Настоящее изобретение направлено на способ получения амида карбоновой кислоты посредством взаимодействия амина формулы (I)
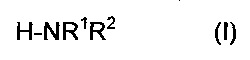
причем целые числа могут быть равными или различными,
R1 выбирают из алкила, содержащего от 1 до 4 атома углерода,
R2 выбирают из водорода и алкила, содержащего от 1 до 4 атома углерода,
причем R1 и R2 комбинируют таким образом, чтобы амин по формуле (I) обладал более низкой точкой кипения, чем вода,
с карбоновой кислотой по меньшей мере с 3 атомами кислорода на молекулу, причем указанная карбоновая кислота необязательно несет по меньшей мере одну спиртовую гидроксильную группу на молекулу,
выбирая молярное соотношение амина согласно формуле (I) к карбоновой кислоте в интервале от 1,5:1 до 1:1,
включающий в себя следующие стадии:
(a) взаимодействие амина согласно формуле (I) с указанной карбоновой кислотой в условиях температуры и давления, при которых вода и амин согласно формуле (I) являются газообразными, в котором взаимодействие (а) осуществляют в единичном реакторе,
(b) отгонку образованной воды вместе с не прореагировавшим амином согласно формуле (I),
(c) отделение не прореагировавшего амина согласно формуле (I) от воды и
(d) повторное введение указанного амина согласно формуле (I) в реакционную смесь на стадии (а).
Алкиламиды и диалкиламиды жирных кислот применяются в различных использованиях, таких как не оказывающие негативного воздействия на окружающую среду растворители и в качестве технологических добавок для полимеров. Способы изготовления подобных амидов известны в технике. Многие из них исходят из карбоновой кислоты или производного, такого как соответствующий галогенид или сложный эфир и алкил- или диалкиламид. Однако можно наблюдать некоторые недостатки. Галогениды карбоновых кислот, однако, являются дорогостоящими, и они имеют тенденцию гидролизовать галогениды водорода в ходе различных случаев, таких как хранение, транспортировка и взаимодействия. Подобные галогениды являются в большой степени коррозийными, и в ходе образования амида их необходимо нейтрализовать или одним эквивалентом амина, или добавлением основания, которое может также вместо этого взаимодействовать с галогенидом карбоновой кислоты.
В ходе образования амидов из сложных эфиров (или лактонов) и аминов будут образовываться спирты, понижая проблему коррозии, описанную выше, см., например, WO 2010/037776. Однако сложные эфиры и лактоны обычно вполне дорогостоящие по сравнению с карбоновыми кислотами.
В US 2009/0062565 раскрывается способ, в котором амиды жирных кислот получают из соответствующей карбоновой кислоты и амина. Раскрытый способ использует систему из двух реакторов. Образованную воду отгоняют вместе с амином и после разделения амин можно повторно использовать посредством введения в кислоту для того, чтобы начать реакцию образования амида. Однако для этого способа обычно необходим избыток амина. Это особенно невыгодно для получения в небольшом масштабе и для периодических процессов.
Следовательно, цель настоящего изобретения заключалась в предоставлении способа получения амидов карбоновых кислот из карбоновых кислот, который не требует большого избытка амина, а дает возможность получать амиды с большим выходом и удовлетворительной чистотой.
Соответственно обнаружен способ, определенный в начале, также далее в данном документе упоминаемый как способ согласно изобретению.
В ходе выполнения способа согласно изобретению карбоновая кислота, также упоминаемая как карбоновая кислота (II), будет взаимодействовать с амином формулы (I)
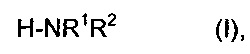
кратко также упоминаемым как амин (I), причем целые числа могут быть различными или предпочтительно идентичными,
причем R1 выбирают из алкилов, содержащих от 1 до 4 атомов углерода, таких как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил, предпочтительно n-алкил, содержащий от 1 до 4 атомов углерода, и особенно метил или этил,
причем R2 выбирают из водорода и алкилов, содержащих от 1 до 4 атомов углерода, таких как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил, предпочтительно n-алкил, содержащий от 1 до 4 атомов углерода, и особенно метил или этил,
где R1 и R2 комбинируют таким образом, чтобы амин по формуле (I) обладал более низкой точкой кипения, чем вода.
