ПРИОРИТЕТ ИЗОБРЕТЕНИЯ
Настоящая заявка испрашивает приоритет предварительной заявки на патент США 61/470803, которая была подана 1 апреля 2011 года, и предварительной заявки на патент США 61/470624, которая была подана 1 апреля 2011 года. Все содержание указанных предварительных заявок настоящим включено посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение в общем относится к фармацевтическим комбинациям соединений с активностью против гиперпролиферативных нарушений, таких как рак, и которые включают соединения, которые ингибируют активность AKT-киназы. Изобретение также относится к способам применения комбинаций для in vitro, in situ и in vivo диагностики или лечения клеток млекопитающих, или связанных патологических состояний.
УРОВЕНЬ ТЕХНИКИ
Протеинкиназы (PK) являются ферментами, которые катализируют фосфорилирование гидроксигрупп на остатках тирозина, серина и треонина в белках путем переноса концевого (гамма) фосфата с АТФ. Через пути передачи сигнала указанные ферменты модулируют рост, дифференцировку и пролиферацию клеток, т.е. фактически все аспекты жизни клетки так или иначе зависят от активности PK (Hardie, G. and Hanks, S. (1995) The Protein Kinase Facts Book. I and II, Academic Press, San Diego, CA). Кроме того, аномальную активность PK связывали с множеством нарушений, в пределах от относительно не опасных для жизни заболеваний, таких как псориаз, до чрезвычайно опасных заболеваний, таких как глиобластома (рак мозга). Протеинкиназы являются важным классом мишеней для терапевтической модуляции (Cohen, P. (2002) Nature Rev. Drug Discovery 1:309).
В опубликованной международной заявке на патент WO 2008/006040 обсуждается ряд ингибиторов AKT формулы I:
В настоящее время сохраняется потребность в улучшенных способах и композициях, которые могут быть использованы для лечения гиперпролиферативных заболеваний, таких как рак.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Было определено, что аддитивные или синергические эффекты при ингибировании роста раковых клеток in vitro и in vivo могут быть достигнуты путем введения соединения формулы I или его фармацевтически приемлемой соли в комбинации с некоторыми другими определенными химиотерапевтическими средствами. Комбинации и способы могут быть применимы в лечении гиперпролиферативных нарушений, таких как рак.
В одном аспекте изобретения предложен способ лечения гиперпролиферативного нарушения у млекопитающего, включающий введение млекопитающему a) соединения формулы I:
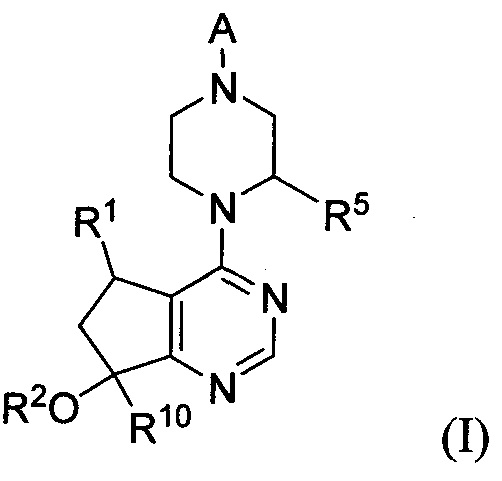
или его фармацевтически приемлемой соли; и b) одного или более средств, выбранных из 5-FU, платиносодержащего средства (карбоплатина, цисплатина, оксалиплатина и т.д.), иринотекана, доцетаксела, доксорубицина, гемцитабина, SN-38, капецитабина, темозоломида, эрлотиниба, PD-0325901, паклитаксела, бевацизумаба, пертузумаба, тамоксифена, рапамицина, лапатиниба, PLX-4032, MDV3100, абиратерона и GDC-0973.
Соединение формулы I или его фармацевтически приемлемая соль и химиотерапевтическое средство могут быть составлены в комбинированной форме для введения в комбинации в качестве фармацевтической композиции, или они могут быть поочередно (последовательно) введены раздельно в виде терапевтической комбинации.
В одном аспекте изобретения предложен способ лечения заболевания или состояния, модулируемого AKT-киназой, у млекопитающего, включающий введение млекопитающему a) соединения формулы I или его фармацевтически приемлемой соли; и b) одного или более средств, выбранных из 5-FU, платиносодержащего средства, иринотекана, доцетаксела, доксорубицина, гемцитабина, SN-38, капецитабина, темозоломида, эрлотиниба, PD-0325901, паклитаксела, бевацизумаба, пертузумаба, тамоксифена, рапамицина, лапатиниба, PLX-4032, MDV3100, абиратерона и GDC-0973.
В одном аспекте изобретения предложена комбинация a) соединения формулы I или его фармацевтически приемлемой соли; и b) одного или более средств, выбранных из 5-FU, платиносодержащего средства, иринотекана, доцетаксела, доксорубицина, гемцитабина, SN-38, капецитабина, темозоломида, эрлотиниба, PD-0325901, паклитаксела, бевацизумаба, пертузумаба, тамоксифена, рапамицина, лапатиниба, PLX-4032, MDV3100, абиратерона и GDC-0973, для лечения гиперпролиферативного нарушения.
В одном аспекте изобретения предложена комбинация a) соединения формулы I или его фармацевтически приемлемой соли; и b) одного или более средств, выбранных из 5-FU, платиносодержащего средства, иринотекана, доцетаксела, доксорубицина, гемцитабина, SN-38, капецитабина, темозоломида, эрлотиниба, PD-0325901, паклитаксела, бевацизумаба, пертузумаба, тамоксифена, рапамицина, лапатиниба, PLX-4032, MDV3100, абиратерона и GDC-0973, для лечения заболевания или состояния, модулируемого AKT-киназой.
В одном аспекте изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения гиперпролиферативного нарушения у млекопитающего, где одно или более средств, выбранных из 5-FU, платиносодержащего средства, иринотекана, доцетаксела, доксорубицина, гемцитабина, SN-38, капецитабина, темозоломида, эрлотиниба, PD-0325901, паклитаксела, бевацизумаба, пертузумаба, тамоксифена, рапамицина, лапатиниба, PLX-4032, MDV3100, абиратерона и GDC-0973, вводят млекопитающему.
В одном аспекте изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения заболевания или состояния, модулируемого AKT-киназой, у млекопитающего, где одно или более средств, выбранных из 5-FU, платиносодержащего средства, иринотекана, доцетаксела, доксорубицина, гемцитабина, SN-38, капецитабина, темозоломида, эрлотиниба, PD-0325901, паклитаксела, бевацизумаба, пертузумаба, тамоксифена, рапамицина, лапатиниба, PLX-4032, MDV3100, абиратерона и GDC-0973, вводят млекопитающему.
В одном аспекте изобретения предложен набор, включающий соединение формулы I или его фармацевтически приемлемую соль, контейнер, а также вкладыш в упаковку или этикетку с указаниями по применению соединения формулы I или его фармацевтически приемлемой соли с одним или более средствами, выбранными из 5-FU, платиносодержащего средства, иринотекана, доцетаксела, доксорубицина, гемцитабина, SN-38, капецитабина, темозоломида, эрлотиниба, PD-0325901, паклитаксела, бевацизумаба, пертузумаба, тамоксифена, рапамицина, лапатиниба, PLX-4032, MDV3100, абиратерона и GDC-0973, для лечения гиперпролиферативного нарушения.
В одном аспекте изобретения предложен продукт, включающий соединение, имеющее формулу I, или его фармацевтически приемлемую соль и химиотерапевтическое средство, выбранное из 5-FU, платиносодержащего средства, иринотекана, доцетаксела, доксорубицина, гемцитабина, SN-38, капецитабина, темозоломида, эрлотиниба, PD-0325901, паклитаксела, бевацизумаба, пертузумаба, тамоксифена, рапамицина, лапатиниба, PLX-4032, MDV3100, абиратерона и GDC-0973; в виде комбинированного препарата для раздельного, одновременного или последовательного применения при лечении гиперпролиферативного нарушения.
В одном аспекте изобретения предложен продукт, включающий соединение, имеющее формулу I, или его фармацевтически приемлемую соль и абиратерон или его фармацевтически приемлемую соль; в виде комбинированного препарата для раздельного, одновременного или последовательного применения при лечении гиперпролиферативного нарушения, такого как рак предстательной железы.
В дополнение к обеспечению улучшенного лечения данного гиперпролиферативного нарушения, введение некоторых комбинаций изобретения может повысить качество жизни пациента по сравнению с качеством жизни, которое имеет тот же пациент, получающий другое лечение. Например, введение пациенту комбинации соединения формулы I или его фармацевтически приемлемой соли и химиотерапевтического средства, как описано в настоящей заявке, может обеспечить повышение качества жизни по сравнению с качеством жизни, которое имел бы тот же пациент, если бы он получал только химиотерапевтическое средство в качестве терапии. Например, комбинированная терапия с использованием комбинации, описанной в настоящей заявке, может снижать дозу необходимых химиосредств, уменьшая тем самым побочные эффекты, связанные с высокой дозой химиотерапевтических средств (например, тошноту, рвоту, потерю волос, высыпание, сниженный аппетит, потерю веса и т.д.). Комбинация также может вызывать уменьшение опухолевой нагрузки и ассоциируемых неблагоприятных явлений, таких как боль, дисфункция органов, потеря веса и т.д. Таким образом, в одном аспекте изобретения предложено соединение формулы I или его фармацевтически приемлемая соль для терапевтического применения в целях повышения качества жизни пациента, который проходит лечение от гиперпролиферативного нарушения с использованием средства, выбранного из 5-FU, платиносодержащего средства, иринотекана, доцетаксела, доксорубицина, гемцитабина, SN-38, капецитабина, темозоломида, эрлотиниба, PD-0325901, паклитаксела, бевацизумаба, пертузумаба, тамоксифена, рапамицина, лапатиниба, PLX-4032, MDV3100, абиратерона и GDC-0973. Соответственно, в другом аспекте изобретения предложено соединение формулы I или его фармацевтически приемлемая соль для терапевтического применения в целях повышения качества жизни пациента, который проходит лечение от гиперпролиферативного нарушения с использованием абиратерона или его фармацевтически приемлемой соли.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На фигуре 1 представлены данные для соединения формулы 1a (GDC-0068), вводимого внутрь, + абиратерон в первичных опухолях предстательной железы LuCaP35V.
На фигуре 2 представлены данные для соединения формулы 1a (GDC-0068), вводимого внутрь, + абиратерон в первичных опухолях предстательной железы DU-145.x1.
ПОДРОБНОЕ ОПИСАНИЕ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ И ОПРЕДЕЛЕНИЙ
Слова "включает" и "включающий", при использовании в настоящем описании и формуле изобретения, служат для определения присутствия установленных свойств, целых чисел, компонентов или стадий, но не исключают присутствие или добавление одного или большего количества других свойств, целых чисел, компонентов, стадий или их групп.
Активность антиандрогенных вариантов терапии, включающих бикалутамид, агонист GnRH и абиратерон, привела к повышению выживаемости пациентов с раком предстательной железы. Однако почти у всех пациентов, у которых имели место поздние стадии гормоночувствительного рака предстательной железы, наблюдали переход в CRPC (КРПЖ), и при этом требовались другие формы терапии. Активация сигнализации PI3K/Akt, часто проявляющаяся в потере PTEN, является частым признаком КРПЖ. Нарушение регуляции данного пути приводит к активации нижестоящих мишеней (например, PRAS40, MTOR, GSK3b, FOXO и т.д.), вовлеченных в выживание, пролиферацию, прогрессию клеточного цикла, рост, миграцию и ангиогенез. Примечательно, что простат-специфическая делеция PTEN в моделях на мышах надежно воспроизводит свойства рака предстательной железы человека, а делеция Akt1 в условной модели с нокаутом Pten существенно уменьшает раковые образования предстательной железы (Chen et al., 2006; Guertin et al., 2009; Nardella et al., 2009). Кроме того, делеция Pten вызывает андрогенонезависимость в линиях клеток и моделях на мышах рака предстательной железы (Gao et al., 2006; Jiao et al., 2007). У больных раком предстательной железы потеря PTEN связана с более высокими баллами по шкале Глисона, рецидивом после простатэктомии, метастазами в костной ткани и прогрессией до кастраторезистентности. Более того, потеря PTEN связана с уменьшением общей выживаемости. В совокупности, представленные результаты указывают, что активация пути PI3K/Akt является важным фактором развития рака предстательной железы.
Недавние неклинические данные указывают, что при PTEN-дефицитном КРПЖ между путями AR и PI3K/Akt присутствует взаимная перекрестная связь. В частности, активация пути PI3K/Akt связана с подавлением андрогенной сигнализации, а ингибирование пути PI3K/Akt восстанавливает AR сигнализацию в PTEN-дефицитных клетках предстательной железы. Предполагаемые механизмы, объясняющие подобные наблюдения, включают ингибирование PI3K/Akt, приводящее к обратной активации AR через апрегуляцию HER-киназ, и ингибирование AR, уменьшающее обратное ингибирование Akt под действием фосфатазы PHLPP (Carver et al., 2011). Данная взаимная кооперативность между путями PI3K/Akt и AR позволяет предположить, что ингибирование только одного пути могло бы привести к недостаточной клинической эффективности. Таким образом, одновременное ингибирование путей AR и PIK3/Akt может привести к более полному подавлению жизнеспособности опухолевых клеток и более надежному благоприятному клиническому результату.
Термин "алкил", используемый в настоящем описании, относится к насыщенному неразветвленному или разветвленному одновалентному углеводородному радикалу, содержащему от одного до двенадцати атомов углерода, где алкильный радикал необязательно может быть независимо замещен одним или более заместителями, описанными ниже. Примеры алкильных групп включают, но не ограничиваются ими, метил (Ме, -CH3), этил (Et,-CH2CH3), 1-пропил (n-pr, н-пропил, -CH2CH2CH3), 2-пропил (i-Pr, изопропил, -CH(CH3)2), 1-бутил (n-Bu, н-бутил, -CH2CH2CH2CH3), 2-метил-1-пропил (i-Bu, изобутил, -CH2CH(CH3)2), 2-бутил (s-Bu, втор-бутил, -CH(CH3)CH2CH3), 2-метил-2-пропил (t-Bu, трет-бутил, -C(CH3)3), 1-пентил (н-пентил, -CH2CH2CH2CH2CH3), 2-пентил (-CH(CH3)CH2CH2CH3), 3-пентил (-CH(CH2CH3)2), 2-метил-2-бутил (-C(CH3)2CH2CH3), 3-метил-2-бутил (-CH(CH3)CH(CH3)2), 3-метил-1-бутил (-CH2CH2CH(CH3)2), 2-метил-1-бутил (-CH2CH(CH3)CH2CH3), 1-гексил (-CH2CH2CH2CH2CH2CH3), 2-гексил (-CH(CH3)CH2CH2CH2CH3), 3-гексил (-CH(CH2CH3)(CH2CH2CH3)), 2-метил-2-пентил (-C(CH3)2CH2CH2CH3), 3-метил-2-пентил (-CH(CH3)CH(CH3)CH2CH3), 4-метил-2-пентил (-CH(CH3)CH2CH(CH3)2), 3-метил-3-пентил (-C(CH3)(CH2CH3)2), 2-метил-3-пентил (-CH(CH2CH3)CH(CH3)2), 2,3-диметил-2-бутил (-C(CH3)2CH(CH3)2), 3,3-диметил-2-бутил (-CH(CH3)C(CH3)3, 1-гептил, 1-октил и т.п.
Термин "алкенил" относится к неразветвленному или разветвленному одновалентному углеводородному радикалу, содержащему от двух до двенадцати атомов углерода, по меньшей мере с одним участком ненасыщенности, т.е. углерод-углеродной, sp2 двойной связью, где алкенильный радикал необязательно может быть независимо замещен одним или более заместителями, описанными в настоящей заявке, и включает радикалы, имеющие "цис" и "транс" ориентации или, альтернативно, "E" и "Z" ориентации. Примеры включают, но не ограничиваются ими, этиленил или винил (-CH=CH2), аллил (-CH2CH=CH2) и т.п.
Термин "алкинил" относится к неразветвленному или разветвленному одновалентному углеводородному радикалу, содержащему от двух до двенадцати атомов углерода, по меньшей мере с одним участком ненасыщенности, т.е. углерод-углеродной, sp тройной связью, где алкинильный радикал необязательно может быть независимо замещен одним или более заместителями, описанными в настоящей заявке. Примеры включают, но не ограничиваются ими, этинил (-C≡CH), пропинил (пропаргил, -CH2C≡CH) и т.п.
Термины "карбоцикл", "карбоциклил", "карбоциклическое кольцо" и "циклоалкил" относятся к одновалентному неароматическому, насыщенному или частично ненасыщенному кольцу, содержащему 3-12 атомов углерода в виде моноциклического кольца или 7-12 атомов углерода в виде бициклического кольца. Бициклические карбоциклы, содержащие 7-12 атомов, могут быть организованы, например, как бицикло [4,5], [5,5], [5,6] или [6,6] система, и бициклические карбоциклы, содержащие 9 или 10 атомов в кольце, могут быть организованы как бицикло [5,6] или [6,6] система, или как мостиковые системы, такие как бицикло[2.2.1]гептан, бицикло[2.2.2]октан и бицикло[3.2.2]нонан. Примеры моноциклических карбоциклов включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, 1-циклопент-1-енил, 1-циклопент-2-енил, 1-циклопент-3-енил, циклогексил, 1-циклогекс-1-енил, 1-циклогекс-2-енил, 1-циклогекс-3-енил, циклогексадиенил, циклогептил, циклооктил, циклононил, циклодецил, циклоундецил, циклододецил и т.п.
"Арил" означает одновалентный ароматический углеводородный радикал, содержащий 6-20 атомов углерода, получаемый за счет удаления одного атома водорода от одного атома углерода исходной ароматической кольцевой системы. Некоторые арильные группы представлены в примерных структурах как "Ar". Арил включает бициклические радикалы, содержащие ароматическое кольцо, конденсированное с насыщенным, частично ненасыщенным кольцом, или ароматическим карбоциклическим или гетероциклическим кольцом. Типичные арильные группы включают, но не ограничиваются ими, радикалы, которые являются производными бензола (фенил), замещенные бензолы, нафталин, антрацен, бифенил, инденил, инданил, 1,2-дигидронафталин, 1,2,3,4-тетрагидронафтил и т.п. Арильные группы необязательно независимо замещены одним или более заместителями, описанными в настоящей заявке.
Термины "гетероцикл", "гетероциклил" и "гетероциклическое кольцо" в настоящем описании используются взаимозаменяемо и относятся к насыщенному или частично ненасыщенному (т.е. содержащему одну или более двойных и/или тройных связей в кольце) карбоциклическому радикалу, содержащему 3-20 атомов в кольце, в котором по меньшей мере один атом в кольце является гетероатомом, выбранным из азота, кислорода и серы, а остальные атомы в кольце представляют собой C, где один или более атомов в кольце необязательно независимо замещены одним или более заместителями, описанными ниже. Гетероцикл может быть моноциклом, содержащим 3-7 членов кольца (2-6 атомов углерода и 1-4 гетероатомов, выбранных из N, O, P и S), или бициклом, содержащим 7-10 членов кольца (4-9 атомов углерода и 1-6 гетероатомов, выбранных из N, O, P и S), например: бицикло [4,5], [5,5], [5,6] или [6,6] системой. Гетероциклы описаны в публикациях Paquette, Leo A.; "Principles of Modern Heterocyclic Chemistry" (W.A. Benjamin, New York, 1968), а именно, в главах 1, 3, 4, 6, 7 и 9; "The Chemistry of Heterocyclic Compounds, A series of Monographs" (John Wiley & Sons, New York, 1950 to present), а именно, в томах 13, 14, 16, 19 и 28; и J. Am. Chem. Soc. (1960) 82:5566. Термин "гетероцикл" включает гетероциклоалкокси. "Гетероциклил" также включает радикалы, в которых гетероциклические радикалы конденсированы с насыщенным, частично ненасыщенным кольцом или ароматическим, карбоциклическим или гетероциклическим, кольцом. Примеры гетероциклических колец включают, но не ограничиваются ими, пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидротиенил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, пиперидино, морфолино, тиоморфолино, тиоксанил, пиперазинил, гомопиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 2-пирролинил, 3-пирролинил, индолинил, 2Н-пиранил, 4Н-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинилимидазолинил, имидазолидинил, 3-азабицикло[3.1.0]гексанил, 3-азабицикло[4.1.0]гептанил, азабицикло[2.2.2]гексанил, 3Н-индолил, хинолизинил и N-пиридил мочевины. Спирогруппы также включены в объем данного определения. Примерами гетероциклической группы, в которой 2 атома углерода в кольце замещены оксогруппами (=O), являются пиримидинонил и 1,1-диоксотиоморфолинил. Гетероциклические группы в настоящем описании необязательно независимо замещены одним или более заместителями, описанными в настоящей заявке.
Термин "гетероарил" относится к одновалентному ароматическому радикалу из 5-, 6- или 7-членных колец и включает конденсированные кольцевые системы (в которых по меньшей мере одно кольцо является ароматическим) из 5-20 атомов, содержащие один или более гетероатомов, независимо выбранных из азота, кислорода и серы. Примерами гетероарильных групп являются пиридинил (включая, например, 2-гидроксипиридинил), имидазолил, имидазопиридинил, пиримидинил (включая, например, 4-гидроксипиримидинил), пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, триазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил и фуропиридинил. Гетероарильные группы необязательно независимо замещены одним или более заместителями, описанными в настоящей заявке.
Гетероциклические или гетероарильные группы могут быть присоединены через углерод (углерод-связанные), азот (азот-связанные) или кислород (кислород-связанные), если это возможно. В качестве неограничивающего примера, углерод-связанные гетероциклы или гетероарилы связаны в положении 2, 3, 4, 5 или 6 пиридина, положении 3, 4, 5 или 6 пиридазина, положении 2, 4, 5 или 6 пиримидина, положении 2, 3, 5 или 6 пиразина, положении 2, 3, 4 или 5 фурана, тетрагидрофурана, тиофурана, тиофена, пиррола или тетрагидропиррола, положении 2, 4 или 5 оксазола, имидазола или тиазола, положении 3, 4 или 5 изоксазола, пиразола или изотиазола, положении 2 или 3 азиридина, положении 2, 3 или 4 азетидина, положении 2, 3, 4, 5, 6, 7 или 8 хинолина или в положении 1, 3, 4, 5, 6, 7 или 8 изохинолина.
В качестве неограничивающего примера, азот-связанные гетероциклы или гетероарилы связаны в положении 1 азиридина, азетидина, пиррола, пирролидина, 2-пирролина, 3-пирролина, имидазола, имидазолидина, 2-имидазолина, 3-имидазолина, пиразола, пиразолина, 2-пиразолина, 3-пиразолина, пиперидина, пиперазина, индола, индолина, 1H-индазола, в положении 2 изоиндола или изоиндолина, положении 4 морфолина и положении 9 карбазола или β-карболина.
Термины "лечить" и "лечение" относятся к терапевтическому воздействию и профилактическим или превентивным мерам, цель которых заключается в предотвращении или замедлении (уменьшении) нежелательного физиологического изменения или нарушения, такого как рост, развитие или распространение рака. В рамках настоящего изобретения выгодные или желательные клинические результаты включают, но не ограничиваются ими, уменьшение проявления симптомов, уменьшение тяжести заболевания, стабилизацию (т.е. не ухудшение) течения заболевания, задержку или замедление прогрессирования заболевания, уменьшение тяжести или ослабление заболевания и ремиссию (частичную или полную), которая может быть обнаружена или не обнаружена. "Лечение" может также означать продление выживания по сравнению с выживанием, ожидаемым без получения лечения. Лица, нуждающиеся в лечении, включают лиц, уже имеющих заболевание или нарушение, а также склонных к развитию заболевания или нарушения, или лиц, у которых необходимо предотвратить развитие заболевания или нарушения.
Фраза "терапевтически эффективное количество" означает количество соединения настоящего изобретения, которое обеспечивает (i) лечение конкретного заболевания, состояния или нарушения, (ii) ослабление, уменьшение тяжести или устранение одного или более симптомов конкретного заболевания, состояния или нарушения, или (iii) предотвращение или задержку начала одного или более симптомов конкретного заболевания, состояния или нарушения, описанных в настоящей заявке. В случае рака, терапевтически эффективное количество лекарственного средства может уменьшать количество раковых клеток; уменьшать размер опухоли; ингибировать (т.е. замедлять в некоторой степени и предпочтительно останавливать) инфильтрацию раковых клеток в периферические органы; ингибировать (т.е. замедлять в некоторой степени и предпочтительно останавливать) метастазирование опухоли; ингибировать, в некоторой степени, рост опухоли; и/или уменьшать до некоторой степени один или более симптомов, связанных с раком. В случае если лекарственное средство может предотвращать рост и/или убивать существующие раковые клетки, оно может быть цитостатическим и/или цитотоксическим. В отношении противоопухолевой терапии эффективность может быть измерена, например, при оценке времени до прогрессирования заболевания (TTP) и/или определении частоты ответа (RR).
Термины "рак" и "раковый" относятся к физиологическому состоянию у млекопитающих, которое обычно характеризуется нерегулируемым ростом клеток, или описывают его. "Опухоль" включает одну или более раковых клеток. Примеры рака включают, но не ограничиваются ими, карциному, лимфому, бластому, саркому и лейкоз или лимфолейкозы. Более конкретные примеры таких раковых опухолей включают плоскоклеточный рак (например, эпителиальный плоскоклеточный рак), рак легкого, включая мелкоклеточный рак легкого, немелкоклеточный рак легкого ("NSCLC"), аденокарциному легкого и плоскоклеточную карциному легкого, перитонеальный рак, гепатоцеллюлярный рак, рак желудка, включая рак желудочно-кишечного тракта, рак поджелудочной железы, глиобластому, рак шейки матки, рак яичников, рак печени, рак мочевого пузыря, гепатому, рак молочной железы, рак толстой кишки, рак прямой кишки, рак толстой и прямой кишки, карциному эндометрия или матки, карциному слюнной железы, рак почки, рак предстательной железы, рак вульвы, рак щитовидной железы, карциному печени, анальную карциному, карциному полового члена, а также злокачественные образования головы и шеи. Рак желудка, как используется в настоящем описании, включает рак, который может развиваться в любой части желудка и распространяться по всему желудку и в другие органы, особенно в пищевод, легкие, лимфатические узлы и печень.
"Химиотерапевтическое средство" является биологическим (высокомолекулярным) или химическим (низкомолекулярным) соединением, применяемым в лечении рака, независимо от механизма действия. Классы химиотерапевтических средств включают, но не ограничиваются ими: алкилирующие средства, антиметаболиты, растительные алкалоиды, являющиеся веретенными ядами, цитотоксические/противоопухолевые антибиотики, ингибиторы топоизомеразы, белки, антитела, фотосенсибилизаторы и ингибиторы киназ. Химиотерапевтические средства включают соединения, используемые в "направленной терапии", и обычную ненаправленную химиотерапию.
Термин "млекопитающее" включает, но не ограничивается ими, людей, мышей, крыс, морских свинок, обезьян, собак, кошек, лошадей, коров, свиней, овец и домашнюю птицу.
Термин "вкладыш в упаковку" используется для обозначения инструкций, обычно включаемых в коммерческие упаковки терапевтических продуктов, которые содержат информацию по показаниям, применению, дозировке, введению, противопоказаниям и/или предупреждения относительно применения таких терапевтических продуктов.
Фраза "фармацевтически приемлемая соль", используемая в настоящем описании, относится к фармацевтически приемлемым органическим или неорганическим солям соединения изобретения. Примеры солей включают, но не ограничиваются ими, сульфат, цитрат, ацетат, оксалат, хлорид, бромид, йодид, нитрат, бисульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат "мезилат", этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат (т.е. 1,1’-метилен-бис-(2-гидрокси-3-нафтоат)). Фармацевтически приемлемая соль может включать другую молекулу, такую как ацетат-ион, сукцинат-ион или другой противоион. Противоион может быть любой органической или неорганической молекулой, которая стабилизирует заряд на исходном соединении. Кроме того, фармацевтически приемлемая соль может иметь более одного заряженного атома в своей структуре. В случаях, когда несколько заряженных атомов являются частью фармацевтически приемлемой соли, она может иметь несколько противоионов. Следовательно, фармацевтически приемлемая соль может иметь один или более заряженных атомов и/или один или более противоионов.
Если соединение является основанием, то требуемая фармацевтически приемлемая соль может быть получена любым подходящим способом, доступным из уровня техники, например, путем обработки свободного основания неорганической кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, метансульфоновая кислота, фосфорная кислота и т.п., или органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, пиранозидиловая кислота, такая как глюкуроновая кислота или галактуроновая кислота, альфа-гидроксикислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глутаминовая кислота, ароматическая кислота, такая как бензойная кислота или коричная кислота, сульфоновая кислота, такая как п-толуолсульфоновая кислота или этансульфоновая кислота, или т.п. Кислоты, которые обычно считают подходящими для получения фармацевтически пригодных или приемлемых солей из основных фармацевтических соединений, обсуждаются, например, в публикациях P. Stahl et al., Camille G. (eds.) Handbook of Pharmaceutical Salts. Properties, Selection and Use. (2002) Zurich: Wiley-VCH; S. Berge et al, Journal of Pharmaceutical Sciences (1977) 66(1) 1 19; P. Gould, International J. of Pharmaceutics (1986) 33 201 217; Anderson et al, The Practice of Medicinal Chemistry (1996), Academic Press, New York; Remington’s Pharmaceutical Sciences, 18th ed., (1995) Mack Publishing Co., Easton PA; и в The Orange Book (Food & Drug Administration, Washington, D.C., на их веб-сайте). Содержание указанных источников включено в настоящее описание посредством ссылки.
Если соединение является кислотой, то требуемая фармацевтически приемлемая соль может быть получена любым подходящим способом, например, путем обработки свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксид щелочного металла или гидроксид щелочноземельного металла, или т.п. Иллюстративные примеры подходящих солей включают, но не ограничиваются ими, органические соли, полученные из аминокислот, таких как глицин и аргинин, аммиака, первичных, вторичных и третичных аминов, и циклических аминов, таких как пиперидин, морфолин и пиперазин, и неорганические соли, полученные из натрия, кальция, калия, магния, марганца, железа, меди, цинка, алюминия и лития.
