ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Настоящее изобретение относится к замещенным амидным соединениям, фармацевтическим композициям, содержащим такие соединения, и применению таких соединений для лечения сердечнососудистого заболевания, включая атеросклероз, гиперлипидемию, гиперхолестеринемию и гипертриглицеридемию, у млекопитающих, включая людей.
Установлено, что атеросклероз, представляющий собой заболевание артерий, является основной причиной смертности в Соединенных Штатах Америки и Западной Европе. Последовательность развития патологического процесса, приводящего к атеросклерозу и окклюзионному поражению сосудов сердца, хорошо известна. Самой ранней стадией в этой последовательности является образование "жировых прожилок" в сонной, коронарных и мозговых артериях и в аорте. Эти повреждения имеют желтый цвет из-за присутствия жировых отложений, обнаруживаемых, главным образом, в клетках гладких мышц и в макрофагах интимы артерий и аорты. Кроме того, можно предположить, что большая часть холестерина, обнаруживаемого в жировых прожилках, в свою очередь приводит к развитию "фиброзной бляшки", которая состоит из аккумулированных интимальных клеток гладких мышц, нагруженных липидом и окруженных внеклеточным липидом, коллагеном, эластином и протеогликанами. Эти клетки вместе с матриксом образуют фиброзное утолщение, которое покрывает более глубокое отложение клеточного дебриса и находящегося над ним внеклеточного липида. Этот липид представляет собой, в основном, свободный и этерифицированный холестерин. Фиброзная бляшка образуется медленно и со временем, вероятно, подвергается кальцификации и некрозу, что приводит к развитию "осложненного патологического повреждения", которое является причиной артериальной окклюзии и тенденции к возникновению пристеночного тромбоза и спазма артериальной мышцы, характерных для прогрессирующего атеросклероза.
Эпидемиологические данные ясно свидетельствуют о том, что гиперлипидемия является главным фактором риска в развитии сердечнососудистых заболеваний (ССЗ), обусловленных атеросклерозом. В последнее время ведущие специалисты в области медицины придают особое значение снижению уровня холестерина в плазме крови, и особенно холестерина липопротеинов низкой плотности, как главной меры в предупреждении ССЗ. В настоящее время известно, что верхние пределы "нормального" уровня холестерина значительно ниже тех уровней, которые были признаны до сих пор. В результате, в настоящее время очевидно, что большая часть населения Западной Европы и Америки находится в группе повышенного риска. Дополнительные независимые факторы риска включают нарушение толерантности к глюкозе, гипертрофию левого желудочка, гипертензию и мужской пол. Сердечно-сосудистые заболевания чаще всего встречаются у больных диабетом, по меньшей мере отчасти из-за наличия у этой группы людей множества независимых факторов риска. Поэтому, успешное лечение гиперлипидемии у основной части населения, и в частности у больных диабетом, имеет исключительно важное медицинское значение.
Несмотря на то, что существует множество соединений с антиатеросклеротическим действием, сердечно-сосудистые заболевания все еще являются основной причиной смертности и, таким образом, в данной области техники существует постоянная потребность и продолжаются постоянные поиски альтернативных средств лечения.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям формулы I
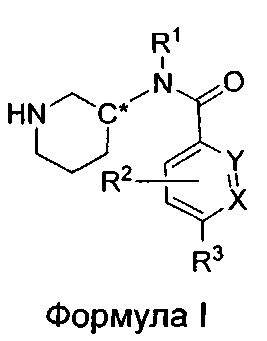
или их фармацевтически приемлемой соли, где
R1 представляет собой пирид-2-ил, изохинолин-1-ил или 1Н-пирроло[2,3-с]пиридин-7-ил;
R1, возможно, моно- или дизамещен хлором или (C1-C4)алкилом;
X и Y независимо представляют собой либо N, либо C(H), при условии, что по меньшей мере один из X или Y представляет собой C(H);
R2 представляет собой Н, фтор, гидроксил или метил;
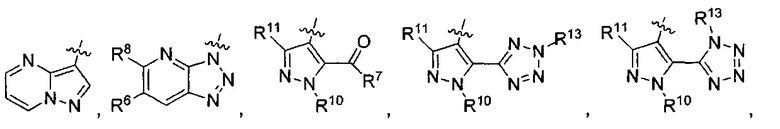
R3 представляет собой
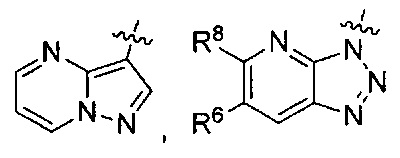 или
или 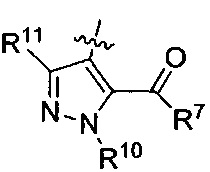 ,
,
где каждый из R6 и R8 независимо представляет собой Н, метил, галоген или (C1-C4)алкилокси, при условии, что только один из R6 и R8 представляет собой галоген;
где каждый из R10 и R11 независимо представляет собой Н, (C1-C4)алкил или (C3-C5)циклоалкил; и
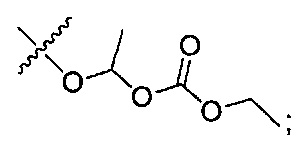
где R7 представляет собой гидроксил, (C1-C4)алкилокси, (C1-C4)алкоксикарбонилокси(C1-C4)алкилокси или (C1-C4)алкилкарбонилокси(С1-С4)алкокси.
Настоящая заявка также относится к способам лечения дислипидемии, гиперхолестеринемии (включая гетерозиготную и гомозиготную семейную гиперхолестеринемию), гипертриглицеридемии, гиперлипидемии, гипо-альфа-липопротеинемии, метаболического синдрома, осложнений диабета, атеросклероза, инсульта, сосудистой деменции, хронической почечной недостаточности, ишемической болезни сердца, коронарной артериальной болезни, ретинопатии, воспаления, тромбоза, заболевания периферических сосудов или застойной сердечной недостаточности у млекопитающего путем введения млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы I или фармацевтически приемлемой соли указанного соединения.
Настоящая заявка также относится к фармацевтическим композициям, содержащим терапевтически эффективное количество соединения формулы I или фармацевтически приемлемой соли указанного соединения и фармацевтически приемлемый носитель, наполнитель или разбавитель.
Кроме того, настоящая заявка относится к фармацевтическим комбинированным композициям, содержащим: терапевтически эффективное количество композиции, содержащей
первое соединение, представляющее собой соединение формулы I или фармацевтически приемлемую соль указанного соединения;
второе соединение, представляющее собой липид-модулирующий агент; и
фармацевтически приемлемый носитель, наполнитель или разбавитель.
Примеры липид-модулирующих агентов включают ингибитор липазы, ингибитор HMG-CoA-редуктазы (гидроксиметилглутарил-кофермент А (СоА)-редуктазы), ингибитор HMG-CoA-синтазы, ингибитор экспрессии гена HMG-CoA-редуктазы, ингибитор экспрессии гена HMGCoA-синтазы, ингибитор секреции МТР/Аро B (микросомального белка, переносящего триглицериды/аполипопротеина В), ингибитор СЕТР (белка, переносящего эфиры холестерина), ингибитор абсорбции желчных кислот, ингибитор абсорбции холестерина, ингибитор синтеза холестерина, ингибитор сквален-синтетазы, ингибитор сквален-эпоксидазы, ингибитор сквален-циклазы, комбинированный ингибитор сквален-эпоксидазы/сквален-циклазы, фибрат, ниацин, комбинацию ниацина и ловастатина, ионообменную смолу, антиоксидант, ингибитор АСАТ (ацил-СоА: холестерин-ацилтрансферазы) и секвестрант желчных кислот.
Другой аспект этого изобретения относится к способу лечения дислипидемии путем введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения, селективно ингибирующего трансляцию м-РНК (матричной рибонуклеиновой кислоты), кодирующей PCSK9 (пропротеинконвертазу субтилизин/кексин тип 9), в белок PCSK9. Предпочтительно соединение вводят путем перорального введения.
Настоящее изобретение также относится к соединениям формулы II
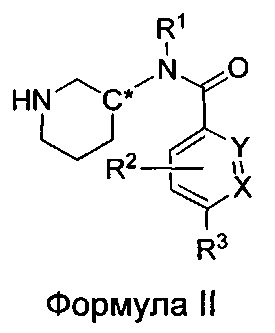
или их фармацевтически приемлемой соли, где
R1 представляет собой пирид-2-ил, изохинолин-1-ил или 1Н-пирроло[2,3-с]пиридин-7-ил;
R1, возможно, моно- или дизамещен хлором или (C1-C4)алкилом;
X и Y независимо представляют собой либо N, либо C(H), при условии, что по меньшей мере один из X или Y представляет собой C(H);
R2 представляет собой Н, фтор, гидроксил или метил;
R3 представляет собой
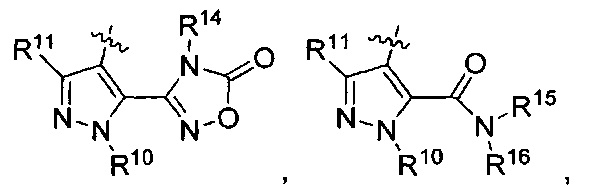
где каждый из R6 и R8 независимо представляет собой Н, метил, галоген или (C1-С4)алкилокси, при условии, что только один из R6 и R8 представляет собой галоген;
где каждый из R10 и R11 независимо представляет собой Н, (С1-С4)алкил или (C3-C5)циклоалкил;
где R7 представляет собой гидроксил, (C1-C4)алкилокси, (C1-C4)алкоксикарбонилокси(C1-C4)алкилокси или (C1-C4)алкилкарбонилокси(C1-C4)алкокси;
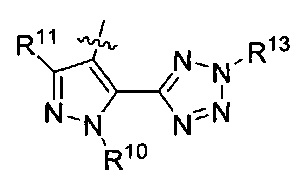
R13 представляет собой Н, (C1-C4)алкил, (C1-C4)алкилкарбонилокси(Сг С4)алкил или (C1-C4)алкоксикарбонилокси(C1-C4)алкил;
R14 представляет собой Н, (C1-C4)алкил, (C1-С4)алкилкарбонилокси(C1-C4)алкил или (C1-C4)алкоксикарбонилокси(C1-C4)алкил;
R15 представляет собой гидроксил, тетразолил, (C1-С4)алкилсульфонил или трифторметилсульфонил; и
R16 представляет собой Н, (C1-C4)алкил, (C1-C4)алкилкарбонилокси(C1-С4)алкил или (C1-C4)алкоксикарбонилокси(C1-C4)алкил.
Настоящая заявка также относится к способам лечения дислипидемии, гиперхолестеринемии (включая гетерозиготную и гомозиготную семейную гиперхолестеринемию), гипертриглицеридемии, гиперлипидемии, гипо-альфа-липопротеинемии, метаболического синдрома, осложнений диабета, атеросклероза, инсульта, сосудистой деменции, хронической почечной недостаточности, ишемической болезни сердца, коронарной артериальной болезни, ретинопатии, воспаления, тромбоза, заболевания периферических сосудов или застойной сердечной недостаточности у млекопитающего путем введения млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы II или фармацевтически приемлемой соли указанного соединения.
Настоящая заявка также относится к фармацевтическим композициям, содержащим терапевтически эффективное количество соединения формулы II или фармацевтически приемлемой соли указанного соединения и фармацевтически приемлемый носитель, наполнитель или разбавитель.
Кроме того, настоящая заявка относится к фармацевтическим комбинированным композициям, содержащим: терапевтически эффективное количество композиции, содержащей
первое соединение, представляющее собой соединение формулы II или фармацевтически приемлемую соль указанного соединения;
второе соединение, представляющее собой липид-модулирующий агент; и
фармацевтически приемлемый носитель, наполнитель или разбавитель.
Следует понимать, что и вышеупомянутое общее описание, и последующее подробное описание служат только в качестве примера и пояснения и не ограничивают заявленное изобретение.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг. 1 представлена картина дифракции рентгеновских лучей на порошке, описывающая кристаллическую форму соединения по Примеру 2 (по вертикальной оси: интенсивность (имп./с); по горизонтальной оси: углы два-тета (градусы)).
На Фиг. 2 представлена картина дифракции рентгеновских лучей на порошке, описывающая кристаллическую форму соединения по Примеру 6 (по вертикальной оси: интенсивность (имп./с); по горизонтальной оси: углы два-тета (градусы)).
На Фиг. 3 представлена картина дифракции рентгеновских лучей на порошке, описывающая кристаллическую форму соединения по Примеру 13 (по вертикальной оси: интенсивность (имп./с); по горизонтальной оси: углы два-тета (градусы)).
На Фиг. 4 представлена картина дифракции рентгеновских лучей на порошке, описывающая кристаллическую форму соединения по Примеру 15b (по вертикальной оси: интенсивность (имп./с); по горизонтальной оси: углы два-тета (градусы)).
На Фиг. 5 представлена рентгеновская кристаллическая структура (диаграмма ORTEP (программа изображения кристаллических структур)) соединения по Примеру 15b.
На Фиг. 6 представлена картина дифракции рентгеновских лучей на порошке, описывающая кристаллическую форму соединения по Примеру 16 (по вертикальной оси: интенсивность (имп./с); по горизонтальной оси: углы два-тета (градусы)).
На Фиг. 7 представлена рентгеновская кристаллическая структура (диаграмма ORTEP) соединения по Примеру 16.
На Фиг. 8 представлена рентгеновская кристаллическая структура (диаграмма ORTEP) соединения по Получению 23а.
На Фиг. 9 представлена картина дифракции рентгеновских лучей на порошке, описывающая кристаллическую форму соединения по Примеру 30 (по вертикальной оси: интенсивность (имп./с); по горизонтальной оси: углы два-тета (градусы)).
На Фиг. 10 представлена картина дифракции рентгеновских лучей на порошке, описывающая кристаллическую форму соединения по Примеру 31 (по вертикальной оси: интенсивность (имп./с); по горизонтальной оси: углы два-тета (градусы)).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение может быть более легко понято с учетом следующего подробного описания примеров осуществления изобретения и включенных в него примеров.
Ссылки на соединения формулы I или тому подобное, как определено здесь, также включают соединения формулы II.
Перед тем, как соединения, композиции и способы по настоящему изобретению будут раскрыты и описаны, следует понять, что это изобретение не ограничено конкретными способами синтеза соединений, которые, разумеется, могут быть модифицированы. Также следует понимать, что используемая здесь терминология предназначена только для описания, а не для ограничения конкретных воплощений.
Предпочтительная группа соединений, обозначаемая как А-группа, содержит такие соединения, которые имеют формулу I, как показано выше, где:
R1 представляет собой пирид-2-ил, и пиперидинил C* имеет (R)-конфигурацию.
Группа соединений, которая является предпочтительной в А-группе соединений, обозначаемая как В-группа, содержит такие соединения, где:
X и Y оба представляют собой C(H), R2 представляет собой Н, и R1, возможно, монозамещен хлором или метилом.
Группа соединений, которая является предпочтительной в В-группе соединений, обозначаемая как С-группа, содержит такие соединения, где
R3 представляет собой
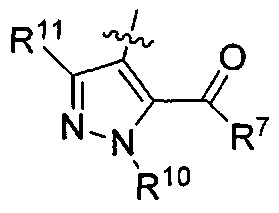
R7 представляет собой гидроксил, (С1-С2)алкилокси или
R10 представляет собой метил; и
R11 представляет собой Н.
Группа соединений, которая является предпочтительной в В-группе соединений, обозначаемая как D-группа, содержит такие соединения, где
R3 представляет собой 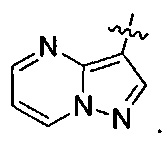
Группа соединений, которая является предпочтительной в В-группе соединений, обозначаемая как Е-группа, содержит такие соединения, где
R3 представляет собой 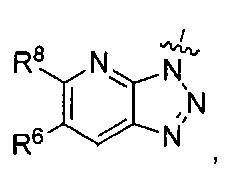
R6 представляет собой Н или метил, и R8 представляет собой Н.
Предпочтительная группа соединений, обозначаемая как F-группа, содержит такие соединения, которые имеют формулу I, как показано выше, где:
R1 представляет собой изохинолин-1-ил; и пиперидинил C* имеет (R)-конфигурацию.
Группа соединений, которая является предпочтительной в F-группе соединений, обозначаемая как G-группа, содержит такие соединения, где
X и Y оба представляют собой C(H), R2 представляет собой Н, гидроксил или метил, и R1, возможно, монозамещен хлором или метилом.
Группа соединений, которая является предпочтительной в G-группе соединений, обозначаемая как Н-группа, содержит такие соединения, где
R3 представляет собой 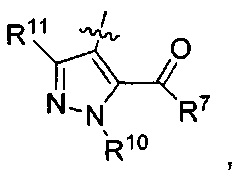
R7 представляет собой гидроксил, (С1-С2)алкилокси или
R10 представляет собой метил; и
R11 представляет собой Н.
Группа соединений, которая является предпочтительной в G-группе соединений, обозначаемая как I-группа, содержит такие соединения, где
R3 представляет собой 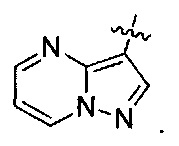
Группа соединений, которая является предпочтительной в G-группе соединений, обозначаемая как J-группа, содержит такие соединения, где
R3 представляет собой 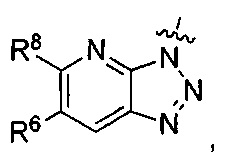
R6 представляет собой Н или метил, и R8 представляет собой Н.
Предпочтительная группа соединений, обозначаемая как К-группа, содержит такие соединения, которые имеют формулу I, как показано выше, где:
R1 представляет собой 1Н-пирроло[2,3-с]пиридин-7-ил, и пиперидинил C* имеет (R)-конфигурацию.
Группа соединений, которая является предпочтительной в К-группе соединений, обозначаемая как L-группа, содержит такие соединения, где
X и Y оба представляют собой C(H), R2 представляет собой Н, гидроксил или метил, и R1, возможно, монозамещен хлором или метилом.
Группа соединений, которая является предпочтительной в L-группе соединений, обозначаемая как M-группа, содержит такие соединения, где
R3 представляет собой
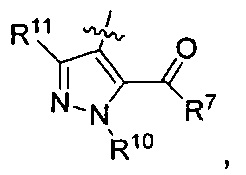
R7 представляет собой гидроксил, (С1-C2)алкилокси или
R10 представляет собой метил; и
R11 представляет собой Н.
Группа соединений, которая является предпочтительной в L-группе соединений, обозначаемая как N-группа, содержит такие соединения, где
R3 представляет собой 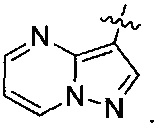
Группа соединений, которая является предпочтительной в L-группе соединений, обозначаемая как O-группа, содержит такие соединения, где
R3 представляет собой 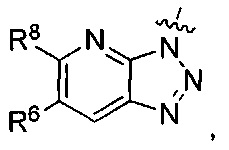 и R6 представляет собой Н или метил, и R8 представляет собой Н.
и R6 представляет собой Н или метил, и R8 представляет собой Н.
Предпочтительная группа соединений, обозначаемая как Р-группа, содержит следующие соединения:
N-(3-метилпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(пиразоло[1,5-а]пиримидин-3-ил)бензамид;
N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(пиразоло[1,5-а]пиримидин-3-ил)бензамид;
N-(3-хлорпиридин-2-ил)-4-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-N-[(3R)-пиперидин-3-ил]бензамид;
4-(4-{изохинолин-1-ил[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоновую кислоту;
N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-5-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиридин-2-карбоксамид;
этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат;
4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоновую кислоту;
4-(4-{(3-хлорпиридин-2-ил)[пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоновую кислоту;
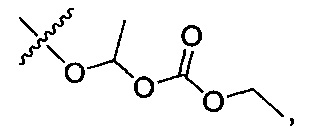
1-[(этоксикарбонил)окси]этил-4-(4-{(3-хлорпиридин-2-ил)[пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат;
1-[(этоксикарбонил)окси]этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат;
(1R)-1-[(этоксикарбонил)окси]этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат;
(1S)-1-[(этоксикарбонил)окси]этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат; или
N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензамид; или
фармацевтически приемлемую соль любого из указанных соединений.
Предпочтительная группа соединений, обозначаемая как Q-группа, содержит следующие соединения:
N-(3-хлорпиридин-2-ил)-5-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-N-[(3R)-пиперидин-3-ил]пиридин-2-карбоксамид;
метил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат;
1-[(этоксикарбонил)окси]этил-4-(4-{изохинолин-1-ил[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат;
1-метил-4-(4-{(1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-карбоновую кислоту;
метил-1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-карбоксилат; или
1-[(этоксикарбонил)окси]этил-1-метил-4-(4-{(1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-карбоксилат; или
фармацевтически приемлемую соль любого из указанных соединений.
Предпочтительная группа соединений, обозначаемая как R-группа, содержит такие соединения, которые имеют формулу II, как показано выше, где:
R1 представляет собой пирид-2-ил, возможно, монозамещенный хлором или метилом;
пиперидинил С* имеет R-конфигурацию;
X и Y оба представляют собой C(H);
R2 представляет собой Н;
R3 представляет собой
 ;
;
R10 представляет собой метил;
R11 представляет собой Н; и
R13 представляет собой (C1-C4)алкилкарбонилокси(C1-C4)алкил.
Предпочтительная группа соединений, обозначаемая как S-группа, содержит такие соединения, которые имеют формулу II, как показано выше, где:
R1 представляет собой пирид-2-ил, возможно, монозамещенный хлором или метилом;
пиперидинил С* имеет R-конфигурацию;
X и Y оба представляют собой C(H);
R2 представляет собой Н; и
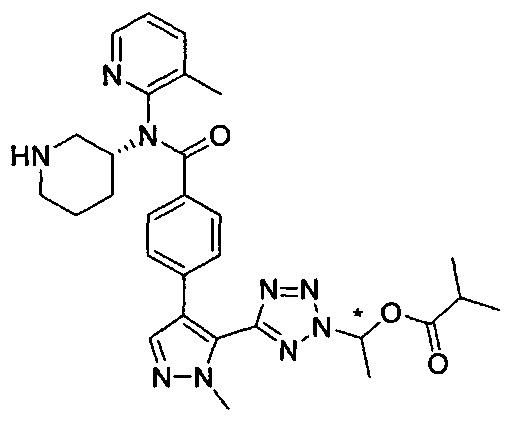
R3 представляет собой 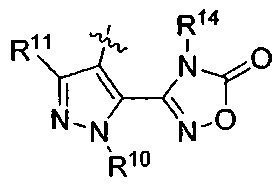 или
или 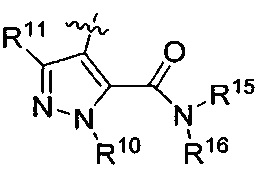 .
.
Предпочтительная группа соединений, обозначаемая как Т-группа, содержит следующие соединения:
4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-N-[(трифторметил)сульфонил]-1Н-пиразол-5-карбоксамид;
N-(3-хлорпиридин-2-ил)-4-[1-метил-5-(2Н-тетразол-5-ил)-1Н-пиразол-4-ил]-N-[(3R)-пиперидин-3-ил]бензамид;
1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-N-(метилсульфонил)-1Н-пиразол-5-карбоксамид;
N-(3-метилпиридин-2-ил)-4-[1-метил-5-(2Н-тетразол-5-ил)-1Н-пиразол-4-ил]-N-[(3R)-пиперидин-3-ил]бензамид; или
этил-1-[{[1-мeтил-4-(4-{(3-мeтилпиpидин-2-ил)[(3R)-пипepидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]карбонил}(метилсульфонил)амино]этил-карбонат;
или фармацевтически приемлемую соль любого из указанных соединений.
Предпочтительная группа соединений, обозначаемая как U-группа, содержит следующие соединения:
этил-1-{5-[1-мeтил-4-(4-{(3-мeтилпиpидин-2-ил)[(3R)-пипepидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-1Н-тетразол-1-ил}этил карбонат;
этил-(1S)-1-{5-[1-мeтил-4-(4-{(3-мeтилпиpидин-2-ил)[(3R)-пипepидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}этил карбонат; или
этил-(1R)-1-{5-[1-метил-4-(4-{(3-метилпиpидин-2-ил)[(3R)-пипepидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}этил-карбонат;
Предпочтительная группа соединений, обозначаемая как V-группа, содержит следующие соединения:
(1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}этил-2-метилпропаноат (диастереомер B; Пример 51)
2-метил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропил-2-метилпропаноат (диастереомер B; Пример 55)
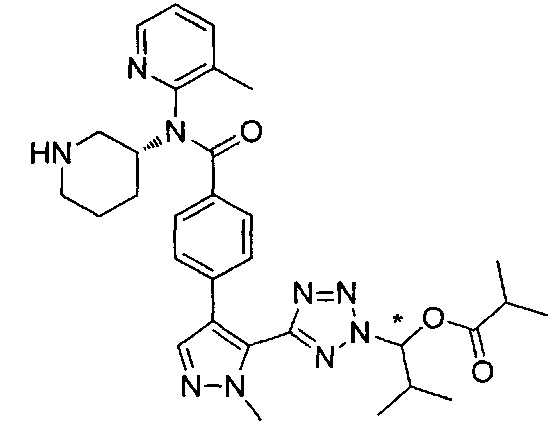 ,
,
или фармацевтически приемлемую соль любого из указанных соединений.
Предпочтительная группа соединений, обозначаемая как W-группа, содержит следующие соединения:
(1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}этил-2-метилпропаноат; или
2-метил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропил-2-метилпропаноат;
или фармацевтически приемлемую соль любого из указанных соединений.
Другую предпочтительную группу соединений составляет каждое из соединений в P- и Q-группах, взятых по отдельности.
Другую предпочтительную группу соединений составляет каждое из соединений в T- и U-группах, взятых по отдельности.
Другую предпочтительную группу соединений составляет каждое из соединений в V- и W-группах, взятых по отдельности.
Также предпочтительно, когда каждое из этих соединений, взятых по отдельности, представляет собой фармацевтически приемлемую соль, и особенно предпочтительно, когда каждое соединение, взятое по отдельности, представляет собой соль присоединения кислоты.
В одном предпочтительном воплощении фармацевтических комбинированных композиций, способов и наборов по настоящему изобретению второе соединение представляет собой ингибитор HMG-CoA-редуктазы или ингибитор СЕТР, например, розувастатин, ривастатин, питавастатин, ловастатин, симвастатин, правастатин, флувастатин, аторвастатин или церивастатин, или пролекарство указанного соединения, или фармацевтически приемлемую соль указанного соединения или пролекарства. Особенно предпочтительно, когда второе соединение представляет собой гемикальциевую соль аторвастатина.
Фармацевтически приемлемые соли соединений формулы I включают соли присоединения кислот и оснований. Подходящие соли присоединения кислот получают из кислот, которые образуют нетоксичные соли. Примеры включают соли ацетат, адипат, аспартат, бензоат, бесилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камсилат, цитрат, цикламат, эдисилат, эсилат, формиат, фумарат, глюцептат, глюконат, глюкуронат, гексофторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглутамат, сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат и ксинафоат.
Подходящие основные соли получают из оснований, которые образуют нетоксичные соли. Примеры включают соли алюминия, аргинина, кальция, холина, диэтиламина, глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка. Также могут быть образованы полусоли кислот и оснований, например гемисульфаты и гемикальциевые соли. Обзор подходящих солей см. в Handbook of Pharmaceutical Salts: Properties, Selection, and Use. Stahl and Wermuth (Wiley-VCH, 2002).
Соединения по изобретению могут существовать как в несольватированных, так и сольватированных формах. Термин "сольват" использован здесь для обозначения молекулярного комплекса, содержащего соединение по изобретению и одну или более чем одну молекулу фармацевтически приемлемого растворителя, например этанола. Такие молекулы растворителя представляют собой молекулы, обычно используемые в области фармации, которые, как известно, безвредны для реципиента, например, воду, этанол и тому подобное. Другие растворители могут быть использованы в качестве промежуточных сольватов при получении более желательных сольватов, такие как метанол, метил-трет-бутиловый эфир, этилацетат, метилацетат, (S)-пропиленгликоль, (R)-пропиленгликоль, 1,4-бутин-диол и тому подобные. Термин "гидрат" используют, когда указанным растворителем является вода. Фармацевтически приемлемые сольваты включают гидраты и другие сольваты, где растворитель для кристаллизации может быть замещенным изотопом, например D2O, d6-ацетон, d6-DMSO. Термин "гидрат" относится к комплексу, в котором молекула растворителя представляет собой воду. Сольваты и/или гидраты предпочтительно существуют в кристаллической форме.
Соединения по изобретению также могут существовать в виде комплексов, таких как клатраты, комплексы включения лекарственное средство-хозяин, где, в отличие от вышеупомянутых сольватов, лекарственное средство и хозяин присутствуют в стехиометрических или нестехиометрических количествах. В объем изобретения также включены комплексы лекарственного средства, содержащие два или более органических и/или неорганических компонента, которые могут находиться в стехиометрических или нестехиометрических количествах. Полученные комплексы могут быть ионизированными, частично ионизированными или неионизированными. Обзор таких комплексов см. в J Pharm Sci, 64 (8), 1269-1288, Haleblian (август 1975).
Соединения по изобретению включают соединения формулы I, как определено выше, полиморфы и их изомеры (включая оптические, геометрические и таутомерные изомеры, включая соединения, проявляющие более одного типа изомерии, и смеси одного или более чем одного из них) и меченые изотопами соединения формулы I. Таким образом, соединения по настоящему изобретению могут существовать в форме различных стереоизомеров, R и S изомеров в зависимости от наличия ассиметрических атомов углерода. Здесь они могут быть названы как "R-конфигурация" или "S-конфигурация" или тому подобное. Настоящим изобретением охватываются как индивидуальные изомеры, так и их смеси, включая рацемические и диастереомерные смеси.
Соединения формулы I, содержащие асимметрический атом углерода, могут существовать в виде двух или более стереоизомеров. Альфа и бета относятся к ориентации заместителя относительно плоскости кольца. Бета расположен выше плоскости кольца, а альфа расположен ниже плоскости кольца.
В тех случаях, когда соединение формулы I содержит алкенильную или алкениленовую группу или циклоалкильную группу, возможны геометрические цис/транс (или Z/E) изомеры. Таким образом, соединения по изобретению существуют в виде цис- или транс-конфигураций и в виде их смесей. Термин "цис" относится к ориентации двух заместителей относительно друг друга и плоскости кольца (либо оба "вверху", либо оба "внизу"). Аналогично, термин "транс" относится к ориентации двух заместителей относительно друг друга и плоскости кольца (заместители находятся на противоположных сторонах кольца).
В тех случаях, когда соединение содержит, например, кето или оксимную группу или ароматическую группировку, может существовать таутомерная изомерия ("таутомерия"). Примером таутомерии в рамках заявленных соединений является случай, когда R3 представляет собой нижеприведенный пиразол, и R10 представляет собой водород.

Настоящее изобретение включает все фармацевтически приемлемые меченые изотопами соединения формулы I, где один или более чем один атом заменен атомами, имеющими такой же атомный номер, но атомная масса или массовое число отличается от атомной массы или массового числа, которое обычно обнаруживается в природе.
Примеры изотопов, подходящих для включения в соединения по изобретению, включают изотопы водорода, такие как 2Н и 3Н, углерода, такие как 11С, 13С и 14С, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32Р, и серы, такие как 35S.
Некоторые меченые изотопами соединения формулы (I), например соединения, включающие радиоактивный изотоп, полезны в исследованиях по распределению лекарственного средства и/или субстрата в тканях. Радиоактивные изотопы тритий, то есть 3Н, и углерод-14, то есть 14С, особенно полезны для этой цели ввиду легкости их введения и наличия готовых средств детектирования.
Замещение более тяжелыми изотопами, такими как дейтерий, то есть 2Н, может давать некоторые терапевтические преимущества, вытекающие из более высокой метаболической стабильности, например увеличенное время полувыведения in vivo или сниженные дозировки, и, следовательно, в некоторых случаях может быть предпочтительным.
Замещение позитрон-излучающими изотопами, такими как 11С, 18F, 15O и 13N, может быть полезно в исследованиях методом позитронной эмиссионной томографии (ПЭТ) для проверки занятости рецепторов субстратом.
Приводимые здесь ссылки на "лечить", "процесс лечения", "лечение" и тому подобное включают куративное, паллиативное и профилактическое лечение.
Используемые здесь выражения "реакционно-инертный растворитель" и "инертный растворитель" относятся к растворителю или его смеси, которые не взаимодействуют с исходными соединениями, реагентами, промежуточными соединениями или продуктами, не оказывая тем самым неблагоприятного влияния на выход целевого продукта.
Под "фармацевтически приемлемым" подразумевается, что носитель, наполнитель или разбавитель и/или соль должны быть совместимы с другими ингредиентами композиции и безвредны для реципиента.
Термин "фармацевтически эффективное количество", который использован здесь, относится к количеству соединения формулы I (или агента комбинации или соединения формулы I в комбинации с агентом комбинации), достаточному для лечения, предупреждения появления или отсрочки появления или ослабления симптомов и физиологических проявлений указанных заболеваний, описанных здесь.
Термин "комнатная температура или температура окружающей среды" означает температуру от 18 до 25°C, "ЖХВД" относится к жидкостной хроматографии высокого давления, "ЖХСД" относится к жидкостной хроматографии среднего давления, "ТСХ" относится к тонкослойной хроматографии, "МС" относится к масс-спектру, или масс-спектроскопии, или масс-спектрометрии, "ЯМР" относится к спектроскопии ядерного магнитного резонанса, "DCM" означает дихлорметан, "DMSO" означает диметилсульфоксид, "DME" означает диметоксиэтан, "EtOAc" означает этилацетат, "МеОН" означает метанол, "Ph" означает фенильную группу, "Pr" означает пропил, "тритил" означает трифенилметильную группу, "ACN" означает ацетонитрил, "DEAD" означает диэтилазодикарбоксилат, и "DIAD" означает диизопропилазодикарбоксилат.
Следует понимать, что если карбоциклическая или гетероциклическая группировка может быть связана или иным способом присоединена к указанному субстрату посредством различных кольцевых атомов без обозначения конкретной точки присоединения, то подразумеваются все возможные точки присоединения, как через атом углерода, так и, например, через трехвалентный атом азота. Например, термин "пиридил" означает 2-, 3-или 4-пиридил, термин "тиенил" означает 2- или 3-тиенил и тому подобное. В общем, соединения по этому изобретению могут быть получены способами, аналогичными известным в области химии, конкретно с учетом описания, приведенного здесь.
Термин "коронарная артериальная болезнь", который использован здесь, выбран из группы, состоящей из атеросклеротической бляшки (например, предупреждение образования, регрессия, стабилизация), нестабильной бляшки (например, предупреждение образования, регрессия, стабилизация), площади нестабильной бляшки (уменьшение), кальцификации артерий (например, кальцинированный аортальный стеноз), повышенного кальциевого индекса коронарных артерий, дисфункциональной сосудистой реактивности, нарушений вазодилатации, спазма коронарных артерий, первичного инфаркта миокарда, повторного инфаркта миокарда, ишемической кардиомиопатии, рестеноза стента, рестеноза после ЧТКА (чрескожная транслюминальная коронарная ангиопластика), рестеноза артерий, рестеноза коронарного шунта, рестеноза сосудистого анастомоза, сниженного времени переносимости нагрузки на тредмиле, стенокардии/боли в груди, нестабильной стенокардии, одышки при физической нагрузке, сниженной способности переносить физическую нагрузку, ишемии (уменьшения времени до наступления), «тихой» ишемии (уменьшения времени до наступления), повышенной тяжести и частоты симптомов ишемии, реперфузии после тромболитической терапии при остром инфаркте миокарда, но не ограничивается ими.
Термин "гипертензия", который использован здесь, выбран из группы, состоящей из нарушений липидного обмена с гипертензией, систолической гипертензии и диастолической гипертензии, но не ограничивается ими.
Термин "заболевание периферических сосудов", который использован здесь, выбран из группы, состоящей из заболевания периферических сосудов и хромоты, но не ограничивается ими.
Термин "диабет", который использован здесь, относится к любому из ряда диабетогенных состояний, включая диабет I типа, диабет II типа, синдром X, метаболический синдром, нарушения липидного обмена, связанные с инсулинорезистентностью, нарушение толерантности к глюкозе, инсулинонезависимый сахарный диабет, микрососудистые диабетические осложнения, снижение скорости нервной проводимости, снижение или потерю зрения, диабетическую ретинопатию, повышенный риск ампутации, снижение функции почек, почечную недостаточность, синдром инсулинорезистентности, плюриметаболический синдром, центральное ожирение (висцеральное) (верхнее), диабетическую дислипидемию, уменьшение уровня инсулиновых сенсибилизаторов, диабетическую ретинопатию/нейропатию, диабетическую нейропатию/микро- и макроангиопатию и микро/макроальбуминурию, диабетическую кардиомиопатию, диабетический гастропарез, ожирение, повышенный уровень гликозилированного гемоглобина (включая HbA1C), усиленный контроль глюкозы, нарушение функции почек (диализ, конечная стадия) и функции печени (легкое, умеренное, тяжелое).
"Метаболический синдром", также известный как "синдром X," относится к общему клиническому нарушению, которое определяют как наличие повышенных концентраций инсулина в сочетании с другими нарушениями, включая висцеральное ожирение, гиперлипидемию, дислипидемию, гипергликемию, гипертензию и потенциально гиперурикемию и нарушение функции почек.
Содержание атомов углерода в различных углеводородных группировках указано с помощью префикса, обозначающего минимальное или максимальное число атомов углерода в группировке, то есть префикс Ci-Cj указывает на группировку, содержащую атомы углерода в количестве от целого числа "i" до целого числа "j". Таким образом, например, C1-C3алкил относится к алкилу, содержащему от одного до трех атомов углерода, или метилу, этилу, пропилу и изопропилу, и всем их изомерным формам и прямым и разветвленным формам.
Под термином "галогено" или "галоген" подразумевается хлор, бром, йод или фтор.
Под термином "алкил" подразумевается насыщенный углеводород с прямой цепью или насыщенный углеводород с разветвленной цепью. Примерами таких алкильных групп (при условии, что данная длина охватывает конкретный пример) являются метил, этил, пропил, изопропил, бутил, втор-бутил, третичный бутил, пентил, изопентил, неопентил, третичный пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, гексил, изогексил, гептил и октил. Этот термин также включает насыщенный углеводород (с прямой цепью или разветвленной), где атом водорода удален с каждого концевого атома углерода.
Упоминаемый здесь "алкенил" может быть линейным или разветвленным, и они также могут быть циклическими (например, циклобутенил, циклопентенил, циклогексенил) или бициклическими или могут содержать циклические группы. Они содержат 1-3 двойные углерод-углеродные связи, которые могут иметь цис- или транс-конфигурацию.
Под термином "алкокси" подразумевается насыщенный алкил с прямой цепью или насыщенный алкил с разветвленной цепью, связанный посредством окси. Примерами таких алкоксигрупп (при условии, что данная длина охватывает конкретный пример) являются метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, третичный бутокси, пентокси, изопентокси, неопентокси, тертичный пентокси, гексокси, изогексокси, гептокси и октокси.
Некоторые способы получения соединений по данному изобретению предложены в качестве дополнительных отличительных признаков изобретения и иллюстрируются с помощью приведенных ниже в качестве примера реакционных схем. Специалистам в данной области техники будет понятно, что для того, чтобы синтезировать соединения по изобретению, могут быть использованы другие пути синтеза. Более подробное описание конкретных реакционных стадий смотри в разделе "Примеры", приведенном ниже. Хотя конкретные исходные вещества и реагенты показаны на схемах и рассмотрены ниже, для обеспечения разнообразия производных и/или реакционных условий они могут быть легко заменены другими исходными веществами и реагентами. Кроме того, многие соединения, полученные способами, описанными ниже, могут быть дополнительно модифицированы с учетом данного раскрытия с использованием традиционной химии, хорошо известной специалистам в данной области техники. В частности, следует отметить, что соединения, полученные согласно этим схемам, могут быть дополнительно модифицированы для получения новых соединений по Примерам в объеме данного изобретения. Кроме того, из подробных описаний, данных в экспериментальной части, будет ясно, что используемые способы получения не ограничены общими методиками, описанными здесь.
Исходные вещества обычно имеются в продаже у таких коммерческих поставщиков, как Aldrich Chemicals (Milwaukee, WI), или легко могут быть получены с использованием способов, известных специалистам в данной области техники (например, получены способами, описанными в общем в Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, New York (1967-1999 ed.), или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая дополнения (также доступно через онлайновую базу данных Beilstein)).
В качестве первого шага при получении соединений по настоящему изобретению следует отметить, что при осуществлении некоторых способов получения, используемых для получения соединений, описанных здесь, может потребоваться защита пространственно удаленной функциональной группы (например, первичной аминогруппы, вторичной аминогруппы, карбоксила в промежуточных соединениях). Необходимость такой защиты будет зависеть от природы пространственно удаленной функциональной группы и условий способов получения и легко может быть определена специалистом в данной области техники. Применение таких способов защиты/снятия защиты также находится в компетенции специалиста в данной области техники. Общее описание защитных групп и их применения см. в T.W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
Например, в реакционных схемах, приведенных ниже, некоторые соединения содержат функциональные группы первичных аминов или карбоновых кислот, которые могут вмешиваться во взаимодействия в других участках молекулы, если они останутся незащищенными. Соответственно, такие функциональные группы могут быть защищены с помощью соответствующей защитной группы, которая может быть удалена на последующей стадии. Подходящие защитные группы для защиты амина и карбоновой кислоты включают те защитные группы, которые обычно используют в пептидном синтезе (такие как N-трет-бутоксикарбонил, бензилоксикарбонил и 9-флуоренилметиленоксикарбонил для аминов и низшие алкилэфирные или бензилэфирные группы для карбоновых кислот), которые обычно не являются химически реакционноспособными в описанных реакционных условиях и обычно могут быть удалены без химического изменения другой функциональной группы в соединении.
Схемы, приведенные ниже, наряду с тем, что они изображают рацемические смеси, могут быть использованы для синтеза индивидуальных энантиомеров, начиная с соответствующих хиральных исходных веществ.
На приведенной ниже реакционной схеме I показаны методики получения соединений формулы I, где в R2, X и Y являются такими, как определено выше, R1 представляет собой изохинолин-1-ил, и R3 представляет собой замещенный триазолопиридин или пиразолопиримидин. В общем, эти соединения получают путем добавления хлорангидрида или активированной арилкарбоновой кислоты с получением соответствующих соединений формулы 3 с последующим включением изохинолинилового гетероцикла. Соединения формулы I получают путем дальнейшей модификации соединений формулы 6 посредством катализируемых металлами реакций арильного сочетания с последующим отщеплением трет-бутоксикарбонильной (ВОС) группы с получением соединений формулы 9.
СХЕМА I
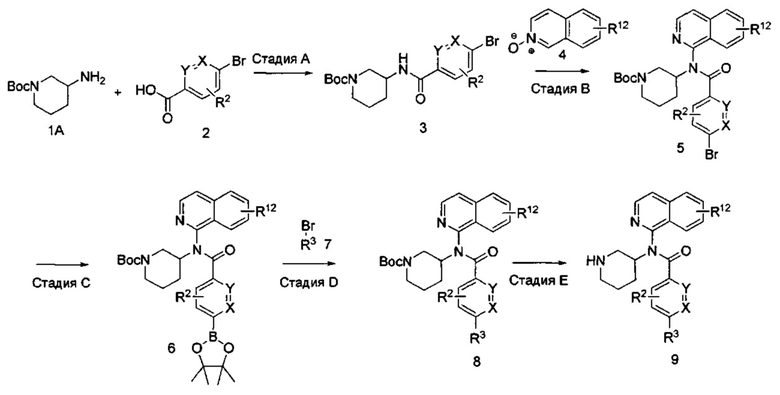
Исходное вещество формулы IA в реакционной схеме I трет-бутил-3-аминопиперидин-1-карбоксилат может быть получен от коммерческих поставщиков. На стадии А амин формулы IA и карбоновую кислоту формулы 2 оставляют для взаимодействия с активирующим агентом, таким как карбонилдиимидазол (CDI) (Lopez-Rodriguez, Maria L. et al, Bioorg. Med. Chem. 2004, 12, 5181-5184), с получением амидных производных формулы 3. В соединении формулы 3 R2 может представлять собой тот же заместитель, который требуется в конечном продукте, или может быть модифицирован после введения с использованием методик, известных в данной области техники, с получением требуемого заместителя. Карбоновая кислота формулы 2 либо может быть получена от коммерческих поставщиков, либо синтезирована специалистами в области химии. Активирующий агент используют в эквимолярных количествах или в небольшом избытке в растворителях, таких как N,N-диметилформамид (DMF) или тетрагидрофуран (THF), с добавлением или без добавления вспомогательных веществ, таких как триэтиламин, 1,8-диазабициклоундец-7-ен или диизопропилэтиламин. Это взаимодействие может быть проведено при температурах от 0°C до 50°C, в зависимости от выбора условий в течение периода времени от примерно 0,5 часа до примерно 18 часов. Альтернативные активирующие агенты включают такие агенты, которые могут активировать карбоновую кислоту до хлорангидрида, например трихлорид фосфора, оксихлорид фосфора (POCl3), тионилхлорид или оксалилхлорид. Альтернативные активирующие агенты могут также включать дегидратирующие агенты, такие как N,N-дициклогексилкарбодиимид (DCC) или гексафторфосфат бензотриазол-1-илокситрис(диметиламино)-фосфония (ВОР).
На стадии B изохинолиновый заместитель вводят в амид формулы 3 с помощью соединения формулы 4, N-оксида изохинолина, с получением соединения формулы 5 (Bilodeau, Mark Т. et al, Org. Lett., 2002, 3127-3129, Londregan, Allyn et al Org. Lett., 2011, 1840-1843). Взаимодействие предпочтительно проводят с активирующим агентом, таким как оксалилхлорид, гексафторфосфат бензотриазол-1-илокси-трис(диметиламино)-фосфония (ВОР), гексафторфосфат бром-трис-пирролидинофосфония (PyBrOP), или подходящим заместителем в растворителях, таких как дихлорметан, 1,4-диоксан, тетрагидрофуран (THF), ацетонитрил и DMF, при температуре от примерно 0°C до примерно 50°C в течение периода времени от примерно 0,5 часа до примерно 24 часов. Кроме того, стадию B проводят в присутствии вспомогательных веществ, таких как диизопропилэтиламин, триэтиламин, 2,6-лутидин или сходные основания.
Стадию С предпочтительно проводят с подходящим боронатным источником, таким как боронат пинакола, в присутствии соединения палладия (например, трис(дибензилиденацетон)дипалладий (Pd2(dba)3), 1,1-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (PdCl2(dppf)2), тетракис(трифенилфосфин)палладий (Pd(PPh3)4)) или другого подходящего катализатора. Взаимодействие проводят при температуре от примерно 23°C до примерно 180°C в течение периода времени от примерно 1 часа до примерно 24 часов. Типичными растворителями для стадии C являются метанол, этанол, вода, ацетонитрил, N,N-диметилформамид (DMF), 1,4-диоксан и тетрагидрофуран (THF). Стадию C проводят в присутствии основания, такого как ацетат калия (КОАс), карбонат цезия (CS2CO3), гидроксид натрия (NaOH), гидроксид калия (КОН), карбонат калия или натрия и бикарбонат натрия (К2СО3, Na2CO3, NaHCO3).
На стадии D вводят R3 посредством реакции кросс-сочетания боронатного эфира формулы 6 и бромида формулы 7 в реакционных условиях, сходных с условиями, применяемыми на стадии С. Заместитель R3 выбран так, чтобы получить требуемый заместитель R3 в соединении формулы I, как определено выше, или заместитель R3 может быть модифицирован после введения посредством использования методик, известных в области химии, с получением альтернативного заместителя R3 (как определено выше).
На стадии E отщепляют трет-бутоксикарбонил (ВОС) с помощью кислот, таких как соляная кислота (HCl), трифторуксусная кислота (TFA), пара-толуолсульфоновая кислота, в водной или безводной среде (например, дихлорметан, тетрагидрофуран, этилацетат, толуол) при температуре от примерно 0°C до примерно 50°C в течение периода времени от примерно 0,5 часа до примерно 18 часов. Специалистам в данной области техники будет понятно, что для отщепления трет-бутоксикарбонильной (ВОС) группы может быть использован ряд других условий. На схеме I R12, возможно, представляет собой моно- или дихлор или (C1-С4)алкил.
СХЕМА II
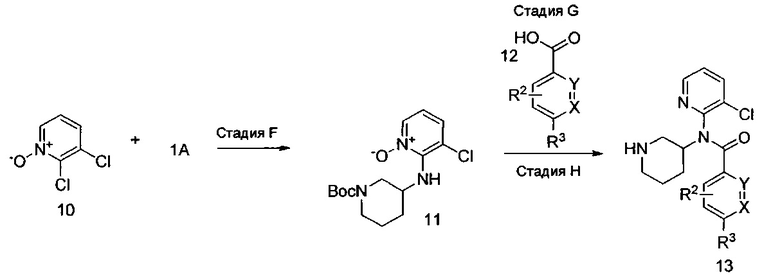
Соединения формулы I, где R2, R3, X и Y являются такими, как определено выше, получают, как показано на схеме II, приведенной выше. На стадии F амин формулы 1А и N-оксид формулы 10 (легко получаемые от коммерческих поставщиков) предпочтительно подвергают взаимодействию в присутствии основания, такого как диизопропилэтиламин, триэтиламин (возможно, со вспомогательным веществом, таким как фторид цезия), ацетат калия, карбонат цезия или другие источники карбоната, в растворителях, таких диметилсульфоксид (DMSO), ацетонитрил или изопропанол, при температуре от примерно 20°C до примерно 160°C в течение периода времени от примерно 1 часа до примерно 24 часов с получением N-оксида формулы 11. На стадии G проводят взаимодействие между карбоновой кислотой формулы 12 и N-оксидом формулы 11 способом, аналогичным используемому на стадии В. заместители R2 и R3 в кислоте формулы 12 выбраны так, чтобы получить требуемые заместители в соединении формулы I, или заместители R2 и R3 могут быть модифицированы после введения посредством использования методик, известных в области химии, с получением альтернативных заместителей R2 и R3 в соединении формулы I. Стадия G включает восстановление N-оксида формулы 11, проводимое в одном сосуде, с последующим отщеплением (стадия Н) трет-бутоксикарбонильной группы (ВОС) способом, аналогичным используемому на стадии Е.
СХЕМА III
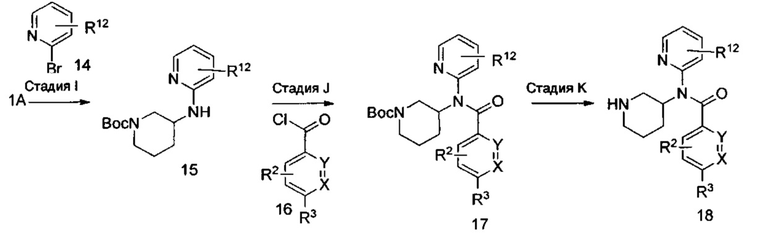
Альтернативно, соединения формулы I, где R2, R3, X и Y являются такими, как определено выше, и R12, возможно, представляет собой моно- или дихлор или (C1-C4)алкил, получают согласно схеме III, приведенной выше. Стадию I предпочтительно проводят с амином формулы 1А и арилбромидом формулы 14 в присутствии палладиевого катализатора, или предкатализатора и лиганда (например, 2-(диметиламинометил)ферроцен-1-ил-палладий(II) хлорид динорборнилфосфин, ацетат палладия (Pd(OAc)2), Brettphos, PEPPSI™, Josiphos, BINAP (2,2'-бис(дифенилфосфино)-1,1'-бинафтил)), или других подходящих катализаторов. Взаимодействие проводят при температуре от примерно 90°C до примерно 150°C в течение периода времени от примерно 1 часа до примерно 24 часов в растворителях, таких как метанол, этанол, вода, ацетонитрил, N,N-диметилформамид (DMF), 1,4-диоксан и THF. Типичным основаниями для этого взаимодействия являются трет-бутоксид калия (KOt-Bu) и карбонат цезия (Cs2CO3). На стадии J соединение формулы 17 синтезируют посредством депротонирования защищенного амина формулы 15 с использованием сильного основания, такого как хлорид метилмагния (MeMgCl), н-бутиллитий (n-BuLi), N,N-диизопропиламин лития, гексаметилдисилилазид лития (LiHMDS) или другие подобные основания, в растворителях, таких как THF, 1,4-диоксан или 1,2-диметоксиэтан (DME), при температуре от примерно -78°C до примерно 23°C в течение периода времени от примерно 1 часа до примерно 4 часов. Введение хлорангидрида формулы 16 при температуре от примерно -10°C до примерно 23°C в течение периода времени от примерно 1 часа до примерно 18 часов дает соединение формулы 17. Хлорангидрид формулы 16 имеется в продаже, или его синтезируют с использованием способов, известных специалистам в области химии. Заместители R2 и R3 в соединении формулы 16 выбраны так, чтобы получить требуемые заместители в соединении формулы I, или заместители R2 и R3 могут быть модифицированы после введения посредством использования методик, известных в области химии, с получением альтернативных заместителей R2 и R3 в соединении формулы I. Стадия K включает отщепление трет-бутоксикарбонильной (ВОС) группы способом, аналогичным используемому на стадиях Е и Н.
СХЕМА IV
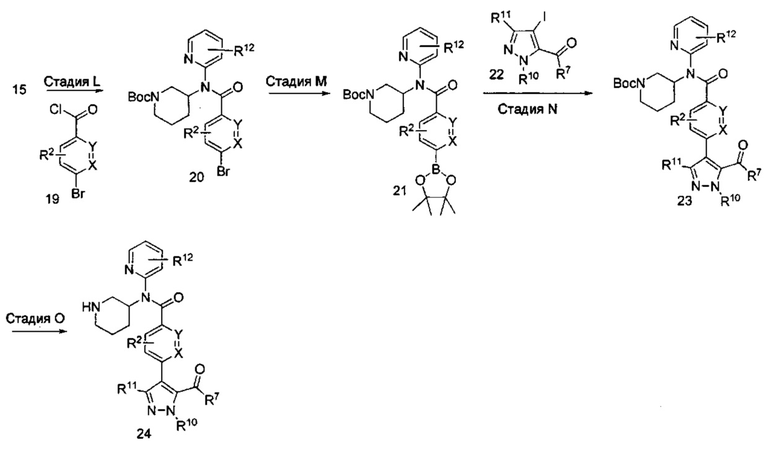
На схеме IV подробно представлен синтез соединений формулы I, где R3 содержит ацилпиразольную терминальную группу (показана в соединении формулы 24). Кроме того, X, Y, R2, R7, R10 и R11 являются такими, как описано выше, и R12, возможно, представляет собой моно- или дихлор или (C1-C4)алкил. Соединения формулы 20 синтезируют путем обработки соединений формулы 15 в уловиях, аналогичных условиям, применяемым на стадии J. На стадии М выделяют боронатный эфир формулы 21, предпочтительно используя катализируемую палладием реакцию кросс-сочетания, аналогичную той, которая описана выше на стадии C или стадии D. На стадии N получают ацилпиразол формулы 23 посредством реакции кросс-сочетания соединения формулы 21 и ацилпиразола формулы 22 в уловиях, аналогичных условиям, применяемым на стадиях C, D или М. Заместители R2, R7, R10 и R11 в ацилпиразоле формулы 22 выбраны так, чтобы получить требуемые заместители в соединении формулы I, или заместители R2, R7, R10 и R11 могут быть модифицированы после введения посредством использования методик, известных в области химии, с получением альтернативных требуемых заместителей R2, R7, R10 и R11 в соединении формулы I. Соединение формулы 24 получают посредством отщепления трет-бутоксикарбонильной группы на стадии O способом, аналогичным используемому выше на стадиях E Н и К.
Аналогично, согласно приведенной ниже схеме V, на стадии P могут быть получены соединения формулы 26 посредством кросс-сочетания соединений формулы 6 и соединений формулы 22 с использованием способов, аналогичных способам, описанным выше на стадиях C, D или М, с последующим отщеплением трет-бутоксикарбонильной (ВОС) группы способом, аналогичным используемому на стадиях E, Н и К.
СХЕМА V
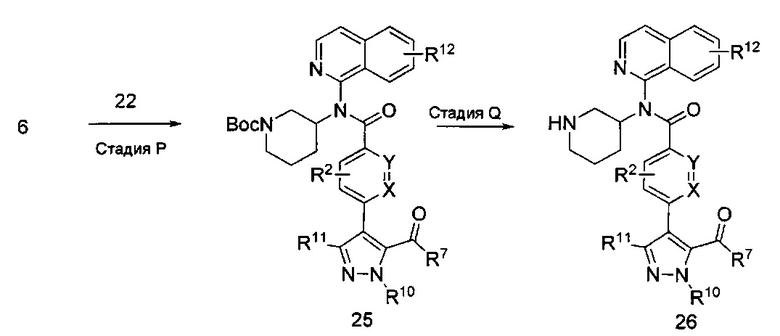
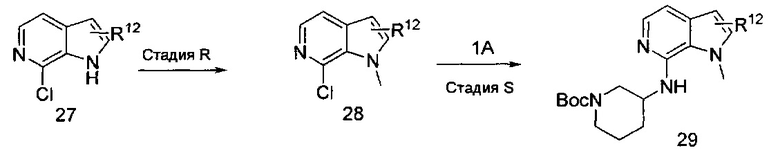
Соединения формулы I, где R2, R3, R12, X и Y являются такими, как описано выше, R1 представляет собой азаиндол-7-ил, получают из защищенного соединения формулы 29, приведенного на схеме VI. Соединение формулы 27 может быть закуплено у коммерческих поставщиков или синтезировано способами, известными специалистам в области химии. На стадии R получают соединение формулы 28 посредством обработки соединения формулы 27 основанием, таким как гидрид натрия (NaH), гидроксид натрия (NaOH), гидроксид калия (КОН) или карбонат калия, возможно, со вспомогательным веществом, таким как бромид тетрабутиламмония, в растворителях, таких как N,N-диметилформамид (DMF), диметилсульфоксид (DMSO) или тетрагидрофуран (THF), с последующим добавлением метилйодида при температуре от примерно 0°C до примерно 50°C в течение периода времени от примерно 1 часа до примерно 18 часов. На стадии S соединение формулы 29 получают посредством реакции кросс-сочетания соединения формулы 28 и соединения формулы 1 способом, аналогичным используемому на стадии I. Соединение формулы 29 может быть дополнительно модифицировано способом, аналогичным модификации соединения формулы 15 в схеме 3, с получением соединений формулы I, где R1 представляет собой азаиндол-7-ил, и R12, возможно, представляет собой моно- или дихлор или (C1-C4)алкил.
СХЕМА VI
На реакционной схеме VII, приведенной ниже, показаны методики получения соединений формулы II, где R2, R10, R11, R12, R15, R16, X и Y являются такими, как определено выше. В общем, эти соединения могут быть получены, начиная с соединений, описанных в схеме IV (например, соединение 23, 24). Соединения формулы 31 могут быть получены путем модификации соединения 30, приведенного на схеме VII, где R7 представляет собой метокси (ОМе) или этокси (OEt), посредством катализируемых реакций амидирования с использованием основания, такого как этилат натрия, метилат натрия и гидроксид калия. Взаимодействия обычно проводят при температурах от примерно 0°C до примерно 23°C, стадия T (Lauder et al, J. Am. Chem. Soc. 39, 659-68; St. Maurice et al, Biochem. 43, 2524-2532).
Альтернативно, соединение формулы 30 (схема VII, где R7 представляет собой гидроксил) может быть обработано активирующим агентом в присутствии амина или обработано активирующим агентом с последующим добавлением амина с получением соединения формулы 31. Аналогично стадии A на схеме I, активирующий агент, такой как карбонилдиимидазол (CDI) (Lopez-Rodriguez, Maria L. et al, Bioorg. Med. Chem. 2004, 12, 5181-5184), может быть использован в эквимолярных количествах или в небольшом избытке в растворителях, таких как N,N-диметилформамид (DMF), дихлорметан (DCM) или тетрагидрофуран (THF), с добавлением или без добавления вспомогательных веществ, таких как триэтиламин, 1,8-диазабициклоундец-7-ен или диизопропилэтиламин. Это взаимодействие может быть проведено при температурах от 0°C до 50°C, в зависимости от выбора условий в течение периода времени от примерно 0,5 часа до примерно 18 часов. Альтернативные активирующие агенты включают трихлорид фосфора, оксихлорид фосфора (POCl3), тионилхлорид или оксалилхлорид. Кроме того, дегидратирующие агенты, такие как N,N-дициклогексилкарбодиимид (DCC) или гексафторфосфат бензотриазол-1-илокситрис(диметиламино)-фосфония (ВОР), могут быть использованы для активации соединения формулы 30.
СХЕМА VII
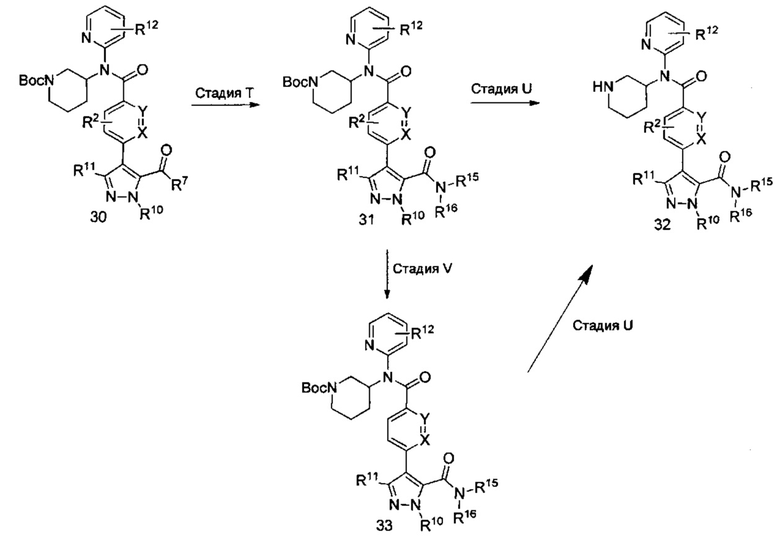
На стадии U, трет-бутоксикарбонил (ВОС) отщепляют с помощью кислот, таких как соляная кислота (HCl), трифторуксусная кислота (TFA), пара-толуолсульфоновая кислота, в водной или безводной среде (например, дихлорметан, тетрагидрофуран, этилацетат или толуол) при температуре от примерно 0°C до примерно 50°C в течение периода времени от примерно 0,5 часа до примерно 18 часов с получением производных (32) соединения формулы II с R15 и R16, как описано выше. Соединения формулы 31 могут быть дополнительно дериватизированы, когда R15 или R16 представляет собой водород, на стадии V способами, известными специалистам в области химии. Обычные условия включают применение алкилхлорида с основанием, таким как триэтиламин или диизопропилэтиламин, возможно, со вспомогательным веществом, таким как бромид тетрабутиламмония, в растворителях, таких как N,N-диметилформамид (DMF), толуол или дихлорметан (DCM), при температуре от примерно 0°C до примерно 50°C в течение периода времени от примерно 1 часа до примерно 18 часов. Как и на стадии U, трет-бутоксикарбонильную группу удаляют с получением производных (32) соединения формулы II с R15 и R16, как описано выше.
СХЕМА VIII
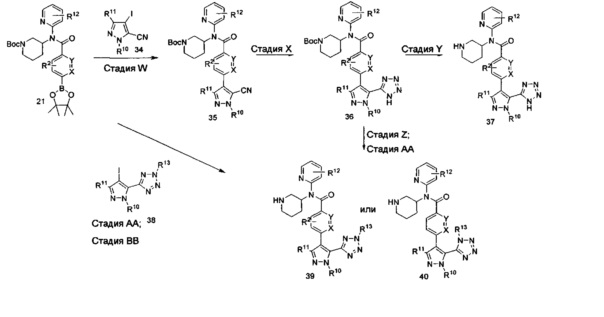
Соединения формулы II также могут быть получены согласно схеме VIII. На стадии W боронат формулы 21 (схема IV) и пиразол формулы 34 объединяют посредством реакции кросс-сочетания в условиях, аналогичных тем, которые используют на стадиях C, D (схема I) или М (схема IV). Заместители R2, R10, R11 и R12 в цианопиразоле формулы 35 выбраны так, чтобы получить требуемые заместители в соединении формулы II, или заместители R2, R10, R11 и R12 могут быть модифицированы после введения посредством использования методик, известных в области химии, с получением альтернативных требуемых заместителей R2, R10, R11 и R12 в соединении формулы II.
