ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке в соответствии с 35 U.S.С. 119 (е) испрашивается приоритет по предварительной заявке U.S. №61/771291, поданной 1 марта 2013 г., полное содержание которой явно включено в настоящее изобретение в качестве ссылки.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
1. Область техники, к которой относится изобретение (и предлагаемые в нем решения)
Настоящее изобретение (и предлагаемые в нем решения) в целом относится к полимерам ацетат-сукцината гидроксипропилметилцеллюлозы (ГПМЦ-АС), обладающим уникальной схемой замещения, к способам получения полимеров и к композициям лекарственных средств, содержащим полимеры и обладающие низкой эффективностью лекарственные средства. Композиции лекарственных средств обладают повышенной эффективностью и/или улучшенной обрабатываемостью.
2. Уровень техники и применимые аспекты настоящего изобретения (и предлагаемых в нем решений)
Для обеспечения определенного необходимого терапевтического воздействия фармацевтические композиции часто содержат полимеры, включая полимеры, предназначенные для применения в качестве агентов для нанесения покрытий, в качестве пленкообразователей, в качестве регулирующих скорость полимеров для обеспечения пролонгированного или регулируемого высвобождения, в качестве стабилизирующих агентов, в качестве суспендирующих агентов, в качестве связующих для таблеток и в качестве агентов, повышающих вязкость.
ГПМЦ-АС был разработан изначально в качестве энтеросолюбильного полимера, предназначенного для фармацевтических дозированных форм и для нанесения слоя на фотографические пленки, предупреждающего образования ореола. Энтеросолюбильными полимерами являются полимеры, которые остаются неизменными в кислой среде желудка; нанесенное на дозированные формы покрытие из таких полимеров, предупреждает инактивацию и разложение лекарственного средства в кислой среде или предупреждает раздражение желудка лекарственным средством. В настоящее время ГПМЦ-АС продаются фирмой Shin-Etsu Chemical (Tokyo, Japan), они известны под торговым названием "AQOAT".
Shin-Etsu производит AQOAT трех марок, которые обладают разной комбинацией содержаний заместителей, обеспечивающие защиту в кишечнике при разных значениях рН. АС марки LF и АС марки LG ("F" обозначает тонкодисперсный и "G" обозначает гранулированный) обеспечивают защиту в кишечнике до значения рН, равного примерно 5,5. АС марки MF и АС марки MG обеспечивают защиту в кишечнике до значения рН, равного примерно 6,0, тогда как АС марки HF и АС марки HG обеспечивают защиту в кишечнике до значения рН, равного примерно 6,8. Фирма Shin-Etsu предоставляет следующие спецификации для полимеров AQOAT этих трех марок:

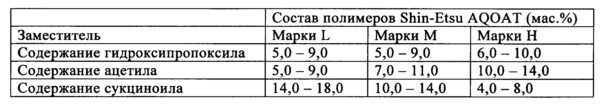
Хотя доказано, что фармацевтические препараты, содержащие обладающие низкой растворимостью лекарственные средства и ГПМЦ-АС, являются эффективными, полимеры AQOAT, выпускающиеся фирмой Shin-Etsu, можно использовать для улучшения растворимости очень небольшой группы лекарственных средств. Кроме того, использование выпускающихся фирмой Shin-Etsu полимеров приводит к затруднениям при обработке лекарственных средств. Необходимо найти новые этерифицированные простые эфиры целлюлозы с целью улучшения растворимости и улучшения обрабатываемости большего количества разных лекарственных средств.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг. 1 приведен график зависимости динамической вязкости Eta* от угловой частоты для образцов ГПМЦ-АС марки Н: Shin-Etsu AQOAT HF, полимера 1, полимера 3 и полимера 7.
На фиг. 2 приведен график зависимости динамической вязкости Eta* от температуры для образцов ГПМЦ-АС марки Н: Shin-Etsu AQOAT HF, полимера 1, полимера 3 и полимера 7.
На фиг. 3 приведен график зависимости динамической вязкости Eta* от угловой частоты для образцов ГПМЦ-АС марки М: Shin-Etsu AQOAT MF, полимера 2, полимера 6 и полимера 9.
На фиг. 4 приведен график зависимости динамической вязкости Eta* от температуры для образцов ГПМЦ-АС марки М: Shin-Etsu AQOAT MF, полимера 2, полимера 6 и полимера 9.
На фиг. 5 приведен график зависимости динамической вязкости Eta* от угловой частоты для образцов ГПМЦ-АС марки L: Shin-Etsu AQOAT LF, полимера 4, полимера 5 и полимера 8.
На фиг. 6 приведен график зависимости динамической вязкости Eta* от температуры для образцов ГПМЦ-АС марки L: Shin-Etsu AQOAT LF, полимера 4, полимера 5 и полимера 8.
На фиг. 7 приведен график зависимости динамической вязкости Eta* от угловой частоты для образцов ГПМЦ-АС марки Н: Shin-Etsu AQOAT HF, полимера 13 и полимера 16.
На фиг. 8 приведен график зависимости динамической вязкости Eta* от температуры для образцов ГПМЦ-АС марки Н: Shin-Etsu AQOAT HF, полимера 13 и полимера 16.
На фиг. 9 приведен график зависимости динамической вязкости Eta* от угловой частоты для образцов ГПМЦ-АС марки Н: Shin-Etsu AQOAT HF, полимера 12 и полимера 17.
На фиг. 10 приведен график зависимости динамической вязкости Eta* от температуры для образцов ГПМЦ-АС марки Н: Shin-Etsu AQOAT HF, полимера 12 и полимера 17.
На фиг. 11 приведен график зависимости динамической вязкости Eta* от угловой частоты для образцов ГПМЦ-АС марки L: Shin-Etsu AQOAT LF, полимера 18 и полимера 19.
На фиг. 12 приведен график зависимости динамической вязкости Eta* от температуры для образцов ГПМЦ-АС марки L: Shin-Etsu AQOAT LF, полимера 18 и полимера 19.
На фиг. 13 приведен график зависимости модулей G' и G" от температуры для полимера 1.
На фиг. 14 приведен график зависимости модулей G' и G" от температуры для полимера 3.
На фиг. 15 приведен график зависимости модулей G' и G" от температуры для полимера 7.
На фиг. 16 приведен график зависимости модулей G' и G" от температуры для Shin-Etsu AQOAT HF.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Прежде чем подробно разъяснять по меньшей мере один вариант осуществления настоящего изобретения (и предлагаемых в нем решений) с помощью типичных чертежей, экспериментов, результатов и лабораторных методик следует понять, что настоящее изобретение (и предлагаемые в нем решения) в своем применении не ограничиваются особенностями строения и расположения компонентов, указанными в последующем описании или проиллюстрированными с помощью чертежей, экспериментов и/или результатов. Для настоящего изобретения (и предлагаемых в нем решений) возможны другие варианты осуществления или его можно осуществить на практике или выполнить разными путями. Само по себе приведенное изложение предназначено для описания наибольшего возможного объема и содержания; и варианты осуществления являются типичными, а не ограничивающими. Также следует понимать, что использующиеся в настоящем изобретении фразеология и терминология предназначены для описания и их не следует рассматривать в качестве ограничивающих.
Если в настоящем изобретении не приведено другое определение, то научные и технические термины, использующиеся в настоящем изобретении (и предлагаемых в нем решениях), обладают значениями, которые обычно понятны специалистам с общей подготовкой в данной области техники. Кроме того, если иное не следует из контекста, термины во множественном числе включают термины в единственном числе и термины в единственном числе включают термины во множественном числе. Обычно использующаяся номенклатура и химические методики хорошо известны и обычно используются в данной области техники. Реакции и методики очистки выполняются в соответствии со спецификациями изготовителя или так, как обычно принято в данной области техники или описано в настоящем изобретении. Обычно использующаяся номенклатура и лабораторные процедуры и методики аналитической химии, синтетической органической химии и медицинской и фармацевтической химии, описанные в настоящем изобретении, хорошо известны и обычно используются в данной области техники. Для химических синтезов, химического анализа, приготовления фармацевтических препаратов, составов и доставки и лечения пациентов используются стандартные методики. Все патенты, опубликованные заявки на патенты и непатентные публикации, указанные в описании, указывают на уровень подготовки специалистов в области техники, к которой относится настоящее изобретение (и предлагаемые в нем решения). Все патенты, опубликованные заявки на патенты и непатентные публикации, указанные в любой части настоящей заявки, явно включены в нее в качестве ссылки во всей своей полноте в такой степени, как если бы специально и по отдельности для каждого отдельного патента или публикации них было указано о включении в качестве ссылки.
Все композиции и/или методики, раскрытые и заявленные в настоящем изобретении, с учетом настоящего раскрытия можно получить и выполнить без чрезмерного количества экспериментальных исследований. Хотя композиции и способы, предлагаемые в настоящем изобретении, описаны с помощью предпочтительных вариантов осуществления, для специалистов в данной области техники должно быть понятно, что без отклонения от основных положений, сущности и объема настоящего изобретения могут быть внесены изменения в композиции и/или способы и в стадии или в последовательность стадий способа, описанного в настоящем изобретении. Предполагается, что все такие аналогичные замены и модификации, очевидные для специалистов в данной области техники, входят в основные положения, сущность и объем настоящего изобретения (и предлагаемых в нем решений), определенные в прилагаемой формуле изобретения.
