ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новому гетероциклическому соединению пиримидин-4(3H)-она для ингибирования или регулирования SHP2 или его фармацевтически приемлемой соли, фармацевтической композиции, содержащей соединение или его фармацевтически приемлемую соль, способу получения соединения или его фармацевтически приемлемой соли, применению соединения или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей соединение или его фармацевтически приемлемую соль, для получения медикамента для лечения и/или предотвращения опосредованных SHP2 заболеваний, в частности, рака, и способу его введения.
УРОВЕНЬ ТЕХНИКИ
Фосфатаза 2, содержащая домен гомологии 2 Src (SHP2), представляет собой нерецепторную протеинтирозинфосфатазу, кодируемую нерецепторным геном протеинтирозинфосфатазы типа 11 (PTPN11). SHP2 содержит два домена гомологии Src (SH2), один домен протеинтирозинфосфатазы (РТР) и один С-концевой хвост. В нормальных условиях SHP2 принимает автоингибирующую конформацию, а его N-SH2 связывается с РТР, блокируя субстратный канал каталитического сайта РТР, тем самым ингибируя активность РТР. Когда SH2 связывается с дифосфотирозиновым пептидом (таким как IRS-1), автоингибирующее взаимодействие SH2-PTP прекращается, и каталитический сайт РТР подвергается воздействию, что позволяет SHP2 находиться в активном состоянии для катализа дефосфорилирования тирозина.
SHP2 широко экспрессируется, что в качестве онкогена опосредует активацию различных сигнальных путей онкогенных клеток, таких как пути RAS-ERK, PI3K-AKT и JAK-STAT, и способствует выживанию и пролиферации раковых клеток. SHP2 может связывать и дефосфорилировать RAS, усиливать ассоциацию RAS-RAF и активировать нижележащие сигналы клеточной пролиферации. SHP2 также опосредует пути компенсаторной активации после ингибирования киназ, таких как MEK, что приводит к лекарственной устойчивости при терапии опухолей (Ruess DA, et al., Nat. Med. 2018, 24, 954-960). Следовательно, активация SHP2 тесно связана с патогенезом различных заболеваний, таких как лейкоз, меланома, рак молочной железы, рак легкого, рак толстой кишки, нейробластома и гепатоцеллюлярная карцинома.
SHP2 также играет важную роль в путях иммунных контрольных точек PD-1 и аттенюатора В- и Т-лимфоцитов (BTLA), что не только ингибирует активацию Т-клеток, но также способствует анергии Т-клеток (Li J, et al., Cancer Res. 2015, 75, 508-518).
Поэтому SHP2 в качестве противоопухолевой мишени привлекает большое внимание. Однако высокая гомология белковой последовательности каталитического сайта РТР и высокая гидрофильность каталитического кармана РТР приводят к плохой селективности, плохой клеточной проницаемости и низкой биодоступности каталитического сайта SHP2. Открытие Novartis аллостерического ингибитора SHP099 (Chen Y, et al., Nature 2016, 535, 148-52) обеспечивает новый канал для разработки высокоспецифичных пероральных ингибиторов SHP2. В настоящее время некоторые патентные заявки на ингибиторы SHP2 были раскрыты множеством компаний, включая WO 2015107495, WO 2016203405, WO 2018057884, WO 2018013597 и WO 2017211303. В настоящем изобретении разработаны соединения, имеющие структуру, представленную общей формулой (I), и было обнаружено, что соединения с такой структурой проявляют превосходный эффект ингибирования активности SHP2.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению, представленному общей формулой (I), в качестве ингибитора SHP2, или его пролекарствам, стабильным изотопным производным, фармацевтически приемлемым солям и изомерам, а также их смесям:
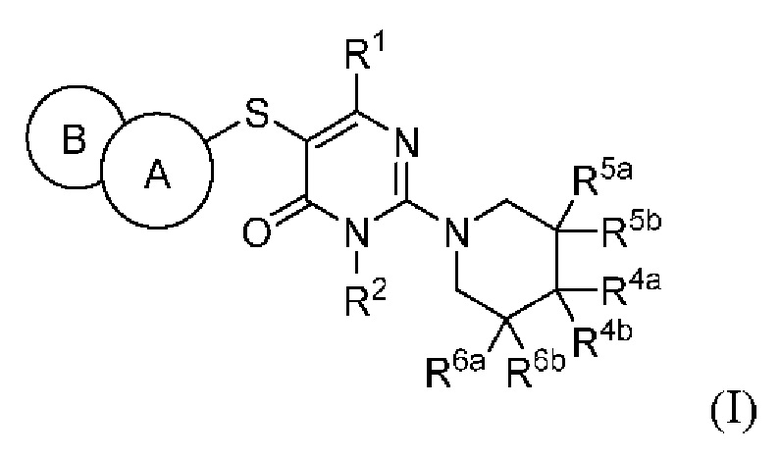
где:
кольцо А выбирается из группы, состоящей из фенила и 6-членного гетероарила, кольцо В выбирается из группы, состоящей из 5-членного гетероарильного кольца, конденсированного с кольцом А, где фенил и гетероарил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D, галогена, циано, оксо, C1-6 алкила, С3-6 циклоалкила, 4-7-членного гетероциклила, 5-6-членного гетероарила, -ORa, -NRaRb, -C(O)Ra, -C(O)NRaRb, -S(O)2Ra, -S(O)2NRaRb, -NRaS(O)2Rb и -Р(O)(СН3)2, где алкил, циклоалкил, гетероциклил, и гетероарил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D, галогена, циано, оксо, C1-6 алкила, С3-6 циклоалкила, 4-7-членного гетероциклила, -ORa, -NRaRb, -C(O)Ra и -C(O)NRaRb;
R1 выбирается из группы, состоящей из Н, D, циано, С1-2 алкила, циклопропила, -ORa, -NRaRb и -C(O)NRaRb, где один или несколько атомов водорода алкила и циклопропила необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D и фтора;
R2 выбирается из группы, состоящей из Н, С1-2 алкила и циклопропила, где один или несколько атомов водорода алкила и циклопропила необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D, фтора и гидроксила;
R4a и R4b каждый независимо выбираются из группы, состоящей из Н, D, галогена, циано, -ORa, -NRaRb, -C(O)Ra, -C(O)NRaRb, C1-6 алкила, C3-6 циклоалкила, 4-7-членного гетероциклила и 5-6-членного гетероарила, при условии, что R4a и R4b одновременно не могут быть выбраны из группы, состоящей из циано, -ORa и -NRaRb, где алкил, циклоалкил, гетероциклил и гетероарил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D, галогена, циано, оксо, C1-6 алкила, С3-6 циклоалкила, 4-7-членного гетероциклила, 5-6-членного гетероарила, -ORa, -NRaRb, -C(O)Ra и -C(O)NRaRb; R4a и R4b необязательно вместе с атомом углерода, к которому присоединены R4a и R4b, образуют С3-7 карбоциклическое кольцо или 4-8-членное гетероциклическое кольцо;
R5a, R5b, R6a и R6b каждый независимо выбирается из группы, состоящей из Н, D, фтора и метила;
Ra и Rb каждый независимо выбирается из группы, состоящей из Н, C1-6 алкила, С3-6 циклоалкила и 4-7-членного гетероциклила, где алкил, циклоалкил и гетероциклил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D, фтора, циано, оксо, гидроксила, -ОСН3 и -NH2.
В варианте выполнения настоящее изобретение относится к соединению, представленному вышеприведенной общей формулой (I), или к его фармацевтически приемлемым солям, пролекарствам, стабильным изотопным производным и изомерам, а также их смесям, представляющему собой соединение, представленное общей формулой (II), или его фармацевтически приемлемые соли, пролекарства, стабильные изотопные производные и изомеры, а также их смеси:
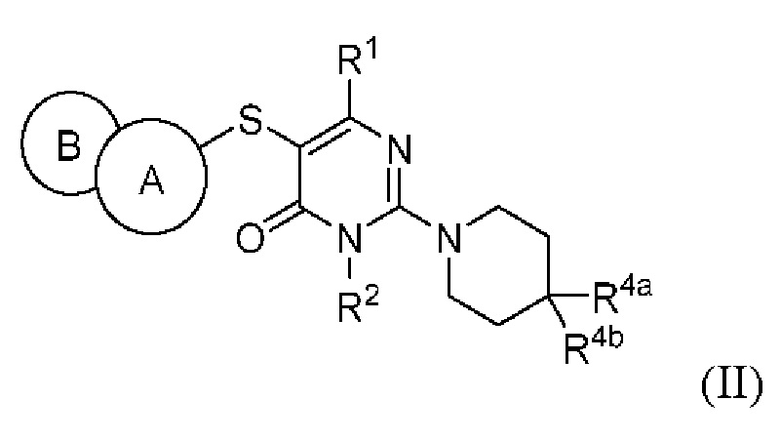
где:
кольцо А выбирается из группы, состоящей из фенила и 6-членного гетероарила, кольцо В выбирается из группы, состоящей из 5-членного гетероарильного кольца, конденсированного с кольцом А, где фенил и гетероарил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D, галогена, циано, оксо, C1-6 алкила, С3-6 циклоалкила, 4-7-членного гетероциклила, 5-6-членного гетероарила, -ORa, -NRaRb, -C(O)Ra, -C(O)NRaRb, -S(O)2Ra, -S(O)2NRaRb и -NRaS(O)2Rb, где алкил, циклоалкил, гетероциклил и гетероарил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D, галогена, циано, оксо, C1-6 алкила, С3-6 циклоалкила, 4-7-членного гетероциклила, -ORa, -NRaRb, -C(O)Ra и -C(O)NRaRb;
R1 выбирается из группы, состоящей из Н, D, циано, C1-2 алкила, -ORa и -NRaRb, где один или несколько атомов водорода алкила необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D и фтора;
R2 выбирается из группы, состоящей из Н и C1-2 алкила, где один или несколько атомов водорода алкила необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D и фтора;
R4a и R4b каждый независимо выбирается из группы, состоящей из Н, D, галогена, циано, -ORa, -NRaRb, -C(O)NRaRb, Ci.6 алкила, Сз_б циклоалкила, 4-7-членного гетероциклила и 5-6-членного гетероарила, при условии, что R4a и R4b не могут быть одновременно выбраны из группы, состоящей из циано, -ORa и -NRaRb, где алкил, циклоалкил, гетероциклил и гетероарил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D, галогена, циано, оксо, C1-6 алкила, С3-6 циклоалкила, 4-7-членного гетероциклила, 5-6-членного гетероарила, -ORa, -NRaRb, -C(O)Ra и -C(O)NRaRb; R4a и R4b необязательно вместе с атомом углерода, к которому присоединены R4a и R4b, образуют С3-7 карбоциклическое кольцо или 4-7-членное гетероциклическое кольцо;
Ra и Rb каждый независимо выбирается из группы, состоящей из Н, C1-6 алкила, С3-6 циклоалкила и 4-7-членного гетероциклила, где алкил, циклоалкил и гетероциклил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D, фтора, циано, оксо, гидроксила, -ОСН3 и -NH2.
В другом варианте выполнения настоящее изобретение относится к соединению, представленному вышеприведенной общей формулой (I), или к его фармацевтически приемлемым солям, пролекарствам, стабильным изотопным производным и изомерам, а также их смесям, представляющему собой соединение, представленное общей формулой (III), или его фармацевтически приемлемые соли, пролекарства, стабильные изотопные производные и изомеры, а также их смеси:
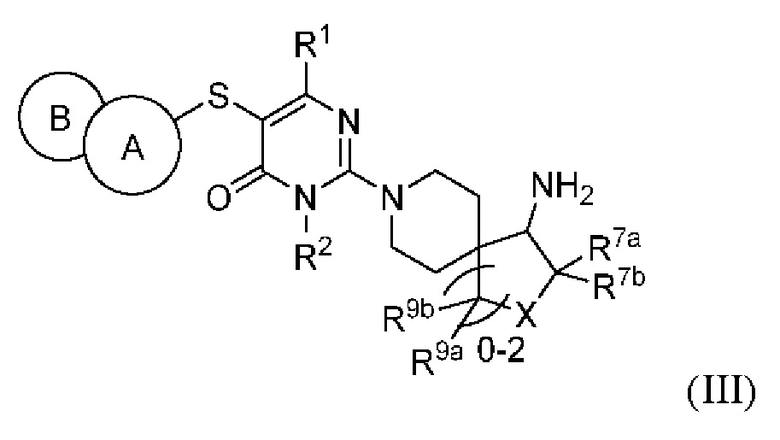
где:
кольцо А выбирается из группы, состоящей из фенила и 6-членного гетероарила, кольцо В выбирается из группы, состоящей из 5-членного гетероарильного кольца, конденсированного с кольцом А, где фенил и гетероарил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D, галогена, циано, оксо, C1-6 алкила, С3-6 циклоалкила, 4-7-членного гетероциклила, 5-6-членного гетероарила, -ORa, -NRaRb, -C(O)Ra и -C(O)NRaRb, где алкил, циклоалкил, гетероциклил и гетероарил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D, галогена, циано, оксо, С1-2 алкила, -ORa и -NRaRb;
X выбирается из группы, состоящей из -О- и -CR8aR8b-;
R1 выбирается из группы, состоящей из Н, D, циано, С1-2 алкила и -NRaRb, где один или несколько атомов водорода алкила необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D и фтора;
R2 выбирается из группы, состоящей из Н и С1-2 алкила, где один или несколько атомов водорода алкила необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D и фтора;
R7a, R7b, R8a, R8b, R9a и R9b каждый независимо выбирается из группы, состоящей из Н, D, галогена, циано, С1-2 алкила и -ORc, где один или несколько атомов водорода алкила необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D, фтора и гидроксила, при условии, что R7a и R7b, R8a и R8b и R9a и R9b необязательно одновременно не могут быть выбраны из группы, состоящей из -ORc;
Ra и Rb каждый независимо выбирается из группы, состоящей из Н, C1-6 алкила, С3-6 циклоалкила и 4-7-членного гетероциклила, где алкил, циклоалкил и гетероциклил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D, фтора, циано, оксо, гидроксила, -ОСН3 и -NH2;
Rc выбирается из группы, состоящей из Н и С1-2 алкила, где один или несколько атомов водорода алкила необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D и фтора.
В другом варианте выполнения настоящее изобретение относится к соединению, представленному вышеприведенной общей формулой (I), или к его фармацевтически приемлемым солям, пролекарствам, стабильным изотопным производным и изомерам, а также их смесям, представляющему собой соединение, представленное общей формулой (IV), или его фармацевтически приемлемые соли, пролекарства, стабильные изотопные производные и изомеры, а также их смеси:
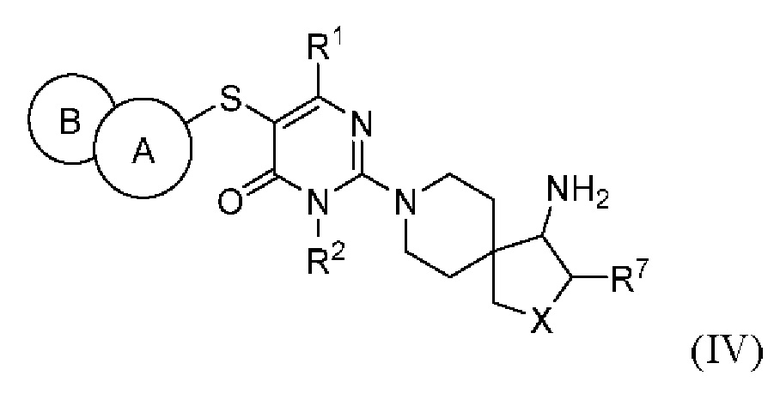
где:
кольцо А выбирается из группы, состоящей из фенила и 6-членного гетероарила, кольцо В выбирается из группы, состоящей из 5-членного гетероарильного кольца, конденсированного с кольцом А, где фенил и гетероарил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D, галогена, циано, оксо, C1-2 алкила, -ORa и -NRaRb, где один или несколько атомов водорода алкила необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D и фтора;
X выбирается из группы, состоящей из -О- и -СН2-;
R1 независимо выбирается из группы, состоящей из Н, D, С1-2 алкила и -NRaRb, где один или несколько атомов водорода алкила необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D и фтора;
R2 выбирается из группы, состоящей из Н и С1-2 алкила, где один или несколько атомов водорода алкила необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D и фтора;
R7 выбирается из группы, состоящей из Н, D и С1-2 алкила, где один или несколько атомов водорода алкила необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D и фтора;
Ra и Rb каждый независимо выбирается из группы, состоящей из Н и С1-2 алкила, где один или несколько атомов водорода алкила необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из D и фтора.
Настоящее изобретение дополнительно относится к соединению, представленному приведенной выше общей формулой (I), где соединение выбирается из группы, состоящей из:
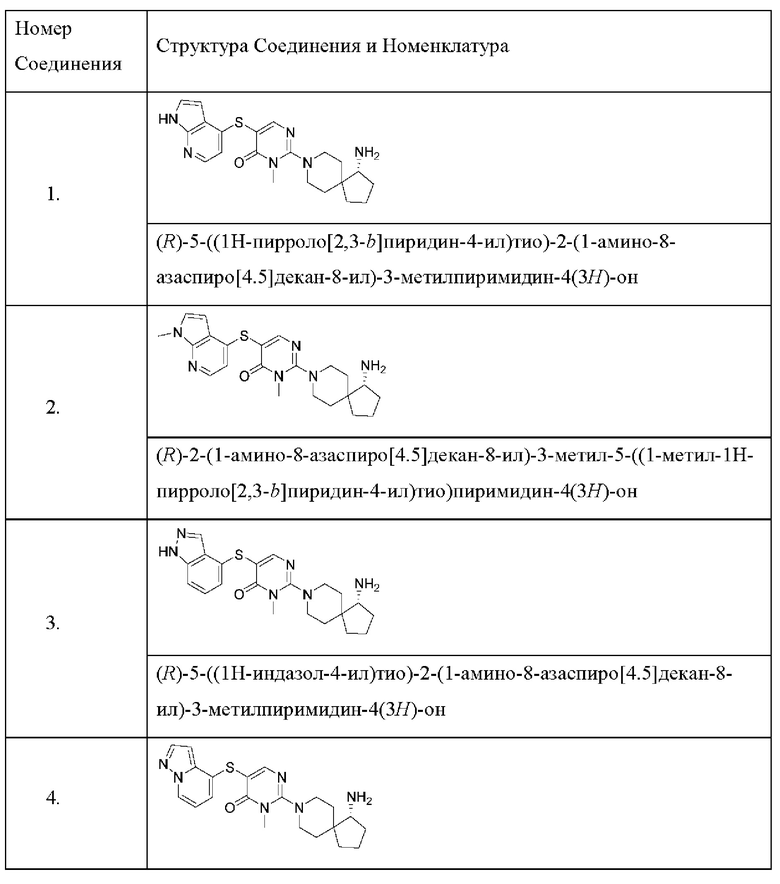
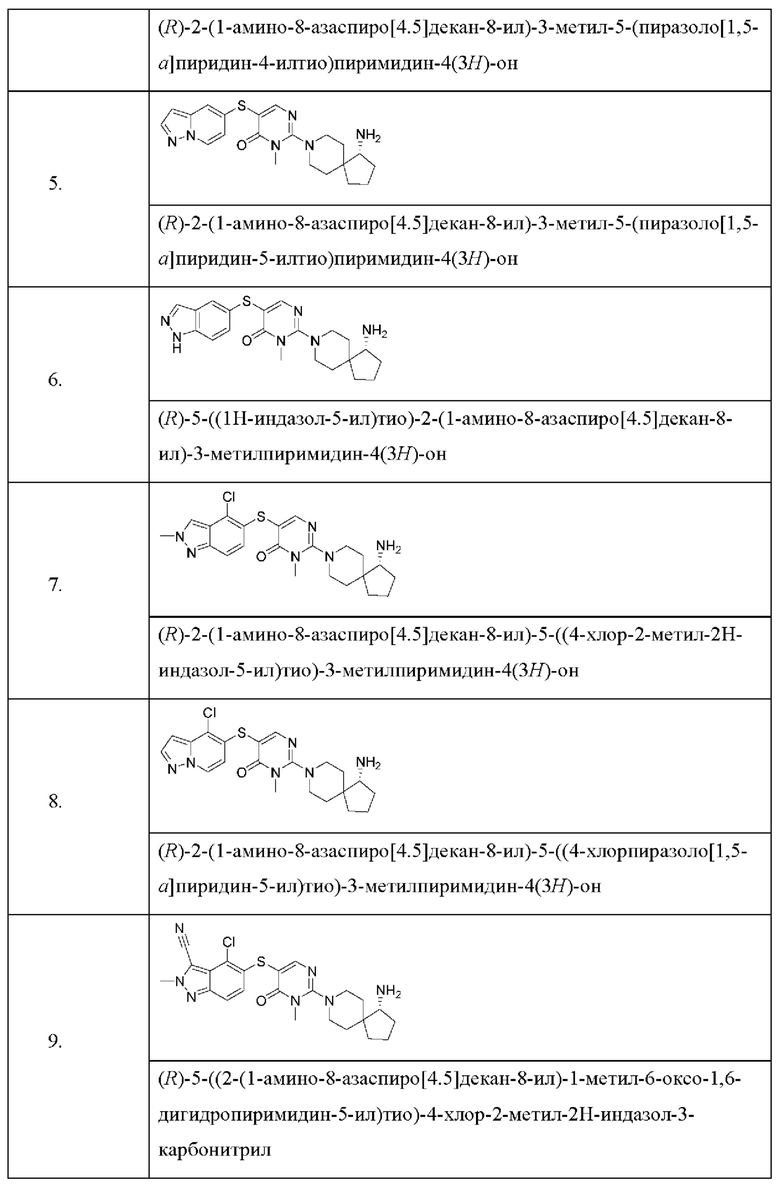
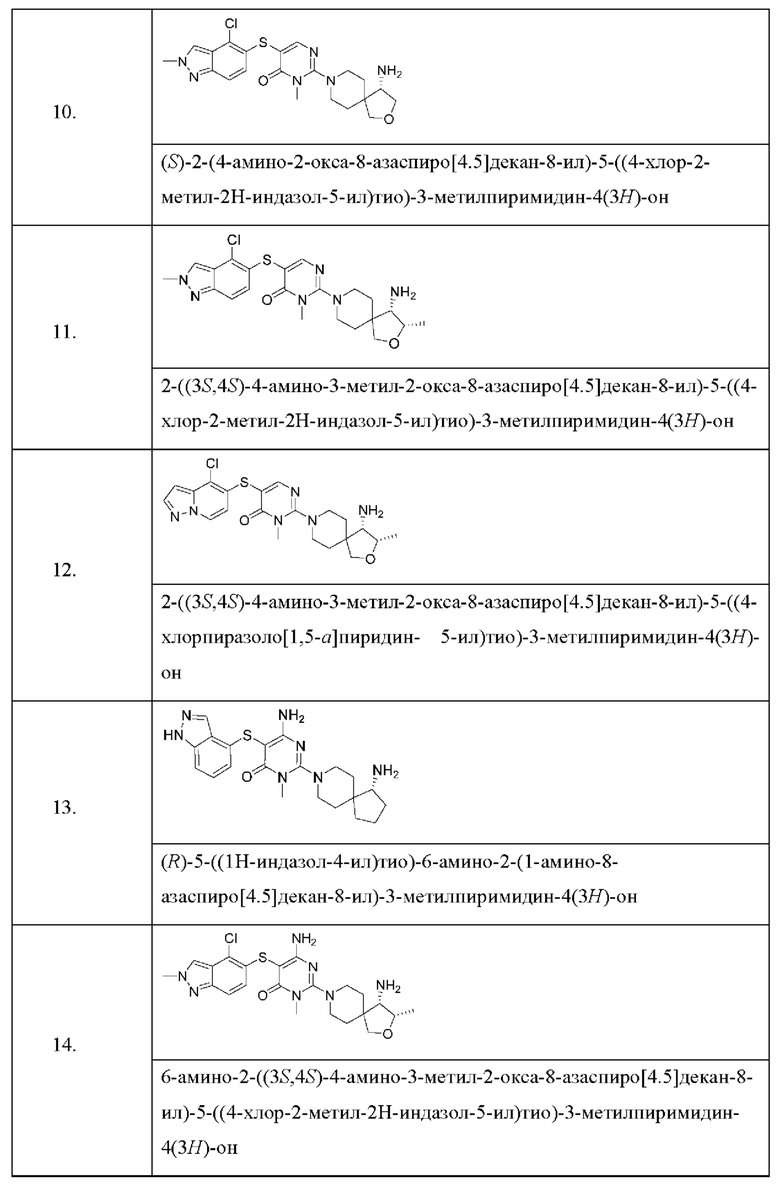
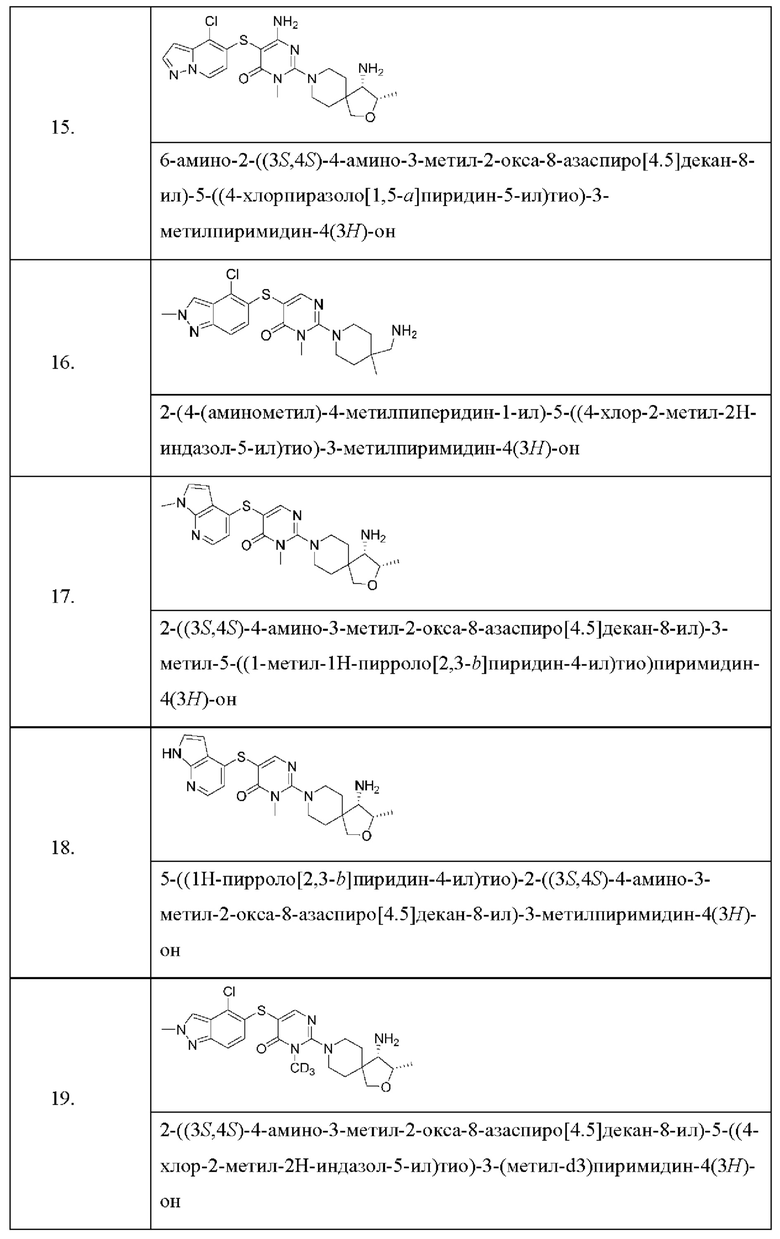
или его фармацевтически приемлемые соли, пролекарства, стабильные изотопные производные и изомеры, а также их смеси.
