Область техники, к которой относится изобретение
Настоящее изобретение относится к новому производному фенола, которое характеризуется высокой концентрацией неизмененного соединения в моче, а также обладает заметным урикозурическим действием, или к его фармацевтически приемлемой соли, или к их гидрату или к их сольвату, и к лекарственному средству, содержащему указанное производное в качестве активного ингредиента.
Предпосылки создания изобретения
Мочевая кислота образуется в результате катаболизма пурина, который образуется в результате расщепления нуклеиновой кислоты и аденозинтрифосфата (АТФ), который представляет собой источник энергии живого организма, с последующим окислением метаболизированного пурина (ксантина) ксантиноксидазой или ксантиндегидрогеназой. У людей мочевая кислота (константа диссоциации pKa=5,75) представляет собой конечный метаболит пурина и присутствует в организме в виде свободной мочевой кислоты или соли.
Мочевая кислота обычно выводится с мочой, и если выработка мочевой кислоты превышает ее выведение, а содержание мочевой кислоты в крови возрастает, то возникает гиперурикемия. Если в течение длительного периода избыток мочевой кислоты в крови превышает верхний предел растворимости (приблизительно 7 мг/дл), то в осадок выпадает кристаллический урат (как правило, натриевая соль).
Кристаллический урат откладывается в хрящевых тканях или суставах с образованием осадка, что приводит, тем самым, к образованию подагрических узлов. При этом индуцируется острый подагрический артрит и развивается хронический подагрический артрит.
Если кристаллический урат выпадает в осадок в моче, то возникает повреждение почек (подагрическая почка), такое как интерстициальный нефрит, мочекаменная болезнь и т.п. С целью коррекции гиперурикемии после затихания приступа острого подагрического артрита медикаментозное лечение осуществляют совместно с поддержанием положительной динамики качества жизни.
С целью профилактики острого подагрического артрита, подагрической почки, мочекаменной болезни и т.п. очень важно осуществлять коррекцию гиперурикемии и подходящим образом регулировать содержание мочевой кислоты.
Считается, что гиперурикемия осложняется заболеваниями, связанными с образом жизни, такими как ожирение, гиперлипидемия, аномальная толерантность к глюкозе и гипертензия с высокими показателями давления (см. непатентный документ 1 (стр. 7-9)). Увеличение концентрации уратов в сыворотке крови характеризуется положительной взаимосвязью с показателем смертности вследствие сердечно-сосудистых заболеваний. Поскольку высокая концентрация уратов в сыворотке крови увеличивает смертность вследствие сердечно-сосудистых заболеваний, предполагается, что увеличение содержания мочевой кислоты в сыворотке крови самостоятельно и значительно связано с риском смерти вследствие сердечно-сосудистых заболеваний (см. непатентный документ 2).
Также предполагается, что концентрация уратов в сыворотке крови представляет собой высокий фактор риска инфаркта миокарда и кровоизлияния в мозг (см. непатентный документ 3). До настоящего момента сообщалось, что гиперурикемия ассоциирована с ожирением, гиперлипидемией, дислипидемией, аномальной толерантностью к глюкозе, сахарным диабетом, метаболическим синдромом, заболеванием почек (например, почечная недостаточность, протеинурия, терминальная стадия почечной недостаточности (ESRD) и т.п.), сердечно-сосудистыми заболеваниями (например, гипертензия, ишемическая болезнь сердца, заболевание сонных артерий, эндотелиальная дисфункция, артериосклероз, гипертрофия сердца, цереброваскулярное заболевание и т.п.) или является фактором риска этих заболеваний (см. непатентные документы 2-11). Также сообщалось, что концентрация мочевой кислоты увеличивается в спинномозговой жидкости при сосудистой деменции (см. непатентный документ 12).
В этих обстоятельствах предполагается, что снижение содержания уратов в крови может отсрочить развитие заболевания почек, а также может снизить риск сердечно-сосудистого заболевания (см. непатентные документы 5, 8, 13 и 14), и сообщается, что лечение также следует применять в случае бессимптомной гиперурикемии (см. непатентный документ 14).
Соответственно считается, что снижение содержания уратов в крови при вышеупомянутых заболеваниях эффективно для лечения или профилактики указанных заболеваний, а также важно с точки зрения предотвращения рецидива инфаркта миокарда и поддержания функции почек.
Основной фактор увеличения содержания уратов в крови включает повышенную продукцию и недостаточное выведение мочевой кислоты. Считается, что способ супрессии продукции мочевой кислоты или ускорения выведения мочевой кислоты является эффективным для снижения содержания уратов в крови. Известно, что обладающее первым механизмом действия лекарство (ингибитор продукции мочевой кислоты) включает аллопуринол, тогда как обладающее последним механизмом действия лекарство (урикозурическое лекарство) включает бензбромарон, пробенецид, соединение по JP-A-2006-176505 (патентный документ 1) и т.п.
Японское руководство по лечению гиперурикемии и подагры в качестве общего правила предписывает, что в случае лечения гиперурикемии урикозурические лекарства применяют у пациентов с недостаточным выведением мочевой кислоты, а ингибиторы продукции мочевой кислоты применяют у пациентов с повышенной продукцией мочевой кислоты, соответственно (см. непатентный документ 1 (стр. 31-32)).
Сообщается, что в Японии пациенты с недостаточным выведением мочевой кислоты составляют приблизительно 60% от всех пациентов с гиперурикемией, а пациенты смешанного типа с недостаточным выведением и повышенной продукцией мочевой кислоты составляют приблизительно 25% от всех пациентов с гиперурикемией (непатентный документ 15). Также сообщается, что недостаточное выведение мочевой кислоты наблюдается приблизительно у 85% пациентов с подагрой, и даже у пациентов с повышенной продукцией мочевой кислоты среднее значение выведения мочевой кислоты значительно ниже, чем у здорового человека, и недостаточное выведение мочевой кислоты предположительно является обычным явлением у всех пациентов с подагрой (непатентный документ 16).
Соответственно, при гиперурикемии (особенно при подагре) считается важным лечение пациентов с недостаточным выведением мочевой кислоты, и значимость существования урикозурического лекарства особенно велика.
Из числа основных урикозурических лекарств пробенецид практически не используется, поскольку он обладает слабым действием и определенно вызывает желудочно-кишечные расстройства и взаимодействует с другими лекарствами; при этом сообщалось о серьезном повреждении печени бензбромароном, который обладает сильным урикозурическим действием и часто используется в Японии в качестве урикозурического лекарства (см. непатентный документ 17).
Бензбромарон или его аналог характеризуется митохондриальной токсичностью, например, ингибированием активности ферментативного комплекса дыхательной цепи митохондрии, разобщающим воздействием, ингибированием дыхания, ингибированием окисления β-жирных кислот, снижением мембранного потенциала митохондрии, апоптозом, продукцией активных форм кислорода и т.п., и митохондриальная токсичность предположительно вовлечена в индукцию повреждения печени (см. непатентные документы 18 и 19). Активный метаболит бензбромарона, 6-гидроксибензбромарон, также характеризуется токсичностью в отношении митохондрий.
Кроме того, бензбромарон обладает ингибирующим действием в отношении цитохрома P450 (CYP), который является ферментом, метаболизирующим лекарства, и характеризуется особенно сильным ингибирующим действием в отношении CYP2C9, и предположительно вызывает фармакокинетическое взаимодействие лекарств (см. непатентные документы 20 и 21).
В JP-A-2006-176505 (патентный документ 1) описано азотсодержащее соединение с конденсированными кольцами, которое обладает ингибирующим действием в отношении URAT1 как типа переносчиков урата, а также имеет структуру, аналогичную структуре соединений по настоящему изобретению. Однако это соединение не обладает достаточным эффектом, и новое применимое на практике урикозурическое лекарство разработано не было.
Недавно были получены результаты, что урикозурическое действие зависит от концентрации лекарства, обладающего таким действием, в моче, то есть урикозурическое лекарство проявляет эффективность лекарства в процессе выведения с мочой (см. патентный документ 2, непатентные документы 22 и 23).
Соответственно, предполагается более мощное, эффективное урикозурическое лекарство, которое выводится с мочой в больших количествах. Однако вышеупомянутые существующие урикозурические лекарства характеризуются очень низкой концентрацией в моче, и нельзя сказать, что достигается удовлетворительная активность.
Что касается выведения лекарства с мочой, то могут быть рассмотрены случай, когда введенное лекарство выводится в виде неизмененного соединения «как есть», и случай, когда лекарство преобразуется до активного метаболита, а затем выводится. В последнем случае существует риск того, что из-за индивидуального различия в продукции активного метаболита может возрасти его количество. Для получения стабильной эффективности лекарства и безопасности более желательным является лекарство, выводимое в виде неизмененного соединения.
Таким образом, желательна разработка лекарственного средства, которое характеризуется высокой концентрацией неизмененного соединения в моче, а также обладает заметным урикозурическим действием и высокой безопасностью по сравнению с существующими урикозурическими лекарствами.
Перечень цитируемых документов
Патентные документы
Патентный документ 1: JP-A-2006-176505
Патентный документ 2: WO 2005/121112
Непатентные документы
Непатентный документ 1: Guidelines for the Management of Hyperuricemia and Gout (First Edition) pp. 7-9, and pp. 31-32, Gout and Nucleic Acid Metabolism, Vol. 26, Supplement 1, 2002, Japanese Society of Gout and Nucleic Acid Metabolism
Непатентный документ 2: JAMA 283: 2404-2410 (2000)
Непатентный документ 3: Stroke 37: 1503-1507 (2006)
Непатентный документ 4: Nephrology 9: 394-399 (2004)
Непатентный документ 5: Semin. Nephrol. 25: 43-49 (2005)
Непатентный документ 6: J. Clin. Hypertens. 8: 510-518 (2006)
Непатентный документ 7: J. Hypertens. 17: 869-872 (1999)
Непатентный документ 8: Curr. Med. Res. Opin. 20: 369-379 (2004)
Непатентный документ 9: Curr. Pharm. Des. 11: 4139-4143 (2005)
Непатентный документ 10: Hypertension 45: 991-996 (2005)
Непатентный документ 11: Arch. Intern. Med. 169: 342-350 (2009)
Непатентный документ 12: J. Neural. Transm. Park Dis. Dement. Sect. 6: 119-126 (1993)
Непатентный документ 13: Am. J. Kidney Dis. 47: 51-59 (2006)
Непатентный документ 14: Hyperuricaemia and Gout 9: 61-65 (2001)
Непатентный документ 15: Nippon Rinsho 54: 3230-3236 (1996)
Непатентный документ 16: Nippon Rinsho 54: 3248-3255 (1996)
Непатентный документ 17: J. Hepatol. 20: 376-379 (1994)
Непатентный документ 18: J. Hepatol. 35: 628-636 (2001)
Непатентный документ 19: Hepatology 41: 925-935 (2005)
Непатентный документ 20: Journal of Saitama Medical University (J. Saitama. Med. School) 30: 187-194 (2004)
Непатентный документ 21: Drug Metab. Dispos. 31: 967-971 (2003)
Непатентный документ 22: Proceedings of the 42nd Annual Meeting of the Japanese Society of Gout and Nucleic Acid Metabolism, p. 59 (2009)
Непатентный документ 23: ACR 2008 Annual Scientific Meeting, No. 28
Краткое описание сущности изобретения
Техническая задача
Целью настоящего изобретения является создание новых соединений и фармацевтического препарата, которые обладают заметным урикозурическим действием.
Решение задачи
Для достижения описанной выше цели изобретения авторы настоящего изобретения провели интенсивные исследования и обнаружили новое производное фенола, обладающее высокой безопасностью и заметным урикозурическим действием, оформив тем самым настоящее изобретение.
Таким образом, настоящее изобретение относится к новому производному фенола, представленному следующей общей формулой (1):
[Химическая формула 1]
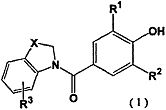 ,
,
где R1 и R2 являются одинаковыми или разными и представляют собой низшую алкильную группу, низшую алкенильную группу, низшую алкинильную группу, низшую алкоксигруппу, галогеналкильную группу, галогеналкоксигруппу, алкилсульфанильную группу, алкилсульфинильную группу, алкилсульфонильную группу, замещенную низшим алкилом карбамоильную группу, насыщенную азотсодержащую гетероциклическую N-карбонильную группу, атом галогена, цианогруппу или атом водорода, R3 представляет собой низшую алкильную группу, галогеналкильную группу, атом галогена, гидроксильную группу или атом водорода, и X представляет собой атом серы, -S(=O)- или -S(=О)2, к его фармацевтически приемлемой соли, и к их гидрату и их сольвату, и к содержащей их фармацевтической композиции.
В настоящем описании «низшая алкильная группа» представляет собой C1-6алкильную группу, и может представлять собой любую из неразветвленной, разветвленной и циклической низших алкильных групп, и алкильную группу, состоящую из их сочетания. Это же должно относиться к алкильным фрагментам заместителей, содержащих алкильный фрагмент [низшая алкоксигруппа, замещенная низшим алкилом карбамоильная группа, алкилсульфанильная группа и т.д.]. Примеры C1-6алкильной группы включают метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, н-пентильную группу, н-гексильную группу, циклопропильную группу, циклобутильную группу и т.п. Примеры низшей алкоксигруппы включают метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, циклопропоксигруппу, н-бутоксигруппу, изобутоксигруппу, трет-бутоксигруппу, н-пентоксигруппу, н-гексилоксигруппу и т.п. Примеры низшей галогеналкоксигруппы включают трифторметоксигруппу и трифторэтоксигруппу. Примеры низшей галогеналкильной группы включают трифторметильную группу, трифторэтильную группу и т.п. Примеры низшей алкилсульфанильной группы включают метилсульфанильную группу, этилсульфанильную группу, изопропилсульфанильную группу и т.п. Примеры атома галогена включают атом фтора, атом хлора, атом брома и атом йода. Примеры замещенной низшим алкилом карбамоильной группы включают метилкарбамоильную группу, этилкарбамоильную группу, диметилкарбамоильную группу, диэтилкарбамоильную группу и т.п. Примеры насыщенной азотсодержащей гетероциклической N-карбонильной группы включают пирролидин-1-илкарбонильную группу, тиазолидин-3-илкарбонильную группу, 1-оксотиазолидин-3-илкарбонильную группу, 1,1-диоксотиазолидин-3-илкарбонильную группу и т.п. Примеры низшей алкенильной группы включают винильную группу, пропенильную группу и т.п. Примеры низшей алкинильной группы включают этинильную группу, пропинильную группу и т.п. Примеры низшей алкилсульфинильной группы включают метилсульфинильную группу, этилсульфинильную группу и т.п. Примеры низшей алкилсульфонильной группы включают метилсульфонильную группу, этилсульфонильную группу и т.п.
Низшая алкильная группа, представленная R1, предпочтительно представляет собой этильную группу, изопропильную группу, н-бутильную группу, трет-бутильную группу, циклопропильную группу или циклобутильную группу. Низшая галогеналкильная группа предпочтительно представляет собой трифторметильную группу. Низшая алкоксигруппа предпочтительно представляет собой метоксигруппу. Низшая галогеналкоксигруппа предпочтительно представляет собой трифторметоксигруппу. Алкилсульфанильная группа предпочтительно представляет собой метилсульфанильную группу, этилсульфанильную группу или изопропилсульфанильную группу. Алкилсульфонильная группа предпочтительно представляет собой метилсульфонильную группу. Атом галогена предпочтительно представляет собой атом фтора или атом хлора. Замещенная низшим алкилом карбамоильная группа предпочтительно представляет собой диметилкарбамоильную группу. Насыщенная азотсодержащая гетероциклическая N-карбонильная группа предпочтительно представляет собой пирролидин-1-илкарбонильную группу, тиазолидин-3-илкарбонильную группу, 1-оксотиазолидин-3-илкарбонильную группу или 1,1-диоксотиазолидин-3-илкарбонильную группу. Низшая алкинильная группа предпочтительно представляет собой этинильную группу. Низшая алкилсульфинильная группа предпочтительно представляет собой метилсульфинильную группу. R2 предпочтительно представляет собой атом фтора, атом хлора, цианогруппу, метилсульфонильную группу или трифторметильную группу. X предпочтительно представляет собой атом серы или -S(=О)2-. R3 предпочтительно представляет собой гидроксильную группу, трифторметильную группу или атом водорода.
Более предпочтительно, в качестве примеров могут быть представлены соединения, в которых X представляет собой -S(=О)2-, R1 представляет собой атом хлора, низшую алкильную группу, низшую алкоксигруппу, трифторметоксигруппу, алкилсульфанильную группу или трифторметильную группу, R2 представляет собой цианогруппу, атом хлора, атом фтора, метилсульфонильную группу или трифторметильную группу, и R3 представляет собой атом водорода или гидроксильную группу.
В частности, соединения предпочтительно представляют собой 3-(3,5-дихлор-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол, 3-(3-хлор-5-циано-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол, 3-(3-циано-4-гидрокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-l,3-бензотиазол, 3-(3-циано-5-этил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол, 3-(3-циано-4-гидрокси-5-метилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол, 3-(3-циано-5-этилсульфанил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол, 3-(3-хлор-4-гидрокси-5-метилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол, 3-(3-хлор-4-гидрокси-5-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол, 3-(3-хлор-5-фтор-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол, 3-(3-хлор-4-гидрокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол, 3-(3-хлор-4-гидрокси-5-трифторметоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол, 3-(5-трет-бутил-4-гидрокси-3-метилсульфонилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол, 3-(3-циано-5-циклопропил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол, 3-(3-циано-5-этинил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол или их фармацевтически приемлемую соль, или их гидрат или их сольват.
Что касается соединений по настоящему изобретению, то могут существовать изомеры. Например, могут существовать геометрические изомеры, оптические изомеры или диастереоизомеры. Любой отдельный изомер из указанных изомеров, произвольные смеси изомеров, рацематы и т.п. подпадают под объем настоящего изобретения.
В зависимости от типа заместителя соединения по настоящему изобретению могут формировать основно-аддитивную соль или кислотно-аддитивную соль. Конкретного ограничения по типу соли не существует, и ее примеры включают без ограничения соли металлов, такие как соли натрия, соли калия и соли кальция; основно-аддитивные соли, такие как соли аммония и соли органических аминов; соли неорганических кислот, такие как гидрохлориды, сульфаты и нитраты; соли органических кислот, такие как пара-толуолсульфонаты, метансульфонаты и соли винной кислоты.
Соединения по настоящему изобретению и их соли могут существовать в виде гидрата или сольвата, и указанные вещества также подпадают под объем настоящего изобретения. Примеры гидратов включают 1/2 гидраты, моногидраты, дигидраты и т.п.
Пролекарство в качестве эквивалентного соединения нового производного фенола, представленного общей формулой (1) по настоящему изобретению, или его фармацевтически приемлемой соли, или их гидрата или их сольвата, также подпадает под объем настоящего изобретения. «Пролекарство» означает соединение, которое преобразуется до соединения (1) по механизму метаболизма in vivo, т.е. соединение, которое ферментативно вызывает окисление, восстановление или гидролиз in vivo, или вызывает гидролиз кислотой желудочного сока, и преобразуется тем самым до соединения общей формулы (1). Примеры пролекарства общей формулы (1) включают соединения, в которых фенольная гидроксильная группа модифицирована ацильной группой, алкильной группой и т.п., например, ацетилированные и пивалоилированные соединения.
Указанные соединения могут быть синтезированы из соединения (1) известным способом. Пролекарство соединения (1) может представлять собой пролекарство, которое преобразуется до соединения (1) при условиях, описанных в «Soyaku Kagaku», pp. 204-208, published in 2004 by Tokyo Kagaku Dojin Co. Ltd.
Конкретного ограничения по способу синтеза соединений по настоящему изобретению не существует, и, например, они могут быть синтезированы в соответствии со следующими стадиями. В этом случае иногда они могут быть получены, эффективно с точки зрения методики синтеза, путем введения подходящей защитной группы в функциональную группу исходного вещества или промежуточного соединения, зависящей от типа функциональной группы. Примеры такой функциональной группы включают аминогруппу, гидроксигруппу, карбоксигруппу и т.п. Если синтез проводят путем введения защитной группы в функциональную группу, то необходимое соединение может быть получено путем удаления подходящим образом защитной группы на соответствующих стадиях синтеза. Примеры типа такой защитной группы и способов защиты и снятия защиты включают таковые, описанные, например, в Greene and Wuts, «Protective Groups in Organic Synthesis (Fourth Edition)», и т.п.
Полезные эффекты настоящего изобретения
Новое производное фенола по настоящему изобретению или его фармацевтически приемлемая соль, или их гидрат или их сольват, характеризуется высокой концентрацией неизмененного соединения в моче, а также обладает превосходным урикозурическим действием и является превосходным в плане безопасности, а потому применимо в качестве лекарственного средства для ускорения выведения мочевой кислоты; лекарственного средства для снижения количества мочевой кислоты и/или концентрации мочевой кислоты в крови и/или ткани; лекарственного средства для применения при профилактике и/или лечении заболевания, ассоциированного с мочевой кислотой в крови и/или ткани; лекарственного средства для применения при профилактике и/или лечении гиперурикемии; и лекарственного средства для применения при профилактике и/или лечении заболевания, ассоциированного с гиперурикемией и/или заболевания, сопровождающегося гиперурикемией.
Краткое описание чертежей
На фиг. 1 представлена общая формула, отражающая новое производное фенола по настоящему изобретению.
Описание вариантов осуществления
Типичный способ синтеза новых производных фенола, представленных следующей общей формулой (1) по настоящему изобретению, будет описан ниже.
<Способ получения>
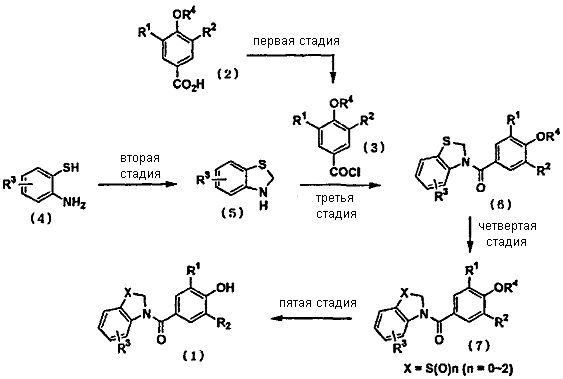
Первая стадия: Хлорангидрид (3) может быть синтезирован из промежуточного соединения карбоновой кислоты (2) в качестве исходного вещества в органическом растворителе с использованием тионилхлорида, пентахлорида фосфора, трихлорида фосфора, оксихлорида фосфора, оксалилхлорида и т.п.
Вторая стадия: 2,3-дигидро-1,3-бензотиазол (5), замещенный R3, может быть получен путем осуществления взаимодействия 2-аминобензолтиола (4), замещенного R3, с водным раствором формалина, или с эквивалентом формальдегида, таким как параформальдегид.
Третья стадия: Соединение амида (6) может быть синтезировано путем конденсации синтезированного на первой стадии хлорангидрида, в котором фенол защищен, и синтезированного на второй стадии 2,3-дигидро-1,3-бензотиазола, замещенного R3, в присутствии общепринятого основания.
Четвертая стадия: Если R1, R2 и R3 соединения амида (6) представляют собой функциональные группы, на которые не влияет процесс окисления, то сульфоксид или сульфон могут быть получены путем общепринятого окисления с использованием органической пероксикислоты, такой как перхлорбензойная кислота или перуксусная кислота, пероксида водорода, и катализатора. Если R1 представляет собой функциональную группу, на которую влияет процесс окисления, например, алкилсульфанильную группу и т.д., то производное сульфона может быть синтезировано с одновременным проведением окисления. В случае синтеза производного, в котором R1 представляет собой алкилсульфанильную группу, производное может быть получено из соединения, в котором R1 представляет собой галоген, такой как йод, с применением реакции сочетания или т.д.
