Настоящее изобретение относится к получению алкоксигидроксибензальдегида ("AHBA") и предпочтительно к его получению с возможно большей чистотой.
Изобретение более предпочтительно относится к получению AHBA из гидроксифенола ("HP").
Изобретение более предпочтительно относится к получению ванилина ("VA") и/или этилванилина ("EVA").
Предшествующий уровень техники
Алкоксигидроксибензальдегиды ("AHBA") представляют собой очень важные соединения, используемые прежде всего в качестве ароматизаторов и отдушек, а также в качестве промежуточных соединений во многих областях, таких как, например, агрохимия, фармацевтика, парфюмерно-косметическая промышленность и другие отрасли промышленности.
В частности, ванилин и этилванилин представляют собой соединения в основном пищевого назначения. Таким образом, важно предложить рынку соединения высокой химической чистоты и хорошего вкусового качества, обеспечивая при этом как можно меньшую стоимость производства.
Таким образом, в способе их синтеза требуется улучшить осуществляемые химические реакции и реализовать эффективные операции разделения и очистки.
Были предложены различные способы синтеза ароматичных альдегидов. Наиболее важные способы основаны на функционализации исходного фенольного соединения, например: фенола, катехола (называемого также как пирокатехин или "PC"), производного соединения катехола, гваякола (или 2-метоксифенола) и/или гветола (или 2-этоксифенола). При этом, в общем случае, используя в способе такого типа фенольное соединение в солевой форме, например, в виде натриевой соли, разными способами присоединяют формильную группу в параположении относительно гидроксигруппы, связанной с бензольным циклом.
Для получения гваякола в некоторых способах в качестве исходного соединения выбирают ортонитрохлорбензол (ONCB).
Эти способы имеют множество недостатков по сравнению со способом с исходным пирокатехином: использование токсичных соединений CMR (канцерогены, мутагены и репротоксины), таких как о-анизидин; множество стадий; экзотермические и опасные реакции, такие как нитрование или гидролиз соли диазония; образование множества отходов; образование примесей, таких как 5-хлоргваякол, который оказывается в ванилине в виде 6-хлорванилина, и т.д.
В то же время существует полностью отличающийся путь получения ванилина, состоящий в том, что в щелочной среде гваякол и глиоксиловую кислоту приводят во взаимодействие, ведущее к получению 4-гидрокси-3-метоксиминдальной кислоты, с последующим окислением соединения, полученного конденсацией.
Синтез гваякола (GA) или гветола (GE) из катехола является существенно более прямым.
Также существуют способы синтеза параметоксифенола (PMP) и/или параэтоксифенола (PEP) из гидрохинона.
Соединения, такие как GA, GE, PMP и PEP, представляют собой алкоксифенолы ("AP").
Присоединение альдегидной группы к алкоксифенолу ("AP") для получения AHBA может быть осуществлено по реакции между AP и глиоксиловой кислотой с последующим окислением образующегося соединения. Однако AHBA, полученный по этому пути, в качестве примеси содержит, в частности, соединение типа алкилалкоксигидроксибензальдегида ("AAHBA"). При этом эту смесь особенно трудно разделять.
В силу этого трудного разделения смесь AHBA/AAHBA разделяют кристаллизацией, что требует применения органических растворителей.
Эта проблема приобретает особое значение при получении ванилина ("VA") или этилванилина ("EVA") из 1,2-дигидроксибензола(катехола или пирокатехина, обозначаемого по настоящему изобретению как "PC"), поскольку метилированная примесь ванилина или этилированная примесь этилванилина очень трудно отделима от VA и/или EVA.
В общем случае применение органических растворителей является маложелательным в силу, в частности, токсичности органических растворителей, приемлемых для применения, дополнительных затрат, порождаемых их применением или обработкой растворителя в целью рецикла или регенерации.
Цели изобретения
Целью настоящего изобретения является, таким образом, решение указанных ранее проблем.
В частности, основной целью настоящего изобретения является улучшение существующих промышленных способов получения алкоксигидроксибензальдегидов ("AHBA"), в частности ванилина ("VA") и/или этилванилина ("EVA"), предпочтительно в отношении чистоты AHBA, в частности ванилина (VA) и/или этилванилина (EVA).
Целью настоящего изобретения является предпочтительно получение этих соединений, в существенной степени свободных от примесей алкилалкоксигидроксибензальдегида ("AAHBA"), в частности 5-метилванилина ("MEVA") и 5-этилэтилванилина ("EEVA"), при производстве по меньшей мере одного соединения типа алкоксигидроксибензальдегида и, как правило, ванилина и/или этилванилина.
Целью настоящего изобретения является также оптимизация таких промышленных способов и, в частности, улучшение выхода существующих способов по AHBA с максимальной чистотой AHBA.
Целью настоящего изобретения является, в частности, разработка способа очистки потока VA и/или EVA в случае, когда VA и/или EVA получают из GA и/или GE.
Целью настоящего изобретения также является, в частности, разработка способа очистки потока VA и/или EVA в случае, когда VA и/или EVA получают из PC.
Целью настоящего изобретения предпочтительно является разработка способов, которые включают реакцию O-алкилирования гидроксифенола в паровой фазе для образования соответствующего алкоксифенола.
Целью настоящего изобретения является получение соединения типа алкоксифенола в промышленном масштабе с хорошим выходом, с низкой степенью образования примесей и/или с эффективным удалением их и с высокой конверсией гидроксифенола. При этом, например, в случае способа, позволяющего получать гваякол, образуется также вератрол, образующийся при дополнительном (последовательном) метилировании гваякола.
Таким образом, целью настоящего изобретения является ограничение содержания примеси диалкоксибензола, образующегося при получении по меньшей мере одного алкоксифенола исходя по меньшей мере из одного гидроксифенола. Целью настоящего изобретения предпочтительно является ограничение содержания вератрола при синтезе гваякола.
Краткое описание изобретения
Настоящее изобретение позволяет решить проблемы, указанные ранее.
Настоящее изобретение позволяет получать соединение типа алкоксигидроксибензальдегида в промышленном масштабе с высокой чистотой, при этом сохраняя в синтезе хороший выход, низкую степень образования примесей и/или эффективное удаление их, высокую конверсию дигидроксибензола и, в частности, эффективное рециклирование применяемых соединений.
Настоящее изобретение относится к способу получения по меньшей мере одного алкоксигидроксибензальдегида ("AHBA") исходя по меньшей мере из одного гидроксифенола ("HP"), причем способ включает стадию образования по меньшей мере одного алкоксифенола ("AP") и алкилалкоксифенола ("AAP") и стадию отделения (S) AP от AAP, причем стадию отделения (S) осуществляют перед получением AHBA.
В силу своих структур AAHBA и AHBA имеют близкие физические характеристики, в частности близкие температуры кипения. Таким образом, специалисты в данной области техники разделяют эти соединения, как правило, кристаллизацией. Для разделения этих соединений очень трудно использовать перегонку, главным образом, в силу близости температур кипения.
Авторами было найдено, что оказалось возможным разработать способ очистки AHBA, полученного из HP, при этом решая проблемы, указанные ранее.
Настоящее изобретение позволяет предпочтительно ограничить образование алкилированной примеси и более предпочтительно ограничить содержание соединения типа алкилалкоксигидроксибензальдегида при производстве по меньшей мере одного соединения типа алкоксигидроксибензальдегида, такого как 5-метилванилин или 5-этилэтилванилин при производстве ванилина и/или этилванилина.
Настоящее изобретение позволяет также удалять диалкоксилированную примесь, также образующуюся при этом, в частности вератрол, при производстве ванилина.
Синтез алкоксифенола из гидроксифенола известен как в паровой фазе, так и в трехфазной среде.
Авторами было найдено, в частности, что в паровой фазе образование примеси типа алкилалкоксигидроксибензальдегида ограничивается при производстве по меньшей мере одного соединения типа алкоксигидроксибензальдегида, если отделение осуществляют перед получением алкоксигидроксибензальдегида.
Подробное описание изобретения
Способ по настоящему изобретению более предпочтительно относится к получению ванилина и этилванилина, но он является приемлемым также в случае других ароматичных альдегидов. Способ по настоящему изобретению относится также к получению гваякола и/или гветола из пирокатехина, но он является приемлемым также в случае других алкоксифенолов.
Под термином "гидроксифенол" понимают по меньшей мере соединение типа бензола, необязательно имеющее заместители и содержащее две гидроксигруппы. Гидроксифенол может иметь, например, один или несколько заместителей, выбранных из гидроксигруппы, алкилов, алкоксигрупп, атомов галогенов, нитрогруппы, арилов, причем группировки, содержащие атомы углерода, могут содержать 1 или несколько гетероатомов, предпочтительно выбранных из атомов O, N, и S. Он более предпочтительно соответствует следующей формуле (I):
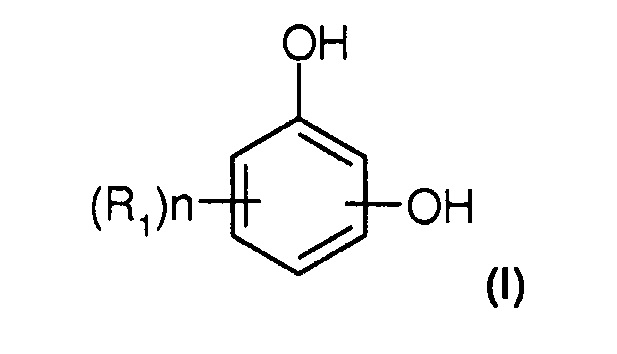
при этом в указанной формуле (I):
- одна или несколько групп R1, являющихся одинаковыми или разными, представляют собой атомы водорода, гидроксигруппы, алкилы или алкоксигруппы;
- n означает число от 0 до 3.
При этом соответствующее положение двух гидроксигрупп в формуле (I) не является фиксированным. Они могут находиться в орто-, мета- или параположении.
Среди соединений формулы (I) можно назвать пирокатехин ("PC" или ортогидроксифенол), резорцин ("RS" или метагидроксифенол) и гидрохинон ("HQ" или парагидроксифенол), которые являются предпочтительными соединениями для способа по настоящему изобретению.
В рамках настоящего изобретения под термином "алкил" понимают линейную или разветвленную углеводородную цепь, содержащую от 1 до 12 атомов углерода и предпочтительно от 1 до 4 атомов углерода. Предпочтительные примеры алкилов представляют собой метил, этил, пропил, изопропил и трет-бутил.
Под термином "алкоксигруппа" понимают -O-алкил, в котором алкил имеет значения, указанные ранее. Предпочтительные примеры алкоксигрупп представляют собой метокси- и этоксигруппы.
Термин "алкоксифенол" (AP) по настоящему изобретению относится к соединению типа отдельного алкоксифенола или к смеси алкоксифенолов. Аббревиатура "G" по настоящему изобретению относится к гваяколу (GA), гветолу (GE) и любой их смеси.
Под термином "алкоксифенол" понимают по меньшей мере соединение типа бензола, содержащее гидроксигруппу и алкоксигруппу (или O-алкил). Оно более предпочтительно соответствует следующей формуле (II):
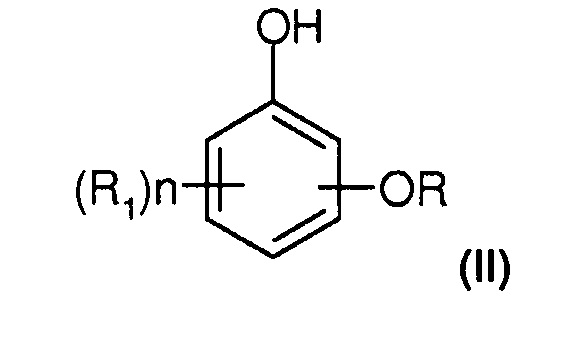
при этом в указанной формуле (II):
- R представляет собой алкил, предпочтительно содержащий от 1 до 4 атомов углерода и ковалентно связанный с атомом кислорода;
- R1 и n имеют определенные ранее значения со ссылкой на соединение формулы (I).
Соединения типа алкоксифенола получают из соответствующего соединения типа гидроксифенола, так что группа R, приносимая агентом O-алкилирования, замещает атом водорода одной из гидроксильных групп исходного соединения типа дигидроксифенола.
Можно назвать, в частности, гваякол, гветол, параметоксифенол (PMP) и параэтоксифенол (PEP).
Термин "алкоксигидроксибензальдегид" (AHBA) по настоящему изобретению относится к соединению типа отдельного алкоксигидроксибензальдегида или смеси алкоксигидроксибензальдегидов.
Под термином "алкоксигидроксибензальдегид" понимают по меньшей мере соединение типа фенола, содержащее гидроксигруппу, алкоксигруппу (или O-алкил) и формильную группу. Оно более предпочтительно соответствует следующей формуле (III):
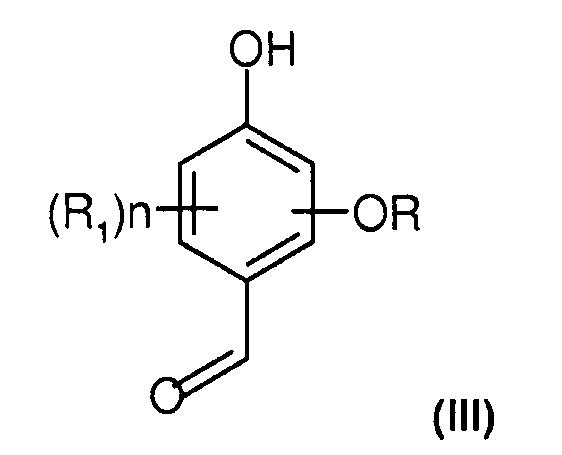
где R, R1 и n имеют определенные ранее значения со ссылкой на соединение формулы (I).
Алкоксигидроксибензальдегид, предпочтительно используемый в способе по настоящему изобретению, соответствует формуле (III), где R представляет собой метил или этил, а n=0.