Фраза "фармацевтически приемлемый" указывает, что вещество или композиция должны быть совместимыми, химически и/или токсикологически, с другими компонентами, составляющими лекарственную форму, и/или с млекопитающим, которое подвергают лечению с использованием данной лекарственной формы.
"Сольват" относится к физической ассоциации или комплексу одной или более молекул растворителя и соединения изобретения. Соединения могут существовать в сольватированных, а также несольватированных формах. Примеры растворителей, которые образуют сольваты, включают, но не ограничиваются ими, воду, изопропанол, этанол, метанол, ДМСО, этилацетат, уксусную кислоту и этаноламин. Термин "гидрат" относится к комплексу, в котором молекулой растворителя является вода. Такая физическая ассоциация включает различные степени ионного и ковалентного связывания, включая образование водородных связей. В некоторых случаях сольват может быть выделен, например, когда одна или более молекул растворителя включены в кристаллическую решетку кристаллического вещества. Получение сольватов является общеизвестным, например, M. Caira et al., J. Pharmaceutical Sci., 93(3), 601 611 (2004). Подобные способы получения сольватов, гемисольватов, гидратов и т.п. описаны в публикациях E.C. van Tonder et al., AAPS PharmSciTech., 5(1), article 12 (2004); и A.L. Bingham et al., Chem. Commun., 603 604 (2001). Типичный, неограничивающий процесс включает растворение соединения изобретения в требуемом количестве нужного растворителя (органического или водного, или их смесей), при температуре выше температуры окружающей среды, и охлаждение раствора со скоростью, достаточной для образования кристаллов, которые затем выделяют стандартными методами. Аналитические методы, такие как, например, ИК-спектроскопия, показывают присутствие растворителя (или воды) в кристаллах в форме сольвата (или гидрата).
Термин "синергический", используемый в настоящем описании, относится к терапевтической комбинации, которая является более эффективной, чем аддитивные эффекты двух или более отдельных средств. Определение синергического взаимодействия между соединением формулы I или его фармацевтически приемлемой солью и одним или более химиотерапевтическими средствами может быть основано на результатах, полученных из анализов, описанных в настоящей заявке. Результаты указанных анализов можно проанализировать, используя комбинацию метода Chou и Talalay и анализ дозы-эффекта с помощью программы CalcuSyn, с получением комбинаторного индекса (Chou and Talalay, 1984, Adv. Enzyme Regul. 22:27-55). Комбинации, предложенные в настоящем изобретении, оценивали в нескольких системах анализа, и полученные данные могут быть проанализированы с использованием стандартной программы для количественного определения синергизма, аддитивизма и антагонизма между противоопухолевыми средствами. Подходящей программой является программа, описанная Chou и Talalay в "New Avenues in Developmental Cancer Chemotherapy", Academic Press, 1987, Chapter 2. Значения комбинаторного индекса меньше 0,8 указывают на синергию, значения больше 1,2 указывают на антагонизм, и значения от 0,8 до 1,2 указывают на аддитивные эффекты. Комбинированная терапия может обеспечивать "синергию" и оказаться "синергической", т.е. достигаемый эффект при использовании активных компонентов вместе превышает сумму эффектов от использования соединений по отдельности. Синергический эффект может быть достигнут, когда активные компоненты: (1) включают в одну композицию и вводят или доставляют одновременно в комбинированной, единичной дозированной лекарственной форме; (2) доставляют последовательно или параллельно в виде отдельных форм; или (3) согласно каким-либо другим схемам. При доставке в последовательной терапии синергический эффект может достигаться, когда соединения вводят или доставляют последовательно, например, при различных инъекциях в отдельных шприцах. Как правило, в процессе последовательной терапии эффективную дозу каждого активного компонента вводят последовательно, т.е. поочередно, тогда как в комбинированной терапии эффективные дозы двух или более активных компонентов вводят вместе. В некоторых примерах комбинационные эффекты оценивали, используя независимую модель BLISS и модель наибольшего отдельного средства (HSA) (Lehar et al., 2007, Molecular Systems Biology 3:80). Оценки BLISS количественно определяют степень потенцирования от отдельных средств, при этом оценка BLISS >0 предполагает превышение простой аддитивности. Оценка HSA >0 указывает, что комбинационный эффект превышает максимум ответов отдельных средств в соответствующих концентрациях.
В одном аспекте изобретения предложен способ лечения гиперпролиферативного нарушения, в котором введение соединения формулы I или его соли и одного или более средств, выбранных из 5-FU, платиносодержащего средства, иринотекана, доцетаксела, доксорубицина, гемцитабина, SN-38, капецитабина, темозоломида, эрлотиниба, PD-0325901, паклитаксела, бевацизумаба, пертузумаба, тамоксифена, рапамицина, лапатиниба, PLX-4032, MDV3100, абиратерона и GDC-0973, обеспечивает синергический эффект при лечении гиперпролиферативного нарушения. В другом аспекте синергический эффект имеет значение комбинаторного индекса менее чем приблизительно 0,8.
В одном аспекте изобретения предложен способ лечения гиперпролиферативного нарушения, в котором введение соединения формулы I или его соли в комбинации с абиратероном или его солью обеспечивает синергический эффект при лечении гиперпролиферативного нарушения. В другом аспекте синергический эффект имеет значение комбинаторного индекса менее чем приблизительно 0,8.
В одном аспекте GDC-0068 или его соль вводят в комбинации с абиратероном или его солью (и необязательно также в комбинации с преднизоном или преднизолоном) для лечения рака. В одном примере рак является раком предстательной железы. В другом примере рак является метастазирующим раком предстательной железы. В одном примере рак является аденокарциномой предстательной железы.
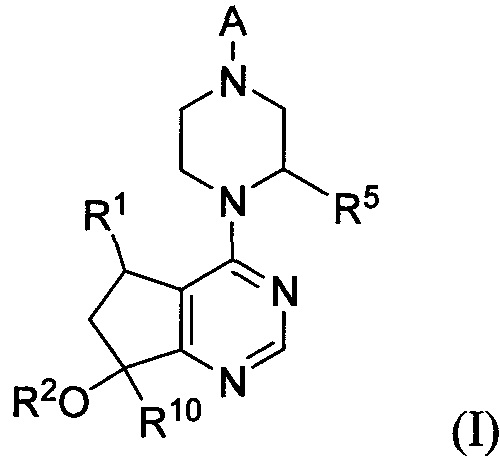
СОЕДИНЕНИЯ ФОРМУЛЫ I
Соединения формулы I включают соединение формулы I:
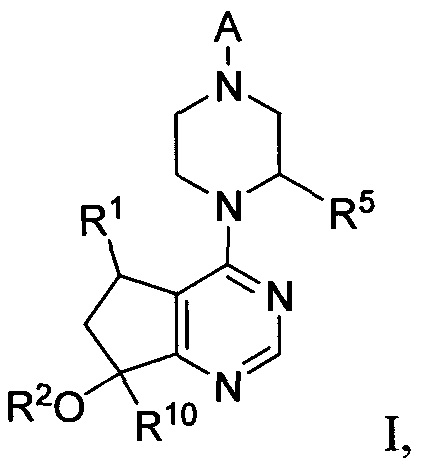 I
I
и его фармацевтически приемлемые соли, где:
R1 представляет собой H, Me, Et, винил, CF3, CHF2 или CH2F;
R2 представляет собой H или Me;
R5 представляет собой H, Me, Et или CF3;
A представляет собой 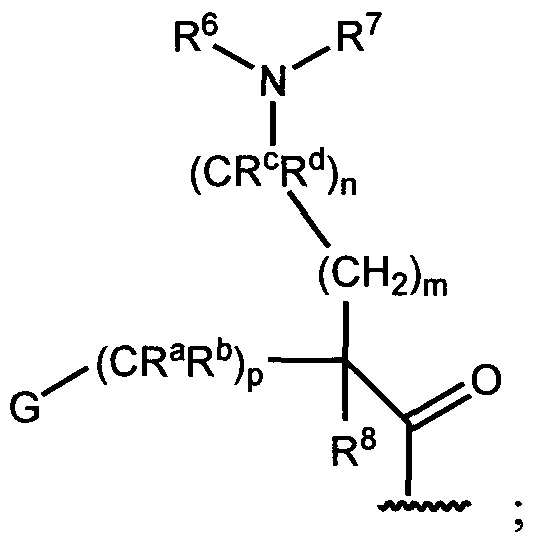
G представляет собой фенил, необязательно замещенный одной-четырьмя группами R9 или 5-6-членным гетероарилом, необязательно замещенным галогеном;
R6 и R7 независимо представляют собой H, OCH3, (C3-C6 циклоалкил)-(CH2), (C3-C6 циклоалкил)-(CH2CH2), V-(CH2)0-1, где V является 5-6-членным гетероарилом, содержащим от одного до двух гетероатомов в кольце, независимо выбранных из N, O и S, W-(CH2)1-2, где W является фенилом, который необязательно замещен F, Cl, Br, I, OMe, CF3 или Me, C3-C6-циклоалкилом, необязательно замещенным C1-C3 алкилом или O(C1-C3 алкилом), гидрокси-(C3-C6-циклоалкилом), фтор-(C3-C6-циклоалкилом), CH(CH3)CH(OH)фенилом, 4-6-членным гетероциклом, который необязательно замещен F, OH, C1-C3-алкилом, циклопропилметилом или C(=O)(C1-C3 алкилом), или C1-C6-алкилом, необязательно замещенным одной или более группами, независимо выбранными из OH, оксо, О(C1-C6-алкила), CN, F, NH2, NH(C1-C6-алкила), N(C1-C6-алкил)2, циклопропила, фенила, имидазолила, пиперидинила, пирролидинила, морфолинила, тетрагидрофуранила, оксетанила или тетрагидропиранила, или R6 и R7 вместе с азотом, к которому они присоединены, образуют 4-7-членное гетероциклическое кольцо, где указанное гетероциклическое кольцо необязательно замещено одной или более группами, независимо выбранными из OH, галогена, оксо, CF3, CH2CF3, CH2CH2OH, О(C1-C3-алкила), C(=O)CH3, NH2, NHMe, N(Me)2, S(O)2CH3, циклопропилметила и C1-C3 алкила;
Ra и Rb представляют собой H,
или Ra представляет собой H, а Rb и R6 вместе с атомами, к которым они присоединены, образуют 5-6-членное гетероциклическое кольцо, содержащее один или два атома азота в кольце;
Rc и Rd представляют собой H или Me,
или Rc и Rd вместе с атомом, к которому они присоединены, образуют циклопропильное кольцо;
R8 представляет собой H, Me, F или OH,
или R8 и R6 вместе с атомами, к которым они присоединены, образуют 5-6-членное гетероциклическое кольцо, содержащее один или два атома азота в кольце;
каждый R9 независимо представляет собой галоген, C1-C6-алкил, C3-C6-циклоалкил, O-(C1-C6-алкил), CF3, OCF3, S(C1-C6-алкил), CN, OCH2-фенил, CH2O-фенил, NH2, NH-(C1-C6-алкил), N-(C1-C6-алкил)2, пиперидин, пирролидин, CH2F, CHF2, OCH2F, OCHF2, OH, SO2C(C1-C6-алкил), C(O)NH2, C(O)NH(C1-C6-алкил) и C(O)N(C1-C6-алкил)2;
R10 представляет собой H или Me; и
m, n и p независимо представляют собой 0 или 1.
Определенное соединение формулы I является соединением, в котором A представляет собой
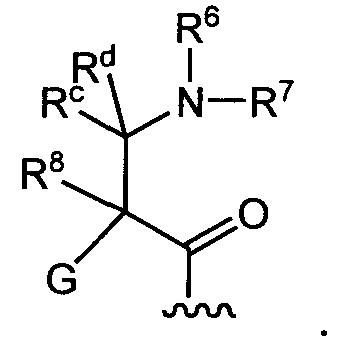
Определенное соединение формулы I является соединением формулы Ia:
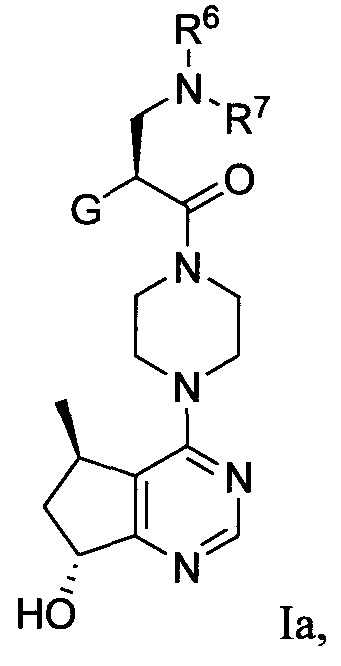
или его фармацевтически приемлемой солью.
В одном аспекте изобретения соединение формулы I не включает соединение (S)-2-(4-хлорфенил)-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-(изопропиламино)пропан-1-он формулы 1a:
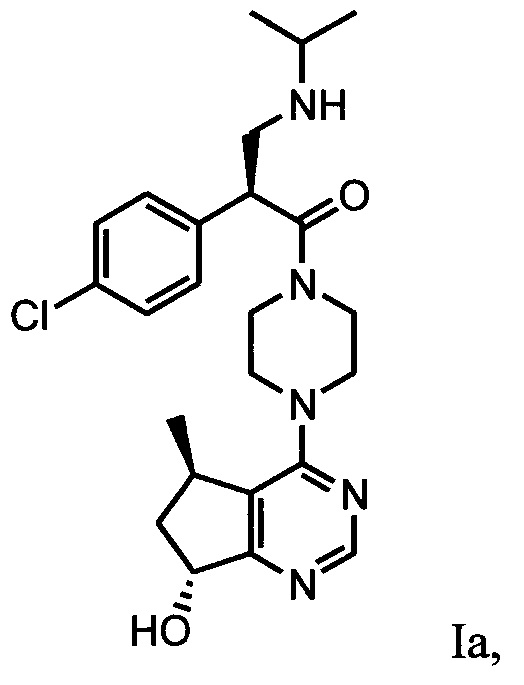
и его фармацевтически приемлемые соли (это соединение может также называться GDC-0068).
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ ФОРМУЛЫ I
Соединения настоящего изобретения могут быть синтезированы способами синтеза, которые включают процессы, аналогичные тем, которые хорошо известны в области химии, в частности, в свете описания, содержащегося в настоящей заявке. Исходные соединения общедоступны из коммерческих источников, таких как Aldrich Chemicals (Milwaukee, WI), или могут быть легко получены с использованием способов, хорошо известных квалифицированным специалистам (например, получены способами, описанными в общих чертах в публикациях Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, N.Y. (1967-1999 ed.) или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая дополнения).
Соединения формулы I могут быть получены по отдельности или в виде библиотек соединений, включающих по меньшей мере 2, например 5-1000 соединений или 10-100 соединений. Библиотеки соединений формулы I могут быть получены методом комбинаторного "расщепления и смешивания" или с помощью множественных параллельных синтезов с использованием жидкофазной или твердофазной химии, способами, известными квалифицированным специалистам. Таким образом, согласно другому аспекту изобретения предложена библиотека соединений, включающая по меньшей мере 2 соединения формулы I или их соли.
В иллюстративных целях на схемах 1-4 и схемах A-J показан общий способ получения соединений настоящего изобретения, а также ключевых промежуточных соединений. Для более подробного описания отдельных стадий реакций см. раздел Примеры, ниже. Квалифицированные специалисты сумеют оценить, что для синтеза соединений изобретения могут быть использованы другие пути синтеза. Хотя определенные исходные соединения и реагенты изображены на схемах и обсуждаются ниже, другие исходные продукты и реагенты могут быть легко использованы вместо них, обеспечивая получение различных производных и/или условий реакции. Кроме того, многие из соединений, полученных описанными ниже способами, могут быть дополнительно изменены в свете настоящего описания с использованием стандартной химии, хорошо известной квалифицированным специалистам.
Схема 1
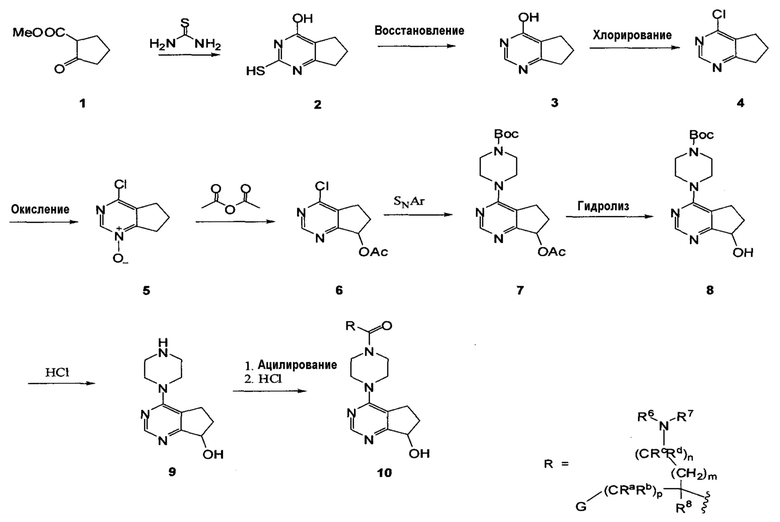
На схеме 1 показан способ получения соединения 10 формулы I, в которой R1 представляет собой H, R2 представляет собой H и R5 представляет собой H. Образование пиримидина 2 может быть выполнено путем реакции кетоэфира 1 с тиомочевиной в присутствии основания, такого как KOH, в соответствующем растворителе, таком как этанол. После восстановления меркаптогруппы соединения 2 в стандартных восстанавливающих условиях (например, Ni Ренея и NH4OH) с получением соединения 3, гидроксипиримидин 3 может быть подвергнут хлорированию в стандартных условиях (например, POCl3 в DIEA/DCE) с получением соединения 4. Затем соединение 4 окисляют в стандартных условиях (например, MCPBA в соответствующем растворителе, таком как CHCl3) с получением пиримидин-оксида 5. Обработка пиримидин-оксида уксусным ангидридом дает продукт перегруппировки 6. Соединение 7 получают путем взаимодействия соединения 6 с подходящим образом замещенным пиперидином в стандартных условиях реакции SNAr с получением соединения 7. Соединение 7 подвергают гидролизу с получением соединения 8, с которого затем снимают защиту, получая промежуточное соединение 9. Ацилирование пиперазинилциклопента[d]пиримидина 9 соответствующей аминокислотой в присутствии сшивающего реагента, такого как HBTU, с последующим снятием защиты в случае необходимости, приводит к соединению 10 формулы I.
Схема 2
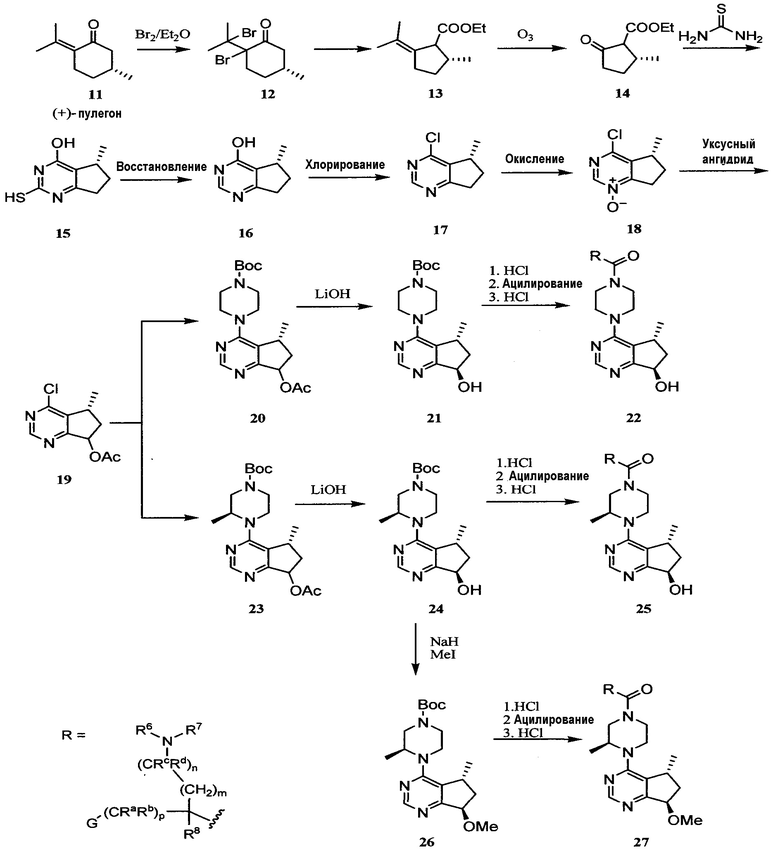
На схеме 2 показан способ получения соединений 22, 25 и 27 формулы I, в которой R1, R2 и R5 представляют собой метил. Согласно схеме 2 бромирование (+)-пулегона 11 бромом дает дибромид 12. Обработка дибромида 12 основанием, таким как этоксид натрия, приводит к пулегенату 13. Озонолиз пулегената 13 дает кетоэфир 14. Обработка кетоэфира 14 тиомочевиной в присутствии основания, такого как KOH в этаноле, с последующим восстановлением меркаптогруппы в нормальных условиях (например, катализатор Ni Ренея в аммиаке) приводит к гидроксипиримидину 16. Хлорирование гидроксипиримидина 16 при нормальных условиях (например, POCl3) дает 4-хлорпиримидин 17. Окисление 4-хлорпиримидина 17 окислителем, таким как MCPBA или пероксид водорода, дает N-оксид 18. Перегруппировка N-оксида 18 с использованием уксусного ангидрида приводит к получению промежуточного соединения 19. Соединение 19 подвергают реакции с требуемым пиперазином согласно методике, описанной на схеме 1, с получением соединения 20, в котором R5 представляет собой H, и 23, в котором R5 представляет собой Me. Соединения 20 и 23 подвергают хиральному разделению, используя ВЭЖХ с хиральной неподвижной фазой, и затем гидролизу при обработке основанием, таким как гидроксид лития, с получением соединений 21 и 24, соответственно. После снятия защиты соединения 21 и 24 подвергают реакции с соответствующей аминокислотой, получая соединения 22 и 25, соответственно.
Альтернативно, 7-гидроксигруппа соединения 24 может быть алкилирована алкилирующим реагентом, таким как алкилгалогенид, в присутствии основания, такого как NaH или KOH, с получением соединения 26, в котором R2 представляет собой Me. После снятия защиты соединение 26 подвергают реакции с соответствующей аминокислотой, получая соединение 27.
Схема 3
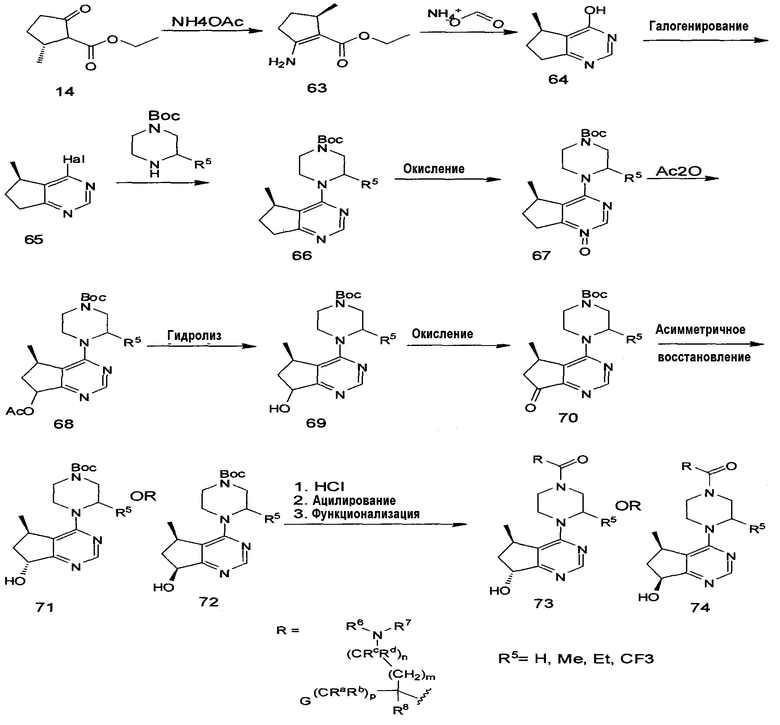
На схеме 3 показан альтернативный способ получения соединений 73 и 74. Согласно схеме 3 аминирование соединения 14 с использованием синтона аммиака дает 63. Образование пиримидина с использованием, например, формиата аммония в присутствии формамида при 50°C-250°C и/или при высоком давлении дает бициклическое звено 64. Активация 64 с использованием, например, POCl3 или SOCl2 дает активированный пиримидин 65. Вытеснение данной уходящей группы с использованием подходящего защищенного/замещенного пиперазина при температуре от 0°C до 150°C дает пиперазин 66. Окисление с использованием, например, м-хлорпероксибензойной кислоты ("MCPBA" или "m-CPBA") или Оксона® при температуре от -20°C до 50°C дает N-оксид 67. Обработка ацилирующим агентом (например, уксусным ангидридом) с последующим нагреванием (от 40°C до 200°C) вызывает перегруппировку с образованием 68. Гидролиз с использованием, например, LiOH или NaOH при температуре от 0°C до 50°C дает спирт 69. Окисление с использованием, например, условий Сверна, MnO4 или комплекса пиридин-SO3 при соответствующих температурах дает кетон 70. Асимметричное восстановление с использованием, например, каталитического количества хирального катализатора в присутствии водорода, катализатора CBS или боргидридного восстановителя в присутствии хирального лиганда приводит к (R) или (S) стереохимии в спирте 71 или 72. Альтернативно, можно использовать нехиральный восстановитель (например, H2, Pd/C), что позволяет метильной группе на циклопентановой группе обеспечить фациальную селективность и, в конечном счете, диастереоселективность. Если восстановление дает более низкую диастереоселективность, то диастереомеры могут быть разделены (например) с помощью хроматографии, кристаллизации или получения производных. Наконец, снятие защитной Boc-группы с использованием, например, кислоты при 0°C-50°C, ацилирование с использованием соответствующего производного аминокислоты и конечная функционализация амина данной аминокислоты (например, удаление любой защитной группы, алкилирование, восстановительное аминирование или ацилирование с введением новых заместителей) приводят к получению конечных соединений 73 и 74.
Схема 4
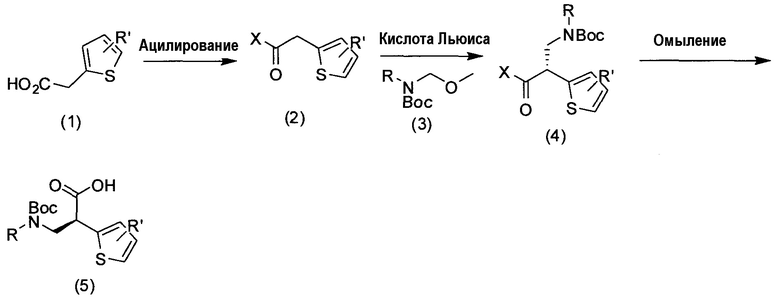
Введение хирального вспомогательного агента (например, оксазолидинона Эванса и т.д.) в соединение 1 может быть достигнуто с помощью стандартных методик ацилирования с получением конъюгата 2. Например, обработка кислоты активирующим агентом (например, COCl2) или образование смешанного ангидрида (например, 2,2-диметилпропаноилхлорида) в присутствии амина в качестве основания при температуре от -20°C до 100°C, с последующей обработкой подходящим хиральным вспомогательным агентом X, дает соединение 2. Стереохимия и выбор хирального вспомогательного агента могут определять стереохимию заново создаваемого хирального центра и диастереоселективность. Обработка соединения 2 кислотой Льюиса (например, TiCl4) при низкой температуре (например, от -20°C до -100°C) и аминным основанием (например, основанием Хунига), с последующим использованием подходящим образом замещенного предшественника иминиевого иона 3 при низкой температуре, приводит затем к получению соединения 4. Температура, кислота Льюиса и хиральный вспомогательный агент, как можно ожидать, будут влиять на диастереоселективность аддукта присоединения. Наконец, омыление в мягких условиях (например, LiOH/H2O при температуре от -10°C до 30°C) приводит к образованию требуемой кислоты 5.
Таким образом, в другом аспекте настоящего изобретения предложен способ получения соединения формулы I, включающий:
взаимодействие соединения, имеющего формулу:
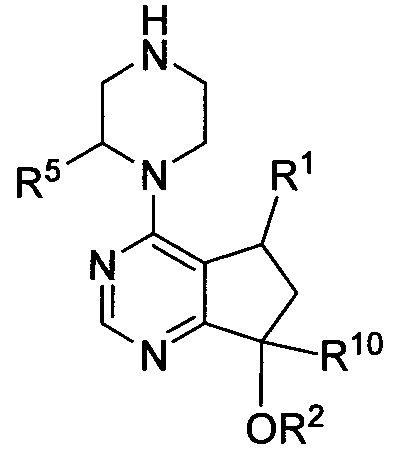
в которой R1, R2, R5 и R10 являются такими, как определено в настоящем описании, с аминокислотой, имеющей формулу:
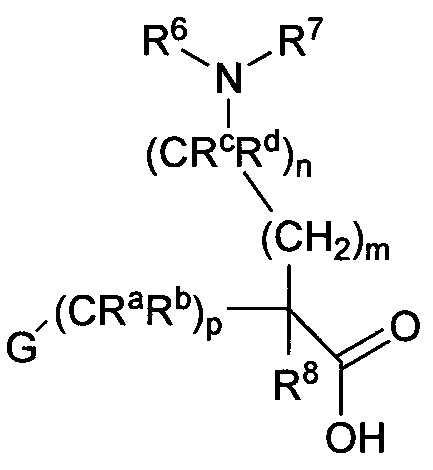
в которой R6, R7, Ra, Rb, Rc, Rd, G, m, n и p являются такими, как определено в настоящем описании.