Пятая стадия: Что касается снятия защиты с защищенной гидроксифенольной группы, то целевой продукт (1) может быть синтезирован, например, в условиях снятия защиты, описанных в «Protective Groups in Organic Synthesis (Fourth Edition)» (written by Greene and Wuts). Например, если защитная группа представляет собой метильную группу, то целевой продукт (1) может быть получен путем нагревания по меньшей мере эквивалентного количества хлорида лития в N,N-диметилформамиде. В случае бензильной группы целевой продукт (1) может быть получен путем проведения каталитического гидрирования в присутствии катализатора, такого как палладий.
Промежуточное соединение карбоновой кислоты (2), используемое на первой стадии, может быть синтезировано из соответствующих исходных веществ путем проведения следующего общепринятого реакционного процесса, представленного на следующей схеме
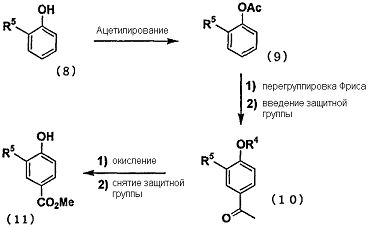
Способ синтеза i) Способ синтеза сложного эфира 4-гидроксибензойной кислоты, замещенного в 3-м положении: Например, что касается соединения, в котором R5 представляет собой трифторметоксигруппу, то соединение (11), в котором R5 представляет собой трифторметоксигруппу и которое представляет собой исходное вещество для способа синтеза ii), может быть синтезировано путем ацетилирования 2-трифторметоксифенола (8) уксусным ангидридом или т.п., с проведением перегруппировки Фриса с использованием трифторметансульфоновой кислоты или т.п., с последующим проведением защиты гидроксильной группы и этерификации путем галоформной реакции.
Способ синтеза ii) При помощи следующей методики соединение, в котором R6 представляет собой атом галогена, и R5 представляет собой цианогруппу, трифторметильную группу или трифторметоксигруппу, может быть синтезировано из сложного эфира 4-гидроксибензойной кислоты (11), замещенной в 3-м положении.
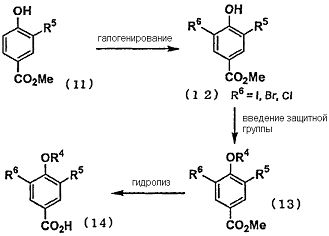
Например, промежуточное соединение сложного эфира (13), в котором R6 представляет собой атом галогена, и R5 представляет собой цианогруппу, может быть синтезировано путем галогенирования сложного эфира 3-циано-4-гидроксибензойной кислоты (11) общепринятым галогенирующим агентом, таким как N-хлорсукцинимид (NCS), N-бромсукцинимид (NBS) или N-йодсукцинимид (NIS), с последующим осуществлением взаимодействия фенольной гидроксигруппы с диметилсерной кислотой, бензилбромидом и т.д. в присутствии общепринятого основания, выполняя тем самым защиту при помощи R4 (метильная группа, бензильная группа или т.п.). Полученное таким образом промежуточное соединение сложного эфира (13) подвергали общепринятой реакции гидролиза с получением промежуточного соединения карбоновой кислоты (14). Например, в следующих условиях гидролиза промежуточное соединение карбоновой кислоты может быть синтезировано путем осуществления взаимодействия при комнатной температуре или при нагревании с обратным холодильником в органическом растворителе, воде или в растворителе, смешанном с органическим растворителем, в присутствии соответствующего для реакции количества кислоты или основания. Примеры кислоты включают соляную кислоту, серную кислоту, бромистоводородную кислоту, трифторуксусную кислоту и т.п., и примеры основания включают гидроксид натрия, гидроксид лития и т.п.
Что касается промежуточного соединения сложного эфира (13-1), в котором R6 представляет собой атом йода, то атом йода может быть преобразован до функциональной группы, которая может быть введена путем обычной реакции сочетания.
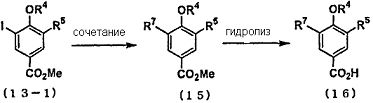
Например, промежуточное соединение сложного эфира (15-1), в котором R7 представляет собой алкильную группу, алкинильную группу или алкилсульфанильную группу, может быть синтезировано из промежуточного соединения сложного эфира (13-1-1), в котором R5 представляет собой цианогруппу, и R6 представляет собой атом йода, в присутствии катализатора, такого как палладий или никель, с применением борорганического соединения, алкина, диалкилдисульфида и т.п. Кроме того, полученное в этом документе производное алкина может быть преобразовано до производного алкена, производного алкила и т.п. путем проведения общепринятого каталитического восстановления с использованием палладиевого катализатора, газообразного водорода и т.п. Кроме того, промежуточное соединение сложного эфира (15-2), в котором R5 представляет собой цианогруппу, и R7 представляет собой трифторметильную группу, может быть синтезировано путем осуществления взаимодействия промежуточного соединения сложного эфира (13-1-1) с метилфторсульфонилдифторацетатом при нагревании в присутствии йодида меди. Кроме того, проведение указанных реакций сочетания возможно в случае соединения (12-1), в котором отсутствует защитная группа R4. Промежуточное соединение сложного эфира (15), в котором защищена фенолгидроксильная группа, подвергают общепринятой реакции гидролиза с получением промежуточного соединения карбоновой кислоты (16).
Способ синтеза iii) Если R8 представляет собой функциональную группу, на которую не влияет последующая реакция, например, алкильную группу, трифторметильную группу, алкоксигруппу или т.п., то промежуточное соединение карбоновой кислоты (23) может быть синтезировано с применением фенола (17), замещенного R8 во 2-м положении, в качестве исходного вещества.
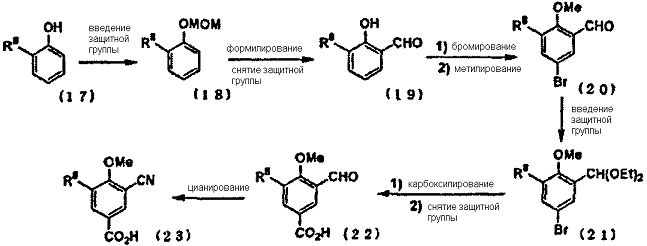
Гидроксильную группу фенола, замещенного R8 во 2-м положении, защищали метоксиметильной группой или т.п., литировали литийорганическим реагентом (н-бутиллитием, втор-бутиллитием, метиллитием и т.д.) и формилировали N,N-диметилформамидом (DMF), а затем снимали защитную группу с гидроксильной группы с получением салицилальдегида (19), замещенного в 3-м положении. Соединение (21) может быть получено путем проведения бромирования гидроксильной группы в пара-положении, путем защиты гидроксильной группы и путем защиты формильной группы. 3-Формил-4-алкоксибензойная кислота (22), замещенная R8 в 5-м положении, может быть синтезирована путем добавления магния и активирующего агента с получением реактива Гриньяра, взаимодействия с диоксидом углерода, и последующим снятием защиты в кислых условиях. Промежуточное соединение карбоновой кислоты (23), замещенное цианогруппой в 3-м положении, может быть синтезировано путем осуществления взаимодействия 3-формил-4-алкоксибензойной кислоты (22) с гидроксиламином с получением оксима, и последующим проведением реакции дегидратации оксима, превращая, тем самым, формильную группу в цианогруппу.
Кроме того, промежуточное соединение карбоновой кислоты (26), замещенное карбамоильной группой в 3-м положении, может быть синтезировано путем преобразования карбоновой кислоты (22) до сложного эфира и окисления формильной группы с получением карбоновой кислоты (24), путем осуществления взаимодействия карбоновой кислоты с амином в присутствии конденсирующего агента и последующего проведения гидролиза сложного эфира.

Синтезированные описанными выше способами соединения общей формулы (1) могут быть выделены и очищены в свободной форме или форме соли путем общепринятых химических операций, таких как экстракция, концентрирование, дистилляция, кристаллизация, фильтрация, перекристаллизация, различные виды хроматографии и т.п. Кроме того, оптические изомеры, стереоизомеры и позиционные изомеры соединений могут быть соответствующим образом выделены методом фракционной перекристаллизации, методом разделения на хиральной колонке, методом образования диастереоизомеров и т.п.
Фармацевтическая композиция, содержащая в качестве активного(ых) ингредиента(ов) вещество(а), выбранное(е) из группы, состоящей из соединения, представленного общей формулой (1), и его фармацевтически приемлемой соли, и их гидрата и их сольвата, может быть использовано «как есть», или может быть использовано в виде состава, содержащего одну, или две, или более фармацевтических добавок. Фармацевтическая композиция может быть использована в любой лекарственной форме и может применяться в виде таблеток, пилюль, капсул, порошков, ожиженных гранул, гранул, растворов, суспензий, сиропов, инъекционных препаратов, наружных препаратов, суппозиториев и т.п.
На типы фармацевтических добавок особых ограничений не существует, если фармацевтическая композиция, содержащая в качестве активных ингредиентов вещество(а), выбранное(е) из группы, состоящей из соединения, представленного общей формулой (1), и его фармацевтически приемлемой соли, и их гидрата и их сольвата, используется в виде вышеупомянутого фармацевтического состава, и возможно применение оснований, наполнителей, смазок, покровных веществ, глазировочных средств, смачивающих веществ, связующих веществ, разрыхлителей, растворителей, солюбилизаторов, растворяющих агентов, способствующих растворению агентов, суспендирующих веществ, диспергирующих веществ, эмульгаторов, поверхностно-активных веществ, изотонических веществ, буферных веществ, регуляторов pH, успокаивающих средств, антисептиков, консервантов, стабилизаторов, антиоксидантов, красителей, подсластителей и т.п., по отдельности или в подходящем сочетании.
Примеры оснований включают каолин, какао-масло, кукурузный крахмал, высушенный гель гидроксида алюминия, кристаллическую целлюлозу, метилцеллюлозу, гидроксипропилцеллюлозу, макрогол и т.п. Примеры наполнителей включают лактозу, сахарозу, крахмал, D-маннит, кукурузный крахмал, кристаллическую целлюлозу, производные целлюлозы (гидроксипропилцеллюлоза, кармелоза кальция, гидроксипропилцеллюлоза с низкой степенью замещения, и т.д.), легкую безводную кремниевую кислоту, фосфорнокислый кальций и т.п. Примеры смазок включают стеарат магния, стеарат кальция, тальк, оксид титана и т.п. Примеры покровных веществ включают кармелозу кальция, оксид титана, стеарат алюминия, тальк и т.п. Примеры глазировочных средств включают сахарозу, лактозу, желатин, парафин, кристаллическую целлюлозу, и т.п. Примеры смачивающих веществ включают глицерин, мочевину, макрогол и т.п. Примеры связующих веществ включают кристаллическую целлюлозу, сахарозу, порошкообразную аравийскую камедь, аргинат натрия, карбоксиметилэтилцеллюлозу, крахмал, сахарозу, очищенный желатин, декстрин, метилцеллюлозу, карбоксиметилцеллюлозу, карбоксиметилцеллюлозу натрия, карбоксиметилэтилцеллюлозу, гидроксиэтилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, пуллулан, поливиниловый спирт, поливинилпирролидон и т.п. Примеры разрыхлителя включают сахарозу, лактозу, крахмал, порошок агара, кроссповидон, карбоксиметилцеллюлозу, карбоксиметилкрахмал натрия, кармелозу, гидроксипропилметилцеллюлозу, лимонный ангидрид, лаурилсульфат натрия, дигидрофосфат кальция и т.п. Примеры растворителей включают очищенную воду, воду для инъекций, этанол, глицерин, пропиленгликоль, макрогол, кунжутное масло, кукурузное масло, соляную кислоту, уксусную кислоту и т.п. Примеры солюбилизаторов включают глицерин, полиоксилстеарат, полисорбат, макрогол и т.п. Примеры растворяющих агентов включают, в дополнение к используемым в качестве вышеупомянутых растворителей, гидроксид натрия, карбонат натрия, меглумин и т.п. Примеры способствующих растворению агентов включают соляную кислоту, уксусную кислоту, лимонную кислоту, цитрат натрия, аспарагиновую кислоту, гидроксид натрия, этанол, пропиленгликоль, D-маннит, бензоант натрия, бензилбензоат, мочевину, триэтаноламин, полисорбат, поливинилпирролидон, макрогол и т.п. Примеры способствующих суспендированию веществ включают аравийскую камедь, бензалкония хлорид, каолин, кармелозу, лаурилсульфат натрия, лауриламинопропионовую кислоту, глицерилмоностеарат, поливиниловый спирт, поливинилпирролидон, карбоксиметилцеллюлозу натрия, метилцеллюлозу, гидроксиметилцеллюлозу, гидроксиэтилцеллюлозу, гидроксипропилцеллюлозу и т.п. Примеры способствующих диспергированию веществ включают цитрат натрия, светлый оксид алюминия, оксид титана, стеарат цинка, полисорбат, макрогол, декстрин, гидроксипропилцеллюлозу с низкой степенью замещения, гидроксипропилцеллюлозу и т.п. Примеры эмульгаторов включают бензалкония хлорид, глицерин, пропиленгликоль, цетанол, лецитин, ланолин, лаурилсульфат натрия и т.п. Примеры поверхностно-активного вещества включают сквален, цетанол, полиоксиэтиленцетиловый эфир, лауромакрогол и т.п. Примеры изотонических веществ включают глюкозу, D-сорбит, хлорид натрия, глицерин, D-маннит и т.п. Примеры буферных веществ включают буферные растворы, такие как фосфатный, ацетатный, карбонатный, цитратный буферы и т.п. Примеры регуляторов pH включают неорганические кислоты, такие как соляная кислота и фосфорная кислота и их соли, органические кислоты, такие как уксусная кислота, лимонная кислота и молочная кислота и их соли и т.п. Примеры успокаивающих средств включают креатинин, бензиловый спирт и т.п. Примеры антисептиков включают сложные эфиры параоксибензойной кислоты, хлорбутанол, бензиловый спирт, фенетиловый спирт, дегидроуксусную кислоту, сорбиновую кислоту и т.п. Примеры консервантов включают бензойную кислоту, сложные эфиры параоксибензойной кислоты, сорбиновую кислоту и т.п. Примеры стабилизаторов включают таурин, аминокислоты, сложные эфиры параоксибензойной кислоты, бензиловый спирт, кристаллическую целлюлозу, макрогол и т.п. Примеры антиоксидантов включают сульфит, аскорбиновую кислоту и т.п. Примеры красителей включают пищевые красители (β-каротин, рибофлавин) и т.п. Примеры подсластителей включают аспартам, сахарозу, D-сорбит, мальтозу и т.п. Примеры ароматизаторов включают горькую эссенцию, горькую основу, и т.п.
Новое производное фенола по настоящему изобретению характеризуется высокой концентрацией неизмененного соединения в моче и обладает заметным урикозурическим действием, а потому новое производное фенола или его фармацевтически приемлемая соль, или их гидрат или их сольват, применимы в качестве лекарственного средства для регуляции реабсорбции мочевой кислоты и ускорения выведения мочевой кислоты; лекарственного средства для снижения количества мочевой кислоты и/или концентрации мочевой кислоты в крови и/или ткани; лекарственного средства для применения при профилактике и/или лечении заболевания, ассоциированного с мочевой кислотой в крови и/или ткани; лекарственного средства для применения при профилактике и/или лечении гиперурикемии; и лекарственного средства для применения при профилактике и/или лечении заболевания, ассоциированного с гиперурикемией, и/или заболевания, сопровождающегося гиперурикемией.
Для терминов «заболевание, ассоциированное с мочевой кислотой в крови и/или ткани» или «заболевание, ассоциированное с гиперурикемией, и/или заболевание, сопровождающееся гиперурикемией» особых ограничений не существует, если только заболевание представляет собой ассоциированное с мочевой кислотой заболевание, вне зависимости от того, является ли связь прямой или косвенной, или если предполагается, что заболевание ассоциировано с мочевой кислотой и/или заболевание осложнено этими заболеваниями. Их примеры включают подагру, мочекаменную болезнь, ожирение, гиперлипидемию, аномальную толерантность к глюкозе, сахарный диабет, метаболический синдром, заболевание почек, кровоизлияние в мозг и/или сердечно-сосудистое заболевание, а также могут быть включены осложнения указанных заболеваний.
Для субъекта с подагрой особого ограничения не существует, если только у него присутствует болезненное состояние, которое соответствует диагностическим критериям или согласуется с ними. Например, включаются субъекты, по меньшей мере, с одним болезненным состоянием из числа подагрического узла, подагрического артрита и подагрической почки. Примеры заболевания почек включают без ограничения почечную недостаточность, альбуминурию, нефрит, уремию, ESRD и т.п. Примеры цереброваскулярного заболевания включают без ограничения острое нарушение мозгового кровообращения, деменцию и т.п. Примеры сердечно-сосудистого заболевания включают без ограничения гипертензию, ишемическую болезнь сердца, заболевание сонных артерий, артериосклероз, гипертрофию сердца, тромбоз, эндотелиальную дисфункцию и/или сердечно-сосудистые заболевания (стенокардия, инфаркт миокарда и т.п.).
Кроме того, новое производное фенола по настоящему изобретению может быть использовано в сочетании с другими лекарственными средствами и/или профилактическими средствами от вышеуказанных заболеваний, и применимы для эффективного лечения указанных заболеваний. Новое производное фенола по настоящему изобретению применимо, поскольку оно может супрессировать увеличение содержания урата в крови при использовании в сочетании с лекарством, которое приводит к увеличению уровня урата в крови (например, антигипертензивный диуретик, противотуберкулезное лекарство, гиполипидемическое лекарство, противовоспалительный анальгетик, противоастматическое средство, иммуносупрессирующее лекарство, антиметаболит, противораковое лекарство и т.п.). Предполагается, что вещество, способное снижать содержание урата в крови (аллопуринол), эффективно от нейродегенеративных заболеваний (болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона, боковой амиотрофический склероз и т.п.), панкреатитов и синдрома апноэ во время сна. Таким образом, новое производное фенола по настоящему изобретению или его фармацевтически приемлемую соль, или их гидрат или их сольват, также можно использовать для профилактики и/или лечения нейродегенеративных заболеваний, заболеваний пищеварительной системы, таких как панкреатиты, и заболеваний дыхательных путей, таких как синдром апноэ во время сна.
Доза и число доз соединений по настоящему изобретению или фармацевтической композиции, содержащей указанные соединения, могут быть соответствующим образом подобраны в зависимости от симптомов, возраста и пола пациента, лекарственной формы и типа лекарства, используемого в сочетании, и т.п. Например, суточная доза для взрослых обычно может быть выбрана в пределах от 0,1 до 1000 мг, предпочтительно от 1 до 500 мг, и вышеупомянутая доза может быть введена однократно в сутки или в несколько приемов в виде отдельных порций. Фармацевтическая композиция по настоящему изобретению может быть введена по отдельности или может быть введена в сочетании с другими лекарственными средствами, обладающими такой же и/или другой эффективностью.
Примеры
Настоящее изобретение будет конкретно описано посредством примеров, но настоящее изобретение не ограничивается последующими примерами.
Значения сокращений, используемых в примерах, являются следующими:
1H-ЯМР: спектр протонного ядерного магнитного резонанса, CDCl3: дейтерированный хлороформ, ДМСО-d6: дейтерированный диметилсульфоксид, CD3OD: дейтерированный метанол, Гц: Герц, J: константа взаимодействия, м: мультиплет, септ: септет, квинт: квинтет, кв: квартет, дт: дублет триплетов, дд: дублет дублетов, ддд: двойной дублет дублетов, т: триплет, д: дублет, с: синглет, ушир.с: уширенный синглет, M: молярная концентрация, н: нормальная концентрация. ЯМР означает спектр ядерного магнитного резонанса при 270 МГц, и в качестве внутреннего стандарта использовали тетраметилсилан (TMS). МС означает масс-спектрометрию, и в качестве способа ионизации использовали аппарат с ионизацией электрораспылением (ESI).
Пример 1: 3-(3,5-дихлор-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез 2,3-дигидро-1,3-бензотиазола
37% формалин (5,2 мл) разбавляли водой (80 мл), и добавляли диизопропиловый эфир (80 мл) и 2-аминобензолтиол (7,84 г), а затем перемешивали смесь при комнатной температуре в течение 30 минут. Органический слой разделяли, и экстрагировали водный слой диизопропиловым эфиром. Органические слои объединяли, промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и использовали полученный остаток для синтеза (c).
(b) Синтез 3,5-дихлор-4-метоксибензоилхлорида
К 3,5-дихлор-4-метоксибензойной кислоте (8,81 г) добавляли толуол (170 мл), N,N-диметилформамид (5 капель) и тионилхлорид (6,0 мл), а затем перемешивали смесь при 60°C в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, полученный остаток подвергали азеотропной перегонке с толуолом, а затем использовали для синтеза (c).
(c) Синтез 3-(3,5-дихлор-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол растворяли в хлороформе (50 мл), добавляли к раствору триэтиламин (17,4 мл) и 3,5-дихлор-4-метоксибензоилхлорид, а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и использовали полученный остаток для синтеза (d).
(d) Синтез 3-(3,5-дихлор-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3,5-Дихлор-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол растворяли в хлороформе (230 мл), к раствору при 0°C добавляли 70% мета-хлорпербензойную кислоту (43,25 г), а затем смесь перемешивали при комнатной температуре в течение 20 часов и гасили добавлением 10% тиосульфата натрия. Растворитель отгоняли в условиях пониженного давления, добавляли 1н раствор гидроксида натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (13,25 г) в виде бесцветного кристаллического вещества.
(e) Синтез 3-(3,5-дихлор-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3,5-Дихлор-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (1,00 г) растворяли в N,N-диметилформамиде (5 мл), добавляли хлорид лития (570 мг), а затем перемешивали смесь при 130°C в течение 2 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из этанола с получением указанного в заголовке соединения (749 мг) в виде бесцветного кристаллического вещества.
1Н-ЯМР δ (ДМСО-d6): 5,35 (2H, с), 7,44 (1H, дд, J=7,6, 7,6 Гц), 7,74 (2H, с), 7,76 (1H, дд, J=8,4, 7,6 Гц), 7,90 (1H, д, J=7,6 Гц), 8,04 (1H, д, J=8,4 Гц), 11,04 (1H, ушир.с). MS (m/z): 356 (M-H)-, 358 (M+2-H)-.
Пример 2: 3-(3,5-дихлор-4-гидроксибензоил)-2,3-дигидро-1,3-бензотиазол
3-(3,5-дихлор-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол (300 мг) растворяли в N,N-диметилформамиде (6 мл), добавляли к раствору хлорид лития (374 мг), а затем перемешивали смесь при 120°C в течение 16 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/этилацетата с получением указанного в заголовке соединения (214 мг) в виде коричневого кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 5,36 (2H, с), 7,03-7,13 (2H, м), 7,31-7,37 (1H, м), 7,50 (1H, ушир.с), 7,65 (2H, с), 10,89 (1H, ушир.с). MS (m/z):324 (M-H)-, 326 (M+2-H)-.
Пример 3: 3-(3,5-дихлор-4-гидроксибензоил)-1-оксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез 3-(3,5-дихлор-4-метоксибензоил)-1-оксо-2,3-дигидро-1,3-бензотиазола
3-(3,5-Дихлор-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол (500 мг) растворяли в хлороформе (10 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (320 мг), и перемешивали смесь при 0°C в течение 10 минут. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 1/1) с получением указанного в заголовке соединения (336 мг) в виде бесцветного кристаллического вещества.
(b) Синтез 3-(3,5-дихлор-4-гидроксибензоил)-1-оксо-2,3-дигидро-1,3-бензотиазола
3-(3,5-Дихлор-4-метоксибензоил)-1-оксо-2,3-дигидро-1,3-бензотиазол (336 мг) растворяли в N,N-диметилформамиде (6 мл), добавляли к раствору хлорид лития (400 мг), а затем перемешивали смесь при 120°C в течение 16 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из этилацетата/метанола с получением указанного в заголовке соединения (220 мг) в виде коричневого кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 5,07 (2H, с), 7,38 (1H, дд, J=7,6, 7,6 Гц), 7,70 (1Н, ддд, J=8,3, 7,6, 0,8 Гц), 7,73 (2H, с), 8,00 (1H, д, J=8,3 Гц), 8,07 (1H, д, J=7,6 Гц), 11,06 (1H, ушир.с). MS (m/z): 340(M-H)-, 342(M+2-H)-.
