Изобретение относится к области радиохимической технологии и может быть использовано в головных операциях процесса переработки облученного ядерного топлива (ОЯТ) при отделении трития.
Тритий является биологически опасным компонентом ОЯТ, представляет собой β-активный радионуклид с излучением в мягкой области спектра. Отделение трития в процессе радиохимической переработки ОЯТ является необходимым с точки зрения экологической безопасности и предполагает его полную изоляцию от биосферы.
В существующих технологических схемах радиохимического производства при проведении кислотного растворения ОЯТ тритий переходит в раствор и распределяется по всем водным продуктам технологической схемы, существенно затрудняя тем самым процесс его утилизации. В этой связи выделение и локализацию трития целесообразно проводить его целенаправленным количественным удалением из поступающего в переработку материала с использование специальных операций перед кислотным растворением ОЯТ. С этой целью ОЯТ подвергают термохимической обработке в окислительной среде (волоксидации), обеспечивающей изменение кристаллической структуры материала, в результате которого происходит отделение из твердой фазы легколетучих компонентов. Содержащиеся в ОЯТ тритий (полностью) и йод (в значимых количествах) удаляются в токе рабочего газа под воздействием температурного фактора. Количественное отделение трития является основным критерием эффективности процесса волоксидации. Окислительная обработка ОЯТ направлена на максимально полное изменение кристаллической структуры исходного материала и сопровождается значительным увеличением объема твердофазного продукта.
Кроме физико-химических факторов на эффективность волоксидации влияет полнота удаления топлива из оболочек. Использование известных способов отделения оболочек, таких как оплавление, удаление с использованием фтор-иона, охрупчивание в результате азотирования и гидрирования, либо существенно осложняет процесс, либо приводит к структурным изменениям материала, затрудняющим последующее кислотное растворение и экстракционную переработку ОЯТ. По этой причине целесообразным оказывается не удаление оболочки, а извлечение топлива из оболочки после операции рубки отработавших тепловыделяющих сборок (ОТВС). Процесс сводится к разделке, фрагментированию, механической деформации фрагментов, извлечению разрушенных таблеток из оболочки, сухой сепарации и последующей волоксидации отделенного от оболочки ОЯТ. Использование указанного способа затрудняется организацией устройств механического воздействия в условиях дистанционно-обслуживаемой зоны, приводит к увеличению количества высокоактивных отходов при утилизации используемых агрегатов в результате выработки их ресурса. Наиболее перспективным является извлечение ОЯТ из оболочки непосредственно в результате окислительного процесса.
Из существующего уровня техники известен способ безводной переработки ОЯТ легководных реакторов, так называемый AIROX-процесс [Recycling of Nuclear Spent Fuel with AIROX Processing, DOE/ID-10423, 1992], включающий извлечение ТВЭЛ из ТВС, фрагментацию ТВЭЛ, перфорацию оболочек фрагментов ТВЭЛ через 2,5 см, циклическую термохимическую обработку фрагментов в среде кислорода при температуре 400°С и водорода в присутствии аргона при температуре 600°С. В указанном способе достигается полнота отделения образующегося порошка от оболочек в течение 2 часов, количественное удаление газообразных продуктов деления, в том числе трития, при превращении диоксида урана в закись-окись урана в результате окислительного процесса, а затем в диоксид урана в результате восстановительного процесса. Образование и отделение порошкообразного продукта происходит в результате разрыва оболочки на стадии окисления в атмосфере кислорода и аргона. Разрыв оболочки ТВЭЛ достигается в результате увеличения на 30% объема твердофазного продукта внутри фрагмента ТВЭЛ, что дополнительно увеличивает площадь реакционной поверхности и ускоряет окислительный процесс. Недостатками способа являются: необходимость перфорации оболочек, сложность организации процесса, невозможность разрыва перфорированной циркониевой оболочки ТВЭЛ на стадии окисления по причине пластичности материала оболочки, необходимость многократного проведения обработки с целью охрупчивания оболочки в результате гидрирования на стадии восстановления. К основным проблемам при реализации указанного способа относится необходимость использования водорода для обеспечения отделения твердофазного продукта (ОЯТ) из оболочки, поскольку извлечение порошкообразного продукта из фрагментов ТВЭЛ происходит в результате изменения объема твердофазного продукта в ходе циклических операций окисления и восстановления.
Известен способ переработки облученного ядерного топлива [Патент RU 2 459 299 C1, G21F 9/30, опубл. 20.08.2012], включающий термическую обработку фрагментов ОЯТ в окислительной атмосфере в две стадии: первую проводят при температуре 400-650°С в воздушной среде, дополнительно содержащей углекислый газ в количестве 1-4 об. % в течение 60-360 мин, вторую проводят при температуре 350-450°С в воздушной или обогащенной по кислороду среде, содержащей водяной пар в количестве точки росы парогазовой смеси при температуре 30-40°С в течение 30-120 мин, при этом обе стадии проводятся при постоянной или периодической механоактивации реакционной массы. Недостатками способа являются: высокая температура окислительного процесса, запирание торцов фрагментов ТВЭЛ в результате уплотнения и спекания образующегося твердофазного продукта, необходимость высокой интенсивности механического воздействия на фрагменты ТВЭЛ, истирание окисленного слоя оболочек в результате вибрационного воздействия при механоактивации, приводящее к образованию тонкодисперсных порошков, генерирующих межфазные взвеси на экстракционном переделе радиохимической переработки ОЯТ. К основным проблемам при реализации указанного способа относится необходимость получения мелких фрагментов ТВЭЛ и снижение эффективности волоксидации при увеличении степени замятия торцов фрагментов ТВЭЛ. Известно, что использование рубки ОТВС устройствами гильотинного типа, как наиболее продуктивного способа фрагментации, неизбежно приводит к деформации сечения среза оболочек и увеличению степени замятия до 30% и более, тем самым, увеличивая тенденцию к запиранию фрагментов ТВЭЛ при волоксидации указанным способом. При этом происходит вздутие оболочки фрагмента в области торцов в результате уплотнения твердофазного продукта и прекращение поступления реагентов к находящемуся внутри фрагмента материалу. Твердофазный продукт при этом не извлекается из оболочки, окислительная трансформация ОЯТ не достигает требуемого уровня, а тритий количественно не удаляется из ОЯТ.