На стадии X цианопиразол формулы 35 превращают в тетразольное производное с использованием методик, известных в области химии. Условия для данного превращения включают взаимодействие цианопроизводного с неорганическим, металлорганическим или кремнийорганическим источником азида с кислотой Льюиса или Бренстеда или без нее, но не ограничиваются ими (Roh et al, Eur. J. Org. Chem. 2012, 6101-6118 и пертинентные ссылки в нем). На стадии Y соединения формулы 36 подвергают воздействию кислотных условий, как описано на стадии U схемы VII, для удаления трет-бутоксикарбонильной (ВОС) группы и получения соединений формулы II, где R13 представляет собой Н. Альтернативно, соединения формулы 36 могут быть дополнительно дериватизированы на стадии Z в условиях, аналогичных используемым на стадии V схемы VII, с последующим отщеплением трет-бутоксикарбонильной группы с получением соединений формулы II с R13, как описано выше. На стадии Z взаимодействия соединения формулы 36 с алкилирующими агентами дают два региоизомера формулы 39 и 40, показанные на схеме VIII. Эти региоизомеры могут быть использованы в качестве единого фармацевтического ингредиента или использованы в качестве двух отдельных и отличающихся друг от друга фармацевтических ингредиентов. Затем на стадии АА трет-бутоксикарбонильную группу удаляют как на стадии U схемы VII с получением соединений формулы II, как описано выше. Соединения формулы 39 и 40 также могут быть получены путем взаимодействия соединений формулы 21 с соединениями формулы 38 на стадии BB с использованием условий, аналогичных условиям на стадии W, с последующей стадией АА с получением двух региоизомеров формулы 39 и 40.
В другом воплощении формулы изобретения соединения формулы II могут быть получены с помощью последовательности реакций, показанной на схеме IX. Соединение формулы 40 может быть получено путем первого взаимодействия соединения формулы 35 на стадии СС с гидроксиламином или соответствующей солью гидроксиламина в растворителях, таких как вода, этанол или метанол, с получением производного гидроксиамидина. Взаимодействия обычно проводят при температуре от примерно 23°C до примерно 100°C. Альтернативно, взаимодействия могут быть проведены с гидроксиламином или соответствующей солью гидроксиламина в присутствии щелочи, такой как метилат натрия, этилат натрия или гидроксид калия, в растворителе, таком как метанол, этанол или вода, при температуре от примерно 0°C до примерно 100°C.
На стадии DD производное гидроксиамидина со стадии СС обрабатывают карбонилдиимидазолом (CDI) (Charton, J. et al, Tetrahedron Lett., 2007, 1479-1483) или другим источником карбонила, таким как фосген, этилхлорформиат, в растворителях, таких как толуол, тетрагидрофуран (THF), или 1,4-диоксан. Взаимодействия, подобные этим, обычно проводят в присутствии оснований, таких как триэтиламин, 1,8-диазабициклоундец-7-ен, или диизопропилэтиламин, при температурах от примерно 0°C до примерно 120°C в зависимости от выбора условий в течение периода времени от примерно 0,5 часа до примерно 18 часов.
СХЕМА IX
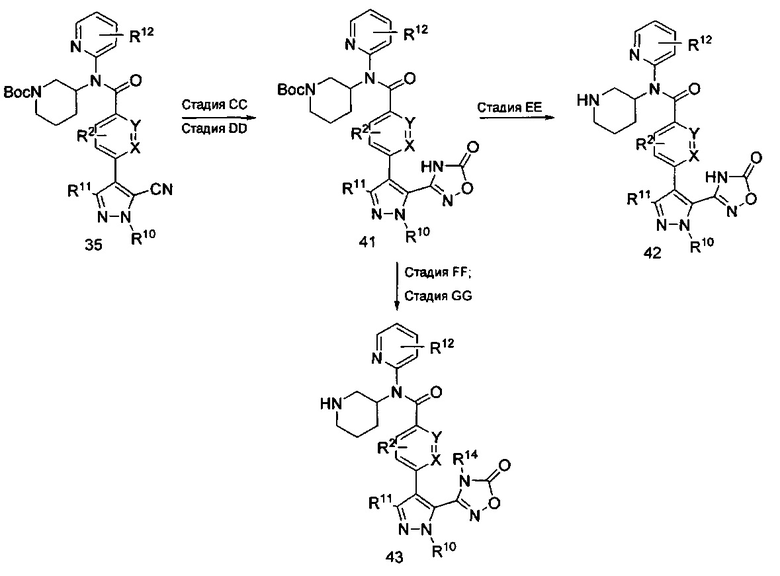
На стадии ЕЕ соединения формулы 41 могут быть помещены в кислотные условия, как описано ранее, для удаления трет-бутоксикарбонила (ВОС) и получения соединений формулы II, где R14 представляет собой Н. Альтернативно, соединения формулы 41 могут быть дополнительно дериватизированы на стадии FF, как показано на стадии V схемы VII и стадии Z схемы VIII, с последующим отщеплением трет-бутоксикарбонильной группы путем проведения стадии GG с получением соединений формулы II с R14, как описано выше.
После завершения взаимодействия требуемое соединение формулы I, представленное на вышеприведенных схемах как соединения формулы 9, 13, 18 и 24, может быть извлечено и выделено способами, известными в данной области техники. Оно может быть выделено посредством упаривания и/или экстракции с использованием методик, известных в данной области техники. Оно может быть, возможно, очищено посредством хроматографии, перекристаллизации, дистилляции или других методик, известных в данной области техники.
Как упомянуто ранее и совершенно очевидно для специалиста в данной области техники, многие из заместителей, которые представляют собой R2, R3, R7, R10 и R11, и возможные заместители по R1 могут подвергаться преобразованиям после получения центральной части соединения формулы I. Например, сульфонильная группировка может быть образована посредством окисления тиоэфира. Такие видоизменения хорошо известны специалистам в данной области техники и должны рассматриваться как часть изобретения.
Соединения формулы I по этому изобретению также могут быть использованы в сочетании с другими фармацевтическими агентами (например, агентами, снижающими уровень холестерина ЛПНП (липопротеинов низкой плотности), агентами, снижающими уровень триглицеридов) для лечения заболевания/состояний, описанных здесь. Например, они могут быть использованы в комбинации с липид-модулирующими агентами, противодиабетическими агентами и сердечно-сосудистыми веществами.
Липид-модулирующие агенты могут быть использованы в качестве агента комбинации в сочетании с соединениями формулы I. Любой ингибитор HMG-СоА-редуктазы может быть использован в комбинации, составляющей аспект настоящего изобретения. Превращение 3-гидрокси-3-метилглутарил-коэнзима A (HMG-CoA) в мевалонат является начальной и лимитирующей скорость реакции стадией в ходе биосинтеза холестерина. Эту стадию катализирует фермент HMG-CoA-редуктаза. Статины ингибируют катализ этого превращения HMG-CoA редуктазой. В следующих параграфах описаны примеры статинов.
Термин "ингибитор HMG-CoA-редуктазы" относится к соединениям, которые ингибируют биопревращение гидроксиметилглутарил-коэнзима A в мевалоновую кислоту, катализируемое ферментом HMG-CoA-редуктазой. Такое ингибирование легко определяется специалистами в данной области техники посредством использования стандартных анализов (например, Meth. Enzymol. 1981; 71: 455-509 и приводимые там ссылки). Ряд таких соединений описан ниже, и на них даны ссылки здесь, однако специалистам в данной области техники известны и другие ингибиторы HMG-CoA-редуктазы. В патенте США №. 4231938 (раскрытие которого включено в данное описание посредством ссылки) раскрыты некоторые соединения, выделенные после культивирования микроорганизма, принадлежащего к роду Aspergillus, такие как ловастатин. Также в патенте США №4444784 (раскрытие которого включено в данное описание посредством ссылки) раскрыты синтетические производные вышеупомянутых соединений, такие как симвастатин. Также в патенте США №4739073 (раскрытие которого включено в данное описание посредством ссылки) раскрыты некоторые замещенные индолы, такие как флувастатин. Также в патенте США №4346227 (раскрытие которого включено в данное описание посредством ссылки) раскрыты ML-236B производные, такие как правастатин. Также в ЕР-491226А (раскрытие которого включено в данное описание посредством ссылки) раскрыты некоторые пиридилдигидроксигептеновые кислоты, такие как церивастатин. Кроме того, в патенте США №5273995 (раскрытие которого включено в данное описание посредством ссылки) раскрыты некоторые 6-[2-(замещенный-пиррол-1-ил)алкил]пиран-2-оны, такие как аторвастатин и любая его фармацевтически приемлемая форма (а именно, LIPITOR®). Дополнительные ингибиторы HMG-СоА-редуктазы включают розувастатин и питавастатин.
Аторвастатин кальций (а именно, аторвастатин гемикальций), раскрытый в патенте США №5273995, который включен в данное описание посредством ссылки, в настоящее время продают как Lipitor®.
Статины также включают такие соединения как розувастатин, раскрытый в US RE37314 E, питивастатин, раскрытый в EP 304063 В1 и патенте США №5011930, симвастатин, раскрытый в патенте США №4444784, который включен в данное описание посредством ссылки; правастатин, раскрытый в патенте США №4346227 который включен в данное описание посредством ссылки; церивастатин, раскрытый в патенте США №5502199, который включен в данное описание посредством ссылки; мевастатин, раскрытый в патенте США №3983140, который включен в данное описание посредством ссылки; велостатин, раскрытый в патенте США №4448784 и №4450171, оба из которых включены в данное описание посредством ссылки; флувастатин, раскрытый в патенте США №4739073, который включен в данное описание посредством ссылки; компактин, раскрытый в патенте США №4804770, который включен в данное описание посредством ссылки; ловастатин, раскрытый в патенте США №4231938, который включен в данное описание посредством ссылки; далвастатин, раскрытый в публикации европейской заявки на патент №738510 А2; флуиндостатин, раскрытый в публикации европейской заявки на патент №363934 А1; и дигидрокомпактин, раскрытый в патенте США №4450171, который включен в данное описание посредством ссылки.
Любой ингибитор HMG-CoA-синтазы может быть использован в комбинации, составляющей аспект настоящего изобретения. Термин "ингибитор HMG-CoA-синтазы" относится к соединениям, которые ингибируют биосинтез гидроксиметилглутарилкоэнзима A из ацетилкоэнзима А и ацетоацетилкоэнзима А, катализируемый ферментом HMG-CoA-синтазой. Такое ингибирование легко определяется специалистами в данной области техники посредством стандартных анализов (Meth. Enzymol. 1975, 35: 155-160; Meth. Enzymol. 1985, 110:19-26 и приводимые там ссылки). Ряд таких соединений описан ниже, и на них даны ссылки ниже, однако специалистам в данной области техники известны и другие ингибиторы HMG-CoA-синтазы. В патенте США №5120729 (раскрытие которого включено в данное описание посредством ссылки) раскрыты некоторые бета-лактамные производные. В патенте США №5064856 (раскрытие которого включено в данное описание посредством ссылки) раскрыты некоторые производные спиролактона, полученные путем культивирования микроорганизма (MF5253). В патенте США №4847271 (раскрытие которого включено в данное описание посредством ссылки) раскрыты некоторые соединения оксетана, такие как производные 11-(3-гидроксиметил-4-оксо-2-оксетаил)-3,5,7-триметил-2,4-ундекадиеновой кислоты.
Любое соединение, которое снижает экспрессию генов HMG-CoA-редуктазы, может быть использовано в комбинации, составляющей аспект настоящего изобретения. Эти агенты могут быть ингибиторами транскрипции HMG-CoA-редуктазы, которые блокируют транскрипцию ДНК, или ингибиторами трансляции, которые предупреждают или снижают трансляцию мРНК, кодирующей HMG-CoA-редуктазу, в белок. Такие соединения либо могут влиять на транскрипцию или трансляцию непосредственно, либо могут быть биотрансформированы в соединения, которые обладают вышеупомянутыми видами активности, под действием одного или нескольких ферментов в каскаде биосинтеза холестерина, либо могут приводить к накоплению изопренового метаболита, обладающего вышеупомянутыми видами активности. Такие соединения могут вызывать этот эффект посредством снижения уровней SREBP (белок, связывающий стеролрегулирующие элементы) путем ингибирования активности сайт-1 протеазы (S1P) или проявления агонизма к рецепторам оксистерола или антагонизма к SCAP белку (белок, активирующий расщепление SREBP). Такая регуляция легко определяется специалистами в данной области техники с использованием стандартных анализов (Meth. Enzymol. 1985; 110: 9-19). Некоторые соединения описаны ниже, и на них даны ссылки ниже, однако специалистам в данной области техники известны и другие ингибиторы экспрессии генов HMG-CoA-редуктазы. В патенте США №5041432 (раскрытие которого включено в данное описание посредством ссылки) раскрыты некоторые производные 15-замещенного ланостерола.
Другие окисленные стеролы, которые подавляют синтез HMG-CoA-редуктазы, рассмотрены E.I. Mercer (Prog. Lip.Res. 1993; 32:357-416).
Любое соединение, обладающее активностью ингибитора СЕТР, может служить в качестве второго соединения в комбинированной терапии, составляющей аспект настоящего изобретения. Термин "ингибитор СЕТР" относится к соединениям, которые ингибируют опосредованный белком-переносчиком холестериновых эфиров (СЕТР) транспорт различных холестериновых эфиров и триглицеридов с HDL (липопротеин высокой плотности) на LDL (липопротеин низкой плотности) и VLDL (липопротеин очень низкой плотности). Такая ингибиторная активность в отношении СЕТР легко определяется специалистами в данной области техники с использованием стандартных анализов (например, патент США №6140343). Ряд ингибиторов СЕТР известен специалистам в данной области техники, например те, которые раскрыты в переуступленном должным образом патенте США №6140343 и переуступленном должным образом патенте США №6197786. Ингибиторы СЕТР также описаны в патенте США №6723752, который включает ряд ингибиторов СЕТР, включая (2R)-3-{[3-(4-хлор-3-этил-фенокси)-фенил]-[[3-(1,1,2,2-тетрафтор-этокси)-фенил]-метил]-амино}-1,1,1-трифтор-2-пропанол. Кроме того, ингибиторы СЕТР, включенные в настоящее изобретение, также описаны в заявке на патент США №10/807838, поданной 23 марта 2004 года. В патенте США №5512548 раскрыты некоторые полипептидные производные, обладающие активностью ингибиторов СЕТР, тогда как некоторые ингибиторы СЕТР, производные розенонлактона и фосфат-содержащие аналоги холестеринового эфира раскрыты в J. Antibiot, 49 (8): 815-816 (1996) и Bioorg. Med. Chem. Lett.; 6: 1951-1954 (1996), соответственно.
Любой модулятор PPAR может быть использован в комбинации, составляющей аспект настоящего изобретения. Термин "модулятор PPAR" относится к соединениям, которые модулируют активность рецепторов, активируемых пролифераторами пероксисом (PPAR), у млекопитающих, особенно у людей. Такая модуляция легко определяется специалистами в данной области техники с использованием стандартных анализов, известных в литературе. Полагают, что такие соединения, модулируя рецептор PPAR, регулируют транскрипцию ключевых генов, вовлеченных в метаболизм липидов и глюкозы, таких как гены, участвующие в окислении жирных кислот и также гены, вовлеченные в сборку липопротеинов высокой плотности (HDL) (например, транскрипцию гена аполипопротеина Al), тем самым снижая общую жировую массу тела и повышая уровень холестерина HDL. В силу своей активности эти соединения также снижают уровни триглицеридов, холестерина VLDL, холестерина LDL и связанных с ними компонентов, таких как аполипопротеин B, в плазме крови у млекопитающих, особенно у людей, а также повышают уровень холестерина HDL и аполипопротеина A1. Поэтому, эти соединения полезны для лечения и коррекции различных наблюдаемых дислипидемий, связанных с развитием и возникновением атеросклероза и сердечно-сосудистого заболевания, включая гипо-альфа-липопротеинемию и гипертриглицеридемию. Ряд таких соединений описан, и на них даны ссылки ниже, однако специалистам в данной области техники известны и другие. В международной публикации № WO 02/064549 и 02/064130 и заявке на патент США 10/720942, поданной 24 ноября 2003 года, и заявке на патент США 60/552114, поданной 10 марта 2004 года (раскрытия которых включены в данное описание посредством ссылки), раскрыты некоторые соединения, которые являются активаторами PPARα.
Любой другой модулятор PPAR может быть использован в комбинации, составляющей аспект настоящего изобретения. В частности, модуляторы PPARβ и/или PPARγ могут быть использованы в комбинации с соединениями по настоящему изобретению. Пример ингибитора PPAR описан в US 2003/0225158 как {5-метокси-2-метил-4-[4-(4-трифторметил-бензилокси)-бензилсульфанил]-фенокси}-уксусная кислота.
Любой ингибитор секреции МТР/Аро B (микросомального белка, переносящего триглицериды и/или аполипопротеина В) может быть использован в комбинации, составляющей аспект настоящего изобретения. Термин "ингибитор секреции МТР/Аро B" относится к соединениям, которые ингибируют секрецию триглицеридов, эфира холестерина и фосфолипидов. Такое ингибирование легко определяется специалистами в данной области техники с использованием стандартных анализов (например, Wetterau, J.R. 1992; Science 258: 999). Ряд ингибиторов секреции МТР/Аро B известен специалистам в данной области техники, включая импутаприд и дополнительные соединения, такие как соединения, раскрытые в WO 96/40640 и WO 98/23593 (две иллюстративные публикации).
Любой ингибитор сквален-синтетазы может быть использован в комбинации, составляющей аспект настоящего изобретения. Термин "ингибитор сквален-синтетазы" относится к соединениям, которые ингибируют конденсацию 2 молекул фарнезилпирофосфата с образованием сквалена, катализируемую ферментом сквален-синтетазой. Такое ингибирование легко определяется специалистами в данной области техники с использованием стандартных анализов (Meth. Enzymol. 1969; 15: 393-454 и Meth. Enzymol. 1985; 110: 359-373 и содержащиеся в них ссылки). Ряд таких соединений описан, и на них даны ссылки ниже, однако специалистам в данной области техники известны и другие ингибиторы сквален-синтетазы. В патенте США №5026554 (раскрытие которого включено в данное описание посредством ссылки) раскрыты продукты ферментации микроорганизма MF5465 (АТСС 74011), включающие зарагозовую кислоту. Был составлен краткий обзор других запатентованных ингибиторов сквален-синтетазы (Curr. Op. Ther. Patents (1993) 861-4).
Любой ингибитор сквален-эпоксидазы может быть использован в комбинации, составляющей аспект настоящего изобретения. Термин "ингибитор сквален-эпоксидазы" относится к соединениям, которые ингибируют биоконверсию сквалена и молекулярного кислорода в сквален-2,3-эпоксид, катализируемую ферментом сквален-эпоксидазой. Такое ингибирование легко определяется специалистами в данной области техники с использованием стандартных анализов (Biochim. Biophys. Acta 1984; 794: 466-471). Ряд таких соединений описан, и на них даны ссылки ниже, однако специалистам в данной области техники известны и другие ингибиторы сквален-эпоксидазы. В патентах США №. 5011859 и 5064864 (раскрытия которых включены в данное описание посредством ссылки) раскрыты некоторые фтораналоги сквалена. В публикации ЕР 395768 A (раскрытие которой включено в данное описание посредством ссылки) раскрыты некоторые замещенные производные аллиламина. В публикации РСТ WO 9312069 A (раскрытие которой включено в данное описание посредством ссылки) раскрыты некоторые производные аминоспиртов. В патенте США №5051534 (раскрытие которого включено в данное описание посредством ссылки) раскрыты некоторые производные циклопропилоксисквалена.
Любой ингибитор сквален-циклазы может быть использован в качестве второго компонента в комбинации, составляющей аспект настоящего изобретения. Термин "ингибитор сквален-циклазы" относится к соединениям, которые ингибируют биоконверсию сквален-2,3-эпоксида в ланостерин, катализируемую ферментом сквален-циклазой. Такое ингибирование легко определяется специалистами в данной области техники с использованием стандартных анализов (FEBS Lett. 1989; 244: 347-350). Кроме того, соединения, описанные и упомянутые ниже, являются ингибиторами сквален-циклазы, однако специалистам в данной области техники известны и другие ингибиторы сквален-циклазы. В публикации РСТ WO 9410150 (раскрытие которой включено в данное описание посредством ссылки) раскрыты некоторые производные 1,2,3,5,6,7,8,8а-октагидро-5,5,8(бета)-триметил-6-изохинолинамина, такие как N-трифторацетил-1,2,3,5,6,7,8,8α-октагидро-2-аллил-5,5,8(бета)-триметил-6(бета)-изохинолинамин. В публикации патента Франции №2697250 (раскрытие которого включено в данное описание посредством ссылки) раскрыты некоторые производные бета, бета-диметил-4-пиперидинэтанола, такие как 1-(1,5,9-триметилдецил)-бета,бета-диметил-4-пиперидинэтанол.
Любой комбинированный ингибитор сквален-эпоксидазы/сквален-циклазы может быть использован в качестве второго компонента в комбинации, составляющей аспект настоящего изобретения. Термин "комбинированный ингибитор сквален-эпоксидазы/сквален-циклазы" относится к соединениям, которые ингибируют биоконверсию сквалена в ланостерин через промежуточное соединение сквален-2,3-эпоксид. В некоторых анализах невозможно найти различия между ингибиторами сквален-эпоксидазы и ингибиторами сквален-циклазы; тем не менее, эти анализы признаны специалистами в данной области техники. Таким образом, ингибирование комбинированными ингибиторами сквален-эпоксидазы/сквален-циклазы легко определяется специалистами в данной области техники посредством вышеупомянутых стандартных анализов для ингибиторов сквален-циклазы или сквален-эпоксидазы. Ряд таких соединений описан, и на них даны ссылки ниже, однако специалистам в данной области техники известны и другие ингибиторы сквален-эпоксидазы/сквален-циклазы. В патентах США №5084461 и 5278171 (раскрытия которых включены в данное описание посредством ссылки) раскрыты некоторые производные азадекалина. В публикации EP 468434 (раскрытие которой включено в данное описание посредством ссылки) раскрыты некоторые производные пиперидилового эфира и тиоэфира, такие как 2-(1-пиперидил)пентилизопентилсульфоксид и 2-(1-пиперидил)этил-этилсульфид. В публикации РСТ WO 9401404 (раскрытие которой включено в данное описание посредством ссылки) раскрыты некоторые ацилпиперидины, такие как 1-(1-оксопентил-5-фенилтио)-4-(2-гидрокси-1-метил)этил)пиперидин. В патенте США №5102915 (раскрытие которого включено в данное описание посредством ссылки) раскрыты некоторые производные циклопропилоксисквалена.
Соединения по настоящему изобретению также могут быть введены в комбинации с природными соединениями, которые действуют, снижая уровни холестерина в плазме. Эти природные соединения обычно называют нутрицевтиками, и они включают, например, экстракт чеснока и ниацин. Имеется форма ниацина с медленным высвобождением, которая известна как ниаспан. Ниацин можно также комбинировать с другими терапевтическими агентами, такими как ловастатин или другой ингибитор HMG-CoA-редуктазы. Этот комбинированный препарат с ловастатином известен как ADVICOR™ (Kos Pharmaceuticals Inc.).
Любой ингибитор абсорбции холестерина может быть использован в качестве дополнительного соединения в комбинации, составляющей аспект настоящего изобретения. Термин "ингибитор абсорбции холестерина" относится к способности соединения предотвращать проникновение холестерина, содержащегося в просвете кишечника, в интестинальные клетки и/или выход из интестинальных клеток в лимфатическую систему и/или кровоток. Такая ингибирующая активность в отношении абсорбции холестерина легко определяется специалистами в данной области техники с использованием стандартных анализов (например, J. Lipid Res. (1993) 34: 377-395). Ингибиторы абсорбции холестерина известны специалистам в данной области техники и описаны, например, в РСТ WO 94/00480. Примером ингибитора абсорбции холестерина является ZETIA™ (эзетимиб) (Schering-Plough/Merck).
Любой ингибитор АСАТ может быть использован в комбинированной терапии, составляющей аспект настоящего изобретения. Термин "ингибитор АСАТ" относится к соединениям, которые ингибируют внутриклеточную этерификацию пищевого холестерина посредством фермента ацил-СоА: холестерин-ацилтрансферазы. Такое ингибирование может быть легко определено специалистом в данной области техники с использованием стандартных анализов, таких как методика Heider et al., описанная в Journal of Lipid Research., 24: 1127 (1983). Ряд таких соединений известен специалистам в данной области техники, например, в патенте США №5510379 раскрыты некоторые карбоксисульфонаты, тогда как и в WO 96/26948, и в WO 96/10559 раскрыты производные мочевины, обладающие ингибирующей активностью в отношении АСАТ. Примеры ингибиторов АСАТ включают соединения, такие как авасимиб (Pfizer), CS-505 (Sankyo) и эфлюцимиб (Eli Lilly и Pierre Fabre).
Ингибитор липазы может быть использован в комбинированной терапии, составляющей аспект настоящего изобретения. Ингибитор липазы представляет собой соединение, которое ингибирует метаболическое расщепление пищевых триглицеридов или фосфолипидов плазмы на свободные жирные кислоты и соответствующие глицериды (например, EL, HL, и так далее). В нормальных физиологических условиях липолиз осуществляется двухстадийным процессом, который включает ацилирование активированной сериновой группировки фермента липазы. Это приводит к получению полуацетального промежуточного соединения жирная кислота-липаза, которое затем расщепляется с высвобождением диглицерида. После дальнейшего деацилирования промежуточное соединение липаза-жирная кислота расщепляется, что в результате дает свободную липазу, глицерид и жирную кислоту. В кишечнике получающиеся в результате свободные жирные кислоты и моноглицериды включаются в мицеллы, покрытые оболочкой из желчной кислоты-фосфолипида, которые затем абсорбируются на уровне щеточной каемки тонкой кишки. Мицеллы в конечном счете попадают в периферический кровоток в виде хиломикронов. Такая ингибирующая активность в отношении липазы легко определяется специалистами в данной области техники с использованием стандартных анализов (например, Methods Enzymol. 286: 190-231).
Панкреатическая липаза опосредует метаболическое расщепление жирных кислот из триглицеридов по 1- и 3-му положениям углерода. В двенадцатиперстной кишке и проксимальном отделе тощей кишки находится первичный сайт метаболизма поглощенных жиров под действием панкреатической липазы, которая обычно секретируется в огромном избытке относительно количеств, необходимых для расщепления жиров в верхнем отделе тонкой кишки. Поскольку панкреатическая липаза представляет собой первичный фермент, требующийся для абсорбции пищевых триглицеридов, ингибиторы являются полезными при лечении ожирения и других родственных состояний. Такая ингибирующая активность в отношении панкреатической липазы легко определяется специалистами в данной области техники с использованием стандартных анализов (например, Methods Enzymol. 286: 190-231).
Желудочная липаза представляет собой иммунологически различимую липазу, которая ответственна за приблизительно 10-40% расщепления пищевых жиров. Желудочная липаза секретируется в ответ на механическую стимуляцию, поглощение пищи, присутствие жирной пищи или с помощью симпатических агентов. Желудочный липолиз поглощенных жиров имеет физиологическое значение при обеспечении жирных кислот, необходимых для инициирования активности панкреатической липазы в кишечнике, и также имеет значение для абсорбции жиров при ряде физиологических и патологических состояний, связанных с недостаточностью поджелудочной железы. См., например, С.К. Abrams, et al., Gastroenterology, 92,125 (1987). Такая ингибирующая активность в отношении желудочной липазы легко определяется специалистами в данной области техники с использованием стандартных анализов (например, Methods Enzymol. 286: 190-231).
Специалисту в данной области техники известен ряд ингибиторов желудочной и/или панкреатической липазы. Предпочтительные ингибиторы липазы представляют собой такие ингибиторы, которые выбраны из группы, состоящей из липстатина, тетрагидролипстатина (орлистата), валилактона, эстерастина, эбелактона A и эбелактона В. Соединение тетрагидролипстатин является особенно предпочтительным. Ингибитор липазы N-3-трифторметилфенил-N'-3-хлор-4'-трифторметилфенилмочевина, а также различные родственные ему производные мочевины раскрыты в патенте США №4405644. Ингибитор липазы эстерацин раскрыт в патентах США №4189438 и 4242453. Ингибитор липазы цикло-O,O'-[(1,6-гександиил)-бис-(иминокарбонил)]диоксим и различные родственные ему бис(иминокарбонил)диоксимы могут быть получены, как описано в Petersen et al., Liebig's Annalen, 562, 205-229 (1949).
Ниже описан ряд ингибиторов панкреатической липазы. Ингибитор панкреатической липазы липстатин, лактон (2S,3S,5S,7Z,10Z)-5-[(S)-2-формамидо-4-метилвалерилокси]-2-гексил-3-гидрокси-7,10-гексадекановой кислоты и тетрагидролипстатин (орлистат), лактон (2S,3S,5S)-5-[(S)-2-формамидо-4-метилвалерилокси]-2-гексил-3-гидрокси-1,3-гексадекановой кислоты и различным образом замещенные N-формиллейциновые производные и их стереоизомеры раскрыты в патенте США №4598089. Например, тетрагидролипстатин получают как описано, например, в патентах США №5274143, 5420305, 5540917 и 5643874. Ингибитор панкреатической липазы FL-386 1-[4-(2-метилпропил)циклогексил]-2-[(фенилсульфонил)окси]-этанон и различным образом замещенные родственные ему сульфонатные производные раскрыты в патенте США №4452813. Ингибитор панкреатической липазы WAY-121898, 4-феноксифенил-4-метилпиперидин-1-илкарбоксилат и различные родственные ему карбаматные эфиры и фармацевтически приемлемые соли раскрыты в патентах США №5512565, 5391571 и 5602151. Ингибитор панкреатической липазы валилактон и способ его получения путем культивирования микроорганизмов рода Actinomyces, штамм MG147-CF2, раскрыты в Kitahara, et al., J. Antibiotics, 40 (11), 1647-1650 (1987). Ингибиторы панкреатической липазы эбелактон A и эбелактон B и способ их получения путем культивирования микроорганизмов рода Actinomyces, штамм MG7-G1, раскрыты в Umezawa, et al., J. Antibiotics, 33, 1594-1596 (1980). Применение эбелактонов A и B в подавлении образования моноглицеридов раскрыто в Japanese Kokai 08-143457, опубликованном 4 июня 1996 года.
Другие соединения, которые предлагаются на рынке для лечения гиперлипидемии, включая гиперхолестеринемию, и которые призваны помочь предотвратить или лечить атеросклероз, включают секвестранты желчных кислот, такие как Welchol®, Colestid®, LoCholest® и Questran®; и производные фиброевой кислоты, такие как Atromid®, Lopid® и Tricor®.
Принимая во внимание взаимосвязь между диабетом и атеросклерозом (например, метаболический синдром) соединения формулы I могут быть введены с противодиабетическими соединениями. Диабет можно лечить путем введения пациенту, страдающему диабетом (в особенности II типа), инсулинорезистентностью, нарушением толерантности к глюкозе, метаболическим синдромом и тому подобным, или любым из осложнений диабета, таких как нейропатия, нефропатия, ретинопатия или катаракты, терапевтически эффективного количества соединения по настоящему изобретению в комбинации с другими агентами (например, инсулином), которые могут использоваться для лечения диабета. Они включают классы описанных здесь противодиабетических агентов (и специфических агентов).
Любой ингибитор гликогенфосфорилазы может быть использован в качестве второго агента в комбинации с соединением по настоящему соединению. Термин "ингибитор гликогенфосфорилазы" относится к соединениям, которые ингибируют биоконверсию гликогена в глюкозо-1-фосфат, которая катализируется ферментом гликогенфосфорилазой. Такая ингибирующая активность в отношении гликогенфосфорилазы легко определяется специалистами в данной области техники с использованием стандартных анализов (например, J. Med. Chem. 41 (1998) 2934-2938). Ряд ингибиторов гликогенфосфорилазы известен специалистам в данной области техники, включая те, которые описаны в WO 96/39384 и WO 96/39385.
Любой ингибитор альдозоредуктазы может быть использован в комбинации с соединением по настоящему соединению. Термин "ингибитор альдозоредуктазы" относится к соединениям, которые ингибируют биоконверсию глюкозы в сорбит, которая катализируется ферментом альдозоредуктазой. Ингибирование альдозоредуктазы легко определяется специалистами в данной области техники с использованием стандартных анализов (например, J. Malone, Diabetes, 29: 861-864 (1980). "Red Cell Sorbitol, an Indicator of Diabetic Control"). Ряд ингибиторов альдозоредуктазы известен специалистам в данной области техники, например, такие, как описано в патенте США №6579879, который включает 6-(5-хлор-3-метил-бензофуран-2-сульфонил)-2Н-пиридазин-3-он.
Любой ингибитор сорбитдегидрогеназы может быть использован в комбинации с соединением по настоящему соединению. Термин "ингибитор сорбитдегидрогеназы" относится к соединениям, которые ингибируют биоконверсию сорбита до фруктозы, которая катализируется ферментом сорбитдегидрогеназой. Такая ингибирующая активность в отношении сорбитдегидрогеназы легко определяется специалистами в данной области техники с использованием стандартных анализов (например, Analyt. Biochem (2000) 280: 329-331). Известен ряд ингибиторов сорбитдегидрогеназы, например, в патентах США №5728704 и 5866578 раскрыты соединения и способ лечения или предупреждения осложнений диабета путем ингибирования фермента сорбитдегидрогеназы.
Любой ингибитор глюкозидазы может быть использован в комбинации с соединением по настоящему соединению. Ингибитор глюкозидазы ингибирует ферментативный гидролиз сложных углеводов гликозидгидролазами, например амилазой или мальтазой, на биологически доступные простые сахара, например глюкозу. Быстрое метаболическое действие глюкозидаз, в особенности после поглощения высоких уровней углеводов, приводит в результате к состоянию алиментарной гипергликемии, которая у субъектов, имеющих излишний жир или страдающих диабетом, приводит к повышенной секреции инсулина, увеличению синтеза жиров и уменьшению разрушения жиров. После таких гипергликемий часто возникает гипогликемия вследствие увеличения уровней присутствующего инсулина. Кроме того, известно, что химус, оставшийся в желудке, способствует выработке желудочного сока, который инициирует развитие или способствует развитию гастрита или язв двенадцатиперстной кишки. Соответственно, известно, что ингибиторы глюкозидазы полезны для ускорения прохождения углеводов через желудок и ингибирования абсорбции глюкозы из кишечника. Кроме того, превращение углеводов в липиды жировой ткани и последующее включение алиментарного жира в отложения жировой ткани соответственно уменьшается или замедляется, при сопутствующей пользе, выражающейся в уменьшении или предупреждении возникающих в результате вредных аномалий. Такая ингибирующая активность в отношении глюкозидазы легко определяется специалистами в данной области техники с использованием стандартных анализов (например, Biochemistry (1969) 8: 4214).
В большинстве случаев предпочтительный ингибитор глюкозидазы включает в себя ингибитор амилазы. Ингибитор амилазы представляет собой ингибитор глюкозидазы, который ингибирует ферментативное расщепление крахмала или гликогена до мальтозы. Такая ингибирующая активность в отношении амилазы легко определяется специалистами в данной области техники с использованием стандартных анализов (например, Methods Enzymol. (1955) 1: 149). Ингибирование такого ферментативного расщепления является полезным в уменьшении количеств биологически доступных Сахаров, включая глюкозу и мальтозу, и возникающих в результате сопутствующих вредных состояний.
Ряд ингибиторов глюкозидазы известен специалисту в данной области техники, и примеры приведены ниже. Предпочтительными ингибиторами глюкозидазы являются такие ингибиторы, которые выбраны из группы, состоящей из акарбозы, адипозина, воглибозы, миглитола, эмиглитата, камиглибозы, тендамистата, трестатина, прадимицина-Q и сальбостатина. Ингибитор глюкозидазы акарбоза и различные родственные ей производные аминосахара раскрыты в патентах США №4062950 и 4174439, соответственно. Ингибитор глюкозидазы адипозин раскрыт в патенте США №4254256. Ингибитор глюкозидазы воглибоза, 3,4-дидезокси-4-[[2-гидрокси-1-(гидроксиметил)этил]амино]-2-С-(гидроксиметил)-D-эпиинозит, и различные родственные ей N-замещенные псевдоаминосахара раскрыты в патенте США №4701559. Ингибитор глюкозидазы миглитол, (2R,3R,4R,5S)-1-(2-гидроксиэтил)-2-(гидроксиметил)-3,4,5-пиперидинтриол, и различные родственные ему 3,4,5-тригидроксипиперидины раскрыты в патенте США №4639436. Ингибитор глюкозидазы эмиглитат, этил-пара-[2-[(2R,3R,4R,5S)-3,4,5-тригидрокси-2-(гидроксиметил)пиперидино]этокси]бензоат, различные родственные ему производные и его фармацевтически приемлемые соли присоединения кислоты раскрыты в патенте США №5192772. Ингибитор глюкозидазы MDL-25637, 2,6-дидезокси-7-O-β-D-глюкопиранозил-2,6-имино-D-глицеро-L-глюкогептитол, различные родственные ему гомодисахариды и его фармацевтически приемлемые соли присоединения кислоты раскрыты в патенте США №4634765. Ингибитор глюкозидазы камиглибоза, сесквигидрат мeтил-6-дeзoкcи-6-[(2R,3R,4R,5S)-3,4,5-тpигидpoкcи-2-(гидpoкcимeтил)-пиперидино]-α-D-глюкопиранозида, родственные ему производные дезоксиноджиримицина, различные их фармацевтически приемлемые соли и синтетические способы их получения раскрыты в патентах США №5157116 и 5504078. Ингибитор глюкозидазы сальбостатин и различные родственные ему псевдосахариды раскрыты в патенте США №5091524.
Ряд ингибиторов амилазы известен специалисту в данной области техники. Ингибитор амилазы тендамистат и различные родственные ему циклические пептиды раскрыты в патенте США №4451455. Ингибитор амилазы А1-3688 и различные родственные ему циклические полипептиды раскрыты в патенте США №4623714. Ингибитор амилазы трестатин, состоящий из смеси трестатина A, трестатина B и трестатина C, и различные родственные ему содержащие трегалозу аминосахара раскрыты в патенте США №4273765.
Дополнительные противодиабетические соединения, которые могут быть использованы в качестве второго агента в комбинации с соединением по настоящему изобретению, включают, например, следующие: бигуаниды (например, метформин), средства, усиливающие секрецию инсулина (например, сульфонилмочевины и глиниды), глитазоны, неглитазоновые агонисты PPARγ, агонисты PPARβ, ингибиторы DPP-IV (дипептидилпептидазы-IV), ингибиторы PDE5 (фосфодиэстеразы 5), ингибиторы GSK-3 (киназы-3 гликогенсинтазы), антагонисты глюкагона, ингибиторы f-1,6-BPase (фруктозо-1,6-бисфосфатазы)(Metabasis/Sankyo), GLP-1 (глюкагоноподобный пептид-1)/аналоги (АС 2993, также известные как эксендин-4), инсулин и миметики инсулина (Merck natural products). Другие примеры могут включать ингибиторы РКС-β (протеинкиназы C-β) и расщепители AGE (конечных продуктов ускоренного гликозилирования).
Соединения по настоящему изобретению также могут быть использованы в комбинации с другими сердечно-сосудистыми веществами, такими как антигипертензивные агенты. Любой антигипертензивный агент может быть использован в качестве второго агента в таких комбинациях, и примеры приведены в данном описании. Такая антигипертензивная активность легко определяется специалистами в данной области техники с использованием стандартных анализов (например, измерения кровяного давления).
Амлодипин и родственные дигидропиридиновые соединения раскрыты в патенте США №4572909, который включен в данное описание посредством ссылки, в качестве сильнодействующих противоишемических и антигипертензивных агентов. В патенте США №4879303, который включен в данное описание посредством ссылки, раскрыта бензолсульфонатная соль амлодипина (называемая также амлодипина бесилатом). Амлодипин и амлодипина бесилат являются сильнодействующими и длительно действующими блокаторами кальциевых каналов. Как таковые амлодипин, амлодипина бесилат, амлодипина малеат и другие фармацевтически приемлемые соли присоединения кислоты амлодипина полезны в качестве антигипертензивных агентов и в качестве противоишемических агентов. Амлодипина бесилат в настоящее время продается как Norvasc®.
Блокаторы кальциевых каналов, которые входят в объем этого изобретения, включают: бепридил, который может быть получен, как раскрыто в патенте США №3962238 или заменяющем патенте США №30577; клентиазем, который может быть получен, как раскрыто в патенте США №4567175; дилтиазем, который может быть получен, как раскрыто в патенте США №3562 фендилин, который может быть получен, как раскрыто в патенте США №3262977; галлопамил, который может быть получен, как раскрыто в патенте США №3261859; мибефрадил, который может быть получен, как раскрыто в патенте США №4808605; прениламин, который может быть получен, как раскрыто в патенте США №3152173; семотиадил, который может быть получен, как раскрыто в патенте США №4786635; теродилин, который может быть получен, как раскрыто в патенте США №3371014; верапамил, который может быть получен, как раскрыто в патенте США №3261859; аранипин, который может быть получен, как раскрыто в патенте США №4572909; барнидипин, который может быть получен, как раскрыто в патенте США №4220649; бенидипин, который может быть получен, как раскрыто в публикации заявки на европейский патент №106275; цилнидипин, который может быть получен, как раскрыто в патенте США №4672068; эфонидипин, который может быть получен, как раскрыто в патенте США №4885284; элгодипин, который может быть получен, как раскрыто в патенте США №4952592; фелодипин, который может быть получен, как раскрыто в патенте США №4264611; исрадипин, который может быть получен, как раскрыто в патенте США №4466972; лацидипин, который может быть получен, как раскрыто в патенте США №4801599; лерканидипин, который может быть получен, как раскрыто в патенте США №4705797; манидипин, который может быть получен, как раскрыто в патенте США №4892875; никардипин, который может быть получен, как раскрыто в патенте США №3985758; нифедипин, который может быть получен, как раскрыто в патенте США №3485847; нилвадипин, который может быть получен, как раскрыто в патенте США №4338322; нимодипин, который может быть получен, как раскрыто в патенте США №3799934; нисолдипин, который может быть получен, как раскрыто в патенте США №4154839; нитрендипин, который может быть получен, как раскрыто в патенте США №3799934; циннаризин, который может быть получен, как раскрыто в патенте США №2882271; флунаризин, который может быть получен, как раскрыто в патенте США №3773939; лидофлазин, который может быть получен, как раскрыто в патенте США №3267104; ломеризин, который может быть получен, как раскрыто в патенте США №4663325; бенциклан, который может быть получен, как раскрыто в патенте Венгрии №151865; этафенон, который может быть получен, как раскрыто в патенте Германии №1265758; и пергексилин, который может быть получен, как раскрыто в патенте Великобритании №1025578, но не ограничиваются ими. Раскрытия всех таких патентов США включены в данное описание посредством ссылки. Примеры имеющихся в настоящее время в продаже продуктов, содержащих антигипертензивные агенты, включают блокаторы кальциевых каналов, такие как Cardizem®, Adalat®, Calan®, Cardene®, Covera®, Dilacor® DynaCirc®, Procardia XL®, Sular®, Tiazac®, Vascor®, Verelan®, Isoptin®, Nimotop®, Norvasc® и Plendil®; ингибиторы ангиотензинпревращающего фермента (АСЕ), такие как Accupril®, Altace®, Captopril®, Lotensin®, Mavik®, Monopril®, Prinivil®, Univasc®, Vasotec® и Zestril®.
Ингибиторы ангиотензинпревращающего фермента (АСЕ-ингибиторы), которые входят в объем этого изобретения, включают: алацеприл, который может быть получен, как раскрыто в патенте США №4248883; беназеприл, который может быть получен, как раскрыто в патенте США №4410520; каптоприл, который может быть получен, как раскрыто в патентах США №4046889 и 4105776; церонаприл, который может быть получен, как раскрыто в патенте США №4452790; делаприл, который может быть получен, как раскрыто в патенте США №4385051; эналаприл, который может быть получен, как раскрыто в патенте США №4374829; фозиноприл, который может быть получен, как раскрыто в патенте США №4337201; имадаприл, который может быть получен, как раскрыто в патенте США №4508727; лизиноприл, который может быть получен, как раскрыто в патенте США №4555502; мовелтоприл, который может быть получен, как раскрыто в патенте Бельгии №893553; периндоприл, который может быть получен, как раскрыто в патенте США №4508729; хинаприл, который может быть получен, как раскрыто в патенте США №4344949; рамиприл, который может быть получен, как раскрыто в патенте США №4587258; спираприл, который может быть получен, как раскрыто в патенте США №4470972; темокаприл, который может быть получен, как раскрыто в патенте США №4699905; и трандолаприл, который может быть получен, как раскрыто в патенте США №4933361, но не ограничиваются ими. Раскрытия всех таких патентов США включены в данное описание посредством ссылки.
Антагонисты рецепторов ангиотензина II (A-II антагонисты), которые входят в объем этого изобретения, включают: кандесартан, который может быть получен, как раскрыто в патенте США №5196444; эпросартан, который может быть получен, как раскрыто в патенте США №5185351; ирбесартан, который может быть получен, как раскрыто в патенте США №5270317; лосартан, который может быть получен, как раскрыто в патенте США №5138069; и валсартан, который может быть получен, как раскрыто в патенте США №5399578, но не ограничиваются ими. Раскрытия всех таких патентов США включены в данное описание посредством ссылки.
Блокаторы бета-адренергических рецепторов (бета- или β-блокаторы), которые входят в объем этого изобретения, включают: ацебутолол, который может быть получен, как раскрыто в патенте США №3857952; алпренолол, который может быть получен, как раскрыто в заявке на патент Нидерландов №6605692; амосулалол, который может быть получен, как раскрыто в патенте США №4217305; аротинолол, который может быть получен, как раскрыто в патенте США №3932400; атенолол, который может быть получен, как раскрыто в патентах США №3663607 или 3836671; бефунолол, который может быть получен, как раскрыто в патенте США №3853923; бетаксолол, который может быть получен, как раскрыто в патенте США №4252984; бевантолол, который может быть получен, как раскрыто в патенте США №3857981; бисопролол, который может быть получен, как раскрыто в патенте США №4171370; бопиндолол, который может быть получен, как раскрыто в патенте США №4,340,541; букумолол, который может быть получен, как раскрыто в патенте США №3663570; буфетолол, который может быть получен, как раскрыто в патенте США №3723476; буфуралол, который может быть получен, как раскрыто в патенте США №3929836; бунитролол, который может быть получен, как раскрыто в патентах США №3940489 и 3961071; бупрандолол, который может быть получен, как раскрыто в патенте США №3309406; бутиридин гидрохлорид, который может быть получен, как раскрыто в патенте Франции №1390056; бутофилолол, который может быть получен, как раскрыто в патенте США №4252825; каразолол, который может быть получен, как раскрыто в патенте Германии №2240599; картеолол, который может быть получен, как раскрыто в патенте США №3910924; карведилол, который может быть получен, как раскрыто в патенте США №4503067; целипролол, который может быть получен, как раскрыто в патенте США №4034009; цетамолол, который может быть получен, как раскрыто в патенте США №4059622; клоранолол, который может быть получен, как раскрыто в патенте Германии №2213044; дилевалол, который может быть получен, как раскрыто в Clifton et al., Journal of Medicinal Chemistry, 1982, 25, 670; эпанолол, который может быть получен, как раскрыто в публикации заявки на европейский патент №41491; инденолол, который может быть получен, как раскрыто в патенте США №4045482; лабеталол, который может быть получен, как раскрыто в патенте США №4012444; левобунолол, который может быть получен, как раскрыто в патенте США №4463176; мепиндолол, который может быть получен, как раскрыто в Seeman et al., Helv. Chim. Acta, 1971, 54, 241; метипранолол, который может быть получен, как раскрыто в заявке на патент Чехословакии №128471; метопролол, который может быть получен, как раскрыто в патенте США №3873600; мопролол, который может быть получен, как раскрыто в патенте США №35017691; надолол, который может быть получен, как раскрыто в патенте США №3935267; надоксолол, который может быть получен, как раскрыто в патенте США №3819702; небивалол, который может быть получен, как раскрыто в патенте США №4654362; нипрадилол, который может быть получен, как раскрыто в патенте США №4394382; окспренолол, который может быть получен, как раскрыто в патенте Великобритании №1077603; пербутолол, который может быть получен, как раскрыто в патенте США №3551493; пиндолол, который может быть получен, как раскрыто в патентах Швейцарии №469002 и 472404; практолол, который может быть получен, как раскрыто в патенте США №3408387; пронеталол, который может быть получен, как раскрыто в патенте Великобритании №909357; пропранолол, который может быть получен, как раскрыто в патентах США №3337628 и 3520919; соталол, который может быть получен, как раскрыто в Uloth et al., Journal of Medicinal Chemistry, 1966, 9, 88; суфиналол, который может быть получен, как раскрыто в патенте Германии №2728641; талиндол, который может быть получен, как раскрыто в патентах США №3935259 и 4038313; тертатолол, который может быть получен, как раскрыто в патенте США №3960891; тилизолол, который может быть получен, как раскрыто в патенте США №4129565; тимолол, который может быть получен, как раскрыто в патенте США №3655663; толипролол, который может быть получен, как раскрыто в патенте США №3432545; и ксибенолол, который может быть получен, как раскрыто в патенте США №4018824, но не ограничиваются ими. Раскрытия всех таких патентов США включены в данное описание посредством ссылки.
Блокаторы альфа-адренергических рецепторов (альфа- или α-блокаторы), которые входят в объем этого изобретения, включают: амосулалол, который может быть получен, как раскрыто в патенте США №4217307; аротинолол, который может быть получен, как раскрыто в патенте США №3932400; дапипразол, который может быть получен, как раскрыто в патенте США №4252721; доксазозин, который может быть получен, как раскрыто в патенте США №4188390; фенспирид, который может быть получен, как раскрыто в патенте США №3399192; индорамин, который может быть получен, как раскрыто в патенте США №3527761; лабетолол, который может быть получен, как раскрыто выше; нафтопидил, который может быть получен, как раскрыто в патенте США №3997666; ницерголин, который может быть получен, как раскрыто в патенте США №3228943; празозин, который может быть получен, как раскрыто в патенте США №3511836; тамсулозин, который может быть получен, как раскрыто в патенте США №4703063; толазолин, который может быть получен, как раскрыто в патенте США №2161938; тримазозин, который может быть получен, как раскрыто в патенте США №3669968; и иохимбин, который может быть выделен из природных источников способами, хорошо известными специалистам в данной области техники, но не ограничиваются ими. Раскрытия всех таких патентов США включены в данное описание посредством ссылки.
Термин "вазодилататор", как он использован здесь, предусматривает включение церебральных вазодилататоров, коронарных вазодилататоров и периферических вазодилататоров. Церебральные вазодилататоры в объеме этого изобретения включают: бенциклан, который может быть получен, как раскрыто выше; циннаризин, который может быть получен, как раскрыто выше; цитиколин, который может быть выделен из природных источников, как раскрыто в Kennedy et al., Journal of the American Chemical Society, 1955, 77, 250, или синтезирован, как раскрыто Kennedy, Journal of Biological Chemistry, 1956, 222, 185; цикланделат, который может быть получен, как раскрыто в патенте США №3663597; циклоникат, который может быть получен, как раскрыто в патенте Германии №1910481; диизопропиламина дихлорацетат, который может быть получен, как раскрыто в патенте Великобритании №862248; эбурнамонин, который может быть получен, как раскрыто в Hermann et al., Journal of the American Chemical Society, 1979, 101, 1540; фасудил, который может быть получен, как раскрыто в патенте США №4678783; фенокседил, который может быть получен, как раскрыто в патенте США №3818021; флунаризин, который может быть получен, как раскрыто в патенте США №3773939; ибудиласт, который может быть получен, как раскрыто в патенте США №3850941; ифенпродил, который может быть получен, как раскрыто в патенте США №3509164; ломеризин, который может быть получен, как раскрыто в патенте США №4663325; нафронил, который может быть получен, как раскрыто в патенте США №3334096; никаметат, который может быть получен, как раскрыто в Blicke et al., Journal of the American Chemical Society, 1942, 64, 1722; ницерголин, который может быть получен, как раскрыто выше; нимодипин, который может быть получен, как раскрыто в патенте США №3799934; папаверин, который может быть получен, как рассмотрено в Goldberg, Chem. Prod. Chem. News, 1954, 17, 371; пентифиллин, который может быть получен, как раскрыто в патенте Германии №860217; тинофедрин, который может быть получен, как раскрыто в патенте США №3563997; винкамин, который может быть получен, как раскрыто в патенте США №3770724; винпоцетин, который может быть получен, как раскрыто в патенте США №4035750; и вихидил, который может быть получен, как раскрыто в патенте США №2500444, но не ограничиваются ими. Раскрытия всех таких патентов США включены в данное описание посредством ссылки.
Коронарные вазодилататоры, которые входят в объем этого изобретения, включают: амотрифен, который может быть получен, как раскрыто в патенте США №3010965; бендазол, который может быть получен, как раскрыто в J. Chem. Soc. 1958, 2426; бенфуродила гемисукцинат, который может быть получен, как раскрыто в патенте США №3355463; бензйодарон, который может быть получен, как раскрыто в патенте США №3012042; хлорацизин, который может быть получен, как раскрыто в патенте Великобритании №740932; хромонар, который может быть получен, как раскрыто в патенте США №3282938; клобенфурал, который может быть получен, как раскрыто в патенте Великобритании №1160925; клонитрат, который может быть получен из пропандиола способами, хорошо известными специалистам в данной области техники, например, см. Annalen, 1870, 155, 165; клорикромен, который может быть получен, как раскрыто в патенте США №4452811; дилазеп, который может быть получен, как раскрыто в патенте США №3532685; дипиридамол, который может быть получен, как раскрыто в патенте Великобритании №807826; дропрениламин, который может быть получен, как раскрыто в патенте Германии №2521113; эфлоксат, который может быть получен, как раскрыто в патентах Великобритании №803372 и 824547; эритритил тетранитрат, который может быть получен посредством нитрования эритрита способами, хорошо известными специалистам в данной области техники; этафенон, который может быть получен, как раскрыто в патенте Германии №1265758; фендилин, который может быть получен, как раскрыто в патенте США №3262977; флоредил, который может быть получен, как раскрыто в патенте Германии №2020464; ганглефен, который может быть получен, как раскрыто в патенте СССР №115905; гексэстрол, который может быть получен, как раскрыто в патенте США №2357985; гексобендин, который может быть получен, как раскрыто в патенте США №3267103; итрамин тозилат, который может быть получен, как раскрыто в патенте Швеции №168308; келлин, который может быть получен, как раскрыто в Baxter et al., Journal of the Chemical Society, 1949, S 30; лидофлазин, который может быть получен, как раскрыто в патенте США №3267104; маннита гексанитрат, который может быть получен путем нитрования маннита способами, хорошо известными специалистам в данной области техники; медибазин, который может быть получен, как раскрыто в патенте США №3119826; нитроглицерин; пентаэритрита тетранитрат, который может быть получен путем нитрования пентаэритрита способами, хорошо известными специалистам в данной области техники; пентринитрол, который может быть получен, как раскрыто в патенте Германии №638422-3; пергексилин, который может быть получен, как раскрыто выше; пимефиллин, который может быть получен, как раскрыто в патенте США №3350400; прениламин, который может быть получен, как раскрыто в патенте США №3152173; пропатилнитрат, который может быть получен, как раскрыто в патенте Франции №1103113; трапидил, который может быть получен, как раскрыто в патенте Восточной Германии №55956; трикромил, который может быть получен, как раскрыто в патенте США №2769015; триметазидин, который может быть получен, как раскрыто в патенте США №3262852; тролнитрат фосфат, который может быть получен путем нитрования триэтаноламина с последующим осаждением фосфорной кислотой способами, хорошо известными специалистам в данной области техники; виснадин, который может быть получен, как раскрыто в патентах США №2816118 и 2980699, но не ограничиваются ими. Раскрытия всех таких патентов США включены в данное описание посредством ссылки.
Периферические вазодилататоры, которые входят в объем этого изобретения, включают: никотинат алюминия, который может быть получен, как раскрыто в патенте США №2970082; баметан, который может быть получен, как раскрыто в Corrigan et al., Journal of the American Chemical Society, 1945, 67, 1894; бенциклан, который может быть получен, как раскрыто выше; бетагистин, который может быть получен, как раскрыто в Walter et al.; Journal of the American Chemical Society, 1941, 63, 2771; брадикинин, который может быть получен, как раскрыто в Hamburg et al., Arch. Biochem. Biophys., 1958, 76, 252; бровинкамин, который может быть получен, как раскрыто в патенте США №4146643; буфенйод, который может быть получен, как раскрыто в патенте США №3542870; буфломедил, который может быть получен, как раскрыто в патенте США №3895030; буталамин, который может быть получен, как раскрыто в патенте США №3338899; цетиедил, который может быть получен, как раскрыто в патенте Франции №1460571; циклоникат, который может быть получен, как раскрыто в патенте Германии №1910481; цинепазид, который может быть получен, как раскрыто в патенте Бельгии №730345; циннаризин, который может быть получен, как раскрыто выше; цикланделат, который может быть получен, как раскрыто выше; диизопропиламина дихлорацетат, который может быть получен, как раскрыто выше; эледоизин, который может быть получен, как раскрыто в патенте Великобритании №984810; фенокседил, который может быть получен, как раскрыто выше; флунаризин, который может быть получен, как раскрыто выше; гепроникат, который может быть получен, как раскрыто в патенте США №3384642; ифенпродил, который может быть получен, как раскрыто выше; илопрост, который может быть получен, как раскрыто в патенте США №4692464; инозитола ниацинат, который может быть получен, как раскрыто в Badgett et al., Journal of the American Chemical Society, 1947, 69, 2907; изоксуприн, который может быть получен, как раскрыто в патенте США №3056836; каллидин, который может быть получен, как раскрыто в Biochem. Biophys. Res. Commun., 1961, 6, 210; калликреин, который может быть получен, как раскрыто в патенте Германии №1102973; моксисилит, который может быть получен, как раскрыто в патенте Германии №905738; нафронил, который может быть получен, как раскрыто выше; никаметат, который может быть получен, как раскрыто выше; ницерголин, который может быть получен, как раскрыто выше; никофураноза, которая может быть получена, как раскрыто в патенте Швейцарии №366,523; нилидрин, который может быть получен, как раскрыто в патентах США №2661372 и 2661373; пентифиллин, который может быть получен, как раскрыто выше; пентоксифиллин, который может быть получен, как раскрыто в патенте США №3422107; пирибедил, который может быть получен, как раскрыто в патенте США №3299067; простагландин Ei, который может быть получен с помощью любого способа, указанного в Merck Index, Twelfth Edition, Budaveri, Ed., New Jersey, 1996, p.1353; сулоктидил, который может быть получен, как раскрыто в патенте Германии №2334404; толазолин, который может быть получен, как раскрыто в патенте США №2161938; и ксантинола ниацинат, который может быть получен, как раскрыто в патенте Германии №1102750 или Korbonits et al., Acta. Pharm. Hung., 1968, 38, 98, но не ограничиваются ими. Раскрытия всех таких патентов США включены в данное описание посредством ссылки.
Термин "диуретик", который входит в объем этого изобретения, предусматривает включение диуретических производных бензотиадиазина, ртутьорганических диуретиков, пуриновых диуретиков, стероидных диуретиков, диуретических производных сульфонамида, урациловых диуретиков и других диуретиков, таких как аманозин, который может быть получен, как раскрыто в патенте Австрии №168063; амилорид, который может быть получен, как раскрыто в патенте Бельгии №639386; арбутин, который может быть получен, как раскрыто в Tschitschibabin, Annalen, 1930, 479, 303; хлоразанил, который может быть получен, как раскрыто в патенте Австрии №168063; этакриновая кислота, которая может быть получена, как раскрыто в патенте США №3255241; этозолин, который может быть получен, как раскрыто в патенте США №3072653; гидракарбазин, который может быть получен, как раскрыто в патенте Великобритании №856409; изосорбид, который может быть получен, как раскрыто в патенте США №3160641; маннит; метохалкон, который может быть получен, как раскрыто в Freudenberg et al., Ber., 1957, 90, 957; музолимин, который может быть получен, как раскрыто в патенте США №4018890; пергексилин, который может быть получен, как раскрыто выше; тикринафен, который может быть получен, как раскрыто в патенте США №3758506; триамтерен, который может быть получен, как раскрыто в патенте США №3081230; и мочевина. Раскрытия всех таких патентов США включены в данное описание посредством ссылки.
Диуретические производные бензотиадиазина, которые входят в объем этого изобретения, включают: алтиазид, который может быть получен, как раскрыто в патенте Великобритании №902658; бендрофлуметиазид, который может быть получен, как раскрыто в патенте США №3265573; бензтиазид, McManus et al., 136th Am. Soc. Meeting (Atlantic City, September 1959), Abstract of papers, pp 13-O; бензилгидрохлортиазид, который может быть получен, как раскрыто в патенте США №3108097; бутиазид, который может быть получен, как раскрыто в патентах Великобритании №861367 и 885078; хлортиазид, который может быть получен, как раскрыто в патентах США №2809194 и 2937169; хлорталидон, который может быть получен, как раскрыто в патенте США №3055904; циклопентиазид, который может быть получен, как раскрыто в патенте Бельгии №587225; циклотиазид, который может быть получен, как раскрыто в Whitehead et al., Journal of Organic Chemistry, 1961, 26, 2814; эпитиазид, который может быть получен, как раскрыто в патенте США №3009911; этиазид, который может быть получен, как раскрыто в патенте Великобритании №861367; фенквизон, который может быть получен, как раскрыто в патенте США №3870720; индапамид, который может быть получен, как раскрыто в патенте США №3565911; гидрохлортиазид, который может быть получен, как раскрыто в патенте США №3164588; гидрофлуметиазид, который может быть получен, как раскрыто в патенте США №3254076; метиклотиазид, который может быть получен, как раскрыто в Close et al., Journal of the American Chemical Society, 1960, 82, 1132; метикран, который может быть получен, как раскрыто в патентах Франции №М2790 и 1365504; метолазон, который может быть получен, как раскрыто в патенте США №3360518; парафлутизид, который может быть получен, как раскрыто в патенте Бельгии №620829; политиазид, который может быть получен, как раскрыто в патенте США №3009911; хинетазон, который может быть получен, как раскрыто в патенте США №2976289; теклотиазид, который может быть получен, как раскрыто в Close et al., Journal of the American Chemical Society, 1960, 82, 1132; и трихлорметиазид, который может быть получен, как раскрыто в deStevens et al., Experientia, 1960, 16, 113, но не ограничиваются ими. Раскрытия всех таких патентов США включены в данное описание посредством ссылки.
Диуретические производные сульфонамида, которые входят в объем этого изобретения, включают: ацетазоламид, который может быть получен, как раскрыто в патенте США №2980679; амбузид, который может быть получен, как раскрыто в патенте США №3188329; азосемид, который может быть получен, как раскрыто в патенте США №3665002; буметанид, который может быть получен, как раскрыто в патенте США №3634583; бутазоламид, который может быть получен, как раскрыто в патенте Великобритании №769757; хлораминофенамид, который может быть получен, как раскрыто в патентах США №2809194, 2965655 и 2965656; клофенамид, который может быть получен, как раскрыто в Olivier, ReC. Trav. Chim., 1918, 37, 307; клопамид, который может быть получен, как раскрыто в патенте США №3459756; клорексолон, который может быть получен, как раскрыто в патенте США №3183243; дисульфамид, который может быть получен, как раскрыто в патенте Великобритании №851287; этоксзоламид, который может быть получен, как раскрыто в патенте Великобритании №795174; фуросемид, который может быть получен, как раскрыто в патенте США №3058882; мефрусид, который может быть получен, как раскрыто в патенте США №3356692; метазоламид, который может быть получен, как раскрыто в патенте США №2783241; пиретанид, который может быть получен, как раскрыто в патенте США №4010273; торасемид, который может быть получен, как раскрыто в патенте США №4018929; трипамид, который может быть получен, как раскрыто в патенте Японии №7305585; и ксипамид, который может быть получен, как раскрыто в патенте США №3567777, но не ограничиваются ими. Раскрытия всех таких патентов США включены в данное описание посредством ссылки.