Пример 4: 3-(3-циано-4-гидрокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез 1-метоксиметокси-2-трифторметилбензола
2-Трифторметилфенол (50,00 г) растворяли в N,N-диметилформамиде (100 мл), добавляли к раствору карбонат калия (85,14 г) и хлорметилметиловый эфир (34,7 мл), а затем перемешивали смесь при охлаждении в течение 1 часа. К реакционному раствору добавляли воду, а затем экстрагировали смесь н-гексаном. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (64,13 г) в виде бесцветного маслянистого вещества.
(b) Синтез 2-гидрокси-3-трифторметилбензальдегида
1-Метоксиметокси-2-трифторметилбензол (64,13 г) растворяли в тетрагидрофуране (500 мл), под струей аргона при -70°C в течение 45 минут добавляли к раствору 2,77M раствор н-бутиллития в н-гексане (123 мл), а затем перемешивали смесь в течение 1 часа. Добавляли N,N-диметилформамид (28,5 мл), а затем перемешивали при комнатной температуре в течение 30 минут. Добавляли 4н соляную кислоту (310 мл), а затем перемешивали при 60°C в течение 19 часов. Органический растворитель отгоняли в условиях пониженного давления, и экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (59,16 г) в виде желтого кристаллического вещества.
(c) Синтез 5-бром-2-гидрокси-3-трифторметилбензальдегида
2-Гидрокси-3-трифторметилбензальдегид (59,16 г) растворяли в ацетонитриле (500 мл), добавляли к раствору N-бромсукцинимид (57,56 г), а затем перемешивали смесь при 0°C в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и промывали полученное кристаллическое вещество н-гексаном (50 мл) с получением указанного в заголовке соединения (63,98 г) в виде бледно-желтого кристаллического вещества.
(d) Синтез 5-бром-2-метокси-3-трифторметилбензальдегида
5-Бром-2-гидрокси-3-трифторметилбензальдегид (63,98 г) растворяли в N,N-диметилформамиде (130 мл), при водяном охлаждении добавляли к раствору карбонат калия (65,79 г) и диметилсерную кислоту (31,6 мл), а затем перемешивали смесь при комнатной температуре в течение 3 часов. К реакционному раствору добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (66,19 г) в виде коричневого кристаллического вещества.
(e) Синтез 5-бром-1-диэтоксиметил-2-метокси-3-трифторметилбензола
5-Бром-2-метокси-3-трифторметилбензальдегид (66,19 г) растворяли в н-гексане (130 мл) и триэтилортоформиате (51 мл), добавляли к раствору Амберлист-15 (6,62 г), а затем нагревали смесь с обратным холодильником в течение 3 часов. Реакционный раствор фильтровали, а затем отгоняли растворитель в условиях пониженного давления с получением указанного в заголовке соединения (82,81 г) в виде коричневого маслянистого вещества.
(f) Синтез 3-формил-4-метокси-5-трифторметилбензойной кислоты
К магнию (5,97 г) добавляли тетрагидрофуран (230 мл) и 5-бром-1-диэтоксиметил-2-метокси-3-трифторметилбензол (31,55 г), а затем перемешивали смесь при комнатной температуре в течение 90 минут. Реакционный раствор охлаждали до 0°C и перемешивали в течение 1 часа в атмосфере углекислого газа, а затем добавляли 2н соляную кислоту (240 мл), и перемешивали смесь при комнатной температуре в течение 16 часов. Органический растворитель отгоняли в условиях пониженного давления, а затем экстрагировали смесь диизопропиловым эфиром. Органический слой экстрагировали добавлением к нему 1н гидроксида натрия (100 мл), а затем дважды промывали водный слой диизопропиловым эфиром. Реакционную смесь подкисляли добавлением к ней 4н соляной кислоты, а затем экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (50,05 г) в виде коричневого твердого вещества.
(g) Синтез 3-циано-4-метокси-5-трифторметилбензойной кислоты
3-Формил-4-метокси-5-трифторметилбензойную кислоту (58,04 г) растворяли в муравьиной кислоте (290 мл), добавляли к раствору гидрохлорид гидроксиламина (17,07 г), и нагревали смесь с обратным холодильником в течение 19 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (15,62 г) в виде коричневого твердого вещества.
1H-ЯМР δ (ДМСО-d6): 4,23 (3H, с), 8,33 (1H, д, J=2,1 Гц), 8,55 (1H, д, J=2,1 Гц). MS (m/z): 244 (M-H)-.
(h) Синтез 3-циано-4-метокси-5-трифторметилбензоилхлорида
К 3-циано-4-метокси-5-трифторметилбензойной кислоте (8,10 г) добавляли толуол (160 мл), N,N-диметилформамид (5 капель) и тионилхлорид (4,80 мл), и перемешивали смесь при 60°C в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, полученный остаток подвергали азеотропной перегонке с толуолом, а затем использовали для синтеза (i).
(i) Синтез 3-(3-циано-4-метокси-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (5,00 г) и 37% формалина (3,0 мл) тем же способом синтеза, что и описанный в примере 1, растворяли в хлороформе (50 мл), добавляли к раствору триэтиламин (11,1 мл) и 3-циано-4-метокси-5-трифторметилбензоилхлорид, а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и использовали полученный остаток для синтеза (j).
(j) Синтез 3-(3-циано-4-метокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-метокси-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазол растворяли в хлороформе (200 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (21,40 г), а затем смесь перемешивали при комнатной температуре в течение 20 часов и гасили добавлением 10% тиосульфата натрия. Растворитель отгоняли в условиях пониженного давления, добавляли 1н раствор гидроксида натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (4,08 г) в виде бледно-желтого твердого вещества.
(k) Синтез 3-(3-циано-4-гидрокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-метокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (4,08 г) растворяли в N,N-диметилформамиде (40 мл), добавляли к раствору хлорид лития (1,74 г), а затем перемешивали смесь при 70°C в течение 2 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/этилацетата с получением указанного в заголовке соединения (2,22 г) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 5,37 (2H, с), 7,44 (1H, дд, J=7,8, 7,8 Гц), 7,77 (1H, ддд, J=7,9, 7,8, 1,3 Гц), 7,91 (1H, дд, J=7,8, 1,3 Гц), 8,09 (1H, д, J=7,9 Гц), 8,10 (1H, д, J=2,1 Гц), 8,27 (1H, д, J=2,1 Гц). MS (m/z): 381 (M-H)-.
Пример 5: 3-(3-Циано-4-гидрокси-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазол
3-(3-Циано-4-метокси-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазол (232 мг) растворяли в N,N-диметилформамиде (3 мл), добавляли к раствору хлорид лития (108 мг), а затем перемешивали смесь при 70°C в течение 1 часа. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/этилацетата с получением указанного в заголовке соединения (131 мг) в виде коричневого кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 5,38 (2H, с), 7,04-7,14 (2H, м), 7,32-7,38 (1H, м), 7,55 (1H, ушир.), 8,05 (1H, д, J=2,1 Гц), 8,22 (1H, д, J=2,1 Гц). MS (m/z): 349 (M-H)-.
Пример 6: 3-(3-Циано-4-гидрокси-5-трифторметилбензоил)-1-оксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез 3-(3-циано-4-метокси-5-трифторметилбензоил)-1-оксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-метокси-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазол (594 мг) растворяли в хлороформе (10 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (433 мг), а затем перемешивали смесь при 0°C в течение 5 минут. Органический растворитель отгоняли в условиях пониженного давления, затем добавляли 1н гидроксид натрия, и промывали выпавшее в осадок кристаллическое вещество 1н гидроксидом натрия и водой с получением указанного в заголовке соединения (619 мг) в виде бесцветного кристаллического вещества.
(b) Синтез 3-(3-циано-4-гидрокси-5-трифторметилбензоил)-1-оксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-метокси-5-трифторметилбензоил)-1-оксо-2,3-дигидро-1,3-бензотиазол (619 мг) растворяли в N,N-диметилформамиде (5 мл), добавляли к раствору хлорид лития (276 мг), и перемешивали смесь при 70°C в течение 3 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (494 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 5,09 (1H, д, J=13,0 Гц), 5,15 (1H, д, J=13,0 Гц), 7,40 (1H, дд, J=7,5, 7,5 Гц), 7,72 (1H, ддд, J=7,5, 7,5, 1,0 Гц), 8,06 (1H, д, J=7,5 Гц), 8,09 (1H, д, J=7,5 Гц), 8,12 (1H, д, J=1,8 Гц), 8,30 (1H, д, J=1,8 Гц). MS (m/z): 365 (M-H)-.
Пример 7: 3-(3-хлор-5-циано-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез метил-3-хлор-5-циано-4-гидроксибензоата
Метил-3-циано-4-гидроксибензоат (2,00 г) растворяли в хлороформе (15 мл) и метаноле (5 мл), добавляли к раствору N-хлорсукцинимид (3,62 г) и 4н соляную кислоту в этилацетате (6,8 мл), а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, добавляли смесь метанола и воды в соотношении 9/1, а затем промывали выпавшее в осадок кристаллическое вещество водой и изопропиловым спиртом с получением указанного в заголовке соединения (1,27 г) в виде бесцветного кристаллического вещества.
(b) Синтез метил-3-хлор-5-циано-4-метоксибензоата
Метил-3-хлор-5-циано-4-гидроксибензоат (1,27 г) растворяли в N,N-диметилформамиде (20 мл), добавляли к раствору карбонат калия (5,00 г) и диметилсерную кислоту (1,70 мл), а затем перемешивали смесь при комнатной температуре в течение 18 часов. Реакционный раствор фильтровали, добавляли воду, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (1,03 г) в виде бесцветного кристаллического вещества.
(c) Синтез 3-хлор-5-циано-4-метоксибензойной кислоты
Метил-3-хлор-5-циано-4-метоксибензоат (1,02 г) растворяли в тетрагидрофуране (15 мл) и воде (6 мл), добавляли к раствору моногидрат гидроксида лития (759 мг), а затем перемешивали смесь при комнатной температуре в течение 90 минут. Органический растворитель отгоняли, и промывали водный слой н-гексаном. Водный слой подкисляли добавлением 1н соляной кислоты, а затем экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (946 мг) в виде бесцветного кристаллического вещества.
1Н-ЯМР δ (ДМСО-d6): 4,43 (3H, с), 8,55 (2H, с), 14,00 (1H, ушир.с). MS (m/z): 210 (M-H)-, 212 (M+2-H)-.
(d) Синтез 3-хлор-5-циано-4-метоксибензоилхлорида
К 3-хлор-5-циано-4-метоксибензойной кислоте (932 мг) добавляли толуол (9,3 мл), N,N-диметилформамид (0,03 мл) и тионилхлорид (0,38 мл), и перемешивали смесь при 60°C в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, а затем проводили азеотропную перегонку с толуолом с получением указанного в заголовке соединения (993 мг) в виде коричневого твердого вещества.
(e) Синтез 3-(3-хлор-5-циано-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (810 мг) и 37% формалина (0,53 мл) тем же способом синтеза, что и описанный в примере 1, растворяли в дихлорметане (15 мл), добавляли к раствору триэтиламин (1,90 мл) и 3-хлор-5-циано-4-метоксибензоилхлорид (993 мг), а затем перемешивали смесь при комнатной температуре в течение 1,5 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 6/1) с получением указанного в заголовке соединения (580 мг) в виде желтого маслянистого вещества.
(f) Синтез 3-(3-хлор-5-циано-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Хлор-5-циано-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол (187 мг) растворяли в дихлорметане (2 мл), и добавляли к раствору 70% мета-хлорпербензойную кислоту (607 мг). После перемешивания смеси при комнатной температуре в течение 5 часов добавляли 1н гидроксид натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (183 мг) в виде бледно-желтого твердого вещества.
(g) Синтез 3-(3-хлор-5-циано-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Хлор-5-циано-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (180 мг) растворяли в N,N-диметилформамиде (2 мл), добавляли к раствору хлорид лития (87 мг), а затем перемешивали смесь при 100°C в течение 1 часа. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/хлороформа с получением указанного в заголовке соединения (146 мг) в виде бледно-желтого кристаллического вещества.
1Н-ЯМР δ (ДМСО-d6): 5,32 (2H, с), 7,44 (1H, ддд, J=8,4, 7,3, 0,8 Гц), 7,75 (1H, ддд, J=8,6, 7,3, 1,4 Гц), 7,88 (1H, дд, J=8,4, 1,4 Гц), 7,99 (1H, д, J=2,2 Гц), 8,00 (1H, д, J=2,2 Гц), 8,06 (1H, д, J=8,6 Гц). MS (m/z): 347 (M-H)-.
Пример 8: 3-(3-хлор-5-циано-4-гидроксибензоил)-2,3-дигидро-1,3-бензотиазол
3-(3-Хлор-5-циано-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол (213 мг) растворяли в N,N-диметилформамиде (2 мл), добавляли к раствору хлорид лития (111 мг), а затем перемешивали смесь при 100°C в течение 2 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, полученный остаток очищали по методу колоночной хроматографии на силикагеле (этилацетат/метанол = 10/1), а затем выкристаллизовывали из н-гексана/хлороформа с получением указанного в заголовке соединения (68 мг) в виде бледно-желтого кристаллического вещества.
1Н-ЯМР δ (ДМСО-d6): 5,37 (2H, с), 7,01-7,16 (2H, м), 7,33 (1H, дд, J=6,5, 2,2 Гц), 7,45 (1H, д, J=7,0 Гц), 7,79 (1H, с), 7,81 (1H, с). MS (m/z): 315 (M-H)-.
Пример 9: 3-(3-трет-бутил-5-циано-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез метил-3-трет-бутил-4-гидроксибензоата
Метил-4-гидроксибензоат (3,00 г) растворяли в метансульфоновой кислоте (15 мл), добавляли к раствору 2-бром-2-метилпропан (11,1 мл), а затем перемешивали смесь при 70°C в течение 16 часов. К полученному реакционному раствору добавляли метанол (20 мл), а затем реакционную смесь перемешивали при 50°C в течение 3 часов. Добавляли 1н гидроксид калия, а затем экстрагировали смесь этилацетатом. Органический слой промывали водным 10% раствором карбоната калия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 3/1) с получением указанного в заголовке соединения (1,83 г) в виде бледно-желтого кристаллического вещества.
(b) Синтез метил-3-трет-бутил-4-гидрокси-5-йодбензоата
Метил-3-трет-бутил-4-гидроксибензоат (1,83 г) растворяли в дихлорметане (24 мл) и метаноле (3 мл), добавляли к раствору N-йодсукцинимид (2,08 г) и трифторметансульфоновую кислоту (3 мл), а затем перемешивали смесь при комнатной температуре в течение 15 минут. К реакционному раствору добавляли воду, а затем разделяли органический слой. Органический слой промывали 10% тиосульфатом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (2,77 г) в виде коричневого кристаллического вещества.
(c) Синтез метил-3-трет-бутил-5-йод-4-метоксибензоата
Метил-3-трет-бутил-4-гидрокси-5-йодбензоат (2,77 г) растворяли в N,N-диметилформамиде (50 мл), добавляли к раствору карбонат калия (12,0 г) и диметилсерную кислоту (4,1 мл), а затем перемешивали смесь при комнатной температуре в течение 16 часов. К реакционному раствору добавляли воду, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (2,77 г) в виде коричневого кристаллического вещества.
(d) Синтез метил-3-трет-бутил-5-циано-4-метоксибензоата
Метил-3-трет-бутил-5-йод-4-метоксибензоат (2,77 г) растворяли в N,N-диметилформамиде (30 мл), добавляли к раствору цианид меди (965 мг), а затем перемешивали смесь при 150°C в течение 2,5 часов. К полученному реакционному раствору добавляли 10% карбонат калия, а затем экстрагировали смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 3/1) с получением указанного в заголовке соединения (1,48 г) в виде желтого маслянистого вещества.
(e) Синтез 3-трет-бутил-5-циано-4-метоксибензойной кислоты
Метил-3-трет-бутил-5-циано-4-метоксибензоат (1,48 г) растворяли в метаноле (20 мл), тетрагидрофуране (5 мл) и воде (5 мл), добавляли к раствору моногидрат гидроксида лития (753 мг), а затем перемешивали смесь при комнатной температуре в течение 2 часов. К полученному реакционному раствору добавляли 10% соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (1,18 г) в виде бледно-желтого кристаллического вещества.
1Н-ЯМР δ (CDCl3): 1,41 (9H, с), 4,26 (3H, с), 8,23 (1H, д, J=2,2 Гц), 8,25 (1H, д, J=2,2 Гц).
(f) Синтез 3-трет-бутил-5-циано-4-метоксибензоилхлорида
К 3-трет-бутил-5-циано-4-метоксибензойной кислоте (586 мг) добавляли толуол (10 мл), N,N-диметилформамид (2 капли) и тионилхлорид (0,27 мл), и перемешивали смесь при 60°C в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, а затем проводили азеотропную перегонку с толуолом с получением указанного в заголовке соединения (630 мг) в виде коричневого маслянистого вещества.
(g) Синтез 3-(3-трет-бутил-5-циано-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (943 мг) и 37% формалина (0,57 мл) тем же способом, что и описанный в примере 1, растворяли в хлороформе (15 мл), добавляли к раствору триэтиламин (1,04 мл) и 3-трет-бутил-5-циано-4-метоксибензоилхлорид (630 мг), а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 6/1) с получением указанного в заголовке соединения (904 мг) в виде желтого маслянистого вещества.
(h) Синтез 3-(3-трет-бутил-5-циано-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-трет-Бутил-5-циано-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол (452 мг) растворяли в хлороформе (9 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (1,02 г), а затем смесь перемешивали при комнатной температуре в течение 16 часов и гасили добавлением 10% тиосульфата натрия. Растворитель отгоняли в условиях пониженного давления, добавляли 1н раствор гидроксида натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (439 мг) в виде бледно-желтого маслянистого вещества.
1Н-ЯМР δ (CDCl3): 1,36 (9H, с), 4,27 (3H, с), 4,93 (2H, с), 7,37 (1H, ддд, J=7,8, 7,1, 1,3 Гц), 7,58 (1H, ддд, J=8,2, 7,1, 1,3 Гц), 7,65 (1H, д, J=2,3 Гц), 7,70 (1H, д, J=2,3 Гц), 7,72 (1H, д, J=2,3 Гц), 7,76-7,80 (1H, м).
(i) Синтез 3-(3-трет-бутил-5-циано-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-трет-Бутил-5-циано-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (364 мг) растворяли в N,N-диметилформамиде (4 мл), добавляли к раствору хлорид лития (401 мг), а затем перемешивали смесь при 120°C в течение 16 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/этилацетата с получением указанного в заголовке соединения (299 мг) в виде бесцветного кристаллического вещества.
1Н-ЯМР δ (ДМСО-d6): 1,37 (9H, с), 5,35 (2H, с), 7,43 (1H, дд, J=7,4, 7,4 Гц), 7,73 (1H, д, J=2,1 Гц), 7,75 (1H, ддд, J=7,4, 7,4, 1,2 Гц), 7,88-7,93 (2H, м), 8,01 (1H, д, J=8,2 Гц), 11,23 (1H, ушир.с). MS (m/z): 369 (M-H)-.
Пример 10: 3-(3-циано-4-гидрокси-5-изопропилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез 5-бром-2-гидрокси-3-изопропилбензальдегида
2-Гидрокси-3-изопропилбензальдегид (20,19 г) растворяли в ацетонитриле (160 мл), при 0°C добавляли к раствору N-бромсукцинимид (17,80 г), а затем перемешивали смесь при комнатной температуре в течение 4 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (25,88 г) в виде желтого маслянистого вещества.
(b) Синтез 5-бром-3-изопропил-2-метоксибензальдегида
5-Бром-2-гидрокси-3-изопропилбензальдегид (25,88 г) растворяли в N,N-диметилформамиде (100 мл), при водяном охлаждении добавляли к раствору карбонат калия (27,64 г) и диметилсерную кислоту (9,5 мл), и перемешивали смесь при комнатной температуре в течение 2 часов. К реакционному раствору добавляли воду, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (26,76 г) в виде коричневого маслянистого вещества.
(c) Синтез 5-бром-1-диэтоксиметил-3-изопропил-2-метоксибензола
5-Бром-3-изопропил-2-метоксибензальдегид (26,76 г) растворяли в н-гексане (50 мл) и триэтилортоформиате (22 мл), добавляли к раствору Амберлист-15 (2,68 г), а затем нагревали смесь с обратным холодильником в течение 4 часов. Реакционный раствор фильтровали, а затем отгоняли растворитель в условиях пониженного давления с получением указанного в заголовке соединения (31,55 г) в виде коричневого маслянистого вещества.
(d) Синтез 3-формил-5-изопропил-4-метоксибензойной кислоты
К магнию (2,43 г) добавляли тетрагидрофуран (100 мл), 5-бром-1-диэтоксиметил-3-изопропил-2-метоксибензол (31,55 г) и 0,97M раствор метилмагнийбромида в тетрагидрофуране (15 мл), а затем перемешивали смесь при комнатной температуре в течение 2 часов. Реакционный раствор охлаждали до 0°C и перемешивали в атмосфере углекислого газа в течение 30 минут, затем добавляли 2н соляную кислоту (100 мл), и перемешивали реакционную смесь при комнатной температуре в течение 16 часов. Органический растворитель отгоняли в условиях пониженного давления, а затем экстрагировали смесь диизопропиловым эфиром. Органический слой экстрагировали добавлением к нему 1н гидроксида натрия (100 мл), а затем водный слой дважды промывали диизопропиловым эфиром. Водный слой подкисляли добавлением к нему 4н соляной кислоты, а затем экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (15,85 г) в виде коричневого твердого вещества.
(e) Синтез 3-циано-5-изопропил-4-метоксибензойной кислоты
3-Формил-5-изопропил-4-метоксибензойную кислоту (15,85 г) растворяли в муравьиной кислоте (80 мл), добавляли к раствору гидрохлорид гидроксиламина (5,45 г), и нагревали смесь с обратным холодильником в течение 19 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (15,62 г) в виде коричневого твердого вещества.
1Н-ЯМР δ (ДМСО-d6): 1,21 (6H, д, J=6,9 Гц), 3,29 (1H, sevent, J=6,9 Гц), 4,04 (3H, с), 8,10 (1H, с). MS (m/z): 218 (M-H)-.
(f) Синтез 3-циано-5-изопропил-4-метоксибензоилхлорида
К 3-циано-5-изопропил-4-метоксибензойной кислоте (658 мг) добавляли толуол (7 мл), N,N-диметилформамид (2 капли) и тионилхлорид (0,33 мл), и перемешивали смесь при 60°C в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, а затем проводили азеотропную перегонку с толуолом с получением указанного в заголовке соединения (710 мг) в виде коричневого маслянистого вещества.
(g) Синтез 3-(3-циано-5-изопропил-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (1,25 г) и 37% формалина (0,83 мл) тем же способом, что и описанный в примере 1, растворяли в хлороформе (7 мл), добавляли к раствору триэтиламин (1,25 мл) и 3-циано-5-изопропил-4-метоксибензоилхлорид (710 мг), а затем перемешивали смесь при комнатной температуре в течение 2 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 6/1) с получением указанного в заголовке соединения (1,02 г) в виде желтого маслянистого вещества.
(h) Синтез 3-(3-циано-5-изопропил-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-5-изопропил-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол (501 мг) растворяли в хлороформе (5 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (996 мг), а затем смесь перемешивали при комнатной температуре в течение 18 часов и гасили добавлением 10% тиосульфата натрия. Растворитель отгоняли в условиях пониженного давления, добавляли 1н раствор гидроксида натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 3/1) с получением указанного в заголовке соединения (439 мг) в виде коричневого аморфного вещества.
