Область техники, к которой относится изобретение
Настоящее изобретение относится к новому циклоалкил-замещенному производному имидазола, обладающему превосходной ингибирующей активностью в отношении TAFIa.
Уровень техники
Когда происходят нарушения in vivo в кровеносных сосудах, тромбоциты и/или каскады коагуляции активируются для предотвращения кровотечения с образованием тромбов, которые, в свою очередь, подавляют кровоизлияние. Тромбин, образованный посредством активации каскада коагуляции, расщепляет фибриноген с образованием нерастворимого фибрина. Фибрин присутствует в форме каркаса в тромбах и работает для придания тромбам прочности. Эта реакция называется коагуляцией. Образовавшийся фибрин затем разлагается посредством in-vivo реакции. Эта реакция называется фибринолизом. При нормальных условиях коагуляция и фибринолиз сбалансированы, и аномальные количества тромбов не накапливаются в кровеносных сосудах. Однако, как только баланс нарушается для ускорения коагуляции, он может войти в состояние, в котором вероятно в кровеносных сосудах образуется тромб, приводя к разнообразным заболеваниям, приписываемым тромбозу. Образование тромба вызывается тремя факторами (Триада Вирчоу: изменение в свойствах сосудистых стенок, изменение в компонентах крови и изменение кровотока). Заболевания, приписываемые образованию тромба, являются одной из наиболее общих причин смерти среди развитых наций.
TAFI (активируемый тромбином ингибитор фибринолиза) представляет собой карбоксипептидазу, которая продуцируется в печени и секретируется в кровь. Этот фермент активируется посредством расщепления N-концевых 92 аминокислотных остатков посредством тромбиновых или тромбин/тромбомодулиновых комплексов. TAFI также называют прокарбоксипептидазой U, прокарбоксипептидазой R или плазменной прокарбоксипептидазой В.
Активированный TAFI называют TAFIa. TAFIa ингибирует фибринолиз посредством удаления С-концевого остатка Lys или Arg фибрина или продуктов деградации фибрина (FDP), которые являются основными компонентами тромбов. Два фермента, tPA (активатор плазминогена тканевого типа) и плазминоген, которые индуцируют и инициируют фибринолиз, связываются с остатком Lys фибрина или FDP через их Lys-связывающие сайты. На поверхности молекулы фибрина tPA последовательно активирует плазминоген и преобразует его в плазмин, инициируя фибринолиз. Плазмин расщепляет фибрин, и остаток Lys или Arg появляется на С-концах образованных FDP. Продолжение фибринолиза дает возможность плазмину и tPA заново связываться с остатками Lys FDP, чтобы дополнительно образовывать плазмин. Это эффективно инициирует фибринолиз (положительный механизм обратной связи для фибринолиза). TAFIa ингибирует активацию плазминогена tPA на молекуле фибрина посредством удаления С-концевых остатков Lys FDP. В результате, эффективного фибринолиза не происходит. TAFIa подавляет механизм положительной обратной связи для фибринолиза. Эти открытия описаны подробно в обзоре по TAFI и его ингибиторам (непатентная литература 1).
Как описано выше, тонкий баланс между коагуляцией и фибринолизом достигается in vivo. Когда коагуляция ускоряется заболеваниями или т.п., вероятно, тромбы начинают образовываться, развивая разнообразные заболевания. Такие заболевания включают инфаркт миокарда, стенокардию, острый коронарный синдром, инфаркт головного мозга, тромбоз глубоких вен, тромбоэмболию, периферическую артериальную окклюзию, сепсис, синдром диссеминированной внутрисосудистой коагуляции и фиброз легких.
До настоящего времени лечение тромбоза было часто нацелено на ферменты в каскадах коагуляции. Эти ферменты включают активированный фактор коагуляции Х (Ха), тромбин и т.п. Ингибиторы против этих ферментов имеют риск потенциальной неблагоприятной реакции, такой как кровотечение. Нельзя ожидать, что гепарин или низкомолекулярный гепарин будут проявлять лекарственную эффективность действия при пероральном введении, и требуется, чтобы их вводили в больницах. Варфарин является перорально вводимым, но требуются периодические тесты крови по причине взаимодействия с другими лекарственными средствами и т.д. Аспирин представляет собой перорально вводимое лекарственное средство, которое ингибирует образование тромба посредством подавления активации тромбоцитов, но имеет неблагоприятную реакцию, такую как желудочное кровотечение. Цель дальнейшего улучшения существующих в настоящее время терапий состоит в предотвращении пролонгации времени кровотечения, при поддержании в то же время высокого терапевтического эффекта при введении лекарственного средства. Предполагают, что ингибиторы TAFIa имеют небольшой риск кровотечения, поскольку они не влияют на процесс гемостаза, включающий коагуляцию и тромбоциты.
При патологиях, где может возникнуть ситуация, что тромб, вероятно, образуется вследствие ускоренных реакций коагуляции, тромбы могут удаляться более быстро посредством придания эффективности фибринолизу через ингибирование TAFIa. Можно ожидать, что при этом будут проявляться превосходные эффекты на лечение/профилактику заболеваний, приписываемых тромбам. До настоящего времени сообщалось о нескольких случаях экспериментов на животных, которые продемонстрировали антитромботический эффект посредством ингибирования TAFIa.
Сообщается, что внутривенное введение мышам TAFIa-ингибирующего полипептида, состоящего из 39 аминокислот (ингибитор карбоксипептидазы картофеля (PCI)), продемонстрировало антитромботический эффект на индуцированных хлоридом железа моделях тромба (непатентная литература 2).
Низкомолекулярный ингибитор TAFIa снижал количество тромбов приблизительно на 35% при внутривенном введении кроличьим моделям венозного тромбоза (непатентная литература 3).
Низкомолекулярное TAFIa-ингибирующее соединение продемонстрировало на крысиных моделях тромбоэмболии снижение количества отложений тромбов в почке вместе с эффектом увеличения D-димерного маркера фибринолиза, а также сравнимый антитромботический эффект при сниженной дозе tPA при комбинированном применении с tPA (непатентная литература 4 и 5).
Источники патентной литературы 1-5 раскрывают соединения, которые проявляют ингибирующую активность в отношении TAFIa.
Перечень ссылок
Патентная литература
Патентная литература 1: Описание опубликованной международной патентной заявки № WO 2002/014285
Патентная литература 2: Описание опубликованной международной патентной заявки № WO 2003/061652
Патентная литература 3: Описание опубликованной международной патентной заявки № WO 2003/061653
Патентная литература 4: Описание опубликованной международной патентной заявки № WO 2005/105781
Патентная литература 5: Описание опубликованной международной патентной заявки № WO 2005/013526
Непатентная литература
Непатентная литература 1: Willemse JL, Journal of Thrombosis and Haemostasis, 2009, 7, 1962-71
Непатентная литература 2: Wang X. et al., Journal of Thrombosis and Haemostasis, 2006, 3, 403-410
Непатентная литература 3: Bunnage ME., et al., Journal of Medicinal Chemistry, 2007, 50, 6095-6103
Непатентная литература 4: Muto, Y., et al., Critical Care Med., 2009, 37, 1744-1749
Непатентная литература 5: Suzuki, K., The Journal of Pharmacology and Experimental Therapeutics, 2004, 309, 607-615
Краткое изложение сущности изобретения
Техническая проблема
Известные в настоящее время соединения, обладающие ингибирующей активностью в отношении TAFIa, являются менее чем удовлетворительными с точки зрения эффективности действия или безопасности, такой как риск кровотечения, и существует высокая потребность в ингибиторе TAFIa, превосходном по безопасности и эффективности действия.
Авторы настоящего изобретения провели разнообразные синтезы и исследования с целью получения терапевтического лекарственного средства против инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких, обладающего превосходной ингибирующей активностью в отношении TAFIa. В результате, авторы настоящего изобретения завершили создание настоящего изобретения, обнаружив, что циклоалкил-замещенное производное имидазола, имеющее конкретную структуру, или его фармакологически приемлемая соль проявляет превосходную ингибирующую активность в отношении TAFIa.
Настоящее изобретение предоставляет циклоалкил-замещенное производное имидазола, имеющее конкретную структуру, или его фармакологически приемлемую соль, которое проявляет превосходную ингибирующую активность в отношении TAFIa, и фармацевтическое лекарственное средство, содержащее его.
Конкретно, настоящее изобретение предоставляет:
(1) соединение, представленное общей формулой (I), или его фармакологически приемлемую соль:
[Формула 1]
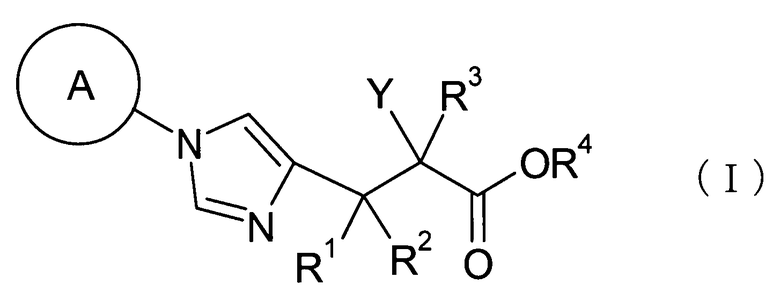
где А представляет С3-С12циклоалкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из группы фтора, гидроксигруппы, С1-С6алкильной группы, С1-С6алкоксигруппы, арилоксигруппы и гетероциклилоксигруппы; R1, R2 и R3, каждый независимо, представляет собой атом водорода, группу фтора или С1-С6алкильную группу; R4 представляет собой атом водорода или группу пролекарства; и Y представляет собой группу: -СН2-CHR5-CH2-NHR6 (где R5 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу, и R6 представляет собой атом водорода или группу пролекарства), -O-CHR7-CH2-NHR8 (где R7 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу, и R8 представляет собой атом водорода или группу пролекарства) или
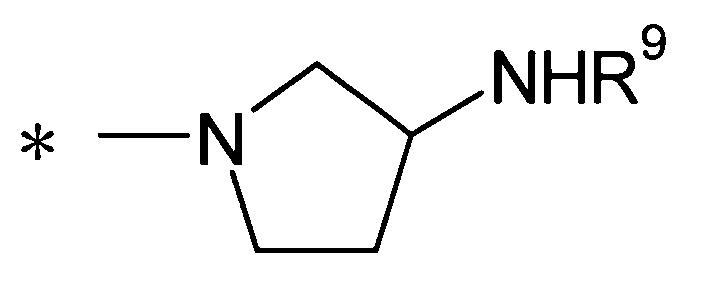
[Формула 2]
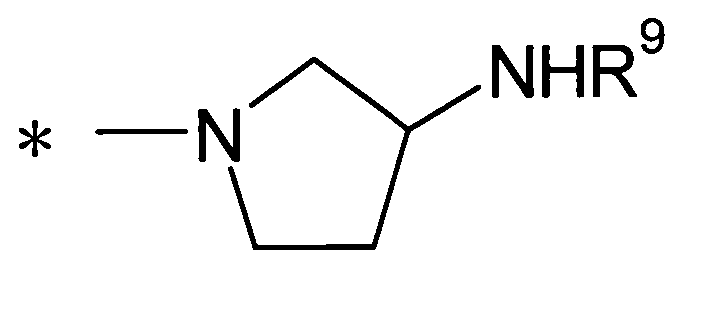
(где R9 представляет собой атом водорода или группу пролекарства, и * представляет собой положение для замещения);
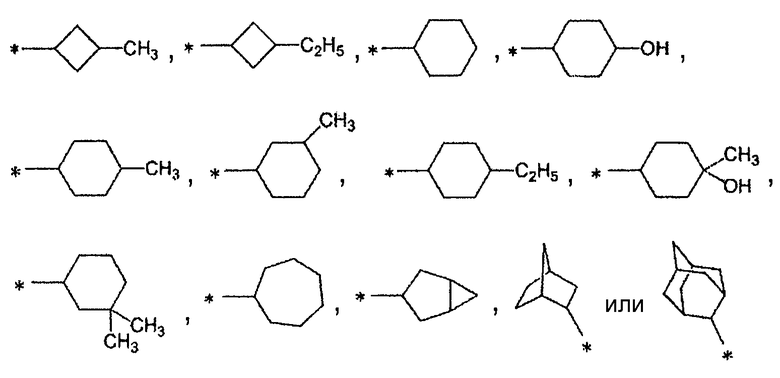
(2) соединение согласно (1) или его фармакологически приемлемую соль, где А представляет собой циклобутильную группу, циклогексильную группу, циклогептильную группу, бицикло[3.1.0]гексильную группу, бицикло[2.2.1]гептильную группу или адамантильную группу, каждая из которых может быть замещена одной-тремя одинаковыми или различными группами, выбранными из группы фтора, гидроксигруппы, С1-С6алкильной группы, С1-С6алкоксигруппы, арилоксигруппы и гетероциклилоксигруппы;
(3) соединение согласно (1) или его фармакологически приемлемую соль, где А представляет собой циклобутильную группу, циклогексильную группу, циклогептильную группу, бицикло[3.1.0]гексильную группу, бицикло[2.2.1]гептильную группу или адамантильную группу, каждая из которых может быть замещена одной-тремя одинаковыми или различными группами, выбранными из гидроксигруппы, метильной группы и этильной группы;
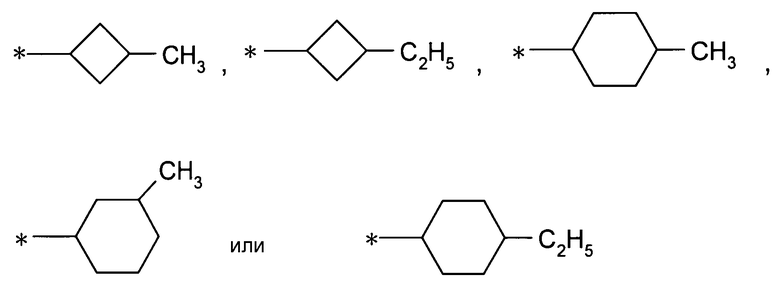
(4) соединение согласно (1) или его фармакологически приемлемую соль, где А представляет собой циклогексильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из группы фтора, гидроксигруппы, С1-С6алкильной группы, С1-С6алкоксигруппы, арилоксигруппы и гетероциклилоксигруппы;
(5) соединение согласно (1) или его фармакологически приемлемую соль, где А представляет собой С3-С12циклоалкильную группу, замещенную одной или двумя одинаковыми или различными С1-С6алкильными группами;
(6) соединение согласно (1) или его фармакологически приемлемую соль, где А представляет собой С3-С12циклоалкильную группу, замещенную метильной группой или этильной группой;
(7) соединение согласно (1) или его фармакологически приемлемую соль, где А представляет собой циклогексильную группу, замещенную одной или двумя одинаковыми или различными С1-С6алкильными группами;
(8) соединение согласно (1) или его фармакологически приемлемую соль, где А представляет собой циклогексильную группу, замещенную метильной группой или этильной группой;
(9) соединение согласно (1) или его фармакологически приемлемую соль, где А представляет собой группу:
[Формула 3]
(где * представляет собой положение для замещения);
(10) соединение согласно (1) или его фармакологически приемлемую соль, где А представляет собой группу:
[Формула 4]
(где * представляет собой положение для замещения);
(11) соединение согласно (1) или его фармакологически приемлемую соль, где А представляет собой группу:
[Формула 5]
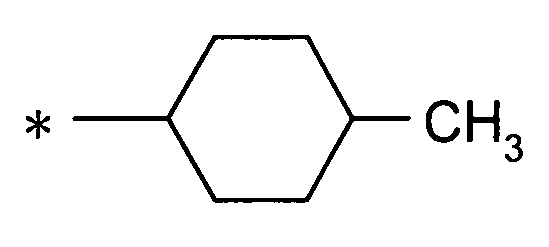
(где * представляет собой положение для замещения);
(12) соединение согласно (1) или его фармакологически приемлемую соль, где А представляет собой группу:
[Формула 6]
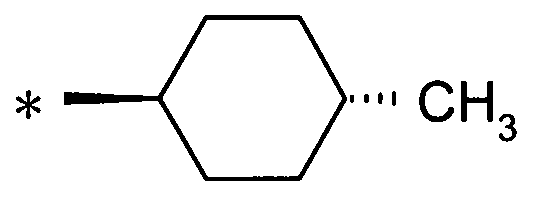
(где * представляет собой положение для замещения);
(13) соединение согласно любому из (1)-(12) или его фармакологически приемлемую соль, где Y представляет собой группу: -СН2-CHR5-CH2-NHR6 (где R5 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу, и R6 представляет собой атом водорода или группу пролекарства);
(14) соединение согласно (13) или его фармакологически приемлемую соль, где R5 представляет собой атом водорода;
(15) соединение согласно (13) или (14) или его фармакологически приемлемую соль, где R6 представляет собой атом водорода;
(16) соединение согласно (13) или (14) или его фармакологически приемлемую соль, где R6 представляет собой группу пролекарства;
(17) соединение согласно (16) или его фармакологически приемлемую соль, где группа пролекарства, представленная R6, представляет собой С1-С6алканоильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из аминогруппы, группы галогена, гидроксигруппы, карбоксигруппы, карбамоильной группы, С1-С6алкоксигруппы, арильной группы и гетероциклильной группы; (С1-С6алкокси)карбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С1-С6алкильной группы, С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкилоксикарбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы;
(18) соединение согласно (16) или его фармакологически приемлемую соль, где группа пролекарства, представленная R6, представляет собой фенилаланильную группу, L-норлейцильную группу, [(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонильную группу, [1-(изобутирилокси)этокси]карбонильную группу, [1-(2,2-диметилпропаноилокси)этокси]карбонильную группу, ({1-[(циклогексилкарбонил)окси]этокси}карбонильную группу или (1-ацетоксиэтокси)карбонильную группу;
(19) соединение согласно (1)-(12) или его фармакологически приемлемую соль, где Y представляет собой группу: -O-CHR7-CH2-NHR8 (где R7 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу, и R8 представляет собой атом водорода или группу пролекарства);
(20) соединение согласно (19) или его фармакологически приемлемую соль, где R7 представляет собой атом водорода;
(21) соединение согласно (19) или (20) или его фармакологически приемлемую соль, где R8 представляет собой атом водорода;
(22) соединение согласно (19) или (20) или его фармакологически приемлемую соль, где R8 представляет собой группу пролекарства;
(23) соединение согласно (22) или его фармакологически приемлемую соль, где группа пролекарства, представленная R8, представляет собой С1-С6алканоильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из аминогруппы, группы галогена, гидроксигруппы, карбоксигруппы, карбамоильной группы, С1-С6алкоксигруппы, арильной группы и гетероциклильной группы; (С1-С6алкокси)карбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С1-С6алкильной группы, С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкилоксикарбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы;
(24) соединение согласно любому из (1)-(12) или его фармакологически приемлемую соль, где Y представляет собой группу:
[Формула 7]
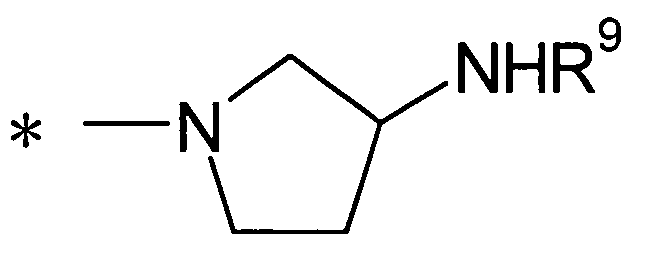
(где R9 представляет собой атом водорода или группу пролекарства, и * представляет собой положение для замещения);
(25) соединение согласно любому из (1)-(12) или его фармакологически приемлемую соль, где Y представляет собой группу:
[Формула 8]
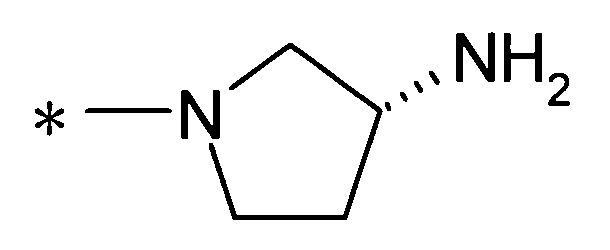
(где * представляет собой положение для замещения);
(26) соединение согласно любому из (1)-(12) или его фармакологически приемлемую соль, где Y представляет собой группу:
[Формула 9]
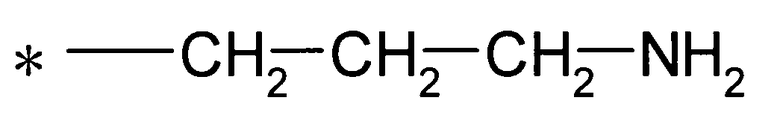
(где * представляет собой положение для замещения);
(27) соединение согласно любому из (1)-(26) или его фармакологически приемлемую соль, где все из R1, R2 и R3 представляют собой атом водорода;
(28) соединение согласно любому из (1)-(27) или его фармакологически приемлемую соль, где R4 представляет собой атом водорода;
(29) соединение согласно любому из (1)-(27) или его фармакологически приемлемую соль, где R4 представляет собой группу пролекарства;
(30) соединение согласно (29) или его фармакологически приемлемую соль, где группа пролекарства, представленная R4, представляет собой С1-С6алкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы;
(31) соединение согласно (29) или его фармакологически приемлемую соль, где группа пролекарства, представленная R4, представляет собой бензильную группу или [(изопропоксикарбонил)окси]этильную группу;
(32) соединение, представленное общей формулой (I-1), или его фармакологически приемлемую соль:
[Формула 10]
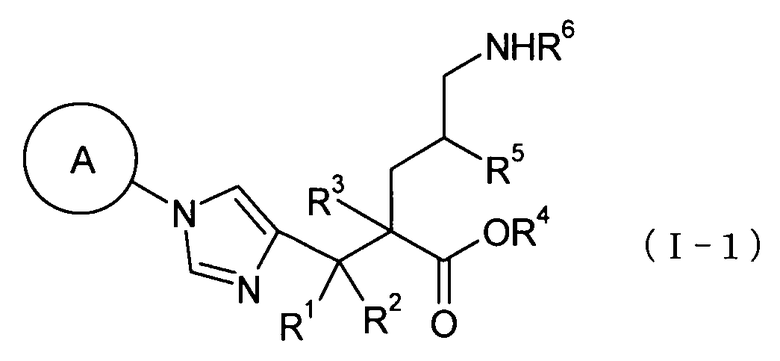
где А представляет С3-С12циклоалкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из группы фтора, гидроксигруппы, С1-С6алкильной группы, С1-С6алкоксигруппы, арилоксигруппы и гетероциклилоксигруппы; R1, R2 и R3, каждый независимо, представляет собой атом водорода, группу фтора или С1-С6алкильную группу; R4 представляет собой атом водорода или группу пролекарства; R5 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу, и R6 представляет собой атом водорода или группу пролекарства;
(33) соединение согласно (32) или его фармакологически приемлемую соль, где А представляет собой циклобутильную группу, циклогексильную группу, циклогептильную группу, бицикло[3.1.0]гексильную группу, бицикло[2.2.1]гептильную группу или адамантильную группу, каждая из которых может быть замещена одной-тремя одинаковыми или различными группами, выбранными из гидроксигруппы, метильной группы и этильной группы; R1, R2 и R3, все представляют собой атом водорода; R4 представляет собой атом водорода; С1-С6алкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из, С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы, R5 представляет собой атом водорода; и R6 представляет собой атом водорода; С1-С6алканоильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из аминогруппы, группы галогена, гидроксигруппы, карбоксигруппы, карбамоильной группы, С1-С6алкоксигруппы, арильной группы и гетероциклильной группы; (С1-С6алкокси)карбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С1-С6алкильной группы, С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкилоксикарбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы;
(34) соединение согласно (32) или его фармакологически приемлемую соль, где А представляет собой циклогексильную группу, замещенную одной или двумя одинаковыми или различными С1-С6алкильными группами; R1, R2 и R3, все представляют собой атом водорода; R4 представляет собой атом водорода, бензильную группу или [(изопропоксикарбонил)окси]этильную группу; R5 представляет собой атом водорода; и R6 представляет собой атом водорода, фенилаланильную группу, L-норлейцильную группу, [(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонильную группу, [1-(изобутирилокси)этокси]карбонильную группу, [1-(2,2-диметилпропаноилокси)этокси]карбонильную группу, ({1-[(циклогексилкарбонил)окси]этокси}карбонильную группу или (1-ацетоксиэтокси)карбонильную группу;
(35) соединение, представленное общей формулой (I-1а), или его фармакологически приемлемую соль:
[Формула 11]
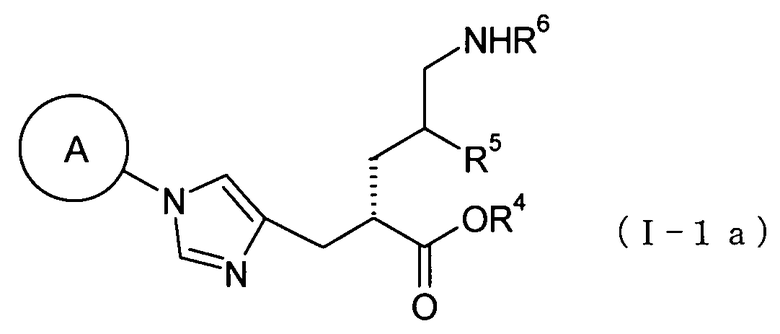
где А представляет собой циклобутильную группу, циклогексильную группу, циклогептильную группу, бицикло[3.1.0]гексильную группу, бицикло[2.2.1]гептильную группу или адамантильную группу, каждая из которых может быть замещена одной-тремя одинаковыми или различными группами, выбранными из гидроксигруппы, метильной группы и этильной группы; R4 представляет собой атом водорода; С1-С6алкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы, R5 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу; и R6 представляет собой атом водорода; С1-С6алканоильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из аминогруппы, группы галогена, гидроксигруппы, карбоксигруппы, карбамоильной группы, С1-С6алкоксигруппы, арильной группы и гетероциклильной группы; (С1-С6алкокси)карбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С1-С6алкильной группы, С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкилоксикарбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы;
(36) соединение согласно (35) или его фармакологически приемлемую соль, где А представляет собой циклогексильную группу, замещенную одной или двумя одинаковыми или различными С1-С6алкильными группами; R1, R2 и R3, все представляют собой атом водорода; R4 представляет собой атом водорода, бензильную группу или [(изопропоксикарбонил)окси]этильную группу; R5 представляет собой атом водорода; и R6 представляет собой атом водорода, фенилаланильную группу, L-норлейцильную группу, [(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонильную группу, [1-(изобутирилокси)этокси]карбонильную группу, [1-(2,2-диметилпропаноилокси)этокси]карбонильную группу, ({1-[(циклогексилкарбонил)окси]этокси}карбонильную группу или (1-ацетоксиэтокси)карбонильную группу;
(37) соединение согласно (35) или его фармакологически приемлемую соль, где А представляет собой циклогексильную группу, замещенную метильной группой или этильной группой; R4, R5 и R6, все представляют собой атом водорода;
(38) соединение, представленное общей формулой (I-2), или его фармакологически приемлемую соль:
[Формула 12]
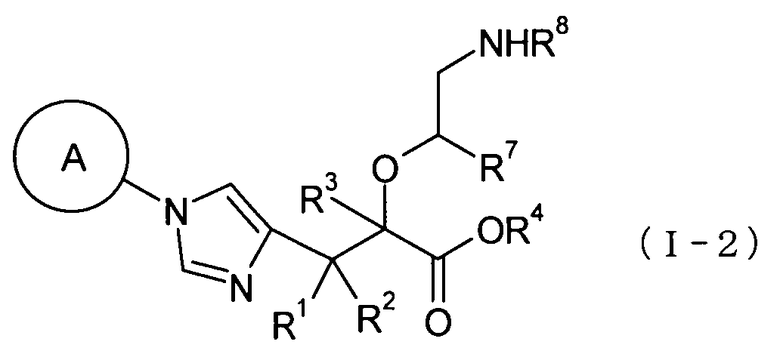
где А представляет собой С3-С12циклоалкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из группы фтора, гидроксигруппы, С1-С6алкильной группы, С1-С6алкоксигруппы, арилоксигруппы и гетероциклилоксигруппы; R1, R2 и R3, каждый независимо представляет собой атом водорода, группу фтора или С1-С6алкильную группу; R4 представляет собой атом водорода или группу пролекарства; R7 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу, и R8 представляет собой атом водорода или группу пролекарства;
(39) соединение согласно (38) или его фармакологически приемлемую соль, где А представляет собой циклобутильную группу, циклогексильную группу, циклогептильную группу, бицикло[3.1.0]гексильную группу, бицикло[2.2.1]гептильную группу или адамантильную группу, каждая из которых может быть замещена одной-тремя одинаковыми или различными группами, выбранными из гидроксигруппы, метильной группы и этильной группы; R1, R2 и R3, все представляют собой атом водорода; R4 представляет собой атом водорода; С1-С6алкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы, R7 представляет собой атом водорода; и R8 представляет собой атом водорода; С1-С6алканоильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из аминогруппы, группы галогена, гидроксигруппы, карбоксигруппы, карбамоильной группы, С1-С6алкоксигруппы, арильной группы и гетероциклильной группы; (С1-С6алкокси)карбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С1-С6алкильной группы, С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкилоксикарбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы;
(40) соединение согласно (38) или его фармакологически приемлемую соль, где А представляет собой циклогексильную группу, замещенную одной или двумя одинаковыми или различными С1-С6алкильными группами; R1, R2 и R3, все представляют собой атом водорода; R4 представляет собой атом водорода, бензильную группу или [(изопропоксикарбонил)окси]этильную группу; и оба из R7 и R8 представляют собой атом водорода;
(41) соединение, представленное общей формулой (I-2а), или его фармакологически приемлемую соль:
[Формула 13]
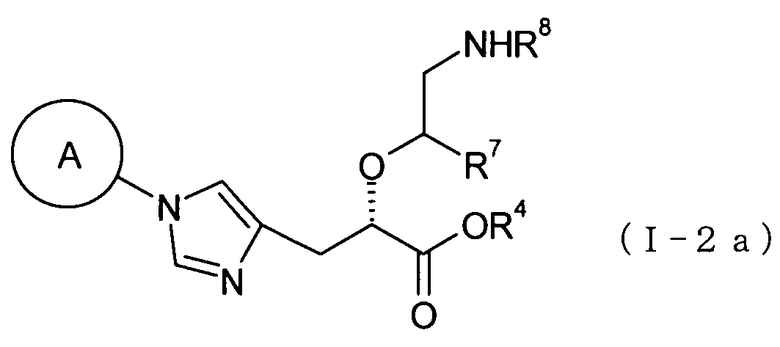
где А представляет собой циклобутильную группу, циклогексильную группу, циклогептильную группу, бицикло[3.1.0]гексильную группу, бицикло[2.2.1]гептильную группу или адамантильную группу, каждая из которых может быть замещена одной-тремя одинаковыми или различными группами, выбранными из гидроксигруппы, метильной группы и этильной группы; R4 представляет собой атом водорода; С1-С6алкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы, R7 представляет собой атом водорода и С1-С6алкильную группу; и R8 представляет собой атом водорода или группу пролекарства;
(42) соединение согласно (41) или его фармакологически приемлемую соль, где А представляет собой циклогексильную группу, замещенную одной или двумя одинаковыми или различными С1-С6алкильными группами; R1, R2 и R3, все представляют собой атом водорода; R4 представляет собой атом водорода, бензильную группу или [(изопропоксикарбонил)окси]этильную группу; R7 представляет собой атом водорода; и R8 представляет собой атом водорода; С1-С6алканоильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из аминогруппы, группы галогена, гидроксигруппы, карбоксигруппы, карбамоильной группы, С1-С6алкоксигруппы, арильной группы и гетероциклильной группы; (С1-С6алкокси)карбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С1-С6алкильной группы, С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы, или гетероциклилалкилоксикарбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы;
(43) соединение согласно (41) или его фармакологически приемлемую соль, где А представляет собой циклогексильную группу, замещенную метильной группой или этильной группой; R4, R7 и R8, все представляют собой атом водорода;
(44) соединение, представленное общей формулой (I-3), или его фармакологически приемлемую соль:
[Формула 14]
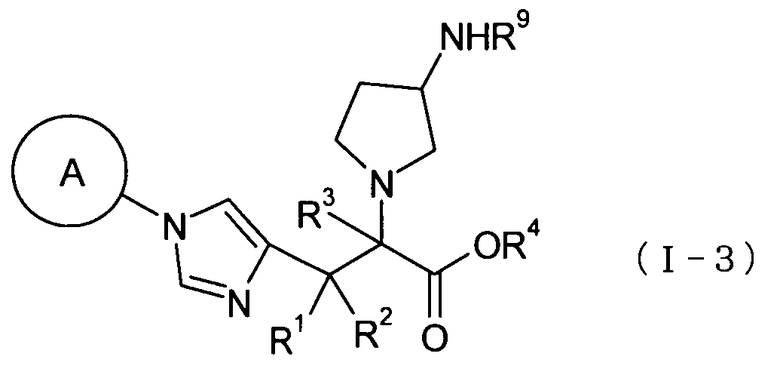
где А представляет собой С3-С12циклоалкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из группы фтора, гидроксигруппы, С1-С6алкильной группы, С1-С6алкоксигруппы, арилоксигруппы и гетероциклилоксигруппы; R1, R2 и R3, каждый независимо представляет собой атом водорода, группу фтора или С1-С6алкильную группу; R4 представляет собой атом водорода или группу пролекарства; и R9 представляет собой атом водорода или группу пролекарства;
(45) соединение согласно (44) или его фармакологически приемлемую соль, где А представляет собой циклобутильную группу, циклогексильную группу, циклогептильную группу, бицикло[3.1.0]гексильную группу, бицикло[2.2.1]гептильную группу или адамантильную группу, каждая из которых может быть замещена одной-тремя одинаковыми или различными группами, выбранными из гидроксигруппы, метильной группы и этильной группы; R1, R2 и R3, все представляют атом водорода; R4 представляет собой атом водорода; С1-С6алкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы; и R9 представляет собой атом водорода; С1-С6алканоильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из аминогруппы, группы галогена, гидроксигруппы, карбоксигруппы, карбамоильной группы, С1-С6алкоксигруппы, арильной группы и гетероциклильной группы; (С1-С6алкокси)карбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С1-С6алкильной группы, С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы, или гетероциклилалкилоксикарбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы;
(46) соединение согласно (44) или его фармакологически приемлемую соль, где А представляет собой циклогексильную группу, замещенную одной или двумя одинаковыми или различными С1-С6алкильными группами; R1, R2 и R3, все представляют собой атом водорода; R4 представляет собой атом водорода, бензильную группу или [(изопропоксикарбонил)окси]этильную группу; и R9 представляет собой атом водорода;
(47) соединение, представленное общей формулой (I-3а), или его фармакологически приемлемую соль:
[Формула 15]
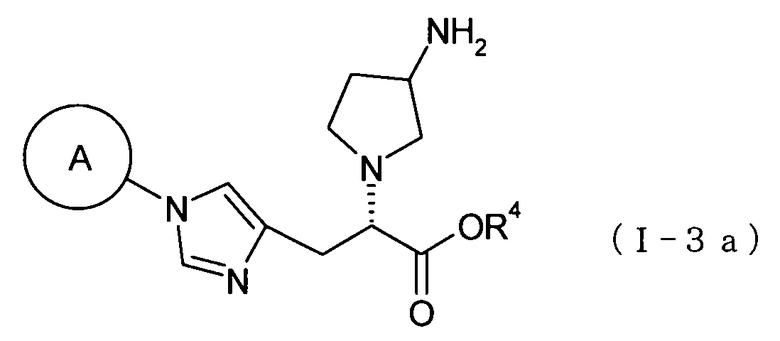
где А представляет собой циклобутильную группу, циклогексильную группу, циклогептильную группу, бицикло[3.1.0]гексильную группу, бицикло[2.2.1]гептильную группу или адамантильную группу, каждая из которых может быть замещена одной-тремя одинаковыми или различными группами, выбранными из гидроксигруппы, метильной группы и этильной группы; и R4 представляет собой атом водорода; С1-С6алкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы;
(48) соединение согласно (47) или его фармакологически приемлемую соль, где А представляет собой циклогексильную группу, замещенную одной или двумя одинаковыми или различными С1-С6алкильными группами; и R4 представляет собой атом водорода, бензильную группу или [(изопропоксикарбонил)окси]этильную группу;
(49) соединение согласно (48) или его фармакологически приемлемую соль, где А представляет собой циклогексильную группу, замещенную метильной группой или этильной группой; и R4 представляет собой атом водорода;
(50) соединение согласно (1) или его фармакологически приемлемую соль, где соединение выбирают из группы, состоящей из
5-амино-2-[(1-циклогексил-1Н-имидазол-4-ил)метил]валериановой кислоты,
5-амино-2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
5-амино-2-{[(1-(4-этилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
5-амино-2-{[(1-(3-этилциклобутил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
5-амино-2-{[(1-(3-метилциклобутил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
5-амино-2-({1-[(1R,3s,5S)-бицикло[3.1.0]гексан-3-ил]-1Н-имидазол-4-ил}метил)валериановой кислоты,
5-амино-2-{[(1-(4-гидроксициклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
5-амино-2-{[(1-(4-гидрокси-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
5-амино-2-{[(1-(3-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
5-амино-2-[(1-циклогептил-1Н-имидазол-4-ил)метил]валериановой кислоты,
5-амино-2-({1-[экзо-бицикло[2.2.1]гепт-2-ил]-1Н-имидазол-4-ил}метил]валериановой кислоты,
5-амино-2-({1-[эндо-бицикло[2.2.1]гепт-2-ил]-1Н-имидазол-4-ил}метил]валериановой кислоты,
2-[(1-адамантан-2-ил-1Н-имидазол-4-ил)метил]-5-аминовалериановой кислоты,
5-амино-2-{[1-(4-феноксициклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
бензил-5-амино-2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерата,
2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-(L-фенилаланиламино)валериановой кислоты,
2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-(L-норлейциламино)валериановой кислоты,
2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-({[(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонил}амино)валериановой кислоты,
5-({[1-(изобутирилокси)этокси]карбонил}амино)-2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
1-[(изопропоксикарбонил)окси]этил-5-({[1-(изобутирилокси)этокси]карбонил}амино)-2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерата,
5-({[1-(2,2-диметилпропаноилокси)этокси]карбонил}амино)-2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
5-[({1-[(циклогексилкарбонил)окси]этокси}карбонил)амино]-2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
2-(2-аминоэтокси)-3-[1-(4-метилциклогексил)-1Н-имидазол-4-ил]пропионовой кислоты,
2-[(1R)-2-амино-1-метилэтокси]-3-[1-(4-метилциклогексил)-1Н-имидазол-4-ил]пропионовой кислоты и
2-[(3S)-3-аминопирролидинил-1-ил]-3-[1-(4-метилциклогексил)-1Н-имидазол-4-ил]пропионовой кислоты;
(51) 5-амино-2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановую кислоту или ее фармакологически приемлемую соль;
(52) 5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановую кислоту или ее фармакологически приемлемую соль;
(53) (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановую кислоту или ее фармакологически приемлемую соль;
(54) фармакологически приемлемую соль соединения согласно любому из (1)-(53), где фармакологически приемлемая соль представляет собой п-толуолсульфонат или бензолсульфонат;
(55) (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановую кислоту;
(56) бензолсульфонат (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты;
(57) п-толуолсульфонат (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты;
(58) ангидрат п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты;
(59) ангидрат п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты согласно (58), который находится в кристаллической форме, проявляющей основные пики при межплоскостных расстояниях d, равных 23,9, 11,9, 4,5, 4,3 и 3,6 ангстрем, при порошковой рентгеновской дифракции, полученной посредством излучения Кα меди;
(60) моногидрат п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты;
(61) моногидрат п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты согласно (60), который находится в кристаллической форме, проявляющей основные пики при межплоскостных расстояниях d, равных 22,9, 5,0, 4,9, 4,7 и 4,0 ангстрем, при порошковой рентгеновской дифракции, полученной посредством излучения Кα меди;
(62) фармацевтическое лекарственное средство, содержащее соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль в качестве активного ингредиента;
(63) ингибитор TAFIa, содержащий соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль в качестве активного ингредиента;
(64) промотор фибринолиза, содержащий соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль в качестве активного ингредиента;
(65) профилактическое или терапевтическое лекарственное средство от заболевания, вызванного при ингибировании фибринолиза, содержащее соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль в качестве активного ингредиента;
(66) профилактическое или терапевтическое лекарственное средство против тромбоза или эмболии или их последствий, включающих: острый коронарный синдром, такой как инфаркт миокарда и стенокардия (стабильная стенокардия и нестабильная стенокардия); венозную тромбоэмболию, такую как тромбоз глубоких вен и тромбоэмболию легких; тромбоз или эмболию, происходящих в сердечно-сосудистой системе после хирургической операции, такой как реваскуляризация сосудов, ангиопластика, установка стента и шунтирующая хирургия; тромбоз или эмболию после операции по замене искусственного сустава, такой как операция замены коленного сустава и операция замены тазобедренного сустава; относящееся к воспалению внутрисосудистое заболевание, такое как сепсис и синдром диссеминированной внутрисосудистой коагуляции (DIC); периферическое сосудистое нарушение, являющееся производным от или относящееся к заболеванию, такое как периферическая артериальная окклюзия (РАО), артериосклероз и сахарный диабет; заболевание, относящееся к опухолевому, такому как солидный рак и рак крови; и нарушение органа, приписываемое тромбу или эмболу, такое как тромбоэмболия легких, инфаркт головного мозга и инфаркт почки, содержащее соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль в качестве активного ингредиента;
(67) профилактическое или терапевтическое лекарственное средство против тромбоза или эмболии, включая: заболевание, вызванное контактом с инородным веществом в организме, инородным веществом, включающим медицинское устройство, такое как протез сустава, применяемый при замене сустава, сосудистый катетер, протез в кровотоке, стент в кровотоке и протез клапана; и заболевание, вызванное контактом между кровью и медицинским устройством вне организма, медицинским устройством, включающим насосный оксигенатор, применяемый при операции на сердце, и медицинским устройством, применяемым при гемодиализе, содержащее соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль в качестве активного ингредиента;
(68) профилактическое или терапевтическое лекарственное средство от заболевания, относящегося к тромбозу или эмболии или сопровождающегося отложением фибрина или фиброзом, включающего: заболевание легких, такое как легочная гипертензия, респираторный дистресс-синдром взрослых, фиброз легких и хроническая тромбоэмболическая легочная гипертензия; заболевание почек, такое как гломерулонефрит (включая острый гломерулонефрит, хронический гломерулонефрит, нефротический нефрит и быстро прогрессирующий гломерулонефрит), инфаркт почек и диабетический нефрит; заболевание печени, такое как фиброз печени, гепатит и цирроз печени; глазное заболевание, ассоциированное с отложением фибрина в глазе; дисфункцию органа после трансплантации или резекции органа; нарушение микроциркуляции, вызванное микротромбом, включая тромботическую микроангиопатию; и заболевание или симптомы, ассоциированные с миграцией раковых клеток или метастазами, содержащее соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль в качестве активного ингредиента;
(69) терапевтическое лекарственное средство против инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких, содержащее соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль в качестве активного ингредиента;
(70) фармацевтическую композицию, содержащую соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль и фармакологически приемлемый носитель;
(71) способ лечения инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких, включающий введение фармацевтической композиции, содержащей соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль в качестве активного ингредиента;
(72) соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль для применения при лечении инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких;
(73) фармацевтическое лекарственное средство для инъекции, содержащее соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль в качестве активного ингредиента;
(74) ингибитор TAFIa для инъекции, содержащий соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль в качестве активного ингредиента;
(75) терапевтическое лекарственное средство для инъекции при инфаркте миокарда, стенокардии, остром коронарном синдроме, инфаркте головного мозга, тромбозе глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсисе, синдроме диссеминированной внутрисосудистой коагуляции или фиброзе легких, содержащее соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль в качестве активного ингредиента;
(76) терапевтическое лекарственное средство против заболевания, происходящего вследствие тромбоэмболии, содержащее соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль в качестве активного ингредиента;
(77) фармацевтическую композицию для инъекции, содержащую соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль и фармакологически приемлемый носитель;
(78) способ лечения инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких, включающий введение фармацевтической композиции для инъекции, содержащей соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль в качестве активного ингредиента;
(79) соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль для применения при лечении инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких посредством инъекции;
(80) фармацевтическую композицию, содержащую соединение согласно любому из (1)-(61) или его фармакологически приемлемую соль и одно или два, или более лекарственных средств, выбранных из антикоагулянта, антитромбоцитарного средства, фермента, имеющего отношение к фибринолизу, противоракового лекарственного средства, противовоспалительного средства, антифиброзного лекарственного средства, гипотензивного лекарственного средства, лекарственного средства против легочной гипертензии и иммуносупрессорного лекарственного средства в качестве активных ингредиентов.
Настоящее изобретение также предоставляет в качестве промежуточных соединений при получении циклоалкил-замещенного производного имидазола, имеющего общую формулу (I), или его фармакологически приемлемой соли:
(81) соединение, представленное следующей общей формулой, или его соль:
[Формула 16]
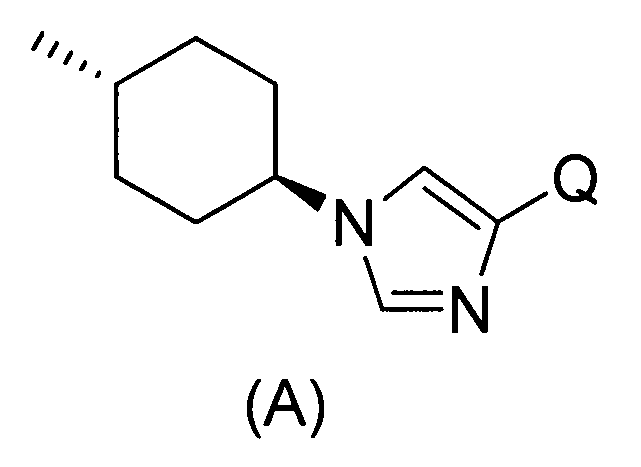
где Q представляет собой группу COOR, гидроксиметильную группу или формильную группу, и R представляет собой С1-С6алкильную группу;
(82) соединение, представленное следующей общей формулой, или его соль:
[Формула 17]
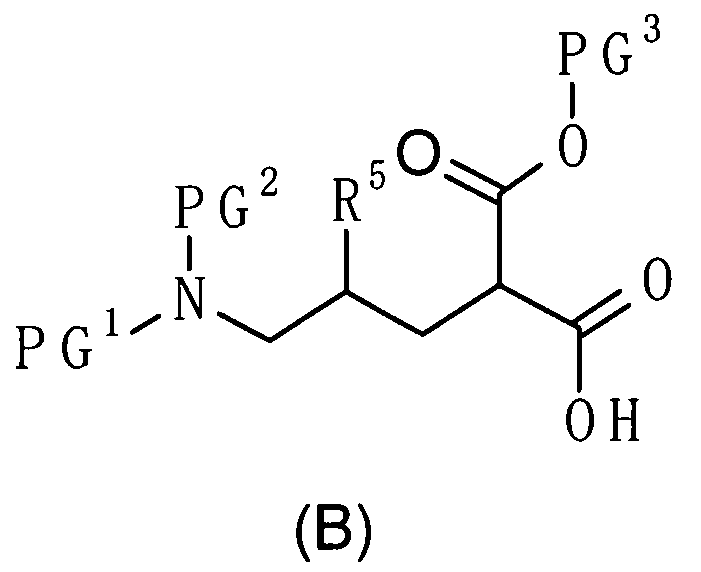
где R5 является таким, как определено выше; PG1 представляет собой защитную группу для аминогруппы; PG2 представляет собой атом водорода или защитную группу для аминогруппы; и PG3 представляет собой защитную группу для карбоксигруппы;
(83) соединение, представленное следующей общей формулой, или его соль:
[Формула 18]
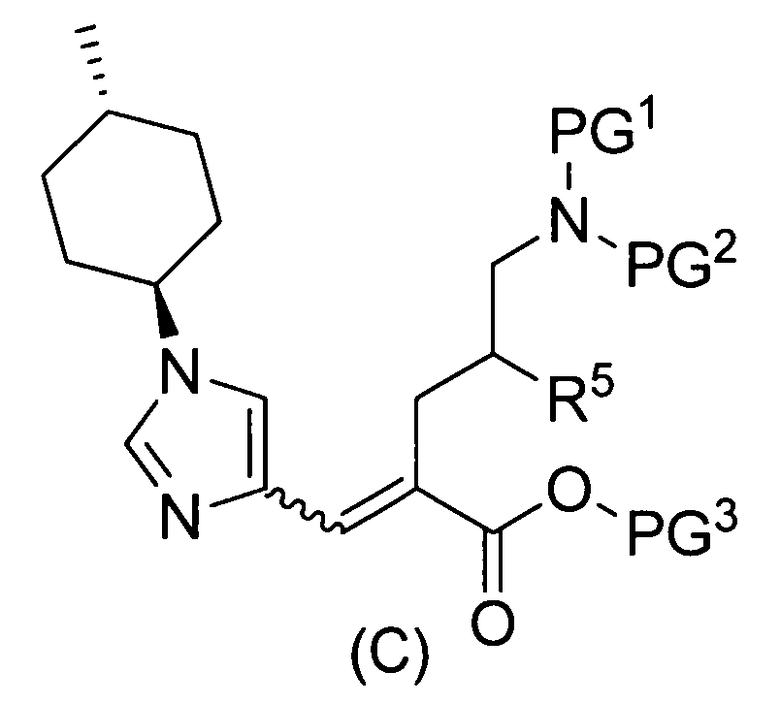
где R5, PG1, PG2 и PG3 являются такими, как определено выше.
Преимущественные эффекты изобретения
Циклоалкил-замещенное производное имидазола настоящего изобретения, представленное общей формулой (I), или его фармакологически приемлемая соль обладают превосходной ингибирующей активностью в отношении TAFIa и проявляют хорошую пероральную всасываемость, концентрацию в плазме и удерживание в крови, и превосходный фармакологический эффект. Более того, соединение общей формулы (I) настоящего изобретения или его фармакологически приемлемая соль являются превосходными с таких позиций, как биораспределение и удерживание в крови, не имеют пролонгирования времени кровотечения, а также являются в высокой степени безопасными.
Следовательно, циклоалкил-замещенное производное имидазола настоящего изобретения, представленное общей формулой (I), или его фармакологически приемлемая соль является применимым в качестве фармацевтического лекарственного средства (конкретно, профилактического или терапевтического лекарственного средства, предпочтительно, терапевтического лекарственного средства против заболевания, вызванного ингибированием фибринолиза), и особенно применимым в качестве профилактического или терапевтического лекарственного средства (предпочтительно, терапевтического лекарственного средства) против тромбоза или эмболии или их последствий, включающих: острый коронарный синдром, такой как инфаркт миокарда и стенокардия (стабильная стенокардия и нестабильная стенокардия); венозную тромбоэмболию, такую как тромбоз глубоких вен и тромбоэмболию легких; тромбоз или эмболию, происходящих в сердечно-сосудистой системе после хирургической операции, такой как реваскуляризация сосудов, ангиопластика, установка стента и шунтирующая хирургия; тромбоз или эмболию после операции по замене искусственного сустава, такой как операция замены коленного сустава и операция замены тазобедренного сустава; относящееся к воспалению внутрисосудистое заболевание, такое как сепсис и синдром диссеминированной внутрисосудистой коагуляции (DIC); периферическое сосудистое нарушение, являющееся производным от или относящееся к заболеванию, такому как периферическая артериальная окклюзия (РАО), артериосклероз и сахарный диабет; заболевание, относящееся к опухолевому, такое как солидный рак и рак крови; и нарушение органа, приписываемое тромбу или эмболу, такое как тромбоэмболия легких, инфаркт головного мозга и инфаркт почки. Более того, соединение настоящего изобретения является применимым в качестве профилактического или терапевтического лекарственного средства (предпочтительно, терапевтического лекарственного средства) против тромбоза или эмболии, включающих: заболевание, вызванное контактом с инородным веществом в организме, например, медицинским устройством, таким как протез сустава, применяемый при замене сустава, сосудистый катетер, протез в кровотоке, стент в кровотоке и протез клапана; и заболевание, вызванное контактом между кровью и медицинским устройством вне организма, например, насосным оксигенатором, применяемым при операциях на сердце, и медицинским устройством, применяемым при гемодиализе. Кроме того, соединение настоящего изобретения является применимым в качестве профилактического или терапевтического лекарственного средства (предпочтительно, терапевтического лекарственного средства) против заболевания, относящегося к тромбозу или эмболии или сопровождающегося отложением фибрина или фиброзом, включающего: заболевание легких, такое как легочная гипертензия, респираторный дистресс-синдром взрослых, фиброз легких и хроническая тромбоэмболическая легочная гипертензия; заболевание почек, такое как гломерулонефрит (включая острый гломерулонефрит, хронический гломерулонефрит, нефротический нефрит и быстро прогрессирующий гломерулонефрит), инфаркт почек и диабетический нефрит; заболевание печени, такое как фиброз печени, гепатит и цирроз печени; глазное заболевание, ассоциированное с отложением фибрина в глазе; дисфункцию органа после трансплантации или резекции органа; нарушение микроциркуляции, вызванное микротромбом, включая тромботическую микроангиопатию; и заболевания или симптомы, ассоциированные с миграцией раковых клеток или метастазами.
Краткое описание фигур
[Фигура 1] На фиг.1 показаны результаты облучения кристаллов типа I ангидрата п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты рентгеновскими лучами Kα Cu 1,54 ангстрем в трансмиссионного типа НТ-совместимом порошковом рентгеновском дифрактометре Bruker, оборудованном двухмерным детектором, D8 DISCOVER с GADDS CST, и данные измерения порошковой рентгеновской дифракции с использованием пленки Mylar. На этой рентгеновской порошковой дифрактограмме ордината представляет интенсивность дифракции, указанную в единицах счета/секунду (cps), и абсцисса представляет углы дифракции, указанные в значениях 2θ. Положение пика находится в интервале 2θ±0,2°.
[Фигура 2] На фиг.2 показаны результаты термического анализа кристаллов типа I ангидрата п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты. При этом термическом анализе (TG/DTA) измерение проводили при скорости нагрева, равной 10°С/мин, в токе азота, составляющем 200 мл/мин сухого азота.
[Фигура 3] На фиг.3 показаны результаты облучения кристаллов типа II моногидрата п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты рентгеновскими лучами Kα Cu 1,54 ангстрем в трансмиссионного типа НТ-совместимом порошковом рентгеновском дифрактометре Bruker, оборудованном двухмерным детектором, D8 DISCOVER с GADDS CST, и данные измерения порошковой рентгеновской дифракции с использованием пленки Mylar. На этой порошковой рентгеновской дифрактограмме ордината представляет интенсивность дифракции, указанную в единицах счета/секунду (cps), и абсцисса представляет углы дифракции, указанные в значениях 2θ. Положение пика находится в интервале 2θ±0,2°.
[Фигура 4] На фиг.4 показаны результаты термического анализа кристаллов типа II моногидрата п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты. При этом термическом анализе (TG/DTA) измерение проводили при скорости нагрева, равной 10°С/мин, в токе азота, составляющем 200 мл/мин сухого азота.
Описание вариантов осуществления
Далее в данном описании будут описаны заместители.
