РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет предварительной заявки США № 61/507347, поданной 13 июля 2011 г., содержание которой включено в настоящий документ в качестве ссылки в полном объеме.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Астма является хроническим воспалительным заболеванием дыхательных путей, характеризующимся хрипами, удушьем, сдавленностью в груди и кашлем. Астма поражает приблизительно 20 миллионов людей в США, и приблизительно 75% пациентов с астмой являются взрослыми людьми. Из числа пациентов с астмой, приблизительно 60% пациентов с астмой страдают легкой формой заболевания, приблизительно 20% страдают умеренной тяжестью заболевания и оставшиеся 20% страдают тяжелой формой заболевания.
Полагают, что интерлейкин-13 (IL-13) является ключевым в патогенезе астмы человека, поскольку повышенные уровни IL-13 присутствуют в легких пациентов с астмой, и эти уровни коррелируют с тяжестью заболевания (фигура 1). Аналогично, повышенное количество IL-13 присутствует как в мокроте, так и в биопсийных пробах пациентов с умеренной до тяжелой формами астмы, которые получают лечение ингалируемыми кортикостироидами (ICS) или системными кортикостероидами и, которые продолжают проявлять симптоматику. Более того, генетические полиморфизмы IL-13 человека связаны с астмой и атопией (аллергической гиперчувствительностью). IL-13 связывается с двумя рецепторами, IL-13Rα1 и IL-13Rα2. IL-13 является хорошо известной мишенью для астмы, так как была продемонстрирована эффективность действия с использованием разнообразных средств антагонизма IL-13 на множественных, доклинических моделях астмы.
Вследствие роли IL-13 человека при разнообразных расстройствах у человека, были разработаны терапевтические подходы для ингибирования или противодействия активности IL-13. В частности, проводится поиск антител, которые связываются с IL-13 и нейтрализуют его, в качестве средства для ингибирования активности IL-13. Однако в данной области существует необходимость в усовершенствованных антителах, способных к связыванию IL-13 для лечения астмы.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам и композициям для лечения астмы, например, легкой или умеренной формы, с использованием антитела против IL-13, или его антигенсвязывающей части.
В одном из аспектов изобретение относится к выделенной композиции, содержащей антитело против IL-13, или его антигенсвязывающую часть, которые, если их вводят внутривенно индивиду при дозе, равной приблизительно 0,3 мг/кг антител или их антигенсвязывающей части, способны к проявлению: (a) площади под кривой (AUC) между приблизительно 1500 и приблизительно 2700 мкгч/мл; (b) объема распределения между приблизительно 65 и 125 мл/кг; (c) пиковой концентрации (Cмакс) между приблизительно 5 и приблизительно 8 мкг/мл; и (d) скорости выведения между приблизительно 0,1 и приблизительно 0,2 мл/ч/кг.
В еще одном аспекте изобретение относится к выделенной композиции, содержащей антитело против IL-13 или его антигенсвязывающую часть, в которой, если композицию вводят внутривенно индивиду при дозе, равной приблизительно 3 мг/кг, антитела или их антигенсвязывающая часть способны к проявлению: (a) площади под кривой (AUC) между приблизительно 21000 и приблизительно 33500 мкгч/мл; (b) объема распределения между приблизительно 55 и приблизительно 100 мл/кг; (c) пиковой концентрации (Cмакс) между приблизительно 55 и приблизительно 90 мкг/мл; и (d) скорости выведения между приблизительно 0,08 и приблизительно 0,15 мл/ч/кг.
В еще одном аспекте изобретение относится к выделенной композиции, содержащей антитела против IL-13, или их антигенсвязывающую часть, в которой, если ее вводят внутривенно индивиду при дозе, равной приблизительно 10 мг/кг, антитела, или их антигенсвязывающая часть, способны к проявлению: (a) площади под кривой (AUC) между приблизительно 75 и приблизительно 100 мкгч/мл; (b) объема распределения между приблизительно 90 и приблизительно l30 мл/кг; (c) пиковой концентрации (Cмакс) между приблизительно 185 и приблизительно 250 мкг/мл; и (d) скорости выведения между приблизительно 0,1 и приблизительно 0,15 мл/ч/кг.
В еще одном аспекте изобретение относится к выделенной композиции, содержащей антитела против IL-13, или их антигенсвязывающую часть, в которой, если ее вводят подкожно индивиду при дозе, равной приблизительно 0,3 мг/кг, антитела или их антигенсвязывающая часть, способны к проявлению: (a) площади под кривой (AUC) между приблизительно 125 и приблизительно 800 мкгч/мл; и (b) пиковой концентрации (Cмакс) между приблизительно 1.0 и приблизительно 6,0 мкг/мл.
В еще одном аспекте изобретение относится к выделенной композиции, содержащей антитело против IL-13, или его антигенсвязывающую часть, в которой, если ее вводят подкожно индивиду при дозе, равной приблизительно 3 мг/кг, антитела или их антигенсвязывающая часть, способны к проявлению: (a) площади под кривой (AUC) между приблизительно 1100 и приблизительно 8500 мкгч/мл; и (b) пиковой концентрации (Cмакс) между приблизительно 12 и приблизительно 60 мкг/мл.
В одном из вариантов осуществления антитело против IL-13, или его антигенсвязывающая часть, представляют собой 13C5.5 или их антигенсвязывающую часть. В еще одном варианте осуществления композиция представляет собой фармацевтическую композицию.
В еще одном аспекте изобретение относится к способам лечения или профилактики астмы у индивида посредством введения композиции по изобретению индивиду, посредством чего происходит лечение или профилактика астмы. В одном из вариантов осуществления композицию вводят однократно. В еще одном варианте осуществления композицию вводят еженедельно. В еще одном другом варианте осуществления композицию вводят в течение приблизительно 3 недель.
В одном из вариантов осуществления астма является астмой в легкой до умеренной форме. В еще одном варианте осуществления индивид является человеком.
В еще одном варианте осуществления способ дополнительно включает введение дополнительного средства. В одном из вариантов осуществления дополнительное средство выбрано из группы, состоящей из терапевтического средства, средства визуализации, цитотоксического средства, ингибитора ангиогенеза, ингибитора киназы, блокатора молекул со-стимуляции, блокатора молекул адгезии, антител против цитокинов или их функционального фрагмента; метотрексата, циклоспорина, рапамицина, FK506, детектируемой метки или репортера, антагониста ФНО, противоревматического средства, мышечного релаксанта, наркотического средства, нестероидного противовоспалительного лекарственного средства (NTHE), анальгезирующего средства, анестезирующего средства, седативного средства, местного анестезирующего средства, нейромышечного блокатора, противомикробного средства, антипсориатического средства, кортикостероида, анаболического стероида, эритропоэтина, средства для иммунизации, иммуноглобулина, иммуносупрессора, гормона роста, средства для гормонозаместительной терапии, радиофармпрепарата, антидепрессанта, антипсихотического средства, стимулятора, медикамента для лечения астмы, бета-агониста, ингалируемого стероида, перорального стероида, эпинефрина или аналога, цитокина и антагониста цитокина.
В еще одном аспекте изобретение относится к способам лечения астмы у индивида посредством введения внутривенно индивиду антител против IL-13, или их антигенсвязывающей части, где по меньшей мере одна фармакокинетическая характеристика выбрана из группы, состоящей из: (a) максимальной сывороточной концентрации (Cмакс) между приблизительно 5 и приблизительно 235 мкг/мл, и (b) площади под кривой концентрация-время для сыворотки (AUC) между приблизительно 1500 и приблизительно 98000 мкгч/мл, достигаемую после введения антител, или их антигенсвязывающей части индивиду.
В одном из вариантов осуществления антитела или их антигенсвязывающую часть, вводят при дозе, равной приблизительно 0,3 мг/кг. В одном из вариантов осуществления Cмакс находится между приблизительно 5 и приблизительно 10 мкг/мл. В одном из вариантов осуществления AUC находится между приблизительно 1500 и приблизительно 2700 мкгч/мл.
В еще одном варианте осуществления антитела или их антигенсвязывающую часть вводят при дозе, равной приблизительно 3 мг/кг. В одном из вариантов осуществления Cмакс находится между приблизительно 55 и приблизительно 90 мкг/мл. В еще одном варианте осуществления AUC находится между приблизительно 20000 и приблизительно 34000 мкгч/мл.
В еще одном варианте осуществления антитела или их антигенсвязывающую часть, вводят при дозе, равной приблизительно 10 мг/кг. В одном из вариантов осуществления Cмакс находится между приблизительно 190 и приблизительно 235 мкг/мл. В одном из вариантов осуществления AUC находится между приблизительно 75000 и приблизительно 100000 мкгч/мл.
В еще одном варианте осуществления значение Cмакс находится между приблизительно 20 и приблизительно 30 (мкг/мл)/(мг/кг) после нормализации дозы. В еще одном варианте осуществления AUC находится между приблизительно 6000 и приблизительно 10000 (мкгч/мл)/(мг/кг) после нормализации дозы.
В еще одном другом аспекте изобретение относится к способам лечения астмы у индивида посредством введения подкожно индивиду антител против IL-13, или их антигенсвязывающей части, где по меньшей мере одна фармакокинетическая характеристика выбрана из группы, состоящей из: (a) максимальной сывороточной концентрации (Cмакс) между приблизительно 1 и приблизительно 60 мкг/мл и (b) площади под кривой концентрация-время для сыворотки (AUC) между приблизительно 125 и приблизительно 8100 мкгч/мл, достигают после введения антитела, или его антигенсвязывающей части индивиду.
В одном из вариантов осуществления антитела или их антигенсвязывающую часть вводят при дозе, равной приблизительно 0,3 мг/кг. В одном из вариантов осуществления Cмакс находится между приблизительно 1 и приблизительно 6 мкг/мл. В еще одном варианте осуществления AUC находится между приблизительно 100 и приблизительно 800 мкгч/мл.
В еще одном варианте осуществления антитела или их антигенсвязывающую часть вводят при дозе, равной приблизительно 3 мг/кг. В одном из вариантов осуществления Cмакс находится между приблизительно 12 и приблизительно 60 мкг/мл. В еще одном варианте осуществления AUC находится между приблизительно 1100 и приблизительно 8100 мкгч/мл.
В одном из вариантов осуществления антитела против IL-13, или их антигенсвязывающая часть представляют собой 13C5.5 или их антигенсвязывающую часть. В еще одном варианте осуществления индивид является человеком. В еще одном варианте осуществления антитела против IL-13 или их антигенсвязывающую часть вводят однократно. В еще одном варианте осуществления антитела против IL-13 или их антигенсвязывающую часть вводят еженедельно. В еще одном другом варианте осуществления антитела против IL-13, или их антигенсвязывающую часть, вводят в течение трех недель.
В одном из вариантов осуществления астма представляет собой астму от легкой до умеренной формы.
В еще одном варианте осуществления способ дополнительно включает введение дополнительного средства. В одном из вариантов осуществления дополнительное средство выбрано из группы, состоящей из терапевтического средства, средства визуализации, цитотоксического средства, ингибитора ангиогенеза, ингибитора киназы, блокатора молекул со-стимуляции, блокатора молекул адгезии, антител против цитокинов или их функционального фрагмента; метотрексата, циклоспорина, рапамицина, FK506, детектируемой метки или репортера, антагониста ФНО, противоревматического средства, мышечного релаксанта, наркотического средства, нестероидного противовоспалительного лекарственного средства (NTHE), анальгезирующего средства, анестезирующего средства, седативного средства, местного анестезирующего средства, нейромышечного блокатора, противомикробного средства, антипсориатического средства, кортикостероида, анаболического стероида, эритропоэтина, средства для иммунизации, иммуноглобулина, иммуносупрессора, гормона роста, средства для гормонозаместительной терапии, радиофармпрепарата, антидепрессанта, антипсихотического средства, стимулятора, медикамента для лечения астмы, бета-агониста, ингалируемого стероида, перорального стероида, эпинефрина или аналога, цитокина и антагониста цитокина.
В еще одном другом аспекте изобретение относится к способам лечения астмы у индивида посредством введения подкожно индивиду антител против IL-13, или их антигенсвязывающей части, при дозе, равной приблизительно 0,3 мг/кг, где по меньшей мере одна фармакокинетическая характеристика выбрана из группы, состоящей из: (a) времени полужизни между приблизительно 24 и 31 днями; (b) Tмакс между от приблизительно 3 до приблизительно 5 днями; и (c) биодоступности, равной по меньшей мере приблизительно 60%, которых достигают после введения антител или их антигенсвязывающей части индивиду. В одном из вариантов осуществления биодоступность составляет по меньшей мере приблизительно 70%.
В еще одном другом аспекте изобретение относится к способам лечения астмы у индивида посредством введения подкожно индивиду антител против IL-13, или их антигенсвязывающей части при дозе, равной приблизительно 3 мг/кг, где по меньшей мере одна фармакокинетическая характеристика выбрана из группы, состоящей из: (a) времени полужизни между приблизительно 23 и 26 днями; (b) Tмакс меньшего чем или равного до приблизительно 5 дней; и (c) биодоступности, равной по меньшей мере приблизительно 60%, достигаемой после введения антител или их антигенсвязывающей части индивиду. В одном из вариантов осуществления биодоступность составляет по меньшей мере приблизительно 70%.
В еще одном аспекте изобретение относится к способам лечения астмы у индивида, включающим введение внутривенно индивиду антител против IL-13, или их антигенсвязывающей части при дозе, равной приблизительно 0,3 мг/кг, где по меньшей мере одна фармакокинетическая характеристика выбрана из группы, состоящей из: (a) скорости выведения между от приблизительно 0,11 до приблизительно 0,19 мл/час/кг; и (b) объема распределения между от приблизительно 70 до приблизительно 130 мл/кг, достигаемых после введения антител, или их антигенсвязывающей части индивиду.
В еще одном аспекте изобретение относится к способам лечения астмы у индивида посредством введения внутривенно индивиду антител против IL-13, или их антигенсвязывающей части при дозе, равной приблизительно 3 мг/кг, где по меньшей мере одна фармакокинетическая характеристика выбрана из группы, состоящей из: (a) скорости выведения между приблизительно 0,08 до приблизительно 0,14 мл/час/кг; и (b) объема распределения между приблизительно 55 до приблизительно 100 мл/кг, достигаемых после введения антител или их антигенсвязывающей части индивиду.
В еще одном аспекте изобретение относится к способам лечения астмы у индивида, включающим введение внутривенно индивиду антител против IL-13, или их антигенсвязывающей части при дозе, равной приблизительно 10 мг/кг, где по меньшей мере одна фармакокинетическая характеристика выбрана из группы, состоящей из: (a) скорости выведения между от приблизительно 0,09 до приблизительно 0,13 мл/час/кг; и (b) объема распределения между от приблизительно 85 до приблизительно 130 мл/кг, которых достигают после введения антител, или их антигенсвязывающей части индивиду.
В одном из вариантов осуществления антитела против IL-13, или их антигенсвязывающая часть представляют собой 13C5.5, или их антигенсвязывающую часть. В еще одном варианте осуществления индивид является человеком. В одном из вариантов осуществления антитела против IL-13 или их антигенсвязывающую часть, вводят однократно. В еще одном варианте осуществления антитела против IL-13, или их антигенсвязывающую часть, вводят еженедельно. В еще одном другом варианте осуществления антитела против IL-13, или их антигенсвязывающую часть, вводят в течение 3 недель.
В одном из вариантов осуществления астма представляет собой астму от легкой до умеренной формы.
В еще одном варианте осуществления способы дополнительно включают введение дополнительного средства. В одном из вариантов осуществления дополнительное средство выбрано из группы, состоящей из терапевтического средства, средства визуализации, цитотоксического средства, ингибитора ангиогенеза, ингибитора киназы, блокатора молекул со-стимуляции, блокатора молекул адгезии, антител против цитокинов или их функционального фрагмента; метотрексата, циклоспорина, рапамицина, FK506, детектируемой метки или репортера, антагониста ФНО, противоревматического средства, мышечного релаксанта, наркотического средства, нестероидного противовоспалительного лекарственного средства (NTHE), анальгезирующего средства, анестезирующего средства, седативного средства, местного анестезирующего средства, нейромышечного блокатора, противомикробного средства, антипсориатического средства, кортикостероида, анаболического стероида, эритропоэтина, средства для иммунизации, иммуноглобулина, иммуносупрессора, гормона роста, средства для гормонозаместительной терапии, радиофармпрепарата, антидепрессанта, антипсихотического средства, стимулятора, медикамента для лечения астмы, бета-агониста, ингалируемого стероида, перорального стероида, эпинефрина или аналога, цитокина и антагониста цитокина.
В одном из вариантов осуществления индивид представляет собой человека.
В еще одном аспекте изобретение относится к выделенной композиции, содержащей антитела против IL-13, или их антигенсвязывающую часть, где, когда ее вводят внутривенно индивиду при дозе, равной приблизительно 0,3 мг/кг, 1 мг/кг, 3 мг/кг или 10 мг/кг, антитела или их антигенсвязывающая часть способны к проявлению какого-либо из фармакокинетического параметра, представленного в описании, Таблицах или на Фигурах.
В еще одном аспекте изобретение относится к выделенной композиции, содержащей антитела против IL-13, или их антигенсвязывающую часть, где, когда ее вводят подкожно индивиду при дозе, равной приблизительно 0,3 мг/кг, 1 мг/кг или 3 мг/кг, антитела или их антигенсвязывающая часть является способными к проявлению какого-либо из фармакокинетического параметров, представленного в описании, Таблицах или на Фигурах.
В еще одном аспекте изобретение относится к способам лечения или профилактики астмы у индивида посредством введения внутривенно индивиду антител против IL-13 или их антигенсвязывающей части при дозе, равной приблизительно 0,3 мг/кг, 1 мг/кг, 3 мг/кг или 10 мг/кг, где по меньшей мере одна из фармакокинетических характеристик, представленных в описании, Таблицах или на Фигурах достигается после введения антител или их антигенсвязывающей части индивиду.
В еще одном другом аспекте изобретение относится к способам лечения или профилактики астмы у индивида посредством введения подкожно индивиду антител против IL-13 или их антигенсвязывающей части при дозе, равной приблизительно 0,3 мг/кг, 1 мг/кг, 3 мг/кг или 10 мг/кг, где по меньшей мере одна из фармакокинетических характеристик представленная в описании, Таблицах или на Фигурах достигается после введения антител, или их антигенсвязывающей части индивиду.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигуре 1 представлен график, отображающий то, что экспрессия IL-13 предшествует дисфункции легких.
На фигуре 2 показано, что 13C5.5, антитела против IL-13, получают из гибридомы с уникальными эпитопами и клеточной линией.
На фигуре 3 показано, что 13C5.5, антитела против IL-13, нейтрализуют IL-13 в легком.
На фигуре 4 показана схема первого дозирования у человека (FIN), применяемого на Фазе I клинических испытаний.
На фигуре 5 показан профиль зависимости средней концентрации 13C5.5 в сыворотке от времени после однократных внутривенных инфузий по 0,3 мг/кг, 1 мг/кг, 3 мг/кг и 10 мг/кг 13C5.5 здоровым индивидам по линейной шкале.
На фигуре 6 показаны фармакокинетические параметры 13C5.5, антител против IL-13.
На фигуре 7 показан профиль зависимости средней концентрации в сыворотке от времени после однократных внутривенных инфузий по 0,3 мг/кг, 1 мг/кг, 3 мг/кг и 10 мг/кг 13C5.5 здоровым индивидам и индивидам с астмой по линейной шкале.
На фигуре 8 показаны фармакокинетические параметры 13C5.5 антител против IL-13.
На фигуре 9 показаны фармакокинетические параметры 13C5.5, антител против IL-13.
На фигуре 10 показаны фармакокинетические параметры 13C5.5 антител против IL-13. Биодоступность после подкожного введения оценивали, как равную приблизительно 70%.
На фигуре 11 показано нормализованные значения Cмакс для средней дозы после трех подкожных инъекций еженедельно по 0,3 мг/кг 13C5.5 (Группа 8) и 3 мг/кг 13C5.5 (Группа 9) (Часть 3 клинического испытания).
На фигуре 12 показано нормализованные значения AUC0-168 для средней дозы после трех подкожных инъекций 13C5.5 еженедельно по 0,3 мг/кг (Группа 8) и 3 мг/кг (Группа 9) (Часть 3 клинического испытания).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам и композициям для лечения астмы, например, легкой или умеренной формы, с использованием антител против IL-13 или их антигенсвязывающей части.
Настоящее изобретение будет более понято после определения некоторых терминов.
Термин "полипептид", как используется в настоящей заявке, относится к любой полимерной цепи из аминокислот. Термины "пептид" и "белок" используют взаимозаменяемо с термином полипептид и также относят к полимерной цепи из аминокислот. Термин "полипептид" охватывает нативные или искусственные белки, фрагменты белка и полипептидные аналоги белковой последовательности. Полипептид может быть мономерным или полимерным.
Термин "выделенный белок" или "выделенный полипептид" означает белок или полипептид, который по своей природе или источнику выделения не связан с природными компонентами, которые находятся вместе с ним в его нативном состоянии; не содержит по существу других белков того же вида; экспрессируется клетками различных видов; или не встречается в природе. Таким образом, полипептид, который химически синтезирован или синтезирован в клеточной системе, отличной от клетки, в которой он существует в природе, будет "выделен" от его природных компонентов. Белок, по существу не содержащий природных компонентов, может также быть получен посредством выделения с использованием способов очистки белка, хорошо известных в данной области.
Термин "выделение" как используют в настоящем описании, относится к способу получения химических типов, таких как полипептид, по существу не содержащих природных компонентов посредством выделения, например, с использованием способов очистки белка, хорошо известных в данной области.
Термины "IL-13" и "IL-13 дикого типа" (сокращенные в настоящем описании как IL-13, IL-13 дт), как используют в настоящем описании, включают цитокин, который секретируется прежде всего Т-хелперными клетками второго типа. Термин включает мономерный белок из полипептида с массой 13 кДа. Структура IL-13 дополнительно описана, например, в статье Moy, Diblasio et al. 2001 J Mol Biol 310 219-30. Подразумевают, что термин IL-13 включает рекомбинантный IL-13 человека (рч IL-13), который может быть получен стандартными способами рекомбинантной экспрессии. Аминокислотная последовательность IL-13 человека, SEQ ID NO:1, является известной в данной области.
Последовательность IL-13 человека - SEQ ID NO: 1
MALLLTTVIALTCLGGFASPGPVPPSTALRELIEELVNITQNQKAPLCNGSMVWS INLTAGMYCAALESLINVSGCSAIEKTQRMLSGFCPHKVSAGQFSSLHVRDTKIE VAQFVKDLLLHLKKLFREGRFN
Термин "вариант IL-13" (сокращенный в настоящем описании как IL-13v), как используют в настоящем описании, включает вариант IL-13, где аминокислотный остаток в положении 130 SEQ ID NO:1 заменен с Аргинина на Глутамин (R130Q).
"Биологическая активность", как используют в настоящем описании, относится ко всем характерным биологическим свойствам цитокина. Биологические свойства IL-13 включают, но ими не ограничены, связывание с рецептором IL-13; (другие примеры включают изотип иммуноглобулина, переходящий в IgE в B-клетках человека и подавляющий продукцию воспалительного цитокина).
Термины "специфическое связывание" или "специфически связываясь с", как используют в настоящем описании, со ссылкой на взаимодействие антитела, белка или пептида со вторым химическим видом, означает, что взаимодействие зависит от присутствия конкретной структуры (например, антигенной детерминанты или эпитопа) у химического вида; например, антитело распознает и связывается с конкретной белковой структурой в большей степени, чем с белками в целом. Если антитело является специфичным для эпитопа "A", присутствие молекулы, содержащей эпитоп (или свободный, немеченный A), в реакции, содержащей меченный "A" и антитело, будет снижать количество меченного А, связанного с антителом.
Термин "антитело", как используют в настоящем описании, в широком смысле относится к любой иммуноглобулиновой (Ig) молекуле, состоящей из четырех полипептидных цепей, двух тяжелых (H) цепей и двух легких (L) цепей, или любому их функциональному фрагменту, мутанту, варианту или деривату, который сохраняет существенные признаки связывания с эпитопом молекулы Ig. Такие форматы мутанта, варианта или производного антитела являются известными в данной области. Их неограничивающие варианты осуществления обсуждаются в настоящем документе. В одном из вариантов осуществления антитело, используемое в композициях и способах по изобретению, представляет собой антитело против IL-13, 13C5.5, описанное в патенте США № 7915388, включенном в настоящее описание в качестве ссылки. В еще одном варианте осуществления антитело, используемое в композициях и способах по изобретению, представляет собой антитело 6A1, 3G4, тралокинумаб, лебрикизумаб, QAZ-576, IMA-638 или IMA-026.
В полноразмерном антителе каждая тяжелая цепь состоит из вариабельной области тяжелой цепи (сокращенной в настоящем описании как HCVR или VH) и константной области тяжелой цепи. Константная область тяжелой цепи состоит из трех доменов, CH1, CH2 и CH3. Каждая легкая цепь состоит из вариабельной области легкой цепи (сокращенной в настоящем описании как LCVR или VL) и константной области легкой цепи. Константная область легкой цепи состоит из одного домена, CL. Области VH и VL могут быть дополнительно подразделены на области гипервариабельности, называемые области, определяющие комплементарность (CDR), перемежающиеся с областями, которые являются более консервативными, называемыми каркасные области (FR). Каждая VH и VL составлена из трех CDR и четырех FR, расположенных от амино-конца к карбокси-концу в следующем порядке: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. Иммуноглобулиновые молекулы могут принадлежать к любому типу (например, IgG, IgE, IgM, IgD, IgA и IgY), классу (например, IgG1, IgG2, IgG3, IgG4, IgA1 и IgA2) или подклассу.
Термин "антигенсвязывающая часть" антитела (или просто "часть антитела"), как используют в настоящем описании, относится к одному или нескольким фрагментам антитела, которые сохраняют способность специфически связываться с антигеном (например, IL-13). Было показано, что антигенсвязывающая функция антитела может осуществляться фрагментами полноразмерного антитела. Такие варианты осуществления антитела могут также иметь биспецифический, с двойной специфичностью или мультиспецифичные форматы; специфически связываясь с двумя или или несколькими различными антигенами. Примеры связывающих фрагментов, охваченные термином "антигенсвязывающая часть" антитела включают (i) Fab-фрагмент, моновалентный фрагмент, состоящий из доменов VL, VH, CL и CH1; (ii) F(ab')2-фрагмент, бивалентный фрагмент, содержащий два Fab фрагмента, связанные посредством дисульфидной области в шарнирной области; (iii) Fd-фрагмент, состоящий из доменов VH и CH1; (iv) Fv-фрагмент, состоящий из доменов VL и VH одиночного плеча антитела, (v) dAb-фрагмент (Ward et al., (1989) Nature 341:544-546, Winter et al., опубликованная заявка PCT WO 90/05144 A1, включенная в настоящее описание в качестве ссылки), которая содержит одиночный вариабельный домен; и (vi) выделенная область, определяющая комплементарность (CDR). Кроме того, несмотря на то, что два домена Fv-фрагмента, VL и VH, кодируются раздельными генами, они могут быть объединены, с использованием рекомбинантных способов, посредством синтетического линкера, который обеспечивает им получение в виде единственной белковой цепи, в которой области VL и VH образуют пару с образованием моновалентных молекул (известных как Fv с одиночной цепью (scFv); см., например, Bird et al. (1988) Science 242:423-426; и Huston et al. (1988) Proc. Natl. Acad. Sci. USA 85:5879-5883). Подразумевают, что такие антитела с одиночной цепью охватываются термином "антигенсвязывающая часть" антитела. Другие формы антител с одиночной цепью, такие как диатела также являются охваченными. Диатела представляют собой бивалентные, биспецифические антитела, в которых домены VH и VL экспрессируются на одиночной полипептидной цепи, но с использованием линкера, который является слишком коротким, чтобы обеспечить возможность спаривания между двумя доменами на той же цепи, посредством этого заставляя домены спариваться с комплементарными доменами еще одной другой цепи и создавать два участка связывания антигена (см., например, Holliger, P., et al. (1993) Proc. Natl. Acad. Sci. USA 90:6444-6448; Poljak, R. J., et al. (1994) Structure 2: 1121-1123). Такие части связывания антител являются известными в данной области (Kontermann and Dubel eds., Antibody Engineering (2001) Springer-Verlag. New York. 790 pp. (ISBN 3-540-41354-5).
Термин "конструкция антитела", как используют в настоящем описании, относится к полипептиду, содержащему одну или несколько антигенсвязывающих частей по изобретению, связанных с линкерным полипептидом или константным доменом иммуноглобулина. Линкерные полипептиды содержат два или несколько аминокислотных остатков, соединенных посредством пептидных связей, и применяют для связывания одной или нескольких антигенсвязывающих частей. Такие линкерные полипептиды являются хорошо известными в данной области (см., например, Holliger, P., et al. (1993) Proc. Natl. Acad. Sci. USA 90:6444-6448; Poljak, R. J., et al. (1994) Structure 2: 1121-1123). Константный домен иммуноглобулина относится к константному домену тяжелой или легкой цепи. Аминокислотные последовательности константного домена тяжелой цепи и легкой цепи IgG человека являются известными в данной области и раскрыты в Таблице 2 Патента США № 7915388, полное содержание которого включено в настоящее описание в качестве ссылки.
Более того, антитело или его антигенсвязывающая часть могут быть частью более крупных молекул иммуноадгезии, образованных посредством ковалентной или нековалентной связи антитела или части антитела с одним или несколькими другими белками или пептидами. Примеры таких молекул иммуноадгезии включают применение центральной области стрептавидина для создания тетрамерной молекулы scFv (Kipriyanov, S. M., et al. (1995) Human Antibodies and Hybridomas 6:93-101) и применение цистеинового остатка, маркерного пептида и C-концевой полигистидиновой метки для получения бивалентных и биотинилированных молекул scFv (Kipriyanov, S. M., et al. (1994) Mol. Immunol. 31: 1047-1058). Части антитела, такие как фрагменты Fab и F(ab')2, могут быть получены из цельных антител с использованием общепринятых способов, таких как расщепление папаином или пепсином, соответственно, цельных антител. Более того, антитела, части антител и молекулы иммуноадгезии могут быть получены с использованием стандартных способов рекомбинантных ДНК, как описано в настоящем описании.
Подразумевают, что термин "выделенное антитело", как используют в настоящем описании, относится к антителу, которое по существу не содержит других антител, имеющих отличные антигенные специфичности (например, выделенное антитело, которое специфически связывается с IL-13, по существу не содержит антитела, которые специфически связываются с антигенами, отличными от IL-13). Выделенное антитело, которое специфически связывается с IL-13, может, однако, иметь перекрестную реактивность по отношению к другим антигенам, таким как молекулы IL-13 от другого вида. Кроме того, выделенное антитело может по существу не содержать другого клеточного материала и/или химикатов.
Подразумевают, что термин "антитело человека", как используют в настоящем описании, включает антитела, имеющие вариабельные и константные области, происходящие от последовательностей иммуноглобулинов человека эмбрионального типа. Антитела человека по изобретению могут включать аминокислотные остатки, некодируемые последовательностями иммуноглобулинов человека эмбрионального типа (например, посредством мутаций, вводимых посредством разупорядоченного или сайт-специфического мутагенеза in vitro или посредством соматической мутации in vivo), например в CDR и, в частности, CDR3. Однако, термин "антитело человека", как используют в настоящем описании, не подразумевает включение антител, в которых последовательности CDR, происходящие от эмбрионального типа еще одного вида млекопитающих, такого как мышь, были привиты на каркасные последовательности человека.
Термин "рекомбинантное антитело человека", как используют в настоящем описании, предназначен для включения всех антител человека, которые получают, экспрессируют, создают или выделяют посредством рекомбинантных средств, таких как антитела, экспрессируемые с использованием рекомбинантного вектора экспрессии, трансфицированного в клетку-хозяина (описанные дополнительно в Патенте США № 7915388, содержание которого включено в настоящем описании в качестве ссылки), антитела, выделенные из рекомбинантных, комбинаторных библиотек антител человека (Hoogenboom H. R., (1997) TIB Tech. 15:62-70; Azzazy H., and Highsmith W. E., (2002) Clin. Biochem. 35:425-445; Gavilondo J. V., and Larrick J. W. (2002) BioTechniques 29: 128-145; Hoogenboom H., and Chames P. (2000) Immunology Today 21:371-378), антитела, выделенные из животного (например, мыши), которое является трансгенным для генов иммуноглобулинов человека (см., например, Taylor, L. D., et al. (1992) Nucl. Acids Res. 20:6287-6295; Kellermann S-A., and Green L. L. (2002) Current Opinion in Biotechnology 13:593-597; Little M. et al (2000) Immunology Today 21:364-370) или антитела, полученные, экспрессируемые, создаваемые или выделенные посредством любого другого средства, которое включает сплайсинг генной последовательности иммуноглобулина человека к другой последовательности ДНК. Такие рекомбинантные антитела человека имеют вариабельные и константные области, происходящие от последовательностей иммуноглобулинов человека эмбрионального типа. В некоторых вариантах осуществления, однако, такие рекомбинантные антитела человека подвергают мутагенезу in vitro (или, когда используют животное, трансгенное для последовательности Ig человека, соматическому мутагенезу in vivo) и, таким образом, аминокислотные последовательности областей VH и VL рекомбинантных антител, представляют собой последовательности, которые, будучи производными от и относящимся к последовательностям VH и VL эмбрионального типа человека, могут естественным образом не существовать в эмбриональном наборе антитела человека in vivo. Один вариант осуществления предоставляет полностью человеческие антитела, способные к связыванию с IL-13 человека, которые можно получать с использованием способов, хорошо известных в данной области, таких как, но ими не ограничиваясь, с использованием библиотек фагов Ig человека, таких как библиотеки, раскрытые в Jermutus et al., опубликованная заявка PCT № WO 2005/007699 A2.
Термин "химерное антитело" относится к антителам, которые содержат последовательности вариабельной области тяжелой и легкой цепи от одного вида и последовательности константной области от еще одного вида, такие как антитела, имеющие вариабельные области тяжелой и легкой цепи мыши, связанные с константными областями человека.
Термин "CDR-привитое антитело" относится к антителам, которые содержат последовательности вариабельной области тяжелой и легкой цепи от одного вида, но в которых последовательности одной или нескольких из областей CDR VH и/или VL заменены на CDR-последовательности еще одного вида, такие как антитела, имеющие мышиные вариабельные области тяжелой и легкой цепи, в которых одну или несколько из CDR мыши (например, CDR3) заменяют на последовательности CDR человека.
Термин "гуманизированное антитело" относится к антителам, которые содержат последовательности вариабельной области тяжелой и легкой цепи от нечеловеческого вида (например, мыши), но в которых по меньшей мере часть последовательности VH и/или VL была изменена, чтобы быть более "человекоподобной", т.е., более сходной с вариабельными последовательностями эмбрионального типа человека. Одним типом гуманизированного антитела является CDR-привитое антитело, в котором последовательности CDR человека вводят в последовательности нечеловеческих VH и VL для замены соответствующих нечеловеческих последовательностей CDR. В одном из вариантов осуществления предоставлены гуманизированные антитела против IL-13 человека и антигенсвязывающие части. Такие антитела получали посредством получения моноклональных антител мыши против IL-13 с использованием традиционной технологии гибридом с последующей гуманизацией с использованием генной инженерии in vitro, такой как, те, что раскрыты в Kasaian et al заявка PCT № WO 2005/123126 A2.