Исходные вещества и реагенты для вышеописанных соединений по настоящему изобретению и агенты комбинации также легко доступны или могут быть легко синтезированы специалистами в данной области техники с использованием традиционных способов органического синтеза. Например, многие соединения, используемые здесь, являются родственными или получены из соединений, которые представляют большой научный интерес и в которых имеется коммерческая необходимость, и, следовательно, многие такие соединения имеются в продаже, или описаны в литературе, или их легко получить из других общедоступных веществ способами, которые описаны в литературе.
Некоторые из соединений или агентов комбинации по настоящему изобретению или промежуточные соединения в синтезе имеют ассиметрические атомы углерода и, поэтому, являются энантиомерами или диастереомерами. Диастереомерные смеси могут быть разделены на индивидуальные диастереомеры на основе их физико-химических различий способами, по существу известными, например, посредством хроматографии и/или фракционной кристаллизации. Энантиомеры могут быть разделены, например, способами хиральной ЖХВД или превращения энантиомерной смеси в диастереомерную смесь путем взаимодействия с соответствующим оптически активным соединением (например, спиртом), разделения диастереомеров и превращения (например, гидролиза) индивидуальных диастереомеров в соответствующие чистые энантиомеры. Также энантиомерные смеси соединений или промежуточного соединения в синтезе, которые содержат кислотную или основную группировку, могут быть разделены на составляющие чистые энантиомеры посредством образования диастереомерной соли с оптически чистым хиральным основанием или кислотой (например, 1-фенил-этиламином или винной кислотой) и разделения диастереомеров посредством фракционной кристаллизации с последующей нейтрализацией для разрушения соли с получением, таким образом, соответствующих чистых энантиомеров. Все такие изомеры, включая диастереомеры, энантиомеры и их смеси, рассматривают как часть настоящего изобретения. Также, некоторые из соединений по настоящему изобретению представляют собой атропоизомеры (например, замещенные биарилы), и их рассматривают как часть настоящего изобретения.
Более конкретно, соединения или агенты комбинации по настоящему изобретению могут быть получены посредством фракционной кристаллизации основного промежуточного соединения с оптически чистой хиральной кислотой с образованием диастереомерной соли. Методы нейтрализации используют для удаления соли и получения энантиомерно чистых соединений. Альтернативно, соединения по настоящему изобретению могут быть получены в энантиомерно обогащенной форме посредством разделения рацемата конечного соединения или промежуточного соединения в синтезе (предпочтительно конечного соединения) с использованием хроматографии (предпочтительно жидкостной хроматографии высокого давления [ЖХВД]) на ассиметрической смоле (предпочтительно Chiralcel™ AD или OD (полученной от Chiral Technologies, Exton, Pennsylvania)) с подвижной фазой, состоящей из углеводорода (предпочтительно гептана или гексана), содержащего от 0 до 50% изопропанола (предпочтительно от 2 до 20%) и от 0 до 5% алкиламина (предпочтительно 0,1% диэтиламина). Концентрирование содержащих продукт фракций дает требуемые вещества.
Некоторые соединения по настоящему изобретению или агенты комбинации являются основными или цвиттерионными и образуют соли с фармацевтически приемлемыми анионами. Все такие соли входят в объем этого изобретения и могут быть получены традиционными способами, такими как объединение кислотных и основных частей, обычно в стехиометрическом соотношении, либо в водной, безводной, либо в частично водной среде, как целесообразно. Соли выделяют либо посредством фильтрации, посредством осаждения с осадителем с последующей фильтрацией, посредством выпаривания растворителя, либо, в случае водных растворов, посредством лиофилизации, как целесообразно. Соединения получают в кристаллической форме согласно методикам, известным в данной области техники, например посредством растворения в соответствующем(их) растворителе(ях), таком(их) как этанол, гексаны или смеси вода/этанол.
Некоторые агенты комбинации по настоящему изобретению являются кислотными и образуют соли с фармацевтически приемлемым катионом. Все такие соли входят в объем настоящего изобретения и могут быть получены традиционными способами, такими как объединение кислотных и основных частей, обычно в стехиометрическом соотношении, либо в водной, безводной, либо в частично водной среде, как целесообразно. Соли выделяют либо посредством фильтрации, посредством осаждения с осадителем с последующей фильтрацией, посредством выпаривания растворителя, либо, в случае водных растворов, посредством лиофилизации, как целесообразно. Соединения могут быть получены в кристаллической форме согласно методикам, известным в данной области техники, например посредством растворения в соответствующем(их) растворителе(ях), таком(их) как этанол, гексаны или смеси вода/этанол.
Некоторые соединения по настоящему изобретению или агенты комбинации могут существовать в более чем одной кристаллической форме (обычно под названием "полиморфы"). Полиморфы могут быть получены посредством кристаллизации в различных условиях, например с использованием различных растворителей или различных смесей растворителей для перекристаллизации; кристаллизации при различных температурах; и/или различных методов охлаждения, начиная от очень быстрого до очень медленного охлаждения во время кристаллизации. Полиморфы также могут быть получены посредством нагревания или плавления соединения по настоящему изобретению с последующим постепенным или быстрым охлаждением. Присутствие полиморфов может быть определено посредством твердотельной ЯМР-спектроскопии, ИК (инфракрасной)-спектроскопии, дифференциальной сканирующей калориметрии, дифракции рентгеновских лучей на порошке или других подобных методик.
Меченые изотопами соединения формулы I или агенты комбинации обычно могут быть получены с использованием традиционных методик, известных специалистам в данной области техники, или способами, аналогичными способам, описанным в прилагаемых примерах и получениях, с использованием соответствующих меченых изотопами реагентов вместо немеченых реагентов, использованных ранее.
Пропротеинконвертаза субтилизин/кексин типа 9, также известный как PCSK9, представляет собой фермент, который у людей кодируется геном PCSK9. Как определено в данном описании и обычно известно специалистам в данной области техники, определение PCSK9 также включает более 50 его мутаций с приобретением и утратой функции, GOF и LOF, соответственно. (http://www.ucI.ac. uk/IdIr/LOVDv.1.1.0/search.php?select_db=PCSK9&srch=all).
Соединения по настоящему изобретению ингибируют трансляцию м-РНК, кодирующей PCSK9, в белок PCSK9.
Полагают, что некоторый(ые) участок(ки) первых 100 кодонов мРНК, которые кодируют белок PCSK9, являются определяющими, будет ли соединение ингибитором трансляции мРНК PCSK9 в белок PCSK9. Таким образом, полагают, что существует взаимодействие между участком(ами) первых 100 кодонов (кодирующих PCSK9), соединением-ингибитором и другими компонентами процесса трансляции PCSK9, что приводит к ингибированию процесса трансляции. Полагают, что эти первые кодоны также являются определяющими для ингибиторной активности соединения, поскольку когда в первых 100 кодонах, которые кодируют ту же аминокислотную последовательность PCSK9, сделаны определенные замены кодонов (смысловые замены), эти замены приводят к потере ингибиторной активности "соединением-ингибитором". Как известно в данной области техники, полноценный секретированный белок не содержит сигнальную последовательность (например, включающий первые 30 аминокислот).
Однако, также полагают, что когда в первых 100 кодонах, которые кодируют аминокислотную последовательность PCSK9, сделаны определенные замены кодонов (несмысловые замены), трансляция по-прежнему подавляется (то есть соединениями сохраняется ингибиторная активность). Это дает основание полагать, что природа растущего пептида (только что образующейся при трансляции аминокислотной последовательности) является по меньшей мере одной детерминантой "ингибиторной" активности соединения.
Ингибирование трансляции PCSK9 дает преимущества над другими способами блокировки активности PCSK9 (например, ингибирование прямого взаимодействия PCSK9 с рецептором LDL). Одним преимуществом ингибирования трансляции PCSK9 является то, что не образуется полноценный PCSK9, и его действие в клетке будет также прекращено. Внутриклеточный PCSK9, как сообщают (Arterioscler Thromb Vasc Biol 2012; 32: 1585-1595), связывается с аполипопротеином В100 (АроВ100) и защищает его от деградации. Таким образом, пониженные уровни внутриклеточного PCSK9 обладают потенциальным преимуществом, снижая секрецию липопротеина очень низкой плотности (VLDL). Такая фармакология вряд ли связана с анти-PCSK9 антителами, поскольку они действуют вне клетки.
Как определено в данном описании, ингибирование трансляции мРНК PCSK9 в белок PCSK9 определяют с помощью "бесклеточного анализа PCSK9", приведенного здесь в описании. Этот "бесклеточный анализ PCSK9" является специфичным для получения белка PCSK9 с помощью мРНК PCSK9 и, поэтому, позволяет выявлять ингибиторы этого трансляционного процесса, а не другие механизмы, посредством которых уровень белка PCSK9 может быть снижен. Любое соединение, которое демонстрирует IC50 (мкМ) ниже примерно 50 мкМ в "бесклеточном анализе PCSK9", считается ингибирующим трансляцию PCSK9. Предпочтительно, чтобы IC50 соединения составляла менее чем примерно 30 мкМ, и особенно предпочтительно, чтобы IC50 соединения составляла менее чем примерно 20 мкМ.
Предпочтительно, чтобы соединение "селективно" ингибировало трансляцию мРНК PCSK9 в белок PCSK9. Термин "селективный" определен как "ингибирование" трансляции менее чем 1 процента белка в типичном глобальном протеомном анализе. Предпочтительным является уровень ниже примерно 0,5% белков, и особенно предпочтительным является уровень ниже примерно 0,1% белков. Обычно в стандартном анализе 1% уровень равен примерно 40 белкам, не являющимся PCSK9, из примерно 4000 белков.
"Ингибирование" конкретного белка в глобальном протеомном анализе (анализ приведен ниже) определено как снижение экспрессии белков в глобальном протеомном анализе до уровня ниже примерно 50% от начального уровня по сравнению с носителем. Это определение "ингибирования", относящееся к глобальному протеомному анализу, не следует путать с приведенным ранее определением "ингибирования", относящимся к бесклеточному анализу PCSK9.
Примеры соединений, полезных в качестве селективных ингибиторов трансляции PCSK9, приведены в данном описании, и другие соединения могут быть определены посредством рутинного скрининга (известного специалистам в данной области техники), например, обычных библиотек соединений с помощью описанного выше бесклеточного анализа PCSK9 и глобального протеомного анализа, приведенных ниже. Таким образом, высокопроизводительный скрининг (HTS) существующих и новых библиотек соединений с использованием вышеуказанных анализов (или их соответствующих модификаций, легко определяемых обычным специалистом в данной области техники) дает соединения, полезные для целей данного изобретения. Идентификация этих соединений также может обеспечить направление рутинных исследований в медицинской химии для идентификации дополнительных соединений, полезных для целей данного изобретения. Такие соединения полезны для способов и применений, описанных здесь ниже, включая лечение дислипидемии. Предпочтительно такие соединения имеют молекулярную массу от примерно 300 до примерно 650.
Соединения по настоящему изобретению, их пролекарства и соли таких соединений и пролекарств все адаптированы для терапевтического применения в качестве агентов, которые проявляют антигонизм в отношении активности внеклеточного белка пропротеинконвертаза субтилизин/кексин типа 9 (PCSK9), включая его взаимодействие с рецептором (LDLR) липопротеина низкой плотности (LDL), у млекопитающих, в особенности людей. Таким образом, считается, как продемонстрировано у человеческих индивидуумов с мутациями с утратой функции (LOF) PCSK9 (например, Hobbs et. al. NEJM, 2006 and Hobbs et. al. Am.J. Hum. Gen., 2006), что соединения по настоящему изобретению, посредством снижения уровней PCSK9, повышают экспрессию рецептора LDL на поверхности клеток и, таким образом, уменьшают уровень холестерина LDL. Следовательно, эти соединения полезны для лечения и коррекции различных наблюдаемых дислипидемии, связанных с развитием и возникновением атеросклероза и сердечно-сосудистого заболевания, включая гипо-альфа-липопротеинемию и гипертриглицеридемию.
Принимая во внимание прямую взаимосвязь уровней холестерина LDL и связанных с ними аполипопротеинов в крови с развитием сердечно-сосудистых, цереброваскулярных заболеваний и заболеваний периферических сосудов, соединения по настоящему изобретению и соли таких соединений благодаря своему фармакологическому действию полезны для предупреждения, прекращения развития и/или регрессии атеросклероза и связанных с ним болезненных состояний. Они включают сердечно-сосудистые нарушения (например, коронарную артериальную болезнь, цереброваскулярное заболевание, коронарную артериальную болезнь, дисфункцию желудочка, нарушение сердечного ритма, легочное сосудистое заболевание, нарушение сосудистого гемостаза, ишемию сердца и инфаркт миокарда), осложнения сердечно-сосудистого заболевания, преходящие церебральные ишемические приступы.
Пользу соединений по настоящему изобретению и солей таких соединений в качестве медицинских агентов в лечении описанных выше заболеваний/состояний у млекопитающих (например, людей: мужчин или женщин) демонстрируют с помощью активности соединений по настоящему изобретению в одном или более чем одном традиционном анализе и анализе in vivo, описанном ниже. Анализы in vivo (с соответствующими модификациями, известными в данной области техники) могут быть использованы для определения активности других агентов, регулирующих уровни липидов и триглицеридов, а также соединений по настоящему изобретению. Таким образом, протоколы, описанные ниже, также могут быть использованы, чтобы продемонстрировать пользу комбинаций агентов (то есть соединений по настоящему изобретению), описанных здесь. Кроме того, такие анализы обеспечивают возможность сравнения активностей соединений по настоящему изобретению и солей таких соединений (или других агентов, описанных здесь) друг с другом и с активностями других известных соединений. Результаты таких сравнений полезны для определения уровней дозы у млекопитающих, включая людей, для лечения таких заболеваний. Следующие протоколы могут быть конечно изменены специалистами в данной области техники.
ALPHALISA АНАЛИЗ PCSK9
СНИЖЕНИЕ PCSK9 B КЛЕТКАХ WT7
In vitro AlphaLISA анализ (Perkin Elmer) разрабатывали для проведения количественного анализа уровня PCSK9, секретируемого в культуральные среды после обработки соединением. Для определения и измерения белка PCSK9 созданное в своей лаборатории анти-PCSK9 антитело связывали с акцепторными гранулами AlphaLISA внешнего поставщика (PerkinElmer), и проводили в своей лаборатории биотинилирование второго анти-PCSK9 антитела внутренней разработки с эпитопом, отличающимся от эпитопа акцепторных гранул. Покрытые стрептавидином донорные гранулы (Perkin Elmer) также вводятся в смесь для анализа, которая затем связывает биотинилированное анти-PCSK9 антитело, и в присутствии PCSK9 этот донорный комплекс и акцепторные гранулы располагаются в непосредственной близости друг от друга. При возбуждении донорных гранул при 680 нм высвобождаются молекулы синглетного кислорода, что запускает каскад передачи энергии в акцепторных гранулах, проявляющейся в виде одиночного пика света, излучаемого при 615 нм. Способность соединения регулировать уровни белка PCSK9 в кондиционированной среде по методу AlphaLISA оценивали на линии клеток гепатоцеллюлярной карциномы человека Huh7, стабильно сверхэкспрессирующей человеческий PCSK9. Эту клеточную линию, обозначаемую WT7, создавали посредством трансфекции клеток Huh7 модифицированным в своей лаборатории pcDNA 3.1 (+) Zeo вектором экспрессии (Life Technologies), содержащим полноразмерную последовательность человеческого белка PCSK9 (идентификатор записей NCBI, NM_174936.3, где метка начала кодирующей последовательности в положении 363) и с-терминальную метку V5 и 6х-His. После трансфекции плазмиды стабильный клон WT7 идентифицировали и поддерживали при селекции с применением зеоцина. Скрининг соединений проводили в 384-луночных планшетах, куда высевали клетки WT7 при плотности 7500 клеток на лунку в 20 мкл среды для выращивания культур тканей, содержащих соединение, в одиннадцати точках разведения, в формате разведения с интервалом 0,5 log при высокой концентрации обработки 20 мкМ в конечном объеме 0,5% DMSO. Помимо этих условий, создаваемых для тестируемых соединений, каждый планшет для скрининга включал лунки, которые содержали 20 мкМ пуромицина в качестве положительного контроля анализа, определяемого как высокопроцентный эффект, НРЕ, а также лунки, содержащие среду в 0,5% DMSO в качестве отрицательного контроля обработки, определяемого как нулевой эффект, ZPE. После инкубирования соединений в течение ночи (16-24 ч) среду для выращивания культур тканей собирали, и аликвоту из каждого образца переносили в отдельные лунки 384-луночного белого планшета Optiplate (Perkin Elmer). Связанные антитела и донорные гранулы добавляли в планшеты для анализов в буфере, состоящем из 30 мМ Трис (трис-(гидроксиметил)-аминометан) (pH 7,4), 0,02% Tween-20 (полиоксиэтиленсорбитан монолаурат) и 0,02% казеина. Добавляли анти-PCSK9 акцепторные гранулы (конечная концентрация 10 мкг/мл) и анти-PCSK9 биотинилированные антитела (конечная концентрация 3 нМ) и инкубировали в течение 30 минут при комнатной температуре с последующим добавлением покрытых стрептавидином донорных гранул (конечная концентрация 40 мкг/мл) в течение еще 60 минут. Кроме того, строили стандартную кривую, в случае, когда реагенты AlphaLISA инкубировали в лунках с введением добавки рекомбинантного человеческого PCSK9, разведенного в среде для выращивания культур тканей от 5000 нг/мл до 0,6 нг/мл. После инкубирования с реагентами AlphaLISA планшеты считывали на планшет-ридере EnVision (Perkin Elmer) при длине волны возбуждения 615 нМ и длине волны излучения/детектирования 610 нМ. Для определения IC50 соединений данные контрольных лунок НРЕ и ZPE сначала анализировали и подсчитывали среднее значение, стандартное отклонение и показатель степени Z для каждого планшета. Данные, полученные для тестируемых соединений, конвертировали в эффект, выраженный в процентах, с использованием контролей ZPE и НРЕ в качестве 0% и 100% активности, соответственно. Уравнение, используемое для конвертирования показания, снятого с каждой лунки, в эффект, выраженный в процентах, представляло собой:
Уравнение 1:
(значение активности в тестируемой лунке - значение активности ZPE) X 100 (значение активности НРЕ - значение активности ZPE)
Затем подсчитывали величину IC50 и регистрировали в виде срединной точки на кривой эффекта, выраженного в процентах, в молях, и величины вносили в таблицу 1 "Биологические данные" в столбец под заголовком "IC50 клеточного PCSK9 (мкМ)". Кроме того, для проверки селективности соединения в отношении PCSK9 измеряли уровень второго секретируемого белка, трансферрина, в такой же кондиционированной среде, обработанной тестируемым соединением по методу AlphaLISA. Конъюгированный с гранулами анти-трансферрин AlphaLISA от PerkinElmer представляет собой мышиный моноклональный иммуноглобулин lgG1 к человеческому трансферрину (клон М10021521; № кат. 10-Т34С; Fitzgerald). Меченое биотином антитело представляет собой аффинно очищенное козье античеловеческое поликлональное антитело (№ кат. А80-128А; Bethyl Laboratories). Для определения и количественной оценки воздействий на трансферрин 0,01 мл культуральной среды переносят в 384-луночный белый планшет Optiplate и добавляют 0,01 мл среды для доведения объема до 0,02 мл. Анти-трансферрин-акцепторные гранулы добавляли до конечной концентрации 10 мкг/мл, биотинилированный анти-трансферрин в концентрации 3 нМ и стрептавидин-донорные гранулы в концентрации 40 мкг/мл. Эффект, выраженный в процентах, и IC50 для трансферрина подсчитывали по методике, аналогичной описанной для PCSK9.
ОПРЕДЕЛЕНИЕ СНИЖЕНИЯ УРОВНЯ PCSK9 И КОНЦЕНТРАЦИИ СОЕДИНЕНИЯ В ЧЕЛОВЕЧЕСКИХ ГЕПАТОЦИТАХ В МНОГОСЛОЙНОЙ КУЛЬТУРЕ (SCHH)
Взаимосвязь параметров фармакокинетики и фармакодинамики тестируемых соединений in vitro оценивали в первичных криоконсервированных человеческих гепатоцитах в многослойной культуре. В этих исследованиях клетки SCHH (BD Biosciences IVT) размораживали при 37°C, затем помещали на лед, после чего клетки добавляли в предварительно согретую (37°C) среду In VitroGRO-HT и центрифугировали при 50×g в течение 3 минут. Клеточный осадок ресуспендировали в концентрации 0,8×106 клеток/мл в среде InVitroGRO-CP для чашек Петри, и жизнеспособность клеток определяли по вытеснению трипанового синего. На 1 сутки суспензии гепатоцитов высевали в 96-луночный планшет BioCoat при плотности 80000 клеток/лунка в объеме 0,1 мл/лунка. После 18-24 часов инкубирования при 37°C в 5% CO2 клетки покрывали слоем 0,25 мг/мл охлажденного на льду матрикса матригеля (BD Matrigel Matrix) без фенолового красного в инкубационной среде при 0,1 мл/лунка. Культуры поддерживали при 37°C в 5% СO2 в InVitroGRO-HI (среда, не содержащая FBS (фетальную бычью сыворотку)), которую заменяли каждые 24 часа, и исследования в динамике начинали на 5 сутки. Перед обработкой соединениями планшеты с клетками 3 раза промывали 0,1 мл/лунка InVitroGRO-HI, и 0,09 мл среды вносили обратно при введении приготовленных на ней соединений. 1 мкл либо DMSO, либо исходных растворов соединений в DMSO в концентрации 30 мМ, 10 мМ, 3 мМ и 1 мМ вносили в 96-луночные полипропиленовые планшеты с V-образным дном. 0,099 мл среды добавляли в планшет с соединениями и тщательно перемешивали перед введением 0,010 мл из промежуточного планшета с соединениями в планшет с клетками. Это приводило к конечной концентрации 0,1% DMSO, при которой соединения оценивали в концентрации 30 мкМ, 10 мкМ, 3 мкМ и 1 мкМ (в некоторых случаях концентрации соединений повышали до 300 мкМ). Клетки инкубировали с соединением в течение 5, 15, 30, 60, 180, 360, 480 и 1440 минут при 37°C в 5% CO2. В указанное время 0,08 мл среды отбирали из планшетов с клетками и замораживали для последующего анализа секретируемого PCSK9 посредством AlphaLISA и для определения уровней лекарственного средства в среде посредством жидкостной хроматографии-тандемной масс-спектрометрии (ЖХ-МС/МС). Оставшуюся среду затем аспирировали и клеточные слои промывали 3x ледяным сбалансированным солевым раствором Хенкса (HBSS) в условиях встряхивания для удаления верхнего слоя матригеля, и планшеты затем хранили при -20°C для последующего определения уровней лекарственного средства в клетках посредством ЖХ-МС/МС. Определение уровней белка PCSK9 посредством AlphaLISA в кондиционированной среде проводили с использованием тех же реагентов и протоколов детектирования, которые описаны выше для клеток WT7. Затем определяли процент снижения PCSK9 в сравнении с клетками, обработанными носителем, для каждой временной точки, и максимальный отклик (и соответствующую концентрацию и время наблюдения) вносили в таблицу 1 "Биологические данные" в столбец под заголовком "Снижение уровня PCSK9 в гепатоцитах в многослойной культуре (SCHH)".
Образцы сред, используемые для определения уровней тестируемых соединений, обрабатывали путем добавления 20 мкл кондиционированной среды к 180 мкл раствора MeOH-IS (метанол с внутренним стандартом) или 20 мкл матрикса с питательной средой, содержащего известные концентрации анализируемого вещества (0-5 мкл), к 180 мкл раствора MeOH-IS. Образцы затем сушили в потоке азота и ресуспендировали в 200 мкл смеси МеОН/H2O (50/50). Анализы ЖХ-МС/МС проводили на тройном квадрупольном масс-спектрометре API-4000 с источником ионизации электрораспылением при атмосферном давлении (MDS SCIEX, Concord, Ontario, Canada), соединенном с двумя насосами Shimadzu LC-20AD, с контроллером СВN-20А. 10 мкл образца вводили в колонку Kinetex С18 (2,6 мкл, 100Å, 30×2,1 мм, Phenomenex, Torrance, СА) и элюировали подвижной фазой при скорости потока 0,5 мл/мин с исходными условиями 10% растворителя B в течение 0,2 минут, с последующим градиентом от 10% растворителя B до 90% растворителя B за 1 минуту (растворитель A: 100% H2O с 0,1% муравьиной кислоты; растворитель B: 100% ацетонитрил с 0,1% муравьиной кислоты), с 90% растворителя B, удерживаемого в течение 0,5 минут, с последующим возвращением к исходным условиям, которые поддерживали в течение 0,75 минуты.
Для определения уровней тестируемого соединения в клетках SCHH планшеты с клетками извлекали из морозильной камеры и клеточные слои лизировали в 0,1 мл метанола, содержащего внутренний стандарт (MeOH-IS), карбамазепин, при встряхивании в течение 20 минут при комнатной температуре. Лизат (90 мкл) затем переносили в новый 96-луночный планшет, сушили в потоке азота и ресуспендировали в 90 мкл смеси МеОН/H2O (50/50). Стандартные кривые строили посредством добавления 0,1 мл MeOH-IS с известными концентрациями анализируемого вещества (0-500 нМ) к обработанным носителем клеточным слоям (матриксный контроль). Все стандарты затем обрабатывали таким же образом, как и неизвестные образцы. Были разработаны способы регистрации при анализе ЖХ-МС/МС для мониторинга множественных реакций (MRM), при этом определяли предварительно заданные переходы для каждого анализируемого вещества и оптимальные потенциалы кластеризации, энергию соударений и выходные потенциалы ячейки соударений для каждого анализируемого вещества при напряжении на капилляре 4,5 кВ, входном потенциале 10 эВ и температуре источника 550°C. Площади пиков анализируемого вещества и внутреннего стандарта подсчитывали с использованием программы Analyst 1.5.2 (MDS SCIEX, Ontario, Canada). Полученные уровни лекарственных средств затем нормировали по отношению к содержанию белка в гепатоцитах на лунку, как определено с помощью набора для анализа белков ВСА Protein Assay Kit (Pierce Biotechnology).
Чтобы устранить барьер проницаемости, присущий клеткам WT7 и клеточным анализам SCHH, для оценки активности соединений также создавали бесклеточную систему. Последовательность, содержащую полноразмерный человеческий PCSK9 (идентификатор записей NCBI, NM_174936.3, где метка начала кодирующей последовательности в положении 363) вместе с 84 дополнительными 3'-нуклеотидами, содержащими метку V5 и полилинкер, за которым следует модифицированный в рамке ген-репортер люциферазы светлячков (соответствующий положениям нуклеотидов 283-1929 в векторе pGL3, идентификатор записей GenBank, JN542721.1) клонировали в вектор экспрессии pT7CFE1 (ThermoScientific). Эту конструкцию затем транскрибировали in vitro с использованием набора MEGAscript Т7 Kit (Life Technologies) и РНК (рибонуклеиновую кислоту) затем очищали с использованием набора MEGAclear Kit (Life Technologies) согласно протоколу производителя. Клеточные лизаты HeLa получали согласно протоколу, описанному Mikami (источником является Cell-Free Protein Synthesis Systems with Extracts from Cultured Human Cells, S. Mikami, T. Kobayashi and H. Imataka; from Methods in Molecular Biology, vol. 607, pages 43-52, Y. Endo et al. (eds.), Humana Press, 2010), с последующими модификациями. Клетки выращивали в среде CD293 (Gibco 11765-054) объемом 20 л с глутамаксом в концентрации, увеличенной до 4 мМ, пенициллином в концентрации 100 Ед/мл и другими добавками, как ранее описано Mikami. Выращивание проводили в одноразовом биореакторе WAVE Biotech на 50 л со скоростью качалки 25 об/мин и углом 6,1 с 5% CO2 и скоростью потока 0,2 л/мин, при этом получали клетки с плотностью 2-2,5×106 клеток/мл. Лизаты дополнительно содержали 1 таблетку полного ингибитора протеаз, содержащего EDTA (этилендиаминтетрауксусную кислоту), от Roche на 50 мл с трис(2-карбоксиэтил)фосфатом (Biovectra) вместо дитиотреитола, и их осветляли посредством дополнительного конечного центрифугирования при 10000 об/мин в роторе Sorvall SS34 при 4°C в течение 10 минут. Скрининг соединений проводили в 384-луночных планшетах в одиннадцати точках, в формате разведения с интервалом 0,5 log при максимальной концентрации тестируемых соединений 100 мкМ в конечном объеме 0,5% DMSO. Помимо этих условий, создаваемых для тестируемых соединений, каждый планшет для скрининга также включал лунки, которые содержали 100 мкМ соединения по Примеру 16, (N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензамид), в качестве положительного контроля анализа, определяемого как высокопроцентный эффект, НРЕ, а также лунки, содержащие среду в 0,5% DMSO в качестве отрицательного контроля обработки, определяемого как нулевой эффект, ZPE. Соединения инкубировали при 30°C в течение 45 минут в растворе, содержащем 0,1 мкг очищенной, транскрибированной in vitro РНК совместно с бесклеточной реакционной смесью (состоящей из солей Mg (1,6 мМ) и К (112 мМ), 4,6 мМ трис(2-карбоксиэтил)фосфина (Biovectra), 5,0 мкл лизата HeLa, 0,2 мкл ингибитора РНКазы RNAsin (Promega) и 1,0 мкл энергетический комплекс (содержащей 1,25 мМ АТФ (аденозинтрифосфата) (Sigma), 0,12 мМ ГТФ 9 (гуанозинтрифосфата) (Sigma), 20 мМ креатинфосфата (Santa Cruz), 60 мкг/мл креатинфосфокиназы (Sigma), 90 мкг/мл тРНК (транспортной РНК) (Sigma) и 20 аминокислот (Life Technologies) в конечных концентрациях, описанных Mikami), и вносили в воду до конечного объема 10 мкл в воде. После завершения анализа из каждого реакционного раствора отбирали 1 мкл и переносили во второй 384-луночный планшет Optiplate (Perkin Elmer), содержащий 24 мкл SteadyGlo (набор для анализа гена люциферазы) (Promega), и интенсивность сигнала измеряли на планшетном анализаторе Envision (Perkin Elmer) с использованием режима усиленной люминесценции. Для определения IC50 соединений данные контрольных лунок НРЕ и ZPE сначала анализировали и подсчитывали среднее значение, стандартное отклонение и показатель степени Z для каждого планшета. Данные, полученные для тестируемых соединений, конвертировали в эффект, выраженный в процентах, с использованием контролей ZPE и НРЕ в качестве 0% и 100% активности, соответственно, используя вышеприведенное уравнение 1. Затем подсчитывали величину IC50 и регистрировали в виде срединной точки на кривой эффекта, выраженного в процентах, в молях, и величины вносили в таблицу 1 "Биологические данные" в столбец под заголовком "IC50 бесклеточного PCSK9 (мкМ)".
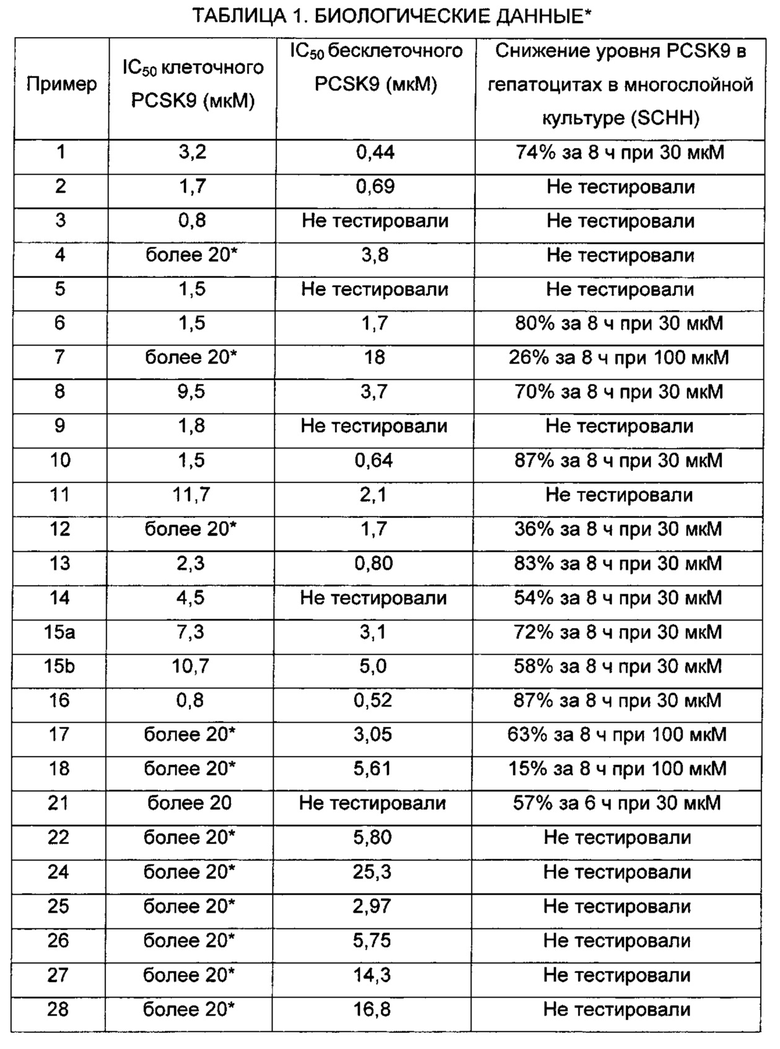
* Некоторые соединения с плохой проницаемостью мембраны при пассивном транспорте имели значения IC50, превышающие максимальную концентрацию в WТ7клеточном анализе (20 мкМ). С использованием бесклеточного анализа действие этих соединений не блокируется клеточной мембраной, и они активны. Также, когда соединения вводят в более высоких концентрациях в анализе SCHH, эти соединения могут демонстрировать активность несмотря на их плохое проникновение в клетку. Активность соединений определяли посредством совмещения трех анализов. Соединения, продемонстрировавшие активность в клеточном анализе, не всегда тестировали в других анализах активности.
Глобальный протеомный анализ - анализ мечения аминокислотами со стабильными изотопами в культуре клеток (SILAC):
Клетки гепатокарциномы человека Huh7 для мечения аминокислотами со стабильными изотопами (SILAC) выращивали в среде RPMI (минус лизин и аргинин) с 10% диализированной фетальной бычьей сывороткой, обогащенной либо немеченым лизином и аргинином (легкая метка), L-аргинином: HCl U-13C6 99% и L-лизином: 2НСl 4,4,5,5-D4, 96-98% (средняя метка) или L-аргинином: HCl U13C6, 99%; U-15N4, 99% и L-лизином: 2НСl U13C6, 99%; U-15N2, 99% (тяжелая метка). Проводили 5-6 пассажей клеток с достижением эффективности включения метки более 95%. Перед началом эксперимента клетки культивировали до достижения стопроцентной конфлюентности для обеспечения синхронизированной клеточной популяции в фазе G0/G1 (анализ клеточного цикла с йодидом пропидия показал, что 75% клеток находится в фазе G0/G1). Затем клетки пересевали на свежую среду, обогащенную 0,5% диализированной фетальной бычьей сывороткой, содержащую либо легкую, среднюю, либо тяжелую форму лизина (Lys) и аргинина (Arg), и добавляли носитель (легкая метка), или соединение по Примеру 16 в концентрации 0,25 мкМ (средняя метка) или 1,30 мкМ (тяжелая метка) в течение либо 1, 4, либо 16 часов. В конце указанных временных точек среду удаляли и добавляли ингибиторы протеаз/фосфатаз перед замораживанием при -80°C. Чтобы разделить клетки, клеточные слои промывали PBS (фосфатно-солевым буфером) перед добавлением буфера для диссоциации клеток, клетки собирали посредством промывания PBS и центрифугировали при 1000 об/мин в течение 5 минут. Клеточный осадок ресуспендировали в PBS для промывки, центрифугировали при 1000 об/мин в течение 5 минут и надосадочную жидкость аспирировали. Клеточный слой затем замораживали при -80°C, и среду, и клеточный осадок подвергали протеомному анализу.
Для протеомного анализа секретируемых белков смешивали равный объем кондиционированной среды из легкой, средней и тяжелой форм клеток с последующим удалением бычьего сывороточного альбумина с помощью анти-BSA агарозных гранул. Полученные белки затем концентрировали с использованием спин-колонок с показателем отсекаемой молекулярной массы (MWCO) 3 кДа, восстанавливали дитиотреитолом и алкилировали йодацетамидом.
Для анализа клеточных белков клеточные осадки лизировали в загрузочном буфере для электрофореза в полиакриламидном геле в присутствии додецилсульфата натрия (SDS-PAGE) в присутствии коктейлей ингибиторов протеаз/фосфатаз. Клеточные лизаты центрифугировали при 12000×g; при 4°C в течение 10 минут. Полученные надосадочные жидкости собирали и концентрации белка измеряли посредством анализа ВСА (с использованием бицинхониновой кислоты). Равное количество белков в легкой, средней и тяжелой форме клеточных лизатов объединяли, восстанавливали дитиотреитолом и алкилировали йодацетамидом.
Белки, полученные из кондиционированной среды и клеточных осадков, затем фракционировали посредством SDS-PAGE. Гели окрашивали кумасси голубым. После обесцвечивания гели были разделены на 12-15 полос. Белки в геле расщепляли трипсином в течение ночи, после чего белки экстрагировали из геля смесью CH3CN:1% муравьиная кислота (1:1, об./об.). Полученные смеси белков обессоливали с помощью наконечников C18 Stage-Tips, сушили на центрифужном концентраторе speedvac и хранили при -20°C до дальнейшего анализа.
Смесь белков восстанавливали в 0,1% муравьиной кислоте. Аликвоту каждого образца загружали на колонку C18 PicoFrit (75 мкм×10 см), соединенную с масс-спектрометром LTQ Orbitrap Velos. Белки разделяли с использованием 2-часового линейного градиента. Инструментальные способы состояли из полного МС сканирования с последующими зависимыми от данных сканированиями CID (СИД) (столкновительная диссоциация ионов) 20 наиболее интенсивных ионов-предшественников, и динамическое исключение активировали, чтобы максимизировать число ионов, подвергаемых фрагментации. Идентификация и относительный количественный анализ белков проводили посредством поиска масс-спектров по базе данных IPI-human (международные индексы белков, International protein indices) с использованием поисковой системы Mascot на платформе программы Proteome Discoverer 1.3. Поиск масс-спектров пептидов, полученных из кондиционированной среды, проводили также и по базе данных IPI-bovine, чтобы отличить белки, перенесенные из фетальной бычьей сыворотки. Параметры поиска учитывали статические модификации при S-карбоксамидометилировании в Cys (цистеине) и обратимые модификации при окислении по Met (метионину) и мечении стабильными изотопами по Lys (лизину) и Arg (аргинину). Для идентификации белков использовали сопоставления пептидных спектров (PSM) при доле ложноположительных результатов 1%. Изменения экспрессии белка при обработке соединениями подсчитывали по относительной интенсивности изотопно-меченых и немеченых пептидов, полученных из этого белка. Точность выбора белков-кандидатов, соответственно идентифицированных с помощью компьютерной программы, с измененной экспрессией (сниженной в 2 раза или на 50% или менее) дополнительно подтверждали посредством проверки вручную МС и МС/МС спектров соответствующих пептидов и устанавливали, что у тех, которые удовлетворяли критериям, после обработки соединениями экспрессия значительно снижена.
Через 16 часов после обработки 0,3 мкМ и 1,5 мкМ (соединения по Примеру 16) уровень экспрессии белка PCSK9 в кондиционированной среде был снижен в 2 и 5 раз, соответственно. Среди приблизительно 900 дополнительных белков, идентифицированных в культуральной среде, только два белка (0,22%), ламин-В1 и тропомиозин альфа-4, демонстрировали значительное снижение на 50% и менее экспрессии белка по сравнению с носителем. Подобным образом, было установлено, что соединение, оказывало крайне незначительные воздействия на белковую экспрессию клеточных белков, где из общей совокупности более чем 3000 белков в присутствии соединения по Примеру 16, ингибитора PCSK9, значительно снижался уровень только 2 дополнительных белков (0,067%), аполипопротеина С3 и кадгерина 1. Взятые вместе, эти исследования SILAC говорят о том, что соединение селективно ингибирует экспрессию белка PCSK9, поскольку только дополнительные 0,098% (4 из 4083) белков демонстрировали значительное снижение экспрессии.
Второе исследование SILAC с использованием соединения по Примеру 15b проводили согласно такому же протоколу, который описан выше, за исключением следующих поправок: использовали одну концентрацию 10 мкМ для обработки наряду с носителем, уменьшали число временных точек анализа до 4 и 16 ч после обработки, и анализ сосредотачивали только на белках, секретируемых в кондиционированную среду. С использованием этих экспериментальных условий и методики анализа было обнаружено в общей сложности приблизительно 1500 белков. Было обнаружено, что наряду с PCSK9 значительно снижался уровень только двух дополнительных белков, аполипопротеина A2 и кадгерина 1, по сравнению с носителем. Эти данные подтверждают селективность этого класса соединений в отношении PCSK9, поскольку только дополнительные приблизительно 0,13% измеренных белков проявляли значительное снижение экспрессии.
Для перорального введения пациентам-людям суточная доза соединений по настоящему изобретению может находиться в пределах от 1 мг до 5000 мг, естественно в зависимости от способа и частоты введения, болезненного состояния и возраста и состояния пациента и так далее. Под пациентом понимают человека, либо мужчину, либо женщину. Пациент может находиться в любой возрастной группе, включая младенцев (до 2 лет), детей (до 12 лет), подростков (в возрасте 13-19 лет), взрослых (в возрасте 20-65 лет), женщин в пременопаузе, женщин в постменопаузе и пожилых людей (старше 65 лет). Терапевтически эффективное количество составляет от примерно 1 мг до примерно 4000 мг в сутки. Предпочтительно терапевтически эффективное количество составляет от примерно 1 мг до примерно 2000 мг в сутки. Особенно предпочтительно, чтобы терапевтически эффективное количество составляло от примерно 50 мг до примерно 500 мг в сутки. Может быть использована пероральная суточная доза в пределах от 3 мг до 2000 мг. Дополнительная перорально вводимая суточная доза находится в пределах от 5 мг до 1000 мг. Для удобства соединения по настоящему изобретению могут быть введены в стандартной лекарственной форме. Если необходимо, могут быть использованы многократные дозы стандартной лекарственной формы в сутки для увеличения общей суточной дозы. Стандартная лекарственная форма, например, может представлять собой таблетку или капсулу, содержащую примерно 0,1, 0,5, 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 125, 150, 175, 200, 250, 500, 1000 или 2000 мг соединения по настоящему изобретению. Общая суточная доза может быть введена в однократной дозе или раздельными дозами и по усмотрению врача может оказаться вне обычных пределов, данных здесь.
Для введения пациентам-людям посредством инфузии суточная доза соединений по настоящему изобретению может находиться в пределах от 1 мг до 2000 мг, естественно в зависимости от способа и частоты введения, болезненного состояния и возраста и состояния пациента и так далее. Дополнительная вводимая посредством инфузии суточная доза находится в пределах от 5 мг до 1000 мг. Общая суточная доза может быть введена в однократной дозе или раздельными дозами и по усмотрению врача может оказаться вне обычных пределов, данных здесь.
Эти соединения также могут быть введены животным, отличным от людей, например по показаниям, описанным выше. Точная вводимая дозировка каждого активного ингредиента будет варьироваться в зависимости от множества факторов, включая тип животного и тип болезненного состояния, требующего лечения, возраст животного и путь(и) введения, но не ограничиваясь ими.
Используют дозировку фармацевтических агентов комбинации, применяемых в сочетании с соединениями формулы I, которая эффективна при указанном заболевании, требующем лечения. Такие дозировки могут быть определены с использованием стандартных анализов, таких как анализы, упомянутые выше и предложенные здесь. Агенты комбинации могут быть введены одновременно или последовательно в любом порядке.
Эти дозировки рассчитаны на среднего человека, имеющего вес примерно от 60 кг до 70 кг. Врач легко сможет определить дозы для субъектов, чей вес выходит за пределы этого диапазона, например младенцев и пожилых людей.
Режимы дозировки могут быть скорректированы для обеспечения оптимального желаемого эффекта. Например, может быть введен отдельный болюс, могут быть введены несколько раздельных доз за определенный период времени или доза может быть пропорционально снижена или увеличена, как диктуют требования терапевтической ситуации. Особенно преимущественно готовить парентеральные композиции в стандартной лекарственной форме для простоты введения и однородности дозирования. Используемый здесь термин "стандартная лекарственная форма" относится к физически дискретным единицам, подходящим в качестве единичных доз для млекопитающих, подвергаемых лечению; причем каждая единица содержит заранее установленное количество активного соединения, рассчитанное так, чтобы обеспечивать требуемый терапевтический эффект, совместно с требуемым фармацевтическим носителем. Специфические особенности стандартных лекарственных форм по изобретению обусловлены и непосредственно зависят от (а) характерных особенностей химиотерапевтического агента и конкретного терапевтического или профилактического эффекта, которого необходимо достичь, и (б) ограничений, характерных для данной области техники, в изготовлении такого активного соединения для лечения чувствительности у индивидуумов.
Таким образом, специалисту в данной области техники будет понятно на основании раскрытия, приведенного здесь, что дозу и режим дозирования регулируют в соответствии со способами, хорошо известными в области терапии. То есть, максимально переносимая доза может быть легко установлена, и также может быть определено эффективное количество, обеспечивающее обнаружимый благоприятный терапевтический эффект у пациента, как и временные требования по введению каждого агента для обеспечения обнаружимого благоприятного терапевтического эффекта у пациента. Следовательно, несмотря на то, что здесь приведены примеры определенных режимов дозирования и введения, эти примеры никак не ограничивают режим дозирования и введения, который может быть предложен пациенту при применении настоящего изобретения на практике.
Следует отметить, что величина дозы может изменяться в зависимости от типа и тяжести состояния, которое нужно облегчить, и может включать однократную или многократные дозы. Следует также понимать, что для любого конкретного субъекта конкретные режимы дозирования должны быть скорректированы с течением времени согласно индивидуальной необходимости и профессиональному мнению субъекта, который вводит или контролирует введение композиций, и что диапазоны дозирования, установленные здесь, приведены только в качестве примера и не предназначены для ограничения объема или применения заявленной композиции. Например, дозы могут быть скорректированы на основании фармакокинетических и фармакодинамических параметров, которые могут включать клинические эффекты, такие как токсические эффекты и/или данные лабораторных анализов. Таким образом, настоящее изобретение включает индивидуальное увеличение дозы у пациента, которое определяет специалист в данной области техники. Определение соответствующих дозировок и режимов введения химиотерапевтического агента хорошо известно в данной области техники и будет понятно специалисту в данной области техники при предоставлении сведений, раскрытых здесь.
Настоящее изобретение дополнительно включает применение соединения формулы I для использования в качестве лекарственного средства (такого как таблетированная лекарственная форма или капсулированная лекарственная форма). В другом воплощении настоящее изобретение включает применение соединения формулы I для изготовления лекарственного средства (такого как таблетированная лекарственная форма или капсулированная лекарственная форма) для лечения одного или более чем одного состояния, определенного ранее в вышеуказанных разделах, описывающих способы лечения.
Фармацевтическая композиция по изобретению может быть приготовлена, упакована или продаваться без упаковки в виде разовой единичной дозы или в виде множества разовых единичных доз. Использованный здесь термин "единичная доза" представляет собой дискретное количество фармацевтической композиции, содержащей заранее установленное количество активного ингредиента. Количество активного ингредиента обычно равно дозе активного ингредиента, которая должна быть введена субъекту, или подходящей части такой дозы, например половины или третьей части такой дозы.
Соединения, описанные здесь, могут быть введены в виде препарата, содержащего фармацевтически эффективное количество соединения формулы I совместно с одним или более чем одним фармацевтически приемлемым эксципиентом, включая носитель, наполнитель и разбавитель. Термин "эксципиент", используемый здесь, означает любое вещество, не являющееся терапевтическим агентом, используемое в качестве разбавителя, адъюванта или носителя для доставки терапевтического агента субъекту или добавляемое в фармацевтическую композицию для улучшения свойств при обращении с ним и хранении или для обеспечения возможности или облегчения образования твердой лекарственной формы, такой как таблетка, капсула, или раствора или суспензии, подходящих для перорального, парентерального, интрадермального, подкожного или местного применения. Эксципиенты могут включать, с целью иллюстрации, а не ограничения, разбавители, разрыхлители, связующие агенты, связывающие вещества, смачивающие вещества, полимеры, смазывающие вещества, скользящие вещества, стабилизаторы и вещества, добавляемые для маскировки или нейтрализации неприятного вкуса или запаха, ароматизаторы, красители, отдушки и вещества, добавляемые для улучшения внешнего вида композиции. Приемлемые эксципиенты включают стеариновую кислоту, стеарат магния, оксид магния, натриевые и кальциевые соли фосфорной или серной кислот, карбонат магния, тальк, желатин, гуммиарабик, альгинат натрия, пектин, декстрин, маннит, сорбит, лактозу, сахарозу, крахмалы, желатин, продукты целлюлозы, такие как эфиры целлюлозы и алкановых кислот и алкиловые эфиры целлюлозы, легкоплавкий воск, какао масло или порошок, полимеры, такие как поливинилпирролидон, поливиниловый спирт и полиэтиленгликоли, и другие фармацевтически приемлемые вещества, но не ограничиваются ими. Примеры эксципиентов и их применения можно найти в Remington's Pharmaceutical Sciences, 20th Edition (LippIncott Williams & Wilkins, 2000). Выбор эксципиента в значительной степени будет зависеть от факторов, таких как конкретный способ введения, влияние эксципиента на растворимость и стабильность и природа лекарственной формы.
Соединения, описанные здесь, могут быть приготовлены в виде препарата для перорального, трансбуккального, интраназального, парентерального (например, внутривенного, внутримышечного или подкожного) или ректального введения или в форме, подходящей для введения посредством ингаляции. Соединения по изобретению также могут быть приготовлены в виде препаратов для продолжительной доставки.
Способы получения различных фармацевтических композиций с некоторым количеством активного компонента известны или будут очевидны с учетом данного раскрытия специалистам в данной области техники. Примеры способов получения фармацевтических композиций см. в Remington's Pharmaceutical Sciences. 20th Edition (Lippincott Williams & Wilkins, 2000).
Фармацевтические композиции по настоящему изобретению могут содержать 0,1%-95% соединения(й) по настоящему изобретению, предпочтительно 1%-70%. Во всяком случае вводимая композиция будет содержать соединение(я) по настоящему изобретению в количестве, эффективном для лечения заболевания/состояния субъекта, подвергаемого лечению.
Поскольку настоящее изобретение имеет аспект, относящийся к лечению заболевания/состояний, описанных здесь, комбинацией активных ингредиентов, которые могут быть введены раздельно, то изобретение также относится к объединению отдельных фармацевтических композиций в набор. Набор содержит две отдельные фармацевтические композиции: соединение формулы I, его пролекарство или соль такого соединения или пролекарства и второе соединение, как описано выше. Набор содержит средство для содержания отдельных композиций, такое, как контейнер, разделенный флакон или разделенный пакет из тонкой пленки. Обычно набор содержит указания по введению отдельных компонентов. Форма набора особенно благоприятна, когда отдельные компоненты предпочтительно вводят в виде разных лекарственных форм (например, пероральной и парентеральной) и с разными интервалами дозирования, или когда лечащий врач требует титрования отдельных компонентов комбинации.
Примером такого набора является так называемая блистерная упаковка. Блистерные упаковки хорошо известны в упаковочной промышленности и широко применяются для упаковки фармацевтических стандартных лекарственных форм (таблеток, капсул и тому подобного). Блистерные упаковки обычно состоят из листа относительно жесткого материала, покрытого тонкой пленкой предпочтительно прозрачного пластического материала. Во время процесса упаковки в пластиковой пленке формируют углубления. Углубления имеют размер и форму таблеток или капсул, подлежащих упаковке. Затем в углубления помещают таблетки или капсулы и к пластиковой пленке герметически прикрепляют лист относительно жесткого материала со стороны пленки, противоположной направлению, в котором были сформированы углубления. В результате таблетки или капсулы оказываются герметически запаянными в углублениях между пластиковой пленкой и листом. Предпочтительно, прочность листа такова, что таблетки или капсулы могут быть извлечены из блистерной упаковки путем надавливания пальцем на углубления, в результате чего на месте углубления в листе образуется отверстие. Затем таблетку или капсулу можно извлечь через указанное отверстие.
Может быть желательным предусмотреть памятку на наборе, например, в виде чисел рядом с таблетками или капсулами, соответствующих суткам приема отмеченных таким образом таблеток или капсул. Другим примером такой памятки является календарь, напечатанный на карточке, например, следующим образом: "Первая неделя: понедельник, вторник и так далее. Вторая неделя: понедельник, вторник и так далее. Легко очевидными являются и другие варианты памяток. "Суточная доза" может представлять собой одну таблетку или капсулу или несколько пилюль или капсул, которые нужно принять за данные сутки. Кроме того, суточная доза соединения формулы I может состоять из одной таблетки или капсулы, тогда как суточная доза второго соединения может состоять из нескольких таблеток или капсул, и наоборот. Памятка должна это отражать.
В другом конкретном воплощении изобретения предусматривают дозирующее устройство, предназначенное для дозирования суточных доз по одной за раз в порядке их заданного применения. Дозирующее устройство предпочтительно снабжают памяткой, чтобы дополнительно облегчить соблюдение режима приема лекарств. Примером такой памятки является механический счетчик, показывающий число суточных доз, которые уже были выданы. Другим примером такой памятки является питаемое от батарейки запоминающее устройство на микрочипе, соединенное с жидкокристаллическим дисплеем или источником звукового напоминающего сигнала, которое считывает данные о том, что последняя суточная доза была принята, и/или напоминает, когда должна быть принята следующая доза.
Также, поскольку настоящее изобретение имеет аспект, относящийся к лечению заболевания/состояний, описанных здесь, комбинацией активных ингредиентов, которые могут быть введены совместно, то изобретение также относится к объединению отдельных фармацевтических композиций в единой лекарственной форме, такой как отдельная таблетка или капсула, двуслойная или многослойная таблетка или капсула, но не ограничивающейся ими, или посредством использования раздельных компонентов или компартментов в пределах одной таблетки или капсулы.
Активный ингредиент может быть доставлен в виде раствора в водном или безводном носителе, с дополнительными растворителями, сорастворителями, эксципиентами или агентами комплексообразования, выбранными из фармацевтически приемлемых разбавителей, эксципиентов, наполнителей или носителей, или без них.
Активный ингредиент может быть приготовлен в виде таблетки или капсулы с немедленным высвобождением или модифицированным высвобождением. Альтернативно, активный ингредиент может быть доставлен в виде одного активного ингредиента в оболочке капсулы без дополнительных эксципиентов.
ОБЩИЕ ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДИКИ
Следующие примеры предложены для того, чтобы с помощью раскрытия и описания дать представление специалистам в данной области техники как получать и оценивать соединения, композиции и способы, заявленные здесь, и они предназначены исключительно для иллюстрации изобретения и не предназначены ограничивать объем того, что авторы изобретения рассматривают как свое изобретение. Если не оговорено особо, процент представляет собой массовый процент данного компонента и общей массы композиции, температура дана в °C или представляет собой температуру окружающей среды, и давление является атмосферным или близким к атмосферному. Имеющиеся в продаже реагенты использовали без дополнительной очистки. Комнатная температура или температура окружающей среды относится к 18-25°C. Все взаимодействия в безводной среде проводили в атмосфере азота для удобства и для обеспечения максимальных выходов. Концентрирование под вакуумом означает, что был использован роторный испаритель. Названия соединений по изобретению генерировали с помощью программного пакета Autonom, PC версия 2.0, от Beilstein Informationssysteme GmbH (ISBN 3-89536-976-4). "DMSO" означает диметилсульфоксид.
Спектроскопию протонного ядерного магнитного резонанса (1Н ЯМР) проводили на спектрометрах при 400 и 500 МГц. Химические сдвиги выражены в миллионных долях слабопольного сдвига относительно тетраметилсилана. Формы пиков обозначены следующим образом: s, синглет; d, дублет; t, триплет; q, квартет; m, мультиплет; br s, уширенный синглет; br m, уширенный мультиплет. Масс-спектрометрический анализ (МС) проводили с помощью источников химической ионизации при атмосферном давлении (ХИАД) или ионизации электронным рассеиванием (ИЭР). Хроматографию на силикагеле проводили первоначально с использованием системы среднего давления с использованием предварительно упакованных колонок от различных коммерческих поставщиков. Микроанализы проводили с помощью аппаратуры Quantitative Technologies Inc. , и они находились в пределах 0,4% от расчетных значений. Термины "концентрировали" и "упаривали" относятся к удалению растворителя при пониженном давлении на роторном испарителе с температурой бани менее 60°C. Аббревиатуры "мин" и "ч" означают "минуты" и "часы", соответственно. Аббревиатура "r" означает граммы. Аббревиатура "мкл" означает микролитры.
Дифракцию рентгеновских лучей на порошке проводили на дифрактометре Bruker AXS - D4 с использованием медного излучения (длина волны: 1,54056 Å). Напряжение и ток трубки устанавливали на 40 кВ и 40 мА, соответственно. Щель расходимости и рассеивающую щель устанавливали на 1 мм, и принимающую щель устанавливали на 0,6 мм. Дифрагированное излучение детектировали с помощью позиционно-чувствительного детектора PSD-Lynx Eye. Использовали размер шага 0,02° и время на шаг 0,3 секунды в диапазоне сканирования от 3,0 до 40° 2θ. Данные собирали и анализировали с использованием программного обеспечения Diffrac Plus (версия 2.6) от Bruker. Образцы готовили, размещая их на индивидуально разработанном держателе и вращали во время сбора данных.
Для проведения измерения рентгеновской дифракции на приборе, работающем по Бреггу-Брентано, аналогично системе Bruker, использованной для измерений, описанных здесь, образец обычно помещали на держатель, который имеет углубление. Порошок образца прижимали предметным стеклом или его эквивалентом для обеспечения случайной поверхности и соответствующей высоты образца. Затем держатель с образцом помещали в прибор. Падающий пучок рентгеновских лучей направляют на образец, в начале под небольшим углом относительно плоскости держателя, а затем перемещают по дуге, которая постоянно увеличивает угол между падающим пучком и плоскостью держателя. Различия в измерениях, связанные с такими рентгеновскими анализами порошка, являются следствием множества факторов, включая: (а) погрешности при приготовлении образца (например, высоты образца), (b) погрешности приборов (например, погрешности плоских образцов), (с) погрешности калибровки, (d) ошибки оператора (включая такие ошибки, которые присутствуют при определении положений пиков) и (е) природу вещества (например, ошибки в предпочтительной ориентации и светопропускании). Погрешности калибровки и погрешности в высоте образца часто приводят к сдвигу всех пиков в том же направлении. Небольшие различия в высоте образца с использованием плоского держателя будут приводить к большим смещениям в положениях пиков XRPD. Систематическое изучение показало, что с использованием дифрактометра Shimadzu XRD-6000 в обычной конфигурации Брегга-Брентано, различие в высоте образца 1 мм приводит к сдвигам пиков вплоть до 1° угла 2θ (Chen et al.; J Pharmaceutical and Biomedical Analysis, 2001; 26, 63). Эти сдвиги могут быть установлены по рентгеновской дифрактограмме и могут быть устранены путем компенсации сдвига (применения систематического поправочного коэффициента для значений положения всех пиков) или повторной калибровки прибора. Как упомянуто выше, имеется возможность корректировки измерений, полученных на различной аппаратуре, с помощью использования систематического поправочного коэффициента для приведения положения пиков в соответствие. Обычно, этот поправочный коэффициент будет приводить измеренные на дифрактометре Bruker положения пиков в соответствие с ожидаемыми положениями пиков и может находиться в диапазоне от 0 до 0,2° угла 2θ.
Аналитическая УЭЖХ (ультраэффективная жидкостная хроматография)-МС (способ 1):
Колонка: Waters Acquity HSS Т3, С18 2,1×50 мм, 1,7 мкм; Т (температура) колонки: 60°C
Градиент: начальные условия: А-95%:В-5%; выдержка при начальных условиях 0,0-0,1 мин; линейное изменение до А-5%:В-95% за 0,1-1,0 мин; выдержка при А-5%:В-95% 1,0-1,1 мин; возвращение к начальным условиям 1,1-1,5 мин
Подвижная фаза А: 0,1% муравьиной кислоты в воде (об./об.)
Подвижная фаза В: 0,1% муравьиной кислоты в ацетонитриле (об./об.)
Скорость потока: 1,25 мл/мин
Аналитическая УЭЖХ-МС (способ 2):
Колонка: Waters Acquity HSS Т3, C18 2,1×50 мм, 1,7 мкм; Т колонки: 60°C
Градиент: начальные условия: А-95%:В-5%; выдержка при начальных условиях 0,0-0,1 мин; линейное изменение до А-5%:В-95% за 0,1-2,6 мин; выдержка при А-5%:В-95% 2,6-2,95 мин; возвращение к начальным условиям 2,95-3,0 мин
Подвижная фаза А: 0,1% муравьиной кислоты в воде (об./об.)
Подвижная фаза В: 0,1% муравьиной кислоты в ацетонитриле (об./об.)
Скорость потока: 1,25 мл/мин
Аналитическая ЖХ-МС (способ 3):
Колонка: Welch Materials Xtimate 2,1 мм×30 мм, 3 мкм
Градиент: 0%-30% (растворитель В) за 0,9 мин и выдержка при 30% в течение 0,6 мин
Подвижная фаза A: 0,0375% TFA в воде
Подвижная фаза B: 0,01875% TFA в ацетонитриле
Скорость потока: 1,2 мл/мин
Аналитическая ЖХ-МС (способ 4):
Колонка: Welch Materials Xtimate 2,1 мм×30 мм, 3 мкм
Градиент: 0-60% (растворитель В) за 2,0 мин
Подвижная фаза A: 0,0375% TFA в воде
Подвижная фаза B: 0,01875% TFA в ацетонитриле
Скорость потока: 1,2 мл/мин
Аналитическая ЖХ-МС (способ 5):
Колонка: Welch Materials Xtimate 2,1 мм×30 мм, 3 мкм
Градиент: 10-80% (растворитель В) за 2,0 мин
Подвижная фаза A: 0,0375% TFA в воде
Подвижная фаза B: 0,01875% TFA в ацетонитриле
Скорость потока: 1,2 мл/мин
Аналитическая ЖХВД (способ 1):
Колонка: Waters ВЕН С8 2,1×100 мм, 1,7 мкм
Градиент: начальные условия: А-95%:В-5%; линейное изменение до А-0%:В-100% за 0,0-8,20 мин; выдержка при А-0%:В-100% 8,2-8,7 мин; возвращение к начальным условиям 8,7-8,8 мин; выдержка при А-95%:В-5% 8,8-10,3 мин
Подвижная фаза А: 0,2% 70%-ной перхлорной кислоты в воде (об./об.)
Подвижная фаза B: ацетонитрил
Скорость потока: 0,5 мл/мин
Детектирование: УФ-210 нм
Аналитическая ЖХВД (способ 2):
Колонка: Waters ВЕН RP С18 2,1×100 мм, 1,7 мкм
Градиент: начальные условия: А-95%:В-5%; линейное изменение до А-0%:В-100% за 0,0-8,20 мин; выдержка при А-0%:В-100% 8,2-8,7 мин; возвращение к начальным условиям 8,7-8,8 мин; выдержка при А-95%:В-5% 8,8-10,3 мин
Подвижная фаза А: 0,1% метансульфоновой кислоты в воде (об./об.)
Подвижная фаза В: ацетонитрил (об./об.)
Скорость потока: 0,5 мл/мин
Детектирование: УФ-210 нм
Аналитическая ЖХВД (способ 3):
Колонка: Waters HSS Т3 2,1×100 мм, 1,8 мкм
Градиент: начальные условия: А-95%:В-5%; линейное изменение до А-0%:В-100% за 0,0-8,20 мин; выдержка при А-0%:В-100% 8,2-8,7 мин; возвращение к начальным условиям 8,7-8,8 мин; выдержка при А-95%:В-5% 8,8-10,3 мин
Подвижная фаза А: 0,1% метансульфоновой кислоты в воде (об./об.)