“Группа галогена” означает группу фтора, хлора, брома или йода, т.е. атом фтора, хлора, брома или йода.
“С1-С6алкильная группа” означает линейную или разветвленную насыщенную углеводородную группу, имеющую 1-6 атомов углерода. Ее примеры включают метил, этил, пропил, изопропил, н-бутил, втор-бутил, трет-бутил, изобутил, н-пентил, н-гексил, 1-этилпропил и 2,2-диметилпропильные группы.
“С1-С6алкоксигруппа” означает линейную или разветвленную алкилоксигруппу, имеющую 1-6 атомов углерода. Ее примеры включают метокси, этокси, пропокси, изопропокси и трет-бутоксигруппы.
“(С1-С6алкокси)карбонильная группа” означает группу, состоящую из С1-С6алкоксигруппы и карбонильной группы. Ее примеры включают метоксикарбонильную, этоксикарбонильную и изопропоксикарбонильную группы.
“С1-С6алканоильная группа” означает линейную или разветвленную алканоильную группу, имеющую 1-6 атомов углерода. Ее примеры включают формильную, ацетильную, пропионильную, бутирильную, изобутирильную, валерильную, изовалерильную, пивалоильную и гексаноильную группы.
“С2-С6алканоилоксигруппа” означает группу, состоящую из линейной или разветвленной алканоильной группы, имеющей 2-6 атомов углерода, и оксигруппы. Ее примеры включают ацетилокси, пропионилокси и гексаноилоксигруппы.
“С3-С12циклоалкильная группа” означает насыщенное углеводородное кольцо, имеющее 3-12 атомов углерода, и охватывает: моноциклоалкильные группы, примерами которых являются циклопропильная, циклобутильная, циклопентильная, циклогексильная, циклогептильная и циклооктильная группы, а также полициклоалкильные группы, например, бициклоалкильные и трициклоалкильные группы. Примеры бициклоалкильной группы включают норборнильные группы, например, экзо-2-норборнильную, эндо-2-норборнильную, 3-пинанильную, бицикло[3.1.0]гексильную, бицикло[2.2.1]гептильную и бицикло[2.2.2]окт-2-ильную группы. Примеры трициклоалкильной группы включают адамантильные группы, например, 1-адамантильную и 2-адамантильную группы.
“(С3-С6циклоалкил)карбонилоксигруппа” означает группу, состоящую из насыщенного углеводородного кольца, имеющего 3-6 атомов углерода, и карбонилоксигруппы. Ее примеры включают циклопропилкарбонилокси и циклогексилкарбонилоксигруппы.
“Арильная группа” означает арильную группу, имеющую 6-14 атомов углерода. Ее примеры включают фенильную, нафтильную, антрильную и фенантрильную группы.
“Гетероциклильная группа” означает моноциклическую или бициклическую 3-10-членную насыщенную или ненасыщенную гетероциклическую группу, содержащую 1-3 атома, выбранных из группы, состоящей из атомов азота, кислорода и серы. Ее примеры включают азиридинильную, азетидинильную, пирролидильную, морфолинильную, пирролильную, фурильную, тиенильную, пиразолильную, имидазолильную, оксазолильную, изотиазолильную, пиранильную, пиридильную, пиридазинильную, пиримидинильную, пиразинильную, бензимидазолильную, бензоксазолильную, хинолильную, пирролинильную, имидазолинильную, пиразолинильную, дигидропиридильную и тетрагидропиридильную группы.
“Арилоксигруппа” означает группу, состоящую из арильной группы и оксигруппы. Ее примеры включают фенокси и нафтилоксигруппы.
“Гетероциклилоксигруппа” означает группу, состоящую из гетероциклильной группы и оксигруппы. Ее примеры включают пирролидинил-3-илокси и пиридин-4-илоксигруппы.
“Гетероциклилалкильная группа” означает группу, состоящую из гетероциклильной группы и С1-С6алкильной группы. Ее примеры включают 1,3-диоксол-4-илметильную группу.
“Гетероциклилалкилоксикарбонильная группа” означает группу, состоящую из гетероциклильной группы, С1-С6алкоксигруппы и карбонильной группы. Ее примеры включают 1,3-диоксол-4-илметоксикарбонильную группу.
“Группа пролекарства” означает группу, которая преобразуется посредством реакции с ферментом, желудочной кислотой или т.п. в физиологических условиях in vivo, с получением соединения (I), служащего в качестве активного ингредиента фармацевтической композиции настоящего изобретения, т.е. группу, которая преобразуется, с получением соединения (I) посредством гидролиза или т.п., вызванного желудочной кислотой или т.п. Ее примеры включают фенилаланильную, L-норлейцильную,
[(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонильную,
[1-(изобутирилокси)этокси]карбонильную,
[1-(2,2-диметилпропаноилокси)этокси]карбонильную,
({1-[(циклогексилкарбонил)окси]этокси}карбонильную,
(1-ацетоксиэтокси)карбонильную, бензильную и
[(изопропоксикарбонил)окси]этильную группы.
Группа пролекарства, представленная R4, представляет собой группу пролекарства для карбоксигруппы и, предпочтительно, представляет собой С1-С6алкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы, более предпочтительно, бензильную группу или [(изопропоксикарбонил)окси]этильную группу. Группа пролекарства, представленная R6, R8 или R9, представляет собой группу пролекарства для аминогруппы и, предпочтительно, представляет собой С1-С6алканоильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из аминогруппы, группы галогена, гидроксигруппы, карбоксигруппы, карбамоильной группы, С1-С6алкоксигруппы, арильной группы и гетероциклильной группы; (С1-С6алкокси)карбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С1-С6алкильной группы, С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкилоксикарбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы, более предпочтительно, фенилаланильную группу,
L-норлейцильную группу,
[(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонильную группу,
[1-(изобутирилокси)этокси]карбонильную группу,
[1-(2,2-диметилпропаноилокси)этокси]карбонильную группу,
({1-[(циклогексилкарбонил)окси]этокси}карбонильную группу или
(1-ацетоксиэтокси)карбонильную группу.
Далее в данном описании соединение общей формулы (I) будет описано подробно.
[Формула 19]
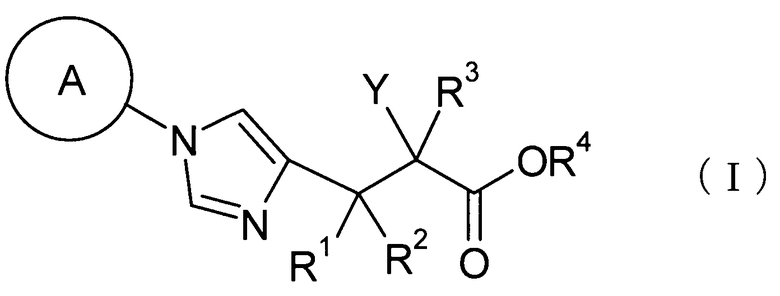
где А представляет собой С3-С12циклоалкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из группы фтора, гидроксигруппы, С1-С6алкильной группы, С1-С6алкоксигруппы, арилоксигруппы и гетероциклилоксигруппы; R1, R2 и R3, каждый независимо представляет собой атом водорода, группу фтора или С1-С6алкильную группу; R4 представляет собой атом водорода или группу пролекарства; и Y представляет собой группу: -СН2-CHR5-CH2-NHR6 (где R5 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу, и R6 представляет собой атом водорода или группу пролекарства), -O-CHR7-CH2-NHR8 (где R7 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу, и R8 представляет собой атом водорода или группу пролекарства) или
[Формулу 20]
(где R9 представляет собой атом водорода или группу пролекарства, и * представляет собой положение для замещения).
А представляет собой С3-С12циклоалкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из группы фтора, гидроксигруппы, С1-С6алкильной группы, С1-С6алкоксигруппы, арилоксигруппы и гетероциклилоксигруппы. А предпочтительно представляет собой циклобутильную группу, циклогексильную группу, циклогептильную группу, бицикло[3.1.0]гексильную группу, бицикло[2.2.1]гептильную группу или адамантильную группу, каждая из которых может быть замещена одной-тремя одинаковыми или различными группами, выбранными из группы фтора, гидроксигруппы, С1-С6алкильной группы, С1-С6алкоксигруппы, арилоксигруппы и гетероциклилоксигруппы; более предпочтительно циклобутильную группу, циклогексильную группу, циклогептильную группу, бицикло[3.1.0]гексильную группу, бицикло[2.2.1]гептильную группу или адамантильную группу, каждая из которых может быть замещена одной-тремя одинаковыми или различными группами, выбранными из гидроксигруппы, метильной группы и этильной группы.
Кроме того, А предпочтительно представляет собой циклогексильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из группы фтора, гидроксигруппы, С1-С6алкильной группы, С1-С6алкоксигруппы, арилоксигруппы и гетероциклилоксигруппы.
Кроме того, А предпочтительно представляет собой С3-С12циклоалкильную группу, замещенную одной или двумя одинаковыми или различными С1-С6алкильными группами, более предпочтительно, С3-С12циклоалкильную группу, замещенную одной С1-С6алкильной группой, даже более предпочтительно, С3-С12циклоалкильную группу, замещенную метильной группой или этильной группой.
Кроме того, А предпочтительно представляет собой циклогексильную группу, замещенную одной или двумя одинаковыми или различными С1-С6алкильными группами, более предпочтительно, циклогексильную группу, замещенную одной С1-С6алкильной группой, даже более предпочтительно, циклогексильную группу, замещенную метильной группой или этильной группой.
Конкретно, А предпочтительно представляет собой группу:
[Формула 21]
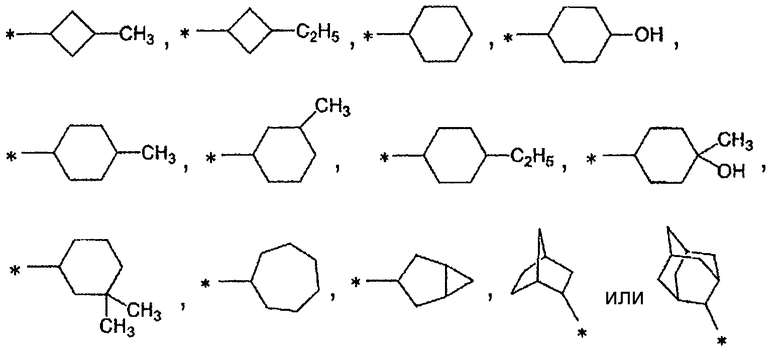 ,
,
более предпочтительно, группу:
[Формула 22]
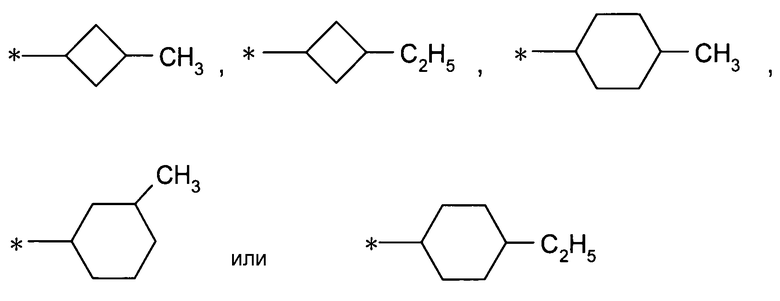 ,
,
даже более предпочтительно, группу:
[Формула 23]
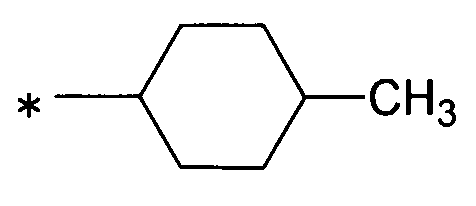 ,
,
особенно предпочтительно, группу:
[Формула 24]
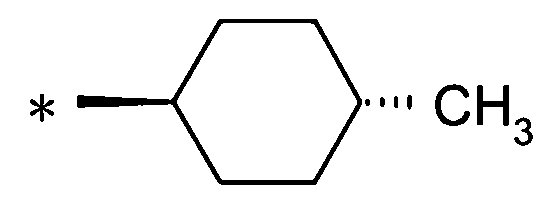 .
.
Y представляет собой группу: -СН2-CHR5-CH2-NHR6 (где R5 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу, и R6 представляет собой атом водорода или группу пролекарства), -O-CHR7-CH2-NHR8 (где R7 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу, и R8 представляет собой атом водорода или группу пролекарства) или
[Формула 25]
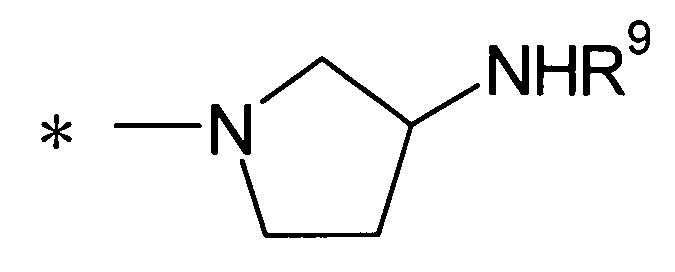
(где R9 представляет собой атом водорода или группу пролекарства, и * представляет собой положение для замещения).
Далее в данном описании случай, где Y представляет собой группу: -СН2-CHR5-CH2-NHR6 (где R5 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу, и R6 представляет собой атом водорода или группу пролекарства), будет описан подробно.
R5 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу, и, предпочтительно, представляет собой атом водорода или метильную группу, более предпочтительно, атом водорода.
R6 представляет собой атом водорода или группу пролекарства. В данном контексте группа пролекарства представляет собой группу пролекарства для аминогруппы и, предпочтительно, представляет собой С1-С6алканоильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из аминогруппы, группы галогена, гидроксигруппы, карбоксигруппы, карбамоильной группы, С1-С6алкоксигруппы, арильной группы и гетероциклильной группы; (С1-С6алкокси)карбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С1-С6алкильной группы, С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкилоксикарбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы, более предпочтительно, фенилаланильную группу, L-норлейцильную группу, [(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонильную группу, [1-(изобутирилокси)этокси]карбонильную группу, [1-(2,2-диметилпропаноилокси)этокси]карбонильную группу, ({1-[(циклогексилкарбонил)окси]этокси}карбонильную группу или (1-ацетоксиэтокси)карбонильную группу.
Y предпочтительно представляет собой группу:
[Формула 26]
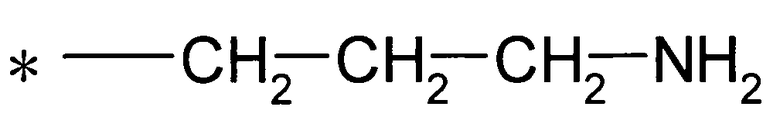
(где * представляет собой положение для замещения).
Далее в данном описании случай, где Y представляет собой группу -O-CHR7-CH2-NHR8 (где R7 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу, и R8 представляет собой атом водорода или группу пролекарства), будет описан подробно.
R7 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу, и, предпочтительно представляет собой атом водорода или метильную группу, более предпочтительно, атом водорода.
R8 представляет собой атом водорода или группу пролекарства. В данном контексте группа пролекарства представляет собой группу пролекарства для аминогруппы и, предпочтительно, представляет собой С1-С6алканоильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из аминогруппы, группы галогена, гидроксигруппы, карбоксигруппы, карбамоильной группы, С1-С6алкоксигруппы, арильной группы и гетероциклильной группы; (С1-С6алкокси)карбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С1-С6алкильной группы, С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкилоксикарбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы, более предпочтительно, фенилаланильную группу, L-норлейцильную группу, [(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонильную группу, [1-(изобутирилокси)этокси]карбонильную группу, [1-(2,2-диметилпропаноилокси)этокси]карбонильную группу, ({1-[(циклогексилкарбонил)окси]этокси}карбонильную группу или (1-ацетоксиэтокси)карбонильную группу. R8 предпочтительно представляет собой атом водорода.
Далее в данном описании будет подробно описан случай, где Y представляет собой группу:
[Формула 27]
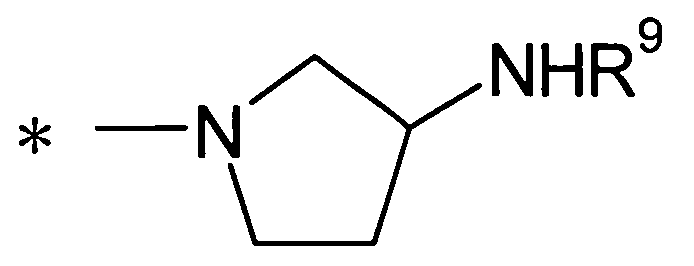
(где R9 представляет собой атом водорода или группу пролекарства, и * представляет собой положение для замещения).
R9 представляет собой атом водорода или группу пролекарства. В данном контексте группа пролекарства представляет собой группу пролекарства для аминогруппы и, предпочтительно, представляет собой С1-С6алканоильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из аминогруппы, группы галогена, гидроксигруппы, карбоксигруппы, карбамоильной группы, С1-С6алкоксигруппы, арильной группы и гетероциклильной группы; (С1-С6алкокси)карбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С1-С6алкильной группы, С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкилоксикарбонильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы, более предпочтительно, фенилаланильную группу, L-норлейцильную группу, [(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонильную группу, [1-(изобутирилокси)этокси]карбонильную группу, [1-(2,2-диметилпропаноилокси)этокси]карбонильную группу, ({1-[(циклогексилкарбонил)окси]этокси}карбонильную группу или (1-ацетоксиэтокси)карбонильную группу. R9 предпочтительно представляет собой атом водорода.
Y предпочтительно представляет собой группу:
[Формула 28]
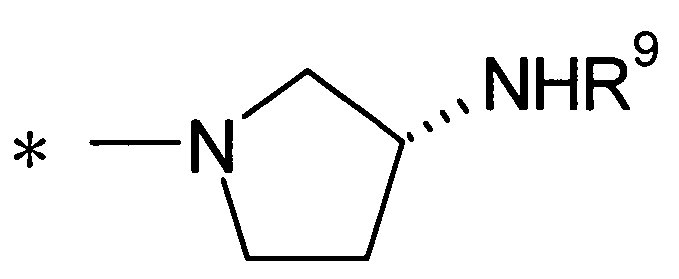
(где R9 представляет собой атом водорода или группу пролекарства, и * представляет собой положение для замещения), более предпочтительно, группу:
[Формула 29]
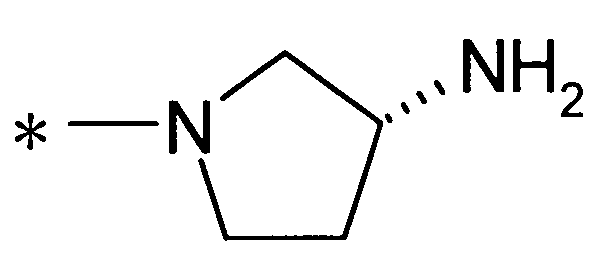
(где * представляет собой положение для замещения)
Y предпочтительно представляет собой группу: -СН2-CHR5-CH2-NHR6 (где R5 представляет собой атом водорода, С1-С6алкильную группу или С1-С6алкоксигруппу, и R6 представляет собой атом водорода или группу пролекарства).
R1, R2 и R3, каждый независимо представляет собой атом водорода, группу фтора или С1-С6алкильную группу. Все из R1, R2 и R3 предпочтительно представляют атом водорода. В данном контексте С1-С6алкильная группа предпочтительно является метильной группой.
R4 представляет собой атом водорода или группу пролекарства. В данном контексте группа пролекарства представляет собой группу пролекарства для карбоксигруппы и, предпочтительно, представляет собой С1-С6алкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из С2-С6алканоилоксигруппы, (С3-С6циклоалкил)карбонилоксигруппы и арильной группы; или гетероциклилалкильную группу, которая может быть замещена одной-тремя одинаковыми или различными группами, выбранными из оксогруппы и С1-С6алкильной группы, более предпочтительно, бензильную группу или [(изопропоксикарбонил)окси]этильную группу. R4 предпочтительно представляет собой атом водорода.
Предпочтительные конкретные примеры соединения, представленного общей формулой (I), включают следующие:
5-амино-2-[(1-циклогексил-1Н-имидазол-4-ил)метил]валериановую кислоту,
5-амино-2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановую кислоту,
5-амино-2-{[(1-(4-этилциклогексил)-1Н-имидазол-4-ил]метил}валериановую кислоту,
5-амино-2-{[(1-(3-этилциклобутил)-1Н-имидазол-4-ил]метил}валериановую кислоту,
5-амино-2-{[(1-(3-метилциклобутил)-1Н-имидазол-4-ил]метил}валериановую кислоту,
5-амино-2-({1-[(1R,3s,5S)-бицикло[3.1.0]гексан-3-ил]-1Н-имидазол-4-ил}метил)валериановую кислоту,
5-амино-2-{[(1-(4-гидроксициклогексил)-1Н-имидазол-4-ил]метил}валериановую кислоту,
5-амино-2-{[(1-(4-гидрокси-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановую кислоту,
5-амино-2-{[(1-(3-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановую кислоту,
5-амино-2-[(1-циклогептил-1Н-имидазол-4-ил)метил]валериановую кислоту,
5-амино-2-({1-[экзо-бицикло[2.2.1]гепт-2-ил]-1Н-имидазол-4-ил}метил]валериановую кислоту,
5-амино-2-({1-[эндо-бицикло[2.2.1]гепт-2-ил]-1Н-имидазол-4-ил}метил]валериановую кислоту,
2-[(1-адамантан-2-ил-1Н-имидазол-4-ил)метил]-5-аминовалериановую кислоту,
5-амино-2-{[1-(транс-4-феноксициклогексил)-1Н-имидазол-4-ил]метил}валериановую кислоту,
бензил-5-амино-2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерат,
2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-(L-фенилаланиламино)валериановую кислоту,
2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-(L-норлейциламино)валериановую кислоту,
2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-({[(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонил}амино)валериановую кислоту,
5-({[1-(изобутирилокси)этокси]карбонил}амино)-2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановую кислоту,
1-[(изопропоксикарбонил)окси]этил-5-({[1-(изобутирилокси)этокси]карбонил}амино)-2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерат,
5-({[1-(2,2-диметилпропаноилокси)этокси]карбонил}амино)-2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановую кислоту,
5-[({1-[(циклогексилкарбонил)окси]этокси}карбонил)амино]-2-{[1-(4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановую кислоту,
2-(2-аминоэтокси)-3-[1-(4-метилциклогексил)-1Н-имидазол-4-ил]пропионовую кислоту,
2-[(1R)-2-амино-1-метилэтокси]-3-[1-(4-метилциклогексил)-1Н-имидазол-4-ил]пропионовую кислоту и
2-[(3S)-3-аминопирролидинил-1-ил]-3-[1-(4-метилциклогексил)-1Н-имидазол-4-ил]пропионовую кислоту.
Далее в данном описании будут описаны конкретные способы получения соединения настоящего изобретения. Однако настоящее изобретение не ограничено этими способами каким-либо образом.
[Способ получения 1]
Соединение, представленное общей формулой (I), или его соль или его сольват могут быть получены, например, следующим способом получения:
[Формула 30]
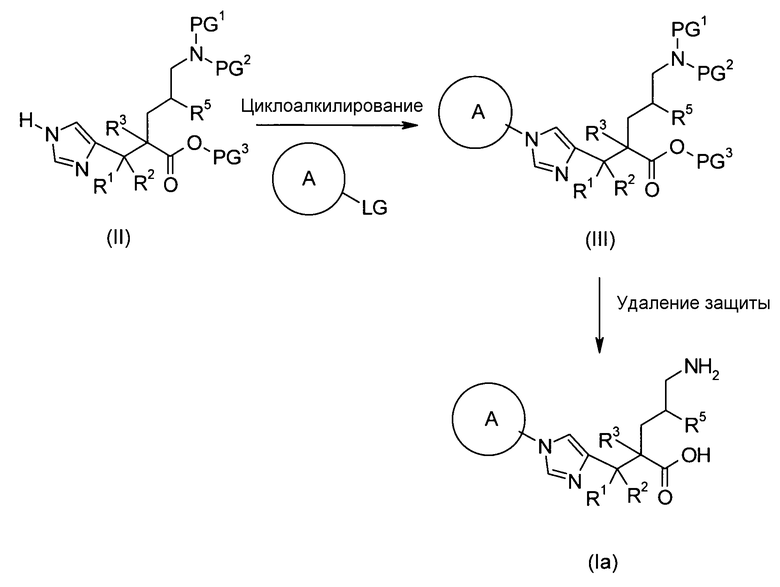
где А, R1, R2, R3 и R5 являются такими, как определено выше; PG1 представляет собой защитную группу для аминогруппы; PG2 представляет собой атом водорода или защитную группу для аминогруппы; PG3 представляет собой защитную группу для карбоксигруппы; и A-LG представляет собой алкилирующий агент или спирт, описанный ниже.
В соответствии с настоящим способом получения атом азота в имидазольном фрагменте соединения (II) циклоалкилируют, с получением соединения (III), и защитные группы в соединении (I) могут быть затем удалены, с получением соединения (Ia).
Реакция циклоалкилирования представляет собой, например, реакцию, посредством которой соединение (III) образуется из соединения (II) и алкилирующего агента A-LG (LG представляет уходящую группу) в присутствии основания. Ациклические, циклические или ароматические углеводороды или полярный ароматический растворитель, например, тетрагидрофуран, N,N-диметилформамид или диэтоксиэтан, или смешанный растворитель из них могут быть использованы в качестве реакционного растворителя. Например, карбонат цезия или гидрид натрия могут быть использованы в качестве основания. Алкилгалогенид (например, A-I или A-Br) или сложный эфир сульфоновой кислоты и спирта (например, A-SO2CH3 или A-SO2CF3) могут быть использованы в качестве алкилирующего агента.
Еще один другой способ реакции циклоалкилирования представляет собой способ, посредством которого соединение (II) и спирт А-LG (LG представляет гидроксигруппу) конденсируют по реакции Мицунобу с образованием соединения (III). Способ с использованием диэтилазодикарбоксилата (DEAD) и трифенилфосфина (Synthesis, 1981, p. 1) является общеизвестным как реакция Мицунобу. В этом случае способ с использованием (цианометилен)трибутилфосфорана (CMBP) или (цианометилен)триметилфосфорана (CMPP) является предпочтительным. Получение может достигаться со ссылкой на следующие документы: 1) Tetrahedron Lett., 1995, Vol. 36, p. 2529; и 2) Tetrahedron Lett., 1996, Vol. 37, p. 2463.
Любая защитная группа, обычно используемая в качестве защитной группы для аминогрупп при синтезе органического соединения, особенно, пептидном синтезе, может использоваться в качестве защитной группы для аминогруппы. Ее конкретные примеры могут включать: алкоксикарбонильные группы, такие как трет-бутоксикарбонильная, метоксикарбонильная и этоксикарбонильная группы; арилметоксикарбонильные группы, такие как бензилоксикарбонильная, пара-метоксибензилоксикарбонильная и пара(или орто)нитробензилоксикарбонильная группы; арилметильные группы, такие как бензильная, 4-метоксибензильная и трифенилметильная группы; алканоильные группы, такие как формильная и ацетильная группы; ароильные группы, такие как бензоильная группа; и арилсульфонильные группы, такие как 2,4-динитробензолсульфонильная и орто-нитробензолсульфонильная группы. Эти защитные группы для аминогруппы могут быть выбраны в соответствии, например, со свойствами соединения, чья аминогруппа подлежит защите. Для удаления защитных групп реагенты или условия могут быть выбраны в соответствии с каждой защитной группой.
Примеры защитной группы для карбоксигруппы включают алкильную, арильную и арилалкильную сложноэфирные группы. Эти защитные группы для карбоксигруппы могут быть выбраны в соответствии, например, со свойствами соединения, чья карбоксигруппа подлежит защите. Для удаления защитных групп реагенты или условия могут быть выбраны в соответствии с каждой защитной группой.
Примеры ссылок на защиту/удаление защиты амино и карбоксигрупп могут включать Greene, T.W., Wuts, P.G.M., Protective Groups in Organic Synthesis (1999), 3rd Ed., Wiley-Interscience.
Соединение (II) может быть получено посредством хорошо известных реакций с использованием коммерчески доступного или известного вещества. Получение может быть достигнуто со ссылкой, например, на J. Med. Chem., 2007, Vol. 50, p. 6095.
[Способ получения 2]
Соединение (I) настоящего изобретения также может быть получено следующим способом:
[Формула 31]
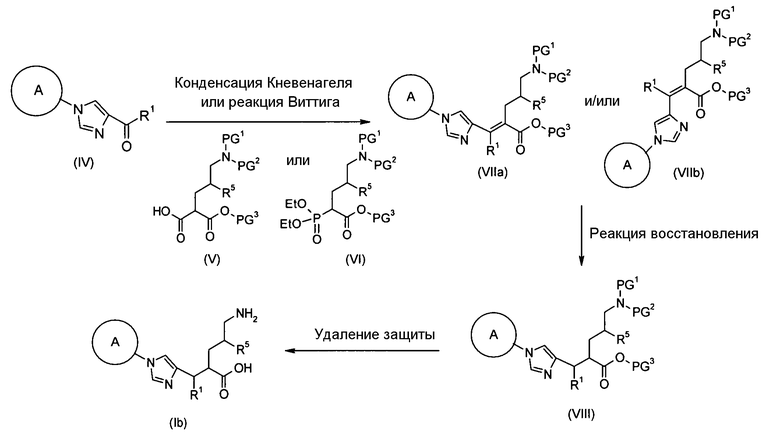
где А, R1, R5, PG1, PG2, PG5 являются такими, как определено выше.
Соединения (VIIa) и/или (VIIb) могут быть синтезированы посредством конденсации Кневенагеля или реакции Виттига с соединением (IV) в качестве исходного вещества. Олефин полученных соединений (VIIa) и/или (VIIb) восстанавливают, чтобы синтезировать соединение (VIII), и защитные группы в соединении (VIII) могут быть удалены, с получением соединения (Ib).
Конденсация Кневенагеля представляет собой, в данном случае, реакцию, посредством которой соединение (V), имеющее активный метилен, и соединение (IV), имеющее карбонильную группу, конденсируют в присутствии аминного катализатора с образованием соединений (VIIa) и/или (VIIb), которые представляют собой α,β-ненасыщенные сложные эфиры. Декарбоксилирование происходит при нагревании до комнатной температуры или 100°С с образованием ненасыщенной карбоновой кислоты. Пиперидин, как правило, используют в качестве катализатора. Получение может быть достигнуто со ссылкой на следующие документы: 1) Org. React. 1967, Vol. 15, p. 204; 2) Comprehensive Organic Synthesis, 1991, Vol. 2, p. 341; и 3) WO 2008/78330.
Реакция Виттига представляет собой, в данном случае, реакцию, посредством которой соединение (VI), имеющее фосфорильную группу, и соединение (IV), имеющее карбонильную группу, подвергают взаимодействию в присутствии основания с образованием соединений (VIIa) и/или (VIIb), которые представляют собой α,β-ненасыщенные сложные эфиры. Гидрид натрия, метоксид натрия, карбонат калия или т.п. могут использоваться в качестве основания. Альтернативно, основание, такое как 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), или триэтиламин могут использоваться в сочетании с хлоридом лития. Спирты, тетрагидрофуран, 1,2-диметоксиэтан, диметилсульфоксид, ацетонитрил или т.п. могут использоваться в качестве растворителя. Температура реакции может быть выбрана как температура, подходящая для субстратов, и реакция может быть осуществлена при от -78°С до условий кипячения с обратным холодильником.
Реакция восстановления представляет собой, в данном случае, реакцию, посредством которой соединения (VIIa) и/или (VIIb) гидрируют до соединения (VIII) с использованием гетерогенного катализатора. Например, вода, метанол, этанол, этилацетат или уксусная кислота могут использоваться в качестве растворителя. Палладий-углерод (Pd/C), катализатор Пирлмана (Pd(OH)2), никель Рэнея, катализатор Адамса (PtO2) или т.п. могут использоваться в качестве катализатора.
Защитные группы и их удаление являются такими, как описано в способе получения 1.
[Способ получения 3]
Соединение (I) настоящего изобретения также может быть получено следующим способом:
[Формула 32]
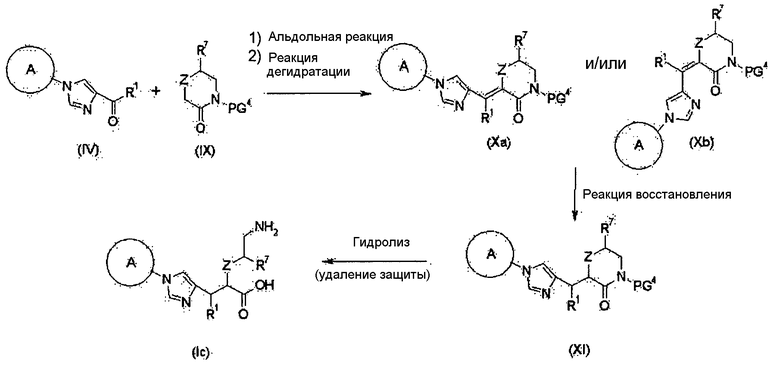
где А, R1 и R7 являются такими, как определено выше; PG4 представляет собой атом водорода или защитную группу для амидной группы; и Z представляет собой атом кислорода или метиленовую группу.
Соединение (IV) и соединение (IX) могут быть подвергнуты альдольной реакции и реакции дегидратации для получения соединений (Xa) и/или (Xb). Олефин полученных соединений (Xa) и/или (Xb) восстанавливают, чтобы синтезировать соединение (XI), которое затем может быть гидролизовано, с получением соединения (Ic).
Примеры защитной группы для амидной группы в соединении (IX) включают аллильную, трет-бутильную, пара-метоксибензильную, бензилоксиметильную, метоксиметильную и трет-бутоксикарбонильную группы. Примеры ссылок на защиту/удаление для этих защитных групп могут включать Greene, T.W., Wuts, P.G.M., Protective Groups in Organic Synthesis (1999), 3rd Ed., Wiley-Interscience.
Альдольная реакция представляет собой, в данном случае, реакцию, посредством которой соединение (IX) в качестве СН-активного соединения и соединение (IV), имеющее карбонильную группу, связываются друг с другом в присутствии сильного основания, с получением β-гидроксикарбонильного соединения. Например, карбонат щелочного металла или щелочноземельного металла (например, карбонат натрия или карбонат калия), алкоксид щелочного металла (например, этоксид натрия или бутоксид калия), гидроксид щелочного металла (например, гидроксид натрия или гидроксид калия), гидрид щелочного металла (например, гидрид натрия или гидрид калия) или органическое основание металла, такое как алкиллитий (например, н-бутиллитий), диалкиламинолитий (например, диизопропиламид лития) или биссилиламин (например, гексаметилдисилазид лития) могут использоваться в качестве сильного основания. Ациклические, циклические или ароматические углеводороды, спирты или полярный апротонный растворитель, например, тетрагидрофуран, N,N-диметилформамид или диэтоксиэтан или их смешанный растворитель могут использоваться в качестве реакционного растворителя. Температура реакции может составлять приблизительно от -78°С до комнатной температуры.
Реакция дегидратации представляет собой реакцию, посредством которой гидроксигруппу в β-гидроксикарбонильном соединении, полученном посредством альдольной реакции, обрабатывают метансульфонилхлоридом или бензолсульфонилхлоридом, или т.п. при от -78°С до 50°С в присутствии триэтиламина в инертном растворителе и затем дополнительно обрабатывают основанием с образованием соединения (X). Примеры инертного растворителя включают: алкилгалогенидные растворители, такие как дихлорметан, хлороформ и тетрахлорид углерода; эфирные растворители, такие как тетрагидрофуран, 1,2-диметоксиэтан и диоксан; ароматические растворители, такие как бензол и толуол; и амидные растворители, такие как N,N-диметилформамид, N,N-диметиласетамид и N-метилпирролидин-2-он. Кроме того, в некоторых случаях могут использоваться сульфоксидные растворители, такие как диметилсульфоксид и сульфолан, кетоновые растворители, такие как ацетон и метилэтилкетон или ацетонитрил или т.п. Основание предпочтительно представляет собой органическое основание, такое как пиридин, 2,6-лутидин, коллидин, 4-диметиламинопиридин, триэтиламин, N-метилморфолин, диизопропилэтиламин или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). Реакция дегидратации, в некоторых случаях, может протекать в условиях альдольной реакции.
Реакция восстановления может быть осуществлена в соответствии со способом, описанным в способе получения 2.
Гидролиз представляет собой реакцию, посредством которой лактамовое кольцо соединения (XI) гидролизуют кислотой, с получением соединения (Ic). Примеры конкретных условий реакции включают нагревание при кипячении с обратным холодильником с использованием концентрированной хлористоводородной кислоты. См. следующую ссылку: J. Org. Chem., 1996, Vol. 61, p. 4990.
Когда PG4 представляет собой защитную группу для амидной группы, которая может быть удалена в кислотных условиях, реакция удаления защиты также может быть достигнута в условиях, показанных выше. Когда она представляет собой защитную группу, которая не может быть удалена в кислотных условиях, реагенты или условия могут быть выбраны в соответствии с защитной группой. Примеры ссылок на них могут включать Greene, T.W., Wuts, P.G.M., Protective Groups in Organic Synthesis (1999), 3rd Ed., Wiley-Interscience.
Соединение (IX) может быть получено посредством хорошо известных реакций с использованием коммерчески доступного или известного вещества. Получение может быть достигнуто со ссылкой на, например, Org. Lett, 2009, Vol. 11, p. 5410.
[Способ получения 4]
Соединение (I) настоящего изобретения также может быть получено следующим способом:
[Формула 33]
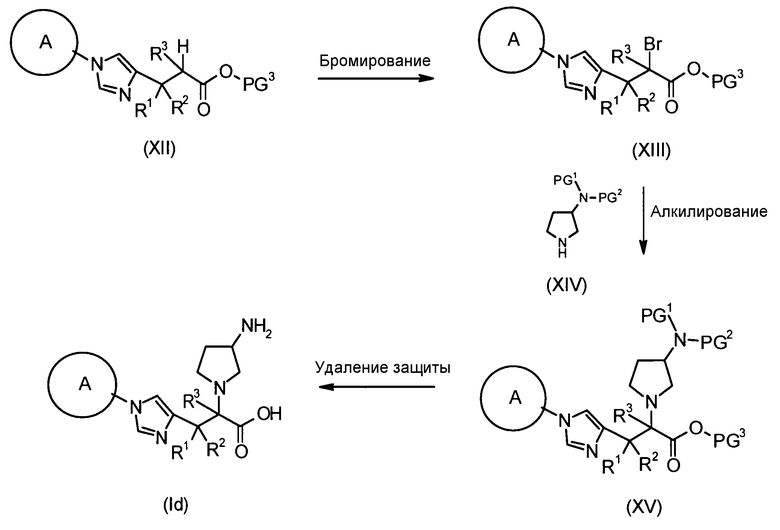
где А, R1, R2, R3, PG1, PG2 и PG3 являются такими, как определено выше.
Соединение (XII) бромируют, чтобы синтезировать соединение (XIII), и соединение (XIV) может быть алкилировано соединением (XIII) в качестве алкилирующего агента, чтобы синтезировать соединение (XV). Защитные группы в полученном соединении (XV) могут быть удалены, с получением соединения (Id).
Реакция бромирования представляет собой реакцию, посредством которой α-положение карбонильной группы в соединении (XII) селективно бромируют, с получением соединения (XIII). Для этой цели соединение (XII) может быть временно преобразовано в силиленоловый эфир и затем обработано бромом или N-бромсукцинимидом (NBS), с получением представляющего интерес соединения. Получение может быть достигнуто со ссылкой на следующий документ: Tetrahedron Asymmetry, 1995, Vol. 6, p. 2291.
Реакция алкилирования представляет собой реакцию, посредством которой соединение (XV) образуется из соединения (XIV) и соединения (XIII) в качестве алкилирующего агента, например, в присутствии основания. Ациклические, циклические или ароматические углеводороды или полярный апротонный растворитель, например, тетрагидрофуран, N,N-диметилформамид или диэтоксиэтан, или их смешанный растворитель, могут быть использованы в качестве реакционного растворителя. Например, органическое основание, такое как пиридин, 2,6-лутидин, коллидин, 4-диметиламинопиридин, триэтиламин, N-метилморфолин, диизопропилэтиламин или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU) могут быть использованы в качестве основания.
Защитные группы и их удаление являются такими, как описано в способе получения 1.
Соединение (XIV) может быть получено посредством хорошо известных реакций с использованием коммерчески доступного или известного вещества.
[Способ получения 5]
Соединение (IV), промежуточное соединение настоящего изобретения, может быть получено, например, следующим способом:
[Формула 34]
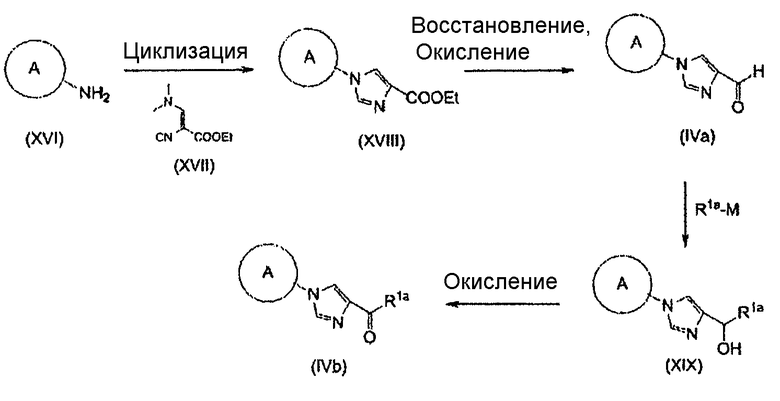
где А является таким, как определено выше; R1a представляет С1-С6алкильную группу; и М представляет Li или MgBr или т.п.
Соединение (XVI), которое является коммерчески доступным или которое синтезируют с использованием хорошо известного способа, может быть подвергнуто взаимодействию с соединением (XVII) (Liebigs Annalen der Chemie, 1979, p. 1444) для построения имидазолового кольца, чтобы синтезировать соединение (XVIII). Получение может быть достигнуто со ссылкой на следующий документ: Org. Lett. 2002, Vol. 4, p. 4133.
Полученное соединение (XVIII) восстанавливают в первичный спирт путем восстановления с использованием гидрида металла в инертном растворителе, и первичный спирт затем может быть окислен в альдегид, с получением соединения (IVa). Примеры гидрида металла включают алюмогидрид лития, боргидрид лития, бис(2-метоксиэтокси)алюмогидрид натрия и боргидрид натрия. Способ окисления, известный в данной области техники, т.е. PCC окисление, PDC окисление, окисление по Сверну, TPAP окисление, окисление по Дессу-Мартину, TEMPO окисление, окисление по Мукаяма или т.п. могут использоваться в качестве способа окисления. Среди них TEMPO окисление является предпочтительным. Получение может быть достигнуто со ссылкой на следующий документ: Org. Lett. 2003, Vol. 5, p. 285.
Альтернативно, соединение (XVIII) также может быть преобразовано непосредственно в соединение (IVa) путем осуществления реакции при низкой температуре с использованием соответствующего гидрида металла. В данном случае, примеры гидрида металла включают диизобутилалюминийгидрид.
Полученное соединение (IVa) может быть обработано литийорганическим или магнийорганическим соединением R1a-M, с получением соединения (XIX). Примеры литийорганического или магнийорганического соединения могут включать: алкиллитий, такой как метиллитий, этиллитий, нормальный пропиллитий, нормальный бутиллитий, изобутиллитий, втор-бутиллитий, трет-бутиллитий, нормальный пентиллитий, изопентиллитий и неопентиллитий; и алкилмагний, такой как метилмагнийбромид, этилмагнийбромид, пропилмагнийбромид, изопропилмагнийбромид, нормальный бутилмагнийбромид, изобутилмагнийбромид, втор-бутилмагнийбромид, трет-бутилмагнийбромид и метилмагниййодид. Ароматические углеводороды (например, толуол или бензол), линейные или циклические алифатические углеводороды (например, пропан, бутан, пентан, гексан, гептан или циклогексан) или эфирный растворитель (например, диэтиловый эфир или тетрагидрофуран) или т.п. могут быть использованы в качестве реакционного растворителя. Температура реакции составляет предпочтительно от -78°С до комнатной температуры. Из полученного соединения (XIX) соединение (IVb) может быть получено посредством способа окисления, известного в данной области. PCC окисление, PDC окисление, окисление по Сверну, TPAP окисление или т.п. могут использоваться в качестве способа окисления. Например, карбонил может быть синтезирован из спирта путем реакции окисления, основанной на TPAP окисление со ссылкой на Synthesis, 1994, p. 639.
[Способ получения 6]
Из соединений (I) настоящего изобретения соединение, содержащее группу пролекарства, введенную в него, может быть получено следующим способом:
[Формула 35]
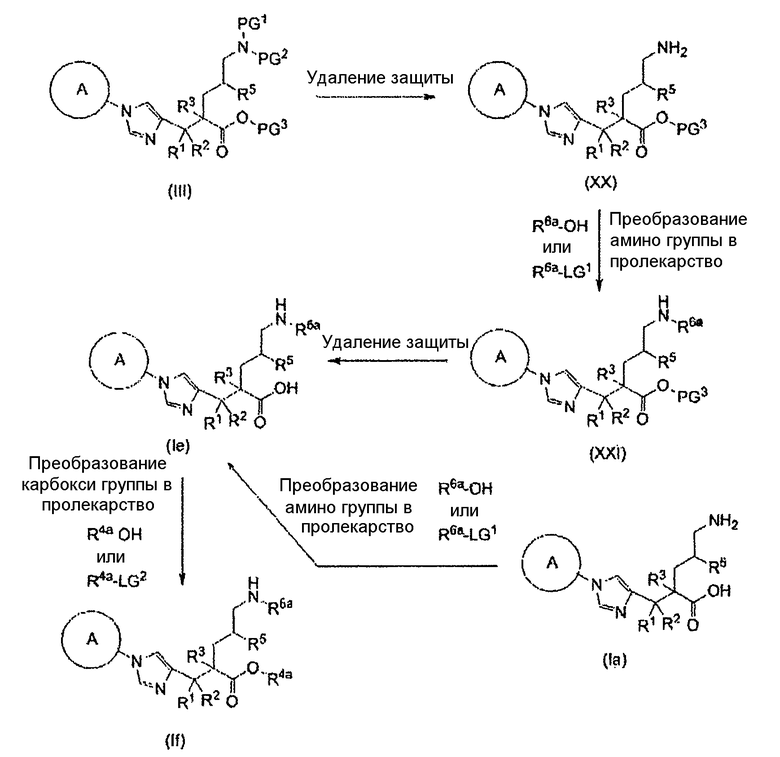
где А, R1, R2, R3, R5, PG1, PG2 и PG3 являются такими, как определено выше; R4a и R6a представляют собой группы пролекарства; и LG1 и LG2 представляют собой уходящие группы.
Защитная группа для аминогруппы в соединении (III), полученном способом получения 1, может быть удалена, с получением соединения (ХХ). Аминогруппу соединения (ХХ) преобразовывают в пролекарство, чтобы синтезировать соединение (XXI), и защитная группа для карбоксигруппы в соединении (XXI) может быть удалена, с получением соединения (Ie) в пролекарственной форме.
Кроме того, соединение (Ie) может быть получено непосредственно преобразованием соединения (Ia) в пролекарство.
Карбоксигруппа полученного соединения (Ie) может быть затем преобразована в пролекарство, с получением соединения (If).
Для защитных групп и их удаления могут быть выбраны защитные группы, как описано в способе получения 1, и реагенты или условия, подходящие для каждой защитной группы, могут быть выбраны для отщепления (удаления) защитных групп.
Преобразование аминогруппы в пролекарство представляет собой реакцию, посредством которой соединение (XXI) получают реакцией конденсации соединения (ХХ) и соединения R6a-OH. Может быть применена любая реакция конденсации, используемая в обычном пептидном синтезе. Примеры конденсирующего агента включают N,N'-дициклогексилкарбодиимид (DCC), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC-HCl), гидрат 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолинийхлорида (DMT-MM), гексафторфосфат (1H-бензотриазол-1-илокси)трис(диметиламино)фосфония (ВОР) и гексафторфосфат 1-[бис(диметиламино)метилен]-1Н-бензотриазолий-3-оксида (HBTU). Получение может быть достигнуто со ссылкой, например, на Tetrahedron, 2004, Vol. 60, p. 2447.
В еще одном другом способе преобразования аминогруппы в пролекарство соединение (ХХ) и активное сложноэфирное соединение R6a-LG1 могут быть конденсированы, с получением соединения (XXI). Примеры LG1 включают п-нитрофенилокси, пентафторфенилоксигруппы и группу хлора. Может быть использован способ реакции конденсации амина и активного сложного эфира, используемый в обычном пептидном синтезе.
Соединение (Ia) также может быть конденсировано с R6a-OH или R6a-LG1 таким же образом, как в приведенном выше способе непосредственного получения соединения (Ie).
Преобразование карбоксигруппы в пролекарство представляет собой реакцию, посредством которой конденсируют соединение (Ie) и спиртовое соединение R4-OH, с получением соединения (If). N,N'-дициклогексилкарбодиимид (DCC), N,N'-диизопропилкарбодиимид (DIC) или т.п. могут быть использованы в качестве конденсирующего агента. Реакционная способность улучшается путем предварительного добавления каталитического количества 4-диметиламинопиридина (DMAP) в систему.
В еще одном другом способе преобразования карбоксигруппы в пролекарство соединение (Ie) и соединение R4a-LG2, которое представляет собой алкилирующий агент, могут взаимодействовать в основных условиях, с получением соединения (If). В этом случае примеры LG2 включают группы йода и брома. Альтернативно, сложный эфир сульфоновой кислоты и спирта (например, R4a-OSO2CH3 или R4a-OSO2CF3) могут быть использованы в качестве R4a-LG2. Вода, тетрагидрофуран, N,N-диметилформамид или диэтоксиэтан, или т.п., или их смешанный растворитель могут быть использованы в качестве реакционного растворителя. Например, карбонат щелочного металла или щелочноземельного металла, такой как карбонат натрия, бикарбонат натрия, карбонат калия или бикарбонат калия, может быть использован в качестве основания.
[Способ получения 7]
Из соединений (I) настоящего изобретения соединение, содержащее группу пролекарства, введенную в него, может быть получено следующим способом:
[Формула 36]
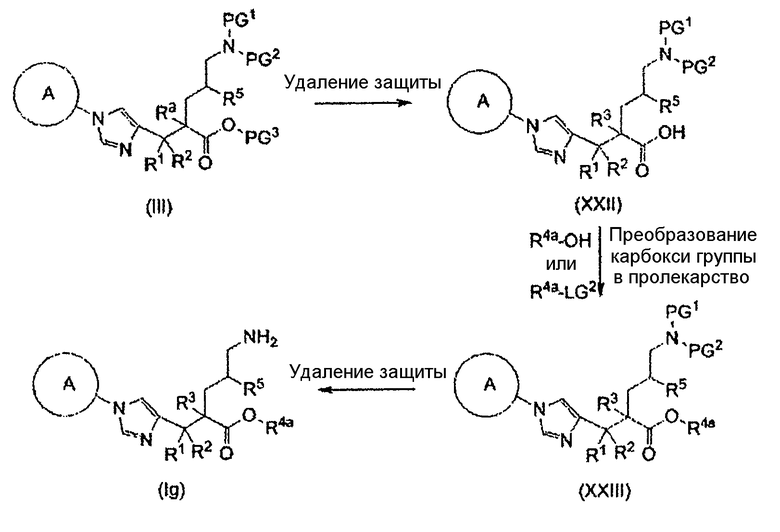
где А, R1, R2, R3, R4a, R5, PG1, PG2, PG3 и LG2 являются такими, как определено выше.
Защитная группа для карбоксигруппы в соединении (III), полученном в способе получения 1, может быть удалена, с получением соединения (XXII). Последовательно группу пролекарства вводят в карбоксигруппу соединения (XXII), и защитная группа его аминогруппы может быть удалена, с получением соединения (Ig) в пролекарственной форме.
Для защитных групп и их удаления могут быть выбраны защитные группы, как описано в способе получения 1, и реагенты или условия, подходящие для каждой защитной группы, могут быть выбраны для отщепления (удаления) защитных групп.
Преобразование карбоксигруппы в пролекарство может осуществляться со ссылкой на способ, описанный в способе получения 6.
[Способ получения 8]
Из соединений (I) настоящего изобретения соединение, содержащее группу пролекарства, введенную в него, может быть получено следующим способом:
[Формула 37]
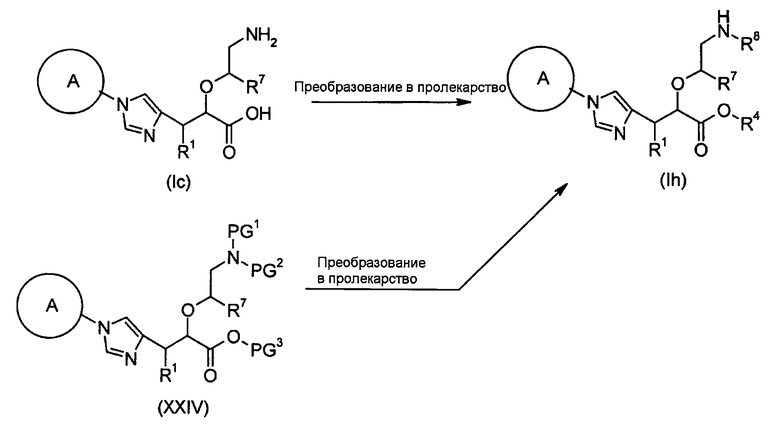
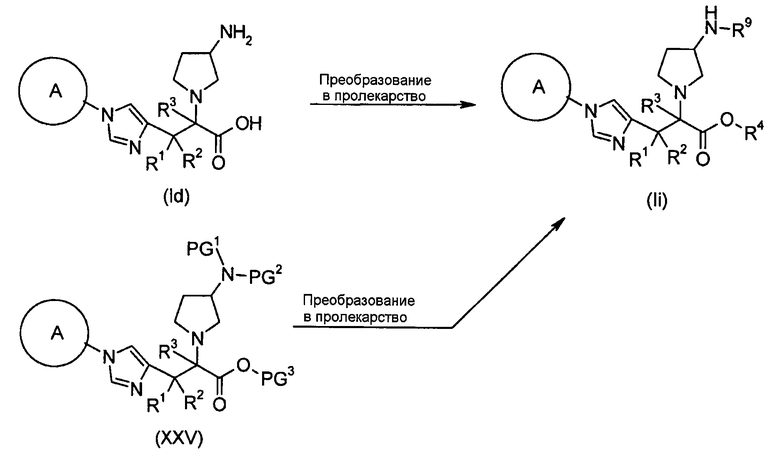
где А, R1, R2, R3, R4, R7, R8, R9, PG1, PG2 и PG3 являются такими, как определено выше, при условии, что R4 и R8 не являются атомом водорода одновременно.
Соединение (Ih) или (Ii) в пролекарственной форме могут быть получены из соединений (Ic), (XXIV), (Id) и (XXV) таким же образом, как в способе получения 6 или 7.
Соединение (XXIV) и соединение (XXV) могут быть получены введением защитной группы в синтетические промежуточные соединения или конечные продукты, продемонстрированные примером в способах получения 3 и 4.
