[0001] Настоящая заявка испрашивает приоритет на основании предварительной заявки на патент США №61/655935, поданной 5 июня 2012 года, и заявки США №13/800202, поданной 13 марта 2013 года, полное содержание которых включено в настоящую заявку посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
[0002] Настоящее изобретение в целом относится к методам органического синтеза для получения антивирусных соединений и их синтетических промежуточных соединений.
[0003] Гепатит С является общепризнанным вирусным заболеванием печени, которое характеризуется заболеванием печени. Несмотря на то, что лекарственные средства, направленные на лечение печени, широко применяются и их эффективность доказана, их практическая применимость ограничена токсичностью и другими побочными эффектами. Ингибиторы вируса гепатита С (HCV) являются подходящими средствами для сдерживания образования и развития инфекции ВГС, а также для диагностических исследований HCV.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
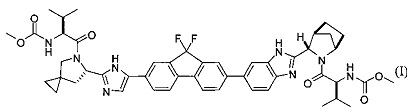
[0004] В одном варианте реализации настоящего изобретения предложен способ получения соединения формулы I:
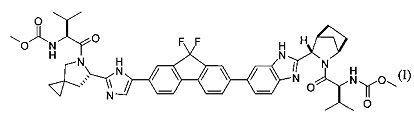 ,
,
или его фармацевтически приемлемой соли или сольвата. Соединение формулы I, также известное как ледиспавир, имеет следующее химическое название: сложный метиловый эфир (1-{3-[6-(9,9-дифтор-7-{2-[5-(2-метоксикарбониламино-3-метилбутирил)-5-азаспиро[2,4]гепт-6-ил]-3Н-имидазол-4-ил}-9Н-флуорен-2-ил)-1Н-бензимидазол-2-ил]-2-азабицикло[2,2,1]гептан-2-карбонил}-2-метилпропил)-карбаминовой кислоты. Способ включает следующие стадии:
(А) реакция сочетания соединения формулы (i)
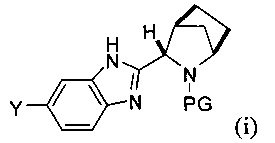
с соединением формулы (ii)
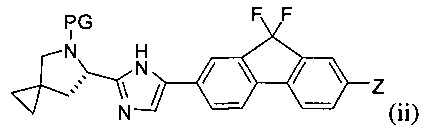
в присутствии металлического катализатора и основания с получением соединения формулы (iii) или его соли:
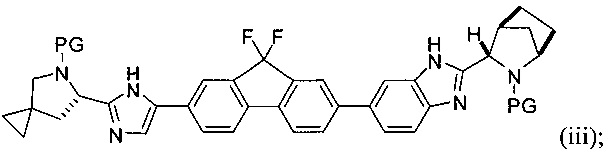
(В) снятие защиты с соединения формулы (iii) с получением соединения формулы (iv):
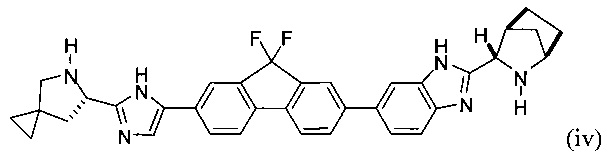
или его соли; и
(С) приведение соединения формулы (iv) в контакт с (S)-2-(метоксикарбониламино)-3-метилбутановой кислотой:
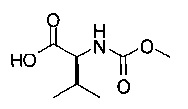
с получением соединения формулы I.
[0005] Каждый PG независимо представляет собой защитную группу амина.
[0006] Заместители Y и Z независимо выбраны из Br и -B(OR)(OR'). В одном варианте реализации в случае, если Y представляет собой -B(OR)(OR'), тогда Z представляет собой Br, и в другом варианте реализации в случае, если Y представляет собой Br, то Z представляет собой -B(OR)(OR').
[0007] Заместители R и R' независимо выбраны из группы, состоящей из водорода и линейного или разветвленного C1-8-алкила, или R и R' вместе представляют собой линейный или разветвленный C1-8-алкилен, С3-8-циклоалкен или С6-12-арилен.
[0008] Любой алкил, алкилен, циклоалкилен или арилен, описанный в настоящей заявке, является возможно замещенным одним или более заместителями, выбранными из группы, состоящей из C1-6-алкила, -С(O)N(С1-6-алкила)2, и -С(O)O(С1-6-алкила).
[0009] Более подробно варианты реализации представлены ниже.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0010] Фиг. 1 представляет собой рентгеновскую порошковую дифрактограмму, полученную для Формы I, кристаллического полиморфа соединения 30, описанного ниже в настоящей заявке.
[0011] Фиг. 2 представляет собой рентгеновскую порошковую дифрактограмму, полученную для Формы II, кристаллического полиморфа соединения 30, описанного ниже в настоящей заявке.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0012] Определения
[0013] Предполагается, что следующие используемые в настоящем описании слова и выражения в основном имеют значения, представленные ниже, за исключением случаев, когда в контексте, в котором они употребляются, указано иначе
[0014] Термин «алкил», используемый в настоящей заявке, относится к насыщенному углеводороду с прямой или разветвленной цепью, содержащему определенное количество атомов углерода. Например, предполагается, что (С1-С8)алкил включает, но не ограничивается указанными, метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, гексил, изогексил и неогексил. Алкильная группа может быть незамещенной или возможно замещенной одним или более заместителей, как описано по всему тексту настоящей заявки.
[0015] Термин «замещенный алкил» относится к:
1) алкильной группе, описанной выше, содержащей 1, 2, 3, 4 или 5 заместителей (в некоторых вариантах реализации изобретения 1, 2 или 3 заместителя), выбранных из группы, состоящей из алкенила, алкинила, алкокси, циклоалкила, циклоалкенила, циклоалкокси, циклоалкенилокси, ацил, ациламино, ацилокси, амино, замещенного амина, аминокарбонила, алкоксикарбониламино, азидо, циано, галогена, гидрокси, кето, тиокарбонила, карбокси, карбоксиалкила, арилтио, гетероарилтио, гетероциклилтио, тиола, алкилтио, арила, арилокси, гетероарила, аминосульфонила, аминокарбониламино, гетероарилокси, гетероциклила, гетероциклоокси, гидроксиамино, алкоксиамино, нитро, -SO-алкила, -SO-циклоалкила, -SO-гетероциклила, -SO-арила, -SO-гетероарила, -SO2-алкила, -SO2-циклоалкила, -SO2-гетероциклила, -SO2-арила и -SO2-гетероарила. Если иное не указано в определении, все заместители могут быть необязательно дополнительно замещены 1, 2 или 3 заместителями, выбранными из алкила, алкенила, алкинила, карбокси, карбоксиалкила, аминокарбонила, гидрокси, алкокси, галогена, CF3, амино, замещенного амино, циано, циклоалкила, гетероциклила, арила, гетероарила и -S(O)nRa, где Ra представляет собой алкил, арил или гетероарил и n равен 0, 1 или 2; или
2) алкильную группу, как определено выше, которая прерывается 1-10 атомами (например, 1, 2, 3, 4 или 5 атомами), независимо выбранными из кислорода, серы и NRa, где Ra выбирают из водорода, алкила, циклоалкила, алкенила, циклоалкенила, алкинила, арила, гетероарила и гетероциклила. Все заместители могут быть необязательно дополнительно замещены алкилом, алкенилом, алкинилом, карбокси, карбоксиалкилом, аминокарбонилом, гидрокси, алкокси, галогеном, CF3, амино, замещенным амино, циано, циклоалкилом, гетероциклилом, арилом, гетероарилом и -S(O)nRa, где Ra представляет собой алкил, арил или гетероарил и n равен 0, 1 или 2, или
3) алкильную группу, как определено выше, которая одновременно содержит 1, 2, 3, 4 или 5 заместителей, как описано выше, и прерывается 1-10 атомами (например, 1, 2, 3, 4 или 5 атомами), как указано выше.
[0016] Термин «алкилен» относится к бирадикалу разветвленной или неразветвленной насыщенной углеводородной цепи, в некоторых вариантах реализации изобретения содержащему от 1 до 20 атомов углерода (например, 1-10 атомов углерода, или 1, 2, 3, 4, 5 или 6 атомов углерода). Указанный термин проиллюстрирован группами, такими как метилен (-СН2-), этилен (-СН2СН2-), изомеры пропилена (например, -СН2СН2СН2- и -СН(СН3)СН2-), и тому подобные.
[0017] Термин «низший алкил» относится к насыщенному углеводороду с насыщенной монорадикальной разветвленной или неразветвленной цепью, содержащему 1, 2, 3, 4, 5 или 6 атомов углерода. Указанный термин проиллюстрирован такими группами, как метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-гексил и тому подобными.
[0018] Термин «замещенный низший алкил» относится к низшему алкилу, определение которого дано выше, содержащему от 1 до 5 заместителей (в некоторых вариантах реализации изобретения 1, 2 или 3 заместителя), как описано выше для замещенного алкила, или к низшей алкильной группе, как описано выше, которая прерывается 1, 2, 3, 4 и 5 атомами, как описано выше для замещенного алкила, или к низшей алкильной группе, как описано выше, которая одновременно содержит 1, 2, 3, 4 или 5 заместителей, как описано выше, и прерывается 1, 2, 3, 4 или 5 атомами, как описано выше.
[0019] Термин «алкилен» относится к бирадикалу разветвленной или неразветвленной насыщенной углеводородной цепи, в некоторых вариантах реализации изобретения содержащему от 1 до 20 атомов углерода (например, 1-10 атомов углерода или 1, 2, 3, 4, 5 или 6 атомов углерода). Указанный термин проиллюстрирован такими группами, как метилен (-СН2-), этилен (-СН2СН2-), изомеры пропилена (например, -СН2СН2СН2- и -СН(СН3)СН2-) и тому подобными.
[0020] Термин «низший алкилен» или «алкилен» относится к бирадикалу разветвленной или неразветвленной насыщенной углеводородной цепи, в некоторых вариантах реализации изобретения содержащему 1, 2, 3, 4, 5 или 6 атомов углерода.
[0021] Термин «замещенный алкилен» относится к алкиленовой группе, как определено выше, содержащей от 1 до 5 заместителей (в некоторых вариантах 1, 2 или 3 заместителя), как определено для замещенного алкила.
[0022] Термин «аралкил» относится к арильной группе, ковалентно связанной с алкиленовой группой, где арил и алкилен определены выше. «Необязательно замещенный аралкил» относится к необязательно замещенной арильной группе, ковалентно связанной с необязательно замещенной алкиленовой группой. Такие аралкильные группы представлены бензилом, фенилэтилом, 3-(4-метоксифенил)пропилом и т.п.
[0023] Термин «аралкилокси» относится к группе -О-аралкил. «Необязательно замещенный аралкилокси» относится к необязательно замещенной аралкильной группе, ковалентно связанной с необязательно замещенной алкиленовой группой. Такие аралкильные группы представлены бензилокси, фенилэтилокси и т.п.
[0024] Термин «алкенил» относится к монорадикалу разветвленной или неразветвленной ненасыщенной углеводородной группы, содержащему от 2 до 20 атомов углерода (в некоторых вариантах реализации изобретения от 2 до 10 атомов углерода, например от 2 до 6 атомов углерода), и содержащему от 1 до 6 углерод-углеродных двойных связей, например, 1, 2 или 3 углерод-углеродные двойные связи. В некоторых вариантах реализации изобретения, алкенильные группы включают этенил (или винил, т.е. -СН=СН2), 1-пропилен (или аллил, т.е., -СН2СН=СН2), изопропилен (-С(СН3)=СН2), и т.п.
[0025] Термин «низший алкенил» относится к алкенилу, как определено выше, содержащему от 2 до 6 атомов углерода.
[0026] Термин «замещенный алкенил» относится к алкенильной группе, как определено выше, содержащей от 1 до 5 заместителей (в некоторых вариантах реализации изобретения 1, 2 или 3 заместителей), как определено для замещенного алкила.
[0027] Термин «алкинил» относится к монорадикалу ненасыщенного углеводорода, в некоторых вариантах реализации изобретения содержащему от 2 до 20 атомов углерода (в некоторых вариантах реализации изобретения от 2 до 10 атомов углерода, например от 2 до 6 атомов углерода), и, содержащему от 1 до 6 углерод-углеродных тройных связей, например, 1, 2 или 3 углерод-углеродных тройных связей. В некоторых вариантах реализации изобретения алкинильные группы включают этинил (-С≡СН), пропаргил (или пропинил, т.е., -С≡ССН3) и тому подобные.
[0028] Термин «замещенный алкинил» относится к алкинильной группе, как определено выше, содержащей от 1 до 5 заместителей (в некоторых вариантах реализации изобретения 1, 2 или 3 заместителя), как определено для замещенного алкила.
[0029] Термин «гидрокси» или «гидроксил» относится к группе -ОН.
[0030] Термин «алкокси» относится к группе R-O-, где R представляет собой алкил или -Y-Z, где Y представляет собой алкилен, и Z представляет алкенил или алкинил, где алкил, алкенил и алкинил являются такими, как определено в настоящей заявке. В некоторых вариантах реализации изобретения алкоксигруппы представляют собой алкил-О- и включают, в качестве примера, метокси, этокси, н-пропокси, изопропокси, н-бутокси, трет-бутокси, втор-бутокси, н-пентокси, н-гексилокси, 1,2-диметилбутокси и т.п.
[0031] Термин «низший алкокси» относится к группе R-O-, где R представляет собой необязательно замещенный низший алкил. Указанный термин представлен группами, такими как метокси, этокси, н-пропокси, изопропокси, н-бутокси, изо-бутокси, трет-бутокси, н-гексилокси и т.п.
[0032] Термин «замещенный алкокси» относится к группе R-O-, где R представляет собой замещенный алкил или -Y-Z, где Y представляет собой замещенный алкилен и Z представляет собой замещенный алкенил или замещенный алкинил, причем замещенный алкил, замещенный алкенил и замещенный алкинил являются такими, как определено в настоящей заявке.
[0033] Термин «циклоалкил» относится к циклическим алкильным группам, содержащим от 3 до 20 атомов углерода, содержащим одно циклическое кольцо или несколько сконденсированных колец. Такие циклоалкильные группы включают, в качестве примера, одиночные кольцевые структуры, такие как циклопропил, циклобутил, циклопентил, циклооктил и т.п., или множественные кольцевые структуры, такие как адамантанил и бицикло[2,2,1]гептанил или циклические алкильные группы, сконденсированные с арильной группой, например инданил и тому подобное, при условии, что точкой присоединения является циклическая алкильная группа.
[0034] Термин «циклоалкенил» относится к циклическим алкильным группам, состоящим из от 3 до 20 атомов углерода, содержащим одно циклическое кольцо или несколько сконденсированных колец и содержащим по меньшей мере одну двойную связь, а в некоторых вариантах реализации изобретения от 1 до 2 двойных связей.
[0035] Термины «замещенный циклоалкил» и «замещенный циклоалкенил» относятся к циклоалкильным или циклоалкенильным группам, содержащим 1, 2, 3, 4 или 5 заместителей (в некоторых вариантах реализации изобретения 1, 2 или 3 заместителей), выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, циклоалкила, циклоалкенила, циклоалкокси, циклоалкенилокси, ацила, ациламино, ацилокси, амино, замещенного амино, аминокарбонила, алкоксикарбониламино, азидо, циано, галогена, гидрокси, кето, тиокарбонила, карбокси, карбоксиалкила, арилтио, гетероарилтио, гетероциклилтио, тиола, алкилтио, арила, арилокси, гетероарила, аминосульфонила, аминокарбониламино, гетероарилокси, гетероциклила, гетероциклоокси, гидроксиамино, алкоксиамино, нитро, -SO-алкила, -SO-циклоалкила, -SO-гетероциклила, -SO-арила, -SO-гетероарила, -SO2-алкила, -SO2-циклоалкила, -SO2-гетероциклила, -SO2-арила и -SO2-гетероарила. Термин «замещенный циклоалкил» включает циклоалкильные группы, в которых к одному или более из кольцевых атомов углерода в циклоалкильной группе присоединена оксогруппа. Если иное не указано в определении, все заместители могут быть необязательно дополнительно замещены 1, 2 или 3 заместителями, выбранными из алкила, алкенила, алкинила, карбокси, карбоксиалкила, аминокарбонила, гидрокси, алкокси, галогена, CF3, амино, замещенного амино, циано, циклоалкила, гетероциклила, арила, гетероарила, и -S(O)nRa, где Ra представляет собой алкил, арил или гетероарил и n равен 0, 1 или 2.
[0036] Термин «циклоалкокси» относится к группе циклоалкил-О-.
[0037] Термин «замещенный циклоалкокси» относится к группе замещенный циклоалкил-О-.
[0038] Термин «циклоалкенилокси» относится к группе циклоалкенил-О-.
[0039] Термин «замещенный циклоалкенилокси» относится к группе замещенный циклоалкенил-О-.
[0040] Термин «арил» относится к ароматической карбоциклической группе, содержащей от 6 до 20 атомов углерода, имеющей одно кольцо (например, фенил) или несколько колец (например, бифенил) или множественные сконденсированные (слитые) кольца (например, нафтил, флуоренил и антрил). В некоторых вариантах реализации изобретения арилы включают фенил, флуоренил, нафтил, антрил и тому подобные.
[0041 Если иное не указано в определении для арильного заместителя, такие арильные группы могут быть необязательно замещены 1, 2, 3, 4 или 5 заместителями (в некоторых вариантах реализации изобретения 1, 2 или 3 заместителями), выбранными из группы, состоящей из алкила, алкенила, алкинила, алкокси, циклоалкила, циклоалкенила, циклоалкокси, циклоалкенилокси, ацила, ациламино, ацилокси, амино, замещенного амино, аминокарбонила, алкоксикарбониламино, азидо, циано, галогена, гидрокси, кето, тиокарбонила, карбокси, карбоксиалкила, арилтио, гетероарилтио, гетероциклилтио, тиола, алкилтио, арила, арилокси, гетероарила, аминосульфонила, аминокарбониламино, гетероарилокси, гетероциклила, гетероциклоокси, гидроксиамино, алкоксиамино, нитро, -SO-алкила, -SO-циклоалкила, -SO-гетероциклила, -SO-арила, -SO-гетероарила, -SO2-алкила, -SO2-циклоалкила, -SO2-гетероциклила, -SO2-арила и -SO2-гетероарила. Если иное не указано в определении, все заместители могут быть необязательно дополнительно замещены 1, 2 или 3 заместителями, выбранными из алкила, алкенила, алкинила, карбокси, карбоксиалкила, гидрокси, алкокси, галогена, CF3, амино, замещенного амино, циано, циклоалкила, гетероциклила, арила, гетероарила и -S(O)nRa, где Ra представляет собой алкил, арил или гетероарил и n равен 0, 1 или 2.
[0042] Термин «арилокси» относится к группе арил-О-, где арильная группа является такой, как определено выше, и включает необязательно замещенные арильные группы, как и описано выше. Термин «арилтио» относится к группе RS-, где R такой, как определено для арила.
[0043] Термин «арилен» в настоящей заявке относится к бирадикалу «арил», как определено выше, который является двухвалентным в связи с удалением соответствующим образом атома водорода из арила.
[0044] Термин «гетероциклил», «гетероцикл» или «гетероциклический» относится к насыщенной монорадикальной группе, содержащей одно кольцо или несколько конденсированных колец, содержащих от 1 до 40 атомов углерода и от 1 до 10 гетероатомов, и от 1 до 4 гетероатомов в кольце, выбранных из азота, серы, фосфора и/или кислорода.
[0045] Если иное не указано в определении для гетероциклического заместителя, такие гетероциклические группы могут быть необязательно замещены от 1 до 5 заместителями (в некоторых вариантах реализации изобретения 1, 2 или 3 заместителями), выбранными из группы, состоящей из алкила, алкенила, алкинила, алкокси, циклоалкила, циклоалкенила, циклоалкокси, циклоалкенилокси, ацила, ациламино, ацилокси, амино, замещенного амино, аминокарбонила, алкоксикарбониламино, азидо, циано, галогена, гидрокси, кето, тиокарбонила, карбокси, карбоксиалкила, арилтио, гетероарилтио, гетероциклилтио, тиола, алкилтио, арила, арилокси, гетероарила, аминосульфонила, аминокарбониламино, гетероарилокси, гетероциклила, гетероциклоокси, гидроксиамино, алкоксиамино, нитро, -SO-алкила, -SO-циклоалкила, -SO-гетероциклила, -SO-арила, -SO-гетероарила, -SO2-алкила, -SO2-циклоалкила, -SO2-гетероциклила, -SO2-арила и -SO2-гетероарила. Если иное не указано в определении, все заместители могут быть необязательно дополнительно замещены 1, 2 или 3 заместителями, выбранными из алкила, алкенила, алкинила, карбокси, карбоксиалкила, аминокарбонила, гидрокси, алкокси, галогена, CF3, амино, замещенного амино, циано, циклоалкила, гетероциклила, арила, гетероарила и -S(O)nRa, где Ra представляет собой алкил, арил или гетероарил и n равен 0, 1 или 2. Примеры гетероциклических групп включают тетрагидрофуранил, морфолино, пиперидинил, и т.п.
[0046] Термин «гетероциклоокси» относится к группе -О-гетероциклила.
[0047] Термин «гетероарил» относится к группе, содержащей одно или несколько колец, содержащих от 1 до 15 атомов углерода и от 1 до 4 гетероатомов, выбранных из кислорода, азота и серы в пределах, по меньшей мере одного кольца. Термин «гетероарил» является общим для терминов «ароматический гетероарил» и «частично насыщенный гетероарил». Термин «ароматический гетероарил» относится к гетероарилу, в котором по меньшей мере одно кольцо является ароматическим, независимо от точки присоединения. Примеры ароматических гетероарилов включают пиррол, тиофен, пиридин, хинолин, птеридин. Термин «частично насыщенный гетероарил» относится к гетероарилу, имеющему структуру, эквивалентную исходному ароматическому гетероарилу, содержащему одну или несколько двойных связей в ароматическом кольце исходного насыщенного ароматического гетероарила. Примеры частично насыщенных гетероарилов включают дигидропиррол, дигидропиридин, хроман, 2-оксо-1,2-дигидропиридин-4-ил, и тому подобные.
[0048] Если иное не указано в определении для гетероарильных заместителей, такие гетероарильные группы могут быть необязательно замещены от 1 до 5 заместителями (в некоторых вариантах реализации изобретения 1, 2 или 3 заместителями), выбранными из группы, состоящей из алкила, алкенила, алкинила, алкокси, циклоалкила, циклоалкенила, циклоалкокси, циклоалкенилокси, ацила, ациламино, ацилокси, амино, замещенного амино, аминокарбонила, алкоксикарбониламино, азидо, циано, галогена, гидрокси, кето, тиокарбонила, карбокси, карбоксиалкила, арилтио, гетероарилтио, гетероциклилтио, тиола, алкилтио, арила, арилокси, гетероарила, аминосульфонила, аминокарбониламино, гетероарилокси, гетероциклила, гетероциклоокси, гидроксиамино, алкоксиамино, нитро, SO-алкила, -SO-циклоалкила, -SO-гетероциклила, -SO-арила, -SO-гетероарила, -SO2-алкила, -SO2-циклоалкила, -SO2-гетероциклила, -SO2-арила и -SO2-гетероарила. Если иное не указано в определении, все заместители могут быть необязательно дополнительно замещены 1, 2 или 3 заместителями, выбранными из алкила, алкенила, алкинила, карбокси, карбоксиалкила, аминокарбонила, гидрокси, алкокси, галогена, CF3, амино, замещенного амино, циано, циклоалкила, гетероциклила, арила, гетероарила, и -S(O)nRa, где Ra представляет собой алкил, арил или гетероарил и n равен 0, 1 или 2. Такие гетероарильные группы могут содержать одно кольцо (например, пиридил или фурил) или несколько сконденсированных колец (например, индолизинил, бензотиазол или бензотиенил). Примеры азотных гетероциклилов и гетероарилов включают, но не ограничиваются ими, пиррол, имидазол, пиразол, пиридин, пиразин, пиримидин, пиридазин, индолизин, изоиндол, индол, индазол, пурин, хинолизин, изохинолин, хинолин, фталазин, нафтилпиридин, хиноксалин, хиназолин, циннолин, птеридин, карбазол, карболин, фенантридин, акридин, фенантролин, изотиазол, феназин, изоксазол, феноксазин, фенотиазин, имидазолидин, имидазолин и т.п., а также N-алкокси-азотсодержащие гетероарильные соединения.
