ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области фармацевтики. В частности настоящее изобретение относится к новому соединению из ряда дейтерированных фениламинопиримидинов или фармацевтическим композициям, которые включают указанное соединение.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
JAK (Янус киназа) является одним из типов семейства не-рецепторных тирозинкиназ, включающим 4 члена, которыми являются JAK1, JAK2, JAK3 и TYK1, соответственно. Они не содержат SH2 или SH3 в структуре, и на их С-концах находятся два соединенных участка киназы. Субстратом JAK является STAT, то есть проводники сигнала и активаторы транскрипции. STAT димеризуется после фосфорилирования с помощью JAK, и проходит через ядерную мембрану в ядро для регуляции экспрессии соответствующих генов, этот путь передачи сигнала называется JAK-STAT путь, и он играет важную роль в обеспечении связи кроветворных клеток и иммунных клеток. Эффективные и специфические ингибиторы четырех JAK-киназ, известные в настоящее время, могут быть использованы для лечения рака, воспаления и других заболеваний. Селективный ингибитор JAK3, тофацитиниб (название продукта: Кселянз, Xeljanz), от Пфайзер, утвержден Управлением по контролю за продуктами питания и лекарственными средствами США для применения в лечении ревматоидного артрита в декабре 2012 года.
Фениламинопиримидиновые соединения и производные являются видом ингибиторов не-рецепторных тирозинкиназ, таких как JAK-киназы. В WO 2008109943 и US 2011212077 раскрыт ряд фениламинопиримидиновых производных, в которых пиримидиновое кольцо было 2,4-бис-замещенным. Среди этих производных, соединение CYT387 является селективным ингибитором JAK1 и JAK2-киназы, с химическим названием N-(цианометил)-4-(2-(4-морфолино-фениламино)пиримидин-4-ил)бензамид, и используется в лечении рака, миелопролиферативных заболеваний и других родственных заболеваний. Это соединение сейчас находится во II фазе клинических испытаний лечения миелопролиферативных заболеваний.
Хотя целевое ингибирование различных протеинкиназ является полезным для лечения заболеваний, связанных с киназой, открытие новых соединений, которые специфически ингибируют определенные протеинкиназы и имеют потенциал к использованию в качестве лекарства, такой как отличная пероральная биодоступность, все еще представляет собой сложности. Кроме того, существуют некоторые побочные эффекты и проблемы лекарственной устойчивости для некоторых из ингибиторов протеинкиназы, доступных в настоящее время на рынке.
Таким образом, в данной области все еще существует потребность в разработке соединений, обладающих ингибирующей JAK-киназу активностью, или имеющих лучшие фармакодинамические свойства.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является предложение класса новых соединений, обладающих ингибирующей JAK киназы активностью и лучшими фармакодинамическими свойствами, и их применение.
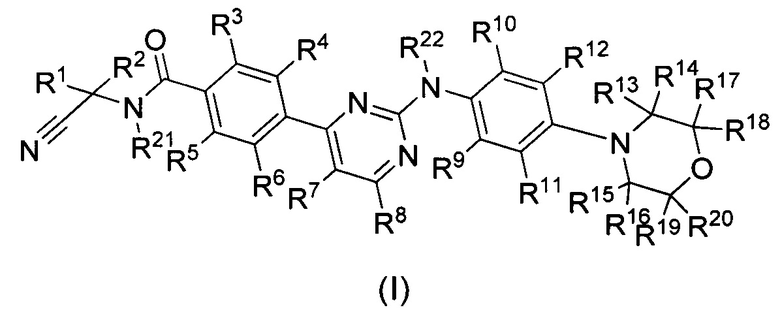
В первом аспекте настоящего изобретения оно предлагает дейтерированный фениламинопиримидин формулы (I), или его кристаллическую форму, фармацевтически приемлемую соль, гидрат или сольват:
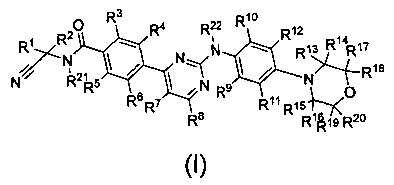
где R1, R2, R13, R14, R15, R16, R17, R18, R19, R20, R21 и R22 представляют собой каждый независимо водород или дейтерий;
R3, R4, R5, R6, R7, R8, R9, R10 и R11 представляют собой каждый независимо водород, дейтерий, галоген, недейтерированный C1-C6 алкил или C1-C6 алкокси, моно- или полидейтерированный или полностью дейтерированный C1-C6 алкил или C1-C6 алкокси, или моно- или полигалогенированный или полностью галогенированный C1-C6 алкил или C1-C6 алкокси;
R12 выбран из группы, состоящей из водорода, дейтерия, галогена, OR23, COOR23, COSR23, CONHR23 или CON(R23)2; причем R23 выбран из группы, состоящей из водорода, дейтерия, замещенного или незамещенного C1-C6-алкил или С3-С8-циклоалкил, при этом заместитель выбран из группы, состоящей из следующих: галоген, циано, C1-C6-алкил или C1-C6-алкокси;
при условии, что по меньшей мере один из R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11, R13, R14, R15, R16, R17, R18, R19, R20, R21 или R22 являются дейтерированным или дейтерием.
В другом предпочтительном варианте реализации содержание изотопа дейтерия в дейтерии в дейтериево-замещенном положении является по меньшей мере выше, чем содержание природного изотопного дейтерия (около 0,015%), предпочтительно на 30%, более предпочтительно 50%, более предпочтительно 75%, более предпочтительно 95%, более предпочтительно 99%.
В другом предпочтительном варианте реализации соединение формулы (I) содержит по меньшей мере один атом дейтерия, более предпочтительно два атома дейтерия, более предпочтительно четыре атома дейтерия, более предпочтительно шесть атомов дейтерия.
В еще одном предпочтительном варианте реализации R1 и R2 представляют собой водород или дейтерий.
В другом предпочтительном варианте реализации, R12 представляют собой водород или дейтерий.
В другом предпочтительном варианте реализации, R13, R14, R15 или R16 представляют собой водород или дейтерий.
В другом предпочтительном варианте реализации, R17, R18, R19 или R20 представляют собой водород или дейтерий.
В другом предпочтительном варианте реализации, R13, R14, R15, R16, R17, R18, R19 или R20 представляют собой дейтерий.
В другом предпочтительном варианте реализации, R3, R4, R5, R6, R7 или R8 каждый независимо выбран из группы, состоящей из водорода, дейтерия, моно- или полидейтерированного или полностью дейтерированного метила или метоксила, или моно- или полидейтерированного или полностью дейтерированного этила или этоксила.
В другом предпочтительном варианте реализации R3, R4, R5, R6, R7 или R8 каждый независимо выбран из группы, состоящей из: монодейтерированного метили, дидейтерированного метила, тридейтерированного метила, монодейтерированного метоксила, дидейтерированного метоксила, тридейтерированного метоксила, монодейтерированного этила, дидейтерированного этила, тридейтерированного этила, тетрадейтерированного этила, пентадейтерированного этила, монодейтерированного этоксила, дидейтерированного этоксила, тридейтерированного этоксила, тетрадейтерированного этоксила и пентадейтерированного этоксила.
В еще одном предпочтительном варианте реализации соединение представляет собой соединение, выбранное из группы, состоящей из следующих веществ или их фармацевтической приемлемой соли:
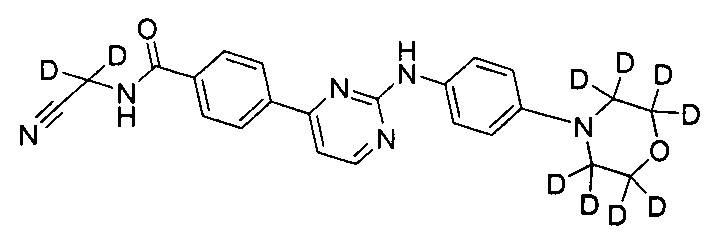
N-(циано(d2-метил))-4-(2-(4-(d8-морфолино)фениламино)пиримидин-4-ил)бензамид;
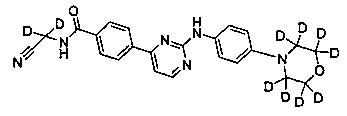
N-(циано(d2-метил))-4-(2-(4-(2',2',6',6'-d4-морфолино)фениламино)пиримидин-4-ил)бензамид;
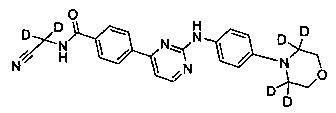
N-(циано(d2-метил))-4-(2-(4-(3',3',5',5'-d4-морфолино)фениламино)пиримидин-4-ил)бензамид;
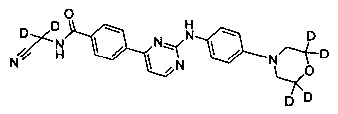
N-(цианометил)-4-(2-(4-(d8-морфолино) фениламино)пиримидин-4-ил)бензамид;
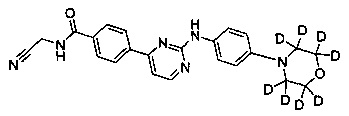
N-(цианометил)-4-(2-(4-(2',2',6',6'-d4-морфолино)фениламино)пиримидин-4-ил)бензамид;
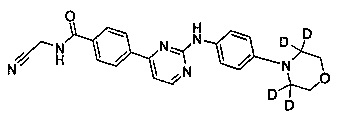
N-(цианометил)-4-(2-(4-(3',3',5',5'-d4-морфолино)фениламино)пиримидин-4-ил)бензамид;
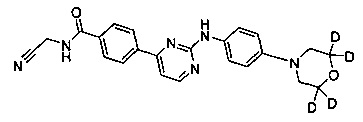
N-(циано(d2-метил))-4-(2-(4-морфолинфениламино)пиримидин-4-ил)бензамид;
В другом предпочтительном варианте реализации, соединение представляет собой
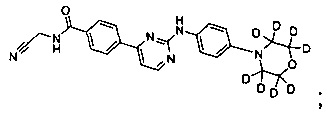
которое имеет следующие характеристики:
MS вычисленное: 422; MS найденное: 423 (М+Н)+, 445 (M+Na)+.
В другом предпочтительном варианте реализации, соединение представляет собой
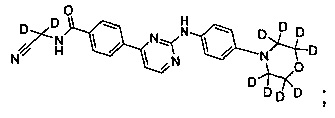
которое имеет следующие характеристики:
MS вычисленное: 424; MS найденное: 425 (М+Н)+, 447 (M+Na)+.
Еще в одном предпочтительном варианте реализации, соединение представляет собой
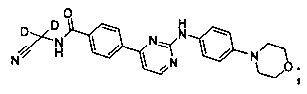
которое имеет следующие характеристики:
MS вычисленное: 416; MS найденное: 417 (М+Н)+, 439 (M+Na)+.
В следующем предпочтительном варианте реализации соединение представляет собой
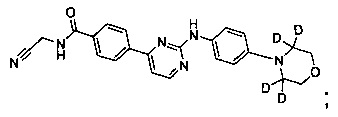
которое имеет следующие характеристики:
MS вычисленное: 418; MS найденное: 419 (М+Н)+, 441 (M+Na)+.
В другом предпочтительном варианте реализации соединение представляет собой
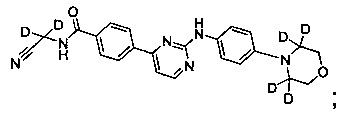
которое имеет следующие характеристики:
MS вычисленное: 420; MS найденное: 421 (М+Н)+, 443 (M+Na)+.
В другом предпочтительном варианте реализации, соединение не содержит недейтерированных соединений.
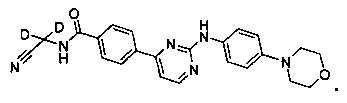
В другом предпочтительном варианте реализации, недейтерированное соединение представляет собой N-(цианометил)-4-(2-(4-морфолинофениламино)пиримидин-4-ил)бензамид.
В другом предпочтительном варианте реализации соединение получают способом, описанным в Примерах 1-8.
Во втором аспекте настоящего изобретения предложен способ получения фармацевтических композиций, включающий следующий этап: смешивание соединения согласно первому аспекту настоящего изобретения, или его кристаллической формы, фармацевтически приемлемой соли, гидрата или сольвата и фармацевтически приемлемого носителя, для получения фармацевтической композиции.
В третьем аспекте настоящего изобретения предлагается фармацевтическая композиция, которая содержит фармацевтически приемлемый носитель и соединение по первому аспекту настоящего изобретения, или его кристаллическую форму, фармацевтически приемлемую соль, гидрат или сольват.
В другом предпочтительном варианте реализации фармацевтическая композиция представляет собой раствор для инъекций, капсулу, таблетку, пилюлю, порошок или гранулу.
В другом предпочтительном варианте реализации фармацевтическая композиция далее содержит дополнительный терапевтический агент, который является лекарством для лечения рака, сердечно-сосудистых заболеваний, воспаления, иммунных заболеваний, миелопролиферативных заболеваний, вирусных болезней, болезней обмена веществ или трансплантации органов.
Более предпочтительно, дополнительный терапевтический агент содержит (но не ограничивается этим): 5-фторурацил, Avastin™ (авастин, бевацизумаб), Bexarotene (бексаротен), бортезомиб (бортезомиб), кальцитриол (кальцитриол), канертиниб (канертиниб), капецитабин (капецитабин), карбоплатин (карбоплатин), целекоксиб (целекоксиб), цетуксимаб (цетуксимаб), цисплатин (цисплатин), дазатиниб (дазатиниб), дигоксин (дигоксин), энзастаурин, Эрлотиниб (Эрлотиниб), этопозид (этопозид), эверолимус (эверолимус), фулвестрант (фулвестрант), гефитиниб (гефитиниб), 2,2-дифтор-дезоксицитидин (гемцитабин), генистеин (генистеин), иматиниб (иматиниб), иринотекан (иринотекан), лапатиниб (лапатиниб), леналидомид (леналидомид), летрозол (летрозол), фолиновую кислоту (лейковорин), матузумаб (матузумаб), оксалиплатин (оксалиплатин), Таксол (паклитаксел), панитумумаб (панитумумаб), пегилированный гранулоцитарный колониестимулирующий фактор (пегфилграстин), пегилированный альфа-интерферон (пегилированный альфа-интерферон), пеметрексед (пеметрексед), Полифенон® Е (Polyphenon® Е), сатраплатин (сатраплатин), сиролимус (сиролимус), сунитиниб (сутент, сунитиниб), сулиндаковую кислоту (сулиндак), Таксотер (таксотер), темозоломид (темодар, темозомоломид), Торисел (Torisel), Темсиролимус (темсиролимус), типифарниб (типифарниб), трастузумаб (трастузумаб), вальпроевую кислоту (вальпроевая кислота), дитартрат (дитартрат), волоциксимаб (Volociximab), вориностат (Vorinostat), сорафениб (Сорафениб), амбрисентан (амбрисентан), CD40- и/или СР154-специфичные антитела, гибридные белки, NF-κВ ингибиторы, нестероидные противовоспалительные препараты, β-агонисты, такие как ингибиторы сальметерол и ингибиторы фактора свертывания крови FXa (например, ривароксабан и т.д.), анти-ФНО (TNF) антитело, препараты простагландина или монтелукаст (монтелукаст).