[Способ получения 9]
[Формула 38]
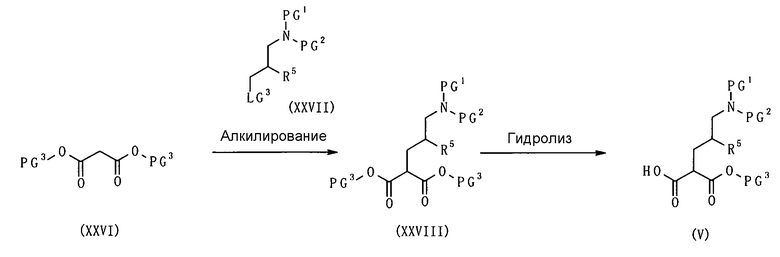
где R5, PG1, PG2 и PG3 являются такими, как определено выше, и LG3 представляет собой уходящую группу.
Реакция алкилирования представляет собой реакцию, посредством которой сложный диэфир малоновой кислоты (XXVI) алкилируют в присутствии основания с использованием соединения (XXVII), которое является коммерчески доступным или может быть получено по хорошо известным реакциям. Например, гидроксид щелочного металла, гидрид щелочного металла, карбонат щелочного металла или щелочноземельного металла или алкоксид щелочного металла (например, карбонат натрия, карбонат калия, этоксид натрия, бутоксид калия, гидроксид натрия, гидроксид калия, гидрид натрия или гидрид калия) или органическое основание металла, такое как алкиллитий (например, н-бутиллитий), диалкиламинолитий (например, диизопропиламид лития) или основание щелочного металла из биссилиламина (например, гексаметилдисилазид лития) могут быть использованы в качестве основания. Кроме того, примеры LG3 могут включать: атомы галогена, такие как хлор, бром и йод; и алкилсульфонилокси или арилсульфонилоксигруппы, такие как мезилат, тозилат и трифлат.
Гидролиз представляет собой реакцию, посредством которой соединение (XXVIII) гидролизуют в присутствии основания, с получением соединения (V). Примеры основания могут включать гидроксид щелочного металла, такой как гидроксид лития, гидроксид натрия и гидроксид калия. Протонный растворитель (например, метанол, этанол или вода), растворитель из апротонного эфира (например, тетрагидрофуран, диоксан или 1,2-диметоксиэтан) или смешанный растворитель из двух или более этих растворителей, комбинируемых при любом соотношении, могут быть использованы в качестве реакционного растворителя.
Соединение (VI) может быть получено посредством хорошо известных реакций с использованием коммерчески доступного или известного вещества. Получение может быть достигнуто со ссылкой, например, на J. Med. Chem., 2007, Vol. 50, p. 6095.
[Способ получения 10]
[Формула 39]
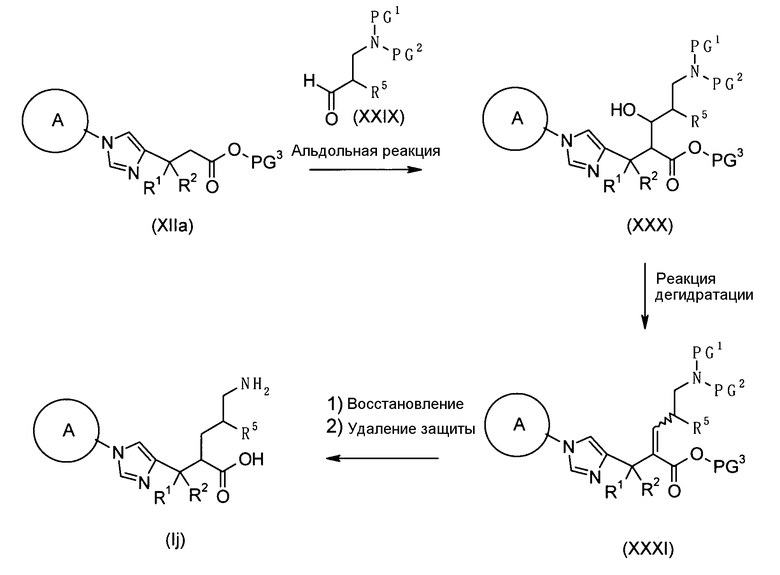
где А, R1, R2, R5, PG1, PG2 и PG3 являются такими, как определено выше.
Соединение (XIIa) и соединение (XXIX) подвергают альдольной реакции, с получением соединения (XXX), и полученное соединение (XXX) может быть подвергнуто реакции дегидратации, с получением соединения (XXXI). Последовательно, олефиновый фрагмент восстанавливают, и защитная группа может быть удалена, с получением соединения (Ij).
Альдольная реакция представляет собой, в данном случае, реакцию, посредством которой соединение (XIIa) в качестве СН-активного соединения и соединение (XXIX), содержащее карбонильную группу, связывают друг с другом в присутствии сильного основания, с получением соединения (XXX). Например, карбонат щелочного металла или щелочноземельного металла (например, карбонат натрия или карбонат калия), алкоксид щелочного металла (например, этоксид натрия или бутоксид калия), гидроксид щелочного металла (например, гидроксид натрия или гидроксид калия), гидрид щелочного металла (например, гидрид натрия или гидрид калия) или органическое основание металла, такое как алкиллитий (например, н-бутиллитий), диалкиламинолитий (например, диизопропиламид лития) или биссилиламин (например, гексаметилдисилазид лития) могут быть использованы в качестве сильного основания. Ациклические, циклические или ароматические углеводороды, спирты или полярный апротонный растворитель, например, тетрагидрофуран, N,N-диметилформамид или диэтоксиэтан или их смешанный растворитель, могут быть использованы в качестве реакционного растворителя. Температура реакции может составлять приблизительно от -78°С до комнатной температуры.
Реакция дегидратации представляет собой реакцию, посредством которой гидроксигруппу в соединении (XXX) преобразовывают в сложный эфир сульфоновой кислоты путем обработки метансульфонилхлоридом или бензолсульфонилхлоридом, или т.п. при от -78°С до 50°С в присутствии триэтиламина в инертном растворителе и затем дополнительно обрабатывают основанием с образованием соединения (XXXI). Примеры инертного растворителя включают: алкилгалогенидные растворители, такие как метиленхлорид, хлороформ и тетрахлорид углерода; эфирные растворители, такие как тетрагидрофуран, 1,2-диметоксиэтан и диоксан; ароматические растворители, такие как бензол и толуол; и амидные растворители, такие как N,N-диметилформамид, N,N-диметиласетамид и N-метилпирролидин-2-он. Кроме того, в некоторых случаях, могут использоваться сульфоксидные растворители, такие как диметилсульфоксид и сульфолан, кетоновые растворители, такие как ацетон и метилэтилкетон или ацетонитрил или т.п. Пиридин, 2,6-лутидин, коллидин, 4-диметиламинопиридин, триэтиламин, N-метилморфолин, диизопропилэтиламин или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU) могут использоваться в качестве основания. В некоторых случаях реакция дегидратации может протекать во время альдольной реакции.
Реакция восстановления может быть осуществлена в соответствии со способом, описанным в способе получения 2. Защитные группы и их удаление могут осуществляться в соответствии со способом, описанным в способе получения 1.
Соединение (Ij) может быть получено из соединения (XXXI) с использованием этих реакций.
Когда соединения, представляющие интерес, или промежуточные соединения в способах получения 1-10 представляют собой изомерные (например, стереоизомерные) смеси, каждый изомер может быть отделен и очищен соответствующим образом препаративной хроматографией среднего давления, ВЭЖХ или т.п. с использованием оптически активной колонки или т.п.
Когда соединение настоящего изобретения, представленное формулой (I), или его фармакологически приемлемая соль или промежуточное соединение при его получении имеет асимметрический углерод, присутствуют их оптические изомеры. Из этих оптических изомеров каждый изомер может быть выделен и очищен посредством общепринятого способа, такого как фракционная кристаллизация (разделение соли) с использованием перекристаллизации из соответствующего растворителя или колоночная хроматография. Примеры ссылок на способ разделения рацемических смесей на оптические изомеры может включать J. Jacques et al., “Enantiomers, Racemates and Resolution, John Wiley And Sons, Inc”.
Циклоалкил-замещенное производное имидазола настоящего изобретения обладает превосходной ингибирующей активностью в отношении TAFIa и имеет хорошую пероральную всасываемость, превосходное распределение, такое как удерживание в крови, и метаболическую стабильность и высокую безопасность. Следовательно, циклоалкил-замещенное производное имидазола настоящего изобретения является полезным в качестве фармацевтического лекарственного средства и особенно полезным в качестве терапевтического лекарственного средства против инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких. Кроме того, оно является полезным в качестве терапевтического лекарственного средства против заболевания, происходящего вследствие эмболии. Более того, оно является полезным в качестве фармацевтического лекарственного средства для улучшения функций органа после трансплантации. Соединение настоящего изобретения является также полезным в качестве терапевтического лекарственного средства против заболеваний коронарных артерий после хирургического вмешательства (чрескожная транслюминальная коронарная ангиопластика), трансплантации или замены сосудистого заменителя (аутологичного или искусственного кровеносного сосуда) или рестеноза/реокклюзии, вызванных имплантацией стента. Более того, оно является полезным для профилактики образования тромба, вызванного сосудистым катетером (внутренним катетером для диализа), экстракорпоральным циркулятором крови и покрытием искусственного кровеносного сосуда или его заполнением раствором ингибитора TAFIa, и для инициации тромболиза. Оно также является полезным в качестве терапевтического лекарственного средства против атеротромбоза или фиброза (фиброза легких, такого как хроническая обструктивная болезнь легких, фиброз после хирургического вмешательства на глазах и т.д.).
Соединение настоящего изобретения, представленное общей формулой (I), имеет основную группу, такую как аминогруппа, и может, следовательно, быть преобразовано в кислотно-аддитивную соль с фармакологически приемлемой кислотой. Примеры такой соли могут включать: гидрогалогениды, такие как гидрофторид, гидрохлорид, гидробромид и гидройодид; соли неорганической кислоты, такие как нитрат, перхлорат, сульфат и фосфат; низшие алкансульфонаты, такие как метансульфонат, трифторметансульфонат и этансульфонат; арилсульфонаты, такие как бензолсульфонат и п-толуолсульфонат; соли органической кислоты, такие как ацетат, малат, фумарат, сукцинат, цитрат, тартрат, оксалат и малеат; и соли аминокислоты, такие как орнитат, глутамат и аспартат. Гидрогалогениды или арилсульфонаты являются предпочтительными; гидрохлорид, бензолсульфонат или п-толуолсульфонат являются более предпочтительными; бензолсульфонат или п-толуолсульфонат являются даже более предпочтительными; и п-толуолсульфонат является особенно предпочтительным.
Кроме того, соединение, представленное общей формулой (I), имеет кислотную группу, такую как карбоксигруппа, и может, следовательно, как правило, образовывать основно-аддитивную соль. Примеры фармакологически приемлемой соли могут включать: соли щелочного металла, такие как соли натрия, соли калия и соли лития; соли щелочноземельного металла, такие как соли кальция и соли магния; неорганические соли, такие как соли аммония; соли органического амина, такие как соли дибензиламина, соли морфолина, соли алкилового сложного эфира фенилглицина, соли этилендиамина, соли N-метилглюкамина, соли диэтиламина, соли триэтиламина, соли циклогексиламина, соли дициклогексиламина, соли N,N'-дибензилэтилендиамина, соли диэтаноламина, соли N-бензил-N-(2-фенилэтокси)амина, соли пиперазина, соли тетраметиламмония и соли трис(гидроксиметил)аминометана; и соли аминокислоты, такие как соли аргинина.
Соединение настоящего изобретения, представленное общей формулой (I), или его фармакологически приемлемая соль могут присутствовать в свободной или сольватированной форме. Эти сольваты также охватываются объемом настоящего изобретения. Сольват не является ограниченным конкретно, поскольку он является фармакологически приемлемым. Особенно предпочтительными являются гидраты, этанолаты или т.п.; и гидраты являются более предпочтительными. Кроме того, соединение настоящего изобретения, представленное общей формулой (I), содержит атом азота. Этот атом азота может находиться в форме N-оксида. Эти сольватированные или N-оксидные формы также являются охваченными объемом настоящего изобретения.
Соединение настоящего изобретения, представленное общей формулой (I), или его фармакологически приемлемая соль и промежуточное соединение при получении соединения настоящего изобретения могут включать разнообразные изомеры, такие как геометрические изомеры (например, цис- и транс-формы) и оптические изомеры (R и S формы), в зависимости от видов или комбинаций заместителей. Соединение настоящего изобретения охватывает все из этих изомеров, стереоизомеров и даже смеси этих изомеров и стереоизомеров при любом соотношении, если не указано иное.
Кроме того, соединение настоящего изобретения или его фармакологически приемлемая соль могут также содержать неприродное соотношение атомных изотопов одного или более из атомов, составляющих такое соединение. Примеры атомных изотопов включают дейтерий (2Н), тритий (3Н), углерод-13 (13С), углерод-14 (14С), азот-15 (15N), хлор-37 (37Cl) и йод-125 (125I). Кроме того, соединение может представлять собой радиоактивно-меченное радиоактивным изотопом, например, тритием (3Н), йодом-125 (125I) или углеродом-14 (14С). Радиоактивно-меченное соединение является полезным в качестве терапевтического или профилактического реагента, исследовательского реагента, например, аналитического реагента и диагностического агента, например, агента для диагностической визуализации in-vivo. Все изотопные варианты соединения настоящего изобретения охватываются объемом настоящего изобретения, независимо от того, являются ли они радиоактивными или нет.
Кроме того, настоящее изобретения также охватывает “фармацевтически приемлемое пролекарственное соединение”, которое преобразуется посредством реакции с ферментом, желудочной кислотой или т.п. в физиологических условиях in-vivo в соединение (I), служащее в качестве активного ингредиента фармацевтической композиции настоящего изобретения, т.е. соединение, которое преобразуется в соединение (I) посредством гидролиза или т.п., вызванного желудочной кислотой или т.п.
Соединение общей формулы (I) настоящего изобретения или его фармакологически приемлемая соль могут образовывать множество кристаллов (кристаллические полиморфы), отличающихся по внутренней структуре и физико-химическим свойствам в зависимости от условий реакции и условий кристаллизации. Каждый из этих кристаллов или их смесь при любом соотношении охватываются настоящим изобретением. Соединение общей формулы (I) или его фармакологически приемлемая соль также могут присутствовать в виде смеси кристаллических твердых веществ и аморфных твердых веществ. Их смесь при любом соотношении охватывается настоящим изобретением. Более конкретно, содержание конкретной кристаллической формы настоящего изобретения составляет предпочтительно 50% или более, более предпочтительно 80% или более, даже более предпочтительно 90% или более, особенно предпочтительно 95% или более, наиболее предпочтительно 97% или более.
В настоящем изобретении кристаллы относятся к твердому веществу, имеющему трехмерные регулярные повторы атомов (или их популяций), составляющих внутреннюю структуру, и отличающемуся от аморфных твердых веществ, которые не имеют такой регулярной внутренней структуры. Является ли определенное твердое вещество кристаллическим или нет, можно исследовать посредством хорошо известного кристаллографического метода (например, порошковой рентгеновской кристаллографией или дифференциальной сканирующей калориметрией). Например, определенное твердое вещество подвергают порошковой рентгеновской кристаллографии с использованием рентгеновских лучей, полученных при излучении Kα меди. Определяют, что твердое вещество является кристаллическим, когда на его рентгеновской дифрактограмме наблюдают различимый пик, или определяют, что оно является аморфным, когда на ней не наблюдают различимого пика. Когда пик может быть считан, но не является различимым (например, пик является широким), определяют, что твердое вещество представляет собой кристаллы, имеющие низкую степень кристалличности. Такие кристаллы, имеющие низкую степень кристалличности, охватываются кристаллами настоящего изобретения.
При рентгеновской кристаллографии порошка с использованием лучей Kα меди образец обычно облучают лучами Kα меди (в которых лучи Kα1 и Kα2 не разделяют). Рентгеновская дифрактограмма может быть получена посредством анализа дифракции, происходящей от лучей Kα, и может быть также получена посредством анализа только дифракции, происходящей от лучей Kα1, собранных от дифракции, происходящей от лучей Kα. В настоящем изобретении порошковая рентгеновская дифрактограмма, полученная посредством излучения Kα, охватывает порошковую рентгеновскую дифрактограмму, полученную посредством анализа дифракции, происходящей от лучей Kα, и порошковую рентгеновскую дифрактограмму, полученную посредством анализа дифракции, происходящей от лучей Kα1, и предпочтительно представляет собой порошковую рентгеновскую дифрактограмму, полученную посредством анализа дифракции, происходящей от лучей Kα1.
Кристаллы типа I ангидрата п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты настоящего изобретения могут представлять собой кристаллы, проявляющие основные пики при межплоскостных расстояниях d, равных 23,9, 11,9, 4,5, 4,3 и 3,6 ангстрем, на порошковой рентгеновской дифрактограмме, полученной посредством излучения Кα меди, например, как показано на фиг.1.
Кристаллы типа II моногидрата п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты настоящего изобретения могут представлять собой кристаллы, проявляющие основные пики при межплоскостных расстояниях d, равных 22,9, 5,0, 4,9, 4,7 и 4,0 ангстрем, на порошковой рентгеновской дифрактограмме, полученной посредством излучения Кα меди, например, как показано на фиг.3.
На порошковой рентгеновской дифрактограмме фиг.1 или 3, приведенных ниже, ордината представляет интенсивность дифракции [счет/секунда (cps)], и абсцисса представляет 2θ углы дифракции (градусы). Кроме того, межплоскостные расстояния d (ангстремы) могут быть рассчитаны в соответствии с формулой 2dsinθ=nλ, где n=1. В этой формуле длина волны λ лучей Кα составляет 1,54 ангстрем, и длина волны λ лучей Кα1 составляет 1,541 ангстрем. Положения и относительная интенсивность пиков при межплоскостных расстояниях d могут до некоторой степени изменяться в зависимости от условий измерения и т.д. Таким образом, идентичность кристаллической формы должна быть распознана соответствующим образом со ссылкой на целую картину спектра, даже когда межплоскостные расстояния d слегка отличаются от ожидаемых расстояний.
Термический анализ (TG/DTA) на фиг.2 и 4 проводили путем измерения при скорости нагрева, равной 10°С, в токе сухого азота, составляющего 200 мл/мин.
Фармацевтическая композиция, содержащая соединение настоящего изобретения, представленное общей формулой (I), или его фармакологически приемлемую соль, может быть получена в соответствии с разнообразными способами составления, обычно применяемыми путем выбора соответствующего получения в соответствии со способом введения.
Фармацевтическая композиция, содержащая соединение настоящего изобретения, представленное общей формулой (I), или его фармакологически приемлемую соль в качестве основного ингредиента, при введении млекопитающему (в частности, человеку) может вводиться системно или местно пероральным или парентеральным путем.
Примеры пероральных форм фармацевтических лекарственных средств включают таблетки, пилюли, порошки, гранулы, капсулы, растворы, суспензии, эмульсии, сиропы и эликсиры. Эти формы фармацевтических лекарственных средств обычно получают в виде фармацевтической композиции, содержащей соединение настоящего изобретения, представленное общей формулой (I), или его фармакологически приемлемую соль в качестве основного ингредиента, смешивают с фармацевтически приемлемыми добавками, такими как разбавители, эксципиенты или носители. Получение фармацевтической композиции может осуществляться в соответствии с общепринятым способом с использованием фармацевтически приемлемых разбавителей, эксципиентов или носителей, или других добавок, выбранных соответствующим образом в соответствии с потребностью из произвольно выбранных фармацевтически приемлемых связующих, дезинтеграторов, лубрикантов, агентов набухания, вспомогательных средств для набухания, средств для покрытия, пластификаторов, стабилизаторов, антисептиков, антиоксидантов, красящих агентов, солюбилизирующих агентов, суспендирующих агентов, эмульгирующих агентов, подсластителей, консервантов, буферов, увлажнителей и т.д.
Примеры парентеральных форм фармацевтических лекарственных средств включают инъекции, мази, гели, кремы, припарки, пластыри, аэрозоли, ингаляторы, спреи, глазные капли, назальные капли и суппозитории. Эти формы фармацевтических лекарственных средств обычно получают в виде фармацевтической композиции, содержащей соединение настоящего изобретения, представленное общей формулой (I), или его фармацевтически приемлемую соль в качестве основного ингредиента, смешивают с фармацевтически приемлемыми добавками, такими как разбавители, эксципиенты или носители. Получение фармацевтической композиции может осуществляться в соответствии с общепринятым способом с использованием фармацевтически приемлемых разбавителей, эксципиентов или носителей, или других добавок, выбранных соответствующим образом в соответствии с потребностью из произвольно выбранных фармацевтически приемлемых стабилизаторов, антисептиков, солюбилизирующих агентов, увлажнителей, консервантов, антиоксидантов, ароматизаторов, гелеобразующих агентов, нейтрализующих агентов, буферов, тонизирующих агентов, поверхностно-активных веществ, окрашивающих агентов, буферных агентов, загустителей, увлажняющих агентов, наполнителей, промоторов всасывания, суспендирующих агентов, связующих и т.д.
Примеры ссылок на фармацевтически приемлемые эксципиенты могут включать “Handbook of Pharmaceutical Excipients, 2nd Edition, (1994), Edited by A. Wade and P.J. Weller”.
Кроме того, примеры ссылок на фармацевтически приемлемые носители или разбавители могут включать “Remington's Pharmaceutical Sciences, Mack Publishing Co. (A.R. Gennaro edit. 1985)”.
Соединение настоящего изобретения, представленное общей формулой (I), или его фармакологически приемлемая соль может применяться в комбинации с дополнительным лекарственным средством. Лекарственные средства, которые могут применяться в комбинации с ним, включают антикоагулянты (варфарин, гепарин, низкомолекулярный гепарин, антитромбиновые лекарственные средства, лекарственные средства против Xa и т.д.), антитромбоцитарные лекарственные средства (аспирин, тиклопидин, клопидогрел, прасугрел, ингибиторы фосфодиэстеразы и т.д.), ферменты, имеющие отношение к фибринолизу (tPA, генетически модифицированный tPA, активаторы плазминогена, такие как урокиназа, стрептокиназа, плазмин и т.д.), противораковые лекарственные средства, противовоспалительные лекарственные средства, антифиброзные лекарственные средства, гипотензивные лекарственные средства, лекарственные средства против легочной гипертензии и иммуносупрессорные лекарственные средства.
Доза соединения настоящего изобретения, представленного общей формулой (I), или его фармакологически приемлемой соли различается в зависимости от симптомов, возраста, массы тела, вида или дозы лекарственного средства, подлежащего введению в комбинации с ним, и т.д. Когда соединение настоящего изобретения, представленное общей формулой (I), или его фармакологически приемлемую соль применяют в качестве фармацевтического лекарственного средства для человеческого организма, его доза находится в интервале от 0,01 мг до 5000 мг, предпочтительно от 0,01 мг до 1000 мг, более предпочтительно от 1 мг до 200 мг, в однократной дозе на взрослого, исходя из количества соединения (I), и находится в интервале от 0,01 мг/кг до 100 мг/кг, предпочтительно от 0,005 мг/кг до 20 мг/кг, более предпочтительно от 0,01 мг/кг до 5 мг/кг соединения формулы (I) исходя из массы тела. Эту суточную дозу вводят системно или местно пероральным или парентеральным путем однократно каждые несколько дней или при одной или нескольких дозировках в день, или непрерывно вводят в вены в течение продолжительности в интервале от 1 часа до 24 часов в день. Кроме того, суточная доза может превышать количество, приведенное выше, если необходимо.
ПРИМЕРЫ
Далее в данном описании настоящее изобретение будет описано конкретно со ссылкой на ссылочные примеры, примеры, тестовые примеры и примеры получения. Однако настоящее изобретение не ограничено этими способами каким-либо образом.
Символы “1Н-ЯМР”, “МС”, “МСВР”, “МСНР” в примерах означают “спектр ядерного магнитного резонанса”, “масс-спектр”, “масс-спектр высокого разрешения”, “масс-спектр низкого разрешения”, соответственно. Соотношение элюирующих растворителей при хроматографическом разделении/очистке представляет собой объемное отношение, если не указано иное. Термины внутри скобок “1Н-ЯМР” представляют растворители для анализа, во всех из которых используют ТМС (тетраметилсилан) в качестве внутреннего стандарта.
Мультиплетность в 1Н-ЯМР означает с=синглет, д=дублет, т=триплет, кв=квартет, м=мультиплет и шир=широкий. Кроме того, в настоящем описании использованы следующие сокращения:
CDCl3: дейтерированный хлороформ;
CD3OD: дейтерированный метанол;
Me: метильная группа;
Et: этильная группа;
tBu: трет-бутильная группа;
Boc: трет-бутоксикарбонильная группа;
Cbz: (бензилокси)карбонильная группа;
TBDMS: трет-бутил(диметил)силильная группа;
TBDPS: трет-бутил(дифенил)силильная группа.
[Ссылочный пример 1] трет-Бутил-5-[(трет-бутоксикарбонил)амино]-2-(диэтоксифосфорил)валерат
[Формула 40]
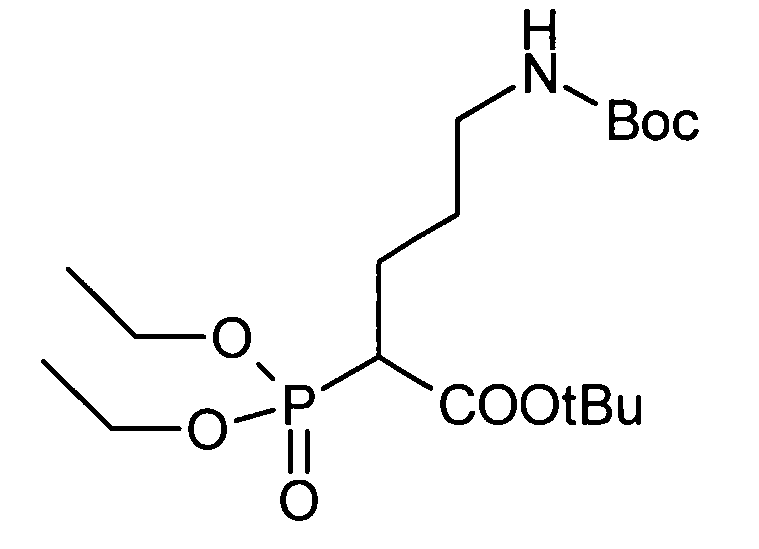
Трет-бутил-диэтилфосфоноацетат (20,0 г) растворяли в тетрагидрофуране (500 мл). К раствору добавляли гидрид натрия (63%, 3,32 г) при 0°С, и смесь перемешивали при 0°С в течение 15 минут и при комнатной температуре в течение 1 часа. К ней медленно добавляли раствор трет-бутил-(3-бромпропил)карбамата (20,0 г) в тетрагидрофуране (20 мл) при комнатной температуре, и смесь перемешивали при комнатной температуре в течение 18 часов. К реакционному раствору добавляли насыщенный водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия, затем сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=1/1-этилацетат), с получением указанного в заголовке соединения (26,6 г).
1H-ЯМР (CDCl3) δ: 1,31-1,36 (6H, м), 1,44 (9H, м), 1,48 (9H, м), 1,51-1,59 (2H, м), 1,78-2,00 (2H, м), 2,83 (1H, ддд, J=22,9, 10,7, 4,4 Гц), 3,06-3,18 (2H, м), 4,10-4,18 (4H, м), 4,58 (1H, шир.).
[Ссылочный пример 2] трет-Бутил-5-[(трет-бутоксикарбонил)амино]-2-(1Н-имидазол-4-илметил)валерат
[Формула 41]
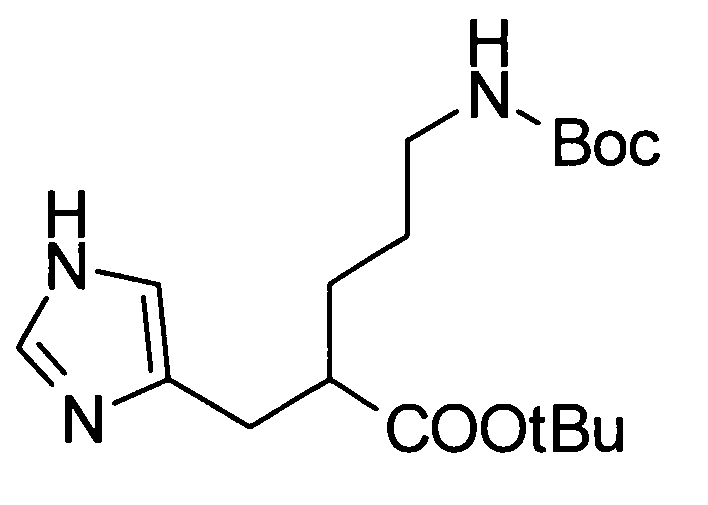
К раствору соединения (8,35 г), полученного в ссылочном примере 1, в ацетонитриле (100 мл), добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (4,58 мл) и хлорид лития (1,30 г) при комнатной температуре. К полученной суспензии добавляли 1-тритил-1Н-имидазол-4-карбальдегид (6,90 г), и смесь перемешивали в течение ночи при комнатной температуре. Растворитель отгоняли при пониженном давлении. К остатку добавляли этилацетат и 10% водную лимонную кислоту. Полученный раствор разделяли на водный и органический слои. Затем органический слой промывали насыщенным раствором хлорида натрия, насыщенным водным бикарбонатом натрия и насыщенным раствором хлорида натрия в указанном порядке. Органический слой сушили над безводным сульфатом натрия, с получением смеси трет-бутил-(2Е)-5-[(трет-бутоксикарбонил)амино]-2-[1-тритил-1Н-имидазол-4-ил)метилен]валерата и трет-бутил-(2Z)-5-[(трет-бутоксикарбонил)амино]-2-[1-тритил-1Н-имидазол-4-ил)метилен]валерата (11,3 г). Полученную смесь суспендировали в метаноле (500 мл). К полученной суспензии добавляли катализатор 10% палладий на углероде (гидратированный, 4 г), и смесь перемешивали при комнатной температуре в течение 3 дней в атмосфере водорода. Катализатор отфильтровывали, и фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метиленхлорид/метанол=9/1), с получением указанного в заголовке соединения (5,60 г).
1H-ЯМР (CDCl3) δ: 1,41 (9H, с), 1,44 (9H, с), 1,48-1,57 (3H, м), 1,57-1,66 (1H, м), 2,58-2,68 (1H, м), 2,73 (1H, дд, J=14,7, 5,3 Гц), 2,89 (1H, дд, J=14,7, 8,4 Гц), 3,02-3,19 (2H, м), 4,67 (1H, шир.с), 6,79 (1H, с), 7,54 (1H, с).
[Ссылочный пример 3] 5-[(трет-бутоксикарбонил)амино]-2-(метоксикарбонил)валериановая кислота
[Формула 42]
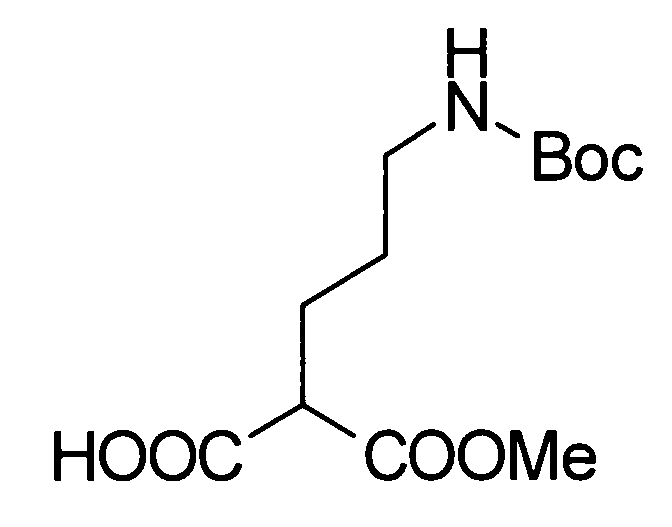
К диметилмалонату (102 мл) добавляли раствор метоксида натрия в метаноле (28%, 90,4 мл) при комнатной температуре, и смесь перемешивали при 60°С в течение 30 минут. Белую суспензию охлаждали до комнатной температуры. Затем к ней сразу же добавляли трет-бутил(3-бромпропил)карбамат (106 г), и смесь перемешивали при комнатной температуре в течение 12 часов. К реакционному раствору добавляли воду, и органическое вещество экстрагировали диэтиловым эфиром. Органический слой промывали 1н. водным гидроксидом натрия и насыщенным раствором хлорида натрия в указанном порядке, затем сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта диметил{3-[(трет-бутоксикарбонил)амино]пропил}малоната. Полученный сложный эфир (94 г) растворяли в метаноле (100 мл). К раствору добавляли раствор моногидрата гидроксида лития (13,6 г) в воде (300 мл) и метанол (300 мл) при 0°С, и смесь перемешивали при комнатной температуре в течение 15 часов. Метанол отгоняли при пониженном давлении, и органическое вещество экстрагировали этилацетатом. К водному слою добавляли 2н. хлористоводородную кислоту (160 мл), с последующей экстракцией этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия, затем сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метиленхлорид-метиленхлорид/метанол=10/1), с получением указанного в заголовке соединения (69,1 г).
1H-ЯМР (CDCl3) δ: 1,44 (9H, м), 1,50-1,60 (2Н, м), 1,86- 2,01 (2Н, м), 3,07-3,20 (2Н, м), 3,43 (1H, м), 3,77 (3H, с), 4,64 (1H, шир.).
[Ссылочный пример 4] 1-(транс-4-метилциклогексил)-1Н-имидазол-4-карбальдегид
[Стадия 1] Этил-1-(транс-4-метилциклогексил)-1Н-имидазол-4-карбоксилат
[Формула 43]
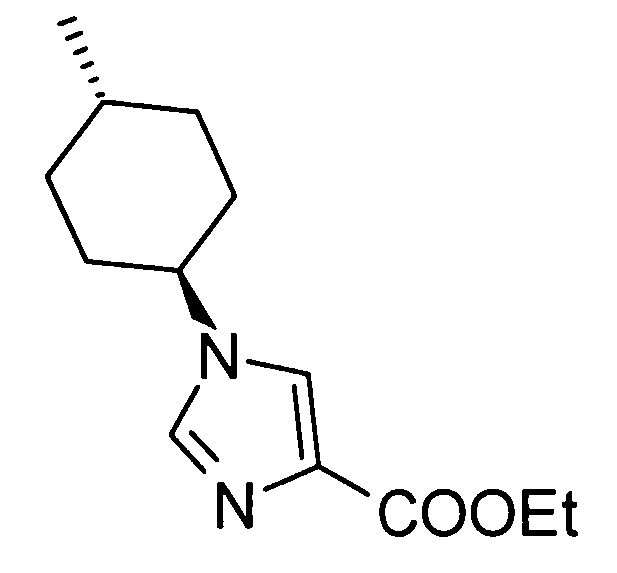
Этил-3-(диметиламино)-2-изоцианоакрилат (Liebigs Annalen der Chemie, 1979, p. 1444) (1,52 г) растворяли в транс-4-метилциклогексиламине (3,07 г), и раствор перемешивали при 70°С в течение 4 часов. К реакционному раствору добавляли насыщенный водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=2/1-1/2), с получением указанного в заголовке соединения (1,90 г).
1H-ЯМР (CDCl3) δ: 0,96 (3H, д, J=6,6 Гц), 1,13 (2H, м), 1,39 (3H, д, J=7,0 Гц), 1,47 (1H, м), 1,68 (2H, м), 1,88 (2H, м), 2,12 (2H, м), 3,91 (1H, тт, J=12,1, 3,9 Гц), 4,36 (2H, кв., J=7,0 Гц), 7,54 (1H, с), 7,66 (1H, с).
[Стадия 2] [1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метанол
[Формула 44]
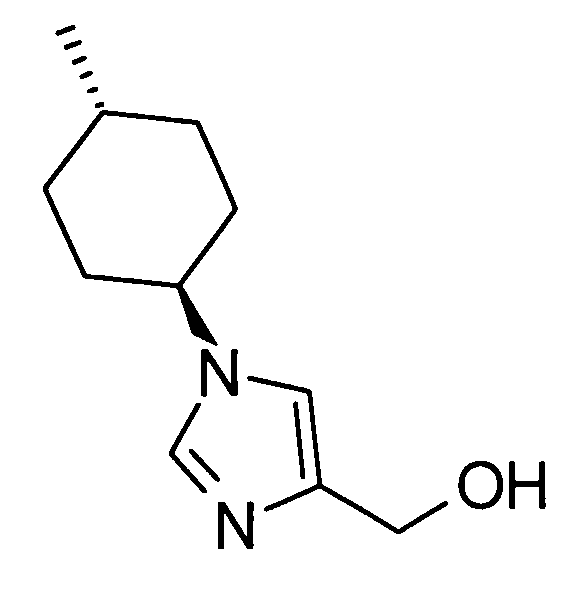
Алюмогидрид лития (92%, 0,31 г) суспендировали в тетрагидрофуране (6 мл). Соединение (1,50 г), полученное на стадии 1 данного ссылочного примера, растворяли в тетрагидрофуране (6 мл), и полученный раствор медленно добавляли по каплям к суспензии при 0°С. После перемешивания при 0°С в течение 30 минут реакционный раствор разбавляли диэтиловым эфиром, и к нему добавляли насыщенный водный сульфат натрия. После перемешивания при комнатной температуре в течение 1 часа образовавшуюся неорганическую соль удаляли фильтрованием через целит. Фильтрат концентрировали при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт промывали смешанным растворителем из гексана и этилацетата (5:1), с получением указанного в заголовке соединения (1,09 г).
1H-ЯМР (CDCl3) δ: 0,95 (3H, д, J=6,6 Гц), 1,04-1,17 (2H, м), 1,44 (1H, м), 1,59-1,73 (2H, м), 1,81-1,89 (2H, м), 2,04-2,13 (2H, м), 2,78 (1H, шир.), 3,84 (1H, тт, J=12,1, 3,9 Гц), 4,59 (2H, с), 6,91 (1H, с), 7,49 (1H, с).
[Стадия 3] 1-(транс-4-метилциклогексил)-1Н-имидазол-4-карбальдегид
[Формула 45]
Соединение (1,04 г), полученное на стадии 2 данного ссылочного примера, растворяли в толуоле (10 мл). К раствору добавляли раствор бикарбоната натрия (1,35 г) в воде (5 мл), йод (2,72 г) и 2,2,6,6-тетраметил-1-пиперидинилокси (84 мг) в указанном порядке, и смесь перемешивали при комнатной температуре в течение 2 часов. К реакционному раствору добавляли насыщенный водный тиосульфат натрия, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=1/1-1/2), с получением указанного в заголовке соединения (0,900 г).
1H-ЯМР (CDCl3) δ: 0,97 (3H, д, J=6,8 Гц), 1,09-1,19 (2H, м), 1,48 (1H, м), 1,65-1,75 (2H, м), 1,87-1,93 (2H, м), 2,11-2,18 (2H, м), 3,95 (1H, тт, J=12,2, 3,9 Гц), 7,62 (1H, с), 7,68 (1H, с), 9,87 (1H, с).
[Ссылочный пример 5] 1-(транс-4-Этилциклогексил)-1Н-имидазол-4-карбальдегид
[Стадия 1] [1-(4-этилциклогексил)-1Н-имидазол-4-ил]метанол
[Формула 46]
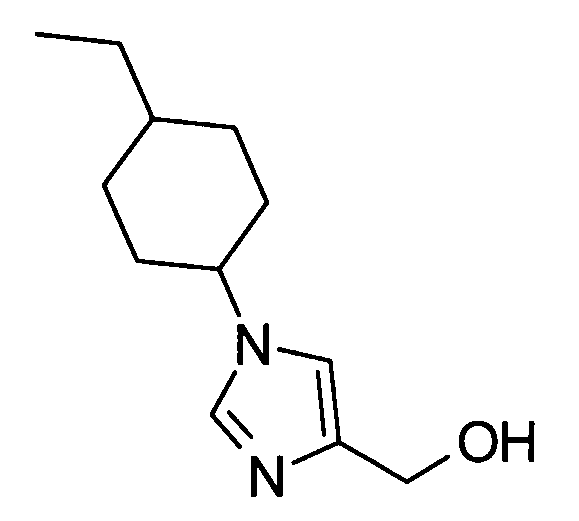
Этил-3-(диметиламино)-2-изоцианоакрилат (2,00 г) растворяли в 4-этилциклогексиламине (3,37 г), и раствор перемешивали при 70°С в течение 4,5 часов. К реакционному раствору добавляли насыщенный водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Алюмогидрид лития (92%, 0,490 г) суспендировали в тетрагидрофуране (12 мл). Полученный неочищенный продукт растворяли в тетрагидрофуране (12 мл), и полученный раствор медленно добавляли по каплям к суспензии при 0°С. После перемешивания при 0°С в течение 30 минут реакционный раствор разбавляли диэтиловым эфиром, и к нему добавляли насыщенный водный сульфат натрия. После перемешивания при комнатной температуре в течение 1 часа образовавшуюся неорганическую соль удаляли фильтрованием через целит. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метиленхлорид-метиленхлорид/метанол=9/1), с получением указанного в заголовке соединения (1,35 г, диастереомерная смесь, транс: цис=4:1).
1H-ЯМР (CDCl3) δ: 0,91 (0,6H, т, J=7,0 Гц), 0,92 (2,4H, т, J=7,0 Гц), 1,01-1,13 (1,6H, м), 1,16-1,40 (2,8H, м), 1,50-1,97 (5H, м), 2,07-2,15 (1,6H, м), 3,85 (0,8H, тт, J=12,1, 3,9 Гц), 3,99 (0,2H, тт, J=8,6, 4,3 Гц), 4,59 (1,6H, с), 4,60 (0,4H, с), 6,91 (0,8H, с), 6,94 (0,2H, с), 7,49 (0,8H, с), 7,53 (0,2H, с).
[Стадия 2] 1-(транс-4-этилциклогексил)-1Н-имидазол-4-карбальдегид
[Формула 47]
Соединение (1,00 г), полученное на стадии 1 данного ссылочного примера, растворяли в толуоле (10 мл). К раствору добавляли раствор бикарбоната натрия (1,21 г) в воде (6 мл), йод (2,19 г) и 2,2,6,6-тетраметил-1-пиперидинилокси (75 мг) в указанном порядке, и смесь перемешивали при комнатной температуре в течение 12 часов. К реакционному раствору добавляли насыщенный водный тиосульфат натрия, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=2/1-1/1), с получением указанного в заголовке соединения (468 мг).
1H-ЯМР (CDCl3) δ: 0,92 (3H, т, J=7,0 Гц), 1,10 (2H, м), 1,19-1,34 (3H, м), 1,68 (2H, м), 1,97 (2H, м), 2,17 (2H, м), 3,95 (1H, тт, J=12,1, 3,5 Гц), 7,62 (1H, с), 7,69 (1H, с), 9,87 (1H, с).
[Ссылочный пример 6] 1-(3-Этилциклобутил)-1Н-имидазол-4-карбальдегид
[Стадия 1] Бензил(3-этилциклобутил)карбамат
[Формула 48]
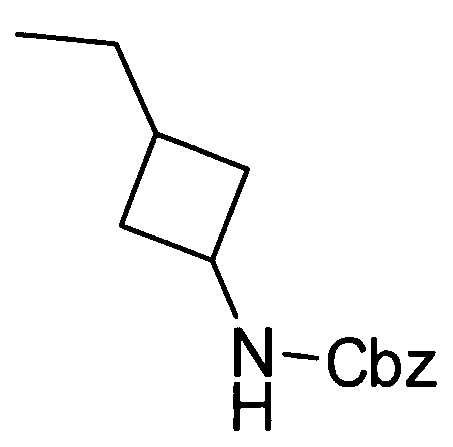
3-Этилциклобутанкарбоновую кислоту (1,67 г) растворяли в толуоле (20 мл), и к нему добавляли диизопропилэтиламин (5,32 мл). Раствор нагревали до 100°С, и к нему по каплям добавляли дифенилфосфорилазид (3,09 мл) в толуоле (10 мл) в течение 40 минут. После перемешивания при 100°С в течение 15 минут добавляли бензиловый спирт (1,48 мл), и смесь дополнительно перемешивали при 100°С в течение 15 минут. Реакционный раствор охлаждали. К нему добавляли 0,2 н. водный гидроксид натрия, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=20/1-10/1), с получением указанного в заголовке соединения (1,81 г, диастереомерная смесь, транс:цис=1:1).
1H-ЯМР (CDCl3) δ: 0,78 (1,5H, т, J=7,4 Гц), 0,81 (1,5H, т, J=7,4 Гц), 1,38 (1H, дкв., J=7,4, 7,4 Гц), 1,46 (1H, дкв., J=7,4, 7,4 Гц), 1,31-1,42 (2H, м), 1,89-2,03 (2H, м), 2,41-2,54 (1H, м), 4,00 (0,5H, м), 4,23 (0,5H, м), 4,75-4,90 (1H, шир.), 5,06 (2H, с), 7,22-7,40 (5H, м).
[Стадия 2] [1-(3-этилциклобутил)-1Н-имидазол-4-ил]метанол
[Формула 49]
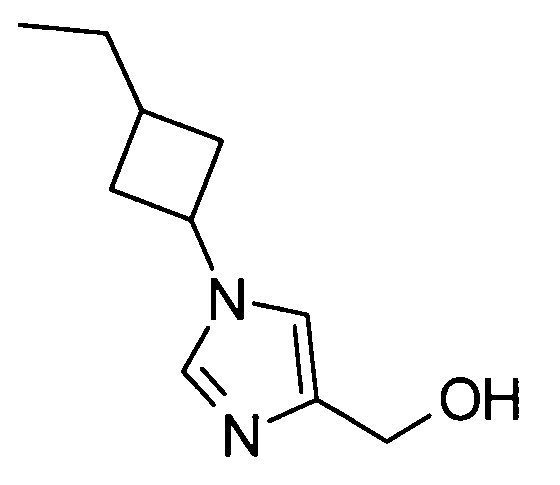
Соединение (1,81 г), полученное на стадии 1 данного ссылочного примера, растворяли в метилацетате (7 мл). К раствору добавляли катализатор, 10% палладий на углероде (гидратированный, 100 мг), и смесь перемешивали при комнатной температуре в течение 8 часов в атмосфере водорода при нормальном давлении. После фильтрования через целит фильтрат концентрировали при пониженном давлении, с получением неочищенного продукта из 3-этилциклобутанамина. Полученный неочищенный продукт и этил-3-(диметиламино)-2-изоцианоакрилат (650 мг) смешивали и перемешивали при 75°С в течение 10 часов в герметизированной пробирке. К реакционному раствору добавляли насыщенный водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=1/1-1/2), с получением этил-1-(3-этилциклобутил)-1Н-имидазол-4-карбоксилата.
Алюмогидрид лития (92%, 80 мг) суспендировали в тетрагидрофуране (4 мл). К полученной суспензии раствор этил-1-(3-этилциклобутил)-1Н-имидазол-4-карбоксилата в тетрагидрофуране (5 мл) медленно добавляли по каплям при 0°С. После перемешивания при 0°С в течение 30 минут реакционный раствор разбавляли диэтиловым эфиром, и к нему добавляли насыщенный водный сульфат натрия. После перемешивания при комнатной температуре в течение 1 часа образовавшуюся неорганическую соль удаляли фильтрованием через целит. Фильтрат концентрировали при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метиленхлорид-метиленхлорид/метанол=5/1), с получением указанного в заголовке соединения (119 мг, диастереомерная смесь, транс:цис=1:1).
1H-ЯМР (CDCl3) δ: 0,86 (1,5H, т, J=7,4 Гц), 0,90 (1,5H, т, J=7,4 Гц), 1,48 (1H, дкв., J=7,4, 7,4 Гц), 1,56 (1H, дкв., J=7,4, 7,4 Гц), 1,84-1,93 (1H, м), 1,96-2,08 (0,5H, м), 2,20-2,32 (1,5H, м), 2,39-2,49 (1H, м), 2,59-2,67 (1H, м), 4,38 (0,5H, тт, J=9,4, 7,8 Гц), 4,59 (1H, с), 4,60 (1H, с), 4,63 (0,5H, тт, J=7,8, 7,4 Гц), 6,93 (0,5H, с), 6,98 (0,5H, с), 7,46 (0,5H, с), 7,49 (0,5H, с).
[Стадия 3] 1-(3-этилциклобутил)-1Н-имидазол-4-карбальдегид
[Формула 50]
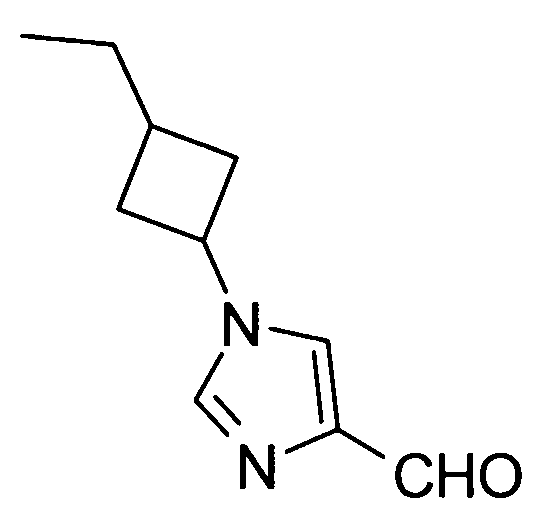
Соединение (119 мг), полученное на стадии 2 данного ссылочного примера, растворяли в толуоле (5 мл). К полученному раствору добавляли раствор бикарбоната натрия (166 мг) в воде (4 мл), йод (305 мг) и 2,2,6,6-тетраметил-1-пиперидинилокси (11 мг) в указанном порядке, и смесь перемешивали при комнатной температуре в течение 12 часов. К реакционному раствору добавляли насыщенный водный тиосульфат натрия, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=1/1-1/2), с получением указанного в заголовке соединения (115 мг, диастереомерная смесь, транс:цис=1:1).
1H-ЯМР (CDCl3) δ: 0,86 (1,5H, т, J=7,3 Гц), 0,90 (1,5H, т, J=7,3 Гц), 1,44 (9H, с), 1,51 (1H, дкв., J=7,4, 7,4 Гц), 1,59 (1H, дкв., J=7,4, 7,4 Гц), 1,87-1,97 (1H, м), 2,04-2,13 (0,5H, м), 2,28-2,38 (1,5H, м), 2,42-2,52 (1H, м), 2,66-2,75 (1H, м), 4,48 (0,5H, тт, J=9,0, 7,8 Гц), 4,72 (0,5H, тт, J=7,8, 7,4 Гц), 7,58 (0,5H, с), 7,61 (0,5H, с), 7,69 (0,5H, с), 7,74 (0,5H, с), 9,87 (0,5H, с), 9,88 (0,5H, с).
[Ссылочный пример 7] 1-(3-Метилциклобутил)-1Н-имидазол-4-карбальдегид
[Формула 51]
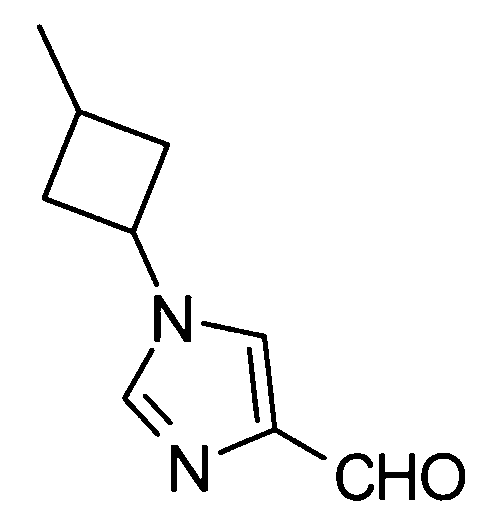
Указанное в заголовке соединение (9,1 мг, диастереомерная смесь, транс:цис=1:1) получали из 3-метилциклобутанкарбоновой кислоты (1,70 г) таким же образом, как в ссылочном примере 6.
1H-ЯМР (CDCl3) δ: 1,18 (1,5H, д, J=6,6 Гц), 1,27 (1,5H, д, J=6,6 Гц), 1,93 (1H, м), 2,22-2,32 (1,5H, м), 2,46-2,60 (1,5H, м), 2,74 (1H, м), 4,46 (0,5H, тт, J=9,4, 7,4 Гц), 4,79 (0,5H, тт, J=7,8, 7,4 Гц), 7,58 (0,5H, с), 7,61 (0,5H, с), 7,70 (0,5H, с), 7,73 (0,5H, с), 9,87 (0,5H, с), 9,88 (0,5H, с)
[Ссылочный пример 8] 1-(транс-4-Гидроксициклогексил)-1Н-имидазол-4-карбальдегид
[Стадия 1] [1-(транс-4-{[трет-бутил(диметил)силил]окси}циклогексил)-1Н-имидазол-4-ил]метанол
[Формула 52]
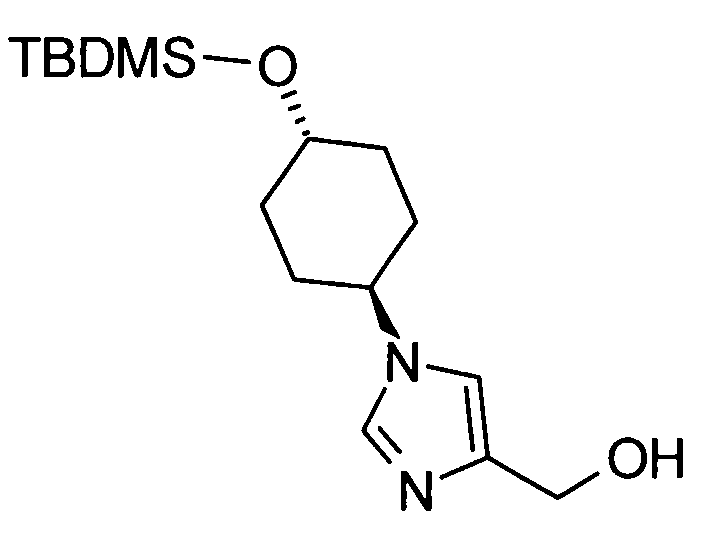
Этил-3-(диметиламино)-2-изоцианоакрилат (300 мг) и транс-4-{[трет-бутил(диметил)силил]окси}циклогексиламин (Synthetic Communications, 1990, Vol. 20, p. 1073) (1,02 г) смешивали и перемешивали при 85°С в течение 12 часов. К реакционному раствору добавляли насыщенный водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=4/1-1/1). Алюмогидрид лития (92%, 105 мг) суспендировали в тетрагидрофуране (8 мл). Полученный неочищенный продукт растворяли в тетрагидрофуране (6 мл), и полученный раствор медленно добавляли по каплям к суспензии при 0°С. После перемешивания при 0°С в течение 1 часа реакционный раствор разбавляли диэтиловым эфиром, и к нему добавляли насыщенный водный сульфат натрия. После перемешивания при комнатной температуре в течение 1 часа образовавшуюся неорганическую соль удаляли фильтрованием через целит. Фильтрат концентрировали при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт промывали смешанным растворителем из гексана и этилацетата (2:1), с получением указанного в заголовке соединения (260 мг).
1H-ЯМР (CD3OD) δ: 0,09 (6H, с), 0,90 (9H, с), 1,50 (2H, м), 1,81 (2H, м), 1,96-2,10 (4H, м), 3,76 (1H, м), 4,05 (1H, м), 4,48 (2H, с), 7,12 (1H, с), 7,64 (1H, с).