Термины "нумерация по Кэбату", "определения по Кэбату” и "мечение по Кэбату" используются в настоящем описании взаимозаменяемо. Эти термины, которые являются признанными в данной области, относятся к системе нумерации аминокислотных остатков, которые являются более вариабельными (т.е. гипервариабельными), чем другие аминокислотные остатки в вариабельных областях тяжелой и легкой цепи антитела или его антигенсвязывающей части (Kabat et al. (1971) Ann. NY Acad, Sci. 190:382-391 и, Kabat, E. A., et al. (1991) Sequences of Proteins of Immunological Interest, Fifth Edition, U.S. Department of Health и Human Services, NIH Publication No. 91-3242). Для вариабельной области тяжелой цепи гипервариабельная область находится в интервале положений аминокислот от 31 до 35 для CDR1, положений аминокислот от 50 до 65 для CDR2, и положений аминокислот от 95 до 102 для CDR3. Для вариабельной области легкой цепи гипервариабельная область находится в интервале положений аминокислот от 24 до 34 для CDR1, положений аминокислот от 50 до 56 для CDR2, и положений аминокислот от 89 до 97 для CDR3.
Как используют в настоящем описании, термины "акцептор" и "акцепторное антитело" относится к последовательности антитела или нуклеиновой кислоты, предоставляющей или кодирующей по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95%, по меньшей мере 96%, по меньшей мере 97%, по меньшей мере 98%, по меньшей мере 99% или 100% аминокислотных последовательностей одной или нескольких каркасных областей. В некоторых вариантах осуществления, термин "акцептор" относится к аминокислотной последовательности антитела или последовательности нуклеиновой кислоты, предоставляющей или кодирующей константные область(области). В еще одном другом варианте осуществления термин "акцептор" относится к аминокислотной последовательности антитела или последовательности нуклеиновой кислоты, предоставляющей или кодирующей одну или несколько каркасных областей и константных области(областей). В конкретном варианте осуществления термин "акцептор" относится к человеческой аминокислотной последовательности антитела или последовательности нуклеиновой кислоты, которая предоставляет или кодирует по меньшей мере 80%, предпочтительно, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95%, по меньшей мере 98% или 100% аминокислотных последовательностей одной или нескольких каркасных областей. В соответствии с этим вариантом осуществления, акцептор может содержать по меньшей мере 1, по меньшей мере 2, по меньшей мере 3, по меньшей мере 4, по меньшей мере 5 или по меньшей мере 10 аминокислотных остатков, которые не встречаются по одному или нескольким конкретным положениям антитела человека. Акцепторная каркасная область и/или акцепторные константные области могут быть, например, произведены или получены из гена антитела эмбрионального типа, гена зрелого антитела, функционального антитела (например, антител, хорошо известных в данной области, разрабатываемых антител или доступных для приобретения антител).
Как используют в настоящем описании, термин "CDR" относится к определяющей комплементарность области в вариабельных последовательностях антитела. В каждой из вариабельных областей тяжелой цепи и легкой цепи находятся три CDR, которые обозначают CDR1, CDR2 и CDR3, для каждой из вариабельных областей. Термин "набор CDR", как используют в настоящем описании, относится к группе из трех CDR, которые находятся в единичной вариабельной области, способной к связыванию с антигеном. Точные границы этих CDR были определены различным образом в соответствии с различными системами. Система, описанная Кэбат (Kabat et al. Sequences of Proteins of Immunological Interest (National Institutes of Health, Bethesda, Md. (1987) and (1991)) не только предоставляет однозначную нумерацию остатка, применимую к любой вариабельной области антитела, но также предоставляет точные границы остатка, определяющие три CDR. Три CDR могут обозначаться как CDR по Кэбату. Chothia и соавторы (Chothia & Lesk, J. Mol. Biol. 196:901-917 (1987) и Chothia et al. Nature 342:877-883 (1989)) обнаружили, что некоторые суб-части в пределах CDR по Кэбату принимают почти идентичные конформации пептидного скелета, несмотря на то, что имеют высокую степень разнообразия на уровне аминокислотной последовательности. Эти суб-части были обозначены как L1, L2 и L3 или H1, H2 и H3, где "L" и "H" обозначает области легкой цепи и тяжелой цепи, соответственно. Эти области могут именоваться как CDR по Хотиа, и имеют границы, которые накладываются на CDR по Кэбату. Другие границы, определяющие CDR, накладывающиеся на CDR по Кэбату были описаны Padlan (FASEB J. 9: 133-139 (1995)) и MacCallum (J Mol Biol 262(5):732-45 (1996)). Хотя другие определения границ CDR могут не жестко следовать одной из указанных выше систем, но тем не менее они будут накладываться на CDR по Кэбату, несмотря на то, что они могут быть укорочены или удлинены в свете предсказания или экспериментальных открытий того, что конкретные остатки или группы остатков или даже полные CDR не имеют значительного воздействия на связывание с антигеном. В способах, используемых в настоящем описании, могут использоваться CDR, определенные в соответствии с любой из этих систем, хотя в предпочтительных вариантах осуществления используют CDR, определенные по Кэбат или Хотиа.
Как используют в настоящем описании, термин "канонический" остаток относится к остатку в CDR или каркасе, который определяет конкретную каноническую структуру CDR, как определено Chothia et al. (J. Mol. Biol. 196:901-907 (1987); Chothia et al., J. Mol. Biol. 227:799 (1992), обе из которых включены в настоящее описание в качестве ссылки). В соответствии с Chothia et al., критические части CDR многих антител имеют почти идентичные конформации пептидного скелета, несмотря на высокую степень разнообразия на уровне аминокислотной последовательности. Каждая каноническая структура устанавливает прежде всего набор торсионных углов пептидного скелета для смежного сегмента аминокислотных остатков, образующих петлю.
Как используют в настоящем описании, термины "донор" и "донорное антитело" относятся к антителу, предоставляющему одну или несколько CDR. В предпочтительном варианте осуществления донорное антитело представляет собой антитело из вида, отличного от антитела, из которого получают или производят каркасные области. В контексте гуманизированного антитела, термин "донорное антитело" относится к нечеловеческому антителу, предоставляющему одну или несколько CDR.
Как используют в настоящем описании, термин "каркас" или "каркасная последовательность" относится к оставшимся последовательностям вариабельной области минус CDR. Поскольку точное определение последовательности CDR может быть определено различными системами, значение каркасной последовательности является предметом соответственно различных интерпретаций. Шесть CDR (CDR-L1, CDR-L2 и CDR-L3 легкой цепи и CDR-H1, CDR-H2 и CDR-H3 тяжелой цепи) также разделяют каркасные области в легкой цепи и тяжелой цепи на четыре подобласти (FR1, FR2, FR3 и FR4) в каждой цепи, в которой CDR1 расположен между FR1 и FR2, CDR2 между FR2 и FR3 и CDR3 между FR3 и FR4. Без установления конкретных подобластей в виде FR1, FR2, FR3 или FR4, каркасная область, как именуется другими, представляет объединенные FR в пределах вариабельной области единичной, природной цепи иммуноглобулина. Как используют в настоящем описании, FR представляет одну из четырех подобластей, и FR представляют две или несколько из четырех подобластей, составляющих каркасную область.
Акцепторные последовательности тяжелой цепи и легкой цепи человека являются известными в данной области. В одном из вариантов осуществления изобретения акцепторные последовательности тяжелой цепи и легкой цепи человека выбраны из последовательностей, описанных в Таблице 3 и Таблице 4, раскрытых в патенте США № 7915388, содержание которого включено в настоящее описание в качестве ссылки.
Как используют в настоящем описании, термин "ген антитела эмбрионального типа" или "фрагмент гена" относится к иммуноглобулиновой последовательности, кодируемой нелимфоидными клетками, которые не претерпели процесс созревания, который приводит к генетической перегруппировке и мутации для экспрессии конкретного иммуноглобулина. (См., например, Shapiro et al. Crit. Rev. Immunol. 22(3): 183-200 (2002); Marchalonis et al. Adv Exp Med. Biol. 484: 13-30 (2001)). Одно из преимуществ, предоставляемых разнообразными вариантами осуществления настоящего изобретения вытекает из признания того, что гены антитела эмбрионального типа более вероятно, чем гены зрелого антитела сохраняют существенные структуры аминокислотной последовательности индивидуумов в виде, следовательно, с меньшей вероятностью могут распознаваться как происходящие из чужеродного источника, когда их применяют терапевтически у данного вида.
Как используют в настоящем описании, термин "ключевые" остатки относится к некоторым остаткам в пределах вариабельной области, которые имеют большее воздействие на специфичность связывания и/или аффинность антитела, в частности, гуманизированного антитела. Ключевой остаток включает, но не ограничен лишь ими, один или несколько из следующих: остаток, который примыкает к CDR, потенциальный сайт гликозилирования (может быть либо N- или O-сайтом гликозилирования), редкий остаток, остаток, способный к взаимодействию с антигеном, остаток, способный к взаимодействию с CDR, канонический остаток, контактный остаток между вариабельной областью тяжелой цепи и вариабельной областью легкой цепи, остаток в пределах зоны Вернье и остаток в области, которая перекрывается между определением по Хотиа вариабельной тяжелой цепи CDR1 и определением по Кэбат первого каркаса тяжелой цепи.
Как используют в настоящем описании, термин "гуманизированное антитело" представляет собой антитело или его вариант, производное, аналог или фрагмент, которые иммуноспецифически связываются с антигеном, представляющим интерес и которые содержат каркасную (FR) область, имеющую по существу аминокислотную последовательность антитела человека и область, определяющую комплементарность (CDR), имеющую по существу аминокислотную последовательность нечеловеческого антитела. Как используют в настоящем описании, термин "по существу" в контексте CDR относится к CDR, имеющей аминокислотную последовательность, которая на по меньшей мере 80%, предпочтительно по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95%, по меньшей мере 96%, по меньшей мере 97%, по меньшей мере 98% или по меньшей мере 99% идентична с аминокислотной последовательности CDR нечеловеческого антитела. Гуманизированное антитело содержит по существу все из по меньшей мере одного, и обычно двух, вариабельных доменов (Fab, Fab', F(ab')2, FabC, Fv), в которых все или по существу все из областей CDR соответствуют таким областям нечеловеческого иммуноглобулина (т.е., донорного антитела) и все или по существу все из каркасных областей являются такими областями консенсусной последовательности иммуноглобулина человека. Предпочтительно, гуманизированное антитело также содержит по меньшей мере часть константной области иммуноглобулина (Fc), обычно такую часть иммуноглобулина человека. В некоторых вариантах осуществления, гуманизированное антитело содержит как легкую цепь, так и по меньшей мере вариабельный домен тяжелой цепи. Антитело также может включать области CH1, шарнирные, CH2, CH3, и CH4 тяжелой цепи. В некоторых вариантах осуществления, гуманизированное антитело содержит только гуманизированную легкую цепь. В некоторых вариантах осуществления, гуманизированное антитело содержит только гуманизированную тяжелую цепь. В конкретных вариантах осуществления гуманизированное антитело содержит только гуманизированный вариабельный домен легкой цепи и/или гуманизированную тяжелую цепь.
В одном из вариантов осуществления изобретения, гуманизированное антитело против IL-13 представляет собой 13C5.5. 13C5.5 имеет последовательности SEQ ID NO:2 (вариабельная область тяжелой цепи) и SEQ ID NO:3 (вариабельная область легкой цепи). См. также патент США № 7915388, полное содержание которого включено в настоящее описание в качестве ссылки.
SEQ ID NO:2 - Вариабельная область тяжелой цепи 13C5.5
EVTLRESGPGLVKPTQTLTLTCTLYGFSLSTSDMGVDWIRQPPGKGLEWLAHIW
WDDVKRYNPALKSRLTISKDTSKNQVVLKLTSVDPVDTATYYCARTVSSGYIY
YAMDYWGQGTLVTVSS
SEQ ID NO:3 - Вариабельная область легкой цепи 13C5.5
DIQMTQSPSSLSASVGDRVTISCRASQDIRNYLNWYQQKPGKAPKLLIFYTSKLH SGVPSRFSGSGSGTDYTLTISSLQPEDIATYYCQQGNTLPLTFGGGTKVEIK
Гуманизированное антитело может быть выбрано из любого класса иммуноглобулинов, включая IgM, IgG, IgD, IgA и IgE и любого изотипа, включая без ограничения, IgG1, IgG2, IgG3 и IgG4. Гуманизированное антитело может содержать последовательности из более чем одного класса или изотипа, и конкретные константные домены могут быть выбраны для оптимизации желательных эффекторных функций с использованием способов, хорошо известных в данной области.
Каркасные и CDR-области гуманизированного антитела необязательно должны соответствовать в точности родительским последовательностям, например, CDR донорного антитела или консенсусный каркас могут быть мутагенезированы посредством замещения, введения/или делеции по меньшей мере одного аминокислотного остатка таким образом, что CDR или каркасный остаток по этому сайту не соответствует либо донорному антителу или консенсусному каркасу. В предпочтительном варианте осуществления такие мутации, однако, не будут обширными. Обычно, по меньшей мере 80%, предпочтительно, по меньшей мере 85%, более предпочтительно, по меньшей мере 90%, и наиболее предпочтительно, по меньшей мере 95% из остатков гуманизированного антитела будут соответствовать таким остаткам родительских FR и последовательностей CDR. Как используют в настоящем описании, термин "консенсусный каркас" относится к каркасной области в консенсусной последовательности иммуноглобулина. Как используют в настоящем описании, термин "консенсусная последовательность иммуноглобулина" относится к последовательности, образованной из наиболее часто встречающихся аминокислот (или нуклеотидов) в семействе родственных последовательностей иммуноглобулинов (См., например, Winnaker, From Genes to Clones (Verlagsgesellschaft, Weinheim, Germany 1987). В семействе иммуноглобулинов, каждое положение в консенсусной последовательности занято аминокислотой, встречающейся наиболее часто в этом положении в семействе. Если две аминокислоты встречаются в равной степени часто, обе они могут быть включены в консенсусную последовательность.
Как используют в настоящем описании, зона "Вернье" относится к подклассу каркасных остатков, которые могут регулировать структуру CDR и тонко регулировать подгонку к антигену, как описано Foote and Winter (1992, J. Mol. Biol. 224:487-499, которая включена в настоящее описание в качестве ссылки). Остатки зоны Вернье из слоя, образуют слой, лежащий в основе CDR, и могут воздействовать на структуру CDR и аффинность антитела.
Термин "белок с мультивалентным связыванием" применяют в настоящем описании для обозначения связывающего белка, содержащего два или несколько участков связывания антигена. Белок с мультивалентным связыванием, предпочтительно, конструируют, чтобы он имел три или несколько участков связывания антигена, и как правило не является природным антителом. Термин "белок с мультиспецифическим связыванием" относится к связывающему белку, способному к связыванию с двумя или несколькими родственными или неродственными мишенями. Связывающие белки с двойственным вариабельным доменом (DVD), как используют в настоящем описании, представляют собой связывающие белки, которые содержат два или несколько участков связывания антигена и белками с тетравалентным или мультивалентным связыванием. Такие DVD могут быть моноспецифическими, т.е. способными к связыванию с одним антигеном или мультиспецифическими, т.е. способными к связыванию с двумя или несколькими антигенами. Связывающие белки с DVD, содержащие полипептиды DVD двух тяжелых цепей и полипептиды DVD двух легких цепей называют DVD Ig. Каждая половина DVD Ig содержит тяжелую цепь DVD полипептида, и легкую цепь DVD полипептида, и два участка связывания антигена. Каждый участок связывания содержит вариабельный домен тяжелой цепи и вариабельный домен легкой цепи, имеющие всего 6 CDR, вовлеченные в связывание антигена, приходящиеся на участок связывания антигена.
Как используют в настоящем описании, термин "нейтрализующий" относится к нейтрализации биологической активности цитокина, когда связывающий белок специфически связывается с цитокином. Предпочтительно, нейтрализующий связывающий белок представляет собой нейтрализующее антитело, чье связывание с IL-13 и/или IL-13 приводит в результате к ингибированию биологической активности IL-13 и/или IL-13. Предпочтительно, нейтрализующий связывающий белок связывается с IL-13 и/или IL-13 и снижает биологически активность IL-13 и/или IL-13 на по меньшей мере приблизительно 20%, 40%, 60%, 80%, 85% или более. Ингибирование биологической активности IL-13 и/или IL-13 посредством нейтрализующего связывающего белка может быть оценено посредством измерения одного или нескольких индикаторов биологической активности IL-13 и/или IL-13, хорошо известных в данной области. Например, может быть измерено ингибирование индуцированного IL-13 человека продуцирования TARC (CCL-17) клетками A-549 (см. Пример 1.1.C Патента США № 7915388, содержание которого включено в настоящее описание в качестве ссылки).
Термин "активность" включает активности, такие как специфичность/аффинность связывания антитела с антигеном, например, антитела против IL-13, которое связывается с антигеном IL-13 и/или нейтрализующая активность антитела, например, антитела против IL-13, чье связывание с IL-13 ингибирует биологическую активность IL-13, например. Например, ингибирование индуцированного IL-13 человека продуцирования TARC (CCL-17) клетками A-549 (см. Пример 1.1.C патента США № 7915388, полное содержание которого включено в настоящее описание в качестве ссылки).
Термин "эпитоп" включает любую полипептидную детерминанту, способную к специфическому связыванию с иммуноглобулином или рецептором T-клетки. В некоторых вариантах осуществления, эпитопные детерминанты включают химически активные поверхностные группировки молекул, таких как аминокислоты, боковые цепи сахаров, фосфорил или сульфонил, и, в некоторых вариантах осуществления, могут иметь специфичные трехмерные структурные характеристики и/или специфичные характеристики заряда. Эпитоп представляет собой область антигена, которая связывается антителом. В некоторых вариантах осуществления заявляют, что антитело специфически связывает антиген, когда оно, предпочтительно, распознает его целевой антиген в сложной смеси белков и/или макромолекул.
Термин "поверхностный плазмонный резонанс", как используют в настоящем описании, относится к оптическому явлению, которое позволяет проводить анализ в режиме реального времени биоспецифических взаимодействий посредством обнаружения изменений концентраций белка в пределах биосенсорной матрицы, например, с использованием системы BIAcore (Pharmacia Biosensor AB, Uppsala, Sweden and Piscataway, N.J.). Для дополнительного описания, см. Jonsson, U., et al. (1993) Ann. Biol. Clin. 51: 19-26; Jonsson, U., et al. (1991) Biotechniques 11:620-627; Johnsson, B., et al. (1995) J. Mol. Recognit. 8: 125-131; и Johnnson, B., et al. (1991) Anal. Biochem. 198:268-277.
Термин "kon", как используют в настоящем описании, предназначен для обозначения константы скорости связи антитела с антигеном с образованием комплекса антитело/антиген, как известно в данной области.
Термин "koff", как используют в настоящем описании, предназначен для обозначения константы скорости диссоциации антитела из комплекса антитело/антиген, как известно в данной области.
Термин “KD” как используют в настоящем описании, предназначен для обозначения константы диссоциации конкретного взаимодействия антитело-антиген, как известно в данной области.
Термин "меченый связывающий белок" как используют в настоящем описании, относится к белку с введенной меткой, который обеспечивает идентификацию связывающего белка. Предпочтительно, метка представляет собой детектируемый маркер, например, введение меченой радиоактивным изотопом аминокислоты или присоединение к полипептиду биотинильных фрагментов, которые могут быть детектированы посредством отмеченного авидина (например, стрептавидина, содержащего флуоресцентный маркер или ферментативной активности, которая может быть детектирована оптическими или колориметрическими способами). Примеры меток для полипептидов включают, но ими не ограничены, следующие: радиоизотопы или радионуклиды (например, 3H, 14C, 35S, 90Y, 99Tc, 111In, 125I, 131I, 177Lu, 166Ho или 153Sm); флуоресцентные метки (например, FITC, родамин, люминофоры на основе комплексов лантанидов), ферментативные метки (например, пероксидаза хрена, люцифераза, щелочная фосфатаза); хемилюминесцентные маркеры; биотинильные группы; предварительно определенные полипептидные эпитопы, распознаваемые вторичным репортером (например, парные последовательности лейциновой “молнии”, участки связывания для вторичных антител, металлсвязывающие домены, эпитопные метки); и магнитные средства, такие как хелаты гадолиния.
Термин "конъюгат антитела" относится к связывающему белку, такому как антитело, химически связанному со вторым химическим фрагментом, таким как терапевтическое или цитотоксическое средство. Термин "средство" используют в настоящем описании для обозначения химического соединения, смеси химических соединений, биологической макромолекулы или экстракта, полученного из биологических материалов. Предпочтительно, терапевтические или цитотоксические средства включают, но не ограничены лишь ими, коклюшный токсин, таксол, цитохалазин B, грамицидин D, этидия бромид, эметин, митомицин, этопозид, тенопозид, винкристин, винбластин, колхицин, доксорубицин, даунорубицин, дигидрокси антрациндион, митоксантрон, митрамицин, актиномицин D, 1-дегидротестостерон, глюкокортикоиды, прокаин, тетракаин, лидокаин, пропранолол и пуромицин и их аналоги или гомологи.
Термины "кристалл" и "кристаллизованное", как используют в настоящем описании, относится к антителу или его антигенсвязывающей части, которые существуют в форме кристалла. Кристаллы представляют собой одну форму твердого состояния вещества, которая отличается от других форм, таких как аморфное твердое состояние или жидкокристаллическое состояние. Кристаллы составлены из регулярных, повторяющихся, трехмерных матриц атомов, ионов, молекул (например, белков, таких как антитела), или молекулярных агрегатов (например, комплексов антиген/антитело). Эти трехмерные матрицы расположены в соответствии с конкретными математическими взаимосвязями, которые являются хорошо изученными в данной области. Фундаментальный блок, или билдинг-блок, который повторяется в кристалле, называют асимметрическим блоком. Повтор асимметрического блока в расположении, который соответствует данной, хорошо определенной кристаллографической симметрии, предоставляет "элементарную ячейку" кристалла. Повтор элементарной ячейки посредством регулярных трансляций во всех трех измерениях предоставляет кристалл. См., Giege, R. and Ducruix, A. Barrett, Crystallization of Nucleic Acids и Proteins, Practical Approach, 2nd ea., pp. 20 1-16, Oxford University Press, New York, N.Y., (1999)."
Термин "полинуклеотид", как используют в настоящем описании, означает полимерную форму из двух или нескольких нуклеотидов, либо рибонуклеотидов или дезоксинуклеотидов или модифицированную форму обоих типов нуклеотидов. Термин включает одно- и двухцепочечные формы ДНК, но предпочтительно относится к двухцепочечной ДНК.
Термин "выделенный полинуклеотид", как используют в настоящем описании, будет означать полинуклеотид (например, геномный, кДНК, или синтетического происхождения или его некоторые комбинации), вследствие своего происхождения, "выделенный полинуклеотид": не является связанным со всем полинуклеотидом или его частью, с которыми "выделенный полинуклеотид" обнаруживается в природе; является функционально связанным с полинуклеотидом, с которым он не связан в природе; или не встречается в природе как часть более крупной последовательности.
Термин "вектор", как используют в настоящем описании, предназначен для обозначения молекулу нуклеиновой кислоты, способной к транспортировке еще одной нуклеиновой кислоты, с которой она была связана. Один тип вектора представляет собой "плазмиду", которая относится к петле круговой двухцепочечной ДНК, в которую могут быть лигированы дополнительные сегменты ДНК. Еще один тип вектора представляет собой вирусный вектор, где дополнительные сегменты ДНК могут быть лигированы в вирусный геном. Некоторые векторы способны к автономной репликации в клетке-хозяине, в которую их вводят (например, бактериальные векторы, имеющие бактериальное происхождение репликации и эписомальные векторы млекопитающих). Другие векторы (например, неэписомальные векторы млекопитающих) могут быть встроены в геном клетки-хозяина при введении в клетку-хозяина, и посредством этого реплицируются наряду с геномом хозяина. Кроме того, некоторые векторы способны к направлению экспрессии генов, с которыми они функционально связаны. Такие векторы называют в настоящем описании "векторами рекомбинантной экспрессии" (или просто, "векторами экспрессии"). В целом, векторы экспрессии, используемые в способах рекомбинантной ДНК, часто находятся в форме плазмид. В настоящем описании термины "плазмида" и "вектор" могут использоваться взаимозаменяемо, так как плазмида является наиболее частой применяемой формой вектора. Однако, подразумевают, что изобретение включает такие другие формы векторов экспрессии, такие как вирусные векторы (например, ретровирусы с дефектом репликации, аденовирусы и аденосвязанные вирусы), которые выполняют эквивалентные функции.
Термин "функционально связанная" относится к непосредственному соседству, где описанные компоненты находятся во взаимосвязи, позволяющей им функционировать предназначенным им образом. Контрольная последовательность, "функционально связанная" с кодирующей последовательностью является лигированной таким образом, что экспрессии кодирующей последовательности достигают при условиях, совместимых с контрольными последовательностями. "Функционально связанные" последовательности включают как последовательности контроля экспрессии, которые являются смежными с интересующим геном, так и последовательности контроля экспрессии, которые действуют при переносе или на расстоянии от контрольного интересующего гена. Термин "последовательность контроля экспрессии", как используют в настоящем описании, относится к полинуклеотидным последовательностям, которые являются необходимыми для воздействия на экспрессию и процессинг кодирующих последовательностей, с которыми они лигированы. Последовательности контроля экспрессии включают соответствующие последовательности инициации транскрипции, терминации, промотерные и энхансерные последовательности; эффективные сигналы процессинга РНК, такие как сигналы сплайсинга и полиаденилирования; последовательности, которые стабилизируют цитоплазматическую мРНК; последовательности, которые усиливают эффективность трансляции (т.е., консенсусная последовательность Козака); последовательности, которые усиливают стабильность белка; и когда желательно, последовательности, которые усиливают секрецию белка. Природа таких контрольных последовательностей различается в зависимости от организма хозяина; у прокариотов, такие контрольные последовательности обычно включают промотор, рибосомальный участок связывания, и последовательность терминации транскрипции; у эукариотов, обычно, такие контрольные последовательности включают промоторы и последовательность терминации транскрипции. Термин "контрольные последовательности" предназначен для включения компонентов, чье присутствие является существенным для экспрессии и процессинга, и могут также включать дополнительные компоненты, чье присутствие является преимущественным, например, лидерные последовательности и партнерные последовательности слияния. Конструкции белка настоящего изобретения могут экспрессироваться и очищаться с использованием векторов экспрессии и клеток-хозяев, известных в данной области, включая кассеты экспрессии, векторы, рекомбинантные клетки-хозяева и способы рекомбинантной экспрессии и протеолитического процессинга рекомбинантных полибелков и пребелков от единичной открытой рамки считывания (например, WO 2007/014162, полное содержание которой включено в настоящее описание в качестве ссылки).
"Трансформация", как определена в настоящем описании, относится к любому процессу, посредством которого экзогенная ДНК попадает в клетку-хозяина. Трансформация может происходить при природных или искусственных условиях с использованием разнообразных способов, хорошо известных в данной области. Трансформация может основываться на любом известном способе внедрения чужеродной последовательности нуклеиновой кислоты внуть прокариотной или эукариотной клетки-хозяина. Способ выбирают, на основании клетки-хозяина, подлежащей трансформации, и он может включать, но ими не ограничиваясь, вирусную инфекцию, электропорацию, липофекцию, и бомбардировку частицами. Такие "трансформированные" клетки включают стабильно трансформированные клетки, в которых внедренная ДНК является способной к репликации либо как автономно реплицирующаяся плазмида или как часть хромосомы хозяина. Они также включают клетки, которые преходящим образом экспрессируют внедренную ДНК или РНК в течение ограниченных периодов времени.
Термин "рекомбинантная клетка-хозяин" (или просто "клетка-хозяин"), как используют в настоящем описании, предназначена для обозначения клетки, в которую была введена экзогенная ДНК. Следует понимать, что такие термины подоазумевают отнесение не только к конкретной клетке-индивиду, но, к потомству такой клетки. Поскольку в последующих поколениях могут происходить определенные модификации вследствие либо мутации или влияний окружающей среды, такое потомство, в действительности может не являться идентичным родительской клетке, но все еще является включенным в пределы охвата термина "клетка-хозяин", как его используют в настоящем описании. Предпочтительно, клетки-хозяева включают прокариотные и эукариотные клетки, выбранные из любого царства живых существ. Предпочтительные эукариотные клетки включают клетки простейших, грибковые, растительные и животные клетки. Наиболее предпочтительно, клетки-хозяева включают, но не ограничены лишь ими, прокариотную клеточную линию E. Coli; клеточные линии млекопитающих CHO, HEK 293 и COS; клеточную линию насекомых Sf9; и грибковые клетки Saccharomyces cerevisiae.
Стандартные способы могут применяться для рекомбинантных ДНК, олигонуклеотидного синтеза, и культуры тканей и трансформации (например, электропорация, липофекция). Ферментативные реакции и способы очистки могут осуществляться в соответствии с описаниями производителя или как обычно осуществляются в данной области или, как описано в настоящем описании. Вышеизложенные способы и методики могут, как правило, осуществляться в соответствии с общепринятыми способами, хорошо известными в данной области, и как описано в разнообразных общих и более конкретных ссылках, которые цитируются и обсуждаются на протяжении всего настоящего описания. См., например, Sambrook et al. Molecular Cloning: Laboratory Manual (2d ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. (1989)), которая включена в настоящее описание в качестве ссылки для любой цели.
"Трансгенный организм", как известно в данной области, и, как используют в настоящем описании, относится к организму, имеющему клетки, которые содержат трансген, где трансген, вводимый в организм (или предок организма) экспрессирует полипептид, не экспрессируемый в природных условиях в организме. "Трансген" представляет собой конструкцию ДНК, которую стабильно и функционально встраивают в геном клетки, из которой трансгенный организм развивается, направляя экспрессию кодируемого генного продукта в одной или нескольких типах клеток или тканей трансгенного организма.
Термин "регулирует" и "модулирует" используют взаимозаменяемо, и, как используют в настоящем описании, они относятся к изменению или перемене активности интересующей молекулы (например, биологической активности IL-13). Модуляция может являться увеличением или снижением по величине определенной активности или функции интересующей молекулы. Примерные активности и функции молекулы включают, но не ограничены лишь ими, характеристики связывания, ферментативную активность, активацию клеточного рецептора и сигнальную трансдукцию.
Соответственно, термин "модулятор", как используют в настоящем описании, относится к соединению, способному к изменению или перемене активности или функции интересующей молекулы (например, биологической активности IL-13). Например, модулятор может вызывать увеличение или снижение по величине определенной активности или функции молекулы по сравнению с величиной активности или функции, наблюдаемой в отсутствие модулятора. В некоторых вариантах осуществления, модулятор представляет собой ингибитор, который снижает величину по меньшей мере одной активности или функции молекулы. Примерные ингибиторы включают, но не ограничены лишь ими, белки, пептиды, антитела, пептид-связанные антитела, углеводы или небольшие органические молекулы. Пептид-связанные антитела описаны, например, в WO01/83525.
Термин "агонист", как используют в настоящем описании, относится к модулятору, который при контакте с интересующей молекулой, вызывает увеличение по величине определенной активности или функции молекулы по сравнению с величиной активности или функции, наблюдаемой в отсутствие агониста. Конкретные агонисты, представляющие интерес, могут включать, но не ограничены лишь ими, IL-13 полипептиды или полипептиды, нуклеиновые кислоты, углеводы или любые другие молекулы, которые связываются с IL-13.
Термин "антагонист" или "ингибитор", как используют в настоящем описании, относится к модулятору, который, при контакте с интересующей молекулой вызывает снижение по величине определенной активности или функции молекулы по сравнению с величиной активности или функции, наблюдаемой в отсутствие антагониста. Конкретные антагонисты, представляющие интерес, включают антагонисты, которые блокируют или модулируют биологическую или иммунологическую активность IL-13 и/или IL-13. Антагонисты и ингибиторы IL-13 и/или IL-13 могут включать, но не ограничены лишь ими, белки; нуклеиновые кислоты, углеводы или любые другие молекулы, которые связываются с IL-13 и/или IL-13.
Термин "ингибирует связывание с рецептором" относится к способности связывающего белка предотвращать связывание IL-13 с одним или несколькими из его рецепторов. Такое ингибирование связывания с рецептором будет приводить к уменьшению или отмене биологической активности, опосредованной связыванием IL-13 с его рецептором или рецепторами.
Как используют в настоящем описании, термин "эффективное количество" относится к количеству терапевтического средства, которое является достаточным, чтобы снизить или уменьшить тяжесть и/или продолжительность расстройства или одного или нескольких его симптомов, предотвратить распространение расстройства, вызвать регрессию расстройства, предотвратить рецидив, развитие, наступление или прогрессию одного или нескольких симптомов, связанных с расстройством, обнаружить расстройство или усилить или улучшить профилактические или терапевтические эффект(ы) еще одной другой терапии (например, профилактического или терапевтического средства).
Термин "образец", как используют в настоящем описании, используют в его самом широком смысле. "Биологический образец", как используют в настоящем описании, включает, но не ограничиваясь лишь ими, любое количество субстанции из живого объекта или ранее бывшего живым объекта. Такие живые объекты включают, но не ограничены лишь ими, людей, мышей, крыс, обезьян, собак, кроликов и других животных. Такие субстанции включают, но не ограничены лишь ими, кровь, сыворотку, мочу, синовиальную жидкость, клетки, органы, ткани, костный мозг, лимфатические узлы и селезенку.
Термин "Cмакс" относится к максимальной или пиковой сывороточной или плазменной концентрации средства, наблюдаемой у индивида после его введения.
В одном из вариантов осуществления антитела против IL-13 или их антигенсвязывающую часть по изобретению (например, гуманизированные антитела против IL-13, такие как 13C5.5 или их антигенсвязывающую часть) вводят внутривенно и они проявляют максимальную сывороточную концентрацию (Cмакс) между приблизительно 5 и приблизительно 235 мкг/мл; пиковую концентрацию (Cмакс) между приблизительно 5 и приблизительно 8 мкг/мл; Cмакс между приблизительно 5 и приблизительно 10 мкг/мл; пиковую концентрацию (Cмакс) между приблизительно 55 и приблизительно 90 мкг/мл; пиковую концентрацию (Cмакс) между приблизительно 185 и приблизительно 250 мкг/мл; Cмакс между приблизительно 190 и приблизительно 235 мкг/мл. В еще одном варианте осуществления Cмакс находится между приблизительно 5 и приблизительно 50, между приблизительно 50 и приблизительно 75, между приблизительно 75 и приблизительно 100, между приблизительно 100 и приблизительно 125, между приблизительно 125 и приблизительно 150, между приблизительно 150 и приблизительно 175, между приблизительно 175 и приблизительно 200, или между приблизительно 200 и приблизительно 235 мкг/мл.
В еще одном варианте осуществления антитела против IL-13 или их антигенсвязывающая часть (например, гуманизированные антитела против IL-13, такие как 13C5.5 или их антигенсвязывающую часть), вводят внутривенно и они проявляют значение Cмакс между приблизительно 20 и приблизительно 30 мкг/мл)/(мг/кг) после нормализации дозы. В еще одном варианте осуществления антитела против IL-13 или их антигенсвязывающую часть вводят внутривенно и они проявляют значения Cмакс около 20, около 21, около 22, около 23, около 24, около 25, около 26, около 27, около 28, около 29 и приблизительно 30 )мкг/мл)/(мг/кг) после нормализации дозы. В еще одном варианте осуществления антитела против IL-13 или их антигенсвязывающую часть вводят внутривенно и они проявляют значение Cмакс между приблизительно 10 и приблизительно 40 (мкг/мл)/(мг/кг) после нормализации дозы.