Снижение температуры процесса и использование дополнительного окислителя в системе позволяет минимизировать негативные воздействия температурного фактора, вызывающего спекание получаемого твердофазного продукта. Наиболее близким к заявленному способу является безводный способ проведения головных операций переработки ОЯТ легководных реакторов [Патент US 8574523 В2, опубл. 05.11.2013] (принятый за прототип), включающий процесс волоксидации в двухзонном аппарате, при котором ОЯТ в первой зоне при температуре 200-450°С подвергается воздействию используемого в качестве газообразного окислителя диоксида азота, с образованием твердофазного продукта, а во второй зоне при температуре 0-80°С происходит регенерация диоксида азота в результате доокисления кислородом образующегося монооксида азота. Недостатками способа являются: сложность организации двустадийного процесса, увеличение длительности процесса при ограничении оболочкой ТВЭЛ реакционной поверхности таблетированного ОЯТ, снижение реакционной способности таблетированного ОЯТ по отношению к окислительному воздействию диоксида азота в результате образования на поверхности прочнофиксированного плотного твердофазного продукта. В способе-прототипе образующийся на поверхности кристаллитов исходного материала твердофазный продукт представляет собой исключительно триоксид урана, что в большей мере и определяет недостатки способа. Процесс в начальной стадии протекает в кинетической области. По мере накопления продукта реакции на реакционной поверхности процесс замедляется и переходит в диффузионную область. Замедление процесса и полное прекращение возникает в результате ограничения поступления к реакционной поверхности газообразного окислителя. Протекание процесса в условиях ограничения (оболочкой ТВЭЛ) реакционного объема приводит к уплотнению твердофазного продукта и деформации фрагментов, что выражается во вздутии оболочки и полном запирании твердофазным продуктом сечения фрагмента. Происходящая деформация оболочки в результате локального увеличения объема возникает по причине пластичности циркония, что позволяет образующемуся продукту не перемещаться во фрагменте в направлении среза, а двигаться, расширяясь, перпендикулярно оси фрагмента, вызывая вздутие оболочки. По этой причине способ-прототип может быть реализован только при интенсивном механическом воздействии на фрагменты ТВЭЛ, необходимом для предотвращения локального уплотнения в оболочке образующегося в результате волоксидации ОЯТ триоксида урана. При этом увеличение степени замятия торцов фрагментов ТВЭЛ до 30% и более делает механоактивацию неэффективной.
Эффективность удаления трития в процессе волоксидации ОЯТ существенно зависит от того, находится ли поступающий на окислительную обработку материал в оболочке или предварительно освобожден от нее. В случае использования фрагментированного оболочечного ОЯТ количественное отделение трития может быть достигнуто только при полном отделении твердофазного продукта из оболочек фрагментов ТВЭЛ. Т.е. полнота удаления трития из оболочечного ОЯТ находится в пропорциональной зависимости от полноты отделения твердофазного продукта окисления ОЯТ из фрагментов ТВЭЛ. В этой связи предпосылкой изобретения является необходимость полного извлечения твердофазного продукта окисления ОЯТ из оболочки в процессе волоксидации.
Задачей изобретения является количественное удаление трития при проведении процесса волоксидации фрагментированного ОЯТ (в оболочке).
Задача решается извлечением твердофазного продукта окисления из фрагментированных ТВЭЛ в процессе волоксидации ОЯТ в результате увеличения реакционной способности окислительной системы, создания условий для получения в качестве твердофазного продукта закиси-окиси урана. Извлечение твердофазного продукта в процессе волоксидации ОЯТ обеспечивается поддержанием в парогазовой фазе заданного соотношения реагентов и каталитической активацией газового потока перед подачей в аппарат-реактор.
Техническим результатом является количественное отделение трития из ОЯТ, количественное извлечение ОЯТ из оболочек в результате окислительного процесса, сокращение длительности процесса, минимизация интенсивности механоактивации.
Технический результат позволяет проводить волоксидацию ОЯТ в оболочке из фрагментов ТВЭЛ длиной более 40 мм с замятием сечения среза более 30% с достижением степени отгонки трития не менее 99,1%.
Для достижения указанного технического результата в способе окислительной обработки (волоксидации) облученного ядерного топлива реакционную смесь получают путем смешения на каталитически активной насадке потока нитрующего компонента - диоксида азота в количестве 15-56% масс. и потока окислительного компонента - кислорода в количестве 41-83% масс., насыщенного парами воды в количестве 1,2-9,5% масс., обработку проводят при температуре 325-375°С при периодической механоактивации.
Сущность изобретения заключается в осуществлении окислительного процесса преимущественно в кинетической области в результате увеличения нитрующей и окислительной способности реакционной системы, обеспечивающей освобождение реакционной поверхности ОЯТ от образующегося твердофазного продукта окисления из-за разнонаправленной трансформации его объема при одновременном воздействии нитрующего и окислительного компонента парогазовой фазы.
В результате нитрующего воздействия (на ОЯТ) приоритетно происходит образование твердофазного азотнокислого продукта, представляющего собой в основе тринитратоуранилат нитрозония, с локальным увеличением объема твердой фазы на реакционной поверхности кристаллитов. Воздействие окислительного компонента парогазовой фазы приводит к окислению образующегося тринитратоуранилата нитрозония до закиси-окиси урана и общей деструкции (разложению) твердофазного азотнокислого продукта, сопровождающейся локальным сокращением объема. Происходящее в результате воздействия компонентов парогазовой фазы видоизменение твердофазного продукта вызывает разрыхление и ослабление фиксации образующейся твердой фазы на реакционной поверхности кристаллитов, приводит к расширению зоны доступа газообразных реагентов к реакционной поверхности.
Возникающее локальное увеличение объема создает избыточное давление в зоне реакции и приводит к беспрепятственному вытеснению образующегося твердофазного продукта из оболочки фрагмента. Рыхлая консистенция трансформированной твердой фазы обеспечивает ее подвижность при прохождении через срез оболочки фрагмента, вне зависимости от величины деформации, полученной в результате рубки. При этом внешнее механическое воздействие на фрагмент направлено на снижение механического напряжения в реакционном фронте, препятствует образованию статичных зон уплотнения и приводит к росту скорости извлечения твердофазного продукта из оболочки фрагмента. Требуемая интенсивность механоактивации находится в диапазоне 0,5-15 импульс./мин
Увеличение нитрующей активности реакционной системы достигается получением в парогазовой фазе азотной кислоты, синтез которой происходит при смешении потоков кислорода, насыщенного парами воды, и диоксида азота на каталитически активной насадке. В частном случае поток диоксида азота получают испарением его конденсированной формы из термостатируемой при температуре 22,2-26,4°С дозирующей емкости. Образование азотной кислоты в парогазовой фазе происходит при прохождении объединенным газовым потоком зоны зернистого слоя насадки при температуре 60-100°С.