Соединение по настоящему изобретению может эффективно ингибировать активность SHP2 с предпочтительной IC50 менее чем 50 нМ. Соединение по настоящему изобретению оказывает значительное ингибирующее действие на пролиферацию клеток NCI-H358 с предпочтительной IC50 менее чем 1000 нМ.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, где фармацевтическая композиция содержит соединение, представленное общей формулой (I), или его фармацевтически приемлемые соли, пролекарства, стабильные изотопные производные и изомеры, а также их смеси, а также фармацевтически приемлемые носители и эксципиенты.
Настоящее изобретение дополнительно относится к фармацевтической композиции, где фармацевтическая композиция содержит соединение, представленное общей формулой (I), или его фармацевтически приемлемые соли, пролекарства, стабильные изотопные производные и изомеры, а также их смеси, и по меньшей мере одно дополнительное лекарственное средство, где по меньшей мере одно дополнительное лекарственное средство включает, но не ограничиваясь ими, химиотерапевтические агенты, таргетные лекарственные средства, ингибиторы синтеза ДНК, лекарственные средства на основе антител, конъюгаты антитело-лекарственное средство, противоопухолевые лекарственные средства и иммунодепрессанты.
Настоящее изобретение дополнительно относится к применению соединения, представленного общей формулой (I), или его фармацевтически приемлемых солей, пролекарств, производных стабильных изотопов и изомеров, а также их смесей, или фармацевтической композиции для получения медикамента, где медикамент показан для лечения или предотвращения заболеваний, опосредованных SHP2, где заболевания включают, но не ограничиваясь ими, лейкоз, синдром Нунан, синдром леопарда, нейробластому, рак легкого, рак молочной железы, рак толстой кишки, рак пищевода, рак желудка и рак головы и шеи.
Согласно настоящему изобретению медикамент может быть в любой дозированной форме, включая, но не ограничиваясь ими, таблетки, капсулы, растворы, лиофилизированные препараты и препараты для инъекций.
Фармацевтическая композиция по настоящему изобретению может быть введена в форме дозированной единицы, содержащей заданную дозу активного фармацевтического ингредиента. В зависимости от подлежащего лечению заболевания, способа введения и возраста, массы тела и состояния пациента такая единица может содержать от 0,5 мг до 1 г, предпочтительно от 1 мг до 700 мг и более предпочтительно от 5 мг до 500 мг соединения по настоящему изобретению. Кроме того, фармацевтический состав можно приготовить способами, известными в области фармацевтики, например, путем смешивания активного фармацевтического ингредиента одним или несколькими эксципиентами и/или адъювантами.
Фармацевтическая композиция по настоящему изобретению пригодна для введения любыми подходящими способами, включая трансоральный (включая пероральный или сублингвальный), трансректальный, трансназальный, топический (включая пероральный, сублингвальный или трансдермальный), вагинальный или парентеральный (включая подкожный, внутримышечный, внутривенный или внутрикожный) подходы.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, следующие термины в описании и в формуле изобретения имеют следующие значения.
«Сх-у» относится к диапазону числа атомов углерода, где × и у оба являются целыми числами, например, С3-8 циклоалкил означает циклоалкил, имеющий 3-8 атомов углерода, а именно 3, 4, 5, 6, 7 или 8 атомов углерода. Кроме того, следует понимать, что «С3-8» дополнительно включает любой поддиапазон, например, С3-7, С3-6, С4-7, С4-6 и С5-6.
«Алкил» относится к насыщенной углеводородной группе с прямой или разветвленной цепью, имеющей от 1 до 20 атомов углерода, например, от 1 до 8, от 1 до 6 или от 1 до 4 атомов углерода. Неограничивающие примеры алкила включают, но не ограничиваясь ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-амил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил и 2-этилбутил.
«Циклоалкильное или карбоциклическое кольцо» относится к насыщенному циклическому углеводородному заместителю, содержащему от 3 до 14 атомов углерода в кольце. Циклоалкил может быть монокарбоциклическим кольцом, обычно содержащим от 3 до 8, от 3 до 7 или от 3 до 6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают, но не ограничиваясь ими, циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Циклоалкил также может представлять собой конденсированное би- или трикарбоциклическое кольцо, например, декагидронафтил, бицикло[2.2.2]октан и спиро[3.3]гептан.
«Гетероциклильное или гетероциклическое кольцо» относится к насыщенной или частично ненасыщенной моно- или полициклической группе, имеющей от 3 до 20 атомов в кольце, например, от 3 до 14, от 3 до 12, от 3 до 10, от 3 до 8, от 3 до 6 или от 5 до 6 атомов в кольце, где один или несколько выбираются из азота, кислорода или S(O)m (где m представляет собой целое число от 0 до 2), за исключением -О-О-, -O-S- или -S-S- в кольцевой структуре, а остальные являются атомами углерода. Предпочтительно имеется от 3 до 12, более предпочтительно от 3 до 10, еще более предпочтительно от 4 до 7, еще более предпочтительно от 4 до 6 и наиболее предпочтительно от 5 до 6 атомов в кольце, где от 1 до 4, более предпочтительно от 1 до 3 и наиболее предпочтительно 1 до 2 являются гетероатомами. Неограничивающие примеры моногетероциклической группы включают, но не ограничиваясь ими, пирролидинил, оксетанил, пиперидинил, пиперазинил, тетрагидрофуранил, тетрагидропиранил, тетрагидротиопиранил, морфолинил, тиоморфолинил, гомопиперазинил и азетидинил. Полигетероциклическая группа включает конденсированный, мостиковый или спирополигетероцикл, например, октагидроциклопентадиено[с]пиррол, октагидропирроло[1,2-а]пиразин, 3,8-диазабицикло[3.2.1]октан, 5-азаспиро[2.4]гептан и 2-окса-7-азаспиро[3.5]нонан.
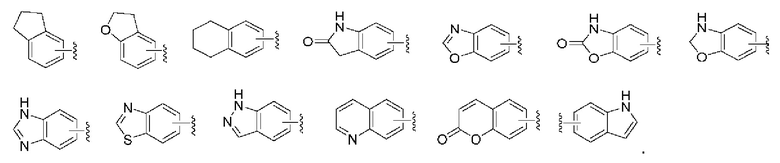
«Арил или арильное кольцо» относится к моноциклической или конденсированной полициклической ароматической группе, имеющей от 6 до 14 атомов углерода, предпочтительно 6-10 членной, например, фенил и нафтил, и наиболее предпочтительно фенил. Ароматическое кольцо может быть конденсировано с гетероарильным, гетероциклическим или циклоалкильным кольцом, где кольцо, присоединенное к исходной структуре, представляет собой арильное кольцо. Неограничивающие примеры включают, но не ограничиваясь:
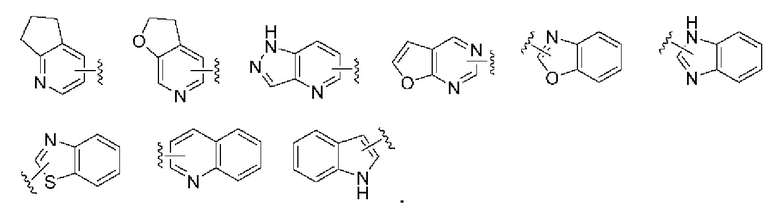
«Гетероарил или гетероарильное кольцо» относится к гетероароматической системе, имеющей от 5 до 14 атомов в кольце, из которых от 1 до 4 выбираются из гетероатомов кислорода, серы и азота. Гетероарил предпочтительно является 5-10-членным и более предпочтительно 5- или 6-членным, например, фурил, тиенил, пиридил, пирролил, пиримидинил, пиразинил, пиразолил, имидазолил, тетразолил, оксазолил, изоксазолил, тиазолил, изотиазолил, хинолинил, изохинолинил, индолил и изоиндолил. Гетероарильное кольцо может быть конденсировано с арильным, гетероциклическим или циклоалкильным кольцом, где кольцо, присоединенное к исходной структуре, представляет собой гетероарильное кольцо. Неограничивающие примеры включают, но не ограничиваясь:
«Галоген» относится к F, Cl, Br или I.
«Циано» относится к -CN.
«Оксо» относится к =O.
«Необязательно» означает, что событие или окружение, описанные ниже, могут, но не должны произойти, и включает случаи, в которых событие или окружение происходит или не происходит. Например, «гетероциклическая группа, необязательно замещенная алкилом» означает, что алкил может присутствовать, но не обязательно присутствует, и это описание включает случаи, когда гетероциклическая группа замещена алкилом, и случаи, когда гетероциклическая группа не замещена алкилом.
«Замещенный» относится к тому, что один или несколько атомов водорода в группе, предпочтительно 5 и более предпочтительно от 1 до 3, независимо замещены соответствующим количеством заместителей. Само собой разумеется, что заместители находятся только в их возможных химических положениях, и специалисты в данной области техники могут определить возможное или невозможное замещение без особых усилий (теоретически или экспериментально). Например, амино или гидроксил со свободным водородом может быть нестабильным, когда связан с атомами углерода, имеющими ненасыщенные связи (напр., олефиновые). Заместители включают, но не ограничиваясь ими, галоген, циано, нитро, оксо, -SF5, С1-4 алкил, С3-7 циклоалкил, 4-7-членный гетероциклил, фенил и 5-6-членный гетероарил.
«Изомер» относится к соединению, имеющему одинаковую молекулярную формулу, но разные свойства связывания, или их порядок, или другое пространственное расположение его атомов. Изомер, имеющий другое пространственное расположение атомов, называется «стереоизомером». Стереоизомеры включают оптический изомер, геометрический изомер и конформер.
Соединения по настоящему изобретению могут существовать в форме оптических изомеров. Оптические изомеры включают энантиомеры и диастереомеры. Энантиомер представляет собой один из двух стереоизомеров, которые являются зеркальным отражением друг друга, но не накладываются друг на друга. Рацемическая смесь или рацемат представляет собой смесь, которая имеет равное количество левых и правых энантиомеров хиральной молекулы. Диастереомеры представляют собой стереоизомеры, которые не являются зеркальным отражением друг друга и не накладываются друг на друга. Когда соединение представляет собой один изомер и имеет определенную абсолютную конфигурацию, его называют изомером «R» или «S» в соответствии с конфигурацией заместителей у хирального атома углерода; когда его абсолютная конфигурация не определена, его называют (+) или (-) изомером в соответствии с измеренным значением оптического вращения. Способы получения и разделения оптических изомеров в данной области техники известны.
Соединения по настоящему изобретению могут также существовать в форме геометрических изомеров, возникающих в результате распределения заместителей вокруг двойных связей углерод-углерод, двойных связей углерод-азот, циклоалкилов или гетероциклических групп. Заместители вокруг углерод-углеродных двойных связей или углерод-азотных двойных связей обозначены как находящиеся в Z- или Е-конфигурации, а заместители вокруг циклоалкильных или гетероциклических колец обозначены как находящиеся в цис- или транс-конфигурации.
Соединения по настоящему изобретению могут дополнительно проявлять таутомерию, например, кето-енольную таутомерию.
Следует отметить, что настоящее изобретение включает любые таутомеры или стереоизомеры, а также их смеси и не ограничивается просто любыми таутомерными или стереоизомерными формами, используемыми в номенклатуре соединений или химической структурной формуле.
«Изотопы» включают все изотопы атомов, присутствующих в соединениях по настоящему изобретению. К изотопам относятся те атомы, которые имеют одинаковые атомные номера, но разную массу. Примерами изотопов, подходящих для включения в соединения по настоящему изобретению, могут быть водород, углерод, азот, кислород, фосфор, сера, фтор и хлор, например, но не ограничиваясь ими, 2Н (D), 3Н, 13С, 14С, 15N, 17O, 18O, 31Р, 32Р, 35S, 18F и 36Cl соответственно. Меченные изотопами соединения по настоящему изобретению, как правило, могут быть получены обычными методиками, известными специалистам в данной области техники, или способами, подобными тем, которые описаны в вариантах выполнения изобретения, с использованием соответствующих реагентов, меченных изотопами, вместо реагентов, не меченных изотопами. Такие соединения имеют различное потенциальное применение, например, в качестве стандартов и реагентов для определения биологической активности. В случае стабильных изотопов такие соединения могут благотворно изменять биологические, фармакологические или фармакокинетические свойства. Дейтерий (D) может быть предпочтительным изотопом в настоящем изобретении. Например, водород в метиле, метилене или метине может быть замещен дейтерием.
Соединения по настоящему изобретению можно вводить в форме пролекарства. «Пролекарства» относятся к производным, которые могут быть преобразованы в биологически активные соединения в физиологических условиях in vivo, таких как окисление, восстановление и гидролиз (каждое из которых может происходить в отсутствие фермента или в его присутствии). Примерами пролекарств могут быть соединения по настоящему изобретению, в которых аминогруппа ацилирована, алкилирована или фосфорилирована, например, эйкозаноиламино, аланиламино и пивалоилоксиметиламино; или гидроксил ацилирован, алкилирован, фосфорилирован или превращается в боронатную соль, например, ацетокси, пальмитоилокси, пивалоилокси, сукцинилокси, фумарилокси и аланилокси; карбоксил этерифицирован или амидирован; тиол образует дисульфидный мостик с молекулой-носителем, такой как пептид, который избирательно доставляет лекарство к мишени и/или цитозолю в клетке. Пролекарства могут быть получены из соединений по настоящему изобретению в соответствии с хорошо известными способами.
«Фармацевтические соли» или «фармацевтически приемлемые соли» относятся к солям, полученным из фармацевтически приемлемых оснований или кислот, включая неорганические основания или кислоты и органические основания или кислоты. Настоящее изобретение дополнительно включает фармацевтически приемлемые соли соединений по настоящему изобретению, которые содержат одну или несколько кислотных или основных групп. Следовательно, соединения по настоящему изобретению, имеющие кислотные группы, могут существовать в форме соли и используются в соответствии с настоящим изобретением, например, в виде солей щелочных металлов, солей щелочноземельных металлов или солей аммония. Более конкретные примеры этих солей могут включать соли натрия, соли калия, соли кальция, соли магния или соли, образованные с аммиаком или органическими аминами, например, этиламином, этаноламином, триэтаноламином или аминокислотами. Соединения по настоящему изобретению, имеющие основные группы, могут существовать в форме соли и используются в соответствии с настоящим изобретением в виде солей, полученных с неорганическими или органическими кислотами. Примеры подходящих кислот могут включать соляную кислоту, бромистоводородную кислоту, фосфорную кислоту, серную кислоту, азотную кислоту, метансульфокислоту, п-толуолсульфокислоту, нафталиндисульфокислоту, щавелевую кислоту, уксусную кислоту, винную кислоту, молочную кислоту, салициловую кислоту, бензойную кислоту, муравьиную кислоту, пропионовую кислоту, триметилуксусную кислоту, пропандиовую кислоту, янтарную кислоту, пимелиновую кислоту, фумаровую кислоту, малеиновую кислоту, яблочную кислоту, сульфаминовую кислоту, фенилпропионовую кислоту, глюконовую кислоту, аскорбиновую кислоту, изоникотиновую кислоту, лимонную кислоту, адипиновую кислоту и другие кислоты, известные специалистам в данной области техники. При условии, что соединения по настоящему изобретению содержат как кислотные, так и основные группы в молекуле, настоящее изобретение дополнительно включает внутренние соли или бетаиновые соли в дополнение к упомянутым формам солей. Каждая соль может быть получена обычными способами, известными специалистам в данной области техники, например, путем смешивания соединения с органической или неорганической кислотой или основанием в растворителе или диспергирующем агенте или путем анионного или катионного обмена с другой солью.
«Фармацевтическая композиция» относится к композиции, содержащей одно или несколько соединений, описанных в настоящем документе, или их фармацевтически приемлемые соли, пролекарства, стабильные изотопные производные и изомеры, а также их смеси и другие компоненты, такие как фармацевтически приемлемые носители и эксципиенты. Фармацевтическая композиция предназначена для легкого введения в организм и лучшего всасывания активных фармацевтических ингредиентов для проявления биологической активности.
Следовательно, когда в настоящей заявке упоминаются «соединение(я)», «соединение(я) по настоящему изобретению» или «соединение(я) по настоящему изобретению», включаются все формы соединений, такие как их фармацевтически приемлемые соли, пролекарства, стабильные изотопные производные и изомеры, а также их смеси.
В настоящем документе термин «терапевтически эффективная доза» относится к дозе соединения по настоящему изобретению, способной эффективно ингибировать функцию SHP2 и/или лечить или предотвращать заболевания.
В настоящем документе термин «пациенты» относится к млекопитающим, в частности, к человеку.
Способы синтеза
Настоящее изобретение дополнительно относится к способу получения соединений. Соединения, представленные общей формулой (I) по настоящему изобретению, могут быть получены следующими типичными способами и примерами, но эти способы и примеры не следует рассматривать как ограничивающие каким-либо образом объем настоящего изобретения. Соединения по настоящему изобретению могут быть дополнительно синтезированы методиками синтеза, известными специалистам в данной области техники, или с использованием комбинации способов, известных в данной области техники, и способов по настоящему изобретению. Продукты, полученные на каждой стадии реакции, могут быть получены методиками разделения, известными в данной области техники, включая, но не ограничиваясь ими, экстракцию, фильтрацию, дистилляцию, кристаллизацию и хроматографическое разделение. Исходные материалы и химические реагенты, используемые для синтеза, могут быть синтезированы традиционным способом в соответствии с литературой (запросы в SciFinder) или приобретены.
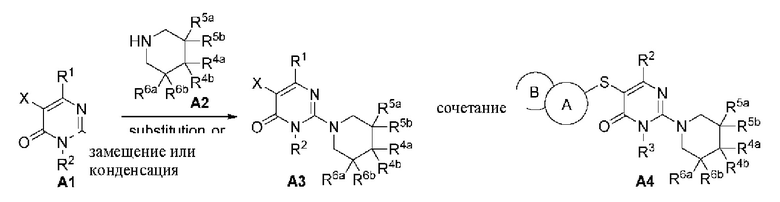
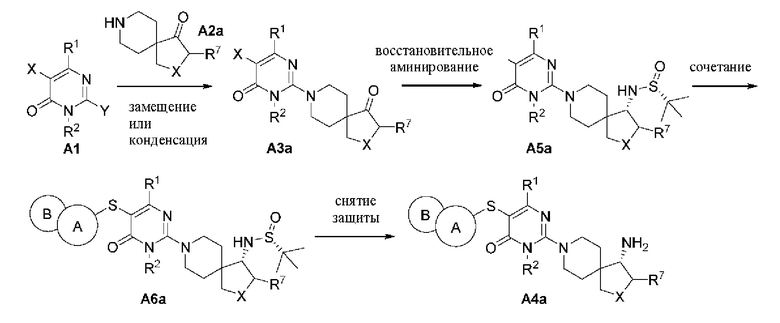
Гетероциклическое соединение, представленное общей формулой (I) по настоящему изобретению, может быть синтезировано в соответствии с маршрутом, показанным в способе А: 1) А1 и NH из пиперидинового соединения А2 подвергаются реакции замещения с помощью основного катализа или реакции конденсации в присутствии конденсирующего агента с получением промежуточного соединения A3; 2) A3 и сульфидрил из 5,6-конденсированного гетероарила подвергаются реакции перекрестного сочетания Бухвальда-Хартвига с образованием продукта А4.