Аминокислоты, используемые в синтезе соединений формулы I, как показано на схемах 1-4 и в примерах, являются либо коммерчески доступными, либо могут быть получены согласно способам, раскрытым в настоящем описании. Например, в некоторых вариантах осуществления аминокислоты, используемые для получения соединений формулы I, включают β-фенилглициновые аминокислоты, имеющие формулу 1A, γ-фенилглициновые аминокислоты, имеющие формулу 2A, β-фенилаланиновые аминокислоты, имеющие формулу 3A, и γ-фенилаланиновые аминокислоты, имеющие формулу 4A.
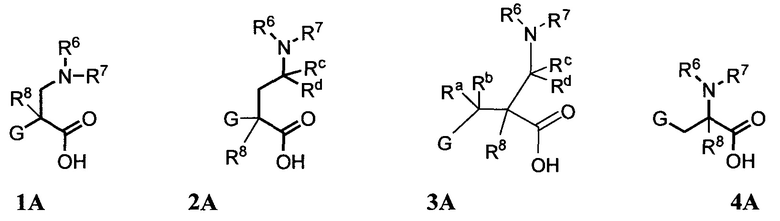
Способы получения аминокислот формул 1A-4A показаны на схемах A-J.
Схема A
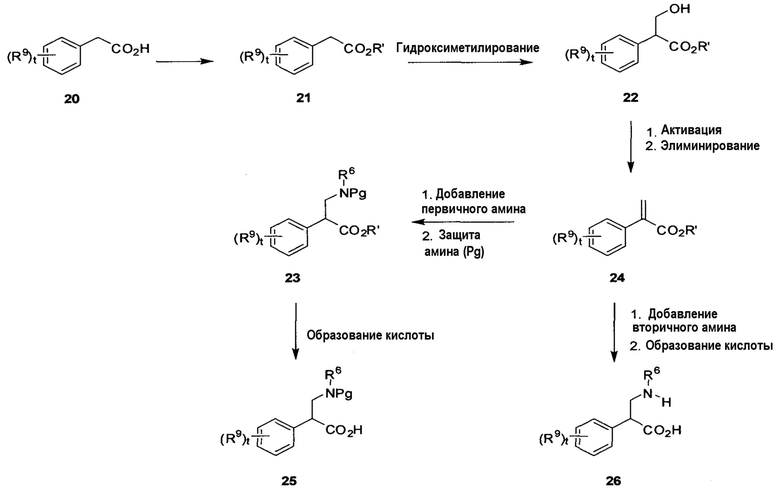
На схеме A показан способ получения необязательно замещенных β-фенилглициновых аминокислот 25 и 26 формулы 1A, в которой R8 представляет собой H, а R6 и R9 определены в настоящем описании, t равно от 0 до 4, и R7 представляет собой H или защитную группу амина. Согласно схеме A кислоту 20 преобразовывают в сложный эфир 21, в котором R’ является алкилом, с использованием стандартных условий, таких как обработка подходящим спиртом (например, MeOH) в присутствии каталитического количества кислоты, такой как концентрированная H2SO4, или сшивающего агента, такого как DCC/DMAP; или, альтернативно, путем обработки подходящим электрофилом (например, MeI, EtBr, BnBr) в присутствии основания, такого как NEt3/DMAP, при соответствующей температуре (например, от -20°C до 100°C). Соответствующий выбор сложного эфира определяется условиями, требуемыми для преобразования кислоты в конце синтеза, при этом многие соответствующие примеры и условия перечислены в ‘Protective Groups in Organic Synthesis’, by Greene and Wuts, Wiley-Interscience, third edition, Chapter 5. Введение гидроксиметильной группы с получением соединения 22 может быть выполнено путем обработки подходящим альдегидом (например, формальдегидом) в присутствии основания, такого как NaOEt, при соответствующей температуре (например, от -20°C до комнатной температуры). Активация спиртовой группы соединения 22 с образованием уходящей группы (например, мезилата, тозилата, галогенида) может быть выполнена путем обработки, например, метансульфонилхлоридом в присутствии избытка основания, такого как NEt3, DIPEA или DBU, при подходящей температуре (например, от -20°C до комнатной температуры). Во многих случаях олефин 24 может быть выделен непосредственно в данной процедуре, в других случаях может потребоваться нагревание (30°C-100°C) или добавление основания (например, DBU в случае галогенида) для завершения элиминирования с получением соединения 24. Активированный олефин 24 можно обработать требуемым первичным амином (например, этиламином) в подходящем растворителе, таком как ТГФ, при подходящей температуре (например, от -20°C до температуры кипения с обратным холодильником) с получением промежуточного аминоэфира. В случае когда соединение 24 содержит богатое электронами ароматическое кольцо или бедный электронами/объемный первичный амин, может потребоваться нагревание (например, 30-240°C в запаянной пробирке) или микроволновая химия. Защита аминогруппы (например, Boc-группой) может быть выполнена с использованием Boc2O в стандартных условиях с получением соединения 23, в котором Pg является защитной группой. Могут быть использованы альтернативные защитные группы, при этом многие соответствующие примеры перечислены в ‘Protective Groups in Organic Synthesis’, by Greene and Wuts, Wiley-Interscience, third edition, Chapter 7. Омыление сложного эфира 23 с образованием защищенной аминокислоты 25 может быть выполнено с использованием условий, подходящих для сложного эфира (например, водный LiOH для метиловых сложных эфиров, гидрирование для бензиловых сложных эфиров, кислота для трет-бутиловых сложных эфиров).
Альтернативно, активированный олефин 24 можно обработать вторичным амином (например, диэтиламином) в подходящем растворителе, таком как ТГФ, при подходящей температуре (например, от -20°C до температуры кипения с обратным холодильником) с получением промежуточного аминоэфира (не показан). В случае, когда соединение 24 содержит богатое электронами ароматическое кольцо или бедный электронами/объемный вторичный амин, может потребоваться нагревание (например, 30-240°C в запаянной пробирке) или микроволновая химия. Омыление сложного эфира с образованием аминокислоты 26 может быть выполнено с использованием условий, подходящих для сложного эфира (например, водный LiOH для метиловых сложных эфиров, гидрирование для бензиловых сложных эфиров, кислота для трет-бутиловых сложных эфиров и т.д.).
В альтернативном варианте схемы A группу Pg можно заменить группой R7 в соединениях 23 и 25.
Схема А1

На схеме А1 показана альтернатива схеме 1, где активированный олефин 24 подвергают реакции с образованием аминокислоты 26A.
Схема B
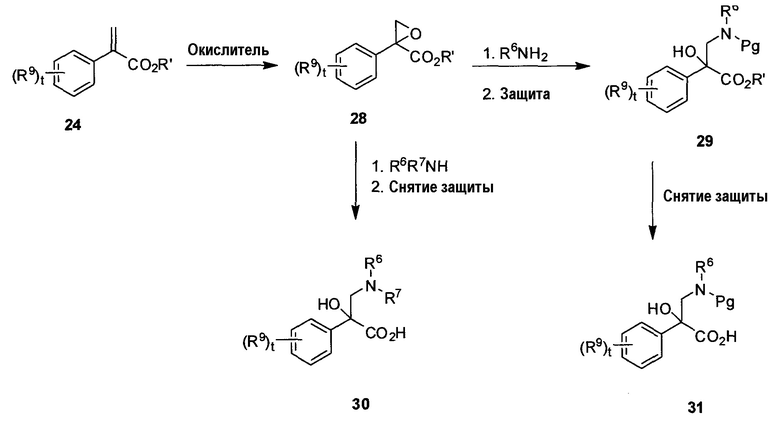
На схеме B показан способ получения необязательно замещенной β-фенилглициновой аминокислоты 30 и 31 формулы 1A, в которой R8 представляет собой OH, а R6 и R9 определены в настоящем описании, t равно от 0 до 4, и R7 определен в настоящем описании или является защитной группой амина. Окисление ненасыщенного сложного эфира 24 (полученного согласно схеме A), в котором t равно 0-4 и R’ является алкилом, с использованием стандартного окислителя, такого как MCPBA, при подходящей температуре (от комнатной температуры до температуры кипения с обратным холодильником) дает промежуточное эпоксидное соединение 28. Промежуточное соединение 28 можно обработать подходящим амином, обычно при высокой температуре (например, 50-300°C) и высоком давлении (например, в запаянной пробирке или баллоне) с получением аминоспирта 29 или 30. Если используется вторичный амин (например, при получении соединения 30), то снятие защиты со сложного эфира можно провести с использованием условий, перечисленных в ‘Protective Groups in Organic Synthesis’, by Greene and Wuts, Wiley-Interscience, third edition, Chapter 5 (например, LiOH для метилового сложного эфира, гидрирование для бензилового сложного эфира и т.д.). При использовании первичного амина (например, при получении соединения 29) защита амина (например, Boc-группой с использованием Boc-ангидрида) с последующим снятием защиты со сложного эфира (с использованием вышеуказанных условий) приводит к гидроксилированной аминокислоте 31.
Схема C
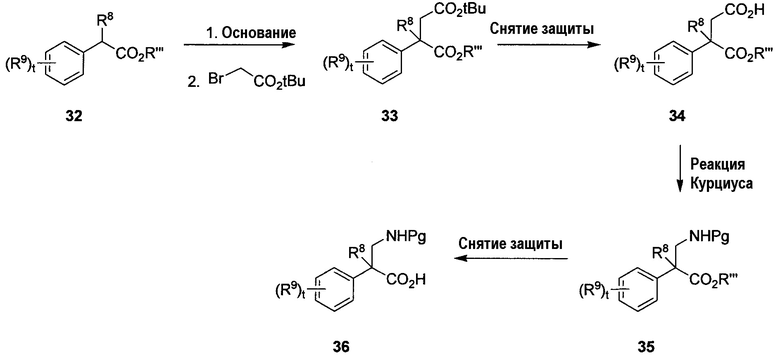
На схеме C показан способ получения необязательно замещенной β-фенилглициновой аминокислоты 36 формулы 1A, в которой R8 представляет собой метил, R6 представляет собой H, R7 представляет собой защитную группу амина, t равно от 0 до 4 и R9 определен в настоящем описании. Сложный эфир 32, в котором R”’ представляет собой алкил, можно обработать основанием (например, NaOtBu) при соответствующей температуре (например, от 0°C до температуры кипения с обратным холодильником) с образованием аниона, с последующим добавлением электрофила (например, трет-бутил-2-бромацетата) при соответствующей температуре (например, от -78°C до комнатной температуры), получая соответствующий сложный эфир 33. Удаление трет-бутилового сложного эфира соединения 33 с использованием соответствующей кислоты, такой как ТФУК или HCl, при подходящей температуре (например, от 0°C до температуры кипения с обратным холодильником) дает соединение 34. Перегруппировка Курциуса соединения 34 с использованием, например, DPPA в присутствии мягкого основания, такого как NEt3, при подходящей температуре (например, от 0°C до температуры кипения с обратным холодильником), с последующей обработкой активного промежуточного соединения спиртом (например, t-BuOH), необязательно в присутствии кислоты Льюиса (например, SnCl2), при более высокой температуре (например, 40-200°C) дает соединение 35, в котором Pg является защитной группой амина. Выбор спирта, используемого для получения соединения 35, определяет защитную группу амина (например, t-BuOH дает Boc-амин). Снятие защиты со сложноэфирной группы соединения 35 с использованием стандартных условий (например, LiOH, если защитная группа представляет собой метиловый сложный эфир, гидрирование в случае бензилового сложного эфира и т.д.) дает кислотное соединение 36.
В одном альтернативном варианте схемы C R8 может быть метилом, H или F.
В другом альтернативном варианте схемы C группу Pg можно заменить группой R7 в соединениях 35 и 36.
Схема D
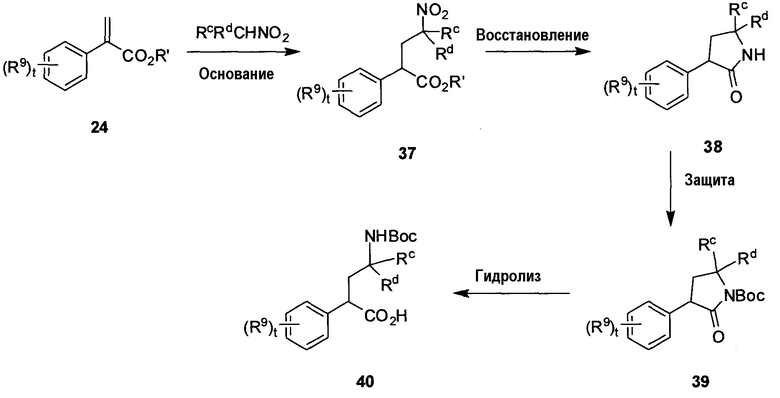
На схеме D показан способ получения необязательно замещенной γ-фенилглициновой аминокислоты 40 формулы 2A, в которой Rc, Rd и R9 определены в настоящем описании, t равно от 0 до 4, R6 представляет собой H и R7 является защитной группой амина, такой как Boc. Исходный ненасыщенный сложный эфир 24, полученный согласно схеме A, можно обработать замещенным производным нитрометана (например, нитроэтаном) в присутствии основания, такого как DBU, при подходящей температуре (например, от 0°C до комнатной температуры) с получением соответствующего аддукта 37. Нитрогруппу соединения 37 можно восстановить, используя стандартные условия (например, гидрирование, цинк/кислота и т.д.), при соответствующей температуре (например, от комнатной температуры до температуры кипения с обратным холодильником), и полученное промежуточное соединение можно циклизовать с получением промежуточного лактама 38. Защита амина, например Boc-группой, с получением соединения 39 может быть выполнена с использованием Boc2O в стандартных условиях.
Могут быть использованы альтернативные защитные группы, при этом многие подходящие примеры перечислены в‘Protective Groups in Organic Synthesis’, by Greene and Wuts, Wiley-Interscience, third edition, Chapter 7. Обработка соединения 39 водным раствором основания, такого как LiOH или KOH, при подходящей температуре (например, от 0 до 100°C) вызывает раскрытие кольца лактама с получением соответствующим образом замещенного, защищенного аминокислотного соединения 40.
В одном альтернативном варианте схемы D группу Boc можно заменить группой R7 в соединениях 39 и 40.
Схема D1
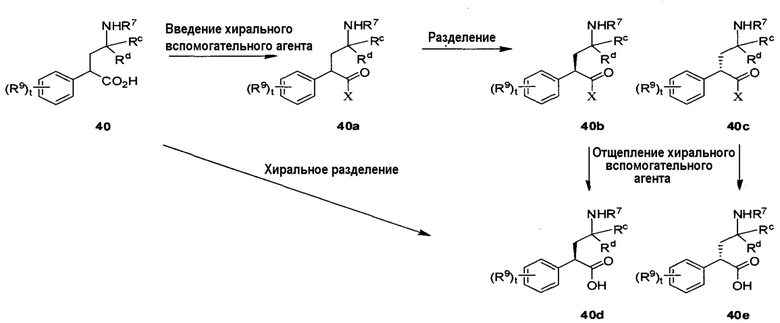
На схеме D1 показаны репрезентативные способы получения отдельных энантиомеров гамма-аминокислот 40d и 40e, где Rc, Rd и R9 определены в настоящем описании, t равно 0-4, R6 представляет собой H и R7 является защитной группой амина, такой как Boc. В одном возможном способе рацемическую аминокислоту подвергают хиральному хроматографическому разделению, используя хиральную неподвижную фазу. Альтернативно, может быть получена диастереомерная смесь, которую можно разделить с помощью общепринятых хроматографических методов. Например, активация соединения 40 (например, COCl2, основание) и введение хирального вспомогательного агента (например, оксазолидинона Эванса) в присутствии основного амина (например, основания Хунига) при температуре от -20°C до 50°C дает диастереомерную смесь соединений 40b и 40c. Эту смесь можно разделить, используя стандартные условия (например, колоночную хроматографию, ВЭЖХ, SFC (сверхкритическую флюидную хроматографию) и т.д.), с получением индивидуальных диастереомеров. Они могут быть преобразованы в требуемые кислоты путем отщепления хирального вспомогательного агента (в случае вспомогательного агента Эванса, путем использования (например) LiOH/HOOH при температуре от -15°C до комнатной) с получением соединений 40d и 40e. Может потребоваться поддерживать как можно более низкую температуру, чтобы предотвратить рацемизацию только что выделенного хирального центра.
Схема E
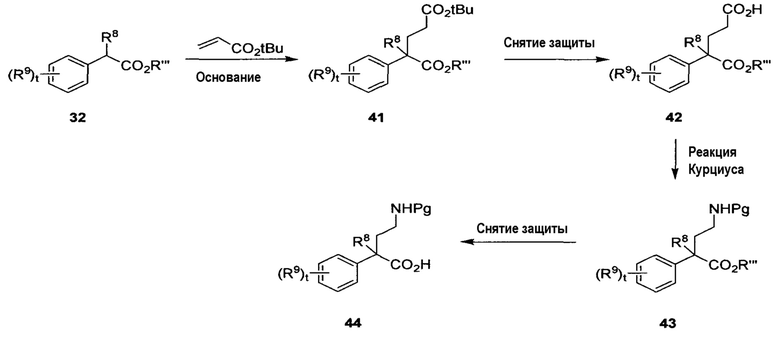
На схеме E показан способ получения необязательно замещенной γ-фенилглициновой аминокислоты 44 формулы 2A, в которой R8 представляет собой метил, R6 представляет собой H, R7 является защитной группой амина, t равно 0-4 и R9 определен в настоящем описании. Сложный эфир 32, в котором R”’ представляет собой алкил и t равно 0-4, можно обработать подходящим основанием, таким как KOtBu, при соответствующей температуре (например, от 0°C до температуры кипения с обратным холодильником), получая анион, с последующим присоединением акрилатной группы (например, трет-бутилакрилата) при температуре в пределах от -78°C до комнатной температуры с получением соответствующего сложного эфира 41. Омыление трет-бутилового эфира соединения 41 путем обработки подходящей кислотой, такой как ТФУК или HCl, при подходящей температуре (например, от 0°C до температуры кипения с обратным холодильником) дает соединение 42. Перегруппировка Курциуса соединения 42 с использованием, например, DPPA в присутствии мягкого основания, такого как NEt3, при соответствующей температуре (например, от 0°C до температуры кипения с обратным холодильником), с последующей обработкой активного промежуточного соединения соответствующим спиртом (например, tBuOH), необязательно в присутствии кислоты Льюиса (например, SnCl2), при повышенной температуре (например, 40-200°C) дает соединение 43. Выбор спирта определяет защитную группу амина соединения 43 (например, tBuOH дает Boc-амин). Снятие защиты со сложного эфира соединения 43 при стандартных условиях (например, LiOH для метилового сложного эфира, гидрирование для бензилового сложного эфира и т.д.) дает кислоту 44.
В одном альтернативном варианте схемы E группу Pg можно заменить группой R7 в соединениях 43 и 44.
Схема F
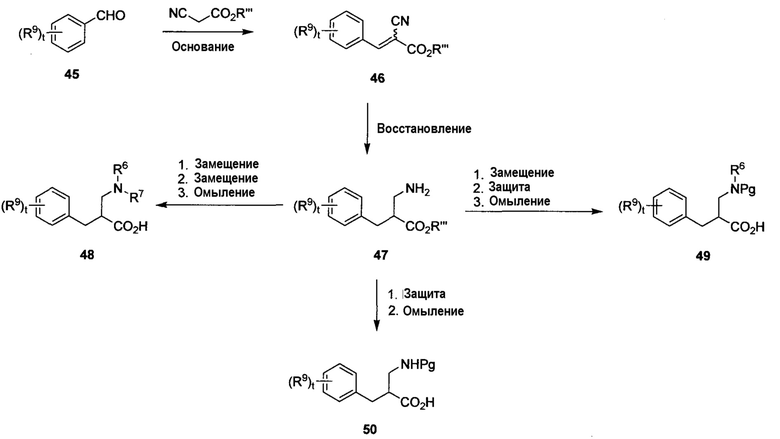
На схеме F показан способ получения необязательно замещенных β-фенилаланиновых аминокислот 48, 49 и 50 формулы 3A, в которой R6 представляет собой H, R7 является защитной группой амина, t равно 0-4 и R9 определен в настоящем описании. Надлежащим образом замещенный альдегид 45 можно обработать цианоацетатом формулы CN-CH2CO2R”’, в которой R”’ представляет собой алкил (например, этил-2-цианоацетат), в присутствии подходящего основания, такого как пиперидин, при соответствующей температуре (например, от комнатной температуры до температуры кипения с обратным холодильником) с получением ненасыщенного сложного эфира 46. Восстановление олефиновых и нитрильных групп соединения 46 с получением соединения 47 может быть выполнено множеством способов. Например, олефин может быть восстановлен любым агентом, который, как известно, производит 1,4-восстановление, таким как NaBH4. Нитрил может быть восстановлен с использованием таких агентов, как LiAlH4 или NaBH4, в присутствии кислоты Льюиса, такой как BF3⋅OEt2, или ТФУК. Могут быть использованы многие альтернативные восстановители, такие как перечисленные в ‘Reductions in Organic Chemistry’, by Hudlicky, ACS monograph, 2nd edition, Chapter 18. При желании, первичный амин 47 можно подвергнуть моноалкилированию или бисалкилированию на данной стадии, используя стандартные условия (например, восстановительное аминирование с использованием соответствующего альдегида, кислоты Льюиса и восстановителя), с получением промежуточных соединений (не показаны) в процессе получения соединений 48 и 49. Для получения первичных и вторичных аминов защита может быть выполнена с использованием любого количества защитных групп (например, ‘Protective Groups in Organic Synthesis’, by Greene and Wuts, Wiley-Interscience, third edition, Chapter 7), например, таких как Boc-группа, c использованием Boc-ангидрида при температуре от 0°C до комнатной. Отщепление сложноэфирной группы с образованием аминокислоты 48, 49 или 50 может быть выполнено с использованием водных растворов оснований, таких как LiOH или KOH, или любого из альтернативных реагентов, перечисленных в вышеуказанном тексте ‘Protective Groups’ (например, гидрирование в случае бензилового эфира).
В одном альтернативном варианте схемы F группу Pg можно заменить группой R7 в соединениях 49 или 50.
Схема G
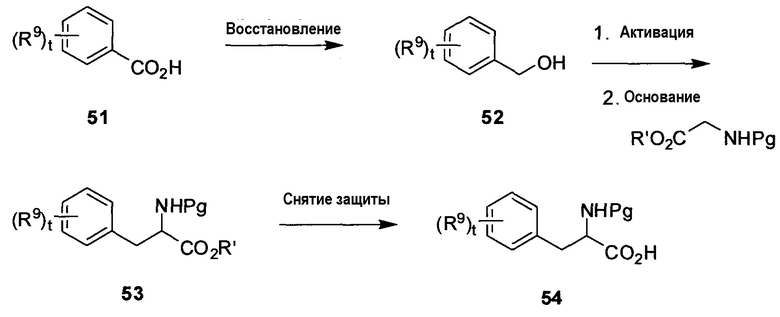
На схеме G показан способ получения необязательно замещенных α-фенилаланиновых аминокислот 54 формулы 4A, в которой R6 представляет собой H, R7 является защитной группой амина, t равно 0-4 и R9 определен в настоящем описании. Надлежащим образом замещенная кислота 51 может быть восстановлена до бензилового спирта 52 с использованием, например LiAlH4, при температуре в пределах от комнатной температуры до температуры кипения с обратным холодильником. Спиртовая группа соединения 52 может быть активирована как уходящая группа (например, галогенид, мезилат и т.д.) с использованием, например, PBr3, MsCl/NEt3 и т.д. Вытеснение данной уходящей группы с использованием защищенного производного глицина, такого как этил-2-(дифенилметиленамино)ацетат, в присутствии сильного основания, такого как LDA, nBuLi, дает промежуточный аминоэфир 53, в котором R1 представляет собой алкил и Pg является защитной группой. Подходящие защитные группы перечислены в ‘Protective Groups in Organic Synthesis’, by Greene and Wuts, Wiley-Interscience. Защитная группа амина может быть изменена на данной стадии, например, для введения Boc-группы. Последующее снятие защиты со сложного эфира 53 (например, с использованием 3 н. HCl, LiOH, гидрирования в случае бензилового эфира и т.д.) при подходящей температуре (например, от 0°C до температуры кипения с обратным холодильником) дает требуемую N-защищенную аминокислоту 54.
В одном альтернативном варианте схемы G группу Pg можно заменить группой R7 в соединении 54 после снятия защиты с соединения 53.
Схема H
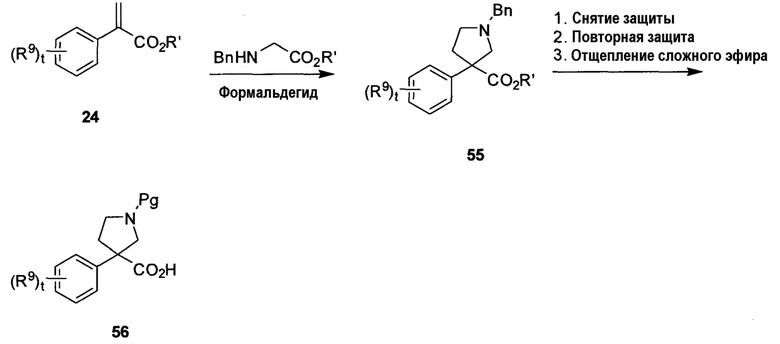
На схеме H показан способ получения необязательно замещенной γ-фенилглициновой аминокислоты 56 формулы 2A, в которой R6 и R8 вместе с атомами, к которым они присоединены, образуют спироциклическое гетероциклическое кольцо, R7 является защитной группой амина, t равно 0-4 и R9 определен в настоящем описании. Согласно схеме H ненасыщенный сложный эфир 24 можно обработать подходящим образом защищенным производным глицина (например, бензилглицином) и формальдегидом в сухих условиях (например, с дополнением молекулярных сит), при подходящей температуре (например, от комнатной температуры до температуры кипения с обратным холодильником) с получением соединения 55. Отщепление бензильной группы, используя стандартные условия (например, гидрирование, 1-хлорэтилформиат и т.д.), с последующим введением защитной группы амина, такой как Boc-группа, и отщеплением сложного эфира в стандартных условиях (например, LiOH в случае метилового сложного эфира, кислота в случае трет-бутилового сложного эфира и т.д., при температуре от 0°C до температуры кипения с обратным холодильником) дает N-защищенную аминокислоту 56.
В одном альтернативном варианте схемы H группу Pg можно заменить группой R7 в соединении 56.
Схема I
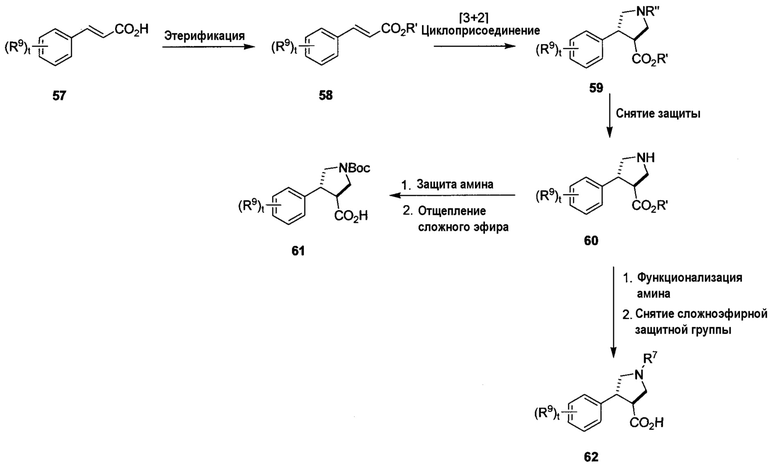
На схеме I показан способ получения необязательно замещенных β-фенилаланиновых аминокислот 61 и 62 формулы 3A, в которой R6 и Rb вместе с атомами, к которым они присоединены, образуют гетероциклическое кольцо, R7 и R9 определены в настоящем описании и t равно 0-4. Кислоту 57 преобразовывают в сложный эфир 58, используя стандартные условия, такие как обработка соответствующим спиртом (например, MeOH) в присутствии каталитической кислоты (например, концентрированной H2SO4 или TMSCl) или сшивающего агента (например, DCC/DMAP); или, альтернативно, путем обработки соответствующим электрофилом (например, MeI, EtBr, BnBr) в присутствии подходящего основания, такого как NEt3/DMAP, при соответствующих температурах (например, от -20°C до 100°C). Выбор подходящего сложного эфира определяется условиями, требуемыми для преобразования кислоты в конце синтеза, как описано в ‘Protective Groups in Organic Synthesis’, by Greene and Wuts, Wiley-Interscience, third edition, Chapter 5. Циклизация соединения 58 с получением соединения 59 может быть достигнута с использованием, например, N-(метоксиметил)(фенил)-N-((триметилсилил)метил)метанамина в присутствии ТФУК. Такой конкретный набор реагентов обеспечивает получение бензиламина, который может быть отщеплен с получением соединения 60 в стандартных условиях, таких как гидрирование при температуре от -20°C до 50°C, или любых других стандартных условиях, таких как перечислено в ‘Protective Groups in Organic Synthesis’, by Greene and Wuts, Wiley-Interscience, third edition, Chapter 7. Защита свободного амина в соединении 60 альтернативной защитной группой (например, Boc) с использованием реагентов, перечисленных в вышеуказанном тексте, таких как Boc-ангидрид, с последующим отщеплением сложного эфира c использованием стандартных условий, подходящих для сложного эфира (например, водный раствор LiOH в случае метиловых сложных эфиров, гидрирование в случае бензиловых сложных эфиров, кислота в случае трет-бутиловых сложных эфиров), приводит к кислотному соединению 61. Альтернативно, свободный амин может быть дополнительно функционализирован (например, с использованием условий алкилирования, восстановительного аминирования или ацилирования) с последующим отщеплением сложного эфира и получением третичного аминокислотного соединения 62.