[Стадия 2] 1-(транс-4-{[трет-бутил(диметилсилил]окси}циклогексил-1Н-имидазол-4-карбальдегид
[Формула 53]
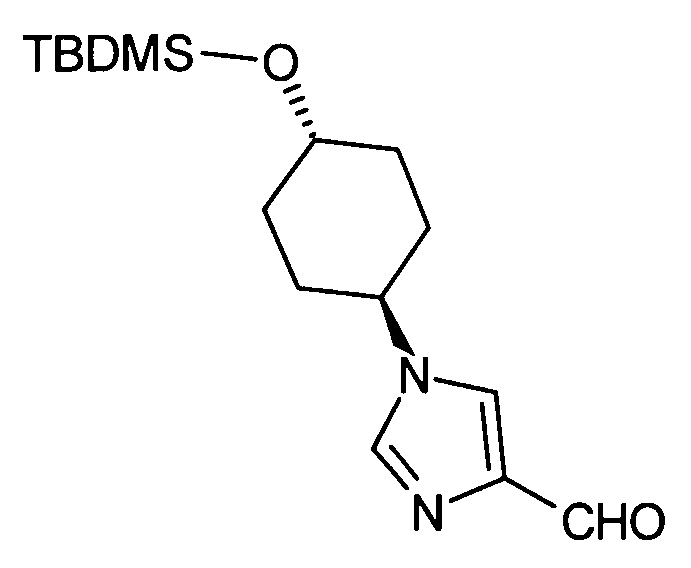
Соединение (260 мг), полученное на стадии 1 данного ссылочного примера, растворяли в толуоле (10 мл) и метиленхлориде (1 мл). К раствору добавляли раствор бикарбоната натрия (210 мг, 2,50 ммоль) в воде (8 мл), йод (370 мг) и 2,2,6,6-тетраметил-1-пиперидинилокси (15 мг) в указанном порядке, и смесь перемешивали при комнатной температуре в течение 12 часов. К реакционному раствору добавляли насыщенный водный тиосульфат натрия, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=2/1-1/1), с получением указанного в заголовке соединения (258 мг).
1H-ЯМР (CDCl3) δ: 0,08 (6H, с), 0,90 (9H, с), 1,52 (2H, м), 1,75 (2H, м), 2,02 (2H, м), 2,16 (2H, м), 3,68 (1H, м), 4,00 (1H, м), 7,62 (1H, с), 7,67 (1H, с), 9,87 (1H, с).
[Стадия 3] 1-(транс-4-гидроксициклогексил)-1Н-имидазол-4-карбальдегид
[Формула 54]
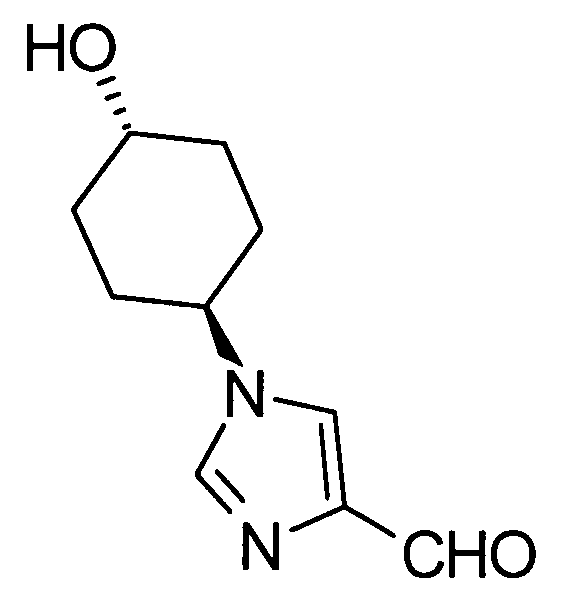
Соединение (540 мг), полученное на стадии 2 данного ссылочного примера, растворяли в тетрагидрофуране (8 мл). К полученному раствору добавляли раствор тетрабутиламмонийфторида в тетрагидрофуране (1,0 М, 2,62 мл), и смесь перемешивали при комнатной температуре в течение 8 часов. К реакционному раствору добавляли насыщенный водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали диол-связанной колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=1/1-этилацетат), с получением указанного в заголовке соединения (250 мг).
1H-ЯМР (CDCl3) δ: 1,52 (2H, м), 1,78 (2Н, м), 2,11-2,25 (4Н, м), 3,76 (1Н, м), 4,03 (1H, м), 7,63 (1H, с), 7,68 (1H, с), 9,87 (1H, с).
[Ссылочный пример 9] 1-(4-Гидрокси-4-метилциклогексил)-1Н-имидазол-4-карбальдегид
[Стадия 1] Бензил(4-гидрокси-4-метилциклогексил)карбамат
[Формула 55]
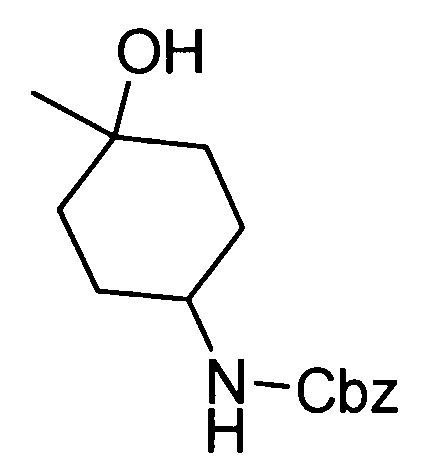
Бензил(4-оксоциклогексил)карбамат (2,00 г) растворяли в тетрагидрофуране (15 мл), и к нему добавляли хлорид церия (5,98 г). Реакционный раствор охлаждали до -78°С. Затем к нему добавляли раствор метиллития в диэтиловом эфире (1,6М, 15,2 мл), и смесь перемешивали при -78°С в течение 1 часа и при 0°С в течение 3 часов. К реакционному раствору добавляли насыщенный водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=9/1-2/1), с получением диастереомерной смеси указанного в заголовке соединения (1,31 г, транс:цис=3:7).
1H-ЯМР (CDCl3) δ: 1,23 (3H, с), 1,44-1,67 (6H, м), 1,81 (2H, м), 3,48 (1H, м), 4,65 (1H, м), 5,08 (2H, с), 7,29-7,41 (5H, м).
[Стадия 2] 1-(4-гидрокси-4-метилциклогексил)-1Н-имидазол-4-карбоксилат
[Формула 56]
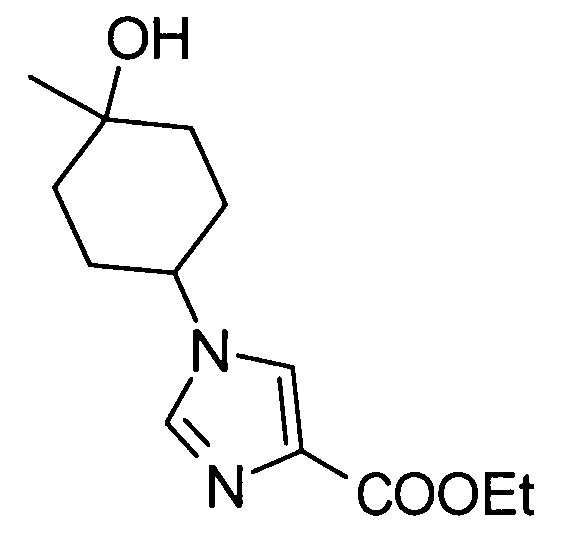
Соединение, полученное на стадии 1 данного ссылочного примера, растворяли в этаноле (12 мл). К раствору добавляли катализатор, 10% палладий на углероде (гидратированный, 400 мг), и смесь перемешивали при комнатной температуре в течение 15 часов в атмосфере водорода при нормальном давлении. После фильтрования через целит фильтрат концентрировали при пониженном давлении, с получением неочищенного продукта из 4-амино-1-метилциклогексанола. Полученный неочищенный продукт и этил-3-(диметиламино)-2-изоцианоакрилат (450 мг) смешивали и перемешивали при 75°С в течение 8 часов. К реакционному раствору добавляли насыщенный водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=1/1-этилацетат), с получением диастереомерной смеси указанного в заголовке соединения (462 мг, транс:цис=1:3).
1H-ЯМР (CDCl3) δ: 1,31 (2,25H, с), 1,34 (0,75H, с), 1,38 (3H, т, J=7,0 Гц), 1,52-1,70 (2H, м), 1,77-1,96 (4H, м), 2,08-2,19 (2H, м), 3,93 (0,75H, тт, J=12,2, 3,9 Гц), 4,06 (0,25H, м), 4,12 (0,5H, кв., J=7,0 Гц), 4,36 (1,5H, кв., J=7,0 Гц), 7,57 (1H, с), 7,68 (0,25H, с), 7,70 (0,75H, с).
[Стадия 3] 1-(4-гидрокси-4-метилциклогексил)-1Н-имидазол-4-карбальдегид
[Формула 57]
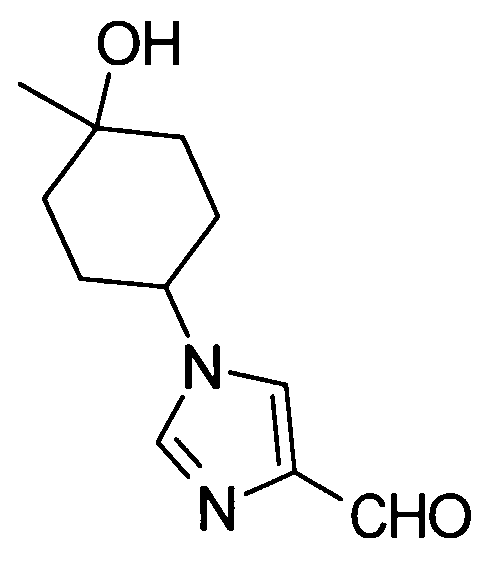
Алюмогидрид лития (92%, 60 мг) суспендировали в тетрагидрофуране (5 мл). К полученной суспензии раствор соединения (455 мг), полученного на стадии 2 данного ссылочного примера, в тетрагидрофуране (5 мл) медленно добавляли по каплям при 0°С. После перемешивания при 0°С в течение 4 часов и при комнатной температуре в течение 30 минут реакционный раствор разбавляли диэтиловым эфиром, и к нему добавляли насыщенный водный сульфат натрия. После перемешивания при комнатной температуре в течение 1 часа образовавшуюся неорганическую соль удаляли фильтрованием через целит. Фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт растворяли в метиленхлориде (8 мл) и хлороформе (4 мл), и к нему добавляли диоксид марганца (2,00 г). После перемешивания при комнатной температуре в течение 15 часов образовавшуюся неорганическую соль удаляли фильтрованием через целит. Фильтрат концентрировали при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=1/1-этилацетат), с получением диастереомерной смеси указанного в заголовке соединения (300 мг, транс:цис=1:3).
1H-ЯМР (CDCl3) δ: 1,32 (2,25H, с), 1,36 (0,75H, с), 1,54-1,73 (2H, м), 1,78-2,00 (4H, м), 2,11-2,23 (2H, м), 3,97 (0,75H, тт, J=12,2, 3,9 Гц), 4,10 (0,25H, м), 7,66 (1H, с), 7,72 (0,25H, с), 7,75 (0,75H, с), 9,86 (0,75H, с), 9,87 (0,25H, с).
[Ссылочный пример 10] 1-[экзо-бицикло[2.2.1]гепт-2-ил]-1Н-имидазол-4-карбальдегид
[Стадия 1] Этил-1-[экзо-бицикло[2.2.1]гепт-2-ил]-1Н-имидазол-4-карбоксилат
[Формула 58]
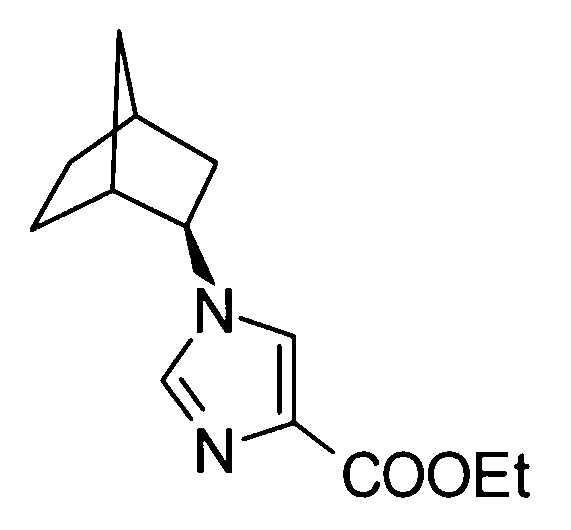
Этил-3-(диметиламино-2-изоцианоакрилат (0,58 г) растворяли в экзо-2-аминонорборнане (0,46 г), и раствор перемешивали при 150°С в течение 1,5 часов. Реакционный раствор очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метиленхлорид-метиленхлорид/метанол=95/5 и этилацетат), с получением указанного в заголовке соединения (0,50 г).
1H-ЯМР (CDCl3) δ: 1,22-1,37 (3H, м), 1,38 (3H, т, J=7,1 Гц), 1,56-1,65 (2H, м), 1,65-1,73 (1H, м), 1,75-1,82 (1H, м), 1,97-2,04 (1H, м), 2,48 (1H, м), 2,52-2,55 (1H, м), 4,04-4,09 (1H, м), 4,37 (2H, кв., J=7,1 Гц), 7,57 (1H, с), 7,67 (1H, с).
[Стадия 2] 1-[экзо-бицикло[2.2.1]гепт-2-ил]-1Н-имидазол-4-карбальдегид
[Формула 59]
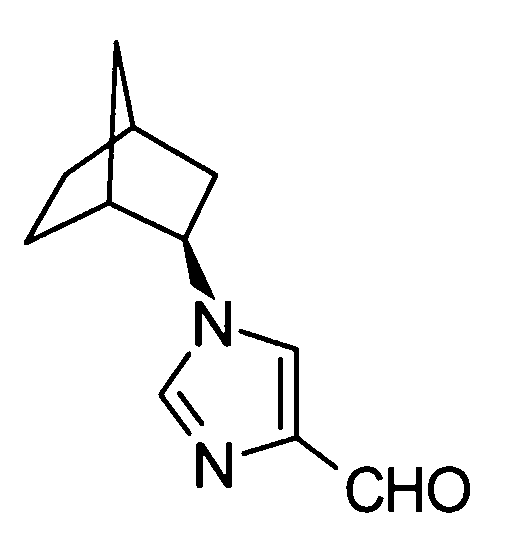
Указанное в заголовке соединение (0,21 г) получали из соединения (0,50 г), полученного на стадии 1 данного ссылочного примера, таким же образом, как на стадиях 2 и 3 ссылочного примера 4.
1H-ЯМР (CDCl3) δ: 1,23-1,41 (3H, м), 1,56-1,66 (2H, м), 1,67-1,75 (1H, м), 1,75-1,82 (1H, м), 2,01-2,07 (1H, м), 2,49 (1H, м), 2,53-2,57 (1H, м), 4,08-4,12 (1H, м), 7,63 (1H, с), 7,69 (1H, с), 9,87 (1H, с).
[Ссылочный пример 11] 1-[эндо-бицикло[2.2.1]гепт-2-ил]-1Н-имидазол-4-карбальдегид
[Стадия 1] Этил-1-[эндо-бицикло[2.2.1]гепт-2-ил]-1Н-имидазол-4-карбоксилат
[Формула 60]
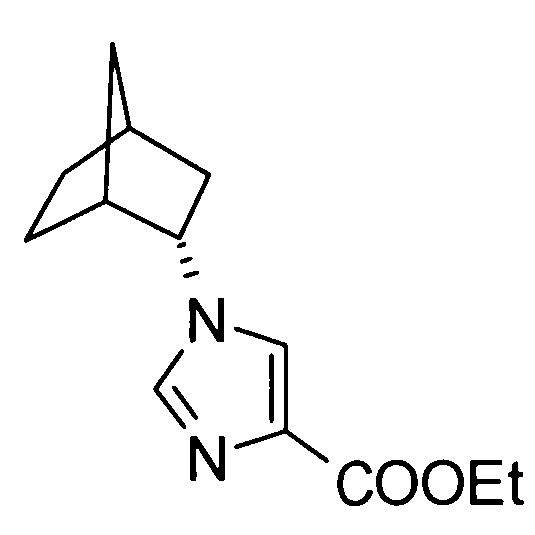
Этил-3-(диметиламино)-2-изоцианоакрилат (0,58 г) и гидрохлорид эндо-2-аминонорборнана (0,61 г) растворяли в н-бутаноле (5,8 мл). Затем к раствору добавляли триэтиламин (0,58 мл) при комнатной температуре, и смесь перемешивали при 150°С в течение 6,5 часов. Реакционный раствор концентрировали и затем очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=50/50-этилацетат), с получением указанного в заголовке соединения (0,13 г).
1H-ЯМР (CDCl3) δ: 1,19-1,71 (7H, м), 1,40 (3H, т, J=7,1 Гц), 2,19-2,27 (1H, м), 2,42 (1H, м), 2,60 (1H, м), 4,38 (2H, кв., J=7,1 Гц), 4,44-4,49 (1H, м), 7,54 (1H, с), 7,65 (1H, с).
[Стадия 2] 1-[эндо-бицикло[2.2.1]гепт-2-ил]-1Н-имидазол-4-карбальдегид
[Формула 61]
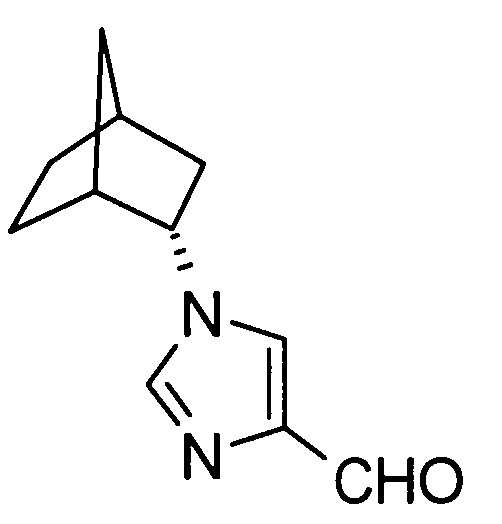
Указанное в заголовке соединение (0,17 г) получали из соединения (0,42 г), полученного на стадии 1 данного ссылочного примера, таким же образом, как на стадиях 2 и 3 ссылочного примера 4.
1H-ЯМР (CDCl3) δ: 1,18-1,25 (1H, м), 1,30-1,37 (1H, м), 1,44-1,73 (5H, м), 2,22-2,30 (1H, м), 2,45 (1H, м), 2,62 (1H, м), 4,47-4,53 (1H, м), 7,61 (1H, с), 7,68 (1H, с), 9,89 (1H, с).
[Ссылочный пример 12] 1-Адамантан-2-ил-1Н-имидазол-4-карбальдегид
[Стадия 1] Этил-1-адамантан-2-ил-1Н-имидазол-4-карбоксилат
[Формула 62]
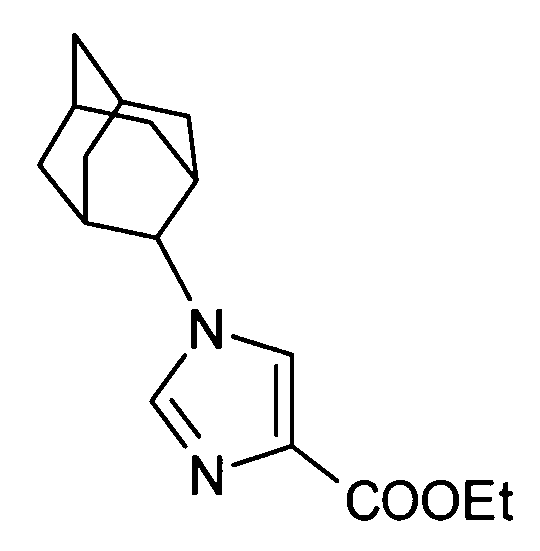
Этил-3-(диметиламино)-2-изоцианоакрилат (0,50 г), 2-аминоадамантан (0,54 г) и н-бутанол (2,5 мл) добавляли и перемешивали при 150°С в течение 13 часов. К реакционному раствору добавляют воду, и органическое вещество экстрагировали этилацетатом. Органический слой концентрировали и затем очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метиленхлорид-метиленхлорид/метанол=95/5 и этилацетат), с получением указанного в заголовке соединения (0,24 г).
1H-ЯМР (CDCl3) δ: 1,40 (3H, т, J=7,2 Гц), 1,61-2,08 (12H, м), 2,52 (2H, м), 4,20 (1H, м), 4,38 (2H, кв., J=7,2 Гц), 7,67 (1H, с), 7,76 (1H, с).
[Стадия 2] 1-адамантан-2-ил-1Н-имидазол-4-карбальдегид
[Формула 63]
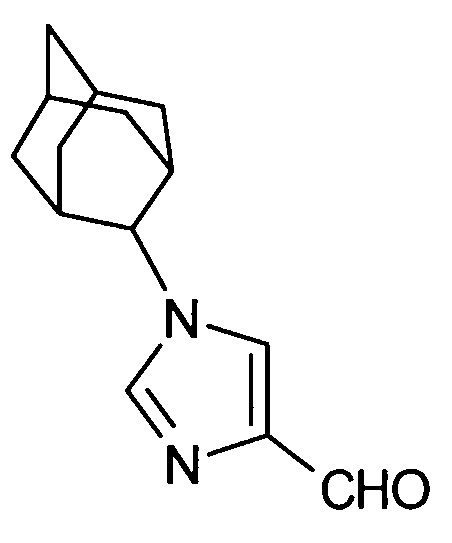
Указанное в заголовке соединение (0,15 г) получали из соединения (0,37 г), полученного на стадии 1 данного ссылочного примера, таким же образом, как на стадиях 2 и 3 ссылочного примера 4.
1H-ЯМР (CDCl3) δ: 1,49-2,10 (12H, м), 2,53 (2H, м), 4,24 (1H, м), 7,74 (1H, с), 7,80 (1H, с), 9,90 (1H, с).
[Ссылочный пример 13] 1-(транс-4-Феноксициклогексил)-1Н-имидазол-4-карбальдегид
[Стадия 1] Трет-бутил(транс-4-феноксициклогексил)карбамат
[Формула 64]
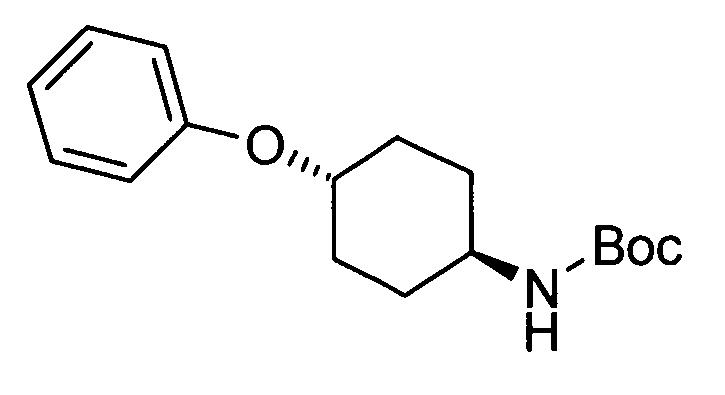
Трет-бутил(цис-4-гидроксициклогексил)карбамат (2,00 г), фенол (1,14 г) и трифенилфосфин (3,17 г) растворяли в тетрагидрофуране (40,0 мл). Затем к раствору по каплям добавляли диизопропилазодикарбоксилат (6,49 мл) при комнатной температуре, и смесь перемешивали при комнатной температуре в течение 63 часов. Реакционный раствор концентрировали и затем очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан-гексан/этилацетат=90/10), с получением указанного в заголовке соединения (1,80 г).
1H-ЯМР (CDCl3) δ: 1,20-1,30 (2H, м), 1,45 (9H, с), 1,51-1,61 (2H, м), 2,05-2,16 (4H, м), 3,47-3,58 (1H, м), 4,17 (1H, м), 6,81-6,95 (3H, м), 7,21-7,29 (2H, м).
[Стадия 2] Гидрохлорид транс-4-феноксициклогексанамина
[Формула 65]
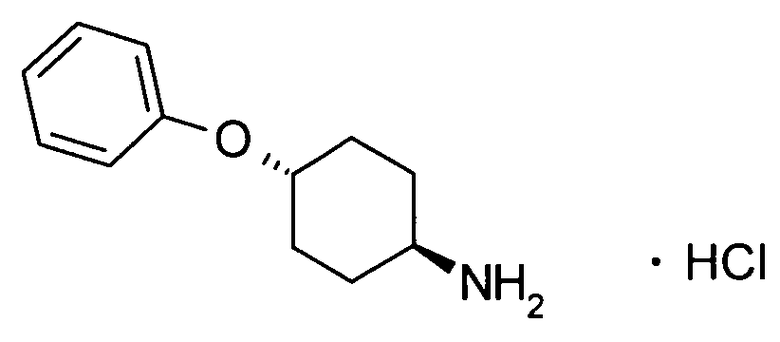
Соединение (1,80 г), полученное на стадии 1 данного ссылочного примера, растворяли в этилацетате (18,0 мл). К полученному раствору добавляли смесь 4М хлористоводородная кислота/этилацетат (18,0 мл) при комнатной температуре, и смесь перемешивали в течение 1 часа. К реакционному раствору добавляли гексан (18,0 мл), и осажденное твердое вещество затем отделяли фильтрованием и промывали смешанным растворителем из гексана и этилацетата (50:50), с получением указанного в заголовке соединения (1,01 г).
1H-ЯМР (CD3OD) δ: 1,48-1,61 (4H, м), 2,07-2,14 (2H, м), 2,18-2,25 (2H, м), 3,13-3,21 (1H, м), 4,28 (1H, м), 6,87-6,94 (3H, м), 7,21-7,28 (2H, м).
[Стадия 3] Этил-1-(транс-4-феноксициклогексил)-1Н-имидазол-4-карбоксилат
[Формула 66]
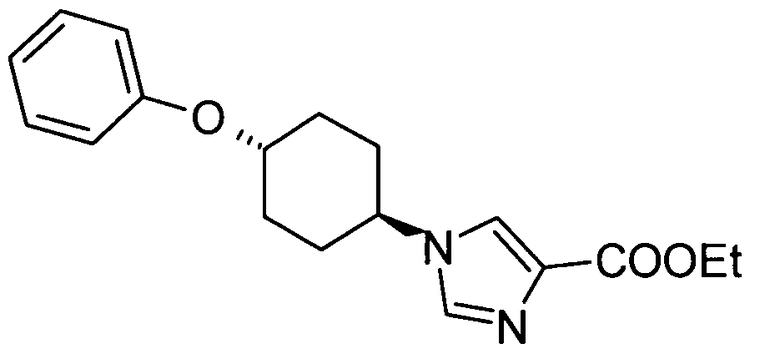
Этил-3-(диметиламино)-2-изоцианоакрилат (0,70 г) и соединение (1,14 г), полученное на стадии 2 данного ссылочного примера, растворяли в н-бутаноле (7,0 мл). Затем к раствору добавляли триэтиламин (0,70 мл) при комнатной температуре, и смесь перемешивали при 150°С в течение 3,25 часов. Реакционный раствор концентрировали и затем очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=50/50-этилацетат), с получением указанного в заголовке соединения (0,28 г).
1H-ЯМР (CDCl3) δ: 1,39 (3H, т, J=7,1 Гц), 1,57-1,71 (2H, м), 1,80-1,90 (2H, м), 2,22-2,37 (4H, м), 4,08 (1H, м), 4,29 (1H, м), 4,37 (2H, кв., J=7,1 Гц), 6,85-7,00 (3H, м), 7,26-7,32 (2H, м), 7,59 (1H, с), 7,69 (1H, с).
[Стадия 4] 1-(транс-4-феноксициклогексил)-1Н-имидазол-4-карбальдегид
[Формула 67]
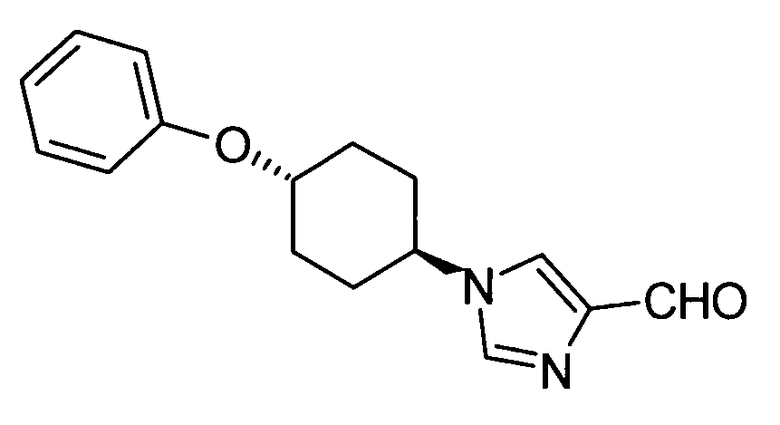
Указанное в заголовке соединение (0,07 г) было получено из соединения (0,28 г), полученного на стадии 3, таким же образом, как на стадиях 2 и 3 ссылочного примера 4.
1H-ЯМР (CDCl3) δ: 1,62-1,73 (2H, м), 1,80-1,91 (2H, м), 2,24-2,38 (4H, м), 4,11 (1H, м), 4,30 (1H, м), 6,88-7,01 (3H, м), 7,26-7,33 (2H, м), 7,66 (1H, с), 7,71 (1H, с), 9,88 (1H, с).
[Ссылочный пример 14] Этил-5-[(трет-бутоксикарбонил)амино]-2-(диэтоксифосфорил)валерат
[Формула 68]
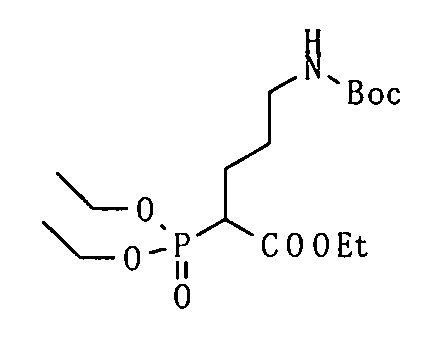
Указанное в заголовке соединение (14,1 г) синтезировали из триэтилфосфоноацетата (10 г) таким же образом, как в ссылочном примере 1.
[Ссылочный пример 15] 1-(3,3-Диметилциклогексил)-1Н-имидазол-4-карбальдегид
[Стадия 1] Этил-1-(3,3-диметилциклогексил)-1Н-имидазол-4-карбоксилат
[Формула 69]
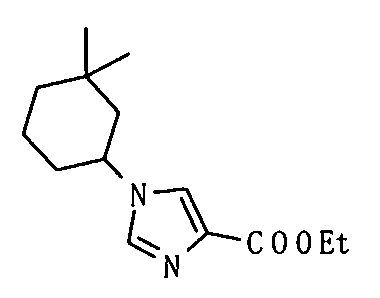
Гидрохлорид гидроксиламина (8,76 г) растворяли в воде (100 мл). К раствору при комнатной температуре добавляли раствор ацетата натрия (17,8 г) и 3,3-диметилциклогексанона (4,55 г) в метаноле (20 мл), и смесь нагревали при кипячении с обратным холодильником в течение 1,5 часов. Органическое вещество экстрагировали этилацетатом и сушили над безводным сульфатом натрия, и растворитель затем отгоняли при пониженном давлении, с получением неочищенного продукта оксима 3,3-диметилциклогексанона.
Алюмогидрид лития (4,11 г) суспендировали в тетрагидрофуране (100 мл). К суспензии по каплям при охлаждении льдом добавляли раствор неочищенного продукта оксима 3,3-диметилциклогексанона, полученного таким образом, в тетрагидрофуране (50 мл), и смесь затем нагревали при кипячении с обратным холодильником в течение 10,5 часов. К реакционному раствору при охлаждении льдом добавляли декагидрат сульфата натрия. Затем к нему добавляли этилацетат, и смесь перемешивали в течение 30 минут. После фильтрования через целит растворитель в фильтрате отгоняли при пониженном давлении, с получением неочищенного продукта 3,3-диметилциклогексиламина.
Полученный неочищенный продукт и этил-3-(диметиламино)-2-изоцианоакрилат (3,04 г) смешивали и перемешивали при 70°С в течение 16 часов. Полученную смесь очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=1/1-1/3), с получением указанного в заголовке соединения (3,51 г).
[Стадия 2] 1-(3,3-диметилциклогексил)-1Н-имидазол-4-карбальдегид
[Формула 70]
Указанное в заголовке соединение (1,23 г) получали из соединения (3,51 г), полученного на стадии 1 данного ссылочного примера, таким же образом, как на стадиях 2 и 3 ссылочного примера 4.
1H-ЯМР (CDCl3) δ: 0,92-0,96 (1H, м), 1,03 (6H, с), 1,18-1,26 (1H, м), 1,46-1,68 (3H, м), 1,76-1,85 (2H, м), 2,11-2,17 (1H, м), 4,11-4,19 (1H, м), 7,62 (1H, с), 7,68 (1H, с), 9,86 (1H, с).
[Ссылочный пример 16] Трет-бутил(2-формилбутил)карбамат
[Стадия 1] Этил-2-метиленбутират
[Формула 71]
Карбонат калия (5,5 г) растворяли в воде (15 мл). К раствору при комнатной температуре добавляли этил-2-(диэтоксифосфорил)бутират (5,0 г) и 37% водный формальдегид (6,2 г), и смесь перемешивали при 85°С в течение 45 минут. Органическое вещество экстрагировали диэтиловым эфиром, сушили над безводным сульфатом натрия и фильтровали, и растворитель в фильтрате отгоняли при пониженном давлении, с получением неочищенного продукта.
1H-ЯМР (CDCl3) δ: 1,08 (3H, т, J=7,4 Гц), 1,31 (3H, т, J=7,0 Гц), 2,30-2,36 (2H, м), 4,21 (2H, кв., J=7,0 Гц), 5,51-5,52 (1H, м), 6,12-6,14 (1H, м).
[Стадия 2] Этил-2-[(бензиламино)метил]бутират
[Формула 72]
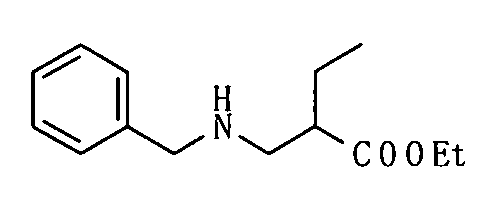
Соединение, полученное на стадии 1, растворяли в этаноле (7 мл). К раствору при комнатной температуре добавляли бензиламин (2,7 мл), и смесь перемешивали при 70°С в течение 17 часов. Растворитель в реакционном растворе отгоняли при пониженном давлении, и полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан-гексан/этилацетат=7/3), с получением указанного в заголовке соединения (2,34 г).
1H-ЯМР (CDCl3) δ: 0,91 (3H, т, J=7,4 Гц), 1,26 (3H, т, J=7,2 Гц), 1,53-1,70 (2H, м), 2,47-2,55 (1H, м), 2,69 (1H, дд, J=11,9, 4,9 Гц), 2,88 (1H, дд, J=11,9, 8,8 Гц), 3,79 (2H, д, J=4,3 Гц), 4,13-4,19 (2H, м), 7,22-7,26 (2H, м), 7,29-7,32 (3H, м).
[Стадия 3] Этил-2-{[(трет-бутоксикарбонил)амино]метил}бутират
[Формула 73]
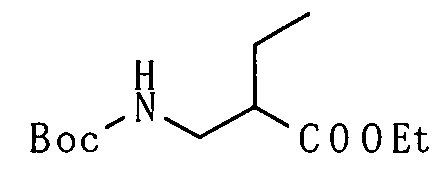
Соединение (2,34 г), полученное на стадии 2, растворяли в этаноле (50 мл). К раствору добавляли катализатор 10% палладий-углерод (гидратированный, 1,17 г), и смесь перемешивали в течение 4 часов в атмосфере водорода. Затем к нему добавляли ди-трет-бутилдикарбонат (2,6 г), и смесь перемешивали в течение 1 часа. Катализатор отфильтровывали, и растворитель в фильтрате отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан-гексан/этилацетат=8/2), с получением указанного в заголовке соединения (1,97 г).
1H-ЯМР (CDCl3) δ: 0,94 (3H, т, J=7,4 Гц), 1,27 (3H, т, J=7,4 Гц), 1,43 (9H, с), 1,49-1,71 (2H, м), 2,48-2,56 (1H, м), 3,21-3,28 (1H, м), 3,32-3,39 (1H, м), 4,11-4,20 (3H, м), 4,86 (1H, шир.с).
[Стадия 4] Этил-2-{[бис(трет-бутоксикарбонил)амино]метил}бутират
[Формула 74]
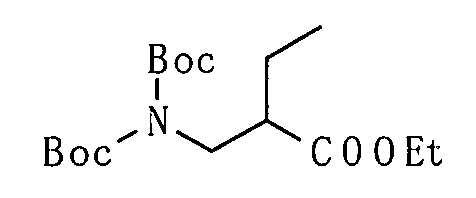
К раствору соединения (578 мг), полученному на стадии 3, в тетрагидрофуране (15 мл) добавляли раствор н-BuLi в гексане (1,65М, 1,57 мл) при -78°С, и смесь перемешивали в течение 1 часа. Затем к ней добавляли ди-трет-бутилдикарбонат (668 мг) при -78°C, и смесь постепенно нагревали и затем перемешивали в течение ночи. К реакционному раствору добавляли водный хлорид аммония, и органическое вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и фильтровали. Растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=98/2-90/10), с получением указанного в заголовке соединения (684 мг).
[Стадия 5] Трет-бутил[2-гидроксиметил)бутил]карбамат
[Формула 75]
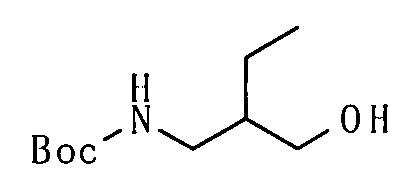
Алюмогидрид лития (153 мг) суспендировали в тетрагидрофуране (20 мл). К суспензии по каплям при охлаждении льдом добавляли раствор соединения, полученного на стадии 4, в тетрагидрофуране (2 мл), и смесь затем перемешивали в течение ночи. К реакционному раствору при охлаждении льдом добавляли декагидрат сульфата натрия. Затем к нему добавляли этилацетат, и смесь перемешивали. После фильтрования через целит, растворитель в фильтрате отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=9/1-3/7), с получением указанного в заголовке соединения (168 мг).
1H-ЯМР (CDCl3) δ: 0,93 (3H, т, J=7,4 Гц), 1,19-1,37 (2H, м), 1,45 (9H, с), 3,06-3,13 (1H, м), 3,28-3,36 (2H, м), 3,37-3,44 (1H, м), 3,56-3,62 (1H, м), 4,78 (1H, шир.с).
[Стадия 6] Трет-бутил(2-формилбутил)карбамат
[Формула 76]
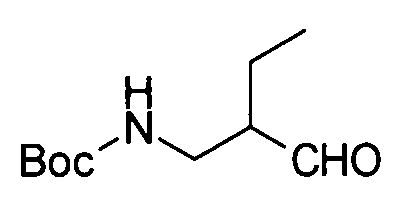
Оксалилхлорид (141 мкл) растворяли в метиленхлориде (1 мл). К раствору по каплям при -78°С добавляли раствор диметилсульфоксида (176 мкл) в метиленхлориде (1 мл), и смесь перемешивали в течение 15 минут. К нему по каплям при -78°С добавляли раствор соединения (168 мг), полученного на стадии 5, в метиленхлориде (2 мл), и смесь перемешивали в течение 2 часов. К нему добавляли триэтиламин (695 мкл), и смесь нагревали до 0°С и затем перемешивали в течение ночи. К реакционному раствору добавляли метиленхлорид, и органический слой промывали водой и насыщенным раствором хлорида натрия, затем сушили над безводным сульфатом натрия и фильтровали. Растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=9/1-7/3), с получением указанного в заголовке соединения (113 мг).
1H-ЯМР (CDCl3) δ: 1,02 (3H, т, J=7,8 Гц), 1,42 (9H, с), 1,48-1,54 (1H, м), 1,70-1,81 (1H, м), 2,43-2,51 (1H, м), 3,27-3,40 (2H, м), 4,82 (1H, шир.с), 9,68-9,69 (1H, м).
[Ссылочный пример 17] 1-(цис-4-{[трет-бутил(дифенил)силил]окси}циклогексил)-1Н-имидазол-4-карбальдегид
[Стадия 1] Трет-бутил(цис-4-{[трет-бутил(дифенил)силил]окси}циклогексил)карбамат
[Формула 77]
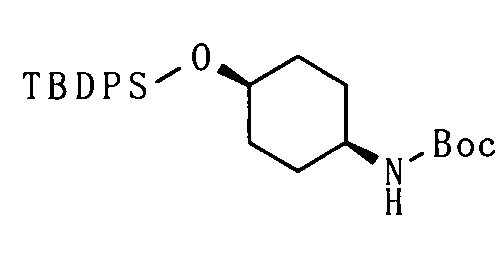
К раствору трет-бутил(цис-4-гидроксициклогексил)карбамата (2,0 г) в диметилформамиде (40 мл) при охлаждении льдом добавляли имидазол (756 мг) и трет-бутилдифенилхлорсилан (2,86 мл), и смесь перемешивали в течение 24 часов. К ней дополнительно добавляли имидазол (226 мг) и трет-бутилдифенилхлорсилан (858 мкл), и смесь перемешивали в течение 6 дней. К реакционному раствору добавляли этилацетат, и органический слой промывали три раза 10% раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Растворитель отгоняли при пониженном давлении. Этот остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=98/2-9/1), с получением указанного в заголовке соединения (5,09 г).
1H-ЯМР (CDCl3) δ: 1,07 (9H, с), 1,45 (9H, с), 1,57-1,71 (8H, м), 3,40-3,49 (1H, м), 3,88-3,92 (1H, м), 4,50-4,57 (1H, м), 7,34-7,44 (6H, м), 7,64-7,66 (4H, м).
[Стадия 2] цис-4-{[трет-бутил(дифенил)силил]окси}циклогексанамин
[Формула 78]
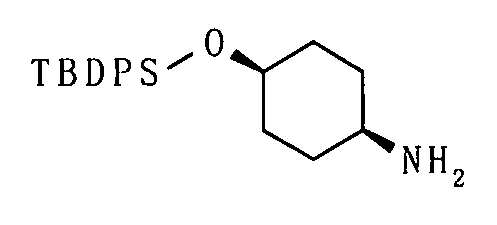
Соединение, полученное на стадии 2, растворяли в метиленхлориде (25 мл). К раствору при охлаждении льдом добавляли трифторуксусную кислоту (5 мл), и смесь перемешивали в течение 45 минут. К ней при охлаждении льдом дополнительно добавляли трифторуксусную кислоту (5 мл), и смесь перемешивали в течение 1 часа. Органический слой промывали водным карбонатом калия, сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением нечищеного продукта указанного в заголовке соединения (4,17 г).
[Стадия 3] Этил-1-(цис-4-{[трет-бутил(дифенил)силил]окси}циклогексил)-1Н-имидазол-4-карбоксилат
[Формула 79]
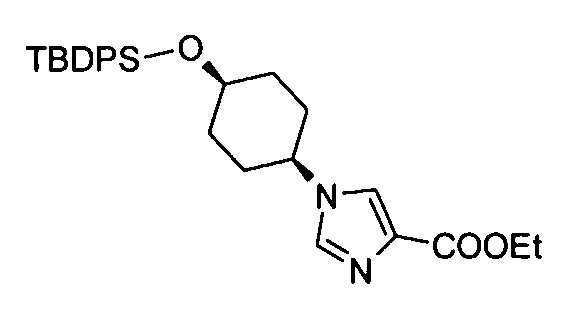
Соединение, полученное на стадии 2, и этил-3-(диметиламино)-2-изоцианоакрилат (1,56 г) смешивали и перемешивали при 70°С в течение 33 часов. Полученную смесь очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=8/2-этилацетат), с получением указанного в заголовке соединения (870 мг).
[Стадия 4] 1-(цис-4-{[трет-бутил(дифенил)силил]окси}циклогексил)-1Н-имидазол-4-карбальдегид
[Формула 80]
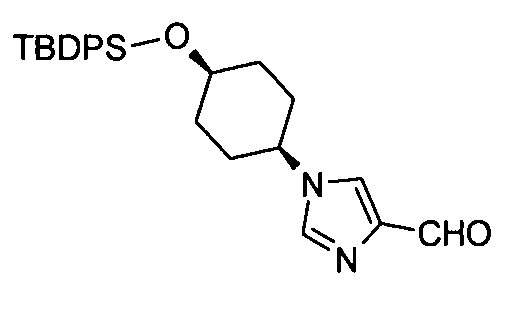
Указанное в заголовке соединение (370 мг) получали из соединения, полученного на стадии 3 данного ссылочного примера, таким же образом, как на стадиях 2 и 3 ссылочного примера 4.
1H-ЯМР (CDCl3) δ: 1,11 (9H, с), 1,42-1,49 (2H, м), 1,81-1,93 (4H, м), 2,24-2,32 (2H, м), 3,95-4,01 (1H, м), 4,07-4,10 (1H, м), 7,37-7,41 (4H, м), 7,43-7,47 (2H, м), 7,65-7,67 (5H, м), 7,75 (1H, с), 9,90 (1H, с).
[Ссылочный пример 18] 1-(цис-4-метилциклогексил)-1Н-имидазол-4-карбальдегид
[Стадия 1] Этил-1-(цис-4-метилциклогексил)-1Н-имидазол-4-карбоксилат
[Формула 81]
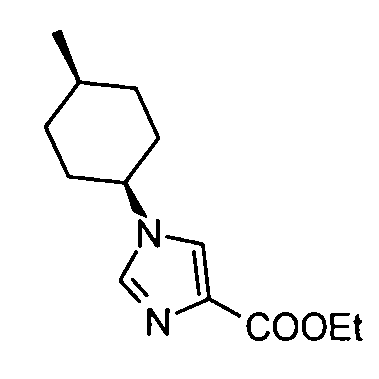
К гидрохлориду цис-4-метилциклогексиламина (5,0 г) добавляли воду и бикарбонат натрия, и органический слой отделяли. Органический слой сушили над безводным сульфатом натрия, и растворитель отгоняли, с получением свободной формы цис-4-метилциклогексиламина (770 мг). К водному слою, полученному выше, дополнительно добавляли 5 н. хлористоводородную кислоту. К нему добавляли PoraPak Rxn CX (ионообменная смола, 30 г), и смесь оставляли при комнатной температуре. Смолу промывали деионизированной водой, с последующим элюированием раствором 0,4н. аммиак/метанол. Элюат концентрировали, с получением свободной формы цис-4-метилциклогексиламина (1,01 г). Полученные свободные формы объединяли (1,78 г) и подвергали взаимодействию таким же образом, как на стадии 1 ссылочного примера 4, с получением указанного в заголовке соединения (1,67 г).
[Стадия 2] 1-(цис-4-метилциклогексил)-1Н-имидазол-4-карбальдегид
[Формула 82]
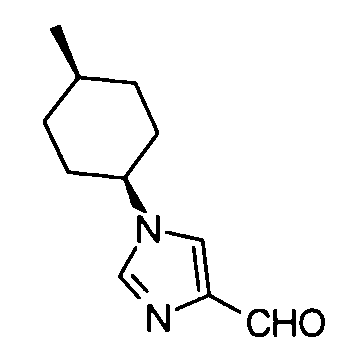
Алюмогидрид лития (0,35 г) суспендировали в тетрагидрофуране (10 мл). К суспензии по каплям при охлаждении льдом добавляли раствор соединения (1,67 г), полученного на стадии 1 данного ссылочного примера, в тетрагидрофуране (10 мл). Смесь перемешивали при 0°С в течение 30 минут, затем при комнатной температуре в течение 2 часов и 40 минут, и к ней при охлаждении добавляли воду (2 мл), 5 н. водный гидроксид натрия (2 мл) и воду (6 мл) в указанном порядке. Смесь перемешивали при комнатной температуре в течение 2 часов. Затем к ней добавляли безводный сульфат натрия, и смесь фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток растворяли в метиленхлориде (20 мл). К раствору добавляли диоксид марганца (21,6 г), и смесь перемешивали при комнатной температуре в течение 17 часов и затем фильтровали через целит. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=50/50-20/80), с получением указанного в заголовке соединения (0,79 г).
1H-ЯМР (CDCl3) δ: 1,00 (3H, д, J=7,0 Гц), 1,45-1,52 (2H, м), 1,64-1,73 (3H, м), 1,85-2,07 (4H, м), 4,06-4,13 (1H, м), 7,67 (1H, д, J=1,2 Гц), 7,74 (1H, д, J=1,2 Гц), 9,89 (1H, с).
[Пример 1] 5-Амино-2-[(1-циклогексил-1Н-имидазол-4-ил)метил]валериановая кислота
[Стадия 1] Трет-бутил-5-[(трет-бутоксикарбонил)амино]-2-[(1-циклогекс-2-ен-1-ил-1Н-имидазол-4-ил)метил]валерат
[Формула 83]
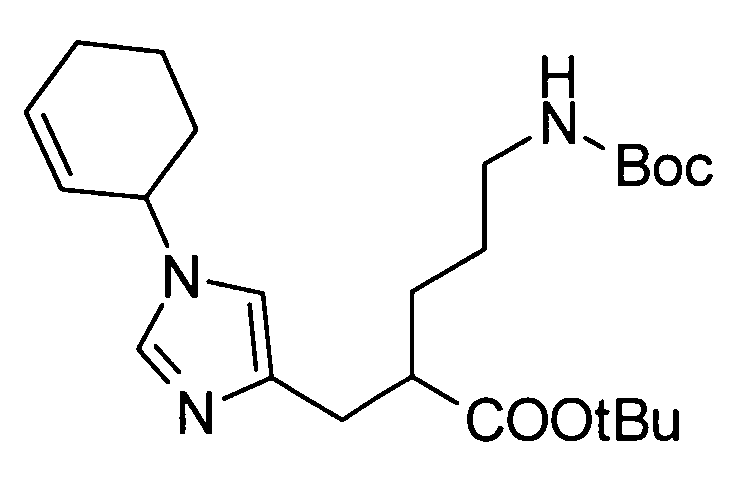
Соединение (200 мг), полученное в ссылочном примере 2, растворяли в N,N-диметилформамиде (3 мл), и к нему при 0°С добавляли гидрид натрия (63%, 43 мг). После перемешивания при 0°С в течение 15 минут и при комнатной температуре в течение 45 минут, к смеси при 0°С добавляли 3-бромциклогексен (90%, 0,150 мл), и смесь перемешивали при комнатной температуре в течение 30 минут. К реакционному раствору добавляли насыщенный водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой промывали водой, затем сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метиленхлорид-метиленхлорид/метанол=10/1), с получением указанного в заголовке соединения (220 мг).
1H-ЯМР (CDCl3) δ: 1,39 (9H, с), 1,44 (9H, с), 1,47-2,15 (10H, м), 2,60-2,70 (2H, м), 2,85 (1H, м), 3,02-3,18 (2H, м), 4,61 (1H, м), 4,76 (1H, шир.), 5,70 (1H, м), 6,05 (1H, м), 6,68 (1H, с), 7,42 (1H, с).
[Стадия 2] Трет-бутил-5-[(трет-бутоксикарбонил)амино]-2-[(1-циклогексил-1Н-имидазол-4-ил)метил]валерат
[Формула 84]
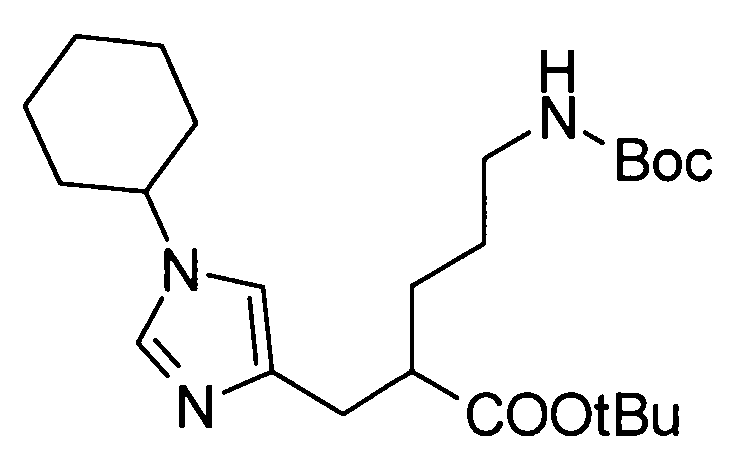
Катализатор 10% палладий-углерод (гидратированный, 200 мг) суспендировали в растворе соединения (250 мг), полученного на стадии 1 данного примера, в этаноле (6 мл). Суспензию перемешивали при комнатной температуре в течение 3 часов в атмосфере водорода при нормальном давлении. Реакционный раствор фильтровали через целит, и фильтрат концентрировали. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метиленхлорид/метанол=20-1-10/1), с получением указанного в заголовке соединения (240 мг).
1H-ЯМР (CDCl3) δ: 1,19-1,36 (4H, м), 1,38 (9H, с), 1,44 (9H, с), 1,48-1,64 (5H, м), 1,73 (1H, м), 1,88 (2H, м), 2,06 (2H, м), 2,59-2,70 (2H, м), 2,84 (1H, м), 3,05-3,16 (2H, м), 3,81 (1H, м), 4,76 (1H, шир.), 6,68 (1H, с), 7,42 (1H, с).
[Стадия 3] 5-амино-2-[(1-циклогексил-1Н-имидазол-4-ил)метил]валериановая кислота
[Формула 85]
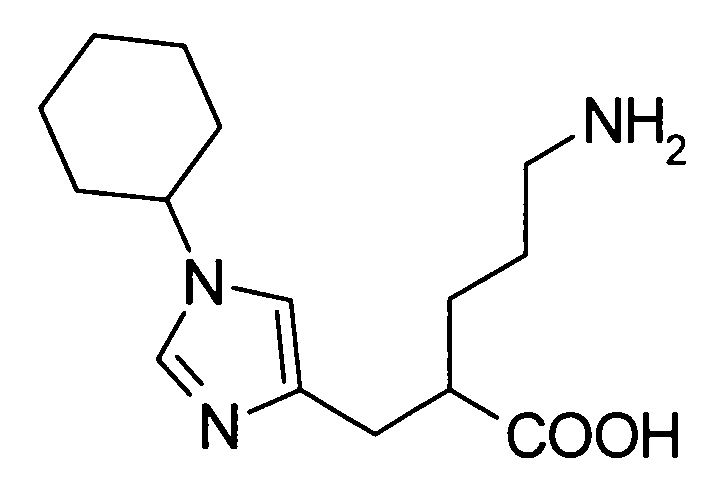
Соединение (100 мг), полученное на стадии 2 данного примера, растворяли в тетрагидрофуране (1 мл), и к нему добавляли 2 н. хлористоводородную кислоту (5 мл). После нагревания при кипячении с обратным холодильником в течение 2,5 часов, растворитель отгоняли при пониженном давлении. Полученный неочищенный гидрохлорид растворяли в воде, и к нему добавляли DOWEX 50WX8-200. Смолу промывали водой с последующим элюированием 4% аммиачной водой. Элюат концентрировали, с получением указанного в заголовке соединения (7,0 мг).
1H-ЯМР (CD3OD) δ: 1,23-1,75 (10H, м), 1,87 (2H, м), 2,04 (2H, м), 2,46-2,59 (2H, м), 2,84-2,95 (3H, м), 3,95 (1H, м), 6,95 (1H, с), 7, 57 (1H, с).
МСВР (ESI): m/z вычислено для C15H25N3NaO2: 302,1845 [M+Na]+; найдено: 302,1835.