(i) Синтез 3-(3-циано-4-гидрокси-5-изопропилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-5-изопропил-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (434 мг) растворяли в N,N-диметилформамиде (5 мл), добавляли к раствору хлорид лития (496 мг), а затем перемешивали смесь при 100°C в течение 20 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/хлороформа с получением указанного в заголовке соединения (351 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 1,18 (6H, д, J=6,8 Гц), 3,35 (1H, sevent, J=6,8 Гц), 5,34 (2H, с), 7,43 (1H, ддд, J=7,8, 7,8, 0,8 Гц), 7,75 (1H, ддд, J=8,4, 7,8, 1,3 Гц), 7,76 (1H, д, J=2,3 Гц), 7,87 (1H, д, J=2,3 Гц), 7,90 (1H, дд, J=7,8, 0,8 Гц), 8,00 (1H, д, J=8,4 Гц). MS (m/z): 355 (M-H)-.
Пример 11: 3-(3-циано-5-циклобутил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
(a) Синтез 1-циклобутил-2-метоксиметоксибензола
2-Циклобутилфенол (871 мг) растворяли в N,N-диметилформамиде (5 мл), и при 0°C добавляли к раствору 60% гидрида натрия (1,30 г). После перемешивания смеси в течение 30 минут добавляли хлорметилметиловый эфир (2,1 мл), и перемешивали смесь в течение 14 часов. К реакционному раствору добавляли воду, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 10/1) с получением указанного в заголовке соединения (1,13 г) в виде бесцветного маслянистого вещества.
(b) Синтез 3-циклобутил-2-гидроксибензальдегида
1-Циклобутил-2-метоксиметоксибензол (1,18 г) растворяли в тетрагидрофуране (11 мл), и под струей аргона при -60°C в течение 15 минут добавляли к раствору 1,01M раствор втор-бутиллития в циклогексане (8,7 мл), а затем перемешивали смесь в течение 2 часов. Добавляли N,N-диметилформамид (0,90 мл), и перемешивали смесь при той же температуре в течение 2 часов. При комнатной температуре добавляли 4н соляную кислоту (15 мл), а затем перемешивали смесь при 60°C в течение 20 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 20/1) с получением указанного в заголовке соединения (0,95 г) в виде бесцветного маслянистого вещества.
(c) Синтез 5-бром-3-циклобутил-2-гидроксибензальдегида
3-Циклобутил-2-гидроксибензальдегид (2,29 г) растворяли в ацетонитриле (30 мл), при 0°C добавляли к раствору N-бромсукцинимид (5,10 г), а затем перемешивали смесь при комнатной температуре в течение 4 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и использовали полученный остаток для синтеза (d).
(d) Синтез 5-бром-3-циклобутил-2-метоксибензальдегида
5-Бром-3-циклобутил-2-гидроксибензальдегид растворяли в N,N-диметилформамиде (30 мл), при водяном охлаждении добавляли к раствору карбонат калия (10,79 г) и диметилсерную кислоту (3,7 мл), а затем перемешивали смесь при комнатной температуре в течение 14 часов. Реакционный раствор фильтровали, добавляли воду, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 20/1) с получением указанного в заголовке соединения (1,27 г) в виде желтого маслянистого вещества.
(e) Синтез 5-бром-3-циклобутил-1-диэтоксиметил-2-метоксибензола
5-Бром-3-циклобутил-2-метоксибензальдегид (1,15 г) растворяли в н-гексане (5 мл) и триэтилортоформиате (0,93 мл), добавляли к раствору Амберлист-15 (115 мг), а затем нагревали смесь с обратным холодильником в течение 3 часов. Реакционный раствор фильтровали, а затем отгоняли растворитель в условиях пониженного давления с получением указанного в заголовке соединения (1,33 г) в виде желтого маслянистого вещества.
(f) Синтез 3-циклобутил-5-формил-4-метоксибензойной кислоты
К магнию (106 мг) добавляли тетрагидрофуран (3,5 мл), 5-бром-3-циклобутил-1-диэтоксиметил-2-метоксибензол (1,33 г) и 0,97M раствор метилмагнийбромида в тетрагидрофуране (1,32 мл), а затем перемешивали смесь при комнатной температуре в течение 1,5 часов. Реакционный раствор охлаждали до 0°C и перемешивали в атмосфере углекислого газа в течение 15 часов, затем добавляли 2н соляную кислоту (10 мл), и перемешивали смесь при комнатной температуре в течение 1 часа. Органический растворитель отгоняли в условиях пониженного давления, а затем экстрагировали диизопропиловым эфиром. Органический слой экстрагировали добавлением к нему 1н гидроксида натрия, а затем водный слой дважды промывали диизопропиловым эфиром. Водный слой подкисляли добавлением к нему 4н соляной кислоты, а затем экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (458 мг) в виде коричневого твердого вещества.
(g) Синтез 3-циано-5-циклобутил-4-метоксибензойной кислоты
3-Циклобутил-5-формил-4-метоксибензойную кислоту (458 мг) растворяли в муравьиной кислоте (2,5 мл), добавляли к раствору гидрохлорид гидроксиламина (163 мг), а затем нагревали смесь с обратным холодильником в течение 19 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (404 мг) в виде коричневого твердого вещества.
1H-ЯМР δ (CDCl3): 1,82-2,48 (6H, м), 3,76 (1H, квинтет, J=8,7 Гц), 4,15 (3H, с), 8,18 (1H, д, J=2,2 Гц), 8,20 (1H, д, J=2,2 Гц). MS (m/z): 230 (M-H)-.
(h) Синтез 3-циано-5-циклобутил-4-метоксибензоилхлорида
К 3-циано-5-циклобутил-4-метоксибензойной кислоте (190 мг) добавляли толуол (2,0 мл), N,N-диметилформамид (1 каплю) и тионилхлорид (0,07 мл), и перемешивали смесь при 60°C в течение 3 часов. Растворитель отгоняли в условиях пониженного давления, а затем проводили азеотропную перегонку с толуолом с получением указанного в заголовке соединения (204 мг) в виде коричневого маслянистого вещества.
(i) Синтез 3-(3-циано-5-циклобутил-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (153 мг) и 37% формалина (0,10 мл) тем же способом, что и описанный в примере 1, растворяли в дихлорметане (3 мл), добавляли к раствору триэтиламин (0,34 мл) и 3-циано-5-циклобутил-4-метоксибензоилхлорид (204 мг), а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и использовали полученный остаток для синтеза (j).
(j) Синтез 3-(3-циано-5-циклобутил-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-5-циклобутил-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол растворяли в дихлорметане (4 мл), и добавляли к раствору 70% мета-хлорпербензойную кислоту (1,62 г). После перемешивания смеси при комнатной температуре в течение 28 часов добавляли 1н гидроксид натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (156 мг) в виде желтого твердого вещества.
(k) Синтез 3-(3-циано-5-циклобутил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(5-Циано-3-циклобутил-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (150 мг) растворяли в N,N-диметилформамиде (1,5 мл), добавляли к раствору хлорид лития (248 мг), а затем перемешивали смесь при 120°C в течение 2,5 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/хлороформа с получением указанного в заголовке соединения (95 мг) в виде коричневого кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 1,72-2,18 (4H, м), 2,25-2,39 (2H, м), 3,77 (1H, квинтет, J=8,7 Гц), 5,35 (2H, с), 7,44 (1H, дд, J=7,6, 7,6 Гц), 7,77 (1H, дд, J=8,4, 7,6 Гц), 7,79 (1H, д, J=2,2 Гц), 7,87 (1H, д, J=2,2 Гц), 7,91 (1H, д, J=7,6 Гц), 8,04 (1H, д, J=8,4 Гц). MS (m/z): 367 (M-H)-.
Пример 12: 3-(3-циано-5-этил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез метил-3-циано-4-метокси-5-триметилсиланилэтинилбензоата
Метил-3-циано-5-йод-4-метоксибензоат (2,13 г) растворяли в тетрагидрофуране (30 мл), добавляли к раствору триэтиламин (10 мл), йодид меди (256 мг), тетракис(трифенилфосфин)палладий (777 мг) и триметилсилилацетилен (858 мг), а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 4/1) с получением указанного в заголовке соединения (2,03 г) в виде коричневого маслянистого вещества.
(b) Синтез метил-3-циано-5-этинил-4-метоксибензоата
Метил-3-циано-4-метокси-5-триметилсиланилэтинилбензоат (2,03 г) растворяли в тетрагидрофуране (20 мл), добавляли к раствору водный 1н раствор гидроксида натрия (8 мл), а затем перемешивали смесь при комнатной температуре в течение 10 минут. Растворитель отгоняли в условиях пониженного давления, добавляли 1н соляную кислоту, и экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (1,37 г) в виде бесцветного кристаллического вещества.
(c) Синтез метил-3-циано-5-этил-4-метоксибензоата
Метил-3-циано-5-этинил-4-метоксибензоат (475 мг) растворяли в тетрагидрофуране (10 мл), добавляли к раствору 5% палладий на углероде (150 мг), а затем перемешивали смесь в атмосфере водорода при комнатной температуре в течение 30 минут. Реакционный раствор фильтровали, затем отгоняли растворитель в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 5/1) с получением указанного в заголовке соединения (480 мг) в виде бесцветного кристаллического вещества.
(d) Синтез 3-циано-5-этил-4-метоксибензойной кислоты
Метил-3-циано-5-этил-4-метоксибензоат (480 мг) растворяли в тетрагидрофуране (6 мл) и воде (2 мл), добавляли к раствору моногидрат гидроксида лития (370 мг), а затем перемешивали смесь при комнатной температуре в течение 5 часов. Растворитель отгоняли в условиях пониженного давления, добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (403 мг) в виде бесцветного кристаллического вещества.
1Н-ЯМР δ (ДМСО-d6): 1,50 (3H, т, J=7,5 Гц), 3,01 (2H, кв., J=7,5 Гц), 4,36 (3H, с), 8,40 (1H, д, J=2,1 Гц), 8,41 (1H, д, J=2,1 Гц). MS (m/z): 204 (M-H)-.
(e) Синтез 3-циано-5-этил-4-метоксибензоилхлорида
К 3-циано-5-этил-4-метоксибензойной кислоте (347 мг) добавляли толуол (3,5 мл), N,N-диметилформамид (0,01 мл) и тионилхлорид (0,15 мл), а затем перемешивали смесь при 60°C в течение 14 часов. Растворитель отгоняли в условиях пониженного давления, а затем проводили азеотропную перегонку с толуолом с получением указанного в заголовке соединения (387 мг) в виде коричневого маслянистого вещества.
(f) Синтез 3-(3-циано-5-этил-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (318 мг) и 37% формалина (0,21 мл) тем же способом, что и описанный в примере 1, растворяли в дихлорметане (6 мл), добавляли к раствору триэтиламин (0,71 мл) и 3-циано-5-этил-4-метоксибензоилхлорид (387 мг), а затем перемешивали смесь при комнатной температуре в течение 1,5 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 6/1) с получением указанного в заголовке соединения (498 мг) в виде желтого маслянистого вещества.
(g) Синтез 3-(3-циано-5-этил-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-5-этил-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол (366 мг) растворяли в дихлорметане (7 мл), и добавляли к раствору 70% мета-хлорпербензойную кислоту (1,20 г). После перемешивания смеси при комнатной температуре в течение 14 часов добавляли 1н гидроксид натрия, и экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (309 мг) в виде бесцветного твердого вещества.
(h) Синтез 3-(3-циано-5-этил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-5-этил-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (309 мг) растворяли в N,N-диметилформамиде (3 мл), добавляли к раствору хлорид лития (443 мг), а затем перемешивали смесь при 120°C в течение 2,5 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/хлороформа с получением указанного в заголовке соединения (257 мг) в виде коричневого кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 1,15 (3H, т, J=7,6 Гц), 2,68 (2H, кв., J=7,6 Гц), 5,34 (2H, с), 7,43 (1H, дд, J=7,6, 7,6 Гц), 7,74 (1H, д, J=2,2 Гц), 7,75 (1H, дд, J=8,4, 7,6 Гц), 7,88 (1H, д, J=2,2 Гц), 7,90 (1H, д, J=7,6 Гц), 8,00 (1H, д, J=8,4 Гц), 11,01 (1H, ушир.с). MS (m/z): 341 (M-H)-.
Пример 13: 3-(3-циано-5-циклопропил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез метил-3-циано-5-циклопропил-4-метоксибензоата
Метил-3-циано-4-гидрокси-5-йодбензоат (1,00 г) растворяли в 1,4-диоксане (15 мл), добавляли к раствору карбонат калия (1,31 г), циклопропилбороновую кислоту (325 мг) и дихлорид [1,3-бис-(2,6-диизопропилфенил)имидазол-2-илиден](3-хлорпиридил)палладия (108 мг), а затем перемешивали смесь под струей аргона при 95°C в течение 22 часов. Реакционный раствор фильтровали, отгоняли растворитель в условиях пониженного давления, а затем очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 4/1) с получением указанного в заголовке соединения (348 мг) в виде бледно-желтого кристаллического вещества.
(b) Синтез 3-циано-5-циклопропил-4-метоксибензойной кислоты
Метил-3-циано-5-циклопропил-4-метоксибензоат (491 мг) растворяли в тетрагидрофуране (7,5 мл) и воде (2,5 мл), добавляли к раствору моногидрат гидроксида лития (359 мг), а затем перемешивали смесь при комнатной температуре в течение 20 часов. Органический растворитель отгоняли в условиях пониженного давления, и промывали водный слой н-гексаном. К водному слою добавляли 1н соляную кислоту, и экстрагировали смесь этилацетатом в кислых условиях. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (394 мг) в виде бледно-коричневого кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 0,74-0,79 (2H, м), 1,03-1,10 (2H, м), 2,13-2,23 (1H, м), 4,06 (3H, с), 7,65 (1H, д, J=2,0 Гц), 8,04 (1H, д, J=2,0 Гц). MS (m/z): 216 (M-H)-.
(c) Синтез 3-циано-5-циклопропил-4-метоксибензоилхлорида
К 3-циано-5-циклопропил-4-метоксибензойной кислоте (200 мг) добавляли толуол (2 мл), N,N-диметилформамид (1 капля) и тионилхлорид (0,10 мл), и перемешивали смесь при 60°C в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, полученный остаток подвергали азеотропной перегонке с толуолом и использовали для синтеза (d).
(d) Синтез 3-(3-циано-5-циклопропил-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (346 мг) и 37% формалина (0,23 мл) тем же способом, что и описанный в примере 1, растворяли в хлороформе (3 мл), добавляли к раствору триэтиламин (0,38 мл) и 3-циано-5-циклопропил-4-метоксибензоилхлорид, а затем перемешивали при комнатной температуре в течение 2 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и использовали полученный остаток для синтеза (e).
(e) Синтез 3-(3-циано-5-циклопропил-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-5-циклопропил-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол растворяли в хлороформе (5 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (422 мг), а затем смесь перемешивали при комнатной температуре в течение 16 часов и гасили добавлением 10% тиосульфата натрия. Растворитель отгоняли в условиях пониженного давления, добавляли 1н раствор гидроксида натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/этилацетата/метанола с получением указанного в заголовке соединения (147 мг) в виде бесцветного кристаллического вещества.
(f) Синтез 3-(3-циано-5-циклопропил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-5-циклопропил-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (142 мг) растворяли в N,N-диметилформамиде (2 мл), добавляли к раствору хлорид лития (163 мг), а затем перемешивали смесь при 100°C в течение 23 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/этилацетата с получением указанного в заголовке соединения (115 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 0,69-0,75 (2H, м), 0,94-1,01 (2H, м), 2,05-2,15 (1H, м), 5,30 (2H, с), 7,41 (1H, д, J=2,1 Гц), 7,43 (1H, дд, J=7,8, 7,8 Гц), 7,75 (1H, ддд, J=8,4, 7,8, 1,2 Гц), 7,84 (1H, д, J=2,1 Гц), 7,90 (1H, д, J=7,8 Гц), 8,02 (1H, д, J=8,4 Гц). MS (m/z): 353 (M-H)-.
Пример 14: 3-(3-циано-5-этинил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез 3-циано-5-этинил-4-метоксибензойной кислоты
Метил-3-циано-5-этинил-4-метоксибензоат (640 мг) растворяли в тетрагидрофуране (6 мл) и воде (3 мл), добавляли к раствору моногидрат гидроксида лития (495 мг), а затем перемешивали смесь при комнатной температуре в течение 3 часов. Растворитель отгоняли в условиях пониженного давления, добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (610 мг) в виде коричневого кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 4,20 (3H, с), 4,72 (1H, с), 8,17 (1H, д, J=2,1 Гц), 8,24 (1H, д, J=2,1 Гц). MS (m/z): 200 (M-H)-.
(b) Синтез 3-циано-5-этинил-4-метоксибензоилхлорида
К 3-циано-5-этинил-4-метоксибензойной кислоте (610 мг) при охлаждении на льду добавляли толуол (6 мл), N,N-диметилформамид (1 каплю) и оксалилхлорид (0,32 мл), и перемешивали смесь при комнатной температуре в течение 1,5 часов. Растворитель отгоняли в условиях пониженного давления, а полученный остаток подвергали азеотропной перегонке с толуолом и использовали для синтеза (c).
(c) Синтез 3-(3-циано-5-этинил-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (570 мг) и 37% формалина (0,38 мл) тем же способом, что и описанный в примере 1, растворяли в дихлорметане (10 мл), добавляли к раствору триэтиламин (1,2 мл) и 3-циано-5-этинил-4-метоксибензоилхлорид, а затем перемешивали смесь при комнатной температуре в течение 2,5 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 5/1) с получением указанного в заголовке соединения (283 мг) в виде желтого маслянистого вещества.
(d) Синтез 3-(3-циано-5-этинил-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-5-этинил-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол (344 мг) растворяли в дихлорметане (5 мл), и добавляли к раствору 70% мета-хлорпербензойную кислоту (1,94 г). После перемешивания смеси при комнатной температуре в течение 16 часов добавляли 1н гидроксид натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (199 мг) в виде желтого маслянистого вещества.
(e) Синтез 3-(3-циано-5-этинил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-5-этинил-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (197 мг) растворяли в N,N-диметилформамиде (2 мл), добавляли к раствору хлорид лития (239 мг), а затем перемешивали смесь при 100°C в течение 1 часа. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, полученный остаток очищали по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 1/1), а затем выкристаллизовывали из н-гексана/ацетона с получением указанного в заголовке соединения (102 мг) в виде бледно-желтого кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 4,58 (1H, с), 5,35 (2H, с), 7,44 (1H, дд, J=7,6, 7,6 Гц), 7,76 (1H, дд, J=8,4, 7,6 Гц), 7,91 (1H, д, J=8,4, Гц), 7,93 (1H, д, J=2,4 Гц), 8,04 (1H, д, J=2,4 Гц), 8,05 (1H, д, J=7,6 Гц). MS (m/z): 337 (M-H),
Пример 15: 3-(3-циано-4-гидрокси-5-метилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез метил-3-бром-4-гидроксибензоата
Метил-4-гидроксибензоат (25,00 г) растворяли в хлороформе (225 мл) и метаноле (25 мл), к раствору по каплям добавляли раствор брома (8,5 мл) в хлороформе (30 мл), а затем перемешивали смесь в течение 2 часов. Реакционный раствор разбавляли хлороформом, промывали водой, водным 10% раствором тиосульфата натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (37,81 г) в виде бесцветного кристаллического вещества.
(b) Синтез метил-3-циано-4-гидроксибензоата
Метил-3-бром-4-гидроксибензоат (37,81 г) растворяли в N,N-диметилформамиде (250 мл), и добавляли к раствору цианид меди (22,03 г). После перемешивания смеси при 150°C в течение 16 часов к раствору при охлаждении на льду добавляли карбонат калия (68,00 г) и хлорметилметиловый эфир (14,8 мл), а затем перемешивали смесь в течение 2 часов. Реакционный раствор фильтровали, добавляли воду, а затем экстрагировали реакционную смесь этилацетатом. После трехкратного повторения добавления воды к органическому слою, перемешивания смеси, фильтрования смеси и разделения органического слоя органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и растворяли полученный остаток в хлороформе (30 мл). К раствору добавляли трифторуксусную кислоту (30 мл), а затем перемешивали смесь при комнатной температуре в течение 2 часов. Растворитель отгоняли в условиях пониженного давления, и промывали полученный остаток смесью н-гексана и этилацетата в соотношении 2/1 с получением указанного в заголовке соединения (6,53 г) в виде бледно-желтого кристаллического вещества.
(c) Синтез метил-3-циано-4-гидрокси-5-йодбензоата
Метил-3-циано-4-гидроксибензоат (6,47 г) растворяли в хлороформе (80 мл) и метаноле (10 мл), добавляли к раствору N-йодсукцинимид (8,63 г) и трифторметансульфоновую кислоту (2,5 мл), а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, и промывали полученный остаток водой с получением указанного в заголовке соединения (11,29 г) в виде бледно-желтого кристаллического вещества.
(d) Синтез метил-3-циано-5-йод-4-метоксибензоата
Метил-3-циано-4-гидрокси-5-йодбензоат (11,29 г) растворяли в N,N-диметилформамиде (230 мл), добавляли к раствору карбонат калия (49,20 г) и диметилсерную кислоту (17,0 мл), а затем перемешивали смесь при комнатной температуре в течение 18 часов. После фильтрования реакционной смеси добавляли воду, и собирали выпавшее в осадок кристаллическое вещество путем фильтрования с получением указанного в заголовке соединения (8,99 г) в виде бледно-желтого кристаллического вещества.
(e) Синтез 3-циано-5-йод-4-метоксибензойной кислоты
Метил-3-циано-5-йод-4-метоксибензоат (8,00 г) растворяли в тетрагидрофуране (100 мл) и воде (50 мл), добавляли к раствору моногидрат гидроксида лития (4,23 г), а затем перемешивали смесь при комнатной температуре в течение 4 часов. Органический растворитель отгоняли в условиях пониженного давления, и промывали водный слой н-гексаном. Водный слой подкисляли добавлением 2н соляной кислоты, а затем экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (7,26 г) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 4,38 (3H, с), 8,58 (1H, д, J=2,0 Гц), 8,86 (1H, д, J=2,0 Гц), 13,89 (1H, ушир.с). MS (m/z):302 (M-1)-.
(f) Синтез 3-циано-5-йод-4-метоксибензоилхлорида
К 3-циано-5-йод-4-метоксибензойной кислоте (512 мг) добавляли толуол (5 мл), N,N-диметилформамид (1 каплю) и тионилхлорид (0,15 мл), и перемешивали смесь при 60°C в течение 15 часов. Растворитель отгоняли в условиях пониженного давления, а затем проводили азеотропную перегонку с толуолом с получением указанного в заголовке соединения (527 мг) в виде бледно-желтого твердого вещества.
(g) Синтез 3-(3-циано-5-йод-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (317 мг) и 37% формалина (0,21 мл) тем же способом, что и описанный в примере 1, растворяли в дихлорметане (6 мл), добавляли к раствору триэтиламин (0,70 мл) и 3-циано-5-йод-4-метоксибензоилхлорид (527 мг), а затем перемешивали смесь при комнатной температуре в течение 14 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 5/1) с получением указанного в заголовке соединения (435 мг) в виде желтого маслянистого вещества.
(h) Синтез 3-(3-циано-5-йод-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-5-йод-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол (196 мг) растворяли в дихлорметане (4 мл), и добавляли к раствору 70% мета-хлорпербензойную кислоту (495 мг). После перемешивания смеси при комнатной температуре в течение 2 часов добавляли 1н гидроксид натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, а затем очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 2/1) с получением указанного в заголовке соединения (106 мг) в виде бледно-желтого маслянистого вещества.
(i) Синтез 3-(3-циано-4-гидрокси-5-йодбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-5-йод-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (106 мг) растворяли в N,N-диметилформамиде (1 мл), добавляли к раствору хлорид лития (40 мг), а затем перемешивали смесь при 100°C в течение 2 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (100 мг) в виде желтого маслянистого вещества.
