ОБЛАСТЬ ТЕХНИКИ
[0001] Настоящее изобретение относится к новым ингибиторам пролилгидроксилазы (далее в настоящем документе также обозначаемой как PHD), в частности к ингибиторам пролилгидроксилазы 2 (далее в настоящем документе также обозначаемой как PHD2).
УРОВЕНЬ ТЕХНИКИ
[0002] Эритроциты в крови отвечают за транспорт кислорода по всему организму и играют важную роль в поддержании постоянных уровней кислорода in vivo. Если за счет кровотечения заболеваний определенных типов, а также несчастных случаев или хирургических операций снижается число эритроцитов или уровень гемоглобина в крови, развивается чувство усталости, головокружение, одышка и другие симптомы анемии. При анемии весь организм будет подвержен дефициту кислорода и при таких гипоксических условиях живой организм использует компенсаторные реакции, при которых, в первую очередь в почках, происходит образование гематопоэтического фактора эритропоэтина (далее в настоящем документе также обозначаемый как EPO), который способствует формированию эритроцитов, чтобы повышать уровни эритроцитов и гемоглобина в крови, таким образом, помогая облегчать анемию. Однако при заболеваниях определенных типов происходит ослабление этого эритропоэтического действия эритропоэтина, а хроническая анемия пепсистирует. Например, известно, что у пациентов с почечной недостаточностью, которые имеют нарушения в почках, описанный выше механизм образования эритропоэтина при гипоксических условиях не может работать должным образом, что вызывает у них анемию определенного типа (почечную анемию), которая отличается сниженными числом эритроцитов и уровнем гемоглобина (см. непатентные документы 1 и 2).
[0003] Лечение почечной анемии и анемии, которая сопутствует химиотерапии злокачественных опухолей или лекарственной терапии пациентов с ВИЧ-инфекцией, в настоящее время осуществляют с помощью стимулирующих эритропоэз средств (ESA), таких как препараты рекомбинантного эритропоэтина человека. ESA в основном способствуют повышению качества жизни пациента посредством повышения числа эритроцитов и уровня гемоглобина в достаточной мере для того, чтобы улучшать симптомы, которые сопутствуют анемии. Однако с другой стороны, все доступные в настоящее время ESA представляют собой биологические средства в форме дорогостоящих инъекций, поэтому желательно разработать перорально вводимое фармацевтическое лекарственное средство для лечения анемии.
[0004] В недавнем исследовании сообщалось о том, что эритропоэтин также оказывает действие для защиты тканей, таких как сердце и головной мозг, находящийся в гипоксических условиях, которые сопутствуют анемии. Следовательно, перорально вводимые препараты ESA потенциально могут найти широкий спектр применений, покрывающих не только почечную, но и анемии других типов, которые возникают в результате различных причин, а также различных ишемических заболеваний (см. непатентный документ 3).
[0005] Веществом, которое может быть отмечено в качестве фактора, который повышает образование эритропоэтина, является индуцируемый гипоксией фактор (далее в настоящем документе также обозначаемый как HIF). HIF представляет собой фактор транскрипции, содержащий α-субъединицу, разрушение которой регулируют изменения концентрации кислорода, и β-субъединицу, которая экспрессируется постоянно. Пролилгидроксилазы (PHD-1, -2 и -3) известны в качестве факторов, которые регулируют разрушение α-субъединицы HIF (HIF-α). В условиях нормального давления кислорода происходит гидроксилирование остатков пролина в HIF-α с помощью этих пролилгидроксилаз и быстрое разрушение HIF-α с помощью протеасом. С другой стороны, в гипоксических условиях активность пролилгидроксилаз снижается, так что разрушение HIF-α подавляется, таким образом, способствуя транскрипции эритропоэтина и других чувствительных к HIF генов. Следовательно, посредством ингибирования пролилгидроксилаз способствуют стабилизации HIF-α, что делает возможным повышение образования эритропоэтина (см. непатентные документы 1, 2 и 4).
[0006] Соединения по настоящему изобретению предоставляют средство для ингибирования активностей этих пролилгидроксилаз для того, чтобы повышать количество эритропоэтина и тем самым лечить анемию. В качестве другого эффекта, не только анемию, но также различные другие ишемические заболевания (например, инсульт головного мозга, инфаркт миокарда и ишемическое нарушение функции почек) и осложнения диабета (нефропатия, ретинопатия и нейропатия) также можно симптоматически лечить или предотвращать или улучшать или облегчать посредством введения соединений по настоящему изобретению (см. непатентный документ 5).
[0007] Обыкновенные ингибиторы PHD, о которых сообщалось на сегодняшний день, включают производные 4-гидроксиизохинолина (см. патентный документ 1), производные 5-гидрокси-3-оксо-2,3-дигидро-1H-пиразола (см. патентный документ 2), производные 4-гидрокси-2-оксо-1,2-дигидрохинолина (см. патентный документ 3), производные 3-гидроксипиридина (см. патентный документ 4), производные 2-оксо-2,3-дигидроиндола (см. патентный документ 5) и т.д., а соединения, имеющие структуры в соответствии с настоящим изобретением, не раскрыты. Также на сегодняшний день сообщалось о производных 6-гидрокси-2,4-диоксо-1,2,3,4-тетрагидропиримидина (см. патентный документ 6), производных 4-гидрокси-6-оксо-1,6-дигидропиримидина (см. патентный документ 7), производных 5-гидрокси-3-оксо-2,3-дигидропиридазина (см. патентный документ 8), производных 6-гидрокси-4-оксо-4H-1,3-диоксина (см. патентный документ 9), производных 4-гидрокси-2-оксо-1,2,5,7-тетрагидрофтор[3,4-b]пиридина (см. патентный документ 10), производных 4-гидрокси-2-оксо-1,2-дигидропиридина (см. патентные документы 11 и 12) и т.д., а соединения, имеющие структуры в соответствии с настоящим изобретением, не раскрыты.
СПИСОК ЦИТИРУЕМОЙ ЛИТЕРАТУРЫ
ПАТЕНТНЫЕ ДОКУМЕНТЫ
[0008]
Патентный документ 1: WO 2004/108681
Патентный документ 2: WO 2006/114213
Патентный документ 3: WO 2007/038571
Патентный документ 4: US 2007/0299086
Патентный документ 5: WO 2008/144266
Патентный документ 6: WO 2007/150011
Патентный документ 7: WO 2008/089051
Патентный документ 8: WO 2008/089052
Патентный документ 9: WO 2009/049112
Патентный документ 10: WO 2009/108496
Патентный документ 11: WO 2009/158315
Патентный документ 12: WO 2010/025087
НЕПАТЕНТНЫЕ ДОКУМЕНТЫ
[0009] Непатентный документ 1: American Journal of Physiology-Renal Physiology, 2010, 299, Fl-13
Непатентный документ 2: American Journal of Physiology-Renal Physiology, 2010, 298, F1287-1296
Непатентный документ 3: The Journal of Physiology, 2011, 589, 1251-1258
Непатентный документ 4: Expert Opinion on Therapeutic Patents, 2010, 20, 1219-1245
Непатентный документ 5: Diabetes, Obesity and Metabolism, 2008, 10, 1-9
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ПРОБЛЕМА
[0010] Цель настоящего изобретения состоит в том, чтобы предоставить превосходящие ингибиторы PHD2.
РЕШЕНИЕ ПРОБЛЕМЫ
[0011] Авторы настоящего изобретения проводили интенсивные исследования с тем, чтобы достичь вышеуказанной цели и в результате обнаружили соединения, представленные с помощью следующей общей формулы (I) или (I'), которые имеют превосходящий ингибирующий PHD2 эффект.
[0012] В кратком изложении, настоящее изобретение направлено на:
[0013] (1) предоставление соединения, представленного следующей общей формулой (I')
[0014]

(где в формуле (I')
W представляет формулу -CR15R16-, формулу -CR11R12CR13R14-, или формулу -CH2CR17R18CH2-;
R15 представляет атом водорода, С1-4 алкил или фенил;
R16 представляет атом водорода или С1-4 алкил;
при условии, что R15 и R16, вместе со смежным углеродным атомом, необязательно образуют C3-8 циклоалкан;
R11 представляет атом водорода, атом фтора, С1-4 алкил или фенил;
R12 представляет атом водорода, атом фтора или С1-4 алкил;
при условии, что R11 и R12, вместе со смежным углеродным атомом, необязательно образуют C3-8 циклоалкан или 4-8-членный насыщенный гетероцикл, который содержит атом кислорода;
R13 представляет атом водорода, карбамоил, С1-4 алкил (С1-4 алкил необязательно замещен одной группой, выбранной из группы, состоящей из гидрокси, С1-3 алкокси, и ди-C1-3 алкиламино), галоген-C1-4 алкил, фенил, пиридил, бензил, или фенэтил;
R14 представляет атом водорода, С1-4 алкил или галоген-C1-4 алкил;
при условии, что R13 и R14, вместе со смежным углеродным атомом, необязательно образуют C3-8 циклоалкан, 4-8-членный насыщенный гетероцикл, который содержит атом кислорода, или 4-8-членный насыщенный гетероцикл, который содержит атом азота (4-8-членный насыщенный гетероцикл, который содержит атом азота, необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из метила, бензила, фенилкарбонила и оксо);
при условии, что указанные R12 и R13, вместе со смежными углеродными атомами, необязательно образуют C3-8 циклоалкан;
R17 представляет атом водорода или С1-4 алкил;
R18 представляет атом водорода или С1-4 алкил;
при условии, что R17 и R18, вместе со смежным углеродным атомом, необязательно образуют C3-8 циклоалкан;
Y представляет одинарную связь или C1-6 алкандиил (C1-6 алкандиил необязательно замещен одной гидрокси, и один из углеродных атомов в C1-6 алкандииле необязательно замещен C3-6 циклоалкан-1,1-диила);
R2 представляет атом водорода, C1-6 алкил, C3-8 циклоалкил {С3-8 циклоалкил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из C1-6 алкила (C1-6 алкил необязательно замещен одним фенилом), фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и галоген-С1-6 алкила), C1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила, фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила), и пиридила (пиридил необязательно замещен одним атомом галогена)], С3-8 циклоалкокси, фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, С3-8 циклоалкила и галоген-C1-6 алкила), и пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, С3-8 циклоалкила и галоген-C1-6 алкила)}, фенил (фенил необязательно замещен от одной до трех групп, которые являются одинаковыми или различными и которые выбирают из группы α3 заместителей), нафтил, инданил, тетрагидронафтил, пиразолил, имидазолил, изоксазолил, оксазолил [пиразолил, имидазолил, изоксазолил, и оксазолил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из C1-6 алкила и фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и С1-6 алкила)], тиазоил [тиазоил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из C1-6 алкила, фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила), и морфолино], пиридил (пиридил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы α5 заместителей), пиридазинил, пиримидинил, пиразинил [пиридазинил, пиримидинил и пиразинил необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, фенила, С1-6 алкокси (С1-6 алкокси необязательно замещен одним С3-8 циклоалкила), и фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и С3-8 циклоалкила)], бензотиофенил, хинолил, метилендиоксифенил (метилендиоксифенил необязательно замещен одним или двумя атомами фтора), 4-8-членный насыщенный гетероциклил, содержащий атом азота [4-8-членный насыщенный гетероциклил, содержащий атом азота, необязательно замещен одной группой, выбранной из группы, состоящей из пиримидинила, фенил-C1-3 алкила, С3-8 циклоалкил-C1-3 алкилкарбонила и фенил-C1-3 алкоксикарбонила] или следующую формулу (I'')
[0015]
-CONR5CH2-R6 (I'')
[где в формуле (I'')
R5 представляет атом водорода или С1-3 алкил, а R6 представляет фенил (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила и фенила)],
группа α3 заместителей состоит из гидрокси, циано, карбокси, атома галогена, C1-6 алкила {C1-6 алкил необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила, фенила, C1-6 алкокси [алкокси необязательно замещен одним С3-8 циклоалкилом (С3-8 циклоалкил необязательно замещен одним C1-6 алкилом)], фенокси (фенокси необязательно замещен одним C1-6 алкилом) и пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкила и галоген-C1-6 алкила)}, галоген-C1-6 алкила, С3-8 циклоалкила (С3-8 циклоалкил необязательно замещен одним или двумя атомами галогена), C3-8 циклоалкенила (С3-8 циклоалкенил необязательно замещен одним или двумя атомами галогена), фенила (фенил необязательно замещен от одной до трех групп, которые являются одинаковыми или различными и которые выбирают из группы α4 заместителей), тиенила (тиенил необязательно замещен одним C1-6 алкилом), пиразолила (пиразолил необязательно замещен одним C1-6 алкилом), изоксазолила, тиазоила (тиазоил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из гидрокси, C1-6 алкила и C1-6 алкокси), пиридила (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из карбокси, гидрокси, амино, атом галогена, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси, галоген-C1-6 алкокси и C1-6 алкилсульфонила), пиримидинила (пиримидинил необязательно замещен одной амино), хинолила, C1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из карбокси, гидрокси, карбамоила, С3-8 циклоалкила (C3-8 циклоалкил необязательно замещен одним C1-6 алкилом), фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из гидрокси, атома галогена, С1-6 алкила, галоген-C1-6 алкила, С1-6 алкокси, галоген-C1-6 алкокси и ди-C1-6 алкиламино), пиридила (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и С1-6 алкила), оксазолила (оксазолил необязательно замещен одним или двумя С1-6 алкилами), пиразолила (пиразолил необязательно замещен одним или двумя C1-6 алкилами), тиазоила (тиазоил необязательно замещен одним C1-6 алкилом), индазолила (индазолил необязательно замещен одним C1-6 алкилом), бензотриазолила, имидазотиазоила и ди-C1-6 алкиламино], галоген-C1-6 алкокси, C2-6 алкенилокси, С3-8 циклоалкокси, фенокси (фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила, C1-6 алкокси и галоген-C1-6 алкокси), пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила и С3-8 циклоалкила), пиримидинилокси, пиперазинила (пиперазинил необязательно замещен одним С1-6 алкилом), моно-C1-6 алкиламинокарбонила (C1-6 алкил в моно-C1-6 алкиламинокарбониле необязательно замещен одной группой, выбранной из группы, состоящей из карбокси, гидрокси, ди-C1-6 алкиламино, пиридила, фенила и 2-оксопирролидинила), ди-C1-6 алкиламинокарбонила (где два C1-6 алкила в ди-C1-6 алкиламинокарбониле, вместе со смежным атомом азота, необязательно образуют 4-8-членный насыщенный гетероцикл, который содержит атом азота), C1-6 алкилсульфанила и C1-6 алкилсульфонила;
группа α4 заместителей состоит из карбокси, циано, гидрокси, сульфамоила, атома галогена, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, фенила, C1-6 алкокси, галоген-C1-6 алкокси, C1-6 алкилкарбонила, ди-C1-6 алкиламинокарбонила, C1-6 алкилсульфонила, моно-C1-6 алкиламиносульфонила (С1-6 алкил в моно-C1-6 алкиламиносульфониле необязательно замещен одной гидрокси) и ди-C1-6 алкиламиносульфонила;
группа α5 заместителей состоит из атома галогена, C1-6 алкила, галоген-C1-6 алкила, C1-6 алкокси [С1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила (С3-8 циклоалкил необязательно замещен одним C1-6 алкилом) и фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила)], галоген-C1-6 алкокси, фенила (фенил необязательно замещен одной группой, выбранной из группы α6 заместителей), пиридила, фенокси [фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, циано, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси (C1-6 алкокси необязательно замещен одним фенилом), и галоген-C1-6 алкокси], пиридилокси (пиридилокси необязательно замещен одним C1-6 алкилом) и фенилсульфанила (фенилсульфанил необязательно замещен одним атомом галогена);
группа α6 заместителей состоит из атома галогена, C1-6 алкила, галоген-C1-6 алкила, C3-8 циклоалкила, C1-6 алкокси и галоген-C1-6 алкокси;
Y4 представляет С1-4 алкандиил;
R3 представляет атом водорода или метил;
R4 представляет -COOH, -CONHOH или тетразолил);
или его фармацевтически приемлемой соли.
[0016] (2) В другом варианте настоящее изобретение направлено на предоставление соединения согласно (1), где в указанной выше общей формуле (I'),
Y4 представляет собой метандиил,
R3 представляет собой атом водорода,
R4 представляет собой -COOH,
или его фармацевтически приемлемой соли.
[0017] (3) В другом варианте настоящее изобретение направлено на предоставление соединения согласно (2), где в указанной выше общей формуле (I')
W представляет собой формулу -CR15R16-, и соединение представлено общей формулой (I'-1):
[0018]
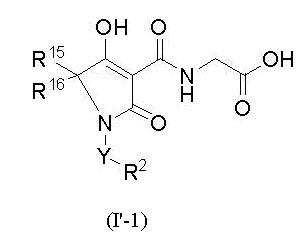
(где в формуле (I'-1)
R15 представляет собой атом водорода, С1-4 алкил или фенил,
R16 представляет собой атом водорода или С1-4 алкил,
при условии, что R15 и R16, вместе со смежным углеродным атомом, необязательно образует C3-8 циклоалкан),
или его фармацевтически приемлемой соли.
[0019] (4) В другом варианте настоящее изобретение направлено на предоставление соединения согласно (2), где в указанной выше общей формуле (I'),
W представляет собой формулу -CR11R12CR13R14-, а соединение представлено общей формулой (I'-2):
[0020]
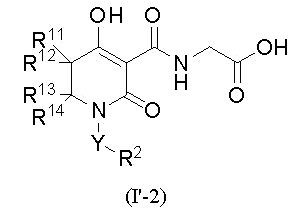
(где в формуле (I'-2)
R11 представляет собой атом водорода, атом фтора, С1-4 алкил или фенил,
R12 представляет собой атом водорода, атом фтора или С1-4 алкил,
при условии, что R11 и R12, вместе со смежным углеродным атомом, необязательно образуют C3-8 циклоалкан или 4-8-членный насыщенный гетероцикл, который содержит атом кислорода;
R13 представляет собой атом водорода, карбамоил, C1-4 алкил (C1-4 алкил необязательно замещен одной группой, выбранной из группы, состоящей из гидрокси, C1-3 алкокси и ди-C1-3 алкиламино), галоген-C1-4 алкил, фенил, пиридил, бензил или фенэтил;
R14 представляет собой атом водорода, С1-4 алкил или галоген-С1-4 алкил,
при условии, что R13 и R14, вместе со смежным углеродным атомом, необязательно образуют C3-8 циклоалкан, 4-8-членный насыщенный гетероцикл, который содержит атом кислорода, или 4-8-членный насыщенный гетероцикл, который содержит атом азота (4-8-членный насыщенный гетероцикл, который содержит атом азота, необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из метила, бензила, фенилкарбонила и оксо),
при условии, что указанные выше R12 и R13, вместе со смежными углеродными атомами, необязательно образуют C3-8 циклоалкан),
или его фармацевтически приемлемой соли.
[0021] (5) В другом варианте настоящее изобретение направлено на предоставление соединения согласно (4), где в указанной выше общей формуле (I'-2), Y представляет собой одинарную связь или C1-6 алкандиил (один из углеродных атомов в C1-6 алкандииле необязательно замещен C3-6 циклоалкан-1,1-диилом),
R2 представляет собой С3-8 циклоалкил {С3-8 циклоалкил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из C1-6 алкила (C1-6 алкил необязательно замещен одним фенилом), фенила (фенил необязательно замещен одним галоген-C1-6 алкилом), C1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила, фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила) и пиридила (пиридил необязательно замещен одним атомом галогена)], С3-8 циклоалкокси, фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, С3-8 циклоалкила и галоген-C1-6 алкила) и пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, С3-8 циклоалкила и галоген-C1-6 алкила)}, фенил (фенил необязательно замещен от одной до трех групп, которые являются одинаковыми или различными и которые выбирают из указанной выше группы α3 заместителей), нафтил, инданил, тетрагидронафтил, пиразолил [пиразолил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из C1-6 алкила и фенила (фенил необязательно замещен одним C1-6 алкилом)], имидазолил (имидазолил необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкила и фенила), изоксазолил [изоксазолил необязательно замещен одним фенилом (фенил необязательно замещен одним атомом галогена)], оксазолил (оксазолил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из C1-6 алкила и фенила), тиазоил (тиазоил необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкила, фенила и морфолино), пиридил (пиридил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из указанной выше группы α5 заместителей), пиридазинил [пиридазинил необязательно замещен одним С1-6 алкокси (C1-6 алкокси необязательно замещен одним С3-8 циклоалкилом)], пиримидинил [пиримидинил необязательно замещен одной группой, выбранной из группы, состоящей из галоген-С1-6 алкила, С3-8 циклоалкила, фенила и фенокси (фенокси необязательно замещен одним C1-6 алкилом)], пиразинил [пиразинил необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкокси (C1-6 алкокси необязательно замещен С3-8 циклоалкилом) и фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и С3-8 циклоалкила)], бензотиофенил, хинолил, или метилендиоксифенил (метилендиоксифенил необязательно замещен одним или двумя атомами фтора),
или его фармацевтически приемлемую соль.
[0022] (6) В другом варианте настоящее изобретение направлено на предоставление соединения согласно (5), где в указанной выше общей формуле (I'-2),
R11 представляет собой атом водорода,
R12 представляет собой атом водорода,
R13 представляет собой атом водорода,
R14 представляет собой атом водорода,
Y представляет собой метандиил,
R2 представляет собой
фенил {фенил замещен одной группой, выбранной из группы, состоящей из фенила [фенил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из карбокси, циано, гидрокси, сульфамоила, атома галогена, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, фенила, C1-6 алкокси, галоген-C1-6 алкокси, C1-6 алкилкарбонила, ди-C1-6 алкиламинокарбонила, C1-6 алкилсульфонила, моно-C1-6 алкиламиносульфонила (C1-6 алкил в моно-C1-6 алкиламиносульфониле необязательно замещен одной гидрокси), и ди-C1-6 алкиламиносульфонила], пиридила (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из карбокси, гидрокси, амино, атома галогена, C1-6 алкила, галоген-C1-6 алкила, C3-8 циклоалкила, C1-6 алкокси и C1-6 алкилсульфонила), фенокси (фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, C1-6 алкила, С1-6 алкокси и галоген-C1-6 алкокси), и пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила и С3-8 циклоалкила), и дополнительно он может быть замещен одним атомом галогена};
пиридил {пиридил замещен одной группой, выбранной из группы, состоящей из фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси и галоген-C1-6 алкокси), пиридила, фенокси [фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, циано, C1-6 алкила, галоген-C1-6 алкила, C3-8 циклоалкила, C1-6 алкокси (C1-6 алкокси необязательно замещен одним фенилом), и галоген-C1-6 алкокси], и пиридилокси (пиридилокси необязательно замещен одним C1-6 алкилом), и дополнительно он может быть замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила}; или
пиразинил, который замещен одним фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и С3-8 циклоалкила),
или его фармацевтически приемлемой соли.
[0023] (7) В другом варианте настоящее изобретение направлено на предоставление следующего соединения согласно (1):
N-{[4-гидрокси-2-оксо-1-(4-феноксибензил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-[(4-гидрокси-1-{[6-(4-метилфенокси)-3-пиридинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-({4-гидрокси-2-оксо-1-[(6-фенокси-3-пиридинил)метил]-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-({1-[4-(4-фторфенокси)бензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-({4-гидрокси-1-[4-(4-метилфенокси)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-[(1-{[6-(4-цианофенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-({4-гидрокси-2-оксо-1-[4-(2-пиримидинилокси)бензил]-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-[(1-{[6-(4-фторфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(4-хлорфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-{[4-гидрокси-2-оксо-1-({6-[4-(трифторметил)фенокси]-3-пиридинил}метил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-[(4-гидрокси-1-{[6-(3-метилфенокси)-3-пиридинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(3-фторфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-({4-гидрокси-1-[4-(3-метилфенокси)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-({1-[4-(3-фторфенокси)бензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-[(1-{[5-(4-фторфенокси)-2-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-1-{[5-(4-метилфенокси)-2-пиридинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-({1-[4-(4-хлорфенокси)бензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-[(4-гидрокси-1-{4-[(6-метил-3-пиридинил)окси]бензил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(2-фторфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-1-{[6-(2-метилфенокси)-3-пиридинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-({1-[4-(2-фторфенокси)бензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-({4-гидрокси-1-[4-(2-метилфенокси)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-[(1-{[6-(3-хлорфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-{[4-гидрокси-2-оксо-1-({6-[3-(трифторметил)фенокси]-3-пиридинил}метил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-({4-гидрокси-1-[4-(3-метоксифенокси)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-{[4-гидрокси-2-оксо-1-({6-[3-(трифторметокси)фенокси]-3-пиридинил}метил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-[(1-{4-[(5-фтор-2-пиридинил)окси]бензил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{4-[(5-хлор-2-пиридинил)окси]бензил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(4-циклопропилфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-1-{4-[(5-метил-2-пиридинил)окси]бензил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-{[4-гидрокси-2-оксо-1-(4-{[5-(трифторметил)-2-пиридинил]окси}бензил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-{[4-гидрокси-1-({5-метил-6-[(6-метил-3-пиридинил)окси]-3-пиридинил}метил)-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-[(1-{[5-(4-хлорфенокси)-2-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-1-{[6-(3-метоксифенокси)-3-пиридинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{4-[(6-хлор-3-пиридинил)окси]бензил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-{[4-гидрокси-2-оксо-1-({5-[4-(трифторметил)фенокси]-2-пиридинил}метил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-{[4-гидрокси-2-оксо-1-(4-{[6-(трифторметил)-3-пиридинил]окси}бензил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-[(1-{[6-(3-хлор-4-метилфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(3-фтор-4-метилфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(4-фтор-3-метилфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(4-этилфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-2-оксо-1-{[6-(4-пропилфенокси)-3-пиридинил]метил}-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-1-{[6-(4-изопропилфенокси)-3-пиридинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-1-{[5-(4-метилфенокси)-2-пиразинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-({1-[4-(3,4-диметилфенокси)бензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-[(1-{[5-хлор-6-(4-метилфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[5-фтор-6-(4-метилфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{4-[(5-циклопропил-2-пиридинил)окси]бензил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-1-{[2-(4-метилфенокси)-5-пиримидинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(4-хлорфенокси)-5-метил-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[5-(4-хлорфенокси)-2-пиразинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин; или
N-[(1-{[5-(4-циклопропилфенокси)-2-пиразинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин,
или его фармацевтически приемлемой соли.
[0024] (8) В другом варианте настоящее изобретение направлено на предоставление соединения, которое имеет указанную выше общую формулу (I'), где
W представляет собой формулу -CR11R12CR13R14-, и соединение представлено общей формулой (I):
[0025]
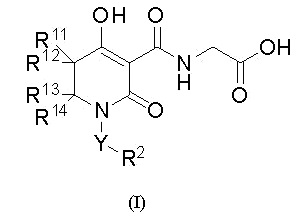
(где в формуле (I)
R11 представляет собой атом водорода, С1-4 алкил или фенил,
R12 представляет собой атом водорода или С1-4 алкил,
при условии, что R11 и R12, вместе со смежным углеродным атомом, необязательно образуют C3-8 циклоалкан или 4-8-членный насыщенный гетероцикл, который содержит атом кислорода;
R13 представляет собой атом водорода, С1-4 алкил, галоген-С1-4 алкил, фенил, бензил или фенэтил,
R14 представляет собой атом водорода или С1-4 алкил,
при условии, что R13 и R14, вместе со смежным углеродным атомом, необязательно образуют C3-8 циклоалкан или 4-8-членный насыщенный гетероцикл, который содержит атом кислорода,
при условии, что указанные выше R12 и R13, вместе со смежными углеродными атомами, необязательно образуют C3-8 циклоалкан;
Y представляет собой одинарную связь или C1-6 алкандиил (один из углеродных атомов в C1-6 алкандииле необязательно замещен C3-6 циклоалкан-1,1-диилом);
R2 представляет собой С3-8 циклоалкил (С3-8 циклоалкил необязательно замещен одной группой, выбранной из группы, состоящей из фенила и бензила), фенил (фенил необязательно замещен от одной до трех групп, которые являются одинаковыми или различными и которые выбирают из группы α1 заместителей), нафтил, инданил, тетрагидронафтил, пиразолил [пиразолил замещен одним фенилом (фенил необязательно замещен одним C1-6 алкилом) и дополнительно можно замещать одним C1-6 алкилом], имидазолил (имидазолил замещен одним фенилом), изоксазолил [изоксазолил замещен одним фенилом (фенил необязательно замещен одним атомом галогена)], оксазолил (оксазолил замещен одним фенилом и дополнительно можно заменить C1-6 алкилом), тиазоил (тиазоил замещен одним фенилом), пиридил [пиридил замещен одной группой, выбранной из группы, состоящей из фенила, фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, циано, С1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси и галоген-C1-6 алкокси) и фенилсульфанила (фенилсульфанил необязательно замещен одним атомом галогена)], пиримидинил (пиримидинил замещен одной группой, выбранной из группы, состоящей из циклогексила и фенила), бензотиофенил, хинолил или метилендиоксифенил (метилендиоксифенил необязательно замещен одним или двумя атомами фтора);
группа α1 заместителей состоит из атома галогена, C1-6 алкила {С1-6 алкил необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила, фенила и C1-6 алкокси [C1-6 алкокси необязательно замещен одним C3-8 циклоалкила (С3-8 циклоалкил необязательно замещен одним C1-6 алкилом)]}, галоген-C1-6 алкила, С3-8 циклоалкила, фенила (фенил необязательно замещен от одной до трех групп, которые являются одинаковыми или различными и которые выбирают из группы α2 заместителей), тиенила, пиразолила (пиразолил необязательно замещен одним C1-6 алкилом), изоксазолила, тиазоила (тиазоил необязательно замещен одним или двумя C1-6 алкилами), пиридила (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкила, галоген-C1-6 алкила, C1-6 алкокси и галоген-C1-6 алкокси), хинолила, С1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила и фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила)], галоген-C1-6 алкокси, C2-6 алкенилокси, С3-8 циклоалкокси, фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила, C1-6 алкокси и галоген-C1-6 алкокси), пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и галоген-C1-6 алкила) и C1-6 алкилсульфанила;
группа α2 заместителей состоит из атома галогена, циано, гидрокси, C1-6 алкила, галоген-C1-6 алкила, фенила, C1-6 алкокси, галоген-C1-6 алкокси, C1-6 алкилкарбонила и ди-C1-6 алкиламиносульфонила),
или его фармацевтически приемлемой соли.
[0026] (9) В другом варианте настоящее изобретение направлено на предоставление лекарственного средства, которое содержит соединение согласно какому-либо одному из с (1) до (8) или его фармацевтически приемлемую соль в качестве активного ингредиента.
[0027] (10) В другом варианте настоящее изобретение направлено на предоставление ингибитора PHD2, который содержит соединение согласно какому-либо одному из с (1) до (8) или его фармацевтически приемлемую соль в качестве активного ингредиента.
[0028] (11) В другом варианте настоящее изобретение направлено на предоставление стимулятор образования EPO, который содержит соединение согласно какому-либо одному из с (1) до (8) или его фармацевтически приемлемую соль в качестве активного ингредиента.
[0029] (12) В другом варианте настоящее изобретение направлено на предоставление лекарственного средства для предотвращения или лечения анемии, которое содержит соединение согласно какому-либо одному из с (1) до (8) или его фармацевтически приемлемую соль в качестве активного ингредиента.
ПОЛЕЗНЫЕ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
[0030] Настоящее изобретение сделало возможным предоставить соединения, которые обладают превосходящим ингибирующим PHD2 эффектом.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0031] Настоящее изобретение предусматривает соединения, которые обладают превосходящим ингибирующим PHD2 эффектом, которые представлены общей формулой (I) или (I'), или их фармацевтически приемлемые соли.
[0032] Далее в описании соединения по настоящему изобретению описаны более подробно, но следует понимать, что настоящее изобретение никаким образом не ограничено следующими иллюстрациями.
[0033] Как используют в настоящем документе, символ «н» относится к нормальному, «в» или «втор» - к вторичному, «т» или «трет» - к третичному, «ц» - к цикло-, «о» - к орто-, «м» - к мета- и «п» - к пара-.
[0034] «Атом галогена» относится к атому фтора, атому хлора, атому брома и атому йода.
[0035] «C1-3 алкил» относится к линейному или разветвленному алкилу, который имеет от одного до трех углеродных атомов. В частности, так обозначают метил, этил, н-пропил и изопропил.
[0036] «C1-4 алкил» относится к линейному или разветвленному алкилу, который имеет от одного до четырех углеродных атомов. В частности, так обозначают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил.
037] «C1-6 алкил» относится к линейному или разветвленному алкилу, которые имеет от одного до шести углеродных атомов, и примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, 2-метилбутил, н-гексил, изогексил и т.д.
[0038] «Галоген-C1-4 алкил» относится к линейному или разветвленному алкилу, который имеет от одного до четырех углеродных атомов, с заменой с помощью атома галогена. Число замен с помощью атома галогена предпочтительно составляет от одной до трех и предпочтительно атом галогена представляет собой атом фтора. Примеры включают монофторметил, дифторметил, трифторметил, 1-фторэтил, 1,1-дифторэтил, 2-фторэтил, 2-фтор-2-метилпропил, 2,2-дифторпропил, 1-фтор-2-метилпропан-2-ил, 1,1-дифтор-2-метилпропан-2-ил и т.д.
[0039] «Галоген-C1-6 алкил» относится к линейному или разветвленному алкилу, который имеет от одного до шести углеродных атомов, с заменой с помощью атома галогена. Число замен с помощью атома галогена предпочтительно составляет от одной до пяти и предпочтительно атом галогена представляет собой атом фтора. Примеры включают монофторметил, дифторметил, трифторметил, 1-фторэтил, 1,1-дифторэтил, 1,1,2,2,2-пентафторэтил, 2-фторэтил, 2-фтор-2-метилпропил, 2,2-дифторпропил, 1-фтор-2-метилпропан-2-ил, 1,1-дифтор-2-метилпропан-2-ил, 1-фторпентил, 1-фторгексил и т.д.
[0040] «C3-6 циклоалкан» относится к циклическому алкану, который имеет от трех до шести углеродных атомов. Примеры включают циклопропан, циклобутан, циклопентан и циклогексан.
[0041] «C3-8 циклоалкан» относится к циклическому алкану, который имеет от трех до восьми углеродных атомов. Примеры включают циклопропан, циклобутан, циклопентан, циклогексан, циклогептан и циклооктан.
[0042] «С3-8 циклоалкил» относится к циклическому алкилу, который имеет от трех до восьми углеродных атомов. Примеры включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил.
[0043] «С3-8 циклоалкенил» относится к циклическому алкенилу, который имеет от трех до восьми углеродных атомов. Примеры включают циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогептенил и циклооктенил.
[0044] «4-8-членный насыщенный гетероцикл, который содержит атом кислорода» относится к 4-8-членному моноциклическому насыщенному гетероциклу, который содержит один атом кислорода в кольце. Примеры включают оксетан, тетрагидрофуран, тетрагидропиран и т.д.
[0045] «4-8-членный насыщенный гетероцикл, который содержит атом азота» относится к 4-8-членному моноциклическому насыщенному гетероциклу, который содержит один атом азота в кольце. Примеры включают азетидин, пирролидин, пиперидин и т.д.
[0046] «4-8-членный насыщенный гетероциклил, который содержит атом азота» относится к 4-8-членной моноциклической насыщенной гетероциклической группе, которая содержит один атом азота в кольце. Примеры включают азетидинил, пирролидинил, пиперидинил и т.д.
[0047] «C1-3 алкокси» относится к линейной или разветвленной алкокси, которая имеет от одного до трех углеродных атомов. В частности, так обозначают метокси, этокси, н-пропокси и изопропокси.
[0048] «C1-6 алкокси» относится к линейной или разветвленной алкокси, которая имеет от одного до шести углеродных атомов. Примеры включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, н-пентилокси, изопентилокси, неопентилокси, 2-метилбутокси, н-гексилокси, изогексилокси и т.д.
[0049] «Галоген-C1-6 алкокси» относится к линейной или разветвленной алкокси, которая имеет от одного до шести углеродных атомов, с заменой с помощью атома галогена. Число замен с помощью атома галогена предпочтительно составляет от одного до пяти, и предпочтительно атом галогена представляет собой атом фтора. Примеры включают монофторметокси, дифторметокси, трифторметокси, 1-фторэтокси, 1,1-дифторэтокси, 1,1,2,2-тетрафторэтокси, 2-фторэтокси, 2,2,2-трифторэтокси, 3,3,3-трифторпропокси, 1,3-дифторпропан-2-илокси, 2-фтор-2-метилпропокси, 2,2-дифторпропокси, 1-фтор-2-метилпропан-2-илокси, 1,1-дифтор-2-метилпропан-2-илокси, 4,4,4-трифторбутокси и т.д.
[0050] «C2-6 алкенилокси» относится к группе такой структуры, которая связана кислородом с линейным или разветвленным алкенилом, который имеет от двух до шести углеродных атомов. Примеры включают этенилокси, (E)-проп-1-ен-1-илокси, (Z)-проп-1-ен-1-илокси, проп-2-ен-1-илокси, (Z)-бут-2-ен-1-илокси. (Z)-пент-3-ен-1-илокси, (Z)-гекс-4-ен-1-илокси, (Z)-гепт-5-ен-1-илокси и (Z)-окт-6-ен-1-илокси и т.д.
[0051] «С3-8 циклоалкокси» относится к циклической алкокси, которая имеет от трех до восьми углеродных атомов. Примеры включают циклопропокси, циклобутокси, циклопентилокси, циклогексилокси, циклогептилокси и циклооктилокси.
[0052] «Ди-C1-3 алкиламино» относится к амино, которая имеет указанный выше «C1-3 алкил» в качестве двух заместителей, которые являются одинаковыми или различающимися. Примеры включают диметиламино, диэтиламино, ди(н-пропил)амино, ди(изопропил)амино, этилметиламино, метил(н-пропил)амино и т.д.
[0053] «Ди-C1-6 алкиламино» относится к амино, которая имеет указанный выше «C1-6 алкил» в качестве двух заместителей, которые являются одинаковыми или различающимися. Примеры включают диметиламино, диэтиламино, ди(н-пропил)амино, ди(изопропил)амино, этилметиламино, метил(н-пропил)амино и т.д.
[0054] «С1-6 алкилкарбонил» относится к группе такой структуры, где карбонил связан с указанным выше «C1-6 алкилом». Примеры включают метилкарбонил, этилкарбонил, н-пропилкарбонил, изопропилкарбонил, н-бутилкарбонил, изобутилкарбонил, втор-бутилкарбонил, трет-бутилкарбонил, н-пентилкарбонил, изопентилкарбонил, неопентилкарбонил, 2-метилбутилкарбонил, н-гексилкарбонил, изогексилкарбонил и т.д.
[0055] «Моно-C1-6 алкиламинокарбонил» относится к группе такой структуры, где карбонил связан с амино, имеющей указанный выше «C1-6 алкил» в качестве одного заместителя. Примеры включают метиламинокарбонил, этиламинокарбонил, н-пропиламинокарбонил, изопропиламинокарбонил, н-бутиламинокарбонил, изобутиламинокарбонил, втор-бутиламинокарбонил, трет-бутиламинокарбонил, н-пентиламинокарбонил, н-гексиламинокарбонил и т.д.
[0056] «Ди-C1-6 алкиламинокарбонил» относится к группе такой структуры, что карбонил связан с амино, имеющей указанный выше «C1-6 алкил» в качестве двух заместителей, которые являются одинаковыми или различающимися. Примеры включают диметиламинокарбонил, ди(н-пропил)аминокарбонил, ди(изопропил)аминокарбонил, этилметиламинокарбонил, метил(н-пропил)аминокарбонил и т.д.
Два C1-6 алкила в ди-C1-6 алкиламинокарбониле, вместе со смежным атомом азота, могут необязательно образовывать 4-8-членный насыщенный гетероцикл, который содержит атом азота.
[0057] «C1-6 алкилсульфанил» относится к группе такой структуры, где сульфанил связан с указанным выше «C1-6 алкилом». Примеры включают метилсульфанил, этилсульфанил, н-пропилсульфанил, изопропилсульфанил, изобутилсульфанил, н-гексилсульфанил и т.д.
[0058] «С1-6 алкилсульфонил» представляет собой группу такой структуры, где сульфонил связан с указанным выше «C1-6 алкилом». Примеры включают метилсульфонил, этилсульфонил, н-пропилсульфонил, изопропилсульфонил, изобутилсульфонил, н-гексилсульфонил и т.д.
[0059] «Моно-C1-6 алкиламиносульфонил» относится к группе такой структуры, что сульфонил связан с амино, которая имеет указанный выше «С1-6 алкил» в качестве одного заместителя. Примеры включают метиламиносульфонил, этиламиносульфонил, н-пропиламиносульфонил, изопропиламиносульфонил, н-бутиламиносульфонил, изобутиламиносульфонил, втор-бутиламиносульфонил, трет-бутиламиносульфонил, н-пентиламиносульфонил, н-гексиламиносульфонил и т.д.
[0060] «Ди-C1-6 алкиламиносульфонил» относится к группе такой структуры, где сульфонил связан с амино, которая имеет указанный выше «C1-6 алкил» в качестве двух заместителей, которые являются одинаковыми или различающимися. Примеры включают диметиламиносульфонил, диэтиламиносульфонил, ди(н-пропил)аминосульфонил, ди(изопропил)аминосульфонил, этилметиламиносульфонил, метил(н-пропил)аминосульфонил, изопропил(метил)аминосульфонил и т.д.
[0061] «C1-4 алкандиил» относится к двухвалентной углеводородной группе такой структуры, где один атом водорода удален из алкильной группы, которая имеет от одного до четырех углеродных атомов. Примеры включают метандиил, этан-1,1-диил, этан-1,2-диил, пропан-1,1-диил, пропан-1,2-диил, пропан-1,3-диил, пропан-2,2-диил, бутан-1,4-диил, 2-метилпропан-1,2-диил и т.д. Среди них метандиил, этан-1,1-диил, этан-1,2-диил, пропан-1,1-диил, пропан-1,2-диил, пропан-1,3-диил, и пропан-2,2-диил представляют собой C1-3 алкандиилы.
«C1-6 алкандиил» относится к двухвалентной углеводородной группе такой структуры, где один атом водорода удален из алкильной группы, которая имеет от одного до шести углеродных атомов. Примеры включают метандиил, этан-1,1-диил, этан-1,2-диил, пропан-1,1-диил, пропан-1,2-диил, пропан-1,3-диил, пропан-2,2-диил, бутан-1,4-диил, 2-метилпропан-1,2-диил, пентан-1,5-диил, гексан-1,6-диил и т.д.
[0062] «C3-6 циклоалкан-1,1-диил» относится к двухвалентной циклической углеводородной группе такой структуры, где один атом водорода удален из циклоалкильной группы, которая имеет от трех до шести углеродных атомов. Примеры включают циклопропан-1,1-диил, циклобутан-1,1-диил, циклопентан-1,1-диил и циклогексан-1,1-диил.
«Фенил-C1-3 алкил» относится к указанному выше «C1-3 алкилу», который имеет фенильную группу в качестве заместителя. Примеры включают бензил, фенэтил и фенилпропил.
«С3-8 циклоалкил-C1-3 алкилкарбонил» относится к группе такой структуры, где указанная выше циклоалкильная группа, которая имеет от трех до восьми углеродных атомов, связывает карбонильную группу через указанный выше С1-3 алкил. Примеры включают циклопропилметилкарбонил, циклопропилэтилкарбонил, циклобутилметилкарбонил, циклопентилметилкарбонил, циклогексилметилкарбонил и т.д.
«Фенил-C1-3 алкоксикарбонил» относится к группе такой структуры, где фенильная группа связывает карбонильную группу через указанную выше С1-3 алкокси. Примеры включают фенилметоксикарбонил, фенилэтоксикарбонил и фенилпропоксикарбонил.
[0063] Предпочтительные варианты соединений по настоящему изобретению представляют собой следующее. Предпочтительным вариантом W является формула -CR15R16- или формула -CR11R12CR13R14-.
[0064] Когда W представляет формулу -CR15R16-,
один предпочтительный вариант R15 представляет собой атом водорода или С1-4 алкил, причем более предпочтительный вариант R15 представляет собой атом водорода или метил и еще более предпочтительный вариант R15 представляет собой атом водорода;
один предпочтительный вариант R16 представляет собой атом водорода или С1-4 алкил, причем более предпочтительный вариант R16 представляет собой атом водорода или метил и еще более предпочтительный вариант R16 представляет собой атом водорода;
другой предпочтительный вариант R15 и R16 является таким, что R15 и R16, вместе со смежным углеродным атомом, образуют C3-8 циклоалкан, причем более предпочтительный вариант R15 и R16 является таким, что R15 и R16, вместе со смежным углеродным атомом, образуют циклобутан, циклопентан или циклогексан.
[0065] Когда W представляет формулу -CR11R12CR13R14-,
один предпочтительный вариант R11 представляет собой атом водорода или С1-4 алкил, причем более предпочтительный вариант R11 представляет собой атом водорода или метил и еще более предпочтительный вариант R11 представляет собой атом водорода;
один предпочтительный вариант R12 представляет собой атом водорода или C1-4 алкил, причем более предпочтительный вариант R12 представляет собой атом водорода или метил и еще более предпочтительный вариант R12 представляет собой атом водорода;
другой предпочтительный вариант R11 и R12 является таким, что R11 и R12, вместе со смежным углеродным атомом, образуют C3-8 циклоалкан или 4-8-членный насыщенный гетероцикл, который содержит атом кислорода, причем более предпочтительный вариант R11 и R12 является таким, что R11 и R12, вместе со смежным углеродным атомом, образуют C3-6 циклоалкан, и еще более предпочтительный вариант R11 и R12 является таким, что R11 и R12, вместе со смежным углеродным атомом, образуют циклопропан;
один предпочтительный вариант R13 представляет собой атом водорода, C1-4 алкил или галоген-C1-4 алкил, причем более предпочтительный вариант R13 представляет собой атом водорода или метил и еще более предпочтительный вариант R13 представляет собой атом водорода;
один предпочтительный вариант R14 представляет собой атом водорода или С1-4 алкил, причем более предпочтительный вариант R14 представляет собой атом водорода или метил и еще более предпочтительный вариант R14 представляет собой атом водорода;
другой предпочтительный вариант R13 и R14 является таким, что R13 и R14, вместе со смежным углеродным атомом, образуют C3-8 циклоалкан, 4-8-членный насыщенный гетероцикл, который содержит атом кислорода, или 4-8-членный насыщенный гетероцикл, который содержит атом азота (где 4-8-членный насыщенный гетероцикл, который содержит атом азота, необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из метила, бензила, фенилкарбонила и оксо), причем более предпочтительный вариант R13 и R14 является таким, что R13 и R14, вместе со смежным углеродным атомом, образуют C3-6 циклоалкан, еще более предпочтительный вариант R13 и R14 является таким, что R13 и R14, вместе со смежным углеродным атомом, образуют циклопропан, циклобутан, циклопентан или циклогексан, и особенно предпочтительный вариант R13 и R14 является таким, что R13 и R14, вместе со смежным углеродным атомом, образуют циклопропан.
[0066] Предпочтительный вариант Y представляет собой одинарную связь или C1-6 алкандиил (один из углеродных атомов в С1-6 алкандииле необязательно замещен C3-6 циклоалкан-1,1-диилом), причем более предпочтительный вариант Y представляет собой одинарную связь, метандиил, этан-1,1-диил, пропан-1,1-диил, пропан-2,2-диил, циклопропан-1,1-диил или этан-1,2-диил, причем еще более предпочтительный вариант Y представляет собой одинарную связь или метандиил и особенно предпочтительный вариант Y представляет собой метандиил.
[0067] Предпочтительные варианты R2 описаны ниже в с (1) до (4).
[0068] (1) Предпочтительный вариант R2 представляет собой С3-8 циклоалкил {С3-8 циклоалкил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из C1-6 алкила (C1-6 алкил необязательно замещен одним фенилом), фенила (фенил необязательно замещен одним галоген-C1-6 алкилом), C1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила, фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила) и пиридила (пиридил необязательно замещен одним атомом галогена)], С3-8 циклоалкокси, фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, С3-8 циклоалкила и галоген-C1-6 алкила) и пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, С3-8 циклоалкила и галоген-C1-6 алкила)}, фенил (фенил необязательно замещен от одной до трех групп, которые являются одинаковыми или различными и которые выбирают из группы α3 заместителей), инданил, изоксазолил [изоксазолил необязательно замещен одним фенилом (фенил необязательно замещен одним атомом галогена)], оксазолил (оксазолил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из C1-6 алкила и фенила), тиазоил (тиазоил необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкила, фенила и морфолино), пиридил (пиридил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из указанной выше группы α5 заместителей), пиримидинил [пиримидинил необязательно замещен одной группой, выбранной из группы, состоящей из галоген-C1-6 алкила, С3-8 циклоалкила, фенила и фенокси (фенокси необязательно замещен одним C1-6 алкилом)], пиразинил [пиразинил необязательно замещен одной группой, выбранной из группы, состоящей из С1-6 алкокси (C1-6 алкокси необязательно замещен одним С3-8 циклоалкилом) и фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и С3-8 циклоалкила)] или бензотиофенил.
[0069] (2) Более предпочтительный вариант R2 представляет собой
С3-8 циклоалкил {С3-8 циклоалкил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из фенила (фенил необязательно замещен одним галоген-C1-6 алкилом), C1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила, фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила) и пиридила (пиридил необязательно замещен одним атомом галогена)], фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, С3-8 циклоалкила и галоген-С1-6 алкила) и пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, С3-8 циклоалкила и галоген-C1-6 алкила)};
фенил (фенил необязательно замещен от одной до трех групп, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, C1-6 алкила {C1-6 алкил необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила, фенила, C1-6 алкокси [C1-6 алкокси необязательно замещен одним С3-8 циклоалкилом (С3-8 циклоалкил необязательно замещен одним C1-6 алкилом)], фенокси (фенокси необязательно замещен одним С1-6 алкилом) и пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкила и галоген-C1-6 алкила)}, галоген-C1-6 алкила, С3-8 циклоалкила (С3-8 циклоалкил необязательно замещен одним или двумя атомами галогена), фенила [фенил необязательно замещен одной-тремя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из карбокси, циано, гидрокси, сульфамоила, атома галогена, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, фенила, С1-6 алкокси, галоген-C1-6 алкокси, C1-6 алкилкарбонила, ди-C1-6 алкиламинокарбонила, C1-6 алкилсульфонила, моно-C1-6 алкиламиносульфонила (C1-6 алкил моно-C1-6 алкиламиносульфонила необязательно замещен одной гидрокси) и ди-C1-6 алкиламиносульфонила], тиенила (тиенил необязательно замещен одним C1-6 алкилом), пиразолила (пиразолил необязательно замещен одним C1-6 алкилом), изоксазолила, тиазоила (тиазоил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из гидрокси, C1-6 алкила и C1-6 алкокси), пиридила (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из карбокси, гидрокси, амино, атома галогена, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, С1-6 алкокси, галоген-C1-6 алкокси и C1-6 алкилсульфонила), пиримидинила (пиримидинил необязательно замещен одним амино), хинолила, С1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила (С3-8 циклоалкил необязательно замещен одним C1-6 алкилом), фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из гидрокси, атома галогена, C1-6 алкила, галоген-C1-6 алкила, C1-6 алкокси, галоген-C1-6 алкокси и ди-C1-6 алкиламино), пиридила (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила), оксазолила (оксазолил необязательно замещен одним или двумя C1-6 алкилами), пиразолила (пиразолил необязательно замещен одним или двумя C1-6 алкилами), тиазоила (тиазоил необязательно замещен одним C1-6 алкилом), индазолила (индазолил необязательно замещен одним C1-6 алкилом), бензотриазолила, имидазотиазоила и ди-C1-6 алкиламино], галоген-C1-6 алкокси, C2-6 алкенилокси, С3-8 циклоалкокси, фенокси (фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила, C1-6 алкокси и галоген-C1-6 алкокси), пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила и С3-8 циклоалкила), C1-6 алкилсульфанила и C1-6 алкилсульфонила);
пиридил {пиридил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, C1-6 алкила, C1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из C3-8 циклоалкила (С3-8 циклоалкил необязательно замещен одним C1-6 алкилом), фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила)], галоген-С1-6 алкокси, фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси и галоген-C1-6 алкокси), пиридила, фенокси [фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, циано, С1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, С1-6 алкокси (C1-6 алкокси необязательно замещен одним фенилом), и галоген-C1-6 алкокси], и пиридилокси (пиридилокси необязательно замещен одним C1-6 алкилом)}; или
пиразинил [пиразинил необязательно замещен одной группой, выбранной из группы, состоящей из С1-6 алкокси (C1-6 алкокси необязательно замещен одним С3-8 циклоалкилом) и фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и С3-8 циклоалкила)].
[0070] (3) Еще более предпочтительный вариант R2 представляет собой
фенил {фенил замещен одной группой, выбранной из группы, состоящей из фенила [фенил необязательно замещен от одной до трех групп, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из карбокси, циано, гидрокси, сульфамоила, атома галогена, С1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, фенила, C1-6 алкокси, галоген-C1-6 алкокси, С1-6 алкилкарбонила, ди-C1-6 алкиламинокарбонила, C1-6 алкилсульфонила, моно-C1-6 алкиламиносульфонила (C1-6 алкил моно-C1-6 алкиламиносульфонила необязательно замещен одной гидрокси) и ди-C1-6 алкиламиносульфонила], пиридила (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из карбокси, гидрокси, амино, атома галогена, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси, галоген-C1-6 алкокси и C1-6 алкилсульфонила), C1-6 алкокси [C1-6 алкокси замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила (С3-8 циклоалкил необязательно замещен одним С1-6 алкилом), фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из гидрокси, атома галогена, C1-6 алкила, галоген-C1-6 алкила, C1-6 алкокси, галоген-C1-6 алкокси и ди-C1-6 алкиламино), пиридила (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила), оксазолила (оксазолил необязательно замещен одним или двумя C1-6 алкилами), пиразолила (пиразолил необязательно замещен одним или двумя C1-6 алкилами), тиазоила (тиазоил необязательно замещен одним C1-6 алкилом), индазолила (индазолил необязательно замещен одним C1-6 алкилом), бензотриазолила, имидазотиазоила и ди-C1-6 алкиламино], галоген-C1-6 алкокси, С3-8 циклоалкокси, фенокси (фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, С1-6 алкила, галоген-C1-6 алкила, С1-6 алкокси и галоген-C1-6 алкокси), пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила и C3-8 циклоалкила), C1-6 алкилсульфанила и C1-6 алкилсульфонила и дополнительно можно замещать одним атомом галогена};
пиридил {пиридил замещен одной группой, выбранной из группы, состоящей из C1-6 алкокси [C1-6 алкокси замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила (С3-8 циклоалкил необязательно замещен одним C1-6 алкилом), фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила)], галоген-C1-6 алкокси, фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, С1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси и галоген-C1-6 алкокси), пиридила, фенокси [фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, циано, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси (C1-6 алкокси необязательно замещен одним фенилом) и галоген-C1-6 алкокси], и пиридилокси (пиридилокси необязательно замещен одним C1-6 алкилом) и дополнительно можно замещать одной группой, выбранной из группы, состоящей из атома галогена и С1-6 алкила}; или
пиразинил [пиразинил замещен одной группой, выбранной из группы, состоящей из C1-6 алкокси (C1-6 алкокси замещен одним C3-8 циклоалкилом) и фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и С3-8 циклоалкила)].
[0071] (4) Особенно предпочтительный вариант R2 представляет собой
фенил [фенил замещен одной группой, выбранной из группы, состоящей из фенокси (фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, С1-6 алкила, C1-6 алкокси и галоген-C1-6 алкокси) и пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила и С3-8 циклоалкила) и дополнительно можно замещать одним атомом галогена];
пиридил {пиридил замещен одной группой, выбранной из группы, состоящей из фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси и галоген-C1-6 алкокси), пиридила, фенокси [фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, циано, C1-6 алкила, галоген-C1-6 алкила, C3-8 циклоалкила, C1-6 алкокси (C1-6 алкокси необязательно замещен одним фенилом) и галоген-C1-6 алкокси], и пиридилокси (пиридилокси необязательно замещен одним С1-6 алкилом) и дополнительно можно замещать одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила}; или
пиразинил, замещен одним фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и С3-8 циклоалкила).
[0072] В приведенном выше варианте предпочтительные группы в группе α3 заместителей представляют собой атом галогена, C1-6 алкил {С1-6 алкил необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила, фенила, C1-6 алкокси [С1-6 алкокси необязательно замещен одним С3-8 циклоалкилом (С3-8 циклоалкил необязательно замещен одним C1-6 алкилом)], фенокси (фенокси необязательно замещен одним C1-6 алкилом) и пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкила и галоген-C1-6 алкила)}, галоген-C1-6 алкил, С3-8 циклоалкил (С3-8 циклоалкил необязательно замещен одним или двумя атомами галогена), фенил (фенил необязательно замещен от одной до трех групп, которые являются одинаковыми или различными и которые выбирают из группы α4 заместителей), тиенил (тиенил необязательно замещен одним C1-6 алкилом), пиразолил (пиразолил необязательно замещен одним C1-6 алкилом), изоксазолил, тиазоил (тиазоил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из гидрокси, C1-6 алкила и C1-6 алкокси), пиридил (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из карбокси, гидрокси, амино, атома галогена, С1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси, галоген-C1-6 алкокси и C1-6 алкилсульфонила), пиримидинил (пиримидинил необязательно замещен одной амино), хинолил, C1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из карбокси, гидрокси, карбамоила, С3-8 циклоалкила (С3-8 циклоалкил необязательно замещен одним C1-6 алкилом), фенил (фенил необязательно замещен одной группой, выбранной из группы, состоящей из гидрокси, атома галогена, C1-6 алкила, галоген-C1-6 алкила, C1-6 алкокси, галоген-C1-6 алкокси и ди-C1-6 алкиламино), пиридил (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила), оксазолил (оксазолил необязательно замещен одним или двумя C1-6 алкилами), пиразолил (пиразолил необязательно замещен одним или двумя C1-6 алкилами), тиазоил (тиазоил необязательно замещен одним C1-6 алкилом), индазолил (индазолил необязательно замещен одним C1-6 алкилом), бензотриазолил, имидазотиазоил и ди-C1-6 алкиламино], галоген-C1-6 алкокси, C2-6 алкенилокси, С3-8 циклоалкокси, фенокси (фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила, C1-6 алкокси и галоген-C1-6 алкокси), пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила и С3-8 циклоалкила), C1-6 алкилсульфанил и C1-6 алкилсульфонил;
в приведенном выше варианте предпочтительные группы в группе α4 заместителей представляют собой карбокси, циано, гидрокси, сульфамоил, атом галогена, C1-6 алкил, галоген-C1-6 алкил, С3-8 циклоалкил, фенил, C1-6 алкокси, галоген-C1-6 алкокси, C1-6 алкилкарбонил, ди-C1-6 алкиламинокарбонил, C1-6 алкилсульфонил, моно-C1-6 алкиламиносульфонил (C1-6 алкил моно-C1-6 алкиламиносульфонила необязательно замещен одним гидрокси) и ди-C1-6 алкиламиносульфонил.
[0073] Предпочтительные группы в группе α5 заместителей представляют собой атом галогена, C1-6 алкил, C1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила (С3-8 циклоалкил необязательно замещен одним C1-6 алкилом), фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила)], галоген-C1-6 алкокси, фенил (фенил необязательно замещен одной группой, выбранной из группы α6 заместителей), пиридил, фенокси [фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, циано, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, С1-6 алкокси (C1-6 алкокси необязательно замещен одним фенилом) и галоген-C1-6 алкокси], пиридилокси (пиридилокси необязательно замещен одним C1-6 алкилом) и фенилсульфанил (фенилсульфанил необязательно замещен одним атомом галогена);
в этом случае, предпочтительные группы в группе α6 заместителей представляют собой атом галогена, C1-6 алкил, галоген-C1-6 алкил, С3-8 циклоалкил, C1-6 алкокси и галоген-C1-6 алкокси.
[0074] Предпочтительный вариант Y4 представляет собой С1-3 алкандиил, причем более предпочтительный вариант Y4 представляет собой метандиил;
предпочтительный вариант R3 представляет собой атом водорода; и
предпочтительный вариант R4 представляет собой -COOH.
[0075] Один предпочтительный вариант соединений по настоящему изобретению представляет собой соединения, представленные указанной ниже формулой (I-c), или их фармацевтически приемлемые соли:
[0076]
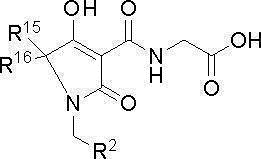
(I-c)
где предпочтительные варианты R15, R16 и R2 представляют собой то, что описано выше.
[0077] В этом случае более предпочтительным вариантом является тот, где R2 представляет собой фенил [фенил замещен одной группой, выбранной из группы, состоящей из фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома фтора, атома хлора и трифторметила), C1-6 алкокси (C1-6 алкокси замещен одним С3-8 циклоалкилом) и пиридилокси (пиридилокси необязательно замещен одним трифторметилом)].
[0078] Другой предпочтительный вариант соединений по настоящему изобретению представляют собой соединения, представленные указанной ниже формулой (I-a), или их фармацевтически приемлемые соли.
[0079]
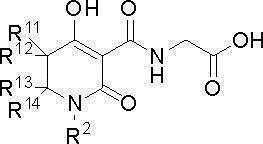
(I-a)
где предпочтительные варианты R11, R12, R13, R14 и R2 представляют собой то, что описано выше.
[0080] В этом случае более предпочтительным вариантом является тот, где R11, R12, R13 и R14 все представляют собой атом водорода и где R2 представляет собой С3-8 циклогексил [С3-8 циклоалкил замещен одним С1-6 алкилом (C1-6 алкил замещен одним фенилом)] или фенил (фенил замещен одной фенокси).
[0081] Другой предпочтительный вариант соединения по настоящему изобретению представляет собой соединения, представленные указанной ниже формулой (I-b), или их фармацевтически приемлемые соли.
[0082]

(I-b)
где предпочтительные варианты R11, R12, R13, R14 и R2 представляют собой то, что описано выше.
[0083] В этом случае более предпочтительным вариантом является тот, где R11, R12, R13 и R14 все представляют собой атом водорода и где R2 представляет собой
фенил {фенил замещен одной группой, выбранной из группы, состоящей из фенила [фенил необязательно замещен от одной до трех групп, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из карбокси, циано, гидрокси, сульфамоила, атома галогена, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, фенила, C1-6 алкокси, галоген-C1-6 алкокси, C1-6 алкилкарбонила, ди-C1-6 алкиламинокарбонила, C1-6 алкилсульфонила, моно-C1-6 алкиламиносульфонила (С1-6 алкил моно-C1-6 алкиламиносульфонила необязательно замещен одной гидрокси) и ди-C1-6 алкиламиносульфонила], пиридила (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из карбокси, гидрокси, амино, атома галогена, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси, галоген-С1-6 алкокси и С1-6 алкилсульфонила), C1-6 алкокси [C1-6 алкокси замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила (C3-8 циклоалкил необязательно замещен одним C1-6 алкилом), фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из гидрокси, атома галогена, C1-6 алкила, галоген-С1-6 алкила, C1-6 алкокси, галоген-C1-6 алкокси и ди-C1-6 алкиламино), пиридила (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила), оксазолила (оксазолил необязательно замещен одним или двумя C1-6 алкилами), пиразолила (пиразолил необязательно замещен одним или двумя С1-6 алкилами), тиазоила (тиазоил необязательно замещен одним C1-6 алкилом), индазолила (индазолил необязательно замещен одним C1-6 алкилом), бензотриазолила, имидазотиазоила и ди-C1-6 алкиламино], галоген-C1-6 алкокси, С3-8 циклоалкокси, фенокси (фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила, C1-6 алкокси и галоген-C1-6 алкокси), пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, С1-6 алкила, галоген-C1-6 алкила, и С3-8 циклоалкила), C1-6 алкилсульфанила и C1-6 алкилсульфонила и дополнительно можно замещать одним атомом галогена};
пиридил {пиридил замещен одной группой, выбранной из группы, состоящей из C1-6 алкокси [C1-6 алкокси замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила (C3-8 циклоалкил необязательно замещен одним C1-6 алкилом) и фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила)], галоген-C1-6 алкокси, фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси и галоген-C1-6 алкокси), пиридила, фенокси [фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, циано, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси (C1-6 алкокси необязательно замещен одним фенилом), и галоген-C1-6 алкокси], и пиридилокси (пиридилокси необязательно замещен одним C1-6 алкилом) и дополнительно можно замещать одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила}; или
пиразинил [пиразинил замещен одной группой, выбранной из группы, состоящей из C1-6 алкокси (C1-6 алкокси замещен одним С3-8 циклоалкилом) и фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и С3-8 циклоалкила)].
[0084] В приведенном выше варианте еще более предпочтительным вариантом является тот, где R11, R12, R13 и R14 все представляют собой атом водорода и где R2 представляет собой
фенил [фенил замещен одной группой, выбранной из группы, состоящей из фенокси (фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, C1-6 алкила, C1-6 алкокси и галоген-C1-6 алкокси) и пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила и С3-8 циклоалкила) и дополнительно можно замещать одним атомом галогена];
пиридил {пиридил замещен одной группой, выбранной из группы, состоящей из фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси и галоген-C1-6 алкокси), пиридила, фенокси [фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, циано, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси (C1-6 алкокси необязательно замещен одним фенилом) и галоген-C1-6 алкокси], и пиридилокси (пиридилокси необязательно замещен одним C1-6 алкилом) и дополнительно можно замещать одной группой, выбранной из группы, состоящей из атома галогена и С1-6 алкила}; или
пиразинил, который замещен одним фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и С3-8 циклоалкила).
[0085] И другие предпочтительные варианты соединения по настоящему изобретению представляют собой то, что описано ниже (эти варианты также применимы к указанным выше формулам (I-c), (I-a) и (I-b)).
Один предпочтительный вариант R11 представляет собой атом водорода или С1-4 алкил, причем более предпочтительный вариант R11 представляет собой атом водорода или метил.
Один предпочтительный вариант R12 представляет собой атом водорода или С1-4 алкил, причем более предпочтительный вариант R12 представляет собой атом водорода или метил.
Другой предпочтительный вариант R11 и R12 представляет собой тот, где R11 и R12, вместе со смежным углеродным атомом, образуют C3-8 циклоалкан или 4-8-членный насыщенный гетероцикл, который содержит атом кислорода, причем более предпочтительный вариант R11 и R12 представляет собой тот, где R11 и R12, вместе со смежным углеродным атомом, образуют C3-6 циклоалкан.
Один предпочтительный вариант R13 представляет собой атом водорода, С1-4 алкил или галоген-С1-4 алкил, причем более предпочтительный вариант R13 представляет собой атом водорода или метил.
Один предпочтительный вариант R14 представляет собой атом водорода или С1-4 алкил, причем более предпочтительный вариант R14 представляет собой атом водорода или метил.
И другой предпочтительный вариант R13 и R14 представляет собой тот, где R13 и R14, вместе со смежным углеродным атомом, образуют С3-8 циклоалкан или 4-8-членный насыщенный гетероцикл, который содержит атом кислорода, причем более предпочтительный вариант R13 и R14 представляет собой тот, где R13 и R14, вместе со смежным углеродным атомом, образуют C3-6 циклоалкан.
[0086] Предпочтительный вариант Y представляет собой одинарную связь или C1-6 алкандиил (один из углеродных атомов в С1-6 алкандииле необязательно замещен C3-6 циклоалкан-1,1-диилом);
более предпочтительный вариант Y представляет собой одинарную связь, метандиил, этан-1,1-диил, пропан-1,1-диил, пропан-2,2-диил, циклопропан-1,1-диил или этан-1,2-диил;
и еще более предпочтительный вариант Y представляет собой одинарную связь или метандиил.
Предпочтительный вариант R2 представляет собой С3-8 циклоалкил (С3-8 циклоалкил необязательно замещен одной группой, выбранной из группы, состоящей из фенила и бензила), фенил (фенил необязательно замещен от одной до трех групп, которые являются одинаковыми или различными и которые выбирают из группы α1 заместителей), нафтил, инданил, тетрагидронафтил, пиразолил [пиразолил замещен одним фенилом (фенил необязательно замещен одним C1-6 алкилом) и дополнительно можно замещать одним C1-6 алкилом], имидазолил [имидазолил замещен одним фенилом], изоксазолил [изоксазолил замещен одним фенилом (фенил необязательно замещен одним атомом галогена)], оксазолил (оксазолил замещен одним фенилом и дополнительно можно замещать одним C1-6 алкилом), тиазоил (тиазоил замещен одним фенилом), пиридил [пиридил замещен одной группой, выбранной из группы, состоящей из фенила, фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, циано, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси и галоген-C1-6 алкокси) и фенилсульфанила (фенилсульфанил необязательно замещен одним атомом галогена)], пиримидинил (пиримидинил замещен одной группой, выбранной из группы, состоящей из циклогексила и фенила), бензотиофенил, хинолил или метилендиоксифенил (метилендиоксифенил необязательно замещен одним или двумя атомами фтора);
в этом случае предпочтительные группы в группе α1 заместителей представляют собой
атом галогена, C1-6 алкил {C1-6 алкил необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила, фенила и C1-6 алкокси [C1-6 алкокси необязательно замещен одним С3-8 циклоалкилом (С3-8 циклоалкил необязательно замещен одним C1-6 алкилом)]}, галоген-C1-6 алкил, С3-8 циклоалкил, фенил (фенил необязательно замещен от одной до трех групп, которые являются одинаковыми или различными и которые выбирают из группы α2 заместителей), тиенил, пиразолил (пиразолил необязательно замещен одним C1-6 алкилом), изоксазолил, тиазоил (тиазоил необязательно замещен одним или двумя C1-6 алкилами), пиридил (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из С1-6 алкила, галоген-C1-6 алкила, C1-6 алкокси и галоген-C1-6 алкокси), хинолил, C1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из C3-8 циклоалкила и фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила)], галоген-C1-6 алкокси, C2-6 алкенилокси, С3-8 циклоалкокси, фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила, C1-6 алкокси и галоген-C1-6 алкокси), пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и галоген-C1-6 алкила) и C1-6 алкилсульфанил;
в этом случае предпочтительные группы в группе α2 заместителей представляют собой атом галогена, циано, гидрокси, C1-6 алкил, галоген-C1-6 алкил, фенил, C1-6 алкокси, галоген-C1-6 алкокси, C1-6 алкилкарбонил и ди-C1-6 алкиламиносульфонил.
[0087] Соединения по настоящему изобретению представляют собой те, которые имеют частично насыщенные, азотсодержащие гетероциклические структуры, и они могут быть в форме их фармацевтически приемлемых солей (оба типа далее в настоящем документе обозначают как «соединения по настоящему изобретению», в зависимости от ситуации).
[0088] Примеры фармацевтически приемлемых солей включают кислотно-аддитивные соли, включая соли неорганических кислот, такие как гидрохлорид, гидробромид, гидройодид, фосфат, сульфат и нитрат; соли сульфоновых кислот, такие как метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат и трифторметансульфонат; соли органических кислот, такие как оксалат, тартрат, цитрат, малеат, сукцинат, ацетат, трифторацетат, бензоат, манделат, аскорбат, лактат, глюконат и малат; соли аминокислот, такие как соль глицина, соль лизина, соль аргинина, соль орнитина, глутамат и аспартат; неорганические соли, такие как соль лития, соль натрия, соль калия, соль кальция и соль магния; и соли с органическими основаниями, такие как соль аммония, соль триэтиламина, соль диизопропиламина и соль циклогексиламина. Термин «соль(и)», как используют в настоящем документе, охватывает гидратную соль(и).
[0089] Соединения по настоящему изобретению имеют асимметричный центр или асимметричные центры в определенных случаях, в которых они дают начало ряду оптических изомеров. Следовательно, соединения по настоящему изобретению могут существовать в виде отдельных оптических изомеров (R) и (S) или в качестве рацемата или (RS) смеси. В случае соединений, имеющих два или более асимметричных центра, они дают начало диастереомера из-за соответствующей им оптической изомерии. Соединения по настоящему изобретению охватывают смеси, которые содержат изомеры всех этих типов в каких-либо пропорциях. Например, диастереомеры можно разделять способами, хорошо известными специалистам в данной области, например, фракционная кристаллизация, а оптически активные формы можно получать способами, которые в органической химии хорошо известны для этой цели. Кроме того, соединения по настоящему изобретению иногда дают начало геометрическим изомерам, таким как цис- и транс-формы. Кроме того, соединения по настоящему изобретению могут иметь таутомерию, что дает начало ряду таутомеров. Соединения по настоящему изобретению охватывают указанные выше изомеры, а также смеси, содержащие эти изомеры в каких-либо пропорциях.
Кроме того, если соединения по настоящему изобретению или их соли образуют гидраты или сольваты, они также включены в объем соединений по настоящему изобретению или их солей.
[0090] Соединения по настоящему изобретению можно вводить или независимо или вместе с фармацевтически приемлемыми носителями или разбавителями.
[0091] Для того, чтобы использовать соединения по настоящему изобретению в качестве лекарственных средств, они могут принимать любые формы, т.е. в виде твердой композиции, жидкой композиции или других композиций, причем оптимальные формы выбирают в зависимости от потребностей. Лекарственные изобретения по настоящему изобретению можно получать посредством включения фармацевтически приемлемых носителей для соединений по настоящему изобретению. Говоря конкретно, можно добавлять широко используемые эксципиенты, наполнители, связующие средства, разрыхлители, покрывающие средства, сахаросодержащие покрывающие средства, модификаторы pH, солюбилизаторы или водные или неводные растворители и т.д., и можно применять широко используемые способы для фармацевтических составов, чтобы получать таблетки, пилюли, капсулы, гранулы, присыпки, порошки, жидкости, эмульсии, суспензии, инъекции и т.д. Примеры эксципиентов и наполнителей включают лактозу, стеарат магния, крахмал, тальк, желатин, агар, пектин, аравийскую камедь, оливковое масло, сезамовое масло, кокосовое масло, этиленгликоль и какие-либо другие вещества, широко используемые в качестве эксципиентов или наполнителей.
[0092] Кроме того, соединения по настоящему изобретению можно формулировать в фармацевтические препараты посредством формирования соединения включения с α, β или γ-циклодекстрином или метилированным циклодекстрином и т.д.
[0093] Если соединения по настоящему изобретению используют в качестве ингибиторов PHD2 и т.п., по существу, их можно вводить или перорально или не перорально. Альтернативно, соединения по настоящему изобретению можно вводить или перорально или не перорально в качестве средств, которые содержат их в качестве активного ингредиента. Пример не перорального введения включает внутривенное, трансназальное, чрескожное, подкожное, внутримышечное и сублингвальное введение.
[0094] Доза соединений по настоящему изобретению варьирует в зависимости от субъекта введения, пути введения, заболевания, подлежащего лечению, симптомов и т.п.; например, если они подлежат введению перорально взрослому пациенту, имеющему анемию, одна доза типично находится в диапазоне от 0,1 мг до 1000 мг, предпочтительно от 1 мг до 200 мг и эту дозу по желанию вводят от одного до трех раз в сутки, или один раз каждые двое или трое суток.
Следует отметить, что соединения по настоящему изобретению обладают свойствами, желаемыми для фармацевтических препаратов. Свойством, которое можно привести в качестве примера, является то, которое позволяет избегать чрезмерного образования эритропоэтина.
[0095] Ингибирующий PHD2 эффект соединений по настоящему изобретению можно оценивать известными способами, такими как способы, описанные в настоящем документе в Тестах.
[0096] Далее в настоящем документе, способы получения соединений по настоящему изобретению описаны подробно, но без конкретного ограничения следующими иллюстрациями. Кроме того, растворители, подлежащие использованию в реакциях, могут относиться к любым типам, которые не будут препятствовать соответствующим реакциям, и они конкретно не ограничены следующим описанием.
[0097] Далее описаны способы получения соединений, представленных формулой (I) или (I'), далее в настоящем документе иногда обозначаемых как соединение (I) или соединение (I').
[0098] Соединение (I) или (I') можно получать способами, известными per se, например, способами с 1 до 10 или их модификаций. Также следует отметить, что в соответствующих способах получения, описанных ниже, начальные соединения можно использовать в форме солей, и примеры таких солей включают указанные выше «фармацевтически приемлемые соли». Кроме того, целевые соединения также можно получать в форме солей, и примеры таких солей включают указанные выше «фармацевтически приемлемые соли».
[0099] Кроме того, получаемые целевые соединения можно использовать на следующей стадии в состоянии, еще подлежащем очистке.
[0100] Соединение (I-9), которое относится к соединению (I) или (I') по настоящему изобретению, можно получать, например, следующим способом получения 1 или его модификаций.
Способ получения 1:
[0101]
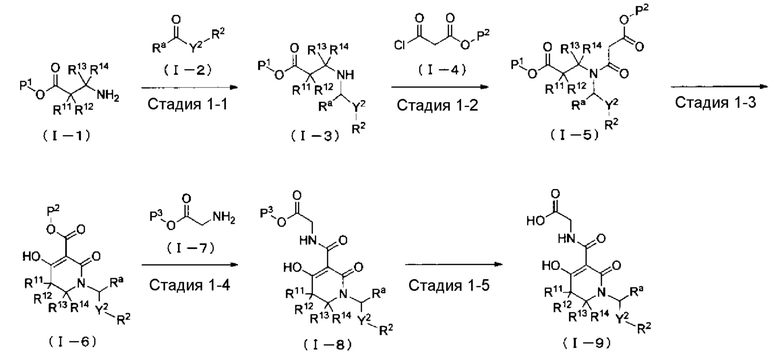
[где R11, R12, R13, R14 и R2 имеют те же значения, которые определены выше; Ra представляет атом водорода, метил или этил; Y2 представляет одинарную связь или С1-5 алкандиил; P1, P2 и P3 представляют обыкновенные защитные группы для карбоновых кислот, как показано на примере с помощью групп, описанных в Protective Groups in Organic Synthesis (3rd Edition, 1999, edited by P. G. M. Wuts and T. W. Greene) и т.д., и конкретными примерами являются C1-6 алкил, бензил, 4-метоксибензил, 2-(триметилсилил)этил и т.д.]
[0102] [Стадия 1-1]
Эта стадия представляет собой способ получения соединения (I-3) посредством осуществления реакции восстановительного аминирования с использованием соединения (I-1) и соединения (I-2).
Восстанавливающие агенты, которые можно использовать в реакции, включают триацетоксиборогидрид натрия, борогидрид натрия, цианоборогидрид натрия, комплекс боран-2-пиколина и т.д. Количество восстанавливающих агентов, подлежащее использованию, находится в диапазоне от одного до трех эквивалентов, предпочтительно от одного до двух эквивалентов, по отношению к одному эквиваленту соединения (I-1).
Растворители, которые можно использовать в реакции, включают, например, спиртовые растворители, такие как метанол и этанол; простые растворители на основе эфиров, такие как тетрагидрофуран и диоксан; растворители на основе галогенированных углеводородов, такие как метиленхлорид и хлороформ; растворители на основе ароматических углеводородов, такие как толуол и ксилол; и апротонные полярные растворители, такие как N,N-диметилформамид.
Реакцию, представляющую интерес, типично можно осуществлять между 0°C и температурой нагревания с обратным холодильником.
Таким образом полученное соединение (I-3) можно выделять и очищать известными средствами разделения/очистки, таких как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0103] [Стадия 1-2]
Эта стадия представляет собой способ получения соединения (I-5) посредством проведения реакции соединения (I-3) с соединением (I-4) в присутствии основания.
Основания, которые можно использовать в реакции, типично включают, например, триэтиламин, пиридин и т.д. Количество оснований, подлежащее использованию, находится в диапазоне от одного до пяти эквивалентов, предпочтительно, от одного до трех эквивалентов, по отношению к одному эквиваленту соединения (I-3).
Растворители, которые можно использовать в реакции, включают, например, простые растворители на основе эфиров, такие как тетрагидрофуран и диоксан; растворители на основе галогенированных углеводородов, такие как метиленхлорид и хлороформ; растворители на основе ароматических углеводородов, такие как толуол и ксилол; и апротонные полярные растворители, такие как этилацетат и N,N-диметилформамид.
Реакцию, представляющую интерес, типично можно осуществлять между 0°C и комнатной температурой.
Таким образом полученное соединение (I-5) можно выделять и очищать известными средствами разделения/очистки, таких как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0104] [Стадия 1-3]
Эта стадия представляет собой способ получения соединения (I-6) посредством замыкания в цикл соединения (I-5) в присутствии основания.
Основания, которые можно использовать в реакции, типично включают, например, этоксид натрия, метоксид натрия, гидрид натрия, трет-бутоксид калия, карбонат калия, карбонат цезия и т.д. Количество оснований, подлежащее использованию, типично находится в диапазоне от одного до пяти эквивалентов, предпочтительно от двух до трех эквивалентов, по отношению к одному эквиваленту соединения (I-5).
Растворители, которые можно использовать в реакции, включают, например, спиртовые растворители, такие как метанол, этанол и пропанол; простые растворители на основе эфиров, такие как тетрагидрофуран и диоксан; растворители на основе галогенированных углеводородов, такие как метиленхлорид и хлороформ; растворители на основе ароматических углеводородов, такие как толуол и ксилол; и апротонные полярные растворители, такие как этилацетат и N,N-диметилформамид.
Реакцию, представляющую интерес, типично можно осуществлять между 0°C и температурой нагревания с обратным холодильником.
Таким образом полученное соединение (I-6) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0105] [Стадия 1-4]
Эта стадия представляет собой способ получения соединения (I-8) из соединения (I-6) и соединения (I-7).
Растворители, которые можно использовать в реакции, включают, например, простые растворители на основе эфиров, такие как 1,2-диметоксиэтан, тетрагидрофуран и диоксан; растворители на основе галогенированных углеводородов, такие как метиленхлорид и хлороформ; растворители на основе ароматических углеводородов, такие как толуол и ксилол; и апротонные полярные растворители, такие как N,N-диметилформамид.
В реакции, представляющей интерес, можно использовать основание в качестве добавки. Примеры основания включают триэтиламин, N,N-диизопропилэтиламин и т.д.
Эту реакцию типично можно осуществлять между комнатной температурой и температурой нагревания с обратным холодильником.
Таким образом полученное соединение (I-8) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0106] [Стадия 1-5]
Эта стадия представляет собой способ получения соединения (I-9) посредством снятия защитных групп с соединения (I-8).
Эту реакцию можно осуществлять, например, способом, описанным в Protective Groups in Organic Synthesis (3rd Edition, 1999, edited by P.G.M. Wuts and T.W. Greene) и т.д., или его модификаций. В частности, если P3 представляет собой трет-бутил, 4-метоксибензил или триметилсилил, соединение (I-9) можно получать с использованием неорганической кислоты, такой как соляная кислота, или органической кислоты, такой как уксусная кислота или трифторуксусная кислота, в растворителе, таком как растворитель на основе простых эфиров, например, тетрагидрофурана или диоксана, растворитель на основе галогенированных углеводородов, например метиленхлорид или хлороформ, или растворитель на основе ароматических углеводородов, например, толуол или ксилол. Если P3 представляет собой бензил или 4-метоксибензил, соединение (I-9) также можно получать посредством гидрогенолиза в растворителе, таком как спиртовой растворитель, например, метанол или этанол, растворитель на основе простых эфиров, например, тетрагидрофуран или диоксан, растворитель на основе галогенированных углеводородов, например, метиленхлорид или хлороформ, или растворитель на основе ароматических углеводородов, например, толуол или ксилол, в присутствии катализатора, такого как палладий-углерод. Если P3 представляет собой 2-(триметилсилил)этил, триметилсилил или трет-бутилдиметилсилил, также возможно получать соединение (I-9) посредством обработки с использованием фторида калия, фторида тетрабутиламмония и т.д. Если P3 представляет собой метил, этил или н-пропил, растворитель, подлежащий использованию, может представлять собой спиртовой растворитель, такой как метанол или этанол, растворитель на основе простых эфиров, такой как тетрагидрофуран или диоксан, растворитель на основе ароматических углеводородов, такой как толуол или ксилол, апротонный полярный растворитель, такой как ацетонитрил или N,N-диметилформамид, вода или тому подобное; эти растворители можно использовать в смеси в подходящих пропорциях, и обработка с использованием основания, такого как гидроксид лития, гидроксид натрия, гидроксид калия, карбонат калия, карбонат цезия и т.д., также позволяет получать соединение (I-9).
Реакцию, представляющую интерес, типично можно осуществлять между комнатной температурой и температурой нагревания с обратным холодильником.
Таким образом полученное соединение (I-9) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0107] Соединение (II-6), которое относится к соединению (I) или (I') по настоящему изобретению, можно получать, например, следующим способом получения 2 или его модификаций.
Способ получения 2:
[0108]