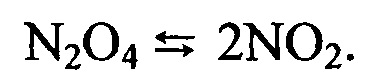

Действие каталитически активной насадки при этом направлено на увеличение полноты смешения движущихся через сечение зернистого слоя ламинарных потоков газообразных реагентов. Полнота диспергирования компонентов газового потока достигается при скорости прохождения 5-5000 колоночных объемов (к.о.) слоя насадки в минуту, обеспечивается размером зерна насадки 0,16-1,2 мм, соотношением диаметра слоя к высоте в диапазоне (1:3)÷(1:10). Использование в качестве носителя каталитически активной насадки оксида алюминия с площадью удельной поверхности 1,8-80 м2/г позволяет максимально сдвинуть протекающую реакцию получения азотной кислоты в сторону образования продукта, увеличить концентрацию азотной кислоты в парогазовой фазе до необходимого уровня активности реакционной системы.
Увеличение окислительной активности реакционной системы достигается за счет создания условий получения в парогазовой фазе безводной азотной кислоты. С этой целью соотношение потоков газообразных реагентов выбирается таким образом, чтобы обеспечить мольное соотношение между диоксидом азота и водой в интервале NO2:Н2O=(2:1)÷(10:1). Направленный синтез безводной азотной кислоты в парогазовой фазе позволяет получить наиболее реакционно-активные ионные формы.
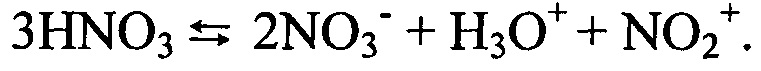
Интенсификация процесса ионизации азотной кислоты в парогазовой фазе осуществляется при прохождении объединенным газовым потоком, состоящим из диоксида азота, кислорода и образовавшейся безводной азотной кислоты, через нагретую до 290-360°С зону зернистого слоя насадки. В частном случае зернистый слой каталитически активной насадки имеет на входе объединенного газового потока температуру 60-100°С, на выходе из зернистого слоя в аппарат-реактор - 290-360°С.
Избыток диоксида азота по отношению к введенной воде позволяет получить газовый поток, состоящий из трех макрокомпонентов - диоксида азота, кислорода, диссоциированной азотной кислоты. Поддержание основного компонента (диоксида азота) на исходном уровне, составляющем 15-56% масс., достигается окислением газообразных продуктов кислородом, имеющем долю в газовом потоке 41-83% масс., непосредственно при температуре реакции основного процесса и не требует организации дополнительного аппаратного объема с пониженным температурным режимом. На количественную диссоциацию образованной в результате введения в парогазовую фазу 1,2-9,5% масс. воды влияет температурный фактор зернистого слоя насадки и природа каталитически активного компонента.
Зерно насадки содержит поверхностный тонкодисперсный металлизированный слой с размером кристаллитов 0,01-25 мкм, представляющий собой взаимно интегрированные структуры металла платиновой группы и неблагородного металла, обладающего собственной каталитической активностью. Количество металла платиновой группы составляет 10-100% от суммы компонентов в каталитически активном слое. При этом количество металла платиновой группы составляет 0,05-0,9% масс. от количества катализатора. В частном случае в качестве металла платиновой группы используется платина или рутений, или композиция платины и рутения. Каталитически активный слой может содержать окисленные формы используемого неблагородного металла в количестве 10-90% от общего содержания неблагородного металла. В частном случае в качестве неблагородного металла используется цирконий либо титан, либо композиция титана и циркония.
Каталитически активный компонент насадки оказывает воздействие, направленное на ионизацию компонентов парогазовой фазы, что позволяет увеличить количество окислительно-активных ионных пар, обладающих максимальной реакционной способностью, а также повысить долю в продукте диссоциации сверхокисленных кислородных соединений азота. В реакционной системе на основе диоксида азота в результате диссоциации самопроизвольно образуются в качестве промежуточных соединений радикалы типа 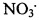 и оксиды типа NO3, N2O6. Обогащение реакционной системы окислительно-активными компонентами при прохождении газового потока через зернистый слой насадки является результатом каталитической активации парогазовой фазы в результате реализации редокс-цикла на каталитически активном компоненте.
и оксиды типа NO3, N2O6. Обогащение реакционной системы окислительно-активными компонентами при прохождении газового потока через зернистый слой насадки является результатом каталитической активации парогазовой фазы в результате реализации редокс-цикла на каталитически активном компоненте.
Наиболее вероятный механизм каталитического взаимодействия зернистого слоя насадки и диоксида азота заключается в окислении образующимся катионом нитрония каталитически активного компонента поверхности зерна насадки, образовании окисленного центра, окислении на поверхности зерна насадки адсорбированного диоксида азота до сверхокисленной формы, восстановлении окислительного центра поверхности кристаллита до исходной металлической формы.


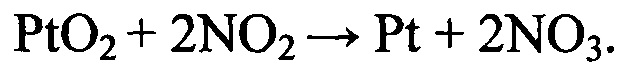
При этом имеет место процесс ассоциации образующегося иона нитрозония и нитрат-иона с образованием ионной пары, переходящей в состав диссоциирующей системы диоксида азота.
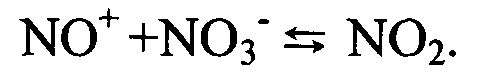
Происходящие на поверхности катализатора процессы приводят к перегруппировке десорбирующихся с поверхности зерна насадки ионных пар без снижения их окислительной активности. Восстановление окислительного центра сопровождается переходом каталитически активного компонента насадки в исходную форму и приводит к регенерации активных окисленных центров. Доступность каталитически активной поверхности для следующего акта позволяет реализовать гетерофазный редокс-цикл.
Отсутствие свободной воды в парогазовой фазе обеспечивает относительную устойчивость образующихся пероксидных соединений азота в газовом потоке реакционной смеси благодаря предотвращению их гидролитического разложения.
Каталитическая активация газового потока является отличительным признаком предлагаемого способа и заключается в количественном получении безводной азотной кислоты, инициировании процесса ее диссоциации с получением кислородных соединений азота, обладающих большей окислительной способностью, чем исходная парогазовая фаза.
После каталитической активации находящаяся при температуре 290-360°С парогазовая фаза со скоростью 2-100 аппаратн. об/ч поступает в аппарат-реактор с предварительно нагретыми до температуры 325-375°С фрагментами ТВЭЛ. Твердофазным реагентом при этом является таблетированное ОЯТ, имеющее в качестве основной фазы диоксид урана, с реакционно-доступной (свободной от оболочки) долей общей площади таблеток в диапазоне от 1 до 10%. Продукты деления находятся в ОЯТ в виде твердого раствора в основной фазе, либо образуют интегрированные в основную самостоятельные фазы отдельных соединений. В частном случае подвергаемый окислительному воздействию материал представляет собой нарубленные фрагменты ТВЭЛ реакторов типа ВВЭР-1000 на основе диоксида урана длинной 20-100 мм со степенью замятия сечения среза 0-100% с одной стороны и 0-50% с другой.
