Настоящее изобретение относится к некоторым 4-(замещенным анилино)-6-O-(замещенным пиперазин-карбонил)хиназолиновым соединениям и их фармацевтическим солям, которые могут быть полезны в лечении или предупреждении заболевания или медицинского состояния, опосредованного активирующими мутированными формами рецептора эпидермального фактора роста (EGFR), например L858R активирующим мутантом и/или активирующими мутантами с делецией в экзоне 19. Такие соединения и их соли могут быть полезны в лечении или предупреждении ряда различных злокачественных новообразований. Изобретение также относится к фармацевтическим композициям, содержащим указанные соединения или их фармацевтически приемлемую соль, кристаллическим формам этих соединений или их фармацевтически приемлемой соли, промежуточным соединениям, полезным при производстве указанных соединений или их фармацевтически приемлемой соли, и к способам лечения заболеваний, опосредованных активирующими мутированными формами EGFR, с использованием указанных соединений или их фармацевтически приемлемой соли.
EGFR (также известный как ErbB1 или HER1) представляет собой трансмембранный белок тирозинкиназу, являющуюся членом семейства erbB рецептора. При связывании лиганда фактора роста, такого как эпидермальный фактор роста (EGF), рецептор может гомодимеризоваться с другой молекулой EGFR или гетеродимеризоваться с другим членом семейства, таким как erbB2 (HER2), erbB3 (HER3) или erbB4 (HER4).
Гомо- и/или гетеро-димеризация erbB рецепторов приводит в результате к фосфорилированию ключевых тирозиновых остатков во внутриклеточном домене и ведет к стимуляции огромного количества внутриклеточных путей передачи сигнала, вовлеченных в клеточную пролиферацию и выживание. Нарушение регуляции передачи сигнала erbB семейства вызывает пролиферацию, инвазию, метастаз, ангиогенез и выживание опухолевых клеток и было описано для многих злокачественных новообразований у человека, в том числе легкого, головы и шеи, и груди.
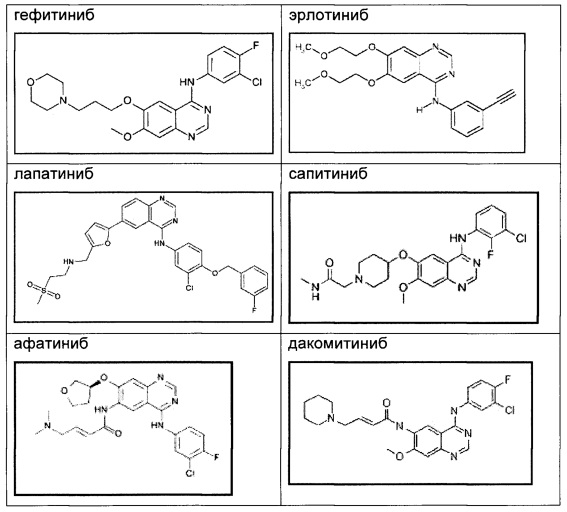
Таким образом, erbB семейство представляет рациональную мишень для разработки противораковых лекарственных средств, и в настоящее время в клинической практике доступен ряд агентов, направленных на EGFR или erbB2, включая гефитиниб (IRESSA™), эрлотиниб (TARCEVA™) и лапатиниб (TYKERB™, TYVERB™). Подробное описание передачи сигнала erbB рецептора и его вовлеченности в онкогенез представлено в New England Journal of Medicine [2008] Vol. 358; 1160-74, и Biochemical and Biophysical Research Communications [2004] Vol. 319: 1-11.
В 2004 году сообщалось (Science [2004] Vol. 304: 1497-500, и New England Journal of Medicine [2004] Vol. 350; 2129-39), что активирующие мутации в EGFR коррелировали с ответом на терапию гефитинибом при немелкоклеточном раке легкого (NSCLC). Приблизительно 90% связанных с NSCLC мутаций EGFR включают две основных мутации EGFR (делеция Е746-А750 в экзоне 19 и мутация с заменой L858R в экзоне 21) (Pao et al. Proceedings of the National Academy of Sciences of the United States of America [2004], Vol. 13: 306-11, и Kosada et al. Cancer Research [2004], Vol. 64: 8919-23). Эти активирующие мутации приводят в результате к увеличению сродства к низкомолекулярным ингибиторам тирозинкиназы, таким как гефитиниб и эрлотиниб, и снижению сродства к аденозинтрифосфату (АТР) по сравнению с EGFR дикого типа (WT).
Однако, о побочных действиях, таких как кожная сыпь и диарея, которые, как считают, связаны с подавлением сигнальных путей EGFR WT в нормальных клетках кожи и кишечника, сообщалось более чем у 60% пациентов с NSCLC, подвергаемых лечению гефитинибом или эрлотинибом (Zhou СС et al. Journal of Clinical Oncology [2011], Vol. 12: 735-42; Mok TS et al. New England Journal of Medicine [2009], Vol. 361: 947-57). Кроме того, и гефитиниб, и эрлотиниб демонстрировали ограниченное воздействие при лечении пациентов с NSCLC с метастазами в головной мозг, поскольку ни один из них эффективно не проникает через гематоэнцефалический барьер (ВВВ) (McKillop D et al. Xenobiotica [2004], Vol. 34: 983-1000; Jackman DM et al. Journal of Clinical Oncology [2006], Vol. 24: 4517-20 Grommes С et al. Neuro-Oncology [2011], Vol. 13: 1364-9), тогда как по некоторым данным видно, что решение проблемы метастазов в головной мозг при раке легкого становится нереализованной потребностью медицины (Gavrilovic et al, Journal of Neurooncology [2005], Vol. 75: 5-14; Barnholtz-Sloan JS et al. Journal of Clinical Oncology [2004], 22: 2865-72; Schouten LJ et al, Cancer [2002], Vol. 94: 2698-705).
Лептоменингеальные метастазы встречаются, когда злокачественная опухоль распространяется на мозговые оболочки, слои ткани, которые покрывают головной и спинной мозг. Метастазы могут распространяться на мозговые оболочки через кровь или они могут перемещаться из метастазов головного мозга, переносимые спинномозговой жидкостью (CSF), которая протекает через мозговые оболочки. Если клетки опухоли проникают в CSF и выживают, они могут перемещаться по центральной нервной системе, вызывая неврологические проблемы (Le Rhun et al. Surg Neurol Int. [2013], Vol. 4: S265-88). Частота возникновения лептоменингеальных метастазов возрастает, отчасти вследствие того, что продолжительность жизни онкологических больных увеличивается, но также потому, что многие виды препаратов для химиотерапии и молекулярной целевой терапии не способны достичь достаточной концентрации в спинномозговой жидкости, чтобы уничтожить клетки опухоли. Лечение обычно неэффективно, а продолжительность жизни измеряется неделями. В компании AstraZeneca исследовали сапитиниб (AZD8931), равноэффективный ингибитор рецепторов EGFR, HER2 и HER3, для применения при раке молочной железы. На сегодняшний день сапитиниб изучается в трех клинических исследованиях стадии II; во-первых, в комбинации с паклитакселом в сравнении с паклитакселом, взятым отдельно, у пациентов с распространенным раком молочной железы, демонстрирующих низкий уровень HER2; во-вторых, в комбинации с анастрозолом в сравнении с анастрозолом, взятым отдельно, при гормон-рецептор положительном распространенном раке молочной железы; и в-третьих, в комбинации с паклитакселом в сравнении с паклитакселом, взятым отдельно, при метастатическом раке желудка или гастроэзофагеального перехода, прогрессирующем после терапии первой линии и не подлежащем лечению трастузумабом из-за HER2 статуса. Соединение по настоящему изобретению структурно отличается от сапитиниба и обладает повышенной способностью проникать в головной мозг, что делает его потенциально полезным в лечении злокачественных новообразований, которые метастазируют в центральную нервную систему [CNS], в частности тех, которые метастазируют в головной мозг, и тех, которые приводят к лептоменингеальным метастазам.
В настоящее время некоторые необратимые хиназолиновые ингибиторы EGFR, такие как афатиниб и дакомитиниб, находятся на стадии клинической разработки. Хотя эти соединения показывали сравнимые с гефитинибом и эрлотинибом воздействия на активирующие мутации в EGFR у пациентов с NSCLC, они демонстрировали более тяжелые побочные действия, такие как кожная сыпь (в более чем 90% - случаи кожной сыпи и диареи) (Zhou СС et al. Journal of Clinical Oncology [2011], Vol. 12: 735-42; Mok TS et al. New England Journal of Medicine [2009], Vol. 361: 947-57; Miller VA et al. Lancet Oncology [2012], Vol. 13: 528-38; Ramalingam SS et al. Journal of Clinical Oncology [2012], Vol. 30: 3337-44). Соединения по настоящему изобретению являются обратимыми ингибиторами, и поэтому ожидается, что они обладают менее выраженными побочными действиями, связанными с EGFR, чем афатиниб и дакомитиниб.
Некоторые хиназолиновые соединения были раскрыты, смотри, например "Получение хиназолиновых производных для лечения опухолей" (US 20080177068 А1), "Получение хиназолиновых производных для лечения опухолей" (US 20080167328 А1), "Получение сахаридных производных хиназолинов в качестве ингибиторов белка тирозинкиназы" (CN 101857618 А), "Получение хлорфторанилинометокси-N-метилкарбамоилметил-пиперидинилоксихиназолиновых производных для применения в качестве противоопухолевых агентов" (WO 2010061208 А2), "Получение 4-аминохиназолиновых производных в качестве антинеопластических агентов (CN 101367793 А)", "Получение пролин-хиназолиновых производных в качестве антипролиферативных агентов (BR 2006002275 А)", "Получение хиназолиновых производных в качестве ингибиторов протеинкиназы" (WO 2005097137 А2), "Получение хиназолиновых производных в качестве ингибиторов протеинкиназы" (WO 2005097134 А2), "Получение хиназолиновых производных в качестве ингибиторов тирозинкиназы EGFR" (WO 2005028469 А1), "Получение фениламино-замещенных хиназолинов в качестве ингибиторов EGF и ErbB-2 киназ" (WO 2005028470 А1), "Получение хиназолиновых производных в качестве ингибиторов тирозинкиназы EGFR" (WO 2005026156 А1), "Получение пиперидил-хиназолиновых производных в качестве ингибиторов тирозинкиназы для лечения опухолей" (WO 2005012290 А1), "Получение 4-анилинохиназолинов в качестве антипролиферативных агентов" (WO 2003082831 А1), "Получение аминохиназолинов в качестве ингибиторов сигнальной трансдукции через рецепторы эпидермального фактора роста" (WO 2002018351 А1), "Получение хиназолинов в качестве ингибиторов Аврора-2 киназы" (WO 2001021594 А1), "Хиназолины и другие бициклические гетероциклы, фармацевтические композиции, содержащие эти соединения, в качестве ингибиторов тирозинкиназ и способы их получения" (WO 2000055141 А1), "Получение хиназолиновых производных и их ингибиторные свойства в отношении рецепторных тирозинкиназ" (WO 9738994 А1), "Хиназолиновые производные в качестве противоопухолевых агентов" (WO 9730034 А1), "Получение галогеноанилинохиназолинов в качестве ингибиторов тирозинкиназы рецептора I класса" (WO 9633980 А1) и "Хиназолиновые производные, полезные при лечении неопластического заболевания" (US 5457105).
Соединения по изобретению или их фармацевтически приемлемая соль при сравнении с другими клинически приемлемыми ингибиторами EGFR проявляют некоторые улучшенные свойства, например более высокий уровень проникновения через ВВВ (что делает их потенциально полезными для лечения злокачественных новообразований, которые метастазируют в CNS, в частности метастазов в головной мозг и лептоменингеальных метастазов); показывают лучшую селективность между EGFR WT и мутированным EGFR (что может приводить к меньшим побочным эффектам от лечения, кожной сыпи и диарее); поддерживая аналогичную или улучшенную активность в отношении активирующего мутированного EGFR (например L858R активирующий мутант EGFR и/или активирующие мутанты с делецией в экзоне 19). Поэтому такие соединения или их фармацевтически приемлемая соль могут быть особенно полезны в лечении болезненных состояний, в которые вовлечены эти активирующие мутации EGFR, например в лечении злокачественного новообразования.
Таким образом, согласно настоящему изобретению предложено соединение формулы (I):
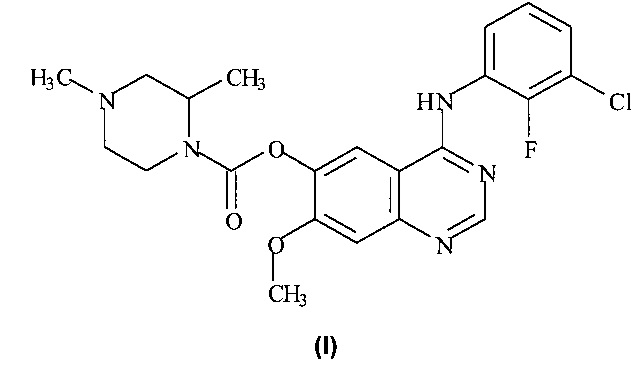
или его фармацевтически приемлемая соль.
Структуры клинических соединений, упомянутых выше, являются следующими:

