Область техники
Перекрестная ссылка на родственные заявки
Настоящая заявка испрашивает приоритет японской патентной заявки №2012-037565 (полное раскрытие которой включено в настоящее описании посредством ссылки), поданной 23 февраля 2012 года.
Настоящее изобретение относится к хинолилпирролпиримидильным конденсированным соединениям, обладающим ингибирующим действием в отношении рецептора эпидермального фактора роста (EGFR), и к фармацевтическим композициям, содержащим их в качестве активного ингредиента.
Уровень техники
EGFR является рецепторным типом тирозинкиназы, проявляющим свою физиологическую функцию в нормальной ткани, когда он связан с эпидермальным фактором роста (EGF), который является лигандом, и способствует росту ткани, ингибированию апоптоза в эпителиальных тканях и т.п. (непатентный документ (NPL) 1).
Кроме того, EGFR является одним из онкогенов, и амплификация гена EGFR и высокая экспрессия или мутация этого белка имеют место при различных типах рака, таких как рак головы и шеи, рак молочной железы, колоректальный рак, рак пищевода, рак поджелудочной железы, рак легких, рак яичников, рак почки, рак мочевого пузыря, рак кожи и опухоль мозга (непатентный документ (NPL) 2). В Японии и западных странах каждый год приблизительно от 170 до 375 на каждые 100000 человек умирают из-за рака, и рак занимает видное место в качестве причины смерти (непатентный документ (NPL) 3). Кроме того, число умерших от рака легких во всем мире достигает приблизительно 1400000 человек в год, и поскольку немелкоклеточный рак легких составляет 80% или более из числа случаев рака легких, то разработка эффективной терапии для этого случая является желательной (непатентный документ (NPL) 4).
За последние годы были определены гены, ответственные за эти виды рака, и мутация в гене EGFR также является одним из факторов, которые приводят к активной мутации белка EGFR. Активный мутированный белок EGFR имеет, например, делецию аминокислот в положениях 746-750 (EGFR (d476-750)), мутацию аминокислоты лейцин в положении 858 на аргинин (EGFR (L858R)) или подобное. Такие мутации выявлены, например, в 20-40% случаях немелкоклеточного рака легких в Японии, и в 10-15% случаях немелкоклеточного рака легких в западных странах. Поскольку немелкоклеточный рак легких при таких мутациях очень восприимчив к лечению гефитинибом (название продукта: Иресса (зарегистрированная торговая марка Iressa)) и эрлотинибом (название продукта: Тарцева (зарегистрированная торговая марка Tarceva)), которые являются химическими агентами (ингибиторами EGFR), которые ингибируют киназную активность EGFR, и эти химические агенты используются в качестве терапевтических агентов в Японии и западных странах. Тем не менее, рак приобретает устойчивость к гефитинибу и эрлотинибу через 6-12 месяцев с начала использования этих препаратов, и их терапевтический эффект становится слабым. Таким образом, эта приобретенная резистентность является серьезной проблемой при лечении немелкоклеточного рака легких с EGFR, высоковосприимчивых к мутациям. Было установлено, что приблизительно 50% случаев приобретенной устойчивости связаны с появлением устойчивых мутированных форм белка EGFR (EGFR (d476-750/T790M) или EGFR (T790M/L858R)), которые имеют вторую мутацию в гене EGFR, в результате которой аминокислота треонин в положении 790 заменяется на метионин. Поэтому важной задачей является создание терапевтического агента, который был бы эффективным против немелкоклеточного рака легких с лекарственной устойчивостью, приобретенной за счет мутированного EGFR (непатентная литература (NPL) 5).
С другой стороны, сообщалось, что аномалии кожи и расстройства желудочно-кишечного тракта являются обычными побочными эффектами при применении ингибиторов EGFR, таких как гефитиниб и эрлотиниб, которые в настоящее время используются в качестве терапевтических агентов в клинической практике, и при применении ингибиторов EGFR, таких как BIBW2992 и т.п., которые проходят клинические испытания. Широко распространено мнение, что эти побочные эффекты ингибиторов EGFR вызваны ингибированием активности не только мутированного EGFR, экспрессируемого при немелкоклеточном раке легких, но и ингибированием активности EGFR дикого типа (EGFR (WT)) экспрессируемого в коже или в желудочно-кишечном тракте (непатентный документ (NPL) 1). С точки зрения уменьшения побочных эффектов, считается предпочтительным иметь слабую ингибирующую активность в отношении EGFR (WT) нормальных тканей.
Таким образом, существует ожидание возможности подавления роста клеток немелкоклеточного рака легких, имеющих лекарственную устойчивость мутированного EGFR, при введении химического агента, имеющего слабую ингибирующую активность в отношении EGFR дикого типа, по сравнению с ингибирующей активностью против мутированного EGFR с лекарственной устойчивостью, в котором аминокислота в положении 790 мутирована в метионин, при вводимой дозе, когда побочный эффект в отношении кожи или желудочно-кишечного тракта в выраженной степени не возникает. Это прогнозируется как вклад в лечение рака, в продление жизни и в улучшение качества жизни больных. Кроме того, если такое химическое вещество будет обладать слабой ингибирующей активностью в отношении EGFR дикого типа, но будет иметь сильную ингибирующую активность не только против мутированного EGFR с лекарственной устойчивостью, но и против высокочувствительных мутированных EGFR, таких как EGFR (d476-750) и EGFR (L858R) и т.п., которые очень восприимчивы к действию гефитиниба и эрлотиниба. Ожидается возможность подавления роста клеток немелкоклеточного рака легких, экспрессирующих высокочувствительные мутированные формы EGFR или мутированные формы EGFR с лекарственной устойчивостью на вводимые дозы, где побочный эффект в отношении кожи или желудочно-кишечного тракта сильно не проявляется, или можно ожидать уменьшение частоты случаев лекарственной устойчивости мутированного EGFR, которая возникает как приобретенная устойчивость клеток в случае немелкоклеточного рака легких, где такие клетки экспрессируют высокочувствительные мутированные формы EGFR. Это предполагает существенный вклад в лечение рака, продление жизни и улучшение качества жизни больных. Кроме того, поскольку экспрессия высокочувствительных мутированных форм EGFR и мутированных форм EGFR с лекарственной устойчивостью может наблюдаться в рамках реальной терапии, и использоваться в качестве индекса для группировки при выборе пациентов, этот факт может быть в значительной степени полезен с этической точки зрения.
Известно соединение, имеющее структуру, аналогичную структуре соединения по настоящему изобретению, а именно, производное N-(3-(4-амино-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)фенил)бензамида, (патентный документ (PTL) 1). Хотя патентный документ 1 описывает использование амидного соединения для лечения заболеваний, опосредованных киназой B-RAF, этот документ не раскрывает специфические тесты и результаты, на основании которых можно подтвердить наличие активности по ингибированию киназы, и, следовательно, такая активность не была подтверждена.
Список цитированных документов
Патентный документ:
PTL 1: описание публикации международной заявки WO 2006/102079.
Непатентные документы:
NPL 1: Nature Rev. Cancer, vol.6, pp. 803-811 (2006).
NPL 2: J. Clin. Oncol., vol.19, 32s-40s (2001).
NPL 3: Ministry of Internal Affairs and Communications Statistics Bureau homepage/statistical data/world statistics “World Statistics 2011” Chapter 14: People′s Life and Social Security, 14-1 Death Rates by Causes Death.
NPL 4: Lung Cancer, vol.69, pp. 1-12 (2010).
NPL 5: Nature Rev. Cancer, vol.10, pp. 760-774 (2010).
Сущность изобретения
Техническая проблема, решаемая изобретением
Как описано выше, ингибиторы EGFR, несмотря на ожидаемую эффективность в терапии рака, в настоящее время клинически не являются достаточно эффективными.
Таким образом, целью настоящего изобретения является создание нового соединения или его соли, которое в высокой степени ингибирует EGFR. Еще одной задачей настоящего изобретения является предоставление: нового соединения, которое ингибирует EGFR (d476-750), EGFR (L858R), EGFR (d476-750/T790M) и EGFR (T790M/L858R), но не ингибирует EGFR (WT); или его соль.
Решение проблемы
Авторы настоящего изобретения провели тщательное исследование для достижения описанной выше цели. В результате они обнаружили, что группа хинолилпирролпиримидильных конденсированных соединений по настоящему изобретению обладает превосходной ингибирующей активностью в отношении EGFR и эти соединения обладают ингибирующей активностью в отношении роста раковых клеток, и они полезны в качестве лекарственного средства для лечения рака. За счет этого открытия авторы выполнили настоящее изобретение.
Таким образом, настоящее изобретение включает следующее.
Пункт 1. Соединение, представленное формулой (I), или его соль:
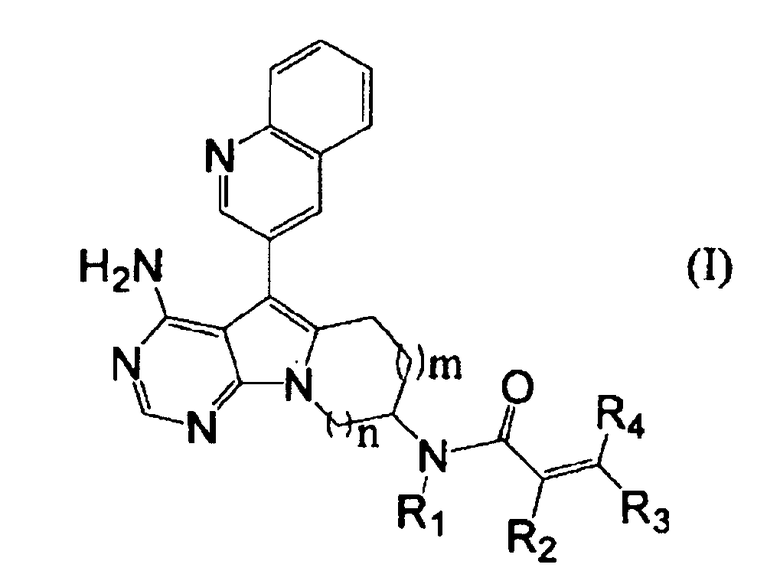 ,
,
где m равно 1 или 2;
n равно 1 или 2;
группа R1 представляет собой атом водорода или C1-C4-алкильную группу; и
группы R2, R3 и R4 могут быть одинаковыми или различными, и каждая из них представляет собой атом водорода, атом галогена, C1-C4-алкильную группу, или группу, представленную формулой (а):
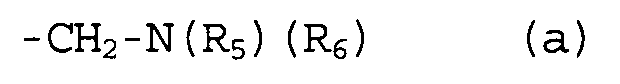 ,
,
где группы R5 и R6 могут быть одинаковыми или различными, и каждая из них представляет собой атом водорода или C1-C4-алкильную группу, или группы R5 и R6 могут образовывать гетероциклоалкильную группу, имеющую 4-6-членное кольцо вместе с атомом азота, связанным с ними.
Пункт 2. Соединение или его соль по п. 1, в котором
m равно 1 или 2;
n равно 1 или 2;
группа R1 представляет собой атом водорода или C1-C4-алкильную группу; и
группы R2, R3 и R4 могут быть одинаковыми или различными, и каждая из них представляет собой атом водорода, атом галогена, C1-C4-алкильную группу, или группу, представленную формулой (а):
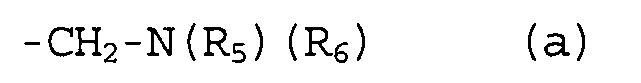 ,
,
где группы R5 и R6 могут быть одинаковыми или различными, и каждая из них представляет собой атом водорода или C1-C4-алкильную группу.
Пункт 3. Соединение или его соль по пункту 1 или 2, в котором
m равно 1 или 2;
n равно 1 или 2;
группа R1 представляет собой атом водорода или метильную группу; и
группы R2, R3 и R4 могут быть одинаковыми или различными, и каждая из них представляет собой атом водорода, атом хлора или диметиламинометильную группу.
Пункт 4. Соединение или его соль по любому из пунктов 1-3, в котором значения m и n представляют собой (m,n)=(1,1), (1,2) или (2,1).
Пункт 5. Соединение или его соль по любому из пунктов 1-4, где соединение выбрано из следующей группы соединений:
(R)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид,
(S)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9- тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид,
N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-N-метилакриламид,
(Е)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-4-(диметиламино)-2-бутенамид,
(S,E)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-3-хлоракриламид,
(S,Z)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-3-хлоракриламид,
(S)-N-(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5] пирроло[1,2-а]азепин-8-ил)акриламид,
(S)-N-(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5] пирроло[1,2-а]азепин-9-ил)акриламид,
(R)-N-(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5]пирроло[1,2-а]азепин-9-ил)акриламид.
Пункт 6. Ингибитор EGFR, содержащий в качестве активного ингредиента соединение или его соль по любому из пунктов 1-5.
Пункт 7. Фармацевтическая композиция, содержащая соединение или его соль по любому из пунктов 1-5.
Пункт 8. Противоопухолевое средство, содержащее в качестве активного ингредиента соединение или его соль по любому из пунктов 1-5.
Пункт 9. Способ лечения или профилактики рака, включающий стадию введения млекопитающему соединения или его соли по любому из пунктов 1-5 в дозе, эффективной для лечения или профилактики рака.
Пункт 10. Применение соединения или его соли по любому из пунктов 1-5 для изготовления противоопухолевого средства.
Пункт 11. Соединение или его соль по любому из пунктов 1-5 для применения при лечении или профилактике рака.
Настоящее изобретение также относится к способу получения промежуточных синтетических соединений для получения из них соединений по настоящему изобретению, указанных ниже.
Пункт 12 Способ получения соединения, представленного формулой (VIII), или его соли, где способ включает стадии:
[I] взаимодействия органического борана с соединением, представленным формулой (VII), или его солью:
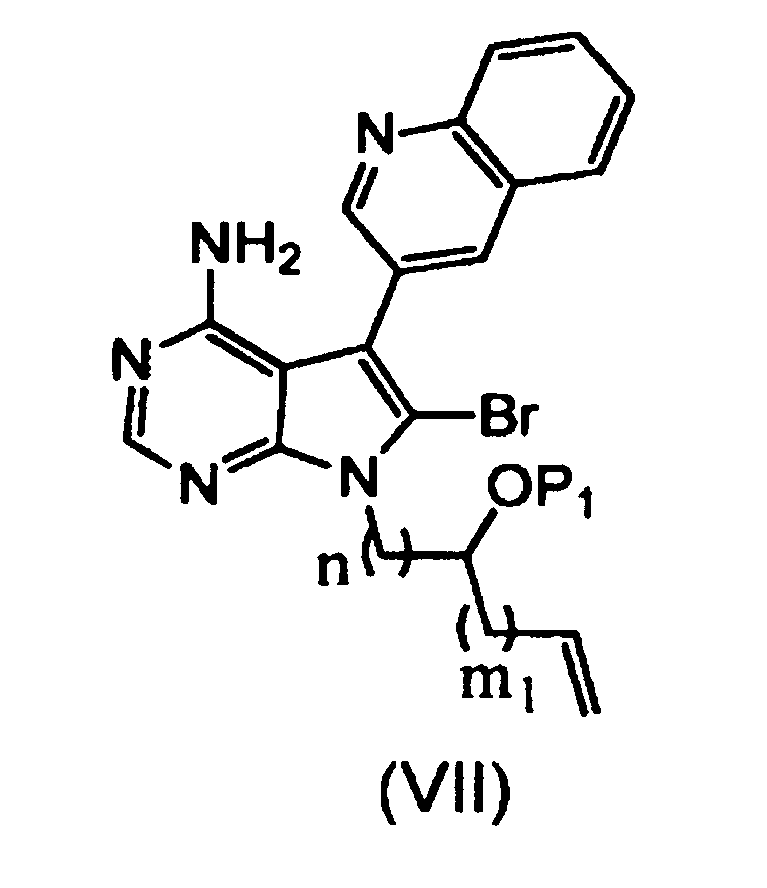 ,
,
где группа P1 представляет собой защитную группу для гидроксильной группы, n равно 1 или 2, и m1 равно 0 или 1; и
[II] внутримолекулярной циклизации продукта реакции со стадии [I] с использованием палладиевого (0) катализатора в присутствии гидроксида щелочного металла, с получением соединения:
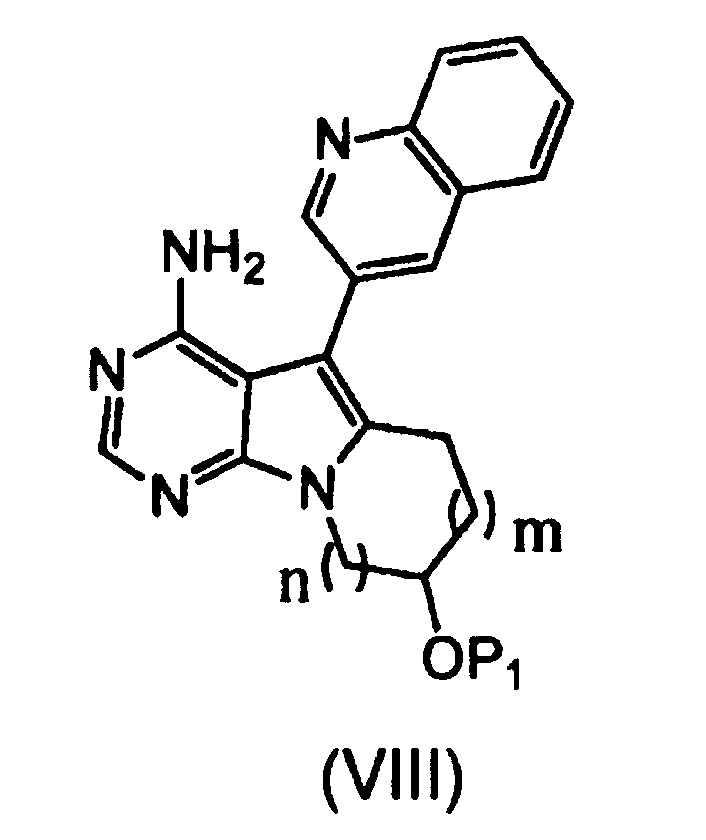 ,
,
где m равно 1 или 2, и P1 и n имеют значения, описанные выше.
Пункт 13. Способ получения соединения формулы (XX) или его соли:
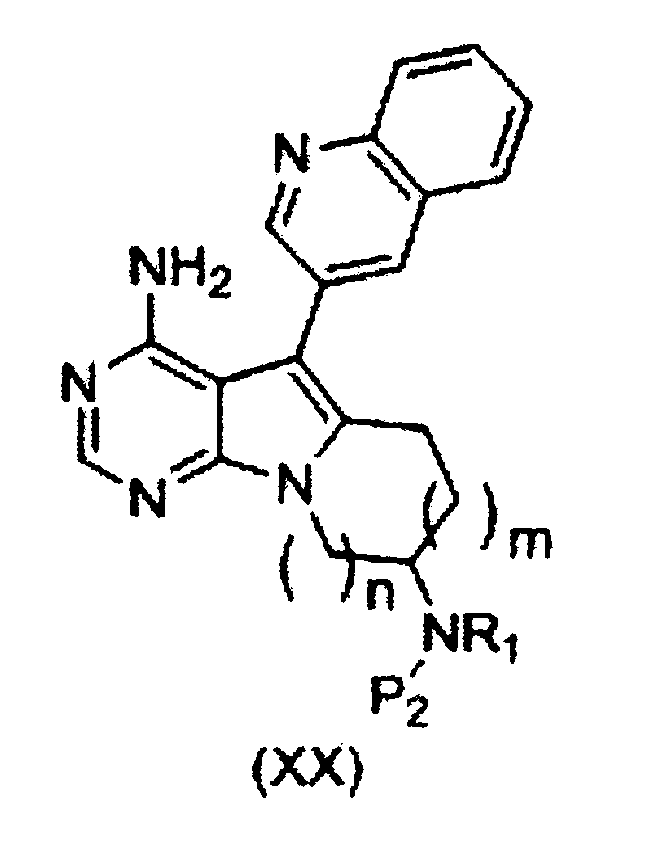 ,
,
где группа R1 представляет собой атом водорода или C1-C4-алкильную группу, группа P2 представляет собой защитную группу аминогруппы, m равно 1 или 2, n равно 1 или 2,
где способ включает стадии:
[I] взаимодействия органического борана с соединением формулы (XIX) или его солью:
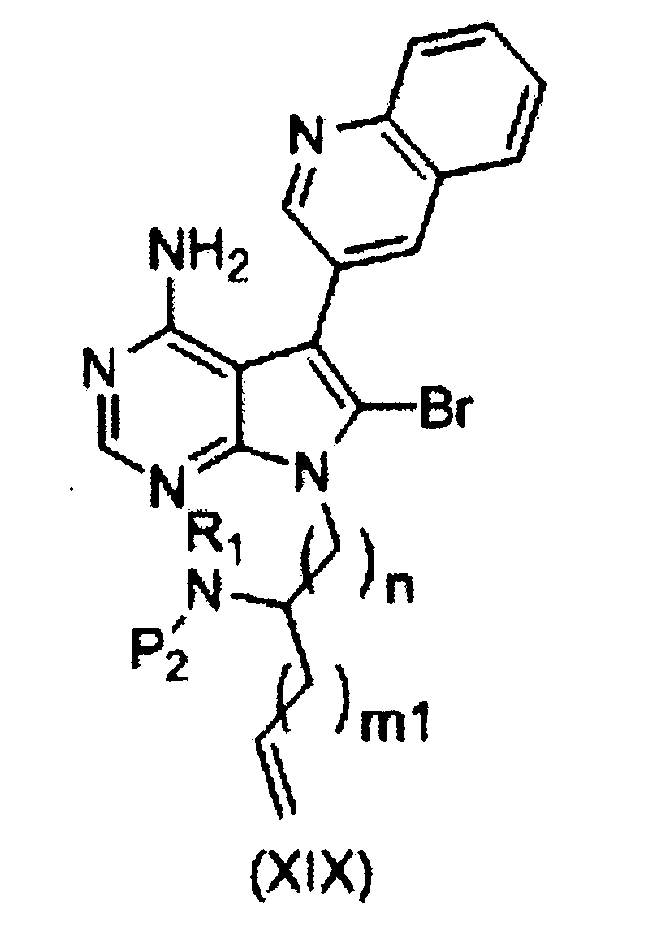 ,
,
где R1, Р2 и n имеют значения, как описано выше, и m1 равно 0 или 1; и
[II] внутримолекулярной циклизации продукта реакции со стадии [I] с использованием палладиевого (0) катализатора в присутствии гидроксида щелочного металла.
Полезные эффекты изобретения
В соответствии с настоящим изобретением предоставляется новое соединение, представленное формулой (I), описанной выше, или его соль, которые полезны в качестве ингибитора EGFR.
Ясно, что соединение по настоящему изобретению или его соль обладает превосходной ингибирующей активностью в отношении EGFR и эффектом подавления роста в отношении клеточных линий рака. Кроме того, соединение или его соль имеет преимущество в том, что оно вызывает небольшие побочные эффекты, поскольку оно обладает превосходной селективностью в отношении EGFR. Следовательно, соединение или его соль по настоящему изобретению является полезным в качестве средства для лечения и/или профилактики рака.
Описание вариантов выполнения изобретения
Соединение формулы (I) по настоящему изобретению представляет собой хинолилпирролпиримидильное конденсированное соединение, которое включает структуру хинолина и структуру α,β-ненасыщенного амида, и, таким образом, оно является новым соединением, которое нигде не раскрыто в любом из вышеуказанных документов уровня техники и т.п.
В частности, соединение конкретно раскрытое в PTL 1 представляет собой производное N-(3-(4-амино-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)фенил)бензамида. Соединение по настоящему изобретению отличается от соединения, описанного в PTL 1, тем, что соединение по настоящему изобретению включает структуру хинолина и структуру α,β-ненасыщенного амида.
В настоящем описании, термин ″C1-C4 алкил″ относится к линейной или разветвленной алкильной группе, имеющей от 1 до 4 атомов углерода. Конкретные примеры такой группы включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил и тому подобное.
В данном описании примеры ″галогена″ включают хлор, бром, фтор и йод.
В данном описании термин ″4-6-членный гетероциклоалкил″ относится к 4-6-членной циклоалкильной группе, имеющей от 1 до 2 атомов азота в кольце. Конкретные примеры такой группы включают азетидинил, пирролидинил, пиперидил, имидазолидинил и тому подобное.
Значения m и n в формуле (I) предпочтительно представляют собой (m,n)=(1,1), (1,2) или (2,1).
Группа R1 в формуле (I) предпочтительно представляет собой водород или метил.
Группы R2, R3 и R4 в формуле (I) могут быть одинаковыми или различными, и каждая из них предпочтительно представляет собой водород, галоген, C1-C4-алкил, или группу, представленную приведенной выше формулой (а). Когда по меньшей мере одна из групп R2, R3 и R4 в формуле (I) представляет собой группу, представленную формулой (а), каждая группа R5 и R6 предпочтительно представляет собой C1-C4-алкил, и более предпочтительно, когда обе группы R5 и R6 представляют собой метил.
Группа R2 в формуле (I) более предпочтительно представляет собой водород.
Группа R3 в формуле (I) более предпочтительно представляет собой водород, хлор или диметиламинометил.
Группа R4 в формуле (I) более предпочтительно представляет собой водород или хлор.
В настоящем изобретении соединение формулы (I), где m равно 1 или 2; n равно 1 или 2; группа R1 представляет собой водород или метил; группы R2, R3 и R4 могут быть одинаковыми или различными и они представляют собой водород, хлор или диметиламинометил; или его соль, является предпочтительным.
Когда m равно 1 и n равно 1, то соединение формулы (I), где группа R1 представляет собой водород или метил; группа R2 представляет собой водород; одна из групп R3 и R4 представляет собой водород, хлор или диметиламинометил, а другая группа представляет собой водород; или его соль, является предпочтительным.
Когда m=1 и n=2 или m=2 и n=1, то соединение формулы (I), где все группы R1, R2, R3 и R4 представляют собой водород; или его соль, является предпочтительной.
Конкретные примеры предпочтительных соединений по настоящему изобретению включают следующие соединения:
(R)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид;
(S)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид;
N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-N-метилакриламид;
(Е)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо [5,4-b]индолизин-8-ил)-4-(диметиламино)-2-бутенамид;
(S,E)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-3-хлоракриламид;
(S,Z)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-3-хлоракриламид;
(S)-N-(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5] пирроло[1,2-а]азепин-8-ил)акриламид;
(S)-N-(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5] пирроло[1,2-а]азепин-9-ил)акриламид; и
(R)-N-(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5]пирроло[1,2-а]азепин-9-ил)акриламид.
Соединения, которые обладают высокой ингибирующей активностью против ферментативной активности EGFR (T790M/L858R), являются предпочтительными, и соединения с ингибирующей активностью, равной 2 нМ или менее, являются более предпочтительными. Соединения, которые обладают высокой ингибирующей активностью против ферментативной активности EGFR (d476-750/T790M), являются предпочтительными, и соединения с ингибирующей активностью, равной 2 нМ или менее, являются более предпочтительными.
Далее будет пояснен способ получения соединения в соответствии с настоящим изобретением.
Соединение (I) по настоящему изобретению может быть получено, например, следующими способами получения, или способами, описанными в примерах. Однако способ получения соединения (I) по настоящему изобретению не ограничивается этими конкретными примерами реакции.
Способ получения 1
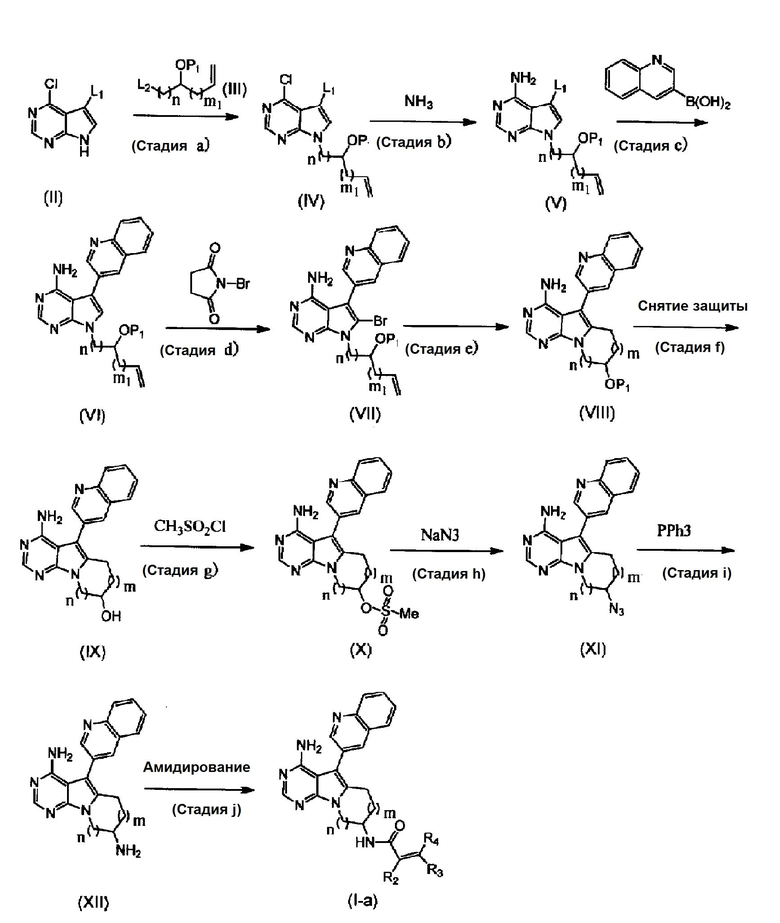
где P1 обозначает защитную группу для гидроксильной группы, L1 и L2 обозначают уходящие группы, m1 равно 0-1, и R2, R3, R4, m и n определены выше.
(Стадия а)
На этой стадии соединения формул (II) и (III) взаимодействуют в присутствии основания с получением соединения формулы (IV).
Примеры уходящей группы, представленной группой L1 в соединении формулы (II), включают атом брома или йода. Соединение формулы (II) может быть коммерчески доступным продуктом, или может быть получено известным способом. Примеры защитной группы для гидроксильной группы, представленной как P1 в формуле (III), включают трет-бутилдиметилсилил, трет-бутилдифенилсилил, триэтилсилил и тому подобное. Примеры уходящих групп, представленных как L2, включают бром, йод, сложный эфир метансульфоновой кислоты, сложный эфир п-толуолсульфоновой кислоты и тому подобное. Соединение формулы (III) может быть коммерчески доступным продуктом, или может быть получено известным способом. Соединение формулы (III) может быть использовано в количестве от 1 до 10 молей, предпочтительно от 1 до 5 моль, из расчета на один моль соединения формулы (II).
Примеры используемых оснований включают неорганические основания, такие как гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид цезия, гидрид натрия и гидрид калия; органические амины, такие как триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, лутидин и коллидин. Такое основание может быть использовано в количестве от 1 до 100 молей, предпочтительно от 1 до 10 молей, из расчета на один моль соединения формулы (II).
Примеры используемых растворителей включают N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, тетрагидрофуран, 1,4-диоксан, N-метилпирролидин-2-он, ацетонитрил и тому подобное. Такие растворители могут быть использованы по-отдельности или в виде смеси. Время реакции составляет от 0,1 до 100 часов, предпочтительно от 0,5 до 24 часов. Реакция протекает при температуре от 0°C до температуры кипения растворителя, предпочтительно от 0 до 100°С.
Полученное таким образом соединение формулы (IV) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
(Стадия b)
На этой стадии соединение формулы (IV) подвергают взаимодействию с аммиаком или его солью, с получением соединения формулы (V).
Количество аммиака или его соли, используемого на этой стадии, как правило, составляет от эквимолярного до избыточного молярного количества на моль соединения формулы (IV).
Для реакции может быть использован любой растворитель, который не оказывает вредного влияния на реакцию. Примеры используемых растворителей для реакции включают воду, метанол, этанол, изопропанол, трет-бутиловый спирт, тетрагидрофуран, 1,2-диметоксиэтан, 1,4-диоксан, N,N-диметилформамид, N-метилпирролидин-2-он, диметилсульфоксид и смеси этих растворителей.
Реакция протекает обычно при температуре от 0 до 200°C и, предпочтительно от комнатной температуры до 150°С. Время реакции обычно составляет от 5 минут до 7 дней, и предпочтительно от 30 минут до 24 часов.
Полученное таким образом соединение формулы (V) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
(Стадия с)
На этой стадии соединение формулы (V) подвергают реакции сочетания с 3-хинолинбороновой кислотой или со сложным эфиром 3-хинолинбороновой кислоты, с получением соединения формулы (VI).
Эта стадия может быть выполнена в соответствии с любым общеизвестным способом (например, см. Chemical Reviews, Vol. 95, p.2457, 1995). В частности, эта стадия может быть осуществлена в присутствии катализатора на основе переходного металла и в присутствии основания в растворителе, не оказывающем вредного влияния на реакцию.
Количество используемой 3-хинолинбороновой кислоты или сложного эфира 3-хинолинбороновой кислоты может составлять от 1 до 10 молей, предпочтительно от 1 до 3 молей, из расчета на один моль соединения формулы (V).