[Пример 2] 5-Амино-2-{[(1-(транс-4-метилциклогексил-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] Трет-бутил-5-[(трет-бутоксикарбонил)амино]-2-{[(1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерат
[Формула 86]
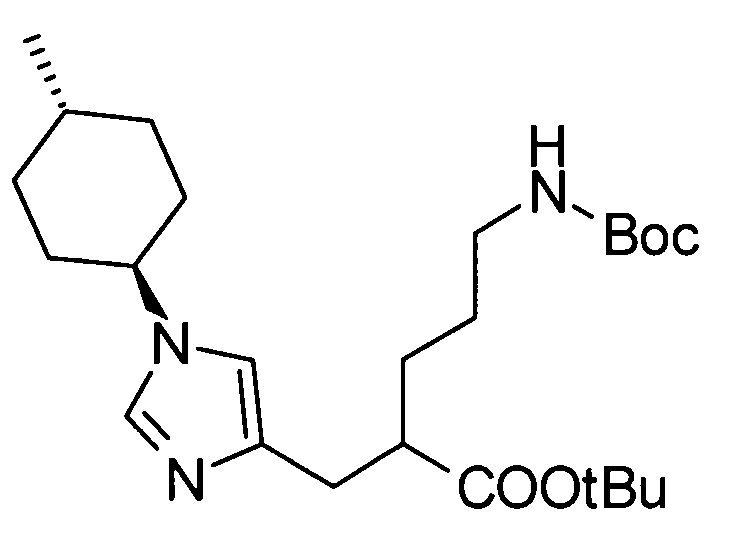
Соединение (970 мг), полученное в ссылочном примере 1, растворяли в ацетонитриле (7 мл), и к нему добавляли хлорид лития (100 мг). После перемешивания при комнатной температуре в течение 1 часа, к смеси добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (0,38 мл). После дополнительного перемешивания при комнатной температуре в течение 30 минут, к ней добавляли раствор соединения (350 мг), полученного в ссылочном примере 4, в ацетонитриле (4 мл), и смесь перемешивали при комнатной температуре в течение 14 часов. Растворитель отгоняли при пониженном давлении. Затем к остатку добавляли воду, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Полученный неочищенный продукт растворяли в этаноле (10 мл). К раствору добавляли катализатор 10% палладий-углерод (гидратированный, 200 мг), и смесь перемешивали при комнатной температуре в течение 9 часов в атмосфере водорода при нормальном давлении. После фильтрования через целит, фильтрат концентрировали при пониженном давлении, с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метанол-метиленхлорид/метанол=20/1), с получением указанного в заголовке соединения (435 мг).
1H-ЯМР (CDCl3) δ: 0,94 (3H, д, J=6,3 Гц), 1,05-1,14 (2H, м), 1,38 (9H, с), 1,41-1,68 (7H, м), 1,44 (9H, с), 1,81-1,87 (2H, м), 2,03-2,08 (2H, м), 2,60-2,69 (2H, м), 2,84 (1H, м), 3,05-3,15 (2H, м), 3,78 (1H, тт, J=11,7, 3,9 Гц), 4,73 (1H, шир.), 6,67 (1H, с), 7,40 (1H, с).
[Стадия 2] 5-амино-2-{[(1-(транс-4-метилциклогексил-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 87]
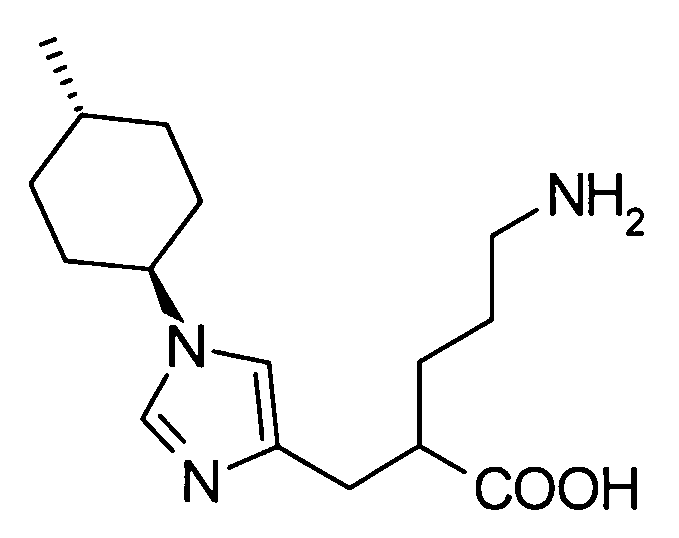
К соединению (430 мг), полученному на стадии 1 данного примера, добавляли 2 н. хлористоводородную кислоту (5 мл), и смесь нагревали при кипячении с обратным холодильником в течение 3 часов. После охлаждения растворитель отгоняли при пониженном давлении. Полученный неочищенный гидрохлорид растворяли в воде, и к нему добавляли DOWEX 50WX8-200. Смолу промывали водой с последующим элюированием 4% аммиачной водой. Элюат концентрировали, и неочищенный продукт промывали ацетоном, с получением указанного в заголовке соединения (90 мг).
1H-ЯМР (CD3OD) δ: 0,95 (3H, д, J=6,6 Гц), 1,07-1,20 (2H, м), 1,38-1,77 (7H, м), 1,79-1,87 (2H, м), 1,97-2,06 (2H, м), 2,43-2,57 (2H, м), 2,81-2,95 (3H, м), 3,92 (1H, тт, J=11,7, 3,5 Гц), 6,93 (1H, с), 7,54 (1H, с).
МСВР (ESI): m/z вычислено для C16H28N3O2: 294,2182 [M+H]+; найдено: 294,2183.
[Пример 3] 5-Амино-2-{[(1-(транс-4-этилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] Метил-5-[(трет-бутоксикарбонил)амино]-2-{[(1-(транс-4-этилциклогексил)-1Н-имидазол-4-ил]метил}валерат
[Формула 88]
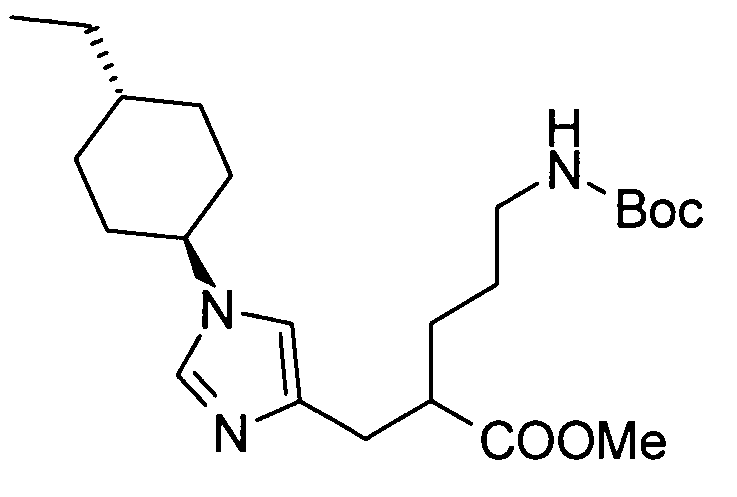
Соединение (100 мг), полученное в ссылочном примере 5, и соединение (267 мг), полученное в ссылочном примере 3, суспендировали в циклогексане (5 мл). К полученной суспензии добавляли раствор пиперидина (0,048 мл) и пропионовой кислоты (0,036 мл) в циклогексане (2 мл), и смесь нагревали при кипячении с обратным холодильником в течение 10 часов. После охлаждения к реакционному раствору добавляли водный карбонат калия, и органическое вещество экстрагировали этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия, затем сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Полученный неочищенный продукт растворяли в метаноле. К раствору добавляли катализатор 10% палладий-углерод (гидратированный, 200 мг), и смесь перемешивали при комнатной температуре в течение 8 часов в атмосфере водорода при нормальном давлении. После фильтрования через целит, фильтрат концентрировали при пониженном давлении, с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=2/1-1/2), с получением указанного в заголовке соединения (185 мг).
1H-ЯМР (CDCl3) δ: 0,91 (3H, т, J=7,0 Гц), 1,06 (2H, м), 1,15-1,68 (9H, м), 1,44 (9H, с), 1,93 (2H, м), 2,09 (2H, м), 2,71 (1H, дд, J=13,7, 5,9 Гц), 2,80 (1H, м), 2,89 (1H, дд, J=13,7, 7,8 Гц), 3,03-3,17 (2H, м), 3,63 (3H, с), 3,81 (1H, тт, J=12,1, 3,9 Гц), 4,76 (1H, шир.), 6,68 (1H, с), 7,47 (1H, с).
[Стадия 2] 5-амино-2-{[(1-(транс-4-этилциклогексил-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 89]
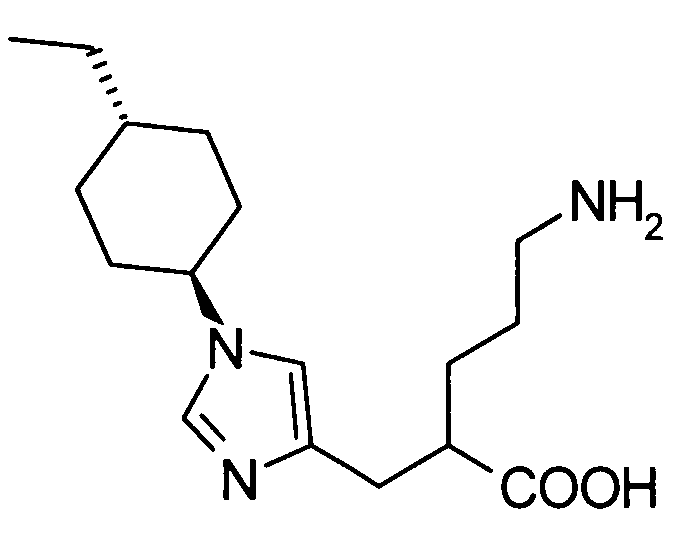
К соединению (180 мг), полученному на стадии 1 данного примера, добавляли 5 н. хлористоводородную кислоту (4 мл), и смесь нагревали при кипячении с обратным холодильником в течение 3 часов. После охлаждения растворитель отгоняли при пониженном давлении. Полученный неочищенный гидрохлорид растворяли в воде, и к нему добавляли DOWEX 50WX8-200. Смолу промывали водой с последующим элюированием 4% аммиачной водой. Элюат концентрировали, и неочищенный продукт промывали ацетоном, с получением указанного в заголовке соединения (53 мг).
1H-ЯМР (CD3OD) δ: 0,92 (3H, т, J=7,0 Гц), 1,10 (2H, м), 1,17-1,33 (3H, м), 1,42-1,75 (6H, м), 1,91 (2H, м), 2,05 (2H, м), 2,43-2,58 (2H, м), 2,79-2,95 (3H, м), 3,93 (1H, тт, J=12,1, 3,5 Гц), 6,94 (1H, с), 7,56 (1H, с).
МСВР (ESI): m/z вычислено для C17H30N3O2: 308,2338 [M+H]+; найдено: 308,2338.
[Пример 4] 5-Амино-2-{[(1-(3-этилциклобутил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] Метил-5-[(трет-бутоксикарбонил)амино]-2-{[(1-(3-этилциклобутил)-1Н-имидазол-4-ил]метил}валерат
[Формула 90]
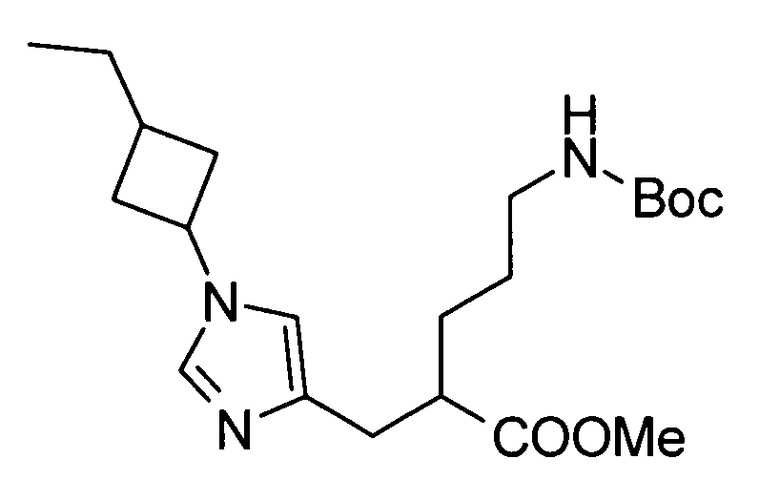
Соединение (115 мг), полученное в ссылочном примере 6, и соединение (355 мг), полученное в ссылочном примере 3, суспендировали в циклогексане (6 мл). К полученной суспензии добавляли раствор пиперидина (0,064 мл) и пропионовой кислоты (0,048 мл) в циклогексане (3 мл), и смесь нагревали при кипячении с обратным холодильником в течение 14 часов. После охлаждения к реакционному раствору добавляли водный карбонат калия, и органическое вещество экстрагировали этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия, затем сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Полученный неочищенный продукт растворяли в этаноле (5 мл). К раствору добавляли катализатор 10% палладий-углерод (гидратированный, 200 мг), и смесь перемешивали при комнатной температуре в течение 8 часов в атмосфере водорода при нормальном давлении. После фильтрования через целит, фильтрат концентрировали при пониженном давлении, с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=1/1-1/2), с получением указанного в заголовке соединения (190 мг, диастереомерная смесь, транс:цис=1:1).
1H-ЯМР (CDCl3) δ: 0,86 (1,5H, т, J=7,3 Гц), 0,90 (1,5H, т, J=7,3 Гц), 1,44 (9H, с), 1,44-1,70 (6H, м), 1,81-1,90 (1H, м), 1,94-2,04 (0,5H, м), 2,18-2,30 (1,5H, м), 2,37-2,47 (1H, м), 2,57-2,64 (1H, м), 2,66-2,73 (1H, м), 2,76-2,83 (1H, м), 2,86-2,93 (1H, м), 3,04-3,17 (2H, м), 3,64 (3H, с), 4,34 (0,5H, тт, J=9,3, 7,8 Гц), 4,58 (0,5H, тт, J=7,8, 7,3 Гц), 4,79 (1H, шир.), 6,68 (0,5H, с), 6,73 (0,5H, с), 7,39 (0,5H, с), 7,42 (0,5H, с).
[Стадия 2] 5-амино-2-{[(1-(3-этилциклобутил-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 91]
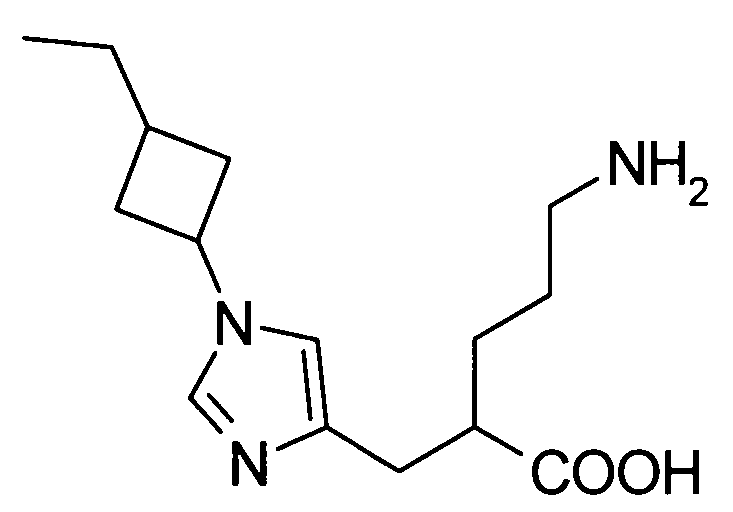
К соединению (185 мг), полученному на стадии 1 данного примера, добавляли 5 н. хлористоводородную кислоту (4 мл), и смесь нагревали при кипячении с обратным холодильником в течение 3 часов. После охлаждения растворитель отгоняли при пониженном давлении. Полученный неочищенный гидрохлорид растворяли в воде, и к нему добавляли DOWEX 50WX8-200. Смолу промывали водой с последующим элюированием 4% аммиачной водой. Элюат концентрировали, и неочищенный продукт промывали ацетоном, с получением указанного в заголовке соединения (51 мг, диастереомерная смесь, транс:цис=1:1).
1H-ЯМР (CD3OD) δ: 0,87 (1,5H, т, J=7,4 Гц), 0,91 (1,5H, т, J=7,4 Гц), 1,45-1,73 (6H, м), 1,85-2,06 (1H, м), 2,17-2,29 (1,5H, м), 2,41-2,64 (4H, м), 2,82-2,95 (3H, м), 4,47 (0,5H, тт, J=9,4, 7,8 Гц), 4,72 (0,5H, тт, J=8,2, 7,8 Гц), 6,97 (0,5H, с), 7,03 (0,5H, с), 7,53 (0,5H, с), 7,56 (0,5H, с).
МСВР (ESI): m/z вычислено для C15H26N3O2: 280,2025 [M+H]+; найдено: 280,2015.
[Пример 5] 5-Амино-2-{[(1-(3-метилциклобутил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 92]
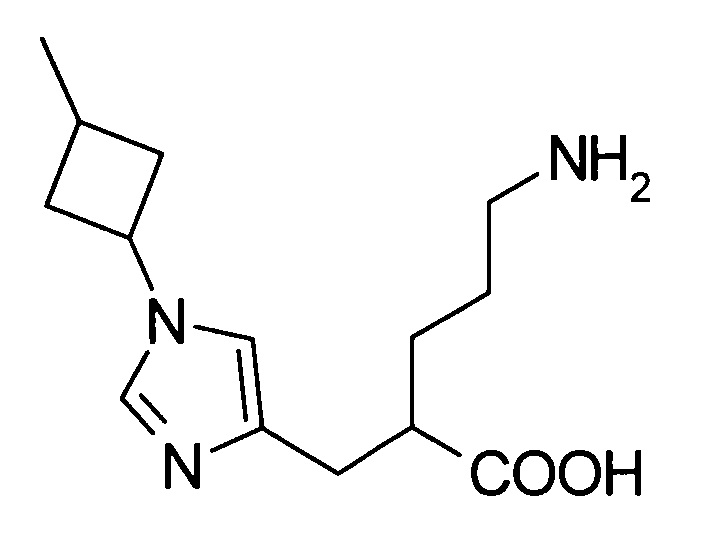
Указанное в заголовке соединение (2,0 мг, диастереомерная смесь, транс:цис=1:1) получали из соединения (10 мг), полученного в ссылочном примере 7, таким же образом, как в примере 4.
1H-ЯМР (CD3OD) δ: 1,15 (1,5H, д, J=6,6 Гц), 1,24 (1,5H, д, J=6,6 Гц), 1,44-1,72 (4H, м), 1,85-1,96 (1H, м), 2,10-2,22 (1,5H, м), 2,41-2,63 (4,5H, м), 2,81-2,95 (3H, м), 4,45 (0,5H, тт, J=9,4, 7,4 Гц), 4,79 (0,5H, тт, J=7,8, 7,8 Гц), 6,98 (0,5H, с), 7,02 (0,5H, с), 7,54 (0,5H, с), 7,57 (0,5H, с).
МСВР (ESI): m/z вычислено для C14H24N3O2: 266,1869 [M+H]+; найдено: 266,1874.
[Пример 6] (2RS)-5-Амино-2-({1-[(1R,3s,5S)-бицикло[3.1.0]гексан-3-ил]-1Н-имидазол-4-ил}метил)валериановая кислота
[Стадия 1] (1R,3r,5S)-бицикло[3.1.0]гексан-3-илметансульфонат
[Формула 93]
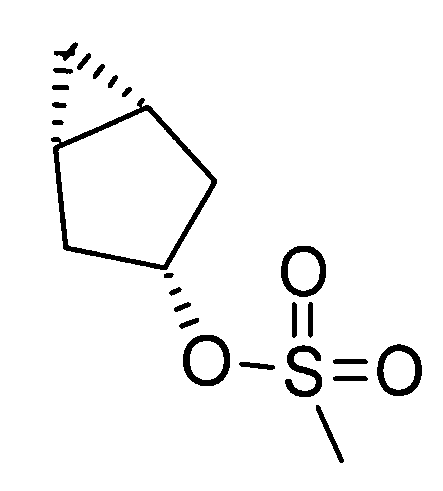
К раствору (1R,3r,5S)-бицикло[3.1.0]гексан-3-ола (1,00 г) в метиленхлориде при 0°С добавляли триэтиламин (1,70 мл) и метансульфонилхлорид (0,94 мл), и смесь перемешивали при комнатной температуре в течение 12 часов. К реакционному раствору добавляли воду, и органическое вещество экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=4/1-2/1), с получением указанного в заголовке соединения (1,34 г).
1H-ЯМР (CDCl3) δ: 0,44 (1H, м), 0,54 (1H, м), 1,35 (2H, м), 2,10 (2H, м), 2,26 (2H, м), 2,96 (3H, с), 5,19 (1H, м).
[Стадия 2] Трет-бутил-(2RS)-2-({1-[(1R,3s,5S)-бицикло[3.1.0]гексан-3-ил]-1Н-имидазол-4-ил}метил)-5-[(трет-бутоксикарбонил)амино]валерат
[Формула 94]
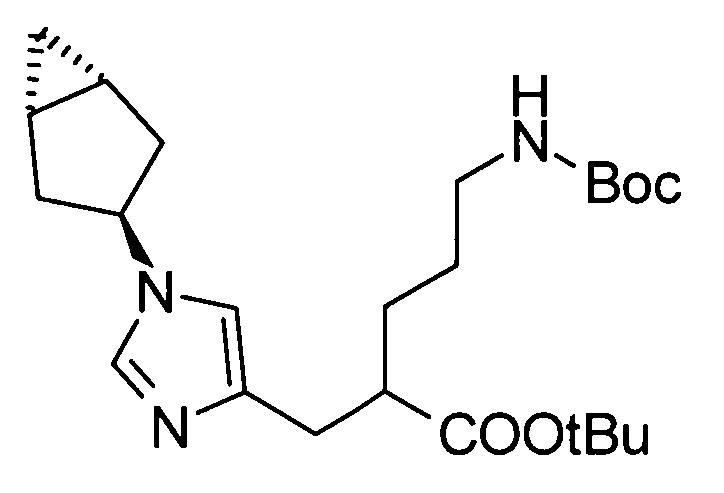
Соединение (250 мг), полученное в ссылочном примере 2, растворяли в N,N-диметилформамиде (4 мл), и к раствору добавляли карбонат цезия (630 мг) и соединение (250 мг), полученное на стадии 1 данного примера. После перемешивания при 110°C в течение 9 часов к реакционному раствору добавляли воду, и органическое вещество экстрагировали диэтиловым эфиром. Органический слой промывали водой, затем сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метиленхлорид-метиленхлорид/метанол-10/1), с получением указанного в заголовке соединения (55 мг).
1H-ЯМР (CDCl3) δ: 0,26 (1H, дт, J=5,7, 3,9 Гц), 0,47 (1H, тд, J=7,8, 5,7 Гц), 1,38 (9H, с), 1,44 (9H, с), 1,35-2,07 (8H, м), 2,26-2,33 (2H, м), 2,58-2,68 (2H, м), 2,83 (1H, м), 3,05-3,15 (2H, м), 4,03 (1H, тт, J=10,2, 7,4 Гц), 4,75 (1H, шир.), 6,65 (1H, с), 7,37 (1H, с).
[Стадия 3] (2RS)-5-амино-2-({1-[(1R,3s,5S)-бицикло[3.1.0]гексан-3-ил]-1Н-имидазол-4-ил}метил)валериановая кислота
[Формула 95]
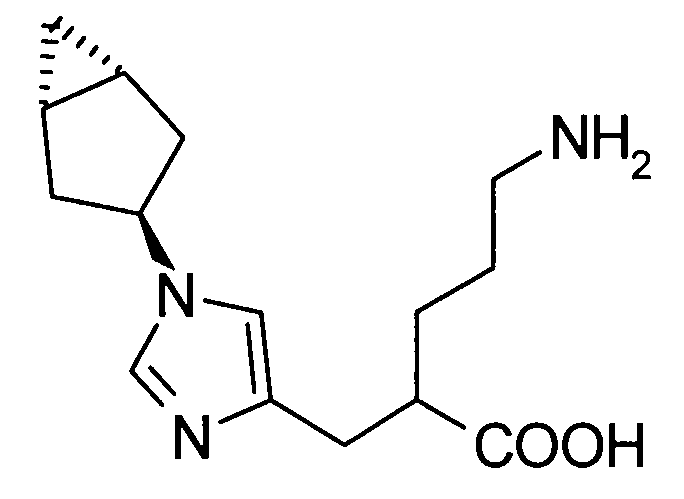
Соединение (55 мг), полученное на стадии 2 данного примера, растворяли в метиленхлориде (2 мл). К раствору добавляли трифторуксусную кислоту (1 мл), и смесь перемешивали при комнатной температуре в течение 5 часов. Затем растворитель отгоняли при пониженном давлении. К остатку добавляли толуол, и растворитель повторно отгоняли при пониженном давлении. Полученный неочищенный трифторацетат растворяли в воде, и к нему добавляли DOWEX 50WX8-200. Смолу промывали метанолом с последующим элюированием 4% аммиачной водой. Элюат концентрировали, и неочищенный продукт промывали ацетоном, с получением указанного в заголовке соединения (30 мг).
1H-ЯМР (CD3OD) δ: 0,34 (1H, дт, J=5,4, 3,9 Гц), 0,45 (1H, тд, J=7,4, 5,4 Гц), 1,38-1,45 (2H, м), 1,46-1,71 (4H, м), 2,03-2,11 (2H, м), 2,23-2,30 (2H, м), 2,44-2,57 (2H, м), 2,82-2,95 (3H, м), 4,21 (1H, тт, J=10,2, 7,4 Гц), 6,92 (1H, с), 7,50 (1H, с).
МСВР (ESI): m/z вычислено для C15H23N3NaO2: 300,1688 [M+Na]+; найдено: 300,1679.
[Пример 7] 5-Амино-2-{[(1-(транс-4-гидроксициклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] Метил-5-[(трет-бутоксикарбонил)амино]-2-{[(1-(транс-4-гидроксициклогексил)-1Н-имидазол-4-ил]метил}валерат
[Формула 96]
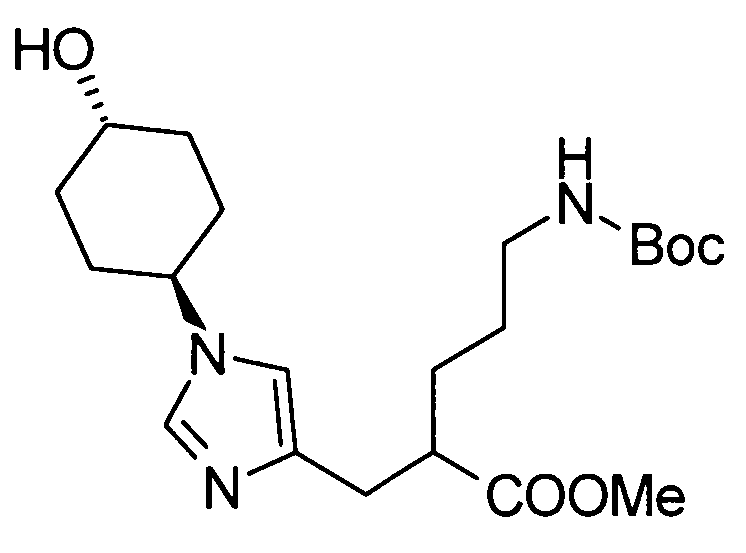
Соединение (185 мг), полученное в ссылочном примере 8, и соединение (524 мг), полученное в ссылочном примере 3, суспендировали в циклогексане (6 мл). К суспензии добавляли раствор пиперидина (0,094 мл) и пропионовой кислоты (0,071 мл) в циклогексане (2 мл), и смесь нагревали при кипячении с обратным холодильником в течение 12 часов. После охлаждения к реакционному раствору добавляли водный карбонат калия, и органическое вещество экстрагировали этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия, затем сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Полученный неочищенный продукт растворяли в метаноле (6 мл). К раствору добавляли катализатор 10% палладий-углерод (гидратированный, 200 мг), и смесь перемешивали при комнатной температуре в течение 7 часов в атмосфере водорода при нормальном давлении. После фильтрования через целит, фильтрат концентрировали при пониженном давлении, с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метиленхлорид-метиленхлорид/метанол=9/1), с получением указанного в заголовке соединения (326 мг).
1H-ЯМР (CDCl3) δ: 1,40-1,88 (8H, м), 1,43 (9H, с), 2,08-2,16 (4H, м), 2,70 (1H, дд, J=14,6, 6,3 Гц), 2,80 (1H, м), 2,89 (1H, дд, J=14,6, 8,3 Гц), 3,03-3,15 (2H, м), 3,63 (3H, с), 3,72 (1H, м), 3,88 (1H, м), 4,73 (1H, шир.), 6,67 (1H, с), 7,47 (1H, с).
[Стадия 2] 5-амино-2-{[(1-(транс-4-гидроксициклогексил-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 97]
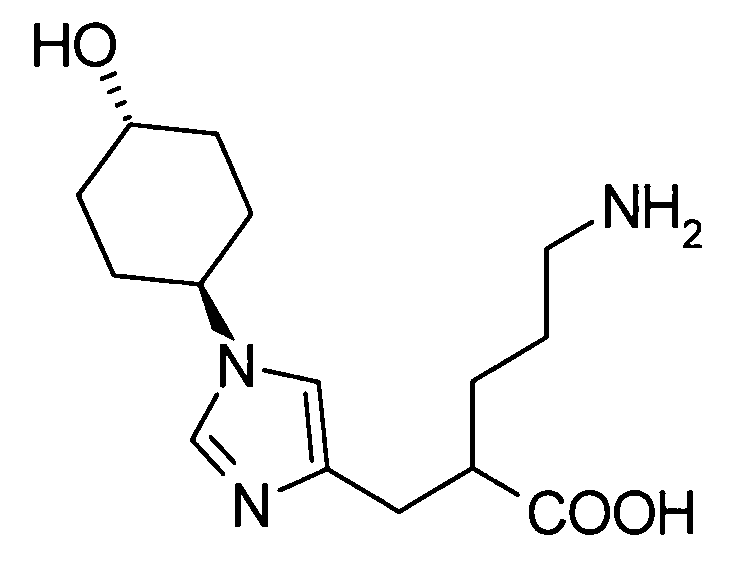
К соединению (246 мг), полученному на стадии 1 данного примера, добавляли 5 н. хлористоводородную кислоту (5 мл), и смесь нагревали при кипячении с обратным холодильником в течение 3 часов. После охлаждения растворитель отгоняли при пониженном давлении. Полученный неочищенный гидрохлорид растворяли в метаноле, и к нему добавляли DOWEX 50WX8-200. Смолу промывали водой с последующим элюированием 4% аммиачной водой. Элюат концентрировали, и неочищенный продукт промывали ацетоном, с получением указанного в заголовке соединения (74 мг).
1H-ЯМР (CD3OD) δ: 1,39-1,87 (8H, м), 2,01-2,13 (4H, м), 2,53-2,69 (2H, м), 2,84-2,97 (3H, м), 3,64 (1H, м), 4,09 (1H, м), 7,10 (1H, с), 8,01 (1H, с).
МСВР (ESI): m/z вычислено для C15H26N3O3: 296,1974 [M+H]+; найдено: 296,1975.
[Пример 8] 5-Амино-2-{[1-(4-гидрокси-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] Трет-бутил-5-[(трет-бутоксикарбонил)амино]-2-{[1-(4-гидрокси-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерат
[Формула 98]
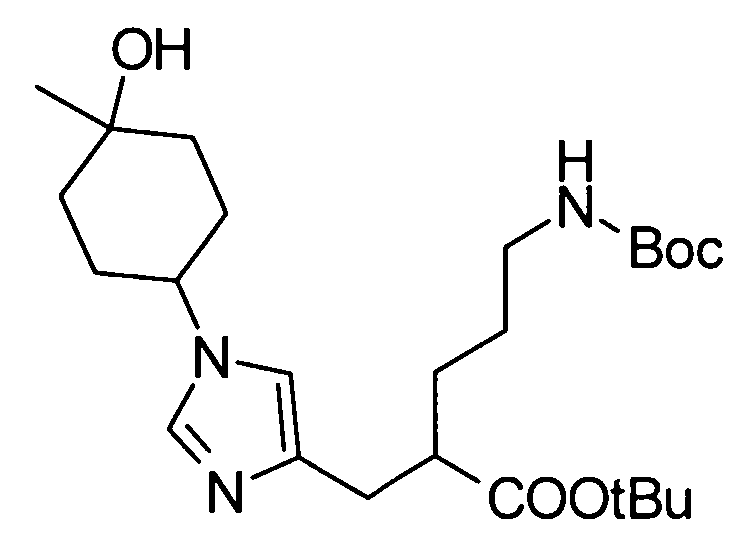
Соединение (796 мг), полученное в ссылочном примере 1, растворяли в ацетонитриле (6 мл), и к нему добавляли хлорид лития (111 мг). После перемешивания при комнатной температуре в течение 1 часа, к смеси добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (0,34 мл). После дополнительного перемешивания при комнатной температуре в течение 1 часа, к ней добавляли раствор соединения (300 мг), полученного в ссылочном примере 9, в ацетонитриле (4 мл), и смесь перемешивали при комнатной температуре в течение 12 часов. Растворитель отгоняли при пониженном давлении. Затем к остатку добавляли воду, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Полученный неочищенный продукт растворяли в этаноле (10 мл). К раствору добавляли катализатор 10% палладий-углерод (гидратированный, 150 мг), и смесь перемешивали при комнатной температуре в течение 9 часов в атмосфере водорода при нормальном давлении. После фильтрования через целит, фильтрат концентрировали при пониженном давлении, с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=2/1-1/3), с получением диастереомерной смеси указанного в заголовке соединения (431 мг, транс:цис=1:3).
1H-ЯМР (CDCl3) δ: 1,29 (2,25H, с), 1,33 (0,75H, с), 1,38 (9H, с), 1,43 (9H, с), 1,47-1,69 (6H, м), 1,75-1,90 (4H, м), 2,05-2,12 (2H, м), 2,61-2,70 (2H, м), 2,80-2,88 (1H, м), 3,04-3,17 (2H, м), 3,81 (0,75H, тт, J=12,2, 3,9 Гц), 3,93 (0,25H, м), 4,74 (1H, шир.), 6,70 (0,25H, с), 6,72 (0,75H, с), 7,44 (0,25H, с), 7,45 (0,75H, с).
[Стадия 2] 5-амино-2-{[1-(4-гидрокси-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 99]
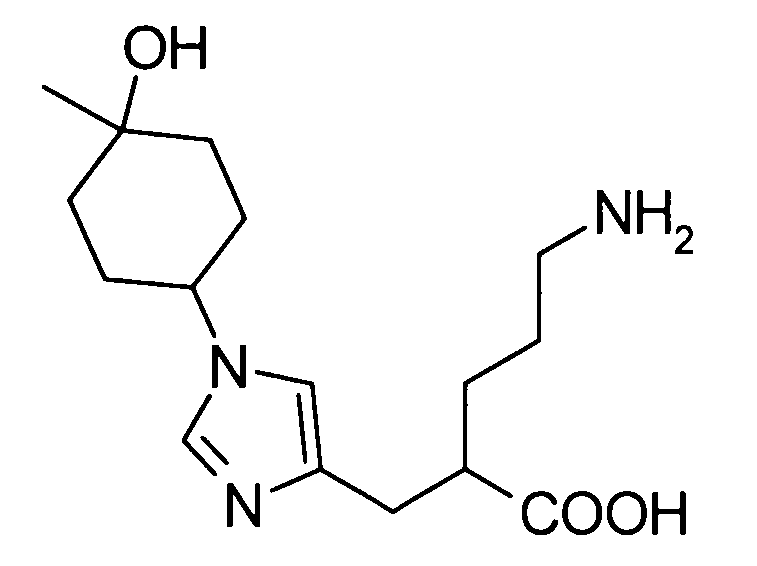
К соединению (306 мг), полученному на стадии 1 данного примера, добавляли 2 н. хлористоводородную кислоту (5 мл), и смесь нагревали при 40°C в течение 3 часов и при 55°C в течение 5 часов. После охлаждения растворитель отгоняли при пониженном давлении. Полученный неочищенный гидрохлорид растворяли в воде, и к нему добавляли DOWEX 50WX8-200. Смолу промывали водой с последующим элюированием 4% аммиачной водой. Элюат концентрировали, и неочищенный продукт промывали ацетоном, с получением указанного в заголовке соединения (50 мг, транс:цис=1:3).
1H-ЯМР (CDCl3) δ: 1,23 (2,25H, с), 1,31 (0,75H, с), 1,47-1,90 (10H, м), 1,97-2,11 (2H, м), 2,46-2,59 (2H, м), 2,83-2,95 (3Н, м), 3,97 (0,75Н, тт, J=12,2, 3,9 Гц), 4,04 (0,25H, м), 6,97 (0,25H, с), 6,99 (0,75H, с), 7,63 (0,25H, с), 7,64 (0,75H, с).
МСВР (ESI): m/z вычислено для C16H28N3O3: 310,2131 [M+H]+; найдено: 310,2123.
[Пример 9] 5-Амино-2-{[1-(3-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 100]
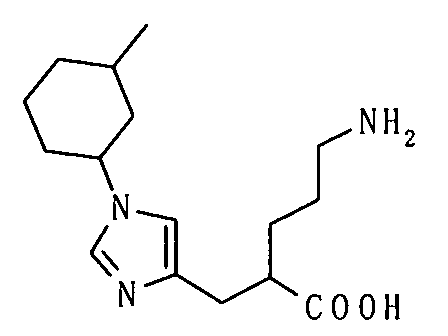
Указанное в заголовке соединение (10 мг) получали таким же образом, как в примере 6, используя 3-метилциклогексанол (1,84 г) вместо (1R,3R,5S)-бицикло[3.1.0]гексан-3-ола.
1H-ЯМР (CD3OD) δ: 1,05 (3H, д, J=6,8 Гц), 1,32-1,40 (1H, м), 1,47-1,55 (1H, м), 1,55-1,76 (7H, м), 1,78-1,87 (1H, м), 1,90-2,05 (3H, м), 2,46-2,58 (2H, м), 2,84-2,95 (3H, м), 4,24 (1H, м), 6,96 (1H, с), 7,57 (1H, с).
МСВР (ESI): m/z вычислено для C16H28N3O3: 294,21815 [M+H]+; найдено: 294,21898.
[Пример 10] 5-Амино-2-[(1-циклогептил-1Н-имидазол-4-ил)метил]валериановая кислота
[Формула 101]
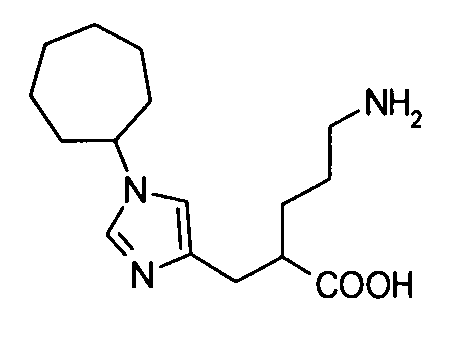
Указанное в заголовке соединение (30 мг) получали таким же образом, как на стадиях 1 и 3 примера 1, используя бромциклогептан (890 мг) вместо 3-бромциклогексена.
1H-ЯМР (CD3OD) δ: 1,46-1,74 (10H, м), 1,74-1,88 (2H, м), 1,85-1,94 (2H, м), 1,99-2,08 (2H, м), 2,45-2,58 (2H, м), 2,82-2,95 (3H, м), 4,16 (1H, м), 6,93 (1H, с), 7,57 (1H, с).
МСВР (ESI): m/z вычислено для C16H28N3O2: 294,21815 [M+H]+; найдено: 294,21863.
[Пример 11] 5-Амино-2-({1-[экзо-бицикло[2.2.1]гепт-2-ил]-1Н-имидазол-4-ил}метил]валериановая кислота
[Формула 102]
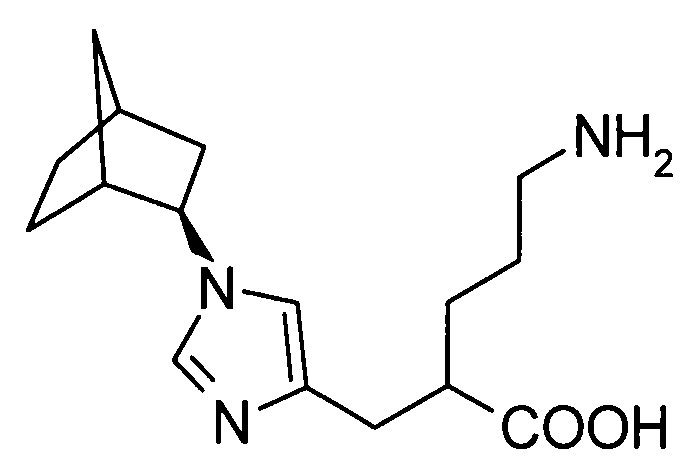
Указанное в заголовке соединение (0,19 г) получали из соединения (0,21 г), полученного в ссылочном примере 10, таким же образом, как в примере 3.
1H-ЯМР (CD3OD) δ: 1,21-1,37 (3H, м), 1,46-1,71 (7H, м), 1,77-1,84 (1H, м), 1,90-1,97 (1H, м), 2,38-2,45 (2H, м), 2,45-2,57 (2H, м), 2,83-2,95 (3H, м), 4,04-4,10 (1H, м), 6,93 (1H, с), 7,56 (1H, с).
МСВР (ESI): m/z вычислено для C16H28N3O2: 292,20250 [M+H]+; найдено: 292,20319.
[Пример 12] 5-Амино-2-({1-[эндо-бицикло[2.2.1]гепт-2-ил]-1Н-имидазол-4-ил}метил]валериановая кислота
[Формула 103]
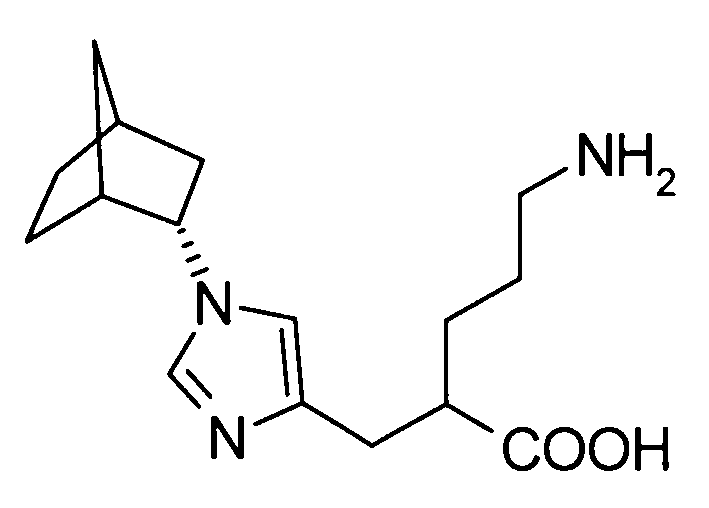
Указанное в заголовке соединение (0,07 г) получали из соединения (0,17 г), полученного в ссылочном примере 11, таким же образом, как в примере 3.
1H-ЯМР (CD3OD) δ: 1,15-1,23 (1H, м), 1,33-1,43 (2H, м), 1,44-1,55 (2H, м), 1,55-1,71 (6H, м), 2,10-2,18 (1H, м), 2,33-2,37 (1H, м), 2,46-2,59 (3H, м), 2,83-2,95 (3H, м), 4,43-4,50 (1H, м), 6,93 (1H, с), 7,57 (1H, с).
МСВР (ESI): m/z вычислено для C16H26N3O2: 292,20250 [M+H]+; найдено: 292,20252.
[Пример 13] 2-[(1-Адамантан-2-ил-1Н-имидазол-4-ил)метил]-5-аминовалериановая кислота
[Формула 104]
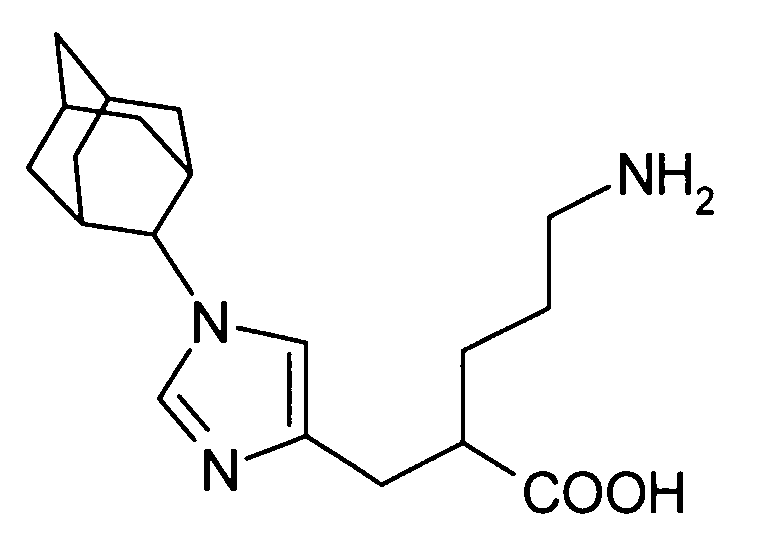
Указанное в заголовке соединение (0,04 г) получали из соединения (0,15 г), полученного в ссылочном примере 12, таким же образом, как в примере 3.
1H-ЯМР (CD3OD) δ: 1,48-1,57 (1H, м), 1,58-1,72 (5H, м), 1,77-1,86 (5H, м), 1,92-1,99 (3H, м), 2,01-2,07 (2H, м), 2,48-2,61 (4H, м), 2,85-2,95 (3H, м), 4,17 (1H, с), 7,03 (1H, с), 7,65 (1H, с).
МСВР (ESI): m/z вычислено для C19H30N3O2: 332,23380 [M+H]+; найдено: 332,23325.
[Пример 14] 5-Амино-2-{[1-(транс-4-феноксициклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 105]
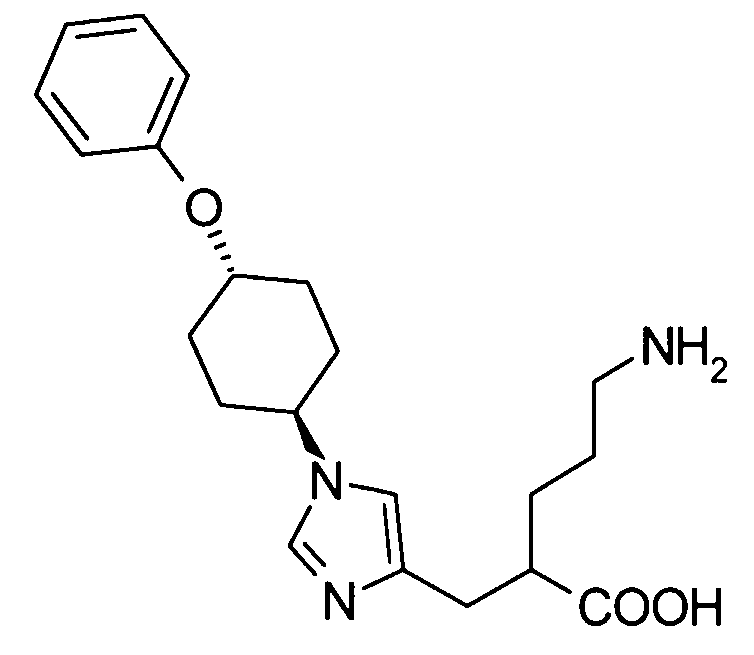
Указанное в заголовке соединение (7 мг) получали из соединения (0,07 г), полученного в ссылочном примере 13, таким же образом, как в примере 3.
1H-ЯМР (CD3OD) δ: 1,47-1,73 (6H, м), 1,84-1,95 (2H, м), 2,08-2,16 (2H, м), 2,21-2,28 (2H, м), 2,46-2,59 (2H, м), 2,84-2,95 (3H, м), 4,09 (1H, м), 4,36 (1H, м), 6,88-6,95 (3H, м), 6,97 (1H, с), 7,23-7,28 (2H, м), 7,59 (1H, с).
МСВР (ESI): m/z вычислено для C21H30N3O3: 372,22872 [M+H]+; найдено: 372,22850.
[Пример 15] (2R)-5-Амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота и (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] Метил-5-[(трет-бутоксикарбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерат
[Формула 106]
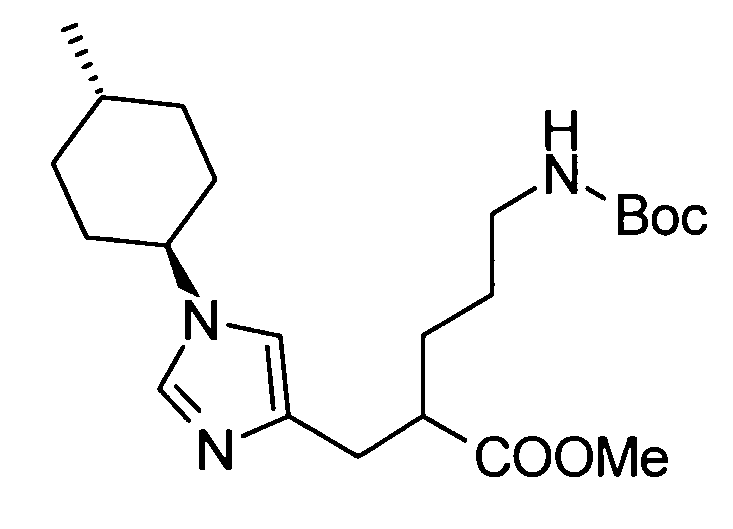
Соединение (300 мг), полученное в ссылочном примере 4, и соединение (860 мг), полученное в ссылочном примере 3, суспендировали в циклогексане (10 мл). К суспензии добавляли раствор пиперидина (0,154 мл) и пропионовой кислоты (0,116 мл) в циклогексане (10 мл), и смесь нагревали при кипячении с обратным холодильником в течение 48 часов. После охлаждения к реакционному раствору добавляли водный карбонат калия, и органическое вещество экстрагировали этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия, затем сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Полученный неочищенный продукт растворяли в этаноле (12 мл). К раствору добавляли катализатор 10% палладий-углерод (гидратированный, 250 мг), и смесь перемешивали в атмосфере водорода при нормальном давлении при комнатной температуре в течение 4 часов и при 60°С в течение 2,5 часов. После фильтрования через целит, фильтрат концентрировали при пониженном давлении, с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=2/1-1/3), с получением указанного в заголовке соединения (562 мг).
1H-ЯМР (CDCl3) δ: 0,94 (3H, д, J=6,6 Гц), 1,02-1,15 (2H, м), 1,34-1,69 (7H, м), 1,43 (9H, с), 1,80-1,87 (2H, м), 1,99-2,09 (2H, м), 2,69 (1H, дд, J=13,7, 6,3 Гц), 2,79 (1H, м), 2,88 (1H, дд, J=13,7, 7,4 Гц), 3,03-3,13 (2H, м), 3,63 (3H, с), 3,79 (1H, тт, J=12,1, 3,9 Гц), 4,76 (1H, шир.), 6,67 (1H, с), 7,47 (1H, с).
[Стадия 2] Метил-(2R)-5-[(трет-бутоксикарбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерат и метил-(2S)-5-[(трет-бутоксикарбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерат
[Формула 107]
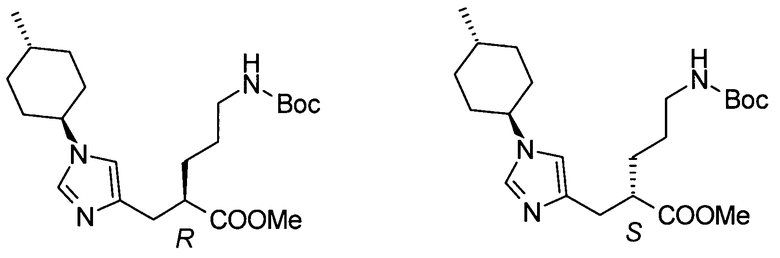
Соединение (40 мг), полученное на стадии 1 данного примера, растворяли в гексане (1,5 мл) и этаноле (0,5 мл) и оптически разделяли высокоэффективной жидкостной хроматографией с использованием полупрепаративной колонки CHIRALPAK IA (2,0 см × 25,0 см). Объемная скорость потока: 15 мл/мин, элюирующий растворитель: гексан/этанол=75/25, длина волны обнаружения: 220 нм.
Растворитель элюата, содержащего оптически активное соединение, отгоняли при пониженном давлении, с получением соответственно каждого энантиомера (15 мг). Аналитической высокоэффективной жидкостной хроматографией было подтверждено, что оба энантиомера являются оптически активными соединениями. Колонка: CHIRALPAK IA (0,46 см × 25,0 см), объемная скорость потока: 1 мл/мин, элюирующий растворитель: гексан/этанол=80/20 <об/об>, длина волны обнаружения: 220 нм, время удерживания: метил-(2R)-5-[(трет-бутоксикарбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерата (7,2 минуты) и метил-(2S)-5-[(трет-бутоксикарбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерата (11,2 минуты).
[Стадия 3] (2R)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота и (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 108]
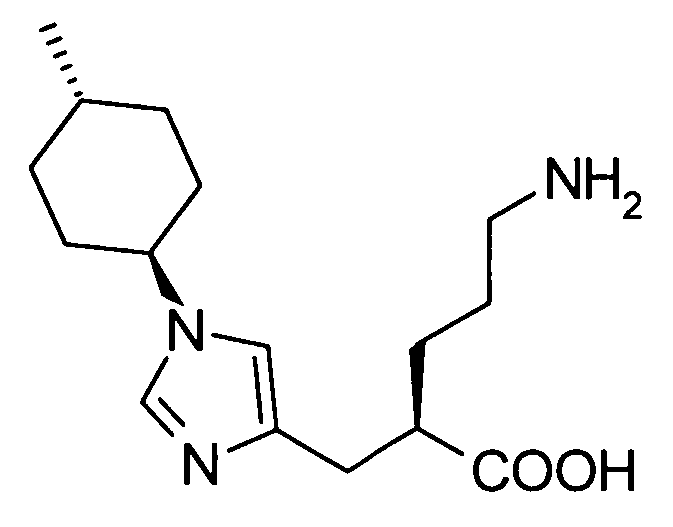
К метил-(2R)-5-[(трет-бутоксикарбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерату (15,0 мг), полученному на стадии 2 данного примера, добавляли 5 н. хлористоводородную кислоту (2 мл), и смесь нагревали при кипячении с обратным холодильником в течение 4 часов. После охлаждения растворитель отгоняли при пониженном давлении. Полученный неочищенный гидрохлорид растворяли в метаноле, и к нему добавляли DOWEX 50WX8-200. Смолу промывали водой с последующим элюированием 4% аммиачной водой. Элюат концентрировали, и неочищенный продукт промывали ацетоном, с получением указанного в заголовке соединения (2,2 мг).