При использовании в настоящем изобретении приведенные ниже термины, если не указано иное, обладают указанными ниже значениями: Использование слова в единственном числе вместе с термином "включающий" в формуле изобретения и/или описании можно означать термин в единственном числе, а также "один или большее количество", "по меньшей мере один" и "один или больше, чем один". Термин "или" в формуле изобретения может означать "и/или", если явно не указаны альтернативы или альтернативы не являются взаимоисключающими, хотя раскрытие включает использование только альтернатив и "и/или." В настоящей заявке термин "примерно" используется для указания того, что значение включает характерную погрешность устройства, методики, использующейся для определения значения, и/или колебания, которые присущи исследуемым объектам. Термин "по меньшей мере один" следует понимать, как включающий один, а также любое количество, большее, чем один, включая, но не ограничиваясь только ими, 2, 3, 4, 5, 10, 15, 20, 30, 40, 50, 100 и т.п. Термин "по меньшей мере один" может означать до 100 или 1000 или более в зависимости от термина, к которому он относится; кроме того, количества 100/1000 не следует считать предельными, поскольку более значительные предельные значения также могут приводить к удовлетворительным результатам. Кроме того, термин "по меньшей мере один из X, Y и Z" следует понимать, как включающий только X, только Y и только Z, а также любую комбинацию X, Y и Z.
При использовании в настоящем описании и в формуле изобретения слова "включающий" (и любой его формы, такой как "включает" и "включают"), "содержащий" (и любой его формы, такой как "содержит" и "содержат") или "состоящий из" (и любой его формы, такой как "состоит из" и "состоят из") являются охватывающими или допускающими изменения и не исключают дополнительные, не указанные элементы или стадии способа.
Термин "или их комбинации" при использовании в настоящем изобретении означает все перестановки и комбинации перечисленных элементов, указанные перед этим термином. Например, "А, В, С или их комбинации" включает по меньшей мере одно из следующих: А, В, С, АВ, АС, ВС или ABC, и, если в конкретном контексте важен порядок, также ВА, СА, СВ, СВА, ВСА, АСВ, ВАС или CAB. Продолжая этот пример, отметим, что явно включены комбинации, которые содержат повторы одного или большего количества элементов или терминов, такие как ВВ, AAA, MB, ВВС, АААВСССС, СВВААА, САВАВВ и т.п. Специалист в данной области техники должен понимать, что обычно не налагаются ограничения на количество элементов или терминов в любой комбинации, если иное не следует из контекста.
Настоящее изобретение (и предлагаемые в нем решения) в целом относится к полимерам ацетат-сукцината гидроксипропилметилцеллюлозы (ГПМЦ-АС), обладающим уникальной схемой замещения, к способам получения полимеров и к композициям лекарственных средств, содержащим полимеры и обладающие низкой эффективностью лекарственные средства. Композиции лекарственных средств обладают повышенной эффективностью и/или улучшенной обрабатываемостью.
ГПМЦ-АС представляет собой замещенный полимер целлюлозы. "Замещенный полимер целлюлозы" означает полимер целлюлозы, который модифицирован путем реакции по меньшей мере части гидроксигрупп, содержащихся в повторяющихся звеньях сахарида, с соединением с образованием заместителя, присоединенного с помощью сложноэфирной или простой эфирной группы.
При использовании в настоящем изобретении и в формуле изобретения "ГПМЦ-АС" означает полимер целлюлозы, содержащий 2-гидроксипропоксигруппы (-ОСН2СН(СН3)ОН), метоксигруппы (-ОСН3), ацетильные группы (-СОСН3) и сукциноильные группы (-СОСН2СН2СООН). Полимер может содержать другие заместители в небольших количествах, при условии, что они не оказывают существенного воздействия на эффективность и характеристики ГПМЦ-АС.
Количество одного любого заместителя, содержащегося в полимере, характеризуется степенью замещения им полимера. "Степень замещения" заместителем и/или группой, содержащейся в полимере, означает среднее количество таких заместителей, включенных в повторяющиеся звенья сахарида, содержащиеся в цепи целлюлозы. Заместитель может быть присоединен непосредственно к повторяющемуся звену сахарида путем замещения любой из трех гидроксигрупп, содержащихся в повторяющемся звене сахарида, как это показано ниже (С2-ОН, С3-ОН и С6-ОН), или он может быть присоединен через замещающую гидроксипропоксигруппу, эта замещающая гидроксипропоксигруппа присоединена к повторяющемуся звену сахарида путем замещения любой из трех гидроксигрупп, содержащихся в повторяющемся звене сахарида, как это показано ниже (СНР-ОН).
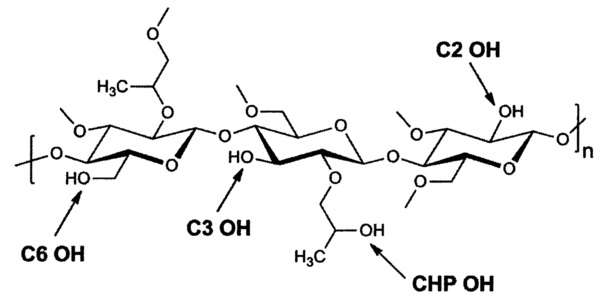
При использовании в настоящем изобретении термины, относящиеся к заместителям, определены следующим образом:
СЗАс = степень замещения (СЗ) ацетилом в пересчете на звено безводной глюкозы (ЗБГ)
СЗSuc = степень замещения (СЗ) сукциноилом в пересчете на звено безводной глюкозы (ЗБГ)
CHP = гидроксигруппа, содержащаяся в боковой цепи ГПМЦ или ГПМЦ-АС - гидроксипропоксигруппе (HP)
С2 = гидроксигруппа, присоединенная ко второму атому углерода (2) основной цепи целлюлозы ГПМЦ или ГПМЦ-АС
С3 = гидроксигруппа, присоединенная к третьему атому углерода (3) основной цепи целлюлозы ГПМЦ или ГПМЦ-АС
С6 = гидроксигруппа, присоединенная к шестому атому углерода (6) основной цепи целлюлозы ГПМЦ или ГПМЦ-АС
СНР СЗАс = СЗ ГПМЦ-АС ацетилом в положении CHP
С2 СЗАс = СЗ ГПМЦ-АС ацетилом в положении С2
С3 СЗАс = СЗ ГПМЦ-АС ацетилом в положении С3
С6 СЗАс = СЗ ГПМЦ-АС ацетилом в положении С6
CHP CЗSuc = СЗ ГПМЦ-АС сукциноилом в положении CHP
С2 СЗSuc = СЗ ГПМЦ-АС сукциноилом в положении С2
С3 СЗSuc = СЗ ГПМЦ-АС сукциноилом в положении С3
C6 С3Suc = СЗ ГПМЦ-АС сукциноилом в положении С6
C6 СЗАс, % = выраженная в процентах полная СЗ ацетилом, расположенным в положении C6
С3 СЗАс, % = выраженная в процентах полная СЗ ацетилом, расположенным в положении С3
C6 СЗSuc, % = выраженная в процентах полная СЗ сукциноилом, расположенным в положении С6
С3 CЗSuc, % = выраженная в процентах полная СЗ сукциноилом, расположенным в положении С3
Выраженное в мас. % количество ацетильных и сукциноильных групп, находящихся в разных положениях замещения, можно определить путем анализа с помощью 13С ЯМР (ядерный магнитный резонанс) и рассчитать по следующей формуле:
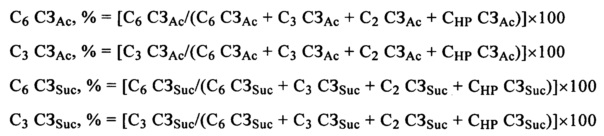
Установлено, что степень замещения в определенном положении сукциноильной и/или ацетильной группой в пересчете на звено безводной глюкозы (ЗБГ) является важной для повышения эффективности лекарственных средств, например, но без наложения ограничений, для улучшения растворимости обладающих низкой растворимостью лекарственных средств и/или для улучшения обрабатываемости лекарственных средств. Точнее, установлено, что замещение сукциноилом и/или ацетилом в положениях С3-ОН и C6-ОН является важным для усиления воздействия лекарственного средства и/или его обрабатываемости.
Полимеры ГПМЦ-АС предшествующего уровня техники, выпускающиеся фирмой Shin-Etsu, обладают приведенными ниже степенями замещения сукциноилом и/или ацетилом в положениях С3-ОН и C6-ОН, где указанные в настоящем изобретении диапазоны включают диапазоны для марок L, Н и М, приобретенных у фирмы Shin-Etsu.

Композиция, предназначенная для повышения эффективности и улучшения обрабатываемости лекарственного средства, предлагаемая в настоящем изобретении (и предлагаемых в нем решениях), содержит полимер и лекарственное средство. Полимер включает ГПМЦ-АС, обладающие разной степенью замещения сукциноильными и/или ацетильными группами в положениях C6-ОН и С3-ОН. Полимер можно получить по методике В.
В методике В уксусный ангидрид и ацетат натрия вводят в реакцию с гидроксипропилметилцеллюлозой при температуре, находящейся в диапазоне от примерно 85 до примерно 115°C, и получают промежуточный продукт. В одном неограничивающем варианте осуществления температуру можно менять в диапазоне, составляющем от примерно 95 до примерно 115°C. В другом неограничивающем варианте осуществления температуру можно менять в диапазоне, составляющем от примерно 95 до примерно 110°C. После того, как внутренняя температура достигает значения, находящегося в указанном выше диапазоне, реакционную смесь перемешивают в течение определенного периода времени, составляющего, например, но без наложения ограничений, от примерно 30 мин до примерно 2,5 ч. Затем добавляют янтарный ангидрид и смесь перемешивают в таком же температурном диапазоне в течение периода времени, составляющего от примерно 2,5 до примерно 23,5 ч. В одном неограничивающем варианте осуществления период времени можно менять в диапазоне, составляющем от примерно 2,5 до примерно 15,5 ч. В другом неограничивающем варианте осуществления период времени можно менять в диапазоне, составляющем от примерно 2,5 до примерно 5,5 ч. Затем реакционную смесь охлаждают до температуры окружающей среды и смешивают с водой и осаждается почти белое твердое вещество. Осадок смешивают с водой и промывают водой и сушат в сушильном устройстве с псевдоожиженным слоем примерно при 65°C.