Примеры катализаторов на основе переходного металла включают палладиевые катализаторы (например, ацетат палладия, хлорид палладия, тетракистрифенилфосфинпалладий, дихлорид 1,1′-бис(дифенилфосфино)ферроцен-палладия(II), и трис(дибензилиденацетон)палладий (0)), никелевые катализаторы (например, хлорид никеля) и тому подобное. Если необходимо, может быть добавлен лиганд (например, трифенилфосфин, три-трет-бутилфосфин или 2-дициклогексилфосфино-2′,4′,6′-триизопропилбифенил), и в качестве сокатализатора может быть использован оксид металла (например, оксид меди или оксид серебра). Количество используемого катализатора на основе переходного металла может варьироваться в зависимости от типа катализатора. Катализатор на основе переходного металла обычно используют в количестве от 0,0001 до 1 моля, предпочтительно от 0,01 до 0,5 моля, из расчета на один моль соединения формулы (V). Количество используемого лиганда составляет, как правило, от 0,0001 до 4 молей, предпочтительно от 0,01 до 2 молей, из расчета на один моль соединения формулы (V). Количество используемого сокатализатора составляет, как правило, от 0,0001 до 4 молей, предпочтительно от 0,01 до 2 молей, из расчета на один моль соединения формулы (V).
Примеры используемых оснований включают органические амины (например, триметиламин, триэтиламин, диизопропилэтиламин, N-метилморфолин, 1,8-диазабицикло[5,4,0]ундец-7-ен, пиридин и N,N-диметиланилин), соли щелочных металлов (например, гидрокарбонат натрия, гидрокарбонат калия, карбонат натрия, карбонат калия, карбонат цезия, фосфат натрия, фосфат калия, гидроксид натрия и гидроксид калия), гидриды металлов (например, гидрид калия и гидрид натрия), алкоксиды щелочных металлов (например, метоксид натрия, этоксид натрия, трет-бутоксид натрия и трет-бутоксид калия), дисилазиды щелочных металлов (например, дисилазид лития, дисилазид натрия и дисилазид калия) и тому подобное. Среди них предпочтительными являются соли щелочных металлов, такие как гидрокарбонат натрия, карбонат калия, карбонат цезия, фосфат натрия и фосфат калия; алкоксиды щелочных металлов, такие как трет-бутоксид натрия и трет-бутоксид калия; и органические амины, такие как триэтиламин и диизопропилэтиламин. Количество используемого основания составляет, как правило, от 0,1 до 10 молей, предпочтительно от 1 до 5 молей, из расчета на один моль соединения формулы (V).
Может быть использован любой растворитель, не оказывающий вредного влияния на реакцию. Примеры используемых растворителей включают углеводороды (например, бензол, толуол и ксилол), галогенированные углеводороды (например, хлороформ и 1,2-дихлорэтан), нитрилы (например, ацетонитрил), простые эфиры (например, 1,2-диметоксиэтан, тетрагидрофуран, и 1,4-диоксан), спирты (например, метанол и этанол), апротонные полярные растворители (например, N,N-диметилформамид, диметилсульфоксид и гексаметилфосфориламид), вода и смеси этих растворителей. Время реакции составляет от 0,1 до 100 часов, предпочтительно от 0,5 до 24 часов. Реакция протекает при температуре от 0°C до температуры кипения растворителя, предпочтительно от 20 до 150°С.
Полученное таким образом соединение формулы (VI) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
(Стадия d)
На этой стадии соединение (VI) подвергают бромированию с помощью N-бромсукцинимида, что приводит к получению соединения (VII).
Галогенирование может быть осуществлено способом, описанным в WO 2006/102079, или с помощью аналогичного метода.
Количество используемого на этой стадии N-бромсукцинимида составляет от 0,5 до 2,0 молей, предпочтительно от 0,9 до 1,2 молей, из расчета на один моль соединения формулы (VI).
В этой реакции может быть использован любой растворитель, который не оказывает вредного влияния на реакцию. Например, предпочтительно могут быть использованы тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид, N-метилпирролидин-2-он или смеси этих растворителей.
Реакция протекает обычно при температуре от -20 до +50°С, предпочтительно от 0°C до комнатной температуры. Время реакции обычно составляет от 1 минуты до 2 дней, и предпочтительно от 5 минут до 12 часов.
Полученное таким образом соединение формулы (VII) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
(Стадия е)
На этой стадии происходит взаимодействие органического борана и соединения формулы (VII) с получением в реакционной системе промежуточного алкилборанового соединения, и затем промежуточное соединение превращают в соединение формулы (VIII) в присутствии основания и катализатора на основе переходного металла.
Эта стадия может быть выполнена любым общеизвестным способом (см., например, WO 2006/102079).
Примеры органических боранов включают 9-BBN (9-борабицикло[3.3.1]нонан), димер 9-BBN (димер 9-борабицикло[3.3.1]нонана), дисиамилборан (бис(1,2-диметилпропил)боран), гексилборан ((1,1,2-триметилпропил)боран) и тому подобное. Органический боран предпочтительно представляет собой 9-BBN (9-борабицикло[3.3.1]нонан) или димер 9-BBN (димер 9-борабицикло [3.3.1]нонана), особенно предпочтителен 9-BBN (9-борабицикло[3.3.1]нонан). Количество используемого органического борана особенно не ограничено, если оно обеспечивает получение алкилборанового промежуточного соединения. Органический боран можно использовать в количестве от 1 до 20 молей на моль соединения формулы (VII). С точки зрения облегчения протекания реакции, количество органического борана предпочтительно составляет от 6 до 10 молей.
В качестве катализатора на основе переходного металла могут быть использованы, например, катализатор на основе двухвалентного палладия (например, ацетат палладия, хлорид палладия, и дихлорид 1,1′-бис(дифенилфосфино)ферроцен-палладия (II)). Если необходимо, может быть использован лиганд (например, трифенилфосфин и три-трет-бутилфосфин). Количество используемого катализатора на основе переходного металла может варьироваться в зависимости от типа катализатора. Катализатор на основе переходного металла обычно используют в количестве от 0,0001 до 1 моль, предпочтительно от 0,01 до 0,5 моль, из расчета на один моль соединения формулы (VII). Лиганд обычно используют в количестве от 0,0001 до 4 молей, предпочтительно от 0,01 до 2 молей, из расчета на один моль соединения формулы (VII).
Альтернативно, может быть использован, например, катализатор на основе палладия с нулевой валентностью. Примеры катализаторов на основе палладия с нулевой валентностью включают тетракистрифенилфосфинпалладий (0), трис(дибензилиденацетон)палладий (0), палладий (0) на угле и тому подобное. Тетракистрифенилфосфинпалладий (0) или трис(дибензилиденацетон)палладий (0) являются более предпочтительными, и тетракистрифенилфосфинпалладий (0) является особенно предпочтительным. Количество используемого катализатора на основе палладия с нулевой валентностью, особенно не ограничено, если оно обеспечивает внутримолекулярную циклизацию, и это количество может варьировать в зависимости от типа катализатора. Катализатор на основе палладия с нулевой валентностью может быть использован в количестве от 0,0001 до 1 моля, предпочтительно от 0,01 до 0,5 моля, из расчета на один моль соединения формулы (VII).
Если необходимо, то к катализатору на основе палладия с нулевой валентностью может быть добавлен лиганд. Примеры таких лигандов включают трифенилфосфин, 1,1′-бис(дифенилфосфино)ферроцен, три-трет-бутилфосфин, трициклогексилфосфин, 2-дициклогексилфосфино-2′,6′-диметоксибифенил, 2-дициклогексилфосфино-2′,4′,6′-триизопропилбифенил, 2-(дитретбутилфосфино)бифенил, 2-дициклогексилфосфино-2′-(N,N-диметиламино)бифенил, 4,5′-бис(дифенилфосфино)-9,9′-диметилксантин и т.п. Когда в качестве палладиевого катализатора с нулевой валентностью используется трис(дибензилиденацетон)палладий (0), в качестве лиганда может быть добавлен трифенилфосфин. Количество используемого лиганда не имеет особых ограничений, поскольку протекает внутримолекулярная реакция циклизации. Лиганд может быть использован в количестве от 0,0001 до 4 молей, предпочтительно от 0,01 до 2 молей, из расчета на один моль соединения формулы (VII).
Примеры оснований включают неорганические основания, такие как гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия, и гидроксидов щелочных металлов. Гидроксиды щелочных металлов являются предпочтительными. Примеры гидроксидов щелочных металлов включают гидроксид лития, гидроксид натрия, гидроксид калия и гидроксид цезия. Гидроксид лития, гидроксид натрия, гидроксид калия или гидроксид цезия, предпочтительно используют. Гидроксид лития или натрия является особенно предпочтительным. Количество используемого основания по мере протекания реакции конкретно не ограничивается. Основание можно использовать в количестве от 1 до 100 молей, предпочтительно от 2 до 20 молей, из расчета на один моль соединения формулы (VII). Гидроксид щелочного металла может быть использован в виде водного раствора гидроксида щелочного металла.
При использовании комбинации органического борана, гидроксида щелочного металла и катализатора на основе палладия с нулевой валентностью, предпочтительно использование комбинации предпочтительного органического борана, предпочтительного гидроксида щелочного металла и предпочтительного катализатора на основе палладия с нулевой валентностью. Особенно предпочтительно использование особенно предпочтительного органического борана, особенно предпочтительного гидроксида щелочного металла и особенно предпочтительного катализатора на основе палладия с нулевой валентностью.
Может быть использован любой растворитель, не оказывающий вредного влияния на реакцию. Примеры таких растворителей включают углеводороды (например, бензол, толуол и ксилол), простые эфиры (например, 1,2-диметоксиэтан, тетрагидрофуран и 1,4-диоксан), апротонные полярные растворители (например, N,N-диметилформамид, диметилсульфоксид и гексаметилфосфориламид), воду и их смеси. Предпочтительно используют 1,2-диметоксиэтан или тетрагидрофуран. Тетрагидрофуран является особенно предпочтительным с точки зрения стабильности органического борана и получения промежуточного алкилборанового соединения. Количество используемого в реакции растворителя конкретно не ограничено. Растворитель может быть использован в количестве, которое от 1 до 300 раз больше, а предпочтительно от 10 до 96 раз больше массы соединения формулы (VII).
Время реакции получения соединения формулы (VIII) особо не ограничено. Время реакции может составлять от 0,1 до 100 часов, предпочтительно от 0,5 до 24 часов.
Температура реакции получения соединения формулы (VIII) не имеет особых ограничений. Температура реакции может составлять от -20°C до температуры кипения растворителя, предпочтительно от 0 до 150°С. В ходе внутримолекулярной реакции циклизации промежуточного алкилборана с использованием катализатора на основе палладия с нулевой валентностью и щелочного водного раствора гидроксида металла, низкая температура реакции имеет тенденцию вызывать побочные реакции, что приводит к низкому выходу. Таким образом, предпочтительная температура реакции составляет 61°С или выше.
Полученное таким образом соединение формулы (VIII) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
Образование алкилборанового промежуточного соединения в реакционной системе на этой стадии может быть подтверждено. Например, в качестве метода подтверждения может быть использован спектр, полученный с помощью LCMS.
(Стадия f)
На этой стадии снимают защиту с защищенной гидроксильной группы в соединении формулы (VIII) для получения соединения формулы (IX).
Удаление защитной группы может быть осуществлено известным способом, например, методом, описанным в Protective Groups in Organic Synthesis, T.W. Greene, John Wiley & Sons (1981); или аналогичным.
Когда в качестве защитной группы используется трет-бутилдиметилсилил, для снятия защиты используется реагент тетрабутиламмонийфторид. Количество используемого реагента предпочтительно составляет от 1 до 10 молей на моль соединения (VIII).
В реакции может быть использован любой растворитель, не оказывающий вредного влияния на реакцию. Примеры используемых растворителей включают простые эфиры (например, 1,2-диметоксиэтан и тетрагидрофуран), апротонные полярные растворители (например, N,N-диметилформамид, диметилсульфоксид и гексаметилфосфориламид) и смеси этих растворителей. Время реакции составляет от 0,1 до 100 часов, предпочтительно от 0,5 до 24 часов. Реакция протекает при температуре от 0 до 80°С, предпочтительно от 0 до 50°С.
Полученное таким образом соединение формулы (IX) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
(Стадия g)
На этой стадии проводят реакцию взаимодействия метансульфонилхлорида с соединением формулы (IX) с получением соединения формулы (X).
Количество используемого метансульфонилхлорида может составлять от 1 до 5 молей, более предпочтительно от 1 до 2 молей, из расчета на один моль соединения формулы (IX).
Примеры используемых оснований включают органические амины, такие как триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, лутидин и коллидин. Такое основание может быть использовано в количестве от 1 до 10 молей, предпочтительно от 1 до 5 молей, из расчета на один моль соединения формулы (IX).
В реакции может быть использован любой растворитель, не оказывающий вредного влияния на реакцию. Примеры используемых растворителей включают углеводороды (например, бензол, толуол и ксилол), галогенированные углеводороды (например, хлороформ и 1,2-дихлорэтан), нитрилы (например, ацетонитрил), простые эфиры (например, 1,2-диметоксиэтан, тетрагидрофуран и 1,4-диоксан), спирты (например, метанол и этанол), апротонные полярные растворители (например, N,N-диметилформамид, диметилсульфоксид и гексаметилфосфорамид) и смеси этих растворителей. Время реакции составляет от 0,1 до 24 часов, предпочтительно от 0,1 до 12 часов. Реакция протекает при температуре от -20°C до температуры кипения растворителя, и предпочтительно от 0°С до комнатной температуры.
Полученное таким образом соединение формулы (X) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
(Стадия h)
На этой стадии азид натрия взаимодействует с соединением формулы (X) с получением соединения формулы (XI).
Количество азида натрия в реакции может составлять от 1 до 10 молей, предпочтительно от 1 до 5 молей, из расчета на один моль соединения формулы (X).
В реакции может быть использован любой растворитель, не оказывающий вредного влияния на реакцию. Примеры используемых растворителей включают N,N-диметилформамид, диметилсульфоксид, гексаметилфосфориламид и смеси этих растворителей. Время протекания реакции составляет от 0,1 до 24 часов, предпочтительно от 0,5 до 12 часов. Реакция протекает обычно при температуре от комнатной до температуры кипения растворителя, предпочтительно от 50 до 100°С.
Полученное таким образом соединение формулы (XI) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
(Стадия i)
На этой стадии соединение формулы (XI) подвергают реакции в присутствии трифенилфосфина в водном растворителе с получением соединения формулы (XII).
Трифенилфосфин может быть традиционно используемым реагентом или реагентом на твердой подложке. Количество используемого в реакции трифенилфосфина может составлять от 1 до 10 молей, предпочтительно от 1 до 5 молей, из расчета на один моль соединения формулы (XI).
В реакции может быть использован любой растворитель, не оказывающий вредного влияния на реакцию. Примеры используемых растворителей включают тетрагидрофуран/вода, 1,4-диоксан/вода и тому подобное. Время реакции составляет от 0,1 до 24 часов, предпочтительно от 0,5 до 12 часов. Реакция протекает при температуре от комнатной до температуры кипения растворителя, предпочтительно от 50°C до температуры кипения растворителя.
Полученное таким образом соединение формулы (XII) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
(Стадия j)
На этой стадии соединение формулы (XII) амидируют с использованием α,β-ненасыщенной карбоновой кислоты или хлорангидрида или бромангидрида α,β-ненасыщенных кислот, с получением соединения формулы (Ia) в соответствии с настоящим изобретением.
При использовании карбоновой кислоты в качестве амидирующего реагента, карбоновую кислоту можно использовать в количестве от 0,5 до 10 молей, предпочтительно от 1 до 3 молей, из расчета на один моль соединения формулы (XII), в присутствии подходящего конденсирующего агента. Карбоновая кислота может быть коммерчески доступным продуктом, или может быть получена известным способом.
В реакции может быть использован любой растворитель, не оказывающий вредного влияния на реакцию. Примеры используемых растворителей включают толуол, бензол, метиленхлорид, хлороформ, тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид, диметилацетамид, N-метилпирролидин-2-он, диметилсульфоксид и смеси этих растворителей. Реакция протекает обычно при температуре от -78 до 200°C, предпочтительно от 0 до 50°С. Время реакции составляет обычно от 5 минут до 3 дней, предпочтительно от 5 минут до 10 часов.
Примеры конденсирующих агентов включают дифенилфосфорилазид, N,N′-дициклогексилкарбодиимид, соли бензотриазол-1-илокси-трисдиметиламинофосфония, хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния, 1-этил-3-(3-диметиламинопропил)карбодиимид, комбинацию 1-этил-3-(3-диметиламинопропил)карбодиимида и 1-гидроксибензотриазола, 2-хлор-1,3-диметилимидазолиний, гексафторфосфат O-(7-азабензотриазоло-1-ил)-N,N,N′,N′-тетраметилгексаурония и тому подобное.
При необходимости, для протекания реакции может быть необязательно добавлено основание. Примеры используемых оснований включают органические основания, такие как триэтиламин, диизопропилэтиламин, пиридин, лутидин, коллидин, 4-(N,N-диметиламино)пиридин, трет-бутират калия, трет-бутират натрия, метоксид натрия, этоксид натрия, гексаметилдисилазид лития, гексаметилдисилазид натрия, гексаметилдисилазид калия и бутиллитий; и неорганические основания, такие как гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия и гидрид натрия. Такое основание может быть добавлено в количестве от 1 до 100 молей, предпочтительно от 1 до 10 молей, из расчета на один моль соединения формулы (XII).
Когда в качестве амидирующего реагента используют хлорангидрид или бромангибрид кислоты, галогенангидрид используется в количестве от 0,5 до 5 молей, предпочтительно от 0,9 до 1,1 моля, из расчета на один моль соединения формулы (XII). Галогенангидрид может быть коммерчески доступным продуктом, или может быть получен известным способом.
В реакции может быть использован любой растворитель, не оказывающий вредного влияния на реакцию. Примеры используемых растворителей включают толуол, бензол, метиленхлорид, хлороформ, тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид, диметилацетамид, N-метилпирролидин-2-он, ацетонитрил, воду и смеси этих растворителей. Реакция протекает обычно при температуре от -78 до 200°С, предпочтительно от 0 до 50°С. Время реакции обычно составляет от 5 минут до 3 дней, предпочтительно от 5 минут до 10 часов.
К реакции, если необходимо, может быть добавлено основание. Примеры используемых оснований включают органические основания, такие как триэтиламин, диизопропилэтиламин, пиридин, лутидин, коллидин, 4-(N,N-диметиламино)пиридин, трет-бутират калия, трет-бутират натрия, метоксид натрия, этоксид натрия, гексаметилдисилазид лития, гексаметилдисилазид натрия, гексаметилдисилазид калия и бутиллитий; и неорганические основания, такие как гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия и гидрид натрия. Такое основание может быть добавлено в количестве от 1 до 100 молей, предпочтительно от 1 до 20 молей, из расчета на один моль соединения формулы (XII).
Полученные таким образом соединение формулы (Ia) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
Способ получения 2
 ,
,
где R1, R2, R3, R4, m и n имеют определения и значения, указанные выше.
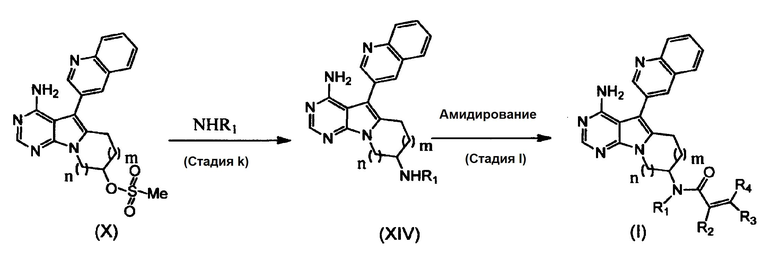
(Стадия k)
На этой стадии алкиламин взаимодействует с соединением формулы (X), с получением соединения формулы (XIV).
Количество используемого алкиламина составляет от 2 молей до избыточного молярного количества из расчета на один моль соединения формулы (X).
В реакции может быть использован любой растворитель, не оказывающий вредного влияния на реакцию. Примеры используемых растворителей включают углеводороды (например, бензол, толуол и ксилол), галогенированные углеводороды (например, хлороформ и 1,2-дихлорэтан), нитрилы (например, ацетонитрил), простые эфиры (например, 1,2-диметоксиэтан, тетрагидрофуран и 1,4-диоксан), апротонные полярные растворители (например, N,N-диметилформамид, диметилсульфоксид и гексаметилфосфориламид), и смеси этих растворителей. Время реакции составляет от 0,1 до 100 часов, предпочтительно от 1 до 24 часов. Реакция протекает при температуре от комнатной до температуры кипения растворителя, и предпочтительно реакция протекает при температуре от 50°C до температуры кипения растворителя.
Полученное таким образом соединение формулы (XIV) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
(Стадия l)
Эта стадия может быть осуществлена аналогично, как и стадия j.
Способ получения 3
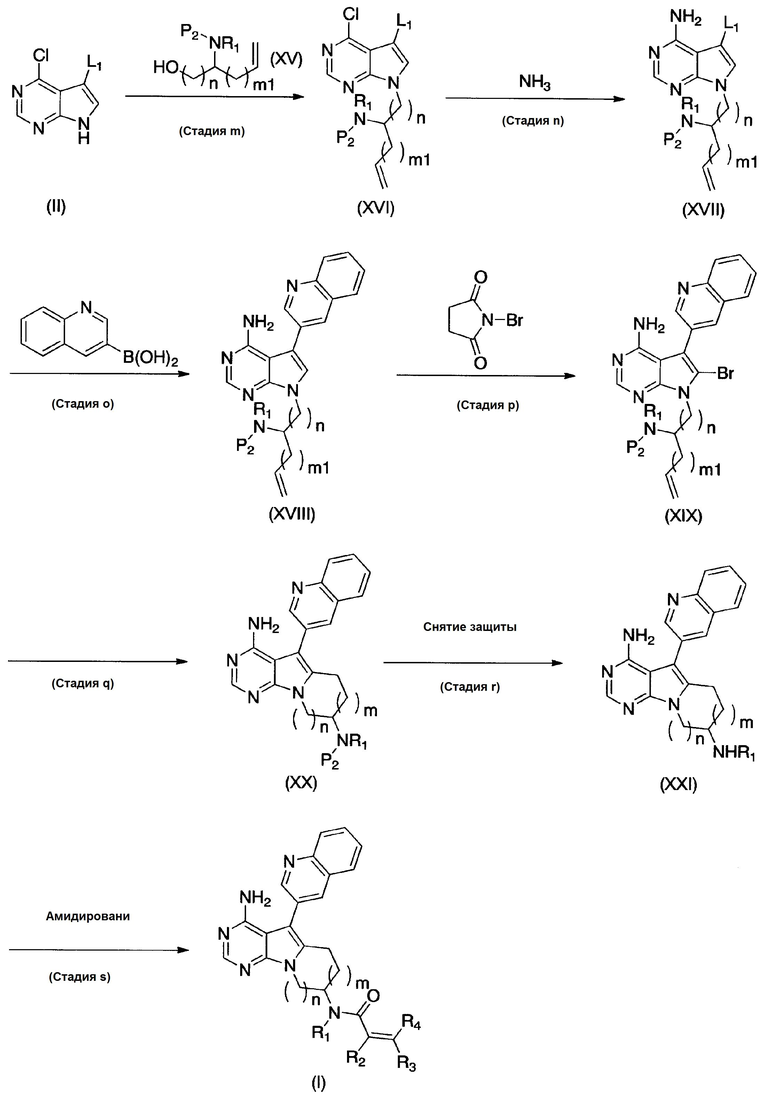
где Р2 обозначает защитную группу для аминогруппы, L1 обозначает уходящую группу, m1 равно 0 или 1; и R1, R2, R3, R4, m и n имеют определения и значения, указанные выше.
(Стадия m)
На этой стадии соединения формул (II) и (XV) вступают в реакцию Мицунобу с получением соединения формулы (XVI).
В соединении формулы (II) уходящая группа L1 может представлять собой, например, бром или иод. Соединение формулы (II) может быть коммерчески доступным продуктом, или оно может быть получено известным способом. В соединении формулы (XV) защитная группа для аминогруппы, представленная как Р2, может представлять собой, например, трет-бутоксикарбонил или бензоил. Соединение формулы (XV) может быть коммерчески доступным продуктом, или оно может быть получено любым известным способом. Соединение формулы (XV) может быть использовано в количестве от 1 до 10 молей, предпочтительно от 1 до 5 молей, из расчета на один моль соединения формулы (II).
Реакция Мицунобу может быть выполнена любым общеизвестным способом (см., например, Synthesis, p. 1, 1981) или аналогичным способом.
Примеры сложных эфиров азодикарбоновой кислоты включают диэтилазодикарбоксилат и диизопропилазодикарбоксилат. Такой сложный эфир азодикарбоновой кислоты может быть использован в количестве от 1 до 10 молей, предпочтительно от 1 до 5 молей, из расчета на один моль соединения формулы (II).
В качестве фосфинового соединения могут быть использованы трифенилфосфин, трибутилфосфин и т.п. Фосфиновое соединение может быть использовано в количестве от 1 до 10 молей, предпочтительно от 1 до 5 молей, из расчета на один моль соединения формулы (II).
В качестве растворителя могут быть использованы по отдельности или в виде смеси тетрагидрофуран, 1,2-диметоксиэтан, 1,4-диоксан, толуол, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, N-метилпирролидин-2-он и тому подобное. Время реакции составляет от 0,1 до 100 часов, предпочтительно от 0,1 до 24 часов. Реакция протекает при температуре от 0°C до температуры кипения растворителя, предпочтительно от 0 до 100°С.
Полученное таким образом соединение формулы (XVI) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
(Стадия n)
На этой стадии соединение формулы (XVI) подвергают взаимодействию с аммиаком или его солью, с получением соединения формулы (XVII).
Эта стадия может быть осуществлена аналогично, как и стадия b.
Полученное таким образом соединение формулы (XVII) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
(Стадия о)
На этой стадии соединение формулы (XVII) подвергают реакции сочетания с 3-хинолинбороновой кислотой или со сложным эфиром 3-хинолинбороновой кислоты, с получением соединения формулы (XVIII).
Эта стадия может быть осуществлена аналогично, как и стадия с.
Полученное таким образом соединение формулы (XVIII) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
(Стадия р)
На этой стадии соединение (XVIII) бромируют с использованием N-бромсукцинимида, с получением соединения формулы (XIX).
Эта стадия может быть осуществлена аналогично, как и стадия d.
Полученное таким образом соединение формулы (XIX) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
(Стадия q)
На этой стадии, после того, как органический боран провзаимодействует с соединением формулы (XIX), и в реакционной системе образуется алкилборановое промежуточное соединение, получают в присутствии катализатора на основе переходного металла и основания соединение формулы (XX).
Эта стадия может быть осуществлена общеизвестными методами (см., например, WO 2006/102079).
Примеры органических боранов включают 9-BBN (9-борабицикло[3.3.1]нонан), димер 9-BBN (димер 9-борабицикло[3.3.1]нонана), дисиамилборан (бис(1,2-диметилпропил)боран), гексилборан ((1,1,2-триметилпропил)боран) и тому подобное. Предпочтительно используют 9-BBN (9-борабицикло[3.3.1]нонан) или димер 9-BBN (димер 9-борабицикло[3.3.1]нонана). Особенно предпочтительным является 9-BBN (9-борабицикло[3.3.1]нонан). Количество используемого органического борана особенно не ограничено, если оно обеспечивает образование алкилборанового промежуточного соединения. Органический боран можно использовать в количестве от 1 до 20 молей на один моль соединения формулы (XIX). В целях облегчения протекания реакции, количество органического борана предпочтительно составляет от 6 до 10 молей на один моль соединения формулы (XIX).
Примеры катализаторов на основе переходного металла включают двухвалентные палладиевые катализаторы (например, ацетат палладия, хлорид палладия и дихлорид 1,1′-бис(дифенилфосфино)ферроцен-палладия (II)). Если необходимо, может быть добавлен лиганд (например, трифенилфосфин и три-трет-бутилфосфин). Количество используемого катализатора на основе переходного металла может варьироваться в зависимости от типа катализатора. Катализатор на основе переходного металла обычно используют в количестве от 0,0001 до 1 моля, предпочтительно от 0,01 до 0,5 молей, из расчета на один моль соединения формулы (XIX). Количество используемого лиганда составляет, как правило, от 0,0001 до 4 молей, предпочтительно от 0,01 до 2 молей, из расчета на один моль соединения формулы (XIX).
В качестве катализатора на основе переходного металла также может быть использован, например, катализатор на основе палладия с нулевой валентностью. Примеры используемых катализаторов на основе палладия с нулевой валентностью включают тетракистрифенилфосфинпалладий (0), трис(дибензилиденацетон)палладий (0) и палладий (0) на углероде. Предпочтительно используют тетракистрифенилфосфинпалладий (0) или трис(дибензилиденацетон) палладий (0). Особенно предпочтительным является тетракистрифенилфосфинпалладий (0). Количество используемого катализатора на основе палладия с нулевой валентностью особенно не ограничено, если оно обеспечивает внутримолекулярную циклизацию. Количество используемого катализатора на основе палладия с нулевой валентностью может варьировать в зависимости от типа катализатора. Катализатор на основе палладия с нулевой валентностью может быть использован в количестве от 0,0001 до 1 моля, предпочтительно от 0,01 до 0,5 моля, из расчета на один моль соединения формулы (XIX).
При необходимости, к катализатору на основе палладия с нулевой валентностью может быть дополнительно добавлен лиганд. Примеры таких лигандов включают трифенилфосфин, 1,1′-бис(дифенилфосфино)ферроцен, три-трет-бутилфосфин, трициклогексилфосфин, 2-дициклогексилфосфино-2′,6′-диметоксибифенил, 2-дициклогексилфосфино-2′,4′,6′-триизопропилбифенил, 2-(ди-трет-бутилфосфино)бифенил, 2-дициклогексилфосфино-2′-(N,N-диметиламино)бифенил, 4,5′-бис(дифенилфосфино)-9,9′-диметилксантин и т.п. Когда в качестве палладиевого катализатора с нулевой валентностью используется трис(дибензилиденацетон)палладий (0), в качестве лиганда может быть добавлен трифенилфосфин. Количество используемого лиганда не имеет особых ограничений, поскольку выполняется внутримолекулярная реакция циклизации. Лиганд может быть использован в количестве от 0,0001 до 4 молей, предпочтительно от 0,01 до 2 моля, из расчета на один моль соединения формулы (XIX).
Примеры оснований включают неорганические основания, такие как гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия и гидроксиды щелочных металлов. Гидроксиды щелочных металлов являются предпочтительными. Примеры гидроксидов щелочных металлов включают гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид цезия и тому подобное. Предпочтительно используют гидроксид лития, гидроксид натрия, гидроксид калия или гидроксид цезия. Особенно предпочтительным является гидроксид лития или натрия. Количество используемого основания при протекании реакции конкретно не ограничивается. Основание можно использовать в количестве от 1 до 100 молей, предпочтительно от 2 до 20 молей, из расчета на один моль соединения формулы (XIX). Гидроксид щелочного металла может быть использован в виде водного раствора гидроксида щелочного металла.
Предпочтительной комбинацией органического борана, гидроксида щелочного металла и катализатора на основе палладия с нулевой валентностью является комбинация из предпочтительного органического борана, предпочтительного гидроксида щелочного металла и предпочтительного катализатора на основе палладия с нулевой валентностью. Особенно предпочтительной комбинацией является комбинация из особенно предпочтительного органического борана, особенно предпочтительного гидроксида щелочного металла и особенно предпочтительного катализатора на основе палладия с нулевой валентностью.
Может быть использован любой растворитель, не оказывающий вредного влияния на реакцию. Примеры таких растворителей включают углеводороды (например, бензол, толуол и ксилол), простые эфиры (например, 1,2-диметоксиэтан, тетрагидрофуран и 1,4-диоксан), апротонные полярные растворители (например, N,N-диметилформамид, диметилсульфоксид, и гексаметил фосфорил амид), воду и их смеси. Предпочтительно использование 1,2-диметоксиэтана или тетрагидрофурана. Тетрагидрофуран является особенно предпочтительным с точки зрения стабильности органического борана и получаемого алкилборанового промежуточного соединения. Количество используемого растворителя в реакции конкретно не ограничено. Растворитель может быть использован в количестве от 1 до 300 раз больше, а предпочтительно от 10 до 96 раз больше массы соединения формулы (XIX).
Время реакции получения соединения формулы (XX) особо не ограничено. Время реакции может составлять от 0,1 до 100 часов, предпочтительно от 0,5 до 24 часов.
Температура реакции получения соединения формулы (XX) не имеет особых ограничений. Температура реакции может быть от -20°C до температуры кипения растворителя, предпочтительно от 0 до 150°С. Низкая температура внутримолекулярной реакции циклизации промежуточного алкилборана, в которой используются катализатор на основе палладия с нулевой валентностью и щелочной водный раствор гидроксида металла, может вызывать побочные реакции, что приводит к низкому выходу целевого продукта. Таким образом, предпочтительная температура составляет 61°С или выше.
Полученное таким образом соединение формулы (XX) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
Образование алкилборанового промежуточного соединения в реакционной системе на этой стадии может быть подтверждено. Например, в качестве метода подтверждения может быть использован спектр, полученный с помощью LCMS.
(Стадия r)
На этой стадии снимают защиту с защищенной аминогруппы соединения формулы (XX), с получением соединения формулы (XXI).
Удаление защитной группы может быть осуществлено любым известным способом, например способом, описанным в Protective Groups in Organic Synthesis, T.W. Greene, John Wiley & Sons (1981); или аналогичным способом.