[0049] Термин «гетероарилокси» относится к группе гетероарил-О-.
[0050] Термин «амино» относится к группе -NH2.
[0051] Термин «замещенный амино» относится к группе -NRR, где каждый R независимо выбран из группы, включающей водород, алкил, циклоалкил, арил, гетероарил и гетероциклил при условии, что обе группы R не являются водородом или группе -YZ, где Y представляет собой необязательно замещенный алкилен и Z представляет собой алкенил, циклоалкенил или алкинил. Если иное не указано в определении, все заместители могут быть необязательно дополнительно замещены 1, 2 или 3 заместителями, выбранными из алкила, алкенила, алкинила, карбокси, карбоксиалкила, аминокарбонила, гидрокси, алкокси, галогена, CF3, амино, замещенного амино, циано, циклоалкила, гетероциклила, арила, гетероарила, и -S(O)nRa, где Ra представляет собой алкил, арил или гетероарил и n равен 0, 1 или 2.
[0052] Термин «алкиламин» относится к R-NH2, где R представляет собой возможно замещенный алкил.
[0053] Термин «диалкиламин» относится к R-NHR, где каждый R независимо представляет собой необязательно замещенный алкил.
[0054] Термин «триалкиламин» относится к группе NR3, где каждый из R независимо представляет собой необязательно замещенный алкил.
[0055] Термин «циано» относится к группе -CN.
[0056] Термин «азидо» относится к группе 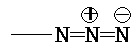 .
.
[0057] Термин «кето» или «оксо» относится к группе =O.
[0058] Термин «карбокси» относится к группе -С(O)-ОН.
[0059] Термин «сложный эфир» или «карбоксиэфир» относится к группе -C(O)OR, где R представляет собой алкил, циклоалкил, арил, гетероарил или гетероциклил, которые возможно могут быть дополнительно замещены алкилом, алкокси, галогеном, CF3, амино, замещенным амино, циано или -S(O)nRa, где Ra представляет собой алкил, арил или гетероарил, а n равен 0, 1 или 2.
[0060] Термин «ацил» означает группу -C(O)R, в которой R приставляет собой водород, возможно замещенный алкил, возможно замещенный циклоалкил, возможно замещенный гетероциклил, возможно замещенный арил или возможно замещенный гетероарил.
[0061] Термин «карбоксиалкил» относится к группам -С(O)O-алкил или -С(O)O-циклоалкил, причем алкил и циклоалкил соответствуют определению, данному в настоящей заявке, и возможно дополнительно могут быть замещены алкилом, алкенилом, алкинилом, карбокси, карбоксиалкилом, аминокарбонилом, гидрокси, алкокси, галогеном, CF3, амино, замещенным амино, циано, циклоалкилом, гетероциклилом, арилом, гетероарилом и -S(O)nRa, где Ra представляет собой алкил, арил или гетероарил и n представляет собой 0, 1 или 2.
[0062] Термин «аминокарбонил» относится к группе -C(O)NRR, где каждый из R независимо представляет собой водород, алкил, циклоалкил, арил, гетероарил, гетероциклил или где обе R группы объединены с образованием гетероциклической группы (например, морфолино). Если иное не указано в определении, все заместители могут быть необязательно дополнительно замещены 1, 2 или 3 заместителями, выбранными из алкила, алкенила, алкинила, карбокси, карбоксиалкила, аминокарбонила, гидрокси, алкокси, галогена, CF3, амино, замещенного амино, циано, циклоалкила, гетероциклила, арила, гетероарила, и -S(O)nRa, где Ra представляет собой алкил, арил или гетероарил и n равен 0, 1 или 2.
[0063] Термин «ацилокси» относится к группам -ОС(O)-алкил, -ОС(O)-циклоалкил, -ОС(O)-арил, -ОС(O)-гетероарил и -ОС(O)-гетероциклил. Если иное не указано в определении, все заместители могут быть необязательно дополнительно замещены 1, 2 или 3 заместителями, выбранными из алкила, алкенила, алкинила, карбокси, карбоксиалкила, аминокарбонила, гидрокси, алкокси, галогена, CF3, амино, замещенного амино, циано, циклоалкила, гетероциклила, арила, гетероарила, и -S(O)nRa, где Ra представляет собой алкил, арил или гетероарил и n равен 0, 1 или 2.
[0064] Термин «ациламино» относится к группе -NRC(O)R, где каждый R независимо представляет собой водород, алкил, циклоалкил, арил, гетероарил или гетероциклил. Все заместители могут быть необязательно дополнительно замещены алкилом, алкенилом, алкинилом, карбокси, карбоксиалкилом, аминокарбонилом, гидрокси, алкокси, галогеном, CF3, амино, замещенным амино, циано, циклоалкилом, гетероциклилом, арилом, гетероарилом, и - S(O)nRa, где Ra представляет собой алкил, арил или гетероарил и n равен 0, 1 или 2.
[0065] Термин «алкоксикарбониламино» относится к группе - N(Rd)C(O)OR, где R представляет собой необязательно замещенный алкил, и Rd представляет собой водород или необязательно замещенный алкил.
[0066] Термин «аминокарбониламино» относится к группе - NRcC(O)NRR, в которой Rc представляет собой водород или необязательно замещенный алкил и каждый R независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, арила, гетероарила и гетероциклила. Если иное не указано в определении, все заместители могут быть необязательно дополнительно замещены 1, 2 или 3 заместителями, выбранными из группы, состоящей из алкила, алкенила, алкинила, алкокси, циклоалкила, циклоалкенила, циклоалкокси, циклоалкенилокси, ацила, ациламино, ацилокси, амино, замещенного амино, аминокарбонила, алкоксикарбониламино, азидо, циано, галогена, гидрокси, кето, тиокарбонила, карбокси, карбоксиалкила, арилтио, гетероарилтио, гетероциклилтио, тиола, алкилтио, арила, арилокси, гетероарила, аминосульфонила, аминокарбониламино, гетероарилокси, гетероциклила, гетероциклоокси, гидроксиамино, алкоксиамино, нитро, -SO-алкила, -SO-циклоалкила, -SO-гетероциклила, -SO-арила, -SO-гетероарила, -SO2-алкила, -SO2-циклоалкила, -SO2-гетероциклила, -SO2-арила и -SO2-гетероарила.
[0067] Термин «тиол» относится к группе -SH.
[0068] Термин «тиокарбонил» относится к группе = S.
[0069] Термин «алкилтио» относится к группе -S-алкил.
[0070] Термин «замещенный алкилтио» относится к группе -S-замещенный алкил.
[0071] Термин «гетероциклотио» относится к группе -S-гетероциклил.
[0072] Термин «арилтио» относится к группе -S-арил.
[0073] Термин «гетероарилтиол» относится к группе -S-гетероарил, причем гетероарильная группа представляет собой группу, описанную выше, включая возможно замещенные гетероарильные группы, которые тоже описаны выше.
[0074] Термин «сульфоксид» относится к группе -S(O)R, где R представляет собой алкил, циклоалкил, гетероциклил, арил или гетероарил. «Замещенный сульфоксид» относится к группе - S(O)R, где R представляет собой замещенный алкил, замещенный циклоалкил, замещенный гетероциклил, замещенный арил или замещенный гетероарил, как определено в настоящей заявке.
[0075] Термин «сульфон» относится к группе -S(O)2R, где R представляет собой алкил, циклоалкил, гетероциклил, арил или гетероарил. «Замещенный сульфон» относится к группе -S(O)2R, где R представляет собой замещенный алкил, замещенный циклоалкил, замещенный гетероциклил, замещенный арил или замещенный гетероарил, как определено в настоящей заявке.
[0076] Термин «аминосульфонил» относится к группе -S(O)2NRR, где каждый R независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, арила, гетероарила и гетероциклила. Если иное не указано в определении, все заместители могут быть необязательно дополнительно замещены 1, 2 или 3 заместителями, выбранными из группы, состоящей из алкила, алкенила, алкинила, алкокси, циклоалкила, циклоалкенила, циклоалкокси, циклоалкенилокси, ацила, ациламино, ацилокси, амино, замещенного амино, аминокарбонила, алкоксикарбониламино, азидо, циано, галогена, гидрокси, кето, тиокарбонила, карбокси, карбоксиалкила, арилтио, гетероарилтио, гетероциклилтио, тиола, алкилтио, арила, арилокси, гетероарила, аминосульфонила, аминокарбониламино, гетероарилокси, гетероциклила, гетероциклоокси, гидроксиамино, алкоксиамино, нитро, -SO-алкила, -SO-циклоалкила, -SO-гетероциклила, -SO-арила, -SO-гетероарила, -SO2-алкила, -SO2-циклоалкила, -SO2-гетероциклила, -SO2-арила и -SO2-гетероарила.
[0077] Термин «гидроксиамино» относится к группе -NHOH.
[0078] Термин «алкоксиамино» относится к группе -NHOR, где R представляет собой возможно замещенный алкил.
[0079] Термин «галоген» или «гало» относится к фтору, брому, хлору и йоду.
[0080] «Необязательный» или «необязательно» означает, что описываемое далее событие или обстоятельство может произойти, а может не произойти, и настоящее описание охватывает случаи, когда указанное событие или обстоятельство происходит или не происходит.
[0081] «Замещенная» группа включает варианты реализации изобретения, в которых монорадикальный заместитель связан с одним атомом замещенной группы (например, формирование разветвления), а также включает варианты реализации изобретения, в которых заместитель может быть дирадикальной мостиковой группой, связанной с двумя смежными атомами замещенной группы, тем самым образуя сконденсированное кольцо на замещенной группе.
[0082] В случае, когда указанная группа (остаток) в настоящей заявке описана как присоединенная ко второй группе, и место присоединения не определено явно, указанная группа может быть присоединена в любом доступном положении указанной группы к любому доступному положению второй группы. Например, «низший алкил-замещенный фенил», где положение присоединения не указано явно, может содержать присоединение любого доступного положения низшей алкильной группы к любому доступному положению фенильной группы. В связи с вышеуказанным «доступное положение» представляет собой положение в группе, в котором водород в указанной группе может быть заменен заместителем.
[0083] Необходимо понимать, что во всех группах заместителей, определенных выше, полимеры, характеризующиеся наличием заместителей, содержащих дополнительные заместители указанных заместителей (например, замещенный арил, содержащий в качестве заместителя замещенную арильную группу, которая также замещена замещенной арильной группой и т.д.) не включены в объем настоящей заявки. Также не включены в объем настоящей заявки бесконечные числа заместителей, независимо от того, являются ли заместители одинаковыми или разными. В таких случаях максимальное количество таких заместителей - три. Каждое из приведенных выше определений ограничено таким образом, что, например, замещенные арильные группы ограничены группами -замещенный арил-(замещенный арил)-замещенный арил.
[0084] Подразумевается, что соединение заданной Формулы (например, соединения Формулы I) охватывает соединения согласно настоящему изобретению, фармацевтически приемлемые соли, фармацевтически приемлемые сложные эфиры, изомеры, таутомеры, сольваты, изотопы, гидраты, и пролекарства таких соединений. Кроме того, соединения согласно настоящему раскрытию могут иметь один или более центров асимметрии и могут быть получены в виде рацемической смеси или в виде отдельных энантиомеров или диастереомеров. Количество стереоизомеров, присутствующих в любом заданном соединении заданной формулы зависит от количества присутствующих центров асимметрии (возможно получение 2n стереоизомеров, где n представляет собой количество центров асимметрии). Отдельные стереоизомеры могут быть получены путем разделения рацмической или нерацемической смеси промежуточного соединения на какой-либо подходящей стадии синтеза, или путем разделения соединения традиционными способами. Отдельные стереоизомеры (включая отдельные энантиомеры и диастереоизомеры) так же, как и рацемические или нерацемические смеси стереоизомеров включены в объем настоящего раскрытия, причем предполагается, что все стереоизомеры в настоящем описании изображены в виде структурных формул, если отдельно не указано иное.
[0085] «Изомеры» представляют собой различные соединения с одинаково молекулярной формулой. Изомеры включают стереоизомеры, энантиомеры и диастереоизомеры.
[0086] «Стереоизомеры» представляют собой изомеры, которые отличаются лишь расположением атомов в пространстве.
[0087] «Энантиомеры» представляют собой пары стереоизомеров, которые являются не совпадающим при наложении зеркальным отображением друг друга. Смесь пары энантиомеров в соотношении 1:1 является «рацемической» смесью. Термин «(±)» применяется при необходимости для обозначения рацемической смеси.
[0088] «Диастереоизомеры» являются стереоизомерами, которые имеют по меньшей мере два асимметрических атома, но которые не являются зеркальным отображением друг друга.
[0089] Абсолютная стереохимия определена в соответствии с системой RS Кана Ингольда Прелога. В случае, когда соединение представляет собой чистый энантиомер, стереохимия каждого хирального атома углерода может быть определена либо как R, либо как S. Разделенные соединения, абсолютная конфигурация которых неизвестна, обозначены (+) или (-), в зависимости от направления (право- или левовращающий), в котором они вращают плоскость поляризованного света при длине волны линии D натриевого спектра.
[0090] Термин «защитная группа амина» также является хорошо понятным специалисту в области органической химии синтеза и относится к группе, которая может быть селективно присоединена и удалена из функциональной группы подходящего амина. Область, относящаяся к методологии защитных групп, перспективна и многие защитные группы амина и способы их применения хорошо известны в уровне техники, как например, описанные в работе уважаемых авторов на эту тему, P.G.М. Wuts and Т.W. Greene, Greene's Protective Groups in Organic Synthesis, 4th Edition (Wiley, 2006).
[0091] Термин «борилирующий агент» также хорошо понятен в области органического синтеза как реагент, подходящий для включения любого из широкого круга боронатных остатков в подходящий субстрат. Неограничивающие примеры борилирующих агентов и связанных с ними методов синтеза представлены в Т. Ishiyama et al., J. Org. Chem. 1995, 60, 7508-7510.
[0092] Термин «гидрокси» или «гидроксил» относится к группе -ОН.
[0093] При наличии несоответствия между изображенной структурой и названием, указанным для структуры, преимущество отдается изображенной структуре. Кроме того, если стереохимия структуры или части структуры не показана, например, жирным шрифтом, курсивом или пунктирными линиями, структуру или часть структуры следует интерпретировать как охватывающую все стереоизомеры указанного соединения.
[0094] Термин «сольват» относится к комплексу, образованному при комбинировании соединения Формулы I или любой другой Формулы, описанной в настоящей заявке, и растворителя.
[0095] Термин «гидрат» относится к комплексу, образованному посредством комбинирования Соединения Формулы I или любой другой формулы, описанной в настоящей заявке, с водой.
[0096] Термин «пролекарство» относится к соединениям Формулы I или любой другой Формулы, описанной в настоящей заявке, содержащим химические группы, которые в условиях in vivo могут превращаться и/или отделяться от остатка молекулы с обеспечением активного лекарственного вещества, их фармацевтически приемлемой соли или биологически активному метаболиту.
[0097] Предполагается также, что любая формула или структура, приведенная в настоящей заявке, включая соединения Формулы I или любой другой Формулы, описанной в настоящей заявке, представляют собой как немеченые, так и меченые изотопами формы указанных соединений. Меченые изотопами соединения имеют структуры, представленные приведенными в настоящей заявке формулами, за исключением того, что один или более атомов заменены атомами с выбранной атомной массой или массовым числом. Примеры изотопов, которые могут быть включены в соединения согласно настоящему раскрытию, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора или хлора, как например, но не ограничиваясь указанными, 2Н (дейтерий, D), 3Н (тритий), 11С, 13С, 14С, 15N, 18F, 31Р, 32Р, 35S, 36Cl и 125I. Различные меченые изотопами соединения согласно настоящему раскрытию, например, такие, в которые включены радиоактивные изотопы, такие как 3Н, 13С и 14С. Такие меченые изотопами соединения могут применяться для проведения метаболических исследований, исследований кинетических реакций, детектирования и методов визуализации, например, позитронно-эмиссионной томографии (PET) или однофотонной эмиссионной компьютерной томографии (SPECT), включая определение распределения лекарственных средств или ткани субстрата, или радиоактивное лечение пациентов.
[0098] Настоящее изобретение также включает Соединение Формулы I или любой другой Формулы, описанной в настоящей заявке, в котором от 1 до "n" атомов водорода, присоединенных к атому углерода, заменены дейтерием, где n представляет собой количество атомов водорода в молекуле. Такие соединения проявляют повышенное сопротивление метаболизму и, таким образом, являются подходящими для увеличения времени полувыведения любого соединения Формулы I при введении млекопитающему. См., например, Foster, "Deuterium Isotope Effects in Studies of Drug Metabolism", Trends Pharmacol. Sci. 5(12): 524-527 (1984). Такие соединения получают способами, хорошо известными в области техники, например, при использовании исходных веществ, в которых один или более атомов водорода заменены дейтерием.
[0099] Меченые дейтерием или замещенные дейтерием терапевтические соединения согласно настоящему изобретению могут обладать улучшенными свойствами DMPK (метаболизм и фармакокинетика лекарственных средств), относящимися к распределению, метаболизму и выделению (ADME). Замещение более тяжелыми изотопами, такими как дейтерий, может обеспечивать определенную терапевтическую выгоду в результате большей метаболической стабильности, например, увеличенное время полувыведения в условиях in vivo или снижение требуемой дозы. Соединение, меченное 18F, может быть подходящим для исследований методом позитронно-эмиссионной томографии (PET) или методом однофотонной эмиссионной компьютерной томографии (SPECT). Соединения, меченные изотопами согласно настоящему описанию и их пролекарства, как правило, могут быть получены путем выполнения методик, описанных в схемах или примерах, а описанные ниже лекарственные препараты - замещением немеченых изотопами реагентов легко доступными мечеными изотопами реагентами. Кроме того, замещение более тяжелыми изотопами, в частности, дейтерием (т.е., 2Н или D) может обеспечивать определенную терапевтическую выгоду в результате большей метаболической стабильности, например, увеличенное время полувыведения в условиях in vivo или снижение требуемой дозы или улучшение терапевтического индекса. Следует понимать, что дейтерий в данном контексте рассматривается как заместитель соединения Формулы I или любой другой Формулы, описанной в настоящей заявке.
[0100] Концентрацию такого более тяжелого изотопа, в частности, дейтерия, можно определить посредством коэффициента обогащения изотопов. Предполагается, что в соединениях согласно настоящему изобретению любой атом, не обозначенный как конкретный изотоп, представляет собой любой стабильный изотоп указанного атома. Если не указано иное, в случае, когда положение конкретно обозначено как «Н» или «водород», следует понимать, что в указанном положении водород содержится в природной изотопической композиции. Соответственно, предполагается, что в соединениях согласно настоящему изобретению любой атом, конкретно обозначенный как дейтерий (D), представляет собой дейтерий.
[0101] Во многих случаях соединения согласно настоящему раскрытию способны образовывать кислотные и/или основные соли в силу наличия амино и/или карбоксильных групп или групп, подобных указанным.
[0102] Термин «фармацевтически приемлемая соль» заданного соединения относится к солям, которые сохраняют биологическую эффективность и свойства заданного соединения и не являются нежелательными с биологической или иной точки зрения. См.: P. Heinrich Stahl and Camille G. Wermuth (Eds.) Pharmaceutical Salts: Properties, Selection, and Use (International Union of Pure and Applied Chemistry), Wiley-VCH; 2oe исправленное издание (16 мая 2011 г.). Фармацевтически приемлемые соли присоединения основания могут быть получены с использованием неорганических или органических оснований. Соли, полученные из неорганических оснований включают, только в качестве примера, соли натрия, калия, лития, аммония, кальция и магния. Соли, полученные из органических оснований, включают, но не ограничиваются указанными, соли первичных, вторичных и третичных аминов, таких как алкиламины, диалкиламины, триалкиламины, замещенные алкиламины, дизамещенные алкиламины, тризамещенные алкиламины, алкениламины, диалкениламины, триалкениламины, замещенные алкениламины, дизамещенные алкениламины, тризамещенные алкениламины, циклоалкиламины, дициклоалкиламины, трициклоалкиламины, замещенные циклоалкиламины, дизамещенные циклоалкиламины, тризамещенные циклоалкиламины, циклоалкениламины, дициклоалкениламины, трициклоалкениламины, замещенные циклоалкениламины, дизамещенные циклоалкениламины, тризамещенные циклоалкениламины, ариламины, диариламины, триариламины, гетероариламины, дигетероариламины, тригетероариламины, гетероциклические амины, дигетероциклические амины, тригетероциклические амины, смешанные ди- и три-амины, где по меньшей мере два заместителя амина являются различными и выбраны из группы, состоящей из алкила, замещенного алкила, алкенила, замещенного алкенила, циклоалкила, замещенного циклоалкила, циклоалкенила, замещенного циклоалкенила, арила, гетероарила, гетероциклила и подобных. Также сюда включены амины, где два или три заместителя совместно с атомом азота аминогруппы образуют гетероциклическую или гетероарильную группу. Амины представляют собой соединения общей структуры N(R30)(R3l)(R32), где два из трех заместителей у атома азота монозамещенных аминов (R30, R31 и R32) представляют собой водород, один из трех заместителей у атома азота дизамещенных аминов (R30, R31 и R32) представляет собой водород, в тризамещенных аминах ни один из трех заместителей у атома азота не является водородом, и R30, R31 и R32 выбраны из множества заместителей, таких как водород, возможно замещенный алкил, арил, гетероарил, циклоалкил, циклоалкенил, гетероциклил и т.п. Упомянутые выше амины относятся к соединениям, в которых один, два или три заместителя у атома азота являются такими, как указано в названии. Например, термин «циклоалкенил-амин» относится к циклоалкенил-NH2, причем «циклоалкенил» является таким, как определено выше. Термин «дигетероариламин» относится к NH(гетероарилу)2, причем «гетероарил» является таким, как описано в настоящей заявке, и т.д..
[0103] Конкретные примеры подходящих аминов включают, только в качестве примера, изопропиламин, триметиламин, диэтиламин, три(изопропил)амин, три(н-пропил)амин, этаноламин, 2-диметиламиноэтанол, трометамин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, N-алкилглюкамины, теобромин, пурины, пиперазин, пиперидин, морфолин, N-этилпиперидин и тому подобные.