Наиболее вероятный механизм воздействия каталитически активированной парогазовой фазы на основной компонент таблетированного ОЯТ (диоксид урана) заключается в интенсификации окислительного процесса на реакционной поверхности, формировании окисленных центров, образовании азотнокислого продукта на окисленных центрах в результате нитрующего воздействия, окислительной деструкции азотнокислого продукта, приводящей к образованию окислительно-активных ионных форм в реакционном фронте.
Протекающая в поступающей в реактор парогазовой фазе реакция между диоксидом азота и диоксидом урана приводит к образованию тринитратоуранилата нитрозония.
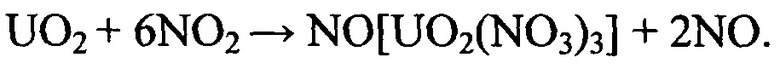
Количественное получение продукта реакции достигается при реализации процесса в две стадии: окисления UO2 до ε-UO3, образования из ε-UO3 тринитратоуранилата нитрозония. Реакционная способность поверхности кристаллитов таблетированного ОЯТ определяется первой стадией процесса. Изменение валентного состояния урана предшествует образованию в волоксидируемом материале его нитратных форм и является лимитирующей фазой преобразования, обусловленной компактностью кристаллической структуры и незначительной площадью поверхности. Однако без дополнительного окислительного воздействия доступная для взаимодействия поверхность диоксида урана является в используемом диапазоне температур реакционно-инертной. Увеличение способности диссоциирующей системы на основе диоксида азота к окислительному воздействию на реакционную поверхность волоксидируемого материала приводит к ускорению лимитирующей стадии процесса за счет каталитической активации поступающего в аппарат-реактор потока парогазовой фазы при увеличении содержания диссоциированных компонентов и сверхокисленных кислородных соединений азота.
Каталитически активированная парогазовая фаза при воздействии на поверхность таблетированного ОЯТ инициирует локальное окисление узлов деформации кристаллитных структур. В результате взаимодействия диоксида урана с окислительно-активными компонентами поступающего в реактор газового потока происходит формирование на реакционной поверхности окисленных центров.
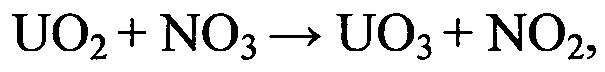
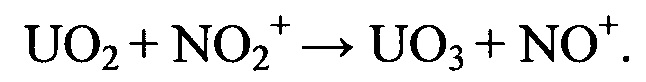
Получение триоксида урана в структуре кристаллитов таблетированного ОЯТ приводит к разрушению кристаллической структуры поверхностного слоя и формированию локальных реакционно-активных зон на основе ε-UO3, которые легко подвергаются нитрующему воздействию диоксида азота.
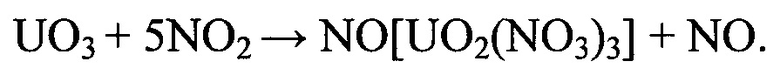
Образующийся на поверхности кристаллитов в локальных реакционно-активных зонах в используемом диапазоне температур 325-375°С тринитратоуранилат нитрозония является промежуточным соединением и подвергается окислению кислородом с образованием закиси-окиси урана.

Протекающий процесс окисления тринитратоуранилата нитрозония при наличии в парогазовой фазе диссоциированной азотной кислоты преобладает над процессом его термического разложения, приводящего к образованию триоксида урана.
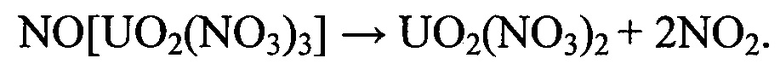
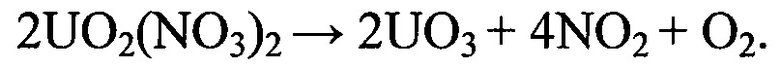
Присутствующий в реакционном фронте катион оксония (в составе ионной пары диссоцированной азотной кислоты) оказывает каталитическое воздействие на процесс трансформации тринитратоуранилата нитрозония по окислительному механизму и, тем самым, обеспечивает преобладание в качестве продуктов деструкции в локальных реакционно-активных зонах закиси-окиси урана и окислительно-активных ионных пар.
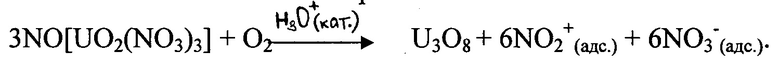
В результате окислительного воздействия кислорода на тринитратоуранилат нитрозония в зоне реакции в непосредственном контакте с непрореагировавшим материалом образуются продукты, содержащие реакционно-активные ионные пары. Окислительное воздействие образующегося в локальных реакционно-активных зонах катиона нитрония распространяется на соседние кристаллические структуры таблетированного ОЯТ и способствует лавинообразной трансформации всей реакционной поверхности в результате интенсификации нитрующего воздействия диоксида азота.

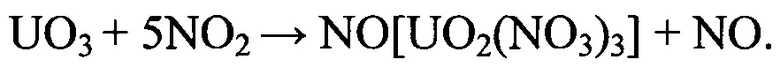
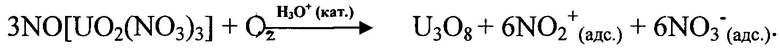
Образование реакционно-активных продуктов в результате окисления кислородом промежуточного соединения (тринитратоуранилата нитрозония) обуславливает возможность поддержания процесса в кинетической области. При этом концентрация кислорода в зоне реакции определяет скорость движения реакционного фронта внутри оболочки ТВЭЛ в условиях минимизации механического воздействия на фрагментированное ОЯТ. В отличие от способа-прототипа вводимое в парогазовую фазу количество кислорода определяется избытком, необходимым для обеспечения окисления образующегося промежуточного азотнокислого продукта в зоне реакции при температуре процесса 325-375°С. Избыток кислорода, необходимый для образования в качестве основного продукта процесса волоксидации закиси-окиси урана, обеспечивается поддержанием концентрации кислорода в поступающей в аппарат-реактор парогазовой фазе на уровне 41-83% масс. при мольном соотношении между диоксидом азота и кислородом в диапазоне от 1:7 до 1:1.
Наличие в парогазовой фазе диссоциированной безводной азотной кислоты в количестве 4,1-56,5% масс. в присутствии более 24,1% кислорода также является условием для получения в качестве основного продукта процесса волоксидации (ОЯТ) закиси-окиси урана.