В одном варианте выполнения настоящего изобретения амин согласно формуле (I) выбирают из метиламина, диметиламина, этиламина, диэтиламина, н-пропиламина, изопропиламина, диизопропиламина, н-бутиламина, изобутиламина, трет-бутиламина, метил-н-пропиламина, н-метил-этиламина и метилизопропиламина. Особенно предпочтительные амины формулы (I) выбирают из диметиламина и диэтиламина.
Карбоновые кислоты, которые будут взаимодействовать согласно способу изобретения, выбирают из карбоновых кислот по меньшей мере с 3 атомами углерода на молекулу, причем указанная кислота необязательно несет по меньшей мере одну спиртовую гидроксильную группу на молекулу.
В одном варианте выполнения настоящего изобретения карбоновую кислоту (II) выбирают из карбоновых кислот, содержащих от 3 до 18 атомов углерода, которые являются разветвленными и не замещенными, таких как изомасляная кислота и изовалериановая кислота.
В одном варианте выполнения настоящего изобретения карбоновую кислоту (II) выбирают из карбоновых кислот, содержащих от 3 до 18 атомов углерода, которые являются предпочтительно с прямой цепью и не замещенными. Примерами являются пропионовая кислота, масляная кислота, валериановая кислота, капроновая кислота (н-C5H11-COOH), каприловая кислота, каприновая кислота, лауриновая кислота, миристиновая кислота, пальмитиновая кислота и стеариновая кислота. Карбоновая кислота (II) может иметь одну или более углерод-углеродных двойных связей, которые не соединены с группой карбоновой кислоты. Предпочтительными являются карбоновые кислоты, которые не имеют углерод-углеродной двойной связи.
В одном частном предпочтительном варианте выполнения настоящего изобретения карбоновую кислоту (II) выбирают из каприловой кислоты, каприновой кислоты и лауриновой кислоты, и амин (I) выбирают из диметиламина и диэтиламина.
В одном варианте выполнения настоящего изобретения карбоновую кислоту (II) выбирают из α-гидроксилкарбоновых кислот, содержащих от 3 до 12 атомов углерода, которые предпочтительно имеют прямую цепь. Особенно предпочтительны α-гидроксилкарбоновые кислоты, содержащие от 3 до 12 атомов углерода, которые не несут дополнительных функциональных групп.
В случае, хиральной карбоновой кислоты (II), например, в качестве карбоновой кислоты (II) выбирают α-гидроксилкарбоновые кислоты, содержащие от 3 до 12 атомов углерода, обнаружено, что стехиометрия не влияет на взаимодействие. Таким образом, в качестве исходного материала можно применять любой энантиомер, а также рацемат.
В одном варианте выполнения настоящего изобретения карбоновую кислоту (II) выбирают из молочной кислоты.
В одном частном предпочтительном варианте выполнения настоящего изобретения карбоновую кислоту (II) выбирают из молочной кислоты, и амин (I) выбирают из диметиламина и диэтиламина.
В процессе согласно изобретению выбирается молярное отношение амина (I) к карбоновой кислоте (II) в интервале от 1,5:1 до 1:1, ссылаясь на общее соотношение исходных материалов, равное предпочтительно от 1,2:1 до 1:1.
Процесс согласно изобретению включает в себя следующие стадии:
(a) взаимодействие амина согласно формуле (I) с карбоновой кислотой (II) в условиях температуры и давления, при которых вода и амин (I) являются газообразными, в котором взаимодействие (а) осуществляют в единичном реакторе,
(b) отгонку образованной воды вместе с непрореагировавшим амином (I),
(c) отделение непрореагировавшего амина согласно формуле (I) от воды и
(d) повторное введение указанного амина (I) в реакционную смесь на стадии (а).
Стадии от (а) до (d) будут обсуждаться ниже более подробно.