Подвижная фаза В: ацетонитрил (об./об.)
Скорость потока: 0,5 мл/мин
Детектирование: УФ-210 нм
Аналитическая УЭЖХ (способ 4):
Колонка: Waters HSS Т3 2,1×100 мм, 1,8 мкм; Т колонки: 45°C
Градиент: начальные условия: А-95%:В-5%; линейное изменение до А-0%:В-100% за 0-8,2 мин; выдержка при А-0%:В-100% 8,2-8,7 мин; возвращение к начальным условиям 8,7-8,8 мин
Подвижная фаза A: 10 мМ бикарбоната аммония в воде
Подвижная фаза B: ацетонитрил
Скорость потока: 0,5 мл/мин
Аналитическая ЖХВД (способ 5):
Колонка: Waters Atlantis dC18 4,6×50, 5 мкм
Градиент: начальные условия А-95%:В-5%; линейное изменение до А-5%:В-95% за 0-4,0 мин; выдержка при А-5%:В-95% 4,0-5,0 мин
Подвижная фаза А: 0,05% TFA в воде (об./об.)
Подвижная фаза В: 0,05% TFA в ацетонитриле (об./об.)
Скорость потока: 2 мл/мин
Хиральная препаративная хроматография (способ 1):
Колонка: Chiralcel-OD-H 20×250 мм, 5 мкм; Т колонки: 40°C
Подвижная фаза 75% сверхкритического жидкого СO2/25% ацетонитрила; изократические условия, рабочее противодавление: 120 бар (12 МПа)
Скорость потока: 65 мл/мин
Хиральная препаративная хроматография (способ 2):
Колонка: Chiralpak AD 5 см×25 см, 20 мкм; Т колонки: температура окружающей среды
Подвижная фаза 100% МеОН; изократические условия
Рабочее противодавление: 120 бар (12 МПа)
Скорость потока: 118 мл/мин
Объем вводимой пробы: 17,5 мл
Концентрация подаваемого вещества 245 г/л
Хиральная препаративная хроматография (способ 3):
Колонка: Chiralpak IC 2,1 см×25 см, 5 мкм
Подвижная фаза: 85/15 СО2/метанол
Скорость потока: 65 мл/мин
Т колонки: температура окружающей среды
Длина волны: 280 нм
Объем вводимой пробы: 2,0 мл
Концентрация подаваемого вещества: 125 г/л
Хиральная препаративная хроматография (способ 4):
Колонка: Chiral Tech IC 500 мм×21,2 мм, 5 мкм; Т колонки: температура окружающей среды
Подвижная фаза: 90% CO2/10% изопропанола; изократические условия
Скорость потока: 80,0 мл/мин
Хиральная препаративная хроматография (способ 5):
Колонка: Chiral Tech AD-H 500 мм×21,2 мм, 5 мкм; Т колонки: температура окружающей среды
Подвижная фаза: 80% СО2/20% изопропанола; изократические условия
Скорость потока: 80,0 мл/мин
Хиральная препаративная хроматография (способ 6):
Колонка: Chiral OD-H 500 мм×21,2 мм, 5 мкм; Т колонки: температура окружающей среды
Подвижная фаза: 90% CO2/10% изопропанола; изократические условия
Скорость потока: 80,0 мл/мин
Хиральная препаративная хроматография (способ 7):
Колонка: Chiral Tech AD-H 500 мм×21,2 мм, 5 мкм; Т колонки: температура окружающей среды
Подвижная фаза: 95% СО2/5% изопропанола; изократические условия
Скорость потока: 80,0 мл/мин
Хиральная препаративная хроматография (способ 8):
Колонка: Chiral Tech IC 250 мм×21,2 мм, 5 мкм; Т колонки: температура окружающей среды
Подвижная фаза: 60% СО2/40% МеОН; изократические условия
Скорость потока: 80,0 мл/мин
Хиральная препаративная хроматография (способ 9):
Колонка: Lux Cellulose-3 500 мм×21,2 мм, 5 мкм; Т колонки: температура окружающей среды
Подвижная фаза: 95% СO2/5% изопропанола; изократические условия
Скорость потока: 80,0 мл/мин
Хиральная препаративная хроматография (способ 10):
Колонка: Lux Amylose-2 500 мм×21,2 мм, 5 мкм; Т колонки: температура окружающей среды
Подвижная фаза: 95% CO2/5% MeOH/MeCN (1:1); изократические условия
Скорость потока: 80,0 мл/мин
Хиральная препаративная хроматография (способ 11):
Колонка: Lux Amylose-2 250 мм×50,0 мм, 5 мкм; Т колонки: температура окружающей среды
Подвижная фаза: 65% СО2/35% МеОН; изократические условия
Скорость потока: 250 мл/мин
Хиральная препаративная хроматография (способ 12):
Колонка: Chiral Tech IC 500 мм×10,0 мм, 5 мкм; Т колонки: температура окружающей среды
Подвижная фаза: 90% СО2/10% изопропанола; изократические условия
Скорость потока: 15 мл/мин
Хиральная препаративная хроматография (способ 13):
Колонка: Lux Amylose IC 500 мм×21,2 мм, 5 мкм; Т колонки: температура окружающей среды
Подвижная фаза: 95% СО2/5% изопропанола; изократические условия
Скорость потока: 80,0 мл/мин
Хиральная препаративная хроматография (способ 14):
Колонка: Chiral Tech OJ-H 500 мм×21,2 мм, 5 мкм; Т колонки: температура окружающей среды
Подвижная фаза: 95% СО2/5% изопропанола; изократические условия
Скорость потока: 80,0 мл/мин
Хиральная аналитическая хроматография (способ 1):
Колонка: Chiral Tech IC 250 мм×4,6 мм, 5 мкм
Градиент: начальные условия: А-95%:В-5%; линейное изменение до А-40%:В-60% за 1,0-9,0 мин; выдержка при А-40%:В-60% 9,0-9,5 мин; линейное изменение до А-95%:В-5% за 9,5-10,0 мин
Подвижная фаза A: CO2
Подвижная фаза B: изопропанол
Скорость потока: 3,0 мл/мин
Детектирование: УФ-210 нм
Хиральная аналитическая хроматография (способ 2):
Колонка: Chiral Tech AD-H 250 мм×4,6 мм, 5 мкм
Градиент: начальные условия: А-95%:В-5%; линейное изменение до А-40%:В-60% за 1,0-9,0 мин; выдержка при А-40%:В-60% 9,0-9,5 мин; линейное изменение до А-95%:В-5% за 9,5-10,0 мин
Подвижная фаза A: CO2
Подвижная фаза B: изопропанол
Скорость потока: 3,0 мл/мин
Детектирование: УФ-210 нм
Хиральная аналитическая хроматография (способ 3):
Колонка: Chiral Tech OD-H 250 мм×4,6 мм, 5 мкм
Градиент: начальные условия: А-95%:В-5%; линейное изменение до А-40%:В-60% за 1,0-9,0 мин; выдержка при А-40%:В-60% 9,0-9,5 мин; линейное изменение до А-95%:В-5% за 9,5-10,0 мин
Подвижная фаза A: CO2
Подвижная фаза B: изопропанол
Скорость потока: 3,0 мл/мин
Детектирование: УФ-210 нм
Хиральная аналитическая хроматография (способ 4):
Колонка: Chiral Tech IC 250 мм×4,6 мм, 5 мкм
Градиент: начальные условия: А-95%:В-5%; линейное изменение до А-40%:В-60% за 1,0-9,0 мин; выдержка при А-40%:В-60% 9,0-9,5 мин; линейное изменение до А-95%:В-5% за 9,5-10,0 мин
Подвижная фаза A: СO2
Подвижная фаза B: МеОН
Скорость потока: 3,0 мл/мин
Детектирование: УФ-210 нм
Хиральная аналитическая хроматография (способ 5):
Колонка: Lux Cellulose-3 250 мм×4,6 мм, 5 мкм
Градиент: начальные условия: А-95%:В-5%; линейное изменение до А-40%:В-60% за 1,0-9,0 мин; выдержка при А-40%:В-60% 90-95 мин; линейное изменение до А-95%:В-5% за 9,5-10,0 мин
Подвижная фаза A: CO2
Подвижная фаза B: изопропанол
Скорость потока: 3,0 мл/мин
Детектирование: УФ-210 нм
Хиральная аналитическая хроматография (способ 6):
Колонка: Chiral Tech OJ-H 250 мм×4,6 мм, 5 мкм
Градиент: начальные условия: А-95%:В-5%; линейное изменение до А-40%:В-60% за 1,0-9,0 мин; выдержка при А-40%:В-60% 9,0-9,5 мин; линейное изменение до А-95%:В-5% за 9,5-10,0 мин
Подвижная фаза A: CO2
Подвижная фаза B: изопропанол
Скорость потока: 3,0 мл/мин
Детектирование: УФ-210 нм
Хиральная аналитическая хроматография (способ 7):
Колонка: Lux Amylose-2 250 мм×4,6 мм, 5 мкм
Градиент: начальные условия: А-95%:В-5%; линейное изменение до А-40%:В-60% за 1,0-9,0 мин; выдержка при А-40%:В-60% 9,0-9,5 мин; линейное изменение до А-95%:В-5% за 9,5-10,0 мин
Подвижная фаза A: CO2
Подвижная фаза B: MeOH/MeCN (1:1)
Скорость потока: 3,0 мл/мин
Детектирование: УФ-210 нм
Хиральная аналитическая хроматография (способ 8):
Колонка: Lux Amylose-2 250 мм×4,6 мм, 5 мкм
Градиент: начальные условия: А-95%:В-5%; линейное изменение до А-40%:В-60% за 1.0-9.0 мин; выдержка при А-40%:В-60% 9,0-9,5 мин; линейное изменение до А-95%:В-5% за 9,5-10,0 мин
Подвижная фаза A: CO2
Подвижная фаза B: изопропанол
Скорость потока: 3,0 мл/мин
Детектирование: УФ-210 нм
ПОЛУЧЕНИЯ
Получение 1: трет-бутил-(3R)-3-[(3-хлорпиридин-2-ил)амино]пиперидин-1-карбоксилат
Смесь 2-бром-3-хлорпиридина (203,8 г; 1,06 моль), трет-амилата натрия (147 г; 1,27 моль), трет-бутил-(3R)-3-аминопиперидин-1-карбоксилата (249,5 г; 1,25 моль) в толуоле (2 л) нагревали до 80°C. В этот раствор добавляли хлор-(ди-2-норборнилфосфино)(2-диметиламиноферроцен-1-ил)палладий(II) (6,1 г; 10,06 ммоль) с последующим нагреванием до 105°C и выдержкой в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры, добавляли 1 л воды, затем двухфазную смесь фильтровали через Celite®. После разделения слоев органическую фазу промывали 1 л воды с последующей обработкой 60 г активированного угля Darco® G-60 при 50°C. Смесь фильтровали через Celite®, и концентрировали до общего конечного объема 450 мл, что приводило к осаждению твердых веществ. К суспензии твердых веществ добавляли 1 л гептана. Твердые вещества собирали посредством фильтрации и затем сушили с получением указанного в заголовке соединения в виде бледно-оранжевого твердого вещества (240,9 г; выход 73%).
1Н ЯМР (CDCl3) δ 8.03 (m, 1Н), 7.45 (m, 1Н), 6.54 (m, 1Н), 5.08 (br s, 1Н), 4.14 (br s, 1H), 3.85-3.30 (m, 4H), 2.00-1.90 (m, 1H), 1.80-1.55 (m, 4H), 1.43 (br s, 9H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,72 мин.
MC (ЭР+) 312,0 (М+Н)+.
Получение 2: трет-бутил-(3R)-3-[(3-метилпиридин-2-ил)амино]пиперидин-1-карбоксилат
В раствор 2-бром-3-метилпиридина (75,0 г; 436 ммоль) и трет-бутил-(3R)-3-аминопиперидин-1-карбоксилата (87,3 г; 436 ммоль) в толуоле (1,2 л) добавляли CS2CO3 (426 г; 1,31 моль), 2-(диметиламинометил)ферроцен-1-ил-палладий(II) хлорид динорборнилфосфин (MFCD05861622) (1,56 г; 4,36 ммоль) and Pd(OAc)2 (0,490 г; 2,18 ммоль) в атмосфере N2. Смесь перемешивали при 110°C в течение 48 ч. Смесь охлаждали до комнатной температуры, затем вливали в воду (500 мл) и экстрагировали EtOAc (3×300 мл). Органические слои сушили над Na2SO4, фильтровали и фильтрат концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения в виде желтого твердого вещества (65 г; 60%).
1Н ЯМР (CDCl3) δ 8.00 (d, 1Н), 7.20 (d, 1Н), 6.51(dd, 1Н), 4.36 (br s, 1Н), 4.16 (br s, 1Н), 3.63 (d, 1Н), 3.52 (br s, 2Н), 3.36-3.30 (m, 1Н), 2.06 (s, 3Н), 1.90 (br s, 1Н), 1.73 (br s, 2H), 1.59 (br s, 1H), 1.38 (br s, 9H).
Получение 3: трет-бутил-(3R)-3-(изохинолин-1-иламино)пиперидин-1-карбоксилат
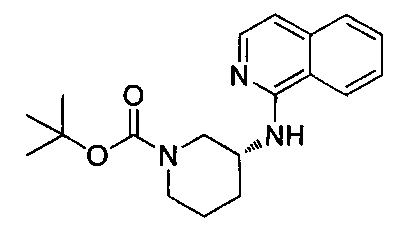
В раствор 1-хлоризохинолина (5,00 г; 30,6 ммоль) и трет-бутил (3R)-3-аминопиперидин-1-карбоксилата (7,34 г; 36,6 ммоль) в безводном толуоле (150 мл) добавляли f-BuOK (10,27 г; 91,7 ммоль), BINAP (951 мг; 1,53 ммоль) и Pd(OAc)2 (343 мг; 1,53 ммоль). Реакционную смесь трижды продували азотом и нагревали при 110°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры и вливали в воду. Смесь затем экстрагировали EtOAc (4×150 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученное темное твердое вещество очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом (10-15%) EtOAc/петролейный эфир, с получением указанного в заголовке соединения (4,89 г; 49%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 7.97 (d, 1Н), 7.72 (d, 1Н), 7.68 (d, 1Н), 7.56 (dd, 1Н), 7.43 (dd, 1Н), 6.92 (d, 1Н), 5.34 (br s, 1Н), 4.33-4.29 (m, 1Н), 3.70-3.55 (m, 3Н), 3.36-3.30 (m, 1Н), 2.04-1.77 (m, 4Н), 1.35 (br s, 9Н).
Получение 4: трет-бутил (3R)-3-[(1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)амино]пиперидин-1-карбоксилат
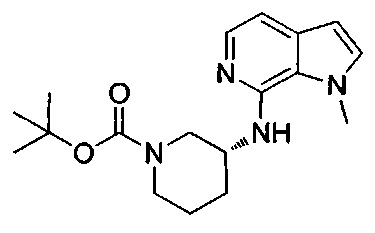
Стадия 1: 7-хлор-1-метил-1Н-пирроло[2,3-с]пиридин
В раствор 7-хлор-1Н-пирроло[2,3-с]пиридина (3,00 г; 19,7 ммоль) в THF (30 мл), охлажденный до 0°C, добавляли 60% NaH в минеральном масле (0,940 г; 23,6 ммоль NaH). Реакционную смесь перемешивали при 0°C в течение 20 минут. Добавляли йодметан (6,20 г; 43,7 ммоль) и реакционную смесь перемешивали при 0°C в течение 1 ч и при 20°C в течение 2 ч. Смесь вливали в воду (20 мл) и экстрагировали EtOAc (3×20 мл). Органические слои сушили над Na2SO4, фильтровали и фильтрат концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом 17-33% EtOAc/петролейный эфир, с получением указанного в заголовке соединения в виде желтого твердого вещества (2,8 г; 85%).
1Н ЯМР (CDCl3) δ 7.96 (d, 1Н), 7.43 (d, 1Н), 7.17 (d, 1Н), 6.50 (d, 1Н), 4.18 (s, 3Н).
Стадия 2: (R)-трет-бутил-3-((1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)амино)пиперидин-1-карбоксилат
Раствор Pd2(dba)3 (1,53 г; 1,68 ммоль), BrettPhos (1,86 г; 3,36 ммоль) в толуоле (40 мл) в атмосфере N2 перемешивали в течение 20 мин. Добавляли NaOtBu (3,23 г; 33,6 ммоль), соединение со стадии 1 7-хлор-1-метил-1Н-пирроло[2,3-с]пиридин (2,80 г; 16,8 ммоль) и трет-бутил-(3R)-3-аминопиперидин-1-карбоксилат (4,04 г; 20,2 ммоль). Реакционную смесь перемешивали при 105°C в течение 3 ч. Реакционную смесь вливали в воду (20 мл) и экстрагировали EtOAc (3×20 мл). Органические слои сушили над Na2SO4, фильтровали и фильтрат концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом 17-50% EtOAc/петролейный эфир, с получением указанного в заголовке соединения в виде коричневого твердого вещества (5,2 г; 94%).
1Н ЯМР (CDCl3) δ 7.69 (d, 1Н), 6.91 (d, 1Н), 6.99 (d, 1Н), 6.31 (d, 1Н), 4.75 (br s, 1Н), 4.33 (brs, 1Н), 4.08 (s, 3Н), 3.89-3.77 (m, 2Н), 3.44 (d, 1Н), 3.14 (dd, 1Н), 1.90 (br s, 1Н), 1.75-1.72 (m, 1Н), 1.42 (br s, 2 Н), 1.22 (br s, 9Н).
Получение 5: 4-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензойная кислота
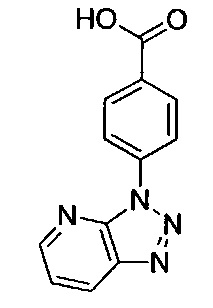
Стадия 1: этил-4-((3-нитропиридин-2-ил)амино)бензоат
Общая методика Е. В раствор 2-хлор-3-нитропиридина (95,0 г; 0,600 моль) и этил-4-аминобензоата (99,0 г; 0,600 моль) в толуоле (3 л) добавляли K2CO3 (166 г; 1,20 моль), BINAP (7,40 г; 11,8 ммоль), Pd(OAc)2 (2,80 г; 12,5 ммоль) и NaI (2,70 г; 18,0 ммоль). Смесь перемешивали при 110°C в течение 6 ч. Реакционную смесь охлаждали до 30°C, фильтровали через Celite® и фильтрат концентрировали под вакуумом. Остаток переносили в делительную воронку с водой (300 мл) и экстрагировали EtOAc (3×300 мл). Органические слои сушили над Na2SO4, фильтровали и фильтрат концентрировали под вакуумом с получением темного твердого остатка, который промывали смесью ацетон/вода (5/1; 100 мл) и фильтровали с получением указанного в заголовке соединения в виде желтого твердого вещества (142 г; 82%).
1Н ЯМР (DMSO-d6) δ 8.60-8.57 (m, 2Н), 7.96-7.94 (m, 2Н), 7.89-7.87 (m, 2Н), 7.12 (dd, 1Н), 4.31 (q, 2Н), 1.33 (t, 3Н).
Стадия 2: этил-4-((3-аминопиридин-2-ил)амино)бензоат
Общая методика F. В раствор соединения со стадии 1 этил-4-((3-нитропиридин-2-ил)амино)бензоата (120 г; 0,417 моль) в этаноле (2,00 л) добавляли Raney-Ni (никель Ренея) (30 г). Реакционную смесь гидрировали в атмосфере Н2 (50 ф/кв дюйм (0,34 МПа)) при 30°C в течение 20 ч. Смесь фильтровали через Celite®. Фильтрат сушили над Na2SO4, фильтровали и фильтрат концентрировали под вакуумом с получением желтого твердого вещества. Желтое твердое вещество промывали DCM с получением указанного в заголовке соединения в виде желтого твердого вещества (75 г; 70%).
1Н ЯМР (DMSO-d6) δ 10.05 (br, 1Н), 7.94 (d, 2Н) 7.54 (d, 1Н), 7.46 (d, 2Н), 7.40 (d, 1Н), 7.09 (dd, 1Н), 4.30 (q, 2Н), 1.31 (t, 3Н).
Стадия 3: этил-4-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензоат
Общая методика G. В раствор соединения со стадии 2 этил-4-((3-аминопиридин-2-ил)амино)бензоата (70,0 г; 0,272 моль) в смеси АсОН (70 мл) и воды (70 мл) добавляли NaNO2 (23,8 г; 0,345 моль) при 0°C. Смесь перемешивали при 30°C в течение 20 мин. Смесь разбавляли DCM (100 мл), промывали водой (3×50 мл). Органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали под вакуумом с получением темного твердого вещества. Твердое вещество промывали ацетоном (30 мл) с получением указанного в заголовке соединения в виде белого твердого вещества (63 г; 86%).
1Н ЯМР (DMSO-d6) δ 8.91 (d, 1Н), 8.91 (dd, 1Н), 8.50 (d, 2Н), 8.24 (d, 2Н), 7.66 (dd, 1Н), 4.37 (q, 2Н), 1.36 (t, 3Н).
Стадия 4: 4-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензойная кислота
Общая методика Н. В раствор соединения со стадии 3 этил-4-(3Н-[1,2,3]триазоло[4,5-L]пиридин-3-ил)бензоата (60,0 г; 0,224 моль) в МеОН (700 мл) добавляли 2 н. NaOH (260 мл; 0,520 ммоль). Смесь перемешивали при 60°C в течение 2 ч. Смесь подкисляли 1 н HCl, так что pH раствора составлял приблизительно pH 1. Реакционную смесь экстрагировали EtOAc (3×100 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и фильтрат концентрировали под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (52 г; 97%).
1Н ЯМР (DMSO-d6) δ 13.23 (br, 1Н), 8.91 (d, 1Н), 8.76 (d, 1Н), 8.48 (d, 2Н), 8.24 (d, 2Н), 7.67 (dd, 1Н).
Получение 6: 4-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензойная кислота
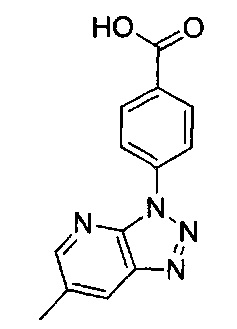
Стадия 1: этил-4-((5-метил-3-нитропиридин-2-ил)амино)бензоат
Получали согласно общей методике Е, начиная с 2-хлор-5-метил-3-нитропиридина (10,0 г; 57,9 ммоль), с получением указанного в заголовке соединения в виде коричневого твердого вещества (16,5 г; 95%).
1Н ЯМР (DMSO-d6) δ 10.01 (s, 1Н), 8.48 (d, 1Н), 8.44 (d, 1Н), 7.93 (d, 2Н), 7.85 (d, 2Н), 4.30 (q, 2Н), 2.33 (s, 3Н), 1.33 (t, 3Н).
Стадия 2: этил-4-((3-амино-5-метилпиридин-2-ил)амино)бензоат
Получали согласно общей методике F, начиная с соединения со стадии 1 этил-4-((5-метил-3-нитропиридин-2-ил)амино)бензоата (16,5 г; 54,8 ммоль), с получением указанного в заголовке соединения в виде черного твердого вещества (13,8 г; 93%).
Стадия 3: этил-4-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензоат
Получали согласно общей методике G, начиная с соединения со стадии 2 этил-4-((3-амино-5-метилпиридин-2-ил)амино)бензоата (13,8 г; 50,8 ммоль), с получением указанного в заголовке соединения в виде черного твердого вещества (13 г; 91%).
Стадия 4: 4-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензойная кислота
Получали согласно общей методике Н, начиная с соединения со стадии 3 этил-4-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензоата (13,0 г; 46,1 ммоль), с получением указанного в заголовке соединения в виде коричневого твердого вещества (10,5 г; 90%).
1Н ЯМР (DMSO-d6) δ 8.78 (s, 1Н), 8.54 (s, 1Н), 8.48 (d, 2Н), 8.24 (d, 2Н), 2.56 (s, 3Н).
Получение 7: 5-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиридин-2-карбоновая кислота
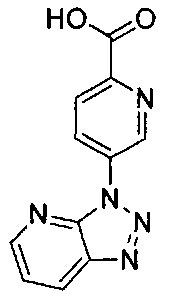
Стадия 1: этил-5-аминопиколинат
В раствор 5-аминопиколиновой кислоты (13,5 г; 97,7 ммоль) в безводном этаноле (200 мл) по каплям добавляли SOCl2 (60,0 мл; 504 ммоль) при 0°C в атмосфере N2. Полученную смесь нагревали при температуре дефлегмации и перемешивали в течение ночи. Реакционную смесь концентрировали под вакуумом. Остаток растворяли в насыщенном водном растворе NaHCO3, так чтобы pH раствора составлял приблизительно pH 9-10. Реакционную смесь экстрагировали EtOAc (8×250 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали под вакуумом с получением указанного в заголовке соединения в виде коричневого твердого вещества (14,3 г; 88%).
1Н ЯМР (CDCl3) δ 8.14 (d, 1Н), 7.94 (d, 1Н), 6.97 (dd, 1Н), 4.41 (q, 2Н), 4.16 (br s, 2Н), 1.39 (t, 3Н).
Стадия 2:
Получали согласно общей методике Е, начиная с соединения со стадии 1 этил-5-аминопиколината (13,3 г; 80,0 ммоль) и 2-хлор-3-нитропиридина (15,2 г; 95,9 ммоль), с получением указанного в заголовке соединения в виде желтого твердого вещества (9,3 г; 40%).
1Н ЯМР (CDCl3) δ 10.34 (s, 1Н), 8.95 (d, 1Н), 8.61-8.55 (m, 2Н), 8.47 (dd, 1Н), 8.16 (d, 1Н), 7.02 (dd, 1Н), 4.48 (q, 2Н), 1.45 (t, 3Н).
Стадия 3: этил-5-((3-аминопиридин-2-ил)амино)пиколинат
Получали согласно общей методике F, начиная с соединения со стадии 2 этил-5-((3-нитропиридин-2-ил)амино)пиколината (9,30 г; 32,3 ммоль), с получением указанного в заголовке соединения в виде желтого твердого вещества (количественный выход).
1Н ЯМР (CDCl3) δ 8.53 (d, 1Н), 8.14 (dd, 1Н), 8.06 (d, 1Н), 7.87 (dd, 1Н), 7.08 (dd, 1Н), 6.87-6.84 (m, 2Н), 4.43 (q, 2Н), 3.54 (br s, 2Н), 1.41 (t, 3Н).
Стадия 4: этил-5-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиколинат
Получали согласно общей методике G, начиная с соединения со стадии 3 этил-5-((3-аминопиридин-2-ил)амино)пиколината (8,70 г; 33,7 ммоль), с получением указанного в заголовке соединения в виде желтого твердого вещества (9,0 г; 99%).
1Н ЯМР (CDCl3) δ 9.88 (d, 1Н), 8.97 (dd, 1Н), 8.82 (dd, 1Н), 8.51 (dd, 1Н), 8.39 (d, 1Н), 7.50 (dd, 1Н), 4.53 (q, 2Н), 1.49 (t, 3Н).
Стадия 5: 5-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиридин-2-карбоновая кислота
Получали согласно общей методике Н, начиная с соединения со стадии 4 этил-5-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиколината (9,00 г; 33,4 ммоль), с получением указанного в заголовке соединения в виде красного твердого вещества (8,1 г; 99%).
1Н ЯМР (CDCl3) δ 13.50 (br s, 1Н), 9.62 (d, 1Н), 8.93-8.87 (m, 2Н), 8.77 (d, 1Н), 8.36 (d, 1Н), 7.68 (dd, 1Н).
Получение 8: 5-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиридин-2-карбоновая кислота
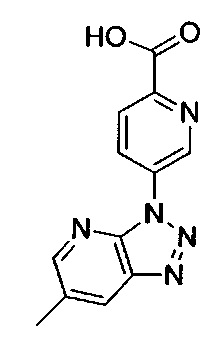
Стадия 1: этил-5-((5-метил-3-нитропиридин-2-ил)амино)пиколинат
Получали согласно общей методике Е, начиная с этил-5-аминопиколината (14,0 г; 84,2 ммоль) и 2-хлор-5-метил-3-нитропиридина (17,4 г; 101 ммоль), с получением указанного в заголовке соединения в виде желтого твердого вещества (13,6 г; 53%).
1Н ЯМР (CDCl3) δ 10.22 (s, 1Н), 8.94 (d, 1Н), 8.46-8.40 (m, 3Н), 8.15 (d, 1Н), 4.47 (q, 2Н), 2.39 (s, 3Н), 1.45 (t, 3Н).
Стадия 2: этил-5-((3-амино-5-метилпиридин-2-ил)амино)пиколинат
Получали согласно общей методике F, начиная с соединения со стадии 1 этил-5-((5-метил-3-нитропиридин-2-ил)амино)пиколината (13,6 г; 45,0 ммоль), с получением указанного в заголовке соединения в виде красного твердого вещества (12,2 г; 99%).
1Н ЯМР (CDCl3) δ 8.43 (d, 1Н), 8.01 (d, 1Н), 7.88 (dd, 1Н), 7.68 (d, 1Н), 6.90 (d, 1Н), 6.83 (s, 1Н), 4.41 (q, 2Н), 3.47 (br s, 2Н), 2.24 (s, 3H), 1.40 (t, 3H).
Стадия 3: этил-5-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-пиколинат
Получали согласно общей методике G, начиная с соединения со стадии 2 этил-5-((3-амино-5-метилпиридин-2-ил)амино)пиколината (12,2 г; 44,8 ммоль), с получением указанного в заголовке соединения в виде красного твердого вещества (12,6 г; 99%).
1Н ЯМР (CDCl3) δ 9.87 (d, 1Н), 8.95 (dd, 1Н), 8.65 (d, 1Н), 8.37 (d, 1Н), 8.25 (s, 1Н), 4.53 (q, 2Н), 2.59 (s, 3H), 1.47 (t, 3H).
Стадия 4: 5-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиридин-2-карбоновая кислота
Получали согласно общей методике Н, начиная с соединения со стадии 3 этил-5-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиколината (12,6 г; 44,5 ммоль), с получением указанного в заголовке соединения в виде красного твердого вещества (10,8 г; 95%).
1Н ЯМР (DMSO-d6) δ 13.50 (br s, 1Н), 9.60 (d, 1Н), 8.87 (dd, 1Н), 8.78 (s, 1Н), 8.55 (s, 1Н), 8.35 (d, 1Н), 2.55 (s, 3H).
Получение 9: трет-бутил-(3R)-3-[(4-бромбензоил)(изохинолин-1-ил)амино1-пиперидин-1-карбоксилат
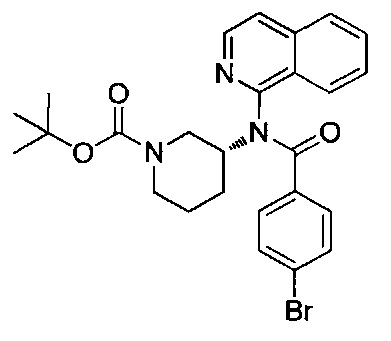
К суспензии 4-бромбензойной кислоты (3,30 г; 16,4 ммоль) в безводном DCM (120 мл) по каплям добавляли оксалилхлорид (6,26 г; 49,3 ммоль) при 0°C с последующим добавлением 3 капель DMF. Полученную смесь перемешивали при комнатной температуре в течение 2 ч. Смесь затем концентрировали под вакуумом с получением 4-бромбензоилхлорида в виде желтого твердого вещества. Твердое вещество растворяли в безводном THF (40 мл) и добавляли раствор соединения по Получению 3 трет-бутил-(3R)-3-(изохинолин-1-иламино)пиперидин-1-карбоксилата (4,89 г; 14,9 ммоль) в безводном THF (50 мл). Полученный раствор обрабатывали, добавляя по каплям бис(триметилсилил)амидом лития (44,8 мл; 44,8 ммоль; 1 М) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь вливали в воду и экстрагировали EtOAc (4×100 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали посредством хроматографии на силикагеле, элюируя градиентом (0-25%) EtOAc/петролейный эфир, с получением указанного в заголовке соединения (4,80 г; 63%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8.42 (s, 1Н), 7.98-7.83 (m, 1Н), 7.73 (d, 1Н), 7.65-7.47 (m, 3H), 7.10-7.06 (m, 4Н), 4.94-4.45 (m, 2Н), 4.26-3.91 (m, 1.5Н), 3.50-3.45 (m, 0.5Н), 2.65-2.15 (m, 2Н), 1.86-1.56 (m, 3H), 1.52 & 1.43 (s, 9Н).
Получение 10: трет-бутил-(3R)-3-[(4-бромбензоил)(3-хлорпиридин-2-ил)-амино]пиперидин-1-карбоксилат
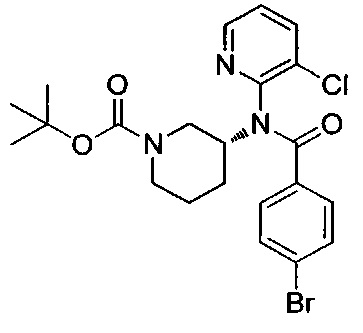
Соединение по Получению 1 трет-бутил-(3R)-3-[(3-хлорпиридин-2-ил)амино]пиперидин-1-карбоксилат (214,4 г; 687,7 ммоль) растворяли в 260 мл THF и полученную суспензию охлаждали до -10°C. Бис(триметилсилил)амид лития (1 моль/л в THF; 687,7 мл; 687,1 ммоль) добавляли в течение 25 мин с последующим нагреванием до 20°C и перемешиванием в течение 1 ч, а затем снова охлаждали до -10°C. Добавляли 4-бромбензоилхлорид (140,0 г; 625,2 ммоль) в виде раствора в 230 мл THF в течение 1,5 ч, поддерживая внутреннюю температуру менее -7°C. После окончания добавления реакционную смесь нагревали до 0°C, при этом ЖХВД показала, что реакция завершена. Добавляли МеОН (101 мл), затем реакционную смесь нагревали до комнатной температуры и концентрировали под вакуумом до небольшого объема. Растворитель затем заменяли на 2-MeTHF. Раствор неочищенного продукта (700 мл в 2-MeTHF) промывали 700 мл полунасыщенного водного NaHCO3 с последующим промыванием 200 мл полунасыщенного рассола. 2-MeTHF раствор концентрировали до небольшого объема с последующим добавлением 400 мл гептана, что приводило к осаждению твердых веществ, которые собирали посредством фильтрации. Собранные твердые вещества сушили с получением указанного в заголовке соединения в виде рыжевато-коричневого порошка (244 г; выход 79%).
1Н ЯМР (ацетонитрил-d3) δ 8.57-8.41 (m, 1Н), 7.85-7.62 (m, 1Н), 7.37 (d, 2Н), 7.31 (dd, 1Н), 7.23 (d, 2Н), 4.63-4.17 (m, 2Н), 4.06-3.89 (m, 1Н), 3.35-3.08 (br s, 0.5Н), 2.67-2.46 (m, 1Н), 2.26-2.10 (br s, 0.5Н), 1.92-1.51 (m, 3Н), 1.46 (s, 9Н), 1.37-1.21 (m, 1Н).
УЭЖХ (УЭЖХ, способ 4): tR составляет 7,03 мин.
Альтернативный способ получения 10:
В раствор соединения по Получению 1 (R)-трет-бутил-3-((3-хлорпиридин-2-ил)амино)пиперидин-1-карбоксилата (100 г; 321 ммоль) и 4-бромбензоилхлорида (73,7 г; 336 ммоль) в безводном THF (500 мл) по каплям добавляли 1 М бис(триметилсилил)амида лития (362 мл; 362 ммоль) при 0°C. Реакционную смесь нагревали и перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили водой и экстрагировали EtOAc (3×1000 мл). Объединенные органические слои промывали рассолом, сушили над Na2SO4, фильтровали и фильтрат концентрировали под вакуумом. Остаток очищали посредством хроматографии на силикагеле с получением указанного в заголовке соединения в виде желтого твердого вещества (100 г; 63%).
1Н ЯМР (CDCl3) δ 8.43 (br s, 1Н), 7.56 (br s, 1H), 7.28-7.14 (m, 5H), 4.48 (br s, 2H), 4.24 (br s, 1H), 4.09 (br s, 1H), 3.28 (br s, 1H), 2.54 (br s, 1H), 2.27 (br s, 1H), 1.63-1.54 (br m, 1H), 1.46 (br s, 10H).
Получение 11: трет-бутил-(3R)-3-[(4-бромбензоил)(1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)амино]пиперидин-1-карбоксилат
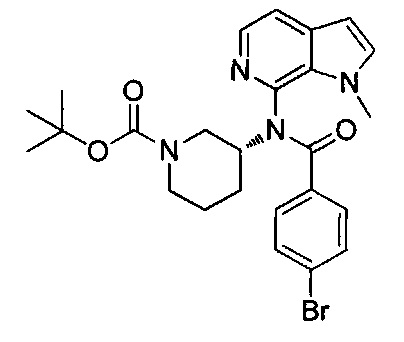
В раствор соединения по Получению 4 (R)-трет-бутил-3-((1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)амино)пиперидин-1-карбоксилата (45,0 г; 136 ммоль) и 4-бромбензоилхлорида (31,3 г; 143 ммоль) в безводном THF (250 мл) по каплям добавляли 1 М бис(триметилсилил)амид лития (163 мл; 163 ммоль) при 0°C. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 15 ч. Реакционную смесь гасили водным NH4Cl и экстрагировали EtOAc (3×500 мл). Объединенные органические слои промывали рассолом, сушили над Na2SO4, фильтровали и фильтрат концентрировали под вакуумом. Остаток очищали посредством перекристаллизации (петролейный эфир : EtOAc; 2:1) с получением указанного в заголовке соединения в виде желтоватого твердого вещества (50 г; 72%).
1Н ЯМР (CDCl3) δ 8.13 (d, 1Н), 7.43 (br s, 1Н), 7.14 (br s, 4Н), 6.99 (br s, 1H), 6.38 (br s, 1H), 4.75-4.65 (br m, 2H), 4.11 (br s, 1H), 3.96-3.82 (br m, 4 H), 3.45 (br 2, 1H), 2.61 (br s, 1H), 2.25 (br s, 1H), 1.78 (br s, 1H), 1.41 (br s, 10H).
Получение 12: трет-бутил-(3R)-3-[(4-бромбензоил)(3-метилпиридин-2-ил)амино]пиперидин-1-карбоксилат
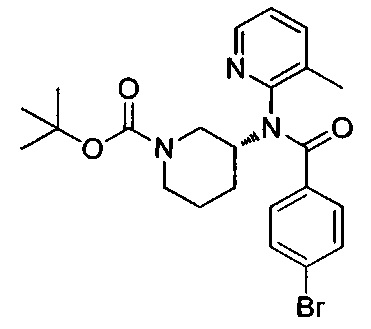
В раствор соединения по Получению 2 (R)-трет-бутил-3-((3-метилпиридин-2-ил)амино)пиперидин-1-карбоксилата (33,3 г; 114 ммоль) и 4-бромбензоилхлорида (26,3 г; 120 ммоль) в безводном THF (300 мл) по каплям добавляли 1 М бис(триметилсилил)амид лития (137 мл; 137 ммоль) при 0°C. Реакционную смесь нагревали и перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь гасили водой и экстрагировали EtOAc (3×1000 мл). Объединенные органические слои промывали рассолом, сушили над Na2SO4, фильтровали и фильтрат концентрировали под вакуумом. Остаток очищали посредством хроматографии на силикагеле с получением указанного в заголовке соединения в виде желтого твердого вещества (27 г; 50%).
1Н ЯМР (CDCl3) δ 8.41 (br s, 1Н), 7.34 (br s, 1Н), 7.25 (d, 2Н), 7.16-7.14 (m, 3H), 4.65 (br s, 1H), 4.48 (br d, 1H), 4.15-4.04 (br m, 2H), 3.39 (br s, 1H), 2.55 (br s, 1H), 2.37 (br s, 1H), 2.01-1.98 (br d, 3H), 1.74 (br s, 1H), 1.47-1.43 (br d, 10H).
Получение 13: этил-4-йод-1-метил-1Н-пиразол-5-карбоксилат
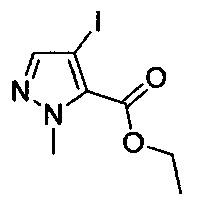
4-Йод-1-метил-1Н-пиразол-5-карбоновую кислоту (301,68 г; 1,2 моль) суспендировали в 1,2 л DCM и DMF (2,3 г; 31 ммоль) с последующим добавлением оксалилхлорида (115 мл; 1,3 моль) в течение 37 минут и затем перемешивали при комнатной температуре в течение 3 ч. К полученному раствору добавляли ЕЮН (750 мл; 12,9 моль) в течение 5 мин с последующим перемешиванием при комнатной температуре в течение 2 ч. Раствор неочищенного продукта концентрировали под вакуумом досуха и затем повторно растворяли в 1,2 л теплого гептана с последующей фильтрацией. Фильтрат концентрировали посредством удаления 500 мл гептана, что приводило к осаждению твердых веществ. Твердые вещества собирали посредством фильтрации и сушили с получением этил-4-йод-1-метил-1Н-пиразол-5-карбоксилата в виде белого твердого вещества (297,6 г; выход 89%).
1Н ЯМР (CDCl3) δ 7.57 (s, 1Н), 4.43 (q, 2Н), 4.21 (s, 3H), 1.47 (t, 3H).
УЭЖХ (УЭЖХ, способ 4): tR составляет 5, 10 мин.
МС (ЭР+) 280,9 (М+Н)+.
Получение 14: трет-бутил-(3R)-3-[(3-метил-1-оксидопиридин-2-ил)амино1-пиперидин-1-карбоксилат
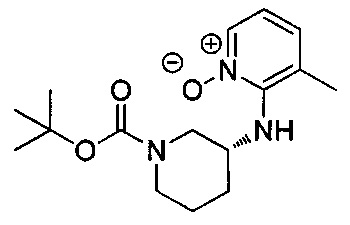
Стадия 1: 1-оксид 2-хлор-3-метилпиридина
В смесь 2-хлор-3-метилпиридина (15 г; 118 ммоль) и пероксида мочевины (44,07 г; 468,5 ммоль) в DCM (300 мл) по каплям добавляли трифторуксусный ангидрид (98,55 г; 469,2 ммоль) при 0°C. Смесь перемешивали при комнатной температуре в течение 20 ч. Смесь затем вливали в воду (150 мл) и слои разделяли. Органический слой промывали насыщенным водным Na2S2O3 (20 мл), сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле (DCM:MeOH; от 100:0 до 10:1) с получением указанного в заголовке соединения (14,5 г; 86%) в виде бесцветного твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 8.41 (d, 1Н), 7.39 (d, 1Н), 7.27 (m, 1Н), 2.50 (s, 3Н).
Стадия 2: 1-оксид (R)-2-(1-(трет-бутоксикарбонил)пиперидин-3-иламино)-3-метилпиридина
В раствор соединения со стадии 1 1-оксида 2-хлор-3-метилпиридина (14,5 г; 101 ммоль) и трет-бутил-(3R)-3-аминопиперидин-1-карбоксилата (24,27 г; 121,2 ммоль) в n-BuOH (120 мл) медленно добавляли диизопропилэтиламин (14,35 г; 111 ммоль) и DMAP (1,22 г; 9,99 ммоль) при 0°C. Полученную смесь нагревали при 100°C в течение 36 ч. Реакционную смесь охлаждали и добавляли воду. Смесь затем экстрагировали EtOAc (3×50 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Неочищенное соединение очищали посредством хроматографии на силикагеле, элюируя градиентом петролейный эфир: EtOAc (от 80:20 до 50:50), с получением указанного в заголовке соединения (7,9 г; 25%) в виде желтого масла.
1Н ЯМР (400 МГц, CDCl3) δ 8.05 (d, 1Н), 7.13 (br s, 1Н), 6.97 (d, 1Н), 6.59 (dd, 1Н), 4.09-3.93 (m, 1Н), 3.90-3.77 (m, 1Н), 3.74-3.63 (m, 1Н), 2.98-2.86 (m, 1Н), 2.85-2.75 (m, 1Н), 2.42 (s, 3H), 2.14-2.06 (m, 1Н), 1.83-1.74 (m, 1Н), 1.57-1.48 (m, 2Н), 1.45 (s, 9Н).
Получение 15: трет-бутил-(3R)-3[(2-оксидоизохинолин-1-ил)амино]пиперидин-1-карбоксилат
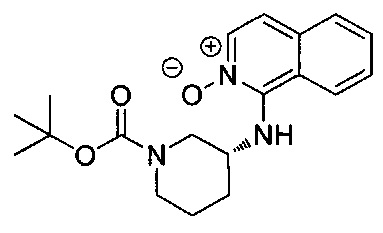
Стадия 1: 2-оксид 1-хлоризохинолина
В раствор 1-хлоризохинолина (3,0 г; 18 ммоль) в DCM (50 мл) добавляли m-СРВА (мета-хлорпербензойную кислоту) (9,5 г; 55 ммоль). Смесь перемешивали при комнатной температуре в течение 48 ч. Смесь разбавляли DCM и затем промывали рассолом, сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной флэш-хроматографии на силикагеле, элюируя градиентом петролейный эфир:EtOAc (от 5:1 до 0:1), с получением указанного в заголовке соединения (1,2 г; 36%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.38 (d, 1Н), 8.05 (d, 2Н), 7.98 (d, 1Н), 7.82 (dd, 1Н), 7.72 (dd, 1Н).
Стадия 2: трет-бутил-(3R)-3-[(2-оксидоизохинолин-1-ил)амино]-пиперидин-1-карбоксилат
В раствор соединения со стадии 2 2-оксида 1-хлоризохинолина (1,2 г; 6,68 ммоль) и трет-бутил-(3R)-3-аминопиперидин-1-карбоксилата (2,0 г; 10,02 ммоль) в n-BuOH (20 мл) добавляли DIPEA (0,95 г; 7,35 ммоль) и DMAP (81 мг; 0,668 ммоль). Смесь перемешивали при 120°C в течение 16 ч. Смесь затем вливали в воду (20 мл) и экстрагировали EtOAc (3×10 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и фильтрат концентрировали под вакуумом. Неочищенный продукт очищали посредством колоночной флэш-хроматографии на силикагеле, элюируя градиентом петролейный эфир: EtOAc (от 1:1 до 0:1), с получением указанного в заголовке соединения (0,8 г; 35%).
1Н ЯМР (400 МГц, DMSO-d6) δ 8.31 (d, 1Н), 8.02 (d, 1Н), 7.92-7.90 (m, 1Н), 7.76-7.70 (m, 2Н), 7.33 (d, 1Н), 4.39 (s, 1Н), 3.91-3.83 (m, 1Н), 3.50-3.37 (m, 3Н), 2.15-2.11 (m, 1Н), 1.84 (s, 2Н), 1.60 (s, 1Н), 1.47-1.32 (m, 9Н).
ЖХ (ЖХ-МС, способ 3), tR составляет 1,15 мин
МС (ЭР+) 344,3 (М+Н)+.
Получение 16: трет-бутил-(3R)-3-{изохинолин-1-ил[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоил]амино}пиперидин-1-карбоксилат
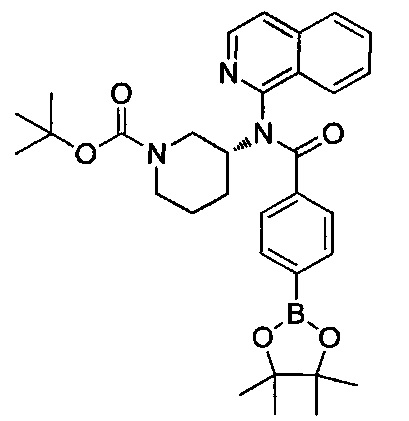
К суспензии соединения по Получению 9 трет-бутил-(3R)-3-[(4-бромбензоил)(изохинолин-1-ил)амино]-пиперидин-1-карбоксилата (3,7 г; 7,2 ммоль), бис(пинаколато)дибора (3,68 г; 14,5 ммоль) и КОАс (2,14 г; 21,8 ммоль) в 1,4-диоксане (25 мл) добавляли PdCl2(dppf) (0,53 г; 0,73 ммоль). Полученную смесь продували N2 и нагревали при 80-90°C в течение 4 ч. Реакционную смесь охлаждали и концентрировали при пониженном давлении. Неочищенное соединение очищали посредством флэш-хроматографии на силикагеле, элюируя градиентом петролейный эфир: EtOAc (от 50:1 до 1,5:1), с получением желтой смолы. Желтую смолу растирали с петролейным эфиром и фильтровали с получением указанного в заголовке соединения (3,8 г; 94%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, MeOH-d4, смесь ротамеров) δ 8.46 (br s, 0.75Н), 8.38 (br s, 0.25H), 8.08 (br s, 0.25H), 7.99 (br s, 0.75H), 7.83 (br s, 1H), 7.79-7.61 (m, 4H), 7.33 (br s, 2H), 7.21 (br s, 2H), 4.65-4.58 (m, 1H), 4.28-4.25 (m, 1H), 4.04-3.96 (m, 1H), 3.45-3.40 (m, 1H), 2.67-2.55 (m, 1H), 2.30-2.09 (m, 1H), 1.87-1.84 (m, 1H), 1.51 (s, 5.4H), 1.42 (s, 3.6H), 1.26 (s, 7.2H), 1.22 (s, 4.8H).
Получение 17: трет-бутил-(3R)-3-{(3-хлорпиридин-2-ил)[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоил]амино}пиперидин-1-карбоксилат
В раствор соединения по Получению 10 (R)-трет-бутил-3-(4-бром-N-(3-хлорпиридин-2-ил)бензамидо)пиперидин-1-карбоксилата (40,0 г; 80,8 ммоль) в 1,4-диоксане (250 мл) добавляли бис(пинаколато)дибор (41,1 г; 162 ммоль), КОАс (23,8 г; 244 ммоль) и PdCl2(dppf) (5,9 г; 8,1 ммоль). Полученную смесь продували N2 и перемешивали при 80-90°C в течение 10 ч. Реакционную смесь охлаждали и фильтровали. Органический раствор концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом 2-25% EtOAc/петролейный эфир, с получением указанного в заголовке соединения в виде желтой смолы. Эту желтую смолу растирали с петролейным эфиром с получением указанного в заголовке соединения в виде белого твердого вещества (30 г; 69%).
1Н ЯМР (MeOH-d4) δ 8.52 (br s, 1Н), 7.74 (br s, 1H), 7.55 (br s, 2H), 7.31 (br s, 3H), 4.53 (br s, 1H), 4.30 (br s, 1H), 4.05-4.02 (br m, 1H), 2.80-2.29 (br m, 2H), 1.95-1.68 (m, 3H), 1.50 (br s, 10 H), 1.32 (br s, 12H).
Получение 18: трет-бутил-3-[(3-хлорпиридин-2-ил){4-[5-(этоксикарбонил)-1-метил-1Н-пиразол-4-ил]бензоил}амино]пиперидин-1-карбоксилат
Соединение по Получению 13 этил-4-йод-1-метил-1Н-пиразол-5-карбоксилат (108,5 г; 387,4 ммоль) растворяли в 1 л THF и полученный раствор охлаждали до -52°C. Хлорид изопропилмагния (2 моль/л раствор в THF; 200 мл; 400 ммоль) добавляли в течение 27 минут, поддерживая внутреннюю температуру ниже -39°C, с последующим добавлением хлорида цинка (1,9 моль/л в 2-MeTHF; 120 мл; 230 ммоль) в течение 15 минут при поддержании внутренней температуры ниже -39°C. Реакционную смесь затем нагревали до 40°C с последующим добавлением соединения по Получению 10 трет-бутил-(3R)-3-[(4-бромбензоил)(3-хлорпиридин-2-ил)амино]пиперидин-1-карбоксилата (158,27 г; 319,9 ммоль) и дихлорида 1,1'-бис(ди-трет-бутилфосфино)-ферроценпалладия (4,60 г; 6,92 ммоль). Реакционную смесь затем нагревали до 55°C и выдерживали в течение 2 ч. После охлаждения до комнатной температуры неочищенную реакционную смесь фильтровали через Celite® и фильтрат концентрировали с получением пены. Это вещество повторно растворяли в 1 л 2-MeTHF и промывали 400 мл воды с последующей фильтрацией двухфазной смеси и разделением фаз. 2-MeTHF удаляли под вакуумом и заменяли на EtOH, затем раствор концентрировали до конечного объема 1,5 л, что приводило к осаждению твердых веществ, которые собирали посредством фильтрации. После сушки указанное в заголовке соединение выделили в виде рыжевато-коричневого твердого вещества (141,5 г; выход 78%).
1Н ЯМР (ацетонитрил-d3) δ 8.56-8.44 (m, 1Н), 7.81-7.64 (m, 1Н), 7.48 (s, 1Н), 7.41-7.31 (m, 2Н), 7.30 (dd, 1Н), 7.29-7.18 (m, 2Н), 4.68-4.23 (m, 2Н), 4.18 (q, 2Н), 4.10 (s, 3H), 4.06-3.90 (m, 1Н), 3.40-3.08 (br s, 0.5Н), 2.69-2.45 (m, 1Н), 2.3-2.08 (br s, 0.5Н), 1.93-1.51 (m, 3Н), 1.47 (s, 9Н), 1.38-1.21 (br s, 1Н), 1.08 (t, 3H).
УЭЖХ (УЭЖХ, способ 4): tR составляет 6,73 мин.
МС (ЭР+) 468,1 (М+Н)+.
Получение 19: 4-(пиразоло[1,5-а]пиримидин-3-ил)бензойная кислота
Стадия 1: этил-4-(пиразоло[1,5-а]пиримидин-3-ил)бензоат
Воду (160 мл) добавляли по каплям при комнатной температуре в смесь 4-этоксикарбонилфенилбороновой кислоты (50 г; 0,26 моль), 3-бромпиразоло[1,5-а]пиримидина (56,25 г; 0,28 моль), Pd(ddpf)Cl2⋅CH2Cl2 (4,25 г; 5,21 ммоль) и CS2CO3 (169,42 г; 0,52 моль) в 1,4-диоксане (1 л). Реакционную смесь затем нагревали при 85°C в течение 4 ч. Реакционную смесь охлаждали, вливали в воду и экстрагировали EtOAc (2×300 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали посредством хроматографии на силикагеле, элюируя градиентом петролейный эфир: EtOAc (от 100:10 до 3:1) с получением указанного в заголовке соединения (64 г; 93%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 8.73 (d, 1Н), 8.63 (d, 1Н), 8.52 (s, 1Н), 8.20-8.09 (m, 4Н), 6.92 (dd, 1Н), 4.41 (q, 2Н), 1.43 (t, 3H).
Стадия 2: 4-(пиразоло[1,5-а]пиримидин-3-ил)бензойная кислота
В смесь соединения со стадии 1 этил-4-(пиразоло[1,5-а]пиримидин-3-ил)бензоата (64 г; 0,24 моль) в МеОН (1 л) добавляли водный раствор гидроксида натрия (600 мл; 1,2 моль; 2 М). Смесь нагревали при 50°C в течение 3 ч. Смесь затем концентрировали для удаления летучих веществ. Полученную суспензию разбавляли водой и по каплям добавляли водный HCl (1 н.) до достижения pH 4. Подкисленную суспензию фильтровали и твердые вещества собирали и сушили с получением указанного в заголовке соединения (57 г; 99%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) δ 12.83 (br, 1Н), 9.21 (d, 1Н), 8.88 (s, 1Н), 8.73 (d, 1Н), 8.30 (d, 2Н), 8.00 (d, 2Н), 7.18-7.16 (m, 1Н).
ЖХ (ЖХ-МС, способ 3) tR составляет 0,78 мин.
МС (ЭР+) 238 (М+Н)+.
Получение 20: 4-йод-1-метил-1Н-пиразол-5-карбоксамид
В круглодонную колбу загружали 4-йод-1-метил-1Н-пиразол-5-карбоновую кислоту (297 г; 1,18 моль), DCM (2,97 л) и 1,1'-карбонилдиимидазол (CDI) (207 г; 97% (мае); 1,24 моль). Реакционную смесь перемешивали при комнатной температуре в течение 45 мин. Добавляли хлорид аммония (189 г; 3,53 моль) и триэтиламин (498 мл; 3,53 моль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали под вакуумом и остаток суспендировали в H2O (примерно 3 л) и гранулировали при комнатной температуре в течение 1 ч. Твердое вещество собирали посредством фильтрации, промывали Н2O и сушили в вакуумной печи с получением 4-йод-1-метил-1Н-пиразол-5-карбоксамида в виде бесцветного твердого вещества (222 г; выход 75%).
1Н ЯМР (CDCl3) δ: 7.53 (s, 1Н), 6.56 (br s, 1Н), 6.01 (br s, 1Н), 4.21 (s, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,15 мин.
МС (ЭР+): 251,1 (М+Н)+.
Получение 21: 4-йод-1-метил-1Н-пиразол-5-карбонитрил
В круглодонную колбу загружали соединение по Получению 20 4-йод-1-метил-1Н-пиразол-5-карбоксамид (222 г; 886 ммоль) и DCM (2,22 л) и реакционную смесь охлаждали до 0°C. Добавляли 2,6-лутидин (310 мл; 2,66 моль) и трифторуксусный ангидрид (253 мл; 1,77 моль). После завершения реакции добавляли насыщенный водный бикарбонат натрия (800 мл) и слои разделяли. Водный слой промывали DCM (800 мл). Органические слои объединяли и промывали насыщенным водным хлоридом аммония (800 мл), 1 н. HCl (800 мл) и рассолом (800 мл). Органический слой сушили над сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток суспендировали в гептанах (примерно 2 л) и гранулировали при 0-5°C в течение 30 мин. Твердое вещество собирали посредством фильтрации и сушили в вакуумной печи с получением 4-йод-1-метил-1Н-пиразол-5-карбонитрила в виде бесцветного твердого вещества (196 г; выход 95%).
1Н ЯМР (CDCl3) δ: 7.60 (s, 1Н), 4.09 (s, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,70 мин.
МС (ЭР+): 233,8 (М+Н)+.
Получение 22: 5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол
Осторожно: В этой реакции образуется азотистоводородная кислота, что требует соблюдения соответствующей техники безопасности.
В реакционный сосуд загружали DMF (1,225 л), соединение по Получению 21 4-йод-1-метил-1Н-пиразол-5-карбонитрил (175 г; 751 ммоль), азид натрия (147 г; 2,25 моль) и хлорид аммония (121 г; 2.25 моль). Медленно добавляли H2O (525 мл), чтобы минимизировать экзотермический эффект. Реакционную смесь нагревали при 100°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры и вливали в смесь H2O (2 л) и льда (1 кг). Добавляли водный раствор NaNO2 (600 мл; 120 г NaNO2; 20% (мас.)) с последующим медленным добавлением водной H2SO4 до доведения pH реакционной смеси до значения 1. Осадок собирали посредством фильтрации, промывали H2O и сушили под вакуумом с получением 5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразола в виде бесцветного твердого вещества (187 г; 90%).
Альтернативный способ получения 22:
В раствор соединения по Получению 21 4-йод-1-метил-1Н-пиразол-5-карбонитрила (500 мг; 2,15 ммоль) в 2-метилтетрагидрофуране (4 мл) добавляли P2S5 (24 мг; 0,11 ммоль) с последующим добавлением гидразина моногидрата (523 мкл; 10,7 ммоль). Реакционную смесь нагревали в герметичном сосуде при 70°C в течение 17 ч. Реакционную смесь медленно добавляли в гептан при интенсивном перемешивании до образования маслянистого осадка. Маточный раствор декантировали и остаток растирали с гептаном и сушили под вакуумом с получением светло-желтого твердого вещества (520 мг). Остаток растворяли в EtOH (5 мл). Добавляли HCl (2,0 мл; 3,0 М водный раствор) с последующим добавлением по каплям NaNO2 (405 мг; 5,88 ммоль), растворенного в Н2O (1,5 мл), чтобы контролировать экзотермический эффект и выделение газа. Реакционную смесь концентрировали под вакуумом до объема примерно 3 мл. Добавляли H2O (20 мл) и DCM (15 мл) с последующим добавлением насыщенного водного NaHCO3 (5 мл) для получения pH раствора больше 7. Реакционную смесь распределяли между слоями и органический слой отбрасывали. Водный слой подкисляли до pH 1 с помощью 6 М HCl. Реакционную смесь экстрагировали EtOAc (2×40 мл). Объединенные органические слои сушили MgSO4 и концентрировали под вакуумом с получением 5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразола в виде желтоватого твердого вещества (390 мг; 66%).
1Н ЯМР (МеОН-d4) δ: 7.69 (s, 1Н), 4.08 (s, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,52 мин.
МС (ЭР+): 276,9 (М+Н)+.
Получение 23: этил-1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил)этилкарбонат
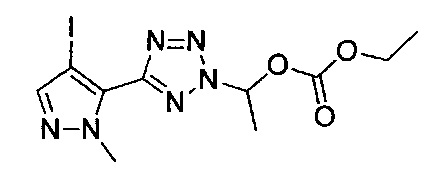
В круглодонную колбу загружали соединение по Получению 22 5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол (191 г; 692 ммоль), 4-диметиламинопиридин (4,27 г; 34,6 ммоль), THF (1,72 л), ацетальдегид (43 мл; 760 ммоль) и триэтиламин (107 мл; 762 ммоль). Реакционный раствор перемешивали и затем добавляли этилхлорформиат (86,2 мл; 97% (мас.); 692 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли EtOAc (965 мл) и H2O (965 мл). Слои разделяли. Водный слой экстрагировали EtOAc (965 мл). Объединенные органические слои сушили над сульфатом магния и концентрировали под вакуумом с получением этил-1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]этилкарбоната в виде бесцветного масла (261 г; 96% выход).
Получение 23а и 23b:
23а: (S)-этил-1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]этилкарбонат
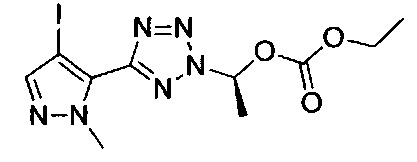
23b: (R)-этил-1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]этилкарбонат
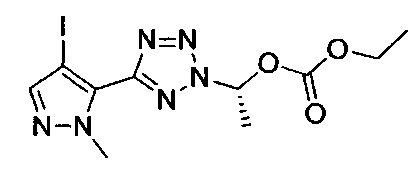
407,5 г соединения по Получению 23 этил-1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]этилкарбоната подвергали обработке с помощью хиральной препаративной хроматографии по способу 3 с последующим концентрированием каждого энантиомера досуха под вакуумом с получением изомера 23а (177,4 г; 99,22%; 99,79% е.е. (энантиомерный избыток); tR составляет 2,12 мин) и изомера 23b (177,74 г; 98,83%; 98,46% е.е; tR составляет 2,59 мин).
1Н ЯМР (MeOH-d4) δ: 7.63 (s, 1Н), 7.28 (q, 1Н), 4.32-4.24 (m, 2Н), 4.23 (s, 3H), 2.10 (d, 3), 1.33 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,87 мин.
МС (ЭР+): 393,0 (М+Н)+.
На Фиг. 8 представлена диаграмма ORTEP (S)-этил-1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]этилкарбоната (23а).
Рентгеноструктурный анализ монокристаллов (S)-этил-1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]этилкарбоната (23а): Сбор данных проводили на дифрактометре Bruker APEX при комнатной температуре. Сбор данных состоял в омега- и фи-сканировании. Структуру устанавливали посредством прямых методов с использованием пакета программ SHELX в пространственной группе P21. Структуру затем уточняли с помощью метода наименьших квадратов в полноматричном приближении. Все неводородные атомы были обнаружены и уточнены с использованием параметров анизотропного замещения.
Все водородные атомы помещали в вычисленные положения и вращали относительно их несущих атомов. Конечное уточнение включало параметры изотропного замещения для всех атомов водорода. Абсолютную конфигурацию определяли путем проверки параметра Флэка. В этом случае параметр равен 0,0396 со стандартным отклонением (ESD) 0,003. Эти значения находятся в пределах определения абсолютной конфигурации.
Конечный R-фактор составлял 3,5%. Конечная разностная карта Фурье не выявила пропусков или смещения электронной плотности.