Полимеры ГПМЦ-АС, полученные по методике В, обладают степенями замещения сукциноильными и ацетильными группами в положениях C6-ОН и С3-ОН, отличающимися от степеней замещения в образцах фирмы Shin-Etsu. В одном неограничивающем варианте осуществления выраженная в процентах полная СЗ сукциноилом в положении C6-ОН составляет менее 12% (C6 СЗSuc, %<12%) и в положении С3-ОН составляет более 53% (С3 СЗSuc, %>53%). Выраженная в процентах полная СЗ ацетилом в положении C6-ОН составляет более 32% (С6 СЗАс, %>32%).
В другом неограничивающем варианте осуществления выраженная в процентах полная СЗ сукциноилом в положении C6-ОН составляет менее 12% (C6 СЗSuc, %>12%) и в положении С3-ОН составляет более 53% (С3 CЗSuc, %>53%). Выраженная в процентах полная СЗ ацетилом в положении C6-ОН составляет более 32% (C6 СЗАс, до %>32%) и в положении С3-ОН составляет менее 27% (С3 СЗАс, %<27%).
В еще одном неограничивающем варианте осуществления выраженная в процентах полная СЗ сукциноилом в положении C6-ОН составляет менее 10C6 СЗSuc, %, %<10%) и в положении С3-ОН составляет более 57% (С3 СЗSuc, %>57%). Выраженная в процентах полная СЗ ацетилом в положении C6-ОН составляет от 33 до 51% (33%<C6 СЗАс, %<51%) и в положении С3-ОН составляет от 16 до 20% (16%<С3 СЗАс, %<20%).
В еще одном неограничивающем варианте осуществления выраженная в процентах полная СЗ сукциноилом в положении C6-ОН меньше или равна 6% (С6 СЗSuc, %≤6%) и в положении С3-ОН составляет от 58 до 84% (58%<С3 CЗSuc, %<84%). Выраженная в процентах полная СЗ ацетилом в положении C6-ОН составляет от 33 до 51% (33%<С6 СЗАс, %<51%) и в положении С3-ОН составляет от 16 до 20% (16%<С3 СЗАс, %<20%).
Также обнаружено, что включающий ГПМЦ-АС полимер, полученный по методике С, также может обеспечить повышение эффективности лекарственного средства. В методике С янтарный ангидрид и ацетат натрия вводят в реакцию с гидроксипропилметилцеллюлозой при температуре, находящейся в диапазоне от примерно 85 до примерно 115°C, и получают промежуточный продукт. В одном неограничивающем варианте осуществления температуру можно менять в диапазоне, составляющем от примерно 95 до примерно 115°C. В другом неограничивающем варианте осуществления температуру можно менять в диапазоне, составляющем от примерно 95 до примерно 110°C. После того, как внутренняя температура достигает значения, находящегося в указанном выше диапазоне, реакционную смесь перемешивают в течение определенного периода времени, составляющего, например, но без наложения ограничений, от примерно 30 мин до примерно 2,5 ч. Затем добавляют уксусный ангидрид и перемешивают в таком же температурном диапазоне в течение периода времени, составляющего от примерно 2,5 до примерно 23,5 ч. В одном неограничивающем варианте осуществления период времени можно менять в диапазоне, составляющем от примерно 2,5 до примерно 15,5 ч. В другом неограничивающем варианте осуществления период времени можно менять в диапазоне, составляющем от примерно 2,5 до примерно 5,5 ч. Затем реакционную смесь охлаждают до температуры окружающей среды и смешивают с водой и осаждается почти белое твердое вещество. Осадок смешивают с водой и промывают водой и сушат в сушильном устройстве с псевдоожиженным слоем примерно при 65°C.
Полимеры ГПМЦ-АС, полученные по методике С, также обладают степенями замещения сукциноильными и ацетильными группами в положениях C6-ОН и С3-ОН, отличающимися от степеней замещения в образцах фирмы Shin-Etsu. В одном неограничивающем варианте осуществления выраженная в процентах полная СЗ сукциноилом ГПМЦ-АС в положении C6-ОН составляет более 18% (C6 СЗSuc, %>18%) и в положении С3-ОН составляет менее 38% (С3 СЗSuc, %<38%). Выраженная в процентах полная СЗ ацетилом в положении C6-ОН составляет менее 26% (C6 СЗАс, %<26%) и в положении С3-ОН составляет более 36% (С3 СЗАс, %>36%).
В другом неограничивающем варианте осуществления выраженная в процентах полная СЗ сукциноилом в положении C6-ОН составляет более 25% (C6 СЗSuc, %>25%) и в положении С3-ОН составляет менее 36% (С3 СЗSuc, %<36%). Выраженная в процентах полная СЗ ацетилом в положении C6-ОН составляет менее 24% (C6 СЗАс, %<24%) и в положении С3-ОН составляет от 38 до 48% (38%<С3 СЗАс, %<48%).
В еще одном неограничивающем варианте осуществления выраженная в процентах полная СЗ сукциноилом в положении C6-ОН составляет от 35 до 45% (35%<С6 СЗSuc, %<45%) и в положении С3-ОН составляет от 30 до 35% (30%<С3 СЗSuc, %<35%). Выраженная в процентах полная СЗ ацетилом в положении С6-ОН составляет от 16 до 20% (16%<С6 СЗАс, %<20%) и в положении С3-ОН составляет от 38 до 48% (38%<С3 СЗАс, %<48%).
Термин "лекарственное средство" является обычно используемым и означает соединение, обладающее полезными профилактическими и/или терапевтическими характеристиками при его введении животному, в особенности людям. В одном неограничивающем варианте осуществления лекарственным средством является "обладающее низкой растворимостью лекарственное средство", это означает, что лекарственное средство обладает минимальной растворимостью в воде при физиологических значениях рН (например, значение рН=1-8), составляющей примерно 0,5 мг/мл или менее. Настоящее изобретение (и предлагаемые в нем решения) лучше всего применять, если растворимость лекарственного средства в воде понижена. Таким образом, композиции, предлагаемые в настоящем изобретении (и предлагаемых в нем решениях), используют для обладающих низкой растворимостью лекарственных средств, обладающих растворимостью в воде, составляющей менее, чем примерно 0,2 мг/мл. В одном неограничивающем варианте осуществления обладающие низкой растворимостью лекарственные средства обладают растворимостью в воде, составляющей менее, чем примерно 0,1 мг/мл. В другом неограничивающем варианте осуществления обладающие низкой растворимостью лекарственные средства обладают растворимостью в воде, составляющей менее, чем примерно 0,05 мг/мл. В еще одном неограничивающем варианте осуществления обладающие низкой растворимостью лекарственные средства обладают растворимостью в воде, составляющей менее, чем примерно 0,01 мг/мл.
Обычно можно сказать, что лекарственное средство обладает отношением доза/растворимость в воде, равным более, чем примерно 10 мл, и чаще равным более, чем примерно 100 мл, где растворимость в воде (мг/мл) является минимальным значением, полученным в любом водном растворе, обладающем физиологическим значением рН (например, в растворах, обладающих значениями рН, равными от 1 до 8), включая искусственные желудочный и кишечный сок, приготовленные в соответствии с ФСША (Фармакопея США), и дозы приведены в мг. Таким образом, отношение доза/растворимость в воде можно рассчитать путем деления дозы (в мг) на растворимость в воде (в мг/мл).
Чтобы настоящее изобретение (и предлагаемые в нем решения) можно было выгодно применять для лекарственного средства, оно не обязательно должно являться обладающим низкой растворимостью лекарственным средством, хотя обладающие низкой растворимостью лекарственные средства представляют собой класс, предпочтительный для применения настоящего изобретения (и предлагаемых в нем решений). Даже для лекарственного средства, которое все же обладает соответствующей растворимостью в воде в необходимой использующейся среде, с помощью настоящего изобретения (и предлагаемых в нем решений) можно успешно повысить его концентрацию в воде и улучшить биологическую доступность, если это приводит к уменьшению величины дозы, необходимого для обеспечения терапевтической эффективности, или приводит к повышению скорости всасывания лекарственного средства в случаях, когда необходимо быстрое начало воздействия лекарственного средства. В таких случаях лекарственное средство может обладать растворимостью в воде, достигающей примерно от 1 до 2 мг/мл, или даже примерно от 20 до 40 мг/мл.