Нет необходимости, чтобы термин "стадии" в контексте настоящего изобретения предполагал, что различные этапы осуществляются последовательно. Например, повторно введенный амин (I) согласно стадии (d) будет снова взаимодействовать с карбоновой кислотой (II), и в то же время большее количество воды будет отгоняться.
Стадия (а) процесса согласно изобретению включает взаимодействие амина (I) с карбоновой кислотой (II). Указанное взаимодействие может быть одностадийным или двухстадийным взаимодействием. Указанное взаимодействие может включать промежуточное образование или соли (карбоксилата аммония), который впоследствии конденсируется с образованием амида, или оно может развиваться в прямом направлении.
Стадию (а) проводят в условиях температуры и давления, при которых вода и амин согласно формуле (I) являются газообразными, что означает, что условия давления и температуры являются до некоторой степени такими, чтобы вода и амин (I) были газообразными. Таким образом, например, если амин (I) выбирают из диэтиламина и давление выбирают, что оно является обычным давлением (атмосферным давлением, 1 бар), температура взаимодействия составляет по меньшей мере 105°C и предпочтительно в интервале от 130 до 230°C.
В одном варианте выполнения настоящего изобретения взаимодействие на стадии (а) осуществляют при температуре в интервале от 130 до 230°C, причем соответственно регулируют давление, предпочтительно в интервале от 150 до 210°C.
В одном варианте выполнения настоящего изобретения взаимодействие на стадии (а) осуществляют при давлении в интервале от 0,5 до 40 бар, предпочтительно от атмосферного давления до 10 бар, причем соответственно регулируют температуру.
На взаимодействие амина (I) с карбоновой кислотой (II) на стадии (а) влияет контактирование амина (I) и карбоновой кислоты (II). Предпочтительно сначала загружать реактор, в котором собираются осуществлять стадию (а), карбоновой кислотой (II). Указанный реактор можно загрузить карбоновой кислотой (II), находящейся предпочтительно в жидкой форме. Однако в зависимости от конкретного случая также возможно загрузить карбоновую кислоту в твердой форме, что может включать стадию расплавления до осуществления стадии (а), и затем вводить амин (I), или расплавлять карбоновую кислоту (II) в присутствии амина (I). В одном варианте выполнения карбоновую кислоту загружают как водный раствор, и прежде всего, будет отгоняться вода из растворителя.
В одном варианте выполнения настоящего изобретения, особенно в вариантах выполнения, где карбоновую кислоту выбирают из α-гидроксилкарбоновых кислот, содержащих от 3 до 12 атомов углерода, карбоновая кислота может содержать в качестве примесей некоторые сложные эфиры. Это могли быть, например, лактид и в случае молочной кислоты олигомеры молочной кислоты.
Предпочтительно взаимодействие на стадии (а) осуществляют при смешении, например, посредством перемешивания или посредством рециркуляции жидкости. Возможно вводить амин (I) в жидкой форме и выполнять выпаривание в сосуде, в котором осуществляется взаимодействие, но предпочтительно вводить амин (I) в газообразном состоянии (в газообразной форме).
Предпочтительно взаимодействие на стадии (а) осуществляют периодически или полупериодически.
Стадию (а) можно проводить в каскадном реакторе, но предпочтительно проводить стадию (а) в одном сосуде, например, в корпусном реакторе. Указанный сосуд и предпочтительно указанный корпусной реактор оснащены - кроме всего прочего - средством для удаления воды в газовом состоянии, средством для введения карбоновой кислоты (II) и для введения амина (I), и средством для повторного введения амина (I) согласно стадии (d), см. ниже.
Предпочтительно указанный сосуд оснащен средством для удаления воды в газовом состоянии, которая обычно содержит некоторое количество амина (I), и средством для разделения воды и амина (I), например, перегонной колонной, ректификационной колонной и/или по меньшей мере одним конденсатором или комбинацией двух или более ректификационных колонн, преимущественно с одним или двумя конденсаторами.
В одном варианте выполнения настоящего изобретения указанный сосуд оснащают двумя ректификационными колоннами и двумя конденсаторами, регулируемыми при двух различных температурах.