Когда в качестве защитной группы используется трет-бутоксикарбонил, то в качестве реагента для снятия защиты может быть использована хлористоводородная кислота, серная кислота, метансульфоновая кислота, трифторуксусная кислота и т.п. Реагент для снятия защиты предпочтительно используют в количестве от 1 до 100 молей из расчета на один моль соединения (XX).
Может быть использован любой растворитель, не оказывающий вредного влияния на реакцию. Примеры используемых растворителей включают воду, метанол, этанол, метиленхлорид, хлороформ и смеси этих растворителей. Время реакции составляет от 0,1 до 100 часов, предпочтительно от 0,5 до 24 часов. Реакция протекает при температуре от 0°C до температуры кипения растворителя.
Полученное таким образом соединение формулы (XXI) может быть использовано на следующей стадии реакции после или без выделения или очистки, которые выполняются с использованием известных средств и способов для разделения и очистки, такими как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, повторное осаждение и хроматография.
(Стадия s)
Эта стадия может быть осуществлена аналогичным образом, как и стадия j.
В приведенных выше способах получения 1-3, в отношении функциональных групп, содержащих активный протон, таких как амино, имино, гидрокси, карбокси, карбонильных и амидных группы, и индола могут быть использованы защитные реагенты, или в такие функциональные группы вводят обычным способом защитную группу, а затем эта защитная группа может быть удалена на соответствующей стадии в каждом способе получения.
″Защитная группа для аминогруппы или защитная группа для иминогруппы″ конкретно не ограничена, если она имеет защитную функцию. Примеры таких защитных групп включают аралкильные группы, такие как бензил, п-метоксибензил, 3,4-диметоксибензил, о-нитробензил, п-нитробензил, бензгидрил, тритил и кумил; низшие алканоильные группы, такие как формил, ацетил, пропионил, бутирил, пивалоил, трифторацетил и трихлорацетил; бензоил; арилалканоильные группы, такие как фенилацетил и феноксиацетил; низшие алкоксикарбонильные группы, такие как метоксикарбонил, этоксикарбонил, пропилоксикарбонил и трет-бутоксикарбонил; аралкилоксикарбонильные группы, такие как п-нитробензилоксикарбонил и фенэтилоксикарбонил; низшие алкилсилильные группы, такие как триметилсилил и трет-бутилдиметилсилил; тетрагидропиранил; триметилсилилэтоксиметил; низшие алкилсульфонильные группы, такие как метилсульфонил, этилсульфонил и трет-бутилсульфонил; низшие алкилсульфинильные группы, например, трет-бутилсульфинил; арилсульфонильные группы, такие как бензолсульфонил и толуолсульфонил; и имидо группы, например, фталимидо. В частности, предпочтительными являются трифторацетил, ацетил, трет-бутоксикарбонил, бензилоксикарбонил, триметилсилилэтоксиметил, кумил и т.п.
″Защитная группа для гидроксильной группы″ особенно не ограничена, если она имеет защитную функцию. Примеры таких защитных групп включают низшие алкильные группы, такие как метил, этил, пропил, изопропил и трет-бутил; низшие алкилсилильные группы, такие как триметилсилил и трет-бутилдиметилсилил; низшие алкоксиметильные группы, такие как метоксиметил и 2-метоксиэтоксиметил; тетрагидропиранил; триметилсилилэтоксиметил; аралкильные группы, такие как бензил, п-метоксибензил, 2,3-диметоксибензил, о-нитробензил, п-нитробензил и тритил; и ацильные группы, такие как формил, ацетил и трифторацетил. В частности, предпочтительными являются метил, метоксиметил, тетрагидропиранил, триметилсилилэтоксиметил, трет-бутилдиметилсилил и ацетил.
″Защитная группа для карбоксильной группы″ особенно не ограничена, если она имеет защитную функцию. Примеры таких защитных групп включают низшие алкильные группы, такие как метил, этил, пропил, изопропил и трет-бутил; группы гало-низшего алкила, такие как 2,2,2-трихлорэтил; низшие алкенильные группы, такие как аллил; триметилсилилэтоксиметил; и аралкильные группы, такие как бензил, п-метоксибензил, п-нитробензил, бензгидрил и тритил. В частности, предпочтительными являются метил, этил, трет-бутил, аллил, бензил, п-метоксибензил, триметилсилилэтоксиметил и тому подобное.
″Защитная группа для карбонильной группы″ особенно не ограничена, если она обладает защитной функцией. Примеры таких защитных групп включают этиленкеталь, триметиленкеталь, диметилкеталь и подобные кетали и ацетали.
Способ удаления такой защитной группы может варьироваться в зависимости от типа защитной группы, стабильности целевого соединения (I) и т.п. Могут быть использованы, например, следующие методы: сольволиз с использованием кислоты или основания, как раскрыто в публикации Protective Groups in Organic Synthesis, third edition, T.W. Green, John Wiley & Sons (1999), или аналогичным с ним способом, где способ включает взаимодействие соединения с 0,01 моля или большим избытком кислоты, предпочтительно трифторуксусной кислоты, муравьиной кислоты или хлористоводородной кислоты, или с эквимолярным или большим избытком основания, предпочтительно гидроксида калия или гидроксида кальция; химическое восстановление с использованием металлгидридного комплекса и т.п.; или каталитическое восстановление с использованием в качестве катализатора палладия на угле, никеля Ренея и т.п.
Соединение по изобретению может быть выделено и очищено обычными средствами и методами выделения и очистки. Примеры таких методов включают экстракцию растворителем, перекристаллизацию, препаративную обращенно-фазовую высокоэффективную жидкостную хроматографию, колоночную хроматографию, препаративную тонкослойную хроматографию и тому подобное.
Когда соединение по настоящему изобретению имеет изомеры, такие как оптические изомеры, стереоизомеры, региоизомеры и поворотные изомеры, любой из изомеров и их смеси входит в объем заявленных соединений по настоящему изобретению. Например, когда соединение имеет оптические изомеры, оптические изомеры, выделенные из рацемической смеси, также входят объем заявленных соединений по настоящему изобретению. Каждый из таких изомеров может быть получен в виде отдельного соединения известными методами синтеза и разделения (например, концентрирование, экстракция растворителем, колоночная хроматография, перекристаллизация и т.п.).
В настоящем изобретении, атом углерода, связанный с заместителем, представленным в формуле (1) как -NR1-(C=O)-CR2=C(R3)R4, представляет собой асимметричный атом углерод; следовательно, соединение по изобретению включает изомеры. Как отмечено выше, если не указано иное, то соединение по настоящему изобретению включает все энантиомеры и их смеси. Соединение по настоящему изобретению может представлять собой смесь R и S энантиомеров. Такая смесь может быть смесью, содержащей 90% или более, 95% или более, 99% или более R-энантиомера; или смесью, содержащей 90% или более, 95% или более, 99% или более S-энантиомера.
Методы хирального разделения включают, например: диастереомерный метод разделения, в котором хиральный разделяющий агент воздействует на соединение настоящего изобретения с образованием соли, и отделение одного из энантиомеров с использованием различия в растворимости и т.п. у полученной соли; метод селективной кристаллизации, в котором в перенасыщенный раствор рацемата вносят один из энантиомеров в качестве затравки для кристаллизации; и колоночная хроматография, такая как ВЭЖХ с использованием хиральной колонки. Хиральный разделяющий агент, который можно использовать в диастереомерном методе разделения, может быть соответственно выбран из, например, кислотных разделяющих агентов, таких как винная кислота, яблочная кислота, молочная кислота, миндальная кислота, 10-камфорсульфоновая кислота и их производных; основных разделяющих агентов, таких как бруцин, стрихнин, хинин и соединений, подобных алкалоидам, аминокислотных производных, цинхонидин; α-метилбензиламина. Кроме того, один из энантиомеров соединения по настоящему изобретению может быть получен не только путем получения соединения по настоящему изобретению в виде смеси энантиомеров, с последующим выполнением указанных выше методов хирального разделения, но также он может быть получен путем использования одного энантиомера соединения по настоящему изобретению, полученного хиральным разделением указанными выше методами и подобными способами, в качестве исходного материала для химического синтеза. Кроме того, способы получения одного из энантиомеров соединения по настоящему изобретению или его исходного соединения включают способ предпочтительного получения одного из энантиомеров путем регулирования условий реакции проведения катализа или т.п. на стадии образования асимметричного атома углерода.
Соединение или его соль по настоящему изобретению могут находиться в кристаллической форме. Монокристаллы и полиморфные смеси включены в объем заявленных соединений или их солей настоящего изобретения. Такие кристаллы могут быть получены путем кристаллизации известными в данной области техники способами кристаллизации. Соединение или его соль по настоящему изобретению могут быть в форме сольвата (например, гидрата) или в не сольватированной форме. Любая из таких форм включена в объем заявленных соединений или их солей настоящего изобретения. Соединения, меченные изотопом (например, 3H, 14C, 35S и 125I), также включены в объем притязаний соединений или их солей настоящего изобретения.
Соль соединения по настоящему изобретению или его промежуточного соединения представляет собой обычную соль, используемую в области органической химии. Примеры таких солей включают основно-аддитивные соли, образованные с карбоксигруппой, когда соединение имеет карбоксильную группу, и кислотно-аддитивные соли с аминогруппой или с основной гетероциклической группой, когда соединение имеет аминогруппу или основную гетероциклическую группу.
Примеры основно-аддитивных солей включают соли щелочных металлов, такие как соли натрия и соли калия; соли щелочноземельных металлов, такие как соли кальция и соли магния; аммонийные соли; и соли органических аминов, такие как соли триметиламина, соли триэтиламина, соли дициклогексиламина, соли этаноламина, соли диэтаноламина, соли триэтаноламина, соли прокаина и соли N,N′-дибензилэтилендиамина.
Примеры кислотно-аддитивных солей включают соли неорганических кислот, такие как гидрохлориды, сульфаты, нитраты, фосфаты и перхлораты; соли органических кислот, такие как ацетаты, формиаты, малеаты, фумараты, тартраты, цитраты, аскорбаты и трифторацетаты; и сульфонаты, такие как метансульфонаты, изетионаты, бензолсульфонаты и п-толуолсульфонаты.
Соединение по изобретению или его соль обладает превосходной ингибирующей активностью в отношении EGFR и полезно в качестве противоопухолевого средства. Кроме того, соединение по изобретению или его соль имеет превосходную селективность по отношению к EGFR, и вызывает, преимущественно, меньшие побочные эффекты, чем эффекты, вызываемые действиями других киназ. Хотя целевой рак конкретно не ограничен, примерами рака являются рак головы и шеи, рак пищевода, рак желудка, рак толстой кишки, рак прямой кишки, рак печени, рак желчного пузыря, холангиокарцинома, рак желчных путей, рак поджелудочной железы, рак легких, рак молочной железы, рак яичников, рак шейки матки, рак эндометрия, рак почки, рак мочевого пузыря, рак предстательной железы, опухоль яичка, остеосаркома, саркома мягких тканей, рак крови, множественная миелома, рак кожи, опухоль мозга и мезотелиома. Предпочтительно, целевой рак представляет собой рак головы и шеи, рак желудка, рак толстой кишки, рак прямой кишки, рак печени, рак поджелудочной железы, рак легких, рак молочной железы, рак яичника, рак почки или рак предстательной железы. Особенно предпочтительным является рак легких.
Когда соединение по настоящему изобретению или его соль используют в виде фармацевтического препарата, то к нему, при необходимости, может быть добавлен фармацевтический носитель, получая тем самым подходящую дозированную форму в соответствии с целями профилактики и лечения. Примеры дозированных форм включают препараты для перорального приема, препараты для инъекций, суппозитории, мази, пластыри и тому подобное. Среди них препараты для перорального приема являются предпочтительными. Такие дозированные формы могут быть получены с помощью обычных способов, известных специалистам в данной области.
В качестве фармацевтического носителя в твердых фармацевтических препаратах могут использоваться различные обычные органические или неорганические вещества, обычно применяемые в твердых фармацевтических препаратах в качестве эксципиента, связующего, дезинтегранта, лубриканта или красящего вещества в твердых препаратах; а в жидких фармацевтических препаратах могут использоваться вещества, обычно используемые в качестве растворителя, солюбилизатора, суспендирующего средства, изотонического агента, буфера или вещества, облегчающего прием препарата. Кроме того, если необходимо, также могут быть использованы фармацевтические добавки, такие как антисептики, антиоксиданты, красители, подсластители и стабилизаторы.
Твердые препараты для перорального приема получают следующим образом. После добавления к соединению по настоящему изобретению эксципиента, необязательно со связующим, дезинтегрантом, лубрикантом, красителем, веществом для маскировки вкуса или флаворантом и т.п., полученную смесь готовят обычными методами в форме таблеток, покрытых таблеток, гранул, порошков, капсул или тому подобного.
Примеры эксципиентов включают лактозу, сахарозу, D-маннит, глюкозу, крахмал, карбонат кальция, каолин, микрокристаллическую целлюлозу и ангидрид кремниевой кислоты. Примеры связующих включают воду, этанол, 1-пропанол, 2-пропанол, простой сироп, жидкую глюкозу, жидкий α-крахмал, жидкий желатин, D-маннит, карбоксиметилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилкрахмал, метилцеллюлозу, этилцеллюлозу, шеллак, фосфат кальция, поливинилпирролидон и тому подобное. Примеры дизентегрантов включают сухой крахмал, альгинат натрия, порошкообразный агар, гидрокарбонат натрия, карбонат кальция, лаурилсульфат натрия, моноглицерид стеариновой кислоты, лактозу и тому подобное. Примеры лубрикантов включают очищенный тальк, натриевую соль стеариновой кислоты, стеарат магния, буру, полиэтиленгликоль и тому подобное. Примеры красителей включают оксид титана, оксид железа и тому подобное. Примеры веществ для маскировки вкуса или флаворантов включают сахарозу, цедру померанца, лимонную кислоту, винную кислоту и тому подобное.
Когда получают жидкий препарат для перорального приема, то к соединению по настоящему изобретению могут быть добавлены вещество для маскировки вкуса, буфер, стабилизатор, флаворант и т.п.; и полученную смесь готовят обычными способами в виде жидкого препарата для перорального приема, сиропа, эликсира и т.п.
В этом случае можно использовать вещество для маскировки вкуса или флаворант, из числа указанных выше. Примером буфера является цитрат натрия, и примеры стабилизатора включают трагакант, гуммиарабик и желатин. При необходимости, на эти препараты для перорального приема, способами, известными в данной области, может быть нанесено энтеросолюбильное покрытие или другое покрытие, с целью, например, сохранения эффектов действия активного вещества. Примеры таких соединений для покрытия включают гидроксипропилметилцеллюлозу, этилцеллюлозу, гидроксиметилцеллюлозу, гидроксипропилцеллюлозу, полиоксиэтиленгликоль и Tween 80 (зарегистрированный товарный знак).
Когда получают препарат для инъекций, к соединению по настоящему изобретению могут быть добавлены регулятор рН, буфер, стабилизатор, изотонический агент, местный анестетик и тому подобное; и смесь может быть приготовлена обычным способом в виде препарата для подкожных, внутримышечных или внутривенных инъекций.
Примеры регулятора рН и буфера, используемых в рамках настоящего изобретения, включают цитрат натрия, ацетат натрия и фосфат натрия. Примеры стабилизатора включают пиросульфит натрия, EDTA, тиогликолевую кислоту и тиомолочную кислоту. Примеры местных анестетиков включают гидрохлорид прокаина и гидрохлорида лидокаина. Примерами изотонического агента являются хлорид натрия, декстроза, D-маннит и глицерин.
Когда изготавливают суппозиторий, то к соединению по настоящему изобретению добавляют известные в данной области фармацевтически приемлемые носители, такие как полиэтиленгликоль, ланолин, масло какао и триглицериды жирных кислот; и, при необходимости могут быть добавлены поверхностно-активные вещества, например, Tween 80 (зарегистрированный товарный знак), и полученная смесь обычным способом формуется в суппозитории.
Когда изготавливают мазь, с соединением по настоящему изобретению смешивают обычно используемую основу, стабилизатор, смачивающее вещество, консервант и тому подобное, как необходимо; и полученная смесь может быть обычным способом перемешана и приготовлена в виде мази.
Примеры основ для мазей включают жидкий парафин, белый вазелин, белый пчелиный воск, октилдодециловый спирт и парафин.
Примеры консервантов включают метилпараоксибензоат, этилпараоксибензоат и пропилпараоксибензоат.
Когда изготавливают пластырь, то описанная выше мазь, крем, гель, паста и т.п., могут быть обычным способом нанесены на обычную подложку.
Для использования в качестве подложки подходящими являются ткани или нетканые материалы на основе хлопка, штапельного волокна или химических волокон; и пленки или листы вспененного материала из мягкого винилхлорида, полиэтилена, полиуретана и т.п.
Количество соединения по настоящему изобретению, которое должно быть включено в каждое из таких дозированных форм, зависит от состояния пациента, которому вводят соединение в дозированной форме, и от т.п. В целом, в случае препарата, принимаемого перорально, количество соединения составляет от 0,05 до 1000 мг в единичной дозированной форме. В случае препарата в форме, предназначенной для инъекций, количество соединения составляет от 0,01 до 500 мг в единичной дозированной форме; и в случае суппозиториев количество соединения составляет от 1 до 1000 мг в единичной дозированной форме.
Суточная доза лекарственного вещества в такой дозированной форме зависит от состояния, массы тела, возраста, пола и других показателей состояния пациента, и она не может быть обобщена. Например, суточная доза для взрослого человека (масса тела 50 кг), обычно может быть от 0,05 до 5000 мг, предпочтительно от 0,1 до 1000 мг; и ее предпочтительно вводят в одной дозе или в двух или трех разделенных дозах в течение дня.
Примеры млекопитающих, которым вводят соединение по настоящему изобретению, включают людей, обезьян, мышей, крыс, кроликов, собак, кошек, коров, лошадей, свиней и овец.
Примеры
Настоящее изобретение подробно разъясняется ниже со ссылкой на примеры. Однако объем притязаний настоящего изобретения не ограничивается этими примерами.
В примерах использовали коммерчески доступные реагенты, если не указано иное.
В качестве колонок для хроматографии на силикагеле использовали снаряженные силикагелевые колонки Purif-Pack (зарегистрированный товарный знак) SI от корпорации Moritex (изготовитель Shoko Scientific Co, Ltd); силикагелевые колонки KP-Sil (зарегистрированный товарный знак), изготовитель Biotage; или HP-Sil (зарегистрированный товарный знак), изготовитель Biotage. В качестве колонок для хроматографии на щелочном силикагеле использовали снаряженные колонки Purif-Pack (зарегистрированный товарный знак) NH от корпорации Moritex (изготовитель Shoko Scientific Co, Ltd); или KP-NH (зарегистрированный товарный знак), изготовитель Biotage.
Для препаративной тонкослойной хроматографии использовали пластины Kieselgel TM 60F 254, Art. 5744, изготовитель Merck, или пластины NH2 Silica Gel 60F254, изготовитель Wako ЯМР-спектр регистрировали с помощью спектрометра AL400 (400 МГц; изготовитель JEOL), Mercury 400 (400 МГц; изготовитель Agilent Technologies, Inc), или с помощью спектрометра Inova 400 (400 МГц; изготовитель Agilent Technologies, Inc) с зондом OMNMR (для поминутного считывания спектра), изготовитель Protasis. Когда дейтерированный растворитель содержал тетраметилсилан, то тетраметилсилан использовали в качестве внутреннего стандарта; и в случае отсутствия тетраметилсилана в качестве внутреннего стандарта использовали растворитель для ЯМР. Все значения показателя дельта представлены с размерностью ″частей на миллион (ppm)″. Для проведения реакций в микроволновой печи использовали печь Discover S-Class, изготовитель CEM Corporation.
Спектры LCMS (ЖХ-МС) получали с использованием установки ACQUITY SQD (квадрупольной), изготовитель Waters Corporation, при следующих условиях.
Колонка: YMC-Triart C18, 2,0×50 мм, 1,9 мкм (изготовитель YMC).
MS-детекция: положительная ионизация с электрораспылением (ESI).
УФ-детекция: при 254 и 210 нм.
Скорость потока в колонке: 0,5 мл/мин.
Подвижная фаза: вода/ацетонитрил (0,1% муравьиная кислота).
Объем впрыска: 1 мкл.
Градиент:
Очистку обращенно-фазовой ВЭЖХ (HPLC) выполняли, используя препаративную систему разделения от Waters Corporation.
Колонка: соединенная колонка YMC-Actus Triart C18, 2050 мм, 5 мкм (изготовитель YMC) и YMC-Actus Triart C18, 20×10 мм, 5 мкм (изготовитель YMC).
УФ-детектирование: 254 нм.
MS-детектирование: положительная ESI.
Скорость потока в колонке: 25 мл/мин.
Подвижная фаза: вода/ацетонитрил (0,1% муравьиная кислота).
Объем вспрыска: от 0,1 до 0,5 мл.
Используемые аббревиатуры и сокращения обозначают следующее.
с: синглет,
д: дублет,
т: триплет,
дд: двойной дублет,
м: мультиплет,
шс: широкий синглет,
DMSO-d6: дейтерированный диметилсульфоксид,
CDCl3: дейтерированный хлороформ,
CD3OD: дейтерированный метанол,
THF: тетрагидрофуран,
DMF: N,N-диметилформамид,
DME: 1,2-диметоксиэтан,
HATU: O-(7-азабензотриазол-1-ил)-N,N,N′,N′-тетраметилуроний.
Пример 1
(R)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид (соединение I-1) и (S)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид (соединение I-2)
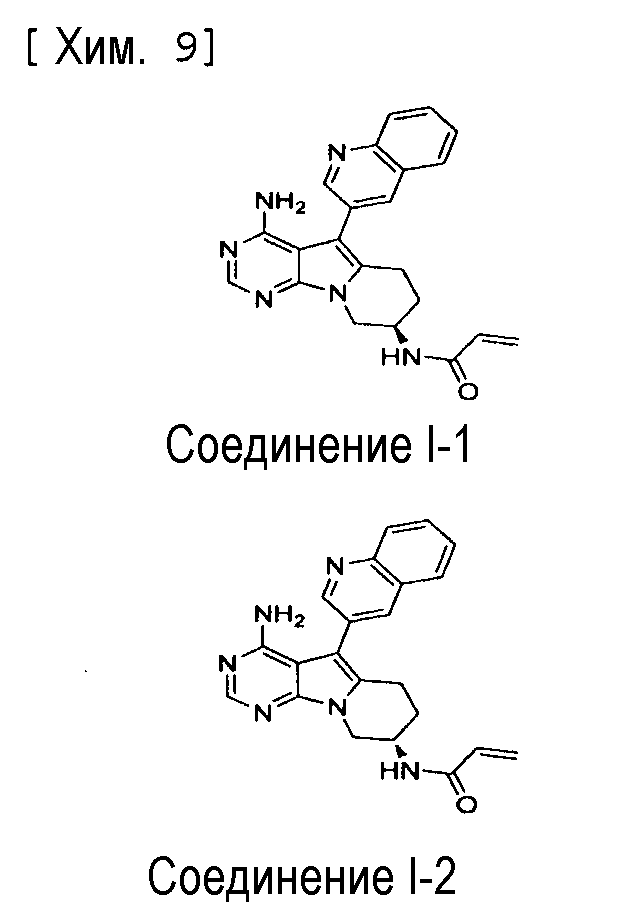
(Стадия 1)
Синтез 4-хлор-5-иод-7H-пирроло[2,3-d]пиримидина
К раствору 4-хлор-7H-пирроло[2,3-d]пиримидина (7,52 г) в DMF (49 мл) при комнатной температуре добавляли N-иодсукцинимид (11,6 г). Смесь перемешивали при той же температуре в течение 1 часа, и к реакционной смеси добавляли воду (150 мл). Полученный осадок собирали фильтрованием, промывали водой и сушили, получая указанное в заголовке соединение в виде светло-желтого твердого вещества (13,57 г).
ESI-MS m/z 280, 282 (МН+).
(Стадия 2)
Синтез 1-бром-2-(трет-бутилдиметилсилилокси)-3-бутена
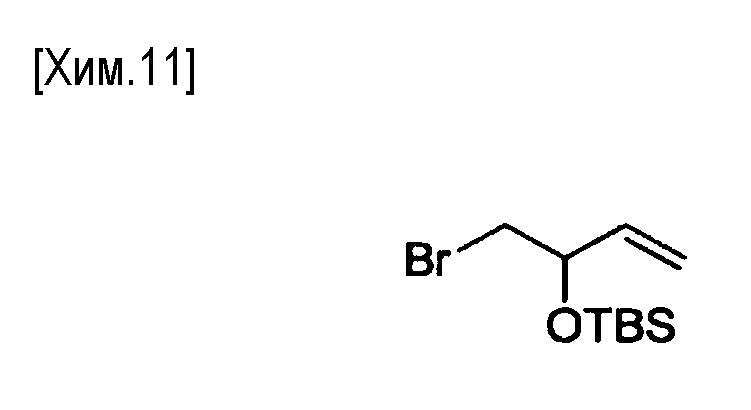
К раствору 1-бром-3-бутен-2-ола (4,5 г) в DMF (30 мл) при комнатной температуре добавляли имидазол (2,25 г) и трет-бутилдиметилсилилхлорид (4,75 г). Смесь перемешивали при той же температуре в течение 16 часов, и к смеси добавляли воду с последующей экстракцией гексаном. Органический слой промывали водой и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении с получением указанного в заголовке соединения в виде светло-желтого маслянистого вещества (7,0 г).
(Стадия 3)
Синтез 7-(2-(трет-бутилдиметилсилилокси)-3-бутенил)-4-хлор-5-иод-7H-пирроло[2,3-d]пиримидина
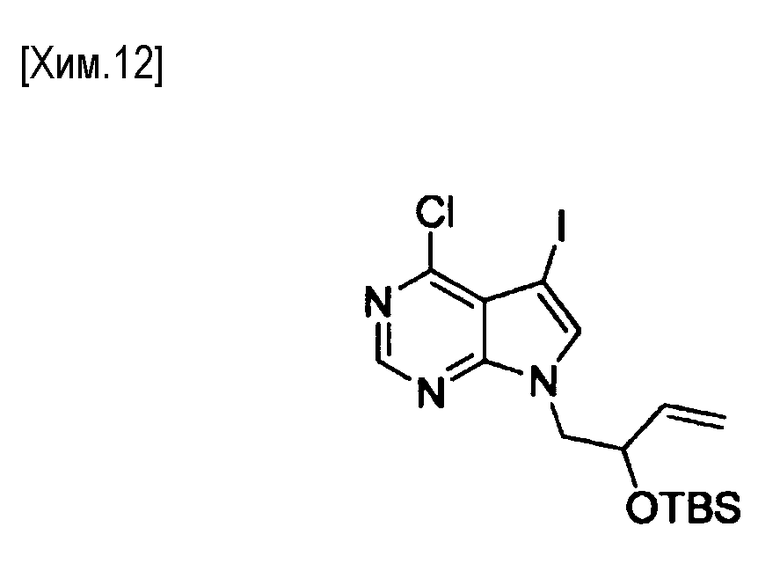
К раствору 4-хлор-5-иод-7H-пирроло[2,3-d]пиримидина (3,7 г), полученного на стадии 1, и 1-бром-2-(трет-бутилдиметилсилилокси)-3-бутена (3,5 г), полученного на стадии 2, в DMF (26 мл) при комнатной температуре добавляли карбонат калия (2,2 г), и смесь перемешивали при 80°С в течение 5 часов. После охлаждения реакционной смеси к ней добавляли воду и этилацетат, и образовавшееся нерастворимое вещество отфильтровывали. Органический слой отделяли, промывали водой и насыщенным раствором хлорида натрия, а затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: гексан/этилацетат) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (2,25 г).
ESI-MS m/z 464, 466 (МН+).
(Стадия 4)
Синтез 7-(2-(трет-бутилдиметилсилилокси)-3-бутенил)-5-иод-7H-пирроло[2,3-d]пиримидин-4-амина
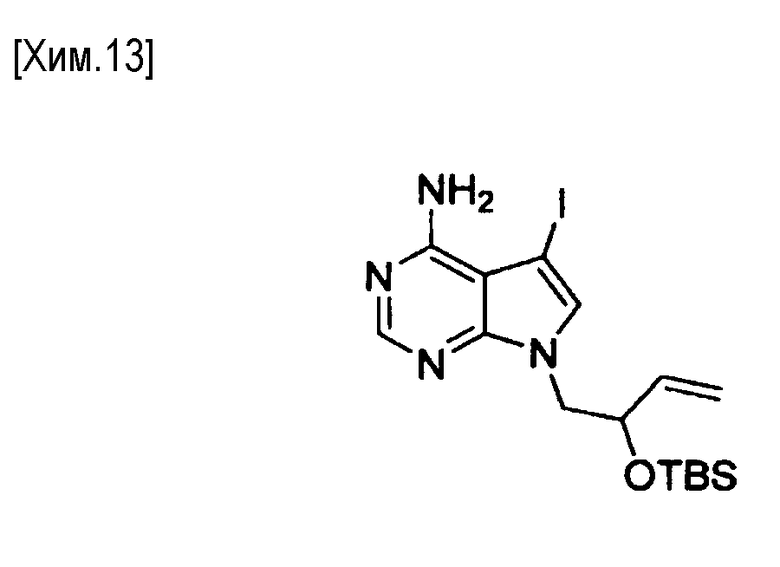
К раствору 7-(2-(трет-бутилдиметилсилилокси)-3-бутенил)-4-хлор-5-иод-7H-пирроло[2,3-d]пиримидина (1,12 г), полученного на стадии 3, в THF (7 мл) добавляли 25% водный раствор аммиака (9 мл). Смесь перемешивали при 120°С в течение 5 часов с использованием микроволнового реактора. Реакционную смесь охлаждали и затем разбавляли водой. Полученный осадок собирали фильтрованием, промывали водой и затем сушили с получением указанного в заголовке соединения в виде белого твердого вещества (1,06 г).
ESI-MS m/z 445 (MH+).
(Стадия 5)
Синтез 7-(2-(трет-бутилдиметилсилилокси)-3-бутенил)-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амина
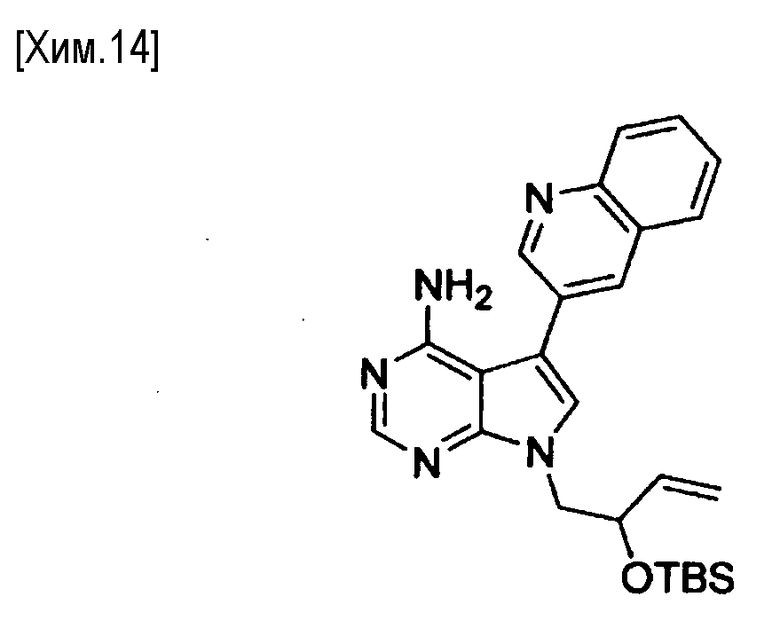
Смесь 7-(2-(трет-бутилдиметилсилилокси)-3-бутенил)-5-иод-7H-пирроло[2,3-d]пиримидин-4-амина (3,62 г), полученного на стадии 4, 3-хинолинбороновой кислоты (1,47 г), карбоната натрия (1,72 г), 2-дициклогексилфосфино-2′,4′,6′-триизопропилбифенила (154 мг), трис(дибензилиденацетон)палладия (0) (148 мг), DME (40 мл) и воды (16 мл) перемешивали в атмосфере азота при 100С в течение 3 часов. После охлаждения реакционную смесь выливали в воду с последующей экстракцией этилацетатом. Органический слой сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: гексан/этилацетат) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (2,13 г).
ESI-MS m/z 446 (MH+).