[где R11, R12, R13, R14, P1, P2 и P3 имеют те же значения, которые определены выше; Rb представляет атом водорода, фенил или бензил; n представляет целое число от 0 до 5].
[0109] [Стадия 2-1]
Эта стадия представляет собой способ получения соединения (II-2) из соединения (I-1) и соединения (II-1).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-1 способа получения 1.
Таким образом полученное соединение (II-2) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0110] [Стадия 2-2]
Эта стадия представляет собой способ получения соединения (II-3) из соединения (II-2) и соединения (I-4).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-2 способа получения 1.
Таким образом полученное соединение (II-3) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0111] [Стадия 2-3]
Эта стадия представляет собой способ получения соединения (II-4) из соединения (II-3).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-3 способа получения 1.
Таким образом полученное соединение (II-4) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0112] [Стадия 2-4]
Эта стадия представляет собой способ получения соединения (И-5) из соединения (II-4) и соединения (I-7).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-4 способа получения 1.
Таким образом полученное соединение (II-5) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0113] [Стадия 2-5]
Эта стадия представляет собой способ получения соединения (II-6) из соединения (II-5). Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-5 способа получения 1.
Таким образом полученное соединение (II-5) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0114] Соединение (III-6), которое относится к соединению (I) или (I') по настоящему изобретению, можно получать, например, следующим способом получения 3 или его модификаций.
Способ получения 3:
[0115]
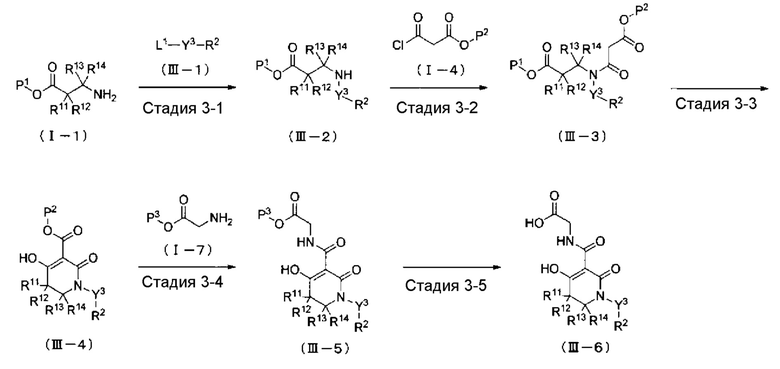
[где R11, R12, R13, R14, R2, P1, P2 и P3 имеют те же значения, которые определены выше; L1 представляет обыкновенную уходящую группу, например атом хлора, атом брома, атом йода, метансульфонилокси, п-толуолсульфонилокси и т.д.; Y3 представляет C1-6 алкандиил].
[0116] [Стадия 3-1]
Эта стадия представляет собой способ получения соединения (III-2) посредством проведения реакции соединения (I-1) с соединением (III-1) в присутствии основания.
Основания, которые можно использовать в реакции, включают, например, гидроксид натрия, трет-бутоксид калия, триэтиламин, пиридин, карбонат цезия, карбонат калия, карбонат натрия, бикарбонат натрия и т.д. Количество оснований, подлежащее использованию, находится в диапазоне от одного до трех эквивалентов по отношению к одному эквиваленту соединения (I-1).
Растворители, которые можно использовать в реакции, включают, например, спиртовые растворители, такие как метанол и этанол; простые растворители на основе эфиров, такие как тетрагидрофуран и диоксан; растворители на основе галогенированных углеводородов, такие как метиленхлорид и хлороформ; растворители на основе ароматических углеводородов, такие как толуол и ксилол; и апротонные полярные растворители, такие как ацетонитрил и N,N-диметилформамид.
Реакцию, представляющую интерес, типично можно осуществлять между 0°C и температурой нагревания с обратным холодильником.
Таким образом полученное соединение (III-2) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0117] [Стадия 3-2]
Эта стадия представляет собой способ получения соединения (III-3) из соединения (III-2) и соединения (I-4).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-2 способа получения 1.
Таким образом полученное соединение (III-3) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0118] [Стадия 3-3]
Эта стадия представляет собой способ получения соединения (III-4) из соединения (III-3).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-3 способа получения 1.
Таким образом полученное соединение (III-4) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0119] [Стадия 3-4]
Эта стадия представляет собой способ получения соединения (III-5) из соединения (III-4) и соединения (I-7).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-4 способа получения 1.
Таким образом полученное соединение (III-5) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0120] [Стадия 3-5]
Эта стадия представляет собой способ получения соединения (III-6) из соединения (III-5).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-5 способа получения 1.
Таким образом полученное соединение (III-6) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0121] Соединение (IV-7), которое относится к соединению (I) или (I') по настоящему изобретению, можно получать, например, следующим способом получения 4 или его модификаций.
Способ получения 4:
[0122]
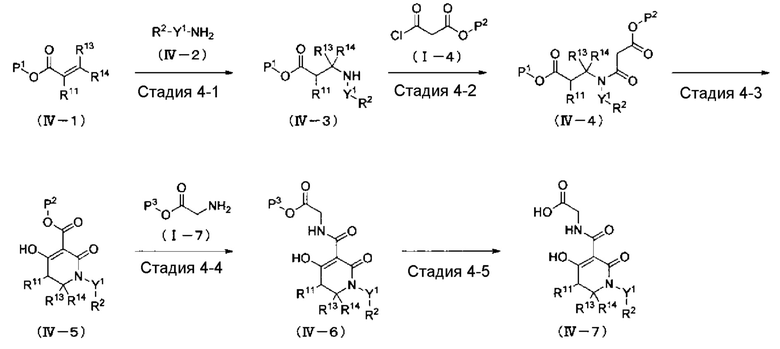
[где R11, R13, R14, R2, P1, P2 и P3 имеют те же значения, которые определены выше; Y1 представляет одинарную связь или C1-6 алкандиил (один из углеродных атомов в C1-6 алкандииле необязательно замещен C3-6 циклоалкан-1,1-диилом)].
[0123] [Стадия 4-1]
Эта стадия представляет собой способ получения соединения (IV-3) посредством проведения реакции соединения (IV-1) с соединением (IV-2).
Растворители, которые можно использовать в реакции, включают спиртовые растворители, такие как метанол и этанол; простые растворители на основе эфиров, такие как тетрагидрофуран и диоксан; растворители на основе галогенированных углеводородов, такие как метиленхлорид и хлороформ; растворители на основе ароматических углеводородов, такие как толуол и ксилол; апротонные полярные растворители, такие как ацетонитрил и N,N-диметилформамид; воду и т.п.; эти растворители можно использовать в смеси в подходящих пропорциях.
В реакции, представляющей интерес, основание или кислоту можно использовать в качестве добавки. Примеры основания включают гидрид натрия, трет-бутоксид калия, триэтиламин, пиридин, карбонат цезия, карбонат калия, карбонат натрия, бикарбонат натрия и т.д. Примеры кислоты включают уксусную кислоту, соляную кислоту, серную кислоту и т.д.
Реакцию, представляющую интерес, типично можно осуществлять между 0°C и температурой нагревания с обратным холодильником; ее даже можно осуществлять при облучении микроволнами.
Таким образом полученное соединение (IV-3) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0124] [Стадия 4-2]
Эта стадия представляет собой способ получения соединения (IV-4) из соединения (IV-3) и соединения (I-4).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-2 способа получения 1.
Таким образом полученное соединение (IV-4) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0125] [Стадия 4-3]
Эта стадия представляет собой способ получения соединения (IV-5) из соединения (IV-4). Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-3 способа получения 1.
Таким образом, полученное соединение (IV-5) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0126] [Стадия 4-4]
Эта стадия представляет собой способ получения соединения (IV-6) из соединения (IV-5) и соединения (I-7).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-4 способа получения 1.
Таким образом, полученное соединение (IV-6) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0127] [Стадия 4-5]
Эта стадия представляет собой способ получения соединения (IV-7) из соединения (IV-6).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-5 способа получения 1.
Таким образом, полученное соединение (IV-7) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0128] Соединение (V-4), которое относится к соединению (I) или (I') по настоящему изобретению, можно получать, например, следующим способом получения 5 или его модификаций.
Способ получения 5:
[0129]
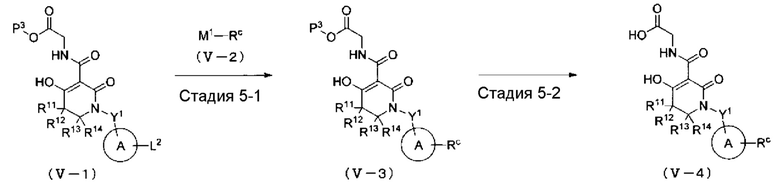
[где R11, R12, R13, R14, Y1 и P3 имеют те же значения, которые определены выше; кольцо A представляет фенил (фенил необязательно замещен от одной до четырех групп, которые являются одинаковыми или различающимися и выбирают из группы, состоящей из атома галогена, С1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси, галоген-C1-6 алкокси и С3-8 циклоалкокси) или пиридил (пиридил необязательно замещен от одной до трех групп, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, C1-6 алкила, C1-6 алкокси и галоген-C1-6 алкокси); L2 представляет обыкновенную уходящую группу, например, атом фтора, атом хлора, атом брома, атом йода, трифторметансульфонилокси и т.д.; M1-Rc представляет металлсодержащее металлорганическое соединение, где M1 представляет бороновую кислоту, сложный эфир бороновой кислоты, бромид магния, хлорид магния и т.д., и Rc представляет C1-6 алкил, галоген-C1-6 алкил, С3-8 циклоалкил, фенил (фенил необязательно замещен от одной до трех групп, которые являются одинаковыми или различными и которые выбирают из группы α2 заместителей), тиенил, пиразолил (пиразолил необязательно замещен одним C1-6 алкилом), изоксазолил, тиазоил (тиазоил необязательно замещен одним или двумя C1-6 алкилами), пиридил (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкила, галоген-C1-6 алкила, C1-6 алкокси и галоген-C1-6 алкокси), хинолил и т.д.; соединение (V-1) можно получать посредством реализации процедур стадий в способах получения с 1 до 4].
[0130] [Стадия 5-1]
Эта стадия представляет собой способ получения соединения (V-3) посредством осуществления реакции сочетания с использованием соединения (V-1) и металлорганического соединения (V-2).
Если M1 представляет собой бороновую кислоту или сложный эфир бороновой кислоты, реакция, представляющая интерес, представляет собой так называемую реакцию сочетания Сузуки-Мияуры, и ее можно осуществлять с помощью документированных способов (Tetrahedron Letters, 1979, 20, 3437-3440; Chemical reviews, 1995, 95, 2457-2483) или их модификаций в присутствии палладиевого катализатора и основания. Если M1 представляет собой реактив Гриньяра, такой как бромид магния или хлорид магния, соединение (V-3) можно получать в присутствии палладиевого катализатора.
В этом случае металлический реактив, такой как хлорид индия, можно добавлять в зависимости от ситуации. Количество соединения (V-2), подлежащее использованию на рассматриваемой стадии, находится в диапазоне от одного до пяти эквивалентов, предпочтительно от одного до трех эквивалентов, по отношению к одному эквиваленту соединения (V-1).
Палладиевые катализаторы, которые можно использовать в реакции сочетания, включают те, которые известны специалистам в данной области, как показано на примере с помощью тетракис(трифенилфосфин)палладия(0), бис(дибензилиденацетон)палладия(0), бис(три-трет-бутилфосфин)палладия(0), дихлорида бис(трифенилфосфин)палладия(II), ацетата бис(трифенилфосфин)палладия(II) и комплекса дихлорметана дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия(II) (1:1) и т.д. При желании, для реакции можно использовать палладиевый(0) катализатор, как образуется в системе с использованием ацетата палладия(II) и фосфинового реактива, такого как трифенилфосфин или три(2-метилфенил)фосфин, в присутствии основания. Количество палладиевого катализатора, подлежащее использованию, типично варьирует от 0,01 до 0,5 эквивалента, предпочтительно от 0,05 до 0,3 эквивалента, по отношению к одному эквиваленту соединения (V-1).
Основания, которые можно использовать, включают карбонат калия, карбонат цезия, карбонат натрия, бикарбонат натрия, фосфат трикалия, фторид калия, фторид цезия, триэтиламин и т.д. Количество оснований, подлежащее использованию, типично варьирует от одного до пяти эквивалентов, предпочтительно от одного до трех эквивалентов, по отношению к одному эквиваленту соединения (V-1).
Растворители, которые можно использовать в реакции, включают спиртовые растворители, такие как метанол, этанол и этиленгликоль; простые растворители на основе эфиров, такие как тетрагидрофуран, диоксан и 1,2-диметоксиэтан; растворители на основе ароматических углеводородов, такие как толуол и ксилол; апротонные полярные растворители, такие как ацетонитрил и N,N-диметилформамид; воду и т.п.; эти растворители можно использовать в смеси в подходящих пропорциях.
В реакции, представляющей интерес, соединение меди можно использовать в качестве добавки. Примеры соединения меди включают йодид меди(I), ацетат меди(II) и т.д.
Реакцию, представляющую интерес, типично можно осуществлять между комнатной температурой и 180°C; ее даже можно осуществлять при облучении микроволнами.
Таким образом полученное соединение (V-3) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0131] [Стадия 5-2]
Эта стадия представляет собой способ получения соединения (V-4) из соединения (V-3).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-5 способа получения 1.
Таким образом, полученное соединение (V-4) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0132] Соединение (VI-2), которое относится к соединению (I) или (I') по настоящему изобретению, можно получать, например, следующим способом получения 6 или его модификаций.
Способ получения 6:
[0133]
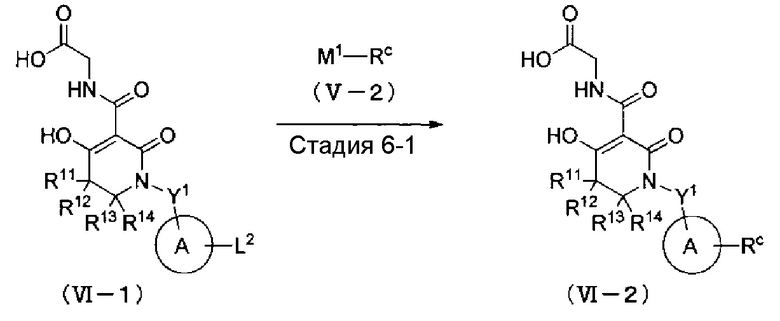
[где R11, R12, R13, R14, Y1, кольцо A, L2, M1 и Rc имеют те же значения, которые определены выше, и соединение (VI-1) можно получать посредством реализации процедур стадий в способах получения с 1 до 4].
[0134] [Стадия 6-1]
Эта стадия представляет собой способ получения соединения (VI-2) из соединения (VI-1) и металлорганического соединения (V-2).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 5-1 способа получения 5.
Таким образом, полученное соединение (VI-2) можно выделять и очищать известных средств разделения/очистки, таких как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0135] Соединение (VII-6), которое относится к соединению (I') по настоящему изобретению, можно получать, например, следующим способом получения 7 или его модификаций.
Способ получения 7:
[0136]

[где R15, R16, Ra, R2, Y2, P1, P2 и P3 имеют те же значения, которые определены выше].
[0137] [Стадия 7-1]
Эта стадия представляет собой способ получения соединения (VII-2) из соединения (VII-1) и соединения (I-2).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-1 способа получения 1.
Таким образом, полученное соединение (VII-2) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0138] [Стадия 7-2]
Эта стадия представляет собой способ получения соединения (VII-3) из соединения (VII-2) и соединения (I-4).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-2 способа получения 1.
Таким образом, полученное соединение (VII-3) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0139] [Стадия 7-3]
Эта стадия представляет собой способ получения соединения (VII-4) из соединения (VII-3).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-3 способа получения 1.
Таким образом, полученное соединение (VII-4) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0140] [Стадия 7-4]
Эта стадия представляет собой способ получения соединения (VII-5) из соединения (VII-4) и соединения (I-7).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-4 способа получения 1.
Таким образом, полученное соединение (VII-5) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0141] [Стадия 7-5]
Эта стадия представляет собой способ получения соединения (VII-6) из соединения (VII-5). Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-5 способа получения 1.
Таким образом, полученное соединение (VII-6) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0142] Соединение (VIII-4), которое относится к соединению (I') по настоящему изобретению, можно получать, например, следующим способом получения 8 или его модификаций.
Способ получения 8:
[0143]

[где R15, R16, Ra, R2, Y2, P1 и P3 имеют те же значения, которые определены выше].
[0144] [Стадия 8-1]
Эта стадия представляет собой способ получения соединения (VIII-2) посредством осуществления реакции конденсации с использованием соединения (VII-2) и соединения (VIII-1).
Реактивы, которые можно использовать в реакции конденсации, включают комбинацию 1-(3-диметиламинопропил)-3-этилкарбодиимида и 1-гидроксибензотриазола, а также 1,1'-карбонилдиимидазола, ангидрида пропилфосфоновой кислоты (циклический тример) и т.д. Количество конденсирующего агента, подлежащее использованию, варьирует от одного до трех эквивалентов, предпочтительно от одного до двух эквивалентов, по отношению к одному эквиваленту соединения (VII-2).
В реакции, представляющей интерес, основание можно использовать в качестве добавки. Примеры основания включают триэтиламин и так далее.
Растворители, которые можно использовать в реакции, включают, например, спиртовые растворители, такие как метанол и этанол; простые растворители на основе эфиров, такие как тетрагидрофуран и диоксан; растворители на основе галогенированных углеводородов, такие как метиленхлорид и хлороформ; растворители на основе ароматических углеводородов, такие как толуол и ксилол; и апротонные полярные растворители, такие как N,N-диметилформамид.
Реакцию, представляющую интерес, типично можно осуществлять между 0°C и температурой нагревания с обратным холодильником.
Таким образом, полученное соединение (VIII-2) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0145] [Стадия 8-2]
Эта стадия представляет собой способ получения соединения (VIII-3) из соединения (VIII-2).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-3 способа получения 1.
Таким образом, полученное соединение (VIII-3) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0146] [Стадия 8-3]
Эта стадия представляет собой способ получения соединения (VIII-4) из соединения (VIII-3).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-5 способа получения 1.
Таким образом, полученное соединение (VIII-4) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0147] Соединение (IX-4), которое относится к соединению (I) или (I') по настоящему изобретению, можно получать, например, следующим способом получения 9 или его модификаций.
Способ получения 9:
[0148]
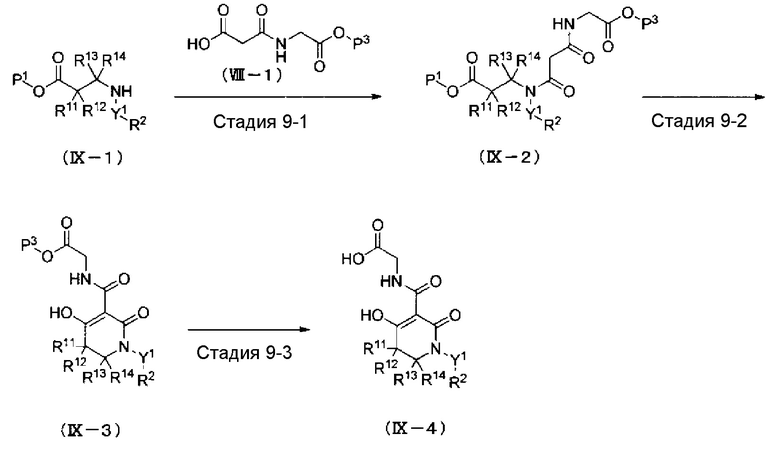
[где R11, R12, R13, R14, R2, Y1, P1 и P3 имеют те же значения, которые определены выше, и соединение (IX-1) можно получать посредством реализации процедур из способов получения с 1 до 4].
[0149] [Стадия 9-1]
Эта стадия представляет собой способ получения соединения (IX-2) посредством осуществления реакции конденсации с использованием соединения (IX-1) и соединения (VIII-1).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 8-1 способа получения 8.
Таким образом, полученное соединение (IX-2) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0150] [Стадия 9-2]
Эта стадия представляет собой способ получения соединения (IX-3) из соединения (IX-2).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-3 способа получения 1.
Таким образом, полученное соединение (IX-3) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0151] [Стадия 9-3]
Эта стадия представляет собой способ получения соединения (IX-4) из соединения (IX-3).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-5 способа получения 1.
Таким образом, полученное соединение (IX-4) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0152] Соединение (X-4), которое относится к соединению (I') по настоящему изобретению, можно получать, например, следующим способом получения 10 или его модификаций.
Способ получения 10:
[0153]
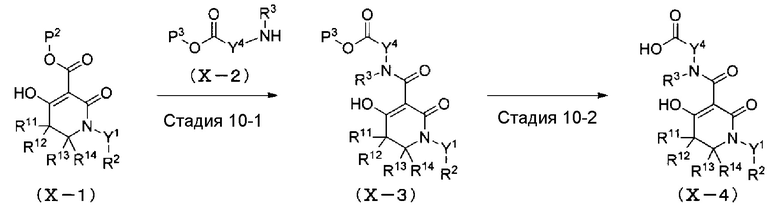
[где R11, R12, R13, R14, R2, R3, Y1, Y4, P2 и P3 имеют те же значения, которые определены выше, и соединение (X-1) можно получать посредством реализации процедур способов получения с 1 до 4].
[0154] [Стадия 10-1]
Эта стадия представляет собой способ получения соединения (X-3) из соединения (X-1) и соединения (X-2).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-4 способа получения 1.
Таким образом, полученное соединение (X-3) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
[0155] [Стадия 10-2]
Эта стадия представляет собой способ получения соединения (X-4) из соединения (X-3).
Реакцию, представляющую интерес, можно осуществлять посредством модификации способа, описанного на стадии 1-5 способа получения 1.
Таким образом, полученное соединение (X-4) можно выделять и очищать известными средствами разделения/очистки, такими как концентрирование, концентрирование при пониженном давлении, повторное осаждение, экстрагирование с растворителем, кристаллизация, хроматография и т.д.
ПРИМЕРЫ
[0156] Настоящее изобретение описано более подробно следующими ссылочными примерами, демонстрационными примерами и тестами, но следует понимать, что они никаким образом не предназначены для того, чтобы ограничивать настоящее изобретение, и их можно изменять в такой мере, которая не будет отступать от объема настоящего изобретения.
[0157] Сокращения, используемые в настоящем документе, имеют следующие значения.
с: синглет
д: дублет
т: триплет
к: квартет
кв: квинтет
септ: септет
дд: двойной дублет
дт: двойной триплет
тд: триплет дублетов
м: мультиплет
ушис: уширенный
J: постоянная взаимодействия
Гц: Герц
ХЛОРОФОРМ-d: дейтерированный хлороформ
ДМСО-d6: дейтерированный диметилсульфоксид
МЕТАНОЛ-d4: дейтерированный метанол
[0158] Спектрометрию протонного ядерного магнитного резонанса (1H-ЯМР) реализовали с помощью следующих ЯМР спектрометров с преобразованием Фурье.
200 МГц: Gemini 2000 (Agilent Technologies)
300 МГц: Inova 300 (Agilent Technologies)
600 МГц: JNM-ECA 600 (JEOL)
Для анализа использовали ACD/SpecManager версии 12.01 (название продукта), ACD/Spectrus Processor™ и т.д. Очень слабые пики, полученные от протонов гидроксигруппы, аминогруппы и других фрагментов, не приведены.
[0159] Масс-спектрометрию (MS) осуществляли с помощью следующих спектрометров.
Platform LC (Waters)
LCMS-2010EV (Shimadzu)
LCMS-IT-TOF (Shimadzu)
GCT (Micromass)
Agilent 6130 (Agilent)
LCQ Deca XP (ThermoFisher Scientific)
Использовали способ ионизации ESI (ионизация электрораспылением), EI (ионизация электронами) или двойную ионизации с использованием ESI и APCI (химическая ионизация при атмосферном давлении). Приведены полученные данные. Обычно наблюдали молекулярные ионные пики, но в случае соединений, которые имеют гидроксигруппу (-OH), иногда наблюдали фрагментарные пики при устраненной H2O. В случае солей обычно наблюдали молекулярные ионные пики или фрагментарные ионные пики их свободных форм.
[0160] Очистку с помощью препаративной высокоэффективной жидкостной хроматографии (препаративная ВЭЖХ) проводили при следующих условиях. Однако следует отметить, что когда трифторуксусную кислоту используют в операции ВЭЖХ в случае соединений, имеющих основные функциональные группы, для получения их свободных форм может требоваться выполнение нейтрализации или других операций.
Аппарат: система Gilson для препаративной ВЭЖХ
Колонка: Waters SunFire™ Prep C18 OBD™ (5 мкм, 30×50 мм)
Скорость потока: 40 мл/мин;
Способ обнаружения: УФ 254 нм
Растворитель: раствор A, 0,1% трифторуксусная кислота, содержащая воду; раствор B, 0,1% трифторуксусная кислота, содержащая ацетонитрил
Градиент: 0 минут (раствор A/раствор В=90/10), 2 минуты (раствор A/раствор В=90/10), 12 минут (раствор A/раствор В=20/80), 13,5 минут (раствор A/раствор В=5/95), 15 минут (раствор A/раствор В=5/95)
[0161] Анализ с помощью оптической высокоэффективной жидкостной хроматографии (оптическая ВЭЖХ) проводили при следующих условиях.
Аппарат: Agilent 1100 (продукт Agilent)
Колонка: DAICEL CHIRALCEL OD-H (5 мкм, 4,6×250 мм)
Скорость потока: 0,5 мл/мин;
Способ обнаружения: УФ 254 нм
Растворитель: 0,1% трифторуксусная кислота, содержащая ацетонитрил
[0162] Очистку с помощью оптической препаративной высокоэффективной жидкостной хроматографии (оптическая препаративная ВЭЖХ) проводили при следующих условиях.
Аппарат: система Gilson для препаративной ВЭЖХ
Колонка: DAICEL CHIRALCEL OD (10 мкм, 20×250 мм)
Скорость потока: 5 мл/мин;
Способ обнаружения: UV 254 нм
Растворитель: 0,1% трифторуксусная кислота, содержащая ацетонитрил
[0163] Для рентгеновской кристаллографии использовали XR-AXIS RAPID II (Rigaku).
[0164] Оптическую чистоту оптически активных форм оценивали в единицах процента энантиомерного избытка (%ee). Этот параметр вычисляли по следующему уравнению с использованием данных, полученных посредством оптической ВЭЖХ.
{Для (R)-формы)
Процент энантиомерного избытка (%ee)=100×[(R)-(S)]/[(R)+(S)]
[где (R) и (S) представляют абсолютные конфигурации соответствующих энантиомеров, а также их площади пиков в оптической высокоэффективной жидкостной хроматографии (ВЭЖХ)].
Процент энантиомерного избытка аналогичным образом определяли для (S)-формы.
Использовали фазовый разделитель Biotage ISOLUTE (зарегистрированный товарный знак) Phase Separator.
[0165] Использовали микроволновый реактор Biotage Initiator.
[0166] Названия соединениям присваивали с помощью ACD/Name (ACD/Labs 12.01, Advanced Chemistry Development Inc.)
[0167] Элементный анализ проводили с использованием следующих аппаратов.
2400II (Perkin Elmer)
vario MICRO cube (Elementar)
MT-6 (Yanaco Analytical Instruments Inc.)
[0168] Ионно-хроматографический анализ проводили с использованием следующих аппаратов.
DX500 (Dionex)
XS100 (Mitsubishi Chemical Corporation)
ICS3000 (Dionex)
[0169] Температуру плавления измеряли с использованием следующего аппарата.
MP-J3 (Yanaco Instrument Development Laboratory)
[0170] В таблицах, приведенных в ссылочных примерах и демонстрационных примерах, для некоторых соединений не представлена информация о солях, что указывает на то, что их получали в форме свободных форм.
Ссылочный пример 1-1
Гидрохлорид метил (4-аминотетрагидро-2H-пиран-4-ил)ацетата
[0171]
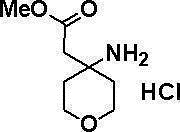
(1) Синтез метил тетрагидро-4H-пиран-4-илиден ацетата
[0172]
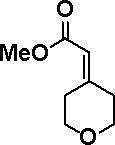
В раствор тетрагидро-4H-пиран-4-она (10,0 г) в толуоле (200 мл), при комнатной температуре добавляли метил (трифенилфофоранилиден)ацетат. После перемешивания при 100°C в течение 15 часов, смесь охлаждали до комнатной температуры. После концентрирования при пониженном давлении добавляли этилацетат (200 мл) и н-гексан (200 мл). После удаления осадка фильтрованием, фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=95:5-50:50) для того, чтобы получать ацетат метил тетрагидро-4H-пиран-4-илидена в виде бесцветного масла (13,9 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,30-2,37 (м, 2H) 2,95-3,05 (м, 2H) 3,70 (с, 3H) 3,71-3,81 (м, 4H) 5,69 (с, 1H).
MS ESI/APCI двойн. положительн.: 157[M+H]+, 179[M+Na]+.
(2) Синтез метил (4-аминотетрагидро-2H-пиран-4-ил)ацетата
[0173]
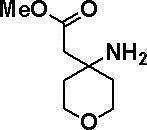
Аммиак-метаноловый раствор 8 моль/л (100 мл) соединения (13,6 г), полученного на приведенной выше стадии (1), перемешивали в герметизированной пробирке при 90°C в течение 4 суток. После охлаждения до комнатной температуры, смесь концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 100:0-90:10) для того, чтобы получать метил (4-аминотетрагидро-2H-пиран-4-ил)ацетат в виде масла желтого цвета (7,09 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,44-1,56 (м, 2H) 1,64-1,79 (м, 2H) 2,45 (с, 2H) 3,63-3,86 (м, 7H).
MS ESI/APCI двойн. положительн.: 174[M+H]+, 196[M+Na]+.
(3) Синтез указанного в заголовке соединения
В этилацетатный раствор (100 мл) соединения (7,09 г) на приведенной выше стадии (2) добавляли этилацетатный раствор хлороводорода 4 моль/л (10,2 мл). После этого добавляли н-гексан и смесь перемешивали при комнатной температуре в течение 5 минут. Осадок собирали с помощью фильтрования для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (5,72 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 1,76-1,85 (м, 4H) 2,90 (с, 2H) 3,49-3,61 (м, 2H) 3,63-3,68 (м, 3H) 3,70-3,83 (м, 2H) 8,35 (ушир. с, 3H).
MS ESI/APCI двойн. положительн.: 174[M+H]+.
Ссылочный пример 1-2
Трет-бутил 4-амино-4-(2-метокси-2-оксоэтил)пиперидин-1-карбоксилат
[0174]
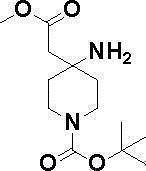
Вместо тетрагидро-4H-пиран-4-она использовали 1-(трет-бутоксикарбонил)-4-пиперидон (5,00 г) и проводили обработку теми же способами, как в ссылочном примере 1-1(1) и (2) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (4,65 г).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,45 (с, 9H) 1,49-1,52 (м, 2H) 1,53-1,60 (м, 2H) 2,41 (с, 2H) 3,28-3,35 (м, 2H) 3,58-3,68 (м, 2H) 3,69 (с, 3H).
MS ESI/APCI двойн. положительн.: 273[M+H]+.
Ссылочный пример 1-3
Трет-бутил 3-амино-3-(2-метокси-2-оксоэтил)азетидин-1-карбоксилат

(1) Синтез трет-бутил 3-(2-метокси-2-оксоэтилиден)азетидин-1-карбоксилата
[0175]
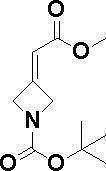
Вместо тетрагидро-4H-пиран-4-она использовали 1-(трет-бутоксикарбонил)-4-азетидинон (4,90 г) и обработку проводили с помощью того же способа, как в ссылочном примере 1-1(1) для того, чтобы получать трет-бутил 3-(2-метокси-2-оксоэтилиден)азетидин-1-карбоксилат в виде бесцветного масла (6,21 г).
1H ЯМР (200 МГц, ДМСО-d6) δ м.д. 1,38-1,41 (м, 9H) 3,61-3,67 (м, 3H) 4,52-4,60 (м, 2H) 4,66-4,73 (м, 2H) 5,84-5,93 (м, 1H)
MS ESI/APCI двойн. отрицательн.: 226[M-H]-.
(2) Синтез указанного в заголовке соединения
В раствор в этаноле (60 мл) соединения (6,04 г), полученного на приведенной выше стадии (1), добавляли 28% раствор аммиака в воде и смесь перемешивали при 80°C в течение 9 часов. После охлаждения до комнатной температуры, реакционную смесь концентрировали при пониженном давлении для того, чтобы получать указанное в заголовке соединение в виде неочищенного продукта (8,14 г). Указанное в заголовке соединение использовали в следующей реакции, поскольку оно оставалось неочищенным продуктом.
Ссылочный пример 2-1
Метил (1-аминоциклобутил)ацетат
[0176]

(1) Синтез 1-азаспиро[3.3]гептан-2-она
[0177]
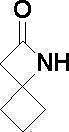
В раствор метиленциклобутана (2,35 г) в диэтиловом эфире (34,9 мл) медленно добавляли хлорсульфонил изоцианат (4,88 г) при охлаждении льдом и смесь перемешивали при комнатной температуре в течение 30 минут. После последовательного добавления 20% раствора тиосульфата натрия в воде (43,0 мл) и 10% раствора гидроксида калия в воде (43,0 мл) при 0°C, смесь перемешивали при этой температуре в течение 2 часов. После подтверждения того, что внутренняя часть реакционной системы была сильно основной, смесь экстрагировали диэтиловым эфиром девять раз. Объединенные органические слои сушили над безводным сульфатом магния и удаляли десикант с помощью фильтрования. Фильтрат концентрировали при пониженном давлении для того, чтобы получать 1-азаспиро[3.3]гептан-2-он в виде масла желтого цвета (2,71 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,63-1,82 (м, 2H) 2,17-2,45 (м, 4H) 2,96-2,99 (м, 2H) 5,90-6,56 (м, 1H).
MS ESI положительн.: 112[M+H]+, 134[M+Na]+.
(2) Синтез указанного в заголовке соединения
В раствор в метаноле (60,0 мл) соединения (2,67 г), полученного на приведенной выше стадии (1), медленно добавляли концентрированную серную кислоту. После нагревания с обратным холодильником в течение часа, смесь охлаждали до комнатной температуры. После концентрирования смеси при пониженном давлении, добавляли этилацетат и смесь экстрагировали с использованием соляной кислоты 1 моль/л два раза. В объединенные водные слои добавляли карбонат калия при 0°C до тех пор, пока не достигали pH >10. После десяти экстрагирований с использованием этилацетата, объединенные органические слои сушили над добавленным безводным сульфатом магния. После удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении для того, чтобы получать указанное в заголовке соединение в виде масла бледно-желтого цвета (2,82 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,59-1,99 (м, 4H) 2,03-2,16 (м, 2H) 2,61 (с, 2H) 3,69 (с, 3H).
MS ESI положительн.: 144[M+H]+, 166[M+Na]+.
Ссылочный пример 2-2
Гидрохлорид метил 3-амино-2,2,3-триметилбутаноата
[0178]
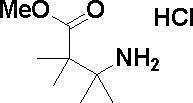
(1) Синтез 3,3,4,4-тетраметилазетидин-2-она
[0179]
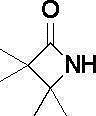
В раствор 2,3-диметил-2-бутена (5,76 г) в толуоле (46,0 мл), добавляли хлорсульфонил изоцианат (5,91 мл) при 0°C. После перемешивания при этой температуре в течение 10 минут, смесь доводили до комнатной температуры. После перемешивания в течение 45 минут смесь разводили добавленным толуолом (69,0 мл). В смесь 25% раствора гидроксида натрия в воде (49,1 мл) и хлорида бензилтриэтиламмония (99,0 мг) добавляли реакционную смесь в течение периода в один час. Получаемую смесь добавляли по каплям в 25% раствор гидроксида натрия в воде, и смесь перемешивали при 50°C в течение часа. После охлаждения до комнатной температуры, смесь экстрагировали толуолом два раза. Объединенные органические слои промывали насыщенным солевым раствором и сушили над добавленным безводным сульфатом магния. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток перекристаллизовывали с использованием толуола для того, чтобы получать 3,3,4,4-тетраметилазетидин-2-он в виде бесцветного твердого вещества (5,52 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,22 (с, 6H) 1,33 (с, 6H) 5,79 (ушир. с, 1H).
MS ESI/APCI двойн. положительн.: 128[M+H]+, 150[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 126[M-H]-.
(2) Синтез указанного в заголовке соединения
В соединение (5,52 г), полученное на приведенной выше стадии (1), добавляли раствор хлороводорода в метаноле 2 моль/л (40,0 мл) и смесь нагревали с обратным холодильником в течение 5 часов. Далее добавляли раствор хлороводорода в метаноле 2 моль/л (20,0 мл) и смесь нагревали с обратным холодильником в течение 6 часов. После охлаждения до комнатной температуры добавляли толуол (40,0 мл) и смесь концентрировали при пониженном давлении. После охлаждения остатка до 0°C, осадок собирали с помощью фильтрования и промывали толуолом. Собранный осадок сушили при пониженном давлении для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (4,67 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,36 (с, 6H) 1,51 (с, 6H) 3,73-3,83 (м, 3H) 8,25-8,83 (м, 3H).
MS ESI/APCI двойн. положительн.: 160[M+H]+, 182[M+Na]+.
Ссылочный пример 2-3
Гидрохлорид метил 3-амино3-этилпентаноата
[0180]
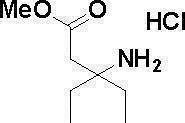
Вместо 2,3-диметил-2-бутена использовали 2-этил-1-бутен (10,0 г) и обработку проводили с помощью того же способа, как в ссылочном примере 2-2 для того, чтобы получать указанное в заголовке соединение в виде бесцветной аморфной массы (12,0 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,06 (т, J=7,5 Гц, 6H) 1,79-2,02 (м, 4H) 2,80 (с, 2H) 3,74 (с, 3H) 8,52 (ушир. с, 3H).
MS ESI/APCI двойн. положительн.: 160[M+H]+, 182[M+Na]+.
Ссылочный пример 3-1
Гидрохлорид этил 3-амино-2,2-дифторпропанoата
[0181]
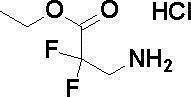
В этанол (12,0 мл) добавляли тионилхлорид (0,587 мл) при 0°C и смесь перемешивали при этой температуре в течение 30 минут. После добавления гидрохлорида 3-амино-2,2-дифторпропановой кислоты (950 мг) при 0°C, смесь нагревали с обратным холодильником в течение 4 часов. После охлаждения до комнатной температуры, смесь концентрировали при пониженном давлении. После добавления этилацетата, получаемый осадок удаляли с помощью фильтрования. Фильтрат концентрировали при пониженном давлении для того, чтобы получать указанное в заголовке соединение в виде масла бледно-коричневого цвета (920 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,36 (т, J=7,0 Гц, 3H) 3,84 (т, J=14,1 Гц, 2H) 4,40 (кв., J=7,0 Гц, 2H) 8,70 (ушир. с, 3H).
MS ESI/APCI двойн. положительн.: 154[M+H]+.
[0182] В следующих ссылочных примерах 3-2 и 3-3, соответствующие соединения β-аланина торгового сорта использовали в качестве исходного материала и обработку проводили с помощью способа, описанного в ссылочном примере 3-1 или его модификации для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены ниже в таблице 1-1.
[0183]

Ссылочный пример 3-4
Гидрохлорид метил (3R)-3-амино-4-гидроксибутаноата
[0184]
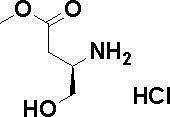
В раствор L-β-гомосерина (1,00 г) в метаноле (8,4 мл) добавляли раствор хлороводорода в 1,4-диоксане 4 моль/л (8,4 мл) и после этого смесь перемешивали при 60°C в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении для того, чтобы получать указанное в заголовке соединение в виде масла бледно-желтого цвета (1,42 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,69 (д, J=6,5 Гц, 2H) 3,34-3,60 (м, 2H) 3,64 (с, 3H) 4,27-4,57 (м, 1H).
MS ESI/APCI двойн. положительн.: 134[M+H]+.
Ссылочный пример 3-5
Гидрохлорид метил (3S)-3-амино-4-гидроксибутаноата
[0185]
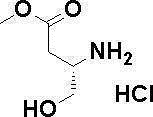
Вместо L-β-гомосерина использовали D-β-гомосерин (1,00 г) и обработку проводили с помощью того же способа, как в ссылочном примере 3-4 для того, чтобы получать указанное в заголовке соединение в виде масла бледно-желтого цвета (1,42 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,70 (д, J=6,7 Гц, 2H) 3,35-3,60 (м, 2H) 3,64 (с, 3H) 4,29-4,55 (м, 1H).
MS ESI/APCI двойн. положительн.: 134[M+H]+.
Ссылочный пример 4-1
Гидрохлорид этил (1-аминоциклопропил)ацетата
[0186]

(1) Синтез 3-(бензилокси)пропаннитрила
[0187]
В суспензию гидрида натрия (60% дисперсия в минеральном масле; 14,6 г) в тетрагидрофуране (281 мл) по каплям добавляли циангидрин этилена (21,0 мл) при 0°C и смесь перемешивали при этой температуре в течение 40 минут. После добавления бензилбромида (44,4 мл) в реакционную смесь, получаемую смесь перемешивали в течение ночи по мере того, как ее доводили до комнатной температуры. Насыщенный водный раствор хлорида аммония добавляли в реакционную смесь, которую затем экстрагировали этилацетатом три раза. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали посредством колоночной хроматографии на NH силикагеле (н-гексан:этилацетат = 100:0-70:30) для того, чтобы получать 3-(бензилокси)пропаннитрил в виде бесцветного масла (24,7 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,63 (т, J=6,4 Гц, 2H) 3,69 (т, J=6,4 Гц, 2H) 4,59 (с, 2H) 7,27-7,41 (м, 5H).
(2) Синтез 1-[2-(бензилокси)этил]циклопропанамина
[0188]

В смесь соединения (24,7 г), полученного на приведенной выше стадии (1), добавляли тетраизопропил ортотитанат (49,4 мл) и метоксициклопентан (306 мл), этилбромид магния (приблизительно 3 моль/л, раствор в диэтиловом эфире, 102 мл) при 0°C и после этого смесь перемешивали при комнатной температуре в течение 3 часов. После добавления комплекса трифторида бора/диэтилового эфира (38,8 мл) при 0°C смесь перемешивали при комнатной температуре в течение 1,5 часа. После добавления воды при 0°C pH корректировали до 12 посредством добавления 10% раствора гидроксида натрия в воде. Реакционную смесь экстрагировали хлороформом три раза. Объединенные органические слои промывали насыщенным солевым раствором и затем пропускали через фазовый разделитель и концентрировали при пониженном давлении. Получаемый остаток очищали посредством колоночной хроматографии на NH силикагеле (н-гексан:этилацетат = 100:0-5:95) и дополнительно очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 90:10-5:95, затем хлороформ:метанол = 100:0-90:10) для того, чтобы получать 1-[2-(бензилокси)этил]циклопропанамин в виде масла бледно-желтого цвета (12,5 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,40-0,45 (м, 2H) 0,53-0,59 (м, 2H) 1,73 (т, J=6,4 Гц, 2H) 3,69 (т, J=6,4 Гц, 2H) 4,52-4,55 (м, 2H) 7,27-7,37 (м, 5H).
(3) Синтез трет-бутил {1-[2-(бензилокси)этил]циклопропил}карбамата
[0189]
В раствор в тетрагидрофуране (130 мл) соединения (12,5 г), полученного на приведенной выше стадии (2), последовательно добавляли водный раствор бикарбоната натрия (7,8%, 106 г) и ди-трет-бутил дикарбонат (22,5 мл) и смесь перемешивали при комнатной температуре в течение 14 часов. После добавления насыщенного солевого раствора смесь экстрагировали этилацетатом три раза. Объединенные органические слои промывали насыщенным солевым раствором и затем пропускали через фазовый разделитель и концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-50:50) для того, чтобы получать трет-бутил {1-[2-(бензилокси)этил]циклопропил}карбамат в виде бесцветного твердого вещества (16,3 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,61-0,80 (м, 4H) 1,41 (с, 9H) 1,84 (т, J=6,3 Гц, 2H) 3,63 (т, J=6,3 Гц, 2H) 4,51 (с, 2H) 4,88 (ушир. с, 1H) 7,27-7,39 (м, 5H).
(4) Синтез трет-бутил [1-(2-гидроксиэтил)циклопропил] карбамата
[0190]

В раствор в этаноле (112 мл) соединения (16,3 г), полученного на приведенной выше стадии (3), добавляли 20% гидроксид палладия/углерод (3,25 г) и смесь перемешивали при 60°C в течение 23 часов в атмосфере водорода. После охлаждения до комнатной температуры реакционную смесь фильтровали через Celite (зарегистрированный товарный знак). Фильтрат концентрировали при пониженном давлении для того, чтобы получать трет-бутил [1-(2-гидроксиэтил)циклопропил]карбамат в виде бесцветного твердого вещества (11,1 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,71-0,78 (м, 2H) 0,79-0,87 (м, 2H) 1,44 (с, 9H) 1,60-1,66 (м, 2H) 3,65-3,78 (м, 2H) 4,83-4,99 (м, 1H).
(5) Синтез {1-[(трет-бутоксикарбонил)амино]циклопропил}уксусной кислоты
[0191]
В раствор в ацетонитриле (275 мл) соединения (11,1 г), полученного на приведенной выше стадии (4), и свободного радикала 2,2,6,6-тетраметилпиперидин-1-оксила (602 мг) добавляли натрий-фосфатный буфер (0,67 моль/л, pH 6,7, 206 мл). После нагревания до 35°C, водный раствор гипохлорита натрия (0,265%, 32,6 мл) и водный раствор хлорита натрия (14,7%), 110 мл) добавляли одновременно в течение периода в 2 часа и смесь перемешивали при этой температуре в течение 55 часов. Смесь охлаждали до комнатной температуры и после добавления воды (400 мл) ее делали основной с использованием 2 моль/л раствора гидроксида натрия в воде. Реакционную смесь выливали в водный раствор тиосульфата натрия (5,75%, 291 мл) при 0°C. После промывания диэтиловым эфиром (750 мл), водный слой добавляли в 2 моль/л соляную кислоту (140 мл) для корректировки pH до 2-3. Смесь экстрагировали диэтиловым эфиром, и этилацетатом и объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток кристаллизовали с использованием жидкой смеси н-гексана/этилацетата для того, чтобы получать {1-[(трет-бутоксикарбонил)амино]циклопропил}уксусную кислоту в виде бесцветного твердого вещества (9,45 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,73-0,83 (м, 2H) 0,89-0,96 (м, 2H) 1,45 (с, 9H) 2,41-2,82 (м, 2H) 5,09-5,49 (м, 1H).
MS ESI/APCI двойн. положительн.: 238[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 214[M-H]-.
(6) Синтез указанного в заголовке соединения
В этанол (34,8 мл) добавляли тионилхлорид (1,51 мл) при 0°C и смесь перемешивали при этой температуре в течение 30 минут. После добавления соединения (1,50 г), полученного на приведенной выше стадии (5), смесь перемешивали при 75°C в течение 4 часов. После охлаждения до комнатной температуры, смесь концентрировали при пониженном давлении для того, чтобы получать указанное в заголовке соединение в виде масла бледно-желтого цвета (1,41 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,73-0,81 (м, 2H) 1,30 (т, J=7,2 Гц, 3H) 1,44-1,51 (м, 2H) 2,74 (с, 2H) 4,23 (кв., J=7,2 Гц, 2H).
MS ESI/APCI двойн. положительн.: 144[M+H]+.
Ссылочный пример 5-1
Этил 1-(аминометил)циклопропанкарбоксилат
[0192]

(1) Синтез этил 1-цианоциклопропанкарбоксилата
[0193]

В раствор этил цианоацетата (11,8 г) в ацетоне (83,0 мл) добавляли карбонат калия (43,1 г) и 1,2-дибромэтан (39,2 г) и смесь нагревали с обратным холодильником в течение 12 часов. После охлаждения до комнатной температуры реакционную смесь фильтровали через Celite (зарегистрированный товарный знак). Фильтрат концентрировали при пониженном давлении и дополнительно сушили при пониженном давлении с нагреванием для того, чтобы получать этил 1-цианоциклопропанкарбоксилат в виде масла красного цвета (14,2 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 1,23 (т, J=7,1 Гц, 3H) 1,57-1,62 (м, 2H) 1,73-1,79 (м, 2H) 4,19 (кв., J=7,1 Гц, 2H).
(2) Синтез указанного в заголовке соединения
В раствор в этаноле (127 мл) соединения (14,0 г), полученного на приведенной выше стадии (1), добавляли катализатор никель Ренея (приблизительно 2,8 г). В атмосфере водорода смесь перемешивали при комнатной температуре в течение 12 часов и дополнительно перемешивали при 40°C в течение 12 часов. После охлаждения до комнатной температуры, реакционную смесь фильтровали через Celite (зарегистрированный товарный знак) и фильтрат концентрировали при пониженном давлении. Получаемый осадок собирали с помощью фильтрования и промывали этанолом. Фильтрат концентрировали для того, чтобы получать указанное в заголовке соединение в виде масла красного цвета (13,4 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 0,82-0,88 (м, 2H) 0,98-1,04 (м, 2H) 1,13-1,21 (м, 3H) 2,63-2,72 (м, 2H) 4,00-4,10 (м, 2H).
[0194] В следующих ссылочных примерах с 5-2 до 5-5 1,2-дибромэтан заменяли на соответствующие дигалогенированные алканы или дигалогенированные алкиловые простые эфиры торговых сортов, которые обрабатывали с помощью способа, описанного в ссылочном примере 5-1 или его модификациях, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены ниже в таблице 2-1.
[0195]
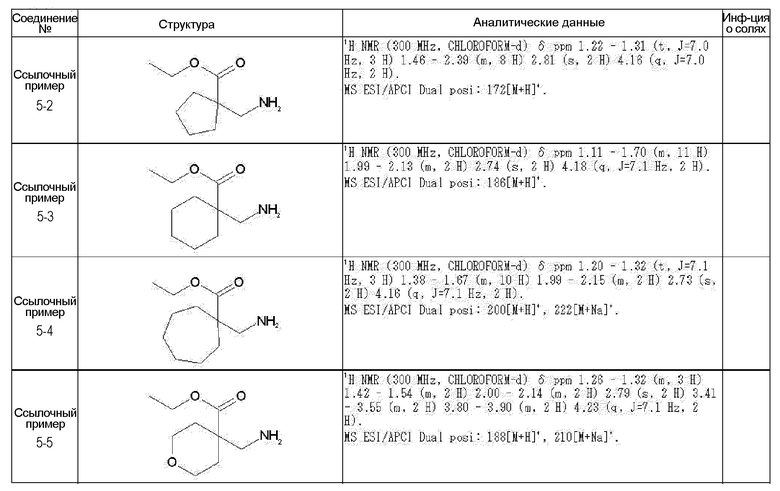
Ссылочный пример 6-1
4-Циклобутилбензальдегид
[0196]
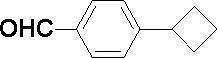
В смесь 4-йодбензальдегида (500 мг) добавляли дихлорид бис(трифенилфосфин)палладия(II) (75,6 мг), йодид меди(I) (24,6 мг) и дегидратированный тетрагидрофуран (10,0 мл), циклобутилбромид цинка (0,5 моль/л, раствор в тетрагидрофуране, 6,46 мл) и смесь перемешивали в герметизированной пробирке при 60°C в течение 14 часов. После охлаждения до комнатной температуры, осадок удаляли с помощью фильтрования через Celite (зарегистрированный товарный знак). Фильтрат концентрировали при пониженном давлении и после этого очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=98:2-90:10) для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (225 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,79-2,28 (м, 4H) 2,29-2,48 (м, 2H) 3,54-3,73 (м, 1H) 7,32-7,39 (м, 2H) 7,74-7,87 (м, 2H) 9,97 (с, 1H).
MS ESI/APCI двойн. положительн.: 161[M+H]+.
Ссылочный пример 6-2
4'-(трифторметил)бифенил-4-карбальдегид
[0197]

Смесь 4-бромбензотрифторида (10,0 г), 4-формилфенилбороновой кислоты (7,33 г), тетракис(трифенилфосфин)палладия(0) (308 мг), карбоната калия (30,7 г), тетрагидрофурана (300 мл) и воды (100 мл) перемешивали при 85°C в течение 2 часов. После охлаждения реакционной смеси до комнатной температуры, в нее добавляли воду, которую затем экстрагировали этилацетатом два раза и объединенные органические слои промывали насыщенным солевым раствором. После добавления безводного сульфата магния в органические слои, десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток растворяли в н-гексане (60 мл) при нагревании и после этого охлаждали до 0°C. Получаемый осадок собирали с помощью фильтрования для того, чтобы получать указанное в заголовке соединение в виде твердого вещества серого цвета (12,1 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 7,71-7,80 (м, 6H) 7,96-8,03 (м, 2H) 10,09 (с, 1H).
MS EI положительн.: 250[M]+.
Ссылочный пример 6-3
4'-фторбифенил-4-карбальдегид
[0198]
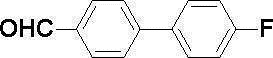
Смесь 4-бромбензальдегида (10,0 г), 4-фторфенилбороновой кислоты (11,3 г), тетракис(трифенилфосфин)палладия(0) (3,12 г), карбоната натрия (28,6 г), толуола (150 мл), этанола (70,0 мл) и воды (70,0 мл) перемешивали при 100°C в течение 12 часов. После охлаждения реакционной смеси до комнатной температуры добавляли воду и два раза проводили экстрагирование толуолом. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 90:10) и, после добавления н-гексана в остаток, смесь перемешивали. Осадок собирали с помощью фильтрования и сушили при пониженном давлении для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (10,4 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 7,12-7,22 (м, 2H) 7,56-7,66 (м, 2H) 7,68-7,75 (м, 2H) 7,93-7,98 (м, 2H) 10,06 (с, 1H).
MS ESI/APCI двойн. положительн.: 201[M+H]+.
Ссылочный пример 6-4
4-Циклопропил-3-(трифторметил)бензальдегид
[0199]
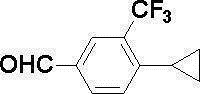
Используя 4-хлор-3-(трифторметил)бензальдегид (1,00 г) и циклопропилбороновую кислоту (1,24 г) в качестве исходных материалов, тот же способ, что и в ссылочном примере 6-3, применяли для того, чтобы получать указанное в заголовке соединение в виде масла бледно-желтого цвета (900 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,85-0,95 (м, 2H) 1,13-1,25 (м, 2H) 2,23-2,37 (м, 1H) 7,13 (д, J=8,1 Гц, 1H) 7,94 (дд, J=8,1, 1,5 Гц, 1H) 8,12 (д, J=1,5 Гц, 1H) 10,00 (с, 1H).
MS ESI/APCI двойн. положительн.: 215[M+H]+.
MS ESI/APCI двойн. отрицательн.: 213[M-H]-.
Ссылочный пример 6-5
3,3'-бипиридин-6-карбальдегид
[0200]
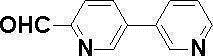
Смесь 5-бром-3-пиридинкарбоксиальдегида (1,00 г), 3-пиридилбороновой кислоты (991 мг), аддукта дихлорметана дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия(II) (220 мг), 2 моль/л раствора карбоната натрия в воде (5 мл) и N,N-диметилформамида (20 мл) перемешивали при 120°C в течение 30 минут при облучении микроволнами.
После охлаждения реакционной смеси до комнатной температуры, в нее добавляли воду, и проводили два экстрагирования этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором и после этого пропускали через фазовый разделитель для концентрирования при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол=100:0-95:5) для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (944 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 7,44-7,54 (м, 1H) 7,93-8,00 (м, 1H) 8,09 (д, J=1,6 Гц, 2H) 8,68-8,79 (м, 1H) 8,90-8,97 (м, 1H) 9,03 (т, J=1,6 Гц, 1H) 10,08-10,19 (м, 1H).
Ссылочный пример 6-6
4-(6-циклопропил-3-пиридинил)бензальдегид
[0201]
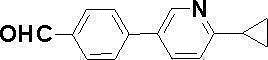
Смесь 3-бром-6-(циклопропил)пиридина (2,00 г), 4-формилфенилбороновой кислоты (1,82 г), ацетата палладия(II) (113 мг), фосфата трикалия (4,50 г) и этиленгликоля (16,8 мл) перемешивали при 80°C в течение 3 часов. После охлаждения реакционной смеси до комнатной температуры, в нее добавляли воду, и проводили два экстрагирования этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором и после этого неочищенный продукт адсорбировали на диатомовой земле, при этом растворитель отгоняли при пониженном давлении. Неочищенный продукт, адсорбированный на диатомовой земле, очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 98:2-50:50) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (1,81 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,00-1,17 (м, 4H) 2,03-2,16 (м, 1H) 7,21-7,30 (м, 1H) 7,66-7,83 (м, 3H) 7,93-8,01 (м, 2H) 8,68-8,76 (м, 1H) 10,06 (с, 1H).
MS ESI/APCI двойн. положительн.: 224[M+H]+.
[0202] В следующих ссылочных примерах 6-7 и 6-11 соответствующие галогенированные пиридины торговых сортов использовали в качестве исходного материала и обработку проводили с помощью способа, описанного в ссылочном примере 6-6, или его модификаций, для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены ниже в таблице 3-1.
[0203]

Ссылочный пример 7-1
4-фенилциклогексанкарбальдегид
[0204]
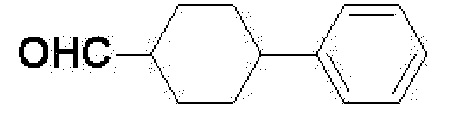
(1) Синтез [4-(метоксиметилиден)циклогексил]бензола
[0205]

В смесь хлорида (метоксиметил)трифенилфосфония (6,14 г) с трет-бутилметиловым простым эфиром (30,0 мл) добавляли трет-бутоксид калия (2,32 г), при этом температуру в системе сохраняли равной -10°C. Смесь перемешивали при -10°C в течение 10 минут и после этого перемешивали при комнатной температуре в течение часа. Добавляли раствор 4-фенилциклогексанона (2,4 0 г) в тетрагидрофуране (10,0 мл), при этом температуру в системе сохраняли равной -10°C. Смесь перемешивали при -10°C в течение 10 минут и после этого перемешивали при комнатной температуре в течение двух часов. После добавления воды смесь экстрагировали этилацетатом. Объединенные органические слои пропускали через фазовый разделитель и после этого концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-60:40) для того, чтобы получать [4-(метоксиметилиден)циклогексил]бензол в виде бесцветного масла (3,59 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,36-1,53 (м, 2H) 1,69-2,23 (м, 5H) 2,56-2,70 (м, 1H) 2,85-2,96 (м, 1H) 3,57 (с, 3H) 5,80-5,83 (м, 1H) 7,12-7,34 (м, 5H).
MS ESI/APCI двойн. положительн.: 203[M+H]+.
(2) Синтез указанного в заголовке соединения
В раствор в тетрагидрофуране (10,0 мл) соединения (3,59 г), полученного на приведенной выше стадии (1), добавляли 3 моль/л соляной кислоты и смесь нагревали с обратным холодильником в течение 4 часов. После охлаждения реакционной смеси до комнатной температуры, в нее добавляли воду и проводили три экстрагирования этилацетатом. Объединенные органические слои промывали водой и затем концентрировали при пониженном давлении для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (2,75 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,13-1,62 (м, 4H) 1,63-2,19 (м, 4H) 2,23-2,40 (м, 1H) 2,42-2,60 (м, 1H) 7,10-7,39 (м, 5H) 9,65-9,82 (м, 1H).
MS EI положительн.: 188[M]+.
Ссылочный пример 8-1
4-(Циклопропилметокси)бензальдегид
[0206]

В смесь 4-гидроксибензальдегида (2,00 г), карбоната калия (4,53 г) и ацетона (50,0 мл) добавляли (бромметил)циклопропан (3,32 г) и смесь нагревали с обратным холодильником в течение 9 часов. После охлаждения реакционной смеси до комнатной температуры, получаемый осадок удаляли с помощью фильтрования через Celite (зарегистрированный товарный знак). Фильтрат концентрировали при пониженном давлении и, после этого, получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 90:10-50:50) для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (2,63 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,34-0,42 (м, 2H) 0,62-0,73 (м, 2H) 1,21-1,37 (м, 1H) 3,89 (д, J=7,0 Гц, 2H) 6,96-7,04 (м, 2H) 7,80-7,86 (м, 2H) 9,88 (с, 1H).
MS ESI/APCI двойн. положительн.: 177[M+H]+, 199[M+Na]+.
[0207] В следующих ссылочных примерах с 8-2 до 8-5 использовали каждый из соответствующих фенолов и галогенированных алканов торговых сортов и обработку проводили с помощью способа, описанного в ссылочном примере 8-1, или его модификаций для того, чтобы получать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблице 4-1.
[0208]

Ссылочный пример 9-1
4-(циклобутилметокси)бензальдегид
[0209]
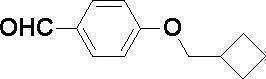
В суспензию гидрида натрия (60% дисперсия в минеральном масле, 0,328 г) в N,N-диметилформамиде (15,0 мл), добавляли раствор 4-гидроксибензальдегида (1,00 г) в N,N-диметилформамиде (5,00 мл) при 0°C и после этого смесь перемешивали при комнатной температуре в течение 30 минут. После добавления (бромметил)циклобутана (1,22 г), смесь перемешивали при 70°C в течение 24 часов. После охлаждения реакционной смеси до комнатной температуры, 0,5 моль/л соляной кислоты добавляли при охлаждении льдом и проводили три экстрагирования этилацетатом. Объединенные органические слои пропускали через фазовый разделитель и затем концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 95:5-40:60) для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (1,19 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,79-2,04 (м, 4H) 2,08-2,26 (м, 2H) 2,72-2,91 (м, 1H) 4,01 (д, J=6,7 Гц, 2H) 6,88-7,07 (м, 2H) 7,77-7,87 (м, 2H) 9,88 (с, 1H).
[0210] В следующих ссылочных примерах 9-2 и 9-3, (бромметил)циклобутан заменяли на соответствующий галогенированный алкан или галогенированный циклоалкан торгового сорта и способ, описанный в ссылочном примере 9-1, или его модификацию применяли для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены ниже в таблице 5-1.
[0211]
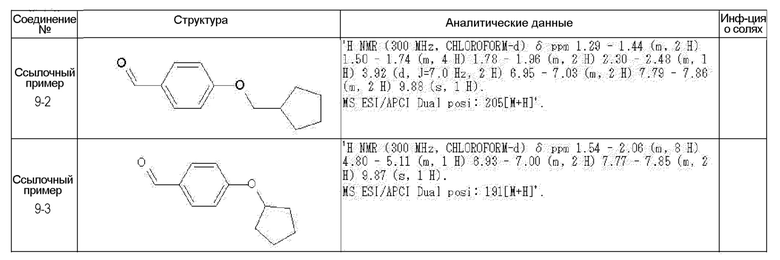
Ссылочный пример 10-1
4-(Циклопропокси)бензальдегид
[0212]

В смесь 4-гидроксибензальдегида (1,20 г), карбоната калия (2,04 г), йодида калия (49,0 мг) и N,N-диметилформамида (9,80 мл) добавляли бромциклопропан (1,02 мл) и смесь перемешивали при 200°C в течение 3 часов при облучении микроволнами. После охлаждения до комнатной температуры, реакционную смесь выливали в воду и экстрагировали диэтиловым эфиром три раза. Объединенные органические слои промывали насыщенным солевым раствором и после этого пропускали через фазовый разделитель для концентрации при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-50:50) для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (510 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,76-0,91 (м, 4H) 3,77-3,88 (м, 1H) 7,12-7,19 (м, 2H) 7,80-7,87 (м, 2H) 9,90 (с, 1H).
MS ESI/APCI двойн. положительн.: 163[M+H]+, 185[M+Na]+.
[0213] В следующих ссылочных примерах с 10-2 до 10-5 бромциклопропан заменяли на соответствующие галогенированные алканы торговых сортов и способ, описанный в ссылочном примере 10-1, или его модификацию применяли для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены ниже в таблице 6-1.
[0214]
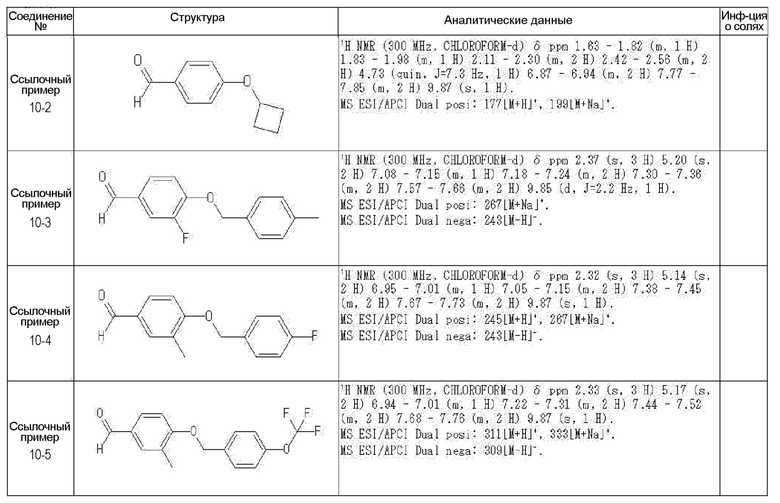
Ссылочный пример 11-1
4-(2-циклопропилэтокси)бензальдегид
[0215]
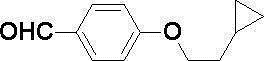
В смесь 4-гидроксибензальдегида (2,84 г), 2-циклопропилэтанола (2,00 г), трифенилфосфина (6,09 г) и тетрагидрофурана (100 мл) добавляли диэтил азодикарбоксилат (2,2 моль/л, раствор в толуоле, 10,5 мл) и смесь перемешивали при комнатной температуре в течение 4 суток. После концентрирования реакционной смеси при пониженном давлении добавляли этилацетат (7,50 мл) и н-гексан (143 мл) и смесь перемешивали при комнатной температуре в течение 15 минут. Осадок удаляли с помощью фильтрования через Celite (зарегистрированный товарный знак). Фильтрат концентрировали при пониженном давлении и, после этого, получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=95:5-50:50) для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (3,34 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,10-0,18 (м, 2H) 0,46-0,55 (м, 2H) 0,78-0,95 (м, 1H) 1,67-1,76 (м, 2H) 4,12 (т, J=6,6 Гц, 2H) 6,97-7,05 (м, 2H) 7,80-7,87 (м, 2H) 9,88 (с, 1H).
MS ESI положительн.: 191[M+H]+.
[0216] В следующих ссылочных примерах с 11-2 до 11-11 использовали соответствующие гидроксибензальдегиды торговых сортов и соответствующие спирты торговых сортов и обработку проводили с помощью способа, описанного в ссылочном примере 11-1, или его модификаций для того, чтобы получать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблицах 7-1 и 7-2.
[0217]

[0218]
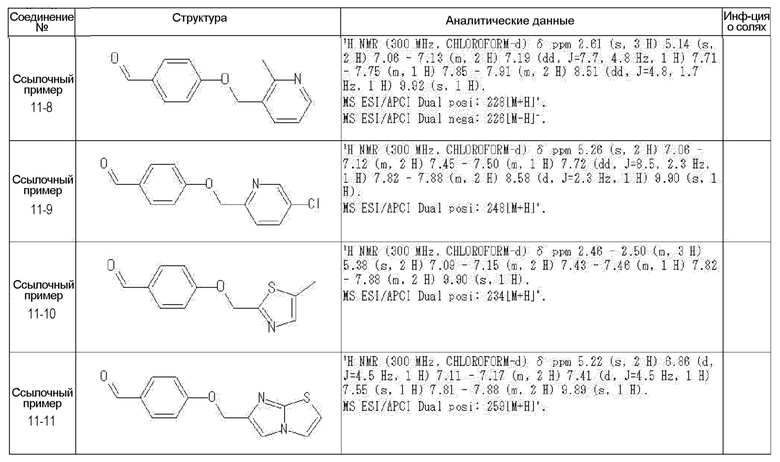
Ссылочный пример 12-1
3-фтор-4-(2,2,2-трифторэтокси)бензальдегид
[0219]
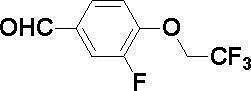
В суспензию гидрида натрия (60% дисперсия в минеральном масле, 0,844 г) в N,N-диметилформамиде (30,0 мл) добавляли 2,2,2-трифторэтанол (2,11 г) при 0°C и после этого смесь перемешивали при комнатной температуре в течение 30 минут. В реакционную смесь добавляли раствор 3,4-дифторбензальдегида (2,00 г) в N,N-диметилформамиде (10,0 мл) и после этого смесь перемешивали при комнатной температуре в течение 30 минут. После добавления 1 моль/л соляной кислоты при 0°C проводили три экстрагирования этилацетатом. Объединенные органические слои пропускали через фазовый разделитель и после этого концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 80:20-30:70) для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (2,50 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 4,52 (кв., J=7,9 Гц, 2H) 7,11-7,19 (м, 1H) 7,63-7,71 (м, 2H) 9,89-9,91 (м, 1H).
MS EI положительн.: 222[M]+.
[0220] В следующих ссылочных примерах с 12-2 до 12-9 использовали соответствующие фторбензальдегиды торговых сортов и соответствующие спирты торговых сортов и обработку проводили с помощью способа, описанного в ссылочном примере 12-1, или его модификаций для того, чтобы получать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблицах 8-1 и 8-2.
[0221]
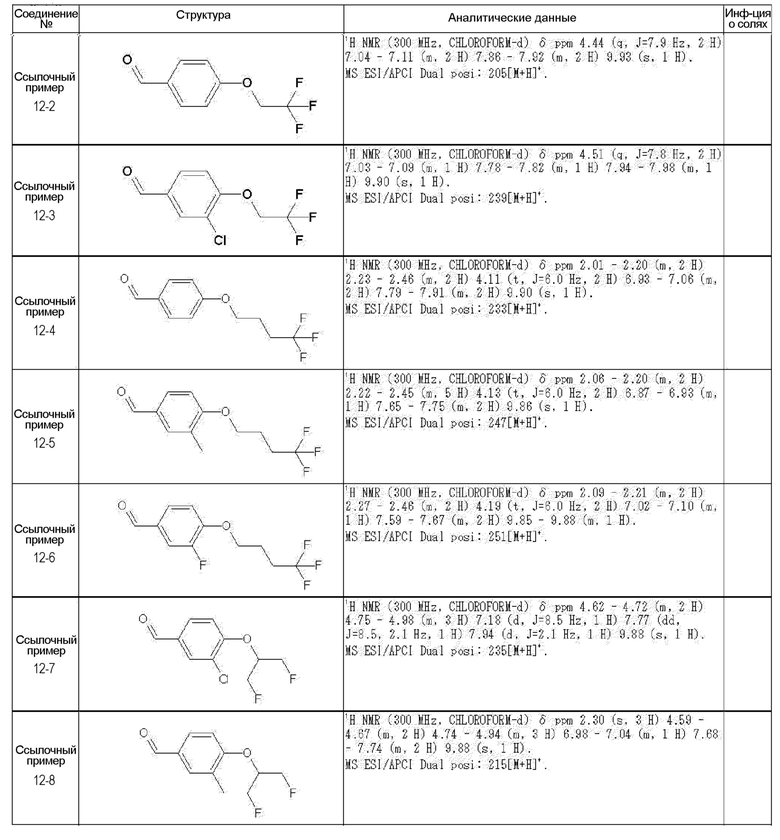
[0222]

Ссылочный пример 13-1
6-[4-(трифторметил)фенокси]пиридин-3-карбальдегид
[0223]