Приведенные концентрационные профили парогазовой фазы по содержанию в ней безводной азотной кислоты и кислорода при длительности процесса волоксидации в интервале 0,2-4,5 ч при температуре 325-375°С позволяют исключить количественное нитрование или окисление до триоксида урана извлеченного в процессе волоксидации из фрагментов ОЯТ твердофазного продукта. Получение твердофазного продукта окисления в диапазоне температуры 325-375°С, имеющего в основе закись-окись урана, является отличительным признаком предлагаемого способа. Образующийся твердофазный продукт представляет собой непрерывный ряд твердых растворов и самостоятельных фаз продуктов деления в закиси-окиси урана в интервале суммарного состава UO2,67-2,78.
Отделение трития из ОЯТ происходит в процессе трансформации объема твердофазного продукта при движении реакционного фронта внутри оболочки ТВЭЛ, а затем из уже отделенного от оболочек продукта непосредственно в аппарате-реакторе при прохождении потока газовой фазы. Удаление трития из аппарата-реактора предполагает его транспортировку в виде паров тритийсодержащей азотной кислоты совместно с отходящим потоком газовой фазы. Отделение йода из ОЯТ происходит в результате окисления его соединений с цезием до молекулярного йода с последующим удалением из аппарата-реактора с отходящим потоком газовой фазы.
Осколочные элементы, обуславливающие более 90% γ-активности, в том числе цезий и редкоземельные элементы (РЗЭ), присутствующие в ОЯТ большей частью в виде твердого раствора в основной фазе материала, в процессе волоксидации подвергаются окислительно-нитрующему воздействию среды совместно с основной фазой. Это воздействие приводит к образованию в качестве промежуточных продуктов тринитратоуранилат производных цезия и РЗЭ, разлагающихся при последующей окислительной деструкции до нитратов под воздействием содержащегося в парогазовой фазе кислорода. Получение нитратных форм осколочных элементов в матрице оксидной формы основной фазы обеспечивается концентрационными кондициями компонентов каталитически активированной парогазовой фазы, а также температурным режимом в диапазоне 325-375°С, что является отличительным признаком предлагаемого способа.
Предлагаемый способ позволяет получать продукт, в котором целевые компоненты находятся в нерастворимой форме, а часть продуктов деления, в т.ч. цезий и РЗЭ в растворимой. Этот отличительный признак предлагаемого способа позволяет проводить последующее за волоксидацией отделение части осколочных элементов при помощи перколяции твердофазного продукта водными растворами от слабокислой до щелочной реакции среды. Перколяция обеспечивает отделение части растворимых в водных растворах γ-активных компонентов ОЯТ от нерастворимого целевого продукта. В частном случае перколяцию твердофазного продукта окисления проводят водой при pН среды 5-7,8, либо водным раствором пероксида водорода с концентрацией 0,001-0,1% масс. В частном случае перколяцию твердофазного продукта окисления проводят раствором гидроксида щелочного металла с концентрацией 0,001-0,01 моль/л. В частном случае перколяцию твердофазного продукта окисления проводят раствором гидроксида аммония с концентрацией 0,001-0,01 моль/л. Перколяция растворами приведенных составов может проводиться последовательно. Так, в частном случае, перколяцию щелочным раствором проводят после перколяции раствором пероксида водорода.
Эффективность процесса перколяции определяется свойствами твердофазного продукта окисления. Предлагаемый способ позволяет получать порошкообразный твердофазный продукт с разрыхленной структурой зерна, выщелачивание из которого растворимых продуктов происходит со значительной скоростью. Использование для перколяции раствора пероксида водорода, растворов гидроксида щелочного металла или гидроксида аммония обеспечивает получение наиболее растворимых в водных растворах форм осколочных элементов ОЯТ.
Вне зависимости от используемых для перколяции реагентов, максимальное влияние на полноту извлечения растворимых компонентов из волоксидированного ОЯТ имеет оказываемое на частицы твердофазного продукта воздействие, обеспечивающее смачивание зерен и проникновение раствора в поры зерна, а также вызывающее разрушение агрегатных структур. Для интенсификации процесса перколяции используют диспергирование суспензии посредством ультразвука. При этом соотношение Т:Ж находится в диапазоне 1:4-1:10. Процесс перколяции проводят при температуре среды 20-80°С. После отделения перколяционного и промывного растворов влажный твердофазный продукт подвергают кислотному растворению известными способами.
В результате обработки порошкообразного твердофазного продукта, полученного предлагаемым способом, в раствор извлекается более 60% γ-активных компонентов ОЯТ, что позволяет проводить предварительную очистку перерабатываемого материала в головных операциях до кислотного растворения ОЯТ, снизить радиационное воздействие на экстрагент, и, в конечном итоге, принимать в переработку ОЯТ с большей глубиной выгорания.
Минимизация интенсивности механического воздействия в процессе волоксидации позволяет сократить (либо полностью исключить) поступление в твердофазный продукт окисления тонкодисперсной фракции окисленного материала оболочек.
Предлагаемый способ позволяет проводить количественное извлечение трития при сокращении длительности процесса до 20-150 минут, обеспечивая тем самым наиболее производительный режим работы оборудования, а также обеспечивает возможность реализации непрерывного процесса в горизонтальном аппарате ротационного типа со шнековым перемещением материала в условиях ограничения габаритных размеров оборудования в дистанционно обслуживаемой зоне.
Предлагаемый способ позволяет проводить количественное отделение трития при проведении волоксидации фрагментов ТВЭЛ до 100 мм с замятием сечения среза более 30%, что предоставляет возможность фрагментации ОЯТ с использованием более производительных устройств с высокой степенью технологической апробации.
Предлагаемый способ имеет более простую организацию процесса, в отличие от способа-прототипа, не требует обеспечения в общем объеме аппарата двух существенно отличающихся температурных зон.
Предлагаемый способ позволяет отделять твердофазный продукт от оболочки непосредственно в процессе волоксидации в сравнении со способом-прототипом, в котором полное отделение твердофазного продукта от оболочки происходит после извлечения его из реактора (волоксидатора), что позволяет обеспечить стабильное получение полноты отделения трития из ОЯТ и не требует операции сухой сепарации после проведения окислительного процесса.
В предлагаемом способе полнота удаления трития обеспечивается формой твердофазного продукта, имеющего в основе закись-окись урана, в кристаллической структуре которого отсутствует связанная вода, в сравнении со способом-прототипом, где конечный продукт окисления представляет собой триоксид урана, имеющий тенденцию к образованию твердофазных гидратных форм.
Пример 1.