(Стадия 6)
Синтез 6-бром-7-(2-(трет-бутилдиметилсилилокси)-3-бутенил)-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амина
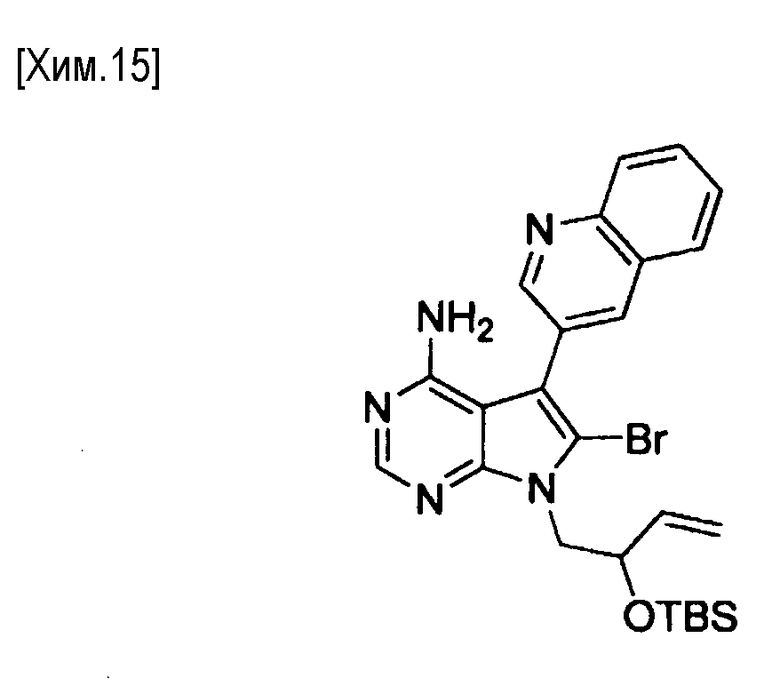
К раствору 7-(2-(трет-бутилдиметилсилилокси)-3-бутенил)-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амина (2,13 г), полученного на стадии 5, в DMF (25 мл) при комнатной температуре добавляли N-бромсукцинимид (894 мг). После перемешивания при той же температуре в течение 30 минут, смесь выливали в насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали водой и насыщенным раствором хлорида натрия и сушили над безводным сульфатом натрия. Затем растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: гексан/этилацетат) с получением указанного в заголовке соединения в виде желтого твердого вещества (2,26 г).
ESI-MS m/z 524, 526 (MH+).
(Стадия 7)
Синтез 8-(трет-бутилдиметилсилилокси)-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4-амина
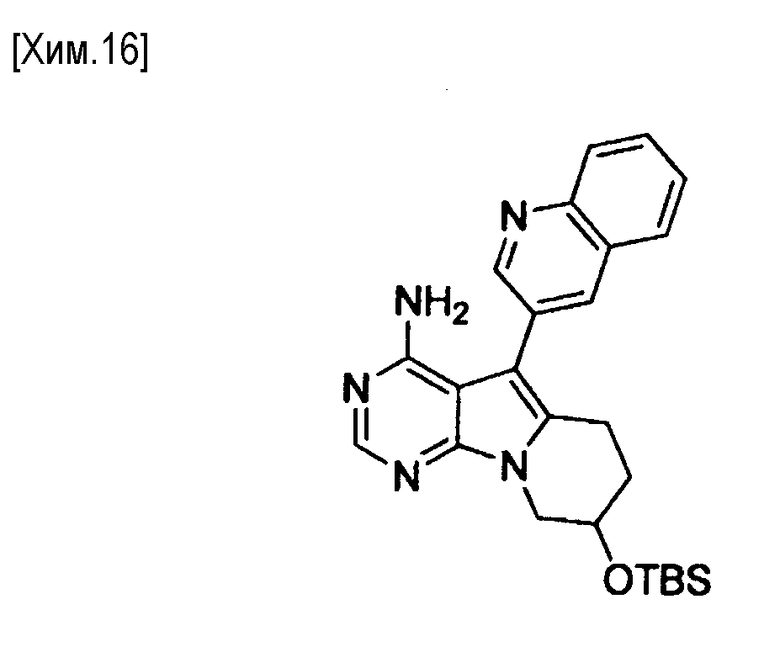
К раствору 6-бром-7-(2-(трет-бутилдиметилсилилокси)-3-бутенил)-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амина (2,26 г), полученного на стадии 6, в THF (30 мл) при охлаждении льдом добавляли 0,5 М раствор 9-борабицикло[3.3.1]нонана в тетрагидрофуране (50 мл). Смесь перемешивали при комнатной температуре в течение 4 часов. После медленного добавления 3 N водного раствора гидроксида натрия (19,5 мл) к реакционной смеси при комнатной температуре, к реакционной смеси добавляли комплекс [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) с дихлорметаном (676 мг). Смесь перемешивали в атмосфере азота при 70°С в течение 4 часов. После охлаждения реакционную смесь выливали в воду с последующей экстракцией этилацетатом. Органический слой сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: гексан/этилацетат) с получением указанного в заголовке соединения в виде желтого маслянистого вещества (778 мг).
ESI-MS m/z 446 (MH+).
(Стадия 8)
Синтез 4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ола
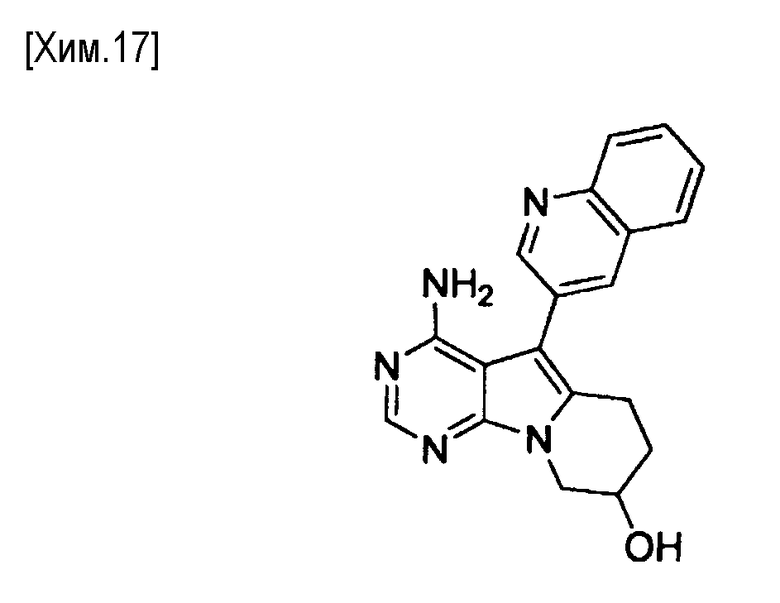
К раствору 8-(трет-бутилдиметилсилилокси)-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4-амина (778 мг), полученного на стадии 7, в THF (20 мл) при комнатной температуре добавляли 1 М раствор тетрабутиламмонийфторида в THF (2,09 мл). Смесь перемешивали при той же температуре в течение 1 часа и растворитель отгоняли при пониженном давлении. Полученный остаток обрабатывали насыщенным водным раствором хлорида аммония с последующей экстракцией этилацетатом. Органический слой сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: хлороформ/метанол) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (580 мг).
ESI-MS m/z 332 (MH+).
(Стадия 9)
Синтез 4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил метансульфоната
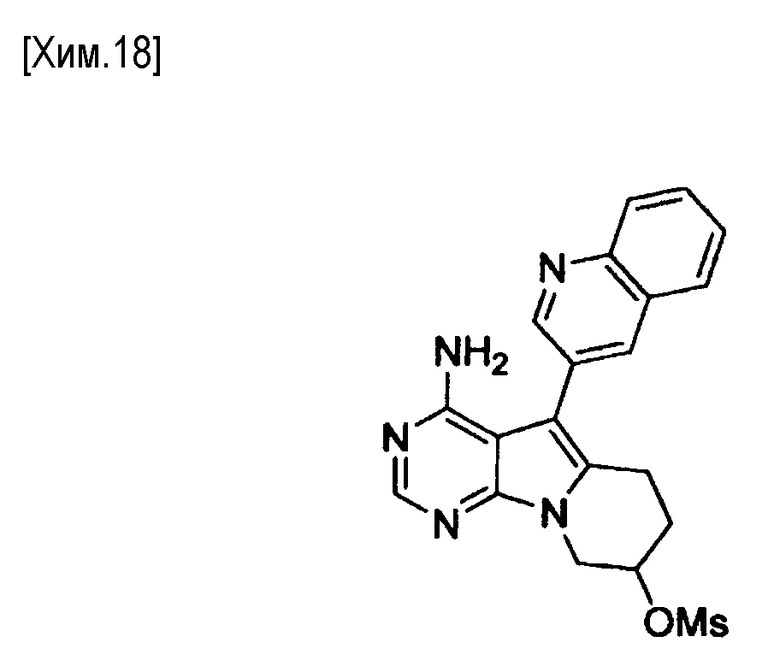
К раствору 4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ола (288 мг), полученного на стадии 8, в THF (5 мл) при охлаждении льдом добавляли триэтиламин (0,157 мл) и метансульфонилхлорид (0,074 мл). После перемешивания при той же температуре в течение 15 минут смесь выливали в насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, получая указанное в заголовке соединение в виде светло-коричневого твердого вещества (615 мг).
ESI-MS m/z 410 (MH+).
(Стадия 10)
Синтез 8-азидо-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4-амина
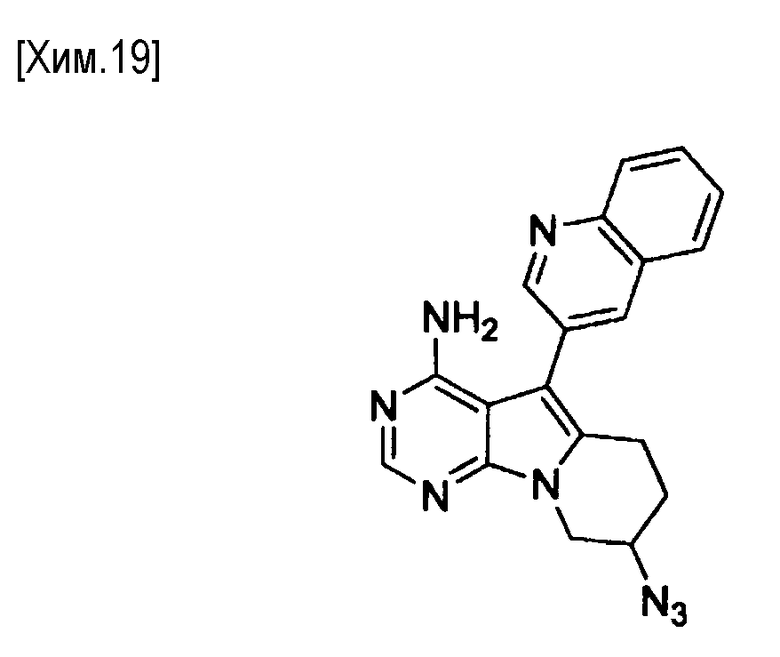
К раствору 4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил метансульфоната (758 мг), полученного на стадии 9, в DMF (9 мл) при комнатной температуре добавляли азид натрия (361 мг), и смесь перемешивали при 80°С в течение 4 часов. После охлаждения реакционную смесь выливали в воду с последующей экстракцией этилацетатом. Органический слой промывали водой и насыщенным раствором хлорида натрия и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении с получением указанного в заголовке соединения в виде желтого твердого вещества (508 мг).
ESI-MS m/z 357 (MH+).
(Стадия 11)
Синтез 5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,8-диамина
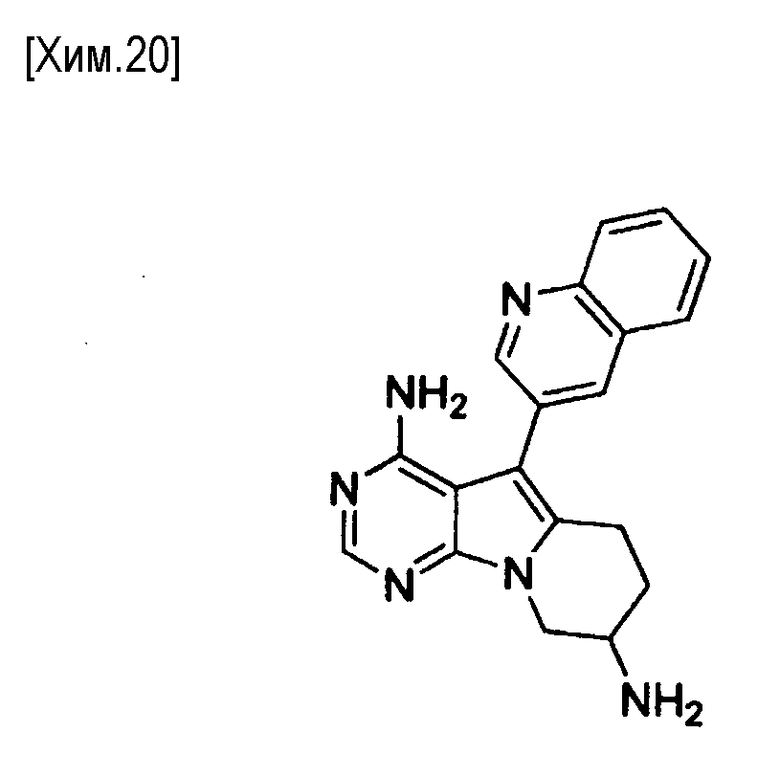
К раствору 8-азидо-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4-амина (508 мг), полученного на стадии 10, в THF (10 мл) и воде (1 мл) при комнатной температуре добавляли трифенилфосфин (~3,0 ммоль/г, 1,42 г) на полимерном носителе. Реакционную смесь нагревали с обратным холодильником в течение 1 часа. После охлаждения реакционную смесь фильтровали через целит, промывали этанолом и растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: хлороформ/метанол) с получением указанного в заголовке соединения в виде желтого твердого вещества (342 мг).
ESI-MS m/z 331 (MH+).
(Стадия 12)
Синтез N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида
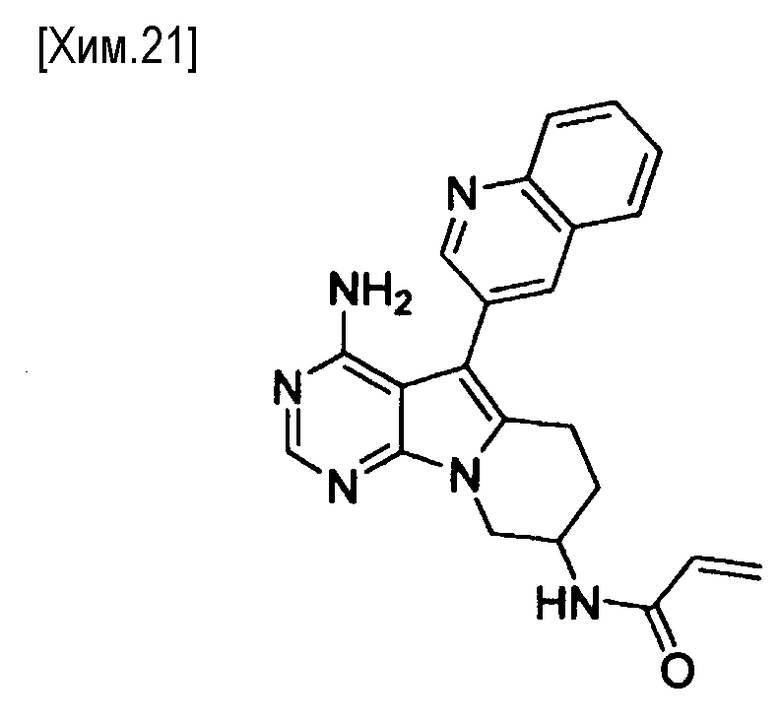
К раствору 5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,8-диамина (68 мг), полученного на стадии 11, в хлороформе (2,5 мл) при охлаждении льдом добавляли N,N-диизопропилэтиламин (0,033 мл) и акрилоилхлорид (0,0154 мл). После перемешивания при той же температуре в течение 15 минут, смесь выливали в насыщенный водный раствор бикарбоната натрия с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: хлороформ/метанол) с получением указанного в заголовке соединения в виде желтого твердого вещества (35,4 мг).
1Н-ЯМР (CDCl3) δ: 2,14 (2H, д, J=5,6 Гц), 3,04 (2H, т, J=6,2 Гц), 4,17 (1Н, дд, J=12,7, 5,8 Гц), 4,46 (1H, дд, J=12,7, 4,5 Гц), 4,70-4,80 (1Н, м), 4,89 (2H, ш.с), 5,71 (1H, д, J=10,2 Гц), 6,21 (1Н, дд, J=16,8, 10,2 Гц), 6,39 (1H, д, J=16,8 Гц), 6,50 (1H, д, J=7,0 Гц), 7,62 (1H, т, J=7,4 Гц), 7,77 (1H, т, J=7,4 Гц), 7,87 (1H, д, J=8,0 Гц), 8,15 (1H, д, J=8,0 Гц), 8,25 (1H, с), 8,32 (1Н, с), 9,00 (1Н, с).
ESI-MS m/z 385 (MH+).
(Стадия 13)
Разделение энантиомера А N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил акриламида и энантиомера B N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида
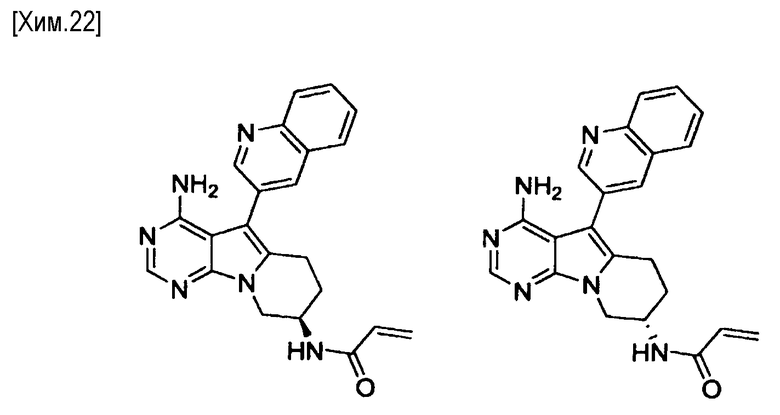
N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид (197 мг), полученный на стадии 12, подвергали оптическому разделению с использованием колонки для оптического разделения (Chiralpak AD-H 20×250 мм, изготовитель Daicel Chemical Industries, Ltd), подвижная фаза: гексан/этанол/триэтиламин, 50:50:0,1; скорость потока 10 мл/мин), с получением 72,4 мг энантиомера А (время удерживания: 15,4 мин, (R)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид (соединение I-1)) и 78,3 мг энантиомера B (время удерживания: 32,5 мин, (S)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид (соединение I-2)) в виде светло- желтого твердого вещества.
Энантиомер А
ESI-MS m/z 385 (MH+).
Энантиомер B
ESI-MS m/z 385 (MH+).
Когда (R)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид, полученный способом, аналогичным способу по Примеру 2, который описан далее, подвергли хроматографии на колонке при таких же условиях, как описано выше, его время удерживания было таким же, как у энантиомера А. Было подтверждено, что энантиомер представляет собой R-изомер, т.е. соединение I-1; и что энантиомер B представляет собой S-изомер, т.е. соединение I-2.
Пример 2
(R)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид (соединение I-1)
(Стадия 1)
Синтез (S)-2-(трет-бутилдиметилсилилокси)-3-бутенил-4-метилбензолсульфоната
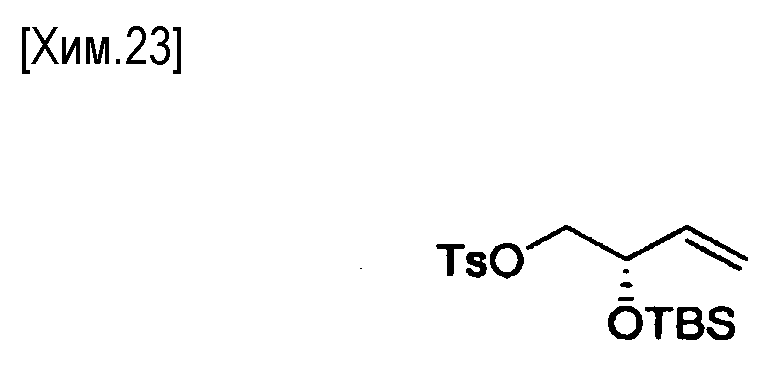
Указанное в заголовке соединение получали способом в соответствии со стадией 2 Примера 1, за исключением того, что вместо 1-бром-3-бутен-2-ола в данном случае использовали (S)-3-бутен-1,2-диол-1-(п-толуолсульфонат), получая указанное соединение в виде бесцветного маслянистого вещества (2,74 г).
(Стадия 2)
Синтез (R)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида
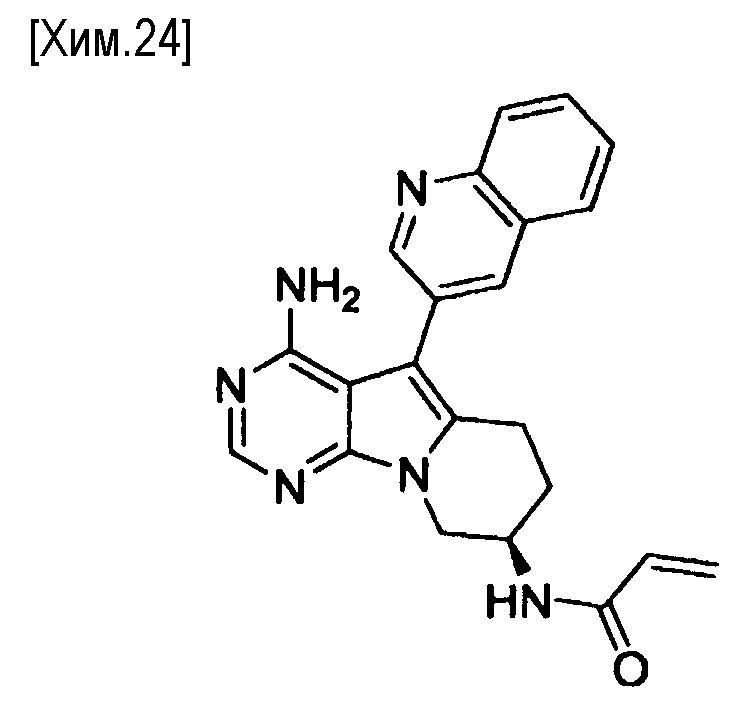
Указанное в заголовке соединение получали способом в соответствии со стадиями 1-12 Примера 1, за исключением того, что вместо 1-бром-2-(трет-бутилдиметилсилилокси)-3-бутена в данном случае использовали (S)-2-(трет-бутилдиметилсилилокси)-3-бутенил-4-метилбензолсульфонат, полученный на стадии 1, получая указанное соединение в виде желтого твердого вещества (13,9 мг).
Пример 3
N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-N-метилакриламид (соединение I-3)
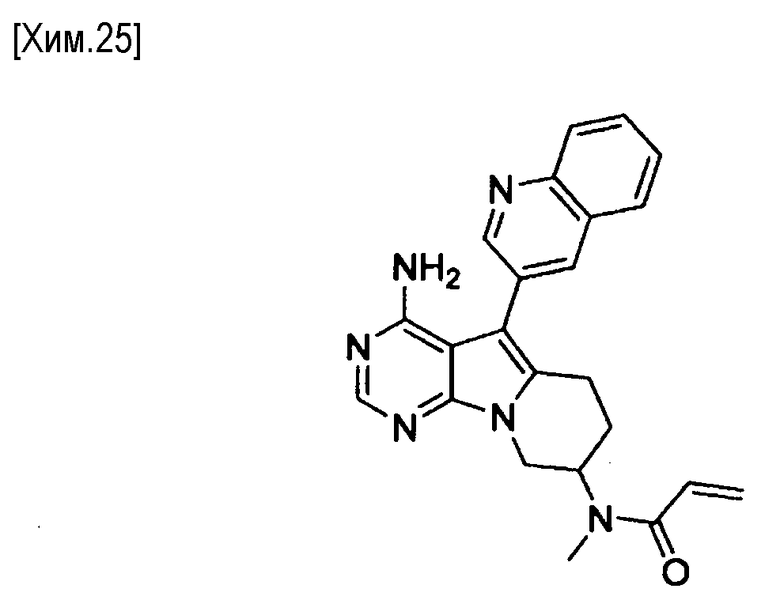
(Стадия 1)
Синтез N8-метил-5-(хинолин-3-ил)-6,7,8,9- тетрагидропиримидо[5,4-b]индолизин-4,8-диамина
В растворе метиламина в 40%-ном метаноле (1 мл) растворяли 4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил метансульфонат (30 мг), полученный на стадии 9 Примера 1. Полученный раствор перемешивали при 60°С в течение 1 часа и при 80°С в течение 22 часов. Реакционную смесь после охлаждения концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: этилацетат/метанол) с получением указанного в заголовке соединения в виде светло-желтого маслянистого вещества (8,1 мг).
ESI-MS m/z 345 (MH+).
(Стадия 2)
Синтез N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-N-метилакриламида
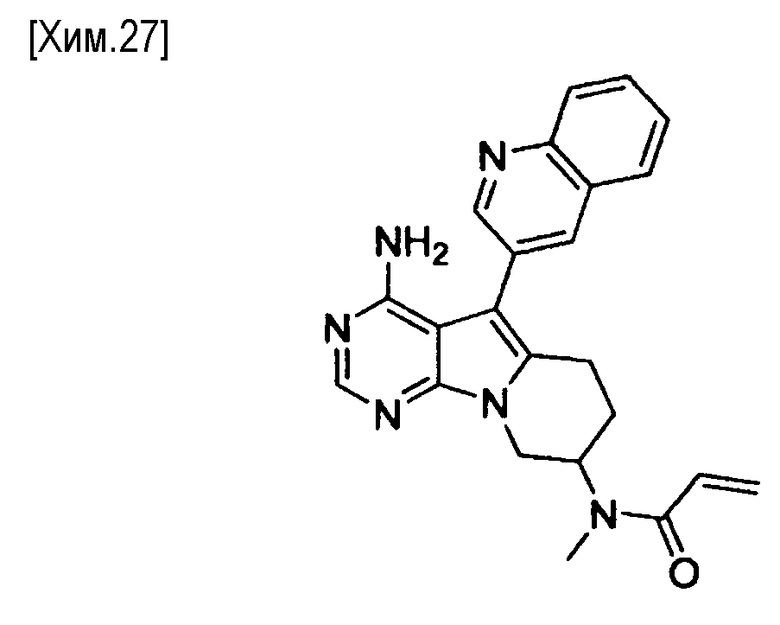
Указанное в заголовке соединение получали способом в соответствии со стадией 12 Примера 1, за исключением того, что вместо 5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,8-диамина, используемого на стадии 12 Примера 1, в данном случае использовали N8-метил-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,8-диамин, полученный на стадии 1, получая указанное соединение в виде светло-желтого твердого вещества (6,2 мг).
1Н-ЯМР (CDCl3) δ: 1,90-2,15 (2H, м), 3,00-3,15 (2Н, м), 3,06 (3Н, с), 3,90-4,03 (1H, м), 4,56-4,64 (1H, м), 5,06 (2H, ш.с), 5,15-5,30 (1Н, м), 5,77 (1H, д, J=10,2 Гц), 6,38 (1H, д, J=16,6 Гц), 6,54-6,70 (1H, м), 7,63 (1H, т, J=7,3 Гц), 7,78 (1H, т, J=7,3 Гц), 7,88 (1H, д, J=8,0 Гц), 8,16 (1H, с), 8,17 (1H, д, J=8,0 Гц), 8,31 (1Н, с), 9,01 (1Н, с).
ESI-MS m/z 399 (MH+).
Пример 4
(E)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-4-(диметиламино)-2- бутенамид (Соединение I-4)
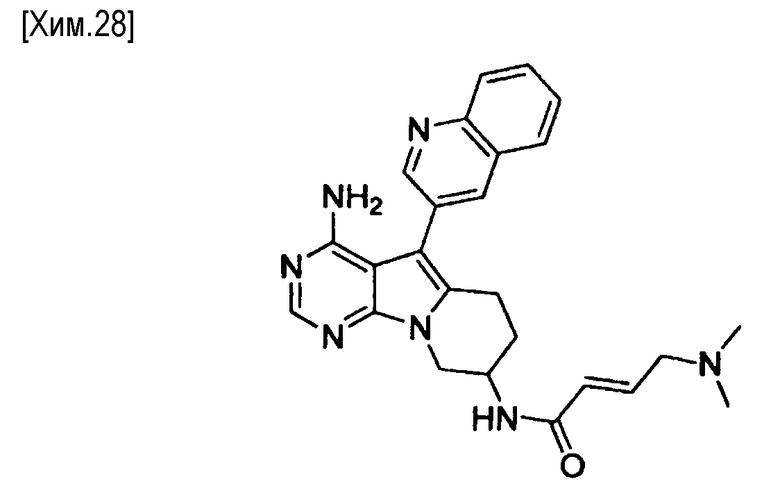
К раствору гидрохлорида транс-4-диметиламинокротоновой кислоты (4,5 мг) в DMFA (0,5 мл) при комнатной температуре добавляли HATU (10,3 мг) и 5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,8-диамин (8,8 мг), полученный на стадии 11 Примера 1. После перемешивания в течение 1 часа при той же температуре, смесь выливали в насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью препаративной тонкослойной хроматографии (пластины силикагель NH2 60, F254, изготовитель Wako, используемый растворитель: хлороформ/метанол) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (6,4 мг).
1Н-ЯМР (CDCl3) δ: 2,05-2,20 (2Н, м), 2,27 (6Н, с), 2,49-2,65 (4H, м), 4,10-4,20 (1H, м), 4,46-5,02 (1H, м), 4,68-4,77 (1H, м), 4,91 (2H, ш.с), 6,06 (1H, д, J=15,4 Гц), 6,39 (1H, д, J=7,0 Гц), 6,85-6,95 (1H, м), 7,62 (1H, т, J=7,4 Гц), 7,77 (1H, т, J=7,4 Гц), 7,87 (1H, д, J=8,0 Гц), 8,15 (1H, д, J=8,0 Гц), 8,17 (1H, с), 8,25 (1Н, с), 9,00 (1Н, с).
ESI-MS m/z 442 (MH+).
Пример 5
(S,E)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-3-хлоракриламид (Соединение I-5)
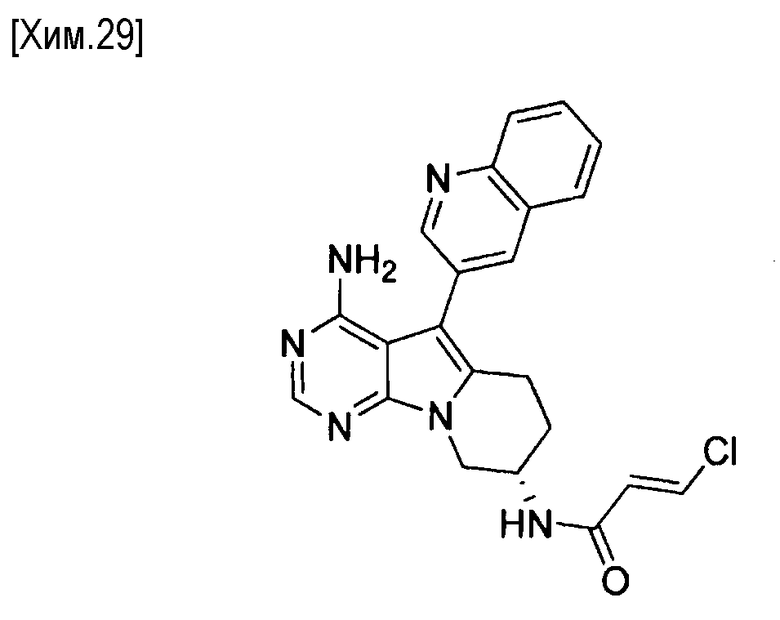
К суспензии 5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,8-диамина (498,0 мг), полученного на стадии 11 Примера 1, в DMF (8 мл) при комнатной температуре добавляли транс-3-хлоракриловую кислоту (399,5 мг). После растворения добавляли при охлаждении льдом 1-(3-диметиламинопропил)-3-этилкарбодиимид (350,1 мг), и смесь перемешивали в течение 1 часа при той же температуре. Реакционную смесь выливали в насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: этилацетат/метанол) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (261,2 мг).
1H-ЯМР (DMSO-d6) δ: 1,84-2,07 (2H, м), 2,92-3,08 (2H, м), 3,88-4,02 (1H, м), 4,27-4,43 (2H, м), 6,07 (2H, ш.с), 6,48 (1H, д, J=13,4 Гц), 7,31 (1H, д, J=13,2 Гц), 7,63 (1H, т, J=7,4 Гц), 7,75 (1H, т, J=7,6 Гц), 8,03 (1H, д, J=10,7 Гц), 8,05 (1H, д, J=10,7 Гц), 8,13 (1Н, с), 8,29 (1H, д, J=2,0 Гц), 8,53 (1H, д, J=6,6 Гц), 8,92 (1H, д, J=2,2 Гц).
ESI-MS m/z 419, 421 (MH+).
Пример 6
(S,Z)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-3-хлоракриламид (Соединение I-6)
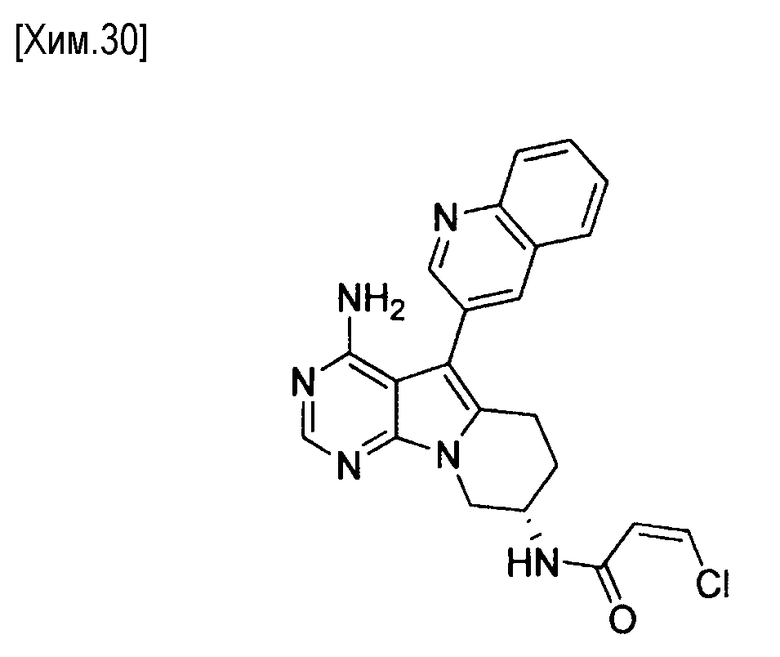
Указанное в заголовке соединение получали способом в соответствии с Примером 5, за исключением того, что вместо транс-3-хлоракриловой кислоты, используемой в Примере 5, использовали цис-3-хлоракриловую кислоту, получая указанное соединение в виде светло-желтого твердого вещества (93 мг).