В раствор 4-гидроксибензотрифторида (505 мг) в N,N-диметилформамиде (5,00 л) добавляли карбонат калия (474 мг) и смесь перемешивали при комнатной температуре в течение 10 минут.
Впоследствии добавляли 6-бром-3-пиридинкарбоксиальдегид (580 мг) и смесь перемешивали при 130°C в течение 2 часов. После охлаждения смеси до комнатной температуры в смесь добавляли воду, которую затем экстрагировали этилацетатом. Объединенные органические слои последовательно промывали водой и насыщенным солевым раствором и после прохождения через фазовый разделитель их концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 95:5-70:30) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (605 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 7,09-7,15 (м, 1H) 7,27-7,33 (м, 2H) 7,66-7,75 (м, 2H) 8,24 (дд, J=8,5, 2,3 Гц, 1H) 8,62 (дд, J=2,3, 0,6 Гц, 1H) 9,99-10,02 (м, 1H).
MS ESI/APCI двойн. положительн.: 268[M+H]+.
[0224] В следующих ссылочных примерах с 13-2 до 13-36 использовали соответствующие фенолы или гидроксипиридины торговых сортов и соответствующие галогенированные бензальдегиды или галогенированные пиридинкарбоксиальдегиды торговых сортов и обработку проводили с помощью способа, описанного в ссылочном примере 13-1, или его модификаций для того, чтобы получать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблицах 9-1 и 9-5.
[0225]
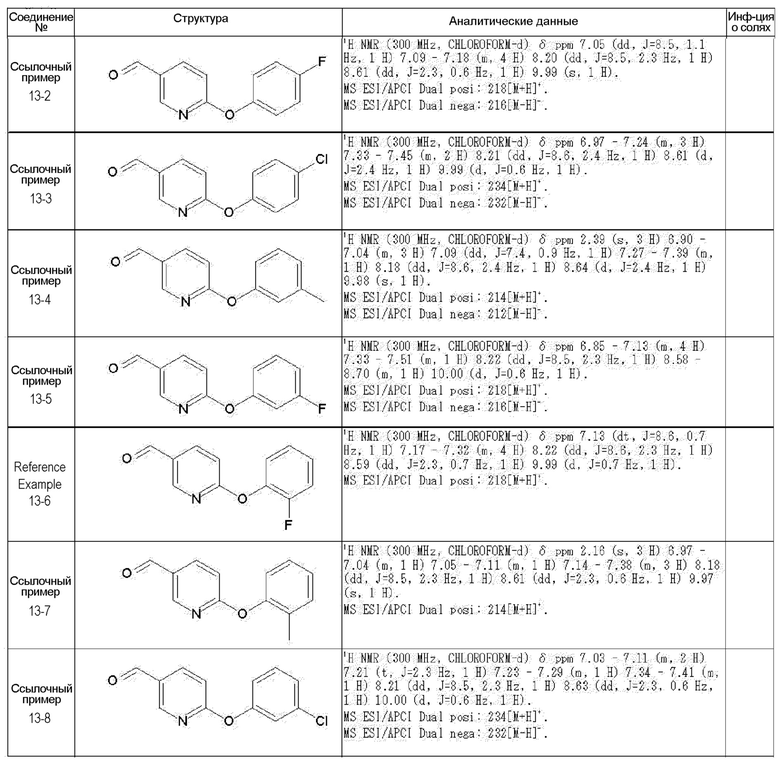
[0226]

[0227]

[0228]
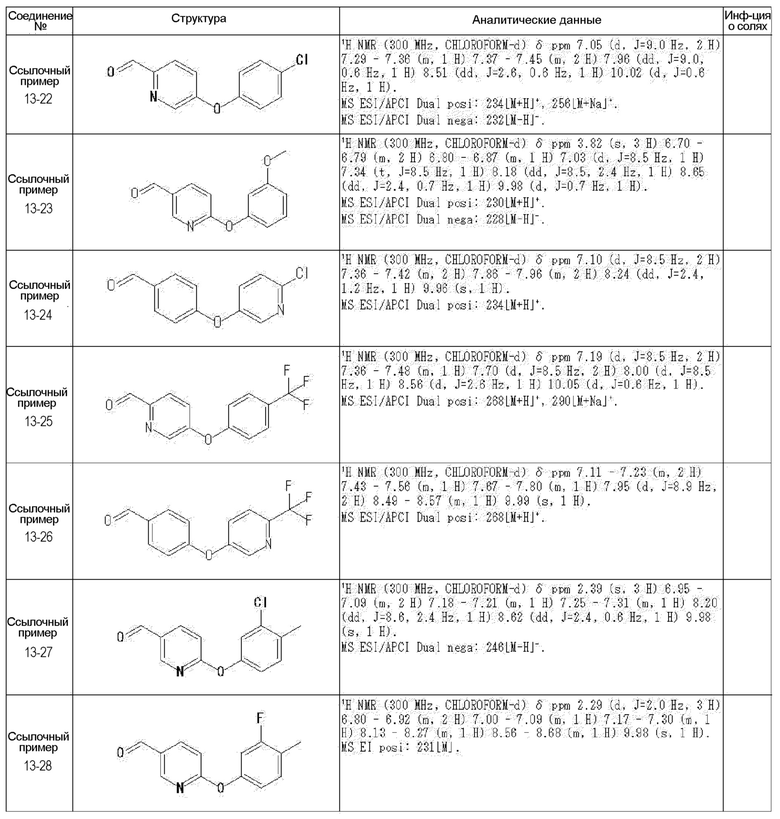
[0229]

Ссылочный пример 13-37
6-(4-циклопропилфенокси)пиридин-3-карбальдегид
[0230]
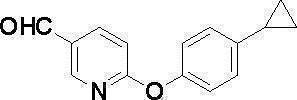
(1) Синтез 6-(4-бромфенокси)пиридин-3-карбальдегида
[0231]
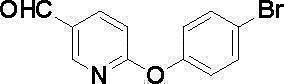
В раствор 4-бромфенола (2,79 г) в N,N-диметилформамиде (25,0 мл) добавляли карбонат калия (2,45 г) и смесь перемешивали при комнатной температуре в течение 10 минут. Впоследствии добавляли 6-бром-3-пиридинкарбоксиальдегид (3,00 г) и смесь перемешивали при 130°C в течение 2,5 часа. После охлаждения реакционной смеси до комнатной температуры, в нее добавляли воду и проводили экстрагирование этилацетатом. Объединенные органические слои последовательно промывали водой и насыщенным солевым раствором и после пропускания через фазовый разделитель их концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-50:50) для того, чтобы получать 6-(4-бромфенокси)пиридин-3-карбальдегид в виде твердого вещества бледно-желтого цвета (3,23 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 7,01-7,13 (м, 3H) 7,51-7,61 (м, 2H) 8,21 (дд, J=8,6, 2,4 Гц, 1H) 8,61 (дд, J=2,4, 0,7 Гц, 1H) 9,99 (д, J=0,7 Гц, 1H).
MS ESI/APCI двойн. положительн.: 278[M+H]+.
(2) Синтез указанного в заголовке соединения
Смесь соединения (3,22 г), полученного на приведенной выше стадии (1), 2-циклопропил-4,4,5,5-тетраметил-1,3,2-диоксаборолана (3,89 г), тетракис(трифенилфосфин)палладия(0) (669 мг), карбоната цезия (11,3 г), толуола (20,0 мл) и воды (10,0 мл) перемешивали при 100°C в течение 6 часов. После охлаждения реакционной смеси до комнатной температуры, осадок удаляли с помощью фильтрования через Celite (зарегистрированный товарный знак). В фильтрат добавляли воду и этилацетат и проводили экстрагирование этилацетатом. Объединенные органические слои последовательно промывали водой и насыщенным солевым раствором и после пропускания через фазовый разделитель их концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-50:50) и дополнительно очищали с помощью препаративной ВЭЖХ. Затем добавляли насыщенный водный раствор бикарбоната натрия и проводили экстрагирование этилацетатом. Объединенные органические слои пропускали через фазовый разделитель и после этого концентрировали при пониженном давлении для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (614 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,65-0,76 (м, 2H) 0,92-1,04 (м, 2H) 1,84-1,98 (м, 1H) 6,94-7,10 (м, 3H) 7,11-7,20 (м, 2H) 8,17 (дд, J=8,6, 2,4 Гц, 1H) 8,60-8,67 (м, 1H) 9,97 (д, J=0,6 Гц, 1H).
MS ESI/APCI двойн. положительн.: 240[M+H]+.
MS ESI/APCI двойн. отрицательн.: 238[M-H]-.
Ссылочный пример 14-1
4-[(5-фторпиридин-2-ил)окси]бензальдегид
[0232]
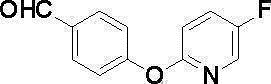
В раствор 4-гидроксибензальдегида (5,00 г) в N,N-диметилацетамиде (60,0 л) добавляли карбонат калия (6,23 г) и смесь перемешивали при комнатной температуре в течение 10 минут. Впоследствии добавляли 2,5-дифторпиридин (4,71 г) и смесь перемешивали при 150°C в течение 64 часов. После охлаждения реакционной смеси до комнатной температуры, в нее добавляли воду и проводили экстрагирование этилацетатом. Объединенные органические слои последовательно промывали водой и насыщенным солевым раствором и после пропускания через фазовый разделитель их концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-70:30) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (3,24 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 6,98-7,05 (м, 1H) 7,21-7,29 (м, 2H) 7,46-7,57 (м, 1H) 7,89-7,96 (м, 2H) 8,05-8,09 (м, 1H) 9,98 (с, 1H).
MS ESI/APCI двойн. положительн.: 218[M+H]+.
[0233] В следующих ссылочных примерах с 14-2 до 14-4 использовали соответствующие галогенированные пиридины торговых сортов и обработку проводили с помощью способа, описанного в ссылочном примере 14-1, или его модификаций для того, чтобы получать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблице 10-1.
[0234]
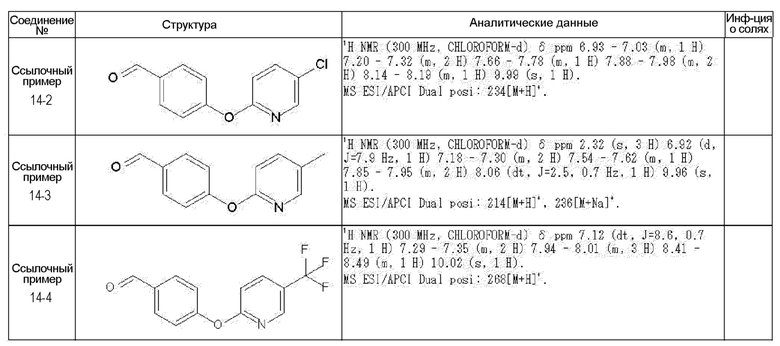
Ссылочный пример 14-5
4-[(5-циклопропилпиридин-2-ил)окси]бензальдегид
[0235]

(1) Синтез 4-[(5-бромпиридин-2-ил)окси]бензальдегида
[0236]

Вместо 2,5-дифторпиридина использовали 2,5-дибромпиридин (13,5 г) и обработку проводили с помощью того же способа, как в ссылочном примере 14-1 для того, чтобы получать указанное в заголовке соединение в виде масла бледно-желтого цвета (12,4 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 6,91-6,97 (м, 1H) 7,24-7,32 (м, 2H) 7,81-7,88 (м, 1H) 7,90-7,98 (м, 2H) 8,23-8,27 (м, 1H) 9,99 (с, 1H).
MS ESI/APCI двойн. положительн.: 277[M+H]+.
(2) Синтез указанного в заголовке соединения
Смесь соединения (5,00 г), полученного на приведенной выше стадии (1), циклопропилбороновой кислоты (2,01 г), ацетата палладия(II) (201 мг), фосфата трикалия (13,4 г), трициклогексилфосфина (0,6 моль/л, раствор в толуоле, 30,0 мл), толуола (95,0 мл) и воды (5,0 мл) перемешивали при 100°C в течение 3 часов. После охлаждения реакционной смеси до комнатной температуры, в нее добавляли воду и проводили два экстрагирования этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором и после этого неочищенный продукт адсорбировали на диатомовой земле, причем растворители отгоняли при пониженном давлении. Неочищенный продукт, адсорбированный на диатомовой земле, очищали колоночной хроматографией на силикагеле (гексан:этилацетат=95:5-63:37) для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (3,89 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,64-0,74 (м, 2H) 0,95-1,07 (м, 2H) 1,82-1,97 (м, 1H) 6,88-6,95 (м, 1H) 7,19-7,26 (м, 2H) 7,37-7,45 (м, 1H) 7,86-7,94 (м, 2H) 8,02-8,09 (м, 1H) 9,96 (с, 1H).
MS ESI/APCI двойн. положительн.: 240[M+H]+.
Ссылочный пример 15-1
4-(2-циклопропилэтил)бензальдегид
[0237]
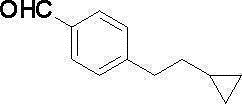
(1) Синтез 4-(циклопропилэтинил)бензальдегида
[0238]
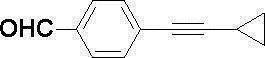
В смесь 4-бромбензальдегида (2,00 г), дихлорида бис(трифенилфосфин)палладия(II) (228 мг), йодида меди(I) (20,6 мг), N,N-диметилформамида (2,00 мл) и триэтиламина (15,1 мл) добавляли циклопропилацетилен и после этого смесь перемешивали в герметизированной пробирке при 110°C в течение одной минуты при облучении микроволнами. После охлаждения до комнатной температуры, реакционную смесь выливали в насыщенный водный раствор хлорида аммония и экстрагировали жидкой смесью н-гексана/этилацетата (1:1) три раза. Объединенные органические слои промывали насыщенным солевым раствором и сушили над добавленным безводным сульфатом магния. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-60:40) для того, чтобы получать 4-(циклопропилэтинил)бензальдегид в виде масла коричневого цвета (1,79 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,80-0,99 (м, 4H) 1,42-1,54 (м, 1H) 7,46-7,54 (м, 2H) 7,75-7,82 (м, 2H) 9,98 (с, 1H).
MS ESI/APCI двойн. положительн.: 171[M+H]+.
(2) Синтез указанного в заголовке соединения
В раствор в этилацетате (22,0 мл) соединения (1,79 г), полученного на приведенной выше стадии (1), добавляли 10% палладий/углерод (179 мг). Смесь перемешивали при комнатной температуре в течение 22 часов в атмосфере водорода. Добавляли дополнительный 10% палладий/углерод (179 мг) и смесь перемешивали при комнатной температуре в течение 3 часов в атмосфере водорода. Нерастворимое вещество удаляли с помощью фильтрования через Celite (зарегистрированный товарный знак). После концентрирования фильтрата при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-70:30) для того, чтобы получать указанное в заголовке соединение в виде неочищенного продукта (1,23 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,01-0,08 (м, 2H) 0,39-0,48 (м, 2H) 0,60-0,77 (м, 1H) 1,50-1,59 (м, 2H) 2,75-2,84 (м, 2H) 7,30-7,39 (м, 2H) 7,74-7,83 (м, 2H) 9,97 (с, 1H).
MS ESI/APCI двойн. положительн.: 175[M+H]+.
MS ESI/APCI двойн. отрицательн.: 173[M-H]-.
Ссылочный пример 16-1
4-[(2,2-диметилпропокси)метил]бензальдегид
[0239]
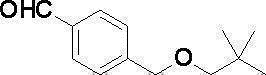
(1) Синтез 4-(хлорметил)-N-метокси-N-метилбензамида
[0240]
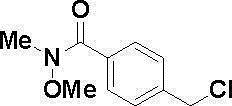
В раствор 4-(бромметил)бензойной кислоты (10,7 г) в хлороформе (200 мл) добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (10,5 г), моногидрат 1-гидроксибензотриазола (8,38 г), гидрохлорид N,O-диметилгидроксиламина (4,85 г) и триэтиламин (6,94 мл). После перемешивания смеси при комнатной температуре в течение 53 часов добавляли хлороформ (200 мл). Смесь последовательно промывали насыщенным водным раствором бикарбоната натрия, насыщенным водным раствором хлорида аммония и насыщенным солевым раствором. После пропускания через фазовый разделитель, промытую смесь концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 95:5-40:60) для того, чтобы получать 4-(хлорметил)-N-метокси-N-метилбензамид в виде бесцветного масла (3,23 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 3,37 (с, 3H) 3,55 (с, 3H) 4,61 (с, 2H) 7,39-7,46 (м, 2H) 7,65-7,72 (м, 2H).
MS ESI/APCI двойн. положительн.: 214[M+H]+, 236[M+Na]+.
(2) Синтез 4-[(2,2-диметилпропокси)метил]-N-метокси-N-метилбензамида
[0241]
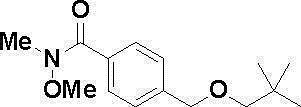
В суспензию гидрида натрия (60% дисперсия в минеральном масле, 286 мг) в N,N-диметилформамиде (23,4 мл) добавляли раствор йодида калия (70,2 мг) и 2,2-диметил-1-пропанола (618 мг) в N,N-диметилформамиде (5,00 мл). После перемешивания смеси при комнатной температуре в течение часа добавляли раствор в тетрагидрофуране (5,00 мл) соединения (1,00 г), полученного на приведенной выше стадии (1). После перемешивания смеси при комнатной температуре в течение 3 часов добавляли насыщенный водный раствор хлорида аммония. Проводили два экстрагирования жидкой смесью н-гексана/этилацетата (1:1) и объединенные органические слои промывали насыщенным солевым раствором. После сушки над безводным сульфатом магния десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-65:35) для того, чтобы получать 4-[(2,2-диметилпропокси)метил]-N-метокси-N-метилбензамид в виде бесцветного масла (304 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,93-0,97 (м, 9H) 3,11-3,16 (м, 2H) 3,36 (д, J=0,3 Гц, 3H) 3,57 (с, 3H) 4,56 (с, 2H) 7,34-7,40 (м, 2H) 7,63-7,69 (м, 2H).
(3) Синтез указанного в заголовке соединения
В раствор в тетрагидрофуране (5,93 мл) соединения (472 мг), полученного на приведенной выше стадии (2), добавляли гидрид диизобутилалюминия (приблизительно 1,0 моль/л, раствор в н-гексане, 2,64 мл) при -78°C. После перемешивания смеси при -78°C в течение 30 минут, при этой температуре добавляли 1 моль/л соляной кислоты (5,00 мл). После перемешивания при комнатной температуре в течение часа, реакционную смесь выливали в соляную кислоту 1 моль/л (20,0 мл). После трех экстрагирований этилацетатом, объединенные органические слои промывали насыщенным солевым раствором. Промытые органические слои пропускали через фазовый разделитель и концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-90:10) для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (302 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,93-0,99 (м, 9H) 3,16 (с, 2H) 4,60 (с, 2H) 7,48-7,53 (м, 2H) 7,82-7,90 (м, 2H) 10,01 (с, 1H).
Ссылочный пример 16-2
4-{[(1-метилциклопропил)метокси]метил} бензальдегид
[0242]

Вместо 2,2-диметил-1-пропанола использовали 1-метилциклопропанметанол и обработку проводили с помощью того же способа, как в ссылочных примерах 16-1(2) и 16-1(3), для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла.
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,32-0,45 (м, 4H) 1,18 (с, 3H) 3,30 (с, 2 Н) 4,61 (с, 2 Н) 7,48-7,54 (м, 2 Н) 7,83-7,90 (м, 2 Н) 10,01 (с, 1 Н).
Ссылочный пример 17-1
4-(1,1-дифторэтил)бензальдегид
[0243]

В раствор 1-бром-4-(1,1-дифторэтил)бензола (1,00 г) в тетрагидрофуране (10,0 мл) добавляли н-бутиллитий (2,69 моль/л, раствор в н-гексане, 1,68 мл) при -80°C и смесь перемешивали при этой температуре в течение 5 минут. Впоследствии N,N-диметилформамид (0,522 мл) добавляли при -80°C и после перемешивания смеси при этой температуре в течение 20 минут добавляли соляную кислоту 2 моль/л (2,50 мл). После приведения реакционной смеси к комнатной температуре проводили два экстрагирования этилацетатом и объединенные органические слои промывали водой. После сушки над безводным сульфатом магния, десикант удаляли с помощью фильтрования; затем фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-90:10) для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (510 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,95 (т, J=18,2 Гц, 3H) 7,64-7,73 (м, 2H) 7,92-7,98 (м, 2H) 10,07 (с, 1H).
MS EI положительн.: 170[M]+.
Ссылочный пример 18-1
4-(дифторметокси)-3,5-диметилбензальдегид
[0244]
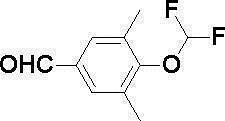
В смесь 4-гидрокси-3,5-диметилбензальдегида (2,00 г), N,N-диметилформамида (54,0 мл) и воды (6,00 мл) добавляли хлордифторацетат натрия (6,09 г) и карбонат калия (3,68 г) и смесь перемешивали при 120°C в течение 4,5 часа. После охлаждения реакционной смеси до комнатной температуры, в нее добавляли воду и проводили два экстрагирования этилацетатом. Объединенные органические слои промывали водой четыре раза и после этого промывали насыщенным солевым раствором. После сушки над безводным сульфатом магния десикант удаляли с помощью фильтрования и затем фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 97:3-88:12) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (2,53 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,35-2,41 (м, 6H) 6,10-6,69 (м, 1H) 7,59-7,64 (м, 2H) 9,93 (с, 1H).
MS ESI/APCI двойн. положительн.: 201[M+H]+.
Ссылочный пример 19-1
[3-(трифторметил)фенил]ацетальдегид
[0245]

В раствор 3-(трифторметил)фенэтилового спирта (2,00 г) в хлороформе (50,0 мл) добавляли периодинан Десса-Мартина (4,70 г) при охлаждении льдом. После доведения до комнатной температуры, реакционную смесь перемешивали в течение часа. Впоследствии добавляли насыщенный водный раствор бикарбоната натрия (25,0 мл) и насыщенный водный раствор тиосульфата натрия (25,0 мл) и смесь энергично перемешивали в течение часа. После разделения фаз органический слой сушили над безводным сульфатом магния, десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 98:2-85:15) для того, чтобы получать указанное в заголовке соединение в виде масла бледно-желтого цвета (1,19 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 3,79 (с, 2H) 7,36-7,62 (м, 4H) 9,77-9,81 (м, 1H).
MS ESI/APCI двойн. отрицательн.: 187[M-H]-.
Ссылочный пример 19-2
[4-(трифторметил)фенил]ацетальдегид
[0246]
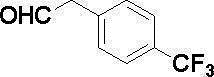
Вместо 3-(трифторметил)фенэтилового спирта использовали 4-(трифторметил)фенэтиловый спирт и обработку проводили с помощью того же способа, как в ссылочном примере 19-1, для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета.
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 3,79 (с, 2H) 7,06-7,75 (м, 4H) 9,77-9,80 (м, 1H).
MS ESI/APCI двойн. отрицательн.: 187[M-H]-.
Ссылочный пример 20-1
3-Циклопропил-4-(трифторметил)бензальдегид
[0247]

(1) Синтез метил-3-циклопропил-4-(трифторметил)бензоата
[0248]

Вместо 6-(4-бромфенокси)пиридин-3-карбальдегида использовали метил 4-(трифторметил)-3-{[(трифторметил)сульфонил]окси}бензоат (см. WO 2007/129745) (2,21 г) и обработку проводили с помощью того же способа, как в ссылочном примере 13-37(2), для того, чтобы получать метил 3-циклопропил-4-(трифторметил)бензоат в виде бесцветного масла (1,42 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,81-0,90 (м, 2H) 1,04-1,14 (м, 2H) 2,16-2,30 (м, 1H) 3,93 (с, 3H) 7,65-7,72 (м, 2H) 7,83-7,93 (м, 1H).
MS ESI/APCI двойн. положительн.: 245[M+H]+.
(2) Синтез [3-циклопропил-4-(трифторметил)фенил]метанола
[0249]

В раствор в дегидратированном тетрагидрофуране (50,0 мл) соединения (1,42 г), полученного на приведенной выше стадии (1), добавляли борогидрид лития(380 мг) и смесь перемешивали при 60°C в течение 4 часов. Впоследствии добавляли соляную кислоту 1 моль/л при охлаждении льдом. После экстрагирования этилацетатом органический слой промывали насыщенным солевым раствором. После сушки над безводным сульфатом магния и удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=98:2-75:25) для того, чтобы получать [3-циклопропил-4-(трифторметил)фенил]метанол в виде бесцветного масла (1,18 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,72-0,83 (м, 2H) 0,97-1,10 (м, 2H) 1,70 (т, J=5,9 Гц, 1H) 2,14-2,28 (м, 1H) 4,71 (д, J=5,9 Гц, 2H) 7,04 (с, 1H) 7,23 (д, J=8,1 Гц, 1H) 7,60 (д, J=8,1 Гц, 1H).
MS ESI/APCI двойн. отрицательн.: 215[M-H]-.
(3) Синтез указанного в заголовке соединения
Соединение (1,18 г), полученное на приведенной выше стадии (2), использовали в качестве исходного материала и обработку проводили с помощью того же способа, как в ссылочном примере 19-1, для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (760 мг).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 0,84-0,90 (м, 2H) 1,08-1,15 (м, 2H) 2,22-2,30 (м, 1H) 7,54 (с, 1H) 7,71-7,76 (м, 1H) 7,77-7,82 (м, 1H) 10,04 (с, 1H).
MS EI положительн.: 214[M]+.
Ссылочный пример 20-2
3-метокси-4-(трифторметил)бензальдегид
[0250]

(1) Синтез метил 3-метокси-4-(трифторметил)бензоата
[0251]
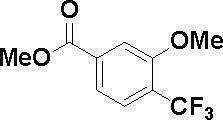
В раствор метил 3-гидрокси-4-(трифторметил)бензоата (см. WO 2007/129745) (2,00 г) в N,N-диметилформамиде (9,00 мл) малыми частями добавляли гидрид натрия (60% дисперсия в минеральном масле, 545 мг) при охлаждении льдом. После перемешивания смеси при комнатной температуре в течение 30 минут добавляли метилйодид (0,849 мл). После перемешивания реакционной смеси при комнатной температуре в течение 2,5 часа, добавляли ледяную воду и проводили два экстрагирования этилацетатом. Объединенные органические слои промывали водой три раза и после этого сушили над безводным сульфатом магния. После удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 95:5-85:15) для того, чтобы получать метил 3-метокси-4-(трифторметил)бензоат в виде бесцветного твердого вещества (2,11 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 3,96 (с, 3H) 3,97 (с, 3H) 7,60-7,72 (м, 3H).
MS EI положительн.: 234[M]+.
(2) Синтез указанного в заголовке соединения
Соединение (2,11 г), полученное на приведенной выше стадии (1), использовали в качестве исходного материала и проводили обработку теми же способами, как в ссылочном примере 20-1(2) и ссылочном примере 19-1, для того, чтобы получать указанное в заголовке соединение в виде масла бледно-желтого цвета (1,26 г).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 3,97-4,01 (м, 3H) 7,49-7,53 (м, 2H) 7,74-7,79 (м, 1H) 10,05 (с, 1H).
MS EI положительн.: 204[M]+.
Ссылочный пример 20-3
3-(дифторметокси)-4-(трифторметил)бензальдегид
[0252]

(1) Синтез метил 3-(дифторметокси)-4-(трифторметил)бензоата
[0253]
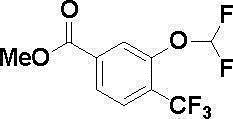
Суспензию метил 3-гидрокси-4-(трифторметил)бензоата (см. WO 2007/129745) (2,00 г), хлордифторацетата натрия (2,08 г) и карбоната калия (2,51 г) в N,N-диметилформамиде (30,0 мл) перемешивали при 100°C в течение 6 часов. После охлаждения реакционной смеси до комнатной температуры, в нее добавляли воду и проводили два экстрагирования этилацетатом. Объединенные органические слои промывали водой три раза и после этого сушили над безводным сульфатом магния. После удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 98:2-85:15) для того, чтобы получать метил 3-(дифторметокси)-4-(трифторметил)бензоат в виде бесцветного масла (1,96 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 3,93-4,01 (м, 3H) 6,28-6,87 (м, 1H) 7,72-7,80 (м, 1H) 7,92-8,02 (м, 2H).
MS EI положительн.: 270[M]+.
(2) Синтез указанного в заголовке соединения
Соединение (1,96 г), полученное на приведенной выше стадии (1), использовали в качестве исходного материала и проводили обработку теми же способами, как в ссылочном примере 20-1(2) и ссылочном примере 19-1 для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (1,35 г).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 6,50-6,79 (м, 1H) 7,79-7,86 (м, 2H) 7,88-7,91 (м, 1H) 10,07 (с, 1H).
MS EI положительн.: 240[M]+.
Ссылочный пример 21-1
4-циклобутил-3-(трифторметил)бензальдегид
[0254]

(1) Синтез 1-[4-бром-2-(трифторметил)фенил]циклобутанола
[0255]
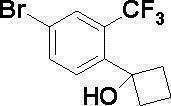
В раствор 5-бром-2-йодобензотрифторида (5,00 г) в дегидратированном тетрагидрофуране (140 мл) добавляли н-бутиллитий (2,69 моль/л, раствор в н-гексане, 5,30 мл) при -78°C и смесь перемешивали при этой температуре в течение 25 минут. После добавления раствора циклобутанона (999 мг) в тетрагидрофуране (5,00 мл), смесь доводили до комнатной температуры и перемешивали в течение 3 суток. После добавления насыщенного водного раствора хлорида аммония при охлаждении льдом, проводили два экстрагирования этилацетатом. Объединенные органические слои промывали водой и после этого сушили над безводным сульфатом магния. После удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 98:2-80:20) для того, чтобы получать 1-[4-бром-2-(трифторметил)фенил]циклобутанол в виде масла бледно-желтого цвета (3,00 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,71-1,88 (м, 1H) 2,20-2,49 (м, 3H) 2,52-2,70 (м, 2H) 7,29-7,35 (м, 1H) 7,62-7,69 (м, 1H) 7,78-7,84 (м, 1H).
MS EI положительн.: 294[M]+.
(2) Синтез 4-бром-1-циклобутил-2-(трифторметил)бензола
[0256]

В раствор в хлороформе (10,0 мл) соединения (1,00 г), полученного на приведенной выше стадии (1), и триэтилсилана (406 мг) добавляли раствор комплекса трифторид бора/диэтиловый эфир (601 мг) в хлороформе (4,00 мл) при -65°C. После доведения до 0°C, смесь перемешивали при этой температуре в течение 30 минут. Впоследствии добавляли карбонат калия (1,08 г) и воду (10,0 мл) и смесь доводили до комнатной температуры. После разделения фаз, органический слой сушили над безводным сульфатом магния. После удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 99:1-90:10) и дополнительно очищали посредством колоночной хроматографии на NH силикагеле (н-гексан) для того, чтобы получать 4-бром-1-циклобутил-2-(трифторметил)бензол в виде бесцветного масла (490 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,78-1,91 (м, 1H) 1,92-2,25 (м, 3H) 2,27-2,42 (м, 2H) 3,83 (кв, J=8,5 Гц, 1H) 7,41-7,47 (м, 1H) 7,59-7,67 (м, 1H) 7,69-7,73 (м, 1H).
MS EI положительн.: 278[M]+.
(3) Синтез указанного в заголовке соединения
Соединение (480 мг), полученное на приведенной выше стадии (2), использовали в качестве исходного материала и обработку проводили с помощью того же способа, как в ссылочном примере 17-1, для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (300 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,82-1,97 (м, 1H) 1,99-2,16 (м, 1H) 2,16-2,33 (м, 2H) 2,33-2,49 (м, 2H) 3,96 (кв, J=8,6 Гц, 1H) 7,74-7,81 (м, 1H) 8,00 -8,08 (м, 1H) 8,09-8,13 (м, 1H) 10,03 (с, 1H).
MS EI положительн.: 228[M]+.
Ссылочный пример 22-1
3-циклобутил-4-(трифторметил)бензальдегид
[0257]

(1) Синтез 1-циклобутил-4-нитро-2-(трифторметил)бензола
[0258]

Смесь 2-йодо-5-нитробензотрифторида (4,62 г), циклобутилбороновой кислоты (4,15 г), дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия(II) (4,15 г), карбоната цезия (22,6 г), толуола (67,0 мл) и воды (33,0 мл) перемешивали в герметизированной пробирке при 80°C в течение 6 часов. После охлаждения реакционной смеси до комнатной температуры, проводили экстрагирование этилацетатом. Объединенные органические слои сушили над безводным сульфатом магния и после удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-94:6) для того, чтобы получать 1-циклобутил-4-нитро-2-(трифторметил)бензол в виде масла бледно-желтого цвета (1,69 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,84-1,99 (м, 1H) 2,00-2,33 (м, 3H) 2,35-2,52 (м, 2H) 3,89-4,05 (м, 1H) 7,74-7,81 (м, 1H) 8,34-8,43 (м, 1H) 8,45-8,50 (м, 1H).
MS ESI/APCI двойн. отрицательн.: 244[M-H]-.
(2) Синтез 4-циклобутил-3-(трифторметил)анилина
[0259]
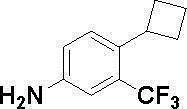
Смесь соединения (1,69 г), полученного на приведенной выше стадии (1), железный порошок (2,14 г), хлорид аммония (442 мг), этанол (26,0 мл) и воду (13,0 мл) перемешивали при 85°C в течение часа. После охлаждения до комнатной температуры, реакционную смесь фильтровали через Celite (зарегистрированный товарный знак). В фильтрат добавляли насыщенный водный раствор бикарбоната натрия и проводили три экстрагирования этилацетатом. Объединенные органические слои сушили над безводным сульфатом магния и после удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=100:0-75:25) для того, чтобы получать 4-циклобутил-3-(трифторметил)анилин в виде масла бледно-желтого цвета (1,34 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,72-1,87 (м, 1H) 1,87-2,21 (м, 3H) 2,21-2,36 (м, 2H) 3,66-3,85 (м, 3H) 6,78-6,86 (м, 1H) 6,88-6,93 (м, 1H) 7,31-7,37 (м, 1H).
MS ESI положительн.: 216[M+H]+.
(3) Синтез 4-циклобутил-2-йодо-5-(трифторметил)анилина
[0260]

В смесь соединения (1,34 г), полученного на приведенной выше стадии (2), бикарбоната натрия (628 мг), хлороформа (32,0 мл) и метанола (8,00 мл) по каплям добавляли раствор монохлорида йода (1,21 г) в хлороформе (8,00 мл) при комнатной температуре в течение периода в 30 минут. Получаемую смесь перемешивали при комнатной температуре в течение двух часов. После добавления 25% раствора метабисульфита натрия в воде (20,0 г) при охлаждении льдом, смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь экстрагировали хлороформом и объединенные органические слои сушили над безводным сульфатом магния. После удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении для того, чтобы получать 4-циклобутил-2-йодо-5-(трифторметил)анилин в виде масла коричневого цвета (2,08 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,71-1,89 (м, 1H) 1,90-2,20 (м, 3H) 2,20-2,37 (м, 2H) 3,60-3,78 (м, 1H) 4,12 (ушир. с, 2H) 6,92 (с, 1H) 7,76 (с, 1H).
MS ESI положительн.: 342[M+H]+.
(4) Синтез 2-циклобутил-4-йодо-1-(трифторметил)бензола
[0261]

В суспензию в ацетонитриле (60,0 мл) соединения (2,08 г), полученного на приведенной выше стадии (3), и нитрита натрия (2,10 г) добавляли концентрированную серную кислоту (6,00 мл) при 0°C в течение периода в 15 минут. После перемешивания смеси при этой температуре в течение часа, добавляли этанол (24,0 мл). После перемешивания при 100°C в течение двух часов, реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь выливали в ледяную воду и проводили два экстрагирования хлороформом. Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия и после этого сушили над безводным сульфатом магния. После удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали два раза посредством колоночной хроматографии на NH силикагеле (с использованием только н-гексана) для того, чтобы получать 2-циклобутил-4-йодо-1-(трифторметил)бензол в виде бесцветного масла (1,25 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,77-1,93 (м, 1H) 1,95-2,26 (м, 3H) 2,27-2,40 (м, 2H) 3,81 (кв, J=8,8 Гц, 1H) 7,26-7,31 (м, 1H) 7,60-7,67 (м, 1H) 7,88 (с, 1H).
(5) Синтез указанного в заголовке соединения
Соединение (1,25 г), полученное на приведенной выше стадии (4), использовали в качестве исходного материала и обработку проводили с помощью того же способа, как в ссылочном примере 17-1, для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (660 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,82-2,17 (м, 2H) 2,17-2,49 (м, 4H) 3,86-4,02 (м, 1H) 7,73-7,83 (м, 2H) 8,09 (с, 1H) 10,11 (с, 1H).
MS EI положительн.: 228[M]+.
Ссылочный пример 23-1
цис-2-фенилциклопропанкарбальдегид
[0262]
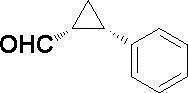
(1) Синтез этил транс-2-фенилциклопропанкарбоксилата и этил цис-2-фенилциклопропанкарбоксилата
[0263]
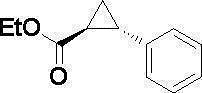
и

В суспензию стирола (3,00 г) и димера ацетата родия(II) (40,0 мг) в 1,2-дихлорэтане (29,0 мл) добавляли раствор этилдиазоацетата (3,03 мл) в 1,2-дихлорэтане (29,0 мл) в течение периода в 4 часа и смесь перемешивали при комнатной температуре в течение 19 часов. Реакционную смесь концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (н-гексан:диэтиловый эфир=20:1) для того, чтобы получать этил транс-2-фенилциклопропанкарбоксилат в виде бесцветного масла (2,42 г) и этил цис-2-фенилциклопропанкарбоксилат в виде бесцветного масла (1,51 г).
Этил транс-2-фенилциклопропанкарбоксилат
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,24-1,36 (м, 4H) 1,54-1,64 (м, 1H) 1,86-1,95 (м, 1H) 2,47-2,58 (м, 1H) 4,17 (кв., J=7,1 Гц, 2H) 7,06-7,13 (м, 2H) 7,16-7,32 (м, 3H).
MS ESI/APCI двойн. положительн.: 191[M+H]+, 213[M+Na]+.
Этил цис-2-фенилциклопропанкарбоксилат
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,97 (т, J=7,1 Гц, 3H) 1,28-1,37 (м, 1H) 1,66-1,76 (м, 1H) 2,02-2,14 (м, 1H) 2,51-2,66 (м, 1H) 3,87 (кв., J=7,1 Гц, 2H) 7,14-7,31 (м, 5H).
MS ESI/APCI двойн. положительн.: 191[M+H]+, 213[M+Na]+.
(2) Синтез (цис-2-фенилциклопропил)метанола
[0264]

В раствор в диэтиловом эфире (12,0 мл) этил цис-2-фенилциклопропанкарбоксилата (1,51 г), полученного на приведенной выше стадии (1), добавляли суспензию гидрида алюминия лития (393 мг) в диэтиловом эфире (12,0 мл) при 0°C. После доведения до комнатной температуры, смесь перемешивали в течение 2 часов. После добавления декагидрата сульфата натрия при 0°C, реакционную смесь доводили до комнатной температуры и перемешивали в течение часа. После удаления нерастворимого вещества с помощью фильтрования, фильтрат концентрировали при пониженном давлении для того, чтобы получать (цис-2-фенилциклопропил)метанол в виде бесцветного масла (1,21 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,84-0,94 (м, 1H) 0,99-1,11 (м, 1H) 1,42-1,58 (м, 1H) 2,20-2,38 (м, 1H) 3,18-3,34 (м, 1H) 3,41-3,55 (м, 1H) 7,11-7,35 (м, 5H).
MS ESI/APCI двойн. положительн.: 171[M+Na]+.
(3) Синтез указанного в заголовке соединения
Соединение (1,21 г), полученное на приведенной выше стадии (2), использовали в качестве исходного материала и обработку проводили с помощью того же способа, как в ссылочном примере 19-1, для того, чтобы получать указанное в заголовке соединение в виде масла бледно-желтого цвета (1,08 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,53-1,65 (м, 1H) 1,84-1,94 (м, 1H) 2,08-2,20 (м, 1H) 2,78-2,89 (м, 1H) 7,19-7,37 (м, 5H) 8,63-8,71 (м, 1H).
MS ESI/APCI двойн. положительн.: 147[M+H]+, 169[M+Na]+.
Ссылочный пример 23-2
транс-2-фенилциклопропанкарбальдегид
[0265]
Этил транс-2-фенилциклопропанкарбоксилат (1,00 г), полученный в ссылочном примере 23-1(1), использовали в качестве исходного материала и проводили обработку теми же способами, как в ссылочном примере 23-1(2) и ссылочном примере 19-1, для того, чтобы получать грубо очищенный продукт (620 мг), содержащий указанное в заголовке соединение.
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,49-1,59 (м, 1H) 1,70-1,78 (м, 1H) 2,13-2,23 (м, 1H) 2,59-2,68 (м, 1H) 7,08-7,16 (м, 2H) 7,18-7,34 (м, 3H) 9,30-9,35 (м, 1H).
MS ESI/APCI двойн. положительн.: 147[M+H]+.
Ссылочный пример 23-3
2-(4-фторфенил)циклопропанкарбальдегид
[0266]
Вместо стирола использовали 4-фторстирол (10,0 г) и проводили обработку теми же способами, как в ссылочном примере 23-1(1) и (2), а также ссылочном примере 19-1, для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (4,48 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,31-1,55 (м, 1H) 1,59-1,78 (м, 1H) 1,80-2,19 (м, 1H) 2,53-2,68 (м, 1H) 6,91-7,16 (м, 4H) 9,34 (д, J=4,5 Гц, 1H).
MS ESI/APCI двойн. положительн.: 165[M+H]+.
Ссылочный пример 24-1 и Ссылочный пример 24-2
транс-3-фенилциклобутанкарбальдегид (Ссылочный пример 24-1) и цис-3-фенилциклобутанкарбальдегид (Ссылочный пример 24-2)
[0267]
и
В раствор диэтил изоцианометилфосфоната (1,47 г) в тетрагидрофуране (45,0 мл) добавляли н-бутиллитий (2,69 моль/л, раствор в н-гексане, 2,99 мл) при -78°C и после этого смесь перемешивали при этой температуре в течение 80 минут. После добавления раствора 3-фенилциклобутанона (1,03 г) в тетрагидрофуране (15,0 мл) при -78°C в течение периода в 30 минут, смесь перемешивали при комнатной температуре в течение 4 часов. После добавления концентрированной соляной кислоты (12,0 мл) при комнатной температуре, смесь перемешивали при этой температуре в течение 18 часов. В реакционную смесь добавляли воду, которую затем экстрагировали этилацетатом два раза. Объединенные органические слои промывали насыщенным солевым раствором и после этого сушили над безводным сульфатом магния. Нерастворимое вещество удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 99:1-96:4) для того, чтобы получать указанное в заголовке соединение из ссылочного примера 24-1 в виде бесцветного масла (160 мг) и указанное в заголовке соединение из ссылочного примера 24-2 в виде бесцветного масла (390 мг).
транс-3-фенилциклобутанкарбальдегид (Ссылочный пример 24-1)
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,30-2,58 (м, 2H) 2,65-2,83 (м, 2H) 3,08-3,28 (м, 1H) 3,47-3,68 (м, 1H) 7,17-7,38 (м, 5H) 9,94-9,97 (м, 1H).
цис-3-фенилциклобутанкарбальдегид (Ссылочный пример 24-2)
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,30-2,49 (м, 2H) 2,51-2,65 (м, 2H) 3,13-3,29 (м, 1H) 3,53-3,68 (м, 1H) 7,17-7,37 (м, 5H) 9,71-9,75 (м, 1H).
Ссылочный пример 24-3
транс-3-(4-фторфенил)циклобутанкарбальдегид
[0268]
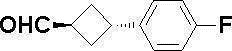
Вместо 3-фенилциклобутанона использовали 3-(4-фторфенил)циклобутанон (4,63 г) и обработку проводили с помощью того же способа, как в ссылочном примере 24-1, для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (720 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,26-2,45 (м, 2H) 2,63-2,81 (м, 2H) 3,09-3,24 (м, 1H) 3,47-3,65 (м, 1H) 6,94-7,07 (м, 2H) 7,12-7,23 (м, 2H) 9,95 (д, J=1,7 Гц, 1H).
MS ESI/APCI двойн. отрицательн.: 177[M-H]-.
Ссылочный пример 25-1
4-бензилциклогексанон
[0269]
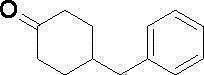
(1) Синтез 9-бензил-3,3-диметил-1,5-диоксаспиро[5.5]ундекан-9-ол
[0270]
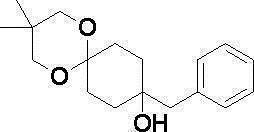
В раствор 1,4-циклогександион моно-2,2-диметилтриметилен кеталя (3,76 г) и хлорида цинка (приблизительно 1,0 моль/л, раствор в диэтиловом эфире, 1,90 мл) в тетрагидрофуране (63,0 мл) добавляли бензилбромид магния (приблизительно 1,0 моль/л, раствор в тетрагидрофуране, 24,7 мл) при 0°C и после этого смесь перемешивали при этой температуре в течение 2,5 часа. В реакционную смесь добавляли насыщенный водный раствор хлорида аммония и проводили три экстрагирования этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором и после этого сушили над безводным сульфатом магния. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 95:5-60:40) для того, чтобы получать 9-бензил-3,3-диметил-1,5-диоксаспиро[5.5]ундекан-9-ол в виде бесцветного твердого вещества (1,28 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,97 (с, 6H) 1,43-1,89 (м, 6H) 1,95-2,13 (м, 2H) 2,76 (с, 2H) 3,41-3,58 (м, 4H) 7,16-7,36 (м, 5H).
MS ESI/APCI двойн. положительн.: 313[M+Na]+.
(2) Синтез указанного в заголовке соединения
В раствор в толуоле (44,0 мл) соединения (1,28 г), полученного на приведенной выше стадии (1), добавляли моногидрат п-толуолсульфоновой кислоты (84,0 мг) и после этого смесь нагревали с обратным холодильником в течение 3 часов с использованием аппарата Дина-Старка. После охлаждения до комнатной температуры, реакционную смесь концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-50:50). Суспензию получаемого очищенного продукта (935 мг) и 20% гидроксида палладия/углерода (93,5 мг) в метаноле (11,3 мл) перемешивали при комнатной температуре в течение 5 часов в атмосфере водорода. Реакционную смесь фильтровали через Celite (зарегистрированный товарный знак). Фильтрат концентрировали при пониженном давлении. В раствор получаемого остатка (938 мг) в тетрагидрофуране (34,3 мл) добавляли соляную кислоту 1 моль/л (9,30 мл) при 0°C и смесь перемешивали при комнатной температуре в течение 14,5 часа. После концентрирования перемешанной смеси при пониженном давлении, проводили три экстрагирования этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором. После пропускания через фазовый разделитель, промытые органические слои концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 90:10-50:50) для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (225 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,36-1,52 (м, 2H) 1,95-2,10 (м, 3H) 2,22-2,45 (м, 4H) 2,62 (д, J=6,7 Гц, 2H) 7,10-7,36 (м, 5H).
MS ESI/APCI двойн. положительн.: 189[M+H]+, 211[M+Na]+.
Ссылочный пример 26-1
1-(бифенил-4-ил)пропан-1-он
[0271]
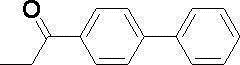
(1) Синтез 1-(бифенил-4-ил)пропан-1-ола
[0272]

В раствор 4-фенилбензальдегида (1,20 г) в диэтиловом эфире (13,2 мл) добавляли этилбромид магния (приблизительно 3,0 моль/л, раствор в диэтиловом эфире, 3,29 мл) при 0°C. После перемешивания смеси при комнатной температуре в течение 3 часов осадок собирали с помощью фильтрования. После растворения собранного осадка в жидкой смеси этилацетата и насыщенного водного раствора хлорида аммония, проводили три экстрагирования этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором. После пропускания через фазовый разделитель, промытые органические слои концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-70:30) для того, чтобы получать 1-(бифенил-4-ил)пропан-1-ол в виде бесцветного твердого вещества (1,27 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,96 (т, J=7,4 Гц, 3H) 1,72-1,98 (м, 2H) 4,59-4,71 (м, 1H) 7,29-7,48 (м, 5H) 7,54-7,63 (м, 4H).
MS EI положительн.: 212[M]+.
(2) Синтез указанного в заголовке соединения
В раствор в диэтиловом эфире (30,0 мл) соединения (1,27 г), полученного на приведенной выше стадии (1), добавляли оксид марганца(IV) (9,57 г) и смесь перемешивали при комнатной температуре в течение 45 часов. После удаления нерастворимого вещества с помощью фильтрования, фильтрат концентрировали при пониженном давлении. В раствор получаемого остатка в ацетоне (60,0 мл) добавляли реактив Джонса {см. Org. Synth., Coll. Vol. VI, 542 (1988)} (1,20 мл) до тех пор, пока не исчезала окраска реактива Джонса. Реакционную смесь концентрировали при пониженном давлении и в остаток добавляли этилацетат и воду. Экстрагирование этилацетатом проводили три раза, и объединенные органические слои промывали насыщенным солевым раствором. После пропускания через фазовый разделитель, промытые органические слои концентрировали при пониженном давлении. Получаемый остаток перекристаллизовывали с н-гексаном для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (921 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,26 (т, J=7,3 Гц, 3H) 3,04 (кв., J=7,3 Гц, 2H) 7,34-7,52 (м, 3H) 7,58-7,73 (м, 4H) 8,00-8,09 (м, 2H).
MS ESI/APCI двойн. положительн.: 211[M+H]+, 233[M+Na]+.
Ссылочный пример 27-1
1-(Бифенил-4-ил)циклопропанамин
[0273]
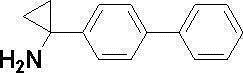
Соединение 4-цианобифенил (2,83 г) использовали в качестве исходного материала и обработку проводили с помощью того же способа, как в ссылочном примере 4-1(2), для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (1,06 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,99-1,07 (м, 2H) 1,08-1,15 (м, 2H) 7,28-7,49 (м, 5H) 7,51-7,62 (м, 4H).
MS ESI/APCI двойн. положительн.: 210[M+H]+.
Ссылочный пример 27-2
1-[4-(трифторметил)фенил]циклопропанамин
[0274]

Соединение 4-(трифторметил)бензонитрил (5,18 г) использовали в качестве исходного материала и обработку проводили с помощью того же способа, как в ссылочном примере 4-1(2) для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (2,92 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,00-1,07 (м, 2H) 1,11-1,19 (м, 2H) 7,31-7,45 (м, 2H) 7,49-7,64 (м, 2H).
MS ESI/APCI двойн. положительн.: 202[M+H]+.
Ссылочный пример 27-3
2-[4-(трифторметил)фенил]пропан-2-амин
[0275]
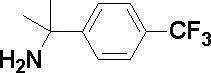
В раствор 4-(трифторметил)бензонитрила (3,01 г) в диэтиловом эфире (88,0 мл) добавляли метилбромид магния (приблизительно 3,0 моль/л, раствор в диэтиловом эфире, 17,6 мл) и смесь перемешивали при комнатной температуре в течение 40 минут. В реакционную смесь медленно добавляли тетраизопропил ортотитанат (5,15 мл) и после этого смесь нагревали с обратным холодильником в течение 6 часов. После охлаждения смеси до 0°C добавляли 20% водный раствор гидроксида натрия и смесь перемешивали при комнатной температуре в течение часа. После разделения фаз, водный слой экстрагировали диэтиловым эфиром два раза. Объединенные органические слои пропускали через фазовый разделитель и после этого концентрировали при пониженном давлении. Получаемый остаток растворяли в 5% соляной кислоте и промывали диэтиловым эфиром два раза. Водный слой делали основным с использованием 20% водного раствора гидроксида натрия и экстрагировали диэтиловым эфиром три раза. Объединенные органические слои промывали насыщенным солевым раствором и после этого пропускали через фазовый разделитель для концентрирования при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=95:5-10:90) для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (1,81 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,51 (с, 6H) 7,54-7,69 (м, 4H).
MS ESI/APCI двойн. положительн.: 204[M+H]+.
Ссылочный пример 28-1
Гидрохлорид метил (3S)-3-амино-4-метоксибутаноата
[0276]
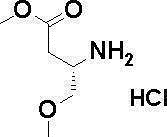
(1) Синтез трет-бутил [(2S)-1-циано-3-гидроксипропан-2-ил]карбамата
[0277]
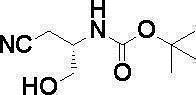
В раствор N-α-(трет-бутоксикарбонил)-β-циано-L-аланина (1,00 г) в тетрагидрофуране (13 мл) последовательно добавляли изобутил хлорформиат (674 мкл) и триэтиламин (716 мкл) при 0°C и смесь перемешивали при этой температуре в течение 2 часов. После удаления осадка с помощью фильтрования, фильтрат концентрировали при пониженном давлении. В смесь получаемого остатка, тетрагидрофуран (13 мл) и воды (4 мл) добавляли борогидрид натрия (530 мг) и смесь перемешивали при комнатной температуре в течение 30 минут. После добавления воды, проводили экстрагирование этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором и после этого пропускали через фазовый разделитель для концентрирования при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=80:20-40:60) для того, чтобы получать трет-бутил [(2S)-1-циано-3-гидроксипропан-2-ил]карбамат в виде бесцветного масла (650 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,46 (с, 9H) 2,10-2,18 (м, 1H) 2,63-2,78 (м, 2H) 3,69-3,88 (м, 2H) 3,89-4,03 (м, 1H) 5,02 (ушир. с, 1H).
MS ESI/APCI двойн. положительн.: 223[M+Na]+.
(2) Синтез трет-бутил [(2S)-1-циано-3-метоксипропан-2-ил]карбамата
[0278]
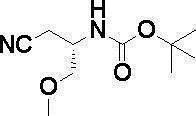
В раствор в тетрагидрофуране (10 мл) соединения (650 мг), полученного на приведенной выше стадии (1), добавляли гидрид натрия (60% дисперсия в минеральном масле, 143 мг) при охлаждении льдом. После перемешивания смеси при той же температуре в течение 10 минут добавляли метилйодид (603 мкл). После перемешивания смеси при той же температуре в течение 30 минут и затем при комнатной температуре в течение часа, добавляли воду и проводили экстрагирование этилацетатом. Объединенные органические слои пропускали через фазовый разделитель и концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 90:10-60:40) для того, чтобы получать трет-бутил [(2S)-1-циано-3-метоксипропан-2-ил]карбамат в виде бесцветного масла (247 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,45 (с, 9H) 2,69 (д, J=6,2 Гц, 2H) 3,39 (с, 3H) 3,41-3,49 (м, 1H) 3,53-3,61 (м, 1H) 4,02 (ушир. с, 1H) 4,96 (ушир. с, 1H).
MS ESI/APCI двойн. положительн.: 237[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 213[M-H]-.
(3) Синтез указанного в заголовке соединения
В раствор в 1,4-диоксане (2,0 мл) соединения (247 мг), полученного на приведенной выше стадии (2), добавляли концентрированную соляную кислоту (3,0 мл) и смесь перемешивали при 100°C в течение часа. После охлаждения до комнатной температуры, реакционную смесь концентрировали при пониженном давлении. Получаемый остаток использовали в качестве исходного материала и обработку проводили с помощью того же способа, как в ссылочном примере 3-4, для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (247 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,60-2,84 (м, 2H) 3,30 (с, 3H) 3,44-3,59 (м, 3H) 3,64 (с, 3H).
MS ESI/APCI двойн. положительн.: 148[M+H]+.
Ссылочный пример 28-2
Гидрохлорид метил (3S)-3-амино-4-(диметиламино)бутаноата
[0279]
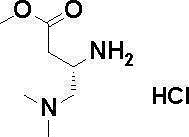
(1) Синтез (2S)-2-[(трет-бутоксикарбонил)амино]-3-цианопропил 4-метилбензолсульфоната
[0280]
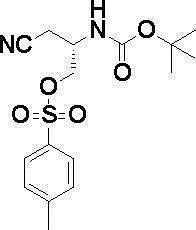
В раствор в хлороформе (20 мл) соединения (820 мг), полученного в ссылочном примере 28-1(1), добавляли хлорид п-толуолсульфонила (1,56 г) и триэтиламин (1,14 мл) и смесь перемешивали при комнатной температуре в течение 3 часов. После добавления дополнительного хлорида п-толуолсульфонила (1,56 г) и триэтиламина (1,14 мл), смесь перемешивали при комнатной температуре в течение 30 минут. Впоследствии добавляли насыщенный водный раствор бикарбоната натрия и проводили экстрагирование хлороформом. Объединенные органические слои пропускали через фазовый разделитель и после этого концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 80:20-40:60) для того, чтобы получать (2S)-2-[(трет-бутоксикарбонил)амино]-3-цианопропил 4-метилбензолсульфонат в виде бесцветного твердого вещества (1,32 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,43 (с, 9H) 2,47 (с, 3H) 2,63-2,71 (м, 2H) 4,02-4,21 (м, 3H) 4,86-5,01 (м, 1H) 7,39 (д, J=8,5 Гц, 2H) 7,81 (д, J=8,5 Гц, 2H).
MS ESI/APCI двойн. положительн.: 377[M+Na]+.
(2) Синтез трет-бутил [(2S)-1-циано-3-(диметиламино)пропан-2-ил]карбамата
[0281]

В раствор в этаноле (20 мл) соединения (1,32 г), полученного на приведенной выше стадии (1), добавляли диметиламин (приблизительно 50%, водный раствор, 3,92 мл) и триэтиламин (519 мкл) и смесь перемешивали при 80°C в течение часа. После охлаждения до комнатной температуры, реакционную смесь концентрировали при пониженном давлении. В получаемый остаток добавляли насыщенный водный раствор бикарбоната натрия и проводили экстрагирование этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором и после пропускания через фазовый разделитель их концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 99:1-95:5) для того, чтобы получать трет-бутил [(2S)-1-циано-3-(диметиламино)пропан-2-ил]карбамата в виде масла бледно-желтого цвета (340 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,45 (с, 9H) 2,25 (с, 6H) 2,31-2,55 (м, 2H) 2,62-2,76 (м, 1H) 2,78-2,91 (м, 1H) 3,74-3,90 (м, 1H) 4,99 (ушир. с, 1H).
MS ESI/APCI двойн. положительн.: 228[M+H]+, 250[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 226[M-H]-.
(3) Синтез указанного в заголовке соединения
Соединение (340 мг), полученное на приведенной выше стадии (2), использовали в качестве исходного материала, и обработку проводили с помощью того же способа, как в ссылочном примере 28-1(3), для того, чтобы получать указанное в заголовке соединение. Следует отметить, что указанное в заголовке соединение использовали в последующей реакции, поскольку оно оставалось неочищенным продуктом.
Ссылочный пример 29-1
Метил (3-амино-5-оксопирролидин-3-ил)ацетат
[0282]
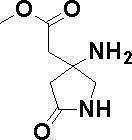
(1) Синтез 2-трет-бутил 1,3-диметил 2-цианопропан-1,2,3-трикарбоксилата
[0283]
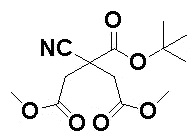
В раствор трет-бутил цианоацетата (10,0 г) в толуоле (100 мл) добавляли гидрид натрия (60% дисперсия в минеральном масле, 2,83 г) при охлаждении льдом. После перемешивания при 80°C в течение часа, смесь охлаждали до 50°C и медленно добавляли метил бромацетат (6,51 мл). После добавления тетрагидрофурана (15 мл), смесь перемешивали при 80°C в течение двух часов. Реакционную смесь охлаждали льдом и затем добавляли гидрид натрия (60% дисперсия в минеральном масле, 2,83 г). После перемешивания при 80°C в течение часа, смесь охлаждали до 50°C и затем медленно добавляли метил бромацетат (6,51 мл). Впоследствии добавляли тетрагидрофуран (15 мл) и смесь перемешивали при 80°C в течение двух часов. После охлаждения реакционной смеси до комнатной температуры добавляли насыщенный водный раствор хлорида аммония и воды и проводили экстрагирование этилацетатом. Объединенные органические слои последовательно промывали водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором затем сушили над безводным сульфатом магния. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=100:0-40:60) для того, чтобы получать 2-трет-бутил 1,3-диметил 2-цианопропан-1,2,3-трикарбоксилат в виде масла бледно-желтого цвета (17,8 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,52 (с, 9H) 2,96-3,12 (м, 4H) 3,74 (с, 6H).
MS ESI/APCI двойн. положительн.: 286[M+H]+, 308[M+Na]+.
(2) Синтез трет-бутил 3-(2-метокси-2-оксоэтил)-5-оксопирролидин-3-карбоксилата
[0284]
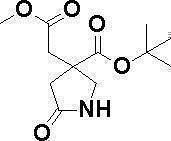
В раствор в метаноле (70 мл) соединения (5,00 г), полученного на приведенной выше стадии (1), добавляли катализатор никель Ренея (приблизительно 7,5 г). Смесь перемешивали при 70°C в течение 8 часов в атмосфере водорода при 0,4 мегапаскаля (мПа). После охлаждения до комнатной температуры, реакционную смесь фильтровали через Celite (зарегистрированный товарный знак) и фильтрат концентрировали при пониженном давлении. В получаемый остаток добавляли диэтиловый эфир (50 мл) и гексан (10 мл) и смесь перемешивали при комнатной температуре в течение 15 минут. Получаемый осадок собирали с помощью фильтрования для того, чтобы получать трет-бутил 3-(2-метокси-2-оксоэтил)-5-оксопирролидин-3-карбоксилат в виде бесцветного твердого вещества (1,75 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,46 (с, 9H) 2,33 (д, J=17,4 Гц, 1H) 2,79-2,90 (м, 3H) 3,35 (дд, J=10,3, 0,8 Гц, 1H) 3,69 (с, 3H) 3,84 (дд, J=10,3, 0,8 Гц, 1H) 5,73 (ушир. с, 1H).
MS ESI/APCI двойн. положительн.: 258[M+H]+, 280[M+Na]+.
(3) Синтез метил (3-{[(бензилокси)карбонил]амино}-5-оксопирролидин-3-ил)ацетата
[0285]

В соединение (3,41 г), полученное на приведенной выше стадии (2), добавляли трифторуксусную кислоту (30 мл) и смесь перемешивали при комнатной температуре в течение 4 часов. После концентрирования при пониженном давлении, в получаемый остаток добавляли хлороформ, который снова концентрировали при пониженном давлении. В раствор получаемого остатка в бензоле (50 мл) последовательно добавляли тетрагидрофуран (12 мл), триэтиламин (3,71 мл), дифенилфосфорил азид (3,73 мл) и бензиловый спирт (1,79 мл) и смесь перемешивали в течение 4 часов при нагревании с обратным холодильником. После охлаждения реакционной смеси до комнатной температуры, в нее добавляли этилацетат, затем ее последовательно промывали водой, водным раствором 10% лимонной кислоты, насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором и сушили над безводным сульфатом магния. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 100:0-85:15) для того, чтобы получать метил (3-{[(бензилокси)карбонил]амино}-5-оксопирролидин-3-ил)ацетат в виде бесцветной смолы (1,91 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,57 (д, J=17,2 Гц, 1H) 2,78 (д, J=17,2 Гц, 1H) 2,91 (д, J=16,2 Гц, 1H) 3,01 (д, J=16,2 Гц, 1H) 3,51 (д, J=10,6 0,8 Гц, 1H) 3,67 (с, 3H) 3,70-3,81 (м, 1H) 5,08 (с, 2H) 5,46 (ушир.с, 1Н) 5,69 (ушир.с, 1Н) 7,27-7,42 (м, 5Н).
MS ESI/APCI двойн. положительн.: 307[M+H]+, 329[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 341 [M+Cl]-.
(4) Синтез указанного в заголовке соединения
В раствор в метаноле (30 мл) соединения (1,91 г), полученного на приведенной выше стадии (3), добавляли 20% гидроксид палладия/углерод (191 мг) и смесь перемешивали при комнатной температуре в течение часа в атмосфере водорода. Реакционную смесь фильтровали через Celite (зарегистрированный товарный знак). Фильтрат концентрировали при пониженном давлении для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (1,12 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,32 (д, J=16,7 Гц, 1H) 2,47 (д, J=16,7 Гц, 1H) 2,61-2,75 (м, 2H) 3,33 (д, J=10,2 Гц, 1H) 3,46 (д, J=10,2 Гц, 1H) 3,72 (с, 3H) 5,72 (ушир. с, 1H).
MS ESI/APCI двойн. положительн.: 173[M+H]+.
Ссылочный пример 29-2
Метил (3-амино-1-метил-5-оксопирролидин-3-ил)ацетат
[0286]

(1) Синтез трет-бутил 3-(2-метокси-2-оксоэтил)-1-метил-5-оксопирролидин-3-карбоксилата
[0287]
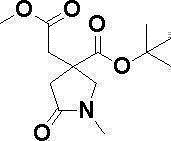
Соединение (4,0 г), полученное в ссылочном примере 29-1(2), использовали в качестве исходного материала и обработку проводили с помощью того же способа, как в ссылочном примере 28-1(2), для того, чтобы получать трет-бутил 3-(2-метокси-2-оксоэтил)-1-метил-5-оксопирролидин-3-карбоксилат в виде масла бледно-желтого цвета (3,74 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,45 (с, 9H) 2,39 (д, J=17,3 Гц, 1H) 2,70 (д, J=16,6 Гц, 1H) 2,79-2,90 (м, 5H) 3,32 (д, J=10,6 Гц, 1H) 3,69 (с, 3H) 3,86 (д, J= 10,6 Гц, 1H).
MS ESI/APCI двойн. положительн.: 272[M+H]+.
(2) Синтез указанного в заголовке соединения
Соединение (3,74 г), полученное на приведенной выше стадии (1), использовали в качестве исходного материала и проводили обработку теми же способами, как в ссылочном примере 29-1(3) и (4), для того, чтобы получать указанное в заголовке соединение.
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,36-2,45 (м, 1H) 2,47-2,56 (м, 1H) 2,59-2,72 (м, 2H) 2,85-2,89 (м, 3H) 3,33 (д, J=10,4 Гц, 1H) 3,47 (д, J=10,4 Гц, 1H) 3,72 (с, 3H).
MS ESI/APCI двойн. положительн.: 187[M+H]+, 209[M+Na]+.
Ссылочный пример 30-1
Метил L-α-аспарагинат
[0288]
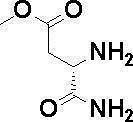
В раствор N-α-(9-флуоренилметоксикарбонил)-L-аспарагинового α-амида (744 мг) в тетрагидрофуране (8 мл) добавляли триметилсилилдиазометан (2,0 моль/л, раствор в диэтиловом эфире, 1,20 мл) и метанол (808 мкл) при охлаждении льдом. После доведения до комнатной температуры, смесь перемешивали в течение 2,5 часа. Затем реакционную смесь концентрировали при пониженном давлении. В раствор получаемого остатка в ацетонитриле (14 мл) добавляли диэтиламин (621 мкл) и смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении и в получаемый остаток добавляли диэтиловый эфир и смесь перемешивали. Получаемый осадок собирали с помощью фильтрования для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (274 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,37 (дд, J=15,6, 8,2 Гц, 1H) 2,62 (дд, J=15,6, 5,1 Гц, 1H) 3,46 (дд, J=8,2, 5,1 Гц, 1H) 3,58 (с, 3H).
MS ESI/APCI двойн. положительн.: 169[M+Na]+.
Ссылочный пример 30-2
Метил D-α-аспарагинат
[0289]
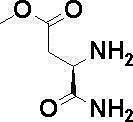
Вместо N-α-(9-флуоренилметоксикарбонил)-L-аспарагинового α-амида использовали N-б-(9-флуоренилметоксикарбонил)-D-аспарагиновый б-амид и обработку проводили с помощью того же способа, как в ссылочном примере 30-1, для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла. Следует отметить, что указанное в заголовке соединение использовали в последующей реакции, поскольку оно оставалось неочищенным продуктом.
Ссылочный пример 31-1
6-[(1-метилциклопропил)метокси]пиридин-3-карбальдегид
[0290]
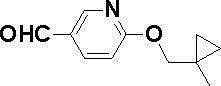
(1) Синтез 6-хлор-N-метокси-N-метилпиридин-3-карбоксамида
[0291]

Вместо 4-(бромметил)бензойной кислоты использовали 6-хлорникотиновую кислоту (6,50 г) и обработку проводили с помощью того же способа, как в ссылочном примере 16-1(1), для того, чтобы получать 6-хлор-N-метокси-N-метилпиридин-3-карбоксамид в виде бесцветного масла (7,55 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 3,39 (с, 3H) 3,56 (с, 3H) 7,39 (дд, J=8,3, 0,7 Гц, 1H) 8,03 (дд, J=8,3, 2,3 Гц, 1H) 8,78 (дд, J=2,3, 0,7 Гц, 1H).
MS ESI/APCI двойн. положительн.: 201[M+H]+.
(2) Синтез н-метокси-N-метил-6-[(1-метилциклопропил)метокси]пиридин-3-карбоксамида
[0292]
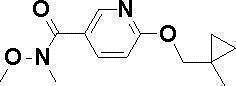
В раствор трет-бутоксида калия (1,68 г) в тетрагидрофуране (30 мл) добавляли раствор 1-метилциклопропанметанола (1,29 г) в тетрагидрофуране (5 мл) и смесь перемешивали при комнатной температуре в течение 10 минут. После реакционную смесь охлаждали до 0°C, добавляли раствор в тетрагидрофуране (5 мл) соединения (3,00 г), полученного на приведенной выше стадии (1), и после доведения до комнатной температуры реакционную смесь перемешивали в течение часа. Затем реакционную смесь концентрировали при пониженном давлении и получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=95:5-50:50) для того, чтобы получать N-метокси-N-метил-6-[(1-метилциклопропил)метокси]пиридин-3-карбоксамид в виде бесцветного масла (2,02 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,38-0,47 (м, 2H) 0,52-0,60 (м, 2H) 1,23 (с, 3H) 3,37 (с, 3H) 3,58 (с, 3H) 4,15 (с, 2H) 6,73-6,84 (м, 1H) 8,00 (дд, J=8,7, 2,3 Гц, 1H) 8,60 (дд, J=2,3, 0,7 Гц, 1H).
MS ESI/APCI двойн. положительн.: 251[M+H]+.
(3) Синтез указанного в заголовке соединения
Использовали соединение (2,00 г), полученное на приведенной выше стадии (2), и обработку проводили с помощью того же способа, как в ссылочном примере 16-1(3), для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (1,51 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,39-0,48 (м, 2H) 0,53-0,62 (м, 2H) 1,23 (с, 3H) 4,20 (с, 2H) 6,83-6,92 (м, 1H) 8,02-8,11 (м, 1H) 8,52-8,63 (м, 1H) 9,91-9,97 (м, 1H).
MS ESI/APCI двойн. положительн.: 192[M+H]+.
Ссылочный пример 31-2
6-(2-циклопропилэтокси)пиридин-3-карбальдегид
[0293]
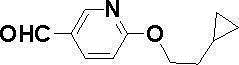
Вместо 1-метилциклопропанметанола использовали 2-циклопропилэтанол (1,29 г) и обработку проводили с помощью того же способа, как в ссылочном примере 31-1, для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (1,25 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,08-0,18 (м, 2H) 0,42-0,54 (м, 2H) 0,75-0,89 (м, 1H) 1,63-1,75 (м, 2H) 4,48 (т, J=6,8 Гц, 2H) 6,77-6,88 (м, 1H) 8,01-8,10 (м, 1H) 8,58-8,67 (м, 1H) 9,92-9,98 (м, 1H).
MS ESI/APCI двойн. положительн.: 192[M+H]+.
Ссылочный пример 32-1
транс-4-(4-хлорфенокси)циклогексанкарбальдегид
[0294]
(1) Синтез цис-4-гидрокси-N-метокси-N-метилциклогексанкарбоксамида
[0295]
Вместо 4-(бромметил)бензойной кислоты использовали цис-4-гидроксициклогексанкарбоновую кислоту (1,45 г) и обработку проводили с помощью того же способа, как в ссылочном примере 16-1(1), для того, чтобы получать цис-4-гидрокси-N-метокси-N-метилциклогексанкарбоксамид в виде масла желтого цвета (0,87 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,49-1,74 (м, 4H) 1,79-2,02 (м, 4H) 2,65-2,79 (м, 1H) 3,19 (с, 3H) 3,70 (с, 3H) 3,99-4,08 (м, 1H).
MS ESI/APCI двойн. положительн.: 188[M+H]+.
(2) Синтез транс-4-(4-хлорфенокси)-N-метокси-N-метилциклогексанкарбоксамида
[0296]
Вместо 2-циклопропилэтанола и 4-гидроксибензальдегида, соответственно, использовали соединение (850 мг), полученное на приведенной выше стадии (1), и п-хлорфенол (700 мг) и обработку проводили с помощью того же способа, как в ссылочном примере 11-1, для того, чтобы получать транс-4-(4-хлорфенокси)-N-метокси-N-метилциклогексанкарбоксамид в виде бесцветного твердого вещества (458 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,37-1,75 (м, 4H) 1,86-1,98 (м, 2H) 2,16-2,29 (м, 2H) 2,65-2,79 (м, 1H) 3,19 (с, 3H) 3,72 (с, 3H) 4,06-4,26 (м, 1H) 6,78-6,88 (м, 2H) 7,18-7,25 (м, 2H).
MS ESI/APCI двойн. положительн.: 298[M+H]+.
(3) Синтез указанного в заголовке соединения
Использовали соединение (455 мг), полученное на приведенной выше стадии (2), и обработку проводили с помощью того же способа, как в ссылочном примере 23-1(2), для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (379 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,38-1,64 (м, 4H) 2,01-2,21 (м, 4H) 2,25-2,39 (м, 1H) 4,03-4,27 (м, 1H) 6,78-6,87 (м, 2H) 7,18-7,26 (м, 2H) 9,66-9,72 (м, 1H).
MS ESI/APCI двойн. отрицательн.: 237[M-H]-.
[0297] В следующих ссылочных примерах с 32-2 до 32-9 использовали соответствующие фенолы торговых сортов и соответствующие спирты торговых сортов и обработку проводили с помощью способа, описанного в ссылочном примере 32-1, или его модификаций для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблице 11-1.
[0298]
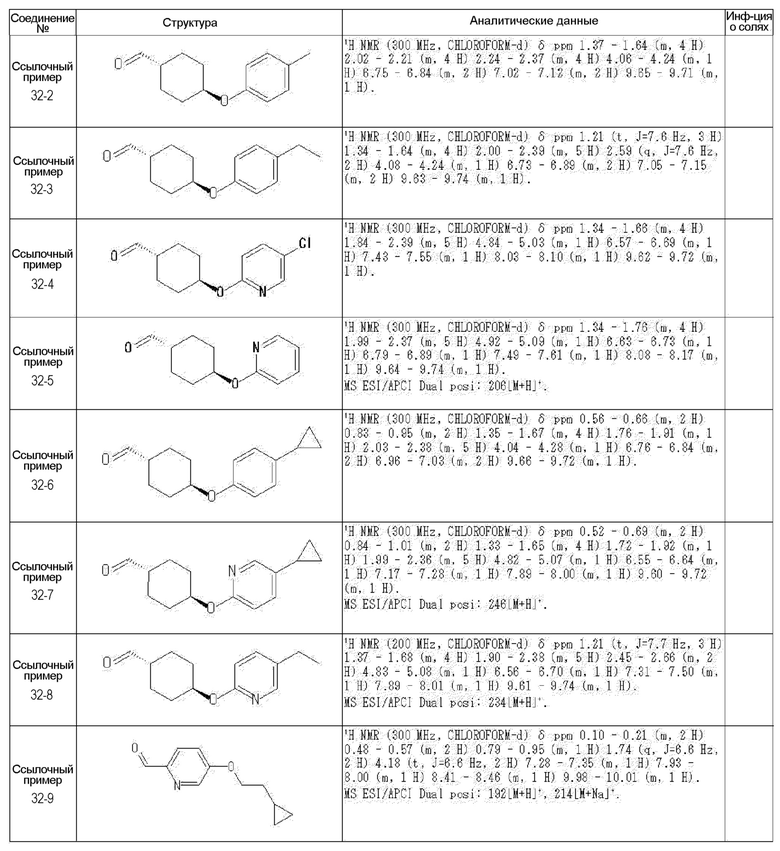
Ссылочный пример 33-1
5-метил-6-(2,2,2-трифторэтокси)пиридин-3-карбальдегид
[0299]

(1) Синтез 5-метил-6-(2,2,2-трифторэтокси)пиридин-3-карбоновой кислоты
[0300]
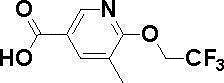
Вместо 3,4-дифторбензальдегида использовали 2-фтор-3-метилпиридин-5-карбоновую кислоту (2,00 г) и обработку проводили с помощью того же способа, как в ссылочном примере 12-1, для того, чтобы получать 5-метил-6-(2,2,2-трифторэтокси)пиридин-3-карбоновую кислоту в виде бесцветного твердого вещества (3,57 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,23 (с, 3H) 5,09 (кв., J=9,0 Гц, 2H) 8,02-8,17 (м, 1H) 8,52-8,63 (м, 1H) 13,11 (ушир. с, 1H).
MS ESI/APCI двойн. отрицательн.: 234[M-H]-.
(2) Синтез н-метокси-N,5-диметил-6-(2,2,2-трифторэтокси)пиридин-3-карбоксамида
[0301]

Использовали соединение (3,57 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в ссылочном примере 16-1(1), для того, чтобы получать N-метокси-N,5-диметил-6-(2,2,2-трифторэтокси)пиридин-3-карбоксамид в виде бесцветного масла (2,98 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,22-2,31 (м, 3H) 3,37 (с, 3H) 3,58 (с, 3H) 4,81 (кв., J=8,5 Гц, 2H) 7,80-7,92 (м, 1H) 8,37-8,49 (м, 1H).
MS ESI/APCI двойн. положительн.: 279[M+H]+.
(3) Синтез указанного в заголовке соединения
Использовали соединение (2,66 г), полученное на приведенной выше стадии (2), и обработку проводили с помощью того же способа, как в ссылочном примере 16-1(3), для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (2,10 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,27-2,34 (м, 3H) 4,76-4,96 (м, 2H) 7,91-8,00 (м, 1H) 8,41-8,51 (м, 1H) 9,97 (с, 1H).
MS ESI/APCI двойн. положительн.: 220[M+H]+.
[0302] В следующих ссылочных примерах с 33-2 до 33-8 использовали соответствующие галогенированные арилы и соответствующие спирты торговых сортов и обработку проводили с помощью способа, описанного в ссылочном примере 33-1, или его модификаций для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблице 12-1.
[0303]
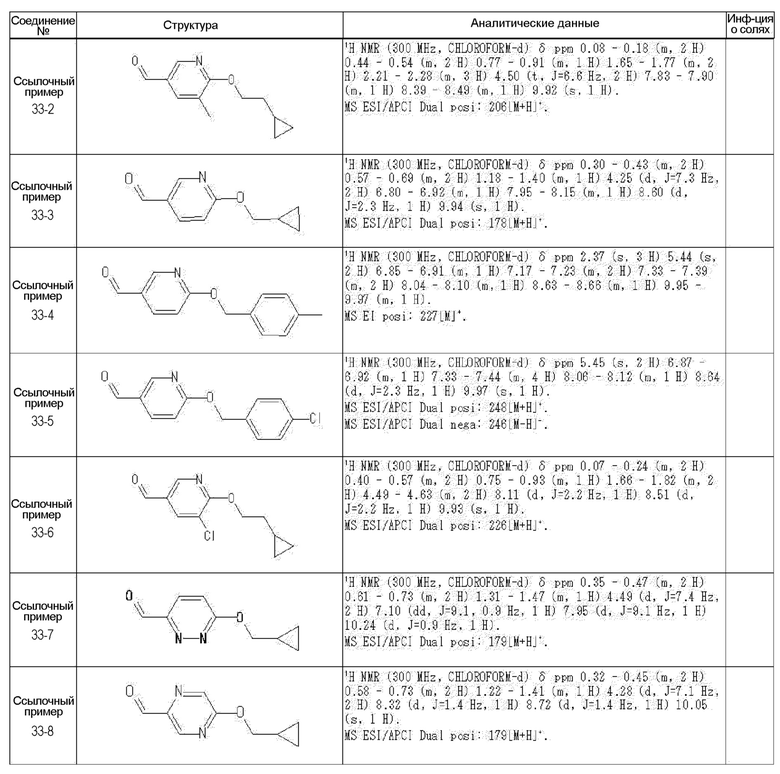
Ссылочный пример 34-1
3-фенилциклопентанкарбальдегид
[0304]
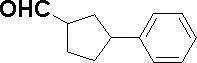
(1) Синтез (3-фенилциклопентил)метанола
[0305]