1H-ЯМР δ (ДМСО-d6): 5,35 (2H, с), 7,43 (1H, дд, J=7,8, 7,3 Гц), 7,76 (1H, дд, J=8,4, 7,3 Гц), 7,90 (1H, д, J=7,8 Гц), 8,02 (1H, д, J=2,2 Гц), 8,04 (1H, д, J=8,4 Гц), 8,27 (1H, д, J=2,2 Гц).
(j) Синтез 3-(3-циано-4-метокси-5-метилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-гидрокси-5-йодбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (334 мг) растворяли в N,N-диметилформамиде (3,5 мл), добавляли к раствору 2,2'-бипиридин (11 мг), цинковый порошок (95 мг), бромид никеля (16 мг) и диметилдисульфид (0,04 мл), а затем перемешивали смесь при 80°C в течение 1 часа. После фильтрования реакционной смеси добавляли 1н соляную кислоту, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и растворяли полученный остаток в N,N-диметилформамиде (3 мл). К раствору добавляли карбонат калия (298 мг) и диметилсерную кислоту (0,13 мл), а затем перемешивали смесь при комнатной температуре в течение 1 часа. К реакционному раствору добавляли воду, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 2/1) с получением указанного в заголовке соединения (142 мг) в виде бледно-желтого маслянистого вещества.
(k) Синтез 3-(3-циано-4-гидрокси-5-метилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-метокси-5-метилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (142 мг) растворяли в N,N-диметилформамиде (1 мл), добавляли к раствору хлорид лития (64 мг), а затем перемешивали смесь при 100°C в течение 1,5 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/хлороформа с получением указанного в заголовке соединения (95 мг) в виде желтого кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 2,46 (3H, с), 5,34 (2H, с), 7,45 (1H, дд, J=7,6, 7,3 Гц), 7,68 (1H, д, J=2,2 Гц), 7,77 (1H, дд, J=8,4, 7,6 Гц), 7,82 (1H, д, J=2,2 Гц), 7,91 (1H, д, J=7,3 Гц), 8,08 (1H, д, J=8,4 Гц). MS (m/z): 359 (M-H)-.
Пример 16: 3-(3-циано-4-гидрокси-5-метилсульфонилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез 3-(3-циано-4-метокси-5-метилсульфонилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-метокси-5-метилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (163 мг) растворяли в дихлорметане (4 мл), и добавляли к раствору 70% мета-хлорпербензойную кислоту (480 мг). После перемешивания смеси при комнатной температуре в течение 18 часов к раствору добавляли 1н гидроксид натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (142 мг) в виде бледно-желтого твердого вещества.
(b) Синтез 3-(3-циано-4-гидрокси-5-метилсульфонилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-метокси-5-метилсульфонилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (138 мг) растворяли в N,N-диметилформамиде (1 мл), добавляли к раствору хлорид лития (58 мг), а затем перемешивали смесь при 70°C в течение 3 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/хлороформа с получением указанного в заголовке соединения (69 мг) в виде коричневого кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 3,26 (3H, с), 5,36 (2H, с), 7,40 (1H, дд, J=7,6 Гц, 7,6 Гц), 7,73 (1H, дд, J=8,1, 7,6 Гц), 7,88 (1H, д, J=8,1 Гц), 7,92 (1H, д, J=7,6 Гц), 8,11 (2H, с). MS (m/z): 391 (M-H)-.
Пример 17: 3-(3-циано-4-гидрокси-5-метилсульфинилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
3-(3-Циано-4-гидрокси-5-метилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (7 мг) растворяли в тетрагидрофуране (0,5 мл) и воде (0,5 мл), добавляли к раствору оксон (6 мг), а затем перемешивали смесь при комнатной температуре в течение 1 часа. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/хлороформа с получением указанного в заголовке соединения (6 мг) в виде бледно-желтого кристаллического вещества.
1H-ЯМР δ (CDCl3): 2,88 (3H, с), 4,96 (2H, с), 7,33 (1H, дд, J=7,6, 7,4 Гц), 7,57 (1H, дд, J=8,0, 7,4 Гц), 7,67 (1H, д, J=8,0 Гц), 7,72 (1H, д, J=7,6 Гц), 7,89 (1H, с), 7,94 (1H, с). MS (m/z): 375 (M-H)-.
Пример 18: 3-(3-хлор-4-гидрокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез метил-3-хлор-4-гидрокси-5-трифторметилбензоата
Метил-4-гидрокси-3-трифторметилбензоат (1,40 г) растворяли в хлороформе (14 мл) и метаноле (3 мл), добавляли к раствору N-хлорсукцинимид (1,70 г) и трифторметансульфоновую кислоту (40 мкл), а затем перемешивали смесь при комнатной температуре в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, добавляли 10% тиосульфат натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (1,26 г) в виде коричневого твердого вещества.
(b) Синтез метил-3-хлор-4-метокси-5-трифторметилбензоата
Метил-3-хлор-4-гидрокси-5-трифторметилбензоат (1,26 г) растворяли в N,N-диметилформамиде (6 мл), добавляли к раствору карбонат калия (3,42 г) и диметилсерную кислоту (1,40 мл), а затем перемешивали смесь при комнатной температуре в течение 2 часов. К реакционному раствору добавляли воду, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 6/1) с получением указанного в заголовке соединения (639 мг) в виде бесцветного маслянистого вещества.
(c) Синтез 3-хлор-4-метокси-5-трифторметилбензойной кислоты
Метил-3-хлор-4-метокси-5-трифторметилбензоат (634 мг) растворяли в тетрагидрофуране (4 мл) и воде (4 мл), добавляли к раствору моногидрат гидроксида лития (396 мг), а затем перемешивали смесь при комнатной температуре в течение 2 часов. Органический растворитель отгоняли в условиях пониженного давления, а затем промывали водный слой н-гексаном. Водный слой подкисляли добавлением 1н соляной кислоты, а затем экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (579 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 3,97 (3H, с), 8,08 (1H, д, J=2,1 Гц), 8,26 (1H, д, J=2,1 Гц), 13,69 (1H, ушир.). MS (m/z): 253 (M-H)-, 255 (M+2-H)-.
(d) Синтез 3-хлор-4-метокси-5-трифторметилбензоилхлорида
К 3-хлор-4-метокси-5-трифторметилбензойной кислоте (300 мг) добавляли толуол (3 мл), N,N-диметилформамид (1 каплю) и тионилхлорид (0,13 мл), и перемешивали смесь при 60°C в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, а полученный остаток подвергали азеотропной перегонке с толуолом и использовали для синтеза (e).
(e) Синтез 3-(3-хлор-4-метокси-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (442 мг) и 37% формалина (0,29 мл) тем же способом, что и описанный в примере 1, растворяли в хлороформе (8 мл), добавляли к раствору триэтиламин (0,49 мл) и 3-хлор-4-метокси-5-трифторметилбензоилхлорид, а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и использовали полученный остаток для синтеза (f).
(f) Синтез 3-(3-хлор-4-метокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Хлор-4-метокси-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазол растворяли в хлороформе (5 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (726 мг), а затем смесь перемешивали при комнатной температуре в течение 18 часов и гасили добавлением 10% тиосульфата натрия. Растворитель отгоняли в условиях пониженного давления, добавляли 1н раствор гидроксида натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 3/1) с получением указанного в заголовке соединения (74 мг) в виде бесцветного кристаллического вещества.
(g) Синтез 3-(3-хлор-4-гидрокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Хлор-4-метокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (74 мг) растворяли в N,N-диметилформамиде (2 мл), добавляли к раствору хлорид лития (77 мг), а затем перемешивали смесь при 70°C в течение 22 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из диэтилового эфира с получением указанного в заголовке соединения (65 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 5,36 (2H, с), 7,44 (1H, ддд, J=7,8, 7,8, 0,8 Гц), 7,77 (1H, ддд, J=8,2, 7,8, 1,3 Гц), 7,86 (1H, д, J=2,1 Гц), 7,91 (1H, дд, J=7,8, 0,8 Гц), 8,06 (1H, д, J=2,1 Гц), 8,07 (1H, д, J=8,2 Гц). MS (m/z): 390 (M-H)-.
Пример 19: 3-(3-хлор-5-фтор-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез 3-хлор-5-фтор-4-метоксибензоилхлорида
К 3-хлор-5-фтор-4-метоксибензойной кислоте (295 мг) добавляли толуол (3 мл), N,N-диметилформамид (1 каплю) и тионилхлорид (0,15 мл), а затем перемешивали смесь при 60°C в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, а полученный остаток подвергали азеотропной перегонке с толуолом и использовали для синтеза (b).
(b) Синтез 3-(3-хлор-5-фтор-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (346 мг) и 37% формалина (0,23 мл) тем же способом, что и описанный в примере 1, растворяли в хлороформе (3 мл), добавляли к раствору триэтиламин (0,38 мл) и 3-хлор-5-фтор-4-метоксибензоилхлорид, а затем перемешивали смесь при комнатной температуре в течение 90 минут. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 8/1) с получением указанного в заголовке соединения (356 мг) в виде бледно-желтого маслянистого вещества.
(c) Синтез 3-(3-хлор-5-фтор-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Хлор-5-фтор-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол (348 мг) растворяли в хлороформе (7 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (739 мг), а затем смесь перемешивали при комнатной температуре в течение 16 часов и гасили добавлением 10% тиосульфата натрия. Растворитель отгоняли в условиях пониженного давления, добавляли 1н раствор гидроксида натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (313 мг) в виде бесцветного кристаллического вещества.
(d) Синтез 3-(3-хлор-5-фтор-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Хлор-5-фтор-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (307 мг) растворяли в N,N-диметилформамиде (6 мл), добавляли к раствору хлорид лития (163 мг), а затем перемешивали смесь при 100°C в течение 16 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 1/1) с получением указанного в заголовке соединения (296 мг) в виде бледно-желтого аморфного продукта.
1H-ЯМР δ (ДМСО-d6): 5,35 (2H, с), 7,43 (1H, дд, J=7,4, 7,4 Гц), 7,59 (1H, дд, J=11,1, 1,8 Гц), 7,61 (1H, с), 7,76 (1H, ддд, J=8,4, 7,4, 1,2 Гц), 7,90 (1H, д, J=7,4 Гц), 8,02 (1H, д, J=8,4 Гц), 11,35 (1H, ушир.с). MS (m/z): 340 (M-H)-, 342 (M+2-H)-.
Пример 20: 3-(3,5-дифтор-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез 3,5-дифтор-4-метоксибензоилхлорида
К 3,5-дифтор-4-метоксибензойной кислоте (310 мг) добавляли толуол (3 мл), N,N-диметилформамид (1 каплю) и тионилхлорид (0,14 мл), а затем перемешивали смесь при 60°C в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, а затем проводили азеотропную перегонку с толуолом с получением указанного в заголовке соединения (347 мг) в виде коричневого маслянистого вещества.
(b) Синтез 3-(3,5-дифтор-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (316 мг) и 37% формалина (0,21 мл) тем же способом, что и описанный в примере 1, растворяли в дихлорметане (3 мл), добавляли к раствору диизопропилэтиламин (0,56 мл) и 3,5-дифтор-4-метоксибензоилхлорид (347 мг), а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и использовали полученный остаток для синтеза (c).
(c) Синтез 3-(3,5-дифтор-4-метоксибензоил)-2,3-дигидро-1,1-диоксо-1,3-бензотиазола
3-(3,5-Дифтор-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол растворяли в дихлорметане (8 мл), и добавляли к раствору 70% мета-хлорпербензойную кислоту (2,01 г). После перемешивания смеси при комнатной температуре в течение 5 часов добавляли 1н гидроксид натрия, и экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (453 мг) в виде бесцветного твердого вещества.
(d) Синтез 3-(3,5-дифтор-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3,5-Дифтор-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (453 мг) растворяли в N,N-диметилформамиде (4 мл), добавляли к раствору хлорид лития (559 мг), а затем перемешивали смесь при 100°C в течение 16 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и полученный остаток очищали по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 1/1), а затем выкристаллизовывали из диэтилового эфира с получением указанного в заголовке соединения (270 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (CD3OD): 5,14 (2H, с), 7,32 (2H, д, J=8,3 Гц), 7,40 (1H, дд, J=7,8, 7,3 Гц), 7,66 (1H, дд, J=8,4, 7,3 Гц), 7,77 (1H, д, J=7,8 Гц), 7,83 (1H, д, J=8,4 Гц). MS (m/z): 324 (M-H)-.
Пример 21: 3-(3-хлор-4-гидрокси-5-метилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез метил-3-хлор-4-гидрокси-5-йодбензоата
Метил-3-хлор-4-гидроксибензоат (12,31 г) растворяли в дихлорметане (100 мл) и метаноле (12 мл), добавляли к раствору N-йодсукцинимид (15,59 г) и трифторметансульфоновую кислоту (2 мл), а затем перемешивали смесь при комнатной температуре в течение 2 часов. Растворитель отгоняли в условиях пониженного давления, и промывали полученный остаток водой (100 мл) с получением указанного в заголовке соединения (20,52 г) в виде бесцветного кристаллического вещества.
(b) Синтез метил-3-хлор-5-йод-4-метоксибензоата
Метил-3-хлор-4-гидрокси-5-йодбензоат (3,00 г) растворяли в N,N-диметилформамиде (20 мл), добавляли к раствору карбонат калия (3,98 г) и диметилсерную кислоту (1,82 мл), а затем перемешивали смесь при комнатной температуре в течение 5 часов. Реакционный раствор фильтровали, добавляли воду, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (2,96 г) в виде бледно-желтого кристаллического вещества.
(c) Синтез 3-хлор-5-йод-4-метоксибензойной кислоты
Метил-3-хлор-5-йод-4-метоксибензоат (2,96 г) растворяли в тетрагидрофуране (23 мл) и воде (7 мл), добавляли к раствору моногидрат гидроксида лития (1,52 г), а затем перемешивали смесь при комнатной температуре в течение 19 часов. После отгонки органического растворителя смесь подкисляли добавлением 1н соляной кислоты и экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (2,74 г) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (CDCl3): 3,95 (3H, с), 8,11 (1H, дд, J=2,2 Гц, 0,5 Гц), 8,42 (1H, дд, J=2,2 Гц, 0,5 Гц). MS (m/z): 311 (M-H)-.
(d) Синтез 3-хлор-5-йод-4-метоксибензоилхлорида
К 3-хлор-5-йод-4-метоксибензойной кислоте (2,74 г) добавляли толуол (27 мл), N,N-диметилформамид (1 каплю) и тионилхлорид (0,76 мл), а затем перемешивали смесь при 60°C в течение 15 часов. Растворитель отгоняли в условиях пониженного давления, а затем проводили азеотропную перегонку с толуолом с получением указанного в заголовке соединения (3,05 г) в виде желтого твердого вещества.
(e) Синтез 3-(3-хлор-5-йод-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (1,65 г) и 37% формалина (1,09 мл) тем же способом, что и описанный в примере 1, растворяли в дихлорметане (15 мл), добавляли к раствору диизопропилэтиламин (3,0 мл) и 3-хлор-5-йод-4-метоксибензоилхлорид (3,05 г), а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из смеси н-гексана и этилацетат в соотношении 1/1 с получением указанного в заголовке соединения (2,44 г) в виде бледно-желтого твердого вещества.
(f) Синтез 3-(3-хлор-5-йод-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Хлор-5-йод-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол (200 мг) растворяли в тетрагидрофуране (5 мл), и добавляли к раствору 70% мета-хлорпербензойную кислоту (462 мг). После перемешивания смеси при комнатной температуре в течение 1,5 часов добавляли 1н гидроксид натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (96 мг) в виде бесцветного маслянистого вещества.
(g) Синтез 3-(3-хлор-4-гидрокси-5-йодбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Хлор-5-йод-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (922 мг) растворяли в N,N-диметилформамиде (6 мл), добавляли к раствору хлорид лития (421 мг), а затем перемешивали смесь при 120°C в течение 2,5 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (1,19 г) в виде коричневого маслянистого вещества.
1H-ЯМР δ (CDCl3): 4,98 (2H, с), 7,30-7,92 (6H, м).
(h) Синтез 3-(3-хлор-4-метокси-5-метилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Хлор-4-гидрокси-5-йодбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (1,19 г) растворяли в N,N-диметилформамиде (9 мл), добавляли к раствору 2,2'-бипиридин (32 мг), цинковый порошок (262 мг), бромид никеля (45 мг) и диметилдисульфид (0,09 мл), а затем перемешивали смесь при 80°C в течение 1,5 часов. После фильтрования реакционной смеси добавляли 1н соляную кислоту, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и растворяли полученный остаток в N,N-диметилформамиде (6 мл). К раствору добавляли карбонат калия (828 мг) и диметилсерную кислоту (0,38 мл), а затем перемешивали смесь при комнатной температуре в течение 14 часов. К реакционному раствору добавляли воду, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 3/1) с получением указанного в заголовке соединения (203 мг) в виде желтого кристаллического вещества.
(i) Синтез 3-(3-хлор-4-гидрокси-5-метилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Хлор-4-метокси-5-метилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (203 мг) растворяли в N,N-диметилформамиде (2 мл), добавляли к раствору хлорид лития (258 мг), а затем перемешивали смесь при 120°C в течение 20 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/диэтилового эфира с получением указанного в заголовке соединения (169 мг) в виде коричневого кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 2,44 (3H, с), 5,34 (2H, с), 7,38 (1H, с), 7,43 (1H, дд, J=7,8, 7,6 Гц), 7,54 (1H, с), 7,76 (1H, дд, J=8,6, 7,8 Гц), 7,90 (1H, д, J=7,6 Гц), 8,02 (1H, д, J=8,6 Гц), 10,54 (1H, с). MS (m/z): 368 (M-H)-.
Пример 22: 3-(3-хлор-4-гидрокси-5-метилсульфонилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез 3-(3-хлор-4-метокси-5-метилсульфонилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Хлор-4-метокси-5-метилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (828 мг) растворяли в дихлорметане (10 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (2,13 г), а затем перемешивали смесь при комнатной температуре в течение 16 часов. Добавляли 1н гидроксид натрия, выпавшее в осадок кристаллическое вещество промывали 1н гидроксидом натрия, водой и метанолом с получением указанного в заголовке соединения (589 мг) в виде бесцветного твердого вещества.
(b) Синтез 3-(3-хлор-4-гидрокси-5-метилсульфонилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Хлор-4-метокси-5-метилсульфонилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (586 мг) растворяли в N,N-диметилформамиде (4 мл), добавляли к раствору хлорид лития (241 мг), а затем перемешивали смесь при 120°C в течение 2 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, выпавшее в осадок кристаллическое вещество промывали водой, а затем выкристаллизовывали из н-гексана/хлороформа с получением указанного в заголовке соединения (310 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 3,34 (3H, с), 5,35 (2H, с), 7,44 (1H, дд, J=7,6, 7,3 Гц), 7,76 (1H, дд, J=8,4, 7,3 Гц), 7,91 (1H, д, J=7,6 Гц), 7,99 (1H, д, J=2,2 Гц), 8,03 (1H, д, J=8,4 Гц), 8,09 (1H, д, J=2,2 Гц). MS (m/z): 400 (M-H)-.
Пример 23: 3-(3-хлор-4-гидрокси-5-метилсульфинилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
3-(3-Хлор-4-гидрокси-5-метилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (51 мг) растворяли в тетрагидрофуране (0,5 мл) и воде (0,5 мл), добавляли к раствору оксон (43 мг), а затем перемешивали смесь при комнатной температуре в течение 1 часа. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/диэтилового эфира с получением указанного в заголовке соединения (45 мг) в виде коричневого кристаллического вещества.
1Н-ЯМР δ (ДМСО-d6): 2,82 (3H, с), 5,35 (2H, с), 7,43 (1H, дд, J=7,6, 7,3 Гц), 7,75 (1H, дд, J=7,8, 7,6 Гц), 7,81 (1H, д, J=1,6 Гц), 7,86-8,02 (2H, м), 7,93 (1H, д, J=1,6 Гц). MS (m/z): 384 (M-H)-.
Пример 24: 3-(4-гидрокси-3-метилсульфонил-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез метил-4-гидрокси-3-йод-5-трифторметилбензоата
Метил-4-гидрокси-3-трифторметилбензоат (916 мг) растворяли в дихлорметане (15 мл), добавляли к раствору N-йодсукцинимид (1,06 г) и трифторуксусную кислоту (5 мл), а затем перемешивали смесь при комнатной температуре в течение 1 часа. После отгонки растворителя в условиях пониженного давления добавляли 10% тиосульфат натрия, и экстрагировали смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (1,44 г) в виде коричневого твердого вещества.
(b) Синтез метил-4-гидрокси-3-метилсульфанил-5-трифторметилбензоата
Метил-4-гидрокси-3-йод-5-трифторметилбензоат (1,26 г) растворяли в N,N-диметилформамиде (12 мл), добавляли к раствору 2,2'-бипиридин (57 мг), цинковый порошок (476 мг), бромид никеля (80 мг) и диметилдисульфид (172 мг), а затем перемешивали смесь при 130°C в течение 1 часа. После фильтрования реакционной смеси добавляли 1н соляную кислоту, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 4/1) с получением указанного в заголовке соединения (276 мг) в виде бесцветного кристаллического вещества.
(c) Синтез метил-4-метокси-3-метилсульфанил-5-трифторметилбензоата
Метил-4-гидрокси-3-метилсульфанил-5-трифторметилбензоат (327 мг) растворяли в N,N-диметилформамиде (6 мл), добавляли к раствору карбонат калия (1,70 г) и диметилсерную кислоту (0,35 мл), а затем перемешивали смесь при комнатной температуре в течение 1 часа. После фильтрования реакционной смеси добавляли 1н соляную кислоту, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (344 мг) в виде бесцветного маслянистого вещества.
(d) Синтез 4-метокси-3-метилсульфанил-5-трифторметилбензойной кислоты
Метил-4-метокси-3-метилсульфанил-5-трифторметилбензоат (303 мг) растворяли в тетрагидрофуране (3 мл) и воде (1,5 мл), добавляли к раствору моногидрат гидроксида лития (215 мг), а затем перемешивали смесь при комнатной температуре в течение 1 часа. После отгонки органического растворителя водный слой подкисляли добавлением 1н соляной кислоты и экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, а затем концентрировали в условиях пониженного давления с получением указанного в заголовке соединения (277 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 2,56 (3H, с), 3,89 (3H, с), 7,90 (1H, д, J=1,7 Гц), 8,00 (1H, д, J=1,7 Гц), 13,49 (1H, ушир.с). MS (m/z): 265 (М-Н)-.
(e) Синтез 4-метокси-3-метилсульфанил-5-трифторметилбензоилхлорида
К 4-метокси-3-метилсульфанил-5-трифторметилбензойной кислоте (277 мг) добавляли толуол (7 мл), N,N-диметилформамид (1 каплю) и тионилхлорид (0,12 мл), а затем перемешивали смесь при 60°C в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, а полученный остаток подвергали азеотропной перегонке с толуолом и использовали для синтеза (f).
(f) Синтез 3-(4-метокси-3-метилсульфанил-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (346 мг) и 37% формалина (0,23 мл) тем же способом, что и описанный в примере 1, растворяли в хлороформе (4 мл), добавляли к раствору триэтиламин (0,43 мл) и 4-метокси-3-метилсульфанил-5-трифторметилбензоилхлорид, а затем перемешивали смесь при комнатной температуре в течение 1 часа. После отгонки растворителя в условиях пониженного давления добавляли воду, и экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и использовали полученный остаток для синтеза (g).
(g) Синтез 3-(4-метокси-3-метилсульфонил-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(4-Метокси-3-метилсульфанил-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазол, синтезированный на стадии (f), растворяли в хлороформе (8 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (1,34 г), а затем смесь перемешивали при комнатной температуре в течение 20 часов и гасили добавлением 10% тиосульфата натрия. После отгонки растворителя в условиях пониженного давления добавляли 1н гидроксид натрия, и экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 2/1) с получением указанного в заголовке соединения (310 мг) в виде бесцветного кристаллического вещества.