Кристаллографические данные, условия сбора данных и параметры уточнения сведены в таблицу 2.
Получение 24: трет-бутил-(3R)-3-{(3-мeтилпиpидин-2-ил)[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоил]амино}пиперидин-1-карбоксилат
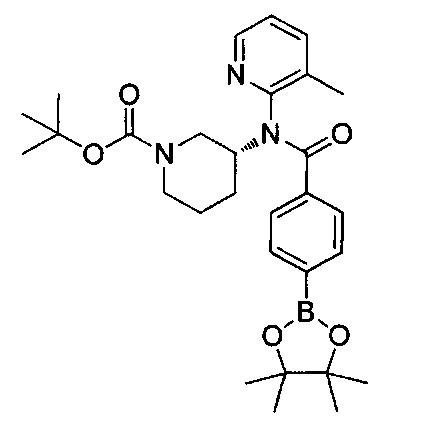
В круглодонную колбу загружали соединение по Получению 12 трет-бутил-(3R)-3-[(4-бромбензоил)(3-метилпиридин-2-ил)амино]пиперидин-1-карбоксилат (150 г; 317 ммоль), бис(пинаколато)дибор (97,8 г; 381 ммоль), ацетат калия (100 г; 1,01 моль) и 2-метилтетрагидрофуран (750 мл). Реакционную смесь нагревали до 75°C. Добавляли комплекс дихлорида 1,1'-бис(дифенилфосфино)ферроцен-палладия (II) с дихлорметаном (Pd(dppf)Cl2⋅CH2Cl2) (5,12 г; 6,21 ммоль) и реакционную смесь нагревали с обратным холодильником в течение 19 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли H2O. Реакционную смесь пропускали через набивку из целита и слои разделяли. Органический слой концентрировали под вакуумом. Коричневый остаток очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом 30-50% EtOAc в гептане. Содержащие продукт фракции концентрировали под вакуумом. Остаток фильтровали через набивку из целита с использованием теплого гептана и DCM для повышения растворимости продукта. Реакционную смесь концентрировали под вакуумом, пока продукт не начинал кристаллизоваться. Твердые вещества гранулировали в течение 16 ч при комнатной температуре, собирали посредством фильтрации и сушили в вакуумной печи с получением трет-бутил-(3R)-3-{(3-метилпиридин-2-ил)[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоил]амино}пиперидин-1-карбоксилата в виде светло-розового твердого вещества (142 г; 86%).
1Н ЯМР (CDCl3) δ: 8.40 (m, 1Н), 7.53-7.27 (m, 5Н), 7.14-6.92 (m, 1Н), 4.75-4.45 (m, 2Н), 4.20-3.90 (m, 1Н), 3.63-3.21 (m, 1Н), 2.84-2.10 (m, 3Н), 2.06-1.88 (m, 3Н), 1.81-1.56 (m, 2Н), 1.53-1.37 (m, 9Н), 1.31 (s, 12Н).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 1,08 мин.
МС (ЭР+): 522,4 (М+Н)+.
Получение 25: трет-бутил-(3R)-3-(4-(1-метил-5-(2H-тетразол-5-ил)-1Н-пиразол-4-ил)-N-(3-метилпиридин-2-ил)бензамидо)пиперидин-1-карбоксилат
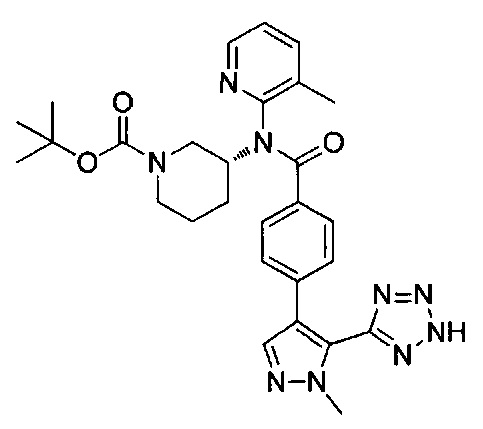
В круглодонную колбу с помещенной в нее магнитной мешалкой добавляли соединение по Получению 24 трет-бутил-(3R)-3-{(3-метилпиридин-2-ил)[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоил]амино}-пиперидин-1-карбоксилат (1,53 г; 2,93 ммоль) и соединение по Получению 22 5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол (674 мг; 2,44 ммоль). В эту же колбу добавляли диоксан (12,2 мл) и водный NaOH (4,07 мл; 3 М раствор; 12,2 ммоль). Реакционную смесь нагревали до 70°C в атмосфере азота в течение 30 мин. В пробирку для микроволновой печи добавляли Pd(OAc)2 (34,3 мг; 0,153 ммоль) и фосфиновый лиганд Catacxium A (123 мг; 0,342 ммоль). Воздушную атмосферу заменяли на азот и добавляли толуол. Катализатор перемешивали при комнатной температуре в течение 15 мин до образования ярко-желтой суспензии. На этом этапе в реакционную смесь добавляли катализатор и температуру доводили до 100°C. Реакционную смесь перемешивали в течение 3 ч при 100°C. После полного исчерпания исходного вещества, реакционную смесь концентрировали при пониженном давлении. Добавляли H2O (10 мл). Реакционную смесь экстрагировали МТВЕ (3×25 мл). Водную фазу охлаждали до 0°C и по каплям добавляли 1М HCl для образования осадка, который собирали посредством фильтрования с отсасыванием и сушили под вакуумом с получением трет-бутил-(3R)-3-(4-(1-метил-5-(2H-тетразол-5-ил)-1Н-пиразол-4-ил)-N-(3-метилпиридин-2-ил)бензамидо)пиперидин-1-карбоксилата (1,2 г; 90%).
1Н ЯМР (MeOH-d4) δ: 8.42 (d, 1Н), 7.80 (s, 1Н), 7.72-7.51 (m, 1Н), 7.30-7.05 (m, 5Н), 4.62-4.44 (m, 2Н), 4.18-3.98 (m, 2Н), 3.95 (s, 3H), 2.75-2.62 (m, 1Н), 2.18-2.04 (m, 2Н), 1.79-1.70 (m, 2Н), 1.53-1.42 (m, 4Н), 1.31 (s, 9Н).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,82 мин.
МС (ЭР+): 544,4 (М+Н)+.
Получение 26: трет-бутил-(3R)-3-{[4-(5-циано-1-метил-1Н-пиразол-4-ил)-бензоил](3-метилпиридин-2-ил)амино}пиперидин-1-карбоксилат
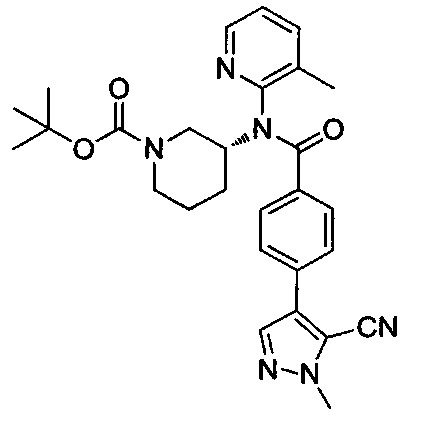
Соединение по Получению 24 трет-бутил-(3R)-3-{(3-метилпиридин-2-ил)[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоил]амино}пиперидин-1-карбоксилат (2,67 г; 5,12 ммоль), соединение по Получению 21 4-йод-1-метил-1Н-пиразол-5-карбонитрил (1,19 г; 5,12 ммоль), Pd2(dba)3 (234 мг; 0,256 ммоль) и XPhos (257 мг; 0,512 ммоль) растворяли в диоксане (27 мл) в атмосфере азота. Добавляли раствор Na2CO3 (1,63 г; 15,4 ммоль) в H2O (3 мл). Реакционную смесь нагревали при 80°C в течение 6 ч, затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли EtOAc (150 мл), промывали 50% рассолом (100 мл). Отделенную органическую фазу сушили над MgSO4 и концентрировали. Остаток очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом 15-100% EtOAc/гептан с получением трет-бутил-(3R)-3-{[4-(5-циано-1-метил-1Н-пиразол-4-ил)бензоил](3-метилпиридин-2-ил)амино}пиперидин-1-карбоксилата в виде бесцветного твердого вещества (1,87 г; выход 73%).
1Н ЯМР (CDCl3) δ: 8.43 (m, 1Н), 7.72 (s, 1Н), 7.40-7.35 (m, 5Н), 7.21-7.02 (m, 1Н), 4.87-4.32 (m, 2Н), 4.07 (s, 3H), 3.49 (d, 1Н), 2.82-2.21 (m, 2Н), 2.05 (s, 3H), 1.80-1.48 (m, 4Н), 1.47 (d, 9Н).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 1,01 мин.
МС (ЭР+): 501,4 (М+Н)+.
Получение 27: трет-бутил-(3R)-3-[{4-[5-(2-{(1S)-1-[(этоксикарбонил)окси]этил}-2Н-тетразол-5-ил)-1-метил-1Н-пиразол-4-ил]бензоил}(3-метилпиридин-2-ил)амино]пиперидин-1-карбоксилат
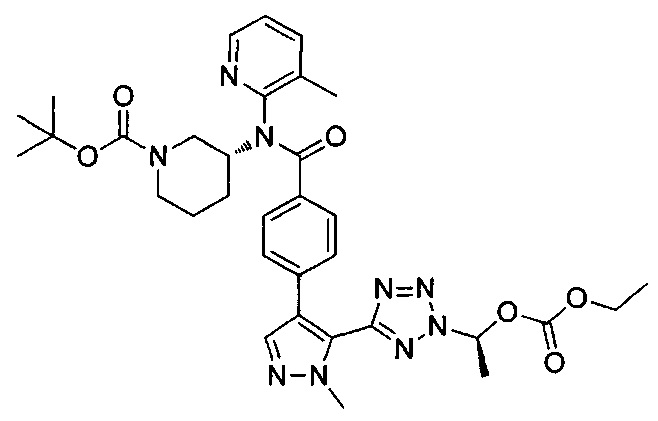
В трехгорлую круглодонную колбу, оснащенную дефлегматором, загружали диоксан (8,5 мл) и водный CsF (7,65 мл; 1 М раствор; 7,65 ммоль). Содержимое нагревали до 80°C в течение 0,5 ч в атмосфере азота. Через 30 мин добавляли соединение по Получению 24 трет-бутил-(3R)-3-{(3-метилпиридин-2-ил)[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоил]-амино}пиперидин-1-карбоксилат (1,66 г; 3,19 ммоль) с последующим добавлением соединения по Получению 23а (S)-этил-1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]этилкарбоната (1,00 г; 2,55 ммоль) и катализатора Pd(amphos)Cl2 (90,6 мг; 0,128 ммоль). Реакционную смесь перемешивали при 80°C в течение 5,5 ч. Добавляли водный раствор хлорида аммония (10 мл) с последующим добавлением EtOAc (10 мл). Слои разделяли и водную фазу дополнительно экстрагировали EtOAc (3×15 мл), сушили MgSO4, пропускали через набивку из диоксида кремния и концентрировали. Неочищенное вещество очищали посредством ЖХСД с использованием градиента 20-70% EtOAc/гептан с получением трет-бутил-(3R)-3-[{4-[5-(2-{(1S)-1-[(этоксикарбонил)окси]этил}-2Н-тетразол-5-ил)-1-метил-1Н-пиразол-4-ил]-бензоил}(3-метилпиридин-2-ил)амино]пиперидин-1-карбоксилата в виде бесцветного твердого вещества (1,05 г; 77%).
1Н ЯМР (CDCl3) δ: 8.41 (d, 1Н), 7.61 (s, 1Н), 7.36-7.32 (m, 1Н), 7.24-7.10 (m, 6Н), 4.76-4.46 (m, 2Н), 4.32 (q, 2Н), 4.07 (s, 3H), 3.47-3.36 (m, 1Н), 2.67-2.14 (m, 3H), 2.06-1.99 (m, 6Н), 1.81-1.59 (m, 3H), 1.47 (s, 9Н), 1.36 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,95 мин.
МС (ЭР+): 660,4 (М+Н)+.
Получение 28: трет-бутил-(3R)-3-[{4-[5-(2-{(1R)-1-[(этоксикарбонил)окси]этил}-2Н-тетразол-5-ил)-1-метил-1Н-пиразол-4-ил]бензоил}(3-метилпиридин-2-ил)амино]пиперидин-1-карбоксилат
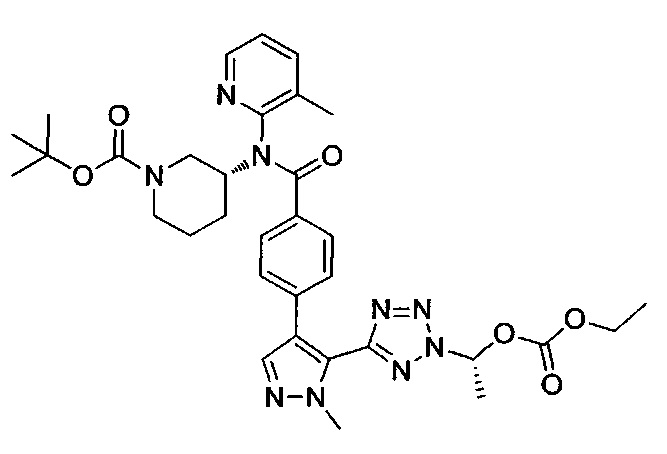
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 23b.
1Н ЯМР (CDCl3) δ: 8.42 (d, 1Н), 7.63 (s, 1Н), 7.42-7.35 (m, 1Н), 7.28-7.03 (m, 6Н), 4.79-4.20 (m, 2Н), 4.30 (q, 2Н), 4.09 (s, 3H), 3.55-3.33 (m, 1Н), 2.73-2.17 (m, 3H), 2.06-2.01 (m, 6Н), 1.84-1.71 (m, 3H), 1.47 (br s, 9Н), 1.36 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,98 мин.
МС (ЭР+): 660,4 (М+Н)+.
Получение 29: 4-(4-{[(3R)-1-(трет-бутоксикарбонил)пиперидин-3-ил](3-метилпиридин-2-ил)карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоновая кислота
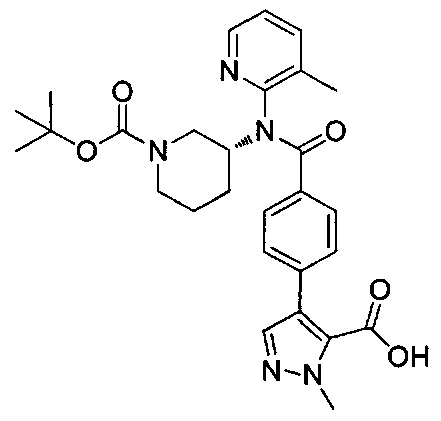
Суспензию продукта стадии 1 ПРИМЕРА 13 трет-бутил-(3R)-3-[{4-[5-(метоксикарбонил)-1-метил-1Н-пиразол-4-ил]бензоил}(3-метилпиридин-2-ил)-амино]пиперидин-1-карбоксилата (0,85 г; 0,59 ммоль) в МеОН (2 мл) и раствор гидроксида калия в МеОН (1,67 мл; 1 М раствор; 1,67 ммоль) нагревали до 70°C в герметично закрытой пробирке для проведения реакций под давлением в течение 2 ч. Реакционную смесь затем концентрировали и в смесь добавляли толуол (10 мл). Растворители выпаривали под вакуумом и сушили под вакуумом в течение 2 ч. Твердое вещество затем суспендировали в метил-трет-бутиловом эфире (15 мл) и гептанах (15 мл) в течение 1 ч, собирали посредством фильтрации и сушили под вакуумом в течение 16 ч с получением 4-(4-{[(3R)-1-(трет-бутоксикарбонил)пиперидин-3-ил](3-метилпиридин-2-ил)-карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоновой кислоты (800 мг; 90%).
1Н ЯМР (DMSO-d6) δ: 8.43 (m, 1Н), 7.76-7.40 (m, 3Н), 7.34-7.21 (m, 2Н), 7.20-6.96 (m, 2Н), 4.76-4.37 (m, 1Н), 4.27 (br s, 1Н), 3.86 (br s, 1Н), 3.73 (s, 3H), 2.23-1.85 (m, 4Н), 1.80-1.52 (m, 2Н), 1.54-1.30 (m, 12Н).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,92 мин.
МС (ЭР+): 520,3 (М+Н)+.
Получение 30: этил-1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]-пропилкарбонат
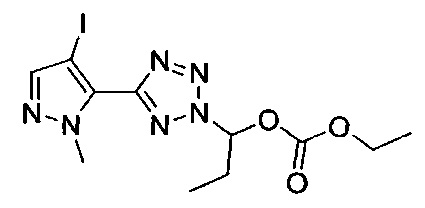
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 23, с использованием соединения по Получению 22, пропиональдегида и этилхлорформиата.
1Н ЯМР (CDCl3) δ: 7.63 (s, 1Н), 7.07 (t, 1Н), 4.33-4.20 (m, 2Н), 4.23 (s, 3H), 2.55-2.40 (m, 2Н), 1.33 (t, 3H), 1.04 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,71 мин.
МС (ЭР+): 407,1 (М+Н)+.
Получение 30а и 30b:
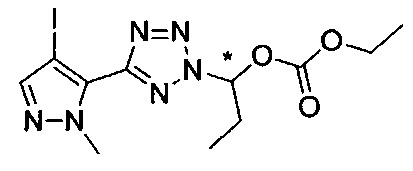
630 мг соединения по Получению 30 подвергали обработке с помощью хиральной препаративной хроматографии по способу 4 с последующим концентрированием каждого энантиомера досуха под вакуумом с получением соединения по Получению 30а (190 мг; 99% е.е., tR составляет 4,22 мин (хиральная аналитическая хроматография, способ 1)) и соединения по Получению 30b (200 мг; 99% е.е., tR составляет 5,01 мин (хиральная аналитическая хроматография, способ 1)). Абсолютную конфигурацию энантиомеров 30а и 30b не определяли. Первый энантиомер, который элюируется из колонки представляет собой соединение по Получению 30а, и второй энантиомер представляет собой соединение по Получению 30b.
Получение 31:
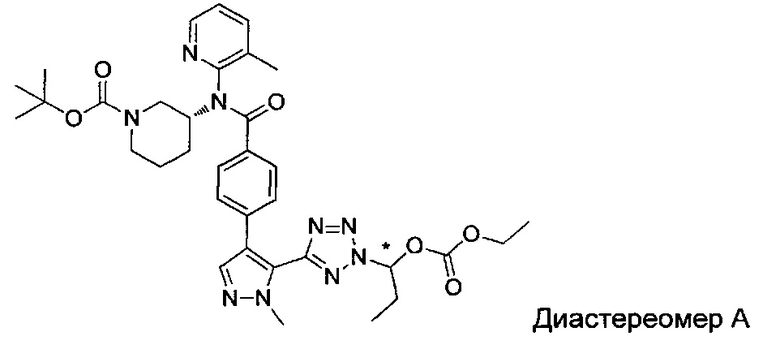
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 30а. Звездочка означает хиральный атом углерода, и поскольку абсолютная конфигурация (например, R/S) диастереомера A не была определена, это соединение по Получению 31 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 30а, из которого соединение по Получению 31 было получено.
1Н ЯМР (CDCl3) δ: 8.56-8.34 (m, 1Н), 7.61 (s, 1Н), 7.41-7.31 (m, 1Н), 7.24-7.20 (m, 2Н), 7.18-7.09 (m, 3H), 6.98 (t, 1Н), 4.80-4.42 (m, 2Н), 4.35-4.19 (m, 2Н), 4.07 (s, 3H), 3.54-3.26 (m, 1Н), 2.73-2.13 (m, 4Н), 2.44-2.28 (m, 3Н), 1.83-1.55 (m, 4Н), 1.51-1.40 (m, 9Н), 1.34 (t, 3H), 0.97 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 2,09 мин.
МС (ЭР+): 674,4 (М+Н)+.
Получение 32:
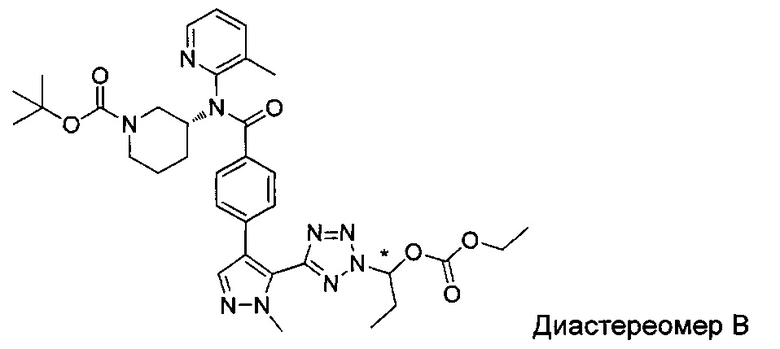
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 30b. Звездочка означает хиральный атом углерода, и поскольку абсолютная конфигурация (например, R/S) диастереомера B не была определена, это соединение по Получению 32 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 30b, из которого соединение по Получению 32 было получено.
1Н ЯМР (CDCl3) δ: 8.40 (d, 1Н), 7.61 (s, 1Н), 7.38-7.33 (m, 1Н), 7.26-7.07 (m, 5Н), 6.98 (t, 1Н), 4.75-4.62 (m, 1Н), 4.54-4.45 (m, 1Н), 4.31-4.22 (m, 2Н), 4.06 (s, 3H), 3.46-3.37 (m, 1Н), 2.68-2.13 (m, 4Н), 2.06-1.95 (m, 3Н), 1.79-1.56 (m, 4Н), 1.41-1.52 (m, 6Н), 1.33 (t, 3H), 0.97 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 1,05 мин.
МС (ЭР+): 674,4 (М+Н)+.
Получение 33а и 33b:
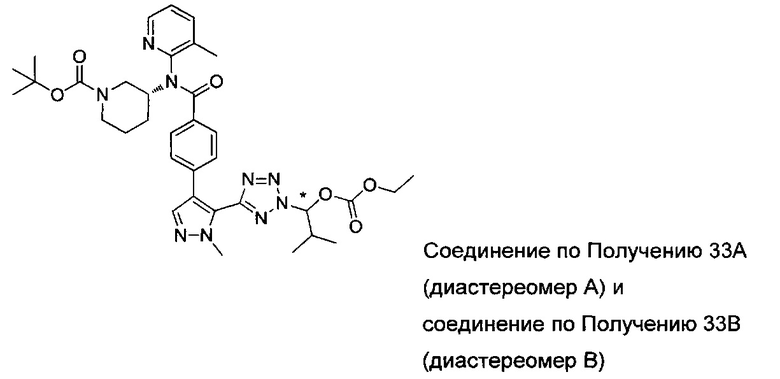
В раствор соединения по Получению 25 (500 мг; 0,920 ммоль) и DABCO (5,6 мг; 0,050 ммоль) в THF (5 мл) добавляли пиперидин (10 мкл; 0,10 ммоль), триэтиламин (0,303 мл; 2,17 ммоль), изобутиральдегид (0,204 мл; 2,17 ммоль) и этилхлорформиат (0,209 мл; 2,12 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли H2O (30 мл) и экстрагировали EtOAc (2×50 мл). Объединенные органические фазы сушили над MgSO4 и концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом 10-60% EtOAc в гептане, с получением указанного в заголовке соединения в виде бесцветного твердого вещества (364 мг; 58%).
364 мг твердого вещества подвергали обработке с помощью хиральной препаративной хроматографии, способ 5, с последующим концентрированием каждого диастереомера досуха под вакуумом с получением соединения по Получению 33а (139 мг; 99% е.е.; tR составляет 6,19 мин (хиральная аналитическая хроматография, способ 2)) и соединения по Получению 33b (143 мг; 94% е.е.; tR составляет 6,48 мин (хиральная аналитическая хроматография, способ 2)). Звездочка означает хиральный атом углерода и, поскольку абсолютная конфигурация (например, R/S) соединений по Получению 33а и 33b не была определена, каждый представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 33а и соединения по Получению 33b. Первый диастереомер, который элюируется из колонки, представляет собой соединение по Получению 33а, и второй диастереомер представляет собой соединение по Получению 33b.
Диастереомер 33а:
1Н ЯМР (CDCl3) δ: 8.41 (br s, 1Н), 7.62 (s, 1Н), 7.38-7.31 (m, 1Н), 7.24-7.19 (m, 2Н), 7.04-7.18 (m, 3H), 6.74 (d, 1Н), 4.83-4.43 (m, 2Н), 4.35-4.18 (m, 2Н), 4.06 (s, 3H), 3.56-3.26 (m, 1Н), 2.74-2.11 (m, 3Н), 2.04-1.92 (m, 3Н), 1.79-1.55 (m, 4Н), 1.52-1.40 (m, 9Н), 1.33 (t, 3H), 1.15 (d, 3H), 0.83 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 2,17 мин.
МС (ЭР+): 688,4 (М+Н)+.
Диастереомер 33b:
1Н ЯМР (CDCl3) δ: 8.41 (br s, 1Н), 7.62 (s, 1Н), 7.38-7.31 (m, 1Н), 7.24-7.19 (m, 2Н), 7.04-7.18 (m, 3Н), 6.74 (d, 1Н), 4.83-4.43 (m, 2Н), 4.35-4.18 (m, 2Н), 4.06 (s, 3H), 3.56-3.26 (m, 1Н), 2.74-2.11(m, 3Н), 2.04-1.92 (m, 3Н), 1.79-1.55 (m, 4Н), 1.52-1.40 (m, 9Н), 1.33 (t, 3H), 1.15 (d, 3H), 0.83 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 2,16 мин.
МС (ЭР+): 688,4 (М+Н)+.
Получение 34: этил-1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]-2,2-диметилпропилкарбонат
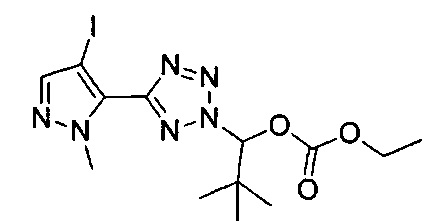
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 23, с использованием соединения по Получению 22, триметилацетальдегида и этилхлорформиата.
1Н ЯМР (CDCl3) δ: 7.66 (s, 1Н), 6.87 (s, 1Н), 4.16-4.36 (m, 5Н), 1.34 (t, 3H), 1.18 (9Н).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,93 мин.
МС (ЭР+): 435,1 (М+Н)+.
Получение 34а и 34b:
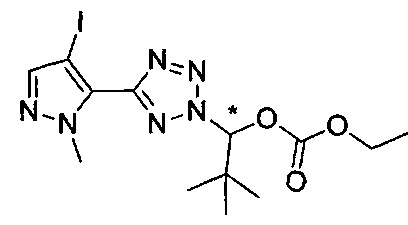
821 мг соединения по Получению 34 подвергали обработке с помощью хиральной препаративной хроматографии по способу 6 с последующим концентрированием каждого энантиомера досуха под вакуумом с получением соединения по Получению 34а (240 мг; 97% е.е.; tR составляет 3,37 мин (хиральная аналитическая хроматография, способ 3)) и соединения по Получению 34b (268 мг; 96% е.е.; tR составляет 3,77 мин (хиральная аналитическая хроматография, способ 3)). Абсолютная конфигурация энантиомеров 34а и 34b не была определена. Первый энантиомер, который элюируется из колонки, представляет собой соединение по Получению 34а, и второй энантиомер представляет собой соединение по Получению 34b.
Получение 35:
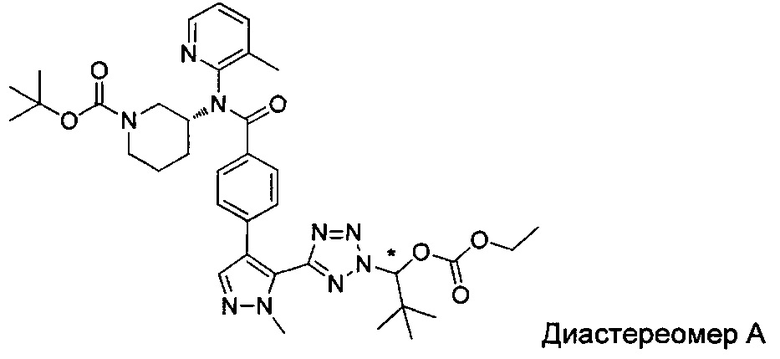
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и Получению 34а. Звездочка означает хиральный атом углерода и поскольку абсолютная конфигурация (например, R/S) диастереомера A не была определена, это соединение по Получению 35 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 34а, из которого соединение по Получению 35 было получено.
1Н ЯМР (CDCl3) δ: 8.45 (d, 1Н), 7.63 (s, 1Н), 7.42-7.33 (m, 1Н), 7.27-7.21 (m, 2Н), 7.20-7.08 (m, 3Н), 6.76 (s, 1Н), 4.58-4.43 (m, 1Н), 4.30-4.18 (m, 2Н), 4.05 (s, 3H), 3.50-3.35 (m, 1Н), 2.71-2.14 (m, 4Н), 2.02 (s, 3H), 1.86-1.65 (m, 3Н), 1.49 (br s, 9Н), 1.33 (m, 3Н), 1.08 (s, 9Н).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 2,23 мин.
МС (ЭР+): 702,5 (М+Н)+.
Получение 36:
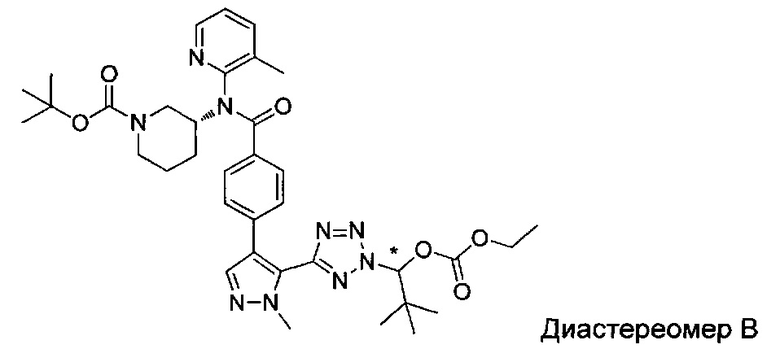
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 34b. Звездочка означает хиральный атом углерода и, поскольку абсолютная конфигурация (например, R/S) диастереомера B не была определена, это соединение по Получению 36 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 34b, из которого соединение по Получению 36 было получено.
1Н ЯМР (CDCl3) δ: 8.40 (br s, 1Н), 7.60 (br s, 1Н), 7.38-7.29 (m, 1Н), 7.24-7.17 (m, 2Н), 7.15-7.08 (m, 3H), 6.73 (s, 1Н), 4.51-4.42 (m, 1Н), 4.22 (q, 2Н), 4.04 (s, 3H), 3.41 (br s, 1Н), 2.67-2.32 (m, 2Н), 2.27-2.11 (m, 1Н), 2.02-1.86 (m, 3H), 1.56 (s, 9Н), 1.53-1.39 (m, 4Н), 1.31 (t, 3H), 1.05 (s, 9Н).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 2,23 мин.
МС (ЭР+): 702,5 (М+Н)+.
Получение 37: изопропил-1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]этилкарбонат
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 23, с использованием соединения по Получению 22, ацетальдегида и изопропилхлорформиата.
1Н ЯМР (CDCl3) δ: 7.64 (s, 1Н), 7.27 (q, 1Н), 4.94 (септет, 1Н), 4.23 (s, 3H), 2.10 (d, 3H), 1.34 (d, 3H), 1.30 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,70 мин.
МС (ЭР+): 407,1 (М+Н)+.
Получение 37а и 37b:
731 мг соединения по Получению 37 подвергали обработке с помощью хиральной препаративной хроматографии, способ 7, с последующим концентрированием каждого энантиомера досуха под вакуумом с получением соединения по Получению 37а (68 мг; 94% е.е.; tR составляет 3,83 мин (хиральная аналитическая хроматография, способ 2)) и соединения по Получению 37b (91 мг; 91% е.е.; tR составляет 4,21 мин (хиральная аналитическая хроматография, способ 2)). Абсолютную конфигурацию энантиомеров 37а и 37b не определяли. Первый энантиомер, который элюируется из колонки, представляет собой соединение по Получению 37а, и второй энантиомер представляет собой соединение по Получению 37b.
Получение 38:
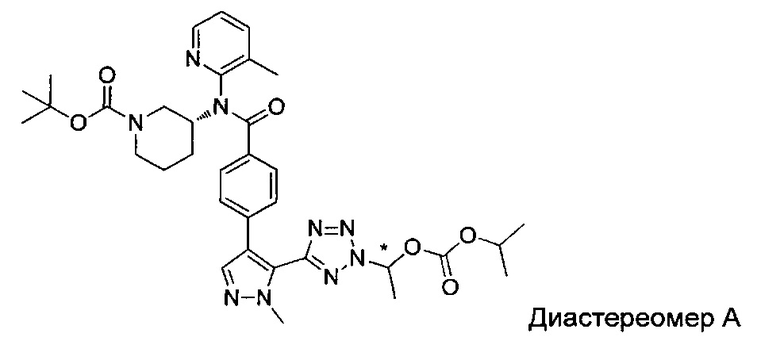
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 37а. Звездочка означает хиральный атом углерода, и поскольку абсолютная конфигурация (например, R/S) диастереомера A не была определена, это соединение по Получению 38 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 37а, из которого соединение по Получению 38 было получено.
1Н ЯМР (CDCl3) δ: 8.48-8.25 (m, 1Н), 7.69-7.57 (m, 1Н), 7.41-7.32 (m, 1Н), 7.22-7.30 (m, 3H), 7.21-7.05 (m, 3Н), 5.07-4.85 (m, 1Н), 4.60-4.39 (m, 1Н), 4.08 (s, 3H), 3.56-3.31 (m, 1Н), 2.73-2.08 (m, 3Н), 2.00-1.98 (m, 6Н), 1.75-1.55 (m, 4Н), 1.47 (br s, 9Н), 1.35 (d, 3H), 1.30 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 2,08 мин.
МС (ЭР+): 674,4 (М+Н)+.
Получение 39:
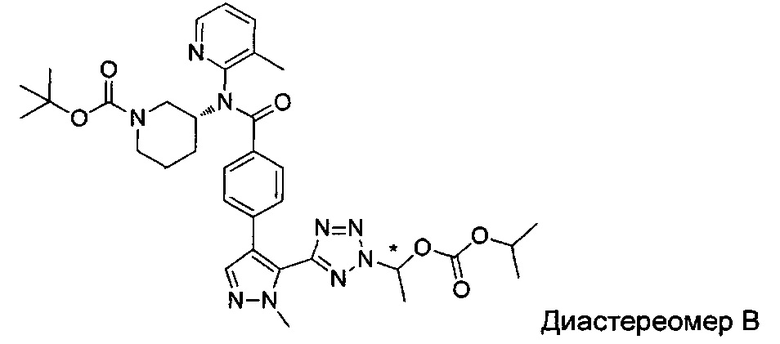
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 37b. Звездочка означает хиральный атом углерода и, поскольку абсолютная конфигурация (например, R/S) диастереомера B не была определена, это соединение по Получению 39 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 37b, из которого соединение по Получению 39 было получено.
1Н ЯМР (МеОН-d4) δ: 8.44-8.34 (m, 1Н), 7.71 (s, 1Н), 7.58-7.47 (m, 1Н), 7.30-7.12 (m, 6Н), 4.89 (септет, 1Н), 4.64-4.45 (m, 2Н), 4.16-4.08 (m, 1Н), 4.07-3.96 (m, 4Н), 2.72-2.47 (m, 1Н), 2.35-2.00 (m, 4Н), 1.94 (br d, 3H), 1.85-1.40 (m, 13Н), 1.31 (d, 3H), 1.26 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 2,08 мин.
МС (ЭР+): 674,4 (М+Н)+.
Получение 40: изопропил-1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]пропилкарбонат
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 23, с использованием соединения по Получению 22, пропиональдегида и изопропилхлорформиата.
1Н ЯМР (CDCl3) δ: 7.64 (s, 1Н), 7.07 (t, 1Н), 4.94 (септет, 1Н), 4.24 (s, 3H), 2.54-2.41 (m, 2Н), 1.35 (d, 3H), 1.31 (d, 3H), 1.04 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,97 мин.
МС (ЭР+): 421,0 (М+Н)+.
Получение 40а и 40b:
631 мг соединения по Получению 40 подвергали обработке с помощью хиральной препаративной хроматографии, способ 7, с последующим концентрированием каждого энантиомера досуха под вакуумом с получением соединения по Получению 40а (174 мг; 92% е.е.; tR составляет 4,10 мин (хиральная аналитическая хроматография, способ 2)) и соединения по Получению 40b (192 мг; 88% е.е.; tR составляет 4,33 мин (хиральная аналитическая хроматография, способ 2)). Абсолютная конфигурация энантиомеров 40а и 40b не была определена. Первый энантиомер, который элюируется из колонки, представляет собой соединение по Получению 40а, и второй энантиомер представляет собой соединение по Получению 40b.
Получение 41:
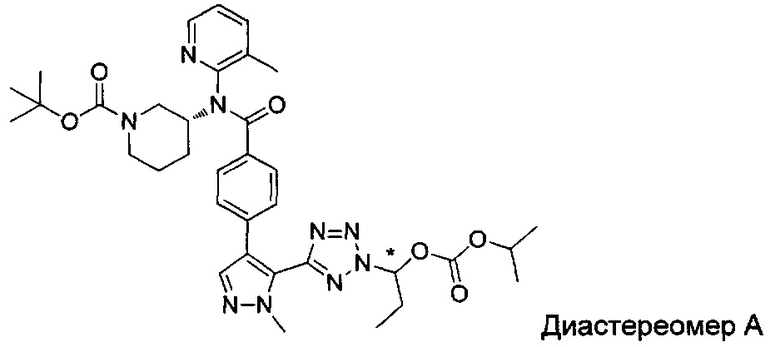
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 40а. Звездочка означает хиральный атом углерода, и поскольку абсолютная конфигурация (например, R/S) диастереомера A не была определена, это соединение по Получению 41 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 40а, из которого соединение по Получению 41 было получено.
1Н ЯМР (CDCl3) δ: 8.42-8.40 (m, 1Н), 7.61 (s, 1Н), 7.40-7.32 (m, 1Н), 7.26-7.22 (m, 3Н), 7.18-7.09 (m, 2Н), 7.01 (dd, 1Н), 4.96-4.98 (m, 1Н), 4.75-4.45 (m, 2Н), 4.22-4.10 (m, 1H), 4.07 (s, 3H), 3.54-3.35 (m, 2H), 2.58-2.47 (m, 3H), 2.39-2.32 (m, 3H), 2.06-1.96 (s, 3H), 1.46 (s, 9H), 1.36 (d, 3H), 1.30 (d, 3H), 1.27-1.25 (m, 2H), 0.98 (m, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,11 мин.
МС (ЭР+): 688,5 (М+Н)+.
Получение 42:
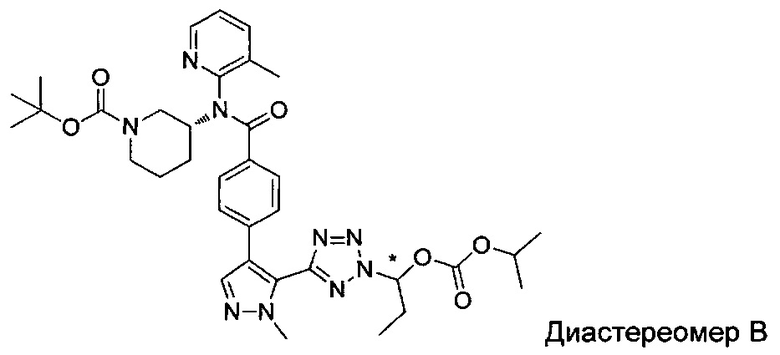
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 40b. Звездочка означает хиральный атом углерода и, поскольку абсолютная конфигурация (например, R/S) диастереомера B не была определена, это соединение по Получению 42 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 40b, из которого соединение по Получению 42 было получено.
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 1,08 мин.
МС (ЭР+): 688,4 (М+Н)+.
Получение 43а и 43b:
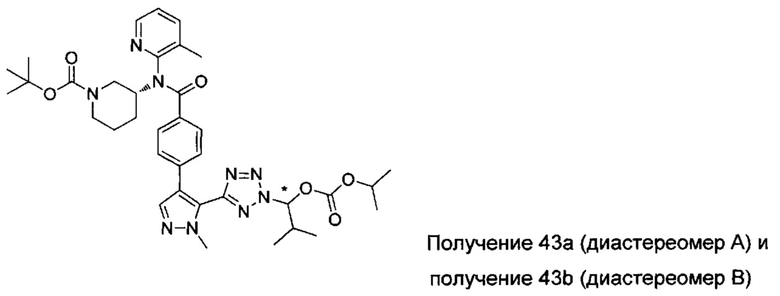
В раствор соединения по Получению 25 (393 мг; 0,723 ммоль) и DMAP (4,9 мг; 0,039 ммоль) в THF (4 мл) добавляли триэтиламин (0,171 мл; 1,23 ммоль), изобутиральдегид (0,113 мл; 1,23 ммоль) и изопропилхлорформиат (1,21 мл; 1,21 ммоль; 1,0 М раствор в PhMe). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли H2O (30 мл) и экстрагировали EtOAc (2×50 мл). Объединенные органические фазы сушили над MgSO4 и концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом 10-60% EtOAc в гептане, с получением смеси диастереомеров в виде бесцветного твердого вещества (192 мг; 38%).
192 мг твердого вещества подвергали обработке с помощью хиральной препаративной хроматографии, способ 8, с последующим концентрированием каждого диастереомера досуха под вакуумом с получением соединения по Получению 43а (55 мг; 97% е.е.; tR составляет 8,70 мин (хиральная аналитическая хроматография, способ 4)) и соединения по Получению 43b (45 мг; 97% е.е.; tR составляет 9,48 мин (хиральная аналитическая хроматография, способ 4)). Звездочка означает хиральный атом углерода и, поскольку абсолютная конфигурация (например, R/S) соединений по Получению 43а и 43b не была определена, каждое соединение представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 43а и соединения по Получению 43b. Первый диастереомер, который элюируется из колонки, представляет собой соединение по Получению 43а, и второй диастереомер представляет собой соединение по Получению 43b.
Диастереомер 43а:
1Н ЯМР (CDCl3) δ: 8.41 (br s, 1Н), 7.61 (s, 1Н), 7.42-7.29 (m, 1Н), 7.26-7.20 (m, 2Н), 7.17-7.09 (m, 3H), 6.74 (d, 1Н), 4.90 (t, 1Н), 4.75-4.08 (m, 2Н), 4.05 (s, 3H), 3.55-3.28 (m, 1Н), 2.77-2.27 (m, 3H), 2.02 (br s, 3H), 1.83-1.54 (m, 4H), 1.52-1.40 (m, 9H), 1.34 (d, 3H), 1.28 (d, 3H), 1.15 (d, 3H), 0.83 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 2,24 мин.
МС (ЭР+): 702,4 (М+Н)+.
Диастереомер 43b:
1Н ЯМР (CDCl3) δ: 8.41 (br. s., 1Н), 7.61 (s, 1H), 7.42-7.29 (m, 1H), 7.26-7.20 (m, 2H), 7.17-7.09 (m, 3H), 6.74 (d, 1H), 4.90 (t, 1H), 4.75-4.08 (m, 2H), 4.05 (s, 3H), 3.55-3.28 (m, 1H), 2.77-2.27 (m, 3H), 2.02 (br. s., 3H), 1.83-1.54 (m, 4H), 1.52-1.40 (m, 9H), 1.34 (d, 3H), 1.28 (d, 3H), 1.15 (d, 3H), 0.83 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 2,24 мин.
MC (ЭР+): 702,4 (М+Н)+.
Получение 44: 1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]этилпропаноат
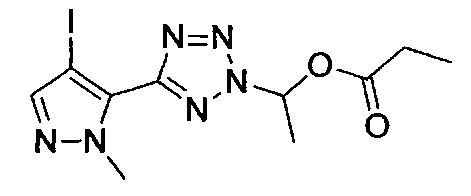
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 23, с использованием соединения по Получению 22, ацетальдегида и пропионилхлорида.
1Н ЯМР (CDCl3) δ: 7.64 (s, 1Н), 7.43 (q, 1Н), 4.24 (s, 3H), 2.50-2.39 (m, 2Н), 2.07 (d, 3H), 1.18 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,88 мин.
MS (АР+): 377,0 (М+Н)+.
Получение 44а и 44b:
916 мг соединения по Получению 44 подвергали обработке с помощью хиральной препаративной хроматографии, способ 9, с последующим концентрированием каждого энантиомера досуха под вакуумом с получением соединения по Получению 44а (228 мг; 96% е.е.; tR составляет 2,97 мин (хиральная аналитическая хроматография, способ 5)) и соединения по Получению 44b (244 мг; 94% е.е.; tR составляет 3,23 мин (хиральная аналитическая хроматография, способ 5)). Абсолютная конфигурация энантиомеров 44а и 44b не была определена. Первый энантиомер, который элюируется из колонки, представляет собой соединение по Получению 44а, и второй энантиомер представляет собой соединение по Получению 44b.
Получение 45:
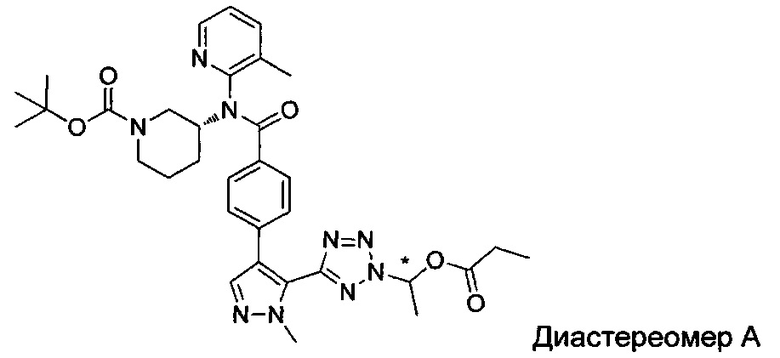
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и содинения по Получению 44а. Звездочка означает хиральный атом углерода, и поскольку абсолютная конфигурация (например, R/S) диастереомера A не была определена, это соединение по Получению 45 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 44а, из которого соединение по Получению 45 было получено.
1Н ЯМР (CDCl3) δ: 8.42 (d, 1Н), 7.62 (s, 1Н), 7.43-7.32 (m, 2Н), 7.30-7.23 (m, 2Н), 7.21-7.07 (m, 3Н), 4.56-4.44 (m, 1Н), 4.08 (s, 3H), 2.52-2.34 (m, 2Н), 2.11-1.99 (m, 4Н), 2.03 (s, 3H), 1.96 (d, 3H), 1.74-1.64 (m, 4Н), 1.48 (d, 9Н), 1.18 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 1,02 мин.
МС (ЭР+): 644,4 (М+Н)+.
Получение 46:
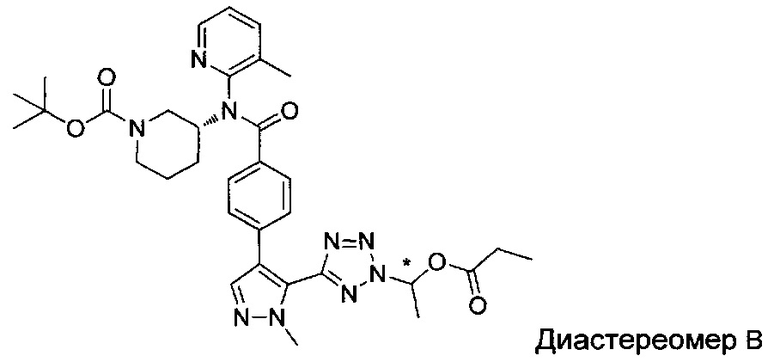
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 44b. Звездочка означает хиральный атом углерода и, поскольку абсолютная конфигурация (например, R/S) диастереомера B не была определена, это соединение по Получению 46 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 44b, из которого соединение по Получению 46 было получено.
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 1,03 мин.
МС (ЭР+): 644,4 (М+Н)+.
Получение 47: 1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]пропилпропаноат
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 23, с использованием соединения по Получению 22, пропиональдегида и пропионового ангидрида.
1Н ЯМР (CDCl3) δ: 7.64 (s, 1Н), 7.23 (t, 1Н), 4.24 (s, 3H), 2.51-2.36 (m, 4Н), 1.18 (t, 3H), 1.02 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,93 мин.
MS(AP+): 391,0 (М+Н)+.
Получение 47а и 47b:
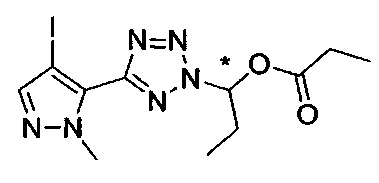
535 мг соединения по Получению 47 подвергали обработке с помощью хиральной препаративной хроматографии, способ 14, с последующим концентрированием каждого энантиомера досуха под вакуумом с получением соединения по Получению 47а (101 мг; 96% е.е.; tR составляет 3,28 мин (хиральная аналитическая хроматография, способ 6)) и соединения по Получению 47b (170 мг; 93% е.е.; tR составляет 3,57 мин (хиральная аналитическая хроматография, способ 6)). Абсолютная конфигурация энантиомеров 47а и 47b не была определена. Первый энантиомер, который элюируется из колонки, представляет собой соединение по Получению 47а, и второй энантиомер представляет собой соединение по Получению 47b.
Получение 48:
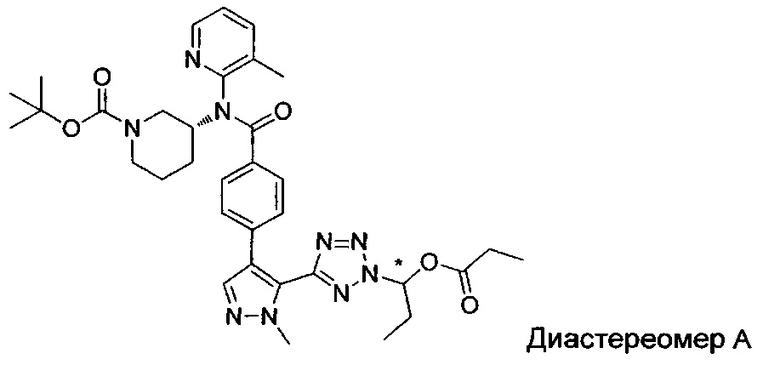
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 47а. Звездочка означает хиральный атом углерода, и поскольку абсолютная конфигурация (например, R/S) диастереомера A не была определена, это соединение по Получению 48 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 47а, из которого соединение по Получению 48 было получено.
1Н ЯМР (CDCl3) δ: 8.48-8.31 (m, 1Н), 7.61 (s, 1Н), 7.43-7.31 (m, 2Н), 7.27-7.20 (m, 2Н), 7.16-7.07 (m, 3H), 4.57-4.30 (m, 1Н), 4.06 (s, 3H), 3.64-3.29 (m, 2Н), 2.72-2.52 (m, 1Н), 2.47-2.25 (m, 5Н), 2.08-1.92 (m, 3H), 1.47 (br s, 9Н), 1.75-1.44 (m, 4H), 1.16 (t, 3H), 0.94 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 2,10 мин.
МС (ЭР+): 658,4 (М+Н)+.
Получение 49:
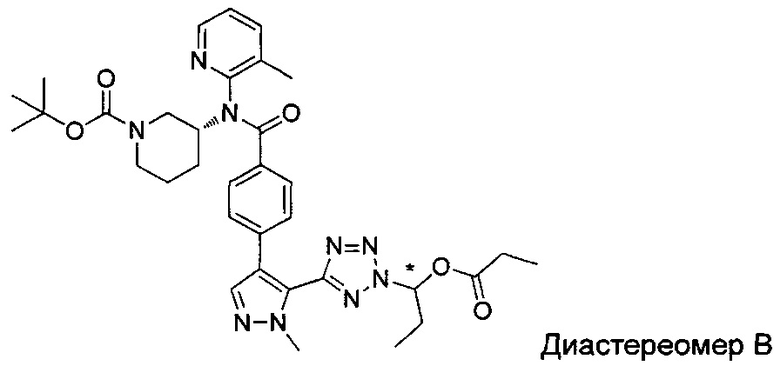
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 47b. Звездочка означает хиральный атом углерода и, поскольку абсолютная конфигурация (например, R/S) диастереомера B не была определена, это соединение по Получению 49 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 47b, из которого соединение по Получению 49 было получено.
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 1,06 мин.
МС (ЭР+): 658,4 (М+Н)+.
Получение 50: 1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]-2-метилпропилпропаноат
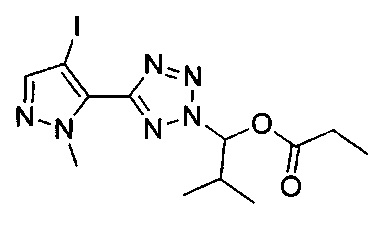
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 23, с использованием соединения по Получению 22, изобутиральдегида и пропионового ангидрида.
1Н ЯМР (CDCl3) δ: 7.63 (s, 1Н), 7.00 (d, 1Н), 4.22 (s, 3H), 2.79-2.73 (m, 1Н), 2.52-2.41 (m, 2Н), 1.18 (t, 3H), 1.14 (d, 3H), 0.92 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,85 мин.
МС (ЭР+): 405,1 (М+Н)+.
Получение 50а и 50b:
475 мг соединения по Получению 50 подвергали обработке с помощью хиральной препаративной хроматографии, способ 9, с последующим концентрированием каждого энантиомера досуха под вакуумом с получением соединения по Получению 50а (148 мг; 97% е.е.; tR составляет 2,77 мин (хиральная аналитическая хроматография, способ 5)) и соединения по Получению 50b (133 мг; 94% е.е.; tR составляет 3,21 мин (хиральная аналитическая хроматография, способ 5)). Абсолютная конфигурация энантиомеров 50а и 50b не была определена. Первый энантиомер, который элюируется из колонки, представляет собой соединение по Получению 50а, и второй энантиомер представляет собой соединение по Получению 50b.
Получение 51:
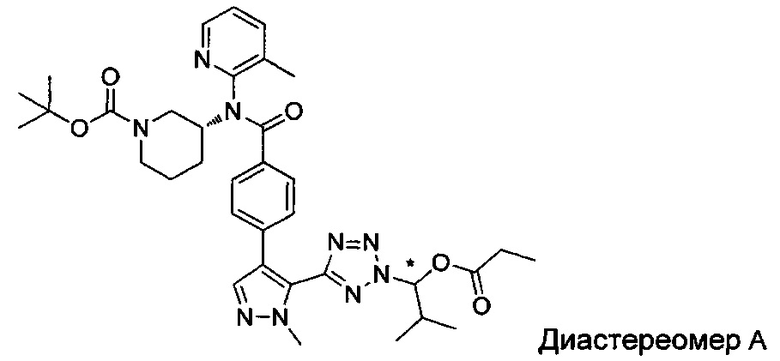
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 50а. Звездочка означает хиральный атом углерода, и поскольку абсолютная конфигурация (например, R/S) диастереомера A не была определена, это соединение по Получению 51 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 50а, из которого соединение по Получению 51 было получено.
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 1,10 мин.
МС (ЭР+): 672,5 (М+Н)+.
Получение 52:
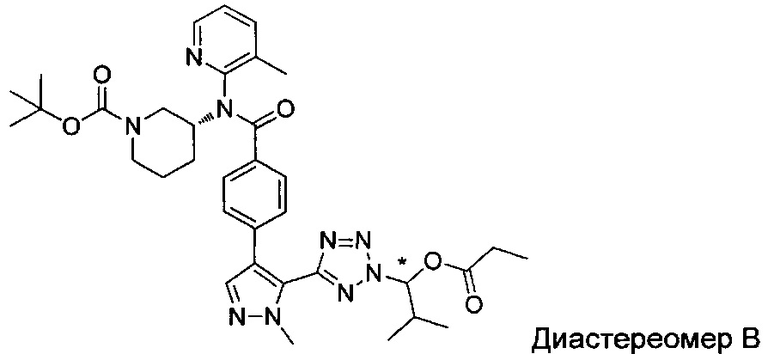
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 50b. Звездочка означает хиральный атом углерода, и поскольку абсолютная конфигурация (например, R/S) диастереомера B не была определена, это соединение по Получению 52 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 50b, из которого соединение по Получению 52 было получено.
1Н ЯМР (CDCl3) δ: 8.40 (br s, 1Н), 7.60 (s, 1Н), 7.34 (br s, 1Н), 7.21 (br s, 2H), 7.16-7.04 (m, 3H), 6.89 (d, 1H), 4.57-4.40 (m, 1H), 4.18-4.09 (m, 2H), 4.04 (s, 3H), 3.41 (br s, 1H), 2.64-2.57 (m, 2H), 2.51-2.32 (m, 2H), 2.06-1.91 (m, 3H), 1.57 (s, 9H), 1.50-1.38 (m, 4H), 1.33-1.21 (m, 3H), 1.15 (t, 3H), 1.08 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 2,18 мин.
МС (ЭР+): 672,4 (М+Н)+.
Получение 53: 1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]этил-2-метилпропаноат
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 23, с использованием соединения по Получению 22, ацетальдегида и изобутирилхлорида.
1Н ЯМР (CDCl3) δ: 7.64 (s, 1Н), 7.40 (q, 1Н), 4.24 (s, 3H), 2.70-2.59 (m, 4Н), 2.05 (d, 3H), 1.22 (2, 3H), 1.19 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,94 мин.
MS (AP+): 391,0 (М+Н)+.
Получение 53а и 53b:
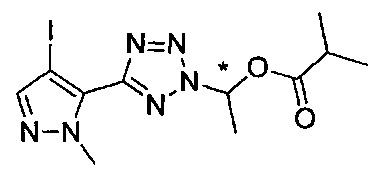
593 мг соединения по Получению 53 подвергали обработке с помощью хиральной препаративной хроматографии, способ 10, с последующим концентрированием каждого энантиомера досуха под вакуумом с получением соединения по Получению 53а (165 мг; 98% е.е.; tR составляет 3,66 мин (хиральная аналитическая хроматография, способ 7)) и соединения по Получению 53b (172 мг; 92% е.е.; tR составляет 3,86 мин (хиральная аналитическая хроматография, способ 7)). Абсолютная конфигурация энантиомеров 53а и 53b не была определена. Первый энантиомер, который элюируется из колонки, представляет собой соединение по Получению 53а, и второй энантиомер представляет собой соединение по Получению 53b.
Альтернативный способ получения 53b:
В раствор соединения по Получению 22 (1,00 г; 3,62 ммоль) и {(2S)-2-{бис[3,5-бис(трифторметил)фенил]гидроксиметил}-1-пирролидинил}[4-(1-пирролидинил)-3-пиридинил]метанона (25,2 мг; 0,036 ммоль) в метил-трет-бутиловом эфире (36 мл) добавляли триэтиламин (0,608 мл; 4,35 ммоль), ацетальдегид (0,244 мл; 4,35 ммоль) и ангидрид изомасляной кислоты (0,782 мл; 4,71 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 64 ч. Реакционную смесь вливали в воду (40 мл) и экстрагировали EtOAc (2×50 мл). Объединенные органические фазы сушили и концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом 0-30% EtOAc в гептане, с получением указанного в заголовке соединения в виде бесцветного масла (904 мг; 64%; 90% е.е.).
Получение 54:
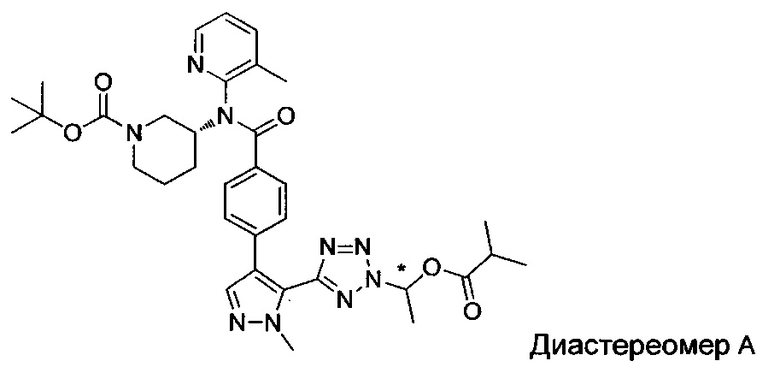
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 53а. Звездочка означает хиральный атом углерода, и поскольку абсолютная конфигурация (например, R/S) диастереомера A не была определена, это соединение по Получению 54 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 53а, из которого соединение по Получению 54 было получено.
1Н ЯМР (CDCl3) δ: 8.40 (br s, 1Н), 7.60 (s, 1Н), 7.35 (br s, 1H), 7.30 (q, 1H), 7.23 (br s, 2H), 7.17-7.08 (br m, 3H), 4.48 (br d, 1H), 4.16-3.95 (br m, 4H), 3.42 (br s, 1H), 2.63-2.50 (m, 2H), 2.39 (br s, 1H), 2.05-1.98 (br m, 3H), 1.94 (d, 3H), 1.67-1.57 (br m, 4H), 1.49-1.43 (br d, 9H), 1.19 (d, 3H), 1.14 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 1,07 мин.
МС (ЭР+): 658,4 (М+Н)+.
Получение 55:
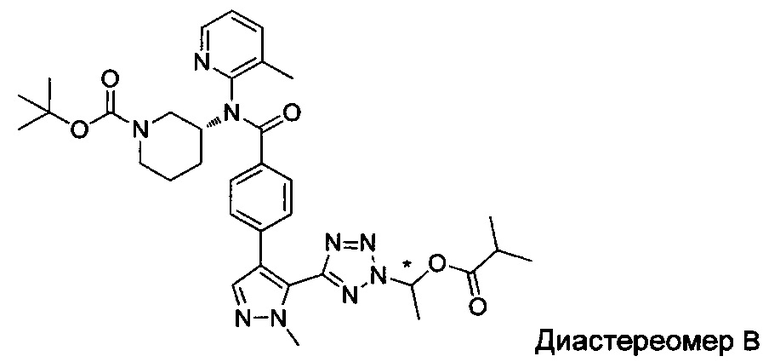
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 53b. Звездочка означает хиральный атом углерода, и поскольку абсолютная конфигурация (например, R/S) диастереомера B не была определена, это соединение по Получению 55 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 53b, из которого соединение по Получению 55 было получено.
1Н ЯМР (CDCl3) δ: 8.40 (br s, 1Н), 7.60 (s, 1Н), 7.34 (br s, 1Н), 7.30 (q, 1Н), 7.23 (br s, 2H), 7.17-7.07 (br m, 3H), 4.48 (br d, 1H), 4.15-3.95 (br m, 4H), 3.42 (br s, 1H), 2.63-2.50 (m, 2H), 2.39 (br s, 1H), 2.05-1.93 (br m, 6H), 1.76-1.62 (br m, 4H), 1.50-1.42 (br m, 9H), 1.19 (d, 3H), 1.14 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 1,06 мин.
МС (ЭР+): 658,3 (М+Н)+.
Альтернативный способ получения 55:
В раствор соединения по Получению 25 (7,50 г; 13,8 ммоль) и {(2S)-2-{бис[3,5-бис(трифторметил)фенил]гидроксиметил}-1-пирролидинил}[4-(1-пирролидинил)-3-пиридинил]метанона (965 мг; 1,38 ммоль) в PhMe (69 мл) добавляли триэтиламин (3,85 мл; 27,6 ммоль), ацетальдегид (1,54 мл; 27,6 ммоль) и ангидрид изомасляной кислоты (4,58 мл; 27,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь концентрировали под вакуумом и остаток очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом 20-100% EtOAc в гептане, с получением указанного в заголовке соединения в виде бесцветного твердого вещества (6,3 г; 69%; 70% е.е.). Твердое вещество подвергали обработке с помощью хиральной препаративной хроматографии, способ 11, с последующим концентрированием досуха под вакуумом с получением соединения по Получению 55 (4,65 г; 99% е.е.).
Получение 56: 1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]пропил-2-метилпропаноат
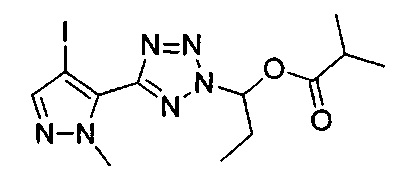
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 23, с использованием соединения по Получению 22, пропиональдегида и изобутирилхлорида.
1Н ЯМР (CDCl3) δ: 7.64 (s, 1Н), 7.21 (t, 1Н), 4.23 (s, 3H), 2.72-2.61 (m, 1Н), 2.51-2.35 (m, 2Н), 1.23 (d, 3H), 1.19 (d, 3H), 1.03 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,99 мин.
