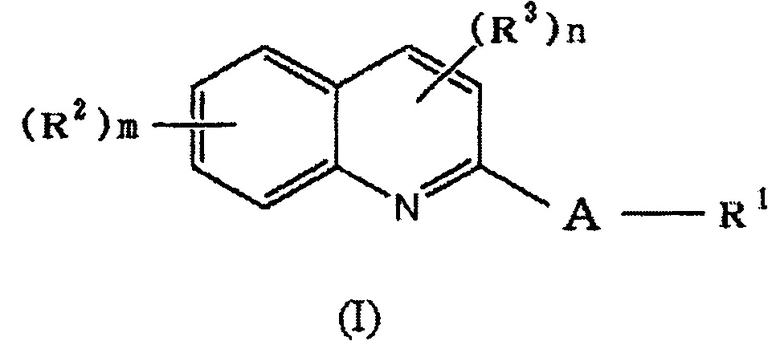
1. Соединение формулы (I):
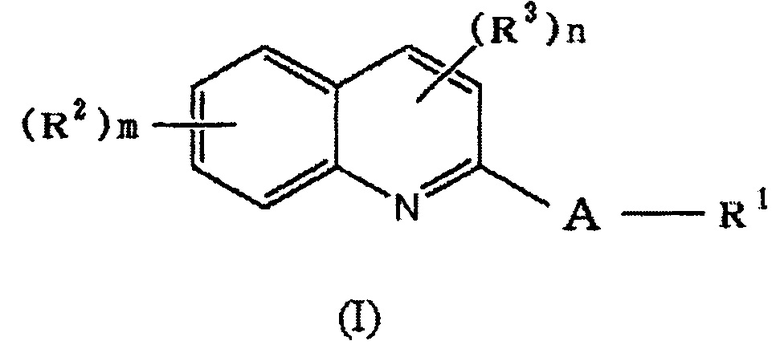 ,
,
где
А обозначает
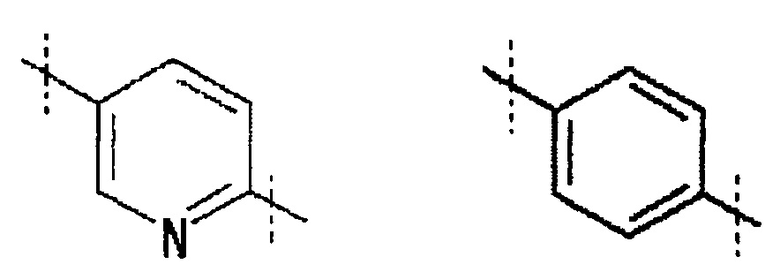
 или
или 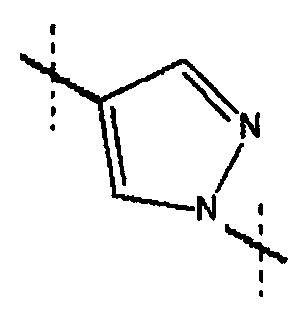 ,
,
R1 обозначает галоген или
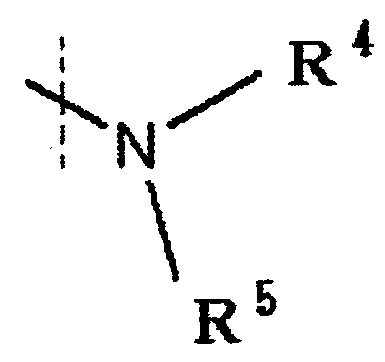 ,
,
в которой
R4 и R5 каждый независимо представляет водород, низшую алкильную группу, или
R4, R5 и атом азота, к которому они присоединены, вместе образуют 6-членное азотсодержащее алифатическое кольцо (один атом углерода, составляющий азотсодержащее алифатическое кольцо, заменен атомом азота, и когда атом углерода заменен атомом азота, то атом азота может быть замещен низшей алкильной группой),
или R4 и атом азота, к которому он присоединен, вместе с кольцом А образуют 10-членное азотсодержащее конденсированное бициклическое кольцо (один атом углерода, составляющий азотсодержащее конденсированное бициклическое кольцо, может быть заменен атомом кислорода), и R5 представляет низшую алкильную группу,
в которой линия, которую пересекает пунктирная линия, означает связь указанной выше общей формулы с другой структурной составляющей,
R2 или R3 каждый независимо представляет NRaRb или -О-низшую алкильную группу (каждая алкильная группа может быть независимо замещена одним или двумя заместителями, выбранными из галогена, гидрокси группы),
кольцо А является незамещенным или замещено R6 , в которой R6 обозначает один заместитель, независимо выбранный из галогена, и -О-низшей алкильной группы (каждая алкильная группа может быть независимо замещена одним или двумя заместителями, выбранными из галогена, гидрокси группы),