[Стадия 4] (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 109]
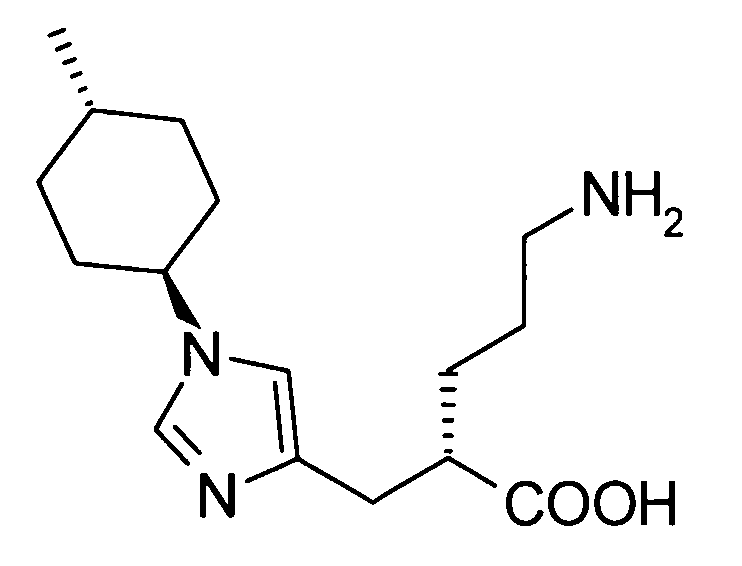
К метил-(2S)-5-[(трет-бутоксикарбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерату (15,0 мг), полученному на стадии 2 данного примера, добавляли 5 н. хлористоводородную кислоту (2 мл), и смесь нагревали при кипячении с обратным холодильником в течение 4 часов. После охлаждения растворитель отгоняли при пониженном давлении. Полученный неочищенный гидрохлорид растворяли в метаноле, и к нему добавляли DOWEX 50WX8-200 (200 мг). Смолу промывали водой с последующим элюированием аммиачной водой (4%, 80 мл). Элюат концентрировали, и неочищенный продукт промывали ацетоном, с получением указанного в заголовке соединения (1,8 мг).
[Пример 16] Гидрохлорид бензил-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерата
[Стадия 1] 5-[(трет-бутоксикарбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 110]
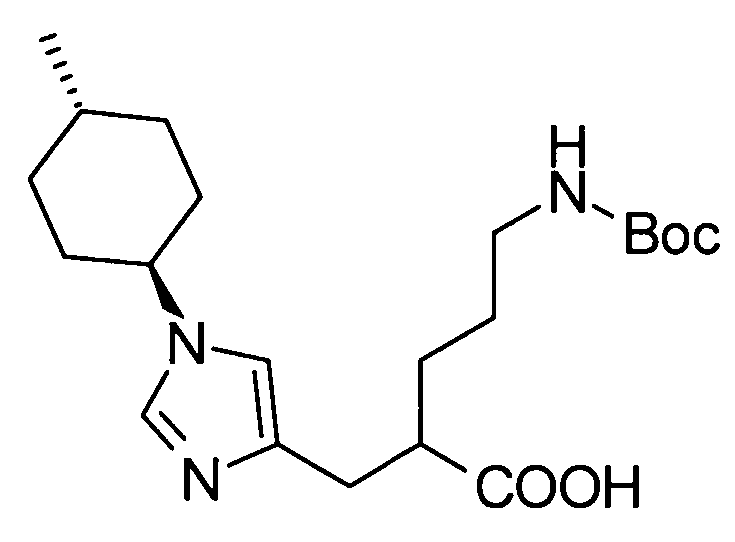
Соединение (7,00 г), полученное на стадии 1 примера 15, растворяли в смешанном растворителе из тетрагидрофурана (70 мл) и воды (14 мл). К раствору при комнатной температуре добавляли моногидрат гидроксида лития (1,26 г), и смесь перемешивали в течение ночи. Реакционный раствор нейтрализовали добавлением 2 н. хлористоводородной кислоты (8,6 мл), и растворитель отгоняли при пониженном давлении. К полученному остатку добавляли метиленхлорид, и смесь сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, с получением неочищенного продукта указанного в заголовке соединения. Неочищенный продукт непосредственно использовали в следующей реакции.
MS (ESI) m/z 394 (M+H)+.
[Стадия 2] Бензил-5-[(трет-бутоксикарбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерат
[Формула 111]
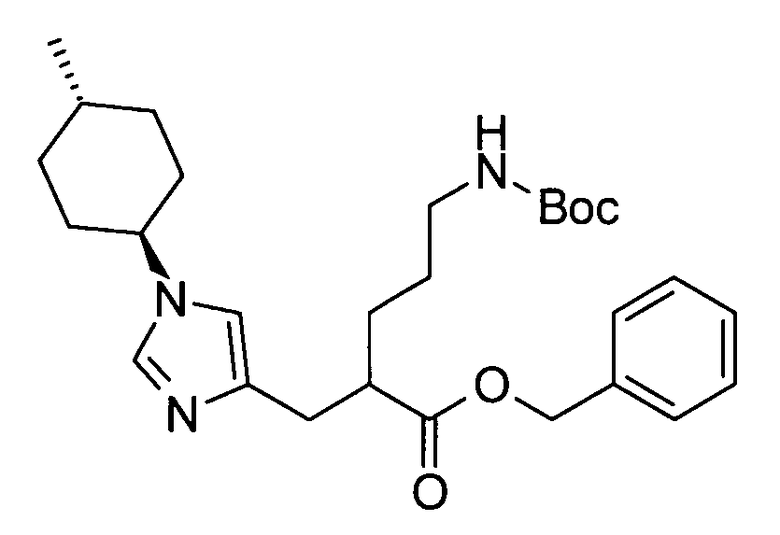
5-[(трет-бутоксикарбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота, полученная на стадии 1 данного примера, растворяли в метиленхлориде (150 мл). К раствору при комнатной температуре добавляли бензиловый спирт (8,85 мл), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (4,95 г) и 4-диметиламинопиридин (3,15 г), и смесь перемешивали в течение 18 часов. Органическое вещество экстрагировали метиленхлоридом и сушили над безводным сульфатом натрия, и растворитель затем отгоняли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=7/3-этилацетат), с получением указанного в заголовке соединения (8,45 г).
1H-ЯМР (CDCl3) δ: 0,94 (3H, д, J=6,3 Гц), 1,01-1,13 (2H, м), 1,38-1,72 (16H, м), 1,79-1,86 (2H, м), 1,97-2,04 (2H, м), 2,71 (1H, дд, J=14,1, 5,9 Гц), 2,80-2,87 (1H, м), 2,91 (1H, дд, J=14,1, 7,8 Гц), 3,07 (2H, шир.с), 3,68-3,76 (1H, м), 4,68 (1H, шир.с), 5,10 (2H, с), 6,57 (1H, с), 7,29-7,40 (6H, м).
МС (ESI) m/z 484 (M+H)+.
[Стадия 3] гидрохлорид бензил-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерата
[Формула 112]
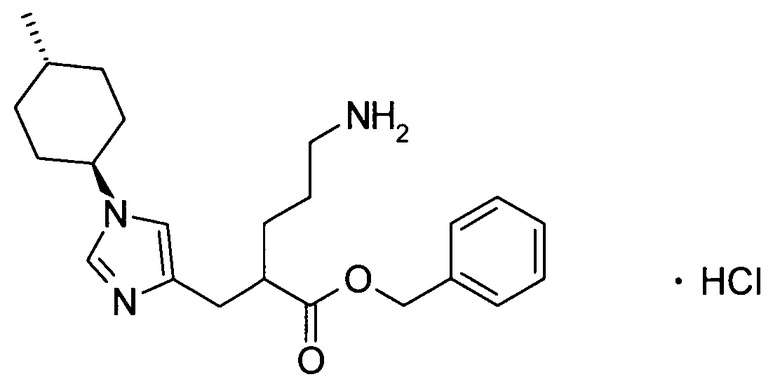
Бензил-5-[(трет-бутоксикарбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерат, полученный на стадии 2 данного примера, растворяли в 1,4-диоксане (40 мл). К раствору по каплям при комнатной температуре добавляли раствор 4 н. хлористоводородной кислоты в 1,4 диоксане (40 мл), и смесь затем перемешивали в течение 24 часов. Растворитель в реакционном растворе отгоняли при пониженном давлении, с получением неочищенного продукта указанного в заголовке соединения (8,04 г).
1H-ЯМР (CD3OD) δ: 0,97 (3H, д, J=6,7 Гц), 1,11-1,22 (2H, м), 1,43-1,54 (1H, м), 1,62-1,89 (8H, м), 1,99-2,06 (2H, м), 2,88-3,04 (5H, м), 4,10 (1H, тт, J=12,1, 3,9 Гц), 5,07 (1H, д, J=12,1 Гц), 5,15 (1H, д, J=12,1 Гц), 7,28-7,37 (6H, м), 8,82 (1H, д, J=1,6 Гц).
МС (ESI) m/z 384 (M+H)+.
[Пример 17] 2-{[1-Транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-(L-фенилаланиламино)валериановая кислота
[Стадия 1] Бензил-5-({N-[(бензилокси)карбонил]-L-фенилаланил}амино-2-{[1-транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерат
[Формула 113]
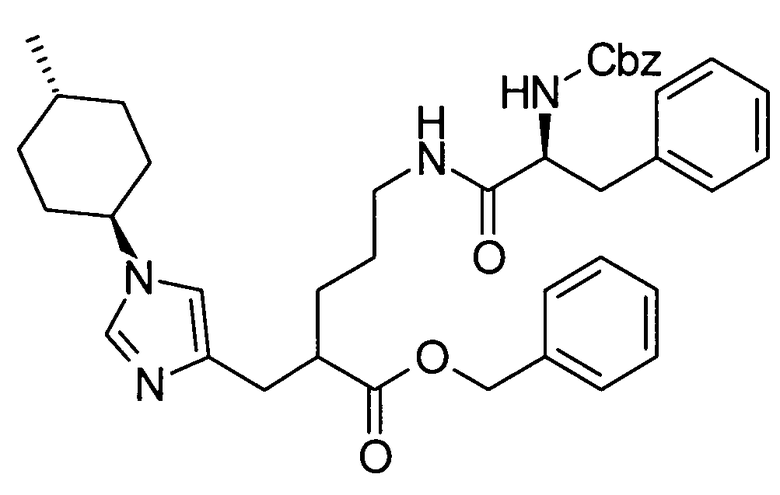
Соединение (200 мг), полученное в примере 16, растворяли в N,N-диметилформамиде (6 мл). К раствору при комнатной температуре добавляли N-[(бензилокси)карбонил]-L-фенилаланин (197 мг), гидрат 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолинийхлорида (DMT-MM, 90%, 182 мг) и триэтиламин (135 мкл), и смесь перемешивали в течение 3 дней. К реакционному раствору добавляли этилацетат, и смесь промывали три раза 10% раствором хлорида натрия и последовательно промывали насыщенным водным бикарбонатом натрия. Полученный органический слой сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=1/1-этилацетат), с получением указанного в заголовке соединения (254 мг).
1H-ЯМР (CDCl3) δ: 0,94 (3H, д, J=6,7 Гц), 1,07 (2H, кв., J=12,9 Гц), 1,43-1,55 (7H, м), 1,80-1,84 (2H, м), 1,97-1,99 (2H, м), 2,67-2,88 (3H, м), 3,08-3,15 (3H, м), 3,68-3,70 (0,5H, м), 4,40-4,41 (0,5H, м), 5,05-5,10 (4H, м), 5,60-5,63 (1Н, м), 6,54-6,56 (2H, м), 7,16-7,21 (4Н, м), 7,29-7,52 (7H, м).
МС (ESI) m/z 665 (M+H)+.
[Стадия 2] 2-{[1-транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-(L-фенилаланиламино)валериановая кислота
[Формула 114]
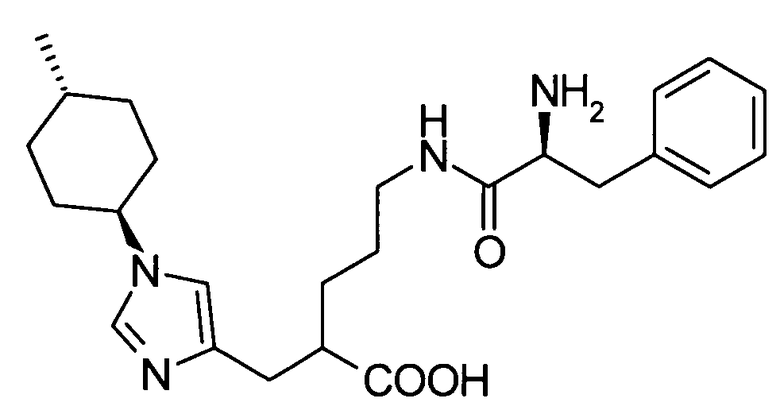
Соединение, полученное на стадии 1 данного примера, растворяли в этаноле (8 мл). К раствору добавляли катализатор 10% палладий-углерод (гидратированный, 85 мг), и смесь перемешивали при комнатной температуре в течение 5 часов в атмосфере водорода при нормальном давлении. Растворитель в реакционном растворе отгоняли при пониженном давлении, и остаток очищали ВЭЖХ с обращенной фазой, с получением указанного в заголовке соединения (128 мг).
1H-ЯМР (CDCl3) δ: 0,95 (3H, д, J=6,7 Гц), 1,07-1,14 (2H, м), 1,41-1,44 (2H, м), 1,59-1,72 (5H, м), 1,84-1,88 (2H, м), 2,07-2,11 (2H, м), 2,71-2,80 (4H, м), 3,23-3,25 (3H, м), 3,62-3,65 (1H, м), 3,82-3,83 (1H, м), 6,75 (1H, с), 7,23-7,30 (5H, м).
МСВР (ESI): m/z вычислено для C25H37N4O3: 441,28656 [M+H]+; найдено: 441,28690.
[Пример 18] 2-{[1-Транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-(L-норлейциламино)валериановая кислота
[Стадия 1] Бензил-5-({N-[(бензилокси)карбонил]-L-норлейцил}амино-2-{[1-транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерат
[Формула 115]
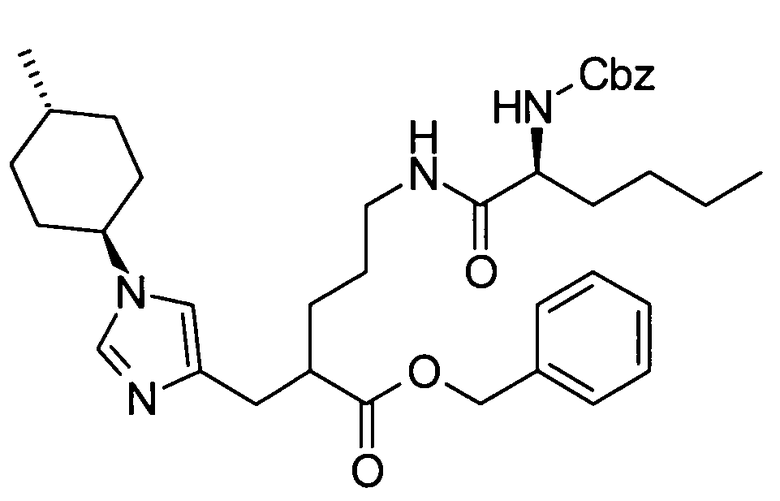
Указанное в заголовке соединение (244 мг) получали из соединения (200 мг), полученного в примере 16, и N-[(бензилокси)карбонил]-L-норлейцина (174 мг) таким же образом, как на стадии 1 примера 17.
1H-ЯМР (CDCl3) δ: 0,83-0,88 (3H, м), 0,94 (3H, д, J=6,7 Гц), 1,02-1,12 (2H, м), 1,23-1,74 (12H, м), 1,78-1,85 (2H, м), 1,96-2,02 (2H, м), 2,73-2,95 (3H, м), 3,17-3,32 (2H, м), 3,67-3,76 (1H, м), 4,10-4,18 (1H, м), 5,09-5,11 м), 5,55-5,58 (1H, м), 6,55 (0,5H, с), 6,57 (0,5H, с), 6,84-6,93 (1H, м), 7,36-7,30 (9H, м), 7,51 (1H, с).
[Стадия 2] 2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-(L-норлейциламино)валериановая кислота
[Формула 116]
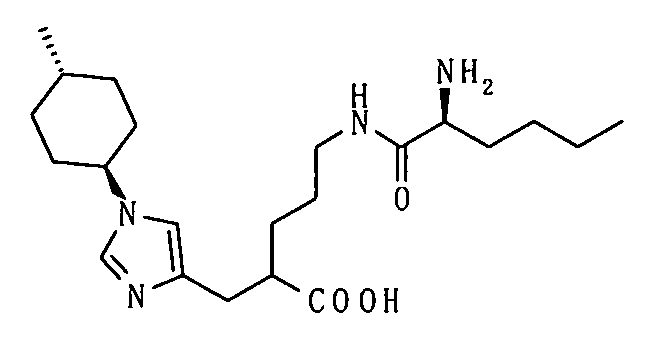
Указанное в заголовке соединение (124 мг) получали из соединения (244 мг), полученного на стадии 1 данного примера, таким же образом, как на стадии 2 примера 17.
1H-ЯМР (CDCl3) δ: 0,84-0,89 (3H, м), 0,94 (3H, д, J=6,3 Гц), 1,04-1,14 (2H, м), 1,26-1,68 (13H, м), 1,79-1,87 (2H, м), 2,03-2,10 (2H, м), 2,58-2,69 (2H, м), 2,85 (1H, дд, J=14,5, 7,4 Гц), 3,11-3,27 (2H, м), 3,45-3,52 (1H, м), 3,77-3,83 (1H, м), 6,72 (1H, с), 7,52 (1H, с), 8,03 (1H, шир.с).
МСВР (ESI): m/z вычислено для C22H39N4O3: 407,30221 [M+H]+; найдено: 407,30257.
[Пример 19] (2S)-2-{[1-(Транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-({[(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонил}амино)валериановая кислота
[Формула 117]
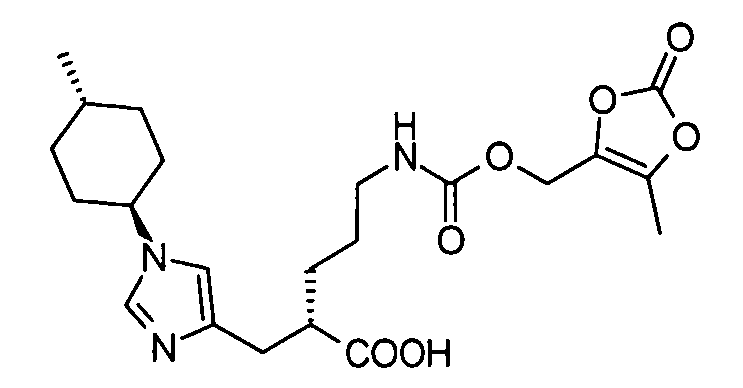
Соединение (200 мг), полученное на стадии 4 примера 15, растворяли в смешанном растворителе из N,N-диметилформамида (2 мл) и воды (1 мл). К раствору при комнатной температуре добавляли (5-метил-2-оксо-1,3-диоксол-4-ил)метил-4-нитрофенилкарбонат (336 мг) (J. Med. Chem., 1996, Vol. 39, p. 480), и смесь перемешивали в течение 4 дней. Растворитель в реакционном растворе отгоняли при пониженном давлении, и остаток затем подвергали тонкослойной хроматографии, с получением указанного в заголовке соединения (100 мг).
1H-ЯМР (CDCl3) δ: 0,96 (3H, д, J=6,5 Гц), 1,08-1,18 (2H, м), 1,40-1,51 (2H, м), 1,55-1,78 (5H, м), 1,82-1,90 (2H, м), 2,07-2,15 (2H, м), 2,18 (3H, с), 2,70-2,84 (3H, м), 3,13-3,20 (2H, м), 3,86-3,95 (1H, м), 4,79 (2H, с), 5,18 (1H, шир.с), 6,78 (1H, с), 7,74 (1H, с).
МСВР (ESI): m/z вычислено для C22H32N3O7: 450,22402 [M+H]+; найдено: 450,22369.
[Пример 20] (2S)-5-({[1-(Изобутирилокси)этокси]карбонил}амино)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] 1-[(хлоркарбонил)окси]этил-2-метилпропионат
[Формула 118]
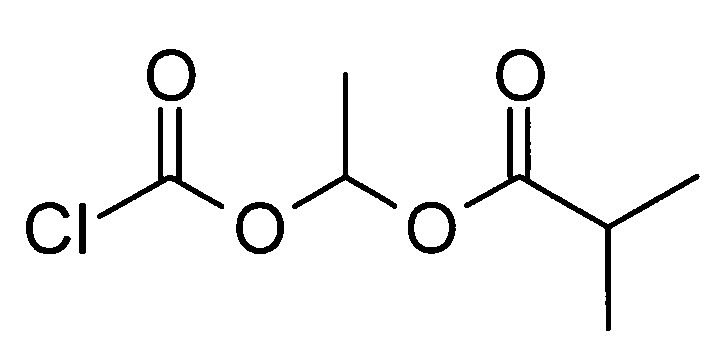
1-{[(этилтио)карбонил]окси}этил-2-метилпропионат (WO2005/66122 (412 мг) охлаждали до -30°С. К нему добавляли сульфурилхлорид (157 мкл), и смесь затем перемешивали в течение 45 минут. Растворитель в реакционном растворе отгоняли при пониженном давлении, с получением неочищенного продукта указанного в заголовке соединения.
[Стадия 2] (2S)-5-({[1-(изобутирилокси)этокси]карбонил}амино)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 119]
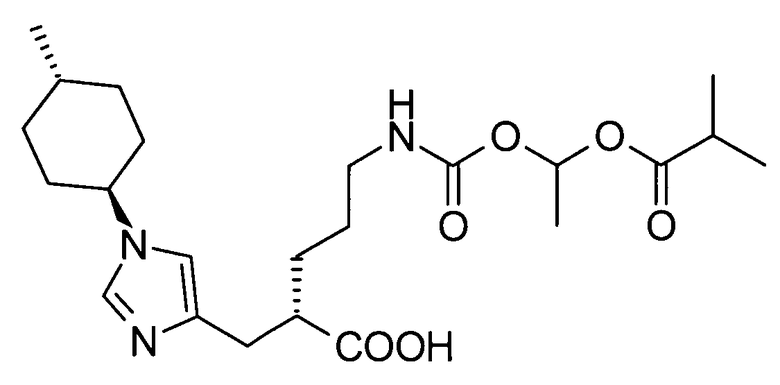
Соединение (500 мг), полученное на стадии 4 примера 15, растворяли в смешанном растворителе из N,N-диметилформамида (6 мл) и воды (2 мл). К раствору при 0°С добавляли раствор соединения, полученного на стадии 1 данного примера, в метиленхлориде (1 мл), и смесь перемешивали в течение 3 дней. Растворитель в реакционном растворе отгоняли при пониженном давлении, и органическое вещество экстрагировали три раза смешанным растворителем этилацетат-метанол (95:5). Органический слой сушили над безводным сульфатом натрия, и растворитель затем отгоняли при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат-метиленхлорид/метанол=95/5), и полученное твердое вещество дополнительно промывали водой, с получением представляющего интерес указанного в заголовке соединения (97 мг).
1H-ЯМР (CDCl3) δ: 0,96 (3H, д, J=6,3 Гц), 1,07-1,13 (2H, м), 1,16 (6H, д, J=7,0 Гц), 1,41-1,49 (5H, м), 1,57-1,78 (5H, м), 1,84-1,90 (2H, м), 2,08-2,14 (2H, м), 2,53 (1H, тт, J=7,0, 7,0 Гц), 2,70-2,85 (3H, м), 3,12-3,20 (2H, м), 3,84-3,92 (1H, м), 4,96 (1H, шир.с), 6,76-6,80 (2H, м), 7,71 (1H, с).
МСВР (ESI): m/z вычислено для C23H38N3O6: 452,27606 [M+Na]+; найдено: 452,27610.
[Пример 21] 1-[(Изопропоксикарбонил)окси]этил-(2S)-5-({[1-(изобутирилокси)этокси]карбонил}амино)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерат
[Стадия 1] 1-иодоэтилизопропилкарбонат
[Формула 120]
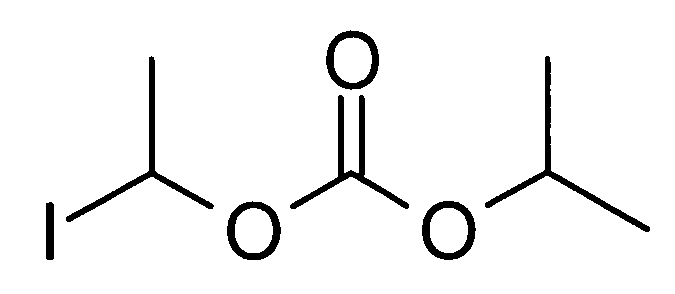
К раствору 1-хлорэтилизопропилкарбоната (1,00 г) в толуоле (30 мл) при комнатной температуре добавляли йодид натрия (2,10 г) и 18-краун-6 (185 мг), и смесь перемешивали при 100°С в течение 5 часов. К реакционному раствору добавляли этилацетат, и смесь промывали водой и 5% водным тиосульфатом натрия в указанном порядке и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, с получением неочищенного продукта указанного в заголовке соединения (1,51 г).
1H-ЯМР (CDCl3) δ: 1,32 (3H, д, J=6,3 Гц), 1,34 (3H, д, J=6,3 Гц), 2,24 (3H, д, J=5,9 Гц), 4,95 (1H, тт, J=6,3, 6,3 Гц), 6,76 (1H, кв., J=5,9 Гц).
[Стадия 2] 1-[(изопропоксикарбонил)окси]этил-(2S)-5-({[1-(изобутирилокси)этокси]карбонил}амино)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерат
[Формула 121]
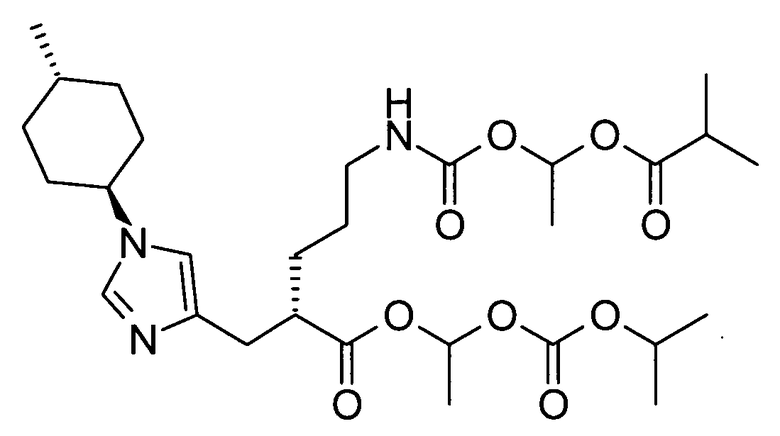
Соединение (97 мг), полученное в примере 20, растворяли в смешанном растворителе из тетрагидрофурана (1 мл) и воды (1 мл). К раствору добавляли бикарбонат натрия (18 мг), и смесь перемешивали при комнатной температуре в течение 3,5 часов. Растворитель в реакционном растворе отгоняли при пониженном давлении. Полученный остаток растворяли в N,N-диметилформамиде (3 мл), и к нему при 0°С добавляли соединение (74 мг), полученное на стадии 1 данного примера. Через три дня, к смеси добавляли соединение (25 мг), полученное на стадии 1 данного примера, и бикарбонат натрия (6 мг), и смесь дополнительно перемешивали в течение 20 часов. Растворитель в реакционном растворе отгоняли при пониженном давлении, и остаток затем очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат-метиленхлорид/метанол=90/10). Полученный неочищенный продукт повторно очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат), с получением указанного в заголовке соединения (43 мг).
1H-ЯМР (CDCl3) δ: 0,95 (3H, д, J=6,3 Гц), 1,05-1,17 (8H, м), 1,30-1,32 (6H, м), 1,42-1,69 (13H, м), 1,82-1,87 (2H, м), 2,05-2,11 (2H, м), 2,49-2,56 (1H, м), 2,68-2,96 (3H, м), 3,10-3,23 (2H, м), 3,76-3,85 (1H, м), 4,85-4,92 (1H, м), 5,23 (0,5H, шир.с), 5,31 (0,5H, шир.с), 6,68-6,73 (2H, м), 6,79 (1H, кв., J=5,5 Гц), 7,45 (0,5H, с), 7,46 (0,5H, с).
МСВР (ESI): m/z вычислено для C29H48N3O9: 582,33905 [M+H]+; найдено: 582,33901.
[Пример 22] (2S)-5-({[1-(2,2-Диметилпропаноилокси)этокси]карбонил}амино)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] S-этил-О-(1-йодэтил)тиокарбонат
[Формула 122]
О-(1-хлорэтил)-S-этилтиокарбонат (Synthesis, 1986, Vol. 8, p. 627) (5,0 г) растворяли в толуоле (100 мл). К раствору при комнатной температуре добавляли йодид натрия (11,6 г) и 18-краун-6 (2,35 г), и смесь перемешивали при 100°С в течение 4 часов. Реакционный раствор охлаждали до комнатной температуры. К нему добавляли этилацетат, и смесь промывали дважды 5% водным тиосульфатом натрия. Органический слой сушили над безводным сульфатом натрия, и растворитель затем отгоняли при пониженном давлении, с получением неочищенного продукта указанного в заголовке соединения. Полученный неочищенный продукт непосредственно использовали в следующей реакции.
1H-ЯМР (CDCl3) δ: 1,31 (3H, т, J=7,4 Гц), 2,18 (3H, д, J=6,3 Гц), 2,84-2,91 (2H, м), 6,89 (1H, кв., J=6,3 Гц).
[Стадия 2] 1-{[(этилтио)карбонил]окси}этилпивалат
[Формула 123]
Пивалиновую кислоту (3,02 г) растворяли в смешанном растворителе из метиленхлорида (100 мл) и воды (50 мл). К раствору при охлаждении льдом добавляли бисульфат тетрабутиламмония (10,0 г) и бикарбонат натрия (4,97 г) в указанном порядке, и смесь затем перемешивали в течение 30 минут. К смеси затем добавляли раствор соединения, полученного на стадии 1 данного примера, в метиленхлориде (5 мл), и смесь перемешивали при комнатной температуре в течение 6 дней. Органический слой отделяли и сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан-гексан/этилацетат=95/5), с получением указанного в заголовке соединения (2,62 г).
1H-ЯМР (CDCl3) δ: 1,20 (9H, с), 1,31 (3H, т, J=7,4 Гц), 1,50 (3H, д, J=5,5 Гц), 2,84-2,90 (2H, м), 6,92 (1H, кв., J=5,5 Гц).
[Стадия 3] (2S)-5-({[1-(2,2-диметилпропаноилокси)этокси]карбонил}амино)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 124]
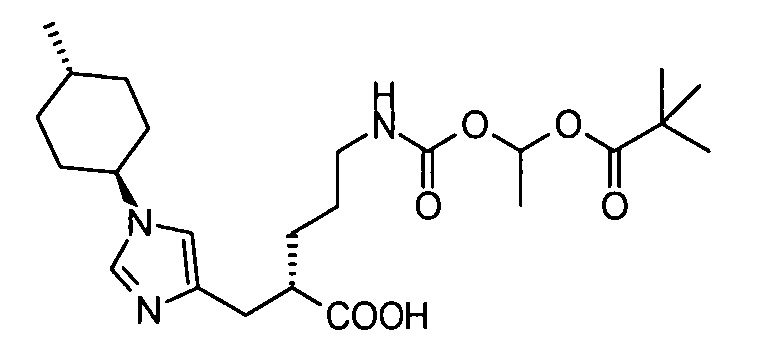
Указанное в заголовке соединение (267 мг) получали из соединения, полученного на стадии 2 данного примера, и соединения (500 мг), полученного на стадии 4 примера 15, таким же образом, как на стадиях 1 и 2 примера 20.
1H-ЯМР (CDCl3) δ: 0,95 (3H, д, J=6,3 Гц), 1,11-1,19 (11H, м), 1,43-1,76 (10H, м), 1,85-1,92 (2H, м), 2,13-2,19 (2H, м), 2,83-2,94 (2H, м), 2,99-3,08 (1H, м), 3,11-3,21 (2H, м), 4,09-4,17 (1H, м), 5,38 (1H, шир.с), 6,75 (1H, кв., J=5,4 Гц), 7,07 (1H, с), 8,79 (1H, с).
МСВР (ESI): m/z вычислено для C24H40N3O6: 466,29171 [M+H]+; найдено: 466,29083.
[Пример 23] (2S)-5-[({1-[(Циклогексилкарбонил)окси]этокси}карбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] 1-{[(этилтио)карбонил]окси}этилциклогексанкарбоксилат
[Формула 125]
Указанное в заголовке соединение (1,62 г) получали из О-(1-хлорэтил)-S-этилтиокарбоната (4,0 г) и циклогексанкарбоновой кислоты (3,04 г) таким же образом, как на стадиях 1 и 2 примера 22.
1H-ЯМР (CDCl3) δ: 1,20-1,28 (3H, м), 1,31 (3H, т, J=7,4 Гц), 1,39-1,48 (2H, м), 1,49 (3H, д, J=5,5 Гц), 1,60-1,66 (1H, м), 1,73-1,77 (2H, м), 1,86-1,93 (2H, м), 2,37-2,27 (1H, м), 2,92-2,82 (2H, м), 6,94 (1H, кв., J=5,5 Гц).
[Стадия 2] (2S)-5-[({1-[(циклогексилкарбонил)окси]этокси}карбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 126]
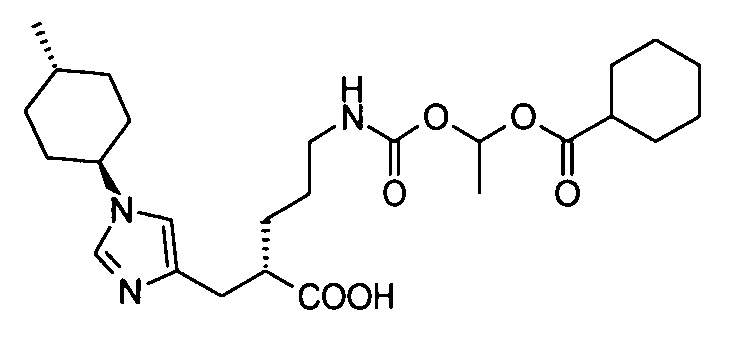
Указанное в заголовке соединение (318 мг) получали из соединения, полученного на стадии 1 данного примера, и соединения (400 мг), полученного на стадии 4 примера 15, таким же образом, как на стадиях 1 и 2 примера 20.
1H-ЯМР (CDCl3) δ: 0,96 (3H, д, J=6,7 Гц), 1,07-1,31 (5H, м), 1,39-1,47 (7H, м), 1,57-1,78 (8H, м), 1,84-1,92 (4H, м), 2,07-2,14 (2H, м), 2,28 (1H, тт, J=11,2, 3,6 Гц), 2,68-2,84 (3H, м), 3,12-3,21 (2H, м), 3,86 (1H, тт, J=12,1, 3,7 Гц), 4,95 (1H, шир.с), 6,76 (1H, с), 6,78 (1H, кв., J=5, 7 Гц), 7, 63 (1H, с).
МСВР (ESI): m/z вычислено для C26H42N3O6: 492,30736 [M+H]+; найдено: 492,30677.
[Пример 24] 2-(2-Аминоэтокси)-3-[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]пропионовая кислота
[Стадия 1] (2Z)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метилен}морфолин-3-он
[Формула 127]
К раствору трет-бутил-3-оксоморфолин-4-карбоксилата (859 мг) в тетрагидрофуране (8 мл) при -78°С добавляли раствор бис(триметилсилил)амида лития в гексане (1,02М, 3,00 мл), и смесь перемешивали при -78°С в течение 30 минут. К полученному реакционному раствору при -78°С добавляли раствор соединения (400 мг), полученного в ссылочном примере 4, в тетрагидрофуране (5 мл). Смесь перемешивали при -78°С в течение 1 часа, затем медленно нагревали до комнатной температуры, и перемешивали в течение 14 часов. К реакционному раствору добавляли насыщенный водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метиленхлорид-метиленхлорид/метанол=10/1), с получением указанного в заголовке соединения (330 мг).
1H-ЯМР (CDCl3) δ: 0,92 (3H, д, J=6,6 Гц), 1,08 (2H, м), 1,43 (1H, м), 1,67 (2H, м), 1,84 (2H, м), 2,09 (2H, м), 3,58 (2Н, м), 3,85 (1Н, тт, J=12,1, 3,9 Гц), 4,24 (2H, м), 6,10 (1H, шир.), 6,93 (1H, с), 7,35 (1H, с), 7,58 (1H, с).
[Стадия 2] 2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}морфолин-3-он
[Формула 128]
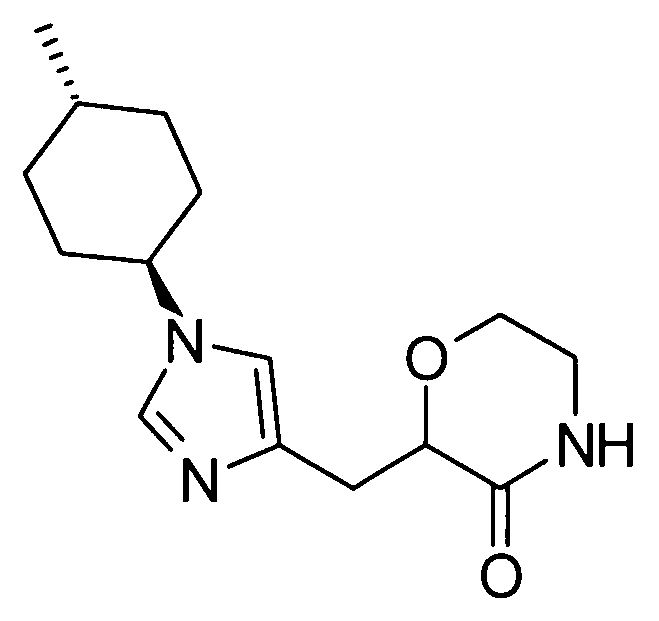
Катализатор 10% палладий-углерод (гидратированный, 300 мг) суспендировали в растворе соединения (330 мг), полученного на стадии 1 данного примера, в этаноле (8 мл). Суспензию перемешивали в атмосфере водорода при нормальном давлении при комнатной температуре в течение 1 часа и при 45°С в течение 1 часа. Реакционный раствор фильтровали через целит, и фильтрат концентрировали. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метиленхлорид/метанол=20/1-10/1), с получением указанного в заголовке соединения (325 мг).
1H-ЯМР (CDCl3) δ: 0,94 (3H, д, J=6,6 Гц), 1,09 (2Н, м), 1,44 (1Н, м), 1,65 (2H, м), 1,84 (2H, м), 2,09 (2H, м), 3,2 (1H, дд, J=15,2, 9,0 Гц), 3,25-3,32 (2H, м), 3,54 (1H, м), 3,75 (1H, м), 3,80 (1H, тт, J=12,1, 3,9 Гц), 4,3 (1H, м), 4,47 (1H, дд, J=9,0, 3,1 Гц), 6,31 (1H, шир.), 6,80 (1H, с), 7,45 (1H, с).
[Стадия 3] 2-(2-аминоэтокси)-3-[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]пропионовая кислота
[Формула 129]
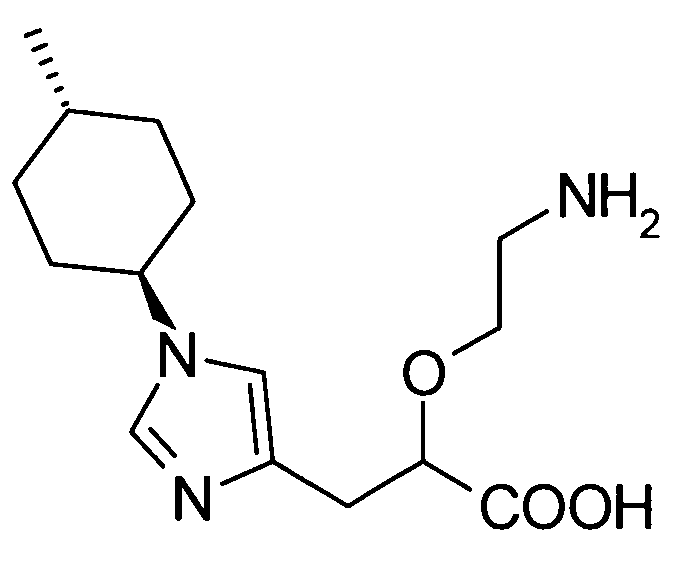
К соединению (300 мг), полученному на стадии 2 данного примера, добавляли концентрированную хлористоводородную кислоту (7 мл), и смесь нагревали при кипячении с обратным холодильником в течение 8 часов. Затем растворитель отгоняли при пониженном давлении. Полученный неочищенный гидрохлорид растворяли в метаноле, и к нему добавляли DOWEX 50WX8-200. Смолу промывали водой с последующим элюированием 4% аммиачной водой. Элюат концентрировали, с получением указанного в заголовке соединения (154 мг).
1H-ЯМР (CD3OD) δ: 0,95 (3H, д, J=6,6 Гц), 1,15 (2Н, м), 1,47 (1Н, м), 1,72 (2H, м), 1,84 (2H, м), 2,04 (2H, м), 2,83-3,07 (4H, м), 3,58-3,68 (2H, м), 3,90-4,01 (2H, м), 6, 98 (1H, с), 7,58 (1H, с).
МСВР (ESI): m/z вычислено для C15H26N3O3: 296,1974 [M+H]+; найдено: 296,1962.
[Пример 25] 2-[(1R)-2-Амино-1-метилэтокси]-3-[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]пропионовая кислота
[Стадия 1] (6R)-4-(метоксиметил)-6-метилморфолин-3-он
[Формула 130]
К гидриду натрия (63%, 4,4 г, 116 ммоль), суспендированному в тетрагидрофуране (100 мл), при охлаждении льдом в течение 30 минут по каплям добавляли раствор (6R)-6-метилморфолин-3-она (ЕР350002) (12,1 г) в тетрагидрофуране (50 мл). Смесь перемешивали при той же температуре в течение 30 минут, и затем дополнительно перемешивали при комнатной температуре в течение 30 минут. При охлаждении льдом в течение 30 минут к смеси по каплям добавляли раствор хлорметилметилового эфира (10 мл) в тетрагидрофуране (50 мл). Смесь перемешивали в течение 30 минут при охлаждении льдом, и затем перемешивали в течение ночи при комнатной температуре. К ней добавляли соответствующее количество воды для разделения, с последующими несколькими экстракциями этилацетатом. Органический слой сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, и полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан-гексан/этилацетат=40/60), с получением указанного в заголовке соединения (7,86 г).
1H-ЯМР (CDCl3) δ: 1,30 (3H, д, J=5,9 Гц), 3,22-3,34 (5H, м), 3,86-3,95 (1H, м), 4,19 (1H, д, J=16,8 Гц), 4,31 (1H, д, J=16,8 Гц), 4,75 (1H, д, J=9,8 Гц), 4,88 (1H, д, J=9,8 Гц).
[Стадия 2] (6R)-4-(метоксиметил)-6-метил-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}морфолин-3-он
[Формула 131]
Диизопропиламин (1,05 мл) растворяли в тетрагидрофуране (10 мл). К раствору при 0°С добавляли раствор н-бутиллития в гексане (1,57М, 4,50 мл), и смесь перемешивали при 0°С в течение 15 минут и при комнатной температуре в течение 5 минут. Реакционный раствор охлаждали до -78°С. Затем к нему добавляли раствор соединения (1,16 г), полученного на стадии 1 данного примере, в тетрагидрофуране (5 мл), и смесь перемешивали при -78°С в течение 1,5 часов. Затем при -78°С к нему добавляли раствор соединения (1,00 г), полученного в ссылочном примере 4, в тетрагидрофуране (5 мл). После перемешивания при -78°С в течение 30 минут, смесь нагревали до комнатной температуры и перемешивали в течение 14 часов. К реакционному раствору добавляли насыщенный водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метиленхлорид-метиленхлорид/метанол=10/1). Полученный неочищенный продукт растворяли в метиленхлориде (10 мл). К раствору добавляли триэтиламин (1,45 мл) и метансульфонилхлорид (0,40 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли насыщенный водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Полученный остаток растворяли в тетрагидрофуране (10 мл). К раствору добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (0,90 мл), и смесь перемешивали при комнатной температуре в течение 3 часов. К реакционному раствору добавляли насыщенный водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Остаток растворяли в этаноле (10 мл), и катализатор 10% палладий-углерод (гидратированный, 300 мг) суспендировали в растворе. Суспензию перемешивали при 50°С в течение 6 часов в атмосфере водорода при нормальном давлении. Реакционный раствор фильтровали через целит, и фильтрат концентрировали. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метиленхлорид-метиленхлорид/метанол=10/1), с получением указанного в заголовке соединения (945 мг).
1H-ЯМР (CD3OD) δ: 0,95 (3H, д, J=6,6 Гц), 1,15 (2H, м), 1,23 (3H, д, J=6,3 Гц), 1,48 (1H, м), 1,71 (2H, м), 1,84 (2H, м), 2,03 (2H, м), 2,97 (1H, дд, J=15,2, 7,0 Гц), 3,14 (1H, м), 3,18 (3H, с), 3,23-3,38 (2H, м), 3,91-3,99 (2H, м), 4,43 (1H, дд, J=7,4, 3,5 Гц), 4,69 (1H, д, J=10,2 Гц), 4,79 (1H, д, J=10,2 Гц), 6,96 (1H, с), 7,58 (1H, с).
[Стадия 3] 2-[(1R)-2-амино-1-метилэтокси]-3-[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]пропионовая кислота
[Формула 132]
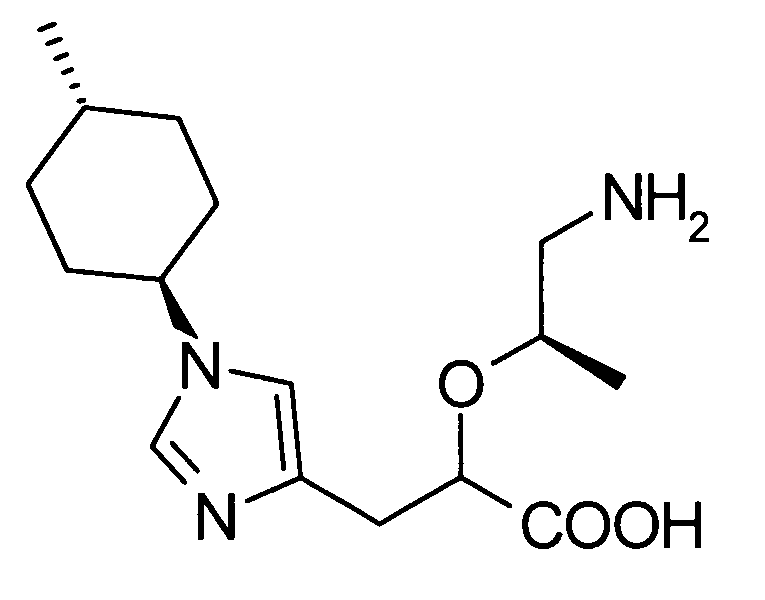
К соединению (100 мг), полученному на стадии 2 данного примера, добавляли концентрированную хлористоводородную кислоту (4 мл), и смесь нагревали при кипячении с обратным холодильником в течение 20 часов. Затем растворитель отгоняли при пониженном давлении. Полученный неочищенный гидрохлорид растворяли в воде, и к нему добавляли DOWEX 50WX8-200. Смолу промывали водой с последующим элюированием 4% аммиачной водой. Элюат концентрировали, с получением указанного в заголовке соединения (35 мг).
1H-ЯМР (CD3OD) δ: 0,93 (3H, д, J=6,3 Гц), 0,95 (3H, д, J=6,8 Гц), 1,16 (2H, м), 1,48 (1H, м), 1,73 (2H, м), 1,84 (2H, м), 2,03 (2H, м), 2,75 (1H, м), 2,77 (1H, дд, J=14,6, 9,8 Гц), 2,95 (1H, м), 3,08 (1H, дд, J=14,6, 3,4 Гц), 3,55 (1H, м), 3,96 (1H, тт, J=12,2, 3,9 Гц), 4,02 (1H, дд, J=9,8, 3,4 Гц), 6,98 (1H, с), 7,59 (1H, с).
МСВР (ESI): m/z вычислено для C16H28N3O3: 310,2131 [M+H]+; найдено: 310,2131.
[Пример 26] 2-[(3S)-3-Аминопирролидинил-1-ил]-3-[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]пропионовая кислота
[Стадия 1] Этил-3-[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]пропионат
[Формула 133]
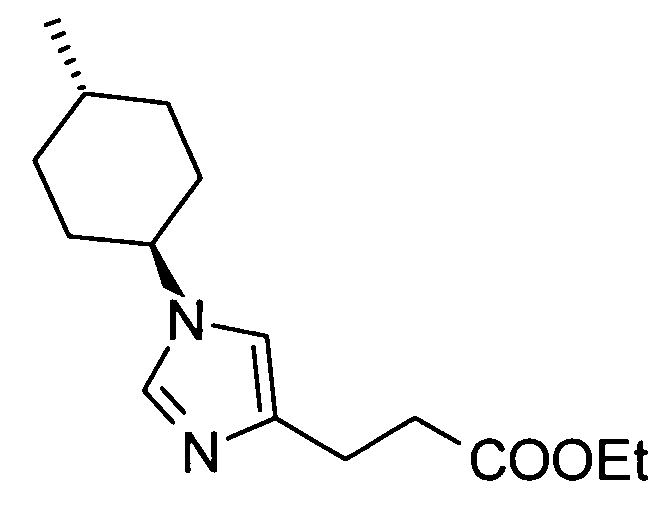
Этилдиэтилфосфоноацетат (1,89 г) растворяли в тетрагидрофуране (15 мл), и к нему при 0°С добавляли гидрид натрия (63%, 321 мг). После перемешивания при 0°С в течение 1 часа, к смеси при 0°С добавляли раствор соединения (1,20 г), полученного в ссылочном примере 4, в тетрагидрофуране (6 мл), и смесь перемешивали при 0°С в течение 1 часа. К реакционному раствору добавляли насыщенный водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Полученный неочищенный продукт растворяли в этаноле (20 мл). К раствору добавляли катализатор 10% палладий-углерод (гидратированный, 500 мг), и смесь перемешивали при 55°С в течение 5 часов в атмосфере водорода при нормальном давлении. После фильтрования через целит, фильтрат концентрировали при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на NH-силикагеле (элюирующий растворитель: гексан/этилацетат=2/1-1/2), с получением указанного в заголовке соединения (1,06 мг).
1H-ЯМР (CDCl3) δ: 0,94 (3H, д, J=6,6 Гц), 1,03-1,15 (2H, м), 1,23 (3H, т, J=7,0 Гц), 1,45 (1H, м), 1,57-1,69 (2H, м), 1,80-1,88 (2H, м), 2,03-2,10 (2H, м), 2,66 (2H, т, J=7,4 Гц), 2,88 (2H, т, J=7,4 Гц), 3,79 (1H, тт, J=12,1, 3,9 Гц), 4,13 (2H, кв., J=7,0 Гц), 6,70 (1H, с), 7,42 (1H, с).
[Стадия 2] Этил-2-{(3S)-3-[(трет-бутоксикарбонил)амино]пирролидинил-1-ил}-3-[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]пропионат
[Формула 134]
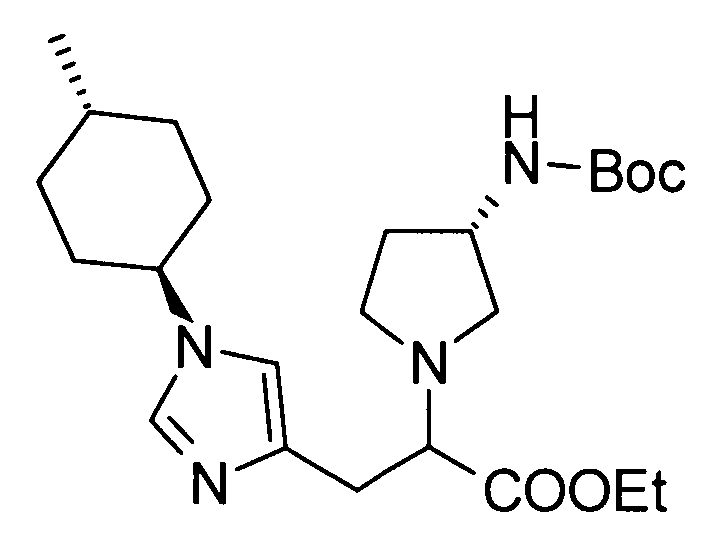
К раствору соединения (400 мг), полученного на стадии 1 данного примера, в тетрагидрофуране (5 мл) при -78°С добавляли раствор бис(триметилсилил)амида лития в гексане (1,02М, 2,00 мл), и смесь перемешивали при -78°С в течение 1 часа. К смеси при -78°С добавляли хлортриметилсилан (0,27 мл) и смесь перемешивали при -78°С в течение 30 минут. Затем при -78°С к ней медленно добавляли по каплям суспензию N-бромсукцинимида (380 мг) в тетрагидрофуране (6 мл). После перемешивания при -78°С в течение 1 часа, потребление реагента было подтверждено, и при -78°С к смеси затем добавляли раствор трет-бутил-(3S)-пирролидин-3-илкарбамата (563 мг) в тетрагидрофуране (3 мл). Смесь нагревали до комнатной температуры и перемешивали в течение 2 часов, и к ней затем добавляли диизопропилэтиламин (0,79 мл). После перемешивания при 50°С в течение 12 часов к реакционному раствору добавляли насыщенный водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=2/1-этилацетат-метиленхлорид/метанол=10/1), с получением указанного в заголовке соединения (269 мг).
1H-ЯМР (CDCl3) δ: 0,95 (3H, д, J=6,3 Гц), 1,05-1,14 (2H, м), 1,18 (1,5H, т, J=7,3 Гц), 1,18 (1,5H, т, J=7,3 Гц), 1,39-1,70 (13H, м), 1,81-1,88 (2H, м), 2,03-2,09 (2H, м), 2,11-2,21 (1H, м), 2,57-2,76 (2H, м), 2,85-3,05 (4H, м), 3,59-3,65 (1H, м), 3,79 (1H, тт, J=12,2, 3,9 Гц), 4,06-4,22 (3H, м), 5,01 (0,5H, шир.), 5,18 (0,5H, шир.), 6,71 (1H, с), 7,43 (1H, с).
[Стадия 3] 2-[(3S)-3-аминопирролидинил-1-ил]-3-[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]пропионовая кислота
[Формула 135]
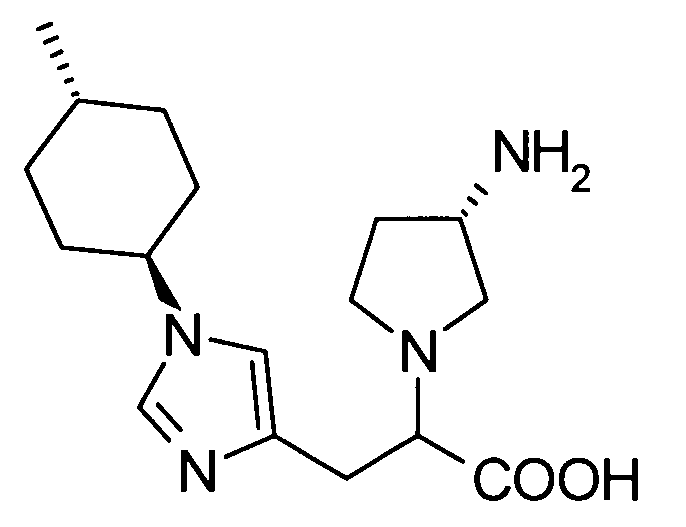
К соединению (160 мг), полученному на стадии 2 данного примера, добавляли концентрированную хлористоводородную кислоту (5 мл), и смесь нагревали при кипячении с обратным холодильником в течение 10 часов. Растворитель отгоняли при пониженном давлении. Затем полученный неочищенный гидрохлорид растворяли в метаноле, и к нему добавляли DOWEX 50WX8-200. Смолу промывали метанолом с последующим элюированием 4% аммиачной водой. Элюат концентрировали, с получением указанного в заголовке соединения (111 мг).