В настоящем изобретении (и предлагаемых в нем решениях) лекарственные средства, подходящие для включения в полимерные системы ГПМЦ-АС, могут включать кислотные, основные, цвиттерионные или нейтральные органические/неорганические биологически активные соединения или их соли. Примеры лекарственных средств могут включать, но не ограничиваются только ими, анальгетики, противосудорожные средства, анестезирующие средства, противодиабетические средства, противоинфекционные средства, противоопухолевые средства, противоревматические средства, средства для лечения сердечно-сосудистых заболеваний, стимуляторы центральной нервной системы (ЦНС), агонисты допаминового рецептора, средства для лечения желудочно-кишечных заболеваний, психотерапевтические средства, средства для лечения заболеваний мочевых путей, гипотензивные средства, седативные средства, антикоагулянты, противосудорожные средства, средства, снижающие содержание глюкозы в крови, противоотечные средства, антигистамины, противокашлевые средства, противоопухолевые средства, бета-блокаторы, противовоспалительные средства, антипсихотические средства, средства, улучшающие познавательную способность, средства, снижающие содержание холестерина, средства, снижающие содержание триглицерида, антиатеросклеротические средства, средства против ожирения, средства для борьбы с аутоиммунными нарушениями, средства против импотенции, антибактериальные и фунгицидные средства, снотворные средства, средства против болезни Паркинсона, средства против болезни Альцгеймера, антибиотики, антидепрессанты, противовирусные средства, ингибиторы гликогенфосфорилазы и ингибиторы белка-переносчика сложного эфира холестерина.
Следует понимать, что каждое указанное лекарственное средство включает любые фармацевтически приемлемые формы лекарственного средства. "Фармацевтически приемлемые формы" означают любые фармацевтически приемлемые производные или модификации, включая стереоизомеры, смеси стереоизомеров, энантиомеры, сольваты, гидраты, изоморфные формы, полиморфные формы, псевдоморфные формы, нейтральные формы, формы солей и пролекарства.
Конкретные примеры гипотензивных средств могут включать празозин, нифедипин, амлодипинбезилат, тримазозин и доксазозин; конкретные примеры средства, снижающего содержание глюкозы в крови, могут включать глипизид и хлорпропамид; конкретным примером средства против импотенции является силденафил и силденафилцитрат; конкретные примеры противоопухолевых средств могут включать хлорамбуцил, ломустин и эхиномицин; конкретным примером противоопухолевого средства имидазольного типа является тубулазол; конкретным примером средства против гиперхолестеринемии является кальциевая соль аторвастатина; конкретные примеры анксиолитиков могут включать гидроксизингидрохлорид и доксепингидрохлорид; конкретные примеры противовоспалительных средств могут включать бетаметазон, преднизолон, аспирин, пироксикам, валдекоксиб, карпрофен, целекоксиб, флурбипрофен и (+)-N-{4-[3-(4-фторфенокси)фенокси]-2-циклопентен-1-ил}-N-гидроксимочевину; конкретным примером барбитурата является фенобарбитал; конкретные примеры противовирусных средств могут включать ацикловир, нелфинавир, делавирдин и виразол; конкретные примеры витаминов/питательных веществ могут включать ретинол и витамин Е; конкретные примеры бета-блокаторов могут включать тимолол и надолол; конкретным примером противорвотного средства является апоморфин; конкретные примеры диуретиков могут включать хлорталидон и спиронолактон; конкретным примером антикоагулянта является дикумарол; конкретные примеры кардиотонических средств могут включать дигоксин и дигитоксин; конкретные примеры андрогенов могут включать 17-метилтестостерон и тестостерон; конкретным примером природного кортикостероида является дезоксикортикостерон; конкретным примером стероидного снотворного/анестезирующего средства является альфаксалон; конкретные примеры анаболических средств могут включать флуоксиместерон и метанстенолон; конкретные примеры антидепрессивных средств могут включать сульпирид, [3,6-диметил-2-(2,4,6-триметилфенокси)пиридин-4-ил]-(1-этилпропил)амин, 3,5-диметил-4-(3'-пентокси)-2-(2',4',6'-триметилфенокси)пиридин, пироксидин, флуоксетин, пароксетин, венлафаксин и сертралин; конкретные примеры антибиотиков могут включать карбенициллининданил-натрий, бакампициллингидрохлорид, тролеандомицин, доксициклингиклат, ампициллин и пенициллин G; конкретные примеры противоинфекционных средств могут включать бензалконийхлорид и хлоргексидин; конкретные примеры средств, расширяющих коронарные сосуды, могут включать нитроглицерин и миофлазин; конкретным примером снотворного средства является этомидат; конкретные примеры ингибиторов карбоангидразы могут включать ацетазоламид и хлорзоламид; конкретные примеры фунгицидных средств могут включать эконазол, терконазол, флуконазол, вориконазол и гризеофульвин; конкретным примером противопротозойного средства является метронидазол; конкретные примеры антигельминтных средств могут включать тиабендазол, оксфендазол и морантел; конкретные примеры антигистаминных средств могут включать астемизол, левокабастин, цетиризин, левоцетиризин, декарбоэтоксилоратадин и циннаризин; конкретные примеры антипсихотических средств могут включать зипрасидон, оланзепин, тиотиксенгидрохлорид, флуспирилен, рисперидон и пенфлуридол; конкретные примеры средств для лечения желудочно-кишечных заболеваний могут включать лоперамид и цисаприд; конкретные примеры антагонистов серотонина могут включать кетансерин и миансерин; конкретным примером анестезирующего средства является лидокаин; конкретным примером средства, снижающего содержание глюкозы в крови, является ацетогексамид; конкретным примером противорвотного средства является дименгидринат; конкретным примером антибактериального средства является котримоксазол; конкретным примером допаминергического средства является L-DOPA; конкретными примерами средств против болезни Альцгеймера могут являться тетрагидро-9-аминоакридин и донепезил; конкретным примером противоязвенного средства/агониста Н2 является фамотидин; конкретные примеры седативных/снотворных средств могут включать хлордиазепоксид и триазолам; конкретным примером сосудорасширяющего средства является алпростадил; конкретным примером ингибитора агрегации тромбоцитов является простациклин; конкретные примеры ингибитора АСЕ/гипотензивных средств могут включать эналаприловую кислоту, хинаприл и лизиноприл; конкретные примеры тетрациклиновых антибиотиков могут включать окситетрациклин и миноциклин; конкретные примеры макролидных антибиотиков могут включать эритромицин, кларитромицин и спирамицин; конкретным примером азалидного антибиотика является азитромицин; конкретные примеры ингибиторов гликогенфосфорилазы могут включать [R-(R*S*)]-5-хлор-N-[2-гидрокси-3-{метоксиметиламино}-3-оксо-1-(фенилметил)пропил-1Н-индол-2-карбоксамид и [(1S)-бензил-(2R)-гидрокси-3-((3R,4S)-дигидроксипирролидин-1-ил-)-3-оксипропил]амид 5-хлор-1Н-индол-2-карбоновой кислоты; и конкретные примеры ингибиторов белка-переносчика сложного эфира холестерина (СЕТР) включают этиловый эфир [2R,4S]-4-[(3,5-бис-трифторметилбензил)метоксикарбониламино]-2-этил-6-трифторметил-3,4-дигидро-2Н-хинолин-1-карбоновой кислоты, также известный, как торцетрапиб.
Ингибиторы СЕТР, в частности, торцетрапиб, и методики получения таких соединений подробно описаны в патентах U.S. №№6197786 и 6313142, в заявках РСТ №№ WO 01/40190 А1, WO 02/088085 А2 и WO 02/088069 А2, раскрытия которых включены в настоящее изобретение в качестве ссылки. Торцетрапиб обладает чрезвычайно низкой растворимостью в водных средах, таких как жидкость, находящаяся в полости ЖК (желудочно-кишечного) тракта человека. Растворимость торцетрапиба в воде составляет менее, чем примерно 0,04 мкг/мл. Торцетрапиб необходимо вводить в ЖК тракт в форме, обладающей улучшенной растворимостью, для обеспечения достаточной концентрации лекарственного средства в ЖК тракте, чтобы обеспечить достаточное всасывание в кровь и добиться необходимого терапевтического воздействия. Ингибиторы СЕТР также описаны в патенте U.S. №6723752, в котором описан ряд ингибиторов СЕТР, включая (2R)-3-{[3-(4-хлор-3-этилфенокси)фенил]-[[3-(1,1,2,2-тетрафторэтокси)фенил]метил]амино}-1,1,1-трифтор-2-пропанол. Кроме того, ингибиторы СЕТР, описанные в нитрованном патенте, также описаны в заявке на патент U.S. №10/807838, поданной 23 марта 2004 г., и в заявке на патент U.S. №60/612863, поданной 23 сентября 2004 г., в которых описан изопропиловый эфир (2R,4R,4aS)-4-[амино-(3,5-бис-(трифторметилфенил)метил]-2-этил-6-(трифторметил)-3,4-дигидрохинолин-1-карбоновой кислоты. Другие ингибиторы СЕТР могут включать JTT-705, также известный, как S-[2-([[1-(2-этилбутил)циклогексил]карбонил]амино)фенил]-2-метилпропантиоат, и соединения, которые раскрыты в заявке РСТ № WO 04/020393, такие как S-[2-([[1-(2-этилбутил)циклогексил]карбонил]амино)фенил]2-метилпропантиоат, транс-4-[[[2-[[[[3,5-бис(трифторметил)фенил]метил](2-метил-2Н-тетразол-5-ил)амино]метил]-4-(трифторметил)фенил]этиламино]метил]циклогексануксусная кислота и транс-4-[[[2-[[[[3,5-бис(трифторметил)фенил]метил](2-метил-2Н-тетразол-5-ил)амино]метил]-5-метил-4-(трифторметил)фенил]этиламино]метил]циклогексануксусная кислота, лекарственные средства, раскрытые в находящихся в совместной собственности заявках на патент U.S. №№09/918127 и 10/066091, раскрытия которых включены в настоящее изобретение в качестве ссылки, и лекарственные средства, раскрытые в следующих патентах и опубликованных заявках, раскрытия которых включены в настоящее изобретение в качестве ссылки: DE 19741400 А1; DE 19741399А1; WO 9914215 А1; WO 9914174; DE 19709125 А1; DE 19704244 А1; DE 19704243 А1; ЕР 818448 А1; WO 9804528 А2; DE 19627431 А1; DE 19627430 А1; DE 19627419 А1; ЕР 796846 А1; DE 19832159; DE 818197; DE 19741051; WO 9941237 А1; WO 9914204 А1; WO 9835937 А1; JP 11049743; WO 0018721; WO 0018723; WO 0018724; WO 0017164; WO 0017165; WO 0017166; WO 04020393; ЕР 992496 и ЕР 987251.