Ra и Rb каждый независимо представляет водород или низшую алкильную группу,
m обозначает целое число от 0 до 1, и
n обозначает целое число от 0 до 1,
где по меньшей мере один из R2, R3 и R6 обозначает -O-низшую алкильную группу, замещенную одной гидрокси группой и одним галогеном,
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где R1 обозначает 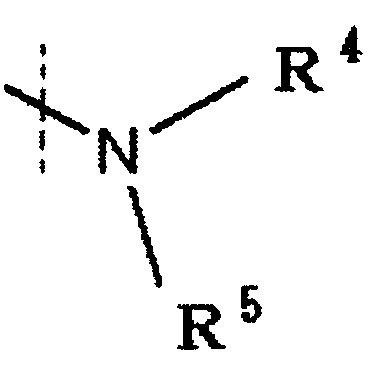 , в которой
, в которой
R4 и R5 каждый независимо представляет водород или низшую алкильную группу, или его фармацевтически приемлемая соль.
3. Соединение по п.1 или 2, где по меньшей мере один из R2, R3 и R6 представлен:
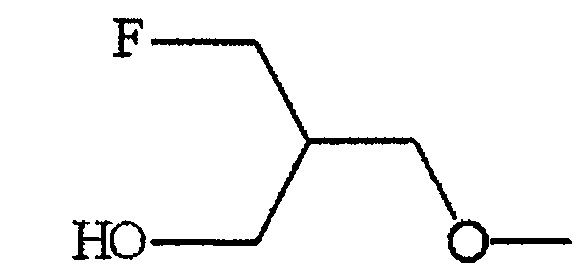 ,
, 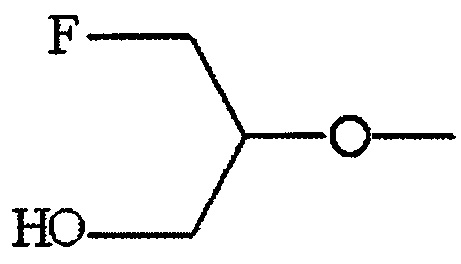 или
или 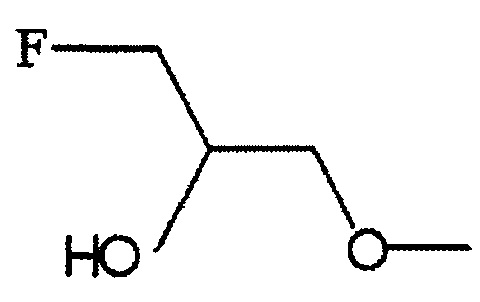 ,
,
или его фармацевтически приемлемая соль.
4. Соединение по п.1 или 2, где по меньшей мере один из R2 и R3 обозначает NRaRb, или его фармацевтически приемлемая соль.
5. Соединение по любому из пп.1 и 2, где соединение является меченым, или его фармацевтически приемлемая соль.
6. Соединение по п.3, где соединение является меченым, или его фармацевтически приемлемая соль.
7. Соединение по п.4, где соединение является меченым, или его фармацевтически приемлемая соль.
8. Фармацевтическая композиция для лечения и/или профилактики конформационного заболевания, содержащая эффективное количество соединения по любому из пп. 1-7 или его фармацевтически приемлемой соли.