[0104] Фармацевтически приемлемые соли присоединения кислоты могут быть получены из неорганических и органических кислот. Соли, полученные из неорганических кислот, включают соляную кислоту, бромистоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и тому подобное. Соли, полученные из органических кислот, включают уксусную кислоту, пропионовую кислоту, гликолевую кислоту, пировиноградную кислоту, щавелевую кислоту, яблочную кислоту, малоновую кислоту, янтарную кислоту, малеиновую кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту, коричную кислоту, миндальную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, п-толуол-сульфоновую кислоту, салициловую кислоту и т.п.
[0105] Кроме того, сокращения, используемые в настоящей заявке, предпочтительно имеют следующие значения:
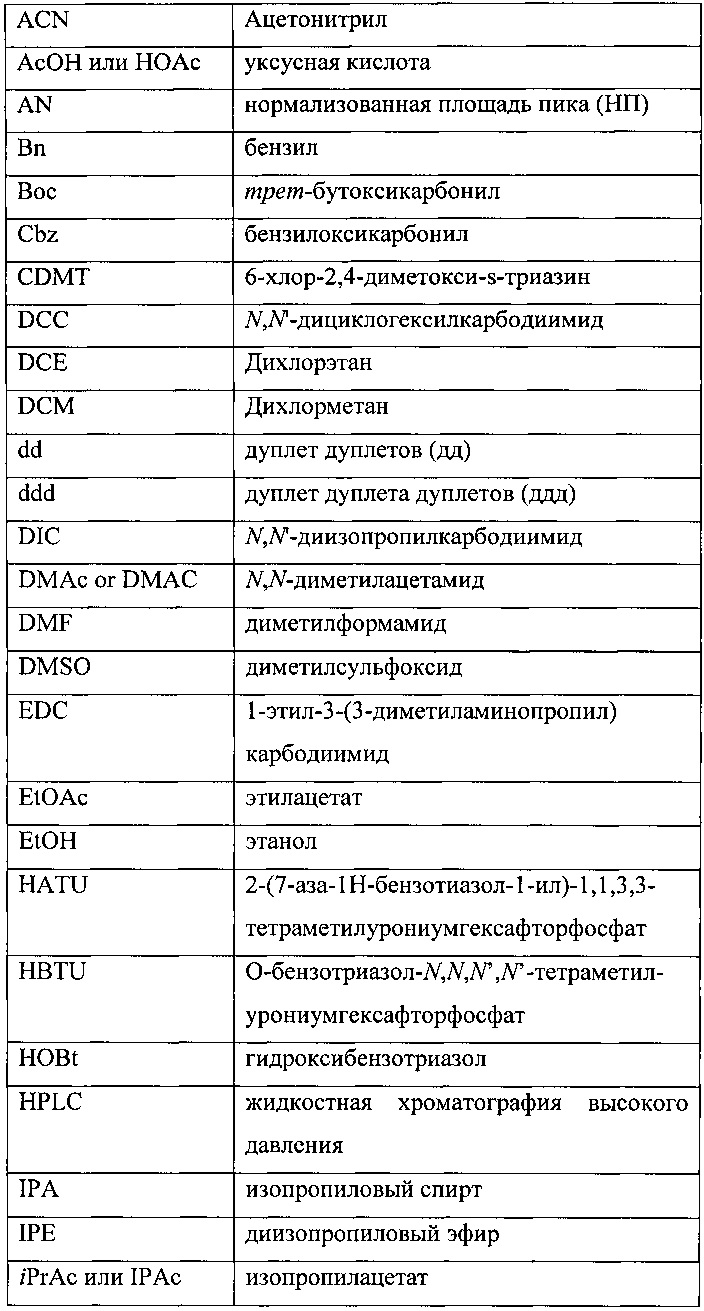

Способы
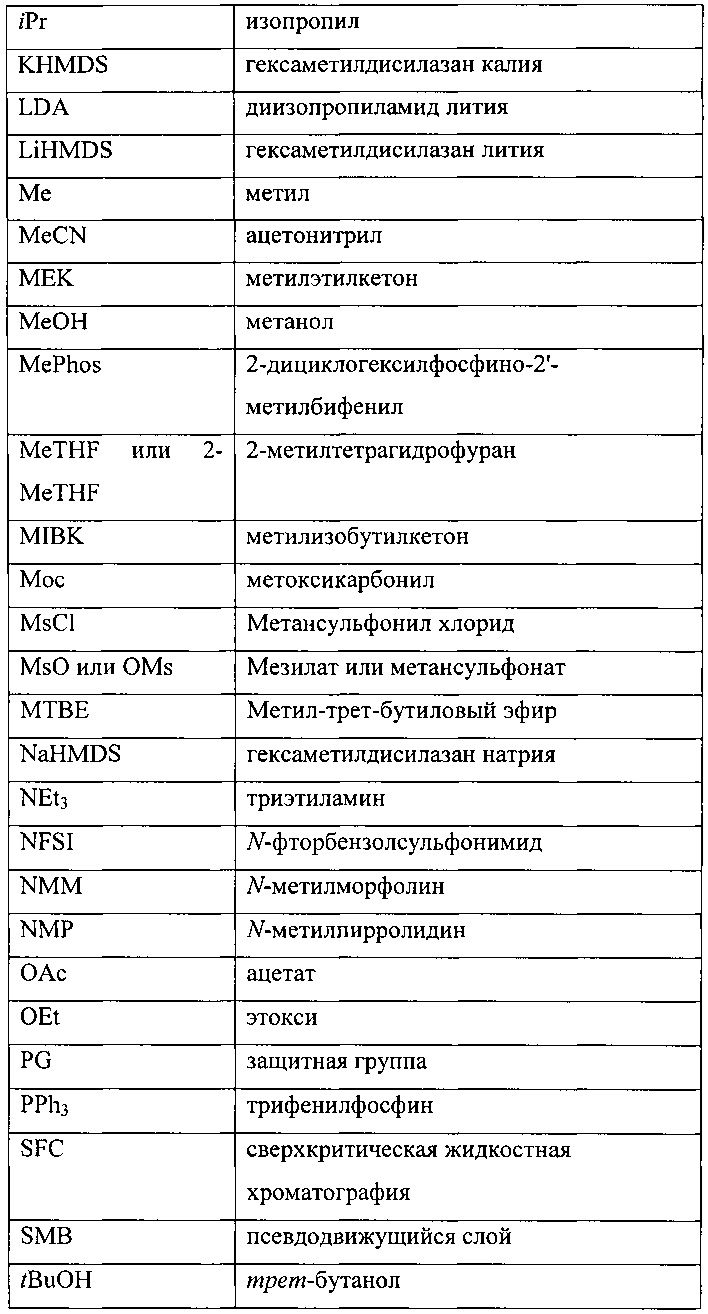
[0106] Как в целом описано выше, в некоторых вариантах реализации настоящего изобретения предложены способы получения соединения формулы I. Стадия А, относящаяся к сочетанию соединений, имеющих формулы (i) и (ii) соответственно, также обеспечивает дополнительные варианты реализации изобретения. Например, в одном варианте реализации изобретения соединение формулы (i) представляет собой боронатное соединение, участвующее в реакции сочетания, где Y представляет собой -B(OR)(OR') и, таким образом, заместитель Z в формуле (ii) представляет собой Br.
[0107] Одним из преимуществ настоящего изобретения является другой вариант реализации изобретения, где соединение формулы (i) получают в условиях in situ, в результате чего, последующая стадия сочетания А происходит в том же реакторе. В указанном варианте реализации изобретения способ включает последовательное приведение соединения формулы (а)
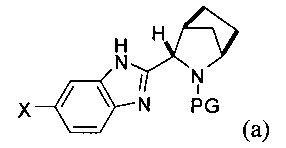
в контакт с источником палладия, а затем с борилирующим агентом, содержащим фрагмент
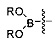 ,
,
в присутствии второго основания, причем соединение формулы (i) образуется в условиях in situ, и
причем PG является таким, как описано выше, X представляет собой галоген, выбранный из О, Br, и I. В некоторых вариантах реализации изобретения борилирующий реагент выбирают из бис(пинаколато)дибора и бис(неопентилгликолято)дибора. Таким образом, в одном варианте реализации настоящего изобретения борилирующий агент представляет собой бис(неопентилгликолято)дибор. В другом варианте реализации настоящего изобретения борилирующий агент представляет собой бис(пинаколато)дибор.
[0108] В другом варианте реализации способа получения соединения формулы (i), галоген X представляет собой Br, а защитная группа PG представляет собой трет-бутоксикарбонил.
[0109] В некоторых вариантах реализации способа, описанного выше, металлический катализатор выбирают из соединений Pd(0) и Pd(II). Конкретная степень окисления Pd неважна, поскольку каталитически активные соединения Pd(0) получают в соответствии с традиционными условиями реакции сочетания Сузуки. Так, например, в некоторых вариантах реализации изобретения металлический катализатор представляет собой PdCl2[P(t-Bu)2Ph]2. В других вариантах реализации изобретения катализатор представляет собой Pd(OAc)2/2-дициклогексилфосфино-2'-метилбифенил. В еще одном варианте реализации изобретения катализатор представляет собой дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладий(II) дихлорметановый аддукт.
[0110] Приемлемы также и другие металлические катализаторы, такие как катализаторы, полученные из Pd или Ni в комбинации с лигандами, добавляемыми в реакционную смесь. Традиционные лиганды включают 1,1'-бис(дифенилфосфин)ферроцен, трифенилфосфин, три(2-метоксифенил)фосфин, трициклогексилфосфин, 2-(дициклогексилфосфино)бифенил, 2-дициклогексилфосфин-2'-метилбифенил, 2-(дициклогексилфосфино)-2'-(N,N-диметиламино)бифенил, 2-(ди-трет-бутил фосфино)-2'-(N,N-циметиламино)бифенил, дициклогексил (2,2-дифенил-1-метилвинил)фосфин, бис(2-дициклогексилфосфинофенил)эфир, дициклогексил(4-(N,N-диметиламино)фенил)фосфин, 2-дициклогексилфосфино-2'-фторбифенил, 2-дициклогексилфосфино-2',6'-дифторфенил.
[0111] В качестве альтернативы можно использовать приготовленные системы металл/лиганд, такие как дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладий(II), бис[ди-трет-бутил(4-диметиламинофенил)фосфино]палладий(II) хлорид и бис[ди-(трет-бутил)(4-трифторметилфенил)фосфин]палладий(II) хлорид. Все указанные возможности включены в настоящее изобретение.
[0112] Процесс борилирования для получения соединения формулы (i), как описано выше, выполняется в присутствии второго основания, которое необязательно является идентичным основанию, использованному на стадии А. Таким образом, в одном варианте реализации изобретения второе основание представляет собой пропионатную соль, такую как пропионат калия. Другие основания также являются подходящими для этих целей и включают ацетаты, такие как ацетат натрия, калия или ацетат цезия, и фосфаты, такие как фосфат натрия или калия.
[0113] Еще в одном варианте реализации изобретения предложен дополнительный способ получения соединения формулы I:
или его фармацевтически приемлемой соли. В указанном варианте реализации изобретения способ включает стадии (1) последовательного приведения соединения формулы (а')
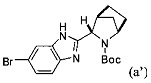
в контакт с каталитически эффективным количеством PdCl2[P(t-Bu)2Ph]2 и бис(неопентилгликолят)дибора в присутствии пропионата калия с получением реакционной смеси, содержащей соединение формулы (ia):
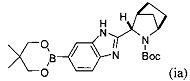 ;
;
[0114] Стадия (2) способа представляет собой приведение реакционной смеси, полученной на стадии (1), в контакт с соединением формулы (ii'):
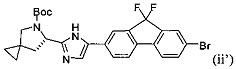
и фосфатом калия с получением соединения формулы (iii'):
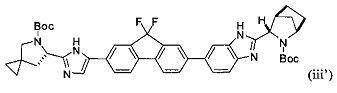 .
.
[0115] В некоторых вариантах реализации изобретения соединение формулы (iii') необязательно приводят в контакт с щавелевой кислотой с получением оксалатной соли согласно формуле (iii''):
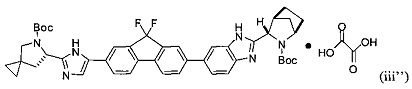 .
.
[0116] Стадия (3) способа представляет собой приведение соединения формулы (iii') или формулы (iii'') в контакт с HCl с получением соединения формулы (iv'):
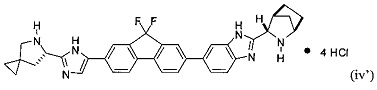 .
.
[0117] И наконец, стадия (4) представляет собой приведение соединения формулы (iv') в контакт с (S)-2-(метоксикарбониламино)-3-метилбутановой кислотой:
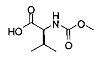
с получением соединения формулы I. В структурных формулах, изображенных выше, «Boc» в каждом случае представляет трет-бутоксикарбонил. В вариациях указанной реакции могут быть использованы другие соли, соответствующие соединению iv', исходя из способов получения солей, описанных в настоящей заявке. В некоторых вариантах реализации изобретения стадия (4) выполняется с использованием свободного основания соединения iv'.
[0118] В альтернативном варианте реализации способ, описанный выше, может быть выполнен с использованием на стадии (1) борилированного промежуточного соединения формулы (ib) вместо (ia):
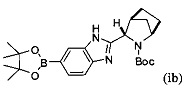 .
.
Соединение (ib) получали посредством реакции соединения (а'), описанного выше, и бис(пинаколато)дибора в присутствии каталитически эффективного количества PdCl2[P(t-Bu)2Ph]2 и пропионата калия.
Соединения
[0119] В других вариантах реализации настоящего изобретения предложены промежуточные соединения, подходящие для применения в способах, описанных в настоящей заявке. Так, например, один вариант реализации представляет собой соединение формулы (ii):
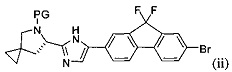
гда PG представляет собой защитную группу амина, описанную выше в настоящей заявке. Типичное соединение формулы (ii) является соединением, в котором PG представляет собой трет-бутоксикарбонил.
[0120] В другом варианте реализации настоящего изобретения предложено соединение формулы (iii'):
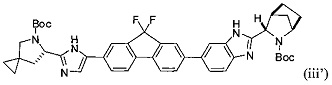
или его оксалатная соль согласно формуле (iii''):
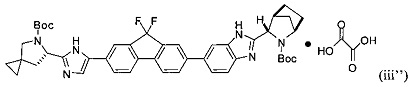 .
.
[0121] В еще одном варианте реализации настоящего изобретения предложено соединение формулы (iv):
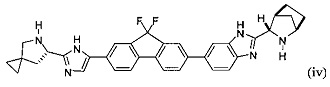
или его соль, гидрат, и/или сольват. Например, конкретный вариант реализации представляет собой соединение формулы (iv'), тетрагидрохлоридную соль:
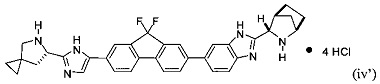 .
.
[0122] Другой вариант реализации представляет собой гидрат соединения формулы (iv') согласно следующей формуле:
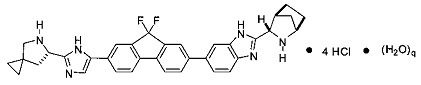 ,
,
где q представляет собой число, дробное или целое, между 0 и 7. Другими словами, гидрат представляет собой соединение, которое может содержать целое или дробное число эквивалентов воды. Например, типичный гидрат представляет собой гидрат, в котором q представляет собой число от 0 до 6, такое как от 2 до 6 или от 5 до 6.
[0123] Объем настоящего изобретения не ограничивается конкретными вариантами реализации, описанными в примерах, которые представлены лишь для иллюстрации нескольких вариантов реализации раскрытия, так же как и настоящее изобретение не ограничивается любыми вариантами реализации, функционально эквивалентными объему настоящего изобретения. В действительности, помимо вариантов, описанных в настоящей заявке, специалистам в области техники будут очевидны различные модификации настоящего изобретения, которые, как предполагается, включены в объем приложенной формулы изобретения. Поэтому необходимо отметить, что один или более атомов водорода или метальных групп могут быть опущены на изображенных структурах в соответствии с принятыми сокращенными обозначениями таких органических соединений, но любой специалист в области органической химии без труда определит наличие таких групп.
ПРИМЕРЫ
I. Получение исходных веществ

А. Циклопропанирование с получением соединения 2
[0124] К раствору DCM (1,5 л) и н-гептана (0,32 л) добавляли диэтилцинк при температуре окружающей среды (800 мл, 1,0 М в н-гептане). Реакционную смесь охлаждали до 0°С и добавляли раствор соединения 1 (45,0 г) в DCM (250 мл) в течение 10 минут. По завершении добавления реакционную смесь охлаждали до -5°С и вносили хлорйодметан посредством шприцевого насоса (176 г) в течение 3,5 часов. Реакционную смесь перемешивали при -5°С в течение дополнительных 16 часов и гасили медленным добавлением (1 час) водной 1N HCl (1,4 л). После нагревания до 20°С, фазы разделяли и водный слой подвергали обратной экстракции DCM (0,5 л). Объединенные органические слои промывали 10% водным NaCl (1,2 л), фазы разделяли и органический слой концентрировали под вакуумом с получением неочищенного масла, которое очищали методом флэш-хроматографии на силикагеле (50% этилацетат/н-гептан). Желательный продукт выделяли в виде смеси соединений 2 и 3 (46,6 г, 67,5 масс. % соединение 2, 62% выход с учетом поправки).
[0125] В альтернативных вариантах реализации изобретения циклопропанирование также может быть достигнуто с использованием диэтилцинка и дийодметана в различных растворителях. Например, для указанной цели подходят дихлорметан, дихлорэтан, толуол, гексан и их комбинации. Кроме того, циклопропанирование можно проводить при температуре от приблизительно -20°С до приблизительно 20°С, хотя типичный температурный диапазон составляет от -5°С до 0°С.
В. Гидролиз/Йодлактониация
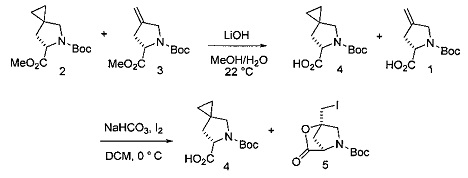
Гидролиз/Йодлактонизация для получения соединения 4:
[0126] Смесь соединений 2 и 3 (161,80 г, 59 масс. % соединения 2) растворяли в МеОН (1,2 л). Добавляли воду, охлаждали смесь до 15°С и добавляли твердый LiOH⋅Н2О (32,8 г). Реакционную смесь нагревали до 25°С и перемешивали в течение 13,5 часов. Затем реакционную смесь концентрировали под вакуумом для удаления МеОН, и добавляли DCM (1 л) и воду (200 мл). Полученную в результате смесь охлаждали до 10°С, и добавляли водную 2N HCl (375 мл). После разделения фаз водный слой экстрагировали DCM (2×500 мл, затем 250 мл). Объединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали под вакуумом до 1,2 л. К раствору добавляли воду (305 мл), NaHCO3 (136 г) и I2 (90,7 г). Реакционную смесь перемешивали при 25°С в течение 40 часов и разбавляли 8% водным NaHCO3 (750 мл), водой (750 мл) и DCM (300 мл). После разделения фаз водный слой экстрагировали водой (1 л). Объединенные водные слои затем промывали изопропилацетатом (300 мл), охлаждали до 0°С и подкисляли добавлением 2N водной HCl (1,1 л). Водную фазу экстрагировали DCM (3×1 л), и объединенный органический слой промывали 10% водным NaHSO3 (2 л) и 10% водным NaCl (1,5 л). Органический слой высушивали над Na2SO4, фильтровали и концентрировали под вакуумом. Полученную в результате твердую фазу дважды растворяли и повторно концентрировали из изопропилацетата (1 л). К твердой фазе добавляли дополнительный изопропилацетат (100 мл), раствор нагревали до 50°С и добавляли н-гептан (800 мл). После охлаждения до 20°С в течение 4 часов суспензию охлаждали до 5°С и выдерживали в течение 2 часов. Продукт собирали посредством фильтрации, промывали р-гептаном (2×150 мл) и высушивали с получением соединения 4 в виде твердого вещества светло-желтого цвета (66,5 г, 74% выход от соединения 2). 1Н ЯМР(400 МГц, d6-DMSO, δ): 12,5 (s, 1Н) 4,23-4,17 (m, 1H), 3,32-3,16 (m, 2H), 2,28-2,22 (m, 1H), 1,74 (dd, J=12,7, 4,3 Гц, 0,6H ротамер1), 1,67 (dd, J=12,7, 3,7 Гц, 0,4H ротамер2), 1,39 (s, 4Н ротамер1), 1,35 (s, 5Н ротамер2), 0,59-0,45 (m, 4Н). 13С ЯМР(100 МГц, d6-DMSO, δ): 173,9, 173,5, 153,4, 153,0, 78,7, 78,6, 59,1, 58,8, 53,7, 53,4, 38,2, 37,5, 28,1, 27,9, 20,5, 19,9, 12,2, 11,5, 8,8, 8,3.
[0127] В некоторых вариантах реализации гидролиз осуществляется при использовании других оснований, таких как гидроксид калия или натрия. В других вариантах реализации для указанной цели подходящими являются комбинации вариантов растворителей, такие как этанол/вода, изопропиловый спирт/вода и THF/вода.
[0128] Гидролиз может осуществляться при температуре от приблизительно 0°С до приблизительно 80°С. Стандартная температура представляет собой температуру окружающей среды, такую как 22°С.
[0129] Для йодлактонизации подходящими являются другие растворители и комбинации растворителей. Например, такие растворители включают дихлорэтан, толуол, эфиры, THF или 2-метил-THF, этилацетат и изопропилацетат.
[0130] Подходящие для йодлактонизации температуры составляют от приблизительно 0°С до приблизительно 50°С. Стандартная традиционная температура составляет 22°C.
[0131] Некоторые варианты реакции йодлактонизации предусматривают использование других оснований, таких как бикарбонат калия (KHCO3), карбонат калия (K2CO3) и карбонат натрия (Na2CO3).
С. Йодирование диола соединения 6
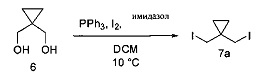
1. Йодирование диола соединения 6 с получением соединения 7а:
[0132] Трифенилфосфин (257,2 г) и имидазол (66,7 г) помещали в реактор. Добавляли DCM (490 мл), начинали перемешивание и охлаждали раствор до 0°С. Йод (249,2 г) добавляли частями в виде твердого вещества в течение одного часа при поддержании внутренней температуры ниже 10°С. По завершении добавления в реактор медленно вносили раствор соединения 6 (50 г) в DCM (113 мл) в течение получаса при поддержании внутренней температуры ниже 10°С. После перемешивания в течение 2,5 часов в реактор вносили водный раствор NaCl (25 г) в воде (225 мл). После разделения фаз нижний органический слой разбавляли н-гептаном (550 мл). Органическую фазу промывали водным раствором сульфита натрия (21 г) в воде (190 мл). После разделения слоев органическую фазу концентрировали до 600 мл с помощью вакуумной дистилляции. Дополнительно вносили н-гептан (550 мл) и смесь повторно концентрировали до 600 мл с помощью вакуумной дистилляции. Полученную в результате суспензию фильтровали через пробку с силикагелем (85 г), суспензионным способом заполненную н-гептаном. Пробку с силикагелем дополнительно промывали н-гептаном (1 л), и затем фильтрат концентрировали с помощью вакуумной дистилляции с получением желаемого продукта 7а в виде бесцветной жидкости (114 г, 70%). 1Н ЯМР(400 МГц, CDCl3) δ 3,33 (s, 2Н), 0,95 (s, 2Н). 13С ЯМР(75 МГц, CDCl3): 19,1, 22,7, 26,0.