Типичные примеры других лекарственных средств, подходящих для применения в настоящем изобретении (и предлагаемых в нем решениях), могут включать, но не ограничиваются только ими, итраконазол, эзетимиб, албутеролсульфат, амоксициллин, бупропионгидрохлорид, карбидопу, цефаклор, натриевую соль диклофенака, эритромицин, лоратидин, карбонат лития, метилфенидат, метапрололтартрат, нифедипин, омепразол, соталолгидрохлорид, верапамилгидрохлорид, албутеролсульфат, амоксициллин, бупропионгидрохлорид, карбидопу, цефаклор, натриевую соль диклофенака, эритромицин, фелодипин, лоратидин, карбонат лития, метилфенидат, метапрололтартрат, нифедипин, омепразол, соталолгидрохлорид, верапамилгидрохлорид или их терапевтически приемлемые комбинации. Приведенный выше перечень лекарственных средств не является исчерпывающим.
В противоположность общепринятой точке зрения, относительное увеличение концентрации в воде и улучшение биологической доступности, обеспечиваемые композициями, предлагаемыми настоящем изобретении (и предлагаемых в нем решениях), обычно обеспечивается для лекарственных средств, обладающих пониженной растворимостью и повышенной гидрофобностью. В действительности, авторы настоящего изобретения обнаружили подкласс гидрофобных лекарственных средств, которые практически нерастворимы в воде, сильно гидрофобны и обладают набором физических характеристик. При приготовлении лекарственных средств этого подкласса, в настоящем изобретении называющихся "гидрофобными лекарственными средствами", с использованием полимеров, предлагаемых в настоящем изобретении (и предлагаемых в нем решениях), обеспечивается значительное повышение их концентрации в воде и биологической доступности. Кроме того, композиции гидрофобных лекарственных средств и полимеров, предлагаемых в настоящем изобретении (и предлагаемых в нем решениях), также могут обладать улучшенной физической стабильностью по сравнению с композициями, содержащими полимеры имеющихся в продаже торговых марок.
Первой характеристикой гидрофобных лекарственных средств является то, что они являются сильно гидрофобными. "Сильно гидрофобные" означает, что значение log Р лекарственного средства может составлять не менее 4,0, не менее 5,0 и даже не менее 5,5. Значение log Р, которое определяют, как десятичный логарифм отношения (1) концентрации лекарственного средства в октанольной фазе к (2) концентрации лекарственного средства в водной фазе, где две фазы находятся в равновесии друг с другом, является общепринятой мерой гидрофобности. Значение log Р можно определить экспериментально или рассчитать по методикам, хорошо известным в данной области техники. Если используют рассчитанное значение log Р, то используют самое большое значение, полученное по общепринятой методике расчета log Р. Рассчитанные значения log Р часто обозначают со ссылкой на методику расчета, например С log Р, A log Р и М log Р. Значение log Р также можно определить по методикам фрагментации, таким как методика фрагментации Криппена (J. Chem. Inf. Comput. Sci. 27, 21-35 (1987)); методика фрагментации Висванадана (J. Chem. Inf. Comput. Sci. 29, 163-172 (1989)) или методика фрагментации Брото (Eur. J. Med. Chem.-Chim. Theor. 19, 71 (1984)). Предпочтительно, если значение Log P рассчитывают с использованием среднего значения, полученного по методикам фрагментации Криппена, Висванадана и Брото.
Второй характеристикой гидрофобных лекарственных средств является то, что они обладают низким значением параметра растворимости. Расчет параметра растворимости раскрыт в US 8207232, полное содержание которого явно включено в настоящее изобретение в качестве ссылки. Значение параметра растворимости может составлять примерно 22 (Дж/см3)1/2 или менее, примерно 21,5 (Дж/см3)1/2 или менее, и даже примерно 21 (Дж/см3)1/2 или менее.
В первую очередь вследствие этих характеристик гидрофобные лекарственные средства обычно обладают очень низкой растворимостью в воде. "Очень низкая растворимость" означает, что минимальная растворимость в воде при физиологических значениях рН (значение рН=1-8) составляет менее, чем примерно 100 мкг/мл, и чаще составляет менее, чем примерно 10 мкг/мл. Кроме того, гидрофобные лекарственные средства обычно обладают высоким отношением доза/растворимость в воде. Очень низкая растворимость в воде часто приводит к недостаточному или медленному всасыванию лекарственного средства из жидкости желудочно-кишечного тракта, если лекарственное средство вводят перорально обычным путем. При увеличении дозы (массы лекарственного средства, вводимого перорально) обладающих очень низкой растворимостью лекарственных средств недостаточное всасывание обычно еще больше ухудшается. Таким образом, второй характеристикой гидрофобных лекарственных средств является очень высокое выраженное в (мл) отношение доза (в мг)/растворимость (в мг/мл). "Очень высокое отношение доза/растворимость" означает, что отношение доза/растворимость может составлять не менее 1000 мл, не менее 5000 мл или даже не менее 10000 мл.
Гидрофобные лекарственные средства обычно обладают очень низкой абсолютной биологической доступностью. Точнее, абсолютная биологическая доступность лекарственных средств этого подкласса при пероральном введении без добавок, а не в виде препарата (т.е. только лекарственного средства) составляет менее, чем примерно 10%, и чаще составляет менее, чем примерно 5%.
В одном неограничивающем варианте осуществления настоящего изобретения (и предлагаемых в нем решений) лекарственным средством может являться чувствительное к воздействию кислоты лекарственное средство, это означает, что лекарственное средство подвергается химическому превращению или другим образом разлагается в присутствии кислых соединений. Чувствительные к воздействию кислоты лекарственные средства часто содержат функциональные группы, которые являются реакционноспособными в кислой среде, такие как сульфонилмочевинные, гидроксамовые, гидроксиамидные, карбаматные, ацетальные, гидроксимочевинные, сложноэфирные и амидные. Лекарственные средства, которые содержат такие функциональные группы, в присутствии кислых соединений могут быть склонны к таким реакциям, как гидролиз, лактонизация или переэтерификация.
Ниже только в качестве примеров приведены типичные примеры чувствительных к воздействию кислоты лекарственных средств. Следует понимать, что каждое указанное лекарственное средство включает нейтральную форму лекарственного средства, фармацевтически приемлемые соли и пролекарства. Примеры чувствительных к воздействию кислоты лекарственных средств могут включать, но не ограничиваются только ими, [4(R)-карбамоил-1(S)-3-фторбензил-2(S)-7-дигидрокси-7-метилоктил]амид хиноксалин-2-карбоновой кислоты; [1-бензил-4-(4,4-дифторциклогексил)-2-гидрокси-4-гидроксикарбамоилбутил]амид хиноксалин-2-карбоновой кислоты; [1-бензил-4-(4,4-дифтор-1-гидроксициклогексил)-2-гидрокси-4-гидроксикарбамоилбутил]амид хиноксалин-2-карбоновой кислоты; (+)-N-{3-[3-(4-фторфенокси)фенил]-2-циклопентен-1-ил}-N-гидроксимочевину; омепразол; этопозид; фамотидин; эритромицин; хинаприл; лансопразол и прогабид.
В фармацевтике известно, что обладающие низкой растворимостью лекарственные средства часто обладают недостаточной биологической доступностью или неравномерным всасыванием, на степень неравномерности влияют такие факторы, как уровень дозы, состояние сытости пациента и форма лекарственного средства.
Вследствие увеличения количества обладающих низкой растворимостью в воде соединений в современных разработанных лекарственных средствах, в качестве средства улучшения биологической доступности используют концепцию перенасыщения, в особенности в области перорального введения лекарственных средств, где ожидается, что повышение внутрипросветной концентрации путем перенасыщения приведет к улучшению всасывания в кишечнике. Для того, чтобы происходило это улучшение всасывания в кишечнике, необходимо обеспечить и поддерживать перенасыщение в среде желудочно-кишечного тракта. После того, как обеспечено термодинамически неустойчивое состояние перенасыщения, его необходимо поддерживать в течение периода времени, достаточного для всасывания в кишечнике. Чрезвычайно важным является поддерживание перенасыщенного состояния.
Установлено, что включение инертных наполнителей, которые задерживают осаждение, может стабилизировать перенасыщение in vitro. Фармацевтические инертные наполнители, использующиеся для этой цели, могут включать полимеры, поверхностно-активные вещества и циклодекстрины. Инертные наполнители обычно называют средствами, предупреждающими зародышеобразование. ГПМЦ-АС, предлагаемый в настоящем изобретении (и предлагаемых в нем решениях), можно использовать в качестве средства, предупреждающего зародышеобразование, для содействия и поддерживания перенасыщения лекарственным средством в случае обладающего низкой растворимостью соединения.