В четвертом аспекте настоящего изобретения предложено применение соединения согласно первому аспекту настоящего изобретения, или его кристаллической формы, фармацевтически приемлемой соли, гидрата или сольвата для приготовления фармацевтической композиции для ингибирования протеинкиназ (например, JAK-киназ).
В другом предпочтительном варианте реализации фармацевтическую композицию применяют для лечения и профилактики следующих заболеваний: рак, миелопролиферативные заболевания, воспаление, иммунные заболевания, трансплантация органов, вирусные заболевания, сердечно-сосудистые заболевания или метаболические заболевания.
В другом предпочтительном варианте реализации рак включает (но не ограничивается этим): немелкоклеточный рак легкого, рак матки, рак прямой кишки, рак толстой кишки, рак мозга, рак головы, рак шеи, рак мочевого пузыря, рак простаты, рак молочной железы, рак почки, лейкоз, рак печени, рак желудка, рак щитовидной железы, рак носоглотки или рак поджелудочной железы.
В другом предпочтительном варианте реализации миелопролиферативные заболевания включают (но не ограничиваются этим): спонтанную тромбоцитемию (ЕТ), идиопатический миелофиброз (МФМ, IMF), хронический миелолейкоз (ХМЛ, CML), первичный миелофиброз, хронический нейтрофильный лейкоз (ХНЛ, CNL) или истинную полицитемию (ИП, PV).
В другом предпочтительном варианте реализации воспаление или иммунные заболевания включают (но не ограничиваются этим): ревматоидный артрит, остеоартрит, ревматоидный спондилит, подагру, астму, бронхит, ринит, хроническое обструктивное заболевание легких, кистозный фиброз (муковисцидоз).
В пятом аспекте настоящего изобретения предложен способ ингибирования протеинкиназы (например, JAK-киназы) или лечения заболеваний (таких как рак, миелопролиферативные заболевания, воспаление, иммунные заболевания, трансплантация органов, вирусные заболевания, сердечнососудистые заболевания или заболевания обмена веществ), причем способ включает следующий этап: введение субъекту, нуждающемуся в этом, соединения по первому аспекту настоящего изобретения, или его кристаллической формы, фармацевтически приемлемой соли, гидрата или сольвата, или введение субъекту, нуждающемуся в этом, фармацевтической композиции по третьему аспекту настоящего изобретения.
Следует понимать, что в пределах объема настоящего изобретения, каждый из технических признаков, конкретно описанных выше и ниже (например, описанные в Примерах), могут быть объединены друг с другом, тем самым составляя новые или предпочтительные технические решения, которые нет необходимости подробно описывать снова в данном документе.
ВАРИАНТЫ РЕАЛИЗАЦИИ ДЛЯ ВЫПОЛНЕНИЯ ИЗОБРЕТЕНИЯ
В ходе исследования изобретатель неожиданно обнаружил, что дейтерированные фениламинопиримидины или их фармацевтически приемлемые соли по настоящему изобретению удивительно превосходят недейтерированные соединения по фармакокинетическим и/или фармакодинамическим свойствам, и, следовательно, больше подходят для использования в качестве ингибиторов JAK-киназы и, таким образом, больше подходят для применения при производстве лекарственных средств для лечения рака и заболеваний, связанных с JAK-киназами. На этой основе было выполнено настоящее изобретение.
ОПРЕДЕЛЕНИЯ
Как здесь использовано, термин "галоген" относится к F, Cl, Br и I. Более предпочтительно атом галогена выбирают из F, Cl и Br.
Как здесь использовано, термин "C1-C6-алкил" относится к прямой или разветвленной алкильной группе, которая включает от 1 до 6 атомов углерода, такой как метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил или аналогичным группам.
Как здесь использовано, термин "C1-C6-алкокси" относится к прямой или разветвленной алкоксигруппе, которая содержит от 1 до 6 атомов углерода, такой как метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, трет-бутокси, или аналогичным группам.
Как здесь использовано, термин "С3-С8-циклоалкил" относится к циклоалкильной группе, которая включает от 3 до 8 атомов углерода, такой как циклопропил, циклобутил, циклопентил, циклогексил, или аналогичным группам.
Как здесь использовано, термин «дейтерированный» означает, что один или несколько водородов в соединении или группе замещены на дейтерий. "Дейтерированный" может быть монозамещенным, дизамещенным, полизамещенным или полностью замещенным. Термин "один или более дейтерированный" и "моно- или полидейтерированный" может быть использован взаимозаменяемо.
Как здесь использовано, термин «недейтерированное соединение» относится к соединению, которое имеет процентное содержание атома дейтерия не выше, чем природное изотопное содержание дейтерия (около 0,015%).
В другом предпочтительном варианте реализации, содержание изотопа дейтерия в дейтерий-замещенном положении больше, чем природный изотопный дейтериевый состав (0,015%), более предпочтительно больше, чем природный изотопный состав дейтерия на 50%, более предпочтительно 75%, более предпочтительно 95%, более предпочтительно 97%, более предпочтительно 99%, более предпочтительно 99,5%.
В другом предпочтительном варианте реализации соединение формулы (I) содержит по меньшей мере 2 атома дейтерия, более предпочтительно 4 атома дейтерия, более предпочтительно 6 атомов дейтерия, более предпочтительно от 8 атомов дейтерия.
Предпочтительно, в соединении формулы (I), N является 14N и/или О является 16O.
В другом предпочтительном варианте реализации в соединении содержание 14N изотопа в положении атома азота составляет ≥95%, предпочтительно ≥99%.
В другом предпочтительном варианте реализации в соединении, содержание 16O изотопа в положении атома кислорода составляет ≥95%, предпочтительно ≥99%.
ОПРЕДЕЛЕНИЯ
Как используется здесь, термин «соединение по настоящему изобретению» относится к соединению формулы (I). Этот термин также включает кристаллические формы, фармацевтически приемлемые соли, гидраты или сольваты соединения формулы (I).
Там, где используется термин "фармацевтически приемлемая соль», он относится к соли, образованной соединением по настоящему изобретению и кислотой или основанием, которое пригодно для применения в качестве лекарственного средства. Фармацевтически приемлемые соли включают неорганические и органические соли. Предпочтительным типом солей являются соли, образованные соединением по настоящему изобретению и кислотой. Подходящие солеобразующие кислоты включают, но не ограничиваются: неорганические кислоты, такие как соляная кислота, бромоводородная кислота, фтороводородная кислота, серная кислота, азотная кислота, фосфорная кислота; органические кислоты, такие как муравьиная кислота, уксусная кислота, трифторуксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, пикриновая кислоты, бензойная кислоты, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, бензолсульфоновая кислота, нафталинсульфокислоты и тому подобное; и аминокислоты, такие как пролин, фенилаланин, аспарагиновая кислота, глутаминовая кислота. Другим предпочтительным типом солей являются соли, образованные соединением по настоящему изобретению и основанием, например, соли щелочных металлов (например, натриевые или калиевые соли), соли щелочно-земельных металлов (например, кальциевые или магниевые соли), аммониевые соли (например, аммониевые соли низших алканолов или другие фармацевтически приемлемые соли аминов), например, соли метиламина, соли этиламина, соли пропиламина, соли диметиламина, соли триметиламина, соли диэтиламина, соли триэтиламина, соли трет-бутил аминов, соли этилендиамина, соли гидроксиэтиламина, соли дигидроксиэтиламина, соли тригидроксиэтиламина и соли аминов, образованных морфолином, пиперазином, и лизином, соответственно.
Термин «сольват» относится к комплексу конкретного соотношения, образованному путем комплексообразования между соединением по настоящему изобретению и молекулой растворителя. Термин «гидрат» относится к комплексу, образованному путем комплексообразования между соединением по настоящему изобретению и водой.
Кроме того, соединения по настоящему изобретению дополнительно включают пролекарства фениламинопиримидиновых соединений формулы (I). Термин "пролекарство" включает класс соединений, которые имеют биологическую активность или отсутствие активности и преобразуются в соединение формулы (I) путем обмена веществ (метаболизма) или химических реакций в организме человека при введении с помощью подходящих методов, или его соль или раствор, образованный соединением формулы (I). Пролекарства включают (но не ограничиваются) сложный эфир карбоновой кислоты, угольной кислоты, фосфат, нитрат, сульфат, сульфоновый эфир, сульфоксид эфиры, аминосоединения, карбаматы, азосоединения, фосфорамиды, глюкозид, эфир, ацеталь соединения, и т.д.
СПОСОБ ПОЛУЧЕНИЯ
Далее более подробно описано получение соединений формулы (I), но такие специфические способы не являются какими-либо ограничениями для настоящего изобретения. Соединения по изобретению могут также быть легко получены с помощью возможного комбинирования различных методов синтеза, приведенных в данном описании или известных в данной области. Такие комбинации могут быть легко выполнены специалистом с обычной квалификацией в данной области, согласно настоящему изобретению.
Способы получения соединений недейтерированного фениламинопиримидина и их физиологически приемлемых солей, используемые в настоящем изобретении, известны в данной области. Получение соответствующих дейтерированных соединений ряда фениламинопиримидина может быть проведено с помощью соответствующего дейтерированного соединения в виде исходного реагента и синтеза аналогичным путем. Например, соединение формулы (I) по настоящему изобретению может быть получено в соответствии со способом, описанным в WO 2008109943, за исключением того, что используют дейтерированный реагент вместо недейтерированного реагента.
Как правило, в процессе подготовки, каждую реакцию обычно проводят в инертном растворителе, при комнатной температуре до температуры дефлегмации (например, 0°C~80°C, предпочтительно от 0°C~50°C). Время реакции обычно составляет от 0,1 часа до 60 часов, предпочтительно от 0,5 до 48 часов.
Следующий общий препаративный путь может быть использован при синтезе компонентов формулы (I) по настоящему изобретению.
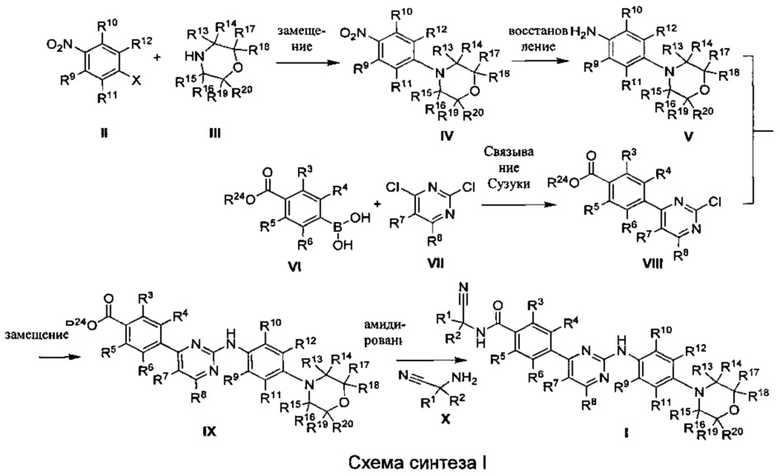
где X выбран из F, Cl, Br, I; определения R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, R13, R14, R15, R16, R17, R18, R19 или R20 такие, как указано выше; R24 выбран из водорода, и C1-C6 алкила.
Как показано на схеме синтеза I, нитробензольное соединение II и соединение морфолина III реагируют с образованием 4-морфолинозамещенных нитробензолов соединения IV, которое затем восстанавливают для получения 4-морфолинозамещенного фениламинового соединения V. Соединение фенилборной кислоты VI и соединение 2,4-дихлорпиримидина VII продуцируют соединение VIII посредством реакции Сузуки. Соединение VIII реагирует с соединением V с образованием фениламинопиримидинового соединения IX. Соединение IX и аминоацетонитрилсоединение X реагируют с получением Соединения I по настоящему изобретению с помощью амидирования. Вышеуказанные реакции проводят в инертных растворителях, таких как дихлорметан, дихлорэтан, ацетонитрил, n-гексан, толуол, тетрагидрофуран, N,N-диметилформамид, диметилсульфоксид, и т.д., и при температуре от 0°C до 200°C.
ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБЫ ВВЕДЕНИЯ
Соединения по настоящему изобретению обладают выдающейся активностью по ингибированию протеинкиназы (такой как JAK-киназы). Таким образом, соединения по настоящему изобретению, и различные кристаллические формы, фармацевтически приемлемые неорганические или органические соли, гидраты или сольваты, и фармацевтическая композиция, содержащая соединение по настоящему изобретению в качестве основного активного ингредиента, может быть использована для лечения, профилактики и облегчения заболеваний, опосредованных протеинкиназами (например, JAK-киназами). На основании предшествующего уровня техники, соединения по настоящему изобретению могут лечить следующие заболевания: рак, миелопролиферативные заболевания, воспаления, иммунные заболевания, трансплантации органов, вирусные заболевания, сердечно-сосудистые заболевания или заболевания обмена веществ и т.д.
Фармацевтическая композиция по настоящему изобретению включает соединение по настоящему изобретению или его фармацевтически приемлемые соли в безопасном и эффективном количестве и фармацевтически приемлемые наполнители или носители. При этом термин "безопасная и эффективная дозировка» относится к количеству соединения, которое является достаточным для улучшения состояния пациента без каких-либо серьезных побочных эффектов. Как правило, фармацевтическая композиция содержит 1-2000 мг соединения по изобретению на дозу, предпочтительно, 10-200 мг соединения по изобретению на дозу (per dose). Предпочтительно "на дозу" («per dose») означает одну капсулу или таблетку.