В еще одном варианте осуществления антитела против IL-13 или их антигенсвязывающую часть по изобретению (например, гуманизированные антитела против IL-13, такие как 13C5.5, или их антигенсвязывающую часть) вводят подкожно и они проявляют максимальную сывороточную концентрацию (Cмакс) между приблизительно 1 и приблизительно 60 мкг/мл; пиковую концентрацию (Cмакс) между приблизительно 1,0 и приблизительно 6,0 мкг/мл; значение Cмакс между приблизительно 6 и приблизительно 12 мкг/мл; пиковую концентрацию (Cмакс) между приблизительно 12 и приблизительно 60 мкг/мл; значение Cмакс между приблизительно 1 и приблизительно 10, между приблизительно 10 и приблизительно 20, между приблизительно 20 и приблизительно 30, между приблизительно 30 и приблизительно 40, между приблизительно 40 и приблизительно 50, между приблизительно 50 и приблизительно 60, между приблизительно 20 и приблизительно 60, или между приблизительно 40 и приблизительно 60 мкг/мл.
Термин "Тмакс” относится к времени, при котором имеет место Cмакс. В одном из вариантов осуществления антитело против IL-13 или его антигенсвязывающую часть по изобретению (например, гуманизированные антитела против IL-13, такие как 13C5.5, или их антигенсвязывающую часть) вводят внутривенно или подкожно и они проявляют Tмакс между приблизительно 1 и приблизительно 5 днями; Tмакс между приблизительно 3 и приблизительно 5 днями; Tмакс меньшее или равное до приблизительно 5 дней; Tмакс около 1 дня, Tмакс около 2 дней, Tмакс около 3 дней, Tмакс около 4 дней, Tмакс около 5 дней, Tмакс около 6 дней, Tмакс около 7 дней, Tмакс около 8 дней, Tмакс около 9 дней или Tмакс около 10 дней.
Термин "биодоступность" или "F%" относится к фракции или процентной доле дозы, которая всасывается и поступает в системную циркуляцию после введения данной дозированной формы. Доза антитела против IL-13 или его антигенсвязывающей части может вводиться любым путем и, предпочтительно, внутривенной или подкожной инъекцией. В одном из вариантов осуществления антитела против IL-13 или их антигенсвязывающую часть по изобретению (например, гуманизированные антитела против IL-13, такие как 13C5.5, или их антигенсвязывающую часть) вводят внутривенно или подкожно и они проявляют биодоступность, равную по меньшей мере приблизительно 60%. В еще одном варианте осуществления антитела или их антигенсвязывающая часть проявляют биодоступность, равную по меньшей мере приблизительно 35%, по меньшей мере около 40%, по меньшей мере около 45%, по меньшей мере около 50%, по меньшей мере около 60%, по меньшей мере около 65%, по меньшей мере приблизительно 70%, по меньшей мере около 75%, по меньшей мере около 80%, по меньшей мере около 85%, по меньшей мере около 90%, по меньшей мере около 95% или по меньшей мере около 100%.
Термин "AUC" или "площадь под кривой" относится к выведению. Более высокая скорость выведения относится к меньшей AUC, и более низкая скорость выведения относится к более высокому значению AUC. Более высокие значения AUC представляют более медленные скорости выведения.
В одном из вариантов осуществления антитела против IL-13 или их антигенсвязывающую часть, по изобретению (например, гуманизированные антитела против IL-13, такие как 13C5.5, или их антигенсвязывающую часть) вводят внутривенно и они проявляют площадь под кривой (AUC) между приблизительно 75 и приблизительно 100000 мкгч/мл; AUC между приблизительно 75 и приблизительно 100; между приблизительно 1500 и приблизительно 2700 мкгч/мл; между приблизительно 1500 и приблизительно 3000; между приблизительно 21000 и приблизительно 33500 мкгч/мл; между приблизительно 1500 и 98000; между приблизительно 20000 и приблизительно 34000 мкгч/мл; между приблизительно 34000 и приблизительно 40000; между приблизительно 40000 и приблизительно 50000; между приблизительно 50000 и приблизительно 60000; между приблизительно 60000 и приблизительно 75000; между приблизительно 75000 и приблизительно 100000 мкгч/мл; около 75, около 100, около 150, около 200, около 250, около 300, около 350, около 400, около 450, около 500, около 550, около 600, около 650, приблизительно 700, около 750, около 800, около 850, около 900, около 950, около 1000; около 1100; около 1200; около 1300; около 1400; около 1500; около 1600; около 1700; около 1800; около 1900; около 2000; около 2250; около 2500; около 2750; около 3000; около 4000; около 5000; около 6000; около 7000; около 8000; около 9000; около 10000; около 12000; около 15000; около 20000; около 25000; около 30000; около 35000; около 40000; около 45000; около 50000; около 55000; около 60000; около 65000; приблизительно 70000; около 75000; около 80000, около 85000; около 90000; около 95000 или приблизительно 100000 мкгч/мл.
В еще одном варианте осуществления AUC находится между приблизительно 6000 и приблизительно 10000 (мкгч/мл)/(мг/кг) после нормализации дозы, между приблизительно 7000 и приблизительно 9000; около 6000; около 6500; около 7000; около 7500; около 8000; около 8500; около 9000; около 9500 или приблизительно 10000 (мкгч/мл)/(мг/кг) после нормализации дозы.
В еще одном варианте осуществления антитела против IL-13, или их антигенсвязывающую часть по изобретению (например, гуманизированные антитела против IL-13, такие как 13C5.5 или их антигенсвязывающую часть) вводят подкожно, и они проявляют площадь под кривой (AUC) между приблизительно 125 и приблизительно 8100 мкгч/мл; между приблизительно 125 и приблизительно 800 мкгч/мл; между приблизительно 800 и приблизительно 1100; между приблизительно 1100 и приблизительно 8100; около 100 и приблизительно 800 мкгч/мл; около 125; около 150; около 175; около 200; около 250; около 300; около 350; около 400; около 450; около 500; около 550; около 600; около 650; приблизительно 700; около 750; около 800; около 850; около 900; около 950; около 1000; около 1100; около 1200; около 1300; около 1400; около 1500; около 1600; около 1700; около 1800; около 1900; около 2000; около 2250; около 2500; около 3000; около 3500; около 4000; около 4500; около 5000; около 5500; около 6000; около 6500; около 7000; около 7500; около 8000 или приблизительно 8100 мкгч/мл.
Как используют в настоящем описании, термин "скорость выведения" относится к AUC или площади под кривой. Более высокая скорость выведения относится к меньшей AUC, и более низкая скорость выведения относится к более высокому значению AUC. Более высокие значения AUC представляют более медленные скорости выведения.
В одном из вариантов осуществления антитела против IL-13 или их антигенсвязывающую часть по изобретению (например, гуманизированные антитела против IL-13, такие как 13C5.5 или их антигенсвязывающую часть) вводят внутривенно и они проявляют скорость выведения между приблизительно 0,08 и приблизительно 0,2 мл/ч/кг, между приблизительно 0,08 и приблизительно 0,15 мл/ч/кг; между приблизительно 0,1 и приблизительно 0,15 мл/ч/кг; между приблизительно 0,11 до приблизительно 0,19 мл/час/кг; между приблизительно 0,08 до приблизительно 0,14 мл/час/кг; между приблизительно 0,09 до приблизительно 0,13 мл/час/кг; около 0,08; около 0,09, около 0,1, около 0,11, около 0,12, около 0,13, около 0,14, около 0,15, около 0,16, около 0,17, около 0,18, около 0,19, около 2,0, около 2,1, около 2,2, около 2,3, около 2,4 или приблизительно 2,5 мл/ч/кг.
Как используют в настоящем описании, термин "объем распределения" представляет собой термин, используемый для количественной оценки распределения лекарственного средства, например, антител против IL-13 или их антигенсвязывающей части, между плазмой и оставшимся организмом после дозирования. Объем распределения представляет собой теоретический объем, в котором общее количество лекарственного средства будет нуждаться в однородном распределении, чтобы продуцировать желательную концентрацию лекарственного средства в крови.
В одном из вариантов осуществления антитела против IL-13 или их антигенсвязывающую часть, по изобретению (например, гуманизированные антитела против IL-13, такие как 13C5.5, или их антигенсвязывающую часть) вводят внутривенно, и они проявляют объем распределения между приблизительно 55 и приблизительно 130 мл/кг; между приблизительно 65 и 125 мл/кг; между приблизительно 55 и приблизительно 100 мл/кг; между приблизительно 90 и приблизительно 130 мл/кг; между от приблизительно 70 до приблизительно 130 мл/кг; между приблизительно 85 до приблизительно 130 мл/кг; между от приблизительно 100 до приблизительно 130; между приблизительно 110 до приблизительно 120; около 55, около 60, около 65, приблизительно 70, около 75, около 80, около 85, около 90, около 95, около 100, около 105, около 110, около 115, около 120, около 125, около 130 или приблизительно 135 мл/кг.
В одном из вариантов осуществления антитела против IL-13 или их антигенсвязывающую часть по изобретению (например, гуманизированные антитела против IL-13, такие как 13C5.5, или их антигенсвязывающую часть) вводят внутривенно или подкожно, и они имеют время полужизни между приблизительно 24 и 31 днями; между приблизительно 23 и 26 днями; между приблизительно 10 и приблизительно 40 днями, между приблизительно 20 и приблизительно 30 днями, около 10 дней, около 15 дней, около 16 дней, около 17 дней, около 18 дней, около 19 дней, около 20 дней, около 21 дней, около 22 дней, около 23 дней, около 24 дней, около 25 дней, около 26 дней, около 27 дней, около 28 дней, около 29 дней, около 30 дней, около 31 дней, около 32 дней, около 33 дней, около 34 дней, около 35 дней, около 36 дней, около 37 дней, около 38 дней, около 39 дней, или приблизительно 40 дней.
Термин "дозирование" или "доза" или "дозировка", как используют в настоящем описании, относится к введению вещества (например, антител против IL-13 или их антигенсвязывающей части), чтобы достичь терапевтической задачи (например, лечения астмы).
В одном из вариантов осуществления композицию по изобретению вводят однократно. В еще одном варианте осуществления композицию по изобретению вводят еженедельно. В еще одном варианте осуществления композицию по изобретению вводят в течение двух недель. В еще одном варианте осуществления композицию по изобретению вводят в течение трех недель. В еще одном варианте осуществления композицию вводят в течение четырех недель, пяти недель, шести недель, семи недель, восьми недель, девяти недель, десяти недель, одиннадцати недель, трех месяцев, четырех месяцев, пяти месяцев, шести месяцев, семи месяцев, восьми месяцев, девяти месяцев, десяти месяцев, одиннадцати месяцев, двенадцати месяцев, тринадцати месяцев, четырнадцати месяцев, пятнадцати месяцев, шестнадцати месяцев, семнадцати месяцев, восемнадцати месяцев, девятнадцати месяцев, двадцати месяцев, двадцати одного месяца, двадцати двух месяцев, двадцати трех месяцев, двух лет, трех лет, четырех лет, пяти лет, десяти лет или в течение жизни индивида.
Термин "комбинация" как во фразе "первое средство в комбинации со вторым средством" включает co-введение первого средства и второго средства, которые, например, могут быть растворены или перемешаны в одном и том же фармацевтически приемлемом носителе, или введение первого средства с последующим вторым средством, или введение второго средства с последующим первым средством. Настоящее изобретение, следовательно, включает способы комбинирования терапевтического лечения и комбинирования фармацевтических композиций.
Термин "сопутствующее", как во фразе "сопутствующее терапевтическое лечение" включает введение средства в присутствии второго средства. Способ сопутствующего терапевтического лечения включает способы, в которых первое, второе, третье или дополнительные средства вводят совместно. Способ сопутствующего терапевтического лечения также включает способы, в которых первое или дополнительные средства вводят в присутстви второго или дополнительных средств, где второе или дополнительные средства, например, могли быть введены прежде. Способ сопутствующего терапевтического лечения может выполняться постадийно различными участниками. Например, один участник может вводить индивиду первое средство, а второй участник может вводить индивиду второе средство, и стадии введения могут выполняться в одно и то же время или почти в одно и то же время или в удаленные моменты времени, поскольку первое средство (и дополнительные средства) находятся после введения в присутствии второго средства (и дополнительных средств). Участник и индивид могут быть одним и тем же объектом (например, человеком).
Термин "комбинированная терапия", как используют в настоящем описании, относится к введению двух или нескольких терапевтических веществ, например, антител против IL-13 и еще одного лекарственного средства. Другие лекарственные средства могут вводиться совместно с введением, перед введением или после введения антител против IL-13.
Термин "набор", как используют в настоящем описании, относится к упакованному продукту, содержащему компоненты, с которыми вводят антитела против IL-13 по изобретению для лечения относящегося к IL-13 расстройству. Набор предпочтительно содержит коробку или контейнер, в которых хранится компоненты набора. Коробка или контейнер заверяют меткой или протоколом, разрешенными к применению Управлением по продовольствию и лекарствам. В коробке или контейнере хранятся компоненты по изобретению, которые предпочтительно содержатся внутри пластиковых, полиэтиленовых, полипропиленовых, этиленовых или пропиленовых сосудов. Сосуды могут представлять собой закрытые крышкой пробирки или бутыли. Набор может также включать инструкции для введения антител против IL-13.
Разнообразные аспекты изобретения описаны более подробно в следующих подразделах.
I. Антитела, которые связываются с IL-13
Настоящее изобретение относится к способам и композициям для использования антител против IL-13, или их антигенсвязывающих частей для лечения астмы. В одном из аспектов настоящее изобретение относится к композициям, которые включают и/или способам, в которых применяются выделенные моноклональные антитела мыши или их антигенсвязывающие части, которые связываются с IL-13 с высокой аффинностью, медленной скоростью диссоциации и высокой нейтрализующей способностью. Во втором аспекте изобретение относится к композициям, которые включают и/или способам, в которых применяются химерные антитела, которые связывают IL-13. В третьем аспекте изобретение относится к композициям, которые включают и/или способам, в которых применяются гуманизированное антитела или их антигенсвязывающие части, которые связывают IL-13. Предпочтительно, антитела или их части представляют собой выделенные антитела. Предпочтительно, антитела представляют собой нейтрализующие антитела против IL-13 и/или гуманизированные или человеческие антитела против IL-13.
A. Способы получения антител против IL-13
Антитела для использования в композициях и/или способах по настоящему изобретению могут быть получены посредством любого из числа способов, известных в данной области. Например, они могут быть получены с использованием способов, раскрытых в патенте США № 7915338, полное содержание которого включено в настоящее описание в качестве ссылки.
1. Моноклональные антитела против IL-13 с использованием технологии гибридом
Моноклональные антитела могут быть получены с использованием широкого разнообразия способов, известных в данной области, включающих применение гибридомы, рекомбинантные технологии и технологии фагового отображения или их комбинацию. Например, моноклональные антитела могут быть получены с использованием способов гибридом, включающих способы, известные в данной области и изложенные, например, в Harlow et al. Antobodies: Laboratory Manual. (Cold Spring Harbor Laboratory Press, 2nd ed. 1988); Hammerling, et al., в: Monoclonal Antobodies и T-Cell Hybridomas 563-681 (Elsevier, N.Y., 1981) (указанные ссылки включены в качестве ссылки в полном объеме). Термин "моноклональное антитело", как используют в настоящем описании, не ограничивается антителами, продуцируемыми посредством технологии гибридом. Термин "моноклональное антитело" относится к антителу, которое получают из одиночного клона, включающего любой эукариотный, прокариотный или фаговый клон, а не к способу, посредством которого его получают.
Способы получения и скрининга конкретных антител с использованием технологии гибридом являются рутинными и хорошо известными в данной области. В одном из вариантов осуществления настоящее изобретение относится к способам получения моноклональных антител, а также антитела, получаемые посредством способа, включающего культивирование клеток гибридомы, секретирующих антитела по изобретению, где, предпочтительно, гибридому получают посредством слияния спленоцитов, выделенных из мышей, иммунизированных антигеном по изобретению, с клетками миеломы и затем, подвергая скринингу гибридомы, получаемые в результате слияния клонов гибридомы, которые секретируют антитела, способные связывать полипептид по изобретению (См. Пример 1.2). Вкратце, мыши могут быть иммунизированы антигеном IL-13. В предпочтительном варианте осуществления антиген IL-13 вводят вместе с адъювантом для стимуляции иммунного ответа. Такие адъюванты включают полный или неполный адъювант Фрейнда, RIBI (мурамилдипептиды) или ISCOM (иммуностимулирующие комплексы). Такие адъюванты могут защищать полипептид от быстрого рассеивания посредством секвестирования его в местном отложении или они могут содержать вещества, которые стимулируют хозяина секретировать факторы, которые являются хемотаксическими для макрофагов и других компонентов иммунной системы. Предпочтительно, если вводят полипептид, средства для схемы иммунизации будут включать два или несколько введений полипептида, разнесенных в течение нескольких недель.
После иммунизации животного антигеном IL-13 антитела и/или продуцирующие антитела клетки могут быть получены из животного. Сыворотку, содержащую антитела против IL-13, получают из животного посредством забора крови или умерщвления животного. Сыворотку можно использовать в том виде, каком ее получают из животного, иммуноглобулиновая фракция может быть получена из сыворотки или антитела против IL-13 могут быть очищены из сыворотки. Сыворотка или иммуноглобулины, полученные таким образом, являются поликлональными, таким образом, имеющие гетерогенный набор свойств.
Как только иммунный ответ обнаруживают, например, антитела, специфические к антигену IL-13, детектируют в сыворотке мыши, селезенку мыши забирают и спленоциты выделяют. Спленоциты затем сливают хорошо известными способами с любыми подходящими клетками миеломы, например клетками из клеточной линии SP20, доступных из ATCC. Гибридомы отбирают и клонируют посредством ограниченного разбавления. Клоны гибридомы затем подвергают аналитическому тестированию посредством способов, известных в данной области для клеток, которые секретируют антитела, способные к связыванию IL-13. Свободная жидкость брюшной полости, которая, как правило, содержит высокие уровни антител, может быть получена посредством иммунизации мышей положительными клонами гибридомы.
В еще одном варианте осуществления продуцирующие антитела иммортализованные гибридомы могут быть получены от иммунизованного животного. После иммунизации, животное умерщвляют и селезеночные B-клетки сливают с иммортализованными клетками миеломы, как является хорошо известным в данной области. См., например, Harlow and Lane, выше. В предпочтительном варианте осуществления клетки миеломы не секретируют иммуноглобулиновые полипептиды (несекреторная клеточная линия). После слияния и селекции с антибиотиками, гибридомы подвергают скринингу с использованием IL-13, или его части или клеток, экспрессирующих IL-13. В предпочтительном варианте осуществления первоначальный скрининг осуществляют с использованием иммуноферментного анализа (ELISA) или радиоиммунологического анализа (RIA), предпочтительно ELISA. Пример скрининга с ELISA представлен в WO 00/37504, включенной в настоящее описание в качестве ссылки.
Гибридомы, продуцирующие антитела против IL-13, отбирают путем селекции, клонируют и дополнительно подвергают скринингу по желательным характеристикам, включающим надежный рост гибридомы, высокая продукция антител и желательные характеристики антитела, как дополнительно обсуждается ниже. Гибридомы могут культивироваться и размножаться in vivo у сингенных животных, у животных с недостатками иммунной системы, например, “голых” мышей или в клеточной культуре in vitro. Способы селекции, клонирования и размножения гибридом являются хорошо известными рядовым специалистам в данной области.
В предпочтительном варианте осуществления гибридомы представляют собой гибридомы мыши, как описано выше. В еще одном предпочтительном варианте осуществления гибридомы продуцируют у видов, не являющихся человеком и мышью, таких как крысы, овцы, свиньи, козы, крупный рогатый скот или лошади. В еще одном варианте осуществления гибридомы являются гибридомами человека, в которых несекреторная миелома человека слита с клетками человека, экспрессирующими антитела против IL-13.
Фрагменты антител, которые распознают специфические эпитопы, можно получать посредством известных способов. Например, Fab и F(ab')2 фрагменты по изобретению могут быть получены посредством протеолитического расщепления иммуноглобулиновых молекул, с использованием ферментов, таких как папаин (для получения Fab-фрагментов) или пепсин (для получения F(ab')2-фрагментов). F(ab')2-фрагменты содержат вариабельную область, константную область легкой цепи и домен CH1 тяжелой цепи.
2. Моноклональные антитела против IL-13 с использованием SLAM
Рекомбинантные антитела для применения в композициях и/или способах по настоящему изобретению можно также получать из одиночных, выделенных лимфоцитов с использованием способа, именуемого в данной области как способ антител выбранных лимфоцитов (SLAM), как описано в патентах США №№ 7915388 и 5627052, публикации PCT WO 92/02551 и Babcock, J. S. et al. (1996) Proc. Natl. Acad. Sci. USA 93:7843-7848, полное содержание каждой из которых включено в настоящее описание в качестве ссылки. Вкратце, одиночные клетки, секретирующие антитела, представляющие интерес, например, лимфоциты, выделенные из любого одного из иммунизированных животных, описанных выше, подвергают скринингу с использованием антиген-специфического анализа на гемолитические бляшки, где антиген IL-13, субъединица IL-13 или их фрагмент, сочетают с овечьими красными кровяными клетками с использованием линкера, такого как биотин, и используют для идентификации одиночных клеток, которые секретируют антитела со специфичностью к IL-13. После идентификации клеток, секретирующих антитела, представляющие интерес, вариабельные области тяжелой и легкой цепи кДНК извлекают из клеток посредством ПЦР с обратной транскриптазой, и эти вариабельные области могут затем экспрессироваться, в контексте соответствующих константных областей иммуноглобулина (например, константных областей человека), в клетках-хозяевах млекопитающих, таких как клетки COS или CHO. Клетки-хозяева, трансфицированные амплифицированными иммуноглобулиновыми последовательностями, полученные из подвергнутых селекции in vivo лимфоцитов, могут затем подвергаться дополнительному анализу и селекции in vitro, например посредством пэннинга трансфицированных клеток для выделения клеток, экспрессирующих антитела против IL-13. С амплифицированными иммуноглобулиновыми последовательностями далее можно проводить манипуляции in vitro, такие как посредством способов созревания аффинности in vitro, таких как способы, описанные в публикации РСТ WO 97/29131 и публикации PCT WO 00/56772.
3. Моноклональные антитела против IL-13 с использованием трансгенных животных
Антитела для применения в композициях и/или способах по настоящему изобретению могут также продуцироваться посредством иммунизации не являющегося человеком животного, содержащего некоторые, или все из локусов иммуноглобулина человека с антигеном IL-13. В предпочтительном варианте осуществления животное, не являющееся человеком, представляет собой трансгенную мышь XENOMOUSE, сконструированный штамм мыши, который содержит крупные фрагменты локусов иммуноглобулинов человека и является дефектным по продуцированию антител мыши. См., например, Green et al. Nature Genetics 7: 13-21 (1994) и Патенты США №№ 5916771, 5939598, 5985615, 5998209, 6075181, 6091001, 6114598 и 6130364. См. также WO 91/10741, опубликованную 25 июля 1991 г, WO 94/02602, опубликованную 3 февраля 1994 г., WO 96/34096 и WO 96/33735, обе из которых опубликованы 31 октября 1996 г., WO 98/16654, опубликованную 23 апреля, 1998, WO 98/24893, опубликованную 11 Июня 1998 г., WO 98/50433, опубликованную 12 ноября 1998 г., WO 99/45031, опубликованную 10 сентября 1999 г., WO 99/53049, опубликованную 21 октября 1999 г., WO 00 09560, опубликованную 24 февраля 2000 г. и WO 00/037504, опубликованную 29 июня 2000 г, полное содержание каждой из которых явным образом включено в настоящее описание в качестве ссылки. Трансгенная мышь XENOMOUSE продуцирует репертуар полностью антител человека взрослого человека и генерирует антиген-специфичные Mab человека. Трансгенная мышь XENOMOUSE содержит приблизительно 80% из репертуара антител человека посредством введения мегабазы, сортированной по конфигурации локусов эмбрионального типа YAC-фрагментов тяжелой цепи и x-локусов легкой цепи человека. См. Mendez et al., Nature Genetics 15: 146-156 (1997), Green and Jakobovits J. Exp. Med. 188:483-495 (1998), раскрытие которых включено в настоящее описание в качестве ссылки.
4. Моноклональные антитела против IL-13 с использованием библиотек рекомбинантных антител
Способы in vitro также могут применяться для получения антител для применения в композициях и/или способах по изобретению, где библиотеку антител подвергают скринингу для идентификации антител, имеющих желательную специфичность связывания. Способы такого скрининга рекомбинантных библиотек антител являются хорошо известными в данной области и включают способы, описанные в, например, Ladner et al. Патент США № 5223409; Kang et al. Публикация РСТ № WO 92/18619; Dower et al. Публикация РСТ № WO 91/17271; Winter et al. Публикация РСТ № WO 92/20791; Markland et al. Публикация РСТ № WO 92/15679; Breitling et al. Публикация РСТ № WO 93/01288; McCafferty et al. Публикация РСТ № WO 92/01047; Garrard et al. Публикация РСТ № WO 92/09690; Fuchs et al. (1991) Bio/Technology 9: 1370-1372; Hay et al. (1992) Hum Antibod Hybridomas 3:81-85; Huse et al. (1989) Science 246: 1275-1281; McCafferty et al., Nature (1990) 348:552-554; Griffiths et al. (1993) EMBO J. 12:725-734; Hawkins et al. (1992) J Mol Biol 226:889-896; Clackson et al. (1991) Nature 352:624-628; Gram et al. (1992) PNAS 89:3576-3580; Garrad et al. (1991) Bio/Technology 9: 1373-1377; Hoogenboom et al. (1991) Nuc Acid Res 19:4133-4137; и Barbas et al. (1991) PNAS 88:7978-7982, Публикация патентной заявки США 20030186374, и Публикация РСТ № WO 97/29131, содержание каждой из которых включено в настоящее описание в качестве ссылки.
Библиотека рекомбинантных антител может быть получена от индивида, иммунизированного IL-13 или IL-13, или частью IL-13 или IL-13. Альтернативно, библиотека рекомбинантных антител может быть получена от нативного индивида, т.е., индивида, который не был иммунизирован с помощью IL-13, такая как библиотека антител человека от человека-индивида, который не был иммунизирован IL-13 человека. Антитела по изобретению подвергают селекции посредством скрининга библиотеки рекомбинантных антител с пептидом, содержащим IL-13 человека, чтобы, таким образом, выбрать такие антитела, которые распознают IL-13. Способы проведения такого скрининга и селекции являются хорошо известными в данной области, такие как способы, описанные в ссылках в предыдущем параграфе. Для выбора антител по изобретению, имеющих конкретные значения аффинностей связывания для IL-13, таких как антитела, которые диссоциируют из IL-13 человека с конкретной koff константой скорости диссоциации, известный в данной области способ поверхностного плазмонного резонанса может применяться для отбора антител, имеющих желательную константу скорости диссоциации. Для отбора антител по изобретению, имеющих конкретную нейтрализующую активность для IL-13, таких как антитела с конкретным IC50, могут применяться стандартные способы, известные в данной области для оценки ингибирования активности IL-13.
В одном из аспектов изобретение имеет отношение к выделенным антителам или их антигенсвязывающей части, которая связывается с IL-13 человека. Предпочтительно, антитело представляет собой нейтрализующее антитело. В разнообразных вариантах осуществления антитело является рекомбинантным антителом или моноклональным антителом.
Например, антитела по настоящему изобретению также можно получать с использованием разнообразных способов отображения фагов, известных в данной области. В способах отображения фагов, домены функционального антитела отображаются на поверхности фаговых частиц, которые несут полинуклеотидные последовательности, кодирующие их. В частности, такой фаг может использоваться для отображения антигенсвязывающих доменов, экспрессируемых из репертуара или комбинаторной библиотеки антител (например, человека или мыши). Фаг, экспрессирующий антигенсвязывающий домен, который связывает антиген, представляющий интерес, может быть отобран или идентифицирован с помощью антигена, например, с использованием меченого антигена или антигена, связанного или соединенного с твердой поверхностью или шариком. Фаг, используемый в этих способах, обычно представляет собой нитевидный фаг, включающий связывающие домены fd и M13, экспрессируемые из фага с Fab, Fv или домены дисульфид-стабилизированного Fv антитела, рекомбинантно слитые либо с фаговым геном III или белком гена VIII. Примеры способов отображения фагов, которые могут применяться для получения антител по настоящему изобретению, включают способы, раскрытые в Brinkman et al., J. Immunol. Methods 182:41-50 (1995); Ames et al., J. Immunol. Methods 184:177-186 (1995); Kettleborough et al., Eur. J. Immunol. 24:952-958 (1994); Persic et al., Gene 187 9-18 (1997); Burton et al., Advances in Immunology 57:191-280 (1994); Заявка РСТ № PCT/GB91/01134; публикации РСТ WO 90/02809; WO 91/10737; WO 92/01047; WO 92/18619; WO 93/11236; WO 95/15982; WO 95/20401; и патентах США №№ 5698426; 5223409; 5403484; 5,580,717; 5427908; 5750753; 5821047; 5571698; 5427908; 5516637; 5780225; 5658727; 5733743 и 5969108; каждый из которых включен в настоящее описание в качестве ссылки во всей полноте.
Как описано в указанных выше ссылках, после селекции фага, области, кодирующие антитело из фага, могут быть выделены и применяться для получения цельных антител, включающих антитела человека или любой другой желательный антигенсвязывающий фрагмент, и экспрессироваться в любом желательном хозяине, включая клетки млекопитающих, клетки насекомых, клетки растений, дрожжей и бактерий, например, как описано подробно ниже. Например, способы для рекомбинантного продуцирования фрагментов Fab, Fab' и F(ab')2, могут также использоваться с использованием способов, известных в данной области, таких как способы, раскрытые в публикации РСТ WO 92/22324; Mullinax et al., BioTechniques 12(6):864-869 (1992); и Sawai et al., AJRI 34:26-34 (1995); и Better et al., Science 240:1041-1043 (1988) (включены в качестве ссылки во всей своей полноте). Примеры способов, которые могут применяться для получения Fv с одиночной цепью и антител, включают способы, описанные в патентах США №№ 4946778 и 5258498; Huston et al., Methods in Enzymology 203:46-88 (1991); Shu et al., PNAS 90:7995-7999 (1993); и Skerra et al. Science 240:1038-1040 (1988).
Как альтернатива скринингу библиотек рекомбинантных антител посредством фагового отображения, другие методологии, известные в данной области для скрининга крупных комбинаторных библиотек могут применяться для идентификации антител по изобретению с двойной специфичностью. Одним типом альтернативной системы экспрессии является система, в которой библиотека рекомбинантных антител экспрессируется в виде слияний РНК-белок, как описано в публикации РСТ № WO 98/31700 Szostak и Roberts, и у Roberts, R. W. и Szostak, J. W. (1997) Proc. Natl. Acad. Sci. USA 94: 12297-12302. В данной системе, ковалентное слияние создается между мРНК и пептидом или белком, которые она кодирует посредством трансляции in vitro синтетических мРНК, которые несут пуромицин, пептидилакцепторный антибиотик, по их 3'-концу. Таким образом, конкретная мРНК может быть извлечена с обогащением из сложной смеси мРНК (например, комбинаторной библиотеки), на основании свойств кодируемого пептида или белка, например, антитела или его части, таких как связывание антитела или его части с антигеном двойной специфичности. Последовательности нуклеиновых кислот, кодирующие антитела, или их части, извлеченные в результате скрининга таких библиотек, могут экспрессироваться рекомбинантными средствами, как описано выше (например, в клетках-хозяевах млекопитающих) и, кроме того, могут быть подвергнуты дополнительному созреванию аффинности посредством либо дополнительных циклов скрининга слияний мРНК-пептид, в которых мутации вводятся в исходно отобранные последовательность(последовательности) или посредством других способов для созревания аффинности in vitro рекомбинантных антител, как описано выше.
При еще одном подходе антитела для применения в композициях и/или способах по настоящему изобретению также можно получать с использованием способов дрожжевого отображения, известных в данной области. В способах дрожжевого отображения, генетические способы применяют для привязывания доменов антитела к клеточной стенке дрожжей и отображения их на поверхности дрожжей. В частности, такие дрожжи могут использоваться для отображения антигенсвязывающих доменов, экспрессируемых из репертуара или комбинаторной библиотеки антител (например, человека или мыши). Примеры способов дрожжевого отображения, которые могут применяться для получения антител по настоящему изобретению, включают способы, раскрытые Wittrup, et al. Патент США № 6699658, включенный в настоящее описание в качестве ссылки.
B. Получение рекомбинантных IL-13 Антител
Антитела для применения в композициях и/или способах по настоящему изобретению могут продуцироваться посредством любого из числа способов, известных в данной области. Например, экспрессии из клеток-хозяев, где вектор(векторы) экспрессии, кодирующие тяжелую и легкие цепи является (являются) трансфицированными в клетку-хозяина посредством стандартных способов. Разнообразные формы термина "трансфекция" предназначены для охвата широкого разнообразия способов, обычно используемых для введения экзогенной ДНК в прокариотную или эукариотную клетку-хозяина, например, электропорацию, осаждение фосфатом кальция, трансфекцию ДЭАЭ-декстрана и т.п. Несмотря на то, что возможно экспрессировать антитела по изобретению либо в прокариотных или эукариотных клетках-хозяевах, экспрессия антител в эукариотных клетках является предпочтительной, и наиболее предпочтительной в клетках-хозяевах млекопитающих, поскольку такие эукариотные клетки (и, в особенности, клетки млекопитающих) более вероятнее, чем прокариотные клетки проходят сборку и секретируют надлежащим образом изогнутые и иммунологически активные антитела.
Предпочтительные клетки-хозяева млекопитающих для экспрессии рекомбинантных антител по изобретению включают клетки яичника китайского хомяка (CHO-клетки) (включая dhfr-CHO-клетки, описанные в Urlaub and Chasin, (1980) Proc. Natl. Acad. Sci. USA 77:4216-4220, применяемые вместе с селектируемым маркером DHFR, например, как описано в R. J. Kaufman and P. A. Sharp (1982) Mol. Biol. 159:601-621), NSO-клетки миеломы, COS-клетки и SP2-клетки. Когда рекомбинантные векторы экспрессии, кодирующие гены антитела, вводят в клетки-хозяева млекопитающих, антитела продуцируются при культивировании клеток-хозяев в течение периода времени, достаточного для обеспечения экспрессии антитела в клетках-хозяевах, или, более предпочтительно, секреции антитела в культуральную среду, в которой клетки-хозяева выращивают. Антитела могут быть извлечены из культуральной среды с использованием стандартных способов очистки белка.
Клетки-хозяева могут также применяться для получения функциональных фрагментов антител, таких как Fab-фрагменты или молекулы scFv. Будет понятным, что вариации указанной выше методики находятся в пределах объема притязаний настоящего изобретения. Например, может быть желательно трансфицировать клетку-хозяина с помощью ДНК, кодирующей функциональные фрагменты либо легкой цепи и/или тяжелой цепи антитела по изобретению. Способ рекомбинантных ДНК может также применяться для удаления некоторых, или всех ДНК, кодирующих любую из двух или обе вместе легкую и тяжелую цепи, что не является необходимым для связывания с антигенами, представляющими интерес. Молекулы, экспрессируемые из таких усеченных молекул ДНК, также охватываются антителами по изобретению. В дополнение, могут продуцироваться бифункциональные антитела, в которых одна тяжелая и одна легкая цепи принадлежат антителам по изобретению, а другие тяжелая и легкая цепи являются специфичными для антигена, отличного от антигенов, представляющих интерес, посредством сшивки антитела по изобретению со вторым антителом посредством стандартных способов химической сшивки.