1H-ЯМР (DMSO-d6) δ: 1,82-1,96 (1H, м), 1,96-2,07 (1H, м), 2,92-3,08 (2H, м), 3,85-3,97 (1H, м), 4,27-4,41 (2H, м), 6,05 (2H, ш.с), 6,39 (1H, д, J=8,0 Гц), 6,77 (1H, д, J=8,0 Гц), 7,63 (1H, т, J=7,4 Гц), 7,75 (1H, т, J=7,4 Гц), 8,02 (1H, д, J=11,4 Гц), 8,04 (1H, д, J=11,4 Гц), 8,13 (1Н, с), 8,29 (1H, д, J=2,0 Гц), 8,50 (1H, д, J=6,3 Гц), 8,92 (1H, д, J=2,0 Гц).
ESI-MS m/z 419, 421 (MH+).
Пример 7
(S)-N-(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5]пирроло[1,2-a]азепин-8-ил)акриламид (Соединение I-7)
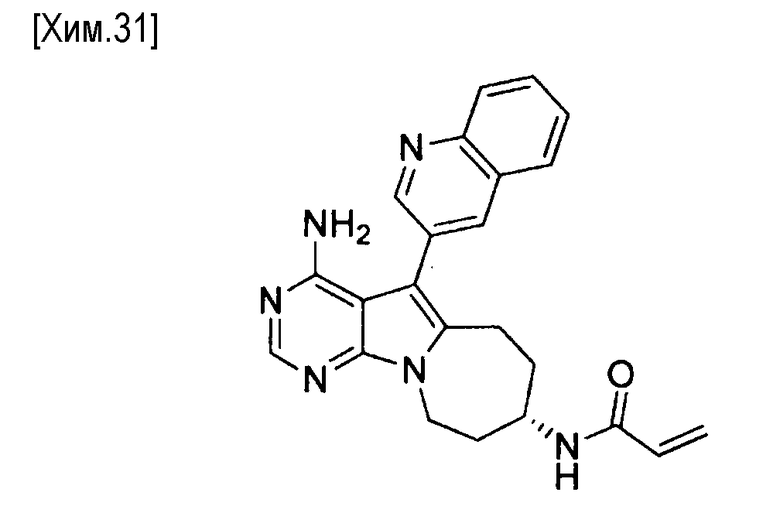
(Стадия 1)
Синтез (R)-трет-бутил-(1-гидрокси-5-(метилтио)пентан-3-ил)карбамата
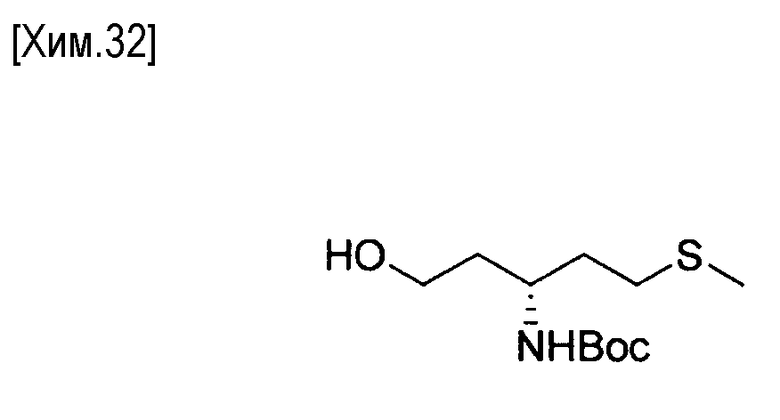
К раствору (R)-3-((трет-бутоксикарбонил)амино)-5-(метилтио)пентановой кислоты (7,92 г) в THF (79,2 мл) при температуре -10°С добавляли N-метилморфолин (3,63 мл) и этилхлорформиат (3,01 мл). После перемешивания при температуре -10°С в течение 15 минут, отфильтровывали образовавшееся нерастворимое вещество. К фильтрату при температуре -10°С добавляли водный раствор боргидрида натрия (1,55 г) (15 мл) и смесь перемешивали при -10°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и смесь перемешивали при комнатной температуре в течение 30 минут. К раствору добавляли этилацетат и отделяли органический слой. Органический слой промывали 0,5 N водным раствором бисульфата калия, водой, 0,5 N водным раствором гидроксида натрия и насыщенным раствором хлорида натрия, а затем сушили над безводным сульфатом натрия. Затем растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: хлороформ/этилацетат) с получением указанного в заголовке соединения в виде светло-желтого маслянистого вещества (7,18 г).
(Стадия 2)
Синтез трет-бутил((3R)-1-гидрокси-5-(метилсульфинил)пентан-3-ил)карбамата
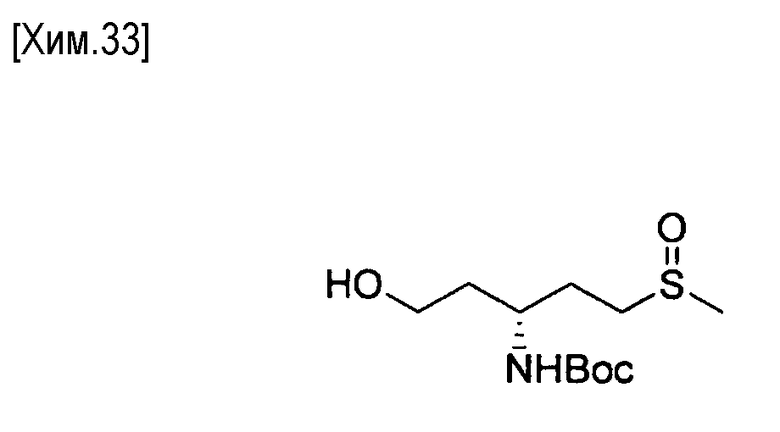
К раствору (R)-трет-бутил-(1-гидрокси-5-(метилтио)пентан-3-ил)карбамата (8,16 г), полученного на стадии 1, в метаноле (98 мл) при температуре 10°С или ниже добавляли суспензию периодата натрия (7,0 г) в воде (32 мл), и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Образовавшееся нерастворимое вещество отфильтровывали, и фильтрат перегоняли при пониженном давлении. Полученный остаток растворяли в насыщенном растворе хлорида натрия, с последующей трехкратной экстракцией хлороформом. Органический слой сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (9,38 г).
(Стадия 3)
Синтез (R)-трет-бутил-5-(гидроксипент-1-ен-3-ил)карбамата
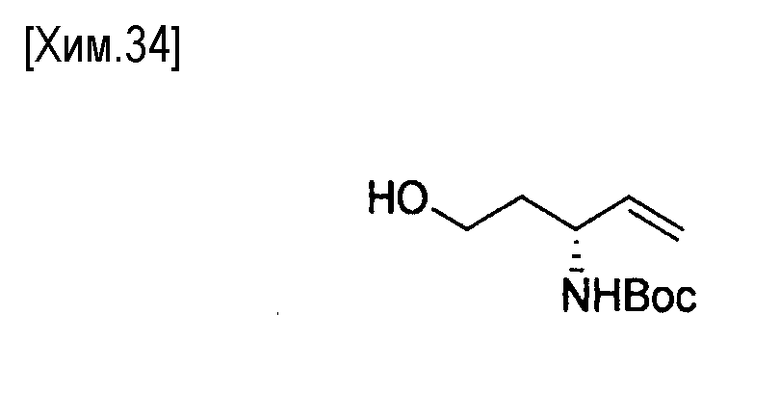
К раствору трет-бутил ((3R)-1-гидрокси-5-(метилсульфинил)пентан-3-ил)карбамата (9,38 г), полученного на стадии 2, в 1,2-дихлорбензоле (140 мл) при комнатной температуре добавляли ацетат натрия (13,45 г). Смесь перемешивали при внутренней температуре 166°С в течение 18 часов. После охлаждения реакционной смеси нерастворимое вещество отфильтровывали, и при пониженном давлении отгоняли 1,2-дихлорбензол. Полученный остаток растворяли в этилацетате, промывали насыщенным водным раствором хлорида натрия, водой и насыщенным раствором хлорида натрия; а затем сушили над безводным сульфатом натрия. Затем растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: гексан/этилацетат), с получением указанного в заголовке соединение (2,50 г) в виде светло-желтого маслянистого вещества.
(Стадия 4)
Синтез (R)-трет-бутил-(5-(4-хлор-5-иод-7H-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата
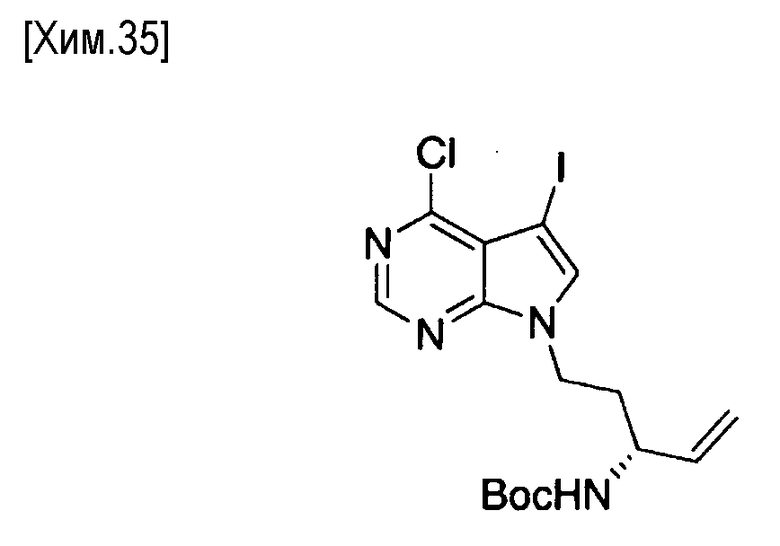
К раствору (R)-трет-бутил-5-(гидроксипент-1-ен-3-ил)карбамата (2,5 г), полученного на стадии 3, и 4-хлор-5-иод-7H-пирроло[2,3-d]пиримидина (2,31 г), полученного на стадии 1 Примера 1, в DME (23 мл) при охлаждении льдом добавляли и растворяли трифенилфосфин (3,25 г). После этого к раствору постепенно добавляли диизопропилазодикарбоксилат (2,44 мл). Реакционную смесь перемешивали при охлаждении льдом в течение 30 минут, а затем при комнатной температуре в течение 1 часа, и растворитель отгоняли при пониженном давлении. Полученный остаток растворяли в этилацетате, промывали водой и сушили над безводным сульфатом натрия. Затем растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: гексан/этилацетат) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (3,49 г).
ESI-MS m/z 463, 465 (MH+).
(Стадия 5)
Синтез (R)-трет-бутил-(5-(4-амино-5-иод-7H-пирроло[2,3- d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата
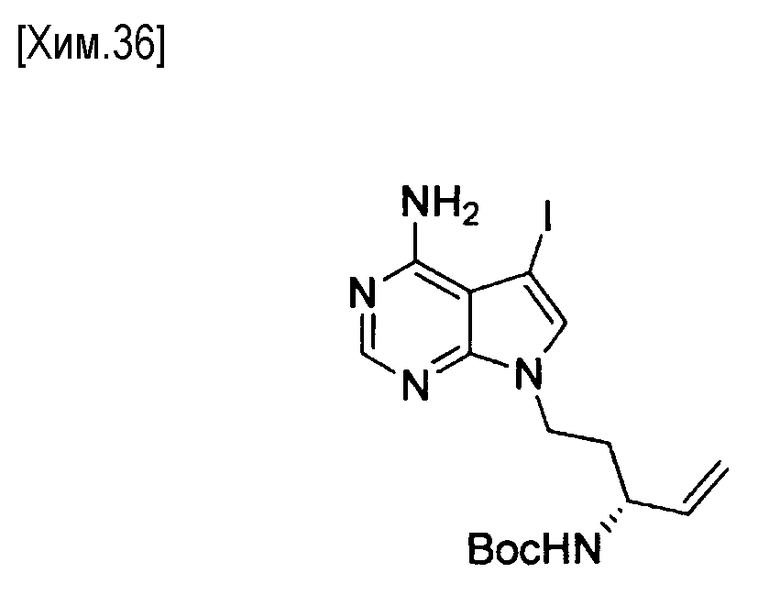
К раствору (R)-трет-бутил-(5-(4-хлор-5-иод-7H-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата (3,49 г), полученного на стадии 4, в DME (17,5 мл) добавляли 28% водный раствор аммиака (17,5 мл), и смесь перемешивали в автоклаве при внутренней температуре 105°С в течение 8 часов. После охлаждения реакционной смеси к ней добавляли воду (70 мл), и смесь перемешивали при комнатной температуре в течение 4 часов. Полученный осадок собирали фильтрованием, промывали водой и сушили, получая указанное в заголовке соединение в виде светло-желтого твердого вещества (3,20 г).
ESI-MS m/z 444 (MH+).
(Стадия 6)
Синтез (R)-трет-бутил-(5-(4-амино-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата
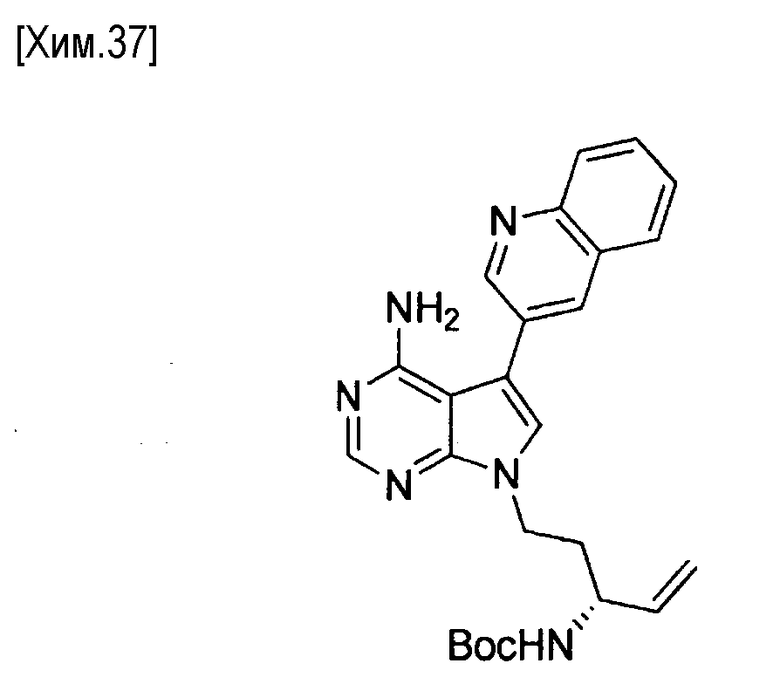
Смесь (R)-трет-бутил-(5-(4-амино-5-иод-7H-пирроло[2,3- d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата (3,2 г), полученного на стадии 5, 3-хинолинбороновой кислоты (1,37 г), карбоната натрия (843 мг), тетракис(трифенилфосфин)палладия (250 мг), DME (32 мл) и воды (32 мл) перемешивали в атмосфере азота при температуре 100°С в течение 6 часов. После охлаждения реакционной смеси к ней добавляли насыщенный водный раствор бикарбоната натрия и этилацетат. Полученную смесь перемешивали при комнатной температуре в течение 30 минут. После отфильтровывания нерастворимого вещества отделяли органический слой и его сушили над безводным сульфатом натрия. Затем растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: гексан/этилацетат, этилацетат/метанол) с получением указанного в заголовке соединения в виде светло-оранжевого твердого вещества (3,21 г).
ESI-MS m/z 445 (MH+).
(Стадия 7)
Синтез (R)-трет-бутил-(5-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата
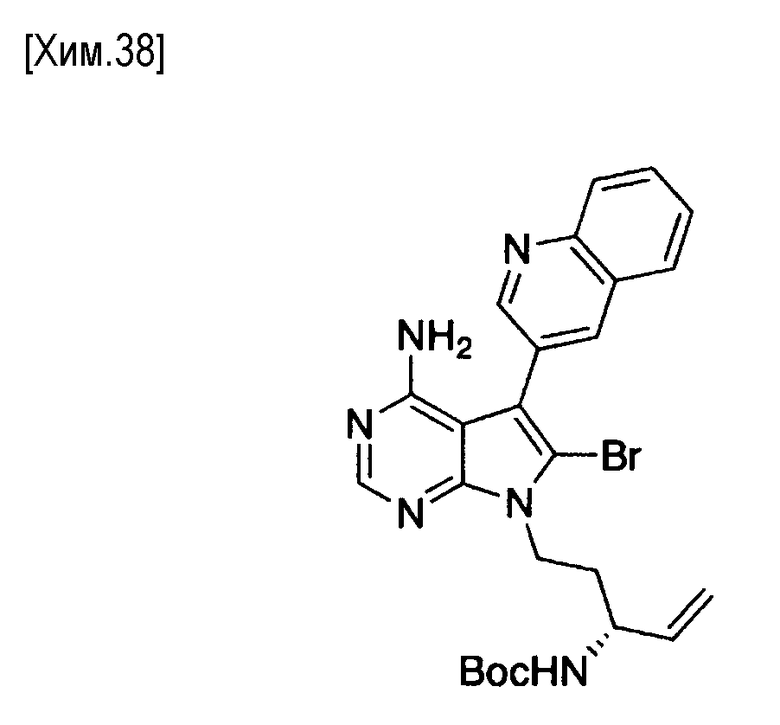
Раствор N-бромсукцинимида (1,35 г) в THF (23 мл) добавляли в течение 30 минут к раствору (R)-трет-бутил-(5-(4-амино-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата (3,21 г), полученного на стадии 6, в THF (26 мл) при охлаждении льдом. Смесь перемешивали при охлаждении льдом в течение 30 минут. После добавления 5% водного раствора тиосульфата натрия, смесь выливали в насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом натрия. Затем растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: гексан/этилацетат, этилацетат/метанол) с получением указанного в заголовке соединения в виде светло-коричневого твердого вещества (3,15 г).
ESI-MS m/z 523, 525 (MH+).
(Стадия 8)
Синтез (S)-трет-бутил (4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5]пирроло[1,2-a]азепин-8-ил)карбамата
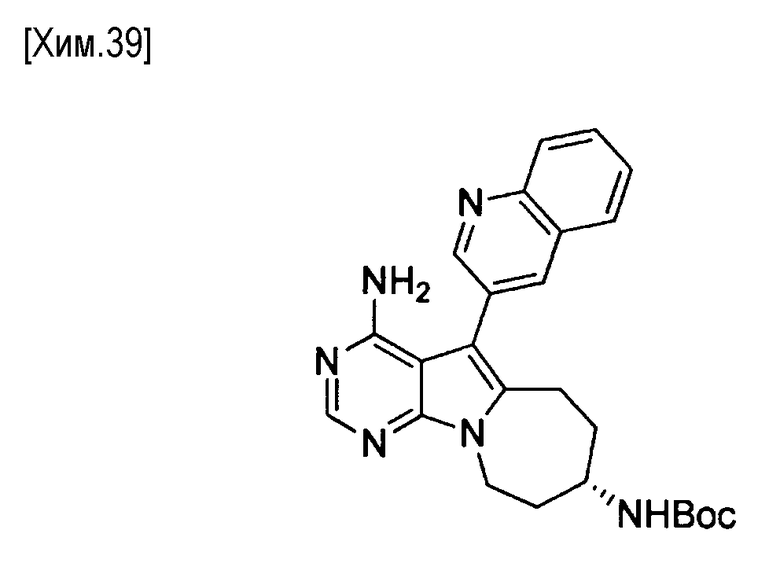
К раствору 9-борабицикло[3.3.1]нонана в 0,5 М THF (22,8 мл) при комнатной температуре добавляли (R)-трет-бутил-(5-(4-амино- 6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамат (994 мг), полученный на стадии 7. Смесь перемешивали при той же температуре в течение 1 часа, и осторожно добавляли к нему 4 N водный раствор гидроксида натрия (5,7 мл). После продувки азотом, смесь нагревали до внутренней температуры 55°С. Добавляли тетракис(трифенилфосфин)палладий (275 мг) и смесь перемешивали при внутренней температуре 66°С в течение 15 часов. После охлаждения реакционной смеси отделяли органический слой и добавляли к нему толуол (7,7 мл) и 20% водный раствор хлорида аммония (5 мл). Органический слой отделяли, промывали 20% солевым раствором, и добавляли силикагель SH (изготовитель Fuji Silysia) (1 г). Смесь перемешивали при внутренней температуре 68°С в течение 1 часа, и к ней добавляли силикагель SH (изготовитель Fuji Silysia) (1 г). Смесь перемешивали при внутренней температуре 68°С в течение 1 часа. После охлаждения силикагель отфильтровывали, и отгоняли растворитель при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: этилацетат/метанол) с получением указанного в заголовке соединения в виде желтого твердого вещества (439 мг).
ESI-MS m/z 445 (MH+).
(Стадия 9)
Синтез (S)-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5]пирроло[1,2-а]азепин-4,8-диамина
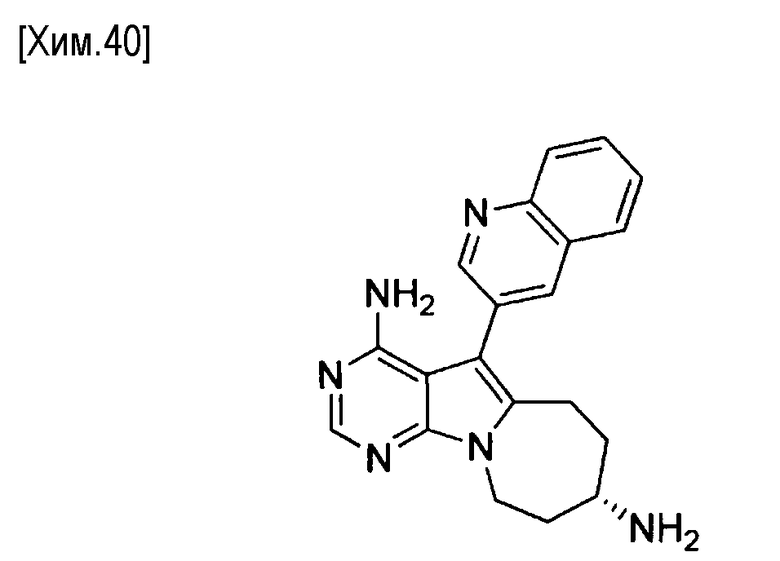
К раствору (S)-трет-бутил(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5]пирроло[1,2-a]азепин-8-ил)карбамата (436 мг), полученного на стадии 8, в этаноле (4 мл) при комнатной температуре добавляли 5 N хлористоводородную кислоту (1 мл). Смесь перемешивали при 60°С в течение 3 часов. Реакционную смесь после охлаждения подщелачивали 5 N водным раствором гидроксида натрия, с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: хлороформ/метанол) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (320 мг).
ESI-MS m/z 345 (MH+).
(Стадия 10)
Синтез (S)-N-(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5]пирроло[1,2-a]азепин-8-ил)акриламида
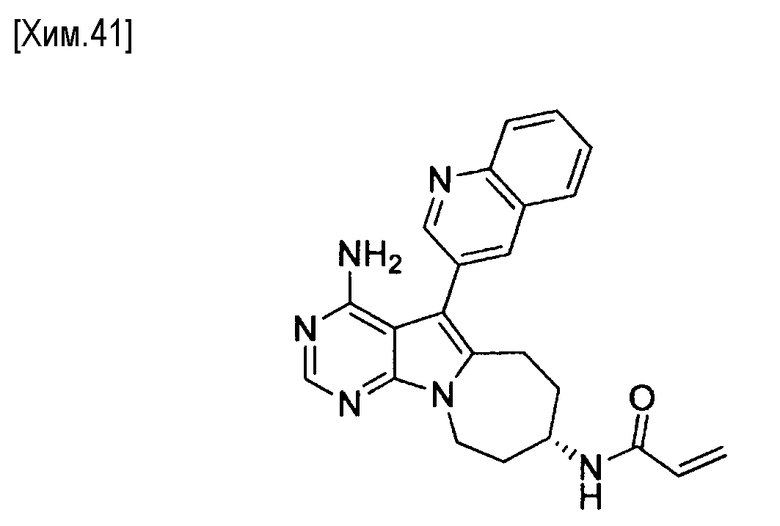
К раствору (S)-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5]пирроло[1,2-а]азепин-4,8-диамина (317 мг), полученного на стадии 9, в смеси ацетонитрила (1,6 мл) и воды (1,6 мл) при охлаждении льдом добавляли N,N-диизопропилэтиламин (0,192 мл) и раствор акрилоилхлорида (83,3 мг) в ацетонитриле (0,83 мл). После перемешивания при той же температуре в течение 15 минут смесь выливали в насыщенный водный раствор бикарбоната натрия с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: этилацетат/метанол) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (226 мг).
1H-ЯМР (DMSO-d6) δ: 1,37-1,56 (2H, м), 1,98-2,20 (2H, м), 2,75-2,83 (1H, м), 2,88-2,97 (1H, м), 3,96-4,18 (2H, м), 4,78-4,90 (1H, м), 5,58 (1Н, дд, J=10,0, 2,2 Гц), 5,93 (2H, ш.с), 6,19 (1Н, дд, J=17,1, 2,2 Гц), 6,21 (1H, дд, J=17,1, 10,0 Гц), 7,64 (1H, т, J=7,4 Гц), 7,77 (1H, т, J=7,4 Гц), 8,01-8,09 (2H, м), 8,14 (1H, с), 8,17 (1H, д, J=7,6 Гц), 8,27 (1H, д, J=2,0 Гц), 8,85 (1H, д, J=2,0 Гц).
ESI-MS m/z 399 (MH+).
Пример 8
(S)-N-(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5]пирроло[1,2-a]азепин-9-ил)акриламид
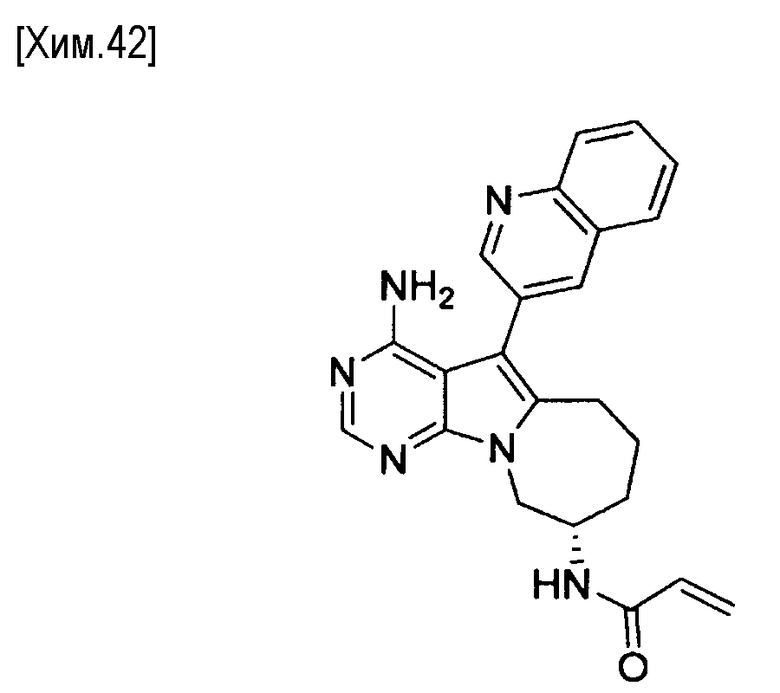
(Стадия 1)
Синтез (S)-трет-бутил-(1-гидроксипент-4-ен-2-ил)карбамата
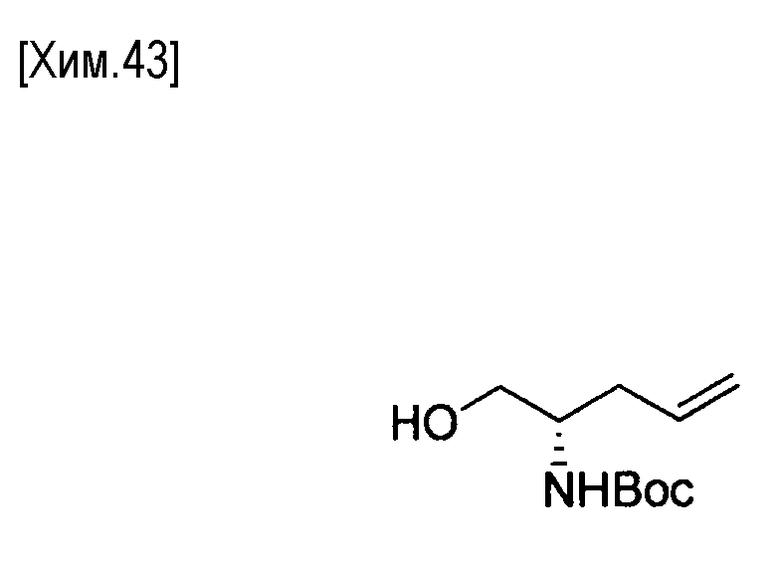
К раствору (S)-2-((трет-бутоксикарбонил)амино)пент-4-еновой кислоты (25,0 г) в THF (250 мл) при температуре -15°С добавляли N-метилморфолин (15,25 мл) и этилхлорформиат (12,60 мл). После перемешивания смеси при -15°С в течение 15 минут отфильтровывали образовавшееся нерастворимое вещество. К фильтрату при температуре -15°С добавляли раствор боргидрида натрия (3,23 г) в воде (32 мл), и смесь перемешивали при температуре -15°С в течение 1 часа. Затем добавляли насыщенный водный раствор хлорида аммония и смесь перемешивали при комнатной температуре в течение 30 минут. К раствору добавляли этилацетат и отделяли органический слой. Органический слой промывали 0,5 N водным раствором бисульфата калия, водой, 0,5 N водным раствором гидроксида натрия и насыщенным раствором хлорида натрия; и затем сушили над безводным сульфатом натрия. Затем при пониженном давлении отгоняли растворитель. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: гексан/этилацетат) с получением указанного в заголовке соединения в виде светло-желтого маслянистого вещества (11,93 г).
(Стадия 2)
Синтез (S)-N-(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5]пирроло[1,2-a]азепин-9-ил)акриламида
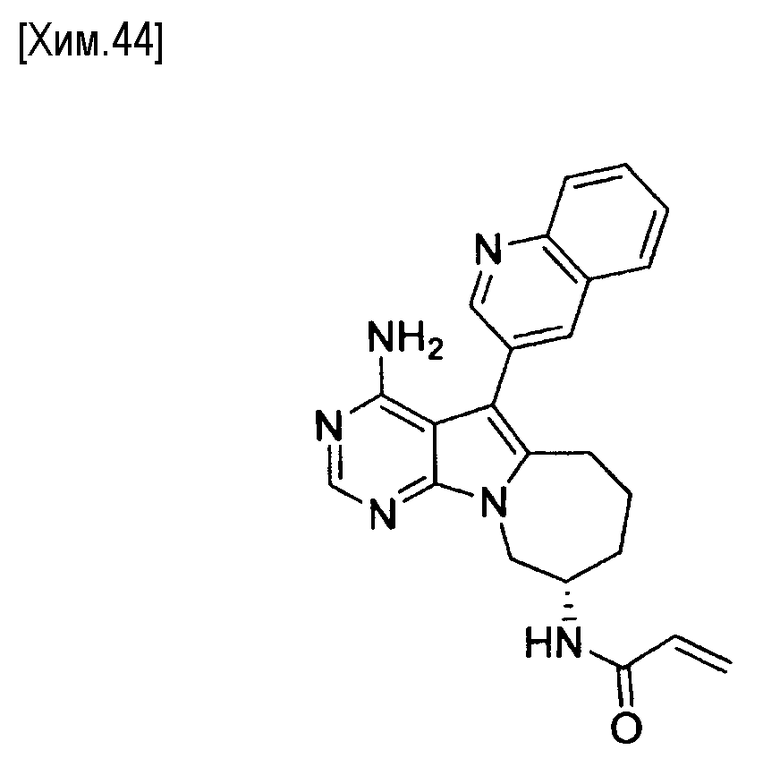
Указанное в заголовке соединение получали способом в соответствии со стадиями 4-10 Примера 7, за исключением того, что вместо (R)-трет-бутил(5-гидроксипент-1-ен-3-ил)карбамата, полученного на стадии 4 примера 7, в данном случае использовали (S)-трет-бутил-(1-гидроксипент-4-ен-2-ил)карбамат, полученный на стадии 1, и указанное соединение получали в виде молочно-белого твердого вещества (400,0 мг).