1H-ЯМР (CD3OD) δ: 0,99 (3H, д, J=6,7 Гц), 1,12-1,25 (2H, м), 1,51 (1H, м), 1,69-1,92 (5H, м), 2,03-2,13 (2H, м), 2,25 (1H, м), 2,65-2,74 (1H, м), 2,83-2,90 (1H, м), 2,91-3,14 (3H, м), 3,19 (0,5Н, м), 3,27 (0,5H, м), 3,33-3,38 (1H, м), 3,74 (1H, м), 4,00 (1H, тт, J=12,1, 3,9 Гц), 7,08 (1H, с), 7,70 (0,5H, с), 7,72 (0,5H, с).
МСВР (ESI): m/z вычислено для C17H29N4O2: 321,2291 [M+H]+; найдено: 321,2283.
[Пример 27] (2S)-5-{[(1-Ацетоксиэтокси)карбонил]амино}-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] 1-{[(этилтио)карбонил]окси}этилацетат
[Формула 136]
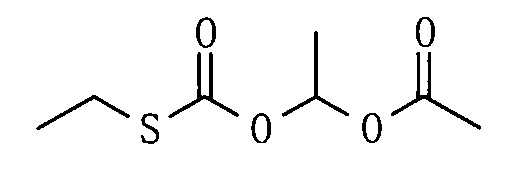
Уксусную кислоту (1,69 мл) растворяли в смешанном растворителе из метиленхлорида (100 мл) и воды (50 мл). К раствору при охлаждении льдом добавляли бисульфат тетрабутиламмония (10,0 г) и бикарбонат натрия (4,97 г) в указанном порядке, и смесь затем перемешивали в течение 1 часа. Затем к ней добавляли соединение, полученное на стадии 1 примера 22, и смесь перемешивали при комнатной температуре в течение 3 дней. Органический слой отделяли и сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан-гексан/этилацетат=95/5), с получением указанного в заголовке соединения (1,67 г).
1H-ЯМР (CDCl3) δ: 1,32 (3H, т, J=7,4 Гц), 1,51 (3H, д, J=5,9 Гц), 2,09 (3H, с), 2,81-2,95 (2Н, м), 6,94 (1Н, кв., J=5,9 Гц).
[Стадия 2] 1-[(хлоркарбонил)окси]этилацетат
[Формула 137]
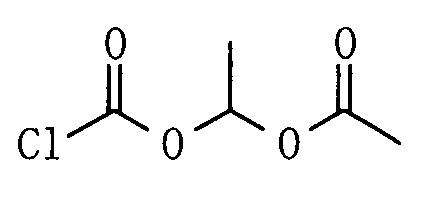
Соединение (394 мг), полученное на стадии 1 данного примера, охлаждали до -30°С. К нему добавляли сульфурилхлорид (175 мкл), и смесь затем перемешивали в течение 30 минут. Растворитель в реакционном растворе отгоняли при пониженном давлении, с получением неочищенного продукта указанного в заголовке соединения.
[Стадия 3] (2S)-5-{[(1-ацетоксиэтокси)карбонил]амино}-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 138]
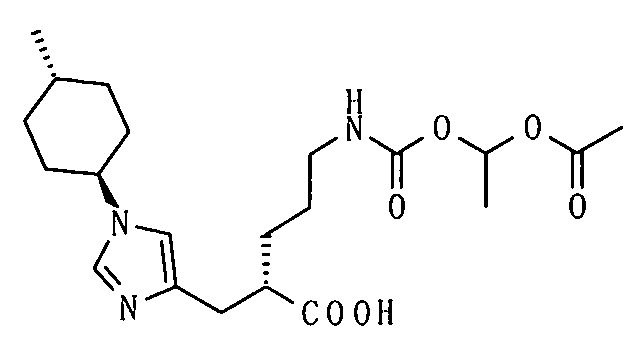
Соединение (400 мг), полученное на стадии 4 примера 15, растворяли в смешанном растворителе из ацетонитрила (12 мл) и воды (3 мл). К раствору при 0°С добавляли раствор соединения, полученного на стадии 2 данного примера, в метиленхлориде (1 мл), и смесь перемешивали в течение 2,5 часов. Растворитель в реакционном растворе отгоняли при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат-метиленхлорид/метанол=90/10). Полученное твердое вещество растворяли в смешанном растворителе этилацетат-ацетон. Нерастворимое вещество отфильтровывали, и растворитель в фильтрате отгоняли при пониженном давлении. Полученный остаток очищали ВЭЖХ с обращенной фазой, с получением указанного в заголовке соединения (185 мг).
1H-ЯМР (CDCl3) δ: 0,96 (3H, д, J=6,3 Гц), 1,09-1,21 (2H, м), 1,45 (3H, д, J=5,5 Гц), 1,52-1,75 (6H, м), 1,86-1,93 (2H, м), 2,06 (3H, с), 2,13-2,19 (2H, м), 2,82-2,91 (2H, м), 2,97-3,05 (1H, м), 3,15-3,21 (2H, м), 4,03-4,11 (1H, м), 5,31 (1H, шир.с), 6,77-6,81 (1H, м), 6,99 (1H, с), 8,97 (1H, с).
[Пример 28] (2S)-2-{[1-(Транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-[({[(2-метилпропаноил)окси]метокси}карбонил)амино]валериановая кислота
[Стадия 1] S-этил-О-(йодметил)тиокарбонат
[Формула 139]
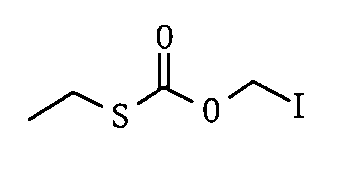
К раствору О-(хлорметил)-S-этилтиокарбоната (10 г) в толуоле (100 мл) добавляли йодид натрия (29,1 г) и 18-краун-6 (5,1 г), и смесь перемешивали при комнатной температуре в течение 19 часов. Поскольку исходные вещества все еще оставались, к смеси дополнительно добавляли йодид натрия (29,1 г) и 18-краун-6 (5,1 г), и смесь перемешивали при комнатной температуре в течение 48 часов и затем при 100°С в течение 5 часов. К ней добавляли этилацетат (100 мл), и органический слой отделяли промывкой 20% водным тиосульфатом натрия. Этилацетат (50 мл) добавляли к водному слою для повторной экстракции. Объединенный органический слой сушили над безводным сульфатом магния, и растворитель затем отгоняли при пониженном давлении. Остаток сушили, с получением указанного в заголовке соединения (12,1 г).
1H-ЯМР (CDCl3) δ: 1,34 (3H, т, J=7,4 Гц), 2,93 (2H, кв., J=7,4 Гц), 5,99 (2H, с).
[Стадия 2] {[(Этилсульфанил)карбонил]окси}метил-2-метилпропаноат
[Формула 140]
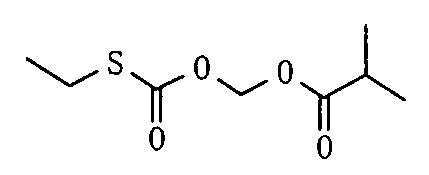
К изомасляной кислоте (2,9 мл), растворенной в смешанном растворителе из метиленхлорида и воды (1:2, 120 мл), при охлаждении льдом добавляли бисульфат тетрабутиламмония (11,0 г) и бикарбонат натрия (5,5 г), и смесь перемешивали при той же температуре в течение 10 минут. К полученному реакционному раствору при комнатной температуре добавляли раствор соединения (4,0 г), полученного на стадии 1 данного примера, в метиленхлориде (10 мл), и смесь перемешивали в течение ночи. Органический слой отделяли, и водный слой затем подвергали несколько раз экстракции метиленхлоридом. Объединенный органический слой сушили над безводным сульфатом магния, и растворитель затем отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан-гексан/этилацетат=95/5), с получением указанного в заголовке соединения (2,8 г).
1H-ЯМР (CDCl3) δ: 1,19 (6H, д, J=7,0 Гц), 1,33 (3H, т, J=7,4 Гц), 2,57-2,64 (1H, м), 2,90 (2H, кв., J=7,4 Гц), 5,81 (2H, с).
[Стадия 3] [(Хлоркарбонил)окси]метил-2-метилпропаноат
[Формула 141]
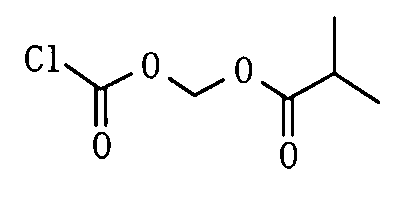
Соединение (400 мг), полученное на стадии 2 данного примера, охлаждали до -30°С. К нему добавляли сульфурилхлорид (159 мкл), и смесь перемешивали при той же температуре в течение 20 минут. Смесь в свою очередь перемешивали в течение 20 минут на бане со льдом и дополнительно при комнатной температуре в течение 1 часа. Растворитель отгоняли при пониженном давлении, и остаток сушили, с получением неочищенного продукта указанного в заголовке соединения.
1H-ЯМР (CDCl3) δ: 1,22 (6H, д, J=7,0 Гц), 2,60-2,70 (1H, м), 5,83 (2H, с).
[Стадия 4] (2S)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-[({[(2-метилпропаноил)окси]метокси}карбонил)амино]валериановая кислота
[Формула 142]
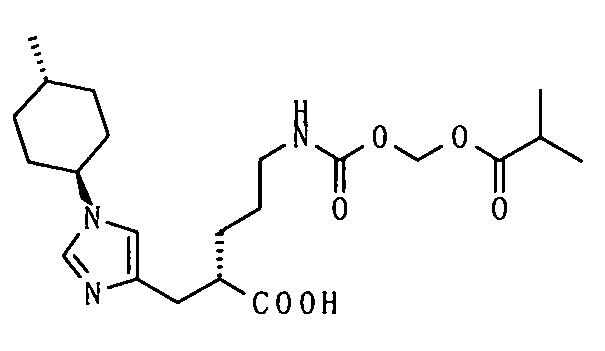
Соединение (400 мг), полученное на стадии 4 примера 15, растворяли в смешанном растворителе из ацетонитрила и воды (1/1, 12 мл). К раствору при охлаждении льдом добавляли триэтиламин (367 мкл). К смеси добавляли раствор соединения, полученного на стадии 3 данного примера, в ацетонитриле (3,0 мл), и смесь перемешивали в течение 1,5 часов при охлаждении льдом и затем при комнатной температуре в течение одного дня и ночи. Растворитель отгоняли при пониженном давлении, и затем к остатку добавляли воду с последующей экстракцией несколько раз этилацетатом. Объединенный органический слой сушили над безводным сульфатом магния, и растворитель затем отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: метиленхлорид-метиленхлорид/метанол=85/15), с получением указанного в заголовке соединения (178 мг).
1H-ЯМР (CDCl3) δ: 0,96 (3H, д, J=6,7 Гц), 1,07-1,17 (2H, м), 1,18 (6H, д, J=7,0 Гц), 1,40-1,51 (2H, м), 1,57-1,81 (5H, м), 1,84-1,91 (2H, м), 2,07-2,14 (2H, м), 2,54-2,64 (1H, м), 2,67-2,75 (1H, м), 2,78-2,89 (2H, м), 3,17-3,22 (2H, м), 3,87 (1H, тт, J=12,1, 3,9 Гц), 5,14 (1H, шир.с), 5,71 (2H, с), 6,75 (1H, с), 7,66 (1H, с).
МСВР (ESI): m/z вычислено для C22H36N3O6: 481,26041 [M+H]+; найдено: 438,26052.
[Пример 29] (2S)-5-[({[(2,2-Диметилпропаноил)окси]метилокси}карбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] {[(Этилсульфанил)карбонил]окси}метил-2,2-диметилпропаноат
[Формула 143]
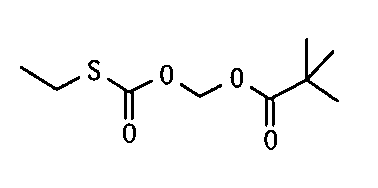
К пивалевой кислоте (4,2 г), растворенной в смешанном растворителе из метиленхлорида и воды (1/2, 120 мл), при охлаждении льдом добавляли бисульфат тетрабутиламмония (11,0 г) и бикарбонат натрия (6,8 г), и смесь перемешивали при той же температуре в течение 10 минут. К полученному реакционному раствору при комнатной температуре добавляли раствор соединения (5,0 г), полученного на стадии 1 примера 28, в метиленхлориде (10 мл), и смесь перемешивали в течение одного дня и ночи. Органический слой отделяли, и водный слой затем дополнительно подвергали несколько раз экстракции метиленхлоридом. Объединенный органический слой сушили над безводным сульфатом магния, и растворитель затем отгоняли при пониженном давлении. Поскольку твердое вещество осаждалось, твердое вещество суспендировали в диэтиловом эфире и отфильтровывали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан-гексан/этилацетат=98/2), с получением указанного в заголовке соединения (3,6 г).
1H-ЯМР (CDCl3) δ: 1,22 (9H, с), 1,33 (3H, т, J=7,4 Гц), 2,89 (2H, кв., J=7,4 Гц), 5,81 (2H, с).
[Стадия 2] (2S)-5-[({[(2,2-диметилпропаноил)окси]метилокси}карбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 144]
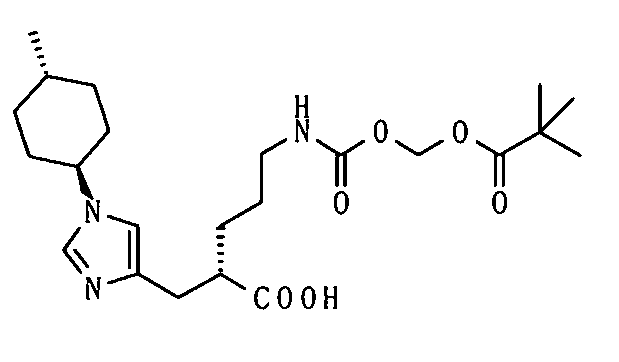
Указанное в заголовке соединение (297 мг) получали из соединения (437 мг), полученного на стадии 1 данного примера, и соединения (400 мг), полученного на стадии 4 примера 15, таким же образом, как на стадиях 3 и 4 примера 28.
1H-ЯМР (CDCl3) δ: 0,96 (3H, д, J=6,3 Гц), 1,07-1,17 (2H, м), 1,21 (9H, с), 1,41-1,50 (2H, м), 1,58-1,78 (5H, м), 1,84-1,90 (2H, м), 2,07-2,14 (2H, м), 2,67-2,74 (1H, м), 2,77-2,89 (2H, м), 3,17-3,22 (2H, м), 3,87 (1H, тт, J=12,1, 3,9 Гц), 5,13 (1H, шир.с), 5,71 (2H, с), 6,75 (1H, с), 7,67 (1H, с).
МСНР (ESI) m/z 452 [M+H]+.
МСВР (ESI): m/z вычислено для C23H38N3O6: 452,27606 [M+H]+; найдено: 452,27619.
[Пример 30] (2S)-5-[({[(Циклогексилкарбонил)окси]метокси}карбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] {[(Этилсульфанил)карбонил]окси}метилциклогексанкарбоксилат
[Формула 145]
Указанное в заголовке соединение (4,1 г) получали из циклогексанкарбоновой кислоты (5,2 г) и соединения (5,0 г), полученного на стадии 1 примера 28, таким же образом, как на стадии 1 примера 29.
1H-ЯМР (CDCl3) δ: 1,20-1,31 (3H, м), 1,33 (3H, т, J=7,4 Гц), 1,40-1,50 (2H, м), 1,60-1,67 (1H, м), 1,72-1,79 (2H, м), 1,87-1,95 (2H, м), 2,36 (1H, тт, J=11,3, 3,5 Гц), 2,89 (2H, кв., J=7,4 Гц), 5,80 (2H, с).
[Стадия 2] (2S)-5-[({[(циклогексилкарбонил)окси]метокси}карбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 146]
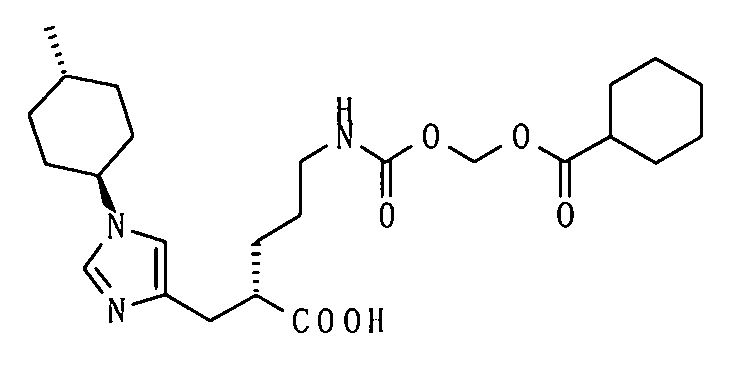
Указанное в заголовке соединение (281 мг) получали в виде белого твердого вещества из соединения (489 мг), полученного на стадии 1 данного примера, и соединения (400 мг), полученного на стадии 4 примера 15, таким же образом, как на стадиях 3 и 4 примера 28.
1H-ЯМР (CDCl3) δ: 0,96 (3H, д, J=6,3 Гц), 1,07-1,18 (2H, м), 1,19-1,33 (3H, м), 1,39-1,50 (4H, м), 1,58-1,78 (8H, м), 1,85-1,94 (4H, м), 2,08-2,14 (2H, м), 2,35 (1H, тт, J=11,3, 3,9 Гц), 2,67-2,74 (1H, м), 2,76-2,90 (2H, м), 3,17-3,22 (2H, м), 3,87 (1H, тт, J=12,1, 3,9 Гц), 5,17 (1H, шир.с), 5,71 (2H, с), 6,76 (1H, с), 7,68 (1H, с).
МСНР (ESI) m/z 478 [M+H]+.
МСВР (ESI): m/z вычислено для C25H40N3O6: 478,29171 [M+H]+; найдено: 478,29145.
[Пример 31] (2S)-5-({[(Ацетилокси]метокси]карбонил}амино)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] {[(Этилсульфанил)карбонил]окси}метилацетат
[Формула 147]
Указанное в заголовке соединение (0,86 г) получали из уксусной кислоты (0,78 г) и соединения (1,6 г), полученного на стадии 1 примера 28, таким же образом, как на стадии 1 примера 29.
1H-ЯМР (CDCl3) δ: 1,34 (3H, т, J=7,4 Гц), 2,14 (3H, с), 2,91 (2Н, кв., J=7,4 Гц), 5,81 (2H, с).
[Стадия 2] (2S)-5-({[(Ацетилокси]метокси]карбонил}амино)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 148]
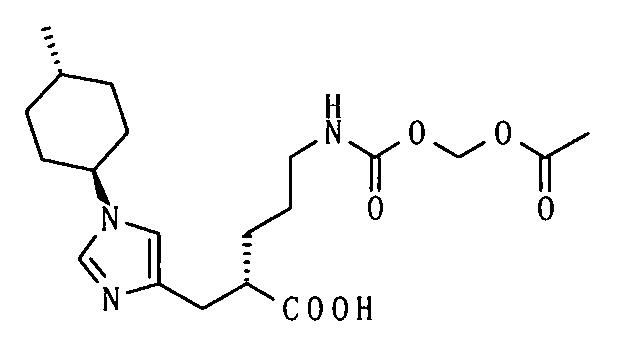
Указанное в заголовке соединение (281 мг) получали в виде белого твердого вещества из соединения (489 мг), полученного на стадии 1 данного примера, и соединения (400 мг), полученного на стадии 4 примера 15, таким же образом, как на стадиях 3 и 4 примера 28.
1H-ЯМР (CDCl3) δ: 0,96 (3H, д, J=6,6 Гц), 1,07-1,18 (2H, м), 1,42-1,52 (2H, м), 1,59-1,80 (5H, м), 1,85-1,91 (2H, м), 2,09-2,14 (2H, м), 2,11 (3H, с), 2,68-2,75 (1H, м), 2,77-2,92 (2H, м), 3,18-3,23 (2H, м), 3,88 (1H, тт, J=12,1, 3,9 Гц), 5,26-5,30 (1H, шир.м), 5,70 (2H, с), 6,78 (1H, с), 7,78 (1H, с).
МСНР (ESI) m/z 410 [M+H]+.
МСВР (ESI): m/z вычислено для C20H32N3O6: 410,22911 [M+H]+; найдено: 410,22892.
[Пример 32] (2S)-5-({[(1R)-1-(Изобутирилокси)этокси]карбонил}амино)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 149]
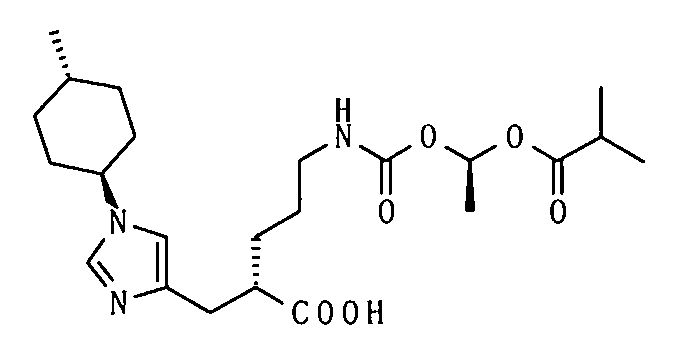
К раствору соединения (0,75 г), полученного на стадии 4 примера 15, в воде (3,13 мл) добавляли раствор (1R)-1-({[(2,5-диоксопирролидин-1-ил)окси]карбонил}окси)этил-2-метилпропионата (0,70 г) в ацетонитриле (12,55 мл), и смесь перемешивали при комнатной температуре в течение 20 часов. К смеси добавляли воду и этилацетат, и органический слой отделяли и сушили над безводным сульфатом магния. К нему добавляли гексан, и осажденное твердое вещество отделяли фильтрованием и сушили при пониженном давлении, с получением указанного в заголовке соединения (0,15 г). Условия анализа: Daicel Chiralpak (зарегистрированный товарный знак) AD-H, 4,6 мм × 250 мм (5 мкм), элюирующий растворитель: гексан/изопропанол (содержащий 0,5% объем. трифторуксусной кислоты и 0,5% объем. диэтиламина)=85/15 (1 мл/мин). Время удерживания: 9,4 мин (изомер А; не наблюдаемый), 11,4 мин (изомер В; не наблюдаемый), 13,6 мин (указанное в заголовке соединение), 15,8 мин (изомер С; не наблюдаемый).
МС (FAB) m/z 452 [M+H]+.
МСВР (ESI): m/z вычислено для C23H38N3O6: 452,27606 [M+H]+; найдено: 452,27582.
[Пример 33] 5-Амино-2-{[1-(3,3-диметилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] Этил-(2Е)-5-[((трет-бутоксикарбонил)амино]-2-{[1-(3,3-диметилциклогексил)-1Н-имидазол-4-ил]метилен}валерат
[Формула 150]
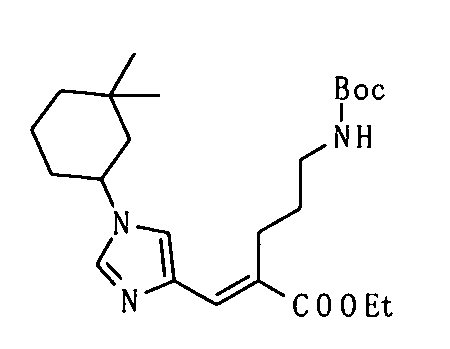
К раствору соединения (553 мг), полученного в ссылочном примере 14, в тетрагидрофуране (15 мл) при комнатной температуре добавляли хлорид лития (61 мг), и смесь перемешивали в течение 5 минут. К полученному реакционному раствору при охлаждении льдом добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (217 мкл), и смесь перемешивали в течение 20 минут. К ней затем при охлаждении льдом добавляли соединение (250 мг), полученное в ссылочном примере 15, и смесь затем перемешивали в течение ночи. К реакционному раствору добавляли водный хлорид аммония с последующей экстракцией этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=3/7-1/1), с получением указанного в заголовке соединения (347 мг).
1H-ЯМР (CDCl3) δ: 1,02 (6H, с), 1,18-1,24 (1H, м), 1,32 (3H, т, J=7,0 Гц), 1,45-1,63 (16H, м), 1,72-1,82 (4H, м), 2,10-2,15 (1H, м), 2,95 (2H, т, J=7,2 Гц), 3,11-3,16 (2H, м), 4,05-4,12 (1H, м), 7,04 (1H, шир.с), 7,15 (1H, с), 7,47 (1H, с), 7,58 (1H, с).
[Стадия 2] Этил-5-[(трет-бутоксикарбонил)амино]-2-{[1-(3,3-диметилциклогексил)-1Н-имидазол-4-ил]метил}валерат
[Формула 151]
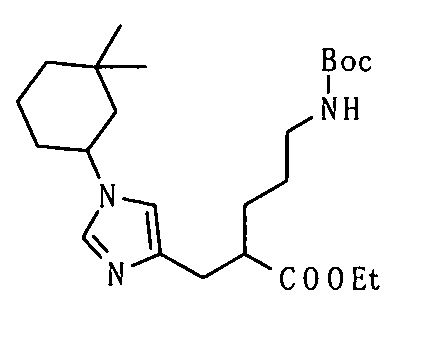
Соединение (347 мг), полученное на стадии 1 данного примера, растворяли в этаноле (10 мл). К раствору добавляли катализатор 10% палладий-углерод (гидратированный, 170 мг), и смесь перемешивали при комнатной температуре в течение 7 часов в атмосфере водорода. Катализатор отфильтровывали, и растворитель в фильтрате отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=1/1-этилацетат), с получением указанного в заголовке соединения (337 мг).
1H-ЯМР (CDCl3) δ: 0,99 (6H, с), 1,14-1,22 (4H, м), 1,41-1,77 (19H, м), 2,04-2,09 (1H, м), 2,68 (1H, дд, J=13,9, 6,5 Гц), 2,73-2,80 (1H, м), 2,88 (1H, дд, J=13,7, 7,4 Гц), 3,04-3,15 (2H, м), 4,00 (1H, тт, J=12,1, 3,8 Гц), 4,10 (2H, кв., J=7,0 Гц), 4,73 (1H, шир.с), 6,67 (0H, с), 7,41 (1H, с).
[Стадия 3] 5-амино-2-{[1-(3,3-диметилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 152]
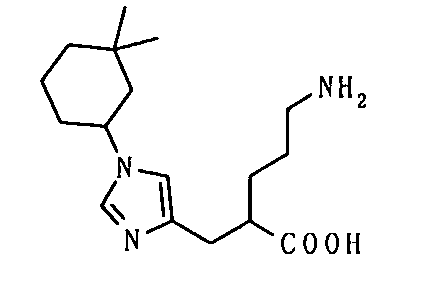
К соединению, полученному на стадии 2 данного примера, добавляли 5 н. хлористоводородную кислоту (10 мл), и смесь нагревали при кипячении с обратным холодильником в течение 6 часов. После отстаивания для охлаждения, растворитель отгоняли при пониженном давлении. Полученный остаток растворяли в деионизированной воде. К раствору добавляли PoraPak Rxn CX (ионообменная смола, 2,5 г). Смолу промывали деионизированной водой с последующим элюированием раствором 2,8% аммиак/метанол (раствор 28% аммиачной воды, разбавленный 10-кратно метанолом). Элюат концентрировали, с получением указанного в заголовке соединения (158 мг).
1H-ЯМР (CD3OD) δ: 0,99 (3H, с), 1,02 (3Н, с), 1,21-1,28 (1Н, м), 1,40-1,44 (1H, м), 1,50-1,78 (9H, м), 2,00-2,05 (1H, м), 2,47-2,58 (2H, м), 2,84-2,94 (2,33H, м), 3,55 (0,66H, т, J=7,1 Гц), 4,13-4,20 (1H, м), 6,94 (0,66H, с), 6,96 (0,33H, с), 7,58 (0,66H, с), 7,62 (0,33H, с).
[Пример 34] (2R,4S)-5-Амино-4-метил-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота и (2S,4S)-5-амино-4-метил-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] (3Е,5S)-5-метил-3-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метилен}пиперидин-2-он
[Формула 153]
Бензил-(5S)-5-метил-2-оксопиперидин-1-карбоксилат (Org. Lett, 2009, Vol. 11, p. 5410) (1,0 г) растворяли в тетрагидрофуране (20 мл). К раствору по каплям при -78°C добавляли гексаметилдисилазид лития (LHMDS, 1 н. раствор в тетрагидрофуране), и смесь перемешивали в течение 20 минут. Последовательно, к ней при -78°C добавляли по каплям раствор соединения (519 мг), полученного на стадии 3 ссылочного примера 4, в тетрагидрофуране (5 мл), и смесь перемешивали в течение ночи. К реакционному раствору добавляли воду, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и затем фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат-этилацетат/метанол=92/8), с получением указанного в заголовке соединения (612 мг).
1H-ЯМР (CDCl3) δ: 0,96 (3H, д, J=6,7 Гц), 1,09 (3H, д, J=6,7 Гц), 1,10-1,17 (2H, м), 1,42-1,52 (1H, м), 1,64-1,73 (2H, м), 1,84-1,91 (2H, м), 2,07-2,14 (3H, м), 2,47 (1H, ддд, J=16,5, 11,1, 2,5 Гц), 3,06-3,12 (1H, м), 3,31-3,36 (1H, м), 3,55-3,61 (1H, м), 3,88 (1H, тт, J=12,1, 3,9 Гц), 5,78 (1H, шир.с), 7,12 (1H, с), 7,57 (1H, с), 7,59 (1H, с).
[Стадия 2] Трет-бутил-(3Е,5S)-5-метил-3-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метилен}-2-оксопиперидин-1-карбоксилат
[Формула 154]
К раствору соединения (612 мг), полученного на стадии 1 данного примера, в тетрагидрофуране (18 мл) при -78°C добавляли 1,57М раствор н-BuLi в гексане (1,49 мл), и смесь перемешивали в течение 45 минут. Затем к ней при -78°C добавляли ди-трет-бутилдикарбонат (605 мг), и смесь постепенно нагревали и затем перемешивали в течение ночи. К реакционному раствору добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия, затем сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=7/3-3/7), с получением указанного в заголовке соединения (833 мг).
1H-ЯМР (CDCl3) δ: 0,96 (3H, д, J=6,7 Гц), 1,11 (3H, д, J=6,7 Гц), 1,11-1,18 (2H, м), 1,42-1,52 (1H, м), 1,55 (9H, с), 1,63-1,73 (2H, м), 1,84-1,91 (2H, м), 2,05-2,14 (3H, м), 2,44 (1H, ддд, J=16,8, 11,0, 2,3 Гц), 3,21 (1H, дд, J=12,5, 10,2 Гц), 3,41-3,47 (1H, м), 3,85-3,93 (2H, м), 7,15 (1H, с), 7,60 (1H, с), 7,68 (1H, с).
[Стадия 3] Трет-бутил-(5S)-5-метил-3-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-2-оксопиперидин-1-карбоксилат
[Формула 155]
Соединение (830 мг), полученное на стадии 2 данного примера, растворяли в этаноле (25 мл). К раствору добавляли катализатор 10% палладий-углерод (гидратированный, 207 мг), и смесь перемешивали в течение 13 часов в атмосфере водорода. Катализатор отфильтровывали, и растворитель в фильтрате отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=1/1-этилацетат), с получением указанного в заголовке соединения (788 мг).
1H-ЯМР (CDCl3) δ: 0,94 (3H, д, J=6,7 Гц), 0,96 (2H, д, J=6,7 Гц), 0,99 (1H, д, J=6,7 Гц), 1,03-1,23 (3H, м), 1,40-1,48 (1H, м), 1,52 (6H, с), 1,53 (3H, с), 1,55-1,68 (3H, м), 1,81-1,87 (2H, м), 1,96-2,10 (3H, м), 2,60-2,91 (2H, м), 3,04-3,20 (2H, м), 3,65-3,97 (2H, м), 6,73 (0,7H, с), 6,76 (0,3H, с), 7,41 (1,0H, с).
[Стадия 4] Трет-бутил-(3R,5S)-5-метил-3-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-2-оксопиперидин-1-карбоксилат и трет-бутил-(3S,5S)-5-метил-3-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-2-оксопиперидин-1-карбоксилат
[Формула 156]
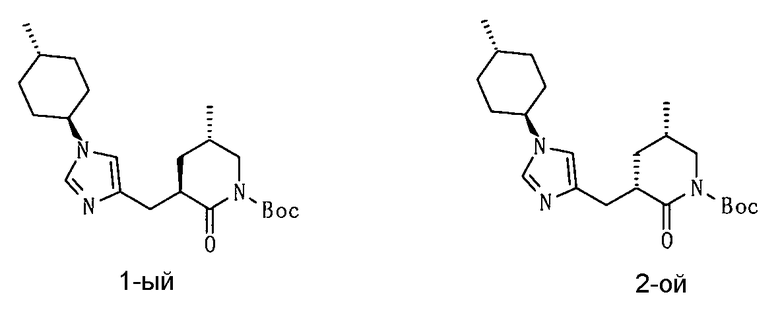
Соединение (788 мг), полученное на стадии 3 данного примера, диастереомерно разделяли высокоэффективной жидкостной хроматографией с использованием полупрепаративной колонки CHIRALPAK AD-H (2,0 см × 25,0 см). Объемная скорость потока: 10 мл/мин, элюирующий растворитель: гексан/изопропанол=88/12, длина волны детектирования: 210 нм. Температура колонки: 25°С.
Растворитель в разделенных растворах отгоняли при пониженном давлении, с получением соответственно обоих диастереомеров ((3R,5S)-форма: 72 мг и (3S,5S)-форма: 371 мг). Аналитической высокоэффективной жидкостной хроматографией подтверждали, что оба диастереомера являются оптически чистыми. Колонка: CHIRALPAK AD (0,46 см × 15,0 см), объемная скорость потока: 1,3 мл/мин, элюирующий растворитель: гексан/изопропанол=80/20, длина волны детектирования: 210 нм, время удерживания: (3R,5S)-форма (4,6 мин), (3S,5S)-форма (5,2 мин).
[Стадия 5] (2R,4S)-5-амино-4-метил-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 157]
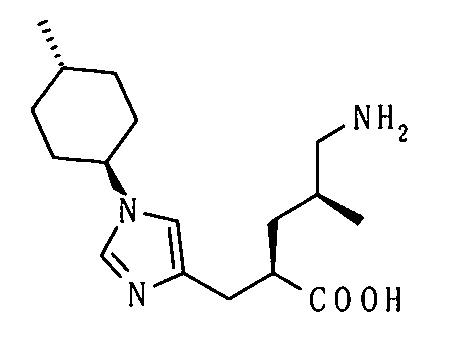
Указанное в заголовке соединение (25 мг) получали из трет-бутил-(3R,5S)-5-метил-3-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-2-оксопиперидин-1-карбоксилата (72 мг), полученного на стадии 4 данного примера, таким же образом, как на стадии 3 примера 33.
1H-ЯМР (CD3OD) δ: 0,95 (3H, д, J=6,8 Гц), 0,97 (3H, д, J=6,8 Гц), 1,10-1,19 (2H, м), 1,37-1,50 (2H, м), 1,64-1,75 (3H, м), 1,81-1,92 (3H, м), 2,00-2,05 (2H, м), 2,51 (1H, дд, J=14,2, 6,3 Гц), 2,54-2,60 (1H, м), 2,71 (1H, дд, J=12,7, 6,3 Гц), 2,92-2,85 (2H, м), 3,93 (1H, тт, J=12,2, 3,9 Гц), 6,93 (1H, с), 7,56 (1H, с).
МСВР (ESI): m/z вычислено для C17H29N3Na1O2: 330,21575 [M+H]+; найдено: 330,21629.
[Стадия 6] (2S,4S)-5-амино-4-метил-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 158]
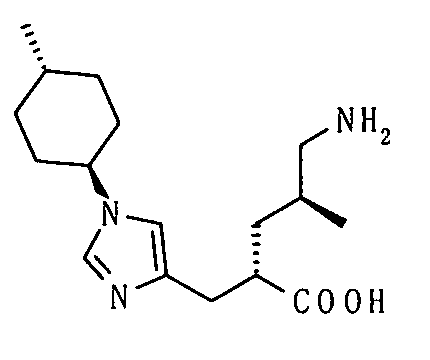
Указанное в заголовке соединение (212 мг) получали из трет-бутил-(3S,5S)-5-метил-3-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-2-оксопиперидин-1-карбоксилата (371 мг), полученного на стадии 4 данного примера, таким же образом, как на стадии 3 примера 33.
1H-ЯМР (CD3OD) δ: 0,95 (3H, д, J=6,8 Гц), 0,98 (3H, д, J=6,8 Гц), 1,10-1,19 (2H, м), 1,22-1,29 (1H, м), 1,43-1,51 (1H, м), 1,64-1,85 (6H, м), 2,01-2,05 (2H, м), 2,53 (1H, дд, J=13,9, 6,6 Гц), 2,55-2,61 (1H, м), 2,77 (2H, д, J=6,8 Гц), 2,88 (1H, дд, J=13,9, 7,1 Гц), 3,93 (1H, тт, J=12,0, 3,9 Гц), 6,94 (1H, с), 7,54 (1H, с).
МСВР (ESI): m/z вычислено для C17H30N3O2: 308,23380 [M+H]+; найдено: 308,23370.
[Пример 35] (2R,4R)-5-Амино-4-метил-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота и (2S,4R)-5-амино-4-метил-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] (3Е,5R)-5-метил-3-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метилен}пиперидин-2-он
[Формула 159]
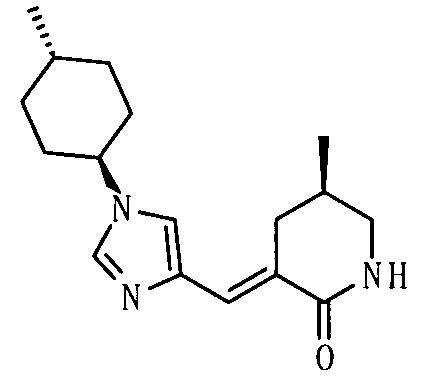
Бензил-(5R)-5-метил-2-оксопиперидин-1-карбоксилат (Org. Lett, 2009, Vol. 11, p. 5410) (772 мг) растворяли в тетрагидрофуране (15 мл). К раствору по каплям при -78°C добавляли гексаметилдисилазид лития (LHMDS, 1 н. раствор в тетрагидрофуране, 3,12 мл), и смесь перемешивали в течение 1 часа. Затем к ней при -78°C добавляли по каплям раствор 1-(транс-4-метилциклогексил)-1Н-имидазол-4-карбальдегида в тетрагидрофуране (5 мл), и смесь перемешивали при 0°С в течение 3 часов. К реакционному раствору добавляли водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и затем фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюирующий растворитель: этилацетат-этилацетат/метанол=92/8), с получением указанного в заголовке соединения (500 мг).
[Стадия 2] Трет-бутил-(3Е,5R)-5-метил-3-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метилен}-2-оксопиперидин-1-карбоксилат
[Формула 160]
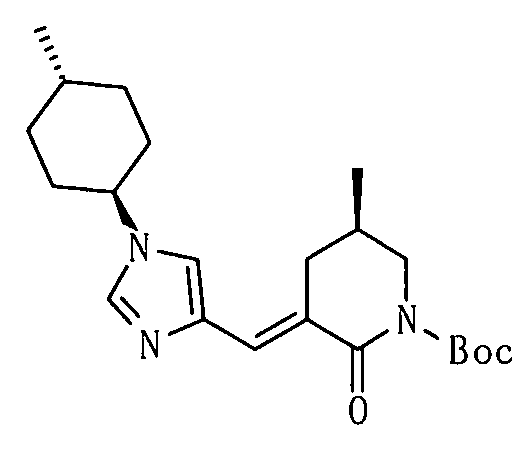
Указанное в заголовке соединение (492 мг) получали из соединения (500 мг), полученного на стадии 1 данного примера, таким же образом, как на стадии 2 примера 34.
[Стадия 3] Трет-бутил-(5R)-5-метил-3-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-2-оксопиперидин-1-карбоксилат
[Формула 161]
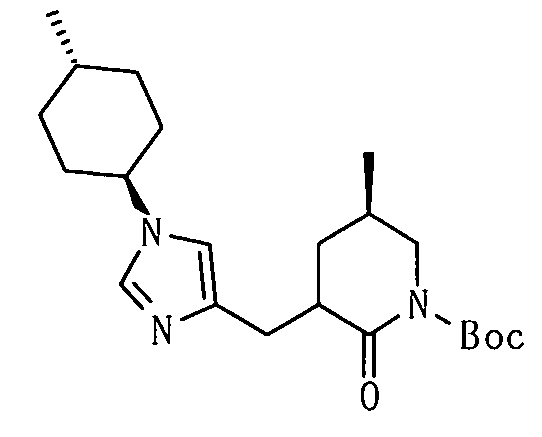
Указанное в заголовке соединение (460 мг) получали из соединения (490 мг), полученного на стадии 2 данного примера, таким же образом, как на стадии 3 примера 34.
[Стадия 4] Трет-бутил-(3R,5R)-5-метил-3-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-2-оксопиперидин-1-карбоксилат и трет-бутил-(3S,5R)-5-метил-3-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-2-оксопиперидин-1-карбоксилат
[Формула 162]
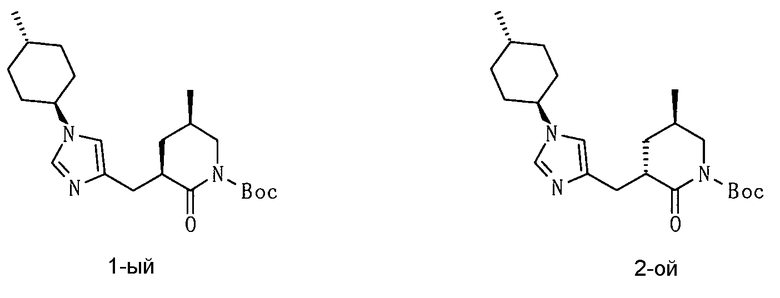
Соединение (460 мг), полученное на стадии 3 данного примера, диастереомерно разделяли высокоэффективной жидкостной хроматографией с использованием полупрепаративной колонки CHIRALPAK AD-H (2,0 см × 25,0 см). Объемная скорость потока: 10 мл/мин, элюирующий растворитель: гексан/изопропанол=90/10, длина волны детектирования: 210 нм. Температура колонки: 25°С.
Растворитель в разделенных растворах отгоняли при пониженном давлении, с получением соответственно обоих диастереомеров ((3R,5R)-форма: 298 мг и (3S,5R)-форма: 109 мг). Аналитической высокоэффективной жидкостной хроматографией подтверждали, что оба диастереомера являются оптически чистыми. Колонка: CHIRALPAK AD (0,46 см × 15,0 см), объемная скорость потока: 1 мл/мин, элюирующий растворитель: гексан/изопропанол=80/20, длина волны детектирования: 210 нм, время удерживания: (3R,5R)-форма (5,8 мин), (3S,5R)-форма (7,6 мин).
[Стадия 5] (2R,4R)-5-амино-4-метил-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 163]
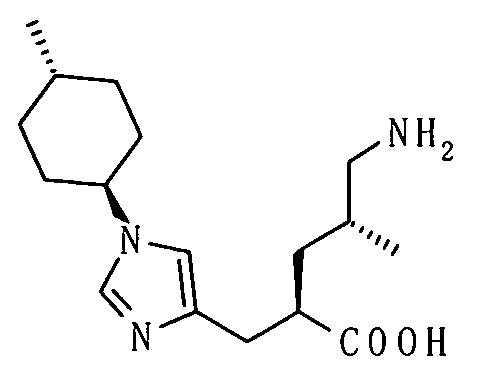
Указанное в заголовке соединение (134 мг) получали из трет-бутил-(3R,5R)-5-метил-3-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-2-оксопиперидин-1-карбоксилата (298 мг), полученного на стадии 4 данного примера, таким же образом, как на стадии 3 примера 33.
1H-ЯМР (CD3OD) δ: 0,95 (3H, д, J=6,7 Гц), 0,98 (3H, д, J=6,7 Гц), 1,09-1,27 (3H, м), 1,43-1,52 (1H, м), 1,63-1,86 (6H, м), 2,00-2,06 (2H, м), 2,53 (1H, дд, J=13,5, 6,5 Гц), 2,56-2,62 (1H, м), 2,77 (2H, д, J=7,0 Гц), 2,88 (1H, дд, J=13,9, 6,8 Гц), 3,93 (1H, тт, J=12,1, 3,9 Гц), 6,95 (1H, с), 7,55 (1H, с).
[Стадия 6] (2S,4R)-5-амино-4-метил-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 164]
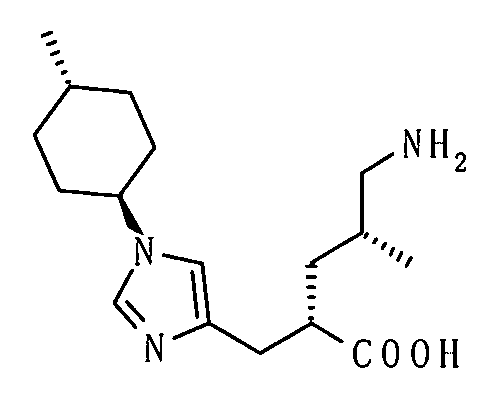
Указанное в заголовке соединение (12 мг) получали из трет-бутил-(3S,5R)-5-метил-3-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-2-оксопиперидин-1-карбоксилата (109 мг), полученного на стадии 4 данного примера, таким же образом, как на стадии 3 примера 33.
1H-ЯМР (CD3OD) δ: 0,95 (3H, д, J=6,7 Гц), 0,97 (3H, д, J=7,0 Гц), 1,09-1,19 (2H, м), 1,36-1,51 (2H, м), 1,63-1,77 (3H, м), 1,80-1,91 (3H, м), 2,00-2,05 (3H, м), 2,51 (1H, дд, J=13,9, 5,7 Гц), 2,54-2,61 (1H, м), 2,71 (1H, дд, J=12,9, 6,3 Гц), 2,84-2,93 (2H, м), 3,93 (1H, тт, J=12,5, 3,5 Гц), 6,93 (1H, с), 7,57 (1H, с).
[Пример 36] 4-(Аминометил)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}гексановая кислота
[Стадия 1] Этил-4-{[(трет-бутоксикарбонил)амино]метил}-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}гекс-2-еноат
[Формула 165]
Соединение (148 мг), полученное на стадии 1 примера 26, растворяли в тетрагидрофуране (2 мл). К раствору по каплям при -78°C добавляли гексаметилдисилазид лития (LHMDS, 1 н. раствор в тетрагидрофуране 561 мкл), и смесь перемешивали в течение 1 часа. Затем к ней при -78°C добавляли по каплям раствор соединения (113 мг), полученного в ссылочном примере 16, в тетрагидрофуране (1 мл), и смесь перемешивали при -78°C в течение 3 часов. К реакционному раствору добавляли водный хлорид аммония, и органическое вещество экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и затем фильтровали, и растворитель отгоняли при пониженном давлении, с получением неочищенного продукта. Полученный неочищенный продукт растворяли в метиленхлориде (5 мл). К раствору при комнатной температуре добавляли метансульфонилхлорид (87 мкл) и триэтиламин (235 мкл), и смесь перемешивали в течение 3 часов. К ней при комнатной температуре добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (251 мкл), и смесь перемешивали в течение ночи. К реакционному раствору добавляли метиленхлорид, и органический слой промывали водой и насыщенным раствором хлорида натрия, затем сушили над безводным сульфатом натрия и фильтровали, и растворитель отгоняли при пониженном давлении. Полученный остаток очищали тонкослойной колоночной хроматографией на силикагеле (проявляющий растворитель: метиленхлорид/метанол=95/5), с получением указанного в заголовке соединения (81 мг).
1H-ЯМР (CDCl3) δ: 0,89 (3H, т, J=7,4 Гц), 0,94 (3H, д, J=6,7 Гц), 1,05-1,16 (2H, м), 1,26 (3H, т, J=7,0 Гц), 1,33-1,40 (2H, м), 1,44 (9H, с), 1,55-1,63 (3H, м), 1,80-1,87 (2H, м), 2,04-2,09 (2H, м), 2,85-2,94 (1H, м), 3,04-3,11 (1H, м), 3,32-3,37 (1H, м), 3,49 (1H, д, J=14,1 Гц), 3,61 (1H, д, J=14,5 Гц), 3,73-3,81 (1H, м), 4,05-4,19 (2H, м), 6,57 (0,5H, с), 6,59 (0,5H, с), 6,76 (1H, с), 7,39 (1H, с), 8,13 (1H, шир.с).
[Стадия 2] Этил-4-{[(трет-бутоксикарбонил)амино]метил}-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}гексаноат
[Формула 166]
Указанное в заголовке соединение (47 мг) получали из соединения (80 мг), полученного на стадии 1 данного примера, таким же образом, как на стадии 3 примера 34.
1H-ЯМР (CDCl3) δ: 0,83 (1H, т, J=7,4 Гц), 0,88 (2H, т, J=7,4 Гц), 0,95 (3H, д, J=6,3 Гц), 1,04-1,15 (2H, м), 1,19 (1H, т, J=7,0 Гц), 1,20 (1H, т, J=7,0 Гц), 1,25-1,34 (3H, м), 1,42-1,48 (11H, м), 1,58-1,68 (3H, м), 1,81-1,86, (2H, м), 2,03-2,09 (2H, м), 2,63-2,72 (1H, м), 2,82-2,99 (2H, м), 3,05-3,18 (2H, м), 3,75-3,83 (1H, м), 4,09 (1H, кв., J=7,0 Гц), 4,10 (1H, кв., J=7,0 Гц), 5,46 (1H, шир.с), 6,68 (1H, с), 7,42 (1H, с).
[Стадия 3] Гидрохлорид 4-(аминометил)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}гексановой кислоты
[Формула 167]
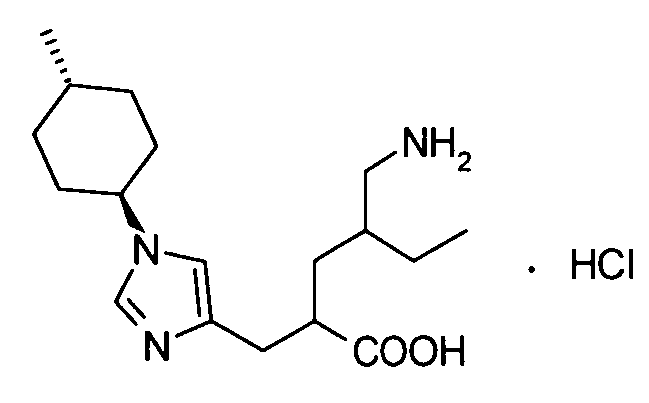
К соединению (47 мг), полученному на стадии 2 данного примера, добавляли 5 н. хлористоводородную кислоту (2 мл), и смесь нагревали при кипячении с обратным холодильником в течение 5 часов. После отстаивания для охлаждения, растворитель отгоняли при пониженном давлении. Полученный остаток растворяли в деионизированной воде. Нерастворимое вещество отфильтровывали через мембранный фильтр, и растворитель отгоняли, с получением представляющего интерес, указанного в заголовке соединения (37 мг).
1H-ЯМР (CD3OD) δ: 0,90-0,98 (6H, м), 1,16-1,24 (2H, м), 1,36-1,57 (4H, м), 1,74-1,92 (6H, м), 2,12-2,16 (2H, м), 2,85-3,03 (5H, м), 4,21-4,27 (1H, м), 7,55 (0,5H, с), 7,56 (0,5H, с), 8,90 (0,5H, с), 8,92 (0,5H, с).
МСВР (ESI): m/z вычислено для C18H32N3O2: 322,24945 [M+H]+; найдено: 322,24948.
[Пример 37] 5-Амино-2-{[1-(цис-4-гидроксициклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] Этил-(2Е)-5-{[(трет-бутоксикарбонил)амино]-2-{[1-(цис-4-{[трет-бутил(дифенил)силил]окси}циклогексил)-1Н-имидазол-4-ил]метилен}валерат
[Формула 168]
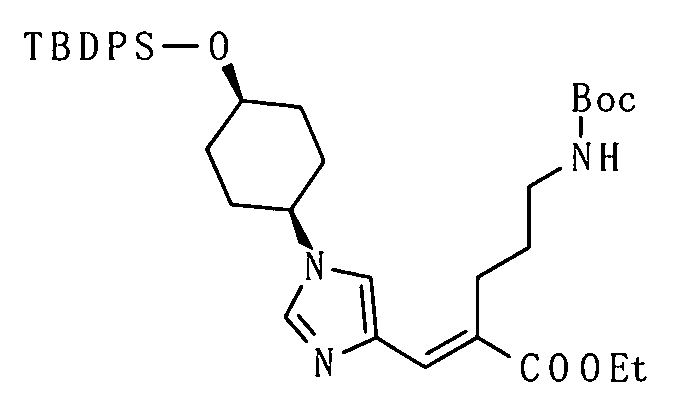
Указанное в заголовке соединение (375 мг) получали с использованием соединения (307 мг), полученного в ссылочном примере 14, таким же образом, как на стадии 1 примера 33.
1H-ЯМР (CDCl3) δ: 1,10 (9H, с), 1,33 (3H, т, J=7,1 Гц), 1,41-1,46 (2H, м), 1,48 (9H, с), 1,74-1,91 (6H, м), 2,22-2,31 (2Н, м), 2,98 (2Н, т, J=7,3 Гц), 3,14-3,17 (2H, м), 3,89-3,95 (1Н, м), 4,06-4,09 (1H, м), 4,24 (2H, кв., J=7,0 Гц), 7,19 (1H, с), 7,37-7,41 (4H, м), 7,43-7,46 (2H, м), 7,49 (1H, с), 7,64-7,67 (5H, м).
[Стадия 2] Этил-(2Е)-5-[(трет-бутоксикарбонил)амино]-2-{[1-(цис-4-гидроксициклогексил)-1Н-имидазол-4-ил]метилен}валерат
[Формула 169]
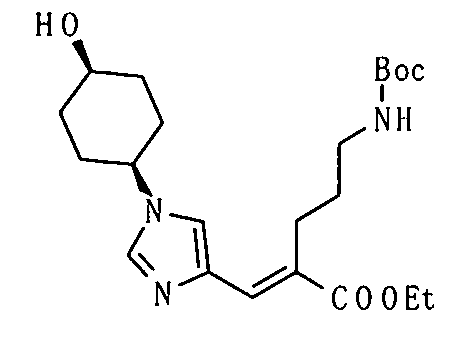
Соединение, полученное на стадии 1 данного примера, растворяли в тетрагидрофуране (10 мл). К раствору при комнатной температуре добавляли раствор тетрабутиламмонийфторида в тетрагидрофуране (1,0М, 682 мкл), и смесь перемешивали в течение ночи. К ней при комнатной температуре дополнительно добавляли раствор тетрабутиламмонийфторида в тетрагидрофуране (1,0М, 204 мкл), и смесь перемешивали в течение 4 дней. Растворитель в реакционном растворе отгоняли при пониженном давлении, и полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=1/1-этилацетат), с получением указанного в заголовке соединения (220 мг).