Схема J
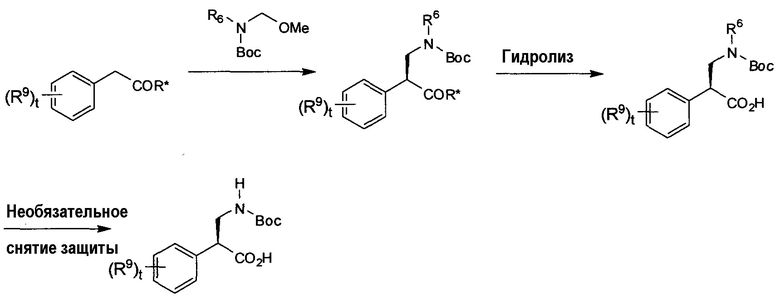
Любой энантиомер b-аминокислот может быть получен с использованием методики, такой как методика, показанная на схеме J. 2-Фенилацетат, связанный с соответствующим хиральным вспомогательным агентом (R*) (например, вспомогательным агентом Эванса или сультамом), с соответствующей стереохимией, имеющий требуемую химию в b-положении аминокислоты, можно обработать имином или синтоном иминиевого иона (например, полученным in situ в присутствии кислоты Льюиса (например, TiCl4) и замещенного требуемым образом алкоксиметанамина или N-(алкоксиметил)амида/карбамата при температуре от -100°C до 50°C). Асимметричное присоединение может потребовать присутствия кислот Льюиса (например, TiCl4), аминных оснований (например, основания Хунига) и пониженной температуры (например, от -100°C до 0°C), чтобы получить наиболее высокие уровни стереохимической индукции. Если d.e. ниже, чем требуется, то отдельные диастереомеры могут быть выделены на данной стадии с помощью (например) хроматографии или кристаллизации. Отщепление хиральных вспомогательных агентов с использованием известных способов для отщепления выбранного вспомогательного агента (например, LiOH/H2O2 при температуре от -50°C до 50°C для вспомогательного агента Эванса) затем приводит к требуемой N-защищенной b-аминокислоте с нужной стереохимией в b-положении. Дополнительно, если R6 также является защитной группой (например, 2,4-диметоксибензилом), то она может быть удалена в присутствии Boc-группы (например, гидрирование или DDQ и т.д.) с получением Boc-аминокислоты, которая после удаления Boc-группы дает первичный амин, который может быть дополнительно функционализирован алкилированием, ацилированием или восстановительным аминированием (до или после сочетания с пиримидин-пиперазиновой группой).
При получении соединений формулы I может быть необходима защита удаленных функциональных групп (например, первичных или вторичных аминов и т.д.) промежуточных соединений. Необходимость такой защиты будет меняться в зависимости от природы удаленных функциональных групп и условий способов получения. Подходящие аминозащитные группы (NH-Pg) включают ацетил, трифторацетил, трет-бутоксикарбонил (BOC), бензилоксикарбонил (CBz) и 9-флуоренилметиленоксикарбонил (Fmoc). Необходимость такой защиты легко может быть определена квалифицированным специалистом. По поводу общего описания защитных групп и их применения см. T. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
МЕТОДЫ РАЗДЕЛЕНИЯ
В любом из способов синтеза для получения соединений формулы I может быть выгодным отделить продукты реакции друг от друга и/или от исходных веществ. Целевые продукты каждой стадии или ряда стадий разделяют и/или очищают до нужной степени гомогенности методами, стандартными в данной области. Обычно такие разделения включают многофазную экстракцию, кристаллизацию из растворителя или смеси растворителей, перегонку, сублимацию или хроматографию. Хроматография может включать любое количество методов, в том числе, например: обращенно-фазовую и с нормальной фазой; гель-фильтрацию; ионный обмен; методы и установки жидкостной хроматографии высокого, среднего и низкого давления; аналитический малый масштаб; моделируемый псевдоожиженный слой (SMB) и препаративную хроматографию в тонком или толстом слое, а также методы мелкомасштабной тонкослойной и флэш-хроматографии.
Другой класс методов разделения включает обработку реакционной смеси реагентом, выбранным с целью связывания или обеспечения иным способом отделения целевого продукта, непрореагировавших исходных веществ, побочного продукта реакции или т.п. Такие реагенты включают адсорбенты или абсорбенты, такие как активированный уголь, молекулярные сита, ионообменные среды или т.п. Альтернативно, реагенты могут быть кислотами в случае основного материала, основаниями в случае кислотного материала, связывающими реагентами, такими как антитела, связывающие белки, селективные хелатообразующие соединения, такие как краун-эфиры, реагенты жидкость/жидкостной ионной экстракции (LIX) или т.п.
Выбор соответствующих методов разделения зависит от природы соответствующих материалов. Например, от температуры кипения и молекулярной массы при перегонке и возгонке, присутствия или отсутствия полярных функциональных групп в хроматографии, стабильности соединений в кислых и основных средах в многофазной экстракции и т.п. Специалист в данной области техники сумеет применить методы так, чтобы гарантировать требуемое разделение.
Диастереомерные смеси могут быть разделены на соответствующие индивидуальные диастереомеры на основе их физико-химических различий методами, известными квалифицированным специалистам, такими как хроматография и/или фракционная кристаллизация. Энантиомеры могут быть разделены путем преобразования энантиомерной смеси в диастереомерную смесь путем реакции с подходящим оптически активным соединением (например, хиральным вспомогательным агентом, таким как хиральный спирт или хлорангидрид кислоты Мошера), разделения диастереомеров и преобразования (например, гидролиза) индивидуальных диастереоизомеров в соответствующие чистые энантиомеры. Кроме того, некоторые соединения настоящего изобретения могут быть атропизомерами (например, замещенными биарилами) и рассматриваются как часть настоящего изобретения. Энантиомеры также могут быть разделены с помощью хиральной ВЭЖХ колонки.
Отдельный стереоизомер, например, энантиомер, по существу отделенный от соответствующего стереоизомера, может быть получен путем разделения рацемической смеси с использованием такого метода, как образование диастереомеров с использованием оптически активных разделяющих агентов (Eliel, E. and Wilen, S. "Stereochemistry of Organic Compounds," John Wiley & Sons, Inc., New York, 1994; Lochmuller, C. H., J. Chromatogr., (1975) 113(3):283-302). Рацемические смеси хиральных соединений изобретения могут быть разделены и выделены любым подходящим способом, включающим: (1) образование ионных, диастереомерных солей с хиральными соединениями и разделение фракционной кристаллизацией или другими методами, (2) образование диастереомерных соединений с хиральными дериватизирующими реагентами, разделение диастереомеров и преобразование в чистые стереоизомеры, и (3) разделение по существу чистых или обогащенных стереоизомеров непосредственно в хиральных условиях. См.: "Drug Stereochemistry, Analytical Methods and Pharmacology", Irving W. Wainer, Ed., Marcel Dekker, Inc., New York (1993).
Согласно способу (1) диастереомерные соли могут быть получены в результате реакции энантиомерно чистых хиральных оснований, таких как бруцин, хинин, эфедрин, стрихнин, α-метил-β-фенилэтиламин (амфетамин) и т.п., с асимметричными соединениями, имеющими кислотные функциональные группы, такими как карбоновые и сульфоновые кислоты. Диастереомерные соли могут быть разделены с помощью фракционной кристаллизации или ионной хроматографии. В случае разделения оптических изомеров аминосоединений, добавление хиральных карбоновых или сульфоновых кислот, таких как камфорсульфоновая кислота, винная кислота, миндальная кислота или молочная кислота, может привести к образованию диастереомерных солей.
Альтернативно, согласно способу (2), разделяемый субстрат подвергают взаимодействию с одним энантиомером хирального соединения с образованием диастереомерной пары (E. and Wilen, S. "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., 1994, p. 322). Диастереомерные соединения могут быть получены путем взаимодействия асимметричных соединений с энантиомерно чистыми хиральными дериватизирующими реагентами, такими как ментиловые производные, с последующим разделением диастереомеров и гидролизом с получением чистого или обогащенного энантиомера. Способ определения оптической чистоты включает получение хиральных сложных эфиров, таких как сложный ментиловый эфир, например, (-)ментилхлорформиат, в присутствии основания, или эфир кислоты Мошера, α-метокси-α-(трифторметил)фенилацетат (Jacob III. J. Org. Chem., (1982) 47:4165), рацемической смеси, и анализ 1H-ЯМР спектра на присутствие двух атропизомерных энантиомеров или диастереомеров. Устойчивые диастереомеры атропизомерных соединений могут быть разделены и выделены хроматографией с нормальной и обращенной фазой согласно способам разделения атропизомерных нафтил-изохинолинов (WO 96/15111). Согласно способу (3) рацемическая смесь двух энантиомеров может быть разделена хроматографией с использованием хиральной неподвижной фазы ("Chiral Liquid Chromatography" (1989) W. J. Lough, Ed., Chapman and Hall, New York; Okamoto, J. of Chromatogr., (1990) 513:375-378). Обогащенные или очищенные энантиомеры можно отличить с помощью методов, используемых для различения других хиральных молекул с асимметричными атомами углерода, таких как оптическое вращение и круговой дихроизм.
ХИМИОТЕРАПЕВТИЧЕСКИЕ СРЕДСТВА
Некоторые химиотерапевтические средства продемонстрировали удивительные и неожиданные свойства в комбинации с соединением формулы I или его фармацевтически приемлемой солью при ингибировании клеточной пролиферации in vitro и in vivo. Такие химиотерапевтические средства включают: 5-FU, платиносодержащее средство, иринотекан, доцетаксел, доксорубицин, гемцитабин, SN-38, капецитабин, темозоломид, эрлотиниб, PD-0325901, паклитаксел, бевацизумаб, пертузумаб, тамоксифен, рапамицин, лапатиниб, PLX-4032, MDV3100, абиратерон и GDC-0973.
5-FU (фторурацил, 5-фторурацил, номер CAS 51-21-8) является ингибитором тимидилатсинтазы и использовался в течение многих десятилетий при лечении рака, включая рак толстой и прямой кишки и рак поджелудочной железы (US 2802005; US 2885396; Duschinsky et al. (1957) J. Am. Chem. Soc. 79:4559; Hansen, R.M. (1991) Cancer Invest. 9:637-642). 5-FU называют 5-фтор-1H-пиримидин-2,4-дионом.
Карбоплатин (номер CAS 41575-94-4) является химиотерапевтическим средством, используемым против карциномы яичников, рака легкого, раковых образований головы и шеи (US 4140707; Calvert et al. (1982) Cancer Chemother. Pharmacol. 9:140; Harland et al. (1984) Cancer Res. 44:1693). Карбоплатин называют азанидом; циклобутан-1,1-дикарбоновой кислотой; платиной.
Цисплатин, цис-платина или цис-диаминдихлорплатина(II) (номер CAS 15663-27-1) является химиотерапевтическим средством, используемым для лечения различных типов раковых образований, включая саркомы, некоторые карциномы (например, мелкоклеточный рак легкого и рак яичников), лимфомы и эмбрионально-клеточные опухоли. Он был первым представителем класса платиносодержащих противоопухолевых средств, который теперь также включает карбоплатин и оксалиплатин. Цисплатин имеет структуру цис-PtCl2(NH3)2.
Оксалиплатин (номер CAS 63121-00-6) представляет собой координационный комплекс, который используется в химиотерапии рака (патент США 4169846). Оксалиплатин сравнивали с другими соединениями платины (цисплатин, карбоплатин) в случае рака на поздних стадиях (рак желудка, яичников). Оксалиплатин обычно вводят с фторурацилом и лейковорином в комбинации, известной как FOLFOX, для лечения рака толстой и прямой кишки.
Иринотекан (номер CAS 97682-44-5) является ингибитором топоизомеразы 1, который препятствует раскручиванию ДНК. Иринотекан активируется при гидролизе в SN-38, ингибитор топоизомеразы I. Ингибирование топоизомеразы I активным метаболитом SN-38 в конечном счете приводит к ингибированию как репликации, так и транскрипции ДНК. Его основным применением является рак толстой кишки, в частности, в комбинации с другими химиотерапевтическими средствами. Это включает схему FOLFIRI, которая состоит из инфузии 5-фторурацила, лейковорина и иринотекана.
Доксорубицин (номер CAS 23214-92-8) является антрациклиновым антибиотиком. Как и все антрациклины, он вызывает интеркаляцию ДНК. Доксорубицин обычно используют при лечении широкого спектра раковых опухолей, включая гемобластозы, множество типов карцином и сарком мягких тканей. Доксорубицин называют (8S,10S)-10-(4-амино-5-гидрокси-6-метилтетрагидро-2H-пиран-2-илокси)-6,8,11-тригидрокси-8-(2-гидроксиацетил)-1-метокси-7,8,9,10-тетрагидротетрацен-5,12-дионом.
Доцетаксел (номер CAS 114977-28-5) используют для лечения рака молочной железы, яичников и NSCLC (US 4814470; US 5438072; US 5698582; US 5714512; US 5750561; Mangatal et al. (1989) Tetrahedron 45:4177; Ringel et al. (1991) J. Natl. Cancer Inst. 83:288; Bissery et al. (1991) Cancer Res. 51:4845; Herbst et al. (2003) Cancer Treat. Rev. 29:407-415; Davies et al. (2003) Expert. Opin. Pharmacother. 4:553-565). Доцетаксел называют (2R,3S)-N-карбокси-3-фенилизосерин-N-трет-бутил-13-5бета,20-эпокси-1,2,4,7,10,13-гексагидрокситакс-11-ен-9-он-4-ацетат-2-бензоата тригидратом (US 4814470; EP 253738; номер CAS 114977-28-5).
Гемцитабин (номер CAS 95058-81-4) является аналогом нуклеозида, который блокирует репликацию ДНК, используется для лечения различных карцином, включая карциному поджелудочной железы, молочной железы, NSCLC и лимфомы (US 4808614; US 5464826; Hertel et al. (1988) J. Org. Chem. 53:2406; Hertel et al. (1990) Cancer Res. 50:4417; Lund et al (1993) Cancer Treat. Rev. 19:45-55). Гемцитабин называют 4-амино-1-[3,3-дифтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил]-1H-пиримидин-2-оном.
SN-38 (номер CAS 86639-52-3) является активным метаболитом иринотекана (см. выше). Он в 200 раз более активен, чем сам иринотекан. Он имеет название 7-этил-10-гидрокси-камптотецин.
Капецитабин (номер CAS 154361-50-9) является пероральным химиотерапевтическим средством, используемым при лечении метастазирующих опухолей молочной железы и толстого кишечника. Капецитабин является пролекарством, которое под действием ферментов преобразуется в 5-фторурацил в опухоли, где он подавляет синтез ДНК и замедляет рост тканей опухоли. Активация капецитабина проходит по пути с тремя ферментативными стадиями и образованием двух промежуточных метаболитов, 5’-дезокси-5-фторцитидина (5’-DFCR) и 5’-дезокси-5-фторуридина (5’-DFUR), с образованием 5-фторурацила. Капецитабин имеет название пентил[1-(3,4-дигидрокси-5-метил-тетрагидрофуран-2-ил)-5-фтор-2-оксо-1H-пиримидин-4-ил]аминометаноат.
Темозоломид (номер CAS 85622-93-1) является алкилирующим средством, которое может использоваться для лечения IV стадии астроцитомы, также известной как мультиформная глиобластома, а также меланомы, одной из форм рака кожи. Темозоломид имеет название 4-метил-5-оксо-2,3,4,6,8-пентазабицикло[4.3.0]нона-2,7,9-триен-9-карбоксамид.
Эрлотиниб (номер CAS 183321-74-6, Тарцева (TARCEVA®, OSI-774, Genentech) используется для лечения немелкоклеточного рака легкого (NSCLC), рака легкого, рака поджелудочной железы и некоторых других типов рака посредством специфичного ингибирования тирозинкиназы рецептора эпидермального фактора роста (EGFR) (US 5747498; US 6900221; Moyer et al. (1997) Cancer Res. 57:4838; Pollack et al. (1999) J. Pharmcol. Exp. Ther. 291:739; Perez-Soler et al. (2004) J. Clin. Oncol. 22:3238; Kim et al. (2002) Curr. Opin. Invest. Drugs 3:1385-1395; Blackhall et al. (2005) Expert Opin. Pharmacother. 6:995-1002). Эрлотиниб имеет название N-(3-этинилфенил)-6,7-бис(метоксиметокси)хиназолин-4-амин (номер CAS 183321-74-6) и следующую структуру:
PD-0325901 (номер CAS 391210-10-9, Pfizer) является не-АТФ-конкурентным, аллостерическим MEK-ингибитором второго поколения для потенциального лечения рака в форме таблеток для перорального приема (US 6960614; US 6972298; US 2004/147478; US 2005/085550). Клинические испытания II фазы проводили для потенциального лечения опухолей молочной железы, опухолей толстого кишечника и меланомы. PD-0325901 имеет название (R)-N-(2,3-дигидроксипропокси)-3,4-дифтор-2-(2-фтор-4-йодофениламино)бензамид и следующую структуру:
Паклитаксел (номер CAS 33069-62-4, Таксол (TAXOL®), Bristol-Myers Squibb Oncology, Princeton NJ) является соединением, выделенным из коры тиса коротколистного, Taxus brevifolia, и используется для лечения рака легкого, яичника, молочной железы и прогрессирующих форм саркомы Капоши (Wani et al (1971) J. Am. Chem. Soc. 93:2325; Mekhail et al. (2002) Expert. Opin. Pharmacother. 3:755-766). Паклитаксел имеет название (2aR-(2a-α,4-β,4a-β,6-β,9-α(α-R*,β-S*),11-α,12-α,12a-α,2b-α))бензолпропановой кислоты β-(бензоиламино)-α-гидрокси-6,12b-бис(ацетилокси)-12-(бензоилокси)-2a,3,4,4a,5,6,9,10,11,12,12a,12b-додекагидро-4,11-дигидрокси-4a,8,13,13-тетраметил-5-оксо-7,11-метано-1H-циклодека(3,4)бенз(1,2-b)оксет-9-иловый эфир и следующую структуру:
Бевацизумаб (номер CAS 216974-75-3, Авастин (AVASTIN®), Genentech) представляет собой рекомбинантное гуманизированное моноклональное антитело против VEGF, фактора роста эндотелия сосудов (US 6054297; Presta et al. (1997) Cancer Res. 57:4593-4599). Его применяют при лечении рака, при этом он подавляет рост опухоли, блокируя образование новых кровеносных сосудов. Бевацизумаб был первым клинически доступным ингибитором ангиогенеза в США, одобренным FDA в 2004 году для применения в комбинации со стандартной химиотерапией при лечении метастазирующего рака толстой кишки и большинства форм метастазирующего немелкоклеточого рака легкого. Несколько последних этапов клинических исследований, находящихся в стадии завершения, направлены на определение его безопасности и эффективности для пациентов с: адъювантным/неметастазирующим раком толстой кишки, метастазирующим раком молочной железы, метастазирующей почечноклеточной карциномой, метастазирующей мультиформной глиобластомой, метастазирующим раком яичников, метастазирующим гормонорезистентным раком предстательной железы и метастазирующим или неоперабельным, локально прогрессирующим раком поджелудочной железы (Ferrara et al. (2004) Nat. Rev. Drug Disc. 3:391-400). Бевацизумаб включает мутированные каркасные области человеческого IgG1 и антигенсвязывающие гипервариабельные области из мышиного моноклонального антитела A4.6.1 против hVEGF, которое блокирует связывание человеческого VEGF со своими рецепторами. Бевацизумаб имеет молекулярную массу приблизительно 149000 Дальтонов и гликозилирован.
Бевацизумаб и другие гуманизированные антитела против VEGF также описаны в US 6884879. Дополнительные антитела против VEGF включают антитела ряда G6 или B20, например, G6-31, B20-4.1, (WO 2005/012359; WO 2005/044853; US 7060269; US 6582959; US 6703020; US 6054297; WO 98/45332; WO 96/30046; WO 94/10202; EP 0666868B1; US 2006/009360; US 2005/0186208; US 2003/0206899; US 2003/0190317; US 2003/0203409; 20050112126; Popkov et al. (2004) Journal of Immunological Methods 288:149-164. "Антитело ряда B20" является антителом против VEGF, которое получено из последовательности антитела B20 или является B20-производным антителом согласно любой из фигур 27-29 из WO 2005/012359, все содержание которой явным образом включено в описание посредством ссылки. В одном варианте осуществления антитело ряда B20 связывается с функциональным эпитопом на VEGF человека, включающим остатки F17, M18, D19, Y21, Y25, Q89, I91, K101, E103 и C104. Другие антитела против VEGF включают антитела, которые связываются с функциональным эпитопом на VEGF человека, включающим остатки F17, M18, D19, Y21, Y25, Q89, I91, K101, E103 и C104 или, альтернативно, включающим остатки F17, Y21, Q22, Y25, D63, I83 и Q89.
Трастузумаб (Герцептин (HERCEPTIN®), huMAb4D5-8, rhuMAb HER2, Genentech) является полученным на основе рекомбинантной ДНК гуманизированного моноклонального антитела IgG1 каппа вариантом мышиного антитела к HER2, которое селективно связывается с высокой аффинностью в клеточном анализе (Kd=5 нМ) с внеклеточным доменом белка рецептора человеческого эпидермального фактора роста 2 типа, HER2 (EbB2) (US 5821337; US 6054297; US 6407213; US 6639055; Coussens L. et al. (1985) Science 230:1132-9; Slamon D.J. et al. (1989) Science 244:707-12). Трастузумаб содержит человеческие каркасные области с гипервариабельными областями мышиного антитела (4D5), которое связывается с HER2. Трастузумаб связывается с антигеном HER2 и таким образом ингибирует рост злокачественных клеток. Было показано, что трастузумаб, в in vitro анализах и у животных, ингибирует пролиферацию человеческих опухолевых клеток, в которых повышена экспрессия HER2 (Hudziak R.M. et al. (1989) Mol Cell Biol 9:1165-72; Lewis G.D. et al. (1993) Cancer Immunol Immunother; 37:255-63; Baselga J. et al. (1998) Cancer Res. 58:2825-2831). Трастузумаб опосредует антителозависимую клеточную цитотоксичность, ADCC (Hotaling T.E. et al. (1996) [abstract], Proc. Annual Meeting Am Assoc Cancer Res; 37:471; Pegram M.D. et al. (1997) [abstract]. Proc Am Assoc Cancer Res; 38:602; Sliwkowski et al. (1999) Seminars in Oncology 26(4), Suppl 12:60-70; Yarden Y. and Sliwkowski M. (2001) Nature Reviews: Molecular Cell Biology, Macmillan Magazines, Ltd., Vol. 2:127-137). HERCEPTIN® был одобрен в 1998 году для лечения пациентов с метастазирующим раком молочной железы с оверэкспрессией ErbB2 (Baselga et al. (1996) J. Clin. Oncol. 14:737-744). FDA одобрило HERCEPTIN® в 2006 году как часть схемы лечения, содержащей доксорубицин, циклофосфамид и паклитаксел для вспомогательной терапии у пациентов с HER2-положительным, узелковым раком молочной железы. Существует значительная клиническая потребность в дальнейшем развитии HER2-направленных противоопухолевых вариантов терапии для пациентов с HER2-оверэкспрессирующими опухолями или другими заболеваниями, связанными с экспрессией HER2, которые не поддаются или плохо поддаются терапии с применением HERCEPTIN®.
Пертузумаб (Омнитарг (OMNITARG™, rhuMab 2C4, Genentech) представляет собой гуманизированное антитело клинической стадии и первое в новом классе средств, известных как ингибиторы димеризации HER (HDI), которые блокируют способность рецептора HER2 взаимодействовать с другими представителями семейства рецепторов HER, т.е. HER1/EGFR, HER3 и HER4 (US 6949245; Agus et al (2002) Cancer Cell 2:127-37; Jackson et al (2004) Cancer Res 64:2601-9; Takai et al (2005) Cancer 104:2701-8). В раковых клетках, нарушая способность HER2 взаимодействовать с другими представителями семейства рецепторов HER, пертузумаб блокирует сигнализацию клеток и, в конечном счете, может привести к ингибированию роста раковой клетки и гибели раковой клетки. HDI, благодаря своему уникальному способу действия, обладают потенциалом действия в целом ряде опухолей, включая опухоли, которые не оверэкспрессируют HER2 (Mullen et al. (2007) Molecular Cancer Therapeutics 6:93-100).
Темозоломид (номер CAS 85622-93-1, темодар (TEMODAR®), темодал (TEMODAL®), Schering Plough) является пероральным химиотерапевтическим средством, одобренным FDA для лечения анапластической астроцитомы, и проходил исследования при других типах опухоли головного мозга, таких как мультиформная глиобластома (US 5260291; Stevens et al. (1984) J. Med. Chem. 27:196; Newlands et al. (1997) Cancer Treat. Rev. 23:35-61; Danson et al. (2001) Expert Rev. Anticancer Ther. 1:13-19). Темозоломид имеет название 4-метил-5-оксо-2,3,4,6,8-пентазабицикло[4.3.0]нона-2,7,9-триен-9-карбоксамид или 3,4-дигидро-3-метил-4-оксоимидазо[5,1-d]-as-тетразин-8-карбоксамид (US 5260291, номер CAS 85622-93-1) и следующую структуру:
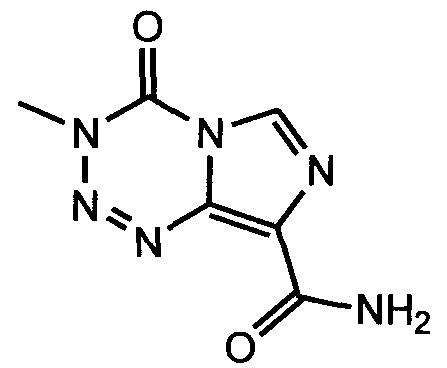
Тамоксифен (номер CAS 10540-29-1, нолвадекс (NOLVADEX®), истубал (ISTUBAL®), валодекс (VALODEX®)) является перорально активным, селективным модулятором эстрогеновых рецепторов (SERM), который используется при лечении рака молочной железы и в настоящее время является наиболее продаваемым в мире препаратом для данного показания. Тамоксифен (Nolvadex®) сначала был одобрен FDA (ICI Pharmaceuticals, теперь AstraZeneca) в 1977 году для лечения метастатического рака молочной железы (Jordan VC (2006) Br J Pharmacol 147 (Suppl 1): S269-76). Тамоксифен в настоящее время используется для лечения раннего и прогрессирующего эстроген-рецептор (ER) положительного рака молочной железы у женщин в пре- и постменопаузальном периоде (Jordan VC (1993) Br J Pharmacol 110(2):507-17). Он также одобрен FDA для предотвращения рака молочной железы у женщин при высоком риске развития заболевания и для уменьшения риска развития контралатерального (в другой груди) рака молочной железы. Тамоксифен имеет название (Z)-2-[4-(1,2-дифенилбут-1-енил)фенокси]-N,N-диметил-этанамин (номер CAS 10540-29-1) и следующую структуру:
Рапамицин (номер CAS 53123-88-9, сиролимус, Рапамун (RAPAMUNE®)) является иммунодепрессантным средством, используемым для предотвращения отторжения при трансплантации органа, и особенно полезен при пересадке почки. Рапамицин представляет собой макролидный антибиотик ("-мицин"), впервые обнаруженный как продукт бактерии Streptomyces hygroscopicus в образце почвы с острова Рапа-Нуи, более известного как остров Пасхи (Pritchard DI (2005). Drug Discovery Today 10 (10): 688-691). Рапамицин ингибирует ответ на интерлейкин-2 (IL-2) и тем самым блокирует активацию T- и B-клеток. Механизм действия рапамицина заключается в связывании цитозольного FK-связывающего белка 12 (FKBP12). Комплекс рапамицин-FKBP12 ингибирует сигнальный путь протеинкиназы mTOR (от англ. mammalian target of rapamycin - мишень рапамицина у млекопитающих) через непосредственное связывание mTOR Комплекса 1 (mTORC1). mTOR также называется FRAP (FKBP-рапамицин ассоциируемый белок) или RAFT (рапамицин и FKBP мишень). Рапамицин имеет название (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-гексадекагидро-9,27-дигидрокси-3-[(1R)-2-[(1S,3R,4R)-4-гидрокси-3-метоксициклогексил]-1-метилэтил]-10,21-диметокси-6,8,12,14,20,26-гексаметил-23,27-эпокси-3H-пиридо[2,1-c][1,4]-оксаазациклогентриаконтин-1,5,11,28,29(4H,6H,31H)-пентон (номер CAS 53123-88-9) и следующую структуру:
Лапатиниб (номер CAS 388082-78-8, тайкерб (TYKERB®), GW572016, Glaxo SmithKline) был одобрен для применения в комбинации с капецитабином (кселодой (XELODA®), Roche) для лечения пациентов с прогрессирующим или метастазирующим раком молочной железы, опухоли у которых овервэкспрессируют HER2 (ErbB2) и которые ранее получали терапию, включающую антрациклин, таксан и трастузумаб. Лапатиниб является АТФ-конкурентным, двойным ингибитором тирозинкиназы рецепторов эпидермального фактора роста (EGFR) и HER2/neu (ErbB-2) (US 6727256; US 6713485; US 7109333; US 6933299; US 7084147; US 7157466; US 7141576), который ингибирует автофосфорилирование и активацию рецепторов, связываясь с АТФ-связывающим карманом домена протеинкиназы EGFR/HER2. Лапатиниб имеет название N-(3-хлор-4-(3-фторбензилокси)фенил)-6-(5-((2-(метилсульфонил)этиламино)метил)фуран-2-ил)хиназолин-4-амин и следующую структуру:
PLX-4032 (номер CAS 1029872-55-5), как было показано, вызывает запрограммированную гибель клеток в линиях клеток меланомы. PLX-4032 прерывает этап B-Raf/MEK пути B-Raf/MEK/ERK в том случае, если B-Raf имеет распространенную мутацию V600E. PLX-4032 действует у пациентов с меланомой, раковые опухоли которых имеют мутацию BRAF V600E (т.е. в аминокислотном положении 600 на белке B-RAF присутствующий в норме валин заменен глутаминовой кислотой). Приблизительно 60% меланом имеют мутацию BRAF V600E. PLX-4032 имеет следующую структуру:
MDV3100 (номер CAS 915087-33-1) является лекарственным средством антагонистом рецепторов андрогенов, разработанным для лечения гормонорезистентного рака предстательной железы. Об уменьшении до 89% уровней простат-специфического антигена в сыворотке сообщали после приема лекарственного средства в течение месяца. В противоположность бикалутамиду, MDV3100 не вызывает транслокацию AR в ядро и, кроме того, предотвращает связывание AR с ДНК и с коактиваторными белками. Было установлено, что MDV3100 активен у больных метастазирующим кастрационно-резистентным раком предстательной железы в продолжающихся испытаниях I и II фазы. MDV3100 имеет название 4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фтор-N-метилбензамид.