"Фармацевтически приемлемый носитель" означает один или несколько совместимых твердых или жидких наполнителей или гелеподобных материалов, которые пригодны для использования человеку, и который должен иметь достаточную чистоту и достаточно низкую токсичность. "Совместимость" здесь означает, что компоненты композиций могут быть смешаны с соединениями по настоящему изобретению или друг с другом, и не будут значительно уменьшать эффективность соединений. Некоторые примеры фармацевтически приемлемых носителей включают целлюлозу и ее производные (например, натрий карбоксиметилцеллюлоза, натрий этилцеллюлоза, ацетатцеллюлозы и т.д.), желатин, тальк, твердые смазки (например, стеариновая кислота, стеарат магния), сульфат кальция, растительные масла (например, соевое масло, кунжутное масло, арахисовое масло, оливковое масло и т.д.), полиолы (такие как пропиленгликоль, глицерин, маннит, сорбит и т.д.), эмульгаторы (например, Твин, Tween®), смачивающий агент (например, додецилсульфат натрия), красящие агенты, ароматизирующие агенты, стабилизаторы, антиоксиданты, консерванты, апирогенная вода, и т.д.
Не существует особых ограничений для режимов введения соединений или фармацевтических композиций по настоящему изобретению, и репрезентативный способ введения включает (но не ограничивается ими): пероральное, внутриопухолевое, ректальное, парентеральное (внутривенное, внутримышечное или подкожное), и местное введение.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых дозированных формах, активные соединения смешивают по меньшей мере с одним из общепринятых инертных наполнителей (или носителей), таких как цитрат натрия или CaHPO4, или смешивают с любым из следующих компонентов: (а) наполнители или агенты, улучшающие совместимость, например, крахмал, лактоза, сахароза, глюкоза, маннит и кремниевая кислота; (b) связующие, например, гидроксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и гуммиарабик; (с) увлажнитель, такой как глицерин; (d) дезинтегрирующие агенты, такие как агар, карбонат кальция, картофельный крахмал или тапиоковый крахмал, альгиновая кислота, некоторые композиционные силикаты, и карбонат натрия; (е) замедляющие растворение агенты, такие как парафин; (f) ускорители абсорбции, например, четвертичные аммониевые соединения; (g) смачивающие агенты, такие как цетиловый спирт, моностеарат глицерина (h) адсорбенты, например, каолин; и (i) смазывающие вещества, такие как тальк, стеарин кальциевый, стеарат магния, твердый полиэтиленгликоль, лаурилсульфат натрия, или их смеси. В капсулах, таблетках и пилюлях, дозированные формы могут также содержать буферные агенты.
Твердые дозированные формы, такие как таблетки, сахарные пилюли, капсулы, пилюли и гранулы, могут быть получены с использованием материалов для покрытий и оболочки, таких как кишечнорастворимые покрытия и любых других материалов, известных в данной области техники. Они могут содержать матирующий агент. Высвобождение активных соединений или соединений в композициях может происходит в замедленном режиме в определенной части желудочно-кишечного тракта. Примеры покрывающих компонентов включают полимеры и воски. При необходимости активные соединения и один или более из указанных выше наполнителей могут образовывать микрокапсулы.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы или настойки. В дополнение к активным соединениям, жидкие лекарственные формы могут содержать любые традиционные инертные разбавители, известные в данной области, такие как вода или другие растворители, солюбилизаторы и эмульгаторы, например, этанол, изопропанол, этиловый карбонат, этилацетат, пропиленгликоль, 1,3-бутандиол, диметилформамид, а также масло, в частности, хлопковое масло, арахисовое масло, кукурузное масло, оливковое масло, касторовое масло и кунжутное масло, или их комбинации.
Помимо этих инертных разбавителей композиции могут также содержать добавки, такие как смачивающие агенты, эмульгаторы и суспендирующие средства, подслащивающие, вкусовые агенты и ароматизаторы.
В дополнение к активным соединениям, суспензия может содержать суспендирующий агент, например, этоксилированный изооктадеканол, полиоксиэтиленсорбит и сложные эфиры сорбита, микрокристаллическую целлюлозу, метанол алюминия и агар, или их комбинации.
Композиции для парентерального введения, могут включать физиологически приемлемые стерильные водные или безводные растворы, дисперсии, суспензии или эмульсии, и стерильные порошки, которые могут быть повторно растворены в стерильных инъекционных растворах или дисперсиях. Подходящие водные и неводные носители, разбавители, растворители или наполнители включают воду, этанол, полиолы и их любые подходящие смеси.
Лекарственные формы для местного введения соединений по настоящему изобретению включают мази, порошки, аэрозоли, пластыри, и ингаляторы. Активные ингредиенты смешивают с физиологически приемлемыми носителями и любыми консервантами, буферами или пропеллентами, в случае необходимости, в стерильных условиях.
Соединения по настоящему изобретению можно вводить отдельно или в сочетании с любыми другими фармацевтически приемлемыми соединениями.
Когда фармацевтические композиции используют, безопасное и эффективное количество соединения по настоящему изобретению вводят млекопитающему (например, человеку), который в этом нуждается, при этом доза введения является фармацевтически эффективной дозой. Для человека, весящего 60 кг, суточная доза обычно составляет от 1 до 2000 мг, предпочтительно от 20 до 500 мг. Конечно, конкретная доза должна зависеть от различных факторов, таких как способ введения, состояния здоровья пациента, что находится в компетенции опытного врача.
По сравнению с недейтерированными соединениями, известными в предшествующем уровне техники, соединения по настоящему изобретению обладают рядом преимуществ. Основные преимущества настоящего изобретения включают:
(1) Соединения по настоящему изобретению обладают превосходной ингибирующей активностью по отношению к протеинкиназе (например, JAK-киназе).
(2) Метаболизм дейтерированных соединений в организме изменяют с помощью технологии дейтерирования, таким образом, придавая соединениям улучшенные фармакокинетические параметры. В этом случае доза может варьироваться и может быть получен препарат длительного действия, с улучшением применимости.
(3) В соединениях водород был замещен дейтерием, лекарственная концентрация соединения у животных может быть повышена за счет влияния дейтериевого изотопа, тем самым повышая эффективность препарата.
(4) Водород в соединениях был замещен дейтерием, и так как некоторые метаболиты подавлены, то безопасность соединений может быть повышена.
Настоящее изобретение будет дополнительно проиллюстрировано ниже со ссылкой на конкретные примеры. Следует понимать, что эти примеры приведены только для иллюстрации настоящего изобретения, но не для ограничения объема изобретения. Экспериментальные методы без конкретных условий, описанные в следующих примерах, как правило, выполняют в соответствии с общепринятыми условиями, или в соответствии с инструкциями производителя. Если не указано иное, то части и проценты вычисляются по весу. В настоящем изобретении "биотин" означает биотин.
Пример 1: получение N-(цианометил)-4-(2-(4-(d8-морфолино)фениламино)пиримидин-4-ил)бензамида (соединение 9)
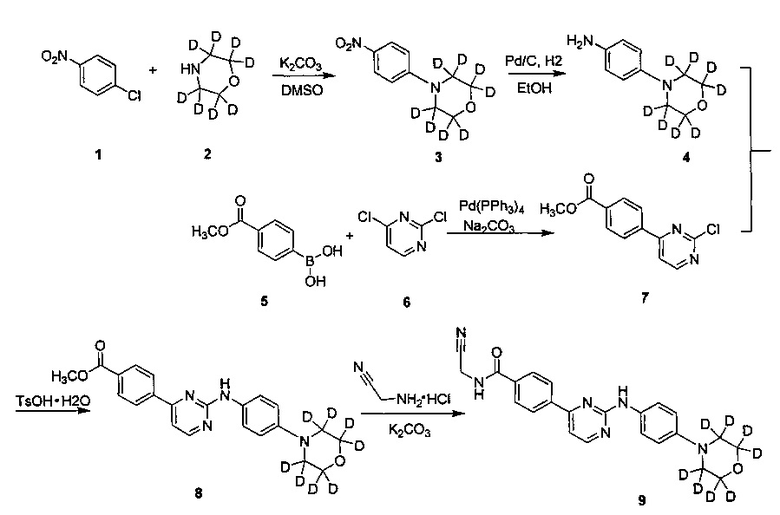
1. Получение 4-(4-нитрофенил)(d8-морфолин) (соединение 3)
Соединение 4-хлорнитробензол (3,53 г, 22,4 ммоль), d8-морфолин (2,35 г, 24,6 ммоль) и карбонат калия (6,07 г, 44 ммоль) последовательно добавляли в колбу, и добавляли диметилсульфоксид (40 мл). После этого смесь нагревали до 100°C и перемешивали в течение 16 ч. ТСХ анализ (этилацетат / петролейный эфир = 1/10) показал, что реакция завершена. Затем ее охлаждали до комнатной температуры, и реакцию гасили добавлением воды (100 мл). Смесь экстрагировали дихлорметаном (100 мл) дважды. Объединенный органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с помощью роторного испарителя с получением сырого продукта. Его кристаллизовали в смешанном растворителе из этилацетата и н-гексана (1/5, об/об, 18 мл), с получением желтого твердого желаемого продукта (3,92 г, выход 81%); MS вычислено: 216; MS найдено: 217 (М+Н)+.
2. Получение 4-(d8-морфолино)фениламин (соединение 4)
4-(4-нитрофенил)(d8-морфолино) (3,80 г, 17,6 ммоль), этанол (30 мл) и воду (3 мл) добавляли в колбу для гидрирования. 10% палладий на угле (0,19 г) добавляли в атмосфере азота. Реакционную систему заменяли водородом 3-4 раза, и перемешивали под давлением 10 атм. давлении водорода, и перемешивают для реагирования при 40°C в течение 10 часов. С помощью ВЭЖХ определяли, что реакция завершена, и затем реакционный раствор охлаждали до комнатной температуры и фильтровали через Цеолит. Остаток промывали этанолом. Фильтрат объединяли и концентрировали под вакуумом с помощью роторного испарителя, получая желаемый продукт, беловатое твердое вещество (3,11 г, выход: 95%). MS вычислено: 186; MS найдено: 187 (М+Н)+.
3. Получение 4-(2-хлорпиримидин-4-ил) метил бензоата (соединение 7)
Соединение 4-(метоксикарбонил)фенилборную кислоту (4,28 г, 23,78 ммоль), 2,4-дихлорпиримидин (3,72 г, 24,97 ммоль), толуол (40 мл) и водный раствор карбоната натрия (2н, 11,9 мл) последовательно добавляли в колбу. Тетракис(трифенилфосфин) палладий (1,10 г, 0,95 ммоль) в атмосфере азота, нагревали до 80°C и перемешивали в течение ночи. После охлаждения до комнатной температуры, воду (10 мл) и этилацетат (50 мл) добавляли для разбавления реакционного раствора, перемешивали в течение 15 мин и расслаивали. Затем этилацетат (30 мл) использовали для экстрагирования водного слоя два раза. Объединенный органический слой промывали водой и солевым раствором. Сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с помощью роторного испарителя с получением сырого продукта. Сырой продукт суспендировали в метаноле (10 мл) и измельчали 30 минут, отфильтровывали и промывали метанолом. Сушили в вакууме с получением желаемого продукта, серого твердого вещества (3,13 г, выход: 53%). MS вычислено: 248; MS найдено: 249 (М+Н)+, 271 (M+Na)+.
4. Получение 4-(2-(4-(d8-морфолино)фениламино)пиримидин-4-ил)метилбензоата (соединение 8)
4-(2-хлорпиримидин-4-ил)метилбензоат (1,24 г, 5,0 ммоль), 4-(d8-морфолино)фениламина (0,84 г, 4,5 ммоль) и 1,4-диоксан (10 мл) последовательно добавляли в колба. Моногидрат п-толуолсульфоновой кислоты (0,88 г, 5,0 ммоль) добавляли в указанную выше суспензию при перемешивании. Нагревали с обратным холодильником в течение 48 ч, затем охлаждали до 30°C, и реакционную смесь концентрировали в вакууме на роторном испарителе. Добавляли этилацетат (20 мл) и водный раствор бикарбоната натрия (20 мл) и расслаивали. Водный слой экстрагировали этилацетатом дважды. Объединенный органический слой промывали водой, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом на роторном испарителе с получением неочищенного продукта. Сырой продукт очищали с помощью колоночной хроматографии с получением желтого твердого вещества, суспендировали в метаноле (5 мл), и измельчали в течение 15 мин. После фильтрования остаток промывали метанолом и сушили под вакуумом с получением желаемого продукта (1,09 г, выход: 61%). MS вычислено: 398; MS найдено: 399 (М+Н)+, 421 (M+Na)+.
5. Получение N-(цианометил)-4-(2-(4-(d8-морфолино)фениламино)пиримидин-4-ил)бензамид (соединение 9)
4-(2-(4-(d8-морфолино)фениламино)пиримидин-4-ил)метилбензоат (0,50 г, 1,25 ммоль), безводного карбоната калия (325 меш, 0,35 г, 2,5 ммоль) и тетрагидрофуран (4 мл) последовательно добавляли к колбе под защитой азота. 2-амино-ацетонитрилгидрохлорид (0,18 г, 1,88 ммоль) добавляли при перемешивании. Затем проводили реакцию в течение 13-17 ч при внутренней температуре реакции, поддерживаемой при 35±2°C, и реакцию контролировали с помощью ВЭЖХ до окончания реакции. Добавляли очищенную воду (10 мл) и реакционный раствор концентрировали под вакуумом на роторном испарителе, затем три раза экстрагировали этилацетатом. Объединенный органический слой промывали водой и солевым раствором. Сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом на роторном испарителе с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии, получая твердый желаемый продукт (0,21 г, выход: 40%). MS вычислено: 422; MS найдено: 423 (М+Н)+, 445 (M+Na)+.