Способ А:
Гетероциклические соединения, представленные общей формулой (I) по настоящему изобретению, также могут быть синтезированы в соответствии с маршрутом, показанным в способе В: 1) А1  вергаются реакции замещения с
вергаются реакции замещения с  (R)-2-метилпропан-2-сульфинамид подвергаются восстановительному аминированию с получением А5а; 3) А5а и сульфидрил из 5,6-конденсированного гетероарила подвергаются реакции перекрестного сочетания Бухвальда-Хартвига с образованием А6а; 4) с А6а снимают защиту в кислой среде с получением продукта А4а.
(R)-2-метилпропан-2-сульфинамид подвергаются восстановительному аминированию с получением А5а; 3) А5а и сульфидрил из 5,6-конденсированного гетероарила подвергаются реакции перекрестного сочетания Бухвальда-Хартвига с образованием А6а; 4) с А6а снимают защиту в кислой среде с получением продукта А4а.
Способ В
Пример
Исходные материалы по настоящему изобретению были синтезированы в соответствии со способами, известными в данной области техники, или приобретены у химических компаний, таких как ABCR GmbH&Co. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc. и Beijing Ouhe Technology Co., Ltd.
Структуры соединений определяли методами ядерного магнитного резонанса (ЯМР) или масс-спектрометрии (MS). Для определения ЯМР использовали ЯМР-спектрометр Bruker Ascend 400 МГц, растворители представляли собой дейтерированный диметилсульфоксид (DMSO-d6), дейтерированный хлороформ (CDCl3) или дейтерированный метанол (CD3OD) и т.д., внутренний стандарт представлял собой тетраметилсилан (TMS), а химические сдвиги были даны в единицах 10-6 (ppm). Для определения MS использовали масс-спектрометр Agilent SQD (ESI) (Agilent 6120).
Для определения методом высокоэффективной жидкостной хроматографии (HPLC) использовали Высокоэффективный Жидкостной Хроматограф Agilent 1260 DAD (колонка Poroshell120 ЕС-С18, 50 × 3,0 мм, 2,7 мкм) или Высокоэффективный Жидкостной Хроматограф Waters Arc (колонка Sunfire С18, 150 × 4,6 мм, 5 мкм).
Если в вариантах выполнения изобретения не указано иное, реакции проводили при комнатной температуре (от 20 до 30°С).
Если в вариантах выполнения изобретения не указано иное, реакции проводили в атмосфере аргона или азота. Атмосфера аргона или азота означает, что реакционная колба соединена с баллоном с аргоном или азотом объемом около 1 л.
Под атмосферой водорода подразумевается, что реакционная колба соединена с баллоном с водородом объемом около 1 л после вакуумирования и заполнения водородом (повторяется три раза).
В микроволновой реакции использовали Микроволновый Реактор СЕМ Discover-SP.
Реакционные процессы в примерах контролировали либо с помощью системы Agilent LC/MS (1260/6120), либо с помощью тонкослойной хроматографии (TLC), в которой пластины силикагеля имели толщину от 0,15 до 0,2 мм (GF254, Qingdao Haiyang Chemical Co., Ltd.).
Соединения очищали с помощью колоночной хроматографии или TLC, где для колоночной хроматографии использовали силикагель от 200 до 300 меш (Qingdao Haiyang Chemical Co., Ltd.), а для TCX использовали пластины силикагеля GF254 (Qingdao Haiyang Chemical Co., Ltd.) с толщиной от 0,4 до 0,5 мм.
Системы растворителей-элюентов для колоночной хроматографии или TLC обычно включали: а) систему дихлорметан-метанол, б) систему петролейный эфир-этилацетат или те системы, которые показаны в примерах. Объемное соотношение растворителей регулировали в соответствии с различной полярностью соединений или дополнительно регулировали путем добавления небольшого количества триэтиламина или других кислотных или основных реагентов.
Соединения альтернативно очищали с помощью Автоматизированной Системы Подготовки Waters MS (детектор масс-спектрометра: SQD2) с обращенно-фазовой колонкой (XBridge-C18, 19×150 мм, 5 мкм) с градиентным элюированием при скорости потока 20 мл/мин в подходящих ацетонитрил/вода (содержащих 0,1% трифторуксусной или муравьиной кислоты или 0,05% водный раствор аммиака) в соответствии с полярностью соединений. В некоторых примерах 1н. разбавленную соляную кислоту добавляли после очистки с помощью автоматизированной системы приготовления с последующим удалением растворителя при пониженном давлении с получением гидрохлоридных солей.
Аббревиатура DMF относится к N,N-диметилформамиду.
Аббревиатура DIPEA относится к N,N-диизопропилэтиламину.
Аббревиатура DBU относится к 1,8-диазабицикло[5.4.0]ундец-7-ену.
Аббревиатура NBS относится к N-бромсукцинимиду.
Аббревиатура NIS относится к N-йодсукцинимиду.
Аббревиатура XantPhos относится к 9,9-диметил-4,5-бис(дифенилфосфино)ксантену.
Аббревиатура Pd2(dba)3 относится к трис(дибензилиденацетон)дипалладию.
Реагент Кастроса относится к бензотриазол-1-илокситрис(диметиламино)фосфония гексафторфосфату.
Пример 1
(R)-5-((1Н-пирроло[2,3-b]пиридин-4-ил)тио)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-3-метилпиримидин-4(3H)-она гидрохлорид
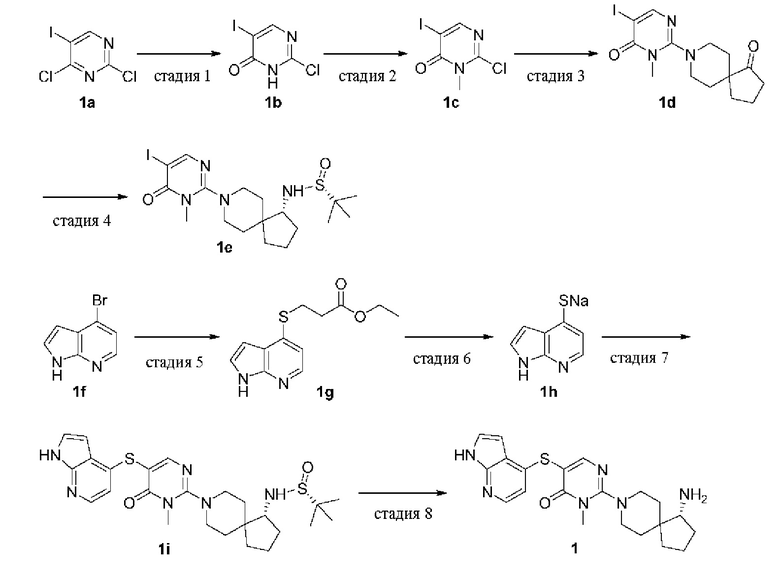
Стадия 1
2-Хлор-5-йодпиримидин-4(3H)-он (1b)
2,4-Дихлор-5-йодпиримидин 1а (8,0 г, 29,1 ммоль) растворяли в тетрагидрофуране (THF) (100 мл) и добавляли раствор гидроксида натрия (1н., 45 мл) при 0°С; реакционную смесь нагревали вплоть до комнатной температуры и перемешивали в течение 16 ч. Значение рН доводили лимонной кислотой до кислого и реакционную смесь экстрагировали этилацетатом (60 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли (30 мл × 3), сушили над безводным сульфатом натрия и удаляли растворитель при пониженном давлении с получением целевого продукта 1b (6,7 г, твердое вещество) с выходом 91%.
MS m/z (ESI): 257 [M+1]
Стадия 2
2-Хлор-5-йод-3-метилпиримидин-4(3H)-он (1с)
1b (5,6 г, 21,7 ммоль) растворяли в THF (100 мл), добавляли йодметан (3,7 г, 26 ммоль) и DIPEA (8,4 г, 65,1 ммоль), нагревали до 60°С и перемешивали в течение 16 ч. Полученную смесь выливали в воду (120 мл) и экстрагировали этилацетатом (80 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли (30 мл × 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 3/7) с получением целевого продукта 1 с (2,2 г, твердое вещество) с выходом 37%.
MS m/z (ESI): 271 [M+1]
Стадия 3
8-(5-Йод-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-8-азаспиро[4.5]декан-1-он (1d)
К раствору 1 с (1,0 г, 3,65 ммоль) в ацетонитриле (25 мл) добавляли 8-азаспиро[4.5]декан-1-он (гидрохлорид, 586 мг, 3,1 ммоль) и карбонат калия (1,51 г, 11 ммоль) и реакционную смесь нагревали до 80°С и перемешивали в течение 16 ч. После охлаждения до комнатной температуры реакционную смесь выливали в воду (50 мл) и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли (20 мл × 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 7/3) с получением целевого продукта 1d (1,2 г, твердое вещество) с выходом 85%.
MS m/z (ESI): 388 [M+1]
Стадия 4
(R)-N-((R)-8-(5-йод-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамид (1е)
(R)-2-метилпропан-2-сульфинамид (750 мг, 6,2 ммоль) и тетраэтилтитанат (2,82 г, 12,4 ммоль) добавляли к раствору 1d (1,2 г, 3,1 ммоль) в THF (30 мл) и реакционную смесь нагревали до 90°С и перемешивали в течение 16 ч. После охлаждения до 0°С к смеси добавляли метанол (10 мл) и боргидрид лития в THF (2,0 М, 1,5 мл, 3 ммоль) и перемешивали в течение 1 ч. Реакцию гасили раствором хлорида аммония и полученную смесь фильтровали. Фильтрат экстрагировали этилацетатом (25 мл × 3); органические фазы объединяли, промывали насыщенным раствором соли (20 мл × 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 2/1) с получением целевого продукта 1е (1,4 г, твердое вещество) с выходом 93%.
MS m/z (ESI): 493 [M+1]
Стадия 5
Этил-3-((1Н-пирроло[2,3-b]пиридин-4-ил)тио)пропионат (1g)
В раствор 4-бром-1Н-пирроло[2,3-b]пиридина 1f (300 мг, 1,52 ммоль) в диоксане (20 мл) добавляли этил 3-меркаптопропаноат (408 мг, 3,04 ммоль), DIPEA (588 мг, 4,56 ммоль), Pd2(dba)3 (140 мг, 0,152 ммоль) и XantPhos (132 мг, 0,228 ммоль). Реакционную смесь нагревали до 100°С в атмосфере азота и проводили реакцию в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 7/3) с получением целевого продукта 1g (200 мг, масло) с выходом 53%.
MS m/z (ESI): 251 [M+1]
Стадия 6
1Н-пирроло[2,3-6]пиридин-4-тиолат натрия (1h)
Метоксид натрия в метаноле (30%, 158 мг, 0,88 ммоль) добавляли к раствору 1g (200 мг, 0,8 ммоль) в метаноле (20 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении с получением целевого продукта 1h (100 мг, твердое вещество) с выходом 73%. Продукт использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 151 [M+1]
Стадия 7
(R)-N-((R)-8-(5-((1Н-пирроло[2,3-b]пиридин-4-ил)тио)-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамид (1i)
1h (55 мг, 0,32 ммоль) растворяли в диоксане (6 мл), добавляли 1е (82 мг, 0,16 ммоль), DIPEA (128 мг, 0,99 ммоль), Pd2(dba)3 (30 мг, 0,033 ммоль) и XantPhos (29 мг, 0,049 ммоль). Реакционную смесь нагревали до 80°С в атмосфере азота и проводили реакцию в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан = 1/9) с получением целевого продукта 1i (30 мг, твердое вещество) с выходом 37%.
MS m/z (ESI): 515 [M+1]
Стадия 8
(R)-5-((1Н-пирроло[2,3-b]пиридин-4-ил)тио)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-3-метилпиримидин-4(3H)-она гидрохлорид (1)
1i (30 мг, 0,058 ммоль) растворяли в метаноле (5 мл), добавляли HCl в диоксане (4,0 М, 5 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой высокоэффективной жидкостной хроматографией (препаративная RP-HPLC) с получением целевого продукта 1 (7,1 мг, твердое вещество) с выходом 31%.
MS m/z (ESI): 411 [M+1]
1Н ЯМР (400 МГц, DMSO-d6) δ 12.68 (s, 1H), 8.24 - 8.14 (m, 5Н), 7.68 (s, 1H), 6.91 - 6.87 (m, 1Н), 6.74 (s, 1H), 3.75 - 3.64 (m, 2H), 3.43 (s, 3H), 3.19 - 3.08 (m, 3H), 2.08 - 2.05 (m, 1H), 1.92 - 1.64 (m, 7H), 1.54 - 1.51 (m, 1H), 1.44 - 1.41 (m, 1H).
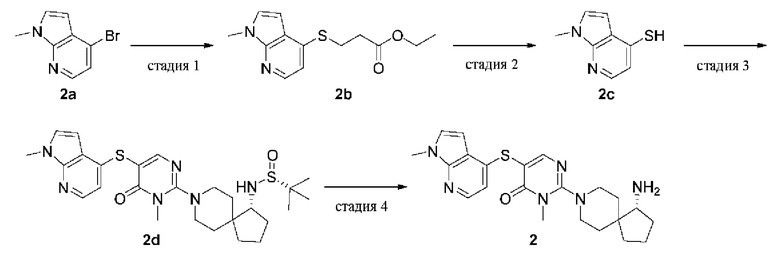
Пример 2
(R)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-3-метил-5-((1-метил-1Н-пирроло[2,3-b]пиридин-4-ил)тио)пиримидин-4(3H)-она формиат
Стадия 1
Этил 3-((1-метил-1Н-пирроло[2,3-b]пиридин-4-ил)тио)пропионат (2b)
Pd2(dba)3 (87 мг, 0,095 ммоль) добавляли к смеси 4-бром-1-метил-1Н-пирроло[2,3-b]пиридина 2а (200 мг, 0,95 ммоль), этил 3-меркаптопропаноата (255 мг, 1,9 ммоль), DIPEA (368 мг, 2,85 ммоль), XantPhos (55 мг, 0,095 ммоль) и диоксана (10 мл) и реакционную смесь нагревали до 100°С в атмосфере азота и проводили реакцию в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, добавляли воду (10 мл) и экстрагировали этилацетатом (20 мл × 3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали для удаления осушителя, из фильтрата удаляли растворитель при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 3/7) с получением целевого продукта 2b (240 мг, твердое вещество) с выходом 96%.
MS m/z (ESI): 265 [M+1]
Стадия 2
1-Метил-1Н-пирроло[2,3-b]пиридин-4-тиол (2с)
К раствору соединения 2b (240 мг, 0,91 ммоль) в THF (5 мл) добавляли трет-бутоксид калия (204 мг, 1,82 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь подкисляли соляной кислотой (6н.) до рН=4 и экстрагировали этилацетатом (20 мл). Органическую фазу сушили над безводным сульфатом натрия, осушитель удаляли фильтрованием, из фильтрата удаляли растворитель при пониженном давлении с получением целевого продукта 2с (130 мг, твердое вещество) с выходом 87%.
MS m/z (ESI): 165 [M+1]
Стадия 3
(R)-2-метил-N-((R)-8-(1-метил-5-((1-метил-1Н-пирроло[2,3-b]пиридин-4-ил)тио)-6-оксо-1,6-дигидропиримидин-2-ил)-8-азаспиро[4.5]декан-1-ил)пропан-2-сульфинамид (2d)
Pd2(dba)3 (20 мг, 0,022 ммоль) добавляли в смесь 2 с (50 мг, 0,24 ммоль), 1е (110 мг, 0,22 ммоль), DIPEA (85 мг, 0,66 ммоль), XantPhos (13 мг, 0,022 ммоль) и диоксана (5 мл) и реакционную смесь нагревали до 100°С в атмосфере азота и проводили реакцию в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан = 1/9) с получением целевого продукта 2d (80 мг, твердое вещество) с выходом 68%.
MS m/z (ESI): 529 [M+1]
Стадия 4
(R)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-3-метил-5-((1-метил-1Н-пирроло[2,3-b]пиридин-4-ил)тио)пиримидин-4(3H)-она формиат (2)
2d (80 мг, 0,14 ммоль) растворяли в метаноле (8 мл), добавляли раствор HCl в диоксане (4,0 М, 2 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 2 (21,7 мг, твердое вещество) с выходом 34%.
MS m/z (ESI): 425 [M+1]
1H ЯМР (400 МГц, DMSO-d6) δ 8.34 (s, 1H), 8.14 (s, 1H), 8.03 (d, J=5.1 Гц, 1H), 7.51 (d, J=3.5 Гц, 1H), 6.58 (d, J=5.1 Гц, 1H), 6.44 (d, J=3.5 Гц, 1H), 3.80 (s, 3H), 3.66-3.58 (m, 2H), 3.41 (s, 3H), 3.07 (t, J=11.5 Гц, 2H), 2.99 - 2.96 (m, 1H), 2.0 - 1.92 (m, 1H), 1.80-1.68 (m,4H), 1.60 - 1.53 (m, 3H), 1.40 (d, J=12.9 Гц, 1H), 1.33 (d, J=13.1 Гц, 1H).
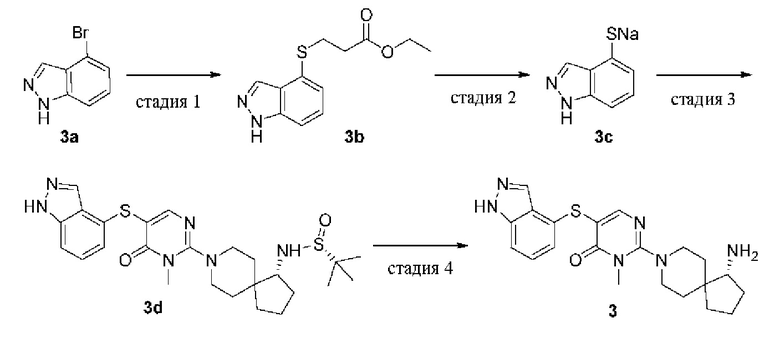
Пример 3
(R)-5-((1Н-индазол-4-ил)тио)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-3-метилпиримидин-4(3H)-она гидрохлорид
Стадия 1
Этил 3-((1Н-индазол-4-ил)тио)пропионат (3b)
В раствор 4-бром-1H-индазола 3а (500 мг, 2,53 ммоль) в диоксане (20 мл) добавляли этил 3-меркаптопропаноат (680 мг, 5,06 ммоль), DIPEA (979 мг, 7,59 ммоль), Pd2(dba)3 (231 мг, 0,253 ммоль) и XantPhos (219 мг, 0,397 ммоль). Реакционную смесь нагревали до 100°С в атмосфере азота и проводили реакцию в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 2,6/1) с получением целевого продукта 3b (560 мг, твердое вещество) с выходом 89%.
MS m/z (ESI): 251 [M+1]
Стадия 2
1H-индазол-4-тиолат натрия (3с)
Метоксид натрия в метаноле (30%, 443,5 мг, 2,46 ммоль) добавляли к раствору 3b (560 мг, 2,24 ммоль) в метаноле (20 мл) и смесь перемешивали при 30°С в течение 1 ч. Из реакционной смеси удаляли растворитель при пониженном давлении с получением целевого продукта 3 с (200 мг, твердое вещество) с выходом 51%. Продукт использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 151 [M+1]
Стадия 3
(R)-N-((R)-8-(5-((1Н-индазол-4-ил)тио)-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамид (3d)
В раствор 3с (70 мг, 0,41 ммоль) в диоксане (6 мл) добавляли 1е (50 мг, 0,10 ммоль), DIPEA (37,2 мг, 0.30 ммоль), Pd2(dba)3 (9,1 мг, 0,01 ммоль) и XantPhos (8,7 мг, 0,015 ммоль). Реакционную смесь нагревали до 80°С в атмосфере азота и проводили реакцию в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан = 1/9) с получением целевого продукта 3d (50 мг, твердое вещество) с выходом 96%.
MS m/z (ESI): 515 [M+1]
Стадия 4
(R)-5-((1Н-индазол-4-ил)тио)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-3-метилпиримидин-4(3H)-она гидрохлорид (3)
3d (50 мг, 0,097 ммоль) растворяли в метаноле (5 мл), добавляли раствор HCl в диоксане (4,0 М, 5 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении, а остаток очищали препаративной RP-HPLC с получением целевого продукта 3(16 мг, твердое вещество) с выходом 41%.
MS m/z (ESI): 411 [M+1]
1H ЯМР (400 МГц, DMSO-d6) δ 8.14 (brs, 3H), 8.06 (s, 1H), 7.97 (s, 1H), 7.40 (d, J=8.4 Гц, 1H), 7.24 (dd, J=8.3, 7.2 Гц, 1H), 6.83 (d, J=6.8 Гц, 1H), 3.60 (d, J=13.5 Гц, 1H), 3.52 (d,J=13.3 Гц, 1H), 3.40 (s, 3H), 3.17 - 3.12 (m, 1H), 3.05 - 2.98 (m, 2H), 2.07 - 2.04 (m, 1H), 1.77- 1.62 (m, 7H), 1.49 (d, J=12.8 Гц, 1H), 1.39 (d, J=13.1 Гц, 1H).
Пример 4
(R)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-3-метил-5-(пиразоло[1,5-a]пиридин-4-илтио)пиримидин-4(3H)-она формиат
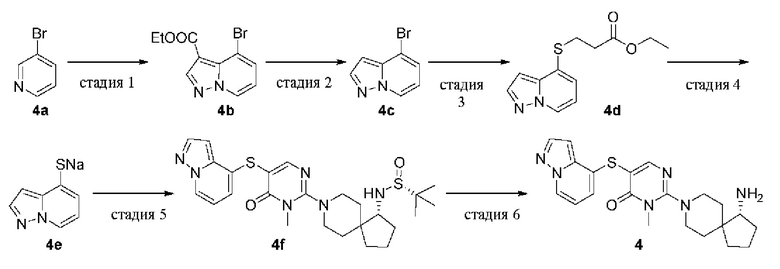
Стадия 1
Этил 4-бромпиразоло[1,5-а]пиридин-3-карбоксилат (4b)
К раствору 3-бромпиридина 4а (5,0 г, 32,0 ммоль) в ацетонитриле (25 мл) добавляли о-(2,4-динитрофенил)гидроксиламин (6,37 г, 32,0 ммоль), и смесь нагревали до 40°С, и перемешивали в течение 16 ч. Из реакционной смеси удаляли растворитель при пониженном давлении, остаток суспендировали в простом эфире и сушили с получением желтого твердого вещества (3,8 г).
Вышеупомянутый продукт (2,0 г, 5,6 ммоль) растворяли в DMF (20 мл), добавляли карбонат калия (12,1 г, 88,1 ммоль) и этилпропиолат (7,1 г, 52,8 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 20 ч. Полученную смесь выливали в воду (50 мл) и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли (20 мл × 3), сушили над безводным сульфатом натрия и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 3/7) с получением целевого продукта 4b (350 мг, твердое вещество) с выходом 8%.
MS m/z (ESI): 269 [M+1]
Стадия 2
4-Бромпиразоло[1,5-a]пиридин (4с)
4b (350 мг, 1,30 ммоль) растворяли в растворе бромистоводородной кислоты (12 мл), нагревали до 100°С и перемешивали в течение 6 ч. Реакционную смесь охлаждали до комнатной температуры, нейтрализовали раствором гидроксида натрия (2н.) и экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли (15 мл × 3), сушили над безводным сульфатом натрия и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 1/4) с получением целевого продукта 4с (200 мг, твердое вещество) с выходом 79%.