В обогреваемый аппарат-реактор (волоксидатор), имеющий горизонтальную ось вращения внутреннего контейнера, помещали фрагментированное ОЯТ ВВЭР-1000 с глубиной выгорания 53 ГВт⋅сут/т урана после 11-летней выдержки. Фрагменты ОЯТ были получены путем рубки устройством гильотинного типа длиной 40-46 мм с замятием сечения среза до 30% с обоих торцов фрагмента. Загруженный в волоксидатор материал нагревали со скоростью 2°С/мин По достижении температуры 310°С в аппарат-реактор через зернистый слой каталитически активной насадки подавали насыщенный парами воды кислород со скоростью 278 ммоль/ч. Насыщение кислорода парами воды проводили путем пропускания диспергированного потока кислорода через вертикальный слой нагретой до заданной температуры воды. Скорость подачи воды в потоке кислорода при этом составляла 63 ммоль/ч. При достижении температуры 350°С приступали к получению газовой смеси на зернистом слое каталитически активной насадки путем внесения в поток насыщенного парами воды кислорода отдельного потока диоксида азота. Заданную скорость подачи диоксида азота 260 ммоль/ч поддерживали путем испарения из дозирующей емкости при температуре 22,9°С его ассоциированной формы.
Массовая доля реагентов в поступающем на каталитическую активацию потоке газовой фазы составила: 5,17% для воды, 53,38% для диоксида азота, 41,45% для кислорода. Мольное соотношение между диоксидом азота и водой в парогазовой фазе, поступающими для смешения на каталитически активной насадке, составило 4,1:1. Мольное соотношение между диоксидом азота и кислородом в парогазовой фазе, поступающими для смешения на каталитически активной насадке, составило 1:1,1. Каталитически активная насадка представляла собой цилиндрический столб (с соотношением диаметра к высоте 1:5) гранулированного оксида алюминия с размером зерна 0,5-0,6 мм, покрытого каталитически активным слоем с содержанием платины 0,1% масс. Температура зернистого слоя насадки на входе составляла 60,9°С, на выходе из насадки - 290°С.
Поток каталитически активированной газовой фазы поступал непосредственно во внутренний объем аппарата со скоростью 6,9 аппаратн.об/ч. С момента подачи газовой смеси в слой каталитически активной насадки производили вращение аппарата со скоростью 9 об/мин, обеспечивая падение фрагментов внутри контейнера.
Через 25 минут с момента подачи каталитически активированной парогазовой фазы без предварительного охлаждения произвели выгрузку содержимого аппарата-реактора (волоксидатора). Твердофазный продукт окисления представлял собой полностью отделенный от оболочек тонкодисперсный не пылящий порошок черного цвета. Оболочки ТВЭЛ не имели признаков вздутия или иной деформации. Признаки формирования окисленного слоя на поверхности металла циркониевого сплава отсутствовали. Цвет оболочек визуально соответствовал исходному. По результатам контрольного травления оболочек в азотной кислоте с концентрацией 14 моль/л извлечение продукта волоксидации из оболочек ТВЭЛ составило 99,9%. Извлечение трития из твердофазного продукта окисления составило 99,12%. Степень волоксидации составила 99,8%.
Пример 2.
Использовали фрагменты ТВЭЛ ОЯТ ВВЭР-1000 из той же партии, что и в примере 1 длиной 40-46 мм с замятием сечения среза с одной стороны фрагмента более 50%, с другой 100%. Загрузку фрагментированного ОЯТ, тип насадки, каталитическую активацию потока газовой фазы и вращение контейнера аппарата-реактора проводили аналогично примеру 1. Скорость подачи кислорода на смешение составила 278 ммоль/ч. Скорость подачи воды составила 46,7 ммоль/ч. Скорость подачи диоксида азота составила 252,4 ммоль/ч. Температурный режим процесса волоксидации поддерживали в интервале 370±5°С.
Массовая доля реагентов в поступающем на каталитическую активацию потоке газовой фазы составила: 3,94% для воды, 54,39% для диоксида азота, 41,67% для кислорода. Мольное соотношение между диоксидом азота и водой в парогазовой фазе, поступающими для смешения на каталитически активной насадке, поддерживали на уровне 5,4:1. Мольное соотношение между диоксидом азота и кислородом в парогазовой фазе, поступающими для смешения на каталитически активной насадке, поддерживали на уровне 1:1,1. Скорость подачи потока газовой фазы в контейнер составила 6,6 аппарата, об/ч.
Длительность процесса волоксидации составила 55 минут. Извлечение продукта волоксидации из оболочек ТВЭЛ составило 99,9%. Извлечение трития из твердофазного продукта окисления составило 99,68% без изменения геометрии и внешнего вида оболочек. Степень волоксидации составила 99,9%.
Пример 3.
Использовали фрагменты ТВЭЛ ОЯТ ВВЭР-1000 из той же партии, что и в примере 1 длиной 28-32 мм с замятием сечения среза до 30% с обоих торцов фрагмента. Загрузку фрагментированного ОЯТ, тип насадки, каталитическую активацию потока газовой фазы и вращение контейнера аппарата-реактора проводили аналогично примеру 1. Температурный режим процесса волоксидации поддерживали в интервале 350±5°С.
Массовая доля реагентов в поступающем на каталитическую активацию потоке газовой фазы составила: 2,0% для воды, 49,5% для диоксида азота, 48,5% для кислорода.
Мольное соотношение между диоксидом азота и водой в парогазовой фазе, поступающими для смешения на каталитически активной насадке, поддерживали на уровне 9,8:1. Мольное соотношение между диоксидом азота и кислородом в парогазовой фазе, поступающими для смешения на каталитически активной насадке, поддерживали на уровне 1:1,4. Скорость подачи потока газовой фазы в контейнер составила 5,8 аппаратн.об/ч.
Длительность процесса волоксидации составила 35 минут.Извлечение продукта волоксидации из оболочек ТВЭЛ составило 99,8%. Извлечение трития из твердофазного продукта окисления составило 99,83% без изменения геометрии и внешнего вида оболочек. Степень волоксидации составила 99,9%.
Полученный продукт волоксидации обрабатывали дистиллятом в соотношении фаз Т:Ж=1:10 при перемешивании реакционного объема при помощи ультразвукового диспергатора в течение 2 часов при его циклическом использовании в интервале 10 минут перемешивания через 5 минут отстаивания. Температуру суспензии поддерживали в диапазоне 40±8°С. По окончании декантировали осветленный раствор, в объеме 87% от исходно взятого. Влажный порошкообразный материал, полученный после операций волоксидации и перколяции, трехкратно промывали дистиллятом в объеме, эквивалентно взятому на перколяцию и передавали далее на операцию кислотного растворения. В результате проведения операции перколяции достигнуто снижение удельной γ-активности материала на 65%. Полученный от операции перколяции объединенный раствор не содержал целевых компонентов, подвергался упариванию и передавался на операции кондиционирования.