В качестве более специфических примеров алкоксигидроксибензальдегидов можно назвать, в частности:
- ванилин;
- этилванилин (3-этокси-4-гидроксибензальдегид);
- изованилин (4-метокси-3-гидроксибензальдегид);
- о-ванилин (3-метокси-2-гидроксибензальдегид);
- сиреневый альдегид (3,5-диметокси-4-гидроксибензальдегид).
Когда по настоящему изобретению дается ссылка на соединение "AHBA", она предпочтительно относится к "VA и/или EVA". Аббревиатуры "VA" и/или "EVA" по настоящему изобретению относятся к ванилину (VA), этилванилину (EVA) и любой их смеси. Аббревиатура "V" по настоящему изобретению относится к соединениям VA и/или EVA.
Термин "алкилалкоксифенол" (AAP) по настоящему изобретению относится к соединению типа отдельного алкилалкоксифенола или к смеси алкилалкоксифенолов. Под термином "алкилалкоксифенол" понимают соединение типа "алкоксифенол" согласно формуле (II), имеющее, к тому же, алкильную группу, связанную с бензольным ядром. Аббревиатура "AG" по настоящему изобретению относится к алкилгваяколу (AGA), в частности к 6-метилгваяколу (MEGA или 2-метокси-6-метилфенолу), к алкилгветолу (AGE), в частности к 6-этилгветолу (EGE или 2-этокси-6-этилфенолу), и к любой их смеси.
Термин "алкилалкоксигидроксибензальдегид" (AAP) по настоящему изобретению относится к соединению типа отдельного алкилалкоксигидроксибензальдегида или к смеси алкилалкоксигидроксибензальдегидов. Аббревиатура "AV" по настоящему изобретению относится к ванилину и/или этилванилину, содержащему алкил в положении 5 фенильной группы, и предпочтительно относится к 5-метилванилину (MEVA или 4-гидрокси-3-метокси-5-метилбензальдегиду) и/или к 5-этилэтилванилину (EEVA или 4-гидрокси-3-этокси-5-этилбензальдегиду).
Любая ссылка на "AHBA" предпочтительно относится к "V". Аналогичным образом, любая ссылка на примесь "AAHBA" предпочтительно относится к примеси "AV", любая ссылка на "AP" предпочтительно относится к "G", а любая ссылка на примесь "AAHB" предпочтительно относится к примеси "AG", если не указано иное.
Реакция O-алкилирования
AAHBA образуется, главным образом, при синтезе в паровой фазе соединения AP из HP, причем синтез включает, например, O-алкилирование гидроксигруппы HP в присутствии по меньшей мере одного O-алкилирующего агента-носителя по меньшей мере одного алкила (называемое в данном случае реакцией O-алкилирования HP).
Синтез алкоксифенола в паровой фазе известен на предшествующем уровне техники. В частности, можно сослаться на патенты EP 0509927, EP 0873786, EP 914854 и EP 0933107. Согласно способу по настоящему изобретению по меньшей мере один гидроксифенол приводят во взаимодействие с O-алкилирующим агентом в паровой фазе при необходимости в присутствии катализатора, который часто находится в твердом состоянии.
Под выражением "паровая фаза" понимают, что различные реагенты в условиях реакции находятся в состоянии пара, но способ не исключает возможного присутствия жидкой среды в очень малом количестве. Реакцию осуществляют в паровой фазе при необходимости в присутствии твердых веществ, таких как один или несколько катализаторов. Согласно предпочтительному варианту способ по настоящему изобретению осуществляют только с реагентами и веществами в паровой фазе, причем только один или несколько катализаторов могут представлять собой твердые вещества или быть нанесены на одно или несколько твердых веществ.
Органические соединения, приемлемые для применения в способе по настоящему изобретению, представляют собой гидроксифенолы. В частности, органические соединения, приемлемые для применения в способе по настоящему изобретению, выбирают из ароматических соединений, определенных по настоящему изобретению, и предпочтительно из катехола, гидрохинона и резорцина.
O-Алкилирующий агент может быть выбран из соединений, связанных по меньшей мере с одним алкилом и предпочтительно по меньшей мере с одним метилом, этилом, пропилом, изопропилом, бутилом, изобутилом, трет-бутилом, амилом, втор-амилом, изоамилом или амилом, причем каждое из соединений предпочтительно имеет температуру кипения в интервале от 50 до 200°C и более предпочтительно в интервале от 60 до 150°C. Среди O-алкилирующих агентов, приемлемых для применения по настоящему изобретению, можно, в частности, назвать спирты, содержащие от 1 до 12 атомов углерода и предпочтительно C1-C4. В качестве примеров O-алкилирующих агентов можно назвать низшие алифатические алканолы, содержащие от 1 до 5 атомов углерода, такие как, например, метанол, этанол, трифторэтанол, пропанол, изопропиловый спирт, бутанол, пентанол, изопентанол, простой моноэтиловый эфир этиленгликоля, а также высшие алифатические спирты, содержащие по меньшей мере от 6 до 12 атомов углерода, такие как, например, гексанол, гептанол, изогептанол, октанол, нонанол, деканол, додеканол. Можно применять циклоалифатические спирты, содержащие от 3 до 12 атомов углерода, такие как, например, циклопентанол, циклогексанол, циклогептанол, циклооктанол, циклододеканол, трипропилциклогексанол, метилциклогексанол и метилциклогептанол. O-алкилирующий агент предпочтительно выбирают из алифатических спиртов. Предпочтительные спирты представляют собой метанол, этанол, изопропанол и трет-бутанол или этиленгликоль. Другие O-алкилирующие агенты выбирают из галогеналкилов, в числе которых предпочтительными являются метил- или этилхлорид, из алкилкарбонатов, таких как метил- или этилкарбонат, или из алкилфосфатов, таких как триметил- или триэтилфосфат.
Таким образом, настоящее изобретение более предпочтительно относится к соединениям AHBA и AP, в которых алкоксигруппа представляет собой группу C1-C4 и предпочтительно метокси- и/или этоксигруппу.
Согласно способу по настоящему изобретению реакцию O-алкилирования осуществляют в паровой фазе, приводя в контакт фенольное соединение формулы (I) и O-алкилирующий агент.
Используемое количество O-алкилирующего агента, предпочтительно алканола, больше или равно стехиометрическому количеству, необходимому для этерификации одной или нескольких гидроксильных групп для образования алкоксигрупп. В общем случае O-алкилирующий агент, предпочтительно алканол, применяют в таком количестве, что соотношение между числом молей O-алкилирующего агента и числом гидроксильных групп фенольного соединения формулы (I) изменяется в интервале от 0,5 до 500 и предпочтительно в интервале от 2 до 20. Согласно одному из вариантов гидроксифенол и O-алкилирующий агент предпочтительно смешивают с массовым соотношением от 0,5:99,5 до 90:10 и более предпочтительно от 1:99 до 80:20. Преимущественно их смешивают с массовым соотношением от 20:80 до 80:20 и более предпочтительно от 30:70 до 70:30.
Для производства алкоксифенола можно использовать катализатор реакции O-алкилирования. Катализаторы, приемлемые для применения, не ограничиваются специфическим типом катализатора, как катализатор, который может ускорять реакцию O-алкилирования гидроксифенола в паровой фазе O-алкилирующим агентом. Катализатор предпочтительно выбирают из катализаторов, содержащих щелочной металл, например, гидроксиды, карбонаты и гидрокарбонаты щелочных металлов, катализаторов на основе п-толуолсульфоновой кислоты и/или фосфата алюминия, катализаторов Al-B-P, катализаторов со щелочноземельными металлами Al-B-P, катализаторов Al-B-P-Si, катализаторов Al-B-P-Ca, катализаторов Al-P-Ti-Si, катализаторов, содержащих активный компонент, полученный из борной и фосфорной кислот, и нанесенных на носитель, содержащий инертный оксид алюминия.
Для производства алкоксифенола из гидроксифенола и предпочтительно из пирокатехина предпочтительно используемыми являются катализаторы Al-P. Катализаторы Al-P содержат катализаторы реакции O-алкилирования типа Al-P-Ti-Si, соответствующие формуле AlaPbTicSidOe (где a, b, c, d и e представляют собой соответственно число атомов Al, P, Ti, Si и O с атомными отношениями: a=1, b=1,0-1,9, c=0,05-0,5, d=0,05-0,2 и e=4,1-7,0) соответственно описанию не прошедшей экспертизу японской заявке № 4-341345. Для производства алкоксифенола из гидроксифенола указанный ранее катализатор Al-P-Ti-Si может быть прибавлен к катализатору, содержащему от 0,004 до 0,015 атома серы на атом алюминия для образования композиционного катализатора. Также можно назвать катализаторы типа AlaPbTicSidXeOf, где X означает Bi, Sb, Bi/Sb или S, с атомными отношениями a:b = от 1:1,0 до 1:1,9; a:c = от 1:0,05 до 1:0,5; a:d = от 1:0,05 до 1:0,2; a:e = от 1:0,01 до 1:0,3, когда X представляет собой по меньшей мере атом Sb или Bi, и a:e = от 1:0,004 до 1:0,015, когда X представляет собой S; и a:f = от 1:4,1 до 1:8,4. Такие катализаторы описаны в патенте FR 0 873 786.
Удельная массовая часовая производительность катализатора (whsv -"weight hourly space velocity" (удельная массовая часовая скорость подачи)) преимущественно составляет от 0,1 до 20 ч-1 и предпочтительно от 0,5 до 5 ч-1: удельная массовая часовая производительность катализатора определяется как массовое отношение часовой подачи фенольного соединения к катализатору.
Катализатор может находиться на поддерживающем носителе. При этом поддерживающий носитель катализатора предпочтительно представляет собой пористое стекло, когда катализатор находится в виде порошка, и решетку в случае, когда он находится в виде экструдированных элементов.
В способе по настоящему изобретению, когда паровая фаза состоит из смеси нескольких компонентов, паровая фаза образуется при испарении жидкости, образованной смесью, содержащей по меньшей мере один гидроксифенол и по меньшей мере один O-алкилирующий агент. Гидроксифенол и O-алкилирующий агент приводят в контакт с катализатором при необходимости в потоке газа-носителя. Газ-носитель используется при необходимости и в общем случае представляет собой газ или смесь газов, являющихся инертными в условиях реакции. Можно назвать такие газы, как азот, воздух, аргон или гелий. Объемное соотношение между газом-носителем и фенольным соединением преимущественно изменяется в интервале от 0 до 10 и предпочтительно в интервале от 0,1 до 2,0.
Давление при реакции может находиться в интервале от 10-2 до 50 бар и предпочтительно равно атмосферному давлению.
Жидкость, образованную смесью нескольких компонентов, предпочтительно вводят в испаритель при температуре приблизительно от 50 до 300°C, предпочтительно от 80 до 250°C и более предпочтительно от 100 до 220°C для предотвращения или ограничения термической деструкции компонентов, особенно компонентов с высокой температурой кипения.
Можно испарять каждый из реагентов (гидроксифенол, O-алкилирующий агент и т.д.) в разных камерах, затем осуществлять их смешивание в камере смешения и подавать образованный газовый поток к катализатору. Газ-носитель может быть введен параллельно с указанным газовым потоком или на уровне камеры смешения.
Другой вариант состоит в получении раствора, содержащего гидроксифенол и O-алкилирующий агент, с последующим испарением указанной смеси и подачей ее к катализатору параллельно с газом-носителем.
Другой вариант практического осуществления настоящего изобретения состоит в том, что гидроксифенол плавят, нагревая его до температуры плавления, и подают в газовый поток, содержащий O-алкилирующий агент. Этот поток насыщают гидроксифенолом и затем приводят в контакт с катализатором.
В конце реакции совокупность газов конденсируют, а непрореагировавшие реагенты отделяют от полученных соединений перегонкой.
В способе по настоящему изобретению, например в случае, когда 100 массовых частей гидроксифенола с температурой кипения в интервале от 150 до 300°C, предпочтительно от 180 до 290°C, при атмосферном давлении и от 80 до 120 массовых частей O-алкилирующего агента с температурой кипения от 50 до 200°C, предпочтительно от 60 до 150°C, и от 100 до 200°C, предпочтительно от 120 до 180°C, при температуре кипения гидроксифенола смешивают между собой, причем образующаяся жидкость может быть испарена и затем введена в реактор O-алкилирования в паровой фазе для получения требуемого алкоксифенола (AP). В частности, можно обратиться к патенту EP 0 914 854 B1 за более подробными сведениями касательно рекомендованных условий и аппаратуры.
Реакцию O-алкилирования в паровой фазе предпочтительно осуществляют в реакторе при температуре от 200 до 400°C, предпочтительно от 220 до 350°C и более предпочтительно от 230 до 300°C при атмосферном или повышенном давлении, например от 98 от 4903 кПа (от 1 до 50 кг/см2) и предпочтительно от 147 до 2942 кПа (от 1,5 до 30 кг/см2).
Время контакта, определенное как отношение между насыпным объемом катализатора и расходом газового потока (включая газ-носитель), может изменяться в широких пределах и наиболее часто находится в интервале от 0,5 до 100 с. Время контакта предпочтительно выбирают в интервале от 1 до 10 с. При практическом осуществлении настоящего изобретения начинают с получения слоя катализатора, который представляет собой катализатор, нанесенный на поддерживающий носитель (например, на пористое стекло, на решетку), что обеспечивает циркуляцию газов без уноса катализатора. Затем подают реагенты, при этом возможно несколько вариантов.
Особенностью способа, являющегося объектом настоящего изобретения, является то, что он позволяет получить по меньшей мере один алкоксифенол с хорошей селективностью реакции.
Способ, являющийся объектом настоящего изобретения, является идеально приемлемым для получения гваякола и/или гветола по реакции O-алкилирования пирокатехина с метанолом и/или этанолом соответственно.
В случае O-метилирования пирокатехина способ по настоящему изобретению является очень привлекательным, поскольку он позволяет управлять производством гваякола или вератрола согласно потребностям рынка, регулируя количество метанола и давление (от 1 до 20 бар).