Подходящая фармацевтически приемлемая соль соединения по изобретению представляет собой, например соль присоединения кислоты, например неорганической или органической кислоты, например соляной, бромистоводородной, серной, фосфорной, лимонной, L-винной, гликолевой, фумаровой, янтарной или малеиновой кислоты, в особенности соляной, бромистоводородной, серной, фосфорной, лимонной, L-винной, гликолевой, фумаровой или малеиновой кислоты. Конкретная фармацевтически приемлемая соль соединения по изобретению представляет собой соль соляной кислоты. Еще одна конкретная фармацевтически приемлемая соль соединения по изобретению представляет собой соль янтарной кислоты. Специалисту будет понятно, что также могут быть приемлемы дополнительные соли присоединения кислот, например те, которые показаны в примерах, но не ограничиваются ими.
Соли соединений формулы (I) могут быть образованы, например, посредством взаимодействия соединения формулы (I) с определенным количеством кислоты в среде, такой как среда, в которой соль выпадает в осадок, или в водной среде с последующей лиофилизацией.
Соединения формулы (I) или их фармацевтически приемлемая соль обладают хиральным центром. Следует понимать, что изобретением охватываются все стереоизомеры (энантиомеры и диастереоизомеры) соединений формулы (I) или их фармацевтически приемлемой соли, которые обладают ингибиторной активностью в отношении активирующего мутированного EGFR. Изобретение также относится к любым возможным таутомерным формам соединений формулы (I) или их фармацевтически приемлемой соли, которые обладают ингибиторной активностью в отношении активирующего мутированного EGFR. В дополнительном аспекте изобретения предложен энантиомер формулы (I) или его фармацевтически приемлемая соль, по существу не содержащие каких-либо других энантиомеров. В дополнительном аспекте изобретения предложен (R)-энантиомер формулы (I) или его фармацевтически приемлемая соль, по существу не содержащие каких-либо других энантиомеров. В дополнительном аспекте изобретения предложен (S)-энантиомер формулы (I) или его фармацевтически приемлемая соль, по существу не содержащие каких-либо других энантиомеров.
В одном воплощении изобретения, когда смесь содержит неравные молярные доли энантиомеров, смесь может иметь энантиомерный избыток, выбранный из более чем 50%, более чем 70%, более чем 90% и более чем 95%. Предпочтительно смесь может иметь энантиомерный избыток более 98%. Более предпочтительно смесь может иметь энантиомерный избыток более 99%. Более предпочтительно смесь может иметь энантиомерный избыток более 99,5%.
Также следует понимать, что некоторые соединения формулы (I) или их фармацевтически приемлемая соль могут существовать в сольватированных, а также несольватированных формах, таких как, например, гидратные формы. Следует понимать, что изобретением охватываются все такие сольватированные формы, которые обладают ингибиторной активностью в отношении активирующего мутированного EGFR.
Также следует понимать, что изобретением охватываются все изотопные формы соединений, описанных в данном описании. Например, водород включает дейтерий, а углерод включает 12С и 13С.
В другом аспекте изобретения конкретные соединения по изобретению представляют собой любое соединение согласно примерам или его фармацевтически приемлемую соль.
В другом аспекте изобретения конкретные соединения по изобретению выбраны из:
4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата;
4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2S)-2,4-диметилпиперазин-1-карбоксилата; и
4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(±)-2,4-диметилпиперазин-1-карбоксилата;
или их фармацевтически приемлемой соли.
В другом аспекте изобретения конкретные соединения по изобретению выбраны из:
4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата;
4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2S)-2,4-диметилпиперазин-1-карбоксилата; и
4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(±)-2,4-диметилпиперазин-1-карбоксилата.
В другом аспекте изобретения конкретное соединение по изобретению выбрано из фармацевтически приемлемой соли 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата.
В другом аспекте изобретения конкретное соединение по изобретению выбрано из гидрохлорида 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата.
В другом аспекте изобретения конкретное соединение по изобретению выбрано из сукцината 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата.
В другом аспекте изобретения конкретное соединение по изобретению выбрано из 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата.
В другом аспекте изобретения конкретное соединение по изобретению выбрано из фармацевтически приемлемой соли 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2S)-2,4-диметилпиперазин-1-карбоксилата.
В другом аспекте изобретения конкретное соединение по изобретению выбрано из 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2S)-2,4-диметилпиперазин-1-карбоксилата.
В другом аспекте изобретения конкретное соединение по изобретению выбрано из фармацевтически приемлемой соли 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(-)-2,4-диметилпиперазин-1-карбоксилата.
В другом аспекте изобретения конкретное соединение по изобретению выбрано из 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(-)-2,4-диметилпиперазин-1-карбоксилата.
В другом аспекте изобретения конкретное соединение по изобретению выбрано из фармацевтически приемлемой соли 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(+)-2,4-диметилпиперазин-1-карбоксилата.
В другом аспекте изобретения конкретное соединение по изобретению выбрано из 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(+)-2,4-диметилпиперазин-1-карбоксилата.
В другом аспекте изобретения конкретное соединение по изобретению выбрано из фармацевтически приемлемой соли 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(±)-2,4-диметилпиперазин-1-карбоксилата.
В другом аспекте изобретения конкретное соединение по изобретению выбрано из 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(±)-2,4-диметилпиперазин-1-карбоксилата.
В тех случаях, когда здесь приведены величины оптического вращения (+) или (-), они конкретно измерены при с10, где с представляет собой концентрацию, выраженную в г/мл, в DMSO (диметилсульфоксиде) при 25°C. Также следует понимать, что некоторые соединения по изобретению или их фармацевтически приемлемая соль могут существовать в определенных кристаллических формах. В частности, 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат идентифицирован как имеющий несколько кристаллических форм - а конкретно форму А, форму Е, форму I и форму J. Кроме того, гидрохлорид 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата также может существовать в кристаллической форме - а конкретно в форме Α1 моногидрохлорида, и сукцинат 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата также может существовать в кристаллической форме - а конкретно в форме А8 сукцината. Следует понимать, что настоящим изобретением охватываются все такие кристаллические формы соединений формулы (I) или их фармацевтически приемлемой соли, которые обладают ингибиторной активностью в отношении активирующего мутированного EGFR.
4-[(3-Хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат в кристаллической форме, форма А
Форма А характеризуется тем, что дает по меньшей мере одно из следующих значений 2θ, измеренных с использованием CuKa излучения: 23,3 и 14,3°. Форма А характеризуется картиной дифракции рентгеновских лучей на порошке по существу такой, как показано на фиг. 1. Десять пиков дифракции рентгеновских лучей на порошке показаны в таблице А.
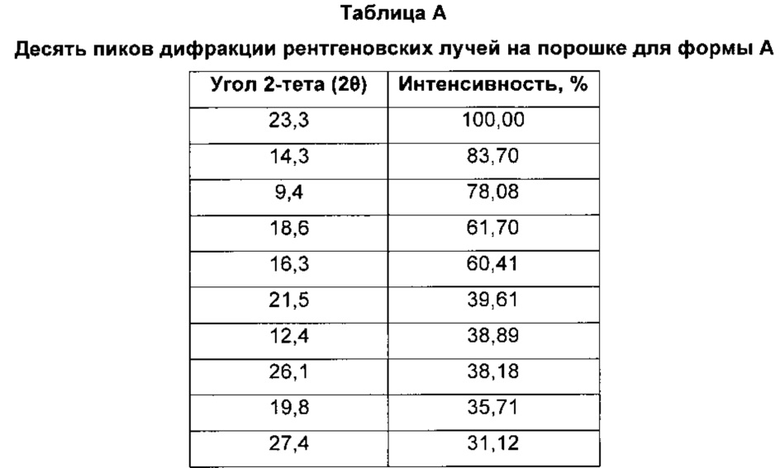
Согласно настоящему изобретению предложена кристаллическая форма, форма А, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при значениях 2-тета, равных примерно 23,3° и 14,3°.
Согласно настоящему изобретению предложена кристаллическая форма, форма А, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при значениях 2-тета, равных примерно 23,3; 14,3; 9,4; 18,6; 16,3; 21,5; 12,4; 26,1; 19,8; 27,4°.
Согласно настоящему изобретению предложена кристаллическая форма, форма А, которая имеет картину дифракции рентгеновских лучей на порошке по существу такую же, как картина дифракции рентгеновских лучей на порошке, показанная на фиг. 1.
Согласно настоящему изобретению предложена кристаллическая форма, форма А, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при значениях 2-тета, равных 23,3° и 14,3°, где указанные значения могут составлять плюс или минус 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, форма А, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при значениях 2-тета, равных 23,3; 14,3; 9,4; 18,6; 16,3; 21,5; 12,4; 26,1; 19,8; 27,4°, где указанные значения могут составлять плюс или минус 0,2° 2-тета.
Анализ формы А посредством DSC (дифференциальной сканирующей калориметрии) показывает эндотерму плавления с началом при 192,4°С и пиком при 195,8°С (фиг. 2).
4-[(3-Хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат в кристаллической форме, форма Ε
Форма Ε характеризуется тем, что дает по меньшей мере одно из следующих значений 2θ, измеренных с использованием CuKa излучения: 7,3 и 13,7°. Форма Ε характеризуется картиной дифракции рентгеновских лучей на порошке по существу такой, как показано на фиг. 3. Девять пиков дифракции рентгеновских лучей на порошке показаны в таблице В.
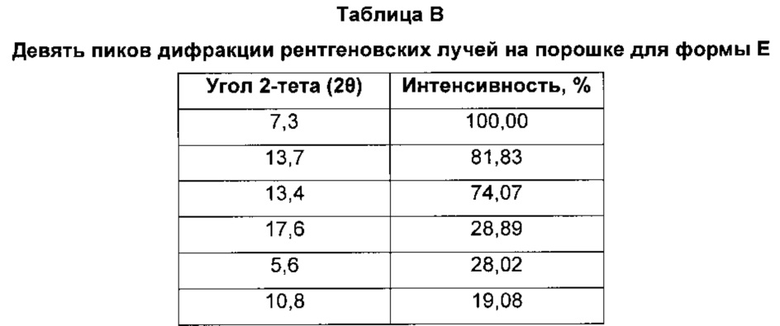

Согласно настоящему изобретению предложена кристаллическая форма, форма Е, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при значениях 2-тета, равных примерно 7,3° и 13,7°.
Согласно настоящему изобретению предложена кристаллическая форма, форма Е, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при значениях 2-тета, равных примерно 7,3; 13,7; 13,4; 17,6; 5,6; 10,8; 21,7; 26,5; 28,4°.
Согласно настоящему изобретению предложена кристаллическая форма, форма Е, которая имеет картину дифракции рентгеновских лучей на порошке по существу такую же, как картина дифракции рентгеновских лучей на порошке, показанная на фиг. 3.
Согласно настоящему изобретению предложена кристаллическая форма, форма Е, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при значениях 2-тета, равных 7,3° и 13,7°, где указанные значения могут составлять плюс или минус 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, форма Е, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при значениях 2-тета, равных 7,3; 13,7; 13,4; 17,6; 5,6; 10,8; 21,7; 26,5; 28,4°, где указанные значения могут составлять плюс или минус 0,2° 2-тета.
Анализ формы Ε посредством DSC показывает эндотерму плавления с началом при 194,2°С и пиком при 196,3°С (фиг. 4).
4-[(3-Хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат в кристаллической форме, форма I
Форма I характеризуется тем, что дает по меньшей мере одно из следующих значений 2θ, измеренных с использованием CuKa излучения: 3,5 и 7,0°. Форма Ε характеризуется картиной дифракции рентгеновских лучей на порошке по существу такой, как показано на фиг. 5. Десять пиков дифракции рентгеновских лучей на порошке показаны в таблице С.

Согласно настоящему изобретению предложена кристаллическая форма, форма I, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере тремя характеристическими пиками при значениях 2-тета примерно 3,5°, 7,0° и 9,5°.
Согласно настоящему изобретению предложена кристаллическая форма, форма I, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при значениях 2-тета, равных примерно 3,5; 7,80; 9,5; 6,4; 14,3; 18,0; 16,4; 15,3; 4,7; 21,3°.
Согласно настоящему изобретению предложена кристаллическая форма, форма I, которая имеет картину дифракции рентгеновских лучей на порошке по существу такую же, как картина дифракции рентгеновских лучей на порошке, показанная на фиг. 5.
Согласно настоящему изобретению предложена кристаллическая форма, форма I, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере тремя характеристическими пиками при значениях 2-тета, равных 3,5°, 7,0° и 9,5°, где указанные значения могут составлять плюс или минус 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, форма I, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при значениях 2-тета, равных 3,5; 7,0; 9,5; 6,4; 14,3; 18,0; 16,4; 15,3; 4,7; 21,3°, где указанные значения могут составлять плюс или минус 0,2° 2-тета.
Анализ формы I посредством DSC показывает эндотерму плавления с началом при 193,3°С и пиком при 195,9°С (фиг. 6).
4-[(3-Хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат в кристаллической форме, Форма J
Форма J характеризуется тем, что дает по меньшей мере одно из следующих значений 2θ, измеренных с использованием CuKa излучения: 7,8 и 7,0°. Form J характеризуется картиной дифракции рентгеновских лучей на порошке по существу такой, как показано на фиг. 7. Десять пиков дифракции рентгеновских лучей на порошке показаны в таблице D.

