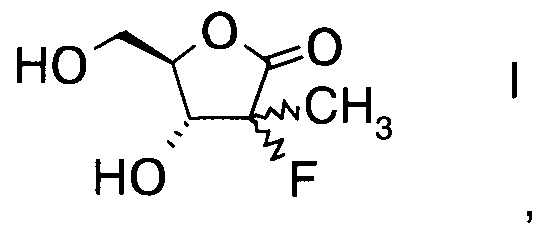
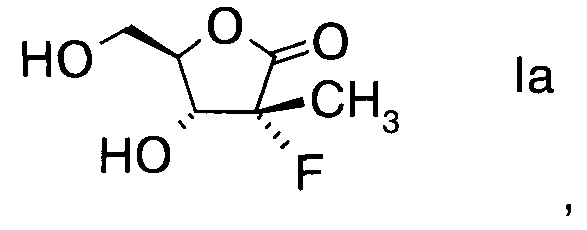
Настоящее изобретение относится к новому способу получения производного фторлакгона формулы
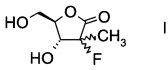 ,
,
и его ацилированного производного формулы
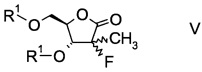 ,
,
в которой R1 обозначает гидрокси-протекторную группу.
Ацилированные фторлактоны формулы V, в частности бензоильное производное, в котором R1 = бензоил, представляют собой важные предшественники для синтеза пролекарственных соединений, которые потенциально являются сильными ингибиторами NS5B-полимеразы Вируса гепатита С (HCV, от англ. Hepatitis С Virus) (РСТ Int. Publ. WO 2007/065829).
Целью настоящего изобретения явился поиск селективного и масштабируемого синтеза получения фторлакгона формулы I и его ацилированных производных формулы V.
Эту цель можно достичь с помощью синтеза по настоящему изобретению, описанного ниже.
Способ по настоящему изобретению включает получение производного фторлакгона формулы
включая следующие стадии:
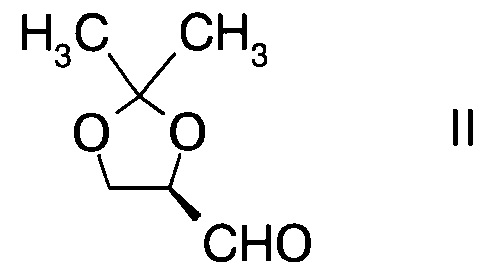
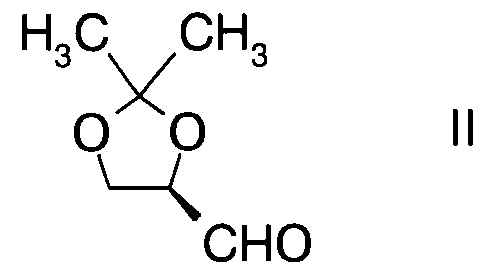
а) введение в реакцию альдегида формулы
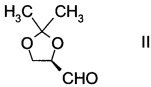
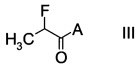
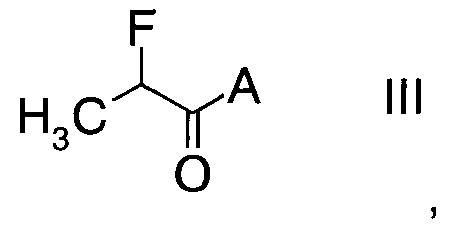
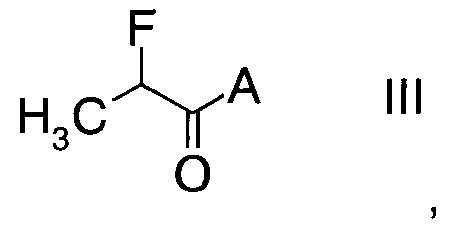
с производным фторпропионата формулы
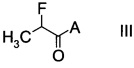 ,
,
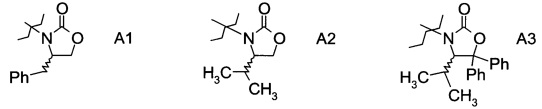
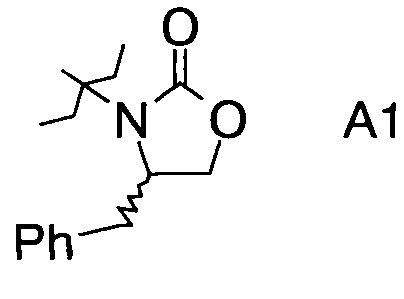
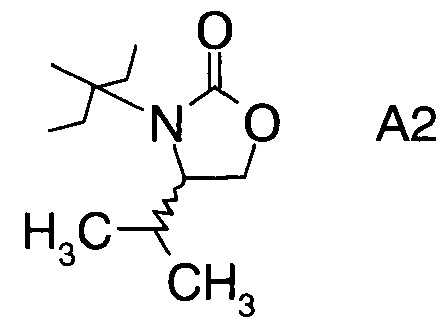
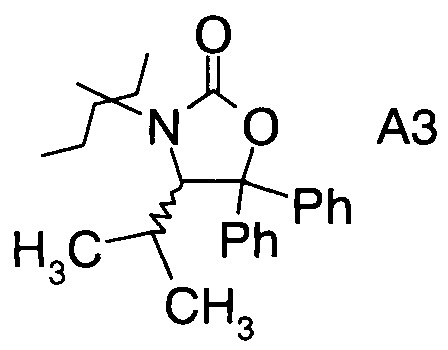
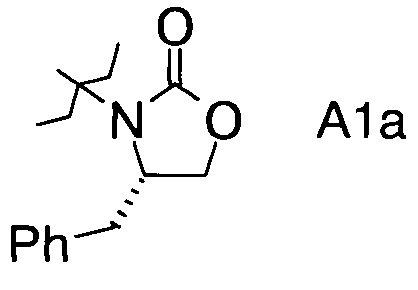
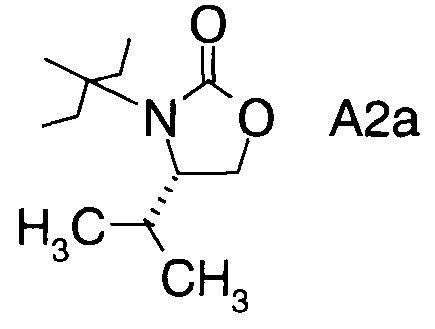
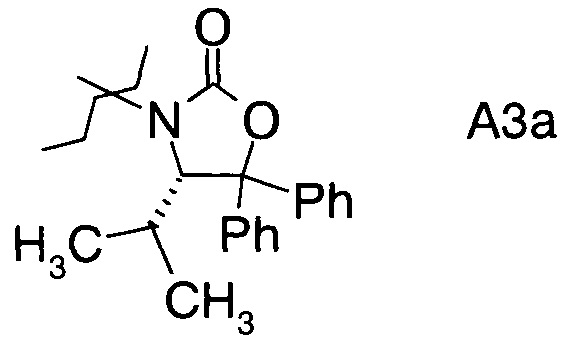
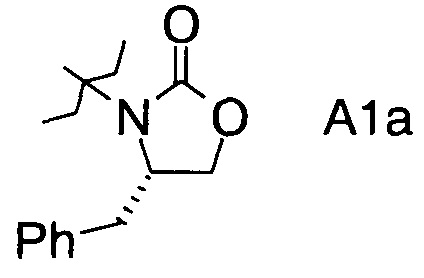
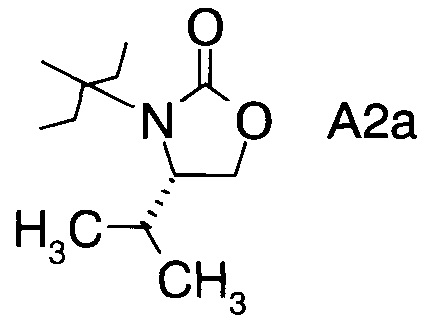
в которой А выбран из хиральных фрагментов
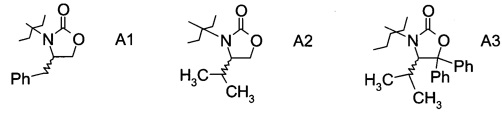 ,
,
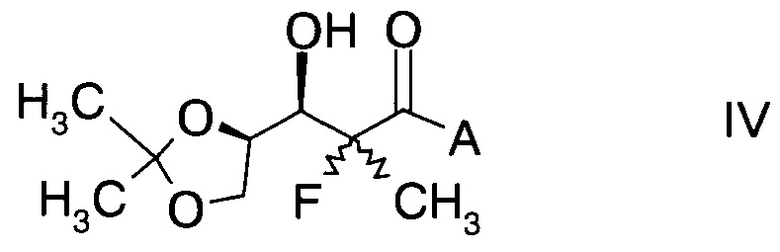
и Ph обозначает фенил, с образованием альдольного аддукта формулы
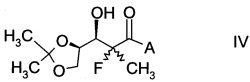 ,
,
в которой А определен выше; и
b) проведение гидролиза альдольного аддукта формулы IV с получением производного фторлакгона формулы I.