MS m/z (ESI): 197 [M+1]
Стадия 3
Этил 3-(пиразоло[1,5-а]пиридин-4-илтио)пропионат (4d)
Этил 3-меркаптопропаноат (680 мг, 5,06 ммоль), DIPEA (394 мг, 3,06 ммоль), Pd2(dba)3 (93,3 мг, 0,102 ммоль) и XantPhos (88,4 мг, 0,153 ммоль) добавляли к раствору 4с (200 мг, 1,02 ммоль) в диоксане (20 мл). Реакционную смесь нагревали до 100°С в атмосфере азота и проводили реакцию в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 3/7) с получением целевого продукта 4d (240 мг, твердое вещество) с выходом 94%.
MS m/z (ESI): 251 [M+1]
Стадия 4
Пиразоло[1,5-а]пиридин-4-тиолат натрия (4е)
Метоксид натрия в метаноле (30%, 182 мг, 1,01 ммоль) добавляли к раствору 4d (230 мг, 0,92 ммоль) в метаноле (20 мл) и реакционную смесь перемешивали при 30°С в течение 4 ч. Из реакционной смеси удаляли растворитель при пониженном давлении с получением целевого продукта 4е (150 мг, твердое вещество) с выходом 95%. Продукт использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 151 [M+1]
Стадия 5
(R)-2-метил-N-((R)-8-(1-метил-6-оксо-5-(пиразоло[1,5-а]пиридин-4-илтио)-1,6-дигидропиримидин-2-ил)-8-азаспиро[4.5]декан-1-ил)пропан-2-сульфинамид (4f)
1e (70 мг, 0,142 ммоль), DIPEA (54,9 мг, 0,426 ммоль), Pd2(dba)3 (12,9 мг, 0,0142 ммоль) и XantPhos (12,3 мг, 0,0213 ммоль) добавляли к 4е (85,3 мг, 0,495 ммоль) в диоксане (6 мл). Реакционную смесь нагревали до 80°С в атмосфере азота и проводили реакцию в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан = 1/13) с получением целевого продукта 4f (40 мг, твердое вещество) с выходом 55%.
MS m/z (ESI): 515 [M+1]
Стадия 6
(R)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-3-метил-5-(пиразоло[1,5-а]пиридин-4-илтио)пиримидин-4(3H)-она формиат (4)
4f (40 мг, 0,078 ммоль) растворяли в метаноле (5 мл), добавляли HCl в диоксане (4,0 М, 5 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 4 (16 мг, твердое вещество) с выходом 51%.
MS m/z (ESI): 411 [M+1]
1H ЯМР (400 МГц, DMSO-d6) δ 8.56 (d, J=6.9 Гц, 1H), 8.39 (s, 1H), 8.09 (s, 1H), 8.03 (d, J=2.2 Гц, 1H), 6.89 (d, J=7.0 Гц, 1H), 6.80 (t, J=7.0 Гц, 1H), 6.63 (d, J=1.7 Гц, 1H), 3.64 - 3.53 (m, 2H), 3.39 (s, 3Н), 3.05 - 2.97 (m, 3Н), 1.98 - 1.95 (m, 1H), 1.80 - 1.67 (m, 4H), 1.64- 1.53 (m, 3Н), 1.40 (d, J=13.0 Гц, 1H), 1.32 (d, J=13.3 Гц, 1Н).
Пример 5
(R)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-3-метил-5-(пиразоло[1,5-а]пиридин-5-илтио)пиримидин-4(3H)-она гидрохлорид
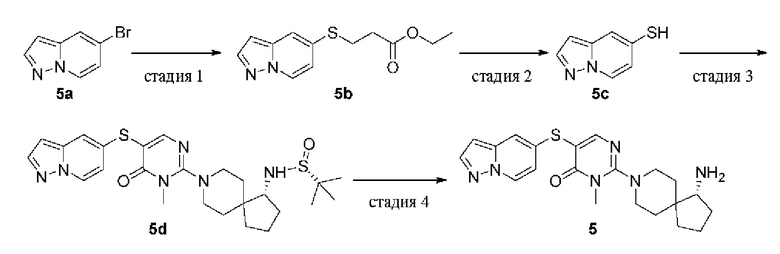
Стадия 1
Этил 3-(пиразоло[1,5-а]пиридин-5-илтио)пропионат (5b)
Этил 3-меркаптопропаноат (326 мг, 2,43 ммоль), DIPEA (627 мг, 4,86 ммоль), Pd2(dba)3 (148 мг, 0,162 ммоль) и XantPhos (140 мг, 0,243 ммоль) добавляли к раствору 5-бромпиразоло[1,5-а]пиридина 5а (320 мг, 1,62 ммоль) в диоксане (20 мл). Реакционную смесь нагревали до 100°С в атмосфере азота и проводили реакцию в течение 6 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 3/1) с получением целевого продукта 5b (400 мг, масло) с выходом 97%.
MS m/z (ESI): 251 [M+1]
Стадия 2
Пиразоло[1,5-а]пиридин-5-тиол (5с)
Метоксид натрия в метаноле (30%, 317 мг, 1,76 ммоль) добавляли к раствору 5b (400 мг, 1,6 ммоль) в метаноле (20 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь выливали в воду (100 мл), доводили до слабокислого рН с помощью лимонной кислоты и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли (20 мл × 3), сушили над безводным сульфатом натрия, фильтровали для удаления осушителя и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 2/3) с получением целевого продукта 5с (100 мг, масло) с выходом 42%.
MS m/z (ESI): 151 [M+1]
Стадия 3
(R)-2-метил-N-((R)-8-(1-метил-6-оксо-5(пиразоло[1,5-а]пиридин-5-илтио)-1,6-дигидропиримидин-2-ил)-8-азаспиро[4.5]декан-1-ил)пропан-2-сульфинамид (5d)
5с (30 мг, 0,20 ммоль) растворяли в диоксане (6 мл) и добавляли 1е (50 мг, 0,10 ммоль), DIPEA (38,7 мг, 0,30 ммоль), Pd2(dba)3 (9,0 мг, 0,010 ммоль) и XantPhos (8,5 мг, 0,015 ммоль). Реакционную смесь нагревали до 80°С в атмосфере азота и проводили реакцию в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан = 1/9) с получением целевого продукта 5d (60 мг, твердое вещество, неочищенный продукт).
MS m/z (ESI): 515 [M+1]
Стадия 4
(R)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-3-метил-5-(пиразоло[1,5-a]пиридин-5-илтио)пиримидин-4(3H)-она гидрохлорид (5)
5d (60 мг, неочищенный продукт) растворяли в метаноле (5 мл), добавляли HCl в диоксане (4,0 М, 5 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 5 (14,1 мг, твердое вещество) с выходом 34% в две стадии.
MS m/z (ESI): 411 [M+1]
1Н ЯМР (400 МГц, DMSO-d6) δ 8.56 (d, J=7.3 Гц, 1H), 8.13 (s, 1H), 8.06 (brs, 3Н), 7.95 (s, 1H), 7.94 (d, J=2.2 Гц, 2H), 7.33 (d,J=1.4 Гц, 1H), 6.68 (dd,J=7.3, 2.1 Гц, 1H), 6.44 (dd, J=2.2, 0.7 Гц, 1H), 3.66 (d, J=13.4 Гц, 1H), 3.58 (d, J=13.3 Гц, 1H), 3.41 (s, 3H), 3.21-3.14 (m, 1H), 3.10-3.02 (m, 2H), 2.07 - 2.03 (m, 1H), 1.87- 1.58 (m, 7H), 1.50 (d, J=12.8 Гц, 1H), 1.41 (d, J=12.9 Гц, 1H).
Пример 6
(R)-5-((1Н-индазол-5-ил)тио)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-3-метилпиримидин-4(3H)-она гидрохлорид
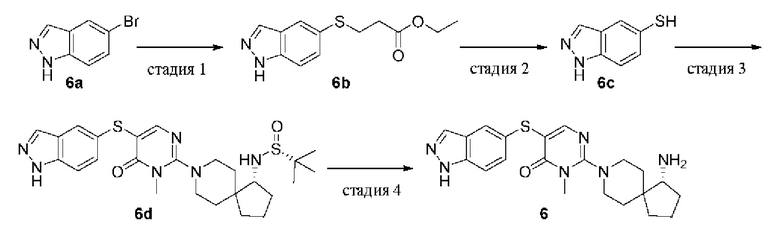
Стадия 1
Этил 3-((1Н-индазол-5-ил)тио)пропионат (6b)
Pd2(dba)3 (243 мг, 0,265 ммоль) добавляли в смесь 5-бром-1H-индазола 6а (520 мг, 2,65 ммоль), этил 3-меркаптопропаноата (533 мг, 3,98 ммоль), DIPEA (1,02 г, 7,95 ммоль), XantPhos (153 мг, 0,265 ммоль) и диоксана (10 мл), а затем реакционную смесь нагревали до 100°С в атмосфере азота и проводили реакцию в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 1/1) с получением целевого продукта 6b (630 мг, твердое вещество) с выходом 95%.
MS m/z (ESI): 251 [M+1]
Стадия 2
1H-Индазол-5-тиол (6c)
К раствору 6b (500 мг, 2,0 ммоль) в THF (10 мл) добавляли трет-бутоксид калия (672 мг, 6,0 ммоль) и смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь подкисляли соляной кислотой (6н.) до pH=4 и экстрагировали дихлорметаном (20 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали для удаления осушителя и удаляли растворитель из фильтрата при пониженном давлении с получением целевого продукта 6с (300 мг, твердое вещество) с выходом 100%.
MS m/z (ESI): 151 [M+1]
Стадия 3
(R)-N-((R)-8-(5-((1Н-индазол-5-ил)тио)-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамид (6d)
Pd2(dba)3 (18 мг, 0,02 ммоль) добавляли в смесь 6 с (60 мг, 0,4 ммоль), 1е (100 мг, 0,2 ммоль), DIPEA (77 мг, 0,6 ммоль), XantPhos (12 мг, 0,02 ммоль) и диоксана (5 мл), а затем реакционную смесь нагревали до 100°С в атмосфере азота и проводили реакцию в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 1/1) с получением целевого продукта 6d (70 мг, твердое вещество) с выходом 67%.
MS m/z (ESI): 515 [M+1]
Стадия 4
(R)-5-((1Н-индазол-5-ил)тио)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-3-метилпиримидин-4(3H)-она гидрохлорид (6)
6d (70 мг, 0,14 ммоль) растворяли в метаноле (5 мл), добавляли HCl в диоксане (4,0 М, 5 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 6 (30 мг, твердое вещество) с выходом 54%.
MS m/z (ESI): 411 [M+1]
1H ЯМР (400 МГц, DMSO-d6) δ 8.28 (s, 3H), 8.05 (s, 1H), 7.79 (s, 1H), 7.69 (s, 1H), 7.55 (d, J=8.7 Гц, 1H), 7.34 (dd, J=8.7, 1.6 Гц, 1H), 3.56 (d, J=13.2 Гц, 1H), 3.49 (d, J=13.5 Гц, 1H), 3.39 (s, 3Н), 3.17 - 3.13 (m, 1Н), 3.06 - 2.96 (m, 2H), 2.07 - 2.01 (m, 1H), 1.90 - 1.61 (m, 7H), 1.52 (d, J=13.1 Гц, 1H), 1.39 (d, J=13.0 Гц, 1H).
Пример 7
(R)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-5-((4-хлор-2-метил-2Н-индазол-5-ил)тио)-3-метилпиримидин-4(3H)-она гидрохлорид
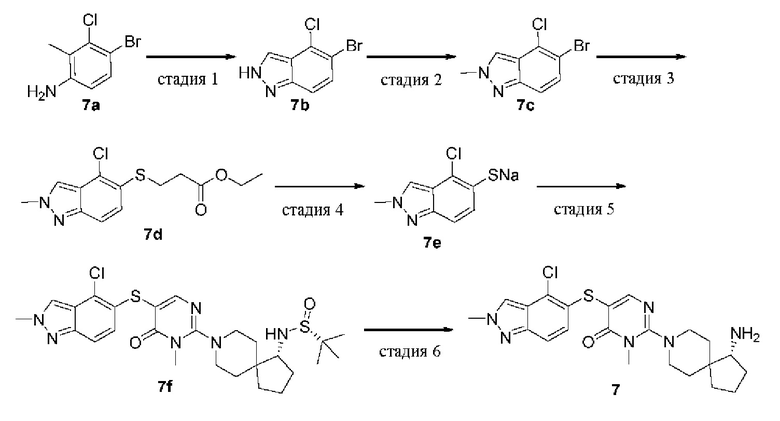
Стадия 1
5-Бром-4-хлор-2Н-индазол (7b)
Водный раствор (4 мл) нитрита натрия (396 мг, 5,73 ммоль) добавляли к 4-бром-3-хлор-2-метиланилину 7а (1,0 г, 4,58 ммоль) в уксусной кислоте (20 мл), охлажденному на ледяной бане, и смесь перемешивали при комнатной температуре в течение 1 ч. Большую часть растворителя удаляли на роторном испарителе, а остаток суспендировали в воде (30 мл) и фильтровали. Осадок с фильтра промывали водой (25 мл × 3) и сушили на воздухе с получением целевого продукта 7b (780 мг, твердое вещество) с выходом 74%. Продукт использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 231 [M+1]
Стадия 2
5-Бром-4-хлор-2-метил-2Н-индазол (7с)
Триметилоксония тетрафторборат (733 мг, 4,95 ммоль) добавляли к 7b (760 мг, 3,3 ммоль) в этилацетате (15 мл), охлажденному на ледяной бане, и реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь разбавляли петролейным эфиром (30 мл), перемешивали 10 мин и фильтровали. Фильтрат добавляли в этилацетат (20 мл) и промывали раствором бикарбоната натрия (30 мл × 3) и насыщенным раствором соли (20 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 1,4/1) с получением целевого продукта 7 с (460 мг, твердое вещество) с выходом 57%.
MS m/z (ESI): 245 [M+1]
Стадия 3
Этил 3-((4-хлор-2-метил-2Н-индазол-5-ил)тио)пропионат (7d)
Этил 3-меркаптопропаноат (483 мг, 3,6 ммоль), DIPEA (697 мг, 5,4 ммоль), Pd2(dba)3 (165 мг, 0,18 ммоль) и XantPhos (156 мг, 0,27 ммоль) добавляли к раствору 7 с (440 мг, 1,8 ммоль) в диоксане (20 мл). Реакционную смесь нагревали до 100°С в атмосфере азота и проводили реакцию в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 1,7/1) с получением целевого продукта 7d (300 мг, твердое вещество) с выходом 59%.
MS m/z (ESI): 299 [M+1]
Стадия 4
4-Хлор-2-метил-2Н-индазол-5-тиолат натрия (7е)
Метоксид натрия в метаноле (30%, 93 мг, 0,52 ммоль) добавляли к раствору 7d (140 мг, 0,47 ммоль) в метаноле (20 мл) и перемешивали при 30°С в течение 32 ч. Из реакционной смеси удаляли растворитель при пониженном давлении с получением целевого продукта 7е (100 мг, твердое вещество) с выходом 96%. Продукт использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 199 [M+1]
Стадия 5
(R)-N-((R)-8-(5-((4-хлор-2-метил-2Н-индазол-5-ил)тио)-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамид (7f)
7е (80 мг, 0,36 ммоль) растворяли в диоксане (8 мл) и добавляли 1е (100 мг, 0,20 ммоль), DIPEA (77 мг, 0,60 ммоль), Pd2(dba)3 (18 мг, 0,020 ммоль) и XantPhos (18 мг, 0,030 ммоль). Реакционную смесь нагревали до 90°С в атмосфере азота и проводили реакцию в течение 2 ч. После охлаждения до комнатной температуры реакционную смесь выливали в смешанный растворитель из этилацетата (15 мл) и петролейного эфира (15 мл) и фильтровали. Осадок с фильтра сушили с получением целевого продукта 7f (61 мг, твердое вещество) с выходом 54%.
MS m/z (ESI): 563 [M+1]
Стадия 6
(R)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-5-((4-хлор-2-метил-2Н-индазол-5-ил)тио)-3-метилпиримидин-4(3H)-она гидрохлорид (7)
7f (61 мг, 0,11 ммоль) растворяли в метаноле (5 мл), добавляли HCl в диоксане (4,0 М, 5 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 7 (16 мг, твердое вещество) с выходом 31%.
MS m/z (ESI): 459 [M+l]
1H ЯМР (400 МГц, DMSO-d6) δ 8.43 (s, 1H), 8.12 (brs, 3H), 7.90 (s, 1H), 7.51 (dd, J=9.0, 0.7 Гц, 1H), 6.99 (d, J=9.0 Гц, 1H), 4.17 (s, 3H), 3.60 (d, J=13.3 Гц, 1H), 3.53 (d, J=13.3 Гц, 1H), 3.40 (s, 3H), 3.16 - 3.13 (m, 1H), 3.06 - 2.98 (m, 2H), 2.07 - 2.04 (m, 1H), 1.87 - 1.60 (m, 7H), 1.49 (d, J=13.0 Гц, 1H), 1.39 (d, J=13.1 Гц, 1H).
Пример 8
(R)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-5-((4-хлорпиразоло[1,5-а]пиридин-5-ил)тио)-3-метилпиримидин-4(3H)-она формиат
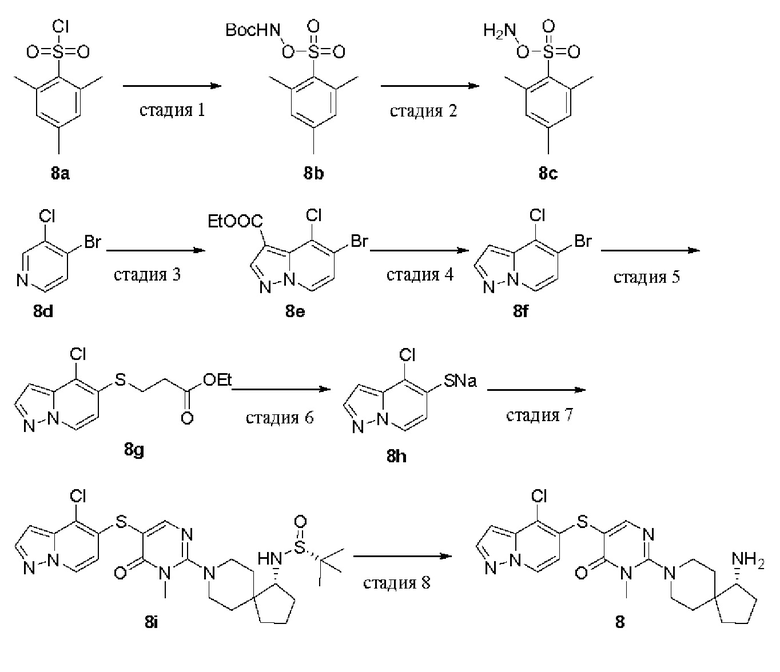
Стадия 1
трет-Бутил [[(2,4,6-триметилфенил)сульфонил]окси]карбамат (8b) 2,4,6-Триметилбензолсульфонилхлорид 8а (2,0 г, 9,15 ммоль) и трет-бутилгидроксикарбамат (1,22 г, 9,15 ммоль) растворяли в трет-бутилметиловом эфире (30 мл) и охлаждали до 0°С, по каплям добавляли триэтиламин (1,4 мл, 10,1 ммоль) и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 4 ч. Реакционную смесь промывали водой, органическую фазу сушили над безводным сульфатом натрия и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=1/5) с получением целевого продукта 8b (2,5 г, твердое вещество) с выходом 87%.
MS m/z (ESI): 338 [М+23], 216 [M+1-100]
Стадия 2
O-(мезитилсульфонил)гидроксиламин (8с)
8b (2,5 г, 7,93 ммоль) добавляли порциями в трифторуксусную кислоту (20 мл) при 0°С и реакционную смесь перемешивали при 0°С в течение 2 ч. Медленно добавляли ледяную воду и смесь перемешивали в течение 15 мин. После фильтрации осадок с фильтра промывали водой до тех пор, пока фильтрат не становился нейтральным. Осадок с фильтра сушили с получением целевого продукта 8с (1,5 г, твердое вещество) с выходом 88%.
MS m/z (ESI): 216 [M+1]
Стадия 3
Этил 5-бром-4-хлорпиразоло[1,5-a]пиридин-3-карбоксилат (8е)
4-Бром-3-хлорпиридин 8d (1,4 г, 7,28 ммоль) растворяли в 25 мл ацетонитрила, добавляли 8 с (1,6 г, 7,28 ммоль) и реакционную смесь оставляли на ночь при перемешивании при 40°С. После фильтрации осадок с фильтра сушили с получением желтого твердого продукта (1,7 г).
Вышеуказанный продукт (1,6 г, 3,92 ммоль) и этилпропиолат (0,42 г, 4,32 ммоль) растворяли в DMF (15 мл), добавляли карбонат калия (1,1 г, 7,84 ммоль) и перемешивали при комнатной температуре в течение 3 ч. Реакцию гасили водой и реакционную смесь экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=1/2) с получением целевого продукта 8е (120 мг, твердое вещество) с выходом 5,8%.
MS m/z (ESI): 303, 305 [M+1]
Стадия 4
5-Бром-4-хлорпиразоло[1,5-а]пиридин (8f)
8е (110 мг, 0,36 ммоль) растворяли в растворе бромистоводородной кислоты (3 мл), нагревали до 100°С и перемешивали в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры, нейтрализовали раствором гидроксида натрия (2 М) и экстрагировали дихлорметаном (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=3/7) с получением целевого продукта 8f (65 мг, твердое вещество) с выходом 77%.
MS m/z (ESI): 231, 233 [M+1]
Стадия 5
Этил 3-((4-хлорпиразоло[1,5-a]пиридин-5-ил)тио)пропионат (8g)
Этил 3-меркаптопропаноат (38 мг, 0,29 ммоль), DIPEA (67 мг, 0,52 ммоль), Pd2(dba)3 (24 мг, 0,026 ммоль) и XantPhos (30 мг, 0,052 ммоль) добавляли к раствору 8f (60 мг, 0,26 ммоль) в диоксане (5 мл). Реакционную смесь нагревали до 100°С в атмосфере азота и проводили реакцию в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/28) с получением целевого продукта 8g (60 мг, твердое вещество) с выходом 81%.
MS m/z (ESI): 285 [M+1]
Стадия 6
4-Хлорпиразоло[1,5-a]пиридин-5-тиолат натрия (8h)
Метоксид натрия в метаноле (30%, 45,5 мг, 0,25 ммоль) добавляли к раствору 8g (60 мг, 0,21 ммоль) в метаноле (3 мл) и смесь перемешивали при 30°С в течение 4 ч. Из реакционной смеси удаляли растворитель при пониженном давлении с получением целевого продукта 8h (40 мг, твердое вещество) с выходом 92%. Продукт использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 185 [M+1]
Стадия 7
(R)-N-((R)-8-(5-((4-хлорпиразоло[1,5-а]пиридин-5-ил)тио)-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамид (8i)
8h (40 мг, 0,19 ммоль) растворяли в диоксане (5 мл) и добавляли 1е (95 мг, 0,19 ммоль), DIPEA (49 мг, 0,38 ммоль), Pd2(dba)3 (17 мг, 0,02 ммоль) и XantPhos (22 мг, 0,04 ммоль). Реакционную смесь нагревали до 100°С в атмосфере азота и проводили реакцию в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/28) с получением целевого продукта 8i (32 мг, масло) с выходом 30%.