Хорошо известно, что всасывание лекарственного средства при пероральном введении зависит от растворимости лекарственного средства в среде желудочно-кишечного (ЖК) тракта и его проницаемости через стенки ЖК. Если для оценки эффективности лекарственного средства in vivo используют исследование растворимости, то чрезвычайно важно как можно точнее воспроизвести условия in vivo в проводимом in vivo исследовании. Обнаружено, что биорелевантная среда может обеспечить более точное моделирование фармакокинетических профилей, чем искусственный желудочный сок или искусственный кишечный сок. Биорелевантной средой, использующейся в настоящем изобретении (и предлагаемых в нем решениях), является порошок SIF®, фосфатный буферный раствор искусственного кишечного сока, соответствующего состоянию натощак (FaSSIF), выпускающийся фирмой Phares Drug Delivery AG, Baselland, Switzerland.
Перенасыщение можно исследовать по методике смещающего воздействия растворителя. В этой методике можно использовать сосуд для растворения, находящийся в водяной бане при 37°C. Размер сосуда для растворения зависит от исследуемого объема. В сосуд можно добавить полимер ГПМЦ-АС и биорелевантную среду и получить смесь и дать установиться равновесию при 37°C. Смесь непрерывно перемешивают с использованием любого оборудования для перемешивания, известного в данной области техники. Получают перенасыщенный раствор лекарственного средства и помещают его в сосуд. В разные моменты времени можно отбирать образцы и центрифугировать. Концентрации лекарственного средства можно определить по любой известной методике анализа, например, но без наложения ограничений, путем анализа с помощью ВЭЖХ (высокоэффективная жидкостная хроматография) с использованием УФ-детектирования.
Индекс текучести расплава (ИТР) является мерой легкости течения расплава термопластичного полимера. Он определен, как выраженная в граммах масса полимера, протекающая за 10 мин через капилляр, обладающий определенными диаметром и длиной, при давлении, прилагаемом с помощью заданных альтернативных гравиметрических масс для альтернативных заданных температур.
Индексы текучести расплавов полимеров можно определить в соответствии со стандартом ASTM D 1238 при условиях, указанных для заданного типа полимера. В таких стандартных методиках указаны геометрические размеры и другие параметры использующегося устройства, а также комбинации условий. Устройством фактически является вертикально расположенный узкий цилиндрический сосуд, снабженный поршнем и съемной (для очистки) диафрагмой на дне. Температуру сосуда регулируют и к поршню прилагают определенную нагрузку для получения заданного усилия и, следовательно, давления, с помощью которого расплав полимера проходит через отверстие. Обычно в сосуд загружают пеллеты полимера и дают установиться температуре, необходимой для проведения испытания, значительно более высокой, чем температура плавления полимера, затем к поршню прилагают нагрузку, обеспечивающую движение полимера через отверстие. Количество экструдата определяют путем простого взвешивания или по объемным методикам (ход поршня) с использованием известной плотности расплава.
Таблетка является фармацевтической дозированной формой. Она содержит смесь активных веществ и инертных наполнителей, обычно в порошкообразной форме, спрессованную из порошкообразной в твердую дозированную форму. Инертные наполнители могут включать разбавители, связующие или гранулирующие агенты, агенты, придающие скользкость (вспомогательные средства, обеспечивающие сыпучесть) и смазывающие вещества для обеспечения эффективного таблетирования; разрыхлители для содействия распаду таблетки в пищеварительном тракте; подсластители или вкусовые добавки для улучшения вкуса и пигменты для придания таблетке привлекательного внешнего вида. Можно использовать целый ряд разных связующих, некоторые обычные включают лактозу, гидрофосфат кальция, сахарозу, кукурузный (маисовый) крахмал, микрокристаллическую целлюлозу, повидон (поливинилпирролидон) и модифицированную целлюлозу (например, но без наложения ограничений, гидроксипропилметилцеллюлозу и гидоксиэтилцеллюлозу).
Часто необходимо, чтобы ингредиент действовал в качестве разрыхлителя для содействия диспергированию таблетки и высвобождению активного фармацевтического ингредиента (АФИ) для всасывания. Некоторые связующие, такие как крахмал и целлюлоза, также являются превосходными разрыхлителями. Стеариновую кислоту можно использовать в качестве вспомогательного средства, обеспечивающего сыпучесть, и смазывающего вещества. В настоящем изобретении (и предлагаемых в нем решениях) полимеры ГПМЦ-АС приготовлены в виде таблеток и измеряют их твердость.
В процедуре прессования таблеток важно, чтобы все ингредиенты были достаточно сухими, представляли собой порошок или гранулы, обладали примерно одинаковым размером частиц и сыпучестью. При проведении производственных операций порошки, обладающие разным размером частиц, вследствие разных плотностей могут сегрегировать, и это приводит к получению таблеток с недостаточно однородным распределением в них лекарственного средства или АФИ, но с помощью гранулирования это можно предупредить. Благодаря однородному распределению каждая таблетка обеспечивает доставку одной и той же дозы АФИ.
Для изготовления таблетки порошок можно загрузить из бункера с порошком в горизонтально расположенное загрузочное устройство. Порошок покрывает часть поверхности матрицы и матрицу. Необходимый насыпаемый объем, называющийся массой таблетки, можно регулировать при первой проверке, называющейся проверкой массы. После соскабливания избытка порошка порошок и матрицу можно сжать вместе путем движения расположенных сверху и снизу пуансонов, которые движутся между обжимными валиками. Таблетку необходимой толщины получают путем продвижения расположенного снизу обжимного валика в направление от укрепленного расположенного сверху обжимного валика или к нему. Сжимающее усилие зависит от комбинации любого набора определенных значений насыпаемого объема (масса) и толщины, которые выбирают для любого определенного активного ингредиента и размера/формы таблетки.
Определение твердости (разрушающего усилия) таблетки играет критически важную роль для выбора дозированной формы, обладающей оптимальными физическими характеристиками, и для исследования того, соответствует ли изготовленная дозированная форма определенными техническим условиям для изготовления. Исследование твердости таблетки проводят не только для того, чтобы убедиться в механической целостности изготовленной таблетки в ходе последующих операций. Поскольку твердость таблетки непосредственно связана со всеми другими физическим параметрами, ее определение является быстрым и эффективным исследованием, которое указывает на то, обладает ли таблетка такими техническими характеристиками, как определенные время распада и ломкость. Поэтому необходимо, чтобы измерение твердости проводилось безошибочно и оборудование, использующееся для определения твердости таблетки, обеспечивало надежные результаты.
Термин "твердость" в настоящем изобретении используют фактически в качестве синонима терминов разрушающее усилие или сопротивление таблетки разрушению. Проще говоря, твердость таблетки означает усилие (нагрузку), необходимое для разрушения таблетки. Стандартной методикой, использующейся для определения твердости таблетки, является исследование на сжатие. Таблетку помещают между двумя зажимами, которые ломают таблетку. Прибор измеряет приложенное к таблетке усилие и регистрирует разрушение таблетки. В настоящем изобретении (и предлагаемых в нем решениях) сжимающую силу измеряют в килоньютонах (кН) и твердость измеряют в килограммах силы (кг-сила).
Получение твердой дисперсии является методикой диспергирования обладающего плохой растворимостью лекарственного средства в полимерной матрице в твердом состоянии. Лекарственное средство может находиться в аморфной или микрокристаллической форме в смеси, которая обеспечивает высокую скорость растворения и/или выраженную растворимость в желудочном и кишечном соках. Для приготовления твердых дисперсий разработано несколько методик, включая совместное осаждение (см., например, патенты U.S. №№5985326 и 6350786), сплавление, распылительную сушку (см., например, патент U.S. №7008640) и экструзию расплава (см., например, патент U.S. №7081255). Все эти методики обеспечивают получение тонко диспергированных частиц лекарственного средства в полимерной матрице, обычно на молекулярном уровне или в виде микрокристаллической фазы.
Твердые дисперсии лекарственного средства в матрице можно получить путем получения однородного раствора или расплава лекарственного средства и матричного материала с последующим отверждением смеси путем охлаждения или удаления растворителя. Такие твердые дисперсии кристаллических лекарственных средств при пероральном введении часто обладают лучшей биологической доступностью, чем композиции для перорального введения, содержащие недиспергированное кристаллическое лекарственное средство.
Высушенная распылительной сушкой твердая дисперсия обладающего умеренной растворимостью лекарственного средства в ГПМЦ-АС, предлагаемая в настоящем изобретении (и предлагаемых в нем решениях), может обладать уникальными характеристиками, что обеспечивает ее широкое применение для приготовления дозированных форм для перорального введения. Если не ограничиваться какой-либо определенной теорией или механизмом, то можно предположить, что для того, чтобы твердая аморфная дисперсия лекарственного средства в матричном материале оптимально обеспечивала улучшение биологической доступности обладающих умеренной растворимостью лекарственных средств, матричный материал обычно должен выполнять следующие функции: 1. диспергировать лекарственное средство и тем самым предупреждать или замедлять кристаллизацию в твердом состоянии, 2. растворяться in vivo и тем самым обеспечивать высвобождение лекарственного средства в желудочно-кишечном тракте, 3. подавлять осаждение или кристаллизацию растворенного в воде лекарственного средства.
Если лекарственное средство не обладает выраженной склонностью к кристаллизации из аморфного твердого состояния, то необходимо выполнение только двух последних функций. После приготовления твердой аморфной дисперсии лекарственного средства в ГПМЦ-АС концентрация лекарственного средства, до или после растворения дисперсии лекарственное средство - ГПМЦ-АС, может стать существенно более высокой, чем при равновесной растворимости самого лекарственного средства. Это означает, что обеспечивается концентрация лекарственного средства, соответствующая перенасыщению, и такая концентрация, соответствующая перенасыщению, поддерживается в течение сравнительно длительного периода времени.