Аналогичным образом данное положение относится к O-метилированию гидрохинона, которое позволяет получать п-метоксифенол и/или п-диметоксибензол. В способе по настоящему изобретению осуществляют O-алкилирование без применения традиционных O-алкилирующих агентов, таких как метиленхлорид, являющийся в некоторой степени токсичным. Кроме того, в его случае не образуются солесодержащие сточные воды, что делает его очень привлекательным в отношении экологических и экономических требований, поскольку он позволяет избавиться от обработки сточных вод.
Реакционная смесь, выходящая из реактора O-алкилирования, содержит алкоксифенол в качестве требуемого вещества, алкилалкоксифенол в качестве примеси, непрореагировавший O-алкилирующий агент, непрореагировавший гидроксифенол и побочные продукты. Следовательно, поток, выходящий из реактора O-алкилирования, содержит смесь M, содержащую AP и примесь AAP.
Предшественники примесей MEVA и EEVA образуются, в частности, при получении O-алкилированием в паровой фазе GA и/или GE из PC.
Таким образом, особенно выгодно отделять AAP от смеси M для получения соединения AP, отделенного от примеси AAP. Это отделение позволяет получать AHBA, в существенной степени свободный от алкилалкоксигидроксибензальдегида (AAHBA). Под выражением "в существенной степени свободный" понимают, что количество примеси AAHBA является достаточно малым и позволяет применять полученный AHBA в пищевой промышленности. Эта количество составляет преимущественно менее 1000 млн-1, предпочтительно менее 500 млн-1 и более предпочтительно менее 200 млн-1 по отношению к образовавшемуся алкоксигидроксибензальдегиду.
Согласно одному из вариантов настоящее изобретение относится к получению потока VA из GA.
Согласно другому варианту настоящее изобретение относится к получению потока EVA из GE.
Согласно другому варианту настоящее изобретение относится к получению потока смеси VA и EVA из смеси GA и GE.
Настоящее изобретение предпочтительно относится к способу очистки потока VA и/или EVA, предпочтительно получаемого из PC. Кроме того, настоящее изобретение предпочтительно относится к предварительному отделению примеси AAP перед его преобразованием в AAHBA и более предпочтительно примеси MEGA или EGE перед его преобразованием в MEVA или EEVA. Таким образом, настоящее изобретение относится к способу очистки VA и/или EVA, получаемого из GA и/или GE, причем способ включает отделение (S) MEVA и/или EEVA перед получением VA и/или EVA.
Стадия отделения (S) по настоящему изобретению позволяет получить соединение AHBA, в частности VA и/или EVA, в значительной степени очищенное от примеси AAHBA (содержание AAHBA менее 100 млн-1 масс. от конечной смеси, содержащей соединение AHBA), в частности от примеси MEVA или EGE.
Согласно одному из вариантов способ по настоящему изобретению включает получение AHBA взаимодействием AP с глиоксиловой кислотой с последующим окислением. Стадию отделения (S) преимущественно осуществляют перед окислением соединения, образующегося при взаимодействии AP с глиоксиловой кислотой. Стадию отделения предпочтительно осуществляют перед взаимодействием AP с глиоксиловой кислотой. Таким образом, предпочтительным является отделение AAP возможно более быстро после его образования при синтезе AHBA из HP.
Как было указано ранее, перегонка смеси VA/MEVA и/или EVA/EEVA является очень затруднительной. Это тем более соответствует истине в случае перегонки смеси GA/MEGA и/или GE/EGE, поскольку температуры кипения являются еще более близкими.
Неожиданно было найдено, что можно отделять GA от MEGA перегонкой и, следовательно, в более общем случае отделять G от AG.
Таким образом, отделение (S) преимущественно реализуют, осуществляя одну или несколько стадий перегонки. По настоящему изобретению указание на перегонку дается независимо от того, входит ли в нее одна или несколько стадий перегонки.
Своим присутствием PC влияет на перегоняемую смесь, содержащую G и AG, так что становится возможным собирать AG в нижней части дистилляционной колонны вместе с PC, в то время как G собирают в верхней части.
Если не указано иное, в смысле настоящего изобретения под термином "колонна (дистилляционная)" предпочтительно понимают аппаратуру для перегонки, включающую (i) устройство цилиндрической формы, образующее дистилляционную колонну, в общем случае оснащенное (ii) в его нижней части нагревательным устройством, называемым испарителем, предпочтительно содержащее также устройство отведения потока или фракции, называемой кубовым остатком, и предпочтительно содержащее устройство для повторной подачи в нижнюю часть колонны по меньшей мере части стекающей испаряемой жидкости, и оснащенное (iii) в его верхней части устройством отведения, при необходимости оснащенного устройством разделения "жидкость-пар", называемого конденсатором, позволяющим отводить газовый поток, называемый флегмой или головным погоном и при необходимости конденсируемый частично или полностью до жидкого состояния, и предпочтительно содержащее также устройство для повторной подачи в верхнюю часть колонны по меньшей мере части сконденсированной жидкости. Нижнюю часть колонны называют также кубом колонны, а верхнюю часть колонны называют головой колонны. Согласно предпочтительному варианту осуществления испаритель представляет собой испаритель со стекающей жидкой пленкой.
Диалкоксилированное соединение HP (DAB или диалкоксибензол) также представляет собой примесь в способе получения V из HP. Эти соединения, как правило, представляют собой вератрол (VER или 1,2-диметоксибензол) и 1,2-диэтоксибензол. DAB имеет температуру кипения более высокую, чем AG, и DAB должен мешать собирать AG в нижней части колонны.
Совершенно неожиданно и в противоположность тому, что ожидали авторы, в случае присутствия DAB в разделяемом потоке, то есть в потоке, содержащем G, AG и PC, PC влияет, в частности, на коэффициенты относительной летучести A/G, DAB/G и AG/G.
Более специфически коэффициенты относительной летучести DAB/G и AG/G могут быть обращены, в частности, в верхней части колоны по отношению к нижней части колонны в присутствии PC, что позволяет собирать очень чистый поток G с очень низкой концентрацией AG, которая может быть менее 1000 млн-1, преимущественно менее 500 млн-1, предпочтительно менее 200 млн-1 и более предпочтительно менее 100 млн-1. Если бы PC отсутствовал, то вератрол в нижней части колонны имел бы коэффициент относительной летучести по отношению к GA меньше коэффициента относительной летучести MEGA/GA, вел бы себя как более "тяжелое" соединение, чем MEGA, и, следовательно, уходил бы более легко с GA. Однако неожиданным образом в присутствии PC коэффициенты относительной летучести в нижней части колонны оказались обращенными, и MEGA по сравнению с вератролом уходит более предпочтительно в нижней части колонны с потоком, содержащим PC. Таким образом, настоящее изобретение предпочтительно относится к способу, включающему перегонку, в случае которой концентрация вератрола, собираемого с GA в верхней части, превышает концентрацию MEGA, собираемого с GA в верхней части, причем PC собирается в нижней части.
Условия перегонки выбирают так, чтобы они задавали такое обращение коэффициентов относительной летучести. Предпочтительно можно принять, что поток, входящий в дистилляционную колонну, содержит по меньшей мере 30% масс., предпочтительно по меньшей мере 35% масс. и более предпочтительно по меньшей мере 40% масс. PC по отношению к общей массе потока.
Способ по настоящему изобретению преимущественно включает получение VA и/или EVA, в существенной степени свободного от 5-метилванилина ("MEVA") и 3-этокси-5-этил-4-гидроксибензальдегида ("EEVA"), из гваякола (GA) и/или гветола (GE), причем способ включает получение смеси (M), содержащей GA и/или GE, в присутствии примеси 6-метилгваякола ("MEGA") и/или 6-этилгветола (2-этокси-6-этилфенола, сокращенно обозначаемого "EGE") и стадию отделения (S) GA и/или GE от MEGA и/или EGE, причем стадию отделения (S) осуществляют перед получением VA и/или EVA.
Согласно одному из вариантов настоящего изобретения соединение AHBA получают присоединением альдегидной группы к соединению AP конденсацией с глиоксиловой кислотой с последующим окислением образующегося соединения, а стадию отделения (S) осуществляют перед окислением с образованием соединения AHBA.
Согласно другому варианту настоящего изобретения стадию отделения осуществляют перед взаимодействием AP с глиоксиловой кислотой.
Эти различные варианты могут комбинироваться между собой.
Стадию отделения (S) преимущественно реализуют, осуществляя одну или несколько стадий перегонки.
Согласно способу по настоящему изобретению перегонка может быть осуществлена в непрерывном или периодическом режиме, но предпочтительно в непрерывном режиме. По настоящему изобретению дистилляционная колонна может быть адаптирована к требуемой степени чистоты и может быть выполнена из материалов, приспособленных к кислым условиям перегоняемой смеси.
Способ по настоящему изобретению предпочтительно включает подачу потока или фракции (E1) смеси M, содержащей GA и/или GE, обозначенные аббревиатурой "G", и MEGA и/или EGE, обозначенные аббревиатурой "AG", в присутствии пирокатехина (PC) в дистилляционной колонне (E) и выделение G в основном в верхней части дистилляционной колонны в виде потока или фракции (E2) и PC и AG в основном в нижней части дистилляционной колонны в виде потока или фракции (E3).
Согласно одному из вариантов смесь M содержит также диалкоксибензол (DAB), причем DAB предпочтительно представляет собой вератрол (VER), при этом соединение DAB выделяют в основном в нижней части дистилляционной колонны в виде потока или фракции (E3).
Согласно одному из вариантов осуществления PC содержится в количестве от 25 до 75%, предпочтительно от 35 до 65% и более предпочтительно от 40 до 45% масс. от общей массы потока или фракции (E1).
Согласно одному из вариантов осуществления G содержится в количестве от 25 до 75%, предпочтительно от 35 до 65% и более предпочтительно от 45 до 50% масс. от общей массы потока или фракции (E1).
Один из вариантов осуществления отличается тем, что G содержится в количестве более 90%, предпочтительно более 95% и более предпочтительно более 99% от общей массы потока или фракции (E2), и предпочтительно отличается тем, что AG содержится в количестве менее 1%, предпочтительно менее 0,5% и более предпочтительно менее 0,2%, масс. по отношению к общей массе потока или фракции (E2).
Поток или фракция (E2) преимущественно содержит от 90 до 100%, предпочтительно от 95 до 100% и более предпочтительно от 99 до 100% G, от 0 до 1%, предпочтительно менее 0,5% и более предпочтительно менее 0,2% AG и от 0 до 1%, предпочтительно менее 0,5% и более предпочтительно менее 0,2% VER, причем процентные доли выражены по массе по отношению к общей массе потока или фракции (E2).
PC преимущественно содержится в количестве более 60%, предпочтительно более 70% и более предпочтительно более 75% от общей массы потока или фракции (E3), а AG предпочтительно содержится в количестве менее 8%, предпочтительно менее 5% и более предпочтительно менее 3% масс. по отношению к общей массе потока или фракции (E3).
Поток или фракция (E3) предпочтительно содержит от 60 до 100%, предпочтительно от 70 до 100% и более предпочтительно от 75 до 100% PC, от 0 до 8%, предпочтительно менее 5% и более предпочтительно менее 3% AG и от 0 до 10%, предпочтительно менее 8% и более предпочтительно менее 5% VER, причем процентные доли выражены по массе по отношению к общей массе потока или фракции (E3).
Настоящее изобретение относится также к способу отделения GA и/или GE от смеси (M), содержащей GA и/или GE и примесь 6-метилгваякола ("MEGA") и/или 6-этилгветола (2-этокси-6-этилфенола, сокращенно обозначаемого "EGE"), в случае которого смесь M подают в дистилляционную колонну в присутствии пирокатехина (PC) и отводят поток или фракцию, содержащую GA и/или GE, из верхней части дистилляционной колонны и поток или фракцию, содержащую PC, MEGA и/или EGE, из нижней части дистилляционной колонны.
Настоящее изобретение относится также к способу синтеза в паровой фазе по меньшей мере одного алкоксифенола, обозначаемого "AP", включающему образование по меньшей мере одного AP по реакции, называемой O-алкилированием, в паровой фазе между по меньшей мере одним гидроксифенолом, обозначаемым "HP", и по меньшей мере одним O-алкилирующим агентом, причем при указанной реакции образуется также по меньшей мере одна примесь типа алкилалкоксифенола, обозначаемая "AAP", причем при указанной реакции образуется смесь M, содержащая AP, AAP и HP, причем указанный способ включает подачу смеси M по меньшей мере в одну дистилляционную колонну и отведение потока или фракции, содержащей AP, из верхней части дистилляционной колонны и потока или фракции, содержащей HP и AAP, из нижней части дистилляционной колонны, причем указанный способ включает при необходимости рециклирование потока или фракции, содержащей AP, после осуществления реакции O-алкилирования и/или рециклирование потока или фракции, содержащей HP, до осуществления реакции O-алкилирования.
Способ преимущественно включает образование по меньшей мере гваякола (GA) и/или гветола (GE) по реакции, называемой O-алкилированием, в паровой фазе между пирокатехином, обозначаемым "PC", и по меньшей мере одним O-алкилирующим агентом, причем при указанной реакции образуется также по меньшей мере одна примесь 6-метилгваякола ("MEGA") и/или 6-этилгветола (2-этокси-6-этилфенола, сокращенно обозначаемого "EGE"), причем при указанной реакции образуется смесь M, содержащая GA и/или GE, MEGA и/или EGE и PC, причем указанный способ включает подачу смеси M по меньшей мере в одну дистилляционную колонну и отведение потока или фракции, содержащей GA и/или GE, из верхней части дистилляционной колонны и потока или фракции, содержащей PC и MEGA и/или EGE, из нижней части дистилляционной колонны, причем указанный способ включает при необходимости рециклирование потока или фракции, содержащей GA и/или GE, после осуществления реакции O-алкилирования и/или рециклирование потока или фракции, содержащей PC, до осуществления реакции O-алкилирования.