Пример 2: Получение N-(цианометил)-4-(2-(4-(d8-морфолино)фениламино)пиримидин-4-ил)бензамида (соединение 9)
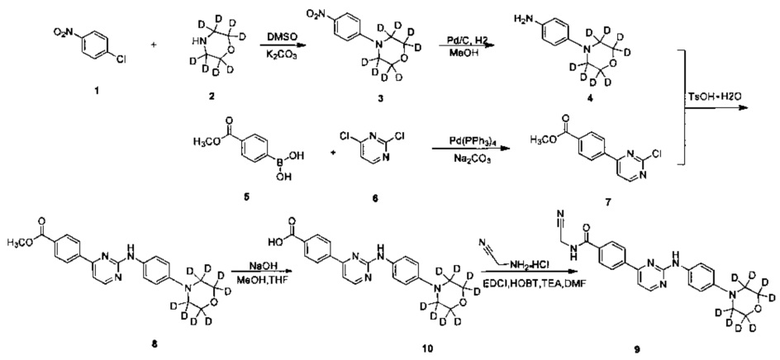
1. Получение 4-(4-нитрофенил)(d8-морфолин) (соединение 3)
4-хлорнитробензол (0,662 г, 4,17 ммоль), d8-морфолин (0,400 г, 4,17 ммоль, приобретенные у Cambridge Isotope Laboratories) и карбонат калия (1,733 г, 12,54 ммоль) последовательно добавляли в колбу. Добавляли диметилсульфоксид (6 мл), затем нагревали до 100°C, и перемешивали в течение 20 часов. Реакционную смесь охлаждали до комнатной температуры, затем добавляли воду (30 мл) для гашения реакции, и желтое твердое вещество выпадало в осадок. После перемешивания в течение 30 мин смесь фильтровали с получением неочищенного продукта. Сырой продукт кристаллизовали в смешанном растворителе из этилацетата и петролейного эфира (1/2,5, объем / объем, 14 мл), с получением желтого твердого желаемого продукта (0,600 г, чистота по ВЭЖХ: 98,6%, выход 67%).
2. Получение 4-(d8-морфолино)фениламина (соединение 4)
4-(4-нитрофенил)(d8-морфолин) (0,600 г, 2.78 ммоль) и метанол (40 мл) добавляли к сосуду для реакции гидрогенизации. 10% палладий на угле (0,060 г) добавляли под защитой азота. Реакционную систему заменяли водородом в 3-4 раза, и перемешивали в атмосфере водорода при давлении, и перемешивали для проведения реакции при 40°C в течение 20 часов. По ВЭЖХ определяли, что реакция завершена, а затем реакционный раствор охлаждали до комнатной температуры и фильтровали через целит. Остаток промывали метанолом. Объединенный фильтрат концентрировали под вакуумом на роторном испарителе, получая желаемый продукт, розовое твердое вещество (0,500 г, чистота по ВЭЖХ: 98,1%, выход: 96,5%).
3. Получение 4-(2-хлорпиримидин-4-ил)метилбензоата (соединение 7)
4-(метоксикарбонил) фенилборной кислоты (4,000 г, 22,23 ммоль), 2,4-дихлорпиримидин (3,150 г, 21.11 ммоль), толуол (40 мл) и водный раствор карбоната натрия (2 Н, 10,5 мл) добавляли в колбу. Тетракис (трифенилфосфин) палладий (0,513 г, 0,45 ммоль) в атмосфере азота, нагревали до 80°C и перемешивали в течение ночи. После охлаждения до комнатной температуры, добавляли воду (10 мл) и этилацетат (50 мл) для разбавления реакционного раствора, перемешивали в течение 15 мин и расслаивали. Водный слой экстрагировали этилацетатом (30 мл) два раза. Объединенный органический слой промывали водой и насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с помощью роторного испарителя с получением сырого продукта. Неочищенный продукт отделяли и очищали колоночной хроматографией на силикагеле (этилацетат / петролейный эфир = 0-30%), сушили под вакуумом, для получения белого твердого вещества желаемого продукта (2,50 г, выход: 48%). 1Н ЯМР (400 МГц, CDCl3) δ 8,71-8,70 (1Н, d), 8,19-8,14 (4Н, м), 7,71-7,70 (1Н, d), 3,97 м.д. (3Н, s).
4. Получение 4-(2-(4-d8-морфолино)фениламино)пиримидин-4-ил)метилбензоата (соединение 8)
4-(2-хлорпиримидин-4-ил)метилбензоат (0,606 г, 2,44 ммоль), 4-d8-морфолино)фениламин (0,500 г, 2.68 ммоль) и 1,4-диоксан (25 мл) добавляли в колбу. П-толуолсульфоновой кислоты моногидрат (0,510 г, 2,68 ммоль) добавляли к суспензии при перемешивании. Нагревали с обратным холодильником в течение 20 ч, затем реакционную смесь концентрировали под вакуумом на роторном испарителе. Добавляли этилацетат (15 мл) и 5% водный раствор бикарбоната натрия (15 мл), и желтое твердое вещество выпадало в осадок. После перемешивания в течение 30 минут, смесь фильтровали с получением неочищенного продукта. Сырой продукт суспендировали в метаноле (5 мл) и измельчали в течение 5 мин, фильтровали и остаток промывали метанолом, сушили под вакуумом для получения желаемого твердого продукта (0,680 г., чистота по ВЭЖХ: 95%, выход: 70%).
5. 4-(2-(4-d8-морфолино)фениламино)пиримидин-4-ил)бензойная кислота (соединение 10)
4-(2-(4-(d8-морфолино)фениламино)пиримидин-4-ил)метилбензоат (0,680 г, 1,706 ммоль), гидроксид натрия (0,137 г, 3,413 ммоль), воду (3,5 мл), и смешанный растворитель из метанола и тетрагидрофурана (12 мл, 3:1) добавляли в колбу, нагревали до 65°C и реакцию проводили в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры. Растворитель удаляли и pH доводили до 3 с помощью 10% разбавленной соляной кислоты, фильтровали и сушили с получением серого твердого вещества. Сырой продукт измельчали в порошок и добавляли метанол (5 мл), измельчали 5 мин, затем фильтровали и сушили с получением твердого желаемого продукта (0,570 г, степень очистки: 98,3% по ВЭЖХ, выход продукта: 87%).
6. Получение N-(цианометил)-4-(2-(4-d8-морфолино)фениламино)пиримидин-4-ил)бензамида (соединение 9)
4-(2-(4-d8-морфолино)фениламино)пиримидин-4-ил)бензойную кислоту (0,300 г, 0,780 ммоль), 1-этил-(3-диметиламинопропил)карбодиимид гидрохлорид (0,180 г, 0,936 ммоль), 1-гидрокси-фенилпропилтриазол (0,127 г, 0,936 ммоль), триэтиламин (0,473 г, 4,682 ммоль) и N,N-диметилформамид (3 мл) добавляли в колбу в атмосфере азота. При перемешивании добавляли 2-амино-ацетонитрилгидрохлорид, и реакция проходила в течение 20 ч при комнатной температуре. Очищенную воду (5 мл) и насыщенный раствор бикарбоната (5 мл) добавляли к реакционной смеси, и желтое твердое вещество выпадало в осадок. После перемешивания в течение 30 минут, смесь фильтровали и промывали чистой водой. Неочищенный продукт был получен после сушки, и его отделяли разделением и очисткой препаративной хроматографией с получением желтого твердого желаемого продукта (0,150 г, чистота по ВЭЖХ: 98,1%, выход продукта: 45%). MS вычислено: 422; MS найдено: 423 (М+Н)+. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,65 (1Н, с), 9,38-9,35 (1Н, м), 8,57-8,56 (1Н, д), 8,29-8,27 (2Н, д), 8,05-8,03 (2Н, д), 7,75-7,72 (2Н, д), 7,46-7,45 (1Н, д), 7,09-7,07 (2Н, м), 4,37-4,36 м.д. (2Н, д).
Пример 3: Получение N-(циано(d2-метил))-4-(2-(4-d8-морфолино)фениламино)пиримидин-4-ил)бензамида (соединение 13)
1. Получение 2-амино-2,2-d2-ацетонитрил гидрохлорида (соединение 12)
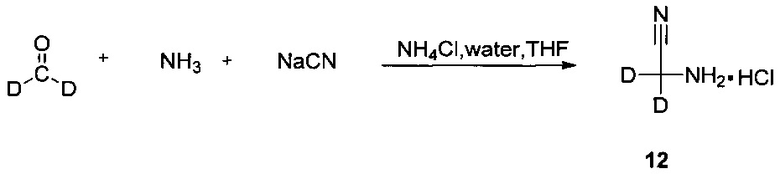
Цианид натрия (1,59 г, 32,46 ммоль) и аммиак (6,80 г, 99,87 ммоль) добавляли в колбу на ледяной бане, внутреннюю температуру контролировали при 5°C и добавляли хлорид аммония (2,27 г, 42,25 ммоль). После 10 мин перемешивания медленно по каплям в течение 10 минут добавляли дейтерированный формальдегид (1,00 г, 31,21 ммоль), и внутреннюю температуру регулировали так, чтобы она составляла 16-20°C. После перемешивания при термической консервации в течение 3 ч, добавляли дихлорметан (100 мл) и перемешивали в течение 45 мин, и жидкость, отделяли для получения органической фазы. Водную фазу дважды экстрагировали дихлорметаном (100 мл × 2), объединенные органические фазы сушили над безводным сульфатом натрия и фильтровали. Раствор хлористого водорода в изопропаноле (15 мл) добавляли к фильтрату на ледяной бане и перемешивают в течение 30 минут и осаждали белое твердое вещество. Фильтровали для получения белого твердого вещества целевого соединения (1,70 г, выход: 58%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,12 м.д. (3Н, с).
2. Получение N-(циано(d2-метил))-4-(2-(4-(d8-морфолино)фениламино)пиримидин-4-ил)бензамида; (соединение 13)
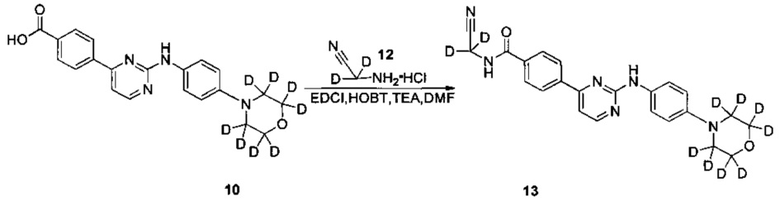
4-(2-(4-(d8-морфолино)фениламино)пиримидин-4-ил)бензойную кислоту (0,260 г, 0,676 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (0,156 г, 0,811 ммоль), 1-гидроксифенилпропил триазол (0,109 г, 0,811 ммоль), триэтиламин (0,410 г, 4,056 ммоль) и N,N-диметилформамид (2,5 мл) добавляли в колбу под защитой азота. 2-амино-2,2-d2-ацетонитрил гидрохлорид (0,192 г, 2,029 ммоль) добавляли при перемешивании. Реакцию проводили в течение 19 ч при комнатной температуре. Очищенную воду (3 мл) и насыщенный раствор бикарбоната (3 мл) добавляли к реакционной смеси, и желтое твердое вещество выпадало в осадок. Через 30 мин перемешивания осадок отфильтровывали и промывали чистой водой и сушили для получения неочищенного продукта. Неочищенный продукт очищали с помощью препаративной хроматографии с получением желтого твердого желаемого продукта (0,192 г, чистота по ВЭЖХ: 98,2%, выход продукта: 67%).
MS вычислено: 424; MS Найдено: 425 (М+Н)+. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,65 (1Н, с), 9,35 (1Н, с), 8.57-8.56 (1Н, д), 8.29-8.27 (2Н, д), 8.05-8.03 (2Н, д), 7.74-7.73 (2Н, д), 7,45 (1Н, с), 7,09 м.д. (2Н, с).
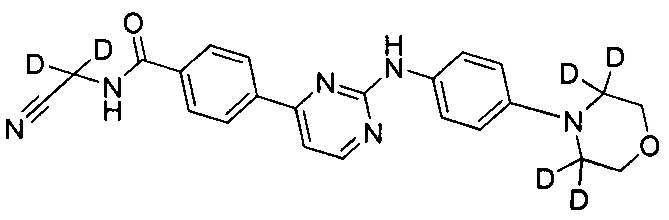
Пример 4: Получение N-(циано(d2-метил))-4-(2-(4-(2',2',6',6'-d4-морфолино)фениламино)пиримидин-4-ил)бензамида (соединение 24)
1. Получение 2,2,6,6-d4-морфолина (соединение 19)
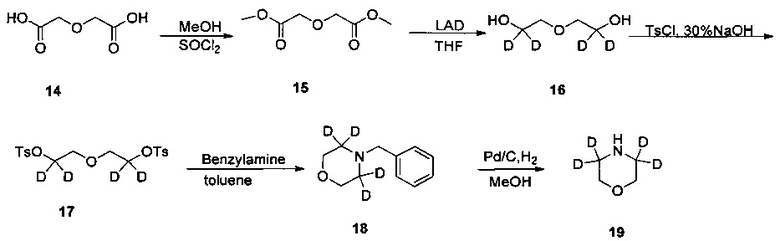
1) Получение дигликолевой кислоты диметилового эфира (соединение 15)
Дигликолевую кислоту (3,00 г, 22,37 ммоль) и безводный метанол (30 мл) добавляли в колбу на ледяной бане и диметилсульфоксид (7,98 г, 67,12 ммоль) медленно добавляли по каплям, после чего нагревали до комнатной температуры и перемешивали в течение 20 час. Реакционную смесь концентрировали с получением бесцветного маслянистого желаемого продукта (2,50 г, чистота по ВЭЖХ: 97,3%, выход 69%). 1Н-ЯМР (400 МГц, CDCl3) δ 4,25 (4Н, с), 3,76 м.д. (6Н, с).
2) Получение 2,2'-окси-бис(1,1-d2-этанол) (соединение 16)
Дейтерированный литий-алюминий гидрид (1,49 г, 35,5 ммоль, приобретен у J&K) и безводный тетрагидрофуран (10 мл) добавляли в колбу под защитой азота, охлаждали до 0°C с помощью ледяной бани и медленно по каплям добавляли раствор диметилового эфира в дигликолевой кислоте (2,5 г, 15,4 ммоль) в тетрагидрофуране (15 мл). После этого смесь нагревали до 65°C, перемешивали в течение 2 ч для проведения реакции, затем охлаждали до комнатной температуры и перемешивали в течение ночи. К реакционной смеси добавляли 5 мл воды, 2,5 мл 15% водного раствора гидроксида натрия, перемешивали в течение 30 мин и фильтровали, промывали тетрагидрофураном (20 мл). Фильтрат концентрировали с получением желтого маслянистого желаемого продукта (1,50 г, чистота по ВЭЖХ: 92%, выход 88%). 1Н-ЯМР (400 МГц, CDCl3) δ 3,59 м.д. (4Н, с).