Согласно настоящему изобретению предложена кристаллическая форма, форма J, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при значениях 2-тета, равных примерно 7,8° и 7,0°.
Согласно настоящему изобретению предложена кристаллическая форма, форма J, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при значениях 2-тета, равных примерно 7,8; 7,0; 4,9; 15,9; 17,7; 3,4; 20,7; 9,8; 13,9; 12,7°
Согласно настоящему изобретению предложена кристаллическая форма, форма J, которая имеет картину дифракции рентгеновских лучей на порошке по существу такую же, как картина дифракции рентгеновских лучей на порошке, показанная на фиг. 7.
Согласно настоящему изобретению предложена кристаллическая форма, форма J, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при значениях 2-тета, равных 7,8° и 7,0°, где указанные значения могут составлять плюс или минус 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, форма J, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при значениях 2-тета, равных 7,8; 7,0; 4,9; 15,9; 17,7; 3,4; 20,7; 9,8; 13,9; 12,7°, где указанные значения могут составлять плюс или минус 0,2° 2-тета.
Анализ формы J посредством DSC показывает эндотерму плавления с началом при 193,3°С и пиком при 195,8°С (фиг. 8).
Соль гидрохлорид 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата в кристаллической форме, соль моногидрохлорид, форма A1
Соль моногидрохлорид, Форма Α1, характеризуется тем, что дает по меньшей мере одно из следующих значений 2θ, измеренных с использованием CuKa излучения: 12,3 и 13,9°. Соль моногидрохлорид, Форма А1, характеризуется картиной дифракции рентгеновских лучей на порошке по существу такой, как показано на фиг. 9. Девять пиков дифракции рентгеновских лучей на порошке показаны в таблице Е.
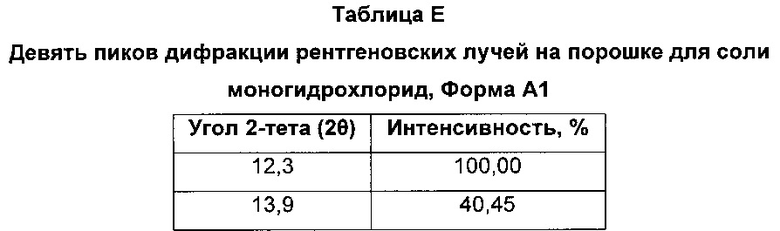
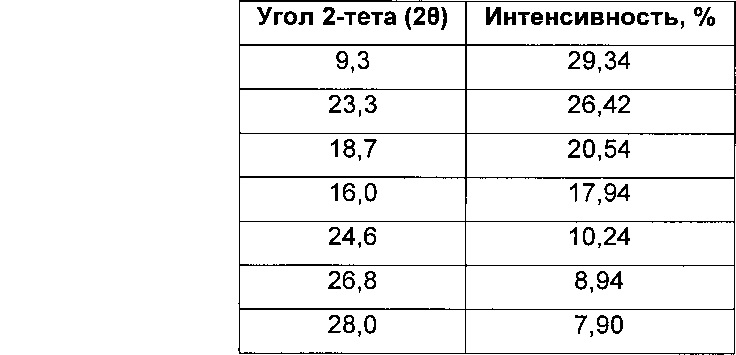
Согласно настоящему изобретению предложена кристаллическая форма, соль моногидрохлорид, Форма А1, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с двумя характеристическими пиками при значениях 2-тета, равных примерно 12,3° и 13,9°
Согласно настоящему изобретению предложена кристаллическая форма, соль моногидрохлорид, Форма А1, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при значениях 2-тета, равных примерно 12,3; 13,9; 9,3; 23,3; 18,7; 16,0; 24,6; 26,8; 28,0°.
Согласно настоящему изобретению предложена кристаллическая форма, соль моногидрохлорид, Форма А1, которая имеет картину дифракции рентгеновских лучей на порошке по существу такую же, как картина дифракции рентгеновских лучей на порошке, показанная на фиг. 9.
Согласно настоящему изобретению предложена кристаллическая форма, соль моногидрохлорид, Форма А1, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при значениях 2-тета, равных 12,3° и 13,9°, где указанные значения могут составлять плюс или минус 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, соль моногидрохлорид, Форма А1, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при значениях 2-тета, равных 12,3; 13,9; 9,3; 23,3; 18,7; 16,0; 24,6; 26,8; 28,0°, где указанные значения могут составлять плюс или минус 0,2° 2-тета.
Анализ соли моногидрохлорид, Форма А1, посредством DSC показывает эндотерму плавления с началом при 259,6°С и пиком при 261,4°С (фиг. 10).
Соль сукцинат 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата в кристаллической форме, соль сукцинат, Форма А8
Соль сукцинат, Форма А8, характеризуется тем, что дает по меньшей мере одно из следующих значений 2θ, измеренных с использованием CuKa излучения: 6,5 и 17,7. Соль сукцинат, Форма A8, характеризуется картиной дифракции рентгеновских лучей на порошке, по существу такой, как показано на фиг. 11. Девять пиков дифракции рентгеновских лучей на порошке показаны в таблице F.
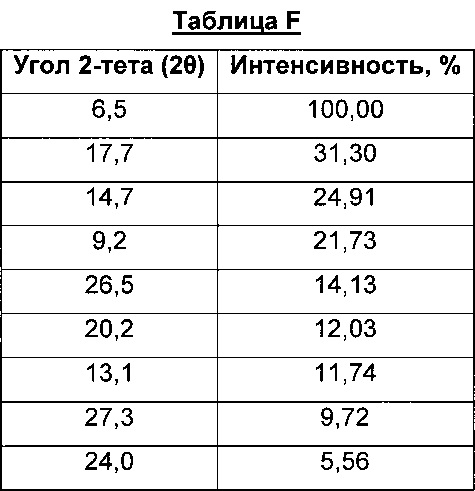
Согласно настоящему изобретению предложена кристаллическая форма, соль сукцинат, Форма А8, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере тремя характеристическими пиками при значениях 2-тета, равных примерно 6,5°, 17,7° и 14,7°.
Согласно настоящему изобретению предложена кристаллическая форма, соль сукцинат, Форма А8, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при значениях 2-тета, равных примерно 6,5; 17,7; 14,7; 9,2; 26,5; 20,2; 13,1; 27,3; 24,0°.
Согласно настоящему изобретению предложена кристаллическая форма, соль сукцинат, Форма А8, которая имеет картину дифракции рентгеновских лучей на порошке по существу такую же, как картина дифракции рентгеновских лучей на порошке, показанная на фиг. 11.
Согласно настоящему изобретению предложена кристаллическая форма, соль сукцинат, Форма А8, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере тремя характеристическими пиками при значениях 2-тета, равных 6,5°, 17,7° и 14,7°, где указанные значения могут составлять плюс или минус 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, соль сукцинат, Форма А8, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при значениях 2-тета, равных 6,5; 17,7; 14,7; 9,2; 26,5; 20,2; 13,1; 27,3; 24,0°, где указанные значения могут составлять плюс или минус 0,2° 2-тета.
Анализ соли сукцинат, Формы А8, посредством DSC показывает эндотермы плавления с началом при 191,8°С и пиком при 194,2°С (фиг. 12).
Краткое описание графических материалов
Фиг. 1: картина дифракции рентгеновских лучей на порошке формы А.
Фиг. 2: термограмма DSC формы А.
Фиг. 3: картина дифракции рентгеновских лучей на порошке формы Е.
Фиг. 4: термограмма DSC формы Е.
Фиг. 5: картина дифракции рентгеновских лучей на порошке формы I.
Фиг. 6: термограмма DSC формы I.
Фиг. 7: картина дифракции рентгеновских лучей на порошке формы J.
Фиг. 8: термограмма DSC формы J.
Фиг. 9: картина дифракции рентгеновских лучей на порошке формы A1 гидрохлорида.
Фиг. 10: термограмма DSC формы Α1 гидрохлорида.
Фиг. 11: картина дифракции рентгеновских лучей на порошке формы А8 сукцината.
Фиг. 12: термограмма DSC формы А8 сукцината.
Когда говорят, что настоящее изобретение относится к кристаллической форме, степень кристалличности составляет предпочтительно более чем примерно 60%, более предпочтительно более чем примерно 80%, предпочтительно более чем примерно 90% и более предпочтительно более чем примерно 95%. Наиболее предпочтительно степень кристалличности составляет более чем примерно 98%.
Следует понимать, что значения 2-тета картины дифракции рентгеновских лучей на порошке могут слегка варьировать в зависимости от оборудования или от образца к образцу, и такие зарегистрированные значения не следует интерпретировать как абсолютные. Известно, что может быть получена картина дифракции рентгеновских лучей на порошке, имеющая одну или более чем одну ошибку измерения в зависимости от условий измерения (таких как используемое оборудование или аппаратура). В частности, в целом известно, что интенсивности в картине дифракции рентгеновских лучей на порошке могут колебаться в зависимости от условий измерения. Таким образом, следует понимать, что полиморфные формы по настоящему изобретению не ограничены кристаллами, которые дают картины дифракции рентгеновских лучей на порошке, идентичные картине дифракции рентгеновских лучей на порошке, показанной на фигурах, и любые кристаллы, дающие картины дифракции рентгеновских лучей на порошке по существу такие же, как те, которые показаны на фигурах, входят в объем настоящего изобретения. Специалист в области рентгеновской порошковой дифракции способен оценить по существу идентичность картин дифракции рентгеновских лучей на порошке.
Специалисты в области рентгеновской порошковой дифракции понимают, что на относительную интенсивность пиков может влиять, например, размер зерен выше 30 мкм и неунитарные аспектные отношения, которые могут влиять на анализ образцов. Специалисты также понимают, что на положение отражений может влиять точная высота, на которую образец устанавливают в дифрактометре, и калибровка нуля дифрактометра. Плоскостность поверхности образца также может оказывать небольшое влияние. Поэтому представляемые данные картин дифракции не следует воспринимать как абсолютные значения (Jenkins, R & Snyder, R.L. 'Introduction to X-Ray Powder Diffractometry' John Wiley & Sons 1996; Bunn, C.W. (1948), Chemical Crystallography, Clarendon Press, London; Klug, H.P. & Alexander, L. E. (1974), X-Ray Diffraction Procedures).
Как правило, ошибка измерений угла дифракции в рентгеновской порошковой дифрактограмме равна приблизительно плюс или минус 0,2° 2-тета, и такую степень ошибки измерений следует принимать во внимание при рассмотрении картин дифракции рентгеновских лучей на порошке, показанных на фигурах и в таблицах. Более того, следует понимать, что интенсивности могут колебаться в зависимости от экспериментальных условий и приготовления образца (предпочтительной ориентации).
Поэтому в дополнительном аспекте изобретения предложен 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат в кристаллической форме.
В дополнительном аспекте изобретения предложена фармацевтически приемлемая соль 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата в кристаллической форме.
В дополнительном аспекте изобретения предложена соль гидрохлорид 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметил-пиперазин-1-карбоксилата в кристаллической форме.
В одном аспекте изобретения 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат в кристаллической форме находится в виде Формы А.
В одном аспекте изобретения 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат в кристаллической форме находится в виде Формы Е.
В одном аспекте изобретения 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат в кристаллической форме находится в виде Формы I.
В одном аспекте изобретения 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат в кристаллической форме находится в виде Формы J.
В одном аспекте изобретения гидрохлорид 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата в кристаллической форме находится в виде соли моногидрохлорид, Форма Α1
В одном аспекте изобретения сукцинат 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата в кристаллической форме находится в виде соли сукцинат, Форма А8.
В одном аспекте изобретения 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат в кристаллической форме находится в виде Формы А и по существу не содержит никаких других форм.
В одном аспекте изобретения 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат в кристаллической форме находится в виде Формы Ε и по существу не содержит никаких других форм.
В одном аспекте изобретения 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат в кристаллической форме находится в виде Формы I и по существу не содержит никаких других форм.
В одном аспекте изобретения 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат в кристаллической форме находится в виде Формы J и по существу не содержит никаких других форм.
В одном аспекте изобретения гидрохлорид 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата в кристаллической форме находится в виде соли моногидрохлорид, Форма А1, и по существу не содержит никаких других форм.
В одном аспекте изобретения сукцинат 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата в кристаллической форме находится в виде соли сукцинат, Форма А8, и по существу не содержит никаких других форм.
Термин "по существу не содержит" относится к менее чем 10% другой формы или форм, энантиомера или энантиомеров, в частности менее чем 5%. В другом аспекте термин "по существу не содержит" относится к менее чем 1% другой формы или форм, энантиомера или энантиомеров. Здесь форма также включает аморфную форму.
Как сказано выше, соединения или их фармацевтически приемлемая соль, определенные в настоящем изобретении, обладают противоопухолевой активностью, которая, как полагают, обусловлена ингибиторной активностью в отношении активирующего мутированного EGFR, и другими свойствами соединений или их фармацевтически приемлемой соли. Эти свойства могут быть определены, например, с помощью методик, изложенных ниже.
Анализ 1: анализ клеточного фосфорилирования
Линию клеток легкого человека NCI-H3255 (L858R) получали из Американской коллекции типовых культур. Клетки NCI-H3255 поддерживали в среде ВЕВМ (Lonza; СС-3171), содержащей 10% фетальной бычьей сыворотки (FBS) (Gibco; 10099-141), с добавлением набора BEGM (для роста бронхиальных эпителиальных клеток) (Lonza; СС-4175). Линию клеток легкого человека РС-9 (EGFR с делецией в экзоне 19) получали из Американской коллекции типовых культур. Клетки РС-9 поддерживали в среде RPMI 1640 (Gibco; 22400-089), содержащей 10% фетальной бычьей сыворотки. Линию клеток легкого человека NCI-H838 (EGFR дикого типа) получали из Американской коллекции типовых культур. Клетки NCI-H838 поддерживали в среде RPMI 1640 (Gibco; 22400-089), содержащей 10% фетальной бычьей сыворотки.
Клетки выращивали в увлажняемом инкубаторе при 37°C с 5% CO2. Анализы для измерения клеточного фосфорилирования эндогенного p-EGFR в клеточных лизатах проводили согласно протоколу, описанному в наборе PathScan® Phospho-EGF Receptor (Tyr1068) Sandwich ELISA (набор для анализа клеточной передачи сигнала, номер по каталогу #7240).
100 мкл клеток высевали (32000 клеток/лунка) в среду RPMI 1640 с 1% фетальной бычьей сыворотки в 96-луночные планшеты для культур клеток Corning Costar и инкубировали при 37°C с 5% СО2 в течение ночи. Клетки акустически дозировали с использованием аппаратуры компании Tecan, при этом соединения серийно разбавляли в 100%-ном DMSO (диметилсульфоксиде). Планшеты с клетками инкубировали в течение дополнительных 4 часов после добавления соединений (для NCI-H838: rhEGF (R&D, номер по каталогу #236-EG) добавляли в планшет для клеток до конечной концентрации 100 нг/мл rhEGF для стимуляции в течение 5 минут), затем после отсасывания среды в каждую лунку добавляли 110 мкл IP буфера для лизиса (IP буфер для лизиса: добавить коктейль ингибиторов фосфатаз 2&3 в разведении 1:100 (Sigma, номер по каталогу Р5726&Р0044), коктейль ингибиторов протеаз в разведении 1:100 (Sigma, номер по каталогу Р8340) в IP буфере для лизиса фирмы Pierce (Thermo, номер по каталогу #87788)). Планшеты размещали при 4°С при вращении 300 об/мин в течение 0,5-1 часа. 100 мкл/лунка клеточного лизата переносили в покрытые планшеты (набор для анализа клеточной передачи сигнала, номер по каталогу #7240) и инкубировали в течение ночи при 4°С при вращении 300 об/мин. Планшеты перемещали из температуры 4°С в 37°С при вращении 300 об/мин в течение 1 часа. После аспирации и промывки планшетов 1× промывочным буфером в каждую лунку добавляли 100 мкл детекторного антитела (набор для анализа клеточной передачи сигнала, номер по каталогу #7240). Планшет герметизировали пленкой и инкубировали в течение 2 часов при 37°С при вращении 300 об/мин. После аспирации и промывки планшетов 1× промывочным буфером в каждую лунку добавляли 100 мкл HRP-связанного (конъюгированного с пероксидазой хрена) вторичного антитела (набор для анализа клеточной передачи сигнала, номер по каталогу #7240). Планшет герметизировали пленкой и инкубировали в течение 1 часа при 37°С при вращении 300 об/мин. После аспирации и промывки планшетов 1× промывочным буфером в каждую лунку добавляли 100 мкл субстрата ТМВ (тетраметилбензидин) (набор для анализа клеточной передачи сигнала, номер по каталогу #7240). Планшет герметизировали пленкой и инкубировали в течение 30 минут при 37°С при 300 об/мин. В планшеты добавляли 100 мкл стоп-реагента (набор для анализа клеточной передачи сигнала, номер по каталогу #7240) и оптическую плотность считывали при 450 нм в течение 30 минут на планшет-ридере SpectraMax М5е.
Данные, полученные с каждым соединением, вводили в подходящий программный пакет (такой как Η-BASE) для проведения аналитической аппроксимации кривых. Исходя из этих данных, определяли значения IC50 посредством расчета концентрации соединения, которая требуется для получения 50% эффекта.
Аналитические данные (мкМ) в анализе 1 для соединений по примерам этой заявки, а также данные, полученные для гефитиниба и эрлотиниба, показаны в таблице ниже (где n представляет собой число повторностей эксперимента).
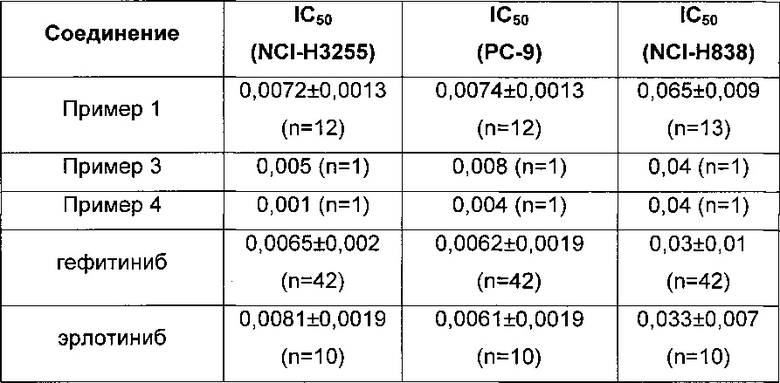
Это показывает, что соединения по примеру 1, примеру 2 и примеру 3 обладают эффективностью, соизмеримой с эффективностью гефитиниба и эрлотиниба.
Анализ 2: анализ проникновения через гемато-энцефалический барьер
Как Kp.uu brain, так и Kp.uu должны быть основными параметрами, измеряемыми и оптимизируемыми при изыскании новых лекарственных средств для CNS (Di L et al., Journal of Medicinal Chemistry [2013], 56: 2-12). Kp.uu brain, отношение между концентрациями несвязанного лекарственного средства в головном мозге и в крови, прогнозирует действие лекарственного средства на метастатические опухоли головного мозга. Лептоменингеальные метастазы (LM) являются следствием метастазирования злокачественной опухоли в мягкую и паутинную оболочки мозга, вызывая дисфункцию центральной нервной системы. Kp.uu CSF представляет собой распределение лекарственного средства в CSF в сравнении с распределением в крови, что определяет реакцию на воздействие лекарственного средства во время лечения лептоменингеальных метастаз.
Анализ связывания в крови и головном мозге in vitro проводили в планшете для НТ-диализа (диализа высокой производительности) (Gales Ferry, СТ) с полупроницаемой мембраной. В разбавленную кровь (1:1 с DPBS (фосфатно-солевой буфер Дульбекко) рН 7,4) и разбавленный гомогенат головного мозга (1:3 с DPBS рН 7,4) добавляли 5 мкМ исследуемого соединения (в трехкратной повторности) и диализировали против равного объема 150 мкл 100 мМ фосфатно-солевого буфера PBS (рН 7,4) при 37°С в течение 4 часов в медленно вращаемом планшете. По окончании инкубирования отбирали аликвоту 50 мкл из приемной камеры и 5 мкл из донорской камеры. Образец 5 мкл дополнительно разбавляли 45 мкл контрольного образца крови или гомогената головного мозга. Парные образцы сопоставляли при помощи матрицы либо с буфером, либо с контрольным образцом крови/гомогенатом головного мозга и смешивали в течение 2 минут, а затем осаждали с помощью 150 мкл холодного ацетонитрила со 100 нг/мл толбутамида в качестве внутреннего стандарта. После центрифугирования при 4000 об/мин в течение 20 минут надосадочную жидкость разбавляли 0,1%-ным водным раствором муравьиной кислоты и анализировали посредством ЖХ/МС/МС (жидкостная хроматография-тандемная масс-спектрометрия) (API 4000, Applied Biosystems, Foster City). Несвязанную фракцию (fu) исследуемого соединения в гомогенате головного мозга и разбавленной крови подсчитывали по соотношению побочной реакции буфера к побочной реакции гомогената головного мозга/разбавленной крови, и несвязанную фракцию (fu.bl и fu.br) исследуемого соединения в неразбавленной крови и тканях подсчитывали исходя из измеренной fu в гомогенате и разбавленной крови с помощью следующего уравнения: fu.bl (fu.br)=(1/D)/[(1/fu-1)+1/D)]. D представляет собой коэффициент разведения.
Модель быстрой пероральной абсорбции (SOA) представляет собой in vivo скрининговую модель для установления проникновения соединения в головной мозг. Шести самцам крыс линии Han Wistar, закупленных у Beijing Vital River, перорально вводили соединение в дозе 2 мкг/кг в 1%-ной метилцеллюлозе. Через 0,25; 0,5; 1; 2; 4 и 7 часов после введения отбирали спинномозговую жидкость (CSF) из мозжечково-мозговой цистерны и отбирали образцы крови (более 60 мкл/временная точка/каждый участок) посредством пункции сердца в отдельные пробирки с EDTA (этилендиаминтетрауксусная кислота) в качестве антикоагулянта и затем сразу же разбавляли 3-кратным объемом воды. Мозговую ткань отбирали для анализа и гомогенизировали в 3× объеме 100 мМ фосфатно-солевого буферного раствора (рН 7,4). Все образцы хранили при приблизительно -70°С до ЖХ/МС/МС анализа.
Стандарты готовили посредством добавления контрольного образца крови, гомогената головного мозга и искусственной CSF, охватывая диапазон от 0,2 до 500 нг/мл. Гомогенизированную мозговую ткань вместе с образцами крови осаждали посредством добавления 3-кратного объема холодного ацетонитрила, содержащего внутренний стандарт (40 нг/мл дексаметазона и 40 нг/мл диклофенака), и образцы CSF по 10 мкл осаждали с помощью 100 мкл холодного ацетонитрила, содержащего внутренний стандарт. После вихревого перемешивания в течение 2 минут и центрифугирования при 14000 об/мин в течение 5 минут надосадочную жидкость анализировали посредством ЖХ/МС/МС (API 4000, Applied Biosystems, Foster City). Строили две группы стандартных кривых в начале и в конце каждой серии по анализу образцов крови. Для образцов ткани головного мозга и CSF анализировали одну стандартную кривую наряду с исследуемыми образцами.
Общие уровни в головном мозге, выраженные в виде соотношения головной мозг/кровь (Kp.brain), измеряли с помощью AUCbrain/AUCblood У грызунов после перорального введения. Свободную фракцию исследуемого соединения в биологической среде определяли посредством in vitro анализа связывания в крови и головном мозге. Kp.uu brain и Kp.uu CSF рассчитывали с помощью следующего уравнения: Kp.uu brain=AUCbrain/AUCblood×(fu.brain/fu.blood) и Kp.uu CSF=AUCCSF/(AUCblood×fu.blood).
Аналитические данные в анализе 2 для соединений по примерам этой заявки, а также данные, полученные для сапитиниба (в форме свободного основания), показаны в таблице ниже.