Взаимодействие карбоновой кислоты (II) с амином (I) можно проводить в присутствии органического растворителя, такого как толуол или ксилол, но предпочтительно проводить стадию (а) без применения какого-либо органического растворителя. В этом случае стадия (b) не будет требовать никакого органического растворителя.
В одном варианте выполнения настоящего изобретения стадию (а) проводят с применением катализатора. В другом варианте выполнения стадия (а) будет проводиться без катализатора.
В одном варианте выполнения настоящего изобретения стадию (а) проводят с применением добавки, например, пеногасителя или противовспенивающего агента или антиоксиданта, такого как, но не ограничивающегося ими, гипофосфита щелочных металлов. В альтернативном варианте выполнения стадия (а) будет проводиться без добавок.
На стадии (b), воду, образованную посредством образования амида, будут отгонять. В ходе стадии (b), воду будут отгонять вместе с непрореагировавшим амином (I). Воду можно отгонять с большим количеством непрореагировавшего амина (I) или со всем избытком амина (I), или ее можно отгонять вместе только с очень небольшим процентным содержанием амина (I). На отгонку будут влиять удаляемые части газовой фазы в сосуде и в частности в корпусном реакторе, в котором проводится стадия (а). Указанное удаление можно проводить, например, посредством открытия входного отверстия или клапана из сосуда в средство для отделения амина (I) от воды. Также возможно иметь постоянно открытое входное отверстие и давать возможность газообразному амину (I) и пару улетучиваться из сосуда, в котором проводят стадию (а), и принуждать его перемещаться в средство для отделения амина (I) от воды.
Течение газообразных материалов (воды, амина (I) можно усилить по меньшей мере одним насосом (например, воздуходувкой).
На стадии (с) разделяют непрореагировавший амин (I) и воду, отогнанную в стадии (b). Указанного разделения можно выгодно достичь с применением одной дистилляционной колонны, двух дистилляционных колонн, одной ректификационной колонны, двух ректификационных колонн, трех или более дистилляционных колонн, трех или более ректификационных колонн или одной или более мембран. Предпочтительно применение одной или более дистилляционных или ректификационных колонн. В частности, предпочтительно применять одну или более дистилляционных колонн в комбинации с одним или более конденсаторов или с одним или более дефлегматоров.
В случае применения одного или более конденсаторов в комбинации с ректификационными или дистилляционными колоннами предпочтительно, чтобы указанный конденсатор (конденсаторы) функционировал таким образом, чтобы по меньшей мере 90 масс. % воды, которая отгоняется, была удалена из газообразного пара, предпочтительно по меньшей мере 95 масс. %. В одном варианте выполнения вода, которую следует отгонять, будет удалена полностью или вплоть до 99,9 масс.% воды.
Предпочтительно удалять воду из смеси на стадии (с) в форме жидкости.
В случае применения одной или более ректификационных колонн предпочтительно применять подобные колонны, выбираемые из тарельчатых колонн и насадочных колонн. Примеры для тарелок в тарельчатых колоннах представляют собой колпачковые тарелки, ситчатые тарелки и клапанные тарелки. Примеры насадок, подходящие для насадочных колонн, представляют собой неупорядоченные насыпные насадки и структурные насадки.
В случае, когда на стадии (с) применяют комбинацию по меньшей мере одной ректификационной колонны или по меньшей мере одной ректификационной колонны по меньшей мере с одним конденсатором или по меньшей мере одним дефлегматором, коэффициент дефлегмации регулируют таким образом, чтобы отток воды в реакционную смесь стадии (а) был настолько небольшим, насколько возможно.
В одном варианте выполнения настоящего изобретения, стадию (с) проектируют таким образом, чтобы ректификационная колонна имела в интервале от 2 до 40 равновесных шагов.
В предпочтительном варианте выполнения настоящего изобретения коэффициент дефлегмации и равновесные ступени колонны (колонн), комбинированных с конденсатором (конденсаторами) или дефлегматором, регулируют таким образом, чтобы воду можно было применять без дополнительной очистки, и амин (I) чистотой 90 масс. % или выше можно было повторно вводить во взаимодействие.