Перегонку в колонне (E) предпочтительно осуществляют при температуре в интервале от 80 до 200°C. Более конкретно температуру в нижней части колонны предпочтительно поддерживают в интервале от 120 до 180°C. Температуру в верхней части колонны предпочтительно поддерживают в интервале от 80 до 120°C. В рамках настоящего изобретения, если не указано иное, температура в нижней части дистилляционной колонны соответствует температуре испарителя.
Согласно одному из вариантов осуществления перегонку в колонне (E) осуществляют при атмосферном давлении. Предпочтительно перегонку осуществляют в вакууме и предпочтительно при давлении в интервале от 1 до 300 мбар (от 0,1 до 30 кПа) и более предпочтительно от 15 до 100 мбар (от 1,5 до 10 кПа).
Согласно одному из вариантов осуществления колонна (E) содержит от 12 до 70 теоретических тарелок и предпочтительно от 20 до 60 теоретических тарелок. Подачу потока или фракции (E1) предпочтительно осуществляют на уровне теоретической тарелки, порядковый номер которой находится в интервале от 5 до 70% и предпочтительно в интервале от 10 до 60% от общего числа теоретических тарелок дистилляционной колонны (E). Ввод подаваемого потока (E1) преимущественно осуществляют в центральной части колонны между теоретическими тарелками №№ 12 и 40 и предпочтительно между теоретическими тарелками №№ 15 и 30.
Согласно одному из вариантов осуществления перегонку в колонне (E) осуществляют при флегмовом числе в интервале от 1 до 10 и предпочтительно в интервале от 2 до 8.
Настоящее изобретение относится, кроме того, к способу очистки GA и/или GE, получаемых из PC. Этот способ предпочтительно включает стадию отделения (S), подобную описанной ранее стадии согласно любому из вариантов осуществления и позволяющую отделять примесь AG от полученного G.
Способ по настоящему изобретению позволяет также рециклировать PC, в частности, перегонкой после стадии отделения (S).
При этом, после отделения примеси AAP от смеси M поток или фракция (E3), содержащая PC и отбираемая в нижней части колонны (E), содержит AAP в качестве примеси: таким образом, PC и AAP содержатся в виде смеси в потоке или фракции (E3) на выходе из нижней части дистилляционной колонны (E). Следовательно, рецикл PC по настоящему изобретению может предпочтительно включать перегонку PC для отделения PC от примеси AAP. Кроме того, в настоящем изобретении предпринята попытка удаления примеси AG, содержащейся в потоке или фракции, содержащей PC, поскольку примесь AG может способствовать образованию различных токсичных производных.
Это отделение является трудным. Таким образом, настоящее изобретение относится к способу отделения AG от смеси, содержащей AG и PC.
В частности, особенно трудно получить PC с содержанием менее 4% и предпочтительно менее 1% масс. AG по отношению к общей массе PC.
Было установлено, что для получения PC с такой чистотой в отношении AG (как правило, MEGA) необходимо осуществлять дополнительную перегонку в колонне (F) потока или фракции из нижней части дистилляционной колонны (E). В общем случае этот поток или фракция (E3) на выходе из дистилляционной колонны (E) содержит также примесь DAB (VER и/или 1,2-диэтоксибензол), также трудно отделяемую от PC. Настоящее изобретение направлено на получение потока или фракции (F6), содержащей PC с суммарным содержанием примесей AG и DAB в количестве менее 10% и предпочтительно менее 8% масс. по отношению к общей массе PC.
С этой целью рекомендуется предпочтительно осуществлять по меньшей мере одну перегонку с боковым отводом потока или фракции (F6), содержащей наибольшее количество PC. Таким образом, согласно предпочтительному варианту осуществления способ включает стадию дополнительной перегонки в колонне (F) потока или фракции (E3), поступающей из нижней части дистилляционной колонны (E), с осуществлением бокового отвода потока или фракции (F6) из колонны (F) для отведения потока или фракции, обогащенной PC. Отсутствие бокового отвода будет проявляться, в частности, в количестве нежелательного DAB и/или AG в отводимых потоке или фракции, преобладающим образом содержащих PC. Этот вариант осуществления позволяет отводить поток или фракцию (F4) из верхней части колонны (F), содержащую также преобладающим образом PC и остаточные количества G, DAB и AG. Поток или фракция (F5) из нижней части колонны (F) содержит в основном продукты осмоления. Поток или фракция (F4) может быть снова направлена в дистилляционную колонну (F2) для сбора потока или фракции (F7) в нижней части колонны (F2), содержащей PC при содержании G, DAB и AG в количестве по массе по меньшей мере в два раза меньше, чем в потоке (F4), и сбора потока или фракции (F8) в верхней части колонны (F2), преобладающим образом содержащей G, DAB и AG. Поток или фракцию (F7), содержащую PC, преимущественно смешивают с потоком или фракцией (F6) предпочтительно для рециклирования на реакцию алкилирования PC. Поток или фракция из верхней части колонны (F2) преимущественно может быть смешана с потоком или фракцией (E1), входящей в дистилляционную колонну (E), для ее рециклирования.
Перегонку в колонне (F) предпочтительно осуществляют при температуре в интервале от 120 до 250°C. Более конкретно температуру в нижней части колонны предпочтительно поддерживают в интервале от 180 до 220°C. Температуру в верхней части колонны предпочтительно поддерживают в интервале от 120 до 180°C.
Согласно одному из вариантов осуществления перегонку в колонне (F) осуществляют при атмосферном давлении. Предпочтительно перегонку осуществляют в вакууме и предпочтительно при давлении в интервале от 1 до 300 мбар (от 0,1 до 30 кПа) и более предпочтительно от 15 до 100 мбар (от 1,5 до 10 кПа).
Согласно одному из вариантов осуществления колонна (F) содержит от 8 до 50 теоретических тарелок и предпочтительно от 10 до 40 теоретических тарелок. Подачу потока или фракции (F1) предпочтительно осуществляют на уровне теоретической тарелки, порядковый номер которой находится в интервале от 5 до 70% и предпочтительно в интервале от 10 до 60% от общего числа теоретических тарелок дистилляционной колонны (F). Ввод подаваемого потока (F1) преимущественно осуществляют в центральной части колонны между теоретическими тарелками №№ 2 и 20 и предпочтительно между теоретическими тарелками №№ 5 и 20.
Согласно одному из вариантов осуществления перегонку в колонне (F) осуществляют при флегмовом числе в интервале от 1 до 10 и предпочтительно в интервале от 1 до 8.
Перегонку в колонне (F2) предпочтительно осуществляют при температуре в интервале от 80 до 200°C. Более конкретно температуру в нижней части колонны предпочтительно поддерживают в интервале от 120 до 180°C. Температуру в верхней части колонны предпочтительно поддерживают в интервале от 80 до 140°C.
Согласно одному из вариантов осуществления перегонку в колонне (F2) осуществляют при атмосферном давлении. Предпочтительно перегонку осуществляют в вакууме и предпочтительно при давлении в интервале от 1 до 300 мбар (от 0,1 до 30 кПа) и более предпочтительно от 15 до 100 мбар (от 1,5 до 10 кПа).
Согласно одному из вариантов осуществления колонна (F2) содержит от 1 до 20 теоретических тарелок и предпочтительно от 2 до 10 теоретических тарелок. Подачу потока или фракции (F4) предпочтительно осуществляют на уровне теоретической тарелки, порядковый номер которой находится в интервале от 5 до 70% и в интервале от 10 до 60% от общего числа теоретических тарелок дистилляционной колонны (F2). Ввод подаваемого потока (F4) преимущественно осуществляют в центральной части колонны между теоретическими тарелками №№ 1 и 15 и предпочтительно между теоретическими тарелками №№ 2 и 10.
Согласно одному из вариантов осуществления перегонку в колонне (F2) осуществляют при флегмовом числе в интервале от 1 до 10 и предпочтительно в интервале от 2 до 8.
Согласно предпочтительному варианту осуществления перегонка включает предварительное отделение предпочтительно перегонкой в колонне (A) более легких соединений от более тяжелых соединений для отвода из верхней части колонны (A) потока или фракции (A1), содержащей легкие соединения, и из нижней части колонны (A) потока или фракции (A2), содержащей тяжелые соединения, в том числе AP. Эта стадия отделения позволяет, в частности, удалять O-алкилирующий агент, не прореагировавший во время реакции с HP, в частности с PC. Согласно одному из вариантов поток или фракцию (A2) используют в качестве входящего потока дистилляционной колонны (E), названного ранее потоком или фракцией (E1).
Перегонку в колонне (A) предпочтительно осуществляют при температуре в интервале от 80 до 250°C. Более конкретно температуру в нижней части колонны предпочтительно поддерживают в интервале от 130 до 220°C. Температуру в верхней части колонны предпочтительно поддерживают в интервале от 80 до 150°C.
Согласно одному из вариантов осуществления перегонку в колонне (A) осуществляют при атмосферном давлении. Предпочтительно перегонку осуществляют в вакууме и предпочтительно при давлении в интервале от 1 до 300 мбар (от 0,1 до 30 кПа) и предпочтительно от 15 до 100 мбар (от 1,5 до 10 кПа).
Согласно одному из вариантов осуществления колонна (A) содержит от 8 до 50 теоретических тарелок и предпочтительно от 10 до 40 теоретических тарелок. Подачу потока или фракции (A1) предпочтительно осуществляют на уровне теоретической тарелки, порядковый номер которой находится в интервале от 5 до 70% и предпочтительно в интервале от 10 до 60% от общего числа теоретических тарелок дистилляционной колонны (A). Ввод подаваемого потока (A1) преимущественно осуществляют в центральной части колонны между теоретическими тарелками №№ 2 и 20 и предпочтительно между теоретическими тарелками №№ 5 и 20.
Согласно одному из вариантов осуществления перегонку в колонне (A) осуществляют при флегмовом числе в интервале от 0,01 до 10 и предпочтительно в интервале от 0,1 до 5.
Колонны могут быть масштабированы согласно сведениям, располагаемым специалистами в данной области техники.
Настоящее изобретение относится также к получению по меньшей мере одного алкоксигидроксибензальдегида из гидроксифенола.
Синтез алкоксигидроксибензальдегида
Способ по настоящему изобретению позволяет получать алкоксигидроксибензальдегид присоединением альдегидной группы к соединению типа алкоксифенола, указанному ранее.
Способ по настоящему изобретению предпочтительно позволяет получать ванилин и/или этилванилин из гваякола и/или гветола и предпочтительно из гваякола и/или гветола, очищенного способом по настоящему изобретению. Смесь "гваякол/гветол" получают с любым соотношением. Соотношение Ga/Ge зависит от условий O-алкилирования. Массовое соотношение Ga/Ge может изменяться, например, от 0,1/100 до 100/1. Массовое соотношение "ванилин/этилванилин" изменяется предпочтительно от 80/20 до 40/60 и более предпочтительно от 80/20 до 45/55. Согласно одному из вариантов осуществления молярное соотношение "ванилин/этилванилин" равно по меньшей мере 2. Массовое содержание ванилина преимущественно находится в интервале от 65 до 72% и предпочтительно в интервале от 60 до 70%, а содержание этилванилина находится в интервале от 28 до 35% и предпочтительно в интервале от 30 до 33% масс.
При этом указания относятся к соответствующим соединениям типа алкоксифенола, гваякола, гветола и алкоксигидроксибензальдегидам независимо от того, находятся ли они в солевой или в протонированной форме. Ясное указание на соль позволяет описать особую характеристику этого варианта. Специалисты в данной области техники в состоянии знать, когда речь идет о солевой или протонированной форме, в частности, с учетом указанных литературных источников и собственных общих знаний.
Присоединение альдегидной группы предпочтительно осуществляют по двум основным реакциям, а именно за счет конденсации соединения типа алкоксифенола с глиоксиловой кислотой и окислением соединения, образующегося при реакции конденсации.
Согласно предпочтительному варианту осуществления способ по настоящему изобретению включает, кроме того, следующие стадии:
- конденсация соединения типа алкоксифенола с глиоксиловой кислотой для получения соответствующего производного соединения миндальной кислоты;
- отделение соединения типа алкоксифенола от раствора, содержащего производное соединение миндальной кислоты, предпочтительно с рециклированием соединения типа алкоксифенола;
- окисление производного соединения миндальной кислоты для получения алкоксигидроксибензальдегида;
- отделение алкоксигидроксибензальдегида экстракцией органическим растворителем с последующим отделением алкоксигидроксибензальдегида от органического растворителя предпочтительно с рециклированием органического растворителя, применяемого для экстракции алкоксигидроксибензальдегида;
- выделение алкоксигидроксибензальдегида при необходимости в твердом состоянии.
В случае, когда термины "производное соединение миндальной кислоты", "ванилин" или "этилванилин" употребляются без уточнения, речь идет о производном соединении параминдальной кислоты, параванилине или этил-параванилине соответственно, если не указано иное.
Производные соединения параминдальной кислоты (формула (IV)), в частности, соединения параванилина, соответствуют следующим формулам:
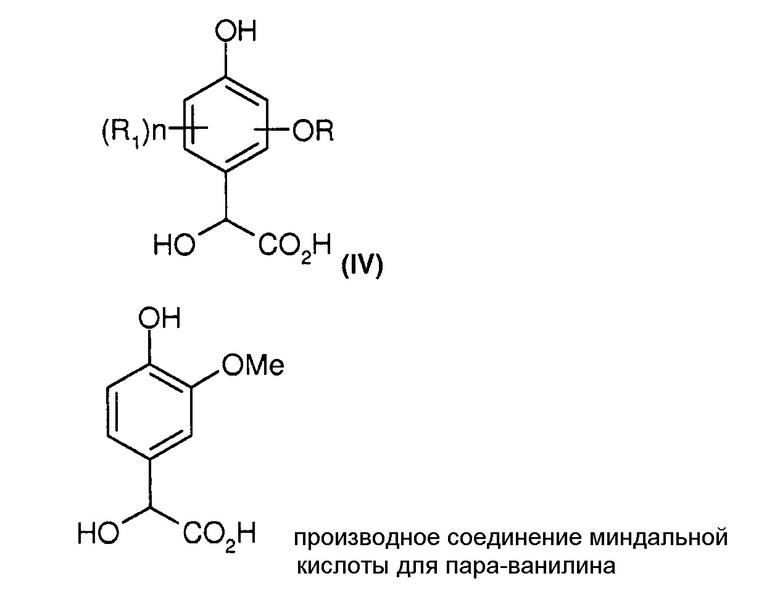
Стадии и их технологическую последовательность предпочтительно осуществляют в непрерывном режиме за исключением некоторых стадий, таких как разделение соединений.