Термин "гидрокси-протекторная группа", используемый для заместителя R1, обозначает любые заместители, стандартно применяемые для блокировки реакционной способности гидроксигруппы. Подходящие гидрокси-протекторные группы описаны в литературе (Green Т., "Protective Groups in Organic Synthesis", Chapter 1, John Wiley and Sons, Inc., 1991, 10-142) и их можно, например, выбирать из бензоила, ацетила, триметилсилила, трет-бутила, трет-бутилдиметилсилила или дигидропиранила, в частности бензоила.
Волнистая линия  обозначает хиральную связь,
обозначает хиральную связь,  или
или  .
.
Стадия а)
Стадия а) требует проведения реакции альдегида формулы II с производным фторпропионата формулы III с образованием альдольного аддукта формулы IV.
D-глицеральдегид ацетонид представляет собой альдегид формулы II и доступен в продаже.
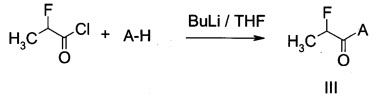
Производные фторпропионата формулы III можно получить в ходе превращения 2-фторпропионилхлорида с помощью, например, бутиллития в тетрагидрофуране, при температуре в интервале от -50°C до -10°C по ниже следующей схеме.
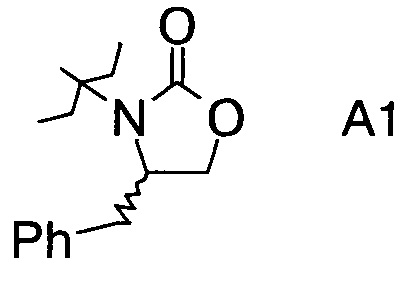
Хиральные амины А-Н, в которых А выбран из хиральных фрагментов

либо имеются в продаже, либо их можно получить в соответствии с ниже следующими схемами:
Схема 1a: (A-H, A=A1)
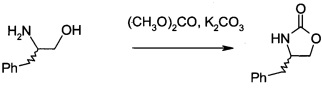
Схема 1b: (A-H, A=A2)
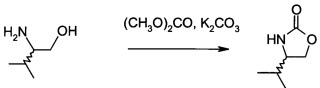
Схема 1c: (A-H, A=3)
В конкретном воплощении используется производное фторпропионата формулы III, в котором А представляет собой A3.
Еще в одном воплощении заместитель А в производном фторпропионата формулы III выбран из хиральных фрагментов.
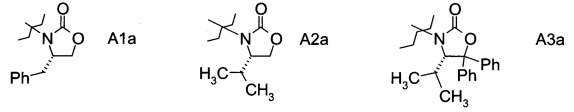
и в частности из А3а.
Производные фторпропионата формулы III
в которых А выбран из хиральных фрагментов
где Ph обозначает фенил, до сих пор не были описаны в данной области техники, и поэтому представляют собой частные воплощения настоящего изобретения.
В частном случае производное фторпропионата формулы III А выбрано из
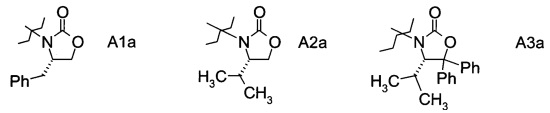
и в частности из А3а.
Реакцию проводят в присутствии катализатора, выбранного из дибутилборона трифторметансульфоната, хлорида титана, трихлорида изопропоксида титана (IV), изопропоксида титана, хлорида магния, трифлата магния или хлорида цинка.
В частности, используют дибутилборон трифторметансульфонат.
Как правило, присутствует основание, такое как третичный амин, выбранный из 2,6-лутидина, 2,3-лутидина, 2,4-лутидина, 2,5-лутидина, 3,4-лутидина, 3,5-лутидина, триэтиламина, диизопропилэтиламина, диэтиламина, пиридина или 1,6-диметилпиридина, и подходящий органический растворитель, такой как метиленхлорид, 1,2-дихлорэтан, хлороформ, ацетонитрил, толуол, ксилол, хлорбензол, тетрагидрофуран, 2-метилтетрагидрофуран или метил-изобутиловый эфир или их смеси.
В частности, подходящий третичный амин представляет собой 2,6-лутидин и в частности подходящий органический растворитель представляет собой метиленхлорид.
Температуру реакции обычно выдерживают в интервале от -78°C до 50°C. Конечный альдольный аддукт формулы IV можно получить из реакционной смеси способами, известными специалисту в данной области техники, в частности путем добавления воды к реакционной смеси, отделения органической фазы и удаления растворителя. Дальнейшую очистку можно проводить путем кристаллизации из раствора метиленхлорида с гексаном.
Альдольные аддукты формулы IV не известны в данной области техники и, таким образом, составляют конкретные воплощения настоящего изобретения.
В частности, альдольные аддукты имеют формулу
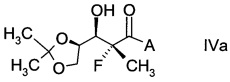 ,
,
в которой хиральный фрагмент А определен выше и выбран из А1, А2 или A3, в частности из А1а, А2а или А3а и в частном случае из А3а.
Стадия b)
Стадия b) требует проведения гидролиза альдольного аддукта формулы IV с получением производного фторлакгона формулы I.
В конкретном воплощении используют альдольный аддукт формулы IVa с предпочтениями, выделенными выше, с получением производного фторлакгона формулы
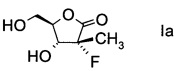 .
.
Гидролиз, как правило, осуществляют с помощью окислителя, выбранного из пероксида водорода, мета-хлорпероксибензойной кислоты, гипохорита натрия, перхлората натрия или этиленоксида, в присутствии гидроксида щелочного металла, выбранного из гидроксида лития, натрия или калия в качестве основания.
Обычно в качестве подходящего окислителя выбирают пероксид водорода, а в качестве основания в частности используют водный раствор гидроксида лития.
Гидролиз обычно проводят при температуре реакции в интервале от -30°С до 50°С.
В частном воплощении настоящего изобретения хиральные фрагменты А отщепляются при гидролизе и их можно извлечь в форме соответствующих хиральных аминов формул А-Н
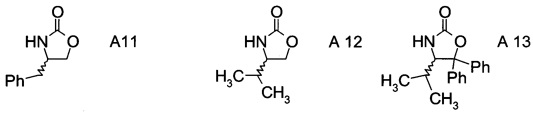
или, в частности, формул
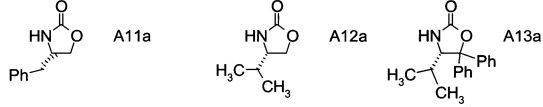 .
.
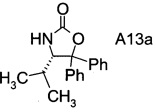
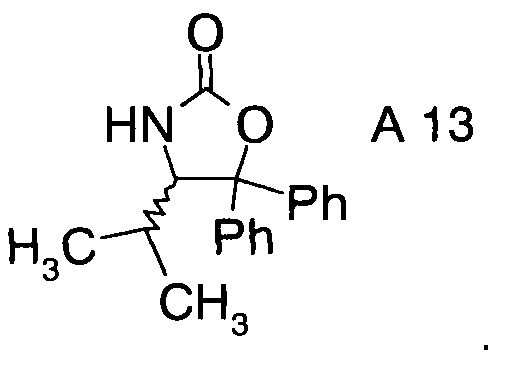
Хиральные амины А-Н можно выделить и получить из реакционной смеси способами, известными специалисту в данной области техники, например в случае хирального амина А13а, простой фильтрацией из реакционной смеси.