В одном варианте выполнения настоящего изобретения для разделения воды и амина применяют мембрану.
Посредством отделения амина (I) от воды амин (I) восстанавливается.
На стадии (d) амин (I), восстановленный согласно стадии (с), будет повторно введен в реакционную смесь на стадии (а). Амин (I) можно повторно вводить в жидкой или газообразной форме. Предпочтительно повторно вводить амин (I) во взаимодействие согласно стадии (а) в газообразной форме.
В одном варианте выполнения настоящего изобретения в качестве средства для повторного введения амина (I) выбирают один или более нагнетателей (компрессоров) особенно вместе с направляющим аппаратом, таким как, например, оросительное кольцо.
В одном варианте выполнения настоящего изобретения, в качестве средства для повторного введения амина (I) выбирают активаторы газификации, предпочтительно активаторы газификации с всасывающей способностью без нагнетателя или в комбинации с нагнетателем.
В одном варианте выполнения настоящего изобретения в качестве средства для повторного введения амина (I) выбирают жидкоструйные форсунки. В этом варианте выполнения реакционный сосуд, обсужденный на стадии (а), может содержать, но не обязательно, мешалку.
В одном варианте выполнения настоящего изобретения для функционирования жидкоструйной форсунки будет применяться жидкая реакционная смесь стадии (а), например, в качестве рабочей жидкости (эжектора).
Процессом согласно изобретению можно оперировать как периодическим процессом, полупериодическим процессом или непрерывным процессом. Предпочтительно оперировать им как периодическим или полупериодическим процессом.
В случае, когда процессом согласно изобретению оперируют как периодическим или полупериодическим процессом, взаимодействие будет обрываться после превращения всего или почти всего количества, такого как от 90 до 99,9 мол. %, карбоновой кислоты (II), предпочтительно 93 мол. % или более.
После прекращения взаимодействия амид карбоновой кислоты (II) и амин (I) можно выделить с превосходным выходом и удовлетворительной чистотой. Для многих использований подобный амид можно применять без дополнительной очистки, но возможно в альтернативе его очищать. Пригодными способами очистки являются перегонка, дезодорирование (отгонка), обесцвечивание, например, с помощью древесного угля, или фильтрация через кремнезем.
В случае, когда карбоновая кислота (II) имеет одну или более спиртовых гидроксильных групп, можно обнаружить только очень небольшое количество побочного продукта, генерированного нуклеофильным замещением спиртовой гидроксильной группы амином (I), если это произойдет, такое как от нуля до 3,0 мол. %, в частности от 0,1 до 1,5 мол. %, от нуля до 1,0 мол. %, в частности от 0,001 до 0,5 мол. %, ссылаясь на общее желательное количество амида. Указанные побочные продукты нуклеофильного замещения обычно обладают очень неприятным запахом и присутствие следов как таковых можно легко обнаружить.
Далее изобретение иллюстрируется примерами.
Части означают массовые части.
Пример 1: Изготовление N,N-диметиллактамида
Применяли следующую схему аппаратуры: корпусной реактор с мешалкой, нагревательная система, на верху выходное отверстие к низу ректификационной колонны ("первой колонны") с упаковкой Зульцера (40 элементов, 40-200 Sulzer M752Y), без дефлегмации с последующей другой ректификационной колонной ("второй колонной") (упаковка Зульцера, 22-250 Sulzer M752Y Elements), питающей на верху и присоединенной к конденсатору (20°C) на верху колонны. Во втором конденсаторе воду конденсировали, но диметиламин оставался в газообразном состоянии. Схема также содержала жидкоструйную форсунку (эжекторный насос) для повторного ввода газообразного диметиламина в корпусной реактор.