Вместо этил цис-2-фенилциклопропанкарбоксилата использовали сложный метиловый эфир 3-фенил-циклопентанкарбоновой кислоты (1,76 г) и обработку проводили с помощью того же способа, как в ссылочном примере 23-1(2), для того, чтобы получать (3-фенилциклопентил)метанол в виде бесцветного масла (1,31 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,26-1,48 (м, 2H) 1,54-2,19 (м, 5H) 2,29-2,49 (м, 1H) 2,97-3,16 (м, 1H) 3,53-3,67 (м, 2H) 7,12-7,35 (м, 5H).
MS EI положительн.: 176[M]+.
(2) Синтез указанного в заголовке соединения
Использовали соединение (0,90 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в ссылочном примере 19-1, для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (0,90 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,56-2,55 (м, 7H) 2,90-3,19 (м, 1H) 7,14-7,40 (м, 5H) 9,65-9,78 (м, 1H).
[0306] В следующих ссылочных примерах 34-2 и 34-3 использовали соответствующие сложные эфиры торговых сортов и обработку проводили с помощью способа, описанного в ссылочном примере 34-1, или его модификации для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблице 13-1.
[0307]
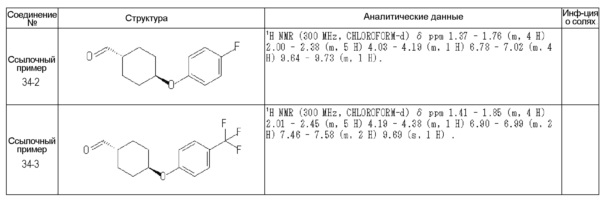
Ссылочный пример 35-1
4-{[(6-метилпиридин-3-ил)окси]метил}бензальдегид
[0308]
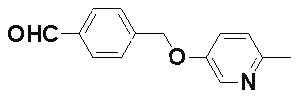
Вместо 4-гидроксибензальдегида и (бромметил)циклобутана соответственно использовали 5-гидрокси-2-метилпиридин (767 мг) и 4-(хлорметил)бензиловый спирт (1,00 г) и обработку проводили с помощью тех же способов, что и в ссылочных примерах 9-1 и 19-1, для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (1,65 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,50 (с, 3H) 5,17 (с, 2H) 7,05-7,11 (м, 1H) 7,14-7,20 (м, 1H) 7,57-7,63 (м, 2H) 7,89-7,94 (м, 2H) 8,25-8,29 (м, 1H) 10,03 (с, 1H).
MS ESI/APCI двойн. положительн.: 228[M+H]+.
MS ESI/APCI двойн. отрицательн.: 226[M-H]-, 262[M+Cl]-.
Ссылочный пример 36-1
цис-4-[(4-хлорбензил)окси]циклогексанкарбальдегид
[0309]

(1) Синтез этил цис-4-[(4-хлорбензил)окси]циклогексанкарбоксилата и этил транс-4-[(4-хлорбензил)окси]циклогексанкарбоксилата
[0310]

и
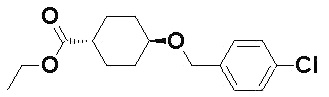
Вместо 4-гидроксибензальдегида и (бромметил)циклобутана соответственно использовали этил 4-гидроксициклогексанкарбоксилат (2,00 г) и 4-хлорбензилбромид (2,86 г) и обработку проводили с помощью того же способа, как в ссылочном примере 9-1, для того, чтобы получать этил цис-4-[(4-хлорбензил)окси]циклогексанкарбоксилат в виде бесцветного масла (0,33 г) и этил транс-4-[(4-хлорбензил)окси]циклогексанкарбоксилат в виде масла бледно-желтого цвета (0,47 г).
Этил цис-4-[(4-хлорбензил)окси]циклогексанкарбоксилат
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,29 (т, J=7,1 Гц, 3H) 1,52-1,78 (м, 4H) 1,83-2,06 (м, 4H) 2,30-2,50 (м, 1H) 3,54-3,66 (м, 1H) 4,17 (кв., J=7,1 Гц, 2H) 4,51 (с, 2H) 7,25-7,40 (м, 4H).
Этил транс-4-[(4-хлорбензил)окси]циклогексанкарбоксилат
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,17-1,57 (м, 7H) 1,95-2,18 (м, 4H) 2,20-2,35 (м, 1H) 3,25-3,41 (м, 1H) 4,11 (кв., J=7,1 Гц, 2H) 4,51 (с, 2H) 7,21-7,37 (м, 4H).
(2) Синтез {цис-4-[(4-хлорбензил)окси]циклогексил}метанола
[0311]
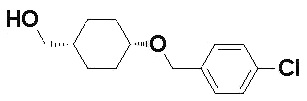
Использовали соединение цис-4-[(4-хлорбензил)окси]циклогексанкарбоксилат (0,33 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в ссылочном примере 23-1(2), для того, чтобы получать {цис-4-[(4-хлорбензил)окси]циклогексил}метанол в виде бесцветного масла (0,29 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,20-1,67 (м, 8H) 1,83-2,02 (м, 2H) 3,49 (д, J=5,9 Гц, 2H) 3,59-3,67 (м, 1H) 4,46 (с, 2H) 7,24-7,35 (м, 4H).
(3) Синтез указанного в заголовке соединения
Использовали соединение (283 мг), полученное на приведенной выше стадии (2), и обработку проводили с помощью того же способа, как в ссылочном примере 19-1, для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (257 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,49-1,99 (м, 8H) 2,20-2,35 (м, 1H) 3,51-3,65 (м, 1H) 4,47 (с, 2H) 7,22-7,35 (м, 4H) 9,59-9,68 (м, 1H).
MS ESI/APCI двойн. положительн.: 275[M+Na]+.
Ссылочный пример 36-2
Транс-4-[(4-хлорбензил)окси]циклогексанкарбальдегид
[0312]

Использовали этил транс-4-[(4-хлорбензил)окси]циклогексанкарбоксилат (856 мг), полученный в ссылочном примере 36-1(1), и проводили обработку теми же способами, как в ссылочном примере 36-1(2) и (3), для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (388 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,23-1,50 (м, 4H) 1,98-2,31 (м, 5H) 3,26-3,41 (м, 1H) 4,52 (с, 2H) 7,22-7,36 (м, 4H) 9,62-9,67 (м, 1H).
Ссылочный пример 36-3
Транс-4-[(5-хлор-2-пиридинил)метокси]циклогексанкарбальдегид
[0313]
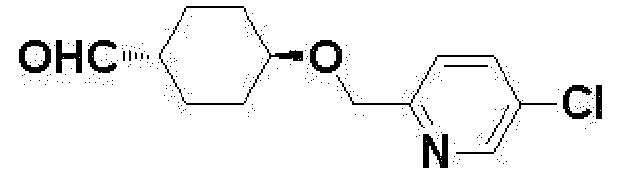
Вместо 4-хлорбензилбромида использовали 2-(бромметил)-5-хлорпиридин (5,23 г) и обработку проводили с помощью того же способа, как в ссылочном примере 36-1, для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (0,25 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,28-1,67 (м, 4H) 1,99-2,31 (м, 5H) 3,33-3,52 (м, 1H) 4,65 (с, 2H) 7,39-7,48 (м, 1H) 7,64-7,75 (м, 1H) 8,48-8,54 (м, 1H) 9,61-9,71 (м, 1H).
Ссылочный пример 37-1
Транс-4-феноксициклогексанкарбальдегид
[0314]
(1) Синтез метил транс-4-феноксициклогексанкарбоксилата
[0315]
Вместо 4-гидроксибензальдегида и 2-циклопропилэтанола соответственно использовали фенол (1,43 г) и метил цис-4-гидроксициклогексанкарбоксилат (2,00 г) и обработку проводили с помощью того же способа, как в ссылочном примере 11-1, для того, чтобы получать метил транс-4-феноксициклогексанкарбоксилат в виде бесцветного масла (1,33 г).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,43-1,65 (м, 4H) 2,03-2,13 (м, 2H) 2,15-2,23 (м, 2H) 2,32-2,44 (м, 1H) 3,68 (с, 3H) 4,15-4,26 (м, 1H) 6,86-6,97 (м, 3H) 7,23-7,31 (м, 2H).
MS ESI/APCI двойн. положительн.: 235[M+H]+.
(2) Синтез (транс-4-феноксициклогексил)метанола
[0316]
Использовали соединение (1,31 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в ссылочном примере 23-1(2), для того, чтобы получать (транс-4-феноксициклогексил)метанол в виде бесцветного масла (897 мг).
1H ЯМР (200 МГц, ХЛОРОФОРМ-d) δ м.д. 0,96-1,70 (м, 6H) 1,81-2,01 (м, 2H) 2,10-2,31 (м, 2H) 3,50 (д, J=6,2 Гц, 2H) 4,04-4,27 (м, 1H) 6,82-7,00 (м, 3H) 7,16-7,35 (м, 2H).
MS ESI/APCI двойн. положительн.: 207[M+H]+.
(3) Синтез указанного в заголовке соединения
Использовали соединение (897 мг), полученное на приведенной выше стадии (2), и обработку проводили с помощью того же способа, как в ссылочном примере 19-1, для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (801 мг).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,42-1,64 (м, 4H) 2,04-2,22 (м, 4H) 2,27-2,37 (м, 1H) 4,16-4,28 (м, 1H) 6,86-6,98 (м, 3H) 7,23-7,33 (м, 2H) 9,69 (с, 1H).
[0317] В следующих ссылочных примерах с 37-2 до 37-8 использовали соответствующие фенолы торговых сортов и соответствующие спирты торговых сортов и обработку проводили с помощью способа, описанного в ссылочном примере 37-1, или его модификаций для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблице 14-1.
[0318]
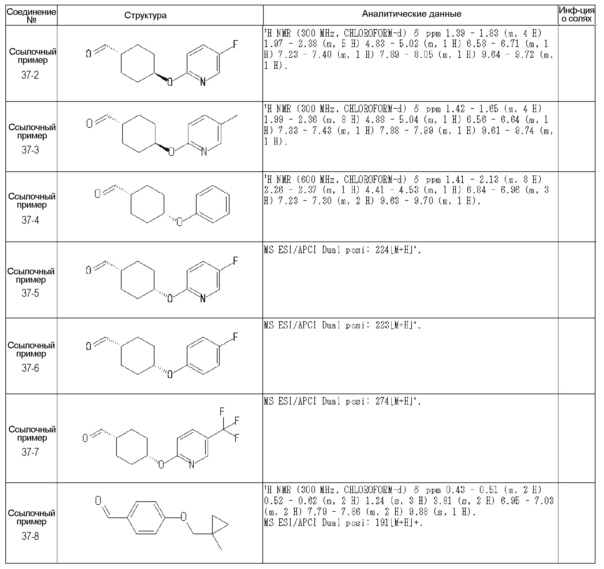
Ссылочный пример 38-1
4-({[6-(трифторметил)пиридин-3-ил]окси}метил)бензальдегид
[0319]
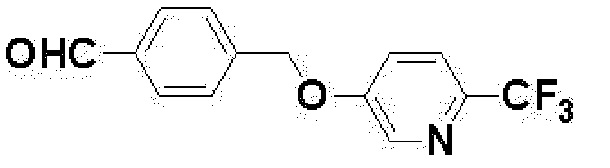
(1) Синтез 4-({[6-(трифторметил)пиридин-3-ил]окси}метил)бензонитрила
[0320]

Вместо 4-гидроксибензальдегида и бромциклопропана соответственно использовали 6-(трифторметил)пиридин-3-ол (1,21 г) и 4-цианобензилбромид (1,45 г) и обработку проводили с помощью того же способа, как в ссылочном примере 10-1, для того, чтобы получать 4-({[6-(трифторметил)пиридин-3-ил]окси}метил)бензонитрил в виде твердого вещества бледно-коричневого цвета (1,99 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 5,23 (с, 2H) 7,34 (дд, J=8,7, 2,8 Гц, 1H) 7,52-7,59 (м, 2H) 7,64 (д, J=8,7 Гц, 1H) 7,69-7,77 (м, 2H) 8,46 (д, J=2,8 Гц, 1H).
MS ESI/APCI двойн. положительн.: 279[M+H]+.
MS ESI/APCI двойн. отрицательн.: 277[M-H]-.
(2) Синтез указанного в заголовке соединения
Использовали соединение (1,99 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в ссылочном примере 16-1(3), для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (980 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 5,26 (с, 2H) 7,35 (дд, J=8,8, 3,0 Гц, 1H) 7,58-7,67 (м, 3H) 7,91-7,98 (м, 2H) 8,48 (д, J=3,0 Гц, 1H) 10,05 (с, 1H).
MS ESI/APCI двойн. положительн.: 282[M+H]+.
Ссылочный пример 38-2
4-({[5-(трифторметил)пиридин-2-ил]окси}метил)бензальдегид
[0321]

Вместо 6-(трифторметил)пиридин-3-ола и 4-цианобензилбромида соответственно использовали 4-(гидроксиметил)бензонитрил (1,87 г) и 2-фтор-5-(трифторметил)пиридин (1,55 г) и обработку проводили с помощью того же способа, как в ссылочном примере 38-1, для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (1,49 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 5,53 (с, 2H) 6,90-6,97 (м, 1H) 7,58-7,65 (м, 2H) 7,82 (дд, J=8,9, 2,3 Гц, 1H) 7,87-7,94 (м, 2H) 8,40-8,47 (м, 1H) 10,03 (с, 1H).
MS ESI/APCI двойн. положительн.: 282[M+H]+.
Ссылочный пример 39-1
транс-4-[(4-фторбензил)окси]циклогексанкарбальдегид
[0322]
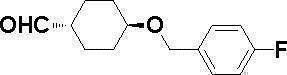
(1) Синтез транс-4-гидрокси-N-метокси-N-метилциклогексанкарбоксамида
[0323]

Вместо 4-(бромметил)бензойной кислоты использовали транс-4-гидроксициклогексанкарбоновую кислоту (7,21 г) и обработку проводили с помощью того же способа, как в ссылочном примере 16-1(1), для того, чтобы получать транс-4-гидрокси-N-метокси-N-метилциклогексанкарбоксамид в виде бесцветного масла (8,52 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,21-1,40 (м, 2H) 1,49-1,68 (м, 2H) 1,78-1,90 (м, 2H) 2,00-2,13 (м, 2H) 2,54-2,73 (м, 2H) 3,18 (с, 3H) 3,57-3,74 (м, 4H).
(2) Синтез транс-4-{[трет-бутил(диметил)силил]окси}-N-метокси-N-метилциклогексанкарбоксамида
[0324]

В раствор в N,N-диметилформамиде (91 мл) соединения (8,52 г), полученного на приведенной выше стадии (1), добавляли имидазол (4,03 г) и трет-бутилдиметилхлорсилан (6,86 г) и смесь перемешивали при комнатной температуре в течение 1,5 часа. Реакционную смесь концентрировали при пониженном давлении и в получаемый остаток добавляли воду. После экстрагирования этилацетатом объединенные органические слои промывали водой и насыщенным солевым раствором. Промытые органические слои сушили над безводным сульфатом магния и после удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=100:0-75:25), для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (10,5 г).
1H ЯМР (200 МГц, ХЛОРОФОРМ-d) δ м.д. 0,06 (с, 6H) 0,88 (с, 9H) 1,15-1,69 (м, 4H) 1,71-2,08 (м, 4H) 2,50-2,73 (м, 1H) 3,17 (с, 3H) 3,48-3,75 (м, 4H).
(3) Синтез транс-4-[(4-фторбензил)окси]-N-метокси-N-метилциклогексанкарбоксамида
[0325]

В раствор в ацетонитриле (11 мл) соединения (1,00 г), полученного на приведенной выше стадии (2), добавляли триэтилсилан (579 мг). Трибромид висмута (104 мг) и 4-фторбензальдегид (617 мг) добавляли при охлаждении льдом и смесь перемешивали при комнатной температуре в течение двух часов. В реакционную смесь добавляли насыщенный водный раствор бикарбоната натрия и этилацетат и удаляли нерастворимое вещество с помощью фильтрования через Celite (зарегистрированный товарный знак). Органический слой в фильтрате отделяли и промывали насыщенным солевым раствором. Промытый органический слой сушили над безводным сульфатом магния и после удаления десиканта с помощью фильтрования фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-50:50) для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (0,61 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,23-1,43 (м, 2H) 1,47-1,65 (м, 2H) 1,79-1,93 (м, 2H) 2,10-2,23 (м, 2H) 2,57-2,74 (м, 1H) 3,18 (с, 3H) 3,29-3,43 (м, 1H) 3,70 (с, 3H) 4,52 (с, 2H) 6,97-7,08 (м, 2H) 7,25-7,36 (м, 2H).
(4) Синтез указанного в заголовке соединения
Использовали соединение (0,59 г), полученное на приведенной выше стадии (3), и обработку проводили с помощью того же способа, как в ссылочном примере 23-1(2), для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (0,46 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,27-1,49 (м, 4H) 1,97-2,34 (м, 5H) 3,25-3,41 (м, 1H) 4,52 (с, 2H) 6,97-7,08 (м, 2H) 7,25-7,36 (м, 2H) 9,62-9,70 (м, 1H).
[0326] В следующих ссылочных примерах с 39-2 до 39-7 использовали соответствующие альдегиды торговых сортов и обработку проводили с помощью способа, описанного в ссылочном примере 39-1, или его модификаций для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблице 15-1.
[0327]
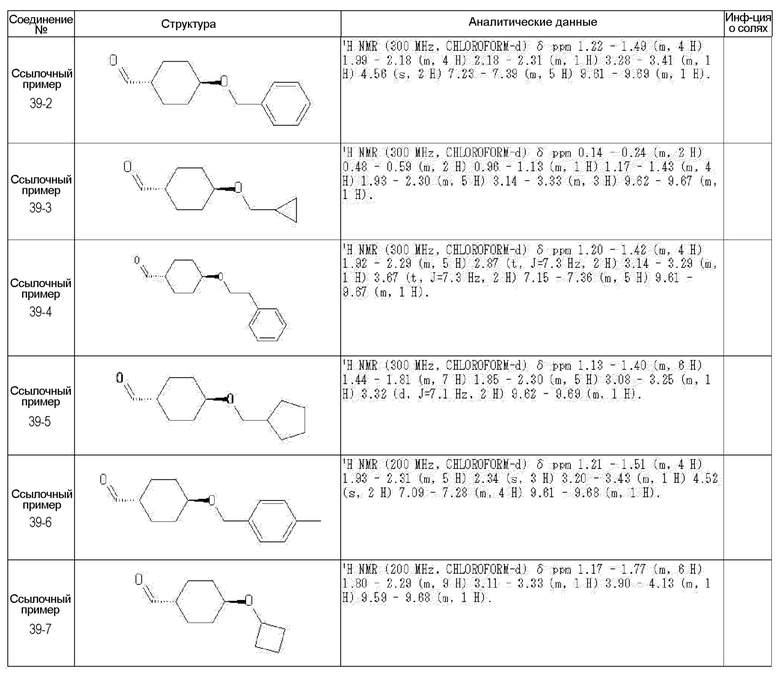
Ссылочный пример 40-1
4-(пиперидин-1-илкарбонил)бензальдегид
[0328]
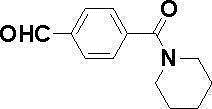
В раствор 4-карбоксибензальдегида (1,06 г) в хлороформе (14,1 мл) добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (2,03 г), моногидрат 1-гидроксибензотриазола (1,62 г) и пиперидин (1,05 мл). После перемешивания смеси при комнатной температуре в течение 15 часов, добавляли насыщенный водный раствор хлорида аммония. После экстрагирования этилацетатом объединенные органические слои промывали насыщенным солевым раствором. Промытые органические слои сушили над безводным сульфатом магния и после удаления десиканта с помощью фильтрования фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=85:15-30:70) для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (1,57 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,50-1,53 (м, 2H) 1,65-1,73 (м, 4H) 3,24-3,35 (м, 2H) 3,68-3,79 (м, 2H) 7,52-7,57 (м, 2H) 7,90-7,96 (м, 2H) 10,05 (с, 1H).
MS ESI/APCI двойн. положительн.: 218[M+H]+.
MS ESI/APCI двойн. отрицательн.: 232[M+Cl]-.
[0329] В следующих ссылочных примерах с 40-2 до 40-8 использовали соответствующе амины торговых сортов и обработку проводили с помощью способа, описанного в ссылочном примере 40-1, его или модификаций для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблице 16-1.
[0330]
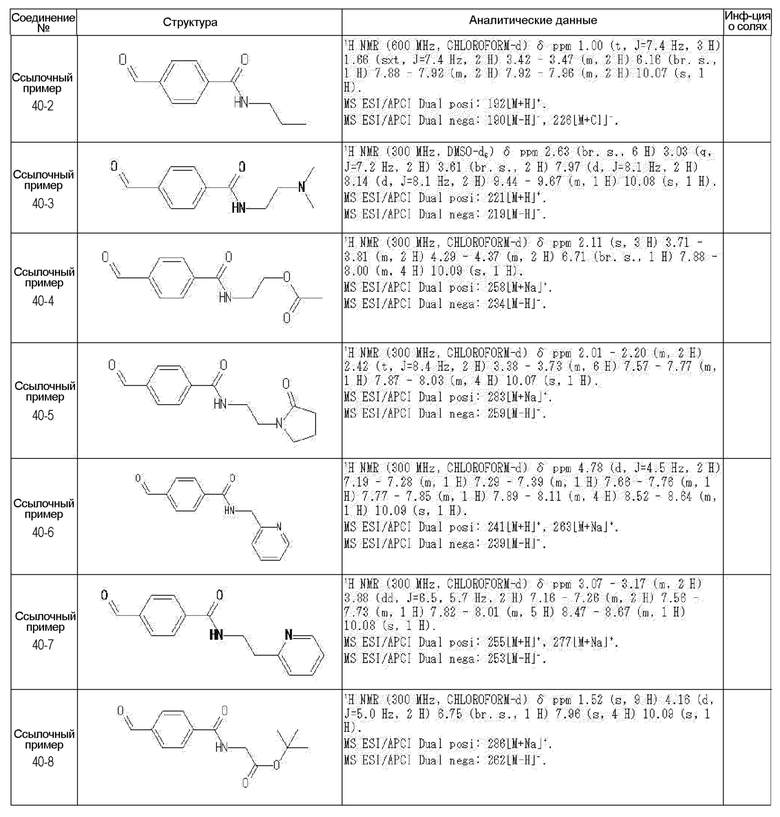
Ссылочный пример 41-1
5-(4-метилфенокси)пиразин-2-карбальдегид
[0331]
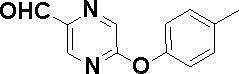
(1) Синтез метил 5-(4-метилфенокси)пиразин-2-карбоксилата
[0332]
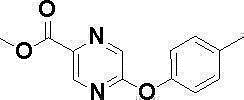
Вместо 4-гидроксибензотрифторида и 6-бром-3-пиридинкарбоксиальдегида соответственно использовали п-крезол (833 мг) и метил 5-хлор-2-пиразинкарбоксилат (1,33 г) и обработку проводили с помощью того же способа, как в ссылочном примере 13-1, для того, чтобы получать метил 5-(4-метилфенокси)пиразин-2-карбоксилат в виде бесцветного твердого вещества (1,36 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,39 (с, 3H) 4,01 (с, 3H) 7,03-7,09 (м, 2H) 7,18-7,33 (м, 2H) 8,48 (д, J=1,2 Гц, 1H) 8,83 (д, J=1,2 Гц, 1H).
MS ESI/APCI двойн. положительн.: 245[M+H]+.
(2) Синтез указанного в заголовке соединения
Использовали соединение (1,36 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в ссылочном примере 16-1(3), для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (1,10 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,40 (с, 3H) 7,02-7,10 (м, 2H) 7,22-7,31 (м, 2H) 8,51 (д, J=1,2 Гц, 1H) 8,71 (д, J=1,2 Гц, 1H) 10,08 (с, 1H).
MS ESI/APCI двойн. положительн.: 215[M+H]+.
Ссылочный пример 41-2
5-(4-хлорфенокси)пиразин-2-карбальдегид
[0333]

Вместо п-крезола использовали 4-хлорфенол (2,23 г) и обработку проводили с помощью того же способа, как в ссылочном примере 41-1, для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (0,45 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 7,08-7,20 (м, 2H) 7,36-7,49 (м, 2H) 8,55 (д, J=1,4 Гц, 1H) 8,67-8,72 (м, 1H) 10,09 (с, 1H).
MS ESI/APCI двойн. положительн.: 235[M+H]+.
Ссылочный пример 41-3
5-(4-циклопропилфенокси)пиразин-2-карбальдегид
[0334]
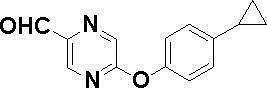
(1) Синтез 5-(4-бромфенокси)пиразин-2-карбальдегида
[0335]
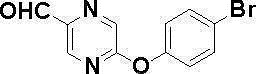
Вместо п-крезола использовали 4-бромфенол (5,01 г) и обработку проводили с помощью того же способа, как в ссылочном примере 41-1, для того, чтобы получать 5-(4-бромфенокси)пиразин-2-карбальдегид в виде твердого вещества бледно-коричневого цвета (1,88 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 7,09 (д, J=9,0 Гц, 2H) 7,58 (д, J=9,0 Гц, 2H) 8,55 (д, J=1,2 Гц, 1H) 8,70 (с, 1H) 10,09 (с, 1H).
MS ESI/APCI двойн. положительн.: 279[M+H]+.
(2) Синтез 2-(4-бромфенокси)-5-(1,3-диоксолан-2-ил)пиразина
[0336]
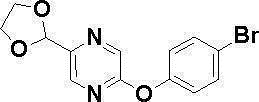
В раствор в толуоле (20 мл) соединения (1,66 г), полученного на приведенной выше стадии (1), добавляли этиленгликоль (1,11 г) и моногидрат п-толуолсульфоновой кислоты (56,6 мг) и после этого смесь перемешивали при 140°C в течение часа с использованием аппарата Дина-Старка. После охлаждения реакционной смеси до комнатной температуры, в нее добавляли насыщенный водный раствор бикарбоната натрия при охлаждении льдом и проводили экстрагирование этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Очистка колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-60:40) давала 2-(4-бромфенокси)-5-(1,3-диоксолан-2-ил)пиразин в виде бесцветного твердого вещества (1,78 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 4,01-4,24 (м, 4H) 5,92 (с, 1H) 7,05 (д, J=9,0 Гц, 2H) 7,48-7,59 (м, 2H) 8,26-8,30 (м, 1H) 8,40-8,44 (м, 1H).
MS ESI/APCI двойн. положительн.: 323[M+H]+.
(3) Синтез 2-(4-циклопропилфенокси)-5-(1,3-диоксолан-2-ил)пиразина
[0337]
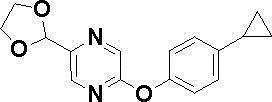
Использовали соединение (1,73 г), полученное на приведенной выше стадии (2), и обработку проводили с помощью того же способа, как в ссылочном примере 14-5(2), для того, чтобы получать 2-(4-циклопропилфенокси)-5-(1,3-диоксолан-2-ил)пиразин в виде масла коричневого цвета (2,46 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,65-0,74 (м, 2H) 0,91-1,02 (м, 2H) 1,84-1,98 (м, 1H) 4,02-4,23 (м, 4H) 5,91 (с, 1H) 6,98-7,07 (м, 2H) 7,09-7,16 (м, 2H) 8,26-8,30 (м, 1H) 8,35-8,42 (м, 1H).
MS ESI/APCI двойн. положительн.: 285[M+H]+.
(4) Синтез указанного в заголовке соединения
В раствор в ацетоне (107 мл) соединения (2,46 г), полученного на приведенной выше стадии (3), добавляли моногидрат п-толуолсульфоновой кислоты (2,04 г) и смесь перемешивали при 50°C в течение двух часов. После охлаждения реакционной смеси до комнатной температуры, в нее добавляли насыщенный водный раствор бикарбоната натрия при охлаждении льдом и проводили два экстрагирования этилацетатом. Объединенные органические слои сушили над безводным сульфатом магния; после этого десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-80:20), для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,67 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,66-0,78 (м, 2H) 0,91-1,05 (м, 2H) 1,87-2,00 (м, 1H) 7,01-7,10 (м, 2H) 7,12-7,20 (м, 2H) 8,51 (с, 1H) 8,71 (с, 1H) 10,08 (с, 1H).
MS ESI/APCI двойн. положительн.: 241[M+H]+.
Ссылочный пример 41-4
2-(4-метилфенокси)пиримидин-5-карбальдегид
[0338]
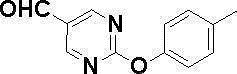
(1) Синтез 2-хлор-5-(1,3-диоксолан-2-ил)пиримидина
[0339]

Использовали соединение 2-хлорпиримидин-5-карбальдегид (1,00 г) и обработку проводили с помощью того же способа, как в ссылочном примере 41-3(2), для того, чтобы получать 2-хлор-5-(1,3-диоксолан-2-ил)пиримидин в виде масла бледно-желтого цвета (290 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 3,96-4,21 (м, 4H) 5,89 (с, 1H) 8,71 (с, 2H).
MS ESI/APCI двойн. положительн.: 187[M+H]+.
(2) Синтез 5-(1,3-диоксолан-2-ил)-2-(4-метилфенокси)пиримидина
[0340]

Использовали соединение (290 мг), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в ссылочном примере 13-1, для того, чтобы получать 5-(1,3-диоксолан-2-ил)-2-(4-метилфенокси)пиримидин в виде бесцветного твердого вещества (380 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,37 (с, 3H) 3,97-4,19 (м, 4H) 5,83 (с, 1H) 7,04-7,10 (м, 2H) 7,19-7,26 (м, 2H) 8,61 (с, 2H).
MS ESI/APCI двойн. положительн.: 259[M+H]+.
(3) Синтез указанного в заголовке соединения
Использовали соединение (380 мг), полученное на приведенной выше стадии (2), и обработку проводили с помощью того же способа, как в ссылочном примере 41-3(4), для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (139 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,39 (с, 3H) 7,09 (д, J=8,3 Гц, 2H) 7,26 (д, J=8,3 Гц, 2H) 9,01 (с, 2H) 9,99-10,09 (м, 1H).
MS ESI/APCI двойн. положительн.: 215[M+H]+.
Ссылочный пример 42-1
1-(пиримидин-2-ил)пиперидин-4-карбальдегид
[0341]
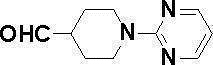
(1) Синтез [1-(пиримидин-2-ил)пиперидин-4-ил]метанола
[0342]
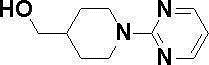
В раствор 4-пиперидинметанола (1,00 г) в диметилсульфоксиде (28,9 мл) добавляли 2-хлорпиримидин (994 мг) и карбонат калия (2,40 г) и смесь перемешивали при 100°C в течение трех часов. После охлаждения реакционной смеси до комнатной температуры, в нее добавляли воду и проводили экстрагирование этилацетатом. Объединенные органические слои сушили над безводным сульфатом натрия, и после удаления десиканта с помощью фильтрования фильтрат концентрировали при пониженном давлении для того, чтобы получать [1-([пиримидин-2-ил)пиперидин-4-ил]метанол в виде бесцветного масла (1,50 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,14-1,32 (м, 2H) 1,72-1,89 (м, 3H) 2,82-2,96 (м, 2H) 3,53 (д, J=5,9 Гц, 2H) 4,74-4,85 (м, 2H) 6,41-6,47 (м, 1H) 8,26-8,33 (м, 2H).
MS ESI/APCI двойн. положительн.: 194[M+H]+.
(2) Синтез указанного в заголовке соединения
Использовали соединение (1,50 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в ссылочном примере 19-1, для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (1,21 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,57-1,72 (м, 2H) 1,94-2,05 (м, 2H) 2,49-2,61 (м, 1H) 3,12-3,24 (м, 2H) 4,53-4,64 (м, 2H) 6,48 (т, J=4,7 Гц, 1H) 8,31 (д, J=4,7 Гц, 2H) 9,70 (д, J=0,9 Гц, 1H).
MS ESI/APCI двойн. положительн.: 192[M+H]+.
Ссылочный пример 42-2
1-(циклопропилацетил)пиперидин-4-карбальдегид
[0343]

(1) Синтез 2-циклопропил-1-[4-(гидроксиметил)пиперидин-1-ил]этанона
[0344]
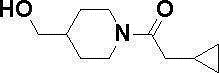
Вместо 4-(бромметил)бензойной кислоты и гидрохлорида N,O-диметилгидроксиламина соответственно использовали циклопропилуксусную кислоту (869 мг) и 4-пиперидинметанол (1,00 г) и обработку проводили с помощью того же способа, как в ссылочном примере 16-1(1), для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (1,25 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,12-0,23 (м, 2H) 0,51-0,60 (м, 2H) 0,96-1,30 (м, 3H) 1,54-1,90 (м, 3H) 2,28 (д, J=6,7 Гц, 2H) 2,47-2,65 (м, 1H) 2,95-3,10 (м, 1H) 3,43-3,59 (м, 2H) 3,80-3,94 (м, 1H) 4,60-4,76 (м, 1H).
MS ESI/APCI двойн. положительн.: 198[M+H]+.
(2) Синтез указанного в заголовке соединения
Использовали соединение (1,25 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в ссылочном примере 19-1, для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (800 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,13-0,22 (м, 2H) 0,52-0,61 (м, 2H) 0,96-1,10 (м, 1H) 1,50-1,71 (м, 2H) 1,88-2,03 (м, 2H) 2,29 (д, J=6,8 Гц, 2H) 2,45-2,59 (м, 1H) 2,92-3,04 (м, 1H) 3,11-3,25 (м, 1H) 3,72-3,84 (м, 1H) 4,29-4,41 (м, 1H) 9,68 (с, 1H).
MS ESI/APCI двойн. положительн.: 196[M+H]+.
Ссылочный пример 43-1
1-(пиримидин-2-ил)азетидин-3-карбальдегид
[0345]

(1) Синтез трет-бутил 3-[метокси(метил)карбамоил]азетидин-1-карбоксилата
[0346]
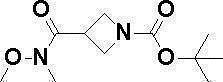
В раствор 1-(трет-бутоксикарбонил)азетидин-3-карбоновой кислоты (5,00 г) в тетрагидрофуране (62,1 мл) добавляли 1,1'-карбонилдиимидазол (6,05 г) и смесь перемешивали при комнатной температуре в течение часа. В реакционную смесь добавляли раствор гидрохлорида N,O-диметилгидроксиламина (3,64 г) и триэтиламина (4,02 г) в ацетонитриле (62,1 мл) и смесь перемешивали при той же температуре в течение 15 часов. Реакционную смесь концентрировали при пониженном давлении и в получаемый остаток добавляли воду. Проводили экстрагирование этилацетатом и объединенные органические слои промывали 5% водным раствором лимонной кислоты и насыщенным солевым раствором. Промытые органические слои сушили над безводным сульфатом натрия и после удаления десиканта с помощью фильтрования фильтрат концентрировали при пониженном давлении для того, чтобы получать трет-бутил 3-[метокси(метил)карбамоил]азетидин-1-карбоксилат в виде масла бледно-желтого цвета (7,30 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,43 (с, 9H) 3,21 (с, 3H) 3,56-3,68 (м, 4H) 4,00-4,09 (м, 2H) 4,09-4,19 (м, 2H).
(2) Синтез н-метокси-N-метил-1-(пиримидин-2-ил)азетидин-3-карбоксамида
[0347]
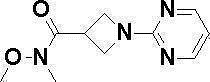
В раствор в хлороформе (24,8 мл) соединения (7,30 г), полученного на приведенной выше стадии (1), добавляли трифторуксусную кислоту (12,4 мл) и смесь перемешивали при комнатной температуре в течение 15 часов и затем концентрировали при пониженном давлении. Использовали получаемый остаток (6,29 г) и обработку проводили с помощью того же способа, как в ссылочном примере 42-1(1), для того, чтобы получать N-метокси-N-метил-1-(пиримидин-2-ил)азетидин-3-карбоксамид в виде бесцветного твердого вещества (810 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 3,23 (с, 3H) 3,70 (с, 3H) 3,80-3,96 (м, 1H) 4,16-4,41 (м, 4H) 6,51-6,59 (м, 1H) 8,28-8,35 (м, 2H).
MS ESI/APCI двойн. положительн.: 223[M+H]+.
(3) Синтез указанного в заголовке соединения
Использовали соединение (810 мг), полученное на приведенной выше стадии (2), и обработку проводили с помощью того же способа, как в ссылочном примере 16-1(3), для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (707 мг).
MS ESI/APCI двойн. положительн.: 164[M+H]+.
Ссылочный пример 44-1
Трет-бутил 4-формил-2-метил-1H-имидазол-1-карбоксилат
[0348]
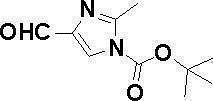
В раствор 2-метил-1H-имидазол-4-карбальдегида (500 мг) в хлороформе (15 мл) добавляли ди-трет-бутил дикарбонат (1,19 г), триэтиламин (949 мкл) и 4-диметиламинопиридин и смесь перемешивали при комнатной температуре в течение 30 минут. В реакционную смесь добавляли насыщенный водный раствор бикарбоната натрия и проводили экстрагирование хлороформом. Объединенные органические слои пропускали через фазовый разделитель и после этого концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (883 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,64 (с, 9H) 2,68 (с, 3H) 7,98 (с, 1H) 9,85 (с, 1H).
MS ESI/APCI двойн. положительн.: 233[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 209[M-H]-.
Ссылочный пример 45-1
Гидрохлорид 1-(4-циклогексил-3-фторфенил)метанамина
[0349]

(1) Синтез 4-циклогексен-1-ил-3-фторбензонитрила
[0350]

В смесь 3-фтор-4-йодобензонитрила (1,53 г), 1-циклогексен-1-ил-бороновой кислоты (938 мг), дихлорида бис(трифенилфосфин)палладия(II) (435 мг) и этанола (9,75 мл) добавляли этоксид натрия (приблизительно 20%, раствор в этаноле, 5,75 мл) и смесь перемешивали при 90°C в течение 15 минут при облучении микроволнами. После охлаждения до комнатной температуры реакционную смесь выливали в воду и проводили три экстрагирования хлороформом. Объединенные органические слои промывали насыщенным солевым раствором и пропускали через фазовый разделитель для концентрирования при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-75:25) для того, чтобы получать указанное в заголовке соединение в виде масла бледно-желтого цвета (980 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,63-1,84 (м, 4H) 2,18-2,29 (м, 2H) 2,30-2,41 (м, 2H) 6,01-6,10 (м, 1H) 7,27-7,42 (м, 3H).
MS ESI/APCI двойн. положительн.: 224[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 236[M+Cl]-.
(2) Синтез указанного в заголовке соединения
В раствор в изопропиловом спирте (24 мл) соединения (980 мг), полученного на приведенной выше стадии (1), добавляли раствор (3,7 мл) хлороводорода в 1,4-диоксане 4 моль/л и 20% гидроксид палладия/углерод (98 мг). Смесь перемешивали при комнатной температуре в течение 4 часов в атмосфере водорода. Реакционную смесь фильтровали через Celite (зарегистрированный товарный знак) и фильтрат концентрировали при пониженном давлении. В раствор получаемого остатка в этаноле (10 мл) добавляли раствор (2,0 мл) хлороводорода в метаноле 2 моль/л и 20% гидроксид палладия/углерод (98 мг). Смесь перемешивали при комнатной температуре в течение 26 часов в атмосфере водорода. Реакционную смесь фильтровали через Celite (зарегистрированный товарный знак) и фильтрат концентрировали при пониженном давлении. В получаемый остаток добавляли этанол (5 мл) и диэтиловый эфир (50 мл) и смесь перемешивали в течение 15 минут. Получаемый осадок собирали с помощью фильтрования для того, чтобы получать указанное в заголовке соединение в виде твердого вещества коричневого цвета (1,06 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 1,14-1,54 (м, 5H) 1,63-1,86 (м, 5H) 2,70-2,87 (м, 1H) 3,99 (с, 2H) 7,22-7,42 (м, 3H) 8,43 (ушир. с, 2H).
MS ESI/APCI двойн. положительн.: 208[M+H]+.
Ссылочный пример 45-2
Гидрохлорид 1-[4-(аминометил)фенил]-4,4-дифторциклогексанола
[0351]

(1) Синтез 4-(4,4-дифтор-1-гидроксициклогексил)бензонитрила
[0352]
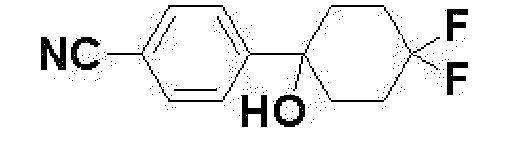
В раствор 4-йодобензонитрила (5,35 г) в тетрагидрофуране (100 мл) по каплям добавляли изопропилбромид магния (приблизительно 1 моль/л, раствор в тетрагидрофуране, 35 мл) при -40°C в атмосфере аргона. После перемешивания смеси при этой температуре в течение часа, по каплям добавляли раствор 4,4-дифторциклогексанона (4,70 г) в простом циклопентилметиловом эфире (10 мл). Смесь доводили до комнатной температуры в течение периода в 5,5 часа и добавляли насыщенный водный раствор хлорида аммония. Проводили три экстрагирования этилацетатом и объединенные органические слои промывали насыщенным солевым раствором и после этого сушили над безводным сульфатом магния. Нерастворимое вещество удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении.
Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 95:5-40:60) для того, чтобы получать 4-(4,4-дифтор-1-гидроксициклогексил)бензонитрил в виде бесцветного твердого вещества (2,19 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,77-1,92 (м, 2H) 2,00-2,45 (м, 6H) 7,58-7,72 (м, 4H).
MS EI положительн.: 237[M]+.
(2) Синтез указанного в заголовке соединения
Использовали соединение (2,19 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в ссылочном примере 45-1(2), для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (1,51 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 1,66-1,79 (м, 2H) 1,85-2,04 (м, 4H) 2,09-2,36 (м, 2H) 3,99 (с, 2H) 5,28 (с, 1H) 7,40-7,48 (м, 2H) 7,49-7,57 (м, 2H) 8,32 (ушир. с, 3H).
MS ESI/APCI двойн. положительн.: 242[M+H]+.
MS ESI/APCI двойн. отрицательн.: 276[M+Cl]-.
Ссылочный пример 46-1
1-[транс-3-(4-хлорфенокси)циклобутил]метанамин
[0353]

(1) Синтез N-бензил-3-оксоциклобутанкарбоксамида
[0354]
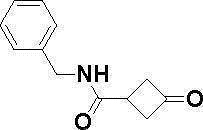
В раствор 3-оксоциклобутанкарбоновой кислоты (13,5 г) в тетрагидрофуране (135 мл) добавляли 1,1'-карбонилдиимидазол (23,0 г) при охлаждении льдом. Смесь доводили до комнатной температуры и перемешивали в течение 90 минут. Добавляли бензиламин (15,5 мл) и смесь перемешивали при этой температуре в течение 14 часов. Неочищенный продукт адсорбировали на диатомовой земле, при этом растворитель отгоняли при пониженном давлении. Неочищенный продукт, адсорбированный на диатомовой земле, очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 100:0-95:5) для того, чтобы получать N-бензил-3-оксоциклобутанкарбоксамид в виде бесцветного твердого вещества (16,9 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,89-3,10 (м, 1H)3,11-3,32 (м, 2H) 3,41-3,67 (м, 2H) 4,50 (д, J=5,8 Гц, 2H) 7,25-7,42 (м, 5H).
MS ESI/APCI двойн. положительн.: 204[M+H]+.
(2) Синтез цис-3-[(бензиламино)метил]циклобутанола
[0355]
Использовали соединение (16,9 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в ссылочном примере 23-1(2), для того, чтобы получать цис-3-[(бензиламино)метил]циклобутанол в виде масла бледно-желтого цвета (16,2 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 1,35-1,57 (м, 2H) 1,66-1,93 (м, 2H) 2,12-2,36 (м, 2H) 2,42-2,53 (м, 2H) 3,69 (с, 2H) 3,78-4,00 (м, 1H) 7,12-7,41 (м, 5H).
MS ESI/APCI двойн. положительн.: 192[M+H]+.
(3) Синтез цис-3-(аминометил)циклобутанола
[0356]
Использовали соединение (16,2 г), полученное на приведенной выше стадии (2), и обработку проводили с помощью того же способа, как в ссылочном примере 29-1(4), для того, чтобы получать цис-3-(аминометил)циклобутанол в виде бесцветного масла (10,4 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 1,29-1,51 (м, 2H) 1,53-1,70 (м, 1H) 2,08-2,29 (м, 2H) 2,41-2,51 (м, 2H) 3,76-3,98 (м, 1H).
(4) Синтез трет-бутил [(цис-3-гидроксициклобутил)метил]карбамата
[0357]
Использовали соединение (10,3 г), полученное на приведенной выше стадии (3), и обработку проводили с помощью того же способа, как в ссылочном примере 44-1, для того, чтобы получать трет-бутил [(цис-3-гидроксициклобутил)метил]карбамат в виде бесцветного твердого вещества (6,08 г).
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 1,37 (с, 9H) 1,37-1,49 (м, 2H) 1,66-1,79 (м, 1H) 2,08-2,23 (м, 2H) 2,87-2,91 (м, 2H) 3,78-3,91 (м, 1H) 4,82-4,94 (м, 1H) 6,76 (т, J=5,4 Гц, 1H).
MS ESI/APCI двойн. положительн.: 224[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 200[M-H]-.
(5) Синтез трет-бутил {[транс-3-(4-хлорфенокси)циклобутил]метил}карбамата
[0358]
Вместо 4-гидроксибензальдегида и 2-циклопропилэтанола соответственно использовали 4-хлорфенол (767 мг) и соединение (1,00 г), полученное на приведенной выше стадии (4), и обработку проводили с помощью того же способа, как в ссылочном примере 11-1, для того, чтобы получать трет-бутил {[транс-3-(4-хлорфенокси)циклобутил]метил}карбамат в виде бесцветного твердого вещества (1,05 г).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,45 (с, 9H) 2,15-2,37 (м, 4H) 2,40-2,65 (м, 1H) 3,12-3,35 (м, 2H) 4,64-4,79 (м, 1H) 6,63-6,76 (м, 2H) 7,15-7,25 (м, 2H).
(6) Синтез указанного в заголовке соединения
В раствор в 1,4-диоксане (30 мл) соединения (0,98 г), полученного на приведенной выше стадии (5), добавляли раствор (25 мл) хлороводорода в 1,4-диоксане 4 моль/л и смесь перемешивали при комнатной температуре в течение 5 часов. После добавления диэтилового эфира (120 мл) смесь перемешивали в течение других двух часов и после этого осадок собирали с помощью фильтрования. Собранный осадок растворяли в водном растворе гидроксида натрия 1 моль/л и хлороформе и проводили два экстрагирования хлороформом. Объединенные органические слои сушили над безводным сульфатом магния и удаляли десикант с помощью фильтрования. Фильтрат концентрировали при пониженном давлении для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (660 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,12-2,48 (м, 5H) 2,74-2,87 (м, 2H) 4,61-4,77 (м, 1H) 6,63-6,80 (м, 2H) 7,12-7,27 (м, 2H).
[0359] В следующих ссылочных примерах с 46-2 до 46-4 использовали соответствующие фенолы торговых сортов и обработку проводили с помощью способа, описанного в ссылочном примере 46-1, или его модификациях для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблице 17-1.
[0360]

Ссылочный пример 46-5
1-{цис-3-[(4-хлорбензил)окси]циклобутил}метанамин
[0361]

(1) Синтез трет-бутил ({цис-3-[(4-хлорбензил)окси]циклобутил}метил)карбамата
[0362]

Вместо 4-гидроксибензальдегида и (бромметил)циклобутана соответственно использовали соединение (1,00 г), полученное в ссылочном примере 46-1(4), и 4-хлорбензилбромид (1,02 г) и обработку проводили с помощью того же способа, как в ссылочном примере 9-1, для того, чтобы получать трет-бутил ({цис-3-[(4-хлорбензил)окси]циклобутил}метил)карбамат в виде бесцветного твердого вещества (750 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,44 (с, 9H) 1,58-1,71 (м, 2H) 1,88-2,10 (м, 1H) 2,25-2,43 (м, 2H) 3,08-3,22 (м, 2H) 3,81-3,95 (м, 1H) 4,36 (с, 2H) 7,20-7,37 (м, 4H).
(2) Синтез указанного в заголовке соединения
Использовали соединение (750 мг), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в ссылочном примере 46-1(6), для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (523 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,52-1,71 (м, 2H) 1,77-1,95 (м, 1H) 2,27-2,45 (м, 2H) 2,72 (д, J=6,7 Гц, 2H) 3,81-4,01 (м, 1H) 4,38 (с, 2H) 7,20-7,39 (м, 4H).
MS ESI/APCI двойн. положительн.: 226[M+H]+.
Ссылочный пример 46-6
1-[цис-3-(4-хлорфенокси)циклобутил]метанамин
[0363]
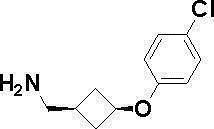
(1) Синтез транс-3-[({[(2-метил-2-пропанил)окси]карбонил}амино)метил]циклобутил 4-нитробензоата
[0364]

В смесь соединения (2,00 г), полученного в ссылочном примере 46-1(4), 4-нитробензойной кислоты (3,32 г), трифенилфосфина (5,21 г) и тетрагидрофурана (50 мл) добавляли диизопропил азодикарбоксилат (1,0 моль/л, раствор в толуоле, 10,5 мл) и смесь перемешивали при комнатной температуре в течение 16 часов. После концентрирования при пониженном давлении, получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=100:0-75:25) для того, чтобы получать транс-3-[({[(2-метил-2-пропанил)окси]карбонил}амино)метил]циклобутил 4-нитробензоат в виде бесцветного твердого вещества (3,88 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,46 (с, 9H) 2,24-2,45 (м, 4H) 2,46-2,68 (м, 1H) 3,21-3,33 (м, 2H) 5,29-5,41 (м, 1H) 8,18-8,25 (м, 2H) 8,26-8,32 (м, 2H).
(2) Синтез трет-бутил[(транс-3-гидроксициклобутил)метил]карбамата
[0365]

В раствор в тетрагидрофуране (100 мл) соединения (3,88 г), полученного на приведенной выше стадии (1), добавляли водный раствор гидроксида натрия 1 моль/л (19,9 мл) и смесь перемешивали при комнатной температуре в течение 4 часов. Проводили экстрагирование этилацетатом и после сушки объединенных органических слоев над безводным сульфатом магния десикант удаляли с помощью фильтрования. При этом растворитель отгоняли при пониженном давлении, неочищенный продукт адсорбировали на диатомовой земле. Неочищенный продукт, адсорбированный на диатомовой земле, очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 99:1-0:100, затем хлороформ:метанол = 100:0-90:10) для того, чтобы получать трет-бутил [(транс-3-гидроксициклобутил)метил]карбамат в виде бесцветного твердого вещества (1,87 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 1,37 (с, 9H) 1,72-2,00 (м, 4H) 2,01-2,22 (м, 1H) 2,85-3,02 (м, 2H) 4,05-4,23 (м, 1H) 4,81-4,95 (м, 1H) 6,72-6,91 (м, 1H).
(3) Синтез указанного в заголовке соединения
Использовали соединение (500 мг), полученное на приведенной выше стадии (2), и обработку проводили с помощью тех же способов, как в ссылочном примере 46-1(5) и (6), для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (460 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,71-1,90 (м, 2H) 1,95-2,13 (м, 1H) 2,50-2,68 (м, 2H) 2,77 (д, J=6,8 Гц, 2H) 4,43-4,60 (м, 1H) 6,66-6,80 (м, 2H) 7,14-7,26 (м, 2H).
MS ESI/APCI двойн. положительн.: 212[M+H]+.
Ссылочный пример 46-7
1-{транс-3-[(4-хлорбензил)окси]циклобутил}метанамин
[0366]
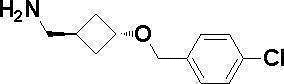
Использовали соединение (1,30 мг), полученное в ссылочном примере 46-6(2), и обработку проводили с помощью того же способа, как в ссылочном примере 46-5, для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (630 smg).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,91-2,34 (м, 5H) 2,72 (д, J=7,3 Гц, 2H) 4,03-4,20 (м, 1H) 4,36 (с, 2H) 7,20-7,38 (м, 4H).
MS ESI/APCI двойн. положительн.: 226[M+H]+.
Ссылочный пример 47-1
2-{[трет-бутил(диметил)силил]окси}-1-(4-йодфенил)этанамин
[0367]

(1) Синтез амино(4-йодфенил)ацетонитрила
[0368]

В раствор 4-йодбензальдегида (10,4 г) в метаноле (36 мл) добавляли тетраизопропил ортотитанат (50,0 мл) и раствор (50 мл) 8 моль/л аммиака в метаноле и смесь перемешивали при комнатной температуре в течение 3,5 часа. Триметилсилилцианид (5,89 мл) медленно добавляли в смесь, которую затем перемешивали при той же температуре в течение 14 часов. Ледяную воду добавляли в реакционную смесь, которую затем фильтровали через Celite (зарегистрированный товарный знак). Фильтрат концентрировали при пониженном давлении, и получаемый остаток экстрагировали этилацетатом. Объединенные органические слои промывали водой и сушили над безводным сульфатом магния. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 99:1-25:75) для того, чтобы получать амино(4-йодфенил)ацетонитрил в виде твердого вещества бледно-желтого цвета (5,05 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 4,87 (с, 2H) 7,27-7,33 (м, 2H) 7,69-7,83 (м, 2H).
(2) Синтез гидрохлорида амино(4-йодфенил)уксусной кислоты
[0369]
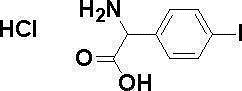
Суспензию соединения (5,05 г), полученного на приведенной выше стадии (1), в 6 моль/л соляной кислоте (65 мл) перемешивали при 105°C в течение 14 часов. После охлаждения до комнатной температуры, суспензию перемешивали при комнатной температуре в течение часа и затем перемешивали в течение 30 минут при охлаждении льдом. Получаемый осадок собирали с помощью фильтрования для того, чтобы получать гидрохлорид амино(4-йодфенил)уксусной кислоты в виде бесцветного твердого вещества (4,32 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 4,18 (с, 2H) 7,08-7,30 (м, 2H) 7,60-7,77 (м, 2H).
MS ESI/APCI двойн. положительн.: 278[M+H]+.
MS ESI/APCI двойн. отрицательн.: 276[M-H]-.
(3) Синтез 2-амино-2-(4-йодфенил)этанола
[0370]
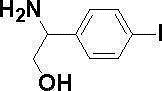
В жидкую смесь борогидрида лития (2,0 моль/л, раствор в тетрагидрофуране, 19,7 мл) и хлортриметилсилана (9,97 мл), малыми частями добавляли соединение (4,95 г), полученное на приведенной выше стадии (2), при комнатной температуре и смесь перемешивали в течение 16 часов. При охлаждении льдом метанол (3,5 мл) добавляли в смесь, которую затем доводили до комнатной температуры и перемешивали в течение 15 минут. В перемешанную смесь последовательно добавляли воду (19,7 мл), этилацетат (39,4 мл), насыщенный солевой раствор (19,7 мл) и гидроксид натрия (1,87 г) и смесь перемешивали при той же температуре в течение 16 часов. Реакционную смесь экстрагировали этилацетатом, и объединенные органические слои сушили над безводным сульфатом магния. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении для того, чтобы получать 2-амино-2-(4-йодфенил)этанол в виде твердого вещества желтого цвета (4,70 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 3,43-3,59 (м, 1H) 3,65-3,77 (м, 1H) 3,94-4,10 (м, 1H) 7,02-7,15 (м, 2H) 7,62-7,75 (м, 2H).
MS ESI/APCI двойн. положительн.: 264[M+H]+.
(4) Синтез указанного в заголовке соединения
В смесь соединения (4,70 г), полученного на приведенной выше стадии (3), 4-диметиламинопиридина (48,2 мг), триэтиламина (4,40 мл) и хлороформа (63,2 мл) по каплям добавляли раствор трет-бутилдиметилхлорсилана (2,38 г) в хлороформе (31,6 мл) при охлаждении льдом, и смесь перемешивали при той же температуре в течение 30 минут, а затем при комнатной температуре в течение трех суток. Реакционную смесь концентрировали при пониженном давлении и затем добавляли воду и этилацетат. Проводили экстрагирование этилацетатом и объединенные органические слои сушили над безводным сульфатом магния. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 100:0-98:2) для того, чтобы получать указанное в заголовке соединение в виде масла бледно-желтого цвета (4,68 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,02 (с, 6H) 0,89 (с, 9H) 3,40-3,56 (м, 1H) 3,62-3,74 (м, 1H) 3,97-4,07 (м, 1H) 7,06-7,20 (м, 2H) 7,56-7,72 (м, 2H).
Ссылочный пример A-1
Метил [4-({[4'-(трифторметил)бифенил-4-ил]метил}амино)тетрагидро-2H-пиран-4-ил]ацетат
[0371]
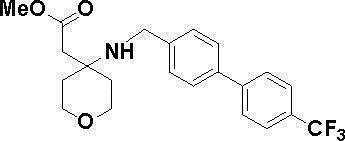
В раствор в хлороформе (20 мл) соединения (500 мг), полученного в ссылочном примере 1-1, добавляли соединение (627 мг), полученное в ссылочном примере 6-2, и смесь перемешивали при комнатной температуре в течение 30 минут. Триацетоксиборогидрид натрия (654 мг) добавляли в смесь, которую дополнительно перемешивали при комнатной температуре в течение 12 часов. При охлаждении льдом насыщенный водный раствор бикарбоната натрия добавляли в смесь, которую затем доводили до комнатной температуры. Проводили три экстрагирования хлороформом. Объединенные органические слои пропускали через фазовый разделитель и после этого концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=80:20-0:100) для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (784 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,66-1,76 (м, 4H) 2,60 (с, 2H) 3,60-3,78 (м, 7H) 3,86-3,99 (м, 2H) 4,77 (с, 1H) 7,45-7,52 (м, 2H) 7,54-7,63 (м, 2H) 7,65-7,73 (м, 4H).
MS ESI/APCI двойн. положительн.: 408[M+H]+, 430[M+Na]+.
[0372] В следующих ссылочных примерах с A-2 до A-485 соединения, полученные в ссылочных примерах с 1-1 до 5-5, ссылочных примерах с 28-1 до 30-2, или соответствующие сложные эфиры β-аланина торговых сортов, а также соединения, полученные в ссылочных примерах с 6-1 до 26-1, ссылочных примерах с 31-1 до 44-1, или соответствующие альдегиды или кетоны торговых сортов использовали в качестве исходных материалов и обработку проводили с помощью способа, описанного в ссылочном примере A-1, или его модификаций для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблицах с 18-1 до 18-69.
[0373]
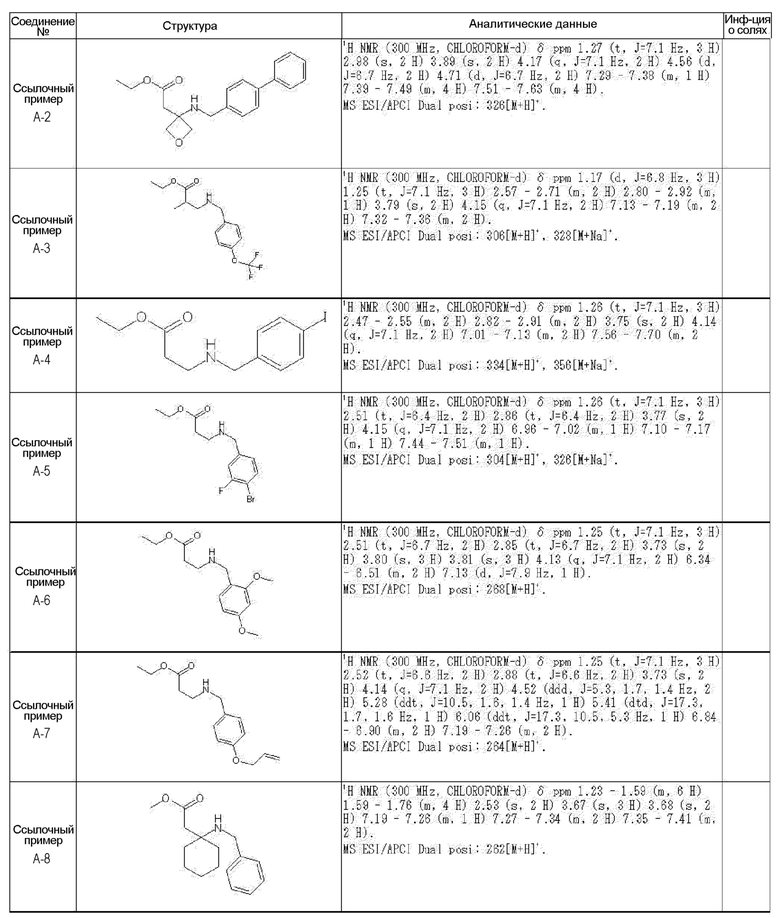
[0374]
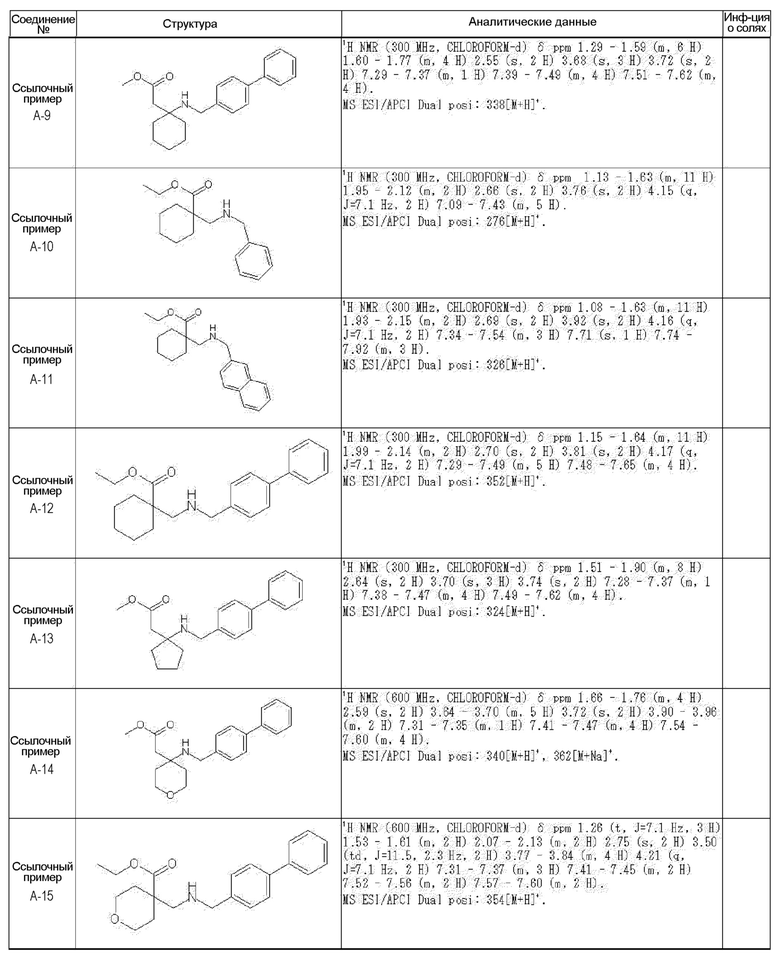
[0375]
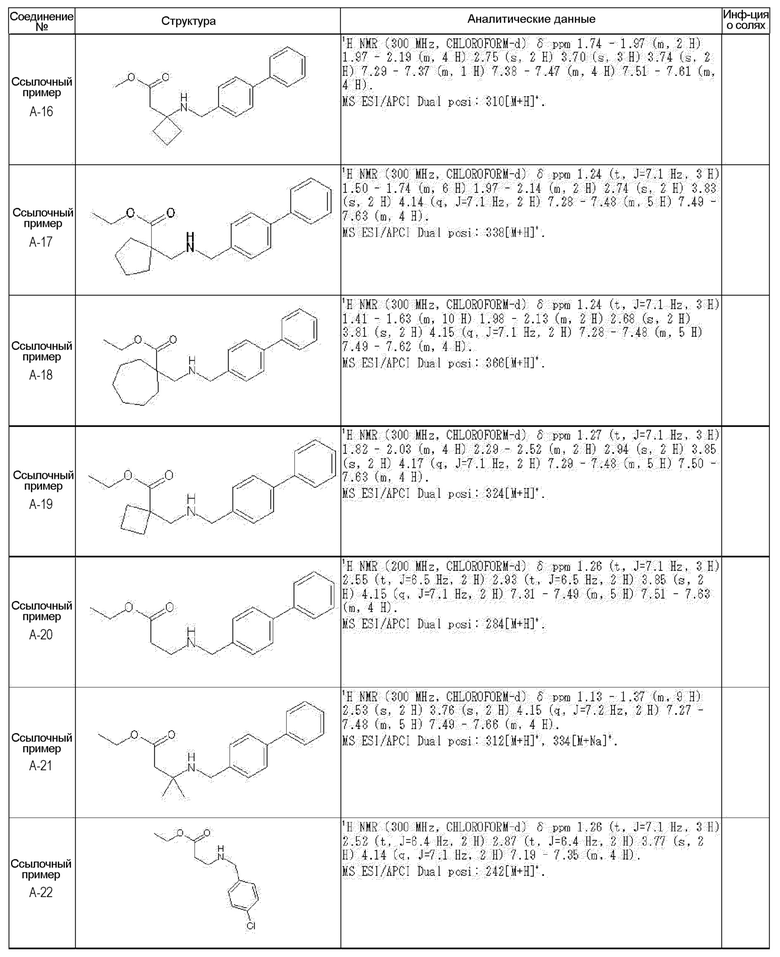
[0376]

[0377]

[0378]

[0379]
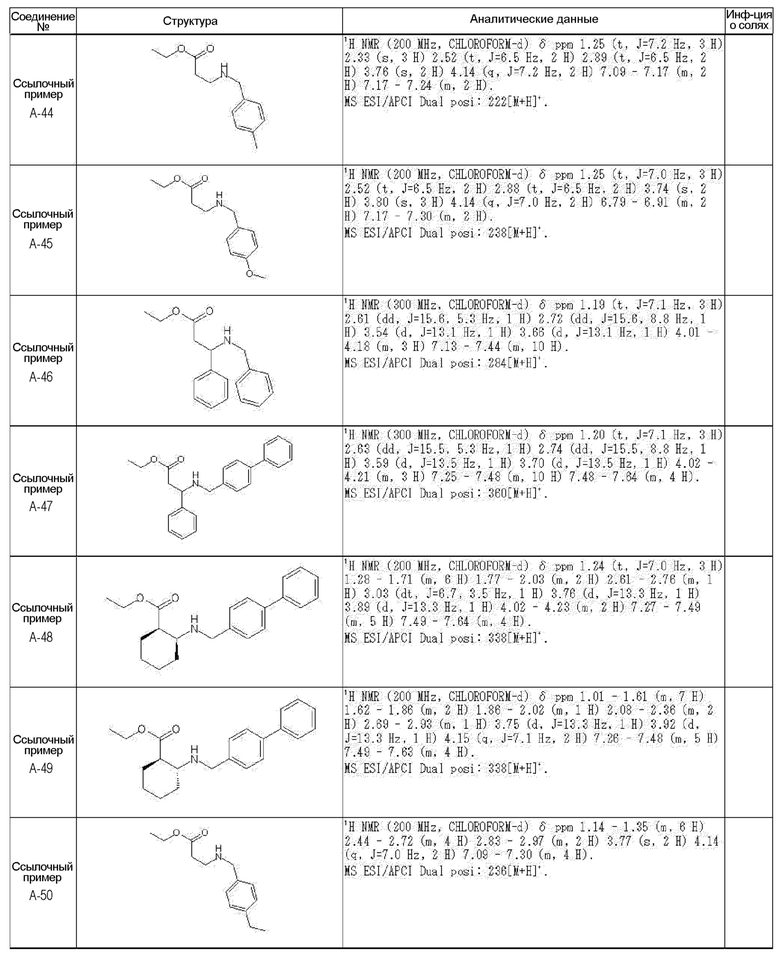
[0380]
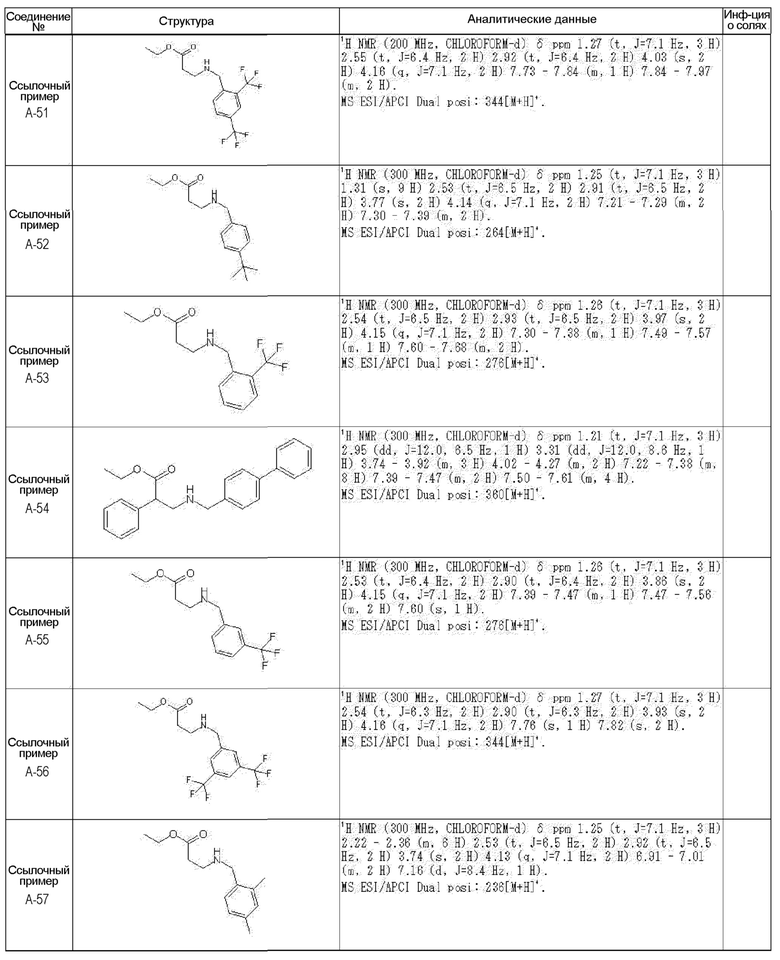
[0381]

[0382]
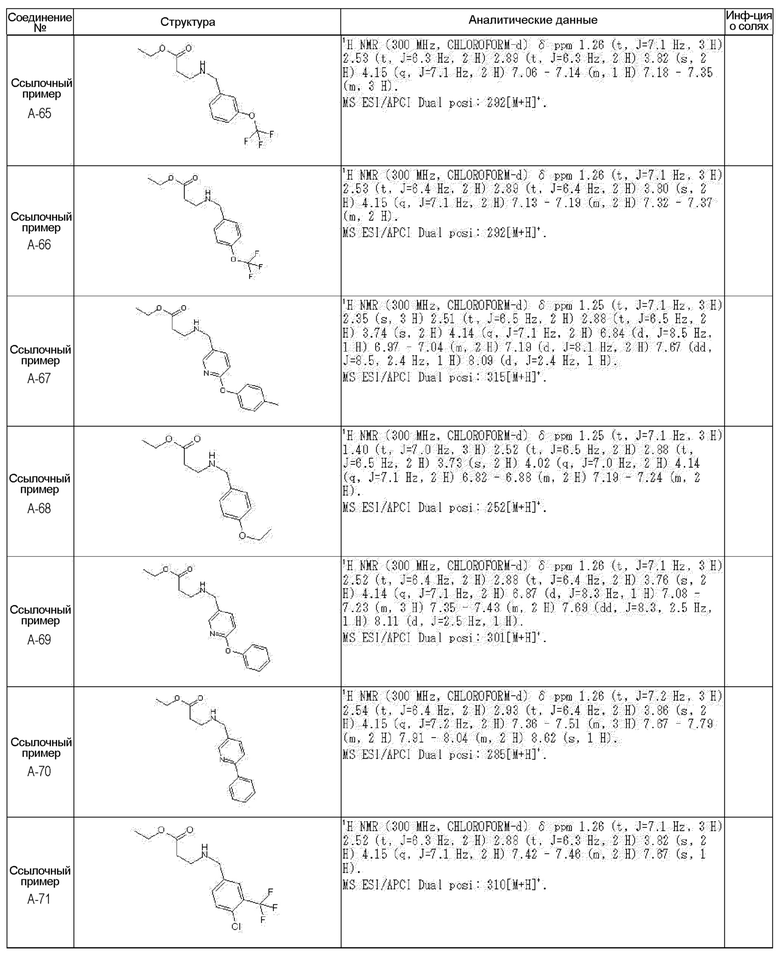
[0383]
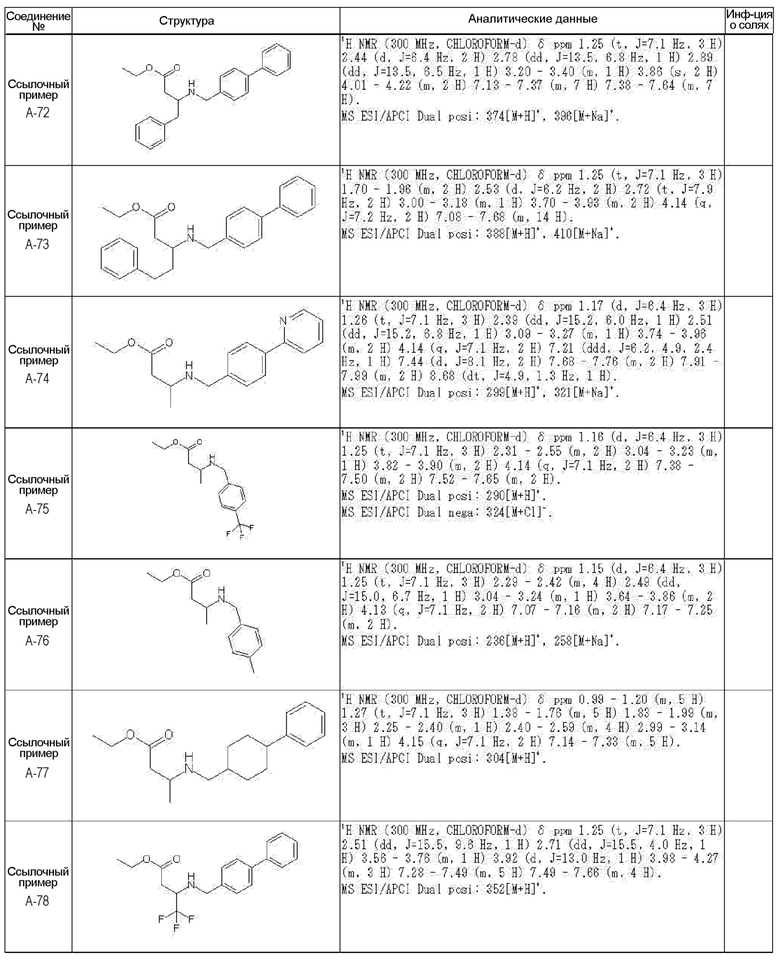
[0384]
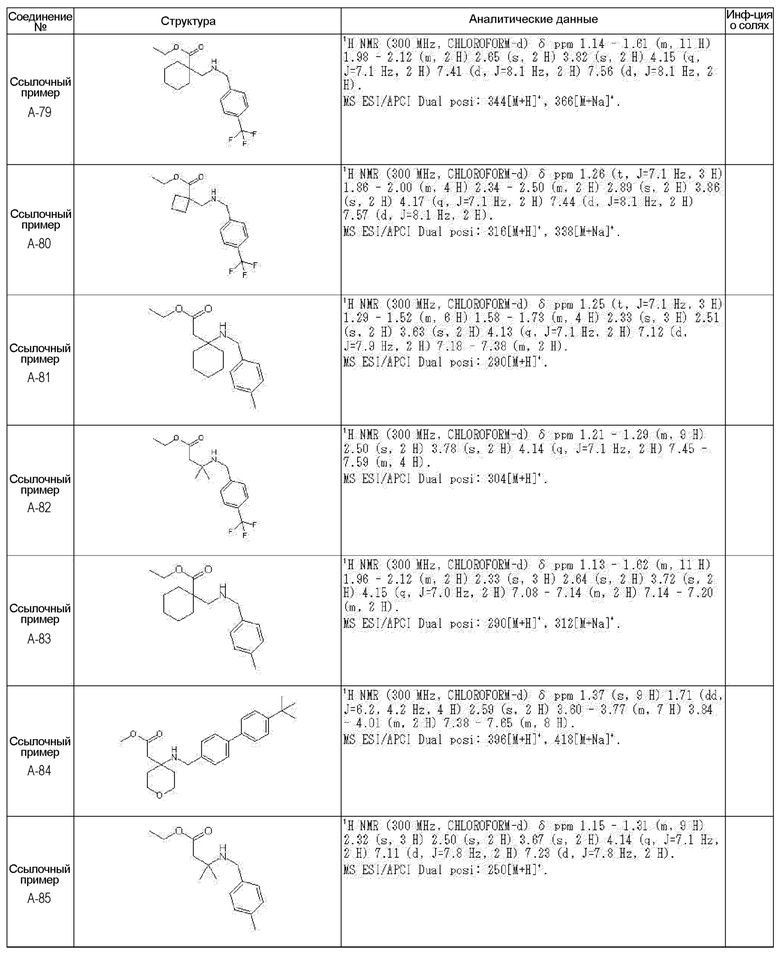
[0385]
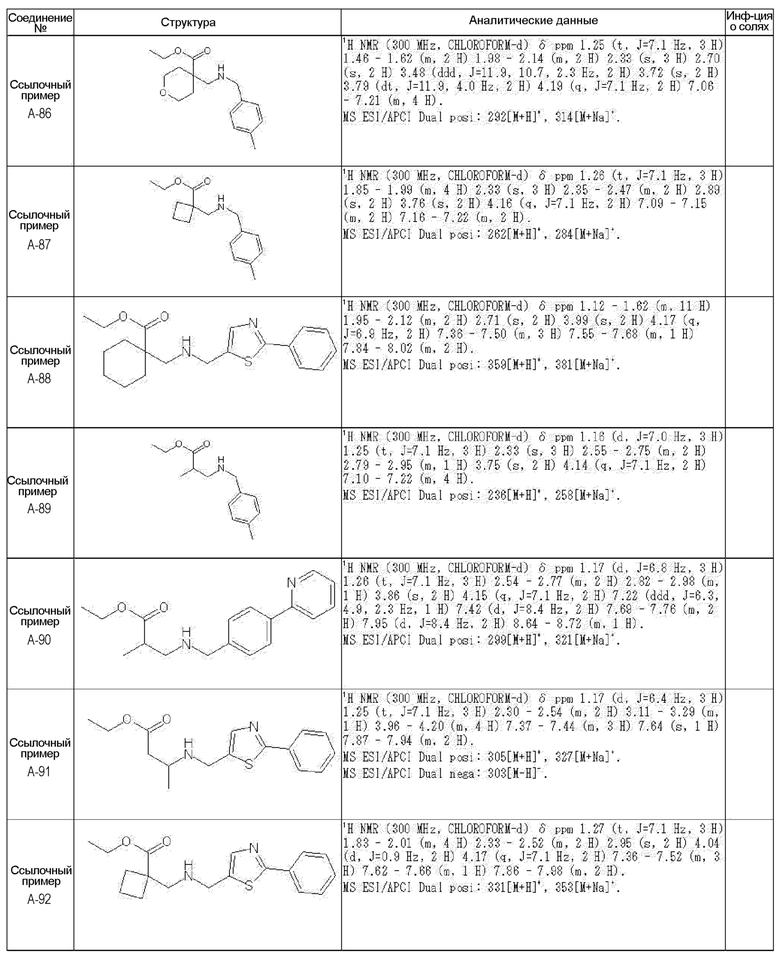
[0386]
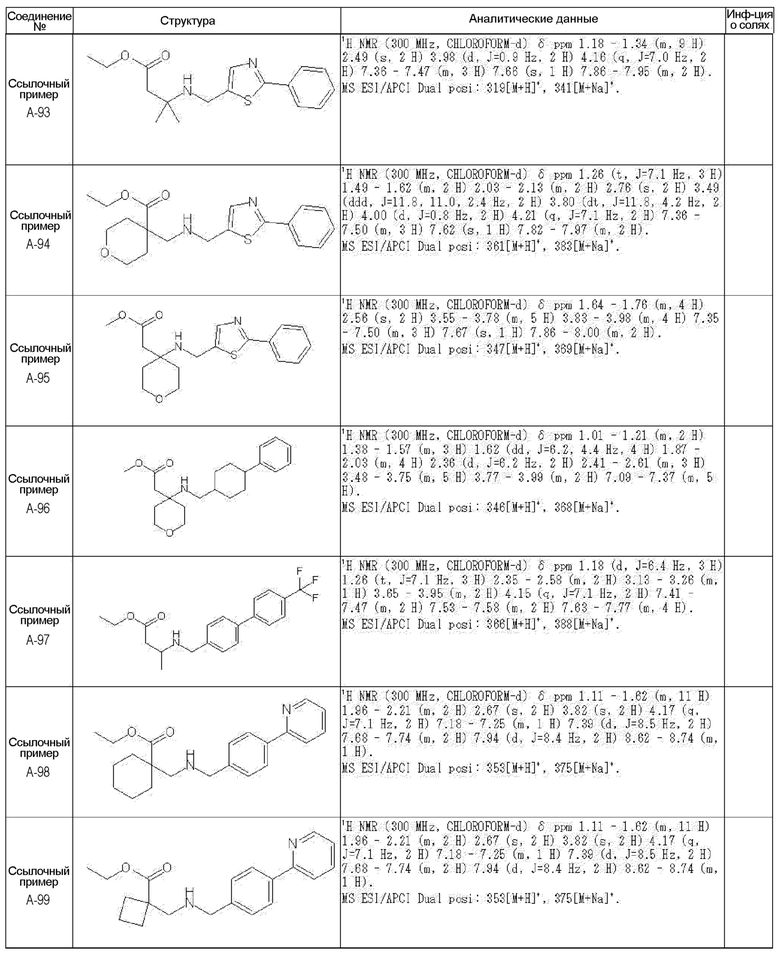
[0387]
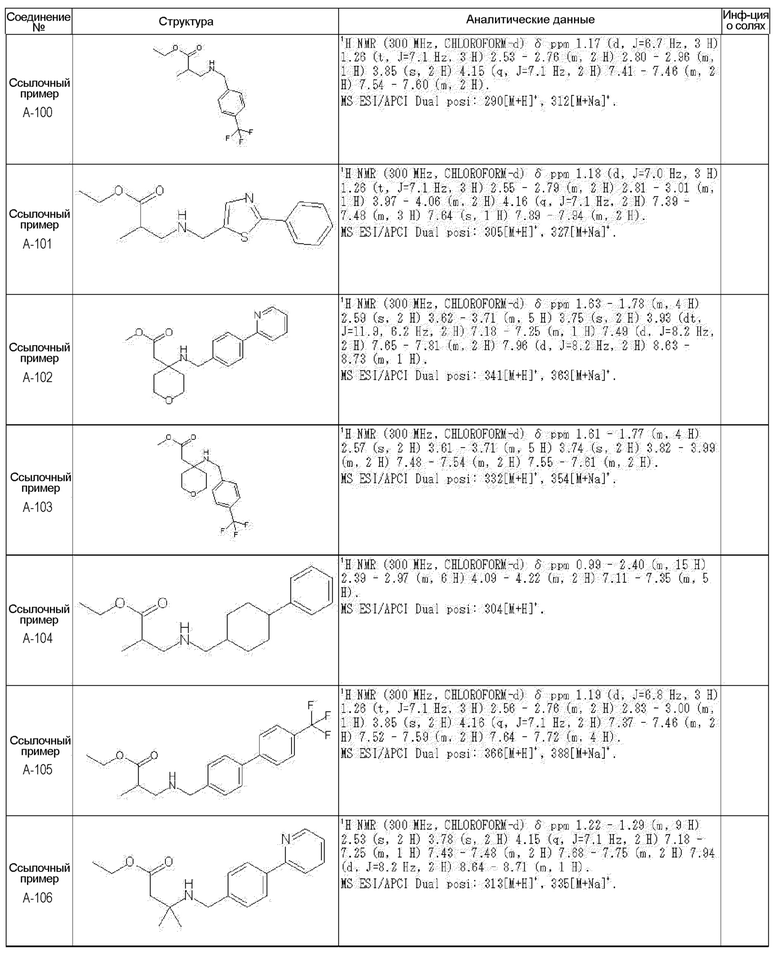
[0388]