MS m/z (ESI): 549 [M+1]
Стадия 8
(R)-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-5-((4-хлорпиразоло[1,5-a]пиридин-5-ил)тио)-3-метилпиримидин-4(3H)-она формиат (8)
8i (32 мг, 0,06 ммоль) растворяли в метаноле (2 мл), добавляли HCl в диоксане (4,0 М, 1 мл) и перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 8(13 мг, твердое вещество) с выходом 47%.
MS m/z (ESI): 445 [M+1]
1H ЯМР (400 МГц, DMSO-d6) δ 8.54 (d, J=7.3 Гц, 1H), 8.35 (s, 1H), 8.19 (s, 1H), 8.06 (d, J=2.2 Гц, 1H), 6.60 (d, J=1.8 Гц, 1H), 6.48 (d, J=7.3 Гц, 1H), 3.69-3.60 (m, 2Н), 3.41 (s, 3Н), 3.09 (t, J=11.7 Гц, 2Н), 3.02-2.99 (m, 1H), 2.00-1.91 (m, 1H), 1.76-1.50 (m, 7Н), 1.42 (d,J=13.2 Гц, 1H), 1.35 (d, J=12.9 Гц, 1H).
Пример 9
(R)-5-((2-(1-амино-8-азаспиро[4.5]декан-8-ил)-1-метил-6-оксо-1,6-дигидропиримидин-5-ил)тио)-4-хлор-2-метил-2Н-индазол-3-карбонитрил
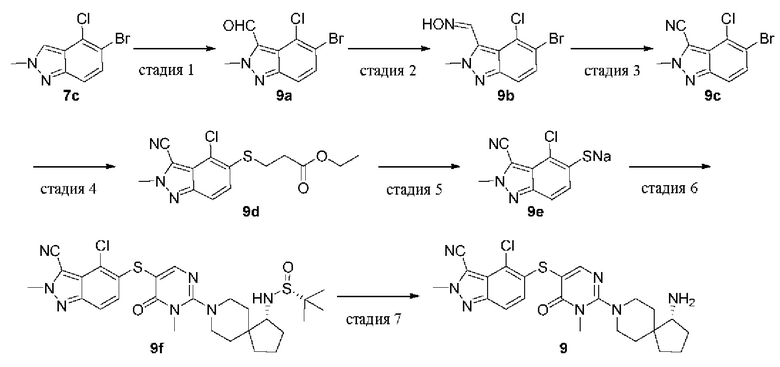
Стадия 1
5-Бром-4-хлор-2-метил-2Н-индазол-3-карбальдегид (9а)
7с (800 мг, 3,28 ммоль) растворяли в THF (20 мл), охлаждали до -78°С, добавляли диизопропиламид лития в THF (2 М, 2,95 мл, 5,9 ммоль), перемешивали в течение 90 мин, нагревали вплоть до 0°С и продолжали перемешивание в течение 30 мин. Затем реакционную смесь охлаждали до -78°С, добавляли по каплям DMF (0,76 мл), перемешивали в течение 0,5 ч, нагревали до комнатной температуры и продолжали перемешивание в течение 2 ч. Реакцию гасили насыщенным раствором хлорида аммония и смесь экстрагировали этилацетатом. Органическую фазу сушили над безводным сульфатом натрия, фильтровали для удаления осушителя и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=3/7) с получением целевого продукта 9а (520 мг, твердое вещество) с выходом 60%.
MS m/z (ESI): 273 [M+1]
Стадия 2
5-Бром-4-хлор-2-метил-2Н-индазол-3-карбоксальдоксим (9а) 9а (490 мг, 1,8 ммоль) растворяли в изопропаноле (8 мл), метаноле (8 мл) и воде (8 мл), добавляли гидроксиламина гидрохлорид (497 мг, 7,2 ммоль) и карбонат натрия (763 мг, 7,2 ммоль), нагревали до 50°С и перемешивали в течение 16 ч. Полученную смесь выливали в воду (60 мл) и экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли (20 мл × 3), сушили над безводным сульфатом натрия и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=3/7) с получением целевого продукта 9b (470 мг, твердое вещество) с выходом 92%.
MS m/z (ESI): 288 [M+1]
Стадия 3
5-Бром-4-хлор-2-метил-2Н-индазол-3-карбонитрил (9с)
9b (470 мг, 1,63 ммоль) растворяли в ацетонитриле (30 мл), добавляли ацетат меди (147 мг, 0,81 ммоль), нагревали до 85°С и перемешивали в течение ночи. Реакционную смесь удаляли при пониженном давлении, остаток выливали в воду (50 мл) и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли (25 мл × 3), сушили над безводным сульфатом натрия и удаляли растворитель при пониженном давлении с получением целевого продукта 9с (390 мг, твердое вещество) с выходом 89%.
MS m/z (ESI): 270 [M+1]
Стадия 4
Этил 3-((4-хлор-3-циано-2-метил-2Н-индазол-5-ил)тио)пропионат (9d) Этил 3-меркаптопропаноат (385 мг, 2,8 ммоль), DIPEA (542 мг, 4,2 ммоль), Pd2(dba)3 (128 мг, 0,14 ммоль) и XantPhos (121 мг, 0,21 ммоль) добавляли к раствору 9с (380 мг, 1,4 ммоль) в диоксане (5 мл). Реакционную смесь нагревали до 100°С в атмосфере азота и проводили реакцию в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=1/1) с получением целевого продукта 9d (350 мг, твердое вещество) с выходом 77%.
MS m/z (ESI): 324 [M+1]
Стадия 5
4-Хлор-3-циано-2-метил-2Н-индазол-5-тиолат натрия (9е)
Метоксид натрия в метаноле (30%, 130 мг, 0,72 ммоль) добавляли к раствору 9d (195 мг, 0,60 ммоль) в метаноле (20 мл) и перемешивали при 30°С в течение 16 ч. Из реакционной смеси удаляли растворитель при пониженном давлении с получением целевого продукта 9е (220 мг, твердое вещество, неочищенный продукт). Продукт использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 224 [M+1]
Стадия 6
(R)-N-((R)-8-(5-((4-хлор-3-циано-2-метил-2Н-индазол-5-ил)тио)-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамид (9f)
9е (89,2 мг, 0,36 ммоль) растворяли в диоксане (6 мл) и добавляли 1е (120 мг, 0,24 ммоль), DIPEA (92,8 мг, 0,72 ммоль), Pd2(dba)3 (21,9 мг, 0,024 ммоль) и XantPhos (20,8 мг, 0,036 ммоль). Реакционную смесь нагревали до 80°С в атмосфере азота и проводили реакцию в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/12) с получением целевого продукта 9f (90 мг, твердое вещество) с выходом 63% в две стадии.
MS m/z (ESI): 588 [M+1]
Стадия 7
(R)-5-((2-(1-амино-8-азаспиро[4.5]декан-8-ил)-1-метил-6-оксо-1,6-дигидропиримидин-5-ил)тио)-4-хлор-2-метил-2Н-индазол-3-карбонитрила формиат (9)
9f (90 мг, 0,15 ммоль) растворяли в метаноле (10 мл), добавляли HCl в диоксане (4,0 М, 10 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 9(14 мг, твердое вещество) с выходом 19%.
MS m/z (ESI): 484 [M+1]
1H ЯМР (400 МГц, DMSO-d6) δ 8.14 (s, 1H), 7.79 (brs, 3Н), 7.75 (d, J=9.0 Гц, 1H), 7.05 (d, J=9.1 Гц, 1H), 4.34 (s, 3H), 3.69 (d,J=14.1 Гц, 1H), 3.61 (d,J=14 Гц, 1H), 3.41 (s, 3H), 3.24-3.16 (m, 1H), 3.13-3.06 (m, 2H), 2.08-2.04 (m, 1H), 1.80-1.64 (m, 7H), 1.49-1.41 (m, 2H).
Пример 10
(S)-2-(4-амино-2-окса-8-азаспиро[4.5]декан-8-ил)-5-((4-хлор-2-метил-2Н-индазол-5-ил)тио)-3-метилпиримидин-4(3Н)-он
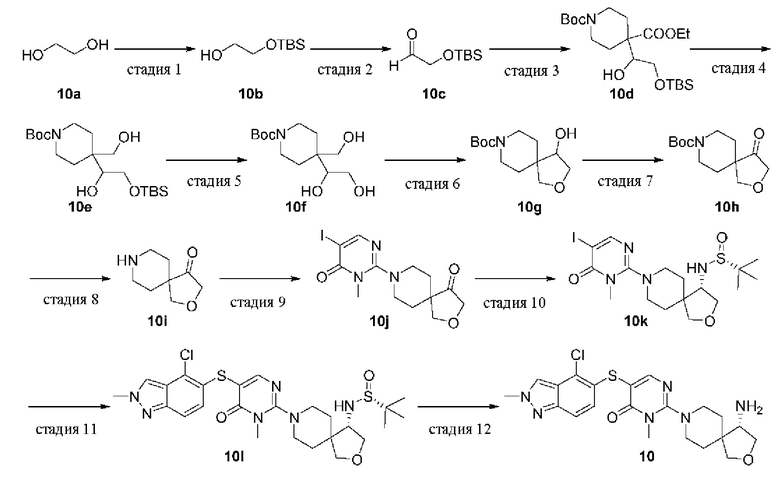
Стадия 1
2-((трет-Бутилдиметилсилил)окси)этан-1-ол (10b)
трет-Бутилдиметилсилилхлорид (50 г, 334 ммоль) добавляли по каплям в смесь этиленгликоля 10а (124 г, 2 моль), имидазола (34 г, 500 ммоль) и дихлорметана (500 мл) при 0°С и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли водой (300 мл) и дихлорметаном (600 мл) и разделяли. Водную фазу экстрагировали дихлорметаном (300 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=1/5) с получением целевого продукта 10b (35 г, масло) с выходом 10%.
MS m/z (ESI): 177 [M+1]
Стадия 2
2-((трет-Бутилдиметилсилил)окси)ацетальдегид (10с)
Оксалилхлорид (27,6 г, 218,9 ммоль) добавляли по каплям в дихлорметан (500 мл) при -30°С, и смесь охлаждали до -78°С, к ней по каплям добавляли диметилсульфоксид (21,7 г, 278,6 ммоль). Реакционную смесь перемешивали при -78°С в течение 30 минут, медленно добавляли раствор 10b (35 г, 199 ммоль) в дихлорметане (100 мл) и перемешивали в течение 1 ч. Добавляли по каплям триэтиламин (100,5 г, 995 ммоль) и смесь перемешивали при -78°С в течение 30 мин, а затем при комнатной температуре в течение ночи. Реакционную смесь последовательно промывали водой (300 мл), разбавленной соляной кислотой (1н., 400 мл × 2), насыщенным раствором бикарбоната натрия (400 мл) и насыщенным раствором соли (400 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением целевого продукта 10 с (34 г, масло) с выходом 98%.
1Н ЯМР (400 МГц, CDCl3) δ 9.60 (s, 1H), 4.11 (s, 2Н), 0.82 (s, 9Н), 0.00 (s, 6Н).
Стадия 3
1-трет-Бутил 4-этил-4-(2-((трет-бутилдиметилсилил)окси)-1-гидроксиэтил)пиперидин-1,4-дикарбоксилат (10 d)
1-трет-Бутил-4-этилпиперидин-1,4-дикарбоксилат (50 г, 197 ммоль) в THF (100 мл) добавляли по каплям к раствору диизопропиламида лития в THF (2 М, 148 мл, 296 ммоль) и THF (400 мл) при -10°С и реакционную смесь перемешивали при 0°С в течение 30 мин. Добавляли 10с (34 г, 197 ммоль) и реакционную смесь перемешивали при 0°С в течение 1 ч и при комнатной температуре в течение 1 ч. Добавляли смесь насыщенного раствора бикарбоната натрия и воды (400 мл, 1/4 об./об.), затем этилацетат (200 мл) и полученную смесь разделяли. Водную фазу экстрагировали этилацетатом (200 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=2/3) с получением целевого продукта 10d (75 г, масло) с выходом 88%.
MS m/z (ESI): 332 [M+1-100]
Стадия 4
трет-Бутил 4-(2-((трет-бутилдиметилсилил)окси)-1-гидроксиэтил)-4-(гидроксиметил)пиперидин-1-карбоксилат (10е)
Боргидрид лития в THF (2 М, 130 мл, 261 ммоль) добавляли к 10d (75 г, 174 ммоль) в THF (600 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. После охлаждения до 0°С реакцию гасили насыщенным раствором бикарбоната натрия и водой (150 мл, 1/2 об./об.), реакционную смесь разбавляли этилацетатом (200 мл) и фильтровали. Фильтрат отделяли и водную фазу экстрагировали этилацетатом (200 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением целевого продукта 10е (55 г, масло) с выходом 81%.
MS m/z (ESI): 290 [M+1-100]
Стадия 5
трет-Бутил 4-(1,2-дигидроксиэтил)-4-(гидроксиметил)пиперидин-1-карбоксилат (10f)
Фторид тетрабутиламмония в THF (1 М, 193 мл, 193 ммоль) добавляли к 10е (55 г, 128,5 ммоль) в THF (400 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли насыщенным раствором бикарбоната натрия и водой (100 мл, 1/2 об./об.), добавляли этилацетат (200 мл) и две фазы разделяли. Водную фазу экстрагировали этилацетатом (200 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = от 1/4 до 1/0) с получением целевого продукта 10f (23 г, масло) с выходом 65%.
MS m/z (ESI): 176 [M+1-100]
Стадия 6
трет-Бутил 4-гидрокси-2-окса-8-азаспиро[4.5]декан-8-карбоксилат (10g)
Гидрид натрия (60%, 11,7 г, 292,6 ммоль) суспендировали в THF (100 мл), охлаждали до 0°С и добавляли по каплям 10f (23 г, 83,6 ммоль) в THF (100 мл) и n-толуолсульфонилхлорид (16 г, 83,6 ммоль) в THF (200 мл). Реакционную смесь перемешивали при 0°С в течение 2 ч. Медленно добавляли насыщенный раствор хлорида аммония (50 мл) и реакционную смесь энергично перемешивали до прекращения образования газа. Добавляли насыщенный раствор хлорида аммония (100 мл) и насыщенный раствор соли (100 мл) и полученную смесь экстрагировали этилацетатом (200 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = от 0/1 до 1/0) с получением целевого продукта 10h (8,5 г, масло) с выходом 39%.
MS m/z (ESI): 158 [M+1-100]
Стадия 7
трет-Бутил 4-оксо-2-окса-8-азаспиро[4.5]декан-8-карбоксилат (10h) Реакционную смесь 10g (8,5 г, 32,9 ммоль), перйодинана Десс-Мартина (27,4 г, 64,56 ммоль) и дихлорметана (200 мл) перемешивали при 0°С в течение 2 ч. Добавляли насыщенный раствор бикарбоната натрия и насыщенный раствор тиосульфата натрия (100 мл, 1/1 об./об.) и полученную смесь энергично перемешивали. Две фазы разделяли и водную фазу экстрагировали дихлорметаном (200 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=2/3) с получением целевого продукта 10h (6,2 г, масло) с выходом 74%.
MS m/z (ESI): 156 [M+1-100]
Стадия 8
2-Окса-8-азаспиро[4.5]декан-4-он (10i)
HCl в диоксане (4 М, 2 мл) добавляли к раствору 10h (500 мг, 1,96 ммоль) в дихлорметане (8 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь удаляли при пониженном давлении с получением целевого продукта 10i (гидрохлорид, 370 мг, твердое вещество) с выходом 99%. Продукт использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 156 [M+1]
Стадия 9
8-(5-Йод-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-2-окса-8-азаспиро[4.5]декан-4-он (10j)
1c (100 мг, 0,37 ммоль) добавляли в смесь 10i (гидрохлорид, 106 мг, 0,555 ммоль), карбоната калия (153 мг, 1,11 ммоль) и ацетонитрила (4 мл) и смесь нагревали до 80°С и перемешивали в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры, смешивали с водой (5 мл) и этилацетатом (20 мл) и фазы разделяли. Водную фазу экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 1/4) с получением целевого продукта 10j (70 мг, твердое вещество) с выходом 49%.
MS m/z (ESI): 390 [M+l]
Стадия 10
(R)-N-((S)-8-(5-йод-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-2-окса-8-азаспиро[4.5]декан-4-ил)-2-метилпропан-2-сульфинамид (10k)
Реакционную смесь 10j (60 мг, 0,15 ммоль), (R)-2-метилпропан-2-сульфинамида (36,3 мг, 0,3 ммоль), тетраэтилтитаната (137 мг, 0,6 ммоль) и THF (5 мл) нагревали до 90°С и перемешивали в течение 4 ч. Смесь охлаждали до 0°С, добавляли метанол (2 мл) и боргидрид лития в THF (2,0 М, 0,15 мл, 0,3 ммоль) и перемешивали в течение 1 ч. Реакционную смесь смешивали с водой (5 мл) и дихлорметаном (20 мл) и разделяли два слоя. Водную фазу экстрагировали дихлорметаном (10 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан = 1/9) с получением целевого продукта 10k (70 мг, твердое вещество) с выходом 92%.
MS m/z (ESI): 495 [M+l]
Стадия 11
(R)-N-((S)-8-(5-((4-хлор-2-метил-2Н-индазол-5-ил)тио)-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-2-окса-8-азаспиро[4.5]декан-4-ил)-2-метилпропан-2-сульфинамид (101)
Pd2(dba)3 (12,8 мг, 0,014 ммоль) добавляли в смесь 10k (70 мг, 0,14 ммоль), 7е (37,4 мг, 0,17 ммоль), DIPEA (54 мг, 0,42 ммоль), XantPhos (8,1 мг, 0,014 ммоль) и диоксана (4 мл) и реакционную смесь нагревали до 100°С в атмосфере азота и проводили реакцию в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, смешивали с водой (5 мл) и дихлорметаном (20 мл) и два слоя разделяли. Водную фазу экстрагировали дихлорметаном (10 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан = 1/9) с получением целевого продукта 101 (50 мг, твердое вещество) с выходом 63%.
MS m/z (ESI): 565 [M+1]
Стадия 12
(S)-2-(4-амино-2-окса-8-азаспиро[4.5]декан-8-ил)-5-((4-хлор-2-метил-2Н-индазол-5-ил)тио)-3-метилпиримидин-4(3H)-он (10)
101 (50 мг, 0,09 ммоль) растворяли в метаноле (8 мл), добавляли HCl в диоксане (4,0 М, 2 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 10 (5,8 мг, твердое вещество) с выходом 14%.
MS m/z (ESI): 461 [M+1]
1H ЯМР (400 МГц, DMSO-d6) δ 8.43 (s, 1H), 7.91 (s, 1H), 7.51 (d, J=9.0 Гц, 1H), 6.99 (d, J=9.0 Гц, 1H), 4.18 (s, 3H), 4.02-3.95 (m, 1H), 3.69 (d, J=8.5 Гц, 1H), 3.63 (d, J=8.5 Гц, 1H), 3.39 (s, 3H), 3.15-3.13 (m, 1H), 3.08-3.00 (m, 2H), 1.84-1.76 (m, 1H), 1.72-1.65 (m, 1H), 1.54-1.43 (m, 2H).
Пример 11
2-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)-5-((4-хлор-2-метил-2Н-индазол-5-ил)тио)-3-метилпиримидин-4(3H)-она формиат
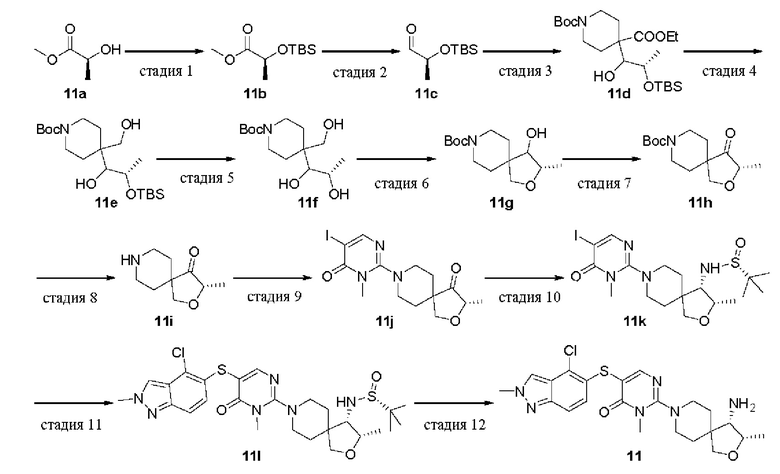
Стадия 1
Метил (S)-2-((трет-бутилдиметилсилил) окси)пропаноат (11b)
Метил (S)-2-гидроксипропаноат 11а (70 г, 672 ммоль) по каплям добавляли к имидазолу (68,5 г, 1008 ммоль) в дихлорметане (800 мл) при 0°С, медленно добавляли трет-бутилдиметилсилилхлорид (120 г, 807 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли водой (600 мл) и экстрагировали дихлорметаном (500 мл × 2). Органические фазы объединяли, промывали разбавленной соляной кислотой (1н., 300 мл × 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением целевого продукта 11b (150 г, масло) с выходом 99%.
1H ЯМР (400 МГц, CDCl3) δ 4.32 (q, J=6.7 Гц, 1H), 3.70 (s, 3Н), 1.38 (d, J=6.8 Гц, 3Н), 0.88 (s, 9Н), 0.08 (s, 3Н), 0.05 (s, 3Н).
Стадия 2
(S)-2-((трет-бутилдиметилсилил)окси)пропиональдегид (11с) 11b (151,2 г, 693 ммоль) в дихлорметане (1200 мл), охлаждали до -78°С, добавляли по каплям гидрид диизобутилалюминия в н-гексане (1 М, 832 мл, 832 ммоль) и реакционную смесь нагревали вплоть до -40°С и перемешивали в течение 1 ч. Реакционную смесь выливали в насыщенный раствор тартрата калия-натрия (140 мл), смешивали с простым эфиром (100 мл) и перемешивали при комнатной температуре в течение 2 ч. Два слоя разделяли и водную фазу экстрагировали простым эфиром. Органические фазы объединяли, промывали насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = 4/1) с получением целевого продукта 11с (62,3 г, масло) с выходом 48%.
1Н ЯМР (400 МГц, CDCl3) δ 9.60 (s, 1Н), 4.09 (q, J=6.8 Гц, 1Н), 1.27 (d, J=6.9 Гц, 3Н), 0.91 (s, 9Н), 0.10 (s, 3Н), 0.08 (s, 3Н).