Абиратерон (номер CAS 154229-19-3; см., патенты США 5604213 и 5618807) и его соль, ацетат абиратерона, является лекарственным средством, находящимся в стадии исследований, для применения при кастрационно-резистентном раке предстательной железы. Он блокирует образование тестостерона, ингибируя фермент CYP17A1 (CYP450c17), также известный как 17α-гидроксилаза/17,20 лиаза. Этот фермент участвует в образовании DHEA (ДГЭА) и андростендиона, который может в конечном счете метаболизироваться в тестостерон. Абиратерон имеет название (3S,8R,9S,10R,13S,14S)-10,13-диметил-17-(пиридин-3-ил)-2,3,4,7,8,9,10,11,12,13,14,15-додекагидро-1H-циклопента[a]фенантрен-3-ол. Его также можно вводить в виде ацетатного пролекарства (3S,8R,9S,10R,13S,14S)-10,13-диметил-17-(пиридин-3-ил)-2,3,4,7,8,9,10,11,12,13,14,15-додекагидро-1H-циклопента[a]фенантрен-3-илацетата.
ZYTIGA® (абиратерона ацетат) (JOHNSON & JOHNSON Corp) является лекарственным продуктом, одобренным в США и предназначенным для применения в комбинации с преднизоном для лечения пациентов с метастазирующим кастрационно-резистентным раком предстательной железы, которые ранее получали химиотерапию, содержащую доцетаксел.
GDC-0973 является селективным ингибитором MEK, также известной как киназа митоген-активируемой протеинкиназы (MAPKK), которая является ключевым компонентом пути RAS/RAF/MEK/ERK, часто активируемого в опухолях у человека. Неадекватная активация пути MEK/ERK способствует росту клеток в отсутствие экзогенных факторов роста. Клиническое испытание I фазы, в котором оценивают действие GDC-0973 против солидных опухолей, продолжается. GDC-0973 может быть получен, как описано в опубликованной международной заявке на патент WO2007044515(А1). GDC-0973 имеет название (S)-(3,4-дифтор-2-(2-фтор-4-йодофениламино)фенил)(3-гидрокси-3-(пиперидин-2-ил)азетидин-1-ил)метанон и следующую структуру:
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Фармацевтические композиции или лекарственные формы настоящего изобретения включают комбинации соединения формулы I, химиотерапевтического средства и одного или более фармацевтически приемлемых носителей, скользящего вещества, разбавителя или эксципиента.
Соединение формулы I и химиотерапевтические средства настоящего изобретения могут существовать как в несольватированной, так и в сольватированной формах, с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п., при этом предполагается, что изобретение охватывает как сольватированные, так и несольватированные формы.
Соединения формулы I и химиотерапевтические средства настоящего изобретения также могут существовать в различных таутомерных формах, при этом изобретение охватывает все такие формы. Термин "таутомер" или "таутомерная форма" относится к структурным изомерам с различными энергиями, которые переходят друг в друга через низкоэнергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимопревращения в результате миграции протонов, такие как кето-енольная и имино-енаминовая изомеризация. Валентные таутомеры включают взаимопревращения путем реорганизации некоторых из связывающих электронов.
Фармацевтические композиции охватывают как недозированные композиции, так и отдельные дозированные единицы, состоящие более чем из одного (например, двух) фармацевтически активного средства, включающего соединение формулы I и химиотерапевтическое средство, выбранное из списков дополнительных средств, описанных в настоящей заявке, в сочетании с любыми фармацевтически неактивными эксципиентами, разбавителями, носителями или скользящими веществами. Недозированная композиция и каждая отдельная дозированная единица могут содержать установленные количества вышеуказанных фармацевтически активных веществ. Недозированная композиция представляет собой материал, из которого еще не были сформированы отдельные дозированные единицы. Примером дозированной единицы является дозированная единица для перорального применения, такая как таблетки, пилюли, капсулы и т.п. Аналогичным образом, предполагается, что описанный в настоящей заявке способ лечения пациента путем введения фармацевтической композиции настоящего изобретения также охватывает введение недозированной композиции и отдельных дозированных единиц.
Фармацевтические композиции также охватывают изотопно-меченные соединения настоящего изобретения, которые идентичны соединениям, представленным в настоящей заявке, за исключением того факта, что один или более атомов в них заменены атомом, имеющим иную атомную массу или массовое число, нежели атомная масса или массовое число, обычно существующие в природе. Все изотопы любого конкретного атома или элемента, как указано, включены в рамки соединений изобретения и их областей применения. Примеры изотопов, которые могут быть включены в соединения, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, хлора и йода, такие как 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 32P, 33P, 35S, 18F, 36Cl, 123I и 125I. Некоторые изотопно-меченные соединения настоящего изобретения (например, соединения, меченные 3H и 14C) применимы в анализах распределения соединения и/или субстрата в тканях. Изотопы трития (3H) и углерода-14 (14C) применимы благодаря легкости их получения и возможности обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий (2H), может обеспечить некоторые терапевтические преимущества, следующие из более высокой метаболической стабильности (например, увеличенного полупериода выведения in vivo или требования уменьшенной дозировки), и, следовательно, может быть предпочтительно при некоторых обстоятельствах. Позитронно-излучающие изотопы, такие как 15O, 13N, 11C и 18F, являются пригодными для позитронно-эмиссионных томографических (ПЭТ) исследований с целью изучения занятости рецепторов субстратом. Изотопно-меченные соединения настоящего изобретения обычно могут быть получены следующими методами, аналогичными тем, которые представлены на схемах и/или в примерах ниже, путем замены не изотопно-меченного реагента изотопно-меченным реагентом.
Соединения формулы I и химиотерапевтические средства включают в лекарственную форму в соответствии со стандартной фармацевтической практикой для применения в терапевтической комбинации для терапевтической обработки (включая профилактическую обработку) гиперпролиферативных нарушений у млекопитающих, включая людей. В изобретении предложена фармацевтическая композиция, содержащая соединение формулы I в сочетании с одним или более фармацевтически приемлемым носителем, скользящим веществом, разбавителем или эксципиентом.
Подходящие носители, разбавители и эксципиенты известны специалистам в данной области техники и включают такие материалы, как углеводы, воски, водорастворимые и/или набухающие полимеры, гидрофильные или гидрофобные вещества, желатин, масла, растворители, воду и т.п. Конкретный используемый носитель, разбавитель или эксципиент будет зависеть от способов и целей, в которых применяется соединение настоящего изобретения. Растворители обычно выбирают из растворителей, которые специалистам в данной области признаны безопасными (GRAS) для введения млекопитающему. Как правило, безопасными растворителями являются нетоксичные водные растворители, такие как вода и другие нетоксичные растворители, которые растворяются в воде или смешиваются с ней. Подходящие водные растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, ПЭГ 400, ПЭГ 300) и т.д., а также их смеси. Лекарственные формы также могут включать один или более буферов, стабилизирующих веществ, поверхностно-активных веществ, смачивающих веществ, смазывающих веществ, эмульгаторов, суспендирующих веществ, консервантов, антиоксидантов, опакеров, скользящих веществ, технологических добавок, красителей, подсластителей, ароматизаторов, вкусовых добавок и других известных добавок, которые обеспечивают хорошие качества выпускаемого лекарственного средства (т.е. соединения настоящего изобретения или его фармацевтической композиции) или облегчают производство фармацевтического продукта (т.е. лекарственного препарата).
Лекарственные формы могут быть получены с использованием стандартных процедур растворения и смешивания. Например, недозированное лекарственное вещество (т.е. соединение настоящего изобретения или стабилизированная форма соединения (например, комплекс с производным циклодекстрина или другим известным комплексообразующим соединением) растворяют в подходящем растворителе в присутствии одного или большего количества эксципиентов, описанных выше. Соединение настоящего изобретения обычно включают в фармацевтические дозированные формы, чтобы обеспечить удобный контроль дозы лекарственного средства и соблюдение пациентом предписанной схемы лечения.
Фармацевтическая композиция (или лекарственная форма) для применения может быть упакована различными способами в зависимости от метода, используемого для введения лекарственного средства. Как правило, продукт для распространения включает контейнер, в котором содержится фармацевтическая композиция в подходящей форме. Подходящие контейнеры известны квалифицированным специалистам и включают такие виды, как флаконы (пластмассовые и стеклянные), пакетики, ампулы, полиэтиленовые пакеты, металлические цилиндры и т.п. Контейнер также может включать приспособление, обеспечивающее индикацию вскрытия, чтобы предотвратить случайный доступ к содержимому упаковки. Кроме того, на контейнер нанесена этикетка, которая описывает содержимое контейнера. Этикетка также может включать соответствующие предупреждения.
Фармацевтические композиции соединений настоящего изобретения могут быть получены для различных путей и типов введения. Например, соединение формулы I, обладающее необходимой степенью чистоты, необязательно может быть смешано с фармацевтически приемлемыми разбавителями, носителями, эксципиентами или стабилизаторами (Remington’s Pharmaceutical Sciences (1995) 18th edition, Mack Publ. Co., Easton, PA), в форме лиофилизированной композиции, измельченного порошка или водного раствора. Лекарственная форма может быть получена путем смешивания при температуре окружающей среды, при соответствующем pH и с требуемой степенью чистоты с физиологически приемлемыми носителями, т.е. носителями, которые являются нетоксичными для реципиентов в применяемых дозах и концентрациях. Показатель pH лекарственной формы зависит главным образом от конкретного применения и концентрации соединения, но может изменяться в пределах от приблизительно 3 до приблизительно 8.
Фармацевтическая композиция предпочтительно является стерильной. В частности, композиции, которые используются для введения in vivo, должны быть стерильными. Такая стерилизация легко достигается путем фильтрования через стерильные фильтрующие мембраны.
Фармацевтическую композицию обычно можно хранить в виде твердой композиции, лиофилизированной композиции или в виде водного раствора.
Фармацевтические композиции дозируют и вводят таким способом, т.е. в количествах, концентрациях, схемах, курсах, средах и путями введения, которые согласуются с надлежащей медицинской практикой. Факторы, учитываемые в данном контексте, включают конкретное нарушение, подвергаемое лечению, конкретное млекопитающее, подвергаемое лечению, клиническое состояние индивидуального пациента, причину нарушения, участок доставки средства, способ введения, схему введения и другие факторы, известные врачам. "Терапевтически эффективное количество" соединения, которое вводят, определяется такими факторами и является минимальным количеством, необходимым для предотвращения, уменьшения тяжести или лечения нарушения, опосредованного фактором свертывания крови. Такое количество предпочтительно ниже количества, которое является токсичным для реципиента или делает реципиента значительно более восприимчивым к кровотечению.
В качестве общей нормы, начальное фармацевтически эффективное количество соединения формулы I, вводимого перорально или парентерально, в одной дозе будет находиться в диапазоне приблизительно 0,01-1000 мг/кг, а именно приблизительно 0,1-20 мг/кг массы тела пациента в день, причем типичный используемый начальный диапазон соединения будет составлять 0,3-15 мг/кг/день. Доза соединения формулы I и доза химиотерапевтического средства, которые вводят, может изменяться для каждого в пределах от приблизительно 1 мг до приблизительно 1000 мг в единичной дозированной форме, или приблизительно от 10 мг до приблизительно 100 мг в единичной дозированной форме. Дозы соединения формулы I и химиотерапевтического средства могут быть введены в соотношении от приблизительно 1:50 до приблизительно 50:1 по массе, или в соотношении от приблизительно 1:10 до приблизительно 10:1 по массе.
Приемлемые разбавители, носители, эксципиенты и стабилизаторы нетоксичны для реципиентов в используемых дозировках и концентрациях и включают такие буферы, как фосфатный, цитратный и другие органические кислоты; антиоксиданты включают аскорбиновую кислоту и метионин; консерванты (такие как хлорид октадецилдиметилбензиламмония; хлорид гаксаметония; хлорид бензалкония, хлорид бензетония; фенол, бутиловый или бензиловый спирт; алкилпарабены, такие как метил или пропилпарабен; катехин; резорцин; циклогексанол; 3-пентанол и м-крезол); низкомолекулярные (менее чем приблизительно 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводороды, включая глюкозу, маннозу или декстрины; хелатообразующие агенты, такие как ЭДТА; сахара, такие как сахароза, маннит, трегалоза или сорбит; образующие соль противоионы, такие как натрий; комплексы металлов (например, Zn-белковые комплексы); и/или неионные поверхностно-активные вещества, такие как TWEEN™, PLURONICS™ или полиэтиленгликоль (ПЭГ). Активные фармацевтические компоненты также могут быть заключены в микрокапсулы, полученные, например, по методикам коацервации или межфазной полимеризации, например, гидроксиметилцеллюлозные или желатиновые микрокапсулы и поли(метилметацилатные) микрокапсулы, соответственно, в системах доставки коллоидных лекарственных средств (например, в липосомах, альбуминовых микросферах, микроэмульсиях, наночастицах и нанокапсулах) или в макроэмульсиях. Такие методики раскрыты в руководстве Remington’s Pharmaceutical Sciences 18th edition, (1995) Mack Publ. Co., Easton, PA.
Могут быть получены препараты соединения формулы I с пролонгированным высвобождением. Подходящие примеры препаратов пролонгированного высвобождения включают полупроницаемые матрицы твердых гидрофобных полимеров, содержащие соединение формулы I, причем указанные матрицы представлены в виде изделий, имеющих определенную форму, например, пленки или микрокапсулы. Примеры матриц пролонгированного высвобождения включают полиэфиры, гидрогели (например, поли(2-гидроксиэтилметакрилат) или поли(виниловый спирт)), полилактиды (US 3773919), сополимеры L-глутаминовой кислоты и гамма-этил-1-глутамата, неразлагаемый этиленвинилацетат, разлагаемые сополимеры молочной кислоты-гликолевой кислоты, такие как LUPRON DEPOT™ (инъецируемые микросферы, состоящие из сополимера молочной кислоты-гликолевой кислоты и ацетата лейпролида) и поли-D(-)3-гидроксимасляная кислота.
Фармацевтические композиции включают композиции, подходящие для путей введения, подробно описанных в настоящей заявке. Композиции для удобства могут быть представлены в единичной дозированной лекарственной форме и могут быть получены любым из способов, известных в области фармации. Методики и композиции обычно можно найти в Remington’s Pharmaceutical Sciences 18th Ed. (1995) Mack Publishing Co., Easton, PA. Такие способы включают стадию объединения активного вещества с носителем, который составляет один или более дополнительных компонентов. Как правило, лекарственные формы получают путем однородного и тщательного объединения активного вещества с жидкими носителями или тонко измельченными твердыми носителями, или и с теми, и с другими, с последующим, в случае необходимости, формованием продукта.
Лекарственные формы соединения формулы I и/или химиотерапевтического средства, подходящие для перорального введения, могут быть получены в виде дискретных единиц, таких как пилюли, твердые или мягкие, например, желатиновые капсулы, облатки, пастилки, леденцы, водные или масляные суспензии, диспергируемые порошки или гранулы, эмульсии, сиропы или эликсиры, каждый из которых содержит предварительно определенное количество соединения формулы I и/или химиотерапевтического средства. Количество соединения формулы I и количество химиотерапевтического средства могут быть включены в пилюлю, капсулу, раствор или суспензию в виде комбинированной композиции. Альтернативно, соединение формулы I и химиотерапевтическое средство могут быть включены отдельно в пилюлю, капсулу, раствор или суспензию для поочередного введения.
Лекарственные формы могут быть получены согласно любому способу, известному в области техники для изготовления фармацевтических композиций, и такие композиции могут содержать одну или более добавок, включающих подсластители, ароматизаторы, красители и консерванты, для получения приятного на вкус препарата. Прессованные таблетки могут быть получены путем прессования в подходящей машине активного вещества в свободно текучей форме, такой как порошок или гранулы, необязательно смешанного со связующим веществом, смазывающим веществом, инертным разбавителем, консервантом, поверхностно-активным веществом или диспергирующим веществом. Формованные таблетки могут быть изготовлены путем формования в подходящей машине смеси порошка активного компонента, увлажненного инертным жидким разбавителем.
Эксципиенты для изготовления таблеток фармацевтической композиции могут включать: наполнитель (или разбавитель) для увеличения общего объема порошкового лекарственного вещества, составляющего таблетку; разрыхлители, вызывающие распад таблетки на мелкие фрагменты, в идеальном варианте отдельные частицы лекарственного средства, при проглатывании и обеспечивающие быстрое растворение и абсорбцию лекарственного средства; связующее вещество, позволяющее формовать гранулы и таблетки с необходимой механической силой и укреплять таблетку после прессования, препятствуя ее размельчению на составляющий ее порошок в процессе упаковки, транспортировки и обычной обработки; скользящее вещество, повышающее текучесть порошка, составляющего таблетку, в процессе производства; смазывающее вещество, гарантирующее, что таблетируемый порошок не будет налипать на оборудование, используемое для прессования таблетки, в процессе изготовления. Они улучшают ток порошковых смесей через прессы и минимизируют трение и разрушение при выходе готовых таблеток из оборудования; антиадгезив, функция которого подобна функции скользящего вещества и состоит в уменьшении адгезии между порошком, составляющим таблетку, и машиной, которая используется для штамповки формы таблетки в процессе изготовления; ароматизатор, включаемый в таблетки для придания им более приятного вкуса или маскирования неприятного вкуса, и краситель, способствующий идентификации и соблюдению пациентом режима лечения.
Таблетки, содержащие активное вещество в смеси с нетоксичным фармацевтически приемлемым эксципиентом, которые являются подходящими для изготовления таблеток, являются приемлемыми. Указанные эксципиенты могут представлять собой, например, инертные разбавители, такие как карбонат кальция или натрия, лактозу, фосфат кальция или натрия; гранулирующие и разрыхляющие вещества, такие как кукурузный крахмал или альгиновая кислота; связующие вещества, такие как крахмал, желатин или гуммиарабик; и смазывающие вещества, такие как стеарат магния, стеариновая кислота или тальк. Таблетки могут не иметь покрытия или могут быть покрыты с использованием известных методов, включая микроинкапсулирование, для задержки разрушения и адсорбции в желудочно-кишечном тракте и, таким образом, обеспечения пролонгированного действия в течение более длительного периода. Например, может использоваться такое задерживающее средство, как глицерилмоностеарат или глицерилдистеарат, отдельно или с воском.
Для лечения глаз или других наружных тканей, например, полости рта и кожи, лекарственные формы предпочтительно применяют в виде наружной мази или крема, содержащих активное вещество(а) в количестве, например, 0,075-20% мас./мас. В случае включения в мазь, активные вещества могут быть использованы или с парафиновой, или со смешивающейся с водой основой мази. Альтернативно, активные вещества могут быть включены в крем на основе эмульсии масло в воде.
При необходимости водная фаза основы крема может включать многоатомный спирт, т.е. спирт, имеющий две или более гидроксильных групп, такой как пропиленгликоль, 1,3-бутандиол, маннит, сорбит, глицерин и полиэтиленгликоль (включая ПЭГ 400), а также их смеси. Лекарственные формы для наружного применения могут при необходимости включать соединение, которое увеличивает абсорбцию или проникновение активного вещества через кожу или другие участки действия. Примеры таких веществ, усиливающих проникновение в кожу, включают диметилсульфоксид и подобные аналоги.
Масляная фаза эмульсий настоящего изобретения может быть составлена из известных компонентов известным способом, включая смесь по меньшей мере одного эмульгатора с жиром или маслом, или и с жиром, и с маслом. Предпочтительно, гидрофильный эмульгатор включают вместе с липофильным эмульгатором, который действует как стабилизатор. В сочетании эмульгатор(ы) со стабилизатором(ами) или без него(них) составляет эмульгирующий воск, а воск вместе с маслом и жиром включает эмульгирующую основу мази, которая образует масляную дисперсную фазу композиций в форме крема. Эмульгаторы и стабилизаторы эмульсии, подходящие для использования в лекарственной форме, включают Tween® 60, Span® 80, цетостеариловый спирт, бензиловый спирт, миристиловый спирт, глицерилмоностеарат и лаурилсульфат натрия.
Водные суспензии фармацевтических композиций содержат активные вещества в смеси с эксципиентами, подходящими для получения водных суспензий. Такие эксципиенты включают суспендирующие вещества, такие как натриевая соль карбоксиметилцеллюлозы, кроскармеллоза, повидон, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и гуммиарабик, и диспергирующие или смачивающие вещества, такие как природный фосфатид (например, лецитин), продукт конденсации алкиленоксида с жирной кислотой (например, полиоксиэтиленстеарат), продукт конденсации этиленоксида с длинноцепочечным алифатическим спиртом (например, гептадекаэтиленоксицетанол), продукт конденсации этиленоксида с неполным сложным эфиром, полученным из жирной кислоты и ангидрида гексита (например, полиоксиэтиленсорбитан моноолеат). Водная суспензия может также содержать один или более консервантов, таких как этил или н-пропил-п-гидроксибензоат, один или более красителей, одну или более вкусовых добавок и один или более подсластителей, таких как сахароза или сахарин.
Фармацевтические композиции могут быть представлены в форме стерильного препарата для инъекций, такого как стерильная водная или масляная суспензия для инъекций. Такая суспензия может быть приготовлена в соответствии с известной из уровня техники методикой, с использованием подходящих диспергирующих или смачивающих веществ, а также суспендирующих веществ, которые были перечислены выше. Стерильный препарат для инъекций может быть раствором или суспензией в нетоксичном для парентерального введения разбавителе или растворителе, таком как 1,3-бутандиол, или может быть приготовлен в виде лиофилизированного порошка. Среди приемлемых носителей и растворителей, которые могут быть использованы, можно указать воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла могут обычно использоваться в качестве растворителя или суспендирующей среды. Для этой цели можно использовать любое жидкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, для приготовления препаратов для инъекций аналогично могут использоваться жирные кислоты, такие как олеиновая кислота.
Количество активного вещества, которое может быть объединено с веществом-носителем для получения единичной дозированной формы, будет изменяться в зависимости от пациента, подвергающегося лечению, и от конкретного способа введения. Например, лекарственная форма с замедленным высвобождением, предназначенная для перорального введения людям, может содержать приблизительно 1-1000 мг активного вещества, смешанного с соответствующим и подходящим количеством вещества-носителя, которое может варьировать примерно от 5 до примерно 95% (в массовом соотношении) от общего состава композиций. Фармацевтическая композиция может быть приготовлена так, чтобы обеспечивать для введения легко измеряемые количества. Например, водный раствор, предназначенный для внутривенной инфузии, может содержать от приблизительно 3 до 500 мкг активного вещества на миллилитр раствора для того, чтобы могла происходить инфузия соответствующего объема со скоростью 30 мл/ч.
Лекарственные формы, подходящие для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостаты и растворы, которые делают лекарственную форму изотонической с кровью предполагаемого реципиента; а также водные и неводные стерильные суспензии, которые могут включать суспендирующие вещества и загустители.
Лекарственные формы, подходящие для наружного введения в глаз, также включают глазные капли, в которых активное вещество растворено или суспендировано в подходящем носителе, особенно в водном растворителе для активного вещества. Активное вещество предпочтительно присутствует в таких лекарственных формах в концентрации приблизительно 0,5-20% мас./мас., например, приблизительно 0,5-10% мас./мас., например, приблизительно 1,5% мас./мас.
Лекарственные формы, подходящие для наружного введения в полость рта, включают леденцы, содержащие активное вещество в ароматизированной основе, обычно сахарозе и гуммиарабике или трагаканте; пастилки, содержащие активное вещество в инертной основе, такой как желатин и глицерин, или сахароза и гуммиарабик; и ополаскиватели для полости рта, содержащие активное вещество в подходящем жидком носителе.
Лекарственные формы для ректального введения могут быть представлены в виде суппозитория с подходящей основой, содержащей, например, масло какао или салицилат.
Лекарственные формы, подходящие для внутрилегочного или назального введения, имеют размер частиц, например, в диапазоне 0,1-500 микронов (включая размеры частиц в диапазоне 0,1-500 микронов с микронными приращениями, например, 0,5, 1, 30 микронов, 35 микронов и т.д.), которые вводят путем быстрой ингаляции через носовые ходы или ингаляцией через рот, достигая альвеолярных мешочков. Подходящие лекарственные формы включают водные или масляные растворы активного вещества. Лекарственные формы, подходящие для аэрозольного введения или введения в виде сухого порошка, могут быть приготовлены согласно обычным способам и могут доставляться с другими терапевтическими средствами, такими как соединения, используемые ранее при лечении или профилактике нарушений, как описано ниже.
Лекарственные формы, подходящие для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или спреев, содержащих в дополнение к активному веществу такие носители, которые, как известно из уровня техники, являются подходящими.
Лекарственные формы могут быть упакованы в контейнерах для единичной дозы или множества доз, например, запаянные ампулы и закрытые флаконы, и могут храниться в сублимационно высушенном (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например, воды, для инъекций непосредственно перед применением. Экстемпоральные растворы и суспензии для инъекций приготавливают из стерильных порошков, гранул и таблеток описанного выше типа. Предпочтительными единичными дозированными формами являются такие формы, которые содержат ежедневную дозу или единичную ежедневную субдозу, как описано выше, или их соответствующую часть, активного вещества.
В изобретении также предложены ветеринарные композиции, содержащие по меньшей мере одно активное вещество, как определено выше, вместе с ветеринарным носителем. Ветеринарные носители являются веществами, применяемыми в целях введения композиции, и могут быть твердыми, жидкими или газообразными веществами, которые в остальных отношениях являются инертными или приемлемыми в области ветеринарии и совместимыми с активным веществом. Такие ветеринарные композиции можно вводить парентерально, перорально или любым другим желательным путем.
КОМБИНИРОВАННАЯ ТЕРАПИЯ
Соединение формулы I или его фармацевтически приемлемую соль можно использовать в комбинации с другими химиотерапевтическими средствами для лечения гиперпролиферативного заболевания или нарушения, включая опухоли, раковые образования и неопластическую ткань, наряду с пренеопластическими и не неопластическими или незлокачественными гиперпролиферативными нарушениями. В некоторых вариантах осуществления соединение формулы I или его фармацевтически приемлемую соль объединяют в схеме введения в виде комбинированной терапии со вторым соединением, которое обладает антигиперпролиферативными свойствами или которое применимо для лечения гиперпролиферативного нарушения. Второе соединение схемы введения предпочтительно обладает взаимодополняющими активностями в отношении соединения формулы I или его фармацевтически приемлемой соли, при этом они не оказывают неблагоприятного действия в отношении друг друга. Такие соединения можно вводить в количествах, которые являются эффективными для предполагаемой цели. В одном варианте осуществления терапевтическую комбинацию вводят по схеме введения, в которой терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемой соли вводят в диапазоне от двух раз в день до одного раза в три недели (q3wk), и терапевтически эффективное количество химиотерапевтического средства вводят в диапазоне от двух раз в день до одного раза в три недели.
Комбинированную терапию можно проводить по одновременной или последовательной схеме. При последовательном введении комбинацию можно вводить в двух или большем количестве введений. Комбинированное введение включает совместное введение с использованием отдельной композиции и последовательное введение в любом порядке, когда предпочтительно существует такой период времени, в течение которого оба активных средства (или все) одновременно проявляют свое биологическое действие.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль можно вводить в течение периода времени продолжительностью от приблизительно 1 до приблизительно 10 дней после начала введения одного или более средств. В другом определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль можно вводить в течение периода времени продолжительностью от приблизительно 1 до 10 дней перед началом введения комбинации. В другом определенном аспекте изобретения введение соединения формулы I или его фармацевтически приемлемой соли и введение химиотерапевтического средства начинается в один и тот же день.
Подходящие дозы для любого из вышеуказанных совместно вводимых средств являются дозами, используемыми на настоящий момент, и они могут быть понижены благодаря комбинированному действию (синергии) недавно идентифицированного средства и других химиотерапевтических средств или вариантов терапии, например, для увеличения терапевтического индекса или уменьшения токсичности, или других побочных эффектов или последствий.
В конкретном варианте осуществления противоопухолевой терапии соединение формулы I или его фармацевтически приемлемую соль можно комбинировать с химиотерапевтическим средством, а также объединять с хирургической терапией и лучевой терапией. Количества соединения формулы I или его фармацевтически приемлемой соли и другого фармацевтически активного химиотерапевтического средства (средств) и относительные интервалы введения будут выбирать таким образом, чтобы достигать требуемого комбинированного терапевтического эффекта.
ВВЕДЕНИЕ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ
Соединения можно вводить любым путем, который соответствует состоянию, подвергаемому лечению. Подходящие пути включают пероральный, парентеральный (включая подкожный, внутримышечный, внутривенный, внутриартериальный, ингаляционный, внутрикожный, интратекальный, перидуральный, а также инфузионные методы), трансдермальный, ректальный, назальный, наружный (включая буккальный и подъязычный), вагинальный, внутрибрюшинный, внутрилегочный и интраназальный. Наружное введение также может включать применение трансдермального введения, например, трансдермальных пластырей или ионтофорезных устройств.
Приготовление форм лекарственных средств обсуждается в руководстве Remington’s Pharmaceutical Sciences, 18th Ed., (1995) Mack Publishing Co., Easton, PA. Другие примеры форм лекарственных средств можно найти в публикации Liberman, H. A. and Lachman, L., Eds., Pharmaceutical Dosage Forms, Marcel Decker, Vol 3, 2nd Ed., New York, NY. В случае местной иммунодепрессивной терапии, соединения можно вводить посредством внутриочагового введения, включая перфузию или иной контакт трансплантата с ингибитором перед трансплантацией. Следует понимать, что предпочтительный путь может изменяться в зависимости, например, от состояния реципиента. В случае, когда соединение вводят перорально, оно может быть составлено в форме пилюли, капсулы, таблетки и т.д. с фармацевтически приемлемым носителем, скользящим веществом или эксципиентом. В случае, когда соединение вводят парентерально, оно может быть включено в лекарственную форму с фармацевтически приемлемой парентеральной основой или разбавителем, и в дозированную единичную форму для инъекций, как подробно описано ниже.