1H-ЯМР (DMSO-d6) δ: 1,57-1,65 (1H, м), 1,78-1,86 (1H, м), 1,93-2,05 (2H, м), 2,77-2,89 (2H, м), 3,98-4,04 (1H, м), 4,21-4,26 (1Н, м), 4,63 (1H, д, J=13,7 Гц), 5,60 (1Н, дд, J=10,0, 2,4 Гц), 5,93 (1H, ш.с), 6,12 (1H, дд, J=17,1, 2,4 Гц), 6,25 (1Н, дд, J=17,1, 10,0 Гц), 7,63-7,67 (1H, м), 7,77-7,81 (1Н, м), 8,07 (1H, т, J=8,8 Гц), 8,12 (1Н, с), 8,15 (1H, д, J=7,6 Гц), 8,28 (1H, д, J=2,2 Гц), 8,87 (1H, д, J=2,2 Гц).
ESI-MS m/z 399 (MH+).
Пример 9
(R)-N-(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5]пирроло[1,2-a]азепин-9-ил)акриламид (Соединение I-9)
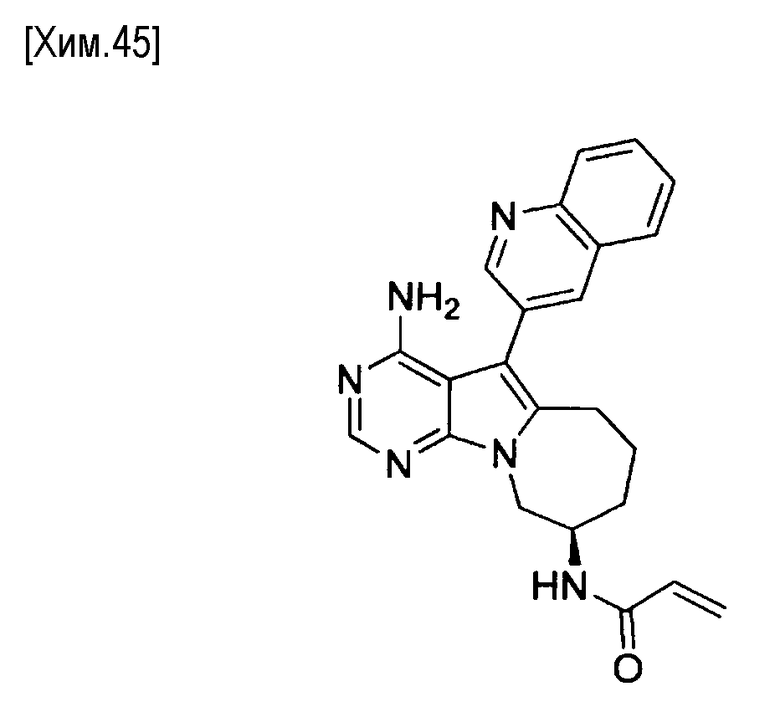
(Стадия 1)
Синтез (R)-трет-бутил-(1-гидроксипент-4-ен-2-ил)карбамата
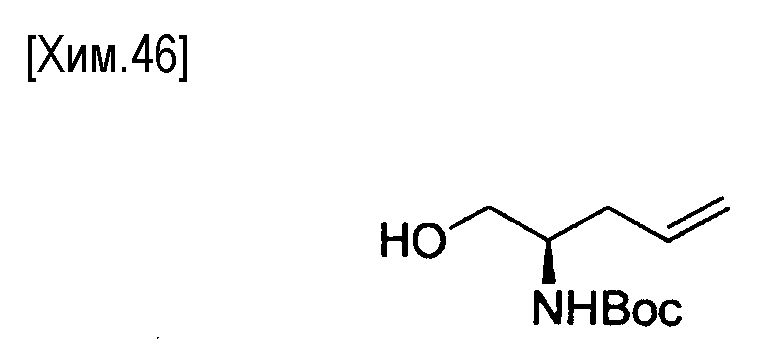
Указанное в заголовке соединение получали способом в соответствии со стадией 1 Примера 8, за исключением того, что вместо (S)-2-((трет-бутоксикарбонил)амино)пент-4-еновой кислоты, использованной на стадии 1 примера 8, в данном случае использовали (R)-2-((трет-бутоксикарбонил)амино)пент-4-еновую кислоту, и указанное соединение получали в виде светло-желтого маслянистого вещества.
(Стадия 2)
Синтез (R)-N-(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5]пирроло[1,2-a]азепин-9-ил)акриламида
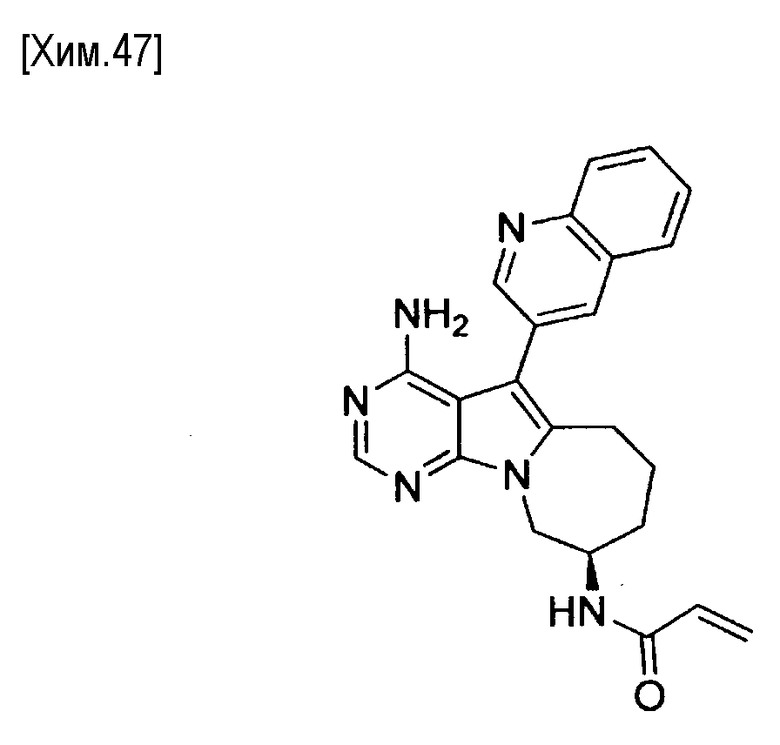
Указанное в заголовке соединение получали способом в соответствии со стадиями 4-10 Примера 7, за исключением того, что вместо (R)-трет-бутил (5-гидроксипент-1-ен-3-ил)карбамата, используемого на стадии 4 Примера 7, в данном случае использовали (R)-трет-бутил-(1-гидроксипент-4-ен-2-ил)карбамат, полученный на стадии 1, и указанное соединение получали в виде молочно-белого твердого вещества (115,5 мг).
1H-ЯМР (DMSO-d6) δ: 1,57-1,65 (1H, м), 1,78-1,86 (1H, м), 1,93-2,05 (2H, м), 2,77-2,89 (2H, м), 3,98-4,04 (1H, м), 4,21-4,26 (1Н, м), 4,63 (1H, д, J=13,7 Гц), 5,60 (1Н, дд, J=10,0, 2,4 Гц), 5,93 (1H, ш.с), 6,12 (1H, дд, J=17,1, 2,4 Гц), 6,25 (1Н, дд, J=17,1, 10,0 Гц), 7,63-7,67 (1H, м), 7,77-7,81 (1Н, м), 8,07 (1H, т, J=8,8 Гц), 8,12 (1Н, с), 8,15 (1H, д, J=7,6 Гц), 8,28 (1H, д, J=2,2 Гц), 8,87 (1H, д, J=2,2 Гц).
ESI-MS m/z 399 (MH+).
Сравнительный пример 1
Синтез N-(3-(4-амино-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)фенил)бензамида
Синтез проводили в соответствии со способом, описанным в WO 2006/102079.
ESI-MS m/z 384 (MH+).
Способы синтеза промежуточных соединений для получения соединений настоящего изобретения описаны ниже. Эти методы не ограничиваются представленными здесь вариантами выполнения.
Ссылочный пример 1
(S)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамат
(Стадия 1)
Синтез (S)-трет-бутил-(1-(4-хлор-5-иод-7H-пирроло[2,3-d]пиримидин-7-ил бут-3-ен-2-ил)карбамата
К раствору трифенилфосфина (13,1 г) в тетрагидрофуране (70 мл) при охлаждении льдом медленно добавляли диизопропилазодикарбоксилат (2,44 мл). Реакционную смесь перемешивали при охлаждении льдом в течение 1 часа, и добавляли раствор (S)-трет-бутил-(1-гидроксибут-3-ен-2-ил)карбамата (7,0 г), синтезированного в соответствии с методом, описанным в непатентной литературе Org. Lett., 2005, vol. 7, No. 5, pp. 847-849, и затем к полученной реакционной смеси медленно добавляли 4-хлор-5-иод-7H-пирроло[2,3-d]пиримидин (6,97 г) в тетрагидрофуране (35 мл). После перемешивания реакционной смеси при комнатной температуре в течение 2 часов, отгоняли растворитель при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: гексан/этилацетат) с получением указанного в заголовке соединения (20,84 г) в виде светло-желтого маслянистого вещества.
ESI-MS m/z 448,450 (MH+).
(Стадия 2)
Синтез (S)-трет-бутил-(1-(4-амино-5-иод-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата
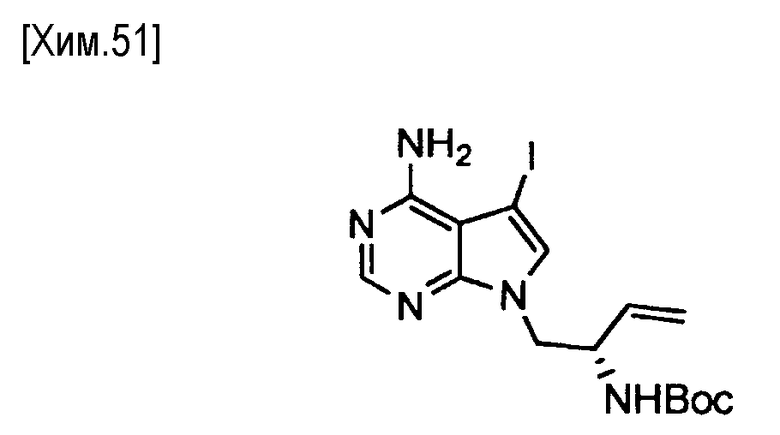
К (S)-трет-бутил-(1-(4-хлор-5-иод-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамату (20,84 г), полученному на стадии 1, добавляли 8 N раствор аммиака в метаноле (89,4 мл) и смесь перемешивали в автоклаве при температуре 120°С в течение 6 часов. Реакционную смесь охлаждали льдом и отгоняли растворитель при пониженном давлении. После этого полученный остаток разбавляли небольшим количеством метанола, этот осадок собирали фильтрованием, промывали холодным метанолом (11 мл), а затем сушили при пониженном давлении с получением указанного в заголовке соединения (8,28 г) в виде молочно-белого твердого вещества.
ESI-MS m/z 430 (MH+).
(Стадия 3)
Синтез (S)-трет-бутил-(1-(4-амино-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата
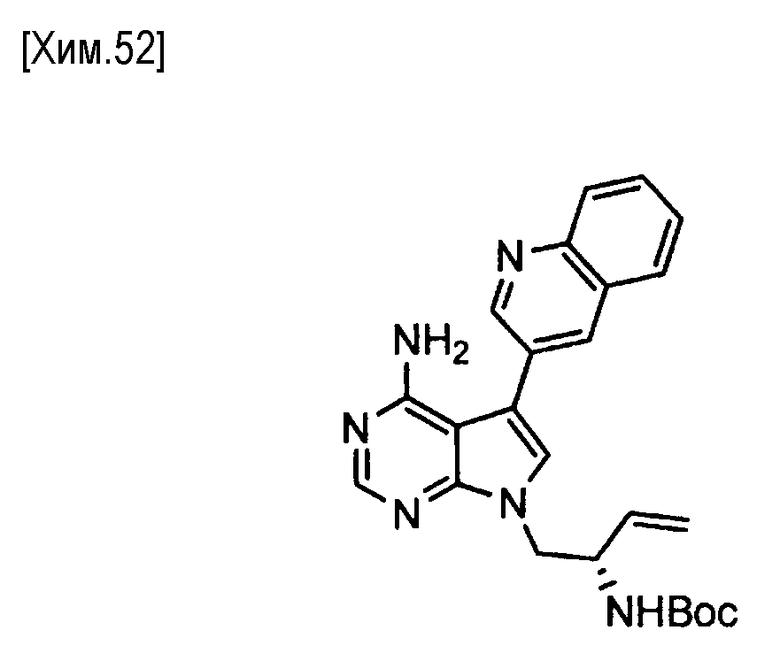
Смесь (S)-трет-бутил-(1-(4-амино-5-иод-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата (8,26 г), полученного на стадии 2, 3-хинолинбороновой кислоты (4,99 г), карбоната цезия (12,54 г), дихлорида 1,1′-бис(дифенилфосфино)ферроцен-палладия (II) (785,6 мг), DME (66 мл) и воды (33 мл) перемешивали в атмосфере азота при 100°С в течение 2 часов. После охлаждения реакционной смеси к ней добавляли воду и этилацетат, и отделяли органический слой. Затем водный слой дважды экстрагировали этилацетатом. Полученный органический слой сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: гексан/этилацетат, этилацетат/метанол) с получением указанного в заголовке соединения (8,0 г) в виде светло-оранжевого твердого вещества.
ESI-MS m/z 431 (MH+).
(Стадия 4)
Синтез (S)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)- 7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата
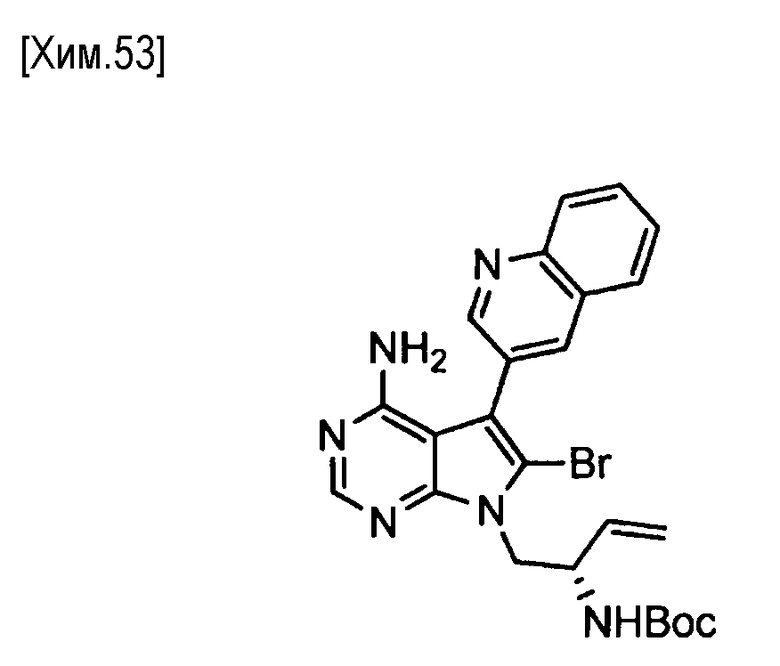
К раствору (S)-трет-бутил-(1-(4-амино-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата (7,98 г), полученного на стадии 3, в DMF (64 мл) при температуре -15°С добавляли N-бромсукцинимид (3,63 г), и смесь перемешивали при температуре -15°С в течение 1 часа. К реакционной смеси добавляли 10% водный раствор тиосульфата натрия и этилацетат, и смесь перемешивали при комнатной температуре в течение 10 минут. Органический слой отделяли, и водный слой дважды экстрагировали этилацетатом. Полученный органический слой дважды промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: этилацетат/метанол) с получением указанного в заголовке соединения (6,30 г) в виде светло-коричневого твердого вещества.
1Н-ЯМР (CDCl3) δ: 1,26 (9Н, с), 4,35-4,39 (1H, м), 4,50-4,56 (1H, м), 4,72 (1H, ш.с), 4,92 (1H, ш.с), 5,26 (2H, д, J=10,5 Гц), 5,33-5,39 (1H, м), 5,92 (1H, дд, J=17,2, 10,6, 5,4 Гц), 7,63-7,67 (1H, м), 7,79-7,83 (1H, м), 7,90-7,92 (1H, м), 8,19 (1H, д, J=8,3 Гц), 8,27 (1H, д, J=1,7 Гц), 8,35 (1H, с), 9,07 (1H, д, J=2,2 Гц).
ESI-MS m/z 509,511 (MH+).
Ссылочный пример 2
(R)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамат
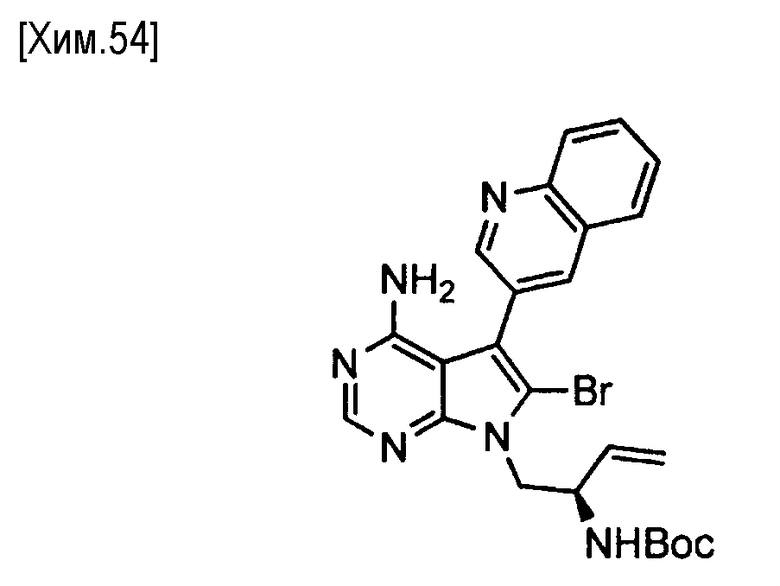
(Стадия 1)
Синтез (R)-трет-бутил-(1-(4-хлор-5-иод-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата
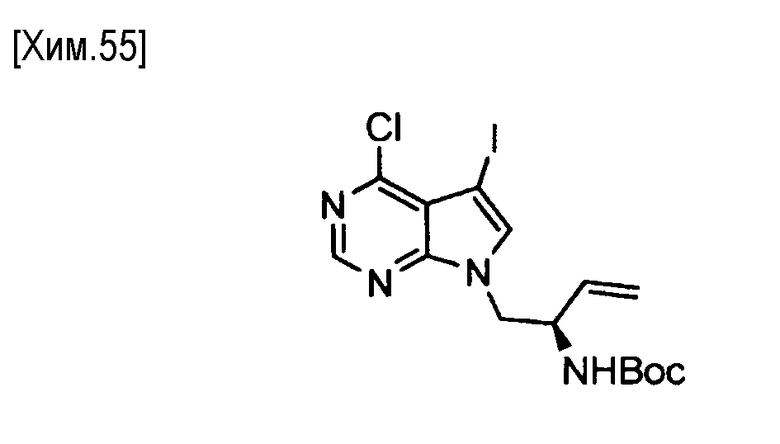
Указанное в заголовке соединение получали способом в соответствии со стадией 1 Ссылочного примера 1, за исключением того, что вместо (S)-трет-бутил(1-гидроксибут-3-ен-2-ил)карбамата, полученного на стадии 1 в Ссылочном примере 1, в данном случае использовали (R)-трет-бутил-(1-гидроксибут-3-ен-2-ил)карбамат (8,74 г), и указанное соединение получали в виде белого твердого вещества (11,05 г).
ESI-MS m/z 448,450 (MH+).
(Стадия 2)
Синтез (R)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата
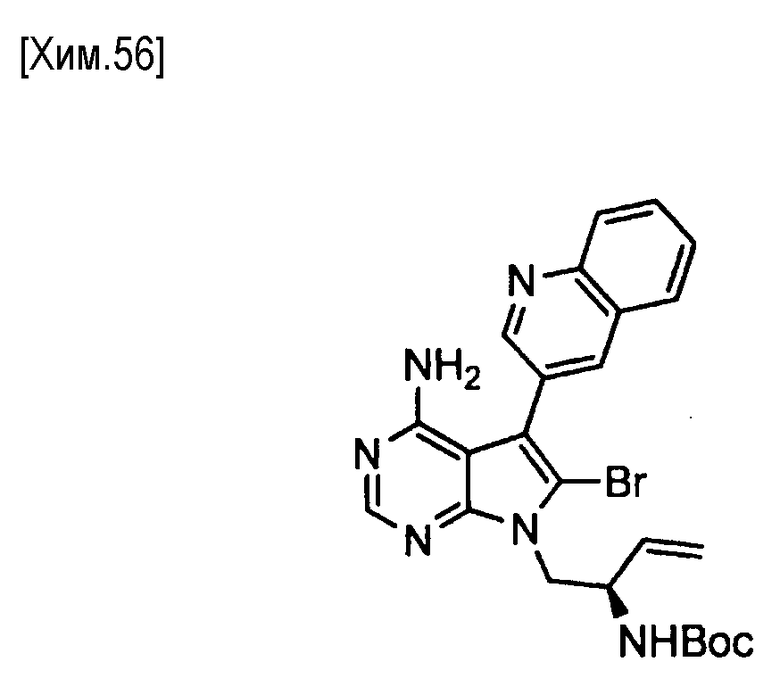
Указанное в заголовке соединение получали способом в соответствии со стадиями 2-4 Ссылочного примера 1, за исключением того, что вместо (S)-трет-бутил-(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата, полученного на стадии 2 в Ссылочном примере 1, в данном случае использовали (R)-трет-бутил-(1-(4-хлор-5-иод-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамат (7,88 г), и указанное соединение получали в виде желтого твердого вещества (6,80 г).
1Н-ЯМР (CDCl3) δ: 1,26 (9Н, с), 4,35-4,39 (1H, м), 4,50-4,56 (1H, м), 4,72 (1H, ш.с), 4,92 (1H, ш.с), 5,26 (2H, д, J=10,5 Гц), 5,33-5,39 (1H, м), 5,92 (1H, дд, J=17,2, 10,6, 5,4 Гц), 7,63-7,67 (1H, м), 7,79-7,83 (1H, м), 7,90-7,92 (1H, м), 8,19 (1H, д, J=8,3 Гц), 8,27 (1H, д, J=1,7 Гц), 8,35 (1H, с), 9,07 (1H, д, J=2,2 Гц).
ESI-MS m/z 509,511 (MH+).
Ссылочный пример 3
(S)-трет-Бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)пента-4-ен-2-ил)карбамат
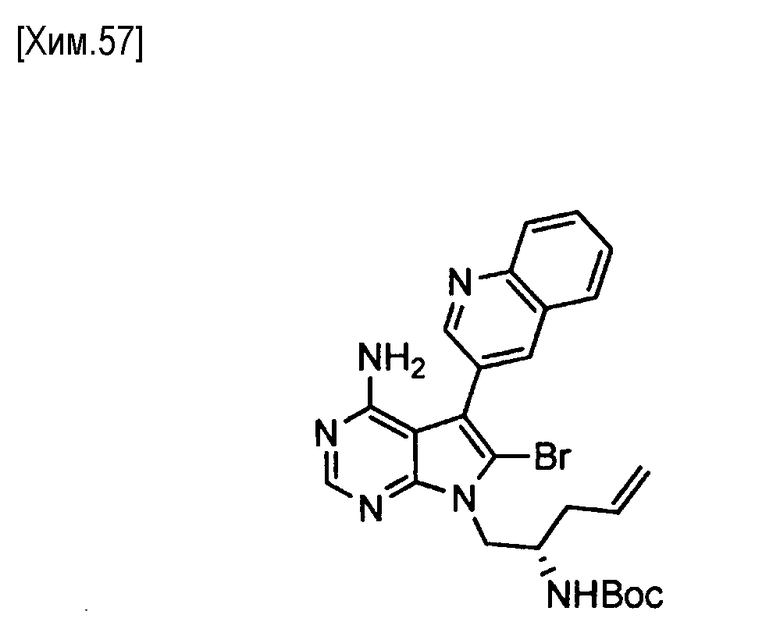
(Стадия 1)
Синтез (S)-трет-бутил-(1-(4-хлор-5-иод-7H-пирроло[2,3-d]пиримидин-7-ил)пента-4-ен-2-ил)карбамата
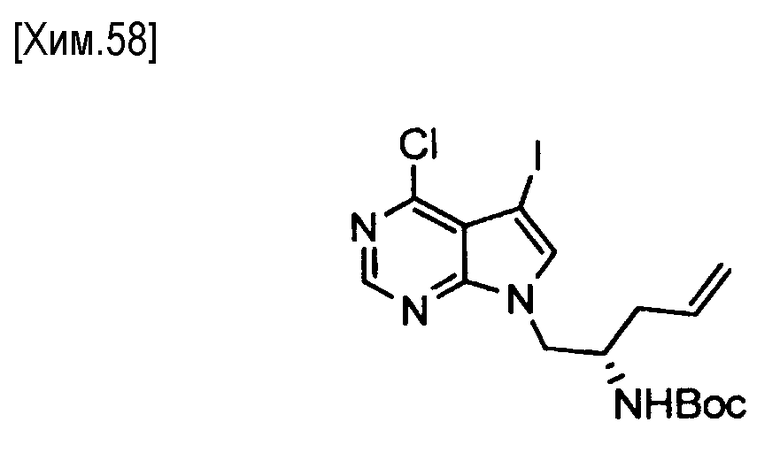
Указанное в заголовке соединение получали способом в соответствии со стадией 1 Ссылочного примера 1, за исключением того, что вместо (S)-трет-бутил(1-гидроксибут-3-ен-2-ил)карбамата, полученного на стадии 1 Ссылочного примера 1, в данном случае использовали (S)-трет-бутил-(1-гидроксипента-4-ен-2-ил)карбамат (11,93 г), и указанное соединение получали в виде желто-коричневого маслянистого вещества (4,96 г).
1Н-ЯМР (CDCl3) δ: 1,35 (9Н, с), 2,18-2,35 (2H, м), 3,97-4,05 (1H, м), 4,27-4,33 (1H, м), 4,40-4,45 (1H, м), 4,63-4,65 (1H, м), 5,14-5,19 (2H, м), 5,76-5,86 (1H, м), 7,42 (1H, ш.с), 8,62 (1H, с).
ESI-MS m/z 462,464 (MH+).
(Стадия 2)
Синтез (S)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)пента-4-ен-2-ил)карбаматп
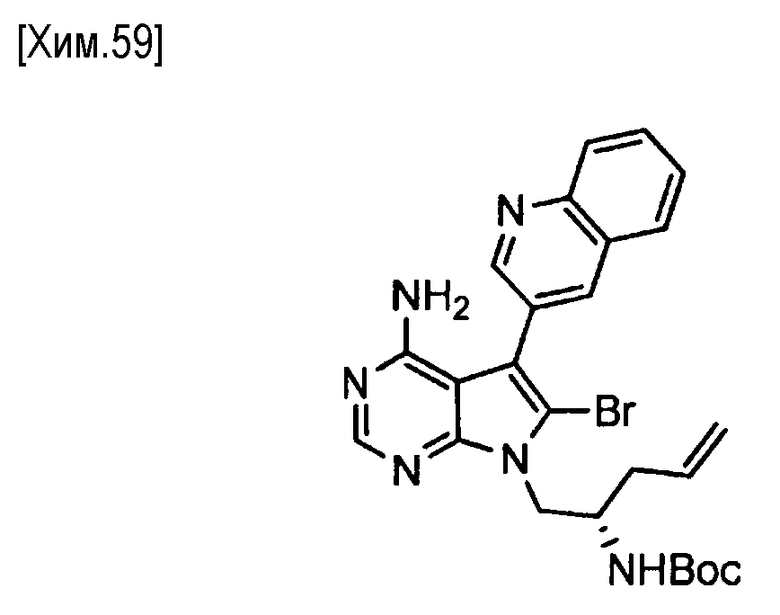
Указанное в заголовке соединение получали способом в соответствии со стадиями 2-4 Ссылочного примера 1, за исключением того, что вместо (S)-трет-бутил-(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата, полученного на стадии 2 Ссылочного примера 1, в данном случае использовали (S)-трет-бутил-(1-(4-хлор-5-иод-7H-пирроло[2,3-d]пиримидин-7-ил)пента-4-ен-2-ил)карбамат (4,90 г), полученный на стадии 1, и указанное соединение получали в виде светло-желтого твердого вещества (3,67 г).
1Н-ЯМР (CDCl3) δ: 1,23 (9Н, с), 2,39-2,42 (2H, м), 4,19-4,27 (1H, м), 4,29-4,34 (1H, м), 4,43-4,50 (1H, м), 4,92 (2H, ш.с), 5,04 (1H, д, J=8,5 Гц), 5,18-5,24 (2H, м), 5,86-5,96 (1H, м), 7,63-7,67 (1H, м), 7,79-7,83 (1H, м), 7,90-7,92 (1H, м), 8,19 (1H, д, J=8,5 Гц), 8,27 (1H, д, J=1,5 Гц), 8,34 (1H, с), 9,07 (1H, д, J=2,0 Гц).
ESI-MS m/z 523,525 (MH+).
Ссылочный пример 4
(R)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)пента-4-ен-2-ил)карбамат
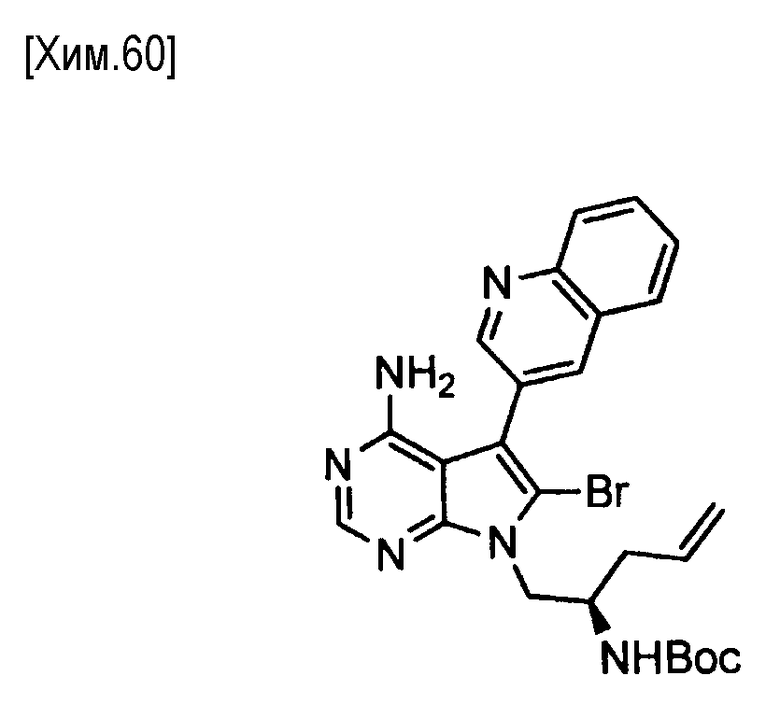
(Стадия 1)
Синтез (R)-трет-бутил-(1-(4-хлор-5-иод-7H-пирроло[2,3-d]пиримидин-7-ил)пента-4-ен-2-ил)карбамата
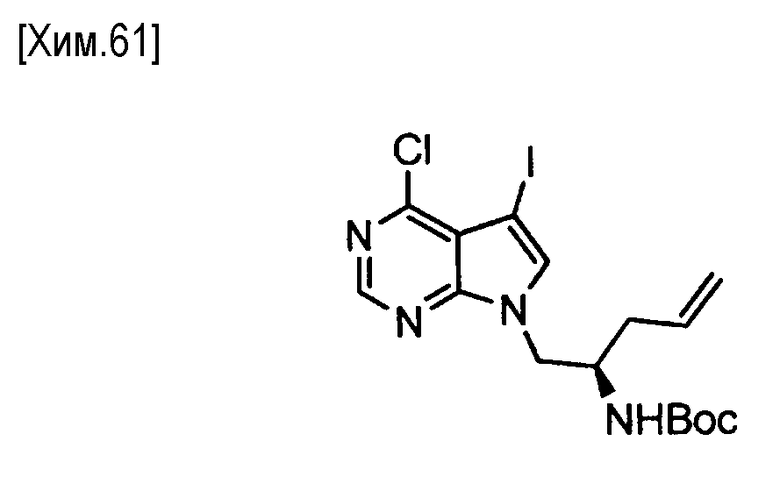
Указанное в заголовке соединение получали способом в соответствии со стадией 1 Ссылочного примера 1, за исключением того, что вместо (S)-трет-бутил-(1-гидроксибут-3-ен-2-ил)карбамата, используемого на стадии 1 Ссылочного примера 1, в данном случае использовали (R)-трет-бутил-(1-гидроксипента-4-ен-2-ил)карбамат (856,4 мг), и указанное соединение получали в виде молочно-белого твердого вещества (1,54 г).
1Н-ЯМР (CDCl3) δ: 1,35 (9Н, с), 2,18-2,35 (2H, м), 3,97-4,05 (1H, м), 4,27-4,33 (1H, м), 4,40-4,45 (1H, м), 4,63-4,65 (1H, м), 5,14-5,19 (2H, м), 5,76-5,86 (1H, м), 7,42 (1H, ш.с), 8,62 (1H, с).
ESI-MS m/z 462,464 (MH+).