Результаты демонстрируют лучшие свойства проникновения через гематоэнцефалический барьер у соединений по настоящему изобретению по сравнению с сапитинибом.
Согласно дополнительному аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено выше, вместе с фармацевтически приемлемым разбавителем или носителем.
Композиция может находиться в форме, подходящей для перорального введения, например в виде таблетки или капсулы, для парентеральной инъекции (включая внутривеннную, подкожную, внутримышечную, внутрисосудистую или инфузию) в виде стерильного раствора, суспензии или эмульсии, для местного введения в виде мази или крема, или ректального введения в виде суппозитория. Предпочтительно композиция может находиться в форме, подходящей для перорального введения.
В большинстве случаев вышеуказанные композиции могут быть получены посредством общепринятых способов при использовании общепринятых эксципиентов.
Соединение формулы (I) или его фармацевтически приемлемую соль обычно вводят теплокровному животному в разовой дозе в диапазоне 0,01-2000 мг/кг, предпочтительно 2,5-1000 мг/кг, предпочтительно 5-500 мг/кг, и это должно обеспечивать терапевтически эффективную дозу. Однако суточная доза при необходимости будет варьироваться в зависимости от организма, подвергаемого лечению, конкретного пути введения и тяжести заболевания, которое лечат. Соответственно, оптимальную дозировку может определить врач, который занимается лечением конкретного пациента.
Согласно дополнительному аспекту настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено выше, для применения при способе лечения человека или животного посредством терапии.
В результате своей ингибиторной активности в отношении активирующего мутированного EGFR соединения формулы (I) или их фармацевтически приемлемая соль, как полагают, являются полезными для лечения заболеваний или медицинских состояний, полностью или частично опосредованных активирующим мутированным EGFR, например рака. Виды рака, которые могут поддаваться лечению с использованием соединений формулы (I) или их фармацевтически приемлемой соли, включают рак яичника, рак шейки матки, колоректальный рак, рак молочной железы, рак поджелудочной железы, глиому, глиобластому, меланому, рак предстательной железы, лейкоз, лимфому, неходжкинскую лимфому, рак легкого, гепатоцеллюлярныи рак, рак желудка, стромальную опухоль желудочно-кишечного тракта, рак щитовидной железы, рак желчных протоков, рак эндометрия, рак почки, анапластическую крупноклеточную лимфому, острый миелоидный лейкоз, множественную миелому, меланому и мезотелиому, но не ограничиваются ими. В конкретном воплощении изобретения вид рака, который может поддаваться лечению с использованием соединения формулы (I) или его фармацевтически приемлемой соли, представляет собой немелкоклеточный рак легкого (NSCLC). В дополнительном конкретном воплощении клетки NSCLC у теплокровного животного несут или, как было показано ранее, несут активирующие мутации в гене EGFR.
Соединение формулы (I) или его фармацевтически приемлемая соль полезны в лечении болезненных состояний, в которые вовлечен активирующий мутированный EGFR. В одном аспекте изобретения, где речь идет об активирующем мутированном EGFR, это относится к одной или более чем одной мутации в АТФ-связывающем сайте (киназный домен) гена EGFR, в частности вблизи экзонов 18-21, например, как описано в WO 2005/094357. В одном аспекте изобретения, где речь идет об активирующем мутированном EGFR, это относится к L858R активирующему мутированному EGFR и/или активирующему мутированному EGFR с делецией в экзоне 19. В одном аспекте изобретения, где речь идет об активирующем мутированном EGFR, это относится к L858R активирующему мутированному EGFR и активирующему мутированному EGFR с делецией в экзоне 19. В одном аспекте изобретения, где речь идет об активирующем мутированном EGFR, это относится к L858R активирующему мутированному EGFR. В другом аспекте изобретения, где речь идет об активирующем мутированном EGFR, это относится к активирующему мутированному EGFR с делецией в экзоне 19.
Предусматривается, что для способов лечения рака, упомянутого здесь, соединения формулы (I) или их фармацевтически приемлемую соль будут вводить млекопитающему, более предпочтительно человеку. Аналогичным образом, при применении соединений формулы (I) или их фармацевтически приемлемой соли для лечения рака, упомянутого здесь, предусматривается, что соединения формулы (I) или их фармацевтически приемлемую соль будут вводить млекопитающему, более предпочтительно человеку.
Поэтому, согласно другому аспекту изобретения предложены соединения формулы (I) или их фармацевтически приемлемая соль, как определено выше, для применения в качестве лекарственного средства.
Согласно дополнительному аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для ингибирования активирующего мутированного EGFR у теплокровного животного, такого как человек.
Согласно этому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для обеспечения противоракового эффекта у теплокровного животного, такого как человек.
Согласно дополнительному аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для использования в лечении рака яичника, рака шейки матки, колоректального рака, рака молочной железы, рака поджелудочной железы, глиомы, глиобластомы, меланомы, рака предстательной железы, лейкоза, лимфомы, неходжкинской лимфомы, рака легкого, гепатоцеллюлярного рака, рака желудка, стромальной опухоли желудочно-кишечного тракта, рака щитовидной железы, рака желчных протоков, рака эндометрия, рака почки, анапластической крупноклеточной лимфомы, острого миелоидного лейкоза, множественной миеломы, меланомы и мезотелиомы.
Согласно дополнительному аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для использования в лечении NSCLC.
Согласно дополнительному аспекту изобретения предложен способ ингибирования активирующего мутированного EGFR у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше.
Согласно дополнительному аспекту изобретения предложен способ получения противоракового эффекта у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше.
Согласно дополнительному аспекту изобретения предложен способ получения противоракового эффекта у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий (1) определение того, несет или нет теплокровное животное активирующую мутацию EGFR в опухолевой клетке и (2) если это так, введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше.
Согласно дополнительному аспекту изобретения предложен способ лечения рака яичника, рака шейки матки, колоректального рака, рака молочной железы, рака поджелудочной железы, глиомы, глиобластомы, меланомы, рака предстательной железы, лейкоза, лимфомы, неходжкинской лимфомы, рака легкого, гепатоцеллюлярного рака, рака желудка, стромальной опухоли желудочно-кишечного тракта, рака щитовидной железы, рака желчных протоков, рака эндометрия, рака почки, анапластической крупноклеточной лимфомы, острого миелоидного лейкоза, множественной миеломы, меланомы и мезотелиомы у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше.
Согласно дополнительному аспекту изобретения предложен способ лечения NSCLC у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше.
Согласно дополнительному аспекту изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено выше, для применения в ингибировании активирующего мутированного EGFR у теплокровного животного, такого как человек.
Согласно этому аспекту изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено выше, для применения для получения противоракового эффекта у теплокровного животного, такого как человек.
Согласно дополнительному аспекту изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено выше, для применения в лечении рака яичника, рака шейки матки, колоректального рака, рака молочной железы, рака поджелудочной железы, глиомы, глиобластомы, меланомы, рака предстательной железы, лейкоза, лимфомы, неходжкинской лимфомы, рака легкого, гепатоцеллюлярного рака, рака желудка, стромальной опухоли желудочно-кишечного тракта, рака щитовидной железы, рака желчных протоков, рака эндометрия, рака почки, анапластической крупноклеточной лимфомы, острого миелоидного лейкоза, множественной миеломы, меланомы и мезотелиомы.
Согласно дополнительной особенности изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено выше, для применения в лечении NSCLC.
В дополнительном аспекте изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено выше, вместе с фармацевтически приемлемым разбавителем или носителем, для применения в ингибировании активирующего мутированного EGFR у теплокровного животного, такого как человек.
В дополнительном аспекте изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено выше, вместе с фармацевтически приемлемым разбавителем или носителем, для применения для получения противоракового эффекта у теплокровного животного, такого как человек.
В дополнительном аспекте изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено выше, вместе с фармацевтически приемлемым разбавителем или носителем, для применения в лечении рака яичника, рака шейки матки, колоректального рака, рака молочной железы, рака поджелудочной железы, глиомы, глиобластомы, меланомы, рака предстательной железы, лейкоза, лимфомы, неходжкинской лимфомы, рака легкого, гепатоцеллюлярного рака, рака желудка, стромальной опухоли желудочно-кишечного тракта, рака щитовидной железы, рака желчных протоков, рака эндометрия, рака почки, анапластической крупноклеточной лимфомы, острого миелоидного лейкоза, множественной миеломы, меланомы и мезотелиомы у теплокровного животного, такого как человек.
В дополнительном аспекте изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено выше, вместе с фармацевтически приемлемым разбавителем или носителем, для применения в лечении NSCLC у теплокровного животного, такого как человек.
В любом из аспектов или воплощений, упомянутых в данном описании, в случаях, когда упоминается рак, указанный рак может быть выбран из рака яичника, рака шейки матки, колоректального рака, рака молочной железы, рака поджелудочной железы, глиомы, глиобластомы, меланомы, рака предстательной железы, лейкоза, лимфомы, неходжкинской лимфомы, рака легкого, гепатоцеллюлярного рака, рака желудка, стромальной опухоли желудочно-кишечного тракта, рака щитовидной железы, рака желчных протоков, рака эндометрия, рака почки, анапластической крупноклеточной лимфомы, острого миелоидного лейкоза, множественной миеломы, меланомы и мезотелиомы.
В любом из аспектов или воплощений, упомянутых в данном описании, в случаях, когда упоминается рак, предпочтительно указанный рак может быть выбран из рака легкого. В дополнительном аспекте предпочтительно указанный рак может быть выбран из немелкоклеточного рака легкого. В дополнительном аспекте предпочтительно указанный рак может быть выбран из неметастатического немелкоклеточного рака легкого. В дополнительном аспекте предпочтительно указанный рак может быть выбран из метастатического немелкоклеточного рака легкого.
Соединение по настоящему изобретению может быть использовано в схемах адъювантного лечения, и/или первой, и/или второй линии лечения пациентов с NSCLC, несущих активирующий мутированный EGFR, с метастазами в CNS или без них, в частности в головной мозг и/или лептоменингеальными метастазами.
В другом аспекте рак находится в неметастатической форме.
В другом аспекте рак находится в метастатической форме.
В частности, в другом аспекте изобретения метастазы представляют собой метастазы в CNS.
В частности, в другом аспекте метастазы в CNS представляют собой метастазы в головной мозг.
В частности, в другом аспекте метастазы в CNS представляют собой лептоменингеальные метастазы. Некоторые пациенты с NSCLC с метастазами в CNS, в частности в головной мозг и/или лептоменингеальными метастазами, демонстрируют симптомы заболевания CNS, такие как головная боль и рвота. Для этих пациентов может быть применена лучевая терапия всего головного мозга (WBRT) для ослабления этих симптомов. Соединение по настоящему изобретени. может быть способно усиливать противоопухолевое действие WBRT, а также дополнительно ослаблять симптомы заболевания CNS при использовании в комбинации c WBRT.
Лечение активности активирующего мутированного EGFR, определенное ранее, можно применять в качестве монотерапии, либо оно может включать, в дополнение к соединению по изобретению, традиционное хирургическое вмешательство, или радиотерапию (например WBRT, как описано выше), или химиотерапию. Такая химиотерапия может включать один или более чем один из следующих противоопухолевых агентов:
1) анти-CTLA-4 (цитотоксический ассоциированный с Т-лимфоцитами протеин-4) антитело;
2) (2-гидрокси-этокси)-амид 6-(4-бром-2-хлор-фениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты (как описано в WO 2007/076245) или его фармацевтически приемлемая соль;
3) анти-PD-L1 антитело;
4) 1-[(1S)-1-(имидазо[1,2-а]пиридин-6-ил)этил]-6-(1-метил-1Н-пиразол-4-ил)-1Н-[1,2,3]триазоло[4,5-b]пиразин (соединение 270 в WO 2011/079804) или его фармацевтически приемлемая соль;
5) анти-PD-1 антитело или
6) агонистическое антитело против ОХ40.
В частности, анти-CTLA-4 антитело представляет собой тремелимумаб (который раскрыт в US 6682736). В другом аспекте изобретения, в частности, анти-CTLA-4 антитело представляет собой ипилимумаб (зарегистрированный Bristol Myers Squib как YERVOY®).
В частности, "(2-гидрокси-этокси)-амид 6-(4-бром-2-хлор-фениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты (который раскрыт в WO 2007/076245) или его фармацевтически приемлемая соль" представляет собой соль гидросульфат (2-гидрокси-этокси)-амида 6-(4-бром-2-хлор-фениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты. Более предпочтительно соль гидросульфат представляет собой 1:1 соединение: H2SO4.
В частности, анти-PD-L1 антитело представляет собой антитело, которое раскрыто в US 20130034559 (Medlmmune). В другом аспекте изобретения, в частности, анти-PD-L1 антитело представляет собой антитело, которое раскрыто в US 2010/0203056 (Genentech/Roche). В другом аспекте изобретения, в частности, анти-PD-L1 антитело представляет собой антитело, которое раскрыто в US 20090055944 (Medarex). В другом аспекте изобретения, в частности, анти-PD-L1 антитело представляет собой антитело, которое раскрыто в US 20130323249 (Sorrento Therapeutics).
В частности, анти-PD-1 антитело представляет собой MRK-3475, которое раскрыто в WO 2009/114335 и US 8168757 (Merck). В другом аспекте изобретения, в частности, Nivolumab представляет собой анти-PD-1 антитело, которое раскрыто в WO 2006/121168 или US 8008449 (Medarex). В другом аспекте изобретения, в частности, анти-PD-1 антитело представляет собой антитело, которое раскрыто в WO 2009/101611 (CureTech). В другом аспекте изобретения, в частности, анти-PD-1 антитело представляет собой антитело, которое раскрыто в WO 2012/145493 (Amplimmune). В другом аспекте изобретения, в частности, анти-PD-1 антитело представляет собой антитело, которое раскрыто в US 7488802 (Wyeth/Medlmmune).
В частности, анти-ОХ40 антитело представляет собой антитело, которое раскрыто в US 20110123552 (Crucell). В другом аспекте изобретения, в частности, анти-PD-1 антитело представляет собой антитело, которое раскрыто в US 20130280275 (Board of Regents, Univ. of Texas). В другом аспекте изобретения, в частности, анти-PD-1 антитело представляет собой антитело, которое раскрыто в WO 99/42585 (Agonox), и WO 95/12673, и WO 95/21915.
Согласно этому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение формулы (I) как определено выше, или его фармацевтически приемлемую соль и любой из противоопухолевых агентов, перечисленных выше в пунктах (1)-(4).
Поэтому, в дополнительном аспекте изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль в комбинации с противоопухолевым агентом, выбранным из перечисленных выше в пунктах (1)-(4). В данном описании, в тех случаях, когда используют термин "комбинация", следует понимать, что это относится к одновременному, раздельному или последовательному введению. В одном аспекте изобретения "комбинация" относится к одновременному введению. В другом аспекте изобретения "комбинация" относится к раздельному введению. В дополнительном аспекте изобретения "комбинация" относится к последовательному введению. В тех случаях, когда введение является последовательным или раздельным, время выдержки перед введением второго компонента должно быть таким, чтобы не утрачивался положительный эффект комбинации.
Согласно дополнительному аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из перечисленных выше в пунктах (1)-(4), вместе с фармацевтически приемлемым разбавителем или носителем.
Согласно дополнительному аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из перечисленных выше в пунктах (1)-(4), вместе с фармацевтически приемлемым разбавителем или носителем, для применения для обеспечения активности в отношении активирующего мутированного EGFR.
Согласно дополнительному аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из перечисленных выше в пунктах (1)-(4), вместе с фармацевтически приемлемым разбавителем или носителем, для применения для получения противоракового эффекта.
Согласно дополнительному аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из перечисленных выше в пунктах (1)-(4), вместе с фармацевтически приемлемым разбавителем или носителем, для применения в лечении рака яичника, рака шейки матки, колоректального рака, рака молочной железы, рака поджелудочной железы, глиомы, глиобластомы, меланомы, рака предстательной железы, лейкоза, лимфомы, неходжкинской лимфомы, рака легкого, гепатоцеллюлярного рака, рака желудка, стромальной опухоли желудочно-кишечного тракта, рака щитовидной железы, рака желчных протоков, рака эндометрия, рака почки, анапластической крупноклеточной лимфомы, острого миелоидного лейкоза, множественной миеломы, меланомы и мезотелиомы.
Согласно дополнительному аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из перечисленных выше в пунктах (1)-(4), вместе с фармацевтически приемлемым разбавителем или носителем для применения в лечении NSCLC.
Согласно дополнительному аспекту настоящего изобретения предложен набор, содержащий соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из перечисленных выше в пунктах (1)-(4).
Согласно дополнительному аспекту настоящего изобретения предложен набор, содержащий:
a) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
b) противоопухолевый агент, выбранный из перечисленных выше в пунктах (1)-(4), во второй стандартной лекарственной форме и
с) контейнерное устройство для вмещения указанных первой и второй лекарственных форм.
Помимо их применения в лечебной медицине соединения формулы (I) или их фармацевтически приемлемая соль также полезны в качестве фармакологических инструментов при разработке и стандартизации in vitro и in vivo тест-систем для оценки эффективности ингибиторной активности в отношении активирующего мутированного EGFR на лабораторных животных, таких как кошки, собаки, кролики, обезьяны, крысы и мыши, при осуществлении поиска новых терапевтических агентов.
В вышеуказанных аспектах других фармацевтических композиций, способов, методов, применений и производства лекарственных средств также используют альтернативные и предпочтительные воплощения соединений по изобретению, раскрытых в данном описании.
Примеры
Теперь изобретение будет проиллюстрировано следующими примерами, в которых, как правило:
(1) за ходом реакций обычно следили посредством жидкостной хроматографии с масс-спектрометрией (ЖХМС) или посредством тонкослойной хроматографии (ТСХ); указанные значения времени протекания реакций необязательно являются минимально достижимыми;
(2) при необходимости органические растворители сушили над безводным сульфатом магния или безводным сульфатом натрия, процедуры выделения проводили с использованием традиционных методик разделения слоев, выпаривание проводили либо посредством ротационного выпаривания при пониженном давлении, либо в испарителе Genevac HT-4/EZ-2;
(3) выходы, там, где они указаны, необязательно являются максимально достижимыми, и при необходимости взаимодействия повторяли, если требовалось большее количество продукта реакции;
(4) обычно структуры конечных продуктов подтверждали посредством ядерного магнитного резонанса (ЯМР) и/или посредством методов масс-спектрального анализа; данные масс-спектрального анализа при ионизации электрораспылением (ИЭР) получали с помощью ЖХ/масс-спектрометра Waters ZMD или Waters ZQ, получая данные как для положительных, так и для отрицательных ионов; как правило, регистрировали только ионы, относящиеся к исходной структуре; величины химических сдвигов в спектрах протонного ЯМР измеряли по шкале дельта при 400 МГц при использовании ЯМР-спектрометра Bruker или ЯМР-спектрометра Varian. Были использованы следующие аббревиатуры: s, синглет; d, дублет; pd, частичный дублет; t, триплет; q, квартет; m, мультиплет; br, уширенный. Способные к обмену протоны не всегда наблюдают или регистрируют в ЯМР конечных продуктов из-за обмена с дейтерированным растворителем или предпочтительно дейтерированной водой в растворителе, или сигнал является слабо выраженным и/или очень широким;
(5) промежуточные соединения не обязательно были полностью очищены, но их структуры и чистоту оценивали посредством ТСХ, аналитической ВЭЖХ и/или ЯМР анализа;
(6) если не оговорено особо, колоночную хроматографию (посредством флэш-метода) и жидкостную хроматографию среднего давления (ЖХСД) проводили на диоксиде кремния Merck Kieselgel (Art. 9385) или посредством использования предварительно упакованных картриджей диоксида кремния на оборудовании для полуавтоматической флэш-хроматографии (ПАФХ) (например CombiFlash Companion); и
(7) были использованы следующие аббревиатуры:
Дифракция рентгеновских лучей на порошке
Аналитический прибор: дифрактометр Empyrean фирмы Panalytical. Рентгеновскую порошковую дифрактограмму определяли путем закрепления образца кристаллического материала на кремниевом монокристаллическом держателе и распределения образца тонким слоем с помощью предметного стекла. 2θ-положение калибровали относительно кремниевого порошкового стандарта Panalytical 640. Образец облучали рентгеновскими лучами, генерируемыми медной длинно-тонкофокусной трубкой, работающей при 45 кВ и 40 мА, с длиной волны Kα1=1,540598 Ангстрем и Kα2=1,544426 Ангстрем (отношение интенсивностей Kα2/Kα1 составляет 0,50). Коллимированный пучок рентгеновских лучей от источника пропускали через автоматически регулируемую щель расходимости, установленную на 10 мм, и отраженное излучение направляли через антирассеивающую щель шириной 5,5 мм. Образец подвергали воздействию в течение 12,7 секунды с шагом 2-тета 0,0167° (непрерывный режим сканирования) в интервале углов 2-тета от 3 градусов до 40 градусов в режиме тета-тета. Время прогона составляло 3 минуты 57 секунд. Прибор был оснащен детектором RTMS (широкодиапазонное измерение в режиме реального времени) (X'Celerator). Контроль и сбор данных осуществляли с помощью Dell Optiplex 780, работающего с программой сбора данных. Специалисты в области рентгеновской порошковой дифракции понимают, что на относительную интенсивность пиков может влиять, например, размер зерен выше 30 мкм и неунитарные аспектные отношения, которые могут влиять на анализ образцов. Специалист также понимает, что на положение отражений может влиять точная высота, на которую образец устанавливают в дифрактометр, и калибровка нуля дифрактометра. Плоскостность поверхности образца также может оказывать небольшое влияние. Поэтому представляемые данные картин дифракции не следует воспринимать как абсолютные значения.
Дифференциальная сканирующая калориметрия
Аналитический прибор: калориметр DSC Q200 или Q2000 фирмы ТА Instruments. Обычно менее 5 мг материала, помещенного в стандартный алюминиевый тигель с крышкой, нагревали в диапазоне температур от 25°С до 300°C с постоянной скоростью нагревания 10°С в минуту. Использовали продувку газообразным азотом со скоростью потока 50 мл в минуту.
Промежуточное соединение 1
5-гидрокси-4-метокси-2-нитробензойная кислота
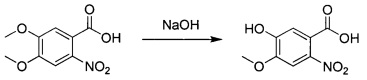
4,5-Диметокси-2-нитробензойную кислоту (145 г; 0,639 моль) растворяли в растворе гидроксида натрия (6 н; 600 мл) и нагревали при 100°С в течение 3 часов. Смесь охлаждали до комнатной температуры и выливали в смесь концентрированной соляной кислоты и дробленого льда (рН менее 2). Смесь фильтровали и осадок на фильтре сушили с получением промежуточного соединения 1 (149 г; неочищенное) в виде желтого твердого вещества, которое использовали без дополнительной очистки. 1Н ЯМР (DMSO-d6, 400 МГц): δ 7.34 (s, 1Н), 6.89 (s, 1Н), 3.80 (s, 3Н).
Промежуточное соединение 2
2-Амино-5-гидрокси-4-метоксибензойная кислота