МС (АР+): 405,0 (М+Н)+.
Получение 56а и 56b:
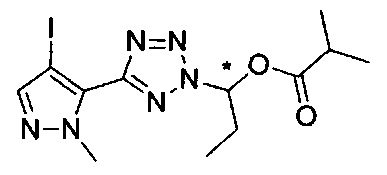
617 мг соединения по Получению 56 подвергали обработке с помощью хиральной препаративной хроматографии, способ 12, с последующим концентрированием каждого энантиомера досуха под вакуумом с получением соединения по Получению 56а (126 мг; 92% е.е.; tR составляет 4,57 мин (хиральная аналитическая хроматография, способ 1)) и соединения по Получению 56b (116 мг; 95% е.е.; tR составляет 5,01 мин (хиральная аналитическая хроматография, способ 1)). Абсолютная конфигурация энантиомеров 56а и 56b не была определена. Первый энантиомер, который элюируется из колонки, представляет собой соединение по Получению 56а, и второй энантиомер представляет собой соединение по Получению 56b.
Получение 57:
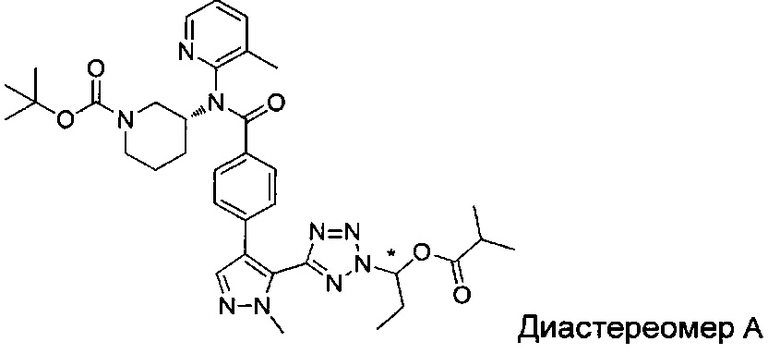
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 56а. Звездочка означает хиральный атом углерода, и поскольку абсолютная конфигурация (например, R/S) диастереомера A не была определена, это соединение по Получению 57 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 56а, из которого соединение по Получению 57 было получено.
1Н ЯМР (CDCl3) δ: 8.41 (s, 1Н), 7.62 (s, 1Н), 7.40-7.32 (m, 1Н), 7.24-7.21 (m, 3Н), 7.13-7.11 (m, 3Н), 4.62-4.45 (m, 2Н), 4.08-4.00 (s, 3H), 3.51-3.36 (m, 1Н), 2.65-2.60 (m, 1Н), 2.58-2.48 (m, 2Н), 2.33-2.29 (m, 1Н), 2.22-2.12 (m, 1Н), 2.03 (s, 3H), 1.80-1.59 (m, 1Н), 1.48 (s, 9Н), 1.32-1.26 (m, 3Н), 1.22 (d, 3H), 1.16 (d, 3H), 0.97 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 2,19 мин.
МС (ЭР+): 672,4 (М+Н)+.
Получение 58:
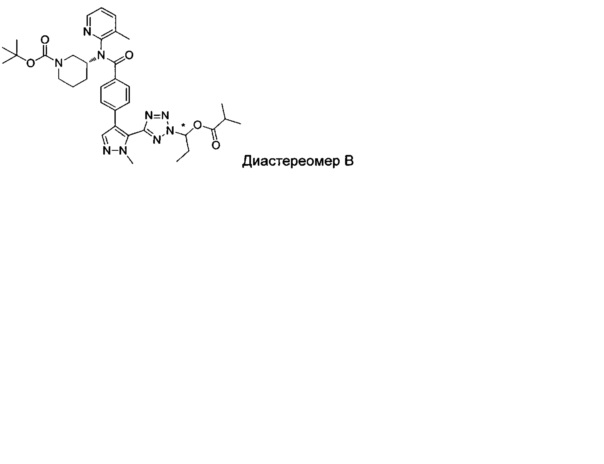
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 56b. Звездочка означает хиральный атом углерода, и поскольку абсолютная конфигурация (например, R/S) диастереомера B не была определена, это соединение по Получению 58 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 56b, из которого соединение по Получению 58 было получено.
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 1,09 мин.
МС (ЭР+): 672,4 (М+Н)+.
Получение 59: 1-[5-(4-йод-1-метил-1Н-пиразол-5-ил)-2Н-тетразол-2-ил]-2-метилпропил-2-метилпропаноат
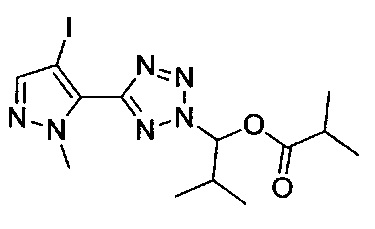
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 23, с использованием соединения по Получению 22, изобутиральдегида и ангидрида изомасляной кислоты.
1Н ЯМР (МеОН-d4) δ: 7.63 (s, 1Н), 6.98 (d, 1Н), 4.22 (s, 3H), 2.79-2.73 (m, 1Н), 2.71-2.64 (m, 1Н), 1.24 (d, 3H), 1.19 (d, 3H), 1.14 (d, 3H), 0.92 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,98 мин.
МС (ЭР+): 419,1 (М+Н)+.
Получение 59а и 59b:
482 мг соединения по Получению 59 подвергали обработке с помощью хиральной препаративной хроматографии, способ 13, с последующим концентрированием каждого энантиомера досуха под вакуумом с получением соединения по Получению 59а (172 мг; более 99% е.е.; tR составляет 3,95 мин (хиральная аналитическая хроматография, способ 8)) и соединения по Получению 59b (168 мг; 98% е.е.; tR составляет 4,07 мин (хиральная аналитическая хроматография, способ 8)). Абсолютная конфигурация энантиомеров 59а и 59b не была определена. Первый энантиомер, который элюируется из колонки, представляет собой соединение по Получению 59а, и второй энантиомер представляет собой соединение по Получению 59b.
Альтернативный способ получения 59b:
В раствор соединения по Получению 22 (5,00 г; 18,1 ммоль) и {(2S)-2-{бис[3,5-бис(трифторметил)фенил]гидроксиметил}-1-пирролидинил}[4-(1-пирролидинил)-3-пиридинил]метанона (507 мг; 0,725 ммоль) в PhMe (181 мл) добавляли триэтиламин (3,04 мл; 21,7 ммоль), изобутиральдегид (1,98 мл; 21,7 ммоль) и ангидрид изомасляной кислоты (3,91 мл; 23,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 65 ч. Реакционную смесь вливали в воду и дважды экстрагировали EtOAC. Объединенные органические фазы сушили и концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом 0-30% EtOAc в гептане, с получением указанного в заголовке соединения в виде бесцветного масла (7,11 г; 90%; 86% е.е.). Эти 7,11 г подвергали обработке с помощью хиральной препаративной хроматографии, способ 13, с последующим концентрированием досуха под вакуумом с получением соединения по Получению 59b (5,8 г; 98% е.е.).
Получение 60:
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 59а. Звездочка означает хиральный атом углерода, и поскольку абсолютная конфигурация (например, R/S) диастереомера A не была определена, это соединение по Получению 60 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 59а, из которого соединение по Получению 60 было получено.
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 1,13 мин.
МС (ЭР+): 686,5 (М+Н)+.
Получение 61:
Указанное в заголовке соединение получали способом, аналогичным способу по Получению 27, с использованием соединения по Получению 24 и соединения по Получению 59b. Звездочка означает хиральный атом углерода, и поскольку абсолютная конфигурация (например, R/S) диастереомера B не была определена, это соединение по Получению 61 представляет собой отдельный диастереомер с характерными свойствами, как показано по уникальности времени удерживания при хиральной хроматографии соединения по Получению 59b, из которого соединение по Получению 61 было получено.
1Н ЯМР (CDCl3) δ: 8.39 (br s, 1Н), 7.60 (s, 1Н), 7.33 (br s, 1Н), 7.21 (br s, 2H), 7.16-7.03 (m, 3H), 6.88 (d, 1H), 4.66 (br s, 1H), 4.55-4.39 (m, 1H), 4.03 (s, 3H), 3.41 (br s, 1H), 2.67-2.58 (m, 1H), 2.05-1.91 (m, 3H), 1.58 (s, 9H), 1.49-1.39 (m, 4H), 1.35-1.22 (m, 4H), 1.21 (d, 3H), 1.14 (d, 3H), 1.09 (d, 3H), 0.83 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 2,26 мин.
МС (ЭР+): 686,5 (М+Н)+.
Общая методика A: образование хлорангидрида;
Общая методика B: опосредованное MeMgCl образование амида;
Общая методика C: опосредованное LiHMDS образование амида;
Общая методика D: снятие защиты Boc с помощью HCl;
Общая методика Е: реакция Бухвальда с 2-хлор-3-нитропиридином;
Общая методика F: восстановление нитро;
Общая методика G: диазотирование/замыкание кольца;
Общая методика Н: гидролиз сложного эфира с получением кислоты;
Общая методика I: гидролиз сложного эфира с получением калиевой соли;
Общая методика J: алкилирование с образованием карбоната.
ПРИМЕР 1: N-(3-метилпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(пиразоло[1,5-а]пиримидин-3-ил)бензамид
Стадия 1: 1-оксид (R)-2-(N-(1-(трет-бутоксикарбонил)пиперидин-3-ил)-4-(пиразоло[1,5-а]пиримидин-3-ил)бензамидо)-3-метилпиридина
В раствор соединения по Получению 14 1-оксида (R)-2-(1-(трет-бутоксикарбонил)пиперидин-3-иламино)-3-метилпиридина (540 мг; 1,76 ммоль) и соединения по Получению 19 4-(пиразоло[1,5-а]пиримидин-3-ил)бензойной кислоты (420 мг; 1,76 ммоль) в DMF (10 мл) добавляли HATU (803 мг; 2,11 ммоль) при 30°C. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Затем по каплям добавляли диизопропилэтиламин (682 мг; 5,28 ммоль) при 0°C. Реакционную смесь перемешивали при 30°C в течение 2 ч. Полученную смесь вливали в воду, затем экстрагировали EtOAc (3×30 мл). Объединенные органические слои промывали рассолом, сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали посредством хроматографии на силикагеле с получением указанного в заголовке соединения (760 мг; 82%) в виде коричневого масла.
1Н ЯМР (400 МГц, CDCl3) δ 8.69 (d, 1Н), 8.57 (s, 1Н), 8.40 (s, 1Н), 8.20-8.14 (m, 1Н), 7.93 (d, 2Н), 7.66-7.56 (m, 2Н), 7.07-6.99 (m, 1Н), 6.94-6.85 (m, 2Н), 2.80-2.63 (m, 1Н), 2.23 (s, 1Н), 2.14 (s, 2Н), 2.02-1.57 (m, 5Н), 1.47 (s, 9Н).
Стадия 2: (R)-трет-бутил-3-(N-(3-метилпиридин-2-ил)-4-(пиразоло[1,5-а]пиримидин-3-ил)бензамидо)пиперидин-1-карбоксилат
В раствор соединения со стадии 1 1-оксида (R)-2-(1-(трет-бутоксикарбонил)-пиперидин-3-иламино)-3-метилпиридина (760 мг; 1,44 ммоль) в ацетонитриле (15 мл) добавляли тетрагидроксид дибора (387 мг; 4,32 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 60°C в течение ночи. Полученную смесь обрабатывали водой, затем экстрагировали EtOAc (3×30 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (760 мг) в виде белой смолы. Это вещество использовали на следующей стадии без дополнительной очистки.
Стадия 3: гидрохлорид N-(3-метилпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(пиразоло[1,5-а]пиримидин-3-ил)бензамида
В раствор соединения со стадии 2 (R)-трет-бутил-3-(N-(3-метилпиридин-2-ил)-4-(пиразоло[1,5-а]пиримидин-3-ил)бензамидо)пиперидин-1-карбоксилата (737 мг; 1,44 ммоль) в 1,4-диоксане (10 мл) по каплям добавляли смесь HCl/1,4-диоксан (7,2 мл 4 М; 28,8 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Полученную смесь концентрировали при пониженном давлении. Неочищенный продукт очищали посредством препаративной ЖХВД с получением гидрохлорида указанного в заголовке соединения (325 мг; 52%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.60 (br s, 1Н), 9.19 (br s, 1H), 9.09 (d, 1H), 8.73-8.60 (m, 2H), 8.45 (br s, 1H), 7.98 (d, 2H), 7.60 (d, 1H), 7.35-7.22 (m, 3H), 7.10 (dd, 1H), 3.60-3.48 (m, 1H), 3.26-3.16 (m, 1H), 2.80-2.66 (m, 1H), 2.08 (s, 3H), 2.02-1.79 (m, 4H).
ЖХ (ЖХ-МС, способ 3): tR составляет 0,838 мин.
МС (ЭР+): 413,1 (M+H)+.
ПРИМЕР 2: N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(пиразоло[1,5-а]пиримидин-3-ил)бензамид
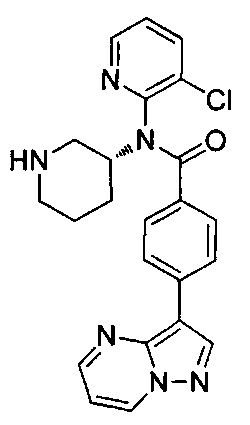
Стадия 1: трет-бутил-(3R)-3-{(3-хлорпиридин-2-ил)[4-(пиразоло[1,5-а]пиримидин-3-ил)бензоил]амино}пиперидин-1-карбоксилат
Общая методика А. Оксалилхлорид (3,86 мл; 44 ммоль) добавляли в раствор соединения по Получению 19 4-(пиразоло[1,5-а]пиримидин-3-ил)бензойной кислоты (10 г; 39,4 ммоль) в DCM (200 мл) и DMF (0,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и затем упаривали. Остаток разбавляли толуолом и упаривали досуха. Эту стадию повторяли 5 раз и остаток использовали на следующей стадии.
Стадия 2:
Общая методика В. Раствор соединения по Получению 1 трет-бутил-(3R)-3-[(3-хлорпиридин-2-ил)амино]пиперидин-1-карбоксилата (12,2 г; 39 ммоль) в дегазированном толуоле (90 мл) обрабатывали хлоридом метилмагния (3 М раствор в THF) (13,1 мл; 39 ммоль) при 0°C. Смесь перемешивали в течение 1 ч при 0°C перед добавлением суспензии твердого хлорангидрида со стадии 1 в THF (150 мл). Полученную смесь перемешивали при 65°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (80 мл) и EtOAc (200 мл). Водный слой экстрагировали EtOAc (2×300 мл) и объединенные органические слои сушили над MgSO4, фильтровали и фильтрат концентрировали. Остаток очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом 25-100% EtOAc/гептан, с получением указанного в заголовке соединения в виде желтого твердого вещества (17 г; 81%).
1Н ЯМР (CDCl3) δ 8.70 (dd, 1H), 8.58 (dd, 1Н), 8.49 (d, 1Н), 8.42 (s, 1Н), 7.91 (d, 2Н), 7.63-7.53 (m, 1Н), 7.45 (d, 2Н), 7.15 (dd, 1Н), 6.88 (dd, 1Н), 4.89-4.24 (m, 3H), 4.12 (m, 1Н), 3.52-3.22 (m, 1Н), 2.77-2.48 (m, 1Н), 2.44-2.26 (m, 1Н), 2.02-1.90 (m, 1Н), 1.82-1.65 (m, 1Н), 1.49 (s, 9Н).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,72 мин.
МС (ЭР+): 533,3 (М+Н)+.
Стадия 3: N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(пиразоло[1,5-а]пиримидин-3-ил)бензамид
Общая методика D. Раствор соединения со стадии 2 трет-бутил-(3R)-3-{(3-хлорпиридин-2-ил)[4-(пиразоло[1,5-а]пиримидин-3-ил)бензоил]амино}-пиперидин-1-карбоксилата (0,929 г; 1,74 ммоль) в DCM (20 мл) и МеОН (15 мл) обрабатывали HCl (4 М раствор в 1,4-диоксане) (4,59 мл; 18,3 ммоль). Смесь затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали. Полученный остаток растирали в диэтиловом эфире с получением гидрохлорида указанного в заголовке соединения в виде светло-желтого твердого вещества (0,702 г; 86%).
1Н ЯМР (МеОН-d4, 400 МГц): δ 8.93 (dd, 1Н), 8.63 (dd, 1Н), 8.60 (s, 1Н), 8.56 (s, 1Н), 8.01 (d, 2Н), 7.87-7.74 (m, 1Н), 7.39 (dd, 3H), 7.07 (dd, 1Н), 5.26-5.01 (m, 1Н), 3.92-3.72 (m, 1Н), 3.68-3.53 (m, 1Н), 3.39 (m, 1Н), 2.92 (m, 1Н), 2.04 (m, 2Н), 1.99-1.78 (m, 1Н), 1.63-1.37 (m, 1Н).
Чистота по ЖХВД (ЖХВД, способ 2): 99,6%, tR составляет 2,533 мин.
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,37 мин.
МС (ЭР+): 433,3 (М+Н)+.
Дифракция рентгеновских лучей на порошке для N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(пиразоло[1,5-а]пиримидин-3-ил)бензамида представлена на Фиг. 1.
ПРИМЕР 3: N-(3-хлорпиридин-2-ил)-4-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-N-[(3R)-пиперидин-3-ил]бензамид
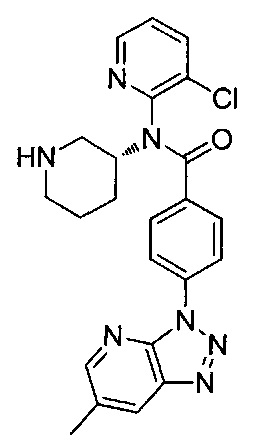
Стадия 1: трет-бутил-(3R)-3-{(3-хлорпиридин-2-ил)[4-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензоил]амино}пиперидин-1-карбоксилат
Оксалилхлорид (0,97 мл; 10,8 ммоль) добавляли в раствор соединения по Получению 6 4-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензойной кислоты (2,5 г; 9,83 ммоль) в DCM (63 мл) и DMF (1,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и затем упаривали. Остаток разбавляли толуолом и упаривали досуха. Эту стадию повторяли 5 раз.
Стадия 2:
Общая методика С. Раствор соединения со стадии 1 и соединения по Получению 1 трет-бутил-(3R)-3-[(3-хлорпиридин-2-ил)амино]пиперидин-1-карбоксилата (3,22 г; 10,3 ммоль) в THF (100 мл) охлаждали до 0°C, затем по каплям добавляли бис(триметилсилил)амид лития (10,3 мл; 10,3 ммоль; 1 М раствор в THF). Полученную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли водой и экстрагировали EtOAc (2×80 мл). Объединенные органические слои сушили над MgSO4, фильтровали и фильтрат концентрировали. Остаток очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом 0-60% EtOAc/гептан, с получением указанного в заголовке соединения в виде желтого твердого вещества (3,56 г; выход 66%).
1Н ЯМР (CDCl3) δ 8.61 (d, 2Н), 8.26 (d, 2Н), 8.21 (dd, 1Н), 7.60 (m, 3H), 7.23-7.12 (m, 1Н), 4.78-4.42 (m, 2Н), 4.39-4.16 (m, 1Н), 3.49-3.26 (m, 1Н), 2.59 (s, 4Н), 2.06-1.95 (m, 1Н), 1.88-1.63 (m, 2Н), 1.50 (s, 9Н), 1.30 (m, 1Н).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 1,04 мин.
МС (ЭР+): 548,2 (М+Н)+.
Стадия 3:
Получали согласно общей методике D, начиная с соединения со стадии 2 трет-бутил-(3R)-3-{(3-хлорпиридин-2-ил)[4-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензоил]амино}пиперидин-1-карбоксилата (3,56 г; 6,5 ммоль), с получением 2,82 г (90%) гидрохлорида указанного в заголовке соединения в виде твердого вещества.
1Н ЯМР (CDCl3) δ 10.19-9.98 (m, 1Н), 9.93-9.70 (m, 1Н), 8.57 (s, 1Н), 8.53-8.39 (m, 1Н), 8.23 (d, 2Н), 8.17 (s, 1Н), 7.65-7.50 (m, 3Н), 7.20 (dd, 1Н), 5.18-4.70 (m, 1Н), 4.03-3.73 (m, 2Н), 3.66-3.38 (m, 1Н), 3.07-2.70 (m, 1Н), 2.57 (s, 3Н), 2.49-2.32 (m, 1Н), 2.30-1.70 (m, 3Н).
Чистота по ЖХВД (ЖХВД, способ 1): 99,5%, tR составляет 3,378 мин.
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,6 мин.
МС (ЭР+): 449,6 (М+Н)+.
ПРИМЕР 4: 4-(4-{изохинолин-1-ил[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоновая кислота
: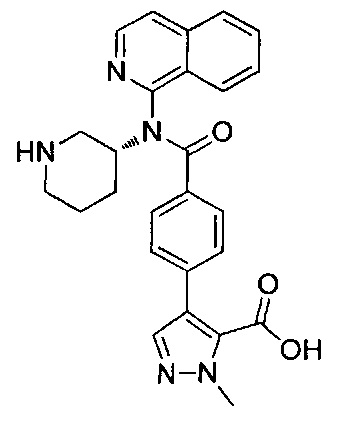
Стадия 1: Суспензию соединения по Получению 16 (50 мг; 0,0897 ммоль), 4-йод-2-метил-2Н-пиразол-3-карбоновой кислоты (MFCD00461121) (23 мг; 0,0897 ммоль), Cs2CO3 (58 мг; 0,179 ммоль) и PdCl2(dppf)2⋅CH2Cl2 (4 мг; 0,00448 ммоль) в 1,4-диоксане (0,8 мл) и H2O (0,2 мл) перемешивали при 80°C в течение 16 ч. Анализ ЖХ-МС показал, что реакция завершена. Реакционную смесь сушили над Na2SO4 и фильтровали. Фильтрат концентрировали досуха и очищали посредством препаративной ЖХВД (петролейный эфир: EtOAc, 1:1) с получением N-BOC-промежуточного соединения (10 мг; 20%) в виде белого твердого вещества. Взаимодействие повторяли для получения дополнительного количества вещества.
Стадия 2:
В раствор объединенных порций вещества со стадии 1 (20 мг; 0,036 ммоль) в DCM (2 мл) при 0°C по каплям добавляли TFA (0,5 мл). Полученную смесь перемешивали при комнатной температуре в течение 30 мин. Анализ ЖХ-МС показал, что реакция завершена. Растворитель удаляли под вакуумом с получением неочищенного вещества (20 мг), которое очищали посредством препаративной ЖХВД с получением указанного в заголовке соединения (5,8 мг; 35,4%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6), Т составляет 80°C) δ 8.45 (d, 1Н), 8.02 (d, 1Н), 7.92 (d, 1Н), 7.82 (d, 1Н), 7.73-7.65 (m, 2Н), 7.41 (s, 1Н), 7.18 (d, 2Н), 7.12 (d, 2Н), 4.99-4.90 (m, 1Н), 3.98 (s, 3H), 3.68 (m, 1Н), 2.78-2.71 (m, 2Н), 2.53 (m, 1Н), 2.16-2.02 (m, 1Н), 1.81-1.72 (m, 2Н), 1.38-1.26 (m, 1Н).
МС (ЭР+): 456,2040 (М+Н)+.
ПРИМЕР 5: N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-5-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиридин-2-карбоксамид
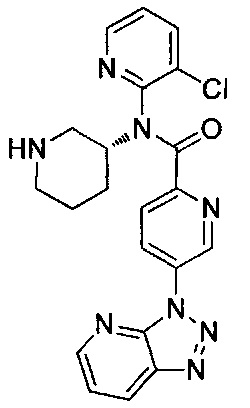
Стадия 1: трет-бутил-(3R)-3-[(3-хлорпиридин-2-ил){[5-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиридин-2-ил]карбонил}амино]пиперидин-1-карбоксилат
Получали с использованием общих методик А и С, начиная с соединения по Получению 1 трет-бутил-(3R)-3-[(3-хлорпиридин-2-ил)амино]пиперидин-1-карбоксилата (3,22 г; 10,3 ммоль) и соединения по Получению 7 5-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиридин-2-карбоновой кислоты (2,37 г; 9,83 ммоль), с получением 2,72 г (52%) продукта, выделенного в виде твердого вещества.
1Н ЯМР (CDCl3) δ: 9.24-9.12 (m, 1Н), 8.92-8.84 (m, 1Н), 8.80 (dd, 1Н), 8.49 (dd, 2Н), 8.32-8.13 (m, 1Н), 7.77-7.58 (m, 1Н), 7.49 (dd, 1H), 7.25-7.16 (m, 1Н), 4.77-4.49 (m, 2Н), 4.45-4.26 (m, 1Н), 3.63-3.32 (m, 1Н), 2.74-2.53 (m, 1Н), 2.48-2.14 (m, 1Н), 2.05-1.96 (m, 1Н), 1.89-1.60 (m, 2Н), 1.54-1.38 (m, 9Н).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,98 мин.
МС (ЭР+): 535,5 (М+Н)+.
Стадия 2: N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-5-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиридин-2-карбоксамид
Получали согласно общей методике D, начиная с соединения со стадии 1 трет-бутил-(3R)-3-[(3-хлорпиридин-2-ил){[5-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиридин-2-ил]карбонил}амино]пиперидин-1-карбоксилата (2,72 г; 5,08 ммоль), с получением 2,35 г (98%) гидрохлорида продукта, выделенного в виде твердого вещества.
1Н ЯМР (DMSO-d6) δ 9.23 (br s, 1Н), 9.02 (br s, 1H), 8.89 (d, 1H), 8.84-8.63 (m, 2H), 8.57 (d, 1H), 8.51-8.41 (m, 1H), 8.19 (d, 1H), 7.96 (d, 1H), 7.66 (dd, 1H), 7.47 (dd, 1H), 5.14-4.51 (m, 1H), 3.61 (m, 1H), 3.53-3.34 (m, 1H), 3.23 (m, 1H), 2.91-2.64 (m, 1H), 2.23-2.07 (m, 1H), 1.98-1.70 (m, 2H), 1.49-1.34 (m, 1H).
Чистота по ЖХВД (ЖХВД, способ 1): 100%, tR составляет 2,880 мин.
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,53 мин.
МС (ЭР+): 436,9 (М+Н)+.
ПРИМЕР 6: этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат
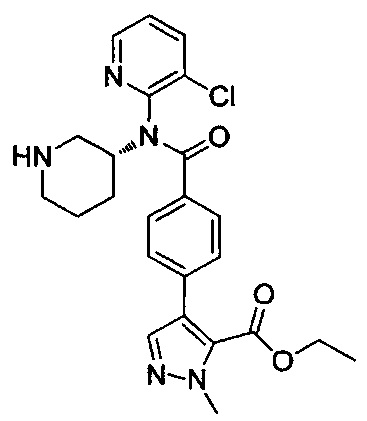
Соединение по Получению 18 (R)-трет-бутил-3-(N-(3-хлорпиридин-2-ил)-4-(5-(этоксикарбонил)-1-метил-1Н-пиразол-4-ил)бензамидо)пиперидин-1-карбоксилат (145,87 г; 256,8 ммоль) растворяли в 1,4 л DCM и затем обрабатывали соляной кислотой (3 моль/л в циклопентилметиловом эфире, 428 мл; 1284 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 19 ч затем добавляли 500 мл DCM с последующим нагреванием до 40°C и последующей фильтрацией через Celite®. Фильтрат концентрировали до общего объема 500 мл под вакуумом и затем доводили до 1,4 л с помощью EtOAc, что приводило к осаждению твердых веществ, которые выделяли посредством фильтрации. После сушки выделили гидрохлорид указанного в заголовке соединения в виде не совсем белого твердого вещества (125,2 г; 97%).
1Н ЯМР (MeOH-d4, 400 МГц) δ 8.57 (br s, 1Н), 7.80 (d, 1Н), 7.48 (s, 1Н), 7.39 (dd, 1Н), 7.32 (d, 2Н), 7.29-7.21 (m, 2Н), 5.49 (s, 1Н), 5.07 (br s, 1Н), 4.18 (q, 2Н), 4.12 (s, 3H), 3.77 (d, 1H), 3.58 (t, 1H), 3.37 (br s, 1H), 2.96-2.82 (m, 1H), 2.45-1.17 (m, 4H), 1.08 (t, 3H).
УЭЖХ (УЭЖХ, способ 4): tR составляет 3,46 мин.
МС (ЭР+) 468,0 (М+Н)+.
Дифракция рентгеновских лучей на порошке для этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилата представлена на Фиг. 2.
ПРИМЕР 7: 4-(4-((3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоновая кислота
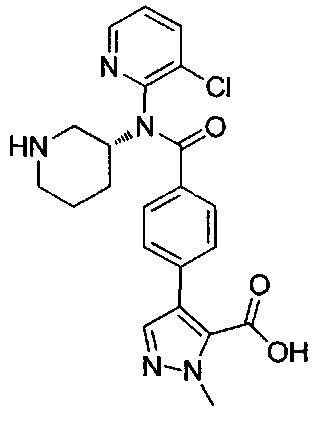
Соединение по Примеру 6 гидрохлорид этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1H-пиразол-5-карбоксилата (7,65 г; 15,2 ммоль) суспендировали в 50 мл THF с последующим добавлением водного NaOH (1 моль/л; 50 мл; 50 ммоль). Добавляли еще 23 мл воды и реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч. Растворитель THF удаляли под вакуумом, затем pH остаточной водной фазы доводили до pH 7 1 М водным HCl, что приводило к осаждению твердых веществ. Твердые вещества собирали посредством фильтрации и после сушки получили гидрохлорид указанного в заголовке соединения в виде не совсем белого твердого вещества (6,49 г; 97%).
1Н ЯМР (400 МГц, DMSO-d6) δ 9.56 (br s, 1Н), 9.12 (d, 1Н), 8.73-8.45 (m, 1Н), 8.06-7.79 (m, 1Н), 7.59 (s, 1Н), 7.43 (dd, 1Н), 7.38-7.11 (m, 4Н), 5.11-4.58 (m, 1Н), 4.03 (s, 3Н), 3.56 (d, 1Н), 3.29 (d, 1Н), 3.18 (d, 1Н), 2.69 (br s, 1H), 2.25-1.18 (m, 4 H).
УЭЖХ (УЭЖХ, способ 4): tR составляет 2,11 мин.
МС (ЭР+) 439,9 (М+Н)+.
ПРИМЕР 8: 1-[(этоксикарбонил)окси]этил-4-(4-((3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат
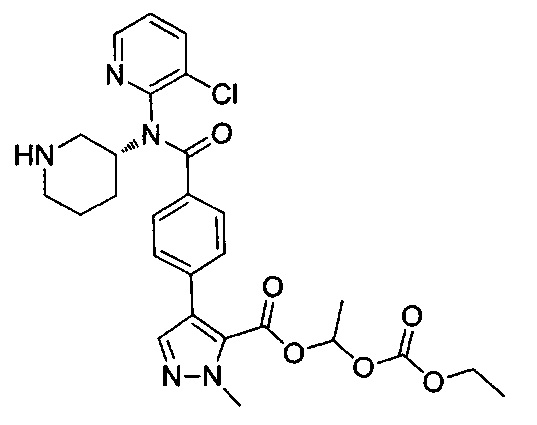
Стадия 1: калиевая соль 4-(4-{[(3R)-1-(трет-бутоксикарбонил)-пиперидин-3-ил](3-хлорпиридин-2-ил)карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоновой кислоты
Общая методика I. Суспензию соединения по Получению 18 трет-бутил-(3R)-3-[(3-хлорпиридин-2-ил){4-[5-(этоксикарбонил)-1-метил-1Н-пиразол-4-ил]-бензоил}амино]пиперидин-1-карбоксилата (34,03 г; 59,9 ммоль) в растворе гидроксида калия в МеОН (1 н; 60 мл; 60 ммоль) нагревали до 85°C в герметично закрытой пробирке на 450 мл для проведения реакций под давлением в течение 1,5 ч. Реакционную смесь затем концентрировали и добавляли в смесь толуол (100 мл). Растворители выпаривали под вакуумом и сушили под глубоким вакуумом в течение 2 ч. Твердое вещество затем суспендировали в метил-трет-бутиловом эфире (150 мл) и гептанах (60 мл) в течение 1 ч. Указанное в заголовке соединение (34,8 г; 100%) получали посредством фильтрации и сушили под глубоким вакуумом в течение 16 ч.
Альтернативное получение:
Соединение по Получению 18 (R)-трет-бутил-3-(N-(3-хлорпиридин-2-ил)-4-(5-(этоксикарбонил)-1-метил-1Н-пиразол-4-ил)бензамидо)пиперидин-1-карбоксилат (62,9 г; 111 ммоль) объединяли с гидроксидом калия (1 моль/л в МеОН; 116 мл; 116 ммоль) и смесь нагревали при температуре дефлегмации и выдерживали в течение 1 ч. Растворитель удаляли под вакуумом и заменяли на толуол с последующим концентрированием до небольшого объема. Добавляли толуол (315 мл) с последующим медленным добавлением гептана (315 мл), что приводило к осаждению твердых веществ. Твердые вещества выделяли посредством фильтрации и сушили с получением указанного в заголовке соединения в виде рыжевато-коричневого твердого вещества (66,0 г). Это вещество могло быть использовано как есть для последующих взаимодействий.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.57 (br s, 1Н), 7.90 (br s, 1Н), 7.55 (d, 2Н), 7.51 (s, 1Н), 7.39 (dd, 1Н), 7.11 (d, 2Н), 4.37 (m, 1Н), 3.15-3.87 (m, 4Н), 3.34 (br s, 3H), 2.55-2.10 (br, m, 2H)1.67 (br s, 2H), 1.41 (br s, 9H).
УЭЖХ-МС (УЭЖХ-МС, способ 1): tR составляет 0,87 мин.
МС (ЭР+): 540,3 (М+Н)+.
Стадия 2: трет-бутил-(3R)-3-[(3-хлорпиридин-2-ил){4-[5-({1-[(этоксикарбонил)окси]этокси}карбонил)-1-метил-1Н-пиразол-4-ил]бензоил}-амино]пиперидин-1-карбоксилат
Общая методика J. К суспензии соединения со стадии 1 калиевой соли 4-(4-{[(3R)-1-(трет-бутоксикарбонил)пиперидин-3-ил](3-хлорпиридин-2-ил)-карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоновой кислоты (51,72 г; 89,46 ммоль) в ацетонитриле (100 мл) по каплям добавляли хлорэтилэтилкарбонат (13,3 мл; 98,5 ммоль) при комнатной температуре в пробирке на 450 мл для проведения реакций под давлением. Реакционную смесь нагревали до 70°C в течение 2 ч или до тех пор, пока посредством ЖХ-МС не было показано израсходование исходных веществ. Хлорид калия отфильтровывали и промывали ацетонитрилом (30 мл). Фильтрат концентрировали и неочищенный продукт очищали посредством комбифлэш-хроматографии, элюируя 0-60% градиентом EtOAc/гептан с получением указанного в заголовке соединения (56,1 г; 96%).
Альтернативное получение:
Соединение со стадии 1 (R)-4-(4-((1-(трет-бутоксикарбонил)пиперидин-3-ил)(3-хлорпиридин-2-ил)карбамоил)фенил)-1-метил-1Н-пиразол-5-карбоксилат калия (62,0 г; 115 ммоль) растворяли в 248 мл ацетонитрила с последующим добавлением 1-хлорэтилэтилкарбоната (21,0 г; 138 ммоль) и смесь нагревали при температуре дефлегмации. Реакционную смесь поддерживали при температуре дефлегмации в течение 6 ч и затем охлаждали до комнатной температуры. После обработки Darco® G60 смесь фильтровали через Celite® и затем концентрировали досуха с получением указанного в заголовке соединения в виде рыжевато-коричневой пены (66,7 г; 88%). Это вещество могло быть кристаллизовано следующим образом: неочищенный продукт (6 г) растворяли в 24 мл iPrOAc, затем медленно добавляли 120 мл гептанов, что приводило к осаждению твердых веществ. Твердые вещества выделяли посредством фильтрации и сушили с получением кристаллизованного указанного в заголовке соединения в виде рыжевато-коричневого твердого вещества (4,5 г; выход 75%).
1Н ЯМР (400 МГц, DMSO-d6) δ 8.57 (br s, 1Н), 7.90 (br s, 1H), 7.66 (s, 1H), 7.41 (m, 1H), 7.24 (br s, 4H), 6.77 (q, 1H), 4.48-4.31 (m, 3H), 4.18 (q, 2H), 4.06 (s, 3H), 3.88 (br s, 2H), 2.14-2.55 (m, 2H), 1.70 (m, 2H), 1.42 (br s, 9H), 1.26 (m, 6H).
УЭЖХ (УЭЖХ, способ 4): tR составляет 7,00 мин.
УЭЖХ-МС (УЭЖХ-МС, способ 2): tR составляет 2,07 мин.
МС (ЭР+): 656,3 (М+Н).
Стадия 3: 1-[(этоксикарбонил)окси]этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат
трет-Бутил-(3R)-3-[(3-хлорпиридин-2-ил){4-[5-({1-[(этоксикарбонил)окси]-этокси}карбонил)-1-метил-1Н-пиразол-4-ил]бензоил}амино]пиперидин-1-карбоксилат, соединение со стадии 2, (45,05 г; 68,66 ммоль) растворяли в DCM (300 мл) и к раствору добавляли HCl в 1,4-диоксане (4,0 М; 172 мл; 687 ммоль) и перемешивали при комнатной температуре в течение 1 ч или до тех пор, пока посредством ЖХ-МС не было показано израсходование исходных веществ. Растворитель удаляли под вакуумом и к остатку добавляли толуол (300 мл×2). Растворитель выпаривали и остаток ресуспендировали в гептане (400 мл×2) и упаривали. Остаток затем растирали с гептаном (500 мл) и перемешивали в гептане в течение 24 ч. Гидрохлорид указанного в заголовке соединения (40,8 г; 100%) получали посредством фильтрации в виде белого порошка.
Альтернативное получение:
(3R)-трет-Бутил-3-(N-(3-хлорпиридин-2-ил)-4-(5-((1-(этоксикарбонилокси)этокси)карбонил)-1-метил-1Н-пиразол-4-ил)бензамидо)пиперидин-1-карбоксилат. Соединение со стадии 2 (6,0 г; 9,1 ммоль) растворяли в 40 мл DCM и затем обрабатывали соляной кислотой (12,1 моль/л в воде; 11 мл; 140 ммоль). Реакционную смесь перемешивали в течение 1 ч при комнатной температуре с последующим добавлением 30 мл воды и разделением фаз. Органическую фазу сушили над MgSO4, фильтровали и фильтрат концентрировали до небольшого объема. Оставшийся DCM заменяли на гептан с получением твердых веществ, которые собирали посредством фильтрации и сушили с получением гидрохлорида указанного в заголовке соединения в виде рыжевато-коричневого твердого вещества (4,95 г; выход 91%).
1Н ЯМР (DMSO-d6) δ 8.58 (s, 1Н), 7.91 (d, 1Н), 7.66 (s, 1Н), 7.44 (m, 1Н), 7.25 (br s, 4Н), 6.77 (q, 1Н), 5.02-4.70 (br m, 1Н), 4.18 (q, 2Н), 4.07 (s, 3H), 3.57-3.26 (m, 4Н), 2.72 (br s, 1Н), 1.67-2.23 (m, 3Н), 1.20-1.34 (m, 6Н).
УЭЖХ-МС (УЭЖХ-МС, способ 2): tR составляет 0,88 мин.
МС (ЭР+): 556,3 (М+Н)+.
ЖХВД (ЖХВД, способ 3): tR составляет 3,66 мин, 99,65%.
Элементный анализ: C27H30ClN5O6 HCl 0,75 H2O. Подсчитано C 53,52; H 5,41; N 11,56; найдено C 53,67; H 5;40; N 11,34.
ПРИМЕР 9: N-(3-хлорпиридин-2-ил)-5-(6-метил-3H-[1,2,3]триазоло[4.5-b]пиридин-3-ил)-N-[(3R)-пиперидин-3-ил]пиридин-2-карбоксамид
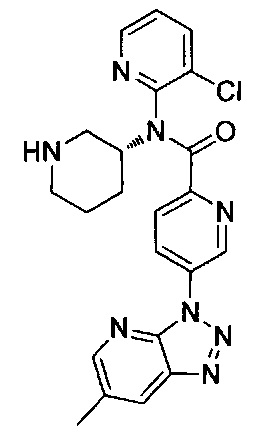
Стадия 1: трет-бутил-(3R)-3-[(3-хлорпиридин-2-ил){[5-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиридин-2-ил]карбонил}амино]пиперидин-1-карбоксилат
Получали согласно общей методике А и С, начиная с соединения по Получению 1 трет-бутил-(3R)-3-[(3-хлорпиридин-2-ил)амино]пиперидин-1-карбоксилата (3,22 г; 10,3 ммоль) и соединения по Получению 8 5-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиридин-2-карбоновой кислоты (2,51 г; 9,83 ммоль), с получением 2,33 г (43%) продукта в виде твердого вещества.
1Н ЯМР (CDCl3) δ 9.27-9.07 (m, 1Н), 8.96-8.78 (m, 1Н), 8.63 (d, 1Н), 8.56-8.37 (m, 1Н), 8.30-8.16 (m, 2Н), 7.79-7.55 (m, 1Н), 7.26-7.12 (m, 1Н), 4.86-4.47 (m, 2Н), 4.45-4.01 (m, 2Н), 3.70-3.29 (m, 1Н), 2.60 (s, 3H), 2.47-1.95 (m, 2H), 1.90-1.55 (m, 2H),1.54-1.33 (s, 9H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 1,02 мин.
МС (ЭР+): 549,4 (М+Н)+.
Стадия 2: N-(3-хлорпиридин-2-ил)-5-(6-метил-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-N-[(3R)-пиперидин-3-ил]пиридин-2-карбоксамид
Получали согласно общей методике D, начиная с соединения со стадии 1 (2,33 г; 4,25 ммоль), с получением 2,03 г (98%) гидрохлорида продукта в виде твердого вещества.
1Н ЯМР (MeOH-d4) δ 9.27-9.09 (m, 1Н), 8.95-8.84 (m, 1Н), 8.72 (s, 1Н), 8.61-8.50 (m, 1Н), 8.49-8.41 (m, 1Н), 8.37 (s, 1Н), 8.22-8.11 (m, 1Н), 8.02-7.80 (m, 1Н), 7.54-7.30 (m, 1Н), 5.17-4.41 (m, 1Н), 3.97-3.60 (m, 2Н), 3.56-3.36 (m, 1Н), 3.10-2.84 (m, 1Н), 2.60 (s, 3H), 2.43-1.80 (m, 3Н), 1.74-1.47 (m, 1Н).
Чистота по ЖХВД (ЖХВД, способ 1): 99,85%, tR составляет 3,215 мин.
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,61 мин.
МС (ЭР+): 449,4 (М+Н)+.
ПРИМЕР 10: метил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат
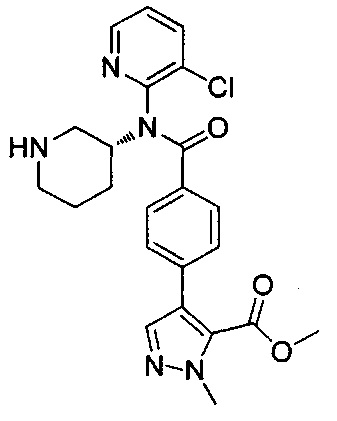
Стадия 1: трет-бутил-3-[(3-хлорпиридин-2-ил){4-[5-(метоксикарбонил)-1-метил-1Н-пиразол-4-ил]бензоил}амино]пиперидин-1-карбоксилат
NaOMe (10,19 мл; 5,09 ммоль; 0,5 М раствор в МеОН) добавляли к суспензии соединения по Получению 18 (2,63 г; 4,6 ммоль) в МеОН (20 мл). Полученную смесь нагревали при температуре дефлегмации в течение 2 ч перед концентрированием под вакуумом с получением смеси метилового эфира в качестве основного компонента с соответствующей кислотой в качестве минорного компонента. Эту неочищенную смесь обрабатывали K2CO3 (320 мг; 2,32 ммоль) и Mel (144 мкл; 2,32 ммоль) в ацетонитриле (20 мл) при температуре дефлегмации в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и экстрагировали EtOAc (3×70 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и фильтрат концентрировали. Остаток очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом 0-70% EtOAc/гептан, с получением указанного в заголовке соединения в виде белого твердого вещества (2,23 г; выход 87%).
1Н ЯМР (CDCl3) δ 8.48 (m, 1Н), 7.59 (m, 1Н), 7.44 (s, 1Н), 7.37-7.25 (m, 2Н), 7.17 (m, 3Н), 4.77-4.27 (m, 2Н), 4.26-3.92 (m, 4Н), 3.70 (s, 3H), 3.35 (m, 1Н), 2.58 (m, 1Н), 2.04-1.64 (m, 2Н), 1.56-1.07 (m, 11Н).
УЭЖХ (УЭЖХ-МС, способ 2), tR составляет 1,86 мин.
МС (ЭР+): 554,3 (М+Н)+.
Стадия 2: метил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]-карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат
Получали согласно общей методике D, начиная с соединения со стадии 2 трет-бутил-3-[(3-хлорпиридин-2-ил){4-[5-(метоксикарбонил)-1-метил-1Н-пиразол-4-ил]бензоил}амино]пиперидин-1-карбоксилата (2,22 г; 4,0 ммоль), с получением 1,82 г (93%) гидрохлорида продукта в виде твердого вещества.
1Н ЯМР (DMSO-d6) δ 9.43-9.12 (m, 1Н), 9.10-8.86 (m, 1Н), 8.68-8.38 (m, 1Н), 7.95 (d, 1Н), 7.65 (s, 1Н), 7.46 (dd, 1Н), 7.24 (m, 4Н), 5.10-4.60 (m, 1Н), 4.05 (s, 3H), 3.78-3.60 (m, 4Н), 3.20-2.90 (m, 2Н), 2.73 (m, 1Н), 2.11-1.84 (m, 3Н), 1.26 (m, 1Н).
Чистота по ЖХВД (ЖХВД, способ 3): 99,44%, tR составляет 2,993 мин.
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,54 мин.
МС (ЭР+): 454,3 (М+Н)+.
ПРИМЕР 11: 1-[(этоксикарбонил)окси]этил-4-(4-{изохинолин-1-ил[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат
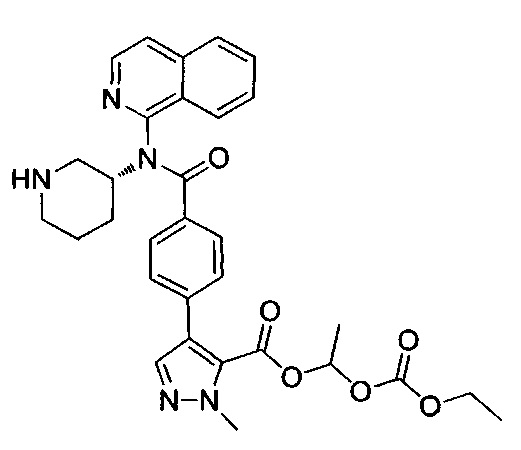
К суспензии соединения по ПРИМЕРУ 4, стадия 1, 4-(4-{изохинолин-1-ил[трет-бутил-(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоновой кислоты (96 мг; 0,17 ммоль) в ацетонитриле (2 мл) добавляли K2CO3 (36 мг; 0,26 ммоль) и хлорэтилэтилкарбонат (46 мкл; 0,35 ммоль). Смесь перемешивали при 65°C в герметичной пробирке в течение 16 ч. Реакционную смесь затем фильтровали через Celite® и концентрировали. Остаток растворяли в DCM (1 мл) и добавляли HCl в 1,4-диоксане (4 М; 1 мл; 4 ммоль). Смесь перемешивали при комнатной температуре в течение 30 мин, затем концентрировали. Остаток сушили под глубоким вакуумом в течение 2 ч, затем суспендировали МТВЕ (1 мл) в течение 16 ч. Гидрохлорид указанного в заголовке соединения (107 мг; 99%) получали посредством фильтрации.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.51 (t, 1Н), 8.05-7.53 (m, 6Н), 7.19 (d, 2Н), 7.08 (d, 2Н), 6.68 (m, 1Н), 5.15 (m, 1Н), 4.17 (q, 2Н), 4.01 (s, 3H), 3.74-3.18 (m, 4Н), 2.68 (br s, 1Н), 2.34-1.78 (m, 3Н), 1.26-1.12 (m, 6Н).
УЭЖХ-МС (УЭЖХ-МС, способ 1): tR составляет 0,66 мин.
MS(ES+): 572,5 (М+Н)+.
ПРИМЕР 12: 1-метил-4-(4-{(1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-карбоновая кислота
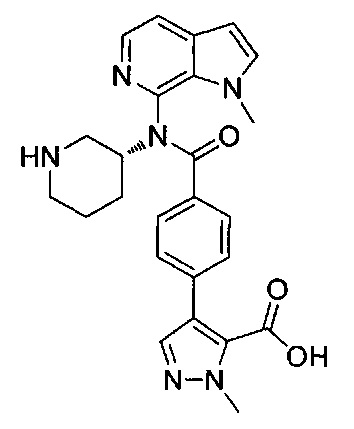
Стадия 1: (R)-трет-бутил-3-(N-(1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензамидо)пиперидин-1-карбоксилат
В раствор соединения по Получению 11 (R)-трет-бутил-3-(4-бром-N-(1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)бензамидо)-пиперидин-1-карбоксилата (0,35 г; 0,68 ммоль) в 1,4-диоксане (10 мл) добавляли бис(пинаколато)дибор (0,35 г; 1,38 ммоль), Pd(dppf)Cl2 (0,05 г; 0,068 ммоль) и КОАс (0,200 г; 2,04 ммоль). Реакционную смесь нагревали при 85°C в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении с получением указанного в заголовке соединения (0,5 г), которое использовали на следующей стадии без очистки.
Стадия 2: (R)-трет-бутил-3-(4-(5-(этоксикарбонил)-1-метил-1Н-пиразол-4-ил)-N-(1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)бензамидо)пиперидин-1-карбоксилат
В раствор соединения со стадии 1 (R)-трет-бутил-3-(N-(1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензамидо)пиперидин-1-карбоксилата (0,5 г; 0,89 ммоль) в 1,4-диоксане (10 мл) добавляли этил-4-бром-1-метил-1Н-пиразол-5-карбоксилат (0,21 г; 0,90 ммоль), Pd(dppf)Cl2 (0,065 г; 0,089 ммоль) и Cs2CO3 (0,58 г; 1,78 ммоль). Реакционную смесь нагревали при 85°C в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении. Полученный неочищенный продукт очищали посредством хроматографии на силикагеле, элюируя градиентом петролейный эфир: EtOAc (от 5:1 до 0:1), с получением указанного в заголовке соединения (0,45 г; 86%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6, Т составляет 80°C) δ 8.09 (d, 0.6Н), 8.07 (d, 0.4Н), 7.54-7.38 (m, 3H), 7.30 (m, 2Н), 7.19 (m, 2Н), 6.46 (d, 0.4Н), 6.44 (d, 0.6H), 4.13-4.07 (m, 2H), 4.03 (s, 3H), 3.94-3.90 (m, 1H), 3.89 (s, 3H), 2.60-2.50 (m, 2H), 1.82-1.48 (m, 3H), 1.45 (s, 6H), 1.38 (s, 3H), 1.03-0.93 (m, 3H).
ЖХ (ЖХ-МС, способ 3): tR составляет 1,12 мин.
МС (ЭР+): 359,1 (M+H)+.
Стадия 3: (R)-4-(4-((1-(трет-бутоксикарбонил)пиперидин-3-ил)(1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)карбамоил)фенил)-1-метил-1Н-пиразол-5-карбоновая кислота
В раствор соединения со стадии 2 (R)-трет-бутил-3-(4-(5-(этоксикарбонил)-1-метил-1Н-пиразол-4-ил)-N-(1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)бензамидо)пиперидин-1-карбоксилата (0,2 г; 0,34 ммоль) в МеОН (10 мл) добавляли водный NaOH (2 н.; 1,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель удаляли при пониженном давлении и pH доводили 1 н. HCl до значения pH 4. Подкисленную смесь экстрагировали EtOAc (3×10 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (0,18 г) в виде желтого масла, которое использовали на следующей стадии без очистки.
Стадия 4: гидрохлорид (R)-1-метил-4-(4-((1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)(пиперидин-3-ил)карбамоил)фенил)-1Н-пиразол-5-карбоновой кислоты
В раствор соединения со стадии 3 (R)-4-(4-((1-(трет-бутоксикарбонил)пиперидин-3-ил)(1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)-карбамоил)фенил)-1-метил-1Н-пиразол-5-карбоновой кислоты (0,18 г; 0,17 ммоль) в 1,4-диоксане (10 мл) добавляли HCl в 1,4-диоксане (10 мл; 4 н.). Смесь перемешивали при комнатной температуре в течение 1 ч. Смесь концентрировали под вакуумом. Полученный остаток растирали с EtOAc с получением гидрохлорида указанного в заголовке соединения (0,12 г; 70% за 2 стадии) в виде коричневого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6), Т составляет 80°C) δ 9.38 (br s, 1Н), 9.11 (br s, 1Н), 8.11 (d, 0.75Н), 8.08 (d, 0.25H), 7.57 (d, 1H), 7.52-7.46 (m, 2H), 7.28-7.17 (m, 4H), 6.61 (d, 0.25H), 6.49 (d, 0.75H), 4.02 (s, 3H), 3.98-3.94 (m, 1H), 3.90 (s, 3H), 3.76-3.67 (m, 1H), 3.61-3.50 (m, 1H), 3.28-3.15 (m, 1H), 2.86-2.64 (m, 2H), 1.90-1.69 (m, 3H).
ЖХ (ЖХ-МС, способ 3): tR составляет 0,937 мин.
МС (ЭР+): 459,0 (M+H)+.
ПРИМЕР 13: метил-1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-карбоксилат
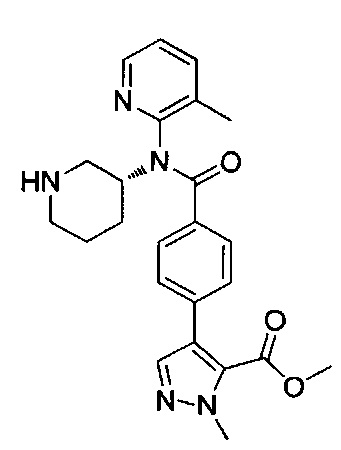
Стадия 1: трет-бутил-(3R)-3-[{4-[5-(метоксикарбонил)-1-метил-1Н-пиразол-4-ил]бензоил}(3-метилпиридин-2-ил)амино]пиперидин-1-карбоксилат
В раствор метилового эфира 4-йод-2-метил-2Н-пиразол-3-карбоновой кислоты (8,1 г; 30,45 ммоль) в THF (безводный, 60 мл) добавляли хлорид изопропилмагния (16,5 мл; 16,5 ммоль) при -45°C. Раствор становился из прозрачного светло-зелено-желтоватым. Перемешивали при этой температуре в течение 30 мин, анализ ГХ-МС (газовая хроматография-масс-спектрометрия) (образец в CD3OD) показал, что обмен галоген-металл полностью завершен. К смеси по каплям добавляли 1,9 н. ZnCl2 в 2-MeTHF (9,62 мл; 18,3 ммоль) и реакционную смесь нагревали до комнатной температуры. Смесь перемешивали в течение 2 ч, за это время она превращалась из прозрачной желтоватой в непрозрачную желтоватую смесь. В эту реакционную смесь добавляли одной порцией соединение по Получению 2 трет-бутил-3-[(4-бромбензоил)(3-метилпиридин-2-ил)амино]пиперидин-1-карбоксилат (12,04 г; 25,37 ммоль) в виде твердого вещества. Температуру реакции затем повышали до 50°C, добавляли дихлорид 1'-бис(ди-трет-бутилфосфино)ферроценпалладия (166 мг; 0,254 ммоль). Реакционную смесь затем перемешивали при 60°C в течение 2 ч, затем охлаждали до комнатной температуры. Реакционную смесь гасили водой и экстрагировали 3×EtOAC. Органический слой сушили и концентрировали. Очистку проводили с помощью колонки ISCO CombiFlash (220 г) 0-75% EtOAc/гептан с выделением требуемого указанного в заголовке соединения (12,8 г; 78%).
1Н ЯМР (DMSO-d6) δ 8.45 (br s, 1Н), 7.54-7.67 (m, 3H), 7.11-7.33 (m, 5Н), 4.27-4.54 (m, 1Н), 4.05 (s, 4Н), 3.80-3.96 (m, 1Н), 3.65 (s, 4Н), 1.92-2.14 (m, 4Н), 1.34-1.56 (m, 13Н).
УЭЖХ-МС (УЭЖХ-МС, способ 1): tR составляет 0,96 мин.
МС (ЭР+): 534,5 (М+Н)+.
Стадия 2: метил-1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-карбоксилат
К соединению, полученному на стадии 1 (31,45 мг; 58,94 ммоль), в МеОН (100 мл) добавляли HCl в 1,4-диоксане (147 мл; 589 ммоль) и перемешивали при комнатной температуре в течение 30 мин. Растворитель удаляли под вакуумом с получением твердого вещества, которое сушили под глубоким вакуумом в течение 2 ч. Добавляли EtOAc (700 мл) и МеОН (20 мл) и смесь перемешивали при комнатной температуре в течение 16 ч. Указанное в заголовке соединение получали посредством фильтрации в виде дигидрохлорида (29,8 г; 99,8%).
1Н ЯМР (DMSO-d6) δ 9.07-9.40 (br m, 1Н), 8.35-8.53 (m, 1Н), 7.55-7.72 (m, 2Н), 7.28-7.37 (m, 1Н), 7.20 (d, 4Н), 4.96 (br m, 1H), 4.05 (s, 3H), 3.65 (s, 3H), 3.59 (d, 1H), 3.39 (d, 1H), 3.20 (d, 1H), 2.68 (br s, 1H), 2.00 (s, 3H), 1.80 (br s, 4H).
УЭЖХ-МС (УЭЖХ-МС, способ 1): tR составляет 0,52 мин.
МС (ЭР+): 434,4 (М+Н)+.
ЖХВД (ЖХВД, способ 2): tR составляет 2,17 мин, 99,88%.
Дифракция рентгеновских лучей на порошке для метил-1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-карбоксилата представлена на Фиг. 3.
ПРИМЕР 14: 1-[(этоксикарбонил)окси]этил-1-метил-4-(4-{(1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-карбоксилат
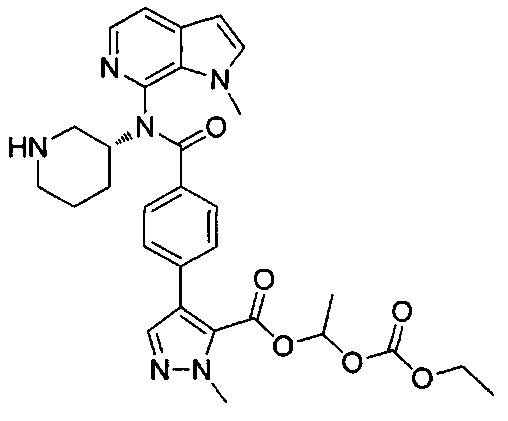
Указанное в заголовке соединение получали с использованием общих методик I, J и D.
1Н ЯМР (DMSO-d6) δ 8.07-8.17 (m, 1Н), 7.58 (br s, 2H), 7.52 (br s, 1H), 7.21 (br s, 2H), 7.09-7.17 (m, 2H), 6.71 (d, 1H), 6.47 (br s, 1H), 4.18 (q, 2H), 4.03 (s, 3H), 3.81-3.96 (m, 3H), 3.63-3.76 (m, 1H), 3.57 (s, 3H), 3.49 (m, 1H), 3.21 (d, 1H), 2.68-2.75 (m, 1H), 1.67-1.84 (m, 2H), 1.22-1.31 (m, 3H), 1.06-1.17 (m, 3H).
УЭЖХ-МС (УЭЖХ-МС, способ 1): tR составляет 0,63 мин.
МС (ЭР+): 575,3 (М+Н)+.
ПРИМЕР 15а и 15b
15а: (1S)-1-[(этоксикарбонил)окси]этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат
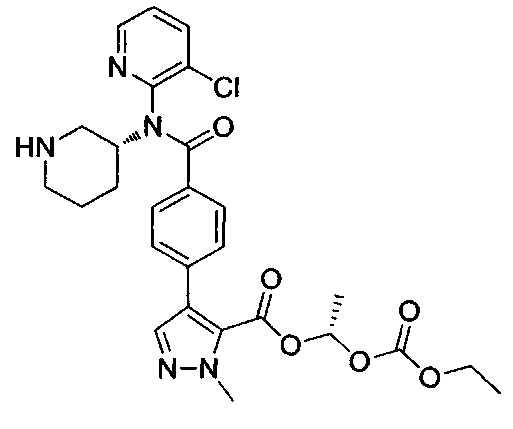
15b: (1R)-1-[(этоксикарбонил)окси]этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат
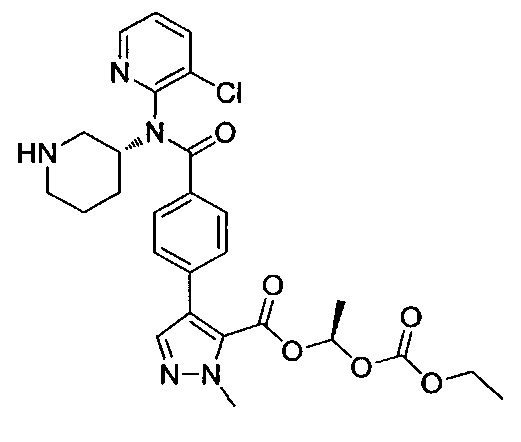
Стадия 1:
50 г соединения по ПРИМЕРУ 8, стадия 2, (3R)-трет-бутил-3-(N-(3-хлорпиридин-2-ил)-4-(5-((1-(этоксикарбонил-окси)этокси)карбонил)-1-метил-1Н-пиразол-4-ил)бензамидо)пиперидин-1-карбоксилата, подвергали обработке с помощью хиральной препаративной хроматографии, способ 1, с последующим концентрированием каждого диастереомера досуха под вакуумом с получением изомера 1 (20,3 г; выход 81%, 100% е.е.; tR составляет 6,6 мин) и изомера 2 (22,0 г; выход 88%, 98,5% е.е; tR составляет 6,9 мин).
Каждый изомер отдельно друг от друга подвергали снятию защиты, как описано ниже.
Стадия 2а:
В раствор изомера 1 (566 мг; 0,863 ммоль) в DCM (3 мл) добавляли 4 М HCl в 1,4-диоксане (3 мл; 12 ммоль) и перемешивали при комнатной температуре в течение 30 мин. Растворитель выпаривали и остаток суспендировали в МТВЕ (10 мл) и гептане (10 мл) в течение 16 ч. Гидрохлорид по Примеру 15а (511 мг; 99%) получали посредством фильтрации с получением белого аморфного порошка.
ПРИМЕР 15а
1Н ЯМР (DMSO-d6) δ 8.45-8.71 (m, 1Н), 7.92 (d, 1Н), 7.67 (br s, 1Н), 7.45 (m, 1Н), 7.25 (br s, 4Н), 6.77 (d, 1Н), 5.02 (br s, 1H), 4.18 (q, 2H), 4.07 (s, 3H), 3.59 (br s, 2H), 3.21 (d, 2H), 2.73 (br s, 1H), 1.75-2.38 (m, 3H), 1.25 (m, 6H).
УЭЖХ-МС (УЭЖХ-МС, способ 1): tR составляет 0,63 мин.
МС (ЭР+): 556,4 (М+Н)+.
Оптическое вращение: [α]20 составляет -107,3° (с составляет 0,87 г/дл, ацетонитрил).