[0389]

[0390]

[0391]
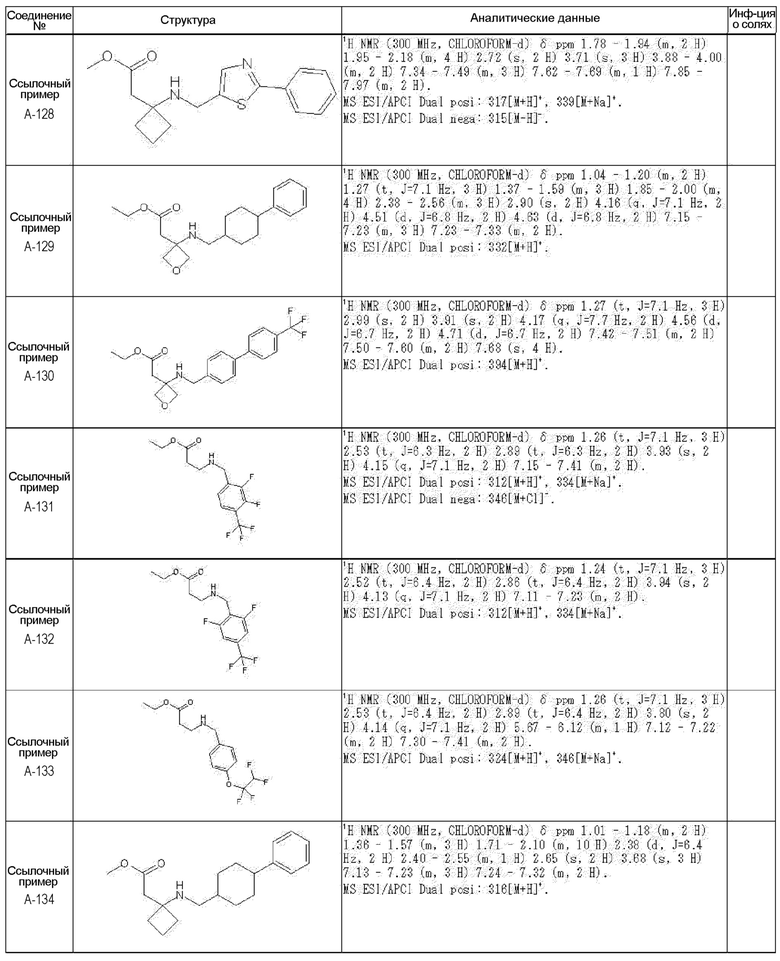
[0392]

[0393]

[0394]
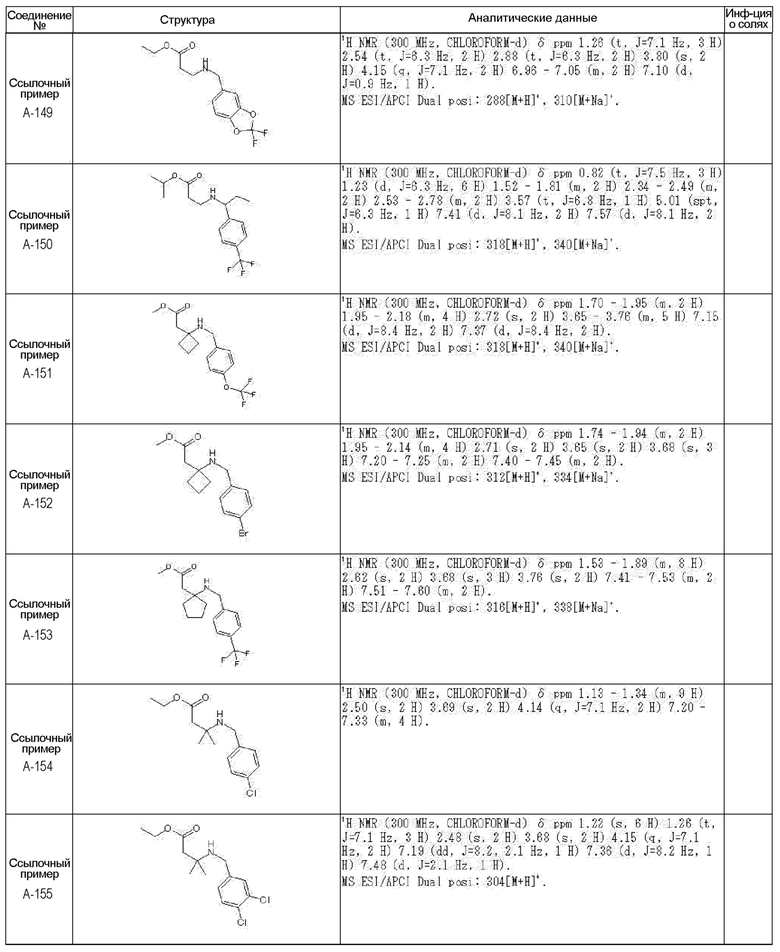
[0395]

[0396]
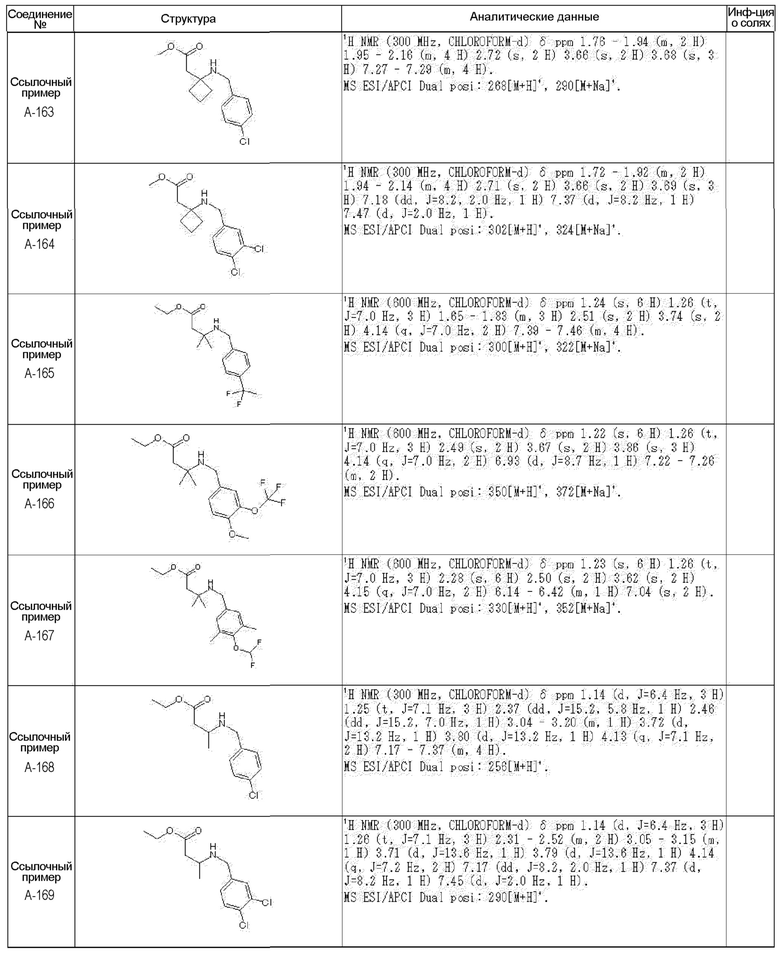
[0397]

[0398]
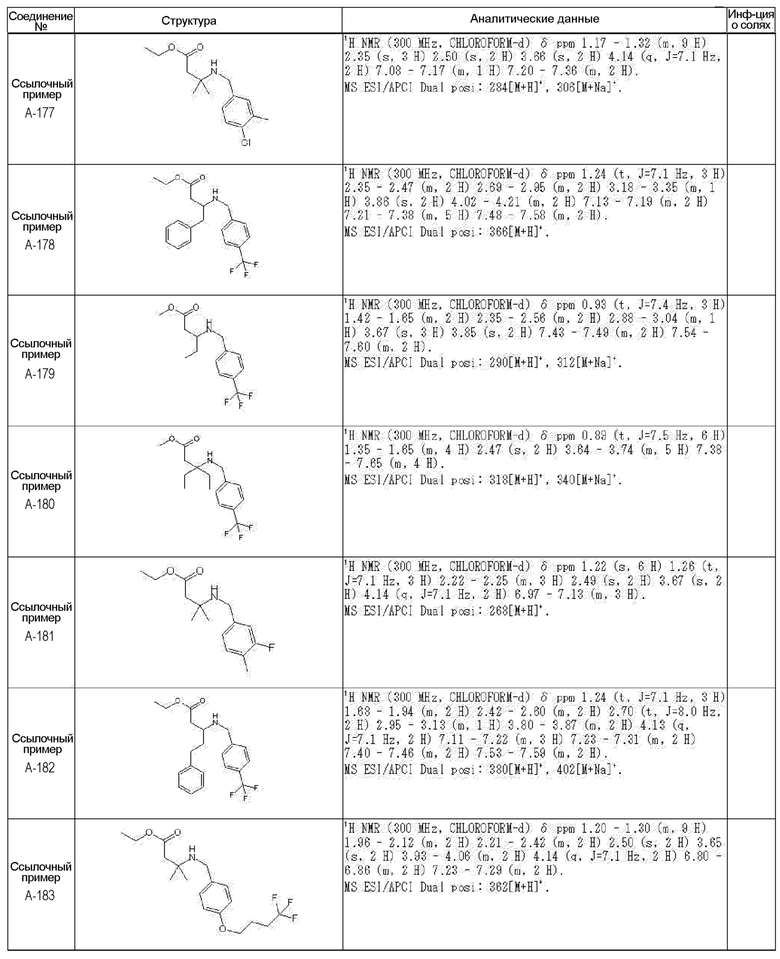
[0399]
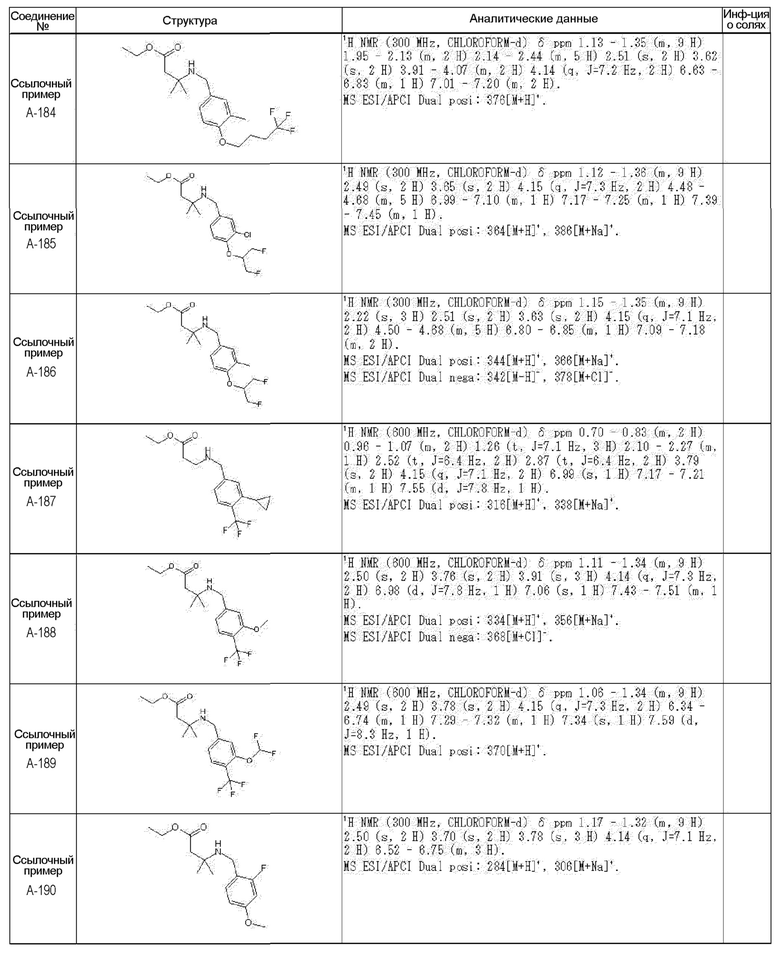
[0400]
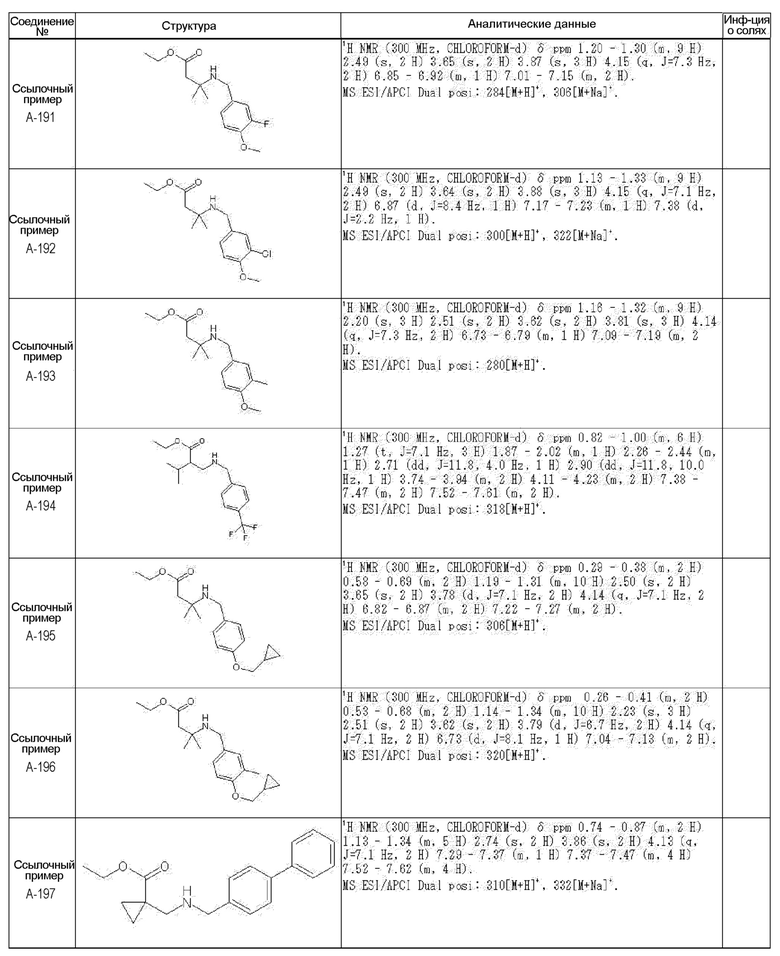
[0401]

[0402]

[0403]
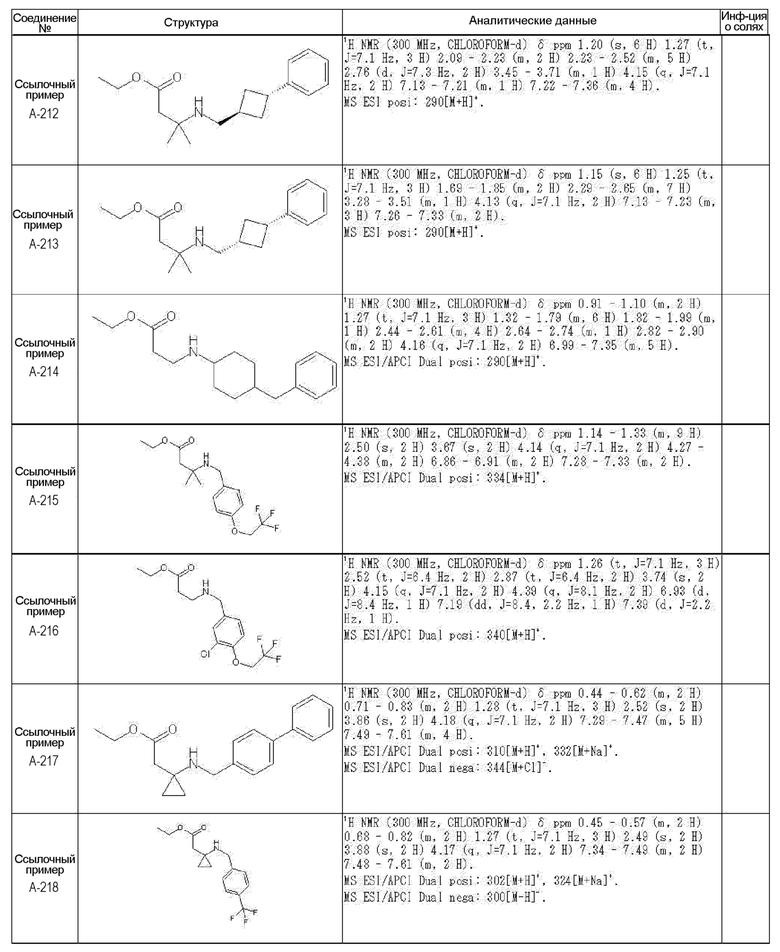
[0404]
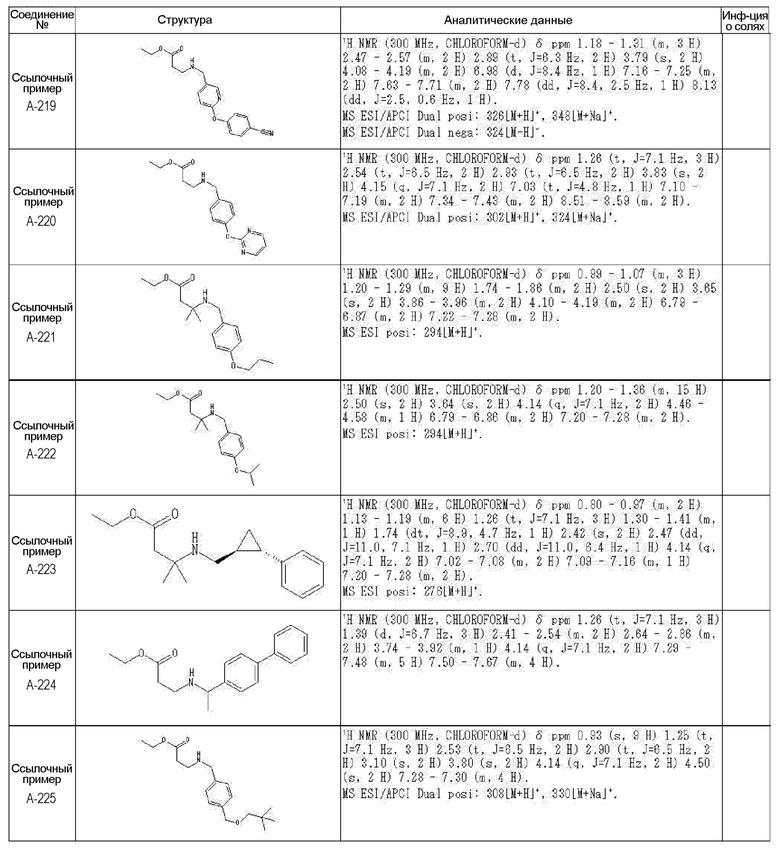
[0405]
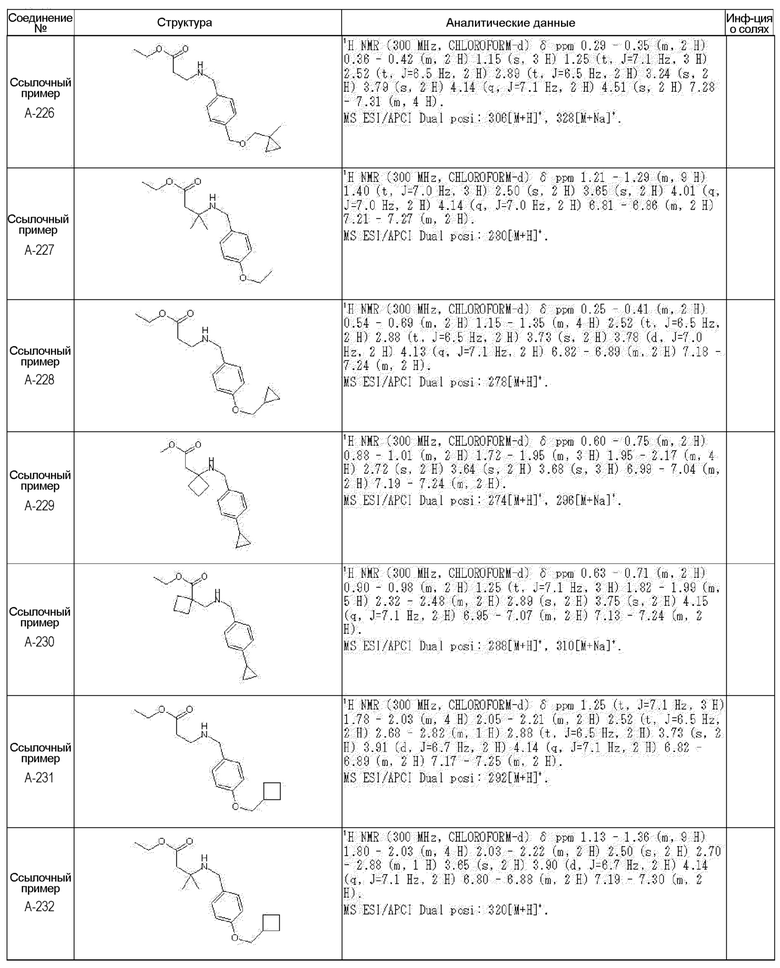
[0406]

[0407]
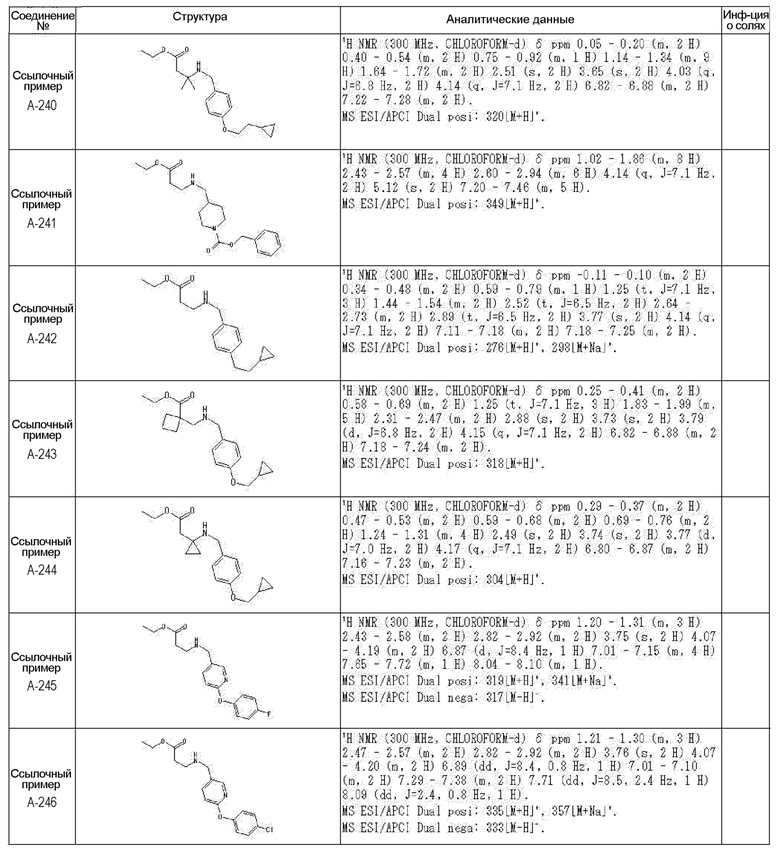
[0408]

[0409]
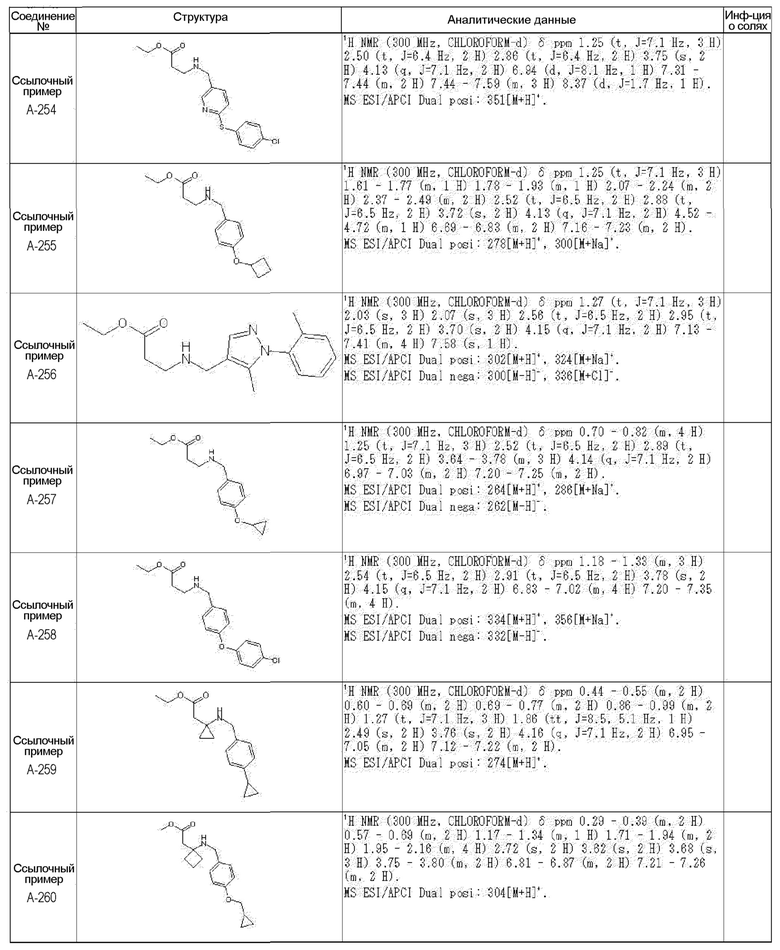
[0410]
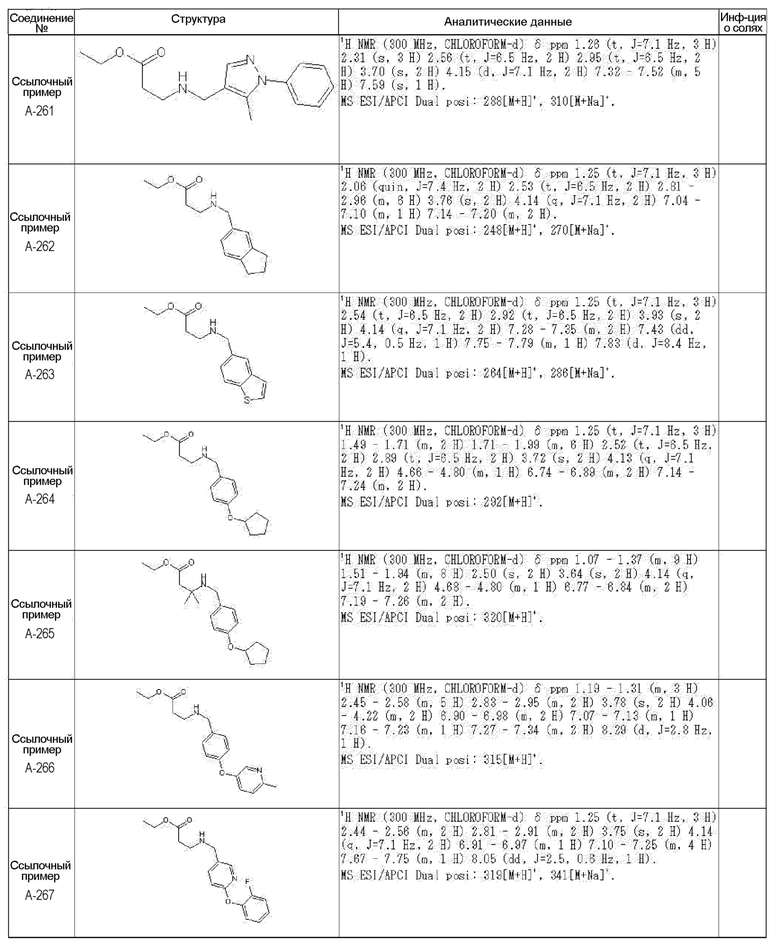
[0411]
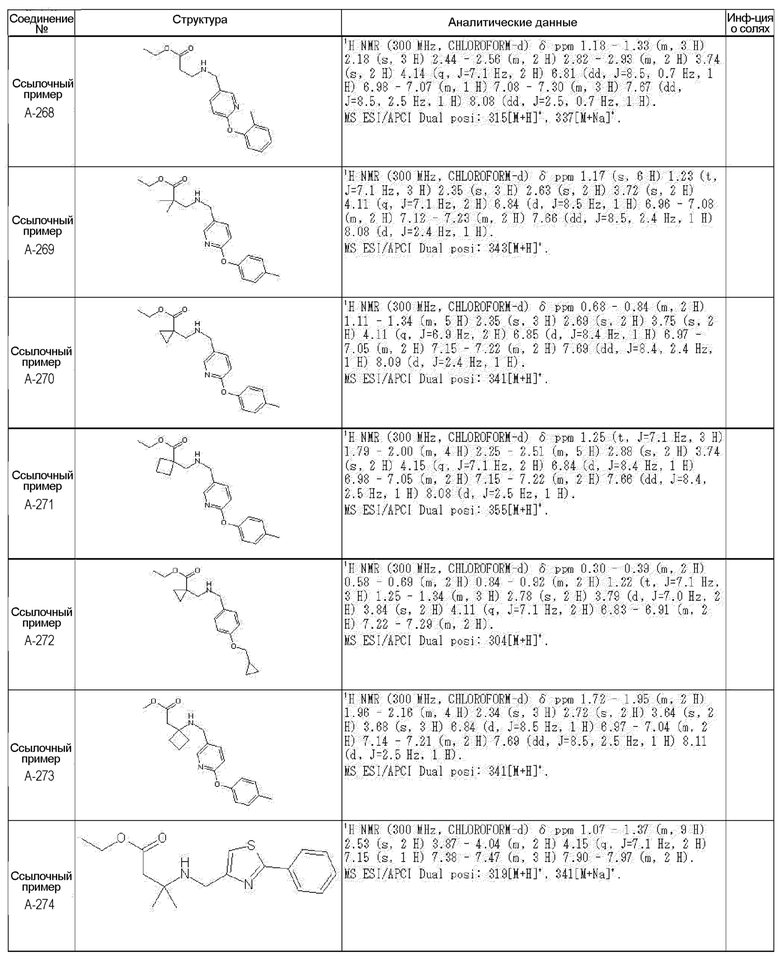
[0412]
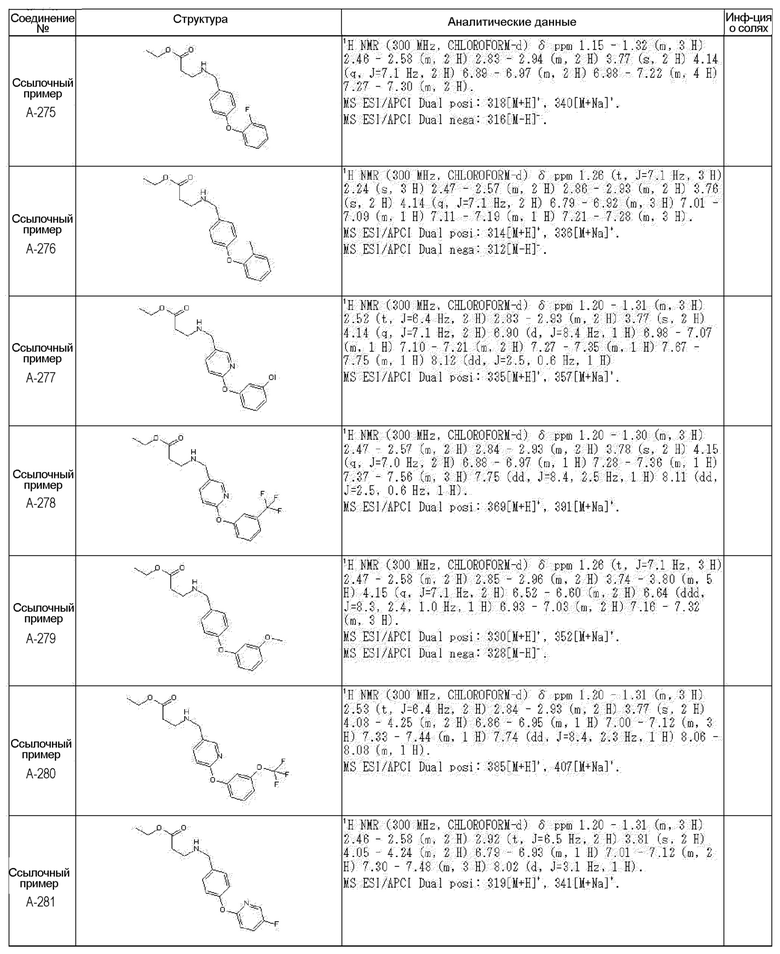
[0413]
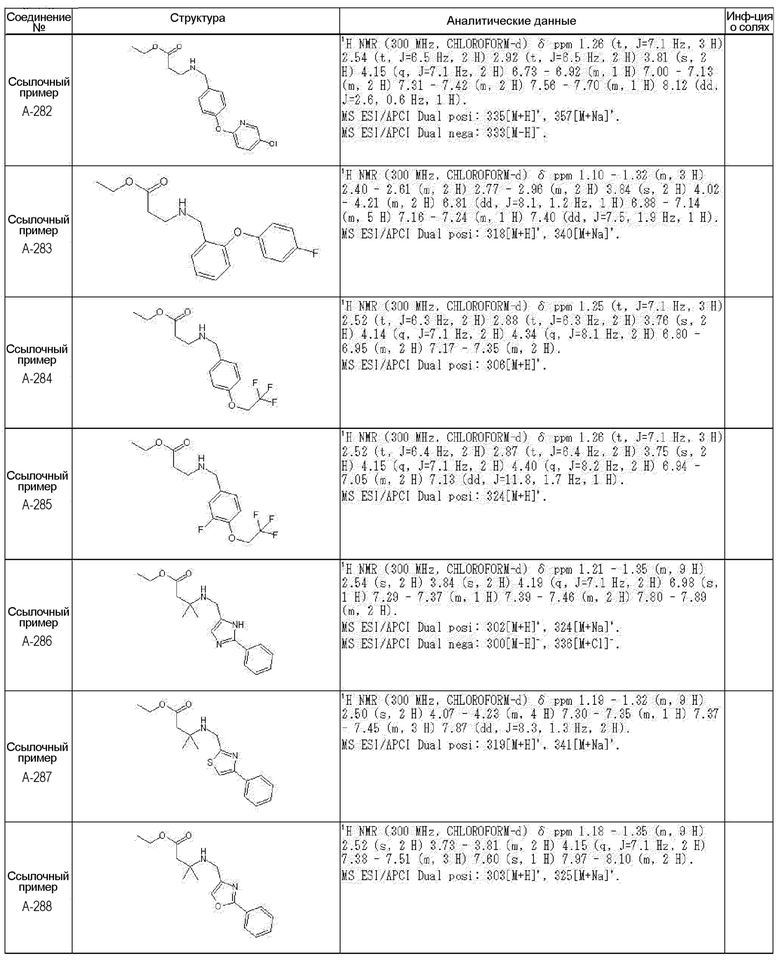
[0414]

[0415]

[0416]

[0417]
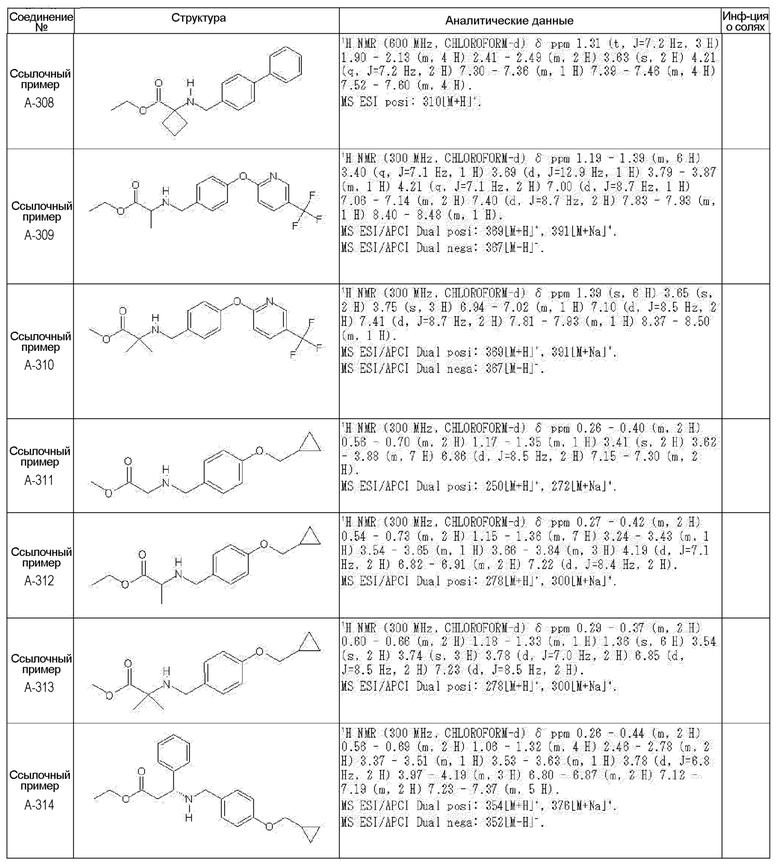
[0418]
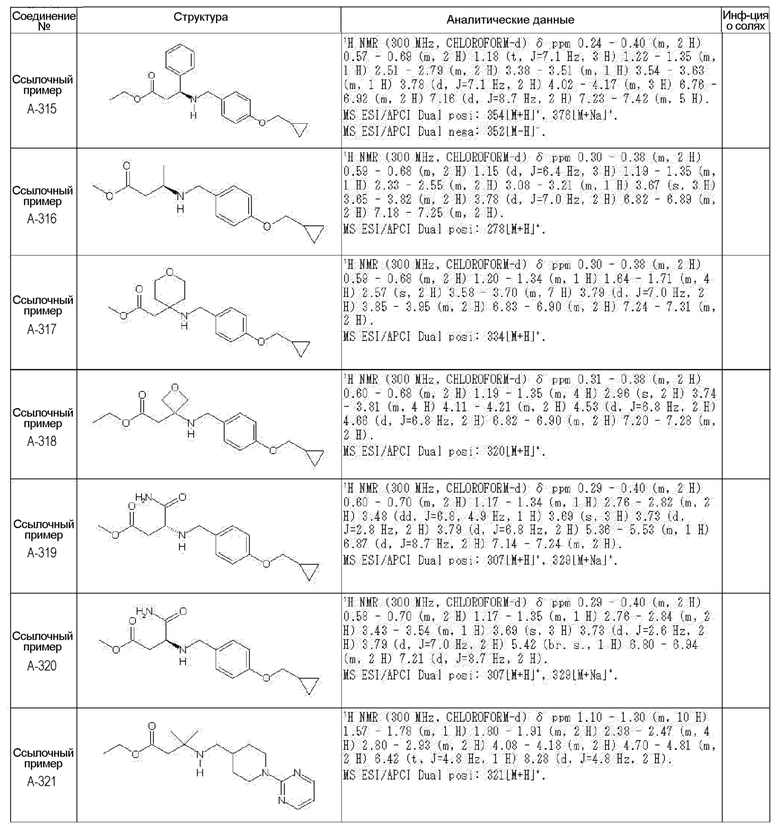
[0419]
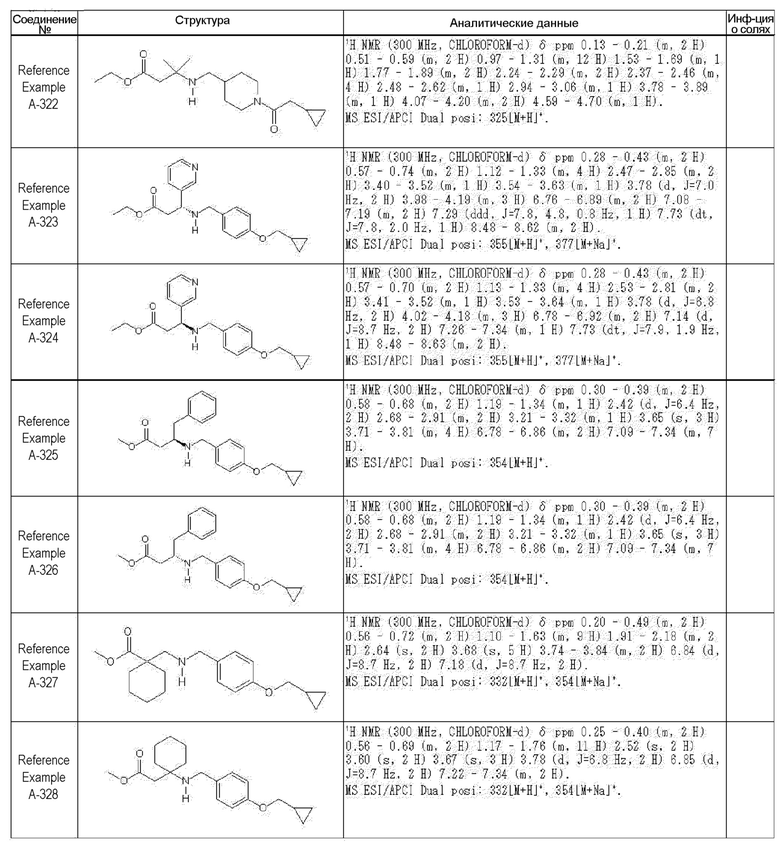
[0420]

[0421]

[0422]

[0423]

[0424]
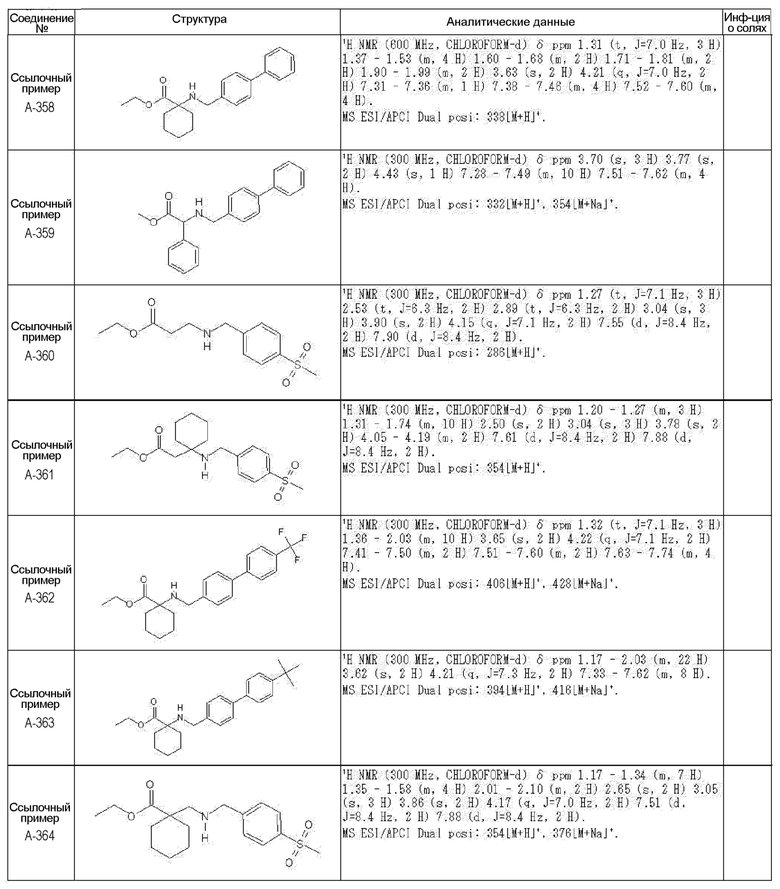
[0425]
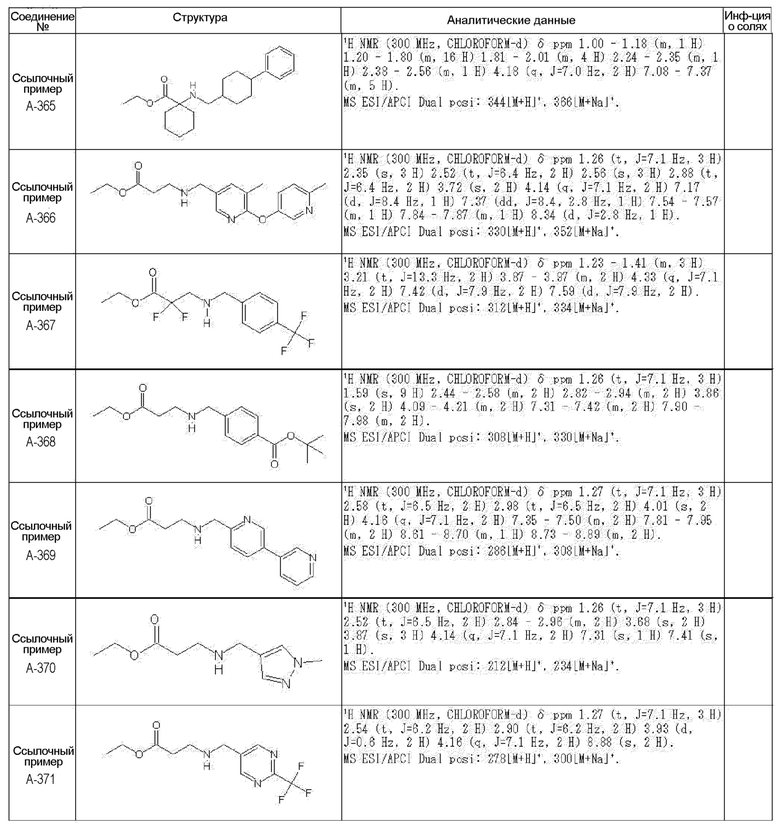
[0426]

[0427]

[0428]
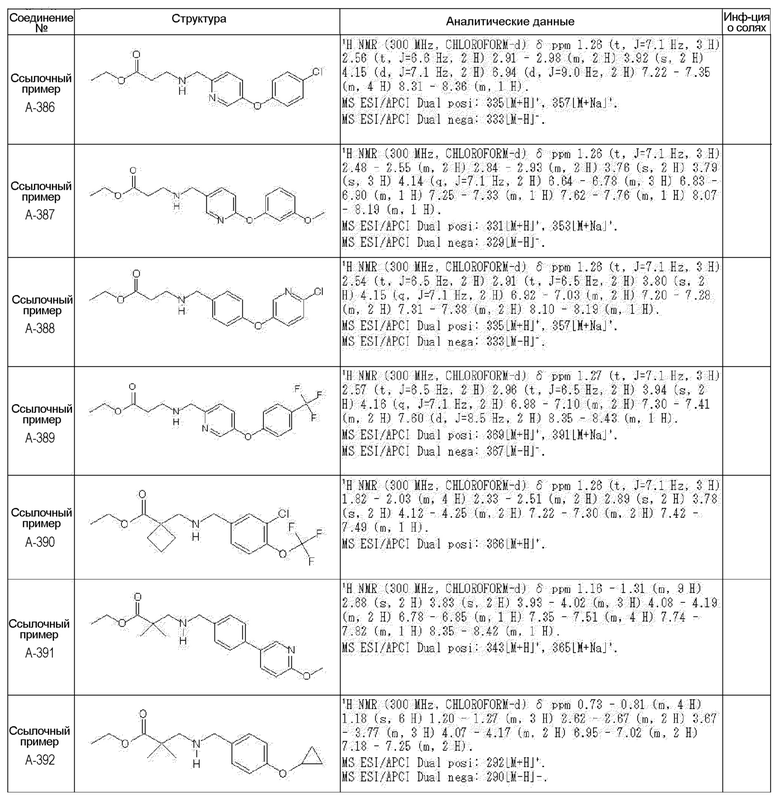
[0429]
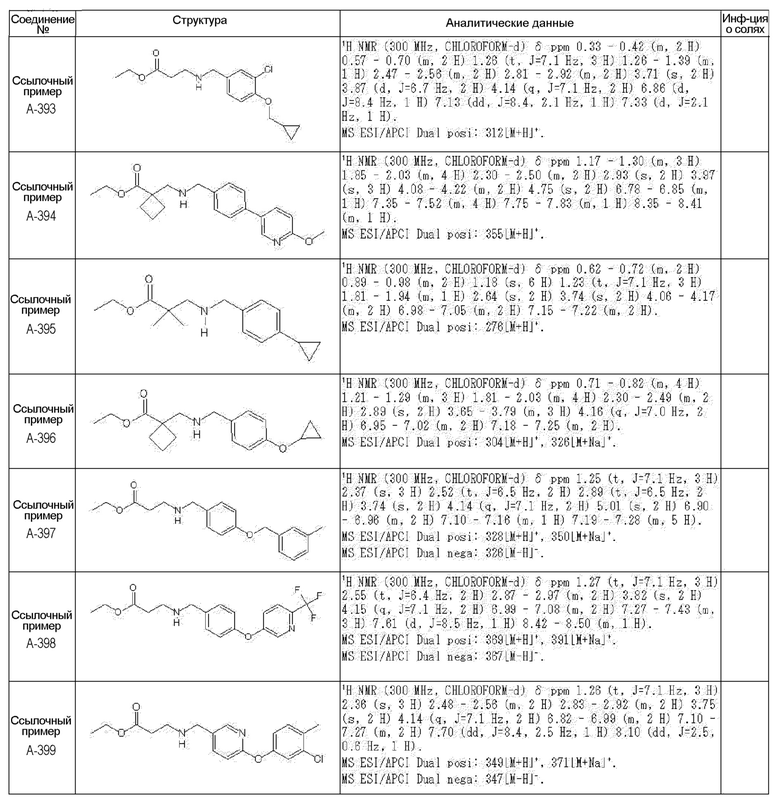
[0430]
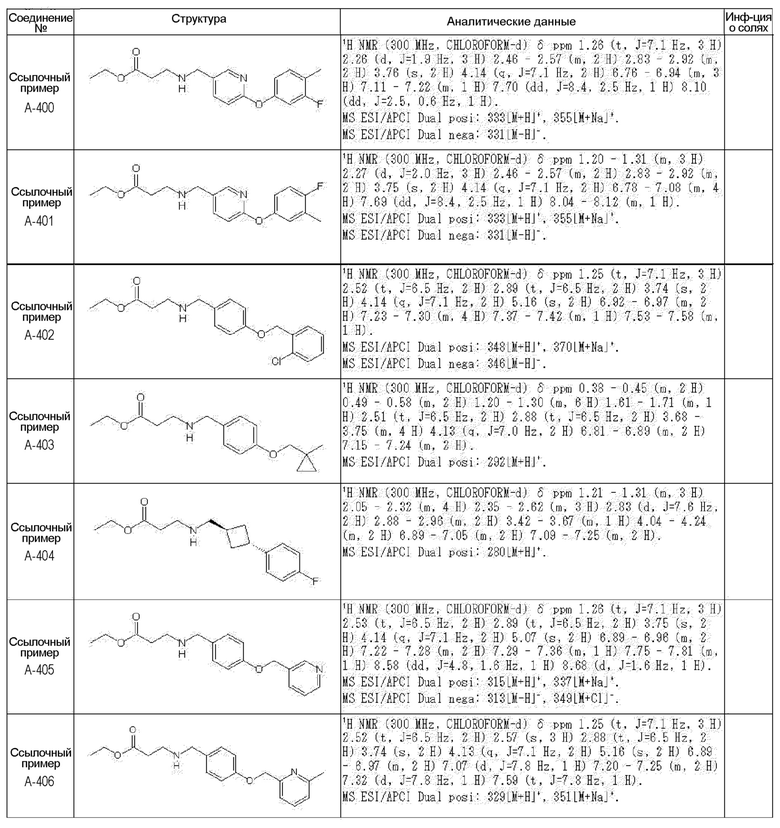
[0431]

[0413]
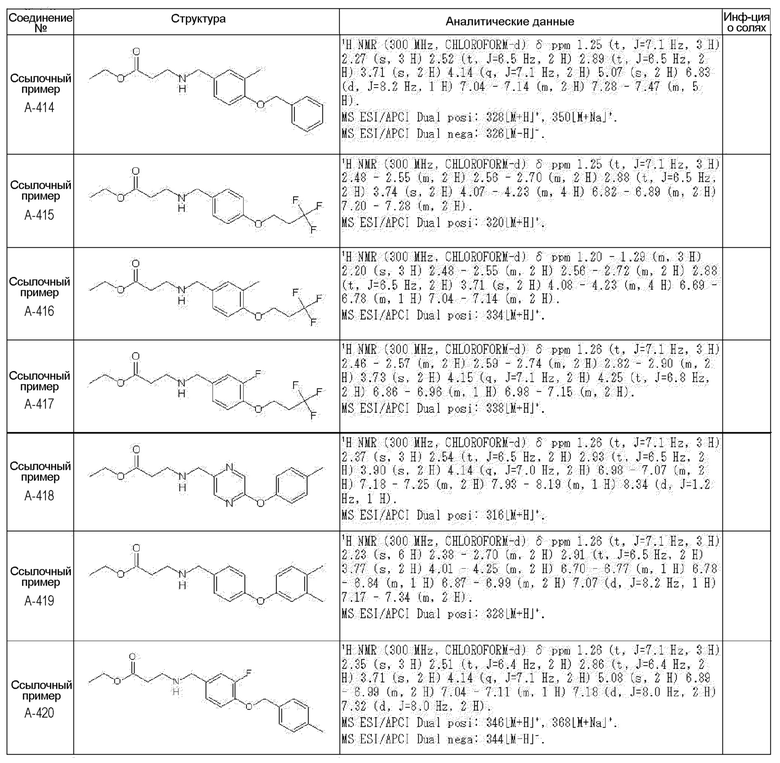
[0433]

[0434]
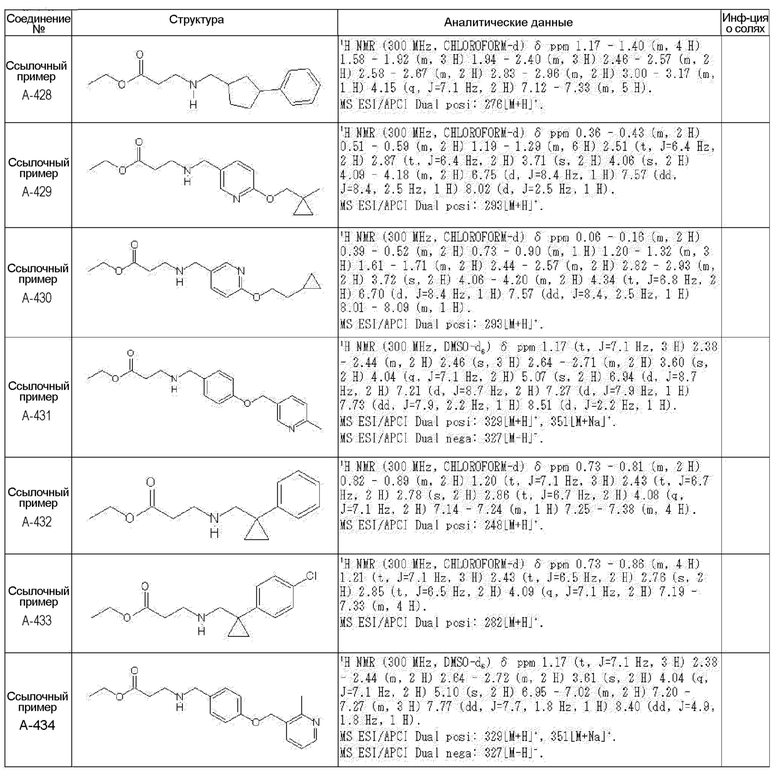
[0435]
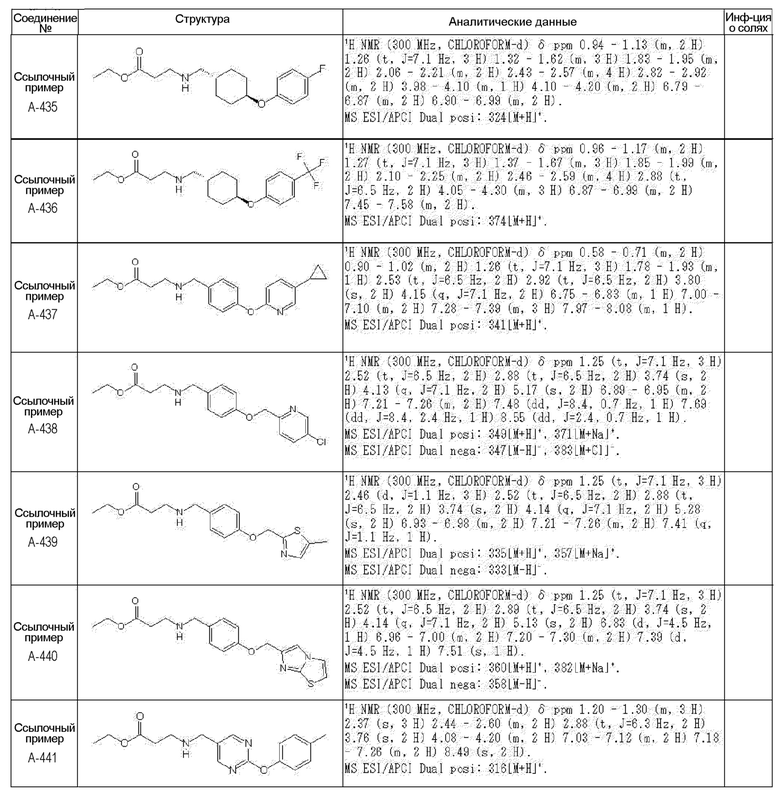
[0436]
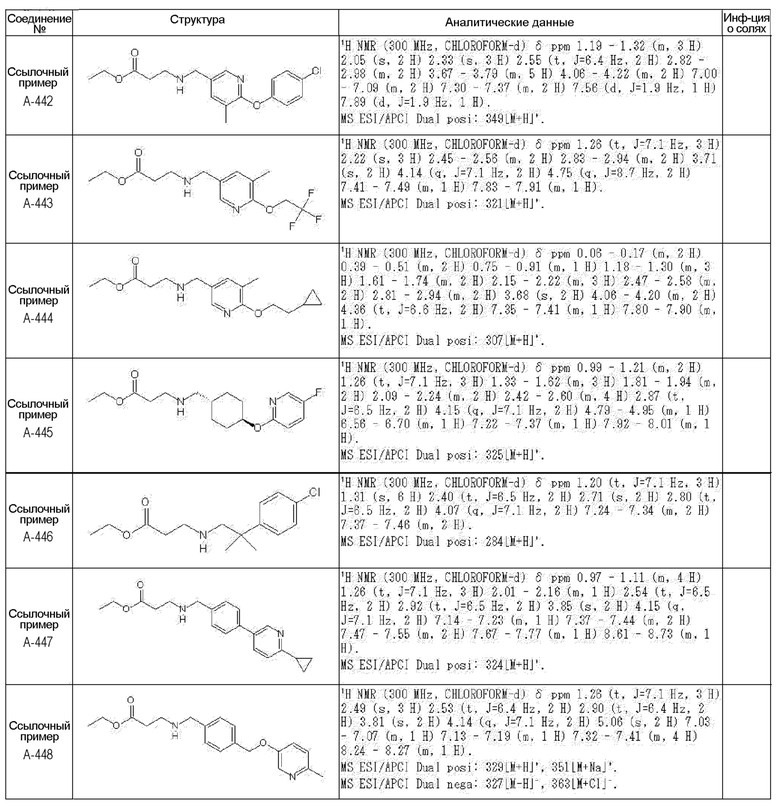
[0437]
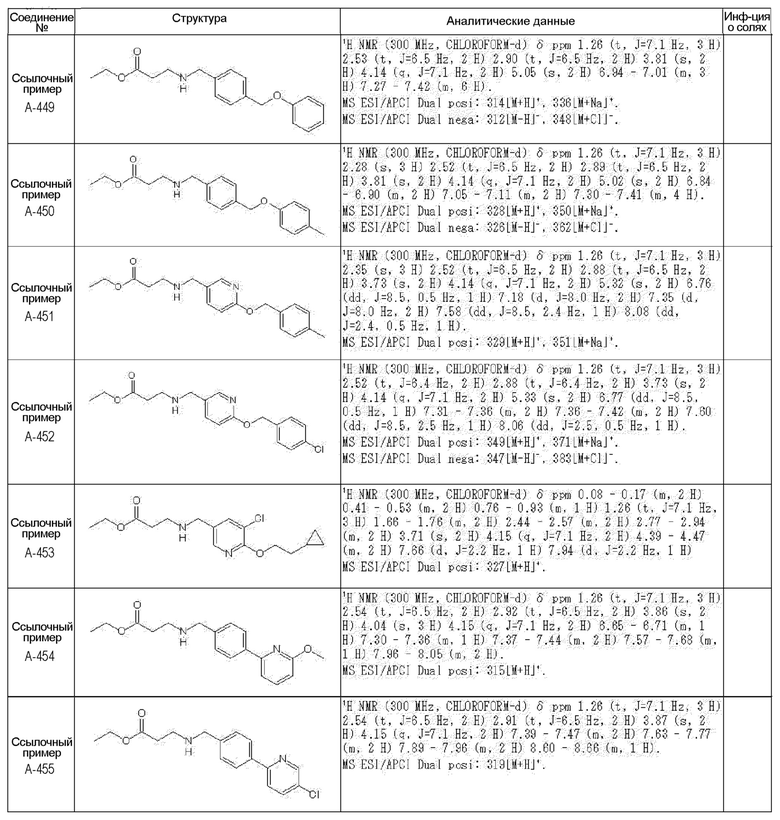
[0438]
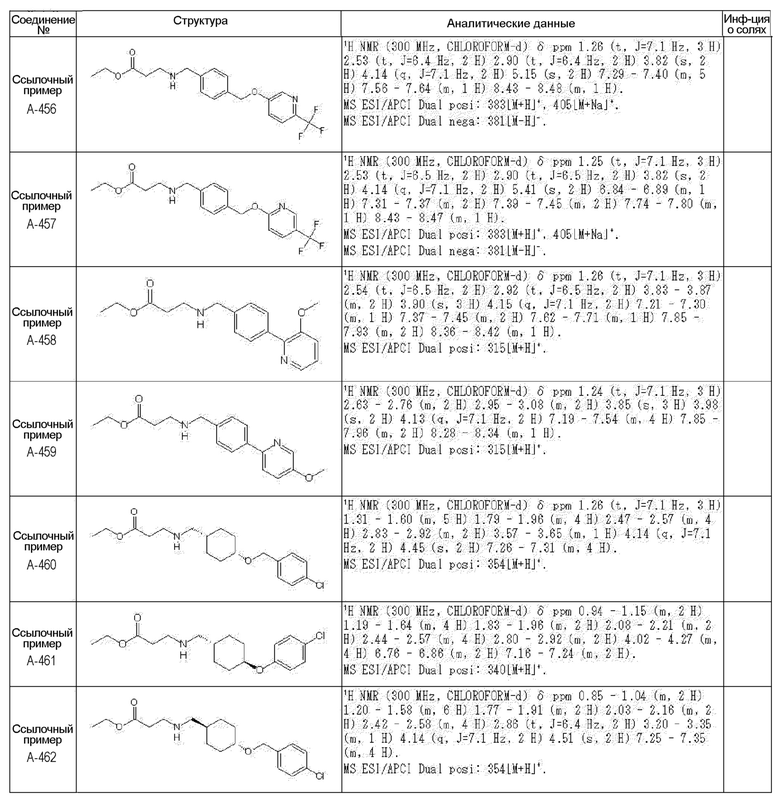
[0439]

[0440]

[0441]
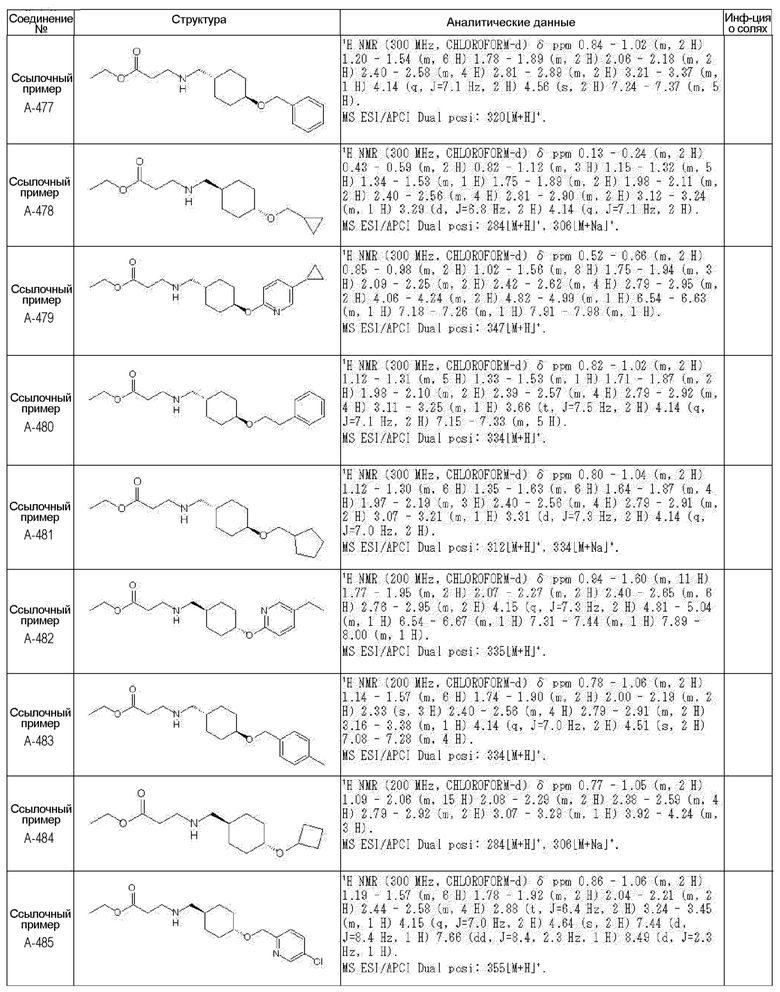
Ссылочный пример A-486
Этил N-[1-(4-хлорфенил)-2-пропанил]-β-аланинат
[0442]
В раствор гидрохлорида сложного β-аланинэтилового эфира (1,00 г) в этаноле (13,5 мл) последовательно добавляли триэтиламин (907 мкл), 4-хлорфенилацетон (1,32 г), уксусную кислоту (1,5 мл) и комплекс боран-2-пиколина (1,39 г) и смесь перемешивали при 60°C в течение 30 минут. После охлаждения до комнатной температуры, смесь концентрировали при пониженном давлении. В получаемый остаток добавляли насыщенный водный раствор бикарбоната натрия, и смесь экстрагировали хлороформом два раза. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол=100:0-95:5) для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (1,70 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,03 (д, J=6,2 Гц, 3H) 1,22 (т, J=7,1 Гц, 3H) 2,40-2,49 (м, 2H) 2,51-2,61 (м, 1H) 2,65-2,76 (м, 1H) 2,79-2,99 (м, 3H) 4,10 (кв, J=7,1 Гц, 2H) 7,06-7,15 (м, 2H) 7,21-7,29 (м, 2H).
MS ESI/APCI двойн. положительн.: 270[M+H]+.
Ссылочный пример B-1
Этил N-{2-[4-(трифторметил)фенил]пропан-2-ил}-β-аланинат
[0443]
В смесь соединения (1,11 г), полученного в ссылочном примере 27-3, метанола (6,00 мл) и воды (3,00 мл) добавляли этилакрилат (0,594 мл) и получаемую смесь перемешивали при 90°C в течение часа при облучении микроволнами. После охлаждения до комнатной температуры реакционную смесь выливали в воду и экстрагировали этилацетатом три раза. Объединенные органические слои промывали насыщенным солевым раствором и после этого пропускали через фазовый разделитель для концентрирования при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=95:5-10:90) для того, чтобы получать указанное в заголовке соединение в виде масла бледно-желтого цвета (957 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,26 (т, J=7,1 Гц, 3H) 1,47 (с, 6H) 2,39-2,49 (м, 2H) 2,53-2,61 (м, 2H) 4,14 (кв., J=7,1 Гц, 2H) 7,58 (с, 4H).
MS ESI/APCI двойн. положительн.: 304[M+H]+.
[0444] В следующих ссылочных примерах с B-2 до B-19 соединения, полученные в ссылочных примерах с 27-1 до 27-3, ссылочных примерах с 45-1 до 47-1, или соответствующие амины торговых сортов, а также соответствующие сложные эфиры акриловой кислоты или сложные эфиры кротоновой кислоты торговых сортов использовали в качестве исходных материалов и обработку проводили с помощью способа, описанного в ссылочном примере B-1, или его модификаций для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблицах 19-1 и 19-2.
[0445]
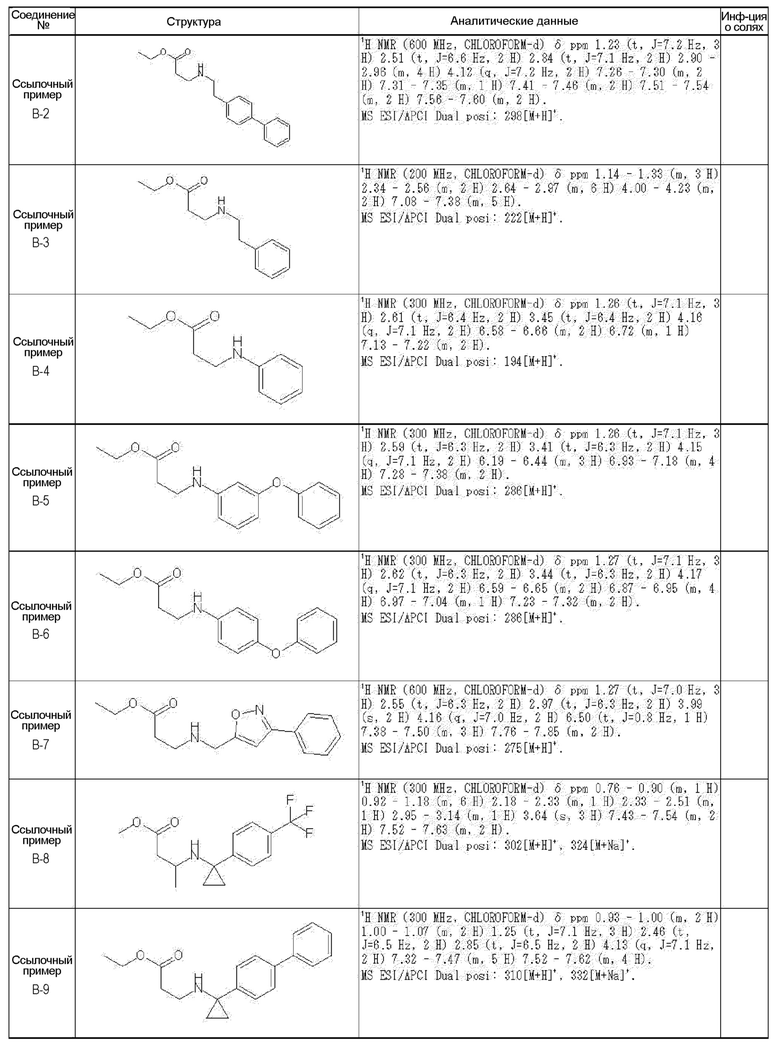
[0446]
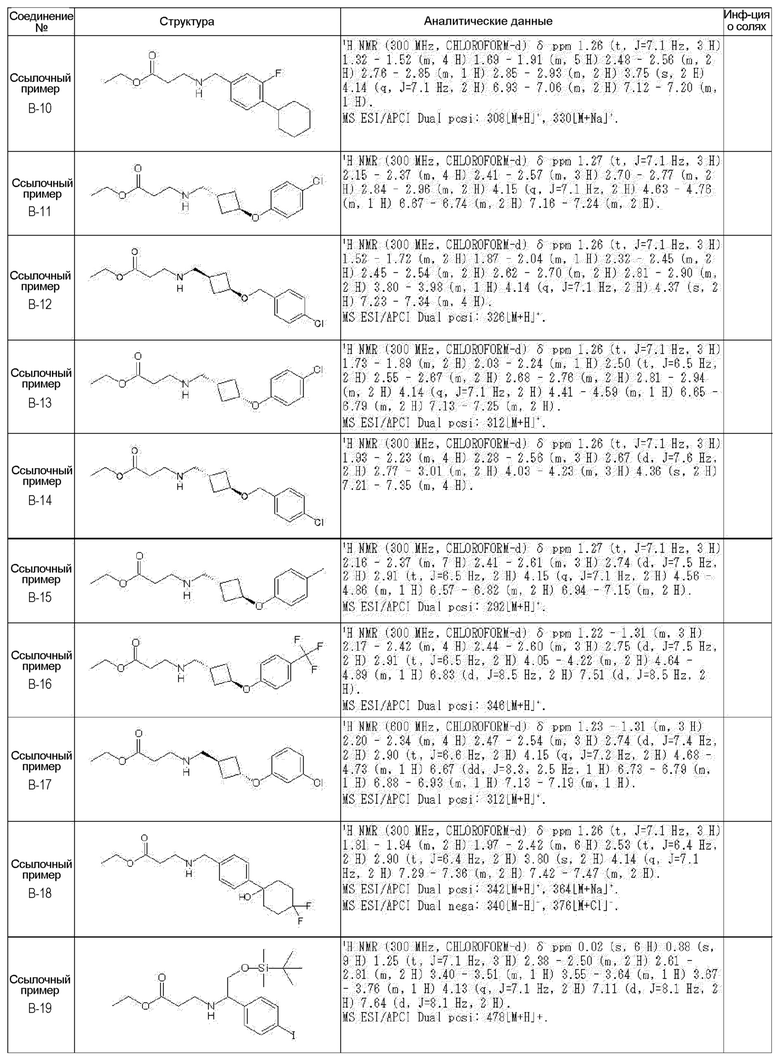
Ссылочный пример C-1
Этил N-(5-фенилпентил)-β-аланинат
[0447]