Доза для лечения больных людей может изменяться в пределах от приблизительно 20 мг до приблизительно 1600 мг соединения формулы I или его фармацевтически приемлемой соли в день. Стандартная доза может составлять от приблизительно 50 мг до приблизительно 800 мг соединения. Дозу можно вводить один раз в день (QD), два раза в день (BID) или более часто, в зависимости от фармакокинетических (PK) и фармакодинамических (PD) свойств, включая абсорбцию, распределение, метаболизм и выведение конкретного соединения. Кроме того, токсические факторы могут влиять на дозировку и схему введения. В случае перорального введения, пилюлю, капсулу или таблетку можно принимать два раза в день, ежедневно или менее часто, например, еженедельно или один раз в две или три недели, в течение указанного периода времени. Схему можно повторять в нескольких циклах терапии.
СПОСОБЫ ЛЕЧЕНИЯ
Терапевтические комбинации: (1) соединения формулы I или его фармацевтически приемлемой соли и (2) химиотерапевтического средства являются пригодными для лечения заболеваний, состояний и/или нарушений, включая, но ими не ограничиваясь, заболевания, модулируемые AKT-киназой у млекопитающего. Раковые образования, которые можно лечить согласно способам настоящего изобретения, включают, но не ограничиваются перечисленными, мезотелиому, рак эндометрия, рак молочной железы, легкого, яичников, предстательной железы (включая кастрационно-резистентный рак предстательной железы “CRPC” ("КРПЖ")), поджелудочной железы, меланому, рак желудка, толстой кишки, глиому, злокачественные образования головы и шеи.
ИЗДЕЛИЯ
В другом варианте осуществления изобретения предложено изделие или "набор", содержащие соединение формулы I или его фармацевтически приемлемую соль, пригодные для лечения заболеваний и нарушений, описанных выше. В одном варианте осуществления набор включает контейнер и соединение формулы I или его фармацевтически приемлемую соль.
Набор может дополнительно включать этикетку или вкладыш в упаковку, на контейнере или приложенный к нему. Термин "вкладыш в упаковку" используется для обозначения инструкций, обычно включаемых в коммерческие упаковки терапевтических продуктов, которые содержат информацию по показаниям, применению, дозировке, введению, противопоказаниям и/или предупреждения относительно применения таких терапевтических продуктов. Подходящие контейнеры включают, например, бутылки, флаконы, шприцы, блистерную упаковку и т.д. Контейнер может быть изготовлен из различных материалов, таких как стекло или пластмасса. Контейнер может содержать соединение формулы I или его фармацевтически приемлемую соль, или их лекарственную форму, которая является эффективной для лечения состояния и может иметь стерильный порт доступа (например, контейнер может быть пакетом или флаконом с раствором для внутривенного введения, которые снабжены пробкой, прокалываемой иглой для подкожной инъекции). По меньшей мере одно активное вещество в композиции является соединением формулы I или его фармацевтически приемлемой солью. Этикетка или вкладыш в упаковку содержат указания о том, что композиция используется для лечения предпочтительного состояния, такого как рак. В одном варианте осуществления на этикетке или вкладыше в упаковку указано, что композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль, может использоваться для лечения нарушения, вызванного аномальным ростом клеток. На этикетке или вкладыше в упаковку также могут содержаться указания, что композиция может использоваться для лечения других нарушений. Альтернативно, или дополнительно, изделие может дополнительно включать второй контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), забуференный фосфатом физиологический раствор, раствор Рингера и раствор декстрозы. Оно может дополнительно включать другие материалы, требуемые с коммерческой и пользовательской позиции, включающие другие буферы, разбавители, фильтры, иглы и шприцы.
Набор может дополнительно включать указания по введению соединения формулы I или его фармацевтически приемлемой соли и, в случае присутствия, вторую фармацевтическую композицию. Например, если набор включает первую композицию, содержащую соединение формулы I или его фармацевтически приемлемую соль, и вторую фармацевтическую композицию, то набор может дополнительно включать указания по одновременному, последовательному или отдельному введению первой и второй фармацевтических композиций пациенту, нуждающемуся в этом.
В другом варианте осуществления наборы подходят для доставки твердых пероральных форм соединения формулы I или его фармацевтически приемлемой соли, таких как таблетки или капсулы. Такой набор предпочтительно включает множество единичных доз. Такие наборы могут включать карту, в которой дозы расположены в порядке их предполагаемого применения. Примером такого набора является "блистерная упаковка". Блистерные упаковки известны в упаковочной промышленности и широко используются для упаковки фармацевтических единичных дозированных форм. При необходимости можно предоставить памятку, например, в форме чисел, букв или других маркировок, или с календарной вставкой, на которой обозначены дни в схеме терапии, по которым можно вводить дозы.
Согласно одному варианту осуществления набор может включать (a) первый контейнер с соединением формулы I или его фармацевтически приемлемой солью, содержащимися в нем; и необязательно (b) второй контейнер со второй фармацевтической композицией, содержащейся в нем, где вторая фармацевтическая композиция содержит второе соединение с антигиперпролиферативной активностью. Альтернативно, или дополнительно, набор может дополнительно включать третий контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), забуференный фосфатом физиологический раствор, раствор Рингера и раствор декстрозы. Он может дополнительно включать другие материалы, требуемые с коммерческой и пользовательской позиции, включающие другие буферы, разбавители, фильтры, иглы и шприцы.
В случаях, когда набор включает композицию соединения формулы I или его фармацевтически приемлемой соли и второго терапевтического средства, т.е. химиотерапевтического средства, набор может включать контейнер для содержания отдельных композиций, например, разделенного флакона или разделенного пакета из фольги, впрочем, отдельные композиции также могут содержаться в одном неразделенном контейнере. Как правило, набор включает указания по введению отдельных компонентов. Форма набора особенно предпочтительна в том случае, когда отдельные компоненты предпочтительно вводят в различных дозированных формах (например, пероральной и парентеральной), вводят в различных интервалах введения, или когда лечащему врачу требуется титрование отдельных компонентов комбинации.
ОПРЕДЕЛЕННЫЕ АСПЕКТЫ ИЗОБРЕТЕНИЯ
В одном определенном аспекте изобретения гиперпролиферативным нарушением является рак.
В одном определенном аспекте изобретения рак ассоциирован с мутацией PTEN.
В одном определенном аспекте изобретения рак ассоциирован с мутацией, оверэкспрессией или амплификацией AKT.
В одном определенном аспекте изобретения рак ассоциирован с мутацией PI3K.
В одном определенном аспекте изобретения рак выбран из рака молочной железы, легкого, яичников, предстательной железы (например, кастрационно-резистентного рака предстательной железы), меланомы, рака желудка, толстой кишки, почки, головы и шеи, и глиомы.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и 5-FU вводят млекопитающему.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль, 5-FU и оксалиплатин вводят млекопитающему, и рак является раком желудка, яичников или толстой кишки.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль, 5-FU и оксалиплатин вводят млекопитающему, и рак является раком желудка, предстательной железы, головы или шеи.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль, 5-FU, оксалиплатин и фолиновую кислоту вводят млекопитающему, и рак является раком желудка, яичников или толстой кишки.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль, 5-FU, оксалиплатин и фолиновую кислоту вводят млекопитающему, и рак является раком желудка, предстательной железы, головы или шеи.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и карбоплатин вводят млекопитающему.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и карбоплатин вводят млекопитающему, и рак является раком молочной железы, легкого или предстательной железы.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и карбоплатин вводят млекопитающему, и рак является раком молочной железы, легкого, предстательной железы, головы или шеи.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и иринотекан вводят млекопитающему.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и иринотекан вводят млекопитающему, и рак является раком толстой кишки.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и доцетаксел вводят млекопитающему.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и доцетаксел вводят млекопитающему, и рак является раком молочной железы, глиомой, раком легкого, меланомой, раком яичников или предстательной железы.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и доцетаксел вводят млекопитающему, и рак является раком молочной железы, яичников или предстательной железы.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и доксорубицин вводят млекопитающему.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и доксорубицин вводят млекопитающему, и рак является раком молочной железы, легкого, яичников, глиомой или раком предстательной железы.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и SN-38 вводят млекопитающему.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и SN-38 вводят млекопитающему, и рак является раком толстой кишки.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и темозоломид вводят млекопитающему.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и темозоломид вводят млекопитающему, и рак является глиомой.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и платиносодержащее средство вводят млекопитающему.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и платиносодержащее средство вводят млекопитающему, и рак является раком яичников.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и GDC-0973 или его фармацевтически приемлемую соль вводят млекопитающему.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и GDC-0973 или его фармацевтически приемлемую соль вводят млекопитающему, и рак является раком поджелудочной железы, предстательной железы, меланомой или раком молочной железы.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и PLX-4032 или его фармацевтически приемлемую соль вводят млекопитающему.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и PLX-4032 или его фармацевтически приемлемую соль вводят млекопитающему, и рак является меланомой.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль и абиратерон или его фармацевтически приемлемую соль вводят млекопитающему, и рак является раком предстательной железы. В одном примере комбинация дополнительно включает введение преднизона.
В одном определенном аспекте изобретения GDC-0068 или его фармацевтически приемлемую соль и абиратерона ацетат вводят млекопитающему, и рак является раком предстательной железы. В одном примере комбинация дополнительно включает введение преднизолона или преднизона.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемую соль вводят перорально.
В одном определенном аспекте изобретения соединение формулы I или его фармацевтически приемлемая соль составлены в форме таблетки.
ОБЩИЕ ПРЕПАРАТИВНЫЕ МЕТОДИКИ
ПРИМЕРЫ
Для пояснения изобретения включены следующие примеры. Однако необходимо понимать, что данные примеры не ограничивают изобретение и предназначены исключительно для предложения способа осуществления изобретения. Специалисты, квалифицированные в данной области, смогут понять, что описанные химические реакции могут быть легко адаптированы для получения множества других ингибиторов AKT изобретения, и альтернативные способы получения соединений настоящего изобретения, как полагают, включены в объем настоящего изобретения. Например, синтез не представленных в качестве примера соединений согласно изобретению может быть успешно выполнен с помощью модификаций, очевидных квалифицированным специалистам, например, путем надлежащей защиты активных групп, путем использования других подходящих реагентов, известных в уровне техники, помимо описанных, и/или путем обычных модификаций условий реакции. Альтернативно, другие реакции, раскрытые в настоящей заявке или известные в уровне техники, будут расцениваться в качестве применимых для получения других соединений изобретения.
Пример 1
Получение дигидрохлорида (S)-3-амино-2-(4-хлорфенил)-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)пропан-1-она
Стадия 1: В круглодонную колбу объемом 1 л добавляли (R)-(+)-пулегон (76,12 г, 0,5 ммоль), безводный NaHCO3 (12,5 г) и безводный эфир (500 мл). Реакционную смесь охлаждали в ледяной бане в атмосфере азота. Бром (25,62 мл, 0,5 ммоль) по каплям добавляли в течение 30 минут. Смесь фильтровали и осторожно добавляли к NaOEt (21%, 412 мл, 1,11 ммоль) в охлаждаемой льдом бане. Смесь перемешивали при комнатной температуре в течение ночи и затем добавляли 1 л 5% HCl и 300 мл эфира. Водную фазу экстрагировали эфиром (2×300 мл). Объединенную органическую фазу промывали водой, сушили и концентрировали. Остаток добавляли к нагретому раствору гидрохлорида семикарбазида (37,5 г) и NaOAc (37,5 г) в воде (300 мл), а затем добавляли кипящий этанол (300 мл), получая прозрачный раствор. Смесь кипятили с обратным холодильником в течение 2,5 часов и затем оставляли на ночь при комнатной температуре и перемешивании. Смесь обрабатывали 1 л воды и 300 мл эфира. Водную фазу экстрагировали эфиром (2×300 мл). Объединенную органическую фазу промывали водой, сушили и концентрировали. Остаток очищали перегонкой в вакууме (73-76°C при 0,8 мм рт.ст.), получая (2R)-этил-2-метил-5-(пропан-2-илиден)циклопентанкарбоксилат (63 г, 64%). 1H-ЯМР (CDCl3, 400 МГц) δ 4,13 (м, 2H), 3,38 (д, J=16 Гц, 0,5H), 2,93 (м, 0,5H), 2,50-2,17 (м, 2H), 1,98 (м, 1H), 1,76 (м, 1H), 1,23 (м, 6H), 1,05 (м, 6H).
Стадия 2: (2R)-Этил-2-метил-5-(пропан-2-илиден)циклопентанкарбоксилат (24 г, 0,122 моль) в этилацетате (100 мл) охлаждали до -68°C сухим льдом/изопропанолом. Озонированный кислород (140-200 л/ч O2) пропускали через раствор в течение 3,5 часов. Реакционную смесь продували азотом при комнатной температуре до тех пор, пока цвет не исчезнет. Этилацетат удаляли в вакууме, остаток растворяли в 150 мл уксусной кислоты и охлаждали водой со льдом, после чего добавляли порошок цинка (45 г). Раствор перемешивали в течение 30 минут и затем фильтровали. Фильтрат нейтрализовывали 2 н. NaOH (1,3 л) и NaHCO3. Водную фазу экстрагировали эфиром (3×200 мл). Органическую фазу объединяли, промывали водой, сушили и концентрировали, получая (2R)-этил-2-метил-5-оксоциклопентанкарбоксилат (20 г, 96%). 1H-ЯМР (CDCl3, 400 МГц) δ 4,21 (и, 2H), 2,77 (д, J=11,2 Гц, 1H), 2,60 (м, 1H), 2,50-2,10 (м, 3H), 1,42 (м, 1H), 1,33 (м, 3H), 1,23 (м, 3H).
Стадия 3: К раствору смеси (2R)-этил-2-метил-5-оксоциклопентанкарбоксилата (20 г, 117,5 ммоль) и тиомочевины (9,2 г, 120,9 ммоль) в этаноле (100 мл) добавляли KOH (8,3 г, 147,9 ммоль) в воде (60 мл). Смесь кипятили с обратным холодильником в течение 10 часов. После охлаждения растворитель удаляли и остаток нейтрализовывали концентрированной HCl (12 мл) при 0°C, а затем экстрагировали DCM (3×150 мл). Растворитель удаляли и остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексан/этилацетат (2:1), с получением (R)-2-меркапто-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ола (12 г, 56%). МС (APCI+) [M+H]+ 183.
Стадия 4: К суспензии (R)-2-меркапто-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ола (12 г, 65,8 ммоль) в дистиллированной воде (100 мл) добавляли никель Ренея (15 г) и NH4OH (20 мл). Смесь кипятили с обратным холодильником в течение 3 часов, затем фильтровали и фильтрат концентрировали, получая (R)-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ол (9,89 г, 99%). МС (APCI+) [M+H]+ 151.
Стадия 5: Смесь (R)-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ола (5,8 г, 38,62 ммоль) в POCl3 (20 мл) кипятили с обратным холодильником в течение 5 минут. Избыток POCl3 удаляли в вакууме и остаток растворяли в DCM (50 мл). Затем смесь добавляли к насыщенному NaHCO3 (200 мл). Водную фазу экстрагировали DCM (3×100 мл) и объединенные органические фазы сушили и концентрировали. Остаток очищали с помощью хроматографии на силикагеле, элюируя этилацетатом, с получением (R)-4-хлор-5-метил-6,7-дигидро-5H-циклопента[d]пиримидина (3,18 г, 49%). 1H-ЯМР (CDCl3, 400 МГц) δ 8,81 (с, 1H), 3,47 (м, 1H), 3,20 (м, 1H), 3,05 (м, 1H), 2,41 (м, 1H), 1,86 (м, 3H), 1,47 (м, 3H).
Стадия 6: К раствору (R)-4-хлор-5-метил-6,7-дигидро-5H-циклопента[d]пиримидина (2,5 г, 14,8 ммоль) в CHCl3 (60 мл) добавляли MCPBA (8,30 г, 37,0 ммоль) тремя порциями. Смесь перемешивали при комнатной температуре в течение 2 дней. Смесь охлаждали до 0°C и по каплям добавляли Na2S2O3 (10 г) в воде (60 мл), а затем Na2CO3 (6 г) в воде (20 мл). Реакционную смесь перемешивали в течение 20 минут. Водную фазу экстрагировали CHCl3 (2×200 мл) и объединенные органические фазы концентрировали при низкой температуре (<25°C). Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетат-DCM/MeOH (20:1), с получением (R)-4-хлор-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-оксида (1,45 г, 53%). 1H-ЯМР (CDCl3, 400 МГц) δ 8,66 (с, 1H), 3,50 (м, 1H), 3,20 (м, 2H), 2,44 (м, 1H), 1,90 (м, 1H), 1,37 (д, J=7,2 Гц, 3H).
Стадия 7: Раствор (R)-4-хлор-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-оксида (1,45 г, 7,85 ммоль) в уксусном ангидриде (20 мл) нагревали до 110°C в течение 2 часов. После охлаждения избыток растворителя удаляли в вакууме. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексан/этилацетат (3:1), с получением (5R)-4-хлор-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-7-илацетата (1,25 г, 70%). 1H-ЯМР (CDCl3, 400 МГц) δ 8,92 (м, 1H), 6,30-6,03 (м, 1H), 3,60-3,30 (м, 1H), 2,84 (м, 1H), 2,40-2,20 (м, 1H), 2,15 (д, J=6 Гц, 2H), 1,75 (м, 2H), 1,47 (д, J=6,8, 2H), 1,38 (д, J=7,2, 1H). МС (APCI+) [M+H]+ 227.
Стадия 8: К раствору (5R)-4-хлор-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-7-илацетата (0,5 г, 2,2 ммоль) в NMP (10 мл) добавляли 1-Boc-пиперазин (0,9 г, 4,8 ммоль). Реакционную смесь нагревали до 110°C в течение 12 часов. После охлаждения реакционную смесь разбавляли этилацетатом (200 мл) и промывали водой (6×100 мл). Органическую фазу сушили и концентрировали. Остаток очищали с помощью хроматографии на силикагеле, элюируя этилацетатом, с получением трет-бутил-4-((5R)-7-ацетокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (0,6 г, 72%). 1H-ЯМР (CDCl3, 400 МГц) δ 8,60 (д, 1H), 6,05-5,90 (м, 1H), 3,80-3,30 (м, 9H), 2,84 (м, 1H), 2,20 (м, 1H), 1,49 (с, 9H), 1,29-1,20 (м, 3H). МС (APCI+) [M+H]+ 377. Полученную смесь диастереомеров очищали с помощью разделения хиральной ВЭЖХ (колонка Chiralcel ODH, 250×20 мм, гексан/EtOH 60:40, 21 мл/мин). Первый пик (RT=3,73 мин) относился к трет-бутил-4-((5R,7R)-7-ацетокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилату (0,144 г, 24%). Второй пик (RT=5,66 мин) относился к трет-бутил-4-((5R,7S)-7-ацетокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилату (0,172 г, 29%). МС (APCI+) [M+H]+ 377.
Стадия 9: К раствору трет-бутил-4-((5R,7R)-7-ацетокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (0,144 г, 0,383 ммоль) в ТГФ (4 мл) добавляли LiOH (3M, 2 мл). Смесь перемешивали при комнатной температуре в течение 6 часов и затем гасили 2 н. HCl (3 мл). Растворитель отгоняли и остаток очищали с помощью хроматографии на силикагеле, элюируя этилацетатом, с получением трет-бутил-4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазине-1-карбоксилата (89 мг, 70%). 1H-ЯМР (CDCl3, 400 МГц) δ 8,52 (с, 1H), 5,48 (уш., 1H), 5,14 (м, 1H), 3,82-3,40 (м, 9H), 2,20 (м, 2H), 1,49 (с, 9H), 1,19 (д, J=6,8 Гц, 3H). МС (APCI+) [M+H]+ 335.
Стадия 10: трет-Бутил-4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилат обрабатывали HCl (4M в диоксане, 2 мл) в DCM (5 мл) в течение 6 часов, получая дигидрохлорид (5R,7R)-5-метил-4-(пиперазин-1-ил)-6,7-дигидро-5H-циклопента[d]пиримидин-7-ола. МС (APCI+) [M+H]+ 235.
Стадия 11: трет-Бутил-2,4-диметоксибензилкарбамат (3,96 г, 14,8 ммоль) растворяли в ТГФ (74 мл) и охлаждали до -78°C. Раствор обрабатывали бутиллитием (7,44 мл, 16,3 ммоль) по каплям в течение пяти минут, получая бледно-желтый раствор. Раствору давали перемешиваться в течение 15 минут перед добавлением хлор(метокси)метана (1,35 мл, 17,8 ммоль) по каплям (чистый). Реакционную смесь перемешивали при -78°C в течение 10 минут, затем давали медленно нагреваться до температуры окружающей среды в течение ночи. Реакционную смесь концентрировали в вакууме, получая желтый гель, который распределяли между полунасыщенным раствором NH4C1 и эфиром. Водный слой однократно экстрагировали и объединяли органические фракции. Органический слой промывали водой, затем солевым раствором, разделяли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. 1H-ЯМР подтвердила присутствие почти чистого (>90%) целевого трет-бутил-2,4-диметоксибензил(метоксиметил)карбамата (4,81 г, выход 104%) в виде бледно-желтого масла, которое использовали без очистки.
Стадия 12: (R)-4-бензил-3-(2-(4-хлорфенил)ацетил)оксазолидин-2-он (3,00 г, 9,10 ммоль) растворяли в DCM (91 мл) и охлаждали до -78°C. К раствору добавляли 1М раствор TiCl4 в толуоле (11,4 мл, 11,4 ммоль), затем DIEA (1,66 мл, 9,55 ммоль), получая темно-фиолетовую реакционную смесь. Смеси давали перемешиваться в течение 15 минут, после чего по каплям добавляли трет-бутил-2,4-диметоксибензил(метоксиметил)карбамат (3,40 г, 10,9 ммоль) в виде раствора в DCM (10 мл). Реакционной смеси давали перемешиваться в течение 15 минут при -78°C, затем давали нагреться до -18°C в ледяной бане с солевым раствором в течение одного часа. Этой реакционной смеси давали медленно нагреваться до 0°C более 2,5 часов. Затем реакцию гасили добавлением насыщенного раствора NH4Cl (100 мл). Слои разделяли и органические слои однократно экстрагировали DCM. Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме, получая желтое масло. Остаток очищали с помощью хроматографии (силикагель, элюирование смесью 4:1 гексаны:этилацетат) с получением чистого продукта в виде бесцветного масла трет-бутил-2,4-диметоксибензил((S)-3-((R)-4-бензил-2-оксооксазолидин-3-ил)-2-(4-хлорфенил)-3-оксопропил)карбамата (4,07 г, выход 73,5%). Полученный трет-бутил-2,4-диметоксибензил((S)-3-((R)-4-бензил-2-оксооксазолидин-3-ил)-2-(4-хлорфенил)-3-оксопропил)карбамат (680 мг, 1,12 ммоль) растворяли в DCM (10,6 мл) и воде (560 мкл; DCM:вода 19:1) при температуре окружающей среды. Раствор обрабатывали DDQ (380 мг, 1,67 ммоль) и оставляли реакционную смесь при перемешивании на один день до завершения реакции согласно ТСХ и ЖХМС анализу. Реакционную смесь разбавляли DCM и дважды промывали полунасыщенным раствором NaHCO3. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме, получая желто-оранжевое масло. Остаток очищали с помощью хроматографии (силикагель, элюирование смесью 9:1 гексаны:этилацетат), получая смесь альдегидного побочного продукта и трет-бутил-(S)-3-((R)-4-бензил-2-оксооксазолидин-3-ил)-2-(4-хлорфенил)-3-оксопропилкарбамата (неразделимую) в виде бледно-желтого масла (общая масса 729 мг). ЖХ/МС (APCI+) m/z 359,1 [M-BOC+H]+.
Стадия 13: К раствору LiOH-H2O (0,0978 г, 2,33 ммоль) в смеси 2:1 ТГФ:H2O (33 мл) добавляли 35% H2O2 (0,240 мл, 2,91 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 35 минут и затем охлаждали до 0°C. Раствор, содержащий смесь трет-бутил-(S)-3-((R)-4-бензил-2-оксооксазолидин-3-ил)-2-(4-хлорфенил)-3-оксопропилкарбамата (0,535 г, 1,17 ммоль) и 2,4-диметоксибензальдегида (0,194 г, 1,17 ммоль) в ТГФ (7 мл), добавляли по каплям с помощью капельной воронки. Ледяной бане давали медленно нагреться и реакционную смесь оставляли на ночь при перемешивании. Затем реакционную смесь охлаждали до 0°C и добавляли 1М Na2SO3 (7 мл). Смесь перемешивали в течение 5 минут и затем нагревали до комнатной температуры и перемешивали еще 20 минут. Затем реакционную смесь переносили в делительную воронку и промывали эфиром (3×). Водный слой подкисляли KHSO4(s) и экстрагировали смесь DCM (2×). Объединенные экстракты сушили (Na2SO4), фильтровали и концентрировали, получая (S)-3-(трет-бутоксикарбониламино)-2-(4-хлорфенил)пропановую кислоту (0,329 г, выход 94,2%) в виде белого остатка. ЖХ/МС (APCI+) m/z 200 [M-BOC+H]+.
Стадия 14: Смесь 4М HCl/диоксан (5,49 мл, 22,0 ммоль) добавляли к раствору (S)-3-(трет-бутоксикарбониламино)-2-(4-хлорфенил)пропановой кислоты (0,329 г, 1,10 ммоль) в смеси 2:1 диоксан:DCM (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи (16 часов), после чего ее концентрировали до 1/3 объема. Полученную мутную смесь разбавляли эфиром и снова концентрировали смесь до 1/3 объема. Смесь снова разбавляли эфиром (20 мл) и твердые частицы отделяли фильтрованием через воронку со стеклянным фильтром средней пористости под давлением азота, промывали эфиром (5×10 мл), сушили под давлением азота и сушили в вакууме, получая гидрохлорид (S)-3-амино-2-(4-хлорфенил)пропановой кислоты (0,199 г, выход 76,8%) в виде белого порошка. Чистота по ВЭЖХ >99% площади. ЖХ/МС (APCI+) m/z 200.
Стадия 15: Boc2O (0,368 г, 1,69 ммоль) добавляли к раствору гидрохлорида (S)-3-амино-2-(4-хлорфенил)пропановой кислоты (0,199 г, 0,843 ммоль) и пентагидрата гидроксида тетраметиламмония (0,382 г, 2,11 ммоль) в смеси 10:1 MeCN:H2O (7,7 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре (12 часов), после чего MeCN удаляли на роторном испарителе. Смесь разбавляли водой и промывали эфиром (2×). Водный слой подкисляли KHSO4(s), смесь экстрагировали DCM и объединенные экстракты сушили (Na2SO4), фильтровали и концентрировали, получая (S)-3-(трет-бутоксикарбониламино)-2-(4-хлорфенил)пропановую кислоту (0,229 г, выход 90,6%) в виде пены. Чистота по ВЭЖХ >99% площади. ЖХ/МС (APCI+) m/z 200 [M-BOC+H]+.
Стадия 16: К раствору дигидрохлорида (5R,7R)-5-метил-4-(пиперазин-1-ил)-6,7-дигидро-5H-циклопента[d]пиримидин-7-ола (88 мг, 0,29 ммоль) и (S)-3-(трет-бутоксикарбониламино)-2-(4-хлорфенил)пропановой кислоты (86 мг, 0,29 ммоль) в DCM (10 мл) и диизопропилэтиламине (0,22 мл, 1,3 ммоль) добавляли HBTU (110 мг, 0,29 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли, остаток растворяли в этилацетате (100 мл) и промывали водой (6×50 мл). Органическую фазу сушили и концентрировали, получая трет-бутил-(S)-2-(4-хлорфенил)-3-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-оксопропилкарбамат (116 мг, 78%). 1H-ЯМР (CDCl3, 400 МГц) δ 8,51 (с, 1H), 7,34-7,20 (м, 4H), 5,15-5,09 (м, 2H), 4,15-4,05 (м, 1H), 3,87-3,85 (м, 2H), 3,78-3,38 (м, 7H), 3,22-3,19 (м, 1H), 2,20-2,10 (м, 2H), 1,48 (с, 9H), 1,41 (с, 9H), 1,14-1,12 (д, J=7,2 Гц, 3H). МС (APCI+) [M+H]+ 516.
Стадия 17: Обработка трет-бутил-(S)-2-(4-хлорфенил)-3-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-оксопропилкарбамата HCl (4M в диоксане, 2 мл) в DCM (5 мл) в течение 6 часов давала дигидрохлорид (S)-3-амино-2-(4-хлорфенил)-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)пропан-1-она. 1H-ЯМР (D2O, 400 МГц) δ 8,38 (с, 1H), 7,37-7,35 (д, J=8,4 Гц, 2H), 7,23-7,21 (д, J=8,4 Гц, 2H), 5,29-5,25 (м, 1H), 4,64 (с, 9H), 4,31-4,28 (м, 1H), 4,11 (м, 1H), 3,88-3,79 (м, 2H), 3,70-3,20 (м, 10H), 2,23-2,17 (м, 1H), 2,07-1,99 (м, 1H), 1,22-1,20 (м, 2H), 0,98-0,96 (д, J=6,8 Гц, 2H). МС (APCI+) [M+H]+ 416.