Смесь промежуточного соединения 1 (50 г; 93,85 ммоль) и 10% Pd/C (5 г) в МеОН (1,2 л) перемешивали в атмосфере H2 (50 фунт-сила/кв. дюйм (0,34 МПа)) при комнатной температуре в течение 4 часов. Смесь фильтровали и промывали МеОН (10×1 л). Объединенные метанольные экстракты концентрировали с получением промежуточного соединения 2 (27,7 г; выход 64%) в виде черного твердого вещества, которое использовали без дополнительной очистки.
Промежуточное соединение 3
7-Метоксихиназолин-4,6-диол

К суспензии промежуточного соединения 2 (88 г; 0,48 моль) в 2-метоксиэтаноле (2 л) добавляли формамидин (101 г; 0,96 моль) и реакционную смесь нагревали с обратным холодильником в течение ночи. Реакционную смесь концентрировали, разбавляли водой (1,5 л) и нейтрализовали (до рН 7) аммиаком. Смесь фильтровали и осадок промывали водой. Осадок сушили при пониженном давлении с получением промежуточного соединения (3) в виде коричневого твердого вещества (62 г; выход 67%). 1Н ЯМР (DMSO-d6, 400 МГц): δ 7.89 (s, 1Н), 7.36 (s, 1Н), 7.08 (s, 1Н), 3.88 (s, 3Н).
Промежуточное соединение 4
4-гидрокси-7-метоксихиназолин-6-илацетат

К суспензии промежуточного соединения 3 (52 г; 0,27 моль) и пиридина (53,6 г; 0,68 моль) в безводном DCM (1 л) по каплям добавляли хлорангидрид уксусной кислоты (52,9 г; 0,68 моль), и смесь перемешивали в течение ночи при комнатной температуре. Смесь выливали в воду (1 л) и несколько раз экстрагировали DCM. Объединенные органические слои промывали рассолом, сушили над Na2SO4, концентрировали с получением промежуточного соединения 4 в виде черного твердого вещества (63,2 г; выход 100%). 1Н ЯМР (DMSO-d6, 400 МГц): δ 8.62 (s, 1Н), 7.88 (s, 1Н), 7.37 (s, 1Н), 3.95 (s, 3Н), 2.74 (s, 3Н).
Промежуточное соединение 5
4-хлор-7-метоксихиназолин-6-илацетат
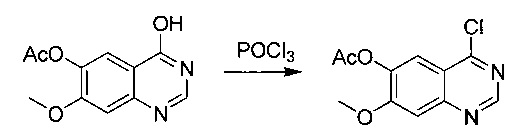
Суспензию промежуточного соединения 4 (75,6 г; 0,323 моль) в POCl3 (287 мл) нагревали до температуры дефлегмации в течение 0,5 часа. Реакционную смесь концентрировали и разбавляли DCM (500 мл), выливали в воду (500 мл), фильтровали и промывали DCM. Объединенные органические слои промывали рассолом, сушили над Na2SO4 и концентрировали. Очистка посредством хроматографии (РЕ/EtOAc, 1/1) давала промежуточное соединение (5) (55 г; выход 67%) в виде белого твердого вещества. 1Н ЯМР (CDCl3, 400 МГц): δ 8.95 (s, 1Н), 7.90 (s, 1Н), 7.43 (s, 1Н), 4.02 (s, 1Н), 2.39 (s, 1Н).
Промежуточное соединение 6
4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-илацетат
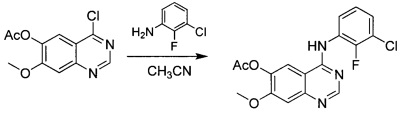
К суспензии промежуточного соединения 5 (100 г; 0,396 моль) в ацетонитриле (4 л) добавляли 2-фтор-3-хлоранилин (60,5 г; 0,416 моль) и реакционную смесь нагревали до 80°С в течение ночи. Осадок собирали посредством фильтрации и сушили под вакуумом с получением промежуточного соединения 6 (181 г; чистота 80%) в виде белого твердого вещества, которое использовали на следующей стадии непосредственно без очистки. 1Н ЯМР (DMSO-d6, 400 МГц): δ 8.93 (s, 1H), 8.82 (s, 1H), 7.67-7.63 (m, 1H), 7.59 (s, 1H), 7.56-7.52 (m, 1H), 7.39-7.35 (m, 1H), 4.02 (s, 3Н), 2.39 (s, 3Н).
Промежуточное соединение 7
4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ол

К раствору промежуточного соединения (6) (181 г; 0,396 моль) в МеОН (2 л) добавляли карбонат калия (138 г; 1 моль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали и твердое вещество промывали МеОН. Фильтрат концентрировали под вакуумом с получением промежуточного соединения 7 (280 г; чистота 60%, содержало карбонат калия). 1Н ЯМР (DMSO-d6, 400 МГц): δ 8.01 (s, 1Н), 7.61-7.58 (m, 1Н), 7.27-7.24 (m, 1Н), 7.17-7.13 (m, 1Н), 6.95 (s, 1Н), 6.83 (s, 1Н), 3.79 (s, 3Н).
Промежуточное соединение 8
трет-Бутил-(3R)-4-(хлоркарбонил)-3-метилпиперазин-1-карбоксилат
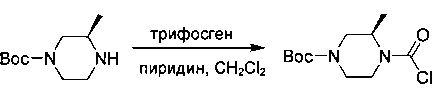
К смеси трифосгена (23 г; 75 ммоль) в безводном DCM (250 мл) по каплям при 0°С добавляли пиридин (18 г; 225 ммоль) с последующим добавлением трет-бутил-(3R)-3-метилпиперазин-1-карбоксилата (15 г; 75 ммоль). Смесь перемешивали в течение ночи при комнатной температуре. Анализ посредством ТСХ показал, что исходное вещество израсходовано. Смесь концентрировали с получением промежуточного соединения 8 в виде желтого твердого вещества, которое использовали без дополнительной очистки.
Промежуточное соединение 9
4-трет-Бутил-1-{4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил}-(2R)-2-метилпиперазин-1,4-дикарбоксилат

Смесь промежуточного соединения 7 (19,2 г; 60 ммоль), промежуточного соединения 8, полученного согласно вышеуказанной процедуре, и карбоната калия (16,6 г; 120 ммоль) в безводном DMF (диметилформамиде) (300 мл) перемешивали в течение ночи при комнатной температуре. Реакционную смесь выливали в воду (250 мл) и фильтровали, и осадок на фильтре сушили под вакуумом с получением промежуточного соединения 9 (25 г; выход 75%) в виде желтого твердого вещества. ВЭЖХ: tR составляет 2,68 мин в 10-80АВ_6 мин хроматографии (Ultimate ХВ-С18, 3,0*50 мм, 3 мкм). ЖХМС: tR составляет 0,792 мин в 5-95АВ_1,5 мин хроматографии (Welch Xtimate С18, 2,1*30 мм, 3 мкм), МС (ИЭР) m/z 546,0 [М+Н]+.
Промежуточное соединение 10
4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2-метилпиперазин-1-карбоксилат
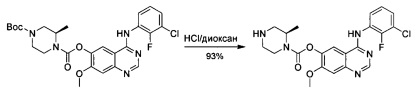
Смесь промежуточного соединения 9 (8,3 г; 15 ммоль) в DCM (100 мл) и смеси HCl/диоксан (10 мл; 4 М) перемешивали в течение 30 минут при комнатной температуре. После фильтрации твердое вещество собирали и повторно растворяли в воде, и затем значение рН доводили до 8 насыщенным NaHCO3. Осадок собирали и промывали CH2Cl2. Твердое вещество сушили под вакуумом с получением промежуточного соединения 10 (8 г; чистота 85%) в виде желтого твердого вещества. Этот неочищенный продукт использовали на следующей стадии без очистки.
Промежуточное соединение 11
(S)-2,4-диметилпиперазин-1-карбонилхлорид
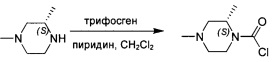
К раствору трифосгена (1,04 г; 3,5 ммоль) в DCM (20 мл) в атмосфере азота при 0°С по каплям добавляли пиридин (2,3 г; 28,0 ммоль) с последующим добавлением (S)-1,3-диметилпиперазина (800 мг; 7,0 ммоль) в DCM (30 мл), реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи, контролируя путем ТСХ (Rf составляет 0,9; РЕ: EtOAc, 1:1). Смесь концентрировали с получением промежуточного соединения 11 (3 г; неочищенное), которое использовали без очистки.
Промежуточное соединение 12
(±)-трет-Бутил-(4-(хлоркарбонил)-3-метилпиперазин-1-карбоксилат

К смеси трифосгена (23 г; 75 ммоль) в безводном DCM (250 мл) при 0°С по каплям добавляли пиридин (18 г; 225 ммоль) с последующим добавлением (±)-трет-бутил-3-метилпиперазин-1-карбоксилата (15 г; 75 ммоль). Смесь перемешивали в течение ночи при комнатной температуре. Анализ посредством ТСХ показал, что исходное вещество израсходовано. Смесь концентрировали с получением промежуточного соединения 12 в виде желтого твердого вещества, которое использовали без дополнительной очистки.
Промежуточное соединение 13
(±)-4-трет-Бутил-1-{4-[(2-фторфенил)амино]-7-метоксихиназолин-6-ил}-2-метилпиперазин-1,4-дикарбоксилат
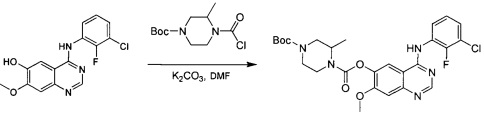
Смесь промежуточного соединения 7 (19,2 г; 60 ммоль), промежуточного соединения 12, полученного согласно вышеописанной процедуре, и карбоната калия (16,6 г; 120 ммоль) в безводном DMF (300 мл) перемешивали в течение ночи при комнатной температуре. Реакционную смесь выливали в воду (250 мл) и фильтровали, и осадок на фильтре сушили под вакуумом с получением промежуточного соединения (13) (25 г; выход 75%) в виде желтого твердого вещества. ВЭЖХ: tR составляет 2,68 мин в 10-80АВ_6 мин хроматографии (Ultimate ХВ-С18, 3,0*50 мм, 3 мкм). ЖХМС: tR составляет 0,792 мин в 5-95АВ_1,5 мин хроматографии (Welch Xtimate С18, 2,1*30 мм, 3 мкм), МС (ИЭР) m/z 546,0 [М+Н]+.
Промежуточное соединение 14:
(±)-4-[(3-Хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-2-метилпиперазин-1-карбоксилат