Стадия 2b:
В раствор изомера 2 (2,8 г; 4,3 ммоль) в DCM (30 мл) добавляли 4 М HCl в 1,4-диоксане (10 мл; 40 ммоль) и перемешивали при комнатной температуре в течение 30 мин. Растворитель затем выпаривали и остаток суспендировали в МТВЕ (10 мл) и гептане (10 мл) в течение 16 ч. Гидрохлорид по Примеру 15b (2,5 г; 99%) получали посредством фильтрации в виде белого аморфного порошка. Аморфный порошок суспендировали в смеси ацетонитрила (20 мл), DCM (10 мл) и МТВЕ (300 мл) и смесь нагревали при температуре дефлегмации в течение 48 ч. Кристаллическое соединение в виде гидрохлорида (2,2 г; 87%) получали посредством фильтрации.
ПРИМЕР 15b
1Н ЯМР (DMSO-d6) δ 8.50-8.66 (m, 1Н), 7.92 (d, 1Н), 7.66 (s, 1Н), 7.40-7.51 (m, 1Н), 7.25 (br s, 4Н), 6.77 (d, 1Н), 5.00 (br s, 1Н), 4.18 (q, 2Н), 4.07 (s, 3H), 3.60 (br s, 2Н), 3.21 (d, 2Н), 2.74 (br s, 1Н), 1.67-2.23 (m, 3Н), 1.20-1.34 (m, 6Н).
УЭЖХ-МС (УЭЖХ-МС, способ 1): tR составляет 0,64 мин.
МС (ЭР+): 556,4 (М+Н)+.
Оптическое вращение: [α]20 составляет -25,0° (с составляет 0,81 г/дл, ацетонитрил).
Элементный анализ: подсчитано для C27H31Cl2N5O6: C 54,74; H 5,27; N 11,82; Cl 11,97; найдено C 54,53; H 5,11; N 11,72; Cl 11,72.
Дифракция рентгеновских лучей на порошке для (1R)-1-[(этоксикарбонил)окси]этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилата представлена на Фиг. 4.
На Фиг. 5 представлена диаграмма ORTEP (1R)-1-[(этоксикарбонил)окси]этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилата.
Рентгеноструктурный анализ монокристаллов для (1R)-1-[(этоксикарбонил)окси]этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилата: Сбор данных проводили на дифрактометре Bruker APEX при комнатной температуре. Сбор данных состоял в омега- и фи-сканировании. Структура определена прямыми методами с использованием пакета программ SHELX в пространственной группе P212121. Структуру затем уточняли методом наименьших квадратов в полноматричном приближении. Все неводородные атомы были обнаружены и уточнены с использованием параметров анизотропного замещения. Водородные атомы, расположенные на азоте, были обнаружены по разностной карте Фурье и уточнены с помощью ограниченных расстояний. Остальные водородные атомы помещали в вычисленные положения и вращали относительно их несущих атомов. Конечное уточнение включало параметры изотропного замещения для всех атомов водорода. В этом случае абсолютную конфигурацию определяли путем проверки параметра Флэка. Здесь параметр равен 0,0308 с ESD 0,0138; в пределах определения абсолютной конфигурации. Конечный R-фактор составлял 3,8%. Конечная разностная карта Фурье не выявила пропусков или смещения электронной плотности.
Кристаллографические данные, условия сбора данных и параметры уточнения сведены в таблицу 3.
Пример 16: N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензамид
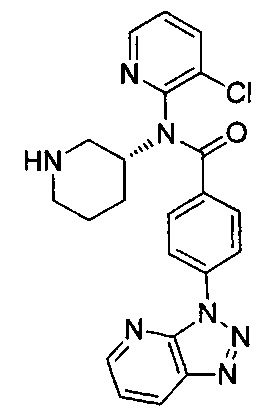
Стадия 1: трет-бутил-(3R)-3-(4-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-N-(3-хлорпиридин-2-ил)бензамидо)пиперидин-1-карбоксилат
Получали согласно общей методике А и С, начиная с соединения по Получению 1 трет-бутил-(3R)-3-[(3-хлорпиридин-2-ил)амино]пиперидин-1-карбоксилата (26,7 г; 85,5 ммоль) и соединения по Получению 5 4-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензойной кислоты (20,1 г; 83,5 ммоль), с получением 35,2 г (79%) продукта в виде твердого вещества.
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 0,75 мин.
МС (ЭР+): 534,3 (М+Н)+.
Стадия 2: N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензамид
Получали согласно общей методике D, начиная с соединения со стадии 1 (33,0 г; 61,8 ммоль), с получением 28,5 г (98%) гидрохлорида продукта в виде твердого вещества.
1Н ЯМР (CDCl3) δ 10.06 (br s, 1Н), 9.78 (br d, 1Н), 8.73 (d, 1Н), 8.35-8.57 (m, 2Н), 8.23 (d, 2Н), 7.49-7.68 (m, 3H), 7.42 (m, 1Н), 7.20 (m, 1Н), 5.14 (br s, 1Н), 3.71-4.07 (m, 2Н), 3.58 (d, 1Н), 2.86 (d, 1Н), 1.67-2.48 (m, 4Н).
Чистота по ЖХВД (аналитическая ЖХВД, способ 1): 99,52%, tR составляет 3,048 мин.
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 0,84 мин.
МС (ЭР+): 434,2 (М+Н)+.
Дифракция рентгеновских лучей на порошке для N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензамида представлена на Фиг. 6.
На Фиг. 7 представлена диаграмма ORTEP N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензамида.
Рентгеноструктурный анализ монокристаллов N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензамида: Сбор данных проводили на дифрактометре Bruker APEX при комнатной температуре. Сбор данных состоял в 3 омега-сканированиях под малым углом и трех под большим углом; каждый с шагом 0,5. Кроме того, проводили 2 фи-сканирования для улучшения качества коррекции поглощения.
Структура определена прямыми методами с использованием пакета программ SHELX в пространственной группе Р1. Структуру затем уточняли с помощью метода наименьших квадратов в полноматричном приближении. Все неводородные атомы были обнаружены и уточнены с использованием параметров анизотропного замещения.
Две молекулы в ассиметричной ячейке, обе с одной и той же стереохимией. Псевдосимметрия (Р-1).
Все водородные атомы помещали в вычисленные положения и позволяли вращаться относительно их несущих атомов. Конечное уточнение включало параметры изотропного замещения для всех атомов водорода.
Анализ абсолютной структуры с использованием методов правдоподобия (Hooft 2008) проводили с использованием программы PLATON (Spek 2010). Результаты показывают, что абсолютная структура определена правильно. С помощью этого метода вычислено, что вероятность того, что структура верна, составляет 100,0. Параметр Хуфта установлен как 0,06 с ESD 0,019.
Конечный R-фактор составлял 3,8%. Конечная разностная карта Фурье не выявила пропусков или смещения электронной плотности.
Кристаллографические данные, условия сбора данных и параметры уточнения сведены в таблицу 4.
ПРИМЕР 17: N-(3-метилпиридин-2-ил)-4-[1-метил-5-(2Н-тетразол-5-ил)-1Н-пиразол-4-ил]-N-[(3R)-пиперидин-3-ил]бензамид
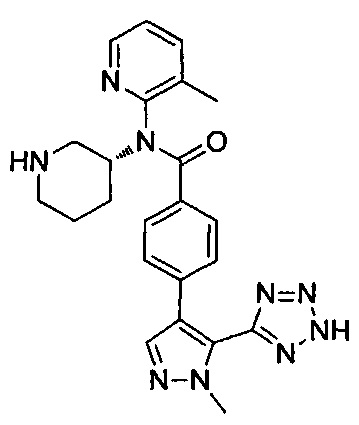
Соединение по Получению 25 трет-бутил-(3R)-3-(4-(1-метил-5-(2Н-тетразол-5-ил)-1H-пиразол-4-ил)-N-(3-метилпиридин-2-ил)бензамидо)-пиперидин-1-карбоксилат (100 мг; 0,185 ммоль) растворяли в метаноле (0,925 мл). Туда добавляли HCI в диоксане (0,694 мл; 4 М раствор; 2,78 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре. Через 2 ч реакционную смесь концентрировали при пониженном давлении и затем концентрировали с толуолом (3×10 мл). Неочищенное вещество растирали с EtOAc/МеОН (20:1) и фильтровали с получением N-(3-метилпиридин-2-ил)-4-[1-метил-5-(2Н-тетразол-5-ил)-1Н-пиразол-4-ил]-N-[(3R)-пиперидин-3-ил]бензамида (89,3 мг; 94%).
1Н ЯМР (МеОН-d4) δ: 8.44 (d, 1Н), 7.78 (s, 1Н), 7.65-7.52 (m, 1Н), 7.34 7.30 (m, 1Н), 7.20-7.13 (m, 2Н), 7.07-7.02 (m, 2Н), 5.12-5.00 (m, 1Н), 4.61-4.48 (m, 1 Н), 3.75-3.71 (m, 1Н), 3.67 (s, 3H), 3.63-3.51 (m, 1Н), 2.99-2.82 (m, 1Н), 2.36-2.23 (m, 1Н), 2.11-2.05 (m, 1Н), 2.00 (s, 3H), 1.97-1.91 (m, 1Н), 1.85-1.80 (m, 2Н), 1.42-1.31 (m, 1Н).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,48 мин.
МС (ЭР+): 444,4 (М+Н)+.
ПРИМЕР 18: 4-[1-метил-5-(5-оксо-4,5-дигидро-1,2,4-оксадиазол-3-ил)-1Н-пиразол-4-ил]-N-(3-метилпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]бензамид
Стадия 1: в раствор соединения по Получению 26 трет-бутил-(3R)-3-{[4-(5-циано-1-метил-1Н-пиразол-4-ил)бензоил](3-метилпиридин-2-ил)амино}-пиперидин-1-карбоксилата (130 мг; 0,26 ммоль) в EtOH (1,2 мл) добавляли NH2OH (70 мг; 1,04 ммоль; 50% (мас./мас.)) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 24 ч. Смесь затем разбавляли водой (30 мл) и экстрагировали DCM (3×30 мл). Объединенные органические слои промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением белого твердого вещества (138 мг), которое использовали без дополнительной очистки.
Стадия 2: в раствор белого твердого вещества (138 мг) в THF (5 мл) добавляли 1,1'-карбонилдиимидазол (64 мг; 0,39 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч и затем концентрировали при пониженном давлении с получением остатка. Полученный остаток растворяли в EtOAc (20 мл) и промывали 1 М NaOH (3×40 мл). Водный слой разбавляли DCM (40 мл), осторожно подкисляли до рН 3 с помощью 1 М HCl при 0°C. Водный слой затем экстрагировали DCM (2×40 мл). Объединенные органические слои промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали с получением желтого масла (138 мг), которое использовали без дополнительной очистки.
Стадия 3: в раствор желтого масла (138 мг) в DCM (5 мл) добавляли HCl в EtOAc (5 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 1 ч и затем концентрировали под вакуумом. Остаток очищали посредством препаративной ЖХВД с получением HCl соли 4-[1-метил-5-(5-оксо-4,5-дигидро-1,2,4-оксадиазол-3-ил)-1Н-пиразол-4-ил]-N-(3-метилпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]бензамида в виде белого твердого вещества (55 мг; 43%). (Условия препаративной ЖХВД: колонка: Agela Durashell С18 (250×21,2 мм×5 мкм); подвижная фаза: градиент от 8% ацетонитрила в H2O (0,1% HCl) до 28% ацетонитрила в Н2O (0,1% HCl)).
1Н ЯМР (DMSO-d6) δ: 9.48 (br s, 1Н), 9.14 (br s, 1H), 8.45 (br s, 0.8H), 8.40 (br s, 0.2H), 7.93 (s, 1H), 7.67-7.53 (m, 1H), 7.36-7.13 (m, 5H), 5.00-4.86 (m, 0.8H), 4.67-4.53 (m, 0.2H), 3.92 (s, 3H), 3.59-3.49 (m, 1H), 3.43-3.29 (m, 1H), 3.23-3.13 (m, 1Н), 2.78-2.63 (m, 1H), 2.07 (s, 0.7H), 1.99 (s, 2.3H), 1.85-1.71 (m, 3H), 1.33-1.16 (m, 1H).
ЖХ (ЖХ-МС способ 4): tR составляет 1,02 мин.
МС (ЭР+): 460,1 (M+H)+.
ПРИМЕР 19: N-(3-хлорпиридин-2-ил)-4-[1-метил-5-(5-оксо-4,5-дигидро-1,2,4-оксадиазол-3-ил)-1Н-пиразол-4-ил]-N-[(3R)-пиперидин-3-ил]бензамид
Указанное в заголовке соединение получали способом, аналогичным способу по Примеру 18. (Условия препаративной ЖХВД: колонка: Agela Durashell С18 (250×21,2 мм×5 мкм); подвижная фаза: градиент от 13% ацетонитрила в H2O (0,1% HCl) до 33% ацетонитрила в H2O (0,1% HCl)).
1Н ЯМР (MeOH-d4) δ: 8.57 (br s, 0.7Н), 8.53 (br s, 0.3H), 7.83-7.77 (m, 2H), 7.42-7.33 (m, 3H), 7.29-7.23 (m, 2H), 5.12-5.03 (m, 1H), 4.00 (s, 3H), 3.82-3.71 (m, 1H), 3.63-3.54 (m, 1H), 3.41-3.36 (m, 1H), 2.96-2.85 (m, 1H), 2.39-2.30 (m, 0.3H), 2.15-1.83 (m, 3H), 1.54-1.42 (m, 0.7H).
ЖХ (ЖХ-МС, способ 4): tR составляет 1,01 мин.
МС (ЭР+): 480,1 (М+Н)+.
ПРИМЕР 20: этил-1-{3-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-5-оксо-1,2,4-оксадиазол-4(5Н)-ил}этил карбонат
Стадия 1: трет-бутил-(3R)-3-[{4-[5-(4-{1-[(этоксикарбонил)окси]этил}-5-оксо-4,5-дигидро-1,2,4-оксадиазол-3-ил)-1-метил-1Н-пиразол-4-ил]бензоил}(3-метилпиридин-2-ил)амино]пиперидин-1-карбоксилат
В раствор продукта стадии 2 примера 18 (133 мг; 0,23 ммоль) в THF (2 мл) добавляли 1-трет-бутил-2,2,4,4,4-пентакис(диметиламино)-2λ5,4λ5-катенади(фосфазен) (240 мкл; 2 М раствор в THF; 0,48 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч с последующим добавлением 1-хлорэтилэтилкарбоната (105 мкл; 0,78 ммоль). Реакционную смесь нагревали до 60°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли насыщенным водным NH4Cl (30 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические фазы сушили над MgSO4 и концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом 0-100% EtOAc/гептан, с получением трет-бутил-(3R)-3-[{4-[5-(4-{1-[(этоксикарбонил)окси]этил}-5-оксо-4,5-дигидро-1,2,4-оксадиазол-3-ил)-1-метил-1Н-пиразол-4-ил]бензоил}(3-метилпиридин-2-ил)амино]пиперидин-1-карбоксилата в виде белого твердого вещества (56 мг; 35%).
1Н ЯМР (CDCl3) δ: 8.41 (br s, 1Н), 7.68 (s, 1Н), 7.46-7.28 (m, 3Н), 7.19-6.96 (m, 3Н), 5.24 (br s, 1Н), 4.77-4.32 (m, 2Н), 4.11 (q, 2Н), 3.97 (s, 3H), 3.40 (d, 1Н), 2.72-2.25 (m, 2Н), 2.16 (d, 1Н), 2.07 (s, 3H), 1.90-1.54 (m, 3Н), 1.47-1.43 (m, 9Н), 1.28-1.18 (m, 3H), 0.99-0.73 (m, 3Н).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 1,00 мин.
МС (ЭР+): 676,4 (М+Н)+.
Стадия 2: трет-бутил-(3R)-3-[{4-[5-(4-{1-[(этоксикарбонил)окси]этил}-5-оксо-4,5-дигидро-1,2,4-оксадиазол-3-ил)-1-метил-1Н-пиразол-4-ил]бензоил}(3-метилпиридин-2-ил)амино]пиперидин-1-карбоксилат (52 мг; 0,08 ммоль) растворяли в DCM (5 мл) и к раствору добавляли HCl в диоксане (0,5 мл; 4 М раствор; 2 ммоль) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали под вакуумом. Полученный остаток растирали в диэтиловом эфире с получением этил-1-{3-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-5-оксо-1,2,4-оксадиазол-4(5Н)-ил}этилкарбоната в виде светло-желтого твердого вещества (40 мг; 80%).
1Н ЯМР (CD3CN) δ: 9.87-9.59 (m, 1Н), 9.52-9.33 (m, 1Н), 9.33-9.02 (m, 1Н), 8.40 (d, 1Н), 7.99-7.75 (m, 2Н), 7.41 (br s, 3H), 7.19 (br s, 2H), 5.52-5.09 (m, 1H), 5.12-4.46 (m, 1H), 4.01 (q, 2H), 3.90 (s, 3H), 3.71-3.55 (m, 1H), 3.31 (d, 1H), 2.82 (br s, 1H), 2.37-2.10 (m, 2H), 1.99-1.94 (m, 1H), 1.92 (s, 3H), 1.85-1.71 (m, 1H), 1.60-1.37 (m, 1H), 1.15 (t, 3H), 0.91-0.78 (m, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,65 мин.
МС (ЭР+): 576,3 (М+Н)+.
ПРИМЕР 21: этил-1-{3-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-5-оксо-1,2,4-оксадиазол-4(5Н)-ил}этил карбонат
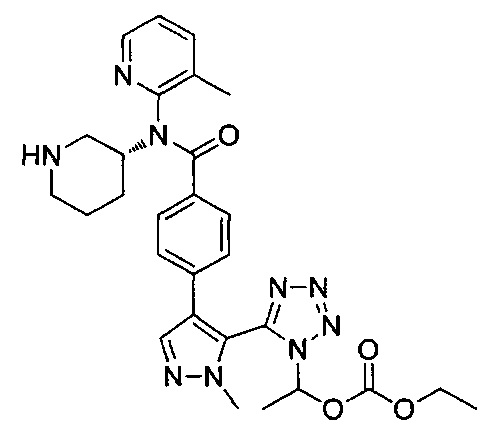
К соединению по Получению 25 трет-бутил-(3R)-3-(4-(1-метил-5-(2H-тетразол-5-ил)-1H-пиразол-4-ил)-N-(3-метилпиридин-2-ил)бензамидо)-пиперидин-1-карбоксилату (310 мг; 0,57 ммоль) в DMF (1 мл) добавляли DIPEA (1 мл; 5,7 ммоль) с последующим добавлением 1-хлорэтилэтилкарбоната (0,46 мл; 3,4 ммоль). Реакционную смесь нагревали при 60°C в течение 16 ч. Реакционную смесь гасили насыщенным водным раствором NH4Cl (10 мл) и экстрагировали EtOAc (2×20 мл). Органические фазы объединяли и сушили над сульфатом натрия, фильтровали и концентрировали под вакуумом с получением неочищенного продукта, который содержит два региоизомера с Вос-защищенным указанным в заголовке соединением в качестве минорного компонента. Остаток очищали посредством колоночной хроматографии на силикагеле, элюируя градиентом 0-80% EtOAc/гептаны, с получением Вос-защищенного указанного в заголовке соединения (24 мг; 6,4%). Вос-защищенное указанное в заголовке соединение (45 мг; 0,068 ммоль) растворяли в DCM (1 мл). К раствору добавляли HCl в диоксане (0,5 мл; 4 М раствор) и смесь перемешивали при комнатной температуре в течение 30 мин, концентрировали под вакуумом с получением HCl соли этил-1-{3-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-5-оксо-1,2,4-оксадиазол-4(5Н)-ил}этил карбоната (40 мг; 95%).
1Н ЯМР (DMSO-d6) δ: 9.35-9.11 (m, 1Н), 9.08-8.83 (m, 1Н), 8.42 (s, 1Н), 7.99 (d, 1Н), 7.68-7.53 (m, 1Н), 7.30 (d, 1Н), 7.17 (d, 2Н), 6.83 (d, 2Н), 6.08-5.95 (m, 1Н), 5.00-4.80 (m, 1Н), 4.43-4.10 (m, 2Н), 4.03-3.87 (m, 2Н), 3.76 (s, 3H), 3.43-3.26 (m, 1Н), 3.26-3.13 (m, 1Н), 2.80-2.61 (m, 1Н), 2.40-2.10 (m, 2Н), 1.97 (d, 3H), 1.92-1.88 (m, 1Н), 1.76 (d, 3H), 1.09 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,61 мин.
МС (ЭР+): 560,4 (М+Н)+.
ПРИМЕР 22: 4-(4-({3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-N-[(трифторметил)сульфонил]-1Н-пиразол-5-карбоксамид
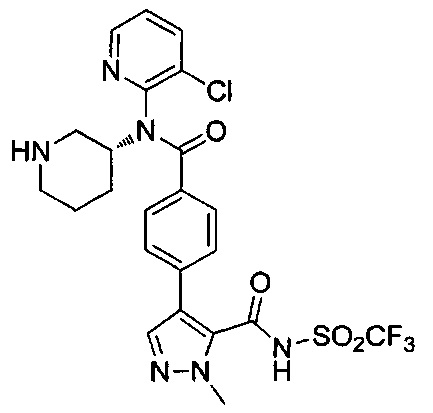
В раствор продукта стадии 1 ПРИМЕРА 8 (0,15 г; 0,28 ммоль) в DCM (2 мл) добавляли соединение трифторметансульфонамид (42 мг; 0,28 ммоль), DMAP (34 мг; 0,28 ммоль) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида EDAP (57 мг; 0,30 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Растворитель концентрировали при пониженном давлении с получением масла, которое использовали без дополнительной очистки. В раствор масла в DCM (2 мл) добавляли трифторуксусную кислоту (2 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 1 ч. Смесь концентрировали под вакуумом и полученный остаток очищали посредством препаративной ЖХВД с получением трифторацетата 4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}-фенил)-1-метил-N-[(трифторметил)сульфонил]-1Н-пиразол-5-карбоксамида в виде белого твердого вещества (80 мг; 42%). (Условия препаративной ЖХВД: колонка: Boston Symmetrix ODS-H (150×30 мм×5 мкм); подвижная фаза: градиент от 15% ацетонитрила в H2O (0,1% TFA) до 35% ацетонитрила в H2O (0,1% TFA).
1Н ЯМР (МеОН-d4) δ: 8.53 (br s, 1Н), 7.90-7.71 (m, 1Н), 7.53 (s, 1Н), 7.45-7.25 (m, 5Н), 5.11- 4.97 (m, 1Н), 3.98 (s, 3H), 3.77-3.63 (m, 1Н), 3.61-3.46 (m, 1Н), 3.39-3.34 (m, 1Н), 2.92-2.80 (m, 1Н), 2.35-2.26 (m, 0.3Н), 2.10-1.93 (m, 2Н), 1.93-1.76 (m, 1Н), 1.58-1.38 (m, 0.7Н).
ЖХ (ЖХ-МС, способ 4): tR составляет 0,89 мин.
МС (ЭР+): 571,0 (М+Н)+.
ПРИМЕР 23: 1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-N-[(трифторметил)сульфонил]-1Н-пиразол-5-карбоксамид
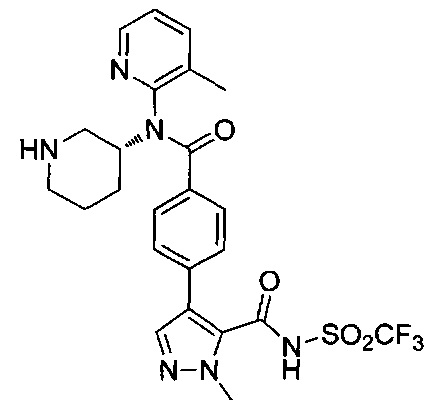
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 22, начиная с соединения по Получению 29.
ЖХВД (ЖХВД, способ 5): tR составляет 1,96 мин.
МС (ЭР+): 551,2 (М+Н)+.
ПРИМЕР 24: 4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-N-(1Н-тетразол-5-ил)-1Н-пиразол-5-карбоксамид
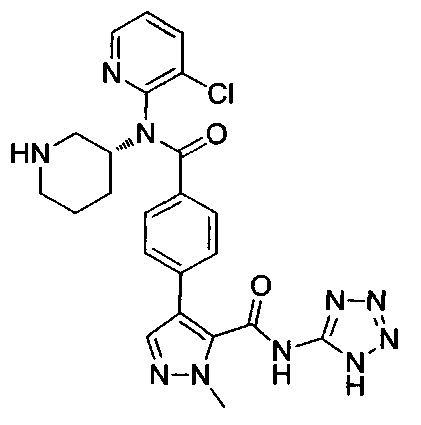
В раствор продукта стадии 1 ПРИМЕРА 8 (200 мг; 0,370 ммоль) в DMF (2 мл) добавляли CDI (78 мг; 0,482 ммоль) и DIPEA (72 мг; 0,556 ммоль). Смесь перемешивали при 80°C в течение 30 мин. В реакционную смесь добавляли 1H-тетразол-5-амин (94 мг; 1,11 ммоль) и полученную смесь перемешивали при 80°C в течение 16 ч. Смесь вливали в воду, подкисляли до pH 3 с помощью 1 н. HCl и экстрагировали EtOAc (10 мл). Органический слой сушили над сульфатом натрия и концентрировали под вакуумом с получением масла, которое использовали без дополнительной очистки. К маслу в DCM (5 мл) при 0°C по каплям добавляли трифторуксусную кислоту (1 мл). Полученную смесь перемешивали при комнатной температуре в течение 30 мин и затем смесь концентрировали под вакуумом. Полученный остаток очищали посредством препаративной ЖХВД с получением трифторацетата 4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-N-(1Н-тетразол-5-ил)-1Н-пиразол-5-карбоксамида в виде белого твердого вещества (45,1 мг; 20%). (Условия препаративной ЖХВД: колонка: DIKMA Diamonsil (2) С18 (200×20 мм×5 мкм); подвижная фаза: градиент от 5% ацетонитрила в H2O (0,1% TFA) до 25% ацетонитрила в H2O (0,1% TFA).)
1Н ЯМР (MeOH-d4) δ: 8.47 (br s, 0.7Н), 8.43 (br s, 0.3H), 7.85-7.67 (m, 2H), 7.40-7.18 (m, 5H), 5.11-4.98 (m, 2H), 4.03 (s, 3H), 3.81-3.66 (m, 1H), 3.64-3.49 (m, 1H), 3.41-3.36 (m, 1H), 2.96-2.82 (m, 1H), 2.16-1.95 (m, 2H), 1.94-1.81 (m, 1H), 1.52-1.39 (m, 1H).
ЖХ (ЖХ-МС, способ 4): tR составляет 0,91 мин.
МС (ЭР+): 507,1 (М+Н)+.
ПРИМЕР 25: 4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-N-гидрокси-1-метил-1Н-пиразол-5-карбоксамид

Металлический натрий (0,55 г; 24,0 ммоль) добавляли в МеОН (10 мл) с последующим добавлением раствора гидрохлорида гидроксиламина (800 мг; 11,0 ммоль) в МеОН (10 мл). Осадок NaCl затем отфильтровывали. К 2 мл полученного таким образом раствора свободного гидроксиламина добавляли соединение по Получению 18 (R)-трет-бутил-3-(N-(3-хлорпиридин-2-ил)-4-(5-(этоксикарбонил)-1-метил-1Н-пиразол-4-ил)бензамидо)пиперидин-1-карбоксилат (200 мг; 0,352 ммоль) и реакционную смесь нагревали при температуре дефлегмации в течение 15 мин. Растворитель удаляли под вакуумом с получением масла, которое использовали без дополнительной очистки. К маслу в DCM (5 мл) при 0°C по каплям добавляли TFA (1 мл). Полученную смесь перемешивали при 30°C в течение 5 мин и затем смесь концентрировали под вакуумом. Полученный остаток очищали посредством препаративной ЖХВД с получением трифторацетата 4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-N-гидрокси-1-метил-1Н-пиразол-5-карбоксамида в виде желтоватого твердого вещества (45 мг; 23%). (Условия препаративной ЖХВД: колонка: YMC-Actus Triart С18 (150×30 мм); подвижная фаза: градиент от 8% ацетонитрила в H2O (0,1% TFA) до 28% ацетонитрила в H2O (0,1% TFA).)
1Н ЯМР (DMSO-d6) δ: 11.29 (br s, 1Н), 8.91 (br s, 1Н), 8.70 (br s, 1H), 8.61 (br s, 0.7H), 8.58 (br s, 0.3H), 8.01-7.81 (m, 2H), 7.48-7.41 (m, 1H), 7.38-7.29 (m, 2H), 7.29-7.17 (m, 2H), 4.98 (br s, 1H), 3.79 (s, 3H), 3.66-3.55 (m, 1H), 3.27-3.14 (m, 1H), 2.87-2.65 (m, 1H), 1.96-1.68 (m, 2H), 1.33-1.22 (m, 1H).
ЖХ (ЖХ-МС, способ 4): tR составляет 0,87 мин.
МС (ЭР+): 455,1 (M+H)+.
ПРИМЕР 26: N-(3-хлорпиридин-2-ил)-4-[1-метил-5-(2Н-тетразол-5-ил)-1Н-пиразол-4-ил]-N-[(3R)-пиперидин-3-ил]бензамид
В раствор (R)-трет-бутил-3-(N-(3-хлорпиридин-2-ил)-4-(5-циано-1-метил-1Н-пиразол-4-ил)бензамидо)пиперидин-1-карбоксилата (полученного способом, аналогичным способу по Получению 26, начиная с соединения по Получению 21 и соединения по Получению 17) (300 мг; 0,58 ммоль) в DMF (5 мл) добавляли азид натрия (142 мг; 2,19 ммоль) и CuBr2 (208 мг; 0,92 ммоль). Реакционную смесь затем нагревали при 120°C в течение 16 ч. Реакционную смесь разбавляли H2O и экстрагировали EtOAc, сушили над сульфатом натрия и концентрировали с получением остатка, который использовали без дополнительной очистки. К остатку в DCM (3 мл) добавляли трифторуксусную кислоту (3 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 1 ч. Смесь концентрировали под вакуумом и полученный остаток очищали посредством препаративной ЖХВД с получением трифторацетата N-(3-хлорпиридин-2-ил)-4-[1-метил-5-(2Н-тетразол-5-ил)-1Н-пиразол-4-ил]-N-[(3R)-пиперидин-3-ил]бензамида в виде белого твердого вещества (162 мг; 49%). (Условия препаративной ЖХВД: колонка: Boston Symmetrix ODS-H (150×30 мм×5 мкм); подвижная фаза: градиент от 15% ацетонитрила в H2O (0,1% TFA) до 35% ацетонитрила в H2O (0,1% TFA)).
1Н ЯМР (DMSO-d6) δ: 9.06 (br s, 1Н), 8.78 (br s, 1Н), 8.58 (br s, 0.7H), 8.54 (br s, 0.3H), 8.01-7.88 (m, 2H), 7.48-7.40 (m, 1H), 7.27-7.04 (m, 4H), 5.07-4.88 (m, 2H), 4.62 (br s, 1H), 3.68-2.95 (m, 4H), 2.84-2.67 (m, 1H), 2.19-2.09 (m, 0.3H), 1.99-1.65 (m, 3H), 1.34-1.19 (m, 0.7H).
ЖХ (ЖХ-МС, способ 5): tR составляет 0,73 мин.
МС (ЭР+): 464,1 (M+H)+.
ПРИМЕР 27: 4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-N-(метилсульфонил)-1Н-пиразол-5-карбоксамид
Стадия 1: в раствор продукта стадии 1 ПРИМЕРА 8 (150 мг; 0,28 ммоль) в DCM (2 мл) добавляли метансульфонамид (26 мг; 0,28 ммоль), DMAP (34 мг; 0,28 ммоль) и EDAP (57 мг; 0,30 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь вливали в EtOAc, промывали рассолом, сушили над сульфатом натрия и концентрировали под вакуумом. Полученный остаток очищали посредством хроматографии на силикагеле, элюируя градиентом 5-10% MeOH/DCM, с получением Вос-защищенного указанного в заголовке соединения в виде бесцветного масла (60 мг; 35%).
Стадия 2: к Вос-защищенному указанному в заголовке соединению (60 мг; 0,098 ммоль) в DCM (2 мл) добавляли TFA (2 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 1 ч. Смесь концентрировали под вакуумом и неочищенный продукт очищали посредством препаративной ЖХВД с получением формиата 4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-N-(метилсульфонил)-1Н-пиразол-5-карбоксамида в виде коричневой смолы (22 мг; 39%). (Условия препаративной ЖХВД: колонка: YMC-Actus Triart С18 (150×30 мм); подвижная фаза: градиент от 15% ацетонитрила в H2O (0,1% муравьиной кислоты) до 35% ацетонитрила в H2O (0,1% муравьиной кислоты).)
1Н ЯМР (DMSO-d6) δ: 8.56 (d, 1Н), 7.92-7.84 (m, 1Н), 7.68 (s, 1Н), 7.46-7.38 (m, 1Н), 7.36-7.24 (m, 4Н), 4.99-4.77 (m, 1Н), 3.87 (s, 3H), 3.66-3.54 (m, 2Н), 2.84-2.73 (m, 1Н), 2.07-1.73 (m, 3H).
ЖХ (ЖХ-МС, способ 4): tR составляет 0,98 мин.
МС (ЭР+): 517,1 (М+Н)+.
ПРИМЕР 28: 1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-N-(метилсульфонил)-1Н-пиразол-5-карбоксамид
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 27, начиная с соединения по Получению 29.
1Н ЯМР (DMSO-d6) δ: 9.41-9.19 (m, 1Н), 9.11-8.85 (m, 1Н), 8.46 (s, 1Н), 7.77 (s, 1Н), 7.68-7.53 (m, 1Н), 7.35-7.27 (m, 1Н), 7.24-7.20 (m, 4Н), 5.05-4.88 (m, 2Н), 3.88 (s, 3H), 3.25-3.14 (m, 1Н), 2.72 (s, 3H), 2.74-2.68 (m, 2Н), 2.22-2.01 (m, 2Н), 1.98 (s, 3H), 1.83-1.75 (m, 2Н).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,5 мин.
МС (ЭР+): 497,2 (М+Н)+.
ПРИМЕР 29: этил-1-[{[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]карбонил}(метилсульфонил)-амино]этилкарбонат
Продукт стадии 1 ПРИМЕРА 28 (81 мг; 0,14 ммоль) обрабатывали KOH (0,14 мл; 1 M в МеОН; 0,14 ммоль) в МеОН (1 мл), перемешивали в течение 1 ч и затем концентрировали. МеОН полностью удаляли посредством совместного испарения с толуолом с получением калиевой соли. Калиевую соль растворяли в ацетонитриле (2 мл), добавляли 1-хлорэтилэтилкарбонат (63 мг; 0,42 ммоль) и смесь нагревали при 50°C в течение 2 суток. Реакционную смесь очищали непосредственно без концентрирования посредством колоночной хроматографии на силикагеле с получением Вос-защищенного указанного в заголовке соединения (34 мг; 36%). Вос-защищенное указанное в заголовке соединение (30 мг; 0,042 ммоль) растворяли в DCM (1 мл), обрабатывали HCl в диоксане (0,5 мл; 4 М раствор) и перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь концентрировали под вакуумом с получением HCl соли этил-1-[{[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]карбонил}-(метилсульфонил)амино]этилкарбоната (27 мг; 94%).
1Н ЯМР (DMSO-d6) δ: 9.06 (br s, 1Н), 8.86 (br s, 1Н), 8.50-8.30 (m, 1Н), 7.76 (s, 1Н), 7.54-7.50 (m, 1Н), 7.31-7.18 (m, 3Н), 7.21-7.09 (m, 2Н), 5.91-5.58 (m, 1Н), 4.94 (br s, 1Н), 4.60-4.20 (m, 1Н), 4.11 (q, 2Н), 3.91 (s, 3H) 3.59-3.55 (m, 1Н), 3.28 (s, 3H), 3.18-3.16 (m, 1Н), 2.95-2.60 (m, 2Н), 2.05-2.01 (m, 2Н), 1.97 (d, 3H),1.77 (s, 3H), 1.45-1.30 (m, 1Н), 1.24 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,64 мин.
МС (ЭР+): 613,2 (М+Н)+.
ПРИМЕР 30: этил-(1S)-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}-этилкарбонат
Соединение по Получению 27 трет-бутил-(3R)-3-[{4-[5-(2-{(1S)-1-[(этоксикарбонил)окси]этил}-2Н-тетразол-5-ил)-1-метил-1Н-пиразол-4-ил]-бензоил}(3-метилпиридин-2-ил)амино]пиперидин-1-карбоксилат (438 мг; 0,664 ммоль) растворяли в ацетонитриле (3,3 мл). Туда по каплям добавляли HCI в диоксане (2,49 мл; 4 М раствор, 15 экв.) при комнатной температуре. Через 1 ч реакционную смесь концентрировали под вакуумом и концентрировали с толуолом (3×15 мл). Неочищенное вещество суспендировали в EtOAc и нагревали до 80°C в течение 16 ч. Фильтрация вещества дала гидрохлорид этил-(1S)-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]-карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}этилкарбоната (242 мг; 65%).
1Н ЯМР (DMSO-d6) δ: 9.46 (br s, 1Н), 9.10 (br s, 1Н), 8.44 (d, 1H), 7.86 (s, 1H), 7.59 (d, 1H), 7.33-7.25 (m, 2H), 7.17-7.10 (m, 4H), 4.96-4.89 (m, 1H), 4.61-4.58 (m, 1H), 4.20 (q, 2H), 3.95 (s, 3H), 3.55-3.52 (m, 1H), 3.19-3.16 (m, 1H), 2.79-2.63 (m, 1H), 2.19-1.97 (m, 1H),1.97 (s, 3H),1.89 (d, 3H), 1.84-1.72 (m, 3H), 1.24 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,02 мин.
МС (ЭР+): 560,4 (М+Н)+.
Элементный анализ: подсчитано для C28H34ClN9O4 C 56,42; H 5,75; N 21,15; Cl 5,95; найдено C 56,17; H 5,81; N 21,14; Cl 6,11.
Дифракция рентгеновских лучей на порошке для примера 30 представлена на Фиг. 9.
ПРИМЕР 31: этил-(1R)-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}-этилкарбонат
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 30, начиная с соединения по Получению 28 трет-бутил-(3R)-3-[{4-[5-(2-{(1R)-1-[(этоксикарбонил)окси]этил}-2Н-тетразол-5-ил)-1-метил-1Н-пиразол-4-ил]бензоил}(3-метилпиридин-2-ил)амино]пиперидин-1-карбоксилата.
1Н ЯМР (DMSO-d6) δ: 9.46 (br s, 1Н), 9.10 (br s, 1Н), 8.44 (d, 1Н), 7.86 (s, 1Н), 7.59 (d, 1Н), 7.33-7.23 (m, 2Н), 7.23-7.05 (m, 4Н), 5.03-4.83 (m, 1Н), 4.61-4.58 (m, 1Н), 4.20 (q, 2Н), 3.95 (s, 3H), 3.61-3.47 (m, 1Н), 3.24-3.12 (m, 1Н), 2.75-2.62 (m, 1Н), 2.20-2.03 (m, 1Н),1.97 (s, 3H), 1.90 (d, 3H), 1.85-1.69 (m, 3H), 1.24 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,05 мин.
МС (ЭР+): 560,3 (М+Н)+.
Элементный анализ: подсчитано для C28H34ClN9O4 C 56,42; Н 5,75; N 21,15; Cl 5,95; найдено C 56,03; H 5,70; N 21,06; Cl 6,17.
Дифракция рентгеновских лучей на порошке для примера 31 представлена на Фиг. 10.
Звездочки в соединениях по примерам 32-55 означают неизвестную абсолютную конфигурацию (R/S) в хиральном центре, где находится звездочка. Каждое соединение по примеру, однако, представляет собой отдельный диастереомер, однозначно установленный, с характерными свойствами, как показано по времени удерживания при хиральной хроматографии конкретных соединений по получениям, из которых соединения по примерам были получены, и уникальности спектра 1Н ЯМР индивидуальных соединений по примерам.
ПРИМЕР 32: этил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропилкарбонат (диастереомер А)
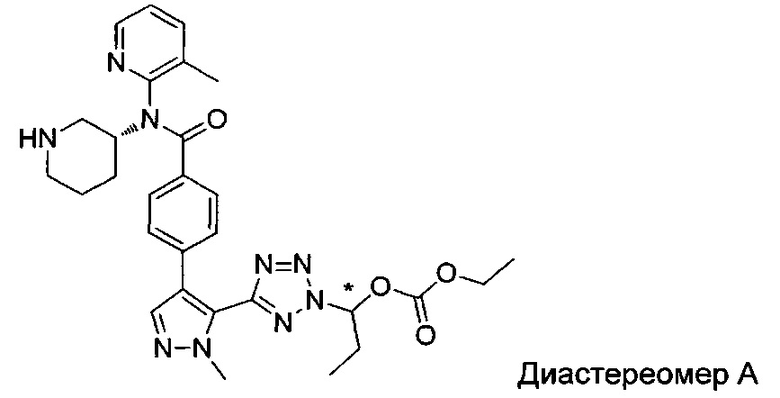
В раствор соединения по Получению 31 (123 мг; 0,183 ммоль) в MeCN (2 мл) добавляли HCl (0,22 мл; 0,88 ммоль; 4,0 М раствор в диоксане). Через 2 ч реакционную смесь концентрировали и сушили под вакуумом с получением гидрохлорида указанного в заголовке соединения в виде бесцветного твердого вещества с количественным выходом.
1Н ЯМР (CD3CN) δ: 9.18 (br s, 1Н), 8.94 (br s, 1Н), 8.38-8.17 (m, 1Н), 7.58 (s, 1Н), 7.47-7.32 (m, 1Н), 7.16-7.03 (m, 5Н), 6.91 (t, 1Н), 5.17-4.55 (m, 1Н), 4.18-4.09 (m, 2Н), 3.91 (s, 3H), 3.60-2.98 (m, 2Н), 2.85-2.64 (m, 1Н), 2.95-2.76 (m, 1Н), 2.28-2,14 (m, 2Н), 2.20-1.98 (m, 1Н), 1.94 (br s, 3H), 1.82-1.74 (m, 1H), 1.59-1.63 (m, 1H), 1.34-1.24 (m, 1H), 1.19 (t, 3H), 0.83 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,22 мин.
МС (ЭР+): 574,3 (М+Н)+.
ПРИМЕР 33: этил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропил карбонат (диастереомер В)
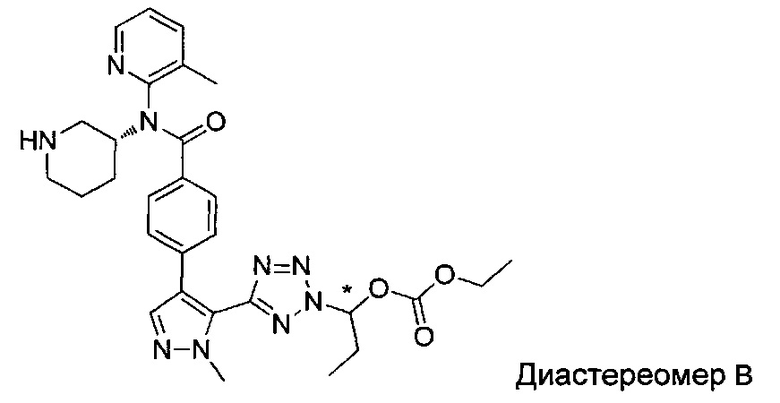
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 32.
1Н ЯМР (CDCl3) δ: 9.99-9.73 (m, 2Н), 8.42 (br s, 1Н), 7.61 (s, 1Н), 7.42 (br s, 1Н), 7.25-7.12 (m, 4Н), 6.98 (t, 1Н), 5.05 (br s, 1Н), 4.33-4.22 (m, 2Н), 4.07 (s, 3H), 3.91-3.67 (m, 2Н), 3.48 (br s, 1Н), 2.91 (br s, 1Н), 2.41-2.32 (m, 2Н), 2.17-1.95 (m, 4Н), 1.84-1.73 (m, 3Н), 1.34 (t, 3H), 0.97 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,17 мин.
МС (ЭР+): 574,4 (М+Н)+.
ПРИМЕР 34: этил-2-метил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропилкарбонат (диастереомер А)
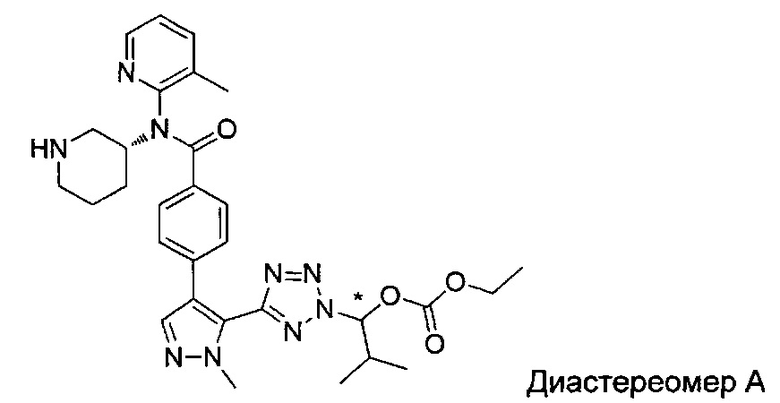
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 33а.
1Н ЯМР (CD3CN) δ: 9.42 (br s, 1Н), 8.96 (br s, 1H), 8.40 (d, 1H), 7.80-7.69 (m, 1H), 7.65 (s, 1H), 7.30-7.23 (m, 1H), 7.23-7.16 (m, 2H), 7.15-7.10 (m, 2H), 6.75 (d, 1H), 5.09-4.90 (m, 1H), 4.25-4.16 (m, 2H), 3.97 (s, 3H), 3.73-3.55 (m, 2H), 3.29-3.22 (m, 1H), 2.86-2.75 (m, 1H), 2.62-2.54 (m, 1H), 2.18-2.08 (m, 3H), 1.85-1.72 (m, 3H), 1.50-1.38 (m, 1H), 1.25 (t, 3H), 1.10 (d, 3H), 0.78 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,26 мин.
МС (ЭР+): 588,4 (М+Н)+.
ПРИМЕР 35: этил-2-метил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропилкарбонат (диастереомер В)
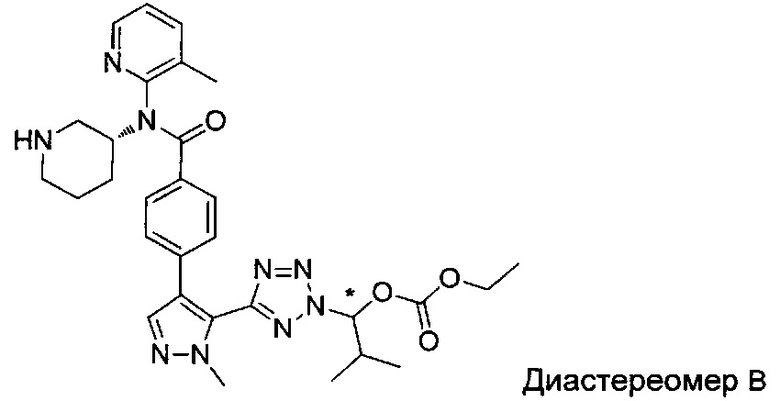
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 33b.
1Н ЯМР (CD3CN) δ: 9.46 (br s, 1Н), 9.17 (br s, 1Н), 8.39 (d, 1Н), 7.64 (s, 1Н), 7.56-7.51 (m, 1Н), 7.29-7.24 (m, 1Н), 7.32-7.22 (m, 2Н), 7.19-7.12 (m, 2Н), 6.75 (d, 1Н), 5.14-5.06 (m, 1Н), 4.25-4.16 (m, 2Н), 3.97 (s, 3H), 3.73-3.51 (m, 2Н), 3.29-3.22 (m, 1Н), 2.82-2.72 (m, 1Н), 2.61-2.54 (m, 1Н), 2.50-2.28 (m, 3H), 1.95-1.72 (m, 3H), 1.45-1.33 (m, 1Н), 1.26 (t, 3H), 1.10 (d, 3H), 0.78 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,26 мин.
МС (ЭР+): 588,4 (М+Н)+.
ПРИМЕР 36: 2,2-диметил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропилэтилкарбонат (диастереомер А)
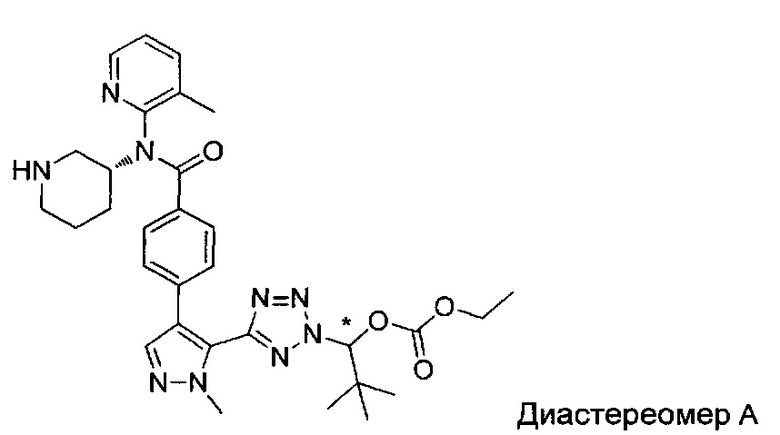
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 35.
1Н ЯМР (DMSO-d6) δ: 9.31 (br s, 1Н), 8.99 (br s, 1H), 8.49-8.34 (m, 1H), 7.87 (s, 1H), 7.58 (d, 1H), 7.32-7.26 (m, 1H), 7.11 (m, 4H), 6.90 (s, 1H), 5.02-4.81 (m, 1H), 4.23-4.11 (m, 2H), 3.91 (s, 3H), 3.61-3.48 (m, 1H), 3.44-3.28 (m, 1H), 3.25-3.11 (m, 1H), 2.76-2.59 (m, 1H), 2.18-1.90 (m, 3H), 1.85-1.69 (m, 4H), 1.21 (t, 3H), 1.00 (s, 9H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,71 мин.
МС (ЭР+): 602,4 (М+Н)+.
ПРИМЕР 37: 2,2-диметил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропилэтилкарбонат (диастереомер В)
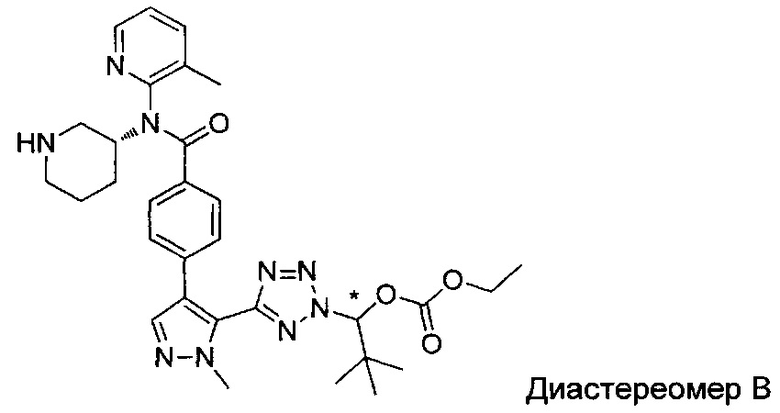
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 36.
1Н ЯМР (DMSO-d6) δ: 9.15 (br s, 1Н), 8.91 (br s, 1Н), 8.48-8.35 (m, 1Н), 7.87 (s, 1Н), 7.66-7.52 (m, 1Н), 7.32-7.27 (m, 1Н), 7.18-7.07 (m, 4Н), 6.91 (s, 1H), 5.02-4.84 (m, 1H), 4.29-4.09 (m, 2H), 3.91 (s, 3H), 3.58-3.51 (m, 1H), 3.45-3.26 (m, 1H), 3.25-3.12 (m, 1H), 2.79-2.59 (m, 1H), 2.18-1.95 (m, 3H), 1.83-1.68 (m, 4H), 1.21 (m, 3H), 1.00 (s, 9H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,22 мин.
МС (ЭР+): 602,4 (М+Н)+.
ПРИМЕР 38: 1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропилпропаноат (диастереомер А)
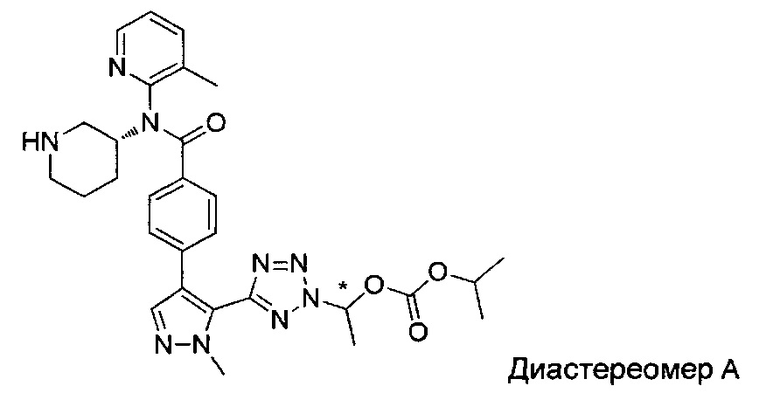
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 38.
1Н ЯМР (CD3CN) δ: 9.71 (br s, 1Н), 9.23 (br s, 1Н), 8.41 (d, 1Н), 8.03-7.73 (m, 1Н), 7.64 (s, 1Н), 7.56-7.39 (m, 1Н), 7.35-7.25 (m, 2Н), 7.22-7.05 (m, 3H), 5.18-4.94 (m, 1Н), 4.88-4.83 (m, 1Н), 3.98 (s, 3H), 3.89-3.54 (m, 2Н), 3.30-3.26 (m, 1Н), 2.88-2.73 (m, 1Н), 2.15 (br. s., 3H), 2.03-1.95 (m, 2H), 1.90 (d, 3H), 1.86-1.74 (m, 1H), 1.59-1.42 (m, 1H), 1.29 (d, 3H), 1.23 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,15 мин.
МС (ЭР+): 574,3 (М+Н)+.
ПРИМЕР 39: 1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}этилпропан-2-илкарбонат (диастереомер В)
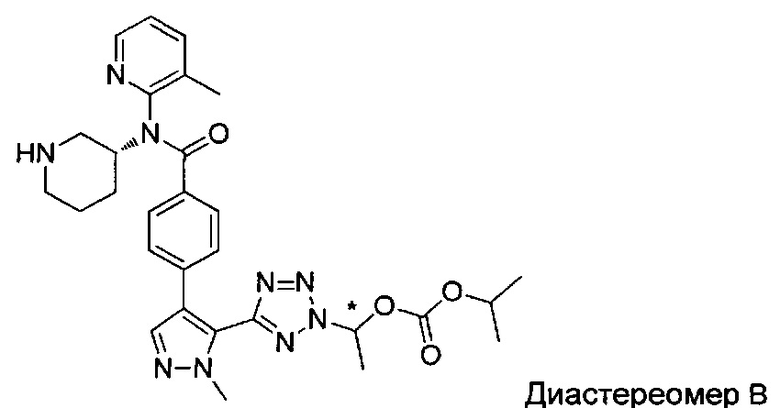
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 39.
1Н ЯМР (DMSO-d6) δ: 8.83-8.53 (m, 2Н), 8.45-8.35 (m, 1Н), 7.84 (s, 1Н), 7.63-7.52 (m, 1Н), 7.33-7.20 (m, 2Н), 7.20-7.03 (m, 4Н), 4.99-4.87 (m, 1Н), 4.80 (септет, 1Н), 3.92 (s, 3H), 3.56 (d, 1Н), 3.37-3.30 (m, 1Н), 3.19 (d, 1Н), 2.80-2.65 (m, 1Н), 2.15-1.64 (m, 10Н), 1.25 (d, 3H), 1.19 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,21 мин.
МС (ЭР+): 574,3 (М+Н)+.
ПРИМЕР 40: 1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропилпропан-2-илкарбонат (диастереомер А)
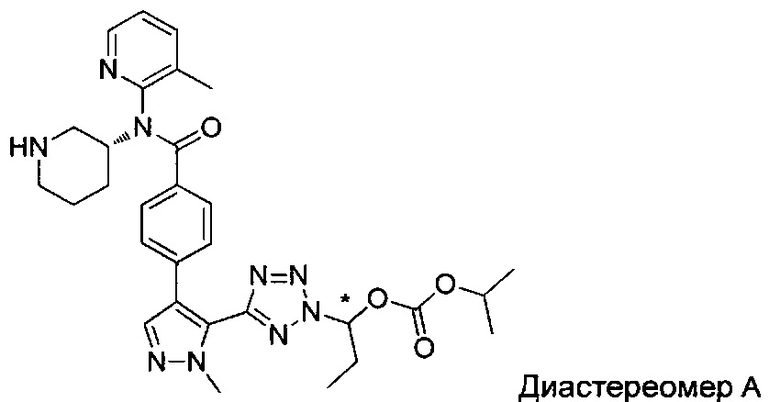
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 41.
1Н ЯМР (DMSO-d6) δ: 9.20-9.17 (m, 1Н), 8.96-8.86 (m, 1Н), 8.44-8.39 (m, 1Н), 7.86 (s, 1Н), 7.66-7.55 (m, 1Н), 7.30-7.27 (m, 1Н), 7.14-7.09 (m, 3Н), 4.95-4.91 (m, 1Н), 4.82-4.78 (m, 2Н), 4.60-4.53 (m, 1Н), 3.92 (s, 3H), 3.54-3.50 (m, 2Н), 3.38-3.32 (m, 1Н), 3.19-3.17 (m, 1Н), 2.81-2.65 (m, 1Н), 2.28-2.20 (m, 2Н), 2.14-2.03 (m, 1Н), 1.95 (s, 3Н, 1.78-1.71 (m, 2Н), 1.27 (d, 3H), 1.20 (d, 3H), 0.86 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,70 мин.
МС (ЭР+): 588,4 (М+Н)+.
ПРИМЕР 41: 1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропилпропан-2-илкарбонат (диастереомер В)
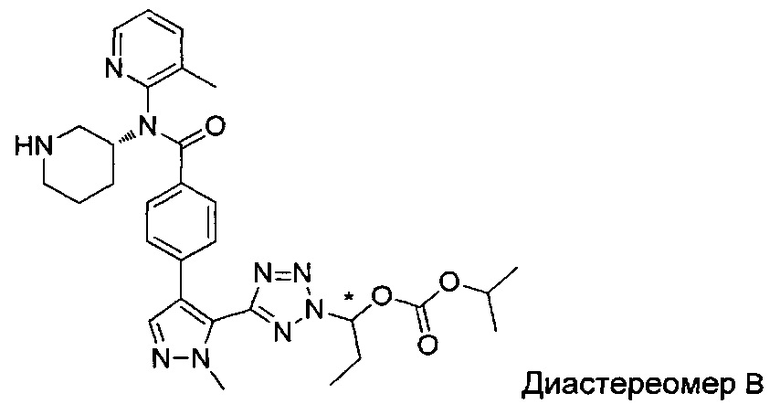
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 42.
1Н ЯМР (CDCl3) δ: 9.93 (br s, 1Н), 9.58 (br s, 1Н), 8.45 (br s, 1H), 7.59 (s, 1H), 7.26-7.15 (m, 5H), 6.97 (t, 1H), 5.14-4.87 (m, 2H), 4.06 (s, 3H), 3.88 (br s, 1H), 3.51 (br s, 1H), 3.13-2.79 (m, 3H), 2.39-1.81 (m, 8H), 1.34 (d, 3H), 1.28 (d, 3H), 0.96 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,67 мин.
МС (ЭР+): 588,4 (М+Н)+.
ПРИМЕР 42: 2-метил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропилпропан-2-илкарбонат (диастереомер А)
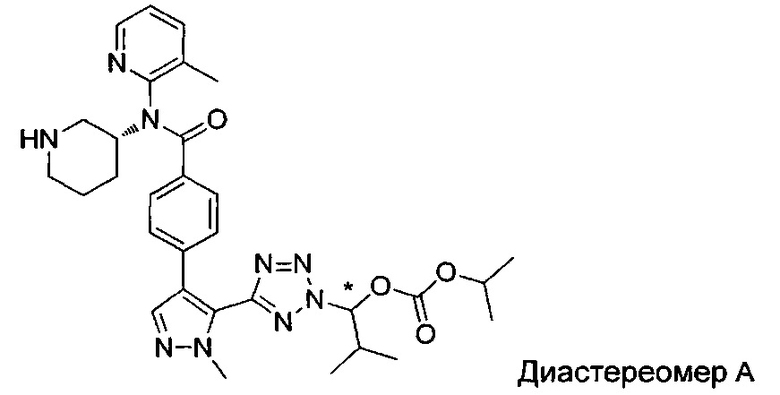
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 43а.
1Н ЯМР (CD3CN) δ: 9.40 (br s, 1H), 9.03 (br s, 1H), 8.39 (d, 1H), 7.70-7.57 (m, 2H), 7.36-7.30 (m, 1H), 7.27-7.21 (m, 2H), 7.14-7.11 (m, 2H), 6.75 (d, 1H), 5.09-5.01 (m, 1H), 4.87-4.81 (m, 1H), 3.97 (s, 3H), 3.78-3.52 (m, 2H), 3.29-3.22 (m, 1H), 2.83-2.75 (m, 1H), 2.61-2.48 (m, 1H), 2.33-2.00 (m, 3H), 1.82-1.72 (m, 3H), 1.52-1.34 (m, 1H), 1.28 (d, 3H), 1.22 (d, 3H), 1.10 (d, 3H), 0.79 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,33 мин.
МС (ЭР+): 602,4 (М+Н)+.
ПРИМЕР 43: 2-метил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропилпропан-2-илкарбонат (диастереомер В)
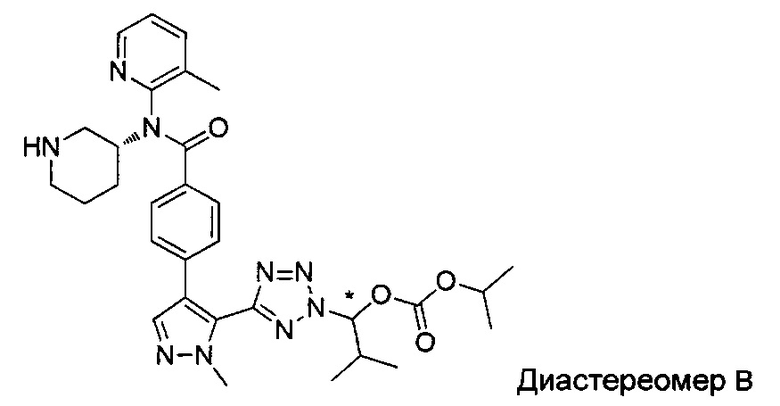
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 43b.
1Н ЯМР (CD3CN) δ: 9.33 (br s, 1Н), 8.95 (br s, 1H), 8.39 (d, 1H), 7.70-7.63 (m, 2H), 7.41-7.34 (m, 1H), 7.28-7.21 (m, 2H), 7.19-7.12 (m, 2H), 6.75 (d, 1H), 5.09-4.99 (m, 1H), 4.87-4.81 (m, 1H), 3.97 (s, 3H), 3.73-3.55 (m, 2H), 3.29-3.22 (m, 1H), 2.83-2.75 (m, 1H), 2.60-2.54 (m, 1H), 2.15-2.05 (m, 3H), 1.82-1.72 (m, 3H), 1.52-1.38 (m, 1H), 1.28 (d, 3H), 1.22 (d, 3H), 1.10 (d, 3H), 0.78 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,33 мин.
МС (ЭР+): 602,4 (М+Н)+.
ПРИМЕР 44: 1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}этилпропаноат (диастереомер А)
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 45.
1Н ЯМР (DMSO-d6) δ: 9.41-9.21 (m, 1Н), 9.13-8.92 (m, 1Н), 8.49-8.35 (m, 1Н), 7.87 (s, 1Н), 7.70-7.52 (m, 1Н), 7.45-7.34 (m, 1Н), 7.34-7.22 (m, 1Н), 7.22-7.09 (m, 4Н), 5.04-4.84 (m, 1Н), 3.94 (s, 3H), 3.61-3.48 (m, 1Н), 3.44-3.31 (m, 1Н), 3.27-3.11 (m, 1Н), 2.79-2.60 (m, 1Н), 2.48-2.32 (q, 2Н), 2.20-2.02 (m, 1Н), 2.02-1.94 (m, 3Н), 1.87 (d, 3H), 1.83-1.72 (m, 3Н), 1.04 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,64 мин.
МС (ЭР+): 544,3 (М+Н)+.
ПРИМЕР 45: 1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}этилпропаноат (диастереомер В)
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 46.
1Н ЯМР (CDCl3) δ: 9.87 (br s, 1Н), 9.59 (br s, 1Н), 8.41 (br s, 1H), 7.59 (s, 1H), 7.47-7.28 (m, 2H), 7.25-7.11 (m, 4H), 5.16-5.03 (m, 1H), 4.07 (s, 3H), 3.90-3.71 (m, 1H), 3.56-3.44 (m, 1H), 2.94-2.79 (m, 1H), 2.47-2.29 (m, 3H), 2.19-1.75 (m, 9H), 1.66-1.48 (m, 1H), 1.15 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,63 мин.
МС (ЭР+): 544,3 (М+Н)+.
ПРИМЕР 46: 1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-илкарбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}этилпропан-2-илкарбонат (диастереомер А)
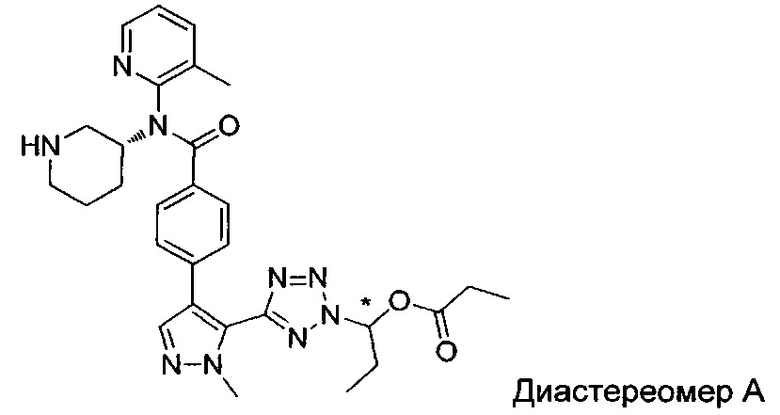
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 48.
1Н ЯМР (CD3CN) δ: 9.38 (br s, 1Н), 8.94 (br s, 1H), 8.39 (d, 1H), 7.83-7.68 (m, 1H), 7.65 (s, 1H), 7.48-7.34 (m, 1H), 7.25 (br s, 2H), 7.20-7.05 (m, 3H), 5.18-4.47 (m, 1H), 3.97 (s, 3H), 3.79-3.51 (m, 2H), 3.36-3.15 (m, 1H), 2.95-2.76 (m, 1H), 2.46-2.31 (m, 4H), 2.27-2,21 (m, 2H), 1.94 (br. s., 3H), 1.89-1.72 (m, 1H), 1.58-1.36 (m, 1H), 1.07 (t, 3H), 0.90 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,06 мин.
МС (ЭР+): 558,9 (М+Н)+.
ПРИМЕР 47: 1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропилпропаноат (диастереомер В)
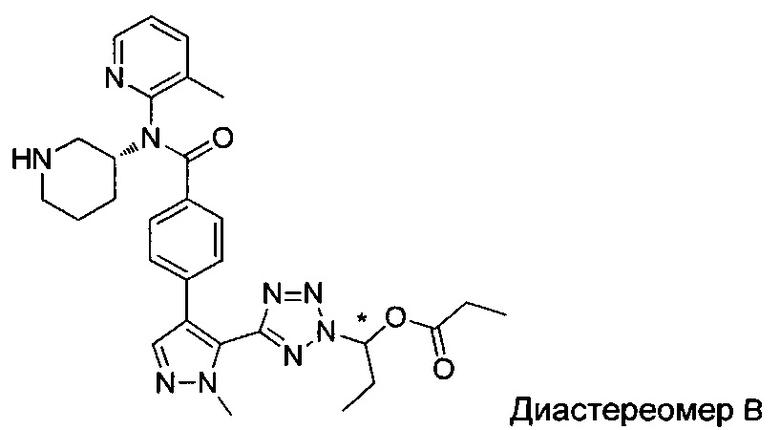
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 49.
1Н ЯМР (CDCl3) δ: 9.91 (br s, 1Н), 9.67 (br s, 1Н), 8.41 (br s, 1H), 7.60 (s, 1H), 7.41 (br s, 1H), 7.25-1.09 (m, 4H), 5.13-5.10 (m, 1H), 4.06 (s, 3H), 3.88-3.75 (m, 1H), 3.54-3.45 (m, 1H), 2.95-2.81 (m, 1H), 2.49-1.56 (m, 13H), 1.15 (t, 3H), 0.96 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,65 мин.
МС (ЭР+): 559,0 (М+Н)+.
ПРИМЕР 48: 2-метил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропилпропаноат (диастереомер А)
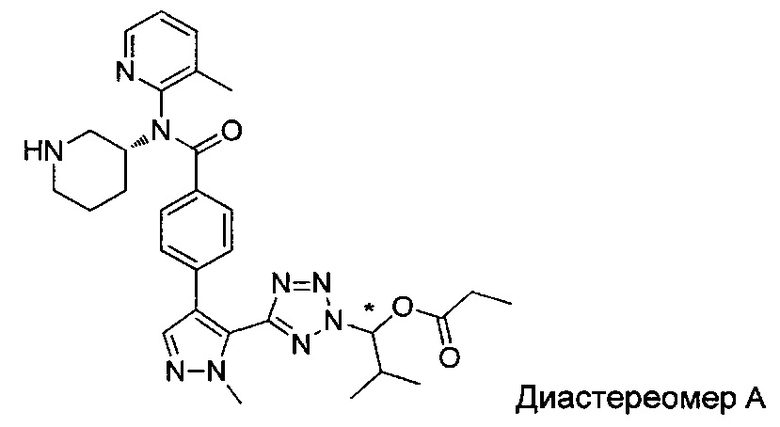
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 51.
1Н ЯМР (CD3CN) δ: 9.63 (br s, 1Н), 9.24 (br s, 1Н), 8.43 (d, 1Н), 7.75 (br s, 1H), 7.67 (s, 1H), 7.43 (br s, 1H), 7.28 (br s, 2H), 7.22-7.16 (br m, 2H), 6.94 (d, 1H), 5.06 (br s, 1H), 3.99 (s, 3H), 3.79 (br s, 1H), 3.64 (br s, 1H), 3.37-3.28 (br m, 1H), 2.89-2.80 (br m, 1H), 2.62-2.54 (m, 1H), 2.52-2.45 (m, 1H), 2.43-2.46 (m, 1H), 2.30-2.12 (br m, 4H), 2.04-1.99 (m, 1H), 1.85 (br s, 1H), 1.51 (br s, 1H), 1.12-1.09 (m, 6H), 0.82 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,25 мин.
МС (ЭР+): 572,4 (М+Н)+.
ПРИМЕР 49: 2-метил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропилпропаноат (диастереомер В)
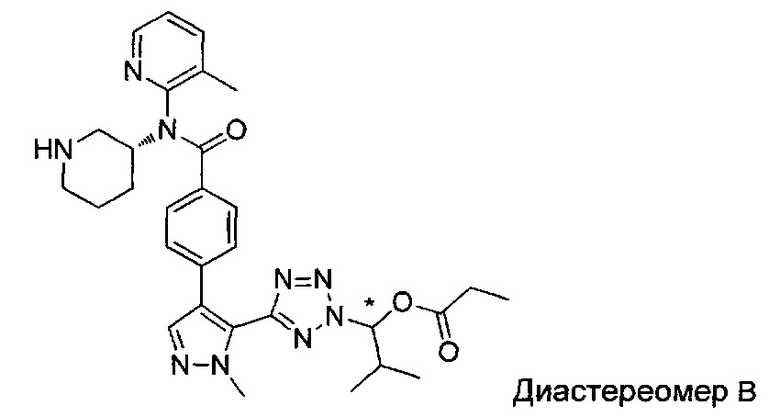
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 52.
1Н ЯМР (CDCl3) δ: 9.09 (br s, 1Н), 8.85 (br s, 1H), 8.46-8.42 (m, 1H), 7.84 (s, 1H), 7.56 (d, 1H), 7.28-7.26 (m, 1H), 7.10-7.08 (m, 3H), 6.99 (d, 1H), 4.97-4.81 (m, 1H), 4.01 (q, 2H), 3.90 (s, 3H), 3.54 (d, 1H), 3.28-3.42 (m, 1H), 3.17 (d, 2H), 2.32-2.44 (m, 2H), 1.94 (br s, 2H), 1.83-1.65 (m, 3H), 1.05-1.00 (m, 6H), 0.74 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,21 мин.
МС (ЭР+): 572,3 (М+Н)+.
ПРИМЕР 50: 1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}этил-2-метилпропаноат (диастереомер А)
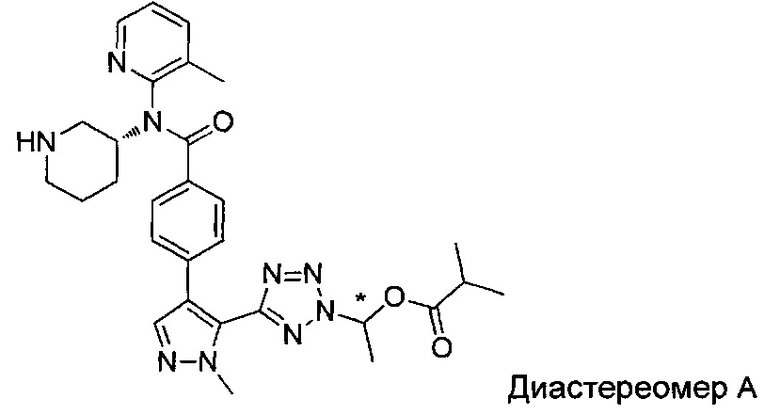
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 54.
1Н ЯМР (CD3CN) δ: 9.58 (br s, 1Н), 9.17 (br s, 1H), 8.44 (d, 1H), 7.78 (br s, 1H), 7.68 (s, 1H), 7.46 (br s, 1H), 7.36-7.29 (m, 3H), 7.22-7.18 (br m, 2H), 5.05 (br s, 1H), 3.99 (s, 3H), 3.80 (br s, 1H), 3.65 (br s, 1H), 3.32 (br d, 1H), 2.92-2.77 (br m, 1H), 2.67-2.57 (m, 1H), 2.18 (br s, 4H), 2.05-2.00 (br m, 1H), 1.92 (d, 3H), 1.87 (br s, 1H), 1.54 (br s, 1H), 1.17 (d, 3H), 1.11 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,19 мин.
МС (ЭР+): 558,4 (М+Н)+.
ПРИМЕР 51: 1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}этил-2-метилпропаноат (диастереомер В)
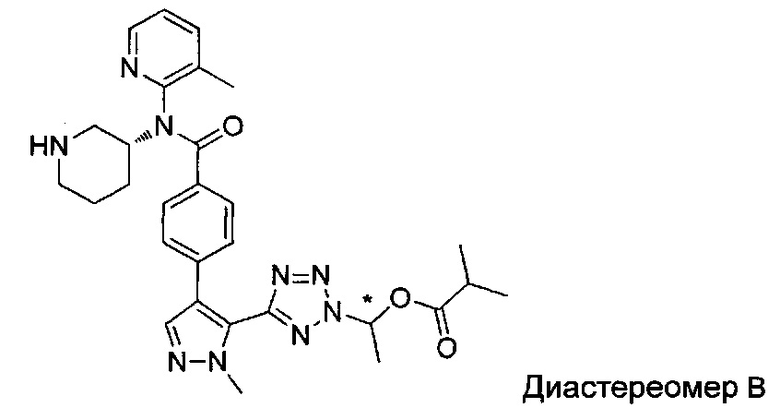
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 55.
1Н ЯМР (CD3CN) δ: 9.75 (br s, 1Н), 9.31 (br s, 1Н), 8.47 (s, 1Н), 7.98 (br s, 1H), 7.68 (s, 1H), 7.60 (br s, 1H), 7.38-7.32 (m, 3H), 7.24-7.20 (br m, 2H), 5.05 (br s, 1H), 3.99 (s, 3H), 3.87 (br s, 1H), 3.68 (br s, 1H), 3.33 (br s, 1H), 2.88 (br s, 1H), 2.64-2.59 (m, 1H), 2.26 (br s, 4H), 2.07-2.01 (m, 1H), 1.94-1.86 (m, 4H), 1.60 (br s, 1H), 1.17 (d, 3H), 1.10 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,19 мин.
МС (ЭР+): 558,4 (М+Н)+.
ПРИМЕР 52: 1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропил-2-метилпропаноат (диастереомер А)
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 57.
1Н ЯМР (DMSO-d6) δ: 9.08-9.03 (m, 1Н), 8.90-8.75 (m, 1Н), 8.49-8.39 (m, 1Н), 7.88 (s, 1Н), 7.66-7.57 (m, 1Н), 7.32-7.27 (m, 1Н), 7.24-7.21 (m, 1Н), 7.15-7.10 (m, 3Н), 5.0-4.98 (m, 1Н), 3.92 (s, 3H), 3.60-3.52 (m, 1Н), 3.44-3.32 (m, 1Н), 3.23-3.15 (m, 1Н), 2.78-2.59 (m, 1Н), 2.34-2.20 (m, 2Н), 2.17-2.04 (m, 1Н), 1.96 (s, 3H), 1.83-1.68 (m, 2Н), 1.28-1.24 (m, 1Н), 1.20-1.16 (m, 1Н), 1.13 (d, 3H), 1.05 (d, 3H), 0.90 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 0,69 мин.
МС (ЭР+): 572,4 (М+Н)+.
ПРИМЕР 53: 1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропил-2-метилпропаноат (диастереомер В)
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 58.
1Н ЯМР (CDCl3) δ: 9.84 (br s, 1Н), 9.53 (br s, 1Н), 8.43 (br s, 1H), 7.59 (s, 1H), 7.43 (br s, 1H), 7.23-7.07 (m, 5H), 5.14-4.98 (m, 1H), 4.04 (s, 3H), 3.92-3.74 (m, 2H), 3.57-3.25 (m, 1H), 2.94-2.78 (m, 1H), 2.68-2.54 (m, 1H), 2.35-2.25 (m, 2H), 2.21-1.78 (m, 5H), 1.67-1.49 (m, 1H), 1.19 (t, 3H), 1.13 (t, 3H), 0.95 (t, 3H).
УЭЖХ (УЭЖХ-МС, способ 1): tR составляет 0,66 мин.
МС (ЭР+): 572,4 (М+Н)+.
ПРИМЕР 54: 2-метил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропил-2-метилпропаноат (диастереомер А)
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 60.
1Н ЯМР (CD3CN) δ: 9.61 (br s, 1Н), 9.20 (br s, 1Н), 8.43 (d, 1Н), 7.75 (br s, 1H), 7.67 (s, 1H), 7.43 (br s, 1H), 7.28 (br s, 2H), 7.17 (br s, 2H), 6.93 (d, 1H), 5.05 (br s, 1H), 3.98 (s, 3H), 3.79 (br s, 1H), 3.64 (br s, 1H), 3.31 (br d, 1H), 2.83 (br s, 1H), 2.69-2.57 (m, 2H), 2.15 (br s, 4H), 2.02 (br s, 1H), 1.84 (br s, 1H), 1.50 (br s, 1H), 1.19 (d, 3H), 1.12-1.11 (m, 6H), 0.84 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,34 мин.
МС (ЭР+): 586,4 (М+Н)+.
ПРИМЕР 55: 2-метил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропил-2-метилпропаноат (диастереомер В)
Указанное в заголовке соединение получали способом, аналогичным способу по ПРИМЕРУ 32, начиная с соединения по Получению 61.
1Н ЯМР (CDCl3) δ: 9.94 (br s, 1Н), 9.61 (br s, 1Н), 8.78-8.44 (m, 1Н), 7.94 (Ьг s, 1Н), 7.60 (s, 1Н), 7.30 (br s, 1Н), 7.18 (br s, 2Н), 6.87 (d, 1Н), 5.03-4.64 (m, 1Н), 4.25 (br s, 1Н), 4.04 (s, 3H), 3.90-3.72 (m, 2Н), 3.54-3.45 (m, 1Н), 2.98-2.88 (m, 1Н), 2.70-2.61 (m, 2H), 2.50-2.06 (m, 4H), 1.31-1.23 (m, 2H), 1.21 (d, 3H), 1.15 (d, 3H), 1.10 (d, 3H), 0.85 (d, 3H).
УЭЖХ (УЭЖХ-МС, способ 2): tR составляет 1,29 мин.
МС (ЭР+): 586,4 (М+Н)+.
В этой заявке даны ссылки на различные публикации. Раскрытия этих публикаций во всей их полноте включены посредством ссылки в этой заявке для любых целей.
Специалистам в данной области техники будет очевидно, что различные модификации и варианты могут быть осуществлены в настоящем изобретении, без отступления от объема или сущности изобретения. Другие воплощения изобретения будут очевидны специалистам в данной области техники из рассмотрения описания и практического осуществления изобретения, раскрытых здесь. Предполагается, что описание и примеры будут рассмотрены только в качестве иллюстраций.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2797295C1 |
| Новое соединение 2,3,5-замещенного тиофена в качестве ингибитора протеинкиназы | 2017 |
|
RU2724957C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| АНТАГОНИСТЫ TLR7/8 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2833035C2 |
| СОЕДИНЕНИЯ ПИРИДАЗИНАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ СЕЛЕЗЕНКИ (SYK) | 2013 |
|
RU2627661C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНОВ | 2011 |
|
RU2554353C2 |
| ПИРАЗОЛО[3,4-С]ПИРИДИНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2638116C2 |
| 6-ГЕТЕРОЦИКЛИЛ-4-МОРФОЛИН-4-ИЛПИРИДИН-2-ОНЫ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РАКА И ДИАБЕТА | 2017 |
|
RU2762968C2 |
| ЗАМЕЩЕННЫЕ [1,2,4]ТРИАЗОЛО[1,5-a]ПИРИМИДИН-7-ИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE2 | 2015 |
|
RU2659070C9 |
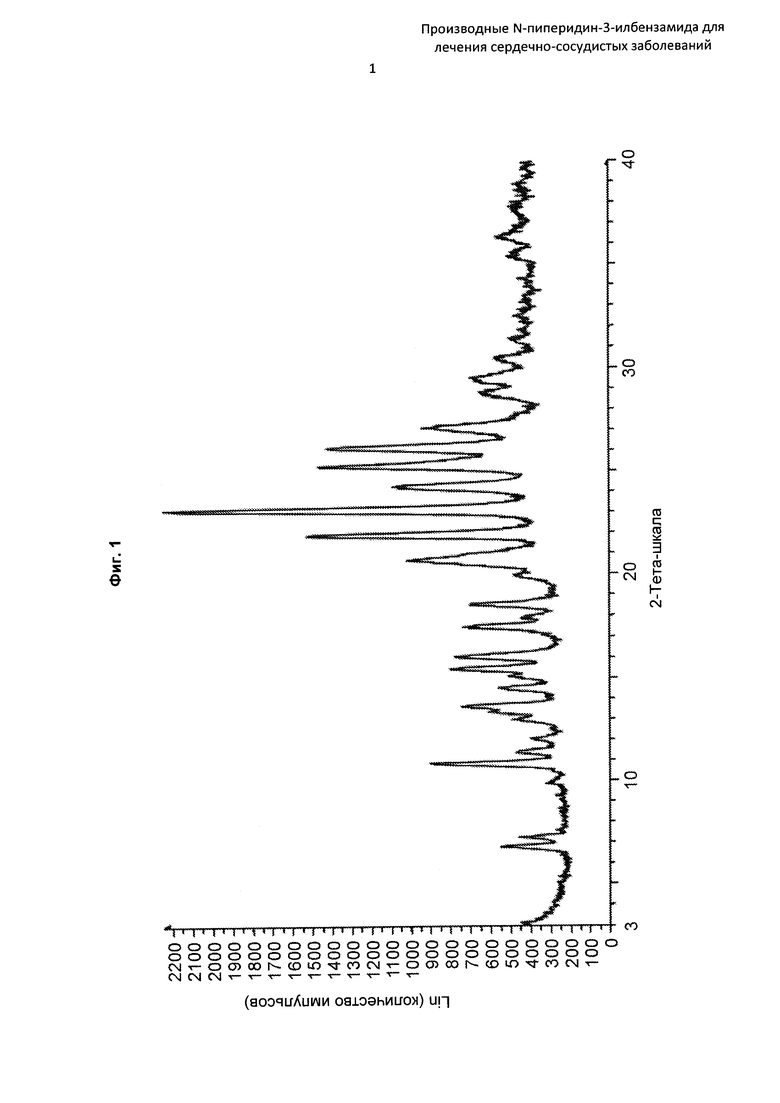
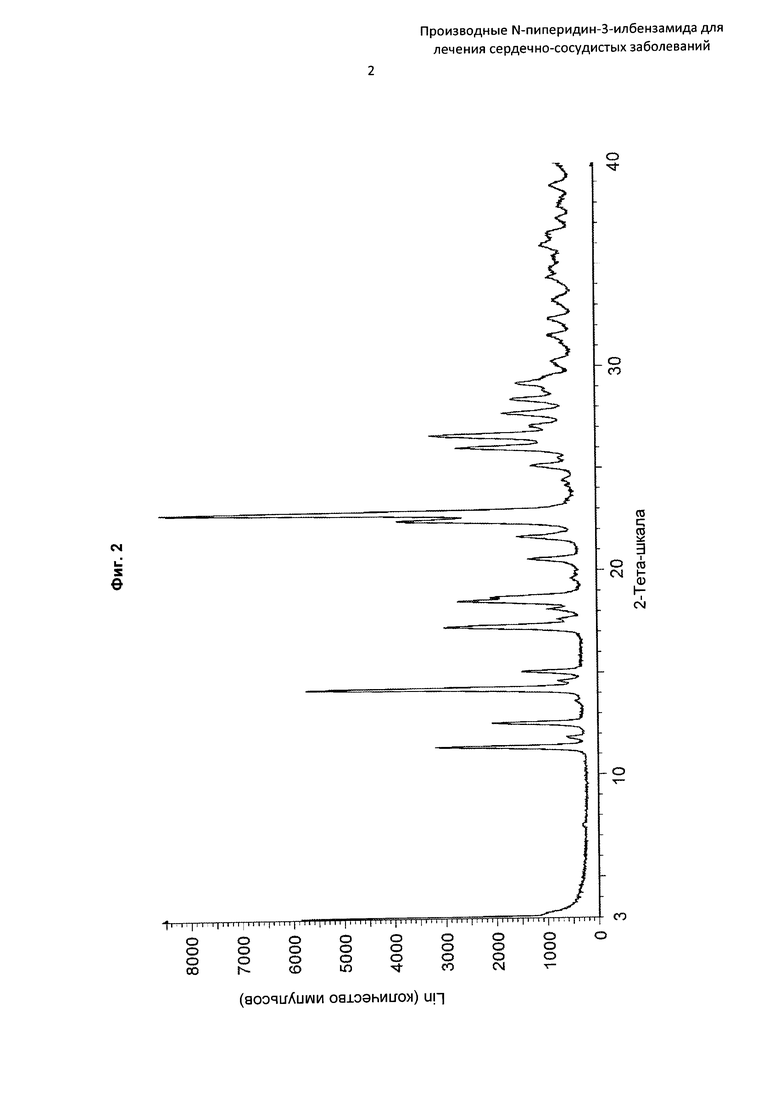
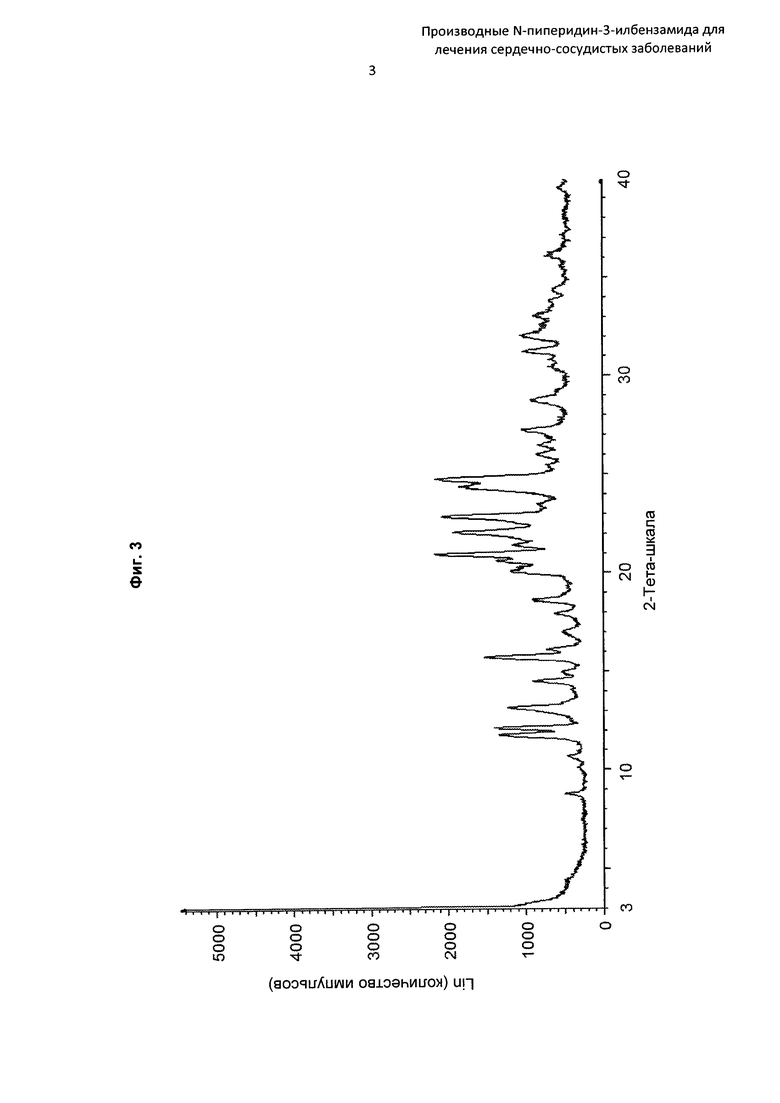
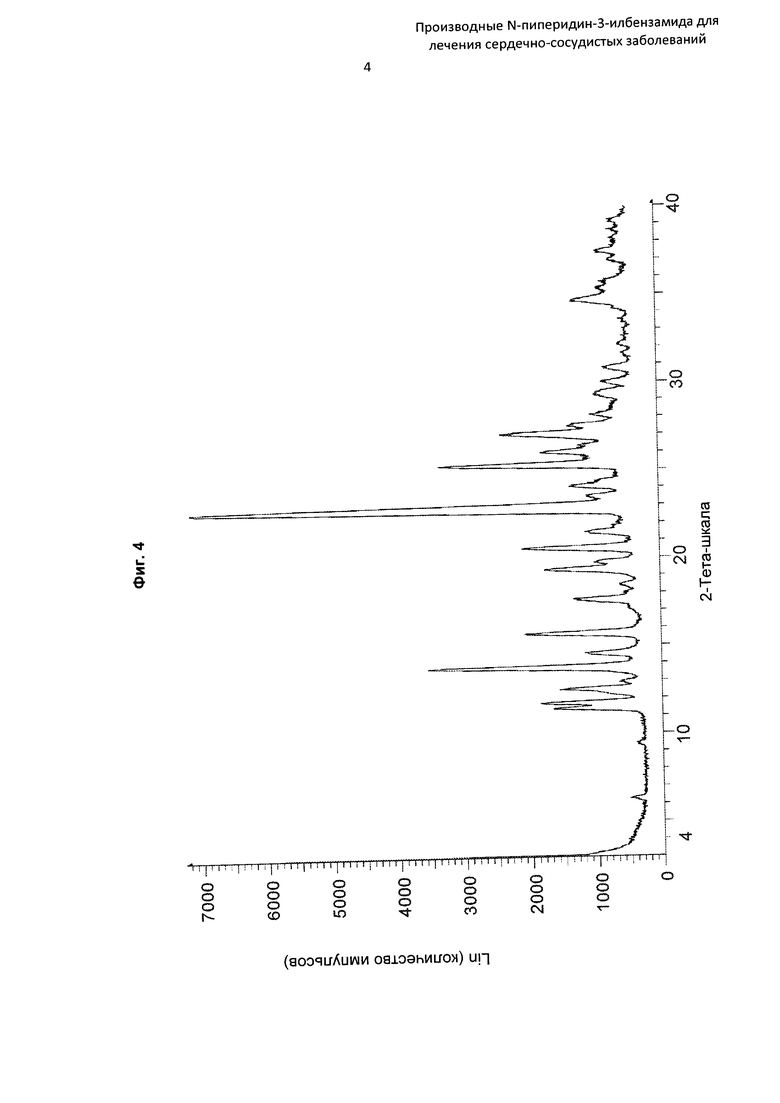
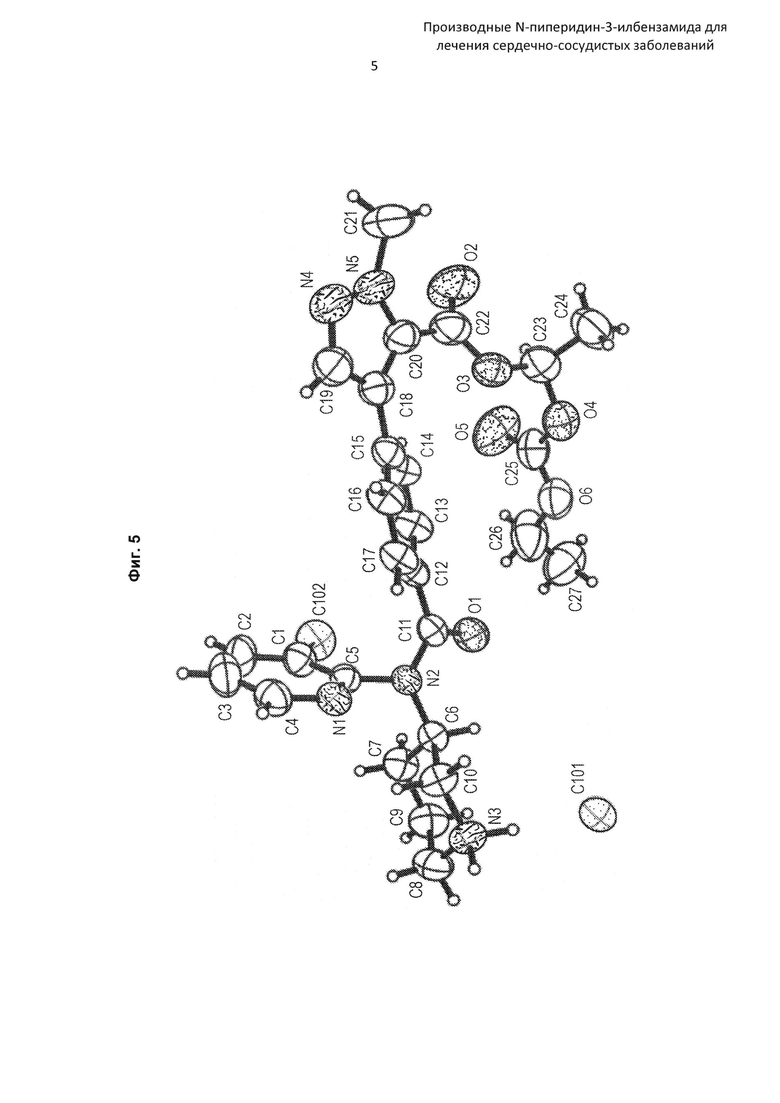
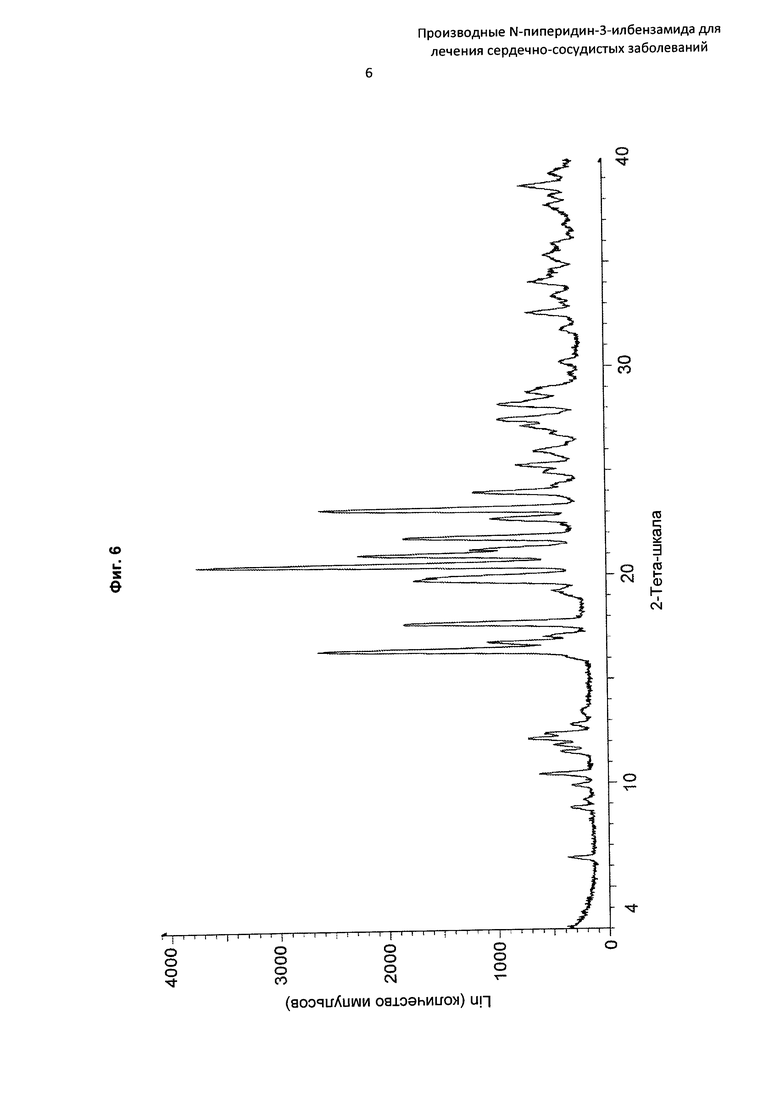
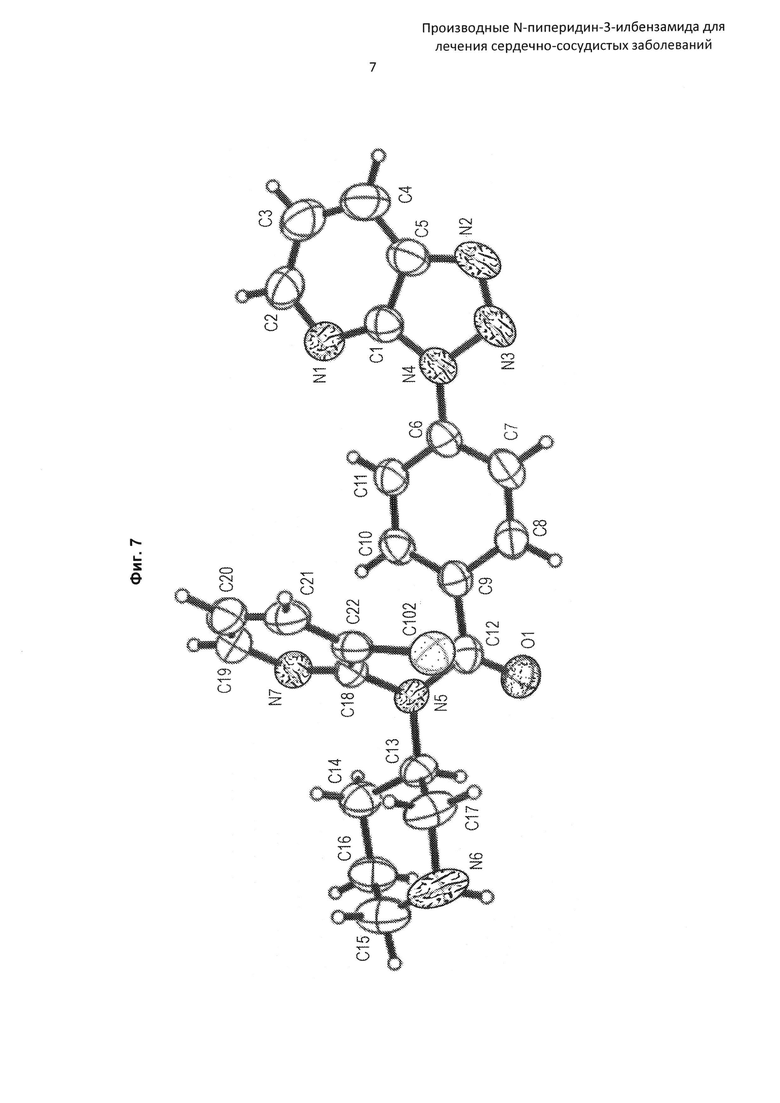
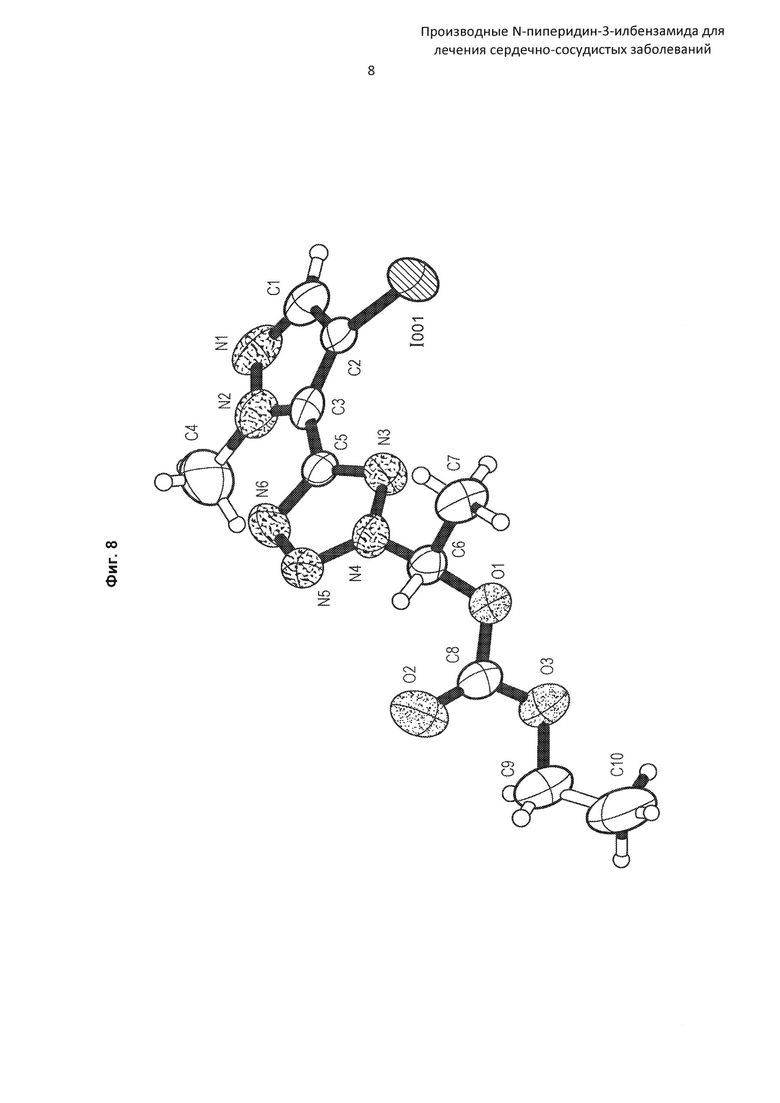
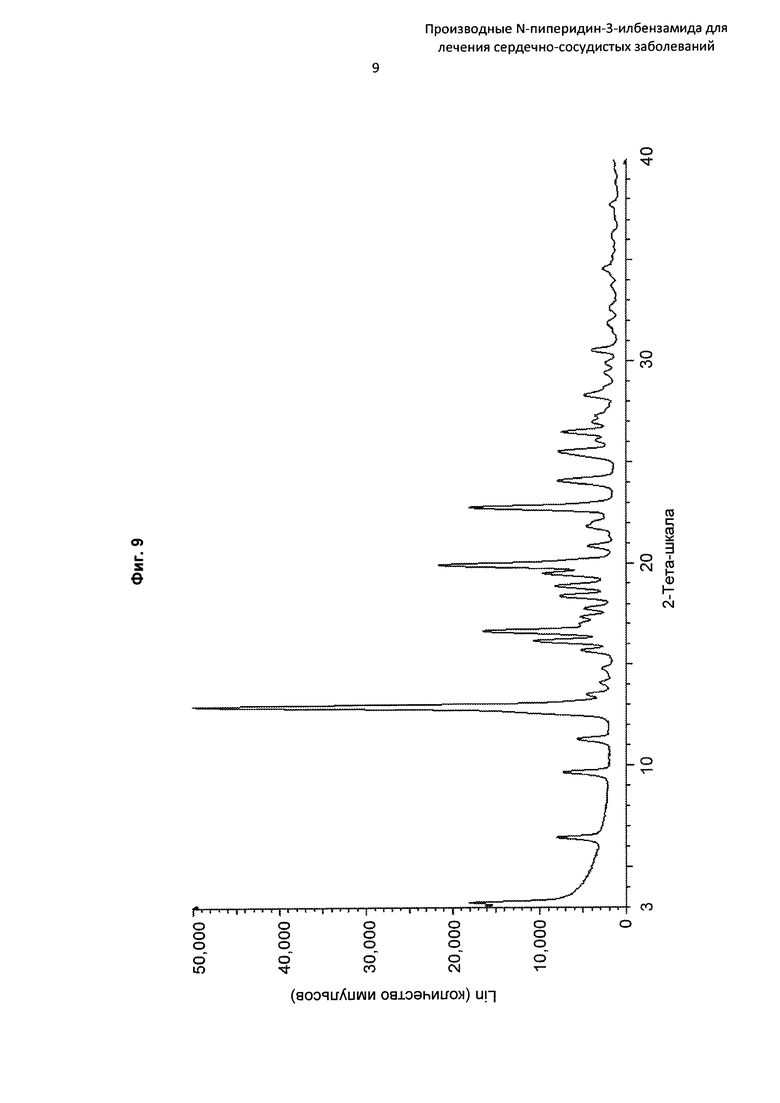
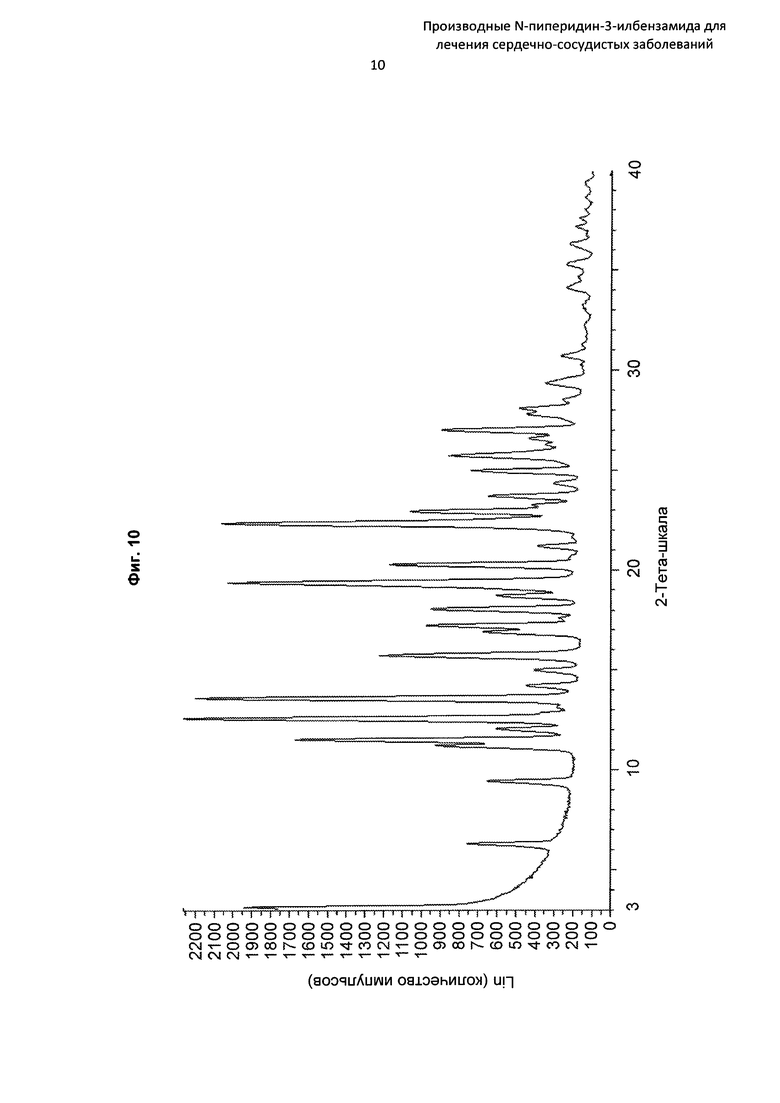
Настоящее изобретение относится к замещенным амидным соединениям формулы (I), фармацевтическим композициям, содержащим такие соединения, и применению таких соединений для снижения в плазме крови уровней липидов, таких как холестерин липопротеинов низкой плотности (LDL-холестерин) и триглицериды, и, таким образом, для лечения заболеваний, которые усугубляются вследствие высоких уровней LDL-холестерина и триглицеридов, таких как атеросклероз и сердечно-сосудистые заболевания, у млекопитающих, включая людей. 13 н. и 29 з.п. ф-лы, 10 ил., 55 пр.
.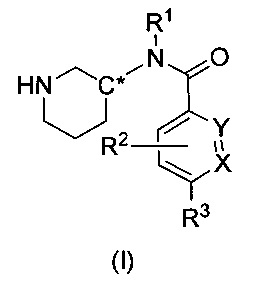
1. Соединение формулы I
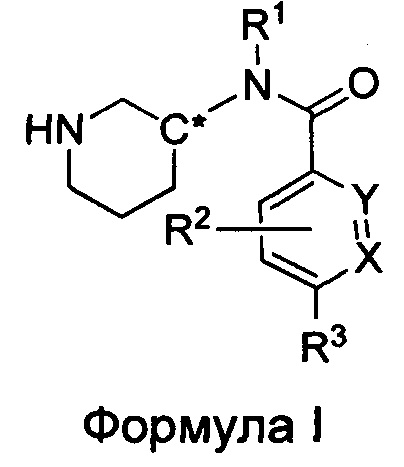
или его фармацевтически приемлемая соль, где
R1 представляет собой пирид-2-ил, изохинолин-1-ил или 1Н-пирроло[2,3-с]пиридин-7-ил;
R1, возможно, монозамещен хлором или (С1-С4)алкилом;
X и Y независимо представляют собой либо N, либо С(Н), при условии, что по меньшей мере один из X или Y представляет собой С(Н);
R2 представляет собой Н;
R3 представляет собой
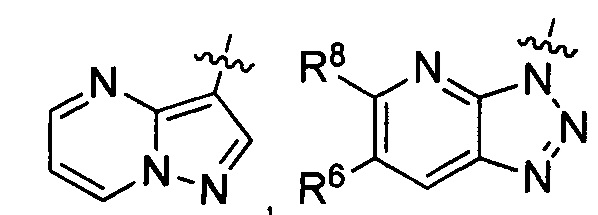 или
или 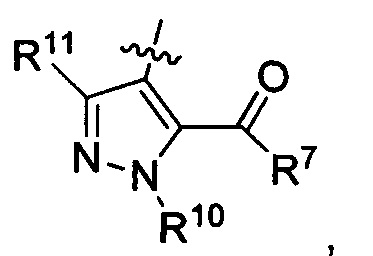
где каждый из R6 и R8 независимо представляет собой Н или метил;
где каждый из R10 и R11 независимо представляет собой Н, (С1-С4)алкил или (С3-С5)циклоалкил;
где R7 представляет собой гидроксил, (С1-С4)алкилокси, (С1-С4)алкоксикарбонилокси(С1-С4)алкилокси или (С1-С4)алкилкарбонилокси(С1-С4)алкокси; и
где пиперидинил С* имеет R-конфигурацию.
2. Соединение по п. 1, где R1 представляет собой пирид-2-ил.
3. Соединение по п. 2, где X и Y оба представляют собой С(Н) и R1, возможно, монозамещен хлором или метилом.
4. Соединение по п. 3, где R3 представляет собой
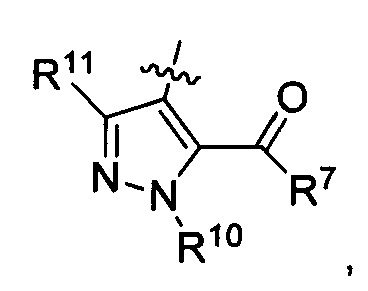
R7 представляет собой гидроксил, (С1-С2)алкилокси или 
R10 представляет собой метил; и
R11 представляет собой Н.
5. Соединение по п. 3, где R3 представляет собой
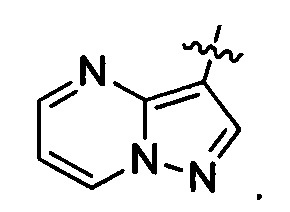
6. Соединение по п. 3, где R3 представляет собой
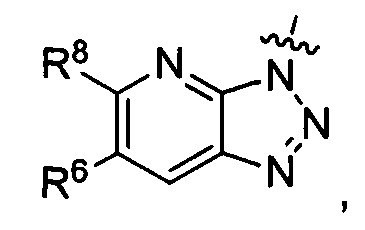
R6 представляет собой Н или метил; и
R8 представляет собой Н.
7. Соединение по п. 1, где R1 представляет собой изохинолин-1-ил.
8. Соединение по п. 7, где X и Y оба представляют собой С(Н) и R1, возможно, монозамещен хлором или метилом.
9. Соединение по п. 8, где R3 представляет собой
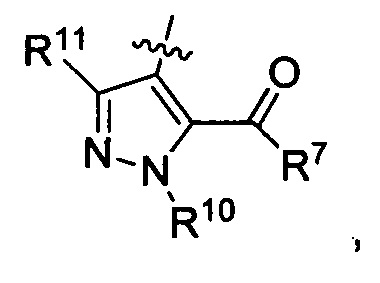
R7 представляет собой гидроксил, (С1-С2)алкилокси или 
R10 представляет собой метил; и
R11 представляет собой Н.
10. Соединение по п. 8, где R3 представляет собой
11. Соединение по п. 8, где R3 представляет собой
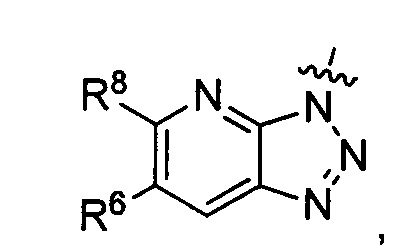
R6 представляет собой Н или метил; и
R8 представляет собой Н.
12. Соединение по п. 1, где R1 представляет собой 1Н-пирроло[2,3-с]пиридин-7-ил.
13. Соединение по п. 12, где X и Y оба представляют собой С(Н) и R1, возможно, монозамещен хлором или метилом.
14. Соединение по п. 13, где R3 представляет собой
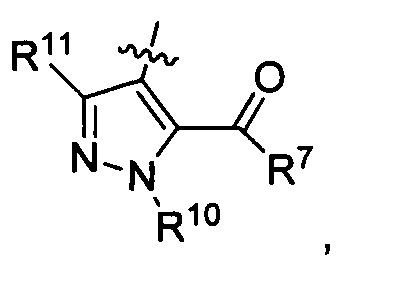
R7 представляет собой гидроксил, (С1-С2)алкилокси или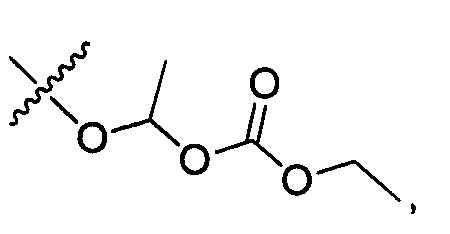
R10 представляет собой метил; и
R11 представляет собой Н.
15. Соединение по п. 13, где R3 представляет собой
16. Соединение по п. 13, где R3 представляет собой
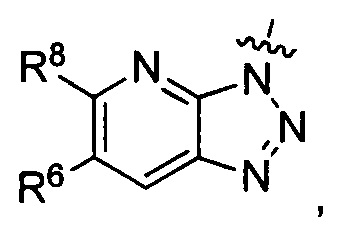
R6 представляет собой Н или метил; и
R8 представляет собой Н.
17. Соединение
N-(3-метилпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(пиразоло[1,5-а]пиримидин-3-ил)бензамид;
N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(пиразоло[1,5-а]пиримидин-3-ил)бензамид;
N-(3-хлорпиридин-2-ил)-4-(6-метил-3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-N-[(3R)-пиперидин-3-ил]бензамид;
4-(4-{изохинолин-1-ил[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоновая кислота;
N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-5-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)пиридин-2-карбоксамид;
этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат;
4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоновая кислота;
1-[(этоксикарбонил)окси]этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат;
(1R)-1-[(этоксикарбонил)окси]этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат;
(1S)-1-[(этоксикарбонил)окси]этил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат; или
N-(3-хлорпиридин-2-ил)-N-[(3R)-пиперидин-3-ил]-4-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)бензамид;
или фармацевтически приемлемая соль любого из указанных соединений.
18. Соединение
N-(3-хлорпиридин-2-ил)-5-(6-метил-3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-N-[(3R)-пиперидин-3-ил]пиридин-2-карбоксамид;
метил-4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат;
1-[(этоксикарбонил)окси]этил-4-(4-{изохинолин-1-ил[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-1Н-пиразол-5-карбоксилат;
1-метил-4-(4-{(1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-карбоновая кислота;
метил-1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-карбоксилат; или
1-[(этоксикарбонил)окси]этил-1-метил-4-(4-{(1-метил-1Н-пирроло[2,3-с]пиридин-7-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-карбоксилат; или
фармацевтически приемлемая соль любого из указанных соединений.
19. Способ лечения дислипидемии, гиперхолестеринемии, гипертриглицеридемии, гиперлипидемии, гипо-альфа-липопротеинемии, метаболического синдрома, осложнений диабета, атеросклероза, инсульта, сосудистой деменции, хронической почечной недостаточности, ишемической болезни сердца, коронарной артериальной болезни, ретинопатии, воспаления, тромбоза, заболевания периферических сосудов или застойной сердечной недостаточности у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по п. 1 или фармацевтически приемлемой соли указанного соединения.
20. Фармацевтическая композиция для ингибирования пропротеинконвертазы субтилизин/кексин типа 9 (PCSK9), содержащая терапевтически эффективное количество соединения по п. 1 или фармацевтически приемлемой соли указанного соединения и фармацевтически приемлемый носитель, наполнитель или разбавитель.
21. Способ лечения дислипидемии, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения по п. 1, которое селективно ингибирует трансляцию мРНК, кодирующей PCSK9 (пропротеинконвертазу субтилизин/кексин типа 9), в белок PCSK9.
22. Способ по п. 21, где соединение вводят путем перорального введения.
23. Способ по п. 22, где соединение имеет молекулярную массу (MW) от примерно 300 до примерно 650.
24. Способ по п. 22, где дислипидемия характеризуется повышенным уровнем LDL (липопротеинов низкой плотности).
25. Способ по п. 23, где терапевтически эффективное количество составляет от примерно 1 мг до примерно 4000 мг в сутки.
26. Способ по п. 23, где терапевтически эффективное количество составляет от примерно 1 мг до примерно 2000 мг в сутки.
27. Способ по п. 23, где терапевтически эффективное количество составляет от примерно 50 мг до примерно 500 мг в сутки.
28. Способ по п. 24, где соединение имеет значение IC50 (полумаксимальной ингибирующей концентрации) ниже чем примерно 50 мкМ в бесклеточном анализе PCSK9.
29. Способ по п. 24, где соединение имеет значение IC50 ниже чем примерно 30 мкМ в бесклеточном анализе PCSK9.
30. Способ по п. 24, где соединение имеет значение IC50 ниже чем примерно 20 мкМ в бесклеточном анализе PCSK9.
31. Способ по п. 23, где селективное ингибирование PCSK9 является таким, что происходит ингибирование менее чем 1% белков, не являющихся PCSK9, в глобальном протеомном анализе.
32. Способ по п. 23, где селективное ингибирование PCSK9 является таким, что происходит ингибирование менее чем 0,5% белков, не являющихся PCSK9, в глобальном протеомном анализе.
33. Способ по п. 23, где селективное ингибирование PCSK9 является таким, что происходит ингибирование менее чем 0,1% белков, не являющихся PCSK9, в глобальном протеомном анализе.
34. Соединение формулы II
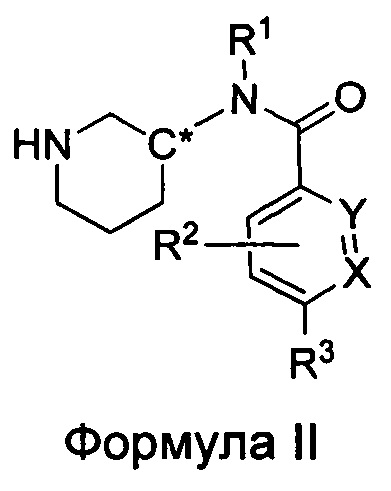
или его фармацевтически приемлемая соль, где
R1 представляет собой пирид-2-ил, изохинолин-1-ил или 1Н-пирроло[2,3-с]пиридин-7-ил;
R1, возможно, монозамещен хлором или (С1-С4)алкилом;
X и Y независимо представляют собой либо N, либо С(Н), при условии, что по меньшей мере один из X или Y представляет собой С(Н);
R2 представляет собой Н;
R3 представляет собой
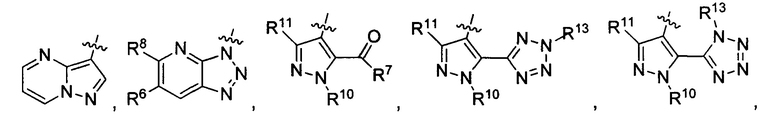
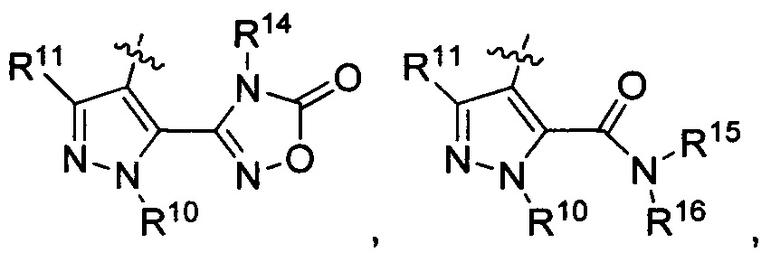
где каждый из R6 и R8 независимо представляет собой Н или метил;
где каждый из R10 и R11 независимо представляет собой Н, (С1-С4)алкил или (С3-С5)циклоалкил;
где R7 представляет собой гидроксил, (С1-С4)алкилокси, (С1-С4)алкоксикарбонилокси(С1-С4)алкилокси или (С1-С4)алкилкарбонилокси(С1-С4)алкокси;
R13 представляет собой Н, (С1-С4)алкил, (С1-С4)алкилкарбонилокси(С1-С4)алкил или (С1-С4)алкоксикарбонилокси(С1-С4)алкил;
R14 представляет собой Н, (С1-С4)алкил, (С1-С4)алкилкарбонилокси(С1-С4)алкил или (С1-С4)алкоксикарбонилокси(С1-С4)алкил;
R15 представляет собой гидроксил, тетразолил, (С1-С2)алкилсульфонил или трифторметилсульфонил;
R16 представляет собой Н, (С1-С4)алкил, (С1-С4)алкилкарбонилокси(С1-С4)алкил или (С1-С4)алкоксикарбонилокси(С1-С4)алкил; и
где пиперидинил С* имеет (R)-конфигурацию.
35. Соединение по п. 34, где
R1 представляет собой пирид-2-ил, возможно, монозамещенный хлором или метилом;
X и Y оба представляют собой С(Н);
R3 представляет собой
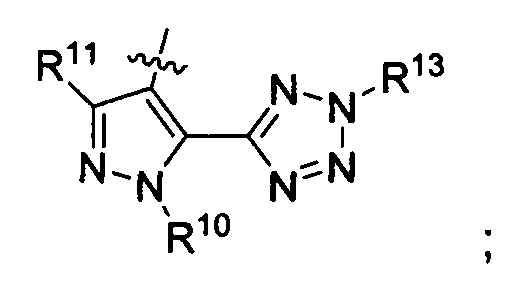
R10 представляет собой метил;
R11 представляет собой Н; и
R13 представляет собой (С1-С4)алкилкарбонилокси(С1-С4)алкил.
36. Соединение по п. 34, где
R1 представляет собой пирид-2-ил, возможно, монозамещенный хлором или метилом;
X и Y оба представляют собой С(Н); и
R3 представляет собой 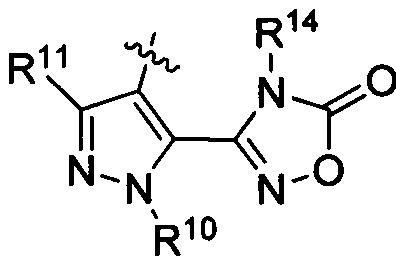 или
или 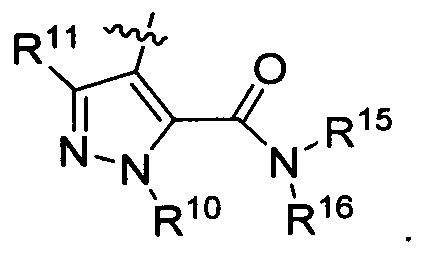
37. Соединение
4-(4-{(3-хлорпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1-метил-N-[(трифторметил)сульфонил]-1Н-пиразол-5-карбоксамид;
N-(3-хлорпиридин-2-ил)-4-[1-метил-5-(2Н-тетразол-5-ил)-1Н-пиразол-4-ил]-N-[(3R)-пиперидин-3-ил]бензамид;
1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-N-(метилсульфонил)-1Н-пиразол-5-карбоксамид;
N-(3-метилпиридин-2-ил)-4-[1-метил-5-(2Н-тетразол-5-ил)-1Н-пиразол-4-ил]-N-[(3R)-пиперидин-3-ил]бензамид; или
этил-1-[{[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]карбонил}(метилсульфонил)амино]этилкарбонат;
или фармацевтически приемлемая соль любого из указанных соединений.
38. Соединение
этил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-1Н-тетразол-1-ил}этилкарбонат;
этил-(1S)-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}этилкарбонат; или
этил-(1R)-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}этилкарбонат;
или фармацевтически приемлемая соль любого из указанных соединений.
39. Соединение
1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}этил-2-метилпропаноат (Диастереомер В; Пример 51)
или
2-метил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропил-2-метилпропаноат (Диастереомер В; Пример 55)
или фармацевтически приемлемая соль любого из указанных соединений.
40. Соединение
1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}этил-2-метилпропаноат; или
2-метил-1-{5-[1-метил-4-(4-{(3-метилпиридин-2-ил)[(3R)-пиперидин-3-ил]карбамоил}фенил)-1Н-пиразол-5-ил]-2Н-тетразол-2-ил}пропил-2-метилпропаноат;
или фармацевтически приемлемая соль любого из указанных соединений.
41. Способ лечения дислипидемии, гиперхолестеринемии, гипертриглицеридемии, гиперлипидемии, гипо-альфа-липопротеинемии, метаболического синдрома, осложнений диабета, атеросклероза, инсульта, сосудистой деменции, хронической почечной недостаточности, ишемической болезни сердца, коронарной артериальной болезни, ретинопатии, воспаления, тромбоза, заболевания периферических сосудов или застойной сердечной недостаточности у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по п. 34 или фармацевтически приемлемой соли указанного соединения.
42. Фармацевтическая композиция для ингибирования пропротеинконвертазы субтилизин/кексин типа 9 (PCSK9), содержащая терапевтически эффективное количество соединения по п. 34 или фармацевтически приемлемой соли указанного соединения и фармацевтически приемлемый носитель, наполнитель или разбавитель.
| Колосоуборка | 1923 |
|
SU2009A1 |
| ЕА 2012270492 А1, 30.11.2012 | |||
| NABIL G SEIDAH, PCSK9 as a therapeutic target of dyslipidemia, Expert Opinion on Therapeutic Targets, 2009, vol.13, N1, pages 19-28 | |||
| ПРОИЗВОДНЫЕ 4-АМИНОПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2266282C2 |