Реакции конденсации алкоксифенолов, в частности гваякола, с глиоксиловой кислотой описаны в литературе. Более предпочтительно можно назвать патенты EP 0578550, WO 99/65853 и WO 09077383, ссылка на которые дается в связи с получением соединений по настоящему изобретению.
Последовательность предыдущих стадий может быть осуществлена согласно следующим реакциям на примере гваякола:
- конденсация гваякола и глиоксиловой кислоты для образования параминдальной кислоты:
гваякол + глиоксиловая кислота + NaOH → гваяколят + манделат (фенолят) + H2O
- подкисление для выделения непрореагировавшего гваякола:
гваяколят + манделат (фенолят) + H2SO4 → гваякол + манделат + Na2SO4
- окисление параминдальной кислоты для образования параванилина:
манделат + NaOH + O2 → ванилат + Na2CO3 + H2O
- подкисление для образования ванилина:
ванилат + Na2CO3 + H2SO4 → VA + CO2 + Na2SO4
При разделении смеси независимо от вида образовавших ее алкоксифенола или алкоксигидроксибензальдегида предпочтительно организуют рецикл, называемый также рециркуляционным контуром. Таким образом, по настоящему изобретению ссылаются на "контур рецикла 1" в случае, когда говорят о рециклировании избытка алкоксифенола во время реакции конденсации, и на "контур рецикла 2" в случае, когда имеют в виду рециклирование растворителя, применяемого для экстракции алкоксигидроксибензальдегида, при реализации стадий разделения.
Алкоксигидроксибензальдегид после отделения может быть обработан для получения в виде очищенного твердого вещества, в данном случае по настоящему изобретению говорят о "блоке получения твердого вещества". Эта обработка может, как правило, включать стадии кристаллизации, отделения от жидкости, сушки или соскабливания. Некоторые из этих стадий, такие как отделение от жидкости, не могут быть осуществлены в непрерывном режиме. Таким образом, способ предпочтительно является непрерывным за исключением стадии отделения от жидкости.
Ванилин и/или этилванилин, полученный таким образом, может быть использован, в частности, в качестве ароматизатора в промышленности, такой, как, например, пищевая, фармацевтическая или парфюмерно-косметическая промышленность, например, для изготовления отдушек.
Ванилин и/или этилванилин может служить также в качестве промежуточного соединения в синтезе, например, для получения вератрового альдегида (3,4-диметоксибензальдегида) и/или 3,4-диэтоксибензальдегида.
В рамках этого варианта, если способ относится к одновременному получения ванилина и этилванилина, можно: синтезировать ванилин и этилванилин в одном и том же потоке, из потока, содержащего гваякол и гветол, в свою очередь полученные из катехола по параллельным реакциям с O-метилирующим и O-этилирующим агентами; раздельно синтезировать гваякол и гветол и затем смешивать их перед стадией конденсации для синтеза ванилина и этилванилина в одном и том же потоке; раздельно синтезировать производные соединения миндальной кислоты по отдельным реакциям гваякола и гветола с глиоксиловой кислотой и затем смешивать соответствующие производные соединения миндальной кислоты перед стадией окисления; раздельно синтезировать ванилин и этилванилин и затем смешивать их, например, перед кристаллизацией.
Таким образом, можно составить схему способа соответственно фиг. 3, предпочтительно включающего следующие стадии после получения AP по настоящему изобретению:
- конденсация соединения типа алкоксифенола (AP) с глиоксиловой кислотой для получения соответствующего производного соединения миндальной кислоты после прибавления воды, глиоксиловой кислоты и основания;
- отделение соединения типа алкоксифенола от раствора, содержащего соответствующее производное соединение миндальной кислоты, после прибавления органического растворителя и кислоты и предпочтительно с рециклированием соединения типа алкоксифенола;
- окисление производного соединения миндальной кислоты взаимодействием с введенным окислителем для получения соответствующего алкоксигидроксибензальдегида;
- отделение алкоксигидроксибензальдегида экстракцией после прибавления органического растворителя и кислоты;
- отделение алкоксигидроксибензальдегида от органического растворителя перегонкой предпочтительно с рециклированием органического растворителя;
- выделение алкоксигидроксибензальдегида (AHBA) при необходимости в твердом состоянии.
Реакция конденсации
Согласно предпочтительному варианту алкоксифенол приводят во взаимодействие с глиоксиловой кислотой в присутствии неорганического или органического основания, предпочтительно в присутствии NaOH или KOH и более предпочтительно гидроксида натрия (NaOH) или гидроксида аммония. Согласно этому варианту получают смесь алкилфенолята и производного соединения типа манделата.
Реакция конденсации между алкоксифенолом (в виде фенолята) и глиоксиловой кислотой позволяет синтезировать соответствующее производное соединение параминдальной кислоты.
Конденсация может быть осуществлена в каскаде реакторов смешения. Согласно одному из вариантов реакцию осуществляют в реакторе вытеснения, оснащенном при необходимости теплообменником. Такой вариант осуществления описан, например, для случая гваякола в заявке WO 09/077383. Реакция конденсации между алкоксифенолом и глиоксиловой кислотой может быть осуществлена в воде в присутствии щелочного металла, причем указанную реакцию проводят в реакторе вытеснения. Она может быть осуществлена в трубчатом реакторе.
Реакция конденсации предпочтительно может быть катализирована гидроксидом четвертичного аммония подобно, например, реакции, описанной в заявке EP 0578550.
Реакция конденсации предпочтительно может быть осуществлена в присутствии дикарбоновой кислоты подобно, например, реакции, описанной в заявке WO 99/65853.
Поскольку алкоксифенол обычно содержится в избытке по отношению к глиоксиловой кислоте, то фракцию непрореагировавшего алкоксифенола предпочтительно выделяют в контуре рецикла 1. Этот избыток уменьшает вероятность образования соединений типа диминдальной кислоты (то есть соединений, образующихся при конденсации двух молекул глиоксиловой кислоты с молекулой гваякола).
В течение 1-го отрезка времени алкоксифенол и гидроксид натрия преимущественно реагируют с образованием алкоксифенолята натрия. Например, в случае гваякола:
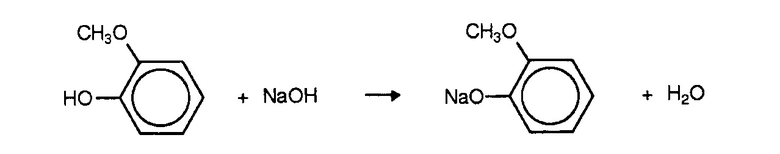
Затем алкоксифенолят взаимодействует с глиоксиловой кислотой с образованием соответствующего параманделата:
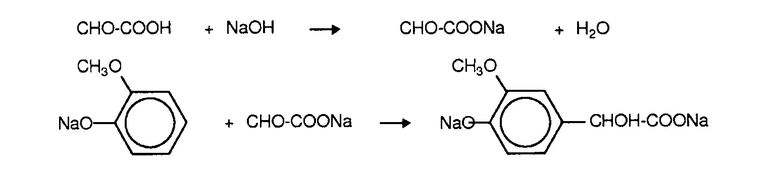
Две эти реакции получения глиоксилата и гваяколята могут быть осуществлены в виде двух раздельных стадий. Альтернативным образом глиоксиловую кислоту приводят в контакт непосредственно с гваяколятом в присутствии основания.
Раствор алкоксифенолята
Согласно предпочтительному варианту получают раствор алкоксифенолята. Этот раствор алкоксифенолята подпитывают алкоксифенолом из расходного бака, в который поступает алкоксифенол, рециклированный в контуре рецикла 1, и свежий алкоксифенол. Раствор алкоксифенолята предпочтительно подают в реактор для осуществления реакции конденсации.
В раствор алкоксифенолята подают общее количество воды (деминерализованной) и неорганического основания (например, гидроксида натрия), необходимых для реакции.
Температуру реактора при получении раствора алкоксифенолята предпочтительно поддерживают равной температуре реакции конденсации.
Контур рецикла 1
Контур рецикла 1 предназначен для выделения алкоксифенола, непрореагировавшего при реакции конденсации. Таким образом, контур 1 относится к отделению соединения типа алкоксифенола от производных соединений миндальной кислоты и предпочтительно включает следующие стадии:
- выделение смеси, содержащей образовавшееся производное соединение миндальной кислоты в виде манделата и непрореагировавший алкоксифенол в виде алкоксифенолята;
- нейтрализация алкоксифенолята с образованием алкоксифенола при сохранении соответствующего манделата в виде конъюгата с основанием (в форме соли);
- отделение алкоксифенола от манделата;
- подача манделата в реактор окисления;
- рециклирование алкоксифенола, не прореагировавшего в качестве реагента, для осуществления реакции конденсации.
Реакционную смесь, образующуюся при конденсации, предпочтительно нейтрализуют неорганической кислотой, предпочтительно серной кислотой, для превращения непрореагировавшего алкоксифенолята в алкоксифенол в условиях сохранения манделата в солевой форме.
Можно отделять непрореагировавший алкоксифенол, содержащийся в органической фракции, от образовавшегося манделата, содержащегося в водной фракции.
Согласно одному из вариантов алкоксифенол, полученный после нейтрализации, можно извлекать экстракцией, прибавляя органический растворитель, который солюбилизирует алкоксифенол, содержащийся в водной фракции. В данном случае можно сослаться, например, на способ, описанный для случая гваякола в патенте FR 2 379 501.
Согласно предпочтительному варианту осуществления алкоксифенол, содержащийся в водной фракции, экстрагируют органическим растворителем. Применяют органический растворитель, который является инертным по отношению к манделату. В качестве растворителей, которые могут быть использованы, можно назвать, в частности, алифатические, циклоалифатические, ароматические углеводороды, в том числе галогенпроизводные, спирты, простые эфиры, кетоны и нитрилы. Более предпочтительно можно назвать гептан, циклогексан, метилциклогексан, бензол, толуол в качестве примеров алифатических, циклоалифатических или ароматических углеводородов; дихлорметан, трихлорметан, дихлорэтан, хлорбензол, дихлорбензолы в качестве примеров алифатических, циклоалифатических или ароматических галогенпроизводных углеводородов; метанол, этанол, пропанол, изопропанол, бутанол в качестве примеров спиртов; диэтиловый эфир, диизопропиловый эфир, метил-трет-бутиловый эфир в качестве примеров простых эфиров; ацетон, метилэтилкетон, метилизобутилкетон, диизобутилкетон, метилизобутилкетон в качестве примеров кетонов; ацетонитрил в качестве примера нитрилов. Можно применять смесь указанных растворителей. Можно использовать растворители пищевого качества.
При этом манделат остается в водной фракции, тогда как алкоксифенол переходит в органическую фракцию.
Алкоксифенол, направляемый на рециклирование, может быть загрязнен остатками органического растворителя. Присутствие органического растворителя в алкоксифеноле ведет к значительному снижению характеристик реакции конденсации. Таким образом, предпочтительным является отделение растворителя, применяемого для экстракции, от алкоксифенола перед повторной подачей алкоксифенола в расходный бак.
После разделения водной и органической фракций смесь "алкоксифенол-органическая фракция" предпочтительно перегоняют для отделения органического растворителя от алкоксифенола, а также от продуктов осмоления, содержащихся в общем случае, для рециклирования органического растворителя. Могут быть предусмотрены также другие способы разделения.
Водную фракцию, содержащую манделат, предпочтительно обрабатывают для удаления остаточного органического растворителя, например, перегонкой. Затем манделат подают на реакцию окисления.
Согласно одному из вариантов непрореагировавший алкоксифенолят можно отделять, используя анионообменные смолы в форме основания или адсорбент, позволяющие селективно фиксировать алкоксифенол и выделять из водного потока содержащиеся производные соединения миндальной кислоты в солевой форме, образующиеся при реакции конденсации. Можно отделять поток алкоксифенола в солевой форме (алкоксифенолят), фиксированный на смоле или адсорбенте, регенерацией смолы или десорбцией. Такие способы разделения описаны, в частности, в заявках WO 09/141280 и WO 11/039331.
Алкоксифенол, не прореагировавший во время реакции конденсации, предпочтительно рециклируют для подачи в раствор алкоксифенолята.
Удаление диалкоксибензола (сокращенно обозначаемого "DAB") в случае его наличия
Как было указано ранее, при реакции O-алкилирования образуется также соединение типа диалкоксибензола. В способе по настоящему изобретению алкоксифенол, полученный по реакции O-алкилирования, находится в смеси с соединением последующего алкоксилирования (диалкоксилирования) типа диалкоксибензола.
В частности, авторами было найдено, что в случае, когда образуется вератрол (1,2-диметоксибензол), в частности в случае, когда гваякол был получен из пирокатехина, то вератрол накапливается в растворе гваякола в силу рециклирования, а также поскольку он не вступает в реакцию конденсации с глиоксиловой кислотой. Концентрация вератрола в расходном баке гваякола (смесь "свежий+рециклированный") может изменяться. Поскольку вератрол не вступает в реакцию конденсации по настоящему изобретению, то, следовательно, вератрол имеет тенденцию накапливаться в растворе гваякола и таким образом понижать содержание гваякола в растворе, подаваемом в реактор конденсации. В способе по настоящему изобретению сделана попытка ограничить накопление вератрола в гваяколе или в растворе гваяколята. Таким образом, согласно предпочтительному варианту осуществления настоящего изобретения образовавшийся диалкоксибензол удаляют. Согласно одному из вариантов способа по настоящему изобретению массовая концентрация вератрола в растворе, содержащем AP, перед реакцией конденсации находится в интервале от 0,01 до 25%, предпочтительно в интервале от 0,01 до 10%, более предпочтительно в интервале от 0,01 до 5% и в общем случае составляет около 2%.