Корпусной реактор загружали 60,0 частями рацемической молочной кислотой (водный раствор 88 мас. %) и 0,51 частями гипофосфита натрия. Из корпусного реактора был выкачан воздух. Диметиламин вводили в корпусной ректор в виде газа (стадия (а.1)). В условиях нагревания в реактор вводили 104 мол. % теоретического количества диметиламина (27,39 частей) за 8,9 ч после начала добавления диметиламина достигались температура, равная 170°C, и давление, равное 2,14 бар (абсолютное). Одновременно из реакционной смеси удаляли воду - вместе с диметиламином (стадия (b.1)) - отгонкой и пропускали через первую колонну. Во второй колонне разделяли воду и диметиламин (стадия (с.1)). Газообразный диметиламин повторно вводили через контур в реактор с помощью жидкоструйной форсунки (стадия (d1)). На протяжении реакции контролировали величину кислотности (DIN 53402).
Взаимодействие продолжалось в течение 37,5 часов, во время которых температуру поддерживали от 166°C до 172°C. Давление в реакторе составляло 1,34 бар (абсолютное) в конце стадии (b.1). Величина кислотности неочищенного продукта взаимодействия составляла в то время 7,8 мг KОН/г.
Неочищенный продукт упаривали в другом сосуде для удаления побочных продуктов с низкой точкой кипения, например, избыток диметиламина. Упаренный неочищенный продукт содержал 95,7% N,N-диметиллактамида (GC-анализ посредством оценки площади газовой хроматограммы).
Пример 2: Изготовление N,N-диметиллактамида
Применяли следующую схему аппаратуры: корпусной реактор с мешалкой, нагревательная система, на верху выходное отверстие к низу ректификационной колонны ("первой колонны") с упаковкой Зульцера (20 элементов, 20-200 Sulzer M752Y), дефлегматор с последующей другой ректификационной колонной ("второй колонной") (упаковка Зульцера, Элементы 22-250 Sulzer M752Y), питающей на верху и присоединенной к конденсатору на верху колонны. Во втором конденсаторе воду конденсировали, но диметиламин оставался в газообразном состоянии. Схема также содержала жидкоструйную форсунку (эжекторный насос) для повторного ввода газообразного диметиламина в корпусной реактор.
Корпусной реактор загружали 120 частями молочной кислоты (водный раствор 88 мас. %) и 0,11 частями гипофосфита натрия. Из корпусного реактора был выкачан воздух. Диметиламин вводили в корпусной ректор в виде газа (стадия (а.1)). Первоначально диметиламин добавляли без нагревания. После того, как было добавлено 66% стехиометрического количества DMA (в течение 7,5 ч), реакционную смесь нагревали до 170°C. При температуре взаимодействия, равном 168-176°C, и давлении, равном 0,5-2,3 бар, добавляли оставшееся количество DMA. В общем в реактор вводили 102 мол. % теоретического количества диметиламина (53,75 частей) за время (окончательно 51 ч после начала загрузки DMA). Одновременно из реакционной смеси удаляли воду - вместе с диметиламином (стадия (b.1)) - отгонкой и пропускали через первую колонну. Во второй колонне разделяли воду и диметиламин (стадия (с.1)). Газообразный диметиламин повторно вводили через контур в реактор с помощью жидкоструйной форсунки (стадия (d.1)). На протяжении реакции контролировали величину кислотности (DIN 53402).
Взаимодействие продолжалось до тех пор, пока величина кислотности неочищенного продукта взаимодействия не достигнет 10 мг KОН/г. Общее время от начала течения DMA до конца составляло 68 ч. Неочищенный продукт содержал 97,3% N,N-диметиллактамида (GC-анализ посредством оценки площади газовой хроматограммы).
Неочищенный продукт очищали посредством продувки азотом для удаления побочных продуктов с низкой точкой кипения, например, избытка диметиламина. Выход очищенного продукта составлял 135,8 частей.
Пример 3: Изготовление N,N-диметил С8/С10 амида
Применяли такое же оборудование, как в примере 2.