1H-ЯМР (CDCl3) δ: 1,32 (3H, т, J=7,3 Гц), 1,47 (9H, с), 1,66-1,78 (4Н, м), 1,88-1,98 (4Н, м), 2,12-2,20 (2Н, м), 2,93(2Н, т, J=7,3 Гц), 3,12-3,16 (2H, м), 3,93-4,00 (1Н, м), 4,12-4,15 (1H, м), 4,23 (2H, кв., J=7,2 Гц), 6,94 (1H, шир.с), 7,21 (1H, с), 7,49 (1H, с), 7,61 (1H, с).
[Стадия 3] Этил-5-[(трет-бутоксикарбонил)амино]-2-{[1-(цис-4-гидроксициклогексил)-1Н-имидазол-4-ил]метил}валерат
[Формула 170]
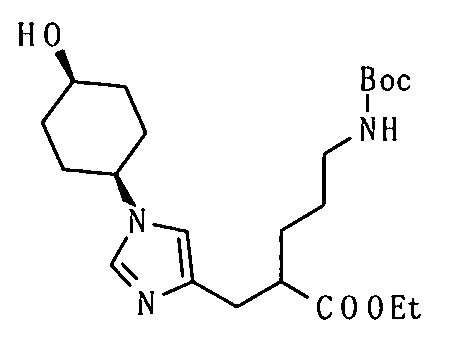
Указанное в заголовке соединение (51 мг) получали из соединения (50 мг), полученного на стадии 2 данного примера, таким же образом, как на стадии 2 примера 33.
1H-ЯМР (CDCl3) δ: 1,20 (3H, т, J=7,4 Гц), 1,43 (9H, с), 1,48-1,71 (6H, м), 1,83-1,94 (4H, м), 2,05-2,15 (2H, м), 2,69 (1H, дд, J=13,9, 6,5 Гц), 2,74-2,81 (1H, м), 2,89 (1H, дд, J=13,7, 7,4 Гц), 3,05-3,14 (2H, м), 3,83-3,90 (1H, м), 4,07-4,13 (3H, м), 4,74 (1H, шир.с), 6,72 (1H, с), 7,45 (1H, с).
[Стадия 4] 5-амино-2-{[1-(цис-4-гидроксициклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
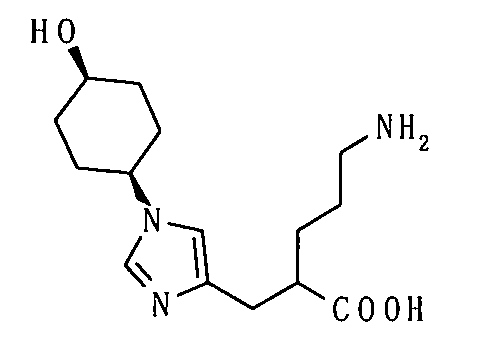
Указанное в заголовке соединение (26 мг) получали из соединения (51 мг), полученного на стадии 3 данного примера, таким же образом, как на стадии 3 примера 33.
1H-ЯМР (CD3OD) δ: 1,61-1,89 (10Н, м), 2,04-2,12 (2H, м), 2,48-2,59 (2Н, м), 2,85-2,92 (3H, м), 3,96-4,02 (2Н, м), 6,96 (1Н, с), 7,59 (1H, с).
[Пример 38] 5-Амино-2-({1-[транс-4-(пиридин-4-илокси)циклогексил]-1Н-имидазол-4-ил}метил)валериановая кислота
[Стадия 1] Этил-(2Е)-5-[(трет-бутоксикарбонил)амино]-2-({1-[транс-4-(пиридин-4-илокси)циклогексил]-1Н-имидазол-4-ил}метилен)валерат
[Формула 172]
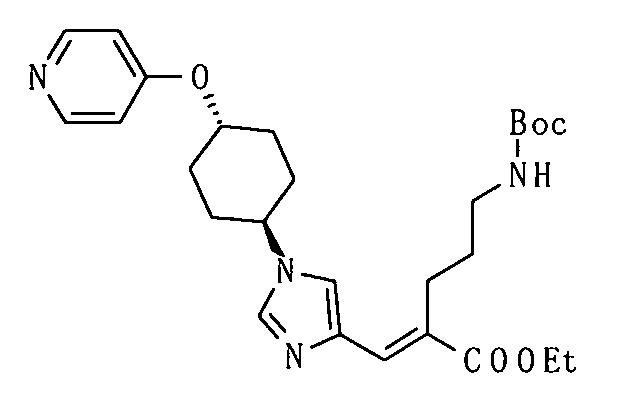
Соединение (170 мг), полученное на стадии 2 примера 37, растворяли в тетрагидрофуране (6 мл). К раствору добавляли трифенилфосфин (137 мг), 4-гидроксипиридин (50 мг) и 40% раствор диизопропилазодикарбоксилата в толуоле (276 мкл), и смесь перемешивали при 55°С в течение 5,5 часов. Растворитель в реакционном растворе отгоняли при пониженном давлении, и полученный остаток очищали колоночной хроматографией на силикагеле (элюирующий растворитель: гексан/этилацетат=7/3-этилацетат), с получением указанного в заголовке соединения (51 мг).
1H-ЯМР (CDCl3) δ: 1,33 (3H, т, J=7,0 Гц), 1,48 (9H, с), 1,68-1,79 (4H, м), 1,84-1,93 (2H, м), 2,25-2,36 (4H, м), 2,90-2,95 (2H, м), 3,12-3,17 (2H, м), 4,04-4,09 (1H, м), 4,24 (2H, кв., J=7,2 Гц), 4,39-4,45 (1H, м), 6,78-6,82 (3H, м), 7,19 (1H, с), 7,49 (1H, с), 7,62 (1H, с), 8,44 (2H, дд, J=5,1, 1,6 Гц).
[Стадия 2] Этил-5-[(трет-бутоксикарбонил)амино]-2-({1-[транс-4-(пиридин-4-илокси)циклогексил]-1Н-имидазол-4-ил}метил)валерат
[Формула 173]
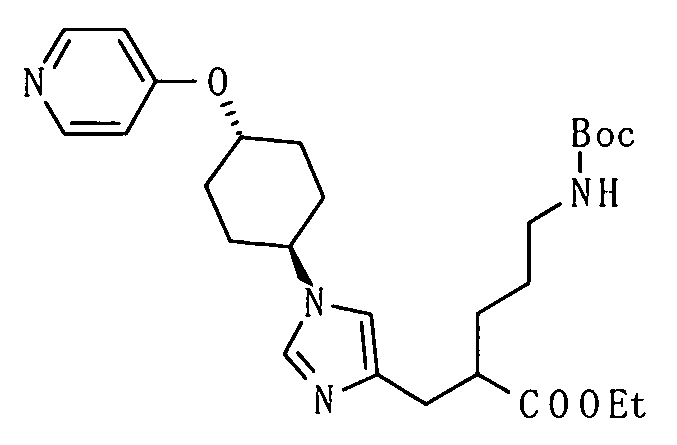
Указанное в заголовке соединение (45 мг) получали из соединения (50 мг), полученного на стадии 1 данного примера, таким же образом, как на стадии 2 примера 33.
1H-ЯМР (CDCl3) δ: 1,20 (3H, т, J=7,0 Гц), 1,44 (9H, с), 1,51-1,70 (6H, м), 1,77-1,87 (2H, м), 2,20-2,32 (4H, м), 2,69 (1H, дд, J=13,7, 6,7 Гц), 2,74-2,81 (1H, м), 2,90 (1H, дд, J=13,7, 7,4 Гц), 3,06-3,14 (2H, м), 3,97 (1H, тт, J=11,7, 3,9 Гц), 4,10 (3H, кв., J=7,0 Гц), 4,38 (1H, тт, J=11,0, 3,9 Гц), 4,70 (1H, шир.с), 6,70 (1H, с), 6,80 (2H, дд, J=4,7, 1,6 Гц), 7,45 (1H, с), 8,43 (2H, дд, J=4,7, 1,6 Гц).
[Стадия 3] Гидрохлорид 5-амино-2-({1-[транс-4-(пиридин-4-илокси)циклогексил]-1Н-имидазол-4-ил}метил)валериановой кислоты
[Формула 174]
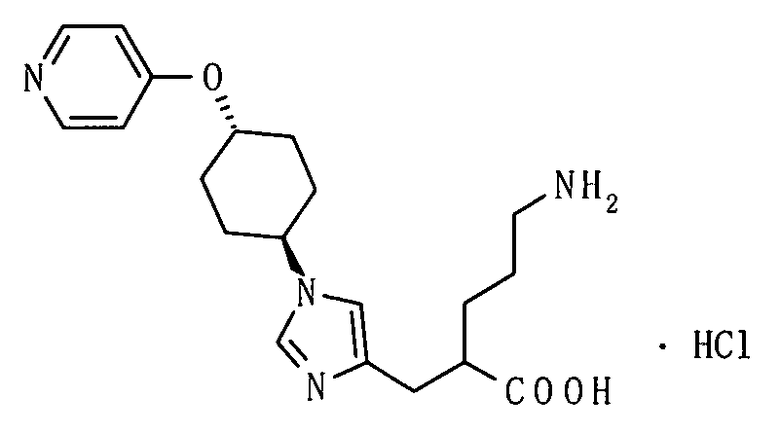
Указанное в заголовке соединение (32 мг) получали из соединения (45 мг), полученного на стадии 2 данного примера, таким же образом, как на стадии 2 примера 36.
1H-ЯМР (CD3OD) δ: 1,69-1,89 (6H, м), 2,06-2,17 (2H, м), 2,28-2,42 (4H, м), 2,79-2,85 (1H, м), 2,90-2,99 (3H, м), 3,04 (2Н, дд, J=15,3, 9,0 Гц), 4,47 (1H, тт, J=12,1, 3,9 Гц), 4,96 (1H, тт, J=11,3, 4,3 Гц), 7,60-7,64 (3H, м), 8,63-8,65 (2H, м), 8,96-8,97 (1H, м).
[Пример 39] 5-Амино-2-{[1-(цис-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Стадия 1] Метил-(2Е)-5-[(трет-бутоксикарбонил)амино]-2-{[1-(цис-4-метилциклогексил)-1Н-имидазол-4-ил]метилен}валерат
[Формула 175]

Указанное в заголовке соединение (1,42 г) получали из соединения (0,79 г), полученного в ссылочном примере 18, таким же образом, как на стадии 1 примера 3.
1H-ЯМР (CDCl3) δ: 1,00 (3H, д, J=7,0 Гц), 1,44-1,53 (3H, м), 1,48 (9H, с), 1,63-1,80 (4H, м), 1,85-2,04 (5H, м), 2,98 (2H, т, J=7,2 Гц), 3,13-3,17 (2H, м), 3,78 (3H, с), 4,00-4,06 (1H, м), 7,19 (1H, с), 7,48 (1H, с), 7,65 (1H, с).
[Стадия 2] Метил-5-[(трет-бутоксикарбонил)амино]-2-{[1-(цис-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерат
[Формула 176]
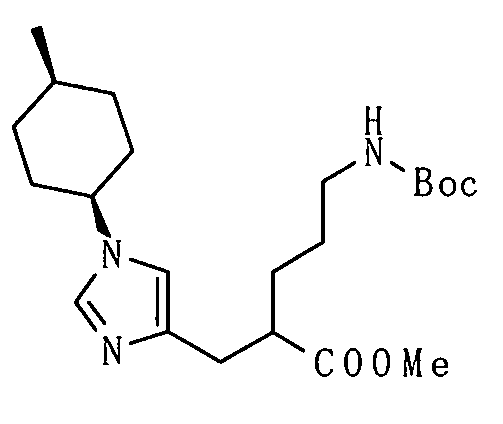
Указанное в заголовке соединение (1,11 г) получали из соединения (1,42 г), полученного на стадии 1 данного примера, таким же образом, как на стадии 2 примера 33.
1H-ЯМР (CDCl3) δ: 0,98 (3H, д, J=6,7 Гц), 1,44 (9H, с), 1,44-1,69 (7H, м), 1,81-1,88 (4H, м), 1,92-2,01 (2H, м), 2,72 (1H, дд, J=13,7, 5,9 Гц), 2,78-2,85 (1H, м), 2,90 (1H, дд, J=13,7, 7,8 Гц), 3,05-3,15 (2H, м), 3,64 (3H, с), 3,90-3,96 (1H, м), 4,76 (1H, шир.с), 6,72 (1H, с), 7,50 (1H, с).
[Стадия 3] 5-амино-2-{[1-(цис-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота
[Формула 177]
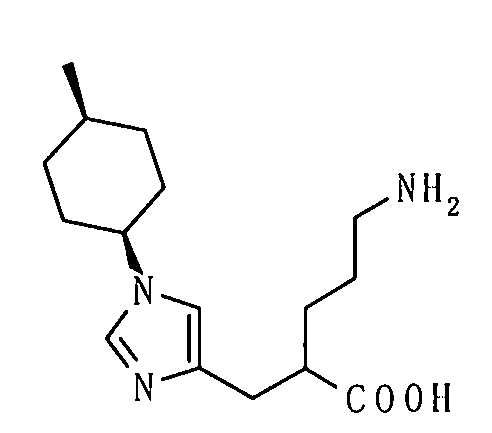
Указанное в заголовке соединение (0,39 г) получали из соединения (1,11 г), полученного на стадии 2 данного примера, таким же образом, как на стадии 3 примера 33.
1H-ЯМР (CD3OD) δ: 1,02 (3H, д, J=7,3 Гц), 1,46-1,55 (3H, м), 1,58-1,72 (5H, м), 1,80-1,87 (3H, м), 1,98-2,06 (2H, м), 2,47-2,58 (2H, м), 2,85-2,94 (3H, м), 3,99-4,04 (1H, м), 6,98 (1H, с), 7,59 (1H, с).
[Пример 40] Ангидрат п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты
[Формула 178]
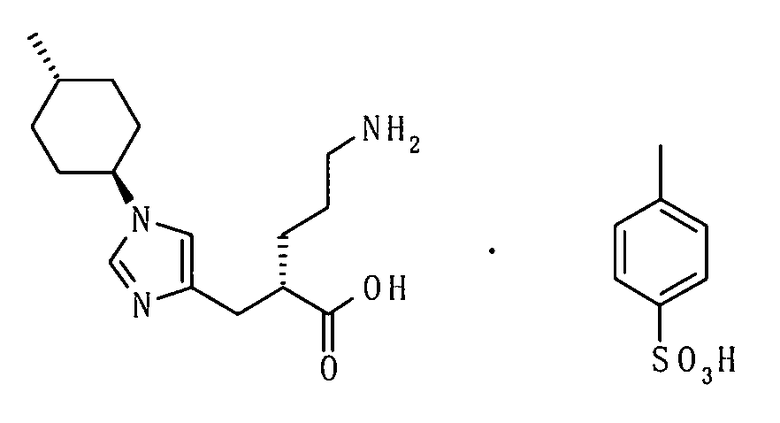
Соединение (2,04 г), полученное на стадии 4 примера 15, суспендировали в тетрагидрофуране (15 мл), и полученную суспензию перемешивали. К ней добавляли моногидрат п-толуолсульфоната (1,32 г), и смесь перемешивали при комнатной температуре в течение 1 дня. Осажденные кристаллы собирали фильтрованием при пониженном давлении и сушили на воздухе в течение 1 дня, с получением указанного в заголовке соединения (3,01 г).
1H-ЯМР (CD3OD) δ: 0,95 (3H, д, J=6,5 Гц), 1,11-1,21 (2H, м), 1,43-1,79 (7H, м), 1,83-1,89 (2H, м), 2,05-2,10 (2H, м), 2,37 (3H, с), 2,57-2,64 (1H, м), 2,70 (1H, дд, J=14,5, 5,5 Гц), 2,85-2,95 (3H, м), 4,07 (1H, тт, J=11,7, 3,9 Гц), 7,18 (1H, с), 7,23 (2H, д, J=7,8 Гц), 7,70 (2H, д, J=8,2 Гц), 8,22 (1H, с).
Анализ: C16H27N3O2·C7H8O3S,
Теоретическое: С; 59,33, H; 7,58, N; 9,02, O; 17,18,S; 6,89,
Найдено: С; 59,09, H; 7,53, N; 8,92, O; 17,22, S; 6,78.
Результаты порошковой рентгеновской дифракции полученного указанного в заголовке соединения показаны на фиг.1 и в таблице 1, и результаты его термического анализа (TG/DTA) показаны на фиг.2. В этом термическом анализе (TG/DTA) измерение проводили при скорости нагревания, равной 10°С/мин, в токе сухого азота, равным 200 мл/мин.
Порошковая рентгеновская дифракция соединения примера 40
2θ (°)
[Пример 41] Моногидрат п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты
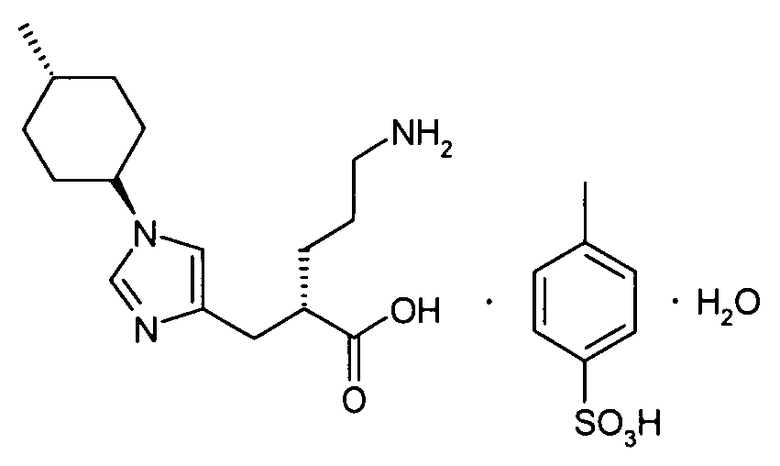
К соединению (101,6 мг), полученному в примере 40, добавляли 6% гидратированный тетрагидрофуран (600 мкл), и соединение растворяли нагреванием при 60°С. Раствор оставляли при комнатной температуре на 1 день, и осажденные кристаллы собирали фильтрованием и сушили на воздухе в течение 1 дня, с получением указанного в заголовке соединения (79,3 мг).
Анализ: C16H27N3O2·C7H8O3S·1H2O,
Теоретическое: С; 57,12, H; 7,71, N; 8,69, O; 19,85,S; 6,63,
Найдено: С; 56,90, H; 7,69, N; 8,67, O; 19,81, S; 6,42.
Результаты порошковой рентгеновской дифракции полученного указанного в заголовке соединения показаны на фиг.3 и в таблице 2, и результаты его термического анализа (TG/DTA) показаны на фиг.4. В этом термическом анализе (TG/DTA) измерение проводили при скорости нагревания, равной 10°С/мин, в токе сухого азота, равным 200 мл/мин.
Порошковая рентгеновская дифракция соединения примера 41
2θ (°)
[Тестовый пример 1] Определение ингибирующей активности в отношении TAFIa
(1) Активация TAFI
При получении реакционного раствора использовали забуфренный HEPES физиологический раствор (20 мМ HEPES, 150 мМ NaCl, рН 7,4; далее здесь называемый HBS). К 12 мкл TAFI с концентрацией 250 мкг/мл добавляли 30 мкл раствора HBS, содержащего 4 Ед/мл человеческого тромбина, 12 Ед/мл тромбомодулина кроличьего легкого и 12 мМ CaCl2, и смесь осторожно перемешивали. Затем TAFI активировали при комнатной температуре. Через десять минут, тромбин нейтрализовали добавлением 10 мкл 100 мкМ РРАСК (ингибитор тромбина), чтобы прекратить активацию TAFI. Образовавшийся TAFIa сохраняли во льду и разводили непосредственно перед использованием при определении 2050 мкл раствора HBS, содержащего BSA (бычий сывороточный альбумин), доведенный до 0,1% в расчете на конечную концентрацию.
(2) Определение ингибирующей активности в отношении TAFIa
Тестируемое вещество растворяли в HBS для получения ряда 10-кратных разведений концентраций оценки. 80 мкл раствора TAFIa и 10 мкл тестируемого вещества добавляли в каждую лунку 96-луночного планшета и смешивали при встряхивании в течение 10 минут. В каждую лунку добавляли 10 мкл фурилакрилоил-аланил-лизина (FAAK), доведенного до 5 мг/мл, и изменение в поглощении этого смешанного раствора при 330 нм считывали в течение 30 минут для определения степени разрушения субстрата.
(3) Вычисление ингибирующей активности IC 50
Степень разрушения субстрата в каждой лунке наносили на стандартную кривую, полученную с использования ряда разведений раствора TAFIa для вычисления активности TAFIa. Концентрацию, ингибирующую 50%, (IC50), вычисляли на основании корреляции между концентрацией тестируемого соединения и активностью TAFIa. Соединение А (соединение примера 7 в описании опубликованной Международной заявки № WO 2002/014285) использовали в качестве контроля. Результаты показаны в таблице 3.
Ингибирующая активность в отношении фермента TAFIa
Соединение настоящего изобретения проявляет превосходную ингибирующую активность в отношении TAFIa и является полезным в качестве фармацевтического лекарственного средства для лечения инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких.
[Тестовый пример 2] Оценка активности по усилению фибринолиза посредством измерения времени лизиса сгустка плазмы
В 96-луночный планшет добавляли 20 мкл/лунка HBS, 50 мкл/лунка нормальной человеческой плазмы, 10 мкл/лунка раствора соединения (раствор соединения получали растворением соединения в HBS с последующим серийным разведением этим буфером) и 10 мкл/лунка tPA (Activacin (Kyowa Hakko Kirin Co., Ltd.) доводили до 600000 Ед/мл раствором для лизиса, включенным в буфер, с последующим разведением HBS), и смесь перемешивали. Затем к ней добавляли 10 мкл/лунка реакционного раствора А (13,8 Ед/мл человеческого тромбина, 170 мМ CaCl2 и 0,9 Ед/мл тромбомодулина), и смесь перемешивали повторно. Поглощение при 405 нм измеряли с использованием планшет-ридера при 30-секундных интервалах, при поддержании температуры при 37°С для измерения степени коагуляции. При изменении поглощения временная отметка, при которой каждая лунка проявляла поглощение, наиболее близкое к среднему (ABS-средн: [(ABS-макс)-(ABS-мин)]/2) от максимального поглощения (ABS-макс) и минимального поглощения (ABS-мин) в процессе фибринолиза, определяли как 1/2 время лизиса (1/2 L/T) и применяли в качестве фибринолитической активности каждой лунки. Концентрацию, которая достигает 50% от 1/2 L/T, вычисляли как ЕС50 от взаимосвязи между концентрацией тестируемого вещества и 1/2 L/T. Соединение А (соединение примера 7 в описании опубликованной Международной заявки № WO 2002/014285) использовали в качестве контроля. Результаты показаны в таблице 4.
Активность по усилению фибринолиза
Соединение настоящего изобретения проявляет превосходную активность по усилению фибринолиза и является полезным в качестве фармацевтического лекарственного средства для лечения инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких.
[Тестовый пример 3] Оценка активности по усилению фибринолиза на крысиных моделях тромбоэмболии
Использовали крыс линии Wistar (приобретены от Japan SLC, Inc.). В произвольный момент времени им перорально вводили тестируемое вещество, полученное вместе с раствором 0,5% метилцеллюлозы, или им внутривенно вводили тестируемое вещество, полученное вместе с физиологическим раствором. Через сорок минут или четыре часа, непрерывно инъецировали реагент РТ (тромбопластин С плюс, Sysmex Corp.), доведенный до 2,25 Ед/мл физиологическим раствором (16,8 мл/кг/час × 20 мин) в яремные вены под анестезией тиопенталом. Группа введения избыточной дозы ингибитора TAFIa была выбрана в качестве группы положительного контроля. Через сорок пять минут после начала обработки реагентом РТ, производили отбор крови из яремных вен с использованием лимонной кислоты для получения плазмы. Количество D-димера, содержащегося в плазме, измеряли с использованием анализатора коагуляции ACL-9000 или ACL-TOP500CTS. Вычисляли его отношение к среднему значению для группы положительного контроля, и ED50 вычисляли как дозу, увеличивающую D-димер на 50%.
Соединение настоящего изобретения проявляет превосходную активность по инициации фибринолиза in vivo и является полезным в качестве фармацевтического лекарственного средства для лечения инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких.
(Пример получения препарата 1) Твердая капсула
Каждую из оболочек стандартной твердой желатиновой капсулы, разделяемых на две части, заполняют 100 мг соединения примера 1 в порошковой форме, 150 мг лактозы, 50 мг целлюлозы и 6 мг стеарата магния, с получением единичных капсул, которые затем промывают и затем сушат.
(Пример получения препарата 2) Мягкая капсула
Смесь соединения примера 2, содержащегося в усваиваемом масляном веществе, например соевом масле, хлопковом масле или оливковом масле, получают и инъецируют в желатин, с использованием поршневого насоса прямого вытеснения, с получением мягких капсул, содержащих 100 мг активного ингредиента. Эти мягкие капсулы промывают и затем сушат.
(Пример получения препарата 3) Таблетка
Каждую таблетку получают в соответствии с общепринятым способом с использованием 100 мг соединения примера 3, 0,2 мг коллоидного диоксида кремния, 5 мг стеарата магния, 275 мг микрокристаллической целлюлозы, 11 мг крахмала и 98,8 мг лактозы.
По желанию, на таблетку наносят покрытие.
(Пример получения препарата 4) Суспензия
Получают 5 мл суспензии, содержащей 100 мг соединения примера 4 в форме микродисперсного порошка, 100 мг натрийкарбоксиметилцеллюлозы, 5 мг бензоата, 1,0 г раствора сорбита (Фармакопея Японии) и 0,025 мл ванилина.
(Пример получения препарата 5) Крем
100 мг соединения примера 5 в форме микродисперсного порошка замешивают в 5 г крема, содержащего 40% белого вазелина, 3% микрокристаллического воска, 10% ланолина, 5% Спан-20, 0,3% Твин-20 и 41,7% воды, с получением крема.
(Пример получения препарата 6) Инъекция
1,5% масс соединения примера 6 замешивают в 10% масс пропиленгликоля, последовательно доводят до данного объема с помощью воды для инъекций и затем стерилизуют, с получением инъекции.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Циклоалкил-замещенное производное имидазола настоящего изобретения, представленное общей формулой (I), или его фармакологически приемлемая соль обладает превосходной ингибирующей активностью в отношении фермента TAFIa и является полезным в качестве фармацевтического лекарственного средства для лечения инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции, фиброза легких или т.п., или в качестве фармацевтического лекарственного средства против заболевания, являющегося следствием тромбоэмболии.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ 5-ГИДРОКСИПИРИМИДИН-4-КАРБОКСАМИДА | 2010 |
|
RU2550693C2 |
| ЦИННАМИДНОЕ СОЕДИНЕНИЕ | 2005 |
|
RU2361872C2 |
| ПРОИЗВОДНОЕ ЦИННАМИДА ТИПА МОРФОЛИНА | 2006 |
|
RU2381225C1 |
| НОВОЕ ПРОИЗВОДНОЕ ТЕТРАГИДРОИЗОХИНОЛИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, ПРИМЕНЕНИЕ ЕГО И СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРЕДОТВРАЩЕНИЯ ЗАБОЛЕВАНИЯ | 2009 |
|
RU2474575C2 |
| АЗАЗАМЕЩЕННЫЕ СПИРОПРОИЗВОДНЫЕ | 2006 |
|
RU2428423C2 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИРУСА ГРИППА, СПОСОБЫ ИХ ПРИМЕНЕНИЯ И ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2737190C2 |
| ИНГИБИТОР СВЯЗЫВАНИЯ СФИНГОЗИН-1-ФОСФАТА | 2007 |
|
RU2395499C2 |
| СОЕДИНЕНИЯ ТРИАЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ СОДЕРЖАЩАЯ, ПРИМЕНЕНИЕ СОЕДИНЕНИЙ ТРИАЗОЛА И СПОСОБ ЛЕЧЕНИЯ ГРИБКОВОЙ ИНФЕКЦИИ | 2002 |
|
RU2276670C2 |
| НОВЫЕ ФИЗИОЛОГИЧЕСКИ АКТИВНЫЕ ВЕЩЕСТВА | 2003 |
|
RU2338741C2 |
| АРИЛ-ЗАМЕЩЕННЫЕ КАРБОКСАМИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ БЛОКАТОРОВ КАЛЬЦИЕВЫХ ИЛИ НАТРИЕВЫХ КАНАЛОВ | 2010 |
|
RU2575168C2 |
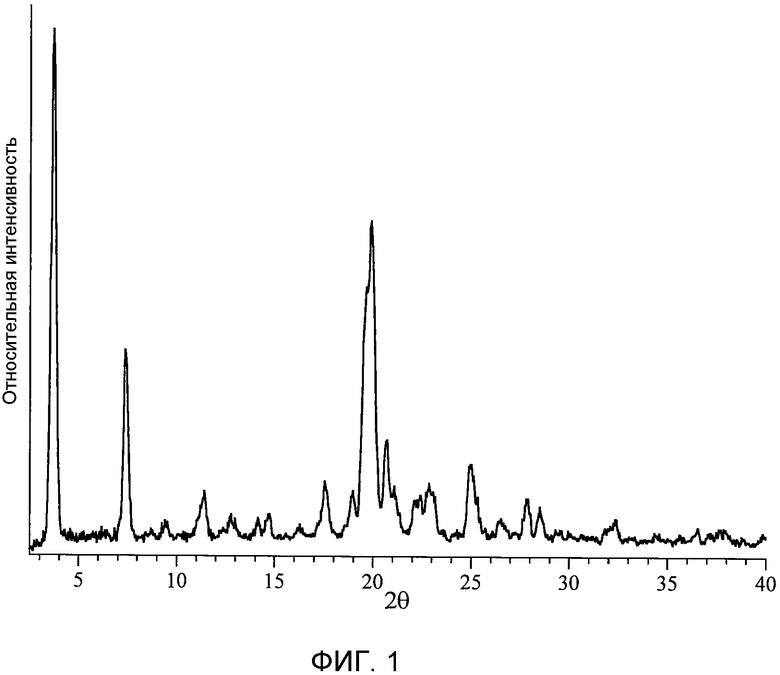
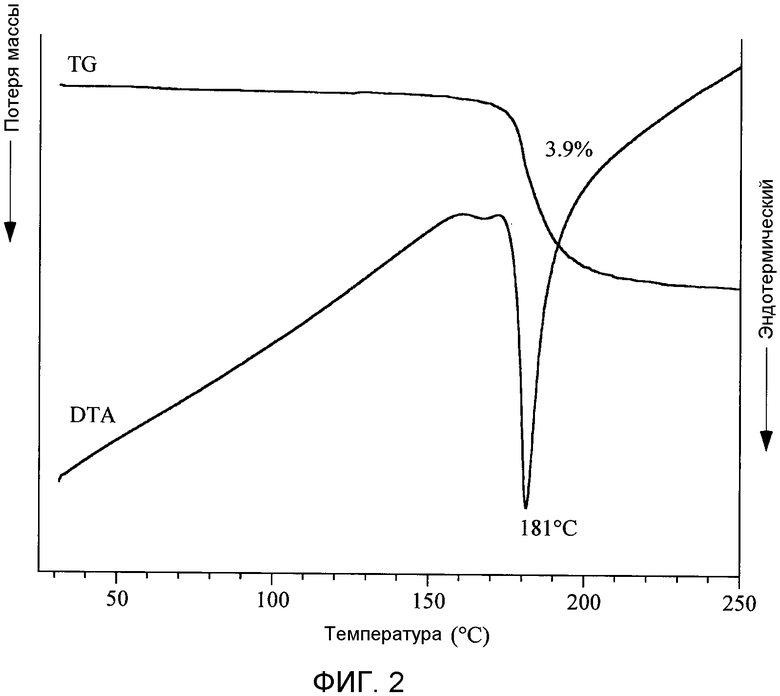
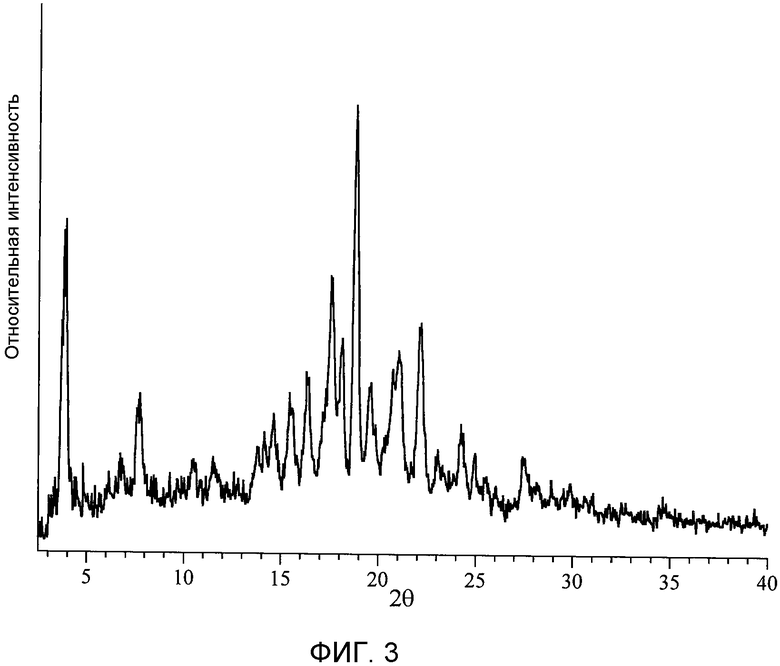
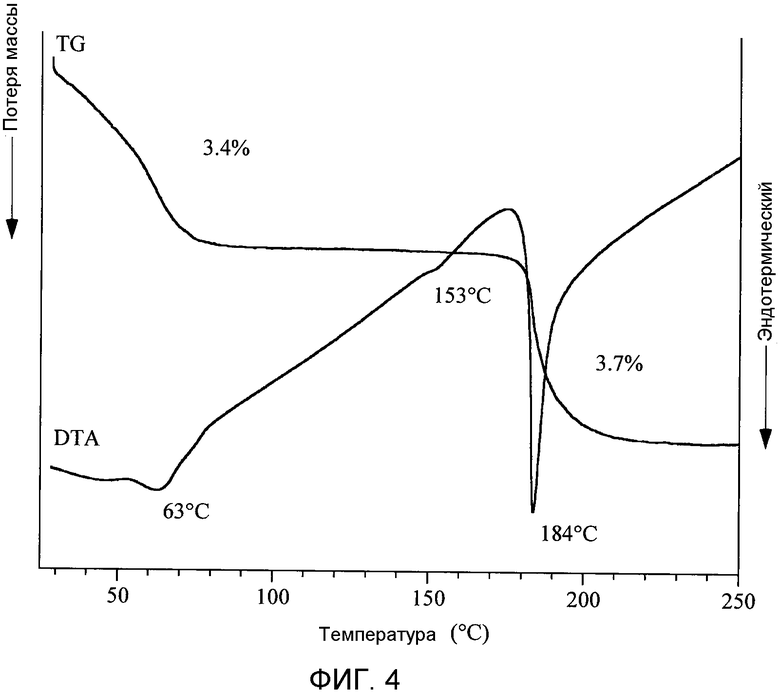
Изобретение относится к области органической химии, а именно к новому производному имидазола общей формулы (I), или к его фармакологически приемлемой соли, где А представляет собой группу: 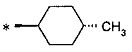 , где * представляет собой положение для замещения; R1, R2 и R3, каждый представляет собой атом водорода; R4 представляет собой атом водорода или группу пролекарства; и Y представляет собой группу: -CH2-CHR5-CH2-NHR6 (где R5 представляет собой атом водорода или С1-С6алкильную группу, и R6 представляет собой атом водорода или группу пролекарства), -O-CHR7-CH2-NHR8 (где R7 представляет собой атом водорода или С1-С6алкильную группу, и R8 представляет собой атом водорода) или
, где * представляет собой положение для замещения; R1, R2 и R3, каждый представляет собой атом водорода; R4 представляет собой атом водорода или группу пролекарства; и Y представляет собой группу: -CH2-CHR5-CH2-NHR6 (где R5 представляет собой атом водорода или С1-С6алкильную группу, и R6 представляет собой атом водорода или группу пролекарства), -O-CHR7-CH2-NHR8 (где R7 представляет собой атом водорода или С1-С6алкильную группу, и R8 представляет собой атом водорода) или 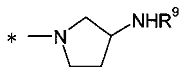 (где R9 представляет собой атом водорода, и * представляет собой положение для замещения); где группа пролекарства, представленная R4, представляет собой [(изопропоксикарбонил)окси]этильную группу или С1-С6алкильную группу, которая замещена одной фенильной группой; и где группа пролекарства, представленная R6, представляет собой С1-С6алканоильную группу, которая замещена одной или двумя одинаковыми или различными группами, выбранными из аминогруппы и фенильной группы; (С1-С6алкокси)карбонильной группы, которая замещена одной группой, выбранной из С2-С6алканоилокси группы и (С3-С6циклоалкил)карбонилокси группы; или 1,3-диоксол-метоксикарбонильной группы, которая замещена двумя разными группами, выбранными из оксогруппы и С1-С6алкильной группы. Также изобретение относится к конкретным соединениям и солям, ингибитору TAFIa, промотору фибринолиза, фармацевтической композиции и профилактическому или терапевтическому лекарственному средству на основе соединения формулы (I), а также к промежуточным соединениям формул (А), (В) и (С). Технический результат: получены новые производные имидазола, обладающие активностью ингибирования TAFIa. 27 н. и 32 з.п. ф-лы, 4 ил., 4 табл., 50 пр.
(где R9 представляет собой атом водорода, и * представляет собой положение для замещения); где группа пролекарства, представленная R4, представляет собой [(изопропоксикарбонил)окси]этильную группу или С1-С6алкильную группу, которая замещена одной фенильной группой; и где группа пролекарства, представленная R6, представляет собой С1-С6алканоильную группу, которая замещена одной или двумя одинаковыми или различными группами, выбранными из аминогруппы и фенильной группы; (С1-С6алкокси)карбонильной группы, которая замещена одной группой, выбранной из С2-С6алканоилокси группы и (С3-С6циклоалкил)карбонилокси группы; или 1,3-диоксол-метоксикарбонильной группы, которая замещена двумя разными группами, выбранными из оксогруппы и С1-С6алкильной группы. Также изобретение относится к конкретным соединениям и солям, ингибитору TAFIa, промотору фибринолиза, фармацевтической композиции и профилактическому или терапевтическому лекарственному средству на основе соединения формулы (I), а также к промежуточным соединениям формул (А), (В) и (С). Технический результат: получены новые производные имидазола, обладающие активностью ингибирования TAFIa. 27 н. и 32 з.п. ф-лы, 4 ил., 4 табл., 50 пр.
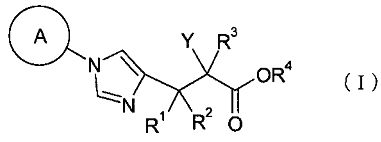 ,
,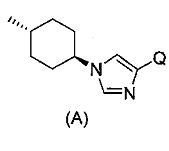 ,
,
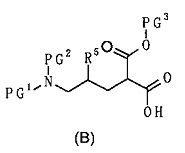 ,
, 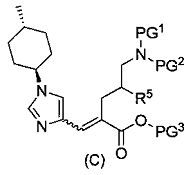
1. Соединение, представленное общей формулой (I), или его фармакологически приемлемая соль:
где А представляет собой группу:
Где * представляет собой положение для замещения; R1, R2 и R3, каждый представляет собой атом водорода; R4 представляет собой атом водорода или группу пролекарства; и Y представляет собой группу: -CH2-CHR5-CH2-NHR6 (где R5 представляет собой атом водорода или С1-С6алкильную группу, и R6 представляет собой атом водорода или группу пролекарства), -O-CHR7-CH2-NHR8 (где R7 представляет собой атом водорода или С1-С6алкильную группу, и R8 представляет собой атом водорода) или
(где R9 представляет собой атом водорода, и * представляет собой положение для замещения);
где группа пролекарства, представленная R4, представляет собой [(изопропоксикарбонил)окси]этильную группу или С1-С6алкильную группу, которая замещена одной фенильной группой; и
где группа пролекарства, представленная R6, представляет собой С1-С6алканоильную группу, которая замещена одной или двумя одинаковыми или различными группами, выбранными из аминогруппы и фенильной группы; (С1-С6алкокси)карбонильной группы, которая замещена одной группой, выбранной из С2-С6алканоилокси группы и (С3-С6циклоалкил)карбонилокси группы; или 1,3-диоксол-метоксикарбонильной группы, которая замещена двумя разными группами, выбранными из оксогруппы и С1-С6алкильной группы.
2. Соединение по п.1 или его фармакологически приемлемая соль, где Y представляет собой группу: -CH2-CHR5-CH2-NHR6.
3. Соединение по п.2 или его фармакологически приемлемая соль, где R6 представляет собой атом водорода.
4. Соединение по п.2 или его фармакологически приемлемая соль, где R6 представляет собой группу пролекарства, как указано в п.1.
5. Соединение по п.4 или его фармакологически приемлемая соль, где группа пролекарства, представленная R6, представляет собой фенилаланильную группу, L-норлейцильную группу,
[(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонильную группу,
[1-(изобутирилокси)этокси]карбонильную группу,
[1-(2,2-диметилпропаноилокси)этокси]карбонильную группу,
{1-[(циклогексилкарбонил)окси]этокси}карбонильную группу или
(1-ацетоксиэтокси)карбонильную группу.
6. Соединение по п.1 или его фармакологически приемлемая соль, где Y представляет собой группу: -O-CHR7-CH2-NHR8.
7. Соединение по п.6 или его фармакологически приемлемая соль, где R7 представляет собой атом водорода.
8. Соединение по п.6 или его фармакологически приемлемая соль, где R8 представляет собой атом водорода.
9. Соединение по п.1 или его фармакологически приемлемая соль, где Y представляет собой группу:
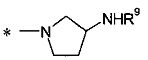
(где R9 представляет собой атом водорода, и * представляет собой положение для замещения).
10. Соединение по п.1 или его фармакологически приемлемая соль, где Y представляет собой группу:
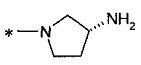
(где * представляет собой положение для замещения).
11. Соединение по п.1 или его фармакологически приемлемая соль, где Y представляет собой группу:
(где * представляет собой положение для замещения).
12. Соединение по п.1 или его фармакологически приемлемая соль, где R4 представляет собой атом водорода.
13. Соединение по п.1 или его фармакологически приемлемая соль, где R4 представляет собой группу пролекарства, как определено в п.1.
14. Соединение по п.13 или его фармакологически приемлемая соль, где группа пролекарства, представленная R4, представляет собой бензильную группу или [(изопропоксикарбонил)окси]этильную группу.
15. Соединение по п.1, представленное общей формулой (I-1), или его фармакологически приемлемая соль:
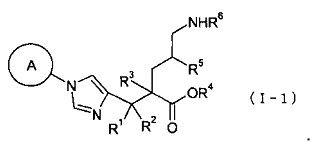
16. Соединение по п.15 или его фармакологически приемлемая соль, где R4 представляет собой атом водорода, бензильную группу или [(изопропоксикарбонил)окси]этильную группу; и R6 представляет собой атом водорода, фенилаланильную группу, L-норлейцильную группу,
[(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонильную группу,
[1-(изобутирилокси)этокси]карбонильную группу,
[1-(2,2-диметилпропаноилокси)этокси]карбонильную группу,
({1-[(циклогексилкарбонил)окси]этокси}карбонильную группу или
(1-ацетоксиэтокси)карбонильную группу.
17. Соединение по п.2, представленное общей формулой (I-1а), или его фармакологически приемлемая соль:
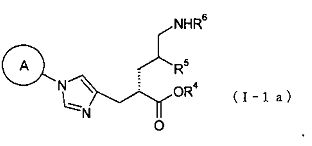
18. Соединение по п.17 или его фармакологически приемлемая соль, где R4 представляет собой атом водорода, бензильную группу или [(изопропоксикарбонил)окси]этильную группу; и R6 представляет собой атом водорода, фенилаланильную группу, L-норлейцильную группу,
[(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонильную группу,
[1-(изобутирилокси)этокси]карбонильную группу,
[1-(2,2-диметилпропаноилокси)этокси]карбонильную группу,
({1-[(циклогексилкарбонил)окси]этокси}карбонильную группу или
(1-ацетоксиэтокси)карбонильную группу.
19. Соединение по п.17 или его фармакологически приемлемая соль, где оба R4 и R6 представляют собой атом водорода.
20. Соединение по п.1, представленное общей формулой (I-2), или его фармакологически приемлемая соль:
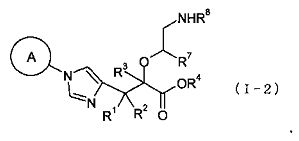
21. Соединение по п.20 или его фармакологически приемлемая соль, где R4 представляет собой атом водорода, бензильную группу или [(изопропоксикарбонил)окси]этильную группу; и оба из R7 и R8 представляют собой атом водорода.
22. Соединение по п.1, представленное общей формулой (I-3), или его фармакологически приемлемая соль:
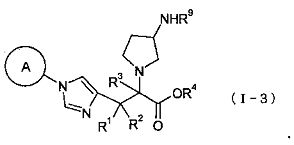
23. Соединение по п.22 или его фармакологически приемлемая соль, где R4 представляет собой атом водорода, бензильную группу или [(изопропоксикарбонил)окси]этильную группу; и R9 представляет собой атом водорода.
24. Соединение по п.1 или его фармакологически приемлемая соль, где соединение выбирают из группы, состоящей из
5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
бензил-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерата,
2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-(L-фенилаланиламино)валериановой кислоты,
2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-(L-норлейциламино)валериановой кислоты,
(2S)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-({[(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонил}амино)валериановой кислоты,
(2S)-5-({[1-(изобутирилокси)этокси]карбонил}амино)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
1-[(изопропоксикарбонил)окси]этил-(2S)-5-({[1-(изобутирилокси)этокси]карбонил}амино)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валерата,
(2S)-5-({[1-(2,2-диметилпропаноилокси)этокси]карбонил}амино)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
(2S)-5-[({1-[(циклогексилкарбонил)окси]этокси}карбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
2-(2-аминоэтокси)-3-[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]пропионовой кислоты,
2-[(1R)-2-амино-1-метилэтокси]-3-[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]пропионовой кислоты
(2S,4S)-5-амино-4-метил-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
(2S,4R)-5-амино-4-метил-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
2-[(3S)-3-аминопирролидин-1-ил]-3-[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]пропионовой кислоты,
(2S)-5-{[(1-ацетоксиэтокси)карбонил]амино}-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
(2S)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}-5-[({[(2-метилпропаноил)окси]метокси}карбонил)амино]валериановой кислоты,
(2S)-5-[({[(2,2-диметилпропаноил)окси]метилокси}карбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
(2S)-5-[({[(циклогексилкарбонил)окси]метокси}карбонил)амино]-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты,
(2S)-5-({[(ацетилокси]метокси]карбонил}амино)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты
и
(2S)-5-({[(1R)-1-(изобутирилокси)этокси]карбонил}амино)-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты.
25. 5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота или ее фармакологически приемлемая соль.
26. (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота или ее фармакологически приемлемая соль.
27. Фармакологически приемлемая соль соединения по п.1, где фармакологически приемлемая соль представляет собой п-толуолсульфонат.
28. (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановая кислота.
29. п-Толуолсульфонат (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты.
30. Ангидрат п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты.
31. Ангидрат п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты по п.30, который находится в кристаллической форме, проявляющей основные пики при межплоскостных расстояниях d, равных 23,9, 11,9, 4,5, 4,3 и 3,6 ангстрем, при порошковой рентгеновской дифракции, полученной посредством излучения Кα меди.
32. Моногидрат п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты.
33. Моногидрат п-толуолсульфоната (2S)-5-амино-2-{[1-(транс-4-метилциклогексил)-1Н-имидазол-4-ил]метил}валериановой кислоты по п.32, который находится в кристаллической форме, проявляющей основные пики при межплоскостных расстояниях d, равных 22,9, 5,0, 4,9, 4,7 и 4,0 ангстрем, при порошковой рентгеновской дифракции, полученной посредством излучения Кα меди.
34. Фармакологически приемлемая соль соединения по любому из пп.1-26, где фармакологически приемлемая соль представляет собой п-толуолсульфонат.
35. Ингибитор TAFIa, содержащий соединение или его фармакологически приемлемую соль по любому из пп.1-34 в качестве активного ингредиента.
36. Ингибитор TAFIa по п.35, дополнительно содержащий фармакологически приемлемый носитель.
37. Промотор фибринолиза, содержащий соединение или его фармакологически приемлемую соль по любому из пп.1-34 в качестве активного ингредиента.
38. Промотор фибринолиза по п.37, дополнительно содержащий фармакологически приемлемый носитель.
39. Профилактическое или терапевтическое лекарственное средство против заболевания, вызываемого посредством ингибирования фибринолиза, содержащее соединение или его фармакологически приемлемую соль по любому из пп.1-34 в качестве активного ингредиента.
40. Профилактическое или терапевтическое лекарственное средство против заболевания, вызываемого посредством ингибирования фибринолиза по п.39, дополнительно содержащее фармакологически приемлемый носитель.
41. Профилактическое или терапевтическое лекарственное средство против тромбоза или эмболии, или их последствий, включающих: острый коронарный синдром, такой как инфаркт миокарда и стенокардия (стабильная стенокардия и нестабильная стенокардия); венозную тромбоэмболию, такую как тромбоз глубоких вен и тромбоэмболию легких; тромбоз или эмболию, происходящих в сердечно-сосудистой системе после хирургической операции, такой как реваскуляризация сосудов, ангиопластика, установка стента и шунтирующая хирургия; тромбоз или эмболию после операции по замене искусственного сустава, такой как операция замены коленного сустава и операция замены тазобедренного сустава; относящееся к воспалению внутрисосудистое заболевание, такое как сепсис и синдром диссеминированной внутрисосудистой коагуляции (DIC); периферическое сосудистое нарушение, являющееся производным от или относящееся к заболеванию, такое периферическая артериальная окклюзия (РАО), артериосклероз и сахарный диабет; заболевание, относящееся к опухолевому, такого как солидный рак и рак крови; и нарушение органа, приписываемое тромбу или эмболу, такое как тромбоэмболия легких, инфаркт головного мозга и инфаркт почки, содержащее соединение или его фармакологически приемлемую соль по любому из пп.1-34 в качестве активного ингредиента.
42. Профилактическое или терапевтическое лекарственное средство против тромбоза или эмболии, включающих: заболевание, вызванное контактом с инородным веществом в организме, инородным веществом, включающим медицинское устройство, такое как протез сустава, применяемый при замене сустава, сосудистый катетер, протез в кровотоке, стент в кровотоке и протез клапана; и заболевание, вызванное контактом между кровью и медицинским устройством вне организма, медицинским устройством, включающим насосный оксигенатор, применяемый при операции на сердце, и медицинским устройством, применяемым при гемодиализе, содержащее соединение или его фармакологически приемлемую соль по любому из пп.1-34 в качестве активного ингредиента.
43. Профилактическое или терапевтическое лекарственное средство против заболевания, относящегося к тромбозу или эмболии, или сопровождающегося отложением фибрина или фиброзом, включающего: заболевание легких, такое как легочная гипертензия, респираторный дистресс-синдром взрослых, фиброз легких и хроническая тромбоэмболическая легочная гипертензия; заболевание почек, такое как гломерулонефрит (включая острый гломерулонефрит, хронический гломерулонефрит, нефротический нефрит и быстро прогрессирующий гломерулонефрит), инфаркт почек и диабетический нефрит; заболевание печени, такое как фиброз печени, гепатит и цирроз печени; глазное заболевание, ассоциированное с отложением фибрина в глазе; дисфункцию органа после трансплантации или резекции органа; нарушение микроциркуляции, вызванное микротромбом, включая тромботическую микроангиопатию; и заболевание или симптомы, ассоциированные с миграцией раковых клеток или метастазами, содержащее соединение или его фармакологически приемлемую соль по любому из пп.1-34 в качестве активного ингредиента.
44. Терапевтическое лекарственное средство против инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких, содержащее соединение или его фармакологически приемлемую соль по любому из пп.1-34 в качестве активного ингредиента.
45. Способ лечения инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких, включающий введение фармацевтической композиции, содержащей соединение или его фармакологически приемлемую соль по любому из пп.1-34 в качестве активного ингредиента.
46. Применение фармацевтической композиции, содержащей соединение или его фармакологически приемлемую соль по любому из пп.1-34 в качестве активного ингредиента, в производстве лекарственного средства для лечения инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких.
47. Соединение или его фармакологически приемлемая соль по любому из пп.1-34 для применения при лечении инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких.
48. Ингибитор TAFIa для инъекции, содержащий соединение или его фармакологически приемлемую соль по любому из пп.1-34 в качестве активного ингредиента.
49. Ингибитор TAFIa для инъекции по п.48, дополнительно содержащий фармакологически приемлемый носитель.
50. Терапевтическое лекарственное средство для инъекции против инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких, содержащее соединение или его фармакологически приемлемую соль по любому из пп.1-34 в качестве активного ингредиента.
51. Терапевтическое лекарственное средство против заболевания, происходящего вследствие тромбоэмболии, содержащее соединение или его фармакологически приемлемую соль по любому из пп.1-34 в качестве активного ингредиента.
52. Терапевтическое лекарственное средство против заболевания, происходящего вследствие тромбоэмболии по п.51, дополнительно содержащее фармакологически приемлемый носитель.
53. Способ лечения инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких, включающий введение фармацевтической композиции для инъекции, содержащей соединение или его фармакологически приемлемую соль по любому из пп.1-34 в качестве активного ингредиента.
54. Применение фармацевтической композиции для инъекции, содержащей соединение или его фармакологически приемлемую соль по любому из пп.1-34 в качестве активного ингредиента в производстве лекарственного средства для лечения инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких.
55. Соединение или его фармакологически приемлемая соль по любому из пп.1-34 для применения при лечении инфаркта миокарда, стенокардии, острого коронарного синдрома, инфаркта головного мозга, тромбоза глубоких вен, тромбоэмболии легких, периферической артериальной окклюзии, сепсиса, синдрома диссеминированной внутрисосудистой коагуляции или фиброза легких посредством инъекции.
56. Фармацевтическая композиция, обладающая активностью ингибирования TAFIa, содержащая соединение или его фармакологически приемлемую соль по любому из пп.1-34 и одно или два, или более лекарственных средств, выбранных из антикоагулянта, антитромбоцитарного средства, фермента, имеющего отношение к фибринолизу, противоракового лекарственного средства, противовоспалительного средства, антифиброзного лекарственного средства, гипотензивного лекарственного средства, лекарственного средства против легочной гипертензии и иммуносуппрессорного лекарственного средства в качестве активных ингредиентов.
57. Соединение, представленное следующей общей формулой, или его соль:
где Q представляет собой группу COOR, гидроксиметильную группу или формильную группу, и R представляет собой С1-С6алкильную группу.
58. Соединение, представленное следующей общей формулой, или его соль:
где R5 представляет собой водород; PG1 представляет собой трет-бутоксикарбонильную группу, метоксикарбонильную группу или этоксикарбонильную группу; PG2 представляет собой атом водорода; и PG3 представляет собой метильную группу.
59. Соединение, представленное следующей общей формулой, или его соль:
где R5 представляет собой водород; PG1 и PG2 представляют собой каждый независимо водород, трет-бутоксикарбонильную группу, метоксикарбонильную группу или этоксикарбонильную группу; и PG3 представляет собой трет-бутильную группу.
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| RU 2140901 C1, 10.11.1999. | |||