[0133] Также в соответствии с другими вариантами реализации изобретения можно оказывать влияние на процесс йодирования с помощью триметилсилилхлорида и йодида натрия в ацетонитриле. Подходящие для указанной реакции температуры составляют от приблизительно -10 до приблизительно 30°С.
2. Вариант способа
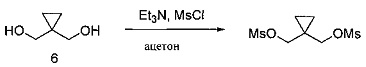
[0134] 1,1-бис(гидроксиметил)циклопропан (40,00 г, 388 ммоль) добавляли в колбу, после чего в колбу добавляли ацетон (400 мл, 15 объемов) и реакцию охлаждали до 0°С. К реакционной смеси добавляли триэтиламин (87,16 г, 861 ммоль, 2,2 эквивалента), а затем медленно добавляли метансульфонилхлорид (98,68 г, 861 ммоль, 2,2 эквивалента) таким образом, чтобы внутренняя температура не превышала 10°С. При добавлении метансульфонилхлорида образовывался белый осадок. По завершении добавления реакцию оставляли для перемешивания при 0°С в течение часа и затем нагревали до 20°С и оставляли для перемешивания в течение 2 часов.
[0135] По завершении реакции добавляли 800 мл (30 объемов) воды и реакцию перемешивали в течение 15 минут. Затем реакционную смесь фильтровали и промывали 100 мл воды. Продукт выделяли на фильтре в виде твердого вещества белого цвета. После высушивания под вакуумом при 20°С, выход составил 85,5 г, 86%. 1Н ЯМР(400 МГц, CDCl3) δ 4,16 (s, 2Н), 3,06 (s, 3Н), 0,83 (s, 2Н).
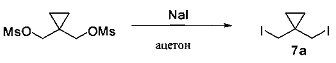
[0136] В круглодонную колбу с верхней мешалкой и датчиком температуры помещали бисмезилатное соединение (26,5 г, 102,5 ммоль) и йодид натрия (46,1 г, 307,5 ммоль, 3 эквивалента), после чего добавляли ацетон (400 мл). Колбу нагревали до внутренней температуры 35°С. Реакционная смесь приобретала желтый/оранжевый цвет и со временем образовывался осадок. Стандартное время реакции составляло 6-7 часов. По завершении реакции реакционную смесь фильтровали и промывали 100 мл ацетона. Раствор концентрировали до ~150 мл и добавляли 300 мл водного 5% раствора сульфита натрия. Добавляли гексан (200 мл) и смесь перемешивали в течение по меньшей мере в течение 15 минут. Оставляли для разделения слоев и верхний органический слой высушивали над сульфатом натрия (20 г). Затем органический слой фильтровали для удаления сульфата натрия и концентрирования масла. Выход 31,0 г, 94%.
D. Алкилирование соединения 8 с получением соединения 9
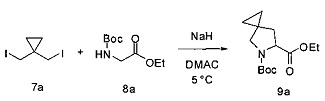
[0137] Гидрид натрия (60,0 г, 3 эквивалента, 60% дисперсия в минеральном масле) и диметилацетамид (600 мл) помещали в колбу и температуру реакции понижали до 0-10°С. Соединение 7а (191,6 г, 1 эквивалент) добавляли в раствор NaH по достижении внутренней температуры приблизительно 5°С. Раствор соединения 8а (121,0 г, 1 эквивалент) в DMAC (600 мл) добавляли в течение 3,5 часов, поддерживая внутреннюю температуру между 0 и 11°С. Раствор перемешивали при 0-10°С и через час отбирали образец для определения конца реакции. Предполагали, что реакция завершена, если оставшееся количество соединения 8а составляло менее 3%. По завершении реакции в течение 2-3 часов медленно добавляли АсОН (50 мл, 1,5 эквивалента), поддерживая температуру в диапазоне 4-9°С. Раствор перемешивали при 0-10°С в течение 12 часов. МТВЕ (1000 мл) и воду (700 мл) добавляли к гашеному раствору. Слои разделяли и водный слой экстрагировали МТВЕ (400 мл). Органические слои объединяли и однократно промывали 15% раствором NaCl (1000 мл), однократно - 5% раствором бикарбоната натрия (900 мл) и однократно солевым раствором (600 мл). Раствор МТВЕ концентрировали до минимального объема. Масло повторно растворяли в ACN (400 мл) и промывали гексаном (200 мл). Фазы разделяли и слой ACN концентрировали до минимального объема и удаляли гексановый слой. Продукт 9а выделяли в виде масла желтого цвета (98 г, 61%). 1Н ЯМР(400 МГц, CDCl3) δ 4,45 (dd, J=8,5, 3,7 Гц, 0,5Н ротамер1), 4,35 (dd, J=8,4, 4,4 Гц, 0,5Н ротамер2), 4,27-4,11 (m, 2Н), 3,44-3,29 (m, 2Н), 2,26 (ddd, J=12,7, 8,4, 4,1 Гц, 1Н), 1,80 (ddd, J=23,5, 12,6, 4,0 Гц, 1Н), 1,58, 1,48-1,40 (m, 9Н), 1,32-1,21 (m, 3Н), 0,68-0,44 (m, 4Н).
[0138] В некоторых вариантах реализации изобретения применяются другие подходящие не нуклеофильные основания. Такие основания включают алкоксиды, такие как трет-бутоксиды лития, натрия или калия.
[0139] Растворители, отличающиеся от DMAC, также приемлемы. Например, такие растворители включают N-метилпирролидин, диметилформамид, и 1,3-диметил-3,4,5,6,-тетрагидро-2-пиримидинон.
[0140] В приведенном выше примере Boc представляет собой защитную группу амина. В некоторых вариантах реализации изобретения, однако, применяется защитная группа, отличная от Boc, такая как метилокси- и изопропилоксикарбонилы. Защитная группа также может представлять собой Cbz.
[0141] Подходящие температуры для выполнения реакции составляют от приблизительно -10°С до приблизительно 40°С.
Альтернативная последовательность алкилирования
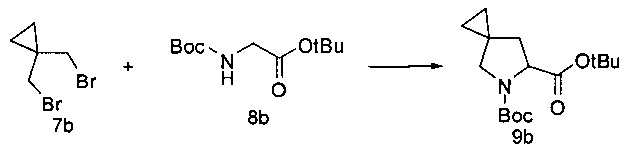
[0142] К охлажденному (до 0°С) раствору N-Boc-глицин-трет-бутилового эфира 8b (10,66 г; 46 ммоль) и дибромида 7b (10,1 г; 44,3 ммоль) в N,N-диметилформамиде (50 мл) добавляли трет-бутилат калия (13,79 г; 122,8 ммоль). Полученную в результате суспензию нагревали до 20°С и перемешивали при указанной температуре в течение 4 часов. Реакционную смесь выливали в раствор MeTHF (100 мл) с водой (100 мл) при перемешивании. Полученный в результате органический раствор последовательно сушили и концентрировали под вакуумом с получением янтарного масла. Неочищенное вещество затем очищали путем хроматографии на силикагеле (90:10 гексан : этилацетат) с получением продукта mpem-бутилового эфира соединения 9b в виде бесцветного масла (5,5 г, 42% выход). Rf: 0,18 (SiO2, 9:1 гексан: этилацетат). 1Н ЯМР(400 МГц, CDCl3): δ 4,33 (ротамер#1: dd, J=8,4, 2,9 Гц, 0,35Н); 4,24 (ротамер#2: dd, J=8,6, 3,3 Гц, 0,65Н); 3,42 (ротамер#2: d, J=10,2 Гц, 0,65Н); 3,36 (ротамер#1: d, J=10,2 Гц, 0,35Н); 3,28 (ротамер#2: d, J=10,2 Гц, 0,65Н); 3,22 (ротамер#1: d, J=10,2 Гц, 0,35Н); 2,31 (ротамер#2: dd, J=12,7, 8,6 Гц, 0,65Н); 2,26 (ротамер#1: dd, J=12,7, 8,6 Гц, 0,35Н); 1,70 (m, 1Н); 1,46 (m, 18Н); 0,54 (m, 4Н). 13С ЯМР (100 МГц, CDCl3, δ): 172,0, 153,9, 80,8, 79,6, 79,4, 60,3, 60,1, 54,1, 53,7, 39,2, 38,3, 28,4, 28,3, 27,9, 20,5, 19,8, 13,4, 13,3, 8,3.
[0143] В некоторых вариантах реализации изобретения использовали другие температуры реакции. Температура реакции может составлять между - 50°С и 50°С.
Е. Гидролиз этилового эфира соединения 9.
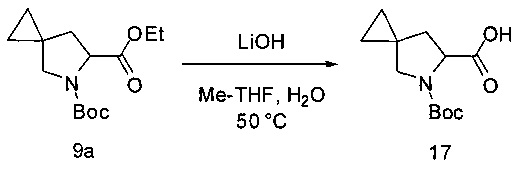
Гидролиз с получением соединения 17
[0144] В колбу, оснащенную верхнеприводной мешалкой, внутренним термометром и линией подачи азота добавляли воду (910 мл), гидроксид лития (284 г, 2,0 эквивалента) и 2-MeTHF (2,0 л). Раствор соединения 9а (911 г) в 2-MeTHF (1,0 л) переносили в колбу, содержащую гидроксид лития. Реакцию нагревали до 50°С до предположительного окончания реакции, которое определяли посредством ВЭЖХ анализа. Затем реакцию охлаждали до 22°С и добавляли к реакционной смеси воду (3,6 л). Слои разделяли и нижний водный слой сохраняли, тогда как верхний органический слой удаляли. 2-MeTHF (4 л) и концентрированную HCl (420 мл) добавляли к водному слою. Слои разделяли и нижний водный слой удаляли. Верхний органический слой концентрировали и выделяли продукт 17 в виде твердого вещества белого цвета (596 г, 71%). Данные, характеризующие соединение 17, совпадают с данными, представленными выше для соединения 4.
[0145] В качестве альтернативы можно применять основания, отличные от LiOH. Таким образом, в некоторых вариантах реализации основание представляет собой гидроксид калия, гидроксид натрия или силанолят калия.
[0146] В других вариантах реализации растворитель также может быть различным. Подходящие растворители включают, например, диалкиловые и циклические эфиры, толуол и дихлорметан.
[0147] Стандартные температуры реакций составляют от приблизительно 0°С до приблизительно 80°С. В приведенном выше примере температура составляла 50°С.
F. Традиционное разделение
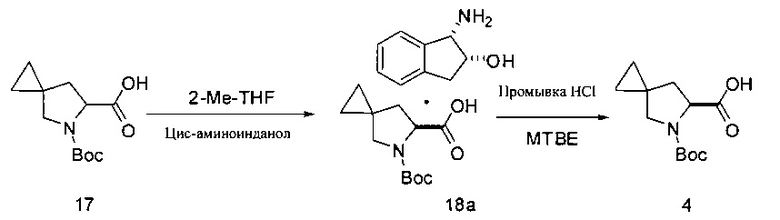
Традиционное разделение с получением Соединения 4:
[0148] Рацемическую карбоновую кислоту 17 (596 г) растворяли в 2-Me-THF (6 л) и затем гомогенный раствор нагревали до 55°С. (1S,2R)-аминоинданол (221 г, 0,6 эквивалента) добавляли в реакционную смесь тремя равными порциями через промежутки времени в 10 минут. После добавления первой порции в раствор вносили затравку в виде соли 18а (0,6 г). После добавления последней порции амина раствор выдерживали при температуре 55°С в течение одного часа. Затем суспензию охлаждали до 22°С со скоростью ~15 градусов в час. По достижении комнатной температуры суспензию фильтровали и отфильтрованный осадок однократно промывали 2-Me-THF (1,2 л). Твердую фазу высушивали при 45°С в вакуумной печи в течение 24 часов. Выделяли Соединение 18а в виде белого твердого вещества (320 г, 33%).
[0149] Твердую фазу 18а растворяли в MeTHF (1,5 л), добавляли 1М HCl (1,0 л) и перемешивали двухфазную смесь в течение 30 минут до полного растворения твердой фазы. Нижний водный слой удаляли и органический слой промывали 1М HCl (1 л), а затем водой (500 мл). Органический слой высушивали над MgSO4 (250 г каждого) в течение 20 минут, фильтровали и промывали отфильтрованный осадок MeTHF. Тот же способ сушки повторяли второй раз, после чего раствор концентрировали до масла с получением соединения 4 (197 г, 100%).
[0150] В других вариантах реализации традиционного способа разделения разделяющие агенты выбраны из (S)-(-)-1-метилбензил амина и (1S,2R)-(+)-норэфедрина, в качестве двух примеров. Альтернативные растворители включают диалкиловые эфиры и циклические эфиры, дихлорметан и алкилацетаты, такие как этилацетат. Подходящие антирастворители включают, например, гексан и гептан. В некоторых вариантах реализации изобретения, температура реакции составляет от приблизительно 0°С до приблизительно 75°С.
Альтернативный пример традиционного разделения:
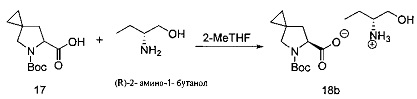
[0151] К раствору рацемической карбоновой кислоты 17 в 2-метилтетрагидрофуране (47 масс% 17; 52,3 г, 217 ммоль) добавляли 2- метилтетрагидрофуран (520 мл). Затем к указанному разбавленному раствору добавляли (R)-2-амино-1-бутанол (13,5 г, 152 ммоль) и полученную в результате суспензию перемешивали при 20°С в течение по меньшей мере 20 часов. Содержимое реакции фильтровали и твердую фазу промывали гептаном (100 мл) и высушивали под вакуумом при 40°С с получением аммоний-карбоксилатного продукта 18b в виде твердого кристаллического вещества белого цвета (22,6 г, 32% выход). 1Н ЯМР(400 МГц, CDCl3, δ): 4,90 (s, широкий, 4Н); 4,25 (ротамер#1: dd, J=8,4, 4,5 Гц, 0,5H); 4,21 (ротамер#2: dd, J=8,2, 5,7 Гц, 1Н); 3,76 (dd, J=11,7, 3,7 Гц, 1,5Н); 3,55 (dd, J=11,7, 6,6 Гц, 1,5Н); 3,43 (ротамер#2: d, J=10,3 Гц, 1Н); 3,34 {ротамер#1: m, 1H); 3,26 (ротамер#2: d, J=10,2 Гц, 1H); 3,10 (dddd, J=6,8, 6,8, 6,8, 3,7 Гц, 1.5Н); 2,19 (ротамер#1: dd, J=12,5, 8,6 Гц, 0,5Н); 2,14 (ротамер#2: dd, J=12,3, 8,2 Гц, 1H); 1,91 (ротамер#2: dd, J=12,5, 5,7 Гц, 1Н); 1,84 (ротамер#1: dd, J=12,5, 4,7 Гц, 0,5Н); 1,67 (m, 3Н); 1,46 (m, 12Н); 1,04 (t, J=7,4 Гц, 4,5Н); 0,58 (m, 6Н). 13С ЯМР(100 МГц, CDCl3, δ): 180,4, 156,5, 80,8, 80,5, 63,5, 63,0, 62,0, 55,9, 55,6, 55,0, 40,8, 40,2, 28,9, 28,8, 23,7, 21,8, 21,4, 12,5, 11,5, 10,9, 10,2, 9,9.
G. Хроматография в псевдодвижущемся слое (ПДС) и гидролиз этилового сложного эфира 19.
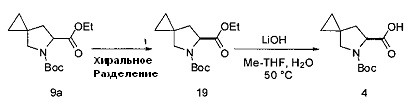
ПДС-хроматография и гидролиз с получением соединения 4
[0152] Соединение 9а отделяли методом хиральной хроматографии с использованием либо Chiralpak® IC, либо Chiralpak® IA с соответствующей подвижной фазой, такой как смесь МТВЕ и гептана. Продукт, полученный в результате разделения в псевдодвижущемся слое концентрировали для получения соединения 19 в виде раствора, который применяли неочищенным на следующей стадии. Выход раствора согласно анализу применяли для определения количества содержащегося в нем продукта. Другие методики хроматографии, известные в области техники, также применимы для выделения соединения 9а. Указанные методики включают различные варианты осуществления высокоэффективной жидкостной хроматографии (ВЭЖХ), такие как прямая и обращенная хиральная хроматография; и колоночная хроматография с нормальными фазами и сверхкритическая жидкостная хроматография (SFC).
[0153] Воду (910 мл), гидроксид лития (284 г, 2,0 эквивалента) и 2-MeTHF (2,0 л) помещали в колбу, оснащенную верхнеприводной мешалкой, внутренним термометром и линией введения азота. Раствор соединения 19 (911 г) в 2-MeTHF (1,0 л) переносили в колбу, содержащую гидроксид лития. Реакцию нагревали до 50°С до предположительного окончания реакции, определяемого посредством анализа ВЭЖХ. Реакцию охлаждали до 22°С и к реакции добавляли воду (3,6 л). Слои разделяли и нижний водный слой оставляли, тогда как верхний органический слой удаляли. К водному слою добавляли 2-MeTHF (4 л) и концентрированную HCl (420 мл). Слои разделяли и нижний слой удаляли. Верхний органический слой концентрировали и продукт выделяли в виде твердого вещества белого цвета (596 г, 71%).
[0154] Подходящие хиральные фазы для стадии SMB, описанной выше, хорошо известны в области техники. Два примера представляют собой Chiralpak® IC и Chiralpak® IA.
[0155] В некоторых вариантах реализации изобретения гидролитический реагент выбирают из вариантов, таких как гидроксид калия, гидроксид натрия и силанолят калия. Системы растворителей также могут быть различны и выбраны из диалкиловых эфиров и циклических эфиров, толуола и дихлорметана, в качестве нескольких примеров. Подходящие температуры реакции составляют от приблизительно 0°С до приблизительно 80°С.
Н. Ферментативное разделение
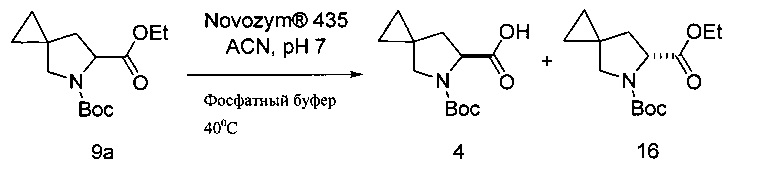
Ферментативное разделение с получением соединения 4
[0156] К раствору 0,2М pH 7 фосфатного буфера (104 г) добавляли Novozym® 435 (4 г), после чего добавляли раствор соединения 9а (10 г, 37,2 ммоль) в MeCN (10 мл). Смесь нагревали до 40°С и pH подводили до необходимого значения с помощью 1,0М водного NaOH для поддержания pH в диапазоне 6,9-7,1. По завершении реакции смесь пропускали через фильтр и отфильтрованный остаток промывали an 5% раствором NaHCO3 (50 г). Собранный фильтрат промывали МТВЕ (18,4 мл). Затем слой МТВЕ подвергали обратно экстракции раствором 5% NaHCO3 (8,8 г). Органический слой удаляли. К объединенным водным слоям добавляли МТВЕ (8,4 мл) и достаточно концентрированную HCl для получения в водной фазе pH of ≤2. После второй экстракции кислой водной фазы МТВЕ (5,1 мл), органические фазы объединяли и суспендировали с MgSO4. Суспензию фильтровали, а затем промывали МТВЕ. Затем фильтрат концентрировали посредством дистилляции. Количество неочищенного выделенного масла составляло 3,40 г (75,9% выход; >99% ее).
[0157] Для проведения разделения могут быть использованы другие ферментные реагенты. Например, для такой трансформации эффективной окажется любая альтернативная форма липазы из Candida Antarctica Липаза В. В некоторых вариантах реализации изобретения предложены различные варианты растворителей, включающих диалкиловые эфиры, циклические эфиры, ацетон, диметилсульфоксид (DMSO). Температуры реакций составляли от приблизительно 22°С до приблизительно 50°С.
[0158] В других вариантах реализации в настоящем раскрытии предложены альтернативы описанных ниже способов получения соединения 4. Схема получения ниже иллюстрирует указанный вариант реализации:
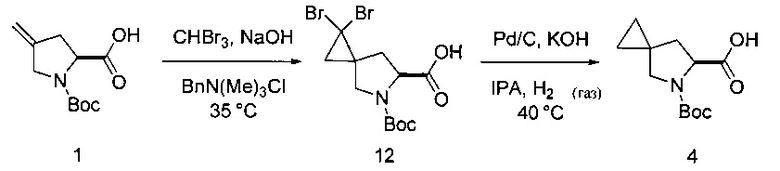
I. Циклопропанирование
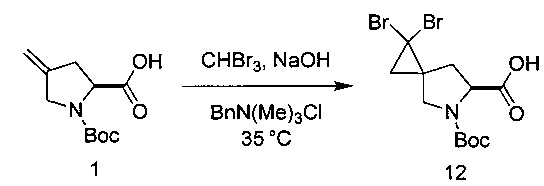
Циклопропанирование с получением соединения 12
[0159] Соединение 1 (40,0 г, 1,0 эквивалент), BnN(Me)3Cl (2,3 г, 0,07 эквивалента), бромоформ (45 мл, 3,0 эквивалента) и DCM (280 мл) добавляли в колбу. Полученный раствор перемешивали при 33°С и добавляли 50% раствор гидроксида натрия (120 мл) в течение 1,5-2 часов (внутренняя температура не превышала 38°С). Раствор выдерживали при 33°С до предполагаемого окончания реакции, которое определяли посредством ВЭЖХ анализа. Содержимое колбы охлаждали до 22°С, добавляли воду (100 мл) и оставляли для разделения слоев в течение 2 часов. Нижний водный слой удаляли, а верхний органический слой промывали 4М HCl (120 мл). Нижний органический слой оставляли, а верхний водный слой удаляли. Затем органический слой промывали водой (80 мл). Нижний органический слой суспендировали с силикагелем (12 г) в течение одного часа. Силикагель отфильтровывали и остаток на фильтре промывали однократно DCM (80 мл). Объем раствора DCM снижали и температуру раствора доводили до 35°С. Гептан вносили в реактор посредством дозирующего насоса в течение 1,5 часов. Затравочные кристаллы соединения 12 добавляли в реактор и суспензию перемешивали при умеренной скорости в течение по меньшей мере 60 минут. Суспензию охлаждали до 20°С (15-25°С) в течение часа и выдерживали при указанной температуре в течение 12 часов. Суспензию фильтровали при 20°С с помощью подходящего фильтра. Отфильтрованный осадок промывали раствором гептана (64 мл) и DCM (16 мл). Продукт высушивали при 40°С с получением соединения 12 в виде твердого вещества светло-коричневого цвета (47 г, 68% в виде смеси диастереомеров 85:15).
[0160] 1Н ЯМР (400 МГц, CDCl3, δ): 4,64-4,53 (m, 1Н), 3,93-3,87 (m, 1Н), 3,50 (d, J=11,1 Гц, 0,4Н), 3,29 (d, J=11,1 Гц, 0,6Н), 2,84 (d, J=9,6 Гц, 0,25Н), 2,66 (dd, J=13,2, 8,8 Гц, 0,75Н), 2,24 (d, J=13,4 Гц, 1H), 2,07-1,69 (m, 2Н), 1,47 (m, 9Н).
[0161] В альтернативном варианте реализации изобретения предложено применение хлороформа, с помощью которого получали дихлор аналог соединения 12, которое затем может быть использовано в последующих стадиях, представленных ниже.