Еще в одном воплощении настоящего изобретения производное фторлакгона формулы I ацилируют с образованием ацилированного фторлакгона формулы
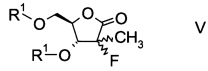
в которой R1 обозначает гидрокси-протекторную группу.
Ацилирование осуществляют с помощью подходящего ацилирующего агента, выбранного из бензоилхлорида, ацетилхлорида, пивалоилхлорида, триметилсилилхлорида, трет-бутилдиметилсилилхлорида или 3,4-дигидро-2Н-пирана, в частности бензоилхлорида, в присутствии третичного амина, такого как триэтиламин, диизопропилэтиламин, 2,3-лутидин, 2,4-лутидин, 2,5-лутидин, 3,4-лутидин, 3,5-лутидин, пиридин, 1,6-диметилпиридин или 1,8-диаза-бицикло[5.4.0]ундец-7-ен или их смеси, в частности триэтиламина.
Обычно в качестве катализатора добавляют 4-(диметиламино)-пиридин.
Может присутствовать подходящий органический растворитель, такой как тетрагидрофуран, 2-метилтетрагидрофуран, дихлорметан, 1,2-дихлорэтан, ацетонитрил, толуол, ксилол, метил-изобутилкетон, метил-трет-бутиловый эфир или ацетон, в частности тетрагидрофуран, и температуру реакции, как правило, поддерживают в интервале от -20°C до 80°C.
В конкретном воплощении ацилированный фторлактон имеет формулу
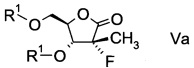 ,
,
в которой R1 обозначает гидрокси-протекторную группу.
Подходящая гидрокси-протекторная группа R1 представляет собой бензил, ацетил, триметилсилил, трет-бутил, трет-бутил-диметилсилил или дигидропиранил, в частности представляет собой бензоил.
Ацилированный фторлактон можно выделить из реакционной смеси способами, известными специалисту в данной области техники, например путем экстракции реакционной смеси подходящим органическим растворителем, таким как метил-трет-бутиловый эфир, и путем удаления растворителя.
ПРИМЕРЫ
Аббревиатура:
Исходные вещества
А. Получение (R)-2,2-диметил-1,3-диоксолан-4-карбальдегида

В сосуд вносили метиленхлорид (400 г), насыщенный раствор NaHCO3 (21 г) и (1S,2S)-1,2-бис(2,2-диметил-1,3-диоксолан-4-ил)этан-1,2-диол (55 г; 0,21 моль). Добавляли NaIO4 (64 г; 0.30 моль) шестью порциями за 60 мин, выдерживая при этом температуру реакции при 20-25°C. Затем реакционную смеси перемешивали при 20-25°C в течение 5 ч, анализ с помощью ТСХ показал завершение реакции. Реакционную смесь фильтровали для удаления твердого остатка, и отделенный водный слой экстрагировали метиленхлоридом (140 г). Объединенные органические слои высушивали над безводным сульфатом магния (40 г) при 0~5°C в течение 3 ч, затем фильтровали для удаления Mg2SO4 и промывали метиленхлоридом (50 г). Фильтрат концентрировали при пониженном давлении досуха. Остаток перегоняли при пониженном давлении и собирали фракцию при 40°C/1 кПа. Получали (R)-2,2-диметил-1,3-диоксолан-4-карбальдегид (23 г; 0,18 моль, выход 42%).
В. Получение хиральных аминов
В1. Получение (S)-4-бензилоксазолидин-2-она
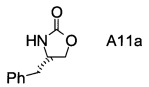
В сосуд вносили (S)-фенилаланол (25 г; 0,17 моль), безводный карбонат калия (2,3 г; 0,017 моль) и диметилкарбонат (30 г; 0,33 моль). Эту смесь нагревали до 80°С. Приемник дистиллята охлаждали на ледяной бане, и из реакционной смеси собирали метанол (ок. 13,5 мл) в течение 4,5 ч. Масляную баню убирали, когда отгонка метанола прекращалась. Полученный светло-желтый остаток охлаждали до комнатной температуры и разбавляли с помощью 125 мл этилацетата. Этот раствор переносили в делительную воронку и промывали водой (125 мл). Органическую фазу высушивали над безводным сульфатом магния, фильтровали и концентрировали на роторном испарителе, с получением белого кристаллического твердого вещества. Это неочищенное твердое вещество вносили в горячий раствор 2:1 этилацетата/гексана (100 мл) и проводили горячую фильтрацию. Фильтрат доводили до комнатной температуры (КТ), и из раствора кристаллизовалось твердое вещество, давая (S)-4-бензилоксазолидин-2-он (А11а) (23 г; выход 78,5%).
В2. Получение (S)-4-изопропилоксазолидин-2-она
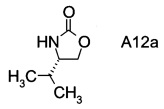
В сосуд вносили L-валинол (5 г; 48,5 ммоль), безводный карбонат калия (0,67 г; 4,85 ммоль) и диметилкарбонат (8,7 г; 96,7 ммоль). Эту смесь нагревали до 80°C. Приемник дистиллята охлаждали на ледяной бане, и из реакционной смеси собирали метанол (ок. 3,8 мл) в течение 4,5 ч. Масляную баню убирали, когда отгонка метанола прекращалась. Полученный светло-желтый остаток охлаждали до комнатной температуры, и разбавляли с помощью 30 мл этилацетата. Этот раствор переносили в делительную воронку и промывали водой (25 мл). Органическую фазу высушивали над безводным сульфатом магния, фильтровали и концентрировали на роторном испарителе с получением белого кристаллического твердого вещества. Это неочищенное твердое вещества вносили в горячий раствор 1:1 этилацетата/гексана (20 мл) и проводили горячую фильтрацию. Фильтрат доводили до КТ, и из раствора кристаллизовалось твердое вещество, давая (S)-4-изопропилоксазолидин-2-он (А12а) (5,0 г; выход 80,0%).
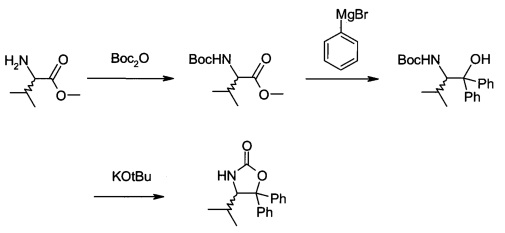
В3. Получение (S)-4-изопропил-5,5-дифенилоксазолидин-2-она
В сосуд вносили L-валин-метиловый эфир гидрохлорид (50 г; 0,30 моль) и 200 г метанола. Вносили триэтиламин (37,5 мл; 0,27 моль) и температуру реакции выдерживали <25°C. Затем вносили ди-трет-бутилдикарбонат (68,3 г; 0,31 моль). После этого вносили по каплям триэтиламин (63,7 мл; 0.46 моль), выдерживая температуру реакции <25°C. Эту смесь перемешивали при 25°C в течение 2 ч до тех пор, пока ТСХ не показала отсутствие исходного вещества. Затем удаляли метанол при пониженном давлении. Вносили метил-изобутиловый эфир (200 мл), а затем добавляли воду (150 мл) и после этого смесь перемешивали в течение 30 мин. Слои разделяли, водную фазу экстрагировали метил-трет-бутиловым эфиром (100 мл). Объединенные органические фазы промывали раствором NaCl (150 мл), высушивали с помощью Na2SO4 (10 г), затем фильтровали и фильтрат концентрировали при пониженном давлении досуха. Boc-L-валин-метиловый эфир (74,5 г) получали в виде неочищенного продукта, который можно было непосредственно использовать в последующей реакции.