Корпусной реактор загружали 91,6 частями С8/С10 жирной кислоты (Edenor V85) и 0,11 частями гипофосфита натрия. Из корпусного реактора был выкачан воздух. Диметиламин (DMA) вводили в корпусной ректор в виде газа (стадия (а.1)) и смесь нагревали до 179°C. Диметиламин добавляли с такой скоростью, чтобы давление оставалось ниже 2,0 бар и т.д. Прикладывали нагревание, чтобы удерживать температуру взаимодействия при 196-198°C. В общем в реактор вводили 101 мол. % теоретического количества диметиламина (28,43 частей) за 9,2 ч. Одновременно из реакционной смеси удаляли воду - вместе с диметиламином (стадия (b.1)) - отгонкой и пропускали через первую колонну. Во второй колонне разделяли воду и диметиламин (стадия (с.1)). Газообразный диметиламин повторно вводили через контур в реактор с помощью жидкоструйной форсунки (стадия (d.1)). На протяжении реакции контролировали величину кислотности (DIN 53402).
Взаимодействие продолжалось до тех пор, пока величина кислотности неочищенного продукта взаимодействия не достигнет 6 мг KОН/г. Общее время взаимодействия от начала течения DMA до конца составляло 12,7 ч.
Неочищенный продукт очищали посредством продувки азотом для удаления побочных продуктов с низкой точкой кипения, например, избытка диметиламина. Выход очищенного продукта N,N-диметил С8/С10 амида составлял 102,4 части.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ НЕПРЕРЫВНОГО РАСТВОРЕНИЯ ТВЕРДОГО ВЕЩЕСТВА В РЕАКЦИОННОЙ СРЕДЕ | 2018 |
|
RU2761042C2 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ АНГИДРИДОВ НЕНАСЫЩЕННОЙ КАРБОНОВОЙ КИСЛОТЫ | 2007 |
|
RU2443673C2 |
| НЕПРЕРЫВНЫЙ СПОСОБ ПОЛУЧЕНИЯ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ ПОСРЕДСТВОМ СОДЕРЖАЩЕГО НИТРОКСИЛЬНЫЙ РАДИКАЛ КАТАЛИЗАТОРА | 2011 |
|
RU2579510C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОЦИАНАТОВ С ИСПОЛЬЗОВАНИЕМ ДИАРИЛКАРБОНАТА | 2008 |
|
RU2523201C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1-ОКТЕНА | 2002 |
|
RU2279420C2 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ ОЛЕФИНОВ | 2003 |
|
RU2327694C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИДА КАРБОНОВОЙ КИСЛОТЫ ИЗ КАРБОНИЛЬНОГО СОЕДИНЕНИЯ И ЦИАНИСТОВОДОРОДНОЙ КИСЛОТЫ | 2009 |
|
RU2552619C9 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ АЛКИЛАМИНО(МЕТ)АКРИЛАМИДОВ | 2010 |
|
RU2546670C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИАМИДОВ ИЗ СОЕДИНЕНИЙ АМИНОКАРБОНОВОЙ КИСЛОТЫ | 1999 |
|
RU2215754C2 |
| СПОСОБ ОЧИСТКИ ЗАГРЯЗНЕННЫХ МЕТАЛЛИЧЕСКИХ ПОВЕРХНОСТЕЙ И ВЕЩЕСТВА, ПРИМЕНИМЫЕ ДЛЯ ТАКОГО СПОСОБА | 2016 |
|
RU2707008C2 |
Изобретение относится к способу получения амида карбоновой кислоты посредством взаимодействия амина формулы (I), который выбирают из метиламина, диметиламина, этиламина, диэтиламина, н-пропиламина, изопропиламина, диизопропиламина, н-бутиламина, изобутиламина, трет-бутиламина, метил-н-пропиламина, н-метил-н-этиламина и метил-изопропиламина, с карбоновой кислотой, которую выбирают из карбоновых кислот, содержащих от 3 до 18 атомов углерода, которые являются разветвленными и незамещенными, из карбоновых кислот, содержащих от 3 до 18 атомов углерода, которые являются предпочтительно с прямой цепью и незамещенными, и из α-гидроксилкарбоновых кислот, содержащих от 3 до 12 атомов углерода, которые предпочтительно имеют прямую цепь. Молярное соотношение амина согласно формуле (I) к карбоновой кислоте выбирают в интервале от 1,5:1 до 1:1. При этом способ включает в себя следующие стадии (а): взаимодействие амина согласно формуле (I) с указанной карбоновой кислотой в условиях температуры