[0162] Также подходящими являются другие основания, помимо NaOH. Такие основания включают, в качестве двух примеров, гидроксид калия и трет-бутилат калия.
[0163] Растворители также могут быть различными. Например, подходящие растворители включают толуол, бензол, диалкиловые эфиры и циклические эфиры.
[0164] Стандартная температура реакций составляет от приблизительно 0°С до приблизительно 60°С.
J. Гидрирование
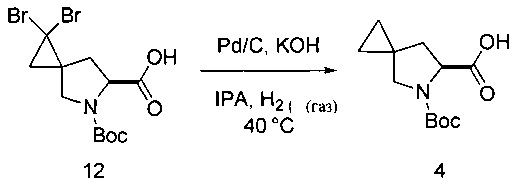
Восстановление до соединения 4
[0165] Соединение 12 (20,0 г) растворяли в изопропиловом спирте (160 мл), а затем гомогенную смесь нагревали до 40°С. Хлопья KOH (17,0 г, 6 эквивалентов) добавляли к раствору и перемешивали до растворения твердых частиц. Раствор продували газом N2, а затем добавляли Pd/C 10% Degussa El01 NE/W (4,0 г). Систему вновь продували газом Н2 и оставляли для перемешивания при 40°С в условиях 1 атм. H2. Окончание реакции определяли с помощью анализа ВЭЖХ. По завершении раствор охлаждали до приблизительно 22°С и продували газом N2. Твердую фазу удаляли фильтрацией через слой целита. Твердую фазу промывали Н2О (100 мл). Чистый раствор затем концентрировали до половины его первоначального объема. К концентрированному раствору добавляли МТВЕ (60 мл) и 4М HCl (60 мл). Смесь перемешивали и затем слои разделяли. Водный слой экстрагировали МТВЕ (40 мл) и затем органическую слои объединяли и промывали водой (40 мл). Раствор концентрировали с получением соединения 4 в виде твердого вещества белого цвета (9,9 г, 82%).
[0166] Варианты условий реакции и реагентов, позволяющих провести гидрирование, хорошо известны специалисту в области химии. Например, специалист может использовать другие источники Pd/C, такие как гидроксид палладия на угле.
Основания также могут быть различны, и, например, представлять собой основание, выбранное из карбоната калия, карбоната натрия, гидроксида натрия, трет-бутилата калия, фосфата натрия и фосфата калия.
[0167] Подходящие варианты растворителей включают метанол, этанол, толуол, бензол и диалкиловые эфиры и циклические эфиры.
[0168] Температура гидрирования может составлять от приблизительно 20°С до приблизительно 80°С.
K. Получение калиевой соли
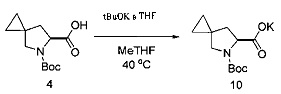
Получение калиевой соли соединения 10
[0169] Карбоновую кислоту 4 (219 г) растворяли в 2-MeTHF (880 мл) и затем указанный раствор нагревали до приблизительно 35°С. Медленно добавляли 1,0 М раствор tBuOK в THF (1,05 л) таким образом, что внутренняя температура не превышала 40°С. Суспензию перемешивали в течение приблизительно 30 минут и затем медленно охлаждали до приблизительно 20°С в течение приблизительно 2 часов. Суспензию выдерживали при 20°С в течение одного часа и затем фильтровали. Отфильтрованный остаток промывали 2-MeTHF (715 мл). Твердую фракцию высушивали в вакуумной печи в течение 24 часов при 40°С. Конечный продукт 10 выделяли в виде белого твердого вещества (212 г, 86%). 1Н ЯМР (400 МГц, CDCl3) δ 4,07 (t, J=7,3 Гц, 1Н), 3,44 (d, J=10,4 Гц, 1H), 3,35 (s, 1Н), 3,10 (d, J=10,4 Гц, 1H), 2,03 (dd, J=12,3, 6,9 Гц, 1H), 1,89 (dd, J=12,3, 8,0 Гц, 1H), 1,38 (s, 9Н), 0,71-0,27 (m, 4Н). 1Н ЯМР (400 МГц, d6-DMSO, δ): 3,89 (dd, J=8,6, 4,1 Гц, 0,4Н ротамер1), 3,85 (dd, J=8,6, 4,3 Гц, 0,6Н ротамер2), 3,21-3,07 (m, 2Н), 2,00-1,92 (m, 1Н), 1,75-1,71 (m, 1Н) 1,36 (s, 4Н ротамер1), 1,32 (s, 5Н ротамер2), 0,46-0,37 (m, 4Н). 13С ЯМР(100 МГц, d6-DMSO) δ 174,5, 174,4, 154,1, 153,4, 77,2, 76,9, 62,3, 62,0, 54,1, 53,8, 38,7, 28,4, 28,3, 20,6, 19,9, 11,8, 11,6, 10,5, 10,2.
[0170] Для целей описанного выше способа получения солей подходят и другие растворители. Например, такие растворители включают диалкиловые эфиры и циклические эфиры.
II. Схема получения промежуточного соединения 22
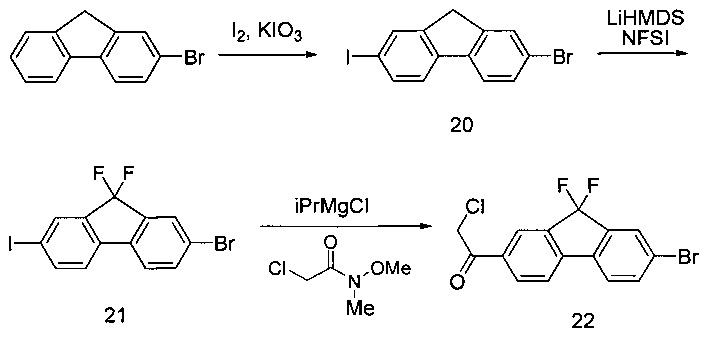
А. Синтез промежуточного соединения 20
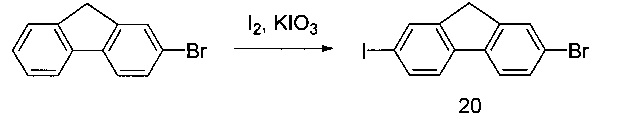
[0171] В трехгорлую колбу вносили 2-бромфлуорен (100 г) и уксусную кислоту (2100 г). Содержимое нагревали до 40-45°С и перемешивали в течение приблизительно 30 минут до получения чистого раствора. После доведения внутренней температуры до 20-30°С, добавляли 20% (об/об) водной H2SO4 (200 г, полученной с помощью 64,0 г H2SO4 и 136 г воды), после чего добавляли I2 (53,0 г, 0,512 мольных эквивалентов), после чего добавляли KIO3 (17,5 г, 0,200 мольных эквивалентов). Суспензию нагревали при 58°С (56-60°С) в течение приблизительно 4 часов. Затем суспензию охлаждали до 20-25°С и вносили в реакционную смесь 9% раствор Na2SO3 (Na2SO3, 47,0 г; вода, 500 г) при внутренней температуре, поддерживаемой на уровне 20-30°С. Суспензию перемешивали при 25°С в течение одного часа и фильтровали. Отфильтрованный остаток промывали 85 масс % НОАс (200 г, приготовленный с помощью 170 г НОАс и 30 г воды), а затем водой (200 г, 2,0 массовых эквивалента). Отфильтрованный остаток вынимали и суспендировали в воде (1500 г) в течение приблизительно одного часа, затем фильтровали и промывали водой до получения pH промывочной воды 6-7, и дополнительно промывали гептанами (200 г). Твердую фракцию высушивали под вакуумом с получением 143 г (выход 95%, чистота 96% НП ВЭЖХ) продукта 20 в виде белого твердого вещества.
[0172] Температуры реакции могут варьировать от приблизительно 20°С до 100°С. Стандартная температура составляет от приблизительно 20°С до приблизительно 60°С.
В. Синтез промежуточного соединения 21
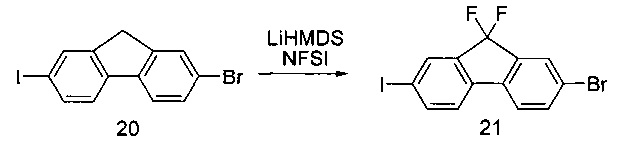
[0173] Исходное вещество (соединение 20, 100 г) и N-фторбензилсульфонимид (NFSI, 251 г, 2,95 мольных эквивалентов) помещали в колбу в виде твердых веществ. К смеси добавляли THF (1000 г) и твердые частицы растворяли при перемешивании. Раствор трижды дегазировали путем медленного приложения вакуума, после чего вакуум компенсировали азотом. Раствор охлаждали на бане при -78°С до внутренней температуры -68°С. После охлаждения образовывалась суспензия от белого до белого с желтоватым оттенком цвета. Раствор основания (1,0М LiHMDS в THF, 720 г, 3,00 мольных эквивалента) добавляли при такой скорости, чтобы внутренняя температура не превышала -55°С. В большинстве случаев при добавлении внутренняя температура составляла <-60°С, общее время добавления составляло 1 час. Окончание реакции отслеживали с помощью анализа ВЭЖХ. Реакцию гасили добавлением NH3/МеОН (7N NH3 в МеОН, 8 г) и удаляли холодную баню. После того, как внутренняя температура поднималась до -20°С, ВЭЖХ анализ показал полное израсходование избыточного N-фторбензилсульфонимида. Внутреннюю температуру доводили до 0°С. Добавляли гептан (342 г) и раствор перемешивали в течение 10 минут. В случае необходимости температуру доводили до 20-25°С. Суспензию фильтровали и твердую фазу дважды промывали смесью THF/гептан (для каждой промывки: THF, 89,0 г; гептан: 205 г). Фильтрат сохраняли при 5°С (2-8°С) в течение приблизительно 20 часов. Затем раствор фильтровали в колбу и концентрировали до 2,5-3,0 объемов под вакуумом с максимальным интервалом температуры 35°С. В суспензию вносили CH2Cl2 (1500 г) и суспензию перемешивали с обратным холодильником (при приблизительно 40°С) в течение 30 минут. После доведения внутренней температуры до 20-25°С, суспензию фильтровали через слой целита, и отфильтрованный остаток промывали DCM (400 г, 4,0 масс, эквивалента). Фильтрат концентрировали до приблизительно 3,0 объемов под вакуумом. Добавляли метанол (600 г) и смесь концентрировали до приблизительно 4,0 объемов, добавляли дополнительный метанол (300 г) и смесь снова концентрировали до приблизительно 4,0 объемов (300 объемов). Суспензию фильтровали и дважды промывали метанолом (для каждой промывки 100 г). Продукт 21 высушивали под вакуумом с получением 90 г (82% выход, 97-98% чистота НП ВЭЖХ) продукта в виде твердого вещества от желтоватого до бледно-желтого цвета. 1Н ЯМР (400 МГц, CDCl3, δ): 7,94 (d, J=1,2 Гц, 1H), 7,81 (d, J=7,8 Гц, 1H), 7,74 (d, J=1,4 Гц, 1H), 7,60 (d, J=8,3 Гц, 1H), 7,41 (d, J=8,1 Гц, 1Н), 7,29 (d, J=8,0 Гц, 1Н). 19F ЯМР (376 МГц, CDCl3) δ - 111,0 (s, 2F).
[0174] В некоторых вариантах реализации в настоящем изобретении предложено применение других оснований для синтеза соединения 21. Такие основания включают, например, гексаметилдисилазан натрия (NaHMDS), KHMDS, и диизопропиламид лития (LDA).
С. Синтез промежуточного соединения 22
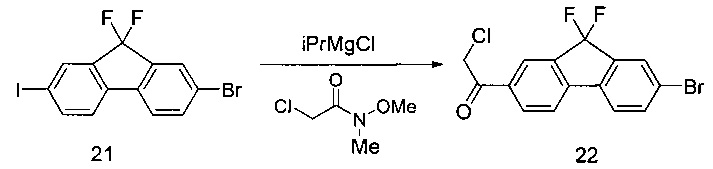
[0175] В трехгорлую колбу помещали соединение 21 (100 г) и THF (800 мл). Раствор трижды дегазировали путем медленного приложения вакуума, после чего вакуум компенчировали азотом. Раствор охлаждали до внутренней температуры -10°С. Раствор 2N i-PrMgCl в THF (125 г, 1,04 мольных эквивалентов) медленно добавляли, поддерживая внутреннюю температуру от -10°С до 0°С. Полученную в результате смесь затем перемешивали в течение 30 минут при -10°С до завершения реакции. Растворяли 2-хлор-N-метокси-N-метилацетамид (40,6 г, 1,20 мольных эквивалентов) в МТВЕ (122 г, 1,22 масс, эквивалентов) и фильтровали через фильтр с размером пор 1 мкм. МТВЕ раствор ацетамида затем медленно добавляли в колбу, поддерживая внутреннюю температуру при от -10°С до 0°С. По завершении добавления внутреннюю температуру доводили до 0°С и перемешивали в течение 2 часов. После завершения реакции медленно добавляли 1N HCl (750 г) таким образом, что внутренняя температура не превышала 20°С. В случае необходимости внутреннюю температуру доводили до 20°С. Слои разделяли и водный слой экстрагировали МТВЕ (410 г). Органические слой объединяли и высушивали над MgSO4. MgSO4 отфильтровывали и промывали THF (200 г). Фильтрат и промывную жидкость концентрировали под вакуумом до 10 объемов (1000 мл). Добавляли изопропанол (785 г), вследствие чего начинали образовываться небольшие количества кристаллов. Указанную суспензию снова концентрировали под вакуумом до 10 объемов (1000 мл). Вновь однократно добавляли изопропанол (785 г) и суспензию концентрировали под вакуумом до 10 объемов (1000 мл). Внутреннюю температуру доводили до 20-25°С и перемешивали в течение приблизительно 30 минут. Суспензию фильтровали и промывали изопропанолом (100 г), затем высушивали под вакуумом с получением 62,28 г (70,8%, 98% чистота по ВЭЖХ) продукта 22 в виде твердого вещества от желтоватого до бледно-желтого цвета. 1Н ЯМР (400 МГц, CDCl3, δ): 8,19 (s, 1Н), 8,12 (d, J=7,8 Гц, 1Н), 7,82 (s, 1H), 7,67 (d, J=8,0 Гц, 2Н), 7,52 (d, J=7,8 Гц, 1H), 4,71 (s, 2Н). 19F ЯМР(376 МГц, CDCl3) δ - 111,4 (s, 2F).
[0176] В некоторых вариантах реализации изобретения растворитель представляет собой 2-MeTHF.
III. Получение промежуточного соединения 24
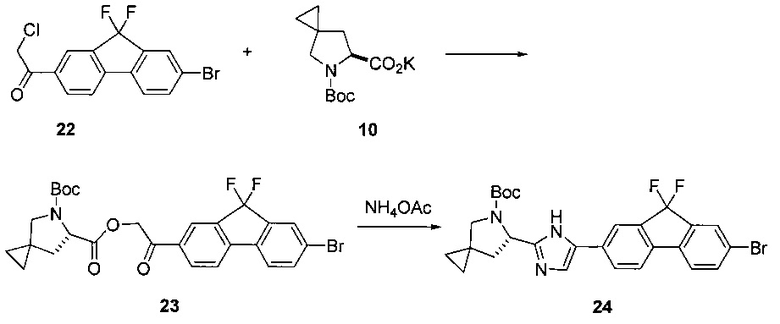
А. Получение соединения 23
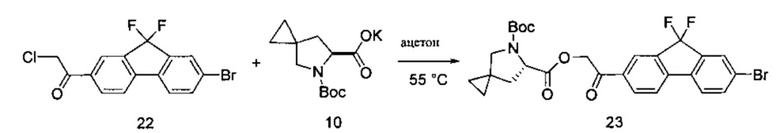
[0177] Соединение 22 (10,8 г, 1,05 эквивалент) и соединение 10 (8,0 г, 1,0 эквивалент) растворяли в ацетоне (106 мл). Гетерогенную смесь нагревали до 55°С и выдерживали до предположительного окончания реакции, определяемого посредством анализа ВЭЖХ. Медленно добавляли воду (22 мл) и выдерживали раствор при 55°С в течение 30 минут. Раствор охлаждали до 50°С и добавляли в качестве затравки кристаллы соединения 23. Медленно добавляли следующую порцию воды (11 мл). Раствор выдерживали при 50°С в течение одного часа, а затем охлаждали до 20°С (15-25°С) в течение 2 часов. Суспензию фильтровали при 20°С (15-25°С) и отфильтрованный остаток промывали смесью ацетона (18 мл) и воды (6 мл). Продукт высушивали с получением соединения 23 в виде твердого вещества желтого цвета (12,8 g, 95%). 1Н ЯМР (400 МГц, CDCl3, смесь ротамеров, δ): 8,13 (s, 1Н), 8,07-7,97 (m, 1Н), 7,79 (s, 1H), 7,67-7,56 (m, 2Н), 7,53-7,44 (m, 1H), 5,61 (d, J=16,3 Гц, 0,5Н), 5,47 (d, J=16,2 Гц, 0,5Н), 5,29 (d, J=16,2 Гц, 0,5Н), 5,15 (d, J=16,3 Гц, 0,5Н), 4,62 (dd, J=8,7, 3,5 Гц, 0,5Н), 4,55 (dd, J=8,7, 4,0 Гц, 0,5Н), 3,48-3,28 (m, 2Н), 2,43-2,35 (m, 1Н), 2,17-2,07 (m, 1H), 1,48 (s, 9Н) 0,77-0,55 (m, 4Н); 13С ЯМР (100 МГц, CDCl3) δ 190,8, 190,3, 172,2, 172,0, 154,4, 153,7, 143,7-143,4 (m), 140,3 (t, J=25,9 Гц), 138,2 (t, J=25,4 Гц), 136,9-136,5 (m), 135,5, 135,4, 134,7, 134,6, 132,4, 127,7, 124,2, 124,1, 123,2, 123,2, 122,7, 121,6 (t, J=244 Гц), 120,8, 120,8, 80,1, 80,0, 66,0, 65,9, 59,4, 59,0, 54,3, 53,7, 38,9, 38,0, 28,4, 28,3,20,7, 20,0, 12,9, 12,3, 8,8, 8,3. 19F ЯМР (376 МГц, CDCl3) δ - 111,41 (s), -111,43 (s).
[0178] В некоторых вариантах реализации изобретения вместо соединения 4 может использоваться соединение 10. В некоторых вариантах реализации изобретения получение согласно способу, описанному выше, выполняется в присутствии основания, такого как выбранное из карбоната калия, карбоната натрия, оснований третичных аминов.
[0179] В другом варианте реализации реакции, схема которой представлена выше, (1S,2R)-аминоинданоловая соль соединения 4 (соединение 18а) или 2-аминобутаноловая соль соединения 4 (соединение 18b) вступает в реакцию непосредственно с соединением 22, в результате чего получают соединение 23.
[0180] В другом варианте реализации реакции растворитель представляет собой ароматический углеводород, такой как толуол или бензол; алифатический эфир, такой как диалкиловый эфир, примером которого может служить диэтиловый эфир; циклические эфиры, такие как тетрагидрофуран; алкилацетаты, такие как этилацетат; полярные гетероциклические растворители, такие как N-метилпирролидон и 1,3-диметил-3,4,5,6-тетрагидро-2-пиримидинон; и полярные апротонные органические растворители, такие как димеилформамид и диметилацетамид.
[0181] Подходящие температуры реакции составляют от приблизительно 20°С до приблизительно 75°С.
В. Получение имидазола 24
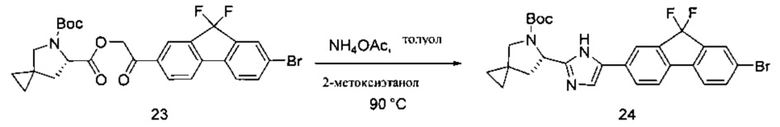
[0182] К соединению 23 (7,0 г) и ацетату аммония (4,8 г, 5,0 эквивалентов) добавляли толуол (62 мл) и 2-метоксиэтанол (3,5 мл). Гетерогенную/двухфазную смесь нагревали до 90°С и выдерживали до момента окончания реакции, что определяли при помощи анализа ВЭЖХ. Затем раствор охлаждали до 55°С и перемешивали до образования суспензии соединения 24 (в случае надобности может быть добавлена затравка). Добавляли гептан (104 мл) при 55°С в течение одного часа и затем суспензию охлаждали до 22°С в течение 3 часов. После того, как суспензия достигала комнатной температуры, ее выдерживали в течение одного часа. Суспензию фильтровали и промывали гептаном (15 мл). Затем твердую фазу растворяли в DMAc (42 мл). Раствор нагревали до 45°С и добавляли воду (7 мл). Температуру раствора повышали до 50°С и добавляли затравочные кристаллы соединения 24. Суспензию выдерживали в течение 30 минут, а затем в течение одного часа добавляли вторую порцию воды (9,1 мл). По завершении суспензию охлаждали до 22°С в течение 3 часов и выдерживали при комнатной температуре в течение одного часа. Твердую фазу фильтровали и промывали раствором DMAc (5 мл) и воды (2 мл). Для удаления DMAc и воды применяли окончательную промывку гептаном (23 мл). Твердую фазу высушивали при 45°С в вакуумной печи. Конечный продукт 24 выделяли в виде твердого вещества коричневого цвета (5,2 г, 77%). 1Н ЯМР (400 МГц, DMSO, смесь ротомеров, δ): 12,31-11,78 (m, 1H), 8,15-8,03 (m, 1H), 8,02-7,84 (m, 2H), 7,84-7,43 (m, 4H), 5,04-4,84 (m, 1H), 3,62-3,21 (m, 2H), 2,42-2,09 (m, 1H), 2,08-1,78 (m, 1H), 1,40 (s, 4H), 1,17 (s, 5H), 0,75-0,31 (m, 4H); 19F ЯМР (376 МГц, CDCl3) δ - 103,85 (s), - 104,03 (s). MS-ESI+: [M+H]+ вычисления для C27H27BrF2N3O2, 542,1, 544,1; найденное значение, 542,1, 544,1.
[0183] В некоторых вариантах реализации изобретения образование имидазола происходило при использовании аммониевых солей карбоксилатов с большей цепью, 
[0184] В других вариантах реализации изобретения растворитель выбирают из толуола, бензола, диалкиловых эфиров и циклических эфиров и алкилацетатов, таких как этилацетат. Растворяющие добавки включают уксусную кислоту и спирты (ROH).
[0185] Подходящие температуры реакции составляют от приблизительно 50°С до приблизительного °С.