В предпочтительной системе для рекомбинантной экспрессии антител или их антигенсвязывающей части по изобретению, рекомбинантный вектор экспрессии, кодирующий как тяжелую цепь антитела, так и легкую цепь антитела вводят в dhfr-CHO клетки посредством опосредованной фосфатом кальция трансфекции. В пределах рекомбинантного вектора экспрессии, каждый из генов тяжелой и легкой цепей антитела функционально связан с регуляторными элементами CMV-энхансера/AdMLP-промотора, чтобы управлять высокими уровнями транскрипции генов. Рекомбинантный вектор экспрессии также несет ген DHFR, который обеспечивает селекцию CHO-клеток, которые были трансфицированы вектором с использованием селекции/амплификации с метотрексатом. Отобранные трансформированные клетки-хозяева культивируют, чтобы обеспечить экспрессию тяжелой и легкой цепей антитела, и интактные антитела извлекают из культуральной среды. Стандартные способы молекулярной биологии используют для получения рекомбинантного вектора экспрессии, трансфекции клеток-хозяев, отбора трансформантов, культивирования клеток-хозяев и извлечения антител из культуральной среды. Еще дополнительно изобретение предоставляет способ синтеза рекомбинантного антитела по изобретению посредством культивирования клетки-хозяина по изобретению в подходящей культуральной среде до завершения синтеза рекомбинантных антител по изобретению. Способ может дополнительно включать выделение рекомбинантных антител из культуральной среды.
1. Антитела против IL-13
Таблица 5 Патента США № 7195388 (содержание которого включено в настоящее описание в качестве ссылки) представляет собой список аминокислотных последовательностей VH и VL областей предпочтительных антител против IL-13 для использования в композициях и/или способах по изобретению. Эти выделенные CDR-последовательности антител против IL-13 составляют семейство IL-13-связывающих белков, выделенных в соответствии с настоящим изобретением, и содержащих полипептиды, которые включают CDR-последовательности, перечисленные в Таблице 6 Патента США № 7195388 (содержание которого включено в настоящее описание в качестве ссылки). Для получения и для селекции CDR по изобретению, имеющие предпочтительные IL-13-связывающую и/или нейтрализующую активность по отношению к IL-13 и/или IL-13, могут применяться стандартные способы, известные в данной области для получения связывающих белков по настоящему изобретению и оценки IL-13 и/или IL-13 связывающих и/или нейтрализующих характеристик этих связывающих белков, включающие, но не ограниченные лишь ими, конкретно описанные в настоящем описании.
В одном из вариантов осуществления антитела, используемые в композициях и/или способах по изобретению представляют собой антитела 13C5.5 (см. Патент США № 7915388, полное содержание которого включено в настоящее описание в качестве ссылки). 13C5.5 является гуманизированным антителом, которое связывается с высокой аффинностью со спиралями А и D интерлейкина 13 (IL-13) (см. Фигуру 1). Последовательности вариабельной области тяжелой и легкой цепей 13C5.5 представлены выше как SEQ NO:2 и SEQ ID NO:3, соответственно.
2. Химерные антитела против IL-13
Химерное антитело представляют собой молекулу, в которой различные части антитела получают из различных видов животных, такое как антитела, имеющие вариабельную область, производную из моноклонального антитела мыши и константной области иммуноглобулина человека. Способы получения химерных антител являются известными в данной области и обсуждаются подробно в Примере 2.1. См., например, Morrison, Science 229:1202 (1985); Oi et al. BioTechniques 4:214 (1986); Gillies et al. (1989) J. Immunol. Methods 125:191-202; патенты США № 5807715; 4816567 и 4816397, которые включены в настоящее описание в качестве ссылки во всей полноте. Дополнительно, могут применяться способы, разработанные для получения "химерных антител" (Morrison et al. 1984, Proc. Natl. Acad. Sci. 81:851-855; Neuberger et al. 1984, Nature 312:604-608; Takeda et al. 1985, Nature 314:452-454, которые включены в настоящее описание в качестве ссылки во всей их полноте) посредством сплайсинга генов из молекулы антитела мыши с соответствующей антигенной специфичностью вместе с генами из молекулы антитела человека с соответствующей биологической активностью.
В одном из вариантов осуществления химерные антитела для применения в композициях и/или способы по изобретению продуцируют посредством замены константной области тяжелой цепи моноклональных антител мыши против IL-13 человека, описанных в разделе 1, на константную область IgG1 человека. В конкретном варианте осуществления химерное антитело по изобретению содержит вариабельную область тяжелой цепи (VH), содержащую аминокислотную последовательность из SEQ ID NO:34; SEQ ID NO:36; SEQ ID NO:41; SEQ ID NO:42; SEQ ID NO:46 и вариабельную область легкой цепи (VL), содержащую аминокислотную последовательность из SEQ ID NO:35; SEQ ID NO:37; SEQ ID NO:40; SEQ ID NO:43; или SEQ ID NO:47, раскрытых в Патенте США № 7195388.
3. Гуманизированные антитела против IL-13
Гуманизированные антитела представляют собой молекулы антител из антител видов, не являющихся человеком, которые связывают желательный антиген, имеющий одну или несколько областей, определяющих комплементарность (CDR) из видов, не являющихся человеком, и каркасные области из молекулы иммуноглобулина человека. Известные последовательности Ig человека раскрыты, например, у Kabat et al. Sequences of Proteins of Immunological Interest, U.S. Dept. Health (1983), включенной в настоящее описание в качестве ссылки в полном объеме. Такие импортированные последовательности могут применяться для снижения иммуногенности или снижения, усиления или модификации связывания, аффинности, скорости связи, скорости диссоциации, авидности, специфичности, времени полужизни или любой подходящей характеристики, как известно в данной области.
Каркасные остатки в каркасных областях человека могут быть замещены соответствующим остатком из CDR донорного антитела для изменения, предпочтительно улучшения, связывания с антигеном. Эти каркасные замещения идентифицируются способами, хорошо известными в данной области, например, посредством моделирования взаимодействий CDR и каркасных остатков для идентификации каркасных остатков, важных для связывания с антигеном и сравнения последовательностей для идентификации необычных каркасных остатков в конкретных положениях. (См., например, Queen et al., Патент США № 5585089; Riechmann et al. Nature 332:323 (1988), которые включены в настоящее описание в качестве ссылки во всей их полноте). Трехмерные модели иммуноглобулинов являются общедоступными и знакомы квалифицированным специалистам в данной области. Доступными являются компьютерные программы, которые иллюстрируют и отображают вероятные трехмерные конформационные структуры отобранных кандидатных последовательностей иммуноглобулинов. Рассмотрение этих отображений позволяет проводить анализ вероятной роли остатков при функционировании кандидатной последовательности иммуноглобулина, т.е., анализ остатков, которые оказывают воздействие на способность кандидатного иммуноглобулина связываться с его антигеном. Таким образом, FR-остатки могут быть отобраны и скомбинированы из консенсусных и импортных последовательностей, так, чтобы достичь желательной характеристики антитела, такой как увеличенная аффинность для целевых антигена(антигенов). В целом, CDR-остатки непосредственно и наиболее существенно вовлечены в оказание воздействия на связывание антигена. Антитела могут быть гуманизированы с использованием разнообразных способов, известных в данной области, таких как, но ими не ограничиваясь, способы, описанные в Jones et al., Nature 321:522 (1986); Verhoeyen et al., Science 239:1534 (1988)), Sims et al., J. Immunol. 151: 2296 (1993); Chothia and Lesk, J. Mol. Biol. 196:901 (1987), Carter et al. Proc. Natl. Acad. Sci. U.S.A. 89:4285 (1992); Presta et al. J. Immunol. 151:2623 (1993), Padlan, Molecular Immunology 28(4/5):489-498 (1991); Studnicka et al. Protein Engineering 7(6):805-814 (1994), Roguska. et al. PNAS 91:969-973 (1994); публикация PCT WO 91/09967, PCT/: US98/16280, US96/18978, US91/09630, US91/05939, US94/01234, GB89/01334, GB91/01134, GB92/01755; WO90/14443, WO90/14424, WO90/14430, EP 229246, EP 592106; EP 519596, EP 239400, патенты США №№ 5565332, 5723323, 5976862, 5824514, 5817483, 5814476, 5763192, 5723323, 5766886, 5714352, 6204023, 6180370, 5693762, 5530101, 5585089, 5225539; 4816567, каждая из которых полностью включена в настоящее описание в качестве ссылки, с включенными ссылками, цитируемыми в них.
C. Получение антител и продуцирующие антитела клеточные линии
Предпочтительно, антитела против IL-13 для применения в композициях и/или способах по настоящему изобретению проявляют высокую способность снижать или нейтрализовать активность IL-13, например, как оценивают посредством одного из некоторых аналитических тестов in vitro и in vivo, известных в данной области (например, см. Пример 1.1.C Патента США № 7195388, содержание которого включено в настоящее описание в качестве ссылки в полном объеме). Например, эти антитела нейтрализуют индуцируемое IL-13 продуцирование TARC клетками A-549 со значениями IC50 в интервале, по меньшей мере, около 10-8 M, около 10-9 M или приблизительно 10-10 M.
В предпочтительных вариантах осуществления, выделенное антитело или его антигенсвязывающая часть связывает IL-13 человека, где антитела или их антигенсвязывающая часть диссоциируют из IL-13 человека с константой скорости диссоциации koff, равной приблизительно 0,1с-1 или менее, как определяют посредством поверхностного плазмонного резонанса, или, которые ингибируют IL-13 человека и/или активность IL-13 человека с IC50, равной приблизительно 1×10-6 M или менее. Альтернативно, антитела или их антигенсвязывающая часть могут диссоциировать от IL-13 человека с константой скорости диссоциации koff, равной приблизительно 1×10-2с-1 М или менее, как определяют посредством поверхностного плазмонного резонанса, или могут ингибировать IL-13 человека и/или активность IL-13 человека с IC50, равной приблизительно 1×10-7 M или менее. Альтернативно, антитела или их антигенсвязывающая часть могут диссоциировать от IL-13 человека с константой скорости диссоциации koff, равной приблизительно 1×10-3с-1 или менее, как определяют посредством поверхностного плазмонного резонанса, или могут ингибировать IL-13 человека и/или активность IL-13 человека с IC50, равной приблизительно 1×10-8 M или менее. Альтернативно, антитела или их антигенсвязывающую часть, могут диссоциировать от IL-13 человека с константой скорости диссоциации koff, равной приблизительно 1×10-4с-1 или менее, как определяют посредством поверхностного плазмонного резонанса, или могут ингибировать IL-13 и/или активность IL-13 человека с IC50, равной приблизительно 1×10-9 M или менее. Альтернативно, антитела или их антигенсвязывающая часть, могут диссоциировать от IL-13 человека с константой скорости диссоциации koff, равной приблизительно 1×10-5с-1 или менее, как определяют посредством поверхностного плазмонного резонанса, или могут ингибировать IL-13 и/или активность IL-13 человека с IC50, равной приблизительно 1×10-10 M или менее. Альтернативно, антитела или их антигенсвязывающая часть, могут диссоциировать от IL-13 человека с константой скорости диссоциации koff, равной приблизительно 1×10-5 с-1 или менее, как определяют посредством поверхностного плазмонного резонанса, или могут ингибировать IL-13 и/или активность IL-13 человека с IC50, равной приблизительно 1×10-11 M или менее.
IL-13 проявляет свои действия посредством связывания с рецептором IL-13 (IL-13R) на поверхности клеток, гетеродимером, состоящим из цепи IL-13Rα1 (IL-13Rα1) и цепи IL-4R (IL-4R). IL-13 связывается с IL-13Rα1 сначала с низкой аффинностью (KD=2-10 нМ) и затем мобилизует IL-4R в комплекс, генерируя рецептор с высокой аффинностью (KD=0,03-0,4 нМ) (Aman, M. J., et al. 1996 J. Biol. Chem. 271, 29265-29270; Miloux, et al. 1997 FEBS Lett. 401, 163-166; Andrews, et al 2002 J. Biol. Chem. 277, 46073-46078). Гетеродимеризация IL-13R вызывает активацию Янус-киназ, TYK2 и JAK1, конститутивно связанных с IL-13Rαl и IL-4R, соответственно, с последующей активацией сигнального трансдуктора и активатора транскрипции 6 (STAT6) (Izuhara, K., and Arima, K. 2004 Drug News Perspect. 17, 91-98). Существует еще одна IL-13-связывающая единица, цепь IL-13Rα2 (IL-13Rα2), которая связывается с IL-13 с высокой аффинностью (0,25-1,2 нМ) (Caput, et al 1996 J. Biol. Chem. 271, 16921-16926; Donaldson et al 1998 J. Immunol. 161, 2317-2324). Неизвестна никакая другая молекула рецептора, вовлеченная в комплекс IL-13IL-13R2. Первоначально полагали, что IL-13R2 действует как несигнальный "рецептор-ловушка". Однако позднее было обнаружено, что он может связываться с IL-13 и сигнализировать через путь AP-1, что приводит к продуцированию TNF-бета у некоторых типов клеток, включающих макрофаги, что, в свою очередь, приводит к фиброзу легких (Fichtner-Feigl, 2006 Nat Med 12:99-106). Следовательно, как путь IL-13Rα1/IL-4Rα, так и путь IL-13Rα2 способствуют общей патофизиологии астмы и других легочных воспалительных состояний. Следовательно, терапевтические антитела против IL-13, которые блокируют связывание IL-13 с обоими рецепторами, будут более эффективными, чем антитела, которые блокируют только один рецептор.
В одном из аспектов настоящее изобретение относится к композициям и/или способам, в которых применяются моноклональные антитела, которые блокируют связывание IL-13 с обоими IL-13Rα1 и IL-13Rα2. Как аналитический тест на связывание с рецептором на основе ELISA и аналитический тест с 125I-меченым IL-13 на поверхности клеток продемонстрировали, что 13C5, как мышиная версия, так и гуманизированная версия (т.е., 13C5.5), были способными к эффективной блокировке связывания IL-13 с обоими рецепторами. Антитела в той же линии клеток 13C5, включающие 25C8 и 33C3, были также способны блокировать связывание IL-13 с обоими рецепторами. Картирование эпитопов 13C5 показало, что его участок (участки) связывания включают область C-концевой Спирали D IL-13 человека (остатки VRDTK IEVAQ FVKDL LLHLK KLFRE GR, соответствующие аминокислотам 104-130 из SEQ ID NO:1). Предположили, что область C-концевой Спирали D вовлечена во взаимодействия с рецептором IL-13 (Zuegg et al 2001 Immunol Cell Biol. 79:332-9). Кристаллическая структура IL-13 человека в комплексе с Fab-частью антитела 13C5.5 указывает на то, что 13C5.5 связывается с областью C-концевой Спирали D, а также областью N-концевой Спирали А IL-13 человека. Предпочтительно антитело или его антигенсвязывающий фрагмент связывает IL-13 человека таким образом, что IL-13 вместе с указанным антителом или его антигенсвязывающим фрагментом, связанные с эпитопом, определяемым топографическими областями Ser26-Thr27-Ala28-Leu29-Arg30-Glu31-Leu32-Ile33-Glu34-Glu35-Leu36-Val37-A-sn38 и Lys123-Lys124-Leu125-Phe126-Arg127-Glu-128-Gly129-Arg130 из SEQ ID NO:1 ингибируются от связывания с рецептором IL-13. Предпочтительно антитело или его антигенсвязывающий фрагмент связывается с IL-13 человека таким образом, что IL-13 с указанным антителом или его антигенсвязывающий фрагмент, связанные с эпитопом, определяемым топографическими областями Arg30-Glu3'-Leu32-Ile33-Glu34-Glu35-Leu36-Val37-Asn38 и Lys123-Lys124-Leu125-Phe126-Arg127 из SEQ ID NO: 1 ингибируются от связывания с рецептором IL-13.альфа.2.
В некоторых вариантах осуществления, антитело содержит константную область тяжелой цепи, такую как константную область IgG1, IgG2, IgG3, IgG4, IgA, IgE, IgM или IgD. Предпочтительно, константная область тяжелой цепи представляет собой константную область тяжелой цепи IgG1 или константную область тяжелой цепи IgG4. Кроме того, антитело может содержать константную область легкой цепи, либо константную область легкой цепи каппа или константную область легкой цепи лямбда. Предпочтительно, антитело содержит константную область легкой цепи каппа. Альтернативно, часть антитела может представлять собой, например, Fab-фрагмент или фрагмент одиночной цепи Fv.
Замены аминокислотных остатков в части Fc для изменения эффекторной функции антитела являются известными в данной области (Winter, et al. Патенты США №№ 5648260; 5624821). Часть Fc антитела опосредует некоторые важные эффекторные функции, например индукцию цитокина, ADCC, фагоцитоз, комплементзависимую цитотоксичность (CDC) и время полужизни/скорость выведения антитела и комплексов антиген-антитело. В некоторых случаях эти эффекторные функции являются желательными для терапевтических антител, но в других случаях может быть ненужной или даже вредной, в зависимости от терапевтических целей. Некоторые изотипы IgG человека, особенно, IgG1 и IgG3, опосредуют ADCC и CDC через связывание с Fc.гамма.R и комплементом Clq, соответственно. Неонатальные рецепторы Fc (FcRn) являются решающими компонентами, определяющими циркуляционное время полужизни антитела. В еще одном другом варианте осуществления по меньшей мере один аминокислотный остаток заменен в константной области антитела, например области Fc антитела, таким образом, что эффекторные функции антитела изменяются.
В одном из вариантов осуществления в способах и композициях по изобретению применяют меченый связывающий белок, где антитело или часть антитела по изобретению дериватизируют или связывают с еще одной функциональной молекулой (например, еще одним пептидом или белком). Например, меченый связывающий белок по изобретению может быть произведен посредством функционального связывания антитела или части антитела по изобретению (посредством химического сочетания, генетической гибридизации, нековалентной связи или иным образом) с одним или несколькими другими молекулярными субстанциями, такими как еще одно антитело (например, биспецифическое антитело или диатело), детектируемый агент, цитотоксическое средство, фармацевтическое средство и/или белок или пептид, которые могут опосредовать связь антитела или части антитела с еще одной другой молекулой (такой как центральная область стрептавидина или полигистидиновая метка).
Применимые детектируемые агенты, с которыми антитело или часть антитела по изобретению могут быть дериватизированы, включают флуоресцентные соединения. Примерные флуоресцентные детектируемые агенты включают флуоресцеин, изотиоцианат флуоресцеина, родамин, 5-диметиламин-1-нафталинсульфонилхлорид, фикоэритрин и т.п. Антитело может также быть дериватизировано с детектируемыми ферментами, такими как щелочная фосфатаза, пероксидаза хрена, глюкозооксидаза и т.п. Когда антитело дериватизируют детектируемым ферментом, его обнаруживают посредством добавления дополнительных реагентов, которые ферменты использует для получения детектируемого продукта реакции. Например, когда присутствует детектируемый агент пероксидаза хрена, добавление пероксида водорода и диаминобензидина приводит к окрашенному продукту реакции, который является обнаруживаемым. Антитело может также быть дериватизировано с биотином, и обнаружено через косвенное измерение связывания авидина или стрептавидина.
В еще одном варианте осуществления в композициях и способах по изобретению применяют кристаллизованный связывающий белок. Предпочтительно изобретение относится к кристаллам цельного антитела против IL-13 и его фрагментов, как раскрыто в настоящем описании, и готовым формам и композициям, содержащим такие кристаллы. В одном из вариантов осуществления кристаллизованный связывающий белок имеет большее время полужизни in vivo чем растворимый аналог связывающего белка. В еще одном варианте осуществления связывающий белок сохраняет биологическую активность после кристаллизации.
Кристаллизованный связывающий белок по изобретению может быть получен в соответствии со способами, известными в данной области и как раскрыто в WO 02072636, включенной в настоящее описание в качестве ссылки.
В еще одном другом варианте осуществления в композициях и/или способах по изобретению применяется гликозилированный связывающий белок, где антитело или его антигенсвязывающая часть содержит один или несколько углеводных остатков. Появляющееся продуцирование белка in vivo может претерпевать дополнительный процессинг, известный как посттрансляционная модификация. В частности, сахарные (гликозильные) остатки могут добавляться ферментативно, в процессе, известном как гликозилирование. Образующиеся в результате белки, несущие ковалентно связанные олигосахаридные боковые цепи, являются известными как гликозилированные белки или гликопротеины. Антитела представляют собой гликопротеины, с одним или несколькими углеводными остатками в Fc-домене, а также вариабельном домене. Углеводные остатки в Fc-домене имеют важное воздействие на эффекторную функцию Fc-домена, с минимальным эффектом на связывание антигена или время полужизни антитела (R. Jefferis, Biotechnol. Prog. 21 (2005), pp. 11-16). Напротив, гликозилирование вариабельного домена может иметь воздействие на антигенсвязывающую активность антитела. Гликозилирование в вариабельном домене может иметь отрицательное воздействие на аффинность связывания антитела, вероятно, вследствие стерической затрудненности (Co, M. S., et al., Mol. Immunol. (1993) 30:1361-1367), или приводить к увеличенной аффинности для антигена (Wallick, S. C, et al., Exp. Med. (1988) 168:1099-1109; Wright, A., et al. EMBO J. (1991) 10:2717 2723).
Один из аспектов настоящего изобретения направлен на получение мутантов по участку гликозилирования, в которых O- или N-связанный участок гликозилирования связывающего белка прошел мутацию. Квалифицированный специалист в данной области может получать такие мутанты с использованием стандартных хорошо известных технологий. Мутанты по участку гликозилирования, которые сохраняют биологическую активность, но имеют увеличенную или сниженную активность связывания, являются еще одной целью настоящего изобретения.
В еще одном другом варианте осуществления гликозилирование антитела или его антигенсвязывающей части по изобретению модифицируют. Например, может быть получено агликозилированное антитело (т.е., антитело с отсутствием гликозилирования). Гликозилирование может изменяться, чтобы, например, увеличить аффинность антитела для антигена. Такие углеводные модификации могут осуществляться посредством, например, изменения одного или нескольких участков гликозилирования в последовательности антитела. Например, могут быть сделаны одна или несколько замен аминокислот, которые приводят к элиминированию одного или нескольких участков гликозилирования вариабельной области, чтобы посредством этого элиминировать гликозилирование на этом участке. Такое агликозилирование может увеличить аффинность антитела для антигена. Такой подход описан более подробно в публикации РСТ WO2003016466A2 и Патентах США №№ 5714350 и 6350861, каждый из которых включен в настоящее описание в качестве ссылки во всей своей полноте.
Дополнительно или альтернативно, может быть получено модифицированное антитело по изобретению, которое имеет измененный тип гликозилирования, такое как гипофукозилированное антитело, имеющее сниженные количества фукозильных остатков, или антитело, имеющее увеличенные биссекторные структуры GlcNAc. Было продемонстрировано, что такие измененные схемы гликозилирования увеличивают способность к ADCC антител. Такие углеводные модификации могут осуществляться посредством, например, экспрессии антитела в клетке-хозяине с измененным механизмом гликозилирования. Клетки с измененным механизмом гликозилирования были описаны в данной области и могут применяться в качестве клеток-хозяев, в которых экспрессируют рекомбинантные антитела по изобретению, чтобы таким образом продуцировать антитело с измененным гликозилированием. См., например, Shields, R. L. et al. (2002) J. Biol. Chem. 277:26733-26740; Umana et al. (1999) Nat. Biotech. 17: 176-1, а также, Европейский Патент №: EP 1176195; Публикации РСТ WO 03/035835; WO 99/54342 80, каждая из которых включена в настоящее описание в качестве ссылки во всей своей полноте.
Гликозилирование белка зависит от аминокислотной последовательности белка, представляющего интерес, а также клетки-хозяина, в которой белок экспрессируется. Различные организмы могут продуцировать различные ферменты гликозилирования (например, гликозилтрансферазы и гликозидазы), и имеют различные доступные субстраты (нуклеотидные сахара). Вследствие таких факторов, профиль гликозилирования белка и состав гликозильных остатков, может различаться в зависимости от системы хозяина, в которой конкретный белок экспрессируется. Гликозильные остатки, применимые в изобретении, могут включать, но ими не ограничены, глюкозу, галактозу, маннозу, фукозу, н-ацетилглюкозамин и сиаловую кислоту. Предпочтительно гликозилированный связывающий белок содержит гликозильные остатки таким образом, что профиль гликозилирования является человеческим.
Квалифицированным специалистам в данной области известно, что различающееся гликозилирование белка может приводить к различающимся характеристикам белка. К примеру, эффективность действия терапевтического белка, продуцируемого в микроорганизме-хозяине, таком как дрожжи, и гликозилированного с использованием дрожжевого эндогенного пути, может быть снижено по сравнению с эффективностью действия такого же белка, экспрессируемого в клетках млекопитающего, таких как клеточная линия CHO. Такие гликобелки могут также быть иммуногенными у людей и показывать сниженное время полужизни in vivo после введения. Специфические рецепторы у людей и других животных могут распознавать конкретные гликозильные остатки и способствовать быстрому выведению белка из кровотока. Другие неблагоприятные эффекты могут включать изменения в фолдинге, растворимости, подверженности действию протеаз, направленной миграции, транспорте, компартментализации, секреции, распознавании другими белками или факторами, антигенности или аллергенности белка. Соответственно, специалист-практик может предпочитать терапевтический белок с конкретным составом и профилем гликозилирования, например составом и профилем гликозилирования, идентичными или по меньшей мере аналогичными составу и профилю гликозилирования в клетках человека или в видоспецифичных клетках предназначенного индивидного животного.
Экспрессия гликозилированных белков, отличных от аналогичных белков клетки-хозяина может достигаться посредством посредством генетической модификации клетки-хозяина для экспрессии гетерологичных гликозилированных ферментов. С использованием способов, известных в данной области, специалист-практик может получать антитела или их антигенсвязывающие части, проявляющие гликозилирование белков человека. Например, штаммы дрожжей были генетически модифицированы для экспрессии неприродных ферментов гликозилирования таким образом, что гликозилированные белки (гликопротеины), продуцируемые в этих штаммах дрожжей, проявляли гликозилирование белка, идентичное гликозилированию в клетках животных, особенно, клетках человека (патентные заявки США 20040018590 и 20020137134 и публикация PCT WO2005100584 A2, содержание каждой из которых включено в настоящее описание в качестве ссылки в полном объеме).
В способах и/или композициях по настоящему изобретению могут также применяться антиидиотипические (анти-Id) антитела, специфичные для таких связывающих белков по изобретению. Антитело анти-Id является антителом, которое распознает уникальные детерминанты, обычно связанные с антигенсвязывающей областью еще одного антитела. Анти-Id могут быть получены посредством иммунизации животного связывающим белком или его CDR-содержащей область. Иммунизированное животное будет распознавать и реагировать на идиотипические детерминанты иммунизирующего антитела и продуцировать анти-Id антитела. Анти-Id антитела могут также применяться в качестве "иммуноген", для индуцирования иммунного ответа у еще одного другого животного, продуцирующего так называемые против-анти-Id антитела.
Далее, квалифицированный специалист в данной области сможет оценить то, что белок, представляющий интерес может экспрессироваться с использованием библиотеки клеток-хозяев, созданной способами генетической инженерии, чтобы экспрессировать разнообразные ферменты гликозилирования, таким образом, что участвующие клетки-хозяева данной библиотеки продуцируют белок, представляющий интерес с вариантными профилями гликозилирования. Специалист-практик может затем отобрать и выделить интересующий белок с конкретно новыми профилями гликозилирования. Предпочтительно, белок, имеющий конкретный отобранный новый профиль гликозилирования, проявляют улучшенные или измененные биологические свойства.
II. Композиции по изобретению
Настоящее изобретение также относится к композициям, например, фармацевтическим композициям по изобретению, содержащим антитела или их антигенсвязывающую часть и фармацевтически приемлемый носитель. Композиции по изобретению содержат антитела против IL-13 или их антигенсвязывающую часть, таким образом, что когда их вводят внутривенно индивиду при дозе, равной приблизительно 0,3 мг/кг, антитела или их антигенсвязывающая часть способны к проявлению: (a) площади под кривой (AUC) между приблизительно 1500 и приблизительно 2700 мкгч/мл; (b) объема распределения между приблизительно 65 и 125 мл/кг; (c) пиковой концентрации (Cмакс) между приблизительно 5 и приблизительно 8 мкг/мл; и (d) скорости выведения между приблизительно 0,1 и приблизительно 0,2 мл/ч/кг.
В еще одном другом аспекте изобретение относится к выделенной композиции, содержащей антитела против IL-13, или их антигенсвязывающую часть, таким образом, что, при введении внутривенно индивиду при дозе, равной приблизительно 3 мг/кг, антитела или их антигенсвязывающая часть способны к проявлению: (a) площади под кривой (AUC) между приблизительно 21000 и приблизительно 33500 мкгч/мл; (b) объема распределения между приблизительно 55 и приблизительно 100 мл/кг; (c) пиковой концентрации (Cмакс) между приблизительно 55 и приблизительно 90 мкг/мл; и (d) скорости выведения между приблизительно 0,08 и приблизительно 0,15 мл/ч/кг.
В еще одном аспекте изобретение относится к выделенной композиции, содержащей антитела против IL-13, или их антигенсвязывающую часть, таким образом, что, при введении внутривенно индивиду при дозе, равной приблизительно 10 мг/кг, антитела или их антигенсвязывающая часть способны к проявлению: (a) площади под кривой (AUC) между приблизительно 75 и приблизительно 100 мкгч/мл; (b) объема распределения между приблизительно 90 и приблизительно l30 мл/кг; (c) пиковой концентрации (Cмакс) между приблизительно 185 и приблизительно 250 мкг/мл; и (d) скорости выведения между приблизительно 0,1 и приблизительно 0,15 мл/ч/кг.
В еще одном аспекте изобретение относится к выделенной композиции, содержащей антитела против IL-13, или их антигенсвязывающую часть, таким образом, что, при введении подкожно индивиду при дозе, равной приблизительно 0,3 мг/кг, антитела или их антигенсвязывающая часть, способны к проявлению: (a) площади под кривой (AUC) между приблизительно 125 и приблизительно 800 мкгч/мл; и (b) пиковой концентрации (Cмакс) между приблизительно 1,0 и приблизительно 6,0 мкг/мл.
В еще одном аспекте изобретение относится к выделенной композиции, содержащей антитела против IL-13, или их антигенсвязывающую часть, таким образом, что, при введении подкожно индивиду при дозе, равной приблизительно 3 мг/кг, антитела или их антигенсвязывающая часть способны к проявлению: (a) площади под кривой (AUC) между приблизительно 1100 и приблизительно 8500 мкгч/мл; и (b) пиковой концентрации (Cмакс) между приблизительно 12 и приблизительно 60 мкг/мл.
В еще одном аспекте изобретение относится к выделенной композиции, содержащей антитела против IL-13, или их антигенсвязывающую часть, таким образом, что, при введении внутривенно индивиду при дозе, равной приблизительно 0,3 мг/кг, 1 мг/кг, 3 мг/кг или 10 мг/кг, антитела или их антигенсвязывающая часть способны к проявлению какого-либо из фармакокинетического параметров, представленных в описании, Таблицах или на Фигурах.
В еще одном аспекте изобретение относится к выделенной композиции, содержащей антитела против IL-13, или их антигенсвязывающую часть, таким образом, что, при введении подкожно индивиду при дозе, равной приблизительно 0,3 мг/кг, 1 мг/кг или 3 мг/кг, антитела или их антигенсвязывающая часть способны к проявлению какого-либо из фармакокинетического параметров, представленных в описании, Таблицах или на Фигурах.
Фармацевтические композиции, содержащие антитела по изобретению, предназначены для применения в, но ими не ограничиваясь, диагностике, обнаружении или мониторинге расстройства, в профилактике, лечении, излечении, или улучшении состояния расстройства или одного или нескольких его симптомов и/или в исследованиях. В конкретном варианте осуществления композиция содержит одно или несколько антител по изобретению. В еще одном варианте осуществления фармацевтическая композиция содержит одно или несколько антител по изобретению и одно или несколько профилактических или терапевтических средств, отличных от свойств антител по изобретению для лечения расстройства, при котором активность IL-13 является вредоносной, такого как астма. Предпочтительно, профилактические или терапевтические средства являются средствами, известными для применения или применявшимися или применяемыми в настоящее время в профилактике, лечении, излечении или улучшении состояния расстройства или одного или нескольких его симптомов. В соответствии с этими вариантами осуществления, композиция может дополнительно содержать носитель, разбавитель или эксципиент.
Обычно фармацевтическая композиция по изобретению содержит антитела против IL-13 или их антигенсвязывающую часть, и фармацевтически приемлемый носитель. Как используют в настоящем описании, термин "фармацевтически приемлемый носитель" включает какие-либо и все растворители, дисперсионные среды, покрытия, антибактериальные и антигрибковые средства, изотонические и замедляющие всасывание средства и т.п. которые являются физиологически совместимыми. Примеры фармацевтически приемлемых носителей включают один или несколько из воды, физраствора, забуференного фосфатом физраствора, декстрозы, глицерина, этанола и т.п., а также их комбинации. Во многих случаях, будет предпочтительно включать в композицию изотонические средства, например, сахара, полиспирты, такие как маннитол, сорбитол или хлорид натрия. Фармацевтически приемлемые носители могут дополнительно содержать минорные количества вспомогательных веществ, таких как смачивающие или эмульгирующие средства, консерванты или буферы, которые увеличивают срок хранения или эффективность антител или части антител.
Разнообразные системы доставки являются известными и могут применяться для введения одного или нескольких видов антител по изобретению или комбинации одну или несколько видов антител по изобретению и профилактического средства или терапевтического средства, применимых для профилактики, излечения, терапии или облегчения расстройства или одного или нескольких его симптомов, например, инкапсулирование в липосомы, микрочастицы, микрокапсулы, рекомбинантные клетки, способные к экспрессии антитела или фрагмента антитела, опосредованному рецептором эндоцитозу (см., например, Wu и Wu, J. Biol. Chem. 262:4429-4432 (1987)), конструкция нуклеиновой кислоты как части ретровирусного или другого вектора и т.д. Способы введения профилактического или терапевтического средства по изобретению включают, но не ограничены лишь ими, парентеральное введение (например, внутрикожное, внутримышечное, внутрибрюшинное, внутривенное и подкожное), эпидуральное введение, внутриопухолевое введение и мукозальное введение (например, интраназальный и пероральный пути). Дополнительно, может использоваться внутрилегочное введение, например, посредством применения ингалятора или распылителя, и готовая форма с аэрозолизирующим средством. См., например, патенты США №№ 6019968, 5985320, 5985309, 5934272, 5874064, 5855913, 5290540 и 4880078; и публикации PCT № WO 92/19244, WO 97/32572, WO 97/44013, WO 98/31346 и WO 99/66903, каждая из которых включена в настоящее описание в качестве ссылки во всей полноте. В одном из вариантов осуществления антитело по изобретению, комбинированную терапию или композицию по изобретению вводят с использованием технологии доставки внутрилегочного лекарственного средства Alkermes AIR™ (Alkermes, Inc., Cambridge, Mass.). В конкретном варианте осуществления профилактические или терапевтические средства по изобретению вводят внутримышечно, внутривенно, внутриопухолево, перорально, интраназально, внутрилегочно или подкожно. Профилактические или терапевтические средства могут вводиться посредством любого общепринятого пути, например, посредством инфузии или болюсной инъекции, посредством всасывания через эпителиальные или слизисто-кожные выстилки (например, слизистую оболочку ротовой полости, ректальную и кишечную слизистую оболочку и т.д.) и могут вводиться вместе с другими биологически активными средствами. Введение может быть системным или местным.