Пример 2
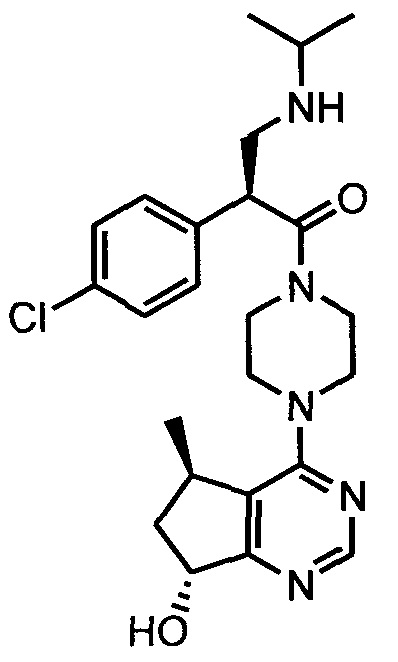
(S)-2-(4-хлорфенил-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-(изопропиламино)пропан-1-он
Стадия 1: Этилпулегенат (130 г, 662 ммоль) в EtOAc (900 мл) охлаждали до -78°C, используя баню с сухим льдом и изопропанолом. Эту смесь подвергали озонолизу до тех пор, пока реакционная смесь не приобретет фиолетовый цвет. В этот момент генерацию озона прекращали и реакционную смесь извлекали из бани с сухим льдом. Через реакционную смесь пропускали кислород до тех пор, пока смесь не становилась желтой. Реакционную смесь концентрировали в вакууме и полученный остаток растворяли в ледяной уксусной кислоте (400 мл). Раствор охлаждали до 0°C и порциями в течение 30 минут добавляли Zn пыль (65 г, 993 ммоль). Затем реакционной смеси давали перемешиваться в течение 2 часов, после чего реакционную смесь фильтровали через слой целита для удаления цинковой пыли. Уксусную кислоту нейтрализовывали до pH 7 водным NaOH и NaHCO3 и экстрагировали эфиром (3×800 мл). Объединенные органические фракции промывали солевым раствором, сушили над MgSO4 и концентрировали, получая (2R)-этил-2-метил-5-оксоциклопентанкарбоксилат в виде коричневой жидкости (107 г, 95%).
Стадия 2: Ацетат аммония (240,03 г, 3113,9 ммоль) добавляли к раствору (R)-этил-2-метил-5-оксоциклопентанкарбоксилата (106,0 г, 622,78 ммоль) в MeOH (1,2 л). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 20 часов, после чего завершение реакции определяли по данным ТСХ и ВЭЖХ. Реакционную смесь концентрировали для удаления MeOH. Полученный остаток растворяли в DCM, дважды промывали H2O, один раз солевым раствором, сушили (Na2SO4), фильтровали и концентрировали, получая (R)-этил-2-амино-5-метилциклопент-1-енкарбоксилат (102 г, выход 97%) в виде оранжевого масла. ЖХ/МС (APCI+) m/z 170 [M+H]+.
Стадия 3: Раствор, содержащий (R)-этил-2-амино-5-метилциклопент-1-енкарбоксилат (161,61 г, 955,024 ммоль) и формиат аммония (90,3298 г, 1432,54 ммоль) в формамиде (303,456 мл, 7640,19 ммоль), нагревали до внутренней температуры 150°C и перемешивали в течение 17 часов. Реакционную смесь охлаждали и переносили в одногорлую колбу объемом 2 л. Затем избыток формамидина удаляли путем перегонки в высоком вакууме. Как только формамидин прекращал поступать, оставшееся в перегонной колбе масло растворяли в DCM и промывали солевым раствором (3×200 мл). Объединенные водные промывки экстрагировали DCM. Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали. Полученное коричневое масло растворяли в минимальном количестве DCM и этот раствор добавляли, используя делительную воронку, в перемешиваемый раствор эфира (приблизительно 5 объемом эфира на объем раствора в DCM), в результате чего образовывалось некоторое количество коричневого осадка. Коричневый осадок удаляли фильтрованием через воронку со стеклянным фильтром средней пористости, промывали эфиром и отбрасывали. Фильтрат концентрировали, остаток растирали с эфиром еще два раза и затем сушили на линии с высоким вакуумом, получая (R)-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ол (93,225 г, выход 65,00%) в виде коричнево-желтого пастообразного вещества. ЖХ/МС (APCI-) m/z 149,2.
Стадия 4: Чистый POCl3 (463,9 мл, 5067 ммоль) медленно добавляли с помощью капельной воронки при 0°C в раствор (R)-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ола (152,2 г, 1013 ммоль) в DCE (1,2 л). По окончании добавления реакционную смесь нагревали до комнатной температуры, затем нагревали до кипения с обратным холодильником и перемешивали в течение 70 минут. Завершение реакции определяли по данным ВЭЖХ. Реакционную смесь охлаждали до комнатной температуры и избыток POCl3 гасили 4 порциями следующим образом: реакционную смесь переносили в делительную воронку и по каплям вливали в мензурку, содержащую лед и насыщенный раствор NaHCO3, охлажденный в ледяной бане. По окончании добавления каждой порции реакционной смеси погашенную смесь перемешивали 30 минут, чтобы гарантировать полное разложение POCl3 до переноса в делительную воронку. Смесь переносили в делительную воронку и дважды экстрагировали DCM. Объединенные экстракты сушили (Na2SO4), фильтровали и концентрировали. Неочищенную смесь очищали на силикагеле следующим образом: силикагель (1 кг) суспендировали в 3 л воронке со стеклянным фильтром в смеси 9:1 гексан:этилацетат, силикагель осаждали в вакууме и покрывали песком. Неочищенную смесь наносили в смеси DCM/гексан и элюировали соединение, используя колбу объемом 1 л с боковой трубкой, в вакууме. Первыми элюировались побочные продукты с высоким Rf, затем (R)-4-хлор-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин (104,4 г, выход 61,09%) в виде коричневого масла. Триэтиламин (93,0 мл, 534 ммоль) и трет-бутил-пиперазин-1-карбоксилат (34,8 г, 187 ммоль) добавляли к раствору (R)-4-хлор-5-метил-6,7-дигидро-5H-циклопента[d]пиримидина (30,0 г, 178 ммоль) в н-BuOH (250 мл). Реакционную смесь нагревали до кипения с обратным холодильником в атмосфере азота и перемешивали в течение ночи (17 часов), после чего смесь концентрировали на роторном испарителе. Полученное масло растворяли в DCM, промывали H2O, сушили (Na2SO4), фильтровали и концентрировали. Полученное коричневое масло очищали на силикагеле, сначала элюируя смесью 2:1 гексан:этилацетат до элюирования чистого продукта, затем градиентом смеси 1:1-1:5 DCM-этилацетат, с получением (R)-трет-бутил-4-(5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (42,0 г, выход 74,1%) в виде бежевого порошка. ЖХ/МС (APCI+) m/z 319,1 [M+H]+.
Стадия 5: Твердую макс. 77% MCPBA (23,9 г, 107 ммоль) порциями добавляли при 0°C к раствору (R)-трет-бутил-4-(5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (20,0 г, 62,8 ммоль) в CHCl3 (310 мл). Реакционную смесь перемешивали в течение 5 минут, затем нагревали до комнатной температуры и перемешивали в течение 90 минут. Результаты ВЭЖХ выглядели такими же через 7,5 часов. Реакционную смесь охлаждали до 0°C, затем добавляли NaHCO3 (13,2 г, 157 ммоль) и еще 0,5 эквивалента m-CPBA. Реакционную смесь перемешивали в течение ночи (14 часов). Реакционную смесь охлаждали до 0°C и по каплям через капельную воронку добавляли раствор Na2S2O3 (29,8 г, 188 ммоль) в H2O (50 мл). После этого через капельную воронку добавляли раствор Na2CO3 (24,6 г, 232 ммоль) в H2O (70 мл) (смесь становилась гомогенной). Реакционную смесь перемешивали в течение 30 минут, затем смесь экстрагировали CHCl3 (3×150 мл). Объединенные экстракты сушили (Na2SO4), фильтровали и концентрировали, получая N-оксид. ЖХ/МС (APCI+) m/z 335,1 [M+H]+.
Стадия 6: Ac2O (77,0 мл, 816 ммоль) добавляли к N-оксиду (21,0 г, 62,8 ммоль) из стадии 5. Реакционную смесь нагревали в атмосфере азота при 90°C в песочной бане и перемешивали в течение 100 минут. Реакционную смесь охлаждали до комнатной температуры и удаляли избыточный уксусный ангидрид на роторном испарителе. Полученное масло растворяли в DCM и полученный раствор осторожно вливали в насыщенный раствор Na2CO3 на льду. Смесь экстрагировали DCM и объединенные экстракты сушили (Na2SO4), фильтровали и концентрировали с получением (5R)-трет-бутил-4-(7-ацетокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (23,6 г, 100%) в виде коричневой пены. ЖХ/МС (APCI+) m/z 377,1 [M+H]+.
Стадия 7: LiOH-H2O (6,577 г, 156,7 ммоль) добавляли при 0°C к раствору (5R)-трет-бутил-4-(7-ацетокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (23,6 г, 62,69 ммоль) в смеси 2:1 ТГФ:H2O (320 мл). Реакционную смесь перемешивали в течение 10 минут и затем нагревали до комнатной температуры. Данные ЖХ/МС выглядели одинаково через 3 часа и 4,5 часа. Реакционную смесь охлаждали до 0°C, после чего к смеси добавляли насыщенный раствор NH4Cl. Смесь перемешивали в течение 5 минут и большую часть ТГФ удаляли на роторном испарителе. Смесь экстрагировали EtOAc (3×250 мл) и объединенные экстракты сушили (Na2SO4), фильтровали и концентрировали. Неочищенную смесь подвергали флэш-хроматографии на Biotage 65M: смесь 4:1 DCM:этилацетат, затем градиент смеси 1:1-1:4 DCM:этилацетат. После того как начинал элюировать продукт, через колонку пропускали этилацетат. Затем смесью 30:1 DCM:MeOH элюировали остальную часть продукта (8,83 г). Смешанные фракции повторно подвергали флэш-хроматографии на Biotage 40M, используя те же условия, с получением еще 2,99 г, что давало общий выход (5R)-трет-бутил-4-(7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (11,82 г, выход 56,38%) в виде коричневой пены. ЖХ/МС (APCI+) m/z 335,1 [M+H]+.
Стадия 8: Раствор ДМСО (5,45 мл, 76,8 ммоль) в DCM (50 мл) по каплям добавляли через капельную воронку при -78°C к раствору оксалилхлорида (3,35 мл, 38,4 ммоль) в DCM (150 мл). Реакционную смесь перемешивали в течение 35 минут и затем через капельную воронку медленно добавляли раствор (5R)-трет-бутил-4-(7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (9,17 г, 27,4 ммоль) в DCM (80 мл). Реакционную смесь перемешивали еще 1 час при -78°C, после чего к смеси добавляли чистый триэтиламин (18,0 мл, 129 ммоль). Затем реакционной смеси давали нагреться до комнатной температуры, после чего смесь перемешивали в течение 30 минут. Добавляли H2O. Смесь экстрагировали DCM (3×200 мл) и объединенные экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенную смесь очищали на силикагеле (Biotage 65M): через колонку пропускали приблизительно 800 мл смеси 4:1 DCM:EtOAc, затем градиент смеси 1:1 DCM:этилацетат до элюирования продукта, после чего продукт элюировали смесью 1:4 DCM:EtOAc, получая (R)-трет-бутил-4-(5-метил-7-оксо-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилат (7,5 г, выход 82,3%) в виде коричневой пены. Пену концентрировали (3×) в смеси DCM/гексаны с получением светло-коричневой пены. ВЭЖХ площадь >95%. ЖХ/МС (APCI+) m/z 333 [M+H]+.
Стадия 9: Триэтиламин (4,33 мл, 31,1 ммоль; дегазированный азотом за 30 минут до использования) и муравьиную кислоту (1,36 мл, 36,1 ммоль; дегазированную азотом за 30 минут до использования) добавляли к раствору (R)-трет-бутил-4-(5-метил-7-оксо-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (9,75 г, 29,3 ммоль) в DCM (210 мл; дегазированном азотом за 30 минут до использования). Смесь перемешивали в течение 5 минут, затем добавляли Ru катализатор (0,0933 г, 0,147 ммоль). Реакционную смесь перемешивали при избыточном давлении азота в течение ночи (18 часов). Реакционную смесь концентрировали досуха и сушили в высоком вакууме. Неочищенный продукт подвергали флэш-хроматографии на Biotage с картриджем 65M, пропуская 500 мл смеси 1:1 DCM:этилацетат, затем смесь 1:4 DCM:этилацетат до появления продукта (2-ое пятно), затем градиент до чистого этилацетата, затем смесью 25:1 DCM:MeOH элюировали остальную часть продукта. Фракции объединяли и концентрировали на роторном испарителе. Остаток снова концентрировали в смеси DCM/гексаны с получением смеси трет-бутил-4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (основного) и трет-бутил-4-((5R,7S)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (второстепенного) (9,35 г, выход 95,3%) в виде бежевой пены. ЖХ/МС (APCI+) m/z 335 [M+H]+. 1H-ЯМР (CDCl3) показал 88% при интегрировании по метиновой группе карбинола.
Стадия 10: 4-Нитробензоилхлорид (4,27 г, 23,0 ммоль) добавляли при 0°C к раствору трет-бутил-4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (7,0 г, 20,9 ммоль) и триэтиламина (4,38 мл, 31,4 ммоль) в DCM (110 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, после чего добавляли насыщенный раствор NaHCO3. Смесь перемешивали 10 минут и затем экстрагировали DCM. Объединенные экстракты сушили (Na2SO4), фильтровали и концентрировали. Неочищенную смесь подвергали флэш-хроматографии на Biotage 65M (нанесение в смеси 3:1 гексаны:этилацетат, затем смесью 2:1 гексаны:этилацетат элюировали трет-бутил-4-((5R,7R)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилат и несколько смешанных фракций). Затем трет-бутил-4-((5R,7S)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилат элюировали, используя смесь 1:2 гексаны:этилацетат. Фракции с продуктом концентрировали на роторном испарителе, получая трет-бутил-4-((5R,7R)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилат (8,55 г, выход 84,5%) в виде желтой пены. ЖХ/МС (APCI+) m/z 484 [M+H]+. 1H-ЯМР (CDCl3) показывал один диастереомер). Фракции с другим диастереомером концентрировали на роторном испарителе, получая трет-бутил-4-((5R,7S)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилат (0,356 г, выход 3,52%) в виде коричневой пены. ЖХ/МС (APCI+) m/z 484 [M+H]+.
Стадия 11: LiOH-H2O (0,499 г, 11,9 ммоль) добавляли при 0°C к раствору трет-бутил-4-((5R,7R)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (2,30 г, 4,76 ммоль) в смеси 2:1 ТГФ:H2O (40 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. ТГФ удаляли на роторном испарителе, добавляли насыщенный раствор NaHCO3 и экстрагировали смесь этилацетатом. Объединенные экстракты промывали (1×) насыщенным раствором NaHCO3, сушили (Na2SO4), фильтровали и концентрировали, получая трет-бутил-4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилат (1,59 г, выход 100,0%) в виде желтой пены. ВЭЖХ после обработки продукта показала чистоту >98% по площади. ЖХ/МС (APCI+) m/z 335 [M+H]+. Трет-бутил-4-((5R,7S)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилат получали, используя аналогичный способ.
Стадия 12: Смесь 4M HCl/диоксан (11,2 мл, 44,9 ммоль) добавляли к раствору трет-бутил-4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (0,600 г, 1,79 ммоль) в диоксане (15 мл). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение ночи (20 часов). Смесь концентрировали досуха и сушили на линии высокого вакуума. Неочищенный продукт суспендировали в эфире, обрабатывали ультразвуком и перемешивали в течение 5 минут. Твердую фазу выделяли фильтрованием через воронку со стеклянным фильтром со средним размером пор под давлением азота, промывали эфиром, сушили под давлением азота и дополнительно сушили на линии высокого вакуума, получая дигидрохлорид (5R,7R)-5-метил-4-(пиперазин-1-ил)-6,7-дигидро-5H-циклопента[d]пиримидин-7-ола (0,440 г, выход 79,8%) в виде желтого порошка. ЖХ/МС (APCI+) m/z 235. Дигидрохлорид (5R,7S)-5-метил-4-(пиперазин-1-ил)-6,7-дигидро-5H-циклопента[d]пиримидин-7-ола получали с использованием аналогичного способа.
Стадия 13: Метил-2-(4-хлорфенил)ацетат (36,7 г, 199 ммоль) и параформальдегид (6,27 г, 209 ммоль) растворяли/суспендировали в ДМСО (400 мл) и обрабатывали NaOMe (537 мг, 9,94 ммоль). Смеси давали перемешиваться при комнатной температуре в течение 2 часов до завершения реакции по данным анализа ТСХ реакционной смеси. Реакционную смесь вливали в ледяную воду (700 мл; белая эмульсия) и нейтрализовывали добавлением 1M раствора HCl. Водный слой экстрагировали этилацетатом (3×) и органические фракции объединяли. Органический слой промывали водой (2×), солевым раствором (1×), отделяли, сушили над MgSO4, фильтровали и концентрировали в вакууме, получая неочищенный продукт в виде желтого масла. Остаток наносили на слой силикагеля на стеклянном фильтре с крупными порами и элюировали смесью 9:1 гексаны:этилацетат, пока не будет собрано исходное вещество/олефин. Затем силикагель элюировали смесью 1:1 гексаны:этилацетат до полного элюирования чистого целевого продукта. Концентрирование чистых фракций давало метил-2-(4-хлорфенил)-3-гидроксипропаноат в виде бесцветного масла (39,4 г, 92%).
Стадия 14: Метил-2-(4-хлорфенил)-3-гидроксипропаноат (39,4 г, 184 ммоль) растворяли в DCM (500 мл) и обрабатывали TEA (64,0 мл, 459 ммоль). Раствор охлаждали до 0°C и медленно обрабатывали MsCl (15,6 мл, 202 ммоль), затем давали перемешиваться в течение 30 минут до завершения реакции по данным ТСХ анализа. Раствор разделяли 1 н. раствором HCl и водный слой однократно экстрагировали DCM. Объединенный органический слой промывали еще раз 1 н. раствором HCl, разделяли, промывали разбавленным раствором NaHCO3 и разделяли. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме с получением оранжевого масла. Остаток наносили на слой силикагеля на стеклянном фильтре с крупными порами и элюировали смесью 9:1 гексаны:этилацетат, получая чистый, по данным ТСХ анализа, целевой продукт. Концентрирование чистых фракций дает метил-2-(4-хлорфенил)акрилат в виде бесцветного масла (30,8 г, 85%). Полученный метил-2-(4-хлорфенил)акрилат (500 мг, 2,54 ммоль) добавляли в виде раствора в ТГФ (1,35 мл) к перемешиваемому раствору i-PrNH2 (217 мкл, 2,54 ммоль) в ТГФ (5,0 мл) при 0°C. Реакционной смеси давали перемешиваться при комнатной температуре в течение ночи до завершения реакции по данным ЖХ/МС анализа. Boc2O (584 мкл, 2,54 ммоль) добавляли пипеткой к перемешиваемому амину. Реакционной смеси давали перемешиваться в течение ночи до завершения реакции по данным ЖХ/МС и ТСХ анализа смеси. Раствор концентрировали в вакууме, получая метил-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорфенил)пропаноат в виде бесцветного масла (854 мг, 94%). ЖХ/МС (APCI+) m/z 256,1 [M-Boc]+.
Стадия 15: Метил-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорфенил)пропаноат (133 г, 374 ммоль) растворяли в ТГФ (1,0 л) и обрабатывали KOTMS (56,0 г, 392 ммоль) при комнатной температуре. Смесь оставляли на ночь при перемешивании до окончания реакции по данным ЖХ/МС анализа смеси. Смесь концентрировали в вакууме с получением влажной пены, которую оставляли сушиться в вакууме на ночь, получая 3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорфенил)пропаноат калия в виде белого твердого вещества (148,7 г, 105%). ЖХ/МС (APCI+) m/z 242,1 [M-Boc-K]+.
Стадия 16: 3-(трет-Бутоксикарбонил(изопропил)амино)-2-(4-хлорфенил)пропаноат калия (77,2 г, 203 ммоль) растворяли в ТГФ (515 мл) и обрабатывали пивалоилхлоридом (26,3 мл, 213 ммоль) при комнатной температуре. Смеси давали перемешиваться в течение 3 часов для образования смешанного ангидрида. (S)-4-бензилоксазолидин-2-он (46,1 г, 260 ммоль) растворяли в ТГФ (600 мл) и охлаждали до -78°C в отдельной колбе. Раствор обрабатывали н-BuLi (102 мл 2,50 M раствор в смеси гексанов, 254 ммоль) и давали перемешиваться в течение одного часа. Полученный раствор ангидрида добавляли через канюлю к перемешиваемому Li-оксазолидинону и смесь оставляли на ночь нагреваться до комнатной температуры. Смесь гасили добавлением насыщенного раствора хлорида аммония, затем распределяли между дополнительным количеством воды и этилацетатом. Водный слой экстрагировали несколько раз и объединяли органические фракции. Органический слой промывали водой, затем солевым раствором, разделяли, сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали/разделяли (диастереомеры) с помощью хроматографии (силикагель, элюирование смесью 4:1 гексаны:этилацетат), получая следующие, полностью разделенные диастереомеры в виде вязких масел: трет-бутил-(R)-3-((S)-4-бензил-2-оксооксазолидин-3-ил)-2-(4-хлорфенил)-3-оксопропил(изопропил)карбамат (12,16 г, 24%, исходя из 1/2 рацемической кислоты) и трет-бутил-(S)-3-((S)-4-бензил-2-оксооксазолидин-3-ил)-2-(4-хлорфенил)-3-оксопропил(изопропил)карбамат (39,14 г, 77%, исходя из 1/2 рацемической кислоты). ЖХ/МС (APCI+) m/z 401,2 [M-Boc]+.
Стадия 17: LiOH-H2O (168 мг, 4,00 ммоль) добавляли к перемешиваемому раствору ТГФ (30 мл) и воды (15 мл) при комнатной температуре до полного растворения. Смесь обрабатывали перекисью водорода (658 мкл 35% мас. раствора в воде, 8,00 ммоль) и давали перемешиваться при комнатной температуре в течение 10 минут. Реакционную смесь охлаждали до 0°C в ледяной бане и по каплям через капельную воронку в течение 10 минут добавляли трет-бутил-(S)-3-((S)-4-бензил-2-оксооксазолидин-3-ил)-2-(4-хлорфенил)-3-оксопропил(изопропил)карбамат (1,00 г, 2,00 ммоль) в виде раствора в ТГФ (15 мл). Смесь оставляли при перемешивании на ночь при комнатной температуре до завершения реакции по данным ЖХ/МС анализа смеси. Реакционную смесь охлаждали до 0°C и затем обрабатывали 1M раствором Na2SO3 (9,00 мл) через капельную воронку в течение десяти минут. После завершения добавления смеси давали нагреться до комнатной температуры в течение 10 минут. Смесь концентрировали для удаления ТГФ, а затем разбавляли водой. Водный слой дважды промывали этилацетатом (который отбрасывали). Водный слой разделяли этилацетатом, затем при перемешивании по каплям обрабатывали 1M HCl, пока pH не достигало значения 2-3. Водный слой дважды экстрагировали этилацетатом и органические фракции объединяли. Органическую фазу промывали солевым раствором, разделяли, сушили над MgSO4, фильтровали и концентрировали в вакууме. Бесцветный маслообразный продукт сушили в высоком вакууме в течение одного часа, получая (S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорфенил)пропановую кислоту в виде вязкого масла/пены (685 мг, 100%). ЖХ/МС (APCI+) m/z 242,1 [M-Boc]+.
Стадия 18: Раствор дигидрохлорида (5R,7R)-5-метил-4-(пиперазин-1-ил)-6,7-дигидро-5H-циклопента[d]пиримидин-7-ола (2,92 г, 9,51 ммоль) и (S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорфенил)пропановой кислоты (3,25 г, 9,51 ммоль) в DCM (40 мл) и DIEA (5,0 мл, 28,7 ммоль) перемешивали при комнатной температуре в течение 10 минут. К смеси добавляли HBTU (3,61 г, 9,51 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли, а остаток растворяли в этилацетате (500 мл) и промывали водой (6×100 мл). Органическую фазу сушили и концентрировали. Остаток очищали с помощью колоночной хроматографии, элюируя смесью EtOAc-DCM/MeOH (20:1), с получением трет-бутил-(S)-2-(4-хлорфенил)-3-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-оксопропил(изопропил)карбамата (3,68 г, 69%) ЖХ/МС (APCI+) m/z 558,2 [M+H]+.
Стадия 19: трет-Бутил-(S)-2-(4-хлорфенил)-3-(4-((5R, 7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-оксопропил(изопропил)карбамат (2,50 г, 4,48 ммоль) растворяли в диоксане (22,4 мл) и обрабатывали 4M HCl в диоксане (22,4 мл, 89,6 ммоль) при комнатной температуре. Полученный раствор оставляли при перемешивании на ночь до окончания реакции по данным ЖХ/МС анализа смеси. Раствор концентрировали в вакууме с получением геля, который растворяли в минимальном количестве метанола (10 мл). Раствор пипеткой переносили в перемешиваемый эфир (300 мл), в результате чего образовывался белый осадок целевого продукта. Добавление выполняли приблизительно наполовину, когда белый осадок переходил в желтый гель. Продукт концентрировали в вакууме с получением желтого геля, который оставляли при пониженном давлении на ночь, получая дигидрохлорид (S)-2-(4-хлорфенил)-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-(изопропиламино)пропан-1-она в виде светло-желтого порошка (2,14 г, 90%).
1H-ЯМР (D20, 400 МГц) δ 8,39 (с, 1H), 7,37-7,35 (д, J=8,4 Гц, 2H), 7,23-7,20 (д, J=8,4 Гц, 2H), 5,29-5,25 (м, 1H), 4,33-4,29 (м, 1H), 4,14-4,10 (м, 1H), 3,89-3,19 (м, 11H), 2,23-2,17 (м, 1H), 2,08-1,99 (м, 1H), 1,20-1,18 (м, 6H), 0,98-0,96 (д, J=6,8 Гц, 3H). МС (APCI+) [M+H]+ 458.
Примеры 3-9, показанные в таблице 1, также могут быть выполнены согласно вышеописанным способам.
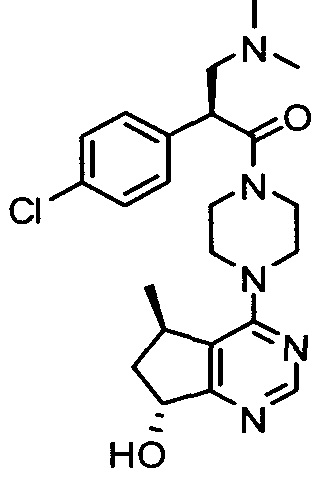
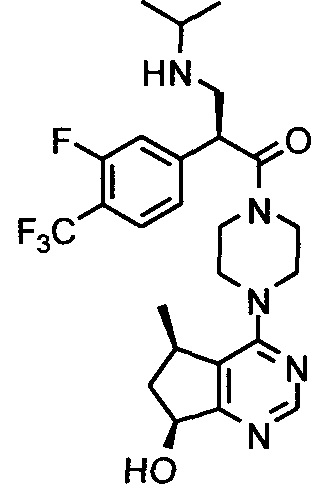
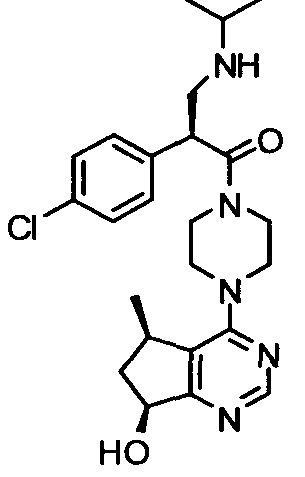
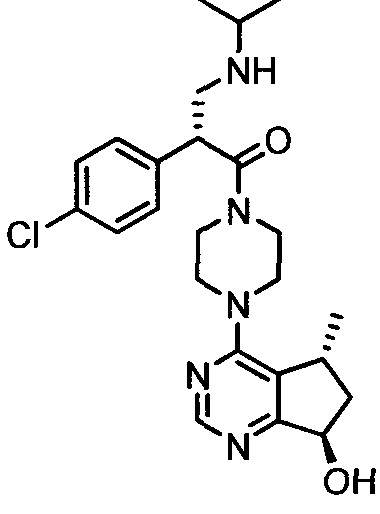
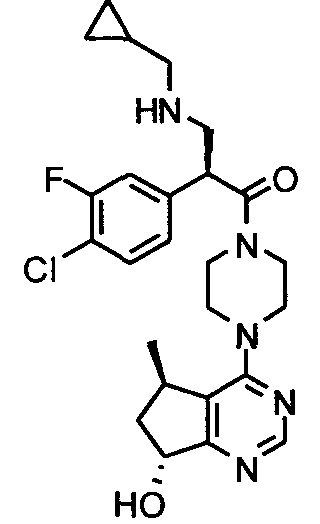
Пример 10
(S)-2-(4-циклопропилфенил-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-2-(S)-пирролидин-2-ил)этанон
Стадия 1: Циклопропилмагнийбромид (64,0 мл, 32,00 ммоль) в ТГФ обрабатывали раствором хлорида цинка (II) (64,00 мл, 32,00 ммоль) в ТГФ. Смесь перемешивали при температуре окружающей среды в течение 20 минут. Добавляли 2-(4-бромфенил)ацетонитрил (5,228 г, 26,67 ммоль) и бис[три-трет-бутилфосфин]палладий (0,6814 г, 1,333 ммоль) в виде раствора в ТГФ (2 мл). Реакционную смесь перемешивали при температуре окружающей среды в атмосфере азота в течение 12 часов. Реакцию гасили насыщенным раствором NH4Cl, разбавляли метиленхлоридом и разделяли. Водный слой промывали метиленхлоридом (2×), а затем объединенные органические слои промывали водой (3×), сушили над Na2SO4 и концентрировали в вакууме. Неочищенный продукт подвергали хроматографии на SiO2, элюируя смесью 25:1 гексаны/этилацетат, с получением 2-(4-циклопропилфенил)ацетонитрила (2,76 г, 66%). 1H-ЯМР (CDCl3, 400 МГц) δ 7,20 (д, J=8,2, 2H), 7,07 (д, J=8,2, 2H), 3,70 (с, 2H), 1,94-1,85 (м, 1H), 1,01-0,95 (м, 2H), 0,71-0,66 (м, 2H).