Стадия 3
1-трет-Бутил 4-этил-4-(2S)-2-((трет-бутилдиметилсилил)окси)-1-гидроксипропил)пиперидин-1,4-дикарбоксилат (11d)
1-трет-Бутил 4-этилпиперидин-1,4-дикарбоксилат (72,8 г, 283,5 ммоль) в THF (50 мл) добавляли по каплям к раствору диизопропиламида лития в THF (1 М, 425 мл, 425 ммоль) и THF (800 мл) при -50°С и реакционную смесь перемешивали при -10°С в течение 1 ч. Добавляли 11с (53,3 г, 283,5 ммоль) и перемешивали при -10°С в течение 1 ч. Реакционную смесь нагревали до 0°С и перемешивали в течение 1 ч, и затем перемешивали при комнатной температуре в течение 1 ч. Добавляли смесь насыщенного раствора бикарбоната натрия и воды (400 мл, 1/4 об./об.), добавляли этилацетат (500 мл) и полученную смесь разделяли. Водную фазу экстрагировали этилацетатом (500 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением целевого продукта 11d (120 г, масло, сырой продукт). Продукт использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 346 [M+1-100]
Стадия 4
трет-Бутил 4-(2S)-2-((трет-бутилдиметилсилил)окси)-1-гидроксипропил)-4-(гидроксиметил)пиперидин-1-карбоксилат (11е)
Боргидрид лития в THF (2 М, 270 мл, 539,2 ммоль) добавляли к 11d (120 г, 269,6 ммоль) в THF (1200 мл) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. После охлаждения до 0°С реакцию гасили насыщенным раствором бикарбоната натрия и водой (600 мл, 1/2 об./об.), смесь разбавляли этилацетатом (800 мл) и фильтровали. Два слоя фильтрата разделяли и водную фазу экстрагировали этилацетатом (500 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли (300 мл × 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением целевого продукта 11е (90 г, масло, сырой продукт). Продукт использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 404 [M+1]
Стадия 5
трет-Бутил 4-((2S)-1,2-дигидроксипропил)-4-(гидроксиметил)пиперидин-1-карбоксилат (11f)
Фторид тетрабутиламмония в THF (1 М, 303 мл, 303 ммоль) добавляли к 11е (90 г, 223 ммоль) в THF (1300 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли насыщенным раствором бикарбоната натрия и водой (800 мл, 1/2 об./об.), добавляли этилацетат (800 мл), разделяли два слоя и водную фазу экстрагировали этилацетатом (500 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли (300 мл × 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=49/1) с получением целевого продукта 11f (24,5 г, масло) с выходом 26% в три стадии.
MS m/z (ESI): 290 [M+1]
Стадия 6
трет-Бутил (3S)-4-гидрокси-3-метил-2-окса-8-азаспиро[4.5]декан-8-карбоксилат (11g)
Гидрид натрия (60%, 10,8 г, 270,0 ммоль) суспендировали в THF (600 мл) и охлаждали до 0°С. Добавляли по каплям 11f (22,3 г, 77,1 ммоль) в THF (40 мл) и добавляли n-толуолсульфонилхлорид (14,6 г, 77,1 ммоль) в THF (50 мл). Реакционную смесь перемешивали при 0°С в течение 6 ч. После охлаждения до -20°С к реакционной смеси медленно добавляли насыщенный раствор хлорида аммония (10 мл) и энергично перемешивали до прекращения образования газа. Смесь экстрагировали этилацетатом (300 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли (150 мл × 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=1/1) с получением целевого продукта 11g (12,1 г, масло) с выходом 58%.
MS m/z (ESI): 172 [M+1-100]
Стадия 7
трет-Бутил (S)-3-метил-4-оксо-2-окса-8-азаспиро[4.5]декан-8-карбоксилат (11h) Периодинан Десс-Мартина (37,8 г, 89,2 ммоль) медленно добавляли к 11g (12,1 г, 44,6 ммоль) в дихлорметане (140 мл) при 0°С и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Добавляли насыщенный раствор бикарбоната натрия, пока раствор не стал нейтральным и раствор экстрагировали дихлорметаном (150 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли (80 мл × 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=3/7) с получением целевого продукта 11h (8,36 г, масло) с выходом 70%.
MS m/z (ESI): 170 [M+1-100]
Стадия 8
(S)-3-метил-2-окса-8-азаспиро[4.5]декан-4-он (11i)
11h (1,2 г, 4,46 ммоль) растворяли в HCl в диоксане (4 М, 20 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь удаляли при пониженном давлении с получением целевого продукта 11i (гидрохлорид, 800 мг, твердое вещество) с выходом 87%.
MS m/z (ESI): 170 [M+1]
Стадия 9
(S)-8-(5-йод-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-он (11j)
11i (гидрохлорид, 108 мг, 0,529 ммоль) и карбонат калия (199 мг, 1,44 ммоль) добавляли к 2-хлор-5-йод-3-метилпиримидин-4(3H)-ону 1 с (130 мг, 0,481 ммоль) в ацетонитриле (20 мл) и реакционную смесь нагревали до 80°С и перемешивали в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры, смешивали с водой (50 мл) и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли (20 мл × 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=1,3/1) с получением целевого продукта 11j (180 мг, твердое вещество) с выходом 92%.
MS m/z (ESI): 404 [M+1]
Стадия 10
(R)-N-((3S,4S)-8-(5-йод-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-ил)-2-метилпропан-2-сульфинамид (11k)
(R)-2-метилпропан-2-сульфинамид (108 мг, 0,892 ммоль) и тетраэтилтитанат (813 мг, 3,59 ммоль) добавляли к раствору 11j (180 мг, 0,446 ммоль) в THF (2 мл), нагревали до 90°С и перемешивали в течение 16 ч. После охлаждения до 0°С к смеси добавляли метанол (5 мл) и боргидрид лития в THF (2,0 М, 0,23 мл, 0,46 ммоль) и перемешивали в течение 1 ч. Реакцию гасили насыщенным раствором хлорида аммония и полученную смесь фильтровали. Фильтрат экстрагировали этилацетатом (25 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли (20 мл × 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/16) с получением целевого продукта 11k (90 мг, твердое вещество) с выходом 39%.
MS m/z (ESI): 509 [M+1]
Стадия 11
(R)-N-((3S,4S)-8-(5-((4-хлор-2-метил-2Н-индазол-5-ил)тио)-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-ил)-2-метилпропан-2-сульфинамид (11l)
7е (60 мг, 0,27 ммоль), DIPEA (60,7 мг, 0,471 ммоль), XantPhos (14,2 мг, 0,023 ммоль) и Pd2(dba)3 (14,4 мг, 0,0157 ммоль) добавляли к 11k (80 мг, 0,157 ммоль) в диоксане (5 мл). Реакционную смесь нагревали до 80°С в атмосфере азота и проводили реакцию в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/13) с получением целевого продукта 11l (55 мг, твердое вещество) с выходом 60%.
MS m/z (ESI): 579 [M+1]
Стадия 12
2-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)-5-((4-хлор-2-метил-2Н-индазол-5-ил)тио)-3-метилпиримидин-4(3H)-она формиат (11)
11l (55 мг, 0,095 ммоль) растворяли в метаноле (5 мл), смешивали с HCl в диоксане (4,0 М, 5 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 11 (16,8 мг, твердое вещество) с выходом 37%.
MS m/z (ESI): 475 [M+1]
1H ЯМР (400 МГц, DMSO-d6) δ 8.43 (s, 1H), 8.21 (s, 1H), 7.91 (s, 1H), 7.51 (d, J=9.1 Гц, 1H), 6.99 (d, J=9.0 Гц, 1H), 4.18 (s, 3H), 4.11-4.05 (m, 1H), 3.68 (d,J=8.6 Гц, 1H), 3.51 (d,J=8.6 Гц, 1H), 3.47-3.43 (m, 2H), 3.41 (s, 3H), 3.20-3.03 (m, 2H), 3.00 (d, J=4.9 Гц, 1H), 1.89-1.82 (m, 1H), 1.77-1.72 (m, 1H), 1.62-1.53 (m, 2H), 1.11 (d, J=6.4 Гц, 3H).
Пример 12
2-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)-5-((4-хлорпиразоло[1,5-a]пиридин-5-ил)тио)-3-метилпиримидин-4(3H)-она формиат
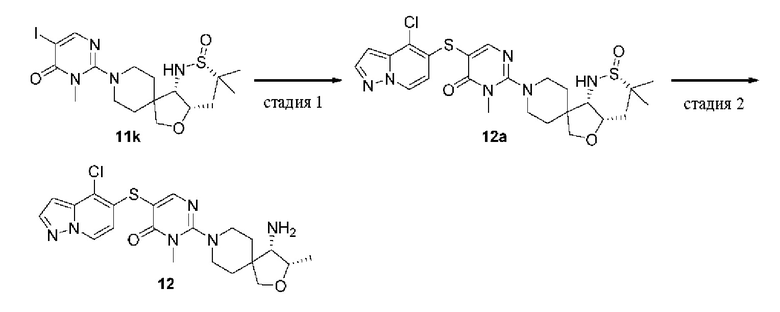
Стадия 1
(R)-N-((3S,4S)-8-(5-((4-хлорпиразоло[1,5-а]пиридин-5-ил)тио)-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-ил)-2-метилпропан-2-сульфинамид (12а)
11k (140 мг, 0,28 ммоль) растворяли в диоксане (16 мл) и добавляли 8h (60 мг, 0,29 ммоль), DIPEA (71,5 мг, 0,56 ммоль), Pd2(dba)3 (25,3 мг, 0,03 ммоль) и XantPhos (32 мг, 0,06 ммоль). Реакционную смесь нагревали до 100°С в атмосфере азота и проводили реакцию в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/25) с получением целевого продукта 12а (100 мг, твердое вещество) с выходом 64%.
MS m/z (ESI): 565 [M+1]
Стадия 2
2-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)-5-((4-хлорпиразоло[1,5-a]пиридин-5-ил)тио)-3-метилпиримидин-4(3H)-она формиат (12)
12а (86 мг, 0,166 ммоль) растворяли в метаноле (5 мл), смешивали с HCl в диоксане (4,0 М, 1 мл) и перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 12 (16,8 мг, твердое вещество) с выходом 19%.
MS m/z (ESI): 461 [M+1]
1H ЯМР (400 МГц, DMSO-d6) δ 8.51 (d, J=7.3 Гц, 1H), 8.26 (s, 1H), 8.17 (s, 1H), 8.04 (d,.7=2.1 Гц, 1H), 6.59 (d,J=1.5 Гц, 1H), 6.48 (d, J=7.3 Гц, 1H), 4.14-4.07 (m, 1H), 3.72 (d,J=8.7 Гц, 1H), 3.57-3.51 (m, 3H), 3.39 (s, 3H), 3.21-3.07 (m, 2H), 3.06 (d, J=4.9 Гц, 1H), 1.88-1.75 (m, 2H), 1.66-1.55 (m, 2H), 1.12 (d, J=6.4 Гц, 3Н).
Пример 13
(R)-5-((1Н-индазол-4-ил)тио)-6-амино-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-3-метилпиримидин-4(3H)-она формиат
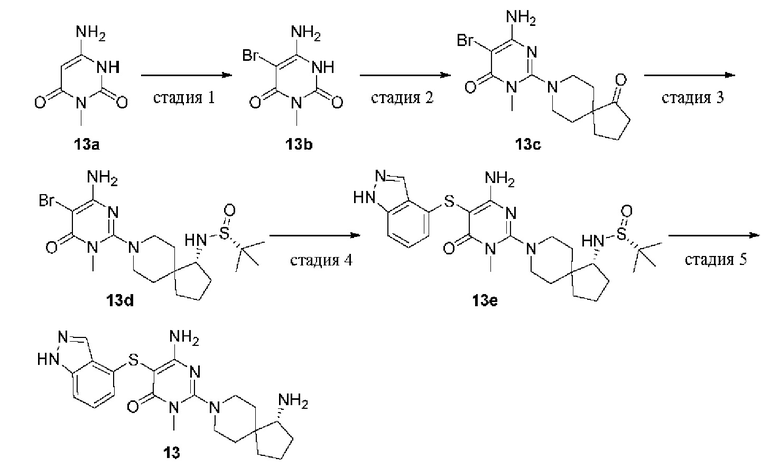
Стадия 1
6-Амино-5-бром-3-метилпиримидин-2,4(1H,3H)-дион (13b)
6-Амино-3-метилпиримидин-2,4(1H,3H)-дион (1,0 г, 7,08 ммоль) растворяли в DMF (6 мл), добавляли NBS (1,38 г, 7,8 ммоль) и перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли водой (20 мл) и фильтровали. Осадок с фильтра промывали водой (5 мл × 2) и сушили в вакууме с получением целевого продукта 13b (1,0 г, твердое вещество) с выходом 64%.
MS m/z (ESI): 220 [M+1]
Стадия 2
8-(4-Амино-5-бром-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-8-азаспиро[4.5]декан-1-он (13с)
13b (200 мг, 0,91 ммоль) растворяли в DMF (5 мл), добавляли 8-азаспиро[4,5]декан-1-он (167 мг, 1,1 ммоль), реагент Кастроса (1,2 г, 2,73 ммоль) и DBU (692 мг, 4,55 ммоль) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли этилацетатом (50 мл) и промывали водой (10 мл × 3) и насыщенным раствором соли (10 мл). Органическую фазу удаляли при пониженном давлении, а остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=7/3) с получением целевого продукта 13с (170 мг, масло) с выходом 53%.
MS m/z (ESI): 355 [M+1]
Стадия 3
(R)-N-((R)-8-(4-амино-5-бром-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамид (13d)
(R)-2-метилпропан-2-сульфинамид (110 мг, 0,9 ммоль) и тетраэтилтитанат (411 мг, 1,8 ммоль) добавляли к 13 с (160 мг, 0,45 ммоль) в THF (15 мл), нагревали до 90°С и перемешивали в течение ночи. После охлаждения до комнатной температуры к смеси добавляли метанол (5 мл) и боргидрид лития в THF (2,0 М, 0,5 мл) и перемешивали в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/20) с получением целевого продукта 13d (200 мг, масло) с выходом 96%.
MS m/z (ESI): 460 [M+1]
Стадия 4
(R)-N-((R)-8-(5-((1Н-индазол-4-ил)тио)-4-амино-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамид (13е)
13d (100 мг, 0,22 ммоль) растворяли в диоксане (10 мл) и добавляли 1H-индазол-4-тиолат натрия 3с (75 мг, 0,44 ммоль), DIPEA (84 мг, 0,66 ммоль), Pd2(dba)3 (40 мг, 0,044 ммоль) и XantPhos (51 мг, 0,088 ммоль). Реакционную смесь нагревали до 100°С в атмосфере азота и перемешивали в течение ночи. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали препаративной тонкослойной хроматографией с получением целевого продукта 13е (30 мг, твердое вещество) с выходом 26%.
MS m/z (ESI): 530 [M+1]
Стадия 5
(R)-5-((1Н-индазол-4-ил)тио)-6-амино-2-(1-амино-8-азаспиро[4.5]декан-8-ил)-3-метилпиримидин-4(3H)-она формиат (13)
13е (30 мг, 0,056 ммоль) растворяли в метаноле (2 мл), добавляли HCl в диоксане (4,0 М, 2 мл) и перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 13 (5 мг, твердое вещество) с выходом 21%.
MS m/z (ESI): 426 [M+1]
1H ЯМР (400 МГц, CD3OD) δ 8.54 (s, 1H), 8.12 (s, 1H), 7.30 (d, J=8.3 Гц, 1H), 7.20 (t, J=7.7 Гц, 1H), 6.78 (d, J=7.0 Гц, 1H), 3.65-3.57 (m, 2H), 3.42 (s, 3H), 3.27-3.25 (m, 1H), 3.09 (t, J=12.3 Гц, 2H), 2.22-2.17 (m, 1H), 1.85-1.72 (m, 7H), 1.54 (t, J=13.5 Гц, 2H).
Пример 14
6-Амино-2-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)-5-((4-хлор-2-метил-2Н-индазол- 5-ил)тио)-3-метилпиримидин-4(3H)-она формиат
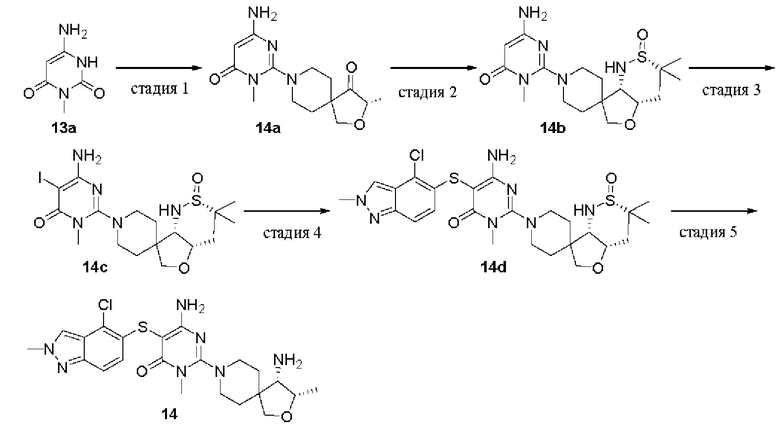
Стадия 1
(S)-8-(4-амино-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-он (14а)
6-Амино-3-метилпиримидин-2,4(1H,3H)-дион 13а (400 мг, 2,8 ммоль) растворяли в DMF (25 мл), добавляли 11i (576 мг, 2,8 ммоль), реактив Кастроса (6,18 г, 14 ммоль) и DBU (1,29 г, 8,5 ммоль) и перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой (60 мл) и реакционную смесь экстрагировали смешанным растворителем из дихлорметана и метанола (10/1 об./об., 50 мл × 3). Органические фазы объединяли, промывали насыщенным раствором соли (20 мл × 3), сушили над безводным сульфатом натрия и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/9) с получением целевого продукта 14а (700 мг, масло) с выходом 84%.
MS m/z (ESI): 293 [M+1]
Стадия 2
(R)-N-((3S,4S)-8-(4-амино-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-ил)-2-метилпропан-2-сульфинамид (14b)
(R)-2-Метилпропан-2-сульфинамид (557 мг, 4,6 ммоль) и тетраэтилтитанат (2,06 г, 9,2 ммоль) добавляли к 14а (700 мг, 2,3 ммоль) в THF (30 мл) и реакционную смесь нагревали до 90°С и перемешивали в течение 16 ч. После охлаждения до 0°С к смеси добавляли метанол (10 мл) и боргидрид лития в THF (2,0 М, 1,35 мл) и перемешивали в течение 1 ч. Реакцию гасили раствором хлорида аммония, смесь фильтровали и фильтрат экстрагировали этилацетатом (25 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/10) с получением целевого продукта 14b (80 мг, твердое вещество) с выходом 8,7%.
MS m/z (ESI): 398 [M+1]
Стадия 3
(R)-N-((3S,4S)-8-(4-амино-5-йод-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-ил)-2-метилпропан-2-сульфинамид (14с)
14b (80 мг, 0,2 ммоль) растворяли в ацетонитриле (10 мл), добавляли NIS (54,2 мг, 0,24 ммоль) и перемешивали при комнатной температуре в течение 1,5 ч. Из реакционной смеси удаляли растворитель при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/12) с получением целевого продукта 14 с (100 мг, твердое вещество) с выходом 95%.
MS m/z (ESI): 524 [M+1]
Стадия 4
(R)-N-((3S,4S)-8-(4-амино-5-((4-хлор-2-метил-2Н-индазол-5-ил)тио)-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-ил)-2-метилпропан-2-сульфинамид (14d)
14с (50 мг, 0,095 ммоль) растворяли в диоксане (6 мл) и добавляли 7е (22,8 мг, 0,114 ммоль), DIPEA (36,7 мг, 0,285 ммоль), Pd2(dba)3 (8,6 мг, 0,0095 ммоль) и XantPhos (8,2 мг, 0,014 ммоль). Реакционную смесь нагревали до 80°С в атмосфере азота и перемешивали в течение 16 ч. После охлаждения до комнатной температуры реакционную смесь разбавляли этилацетатом (100 мл) и промывали насыщенным раствором соли. Органическую фазу сушили над безводным сульфатом натрия и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/14) с получением целевого продукта 14d (46 мг, твердое вещество) с выходом 82%.
MS m/z (ESI): 594 [M+1]
Стадия 5
6-Амино-2-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)-5-((4-хлор-2-метил-2Н-индазол-5-ил)тио)-3-метилпиримидин-4(3H)-она формиат (14)
14d (46 мг, 0,077 ммоль) растворяли в метаноле (5 мл), добавляли HCl в диоксане (4,0 М, 5 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 14 (6,5 мг, твердое вещество) с выходом 17%. MS m/z (ESI): 490 [M+1]
1H ЯМР (400 МГц, DMSO-d6) δ 8.33 (s, 1H), 8.20 (s, 1H), 7.46 (d, J=9.0 Гц, 1H), 6.76 (d, J=9.0 Гц, 1H), 4.15 (s, 3H), 4.10-4.06 (m, 1H), 3.69 (d, J=8.5 Гц, 1H), 3.52 (d, J=8.5 Гц, 1H), 3.39-3.37 (m, 2H), 3.29 (s, 3H), 3.11-2.97 (m, 3H), 1.85-1.71 (m, 2H), 1.63-1.54 (m, 2H), 1.11 (d, J=6.4 Гц, 3Н).
Пример 15
6-Амино-2-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)-5-((4-хлорпиразоло[1,5-a]пиридин-5-ил)тио)-3-метилпиримидин-4(3H)-она формиат
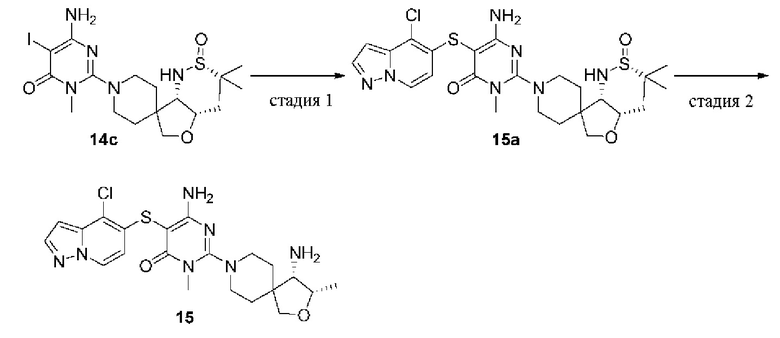
Стадия 1
(R)-N-((3S,4S)-8-(4-амино-5-((4-хлорпиразоло[1,5-a]пиридин-5-ил)тио)-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-ил)-2-метилпропан-2-сульфинамид (15а)
14с (40 мг, 0,076 ммоль) растворяли в диоксане (4 мл) и добавляли 8h (31,3 мг, 0,152 ммоль), DIPEA (29,4 мг, 0,228 ммоль), Pd2(dba)3 (6,9 мг, 0,0076 ммоль) и XantPhos (6,5 мг, 0,0114 ммоль). Реакционную смесь нагревали до 80°С в атмосфере азота и перемешивали в течение 16 ч. После охлаждения до комнатной температуры реакционную смесь разбавляли этилацетатом (100 мл) и промывали насыщенным раствором соли. Органическую фазу сушили над безводным сульфатом натрия и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/17) с получением целевого продукта 15а (35 мг, твердое вещество) с выходом 79%.
MS m/z (ESI): 580 [M+1]
Стадия 2
6-Амино-2-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)-5-((4-хлорпиразоло[1,5-a]пиридин-5-ил)тио)-3-метилпиримидин-4(3H)-она формиат (15)
15а (35 мг, 0,06 ммоль) растворяли в метаноле (5 мл), добавляли HCl в диоксане (4,0 М, 5 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 15 (4,2 мг, твердое вещество) с выходом 15%.
MS m/z (ESI): 476 [M+1]
1H ЯМР (400 МГц, CD3OD) δ 8.52 (s, 1H), 8.28 (d, J=8.0 Гц, 1H), 7.93 (d, J=2.3 Гц, 1H), 6.55 (d,J=1.7 Гц, 1H), 6.45 (d, J=7.3 Гц, 1H), 4.28-4.23 (m, 1H), 3.89 (d,J=8.9 Гц, 1H), 3.77 (d, J=8.9 Гц, 1H), 3.63-3.56 (m, 2H), 3.43 (s, 3H), 3.24-3.02 (m, 3H), 1.97-1.90 (m, 2H), 1.79-1.69 (m, 2H), 1.26 (d, J=6.5 Гц, 3H).