В конкретном варианте осуществления может быть желательным вводить композиции по изобретению местно в область, нуждающуюся в лечении; это может достигаться посредством, например, и неограничивающим образом, местной инфузии, посредством инъекции или посредством имплантата, причем указанный имплантат может состоять из пористого или непористого материала, включая мембраны и матрицы, такие как сиаластиковые мембраны, полимеры, волокнистые матрицы (например, Tissuel™) или коллагеновые матрицы. В одном из вариантов осуществления эффективное количество одного или нескольких антител антагонистов по изобретению вводят местно в пораженную область индивиду для профилактики, терапии, излечения и/или облегчения расстройства или его симптома. В еще одном варианте осуществления эффективное количество одного или нескольких видов антител по изобретению вводят местно в пораженную область в комбинации с эффективным количеством одной или нескольких терапий (например, одного или нескольких профилактических или терапевтических средств), отличных от антител по изобретению, индивиду для профилактики, терапии, излечения и/или облегчения расстройства или одного или нескольких его симптомов.
В еще одном варианте осуществления профилактическое или терапевтическое средство по изобретению может доставляться в системе с регулируемым высвобождением или системе с замедленным высвобождением. В одном из вариантов осуществления для достижения регулируемого или замедленного высвобождения может применяться насос (см. Langer, выше; Sefton, 1987, CRC Crit. Ref. Biomed. Eng. 14:20; Buchwald et al. 1980, Surgery 88:507; Saudek et al. 1989, N. Engl. J. Med. 321:574). В еще одном варианте осуществления могут применяться полимерные материалы для достижения регулируемого или замедленного высвобождения терапевтических средств по изобретению (см., например, Medical Applications of Controlled Release, Langer and Wise (eds.), CRC Pres., Boca Raton, Fla. (1974); Controlled Drug Bioavailability, Drug Product Design and Performance, Smolen и Ball (eds.), Wiley, New York (1984); Ranger and Peppas, 1983, J., Macromol. Sci. Rev. Macromol. Chem. 23:61; См. также Levy et al. 1985, Science 228:190; During et al. 1989, Ann. Neurol. 25:351; Howard et al. 1989, J. Neurosurg. 7 1:105); Патент США № 5679377; 5916597; 5912015; 5989463; 5128326; Публикация РСТ № WO 99/15154; и Публикация РСТ № WO 99/20253. Примеры полимеров, используемых в готовых лекарственных формах замедленного высвобождения, включают, но не ограничены лишь ими, поли(2-гидроксиэтилметакрилат), поли(метилметакрилат), поли(акриловую кислоту), поли(этилен-co-винилацетат), поли(метакриловую кислоту), полигликолиды (PLG), полиангидриды, поли(N-винилпирролидон), поли(виниловый спирт), полиакриламид, поли(этиленгликоль), полилактиды (PLA), поли(лактид-co-гликолиды) (PLGA) и сложные полиортоэфиры. В предпочтительном варианте осуществления полимер, используемый в лекарственной форме замедленного высвобождения, является инертным, не содержащим вымываемых примесей, стабильным при хранении, стерильным и биодеградируемым. В еще одном другом варианте осуществления система с регулируемым или замедленным высвобождением может быть размещена вблизи от профилактической или терапевтической мишени, таким образом, требуя только фракцию системной дозы (см., например, Goodson, in Medical Applications of Controlled Release, выше, vol. 2, pp. 115-138 (1984)).
Системы с регулируемым высвобождением обсуждаются в обзоре Langer (1990, Science 249: 1527-1533). Любой способ, известный квалифицированному специалисту в данной области, может применяться для получения лекарственных форм с замедленным высвобождением, содержащих одно или несколько терапевтических средств по изобретению. См., например, Патент США № 4526938, Публикация PCT WO 91/05548, Публикация PCT WO 96/20698, Ning et al. 1996, "Intratumoral Radioimmunotheraphy of Human Colon Cancer Xenograft Using a Sustained-Release Gel, "Radiotherapy & Oncology 39: 179-189, Song et al. 1995, "Antibody Mediated Lung Targeting of Long-Circulating Emulsions," PDA Journal of Pharmaceutical Science & Technology 50:372-397, Cleek et al. 1997, "Biodegradable Polymer Carriers for bFGF Antibody for Cardiovascular Application," Pro. Int'l. Symp. Control. Rel. Bioact. Mater. 24:853-854, and Lam et al. 1997, "Microencapsulation of Recombinant Humanized Monoclonal Antibody for Local Delivery," Proc. Int'l. Symp. Control Rel. Bioact. Mater. 24:759-760, каждая из которых включена в настоящее описание в качестве ссылки во всей их полноте.
Фармацевтическую композицию по изобретению составляют, чтобы она была совместимой со своим назначенным путем введения. Примеры путей введения включают, но не ограничены лишь ими, парентеральный, например, внутривенное, внутрикожное, подкожное, пероральное, интраназальное (например, ингаляционное), трансдермальное (например, местное), трансмукозальное и ректальное введение. В конкретном варианте осуществления композицию составляют в соответствии с рутинными методиками, как фармацевтическую композицию, адаптированную для внутривенного, подкожного, внутримышечного, перорального, интраназального или местного введения людям. Обычно, композиции для внутривенного введения представляют собой растворы в стерильном изотоническом водном буфере. Там, где необходимо, композиция может также включать солюбилизатор и местное анестезирующее средство, такое как лидокаин, чтобы облегчить боль на участке инъекции.
Если композиции по изобретению предназначены для местного введения, композиции могут быть составлены в форме мази, крема, трансдермального пластыря, лосьона, геля, шампуня, спрея, аэрозоля, раствора, эмульсии или другой формы, хорошо известных квалифицированному специалисту в данной области. См., например, Remington's Pharmaceutical Sciences and Introduction to Pharmaceutical Dosage Forms, 19th ed., Mack Pub. Co., Easton, Pa. (1995). Для нераспыляемых местных дозированных форм обычно используют от вязких до полутвердых или твердые формы, содержащие носитель или один или несколько эксципиентов, совместимые с местным применением и имеющие динамическую вязкость, предпочтительно, более высокую, чем у воды. Подходящие лекарственные формы включают, без ограничения, растворы, суспензии, эмульсии, кремы, мази, порошки, линименты, бальзамы, и т.п., которые, если желательно, стерилизуют или смешивают со вспомогательными средствами, (например, консервантами, стабилизаторами, смачивающими средствами, буферами или солями) для влияния на разнообразные свойства, такие как, например, осмотическое давление. Другие подходящие местные дозированные формы включают распыляемые аэрозольные препараты, где активный ингредиент, предпочтительно, в комбинации с твердым или жидким инертным носителем, пакуют в смеси с находящимся под давлением летучим веществом (например, газообразным пропеллентом, таким как фреон) или пластиковую бутыль. Увлажнители или смачивающие средства могут также добавляться к фармацевтическим композициям и дозированным формам, если желательно. Примеры таких дополнительных ингредиентов являются хорошо известными в данной области.
Для интраназального введения композиция по изобретению композиция может быть составлена в форме аэрозоля, спрея, водного аэрозоля или в форме капель. В частности, профилактические или терапевтические средства для применения в соответствии с настоящим изобретением могут общепринятым образом поставляться в форме презентации аэрозольного спрея из находящихся под давлением упаковок или распылителя, с применением подходящего пропеллента (например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа). В случае находящегося под давлением аэрозоля дозированная единица может определяться посредством обеспечения клапаном для доставки отмеренных количеств. Капсулы и картриджи (составленные из, например, желатина) для применения в ингаляторе или инсуффляторе могут быть составлены вместе с содержащими порошковую смесь соединениями и подходящей порошковой основой, такой как лактоза или крахмал.
Для перорального введения, композиции по изобретению могут быть составлены перорально в форме таблеток, капсул, облаток, желатиновых капсул, растворов, суспензий, и т.п. Таблетки или капсулы могут быть получены общепринятыми средствами с фармацевтически приемлемыми эксципиентами, такими как связующие средства (например, прежелатинизированный кукурузный крахмал, поливинилпирролидон, или гидроксипропилметилцеллюлоза); наполнителями (например, лактоза, микрокристаллическая целлюлоза или гидрофосфат кальция); смазывающими средствами (например, стеарат магния, тальк или оксид кремния); разрыхлителями (например, картофельный крахмал или крахмалгликолят натрия); или смачивающими средствами (например, лаурилсульфат натрия). Таблетки могут быть покрыты способами, хорошо известными в данной области. Жидкие препараты для перорального введения могут принимать форму, но не ограничиваться лишь ими, растворов, сиропов или суспензий или они могут быть представлены в виде сухого продукта для восстановления с водой или другой подходящей средой перед применением. Такие жидкие препараты могут быть получены общепринятыми средствами с фармацевтически приемлемыми добавками, такими как суспендирующие средства (например, сироп сорбитола, производные целлюлозы или гидрированные пищевые жиры); эмульгаторы (например, лецитин или акация); неводные среды (например, миндальное масло, масляные сложные эфиры, этиловый спирт или фракционированные растительные масла); и консерванты (например, метил- или пропил-п-гидроксибензоаты или сорбиновая кислота). Препараты могут также содержать буферные соли, ароматизатор, краситель и подсластитель, как принято. Препараты для перорального введения могут быть подходящим образом составлены для медленного высвобождения, регулируемого высвобождения или замедленного высвобождения профилактических или терапевтических средств(средства).
Способы по изобретению могут включать внутрилегочное введение, например, посредством применения ингалятора или распылителя композиции, составленной вместе с аэрозолизирующим средством. См., например, Патенты США №№ 6019968, 5985320, 5985309, 5934272, 5874064, 5855913, 5290540, и 4880078; и Публикации PCT №№ WO 92/19244, WO 97/32572, WO 97/44013, WO 98/31346, и WO 99/66903, каждая из которых включена в настоящее описание в качестве ссылки во всей полноте. В конкретном варианте осуществления антитело по изобретению, комбинированную терапию и/или композицию по изобретению вводят с использованием технологии доставки внутрилегочного лекарственного средства Alkermes AIR™ (Alkermes, Inc., Cambridge, Mass.).
Способы по изобретению могут также включать введение композиции, составленной для парентерального введения посредством инъекции (например, посредством болюсной инъекции или непрерывной инфузии). Готовые формы для инъекций могут быть представлены в виде единичной дозированной формы (например, в ампулах или в мультидозовых контейнерах) с добавленным консервантом. Композиции могут принимать такие формы как суспензии, растворы или эмульсии в масляных или водных средах и могут содержать вспомогательные вещества, такие как суспендирующие, стабилизирующие и/или диспергирующие средства. Альтернативно, активный ингредиент может находиться в форме порошка для создания жидкой фазы с подходящей средой (например, стерильной апирогенной водой) перед применением.
Способы по изобретению могут дополнительно включать введение композиций, составленных как депо-препараты. Такие готовые формы пролонгированного действия могут вводиться посредством имплантации (например, подкожно или внутримышечно) или посредством внутримышечной инъекции. Таким образом, например, композиции могут быть составлены с подходящими полимерными или гидрофобными материалами (например, как эмульсия в приемлемом масле) или ионо-обменными смолами, или в виде слаборастворимых производных (например, как слаборастворимая соль).
Способы по изобретению охватывают введение композиций, составленных в виде нейтральных или солевых форм. Фармацевтически приемлемые соли включают соли, образованные с анионами, такими как соли, являющиеся производными хлористоводородной, фосфорной, уксусной, щавелевой, винной кислот и т.д., и соли, образованные с катионами, такими как соли, являющиеся производными натрия, калия, аммония, кальция, гидроксидов железа, изопропиламина, триэтиламина, 2-этиламиноэтанола, гистидина, прокаина и т.д.
Обычно ингредиенты композиций поставляют либо раздельно или смешанными вместе в единичной дозированной форме, например, в виде сухого лиофилизированного порошка или безводного концентрата в герметично закупоренном контейнере, таком как ампула или саше, с указанием количества активного агента. Там, где режимом введения является инфузия, композиция может быть распределена с помощью бутыли для инфузии, содержащей стерильную воду фармацевтической категории или физраствор. Там, где режимом введения является инъекция, может быть предоставлена ампула со стерильной водой для инъекции или физраствором таким образом, чтобы ингредиенты могли быть смешаны перед введением.
Композиции по изобретению могут быть упакованы в герметично закупоренный контейнер, такой как ампула или саше с указанием количества средства. В одном из вариантов осуществления композиции по изобретению могут поставляться в виде сухого стерилизованного лиофилизованного порошка или безводного концентрата в герметично закупоренном контейнере и могут быть восстановлены (например, с водой или физраствором) до соответствующей концентрации для введения индивиду. Предпочтительно, композиции по изобретению поставляют в виде сухого стерильного лиофилизованного порошка в герметично закупоренном контейнере при единичной дозировке, равной по меньшей мере 5 мг, более предпочтительно, по меньшей мере 10 мг, по меньшей мере 15 мг, по меньшей мере 25 мг, по меньшей мере 35 мг, по меньшей мере 45 мг, по меньшей мере 50 мг, по меньшей мере 75 мг или по меньшей мере 100 мг. Лиофилизованные профилактические или терапевтические средства, или фармацевтические композиции по изобретению должны храниться в интервале между 2°C и 8°C в их исходном контейнере, и профилактические или терапевтические средства или фармацевтические композиции по изобретению должны вводиться в пределах 1 недели, предпочтительно, в пределах 5 дней, в пределах 72 часов, в пределах 48 часов, в пределах 24 часов, в пределах 12 часов, в пределах 6 часов, в пределах 5 часов, в пределах 3 часов или в пределах 1 часа после восстановления. В альтернативном варианте осуществления одно или несколько профилактических или терапевтических средств или фармацевтических композиций по изобретению поставляют в жидкой форме в герметично закупоренном контейнере с указанием количества и концентрации агента. Предпочтительно, жидкую форму вводимой композиции поставляют в герметично закупоренном контейнере при по меньшей мере 0,25 мг/мл, более предпочтительно по меньшей мере 0,5 мг/мл, по меньшей мере 1 мг/мл, по меньшей мере 2,5 мг/мл, по меньшей мере 5 мг/мл, по меньшей мере 8 мг/мл, по меньшей мере 10 мг/мл, по меньшей мере 15 мг/кг, по меньшей мере 25 мг/мл, по меньшей мере 50 мг/мл, по меньшей мере 75 мг/мл или по меньшей мере 100 мг/мл. Жидкая форма должна храниться в интервале между 2°C и 8°C в своем исходном контейнере.
Композиции по изобретению являются, предпочтительно, подходящими для парентерального введения. Например, композиции по изобретению могут быть получены в виде раствора для инъекций, содержащего 0,1-250 мг/мл антитела. Раствор для инъекций может быть составлен из либо жидкой или лиофилизованной дозированной формы во флаконе из бесцветного или темно-желтого стекла, ампуле или предварительно заполненном шприце. Буфер может представлять собой L-гистидин (1-50 мМ), оптимально 5-10 мМ, при pH от 5,0 до 7,0 (оптимально pH 6,0). Другие подходящие буферы включают, но ими не ограничены, сукцинат натрия, цитрат натрия, фосфат натрия или фосфат калия. Хлорид натрия может применяться для модификации токсичности раствора при концентрации 0-300 мМ (оптимально 150 мМ для жидкой дозированной формы). Криопротекторы могут включаться для лиофилизованной дозированной формы, в основном, 0-10% сахароза (оптимально 0,5-1,0%). Другие подходящие криопротекторы включают трегалозу и лактозу. Объемообразующие средства могут быть включены для лиофилизованной дозированной формы, в основном, 1-10% маннитола (оптимально 24%). Стабилизаторы могут применяться как в жидких, так и лиофилизованных дозированных формах, в основном 1-50 мМ L-Метионина (оптимально 5-10 мМ). Другие подходящие объемообразующие средства включают глицин, аргинин, они могут быть включены в виде 0-0,05% полисорбата-80 (оптимально 0,005-0,01%). Дополнительные поверхностно-активные средства включают, но не ограничены лишь ими, поверхностно-активные средства полисорбат 20 и BRIJ. Фармацевтическая композиция, содержащая антитела и части антител по изобретению, полученные в виде раствора для инъекций для парентерального введения, может дополнительно содержать средство, применимое в качестве адъюванта, такое как средства, применяемые для увеличения всасывания или дисперсию терапевтического белка (например, антитела). Особенно применимым адъювантом является гиалуронидаза, такая как Hylenex™ (рекомбинантная гиалуронидаза человека). Добавление гиалуронидазы в раствор для инъекций улучшает биодоступность для человека после парентерального введения, особенно подкожного введения. Оно также обеспечивает более высокие объемы на участке инъекции (т.е. более 1 мл) при меньшей боли и дискомфорте и минимальную частоту возникновения реакций на участке инъекции (см. WO2004078140, US2006104968, включенные в настоящее описание в качестве ссылки).
Композиции по настоящему изобретению могут находиться в разнообразных формах. Они включают, например, жидкие, полутвердые и твердые дозированные формы, такие как жидкие растворы (например, растворы для инъекций и инфузий), дисперсии или суспензии, таблетки, пилюли, порошки, липосомы и суппозитории. Предпочтительная форма зависит от намеченного режима введения и терапевтического применения. Обычные предпочтительные композиции находятся в форме растворов для инъекций и инфузий, такие как композиции, аналогичные композициям, используемым для пассивной иммунизации людей другими антителами. Предпочтительным режимом введения является парентеральный (например, внутривенный, подкожный, внутрибрюшинный, внутримышечный). В предпочтительном варианте осуществления антитело вводят посредством внутривенной инфузии или инъекции. В еще одном предпочтительном варианте осуществления антитело вводят посредством внутримышечной или подкожной инъекции.
Терапевтические композиции обычно должны быть стерильными и устойчивыми в условиях производства и хранения. Композиция может быть составлена в виде раствора, микроэмульсии, дисперсии, липосом или другой упорядоченной структуры, подходящей для высокой концентрации лекарственного средства. Стерильные растворы для инъекций могут быть получены посредством введения активного соединения (т.е., антитела или части антитела) в требуемом количестве в соответствующем растворителе с одним или комбинацией из ингредиентов, перечисленных выше, как требуется, с последующей фильтрующей стерилизацией. Как правило, дисперсии получают посредством введения активного соединения в стерильную среду, которая содержит основную дисперсионную среду и требуемые другие ингредиенты из тех, что перечислены выше. В случае стерильных, лиофилизованных порошков для получения стерильных растворов для инъекций, предпочтительными способами получения являются сушка в вакууме и распылительная сушка, которая позволяет получить порошок активного ингредиента плюс любой дополнительный желательный ингредиент из ранее его стерильно фильтруемого раствора. Необходимая текучесть раствора может поддерживаться, например, посредством покрытия, такого как лецитин, посредством поддержания требуемого размера частиц в случае дисперсии и посредством применения поверхностно-активных средств. Пролонгированное всасывание инъецируемых композиций может придаваться посредством включения в композицию средства, которое замедляет всасывание, например, моностеаратных солей и желатина.
Композиции по настоящему изобретению могут вводиться посредством разнообразных способов, известных в данной области, хотя для многих терапевтических применений предпочтительным путем/режимом введения является подкожная инъекция, внутривенная инъекция или инфузия. Как сможет оценить квалифицированный специалист в данной области, путь и/или режим введения будет варьировать в зависимости от желательных результатов. В некоторых вариантах осуществления, активное соединение может быть получено с носителем, который будет защищать соединение от быстрого высвобождения, таких как в случае готовой формы с регулируемым высвобождением, включая имплантаты, трансдермальные пластыри и микроинкапсулированные системы доставки. Могут применяться биодеградируемые биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, сложные полиортоэфиры и полимолочная кислота. Многие способы получения таких готовых форм запатентованы или, как правило, являются известными квалифицированным специалистам в данной области. См., например, Sustained and Controlled Release Drug Delivery Systems, J. R. Robinson, ed., Marcel Dekker, Inc., New York, 1978.
В некоторых вариантах осуществления, композиции по изобретению могут вводиться перорально, например, с инертным разбавителем или усваиваемым пищевым носителем. Соединение (и другие ингредиенты, если желательно) могут также быть заключены в желатиновую капсулу с твердой или мягкой оболочкой, спрессованы в таблетки или включены непосредственно в диету индивида. Для перорального терапевтического введения, соединения могут быть включены вместе с эксципиентами и использованы в форме таблеток для глотания, буккальных таблеток, лепешек, капсул, эликсиров, суспензий, сиропов, капсул-имплантатов и т.п.. Для введения соединений по изобретению другим образом, отличным от парентерального введения может быть необходимым покрывать соединение материалом для предотвращения его инактивации или совместно вводить с ним соединение.
Дополнительные активные соединения могут также вводиться в композиции. В некоторых вариантах осуществления, антитело или часть антитела по изобретению составляют совместно с и/или совместно вводят с одним или несколькими дополнительными терапевтическими средствами, которые являются применимыми для лечения расстройств, при которых активность IL-13 является вредной. Например, антитело против IL-13 или часть антитела по изобретению могут быть совместно составлены и/или совместно вводиться с одним или несколькими дополнительными антителами, которые связываются с другими мишенями (например, антитела, которые связываются с другими цитокинами или которые связываются с молекулами клеточной поверхности). Кроме того, одно или несколько антител по изобретению могут применяться в комбинации с двумя или несколькими из вышеприведенных терапевтических средств. При таких комбинационных терапиях могут преимущественно использоваться более низкие дозировки вводимых терапевтических средств, что, таким образом, позволяет избежать возможные токсичности или осложнения, связанные с разнообразными монотерапиями.
В некоторых вариантах осуществления, антитела против IL-13, или их антигенсвязывающую часть, связывают со средой, продлевающей время полужизни, известной в данной области. Такие среды включают, но не ограничены лишь ими, Fc-домен, полиэтиленгликоль и декстран. Такие среды описаны, например, в Заявке США Сер. № 09/428082 и опубликованной Заявке РСТ № WO 99/25044, которые посредством этого включены в настоящее описание в качестве ссылки для любой цели.
Фармацевтические композиции по изобретению могут включать термины "терапевтически эффективное количество" или "профилактически эффективное количество" антитела или части антитела по изобретению. "Терапевтически эффективное количество" относится к количеству, эффективному, при дозировках и в течение периодов времени, необходимых для достижения желательного терапевтического результата. Терапевтически эффективное количество антитела или части антитела может определяться специалистом в данной области и может варьировать в соответствии с факторами, такими как состояние заболевания, возраст, пол и масса тела индивидуума и способности антитела или части антитела вызывать желательный ответ у индивидуума. Терапевтически эффективное количество является также количеством, при котором терапевтически благоприятные эффекты антитела или части антитела преобладают над какими-либо их токсическими или вредными эффектами. "Профилактически эффективное количество" относится к количеству, эффективному при дозировках и в течение периодов времени, необходимых для достижения желательного профилактического результата. Обычно, поскольку профилактическую дозу применяют у индивидов перед заболеванием или на более ранней стадии заболевания, профилактически эффективное количество будет составлять меньше, чем терапевтически эффективное количество.
Режимы дозирования могут регулироваться для обеспечения оптимального желательного ответа (например, терапевтического или профилактического ответа). Например, может вводиться единичный болюс, могут вводиться несколько разделенных доз в течение времени или доза может быть пропорционально снижена или увеличена, как показывают потребности терапевтической ситуации. Особенно преимущественным является составлять парентеральные композиции в единичной (однократной) дозированной форме для простоты введения и однородности дозировки. Единичная дозированная форма, как используют в настоящем описании, относится к физически дискретным единицам, приспособленным для единичных дозировок для млекопитающих индивидов, подлежащих лечению; причем каждая единица содержит предварительно определенное количество активного соединения, рассчитанное для оказания желательного терапевтического эффекта в связи с требуемым фармацевтическим носителем. Спецификация для единичных дозированных форм по изобретению диктуется и является прямо зависимыми от (a) уникальных характеристик активного соединения и конкретного терапевтического или профилактического эффекта, которого нужно достичь, и (b) ограничений, характерных для области составления такого активного соединения для лечения чувствительности у индивидуумов.
Примерный, неограничивающий интервал терапевтически или профилактически эффективного количества антител или части антител по изобретению составляет 0,1-20 мг/кг, более предпочтительно, 1-10 мг/кг. В одном из вариантов осуществления терапевтически или профилактически эффективное количество антител или их антигенсвязывающей части составляет 0,3 мг/кг. В еще одном варианте осуществления терапевтически или профилактически эффективное количество антител или их антигенсвязывающей части составляет 3 мг/кг. В еще одном варианте осуществления терапевтически или профилактически эффективное количество антитела или их антигенсвязывающей части составляет 10 мг/кг. В еще одном другом варианте осуществления терапевтически или профилактически эффективное количество антител или их антигенсвязывающей части составляет 0,1 мг/кг, 0,2 мг/кг, 0,3 мг/кг, 0,4 мг/кг, 0,5 мг/кг, 0,6 мг/кг, 0,7 мг/кг, 0,8 мг/кг, 0,9 мг/кг, 1 мг/кг, 1,25 мг/кг, 1,5 мг/кг, 1,75 мг/кг, 2 мг/кг, 2,5 мг/кг, 3 мг/кг, 3,5 мг/кг, 4 мг/кг, 4,5 мг/кг, 5 мг/кг, 5,5 мг/кг, 6 мг/кг, 6,5 мг/кг, 7 мг/кг, 7,5 мг/кг, 8 мг/кг, 8,5 мг/кг, 9 мг/кг, 9,5 мг/кг, 10 мг/кг, 11 мг/кг, 12 мг/кг, 13 мг/кг, 14 мг/кг, 15 мг/кг, 16 мг/кг, 17 мг/кг, 18 мг/кг, 19 мг/кг или 20 мг/кг. Следует отметить, что эти значения дозировки могут варьировать вместе с типом и тяжестью состояния, подлежащего улучшению. Дополнительно следует понимать, что для любого конкретного индивида, конкретные режимы дозировки должны регулироваться в течение времени в соответствии с индивидуальной потребностью и профессиональным заключением лица, вводящего или предписывающего введение композиций, и то, что интервалы дозировки, приведенные в настоящем описании выше, являются только примерными и не предназначены для ограничения объема притязаний по или практического использования заявленной композиции.
III. Способы по изобретению
В еще одном другом аспекте, настоящая заявка относится к способу лечения (например, излечения, подавления, улучшения состояния, замедления или предотвращения наступления, или предотвращения рецидива или обострения) или профилактики IL-13-связанного расстройства, такого как астма, у индивида.
Способ включает: введение индивиду средства, связывающего IL-13 (особенно, антагониста), например, антител против IL-13 или их антигенсвязывающей части, как описано в настоящем описании, в количестве, достаточном для лечения или профилактики IL-13-связанного расстройства, такого как астма. Антагонист IL-13, например, антитела против IL-13 или их антигенсвязывающая часть, могут вводиться индивиду, по отдельности или в комбинации с другими терапевтическими модальностями, как описано в настоящем описании.
В одном из вариантов осуществления индивид является млекопитающим, например, человеком, страдающим от одного или нескольких IL-13-связанных расстройств, включающих, например, респираторные расстройства (например, астму (например, аллергическую и неаллергическую астму или легкую астму, или умеренную астму), хроническую обструктивную болезнь легких (ХОБЛ), и другие состояния, включающие воспаление дыхательных путей, эозинофилию, фиброз и избыточную секрецию слизи; атопические расстройства (например, атопический дерматит и аллергический ринит). Соответственно, раскрытие включает применение средства, связывающего IL-13, (такого как антитела против IL-13 или их фрагменты, описанные в настоящем описании) для лечения, описанного в настоящем описании и применения средства, связывающего IL-13 (такого как антитела против IL-13 или их фрагменты, описанные в настоящем описании) для получения лекарственного средства для лечения, описанного в настоящем описании.
Примеры IL-13-связанных расстройств включают, но ими не ограничены, расстройство, выбранное из одного или нескольких из следующих: респираторные расстройства, например, астма (например, аллергическая и неаллергическая астма (например, астма вследствие инфицирования, например, респираторно-синцитиальным вирусом (RSV), например, у детей младшего возраста)), хроническая обструктивная болезнь легких (ХОБЛ), и другие состояния, включающие воспаление дыхательных путей, эозинофилия, фиброз и избыточная секреция слизи, например, кистозный фиброз и фиброз легких.
В других вариантах осуществления, настоящая заявка предоставляет способ лечения (например, снижения, облегчения) или профилактики одного или нескольких симптомов, связанных с респираторным расстройством, например, астмой (например, аллергической и неаллергической астмой); аллергией; хроническая обструктивная болезнь легких (ХОБЛ); состоянием, включающим воспаление дыхательных путей, эозинофилией, фиброзом и избыточной секрецией слизи, например, кистозным фиброзом и фиброзом легких. Например, симптомы астмы включают, но не ограничены лишь ими, хрип, затруднение дыхания, бронхоконстрикцию, гиперреактивность дыхательных путей, сниженную жизненную емкость легких, фиброз, воспаление дыхательных путей и секрецию слизи. Способ включает введение индивиду антагониста IL-13, например, антител против IL-13 или их фрагмента, в количестве, достаточном для лечения (например, снижения, облегчения) или профилактики одного или нескольких симптомов. Антитела против IL-13 могут вводиться терапевтически или профилактически или для двойной цели. Антагонист IL-13, например, антитела против IL-13 или их антигенсвязывающая часть могут вводиться индивиду по отдельности или в комбинации с другими терапевтическими модальностями, как описано в настоящем описании. Предпочтительно, индивид является млекопитающим, например, человеком, страдающим от IL-13-связанного расстройства, такого как астма, как описано в настоящем описании.
В еще одном аспекте настоящая заявка относится к способу обнаружения присутствия IL-13 в образце in vitro (например, биологическом образце, таком как сыворотка, плазма, ткань, биопсийная проба). Этот способ может применяться для диагностики расстройства, например, астмы, например, легкой или умеренной астмы. Способ включает: (i) приведения в контакт образца или контрольного образца с антителами против IL-13 или их фрагментом, как описано в настоящем описании; и (ii) обнаружение образования комплекса между антителами против IL-13 или их фрагментом и образцом или контрольным образцом, где статистически достоверное изменение при образовании комплекса в образце относительно контрольного образца указывает на присутствие IL-13 в образце.
В еще одном аспекте настоящая заявка относится к способу обнаружения присутствия IL-13 in vivo (например, визуализация in vivo у индивида). Этот способ может применяться для диагностики расстройства, например, IL-13-связанного расстройства, например, астмы, например, легкой или умеренной формы астмы. Способ включает: (i) введение антител против IL-13 или их фрагмента, как описано в настоящем описании, индивиду или контрольному индивиду в условиях, которые обеспечивают связывание антител или фрагмента с IL-13; и (ii) обнаружение образования комплекса между антителом или фрагментом и IL-13, где статистически достоверное изменение при образовании комплекса у индивида относительно контрольного индивида указывает на присутствие IL- 13.
Антитела по изобретению или их антигенсвязывающие части могут применяться по отдельности или в комбинации для лечения таких заболеваний. Следует понимать, что антитела по изобретению или их антигенсвязывающая часть могут применяться по отдельности или в комбинации с дополнительным средством, например, терапевтическим средством, причем указанное дополнительное средство является выбранным квалифицированным специалистом в данной области для своей предназначенной цели. Например, дополнительное средство может представлять собой терапевтическое средство, признанное в данной области, как являющееся применимым для лечения заболевания или состояния, подвергающегося лечению антителами по настоящему изобретению. Дополнительное средство также может представлять собой средство, которое придает благоприятную характеристику терапевтической композиции, например, средством, которое воздействует на вязкость композиции.
Дополнительно, следует понимать, что комбинации, которые подлежат включению в настоящее изобретение, являются такими комбинациями, которые применимы для их предназначенной цели. Средства, приведенные ниже, являются иллюстративными для целей и не предусмотрены для ограничений. Комбинации, которые составляют часть настоящего изобретения, могут представлять собой антитела по настоящему изобретению и по меньшей мере одно дополнительное средство, выбранное из перечней, приведенных ниже. Комбинация может также включать более одного дополнительного средства, например, два или три дополнительных средства, если комбинация является такой, что образованная композиция может осуществлять свою предназначенную функцию.
Комбинированная терапия может включать один или несколько из антагонистов IL-13, например, антитела против IL-13 или их фрагменты, составляемые совместно с и/или вводимые совместно с одним или несколькими дополнительными терапевтическими средствами, например, одним или несколькими цитокинами и ингибиторами факторов роста, иммунодепрессантами, противовоспалительными средствами (например, системными противовоспалительными средствами), противофиброзными средствами, метаболическими ингибиторами, ингибиторами ферментов и/или цитотоксическими или цитостатическими средствами, как описано в настоящем описании более подробно.
Примеры предпочтительных дополнительных терапевтических средств, которые могут совместно вводиться и/или быть совместно составлены с одним или несколькими антагонистами IL-13, например, антителами против IL-13 или их фрагментами, включают, но не ограничены лишь ими, один или несколько из: ингалируемых стероидов; бета-агонистов, например, бета-агонистов кратковременного или долговременного действия; антагонистов лейкотриенов или лейкотриеновых рецепторов; комбинированных лекарственных средств, таких как ADVAIR; ингибиторов IgE, например, антител против IgE (например, XOLAIR); ингибиторов фосфодиэстераз (например, ингибиторов PDE4); ксантинов; антихолинергических лекарственных средств; средств, стабилизирующих тучные клетки, таких как кромолин; ингибиторов IL-4; ингибиторов IL-5; ингибиторов эотаксина/CCR3; антагонистов гистамина или его рецепторов, включая H1, H2, H3, и H4, и антагонистов простагландина D или его рецепторов (DP1 и CRTH2). Такие комбинации могут применяться для лечения астмы и других респираторных расстройств. Дополнительные примеры терапевтических средств, которые могут совместно вводиться и/или быть совместно составлены с одним или несколькими антителами против IL-13 или их фрагментами, включают один или несколько из следующих: антагонисты ФНО (например, растворимый фрагмент рецептора ФНО, например, p55 или p75 рецептора человека ФНО или их производные, например, 75 кД TNFR-IgG (75 кД ФНО рецептор-IgG слитый белок, ENBREL)); антагонисты фермента ФНО, например, ингибиторы ФНО-преобразующего фермента (TACE); антагонисты мускаринового рецептора; TGF-бета антагонисты; интерферон-гамма; перфенидон; химиотерапевтические средства, например, метотрексат, лефлуномид, или сиролимус (рапарницин) или их аналог, например, ингибиторы CCI-779; COX2 и cPLA2; NSAID; иммуномодуляторы; ингибиторы p38, ингибиторы TPL-2, MK-2 и NFkB, среди прочих.
Другие предпочтительные комбинации представляют собой цитокин-подавляющие противовоспалительные лекарственные средства (средство) (CSAID); антитела против или антагонисты других человеческих цитокинов или факторов роста, например, IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-15, IL-16, IL-18, IL-21, IL-31, интерфероны, EMAP-II, GM-CSF, FGF, EGF, PDGF и эдотелин-1, а также рецепторов этих цитокинов и факторов роста. Антитела по изобретению или их антигенсвязывающие части могут комбинироваться с антителами против молекул поверхности клеток, таких как CD2, CD3, CD4, CD8, CD25, CD28, CD30, CD40, CD45, CD69, CD80 (B7.1), CD86 (B7.2), CD90, CTLA или их лигандов, включая CD154 (gp39 или CD40L).