Следовательно, настоящее изобретение относится к способу получения, который может быть независимо патентоспособным, по меньшей мере одного соединения типа алкоксигидроксибензальдегида исходя по меньшей мере из одного соединения типа гидроксифенола, причем указанный способ отличается тем, что он включает:
(i) - синтез по меньшей мере одного алкоксифенола исходя по меньшей мере из одного гидроксифенола в присутствии по меньшей мере одного O-алкилирующего агента;
(ii) - выделение по меньшей мере одного алкоксифенола и примесей, в число которых входит диалкилированное соединение типа диалкоксибензола (сокращенно обозначаемое "DAB");
(iii) - отделение диалкоксибензола от алкоксифенола;
(iv) - конденсация алкоксифенола с глиоксиловой кислотой и получение соответствующего производного соединения миндальной кислоты;
(v) - окисление производного соединения миндальной кислоты с получением соответствующего алкоксигидроксибензальдегида;
и
(vi) - выделение алкоксигидроксибензальдегида при необходимости в твердом состоянии.
Настоящее изобретение предпочтительно относится к способу получения из пирокатехина по меньшей мере одного алкоксигидроксибензальдегида, выбранного из ванилина (4-гидрокси-3-метоксибензальдегида), этилванилина (3-этокси-4-гидроксибензальдегида) и любой их смеси.
Согласно предпочтительному варианту осуществления стадию (i) осуществляют в трехфазной среде соответственно описанию настоящего изобретения, включая любые варианты и способы осуществления, описанные без каких-либо ограничений.
Согласно предпочтительному варианту осуществления стадия (iii) отделения диалкоксибензола от алкоксифенола включает , кроме того, получение водного раствора алкоксифенола в солевой форме (фенолята) и отделение остаточного диалкоксибензола из водной смеси, содержащей алкоксифенолят.
Согласно предпочтительному варианту осуществления, поясненному, например, фиг. 4, способ включает следующие стадии:
- получение раствора алкоксифенола (AP), например, из смеси, образованной свежим алкоксифенолом и алкоксифенолом, поступающим от рециклирования, причем указанный раствор алкоксифенола содержит также примесь типа диалкоксибензола (DAB);
- получение раствора алкоксифенолята прибавлением воды и по меньшей мере одного основания к раствору алкоксифенола;
- отделение диалкоксибензола от алкоксифенола;
- возможное экономически эффективное использование диалкоксибензола;
- конденсация алкоксифенола с глиоксиловой кислотой и получение соответствующего производного соединения миндальной кислоты, например, в форме соли (манделата).
Согласно одному из вариантов отделение диалкоксибензола от алкоксифенола осуществляют перегонкой или декантацией.
Производные соединения миндальной кислоты преимущественно получают в солевой форме.
Таким образом, способ по настоящему изобретению включает стадию нейтрализации производного соединения миндальной кислоты в форме соли с образованием производного соединения миндальной кислоты в форме кислоты.
Согласно одному из вариантов гидроксифенол представляет собой пирокатехин, алкоксифенол представляет собой гваякол, полученный на стадии (i) с метилирующим агентом, а диалкоксибензол представляет собой вератрол, при этом способ включает также отделение на стадии (iii) пирокатехина от смеси гваякола и вератрола.
Согласно одному из вариантов гидроксифенол представляет собой пирокатехин, алкоксифенол представляет собой гветол, полученный на стадии (i) с этилирующим агентом, а диалкоксибензол представляет собой 1,2-диэтоксибензол, при этом способ включает также отделение на стадии (iii) пирокатехина от смеси гветола и 1,2-диэтоксибензола.
Часть алкоксифенола (алкоксифенолята) предпочтительно направляют в дистилляционную колонну, предназначенную для ограничения содержания диалкоксибензола в гваяколе перед реакцией конденсации. Диалкоксибензол отгоняется из верхней части колонны в виде гетероазеотропной смеси "диалкоксибензол-вода". Температура в нижней части дистилляционной колонны предпочтительно находится в интервале от 90 до 120°C. Этот способ позволяет, например, в случае гваякола ограничить значением 0,01% массовую концентрацию вератрола в гваяколе (или в гваяколяте), используемом для осуществления реакции конденсации.
Затем гетероазеотропную смесь "диалкоксибензол-вода" предпочтительно разделяют декантацией или другим способом разделения.
Поток из нижней части колонны (содержащий алкоксифенолят) предпочтительно направляют на подачу в реактор конденсации для осуществления конденсации алкоксифенола (алкоксифенолята) и глиоксиловой кислоты.
Окисление производных соединений миндальной кислоты с образованием алкоксигидроксибензальдегида
Стадия окисления позволяет преобразовывать производные соединения миндальной кислоты в требуемые алкоксигидроксибензальдегиды. Производное соединение параминдальной кислоты более предпочтительно соответствует приведенной ранее формуле (IV). В частности, можно получать параванилин (в данном случае называемый ванилином) в виде ванилата окислением соответствующего производного соединения параминдальной кислоты.
Алкоксигидроксибензальдегид получают окислением соответствующего производного соединения миндальной кислоты предпочтительно в атмосфере окислителя, такого, как O2 или воздух.
Согласно одному из вариантов реакционная среда представляет собой щелочную водную среду, предпочтительно неорганического основания и более предпочтительно гидроксида натрия или калия, для образования соответствующего фенолята и для связывания выделяющегося CO2 в виде карбоната.
Реакцию можно осуществлять в непрерывном или в периодический режиме, например, в среде, сильно разбавленной водой.
Реакцию можно катализировать. Катализатор этой реакции окисления может быть выбран из катализаторов, содержащих по меньшей мере один металлический элемент, выбранный из группы, в которую входят медь, никель, кобальт, железо, марганец и любые их смеси. В качестве примеров неорганических или органических соединений меди предпочтительно можно назвать такие соединения меди, как бромид меди (I) и меди (II); иодид меди (I); хлорид меди (I) и меди (II); основной карбонат меди (II); нитрат меди (I) и меди (II); сульфат меди (I) и меди (II); сульфит меди (I); оксид меди (I) и меди (II); гидроксид меди (II); ацетат меди (I) и меди (II); трифторметилсульфонат меди (II). В качестве специфических примеров производных никеля, можно назвать галогениды никеля (II), такие как хлорид, бромид или иодид никеля (II); сульфат никеля (II); карбонат никеля (II); соли органических кислот, содержащих от 1 до 18 атомов углерода, такие как, в частности, ацетат, пропионат; комплексы никеля (II), такие как ацетилацетонат никеля (II), дихлоро-бис-(трифенилфосфин)никель (II), дибромо-бис(бипиридин)никель (II); комплексы никеля (0), такие как бис(циклоокта-1,5-диен)никель (0), бис(дифенилфосфиноэтан)никель (0). В качестве примеров соединений на основе кобальта можно назвать, в частности, галогениды кобальта (II) и (III), такие как хлорид, бромид или иодид кобальта (II), хлорид, бромид или иодид кобальта (III); сульфат кобальта (II) и кобальта (III); карбонат кобальта (II), основной карбонат кобальта (II); ортофосфат кобальта (II); нитрат кобальта (II); оксид кобальта (II) и кобальта (III); гидроксид кобальта (II) и кобальта (III); соли органических кислот, содержащих от 1 до 18 атомов углерода, такие как, в частности, ацетат кобальта (II) и кобальта (III), пропионат кобальта (II); комплексы кобальта (II), такие как хлорид гексаминокобальта (II) или (III), сульфат гексаминокобальта (II) или (III), хлорид пентаминокобальта (III), хлорид триэтилендиаминокобальта (III). Также можно назвать каталитические системы на основе железа в общем случае в форме оксидов, гидроксидов или солей, таких как хлорид, бромид, иодид, фторид железа (II) и железа (III); сульфат железа (II) и железа (III); нитрат железа (II) и железа (III); оксид железа (II) и железа (III). Реакцию окисления можно катализировать, например, каталитической системой, содержащей два металлических элемента, выбранных из группы, в которую входят медь, никель, кобальт, железо, марганец и любые их смеси. Настоящее изобретение предпочтительно относится к реакциям, описанным в заявке WO 08/148760.
В течение 1-го отрезка времени соединение в форме манделата взаимодействует с основанием (предпочтительно с гидроксидом натрия) для перевода в солевую форму фенолятной группы соединения в форме манделата. Затем при окислении в среде окислителя (предпочтительно на воздухе) образуется ванилат и CO2 (уловленный в виде карбоната).
Окисление производного соединения параминдальной кислоты в параванилин происходит почти полностью с очень хорошей селективностью.
После реакции окисления получают предшественник алкоксигидроксибензальдегида (ванилина и/или этилванилина) с гидроксигруппой в солевой форме (в ионной форме) и различные примеси, в число которых входят продукты осмоления.
На следующей стадии осуществляют подкисление алкоксигидроксибензальдегида (ванилина и/или этилванилина) в реакционной смеси сильной кислотой, например серной кислотой. Затем выделяют требуемое соединение, а именно алкоксигидроксибензальдегид (ванилин и/или этилванилин), в присутствии продуктов осмоления способом, который позволяет сохранять целостность соединений, подлежащих разделению, и при этом является экономичным и легко осуществимым в промышленных устройствах и более предпочтительно в устройствах, работающих в непрерывном режиме. Известный способ отделения алкоксигидроксибензальдегида (ванилина и/или этилванилина) от смеси неочищенных продуктов реакции представляет собой экстракция органическим растворителем.
Этот способ предпочтительно включает:
- отделение алкоксигидроксибензальдегида от реакционной смеси экстракцией органическим растворителем;
- выделение и рециклирование органического растворителя, применяемого для экстракции.
Контур рецикла 2
Контур рецикла 2 относится к преобразованию гидроксилата алкоксибензальдегида (ванилата и/или этилванилата), полученного по реакции окисления с образованием соответствующего алкоксигидроксибензальдегида, и к выделению и рециклированию растворителя, применяемого для экстракции алкоксигидроксибензальдегида (ванилина и/или этилванилина) из смеси, образующейся при реакции окисления. Настоящее изобретение предпочтительно относится к получению ванилина и/или этилванилина.
Реакционную смесь, образующуюся при окислении, подкисляют кислой средой, предпочтительно содержащей неорганическую кислоту, например серную или соляную кислоту, для преобразования соли алкоксигидроксибензальдегида в алкоксигидроксибензальдегид.
Согласно предпочтительному варианту осуществления алкоксигидроксибензальдегид, содержащийся в водной среде, экстрагируют органическим растворителем, который солюбилизирует алкоксигидроксибензальдегид, содержащийся в водной среде. Применяют органический растворитель, который является инертным по отношению к алкоксигидроксибензальдегиду. В качестве растворителей, которые могут быть использованы, можно назвать, в частности, алифатические, циклоалифатические, ароматические углеводороды, в том числе галогенпроизводные, спирты, простые эфиры, кетоны и нитрилы. Более предпочтительно можно назвать гептан, циклогексан, метилциклогексан, бензол, толуол в качестве примеров алифатических, циклоалифатических или ароматических углеводородов; дихлорметан, трихлорметан, дихлорэтан, хлорбензол, дихлорбензолы в качестве примеров алифатических, циклоалифатических или ароматических галогенпроизводных углеводородов; метанол, этанол, пропанол, изопропанол, бутанол в качестве примеров спиртов; диэтиловый эфир, диизопропиловый эфир, метил-трет-бутиловый эфир в качестве примеров простых эфиров; ацетон, метилэтилкетон, метилизобутилкетон, диизобутилкетон в качестве примеров кетонов; ацетонитрил в качестве примера нитрилов. Можно применять смесь указанных растворителей.
Операцию экстракции осуществляют при температуре, которая зависит от вида растворителя.
Стадия экстракции может быть осуществлена одновременно с подкислением. Однако предпочтительным является вариант, когда сначала смешивают реакционную смесь и органический растворитель, а затем прибавляют кислоту.
Для выделения алкоксигидроксибензальдегида (ванилина и/или этилванилина) из растворителя, применяемого для экстракции, можно осуществлять отделение перекристаллизацией, но предпочтительно осуществляют перегонку указанной смеси, позволяющую получить, например, из верхней части дистилляционной колонны растворитель для экстракции (если он является более летучим соединением в смеси), а из нижней части дистилляционной колонны получить "сырой алкоксигидроксибензальдегид" ("сырые" ванилин и/или этилванилин), а именно смесь, содержащую в основном алкоксигидроксибензальдегид в сочетании с тяжелыми примесями, называемыми "продуктами осмоления", и небольшим количеством легких примесей. Касательно этой стадии предпочтительно можно сослаться на способ, описанный в заявке WO 2010/007161.
Использованный органический растворитель предпочтительно рециклируют. Он может быть вновь введен в технологический процесс для экстрагирования алкоксигидроксибензальдегида из водной среды при подкислении и/или использован для выделения алкоксигидроксибензальдегида из промывных вод.
При необходимости алкоксигидроксибензальдегид может быть обработан для расфасовки в твердом состоянии. Алкоксигидроксибензальдегид предпочтительно очищают перегонкой с последующей кристаллизацией (при применении одного или нескольких растворителей или по технологии соскабливания), например, на следующих стадиях:
- растворение алкоксигидроксибензальдегида в смеси "вода-спирт" (содержащей, например, метанол или этанол);
- кристаллизация в вакууме;
- отделение от жидкости в периодическом режиме;
- сушка алкоксигидроксибензальдегида при температуре 25°C, то есть при температуре ниже температуры плавления алкоксигидроксибензальдегида, предпочтительно в атмосфере инертного газа (как правило, азота).