Пример 4.
Использовали ОЯТ с длиной фрагментов и степенью замятия как в примере 2. Загрузку фрагментированного ОЯТ, тип насадки, каталитическую активацию потока газовой фазы и вращение контейнера аппарата-реактора проводили аналогично примеру 1. Температурный режим процесса волоксидации поддерживали в интервале 350±5°С.
Массовая доля реагентов в поступающем на каталитическую активацию потоке газовой фазы составила: 4,5% для воды, 23,0% для диоксида азота, 72,5% для кислорода.
Мольное соотношение между диоксидом азота и водой в парогазовой фазе, поступающими для смешения на каталитически активной насадке, поддерживали на уровне 2,0:1. Мольное соотношение между диоксидом азота и кислородом в парогазовой фазе, поступающими для смешения на каталитически активной насадке, поддерживали на уровне 1:4,5. Скорость подачи потока газовой фазы в контейнер составила 15,7 аппаратн.об/ч.
Длительность процесса волоксидации составила 135 минут.Извлечение продукта волоксидации из оболочек ТВЭЛ составило 99,7%. Извлечение трития из твердофазного продукта окисления составило 99,92% без изменения геометрии и внешнего вида оболочек. Степень волоксидации составила 99,9%.
Полученный продукт волоксидации перколировали раствором пероксида водорода 0,1% масс. в соотношении фаз Т:Ж=1:5 при перемешивании реакционного объема с помощью ультразвукового диспергатора в течение 40 минут при его циклическом использовании в интервале 10 минут перемешивания через 5 минут отстаивания. Температуру суспензии поддерживали в диапазоне 25±5°С. По окончании перекисной перколяции декантировали осветленный раствор и однократно промывали порошкообразный материал дистиллятом в объеме эквивалентно взятому на перколяцию. Далее проводили щелочную перколяцию раствором гидроксида натрия с концентрацией 0,005 моль/л в соотношении фаз Т:Ж=1:8 при перемешивании реакционного объема при помощи ультразвукового диспергатора в течение 3 часов в интервале 5 минут перемешивания через 10 минут отстаивания. Промывку перколированного продукта проводили аналогично примеру 3.
В результате проведения операции перколяции достигнуто снижение удельной γ-активности материала на 90%. Полученный от операции перколяции объединенный раствор не содержал целевых компонентов, подвергался упариванию и передавался на операции кондиционирования.
Пример 5.
Использовали ОЯТ с длиной фрагментов и степенью замятия аналогично примеру 2. Загрузку фрагментированного ОЯТ, тип насадки, температурный режим процесса, последовательность смешения реагентов на каталитически активной насадке и скорость подачи потока газовой фазы в контейнер соответствовали примеру 1. Вращение контейнера в аппарате-реакторе осуществляли со скоростью 2 об/мин, обеспечивая падение фрагментов внутри контейнера.
Массовая доля реагентов в поступающем на каталитическую активацию потоке газовой фазы составила: 7,3% для воды, 47,5% для диоксида азота, 45,2% для кислорода.
Мольное соотношение между диоксидом азота и водой в парогазовой фазе, поступающими для смешения на каталитически активной насадке, поддерживали на уровне 2,56:1. Мольное соотношение между диоксидом азота и кислородом в парогазовой фазе, поступающими для смешения на каталитически активной насадке, поддерживали на уровне 1:1,37.
Длительность процесса волоксидации составила 110 минут. Извлечение продукта волоксидации из оболочек ТВЭЛ составило 99,7%. Извлечение трития из твердофазного продукта окисления составило 99,87% без изменения геометрии и внешнего вида оболочек. Степень волоксидации составила 99,9%.
Пример 6.
Использовали ОЯТ с длиной фрагментов и степенью замятия аналогично примеру 2. Загрузку фрагментированного ОЯТ, вращение контейнера аппарата-реактора температурный режим процесса, последовательность смешения реагентов на каталитически активной насадке и скорость потока газовой фазы в контейнер соответствовали примеру 1.
Каталитически активная насадка представляла собой цилиндрический столб (с соотношением диаметра к высоте 1:3) гранулированного оксида алюминия с размером зерна 0,3-0,4 мм, покрытого каталитически активным слоем, содержащего платину в количестве 0,8% масс., рутений в количестве 0,01% масс., цирконий в количестве 0,5% масс.
Массовая доля реагентов в поступающем на каталитическую активацию потоке газовой фазы составила: 8,7% для воды, 48,9% для диоксида азота, 42,4% для кислорода.
Мольное соотношение между диоксидом азота и водой в парогазовой фазе, поступающими для смешения на каталитически активной насадке, поддерживали на уровне 2,2:1. Мольное соотношение между диоксидом азота и кислородом в парогазовой фазе, поступающими для смешения на каталитически активной насадке, поддерживали на уровне 1:1,25.
Длительность процесса волоксидации составила 35 минут. Извлечение продукта волоксидации из оболочек ТВЭЛ составило 99,9%. Извлечение трития из твердофазного продукта окисления составило 99,57% без изменения геометрии и внешнего вида оболочек. Степень волоксидации составила 99,8%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОКИСЛИТЕЛЬНОЙ ОБРАБОТКИ (ВОЛОКСИДАЦИИ) ОБЛУЧЕННОГО ЯДЕРНОГО ТОПЛИВА | 2016 |
|
RU2619583C1 |
| СПОСОБ ПЕРЕРАБОТКИ ОБЛУЧЕННОГО ЯДЕРНОГО ТОПЛИВА | 2015 |
|
RU2579753C1 |
| СПОСОБ ПЕРЕРАБОТКИ ОБЛУЧЕННОГО ЯДЕРНОГО ТОПЛИВА | 2011 |
|
RU2459299C1 |
| СПОСОБ РЕГЕНЕРАЦИИ АЗОТНОЙ КИСЛОТЫ ИЗ ТРИТИЙСОДЕРЖАЩЕГО ГАЗОВОГО ПОТОКА | 2017 |
|
RU2664127C1 |
| СПОСОБ ПЕРЕРАБОТКИ ОБЛУЧЕННОГО ЯДЕРНОГО ТОПЛИВА | 2014 |
|
RU2556108C1 |
| СПОСОБ ПЕРЕРАБОТКИ ОБЛУЧЕННОГО ЯДЕРНОГО ТОПЛИВА | 2015 |
|
RU2591215C1 |
| СПОСОБ РАСТВОРЕНИЯ ВОЛОКСИДИРОВАННОГО ОБЛУЧЕННОГО ЯДЕРНОГО ТОПЛИВА | 2016 |
|
RU2626764C1 |
| СПОСОБ ПЕРЕРАБОТКИ ОБЛУЧЁННОГО ЯДЕРНОГО ТОПЛИВА | 2015 |
|
RU2603019C1 |
| СПОСОБ РАСЧЕХЛОВКИ ТЕПЛОВЫДЕЛЯЮЩИХ ЭЛЕМЕНТОВ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2017 |
|
RU2658295C1 |
| СПОСОБ РАСТВОРЕНИЯ ОБЛУЧЕННОГО ЯДЕРНОГО ТОПЛИВА | 2016 |
|
RU2626763C1 |
Изобретение относится к способам переработки облученного ядерного топлива (ОЯТ) и предназначено для использования в головных операциях радиохимической технологии переработки ОЯТ реакторов ВВЭР-1000 с целью отделения трития. Фрагментированное ОЯТ загружают в горизонтальный аппарат-реактор и подвергают окислению в потоке активированной парогазовой фазы. Активацию газового потока осуществляют путем смешения потоков диоксида азота в количестве 15-56 мас.% и кислорода в количестве 41-83 мас.%, насыщенного парами воды в количестве 1,2-9,5 мас.% на зернистом слое каталитически активной насадки. При одновременном воздействии нитрующего и окислительного компонента на реакционную поверхность ОЯТ происходит извлечение топлива из оболочек в результате разнонаправленной трансформации его объема. Изобретение позволяет количественно отделять тритий из находящегося в оболочке ОЯТ в процессе волоксидации. 22 з.п. ф-лы.