Предпочтительные комбинации терапевтических средств могут оказывать воздействия на различные позиции воспалительного каскада; предпочтительные примеры включают антагонисты ФНО, такие как химерные, гуманизированные антитела или антитела против ФНО человека, D2E7, (публикация РСТ № WO 97/29131), CA2 (Ремикейд™), CDP 571 и растворимые p55 или p75 рецепторы ФНО, их производные (p75TNFR1gG (Энбрел™) или p55TNFR1gG (Ленерцепт), и также ингибиторы ФНО-преобразующего фермента (TACE); аналогично, ингибиторы IL-1 (ингибиторы Интерлейкин-1-преобразующего фермента, IL-1RA и т.д.) могут быть эффективными по той же причине. Другие предпочтительные комбинации включают интерлейкин 4. Еще другой предпочтительной комбинацией является другие ключевые факторы астматического ответа, которые могут действовать параллельно, в зависимости от или во взаимодействии с функцией IL-13; особенно предпочтительными являются антагонисты IL-9, включающие антитела против IL-9. Было показано, что IL-13 и IL-9 имеют накладывающиеся, но различимые функции и комбинация антагонистов к ним обоим может быть наиболее эффективной. Еще одной другой предпочтительной комбинацией являются антитела против IL-5. Еще другие предпочтительные комбинации включают антагонисты хемокинов, включающие MCP-1, MCP-4, эотаксины, RANTES, MDC, CCL-12 и CCL-17 (TARC), и хемокиновых рецепторов, включающие CCR2, CCR3, CCR4 и CXCR4. Еще комбинации могут включать антагонисты медиаторов астмы, включающих кислую хитиназу млекопитающих, CRHT2, химазу, S1P1, S1P2, Tyk2, ROCKII, Stat6, p38, NFkB, фосфодиэстеразу 4 (PDE-4), тритазу тучных клеток, NO, аденозин, IKK2, GATA3, ICAM-1, VCAM-1 и ICOS.
Как используют в настоящем описании, термин "расстройство, при котором активность IL-13 является вредной" подразумевает включение заболеваний и других расстройств, при которых присутствие IL-13 у индивида, страдающего от расстройства, было продемонстрировано или подозревается в том, что является либо ответственным за патофизиологию расстройства или фактор который способствует ухудшению расстройства. В одном из вариантов осуществления расстройство при котором активность IL-13 является вредной, представляет собой астму, например, легкую форму астмы или умеренную форму астмы. Соответственно, расстройство, при котором активность IL-13 является вредной, является расстройством, при котором ожидают, что снижение активности IL-13 будет облегчать симптомы и/или развитие расстройства. Свидетельством таких расстройств может быть, например, увеличение концентрации IL-13 в биологической жидкости индивида, страдающего от расстройства (например, увеличение концентрации IL-13 в сыворотке, плазме, синовиальной жидкости, и т.д. индивида), которое может быть обнаружено, например, с использованием антител против IL-13, как описано выше. Неограничивающие примеры расстройств, которые могут подвергаться лечению антителами по изобретению включают астму, например, легкую или умеренную формы астмы, а также такие расстройства, обсуждаемые в разделе ниже, имеющем отношение к фармацевтическим композициям антител по изобретению.
IL-13 вовлечен, в качестве фактора, имеющего ключевую роль, в процесс, приводящий к патологическим ответам, связанным с астмой. Однако другие медиаторы различных иммунологических путей являются также вовлеченными в патогенез астмы, и блокирование этих медиаторов, дополнительно к IL-13, может предложить дополнительный терапевтический благоприятный эффект. Таким образом, связывающие белки по изобретению могут быть включены в белки DVD-Ig, где DVD является способным к связыванию целевых пар, включающих, но не ограниченных лишь ими, IL-13 и провоспалительный цитокин, такой как фактор некроза опухолей-α (TNF-α). TNF-α может усиливать воспалительный ответ при астме и может быть связан с тяжестью заболевания (McDonnell, et al. Progress in Respiratory Research (2001), 31(New Drugs for Asthma, Allergy and COPD), 247-250). Данное обстоятельство позволяет предположить, что блокирование как IL-13, так и TNF-α может иметь благоприятные эффекты, особенно при тяжелом заболевании дыхательных путей. В предпочтительном варианте осуществления DVD-Ig по изобретению связывает мишени IL-13 и TNF-б и применяется для лечения астмы.
Настоящее дополнительно иллюстрируется следующими примерами, которые не должны рассматриваться как ограничивающие каким-либо образом. Содержание всех цитируемых ссылок, включая литературные ссылки, выданные патенты и опубликованные патентные заявки, как они цитируются на протяжении настоящей заявки, выраженным образом включены в настоящее описание в качестве ссылки. Следует дополнительно понимать, что содержание всех чертежей и таблиц, прилагаемых к настоящему документу, а также полное содержание патента США № 7915388 выраженным образом включено в настоящее описание в качестве ссылки.
ПРИМЕРЫ
Введение к примерам
Астма является хроническим воспалительным заболеванием дыхательных путей, характеризуемым хрипом, удушьем, сдавленностью в груди и кашлем. Астма поражает приблизительно 20 миллионов людей в США, и приблизительно 75% пациентов с астмой являются взрослыми людьми. Из числа пациентов с астмой, приблизительно 60% пациентов с астмой имеют легкую форму заболевания, приблизительно 20% имеют умеренную форму заболевания и оставшиеся 20% имеют тяжелую форму заболевания.
Полагают, что интерлейкин-13 (IL-13) играет ключевую роль в патогенезе астмы у человека, поскольку повышенные уровни IL-13 присутствуют в легких пациентов с астмой, и эти уровни коррелируют с тяжестью заболевания (фигура 1). Аналогично, увеличенный IL-13 присутствует как в мокроте, так и в биопсийных образцах легких пациентов с умеренной до тяжелой формой астмы, которые лечат ингалируемыми кортикостероидами (ICS) или системными кортикостероидами, и продолжает быть симптоматическим. Кроме того, генетические полиморфизмы IL-13 человека связывают с астмой и атопией (аллергической гиперчувствительностью). IL-13 связывается с двумя рецепторами, IL-13Rα1 и IL-13Rα2. IL-13 является хорошо подтвержденной мишенью для астмы, поскольку эффективность действия была продемонстрирована с использованием разнообразных средств антагонизма IL-13 на множественных, доклинических моделях астмы.
13C5.5 является гуманизированным рекомбинантным иммуноглобулиновым IgG1 (IgG1, κ) моноклональным антителом (mAb), специфичным для IL-13 человека, дикого типа и вариантного. 13C5.5 распознает уникальный эпитоп на IL-13, который блокирует его связывание с обоими рецепторами α1 и α2 IL-13 (Фигура 2); другие сходные IL-13-связывающие антитела, проанализированные до настоящего времени, только блокируют α1 рецептор, как определено посредством кристаллографии и биохимической характеризации. 13C5.5 является селективным для IL-13, и не распознает другие цитокины. Два остатка (L240A и L241A) в тяжелой цепи были подвергнуты мутации для предотвращения рецептора Fc-гамма и связывания комплемента. Аналогично, 13C5.5 не связывает или стимулирует цельные кровяные клетки человека для высвобождения цитокинов. Вариабельные области тяжелой и легкой цепей приведены выше как SEQ ID NO:2 и SEQ ID NO:3, соответственно.
13C5.5 связывается с IL-13 человека, как дикого типа, так и вариантным, с высокой аффинностью и активностью; это является важным, поскольку вариантный IL-13 присутствует у ~20% людей-астматиков. 13C5.5 не взаимодействует перекрестно с IL-13 мыши, собаки или овцы. 13C5.5 взаимодействует перекрестно с rIL-13 обезьян-макак с 51-кратной более низкой активностью in vitro и с крысиным rIL-13 со 155-кратной более низкой активностью in vitro по сравнению с rIL-13 человека. Несмотря на то, что аффинность и активность 13C5.5 является более низкой против rIL-13 обезьяны или крысы in vitro, 13C5.5 полностью нейтрализуют rIL-13 обезьян-макак in vitro с половинной максимальной ингибиторной концентрацией (IC50), равной 4,1 нМ и крысиный IL-13 с IC50, равной 12,4 нМ.
Первоначальный фармакокинетический анализ после введения однократной дозы 13C5.5 проводили на крысах Спрага-Доули и обезьянах-макаках (Фигура 3). 13C5.5 в целом проявлял низкое выведение, низкий объем распределения и высокую биодоступность. Время полужизни варьировало от приблизительно 12 до 16 дней у крыс и 6-11 дней у обезьян. Фармакокинетика 13C5.5 была в целом сходной при сравнении между самками и самцами обезьян и крыс.
13C5.5 хорошо переносились при токсикологических исследованиях у 4-недельных крыс и 2-недельных обезьян и имели достаточные профили безопасности относительно рекомендованной начальной дозы для клинических испытаний на людях. Уровни отсутствия наблюдаемых нежелательных эффектов (NOAEL) во время токсикологических исследований с повтором дозы составляли 1500 мг/кг/доза при внутривенном (ВВ) введении у крыс и обезьян-макак и 200 мг/кг/доза при подкожном (ПК) введении у обезьян-макак.
Данное исследование представляло собой Стадию 1 клинических испытаний (Фазу 1) впервые на человеке на взрослых индивидах с легкой до умеренной контролируемой формой астмы или без нее. В этом плацебо-контролируемом исследовании оценивали 4 однократные возрастающие дозы (0,3, 1,0, 3,0 и 10,0 мг/кг) 13C5.5, вводимых ВВ, и 2 дозы (0,3 и 3,0 мг/кг) 13C5.5, вводимые как 3 еженедельные ПК дозы.
Исследование 13C5.5, проведенное на здоровых индивидах и индивидах с контролируемой астмой от легкой до умеренной формы обеспечило сбор стандартных данных, касающихся фармакокинетики и биодоступности 13C5.5 у человека, а также переносимости, безопасности и иммуногенности при нарастающих однократных и множественных дозах 13C5.5 при целевом назначении для лечения заболевания.
Протокол эксперимента
Данное исследование представляло собой первую стадию клинических испытаний (Фазу 1), с возрастающей однократной и множественной дозой, плацебо-контролируемое, двойное слепое, рандомизированное, из 3-х частей исследование, которое было проведено в соответствии с последовательным дизайном. Взрослые люди с хорошим общим состоянием здоровья (n=20) и взрослые люди с контролируемой астмой от легкой до умеренной формы (n=27) были отобраны для участия в исследовании в соответствии с критериями отбора. Не более одного индивида подвергали дозированию в когорте в день.
Часть 1 исследования состояла из четырех групп (Группы 1-4), с 5 индивидами в каждой группе. Для Части 1, после соответствия критериям отбора, взрослые люди с общим хорошим состоянием здоровья (n=20) были отнесены к одной из следующих четырех групп однократной дозы:
Группа 1 (внутривенная инфузия 0,3 мг/кг 13C5.5 или плацебо),
Группа 2 (внутривенная инфузия 1,0 мг/кг 13C5.5 или плацебо),
Группа 3 (внутривенная инфузия 3,0 мг/кг 13C5.5 или плацебо), или
Группа 4 (внутривенная инфузия 10,0 мг/кг 13C5.5 или плацебо).
Внутри каждой группы, четыре индивида были рандомизированы для приема 13C5.5 и один индивид был рандомизирован для приема плацебо. Нарастание дозы происходило для новой когорты, после того, как все индивиды в дозовой когорте удовлетворительно проходили через по меньшей мере минимальные однонедельные оценки безопасности.
Часть 2 исследования состояла из трех групп (Группы 5-7), с 5 индивидами, рандомизированными в каждую из Групп 5, 6 и 7. Для Части 2, после установления соответствия критериям отбора, взрослые люди с контролируемой астмой от легкой до умеренной (n=15) были отнесены к одной из трех групп однократной дозы:
Группа 5 (внутривенная инфузия 0,3 мг/кг 13C5.5 или плацебо),
Группа 6 (внутривенная инфузия 3,0 мг/кг 13C5.5 или плацебо), или
Группа 7 (внутривенная инфузия 10,0 мг/кг 13C5.5 или плацебо).
Внутри Групп 5, 6 и 7, четыре индивида были рандомизированы для приема 13C5.5 и один индивид был рандомизирован для приема плацебо.
Как показано на Фигуре 4, дозирование для когорты в Части 2 разрешалось начинать после того, как все индивиды одинакового дозового уровня в Части 1 и все более низких дозовых уровней в Части 2 удовлетворительно завершали прохождение по меньшей мере минимальных однонедельных оценок безопасности после последней дозы.
Часть 3 исследования состояла из двух групп (Группы 8 и 9), с 6 индивидами, рандомизированными в каждую из Групп 8 и 9. Для Части 3, после установления соответствия критериям отбора, взрослые индивиды с легкой до умеренной контролируемой астмой (n=12) были отнесены к одной из двух групп:
Группа 8 (подкожная инъекция 0,3 мг/кг 13C5.5 или плацебо, три еженедельных дозы) или
Группа 9 (подкожная инъекция 3,0 мг/кг 13C5.5 или плацебо, три еженедельных дозы).
Внутри Групп 8 и 9, четыре индивида были рандомизированы для приема 13C5.5 и два индивида были рандомизированы для приема плацебо.
Дозирование для когорт в Части 3 разрешалось начинать после того, как все индивиды одинакового дозового уровня в Части 2, и одного дозового уровня более высокого чем в Части 1 и всех более низких дозовых уровней в Части 3 удовлетворительно завершали прохождение по меньшей мере минимальных однонедельных оценок безопасности после последней дозы. (Фигура 4).
Исследование проводили двойным слепым образом, так, что исследователи и индивиды были не осведомлены об отнесениях для обработки в пределах каждой группы. Для оценки безопасности медицинский монитор был осведомлен об отнесениях для обработки. Диаграмма групп обработки показана в Таблице 1.
Здоровые индивиды
Легкая/Умеренная контролируемая астма
Легкая/Умеренная контролируемая астма
# Группу 5 подвергали дозированию после окончания испытаний для Группы 1 и оценки надлежащей безопасности для этой когорты дозирования (минимальные однонедельные оценки безопасности). Аналогичный подход применяли для Группы 6 после окончания испытаний для Групп 3 и 5 и Группы 7 после окончания испытаний для Групп 4 и 6.
$ Группу 8 подвергали дозированию после окончания испытаний для Групп 2 и 5 и оценки надлежащей безопасности для этой когорты дозирования (минимальные однонедельные оценки безопасности). Аналогичный подход применяли для Группы 9 после окончания испытаний для Групп 4, 6 и 8.
Для Частей 1 и 2, исследуемое лекарственное средство или плацебо вводили в день исследования 1 для каждой дозовой группы. Для Части 3, исследуемое лекарственное средство или плацебо вводили в дни исследования 1, 8, и 15 для каждой дозовой группы. Введение исследуемого лекарственного средства в дни исследования 8 и 15 могло производиться в период 24-часового периода запланированной дозы в когортах 8 и 9. Минимально одна неделя разделяла различные уровни дозы.
Индивиды были ограничены местом проведения клинического исследования и наблюдались в течение приблизительно от 4 до 6 дней, если они находились в группе ВВ дозы (Части 1 и 2) или приблизительно от 6 до 8 дней, если они находились в группе ПК дозы (Часть 3). Ограничение начинали в полдень Исходного дня, который мог иметь место в любое время в пределах 3-дневного окна (т.е., День Исследования -3 - День Исследования -1) перед дозированием в День Исследования 1, и заканчивали после сбора 48-часовых образцов крови (День Исследования 3) для индивидов, внесенных в группы ВВ дозирования, или после сбора 96-часового образца крови (День Исследования 5) для индивидов, внесенных в группы ПК дозирования. Физическая нагрузка во время ограничения не разрешалась.
Дополнительно, индивиды в Части 3 были ограничены местом проведения клинического исследования в дни исследования 8 и 15 в течение 24 часов. Во время ограничения индивиды получали ПК дозу либо 13C5.5 или плацебо и оценивались их основные показатели состояния организма, проводились оценки безопасности, лабораторные оценки, фармакокинетика и ADA, отбор проб на биомаркеры, электрокардиограммы (ЭКГ) и тесты на легочную функцию (PFT).
Индивиды получали стандартизованную диету, с обеспечением приблизительно 30% суточных калорий из жира, не более 45% суточных калорий из углеводов и с обеспечением приблизительно 1900 калорий/день для всех видов пищи во время ограничения. Композиция (белок, жир, углевод и общие калории) всех видов пищи определялись диетологом, и запись о регистрации сохраняли вместе с первичной документацией. Во время ограничения на каждой Части исследования индивиды потребляли только запланированные виды пищи, обеспеченные при исследовании, и воду для утоления жажды. Индивиды воздерживались от всех других видов пищи и напитков.
Для Частей 1 и 2, никакой пищи или питья, за исключением воды для утоления жажды, не разрешалось в День Исследования -1, от 8 часов перед дозированием до приблизительно 2 часов после завершения дозирования.
Для Части 3, никакой пищи или питья, за исключением воды для утоления жажды, не разрешалось в дни исследования 1, 8 и 15 от 8 часов перед дозированием до приблизительно 2 часов после завершения дозирования в День Исследования 1.
Критерии включения
Индивид считался подходящим для участия в исследовании, если он/она соответствовал следующим критериям:
1. Мужчины или женщины, и возраст составлял между 18 и 55 годами, включительно.
2. Для Частей 2 и 3, диагноз астма в течение по меньшей мере 6 месяцев перед Скринингом:
индивиды с легкой до умеренной контролируемой астмой, как определено ниже;
применение прописанных бета-агонистов кратковременного действия при не более чем 4 впрыска/неделю для симптоматического контроля астмы;
бета-агонисты долговременного действия (LABA) могут приниматься по назначению, если применяются одновременно с ингалируемыми кортикостероидами (ICS). LABA не вводили в течение 12 часов перед осуществлением скрининга PFT и их прием мог быть возобновлен при завершении тестирования;
в случае ICS могли применяться вплоть до средней дозы (определенной как эквивалент флутиказона MDS/HFA вплоть до 440 мкг ежедневно или флутиказона DPT вплоть до 500 мкг ежедневно).
3. Для Частей 2 и 3, принудительный объем выдоха 1 (FEV1) >70% при Скрининге и Основных визитах.
4. Для частей 2 и 3, в случае приема ICS, стабильная доза ICS в течение >4 недель перед Днем Исследования 1, и ожидалось, что доза остается стабильной на протяжении всего исследования. Должны были применять ICS в течение по меньшей мере 4 недель перед скрининговым визитом.
5. Для частей 2 и 3, индивиды с астмой имели положительный результат теста со стимуляцией метахолина, доступный из истории болезни (в пределах прошедших 12 месяцев) или демонстрировали обратимость дыхательных путей при измерениях легочной функции посредством демонстрации по меньшей мере 12% увеличения FEV1 от наилучшей попытки при тестировании по меньшей мере через 30 минут после двух-четырех ингаляций ингалируемого альбутерола или распыляемого альбутерола (или эквивалентного бета-агониста кратковременного действия). До 2 попыток могло быть сделано при любой одной сессии для демонстрации обратимости. Индивиды могли возвращаться на другой день для повтора тестирования, если необходимо. Обратимость или тест на стимуляцию метахолином, подтверждающий обратимость могли быть задокументированы посредством медицинской регистрации, если их проводили в пределах прошедших 12 месяцев. Положительная стимуляция метахолином определялась как PC20 при дозе метахолина, равной <8,0 мг/мл с использованием стандартов Американского Торакального Общества для тестирования легочной функции (см. Инструкции по Methacholine и Exercise Challenge Testing 1999. Am J. Respir. Crit. Care Med., Vol. 161, pp. 309-329, 2000).
6. Для Частей 2 и 3, индивиды имели хорошо контролируемую астму, в течение по меньшей мере 4 недель перед Днем Исследования 1, как определялось по:
симптомам астмы <2 дней/неделю (т.е., кашель, хрип, затруднение дыхания);
пробуждения в ночное время <2 раз во время предшествующего 4-недельного периода;
никаких воздействий на нормальную активность;
не более, чем 4 ингаляции/в неделю бета-агонистов кратковременного действия для контроля симптомов астмы (исключая профилактическое применение для предотвращения бронхспазма, индуцированного физической нагрузкой).
7. Для женщин индивид соответствовал одному из следующих критериев:
постменопауза в течение по меньшей мере двух лет,
хирургически стерилизована (двусторонняя овариэктомия, двусторонняя перевязка маточных труб или гистерэктомия). От женщин, перенесших перевязку маточных труб, требовалось согласие на применение второй формы контрацепции, начиная с первого дня ограничения до 160 дней после обработки исследуемым лекарственным средством, которая включала:
внутриматочные (ВМС) спирали;
барьерные способы (диафрагма со спермицидом или презерватив со спермицидом);
инъецированные, пероральные, трансдермальные, вагинальные или имплантационные способы гормональных контрацептивов.
8. Женщины имели отрицательные результаты тестов на беременность, осуществляемые:
при Скрининге на образце сыворотки, полученном в интервале 28 дней перед Днем Исследования 1 и в день Основного Визита, которые могли быть проведены в любое время в пределах 3-дневного окна (т.е., День Исследования -3 - День Исследования -1) перед дозированием в День Исследования 1.
9. Для мужчин, индивид был хирургически стерилизован или практикующим по меньшей мере 1 из следующих способов контроля за рождаемостью во время исследования и в течение 160 дней после последнего введения исследуемого лекарственного средства:
индивид применял презерватив и партнер(ы) применяли ВМС;
индивид применял презерватив и партнер(ы) применяли пероральные, имплантационные или имплантационные способы гормональных контрацептивов;
индивид применял презерватив и партнер(ы) применяли барьерный способ (контрацептивная губка, диафрагма или контрацептивный влагалищный колпачок со спермицидными желе или кремами);
дополнительно, индивиды-мужчины давали согласие не быть донорами спермы во время исследования и в течение 160 дней после последней дозы исследуемого лекарственного средства.
10. Для Части 1, Индекс Массы Тела (BMI) составлял от 18 до 29, включительно. Для Части 2 и 3, BMI составлял от 18 до 34, включительно. BMI рассчитывали как массу телу в кг, разделенную на площадь высоты в метрах. Масса тела не превышала 120 кг.
11. Хорошее общее состояние здоровья (отличное от легкой-умеренной контролируемой астмы и связанных медицинских состояний, такие как легкий-умеренный аллергический ринит, атопический дерматит, и заболевание гастроэзофагеального рефлюкса), на основании результатов истории болезни, физической нагрузки, основных показателей состояния организма, лабораторного профиля и электрокардиограммы с 12 отведениями (ЭКГ).
12. Подписанное добровольное участие и информированное согласие с проставленной датой, одобренное IRB, перед проведением любого скрининга или специфичных для исследований процедур.
Критерии исключения
Индивид не допускался для участия в исследовании, если он/она соответствовал любому из следующих критериев:
1. Индивиды применяли LABA терапию без одновременного применения ICS.
2. Для Частей 2 и 3, обострение астмы в пределах 8 недель от Дня Исследования 1.
3. Для Частей 2 и 3 инфекция верхних дыхательных путей в интервале 4 недель от Дня Исследования 1.
4. Для Частей 2 и 3 обострение астмы, требующее системных кортикостероидов или любой другой причины для требования системных кортикостероидов в интервале 6 месяцев от Дня Исследования 1.
5. Для Частей 2 и 3, обострение астмы, требующее посещения отделения неотложной медицинской помощи (ER), госпитализации, или медицинского вмешательства в интервале 6 месяцев от Дня Исследования 1.
6. История клинически достоверной аллергической реакции, т.е. анафилаксии (по мнению исследователя) на любое лекарственное средство, биологический агент, пищу или вакцину.
7. История основной иммунологической реакции на любое средство, содержащее IgG.
8. История атопического дерматита, включающего >10% площади поверхности тела, или требующего медицинского лечения, отличного от применения местных кортикостероидов с низкой-средней активностью или безрецептурных эмолентов в пределах 6 месяцев от Дня Исследования 1.
9. История диабета (Типа I или Типа II) или снижения сывороточного уровня глюкозы, предполагающего диабет (снижение сывороточной глюкозы >126 мг/дл) при Скрининге.
10. История аллергической реакции или существенной чувствительности к компонентам исследуемого лекарственного средства.
11. История туберкулеза (ТБ) или листериоза.
12. История трудноизлечимых хронических или активных инфекций, которые требовали госпитализации или лечения ВВ антибиотиками, ВВ противовирусными средствами или ВВ антигрибковыми средствами в пределах 30 дней Скрининга или пероральными антибиотиками/противовирусными средствами в интервале 14 дней перед Днем Исследования 1.
13. Индивидов оценивали на латентные инфекции ТБ. Индивиды демонстрировали отсутствие инфекции ТБ или ее воздействия, что подтверждалось по отрицательному результату рентгеновского исследования грудной клетки и отрицательному результату кожного теста на очищенное производное белка (PPD).
14. Положительный результат теста на поверхностный антиген гепатита B (HBsAg), антитела против вируса гепатита С (HCV Ab), или антитела против ВИЧ (HIV Ab).
15. Положительный результат теста серологии на шистосомоз.
16. История необъяснимой диареи или болей в брюшной полости с длительностью более 2 недель.
17. История не подвергавшихся лечению паразитарных инфекций.
18. История генетического или приобретенного иммунодефицита.
19. Для Частей 2 и 3, прием любых заменимых лекарств, витаминов и/или растительных добавок в интервале 2-недельного периода перед введением исследуемого лекарственного средства. Для Части 1, принимались любые назначаемые, безрецептурные лекарства или растительные добавки в интервале 2 недель перед введением исследуемого лекарственного средства.
20. Для Частей 2 и 3, индивид принимал Xolair® в интервале 5 месяцев от Дня Исследования 1.
21. Для Частей 2 и 3, индивид имел изменения по иммунотерапевтической дозе в течение 3 месяцев перед Основным визитом.
22. Принимал любое лекарственное средство посредством инъекции в интервале 30 дней или 5 периодов полужизни (независимо от продолжительности) перед введением исследуемого лекарственного средства.
23. Принимал любой исследовательский продукт в интервале 30 дней или 5 периодов полужизни (независимо от продолжительности) перед введением исследуемого лекарственного средства.
24. История злокачественного образования или лимфопролиферативного заболевания, отличного от успешно излеченного неметастазирующего рака кожи сквамозных клеток или базальной клеточной карциномы.
25. История эпилепсии, любого клинически достоверного сердечного, респираторного (за исключением легкой - умеренной астмы), почечного, печеночного, желудочно-кишечного, гематологического, ревматологического или психиатрического заболевания или расстройства, незаживающих ран или рецидивирующего плохого заживления ран или любой неконтролируемой медицинской болезни. Дополнительно, исключали следующее:
Хроническая обструктивная болезнь легких (ХОБЛ), застойная сердечная недостаточность, тромбоэмболия легочных артерий, инфильтрация легких с эозинофилией, имеющийся кашель, вторичный от приема лекарств, дисфункции голосовых связок.
26. История пневмокардиальной Внезапной Смерти у любого родственника первой степени.
27. Беременность или грудное вскармливание у женщины.
28. Недавняя (6-месячная) история нарко- или алкогольной зависимости.
29. Прием любой живой вакцины в интервале 3 месяцев перед введением исследуемого лекарственного средства.
30. Положительный результат скрининга на наркотическую зависимость, алкоголь или котинин при Скрининге или при госпитализации.
31. Рентгеновское исследование при Скрининге, указывающее на какую-либо клинически достоверную аномалию (включая кальцифицирующую гранулему и/или плевральное сморщивание) по оценке соответствующего медицинского персонала.
32. Лихорадочное заболевание в пределах 14 дней перед дозированием.
33. Индивиды с исходным интервалом QTc с коррекцией по формуле Фридерика (QTcF) > 450 мсек для женщин и >430 мсек для мужчин.
34. Донорский или потерянный значительный объем крови (включая плазмаферез) или полученная трансфузия любого продукта крови в пределах 8 недель перед введением исследуемого лекарственного средства.
35. Индивид был курильщиком, или имел историю курения в интервале 6-месячного периода, предшествующего введению исследуемого лекарственного средства.
36. Участие в рассматриваемое время в еще одном клиническом исследовании.
37. Предыдущее участие в данном исследовании.
38. С учетом мнения исследователя или медицинского монитора, по любой причине являлся неподходящим кандидатом для приема 13C5.5.
Проводимые лечения
Для Частей 1 и 2, однократную дозу 13C5.5 или плацебо для 13C5.5 (0,3, 1,0, 3,0 или 10,0 мг/кг) вводили внутривенно каждому индивиду утром в День Исследования 1. Для Части 3, всего 3 дозы 13C5.5 или 13C5.5-плацебо (0,3 или 3,0 мг/кг) вводили подкожно утром в дни исследования 1, 8 и 15. Для ВВ инфузии, постоянный катетер вводили в вену перед дозированием и быстро пропускали 1 мл 13C5.5-плацебо. 13C5.5 или 13C5.5-плацебо вводили внутривенно посредством непрерывной инфузии в течение приблизительно 120 минут индивидам в положении лежа на спине. После инфузии, вводили 1 мл промывки 13C5.5-плацебо, и линию поддерживали в течение минимально 2 часов после завершения инфузии 0,9% изотоническим физраствором. Индивиды оставались в положении лежа на спине в течение по меньшей мере 5 минут перед инфузией и до 30 минут после окончания инфузии.
Для ПК дозы исследуемое лекарственное средство вводили подкожно в левый верхний квадрант брюшной полости, избегая любых кровеносных сосудов, утолщения или болезненности кожи, рубцов, фиброзной ткани, растяжек, синяков, красноты, родинок, или других неоднородностей кожи. Индивид оставался в положении лежа на спине в течение по меньшей мере 30 минут после каждой инъекции.
Индивиды были отнесены в одну из девяти дозовых групп (Таблица 2). Внутри каждой группы ВВ инфузии, четырех индивидов рандомизировали для приема 13C5.5 и один индивид получал плацебо. Внутри каждой ПК группы четыре индивида получали 13C5.5 и два получали плацебо. Наращивание дозы происходило для новой когорты только после того, как все индивиды в дозовой когорте успешно завершали прохождение минимальной однонедельной оценки безопасности.
Для ВВ дозирования, исследуемый медикамент в PFS дополнительно разбавляли 13C5.5-плацебо и смешивали в шприце для инъекции для введения. Для ПК дозирования, никакого разбавления не требовалось, однако материал переносили в инъекционные шприцы для введения. Исследуемое лекарственное средство хранили при от 2° до 8°C/от 36° до 46°F, защищали от света, и не замораживали.
Способ включения индивидов в группы лечебной обработки
По мере включения в исследование, здоровым индивидам в Части 1 исследования были присвоены уникальные последовательные номера, начинающиеся с 1101, 1201, 1301 и 1401 для Групп 1, 2, 3 и 4, соответственно. Индивидам с легкой-умеренной контролируемой астмой в Части 2 были присвоены уникальные последовательные номера, начинающиеся с 2501, 2601 и 2701 для Групп 5, 6 и 7, соответственно. Индивидам с легкой-умеренной контролируемой астмой в Части 3 были присвоены уникальные последовательные номера, начинающиеся с 3801 и 3901 для Групп 8 и 9, соответственно. Индивиды были статистически отнесены для приема 13C5.5 или плацебо. Схема рандомизации была компьютер-генерированной перед началом исследования.
Подбор доз в исследовании
Максимальную рекомендованную начальную дозу (MRSD) для испытания впервые на людях (FIH) рассчитывали в соответствии с Руководством для промышленности Администрации США по пищевым продуктам и лекарственным веществам (FDA) "Estimating safe starting dose in clinical trials for therapeutics in adult healthy volunteers." По руководству, MRSD для белков с молекулярной массой (ММ) >100000 Дальтон, которые вводят ВВ, должна оцениваться посредством нормализации через виды в мг/кг, в большей степени, чем с использованием масштабирования площади поверхности тела. Дополнительно, 13C5.5 по данным исследований in vitro и in vivo для людей по сравнению с rIL-13 обезьян-макак или крысиными rIL-13 демонстрируют ~8-155-кратный сдвиг активности, соответственно, который включали в окончательные оценки MRSD.
На основании уровня отсутствия события неблагоприятного эффекта (NOAEL) по результатам 2-недельного исследования с повторной дозой на обезьянах-макаках, фактора безопасности 10, и 8-кратного сдвига активности между rIL-13 обезьяны или крысы по данным фармакологии in vivo, MRSD была определена равной 19 мг/кг. Аналогично, на основании NOAEL по результатам 4-недельного исследования с повторной дозой на крысах, фактора безопасности 10 и 52-кратного сдвига активности между rIL-13 крысы и человека по данным фармакологии in vivo, MRSD была определена, равной 3 мг/кг. Кроме того, когда применяли 155-кратный сдвиг активности с использованием данных фармакологии in vitro, MRSD человека была определена равной 1 мг/кг.
Интервал доз оценивали при данных испытаниях для установления линейности дозы, причем низкую дозу подбирали для обеспечения соответствующего профиля безопасности, в то же время минимизируя правдоподобие иммуногенности. Высокую дозу выбирали для оценки профиля безопасности высокой дозы, как у здоровых людей, так и у популяции с контролируемой астмой перед вхождением в Фазу 2, чтобы обеспечить соответствующую схему ранжирования дозы при экспериментальной проверке концепции исследования. Безопасность и переносимость устанавливали после однократного наращивания дозы перед прохождением множественного дозирования. Подкожное множественное дозирование было важным для оценки биодоступности, потенциальных взаимодействий воздействие-ответ у астматической популяции и иммуногенности.
Предшествующая и сопутствующая терапия
Для Части 1, сопутствующие медикации не были разрешены на протяжении всего исследования.
Для Частей 2 и 3 применение бета-агониста кратковременного действия в качестве резервного лечения разрешалось во время исследования. Применение от низкой до средней дозы ICS и применение назального кортикостероида разрешалось во время исследования. Применение бета-агониста долговременного действия было позволено для пациентов, по назначению, если LABA применяли одновременно с ICS при Скрининге. Не должно было существовать плана замены медикаментов для лечения астмы в течение продолжительности исследования.
Разрешалось применение системного кортикостероида по необходимости для контроля обострения астмы, но индивиды прерывали любое дополнительное лечение 13C5.5. Индивиды, которые требовали применение системных кортикостероидов оставались в исследовании и следовали на оценку безопасности.
Только незаменимые медикаменты, необходимые для лечения существующих медицинских состояний были разрешены во время исследования. Они включали липид-понижающие средства, антигипертензивные средства, антациды, антагонисты рецептора гистамина-2, ингибиторы протонового насоса и болеутоляющие средства, такие как аспирин или нестероидные противовоспалительные средства (NSAID). Применение антигистаминов, безрецептурный прием эмолиентов и/или применение местного (Класс IV, V, VI или VII) кортикостероида с активностью от низкой до средней разрешалось индивидам с атопическим дерматитом, соответствующим критериям ввода. Применение назального кортикостероида разрешалось индивидам с аллергическим ринитом. Индивиды находились на стабильной дозе разрешенных медикаментов в течение минимально 8 недель перед Днем Исследования 1 и оставались на стабильной дозе на протяжении исследования. Применение ацетаминофена в количестве 2 г или менее в день разрешалось на чередующейся основе с прерыванием исследователем.
Применение заменимых медикаментов не приветствовалось, но если индивид сообщал о приеме каких-либо безрецептурных или назначенных медикаментов, витаминов и/или растительных добавок, или, если введение какого-либо из них становилось необходимым от 30 дней перед введением исследуемого лекарственного средства до конца исследования, наименование медикамента, информация о дозировке, включая дозу и частоту, дате(датам) введения, включая даты начала и конца и причину применения регистрировали, и медицинский монитор был информирован.