3) Получение 2,2'-окси-бис-(1,1-d2-этан-2,1-диил)бис(4-п-толуолсульфонат) (соединение 17)
2,2'-окси-бис-(1,1-d2-этанол) (1,49 г, 35,5 ммоль) и 30% водный раствор гидроксида натрия (10 мл) добавляли в колбу на ледяной бане, и медленно по каплям добавляли п-толуолсульфонилхлорид (2,5 г, 15,4 ммоль) в тетрагидрофуране (15 мл). После этого смесь нагревали до комнатной температуры и перемешивали в течение ночи. К реакционной смеси добавляли воду и экстрагировали этилацетатом три раза, и органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением белого твердого вещества целевого соединения (4,2 г, выход 79%); 1Н-ЯМР (400 МГц, CDCl3) δ 7,82-7,79 (4Н, д), 7,38-7,36 (4Н, д), 3,62 (4Н, с), 2,48 м.д. (6Н, с).
4) Получение 4-бензил-2,2,6,6-04-морфолина (соединение 18)
2,2-окси-бис-(1,1-d2-этан-2,1-диил)бис(4-п-толуолсульфонаты) (4,20 г, 10,04 ммоль), бензиламин (5,37 г, 50,17 ммоль) и толуол (40 мл) добавляли в колбу на ледяной бане, нагревали с обратным холодильником и проводили реакцию в течение 18 ч. К реакционной смеси добавляли воду и экстрагировали этилацетатом три раза, и органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением неочищенного продукта. Сырой продукт отделяли и очищали с помощью колоночной хроматографии с получением белого твердого вещества целевого соединения (1,3 г, чистота по ВЭЖХ: 91%, выход 72%); 1Н-ЯМР (400 МГц, CDCl3) δ 7,36-7,28 (5Н, м), 3,73 (4Н, с), 3,53 м.д. (2Н, с).
5) Получение 2,2,6,6-d4-морфолина (соединение 19)
4-бензил-2,2,6,6-d4-морфолин (1,30 г, 7,17 ммоль), гидроксид палладия (0,26 г, 20%) и метанол (13 мл) добавляли в колбу в атмосфере водорода и нагревали до 40°C. После 3 дней реакции, реакционную смесь охлаждали до комнатной температуры, фильтровали и концентрировали с получением желтого маслянистого желаемого продукта (0,4 г, выход 61%).
2. Получение N-(циано(d2-метил))-4-(2-(4-(2',2',6',6'-d4-морфолино)фениламино)пиримидин-4-ил)бензамида (соединение 24)
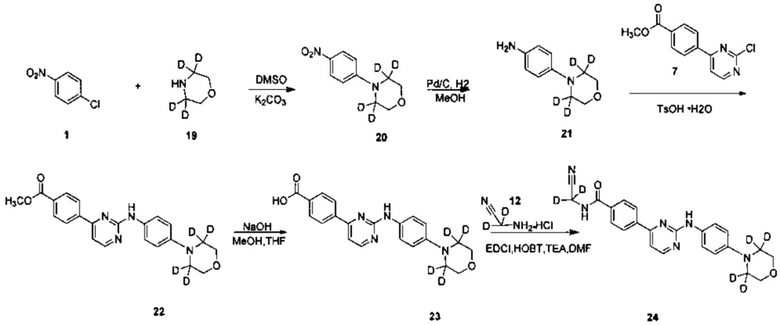
1) Получение 4-(4-нитрофенил)(2',2',6',6'-d4-морфолино) (соединение 20)
4-хлорнитробензол (0,402 г, 4,411 ммоль), 3,3,5,5-d4-морфолин (0,694 г, 4,411 ммоль) и карбонат калия (1,830 г, 13,233 ммоль) добавляли в колбу. Добавляли диметилсульфоксид (6 мл), после этого нагревали до 100°C и перемешивали в течение 18 ч. За ходом реакции следили с помощью ВЭЖХ. После окончания реакции, реакционную смесь охлаждали до комнатной температуры, и смесь гасили добавлением воды (30 мл). Желтое твердое вещество выпадало в осадок, его фильтровали с получением неочищенного продукта после перемешивания в течение 30 минут. Сырой продукт кристаллизовали в смешанном растворителе из этилацетата и петролейного эфира (1 / 2,5, объем/объем, 11 мл) с получением желтого твердого желаемого продукта (0,600 г, чистота по ВЭЖХ: 99,5%, выход 64%).
2) Получение 4-(2',2',6',6'-d4- морфолино) фениламина (соединение 21)
4-(4-нитрофенил) (2',2',6 ',6'-d4-морфолино) (0,600 г, 2,827 ммоль) и метанол (60 мл) добавляли к сосуду для реакции гидрогенизации. 10% палладий на угле (0,060 г) добавляли в атмосфере азота. В реакционной системе заменяли водород 3-4 раза, и перемешивали под давлением водорода для проведения реакции в течение 20 часов при 40°C. Анализ ВЭЖХ показал, что реакция завершена, и затем реакционный раствор охлаждали до комнатной температуры и фильтровали через целит. Остаток промывали метанолом. Объединенный органический слой концентрировали под вакуумом на роторном испарителе, получая твердое целевое соединение (0,420 г, чистота по ВЭЖХ: 95%, выход продукта: 82%).
3) Получение 4-(2-(4-(2',2',6',6'-d4-морфолино)фениламино)пиримидин-4-ил)метил бензоата (соединение 22)
4-(2-хлорпиримидин-4-ил)метил бензоат (0,521 г, 2,096 ммоль), 4-(2',2',6',6'-d4-морфолино)фениламин (0,420 г, 2,306 ммоль) и 1,4-диоксан (20 мл) добавляли в колбу. Моногидрат п-толуолсульфоновой кислоты (0,439 г, 2,306 ммоль) добавляли к суспензии при перемешивании. После нагревали с обратным холодильником в течение 20 ч, реакционную смесь концентрировали под вакуумом на роторном испарителе. Добавляли этилацетат (5 мл) и 5% водный раствор бикарбоната натрия (5 мл), желтое твердое вещество осаждали, после перемешивали в течение 30 мин, фильтровали, получая сырой продукт. Сырой продукт суспендировали в метаноле (5 мл), измельчали в течение 5 мин и фильтровали. Остаток промывали метанолом, сушили под вакуумом для получения твердого целевого продукта (0,400 г, чистота по ВЭЖХ: 92%, выход: 48%).
4) 4-(2-(4-(2',2',6',6'-d4-морфолино)фениламино)пиримидин-4-ил)бензойная кислота (соединение 23)
4-(2-(4-(2',2',6',6'-d4-морфолино)фениламино)пиримидин-4-ил)метил бензоат (0,400 г, 1,01 ммоль), гидроксид натрия (0,008 г, 2,02 ммоль), воду (3 мл), смешанный растворитель из метанола и тетрагидрофурана (12 мл, 3:1) добавляли в колбу, нагревали до 65°C, и реакцию проводили в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, растворитель удаляли, и pH доводили до 3 с помощью 10% разбавленной соляной кислоты, фильтровали и сушили с получением твердого серого вещества. Сырой продукт измельчали в порошок. Добавляли метанол (5 мл) и измельчали в течение 5 мин, а затем фильтровали и сушили с получением твердого желаемого продукта (0,340 г, чистота по ВЭЖХ: 92%, выход: 88%).
5) Получение N-(циано(d2-метил)-4-(2-(4-(2',2',6',6'-d4-морфолино)фениламино)пиримидин-4-ил)бензамида (соединение 24)
4-(2-(4-(2',2',6',6'-d4-морфолино)фениламино)пиримидин-4-ил)бензойную кислоту (0,170 г, 0,447 ммоль), 1-этил-(3-диметиламинопропил) карбодиимид гидрохлорид (0,103 г, 0,536 ммоль), 1-гидроксифенилпропилтриазол (0,724 г, 0,536 ммоль), триэтиламин (0,271 г, 2,682 ммоль) и N,N-диметилформамид (2 мл) добавляли в колбу в атмосфере азота. При перемешивании добавляли 2-амино-2,2-d2-ацетонитрилгидрохлорид (0,124 г, 1,341 ммоль), и реакцию проводили в течение 20 ч при комнатной температуре. Очищенную воду (2 мл) и насыщенный раствор бикарбоната (2 мл) добавляли к реакционной смеси, и в осадок выпадало желтое твердое вещество. После перемешивания в течение 30 минут, смесь фильтровали и промывали чистой водой. Неочищенный продукт получали после сушки, и отделяли и очищали с помощью препаративной хроматографии для получения желтого твердого желаемого продукта (0,112 г, степень очистки по ВЭЖХ: 98,9%, выход продукта: 60%). MS вычислено: 420; MS найдено: 421 (М+Н)+. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,56 (1Н, с), 9,35 (1Н, с), 8,57-8,56 (1Н, д), 8,29-8,27 (2Н, д), 8,05-8,03 (2Н, д), 7,70-7,68 (2Н, д), 7,44-7,42 (1Н, д), 6,99 (2Н, с), 3,76 м.д. (4Н, с).
Пример 5 Получение N-(цианометил)-4-(2-(4-(2',2',6',6'-d4-морфолино)фениламино)пиримидин-4-ил)бензамид (соединение 25)
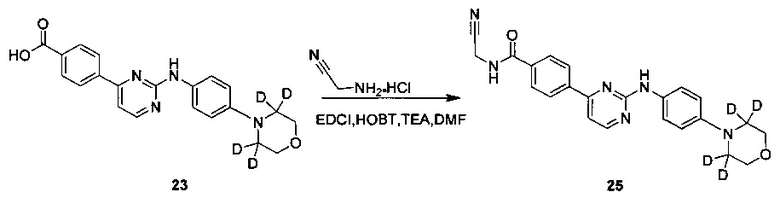
4-(2-(4-(2',2',6',6'-d4-морфолино)фениламино)пиримидин-4-ил)бензойную кислоту (0,170 г, 0,447 ммоль), 1-этил-(3-диметиламинопропил)карбодиимид гидрохлорид (0,103 г, 0,536 ммоль), 1-гидрокси-фенилпропил триазол (0,724 г, 0,536 ммоль), триэтиламин (0,271 г, 2,682 ммоль) и N,N-диметилформамид (2 мл) добавляли в колбу в атмосфере азота. При перемешивании добавляли 2-амино-ацетонитрил гидрохлорид (0,124 г, 1,341 ммоль), и реакцию проводили в течение 20 ч при комнатной температуре. Очищенную воду (2 мл) и насыщенный раствор бикарбоната (2 мл) добавляли к реакционной смеси, и в осадок выпадало желтое твердое вещество. После перемешивания в течение 30 минут, смесь фильтровали и промывали чистой водой. Неочищенный продукт получали после сушки, и отделяли разделением и очисткой с помощью препаративной хроматографии с получением желтого твердого желаемого продукта (0,105 г, чистота по ВЭЖХ: 98,1%, выход: 56%); MS вычислено: 418; MS найдено: 419 (М+Н)+; 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,65 (1Н, с), 9,38-9,36 (1Н, м), 8,58-8,56 (1Н, д), 8,29-8,27 (2Н, д), 8,05-8,03 (2Н, д), 7,75-7,73 (2Н, д), 7,46-7,45 (1Н, д), 7,09 (2Н, с), 4,37-4,36 (2Н, д), 3,79 м.д. (4Н, с).
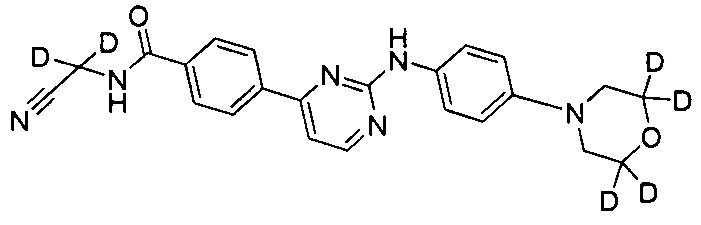
Пример 6 Получение N-(циано(d2-метил))-4-(2-(4-(3',3',5',5'-d4-морфолино-фениламино)пиримидин-4-ил)бензамида (соединение 34)
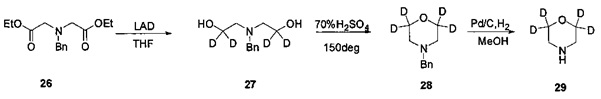
1. Получение 3,3,5,5-d4-морфолина (соединение 29)
1) Получение 2,2'-(бензилимино)бис(1,1-d2-этанол) (соединение 27)
Бензил иминодиуксусной кислоты диэтиловый эфир (3,5 г, 12,53 ммоль) и безводный тетрагидрофуран (35 мл) добавляли в колбу в атмосфере азота, порциями добавляли дейтерированный лития-алюминия гидрид (1,49 г, 35,5 ммоль, приобретен у J&K), и внутренняя температура поддерживали ниже 10°C. Все нагревали до комнатной температуры и перемешивали для проведения реакции в течение ночи. За ходом реакции следили с помощью ВЭЖХ. После того как реакция окончилась, к реакционной смеси добавляли 7 мл воды и 3,5 мл 15% водного раствора гидроксида натрия на ледяной бане и перемешивали в течение 30 мин, затем фильтровали и промывали тетрагидрофураном (20 мл). Фильтрат концентрировали с получением желтого маслянистого желаемого продукта (2,4 г, чистота по ВЭЖХ: 100%, выход: 96%); 1Н-ЯМР (400 МГц, CDCl3) δ 7,37-7,27 (5Н, м), 3,72 (2Н, с), 2,75 (2Н, с), 2,72 м.д. (4Н, с).
2) Получение N-бензил-3,3,5,5-d4-морфолина (соединение 28)
2,2'-(бензил)бис(1,1-d2-этанол) (1,49 г, 35,5 ммоль) и концентрированную серную кислоту (70%, 2 мл) добавляют в колбу. Все нагревали до 150°C, для проведения реакции в течение 16 ч. За ходом реакции следили с помощью ВЭЖХ. После окончания реакции реакционную смесь охлаждали до комнатной температуры, и выливали в 25 мл воды со льдом, а pH нейтрализовали до приблизительно 9 путем добавления твердого карбоната калия, добавляли дихлорметан (2 мл) и перемешивали в течение 30 мин. После фильтрования, фильтрат три раза экстрагировали дихлорметаном, органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением желтого маслянистого желаемого продукта (1,4 г, чистота по ВЭЖХ: 98,9%, выход 64%). 1Н-ЯМР (400 МГц, CDCl3) δ 7,36-7,28 (5Н, м), 3,53 (2Н, с), 2,46 м.д. (4Н, с).