Можно предусматривать несколько вариантов кристаллизации: например, периодическая кристаллизация (партиями), например, охлаждением или кристаллизация в непрерывном режиме, например, в вакууме.
Полученное вещество при необходимости может быть измельчено для получения более мелких гранул.
Другие цели, характеристики и преимущества настоящего изобретения могут быть выяснены специалистами в данной области техники при чтении пояснительного описания, относящегося к примерам, которые приведены только в порядке пояснения и которые никаким образом не могут ограничивать объем патентной охраны настоящего изобретения.
Примеры составляют неотъемлемую часть настоящего изобретения, и любая характеристика, оказывающаяся новой по сравнению с предшествующим уровнем техники, из описания в целом, включая примеры, также составляет неотъемлемую часть настоящего изобретения в его функциональном назначении и общем значении.
Таким образом, любой пример характеризуется общим объемом патентной охраны.
Примеры
Процентные доли приведены по массе по отношению к общей массе композиции. Значения температуры приведены в градусах Цельсия (°C).
Пример 1
После испарения катехол с метанолом в непрерывном режиме приводят во взаимодействие в паровой фазе. Полученная смесь содержит катехол, гваякол, вератрол, метил-6-гваякол, непрореагировавший метанол и продукты осмоления (соединения с большой молекулярной массой по сравнению с другими содержащимися соединения, например, с молекулярной массой, большей в 2 или более раз). В зависимости от заданных рабочих условий содержание вератрола, метил-6-гваякола и продуктов осмоления в конденсированном жидком потоке, выходящем из зоны реакции, может быть более или менее значительным. Как правило, эта реакция может быть осуществлена согласно патенту EP 914854 B1.
Полученную смесь подвергали перегонке в непрерывном режиме, используя показанную на фиг. 1 систему перегонки, включающую первую дистилляционную колонну 10 (A), вторую дистилляционную колонну 20 (E) и третью дистилляционную колонну 30 (F), соединенные последовательно.
Жидкую смесь 1, содержавшую катехол, гваякол, вератрол, метил-6-гваякол, непрореагировавший метанол и продукты осмоления, подавали в центральную часть первой дистилляционной колонны 10, содержавшей в нижней части 12 испаритель со стекающей жидкой пленкой. Температура испарения в среднем составляла 242°C. Первой дистилляционной колонне 10 давали работать в традиционных условиях для разделения на два разных потока или фракции: головной поток или фракцию 15, отбираемую из верхней части 14 колонны 10 и содержащую в основном метанол, и нижний поток или фракцию 21, отбираемую из нижней части 12 колонны 10 и содержащую в основном другие более тяжелые соединения. Головной поток или фракцию 15 из верхней части 14 колонны 10 охлаждали в холодильнике для конденсации по меньшей мере частично головного потока или фракции 15. Часть головного потока или фракции 15 может быть подана в виде потока или фракции 17 в верхнюю часть 14 колонны 10. Поток 15, выходящий из верхней части 14 колонны 10, может быть очищен раздельно, например, последующей перегонкой, в которой используют одну или несколько дистилляционных колонн. Непрореагировавший метанол после очистки преимущественно рециркулируют в реагенты для реакции O-алкилирования. Часть нижнего потока или фракции 21, отбираемой из нижней части 12, может быть вновь подана частично в нижнюю часть колонны 10 в виде потока или фракции 23.
Таким образом, можно, как правило, получать нижний поток или фракцию 21, выходящую из нижней части 12 первой дистилляционной колонны 10 и содержащую:
катехол (PC): от 400 до 600 кг;
гваякол (GA): от 400 до 600 кг;
вератрол (VER): от 10 до 50 кг;
метил-6-гваякол (MEGA): от 5 до 50 кг;
продукты осмоления: от 5 до 100 кг.
Нижний поток 21, поступавший из нижней части 12 первой дистилляционной колонны 10 и имевший температуру приблизительно 115°C, подавали в центральную часть 28 второй дистилляционной колонны 20 и испаритель со стекающей жидкой пленкой.
Во второй дистилляционной колонне 20 получали головную фракцию 25, содержавшую в основном гваякол, и нижнюю фракцию 31, содержавшую в основном катехол, из поступавшего жидкого потока 21. Температура испарения в испарителе составляла в среднем 155°C, а температура в верхней части 24 колонны 20 составляла приблизительно 110°C. Часть нижнего потока или фракции 31, отбираемой из нижней части 22, может быть вновь подана в нижнюю часть колонны 20 в виде потока или фракции 33. Часть отбираемого головного потока 25 может быть введена в виде потока или фракции 27 в верхнюю часть 24 второй колонны 20.
Перегонку в колонне 20 осуществляли для отвода из верхней части 24 колонны максимального количества гваякола и из нижней части максимального количества катехола, вератрола и MEGA-6. Нижний жидкий поток 31, отобранный из нижней части 22 второй дистилляционной колонны 20, отводили со средним расходом около 650 кг/ч. Головной поток 25, отобранный из верхней части 24 второй дистилляционной колонны 20, отводили со средним расходом около 570 кг/ч.
Головной поток 25, отводимый из верхней части 24 колонны 20, предпочтительно содержит:
катехол (PC): около 0 кг;
гваякол (GA): от 400 до 600 кг;
вератрол (VER): от 0 до 5 кг;
метил-6-гваякол (MEGA): от 0 до 5 кг;
продукты осмоления: около 0 кг.
Нижний поток 31, отводимый из нижней части 22 колонны 20, предпочтительно содержит:
катехол (PC): от 400 до 600 кг;
гваякол (GA): от 0 до 50 кг;
вератрол (VER): от 5 до 50 кг;
метил-6-гваякол (MEGA): от 0 до 50 кг;
продукты осмоления: от 5 до 100 кг.
Нижний поток 31, поступавший из нижней части 22 второй дистилляционной колонны 20, подавали в центральную часть 38 третьей дистилляционной колонны 30 и испаритель со стекающей жидкой пленкой.
Перегонку в третьей колонне 30 осуществляли для отвода из верхней части 34 колонны головной фракции 35, содержавшей гваякол, вератрол, MEGA-6 и катехол, и для отвода из центральной части 38 бокового потока 36, содержавшего в основном катехол, и для отвода из нижней части 32 нижнего потока 41, содержавшего максимальное количество продуктов осмоления.
Головной поток 35, преобладающим образом содержавший катехол и имевший температуру около 140°C, отводили со средним расходом около 95 кг/ч из верхней части 34 третьей дистилляционной колонны 30.
Часть нижнего потока или фракции 41, отбираемой из нижней части 32 колонны 30, может быть вновь подана в нижнюю часть колонны 32 в виде потока или фракции 33. Часть головного потока или фракции 35, отбираемой из верхней части 34 колонны 30, может быть вновь подана в верхнюю часть колонны 34 в виде потока или фракции 37.
Нижний поток 41, преобладающим образом содержавший концентрированный катехол и имевший температуру около 210°C, отводили со средним расходом около 68 кг/ч из нижней части 32 третьей дистилляционной колонны 30.
Жидкий поток 36, преобладающим образом содержавший концентрированный катехол и имевший температуру около 155°C, отводили со средним расходом около 485 кг/ч из центральной части 38 третьей дистилляционной колонны 30.
Таким образом, боковой поток 36, отводимый из центральной части 38 колонны 30, преимущественно содержит:
катехол (PC): от около 400 до 600 кг;
гваякол (GA): от 0 до 20 кг;
вератрол (VER): от 0 до 50 кг;
метил-6-гваякол (MEGA): от 0 до 10 кг;
продукты осмоления: около 0 кг.
Таким образом, головной поток 35, отводимый из верхней части 34 колонны 30, предпочтительно содержит:
катехол (PC): от 0 до 100 кг;
гваякол (GA): от 0 до 50 кг;
вератрол (VER): от 0 до 50 кг;
метил-6-гваякол (MEGA): от 0 до 50 кг;
продукты осмоления: около 0 кг.
Таким образом, нижний поток 41, отводимый из нижней части 32 колонны 30, предпочтительно содержит:
катехол (PC): от 0 до 10 кг;
гваякол (GA): около 0 кг;
вератрол (VER): около 0 кг;
метил-6-гваякол (MEGA): около 0 кг;
продукты осмоления: от 5 до 100 кг.
Дистилляционные колонны 20 и 30 предпочтительно позволяют отделить, с одной стороны, головной поток 25, выходящий из верхней части второй колонны 20 в виде конденсируемого при необходимости пара и преобладающим образом содержащий гваякол, и боковой поток 36, выходящий из третьей дистилляционной колонны 30 в виде конденсируемого при необходимости пара и преобладающим образом содержащий катехол.
Таким образом, вератрол и примесь метил-6-гваякола удовлетворительным образом могут быть отделены от потока, содержащего гваякол, для его дальнейшего использования для получения ароматичных соединений, применяемых предпочтительно в пищевой промышленности, таких как ванилин. Вератрол и примесь метил-6-гваякола также могут быть отделены от потока, содержащего концентрированный катехол, так что может быть предусмотрено промышленное рециклирование катехола. Способ по настоящему изобретению позволяет рециклировать гваякол, а также катехол и, в частности, благодаря эффективности отделения дает значительное экономическое преимущество в промышленном масштабе как в отношении чистоты и выхода дистиллятов, так и рентабельности.
Пример 2
Головной поток или фракцию 35, отбираемую из третьей колонны 30 по примеру 1, можно подавать в четвертую дистилляционную колонну 40 (F2) соответственно фиг. 2.
В четвертой дистилляционной колонне 40 получали нижний поток или фракцию 51, содержавшую в основном катехол, и головной поток или фракцию 45, содержавшую в основном смесь гваякола, вератрола и MEGA, из поступавшего головного потока 35.
Перегонку в четвертой колонне 40 осуществляли для отвода из верхней части 44 колонны головной фракции 45, содержавшей гваякол, вератрол и преобладающим образом MEGA, и для отвода из нижней части 42 нижнего потока 51, содержавшего катехол, вератрол и в небольшом количестве MEGA.
Часть нижнего потока или фракции 51, отбираемой из нижней части 42 колонны 40, может быть вновь подана в нижнюю часть колонны 42 в виде потока или фракции 43. Часть головного потока или фракции 45, отбираемой из верхней части 44 колонны 40, может быть вновь подана в верхнюю часть колонны 44 в виде потока или фракции 47 (не показано).
Головной поток 45, имевший температуру около 115°C, отводили со средним расходом около 22 кг/ч из верхней части 24 второй дистилляционной колонны 20.
Нижний поток 51, имевший температуру около 145°C, отводили со средним расходом около 69 кг/ч из верхней части 44 четвертой дистилляционной колонны 40.
Таким образом, нижний поток 51, отводимый из нижней части 42 колонны 40, предпочтительно содержит:
катехол (PC): от 40 до 100 кг;
гваякол (GA): от 0 до 5 кг;
вератрол (VER): от 0 до 10 кг;
метил-6-гваякол (MEGA): от 0 до 5 кг;
продукты осмоления: около 0 кг.
Поток 51 предпочтительно может быть прибавлен частично или полностью к боковому потоку 36 колонны 30 перед рециклированием до осуществления реакции O-алкилирования.
Головной поток 45, отводимый из верхней части 44 колонны 40, предпочтительно содержит:
катехол (PC): около 0 кг;
гваякол (GA): от 0 до 20 кг;
вератрол (VER): от 0 до 10 кг;
метил-6-гваякол (MEGA): от 5 до 50 кг;
продукты осмоления: около 0 кг.
Пример 3
Поток или фракцию (25), содержащую гваякол, можно использовать для получения ванилина высокой чистоты в отношении примеси MEGA. Для синтеза ванилина из гваякола можно осуществлять стадии конденсации и окисления. Эти стадии пояснены далее в рамках синтеза гваякола, полученного в трехфазной среде (G/L/L), но могут быть непосредственно адаптированы для получения ванилина из гваякола, полученного в паровой фазе.
Конденсация
В первый реактор из нержавеющей стали 316L вместимостью 5 л, снабженный рубашкой, устройством механического перемешивания, электродом pH, системой охлаждения и подачи инертного газа в непрерывном режиме подают:
- 64,8 кг/ч деминерализованной воды;
- 9,3 кг/ч (116 моль/ч) водного раствора гидроксида натрия с концентрацией 50% масс.;
- 10,1 кг/ч (81,5 моль/ч) гваякола (свежего и рециклированного, отделенного полностью или частично от вератрола).
Эту реакционную смесь выдерживают при температуре 35°C. Затем эту предварительную смесь подают в первый реактор системы из 3 расположенных каскадом реакторов с водным раствором глиоксиловой кислоты с концентрацией 50% масс. (6,04 кг/ч или 40,8 моль/ч).
3 реактора идеального смешения выполнены из нержавеющей стали 316L и имеют вместимость по 80 л каждый; при этом рабочая температура составляет 35°C.
Общее время пребывания составляет 2 часа.
На выходе из последнего реактора отбирают пробу реакционной смеси, и соединения, содержащиеся в смеси, количественно анализируют жидкофазной хроматографией.
Были получены следующие результаты:
- конверсия гваякола (GA): TT = 47%;
- 4-гидрокси-3-метоксиминдальная кислота (APM):
RT (APM)/GA = 86%;
- 2-гидрокси-3-метоксиминдальная кислота:
RT (AOM)/GA = 5%;
- 2-гидрокси-3-метокси-1,5-диминдальная кислота:
RT (ADM/GA) = 9%.
Реакционную смесь направляют в контур рецикла 1 для отделения избытка гваякола от производных соединений миндальной кислоты.