IV. Получение промежуточного соединения 28
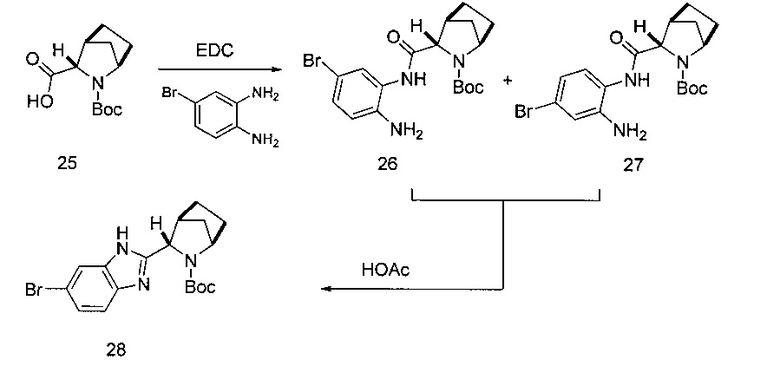
А. Получение соединения 25
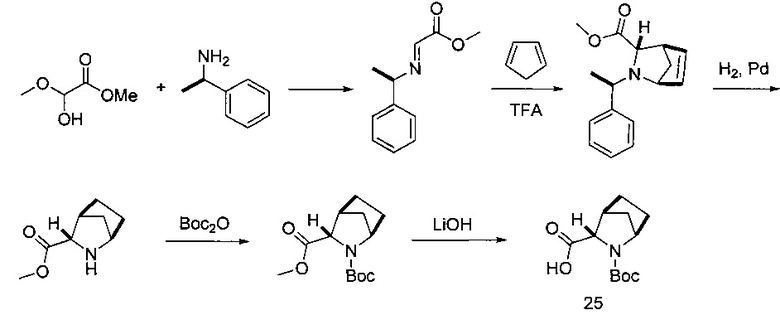
В. Получение соединений 26 и 27
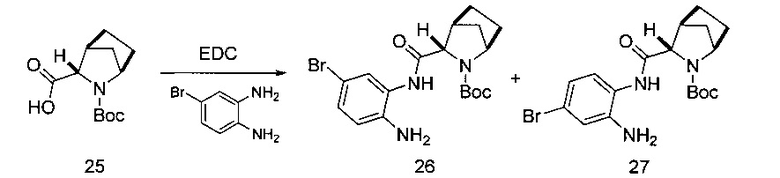
[0186] В колбу помещали соединение 25 (20,00 г, 0,083 моль), 4-бром-1,2-бензилдиамин (16,74 г, 0,089 моль, 1,08 эквивалента), гидроксибензотриазол (HOBt) (13,96 г, 0,091 моль, 1,1 эквивалента.), и 1-этил-3-(3-диметиламинопропил)карбодиимин HCl (EDC-HCl) (17,48 г, 0,091 моль, 1,1 эквивалента). Колбу охлаждали на ледяной бане и добавляли N,N-диметилацетамид (DMAC, 80 мл). Реакцию оставляли для охлаждения до приблизительно 10°С при перемешивании. В течение 5 минут добавляли N-метилморфолин (NMM) (27,34 мл, 0,249 моль, 3 эквивалента), сохраняя внутреннюю температуру ниже 20°С. Реакцию перемешивали при комнатной температуре в течение 20 часов. По завершении реакции реакционную смесь добавляли к МТВЕ (200 мл) и воде (600 мл) в отдельную воронку и аккуратно перемешивали. Выжидали разделения слоев и отделяли водный слой. Указанный водный слой дважды экстрагировали МТВЕ (50 мл), и объединяли органические экстракты. Объединенные органические экстракты затем экстрагировали водой (500 мл), в результате чего образовывалась смесь без четкого разделения фракций. Смесь фильтровали с применением подходящей твердой подложки и разделяли слои. Органическую фазу концентрировали под вакуумом и полученный остаток растворяли в диизопропиловом эфире (100 мл). Раствор охлаждали до приблизительно 5°С при перемешивании. Медленно добавляли уксусную кислоту (5,22 мл, 0,091 моль, 1,1 эквивалента) с сохранением внутренней температуры ниже 10°С, и полученную в результате суспензию перемешивали при 5°С в течение 2 часов. Затем сгущенную суспензию фильтровали и твердый остаток промывали изопропиловым эфиром (100 мл), а затем гептаном (100 мл). Остаток высушивали под вакуумом для получения конечного продукта в виде светло-бежевого твердого вещества, представляющего собой смесь региоизомеров 26 и 27 (28,19 г, 72%, >99% НП). 1Н ЯМР (400 МГц, DMSO) смесь 26 и 27 (данные представлены для двух ротамеров основного региоизомера): δ 9,25 (s, 0,5Н), 9,13 (s, 0,5Н), 7,08 (d, J=8,3 Гц, 0,5Н); 7,06 (d, J=8,2 Гц, 0,5Н), 6,92 (d, J=2,2 Гц, 0,5Н), 6,89 (d, J=2,1 Гц, 0,5Н), 6,71 (dd, J=8,4, 2,2, 0,5Н), 6,66 (dd, J=8,4, 2,2, 0,5Н), 5,10 (br s, 1H), 5,05 (br s, 1H), 4,15 (br s, 0,5H), 4,10 (br s, 0,5H), 3,76 (s, 1H), 2,64 (br s, 1H), 1,96-1,88 (m, 1H), 1,77-1,67 (m, 1H), 1,67-1,19 (m, 4H), 1,41 (s, 4,5H), 1,33 (s, 4,5H). MS-ESI+: [M+H]+ вычисления для C18H25BrO3N3, 410,1, 412,1; найденное значение, 410,0, 412,0
[0187] В некоторых вариантах реализации в настоящем раскрытии предложено применение других реагентов сочетания. Такие реагенты включают, но не ограничиваются указанными, N,N'-дициклогексилкарбодиимид (DCC), N,N'-диизопропилкарбодиимид (DIC), 6-хлор-2,4-диметокси-s-триазин (CDMT), О-бензотиазол-N,N,N',N'-тетраметилурониумгексафторфосфат (HBTU), и 2-(7-аза-1Н-бензотиазол-1-ил)-1,1,3,3-тетраметилурониумгексафторфосфат (HATU).
[0188] Аминное основание также может быть различным или полностью отсутствовать. Например, амин выбирают из третичных аминов (R3N), 2,6-лютидина, пиридина, диуиклогексилметиламина и N-метилморфолина (NMM).
[0189] Подходящие альтернативные растворители выбирают из DMF, NMP, диалкилового и циклического эфиров R2O, THF, 2-MeTHF, DCM, DCE, толуола, EtOAc, IP Ас, ацетона, MIBK и MEK.
[0190] Подходящие температуры реакции составляют от приблизительно -20°С до 80°С.
С. Получение промежуточного Соединения 28
[0191] Смесь 26/27 загружали в реактор (50,0 г, 0,106 моль). Добавляли МТВЕ (200 мл, 4V) и к суспензии добавляли ледяную уксусную кислоту (30,4 мл, 0,532 моль, 5 эквивалентов). Смесь нагревали до 55°С, в результате чего образовывался гомогенный раствор коричневого цвета, и перемешивали при указанной температуре в течение 18 часов. По завершении реакции, что было определено с помощью анализа ВЭЖХ, раствор охлаждали до приблизительно 10°С и затем гасили водным KOH (35 г в 200 мл Н2О) с сохранением внутренней температуры ниже 20°С. Двухфазную смесь энергично перемешивали в течение 15 минут. Прекращали перемешивание и выжидали разделения слоев. Водный слой сливали и проводили повторную экстракцию МТВЕ (50 мл). Органические экстракты смешивали, добавляли Н2О (300 мл) и двухфазную смесь энергично перемешивали в течение 15 минут. Прекращали перемешивание и выжидали разделения слоев. Водный слой сливали и проводили окончательное фильтрование органического слоя светло-коричневого цвета. Растворитель отгоняли до объема приблизительно 50 мл. Добавляли диизопропиловый эфир (IPE, 150 мл) при поддержании внутренней температуры выше 48°С и перегнанный раствор до общего объема приблизительно 80 мл. Снова добавляли IPE (150 мл) и перегнанный раствор до приблизительно 120 мл. Указанный процесс повторяли до тех пор, пока растворитель не представлял собой в основном диизопропиловый эфир, о чем свидетельствовала внутренняя температура приблизительно 69°С во время перегонки или как было определено с помощью 1Н ЯМР. Общий объем к тому времени доводили до приблизительно 120 мл, и раствор оставляли для медленного охлаждения (10°С/час) в течение ночи до 0°С, в результате чего образовывалась суспензия. Затем суспензию фильтровали и промывали холодным IPE (100 мл). Твердую фазу собирали и высушивали в вакуумной печи с получением соединения 28 (39,23 g, 94% выход, >99,5% НП). 1Н ЯМР (400 МГц, CDCl3, δ): 10,70 (s, 1H), 7,86 (s, 0,5Н), 7,58 (d, J=8,6 Гц, 0,5Н), 7,54 (s, 0,5Н), 7,30 (d, 8,3 Гц, 1H), 7,25 (d, J=8,0 Гц, 0,5Н), 4,52 (d, J=3,6 Гц, 1H), 4,15 (s, 1Н), 3,43 (d, J=3,2 Гц, 1Н), 2,03-1,94 (m, 1H), 1,93-1,81 (m, 1H), 1,80-1,55 (m, 4Н), 1,52 (s, 9Н). MS-ESI+: [М+Н]+ вычисления для C18H23BrO2N3, 392,1, 394,1; найденное значение, 392,1, 393,9.
[0192] Стандартная температура реакции составляла от приблизительно 20°С до 100°С.
[0193] В одном варианте реализации изобретения толуол заменен на IPE и/или МТВЕ.
V. Получение Соединения формулы I (Соединение 31)
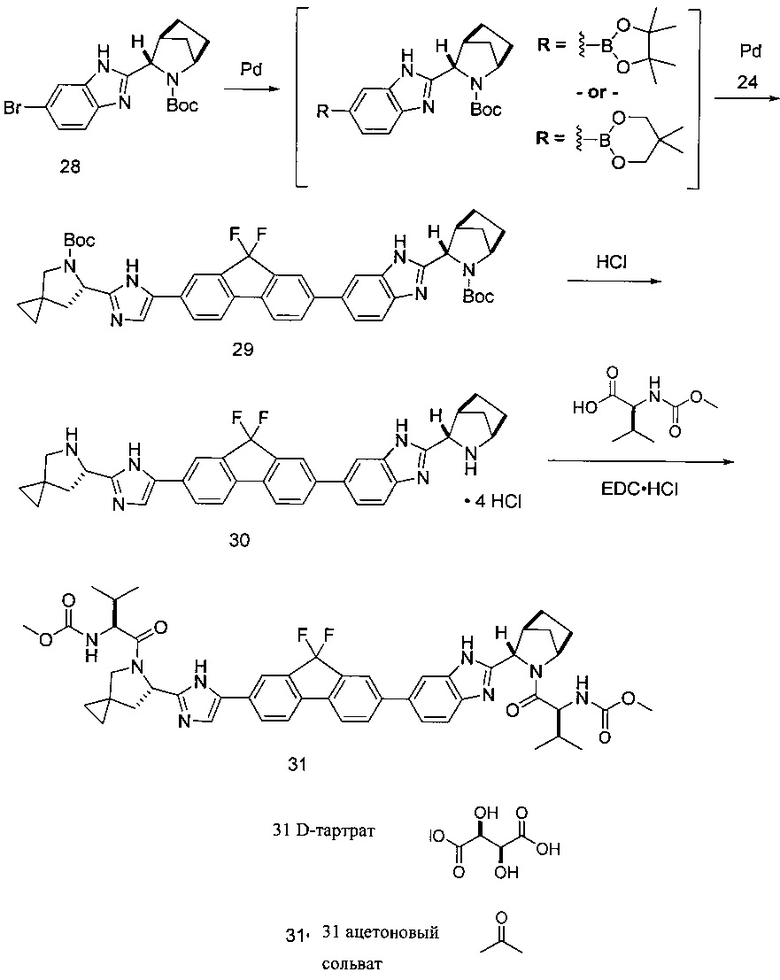
А. Образование Соединения 29
1a. PdCl2[P(f-Bu)2Ph]2 Пример с использованием бис(пинаколато)дибора
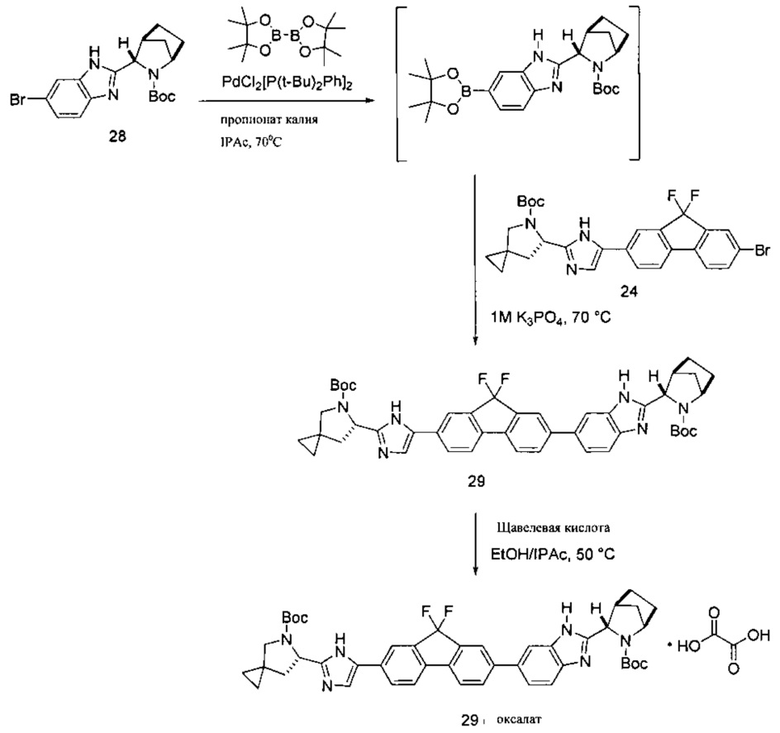
[0194] Соединение 28 (24,98 г), бис(пинаколато)дибор (19,40 г), пропионат калия (21,40 г) и PdCl2[P(t-Bu)2Ph]2 (2,04 г) помещали в реактор, после чего в реакторе создавали инертную среду. Добавляли изопропиловый ацетат (250 мл), начинали перемешивание, и в реакторе повторно создавали инертную среду. Реакционную смесь нагревали до 75°С и перемешивали в течение 3,5 часов. После охлаждения до 25°С в реакционную смесь добавляли соединение 24 (29,31 г) и создавали в реакторе инертную среду. Дегазированный водный 1 М K3PO4 (223 мл) помещали в реактор и реакционную смесь нагревали до 75°С. Реакционную смесь выдерживали при указанной температуре в течение одного часа и затем охлаждали до 35-40°С. Добавляли N-ацетил-L-цистеин (6,27 г) и перемешивали смесь при 35-40°С в течение 15 часов. Реакционную смесь охлаждали до 20°С, прекращали перемешивание и оставляли для разделения слоев. Фазы разделяли и добавляли к органическому слою N-ацетил-L-цистеин (6,27 г). Реакционную смесь нагревали до 45-50°С. После перемешивания смеси при 45-50°С в течение 2 часов, реакцию охлаждали до 20°С и добавляли 5% водный NaOH (250 мл). Фазы разделяли и органический слой промывали 5% водным NaCl (125 мл). Затем органическую фазу обрабатывали 5% водным NaCl (125 мл) и переносили в отдельную воронку посредством фильтрования через фильтровальную бумагу. Слои разделяли. Органическую фазу переносили в реактор и концентрировали до приблизительно 160 мл посредством вакуумной дистилляции. Вносили iPrOAc (20 мл), для того чтобы довести конечный объем до приблизительно 180 мл. Вносили этанол (100 мл) и нагревали содержимое до приблизительно 50°С. Затем к смеси добавляли раствор щавелевой кислоты (9,3 г) в этаноле (40 мл). К раствору в качестве затравки добавляли оксалат соединения 29 (200 мг) и выдерживали при 50°С в течение 72 часов. Изопропиловый ацетат (240 мл) добавляли в течение 5 часов, и суспензию охлаждали до 15°С в течение 4 часов и перемешивали при указанной температуре в течение 20 часов. Продукт собирали посредством фильтрации, промывали раствором этанола в изопропилацетате (48 мл EtOH, 191 мл iPrAc) и высушивали под вакуумом при 45°С с получением оксалата соединения 29 в виде твердого вещества желтовато-белого цвета (41,46 г, 81% выход). 1Н ЯМР (400 МН, DMSO-d6, δ) 11,80 (br s, 4Н), 8,11 (d, J=1,2 Гц, 1H), 8,00 (d, J=9,2 Гц, 1H), 7,98 (s, 1H), 7,90 (s, 2H), 7,87, (d, J=9,2 Гц, 1H), 7,85 (s, 1H), 7,80 (s, 1H), 7,60 (dd, J=8,4, 1,2 Гц, 1H), 7,56 (dd, J=7,6, 1,6 Гц, 1H), 5,03 (m, 0,5H), 4,99 (m, 0,5H), 4,52 (s, 0,5H), 4,50 (s, 0,5H), 4,28 (br s, 0,5H), 4,19 (br s, 0,5H), 3,48 (m, 1H), 3,34 (m, 1H), 2,66 (br d, J=12,7 Гц, 1H), 2,38 (m, 0,5H), 2,26 (m, 0,5H), 2,04 (m, 1H), 1,96 (m, 0,5H), 1,86 (d, J=11,6 Гц, 0,5H), 1,77 (m, 1H), 1,70 (m, 1H), 1,64 (2H, m), 1,43 (s, 6H) 1,41 (s, 3H), 1,35 (m, 1H), 1,19 (s, 5H), 1,14 (s, 4H), 0,65 (m, 2H) 0,54 (m, 1H), 0,42 (m, 1H). HRMS-ESI+: [M+H]+ вычисления для C45H49O4N6F2, 775,3778; найденное значение, 775,3773.
1b. PdCl2[P(t-Bu)2Ph]2 Пример с использованием бис(неопентилгликолято)дибора
[0195] Соединение 28 (20,1 г), бис(неопентилгликолято)дибора (13,2 г), пропионат калия (17,1 г) и PdCl2[P(t-Bu)2Ph]2 (1,6 г) помещали в реактор, после чего в реакторе создавали инертную среду. Добавляли изопропил ацетат (200 мл), начинали перемешивание и в реакторе повторно создавали инертную среду. Реакционную смесь нагревали до 72°С и перемешивали в течение 2 часов. После охлаждения до 20°С, к реакционной смеси добавляли соединение 24 (24,9 г) и в реакторе создавали инертную среду. Дегазированный водный 1 М K3PO4 (186 мл) помещали в реактор и реакционную смесь нагревали до 72°С. Реакционную смесь выдерживали при указанной температуре в течение одного часа и затем охлаждали до 20°С. Перемешивание прекращали и разделяли фазы. Органический слой промывали 5% водным NaCl (300 мл). Добавляли N-ацетил-L-цистеин (6 г) и перемешивали смесь при 20°С в течение 16 часов. Добавляли Celite® (5,6 г), а затем 5% водного NaOH (100 мл). Смесь фильтровали и разделяли фазы. К органическому слою добавляли N-ацетил-L-цистеин (6 г). После перемешивания смеси при 20°С в течение 12 часов добавляли 5% водный NaOH (100 мл). Фазы разделяли и органический слой промывали 5% водным NaCl (100 мл). Фазы разделяли и органический слой промывали другой порцией 5% водного NaCl (100 мл). Органическую фазу переносили в чистый реактор и концентрировали до приблизительно 150 мл путем вакуумной дистилляции. Добавляли этанол (101 мл) и содержимое реактора нагревали до приблизительно 50°С. Затем к смеси добавляли раствор щавелевой кислоты (4,7 г) в этаноле (34 мл). В раствор в качестве затравки добавляли оксалат соединения 29 (160 мг) и выдерживали при 50°С в течение 20 часов. Изопропилацетат (200 мл) добавляли в течение 2 часов, суспензию выдерживали в течение одного часа, и затем охлаждали до 15°С в течение 4 часов и перемешивали при указанной температуре в течение 20 часов. Продукт собирали посредством фильтрации, промывали раствором этанола в изопропилацетате (40 мл EtOH, 162 мл IPAc) и высушивали под вакуумом при 45°С с получением оксалата соединения 29 в виде твердого вещества желтовато-белого цвета (33,0 г, 87% выход).
2. Pd(OAc)2/MePhos Пример
[0196] Соединение 28 (69,96 г), бис(пинаколато)дибор (45,33 г), ацетат калия (69,96 г) и MePhos (2-дициклогексилфосфино-2'-метилфенил, 6,53 г) вносили в реактор с рубашкой и создавали в реакторе инертную среду. Добавляли дегазированный непосредственно перед добавлением t-амиловый спирт (700 мл) и начинали перемешивание. Ацетат палладия (1,99 г) вносили однократно в виде твердого вещества и перемешивали реакционную смесь при температуре окружающей среды в течение получаса, нагревали до 85°С и выдерживали в течение часа. После охлаждения до 25°С, добавляли соединение 24 (82,27 г) и дегазированный водный K3PO4 (625 мл, 1,0М в Н2О). В реакционной емкости создавали инертную среду и нагревали реакционную смесь до 85°С. После перемешивания при 85°С в течение часа реакционную смесь охлаждали до 20°С. После разделения фаз органический слой промывали 5% водным NaCl (2×700 мл) и концентрировали под вакуумом с получением масла, растворенного в изопропилацетате (1,62 л). Вакуумную дистилляцию продолжали до тех пор, пока не достигался минимальный регулируемый объем (приблизительно 300 мл). Добавляли дополнительный изопропилацетат (700 мл) и полученную в результате суспензию фильтровали через слой целита (28 г). После промывания остатка изопропилацетатом (500 мл), фильтрат обрабатывали N-ацетил-L-цистеином (17,5 г), и смесь перемешивали в течение 3,5 часов при температуре окружающей среды. Смесь охлаждали до 15°С, и добавляли 5% водный NaOH (700 мл). После нагревания до 25°С смесь фильтровали и разделяли фазы. Органический слой промывали 5% водным NaOH (700 мл) и 5% водным NaCl (2×700 мл). Полученную в результате органическую фазу обрабатывали N-ацетил-L-цистеином (17,5 г), и суспензию перемешивали в течение 14 часов при температуре окружающей среды. Смесь охлаждали до 15°С, и добавляли 5% водный NaOH (700 мл). После нагревания до 25°С фазы разделяли; органический слой фильтровали. Фильтр промывали изопропилацетатом (160 мл), и фильтрат промывали 5% водным NaOH (700 мл) и 5% водным NaCl (2×700 мл). Органическую фазу фильтровали и концентрировали с помощью вакуумной дистилляции до 500 мл. Добавляли дополнительный изопропилацетат (250 мл) и дистилляцию продолжали до достижения конечного объема 500 мл. Добавляли этанол (335 мл) и раствор нагревали до 50°С. Добавляли раствор щавелевой кислоты (24,51 г, 136 ммоль) в этаноле (110 мл) в течение 15 минут. Добавляли этанол для промывки (25 мл). В раствор добавляли затравку в виде оксалата соединения 29 (527 мг). Суспензию выдерживали при 50°С в течение 20 часов. Изопропилацетат (620 мл) добавляли в течение 3 часов и суспензию охлаждали до 15°С в течение 3 часов. Твердую фракцию собирали посредством фильтрации и отфильтрованный продукт промывали изопропилацетатом (2×300 мл). После высушивания оксалат соединения 29 выделяли в виде твердого вещества светло-желтого цвета (117,53 г, 76,9% выход).