3) Получение 3,3,5,5-d4-морфолина (соединение 29)
N-бензил-3,3,5,5-d4-морфолин (1,40 г, 7,72 ммоль), гидроксид палладия (0,28 г, 20%) и метанол (14 мл) добавляли в колбу в атмосфере водорода, нагревали до 40°C, и реакцию проводили в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали с получением желтого маслянистого желаемого продукта (0,4 г, чистота по ВЭЖХ: 90%, выход 57%).
2. Получение N-(циано(d2-метил))-4-(2-(4-(3',3',5',5'-d4-морфолино)фениламино)пиримидин-4-ил)бензамид (соединение 34)
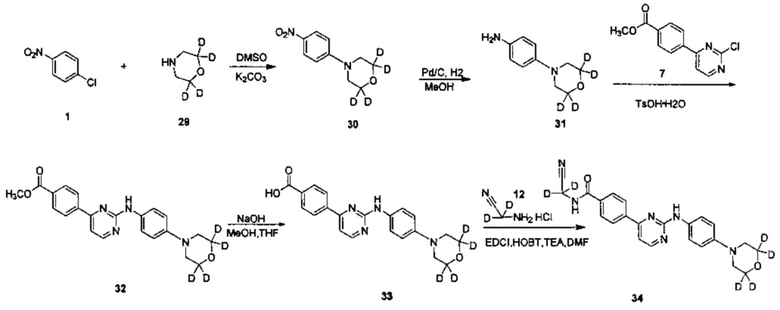
1) Получение 4-(4-нитрофенил)(3',3',5',5'-d4-морфолино) (соединение 30)
4-хлорнитробензол (1,210 г, 7,68 ммоль), 3,3,5,5-морфолин-d4 (0,700 г, 7,68 ммоль), карбонат калия (3,18 г, 23,04 ммоль) и диметилсульфоксид (12 мл) добавляли в колбу и нагревали до 100°C и перемешивали в течение 18 ч после этого. За ходом реакции следили с помощью ВЭЖХ. После окончания реакции реакционную смесь охлаждали до комнатной температуры, и смесь гасили добавлением воды (50 мл). Желтое твердое вещество осаждали, фильтровали с получением неочищенного продукта после перемешивания в течение 30 минут. Сырой продукт кристаллизовали в смешанном растворителе из этилацетата и петролейного эфира (1/2,5, объем/объем, 15 мл), для получения твердого целевого соединения (0,500 г, чистота по ВЭЖХ: 96%, выход: 30%).
2) Получение 4-(3',3',5',5'-d4-морфолино)фениламина (соединение 31)
4-(4-нитрофенил)(3',3',5',5'-d4-морфолино) (0,500 г, 2,356 ммоль) и метанол (50 мл) добавляли к сосуду для реакции гидрогенизации. 10% палладий на угле (0,060 г) добавляли под защитой азота. Реакционную систему заменяли водородом 3-4 раза, и перемешивали под давлением водорода для проведения реакции в течение 20 часов при 40°C. По данным ВЭЖХ определяли, что реакция завершена, а затем реакционный раствор охлаждали до комнатной температуры и фильтровали через целит. Остаток промывали метанолом. Объединенный фильтрат концентрировали под вакуумом на роторном испарителе, получая неочищенный продукт, и неочищенный продукт выделяли и очищали с помощью колоночной хроматографии с получением твердого желаемого продукта (0,170 г, чистота ВЭЖХ: 95%, выход продукта: 40%).
3) Получение 4-(2-(4-(3',3',5',5'-d4-морфолино)фениламина)пиримидин-4-ил)метил бензоата (соединение 32)
4-(2-хлорпиримидин-4-ил)метил бензоат (0,255 г, 1,026 ммоль), 4-(3',3',5',5'-d4-морфолино)фениламин (0,170 г, 0,933 ммоль) и 1,4-диоксан (14 мл) добавляли в колбу. Моногидрат п-толуолсульфоновой кислоты (0,195 г, 1,026 ммоль) добавляли в суспензию при перемешивании. Реакционную смесь нагревали с обратным холодильником в течение 20 ч, и наблюдали с помощью ВЭЖХ. После окончания реакции реакционную смесь концентрировали под вакуумом на роторном испарителе. Этилацетат (5 мл) и 5% водный раствор бикарбоната натрия (5 мл) добавляли, желтое твердое вещество выпадало в осадок после перемешивания в течение 30 мин, фильтровали, получая сырой продукт. Сырой продукт суспендировали в метаноле (5 мл), измельчали в течение 5 мин и фильтровали. Остаток промывали метанолом, сушили под вакуумом для получения твердого целевого продукта (0,200 г, чистоту по ВЭЖХ: 92%, выход: 55%).
4) Получение 4-(2-(4-(3',3',5',5'-d4-морфолино)фениламино)пиримидин-4-ил)бензоата (соединение 33)
4-(2-(4-(3',3',5',5'-d4-морфолино)фениламино)пиримидин-4-ил)метил бензоат (0,200 г, 0,507 ммоль), гидроксид натрия (0,041 г, 1,014 ммоль), воду (1 мл), смешанный растворитель из метанола и тетрагидрофурана (4 мл, 3:1) добавляли в колбу, нагревали до 65°C, и реакцию проводили в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, растворитель удаляли, и pH доводили до 3 10% разбавленной соляной кислотой, фильтровали и сушили с получением серого твердого вещества. Сырой продукт измельчали в порошок. Добавляли метанол (2 мл) и измельчали в течение 5 мин, а затем фильтровали и сушили с получением твердого желаемого продукта (0,200 г, выход: 86%).
5) Получение N-(циано(d2-метил)-4-(2-(4-(3',3',5',5'-d4-морфолино)фениламино)пиримидин-4-ил)бензамида (соединение 34)
4-(2-(4-(3',3',5',5'-d4-морфолино)фениламино)пиримидин-4-ил)бензойную кислоту (0,100 г, 0,263 ммоль), 1-этил-(3-диметиламинопропил)карбодиимид дигидрохлорид (0,061 г, 0,316 ммоль), 1-гидрокси-фенилпропил триазол (0,043 г, 0,316 ммоль), триэтиламин (0,159 г, 1,578 ммоль) и N,N-диметилформамид (2 мл) добавляли в колбу в атмосфере азота. 2-амино-2,2-d2-ацетонитрил гидрохлорид (0,073 г, 0,789 ммоль) добавляли при перемешивании, и реакцию проводили в течение 20 ч при комнатной температуре. Очищенную воду (2 мл) и насыщенный раствор бикарбоната (2 мл) добавляли к реакционной смеси, и в осадок выпадало желтое твердое вещество. После перемешивания в течение 30 минут, смесь фильтровали и промывали чистой водой. Неочищенный продукт получали после сушки, и отделяли и очищали препаративной хроматографией с получением желтого твердого желаемого продукта (0,050 г, чистота ВЭЖХ: 98,9%, выход продукта: 45%). MS вычислено: 420; MS найдено: 421 (М+Н)+. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,57 (1Н, с), 9,35 (1Н, с), 8,56-8,55 (1Н, д), 8,29-8,27 (2Н, д), 8,04-8,02 (2Н, д), 7,70-7,68 (2Н, д), 7,44-7,42 (1Н, д), 6,99-6,98 (2Н, д), 3,08 м.д. (4Н, с).
Пример 7. Получение N-(цианометил)-4-(2-(4-(3',3',5',5'-d4-морфолино)фениламино)пиримидин-4-ил)бензамида (соединение 35);
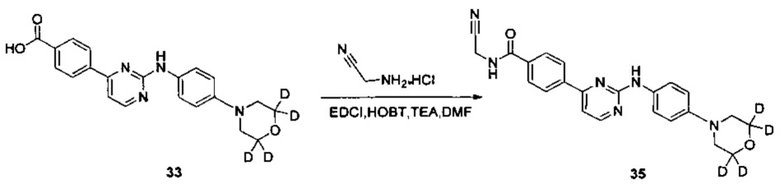
4-(2-(4-(3',3',5',5'-d4-морфолино)фениламино)пиримидин-4-ил)бензойную кислоту (0,100 г, 0,263 ммоль), 1-этил-(3-диметиламинопропил)карбодиимид гидрохлорид (0,061 г, 0,316 ммоль), 1-гидрокси-фенилпропил триазол (0,043 г, 0,316 ммоль), триэтиламин (0,159 г, 1,578 ммоль) и N,N-диметилформамид (2 мл) добавляли в колбу в атмосфере азота. При перемешивании добавляли 2-амино-ацетонитрил гидрохлорид (0,073 г, 0,789 ммоль), и реакцию проводили в течение 20 ч при комнатной температуре. К реакционной смеси добавляли очищенную воду (2 мл) и насыщенный раствор бикарбоната (2 мл), и желтое твердое вещество выпадало в осадок. После перемешивания в течение 30 минут, смесь фильтровали и промывали чистой водой. Неочищенный продукт получали после сушки, и отделяли и очищали препаративной хроматографией с получением желтого твердого желаемого продукта (0,060 г, чистота по ВЭЖХ: 98,4%, выход: 55%); MS вычислено: 418; MS найдено: 419 (М+Н)+. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,62 (1Н, с), 9,38-9,35 (1Н, м), 8,57-8,56 (1Н, д), 8,29-8,27 (2Н, д), 8,04-8,02 (2Н, д), 7,73-7,70 (2Н, д), 7,45-7,44 (1Н, д), 7,05 (2Н, с), 4,37-4,36 (2Н, д), 3,14 м.д. (4Н, с).
Пример 8. Получение N-(циано(d2-метил))-4-(2-(4-морфолинофениламино)пиримидин-4-ил)бензамида (соединение 41)
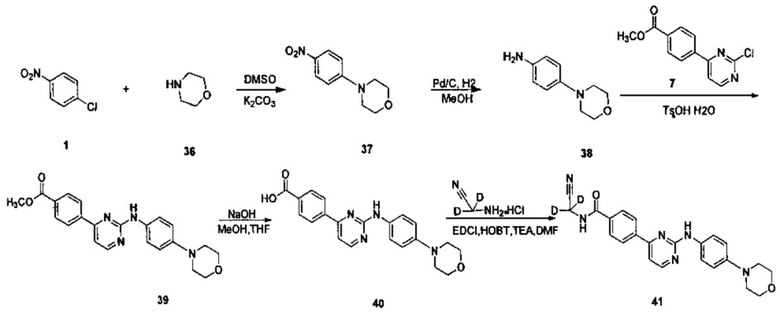
1. Получение 4-(4-нитрофенил)(морфолин) (соединение 37)
4-хлорнитробензола (1,00 г, 6,35 ммоль), морфолина (0,608 г, 7,68 ммоль), карбоната калия (1,76 г, 12,7 ммоль), диметилсульфоксида (11 мл) добавляли в колбу. После этого смесь нагревали до 100°C и перемешивали в течение 18 ч. За ходом реакции следили с помощью ВЭЖХ. После окончания реакции реакционную смесь охлаждали до комнатной температуры, и смесь гасили добавлением воды (50 мл). Желтое твердое вещество осаждали, фильтровали с получением неочищенного продукта после перемешивания в течение 30 минут. Сырой продукт кристаллизовали в смешанном растворителе из этилацетата и петролейного эфира (1/2,5, объем/объем, 16 мл) с получением желтого твердого желаемого продукта (0.800 g, чистота по ВЭЖХ: 98,9%, выход 60%). 1Н ЯМР (400 МГц, CDCl3) δ 8,18-8,15 (2Н, д), 6,87-6,84 (2Н, д), 3,90-3,88 (4Н, т), 3,41-3,38 м.д. (4Н, т).
2. Получение 4-(морфолино)фениламина (соединение 38)
4-(4-нитрофенил)морфолин (0,600 г, 2,882 ммоль) и метанол (60 мл) добавляли к сосуду для реакции гидрогенизации. 10% палладий на угле (0,060 г) добавляли под защитой азота. Реакционную систему замещали водородом 3-4 раза, и перемешивали под давлением водорода для проведения реакции в течение 18 часов при 40°C. По данным ВЭЖХ определяли, что реакция завершена, и затем реакционный раствор охлаждали до комнатной температуры и фильтровали через целит. Остаток промывали метанолом. Объединенный фильтрат концентрировали под вакуумом на роторном испарителе, получая твердое целевое соединение (0,500 г, чистота по ВЭЖХ: 97,1%, выход продукта: 97%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 6.83-6,80 (2Н, д), 6,71-6,68 (2Н, д), 3,88-3,86 (4Н, т), 3,46 (2Н, с), 3,06-3,03 м.д. (4Н, т).
3. Получение 4-(2-((4-морфолино)фениламино)пиримидин-4-ил)метил бензоата (соединение 39)
4-(2-хлорпиримидин-4-ил)метилбензоат (0,651 г, 2,617 ммоль), 4-(морфолино)фениламин (0,513 г, 2,878 ммоль) и 1,4-диоксан (16 мл) добавляли в колбу. Моногидрат п-толуолсульфоновой кислоты (0,547 г, 2,878 ммоль) добавляли к суспензии при перемешивании. После нагревания с обратным холодильником в течение 20 ч реакцию определяли с помощью ВЭЖХ. После окончания реакции реакционную смесь охлаждали до 30°C и концентрировали под вакуумом на роторном испарителе. Добавляли этилацетат (5 мл) и 5% водный раствор бикарбоната натрия (5 мл), желтое твердое вещество выпадало в осадок после перемешивания в течение 30 мин, фильтровали с получением сырого продукта. Сырой продукт суспендировали в метаноле (8 мл), измельчали в течение 5 мин и фильтровали. Остаток промывали метанолом, сушили под вакуумом с получением твердого целевого продукта реакции (0.860 г, чистота по данным ВЭЖХ: 97.6%, выход: 84%); 1Н ЯМР (400 МГц, CDCl3) δ 8,50-8,49 (1Н, д), 8,17-8,13 (4Н, м), 7,60-7,58 (2Н, д), 7,17-7,16 (2Н, д), 6,99-6,97 (2Н, д), 3,98 (3Н, с), 3,92-3,89 (4Н, т), 3,18-3,15 м.д. (4Н, т).
4. 4-(2-(4-(морфолино)фениламино)пиримидин-4-ил)бензойная кислота (соединение 40)
4-(2-(4-(морфолино)фениламино)пиримидин-4-ил)метилбензоат (0,860 г, 2,203 ммоль), гидроксид натрия (0,176 г, 4,405 ммоль), воду (3,5 мл), смешанный растворитель из метанола и тетрагидрофурана (12 мл, 3:1) добавляли в колбу, нагревали до 65°C, и реакцию проводили в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, растворитель удаляли, и pH доводили до 3 с помощью 10% разбавленной соляной кислоты, фильтровали и сушили с получением серого твердого вещества. Сырой продукт измельчали в порошок. Добавляли метанол (10 мл) и измельчали в течение 5 мин, а затем фильтровали и сушили с получением твердого желаемого продукта (0,730 г, чистота по данным ВЭЖХ: 97,7%, выход продукта: 88%).