(h) Синтез 3-(4-гидрокси-3-метилсульфонил-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(4-Метокси-3-метилсульфонил-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (305 мг) растворяли в N,N-диметилформамиде (3 мл), добавляли к раствору хлорид лития (288 мг), а затем перемешивали смесь при 70°C в течение 1 часа. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали водой, 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (288 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 5,34 (2H, с), 5,71 (3H, ушир.с), 7,43 (1H, дд, J=7,6, 7,6 Гц), 7,75 (1H, дд, J=8,4, 7,6 Гц), 7,90 (1H, д, J=7,6 Гц), 8,02 (1H, д, J=8,4 Гц), 8,14 (1H, д, J=1,9 Гц), 8,25 (1H, д, J=1,9 Гц). MS (m/z): 434(M-H)-.
Пример 25: 3-(5-трет-бутил-4-гидрокси-3-метилсульфонилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез метил-3-трет-бутил-4-гидрокси-5-метилсульфанилбензоата
Метил-3-трет-бутил-4-гидрокси-5-йодбензоат (1,00 г) растворяли в N,N-диметилформамиде (10 мл), добавляли к раствору 2,2'-бипиридин (47 мг), цинковый порошок (391 мг), бромид никеля (66 мг) и диметилдисульфид (142 мг), а затем перемешивали смесь при 130°C в течение 1 часа. После фильтрования реакционной смеси добавляли 1н соляную кислоту, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 12/1) с получением указанного в заголовке соединения (382 мг) в виде бледно-желтого маслянистого вещества.
(b) Синтез метил-3-трет-бутил-4-метокси-5-метилсульфанилбензоата
Метил-3-трет-бутил-4-гидрокси-5-метилсульфанилбензоат (377 мг) растворяли в N,N-диметилформамиде (7 мл), добавляли к раствору карбонат калия (818 мг) и диметилсерную кислоту (0,32 мл), а затем перемешивали смесь при комнатной температуре в течение 20 часов. К реакционному раствору добавляли воду, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (344 мг) в виде бледно-желтого маслянистого вещества.
(c) Синтез метил-3-трет-бутил-4-метокси-5-метилсульфонилбензоата
Метил-3-трет-бутил-4-метокси-5-метилсульфанилбензоат (344 мг) растворяли в хлороформе (7 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (884 мг), а затем перемешивали смесь при комнатной температуре в течение 3 часов. После отгонки растворителя в условиях пониженного давления добавляли 1н гидроксид натрия, и экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (387 мг) в виде бесцветного кристаллического вещества.
(d) Синтез 3-трет-бутил-4-метокси-5-метилсульфонилбензойной кислоты
Метил-3-трет-бутил-4-метокси-5-метилсульфонилбензоат (382 мг) растворяли в тетрагидрофуране (4 мл) и воде (2 мл), добавляли к раствору моногидрат гидроксида лития (320 мг), а затем перемешивали смесь при комнатной температуре в течение 5 часов. После отгонки органического растворителя водный слой подкисляли добавлением 1н соляной кислоты, а затем экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, а затем концентрировали в условиях пониженного давления с получением указанного в заголовке соединения (372 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (CDCl3): 1,43 (9H, с), 3,32 (3H, с), 3,99 (3Н, с), 8,26 (1Н, д, J=2,2 Гц), 8,32 (1H, д, J=2,2 Гц). MS (m/z): 285 (M-H)-.
(e) Синтез 3-трет-бутил-4-метокси-5-метилсульфонилбензоилхлорида
К 3-трет-бутил-4-метокси-5-метилсульфонилбензойной кислоте (200 мг) добавляли толуол (4 мл), N,N-диметилформамид (1 каплю) и тионилхлорид (80 мкл), а затем перемешивали смесь при 60°C в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, а полученный остаток подвергали азеотропной перегонке с толуолом и использовали для синтеза (f).
(f) Синтез 3-(3-трет-бутил-4-метокси-5-метилсульфонилбензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (182 мг) и 37% формалина (87 мкл) тем же способом, что и описанный в примере 1, растворяли в хлороформе (4 мл), добавляли к раствору триэтиламин (0,29 мл) и 3-трет-бутил-4-метокси-5-метансульфонилбензоилхлорид, а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 4/1) с получением указанного в заголовке соединения (246 мг) в виде бледно-желтого аморфного продукта.
(g) Синтез 3-(3-трет-бутил-4-метокси-5-метилсульфонилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-трет-Бутил-4-метокси-5-метилсульфонилбензоил)-2,3-дигидро-1,3-бензотиазол (241 мг) растворяли в хлороформе (5 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (410 мг), а затем смесь перемешивали при комнатной температуре в течение 16 часов и гасили добавлением 10% тиосульфата натрия. После отгонки растворителя в условиях пониженного давления добавляли 1н гидроксид натрия, и экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (236 мг) в виде бесцветного аморфного продукта.
(h) Синтез 3-(3-трет-бутил-4-гидрокси-5-метилсульфонилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-трет-Бутил-4-метокси-5-метилсульфонилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (232 мг) растворяли в N,N-диметилформамиде (5 мл), добавляли к раствору хлорид лития (225 мг), а затем перемешивали смесь при 130°C в течение 2 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали водой, 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 1/1) с получением указанного в заголовке соединения (201 мг) в виде бесцветного аморфного продукта.
1Н-ЯМР δ (ДМСО-d6): 1,40 (9H, с), 3,42 (3H, с), 5,33 (2H, с), 7,43 (1H, дд, J=7,6, 7,6 Гц), 7,75 (1H, ддд, J=8,3, 7,6, 1,3 Гц), 7,80 (1H, д, J=2,2 Гц), 7,91 (1H, д, J=7,6 Гц), 7,97 (1H, д, J=8,3 Гц), 7,99 (1H, д, J=2,2 Гц), 10,06 (1H, ушир.с). MS (m/z): 422 (M-H)-.
Пример 26: 3-(4-гидрокси-3-метокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез этил-4-бензилокси-3-метокси-5-трифторметилбензоата
Этил-4-гидрокси-3-метокси-5-трифторметилбензоат (583 мг) растворяли в N,N-диметилформамиде (5 мл), и при 0°C добавляли к раствору 60% гидрид натрия (132 мг). После перемешивания смеси в течение 30 минут к раствору добавляли бензилбромид (0,31 мл), а затем перемешивали смесь в течение 14 часов. К реакционному раствору добавляли воду, а затем экстрагировали реакционную смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 7/1) с получением указанного в заголовке соединения (704 мг) в виде желтого маслянистого вещества.
(b) Синтез 4-бензилокси-3-метокси-5-трифторметилбензойной кислоты
Этил-4-бензилокси-3-метокси-5-трифторметилбензоат (704 мг) растворяли в тетрагидрофуране (6 мл) и воде (2 мл), добавляли к раствору моногидрат гидроксида лития (333 мг), а затем перемешивали смесь при 60°C в течение 3 часов. После отгонки органического растворителя смесь подкисляли добавлением 1н соляной кислоты, а затем экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (606 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (CDCl3): 4,00 (3H, с), 5,21 (2H, с), 7,33-7,54 (5H, м), 7,85 (1H, с), 8,00 (1H, с). MS (m/z): 325 (M-H)-.
(c) Синтез 4-бензилокси-3-метокси-5-трифторметилбензоилхлорида
К 4-бензилокси-3-метокси-5-трифторметилбензойной кислоте (601 мг) добавляли толуол (6 мл), N,N-диметилформамид (1 каплю) и тионилхлорид (0,16 мл), а затем перемешивали смесь при 60°C в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, а затем проводили азеотропную перегонку с толуолом с получением указанного в заголовке соединения (657 мг) в виде желтого маслянистого вещества.
(d) Синтез 3-(4-бензилокси-3-метокси-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (346 мг) и 37% формалина (0,23 мл) тем же способом, что и описанный в примере 1, растворяли в дихлорметане (3 мл), добавляли к раствору диизопропилэтиламин (0,63 мл) и 4-бензилокси-3-метокси-5-трифторметилбензоилхлорид (657 мг), а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 6/1) с получением указанного в заголовке соединения (578 мг) в виде желтого маслянистого вещества.
(e) Синтез 3-(4-бензилокси-3-метокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(4-Бензилокси-3-метокси-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазол (578 мг) растворяли в дихлорметане (10 мл), и добавляли к раствору 70% мета-хлорпербензойную кислоту (1,94 г). После перемешивания смеси при комнатной температуре в течение 4 часов добавляли 1н гидроксид натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (574 мг) в виде бесцветного маслянистого вещества.
(f) Синтез 3-(4-гидрокси-3-метокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(4-Бензилокси-3-метокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (574 мг) растворяли в тетрагидрофуране (6 мл), добавляли к раствору 5% палладий на углероде (310 мг), а затем перемешивали смесь при комнатной температуре в течение 22 часов в атмосфере водорода. Реакционный раствор фильтровали, затем отгоняли растворитель в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/хлороформа с получением указанного в заголовке соединения (353 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 3,93 (3H, с), 5,35 (2H, с), 7,43 (1H, дд, J=8,1, 7,3 Гц), 7,47 (1H, с), 7,54 (1H, с), 7,76 (1H, дд, J=7,3, 7,3 Гц), 7,90 (1H, д, J=7,3 Гц), 8,02 (1H, д, J=8,1 Гц), 10,68 (1H, с). MS (m/z): 386 (M-H)-.
Пример 27: 3-(3-диметилкарбамоил-4-гидрокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез метил-3-формил-4-метокси-5-трифторметилбензоата
3-Формил-4-метокси-5-трифторметилбензойную кислоту (5,00 г) растворяли в метаноле (30 мл), добавляли к раствору гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (4,25 г), а затем перемешивали смесь при комнатной температуре в течение 90 минут. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 4/1) с получением указанного в заголовке соединения (2,98 г) в виде бесцветного кристаллического вещества.
(b) Синтез сложного 1-метилового эфира 4-метокси-5-трифторметилизофталевой кислоты
Метил-3-формил-4-метокси-5-трифторметилбензоат (1,50 г) растворяли в ацетонитриле (15 мл) и водном 5% растворе лимонной кислоты, добавляли к раствору 2-метил-2-бутен (2,00 г) и хлорит натрия (776 мг), а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 10% тиосульфатом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и промывали полученное кристаллическое вещество н-гексаном с получением указанного в заголовке соединения (1,15 г) в виде бесцветного кристаллического вещества.
(c) Синтез метил-3-диметилкарбамоил-4-метокси-5-трифторметилбензоата
Сложный 1-метиловый эфир 4-метокси-5-трифторметилизофталевой кислоты (500 мг) растворяли в дихлорметане (10 мл), добавляли к раствору гидрохлорид диметиламина (440 мг), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (1,56 г) и триэтиламин (3,00 мл), а затем перемешивали смесь при комнатной температуре в течение 4 часов. Растворитель отгоняли в условиях пониженного давления, добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (356 мг) в виде бледно-желтого кристаллического вещества.
(d) Синтез 3-диметилкарбамоил-4-метокси-5-трифторметилбензойной кислоты
Метил-3-диметилкарбамоил-4-метокси-5-трифторметилбензоат (348 мг) растворяли в тетрагидрофуране (3 мл) и воде (1,5 мл), добавляли к раствору моногидрат гидроксида лития (191 мг), а затем перемешивали смесь при комнатной температуре в течение 1 часа. Органический растворитель отгоняли в условиях пониженного давления и подкисляли добавлением 1н соляной кислоты, а затем экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (341 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (CDC13): 2,91 (3H, с), 3,18 (3H, с), 3,96 (3H, с), 8,22 (1H, д, J=2,3 Гц), 8,35 (1H, дд, J=2,3, 0,6 Гц). MS (m/z): 290 (M-H)-.
(e) Синтез 3-диметилкарбамоил-4-метокси-5-трифторметилбензоилхлорида
К 3-диметилкарбамоил-4-метокси-5-трифторметилбензойной кислоте (333 мг) добавляли толуол (3 мл), N,N-диметилформамид (1 каплю) и тионилхлорид (0,13 мл), а затем перемешивали смесь при 60°C в течение 6 часов. Растворитель отгоняли в условиях пониженного давления, а полученный остаток подвергали азеотропной перегонке с толуолом и использовали для синтеза (f).
(f) Синтез 3-(3-диметилкарбамоил-4-метокси-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (214 мг) и 37% формалина (0,14 мл) тем же способом, что и описанный в примере 1, растворяли в хлороформе (4 мл), добавляли к раствору триэтиламин (0,47 мл) и 3-диметилкарбамоил-4-метокси-5-трифторметилбензоилхлорид, а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и использовали полученный остаток для синтеза (g).
(g) Синтез 3-(3-диметилкарбамоил-4-метокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Диметилкарбамоил-4-метокси-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазол растворяли в хлороформе (8 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (718 мг), а затем смесь перемешивали при комнатной температуре в течение 20 часов и гасили добавлением 10% тиосульфата натрия. Растворитель отгоняли в условиях пониженного давления, добавляли 1н раствор гидроксида натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 2/1) с получением указанного в заголовке соединения (298 мг) в виде бледно-желтого аморфного продукта.
(h) Синтез 3-(3-диметилкарбамоил-4-гидрокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Диметилкарбамоил-4-метокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (291 мг) растворяли в N,N-диметилформамиде (3 мл), добавляли к раствору хлорид лития (279 мг), а затем перемешивали смесь при 120°C в течение 2 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (257 мг) в виде бесцветного кристаллического вещества.
1Н-ЯМР δ (ДМСО-d6): 2,96 (6H, с), 5,39 (2H, с), 7,43 (1H, дд, J=7,6, 7,6 Гц), 7,55 (1H, ддд, J=8,4, 7,6, 1,3 Гц), 7,79 (1H, д, J=2,1 Гц), 7,90 (1H, д, J=7,6 Гц), 7,93 (1H, д, J=2,1 Гц), 8,02 (1H, д, J=8,4 Гц), 11,27 (1H, с). MS (m/z): 427 (M-H)-.
Пример 28: 3-(4-гидрокси-3-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез сложного 2-трифторметилфенилового эфира уксусной кислоты
2-Трифторметилфенол (20,00 г) растворяли в хлороформе (160 мл), добавляли к раствору триэтиламин (34,0 мл) и уксусный ангидрид (12,4 мл), а затем перемешивали смесь при комнатной температуре в течение 3 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (23,76 г) в виде бледно-желтого маслянистого вещества.
(b) Синтез 1-(4-гидрокси-3-трифторметилфенил)этанона
Сложный 2-трифторметилфениловый эфир уксусной кислоты (10,00 г) растворяли в трифторметансульфоновой кислоте (10,0 мл), и перемешивали раствор при комнатной температуре в течение 16 часов. Реакционный раствор вливали в воду со льдом, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и промывали полученное кристаллическое вещество н-гексаном с получением указанного в заголовке соединения (4,47 г) в виде бесцветного кристаллического вещества.
(c) Синтез 1-(4-метоксиметокси-3-трифторметилфенил)этанона
1-(4-Гидрокси-3-трифторметилфенил)этанон (2,01 г) растворяли в N,N-диметилформамиде (20 мл), добавляли к раствору карбонат калия (2,70 г) и хлорметилметиловый эфир (1,10 мл), а затем перемешивали смесь при комнатной температуре в течение 1 часа. К реакционному раствору добавляли воду, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (2,33 г) в виде бледно-желтого маслянистого вещества.
(d) Синтез метил-4-гидрокси-3-трифторметоксибензоат
1-(4-Метоксиметокси-3-трифторметилфенил)этанон (2,33 г) растворяли в метаноле (20 мл), добавляли к раствору 5M раствор метоксида натрия в метаноле (9,40 мл) и N-бромсукцинимид (5,10 г), а затем перемешивали смесь при комнатной температуре в течение 30 минут. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 10% тиосульфатом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, добавляли 4н соляную кислоту в этилацетате (20 мл), а затем перемешивали смесь при комнатной температуре в течение 30 минут. Реакционный раствор промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (1,42 г) в виде бесцветного кристаллического вещества.
(e) Синтез метил-4-бензилокси-3-трифторметилбензоата
Метил-4-гидрокси-3-трифторметоксибензоат (936 мг) растворяли в N,N-диметилформамиде (10 мл), добавляли к раствору карбонат калия (1,29 г) и бензилбромид (0,58 мл), а затем перемешивали смесь при комнатной температуре в течение 24 часов. К реакционному раствору добавляли воду, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 4/1) с получением указанного в заголовке соединения (1,70 г) в виде желтого кристаллического вещества.
(f) Синтез 4-бензилокси-3-трифторметилбензойной кислоты
Метил-4-бензилокси-3-трифторметилбензоат (1,38 г) растворяли в тетрагидрофуране (10 мл) и воде (5 мл), добавляли к раствору моногидрат гидроксида лития (1,49 г), а затем перемешивали смесь при комнатной температуре в течение 20 часов. Растворитель отгоняли в условиях пониженного давления, добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (1,25 г) в виде бледно-желтого кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 5,38 (2H, с), 7,31-7,51 (6H, м), 8,12 (1H, д, J=2,1 Гц), 8,19 (1H, д, J=8,6, 2,1 Гц), 13,12 (1H, ушир.с). MS (m/z): 269 (M-H)-.
(g) Синтез 4-бензилокси-3-трифторметилбензоилхлорида
К 4-бензилокси-3-трифторметилбензойной кислоте (444 мг) добавляли толуол (5 мл), N,N-диметилформамид (2 капли) и тионилхлорид (0,16 мл), а затем перемешивали смесь при 60°C в течение 20 часов. Растворитель отгоняли в условиях пониженного давления, а полученный продукт подвергали затем азеотропной перегонке с толуолом и использовали для синтеза (h).
(h) Синтез 3-(4-бензилокси-3-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (282 мг) и 37% формалина (0,19 мл) тем же способом, что и описанный в примере 1, растворяли в хлороформе (6 мл), добавляли к раствору триэтиламин (0,62 мл) и 4-бензилокси-3-трифторметилбензоилхлорид, а затем перемешивали смесь при комнатной температуре в течение 1,5 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 4/1) с получением указанного в заголовке соединения (356 мг) в виде бледно-желтого маслянистого вещества.
(i) Синтез 3-(4-бензилокси-3-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(4-Бензилокси-3-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазол (600 мг) растворяли в хлороформе (10 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (1,04 г), а затем смесь перемешивали при комнатной температуре в течение 20 часов и гасили добавлением 10% тиосульфата натрия. Растворитель отгоняли в условиях пониженного давления, добавляли 1н гидроксид натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 2/1) с получением указанного в заголовке соединения (495 мг) в виде бесцветного кристаллического вещества.
(j) Синтез 3-(4-гидрокси-3-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(4-Бензилокси-3-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (490 мг) растворяли в тетрагидрофуране (5 мл) и метаноле (5 мл), добавляли к раствору 20% гидроксид палладия на угле (100 мг), а затем перемешивали смесь при комнатной температуре в течение 6 часов в атмосфере водорода. После фильтрования реакционной смеси отгоняли растворитель в условиях пониженного давления, а затем выкристаллизовывали полученный остаток из диэтилового эфира с получением указанного в заголовке соединения (397 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 5,29 (2H, с), 7,14 (1H, д, J=8,4 Гц), 7,41 (1H, ддд, J=8,2, 7,8, 0,9 Гц), 7,72 (1H, ддд, J=8,5, 7,3, 1,3 Гц), 7,80 (1H, дд, J=8,4, 2,2 Гц), 7,84-7,88 (2H, м), 7,91 (1H, д, J=8,2 Гц), 11,35 (1H, ушир.с). MS (m/z): 356 (M-H)-.
Пример 29: 3-(3-хлор-4-гидрокси-5-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез 4-бензилокси-3-хлор-5-метоксибензоилхлорида
К 4-бензилокси-3-хлор-5-метоксибензойной кислоте (541 мг) добавляли толуол (5,4 мл), N,N-диметилформамид (1 каплю) и тионилхлорид (0,16 мл), а затем перемешивали смесь при 60°C в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, а затем проводили азеотропную перегонку с толуолом с получением указанного в заголовке соединения (578 мг) в виде желтого твердого вещества.
(b) Синтез 3-(4-бензилокси-3-хлор-5-метоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (347 мг) и 37% формалина (0,23 мл) тем же способом, что и описанный в примере 1, растворяли в дихлорметане (3 мл), добавляли к раствору диизопропилэтиламин (0,63 мл) и 4-бензилокси-3-хлор-5-метоксибензоилхлорид (578 мг), а затем перемешивали смесь при комнатной температуре в течение 1,5 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 6/1) с получением указанного в заголовке соединения (498 мг) в виде бледно-желтого маслянистого вещества.
(c) Синтез 3-(4-бензилокси-3-хлор-5-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(4-Бензилокси-3-хлор-5-метоксибензоил)-2,3-дигидро-1,3-бензотиазол (498 мг) растворяли в дихлорметане (10 мл), и добавляли к раствору 70% мета-хлорпербензойную кислоту (1,22 г). После перемешивания смеси при комнатной температуре в течение 16 часов добавляли 1н гидроксид натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (521 мг) в виде бледно-желтого маслянистого вещества.
(d) Синтез 3-(3-хлор-4-гидрокси-5-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(4-Бензилокси-3-хлор-5-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (519 мг) растворяли в тетрагидрофуране (5 мл), добавляли к раствору 20% гидроксид палладия на угле (101 мг), а затем перемешивали смесь при комнатной температуре в течение 21 часов в атмосфере водорода. После фильтрования реакционной смеси растворитель отгоняли в условиях пониженного давления, а затем выкристаллизовывали полученный остаток из хлороформа с получением указанного в заголовке соединения (185 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 3,88 (3H, с), 5,34 (2H, с), 7,27 (1H, с), 7,35 (1H, с), 7,43 (1H, дд, J=7,6 Гц, 7,6 Гц), 7,75 (1H, дд, J=8,4, 7,6 Гц), 7,90 (1H, д, J=7,6 Гц), 7,99 (1H, д, J=8,4 Гц). MS (m/z): 352 (M-H)-.
Пример 30: 3-[4-гидрокси-3-(пирролидин-1-карбонил)-5-трифторметилбензоил]-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез метил-4-метокси-3-(пирролидин-1-карбонил)-5-трифторметилбензоата
Сложный 1-метиловый эфир 4-метокси-5-трифторметилизофталевой кислоты (4,35 г) растворяли в дихлорметане (50 мл), добавляли к раствору пирролидин (1,10 г) и гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (2,99 г), а затем перемешивали смесь при комнатной температуре в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 2/3) с получением указанного в заголовке соединения (1,33 г) в виде коричневого маслянистого вещества.
(b) Синтез 4-метокси-3-(пирролидин-1-карбонил)-5-трифторметилбензойной кислоты
Метил-4-метокси-3-(пирролидин-1-карбонил)-5-трифторметилбензоат (1,33 г) растворяли в тетрагидрофуране (8 мл) и воде (4 мл), добавляли к раствору моногидрат гидроксида лития (708 мг), а затем перемешивали смесь при комнатной температуре в течение 1 часа. Органический растворитель отгоняли в условиях пониженного давления, и промывали водный слой диизопропиловым эфиром. Водный слой подкисляли добавлением 1н соляной кислоты, а затем экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (790 мг) в виде бесцветного аморфного продукта.
(c) Синтез 4-метокси-3-(пирролидин-1-карбонил)-5-трифторметилбензоилхлорида
К 4-метокси-3-(пирролидин-1-карбонил)-5-трифторметилбензойной кислоте (785 мг) добавляли толуол (8 мл) и оксалилхлорид (0,64 мл), а затем перемешивали смесь при комнатной температуре в течение 22 часов. Растворитель отгоняли в условиях пониженного давления, и использовали полученный продукт для синтеза (d).
(d) Синтез 3-[4-метокси-3-(пирролидин-1-карбонил)-5-трифторметилбензоил]-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (464 мг) и 37% формалина (0,31 мл) тем же способом, что и описанный в примере 1, растворяли в хлороформе (10 мл), добавляли к раствору триэтиламин (1,03 мл) и 4-метокси-3-(пирролидин-1-карбонил)-5-трифторметилбензоилхлорид, а затем перемешивали смесь при комнатной температуре в течение 2 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 1/2) с получением указанного в заголовке соединения (778 мг) в виде бледно-желтого аморфного продукта.