Пример 16
2-(4-(Аминометил)-4-метилпиперидин-1-ил)-5-((4-хлор-2-метил-2Н-индазол-5-ил)тио)-3-метилпиримидин-4(3H)-она формиат
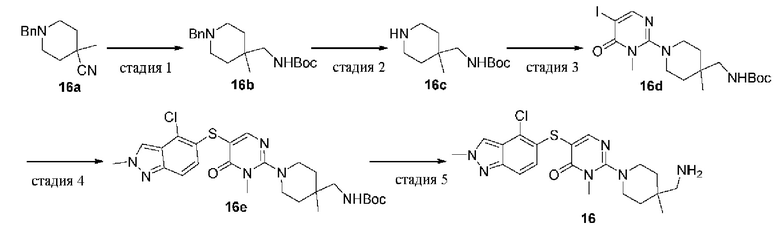
Стадия 1
трет-Бутил ((1-бензил-4-метилпиперидин-4-ил)метил)карбамат (16b) Реакционную смесь 1-бензил-4-метилпиперидин-4-карбонитрила 16а (2 г, 9,3 ммоль), ди-трет-бутилдикарбоната(6.1 г, 27,9 ммоль), гексагидрата хлорида никеля (2,2 г, 9,3 ммоль) и метанола (50 мл) перемешивали при комнатной температуре в течение 15 мин. После охлаждения до 0°С реакционную смесь смешивали с боргидридом натрия (1,77 г, 46,5 ммоль), нагревали до комнатной температуры и перемешивали в течение 8 ч. Из реакционной смеси удаляли растворитель при пониженном давлении и остаток суспендировали в дихлорметане и фильтровали. Из фильтрата удаляли растворитель при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=1/1) с получением целевого продукта 16b (1,2 г, масло) с выходом 40%.
MS m/z (ESI): 319 [M+1]
Стадия 2
трет-Бутил ((4-метилпиперидин-4-ил)метил)карбамат (16с)
16b (1,2 г, 3,77 ммоль) растворяли в метаноле (20 мл), добавляли палладий на угле (10%, содержание воды 55%, 1,2 г) и перемешивали при комнатной температуре в течение 3 ч в атмосфере водорода. Реакционную смесь фильтровали, осадок с фильтра промывали метанолом, фильтрат концентрировали с получением целевого продукта 16с (810 мг, масло) с выходом 94%.
MS m/z (ESI): 229 [M+l]
Стадия 3
трет-Бутил ((1-(5-йод-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-4-метилпиперидин-4-ил)метил)карбамат (16d)
150 мг (0,55 ммоль) 1 с добавляли к смеси 16с (150 мг, 0,66 ммоль), карбоната калия (228 мг, 1,65 ммоль) и ацетонитрила (10 мл), нагревали до 90°С и перемешивали в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/9) с получением целевого продукта 16d (150 мг, твердое вещество) с выходом 58%.
MS m/z (ESI): 463 [M+1]
Стадия 4
трет-Бутил ((1-(5-((4-хлор-2-метил-2Н-индазол-5-ил)тио)-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-4-метилпиперидин-4-ил)метил)карбамат (16е)
Реакционную смесь 16d (150 мг, 0,32 ммоль), 7е (86 мг, 0,39 ммоль), DIPEA (124 мг, 0,96 ммоль), Pd2(dba)3 (29 мг, 0,032 ммоль), XantPhos (18,5 мг, 0,032 ммоль) и диоксана (4 мл) нагревали до 90°С в атмосфере азота и перемешивали в течение 3 ч. Из реакционной смеси удаляли растворитель при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/9) с получением целевого продукта 16е (130 мг, твердое вещество) с выходом 75%.
MS m/z (ESI): 533 [M+1]
Стадия 5
2-(4-(Аминометил)-4-метилпиперидин-1-ил)-5-((4-хлор-2-метил-2Н-индазол-5-ил)тио)-3-метилпиримидин-4(3H)-она формиат (16)
16е (130 мг, 0,24 ммоль) растворяли в дихлорметане (5 мл), добавляли трифторуксусную кислоту (1 мл) и перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 16 (32,4 мг, твердое вещество) с выходом 31%.
MS m/z (ESI): 433 [M+1]
1H ЯМР (400 МГц, DMSO-d6) δ 8.43 (s, 1H), 8.37 (s, 1H), 7.91 (s, 1H), 7.51 (d, J=9.0 Гц, 1H), 6.99 (d, J=9.0 Гц, 1H), 4.18 (s, 3H), 3.40-3.36 (m, 5H), 3.17 (t, J=10.2 Гц, 2H), 2.65 (s, 2H), 1.63-1.57 (m, 2H), 1.42 (d, J=11.1 Гц, 2H), 1.01 (s, 3H).
Пример 17
2-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)-3-метил-5-((1-метил-1H-пирроло[2,3-b]пиридин-4-ил)тио)пиримидин-4(3H)-она формиат
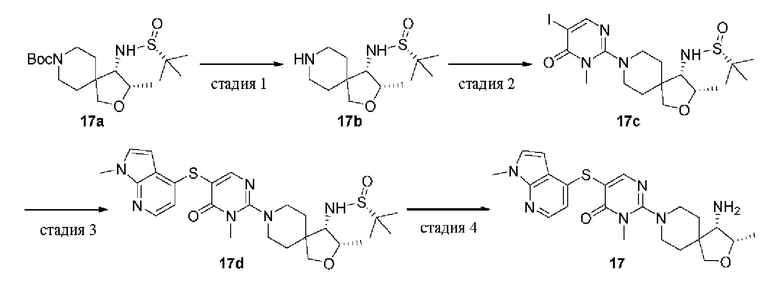
Стадия 1
(R)-2-метил-N-((3S,4S)-3-метил-2-окса-8-азаспиро[4.5]декан-4-ил)пропан-2-сульфинамид (17b)
трет-Бутил (3S,4S)-4-((((R)-трет-бутилсульфинил)амино)-3-метил-2-окса-8-азаспиро[4.5]декан-8-карбоксилат 17а (300 мг, 0,8 ммоль) растворяли в дихлорметане (5 мл), добавляли трифторуксусную кислоту (1 мл) и перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении с получением целевого продукта 17b (350 мг, неочищенный продукт). Продукт использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 275 [M+1]
Стадия 2
(R)-N-((3S,4S)-8-(5-йод-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-ил)-2-метилпропан-2-сульфинамид (17с)
Реакционную смесь 1с (216 мг, 0,8 ммоль), 17b (неочищенный продукт, 350 мг, 0,8 ммоль), карбоната калия (562 мг, 4,05 ммоль) и ацетонитрила (10 мл) нагревали до 80°С и перемешивали в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/9) с получением целевого продукта 17с (360 мг, твердое вещество) с выходом 89%.
MS m/z (ESI): 509 [M+1]
Стадия 3
(R)-2-метил-N-((3S,4S)-3-метил-8-(1-метил-5-((1-метил-1Н-пирроло[2,3-b]пиридин-4-ил)тио)-6-оксо-1,6-дигидропиримидин-2-ил)-2-окса-8-азаспиро[4.5]декан-4-ил)пропан-2-сульфинамид (17d)
Реакционную смесь 17с (180 мг, 0,35 ммоль), 2 с (70 мг, 0,42 ммоль), DIPEA (135 мг, 1,05 ммоль), Pd2(dba)3 (32 мг, 0,035 ммоль), XantPhos (20 мг, 0,035 ммоль) и диоксана (5 мл) нагревали до 90°С в атмосфере азота и перемешивали в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/9) с получением целевого продукта 17d (160 мг, твердое вещество) с выходом 83%.
MS m/z (ESI): 545 [M+1]
Стадия 4
2-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)-3-метил-5-((1-метил-1Н-пирроло[2,3-6]пиридин-4-ил)тио)пиримидин-4(3H)-она формиат (17)
17d (160 мг, 0,29 ммоль) растворяли в метаноле (4 мл), добавляли HCl в диоксане (4,0 М, 2 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 17 (36 мг, твердое вещество) с выходом 28%.
MS m/z (ESI): 441 [M+1]
1H ЯМР (400 МГц, DMSO-d6) δ 8.22 (s, 1H), 8.14 (s, 1H), 8.03 (d, J=5.1 Гц, 1H), 7.51 (d, J=3.5 Гц, 1H), 6.58 (d,J=5.1 Гц, 1H), 6.44 (d, J=3.5 Гц, 1H), 4.12-4.04 (m, 1H), 3.80 (s, 3H), 3.70 (d, J=8.6 Гц, 1H), 3.53-3.48 (m, 3H), 3.40 (s, 3H), 3.19-3.08 (m, 2H), 3.02 (d, J=4.9 Гц, 1H), 1.88-1.84 (m, 2H), 1.64-1.54 (m, 2H), 1.11 (d, J=6.4 Гц, 3Н).
Пример 18
5-((1Н-пирроло[2,3-b]пиридин-4-ил)тио)-2-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)-3-метилпиримидин-4(3H)-она формиат
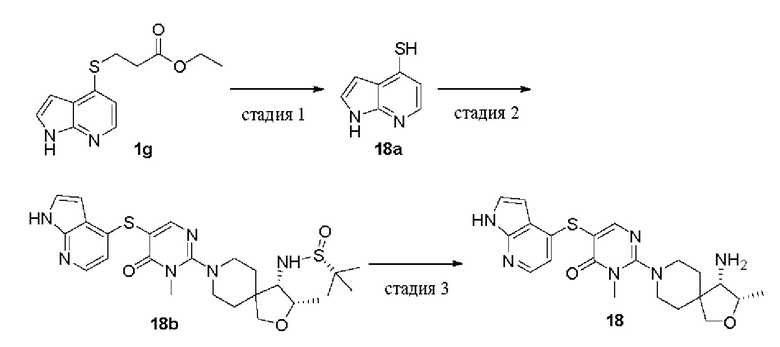
Стадия 1
1Н-пирроло[2,3-b]пиридин-4-тиол (18а)
К 1 г (250 мг, 1 ммоль) в THF (5 мл) добавляли трет-бутоксид калия (224 мг, 2 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакцию гасили водой и полученную смесь промывали этилацетатом. Водную фазу подкисляли соляной кислотой (6н.) до рН=4 и экстрагировали этилацетатом. Органическую фазу сушили над безводным сульфатом натрия, осушитель удаляли фильтрованием, из фильтрата удаляли растворитель при пониженном давлении с получением целевого продукта 18а (100 мг, твердое вещество) с выходом 67%.
MS m/z (ESI): 151 [M+1]
Стадия 2
(R)-N-((3S,4S)-8-(5-((1Н-пирроло[2,3-b]пиридин-4-ил)тио)-1-метил-6-оксо-1,6-дигидропиримидин-2-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-ил)-2-метилпропан-2-сульфинамид (18b)
Реакционную смесь 17с (180 мг, 0,35 ммоль), 18а (63 мг, 0,42 ммоль), DIPEA (135 мг, 1,05 ммоль), Pd2(dba)3 (32 мг, 0,035 ммоль), XantPhos (20 мг, 0,035 ммоль) и диоксана (5 мл) нагревали до 90°С в атмосфере азота и перемешивали в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/9) с получением целевого продукта 18b (150 мг, твердое вещество) с выходом 80%.
MS m/z (ESI): 531 [M+1]
Стадия 3
5-((1Н-пирроло[2,3-6]пиридин-4-ил)тио)-2-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)-3-метилпиримидин-4(3H)-она формиат (18)
18b (150 мг, 0,28 ммоль) растворяли в метаноле (4 мл), добавляли HCl в диоксане (4,0 М, 2 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали препаративной RP-HPLC с получением целевого продукта 18 (37,9 мг, твердое вещество) с выходом 31%.
MS m/z (ESI): 427 [M+1]
1Н ЯМР (400 МГц, DMSO-d6) δ 11.73 (s, 1H), 8.25 (s, 1H), 8.14 (s, 1H), 7.99 (d,J=5.1 Гц, 1H), 7.47-7.45 (m, 1H), 6.54 (d, J=5.1 Гц, 1H), 6.42 (d, J=2.3 Гц, 1H), 4.15-4.05 (m, 1H), 3.71 (d, J=8.5 Гц, 1H), 3.52-3.48 (m, 3Н), 3.41 (s, 3Н), 3.24-3.05 (m, 3Н), 1.89-1.75 (m, 2Н), 1.65-1.55 (m, 2Н), 1.12 (d, J=6.2 Гц, 3Н).
Пример 19
2-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)-5-((4-хлор-2-метил-2Н-индазол- 5-ил)тио)-3-(метил-d3)пиримидин-4(3H)-он
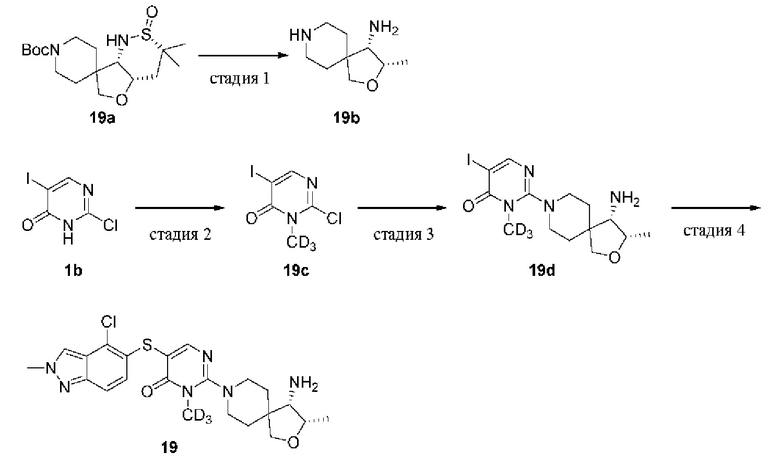
Стадия 1
(3S,4S)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин (19b)
трет-Бутил (3S,4S)-4-((((R)-трет-бутилсульфинил)амино)-3-метил-2-окса-8-азаспиро[4.5]декан-8-карбоксилат 19а (95,0 г, 254 ммоль) и HCl в метаноле (4 М, 1 л) перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в метаноле (500 мл) и к нему медленно добавляли смешанный растворитель из петролейного эфира и этилацетата (1/1, 4 л). Полученную смесь перемешивали при комнатной температуре в течение 2 ч. После фильтрации осадок с фильтра сушили с получением целевого продукта 19b (64,1 г, твердое вещество, дигидрохлорид) с выходом >100%.
MS m/z (ESI): 171 [M+1]
1H ЯМР (400 МГц, DMSO-d6) δ 9.12 (brs, 2Н), 8.33 (s, 3Н), 4.25-4.19 (m, 1Н), 3.82 (d, J=9.1 Гц, 1H), 3.64 (d, J=9.1 Гц, 1H), 3.47 (brs, 1H), 3.27-3.24 (m, 1H), 3.19-3.16 (m, 1H), 2.94-2.85 (m, 2H), 2.03-1.85 (m, 2H), 1.79-1.67 (m, 2H), 1.23 (d, J=6.5 Гц, 3Н).
Стадия 2
2-Хлор-5-йод-3-(метил-d-3)пиримидин-4(3H)-он (19с)
К 5,0 г (19,5 ммоль) 2-хлор-5-йодпиримидин-4(3H)-она 1b в THF (100 мл) добавляли DBU (3,85 г, 25,3 ммоль), полученную смесь охлаждали до 0°С и дополняли йодметаном-d-3 (3,39 г, 23,4 ммоль). Реакционную смесь нагревали до 50°С и перемешивали в течение 6 ч. Из реакционной смеси удаляли растворитель при пониженном давлении. Остаток смешивали с этилацетатом (3,5 л) и промывали разбавленной хлористоводородной кислотой (0,5н., 1 л × 2) и раствором соли (800 мл × 2). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир = от 1/19 до 1/4) с получением целевого продукта 19с (3,3 г, твердое вещество) с выходом 62%.
MS m/z (ESI): 274[М+1]
Стадия 3
2-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)-5-йод-3-(метил-d3)пиримидин-4(3H)-он (19d)
Реакционную смесь 19с (1,8 г, 6,5 ммоль), 19b (1,67 г, 6,9 ммоль, дигидрохлорид), карбоната цезия (9,5 г, 29,3 ммоль) и ацетонитрила (60 мл) перемешивали при комнатной температуре в течение 3 ч в атмосфере азота. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/петролейный эфир=9/1) с получением целевого продукта 19d (2,4 г, твердое вещество) с выходом 91%.
MS m/z (ESI): 408[М+1]
Стадия 4
2-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)-5-((4-хлор-2-метил-2Н-индазол-5-ил)тио)-3-(метил-d-3)пиримидин-4(3H)-он (19)
Реакционную смесь 7е (1,9 г, 9,0 ммоль), 19d (2,3 г, 5,65 ммоль), йодида меди (430 мг, 2,26 ммоль), 1,10-фенантролина (814 мг, 4,52 ммоль), фосфата калия (2,3 г, 11,3 ммоль) и диоксана (50 мл) нагревали до 100°С в атмосфере азота и перемешивали в течение ночи. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (метанол/дихлорметан=1/9). Полученный неочищенный продукт (2,5 г) смешивали с ацетонитрилом (25 мл) и перемешивали при 90°С в течение 3 ч и при комнатной температуре в течение 16 ч. После фильтрации осадок с фильтра сушили в вакууме при 40°С в течение 4 ч с получением целевого продукта 19 (1,47 г, твердое вещество) с выходом 54%.
MS m/z (ESI): 478[М+1]
1H ЯМР (400 МГц, DMSO-d6) δ 8.42 (s, 1H), 7.91 (s, 1H), 7.50 (dd, J=9.0, 0.9 Гц, 1H), 6.98 (d, J=9.0 Гц, 1H), 4.17 (s, 3Н), 4.05-4.02 (m, 1H), 3.64 (d, J=8.5 Гц, 1H), 3.47 (d,J=8.5 Гц, 1H), 3.48-3.37 (m, 2Н), 3.15-3.05 (m, 2Н), 2.90 (d, J=5.1 Гц, 1H), 1.86-1.80 (m, 1H), 1.74-1.68 (m, 1Н), 1.59-1.50 (m, 2Н), 1.38 (brs, 2Н), 1.07 (d, J=6,4 Гц, 3Н).
Биологические эксперименты
Измерение ингибирования активности SHP2
Влияние соединения по настоящему изобретению на активность SHP2 оценивают способом быстрой флуоресцентной детекции с использованием суррогатного субстрата DiFMUP.
Экспериментальный способ излагается следующим образом: Рекомбинантный полноразмерный белок SHP2 человека экспрессировали и очищали с помощью платформы для очистки и идентификации белков Университета Цинхуа; дифосфопептид (Н2М-LN(pY)IDLDLV-(dPEG8)LST(pY)ASINFQK-амид) был синтезирован в Nanjing GenScript Biotechnology Co., Ltd.; суррогатный субстрат DiFMUP был приобретен у Thermo Fisher Scientific (Кат. номер D6567); номер D5879) реакционный буфер содержал следующие компоненты: 60 мМ HEPES (рН 7,2), 75 мМ NaCl, 75 мМ KCl, 1 мМ ЭДТА, 0,05% TWEEN 20 и 5 мМ DTT.
Соединение растворяли в DMSO (Sigma, Кат. номер D5879) и разводили до 100 мкМ. Полученный раствор серийно разводили в 4 раза DMSO до минимальной концентрации 6,1 нМ, и каждую точку концентрации разводили в 25 раз реакционным буфером.
10 мкл раствора соединения и 10 мкл 0,25 нМ раствора белка SHP2 добавляли в 384-луночный микропланшет (Corning, Кат. номер 3575), хорошо перемешивали и инкубировали при комнатной температуре в течение 30 мин. Добавляли 10 мкл 0,5 мкМ дифосфопептида (растворенного в реакционном буфере), хорошо перемешивали и инкубировали при комнатной температуре в течение от 30 до 60 мин. Затем добавляли 10 мкл 60 мкМ раствора DiFMUP (растворенного в реакционном буфере), хорошо перемешивали и оставляли стоять при комнатной температуре. Через 30 мин измеряли сигналы флуоресценции при 340 нм (длина волны возбуждения)/450 нм (длина волны испускания) с использованием устройства для считывания микропланшетов (EnSpire, Perkin Elmer). Значение интенсивности флуоресценции положительно коррелировало со степенью дефосфорилирования субстрата, что отражало каталитическую активность SHP2. В эксперименте группа без белка использовалась как группа со 100% ингибированием, а группа с добавлением белка, но без соединений, использовалась как группа с 0% ингибирования. С помощью программного обеспечения XLfit строили кривую ингибирования соединения и рассчитывали ингибирующее значение IC50. Экспериментальные результаты представлены в таблице ниже.
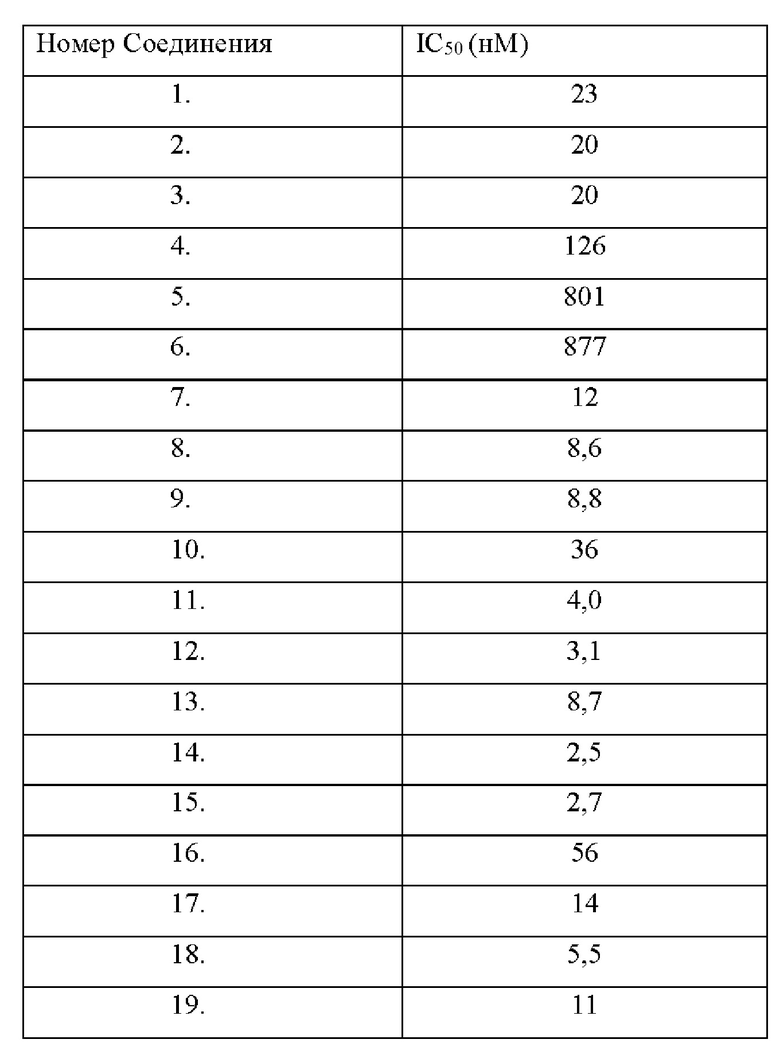
Соединения в вариантах выполнения настоящего изобретения обладают ингибирующим действием на активность SHP2 с предпочтительным значением IC50 менее чем 50 нМ.