Полимер ГПМЦ-АС, предлагаемый в настоящем изобретении (и предлагаемых в нем решениях), прекрасно выполняет все три функции, описанные выше, поэтому он является единственным из числа известных матричных материалов, способных препятствовать дальнейшему осаждению или кристаллизации большого количества обладающих умеренной растворимостью лекарственных средств из перенасыщенного раствора, и, кроме того, если не ограничиваться теоретическими соображениями, то можно предположить, что распылительная сушка обеспечивает быстрое удаление растворителя таким образом, что в отличие от других методик получения дисперсий, таких как выпаривание в роторном испарителе, с ее помощью кристаллизацию лекарственного средства и полимера ГПМЦ-АС можно в значительной степени предотвратить или по меньшей мере свести к минимуму. Кроме того, во многих случаях распылительная сушка обеспечивает достаточно быстрое удаление растворителя и поэтому даже можно в значительной степени предотвратить или свести к минимуму разделение фаз лекарственного средства и полимера ГПМЦ-АС. Таким образом, полимер ГПМЦ-АС, предлагаемый в настоящем изобретении (и предлагаемых в нем решениях), и распылительная сушка могут обеспечить получение лучшей, более однородной дисперсии, в которой лекарственное средство более эффективно диспергировано в полимере. По данным проводимых in vitro исследований повышенная эффективность дисперсии, полученной распылительной сушкой, по сравнению с дисперсиями, полученными по другим методикам, обеспечивает более высокую концентрацию лекарственного средства.
Хотя основными ингредиентами, содержащимися в твердых аморфных композициях, предлагаемых в настоящем изобретении (и предлагаемых в нем решениях), являются просто лекарственное средство, которое необходимо доставить, и ГПМЦ-АС, может оказаться полезным включение в дисперсию других инертных наполнителей. Например, но без наложения ограничений, вместе ГПМЦ-АС в композицию можно включить другие полимеры, отличающиеся от ГПМЦ-АС, которые растворимы в водных растворах по меньшей мере в части диапазона значений рН, составляющего от 1,0 до 8,0. Примеры других полимеров могут включать, но не ограничиваются только ими, поливинилпирролидон (ПВП), гидроксипропилцеллюлоза (ГПЦ) или ГПМЦ. Основным преимуществом ГПМЦ-АС может являться подавление осаждения или кристаллизации лекарственного средства из перенасыщенного раствора, когда лекарственное средство является кристаллическим или аморфным. В одном неограничивающем варианте осуществления лекарственное средство, ГПМЦ-АС и один или большее количество дополнительных полимеров можно подвергнуть совместной распылительной сушке, где количество лекарственного средства и ГПМЦ-АС может составлять не более, чем примерно 75% в пересчете на дисперсию.
Другим типом инертного наполнителя, применимого в качестве компонента дисперсий, предлагаемых в настоящем изобретении, является поверхностно-активное вещество, такое как жирная кислота и алкилсульфонат. Таким вещества можно успешно использовать для повышения скорости растворения вследствие облегчения смачивания и, таким образом, увеличить максимальную концентрацию лекарственного средства и обеспечивающуюся степень перенасыщения, а также для подавление осаждения или кристаллизации лекарственного средства вследствие взаимодействия с растворенным лекарственным средством по таким механизмам, как комплексообразование, образование комплексов включения, образование мицелл, или вследствие адсорбции на поверхности лекарственного средства, кристаллического или аморфного. Количество этих поверхностно-активных веществ может составлять примерно до 25% в пересчете на высушенную распылительной сушкой дисперсию.
Также может быть полезно добавлять модификаторы рН, такие как кислоты, основания или буферы. Модификаторы рН могут эффективно способствовать замедлению растворения дисперсии (например, кислоты, такие как лимонная кислота или янтарная кислота) или, альтернативно, повышать скорость растворения дисперсии (например, основания, такие как ацетат натрия или амины). Обычные матричные материалы, поверхностно-активные вещества, наполнители, разрыхлители или связующие можно добавить в виде части самой дисперсии, можно добавить с помощью влажного или механического гранулирования или другим образом. Если такие добавки включают в виде части самой дисперсии, их можно смешать с лекарственным средством и ГПМЦ-АС в растворителе, использующемся для распылительной сушки, и можно растворить вместе с лекарственным средством и ГПМЦ-АС до приготовления дисперсии путем распылительной сушки, или не проводить растворение. Количество этих материалов может составлять примерно до 25% в пересчете на дисперсию лекарственное средство/ГПМЦ-АС/добавка.
В дополнение к лекарственному средству и ГПМЦ-АС (и другим полимерам, описанным выше) в композициях, предлагаемых в настоящем изобретении (и предлагаемых в нем решениях), можно использовать другие инертные наполнители, обычно использующиеся для приготовления препаратов, включая инертные наполнители, хорошо известные в данной области техники. Обычно инертные наполнители, такие как наполнители, разрыхляющие агенты, пигменты, связующие, смазывающие вещества, ароматизаторы и т.п. можно использовать для обычных целей и в типичных количествах, не оказывая воздействия на характеристики композиций. Эти инертные наполнители используют после того, как образовалась дисперсия ГПМЦ-АС/лекарственное средство, чтобы приготовить препарат дисперсии в виде таблеток, капсул, суспензий, порошков для суспензий, кремов, чрескожных пластырей и т.п.
Термин "распылительная сушка" используют обычным образом, и он в широком смысле означает процедуру, включающую разделение жидких смесей на маленькие капли (распыление) и быстрое удаление из смеси растворителя, проводимое в баке (аппарат для распылительной сушки), в котором обеспечена большая движущая сила, обеспечивающая выпаривание растворителя из капель. Большую движущую силу, обеспечивающую выпаривание растворителя, обычно создают путем поддерживания в аппарате для распылительной сушки парциального давления растворителя, которое значительно ниже давления насыщенных паров растворителя при температуре сушки капель. Это осуществляют путем (1) поддерживания в аппарате для распылительной сушки давления, соответствующего частичному вакууму (например, от 0,01 до 0,50 атм.); (2) смешивания капель жидкости с теплым осушающим газом; или (3) использования обеих вариантов. Например, но без наложения ограничений, раствор лекарственного средства и ГПМЦ-АС в ацетоне можно соответствующим образом высушить распылительной сушкой путем распыления раствора при температуре, равной примерно 50°C, в камере, в которой полное давление, равное от примерно 0,01 до примерно 0,2 атм., поддерживают путем соединения выпускного отверстия с вакуумным насосом. Альтернативно, раствор в ацетоне можно распылить в камере, где его смешивают с азотом или другим инертным газом при температуре, равной от примерно 80 до примерно 180°C, и давлении, равном от примерно 1,0 до примерно 1,2 атм.
Обычно температуру и скорость потока осушающего газа можно выбрать таким образом, что капли раствора ГПМЦ-АС/лекарственное средство были достаточно сухими к тому моменту, когда они оказываются на стенке аппарата, и что они являются в основном твердыми и, таким образом, образуют мелкодисперсный порошок и не прилипают к стенке аппарата. Период времени, необходимый для обеспечения этой степени сухости, зависит от размера капель. Диаметр частиц обычно находится в диапазоне от примерно 1 до примерно 500 мкм, более типичным является диаметр, находящийся в диапазоне от примерно 5 до примерно 100 мкм. Высокое отношение величины площади частиц к их объему и большая движущая сила, предназначенная для выпаривания растворителя, обеспечивают фактическое время сушки, составляющее несколько секунд или менее. Это быстрое высушивание является критически важным для поддержания частиц в виде однородной композиции, а не разделения на обогащенную лекарственным средством и обогащенную полимером фазы.
Такие дисперсии, которые обладают однородным составом, можно считать твердыми растворами и они могут быть перенасыщены лекарственным средством. Такие однородные дисперсии являются предпочтительными, поскольку обеспечиваемое при введении большого количества лекарственного средства максимальное значение концентрации перенасыщения (МКПН) может быть выше, чем в случае дисперсий, в которых по меньшей мере часть лекарственного средства находится в виде обогащенной лекарственным средством аморфной или кристаллической фазы. Время затвердевания может составлять менее, чем примерно 20 с. В одном неограничивающем варианте осуществления время затвердевания может составлять менее, чем примерно 5 с. В другом неограничивающем варианте осуществления время затвердевания может составлять менее, чем примерно 2 с. Обычно для обеспечения такого быстрого затвердевания раствора лекарственное средство/полимер диаметр частиц, образовавшихся во время проведения процедуры распылительной сушки, составляет менее, чем 100 мкм. В одном неограничивающем варианте осуществления диаметр капель составляет менее, чем примерно 50 мкм. В другом неограничивающем варианте осуществления диаметр капель составляет менее, чем примерно 25 мкм. Полученные таким образом твердые частицы обычно обладают диаметром, равным менее, чем примерно 100 мкм. В одном неограничивающем варианте осуществления полученные твердые частицы обладают диаметром, равным менее, чем примерно 50 мкм. В другом неограничивающем варианте осуществления полученные твердые частицы обладают диаметром, равным менее, чем примерно 25 мкм.
После затвердевания твердый порошок можно выдерживать в камере для распылительной сушки в течение от примерно 5 до примерно 50 с для дальнейшего выпаривания растворителя из твердого порошка. Конечное содержание растворителя в твердой дисперсии, в таком виде, в котором ее извлекают из устройства для сушки, может быть низким, поскольку это уменьшает подвижность молекул лекарственного средства в дисперсии, что улучшает ее стабильность. Обычно содержание в дисперсии остаточного растворителя может составлять менее, чем примерно 10 мас. %. В одном неограничивающем варианте осуществления содержание в дисперсии остаточного растворителя может составлять менее, чем примерно 2 мас. %.