9. Композиция для диагностики, лечения и/или профилактики конформационного заболевания, содержащая соединение по любому из пп.1-7 или его фармацевтически приемлемую соль.
10. Набор для диагностики конформационного заболевания или выявления или окрашивания белка бета-листовой структуры, содержащий соединение по любому из пп.1-7 или его фармацевтически приемлемую соль в качестве активного ингредиента.
11. Способ лечения и/или профилактики конформационного заболевания или диагностики конформационного заболевания у пациента, который включает введение пациенту соединения по любому из пп.1-7 или его фармацевтически приемлемой соли.
12. Способ выявления или окрашивания белка β-листовой структуры в образце, который включает окрашивание образца с использованием соединения по любому из пп.1-5 или его фармацевтически приемлемой соли.
13. Способ получения соединения формулы (I), который включает следующие стадии:
(i) взаимодействие соединения формулы (II):
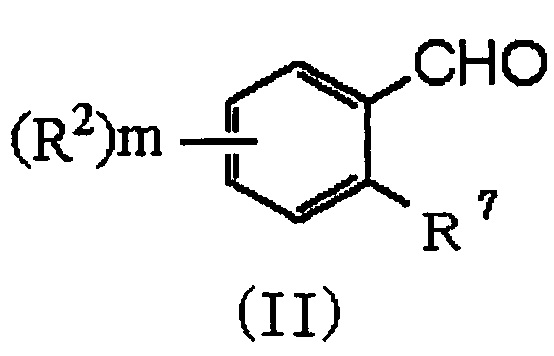 ,
,
в которой R2 и m обозначают, как определено в формуле (I), и R7 представляет NH2 или NO2, с соединением формулы (III):
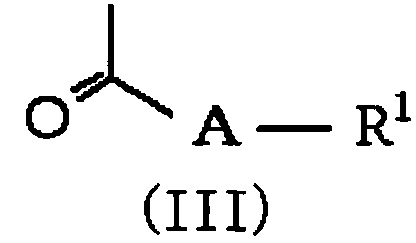 ,
,
в которой А и R1 обозначают, как определено в формуле (I), с получением соединения формулы (IV):
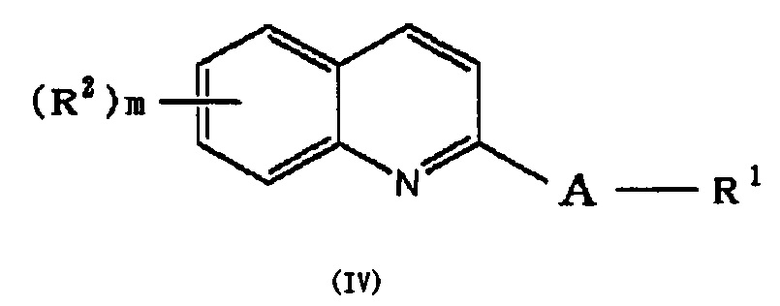 ,
,
и выделение этого соединения в виде соединения формулы (I) в присутствии основания.