Смесь промежуточного соединения 13 (25 г; 46 ммоль) в виде раствора в смеси HCl/диоксан (250 мл; 4 М) перемешивали в течение 30 минут при комнатной температуре. Полученное твердое вещество собирали и повторно растворяли в воде, и затем значение рН доводили до 8 насыщенным NaHCO3. Осадок собирали и промывали CH2Cl2. Твердое вещество сушили под вакуумом с получением продукта (19 г; выход 93%) в виде желтого твердого вещества. ВЭЖХ: tR составляет 1,58 мин в 10-80АВ_6 мин хроматографии (Ultimate ХВ-С18, 3,0*50 мм, 3 мкм). ЖХМС: tR составляет 0,638 мин в 5-95АВ_1,5 мин хроматографии (Welch Xtimate С18, 2,1*30 мм, 3 мкм), МС (ИЭР) m/z 445.1 [М+Н]+. 1Н ЯМР (CD3OD, 400 МГц): δ 8.44 (s, 1Н), 8.08 (s, 1Н), 7.60 (t, 1Н), 7.39 (t, 1Н), 7.27-7.20 (m, 2Н), 4.41 (s, 1Н), 4.00 (s, 3Н), 3.08-2.79 (m, 4Н), 1.43 (brs, 3Н).
Пример 1
4-[(3-Хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат
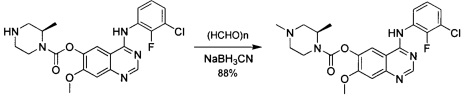
К смеси промежуточного соединения 10 (8 г; 15 ммоль; чистота 85%) и параформальдегида (1 г; 32 ммоль) в МеОН (100 мл) добавляли цианоборогидрид натрия (2 г; 32 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали под вакуумом, остаток разбавляли водой и экстрагировали EtOAc (3×100 мл). Объединенные органические слои промывали рассолом, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством препаративной ВЭЖХ с обращенной фазой (колонка: Synergi 77*250, 10 мкм, градиент: 5-35% В (А - вода/0,05% муравьиная кислота, В - ацетонитрил), скорость потока: 140 мл/мин). Фракцию, содержащую требуемый продукт, нейтрализовали насыщенным карбонатом калия и экстрагировали EtOAc. Объединенный органический слой концентрировали под вакуумом и подвергали лиофильной сушке с получением соединения по примеру 1 (4 г; выход 58% за 2 стадии) в виде белого твердого вещества.
ЖХ-МС: tR составляет 1,406 мин в 4 мин хроматографии, МС (ИЭР) m/z 460,0 [М+Н]+
ПАФХ: tR составляет 1,637 мин в 3 мин хроматографии (Chiralpak AD-3 50*4,6 мм внутр. диам., 3 мкм), МС (ИЭР) m/z 460,1 [М+Н]+
1Н ЯМР (CDCl3, 400 МГц): δ 8.76 (s, 1Н), 8.53-8.48 (m, 1Н), 7.65 (s, 1Н), 7.44 (brs, 1Н), 7.34 (s, 1Н), 7.19-7.15 (m, 2Н), 4.51-4.50 (m, 1Н), 4.20-4.05 (m, 1Н), 3.99 (s, 3Н), 3.50-3.30 (m, 1Н), 2.87 (d, 1Н), 2.73 (d, 1Н), 2.35 (s, 3Н), 2.35-2.25 (m, 1Н), 2.13-2.11 (m, 1Н), 1.47 (s, 3Н).
Пример 1, Форма А
Вещество в Форме А получали посредством нагревания соединения по примеру 1 до 140°С. Приблизительно 10 мг соединения по примеру 1 помещали в алюминиевый тигель. Тигель нагревали до 140°С при скорости нагревания 10°С/мин при использовании дифференциальной сканирующей калориметрии (ДСК) и затем охлаждали до комнатной температуры в атмосфере азота.
Вещество в Форме А также получали посредством медленного упаривания соединения по примеру 1 в IPA. Приблизительно 10 мг соединения по примеру 1 взвешивали во флаконе объемом 3 мл, добавляли 0,25 мл IPA для растворения твердого вещества. После упаривания при комнатной температуре в течение 24 часов получали соединение по примеру 1 (Форма А).
Вещество в Форме А также получали посредством суспендирования соединения по примеру 1 в МТВЕ в течение 24 часов при 50°С. Приблизительно 10 мг соединения по примеру 1 взвешивали во флаконе объемом 3 мл, добавляли 1 мл МТВЕ, и затем суспензию перемешивали в течение 24 часов при 50°C с получением соединения по примеру 1 (Форма А).
Вещество в Форме А также получали посредством добавления антирастворителя, смеси EtOAc/гептан. Приблизительно 10 мг соединения по примеру 1 взвешивали во флаконе объемом 5 мл, добавляли 1 мл EtOAc для растворения твердого вещества и медленно добавляли во флакон 4 мл антирастворителя - гептана. Смесь перемешивали в течение 24 часов при комнатной температуре с получением соединения по примеру 1 (форма А).
Спектры дифракции рентгеновских лучей на порошке для соединения по примеру 1 (Форма А) показали, что вещество является кристаллическим. Вещество имело точку плавления 192,4°С (начало).
Пример 1, Форма Ε
Приблизительно 10 мг соединения по примеру 1 взвешивали во флаконе объемом 5 мл, добавляли 0,25 мл THF для растворения твердого вещества, затем во флакон добавляли 4 мл антирастворителя - гептана, и смесь перемешивали в течение 24 часов при комнатной температуре перед выделением твердого вещества. Образец (Форма Е), как было определено посредством ДРЛП (дифракция рентгеновских лучей на порошке), являлся кристаллическим и имел точку плавления 194,2°С (начало).
Пример 1, Форма I
Приблизительно 10 мг соединения по примеру 1 взвешивали во флаконе объемом 3 мл, добавляли во флакон 1 мл H2O и суспензию перемешивали в течение 24 часов при 50°С перед выделением твердого вещества. Образец (Форма I), как было определено посредством ДРЛП, являлся кристаллическим и имел точку плавления 193,3°С (начало).
Пример 1. Форма J
Приблизительно 10 мг соединения по примеру 1 взвешивали во флаконе объемом 3 мл, добавляли во флакон 1 мл Н2О, и суспензию перемешивали в течение 24 часов при комнатной температуре перед выделением твердого вещества. Образец (Форма J), как было определено посредством ДРЛП, являлся кристаллическим и имел точку плавления 193,3°С (начало).
Пример 2
Гидрохлорид 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата
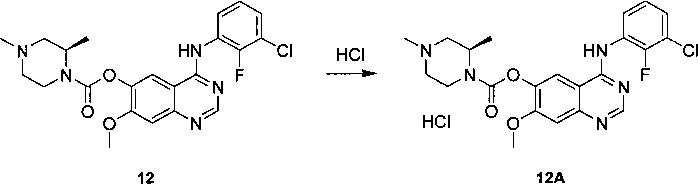
Соединение по примеру 1 (1,8 г) растворяли в ацетонитриле (5 мл), затем медленно добавляли 1 н HCl (5 мл), раствор сушили посредством лиофилизации с получением соединения по примеру 2 (1,93 г) в виде желтого твердого вещества. ЖХ-МС: tR составляет 1,355 мин в 4 мин хроматографии, МС (ИЭР) m/z 460.1 [М+Н]+. ПАФХ: tR составляет 1,773 мин в 3 мин хроматографии (Chiralpak AD-3 50*4,6 мм внутр. диам., 3 мкм), МС (ИЭР) m/z 460.1 [М+Н]+. 1Н ЯМР (CD3OD, 400 МГц): δ 8.55 (s, 1Н), 8.33-8.16 (m, 1Н), 7.56 (t, 1Н), 7.45 (t, 1Н), 7.33 (s, 1Н), 7.28-7.20 (m, 1Н), 4.81-4.59 (m, 1Н), 4.52-4.15 (m, 1Н), 4.10-3.95 (m, 3Н), 3.74-3.48 (m, 3Н), 3.35 (br. s., 1Н), 3.24-3.09 (m, 1Н), 2.97 (s, 3Н), 1.54 (br. s., 3Н). [α]D25=-14.96 (с10, DMSO).
Образование соли моногидрохлорида соединения по примеру 2, Формы A1
К приблизительно 10 мг соединения по примеру 1 добавляли 0,35 мл IPA с последующим добавлением 0,217 мл соляной кислоты. Раствор герметично закрывали крышкой и оставляли для перемешивания на магнитной мешалке. Во время перемешивания наблюдали некоторое количество белого осадка. Приблизительно через 24 часа образец отделяли и сушили при комнатной температуре под вакуумом. Эта форма (соль моногидрохлорид, Форма A1), как было определено посредством ДРЛП, являлась кристаллической и имела точку плавления 259,6°С (начало).
Соль моногидрохлорид, Форму A1, также получали посредством реакции кристаллизации соединения по примеру 1 и соляной кислоты в EtOH при комнатной температуре. К приблизительно 10 мг соединения по примеру 1 добавляли 0,35 мл EtOH для растворения твердого вещества, затем в раствор добавляли 0,217 мл соляной кислоты. Раствор герметично закрывали крышкой и оставляли для перемешивания на магнитной мешалке. Во время перемешивания наблюдали некоторое количество белого осадка. Приблизительно через 24 часа образец отделяли и сушили при комнатной температуре под вакуумом. Эта форма (соль моногидрохлорид, Форма Α1), как было определено посредством ДРЛП, являлась кристаллической и имела точку плавления 259,6°С (начало).
Соль моногидрохлорид, Форму A1, также получали посредством реакции кристаллизации соединения по примеру 1 и соляной кислоты в ацетоне при комнатной температуре. К приблизительно 10 мг соединения по примеру 1 добавляли 0,35 мл ацетона для растворения твердого вещества с последующим добавлением 0,217 мл соляной кислоты. Раствор герметично закрывали крышкой и оставляли для перемешивания на магнитной мешалке. Во время перемешивания наблюдали некоторое количество белого осадка. Приблизительно через 24 часа образец отделяли и сушили при комнатной температуре под вакуумом. Эта форма (соль моногидрохлорид, Форма A1), как было определено посредством ДРЛП, являлась кристаллической и имела точку плавления 259,6°С (начало).
Соль моногидрохлорид, Форму A1, также получали посредством реакции кристаллизации соединения по примеру 1 и соляной кислоты в THF при комнатной температуре. К приблизительно 10 мг соединения по примеру 1 добавляли 0,35 мл THF для растворения твердого вещества, затем добавляли 0,217 мл соляной кислоты. Раствор герметично закрывали крышкой и оставляли для перемешивания на магнитной мешалке. Во время перемешивания наблюдали некоторое количество белого осадка. Приблизительно через 24 часа образец отделяли и сушили при комнатной температуре под вакуумом. Эта форма (соль моногидрохлорид, Форма A1), как было определено посредством ДРЛП, являлась кристаллической и, как было видно, отличалась от ранее рассмотренных форм. Это вещество имело точку плавления 259,6°С (начало).
Пример 3
4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2S)-2,4-диметилпиперазин-1-карбоксилат

Раствор промежуточного соединения 7 (150 мг; 0,47 ммоль), промежуточного соединения 11 (1 г; неочищенное) и K2CO3 (130 мг; 0,94 ммоль) в N,N-диметилформамиде (10 мл) перемешивали при 30°С в течение ночи, контролируя посредством ЖХМС. Раствор фильтровали и очищали посредством препаративной ВЭЖХ с обращенной фазой (колонка: ASВ 150*25 мм*5 мкм, градиент: 3-28% В (А - вода/0,05% HCl, В - ацетонитрил), скорость потока: 30 мл/мин) с получением соединения по примеру 3 (21,0 мг). ЖХ-МС tR составляет 1,156 мин в 4 мин хроматографии, МС (ИЭР) m/z 460,0 [М+Н]+ ПАФХ: tR составляет 2,084 мин в 3 мин хроматографии (Chiralpak AD-3 50*4,6 мм внутр. диам., 3 мкм), МС (ИЭР) m/z 460,1 [М+Н]+; 1Н ЯМР (CD3OD, 400 МГц): δ 8.77 (s, 1Н), 8.43 (s, 1Н), 7.57-7.50 (m, 2Н), 7.38 (s, 1Н), 7.32-7.28 (m, 1Н), 4.51-4.21 (m, 1Н), 4.10 (s, 3Н), 3.77-3.35 (m, 5Н), 3.27-3.17 (m, 1Н), 2.99 (s, 3Н), 1.58-1.49 (m, 3Н).
Пример 4
4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(±)-2,4-диметилпиперазин-1-карбоксилат
Смесь промежуточного соединения 14 (1,0 г; 2,0 ммоль; чистота 96%), параформальдегида (200 мг; 6,6 ммоль), уксусной кислоты (400 мг; 6,6 ммоль) в МеОН (15 мл) перемешивали в течение 2 часов при комнатной температуре. Добавляли цианоборогидрид натрия (400 мг; 6,6 ммоль). Полученную реакционную смесь перемешивали в течение еще 2 часов. Смесь обрабатывали и очищали посредством препаративной ВЭЖХ с обращенной фазой (колонка: ASB, градиент: 5-30% В (А - вода/0,05% HCl, В - ацетонитрил), скорость потока: 30 мл/мин) с получением соединения по примеру 4 (300 мг; 27%) в виде белого твердого вещества. ЖХ-МС tR составляет 1,099 мин в 4 мин хроматографии, МС (ИЭР) m/z 460,1 [М+Н]+; 1Н ЯМР (CD3OD, 400 МГц): δ 8.79 (s, 1Н), 8.51 (s, 1Н), 7.58-7.52 (dd, 2Н), 7.45 (s, 1Н), 7.34-7.30 (t, 1Н), 4.71-4.30 (m, 2Н), 4.13 (s, 3Н), 3.75-3.58 (m, 3Н), 3.55-3.42 (m, 1Н), 3.27 (s, 1Н), 3.02 (s, 3Н), 1.62-1.53 (m, 3Н).
Пример 5
Альтернативные кристаллические формы 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата

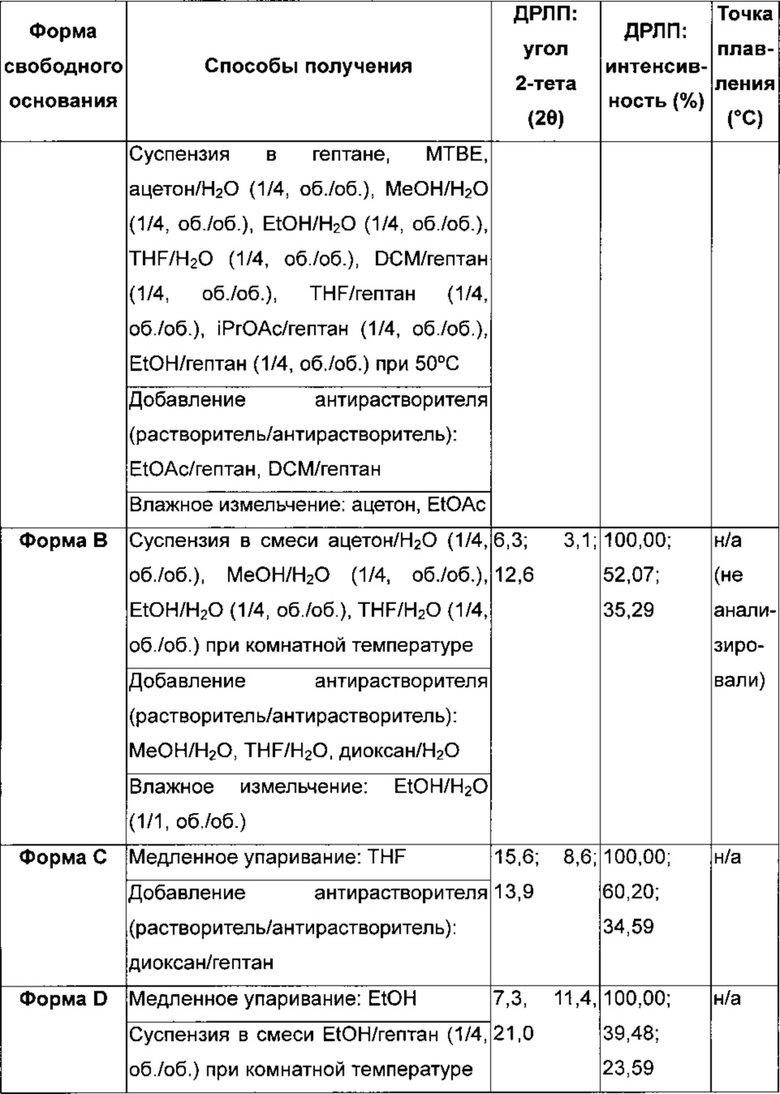
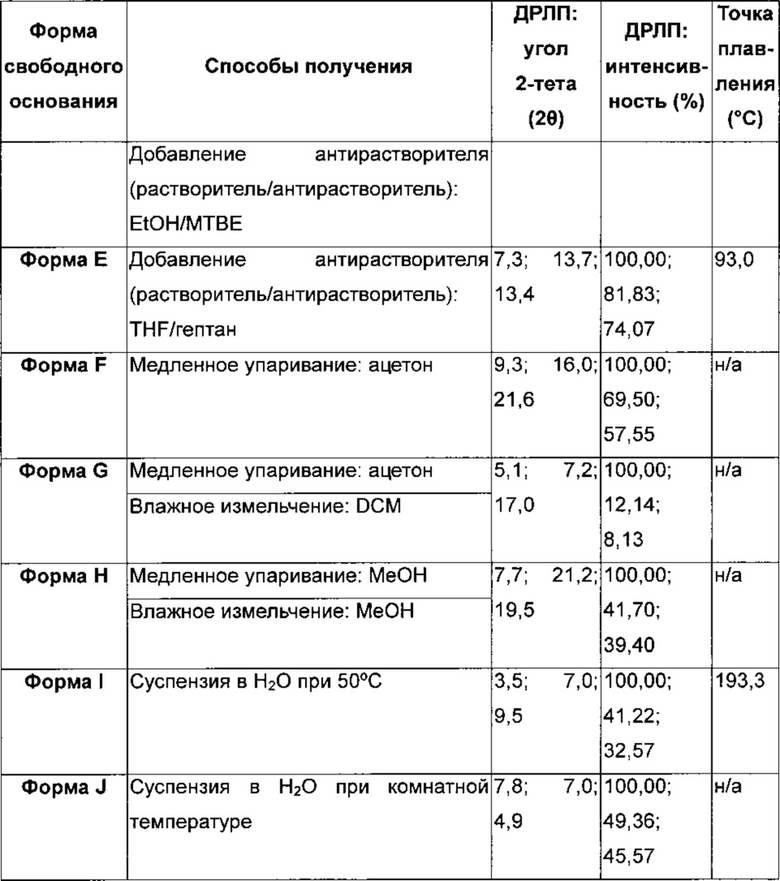
Пример 6
Альтернативные формы солей 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата
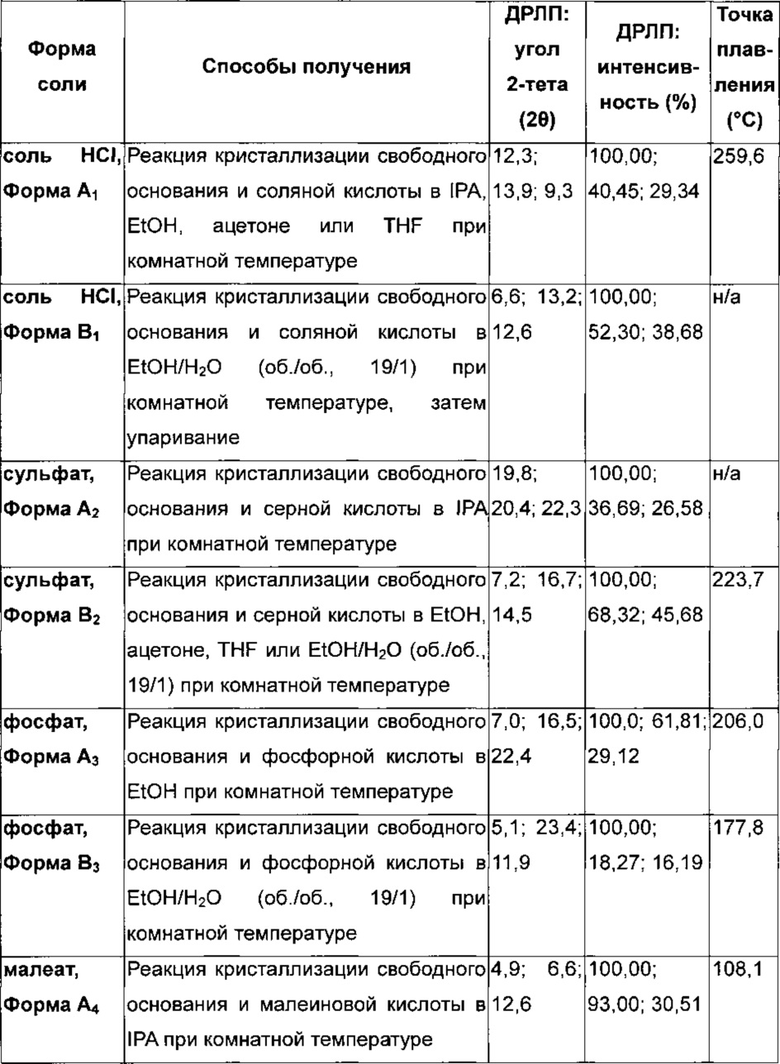
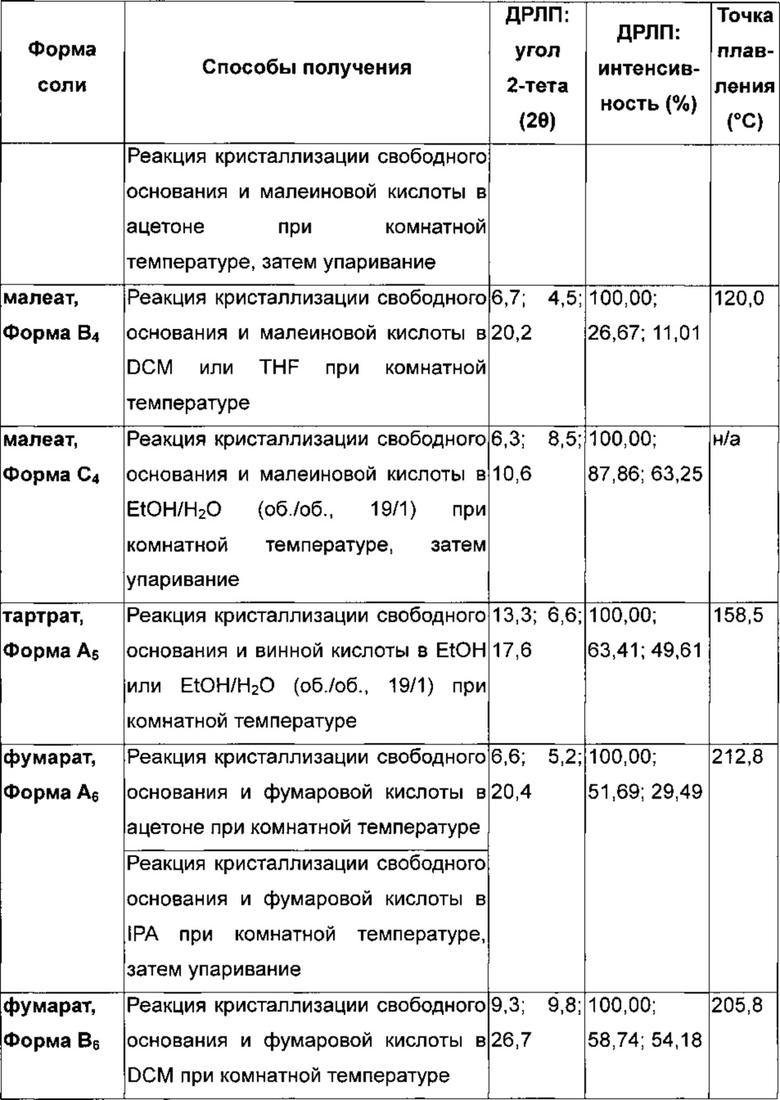

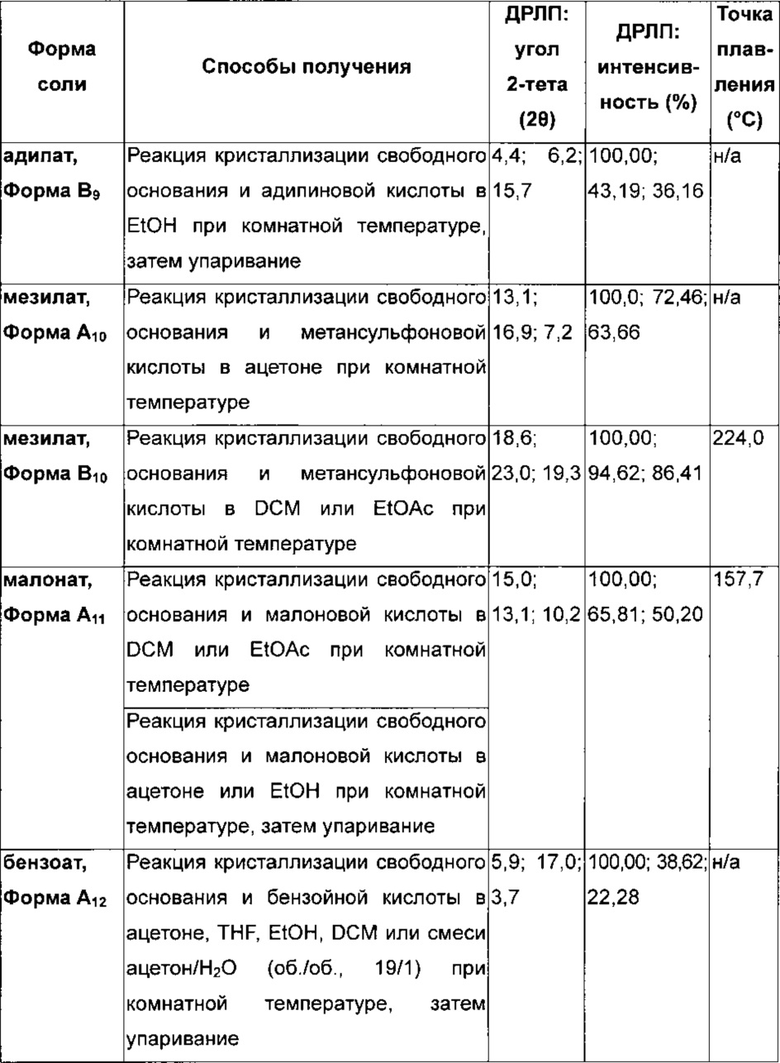

Пример 7
Сукцинат 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата
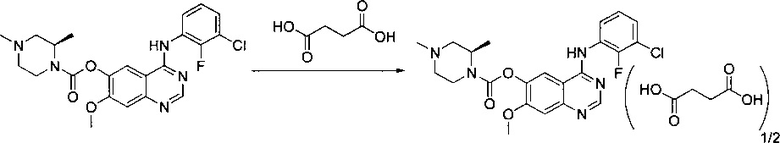
Соединение по примеру 1 (10 мг; 0,022 ммоль) растворяли в 0,44 мл ацетона во флаконе. В раствор добавляли янтарную кислоту (2,57 мг; 0,022 ммоль). Полученную смесь герметично закрывали крышкой и оставляли для перемешивания на магнитной мешалке. Во время перемешивания наблюдали некоторое количество белого осадка. Приблизительно через 24 часа белое твердое вещество отделяли и сушили при комнатной температуре под вакуумом. 1Н ЯМР (400 МГц, DMSO-d6) 9.74 (s, 1Н), 8.47 (s, 1Н), 8.22 (s, 1Н), 7.51-7.47 (m, 2Н), 7.34 (s, 1Н), 7.30-7.26 (m, 1Н), 4.4-4.2 (br, 1Н), 3.95 (s, 3Н), 3.9-3.7 (br, 1Н), 2.82-2.80 (d, 1Н), 2.70-2.67 (s, 1Н), 2.42 (s, 2Н), 2.21 (s, 3Н), 2.12-2.10 (m, 1Н), 1.94-1.89 (m, 1Н), 1.34 (s, 3Н).
Образование соединения по примеру 7, соль сукцинат, форма A8
Соль сукцинат, Форму A8, получали посредством процедуры, описанной выше. Эта форма (соль сукцинат, форма А8), как было определено посредством ДРЛП, являлась кристаллической и имела точку плавления 191,8°С (начало).
Соль сукцинат, Форму A8, также получали посредством реакции кристаллизации соединения по примеру 1 и янтарной кислоты в EtOH при комнатной температуре. К приблизительно 10 мг соединения по примеру 1 добавляли 0,59 мл EtOH для растворения твердого вещества, затем в раствор добавляли 2,57 мг янтарной кислоты. Раствор герметично закрывали крышкой и оставляли для перемешивания на магнитной мешалке. Во время перемешивания наблюдали некоторое количество белого осадка. Приблизительно через 24 часа перемешивания раствор упаривали досуха при комнатной температуре. Эта форма (соль сукцинат, форма A8), как было определено посредством ДРЛП, являлась кристаллической и имела точку плавления 191,8°С (начало).
Соль сукцинат, Форму А8, также получали посредством реакции кристаллизации соединения по примеру 1 и янтарной кислоты в DCM при комнатной температуре. К приблизительно 10 мг соединения по примеру 1 добавляли 0,25 мл DCM для растворения твердого вещества, затем добавляли 2,57 мг янтарной кислоты. Раствор герметично закрывали крышкой и оставляли для перемешивания на магнитной мешалке. Во время перемешивания наблюдали некоторое количество белого осадка. Приблизительно через 24 часа образец отделяли и сушили при комнатной температуре под вакуумом. Эта форма (соль сукцинат, форма А8), как было определено посредством ДРЛП, являлась кристаллической и имела точку плавления 191,8°С (начало).
Соль сукцинат, Форму А8, также получали посредством реакции кристаллизации соединения по примеру 1 и янтарной кислоты в EtOAc при комнатной температуре. К приблизительно 10 мг соединения по примеру 1 добавляли 0,25 мл EtOAc для растворения твердого вещества, затем добавляли 2,57 мг янтарной кислоты. Раствор герметично закрывали крышкой и оставляли для перемешивания на магнитной мешалке. Во время перемешивания наблюдали некоторое количество белого осадка. Приблизительно через 24 часа образец отделяли и сушили при комнатной температуре под вакуумом. Эта форма (соль сукцинат, форма A8), как было определено посредством ДРЛП, являлась кристаллической и имела точку плавления 191,8°С (начало).
| название | год | авторы | номер документа |
|---|---|---|---|
| ХРОМЕНОНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ РI3-КИНАЗЫ ДЛЯ ЛЕЧЕНИЯ РАКА | 2012 |
|
RU2598028C2 |
| Соединения и способы для ингибирования JAK | 2016 |
|
RU2760359C2 |
| СОЛИ ПРОИЗВОДНОГО ХИНАЗОЛИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2016 |
|
RU2720810C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ | 2014 |
|
RU2644769C2 |
| НОВЫЕ СОЕДИНЕНИЯ 951: БИФЕНИЛОКСИПРОПАНОВАЯ КИСЛОТА В КАЧЕСТВЕ МОДУЛЯТОРА CRTh2-РЕЦЕПТОРА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2472785C2 |
| ПОЛИМОРФЫ С-MET/HGFR ИНГИБИТОРА | 2006 |
|
RU2387650C2 |
| ПОЛИКРИСТАЛЛИЧЕСКАЯ ФОРМА СВОБОДНОГО ОСНОВАНИЯ ИЛИ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТЫ ИНГИБИТОРА EGFR, СПОСОБ ЕЁ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2733412C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОПИРИДО[3,4-b]ИНДОЛА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ РАКА | 2014 |
|
RU2664109C2 |
| НОВАЯ ГИДРОСУЛЬФАТНАЯ СОЛЬ | 2006 |
|
RU2418790C2 |
| Соединения циклопентана | 2019 |
|
RU2794997C2 |
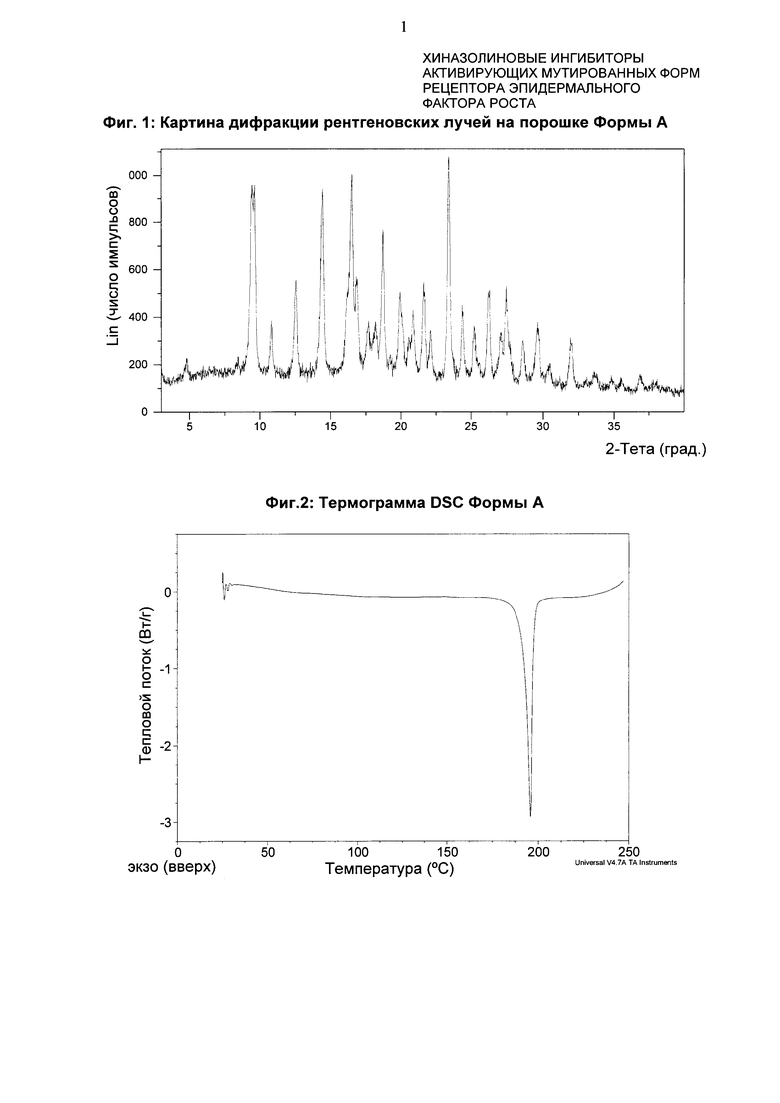
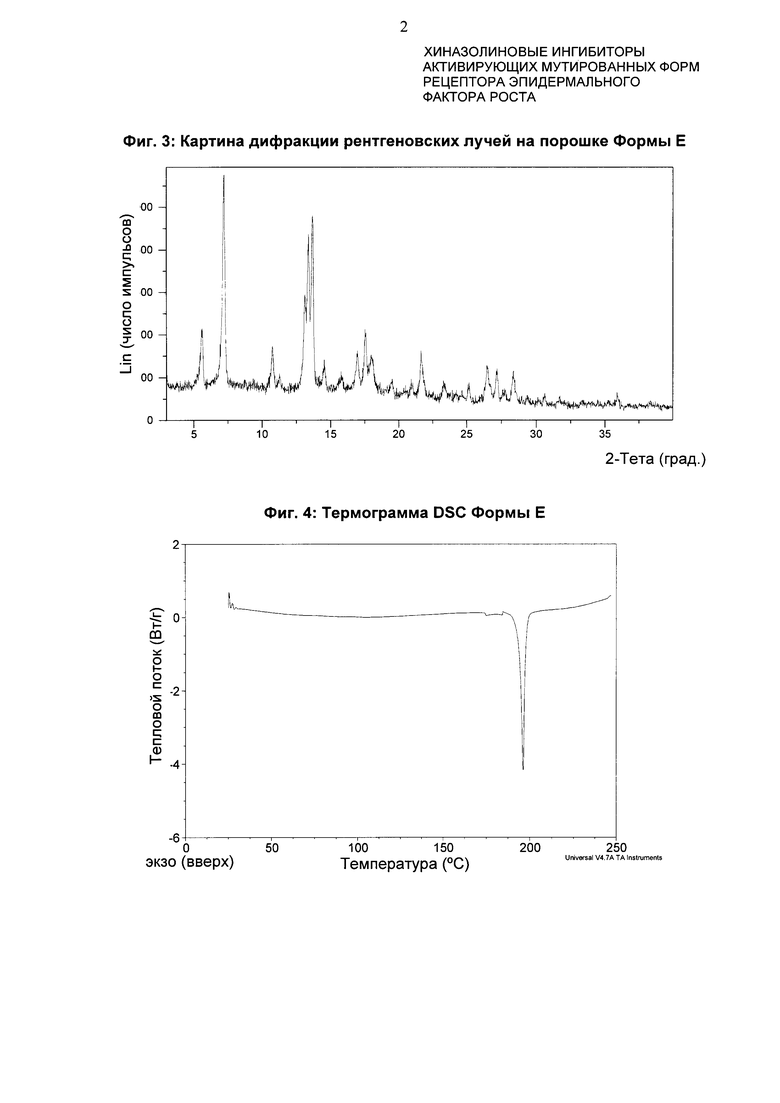