Стадия 2: Метанол (65 мл) охлаждали до 0°C и насыщали HCl (г). Этот раствор обрабатывали раствором 2-(4-циклопропилфенил)ацетонитрила (2,76 г, 17,56 ммоль) в метаноле (6 мл). Реакционную смесь нагревали до кипения с обратным холодильником в течение ночи под осушающей трубкой, содержащей CaSO4. Реакционную смесь охлаждали и концентрировали в вакууме. Неочищенную смесь ресуспендировали в этилацетате и воде, а затем разделяли. Органический слой промывали насыщенным NaHCO3, насыщенным NaCl, сушили над Na2SO4 и концентрировали в вакууме, получая метил-2-(4-циклопропилфенил)ацетат в виде масла (3,10 г, 93%). 1H-ЯМР (CDCl3, 400 МГц) δ 7,16 (д, J=8,3, 2H), 7,02 (д, 2H), 3,68 (с, 3H), 3,58 (с, 2H), 1,92-1,83 (м, 1H), 0,97-0,91 (м, 2H), 0,70-0,64 (м, 2H).
Стадия 3: Метил-2-(4-циклопропилфенил)ацетат (3,10 г, 16,30 ммоль) растворяли в смеси ТГФ/MeOH/вода (2:2:1, 80 мл) и раствор обрабатывали гидратом гидроксида лития (0,8548 г, 20,37 ммоль). Затем смесь перемешивали при температуре окружающей среды в течение 4 часов. Реакционную смесь нейтрализовывали до pH 4 с помощью 3 н. HCl и концентрировали в вакууме. Твердую фазу повторно растворяли в этилацетате и воде. Уровень pH снова доводили приблизительно до pH 3-4 с помощью 3 н. HCl. Затем слои разделяли. Водный слой промывали этилацетатом (2×). После этого объединенные органические слои промывали насыщенным NaCl, сушили над Na2SO4 и концентрировали, получая 2-(4-циклопропилфенил)уксусную кислоту (2,82 г, 98%). 1H-ЯМР (CDCl3, 400 МГц) δ 7,16 (д, J=8,2, 2H), 7,03 (д, 2H), 3,60 (с, 2H), 1,92-1,83 (м, 1H), 098-0,91 (м, 2H), 0,70-0,64 (м, 2H).
Стадия 4: 2-(4-Циклопропилфенил)уксусную кислоту (2,82 г, 16,003 ммоль) объединяли с (R)-4-бензилоксазолидин-2-оном (3,4030 г, 19,204 ммоль) в толуоле (14 мл). Суспензию обрабатывали триэтиламином (6,6917 мл, 48,010 ммоль) и затем нагревали до 80°C. Раствор по каплям обрабатывали раствором пивалоилхлорида (1,9893 мл, 16,003 ммоль) в толуоле (3,5 мл). Реакционную смесь нагревали при 80°C в течение ночи. Реакционную смесь охлаждали и промывали 2 н. HCl и затем разделяли. Водный слой промывали толуолом и объединенные органические фракции затем промывали 2 н. HCl, водой, насыщенным NaHCO3 (2×), насыщенным NaCl, сушили над Na2SO4 и концентрировали в вакууме. Неочищенный продукт подвергали хроматографии на SiO2, элюируя смесью 9:1 гексаны/этилацетат, с получением (R)-4-бензил-3-(2-(4-циклопропилфенил)ацетил)оксазолидин-2-она (3,43 г, 64%). 1H-ЯМР (CDCl3, 400 МГц) δ 7,33-7,20 (м, 5H), 7,16-7,11 (м, 2H), 7,05 (д, J=8,2, 2H), 4,70-4,63 (м, 1H), 4,32-4,14 (м, 4H), 3,26 (дд, J1=3,2, J2=13,3, 1H), 2,75 (дд, J1=9,5, J2=13,3, 1H), 1,93-1,85 (м, 1H), 0,98-0,92 (м, 2H), 0,72-0,66 (м, 2H).
Стадия 5: (S)-2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-ил)-2-(4-циклопропилфенил)уксусную кислоту получали согласно методике, описанной в примере 1, с использованием (R)-4-бензил-3-(2-(4-циклопропилфенил)ацетил)оксазолидин-2-она (0,287 г, 26%). МС (ESI+) [M+H] 345,7.
Стадия 6: (S)-трет-бутил-2-((S)-1-(4-циклопропилфенил)-2-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-2-оксоэтил)пирролидин-1-карбоксилат получали согласно методике, описанной в примере 3, с использованием (S)-2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-ил)-2-(4-циклопропилфенил)уксусной кислоты (0,199 г, 94%). МС (ESI+) [M+H] 562,1.
Стадия 7: (S)-2-(4-циклопропилфенил)-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-2-((S)-пирролидин-2-ил)этанон получали согласно методике, описанной в примере 3, с использованием (S)-трет-бутил-2-((S)-1-(4-циклопропилфенил)-2-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-2-оксоэтил)пирролидин-1-карбоксилата (0,145 г, 77%). МС (ESI+) [M+H] 462,2. 1H-ЯМР (CD3OD, 400 МГц) δ 8,56 (с, 1H), 7,26 (д, 2H), 7,13 (д, 2H), 5,29 (дд, 1H), 5,32-5,26 (дд, 1H), 4,32 (д, 1H), 4,29-4,18 (м, 1H), 4,12-3,95 (м, 2H), 3,88-3,61 (м, 6H), 3,51-3,38 (м, 1H), 3,35-3,30 (м, 1H), 2,32-2,24 (м, 1H), 2,22-2,03 (м, 2H), 1,95-1,85 (м, 2H), 1,82-1,73 (м, 2H), 1,40-1,34 (м, 1H), 1,16 (д, 3H), 1,01-0,95 (м, 2H), 0,69-0,64 (м, 2H).
Примеры, показанные в таблице 2, также могут быть выполнены согласно вышеописанным способам.
Пример 14 Анализ пролиферации клеток in vitro
Активность in vitro комбинаций соединения примера 2 с некоторыми определенными химиотерапевтическими средствами может быть измерена с использованием анализа CellTiter-Glo® Luminescent Cell Viability, коммерчески доступного от Promega Corp., Madison, WI. Данный метод гомогенного анализа основан на рекомбинантной экспрессии люциферазы Coleoptera (US 5583024; US 5674713; US 5700670) и позволяет определить количество живых клеток в культуре на основе количественного измерения присутствующей АТФ, индикатора метаболически активных клеток (Crouch et al. (1993) J. Immunol. Meth. 160:81-88; US 6602677). Анализ CellTiter-Glo® может быть проведен в 96- или 384-луночном формате, что позволяет использовать его для автоматизированного высокопроизводительного скрининга (HTS) (Cree et al. (1995) Anticancer Drugs 6:398-404). Методика гомогенного анализа включает добавление одного реагента (реагента CellTiter-Glo®) непосредственно к клеткам, культивируемым в среде с добавлением сыворотки. Стадии промывки клеток, удаления среды и множество стадий пипетирования выполнять не требуется. Система позволяет детектировать лишь 15 клеток/лунка в 384-луночном формате через 10 минут после добавления реагента и перемешивания.
Формат гомогенного "добавления-смешивания-измерения" приводит к лизису клеток и генерации люминесцентного сигнала, пропорционального количеству присутствующей АТФ. Количество АТФ прямо пропорционально числу клеток, присутствующих в культуре. Анализ CellTiter-Glo® генерирует люминесцентный сигнал по типу "свечения", которое образуется в результате реакции люциферазы, которая имеет полупериод существования, как правило, более пяти часов, в зависимости от типа используемых клеток и среды. Живые клетки выражаются в относительных единицах люминесценции (RLU). Субстрат, люциферин жука-светляка, подвергается окислительному декарбоксилированию под действием рекомбинантной люциферазы жука-светляка с сопутствующим преобразованием АТФ в АМФ и испусканием фотонов. Увеличенный полупериод существования устраняет необходимость в использовании инжекторов реагента и обеспечивает гибкость при обработке в непрерывном или периодическом режиме множества планшетов. Этот анализ пролиферации клеток может быть использован с различными многолуночными форматами, например, 96- или 384-луночным форматом. Данные можно регистрировать с помощью люминометра или визуализационного устройства с CCD-камерой. Количество испускаемой люминесценции выражается в относительных световых единицах (RLU), измеряемых в зависимости от времени.
Антипролиферативные эффекты комбинаций соединения примера 2 и некоторых химиотерапевтических средств измеряли с использованием анализа CellTiter-Glo®. Значения EC50 определяли для тестируемых соединений и комбинаций. Диапазон in vitro активностей в клетках составлял от приблизительно 100 нМ до приблизительно 10 мкМ.
Пример 15 Эффективность в отношении ксенотрансплантатов опухолей in vivo
Эффективность репрезентативных комбинаций изобретения измеряли in vivo путем имплантации аллотрансплантатов или ксенотрансплантатов раковых клеток грызунам и обработки животных с опухолями комбинациями. Следовало ожидать переменных результатов в зависимости от линии клеток, присутствия или отсутствия определенных мутаций в опухолевых клетках, последовательности введения соединения примера 2 и химиотерапевтического средства, схемы введения и других факторов. Используемым мышам вводили лекарственное средство (средства) или контроль (среду) и наблюдали в течение нескольких недель или более с целью определения времени до увеличения опухоли в два раза, логарифма гибели клеток и ингибирования роста опухоли.
Результаты для репрезентативных комбинаций изобретения, которые исследовали в данной модели, представлены на фигурах 1-2.
Данные на фигурах демонстрируют, что репрезентативные комбинации обеспечивают улучшенные результаты по сравнению с введением соответствующих средств по отдельности.
Было определено, что некоторые комбинации изобретения обеспечивают улучшенные эффекты против некоторых фенотипов рака. Например, некоторые комбинации изобретения обеспечивают улучшенные эффекты против раковых опухолей, связанных с мутацией PTEN, мутацией AKT (например, оверэкспрессией или амплификацией), мутацией PI3K или амплификацией Her2/ErbB2. Таким образом, некоторые комбинации, описанные в настоящей заявке, могут быть особенно полезными против указанных типов раковых опухолей. Например, при раке желудка, потеря PTEN предсказывает лучшую эффективность с некоторыми комбинациями изобретения (например, соединение формулы I с 5-FU/цисплатином), а при раке предстательной железы более сильный эффект отмечали для комбинации соединения формулы I и доцетаксела в PTEN нулевых линиях.
Статус PTEN может быть измерен любыми подходящими способами, как известно из уровня техники. В одном примере используется IHC (ИГХ). Альтернативно, может быть использован Вестерн-блот анализ. Антитела к PTEN коммерчески доступны (Cell Signaling Technology, Beverly, MA, Cascade Biosciences, Winchester, MA). Примеры методик для ИГХ и Вестерн-блот анализа статуса PTEN описаны в публикациях Neshat, M.S. et al., Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR, Proc. Natl Acad. Sci. USA 98, 10314-10319 (2001) и Perren, A., et. al. Immunohistochemical Evidence of Loss of PTEN Expression in Primary Ductal Adenocarcinomas of the Breast, American Journal of Pathology, Vol. 155, No. 4, October 1999. Кроме того, раковые образования, связанные с мутацией AKT, мутацией PI3K и с амплификацией или мутацией Her2/ErbB2, могут быть идентифицированы с использованием методов, известных из уровня техники. В одном примере PTEN статус пациента или образца ткани определяют с использованием ИГХ, и гисто-показатель или HScore присваивают образцу или пациенту. В примерном способе вычисления HScore используется формула: HScore=(%1+клеток × 1)+(%2+клеток × 2)+(%3+клеток × 3) (см. Shoman, N, et al., Mod Path (2005) 18, 250-259). Средний PTEN HScore незлокачественной ткани того же пациента или группы пациентов может быть использован для определения, являются ли показатели HScore пациента или образца низкими или нулевыми. В одном примере показатели HScore менее чем приблизительно 200 считаются низкими и соответствует низкому PTEN, и HScore приблизительно 0 считаются нулевыми.
Один аспект включает способ ингибирования роста опухоли (TGI) у пациента, страдающего раком, включающим мутацию PTEN, мутацию AKT (например, оверэкспрессию или амплификацию), мутацию PI3K или амплификацию или мутацию Her2/ErbB2, включающий введение GDC-0068 или его фармацевтически приемлемой соли и абиратерона или его фармацевтически приемлемой соли пациенту. В некоторых вариантах осуществления комбинация является синергической. В некоторых вариантах осуществления TGI комбинации превышает TGI одного GDC-0068 или одного химиотерапевтического средства. В некоторых вариантах осуществления TGI комбинации приблизительно на 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70 или на 75 процентов превышает TGI одного GDC-0068 или одного химиотерапевтического средства.
Способы измерения TGI известны из уровня техники. В одном примерном способе средние объемы опухоли определяют и сравнивают у пациента до и после лечения. Объемы опухоли могут быть измерены по двум измерениям (длина и ширина) с использованием любого способа из уровня техники, например, с помощью электронного штангенциркуля Ultra-Cal IV (Fred V. Fowler Company) или ПЭТ (позитронно-эмиссионной томографии), или каким-либо другим методом. Можно использовать следующую формулу: Объем опухоли (мм3)=(длина × ширина2)×0,5. Измерение объемов опухоли в течение множества периодов времени может быть выполнено с использованием метода линейного моделирования смешанных эффектов (LME) (Pinheiro et al., 2009). Данный метод можно применять к обоим повторенным измерениям (и к множеству пациентов). Кубические сплайны регрессии могут быть использованы для аппроксимации нелинейного профиля с временными зависимостями объема опухоли при каждом уровне дозы. Указанные нелинейные профили затем могут быть соотнесены с дозой в рамках смешанной модели. Ингибирование роста опухоли в процентах для среды может быть вычислено как относительная площадь под аппроксимированной кривой (AUC) в день относительно среды с использованием следующей формулы:
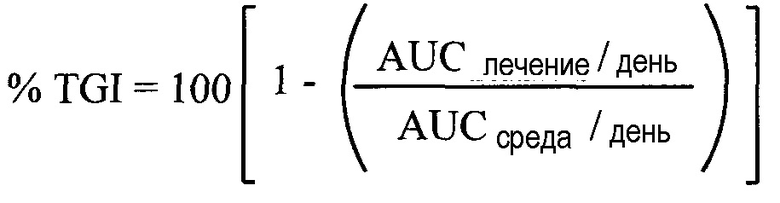
При использовании данной формулы значение TGI 100% указывает на прекращение роста опухоли, более чем приблизительно 1%, но менее чем приблизительно 100% указывает на ингибирование роста опухоли, и более чем приблизительно 100% указывает на регрессию опухоли.
В некоторых вариантах осуществления рак включает одну или более мутаций AKT, PI3k, PTEN и HER2 или нарушенную сигнализацию AKT, PI3k, PTEN или HER2. В одном примере рак является раком желудка, включающим высокую активность pAKT и низкий или нулевой статус PTEN.
В одном определенном аспекте изобретения предложен способ лечения пациента, имеющего рак, который ассоциирован с мутацией или потерей экспрессии PTEN, мутацией или амплификацией AKT, мутацией или амплификацией PI3K, или амплификацией Her2/ErbB2, включающий введение комбинации изобретения пациенту. В другом аспекте изобретения предложен способ идентификации пациента, имеющего рак, который можно лечить комбинацией изобретения, включающий определение, ассоциирован ли рак у пациента с мутацией или потерей экспрессии PTEN, мутацией или амплификацией AKT, мутацией или амплификацией PI3K, или амплификацией Her2/ErbB2, где ассоциация рака у пациента с мутацией или потерей экспрессии PTEN, мутацией или амплификацией AKT, мутацией или амплификацией PI3K, или амплификацией Her2/ErbB2 является показателем наличия рака, который можно лечить комбинацией изобретения. В другом аспекте изобретения предложен способ, дополнительно включающий лечение пациента, идентифицированного с использованием комбинации изобретения. В другом примере рак, подвергаемый лечению, связан с положительным, низким или нулевым статусом PTEN в комбинации с положительным или отрицательным статусом HER2. Примеры включают рак желудка, который является (i) PTEN отрицательным (HScore менее чем приблизительно 10, или 0) и Her2 отрицательным, (ii) PTEN низким (HScore менее чем приблизительно 200) и Her2 отрицательным, (iii) PTEN отрицательным и Her2 положительным, или (iv) PTEN положительным и Her2 отрицательным. В данном примере рак можно лечить с использованием комбинации соединения формулы I, например, GDC-0068, или его соли и абиратерона или его фармацевтически приемлемой соли.
Кроме того, поскольку многочисленные модификации и изменения будут очевидны квалифицированным специалистам, изобретение не желательно ограничивать конкретной показанной конструкцией и способом, как описано выше. Таким образом, все подходящие модификации и эквиваленты можно считать включенными в объем изобретения, определяемого в соответствии с формулой, следующей ниже.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГИДРОКСИЛИРОВАННЫЙ ПИРИМИДИЛ ЦИКЛОПЕНТАН В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗЫ (АКТ) | 2009 |
|
RU2520735C2 |
| ГИДРОКСИЛИРОВАННЫЕ ПИРИМИДИЛЦИКЛОПЕНТАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ (АКТ) | 2009 |
|
RU2504542C2 |
| ЗАМЕЩЕННЫЕ 1,2-ДИГИДРО-3Н-ПИРАЗОЛО[3,4-D]ПИРИМИДИН-3-ОНЫ | 2019 |
|
RU2812726C2 |
| БИОМАРКЕРЫ ДЛЯ ПРОГНОЗИРОВАНИЯ ЧУВСТВИТЕЛЬНОСТИ К ПРОТИВООПУХОЛЕВОЙ ТЕРАПИИ | 2012 |
|
RU2635193C2 |
| БИЦИКЛИЧЕСКИЕ НИТРОИМИДАЗОЛЫ, КОВАЛЕНТНО СОЕДИНЕННЫЕ С ЗАМЕЩЕННЫМИ ФЕНИЛОКСАЗОЛИДИНОНАМИ | 2009 |
|
RU2504547C2 |
| НЕКОТОРЫЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ | 2015 |
|
RU2671494C2 |
| Бициклические гетероциклические соединения и их применения в терапии | 2012 |
|
RU2662827C2 |
| ФОРМЫ И СОСТАВЫ ПИРИМИДИНИЛЦИКЛОПЕНТАНОВОГО СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ, ОТНОСЯЩИЕСЯ К НИМ | 2013 |
|
RU2650511C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛИРОВАННЫХ ЦИКЛОПЕНТАПИРИМИДИНОВЫХ СОЕДИНЕНИЙ И ИХ СОЛЕЙ | 2013 |
|
RU2642311C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE10 | 2011 |
|
RU2543386C2 |
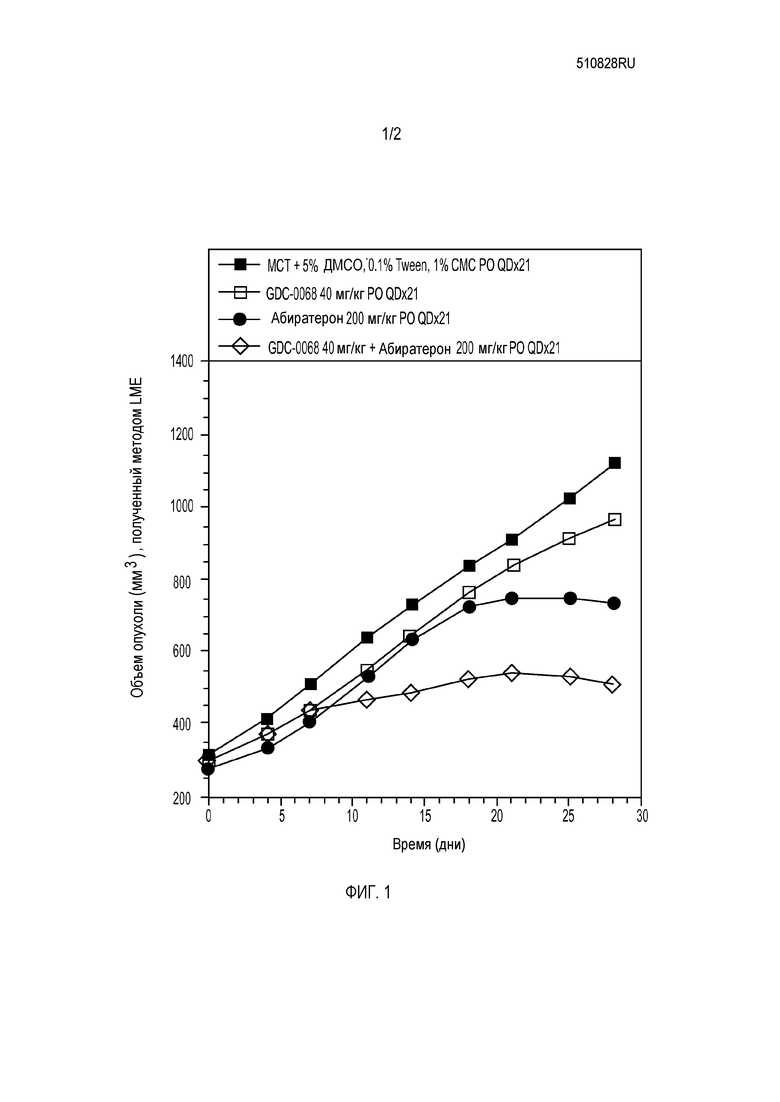
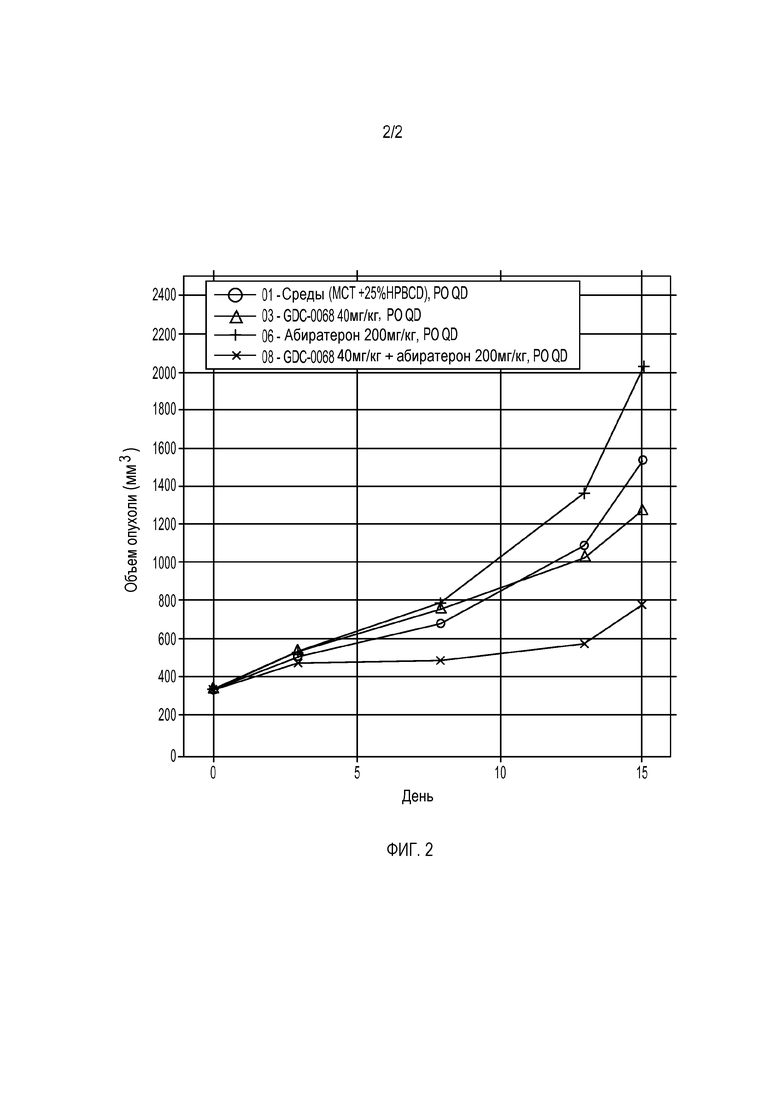
Изобретение относится к новой комбинации а) соединения формулы Ia или его фармацевтически приемлемой соли и b) абиратерона или его фармацевтически приемлемой соли, взятых в терапевтически активном количестве. Комбинация может быть использована для лечения рака предстательной железы. Изобретение также относится к способу лечения рака предстательной железы с использованием указанной комбинации. Соединение формулы Ia соответствует структурной формуле
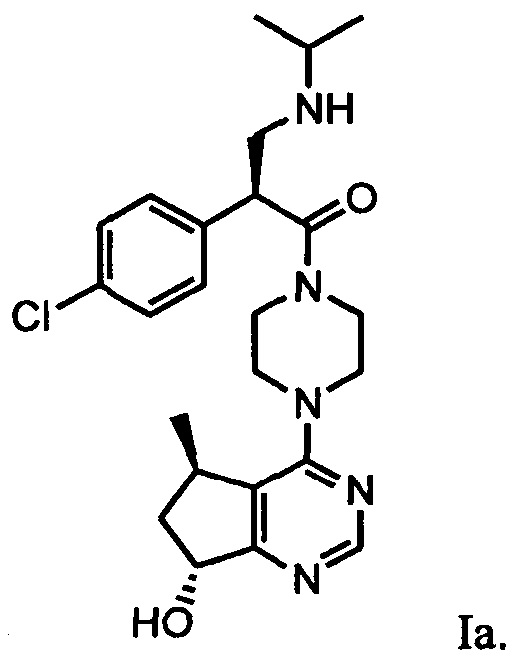
Предпочтительна комбинация, где соединение формулы Ia и абиратерон находятся в соотношении 1:5 (мг/кг). В качестве фармацевтически приемлемой соли абиратерона желательно использовать ацетат абиратерона. Комбинация применима в случае, когда рак ассоциирован с мутацией PTEN или со сверэкспрессией или амплификацией AKT, с мутацией PI3K, с мутацией Her2/ErbB2. Предпочтительно использование комбинации, в случае когда рак, ассоциированный с мутацией PTEN, представляет собой рак предстательной железы с нулевым статусом PTEN. Указанная комбинация обладает повышенной активностью и позволяет значительно снизить объем опухоли по сравнению с контрольным образцом. 4 н. и 10 з.п. ф-лы, 2 ил., 2 табл., 15 пр.
1. Комбинация а) соединения формулы Ia
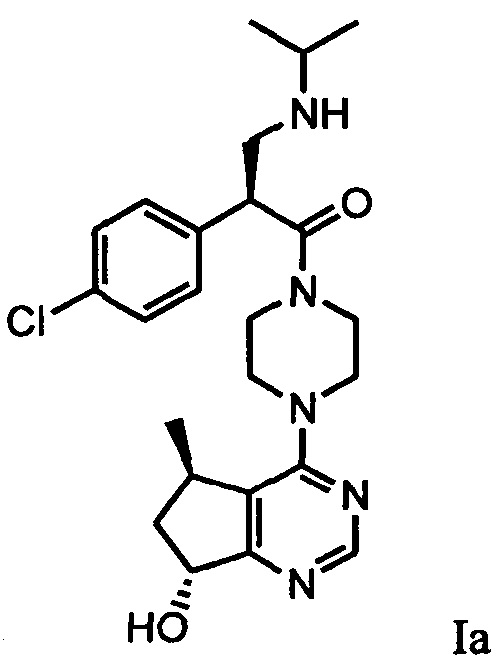
или его фармацевтически приемлемой соли и b) абиратерона или его фармацевтически приемлемой соли, взятых в терапевтически активном количестве, для терапевтического лечения рака предстательной железы.
2. Комбинация по п. 1, где фармацевтически приемлемая соль абиратерона представляет собой ацетат абиратерона.
3. Комбинация по п. 1, где рак ассоциирован с мутацией PTEN.
4. Комбинация по п. 1, где рак ассоциирован с мутацией, сверэкспрессией или амплификацией AKT.
5. Комбинация по п. 1, где рак ассоциирован с мутацией PI3K.
6. Комбинация по п. 1, где рак ассоциирован с мутацией Her2/ErbB2.
7. Комбинация по п. 3, где рак, ассоциированный с мутацией PTEN, представляет собой рак предстательной железы с нулевым статусом PTEN.
8. Комбинация по любому из пп. 1-6, где соединение формулы Ia и абиратерон находятся в соотношении 1:5 (мг/кг).
9. Способ лечения рака предстательной железы у млекопитающего, включающий введение млекопитающему комбинации а) соединения формулы Ia
или его фармацевтически приемлемой соли; и b) абиратерона или его фармацевтически приемлемой соли, взятых в терапевтически эффективном количестве для лечения рака предстательной железы.
10. Способ по п. 9, где фармацевтически приемлемая соль абиратерона представляет собой ацетат абиратерона.
11. Применение соединения формулы Ia или его фармацевтически приемлемой соли, как определено в п. 1, для получения лекарственного средства для лечения рака предстательной железы, где пациенту вводят абиратерон или его фармацевтически приемлемую соль.
12. Применение по п. 11, где фармацевтически приемлемая соль абиратерона представляет собой ацетат абиратерона.
13. Применение соединения формулы Ia или его фармацевтически приемлемой соли, как определено в п. 1, для получения лекарственного средства для лечения рака предстательной железы, модулируемого AKT-киназой, где пациенту вводят абиратерон или его фармацевтически приемлемую соль.
14. Применение по п. 13, где фармацевтически приемлемая соль абиратерона представляет собой ацетат абиратерона.
| US 2008051399 A1, 28.02.2008 | |||
| US 2010069357 A1, 18.03.2010 & RU 2473549 C2 | |||
| US 2009258364 A1, 15.10.2009 | |||
| US 2009137595 A1, 28.05.2009 | |||
| WO 2008006040 A1, 10.01.2008 & RU 2478632 | |||
| RU 2013148814 A1, 10.05.2015. |