В сосуд вносили бромбензол (158,6 г; 1,0 моль) и THF (500 мл) для приготовления раствора бромбензол/THF и заполняли им капельную воронку. В колбу вносили Mg (27,1 г; 1,1 моль) и THF (100 мл), затем 1/10 раствора бромбензола/THF, вносили небольшую гранулу I2 и нагревали до 60°C для инициации реакции. Вносили оставшийся раствор бромбензола/THF при такой скорости, чтобы поддерживать мягкий отток флегмы реакционной смеси. Продолжали нагревать с обратным холодильником еще в течение 1 ч, затем реакционную смесь охлаждали до 0°C. Далее добавляли по каплям раствор Вос-L-валин-метилового эфира (74,5 г; 0,30 моль) в THF (75 мл), выдерживая температуру реакции <3°C. По окончании внесения раствор нагревали до 20°C в течение 1 ч и выдерживали при 20°C в течение 15 ч. После охлаждения до 0°C вносили насыщенный раствор NH4Cl (200 мл), эту смесь затем перемешивали в течение 30 мин, а затем разделяли фазы. Водную фазу экстрагировали этилацетатом (2×250 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (150 мл), высушивали над Na2SO4 (20 г) и фильтровали. Фильтрат концентрировали досуха при пониженном давлении. К остатку добавляли этилацетат (250 мл) и смесь нагревали до температуры флегмы для растворения твердого вещества. Добавляли гексан (250 мл) при >60°C и эту смесь затем охлаждали до 0°C за 2 ч и перемешивали при 0°C в течение 1 ч. Твердое вещество затем отделяли фильтрацией, промывали с помощью 70 мл гексана и высушивали в вакууме с получением Вос-аминол-((S)-2-(Вос-амино)-3-метил-1,1-дифенил-1-бутанола) в виде белого твердого вещества (84 г). Маточный раствор концентрировали примерно до 70 г. Затем вносили гексан (70 мл) для кристаллизации. Вторую партию твердого вещества собирали фильтрацией и промывали небольшим количеством гексана. Твердое вещество высушивали в вакууме и получали 12,5 г твердого вещества.
В сосуд вносили Вос-аминол (96 г; 0,27 моль) и THF (1500 мл) и эту смесь охлаждали до 0°C. Вносили одной порцией трет-бутоксид калия (36,3 г; 0,32 моль) и смесь перемешивали при 0°C в течение 3 ч. По завершении реакции (анализ ТСХ) смесь вливали в 10% раствор NH4Cl (2000 мл) и перемешивали в течение 10 мин. Твердое вещество отфильтровывали, промывали водой (4×400 мл) и затем растворяли в метаноле (500 мл). Раствор нагревали до температуры флегмы в течение 1 ч, затем охлаждали до 15~20°C и перемешивали в течение 1 ч. Эту суспензию фильтровали и осадок на фильтре промывали метанолом (100 мл). Полученное твердое вещество высушивали в вакууме при 38°C и получали 75 г (S)-4-изопропил-5,5-дифенилоксазолидин-2-она (А13а).
С. Получение фторпропионатов формулы III
С1. (4S)-3-(2-Фторпропаноил)-4-бензилоксазолидин-2-он (Формула III, А = А1а)
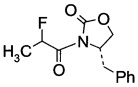
В сосуд вносили (S)-4-бензилоксазолидин-2-он (10 г; 0,056 моль) и THF (200 мл). Раствор охлаждали до -70°C, затем добавляли по каплям 2,7 M раствор н-бутиллития (27,5 мл; 0,074 моль), поддерживая при этом температуру реакции <-70°C. Реакционную смесь затем перемешивали в течение 30 мин, затем добавляли по каплям 2-фторпропионилхлорид (10,1 г; 0,095 моль), поддерживая при этом температуру реакции при <-70°C. Затем реакционную смесь нагревали до 15~20°C в течение 3 ч, а затем перемешивали при 15-20°C еще в течение 60 мин. По завершении реакции добавляли 10% раствор NH4Cl (60 мл) и эту смесь перемешивали в течение 30 мин. Фазы разделяли и водную фазу экстрагировали трет-бутиловым эфиром (30 мл). Объединенные органические слои промывали насыщенным раствором NaCl, высушивали с помощью Na2SO4, фильтровали и выпаривали при пониженном давлении для удаления растворителей. Остаток очищали хроматографией на колонке (элюент: НЕ/ЕА=3/1 об/об.) с получением (4S)-3-(2-фторпропаноил)-4-бензилоксазолидин-2-она (12,1 г; выход 85%) в виде светло-желтого масла.
1Н-ЯМР (CDCl3, 400 МГц): δ 7,28 (m, 5Н), 6,05 (m, 1Н), 4,74 (m, 1Н), 4,31 (m, 2Н), 4,39 (m, 1Н), 2,87 (m, 1Н), 1,65 (m, 3Н).
С2. (4S)-3-(2-Фторпропаноил)-4-изопропилоксазолидин-2-он (Формула III, А = А2а)

В сосуд вносили (S)-4-изопропилоксазолидин-2-он (3,8 г; 0,029 моль) и THF (75 мл). Этот раствор охлаждали до -60°C, затем добавляли по каплям 2,7 М раствор н-бутиллития (18 мл; 0,049 моль), поддерживая при этом температуру реакции <-50°C. Реакционную смесь затем перемешивали при -50°C в течение 30 мин, затем добавляли по каплям 2-фторпропионилхлорид (6,3 г; 0,057 моль), поддерживая при этом температуру реакции <-50°C. Температуру реакции затем повышали до 15~20°C в течение 3 ч, и после этого перемешивали при 15~20°C еще в течение 60 мин. По завершении реакции вносили 10% раствор NH4Cl (30 мл) и реакционную смесь перемешивали в течение 30 мин. Фазы разделяли и водную фазу экстрагировали метил-трет-бутиловым эфиром (30 мл). Объединенные органические слои промывали насыщенным раствором NaCl (30 мл). Органическую фазу высушивали над Na2SO4, фильтровали и выпаривали при пониженном давлении для удаления растворителей. Остаток очищали хроматографией на колонке (элюент: НЕ/ЕА=3/1 об/об.) с получением (4S)-3-(2-фторпропаноил)-4-изопропилоксазолидин-2-она (2,9 г; выход 48%) в виде светло-желтого масла.
1H-ЯМР (CDCl3, 400 МГц): δ 6,00 (m, 1Н), 4,39 (m, 3Н), 2,43 (m, 1Н), 1,61 (m, 3Н), 0,91 (m, 6Н).
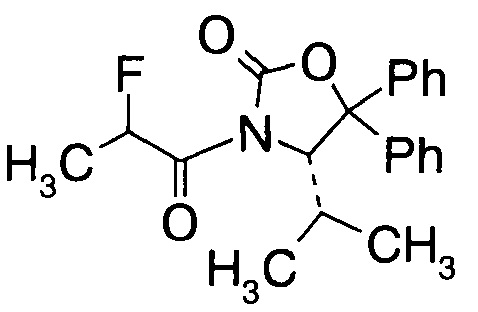
С3. (4S)-3-(2-Фторпропаноил)-4-изопропил-5,5-дифенилоксазолидин-2-он (Формула III, А = А3а)
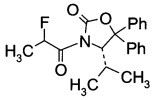
В сосуд вносили (S)-4-изопропил-5,5-дифенилоксазолидин-2-он (70 г; 0,25 моль) и THF (500 мл). Этот раствор охлаждали до -50°C, затем добавляли по каплям 2,5 М раствор н-бутиллития (120 мл; 0,30 моль) поддерживая при этом температуру реакции <-40°C. По окончании внесения температуру реакции повышали до -10°C в течение 3 ч, и перемешивали при -10°C в течение 30 мин. Реакционную смесь охлаждали до -78°C, добавляли по каплям 2-фторпропионилхлорид (41 г; 0,37 моль) при такой скорости, чтобы температуру реакции выдерживать <-60°C. По окончании внесения реакционную смесь нагревали до 15~20°C в течение 3 ч и перемешивали при 15~20°C в течение 60 мин. Вносили 10% раствор NH4Cl (350 мл) и смесь перемешивали в течение 30 мин. Фазы разделяли и водный слой экстрагировали метил-трет-бутиловым эфиром (500 мл). Объединенные органические слои промывали насыщенным раствором NaCl (150 мл). Органическую фазу высушивали над Na2SO4, фильтровали и выпаривали при пониженном давлении для удаления растворителей. Остаток растворяли в метиленхлориде (70 мл) нагреванием смеси, затем добавляли 210 мл гексана в течение 30 мин. Полученную суспензию охлаждали до 0°C за 2 ч и перемешивали при 0°C в течение 60 мин. Суспензию фильтровали и твердое вещество высушивали в вакууме. Получали (4S)-3-(2-фторпропаноил)-4-изопропил-5,5-дифенилоксазолидин-2-он (74,5 г; выход 84,2%) в виде светло-желтого твердого вещества.