В раствор гидрохлорида β-аланин этила (2,00 г) в N,N-диметилформамиде (65,0 мл) добавляли гидрид натрия (60% дисперсия в минеральном масле, 1,15 г) и смесь перемешивали при комнатной температуре в течение часа. После добавления (5-бромпентил)бензола (2,52 мл) смесь перемешивали при 80°C в течение 4 часов. После охлаждения до комнатной температуры, реакционную смесь разводили этилацетатом и промывали насыщенным солевым раствором. Органический слой отделяли и сушили над безводным сульфатом магния; после удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 100:0-85:15) для того, чтобы получать указанное в заголовке соединение в виде масла бледно-желтого цвета (480 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,19-1,30 (м, 3H) 1,31-1,70 (м, 6H) 2,42-2,54 (м, 2H) 2,56-2,67 (м, 4H) 2,81-2,92 (м, 2H) 4,14 (кв., J=7,1 Гц, 2H) 7,10-7,22 (м, 3H) 7,23-7,32 (м, 2H).
MS ESI/APCI двойн. положительн.: 264[M+H]+.
Ссылочный пример D-1
Этил 3-({[3-(4-хлорфенил)изоксазол-5-ил]метил}амино)-3-метилбутаноат
[0448]

(1) Синтез 1-(4-хлорфенил)-N-гидроксиметанимина
[0449]
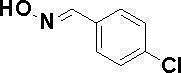
В раствор 4-хлорбензальдегида (10,0 г) в хлороформе (350 мл) добавляли гидрохлорид гидроксиламина (10,2 г) и смесь перемешивали при комнатной температуре в течение 18 часов в атмосфере аргона. После добавления соляной кислоты 2 моль/л (200 мл), проводили три экстрагирования хлороформом. Объединенные органические слои промывали насыщенным солевым раствором и после этого сушили над безводным сульфатом магния. После удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. Получаемый остаток кристаллизовали с использованием жидкой смеси н-гексана и хлороформа для того, чтобы получать 1-(4-хлорфенил)-N-гидроксиметанимин в виде бесцветного твердого вещества (9,27 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 7,34-7,39 (м, 2H) 7,48-7,54 (м, 2H) 8,10 (с, 1H).
MS ESI/APCI двойн. положительн.: 156[M+H]+.
MS ESI/APCI двойн. отрицательн.: 154[M-H]-.
(2) Синтез 5-(бромметил)-3-(4-хлорфенил)изоксазола
[0450]
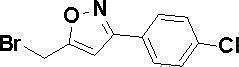
В раствор в хлороформе (28,5 мл) соединения (1,85 г), полученного на приведенной выше стадии (1), добавляли пропаргил бромид (1,07 мл) и триэтиламин (1,99 мл) и затем по каплям добавляли 5% гипохлорит натрия в водном растворе (57,0 мл) при 0°C в течение периода в 30 минут. Реакционную смесь доводили до комнатной температуры и перемешивали в течение 5 часов. После отделения водного слоя, проводили два экстрагирования хлороформом. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. После удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 99:1-85:15) и восстанавливали до порошка с использованием н-гексана, таким образом получая 5-(бромметил)-3-(4-хлорфенил)изоксазол в виде бесцветного твердого вещества (1,01 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 4,51 (с, 2H) 6,58-6,64 (м, 1H) 7,38-7,52 (м, 2H) 7,68-7,79 (м, 2H).
MS ESI/APCI двойн. положительн.: 271[M+H]+.
(3) Синтез указанного в заголовке соединения
В раствор гидрохлорида этил 3-амино-3-метилбутирата (333 мг) в тетрагидрофуране (4,00 мл) добавляли раствор в тетрагидрофуране (2,00 мл) соединения (100 мг), полученного на приведенной выше стадии (2), и карбонат калия (406 мг) и смесь перемешивали при 60°C в течение трех суток. После пропускания реакционной смеси через Celite (зарегистрированный товарный знак), фильтрат концентрировали при пониженном давлении и получаемый остаток грубо очищали с помощью препаративной ВЭЖХ. В получаемый грубо очищенный продукт добавляли насыщенный водный раствор бикарбоната натрия и проводили экстрагирование хлороформом. Объединенные органические слои сушили над безводным сульфатом магния и после удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 98:2-50:50) для того, чтобы получать указанное в заголовке соединение в виде бесцветной аморфной массы.
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,20-1,32 (м, 9H) 2,48 (с, 2H) 3,92 (д, J=0,9 Гц, 2H) 4,15 (кв., J=7,1 Гц, 2H) 6,48 (т, J=0,9 Гц, 1H) 7,37-7,45 (м, 2H) 7,69-7,77 (м, 2H).
MS ESI положительн.: 337[M+H]+.
MS ESI отрицательн.: 335[M-H]-.
Ссылочный пример E-1
2-(триметилсилил)этил глицинат
[0451]
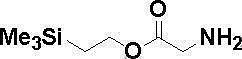
(1) Синтез 2-(триметилсилил)этил N-[(бензилокси)карбонил]глицината
[0452]

В смесь N-[(бензилокси)карбонил]глицина (5,33 г), гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (5,86 г), моногидрата 1-гидроксибензотриазола (4,68 г) и хлороформ (51,0 мл) добавляли 2-(триметилсилил)этанол (4,36 мл) и смесь перемешивали при комнатной температуре в течение 8 часов. В реакционную смесь добавляли 2-(триметилсилил)этанол (3,00 мл), гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимидf (2,93 г) и 4-диметиламинопиридин (312 мг) и смесь перемешивали при комнатной температуре в течение 65 часов. После выливания реакционной смеси в насыщенный водный раствор хлорида аммония, проводили три экстрагирования этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. После удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 95:5-80:20) для того, чтобы получать 2-(триметилсилил)этил N-[(бензилокси)карбонил]глицинат в виде бесцветного масла (6,64 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,04 (с, 9H) 0,96-1,06 (м, 2H) 3,96 (д, J=5,6 Гц, 2H) 4,20-4,30 (м, 2H) 5,13 (с, 2H) 5,19-5,28 (м, 1H) 7,28-7,41 (м, 5H).
(2) Синтез указанного в заголовке соединения
В раствор в этилацетате (20,0 мл) соединения (634 мг), полученного на приведенной выше стадии (1), добавляли 20% гидроксид палладия/углерод (63,0 мг). Смесь перемешивали при комнатной температуре в течение часа в атмосфере водорода. После пропускания реакционной смеси через Celite (зарегистрированный товарный знак), фильтрат концентрировали при пониженном давлении для того, чтобы получать указанное в заголовке соединение в виде масла бледно-желтого цвета (320 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,05 (с, 9H) 0,89-1,07 (м, 2H) 3,40 (с, 2H) 4,13-4,29 (м, 2H).
Ссылочный пример F-1
Этил н-[4-(1H-бензотриазол-1-илметокси)бензил]-β-аланинат
[0453]
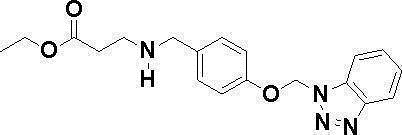
В смесь 1H-бензотриазол-1-метанола (1,22 г), 4-гидроксибензальдегида (1,00 г), трифенилфосфина (2,26 г) и хлороформа (27 мл), добавляли диизопропил азодикарбоксилат (1,9 моль/л, раствор в толуоле, 4,53 мл) при охлаждении льдом. После доведения до комнатной температуры, смесь перемешивали в течение 2,5 часа. После добавления трифенилфосфина (1,13 г) и диизопропил азодикарбоксилата (1,9 моль/л, раствор в толуоле, 2,27 мл), смесь перемешивали в течение дополнительных 40 минут. В реакционную смесь добавляли метанол (165 мкл) и уксусную кислоту (750 мкл) и получаемую смесь перемешивали при той же температуре в течение 20 минут. В реакционную смесь добавляли β-аланин этил гидрохлорид (1,38 г), триэтиламин (1,26 мкл) и триацетоксиборогидрид натрия (2,60 г) и смесь перемешивали при комнатной температуре в течение 1,5 часа. После добавления соляной кислоты 1 моль/л, смесь промывали диэтиловым эфиром. В водный слой добавляли водный раствор гидроксида натрия 2 моль/л для того, чтобы обеспечивать основный pH. После экстрагирования хлороформом, объединенные органические слои промывали насыщенным солевым раствором и после этого пропускали через фазовый разделитель для концентрирования при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-50:50) для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (1,13 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,24 (т, J=7,1 Гц, 3H) 2,47-2,52 (м, 2H) 2,81-2,87 (м, 2H) 3,71 (с, 2H) 4,12 (кв., J=7,1 Гц, 2H) 6,54 (с, 2H) 6,99-7,06 (м, 2H) 7,19-7,25 (м, 2H) 7,40 (ддд, J=8,3, 7,0, 1,0 Гц, 1H) 7,53 (ддд, J=8,3, 7,0, 1,0 Гц, 1H) 7,70 (дт, J=8,3, 1,0 Гц, 1H) 8,07 (дт, J=8,3, 1,0 Гц, 1H).
MS ESI/APCI двойн. положительн.: 355[M+H]+, 377[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 353[M-H]-, 389[M+Cl]-.
[0454] В следующих ссылочных примерах F-2 и F-3 соответствующие спирты торговых сортов использовали в качестве исходного материала и проводили обработку с помощью способа, описанного в ссылочном примере F-1, или его модификации для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблице 20-1.
[0455]
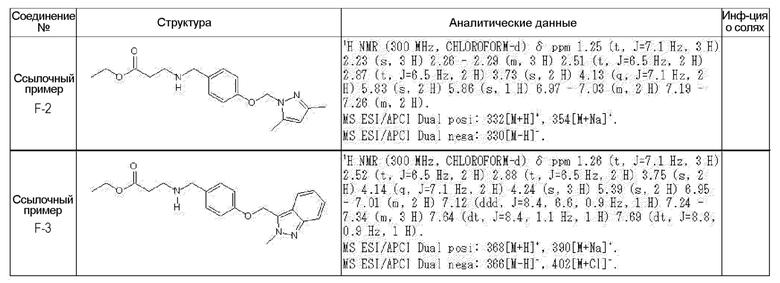
Ссылочный пример G-1
3-({2-[(2-метил-2-пропанил)окси]-2-оксоэтил}амино)-3-оксопропановая кислота
[0456]

(1) Синтез бензил 3-({2-[(2-метил-2-пропанил)окси]-2-оксоэтил}амино)-3-оксопропанoата
[0457]

В раствор монобензилмалоната (2,00 г) в N,N-диметилформамиде (51,5 мл), добавляли глицин трет-бутил гидрохлорид (2,07 г), триэтиламин (3,13 г) и ангидрид пропилфосфоновой кислоты (циклический тример) (48%, раствор в N,N-диметилформамиде, 8,22 г) и смесь перемешивали при комнатной температуре в течение часа. В реакционную смесь добавляли воду и проводили экстрагирование этилацетатом. Объединенные органические слои сушили над безводным сульфатом натрия. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 67:33-50:50) для того, чтобы получать бензил 3-({2-[(2-метил-2-пропанил)окси]-2-оксоэтил}амино)-3-оксопропанoат в виде бесцветного масла (1,46 г).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,48 (с, 9H) 3,40 (с, 2H) 3,97 (д, J=5,0 Гц, 2 Н) 5,19 (с, 2Н) 7,31-7,40 (м, 5Н) 7,47-7,57 (м, 1Н).
MS ESI положительн.: 330[M+Na]+.
MS ESI отрицательн.: 306[M-H]-.
(2) Синтез указанного в заголовке соединения
Использовали соединение (1,46 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в ссылочном примере E-1(2), для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (1,03 г).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,48 (с, 9H) 3,40 (с, 2H) 3,99 (д, J=5,0 Гц, 2H) 7,28-7,41 (м, 1H).
MS ESI положительн.: 240[M+Na]+.
MS ESI отрицательн.: 216[M-H]-.
Ссылочный пример G-2
3-({3-[(2-метил-2-пропанил)окси]-3-оксопропил}амино)-3-оксопропановая кислота
[0458]

Вместо глицин трет-бутил гидрохлорида использовали β-аланин трет-бутил гидрохлорид (1,82 г) и обработку проводили с помощью того же способа, как в ссылочном примере G-1, для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (1,03 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,46 (с, 9H) 2,43-2,54 (м, 2H) 3,32 (с, 2H) 3,55 (кв., J=6,1 Гц, 2H) 7,02-7,18 (м, 1H).
MS ESI/APCI двойн. положительн.: 254[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 230[M-H]-.
Ссылочный пример G-3
3-[(2-этокси-2-оксоэтил)амино]-3-оксопропановая кислота
[0459]

В раствор кислоты Мельдрума (10,0 г) в ацетонитриле (231 мл) добавляли глицин этил гидрохлорид (14,5 г) и триэтиламин (14,1 г) и смесь перемешивали при 60°C в течение 5 часов. После охлаждения смеси до комнатной температуры добавляли этилацетат и проводили экстрагирование насыщенным водным раствором бикарбоната натрия. В объединенные водные слои добавляли соляную кислоту 1 моль/л и проводили экстрагирование этилацетатом. Объединенные органические слои сушили над безводным сульфатом натрия. После удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (7,95 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,30 (т, J=7,2 Гц, 3H) 3,44 (с, 2H) 4,10 (д, J=5,3 Гц, 2H) 4,25 (кв., 3=72 Гц, 2H).
MS ESI/APCI двойн. положительн.: 190[M+H]+.
MS ESI/APCI двойн. отрицательн.: 188[M-H]-.
Пример 1-1
N-[(4-гидрокси-2-оксо-1-{[4'-(трифторметил)бифенил-4-ил]метил}-9-окса-1-азаспиро[5.5]ундец-3-ен-3-ил)карбонил]глицин
[0460]

(1) Синтез этил 3-([4-(2-этокси-2-оксоэтил)тетрагидро-2H-пиран-4-ил]{[4'-(трифторметил)бифенил-4-ил]метил}амино)-3-оксопропанoата
[0461]
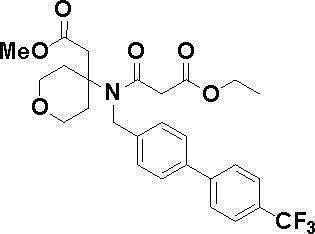
В раствор в этилацетате (11,6 мл) соединения (770 мг), полученного в ссылочном примере A-1, и триэтиламина (287 мг) добавляли этилмалонил хлорид (341 мг) при 0°C и смесь перемешивали при комнатной температуре в течение 30 минут. Дополнительный триэтиламин (95,7 мг) добавляли и после добавления этилмалонил хлорида (114 мг) при 0°C, смесь перемешивали при комнатной температуре в течение 30 минут. После добавления соляной кислоты 1 моль/л проводили два экстрагирования этилацетатом. Объединенные органические слои пропускали через фазовый разделитель и после этого концентрировали при пониженном давлении для того, чтобы получать смесь (1,44 г), которая содержит этил 3-([4-(2-этокси-2-оксоэтил)тетрагидро-2H-пиран-4-ил]{[4'-(трифторметил)бифенил-4-ил]метил}амино)-3-оксопропанoат.
MS ESI/APCI двойн. положительн.: 522[M+H]+, 544[M+Na]+.
(2) Синтез 3-(этоксикарбонил)-2-оксо-1-{[4'-(трифторметил)бифенил-4-ил}метил}-9-окса-1-азаспиро[5.5]ундец-3-ен-4-олата натрия
[0462]

В раствор в этаноле (24,7 мл) смеси (1,43 г), полученной на приведенной выше стадии (1), добавляли этоксид натрия (приблизительно 20%, раствор в этаноле, 1,30 мл) и получаемую смесь перемешивали при внешней температуре 90°C в течение 4 часов. После охлаждения реакционной смеси до комнатной температуры, осадок собирали с помощью фильтрования для того, чтобы получать 3-(этоксикарбонил)-2-оксо-1-{[4'-(трифторметил)бифенил-4-ил]метил}-9-окса-1-азаспиро[5.5]ундец-3-ен-4-олат натрия в виде твердого вещества коричневого цвета (534 мг).
1H NMR (300 МГц, ДМСО-d6) δ м.д. 1,14 (т, J=7,1 Гц, 3H) 1,40-1,55 (м, 2H) 1,74-1,92 (м, 2H) 2,42 (с, 2H) 3,37-3,46 (м, 2H) 3,61-3,71 (м, 2H) 3,94 (кв., J=6,9 Гц, 2H) 4,70 (ушир.с, 2H) 7,40 (д, J=7,9 Гц, 2H) 7,65 (д, J=8,1 Гц, 2H) 7,75-7,82 (м, 2H) 7,84-7,92 (м, 2H).
MS ESI/APCI двойн. положительн.: 512[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 488[M-H]-.
(3) Синтез трет-бутил N-[(4-гидрокси-2-оксо-1-{[4'-(трифторметил)бифенил-4-ил]метил}-9-окса-1-азаспиро[5.5]ундец-3-ен-3-ил)карбонил]глицината
[0463]
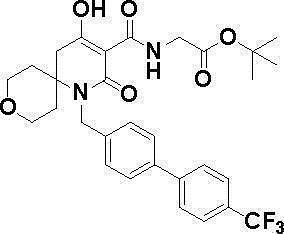
В раствор в 1,2-диметоксиэтане (10,2 мл) соединения (508 мг), полученного на приведенной выше стадии (2), добавляли триэтиламин (100 мг) и глицин трет-бутил гидрохлорид (200 мг) и смесь перемешивали при внешней температуре 90°C в течение двух часов. После охлаждения до комнатной температуры, реакционную смесь концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=85:15-20:80) для того, чтобы получать трет-бутил N-[(4-гидрокси-2-оксо-1-{[4'-(трифторметил)бифенил-4-ил]метил}-9-окса-1-азаспиро[5.5]ундец-3-ен-3-ил)карбонил]глицинат в виде бесцветной аморфной массы (329 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,47-1,53 (м, 9H) 1,59-1,68 (м, 2H) 1,99-2,16 (м, 2H) 2,83-2,98 (м, 2H) 3,44-3,60 (м, 2H) 3,79-3,93 (м, 2H) 3,99-4,08 (м, 2H) 4,84 (ушир.с, 2H) 7,31-7,41 (м, 2H) 7,50-7,59 (м, 2H) 7,62-7,72 (м, 4H) 10,12-10,45 (м, 1H).
MS ESI/APCI двойн. положительн.: 575[M+H]+.
MS ESI/APCI двойн. отрицательн.: 573[M-H]-.
(4) Синтез указанного в заголовке соединения
К соединению (319 мг), полученном на приведенной выше стадии (3), добавляли раствор (6,4 мл) хлороводорода в 1,4-диоксане 4 моль/л и смесь перемешивали при комнатной температуре в течение 18 часов. После концентрирования при пониженном давлении, этилацетат (5,00 мл) добавляли к остатку и при постоянном перемешивании добавляли н-гексан (5,00 мл). После перемешивания реакционной смеси при комнатной температуре в течение 30 минут, осадок собирали с помощью фильтрования для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (245 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 1,46-1,61 (м, 2H) 1,89-2,10 (м, 2H) 2,89-3,14 (м, 2H) 3,41-3,56 (м, 2H) 3,65-3,78 (м, 2H) 3,96-4,16 (м, 2H) 4,74-4,93 (м, 2H) 7,40-7,51 (м, 2H) 7,63-7,74 (м, 2H) 7,75-7,93 (м, 4H) 9,93-10,27 (м, 1H) 12,76-12,95 (м, 1H).
MS ESI/APCI двойн. положительн.: 519[M+H]+, 541[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 517[M-H]-.
Пример 1-2
N-{[5-(бифенил-4-илметил)-8-гидрокси-6-оксо-2-окса-5-азаспиро[3.5]нон-7-ен-7-ил]карбонил}глицин
[0464]
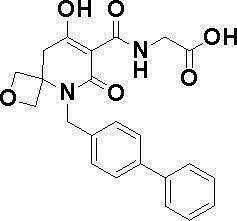
(1) Синтез трет-бутил N-{[5-(бифенил-4-илметил)-8-гидрокси-6-оксо-2-окса-5-азаспиро[3.5]нон-7-ен-7-ил]карбонил}глицинат
[0465]

Вместо соединения, полученного в ссылочном примере A-1, использовали соединение (1,90 г), полученное в ссылочном примере A-2, и обработку проводили с помощью тех же способов, как в примере с 1-1(1) до (3), для того, чтобы получать трет-бутил N-{[5-(бифенил-4-илметил)-8-гидрокси-6-оксо-2-окса-5-азаспиро[3.5]нон-7-ен-7-ил]карбонил}глицинат в виде твердого вещества бледно-коричневого цвета (2,67 г).
1H ЯМР (200 МГц, ХЛОРОФОРМ-d) δ м.д. 1,43-1,52 (м, 9H) 3,01-3,23 (м, 2H) 3,94-4,11 (м, 2H) 4,37-4,62 (м, 2H) 4,74-4,93 (м, 2H) 5,06-5,21 (м, 2H) 7,24-7,50 (м, 5H) 7,51-7,62 (м, 4H) 9,95-10,57 (м, 1H).
MS ESI/APCI двойн. положительн.: 501[M+Na]+.
(2) Синтез указанного в заголовке соединения
В раствор в хлороформе (20,0 мл) соединения (2,60 г), полученного на приведенной выше стадии (1), добавляли трифторуксусную кислоту (8,00 мл) и смесь перемешивали при комнатной температуре в течение 12 часов. После концентрирования при пониженном давлении, в остаток добавляли этилацетат. При продолжающемся перемешивании добавляли н-гексан и осадок собирали с помощью фильтрования для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-коричневого цвета (2,18 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 3,08-3,28 (м, 2H) 3,93-4,12 (м, 2H) 4,45 (д, J=7,1 Гц, 2H) 4,74 (д, J=7,1 Гц, 2H) 5,01-5,18 (м, 2H) 7,26-7,52 (м, 5H) 7,57-7,71 (м, 4H) 9,80-10,34 (м, 1H) 12,89 (ушир. с, 1H).
MS ESI/APCI двойн. положительн.: 423[M+H]+.
Пример 1-3
N-({4-гидрокси-5-метил-2-оксо-1-[4-(трифторметокси)бензил]-1,2,5,6-тетрагидропиридин-3-ил}карбонил)глициновая соль натрия
[0466]

(1) Синтез трет-бутил N-({4-гидрокси-5-метил-2-оксо-1-[4-(трифторметокси)бензил]-1,2,5,6-тетрагидропиридин-3-ил}карбонил)глицината
[0467]
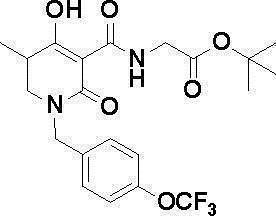
Вместо соединения, полученного в ссылочном примере A-1, использовали соединение (1,12 г), полученное в ссылочном примере A-3, и обработку проводили с помощью тех же способов, как в примере с 1-1(1) до (3), для того, чтобы получать трет-бутил N-({4-гидрокси-5-метил-2-оксо-1-[4-(трифторметокси)бензил]-1,2,5,6-тетрагидропиридин-3-ил}карбонил)глицинат в виде бесцветного твердого вещества (704 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,04-1,16 (м, 3H) 1,42-1,56 (м, 9H) 2,61-2,76 (м, 1H) 2,93-3,07 (м, 1H) 3,26-3,38 (м, 1H) 3,97-4,04 (м, 2H) 4,50-4,68 (м, 2H) 7,11-7,33 (м, 4H) 10,13-10,57 (м, 1H).
MS ESI/APCI двойн. положительн.: 481[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 457[M-H]-.
(2) Синтез N-({4-гидрокси-5-метил-2-оксо-1-[4-(трифторметокси)бензил]-1,2,5,6-тетрагидропиридин-3-ил}карбонил)глицина
[0468]

В соединение (1,12 г), полученное на приведенной выше стадии (1), добавляли раствор (10,0 мл) гидрохлорида в 1,4-диоксане 4 моль/л и смесь перемешивали при комнатной температуре в течение 16 часов. После концентрирования при пониженном давлении, реакционную смесь очищали с помощью препаративной ВЭЖХ для того, чтобы получать N-({4-гидрокси-5-метил-2-оксо-1-[4-(трифторметокси)бензил]-1,2,5,6-тетрагидропиридин-3-ил}карбонил)глицин в виде бесцветной аморфной массы (482 мг).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,07-1,19 (м, 3H) 2,53-2,78 (м, 1H) 2,98-3,10 (м, 1H) 3,33-3,41 (м, 1H) 4,15-4,23 (м, 2H) 4,56-4,70 (м, 2H) 7,15-7,23 (м, 2H) 7,27-7,34 (м, 2H) 10,13-10,50 (м, 1H).
MS ESI/APCI двойн. положительн.: 403[M+H]+, 425[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 401 [M-H]-.
(3) Синтез указанного в заголовке соединения
В раствор в метаноле (3,00 мл) соединения (321 мг), полученного на приведенной выше стадии (2), добавляли гидроксид натрия в водном растворе 1 моль/л (0,798 мл) и смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении. В получаемый остаток добавляли изопропиловый спирт и после этого смесь перемешивали в течение ночи при комнатной температуре. Осадок собирали с помощью фильтрования для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (205 мг).
1H ЯМР (300 МГц, METHANOL-d4) δ м.д. 1,10 (д, J=7,0 Гц, 3H) 2,56-2,73 (м, 1H) 3,06 (дд, J=12,6, 7,9 Гц, 1H) 3,44 (дд, J=12,6, 5,7 Гц, 1H) 3,89 (с, 2H) 4,58 (д, J=14,9 Гц, 1H) 4,70 (д, J=14,9 Гц, 1H) 7,16-7,30 (м, 2H) 7,36-7,47 (м, 2H).
MS ESI/APCI двойн. положительн.: 425[M+Na]+.
[0469]
Пример 1-4
N-[(1-{[6-(4-хлорфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицинат натрия
[0470]
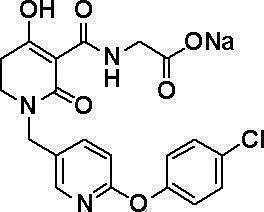
(1) Синтез 2-метил-2-пропанил N-[(1-{[6-(4-хлорфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицината
[0471]
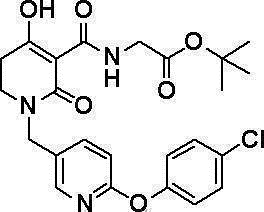
Вместо соединения, полученного в ссылочном примере A-1, использовали соединение (1,15 г), полученное в ссылочном примере A-246, и обработку проводили с помощью тех же способов, как в примере с 1-1(1) до (3), для того, чтобы получать 2-метил-2-пропанил N-[(1-{[6-(4-хлорфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицинат в виде смолы бледно-желтого цвета (1,02 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,49 (с, 9H) 2,50-2,67 (м, 2H) 3,27-3,38 (м, 2H) 3,95-4,07 (м, 2H) 4,56 (с, 2H) 6,88-6,94 (м, 1H) 7,04-7,14 (м, 2H) 7,31-7,41 (м, 2H) 7,63-7,74 (м, 1H) 8,04-8,09 (м, 1H) 10,07-10,51 (м, 1H).
MS ESI/APCI двойн. положительн.: 510[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 486[M-H]-.
(2) Синтез N-[(1-{[6-(4-хлорфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицина
В соединение (1,02 г), полученное на приведенной выше стадии (1), добавляли раствор (10,0 мл) хлороводорода в 1,4-диоксане 4 моль/л и смесь перемешивали в течение ночи при комнатной температуре. Получаемый осадок собирали с помощью фильтрования и получаемое твердое вещество нагревали при добавлении этилацетата. После добавления ацетонитрила, смесь охлаждали до комнатной температуры и перемешивали при этой температуре. Получаемый осадок собирали с помощью фильтрования для того, чтобы получать бесцветное твердое вещество (645 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,50-2,74 (м, 2H) 3,37-3,49 (м, 2H) 3,96-4,08 (м, 2H) 4,48-4,62 (м, 2H) 7,05 (д, J=8,4 Гц, 1H) 7,14-7,20 (м, 2H) 7,42-7,49 (м, 2H) 7,79 (дд, J=8,4, 2,5 Гц, 1H) 8,07-8,14 (м, 1H) 9,94-10,26 (м, 1H) 12,87 (ушир.с, 1H).
MS ESI/APCI двойн. положительн.: 432[M+H]+.
MS ESI/APCI двойн. отрицательн.: 430[M-H]-.
(3) Кристаллизация N-[(1-{[6-(4-хлорфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицина
В соединение (30 мг), полученное на приведенной выше стадии (2), добавляли жидкую смесь воды и этанола (5:4) и получаемую смесь нагревали на горячей водяной бане при 80°C до превращения в раствор, который после этого оставляли стоять в течение ночи при комнатной температуре. Растворитель отгоняли под потоком азота для того, чтобы получать бесцветное твердое вещество (30 мг). темп. плавления: 191°С
(4) Синтез указанного в заголовке соединения
В раствор в ацетоне соединения (645 мг), полученного на приведенной выше стадии (2), добавляли гидроксид натрия в водном растворе 1 моль/л (1,50 мл) и смесь перемешивали при комнатной температуре в течение 30 минут. Получаемый осадок собирали с помощью фильтрования для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (543 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,53-2,59 (м, 2H) 3,34 (т, J=7,1 Гц, 2H) 3,48-3,60 (м, 2H) 4,52 (с, 2H) 7,04 (д, J=8,4 Гц, 1H) 7,12-7,21 (м, 2H) 7,36-7,50 (м, 2H) 7,79 (дд, J=8,4, 2,5 Гц, 1H) 8,09 (д, J=2,5 Гц, 1H) 10,08 (ушир.с, 1H).
MS ESI положительн.: 432[M+H]+.
MS ESI отрицательн.: 430[M-H]-.
[0472] В следующих примерах с 1-5 до 1-464 в качестве исходных материалов использовали соединения, полученные в ссылочных примерах с A-8 до A-245, с A-247 до A-300, с A-343 до A-486, ссылочных примерах с B-1 до B-17, ссылочном примере C-1, ссылочном примере D-1, ссылочных примерах с F-1 до F-3, или соответствующие амины торговых сортов и обработку проводили с помощью способов, описанных в примерах с 1-1 до 1-4, или их модификаций для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблицах с 21-1 до 21-67.
[0473]

[0474]
[0475]

[0476]
[0477]
[0478]

[0479]

[0480]
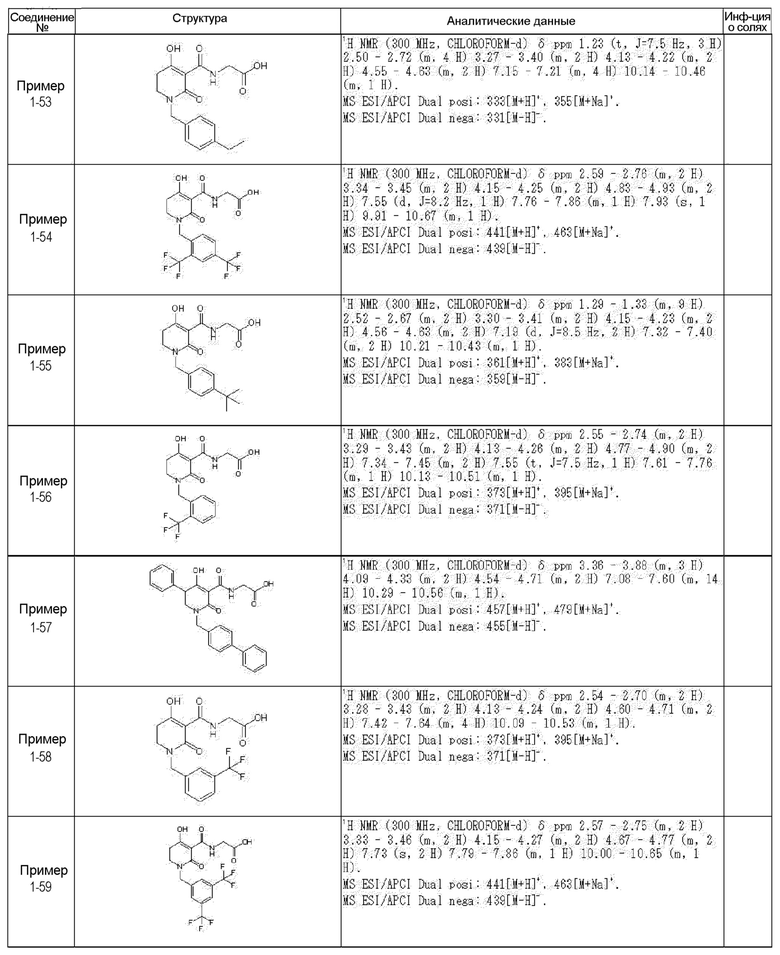
[0481]

[0482]

[0483]
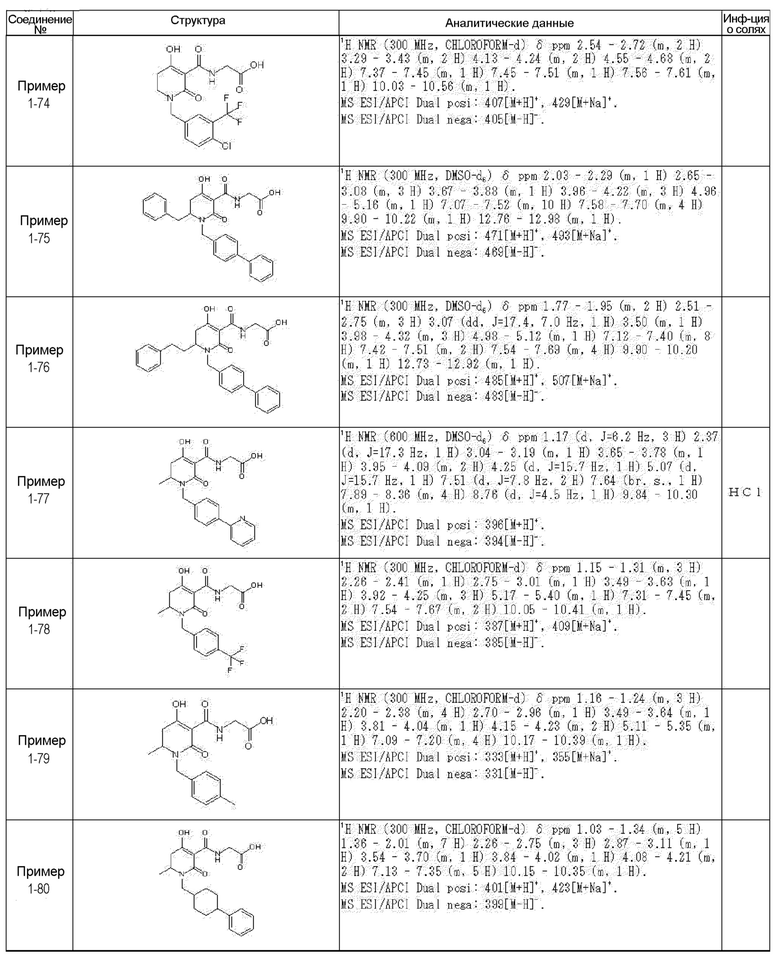
[0484]
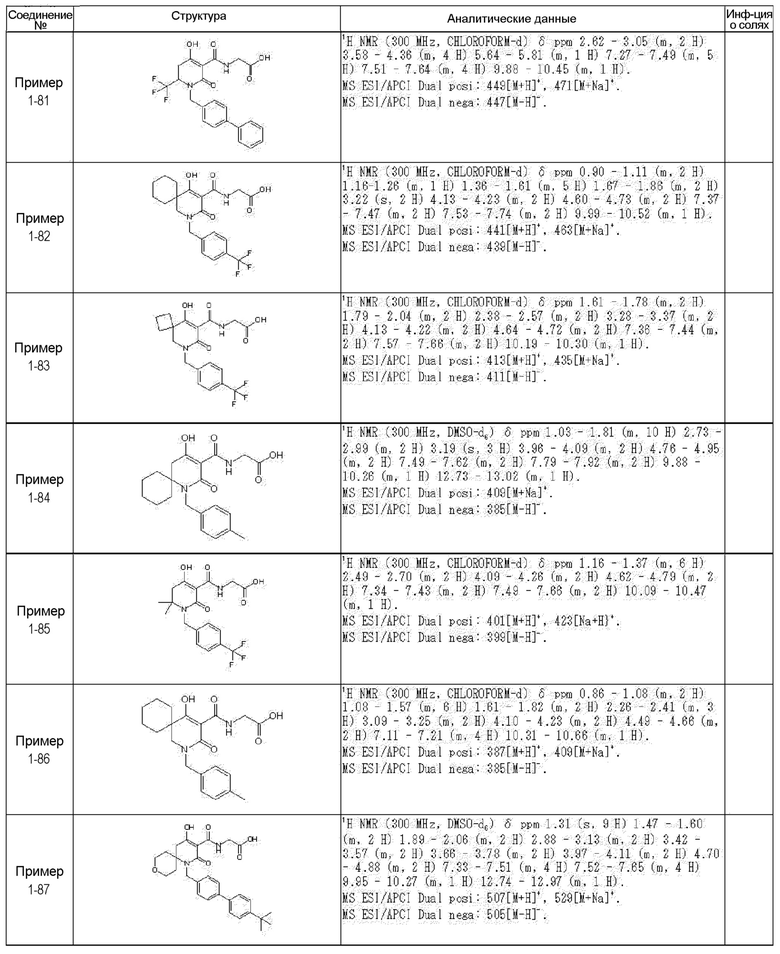
[0485]

[0486]
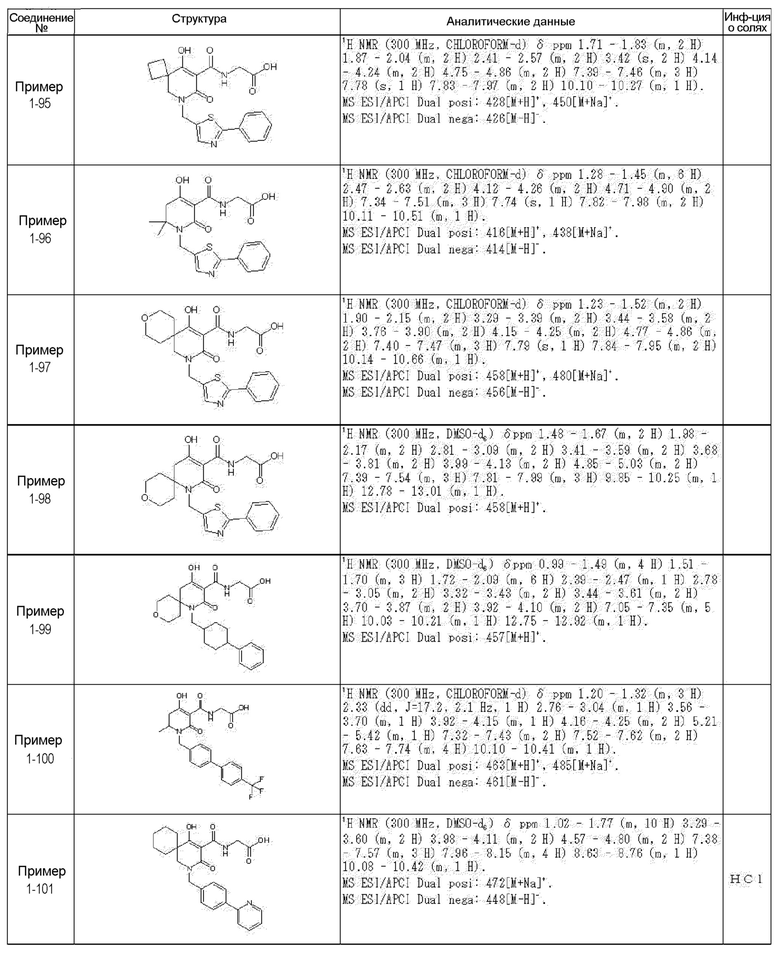
[0487]
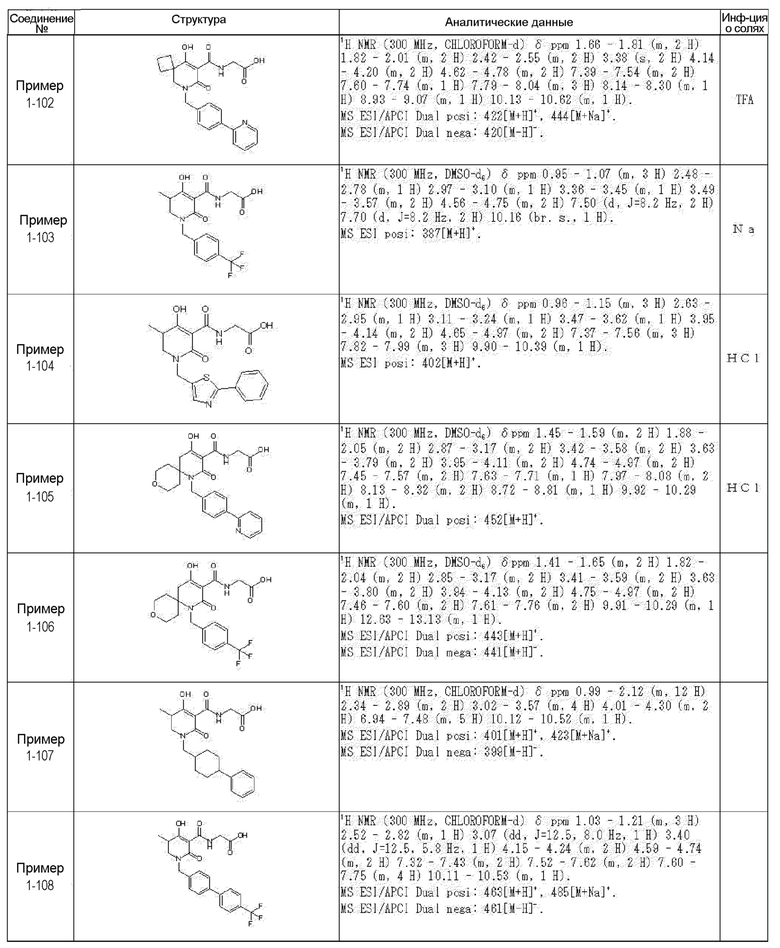
[0488]

[0489]
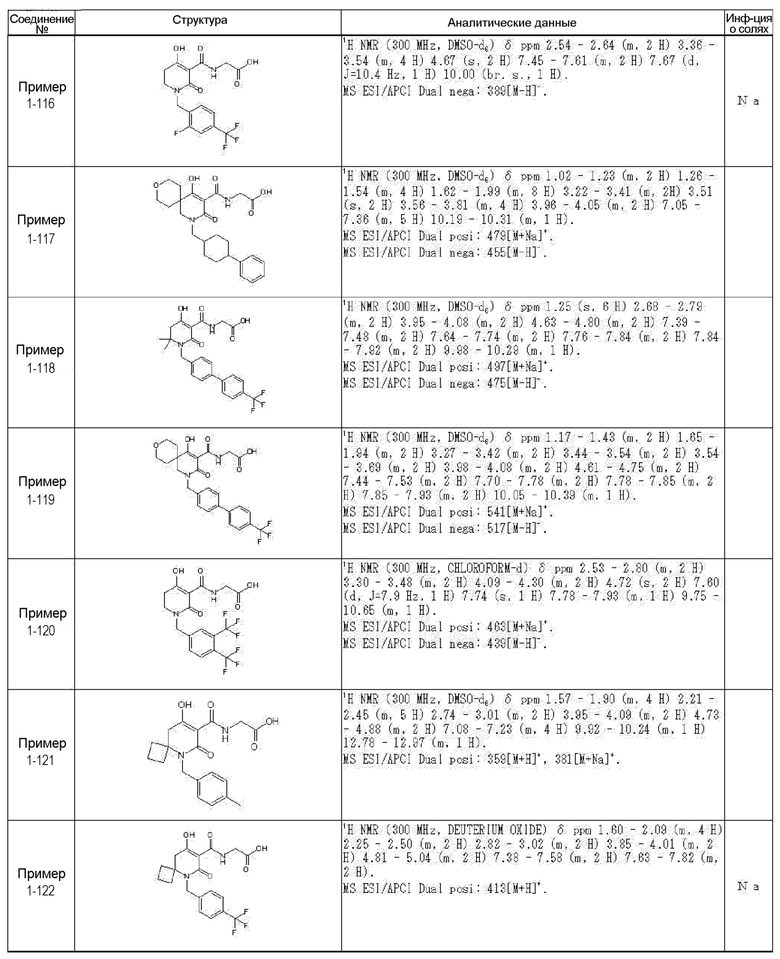
[0490]
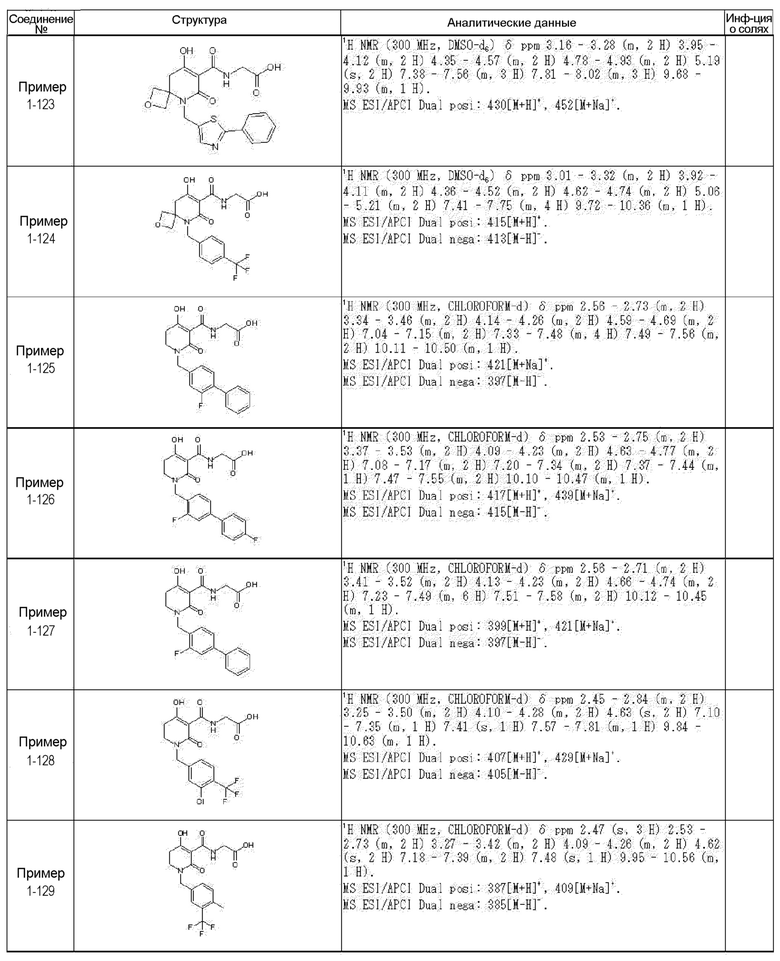
[0491]
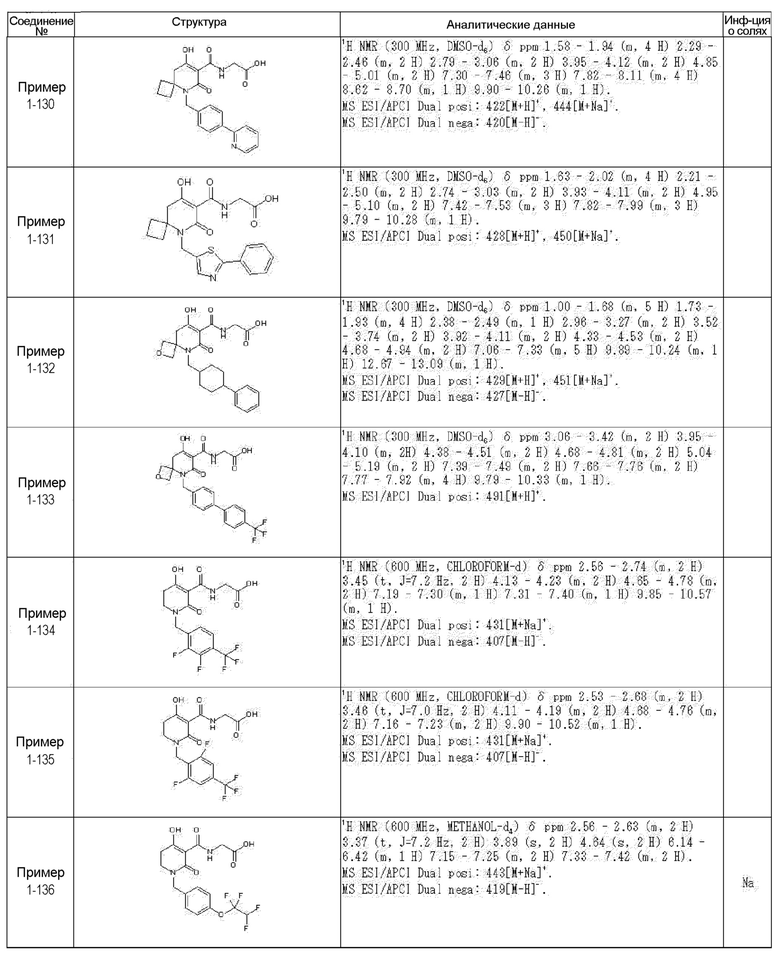
[0492]
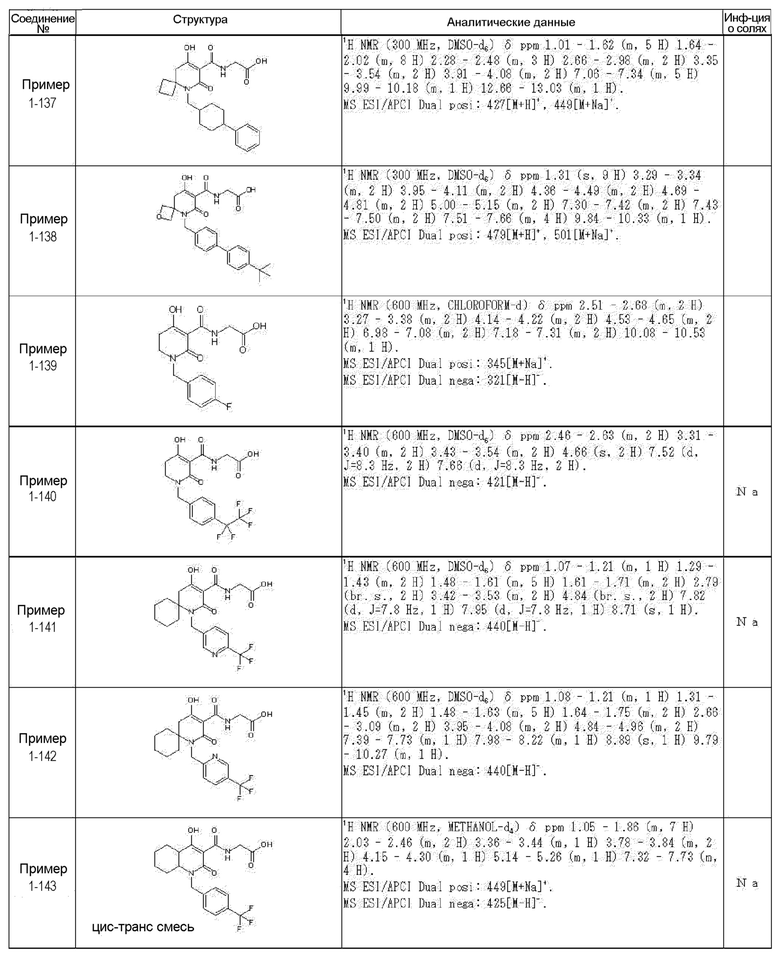
[0493]
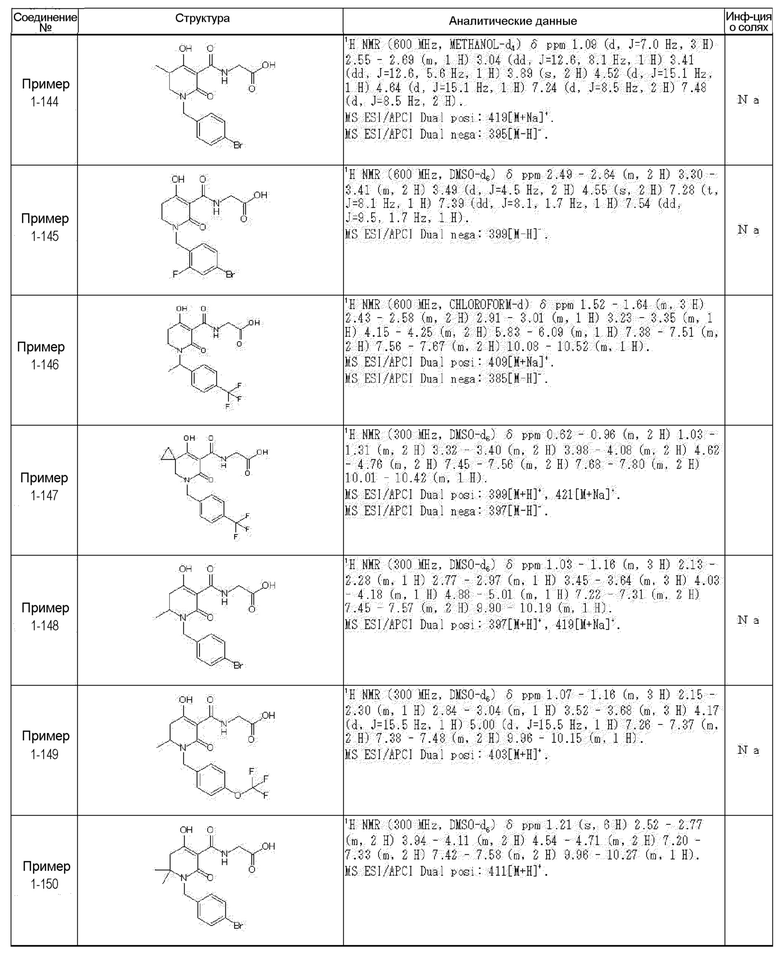
[0494]

[0495]

[0496]

[0497]
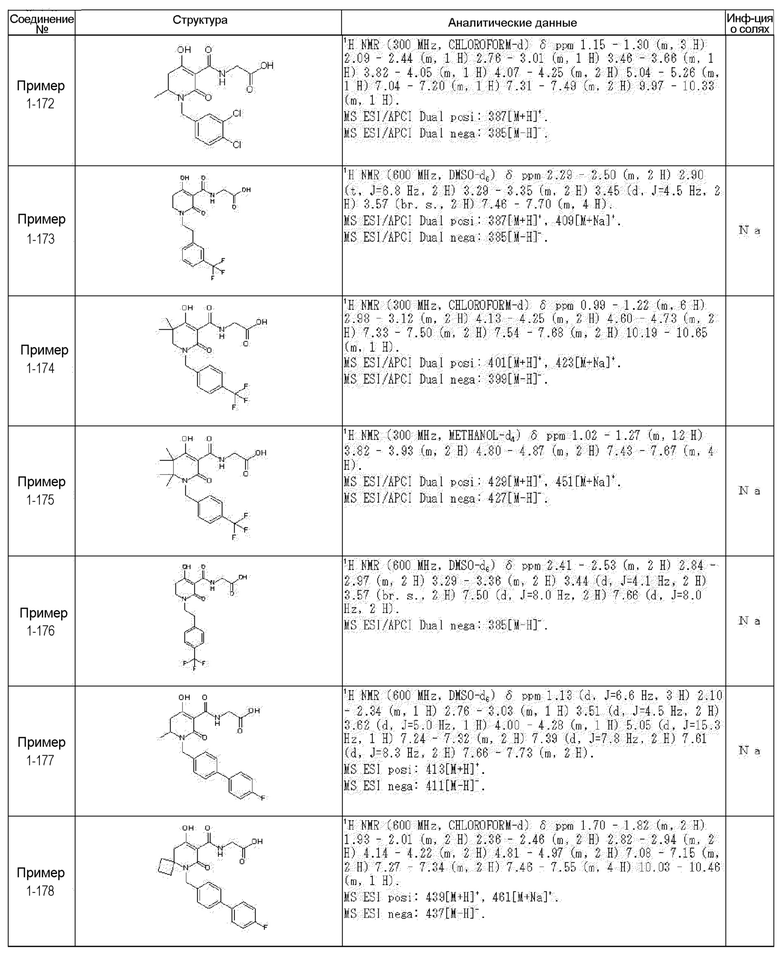
[0498]

[0499]

[0500]
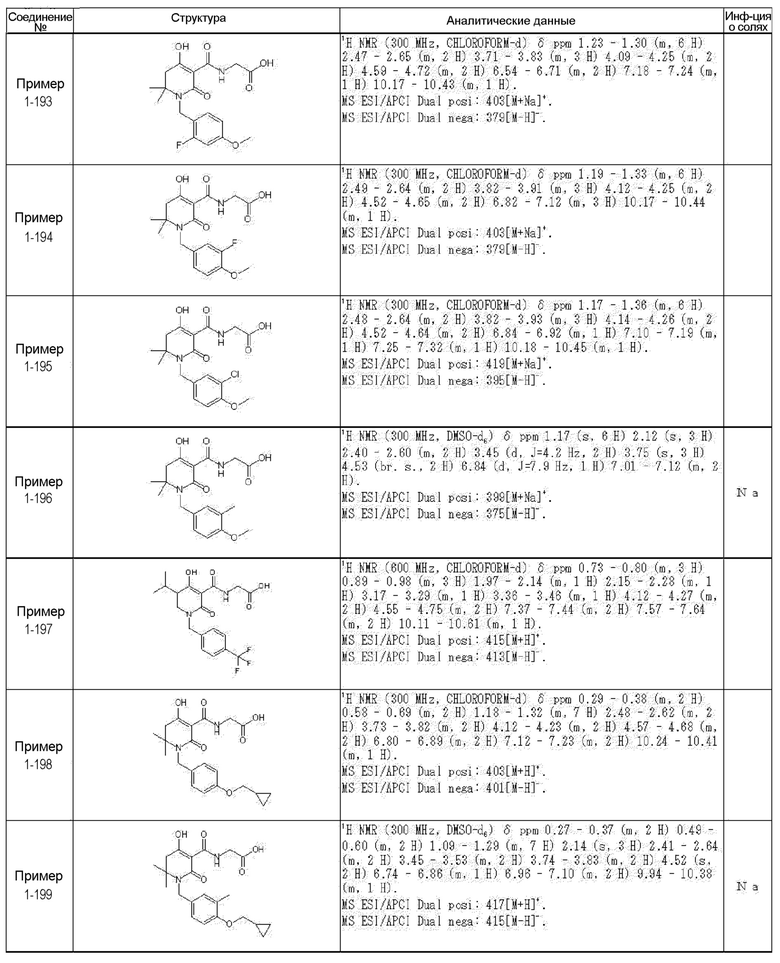
[0501]
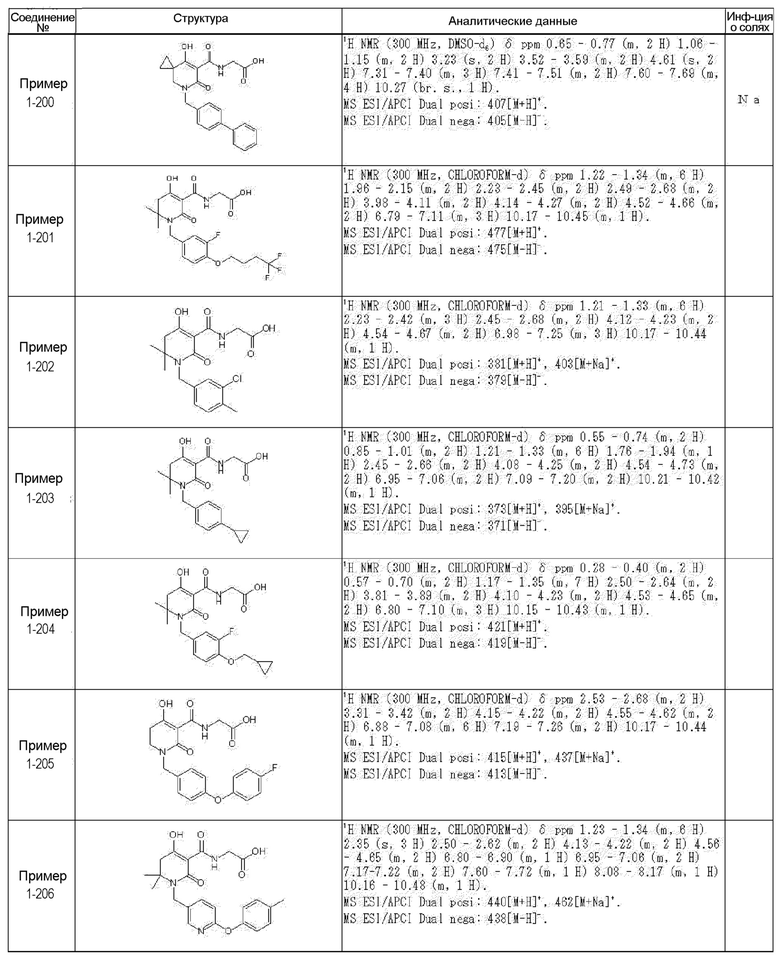
[0502]
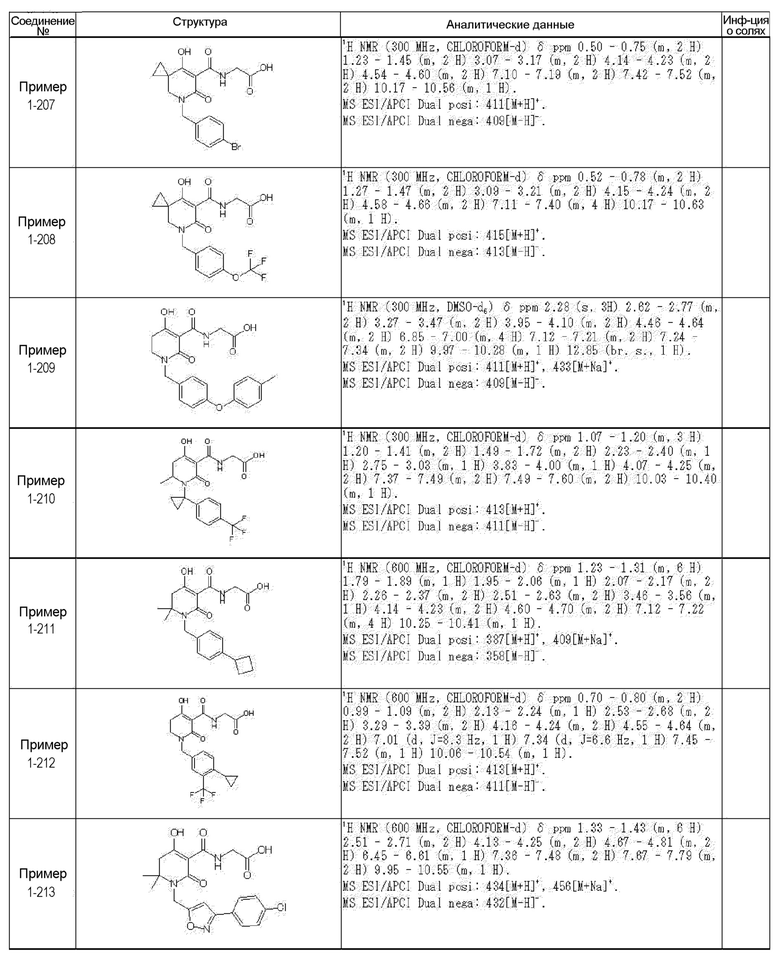
[0503]

[0504]

[0505]
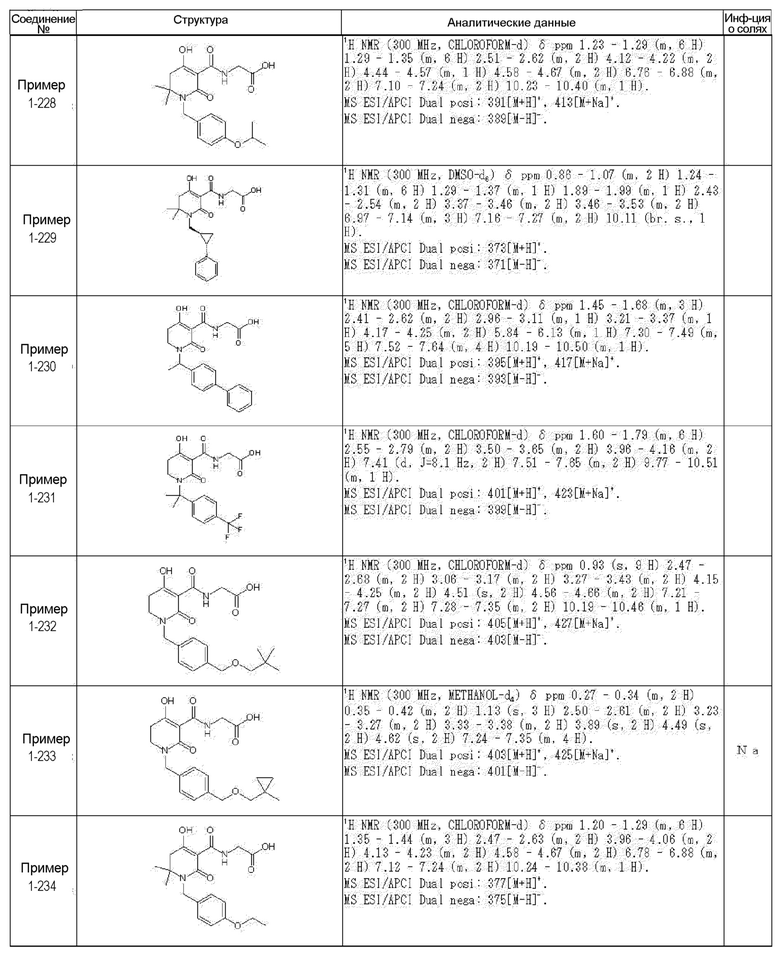
[0506]
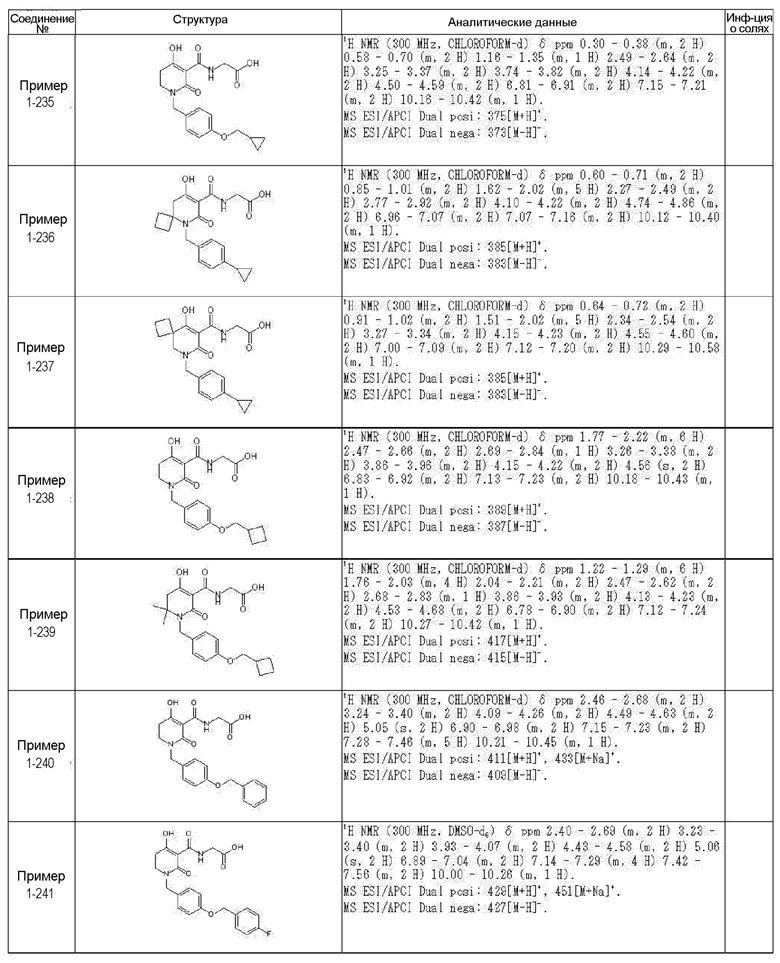
[0507]
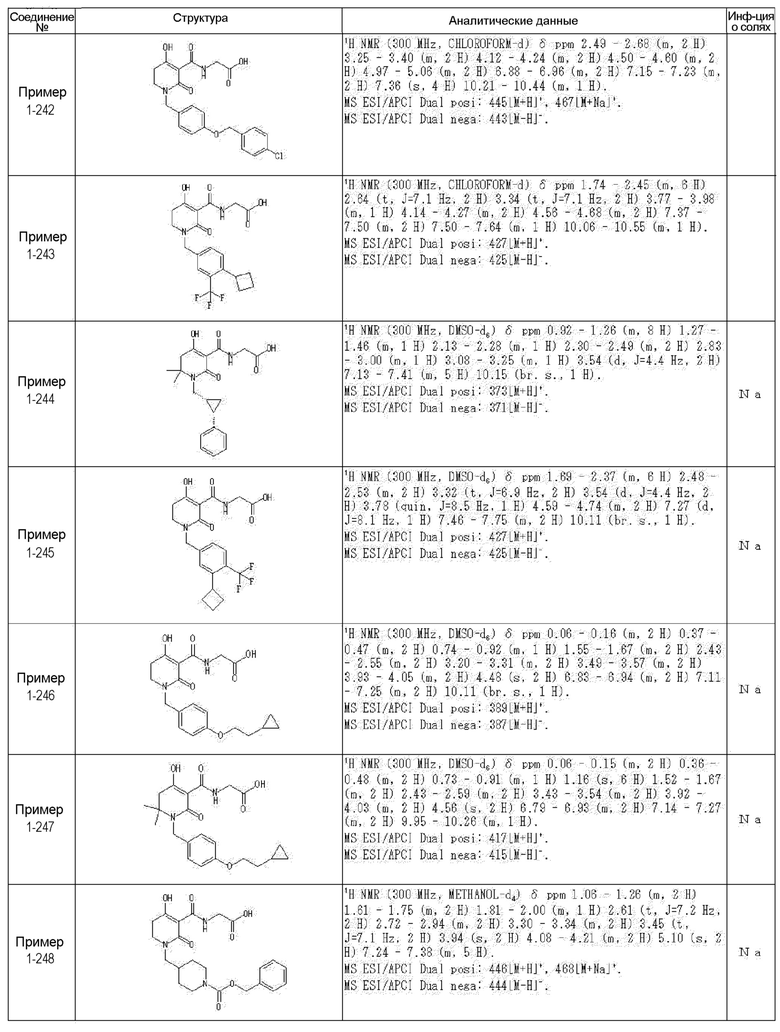
[0508]
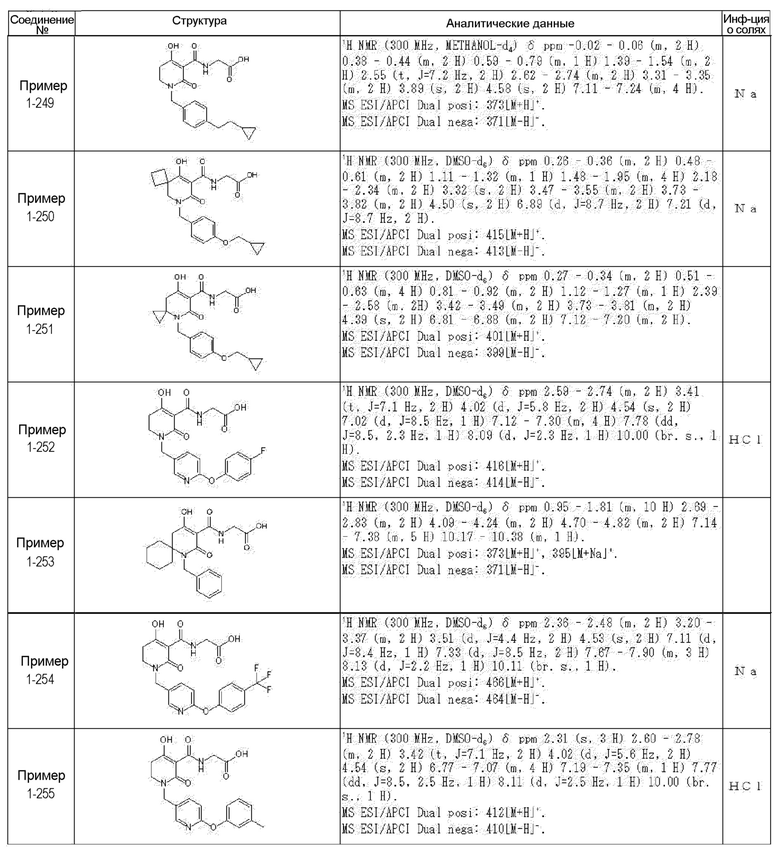
[0509]

[0510]

[0511]

[0512]
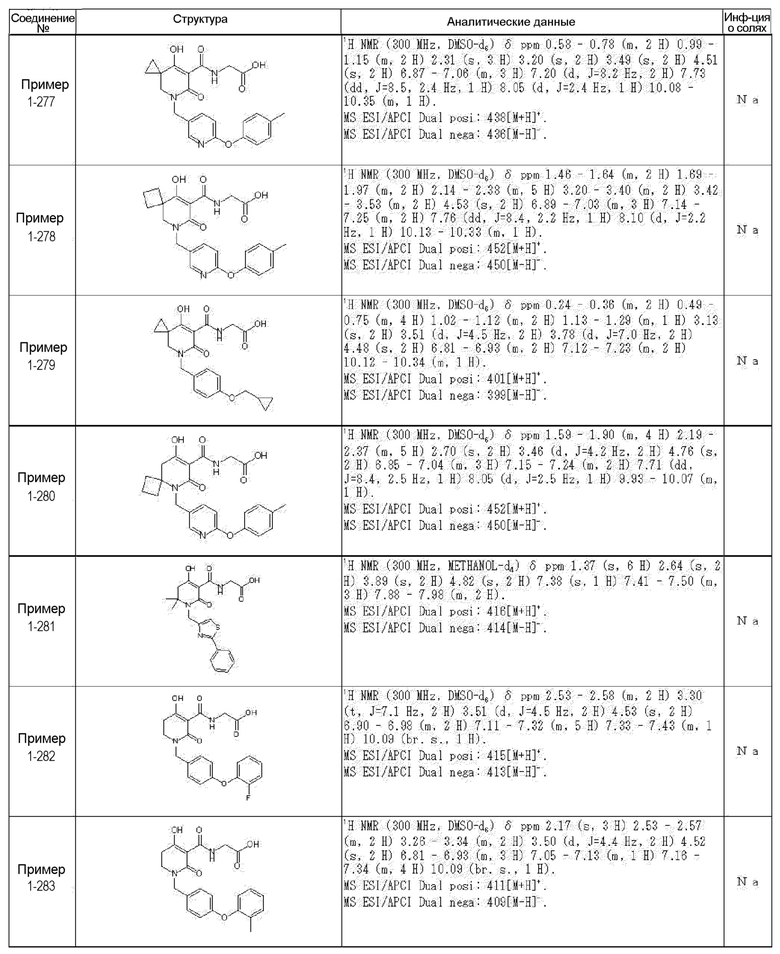
[0513]

[0514]

[0515]
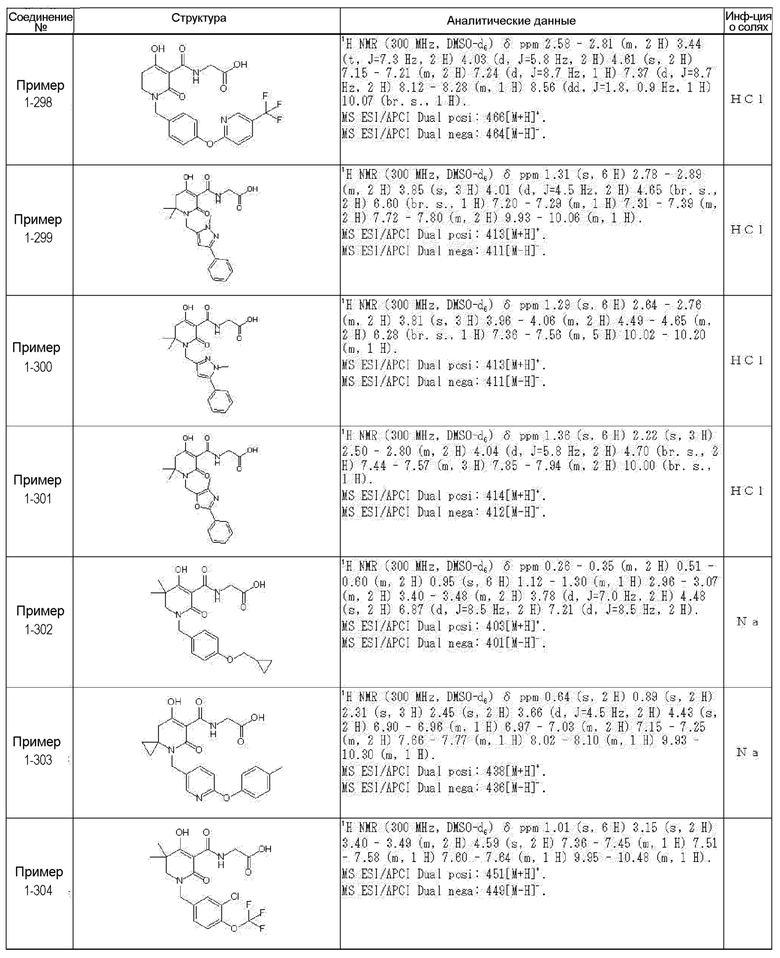
[0516]
[0517]

[0518]

[0519]
[0520]
[0521]

[0522]

[0523]