(Стадия 2)
Синтез (R)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)пента-4-ен-2-ил)карбамата
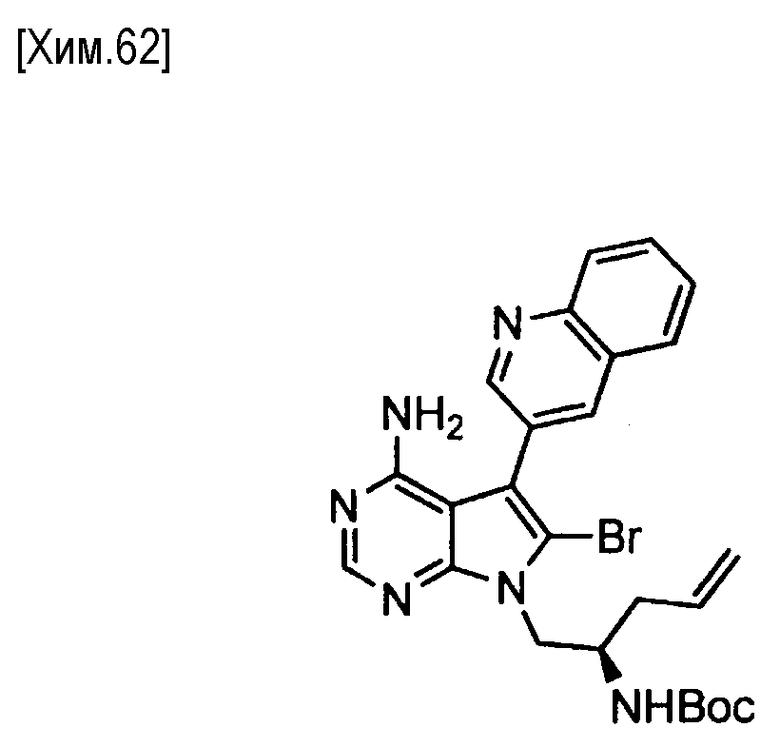
Указанное в заголовке соединение получали способом в соответствии со стадиями 2-4 Ссылочного примера 1, за исключением того, что вместо (S)-трет-бутил-(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата, полученного на стадии 2 Ссылочного примера 1, в данном случае использовали (R)-трет-бутил-(1-(4-хлор-5-иод-7H-пирроло[2,3-d]пиримидин-7-ил)пента-4-ен-2-ил)карбамат (974,9 мг), полученный на стадии 1, и указанное соединение получали в виде светло-коричневого твердого вещества (1,02 г).
1Н-ЯМР (CDCl3) δ: 1,23 (9Н, с), 2,39-2,42 (2H, м), 4,19-4,27 (1H, м), 4,29-4,34 (1H, м), 4,43-4,50 (1H, м), 4,92 (2H, ш.с), 5,04 (1H, д, J=8,5 Гц), 5,18-5,24 (2H, м), 5,86-5,96 (1H, м), 7,63-7,67 (1H, м), 7,79-7,83 (1H, м), 7,90-7,92 (1H, м), 8,19 (1H, д, J=8,5 Гц), 8,27 (1H, д, J=1,5 Гц), 8,34 (1H, с), 9,07 (1H, д, J=2,0 Гц).
ESI-MS m/z 523,525 (MH+).
Ссылочный пример 5
(R)-трет-бутил-(5-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)пента-1-ен-3-ил)карбамат
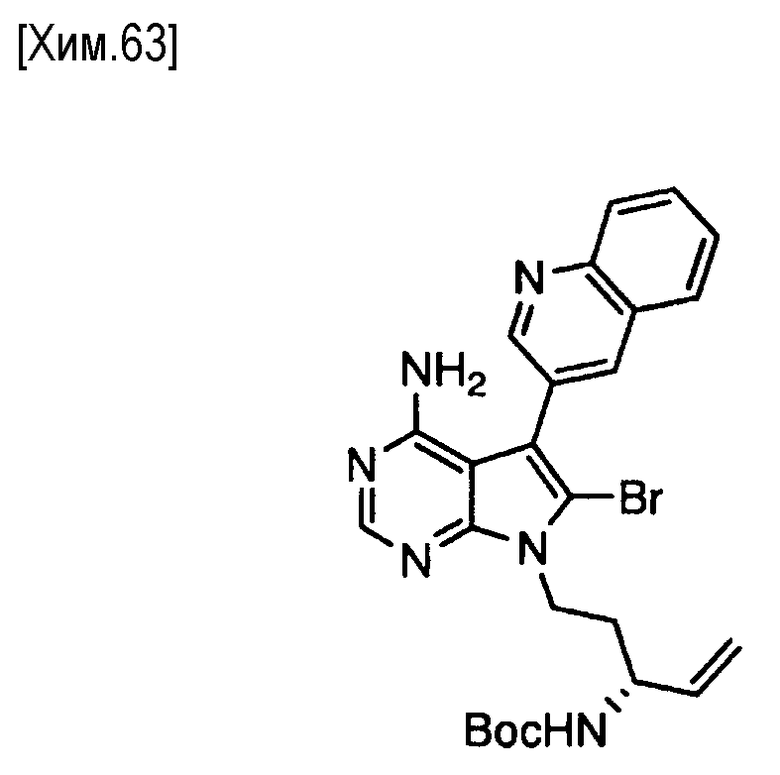
(Стадия 1)
Синтез (R)-трет-бутил-(5-(4-хлор-5-иод-7H-пирроло[2,3-d]пиримидин-7-ил)пента-1-ен-3-ил)карбамата
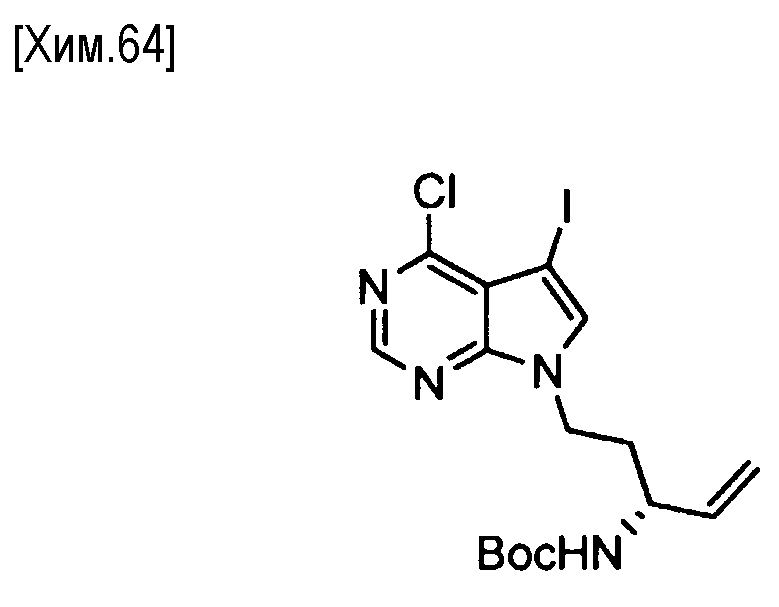
Указанное в заголовке соединение получали способом в соответствии со стадией 1 Ссылочного примера 1, за исключением того, что вместо (S)-трет-бутил(1-гидроксибут-3-ен-2-ил)карбамата, полученного на стадии 1 Ссылочного примера 1, в данном случае использовали (S)-трет-бутил-5-(гидроксипента-1-ен-3-ил)карбамат (2,5 г), и указанное соединение получали в виде светло-желтого твердого вещества (3,49 г).
ESI-MS m/z 463,465 (MH+).
(Стадия 2)
Синтез (R)-трет-бутил(5-(4-амино-6-бром-5-(хинолин-3-ил)- 7H-пирроло[2,3-d]пиримидин-7-ил)пента-1-ен-3-ил)карбамата
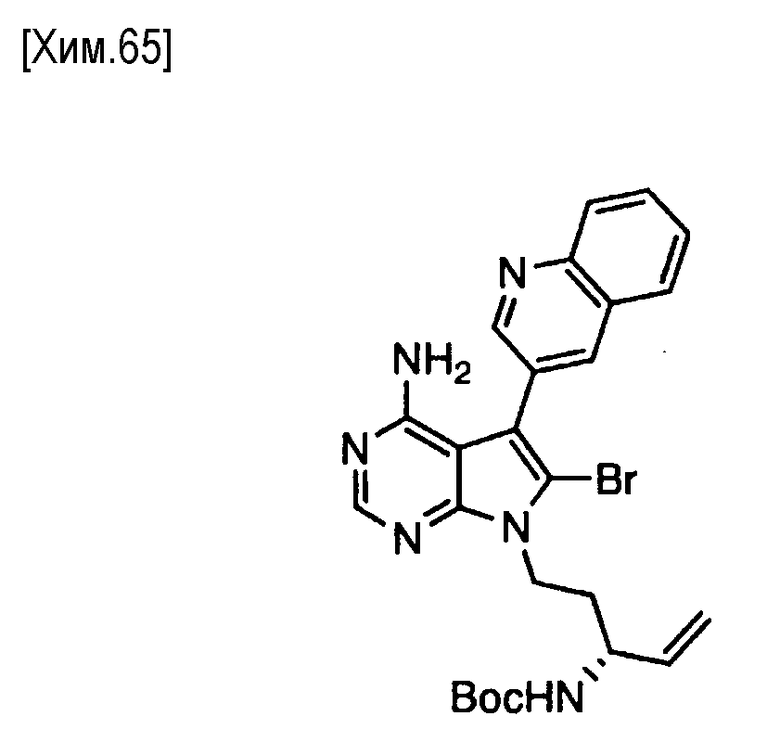
Указанное в заголовке соединение получали способом в соответствии со стадиями 2-4 Ссылочного примера 1, за исключением того, что вместо (S)-трет-бутил-(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата, полученного на стадии 2 Ссылочного примера 1, в данном случае использовали (R)-трет-бутил-(5-(4-хлор-5-иод-7H-пирроло[2,3-d]пиримидин-7-ил)пента-1-ен-3-ил)карбамат (3,21 г), и указанное соединение получали в виде светло-коричневого твердого вещества (3,15 г).
1Н-ЯМР (CDCl3) δ: 1,46 (9H, с), 2,02-2,21 (2H, м), 4,26-4,53 (3H, м), 4,90 (2H, ш.с), 5,07 (1H, д, J=12,4 Гц), 5,15 (1H, д, J=17,2 Гц), 5,15-5,23 (1H, м), 5,78 (1H, дд, J=17,2, 12,4, 5,2 Гц), 7,61-7,67 (1H, м), 7,78-7,83 (1H, м), 7,88-7,93 (1H, м), 8,17-8,21 (1H, м), 8,26 (1H, д, J=2,2 Гц), 8,35 (1H, с), 9,06 (1H, д, J=2,2 Гц).
ESI-MS m/z 523,525 (MH+).
Ссылочный пример 6
(R)-6-бром-7-(2-((трет-бутилдиметилсилил)окси)бут-3-ен-1-ил)-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амин
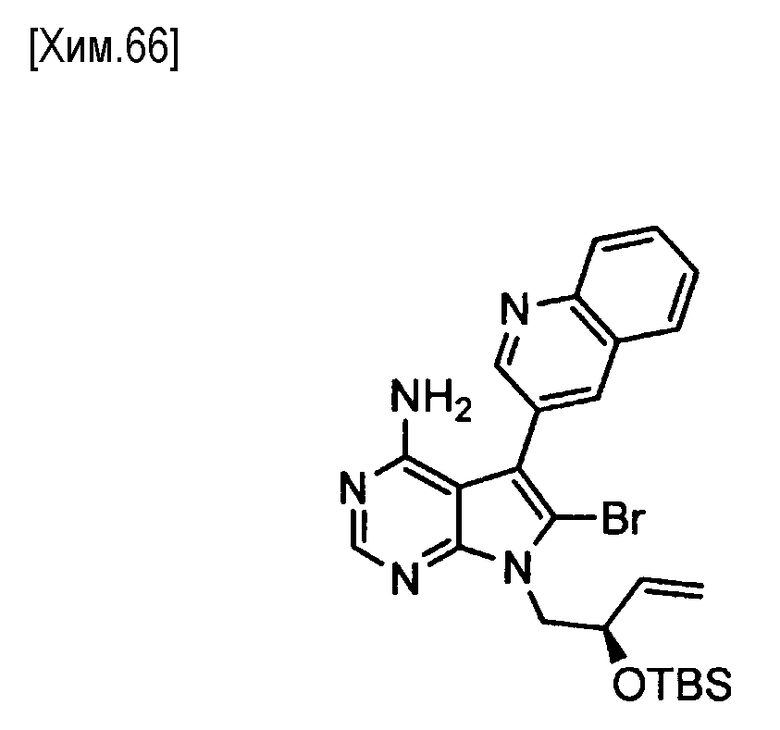
(Стадия 1)
Синтез 4-хлор-5-иод-7-((2-(триметилсилил)этокси)метил)-7H-пирроло[2,3-d]пиримидина
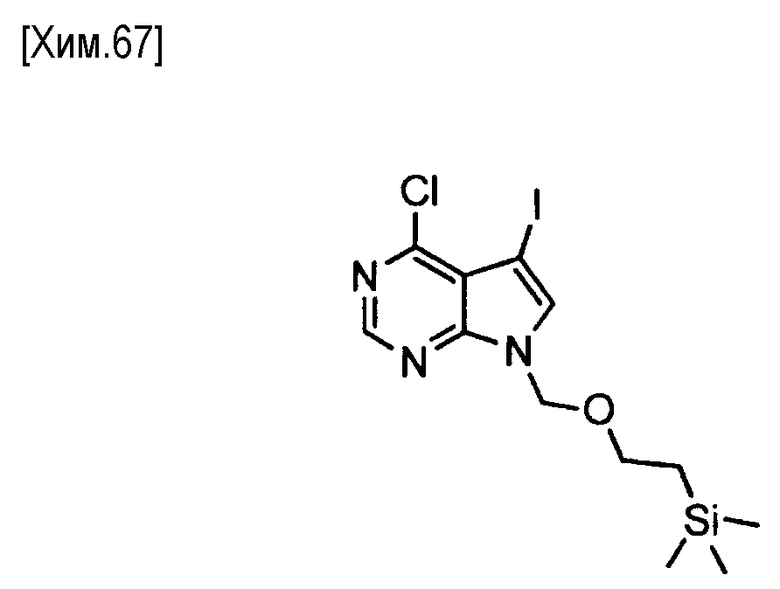
К раствору гидрида натрия (3,4 г) в DMFA (190 мл) при охлаждении льдом медленно добавляли раствор 4-хлор-5-иод-7H-пирроло[2,3-d]пиримидина (20,0 г) в DMF (50 мл). После этого добавляли 2-(триметилсилил)этоксиметилхлорид (13,3 мл) и перемешивали при той же температуре в течение 2 часов. К реакционной смеси дополнительно добавляли 2-(триметилсилил)этоксиметилхлорид (1,3 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь выливали в воду (600 мл) и перемешивали при комнатной температуре в течение 15 минут. Полученный осадок собирали фильтрованием и промывали водой и диизопропиловым эфиром с последующим растворением в этилацетате. Нерастворимое вещество отфильтровывали. Растворитель из фильтрата отгоняли при пониженном давлении. К полученному остатку добавляли гептан, и осадок собирали фильтрованием. Осадок промывали гептаном и сушили при пониженном давлении с получением указанного в заголовке соединения в виде белого твердого вещества (21,2 г).
ESI-MS m/z 409,411 (MH+).
(Стадия 2)
Синтез 5-иод-7-((2-(триметилсилил)этокси)метил)-7H-пирроло[2,3-d]пиримидин-4-амина
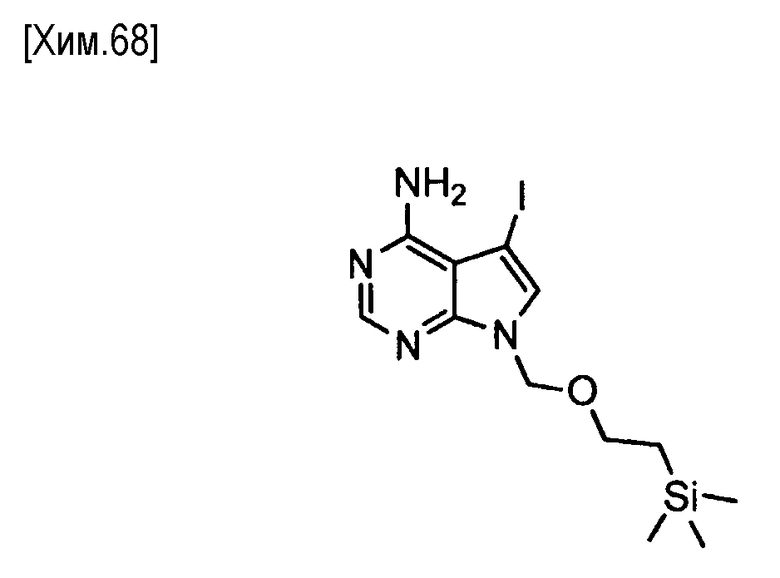
К 4-хлор-5-иод-7-((2-(триметилсилил)этокси)метил)-7H-пирроло[2,3-d]пиримидину (20,0 г), полученному на стадии 1, добавляли 8 N раствор аммиака в метаноле (120 мл), и смесь перемешивали в микроволновом реакторе при температуре 120°С в течение 1 часа. После охлаждения реакционной смеси ее разбавляли метанолом (65 мл) и водой (185 мл). Полученный осадок собирали фильтрованием, промывали водой и сушили при пониженном давлении, получая указанное в заголовке соединение в виде белого твердого вещества (15,2 г).
ESI-MS m/z 391 (MH+).
(Стадия 3)
Синтез 5-(хинолин-3-ил)-7-((2-(триметилсилил)этокси)метил)-7H-пирроло[2,3-d]пиримидин-4-амина
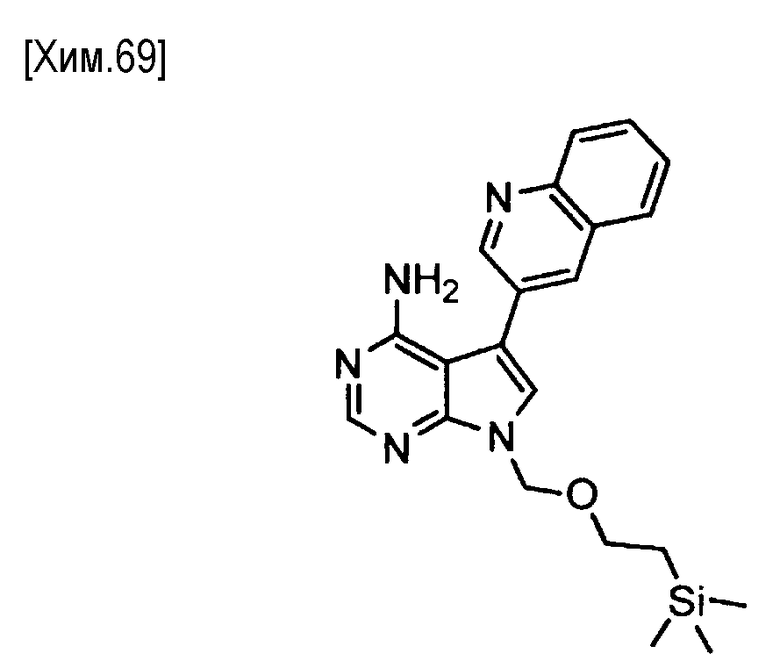
К раствору 5-иод-7-((2-(триметилсилил)этокси)метил)-7H-пирроло[2,3-d]пиримидин-4-амина (15,0 г), полученного на стадии 2, 3-хинолинбороновой кислоты (8,6 г) и тетракистрифенилфосфинпалладию (0) (2,2 г) в DME (270 мл) добавляли 2 М водный раствор карбоната натрия (38 мл), и перемешивали в атмосфере азота при 90°С в течение 6 часов. После этого реакционную смесь охлаждали и добавляли к ней воду (300 мл). Полученный осадок собирали фильтрованием, затем промывали водой и диизопропиловым эфиром и сушили при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: метанол/хлороформ) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (10,17 г).
ESI-MS m/z 392 (MH+).
(Стадия 4)
Синтез гидрохлорида 5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амина
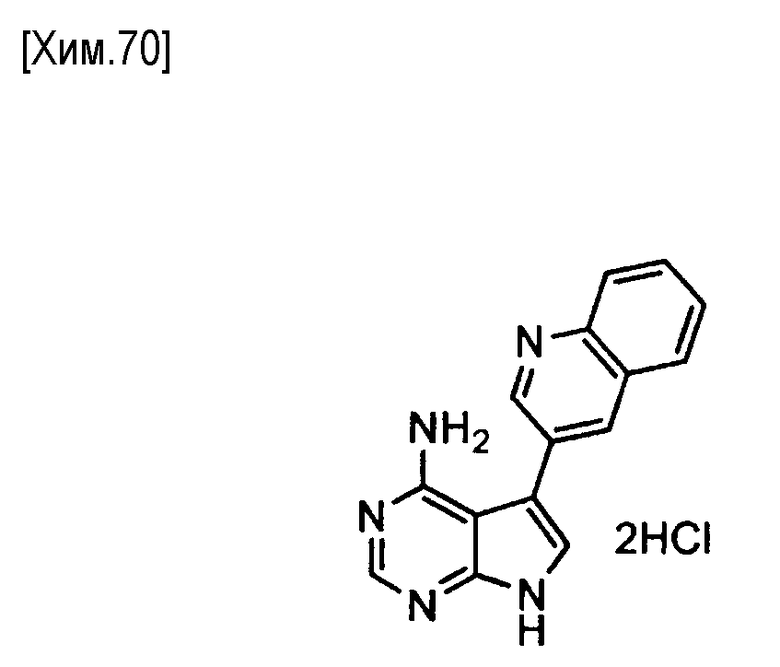
К раствору 5-(хинолин-3-ил)-7-((2-(триметилсилил)этокси)метил)-7H-пирроло[2,3-d]пиримидин-4-амина (10,0 г), полученного на стадии 3, в этаноле (200 мл) при температуре 90°C добавляли концентрированную хлористоводородную кислоту (20 мл), и смесь перемешивали при той же температуре в течение 25 минут. Затем дополнительно добавляли концентрированную хлористоводородную кислоту (30 мл), и смесь перемешивали при той же температуре в течение 75 минут. После охлаждения реакционной смеси к ней добавляли этанол (100 мл) и смесь перемешивали при температуре 95°С в течение 90 минут. После этого к смеси добавляли этанол (100 мл) и концентрированную хлористоводородную кислоту (25 мл), и смесь перемешивали при той же температуре в течение 4 дней. После охлаждения реакционной смеси к ней добавляли этилацетат. Полученный осадок собирали фильтрованием, промывали этилацетатом и сушили при пониженном давлении с получением указанного в заголовке соединения в виде желтого твердого вещества (4,4 г).
ESI-MS m/z 335 (MH+).
(Стадия 5)
Синтез (R)-7-(2-((трет-бутилдиметилсилил)окси)бут-3-ен-1-ил)-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амина
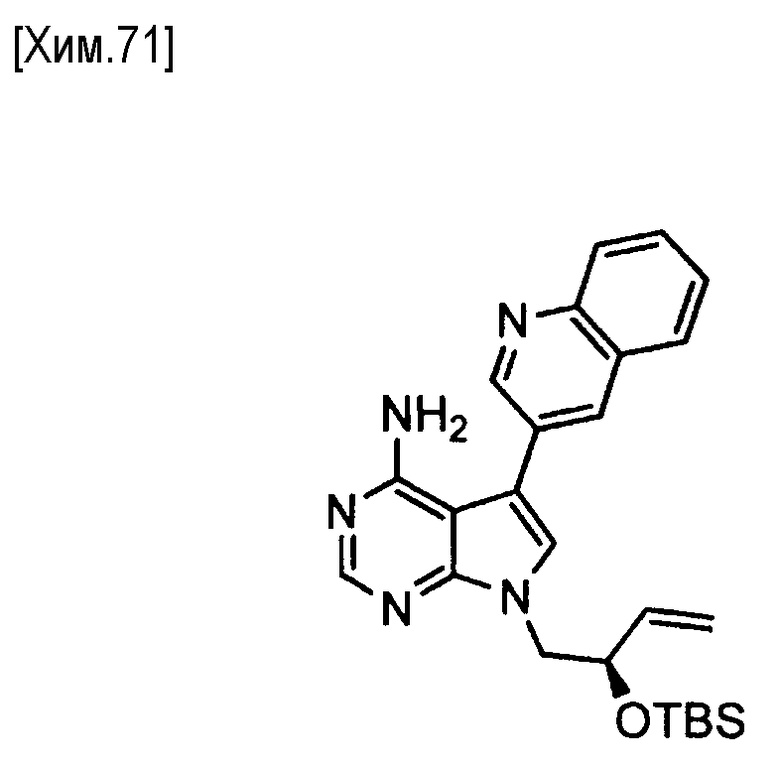
К раствору 5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амина (1,22 г), полученного на стадии 4, в DMF (12,2 мл) при комнатной температуре добавляли карбонат калия (4,0 г) и (R)-2-((трет-бутилдиметилсилил)окси)бут-3-ен-1-ил-4-метилбензолсульфонат (1,43 г), и смесь перемешивали при температуре 90°С в течение 20 часов. После охлаждения реакционной смеси к ней добавляли воду (49 мл), и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Полученный осадок собирали фильтрованием, промывали водой и сушили при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: метанол/этилацетат) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (1,31 г).
1Н-ЯМР (CDCl3) δ: -0,32 (3H, с), -0,11 (3H, с), 0,80 (9Н, с), 4,06 (1Н, дд, J=13,9, 8,5 Гц), 4,46 (1H, дд, J=13,9, 3,2 Гц), 4,59-4,64 (1H, м), 5,06 (2H, ш.с), 5,22 (1H, д, J=10,5 Гц), 5,40 (1H, д, J=16,8 Гц), 5,89-5,97 (1H, м), 7,21 (1H, с), 7,61- 7,65 (1H, м), 7,74-7,78 (1H, м), 7,89 (1H, д, J=8,1 Гц), 8,17 (1H, д, J=8,3 Гц), 8,23 (1H, д, J=2,2 Гц), 8,40 (1H, с), 9,10 (1H, д, J=2,0 Гц).
ESI-MS m/z 446 (MH+).
(Стадия 6)
Синтез (R)-6-бром-7-(2-((трет-бутилдиметилсилил)окси)бут-3-ен-1-ил)-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амина
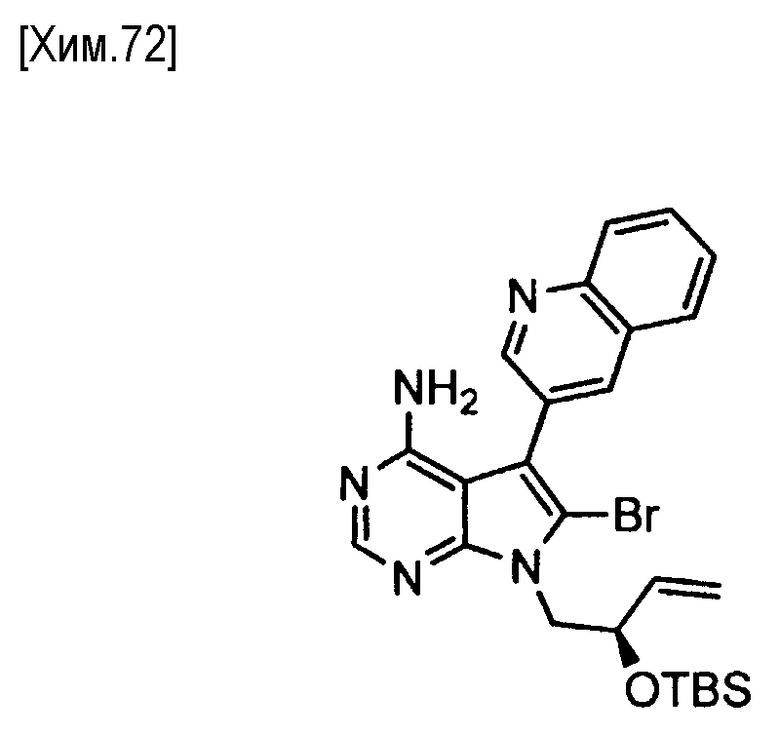
Указанное в заголовке соединение получали способом в соответствии со стадией 4 Ссылочного примера 1, за исключением того, что вместо (S)-трет-бутил-(1-(4-амино-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата, используемого на стадии 4 Ссылочного примера 1, в данном случае использовали (R)-7-(2-((трет-бутилдиметилсилил)окси)бут-3-ен-1-ил)-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амин (1,30 г), полученный на стадии 5, и указанное соединение получали в виде желтого твердого вещества (1,44 г).
1Н-ЯМР (CDCl3) δ: -0,34 (3H, с), -0,12 (3H, с), 0,75 (9Н, с), 4,33-4,40 (2H, м), 4,74-4,79 (1H, м), 4,91 (2H, ш.с), 5,21-5,24 (1H, м), 5,36-5,41 (1H, м), 5,92-6,01 (1H, м), 7,63-7,67 (1H, м), 7,79-7,83 (1H, м), 7,92 (1H, д, J=7,8 Гц), 8,20 (1H, д, J=8,5 Гц), 8,24 (1H, д, J=2,2 Гц), 8,37 (1H, с), 9,06 (1H, д, J=2,2 Гц).
ESI-MS m/z 524,526 (MH+).
Пример 10
(S)-трет-Бутил-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамат
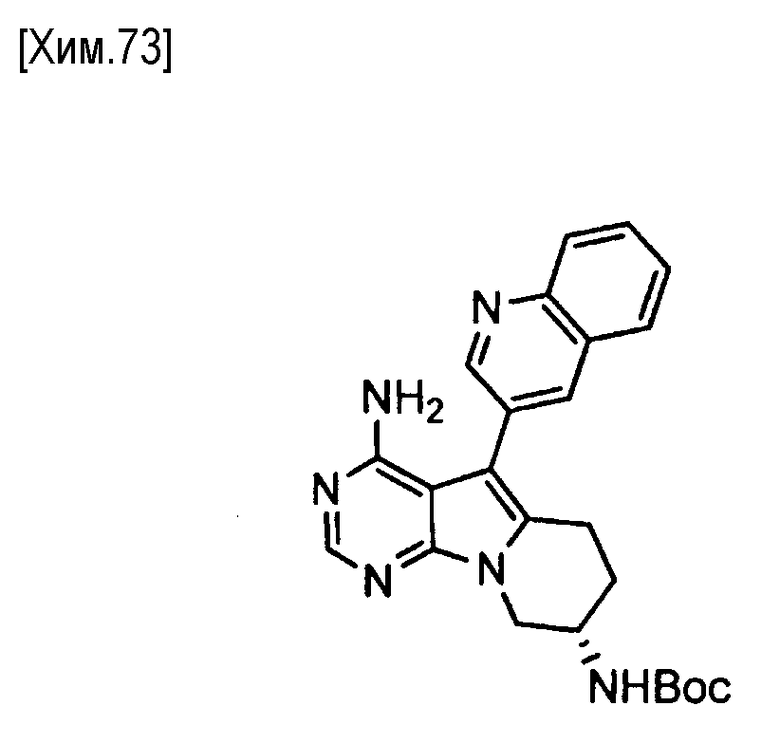
К раствору (S)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата (6,0 г), полученного в Ссылочном примере 1, в тетрагидрофуране (42 мл) при комнатной температуре под атмосферой азота добавляли 0,5 М раствор 9-борабицикло[3.3.1]нонана в тетрагидрофуране (141,3 мл), и полученную смесь перемешивали при комнатной температуре в течение 2 часов. К реакционной смеси при комнатной температуре медленно добавляли 2 N водный раствор гидроксида натрия (84,8 мл), и смесь дегазировали при пониженном давлении. К этой смеси в атмосфере азота добавляли (тетракистрифенилфосфин)палладий (0) (1,70 г), и смесь перемешивали при 66°С в течение 12 часов. После охлаждения реакционной смеси отделяли органический слой и его промывали 20% водным раствором хлорида аммония (60 мл). К органическому слою добавляли силикагель SH (6,0 г), и полученную смесь перемешивали в атмосфере азота при температуре 50°С в течение 14 часов, а затем фильтровали. К фильтрату снова добавляли силикагель SH (изготовитель Fuji Silysia Chemical Ltd) (6,0 г), полученную смесь перемешивали в атмосфере азота при 50°С в течение 14 часов, а затем фильтровали. Растворитель отгоняли из фильтрата при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: этилацетат/метанол) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (4,46 г), (выход: 88%).
1Н-ЯМР (CDCl3) δ: 1,48 (9Н, с), 1,91-2,00 (1H, м), 2,12-2,19 (1H, м), 2,98-3,11 (2H, м), 4,00 (1Н, дд, J=12,7, 7,1 Гц), 4,32 (1H, ш.с), 4,55 (1Н, дд, J=12,7, 4,6 Гц), 4,81-4,83 (1H, м), 4,90 (2H, ш.с), 7,61-7,65 (1Н, м), 7,75-7,80 (1H, м), 7,88 (1H, д, J=8,0 Гц), 8,16-8,18 (2H, м), 8,33 (1Н, с), 9,02 (1H, д, J=2,2 Гц).
ESI-MS m/z 431 (MH+).