1Н-ЯМР (CDCl3, 400 МГц): δ 7,36 (m, 10Н), 5,96 (m, 1Н), 5.49 (d, J=3,2 Гц, 0,5Н), 5,31 (d, J=3,2 Гц, 0,5Н), 2,02 (m, 1Н), 1,73 (dd, J=23,6, 6,8 Гц, 1,5Н), 1,15 (dd, J=23,6, 6,8 Гц, 1,5Н), 0,83 (m, 6Н).
D. Получение альдольного аддукта формулы IV
D1. (S)-3-((2R,3R)-3-((R)-2,2-диметил-1,3-диоксолан-4-ил)-2-фтор-3-гидрокси-2-метилпропаноил)-4-бензилоксазолидин-2-он (Формула IV, А = А1а)
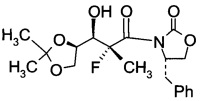
В сосуд вносили (4S)-3-(2-фторпропаноил)-4-бензилоксазолидин-2-он (2,9 г; 0,0115 моль) и метиленхлорид (20 мл) и эту смесь охлаждали до 0°C. Вносили дибутилборон трифторметансульфонат (Bu2BOTf, 1М в CH2Cl2), (17,3 мл; 0,0173 моль) и реакционную смесь перемешивали при 0°C в течение 10 мин. Затем добавляли 2,6-лутидин (2.47 г; 0,0231 моль), поддерживая при этом температуру реакции при 0°C в течение 15 мин. После этого реакционную смесь нагревали до 15~20°C и перемешивали в течение 4 ч. Эту смесь затем охлаждали до -78°C, после чего добавляли по каплям 2,3-О-изопропилиден-D-глицеральдегид (2,25 г; 0,0173 моль), поддерживая при этом температуру реакции <-65°C. Затем эту смесь нагревали до 0°C в течение 5 ч и далее перемешивали при 0°C еще в течение 1 ч. Добавляли воду (30 мл), эту смесь перемешивали в течение 30 мин, затем разделяли слои. Органический слой промывали водой (30 мл), высушивали над Na2SO4 (10 г), фильтровали и концентрировали при пониженном давлении для удаления растворителя. Остаток очищали хроматографией на колонке (элюент: НЕ/ЕА=3/1 об/об ), с получением указанного в заголовке продукта (S)-3-((2R,3R)-3-((R)-2,2-диметил-1,3-диоксолан-4-ил)-2-фтор-3-гидрокси-2-метилпропаноил)-4-бензилоксазолидин-2-она (3,5 г; выход 79,5%) в виде светло-желтого масла.
1Н-ЯМР (CDCl3, 400 МГц): δ 7,29 (m, 5Н), 4,63 (m, 2Н), 4,22 (m, 4Н), 4,04 (dd, J=8, 6,4 Гц, 1Н), 3,56 (dd, J=13,2, 2,8 Гц, 1Н), 2,65 (dd, J=13,2, 10,8 Гц, 1Н), 1,86 (d, J=23,2 Гц, 3Н), 1,42 (s, 3Н), 1,33 (s, 3Н).
D2. (S)-3-((2R,3R)-3-((R)-2,2-диметил-1,3-диоксолан-4-ил)-2-фтор-3-гидрокси-2-метилпропаноил)-4-изопропилоксазолидин-2-он (Формула IV, А = А2а)
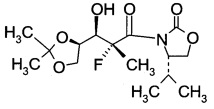
В сосуд вносили (4S)-3-(2-фторпропаноил)-4-изопропилоксазолидин-2-он (1,5 г; 7,4 ммоль) и метиленхлорид (20 мл) и эту смесь охлаждали до 0°C. Вносили дибутилборон трифторметансульфонат (Bu2BOTf, 1 M в CH2Cl2), (11,0 мл; 11 ммоль) и реакционную смесь перемешивали при 0°C в течение 10 мин. Затем добавляли 2,6-лутидин (1,6 г; 14,9 ммоль), поддерживая при этом температуру реакции при 0°C в течение 15 мин. Далее реакционную смесь нагревали до 15-20°C и перемешивали в течение 4 ч. Эту смесь затем охлаждали до -78°C, после чего добавляли по каплям 2,3-O-изопропилиден-D-глицеральдегид (1,5 г; 11,5 моль), поддерживая при этом температуру реакции <-65°С. Затем эту смесь нагревали до 0°C в течение 5 ч и далее перемешивали при 0°C еще в течение 1 ч. Добавляли воду (16 мл), эту смесь перемешивали в течение 30 мин, затем слои разделяли. Органический слой промывали водой (16 мл), высушивали над Na2SO4 (10 г), фильтровали и концентрировали при пониженном давлении для удаления растворителя. Остаток очищали хроматографией на колонке (элюент: НЕ/ЕА=3/1 об/об.) с получением указанного в заголовке продукта (S)-3-((2R,3R)-3-((R)-2,2-диметил-1,3-диоксолан-4-ил)-2-фтор-3-гидрокси-2-метилпропаноил)-4-изопропилоксазолидин-2-она (2,2 г; выход 89,8%) в виде светло-желтого масла.
1Н-ЯМР (CDCl3, 400 МГц): δ 4,50 (m, 1Н), 4,46 (m, 1Н), 4,35 (m, 1Н), 4,28 (m, 1Н), 4,20 (m, 1Н), 4,10 (m, 1Н), 4,01 (m, 1Н), 2,94 (d, J=6,4Гц, 1Н), 2,45 (m, 1Н), 1,81 (d, J=23,2 Гц, 3Н), 1,40 (s, 3Н), 1,35 (s, 3Н), 0,96 (m, 6Н).
D3. (S)-3-((2R,3R)-3-((R)-2,2-диметил-1,3-диоксолан-4-ил)-2-фтор-3-гидрокси-2-метил-пропаноил)-4-изопропил-5,5-дифенилоксазолидин-2-он (Формула IV, А = А3а)
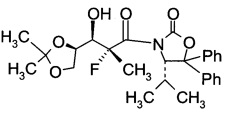
В сосуд вносили (4S)-3-(2-фторпропаноил)-4-изопропил-5,5-дифенил-оксазолидин-2-он (90 г; 0,25 моль) и метиленхлорид (720 мл) и эту смесь охлаждали до 0°C. Вносили дибутилборон трифторметансульфонат (Bu2BOTf, 1 M в CH2Cl2), (378 мл; 0,38 моль) и реакционную смесь перемешивали при 0°C в течение 10 мин. Затем добавляли 2,6-лутидин (55,8 г; 0,52 моль), поддерживая при этом температуру реакции при 0°C в течение 15 мин. Далее реакционную смесь нагревали до 15~20°C и перемешивали при этой температуре в течение 24 ч. Эту смесь затем охлаждали до -78°C, после чего добавляли по каплям 2,3-О-изопропилиден-D-глицеральдегид (54,6 г; 0,42 моль), поддерживая при этом температуру реакции <-65°C. Затем эту смесь нагревали до 0°C в течение 5 ч и перемешивали при 0°C еще в течение 1 ч. Добавляли воду (450 мл), эту смесь перемешивали в течение 30 мин, затем слои разделяли. Органический слой промывали водой (450 мл), высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении для удаления растворителя. Остаток растворяли в метиленхлориде (90 мл) при нагревании до температуры флегмы. Затем добавляли по каплям гексан (270 мл) для инициации кристаллизации. Полученную суспензию охлаждали до 10°C в течение 2 ч, перемешивали при 10°C в течение 1 ч, фильтровали и осадок на фильтре промывали гексаном (90 мл) и высушивали в вакууме. Получали (S)-3-((2R,3R)-3-((R)-2,2-диметил-1,3-диоксолан-4-ил)-2-фтор-3-гидрокси-2-метилпропаноил)-4-изопропил-5,5-дифенилоксазолидин-2-он с выходом 79 г (64,3%).