[0197] В соответствии с другим вариантом реализации изобретения соединение 29 получали, используя противоположный порядок реакций, как показано на схеме ниже:
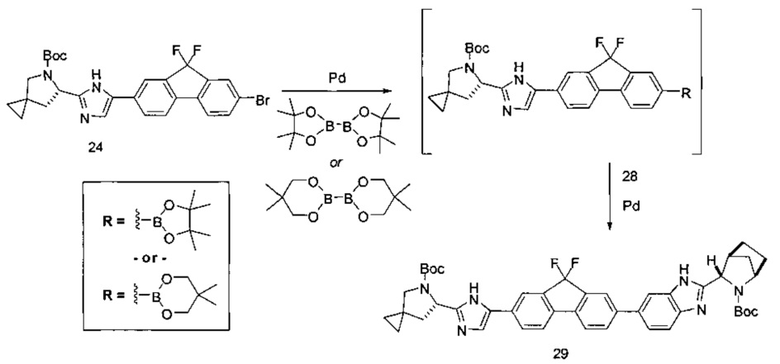
В. Снятие бис-Вос защиты Соединения 29
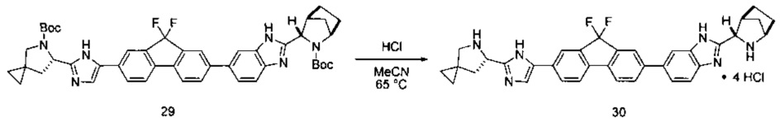
[0198] К раствору соединения 29 (92,5 г, 119 ммоль) в MeCN (324 мл) при 65°С добавляли 1,5 N водный раствор HCl (398 мл, 5,0 мольных эквивалентов). Реакционную смесь перемешивали в течение приблизительно 2 часов при 65°С и наблюдали окончание реакции посредством ВЭЖХ анализа. После того, как было определено, что все исходные вещества израсходованы, температуру реакционной смеси доводили до 45°С. Ацетонитрил (648 мл) добавляли в течение ≥30 минут для поддержания внутренней температуры на уровне 40-50°С. По завершении добавления указанного антирастворителя добавляли затравку в виде кристаллов гидрохлоридной соли соединения 30 (0,103 г). Суспензию выдерживали при 45°С в течение ≥1 часа. Добавляли дополнительный MeCN (1480 мл) в течение ≥30 минут, для того чтобы поддерживать внутреннюю температуру на уровне 40-50°С. Суспензию охлаждали до 20°С в течение ≥2 часов и затем фильтровали. Влажный остаток высушивали для получения 84,6 г соединения 30 (в виде тетра-HCl соли, включая содержание воды ~6%, выход 80,4%). Обычно содержание воды составляет от приблизительно 4 до приблизительно 13%. 1Н ЯМР (400 МГц, DMSO-d6, δ): 10,83 (br s, 2Н), 10,44 (br s, 2H), 10,33 (br s, 1H), 9,33 (br s, 1H), 8,37 (s, 1H), 8,36 (s, 1H), 8,26 (d, J=8,0 Гц, 1H), 8,08 (d, J=0,8 Гц, 1H), 8,06 (d, J=8,0 Гц, 1H), 8,03 (d, J=0,8 Гц, 1H), 8,01 (d, J=8,4 Гц, 1H), 7,98 (dd, J=8,0, 1,2 Гц, 1H), 7,79 (dd, J=8,4, 0,4 Гц, 1H), 7,75 (dd, J=8,4, 1,2 Гц, 1H), 5,29 (dd, J=8,0, 7,6 Гц, 1H), 4,82 (d, J=3,6 Гц, 1H), 4,19 (s, 1H), 3,65 (d, J=10,8 Гц, 1H), 3,14 (s, 1H), 3,12 (d, J=10,8 Гц, 1H), 2,85 (dd, J=13,2, 9,6 Гц, 1H), 2,23 (dd, J=12,8, 7,6 Гц, 1H), 2,11 (m, 1H), 1,99 (d, J=11,2 Гц, 1H), 1,83 (m, 1H), 1,76 (m, 1H), 1,71 (d, J=10,8 Гц, 1H), 1,67 (m, 1H), 0,84 (m, 2H), 0,70 (m, 2H). HRMS-ESI+: [M+H]+ вычисления для C35H33N6F2, 575,2729; обнаружено, 575,2729.
[0199] Соединение 30 выделяли из смеси CH3CN и водной HCl в виде кристаллического твердого вещества. В одном варианте реализации изобретения Соединение 30 представляет собой кристаллический полиморф, Форму I, которая была охарактеризована посредством рентгеновской порошковой дифракции (XRPD). Рентгеновская порошковая дифрактограмма представлена на Фиг. 1.
[0200] В одном варианте реализации изобретения Форма I характеризуется пиками XRPD, включающими 7,1, 8,2, 10,8°2θ±0,2°2θ, полученные на диффрактометре при 25°С с использованием излучения Cu-Kα при 1,54060 Å.
[0201] В другом варианте реализации изобретения Форма I характеризуется пиками XRPD, включающими 7,1, 8,2, 10,8, 11,1, 12,8, 14,1, 14,8, 16,1, 18,9, 24,5, 24,9, и 25,9°2θ±0,2°2θ.
[0202] В одном варианте реализации изобретения соединение 30 представляет собой кристаллический полиморф, Форму II, которая была охарактеризована посредством рентгеновской порошковой дифракции (XRPD). Рентгеновская порошковая дифрактограмма представлена на Фиг. 2.
[0203] В одном варианте реализации изобретения Форма II характеризуется пиками XRPD, включающими 7,4, 9,4, 11,6°2θ±0.2°2θ, полученные на диффрактометре при 25°С с использованием излучения Cu-Kα при 1,54060 Å.
[0204] В другом варианте реализации изобретения Форма II характеризуется пиками XRPD, включающими 7,4, 7,5, 9,4, 11,6, 14,9, 15,2, 22,5, 23,2, и 26,3°2θ±0,2°2θ.
[0205] В соответствии с другими вариантами реализации изобретения снятие защиты может осуществляться с использованием других реагентов. Такие реагенты включают, но не ограничиваются указанными, HCl, HBr, фосфорную кислоту, p-толуолсульфоновую кислоту, серную кислоту, бензолсульфоновую кислоту и TFA.
[0206] Подходящие варианты растворителей включают спирты, такие как изопропиловый спирт, этанол и н-бутанол, полярные апротонные органические растворители, такие как N,N-диметилацетамид; полярные гетероциклические растворители, такие как N-метилпирролидон; циклические эфиры, такие как тетрагидрофуран и 2-метил-тетрагидрофуран; алифатические эфиры, такие как диэтиловый эфир и диизопропиловый эфир; алкилацетаты, такие как этилацетат и изопропилацетат и ароматические углеводороды, такие как бензол и толуол.
[0207] Стандартная температура реакции составляла от приблизительно 20°С до приблизительно 85°С.
С. Реакция амидного сочетания
[0208] EDC-HCl (4,39 г), HOBt (2,06 г), Мос-валин (4,02 г), и DMF (50 мл) вносили в колбу. Реакционную смесь перемешивали в течение 20 минут при 23°С. Затем раствор охлаждали до 0°С. Соль 30-HCl (5,0 г) и N-метилморфолин (5,03 мл) вносили в реакционную смесь. Содержимое нагревали до комнатной температуры и перемешивали в течение 4 часов при 23°С. К реакционной смеси добавляли воду (2,5 мл) и содержимое перемешивали в течение 15 часов при 23°С. Добавляли EtOAc (70 мл) и воду (100 мл) и слои разделяли.. К органическому слою добавляли EtOAc (50 мл) и воду (50 мл), слои перемешивали и разделяли. Органический слой промывали 5% NaHCO3 (50 мл) и водой (2×25 мл). Органический слой перегоняли до 2,5 объемов (12,5 мл) и охлаждали до 23°С. К органическому слою добавляли ацетон (70 мл). К реакционной смеси добавляли в качестве затравки соединение 31 (ацетоновый сольват) и перемешивали в течение 15 часов. Содержимое фильтровали и влажный осадок промывали ацетоном (5 мл), и отфильтрованный осадок высушивали с получением 4,78 г соединения 31 в виде ацетонового сольвата (73%). 1H ЯМР (400 МГц, DMSO-d6, δ): 12,29 (s, 0,1Н), 12,19 (d, J=4,0 Гц, 1H), 12,14 (s, 0,2Н), 11,85 (s, 1Н), 8,10 (s, 0,1Н), 8,08 (s, 1Н), 8,01 (s, 0,1Н), 7,963 (m, 1H), 7,955 (s, 1Н), 7,89 (d, J=6,4 Гц, 1H), 7,87 (s, 1H), 7,83 (dd, J=8,4, 2,4 Гц, 1Н), 7,79 (dd, J=7,2, 2,8 Гц, 1H), 7,78-7,90 (misc., 0,9Н), 7,70 (s, 1H), 7,61 (d, J=8,4 Гц, 1H), 7,55 (s, 1Н), 7,51 (dd, J=8,8, 1,6 Гц, 1Н), 7,44 (m, 0,1Н), 7,31 (d, J=8,4 Гц, 1Н), 7,21 (d, J=8,4 Гц, 1H), 6,91 (d, J=8,0 Гц, 0,1Н), 6,77 (m, 0,2Н), 5,34 (d, J=7,6 Гц, 0,1Н), 5,20 (dd, J=8,0, 5,2 Гц, 1Н), 5,18 (m, 0,1Н), 4,88 (s, 0,1Н), 4,67 (d, J=6,4 Гц, 1Н), 4,55 (s, 1H), 4,17 (dd, J=8,0, 8,0 Гц, 1Н), 4,10 (m, 0,2Н), 4,01 (dd, J=8,4, 8,0 Гц, 1Н), 3,97 (m, 0,1Н), 3,82 (d, J=9,6 Гц, 1Н), 3,77 (s, 0,2Н), 3,71 (d, J=9,6 Гц, 1Н), 3,554 (s, 3Н), 3,548 (s, 3Н), 3,43 (s, 0,4Н), 3,20 (d, J=7,6 Гц, 0,3Н), 2,77 (s, 0,1Н), 2,66 (s, 1H), 2,41 (d, J=8,8 Гц, 1Н), 2,22 (dd, J=12,4, 8,0 Гц, 1H), 2,13 (m, 0,4Н), 2,08 (s, 6Н), 2,05 (dd, J=13,2, 5,2 Гц, 1H), 1,99 (m, 2Н), 1,92 (m, 1Н), 1,77 (m, 2Н), 1,61 (m, 0,3Н), 1,56 (m, 1Н), 1,46 (d, J=9,2 Гц, 1Н), 1,33 (d, J=10,0 Гц, 0,1Н), 0,97 (dd, J=6,4, 2,0 Гц, 3Н), 0,93 (d, J=6,8 Гц, 3Н), 0,88 (d, J=6,4 Гц, 3Н), 0,87 (d, J=6,4 Гц, 3Н), 0,80-1,05 (смеш., 2Н), 0,70 (m, 1Н), 0,59 (m, 2Н), 0,54 (m, 1Н), 0,33 (m, 0,1Н). HRMS-ESI+: [М+Н]+ вычисления для C49H55O6N8F2, 889,4207; найденное значение, 889,4205.
[0209] В некоторых вариантах реализации изобретения сочетающий агент представляет собой агент, выбранный из DCC, DIC, CDMT, HBTU и HATU.
[0210] Подходящие основания согласно другим вариантам реализации включают третичные амины R3N, 2,6-лютидин, пиридин, дициклогексилметиламин и NMM.
[0211] Альтернативные растворители, подходящие для реакции сочетания, описанной выше, включают DMAc, ACN, EtOAc, изопропилацетат (IPAc), MeTHF, IPA и t-BuOH.
[0212] Стандартные температуры реакции сочетания составляют от приблизительно - 30°С до приблизительно 50°С.
D. Образование тартратной соли
[0213] Соединение 31 (в виде ацетонового сольвата, 4,8 г) добавляли в колбу, после чего добавляли EtOAc (36 мл) и нагревали до 50°С. Затем добавляли D-винную кислоту (816 мг) в EtOH (35 мл). В раствор в качестве затравки добавляли кристаллы 31⋅D-винной кислоты и перемешивали при 50°С в течение 16 часов. Раствор охлаждали до 23°С в течение 3 часов и затем фильтровали. Влажный остаток на фильтре промывали раствором EtOAc : EtOH в соотношении 1:1 (9 мл) и твердую фаз высушивали с получением 4,33 г (82%) соединения 31 в виде D-тартрата. 1Н ЯМР (400 МГц, DMSO-d6, δ): 12,2 (br s, 2Н), 8,08 (s, 1H), 7,97 (s, 1H), 7,95 (d, J=8,4, 1H), 7,89 (d, J=8,4, 1H), 7,88 (s, 1H), 7,85 (d, J=8,4, 1H), 7,82 (d, J=8,0, 1H), 7,68 (s, 1H), 7,58 (d, J=8,4, 1H), 7,53 (d, J=8,4, 1H), 7,30 (d, J=8,8, 1H), 7,20 (d, J=8,4, 1H), 5,21 (dd, J=8,0, 5,2, 1H), 4,67 (s, 1H), 4,55 (s, 1H), 4,33 (s, 2H), 4,17 (dd, J=8,0, 8,4, 1H), 4,01 (dd, J=8,0, 8,4, 1H), 3,82 (d, J=10,0, 1H), 3,72 (d, J=9,6, 1H), 3,55 (s, 3H), 2,67 (s, 1H), 2,41 (d, J=9,2, 1H), 2,21 (dd, J=12,4, 8,0, 1H), 2,05 (dd, J=12,4, 5,2, 1H), 1,98 (m, 2H), 1,92 (m, 1H), 1,77 (m, 2H), 1,56 (m, 1H), 1,46 (d, J=9,2, 1H), 0,97 (d, J=6,8, 3H), 0,93 (d, J=6,4, 3H), 0,88 (d, J=6,4, 3H), 0,86 (d, J=6,4, 3H), 0,70 (m, 1H), 0,54 (m, 1H), 0,55-0,62 (m, 2H). HRMS-ESI+: [M+H]+ вычисления для C49H55O6N8F2, 889,4207; найденной значение, 889,4229.
[0214] В другом варианте реализации изобретения тартрат получают из соединения 31 в форме, не содержащей растворителей.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПИРОИНДОЛИНОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ РЕСПИРАТОРНО-СИНЦИТИАЛЬНОГО ВИРУСА (RSV) | 2015 |
|
RU2734248C2 |
| КОНДЕНСИРОВАННЫЕ ГЕТЕРОАРИЛЫ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2552114C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛИРОВАННЫХ ЦИКЛОПЕНТИЛПИРИМИДИНОВЫХ СОЕДИНЕНИЙ | 2013 |
|
RU2643811C2 |
| ПРОИЗВОДНЫЕ ТИЕНОПИРРОЛА ДЛЯ ПРИМЕНЕНИЯ ДЛЯ НАЦЕЛИВАНИЯ НА БЕЛКИ, КОМПОЗИЦИИ С УКАЗАННЫМИ ПРОИЗВОДНЫМИ, СПОСОБЫ И ПРИМЕНЕНИЯ | 2017 |
|
RU2771166C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ПОСТТРАВМАТИЧЕСКОГО СТРЕССОВОГО РАССТРОЙСТВА | 2011 |
|
RU2775783C2 |
| УГЛЕВОДНЫЕ ЛИГАНДЫ, КОТОРЫЕ СВЯЗЫВАЮТСЯ С IgM АНТИТЕЛАМИ ПРОТИВ МИЕЛИН-АССОЦИИРОВАННОГО ГЛИКОПРОТЕИНА | 2015 |
|
RU2760005C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2015 |
|
RU2742773C2 |
| ИНГИБИТОР, ПРЕДСТАВЛЯЮЩИЙ СОБОЙ ПРОИЗВОДНОЕ ПИРИДАЗИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2807611C2 |
| ПИРИМИДО[5,4-b]ИНДОЛИЗИНОВОЕ ИЛИ ПИРИМИДО[5,4-b]ПИРРОЛИЗИНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2017 |
|
RU2721774C1 |
| ИНГИБИТОРЫ MEK И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2812929C1 |
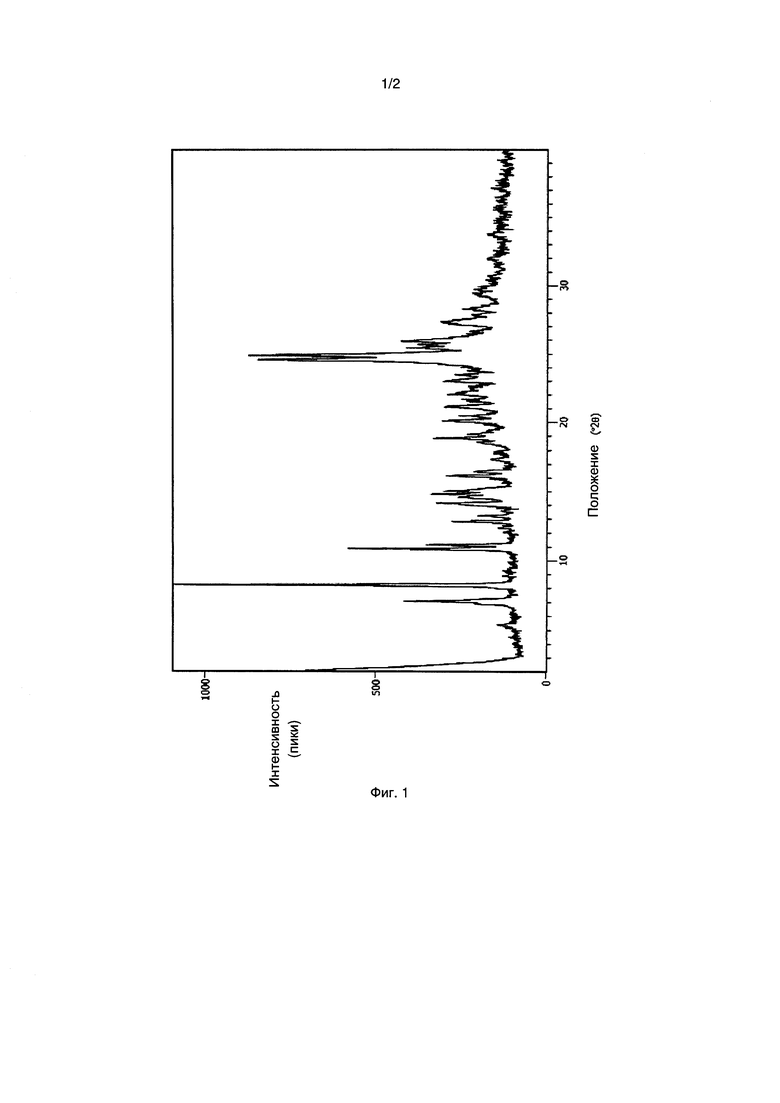
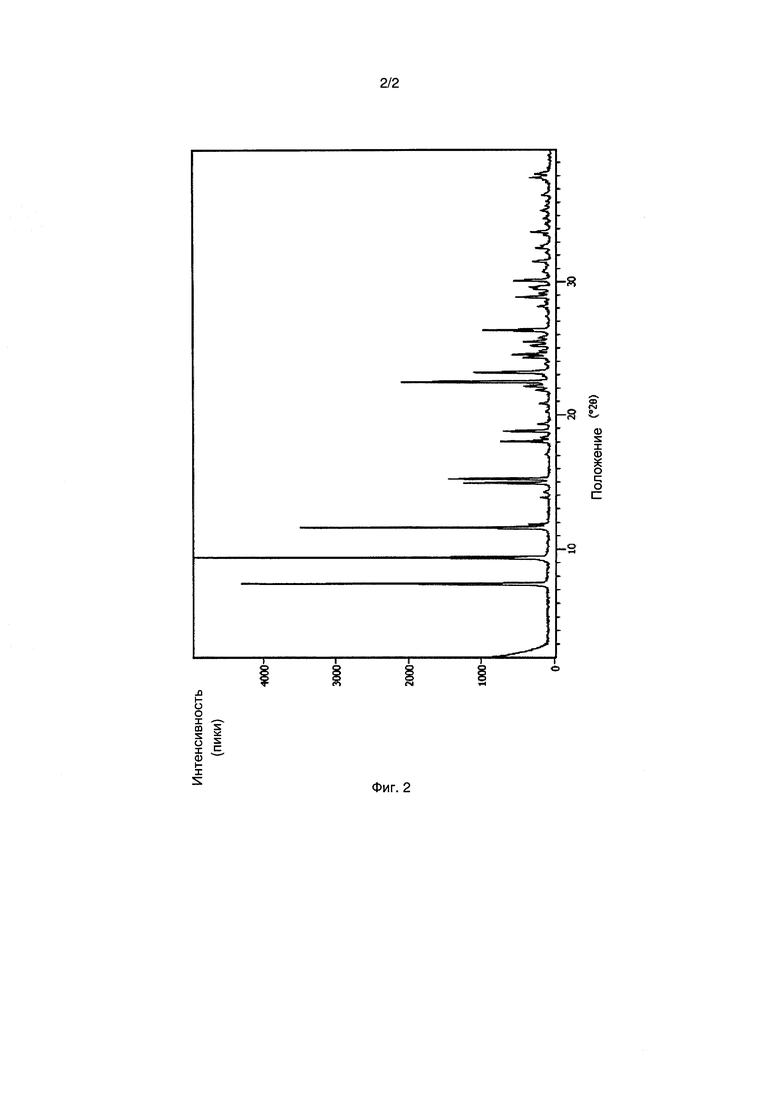
Изобретение относится к области органической химии, а именно к способу получения соединения формулы I или его фармацевтически приемлемой соли, включающему (А) реакцию сочетания соединения формулы (i) с соединением формулы (ii) в присутствии металлического катализатора и основания с получением соединения формулы (iii) или ее соли; (В) снятие защиты с соединения формулы (iii) с получением соединения формулы (iv) или его соли; (С) приведение соединения формулы (iv) в контакт с (S)-2-(метоксикарбониламино)-3-метилбутановой кислотой с получением соединения формулы I; причем каждый из PG независимо представляет собой трет-бутоксикарбонильную, бензилоксикарбонильную или метокси- или изопропилоксикарбонильную защитную группу; Y и Z независимо выбирают из Br и -B(OR)(OR'), причем когда Y представляет собой -B(OR)(OR'), тогда Z представляет собой Br, и Y представляет собой Br, тогда Z представляет собой -B(OR)(OR'); и R и R' независимо выбирают из группы, состоящей из водорода и прямого или разветвленного C1-8-алкила, или R и R' совместно представляют собой C3-8-циклоалкилен, причем любой алкил или циклоалкилен является необязательно замещенным одной или более C1-6-алкильными группами. Также изобретение относится к промежуточным соединениям и их кристаллическим формам. Технический результат: разработан новый способ получения соединения формулы (I), полезного при лечении гепатита С. 8 н. и 18 з.п. ф-лы, 2 ил., 2 пр.