(e) Синтез 3-[4-метокси-3-(пирролидин-1-карбонил)-5-трифторметилбензоил]-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-[4-Метокси-3-(пирролидин-1-карбонил)-5-трифторметилбензоил]-2,3-дигидро-1,3-бензотиазол (773 мг) растворяли в хлороформе (15 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (1,22 г), а затем смесь перемешивали при комнатной температуре в течение 16 часов и гасили добавлением 10% тиосульфата натрия. После отгонки растворителя в условиях пониженного давления добавляли 1н гидроксид натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (720 мг) в виде белого аморфного продукта.
(f) Синтез 3-[4-гидрокси-3-(пирролидин-1-карбонил)-5-трифторметилбензоил]-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-[4-Метокси-3-(пирролидин-1-карбонил)-5-трифторметилбензоил]-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (715 мг) растворяли в N,N-диметилформамиде (7 мл), добавляли к раствору хлорид лития (649 мг), а затем перемешивали смесь при 120°C в течение 2 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (654 мг) в виде бесцветного аморфного продукта.
1H-ЯМР δ (ДМСО-d6): 1,86 (4H, ушир.), 3,45-3,60 (4H, т, J=6,5 Гц), 5,37 (2H, с), 7,43 (1H, ддд, J=7,8, 7,8, 0,8 Гц), 7,76 (1H, ддд, J=8,4, 7,8, 1,3 Гц), 7,90 (2H, ушир.), 8,03 (1H, д, J=8,4 Гц), 12,29 (1H, с). MS (m/z): 453 (M-H)-.
Пример 31: 3-[4-гидрокси-3-(1,3-тиазолидин-3-карбонил)-5-трифторметилбензоил]-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез 3-формил-4-метокси-5-трифторметилбензоилхлорида
К 3-формил-4-метокси-5-трифторметилбензойной кислоте (2,05 г) добавляли толуол (20 мл), N/N-диметилформамид (2 капли) и тионилхлорид (0,70 мл), а затем перемешивали смесь при 60°C в течение 6,5 часов. Растворитель отгоняли в условиях пониженного давления, а затем проводили азеотропную перегонку с толуолом с получением указанного в заголовке соединения (2,42 г) в виде коричневого маслянистого вещества.
(b) Синтез 3-(3-формил-4-метокси-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (1,55 г) и 37% формалина (1,0 мл) тем же способом, что и описанный в примере 1, растворяли в дихлорметане (15 мл), добавляли к раствору диизопропилэтиламин (2,7 мл) и 3-формил-4-метокси-5-трифторметилбензоилхлорид (2,42 г), а затем перемешивали смесь при комнатной температуре в течение 14 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 5/1) с получением указанного в заголовке соединения (1,44 г) в виде желтого маслянистого вещества.
(c) Синтез 3-(3-диэтоксиметил-4-метокси-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазола
3-(3-Формил-4-метокси-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазол (277 мг) растворяли в этаноле (1,5 мл) и триэтилортоформиате (0,16 мл), добавляли к раствору Амберлист-15 (27 мг), а затем смесь нагревали с обратным холодильником в течение 3,5 часов. Реакционный раствор фильтровали, а затем отгоняли растворитель в условиях пониженного давления с получением указанного в заголовке соединения (326 мг) в виде желтого маслянистого вещества.
(d) Синтез 3-(3-формил-4-метокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Формил-4-метокси-5-трифторметилбензоил)-2,3-дигидро-1,3-бензотиазол (326 мг) растворяли в дихлорметане (6 мл), и добавляли к раствору 70% мета-хлорпербензойную кислоту (754 мг). После перемешивания смеси при комнатной температуре в течение 2 часов добавляли 1н гидроксид натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, полученный остаток растворяли в этилацетате (3 мл), добавляли к раствору 4н соляную кислоту в этилацетате (0,74 мл), а затем перемешивали смесь при комнатной температуре в течение 2 часов. Реакционный раствор промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (248 мг) в виде бледно-желтого маслянистого вещества.
(e) Синтез 5-(1,1-диоксо-2,3-дигидро-1,3-бензотиазол-3-карбонил)-2-метокси-3-трифторметилбензойной кислоты
3-(3-Формил-4-метокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (248 мг) растворяли в метаноле (2,5 мл) и водном 10% растворе лимонной кислоты (2,5 мл), добавляли к раствору 2-метил-2-бутен (0,33 мл) и хлорит натрия (84 мг), а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали водным 10% раствором тиосульфата натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (277 мг) в виде бледно-желтого маслянистого вещества.
(f) Синтез 3-[4-метокси-3-(1,3-тиазолидин-3-карбонил)-5-трифторметилбензоил]-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
5-(1,1-Диоксо-2,3-дигидро-1,3-бензотиазол-3-карбонил)-2-метокси-3-трифторметилбензойную кислоту (277 мг) растворяли в дихлорметане (3 мл), добавляли к раствору тиазолидин (0,10 мл) и гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (238 мг), а затем перемешивали смесь при комнатной температуре в течение 19 часов. Растворитель отгоняли в условиях пониженного давления, добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 1/1) с получением указанного в заголовке соединения (138 мг) в виде бесцветного маслянистого вещества.
(g) Синтез 3-[4-гидрокси-3-(1,3-тиазолидин-3-карбонил)-5-трифторметилбензоил]-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-[4-Метокси-3-(1,3-тиазолидин-3-карбонил)-5-трифторметилбензоил]-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (138 мг) растворяли в N,N-диметилформамиде (2 мл), добавляли к раствору хлорид лития (118 мг), а затем перемешивали смесь при 120°C в течение 1,5 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/хлороформа с получением указанного в заголовке соединения (47 мг) в виде бледно-желтого кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 3,06 (2H, ушир.с), 3,77 (2H, ушир.с), 4,58 (2H, ушир.с), 5,38 (2H, с), 7,43 (1H, дд, J=7,6, 7,3 Гц), 1,16 (1H, дд, J=8,4, 7,3 Гц), 7,82-8,00 (2H, м), 7,97 (1H, с), 8,04 (1H, д, J=8,4 Гц), 11,52 (1H, ушир.с). MS (m/z):471 (M-H)-.
Пример 32: 3-[4-гидрокси-3-(1-оксо-1,3-тиазолидин-3-карбонил)-5-трифторметилбензоил]-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
3-[4-Гидрокси-3-(1,3-тиазолидин-3-карбонил)-5-трифторметилбензоил]-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (67 мг) растворяли в тетрагидрофуране (0,5 мл) и воде (0,5 мл), добавляли к раствору оксон (45 мг), а затем перемешивали смесь при комнатной температуре в течение 1 часа. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/хлороформа с получением указанного в заголовке соединения (46 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 2,98-3,15 (2H, м), 3,94-4,06 (1H, м), 4,22 (1Н, ушир.с), 4,61 (2H, с), 5,32 (2H, с), 7,42 (1H, дд, J=7,6, 7,6 Гц), 7,74 (1H, дд, J=8,4, 7,6 Гц), 7,86 (1H, д, J=7,6 Гц), 7,88 (1H, д, J=2,2 Гц), 7,98 (1H, д, J=2,2 Гц), 8,01 (1H, д, J=8,4 Гц). MS (m/z): 487 (M-H)-.
Пример 33: 1,1-диоксо-3-[3-(1,1-диоксо-1,3-тиазолидин-3-карбонил)-4-гидрокси-5-трифторметилбензоил]-2,3-дигидро-1,3-бензотиазол
(a) Синтез 1,1-диоксо-3-[3-(1,1-диоксо-1,3-тиазолидин-3-карбонил)-4-метокси-5-трифторметилбензоил]-2,3-дигидро-1,3-бензотиазола
3-[4-Метокси-3-(1,3-тиазолидин-3-карбонил)-5-трифторметилбензоил]-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (300 мг) растворяли в хлороформе (10 мл), и добавляли к раствору 70% мета-хлорпербензойную кислоту (1,21 г). После перемешивания смеси при комнатной температуре в течение 20 часов добавляли 1н гидроксид натрия, и экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (266 мг) в виде бесцветного маслянистого вещества.
(b) Синтез 1,1-диоксо-3-[3-(1,1-диоксо-1,3-тиазолидин-3-карбонил)-4-гидрокси-5-трифторметилбензоил]-2,3-дигидро-1,3-бензотиазола
1,1-Диоксо-3-[3-(1,1-диоксо-1,3-тиазолидин-3-карбонил)-4-метокси-5-трифторметилбензоил]-2,3-дигидро-1,3-бензотиазол (260 мг) растворяли в N,N-диметилформамиде (2,5 мл), добавляли к раствору хлорид лития (200 мг), а затем перемешивали смесь при 120°C в течение 1,5 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/хлороформа с получением указанного в заголовке соединения (120 мг) в виде бледно-желтого кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 3,50 (2H, т, J=6,8 Гц), 3,86-4,14 (2H, м), 4,64 (2H, с), 5,39 (2H, с), 7,44 (1H, дд, J=8,1, 7,6 Гц), 7,77 (1H, дд, J=8,1, 7,6 Гц), 7,88 (1H, с), 7,91 (1H, д, J=8,1 Гц), 8,01 (1H, с), 8,06 (1H, д, J=8,1 Гц). MS (m/z): 503 (M-H)-.
Пример 34: 3-(3-циано-5-этилсульфанил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез метил-3-циано-4-метоксибензоата
Метил-3-циано-4-гидроксибензоат (1,00 г) растворяли в N,N-диметилформамиде (5 мл), добавляли к раствору карбонат калия (1,56 г) и диметилсерную кислоту (0,70 мл), а затем перемешивали смесь при комнатной температуре в течение 1 часа. После фильтрования реакционной смеси добавляли воду, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (923 мг) в виде желтого твердого вещества.
(b) Синтез 3-циано-4-метоксибензойной кислоты
Метил-3-циано-4-метоксибензоат (879 мг) растворяли в тетрагидрофуране (8 мл) и воде (4 мл), добавляли к раствору моногидрат гидроксида лития (772 мг), а затем перемешивали смесь при комнатной температуре в течение 1 часа. Органический растворитель отгоняли в условиях пониженного давления и подкисляли добавлением 2н соляной кислоты, а затем собирали выпавшее в осадок кристаллическое вещество путем фильтрования с получением указанного в заголовке соединения (754 мг) в виде бесцветного кристаллического вещества.
1Н-ЯМР δ (ДМСО-d6): 4,00 (3H, с), 7,36 (1H, дд, J=6,6, 3,0 Гц), 8,18 (1H, д, J=3,0 Гц), 8,20 (1H, дд, J=6,6, 2,1 Гц), 13,17 (1H, ушир.с). MS (m/z): 176 (M-H)-.
(c) Синтез 3-циано-4-метоксибензоилхлорида
К 3-циано-4-метоксибензойной кислоте (1,78 г) добавляли толуол (20 мл), N,N-диметилформамид (3 капли) и тионилхлорид (1,14 мл), а затем перемешивали смесь при 60°C в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, а полученный остаток подвергали азеотропной перегонке с толуолом и использовали для синтеза (d).
(d) Синтез 3-(3-циано-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (1,89 г) и 37% формалина (1,25 мл) тем же способом, что и описанный в примере 1, растворяли в хлороформе (20 мл), добавляли к раствору триэтиламин (2,08 мл) и 3-циано-4-метоксибензоилхлорид, а затем перемешивали смесь при комнатной температуре в течение 2 часов. Органический растворитель отгоняли в условиях пониженного давления и экстрагировали этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и использовали полученный остаток для синтеза (e).
(e) Синтез 3-(3-циано-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-метоксибензоил)-2,3-дигидро-1,3-бензотиазол растворяли в хлороформе (50 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (9,19 г), а затем перемешивали смесь при комнатной температуре в течение 20 часов и гасили добавлением 10% тиосульфата натрия. Растворитель отгоняли в условиях пониженного давления, добавляли 1н гидроксид натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (2,30 г) в виде бледно-желтого твердого вещества.
(f) Синтез 3-(3-циано-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-метоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (2,30 г) растворяли в N,N-диметилформамиде (25 мл), добавляли к раствору хлорид лития (2,97 г), а затем перемешивали смесь при 130°C в течение 12 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и использовали полученный остаток для синтеза (g).
(g) Синтез 3-(3-циано-4-гидрокси-5-йодбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол растворяли в дихлорметане (27 мл) и метаноле (3 мл), добавляли к раствору N-йодсукцинимид (1,79 г) и трифторметансульфоновую кислоту (5 капель), а затем перемешивали смесь при комнатной температуре в течение 15 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/этилацетата с получением указанного в заголовке соединения (1,30 г) в виде бесцветного кристаллического вещества.
(h) Синтез 3-(3-циано-5-этилсульфанил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-гидрокси-5-йодбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (500 мг) растворяли в N,N-диметилформамиде (5 мл), добавляли к раствору 2,2'-бипиридин (18 мг), цинковый порошок (149 мг), бромид никеля (25 мг) и диэтилдисульфид (70 мг), а затем перемешивали смесь при 110°C в течение 1 часа. После фильтрования реакционной смеси добавляли 1н соляную кислоту, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, полученный остаток очищали по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 2/1), а затем выкристаллизовывали из диэтилового эфира/этилацетата с получением указанного в заголовке соединения (63 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 1,22 (3H, т, J=7,3 Гц), 2,96 (2H, кв., J=7,3 Гц), 5,35 (2H, с), 7,44 (1H, дд, J=7,6, 7,6 Гц), 7,77 (1H, ддд, J=8,3, 7,6, 1,3 Гц), 7,79 (1H, д, J=1,8 Гц), 7,88-7,94 (2H, м), 8,07 (1Н, д, J=8,3 Гц). MS (m/z): 373 (M-H)-.
Пример 35: 3-(3-циано-4-гидрокси-5-изопропилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
3-(3-Циано-4-гидрокси-5-йодбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (1,03 мг) растворяли в N,N-диметилформамиде (10 мл), добавляли к раствору 2,2'-бипиридин (37 мг), цинковый порошок (306 мг), бромид никеля (52 мг) и диизопропилдисульфид (176 мг), а затем перемешивали смесь при 110°C в течение 1 часа. После фильтрования реакционной смеси добавляли 1н соляную кислоту, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 2/1) с получением указанного в заголовке соединения (153 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 1,23 (6H, д, J=6,6 Гц), 3,46 (1H, sevent, J=6,6 Гц), 5,35 (2H, с), 7,44 (1H, дд, J=7,4, 7,4 Гц), 7,76 (1H, ддд, J=8,4, 7,4, 1,3 Гц), 7,90 (1H, д, J=2,1 Гц), 7,91 (1H, д, J=7,4 Гц), 7,98 (1H, д, J=2,1 Гц), 8,04 (1H, д, J=8,4 Гц), 11,32 (1H, ушир.с). MS (m/z): 387 (M-H)-.
Пример 36: 3-(3-циано-4-гидрокси-5-трифторметоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез 1-метоксиметокси-2-трифторметоксибензола
2-Трифторметоксифенол (10,00 г) растворяли в N,N-диметилформамиде (50 мл), добавляли к раствору карбонат калия (15,52 г) и хлорметилметиловый эфир (4,70 мл), а затем перемешивали смесь при комнатной температуре в течение 1 часа. К реакционному раствору добавляли воду, а затем экстрагировали смесь н-гексаном. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (12,21 г) в виде бесцветного маслянистого вещества.
(b) Синтез 2-гидрокси-3-трифторметоксибензальдегида
1-Метоксиметокси-2-трифторметоксибензол (12,21 г) растворяли в тетрагидрофуране (120 мл), под струей аргона при -60°C в течение 15 минут добавляли к раствору 1,59M раствор н-бутиллития в н-циклогексане (40 мл), а затем перемешивали смесь в течение 1 часа. К раствору добавляли N,N-диметилформамид (6,30 мл), а затем перемешивали смесь при комнатной температуре в течение 30 минут. К раствору добавляли 2н соляную кислоту (100 мл), а затем перемешивали смесь при 60°C в течение 15 часов. Органический растворитель отгоняли в условиях пониженного давления, а затем экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (10,38 г) в виде бесцветного кристаллического вещества.
(c) Синтез 5-бром-2-гидрокси-3-трифторметоксибензальдегида
2-Гидрокси-3-трифторметоксибензальдегид (10,38 г) растворяли в дихлорметане (100 мл), добавляли к раствору N-бромсукцинимид (9,41 г), а затем перемешивали смесь при комнатной температуре в течение 30 минут. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 10% тиосульфатом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (14,27 г) в виде коричневого твердого вещества.
(d) Синтез 5-бром-2-метокси-3-трифторметоксибензальдегида
5-Бром-2-гидрокси-3-трифторметоксиальдегид (5,00 г) растворяли в N,N-диметилформамиде (25 мл), добавляли к раствору карбонат калия (4,85 г) и диметилсерную кислоту (2,5 мл), а затем перемешивали смесь при комнатной температуре в течение 16 часов. К реакционному раствору добавляли воду, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (5,22 г) в виде коричневого маслянистого вещества.
(e) Синтез 5-бром-1-диэтоксиметил-2-метокси-3-трифторметоксибензола
5-Бром-2-метокси-3-трифторметоксибензальдегид (5,22 г) растворяли в н-гексане (15 мл) и триэтилортоформиате (4,4 мл), добавляли к раствору Амберлист-15 (522 мг), а затем нагревали смесь с обратным холодильником в течение 3 часов. Реакционный раствор фильтровали, а затем отгоняли растворитель в условиях пониженного давления с получением указанного в заголовке соединения (6,19 г) в виде коричневого маслянистого вещества.
(f) Синтез 3-формил-4-метокси-5-трифторметоксибензойной кислоты
К магнию (403 мг) добавляли тетрагидрофуран (16 мл), 5-бром-1-диэтоксиметил-2-метокси-3-трифторметоксибензол (6,19 г) и 0,97M раствор метилмагнийбромида в тетрагидрофуране (0,42 мл), а затем перемешивали смесь при комнатной температуре в течение 1 часа. Реакционный раствор охлаждали до 0°C и перемешивали в течение 45 минут в атмосфере углекислого газа. Добавляли 4н соляную кислоту (25 мл), а затем перемешивали смесь при комнатной температуре в течение 30 минут. Органический растворитель отгоняли в условиях пониженного давления, и экстрагировали смесь диизопропиловым эфиром. Органический слой экстрагировали добавлением к нему 1н гидроксида натрия (100 мл), а водный слой дважды промывали диизопропиловым эфиром. Водный слой подкисляли добавлением 4н соляной кислоты, а затем экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (4,22 г) в виде коричневого твердого вещества.
(g) Синтез 3-циано-4-метокси-5-трифторметоксибензойной кислоты
3-Формил-4-метокси-5-трифторметоксибензойную кислоту (4,22 г) растворяли в муравьиной кислоте (20 мл), добавляли к раствору гидрохлорид гидроксиламина (1,22 г), а затем нагревали смесь с обратным холодильником в течение 16 часов. Растворитель отгоняли в условиях пониженного давления, добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (3,97 г) в виде коричневого твердого вещества.
1Н-ЯМР δ (ДМСО-d6): 4,17 (3H, с), 8,10-8,14 (1H, м), 8,28 (1H, д, J=2,0 Гц), 13,73 (1H, ушир.с). MS (m/z): 260 (M-H)-.
(h) Синтез 3-циано-4-метокси-5-трифторметоксибензоилхлорида
К 3-циано-4-метокси-5-трифторметоксибензойной кислоте (500 мг) добавляли толуол (10 мл), N,N-диметилформамид (3 капли) и тионилхлорид (0,28 мл), а затем перемешивали смесь при 60°C в течение 6 часов. Растворитель отгоняли в условиях пониженного давления, а полученный остаток подвергали азеотропной перегонке с толуолом и использовали для синтеза (i).
(i) Синтез 3-(3-циано-4-метокси-5-трифторметоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиол (359 мг) и 37% формалина (0,24 мл) тем же способом, что и описанный в примере 1, растворяли в хлороформе (10 мл), добавляли к раствору триэтиламин (0,80 мл) и 3-циано-4-метокси-5-трифторметоксибензоилхлорид, а затем перемешивали смесь при комнатной температуре в течение 17 часов. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и использовали полученный остаток для синтеза (j).
(j) Синтез 3-(3-циано-4-метокси-5-трифторметоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-метокси-5-трифторметоксибензоил)-2,3-дигидро-1,3-бензотиазол растворяли в хлороформе (15 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (1,98 г), а затем смесь перемешивали при комнатной температуре в течение 6 часов и гасили добавлением 10% тиосульфата натрия. Растворитель отгоняли в условиях пониженного давления, добавляли 1н раствор гидроксида натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (631 мг) в виде бледно-желтого твердого вещества.
(k) Синтез 3-(3-циано-4-гидрокси-5-трифторметоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-метокси-5-трифторметоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (300 мг) растворяли в N,N-диметилформамиде (4 мл), добавляли к раствору хлорид лития (309 мг), а затем перемешивали смесь при 100°C в течение 1 часа. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/этилацетата с получением указанного в заголовке соединения (184 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 5,37 (2H, с), 7,42 (1H, ддд, J=7,3, 7,3, 0,7 Гц), 7,77 (1H, ддд, J=8,4, 7,3, 1,3 Гц), 7,88-7,96 (2H, м), 8,04-8,11 (2H, м). MS (m/z): 397 (M-H)-.
Пример 37: 3-(3-хлор-4-гидрокси-5-трифторметоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
(a) Синтез сложного 2-трифторметоксифенилового эфира уксусной кислоты
2-Трифторметоксифенол (10,00 г) растворяли в хлороформе (30 мл), добавляли к раствору триэтиламин (6,11 мл) и уксусный ангидрид (6,37 мл), а затем перемешивали смесь при комнатной температуре в течение 2 часов. К полученному реакционному раствору добавляли 10% карбонат калия, а затем экстрагировали смесь хлороформом. Органический слой промывали водным 5% раствором лимонной кислоты и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (11,39 г) в виде бесцветного маслянистого вещества.
(b) Синтез 1-(4-гидрокси-3-трифторметоксифенил)этанона
Сложный 2-трифторметоксифениловый эфир уксусной кислоты (11,39 г) растворяли в трифторметансульфоновой кислоте (10 мл), а затем перемешивали раствор при комнатной температуре в течение 2,5 часов. Реакционный раствор вливали в воду со льдом, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 3/1) с получением указанного в заголовке соединения (4,55 г) в виде бесцветного кристаллического вещества.
(c) Синтез 1-(4-метоксиметокси-3-трифторметоксифенил)этанона
1-(4-Гидрокси-3-трифторметоксифенил)этанон (1,55 г) растворяли в N,N-диметилформамиде (20 мл), добавляли к раствору карбонат калия (1,46 г) и хлорметилметиловый эфир (0,64 мл), а затем перемешивали смесь при комнатной температуре в течение 1 часа. К реакционному раствору добавляли воду, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (1,54 г) в виде бесцветного маслянистого вещества.
(d) Синтез метил-4-метоксиметокси-3-трифторметоксибензоата
1-(4-Метоксиметокси-3-трифторметоксифенил)этанон (540 мг) растворяли в метаноле (30 мл), добавляли к раствору метоксид натрия (1,10 г) и N-бромсукцинимид (1,09 г), а затем перемешивали смесь при комнатной температуре в течение 30 минут. К реакционному раствору добавляли воду, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (590 мг) в виде желтого кристаллического вещества.
(e) Синтез метил-4-гидрокси-3-трифторметоксибензоата
Метил-4-метоксиметокси-3-трифторметоксибензоат (590 мг) растворяли в хлороформе (10 мл), добавляли к раствору трифторуксусную кислоту (5 мл), а затем перемешивали смесь при комнатной температуре в течение 1 часа. К раствору добавляли воду, а затем экстрагировали смесь хлороформом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и промывали полученный остаток н-гексаном с получением указанного в заголовке соединения (346 мг) в виде бесцветного кристаллического вещества.