и давления, при которых вода и амин согласно формуле (I) являются газообразными, в котором взаимодействие (а) осуществляют в единичном реакторе, (b) отгонку образованной воды вместе с непрореагировавшим амином согласно формуле (I), (c) отделение непрореагировавшего амина согласно формуле (I) от воды и (d) повторное введение указанного амина согласно формуле (I) в реакционную смесь на стадии (а), причем стадии (а) и (b) осуществляют без применения какого-либо органического растворителя. Взаимодействие на стадии (а) осуществляют при температуре в интервале от 130 до 230°С. Кроме того, стадию (а) осуществляют в одном реакторе, который присоединен к ректификационной колонне и конденсатору или в одном реакторе, который присоединен к комбинации двух ректификационных колонн и двух конденсаторов. Повторное введение амина (I) на стадии (d) осуществляют посредством жидкоструйных форсунок. Технический результат – получение амидов карбоновых кислот с большим выходом и удовлетворительной чистотой, не требующее большого избытка амина. 5 з.п. ф-лы, 3 пр.
1. Способ получения амида карбоновой кислоты посредством взаимодействия амина формулы (I), который выбирают из метиламина, диметиламина, этиламина, диэтиламина, н-пропиламина, изопропиламина, диизопропиламина, н-бутиламина, изобутиламина, трет-бутиламина, метил-н-пропиламина, н-метил-н-этиламина и метил-изопропиламина,
с карбоновой кислотой, которую выбирают из карбоновых кислот, содержащих от 3 до 18 атомов углерода, которые являются разветвленными и незамещенными, из карбоновых кислот, содержащих от 3 до 18 атомов углерода, которые являются предпочтительно с прямой цепью и незамещенными, и из α-гидроксилкарбоновых кислот, содержащих от 3 до 12 атомов углерода, которые предпочтительно имеют прямую цепь,
выбирая молярное соотношение амина согласно формуле (I) к карбоновой кислоте в интервале от 1,5:1 до 1:1,
включающий в себя следующие стадии:
(а) взаимодействие амина согласно формуле (I) с указанной карбоновой кислотой в условиях температуры и давления, при которых вода и амин согласно формуле (I) являются газообразными, в котором взаимодействие (а) осуществляют в единичном реакторе,
(b) отгонку образованной воды вместе с непрореагировавшим амином согласно формуле (I),
(c) отделение непрореагировавшего амина согласно формуле (I) от воды и
(d) повторное введение указанного амина согласно формуле (I) в реакционную смесь на стадии (а), причем
стадии (а) и (b) осуществляют без применения какого-либо органического растворителя.
2. Способ по п. 1, отличающийся тем, что взаимодействие на стадии (а) осуществляют при температуре в интервале от 130 до 230°С.
3. Способ по п. 1, отличающийся тем, что указанную карбоновую кислоту выбирают из молочной кислоты.
4. Способ по п. 1, отличающийся тем, что стадию (а) осуществляют в одном реакторе, который присоединен к ректификационной колонне и конденсатору.
5. Способ по по п. 1, отличающийся тем, что стадию (а) осуществляют в одном реакторе, который присоединен к комбинации двух ректификационных колонн и двух конденсаторов.
6. Способ по любому из пп. от 1 до 5, отличающийся тем, что повторное введение амина (I) на стадии (d) осуществляют посредством жидкоструйных форсунок.
| Колосоуборка | 1923 |
|
SU2009A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| LAXMIKANTH RAO J | |||
| et al.: "Cathodically electrodepositable novel coating system from castor oil", JOURNAL OF APPLIED POLYMER SCIENCE, 1992, v.44, no.11, p.1873-1881 | |||
| Способ получения -формилированных соединений | 1974 |
|
SU677657A3 |