1Н-ЯМР (CDCl3, 400 МГц): δ 7,38 (m, 10Н), 5,30 (d, J=3,2 Гц, 1Н), 4,63 (m, 1Н), 4,00 (dd, J=12,8, 6,4 Гц, 1Н), 3,68 (m, 1Н), 3,70 (m, 1Н), 2,20 (d, J=6,8 Гц, 1Н), 1,47 (d, J=23,2 Гц, 3Н), 1,39 (s, 3Н), 1,33 (s, 3Н), 0,98 (d, J=7,2 Гц, 3Н), 0,86 (d, J=6,8 Гц, 3H).
Пример 1
Получение (3R,4R)-3-фтор-4-гидрокси-5-(гидроксиметил)-3-метилдигидро-фуран-2(3H)-она (из альдольного аддукта в Примере D1)
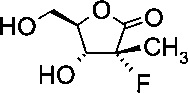
В сосуд вносили (S)-3-((2R,3R)-3-((R)-2,2-диметил-1,3-диоксолан-4-ил)-2-фтор-3-гидрокси-2-метилпропаноил)-4-бензилоксазолидин-2-он (3,3 г; 0,0086 моль), THF (10 мл) и воду (2 мл) и этот раствор охлаждали до 0°C. Добавляли пероксид водорода 50% (2,36 г; 0,035 моль), а затем раствор гидроксида лития моногидрата (0,3 г; 0,014 моль) в воде (15 мл), поддерживая при этом температуру реакции <5°C. Эту смесь затем перемешивали при 0~5°C в течение 1 ч. По завершении реакции (анализ ТСХ) добавляли раствор сульфита натрия (5,50 г) в воде (60 мл) при <10°C. Значение рН реакционной смеси доводили до 6,5-7,0 добавлением 10% водного раствора HCl. Затем THF удаляли при пониженном давлении и к остатку добавляли метиленхлорид (20 мл). Слои разделяли, водный слой экстрагировали метиленхлоридом (20 мл) и водный слой выпаривали при пониженном давлении досуха. К остатку добавляли THF (20 мл) и твердое вещество фильтровали. Осадок на фильтре промывали с помощью THF (10 мл) и объединенные фильтраты концентрировали досуха. Остаток снова разводили в THF (20 мл) и эту смесь перемешивали в течение 30 мин и фильтровали. Осадок на фильтре снова промывали с помощью THF (10 мл) и объединенные фильтраты концентрировали при пониженном давлении досуха. Затем к остатку добавляли этанол (15 мл) и 0,3 мл HCl (0,3 мл) и эту смесь нагревали до 70°C и перемешивали в течение 5 ч. Эту смесь концентрировали досуха и очищали хроматографией на колонке (элюент: CH2Cl2/MeOH = 1/1 об/об), указанный в заголовке продукт собирали (1,2 г) и кристаллизовали из CH2Cl2/MeOH=20/1 об/об. с получением чистого указанного в заголовке продукта (3R,4R)-3-фтор-4-гидрокси-5-(гидроксиметил)-3-метил-дигидрофуран-2(3Н)-она (1,0 г; выход 70%).
1Н-ЯМР (D2O, 400 МГц): δ 4,55 (m, 1Н), 4,19 (dd, J=21,2, 7,6 Гц, 1Н), 4,04 (dd, J=13,2, 1,6 Гц, 1Н), 3,81 (dd, J=13,2, 4,8 Гц, 1Н), 1,62(d, J=24,4 Гц, 3Н).
Пример 2
Получение (3R,4R)-3-фтор-4-гидрокси-5-(гидроксиметил)-3-метилдигидро-фуран-2(3Н)-она (из альдольного аддукта примера D2)
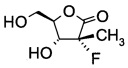
В сосуд вносили (S)-3-((2R,3R)-3-((R)-2,2-диметил-1,3-диоксолан-4-ил)-2-фтор-3-гидрокси-2-метилпропаноил)-4-изопропилоксазолидин-2-он (2,2 г; 0,0066 моль, THF (10 мл) и воду (2 мл) и этот раствор охлаждали до 0°C. Добавляли пероксид водорода 50% (1,75 г; 0,026 моль), а затем раствор лития гидроксида моногидрата (0,2 г; 0,011 моль) в воде (10 мл), поддерживая при этом температуру реакции <5°C. Эту смесь затем перемешивали при 0~5°С в течение 1 ч. По завершении реакции (анализ ТСХ) добавляли раствор сульфита натрия (3,6 г) в воде (35 мл) при <10°C. Значение рН реакционной смеси доводили до 6,5-7,0 добавлением 10% водного раствора HCl. Затем THF удаляли при пониженном давлении и к остатку добавляли метиленхлорид (15 мл). Слои разделяли, водный слой экстрагировали метиленхлоридом (20 мл) и этот водный слой выпаривали при пониженном давлении досуха. К остатку добавляли THF (15 мл) и твердое вещество фильтровали. Остаток на фильтре промывали с помощью THF (10 мл) и объединенные фильтраты концентрировали досуха. Остаток снова разводили в THF (20 мл) и эту смесь перемешивали в течение 30 мин и фильтровали. Остаток на фильтре снова промывали с помощью THF (10 мл) и объединенные фильтраты концентрировали при пониженном давлении досуха. Затем к остатку добавляли этанол (15 мл) и 0,3 мл HCl (0,3 мл) и эту смесь нагревали до 70°C и перемешивали в течение 5 ч. Эту смесь концентрировали досуха, и очищали хроматографией на колонке (элюент: CH2Cl2/MeOH=1/1 об/об), собирали указанный в заголовке продукт (3R,4R)-3-фтор-4-гидрокси-5-(гидроксиметил)-3-метилдигидрофуран-2(3Н)-он (0,72 г; выход 67%).
Пример 3
Получение (3R,4R)-3-фтор-4-гидрокси-5-(гидроксиметил)-3-метилдигидро-фуран-2(3Н)-она (из альдольного аддукта примера D3)
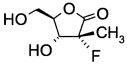
В сосуд вносили (S)-3-((2R,3R)-3-((R)-2,2-диметил-1,3-диоксолан-4-ил)-2-фтор-3-гидрокси-2-метилпропаноил)-4-изопропил-5,5-дифенилоксазолидин-2-он (75 г; 0,155 моль) THF (375 мл) и воду (95 г) и этот раствор охлаждали до 0°С. Добавляли пероксид водорода 50% (42 мл; 0,62 моль), а затем раствор гидроксида моногидрата лития (10,39 г; 0,25 моль) в воде (100 мл), поддерживая при этом температуру реакции <5°С. Эту смесь затем перемешивали при 0-5°С в течение 1 ч. По завершении реакции (анализ ТСХ) добавляли раствор сульфита натрия (120 г; 0,95 моль) в воде (600 мл) при <10°С. Значение рН реакционной смеси доводили до 6,5-7,0 добавлением 10% водного раствора HCl. Реакционную смесь фильтровали и осадок на фильтре (= хиральный амин формулы А13а; см. пример выделения ниже) промывали водой (75 мл). Затем из фильтрата удаляли THF при пониженном давлении, к остатку добавляли метиленхлорид (375 мл) и эту смесь перемешивали в течение 30 мин. Слои разделяли, водный слой экстрагировали метиленхлоридом (375 мл) и этот водный слой выпаривали при пониженном давлении досуха (способ обработки органического слоя см. пример выделения ниже). Остаток разводили в этаноле (150 мл), эту смесь перемешивали в течение 30 мин и фильтровали. Осадок на фильтре промывали этанолом (25 мл) и объединенные фильтраты концентрировали досуха. Остаток снова разводили в этаноле (75 мл), эту смесь перемешивали в течение 30 мин и фильтровали. Осадок на фильтре промывали этанолом (15 мл) и объединенные фильтраты концентрировали при пониженном давлении досуха. Остаток затем растворяли в THF (75 мл), нерастворимое твердое вещество отфильтровывали и фильтрат концентрировали досуха. Эту процедуру повторяли три раза. Получали (3R,4R)-3-фтор-4-гидрокси-5-(гидроксиметил)-3-метилдигидрофуран-2(3Н)-он (25,5 г; выход 100%), который без дополнительной очистки использовали на стадии бензоилирования.