(f) Синтез метил-3-хлор-4-гидрокси-5-трифторметоксибензоата
Метил-4-гидрокси-3-трифторметоксибензоат (2,05 г) растворяли в хлороформе (20 мл) и метаноле (3 мл), добавляли к раствору N-хлорсукцинимид (1,39 г) и трифторметансульфоновую кислоту (0,05 мл), а затем перемешивали смесь при 50°C в течение 19 часов. Растворитель отгоняли в условиях пониженного давления, добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (2,64 г) в виде желтого твердого вещества.
(g) Синтез метил-3-хлор-4-метокси-5-трифторметоксибензоата
Метил-3-хлор-4-гидрокси-5-трифторметоксибензоат (2,64 г) растворяли в N,N-диметилформамиде (15 мл), добавляли к раствору карбонат калия (3,60 г) и диметилсерную кислоту (1,64 мл), а затем перемешивали смесь при комнатной температуре в течение 1 часа. К реакционному раствору добавляли воду, и экстрагировали реакционную смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 9/1) с получением указанного в заголовке соединения (1,70 г) в виде бесцветного маслянистого вещества.
(h) Синтез 3-хлор-4-метокси-5-трифторметоксибензойной кислоты
Метил-3-хлор-4-метокси-5-трифторметоксибензоат (1,70 г) растворяли в тетрагидрофуране (12 мл) и воде (6 мл), добавляли к раствору моногидрат гидроксида лития (1,00 г), и перемешивали смесь при комнатной температуре в течение 2 часов. Растворитель отгоняли в условиях пониженного давления, добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (1,51 г) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (CDCl3): 4,04 (3H, с), 7,93 (1H, с), 8,10 (1H, д, J=1,9 Гц). MS (m/z): 269 (M-H)-.
(i) Синтез 3-хлор-4-метокси-5-трифторметоксибензоилхлорида
К 3-хлор-4-метокси-5-трифторметоксибензойной кислоте (401 мг) добавляли толуол (4 мл), N,N-диметилформамид (1 каплю) и тионилхлорид (0,13 мл), а затем перемешивали смесь при 60°C в течение 13 часов. Растворитель отгоняли в условиях пониженного давления, а затем проводили азеотропную перегонку с толуолом с получением указанного в заголовке соединения (436 мг) в виде коричневого маслянистого вещества.
(j) Синтез 3-(3-хлор-4-метокси-5-трифторметоксибензоил)-2,3-дигидро-1,3-бензотиазола
2,3-Дигидро-1,3-бензотиазол, синтезированный из 2-аминобензолтиола (278 мг) и 37% формалина (0,18 мл) тем же способом, что и описанный в примере 1, растворяли в дихлорметане (6,5 мл), добавляли к раствору диизопропилэтиламин (0,50 мл) и 3-хлор-4-метокси-5-трифторметоксибензоилхлорид (436 мг), а затем перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель отгоняли в условиях пониженного давления, добавляли воду, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и использовали полученный остаток для синтеза (k).
(k) Синтез 3-(3-хлор-4-метокси-5-трифторметоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Хлор-4-метокси-5-трифторметоксибензоил)-2,3-дигидро-1,3-бензотиазол растворяли в хлороформе (10 мл), добавляли к раствору 70% мета-хлорпербензойную кислоту (2,57 г), а затем перемешивали смесь при комнатной температуре в течение 22 часов. Добавляли 1н гидроксид натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 4/1) с получением указанного в заголовке соединения (420 мг) в виде бесцветного маслянистого вещества.
(l) Синтез 3-(3-хлор-4-гидрокси-5-трифторметоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазола
3-(3-Хлор-4-метокси-5-трифторметоксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (420 мг) растворяли в N,N-диметилформамиде (5 мл), добавляли к раствору хлорид лития (421 мг), а затем перемешивали смесь при 120°C в течение 14 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/этилацетата с получением указанного в заголовке соединения (182 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 5,36 (2H, с), 7,44 (1H, дд, J=7,6, 7,0 Гц), 7,67 (1H, ушир.с), 7,76 (1H, дд, J=8,1, 7,6 Гц), 7,82 (1H, д, J=2,2 Гц), 7,91 (1H, д, J=7,0 Гц), 8,03 (1H, д, J=8,1 Гц), 11,52 (1H, ушир.с). MS (m/z): 406 (M-H)-.
Пример 38: 3-(3-циано-4-гидрокси-5-трифторметилбензоил)-1,1-диоксо-5-трифторметил-2,3-дигидро-1,3-бензотиазол
(a) Синтез 5-трифторметил-2,3-дигидро-1,3-бензотиазола
37% формалин (0,22 мл) разбавляли водой (6 мл), добавляли к раствору диэтиловый эфир (6 мл), триэтиламин (0,37 мл) и гидрохлорид 2-амино-4-трифторметилбензолтиола (611 мг), а затем перемешивали смесь при комнатной температуре в течение 30 минут. Органический слой разделяли, и экстрагировали водный слой диэтиловым эфиром. Органические слои объединяли, промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и использовали полученный остаток для синтеза (b).
(b) Синтез 3-(3-циано-4-метокси-5-трифторметилбензоил)-5-трифторметил-2,3-дигидро-1,3-бензотиазола
5-Трифторметил-2,3-дигидро-1,3-бензотиазол растворяли в дихлорметане (5 мл), добавляли к раствору триэтиламин (0,56 мл) и 3-циано-4-метокси-5-трифторметилбензоилхлорид (839 мг), а затем перемешивали смесь при комнатной температуре в течение 1,5 часов. Добавляли воду, и экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 8/1) с получением указанного в заголовке соединения (479 мг) в виде бледно-желтого аморфного продукта.
(c) Синтез 3-(3-циано-4-метокси-5-трифторметилбензоил)-1,1-диоксо-5-трифторметил-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-метокси-5-трифторметилбензоил)-5-трифторметил-2,3-дигидро-1,3-бензотиазол (520 мг) растворяли в хлороформе (8 мл), и при 0°C добавляли к раствору 70% мета-хлорпербензойную кислоту (2,37 г). После перемешивания смеси при комнатной температуре в течение 40 часов добавляли 1н гидроксид натрия, выпавшее в осадок кристаллическое вещество затем собирали путем фильтрования и промывали 1н гидроксидом натрия и водой. Затем фильтрат экстрагировали этилацетатом, органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, полученный продукт объединяли с кристаллическим веществом, собранным путем фильтрования, а затем промывали смесь смесью н-гексана и этилацетата в соотношении 1/1 с получением указанного в заголовке соединения (459 мг) в виде бесцветного кристаллического вещества.
(d) Синтез 3-(3-циано-4-гидрокси-5-трифторметилбензоил)-1,1-диоксо-5-трифторметил-2,3-дигидро-1,3-бензотиазола
3-(3-Циано-4-метокси-5-трифторметилбензоил)-1,1-диоксо-5-трифторметил-2,3-дигидро-1,3-бензотиазол (459 мг) растворяли в N,N-диметилформамиде (4,5 мл), добавляли к раствору хлорид лития (169 мг), а затем перемешивали смесь при 70°C в течение 1 часа. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/хлороформа с получением указанного в заголовке соединения (411 мг) в виде бесцветного кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 5,47 (2H, с), 7,81 (1H, д, J=8,1 Гц), 8,10 (1H, с), 8,21 (1H, д, J=8,1 Гц), 8,28 (1H, с), 8,43 (1H, с). MS (m/z): 449 (M-H)-.
Пример 39: 3-(3,5-дихлор-4-гидроксибензоил)-1,1-диоксо-5-трифторметил-2,3-дигидро-1,3-бензотиазол
(a) Синтез 3-(3,5-дихлор-4-метоксибензоил)-5-трифторметил-2,3-дигидро-1,3-бензотиазола
Гидрохлорид 2-амино-4-трифторметилбензолтиола (502 мг) и 5-трифторметил-2,3-дигидро-1,3-бензотиазол, синтезированный из 37% формалина (0,18 мл) и триэтиламина (0,30 мл) тем же способом, что и описанный в примере 38, растворяли в дихлорметане (5 мл), добавляли к раствору триэтиламин (0,30 мл) и 3,5-дихлор-4-метоксибензоилхлорид (354 мг), а затем перемешивали смесь при комнатной температуре в течение 2 часов. Добавляли воду, и экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой, 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 8/1) с получением указанного в заголовке соединения (223 мг) в виде бесцветного маслянистого вещества.
(b) Синтез 3-(3,5-дихлор-4-метоксибензоил)-1,1-диоксо-5-трифторметил-2,3-дигидро-1,3-бензотиазола
3-(3,5-Дихлор-4-метоксибензоил)-5-трифторметил-2,3-дигидро-1,3-бензотиазол (223 мг) растворяли в хлороформе (5 мл), и при 0°C добавляли к раствору 70% мета-хлорпербензойную кислоту (805 мг). После перемешивания смеси при комнатной температуре в течение 20 часов добавляли 1н гидроксид натрия, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н гидроксидом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления с получением указанного в заголовке соединения (211 мг) в виде бледно-желтого кристаллического вещества.
(c) Синтез 3-(3,5-дихлор-4-гидроксибензоил)-1,1-диоксо-5-трифторметил-2,3-дигидро-1,3-бензотиазола
3-(3,5-Дихлор-4-метоксибензоил)-1,1-диоксо-5-трифторметил-2,3-дигидро-1,3-бензотиазол (209 мг) растворяли в N,N-диметилформамиде (2 мл), добавляли к раствору хлорид лития (106 мг), а затем перемешивали смесь при 120°C в течение 2 часов. К полученному реакционному раствору добавляли 1н соляную кислоту, а затем экстрагировали смесь этилацетатом. Органический слой промывали 1н соляной кислотой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, и выкристаллизовывали полученный остаток из н-гексана/хлороформа с получением указанного в заголовке соединения (167 мг) в виде бледно-желтого кристаллического вещества.
1H-ЯМР δ (ДМСО-d6): 5,46 (2H, с), 7,76 (2H, с), 7,80 (1H, д, J=8,1 Гц), 8,20 (1H, д, J=8,1 Гц), 8,41 (1H, с), 11,11 (1H, ушир.с). MS (m/z): 424 (M-H)-, 426 (M+2-H)-.
Пример 40: 3-(3-хлор-4-гидрокси-5-трифторметилбензоил)-5 или 6-гидрокси-1,1-диоксо-2,3-дигидро-1,3-бензотиазол
3-(3-Хлор-4-гидрокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол (941 мг), полученный в примере 18, суспендировали в 15,7 мл 0,5% раствора метилцеллюлозы (0,5% MC), вводили суспензию восьми самцам крыс Wistar/ST в количестве 1,8 мл каждому, и сразу после введения собирали мочу в течение 6,5 часов. Полученную мочу (28 мл) подкисляли добавлением соляной кислоты, а затем экстрагировали этилацетатом. Полученный органический слой промывали солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли в условиях пониженного давления, полученный остаток очищали по методу колоночной хроматографии на силикагеле (н-гексан/этилацетат = 1/1), а затем выкристаллизовывали из н-гексана/этилацетата с получением указанного в заголовке соединения (36 мг).
1H-ЯМР δ (ДМСО-d6): 5,34 (2H, с), 7,09 (1H, д, J=2,6 Гц), 7,15 (1H, дд, J=9,0, 2,6 Гц), 7,82 (1H, д, J=1,9 Гц), 7,90 (1H, д, J=9,0 Гц), 8,02 (1H, д, J=1,9 Гц), 10,34 (1H, с), 11,38 (1H, ушир.с). MS (m/z): 406 (M-H)-, 408 (M+2-H)-.
В последующей таблице 1 перечислены значения R1, R2, R3 и X, соответствующие представленным выше примерам, применительно к соединению, представленному общей формулой (1).
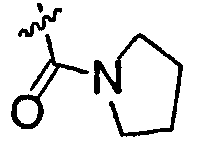
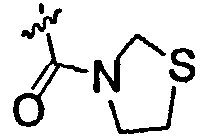
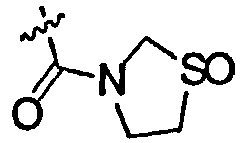
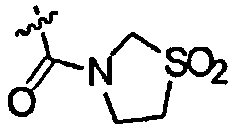
Тестовый пример 1: Урикозурическое действие в модели с пиразинамидом на крысах
7-8-недельным самцам крыс Wistar/ST (по 4 крысы в группе), не получавшим пищу в течение 16 часов, перорально вводили пиразинамид, суспендированный в 0,5% растворе метилцеллюлозы (0,5% МС), в дозе 400 мг/кг. Через 30 минут перорально вводили тестируемое вещество, суспендированное в 0,5% МС, в дозе 30 мг/кг, и собирали мочу в течение 1 часа спустя 2-3 часа после введения. В начале сбора мочи и после окончания сбора мочи у крыс принудительно вызывали мочеиспускание путем нажатия на брюшко крысы. С помощью набора измеряли концентрацию мочевой кислоты и креатинина в моче, и использовали соотношение концентрации мочевой кислоты к концентрации креатинина в качестве индикатора урикозурического действия. Действие каждого тестируемого вещества выражали в виде процентного отношения к контролю.
Тестовый пример 2: Концентрация неизмененного соединения в моче крыс
Двум самцам крыс Wistar/ST, не получавшим пищу в течение 16 часов, перорально вводили тестируемое вещество, суспендированное в 0,5% МС, в дозе 3 мг/кг. Сразу после введения собирали мочу в течение 4 часов. После окончания сбора мочи оставшуюся в мочевом пузыре мочу полностью экскретировали путем нажатия на брюшко крысы. Концентрацию неизмененного соединения в моче измеряли по методу ВЭЖХ и представляли в виде молярной концентрации (мкМ).
Результаты приведенного выше теста представлены ниже в таблице 2.
171 (100 мг/кг)
Как описано выше, новое производное фенола, представленное общей формулой (1), его фармацевтически приемлемая соль, и их гидрат и их сольват, характеризуются урикозурическим действием, равным 20%-108%, а также обладают превосходной эффективностью по сравнению с существующим лекарством, которое характеризуется урикозурическим действием, равным 11%-14%. Новое производное фенола, представленное общей формулой (1), его фармацевтически приемлемая соль, и их гидрат и их сольват, превосходны тем, что по сравнению с существующим лекарством, для которого выведение неизмененного соединения с мочой не обнаруживается, неизмененное соединение по настоящему изобретению проявляет эффективность лекарства, поскольку неизмененное соединение явственно выводится с мочой. Соответственно, новое производное фенола, представленное общей формулой (1), его фармацевтически приемлемая соль, и их гидрат и их сольват, характеризуются высокой концентрацией неизмененного соединения в моче, а также обладают превосходным урикозурическим действием и являются исключительно безопасными, и таким образом применимы в качестве лекарственного средства для ускорения выведения мочевой кислоты; лекарственного средства для снижения количества мочевой кислоты и/или концентрации мочевой кислоты в крови и/или ткани; лекарственного средства для применения при профилактике и/или лечении заболевания, ассоциированного с мочевой кислотой в крови и/или ткани; лекарственного средства для применения при профилактике и/или лечении гиперурикемии; и лекарственного средства для применения при профилактике и/или лечении заболевания, ассоциированного с гиперурикемией и/или заболевания, сопровождающегося гиперурикемией.
После взвешивания вышеуказанных компонентов в соотношении, соответствующем составу, получают порошок для прессования путем способа влажной грануляции. Для получения таблеток этот порошок спрессовывают так, чтобы в каждой таблетке содержалось 5 мг соединения примера 1.
Соединения по настоящему изобретению характеризуются высокой концентрацией неизмененного соединения в моче, а также обладают превосходным урикозурическим действием и являются превосходным в плане безопасности, а потому применимы в качестве лекарственного средства для ускорения выведения мочевой кислоты; лекарственного средства для снижения количества мочевой кислоты и/или концентрации мочевой кислоты в крови и/или ткани; лекарственного средства для применения при профилактике и/или лечении заболевания, ассоциированного с мочевой кислотой в крови и/или ткани; лекарственного средства для применения при профилактике и/или лечении гиперурикемии; и лекарственного средства для применения при профилактике и/или лечении заболевания, ассоциированного с гиперурикемией и/или заболевания, сопровождающегося гиперурикемией.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНАЦИОННОЕ ЛЕКАРСТВО | 2003 |
|
RU2328280C2 |
| ПРОИЗВОДНЫЕ АМИНОМЕТИЛПИРРОЛИДИНА, ИМЕЮЩИЕ АРОМАТИЧЕСКИЕ ЗАМЕСТИТЕЛИ | 2000 |
|
RU2255938C2 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ИНГИБИТОР JAK | 2015 |
|
RU2674262C2 |
| ПРОИЗВОДНЫЕ 1,3-ДИОКСОИНДЕНА, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ АНТИВИРУСНАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2012 |
|
RU2566761C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА И ИНДАЗОЛА, ОБЛАДАЮЩИЕ КОНСЕРВИРУЮЩИМ ДЕЙСТВИЕМ ПО ОТНОШЕНИЮ К КЛЕТКАМ, ТКАНЯМ И ОРГАНАМ | 2009 |
|
RU2460525C2 |
| 6,7-ДИГИДРО-5H-ПИРИДО[2,3-C]ПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛКОВ BCLXL И ПРОАПОПТОТИЧЕСКИХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ | 2020 |
|
RU2832191C2 |
| МАКРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2798235C2 |
| ТЕТРАЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2010 |
|
RU2585622C2 |
| ТЕТРАЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2010 |
|
RU2725140C2 |
| ТИЕНОПИРИМИДИНДИОНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2294937C9 |
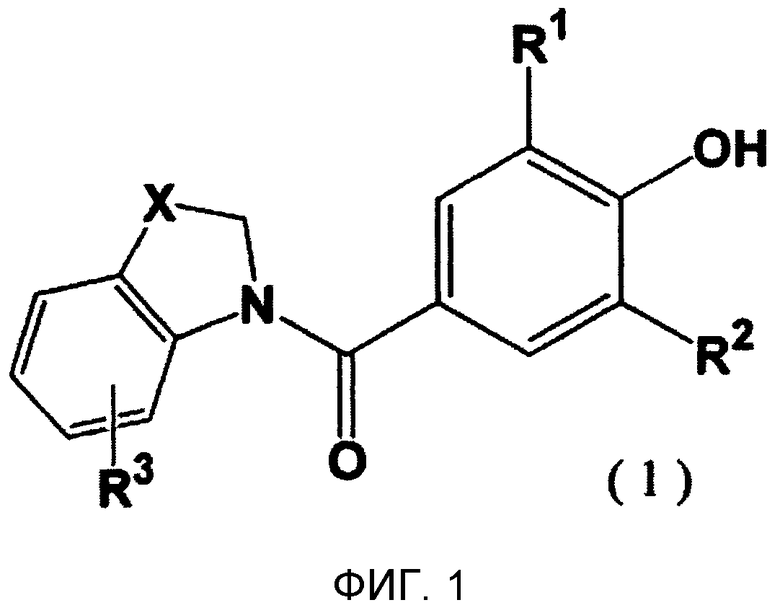
Изобретение относится к производным фенола формулы (1), где R1 представляет собой С1-С6 алкильную группу, С1-С6 алкинильную группу, С1-С6 галогеналкильную группу, С1-С6 алкилсульфанильную группу или атом галогена, R2 представляет собой циано группу или атом галогена, R3 представляет собой атом водорода, и Х представляет собой -S(=O)2. Также изобретение относится к лекарственному средству, содержащему в качестве активного ингредиента соединение формулы (1). Технический результат - производные фенола формулы (1), которые характеризуются высокой концентрацией неизменного соединения в моче и обладают урикозурическим действием. 2 н. и 9 з.п. ф-лы, 1 ил., 2 табл., 42 пр.
1. Новое производное фенола, представленное следующей общей формулой (1):
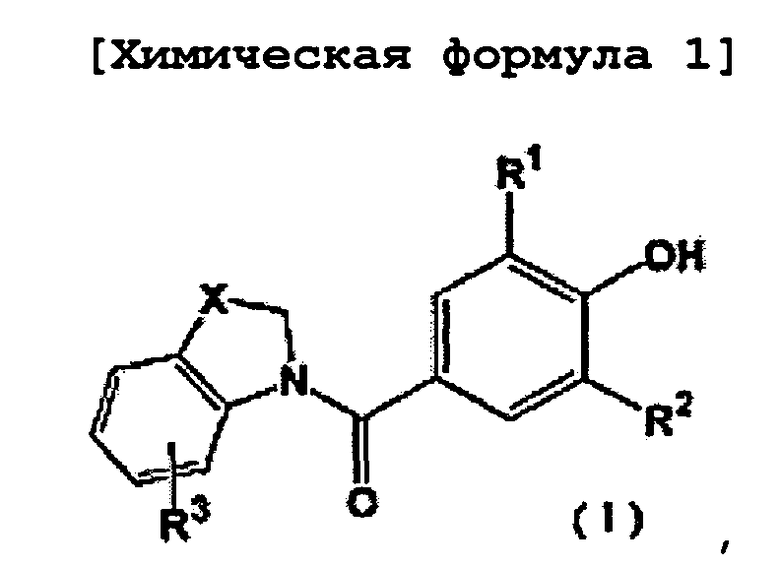
где R1 представляет собой С1-С6 алкильную группу, С1-С6 алкинильную группу, С1-С6 галогеналкильную группу, С1-С6 алкилсульфанильную группу, или атом галогена,
R2 представляет собой циано группу или атом галогена,
R3 представляет собой атом водорода, и
Х представляет собой -S(=O)2,
характеризующееся тем, что соединение выбирают из группы, состоящей из:
3-(3,5-дихлор-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол,
3-(3-циано-4-гидрокси-5-трифторметилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол,
3-(3-хлор-5-циано-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол,
3-(3-циано-5-этил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол,
3-(3-циано-5-циклопропил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол,
3-(3-циано-5-этинил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол,
3-(3-циано-4-гидрокси-5-метилсульфанилбензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол,
3-(3-циано-5-этилсульфанил-4-гидроксибензоил)-1,1-диоксо-2,3-дигидро-1,3-бензотиазол,
или его фармацевтически приемлемая соль.
2. Лекарственное средство, характеризующееся высокой концентрацией неизменного соединения в моче и обладающего урикозурическим действием, содержащее в качестве активного ингредиента соединение по п.1 или его фармацевтически приемлемой соли.
3. Лекарственное средство по п.2, которое представлено в форме фармацевтической композиции, содержащей одну, две или более добавок для приготовления состава.
4. Лекарственное средство по п.2 или 3 для ускорения выведения мочевой кислоты.
5. Лекарственное средство по п.2 или 3 для снижения количества мочевой кислоты и/или концентрации мочевой кислоты в крови и/или ткани.
6. Лекарственное средство по п.2 или 3 для применения при профилактике и/или лечении заболевания, ассоциированного с мочевой кислотой в крови и/или ткани.
7. Лекарственное средство по п.2 или 3 для применения при профилактике и/или лечении гиперурикемии, заболевания, ассоциированного с гиперурикемией и/или заболевания, сопровождающегося гиперурикемией.
8. Лекарственное средство по п.2 или 3 для применения при профилактике и/или лечении подагры, мочекаменной болезни, ожирения, гиперлипидемии, дислипидемии, аномальной толерантности к глюкозе, сахарного диабета, метаболического синдрома, заболевания почек и/или сердечно-сосудистого заболевания.
9. Лекарственное средство по п.8, где подагра включает в качестве болезненного состояния по меньшей мере одно из подагрического узелка, подагрического артрита и подагрической почки.
10. Лекарственное средство по п.8, где сердечно-сосудистое заболевание представляет собой гипертензию, болезнь сонных артерий, артериосклероз, тромбоз, эндотелиальную дисфункцию, цереброваскулярное заболевание и/или заболевание сердца.
11. Лекарственное средство по п.2 или 3 для применения при профилактике и/или лечении осложнения заболевания по любому из пп.6-10.
| УСТРОЙСТВО ДЛЯ ЭЛЕКТРОФОРЕЗА | 1987 |
|
SU1820515A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Пусковое приспособление для двигателей внутреннего горения | 1927 |
|
SU7718A1 |