13C5.5 и Анализ на ADA
Образцы крови для анализа на 13C5.5 и антитела против лекарственного средства (ADA) получали на протяжении исследования, как указано в Таблице 3. Время, в которое собирали каждый образец крови, регистрировали до ближайшей минуты в исходном документе и в соответствующей форме электронного отчета (eCRF). Временной график сбора крови имел приоритет над всеми другими запланированными активностями исследования, за исключением для дозирования.
Дополнительные образцы крови для измерения содержания лекарственного средства могли быть собраны от индивидов, если они прерывали исследование вследствие неблагоприятных событий; точное время и данные взятого образца подлежали регистрации.
Образцы должны были собираться в интервале + 5 минут от запланированных временных отметок в день исследования 1, в интервале + 1 час от запланированных временных отметок в дни исследования 2-6, и в интервале + 3 часов от запланированных временных отметок в дни исследования 7-127.
Для данного исследования сообщают о результатах MSD-анализа на 13C5.5 и скринингового анализа на ADA.
Образцы крови для анализа на 13C5.5 собирали посредством венепункции в соответствующим образом помеченные вакуумированные пробирки для сбора сыворотки без гель-сепаратора. Для ВВ введения исследуемого лекарственного средства (Части 1 и 2), образцы для анализа на 13C5.5 собирали при 0 час (до приема дозы) и при 2, 3, 4, 6, 10, 14, 24, 48, 72, 96, 120, 168, 336, 504, 672, 1008, 1344, 2016 и 2688 часах после начала введения исследуемого лекарственного средства в день исследования 1. Для ПК введения исследуемого лекарственного средства (Часть 3), образцы для анализа на 13C5.5 собирали при 0 час (до приема дозы) и при 8, 24, 48, 72, 96, 120, 168, 336, 360, 384, 408, 432, 456, 504, 672, 1008, 1344, 2016, 2352 и 3024 часах после введения исследуемого лекарственного средства в день исследования 1. Образцы для часов 168 и 336 собирали непосредственно перед дозированием в дни исследования 8 и 15, соответственно. Образец крови для анализа на 13C5.5 получали при визите досрочного завершения исследования, если это было применимым. Объем сыворотки, необходимый для каждого образца, приведен в Таблице 3. Крови давали возможность свернуться в течение 30 минут при комнатной температуре перед центрифугированием.
Образцы крови для анализа на ADA собирали посредством венепункции в соответствующим образом помеченные вакуумированные пробирки для сбора сыворотки без гель-сепаратора. Для ВВ введения исследуемого лекарственного средства, образцы крови для анализа на ADA собирали в День исследования 1 при 0 час (до приема дозы) и в дни исследования 15, 29, 57, 85 и 113. Для ПК введения исследуемого лекарственного средства, образцы крови для анализа на ADA собирали в дни исследования 1 и 15 при 0 часов (до приема дозы) и в дни исследования 29, 43, 57, 85, 99 и 127. Образец крови для анализа на ADA также получали при визите досрочного завершения исследования, если это было применимым. Объем сыворотки, необходимый для каждого образца, приведен в Таблице 3. Крови давали свернуться в течение 30 минут при комнатной температуре перед центрифугированием.
Фармакокинетические переменные
Значения фармакокинетических параметров 13C5.5 после ВВ дозирования оценивали с использованием некомпартментных способов.
Максимальная наблюдаемая сывороточная концентрация (Cмакс) и время до Cмакс (пиковое время, Tмакс) определялась непосредственно по данным сывороточная концентрация-время.
Значение константы кажущейся скорости элиминирования терминальной фазы (β, Бета) получали по углу наклона кривой профиля зависимости сывороточной концентрации от времени в терминальной log-линейной фазе по способу наименьших квадратов линейной регрессии логарифмов. Терминальную log-линейную фазу идентифицировали с использованием Phoenix™ WinNonlin®Version 6.1 (Pharsight Corporation, Mountain View, CA) и визуального осмотра. Для определения β использовали данные минимально для трех точек на кривой концентрация-время. Реальные значения времени, используемые для каждого индивида можно обнаружить в таблицах рассчитанных фармакокинетических параметров. Время полужизни элиминирования на терминальной фазе (t1/2) рассчитывали как ln(2)/β.
Площадь под кривой зависимости плазменная концентрация-время (AUC) от времени 0 до времени последней измеряемой концентрации (AUCt) рассчитывали по линейному способу трапеций. AUC экстраполировали до неопределенного времени посредством деления последней измеряемой плазменной концентрации (Ct) на β. Обозначая экстраполированную часть AUC как AUCext, AUC от времени 0 до неопределенного времени (AUC∞, AUCinf) рассчитывали следующим образом:
AUC∞= AUCt+ AUCext
Процентную долю вклада экстраполированной AUC (AUCext) в суммарную AUC∞ рассчитывали посредством деления AUCext на AUC∞ и умножения этого частного на 100.
Значение выведения (CL) рассчитывали посредством деления вводимой дозы на AUC∞. Значение объема распределения (Vdβ, VDB) рассчитывали посредством деления CL на β. Также представлена оценка объема распределения в устойчивом состоянии (Vss).
Дозо-нормализованные Cмакс, AUCt и AUC∞ также рассчитывали для всех групп в Частях 1 и 2.
Для групп ПК дозы в Части 3, Cмакс, Tмакс и AUC от 0 до 168 часов после дозы (AUC0-168) оценивали после первой и третьей доз. Бета и t1/2 оценивали после введения третьей ПК дозы. Дополнительно, дозо-нормализованные Cмакс и AUC0-168 рассчитывали для Групп 8 и 9 в Части 3. Отношение накопления (Rac) для AUC0-168 для Дня Исследования 15 относительно AUC0-168 для Дня Исследования 1 также рассчитывали.
Фармакокинетика
Для каждой из Частей 1, 2 и 3, сывороточные концентрации 13C5.5 и ADA и значения фармакокинетических параметров заносились в таблицу для каждого индивида и каждой дозовой группы, и суммарную статистику вычисляли для каждого времени отбора образцов и каждого параметра.
Для всех режимов (групп) однократной дозы 13C5.5 у здоровых индивидов (Часть 1), анализы проводили по дозо-нормализованным Cмакс, дозо-нормализованным AUC, Tмакс и β для решения вопроса о линейной фармакокинетике и пропорциональности дозы. Логарифмическая трансформация использовалась для Cмакс и AUC. Для каждого параметра проводили однофакторный дисперсионный анализ (ANOVA). Индивидов классифицировали по уровню дозы. Массу тела или другую размерность включали как ковариат в модели для Cмакс и AUC, если коэффициент регрессии был статистически достоверным на уровне 0,10. В пределах каркаса конечной модели, самую высокую дозу сравнивали с самой низкой дозой. Для логарифмов Cмакс и AUC, 95% доверительный интервал, а также точечная оценка, обеспечивали определение отношения центрального значения самой высокой дозы относительно центрального значения самой низкой дозы. Точечную оценку и 95% доверительный интервал получали посредством потенциирования соответствующей оценки и доверительных пределов для различия средних логарифмических. Если, исследовали по меньшей мере четыре дозовых уровня 13C5.5, тест также проводили при уровне значимости 0,05 в противоположность средним уровням дозы, с контрастом, выбранным, чтобы быть чувствительным к приблизительно линейной тенденции с логарифмом дозы.
ANOVA также проводили для режимов с однократной дозой у индивидов с легкой до умеренной контролируемой астмой (Часть 2). В пределах каркаса модели, тестировали гипотезу об отсутствии различия между самой высокой и самой низкой дозами. Анализы могли осуществляться совместно или не осуществляться совместно (анализы не проводили совместно) для двух популяций (здоровые индивиды и индивиды с легкой-умеренной контролируемой астмой). Если анализы проводили совместно для двух популяций, тогда только данные от дозовых уровней, которые две популяции имели в общем, подлежали включению. В этом случае, модель могла бы оказывать эффекты на популяцию, уровень дозы и взаимодействие популяции по уровню дозы. Масса тела или еще одна характеристика размера могла бы быть включена в модель для Cмакс и AUC. Если статистика по взаимодействию популяция-доза была значимой на уровне 0,10, тогда оценки и другие факторы влияния могли бы быть предоставлены для каждой популяции раздельно. В ином случае, оценки и другие факторы влияния должны были быть основаны на основных эффектах уровня дозы.
ANOVA также проводили для режимов с множеством доз (Часть 3) для определения фармакокинетических параметров, соответствующих параметрам режимов с однократной дозой. Решение включать или не включать показатель размера в качестве ковариата было основано частично на результатах для Частей 1 и 2. Для Cмакс и AUC, точечные оценки и 95% доверительные интервалы были предоставлены для отношения центральных значений двух доз, как объясняется для Части 1.
Пропущенные значения и нарушения модели
Если имели место пропущенные значения вследствие преждевременного прекращения участия в исследовании, которые возможно относились к исследуемому лекарственному средству, следовало бы учитывать возможность отклонения в результате пропущенных значений.
Значения фармакокинетических переменных (Cмакс, AUC и т.д.) обычно определяют без замены пропущенных индивидуальных значений концентрации, просто с использованием доступных данных, и, если необходимо, выполняя анализ с некоторыми пропущенными значениями для фармакокинетических переменных. Однако, пропущенные значения концентрации для выделенных индивидуальных образцов сыворотки могли бы быть заменены (условно начислены), если бы они могли воздействовать на выводы исследования или точечные оценки значительного воздействия.
Если распределение вероятности переменной было несимметричным в той степени, в какой оно могло бы влиять на заключения от ANOVA (ANOVA проводили для всех частей в большей степени, чем ANCOVA) или точечные оценки вводили в заблуждение, тогда трансформация (альтернатива логарифма в случае Cмакс и AUC), которая давала приблизительно симметричное распределение, должна предполагаться. Если удовлетворительная трансформация не смогла быть найдена или, если очевидно, что оба хвоста вероятностного распределения были довольно длинными, мог быть выполнен непараметрический анализ. Если дозовые уровни имели неравные переменные для расширения, которое могло бы повлиять на выводы, тогда могли бы использоваться аппроксимационные способы, которые обеспечивают обработку неравных переменных.
Пример 1: Нарастающие однократные дозы у здоровых индивидов и индивидов с астмой (Группы 1-7)
Профили зависимости концентрации в сыворотке от времени после однократной ВВ инфузии от 0,3 мг/кг до 10 мг/кг 13C5.5 здоровым индивидам (среднее значение+стандартное отклонение (СО)) (Группы 1-4) представлены на фигурах 5 и 6 в линейной и log-линейной шкалах, соответственно. Профили зависимости концентрации в сыворотке от времени после однократной ВВ инфузии 0,3 мг/кг, 3,0 мг/кг или l0 мг/кг 13C5.5 здоровым индивидам и индивидам с астмой от легкой до умеренной формы (среднее значение+СО) (Группы 1, 3-7) представлены на фигурах 7 и 8 в линейной и log-линейной шкалах, соответственно.
Среднее+СО фармакокинетические параметры 13C5.5 после однократной ВВ инфузии 13C5.5 здоровым индивидам и индивидам с астмой показаны в Таблице 4.
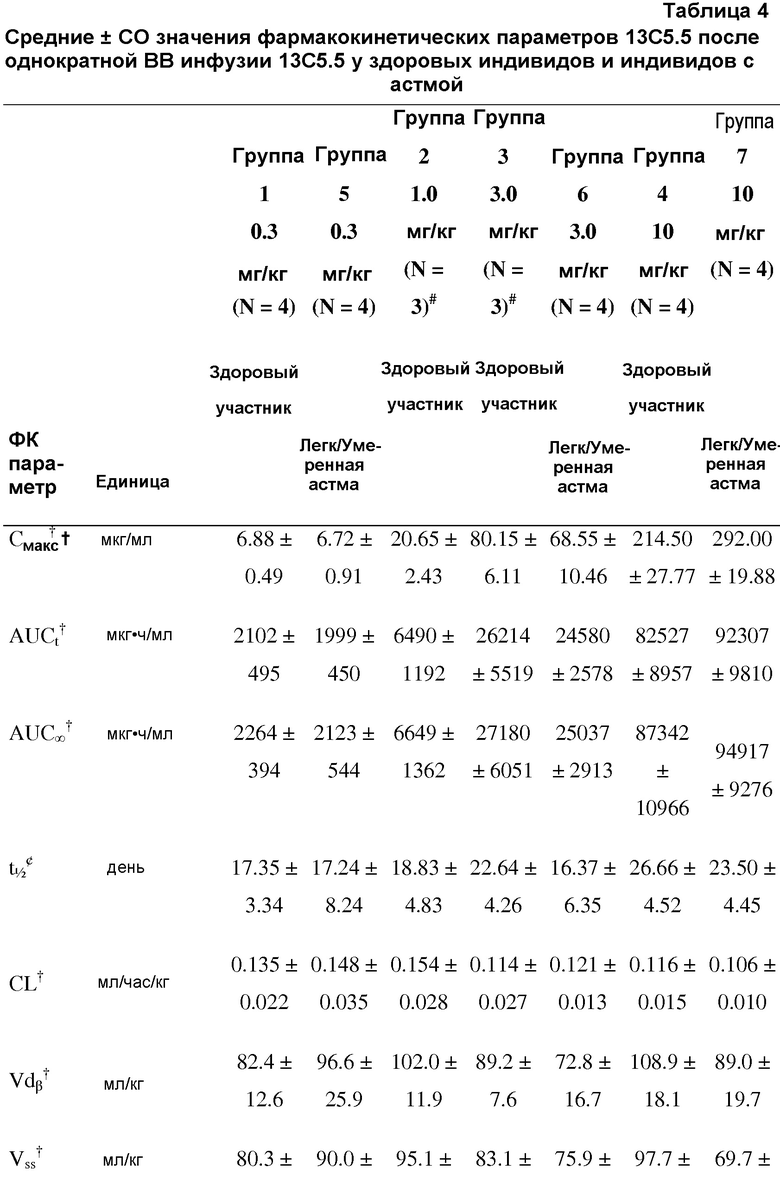
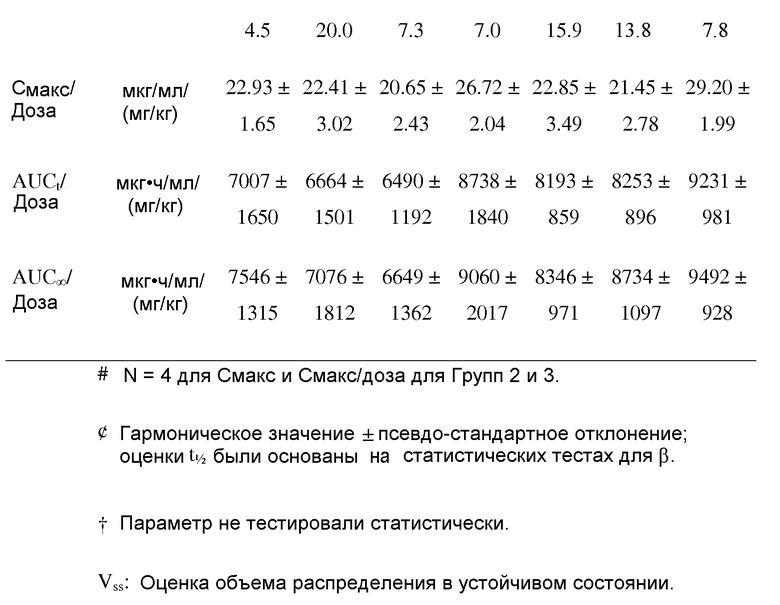
После ВВ введения, воздействия, как определяют посредством AUC и Cмакс, очевидно, увеличивались дозозависимым образом в интервале от 0,3 мг/кг до 10 мг/кг. Воздействия (AUC и Cмакс) от 13C5.5 от здоровых и астматических индивидов были аналогичными при тестируемых дозах (0,3 мг/кг, 3,0 мг/кг, и 10,0 мг/кг ВВ). Фармакокинетика 13C5.5 была сходной с фармакокинетикой типичного иммуноглобулина G1 (IgG1) с небольшим объемом распределения и длительным временем полужизни. Гармоническое значение + псевдо СО значения полужизни 13C5.5 находились в интервале 16,4+6,35 дней до 26,7+4,52 дней, и среднее Vdβ находилось в интервале от 72,8 до 108,9 мл/кг после ВВ инфузий в пределах от 0,3 мг/кг до 10,0 мг/кг дозового интервала.
Суммарные вариабельности Cмакс, AUCt и AUC∞ для 13C5.5, выражаемые в виде процента CV для нарастающих однократных BB инфузий 13C5.5 у здоровых и астматических индивидов показаны в Таблице 5.
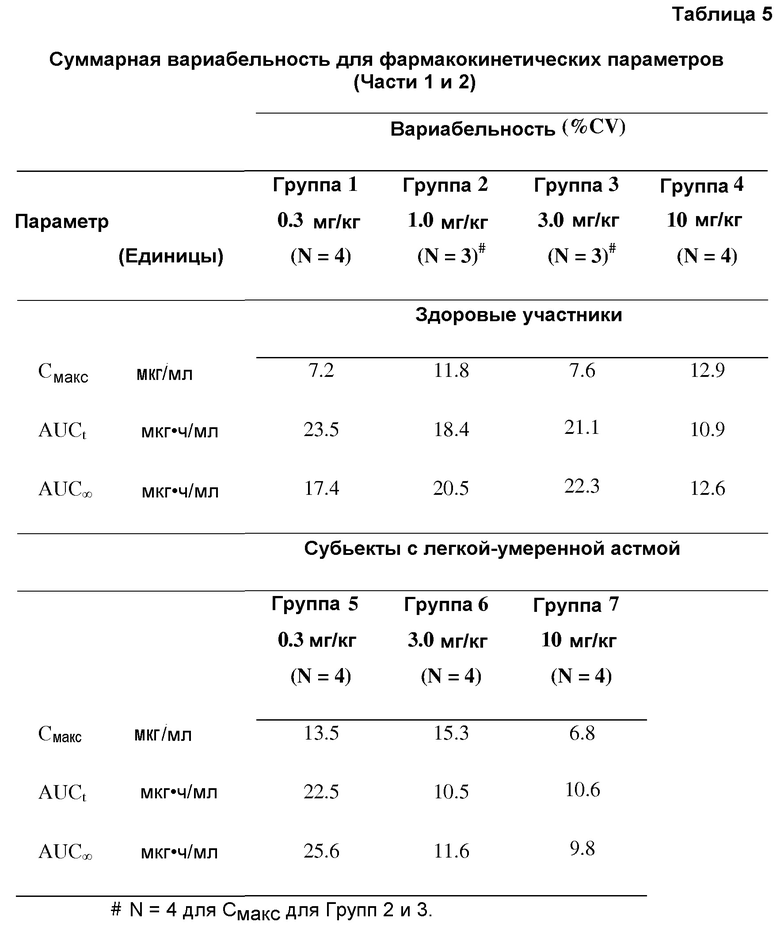
Пример 2: Пропорциональность дозы и фармакокинетическая линейность у здоровых и астматических индивидов (Части 1 и 2)
Средние + СО дозо-нормализованные значения Cмакс и AUC∞ для 13C5.5 после введения однократных ВВ инфузий 13C5.5 в пределах дозового интервала от 0,3 до 10 мг/кг представлены в Таблице 4. Средние + СО дозо-нормализованные значения Cмакс и AUC∞ для 13C5.5 от уровня дозы представлены на Фигуре 9.
Для решения вопросов о фармакокинетической линейности и пропорциональности дозы у здоровых индивидов и индивидов с астмой от легкой до умеренной формы, проводили ANOVA по фармакокинетическим параметрам. Индивидов классифицировали по группе лечебной обработки (режим).
У здоровых индивидов, фармакокинетика 13C5.5 имела линейную зависимость от дозы по Cмакс и AUC. Средние значения Cмакс, AUCt или AUC∞ были сходными между самой высокой дозой (10 мг/кг) и самой низкой дозой (0,3 мг/кг). Статистически достоверные тенденции (p>0,05) отсутствовали у 13C5.5 для изменений по дозо-нормализованной Cмакс или дозо-нормализованной AUC в дозовом интервале (от 0,3 до 10 мг/кг). Однако, мощность тестов была низкой вследствие небольшого числа индивидов в каждой из дозовых групп. На основании статистических тестов для β, значение t1/2 для самой высокой дозы (26,7+4,52 дней, 10 мг/кг) было статистически достоверней, чем значение 17,4+3,34 дней для самой низкой дозы, равной 0,3 мг/кг (Таблица 4). Результаты также указывали на то, что существовали статистически достоверные тенденции (p<0,05) для увеличений t1/2 для 13C5.5 на протяжении дозового интервала от 0,3 до 10 мг/кг.
У индивидов с астмой от легкой до умеренной формы, дозо-нормализованные Cмакс самой высокой дозы (10 мг/кг) были более статистически достоверными, чем для самой низкой дозы, равной 0,3 мг/кг. Дозо-нормализованные значения AUCt и AUC∞ самой высокой дозы были значительно более статистически достоверными (p<0,05), чем для самой низкой дозы. Однако, наблюдаемое отклонение от пропорциональности дозы было небольшим по оценке отношения центральных значений, равных 1,3 для Cмакс и 1,4 для AUC в интервале 33-кратного интервала доз.
Пример 3: Три еженедельных ПК инъекции у астматических индивидов (группы 8 и 9)
Профили зависимости среднего + СО значения сывороточной концентрации от времени для 13C5.5 после 3 еженедельных ПК доз 13C5.5 для Группы 8 (0,3 мг/кг) и Группы 9 (3,0 мг/кг) представлены на Фигуре 10 по линейной и log-линейным шкалам.
Средние значения + СО фармакокинетических параметров 13C5.5 после трех еженедельных доз, равных 0,3 мг/кг или 3,0 мг/кг 13C5.5 ПК показаны в Таблице 6. Накопление после 3 еженедельных доз 13C5.5 очевидно является линейным, так как значения Cмакс и AUC составляют приблизительно в 3 раза выше после трех еженедельных доз 13C5.5 по 0,3 мг/кг или 3,0 мг/кг. Tмакс очевидно ведет себя аналогично после первой и третьей доз 13C5.5 по 0,3 и 3,0 мг/кг. Гармоническое среднее значение ± псевдо-СО времени полужизни для 13C5.5 было равно 27,29±3,33 и 24,30±1,23 дней после ПК введения по 0,3 и 3,0 мг/кг в течение трех дней, соответственно. С использованием одновременного фармакокинетического моделирования данных для ВВ и ПК введения, оцениваемая биодоступность 13C5.5 была равна приблизительно 70%.
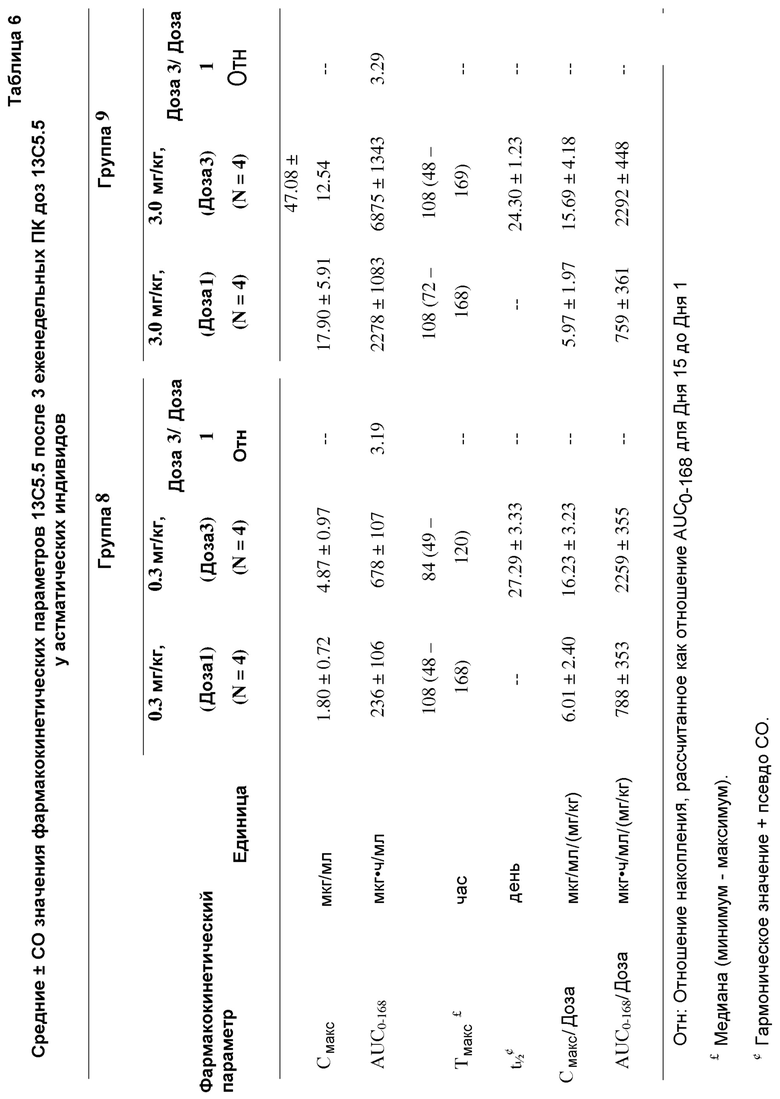
Суммарные вариабельности Cмакс и AUC0-168 для 13C5.5 выражают как процент CV после введения трех еженедельных ПК доз 13C5.5 у индивидов с легкой до умеренной астмы, показаны в Таблице 7.
Пример 4: Пропорциональность дозы и фармакокинетическая линейность (Часть 3)
Средние + СО дозо-нормализованные значения Cмакс и AUC∞ для 13C5.5 после введения 3 еженедельных ПК доз 13C5.5 по 0,3 или 3,0 мг/кг представлены в Таблице 6. Средние + СО дозо-нормализованные значения Cмакс и AUC∞ для 13C5.5 по отношению к дозовому уровню представлены на Фигурах 11 и 12, соответственно.
Для решения вопросов о фармакокинетической линейности и пропорциональности дозы проводили ANOVA по фармакокинетическим параметрам. Индивидов классифицировали по группе лечебной обработки (режим).
У индивидов с астмой от легкой до умеренной формы, которым вводили 3 еженедельных ПК дозы, фармакокинетика 13C5.5 была линейно зависимой от дозы по Cмакс и AUC. Средние значения Tмакс, Cмакс и AUC0-168 были сходными между самой высокой дозой (3,0 мг/кг) и самой низкой дозой (0,3 мг/кг) как на День 1, так и на День 15. Следует отметить, что мощность тестов была низкой вследствие небольшого числа индивидов в каждой из дозовых групп.
Статистика/Анализ для Примеров 1-4
Корректировку для ковариатов не осуществляли.
Индивиды 1202 и 1304 преждевременно прервали участие в исследовании, и их последний анализ образцов крови собирали при Часе 672 и Часе 96, соответственно. Для этих двух индивидов, фармакокинетические параметры не рассчитывали за исключением Cмакс и Tмакс. Индивид 1103 преждевременно прервал участие в исследовании и его последний анализ образца крови собирали при приблизительно 894 часах после дозы. Для данного индивида фармакокинетические параметры были рассчитаны. Несколько случаев пропуска концентраций для индивидуального отбора образца не препятствовали определению значений фармакокинетических параметров, которые считают достоверными.
Индивиды, для которых доступные данные обеспечивали определение значений фармакокинетических параметров, были включены в статистический анализ. Данное исследование проводили в единственном исследовательском центре; следовательно, никакие рассмотрения множественности центров не были необходимыми.
Заключение для Примеров 1-4
Фармакокинетика 13C5.5 после однократного введения дозы соответствовала фармакокинетике IgG1 с долгим временем полужизни и небольшим объемом распределения. Для индивидов, которым вводили однократную ВВ инфузию 13C5.5, системное воздействие (AUC и Cмакс) от 13C5.5 возрастала дозопропорциональным образом в интервале дозы от 0,3 до 10 мг/кг для здоровых индивидов; однако, для индивидов с легкой-умеренной контролируемой астмой, AUC и Cмакс возрастали в несколько большей степени (30-40%), чем при пропорциональности дозы в интервале такого же 33-кратного интервала дозы. Для индивидов с легкой до умеренной астмы, которым вводили 3 еженедельные ПК дозы, фармакокинетика 13C5.5 была линейно зависимой от дозы как для AUC, так и Cмакс между 0,3 и 3,0 мг/кг дозы. Накопление 13C5.5 было, как ожидалось, 3-кратным после трех еженедельных ПК доз.
Во время исследования, 13C5.5 хорошо переносился и был безопасным, как в случае однократной дозы до 10 мг/кг ВВ или как в случае множественных доз, равных 0,3 и 3 мг/кг ПК. Профиль неблагоприятных событий у здоровых взрослых людей был аналогичен такому профилю, наблюдаемому у индивидов с астмой. Никаких относящихся к дозе возрастаний или специфичных для введения тенденций при лечении неотложных неблагоприятных событий не было оценено. В каждой части исследования, составная доля индивидов, сообщающих об инфекции верхних дыхательных путей или вирусной инфекции верхних дыхательных путей, была выше среди тех, кто получал 13C5.5 по сравнению с теми, кто получал плацебо. Все из этих событий имели легкую или умеренную тяжесть, и ни одно из них по заключению исследователя возможно или вероятно не относилось к исследуемому лекарственному средству. Однако частота наступления респираторных инфекций будет подвергаться пристальному мониторингу при будущих исследованиях. Сообщалось о множественных событиях повышенных CPK крови, включая одного индивида с плацебо; эти события происходили после первоначального ограничения на месте исследования и были связаны в каждом случае с преходящими повышениями CPK и историей повышенной физической активности.
Один индивид в группе обработки 0,3 мг/кг 13C5.5 перенес серьезное неблагоприятное событие госпитализации по поводу бурсэктомии, которое было оценено исследователем, как не относящееся к введению исследуемого лекарственного средства. Ни один из индивидов не прервал прием исследуемого лекарственного средства вследствие неотложного лечения неблагоприятного события. За исключением сообщения о боли на месте инфузии у одного индивида, не сообщалось о неблагоприятных событиях, относящихся к инфузии реакциях, и обзор событий, которые могли бы представлять ухудшение состояния при астме и данные спирометрии не позволяют предположить ухудшение основополагающего заболевания у астматических индивидов.
Никаких клинически достоверных тенденций не было обнаружено при других анализах безопасности, включая основные показатели состояния организма, переменные ЭКГ и лабораторные измерения.