[0524]
[0525]
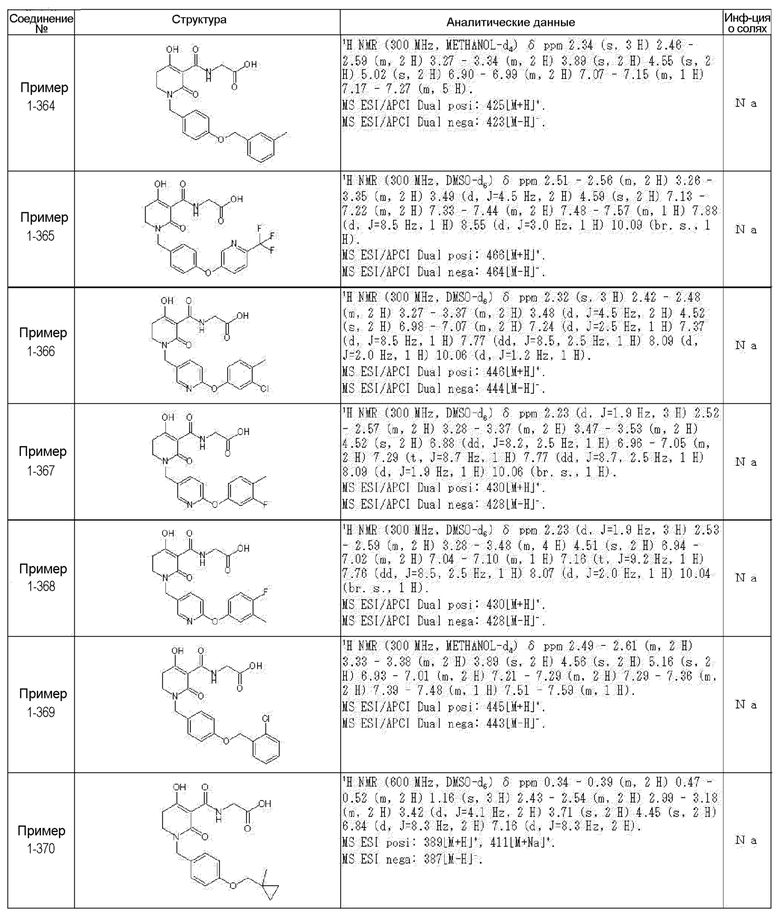
[0526]

[0527]
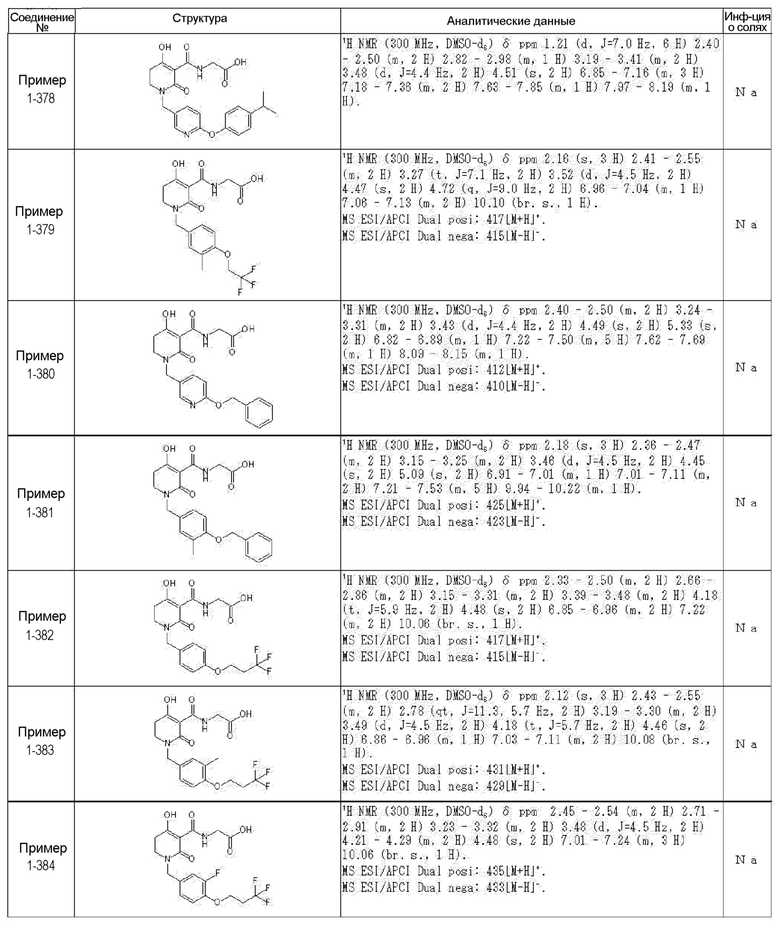
[0528]

[0529]

[0530]
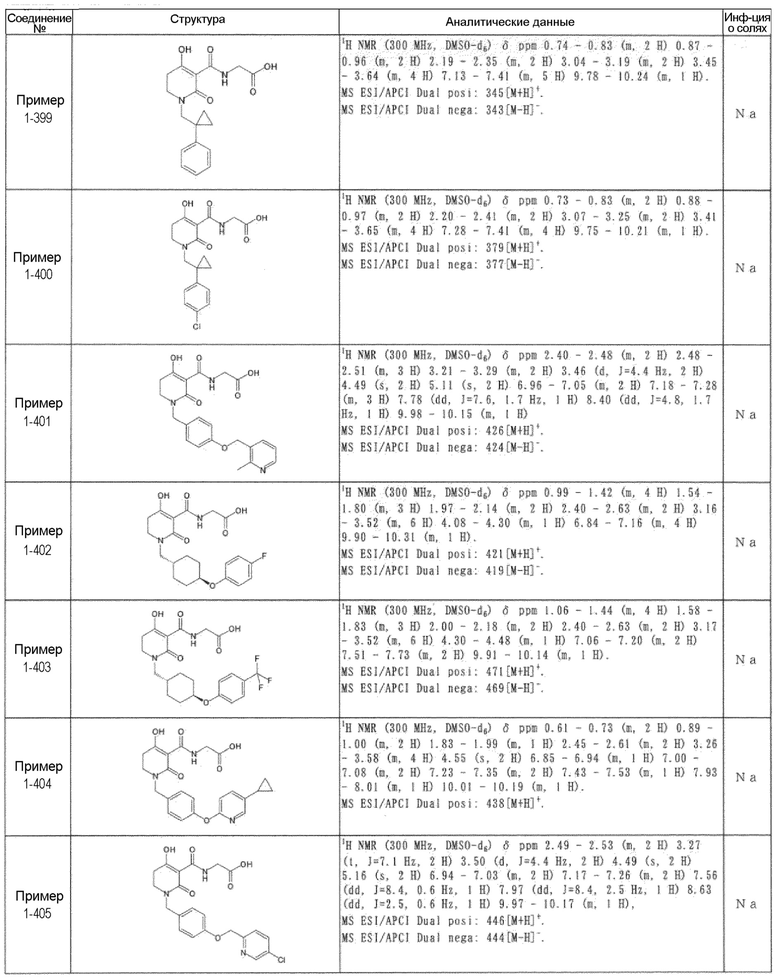
[0531]
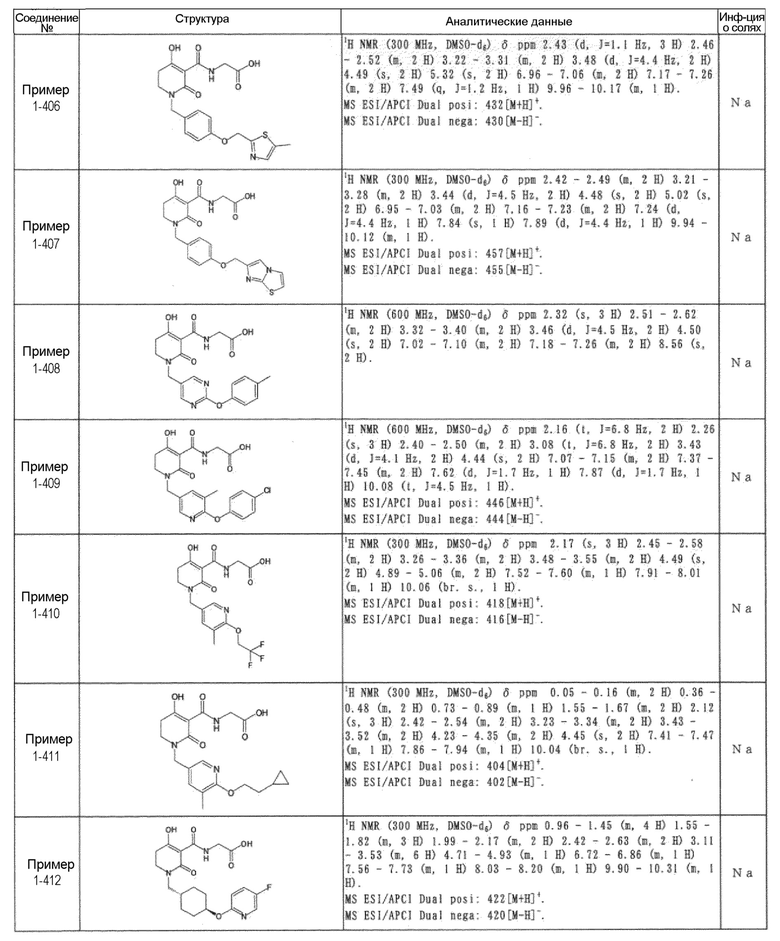
[0532]

[0533]
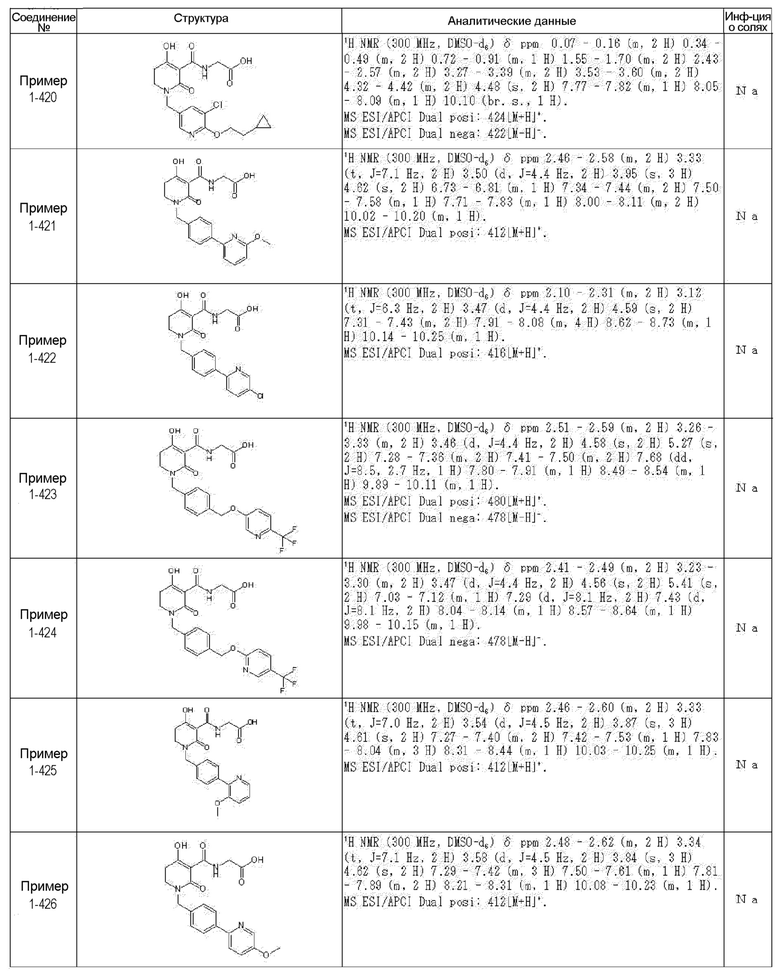
[0534]

[0535]
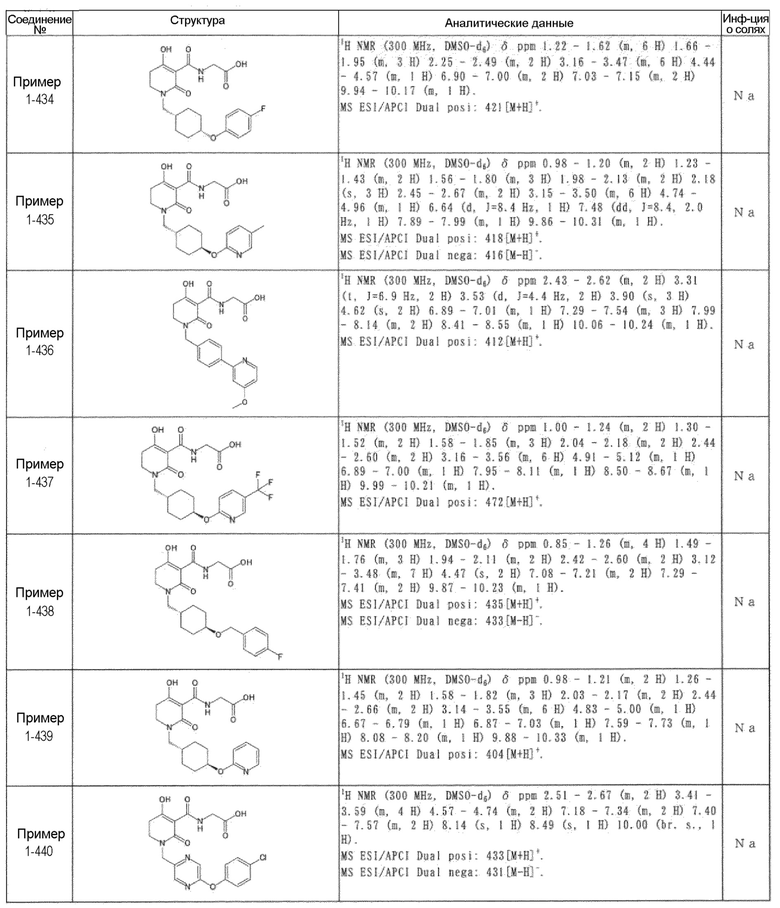
[0536]

[0537]
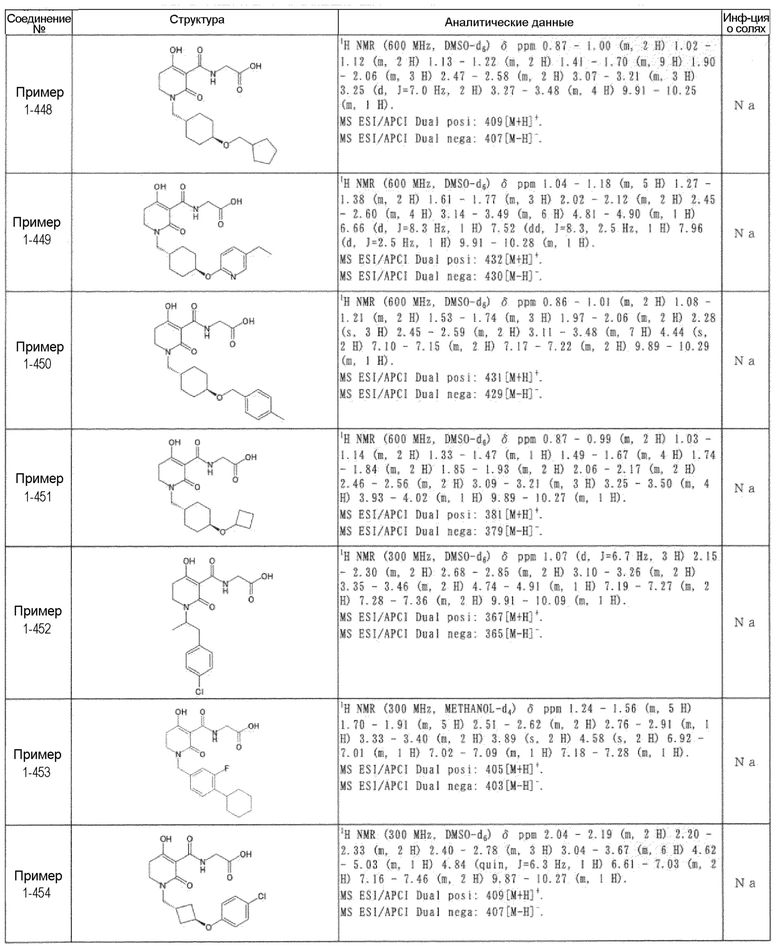
[0538]
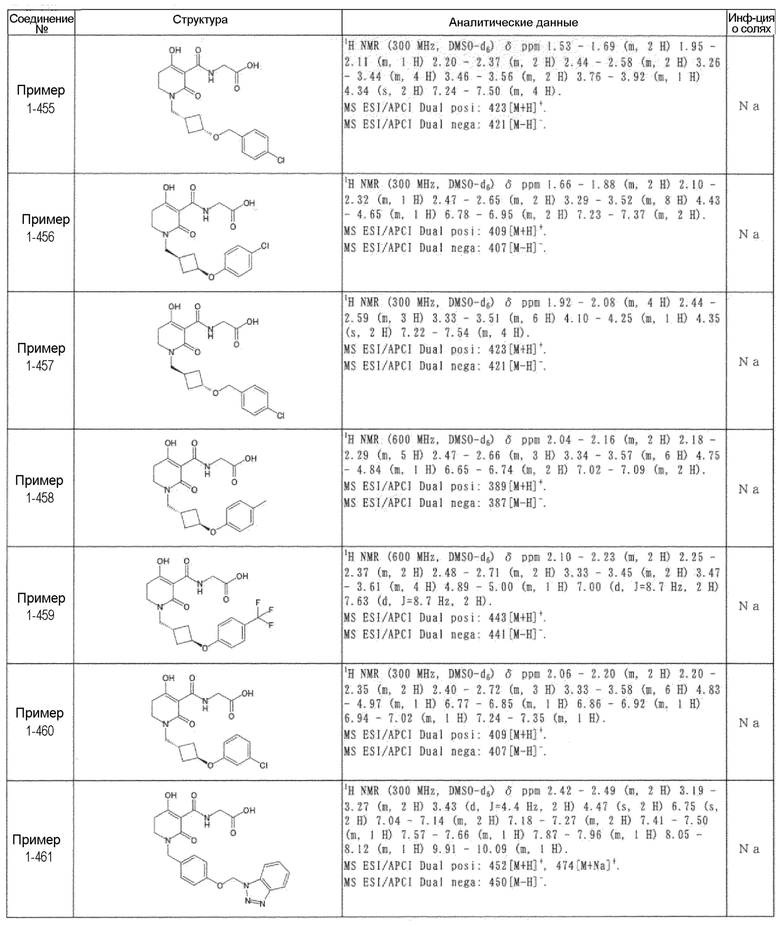
[0539]
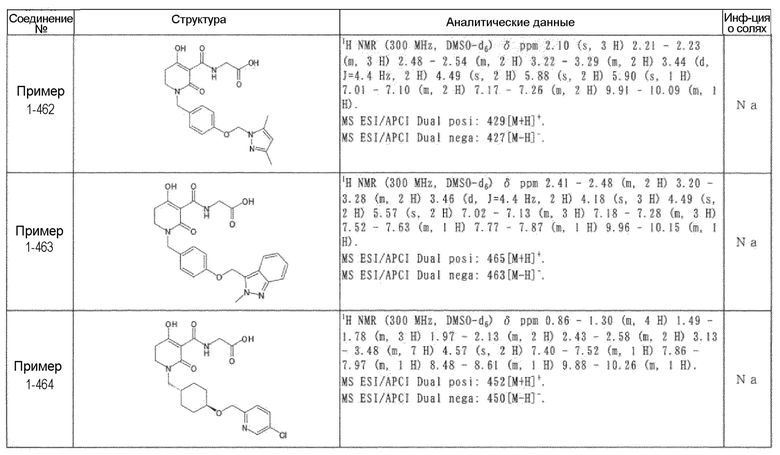
Пример 1-465
N-{[1-(4-бифенилилметил)-6-(хлорметил)-4-гидрокси-6-(гидроксиметил)-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин
[0540]
Использовали соединение (1,70 г), полученное в примере 1-2(1), и обработку проводили с помощью того же способа, как в примере 1-1(4), для того, чтобы получать указанное в заголовке соединение в виде аморфной массы бледно-коричневого цвета (363 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,61-3,08 (м, 2H) 3,45-3,66 (м, 2H) 3,70-3,93 (м, 2H) 3,96-4,10 (м, 2H) 4,69-4,90 (м, 2H) 5,46 (ушир.с, 1H) 7,28-7,51 (м, 5H) 7,54-7,70 (м, 4H) 9,89-10,31 (м, 1H) 12,73-13,04 (м, 1H).
MS ESI/APCI двойн. положительн.: 459[M+H]+.
Пример 1-466
N-{[1-(3-хлор-4-гидроксибензил)-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин динатрия
[0541]

(1) Синтез 2-метил-2-пропанил N-[(1-{3-хлор-4-[(3,5-диметил-1,2-оксазол-4-ил)метокси]бензил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицинат
[0542]

Вместо соединения, полученного в ссылочном примере A-1, использовали соединение (1,36 г), полученное в ссылочном примере A-301, и обработку проводили с помощью тех же способов, как в примере с 1-1(1) до (3), для того, чтобы получать 2-метил-2-пропанил N-[(1-{3-хлор-4-[(3,5-диметил-1,2-оксазол-4-ил)метокси]бензил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицинат в виде смолы бедно-желтого цвета (1,07 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,49 (с, 9H) 2,32 (с, 3H) 2,41 (с, 3H) 2,51-2,66 (м, 2H) 3,27-3,39 (м, 2H) 4,01-4,06 (м, 2H) 4,54 (с, 2H) 4,84-4,88 (м, 2H) 6,90-6,97 (м, 1H) 7,16 (дд, J=8,4, 2,2 Гц, 1H) 7,29-7,34 (м, 1H) 10,13-10,51 (м, 1H).
MS ESI/APCI двойн. положительн.: 520[M+H]+, 542[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 518[M-H]-.
(2) Синтез указанного в заголовке соединения
Использовали соединение (1,07 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью тех же способов, что и в примере 1-1(4) и примере 1-3(3), для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (454 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,12-2,22 (м, 2H) 3,02-3,10 (м, 2H) 3,46 (д, J=4,2 Гц, 2H) 4,37 (с, 2H) 6,85 (д, J=8,4 Гц, 1H) 6,96 (дд, J=8,4, 2,0 Гц, 1H) 7,12 (д, J=2,0 Гц, 1H) 10,04-10,16 (м, 1H).
MS ESI/APCI двойн. положительн.: 355[M+H]+.
MS ESI/APCI двойн. отрицательн.: 353[M-H]-.
Пример 1-467
N-({1-[4-(4,4-дифтор-1-циклогексен-1-ил)бензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глициновая соль натрия
[0543]

(1) Синтез 2-метил-2-пропанил N-({1-[4-(4,4-дифтор-1-гидроксициклогексил)бензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицината
[0544]

Вместо соединения, полученного в ссылочном примере A-1, использовали соединение (583 мг), полученное в ссылочном примере B-18, и обработку проводили с помощью тех же способов, что и в примерах с 1-1(1) до (3), для того, чтобы получать 2-метил-2-пропанил N-({1-[4-(4,4-дифтор-1-гидроксициклогексил)бензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицинат в виде бесцветного твердого вещества (226 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,49 (с, 9H) 1,82-1,94 (м, 2H) 2,07-2,42 (м, 6H) 2,62 (с, 2H) 3,30-3,39 (м, 2H) 4,01-4,06 (м, 2H) 4,61 (с, 2H) 7,25-7,30 (м, 2H) 7,43-7,50 (м, 2H).
MS ESI/APCI двойн. положительн.: 517[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 493[M-H]-, 529[M+Cl]-.
(2) Синтез указанного в заголовке соединения
Использовали соединение (226 мг), полученное на приведенной выше стадии (1), и обработку проводили с помощью тех же способов, что и в примере 1-1(4) и примере 1-3(3), для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (129 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,04-2,30 (м, 4H) 2,59-2,80 (м, 4H) 3,24-3,32 (м, 2H) 3,47 (д, J=4,4 Гц, 2H) 4,55 (с, 2H) 5,95-6,04 (м, 1H) 7,25 (д, J=8,3 Гц, 2H) 7,41 (д, J=8,3 Гц, 2H).
MS ESI/APCI двойн. положительн.: 421 [M+H]+.
MS ESI/APCI двойн. отрицательн.: 419[M-H]-.
Пример 1-468
N-({1-[4-(циклопропилметокси)-3-метилбензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глициновая соль натрия
[0545]

(1) Синтез этил N-({1-[4-(циклопропилметокси)-3-метилбензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицината
[0546]
Вместо соединения, полученного в ссылочном примере A-1, и глицин трет-бутил гидрохлорида соответственно использовали соединение (2,10 г), полученное в ссылочном примере A-302, и глицин этил гидрохлорид (956 мг) и обработку проводили с помощью тех же способов, как в примере с 1-1(1) до (3), для того, чтобы получать этил N-({1-[4-(циклопропилметокси)-3-метилбензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицинат в виде масла желтого цвета (2,01 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,29-0,40 (м, 2H) 0,54-0,68 (м, 2H) 1,19-1,37 (м, 4H) 2,16-2,26 (м, 3H) 2,44-2,64 (м, 2H) 3,23-3,35 (м, 2H) 3,75-3,85 (м, 2H) 4,05-4,17 (м, 2H) 4,24 (кв., J=7,1 Гц, 2H) 4,47-4,54 (м, 2H) 6,67-6,79 (м, 1H) 6,98-7,08 (м, 2H) 10,11-10,60 (м, 1H).
MS ESI/APCI двойн. положительн.: 417[M+H]+.
(2) Синтез указанного в заголовке соединения
В раствор в этаноле (37,8 мл) соединения (1,97 г), полученного на приведенной выше стадии (1), добавляли гидроксид натрия в водном растворе 0,5 моль/л (18,9 мл) при охлаждении льдом и смесь доводили до комнатной температуры, при которой ее перемешивали в течение 15 минут. После реакционную смесь концентрировали при пониженном давлении, получаемый остаток очищали посредством колоночной хроматографии DIAION HP20 (зарегистрированный товарный знак) (с элюированием метанолом). Элюированную фракцию концентрировали при пониженном давлении и в раствор получаемого соединения в воде (2 мл) добавляли ацетон (100 мл) и смесь перемешивали при комнатной температуре в течение часа. Осадок собирали с помощью фильтрования для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (1,60 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 0,25-0,38 (м, 2H) 0,47-0,61 (м, 2H) 1,11-1,28 (м, 1H) 2,13 (с, 3H) 2,34-2,50 (м, 2H) 3,15-3,29 (м, 2H) 3,51 (д, J=4,5 Гц, 2H) 3,73-3,86 (м, 2H) 4,44 (с, 2H) 6,84 (д, J=9,0 Гц, 1H) 7,00-7,06 (м, 2H) 10,12 (ушир.с, 1H).
MS ESI/APCI двойн. положительн.: 389[M+H]+.
MS ESI/APCI двойн. отрицательн.: 387[M-H]-.
Пример 2-1
N-({4-гидрокси-1-[(4'-метилбифенил-4-ил)метил]-2-оксо-1,2,5,6-тетрагидропиридин-3-ил}карбонил)глицин
[0547]
(1) Синтез трет-бутил N-{[4-гидрокси-1-(4-йодбензил)-2-оксо-1,2,5,6-тетрагидропиридин-3-ил]карбонил}глицината
[0548]

Вместо соединения, полученного в ссылочном примере A-1, использовали соединение (6,50 г), полученное в ссылочном примере A-4, и обработку проводили с помощью тех же способов, как в примере с 1-1(1) до (3), для того, чтобы получать трет-бутил N-{[4-гидрокси-1-(4-йодбензил)-2-оксо-1,2,5,6-тетрагидропиридин-3-ил]карбонил}глицинат в виде бесцветного твердого вещества (7,01 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,48 (с, 9H) 2,49-2,65 (м, 2H) 3,24-3,42 (м, 2H) 3,97-4,08 (м, 2H) 4,56 (с, 2H) 6,98-7,08 (м, 2H) 7,57-7,75 (м, 2H) 10,13-10,51 (м, 1H).
MS ESI/APCI двойн. положительн.: 487[M+H]+.
MS ESI/APCI двойн. отрицательн.: 485[M-H]-.
(2) Синтез трет-бутил N-({4-гидрокси-1-[(4'-метилбифенил-4-ил)метил]-2-оксо-1,2,5,6-тетрагидропиридин-3-ил}карбонил)глицината
[0549]
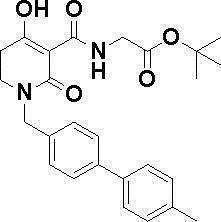
Смесь соединения (300 мг), полученного на приведенной выше стадии (1), 4-метилфенилбороновой кислоты (168 мг), ацетата палладия(II) (6,93 мг), три(2-метилфенил)фосфина (18,8 мг), фосфата трикалия (393 мг), этанола (8,00 мл) и толуола (4,00 мл) перемешивали при 90°C в течение часа. После охлаждения реакционной смеси до комнатной температуры добавляли насыщенный водный раствор хлорида аммония и получаемую смесь концентрировали при пониженном давлении. Добавляли воду и проводили три экстрагирования хлороформом. Объединенные органические слои пропускали через фазовый разделитель и после этого концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-50:50, затем хлороформ:метанол=100:0-90:10) для того, чтобы получать трет-бутил N-({4-гидрокси-1-[(4'-метилбифенил-4-ил)метил]-2-оксо-1,2,5,6-тетрагидропиридин-3-ил}карбонил)глицинат в виде твердого вещества бледно-желтого цвета (485 мг).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,49 (с, 9H) 2,39 (с, 3H) 2,53-2,65 (м, 2H) 3,37 (м, 2H) 4,00-4,08 (м, 2H) 4,66 (с, 2H) 7,25 (д, J=7,8 Гц, 2H) 7,32 (д, J=7,8 Гц, 2H) 7,44-7,50 (м, 2H) 7,51-7,58 (м, 2H) 10,22-10,49 (м, 1H).
MS ESI/APCI двойн. положительн.: 473[M+Na]+.
(3) Синтез указанного в заголовке соединения
Использовали соединение (147 мг), полученное на приведенной выше стадии (2), и обработку проводили с помощью того же способа, как в примере 1-1(4), для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (111 мг).
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 2,33 (с, 3H) 2,46-2,72 (м, 2H) 3,36-3,46 (м, 2H) 3,98-4,07 (м, 2H) 4,57-4,67 (м, 2H) 7,23-7,29 (м, 2H) 7,32-7,40 (м, 2H) 7,50-7,57 (м, 2H) 7,58-7,65 (м, 2H) 10,01-10,25 (м, 1H).
MS ESI/APCI двойн. положительн.: 395[M+1]+, 417[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 393[M-1]-.
Пример 2-2
N-({4-гидрокси-1-[4-(3-метокси-4-пиридинил)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глициновая соль натрия
[0550]

(1) Синтез 2-метил-2-пропанил N-({4-гидрокси-1-[4-(3-метокси-4-пиридинил)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицината
[0551]
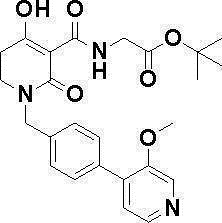
Смесь соединения (1,50 г), полученного в примере 2-1(1), сложного пинаколового эфира 3-метокси-4-пиридинбороновой кислоты (870 мг), комплекса дихлорметана дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия(II) (1:1) (126 мг), карбоната натрия в водном растворе 2 моль/л (3,4 мл) и N,N-диметилформамида (12,3 мл) перемешивали при 120°C в течение 20 минут при облучении микроволнами. После охлаждения реакционной смеси до комнатной температуры, добавляли насыщенный водный раствор бикарбоната натрия и этилацетат и осадок собирали с помощью фильтрования через Celite (зарегистрированный товарный знак). Проводили экстрагирование этилацетатом и объединенные органические слои концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 20:80-0:100, затем хлороформ:метанол=100:0-80:20) для того, чтобы получать 2-метил-2-пропанил N-({4-гидрокси-1-[4-(3-метокси-4-пиридинил)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицинат в виде аморфной массы желтого цвета (1,24 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,49 (с, 9H) 2,52-2,69 (м, 2H) 3,32-3,45 (м, 2H) 3,92 (с, 3H) 4,00-4,07 (м, 2H) 4,67 (с, 2H) 7,22-7,26 (м, 2H) 7,32-7,38 (м, 2H) 7,50-7,60 (м, 2H) 8,28-8,35 (м, 1H) 10,18-10,50 (м, 1H).
MS ESI/APCI двойн. положительн.: 468[M+H]+.
(2) Синтез указанного в заголовке соединения
Использовали соединение (1,24 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью тех же способов, что и в примере 1-2(2) и примере 1-3(3), для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (730 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,46-2,61 (м, 2H) 3,35 (т, J=7,1 Гц, 2H) 3,56 (д, J=4,5 Гц, 2H) 3,89 (с, 3H) 4,61 (с, 2H) 7,29-7,44 (м, 3H) 7,50-7,60 (м, 2H) 8,22-8,33 (м, 1H) 8,45 (с, 1H) 10,08-10,21 (м, 1H).
MS ESI/APCI двойн. положительн.: 412[M+H]+.
Пример 2-3
N-({4-гидрокси-1-[4-(2-метокси-3-пиридинил)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глициновая соль натрия
[0552]

(1) Синтез 2-метил-2-пропанил N-({4-гидрокси-1-[4-(2-метокси-3-пиридинил)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицината
[0553]
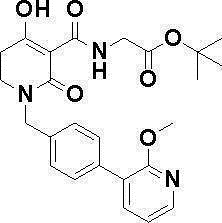
Смесь соединения (1,00 г), полученного в примере 2-1(1), 2-метоксипиридин-3-бороновой кислоты (426 мг), ацетата палладия(II) (25,0 мг), фосфата трикалия (987 мг) и этиленгликоля (12 мл) перемешивали в герметизированной пробирке при 80°C в течение 4 часов. После охлаждения реакционной смеси до комнатной температуры, добавляли воду и этилацетат и осадок собирали с помощью фильтрования через Celite (зарегистрированный товарный знак). Проводили экстрагирование этилацетатом и объединенные органические слои концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-20:80) для того, чтобы получать 2-метил-2-пропанил N-({4-гидрокси-1-[4-(2-метокси-3-пиридинил)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицинат в виде масла желтого цвета (1,10 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,49 (с, 9H) 2,52-2,68 (м, 2H) 3,33-3,42 (м, 2H) 3,97 (с, 3H) 4,00-4,08 (м, 2H) 4,66 (с, 2H) 6,93-7,01 (м, 1H) 7,29-7,36 (м, 2H) 7,46-7,65 (м, 3H) 8,09-8,21 (м, 1H) 10,15-10,53 (м, 1H).
MS ESI/APCI двойн. положительн.: 490[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 466[M-H]-.
(2) Синтез указанного в заголовке соединения
Использовали соединение (1,10 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью тех же способов, что и в примере 1-2(2) и примере 1-3(3), для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (461 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,41-2,61 (м, 2H) 3,40-3,49 (м, 2H) 3,87 (с, 3H) 4,60 (с, 2H) 7,05-7,12 (м, 1H) 7,29-7,39 (м, 2H) 7,48-7,57 (м, 2H) 7,70-7,77 (м, 1 Н) 8,13-8,20 (м, 1 Н).
MS ESI/APCI двойн. положительн.: 412[M+H]+.
MS ESI/APCI двойн. отрицательн.: 410[M-H]-.
Пример 2-4
N-({4-гидрокси-1-[4-(6-метил-3-пиридинил)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глициновая соль натрия
[0554]

(1) Синтез 2-метил-2-пропанил N-({4-гидрокси-1-[4-(6-метил-3-пиридинил)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицината
[0555]
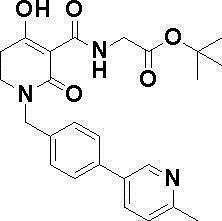
Смесь соединения (1,00 г), полученного в примере 2-1(1), сложного пинаколового эфира 2-пиколин-5-бороновой кислоты (541 мг), тетракис(трифенилфосфин)палладия(0) (238 мг), карбоната калия (569 мг), толуола (10 мл), этанола (2 мл) и воды (2 мл) перемешивали при 80°C в течение 4 часов. После охлаждения реакционной смеси до комнатной температуры, осадок удаляли посредством пропускания через Celite (зарегистрированный товарный знак) и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-30:70) для того, чтобы получать 2-метил-2-пропанил N-({4-гидрокси-1-[4-(6-метил-3-пиридинил)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицинат (560 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,49 (с, 9H) 2,51-2,69 (м, 5H) 3,31-3,44 (м, 2H) 4,00-4,07 (м, 2H) 4,67 (с, 2H) 7,17-7,26 (м, 1H) 7,37 (д, J=8,2 Гц, 2H) 7,48-7,59 (м, 2H) 7,72-7,81 (м, 1H) 8,67-8,77 (м, 1H) 10,15-10,53 (м, 1H).
MS ESI/APCI двойн. положительн.: 452[M+H]+, 474[M+H]+.
MS ESI/APCI двойн. отрицательн.: 450[M-H]-.
(2) Синтез указанного в заголовке соединения
Использовали соединение (560 мг), полученное на приведенной выше стадии (1), и обработку проводили с помощью тех же способов, что и в примере 1-2(2) и примере 1-3(3), для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (254 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,36-2,55 (м, 5H) 3,21-3,34 (м, 2H) 3,50 (д, J=4,5 Гц, 2H) 4,60 (с, 2H) 7,26-7,45 (м, 3H) 7,59-7,71 (м, 2H) 7,89-7,99 (м, 1H) 8,73 (д, J=2,0 Гц, 1H) 10,01-10,22 (м, 1H).
MS ESI/APCI двойн. положительн.: 396[M+H]+.
MS ESI/APCI двойн. отрицательн.: 394[M-H]-.
Пример 2-5
N-({4-гидрокси-1-[4-(5-метил-2-пиридинил)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глициновая соль натрия
[0556]

(1) Синтез 2-метил-2-пропанил N-({4-гидрокси-1-[4-(5-метил-2-пиридинил)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицината
[0557]
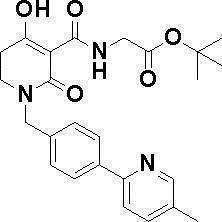
Смесь соединения (2,00 г), полученного в примере 2-1(1), сложного N-фенилдиэтаноламинового эфира 5-метилпиридин-2-бороновой кислоты (2,32 г), карбоната калия (1,14 г), йодида меди(I) (313 мг), три(2-метилфенил)фосфина (250 мг), ацетата палладия(II) (46 мг) и тетрагидрофурана (28,8 мл) перемешивали при 95°C в течение 4 часов. После охлаждения реакционной смеси до комнатной температуры, добавляли этилацетат и осадок удаляли посредством прохождения через Celite (зарегистрированный товарный знак). Фильтрат концентрировали при пониженном давлении и получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=80:20-0:100) для того, чтобы получать 2-метил-2-пропанил N-({4-гидрокси-1-[4-(5-метил-2-пиридинил)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицинат в виде бесцветной аморфной массы (1,00 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,49 (с, 9H) 2,37 (с, 3H) 2,47-2,68 (м, 2H) 3,25-3,40 (м, 2H) 4,00-4,07 (м, 2H) 4,67 (с, 2H) 7,31-7,42 (м, 2H) 7,52-7,66 (м, 2H) 7,87-7,99 (м, 2H) 8,45-8,59 (м, 1H) 10,14-10,53 (м, 1H).
MS ESI/APCI двойн. положительн.: 452[M+H]+.
(2) Синтез указанного в заголовке соединения
Использовали соединение (1,00 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью тех же способов, что и в примере 1-2(2) и примере 1-3(3), для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (505 мг).
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 2,29 (с, 3H) 2,43-2,59 (м, 2H) 3,22-3,56 (м, 4H) 4,58 (с, 2H) 7,28-7,39 (м, 2H) 7,60-7,69 (м, 1H) 7,75-7,85 (м, 1H) 7,94-8,04 (м, 2H) 8,45 (с, 1H) 9,87-10,31 (м, 1H).
MS ESI/APCI двойн. положительн.: 396[M+H]+.
[0558] в следующих примерах с 2-6 до 2-54 в качестве исходного материала использовали соединение, полученное в ссылочных примерах A-4, A-5, A-303, A-304 или A-305, которое вместе с соответствующими бороновыми кислотами или сложными эфирами бороновой кислоты торговых сортов обрабатывали с помощью способов, описанных в примере 2-1(1) и (2) или на стадии (1) каждого из примеров с 2-2 до 2-5, или их модификаций, и полученные таким образом соединения дополнительно обрабатывали с помощью способа, описанного в примере 1-1(4), 1-2(2) или 1-3(3), или его модификаций для того, чтобы синтезировать предполагаемые соединения. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблицах с 22-1 до 22-8. Номера, указанные в колонке «Пример» в каждой таблице, указывают на то, какие из приведенных выше примеров с 2-1 до 2-5 повторяли или модифицировали для того, чтобы синтезировать соединение, представляющее интерес.
[0559]
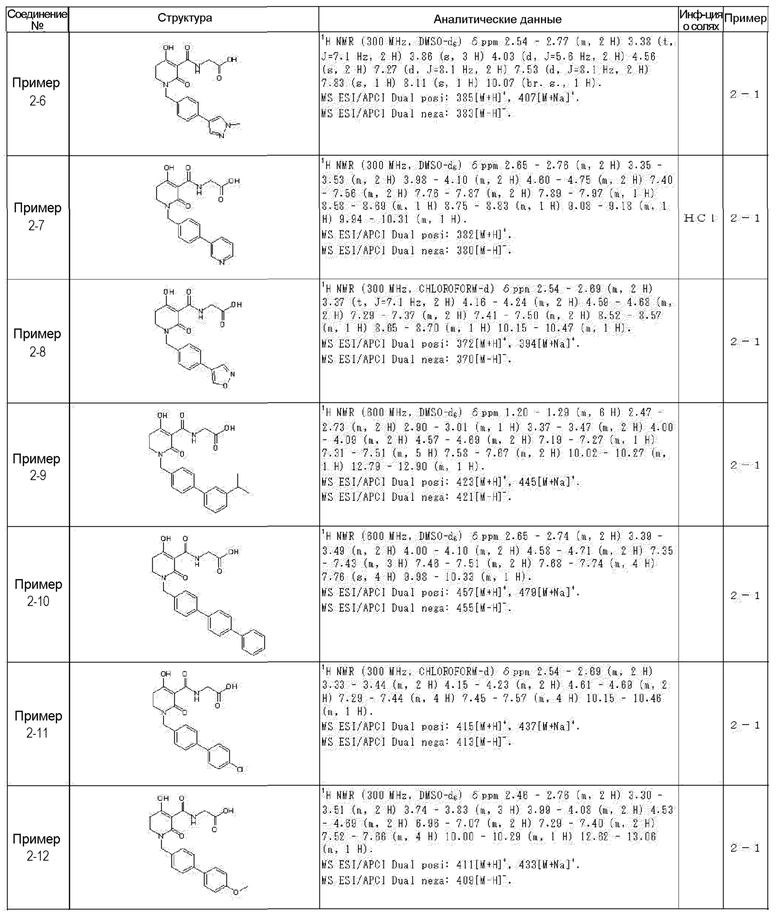
[0560]

[0561]

[0562]

[0563]
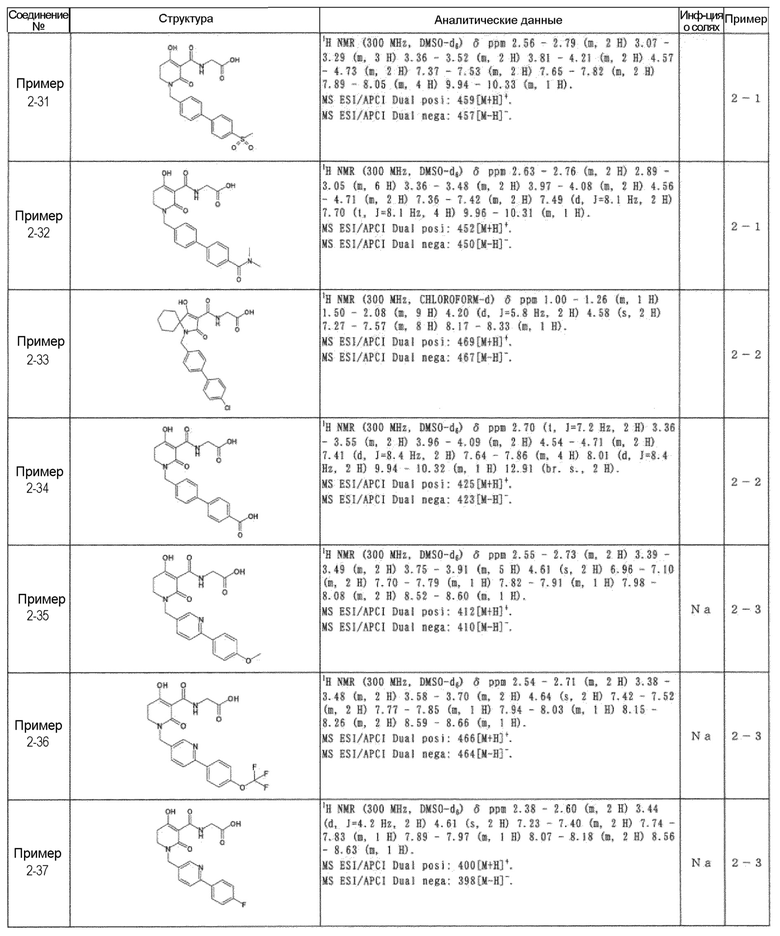
[0564]

[0565]
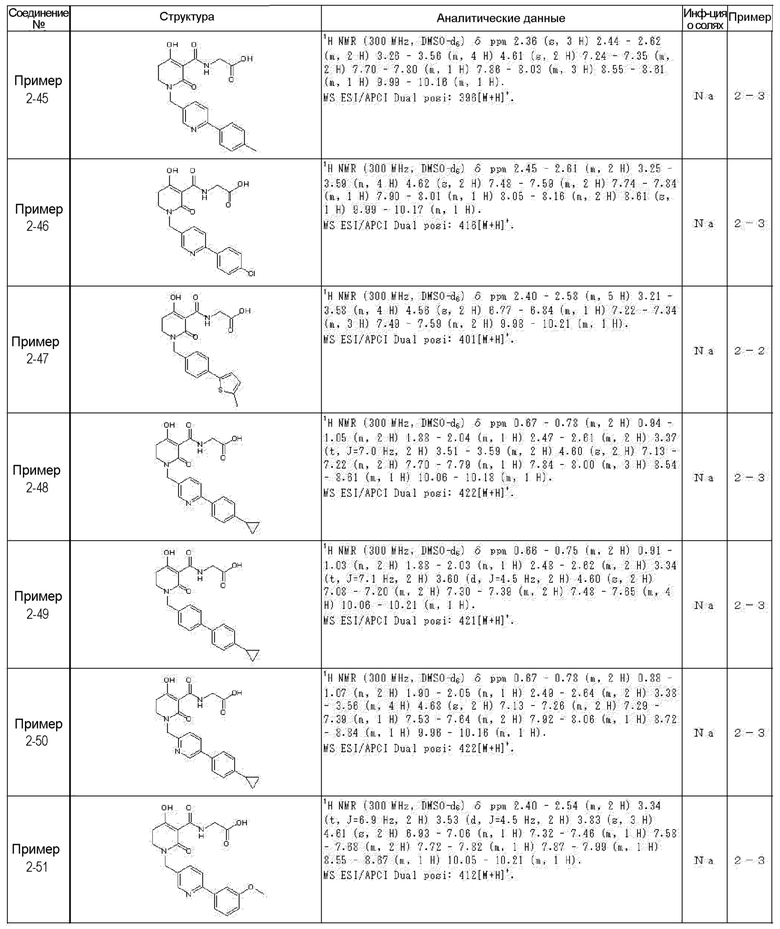
[0566]
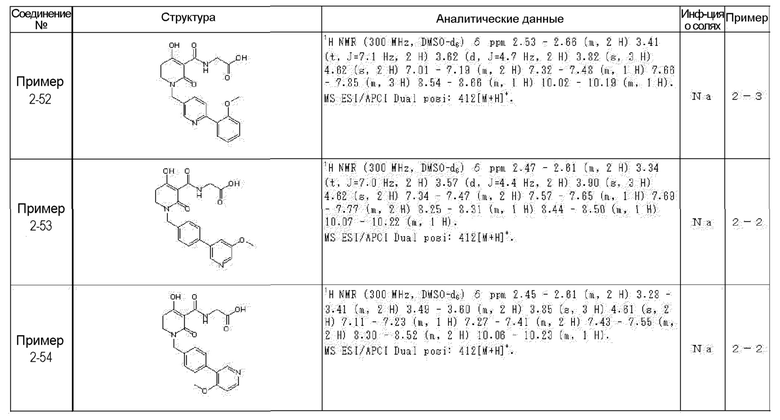
Пример 2-55
N-({4-гидрокси-2-оксо-1-[(4'-сульфамоил-4-бифенилил)метил]-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин
[0567]
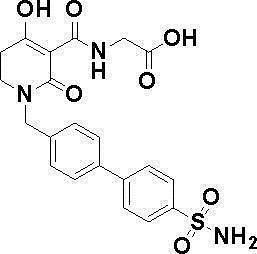
(1) Синтез N-{[4-гидрокси-(4-йодбензил)-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицина
[0568]

В соединение (5,00 г), полученное в примере 2-1(1), добавляли раствор (50 мл) хлороводорода в 1,4-диоксане 4 моль/л и смесь перемешивали при 70°C в течение трех часов. Реакционную смесь охлаждали льдом и получаемый осадок собирали с помощью фильтрования для того, чтобы получать N-{[4-гидрокси-1-(4-йодбензил)-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин в виде бесцветного твердого вещества (3,37 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,65 (ушир. с, 2H) 3,37 (т, J=7,1 Гц, 2H) 4,02 (д, J=5,8 Гц, 2H) 4,53 (с, 2H) 7,11 (д, J=8,4 Гц, 2H) 7,62-7,81 (м, 2H) 10,03 (ушир. с, 1H).
MS ESI/APCI двойн. положительн.: 453[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 429[M-H]-.
(2) Синтез указанного в заголовке соединения
Использовали соединение (100 мг), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в примере 2-2(1), для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (45,9 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,65-2,75 (м, 2H) 3,37-3,49 (м, 2H) 3,96-4,11 (м, 2H) 4,59-4,71 (м, 2H) 7,33-7,44 (м, 4H) 7,68-7,76 (м, 2H) 7,81-7,93 (м, 4H) 9,88-10,33 (м, 1H) 12,85 (ушир. с, 1H).
MS ESI/APCI двойн. положительн.: 460[M+H]+.
MS ESI/APCI двойн. отрицательн.: 458[M-H]-.
[0569] Соединения из следующих примеров с 2-56 до 2-61 синтезировали из соответствующих аналогов бороновых кислот торговых сортов способом, описанным в примере 2-55, или его модификаций. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблице 22-9.
[0570]

Пример 3-1
N-{[1-(4-циклопентилбензил)-4-гидрокси-2-оксо-1,2,5,6-тетрагидропиридин-3-ил]карбонил}глициновая соль натрия
[0571]

(1) Синтез трет-бутил N-{[1-(4-циклопентилбензил)-4-гидрокси-2-оксо-1,2,5,6-тетрагидропиридин-3-ил]карбонил}глицината
[0572]
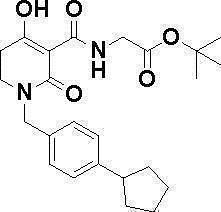
Хлорид индия(III) (768 мг) сушили при пониженном давлении и при нагревании тепловой пушкой. После того, как оставляли остывать до комнатной температуры, реакционную систему изнутри продували аргоном. В реакционную систему добавляли дегидратированный тетрагидрофуран (20,0 мл) и при перемешивании при -80°C по каплям добавляли бромид циклопентилмагния (2,0 моль/л, раствор в диэтиловом эфире, 5,23 мл). После перемешивания при этой температуре в течение 30 минут, реакционную смесь доводили до комнатной температуры и перемешивали в течение 45 минут. Добавляли соединение (1,50 г), полученное в примере 2-1(1), и бис(три-трет-бутилфосфин)палладий(0) (788 мг) и смесь перемешивали при внешней температуре 75°C в течение двух часов. После того, как оставляли остывать до комнатной температуры, добавляли метанол (5,00 мл) и смесь перемешивали в течение 30 минут. После концентрирования при пониженном давлении, добавляли хлороформ (30,0 мл) и нерастворимое вещество удаляли посредством пропускания через Celite (зарегистрированный товарный знак). Фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали с помощью препаративной ВЭЖХ и дополнительно очищали два раза колоночной хроматографией на силикагеле (хлороформ:этилацетат = 100:0-85:15) для того, чтобы получать трет-бутил N-{[1-(4-циклопентилбензил)-4-гидрокси-2-оксо-1,2,5,6-тетрагидропиридин-3-ил]карбонил}глицинат в виде масла бледно-коричневого цвета (600 мг).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,49 (с, 9H) 1,51-1,61 (м, 2H) 1,64-1,73 (м, 2H) 1,75-1,84 (м, 2H) 2,01-2,10 (м, 2H) 2,48-2,62 (м, 2H) 2,93-3,03 (м, 1H) 3,28-3,38 (м, 2H) 3,97-4,06 (м, 2H) 4,58 (с, 2H) 7,14-7,24 (м, 4H) 10,19-10,46 (м, 1H).
MS ESI/APCI двойн. положительн.: 451[M+Na]+.
(2) Синтез указанного в заголовке соединения
В качестве исходного материала использовали соединение (590 мг), полученное на приведенной выше стадии (1), и обработку проводили с помощью тех же способов, что и в примере 1-3(2) и (3), для того, чтобы получать указанное в заголовке соединение в виде твердого вещества оранжевого цвета (118 мг).
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 1,46-1,55 (м, 2H) 1,58-1,68 (м, 2H) 1,70-1,80 (м, 2H) 1,93-2,03 (м, 2H) 2,48-2,60 (м, 2H) 2,89-2,98 (м, 1H) 3,27-3,34 (м, 2H) 3,62-3,68 (м, 2H) 4,52 (с, 2H) 7,13-7,25 (м, 4H) 10,01-10,22 (м, 1H).
MS ESI положительн.: 373[M+H]+.
MS ESI отрицательн.: 371 [M-H]-.
Пример 3-2
N-{[1-(4-циклопропилбензил)-4-гидрокси-2-оксо-1,2,5,6-тетрагидропиридин-3-ил]карбонил}глициновая соль натрия
[0573]
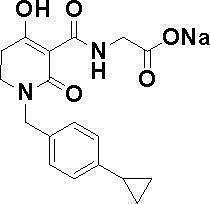
Вместо бромида циклопентилмагния (2,0 моль/л, раствор в диэтиловом эфире) использовали бромид циклопропилмагния (приблизительно 0,7 моль/л, раствор в тетрагидрофуране) и обработку проводили с помощью того же способа, как в примере 3-1, для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-серого цвета (527 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 0,60-0,67 (м, 2H) 0,88-0,96 (м, 2H) 1,81-1,95 (м, 1H) 2,47-2,54 (м, 2H) 3,20-3,30 (м, 2H) 3,47-3,54 (м, 2H) 4,50 (с, 2H) 6,97-7,08 (м, 2H) 7,10-7,19 (м, 2H) 10,08 (ушир.с, 1H).
MS ESI положительн.: 345[M+H]+, 367[M+Na]+.
Пример 4-1
N-{[1-(2,4-диметоксибензил)-4-гидрокси-2-оксо-1,2,5,6-тетрагидропиридин-3-ил]карбонил}глицин
[0574]
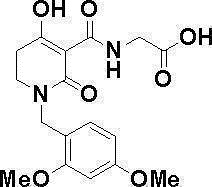
(1) Синтез этил N-(2,4-диметоксибензил)-N-(3-этокси-3-оксопропаноил)-β-аланината
[0575]
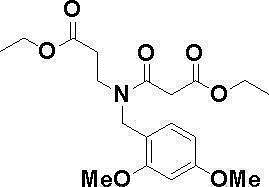
Вместо соединения, полученного в ссылочном примере A-1, в качестве исходного материала использовали соединение, полученное в ссылочном примере A-6 (8,05 г), и обработку проводили с помощью того же способа, как в примере 1-1(1), для того, чтобы получать смесь (13,0 г), содержащую этил N-(2,4-диметоксибензил)-N-(3-этокси-3-оксо-пропаноил)-β-аланинат.
MS ESI/APCI двойн. положительн.: 382[M+H]+, 404[M+Na]+.
(2) Синтез этил 1-(2,4-диметоксибензил)-4-гидрокси-2-оксо-1,2,5,6-тетрагидропиридин-3-карбоксилата
[0576]
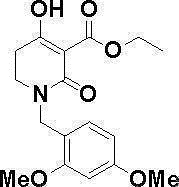
В раствор в этаноле (160 мл) смеси (13,0 г), полученной на приведенной выше стадии (1), добавляли этоксид натрия (приблизительно 20%, раствор в этаноле, 20,8 мл) и получаемую смесь перемешивали при внешней температуре 85°C в течение двух часов. После охлаждения до комнатной температуры добавляли этилацетат и соляную кислоту 2 моль/л. Проводили экстрагирование этилацетатом и объединенные органические слои промывали насыщенным солевым раствором. После сушки над безводным сульфатом магния и удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 95:5-5:95) для того, чтобы получать этил 1-(2,4-диметоксибензил)-4-гидрокси-2-оксо-1,2,5,6-тетрагидропиридин-3-карбоксилата в виде масла коричневого цвета (8,20 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,40 (т, J=7,1 Гц, 3H) 2,55 (т, J=6,6 Гц, 2H) 3,33-3,44 (м, 2H) 3,75-3,85 (м, 6H) 4,38 (кв., J=7,1 Гц, 2H) 4,57 (с, 2H) 6,37-6,50 (м, 2H) 7,20-7,26 (м, 1H).
MS ESI/APCI двойн. положительн.: 336[M+H]+.
MS ESI/APCI двойн. отрицательн.: 334[M-H]-.
(3) Синтез 2-(триметилсилил)этил N-{[1-(2,4-диметоксибензил)-4-гидрокси-2-оксо-1,2,5,6-тетрагидропиридин-3-ил]карбонил}глицината
[0577]
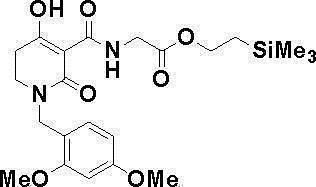
В раствор 1,2-диметоксиэтана (4,60 мл) соединения (311 мг), полученного на приведенной выше стадии (2), добавляли соединение (195 мг), полученное в ссылочном примере E-1, и смесь нагревали с обратным холодильником в течение двух часов. После охлаждения до комнатной температуры, реакционную смесь концентрировали при пониженном давлении. Очистка колоночной хроматографией на силикагеле (н-гексан:этилацетат = 95:5-65:35) давала 2-(триметилсилил)этил N-{[1-(2,4-диметоксибензил)-4-гидрокси-2-оксо-1,2,5,6-тетрагидропиридин-3-ил]карбонил}глицинат в виде масла бледно-желтого цвета (283 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. -0,03-0,01 (м, 9H) 0,97-1,07 (м, 2H) 2,48-2,62 (м, 2H) 3,33-3,43 (м, 2H) 3,76-3,84 (м, 6H) 4,03-4,13 (м, 2H) 4,22-4,29 (м, 2H) 4,52-4,59 (м, 2H) 6,41-6,50 (м, 2H) 7,11-7,23 (м, 1H).
(4) Синтез указанного в заголовке соединения
В раствор в тетрагидрофуране (3,00 мл) соединения (283 мг), полученного на приведенной выше стадии (3), добавляли гидрат фторида тетрабутиламмония (1 моль/л, раствор в тетрагидрофуране, 0,609 мл) и смесь перемешивали при комнатной температуре в течение 12 часов. Добавляли дополнительный гидрат фторида тетрабутиламмония (1 моль/л, раствор в тетрагидрофуране, 0,609 мл) и смесь перемешивали при комнатной температуре в течение 24,5 часа. Растворитель концентрировали при пониженном давлении и получаемый остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 100:0-80:20). В раствор получаемого остатка в этаноле добавляли воду и смесь перемешивали в течение 60 часов. После охлаждения до 0°C, осадок собирали с помощью фильтрования для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (163 мг).
1H ЯМР (200 МГц, ДМСО-d6) δ м.д. 2,36-2,74 (м, 2H) 3,22-3,43 (м, 2H) 3,75 (с, 3H) 3,79 (с, 3H) 3,99 (д, J=5,7 Гц, 2H) 4,37-4,52 (м, 2H) 6,35-6,63 (м, 2H) 7,07 (д, J=7,5 Гц, 1H) 9,91-10,22 (м, 1H).
MS ESI/APCI двойн. положительн.: 365[M+H]+, 387[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 363[M-H]-.
Пример 4-2
N-({4-гидрокси-2-оксо-1-[4-(проп-2-ен-1-илокси)бензил]-1,2,5,6-тетрагидропиридин-3-ил}карбонил)глицин
[0578]

Вместо соединения, полученного в ссылочном примере A-6, в качестве исходного материала использовали соединение, полученное в ссылочном примере A-7 (1,34 г), и обработку проводили с помощью того же способа, как в примере 4-1, для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (115 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,48-2,69 (м, 2H) 3,24-3,40 (м, 2H) 4,12-4,24 (м, 2H) 4,47-4,61 (м, 4H) 5,24-5,34 (м, 1H) 5,35-5,47 (м, 1H) 6,05 (ддт, J=17,3, 10,5, 5,2 Гц, 1H) 6,84-6,93 (м, 2H) 7,14-7,23 (м, 2H) 10,06-10,49 (м, 1H).
MS ESI/APCI двойн. положительн.: 361[M+H]+, 383[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 359[M-H]-.
Пример 4-3
N-{[4-гидрокси-1-(4-гидроксибензил)-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин
[0579]
(1) Синтез 2-(триметилсилил)этил N-({1-[4-(аллилокси)бензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицината
[0580]

Вместо соединения, полученного в ссылочном примере A-6, в качестве исходного материала использовали соединение (1,34 г), полученное в ссылочном примере A-7, и обработку проводили с помощью тех же способов, что и в примере с 4-1(1) до (3), для того, чтобы получать 2-(триметилсилил)этил N-({1-[4-(аллилокси)бензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицинат в виде масла бледно-желтого цвета (1,16 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,05 (с, 9H) 0,97-1,09 (м, 2H) 2,45-2,64 (м, 2H) 3,25-3,37 (м, 2H) 4,05-4,16 (м, 2H) 4,23-4,32 (м, 2H) 4,50-4,56 (м, 4H) 5,24-5,34 (м, 1H) 5,35-5,47 (м, 1H) 6,05 (ддт, J=17,3, 10,6, 5,3, 5,3 Гц, 1H) 6,83-6,93 (м, 2H) 7,14-7,22 (м, 2H) 10,19-10,53 (м, 1H).
MS ESI/APCI двойн. положительн.: 461[M+H]+.
MS ESI/APCI двойн. отрицательн.: 459[M-H]-.
(2) Синтез 2-(триметилсилил)этил N-{[4-гидрокси-1-(4-гидроксибензил)-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицината
[0581]

В раствор в тетрагидрофуране (3,8 мл) соединения (520 мг), полученного на приведенной выше стадии (1), добавляли тетракис(трифенилфосфин)палладий(0) (261 мг) и морфолин (492 мкл) и смесь перемешивали при комнатной температуре в течение 22,5 часа. После концентрирования при пониженном давлении, получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 85:15-55:45) для того, чтобы получать 2-(триметилсилил)этил N-{[4-гидрокси-1-(4-гидроксибензил)-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицинат в виде масла бледно-желтого цвета (362 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,05 (с, 9H) 0,96-1,12 (м, 2H) 2,46-2,66 (м, 2H) 3,24-3,37 (м, 2H) 4,05-4,16 (м, 2H) 4,22-4,35 (м, 2H) 4,54 (с, 2H) 6,74-6,86 (м, 2H) 7,10-7,19 (м, 2H) 10,16-10,49 (м, 1H).
MS ESI/APCI двойн. положительн.: 421[M+H]+, 443[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 419[M-H]-, 455[M+Cl]-.
(3) Синтез указанного в заголовке соединения
Использовали соединение (362 мг), полученное на приведенной выше стадии (2), и обработку проводили с помощью того же способа, как в 4-1(4), для того, чтобы получать указанное в заголовке соединение в виде бесцветной аморфной массы (138 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,38-2,71 (м, 2H) 3,25-3,36 (м, 2H) 3,96-4,09 (м, 2H) 4,39-4,54 (м, 2H) 6,65-6,79 (м, 2H) 7,04-7,15 (м, 2H) 9,25-9,46 (м, 1H) 10,01-10,26 (м, 1H).
MS ESI/APCI двойн. положительн.: 343[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 319[M-H]-.
Пример 5-1 и Пример 5-2
N-{[(5S)-1-(бифенил-4-илметил)-4-гидрокси-5-метил-2-оксо-1,2,5,6-тетрагидропиридин-3-ил]карбонил}глициновая соль натрия (пример 5-1)
N-{[(5R)-1-(бифенил-4-илметил)-4-гидрокси-5-метил-2-оксо-1,2,5,6-тетрагидропиридин-3-ил]карбонил}глициновая соль натрия (пример 5-2)
[0582]

и
Соединение (2,00 г), полученное в примере 1-22, выделяли и очищали с помощью оптической препаративной ВЭЖХ для того, чтобы получать менее полярный изомер (956 мг, 99,9%ee) и более полярный изомер (861 мг, 94,1%ee). В раствор получаемого менее полярного изомера (956 мг) в ацетоне добавляли гидроксид натрия в водном растворе 1 моль/л (2,42 мл). Получаемый осадок собирали с помощью фильтрования для того, чтобы получать указанное в заголовке соединение из примера 5-1 в виде бесцветного твердого вещества (560 мг, 99,9%ee). Указанный выше более полярный изомер (861 мг) аналогичным образом обрабатывали с использованием гидроксида натрия в водном растворе 1 моль/л (2,18 мл) для того, чтобы получать указанное в заголовке соединение из примера 5-2 в виде бесцветного твердого вещества (393 мг, 98,7%ee). Соединения из примеров 5-1 и 5-2 соответственно превращали в соединения, описанные в ссылочных примерах X-1 и X-2, которые приведены далее в настоящем документе, и с помощью рентгеновской кристаллографии определяли абсолютную конфигурацию в положении 5 2-оксо-1,2,5,6-тетрагидропиридинового кольца каждого изомера.
N-{[(5S)-1-(бифенил-4-илметил)-4-гидрокси-5-метил-2-оксо-1,2,5,6-тетрагидропиридин-3-ил]карбонил}глициновая соль натрия (пример 5-1)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 1,03 (д, J=6,8 Гц, 3H) 2,55-2,74 (м, 1H) 2,96-3,12 (м, 1H) 3,37-3,47 (м, 1H) 3,53 (д, J=4,5 Гц, 2H) 4,45-4,73 (м, 2H) 7,29-7,40 (м, 3H) 7,41-7,50 (м, 2H) 7,59-7,70 (м, 4H) 10,18 (ушир.с, 1H).
MS ESI/APCI двойн. положительн.: 395[M+H]+.
MS ESI/APCI двойн. отрицательн.: 393 [M-H]-.
Время удержания в оптической ВЭЖХ: 9,136 мин.
N-{[(5R)-1-(бифенил-4-илметил)-4-гидрокси-5-метил-2-оксо-1,2,5,6-тетрагидропиридин-3-ил]карбонил}глициновая соль натрия (пример 5-2)
1H ЯМР (300 МГц, ДМСО-d6) д м.д. 1,03 (д, J=6,8 Гц, 3H) 2,56-2,74 (м, 1H) 2,97-3,13 (м, 1H) 3,37-3,47 (м, 1H) 3,53 (д, J=4,5 Гц, 2H) 4,45-4,73 (м, 2H) 7,29-7,40 (м, 3H) 7,40-7,50 (м, 2H) 7,59-7,72 (м, 4H) 10,18 (ушир.с, 1H).
MS ESI/APCI двойн. положительн.: 395[M+H]+.
MS ESI/APCI двойн. отрицательн.: 393[M-H]-.
Время удержания в оптической ВЭЖХ: 9,705 мин.
Пример 6-1
N-{[1-(4-бифенилилметил)-4-гидрокси-2-оксо-1-азаспиро[4,4]нон-3-ен-3-ил]карбонил}глицин
[0583]

(1) Синтез метил 1-{(4-бифенилилметил)[3-({2-[(2-метил-2-пропанил)окси]-2-оксоэтил}амино)-3-оксопропаноил]амино}циклопентанкарбоксилата

Вместо монобензилмалоната и глицин трет-бутил гидрохлорида соответственно использовали соединение (300 мг), полученное в ссылочном примере G-1, и соединение (513 мг), полученное в ссылочном примере A-306, и обработку проводили с помощью того же способа, как в ссылочном примере G-1(1), для того, чтобы получать метил 1-{(4-бифенилилметил)[3-({2-[(2-метил-2-пропанил)окси]-2-оксоэтил}амино)-3-оксопропаноил]амино}циклопентанкарбоксилат в виде бесцветного твердого вещества (478 мг).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,47 (с, 9H) 1,60-1,68 (м, 2H) 1,71-1,80 (м, 2H) 1,84-1,92 (м, 2H) 2,44-2,50 (м, 2H) 3,32 (с, 2H) 3,76 (с, 3H) 3,93 (д, J=5,8 Гц, 2H) 4,73 (с, 2H) 7,33-7,47 (м, 5H) 7,56-7,63 (м, 4H) 7,81-7,88 (м, 1H).
MS ESI положительн.: 531[M+Na]+.
(2) Синтез указанного в заголовке соединения
В раствор в этаноле (5,31 мл) соединения (270 мг), полученного на приведенной выше стадии (1), добавляли карбонат цезия (346 мг) и смесь перемешивали при комнатной температуре в течение часа. Реакционную смесь концентрировали при пониженном давлении и в получаемый остаток добавляли соляную кислоту 1 моль/л. Проводили экстрагирование этилацетатом и объединенные органические слои сушили над безводным сульфатом натрия. После удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. В раствор получаемого концентрата (253 мг) в хлороформе (4,88 мл) добавляли трифторуксусную кислоту (2,44 мл) и смесь перемешивали при комнатной температуре в течение 15 часов. После концентрирования при пониженном давлении, остаток перекристаллизовывали с жидкой смесью N-гексана и этилацетата и осадок собирали с помощью фильтрования для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (335 г).
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 1,67-1,84 (м, 6H) 1,84-1,92 (м, 2H) 3,97-4,02 (м, 2H) 4,58 (с, 2H) 7,33-7,40 (м, 3H) 7,43-7,48 (м, 2H) 7,61-7,67 (м, 4H) 8,34-8,41 (м, 1H).
MS ESI/APCI двойн. положительн.: 421[M+H]+.
MS ESI/APCI двойн. отрицательн.: 419[M-H]-.
Пример 6-2
N-({(6S)-1-[4-(циклопропилметокси)бензил]-4-гидрокси-6-метил-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глициновая соль натрия
[0584]
В раствор соединения (400 мг) из ссылочного примера G-3 в этилацетате (7,07 мл) добавляли соединение из ссылочного примера A-307 (392 мг), триэтиламин (428 мг) и ангидрид пропилфосфоновой кислоты (циклический тример) (50%, раствор в этилацетате, 1,35 г) и смесь перемешивали при комнатной температуре в течение двух часов. В реакционную смесь добавляли насыщенный водный раствор бикарбоната натрия и проводили экстрагирование этилацетатом. Объединенные органические слои сушили над безводным сульфатом натрия. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. В раствор получаемого остатка (708 мг) в этаноле (7,07 мл) добавляли карбонат цезия (919 мг) и смесь перемешивали при комнатной температуре в течение двух часов. Реакционную смесь концентрировали при пониженном давлении и в получаемый остаток добавляли соляную кислоту 1 моль/л. Проводили экстрагирование хлороформом и объединенные органические слои сушили над безводным сульфатом натрия. После удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. В раствор получаемого остатка (650 мг) в метаноле (2,82 мл) добавляли гидроксид натрия в водном растворе 2 моль/л (1,41 мл) и смесь перемешивали при комнатной температуре в течение 15 часов. В реакционную смесь добавляли соляную кислоту 2 моль/л и после получаемую смесь растворяли в N,N-диметилформамиде, очистку проводили с помощью препаративной ВЭЖХ. В раствор получаемого очищенного продукта (170 мг) в ацетоне (4,37 мл) добавляли гидроксид натрия в водном растворе 1 моль/л (437 мкл) и смесь перемешивали при комнатной температуре в течение 10 минут. Реакционную смесь концентрировали при пониженном давлении и восстанавливали до порошка с использованием н-гексана, что давало указанное в заголовке соединение в виде бесцветного твердого вещества (180 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 0,26-0,35 (м, 2H) 0,51-0,61 (м, 2H) 1,08 (д, J=6,5 Гц, 3H) 1,13-1,28 (м, 1H) 2,12-2,26 (м, 1H) 2,75-2,93 (м, 1H) 3,51-3,60 (м, 1H) 3,60-3,66 (м, 2H) 3,78 (д, J=7,0 Гц, 2H) 4,05 (д, J=14,9 Гц, 1H) 4,92 (д, J=14,9 Гц, 1H) 6,84-6,92 (м, 2H) 7,17-7,26 (м, 2H) 10,07 (ушир.с, 1H).
MS ESI/APCI двойн. положительн.: 389[M+H]+.
MS ESI/APCI двойн. отрицательн.: 387[M-H]-.
[0585] Соединения из следующих примеров с 6-3 до 6-36 синтезировали из соединений из ссылочных примеров с A-308 до A-340 или A-356 и ссылочного примера G-1, G-2 или G-3 с помощью способа, описанного в примере 6-1 или 6-2 или его модификаций. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблицах с 23-1 до 23-5.
[0586]

[0587]
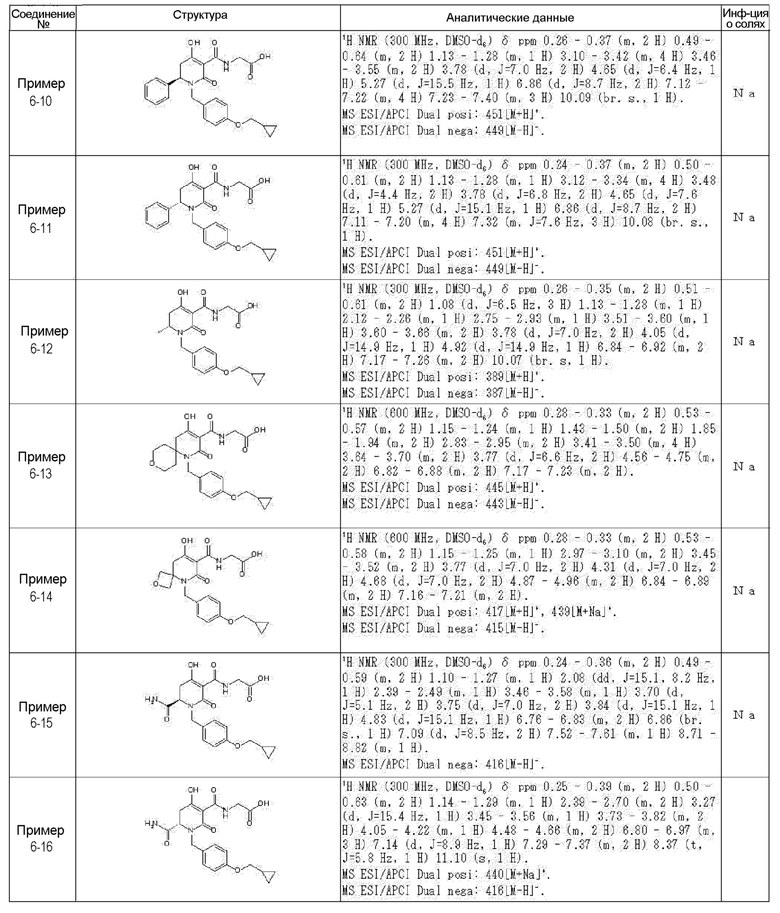
[0588]
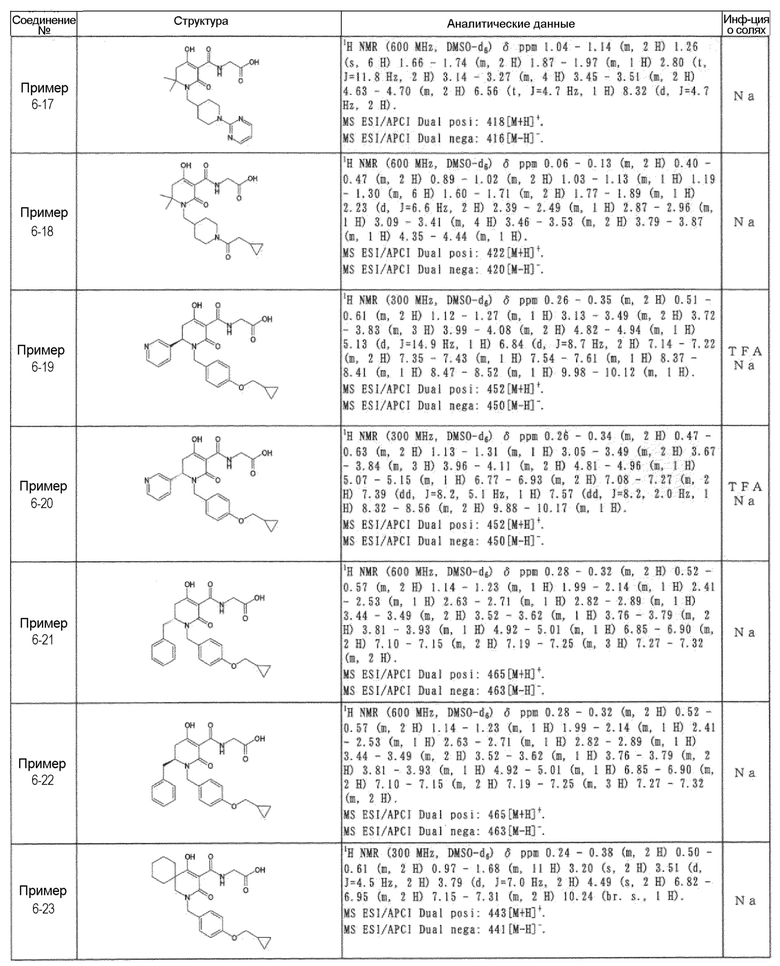
[0589]
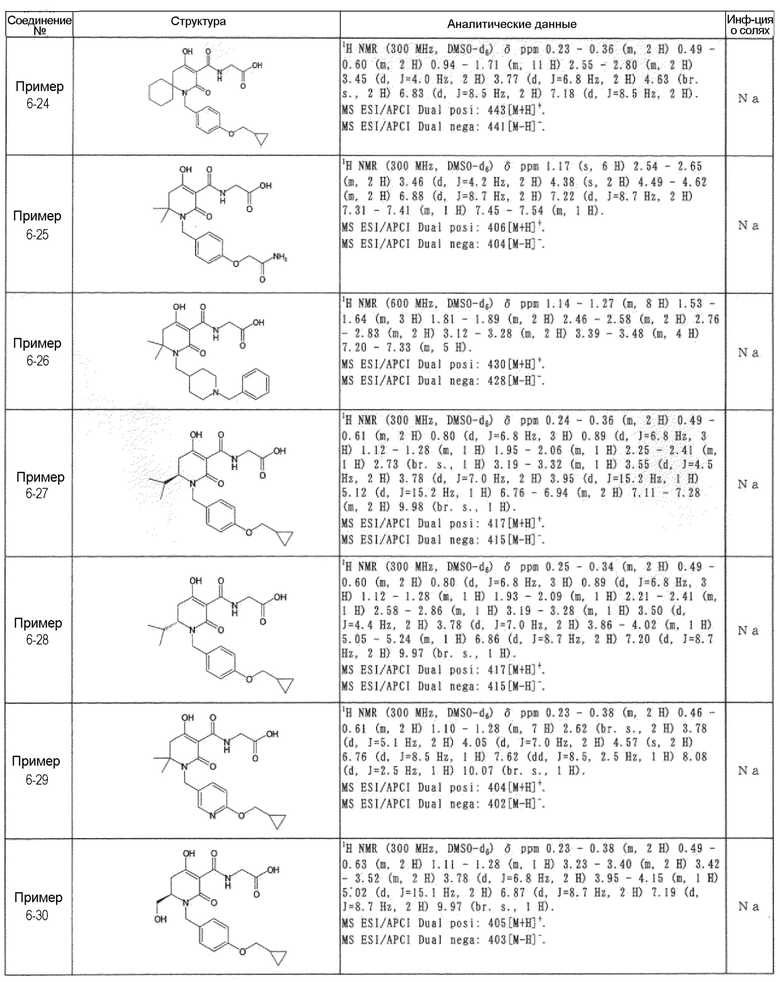
[0590]
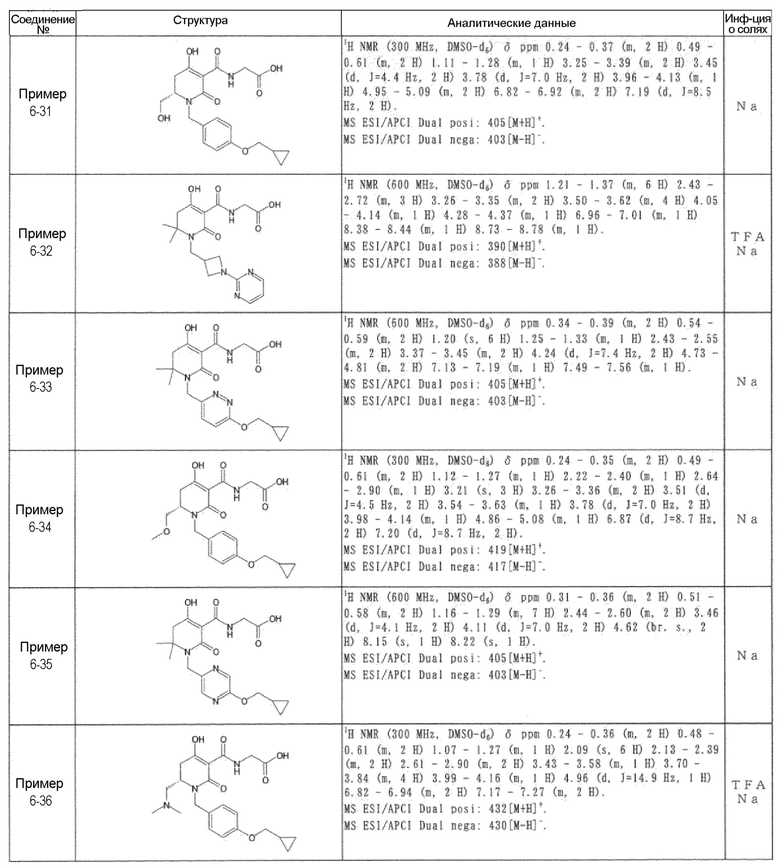
Пример 7-1
N-{[9-бензоил-1-(4-бифенилилметил)-4-гидрокси-2-оксо-1,9-диазаспиро[5.5]ундец-3-ен-3-ил]карбонил}глицин
[0591]