Окисление
В реактор окисления из нержавеющей стали 316L, оснащенный самовсасывающим перемешивающим устройством кавитационного типа ("кавитатор") или типа Раштона и рубашкой, обеспечивающей эффективное охлаждение, в непрерывном режиме подают:
- смесь сокатализаторов и водного раствора производных соединений миндальной кислоты, поступающего из контура рецикла 1, в том числе:
77 кг/ч реакционной смеси, которая образуется при реакции конденсации и из которой удаляют избыток гваякола (и затем рециклируют) в контуре рецикла 1. Эту смесь образуют приблизительно 7,2 кг/ч 4-гидрокси-3-метоксиминдальной кислоты, 0,5 кг/ч 2-гидрокси-3-метоксиминдальной кислоты и 0,9 кг/ч 2-гидрокси-3-метокси-1,5-диминдальной кислоты;
100 г/ч водного раствора CoCl2 и CuSO4 в количестве, выраженном в молярных процентах для каждого из металлов по отношению к сумме в молях производных соединений миндальных кислот и равном 0,04%;
- надлежащее количество водного раствора гидроксида натрия с концентрацией 50% масс., соответствующее по меньшей мере количеству, требуемому по стехиометрическим соотношениям реакции окисления (рассматриваемых в целом);
- количество кислорода при атмосферном давлении, достаточное для достижения почти полной конверсии производных соединений миндальных кислот. Окислитель может представлять собой кислород при атмосферном давлении или воздух под давлением.
Эту реакцию осуществляют при 80°C. На выходе из реактора отбирают пробу реакционной смеси, и соединения, содержащиеся в смеси, количественно анализируют жидкофазной хроматографией.
Были получены следующие результаты:
- конверсия 4-гидрокси-3-метоксиминдальной кислоты: TT > 99,5%;
- выход ванилина VA:
RT (VA)/APM = 98%.
Реакционную смесь направляют в контур экстракции ванилата натрия из водной среды (контур рецикла 2).
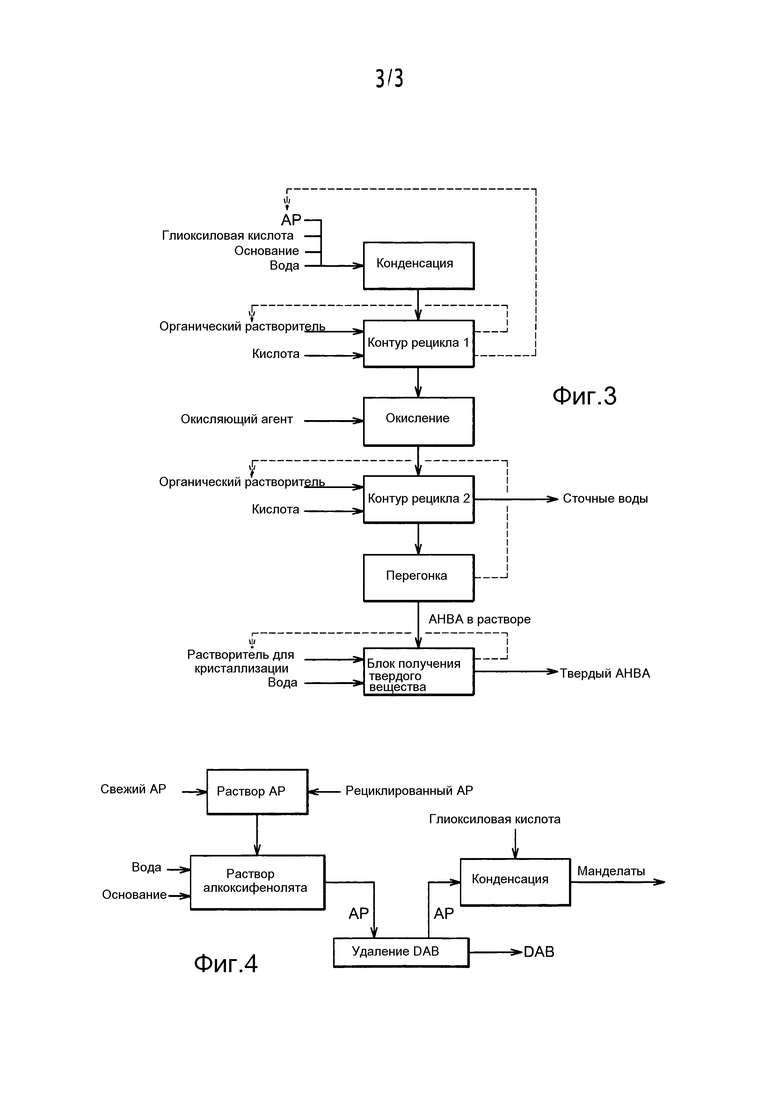
Настоящее изобретение относится к способу получения по меньшей мере одного алкоксигидроксибензальдегида ("AHBA"), который может быть использован в качестве ароматизатора или отдушки, а также в качестве промежуточного соединения из по меньшей мере одного гидроксифенола ("HP"). Способ включает стадию образования по меньшей мере одного алкоксифенола ("AP") и алкилалкоксифенола ("AAP") и стадию отделения (S) AP от AAP, причем стадию отделения (S) осуществляют перед получением AHBA, соединение AHBA получают присоединением альдегидной группы к соединению AP конденсацией с глиоксиловой кислотой с последующим окислением образующегося соединения, а стадию отделения (S) осуществляют перед окислением с образованием соединения AHBA. Предлагаемый способ позволяет получить АНВА с высокой чистотой и хорошим выходом при высокой конверсии НР. Также предлагаемое изобретение относится к способу отделения гваякола (GA) и/или гветола (GE) от смеси, содержащей GA и/или GE, и к способу синтеза в паровой фазе по меньшей мере одного АР. 3 н. и 14 з.п. ф-лы, 4 ил., 3 пр.
1. Способ получения по меньшей мере одного алкоксигидроксибензальдегида ("AHBA") из по меньшей мере одного гидроксифенола ("HP"), отличающийся тем, что он включает стадию образования по меньшей мере одного алкоксифенола ("AP") и алкилалкоксифенола ("AAP") и стадию отделения (S) AP от AAP, причем стадию отделения (S) осуществляют перед получением AHBA, и отличающийся тем, что соединение AHBA получают присоединением альдегидной группы к соединению AP конденсацией с глиоксиловой кислотой с последующим окислением образующегося соединения, а стадию отделения (S) осуществляют перед окислением с образованием соединения AHBA.
2. Способ получения по предыдущему пункту, отличающийся тем, что алкоксигидроксибензальдегид (AHBA) представляет собой ванилин (VA), этилванилин (EVA) или любую их смесь.
3. Способ получения по п. 2, отличающийся тем, что он включает получение VA и/или EVA, в существенной степени свободного от 5-метилванилина ("MEVA") и 3-этокси-5-этил-4-гидроксибензальдегида ("EEVA"), из гваякола (GA) и/или гветола (GE), причем способ включает получение смеси (M), содержащей GA и/или GE, в присутствии примеси 6-метилгваякола ("MEGA") и/или 6-этилгветола (2-этокси-6-этилфенола, сокращенно обозначаемого "EGE") и стадию отделения (S) GA и/или GE от MEGA и/или EGE, причем стадию отделения (S) осуществляют перед получением VA и/или EVA, причем количество MEVA и EEVA меньше 1000 млн-1 по отношению к количеству образовавшегося VA и/или EVA.
4. Способ по любому из предыдущих пунктов, отличающийся тем, что стадию отделения осуществляют перед взаимодействием AP с глиоксиловой кислотой.
5. Способ по любому из пп. 1-3, отличающийся тем, что стадию отделения реализуют, осуществляя одну или несколько стадий перегонки.
6. Способ по любому из пп. 1-3, отличающийся тем, что он включает подачу потока или фракции (E1) смеси M, содержащей гваякол (GA) и/или гветол (GE), обозначенные аббревиатурой "G", и 6-метилгваякол (MEGA) и/или 6-этилгветол (EGE), обозначенные аббревиатурой "AG", в присутствии пирокатехина (PC) в дистилляционную колонну (E) и отбор G в основном из верхней части дистилляционной колонны в виде потока или фракции (E2) и PC и AG в основном из нижней части дистилляционной колонны в виде потока или фракции (E3).
7. Способ по п. 6, отличающийся тем, что смесь M содержит также диалкоксибензол (DAB), причем DAB предпочтительно представляет собой вератрол (VER), а DAB отбирают в основном из нижней части дистилляционной колонны в виде потока или фракции (E3).
8. Способ по п. 6, отличающийся тем, что PC содержится в количестве от 25 до 75%, предпочтительно от 35 до 65% и более предпочтительно от 40 до 45% от общей массы потока или фракции (E1).
9. Способ по п. 6, отличающийся тем, что G присутствует в количестве от 25 до 75%, предпочтительно от 35 до 65% и более предпочтительно от 45 до 50% от общей массы потока или фракции (E1).
10. Способ по п. 6, отличающийся тем, что G содержится в количестве более 90 мас.%, предпочтительно больше 95 мас.% и более предпочтительно более 99 мас.% от общей массы потока или фракции (E2), а AG предпочтительно содержится в количестве менее 1 мас.%, предпочтительно менее 0,5 мас.% и более предпочтительно менее 0,2 мас.%, по отношению к общей массе потока или фракции (E2).
11. Способ по п. 6, отличающийся тем, что поток или фракция (E2) содержит от 90 до 100%, предпочтительно от 95 до 100% и более предпочтительно от 99 до 100% G, от 0 до 1%, предпочтительно менее 0,5% и более предпочтительно менее 0,2% AG и от 0 до 1%, предпочтительно менее 0,5% и более предпочтительно менее 0,2% VER, причем процентные доли выражены по массе по отношению к общей массе потока или фракции (E2).
12. Способ по п. 6, отличающийся тем, что PC присутствует в количестве больше 60 мас.%, предпочтительно больше 70 мас.% и более предпочтительно больше 75 мас.% от общей массы потока или фракции (E3), а AG предпочтительно содержится в количестве менее 8 мас.%, предпочтительно менее 5 мас.% и более предпочтительно менее 3 мас.% по отношению к общей массе потока или фракции (E3).
13. Способ, по п. 6, отличающийся тем, что поток или фракция (E3) содержит от 60 до 100%, предпочтительно от 70 до 100% и более предпочтительно от 75 до 100% PC и от 0 до 8%, предпочтительно менее 5% и более предпочтительно менее 3% AG и от 0 до 10%, предпочтительно менее 8% и более предпочтительно менее 5% VER, причем процентные доли выражены по массе по отношению к общей массе потока или фракции (E3).
14. Способ по п. 6, отличающийся тем, что он включает стадию дополнительной перегонки в колонне (F) потока или фракции (E3), поступающей из нижней части дистилляционной колонны (E) с осуществлением бокового отвода потока или фракции (F6) из колонны (F) для отведения потока или фракции, обогащенной PC.
15. Способ отделения GA и/или GE от смеси (M), содержащей GA и/или GE и примесь 6-метилгваякола ("MEGA") и/или 6-этилгветола (2-этокси-6-этилфенола, сокращенно обозначаемого "EGE"), причем способ отличается тем, что смесь M подают в дистилляционную колонну в присутствии пирокатехина (PC) и отбирают поток или фракцию, содержащую GA и/или GE, из верхней части дистилляционной колонны и поток или фракцию, содержащую PC, MEGA и/или EGE, из нижней части дистилляционной колонны.
16. Способ синтеза в паровой фазе по меньшей мере одного алкоксифенола, обозначаемого "AP", отличающийся тем, что он включает образование по меньшей мере одного AP по реакции, называемой O-алкилированием, в паровой фазе между по меньшей мере одним гидроксифенолом, обозначаемым "HP", и по меньшей мере одним O-алкилирующим агентом, причем при указанной реакции образуется также по меньшей мере одна примесь типа алкилалкоксифенола, обозначаемая "AAP", причем при указанной реакции образуется смесь M, содержащая AP, AAP и HP, причем способ включает подачу смеси M по меньшей мере в одну дистилляционную колонну и отбор потока или фракции, содержащей AP, из верхней части дистилляционной колонны и потока или фракции, содержащей HP и AAP, из нижней части дистилляционной колонны, причем способ включает при необходимости рециклирование потока или фракции, содержащей AP, после осуществления реакции O-алкилирования и/или рециклирование потока или фракции, содержащей HP, до осуществления реакции O-алкилирования.
17. Способ по п. 16, отличающийся тем, что он включает образование по меньшей мере гваякола (GA) и/или гветола (GE) по реакции, называемой O-алкилированием, в паровой фазе между пирокатехином, обозначаемым "PC", и по меньшей мере одним O-алкилирующим агентом, причем при указанной реакции образуется также по меньшей мере одна примесь 6-метилгваякола ("MEGA") и/или 6-этилгветола (2-этокси-6-этилфенола, сокращенно обозначаемого "EGE"), причем при указанной реакции образуется смесь M, содержащая GA и/или GE, MEGA и/или EGE и PC, причем способ включает подачу смеси M по меньшей мере в одну дистилляционную колонну и отбор потока или фракции, содержащей GA и/или GE, из верхней части дистилляционной колонны и потока или фракции, содержащей PC и MEGA и/или EGE, из нижней части дистилляционной колонны, причем способ включает при необходимости рециклирование потока или фракции, содержащей GA и/или GE, после осуществления реакции O-алкилирования и/или рециклирование потока или фракции, содержащей PC, до осуществления реакции O-алкилирования.
| CN 101492353 А, 29.07.2009 | |||
| Устройство с гидроосциллятором для тушения пожара и пожаровзрывопредотвращения пеной низкой и средней кратности | 2017 |
|
RU2664266C1 |
| H.-R | |||
| Bjorsvik et al., High Selectivity in the Oxidation of Mandelic Acid Derivatives and in O-Methylation of Protocatechualdehyde: New Processes for Synthesis of Vanillin, iso-Vanillin, and Heliotropin | |||
| Organic Process Research & Development, 2000, 4(6), 534-543 | |||
| Способ получения ванилина | 1938 |
|
SU55779A1 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТИЛОВОГО И ЭТИЛОВОГО МОНОЭФИРОВ ПИРОКАТЕХИНА | 1983 |
|
SU1181260A1 |