5. Получение N-(циано(d2-метил))-4-(2-(4-(морфолино)фениламино)пиримидин-4-ил)бензамида (соединение 41)
4-(2-(4-(морфолино)фениламино)пиримидин-4-ил)бензойную кислоту (0,230 г, 0,611 ммоль), 1-этил-(3-диметиламинопропил)карбодиимид гидрохлорид (0,141 г, 0,733 ммоль), 1-гидрокси-фенилпропилтриазол (0,099 г, 0,733 ммоль), триэтиламин (0,371 г, 3,666 ммоль) и N,N-диметилформамид (2,5 мл) добавляли в колбу под защитой азота. При перемешивании добавляли 2-амино-2, 2-d2-ацетонитрил гидрохлорид (0,173 г, 1,833 ммоль), и подвергали взаимодействию в течение 20 ч при комнатной температуре. К реакционной смеси добавляли очищенную воду (3 мл) и насыщенный раствор бикарбоната (3 мл), и желтое твердое вещество выпадало в осадок. После перемешивания в течение 30 минут, смесь фильтровали и промывали чистой водой. Неочищенный продукт, полученный после сушки, отделяли и очищали препаративной хроматографией с получением желтого твердого желаемого продукта (0.183 г, чистота по данным ВЭЖХ: 99%, выход: 72%); МС вычислено: 416; МС найдено: 417 (М+Н)+; 1Н NMR (400 МГц, ДМСО-d6) δ 9,65(1Н, с), 9,35 (1Н, с), 8,57-8,56 (1Н, д), 8,29-8,27 (2Н, д), 8,05-8,03 (2Н, д), 7,75-7,73 (2Н, д), 7,46-7,45 (1Н, д), 7,09-7,07 (2Н, д), 3,79-4,36 (4Н, т), 3,17 м.д. (4Н, с).
Пример 9. Получение N-(цианометил)-4-(2-(4-морфолинофениламино)пиримидин-4-ил)бензамида (соединение 42, контрольное соединение)
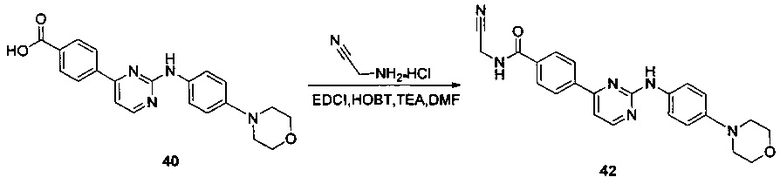
4-(2-(4-(морфолино)фениламино)пиримидин-4-ил)бензойную кислоту (0,500 г, 1,328 ммоль), 1-этил-(3-диметиламинопропил)карбодиимид гидрохлорид (0,305 г, 1,594 ммоль), 1-гидрокси-фенилпропил триазол (0,215 г, 1,594 ммоль), триэтиламин (0,805 г, 7,968 ммоль) и N,N-диметилформамид (5 мл) добавляли в колбу в атмосфере азота. Добавляли 2-амино-ацетонитрил гидрохлорид (0,368 г, 3,985 ммоль) при перемешивании, и подвергали взаимодействию в течение 20 ч при комнатной температуре. К реакционной смеси добавляли очищенную воду (5 мл) и насыщенный раствор бикарбоната натрия (5 мл), и желтое твердое вещество выпадало в осадок. После перемешивания в течение 30 минут, смесь фильтровали и промывали чистой водой. Неочищенный продукт получали после сушки, его отделяли и очищали препаративной хроматографией с получением желтого твердого желаемого продукта (0.130 г, чистота по данным ВЭЖХ: 98.3%, выход: 24%); MS вычислено: 414; MS найдено: 415 (М+Н)+; 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,63 (1Н, с), 9,38-9,35 (1Н, м), 8,57-8,56 (1Н, д), 8,29-8,27 (2Н, д), 8,05-8,03 (2Н, д), 7,74-7,71 (2Н, д), 7,46-7,44 (1Н, д), 7,07-7,05 (2Н, д), 4,37-4,36 (2Н, д), 3,80-3,78 (4Н, т), 3,15 м.д. (4Н, с).
Пример 10: Фармакокинетическая оценка у крыс
12 самцов крыс Sprague-Dawley, возраст 7-8 недель, массой тела примерно 200 г, были разделены на три группы, по четыре в каждой группе. Доза 3 мг/кг (а) контрольного соединения, полученного в примере 9: N-(цианометил)-4-(2-(4-морфолинофениламино)пиримидин-4-ил)бензамида и (б) соединений по настоящему изобретению: соединения, полученные в примерах 1-8, каждый раз вводили с помощью желудочного зонда через рот, и оценивали их фармакокинетическое различие.
Крыс кормили стандартным кормом и давали воду, и переставали давать еду за 16 часов перед испытанием. Препарат растворяли в ПЭГ400 и диметилсульфоксиде. Проводили орбитальный сбор крови (из глазной орбиты мыши/крысы) и времени отбора крови составляло 0,25 часа, 0,5 часа, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 12 часов, 24 часа, 36 часов и 48 часов после введения.
После вдыхания эфира крыс быстро анестезировали; 300 мкл крови собирали с орбиты в пробирку. В пробирке находились 30 мкл 1% солевых растворов гепарина. Перед использованием пробирки сушили в течение ночи при 60°C. После отбора образца крови в подходящий момент времени, крыс анестезировали эфиром и умерщвляли.
После забора крови пробирку немедленно плавно переворачивали по меньшей мере 5 раз, чтобы обеспечить адекватное перемешивание, и помещали на лед. Образцы крови центрифугировали при 5000 об/мин при 4°C в течение 5 минут для отделения плазмы и красных кровяных клеток. 100 мкл плазмы отбирали пипеткой в чистую пластиковую центрифужную пробирку, и указывали название соединений и временную точку. Плазму хранили в -80°C до использования для анализа. Концентрацию соединения по настоящему изобретению в плазме определяли с LC-MS/MS. Фармакокинетические параметры рассчитывали на основе концентрации лекарственного средства в крови каждого животного в различные моменты времени. По результатам можно видеть, что по сравнению с контрольным соединением примера 9, соединения по настоящему изобретению (соединения примеров 1-8) имели лучшую фармакокинетику у животных (см Таблицы 1-3), и, таким образом, имели лучшие фармакодинамический эффект и эффект при лечении.
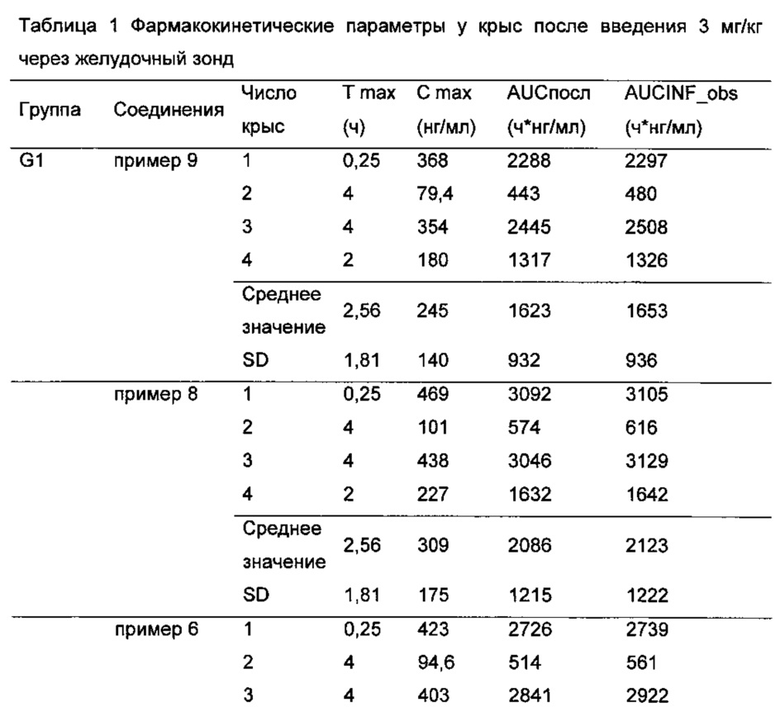

Результат показал, что Cmax для соединения примера 8 (309±175 нг/мл) значительно выше, чем для контрольного соединения примера 9 (245±140 нг/мл); AUCпосл соединения примера 8 (2086±1215 нг⋅ч/мл) значительно выше, чем для для контрольного соединения примера 9 (1623±932 нг⋅ч/мл).
Cmax для соединения примера 6 (282±158 нг/мл) значительно выше, чем для контрольного соединения примера 9 (245±140 нг/мл); AUCпосл для соединения примера 6 (1900±1101 нг⋅ч/мл) значительно выше, чем для контрольного соединения примера 9 (1623±932 нг⋅ч/мл).
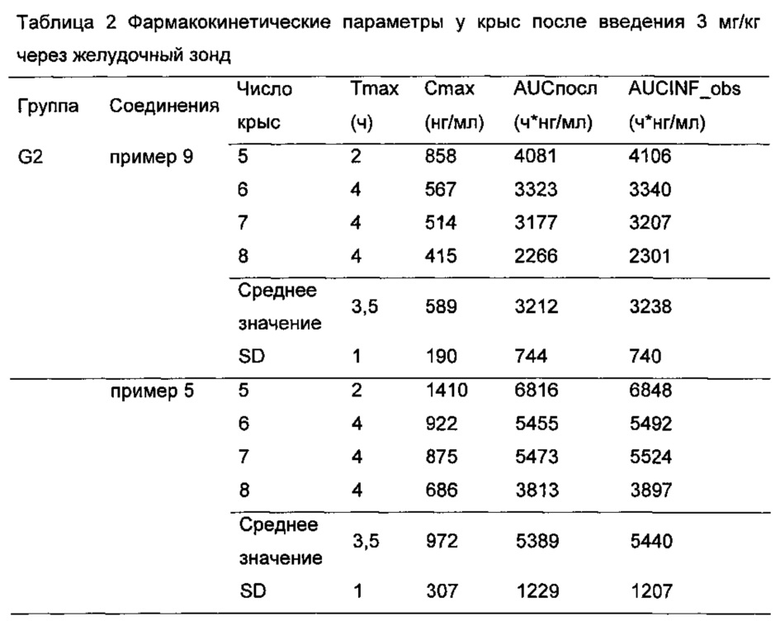
Результат показал, что Cmax для соединения примера 5 (972±307 нг/мл) значительно выше, чем для контрольного соединения примера 9 (589±190 нг/мл); AUCпосл для соединения примера 5 (5389±+1229 нг⋅ч/мл) значительно выше, чем для контрольного соединения примера 9 (3212±744 нг⋅ч/мл).
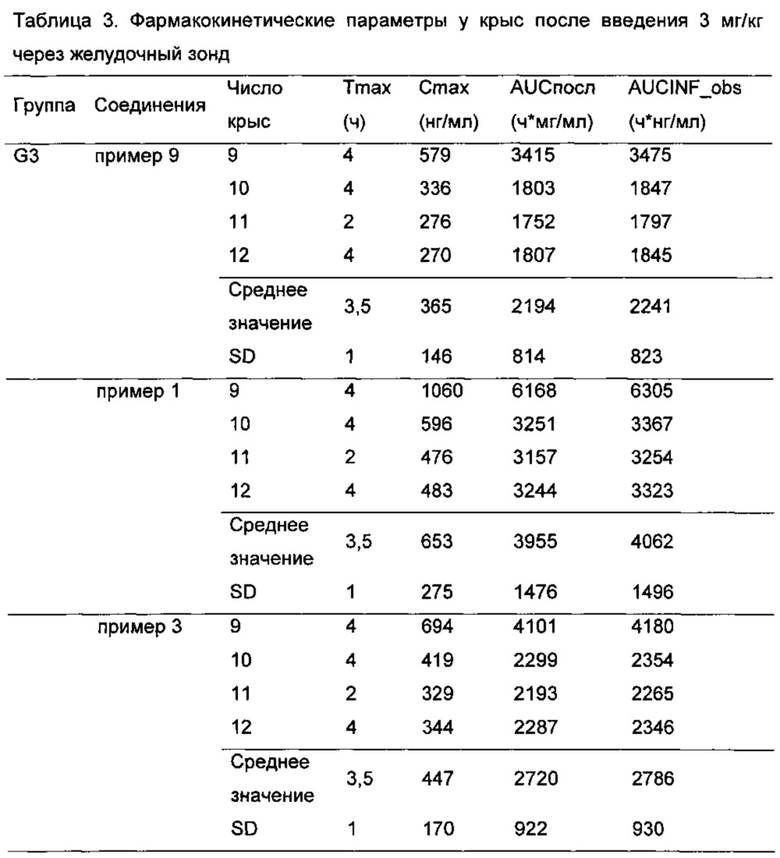
Результат показал, что Cmax для соединения примера 1 (653±275 нг/мл) значительно выше, чем для контрольного соединения примера 9 (365±146 нг/мл); AUCпосл для соединения примера 8 (3955±1476 нг⋅ч/мл) значительно выше, чем для контрольного соединения примера 9 (2 194±814 нг⋅ч/мл).
Cmax для соединения примера 3 (447±170 нг/мл) значительно выше, чем для контрольного соединения примера 9 (365±146 нг/мл); AUCпосл для соединения примера 3 (2720±922 нг⋅ч/мл) значительно выше, чем для контрольного соединения примера 9 (2194±814 нг⋅ч/мл).
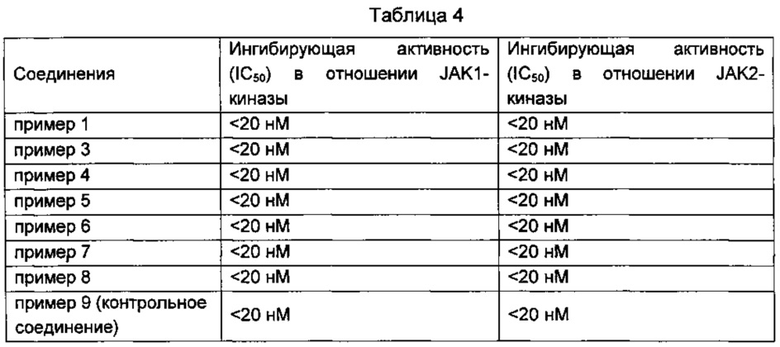
Пример 11: Фармакодинамическая оценка in vitro соединений по изобретению для JAK-киназ
Определение ингибирующего эффекта IC50 на киназы JAK1 и JAK2.