Выделение (S)-4-изопропил-5,5-дифенилоксазолидин-2-она (= хиральный амин формулы А13а
Влажный осадок (см. выше) разводили в воде (375 мл), эту смесь перемешивали в течение 30 мин, фильтровали и осадок на фильтре промывали дважды водой (100 мл × 2). Влажный осадок высушивали в вакууме при 50°С в течение 24 ч с получением белого твердого вещества (35,2 г). Органический слой (см. выше) выпаривали при пониженном давлении досуха и остаток разводили в метаноле (25 мл), фильтровали и промывали метанолом (5 мл). Влажный осадок высушивали в вакууме при 50°С в течение 24 ч с получением белого твердого вещества (6,3 г). Суммарный выход хирального амина составил 41,5 г, что соответствует 95% выходу очистки.
Пример 4
Получение ((3R,4R)-3-(бензоилокси)-4-фтор-4-метил-5-оксотетрагидро-фуран-2-ил)метилбензоата
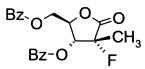
(3R,4R)-3-Фтор-4-гидрокси-5-(гидроксиметил)-3-метилдигидрофуран-2(3Н)-он (25,5 г; 0,155 моль), полученный в примере 3, растворяли в 200 мл THF. Вносили 4-(диметиламино)-пиридин (8,3 г; 0,067 моль) и триэтиламин (35 г; 0,35 моль) и реакционную смесь охлаждали до 0°C. Добавляли бензоилхлорид (46,7 г; 0,33 моль) и эту смесь нагревали до 35~40°C в течение 2 ч. По завершении реакции (анализ ТСХ) вносили воду (100 мл) и эту смесь перемешивали в течение 30 мин. Фазы разделяли и к водной фазе добавляли метил-трет-бутиловый эфир (100 мл) и эту смесь перемешивали в течение 30 мин. Фазы разделяли и органическую фазу промывали насыщенным раствором NaCl (100 мл). Объединенные органические фазы высушивали над Na2SO4 (20 г) фильтровали и фильтрат выпаривали досуха. Остаток разводили в изопропиловом спирте (250 мл), эту смесь нагревали до 50°С и перемешивали в течение 60 мин, затем охлаждали до 0°С и далее перемешивали в течение 60 мин. Твердое вещество фильтровали и влажный осадок промывали изопропиловым спиртом (50 мл) и затем высушивали в вакууме. Получали указанное в заголовке соединение ((3R,4R)-3-(бензоилокси)-4-фтор-4-метил-5-оксотетрагидрофуран-2-ил)метилбензоат (48,3 г; выход 83,9%).
1Н-ЯМР (CDCl3, 400 МГц): δ 8,10 (d, J=7,6 Гц, 2Н), 8,00 (d, J=7,6 Гц, 2Н), 7,66 (t, J=7,6 Гц, 1Н), 7,59 (t, J=7,6 Гц, 1Н), 7,50 (m, 2Н), 7,43 (m, 2Н), 5,53 (dd, J=17,6, 5,6 Гц, 1Н), 5,02 (m, 1Н), 4,77 (dd, J=12,8, 3,6 Гц, 1Н), 4,62 (dd, J=12,8, 5,2 Гц, 1Н), 1,77(d, J=23,2 Гц, 3Н).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЭПОТИЛОНА, НОВЫЕ ПРОИЗВОДНЫЕ ЭПОТИЛОНА, А ТАКЖЕ НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ РЕАЛИЗАЦИИ СПОСОБА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2404985C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЭПОТИЛОНА | 2003 |
|
RU2343155C2 |
| ПРОИЗВОДНОЕ БЕНЗОПИПЕРИДИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2727705C1 |
| СПОСОБ ПОЛУЧЕНИЯ АНТАГОНИСТОВ РЕЦЕПТОРОВ CGRP | 2013 |
|
RU2672056C2 |
| СПОСОБЫ СИНТЕЗА ПРОИЗВОДНЫХ ДИГИДРОПИРИДОФТАЛАЗИНОНА | 2011 |
|
RU2561732C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2625309C2 |
| Производные N-пиперидин-3-илбензамида для лечения сердечно-сосудистых заболеваний | 2014 |
|
RU2618628C1 |
| ПРОИЗВОДНЫЕ АДАМАНТИЛА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ JNK-ОПОСРЕДОВАННОГО РАССТРОЙСТВА | 2012 |
|
RU2626890C2 |
| ПРОИЗВОДНЫЕ БЕНЗИЛФЕНИЛЦИКЛОГЕКСАНА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2505521C2 |
Изобретение относится к области органической химии, а именно к способу получения производного фторлактона формулы I, включающий а) взаимодействие альдегида формулы II с производным фторпропионата формулы III, где А выбран из хиральных фрагментов A1-A3 и Ph обозначает фенил, в присутствии катализатора, основания и органического растворителя при температуре реакции в интервале от -78°С до 20°С, с образованием альдольного аддукта формулы IV, где А определен выше; и b) проведение гидролиза альдольного аддукта формулы IV с помощью окислителя в присутствии основания - гидроксида щелочного металла при температуре реакции в интервале от 0 до 10°С с получением производного фторлактона формулы I. Также изобретение относится к промежуточным соединениям, полученным в ходе синтеза. Технический результат: разработан новый способ получения производного фторлактона, используемого в синтезе соединений, которые потенциально представляют собой сильные ингибиторы NSSB-полимеразы Вируса гепатита С (HCV). 3 н. и 14 з.п. ф-лы, 4 пр.
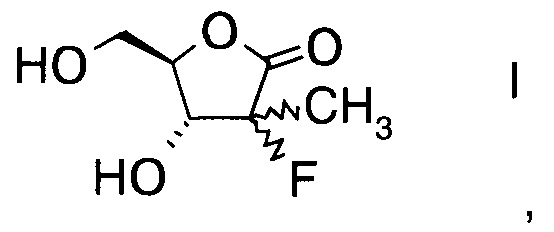
1. Способ получения производного фторлактона формулы
включающий стадии:
а) взаимодействие альдегида формулы
с производным фторпропионата формулы
где А выбран из хиральных фрагментов
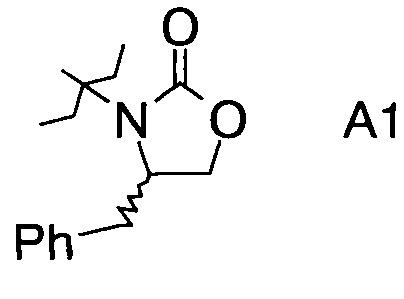
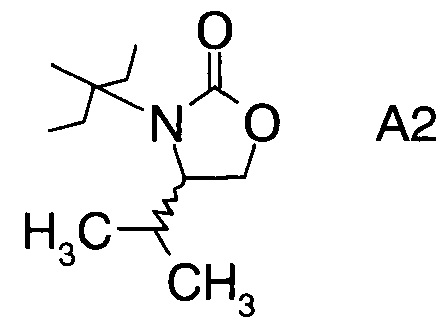
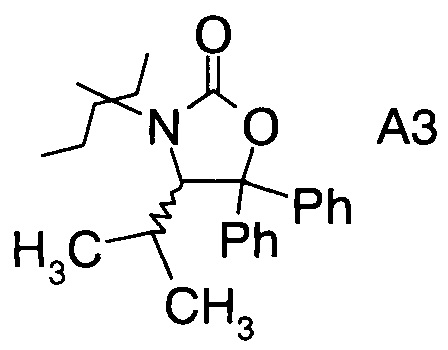
и Ph обозначает фенил, в присутствии катализатора, основания и органического растворителя при температуре реакции в интервале от -78°С до 20°С, с образованием альдольного аддукта формулы
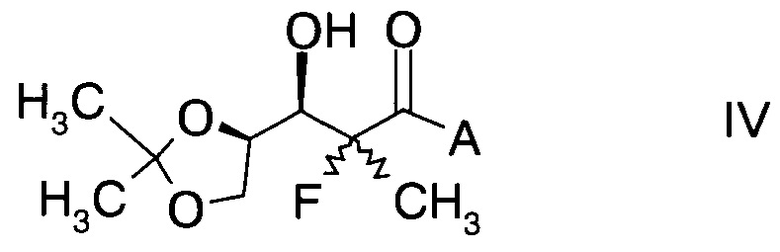
где А определен выше; и
b) проведение гидролиза альдольного аддукта формулы IV с помощью окислителя в присутствии основания - гидроксида щелочного металла при температуре реакции в интервале от 0 до 10°С с получением производного фторлактона формулы I.