1. Способ окислительной обработки (волоксидации) облученного ядерного топлива, включающий термическую обработку фрагментов ОЯТ в окислительной атмосфере, отличающийся тем, что реакционную смесь получают путем смешения на каталитически активной насадке потока нитрующего компонента - диоксида азота в количестве 15-56 мас.%, и потока окислительного компонента - кислорода в количестве 41-83 мас.%, насыщенного парами воды в количестве 1,2-9,5 мас.%, обработку проводят при температуре 325-375°С при периодической механоактивации.
2. Способ по п. 1, отличающийся тем, что используемое ОЯТ представляет собой фрагменты ТВЭЛ на основе диоксида урана без предварительного удаления оболочки.
3. Способ по п. 2, отличающийся тем, что используемое ОЯТ представляет собой нарубленные фрагменты ТВЭЛ реакторов типа ВВЭР-1000 длиной 20-80 мм и степенью замятия сечения среза 0-100% с одной стороны и 0-50% с другой.
4. Способ по п. 1, отличающийся тем, что каталитически активная насадка представляет собой зернистый слой носителя с нанесенным на него каталитически активным слоем, имеющим соотношение диаметра к высоте слоя в диапазоне 1:3÷1:10.
5. Способ по п. 4, отличающийся тем, что в используемой каталитически активной насадке в качестве инертного носителя используется оксид алюминия с размером зерна в диапазоне 0,16-1,2 мм.
6. Способ по п. 4, отличающийся тем, что каталитически активный слой насадки содержит металл платиновой группы в количестве, составляющем 0,05-0,9% от всего количества катализатора и одновременно 10-100% от суммы компонентов в каталитически активном слое.
7. Способ по п. 6, отличающийся тем, что каталитически активный слой насадки в качестве металла платиновой группы содержит платину.
8. Способ по п. 4, отличающийся тем, что слой каталитически активной насадки имеет на входе газового потока зону с температурой 60-100°С, на выходе газового потока зону с температурой 290-360°С.
9. Способ по п. 1, отличающийся тем, что получаемый после каталитической активации поток реагентов в парогазовой фазе поступает непосредственно в рабочий объем аппарата-реактора.
10. Способ по п. 1, отличающийся тем, что скорость потока каталитически активированной парогазовой фазы составляет 2-100 аппаратн. об/ч.
11. Способ по п. 1, отличающийся тем, что частота механоактивации составляет 0,5-15 импульс/мин.
12. Способ по п. 1, отличающийся тем, что в парогазовой фазе, поступающей для смешения на каталитически активной насадке, мольное соотношение между диоксидом азота и кислородом составляет от 1:7 до 1:1.
13. Способ по п. 1, отличающийся тем, что в парогазовой фазе, поступающей для смешения на каталитически активной насадке, мольное соотношение между диоксидом азота и водой составляет от 2:1 до 10:1.
14. Способ по п. 1, отличающийся тем, что полученный после окислительной обработки порошкообразный продукт волоксидированного ОЯТ подвергают перколяции водными растворами при соотношении Т:Ж в диапазоне 1:4-1:10 и температуре среды в 20-80°С.
15. Способ по п. 14, отличающийся тем, что полученный после окислительной обработки порошкообразный продукт волоксидированного ОЯТ подвергают перколяции водой при рН среды 5-7,8.
16. Способ по п. 14, отличающийся тем, что полученный после окислительной обработки порошкообразный продукт волоксидированного ОЯТ подвергают перекисной перколяции с концентрацией пероксида водорода 0,001-0,1 мас.%.
17. Способ по п. 14, отличающийся тем, что полученный после окислительной обработки порошкообразный продукт волоксидированного ОЯТ подвергают щелочной перколяции раствором гидроксида щелочного металла с концентрацией 0,001-0,01 моль/л.
18. Способ по п. 14, отличающийся тем, что полученный после окислительной обработки порошкообразный продукт волоксидированного ОЯТ подвергают щелочной перколяции раствором гидроксида аммония с концентрацией 0,001-0,01 моль/л.
19. Способ по п. 14, отличающийся тем, что процесс перколяции интенсифицируют диспергированием суспензии посредством ультразвука.
20. Способ по п. 14, отличающийся тем, что перколяцию щелочным раствором проводят после перколяции раствором пероксида водорода.
21. Способ по п. 1, отличающийся тем, что смешение компонентов парогазовой фазы проводят на насадке, не содержащей каталитически активный компонент.
22. Способ по п. 14, отличающийся тем, что смешение компонентов парогазовой фазы проводят в отдельном аппарате.
| СПОСОБ ОКИСЛИТЕЛЬНОЙ ОБРАБОТКИ (ВОЛОКСИДАЦИИ) ОБЛУЧЕННОГО ЯДЕРНОГО ТОПЛИВА | 2016 |
|
RU2619583C1 |
| СПОСОБ РАСТВОРЕНИЯ ЯДЕРНОГО ТОПЛИВА В ВИДЕ ИЗМЕЛЬЧЕННЫХ ТЕПЛОВЫДЕЛЯЮЩИХ СБОРОК АТОМНЫХ РЕАКТОРОВ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2007 |
|
RU2371791C2 |
| US 8574523 B2,05.11.2013 | |||
| US 4279875 A1,21.07.1981. | |||