Определение ингибирования пролиферации клеток NCI-H358
Влияние соединения по настоящему изобретению на пролиферацию клеточной линии немелкоклеточного рака легкого человека NCI-H358 оценивают с помощью люминесцентного анализа жизнеспособности клеток.
Экспериментальный способ излагается следующим образом:
Соединение растворяли в DMSO (Sigma, Кат. номер D5879) и разводили до 5 мМ. Полученный раствор серийно разбавляли в 4 раза DMSO до минимальной концентрации 0,31 мкМ, и каждую точку концентрации разбавляли в 50 раз средой RPMI 1640 (Thermo Fisher Scientific, Кат. номер 11995073). Когда значение IC50 соединения было низким, начальную концентрацию соединения дополнительно снижали.
Клетки NCI-H358 (АТСС, Кат. номер CRL-5807) культивировали в Полной Среде RPMI 1640 [среда RPMI 1640 была дополнена 10% FBS (GBICO, Кат. номер 10099-141) и 100 единиц/мл Пенициллина-Стрептомицина (Thermo Fisher Scientific, Кат. номер 15140122)]. Клетки (15000 клеток/мл) высевали в 90 мкл Полной Среды на 96-луночный планшет. После культивирования в течение ночи в каждую лунку добавляли 10 мкл раствора соединения и культивировали при 37°С в инкубаторе с 5% СО2 в течение 5 дней. Согласно инструкции из набора CellTilter-Glo (CTG) (Promega, Кат. номер G7572) планшет с клеточной культурой извлекали из инкубатора и уравновешивали с комнатной температурой. Клетки полностью лизировали 50 мкл реагента CTG и выдерживали 10 мин при комнатной температуре. Сигнал люминесценции считывали устройством для считывания микропланшетов (EnVision, Perkin Elmer). В эксперименте группу из 10 мкМ положительного контроля RMC-4550 использовали в качестве отрицательного контроля (100% ингибирование), а группу, содержащую 0,2% DMSO, использовали в качестве положительного контроля (0% ингибирования). С помощью программного обеспечения XLfit строили кривую ингибирования соединения и рассчитывали ингибирующее значение IC50. Экспериментальные результаты представлены в таблице ниже.
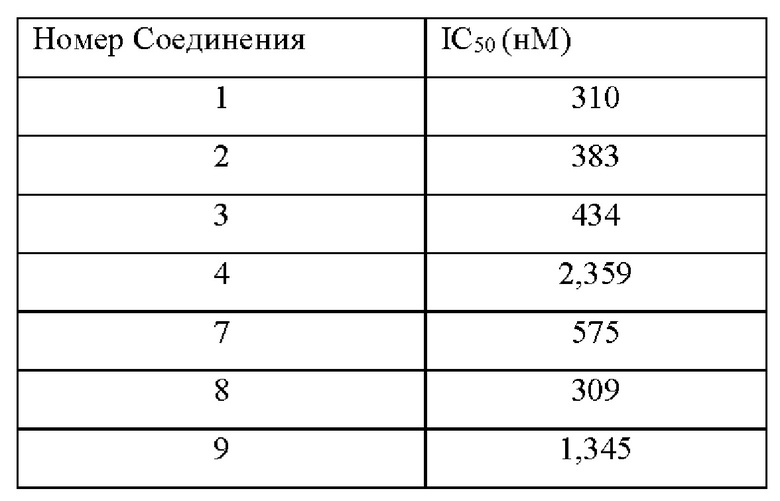
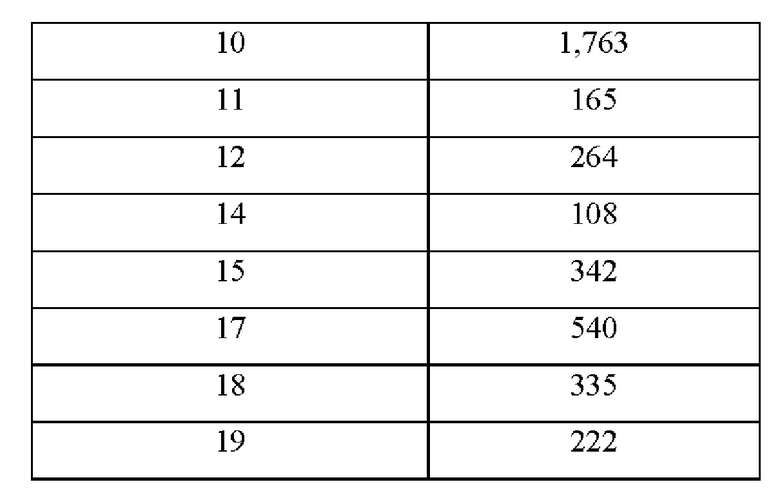
Соединения в вариантах выполнения настоящего изобретения оказывают ингибирующее действие на пролиферацию клеток с предпочтительным значением IC50 менее чем 1000 нМ.
Определение блокады на калиевый канал hERG
Влияние соединения по настоящему изобретению на возможную аритмию оценивают путем определения блокады калиевого канала hERG.
Экспериментальный способ излагается следующим образом:
Внеклеточный раствор: 140 мМ NaCl, 3,5 мМ KCl, 1 мМ MgCh, 2 мМ CaCl2, 10 мМ D-глюкозы, 10 мМ HEPES и 1,25 мМ NaH2PO4, рН=7,4.
Раствор электрода: 20 мМ KCl, 115 мМ K-аспартат, 1 мМ MgCl2, 5 мМ EGTA, 10 мМ HEPES и 2 мМ Na2-АТФ, рН=7,2.
Раствор соединения: тестируемое соединение растворяли в DMSO в виде исходного раствора 10 мМ, который разбавляли DMSO до 3 мМ, а затем внеклеточным раствором до 3 мкМ раствора для последующего использования.
Культура клеток: клеточную линию HEK293, стабильно экспрессирующую калиевый канал hERG (Creacell, Кат. номер А-0320) культивировали в Модифицированной по Дульбекко Среде Игла (DMEM, Gibco, Кат. номер 11995-065) с добавлением 10% фетальной бычьей сыворотки (Gibco, Кат. номер 1428478) и 0,8 мг/мл G418 (Amresco, Кат. номер E859-5G), где температура культивирования составляла 37°С, а концентрация углекислого газа составляла 5%. Использованную среду удаляли и клеточную линию один раз промывали PBS (Gibco, Кат. номер 1009-141). Добавляли 1 мл TrypLE™ Express (Gibco, Кат. номер 12604021) и инкубировали клеточную линию при 37°С в течение 30 с. Когда клетки отделяли от дна чашки для культивирования клеток, добавляли 5 мл Полной Среды, предварительно нагретой до 37°С, клеточную суспензию переносили в стерильную центрифужную пробирку и центрифугировали при 1000 об/мин в течение 5 мин для сбора клеток. Клетки высеивали в чашки для культивирования клеток диаметром 6 см, в каждую из которых высеивали по 2,5*105 клеток (конечный объем: 5 мл). Перед экспериментом пэтч-клэмп 3*103 клеток распределяли на покровном стекле, культивировали на 24-луночном планшете (конечный объем: 500 мкл) и детектировали через 18 ч.
Способ стимуляции напряжением для регистрации тока калия hERG в цельных клетках с помощью пэтч-клэмпа для цельных клеток был следующим: клеточную мембрану зажимали при напряжении -80 мВ после образования цельноклеточного уплотнения. Напряжение фиксатора деполяризовали с -80 мВ до -50 мВ и удерживали в течение 0,5 с (для обнаружения тока утечки), повышали до 30 мВ и удерживали в течение 2,5 с, быстро восстанавливали до -50 мВ и удерживали в течение 4 с для возбуждения пикового хвостового тока канала hERG, в котором калиевый ток hERG регистрировали каждые 10 с. Экспериментальные данные получали с помощью усилителя ЕРС-10 USB Patch Clamp Amplifier (HEKA) и сохраняли в программном обеспечении PatchMaster (НЕКА v2x73).
Измерение: стеклянная капиллярная трубка (Sutter Instruments) была превращена в регистрирующий электрод с помощью съемника микроэлектрода (Sutter Instruments). Покровное стекло с клетками, взятыми из 24-луночного планшета, помещенного в инкубатор, помещали под инвертированный микроскоп. Электродный раствор заливали в регистрирующий электрод и манипулировали съемником микроэлектрода (Sutter Instruments), чтобы позволить записывающему электроду соприкоснуться с поверхностью клеток и обеспечить всасывание с отрицательным давлением для образования уплотнения GΩ. Была предусмотрена быстрая компенсация емкости для непрерывного всасывания отрицательного давления до тех пор, пока клеточная мембрана не была разрушена, формируя режим записи всей клетки. В режиме регистрации целых клеток проводилась медленная компенсация емкости, при этом регистрировались емкость мембраны и последовательное сопротивление, при которых не обеспечивалась компенсация тока утечки. Когда пиковый хвостовой ток hERG в записи целых клеток был стабилен в течение 3-5 минут, 8 мл внеклеточного раствора, не содержащего соединения (холостой контроль), и 8 мл 3 мкМ раствора тестируемого соединения перфузировали под действием силы тяжести и пропускали через регистрирующую ванну последовательно для воздействия на клетки в течение 5 мин (или до тех пор, пока ток не станет стабильным). Ток каждой клетки, измеренный во внеклеточном растворе, не содержащем соединения, использовали в качестве отдельной контрольной группы. Независимо от 2 до 3 клеток измеряли повторно. Все электрофизиологические эксперименты проводились при комнатной температуре.
Анализ данных: Прежде всего, нормализовали ток после воздействия тестируемого соединения и ток холостого контроля 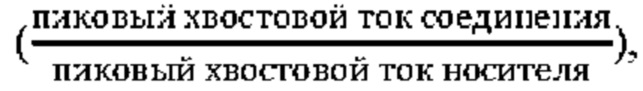 и рассчитывали процент ингибирования, соответствующий концентрации соединения
и рассчитывали процент ингибирования, соответствующий концентрации соединения 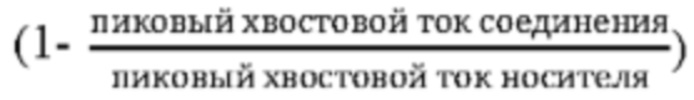 .
.
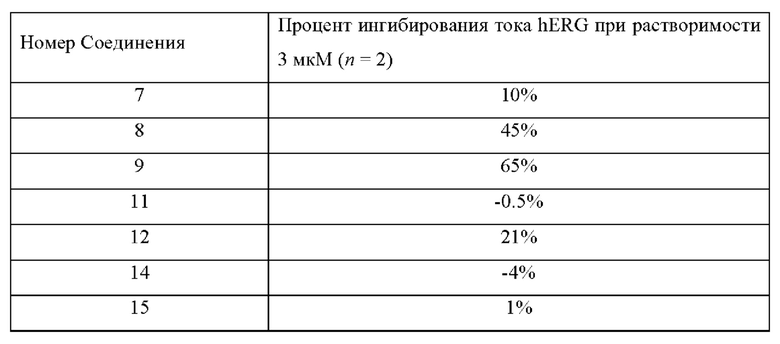
| название | год | авторы | номер документа |
|---|---|---|---|
| АЗАСПИРОПРОИЗВОДНЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ TRPM8 | 2015 |
|
RU2683309C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКОГО ЦИАНОЕНОНА КАК МОДУЛЯТОРЫ KEAP1 | 2021 |
|
RU2822828C1 |
| СПИРО-КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ПИПЕРИДИНА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КАЛИЕВОГО КАНАЛА НАРУЖНОГО МЕДУЛЛЯРНОГО СЛОЯ | 2013 |
|
RU2642066C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2685417C1 |
| ИНГИБИТОРЫ ПРОТЕИНТИРОЗИНФОСФАТАЗЫ | 2020 |
|
RU2799449C2 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АЛЛОСТЕРИЧЕСКИХ ИНГИБИТОРОВ SHP2 | 2018 |
|
RU2776846C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ПУТИ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2673458C1 |
| Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака | 2015 |
|
RU2607371C1 |
| БЕНЗОКСАЗИН-ОКСАЗОЛИДИНОНОВОЕ СОЕДИНЕНИЕ, ЗАМЕЩЕННОЕ АЗОТСОДЕРЖАЩИМ ГЕТЕРОЦИКЛОМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2018 |
|
RU2744784C1 |
| СПИРО-АМИНОСОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ СНА И ЛЕКАРСТВЕННОГО ПРИВЫКАНИЯ | 2010 |
|
RU2562609C2 |
Изобретение относится к соединению, представленному общей формулой (II), или его фармацевтически приемлемым солям, стереоизомерам, стабильным изотопным производным и их смесям, которые могут найти применение для лечения и/или предотвращения заболеваний, связанных с аномальной активностью SHP2. В формуле (II) кольцо А выбрано из группы, состоящей из фенильного кольца и 6-членного гетероарильного кольца, содержащего один гетероатом, выбранный из N; кольцо В выбрано из группы, состоящей из 5-членного гетероарильного кольца, конденсированного с кольцом А, где 5-членное гетероарильное кольцо содержит один или два гетероатома, выбранных из N; и где фенильное кольцо и гетероарильное кольцо необязательно замещены одним или более заместителями, выбранными из группы, состоящей из D, галогена, циано, C1-6 алкила, -ORa и -NRaRb; R1 выбран из группы, состоящей из H, D, циано, C1-2 алкила и -NRaRb; R2 выбран из группы, состоящей из H и C1-2 алкила, где один или более атомов водорода алкила необязательно замещены одним или более заместителями, выбранными из группы, состоящей из D и фтора; R4a и R4b каждый независимо выбран из группы, состоящей из C1-6 алкила, где алкил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из D или -NRaRb; R4a и R4b необязательно вместе с атомом углерода, к которому присоединены R4a и R4b, образуют C3-7 карбоциклическое кольцо или 4-7-членное гетероциклическое кольцо, содержащее один кислород; Ra и Rb каждый независимо представляет собой H. Изобретение относится также к соединению, представленному общей формулой (IV), в котором X выбран из группы, состоящей из -O- и -CH2-; R7 выбран из группы, состоящей из H, D и C1-2 алкила, где один или несколько атомов водорода алкила необязательно замещены одним или более заместителями, выбранными из группы, состоящей из D и фтора; значения А, В, R1 и R2 определены выше. Изобретение относится также к конкретным соединениям, фармацевтической композиции, содержащей указанные выше соединения, и их применению для получения лекарственного средства для лечения и/или предотвращения заболеваний, связанных с аномальной активностью SHP2, и для лечения и/или предотвращения заболеваний, связанных с SHP2, при этом заболевания представляют собой рак, выбранный из лейкоза, синдрома Нунан, синдрома леопарда, нейробластомы, меланомы, рака легкого, рака молочной железы, рака пищевода, рака толстой кишки, рака головы и шеи и рака желудка. 6 н.п. ф-лы, 3 табл., 19 пр.
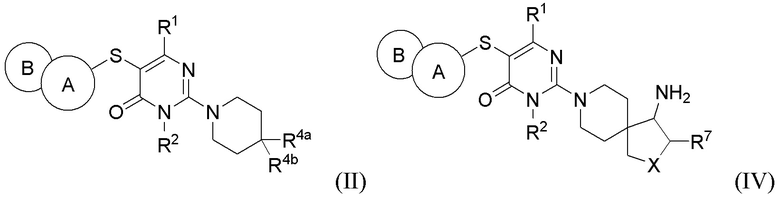
1. Соединение, представленное общей формулой (II), показанной ниже, или его фармацевтически приемлемые соли, стереоизомеры, стабильные изотопные производные, их смеси
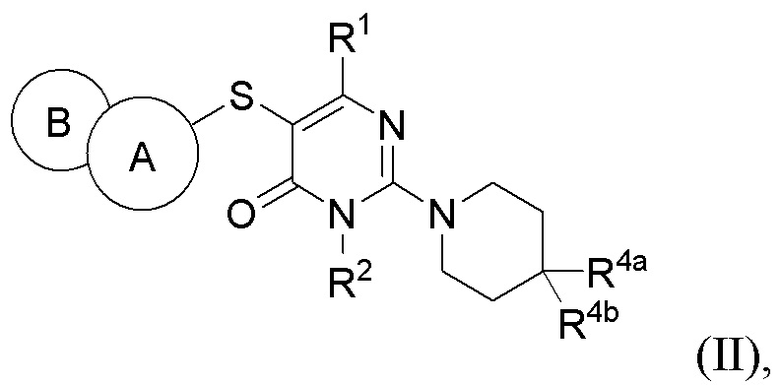
где кольцо А выбрано из группы, состоящей из фенильного кольца и 6-членного гетероарильного кольца, содержащего один гетероатом, выбранный из N;
кольцо В выбрано из группы, состоящей из 5-членного гетероарильного кольца, конденсированного с кольцом А, где 5-членное гетероарильное кольцо содержит один или два гетероатома, выбранных из N; и
где фенильное кольцо и гетероарильное кольцо необязательно замещены одним или более заместителями, выбранными из группы, состоящей из D, галогена, циано, C1-6 алкила, -ORa и -NRaRb;
R1 выбран из группы, состоящей из H, D, циано, C1-2 алкила и -NRaRb;
R2 выбран из группы, состоящей из H и C1-2 алкила, где один или более атомов водорода алкила необязательно замещены одним или более заместителями, выбранными из группы, состоящей из D и фтора;
R4a и R4b каждый независимо выбран из группы, состоящей из C1-6 алкила, где алкил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из D или -NRaRb;
R4a и R4b необязательно вместе с атомом углерода, к которому присоединены R4a и R4b, образуют C3-7 карбоциклическое кольцо или 4-7-членное гетероциклическое кольцо, содержащее один кислород;
Ra и Rb каждый независимо представляет собой H.
2. Соединение, представленное общей формулой (IV), показанной ниже, или его фармацевтически приемлемые соли, стабильные изотопные производные, стереоизомеры и их смеси
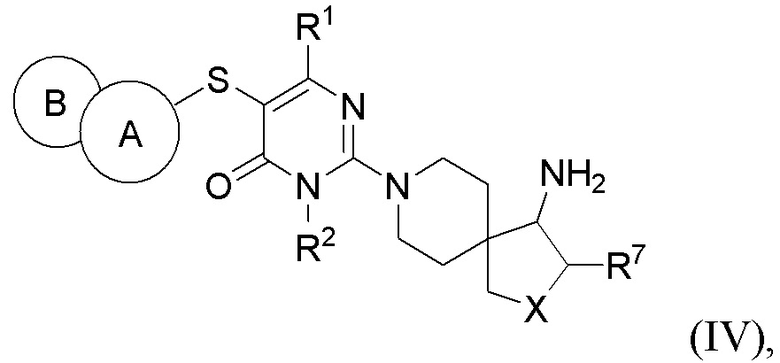
где кольцо А выбрано из группы, состоящей из фенильного кольца и 6-членного гетероарильного кольца, содержащего один гетероатом, выбранный из N;
кольцо В выбрано из группы, состоящей из 5-членного гетероарильного кольца, конденсированного с кольцом А, где 5-членное гетероарильное кольцо содержит один или два гетероатома, выбранных из N; и
где фенильное кольцо и гетероарильное кольцо необязательно замещены одним или более заместителями, выбранными из группы, состоящей из D, галогена, циано, C1-2 алкила, -ORa и -NRaRb;
X выбран из группы, состоящей из -O- и -CH2-;
R1 выбран из группы, состоящей из H, D, C1-2 алкила и -NRaRb;
R2 выбран из группы, состоящей из H и C1-2 алкила, где один или несколько атомов водорода алкила необязательно замещены D и фтором;
R7 выбран из группы, состоящей из H, D и C1-2 алкила, где один или несколько атомов водорода алкила необязательно замещены одним или более заместителями, выбранными из группы, состоящей из D и фтора;
Ra и Rb каждый независимо представляет собой H.
3. Соединение или его фармацевтически приемлемые соли, стабильные изотопные производные, стереоизомеры и их смеси, где соединение выбрано из группы, состоящей из: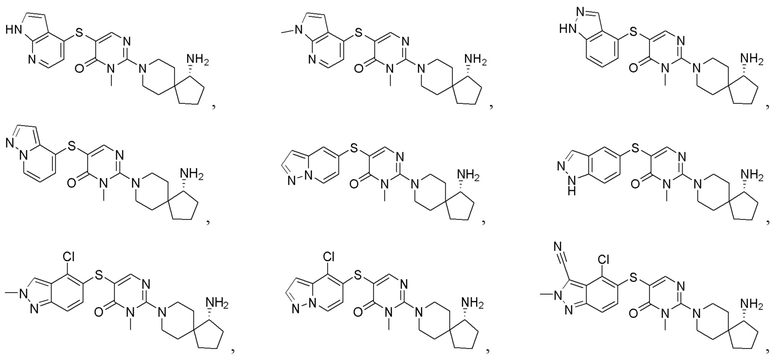
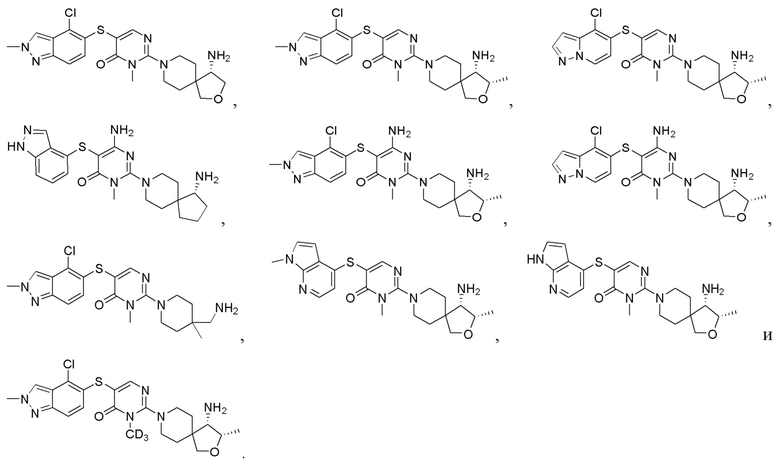
4. Фармацевтическая композиция для лечения и/или предотвращения заболеваний, связанных с аномальной активностью SHP2, содержащая терапевтически эффективную дозу соединения по любому из пп. 1-3 или его фармацевтически приемлемую соль, стабильное изотопное производное, и стереоизомер, и их смесь, и один или более фармацевтически приемлемых носителей и эксципиентов.
5. Применение соединения по любому из пп. 1-3 или его фармацевтически приемлемой соли, стабильного изотопного производного и стереоизомера, а также их смеси или фармацевтической композиции по п. 4 для получения лекарственного средства для лечения и/или предотвращения заболеваний, связанных с аномальной активностью SHP2, при этом заболевания представляют собой рак, выбранный из лейкоза, синдрома Нунан, синдрома леопарда, нейробластомы, меланомы, рака легкого, рака молочной железы, рака пищевода, рака толстой кишки, рака головы и шеи и рака желудка.
6. Применение соединения по любому из пп. 1-3 или его фармацевтически приемлемой соли, стабильного изотопного производного и стереоизомера, а также их смеси или фармацевтической композиции по п. 4 для лечения и/или предотвращения заболеваний, связанных с SHP2, при этом заболевания представляют собой рак, выбранный из лейкоза, синдрома Нунан, синдрома леопарда, нейробластомы, меланомы, рака легкого, рака молочной железы, рака пищевода, рака толстой кишки, рака головы и шеи и рака желудка.
| Токарный резец | 1924 |
|
SU2016A1 |
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Пинцет для выдавливания трахоматозных зерен | 1932 |
|
SU31573A1 |