14. Соединение формулы (I'):
 ,
,
в которой
А обозначает
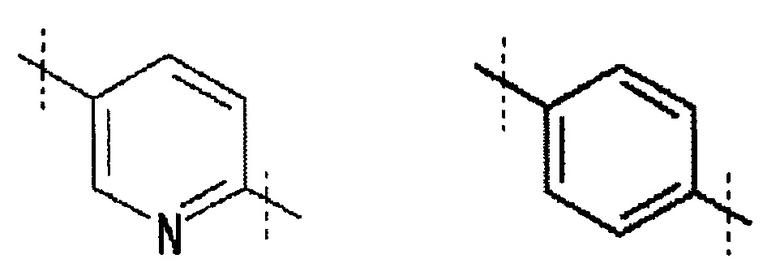 ,
,
R1 обозначает  ,
,
в которой
R4 и R5 каждый независимо представляет водород, низшую алкильную группу,
или R4 и атом азота, к которому он присоединен, вместе с кольцом А образуют 10-членное азотсодержащее конденсированное бициклическое кольцо (один атом углерода, составляющий азотсодержащее конденсированное бициклическое кольцо, заменен атомом кислорода), и R5 обозначает низшую алкильную группу,
в которой линия, которую пересекает пунктирная линия, означает связь представленной выше общей формулы с другой структурной составляющей,
R2 независимо представляет -O-низшую алкильную группу (низшая алкильная группа замещена одной или двумя группами, выбранными из п-толуолсульфонилокси группы или 2-тетрагидропиранилокси группы, а также может быть замещена одной гидрокси группой),
кольцо А является незамещенным или замещено R6 (в которой R6 обозначает один заместитель, выбранный независимо из -O-низшей алкильной группы (низшая алкильная группа каждая независимо может быть замещена двумя заместителями, выбранными из п-толуолсульфонилокси группы или 2-тетрагидропиранилокси группы),
Ra и Rb независимо представляют низшую алкильную группу,
m обозначает целое число 1, и
n обозначает 0,
при условии, что по меньшей мере один из R2 и R6 представляет -O-низшую алкильную группу (низшая алкильная группа замещена п-толуолсульфонилокси группой (тозилокси группой, TsO) или 2-тетрагидропиранилокси (ОТНР), и может быть также замещена одним заместителем, выбранным из гидрокси группы),
или его фармацевтически приемлемая соль.
15. Соединение по п.14, где по меньшей мере один из R2 и R6 обозначает группу формулы:
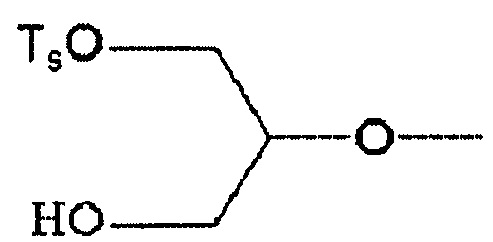 ,
, 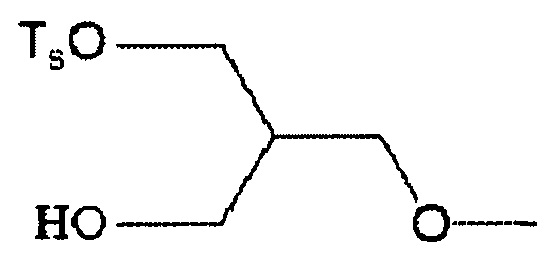 ,
,
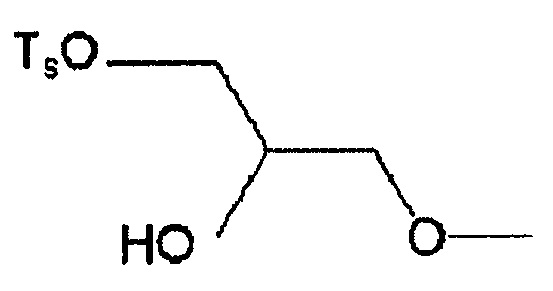 или
или 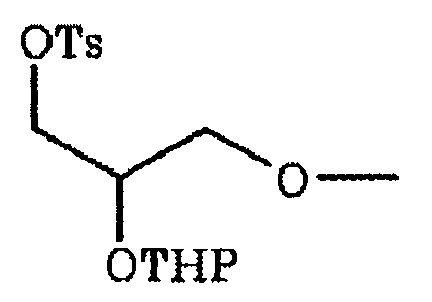 .
.
16. Набор для получения меченого соединения по пп.5-7 или его фармацевтически приемлемой соли, причем указанный набор включает:
соединение по п. 14 или 15 или его фармацевтически приемлемую соль,
метящий агент и, необязательно, инструкции по проведению мечения.
17. Способ получения меченого соединения по пп.5-7, который включает стадию взаимодействия соединения по п.14 или 15 с метящим агентом.
| WO 2010111303 A2, 30.09.2010 | |||
| WO 2008124703 A2, 16.10.2008 | |||
| RU 2005139393 A, 27.07.2006. |