ЭКВИВАЛЕНТЫ
Квалифицированные специалисты в данной области смогут признать или будут в состоянии установить с использованием не более чем рутинного экспериментирования множество эквивалентов конкретных вариантов осуществления изобретения, описанного в настоящем описании. Подразумевают, что такие эквиваленты охвачены посредством следующей формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНТЕРЛЕЙКИН-13-СВЯЗЫВАЮЩИЕ БЕЛКИ | 2012 |
|
RU2605327C2 |
| ИНТЕРЛЕЙКИН-13-СВЯЗЫВАЮЩИЕ БЕЛКИ | 2016 |
|
RU2650767C2 |
| ИНТЕРЛЕЙКИН-13-СВЯЗЫВАЮЩИЕ БЕЛКИ | 2007 |
|
RU2472807C2 |
| СПОСОБ ЛЕЧЕНИЯ ПСОРИАЗА (ВАРИАНТЫ) | 2009 |
|
RU2497545C2 |
| МОЛЕКУЛЫ АНТИТЕЛ ЧЕЛОВЕКА К IL-13 | 2004 |
|
RU2387667C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ АСТМЫ ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2020 |
|
RU2832774C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ АСТМЫ ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2013 |
|
RU2690675C2 |
| КОМПОЗИЦИИ, ИЗДЕЛИЯ И СПОСОБЫ, СВЯЗАННЫЕ С ДОЗИРОВАНИЕМ В КЛЕТОЧНОЙ ТЕРАПИИ | 2018 |
|
RU2776823C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ПОЛИПОЗА НОСА ПУТЕМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2014 |
|
RU2674680C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ АСТМЫ ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2015 |
|
RU2713406C2 |



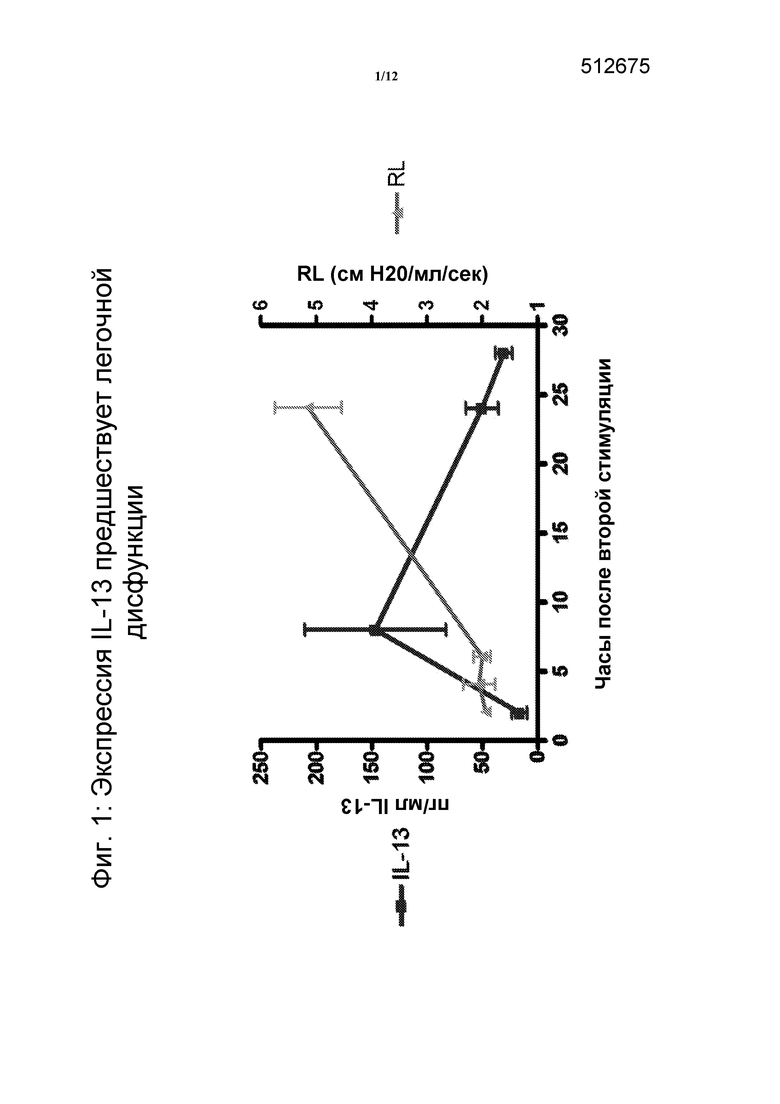
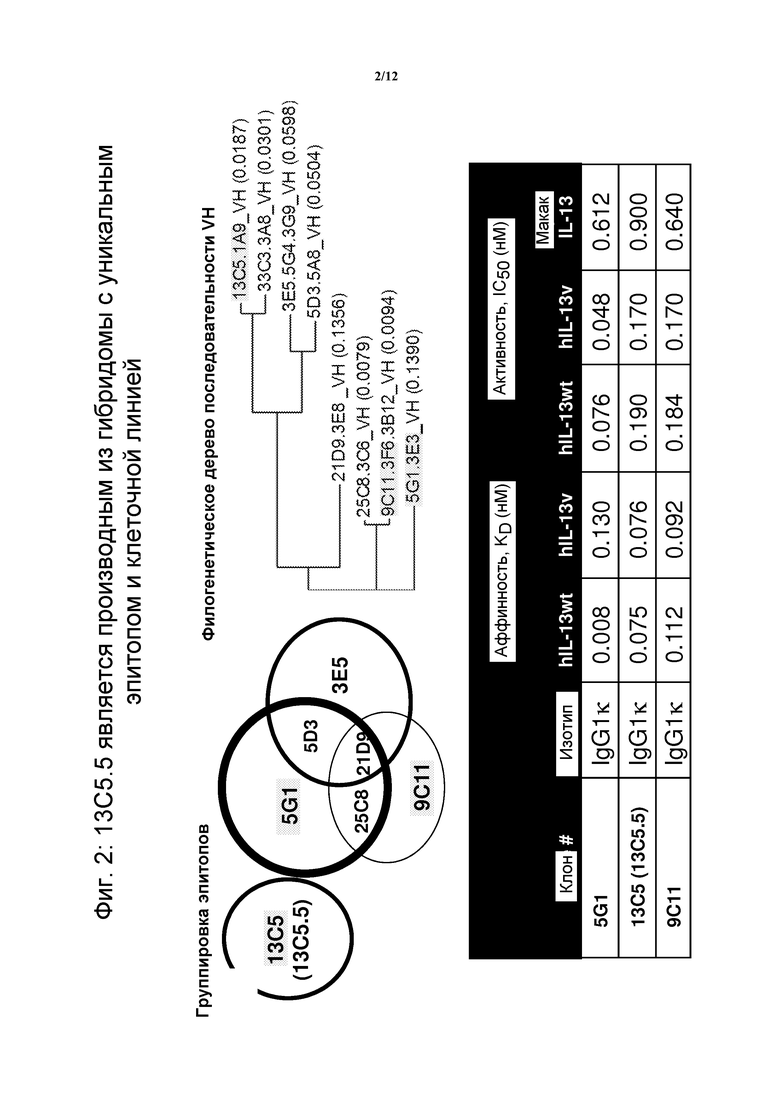
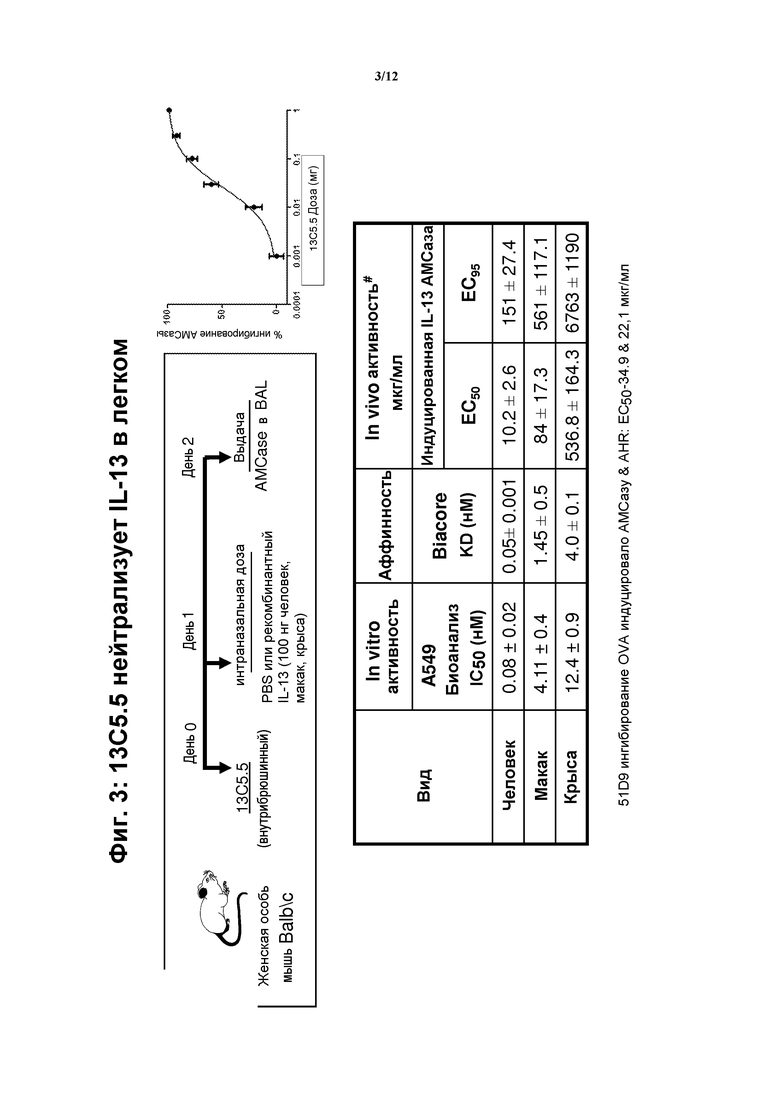
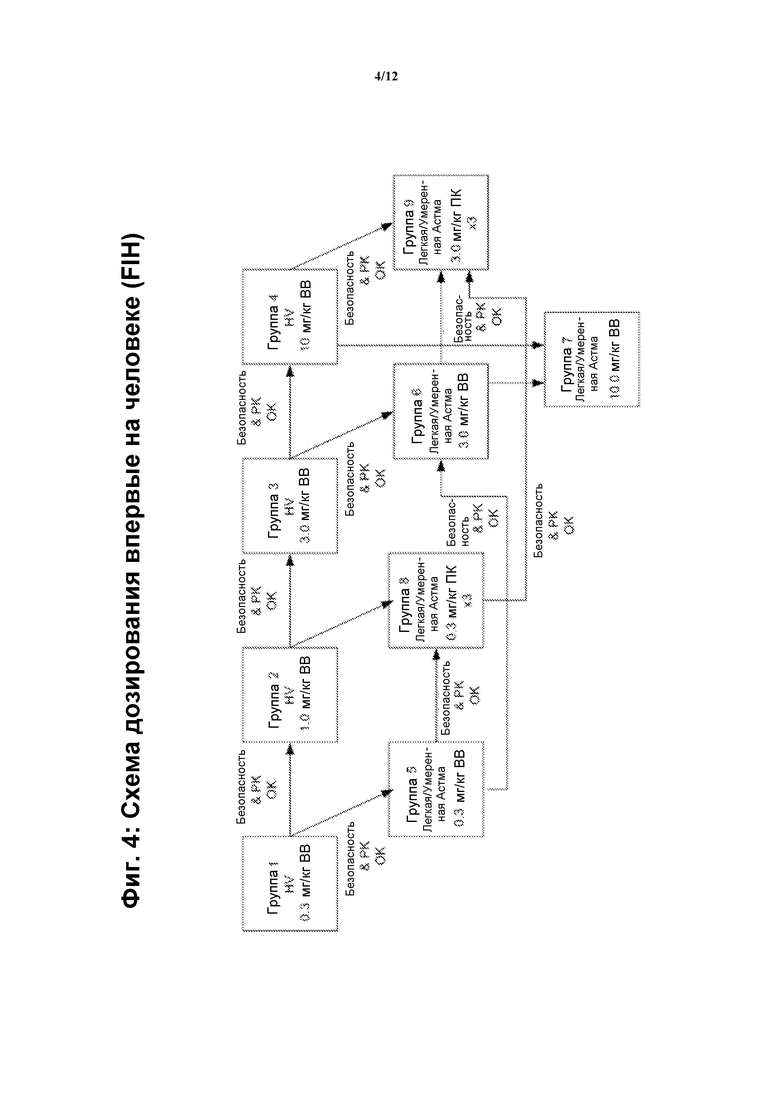
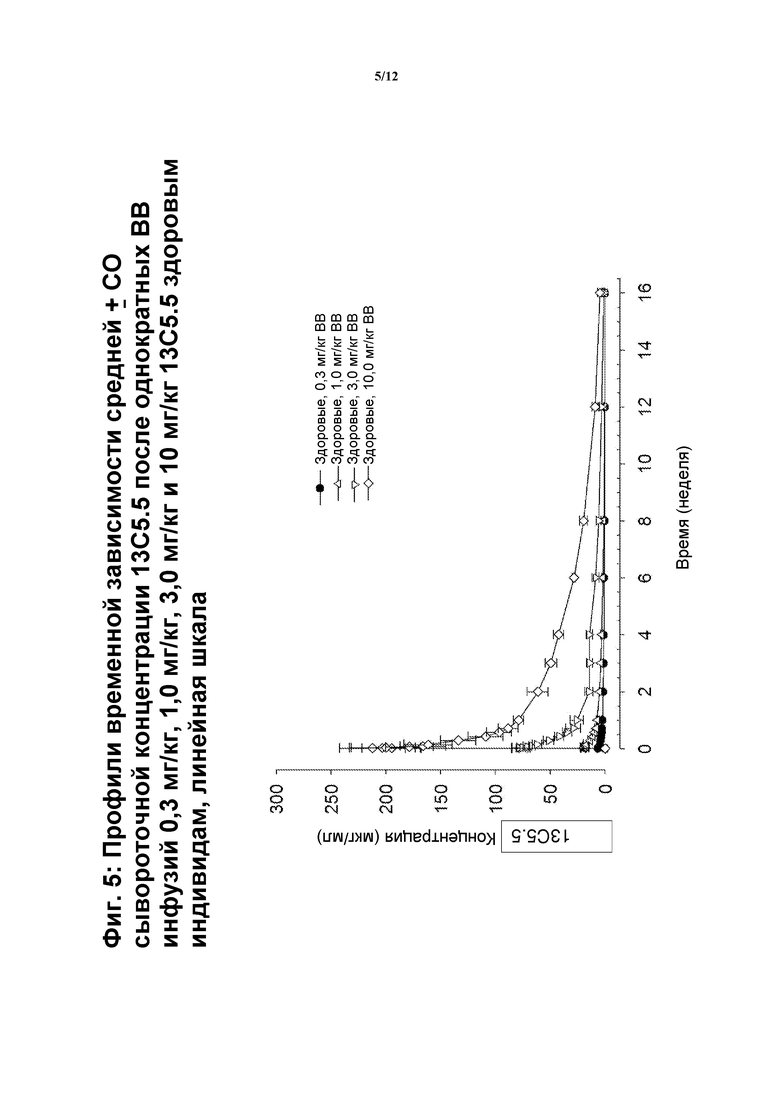
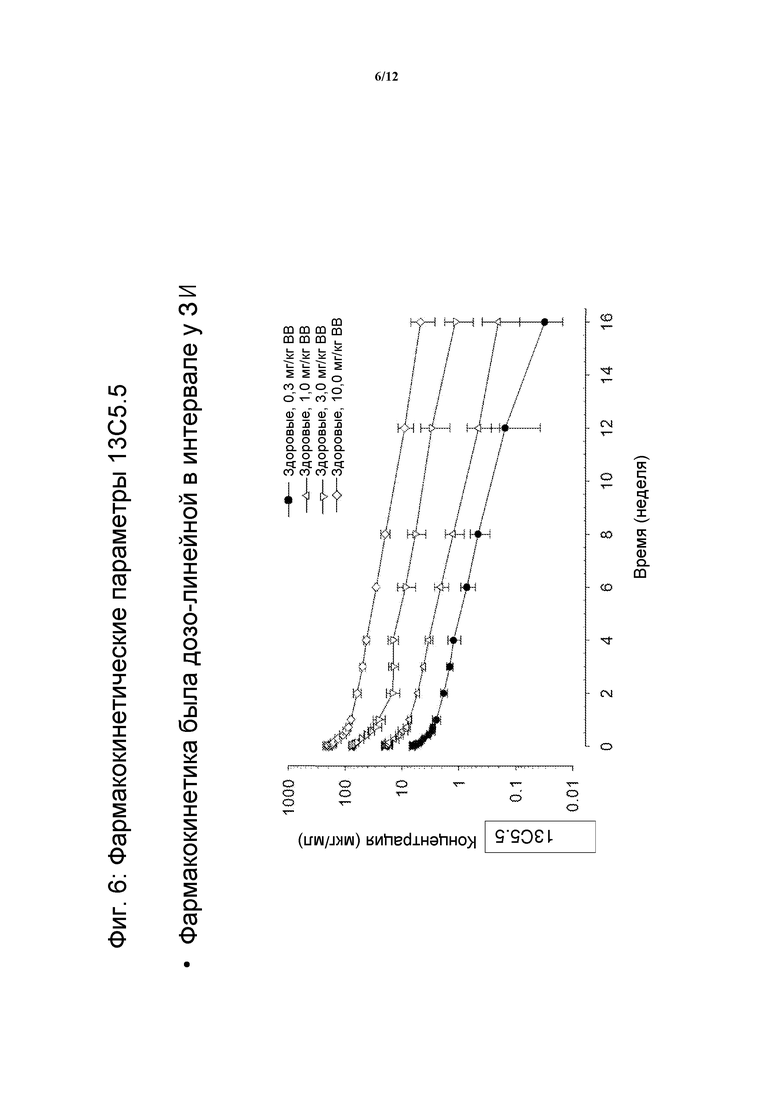
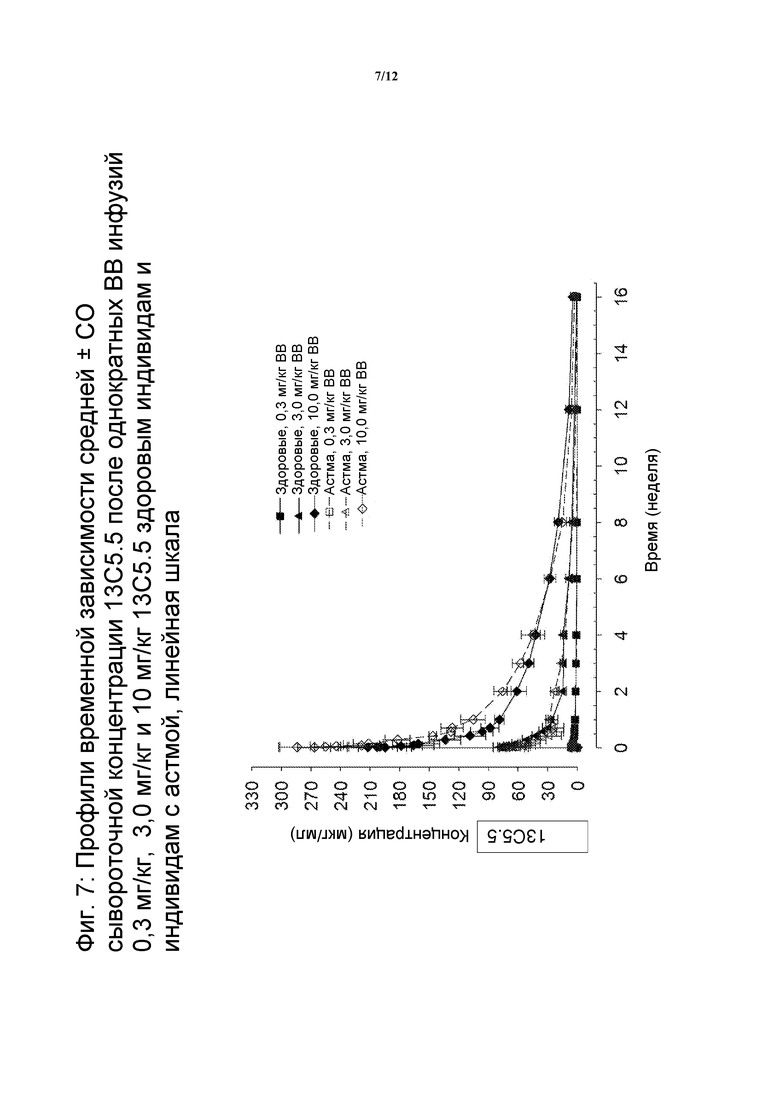
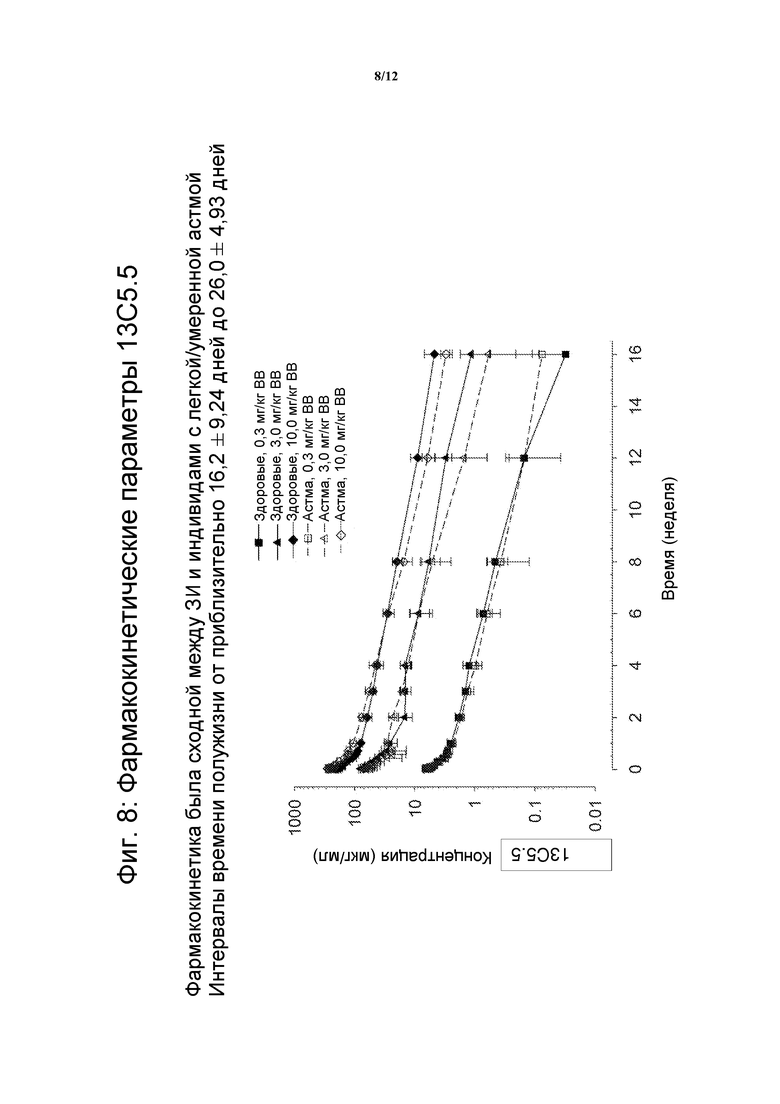
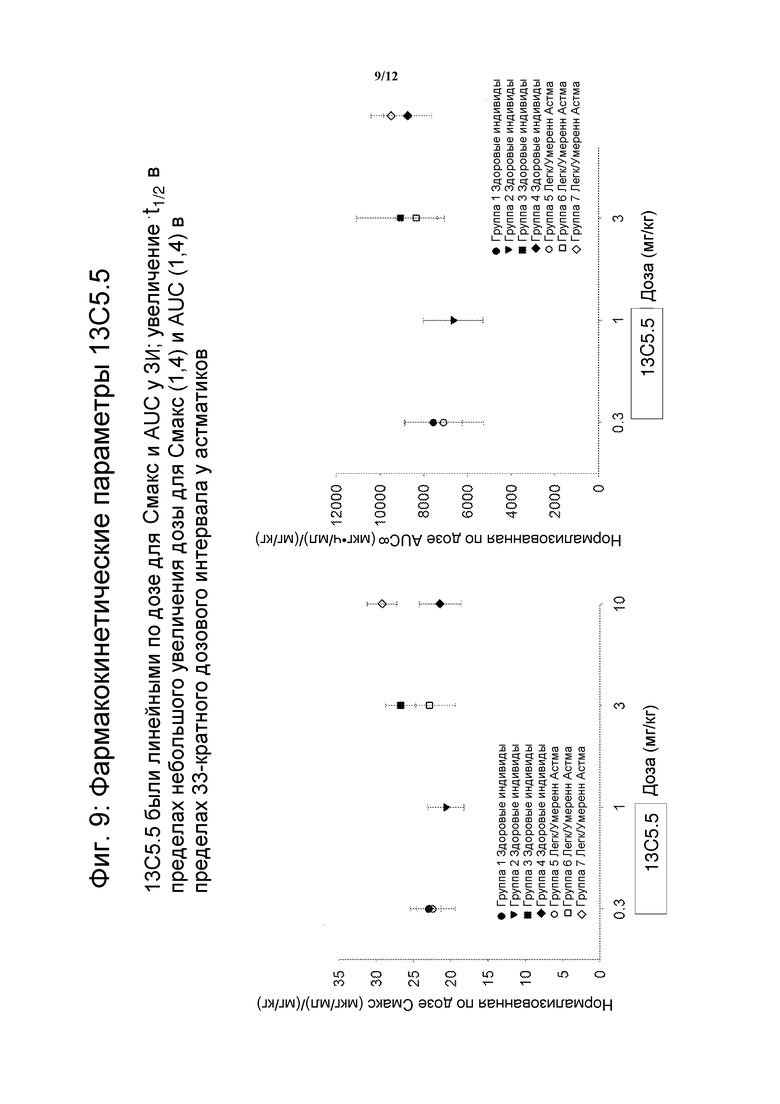
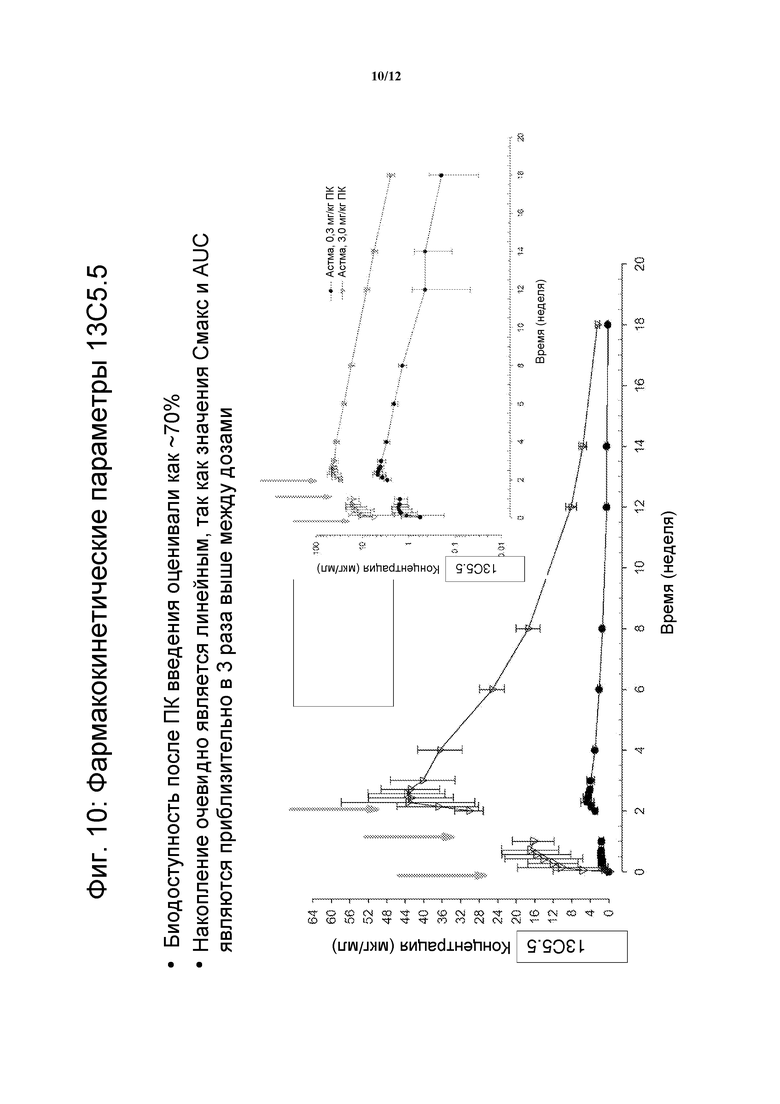
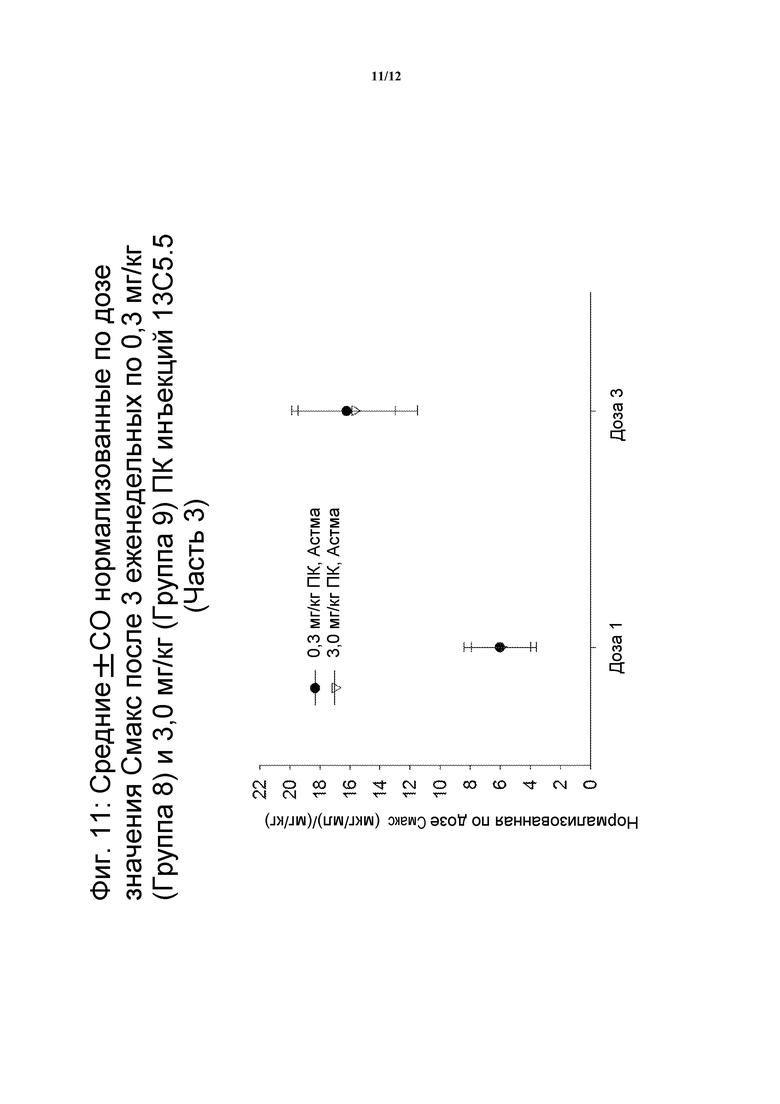
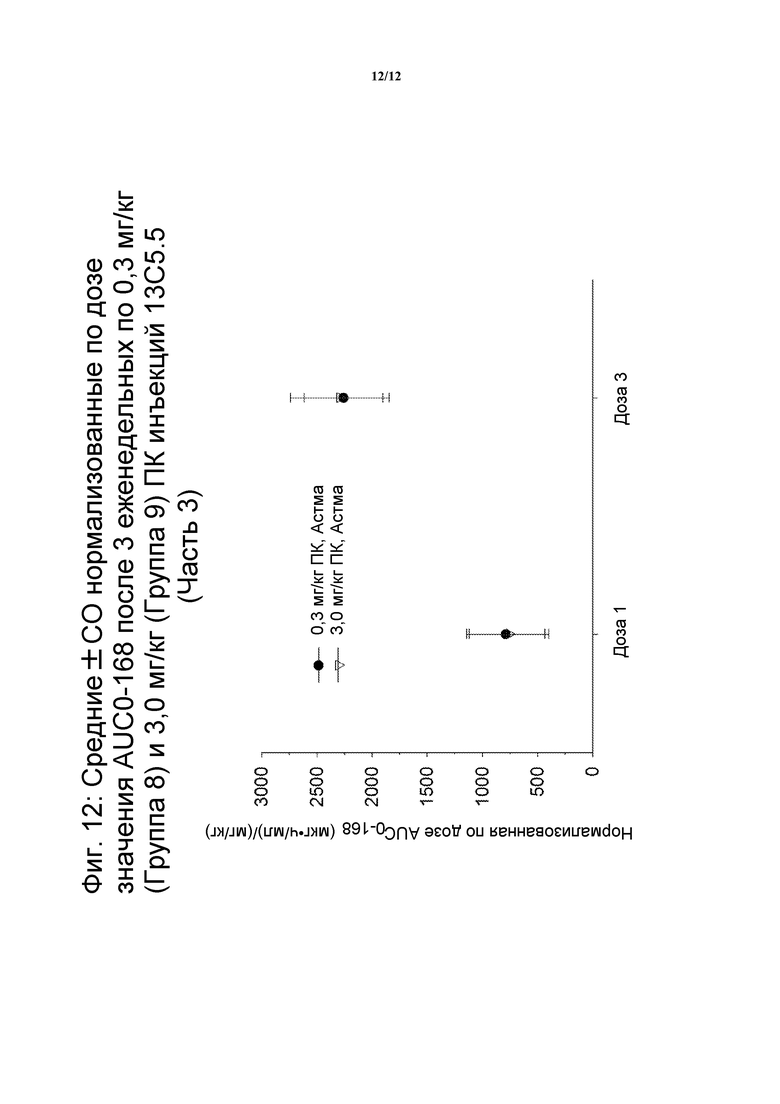
Группа изобретений относится к медицине, а именно к иммунологии, и может быть использована для лечения астмы. Способ лечения астмы у индивида, являющегося человеком, включает внутривенное введение индивиду антитела против IL-13 или его антигенсвязывающей части в дозе, равной приблизительно 0,3 мг/кг. Антитело против IL-13 или его антигенсвязывающая часть содержат вариабельную область тяжелой цепи SEQ ID NO: 2 и вариабельную область легкой цепи SEQ ID NO: 3, где по меньшей мере одна фармакокинетическая характеристика, выбранная из группы, состоящей из: (a) максимальной сывороточной концентрации (Смакс), равной от приблизительно 55 до приблизительно 90 мкг/мл, и (b) площади под кривой концентрация-время для сыворотки (AUC), равной от приблизительно 20000 до приблизительно 34000 мкгч/мл, достигается после введения антитела или его антигенсвязывающей части указанному индивиду. Группа изобретений относится также к вариантам способа лечения бронхиальной астмы от легкой до умеренной формы. Использование данной группы изобретений позволяет подобрать оптимальную дозировку и схему введения вышеуказанного антитела в дозировке 0,3 мг/кг и 3 мг/кг с получением определенных фармакокинетических характеристик. 4 н. и 5 з.п. ф-лы, 4 пр., 7 табл., 12 ил.
1. Способ лечения астмы у индивида, являющегося человеком, включающий внутривенное введение индивиду антитела против IL-13 или его антигенсвязывающей части в дозе, равной приблизительно 3 мг/кг, где антитело против IL-13 или его антигенсвязывающая часть содержат вариабельную область тяжелой цепи SEQ ID NO: 2 и вариабельную область легкой цепи SEQ ID NO: 3, и
где по меньшей мере одна фармакокинетическая характеристика, выбранная из группы, состоящей из:
(a) максимальной сывороточной концентрации (Смакс), равной от приблизительно 55 до приблизительно 90 мкг/мл, и
(b) площади под кривой концентрация-время для сыворотки (AUC), равной от приблизительно 20000 до приблизительно 34000 мкгч/мл,
достигается после введения антитела или его антигенсвязывающей части указанному индивиду.
2. Способ лечения астмы у индивида, являющегося человеком, включающий подкожное введение индивиду антитела против IL-13 или его антигенсвязывающей части в дозе, равной приблизительно 3 мг/кг, где антитело против IL-13 или его антигенсвязывающая часть содержат вариабельную область тяжелой цепи SEQ ID NO: 2 и вариабельную область легкой цепи SEQ ID NO: 3, и где по меньшей мере одна фармакокинетическая характеристика, выбранная из группы, состоящей из:
(a) максимальной сывороточной концентрации (Смакс), равной от приблизительно 12 до приблизительно 60 мкг/мл, и
(b) площади под кривой концентрация-время для сыворотки (AUC), равной от приблизительно 1100 до приблизительно 8100 мкгч/мл,
которая достигается после введения антитела или его антигенсвязывающей части указанному индивиду.
3. Способ лечения астмы у индивида, являющегося человеком, включающий подкожное введение индивиду антитела против IL-13 или его антигенсвязывающей части в дозе, равной приблизительно 3 мг/кг, где антитело против IL-13 или его антигенсвязывающая часть содержат вариабельную область тяжелой цепи SEQ ID NO: 2 и вариабельную область легкой цепи SEQ ID NO: 3, и
где по меньшей мере одна фармакокинетическая характеристика выбрана из группы, состоящей из:
(a) времени полужизни между приблизительно 23 и 26 днями;
(b) Тмакс, меньшего чем или равного до приблизительно 5 дней; и
(c) биодоступности, равной по меньшей мере приблизительно 60%,
достигается после введения антитела или его антигенсвязывающей части указанному индивиду.
4. Способ лечения астмы у индивида, являющегося человеком, включающий внутривенное введение индивиду антитела против IL-13 или его антигенсвязывающей части в дозе, равной приблизительно 3 мг/кг, где антитело против IL-13 или его антигенсвязывающая часть содержат вариабельную область тяжелой цепи SEQ ID NO: 2 и вариабельную область легкой цепи SEQ ID NO: 3, и
где по меньшей мере одна фармакокинетическая характеристика выбрана из группы, состоящей из:
(а) скорости выведения, равной от приблизительно 0,08 до приблизительно 0,14 мл/ч/кг; и
(b) объема распределения, равного от приблизительно 55 до приблизительно 100 мл/кг,
достигается после введения антитела или его антигенсвязывающей части указанному индивиду.
5. Способ по любому из пп. 1-4, где антитело против IL-13 или его антигенсвязывающая часть представляет собой 13С5.5 или его антигенсвязывающую часть.
6. Способ по любому из пп. 1-4, где антитело против IL-13 или его антигенсвязывающую часть вводят однократно.
7. Способ по любому из пп. 1-4, где антитело против IL-13 или его антигенсвязывающую часть вводят еженедельно, необязательно, где антитело или его антигенсвязывающую часть вводят в течение 3 недель.
8. Способ по любому из пп. 1-4, дополнительно включающий введение дополнительного средства указанному индивиду, необязательно, где указанное дополнительное средство выбрано из группы, состоящей из терапевтического средства, средства визуализации, цитотоксического средства, ингибитора ангиогенеза, ингибитора киназы, блокатора молекул со-стимуляции, блокатора молекул адгезии, антител против цитокинов или их функционального фрагмента; метотрексата, циклоспорина, рапамицина, FK506, обнаруживаемой метки или репортера, антагониста ФНО, противоревматического средства, мышечного релаксанта, наркотического средства, нестероидного противовоспалительного лекарственного средства (NSAID), анальгезирующего средства, анестезирующего средства, седативного средства, местного анестезирующего средства, нейромышечного блокатора, противомикробного средства, антипсориатического средства, кортикостероида, анаболического стероида, эритропоэтина, средства для иммунизации, иммуноглобулина, иммуносупрессора, гормона роста, средства для гормонозаместительной терапии, радиофармпрепарата, антидепрессанта, антипсихотического средства, стимулятора, медикамента для лечения астмы, бета-агониста, ингалируемого стероида, перорального стероида, эпинефрина или аналога, цитокина и антагониста цитокина.
9. Способ по любому из пп. 1-4, где астма является астмой от легкой до умеренной формы.
| US 2011165066 A1, 07.07.2011 | |||
| WO 2011032148 A1, 17.03.2011 | |||
| US 2009068195 A1, 12.03.2009 | |||
| SINGH DAVE et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