2. Способ по п.1, где хиральный фрагмент А в производном фторпропионата формулы III представляет собой A3.
3. Способ по п.1, где производное фторлактона имеет формулу
где А выбран из хиральных фрагментов
и Ph представляет собой фенил.
4. Способ по п.3, где хиральный фрагмент А в производном фторпропионата формулы III представляет собой А3а.
5. Способ по п.1, где производное фторлактона формулы I ацилируют с образованием ацилированного фторлактона формулы
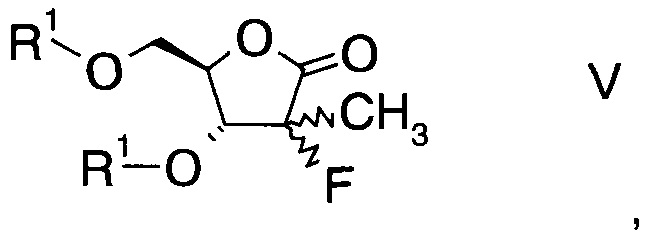
где R1 обозначает гидрокси-протекторную группу.
6. Способ по п.5, где ацилированный фторлактон имеет формулу
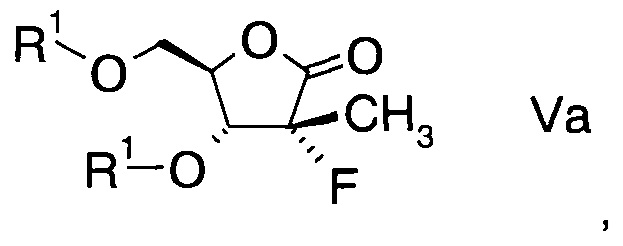
где R1 обозначает гидрокси-протекторную группу.
7. Способ по п.5 или 6, где гидрокси-протекторная группа R1 обозначает бензоил.
8. Способ по п.1, где хиральные фрагменты А можно выделять в виде соответствующих хиральных аминов А-Н формул
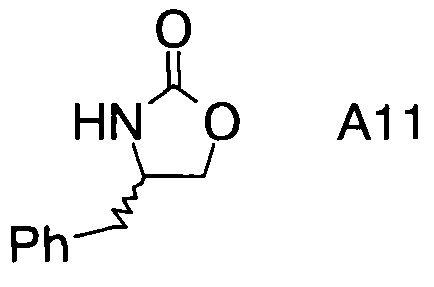
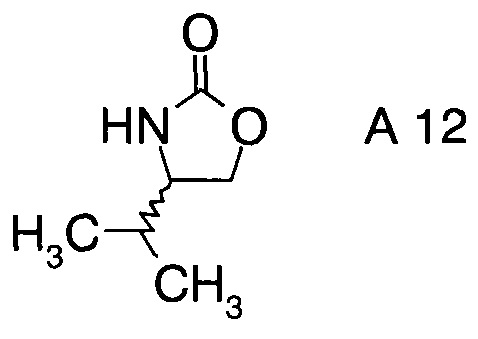
9. Способ по п.1, где катализатор представляет собой дибутилборон трифторметансульфонат.
10. Способ по п.1, где гидролиз на стадии (b) осуществляют с помощью пероксида водорода в качестве окислителя и гидроксида лития в качестве основания - гидроксида щелочного металла.
11. Способ по п.5, где ацилирование осуществляют в присутствии третичного амина при температуре реакции в интервале от 0 до 40°С.
12. Способ по п.5, где ацилирующий агент представляет собой бензоилхлорид.
13. Производное фторпропионата формулы
14. Альдольный аддукт формулы
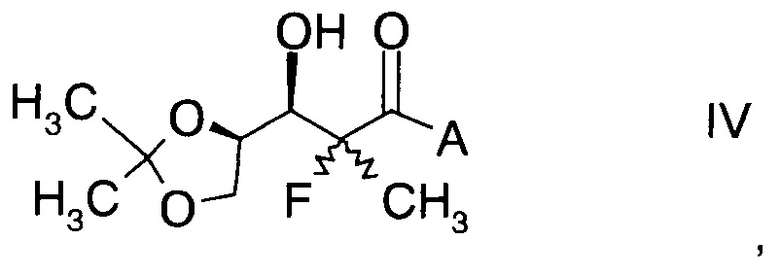
где А выбран из хиральных фрагментов
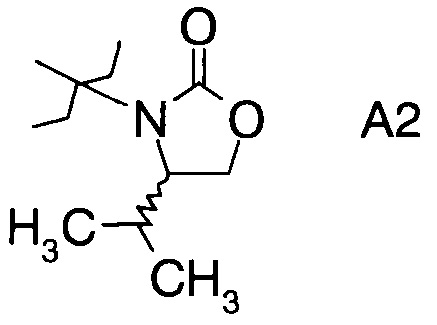
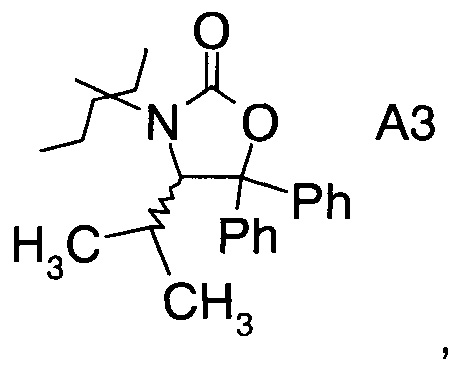
где Ph обозначает фенил.
15. Альдольный аддукт по п.14 формулы
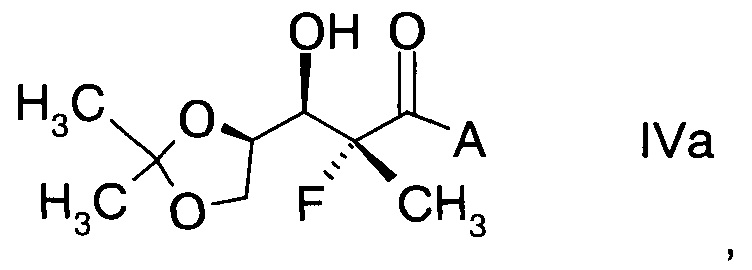
где А определен выше.
16. Альдольный аддукт по п.14 или 15, где А представляет собой
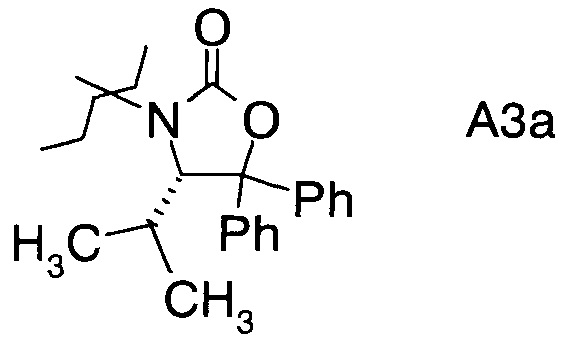
и где Ph обозначает фенил.
17. Альдольный аддукт по п.16, где А представляет собой А3а.
| Vincent A.Brunet и др.: "Titanium mediated asymmetric aldol reaction with α-fluoropropionimide enolates", Journal of Fluorine Chemistry, 128(10), октябрь 2007, стр.1271-1279 | |||
| Victoria Peddie и др.: "Synthesis and Conformation of Fluorinated β-Peptidic Compounds", Chemistry - A European Journal, 18(21), 21 мая 2012, стр | |||
| СТЕНА ИЗ ПОЛЫХ СТЕКЛЯННЫХ ТЕЛ | 1927 |
|
SU6655A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |