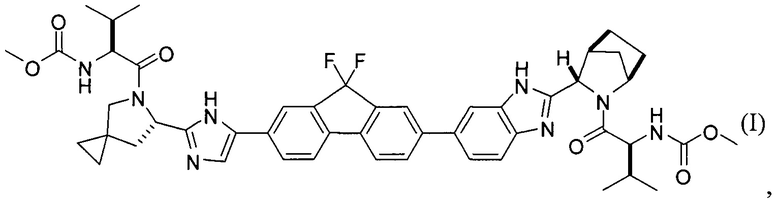
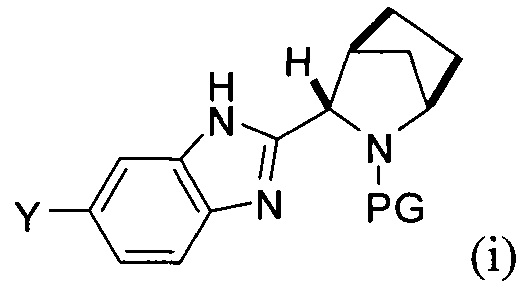
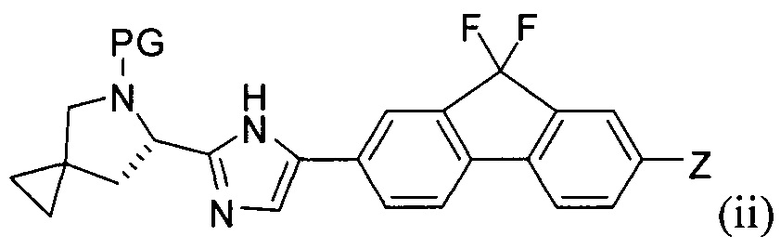
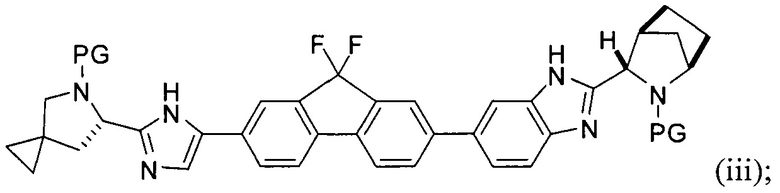
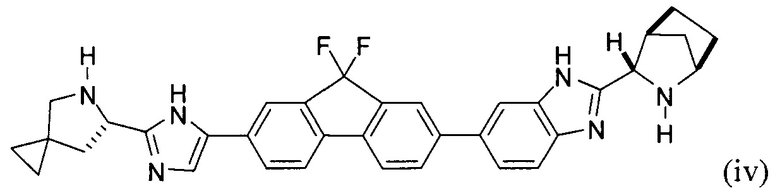
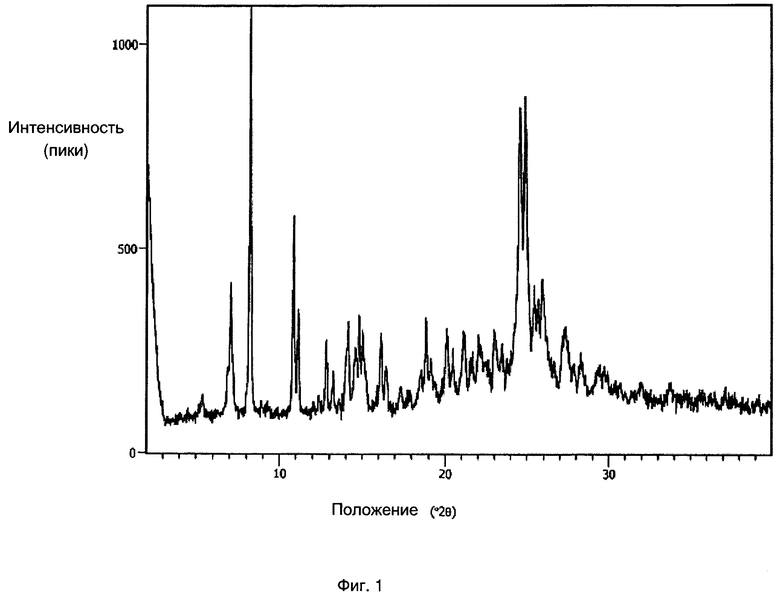
1. Способ получения соединения формулы I:
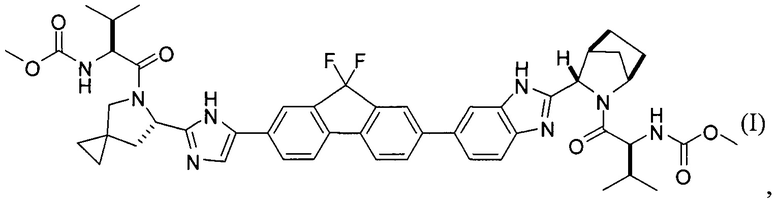
или ее фармацевтически приемлемой соли, включающий
(А) реакцию сочетания соединения формулы (i)
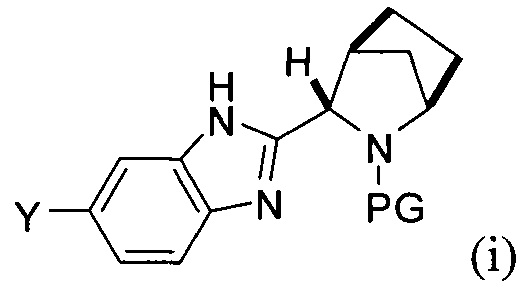
с соединением формулы (ii)
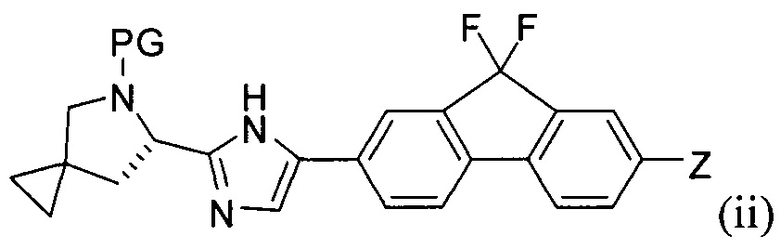
в присутствии металлического катализатора и основания с получением соединения формулы (iii) или ее соли:
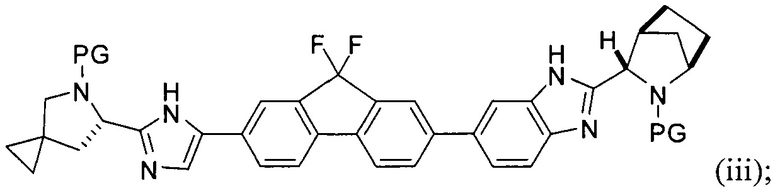
(В) снятие защиты с соединения формулы (iii) с получением соединения формулы (iv):
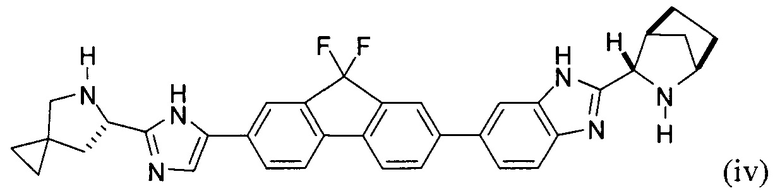
или его соли;
(С) приведение соединения формулы (iv) в контакт с (S)-2-(метоксикарбониламино)-3-метилбутановой кислотой:
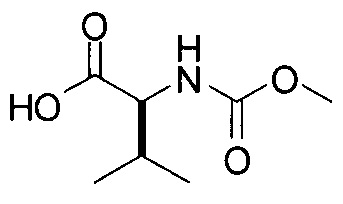
с получением соединения формулы I; причем
каждый из PG независимо представляет собой трет-бутоксикарбонильную, бензилоксикарбонильную или метокси- или изопропилоксикарбонильную защитную группу;
Y и Z независимо выбирают из Br и -B(OR)(OR'), причем когда
Y представляет собой -B(OR)(OR'), тогда Z представляет собой Br, и
Y представляет собой Br, тогда Z представляет собой -B(OR)(OR'); и
R и R' независимо выбирают из группы, состоящей из водорода и прямого или разветвленного C1-8-алкила,
или R и R' совместно представляют собой C3-8-циклоалкилен,
причем любой алкил или циклоалкилен является необязательно замещенным одной или более C1-6-алкильными группами.
2. Способ по п.1, отличающийся тем, что Y представляет собой -B(OR)(OR') и Z представляет собой Br.
3. Способ по п.2, дополнительно включающий стадию образования соединения формулы (i) в условиях in situ
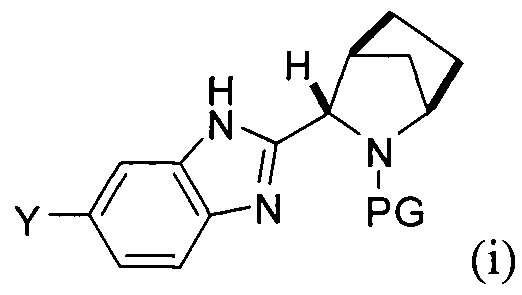 ,
,
включающий последовательное приведение соединения формулы (а)
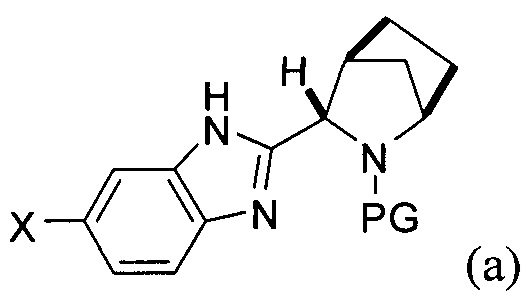
в контакт с источником палладия, а затем с борилирующим агентом, содержащим группу:
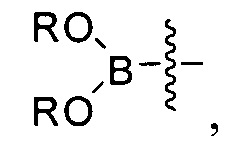
в присутствии второго основания, при условии, что соединение формулы (i) образуется в условиях in situ и X представляет собой галогенид, выбранный из Cl, Br и I.
4. Способ по п.3, отличающийся тем, что стадия образования соединения формулы (i) в условиях in situ и стадия (А) выполняются в одном реакторе последовательно.
5. Способ по п.3, отличающийся тем, что борилирующий агент выбирают из бис(пинаколато)дибора и бис(неопентилгликолято)дибора.
6. Способ по п.5, отличающийся тем, что борилирующий агент представляет собой бис(пинаколато)дибор.
7. Способ по п. 5, отличающийся тем, что борилирующий агент представляет собой бис(неопентилгликолято)дибор.
8. Способ по п.4, отличающийся тем, что X представляет собой Br и PG представляет собой трет-бутоксикарбонил.
9. Способ по п.2, отличающийся тем, что металлический катализатор представляет собой соединение Pd(0) или Pd(II).
10. Способ по п.9, отличающийся тем, что металлический катализатор представляет собой PdCl2[P(t-Bu)2Ph]2.
11. Способ по п.9, отличающийся тем, что металлический катализатор представляет собой Ре(ОАс)2/2-дициклогексилфосфино-2'-метилбифенил.
12. Способ по п.9, отличающийся тем, что металлический катализатор представляет собой аддукт дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладий(II) дихлорметан.
13. Способ по п.3, отличающийся тем, что второе основание представляет собой пропионатную соль.
14. Способ по п.13, отличающийся тем, что пропионатная соль представляет собой пропионат калия.
15. Способ получения соединения формулы I:

или его фармацевтически приемлемой соли, включающий:
(1) последовательное приведение соединения формулы (а')
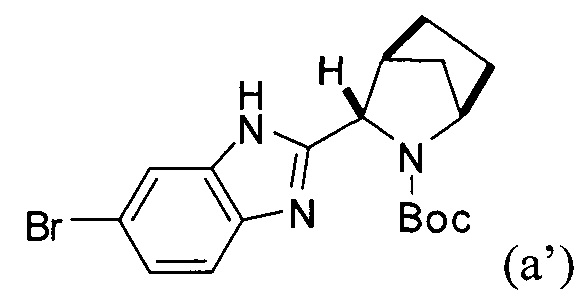
в контакт с каталитически эффективным количеством PdCl2[P(t-Bu)2Ph]2 и бис(неопентилгликолято)дибором в присутствии пропионата калия с получением реакционной смеси, содержащей соединение формулы (ia):
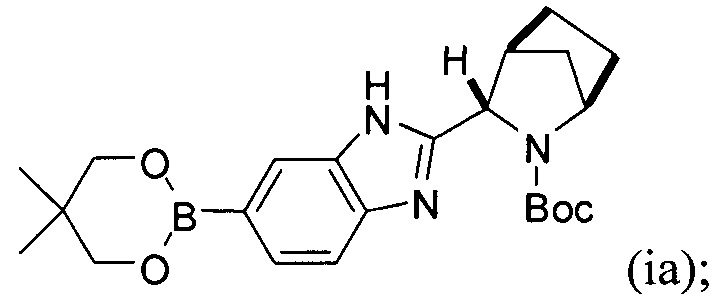
(2) приведение реакционной смеси, полученной на стадии (1), в контакт с соединением формулы (ii'):
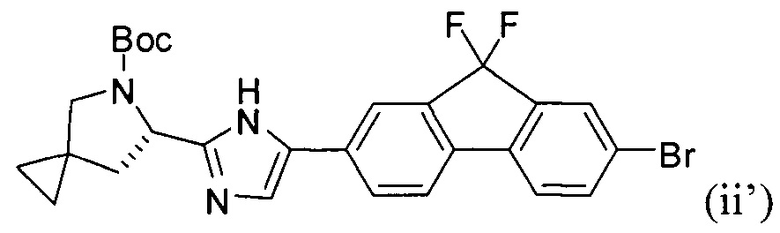
и фосфатом калия с получением соединения формулы (iii'):
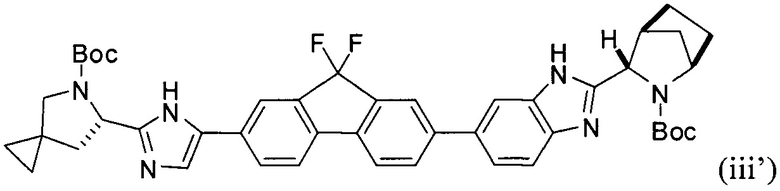 и
и
возможно приведение соединения формулы (iii') в контакт со щавелевой кислотой с получением оксалатной соли формулы (iii''):
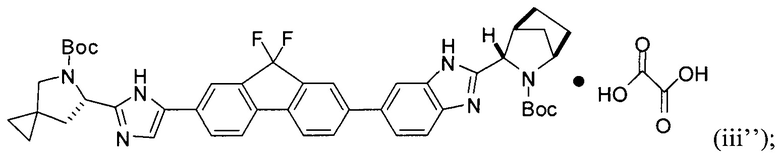
(3) приведение соединения формулы (iii') или формулы (iii'') в контакт с HCl с получением соединения формулы (iv'):
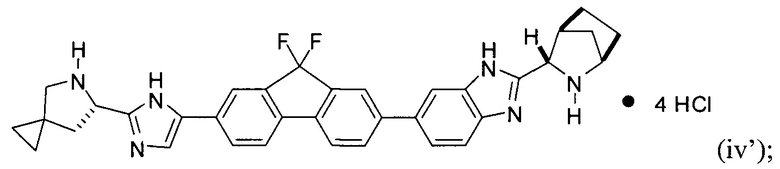
(4) приведение соединения формулы (iv') в контакт с (S)-2-(метоксикарбониламино)-3-метилбутановой кислотой:
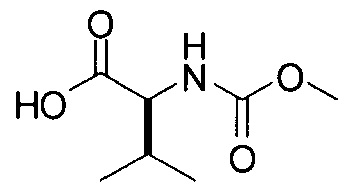
с получением соединения формулы I;
причем Воc в каждом случае представляет собой трет-бутоксикарбонил.
16. Способ получения соединения формулы I:
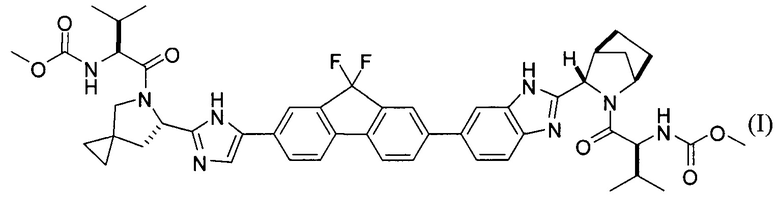
или его фармацевтически приемлемой соли или сольвата, включающий:
(1) последовательное приведение соединения формулы (а')
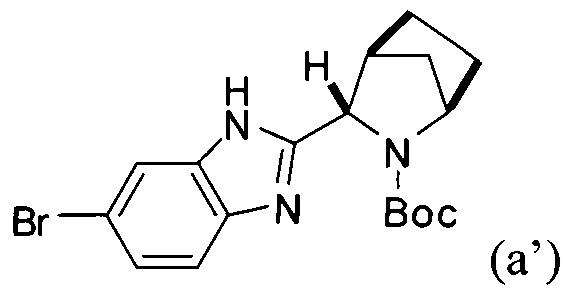
в контакт с каталитически эффективным количеством PdCl2[P(t-Bu)2Ph]2 и бис(пинаколато)дибором в присутствии пропионата калия с получением реакционной смеси, содержащей соединение формулы (ib):
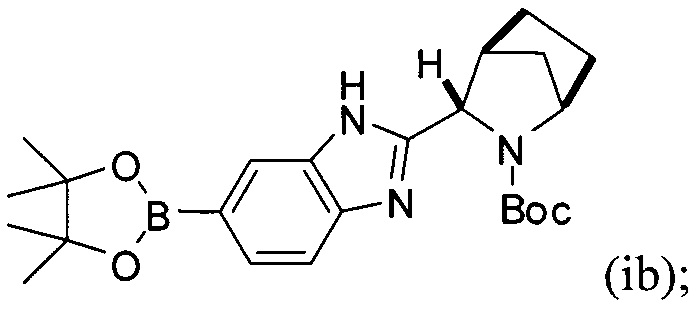
(2) приведение реакционной смеси, полученной на стадии (1), в контакт с соединением формулы (ii'):
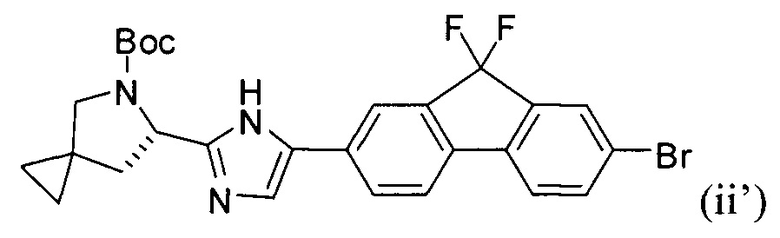
и фосфатом калия с получением соединения формулы (iii'):
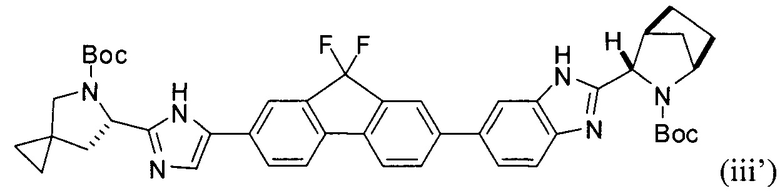 и
и
возможно приведение соединения формулы (iii') в контакт с щавелевой кислотой с получением оксалатной соли формулы (iii''):
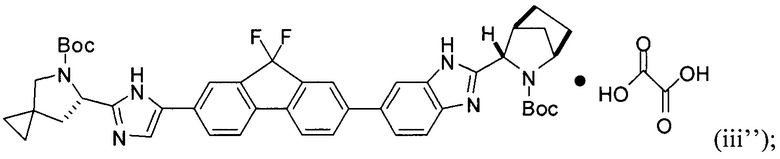
(3) приведение соединения формулы (iii') или формулы (iii'') в контакт с HCl с получением соединения формулы (iv'):
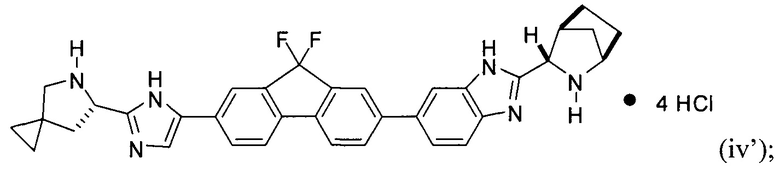
(4) приведение соединения формулы (iv') в контакт с (S)-2-(метоксикарбониламино)-3-метилбутановой кислотой:
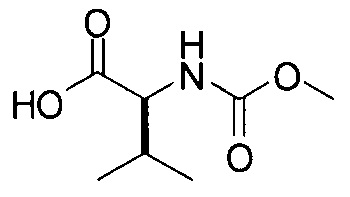
с получением соединения формулы I;
причем Воc в каждом случае представляет собой трет-бутоксикарбонил.
17. Соединение формулы (iii'):
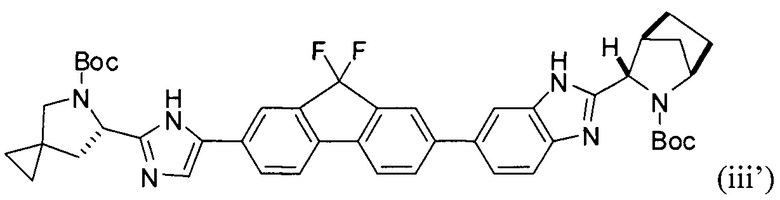
или его оксалат, соответствующий формуле (iii''):
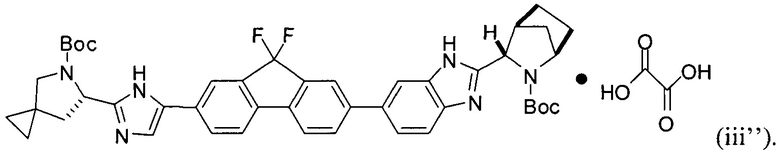
18. Соединение формулы (iv'):
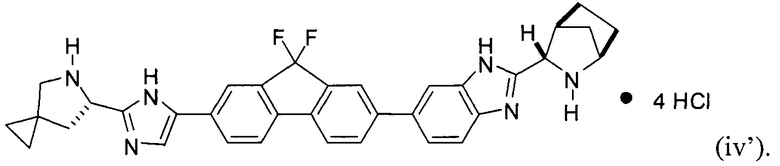
19. Соединение, представляющее собой гидрат согласно формуле:
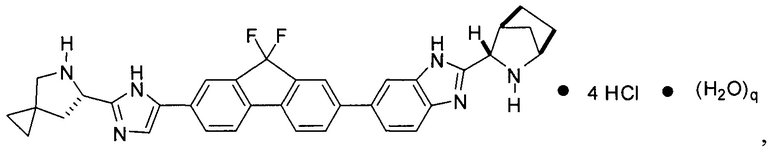
причем q представляет собой дробное или целое число от 0 до 7.
20. Соединение по п. 19, отличающееся тем, что q представляет собой число от 0 до 6 включительно.
21. Кристаллическое соединение формулы iv' (Форма I) по п.18, характеризующееся рентгеновской порошковой дифрактограммой, содержащей следующие пики: 7,1, 8,2, 10,8°2θ±0,2°2θ, определенные на дифрактометре с использованием излучения Cu-Kα при длине волны 1,54060  .
.
22. Кристаллическая Форма I по п.21, отличающаяся тем, что дифрактограмма дополнительно содержит пики при: 11,1, 12,8, 14,1, 14,8, 16,1, 18,9, 24,5, 24,9 и 25,9°2θ±0,2°2θ.
23. Кристаллическая Форма I по п.21, отличающаяся тем, что дифрактограмма по существу представляет собой дифрактограмму, представленную на Фиг. 1.
24. Кристаллическое соединение формулы iv' (Форма II) по п.18, характеризующееся рентгеновской порошковой дифрактограммой, содержащей следующие пики: 7,4, 9,4, и 11,6 2θ±0,2°2θ, определенные на дифрактометре при 25°С с использованием излучения Cu-Kα при длине волны 1,54060 .
25. Кристаллическая Форма II по п.24, отличающаяся тем, что дифрактограмма дополнительно содержит пики при: 7,5, 14,9, 15,2, 22,5, 23,2 и 26,3°2θ±0,2°2θ.
26. Кристаллическая Форма II по п.24, отличающаяся тем, что дифрактограмма по существу представляет собой дифрактограмму, представленную на Фиг. 2.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| ЕА 201001274 А1, 30.06.2011. | |||