Пример 11
К раствору (S)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата (0,3 г), полученного в Ссылочном примере 1, в тетрагидрофуране (4,5 мл) при комнатной температуре в атмосфере азота добавляли димер 9-борабицикло[3.3.1]нонана (0,431 г), и смесь перемешивали при комнатной температуре в течение 2 часов. Затем к реакционной смеси при комнатной температуре медленно добавляли 4 N водный раствор гидроксида натрия (2,12 мл), и дегазировали при пониженном давлении. К полученной смеси добавляли в атмосфере азота (тетракистрифенилфосфин)палладий (0) (0,136 г), и перемешивали при температуре 64°С в течение 12 часов. Затем реакционную смесь охлаждали, разбавляли этилацетатом и добавляли к ней насыщенный водный раствор хлорида аммония. После этого фильтрованием удаляли образовавшиеся на этой стадии нерастворимые продукты, и отделяли органический слой. Полученный органический слой сушили над безводным сульфатом натрия, затем фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: этилацетат/метанол) с получением (S)-трет-бутил (4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо-[5,4-b]индолизин-8-ил)карбамата в виде светло-желтого твердого вещества (203 мг), (выход: 80%).
Пример 12
К раствору (S)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата (300 мг), полученного в Ссылочном примере 1, в 1,2-диметоксиэтане (4,5 мл) при комнатной температуре в атмосфере азота добавляли димер 9-борабицикло[3.3.1]нонана (431 мг), и смесь перемешивали при температуре 48°С в течение 40 минут. После этого смеси давали остыть до комнатной температуры, и к реакционной смеси при комнатной температуре медленно добавляли 4 N водный раствор гидроксида натрия (2,1 мл), и дегазировали при пониженном давлении. К смеси в атмосфере азота добавляли (тетракистрифенилфосфин)палладий (0) (136 мг), и смесь перемешивали при 79°С в течение 5 часов. Затем реакционную смесь охлаждали, разбавляли этилацетатом и добавляли насыщенный водный раствор хлорида аммония. Нерастворимое вещество, образовавшееся на этой стадии, удаляли фильтрацией и отделяли органический слой. Полученный органический слой сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (используемый растворитель: этилацетат/метанол) с получением (S)-трет-бутил (4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо-[5,4-b]индолизин-8-ил)карбамата в виде светло-желтого твердого вещества (0,190 г), (выход: 75%).
Пример 13
(S)-трет-бутил-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамат получали способом в соответствии с Примером 10, за исключением того, что вместо водного раствора гидроксида натрия, использованного в Примере 10, в данном случае использовали 4 N водный раствор гидроксида лития (1,8 мл), и указанное соединение получали в виде светло-желтого твердого вещества (224 мг), (выход: 88%).
Пример 14
(S)-трет-бутил-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамат получали способом в соответствии с Примером 10, за исключением того, что вместо водного раствора гидроксида натрия, использованного в Примере 10, в данном случае использовали 4 N водный раствор гидроксида калия (1,8 мл), и указанное соединение получали в виде светло-желтого твердого вещества (198 мг), (выход: 78%).
Пример 15
(S)-трет-бутил-(4-амино-5-(хинолин-3-ил)-6,7,8,9- тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамат получали способом в соответствии с Примером 10, за исключением того, что вместо водного раствора гидроксида натрия, использованного в Примере 10, в данном случае использовали 4 N водный раствор гидроксида цезия (1,8 мл), и указанное соединение получали в виде светло-желтого твердого вещества (202 мг), (выход: 80%).
Пример 16
(S)-трет-бутил(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамат получали способом в соответствии с Примером 10, за исключением того, что вместо (тетракистрифенилфосфин)палладия (0), используемого в Примере 10, в данном случае использовали трис(дибензилиденацетон)палладий (0) (34 мг) и трифенилфосфин (39 мг), и указанное соединение получали в виде светло-желтого твердого вещества (194 мг), (выход: 76%).
Пример 17
(R)-трет-бутил-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамат
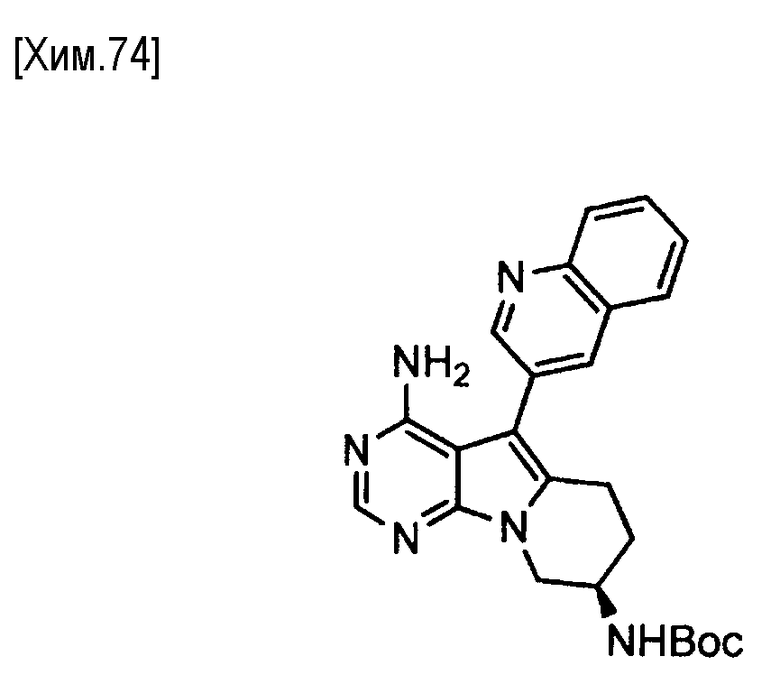
(R)-трет-бутил-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамат получали способом в соответствии с Примером 10, за исключением того, что вместо (S)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата, полученного в Примере 10, в данном случае использовали (R)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамат (6,70 г), полученный в Ссылочном примере 2, и указанное соединение получали в виде светло-желтого твердого вещества (4,76 г), (выход: 84%).
1Н-ЯМР (CDCl3) δ: 1,48 (9Н, с), 1,91-2,00 (1H, м), 2,12-2,19 (1H, м), 2,98-3,11 (2H, м), 4,00 (1Н, дд, J=12,7, 7,1 Гц), 4,32 (1H, ш.с), 4,55 (1Н, дд, J=12,7, 4,6 Гц), 4,81-4,83 (1H, м), 4,90 (2H, ш.с), 7,61-7,65 (1Н, м), 7,75-7,80 (1H, м), 7,88 (1H, д, J=8,0 Гц), 8,16-8,18 (2H, м), 8,33 (1Н, с), 9,02 (1H, д, J=2,2 Гц).
ESI-MS m/z 431 (MH+).
Пример 18
(S)-трет-бутил(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5]пирроло[1,2-a]азепин-9-ил)карбамат
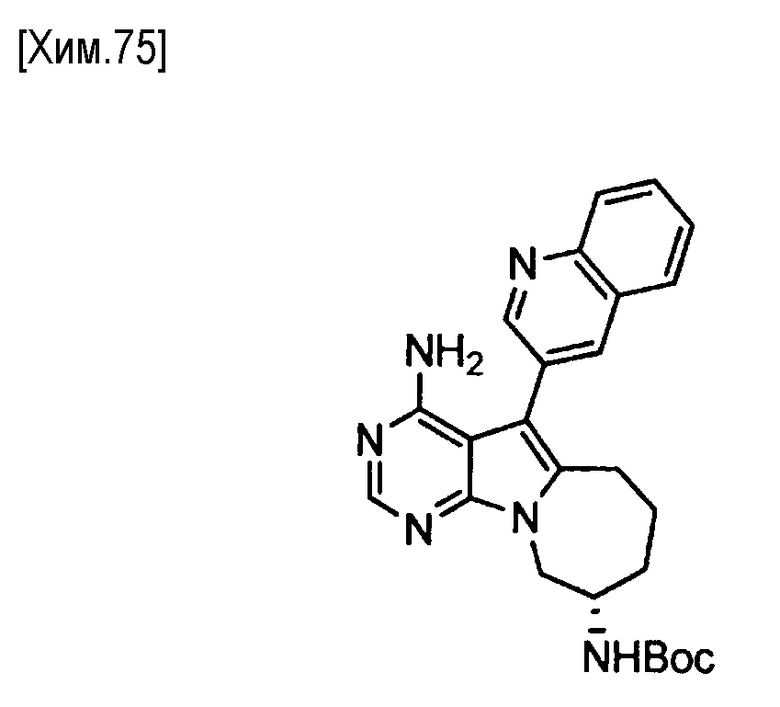
Указанное в заголовке соединение получали способом в соответствии с Примером 10, за исключением того, что вместо (S)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7Н-пирроло[2,3- d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата, полученного в Примере 10, в данном случае использовали (S)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)пента-4-ен-2-ил)карбамат (2,0 г), полученный в Ссылочном примере 8, и указанное соединение получали в виде светло-желтого твердого вещества (1,56 г), (выход: 92%).
1Н-ЯМР (CDCl3) δ: 1,43 (9H, с), 1,77-1,89 (2H, м), 1,95-2,14 (2H, м), 2,71-2,84 (1H, м), 2,86-3,00 (1H, м), 4,00-4,15 (1H, м), 4,24-4,40 (1H, м), 4,40-4,50 (1H, м), 4,84 (3Н, ш.с), 7,62-7,66 (1H, м), 7,77-7,81 (1H, м), 7,89-7,91 (1H, м), 8,18-8,20 (2Н, м), 8,33 (1H, с), 8,98 (1H, д, J=1,5 Гц).
ESI-MS m/z 445 (MH+).
Пример 19
(R)-трет-бутил(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5]пирроло[1,2-a]азепин-9-ил)карбамат
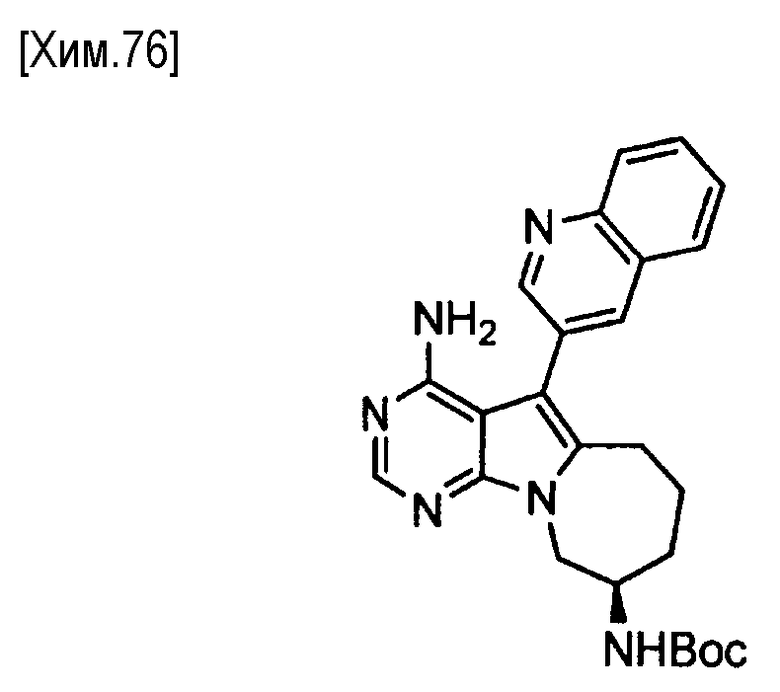
Указанное в заголовке соединение получали способом в соответствии с Примером 10, за исключением того, что вместо (S)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата, полученного в Примере 10, в данном случае использовали (R)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)пента-4-ен-2-ил)карбамат (690 мг), полученный в Ссылочном примере 9, и указанное соединение получали в виде желтого твердого вещества (429 мг), (выход: 73%).
1Н-ЯМР (CDCl3) δ: 1,43 (9H, с), 1,77-1,89 (2H, м), 1,95-2,14 (2H, м), 2,71-2,84 (1H, м), 2,86-3,00 (1H, м), 4,00-4,15 (1H, м), 4,24-4,40 (1H, м), 4,40-4,50 (1H, м), 4,84 (3Н, ш.с), 7,62-7,66 (1H, м), 7,77-7,81 (1H, м), 7,89-7,91 (1H, м), 8,18-8,20 (2Н, м), 8,33 (1H, с), 8,98 (1H, д, J=1,5 Гц).
ESI-MS m/z 445 (MH+).
Пример 20
(S)-трет-бутил(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6H-пиримидо[5′,4′:4,5]пирроло[1,2-a]азепин-8-ил)карбамат
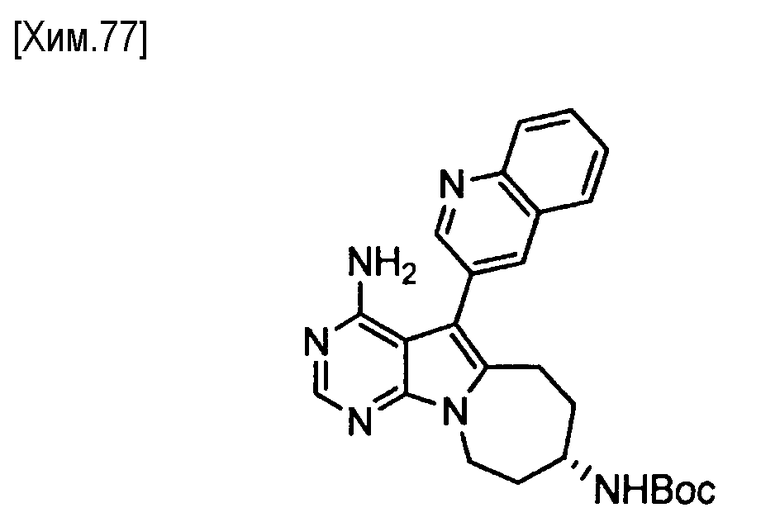
Указанное в заголовке соединение получали способом в соответствии с Примером 10, за исключением того, что вместо (S)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата, полученного в Примере 10, в данном случае использовали (R)-трет-бутил-(5-(4-амино-6-бром-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-7-ил)пента-1- ен-3-ил)карбамат (994 мг), полученный в Ссылочном примере 10, и указанное соединение получали в виде желтого твердого вещества (439 мг) (выход: 52%).
1Н-ЯМР (CDCl3) δ: 1,46 (9H, с), 2,18-2,28 (1H, м), 2,32-2,42 (1H, м), 2,65-2,77 (1H, м), 2,99-3,08 (1H, м), 3,80-3,97 (2H, м), 4,53-4,62 (1H, м), 4,80 (2H, ш.с), 4,97-5,11 (1H, м), 7,61-7,66 (1H, м), 7,76-7,81 (1H, м), 7,88 (1H, д, J=8,0 Гц), 8,15-8,20 (2H, м), 8,33 (1Н, с), 8,97 (1H, д, J=2,2 Гц).
ESI-MS m/z 445 (MH+).
Пример 21
(R)-8-((трет-бутилдиметилсилил)окси)-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4-амин
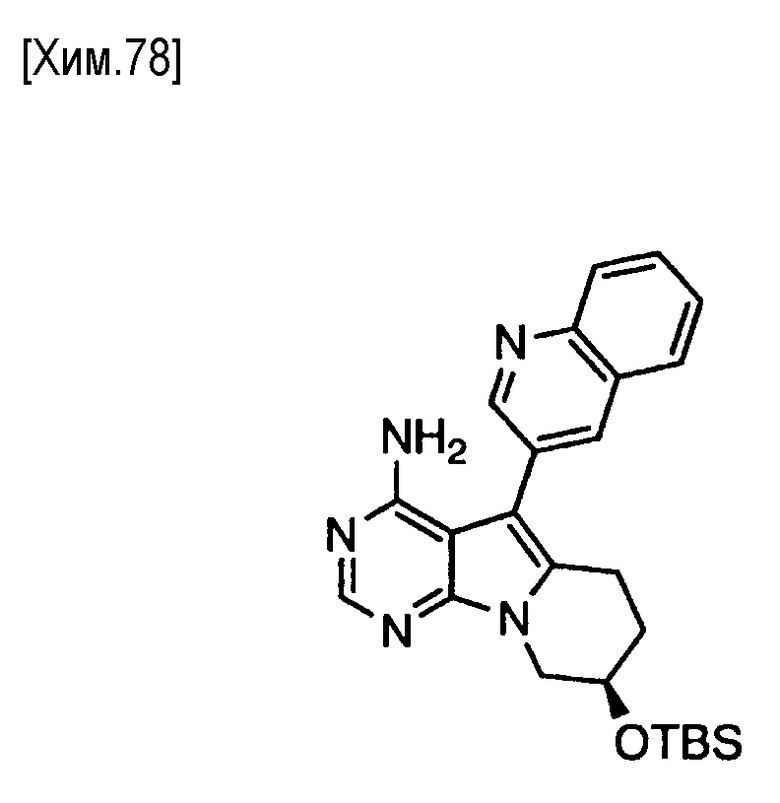
Указанное в заголовке соединение получали способом в соответствии с Примером 10, за исключением того, что вместо (S)-трет-бутил-(1-(4-амино-6-бром-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата, полученного в Примере 10, в данном случае использовали (R)-6-бром-7-(2-((трет-бутилдиметилсилил)окси)бут-3-ен-1-ил)-5-(хинолин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амин (1,0 г), полученный в Ссылочном примере 11. Получили 546 мг целевого вещества (выход: 64%).
1Н-ЯМР (CDCl3) δ: 0,14 (3H, с), 0,15 (3Н, с), 0,91 (9Н, с), 1,97-2,02 (2H, м), 2,85-2,92 (2H, м), 3,14-3,22 (1H, м), 4,11-4,18 (1H, м), 4,28-4,33 (1H, м), 4,41-4,46 (1H, м), 4,95 (2H, ш.с), 7,61-7,65 (1Н, м), 7,75-7,79 (1H, м), 7,88-7,90 (1H, м), 8,16-8,18 (2H, м), 8,35 (1H, с), 9,04 (1H, д, J=2,0 Гц).
ESI-MS m/z 446 (MH+).
Ссылочный пример 7
Соединение (S)-трет-бутил(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамат получали способом в соответствии с Примером 10, за исключением того, что вместо (тетракистрифенилфосфин)палладия (0), используемого в Примере 10, в данном случае использовали дихлорид 1,1′-бис(дифенилфосфино)ферроценпалладия (II) (32 мг). Получили 21 мг целевого соединения (выход: 25%) в виде светло-желтого твердого вещества.
Ссылочный пример 8
Соединение (S)-трет-бутил-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамат получали способом в соответствии с Примером 10, за исключением того, что вместо водного раствора гидроксида натрия, используемого в Примере 10, в данном случае использовали карбонат цезия (2,3 г) и воду (1,8 мл). Получили 88 мг целевого соединения (выход: 35%) в виде светло-желтого твердого вещества.
Примеры испытаний
Соединения по настоящему изобретению были испытаны с помощью следующих методов.
Пример испытаний 1
Измерение ингибирования киназной активности различных EGFR (in vitro)
1) Измерение ингибирования киназной активности EGFR (T790M/L858R)
Были измерены активности соединений I-1, I-2, I-3, I-4, I-5, I-6, I-7, I-8 и I-9 в отношении ингибирования киназной активности EGFR (T790M/L858R).
Были использованы следующие материалы. Пептидный субстрат представлял собой синтезированный биотинилированный пептид (биотин-EEPLYWSFPAKKK), с аминокислотной последовательностью пептида FL-22 (реагент для набора LabChip (зарегистрированный товарный знак); Caliper Life Sciences, Inc.). EGFR (T790M/L858R) представлял собой очищенный рекомбинантный белок EGFR (T790M/L858R) человека от Carna Biosciences, Inc.
Способ измерения состоял в следующем. Каждое из соединений I-1, I-2, I-3, I-4, I-5, I-6, I-7, I-8 и I-9 растворяли в диметилсульфоксиде (DMSO), а затем каждый раствор серийно разбавляли DMSO. Затем к раствору киназного реакционного буфера (Carna Biosciences, Inc) добавляли пептидный субстрат, который представлял собой белок EGFR (T790M/L858R), (конечная концентрация: 250 нМ), хлорид магния (конечная концентрация: 10 мМ), хлорид марганца (конечная концентрация: 10 мМ), АТФ (конечная концентрация: 1 мкМ) и раствор каждого соединения в DMSO (конечная концентрация DMSO 2,5%). Смеси инкубировали при температуре 25°С в течение 120 минут для протекания киназной реакции. Для прекращения реакции к смесям добавляли EDTA до конечной концентрации 24 мМ. Добавляли раствор для детектирования, содержащий антитело PT66 (PerkinElmer) к фосфорилированному тирозину, меченное европием (Eu), и SureLight APC-SA (PerkinElmer). Полученную смесь оставляли при комнатной температуре на время 2 часа или более. Затем с помощью прибора PHERAstar FS (BMG) LabTech регистрировали интенсивность флуоресценции на двух длинах волн 620 нм и 665 нм при облучении светом с длиной волны 337 нм. Уровень фосфорилирования каждого испытуемого образца рассчитывали по отношению интенсивности флуоресценции на двух длинах волн в контроле (с DMSO) и в тестируемом образце, и концентрации соединения, при которой фосфорилирование ингибируется на 50%, было определено как значение IC50 (нМ) для каждого соединения.
2) Измерение ингибирования киназной активности EGFR (d476-750/T790M)
Были измерены активности соединений I-1, I-2, I-3, I-4, I-5, I-6, I-7, I-8 и I-9 в отношении ингибирования киназной активности EGFR (d476-750/T790M).
Были использованы следующие материалы. EGFR (d476-750/T790M) представлял собой очищенный рекомбинантный белок EGFR (d476-750/T790M) человека от Carna Biosciences, Inc. Конечная концентрация АТФ составляла 1,5 мкM. Другие условия, так же как материалы и методы, которые использовались при измерении ингибирования киназной активности EGFR (T790M/L858R), были использованы для определения значения IC50 (нМ) каждого соединения.
3) Измерение ингибирования киназной активности EGFR (L858R)
Были измерены активности соединений I-1, I-2, I-3, I-4, I-5, I-6, I-7, I-8 и I-9 в отношении ингибирования киназной активности EGFR (L858R).
Были использованы следующие материалы. EGFR (L858R) представлял собой очищенный рекомбинантный белок EGFR (L858R) человека от Carna Biosciences, Inc. Конечная концентрация АТФ составляла 4 мкM. Другие условия, так же как материалы и методы, которые использовались при измерении ингибирования киназной активности EGFR (T790M/L858R), были использованы для определения значения IC50 (нМ) каждого соединения.
4) Измерение ингибирования киназной активности EGFR (d476-750
Были измерены активности соединений I-1, I-2, I-3, I-4, I-5, I-6, I-7, I-8 и I-9 в отношении ингибирования киназной активности EGFR (d476-750).
Были использованы следующие материалы. EGFR (d476-750) представлял собой очищенный рекомбинантный белок EGFR (d476-750) человека от Carna Biosciences, Inc. Конечная концентрация АТФ составляла 5 мкM. Другие условия, так же как материалы и методы, которые использовались при измерении ингибирования киназной активности EGFR (T790M/L858R), были использованы для определения значения IC50 (нМ) каждого соединения.
5) Измерение ингибирования киназной активности EGFR дикого типа (WT)
Были измерены активности соединений I-1, I-2, I-3, I-4, I-5, I-6, I-7, I-8 и I-9 в отношении ингибирования киназной активности EGFR (WT).
Были использованы следующие материалы. EGFR (WT) представлял собой цитоплазматический домен человеческого EGFR (WT), в котором маркер FLAG был слит с N-концом молекулы в бакуловирусной системе экспрессии с использованием клеток насекомых Sf9, и который был очищен с помощью антитела против FLAG на агарозе (Sigma-Aldrich). Конечная концентрация пептидного субстрата составляла 500 нМ, и конечная концентрация АТФ составляла 5 мкM. Другие условия, так же как материалы и методы, которые использовались при измерении ингибирования киназной активности EGFR (T790M/L858R), были использованы для определения значения IC50 (нМ) каждого соединения.
Полученные результаты показаны в Таблице 1.
Было подтверждено, что соединения I-1, I-2, I-3, I-4, I-5, I-6, I-7, I-8 и I-9 демонстрируют высокую ингибирующую активность не только в отношении EGFR (L858R) и EGFR (d476-750), но и в отношении EGFR (T790M/L858R) и EGFR (d476-750/T790M). Было также подтверждено, что их ингибирующая активность в отношении EGFR (WT) была ниже, чем активность в отношении вышеуказанных мутированных белков EGFR.
В противоположность этому, было подтверждено, что N-(3-(4-амино-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)фенил)бензамид (см. PTL 1), который представляет собой соединение со структурой, аналогичную структуре соединений настоящего изобретения, практически не ингибирует киназную активность мутированных форм EGFR.
Результаты примера испытаний 1
(T790M/L858R)
(d476-750/T790M)
(L855R)
(d476-750)
(WT)
Пример испытаний 2
Измерение ингибирования роста клеточных линий, экспрессирующих EGFR дикого типа и мутированных EGFR (in vitro)
В среде, рекомендованной АТСС, суспендировали следующие линии клеток: 1) линия клеток аденокарциномы легких NCI-H1975, экспрессирующих EGFR (T790M/L858R), 2) линия клеток аденокарциномы легких HCC827, экспрессирующих EGFR (d476-750), и 3) линия клеток плоскоклеточного рака человека А431, экспрессирующих EGFR (WT). Суспензию клеток высевали в каждую лунку 384-луночного плоского микропланшета или 96-луночного плоского планшета, и культивировали в инкубаторе в атмосфере с 5% углекислого газа при температуре 37°С в течение одного дня. Соединения по настоящему изобретению и сравнительные соединения растворяли в DMSO, а затем этот раствор каждого испытуемого соединения разбавляли с помощью DMSO до концентрации в 200 раз выше, чем конечная концентрация. Разбавленный раствор в DMSO исследуемого соединения далее разбавляли средой, используемой для суспендирования каждой клеточной линии, и полученный разбавленный раствор испытуемого соединения добавляли в каждую лунку с клетками в планшете, из расчета получения конечной концентрации DMSO 0,5%. Затем клетки культивировали в инкубаторе в атмосфере с 5% углекислого газа при температуре 37°С в течение трех дней. Количество клеток измеряли в начальный момент и в конце культивирования с помощью анализа CellTiter-Glo (изготовитель Promega) в соответствии с протоколом, рекомендованным изготовителем. Степень ингибирования роста клеток рассчитывали по нижеприведенной формуле, и определяли концентрацию испытуемого соединения, при которой рост клеток ингибируется на 50% (GI50 (нм)).
Степень ингибирования роста (%)=(C-T)/(C-C0)×100,
где:
T: интенсивность люминесценции лунки, в которой добавлено исследуемое соединение,
С: интенсивность люминесценции лунки, в которую исследуемое соединение не было добавлено,
C0: интенсивность люминесценции лунки, измеренная перед добавлением исследуемого соединения.
Полученные результаты показаны в Таблице 2.
Было подтверждено, что соединения I-2 и I-3 демонстрируют высокую ингибирующую активность не только в отношении клеток, экспрессирующих EGFR (d476-750), но также и в отношении клеток, экспрессирующих EGFR (T790M/L858R). Кроме того, было также подтверждено, что соединения I-2 и I-3 показывают более слабую ингибирующую активность в отношении клеток, экспрессирующих EGFR (WT), чем в отношении вышеуказанных клеток, экспрессирующих мутированные EGFR.
Результаты примера испытаний 2
(T790M/L858R)
(d476-750)
(WT)
Настоящее изобретение относится к новому соединению формулы (I) или его фармацевтически приемлемой соли, которые обладают ингибирующей активностью в отношении EGFR и действует как ингибитор роста клеток. Соединения могут использоваться для получения фармацевтической композиции, полезной для профилактики и/или лечения рака, основанной на ингибирующем эффекте соединения в отношении EGFR. В формуле (I):
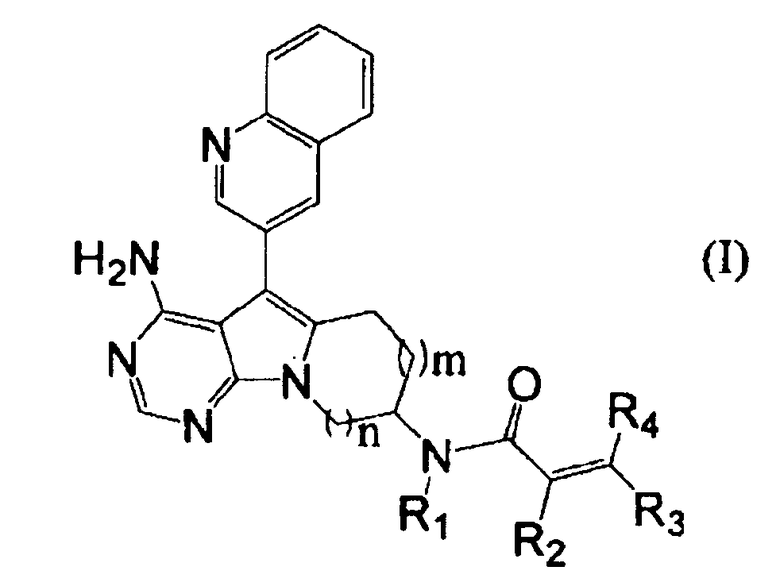
m равно 1 или 2; n равно 1 или 2; группа R1 представляет собой атом водорода или C1-C4-алкильную группу; и группы R2, R3 и R4 могут быть одинаковыми или различными, и каждая из них представляет собой атом водорода, атом галогена или группу, представленную формулой (а) 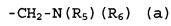 , где группы R5 и R6 могут быть одинаковыми или различными, и каждая из них представляет собой С1-С4-алкильную группу. 6 н. и 4 з.п. ф-лы, 2 табл., 25 пр.
, где группы R5 и R6 могут быть одинаковыми или различными, и каждая из них представляет собой С1-С4-алкильную группу. 6 н. и 4 з.п. ф-лы, 2 табл., 25 пр.
1. Соединение, представленное следующей формулой (I)
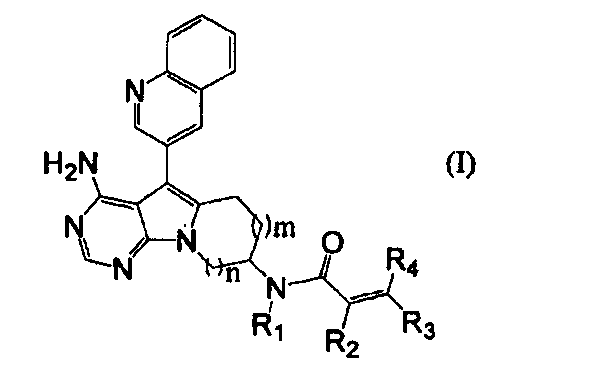
или его фармацевтически приемлемая соль,
где m равно 1 или 2;
n равно 1 или 2;
группа R1 представляет собой атом водорода или C1-C4-алкильную группу; и
группы R2, R3 и R4 могут быть одинаковыми или различными, и каждая из них представляет собой атом водорода, атом галогена или группу, представленную формулой (а)
,
где группы R5 и R6 могут быть одинаковыми или различными, и каждая из них представляет собой С1-С4-алкильную группу.
2. Соединение или его фармацевтически приемлемая соль по п. 1, в котором
m равно 1 или 2;
n равно 1 или 2;
группа R1 представляет собой атом водорода или метильную группу; и
группы R2, R3 и R4 могут быть одинаковыми или различными, и каждая из них представляет собой атом водорода, атом хлора или диметиламинометильную группу.
3. Соединение или его фармацевтически приемлемая соль по п. 1, в котором значения m и n представляют собой (m,n)=(1,1), (1,2) или (2,1).
4. Соединение или его фармацевтически приемлемая соль по п. 1, где соединение выбрано из следующей группы соединений:
(R)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид,
(S)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид,
N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-N-метилакриламид,
(Е)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-4-(диметиламино)-2-бутенамид,
(S,E)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-3-хлоракриламид,
(S,Z)-N-(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-3-хлоракриламид,
(S)-N-(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6Н-пиримидо[5′,4′:4,5]пирроло[1,2-а]азепин-8-ил)акриламид,
(S)-N-(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6Н-пиримидо[5′,4′:4,5]пирроло[1,2-а]азепин-9-ил)акриламид, и
(R)-N-(4-амино-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6Н-пиримидо[5′,4′:4,5]пирроло[1,2-а]азепин-9-ил)акриламид.
5. Ингибитор EGFR, представляющий собой соединение или его
Фармацевтически приемлемую соль по любому из пп. 1-4.
6. Фармацевтическая композиция, обладающая свойствами ингибитора EGFR, содержащая соединение или его фармацевтически приемлемую соль по любому из пп. 1-4 в эффективной дозе.
7. Противоопухолевое средство, представляющее собой соединение или его фармацевтически приемлемую соль по любому из пп. 1-4.
8. Способ лечения или профилактики рака, где способ включает стадию введения млекопитающему соединения или его фармацевтически приемлемой соли по любому из пп. 1-4 в дозе, эффективной для лечения или профилактики рака, опосредованного киназной активностью EGFR.
9. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-4 для изготовления противоопухолевого средства.
10. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-4 для применения при лечении или профилактике рака, опосредованного киназной активностью EGFR.
| US 7718662B2,18.05.2010 | |||
| US 7825118 B2,02.11.2010 | |||
| US 7732454 B2,08.06.2010 | |||
| US 7585868 B2,08.09.2009 | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| ZAPF C.W | |||
| et al.,Covalent Inhibitors in Interleukin-2 Inducible T Cell Kinase(Itk) witw Nanomolar Potency in Whole-Blood Assay, Journal of Medicinal Chemistry,2012,11,v.55,no.22,pp.10047-10063 | |||
| Многовольтовая аккумуляторная батарея | 1928 |
|
SU12873A1 |