Тест на ингибирующую JAK-киназу активность проводили в 96-луночных планшетах. Тестируемые соединения (любое одно из соединений, полученных в Примере 1-7) растворяли в ДМСО, и подготавливали в тестируемой жидкости в различных концентрациях. Их инкубировали с JAK1 или JAK2-тирозинкиназой в буфере для анализа фосфотирозина (5 мМ HEPES, pH 7,5, 50 мМ MgCl 2, 50 мМ NaCl, 100 мМ ванадата натрия и 0,1% Твин-20) в течение 20 минут. Субстрат добавляли (например: биотин- EGPWLEEEEEAYGWMDF-NH2 или биотин-EQEDEPEGDYFEWLEPE), и инкубировали в течение 60 мин. В каждую реакционную лунку добавляли гранулы, покрытые стрептавидином, при облучении светом и инкубировали в течение 150 минут. Планшет считывали после промывания, и значения IC50 рассчитывали на основании этих данных.
Результаты показаны в Таблице 4. Можно видеть, что соединения по настоящему изобретению имеют превосходную ингибирующую активность в отношении обоих JAK1 и JAK2-киназ.
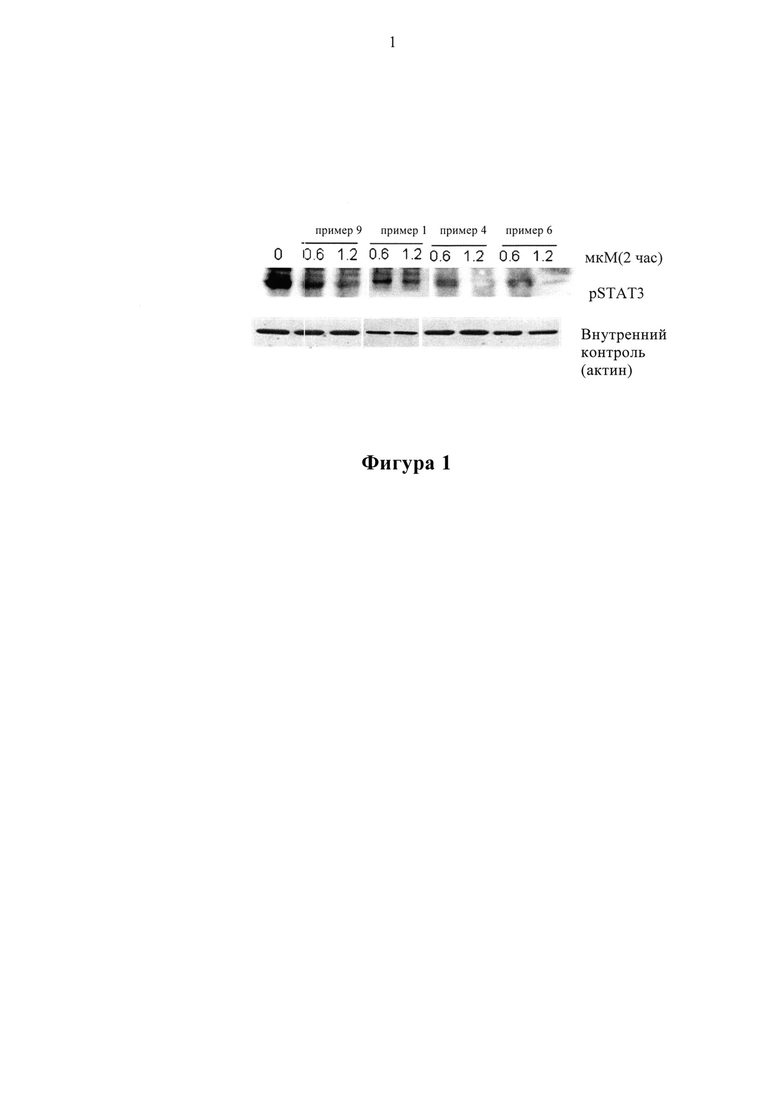
Пример 12: Фармакодинамическая оценка in vitro соединений по настоящему изобретению по ингибированию фосфорилирующей STAT-активности
Реагенты:
культуральная среда DMEM, приобретены у Mediatech Company
антитела анти-фосфоSTAT3, приобретены у Cell Signaling Technology Company
Анти-актиновые антитела, приобретены у Sigma Company
Методика эксперимента:
Клетки НСТ116 (от АТСС) культивировали в культуральной среде DMEM, содержащей 10% фетальной бычьей сыворотки, 2 мМ глутамина, 100 ед/мл пенициллина и 100 мг/мл стрептомицина. Соединения растворяли в ДМСО. Соединения в различных концентрациях (0,6 мкм, 1,2 мкм) добавляли в культуральную среду, и ДМСО использовали в качестве отрицательного контроля, а соединение примера 9 (0,6 мкм, 1,2 мкм) использовали в качестве положительного контроля. Клетки обрабатывали в течение 2 часов, а затем белки собирали для анализа методом Вестерн-блоттинга.
Результаты эксперимента
На Фигуре 1 показано, что соединения по настоящему изобретению имеют активностью, ингибирующую JAK-STAT сигнальный путь, и активность, ингибирующая STAT3-фосфорилирование, аналогична или лучше, чем активность контрольного соединения примера 9.
Пример 13 фармацевтическая композиция
Вышеуказанные материалы смешивали обычными способами и вводили в обычные желатиновые капсулы для получения 1000 капсул.
Все литературные данные, упомянутые в настоящей заявке, включены в настоящее описание в качестве ссылки, как если бы каждая из них была по отдельности включена в качестве ссылки. Кроме того, следует понимать, что после прочтения вышеизложенного, специалисты в данной области техники могут сделать различные изменения и модификации настоящего изобретения. Эти эквиваленты также попадают в рамки, определенные прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТИАЗОЛОПИРИМИДИНЫ | 2012 |
|
RU2610840C2 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ИНГИБИРОВАНИЯ ТИРОЗИНКИНАЗНОЙ АКТИВНОСТИ | 2011 |
|
RU2598852C2 |
| ФЕНИЛАМИНОПИРИМИДИН ИЛИ ПОЛИМОРФНАЯ ФОРМА СОЛИ ФЕНИЛАМИНОПИРИМИДИНА | 2016 |
|
RU2712226C2 |
| ПРОИЗВОДНЫЕ 2-ФЕНИЛАМИНОПИРИМИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ БОГАТОЙ ЛЕЙЦИНОМ ПОВТОРНОЙ КИНАЗЫ 2 (LRRK2) ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2012 |
|
RU2661197C2 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ИНГИБИРОВАНИЯ ПУТИ JAK | 2011 |
|
RU2672100C2 |
| ДЕЙТЕРИРОВАННЫЕ ДИАМИНОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ТАКИЕ СОЕДИНЕНИЯ | 2014 |
|
RU2632907C2 |
| ДИАМИНОТРИАЗОЛЫ, ПРИГОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2003 |
|
RU2350606C2 |
| АЗАИНДОЛИЛПИРИДОНЫ И ДИАЗАИНДОЛИЛПИРИДОНЫ | 2018 |
|
RU2788659C2 |
| ПИРИДИНАМИНПИРИДОНЫ И ПИРИМИДИНАМИНПИРИДОНЫ | 2018 |
|
RU2804638C2 |
Изобретение относится к дейтерированному фениламинопиримидину формулы (I) или его фармацевтически приемлемым солям:
 .
.
В формуле (I) R1, R2, R13, R14, R15, R16, R17, R18, R19, R20, R21 и R22 представляют собой каждый независимо водород или дейтерий; R3, R4, R5, R6, R7, R8, R9, R10 и R11 представляют собой каждый независимо водород; R12 представляет собой водород; при условии, что по меньшей мере один из R1, R2, R13, R14, R15, R16, R17, R18, R19, R20, R21 или R22 являются дейтерированным или дейтерием. Изобретение также относится к фармацевтической композиции для ингибирования JAK-киназы и к применению соединений для получения фармацевтической композиции. Технический результат: получены новые соединения формулы (I), обладающие свойствами ингибитора JAK-киназы. 3 н. и 6 з.п. ф-лы, 1 ил., 4 табл., 13 пр.
1. Дейтерированный фениламинопиримидин формулы (I) или его фармацевтически приемлемая соль:
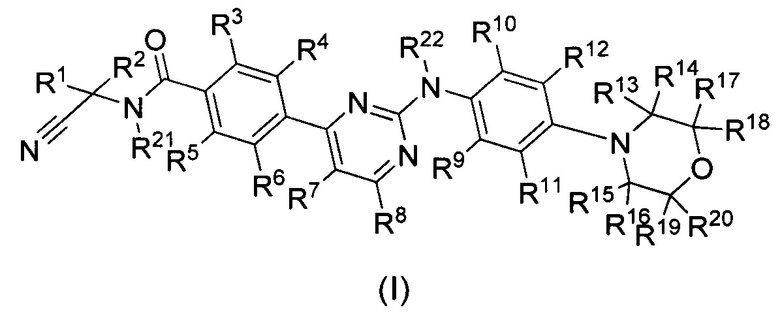
где:
R1, R2, R13, R14, R15, R16, R17, R18, R19, R20, R21 и R22 представляют собой каждый независимо водород или дейтерий;
R3, R4, R5, R6, R7, R8, R9, R10 и R11 представляют собой каждый независимо водород;
R12 представляет собой водород;
при условии, что по меньшей мере один из R1, R2, R13, R14, R15, R16, R17, R18, R19, R20, R21 или R22 являются дейтерированным или дейтерием.
2. Соединение по п. 1, отличающееся тем, что R1 и R2 представляют собой дейтерий.
3. Соединение по п. 1, отличающееся тем, что R13, R14, R15 и R16 представляют собой дейтерий.
4. Соединение по п. 1, отличающееся тем, что R17, R18, R19 и R20 представляют собой дейтерий.
5. Соединение по п. 1, отличающееся тем, что R13, R14, R15, R16, R17, R18, R19 и R20 представляют собой дейтерий.
6. Соединение по п. 1, отличающееся тем, что соединение представляет собой одно из следующих соединений, или их фармацевтически приемлемых солей:
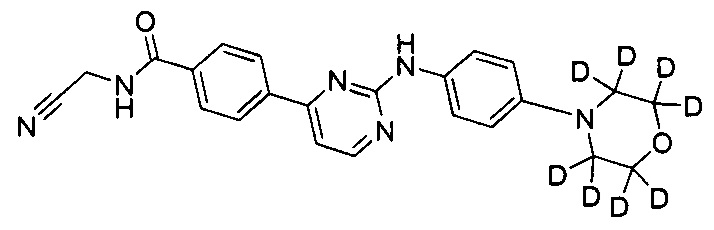
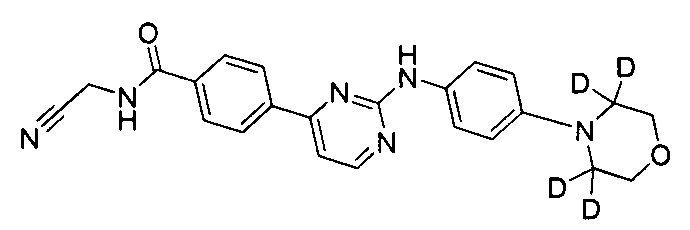
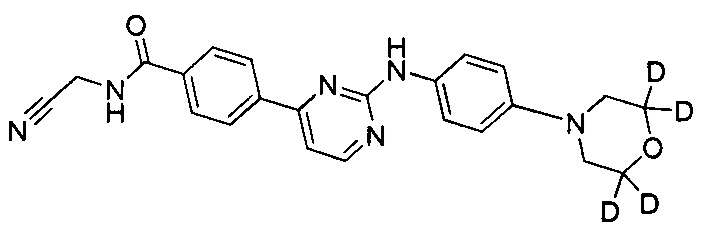
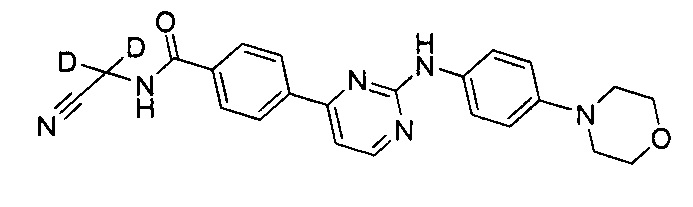 .
.
7. Фармацевтическая композиция для ингибирования JAK-киназы, содержащая фармацевтически приемлемый носитель и соединение по п. 1 или его фармацевтически приемлемую соль в безопасном и эффективном количестве.
8. Применение соединения по п. 1, или его фармацевтически приемлемой соли для получения фармацевтической композиции для ингибирования JAK-киназы.
9. Применение по п. 8, отличающееся тем, что указанную фармацевтическую композицию используют для лечения и профилактики следующих заболеваний: рак, миелопролиферативные заболевания, воспаление, иммунные заболевания,
где рак выбран из группы, состоящей из немелкоклеточного рака легкого, рака матки, рака прямой кишки, рака толстой кишки, рака мозга, рака головы, рака шеи, рака мочевого пузыря, рака простаты, рака молочной железы, рака почки, лейкоза, рака печени, рака желудка, рака щитовидной железы, рака носоглотки и рака поджелудочной железы;
миелопролиферативные заболевания выбраны из группы, состоящей из спонтанной тромбоцитемии (ЕТ), идиопатического миелофиброза (МФМ, IMF), хронического миелолейкоза (ХМЛ, CML), первичного миелофиброза, хронического нейтрофильного лейкоза (ХНЛ, CNL) и истинной полицитемии (ИП, PV); и
воспаление или иммунные заболевания выбраны из группы, состоящей из ревматоидного артрита, остеоартрита, ревматоидного спондилита, подагры, астмы, бронхита, ринита, хронического обструктивного заболевания легких и кистозного фиброза.
| RU 2009137363 A, 20.04.2011 | |||
| WO 2009029998 A1, 12.03.2009 | |||
| WO 2010040826 A1, 15.05.2010 | |||
| US 20100291026 A1, 18.11.2010 | |||
| WO 2012071612 A1, 07.06.2012. |