(1) Синтез 2-метил-2-пропанил 4-[(4-бифенилилметил)(3-этокси-3-оксопропаноил)амино]-4-(2-метокси-2-оксоэтил)-1-пиперидинкарбоксилата
[0592]
В раствор соединения из ссылочного примера A-344 (3,14 г) и триэтиламина (1,50 мл) в этилацетате (100 мл) добавляли этилмалонил хлорид (1,22 мл) при 0°C и после этого смесь перемешивали при комнатной температуре в течение 30 минут. Нерастворимое вещество удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 90:10-50:50) для того, чтобы получать 2-метил-2-пропанил 4-[(4-бифенилилметил)(3-этокси-3-оксопропаноил)амино]-4-(2-метокси-2-оксоэтил)-1-пиперидинкарбоксилат в виде бесцветной аморфной массы (3,36 г).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,26 (т, J=7,2 Гц, 3H) 1,38 (с, 9H) 1,68-1,79 (м, 2H) 2,69-2,81 (м, 2H) 2,82-2,96 (м, 2H) 3,29-3,37 (м, 2H) 3,39 (с, 2H) 3,72 (с, 3H) 3,80-4,04 (м, 2H) 4,17 (кв., J=7,2 Гц, 2H) 4,73 (с, 2H) 7,27-7,31 (м, 2H) 7,34-7,38 (м, 1H) 7,43-7,47 (м, 2H) 7,56-7,63 (м, 4H).
MS ESI/APCI двойн. положительн.: 575[M+Na]+.
(2) Синтез 3-этил 9-(2-метил-2-пропанил) 1-(4-бифенилилметил)-4-гидрокси-2-оксо-1,9-диазаспиро[5.5]ундец-3-ен-3,9-дикарбоксилата
[0593]
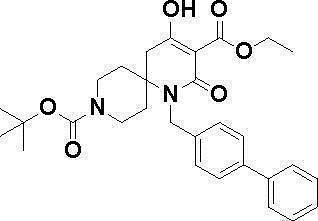
В раствор в тетрагидрофуране (70 мл) смеси (3,35 г), полученной на приведенной выше стадии (1), добавляли этоксид натрия (приблизительно 20%, раствор в этаноле, 2,5 мл) и смесь перемешивали при нагревании с обратным холодильником в течение 4 часов. После охлаждения до комнатной температуры добавляли 2 моль/л соляную кислоту и проводили экстрагирование этилацетатом. Объединенные органические слои сушили над безводным сульфатом магния. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 90:10-40:60) для того, чтобы получать 3-этил 9-(2-метил-2-пропанил) 1-(4-бифенилилметил)-4-гидрокси-2-оксо-1,9-диазаспиро[5.5]ундец-3-ен-3,9-дикарбоксилата в виде бесцветной аморфной массы (1,87 г).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,41-1,46 (м, 12H) 1,57 (с, 2H) 1,64-1,75 (м, 2H) 1,83-1,99 (м, 2H) 2,75-2,88 (м, 4H) 3,91-4,18 (м, 2H) 4,39-4,45 (м, 2H) 4,64-4,95 (м, 2H) 7,30-7,35 (м, 3H) 7,40-7,44 (м, 2H) 7,49-7,53 (м, 2H) 7,55-7,58 (м, 2H) 14,00-14,18 (м, 1H).
MS ESI/APCI двойн. положительн.: 521[M+H]+, 543[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 519[M-H]-.
(3) Синтез гидрохлорида этил 1-(4-бифенилилметил)-4-гидрокси-2-оксо-1,9-диазаспиро[5.5]ундец-3-ен-3-карбоксилата
[0594]
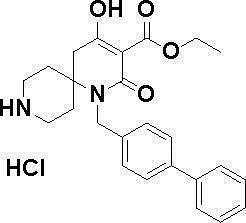
В соединение (1,70 г), полученное на приведенной выше стадии (2), добавляли раствор (10 мл) хлороводорода в 1,4-диоксане 4 моль/л и смесь перемешивали при комнатной температуре в течение 68 часов. После концентрирования при пониженном давлении, получаемый остаток кристаллизовали с использованием жидкой смеси диэтилового эфира и этилацетата для того, чтобы получать гидрохлорид этил 1-(4-бифенилилметил)-4-гидрокси-2-оксо-1,9-диазаспиро[5.5]ундец-3-ен-3-карбоксилата в виде бесцветного твердого вещества (983 мг).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,40 (т, J=7,1 Гц, 3H) 1,68-1,82 (м, 2H) 2,55-2,66 (м, 2H) 2,79-2,87 (м, 2H) 2,95-3,12 (м, 2H) 3,36-3,43 (м, 2H) 4,39 (кв., J=7,0 Гц, 2H) 4,69-5,04 (м, 2H) 7,28-7,32 (м, 1H) 7,33-7,37 (м, 2H) 7,37-7,41 (м, 2H) 7,46-7,53 (м, 4H) 9,30-9,44 (м, 1H) 9,44-9,57 (м, 1H).
MS ESI/APCI двойн. положительн.: 421[M+H]+, 443[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 419[M-H]-.
(4) Синтез этил 9-бензоил-1-(4-бифенилилметил)-4-гидрокси-2-оксо-1,9-диазаспиро[5.5]ундец-3-ен-3-карбоксилата
[0595]
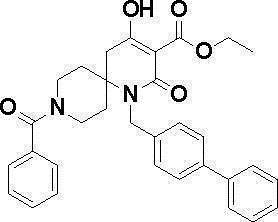
В раствор в тетрагидрофуране (6 мл) соединения (160 мг), полученного на приведенной выше стадии (3), добавляли бензоилхлорид (59 мг) и триэтиламин (120 мг) и смесь перемешивали при комнатной температуре в течение 1,5 часа. В реакционную смесь добавляли насыщенный водный раствор бикарбоната натрия и проводили экстрагирование этилацетатом. Объединенные органические слои сушили над безводным сульфатом магния. Десикант удаляли с помощью фильтрования, и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали посредством колоночной хроматографии на NH силикагеле (хлороформ:метанол = 98:2-80:20) для того, чтобы получать этил 9-бензоил-1-(4-бифенилилметил)-4-гидрокси-2-оксо-1,9-диазаспиро[5.5]ундец-3-ен-3-карбоксилат в виде бесцветного твердого вещества (56 мг).
MS ESI/APCI двойн. положительн.: 525[M+H]+.
MS ESI/APCI двойн. отрицательн.: 523 [M-H]-.
(5) Синтез 2-метил-2-пропанил N-{[9-бензоил-1-(4-бифенилилметил)-4-гидрокси-2-оксо-1,9-диазаспиро[5.5] ундец-3-ен-3-ил]карбонил}глицината
[0596]

В раствор в N,N-диметилформамиде (4,0 мл) соединения (52 мг), полученного на приведенной выше стадии (4), добавляли глицин трет-бутил гидрохлорид (29 мг) и смесь перемешивали при 90°C в течение двух часов. После охлаждения до комнатной температуры, реакционную смесь концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле для того, чтобы получать 2-метил-2-пропанил N-{[9-бензоил-1-(4-бифенилилметил)-4-гидрокси-2-оксо-1,9-диазаспиро[5.5]ундец-3-ен-3-ил]карбонил}глицинат в виде бесцветного масла (14 мг).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,45-1,50 (м, 9H) 1,61-1,90 (м, 3H) 2,02-2,17 (м, 1H) 2,75-2,95 (м, 3H) 3,02-3,20 (м, 1H) 3,55-3,75 (м, 1H) 4,00-4,06 (м, 2H) 4,68-4,90 (м, 3H) 7,27-7,46 (м, 10H) 7,51-7,59 (м, 4H) 10,14-10,41 (м, 1H),
MS ESI/APCI двойн. положительн.: 610[M+H]+, 632[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 608[M-H]-.
(6) Синтез указанного в заголовке соединения
Использовали соединение (14 мг), полученное на приведенной выше стадии (5), и обработку проводили с помощью того же способа, как в примере 1-2(2), для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (6 мг).
1H ЯМР (500 МГц, ХЛОРОФОРМ-d) δ м.д. 1,62-1,86 (м, 3H) 2,09 (с, 1H) 2,79-2,96 (м, 3H) 3,04-3,17 (м, 1H) 3,57-3,74 (м, 1H) 4,14-4,22 (м, 2H) 4,63-4,95 (м, 3H) 7,27-7,47 (м, 10H) 7,50-7,61 (м, 4H) 10,16-10,37 (м, 1H),
MS ESI/APCI двойн. положительн.: 554[M+H]+, 576[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 552[M-H]-.
Пример 7-2
Трифторацетат N-{[9-бензил-1-(4-бифенилилметил)-4-гидрокси-2-оксо-1,9-диазаспиро[5.5]ундец-3-ен-3-ил]карбонил}глицина
[0597]

(1) Синтез этил 9-бензил-1-(4-бифенилилметил)-4-гидрокси-2-оксо-1,9-диазаспиро[5.5]ундец-3-ен-3-карбоксилата
[0598]

В раствор в хлороформе (3 мл) соединения (128 мг), полученного в примере 7-1(3), последовательно добавляли триэтиламин (77 мг), N,N-диметилформамид (1 мл) и бензилбромид (63 мг) и смесь перемешивали при 50°C в течение двух часов. После охлаждения реакционной смеси до комнатной температуры, в нее добавляли воду. Проводили экстрагирование хлороформом и объединенные органические слои концентрировали при пониженном давлении. Получаемый остаток очищали посредством колоночной хроматографии на NH силикагеле (хлороформ:метанол = 97:3-80:20) для того, чтобы получать этил 9-бензил-1-(4-бифенилилметил)-4-гидрокси-2-оксо-1,9-диазаспиро[5.5]ундец-3-ен-3-карбоксилат в виде бесцветного твердого вещества (32 мг).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,41 (т, J=7,1 Гц, 3H) 1,60-1,69 (м, 2H) 2,02-2,15 (м, 4H) 2,71-2,85 (м, 4H) 3,48 (с, 2H) 4,39 (кв., J=7,1 Гц, 2H) 4,72-4,96 (м, 2H) 7,23-7,28 (м, 3H) 7,28-7,34 (м, 5H) 7,39-7,43 (м, 2H) 7,46-7,58 (м, 4H).
MS ESI/APCI двойн. положительн.: 511[M+H]+.
MS ESI/APCI двойн. отрицательн.: 509[M-H]-.
(2) Синтез 2-метил-2-пропанил N-{[9-бензил-1-(4-бифенилилметил)-4-гидрокси-2-оксо-1,9-диазаспиро[5.5]ундец-3-ен-3-ил]карбонил}глицината
[0599]
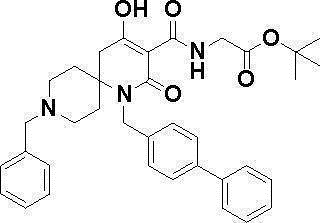
Использовали соединение (32 мг), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в примере 7-1(5), для того, чтобы получать 2-метил-2-пропанил N-{[9-бензил-1-(4-бифенилилметил)-4-гидрокси-2-оксо-1,9-диазаспиро[5.5]ундец-3-ен-3-ил]карбонил}глицинат в виде бесцветного масла (32 мг).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,46 (с, 9H) 1,60-1,67 (м, 2H) 1,99-2,08 (м, 2H) 2,09-2,19 (м, 2H) 2,70-2,82 (м, 4H) 3,30 (с, 2H) 3,98-4,06 (м, 2H) 4,75-4,86 (м, 2H) 7,22-7,27 (м, 3H) 7,27-7,35 (м, 5H) 7,39-7,44 (м, 2H) 7,48-7,58 (м, 4H) 10,18-10,40 (м, 1H).
MS ESI/APCI двойн. положительн.: 596[M+H]+.
MS ESI/APCI двойн. отрицательн.: 594[M-H]-.
(3) Синтез указанного в заголовке соединения
Использовали соединение (27 мг), полученное на приведенной выше стадии (2), и обработку проводили с помощью того же способа, как в примере 1-2(2), для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (31 мг).
1H ЯМР (600 МГц, METHANOL-d4) δ м.д. 1,95-2,03 (м, 2H) 2,18-2,28 (м, 2H) 3,00-3,27 (м, 4H) 3,33-3,39 (м, 2H) 3,58 (с, 2H) 4,07-4,13 (м, 2H) 4,26 (с, 2H) 7,27-7,37 (м, 3H) 7,37-7,41 (м, 2H) 7,42-7,49 (м, 5H) 7,53-7,59 (м, 4H).
MS ESI/APCI двойн. положительн.: 540[M+H]+.
Пример 7-3
N-[(9-бензоил-1-бензил-4-гидрокси-2-оксо-1,9-диазаспиро[5.5]ундец-3-ен-3-ил)карбонил]глицин
[0600]
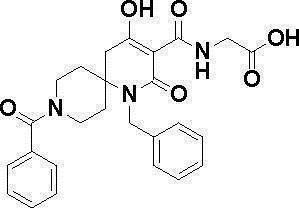
Вместо соединения, полученного в ссылочном примере A-344, использовали соединение (1,08 г), полученное в ссылочном примере A-341, и обработку проводили с помощью того же способа, как в примере 7-1, для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (25 мг).
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 1,41-1,53 (м, 1H) 1,57-1,70 (м, 1H) 1,74-1,93 (м, 2H) 2,46-2,47 (м, 2H) 2,79-2,92 (м, 2H) 2,98-3,09 (м, 2H) 3,89-4,03 (м, 2H) 4,70-4,82 (м, 2H) 7,21-7,35 (м, 7H) 7,35-7,42 (м, 3H) 9,92-10,20 (м, 1H),
MS ESI/APCI двойн. положительн.: 478[M+H]+, 500[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 476[M-H]-.
Пример 8-1
4-гидрокси-N-[2-(гидроксиамино)-2-оксоэтил]-1-(4-метоксибензил)-2-оксо-1,2,5,6-тетрагидро-3-пиридинкарбоксамид
[0601]

Вместо монобензилмалоната и глицин трет-бутил гидрохлорида соответственно использовали соединение (100 мг), полученное в примере 1-48, и гидрохлорид гидроксиламина (31 мг) и обработку проводили с помощью того же способа, как в примере G-1(1), для того, чтобы получать указанное в заголовке соединение в виде бесцветной аморфной массы (62,2 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,47-2,67 (м, 2H) 3,26-3,37 (м, 2H) 3,79 (с, 3H) 4,00-4,18 (м, 2H) 4,47-4,59 (м, 2H) 6,80-6,93 (м, 2H) 7,14-7,24 (м, 2H) 10,16-10,28 (м, 1H).
MS ESI/APCI двойн. положительн.: 350[M+H]+.
Пример 9-1
N-{[4-гидрокси-1-(4-метоксибензил)-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}-β-аланин
[0602]
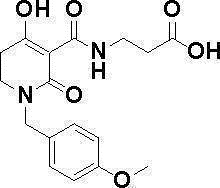
(1) Синтез 5-(этоксикарбонил)-1-(4-метоксибензил)-6-оксо-1,2,3,6-тетрагидро-4-пиридинолата натрия
[0603]

Вместо соединения, полученного в ссылочном примере A-1, использовали соединение (12,2 г), полученное в ссылочном примере A-45, и обработку проводили с помощью тех же способов, что и в примере 1-1(1) и (2), для того, чтобы получать 5-(этоксикарбонил)-1-(4-метоксибензил)-6-оксо-1,2,3,6-тетрагидро-4-пиридинолат натрия в виде твердого вещества (12,5 г).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 1,10-1,18 (м, 3H) 2,03 (т, J=6,5 Гц, 2H) 3,03 (т, J=6,5 Гц, 2H) 3,72 (с, 3H) 3,94 (кв., J=7,0 Гц, 2H) 4,40 (с, 2H) 6,79-6,93 (м, 2H) 7,04-7,27 (м, 2H).
MS ESI/APCI двойн. положительн.: 306[M+H]+, 328[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 304[M-H]-.
(2) Синтез 2-метил-2-пропанил N-{[4-гидрокси-1-(4-метоксибензил)-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}-β-аланинат
[0604]

Соединение, полученное на приведенной выше стадии (1), и β-аланин трет-бутил гидрохлорид (334 мг) использовали вместо глицин трет-бутил гидрохлорида и обработку проводили с помощью того же способа, как в примере 1-1(3), для того, чтобы получать 2-метил-2-пропанил N-{[4-гидрокси-1-(4-метоксибензил)-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}-β-аланинат в виде масла (518 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,48 (с, 9H) 2,46-2,62 (м, 4H) 3,22-3,37 (м, 2H) 3,48-3,72 (м, 2H) 3,80 (с, 3H) 4,47-4,61 (м, 2H) 6,78-6,93 (м, 2H) 9,95-10,41 (м, 2H).
MS ESI/APCI двойн. положительн.: 427[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 403[M-H]'.
(3) Синтез указанного в заголовке соединения
Использовали соединение (518 мг), полученное на приведенной выше стадии (2), и обработку проводили с помощью того же способа, как в примере 1-1(4), для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (283 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,48-2,77 (м, 4H) 3,21-3,37 (м, 2H) 3,58-3,72 (м, 2H) 3,80 (с, 3H) 4,46-4,62 (м, 2H) 6,79-6,94 (м, 2H) 7,11-7,24 (м, 2H) 9,95-10,50 (м, 1H).
MS ESI/APCI двойн. положительн.: 349[M+H]+, 371[M+Na]+.
[0605] Соединения из следующих примеров с 9-2 до 9-4 синтезировали с использованием соответствующих аминов торговых сортов с помощью способа, описанного в примере 9-1, или его модификаций. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблице 24-1.
[0606]

Пример 9-5
N-{[4-гидрокси-1-(4-метоксибензил)-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}-N-метилглицин
[0607]
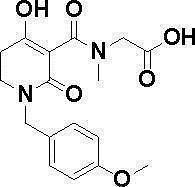
(1) Синтез 2-метил-2-пропанил N-{[4-гидрокси-1-(4-метоксибензил)-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}-N-метилглицината
[0608]

В раствор в 1,2-диметоксиэтане (5,0 мл) соединения (500 мг), полученного в примере 9-1(1), добавляли триэтиламин (215 мг) и саркозин трет-бутил гидрохлорид (345 мг) и смесь перемешивали при 50°C в течение 6 часов. После охлаждения реакционной смеси до комнатной температуры, нерастворимое вещество удаляли посредством пропускания через Celite (зарегистрированный товарный знак). Фильтрат концентрировали при пониженном давлении и получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 100:0-0:100) для того, чтобы получать 2-метил-2-пропанил N-{[4-гидрокси-1-(4-метоксибензил)-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}-N-метилглицинат в виде масла желтого цвета (347 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,48 (с, 9H) 2,49 (т, J=6,7 Гц, 2H) 3,06 (с, 3H) 3,31 (т, J=6,7 Гц, 2H) 3,80 (с, 3H) 4,00 (с, 2H) 4,57 (с, 2H) 6,81-6,91 (м, 2H) 7,14-7,25 (м, 2H).
(2) Синтез указанного в заголовке соединения
Использовали соединение (347 мг), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в примере 1-1(4), для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (116 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,18-2,31 (м, 2H) 2,80 (ушир. с, 2H) 3,14 (т, J=6,9 Гц, 2H) 3,73 (с, 3H) 4,39 (с, 2H) 6,84-6,90 (м, 2H) 7,12-7,23 (м, 2H).
Пример 10-1
N-[(1-{2-[(4-бифенилилметил)амино]-2-оксоэтил}-4-гидрокси-6,6-диметил-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глициновая соль натрия
[0609]

(1) Синтез 2-метил-2-пропанил N-{[1-(2-{[диметил(2-метил-2-пропанил)силил]окси}этил)-4-гидрокси-6,6-диметил-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицината
[0610]

Вместо соединения, полученного в ссылочном примере A-1, использовали соединение (19,7 г), полученное в ссылочном примере A-342, и обработку проводили с помощью тех же способов, как в примере с 1-1(1) до (3) для того, чтобы получать 2-метил-2-пропанил N-{[1-(2-{[диметил(2-метил-2-пропанил)силил]окси}этил)-4-гидрокси-6,6-диметил-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицинат в виде твердого вещества бледно-коричневого цвета (10,9 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,04-0,09 (м, 6H) 0,86-0,93 (м, 9H) 1,31-1,37 (м, 6H) 1,48 (с, 9H) 2,40-2,63 (м, 2H) 3,40-3,58 (м, 2H) 3,64-3,84 (м, 2H) 3,94-4,09 (м, 2H) 10,14-10,39 (м, 1H).
MS ESI/APCI двойн. положительн.: 457[M+H]+.
(2) Синтез 2-метил-2-пропанил N-{[4-гидрокси-1-(2-гидроксиэтил)-6,6-диметил-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицината
[0611]
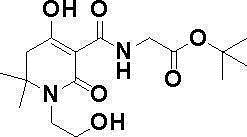
Использовали соединение (5,25 г), полученное на приведенной выше стадии (1), и обработку проводили с помощью того же способа, как в примере 4-1(4), для того, чтобы получать 2-метил-2-пропанил N-{[4-гидрокси-1-(2-гидроксиэтил)-6,6-диметил-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицинат в виде масла бледно-желтого цвета (3,90 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,34 (с, 6H) 1,49 (с, 9H) 2,49-2,64 (м, 2H) 3,55-3,66 (м, 2H) 3,71-3,84 (м, 2H) 3,98-4,06 (м, 2H) 9,92-10,55 (м, 1H).
MS ESI/APCI двойн. положительн.: 343[M+H]+.
MS ESI/APCI двойн. отрицательн.: 341 [M-H]-.
(3) Синтез 2-метил-2-пропанил N-{[4-гидрокси-6,6-диметил-2-оксо-1-(2-оксоэтил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицината
[0612]
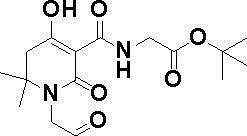
Использовали соединение (3,90 г), полученное на приведенной выше стадии (2), и обработку проводили с помощью того же способа, как в ссылочном примере 19-1, для того, чтобы получать 2-метил-2-пропанил N-{[4-гидрокси-6,6-диметил-2-оксо-1-(2-оксоэтил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицинат в виде твердого вещества бледно-желтого цвета (2,55 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,27-1,33 (м, 6H) 1,43-1,51 (м, 9H) 2,54-2,71 (м, 2H) 3,95-4,18 (м, 4H) 9,52-9,61 (м, 1H) 9,90-10,03 (м, 1H).
MS ESI/APCI двойн. положительн.: 341[M+H]+.
MS ESI/APCI двойн. отрицательн.: 339[M-H]-.
(4) Синтез [4-гидрокси-2,2-диметил-5-({2-[(2-метил-2-пропанил)окси]-2-оксоэтил}карбамоил)-6-оксо-3,6-дигидро-1(2H)-пиридинил]уксусной кислоты
[0613]
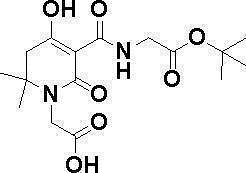
В суспензию в этаноле (40 мл) соединения (1,93 г), полученного на приведенной выше стадии (3), и нитрата серебра (1,93 г) по каплям добавляли раствор гидроксида натрия (907 мг) в воде (26 мл) при охлаждении льдом и смесь перемешивали при той же температуре в течение 20 минут. В реакционную смесь добавляли этилацетат и соляную кислоту 2 моль/л и получаемую смесь доводили до комнатной температуры. Проводили экстрагирование хлороформом и объединенные органические слои сушили над безводным сульфатом магния. После удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 100:0-85:15, затем н-гексан:этилацетат = 98:2-25:75) для того, чтобы получать [4-гидрокси-2,2-диметил-5-({2-[(2-метил-2-пропанил)окси]-2-оксоэтил}карбамоил)-6-оксо-3,6-дигидро-1(2H)-пиридинил]уксусную кислоту в виде бесцветной аморфной массы (1,79 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,31-1,38 (м, 6H) 1,45-1,52 (м, 9H) 2,54-2,70 (м, 2H) 3,97-4,06 (м, 2H) 4,11-4,22 (м, 2H) 9,79-10,54 (м, 1H).
MS ESI/APCI двойн. отрицательн.: 355[M-H]-.
(5) Синтез 2-метил-2-пропанил N-[(1-{2-[(4-бифенилилметил)амино]-2-оксоэтил}-4-гидрокси-6,6-диметил-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицината
[0614]
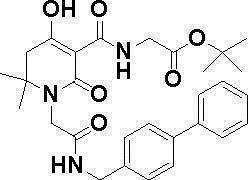
Вместо монобензилмалоната и глицин трет-бутил гидрохлорида соответственно использовали соединение (245 мг), полученное на приведенной выше стадии (4), и 4-фенилбензиламин (189 мг) и обработку проводили с помощью того же способа, как в ссылочном примере G-1(1), для того, чтобы получать 2-метил-2-пропанил N-[(1-{2-[(4-бифенилилметил)амино]-2-оксоэтил}-4-гидрокси-6,6-диметил-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицинат в виде бесцветного твердого вещества (285 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,32-1,37 (м, 6H) 1,45-1,50 (м, 9H) 2,51-2,63 (м, 2H) 3,98-4,06 (м, 2H) 4,09-4,17 (м, 2H) 4,44-4,54 (м, 2H) 6,80-6,90 (м, 1H) 7,29-7,38 (м, 3H) 7,39-7,48 (м, 2H) 7,51-7,62 (м, 4H) 9,85-10,62 (м, 1H).
MS ESI/APCI двойн. положительн.: 544[M+Na]+.
MS ESI/APCI двойн. отрицательн.: 520[M-H]-.
(6) Синтез указанного в заголовке соединения
Использовали соединение (285 мг), полученное на приведенной выше стадии (5), и обработку проводили с помощью тех же способов, что и в примере 1-3(2) и (3), для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (237 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 1,23 (с, 6H) 2,54-2,71 (м, 2H) 3,46 (д, J=4,4 Гц, 2H) 4,01 (с, 2H) 4,33 (д, J=6,1 Гц, 2H) 7,29-7,39 (м, 3H) 7,40-7,50 (м, 2H) 7,56-7,70 (м, 4H) 8,27-8,44 (м, 1H) 9,86-10,12 (м, 1H).
[0615] Соединения из следующих примеров 10-2 и 10-4 синтезировали с использованием соответствующих аминов торговых сортов с помощью способа, описанного в примере 10-1, или его модификации. Структуры синтезированных соединений и их ЯМР и MS данные представлены в таблице 25-1.
[0616] [Таблица 25-1]
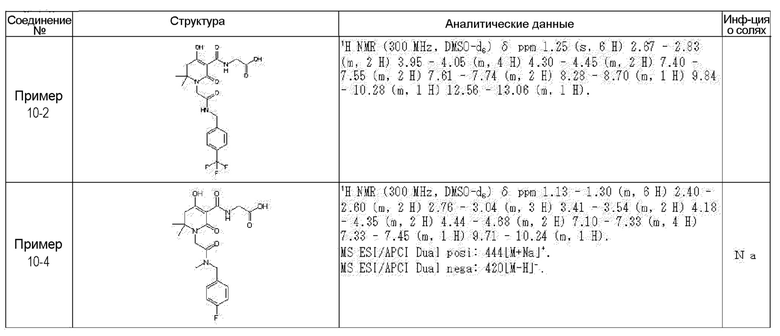
Пример 11-1
N-({1-[1-(4'-фтор-4-бифенилил)-2-гидроксиэтил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин
[0617]

(1) Синтез метил N-({1-[2-{[диметил(2-метил-2-пропанил)силил]окси}-1-(4-йодфенил)этил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицината
[0618]
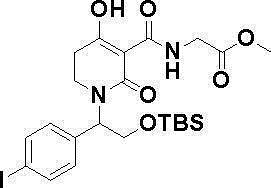
Вместо соединения, полученного в ссылочном примере A-1, и глицин трет-бутил гидрохлорида, соответственно использовали соединение (1,40 г), полученное в ссылочном примере B-19, и глицин метил гидрохлорид (442 мг) и обработку проводили с помощью тех же способов, как в примере с 1-1(1) до (3), для того, чтобы получать метил N-({1-[2-{[диметил(2-метил-2-пропанил)силил]окси}-1-(4-йодфенил)этил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицинат в виде твердого вещества бледно-коричневого цвета (1,40 г).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 0,09 (с, 6H) 0,87 (с, 9H) 2,39-2,62 (м, 2H) 3,05-3,21 (м, 1H) 3,33-3,50 (м, 1H) 3,78 (с, 3H) 3,99-4,21 (м, 4H) 5,55-5,78 (м, 1H) 7,04-7,16 (м, 2H) 7,61-7,74 (м, 2H).
MS ESI/APCI двойн. положительн.: 589[M+H]+.
MS ESI/APCI двойн. отрицательн.: 587[M-H]-.
(2) Синтез метил N-({4-гидрокси-1-[2-гидрокси-1-(4-йодфенил)этил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицината
[0619]
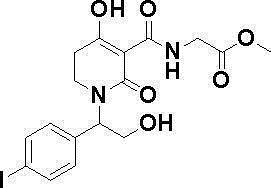
В раствор в этилацетате (67 мл) соединения (1,32 г), полученного на приведенной выше стадии (1), добавляли раствор (12,3 мл) хлороводорода в 1,4-диоксане 4 моль/л и смесь перемешивали при комнатной температуре в течение часа. Затем добавляли воду при охлаждении льдом. Проводили экстрагирование этилацетатом и органический слой промывали насыщенным солевым раствором. Промытый органический слой сушили над безводным сульфатом магния и после удаления десиканта с помощью фильтрования, фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 99:1-0:100) для того, чтобы получать метил N-({4-гидрокси-1-[2-гидрокси-1-(4-йодфенил)этил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицинат в виде аморфной массы бледно-желтого цвета (880 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,35-2,62 (м, 2H) 2,99-3,18 (м, 1H) 3,26-3,46 (м, 1H) 3,77 (с, 3H) 3,94-4,22 (м, 4H) 5,58-5,80 (м, 1H) 6,95-7,13 (м, 2H) 7,57-7,78 (м, 2H) 9,95-10,56 (м, 1H).
MS ESI/APCI двойн. положительн.: 475[M+H]+.
MS ESI/APCI двойн. отрицательн.: 473 [M-H]-.
(3) Синтез метил N-{[7-гидрокси-3-(4-йодфенил)-2,3,6,8a-тетрагидро-5H-[1,3]оксазоло[3,2-a]пиридин-8-ил]карбонил}глицината
[0620]
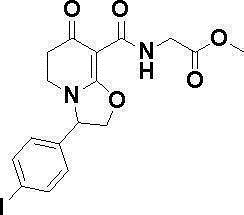
В раствор в ацетонитриле (14,8 мл) соединения (880 мг), полученного на приведенной выше стадии (2), добавляли ангидрид пропилфосфоновой кислоты (циклический тример) (50%, раствор в этилацетате, 5,91 г) и растворитель незамедлительно концентрировали при пониженном давлении. В получаемый остаток добавляли ацетонитрил (14,8 мл) и смесь перемешивали при 90°C в течение трех часов. Реакционную смесь концентрировали при пониженном давлении и в концентрат добавляли хлороформ. При этом растворитель отгоняли при пониженном давлении, неочищенный продукт адсорбировали на диатомовой земле. Неочищенный продукт, адсорбированный на диатомовой земле, очищали посредством колоночной хроматографии на NH силикагеле (хлороформ:метанол = 100:0-95:5) для того, чтобы получать метил N-{[7-гидрокси-3-(4-йодфенил)-2,3,6,8a-тетрагидро-5H-[1,3]оксазоло[3,2-a]пиридин-8-ил]карбонил}глицинат в виде бесцветного твердого вещества (690 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,51-2,74 (м, 2H) 3,11-3,37 (м, 2H) 3,74 (с, 3H) 4,12 (д, J=5,6 Гц, 2H) 4,53 (дд, J=9,2, 8,5 Гц, 1H) 4,75 (т, J=8,5 Гц, 1H) 4,97-5,22 (м, 1H) 7,02-7,12 (м, 2H) 7,73-7,83 (м, 2H) 9,59 (т, J=5,4 Гц, 1H).
MS ESI/APCI двойн. положительн.: 457[M+H]+, 479[M+Na]+.
(4) Синтез метил N-{[3-(4'-фтор-4-бифенилил)-7-гидрокси-2,3,6,8a-тетрагидро-5H-[1,3]оксазоло[3,2-a]пиридин-8-ил]карбонил}глицината
[0621]
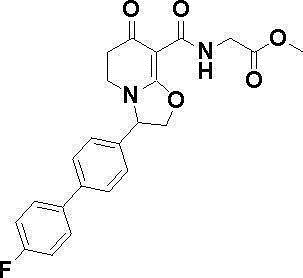
Смесь соединения (100 мг), полученного на приведенной выше стадии (3), 4-фторфенилбороновой кислоты (66 мг), ацетата палладия(II) (6,6 мг), три(2-метилфенил)фосфина (26 мг), карбоната калия (186 мг), метанола (4,4 мл) и толуола (2,2 мл) перемешивали в герметизированной пробирке при 90°C в течение 70 минут. После охлаждения до комнатной температуры реакционную смесь концентрировали при пониженном давлении. Получаемый остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 99:1-0:100, затем хлороформ:метанол = 100:0-85:15) для того, чтобы получать метил N-{[3-(4'-фтор-4-бифенилил)-7-гидрокси-2,3,6,8a-тетрагидро-5H-[1,3]оксазоло[3,2-a]пиридин-8-ил]карбонил}глицинат в виде твердого вещества бледно-коричневого цвета (100 мг).
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 2,56-2,76 (м, 2H) 3,18-3,50 (м, 2H) 3,74 (с, 3H) 4,10-4,17 (м, 2H) 4,54-4,68 (м, 1H) 4,88 (т, J=8,5 Гц, 1H) 5,06-5,22 (м, 1H) 7,09-7,22 (м, 2H) 7,36-7,47 (м, 2H) 7,50-7,66 (м, 4H) 9,69 (т, J=5,5 Гц, 1H).
MS ESI/APCI двойн. положительн.: 425[M+H]+, 447[M+Na]+.
(5) Синтез указанного в заголовке соединения
В раствор в тетрагидрофуране (2,4 мл) и метаноле (2,4 мл) соединения (100 мг), полученного на приведенной выше стадии (4), добавляли гидроксид натрия в водном растворе 1 моль/л (471 мкл) и смесь перемешивали при комнатной температуре в течение 13 часов. Осадок собирали с помощью фильтрования и растворяли в этилацетате и соляной кислоте 4 моль/л. Органический слой концентрировали при пониженном давлении и получаемый остаток очищали с помощью препаративной ВЭЖХ. В раствор получаемого очищенного продукта (45,0 мг) в ацетоне (2 мл) добавляли гидроксид натрия в водном растворе 1 моль/л (105 мкл) и смесь перемешивали при комнатной температуре. Осадок собирали с помощью фильтрования для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (40 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 2,40-2,50 (м, 2H) 3,00-3,22 (м, 1H) 3,37-3,51 (м, 2H) 3,83-4,03 (м, 2H) 5,11 (ушир. с, 1H) 5,68 (ушир. с, 1H) 7,21-7,35 (м, 2H) 7,35-7,48 (м, 2H) 7,55-7,76 (м, 4H).
MS ESI/APCI двойн. положительн.: 429[M+H]+.
MS ESI/APCI двойн. отрицательн.: 427[M-H]-.
Ссылочный пример X-1
Синтез (5S)-1-(бифенил-4-илметил)-N-{2-[(4-бромфенил)амино]-2-оксоэтил}-4-гидрокси-5-метил-2-оксо-1,2,5,6-тетрагидропиридин-3-карбоксамида и определение его абсолютной конфигурации с помощью рентгеновской кристаллографии.
[0622]
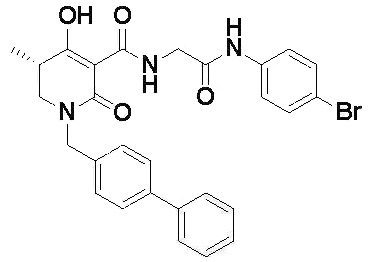
В раствор в N,N-диметилформамиде (1,00 мл) соединения (60,8 мг), полученного в примере 5-1, добавляли 4-броманилин (39,0 мг), ангидрид пропилфосфоновой кислоты (циклический тример) (50%, раствор в N,N-диметилформамиде, 135 мкл) и триэтиламин (64,0 мкл) и смесь перемешивали в течение ночи при комнатной температуре. В реакционную смесь дополнительно добавляли 4-броманилин (39,0 мг), ангидрид пропилфосфоновой кислоты (циклический тример) (50%, раствор в N,N-диметилформамиде, 135 мкл) и триэтиламин (64,0 мкл) и смесь перемешивали при комнатной температуре в течение трех часов. Реакционную смесь очищали с помощью препаративной ВЭЖХ для того, чтобы получать остаток (43,2 мг). Получаемый остаток кристаллизовали с использованием жидкой смеси н-гексана, диэтилового эфира и этилацетата для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (25,8 мг, 95,5%ee). Часть получаемого твердого вещества перекристаллизовывали с использованием жидкой смеси хлороформа и метанола и получаемый игловидный кристалл использовали в рентгеновской кристаллографии. Как результат, определяли, что 2-оксо-1,2,5,6-тетрагидропиридиновое кольцо кристалла имеет абсолютную конфигурацию (S) в положении 5.
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,09-1,24 (м, 3H) 2,69-2,86 (м, 1H) 3,10 (дд, J=12,5, 8,2 Гц, 1H) 3,44 (дд, J=12,5, 5,8 Гц, 1H) 4,11-4,18 (м, 2H) 4,60-4,73 (м, 2H) 7,30-7,39 (м, 3H) 7,40-7,48 (м, 6H) 7,53-7,62 (м, 4H) 8,14-8,24 (м, 1H) 10,46-10,58 (м, 1H).
MS ESI/APCI двойн. положительн.: 548[M+H]+.
MS ESI/APCI двойн. отрицательн.: 546[M-H]-.
Время удержания в оптической ВЭЖХ: 11,041 мин.
Ссылочный пример X-2
Синтез (5R)-1-(бифенил-4-илметил)-N-{2-[(4-бромфенил)амино]-2-оксоэтил}-4-гидрокси-5-метил-2-оксо-1,2,5,6-тетрагидропиридин-3-карбоксамида и определение его абсолютной конфигурации с помощью рентгеновской кристаллографии.
[0623]
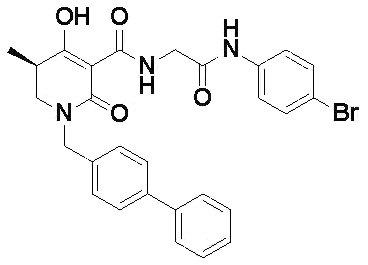
Использовали соединение (54,8 мг), полученное в примере 5-2, и обработку проводили с помощью того же способа, как в ссылочном примере X-1, для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (33,1 мг, 93,8%ee). С помощью последующей рентгеновской кристаллографии определяли, что 2-оксо-1,2,5,6-тетрагидропиридиновое кольцо кристалла имеет абсолютную конфигурацию (R) в положении 5.
1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ м.д. 1,09-1,24 (м, 3H) 2,55-2,82 (м, 1H) 3,10 (дд, J=12,5, 8,1 Гц, 1H) 3,44 (дд, J=12,5, 5,9 Гц, 1H) 4,12-4,18 (м, 2H) 4,59-4,73 (м, 2H) 7,31-7,40 (м, 3H) 7,40-7,49 (м, 6H) 7,54-7,62 (м, 4H) 7,97-8,23 (м, 1H) 10,46-10,59 (м, 1H).
MS ESI/APCI двойн. положительн.: 548[M+H]+.
MS ESI/APCI двойн. отрицательн.: 546[M-H]-.
Время удержания в оптической ВЭЖХ: 12,096 мин.
[0624] Ингибирующие PHD2 активности соединений по настоящему изобретению определяли в соответствии с тестами 1 и 2, описанными ниже.
Тест 1
[0625] (1) Экспрессия и получение PHD2 человека
PHD2 человека экспрессировали в клетках насекомых (клетки HighFive). Зарегистрированную последовательность PHD2 человека (NM_022051) вводили в вектор pFastBac1 (Invitrogen) и верифицировали последовательность. Вектор вводили в клетки насекомых Sf9 (Invitrogen) для того, чтобы получать бакуловирус с PHD2 человека. Клетки насекомых HighFive (Invitrogen) инфицировали этим рекомбинантным вирусом и культивировали при 27°C в течение 72 часов; после этого добавляли раствор для лизиса клеток, содержащий различные ингибиторы протеаз, и разрушали клетки для того, чтобы формировать суспензию. Суспензию разрушенных клеток центрифугировали при 4°C и 100000 ×g в течение 30 минут и собирали супернатант в качестве лизата клеток. Анализ с помощью вестерн-блоттинга подтверждал, что экспрессия белка PHD2 человека происходила только в лизате клеток, инфицированных PHD2 бакуловирусом.
(2) Измерение ингибирующей PHD2 человека активности
Активность фермента PHD2 человека измеряли с использованием субстрата, который представляет собой фрагмент пептида в 19 остатков, основанный на последовательности HIF-1α. В частности, использовали превращение 2-оксоглутарата в янтарную кислоту, которое происходит одновременно с гидроксилированием остатка пролина в пептиде с использованием фермента PHD2. Более конкретно, [14C]-2-оксоглутарат добавляли в реакционную систему для того, чтобы инициировать ферментативную реакцию, а [14C]-2-оксоглутарат, остающийся после реакции, связывался с 2,4-динитрофенилгидразином (DNPH), причем получаемый осадок удаляли посредством пропускания через фильтр. Впоследствии, проводили измерение излучения от получаемой [14C]-янтарной кислоты.
И фермент и субстрат разводили в 20 мМ буфере с трис-соляной кислотой (pH 7,5), содержащем 6,67 мМ KCl, 2 мМ MgCl2, 13,3 мкМ сульфата магния, 2,67 мМ аскорбиновой кислоты и 1,33 мМ DTT, а каждое тестируемое соединение разводили в диметилсульфоксиде (ДМСО).
Тестируемое соединение, пептид HIF-1α и [14C]-2-оксоглутарат предварительно добавляли в 96-луночные планшеты и инициировали реакцию посредством добавления раствора фермента PHD2 человека (4 мкг/лунка). После 15 минут инкубации при 37°C добавляли DNPH-содержащий гасящий раствор и смесь оставляли стоять при комнатной температуре в течение 30 минут. После этого добавляли избыток 2-оксоглутарата без радиоактивной метки и смесь оставляли стоять при комнатной температуре в течение 60 минут. Получаемый осадок удаляли посредством пропускания через фильтр и проводили количественное определение счетов излучения от [14C]-янтарной кислоты (с использованием MicroBeta). Счет излучения проводили для каждой лунки, и ингибирующую PHD2 человека активность каждого тестируемого соединения вычисляли на основе значений для группы, не содержащей субстрат, и группы, не содержащей тестируемое соединение.
[0626]
(3) Результаты
Данные ингибирования PHD2 человека для тестируемых соединений (%, концентрация тестируемого соединения составляла 1 мкМ) представлены в следующих таблицах с 26-1 до 26-3.
[0627]
[0628]
[0629]
Тест 2
(1) Экспрессия и получение PHD2 человека
PHD2 человека экспрессировали в клетках человека (клетки 293FT). Зарегистрированную последовательность PHD2 человека (NM_022051) вводили в вектор pcDNA3.1/Hygro(+) (Invitrogen) и верифицировали последовательность. Вектор вводили в клетки 293FT (Invitrogen), которые культивировали при 37°C в присутствии 5% газообразного CO2 в течение 48 часов; после этого добавляли раствор для лизиса клеток, содержащий различные ингибиторы протеаз, и клетки разрушали для того, чтобы формировать суспензию. Суспензию из разрушенных клеток центрифугировали при 4°C и 100000 ×g в течение 30 минут и собирали супернатант в качестве лизата клеток. Анализ с помощью вестерн-блоттинга подтверждал, что в лизате клеток экспрессирован белок PHD2 человека.
[0630] (2) Измерение ингибирующей PHD2 человека активности
Активность фермента PHD2 человека измеряли с использованием субстрата, который представляет собой фрагмент пептида в 19 остатков, основанный на последовательности HIF-1α; в частности, гидроксилирование остатка пролина в пептиде измеряли посредством FP (флуоресцентная поляризация).
И фермент и субстрат разводили в 50 мМ буфере с трим-соляной кислотой (pH 7,5), содержащем 12,5 мМ KCl, 3,75 мМ MgCl2, 25 мкМ сульфата железа, 5 мМ аскорбиновой кислоты и 2,5 мМ DTT, тогда как каждое тестируемое соединение разводили в диметилсульфоксиде (ДМСО).
Тестируемое соединение и раствор субстрата предварительно добавляли в 384-луночные планшеты, а реакцию инициировали посредством добавления раствора фермента PHD2 человека (40 нг/лунка). После 20 минут инкубации при 30°C, добавляли ЭДТА-содержащий гасящий раствор и с помощью флуоресцентной поляризации определяли количество гидроксилированных остатков пролина через связывание с добавленным раствором антитела HIF-OH.
Измеряли флуоресцентную поляризацию каждой лунки и ингибирующую PHD2 человека активность каждого тестируемого соединения вычисляли на основе значений для группы, не содержащей тестируемое соединение.
[0631]
(3) Результаты
Данные ингибирования PHD2 человека для тестируемых соединений (%, концентрация тестируемого соединения составляла 1 мкМ) представлены в следующих таблицах с 27-1 до 27-4. Для репрезентативных соединений их значения IC50 (нМ) представлены в следующей таблице 28-1.
[0632]
[0633]
[0634]
[0635]
[0636]
(нМ)
(нМ)
(нМ)
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
[0637] Соединения по настоящему изобретению обладают превосходящим ингибирующим PHD2 эффектом и с помощью настоящего изобретения становится возможным предоставлять фармацевтические средства, которые эффективны для предотвращения или лечения заболеваний, обусловленных анемией и т.п., а также ожидают, что они уменьшат бремя пациентов и, таким образом, внесут вклад в развитие фармацевтической промышленности.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФЕНИЛПИРАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ | 2008 |
|
RU2480456C2 |
| ПРОИЗВОДНОЕ 1,2,4-ТРИАЗОЛОНА | 2011 |
|
RU2566754C2 |
| АЗОЛЗАМЕЩЕННОЕ СОЕДИНЕНИЕ ПИРИДИНА | 2017 |
|
RU2730498C2 |
| НОВОЕ ПРОИЗВОДНОЕ ГИДРОКСАМОВОЙ КИСЛОТЫ | 2011 |
|
RU2575129C2 |
| ПРОИЗВОДНЫЕ ЗАМЕЩЕННОГО ТРИАЗОЛДИАМИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2001 |
|
RU2274639C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗ ДЛЯ УСИЛЕНИЯ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ СНИЖЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2822216C2 |
| ИНГИБИТОРЫ ГЛИКОЛАТОКСИДАЗЫ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2805308C2 |
| ИНГИБИТОРЫ КАТЕПСИН-ЦИСТЕИНПРОТЕАЗЫ | 2003 |
|
RU2312861C2 |
| 3-АЛКИЛИДЕНГИДРАЗИНО-ЗАМЕЩЕННЫЕ ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АКТИВАТОРОВ РЕЦЕПТОРА ТРОМБОПОЭТИНА | 2004 |
|
RU2358970C2 |
| СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ TRPM8 | 2012 |
|
RU2563030C2 |
Изобретение относится к соединению формулы (I')

(где W представляет формулу -CR11R12CR13R14-; R11 представляет атом водорода, атом фтора, С1-4 алкил или фенил; R12 представляет атом водорода, атом фтора или С1-4 алкил; при условии, что R11 и R12, вместе со смежным углеродным атомом, необязательно образуют С3-8 циклоалкан или тетрагидропиран; R13 представляет атом водорода, карбамоил, С1-4 алкил (С1-4 алкил необязательно замещен одной группой, выбранной из группы, состоящей из гидрокси, C1-3 алкокси и ди-С1-3 алкиламино), галоген-С1-4 алкил, фенил, пиридил, бензил или фенэтил; R14 представляет атом водорода, C1-4 алкил или галоген-С1-4 алкил; Y представляет одинарную связь или C1-6 алкандиил (C1-6 алкандиил необязательно замещен одной гидроксигруппой, и один из углеродных атомов в C1-6 алкандииле необязательно замещен циклоалкпропан-1,1-диилом); R2 представляет атом водорода, C1-6 алкил, С3-8 циклоалкил {С3-8 циклоалкил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и выбраны из группы, состоящей из C1-6 алкила (C1-6 алкил необязательно замещен одной группой фенила), фенила (фенил необязательно замещен одним атомом галогена), C1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила, фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила) и пиридила (пиридил необязательно замещен одним атомом галогена)], С3-8 циклоалкокси, фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, С3-8 циклоалкила и галоген-C1-6 алкила) и пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, С3-8 циклоалкила и галоген-С1-6 алкила)}, фенил (фенил необязательно замещен одной-тремя группами, которые являются одинаковыми или различными и которые выбирают из группы α3 заместителей), нафтил, инданил, тетрагидронафтил, пиразолил [пиразолил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и выбраны из группы, состоящей из C1-6 алкила и фенила (фенил необязательно замещен одним C1-6 алкилом)], имидазолил [имидазолил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и выбраны из группы, состоящей из C1-6 алкила и фенила], изоксазолил [изоксазолил необязательно замещен одной группой фенила (фенил необязательно замещен одним атомом галогена)], оксазолил [оксазолил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбраны из группы, состоящей из C1-6 алкила и фенила], тиазолил [тиазолил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из C1-6 алкила, фенила и морфолино], пиридил (пиридил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы α5 заместителей), пиридазинил [пиридазинил необязательно замещен одной группой C1-6 алкокси (C1-6 алкокси необязательно замещен одной группой С3-8 циклоалкила)], пиримидинил [пиримидинил необязательно замещен одной группой, выбранной из группы, состоящей из галоген-С1-6 алкила, С3-8 циклоалкила, фенила и фенокси (фенокси необязательно замещен одной группой C1-6 алкила)], пиразинил [пиразинил необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкокси (C1-6 алкокси необязательно замещен одним С3-8 циклоалкилом), и фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и С3-8 циклоалкила)], бензотиофенил, хинолил, метилендиоксифенил (метилендиоксифенил необязательно замещен одним или двумя атомами фтора), азетидинил (азетидинил необязательно замещен одной группой пиримидинила), пиперидинил (пиперидинил необязательно замещен одной группой, выбранной из группы, состоящей из пиримидинила, фенил-С1-3 алкила, С3-8циклоалкил-C1-3алкилкарбонила и фенил-С1-3алкоксикарбонила) или следующую формулу (I'') -CONR5CH2-R6 (I'') [где в формуле (I'') R5 представляет атом водорода или C1-3 алкил и R6 представляет фенил (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, галоген-С1-6 алкила и фенила)], Y4 представляет С1-4 алкандиил; R3 представляет атом водорода или метил; R4 представляет -СООН или -CONHOH), обладающему превосходящим ингибирующим PHD2 эффектом. 5 н. и 11 з.п. ф-лы, 28 табл., 11 пр.
1. Соединение, представленное следующей общей формулой (I')

(где в формуле (I')
W представляет формулу -CR11R12CR13R14-;
R11 представляет атом водорода, атом фтора, С1-4 алкил или фенил;
R12 представляет атом водорода, атом фтора или С1-4 алкил;
при условии, что R11 и R12, вместе со смежным углеродным атомом, необязательно образуют С3-8 циклоалкан или тетрагидропиран;
R13 представляет атом водорода, карбамоил, С1-4 алкил (С1-4 алкил необязательно замещен одной группой, выбранной из группы, состоящей из гидрокси, C1-3 алкокси и ди-С1-3 алкиламино), галоген-С1-4 алкил, фенил, пиридил, бензил или фенэтил;
R14 представляет атом водорода, C1-4 алкил или галоген-С1-4 алкил;
при условии, что R13 и R14, вместе со смежным углеродным атомом, необязательно образуют С3-8 циклоалкан, оксетан, тетрагидропиран, азетидин, пирролидин (пирролидин необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и выбраны из группы, состоящей из метила и оксо) или пиперидин (пиперидин необязательно замещен одной группой, выбранной из группы, состоящей из бензила и фенилкарбонила);
при условии, что указанные R12 и R13, вместе со смежными углеродными атомами, необязательно образуют циклогексан;
Y представляет одинарную связь или C1-6 алкандиил (C1-6 алкандиил необязательно замещен одной гидроксигруппой, и один из углеродных атомов в C1-6 алкандииле необязательно замещен циклоалкпропан-1,1-диилом);
R2 представляет атом водорода, C1-6 алкил, С3-8 циклоалкил {С3-8 циклоалкил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и выбраны из группы, состоящей из C1-6 алкила (C1-6 алкил необязательно замещен одной группой фенила), фенила (фенил необязательно замещен одним атомом галогена), C1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила, фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила) и пиридила (пиридил необязательно замещен одним атомом галогена)], С3-8 циклоалкокси, фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, С3-8 циклоалкила и галоген-C1-6 алкила) и пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, С3-8 циклоалкила и галоген-С1-6 алкила)}, фенил (фенил необязательно замещен одной-тремя группами, которые являются одинаковыми или различными и которые выбирают из группы α3 заместителей), нафтил, инданил, тетрагидронафтил, пиразолил [пиразолил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и выбраны из группы, состоящей из C1-6 алкила и фенила (фенил необязательно замещен одним C1-6 алкилом)], имидазолил [имидазолил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и выбраны из группы, состоящей из C1-6 алкила и фенила], изоксазолил [изоксазолил необязательно замещен одной группой фенила (фенил необязательно замещен одним атомом галогена)], оксазолил [оксазолил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбраны из группы, состоящей из C1-6 алкила и фенила], тиазолил [тиазолил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из C1-6 алкила, фенила и морфолино], пиридил (пиридил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы α5 заместителей), пиридазинил [пиридазинил необязательно замещен одной группой C1-6 алкокси (C1-6 алкокси необязательно замещен одной группой С3-8 циклоалкила)], пиримидинил [пиримидинил необязательно замещен одной группой, выбранной из группы, состоящей из галоген-С1-6 алкила, С3-8 циклоалкила, фенила и фенокси (фенокси необязательно замещен одной группой C1-6 алкила)], пиразинил [пиразинил необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкокси (C1-6 алкокси необязательно замещен одним С3-8 циклоалкилом), и фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и С3-8 циклоалкила)], бензотиофенил, хинолил, метилендиоксифенил (метилендиоксифенил необязательно замещен одним или двумя атомами фтора), азетидинил (азетидинил необязательно замещен одной группой пиримидинила), пиперидинил (пиперидинил необязательно замещен одной группой, выбранной из группы, состоящей из пиримидинила, фенил-С1-3 алкила, С3-8циклоалкил-C1-3алкилкарбонила и фенил-С1-3алкоксикарбонила) или следующую формулу (I'')
-CONR5CH2-R6 (I'')
[где в формуле (I'')
R5 представляет атом водорода или C1-3 алкил и R6 представляет фенил (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, галоген-С1-6 алкила и фенила)],
группа α3 заместителей состоит из гидрокси, карбокси, атома галогена, C1-6 алкила {C1-6 алкил необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила, фенила, C1-6 алкокси [C1-6 алкокси необязательно замещен одним С3-8 циклоалкилом (С3-8 циклоалкил необязательно замещен одним C1-6 алкилом)], фенокси (фенокси необязательно замещен одним C1-6 алкилом) и пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкила и галоген-C1-6 алкила)}, галоген-С1-6 алкила, С3-8 циклоалкила, С3-8 циклоалкенила (С3-8 циклоалкенил необязательно замещен одним или двумя атомами галогена), фенила (фенил необязательно замещен одной-тремя группами, которые являются одинаковыми или различными и которые выбирают из группы α4 заместителей), тиенила (тиенил необязательно замещен одним C1-6 алкилом), пиразолила (пиразолил необязательно замещен одним C1-6 алкилом), изоксазолила, тиазолила (тиазолил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из гидрокси, C1-6 алкила и C1-6 алкокси), пиридила (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из карбокси, гидрокси, амино, атома галогена, C1-6 алкила, галоген-С1-6 алкила, С3-8 циклоалкила, C1-6 алкокси и C1-6 алкилсульфонила), пиримидинила (пиримидинил необязательно замещен одной амино), хинолила, C1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из карбокси, гидрокси, карбамоила, С3-8 циклоалкила (С3-8 циклоалкил необязательно замещен одним C1-6 алкилом), фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-C1-6 алкила, C1-6 алкокси и галоген-С1-6 алкокси), пиридила (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила), оксазолила (оксазолил необязательно замещен одним или двумя C1-6 алкилами), пиразолила (пиразолил необязательно замещен одним или двумя C1-6 алкилами), тиазолила (тиазолил необязательно замещен одним C1-6 алкилом), индазолила (индазолил необязательно замещен одним C1-6 алкилом), бензотриазолила, имидазотиазолила и ди-С1-6 алкиламино], галоген-С1-6 алкокси, С2-6 алкенилокси, С3-8 циклоалкокси, фенокси (фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, C1-6 алкила и C1-6 алкокси), пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-С1-6 алкила и С3-8 циклоалкила), пиримидинилокси, пиперазинила (пиперазинил необязательно замещен одним C1-6 алкилом), моно-C1-6 алкиламинокарбонила (C1-6 алкил в моно-C1-6 алкиламинокарбониле необязательно замещен одной группой, выбранной из группы, состоящей из карбокси, гидрокси, ди-С1-6 алкиламино, пиридила и 2-оксопирролидинила), ди-С1-6 алкиламинокарбонила (где два C1-6 алкила в ди-С1-6 алкиламинокарбониле, вместе со смежным атомом азота, необязательно образуют пиперидин), C1-6 алкилсульфанила и C1-6 алкилсульфонила;
группа α4 заместителей состоит из карбокси, циано, гидрокси, сульфамоила, атома галогена, C1-6 алкила, C1-6 алкилкарбонила, ди-С1-6 алкиламинокарбонила, C1-6 алкилсульфонила, моно-C1-6 алкиламиносульфонила (C1-6 алкил в моно-C1-6 алкиламиносульфониле необязательно замещен одним гидрокси) и ди-C1-6 алкиламиносульфонила;
группа α5 заместителей состоит из атома галогена, C1-6 алкила, галоген-С1-6 алкила, C1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила (С3-8 циклоалкил необязательно замещен одним C1-6 алкилом) и фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила)], галоген-С1-6 алкокси, фенила (фенил необязательно замещен одной группой, выбранной из группы α6 заместителей), пиридила, фенокси [фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, циано, C1-6 алкила, галоген-С1-6 алкила, С3-8 циклоалкила, C1-6 алкокси и галоген-С1-6 алкокси], пиридилокси (пиридилокси необязательно замещен одним C1-6 алкилом) и фенилсульфанила (фенилсульфанил необязательно замещен одним атомом галогена);
группа α6 заместителей состоит из атома галогена, C1-6 алкила, галоген-С1-6 алкила, С3-8 циклоалкила, C1-6 алкокси и галоген-С1-6 алкокси;
Y4 представляет С1-4 алкандиил;
R3 представляет атом водорода или метил;
R4 представляет -СООН или -CONHOH);
или его фармацевтически приемлемая соль.
2. Соединение по п. 1, где в указанной выше общей формуле (I'),
Y4 представляет собой метандиил,
R3 представляет собой атом водорода,
R4 представляет собой -СООН,
или его фармацевтически приемлемая соль.
3. Соединение по п. 2, где соединение представлено общей формулой (I'-2):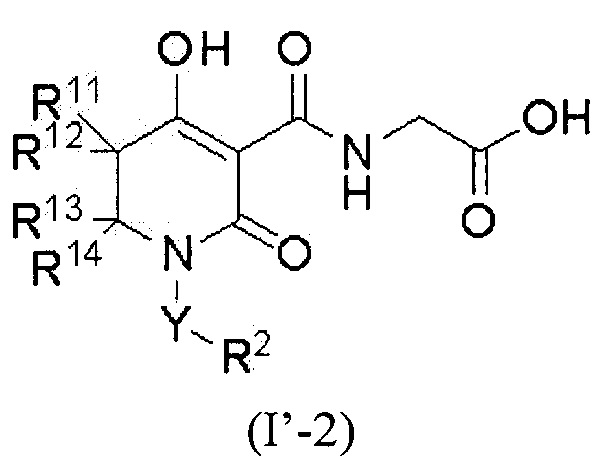
(где в формуле (I'-2)
R11 представляет собой атом водорода, атом фтора, C1-4 алкил или фенил,
R12 представляет собой атом водорода, атом фтора или C1-4 алкил,
при условии, что R11 и R12, вместе со смежным углеродным атомом, необязательно образуют С3-8 циклоалкан или тетрагидропиран;
R13 представляет собой атом водорода, карбамоил, C1-4 алкил (C1-4 алкил необязательно замещен одной группой, выбранной из группы, состоящей из гидрокси, C1-3 алкокси и ди-С1-3 алкиламино), галоген-С1-4 алкил, фенил, пиридил, бензил или фенэтил;
R14 представляет собой атом водорода, C1-4 алкил или галоген-С1-4 алкил,
при условии, что R13 и R14, вместе со смежным углеродным атомом, необязательно образуют С3-8 циклоалкан, оксетан, тетрагидропиран, азетидин, пирролидин (пирролидин необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и выбраны из группы, состоящей из метила и оксо) или пиперидин (пиперидин необязательно замещен одной группой, выбранной из группы, состоящей из бензила и фенилкарбонила),
при условии, что указанные выше R12 и R13, вместе со смежными углеродными атомами, необязательно образуют циклогексан),
или его фармацевтически приемлемая соль.
4. Соединение по п. 3, где в указанной выше общей формуле (I'-2)
Y представляет собой одинарную связь или C1-6 алкандиил (один из углеродных атомов в C1-6 алкандииле необязательно замещен циклопропан-1,1-диилом),
R2 представляет собой С3-8 циклоалкил {С3-8 циклоалкил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из C1-6 алкила (C1-6 алкил необязательно замещен одним фенилом), фенила, C1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила, фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила) и пиридила (пиридил необязательно замещен одним атомом галогена)], С3-8 циклоалкокси, фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, С3-8 циклоалкила и галоген-С1-6 алкила) и пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, С3-8 циклоалкила и галоген-С1-6 алкила)}, фенил (фенил необязательно замещен одной-тремя группами, которые являются одинаковыми или различными и которые выбирают из указанной выше группы α3 заместителей), нафтил, инданил, тетрагидронафтил, пиразолил [пиразолил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из C1-6 алкила и фенила (фенил необязательно замещен одним C1-6 алкилом)], имидазолил (имидазолил необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкила и фенила), изоксазолил [изоксазолил необязательно замещен одним фенилом (фенил необязательно замещен одним атомом галогена)], оксазолил (оксазолил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из C1-6 алкила и фенила), тиазолил (тиазолил необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкила, фенила и морфолино), пиридил (пиридил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из указанной выше группы α5 заместителей), пиридазинил [пиридазинил необязательно замещен одной C1-6 алкокси (C1-6 алкокси необязательно замещен одним С3-8 циклоалкилом)], пиримидинил [пиримидинил необязательно замещен одной группой, выбранной из группы, состоящей из галоген-С1-6 алкила, С3-8 циклоалкила, фенила и фенокси (фенокси необязательно замещен одним C1-6 алкилом)], пиразинил [пиразинил необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкокси (C1-6 алкокси необязательно замещен одним С3-8 циклоалкилом) и фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и С3-8 циклоалкила)], бензотиофенил, хинолил или метилендиоксифенил (метилендиоксифенил необязательно замещен одним или двумя атомами фтора),
или его фармацевтически приемлемая соль.
5. Соединение по п. 4, где в указанной выше общей формуле (I'-2)
R11 представляет собой атом водорода,
R12 представляет собой атом водорода,
R13 представляет собой атом водорода,
R14 представляет собой атом водорода,
Y представляет собой метандиил,
R2 представляет собой
фенил {фенил замещен одной группой, выбранной из группы, состоящей из фенила [фенил необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из карбокси, циано, гидрокси, сульфамоила, атома галогена, C1-6 алкила, галоген-С1-6 алкила, С3-8 циклоалкила, фенила, C1-6 алкокси, C1-6 алкилкарбонила, ди-С1-6 алкиламинокарбонила, C1-6 алкилсульфонила, моно-C1-6 алкиламиносульфонила (C1-6 алкил в моно-C1-6 алкиламиносульфониле необязательно замещен одним гидрокси) и ди-C1-6 алкиламиносульфонила], пиридила (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из карбокси, гидрокси, амино, атома галогена, C1-6 алкила, галоген-С1-6 алкила, С3-8 циклоалкила, C1-6 алкокси и C1-6 алкилсульфонила), фенокси (фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, C1-6 алкила и C1-6 алкокси) и пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-С1-6 алкила и С3-8 циклоалкила), и дополнительно может быть замещен одним атомом галогена};
пиридил {пиридил замещен одной группой, выбранной из группы, состоящей из фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила, галоген-С1-6 алкила, С3-8 циклоалкила, C1-6 алкокси и галоген-С1-6 алкокси), пиридила, фенокси [фенокси необязательно замещен одной или двумя группами, которые являются одинаковыми или различными и которые выбирают из группы, состоящей из атома галогена, циано, C1-6 алкила, галоген-С1-6 алкила, С3-8 циклоалкила, C1-6 алкокси и галоген-С1-6 алкокси] и пиридилокси (пиридилокси необязательно замещен одним C1-6 алкилом), и дополнительно может быть замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила}; или
пиразинил, который замещен одним фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и С3-8 циклоалкила),
или его фармацевтически приемлемая соль.
6. Следующее соединение по п. 1:
N-{[4-гидрокси-2-оксо-1-(4-феноксибензил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-[(4-гидрокси-1-{[6-(4-метилфенокси)-3-пиридинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-({4-гидрокси-2-оксо-1-[(6-фенокси-3-пиридинил)метил]-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-({1-[4-(4-фторфенокси)бензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-({4-гидрокси-1-[4-(4-метилфенокси)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-[(1-{[6-(4-цианофенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-({4-гидрокси-2-оксо-1-[4-(2-пиримидинилокси)бензил]-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-[(1-{[6-(4-фторфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(4-хлорфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-{[4-гидрокси-2-оксо-1-({6-[4-(трифторметил)фенокси]-3-пиридинил}метил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-[(4-гидрокси-1-{[6-(3-метилфенокси)-3-пиридинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(3-фторфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-({4-гидрокси-1-[4-(3-метилфенокси)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-({1-[4-(3-фторфенокси)бензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-[(1-{[5-(4-фторфенокси)-2-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-1-{[5-(4-метилфенокси)-2-пиридинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-({1-[4-(4-хлорфенокси)бензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-[(4-гидрокси-1-{4-[(6-метил-3-пиридинил)окси]бензил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(2-фторфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-1-{[6-(2-метилфенокси)-3-пиридинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-({1-[4-(2-фторфенокси)бензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-({4-гидрокси-1-[4-(2-метилфенокси)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-[(1-{[6-(3-хлорфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-{[4-гидрокси-2-оксо-1-({6-[3-(трифторметил)фенокси]-3-пиридинил}метил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-({4-гидрокси-1-[4-(3-метоксифенокси)бензил]-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-{[4-гидрокси-2-оксо-1-({6-[3-(трифторметокси)фенокси]-3-пиридинил}метил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-[(1-{4-[(5-фтор-2-пиридинил)окси]бензил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{4-[(5-хлор-2-пиридинил)окси]бензил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(4-циклопропилфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-1-{4-[(5-метил-2-пиридинил)окси]бензил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-{[4-гидрокси-2-оксо-1-(4-{[5-(трифторметил)-2-пиридинил]окси}бензил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-{[4-гидрокси-1-({5-метил-6-[(6-метил-3-пиридинил)окси]-3-пиридинил}метил)-2-оксо-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-[(1-{[5-(4-хлорфенокси)-2-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-1-{[6-(3-метоксифенокси)-3-пиридинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{4-[(6-хлор-3-пиридинил)окси]бензил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-{[4-гидрокси-2-оксо-1-({5-[4-(трифторметил)фенокси]-2-пиридинил}метил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-{[4-гидрокси-2-оксо-1-(4-{[6-(трифторметил)-3-пиридинил]окси}бензил)-1,2,5,6-тетрагидро-3-пиридинил]карбонил}глицин;
N-[(1-{[6-(3-хлор-4-метилфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(3-фтор-4-метилфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(4-фтор-3-метилфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(4-этилфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-2-оксо-1-{[6-(4-пропилфенокси)-3-пиридинил]метил}-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-1-{[6-(4-изопропилфенокси)-3-пиридинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-1-{[5-(4-метилфенокси)-2-пиразинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-({1-[4-(3,4-диметилфенокси)бензил]-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил}карбонил)глицин;
N-[(1-{[5-хлор-6-(4-метилфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[5-фтор-6-(4-метилфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{4-[(5-циклопропил-2-пиридинил)окси]бензил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(4-гидрокси-1-{[2-(4-метилфенокси)-5-пиримидинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[6-(4-хлорфенокси)-5-метил-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин;
N-[(1-{[5-(4-хлорфенокси)-2-пиразинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин; или
N-[(1-{[5-(4-циклопропилфенокси)-2-пиразинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин,
или его фармацевтически приемлемая соль.
7. Соединение по п. 1, которое представляет собой N-[(1-{[6-(4-хлорфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин, или его фармацевтически приемлемая соль.
8. Соединение по п. 1, которое представляет собой N-[(1-{[6-(4-циклопропилфенокси)-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин, или его фармацевтически приемлемая соль.
9. Соединение по п. 1, которое представляет собой N-[(4-гидрокси-1-{[6-(3-метилфенокси)-3-пиридинил]метил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин, или его фармацевтически приемлемая соль.
10. Соединение по п. 1, которое представляет собой N-[(1-{[6-(3-фторфенокси-3-пиридинил]метил}-4-гидрокси-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин, или его фармацевтически приемлемая соль.
11. Соединение по п. 1, которое представляет собой N-[(4-гидрокси-1-{4-[(6-метил-3-пиридинил)окси]бензил}-2-оксо-1,2,5,6-тетрагидро-3-пиридинил)карбонил]глицин, или его фармацевтически приемлемая соль.
12. Соединение по п. 1, имеющее указанную выше общую формулу (I'), где
соединение представлено общей формулой (I):
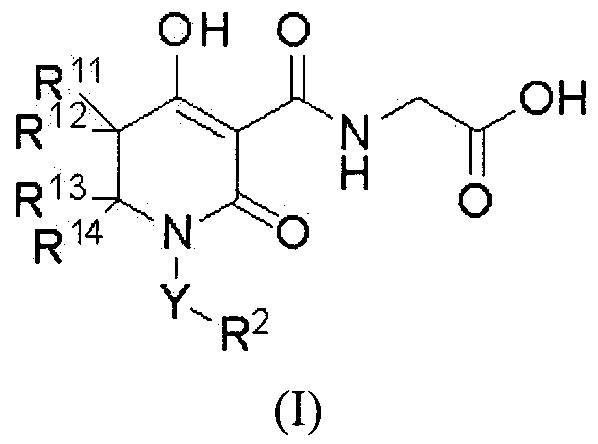
(где в формуле (I)
R11 представляет собой атом водорода, C1-4 алкил или фенил,
R12 представляет собой атом водорода или C1-4 алкил,
при условии, что R11 и R12, вместе со смежным углеродным атомом, необязательно образуют С3-8 циклоалкан или тетрагидропиран;
R13 представляет собой атом водорода, C1-4 алкил, галоген-С1-4 алкил, фенил, бензил или фенэтил,
R14 представляет собой атом водорода или C1-4 алкил,
при условии, что R13 и R14, вместе со смежным углеродным атомом, необязательно образуют С3-8 циклоалкан оксетан или тетрагидропиран,
при условии, что указанные выше R12 и R13, вместе со смежными углеродными атомами, необязательно образуют циклогексан;
Y представляет собой одинарную связь или C1-6 алкандиил (один из углеродных атомов в C1-6 алкандииле необязательно замещен циклопропан-1,1-диилом);
R2 представляет собой С3-8 циклоалкил (С3-8 циклоалкил необязательно замещен одной группой, выбранной из группы, состоящей из фенила и бензила), фенил (фенил необязательно замещен одной-тремя группами, которые являются одинаковыми или различными и которые выбирают из группы α1 заместителей), нафтил, инданил, тетрагидронафтил, пиразолил [пиразолил замещен одним фенилом (фенил необязательно замещен одним C1-6 алкилом) и дополнительно может быть замещен одним C1-6 алкилом], имидазолил (имидазолил замещен одним фенилом), изоксазолил [изоксазолил замещен одним фенилом (фенил необязательно замещен одним атомом галогена)], оксазолил (оксазолил замещен одним фенилом и дополнительно может быть замещен одним C1-6 алкилом), тиазолил (тиазолил замещен одним фенилом), пиридил [пиридил замещен одной группой, выбранной из группы, состоящей из фенила, фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, циано, C1-6 алкила, галоген-C1-6 алкила, С3-8 циклоалкила, C1-6 алкокси и галоген-С1-6 алкокси) и фенилсульфанила (фенилсульфанил необязательно замещен одним атомом галогена)], пиримидинил (пиримидинил замещен одной группой, выбранной из группы, состоящей из циклогексила и фенила), бензотиофенил, хинолил или метилендиоксифенил (метилендиоксифенил необязательно замещен одним или двумя атомами фтора);
группа α1 заместителей состоит из атома галогена, C1-6 алкила {C1-6 алкил необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила, фенила и C1-6 алкокси [C1-6 алкокси необязательно замещен одним С3-8 циклоалкилом (С3-8 циклоалкил необязательно замещен одним C1-6 алкилом)]}, галоген-C1-6 алкила, С3-8 циклоалкила, фенила (фенил необязательно замещен одной-тремя группами, которые являются одинаковыми или различными и которые выбирают из группы α2 заместителей), тиенила, пиразолила (пиразолил необязательно замещен одним C1-6 алкилом), изоксазолила, тиазолила (тиазолил необязательно замещен одним или двумя C1-6 алкилами), пиридила (пиридил необязательно замещен одной группой, выбранной из группы, состоящей из C1-6 алкила, галоген-С1-6 алкила и C1-6 алкокси), хинолила, C1-6 алкокси [C1-6 алкокси необязательно замещен одной группой, выбранной из группы, состоящей из С3-8 циклоалкила и фенила (фенил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена и C1-6 алкила)], галоген-С1-6 алкокси, С2-6 алкенилокси, С3-8 циклоалкокси, фенокси (фенокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и C1-6 алкокси), пиридилокси (пиридилокси необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена, C1-6 алкила и галоген-С1-6 алкила) и C1-6 алкилсульфанила;
группа α2 заместителей состоит из атома галогена, циано, гидрокси, C1-6 алкила, галоген-С1-6 алкила, фенила, C1-6 алкокси, C1-6 алкилкарбонила и ди-С1-6 алкиламиносульфонила),
или его фармацевтически приемлемая соль.
13. Лекарственное средство, обладающее ингибирующим действием в отношении PHD2, которое содержит соединение по любому из пп. 1-12 или его фармацевтически приемлемую соль в качестве активного ингредиента.
14. Ингибитор PHD2, который содержит соединение по любому из пп. 1-12 или его фармацевтически приемлемую соль в качестве активного ингредиента.
15. Стимулятор образования ЕРО, который содержит соединение по любому из пп. 1-12 или его фармацевтически приемлемую соль в качестве активного ингредиента.
16. Лекарственное средство для предотвращения или лечения анемии, которое содержит соединение по любому из пп. 1-12 или его фармацевтически приемлемую соль в качестве активного ингредиента.
| WO 2010025087 A1, 04.03.2010 | |||
| WO 2009158315 A1, 30.12.2009 | |||
| RU 2009106214 A, 10.09.2010 | |||
| WO 2008089051 A1, 24.07.2008. |