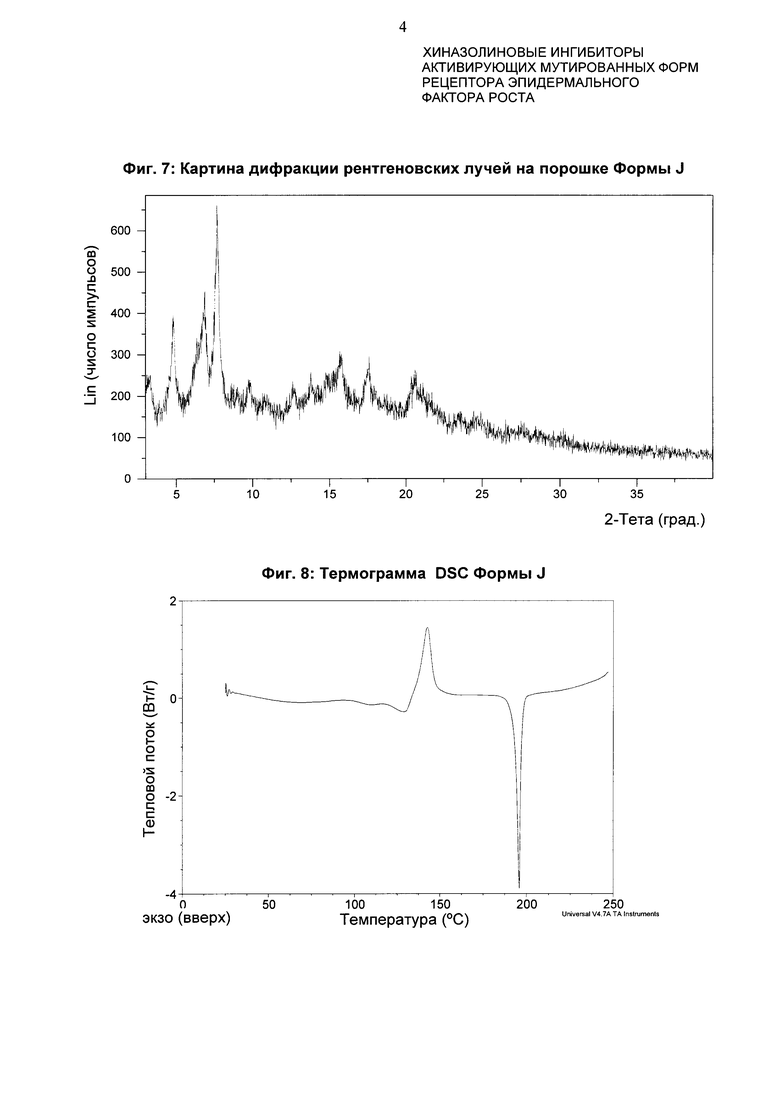
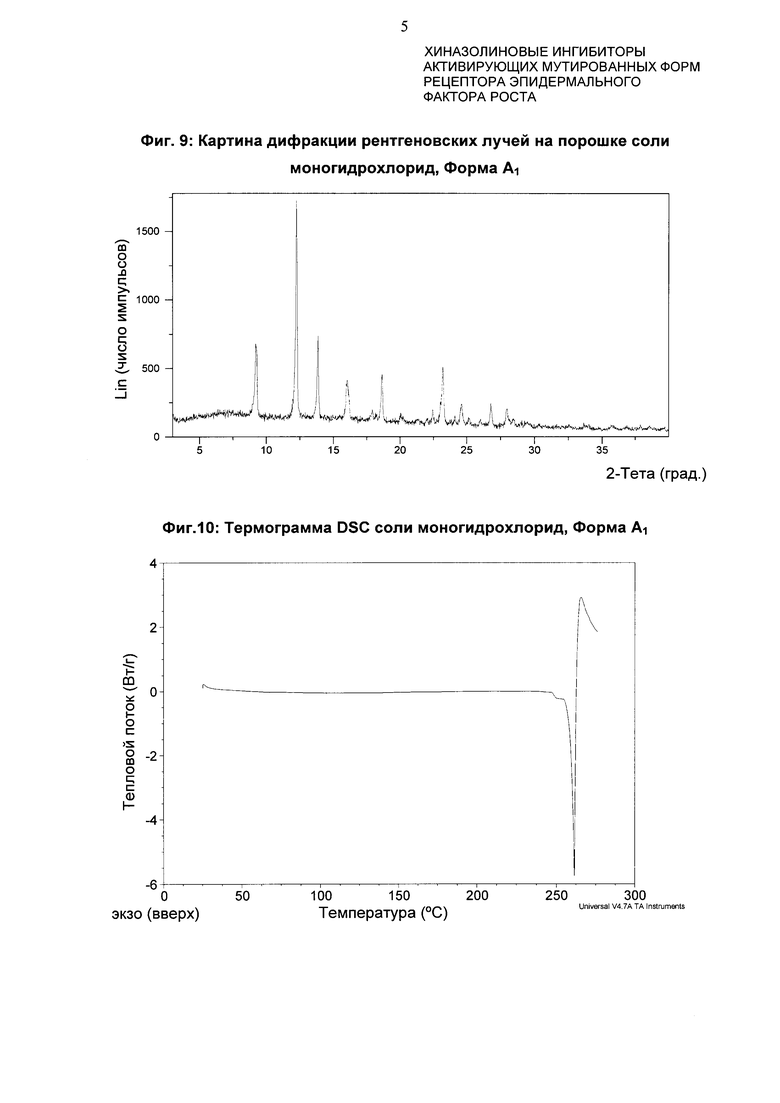

Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы (I) или его фармацевтически приемлемой соли. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I) или его фармацевтически приемлемой соли, их применению и способу лечения рака. Технический результат: получены новое соединение формулы (I) и его фармацевтически приемлемые соли, которые обладают ингибиторной активностью в отношении активирующих мутированных форм EGFR и полезны при лечении рака. 6 н. и 10 з.п. ф-лы, 10 табл., 12 ил., 7 пр.
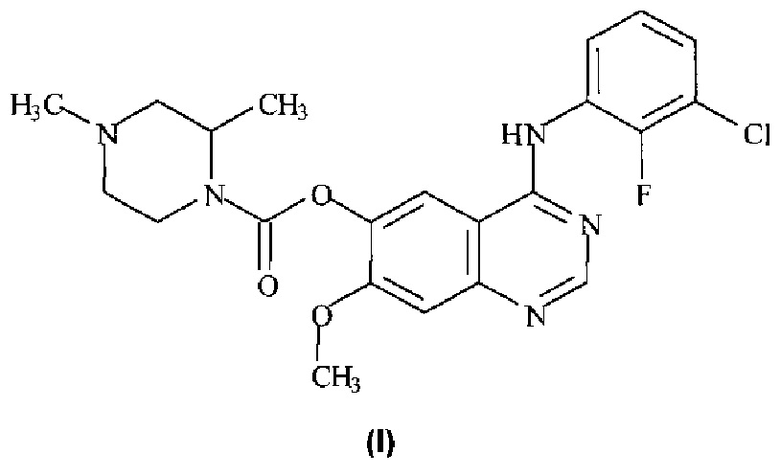
1. Соединение формулы (I):
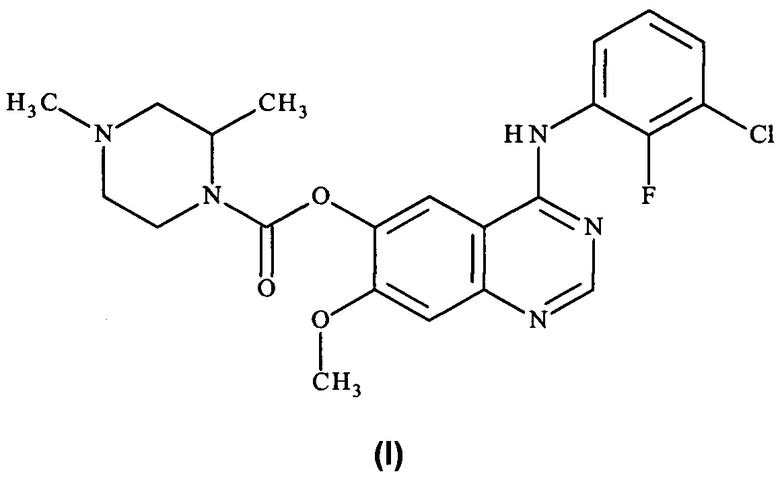
или его фармацевтически приемлемая соль.
2. Соединение формулы (I) по п.1, которое представляет собой 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилат.
3. Фармацевтически приемлемая соль соединения формулы (I):
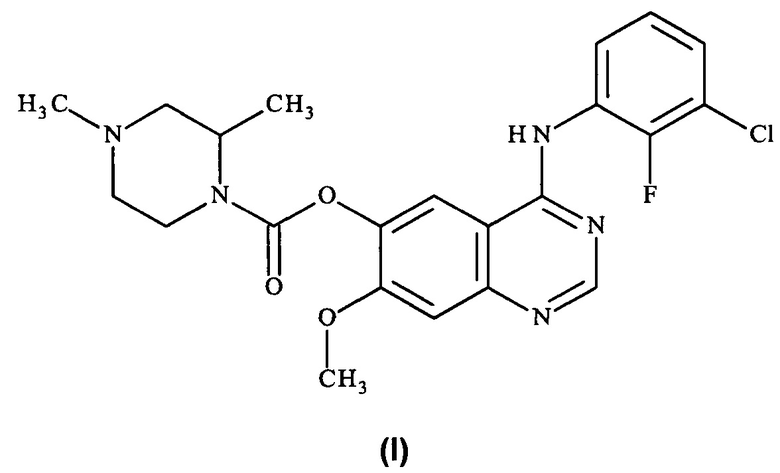
которая представляет собой гидрохлорид 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата.
4. Фармацевтически приемлемая соль соединения формулы (I):
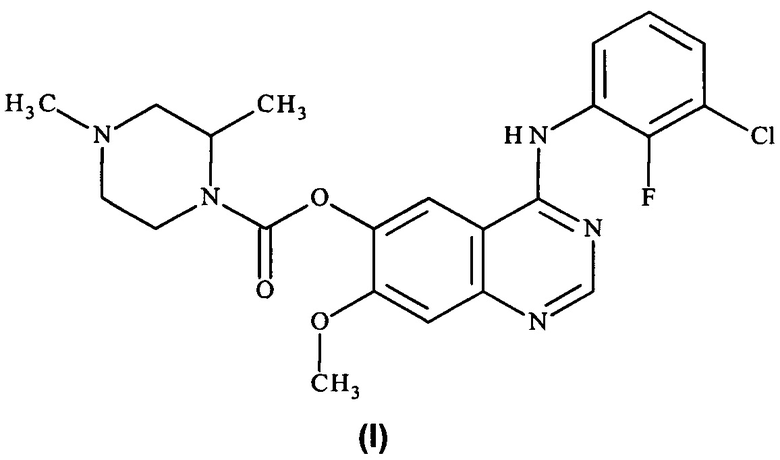
которая представляет собой сукцинат 4-[(3-хлор-2-фторфенил)амино]-7-метоксихиназолин-6-ил-(2R)-2,4-диметилпиперазин-1-карбоксилата.
5. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп.1-4 в кристаллической форме.
6. Фармацевтически приемлемая соль соединения формулы (I) по п.3 в кристаллической форме, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с двумя характеристическими пиками при значениях 2-тета, примерно равных 12,3 и 13,9°, где фармацевтически приемлемая соль представляет собой соль моногидрохлорид.
7. Фармацевтически приемлемая соль соединения формулы (I) по п.3 в кристаллической форме, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при значениях 2-тета, примерно равных 12,3; 13,9; 9,3; 23,3; 18,7; 16,0; 24,6; 26,8; 28,0°, где фармацевтически приемлемая соль представляет собой соль моногидрохлорид.
8. Фармацевтически приемлемая соль соединения формулы (I) по п.4 в кристаллической форме, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с тремя характеристическими пиками при значениях 2-тета, примерно равных 6,5; 17,7 и 14,7°, где фармацевтически приемлемая соль представляет собой соль сукцинат.
9. Фармацевтически приемлемая соль соединения формулы (I) по п.4 в кристаллической форме, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при значениях 2-тета, примерно равных 6,5; 17,7; 14,7; 9,2; 26,5; 20,2; 13,1; 27,3; 24,0°, где фармацевтически приемлемая соль представляет собой соль сукцинат.
10. Фармацевтическая композиция, обладающая ингибиторной активностью в отношении активирующих мутированных форм EGFR (рецептор эпидермального фактора роста), содержащая соединение формулы (I) или его фармацевтически приемлемую соль по любому из пп.1-9 вместе с фармацевтически приемлемым разбавителем или носителем.
11. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп.1-9 для применения в качестве лекарственного средства, обладающего ингибиторной активностью в отношении активирующих мутированных форм EGFR.
12. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп.1-9 в изготовлении лекарственного средства для ингибирования активирующего мутированного EGFR (рецептор эпидермального фактора роста) у теплокровного животного, такого как человек.
13. Способ лечения рака, опосредованного активирующим мутированным EGFR, у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп.1-9.
14. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп.1-9 для применения в лечении немелкоклеточного рака легкого.
15. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп.1-9 для применения в лечении метастатического немелкоклеточного рака легкого.
16. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп.1-9 в комбинации с противоопухолевым агентом, выбранным из:
(1) анти-CTLA-4 антитела;
(2) (2-гидрокси-этокси)-амида 6-(4-бром-2-хлор-фениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты или его фармацевтически приемлемой соли;
(3) анти-PD-L1 антитела;
(4) 1-[(1S)-1-(имидазо[1,2-а]пиридин-6-ил)этил]-6-(1-метил-1H-пиразол-4-ил)-1Н-[1,2,3]триазоло[4,5-b]пиразина или его фармацевтически приемлемой соли;
(5) анти-PD-1 антитела или
(6) агонистического антитела против ОХ40.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Патрон для электрической лампы с несколькими нитями накаливания | 1925 |
|
SU9300A1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ДОСТИЖЕНИЯ АНТИПРОЛИФЕРАТИВНОГО ЭФФЕКТА | 1996 |
|
RU2153495C2 |