Затем дисперсии можно подвергнуть последующей обработке и приготовить их для введения по методикам, известным в данной области техники, таким как вальцовое прессование, агломерация в псевдоожиженном слое или нанесение покрытия распылением.
Процедуры распылительной сушки и оборудование для распылительной сушки в общем описаны в публикациях Perry's Chemical Engineers' Handbook, Sixth Edition (R.H. Perry, D.W. Green, J.O. Maloney, eds.) McGraw-Hill Book Co. 1984, page 20-54 to 20-57, и "Atomization and Spray-Drying", Chem. Eng. Prog. Monograph. Series, 50 (1954) No. 2), полные содержания которых явно включены в настоящее изобретение в качестве ссылки.
Раствор, который сушат распылительной сушкой для получения дисперсии ГПМЦ-АС/лекарственное средство, может содержать только лекарственное средство и ГПМЦ-АС в растворителе. Обычно отношение количества лекарственного средства к количеству ГПМЦ-АС в растворе находится в диапазоне от примерно 1:0,2 до примерно 1:100. В одном неограничивающем варианте осуществления отношение количества лекарственного средства к количеству ГПМЦ-АС находится в диапазоне от примерно 1:0,4 до примерно 1:20. Минимальное значение отношения лекарственное средство : полимер, которое обеспечивает удовлетворительные результаты, может меняться при переходе от одного лекарственного средства к другому лекарственному средству и его лучше всего определять в проводимых in vitro исследованиях растворения.
Фактически, растворителями, подходящими для распылительной сушки, могут являться любые органические соединения, в которых растворимы и лекарственное средство, и ГПМЦ-АС. В одном неограничивающем варианте осуществления растворитель также является летучим и обладает температурой кипения, равной 150°C или менее. Примеры растворителей могут включать, но не ограничиваются только ими, спирты, такие как метанол, этанол, н-пропанол, изопропанол и бутанол; кетоны, такие как ацетон, метилэтилкетон и метилизобутилкетон; сложные эфиры, такие как этилацетат и пропилацетат; и разные другие растворители, такие как ацетонитрил, метиленхлорид, толуол и 1,1,1-трихлорэтан. Также можно использовать обладающие более низкой летучестью растворители, такие как диметилацетамид или диметилсульфоксид. Также можно использовать смеси растворителей, которыми могут быть смеси с водой, если полимер и ГПМЦ-АС обладают достаточной растворимостью для обеспечения практического осуществления процедуры распылительной сушки.
Растворы, которые сушат распылительной сушкой, и полученные дисперсии также могут содержать различные добавки, которые способствуют стабильности, растворению, таблетированию или обработке дисперсии. Как указано выше, примеры таких добавок могут включать, но не ограничиваются только ими, поверхностно-активные вещества, вещества, регулирующие рН (например, кислоты, основания и буферы), наполнители, разрыхлители или связующие. Такие добавки можно добавить непосредственно в раствор, предназначенный для распылительной сушки, например, чтобы растворить или суспендировать добавку в растворе в виде взвеси. Альтернативно, такие добавки можно добавить после проведения процедуры распылительной сушки для содействия формованию в конечную дозированную форму.
Экструзия расплава (ЭР) является методикой, широко использующейся для получения аморфной твердой дисперсии. При использовании в настоящем изобретении экструзия расплава означает процедуру смешивания двух или большего количества компонентов с использованием большого сдвигового усилия и при возможности регулирования температуры в экструдере. Экструдер для расплава состоит из четырех основных частей: мотора, который регулирует вращение шнеков, шнеков (основной источник сдвигового усилия и движения материала), барабанов, в которые вставлены шнеки и которые обеспечивают регулирование температуры, и мундштука (выходного отверстия), которое регулирует размер и форму экструдатов. Порошкообразный материал (в форме гранулята или порошка) обычно загружают в загрузочное отверстие реактора с регулируемой скоростью при вращении шнеков экструдера. Затем материал перемещают вперед посредством вращения шнека и трения материала о поверхность барабана. В зависимости от типа экструдера можно использовать один шнек или двойной шнек для работы вращением в одном или противоположных направлениях. Шнеки могут быть сконструированы соответствующим образом для обеспечения необходимой степени перемешивания. Обычно барабаны разделены на сегменты, чтобы обеспечить регулирование температуры в каждой зоне по всей длине шнека. С помощью выходного отверстия (система мундштука) регулируют форму и размер экструдатов.
Затем экструдат охлаждают и формуют путем каландрования или гранулирования и размалывают с получением частиц необходимого размера. Затем конечный размолотый экструдат обычно смешивают с дополнительными инертными наполнителями и прессуют. Процедуру экструзии проводят при температурах, равных выше Tg полимера, и достаточно высоких для того, чтобы АФИ плавился и/или растворялся в полимерной матрице.
ЭР может обеспечить возможность получения сложных многослойных и многофункциональных композитов путем формирования и объединения нескольких потоков расплава в одной полностью согласованной технологии производства. Таким образом, одно или большее количество активных лекарственных веществ могут быть диспергированы в одной или большем количестве полимерных матриц.
Раствор лекарственного средства и ГПМЦ-АС можно получить в растворителе, таком как ацетон. Раствор в ацетоне по каплям добавляют в подкисленную воду для совместного осаждения смеси лекарственное средство/полимер. Затем осадок отделяют фильтрованием и промывают подкисленной водой, затем сушат. Высушенный порошок просеивают через сито и получают частицы, обладающие одинаковым размером. Затем порошкообразную смесь пропускают через экструдер для расплава, температуру барабанов которого устанавливают равной примерно 70-140°C, и получают экструдированные стержни. Затем экструдированные стержни охлаждают до комнатной температуры и размалывают по методикам механического размола.
Приведенные ниже примеры иллюстрируют настоящее изобретение (и предлагаемые в нем решения), содержания в частях и процентах являются массовыми, если не указано иное. Каждый пример приведен для разъяснения настоящего изобретения (и предлагаемых в нем решений), а не для ограничения настоящего изобретения (и предлагаемых в нем решений). В действительности, для специалистов в данной области техники должно быть понятно, что без отклонения от объема или сущности настоящего изобретения в настоящее изобретение (и предлагаемые в нем решения) можно внести различные модификации и изменения. Например, отличительные признаки, проиллюстрированные или описанные в качестве части одного варианта осуществления, можно использовать в другом варианте осуществления и получить еще один вариант осуществления. Таким образом, следует понимать, что настоящее изобретение (и предлагаемые в нем решения) включает такие модификации и изменения, которые входят в объем прилагаемой формулы изобретения и ее эквивалентов.
ПРИМЕРЫ
Методика определения распределения заместителей, находящихся в определенных положениях ГПМЦ-АС
Как описано выше, ГПМЦ можно заместить ацетатной и сукцинатной группой и получить ГПМЦ-АС. Для каждого заместителя существует 4 возможных положения или позиций замещения, т.е. непосредственно в положениях C2, C3 и/или C6 кольца целлюлозы, а также в концевой группе ОН, содержащейся в гидроксипропоксильной цепи. Для определения положения замещения кольца ангидроглюкозы и ацетатной, и сукцинатной группой можно использовать спектроскопию 13С ЯМР. Определение распределения заместителей, находящихся в определенных положениях простого эфира целлюлозы, с использованием 13С ЯМР подробно описано в публикациях Makromol. Chem., Vol. 191, 681-691 (1990), и Macromolecules, Vol. 20, 2413-2418 (1987), полные содержания которых явно включены в настоящее изобретение в качестве ссылки.
Все спектры ЯМР для образцов ГПМЦ-АС снимали с использованием ЯМР спектрометра Bruker AVIII 500 МГц, снабженного датчиком 10 мм с градиентом по оси z ВВО. Образцы ГПМЦ-АС растворяли в ДМСО-d6 (ДМСО - диметилсульфоксид). В области спектра, соответствующей карбонилу (168,0-174,0 част./млн), наблюдались пики, соответствующие атомам углерода, содержащимся во фрагменте СО ацетила и фрагменте СО сукциноила, как это показано ниже. Для сукциноильного заместителя наблюдались два набора пиков вследствие наличия карбонильных атомов углерода, содержащихся в кислотной и сложноэфирной группе. Для карбонильных атомов углерода, содержащихся сложноэфирной группе, наблюдается один пик (пики), положение которого зависит от положения заместителей в кольце ангидроглюкозы и в гидроксипропоксильной цепи. Отнесение пиков проводили на основании приведенных в литературе значений, модельного соединения и анализа с помощью 2D ЯМР.
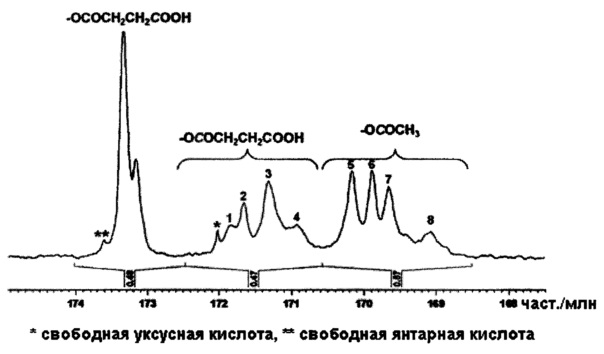
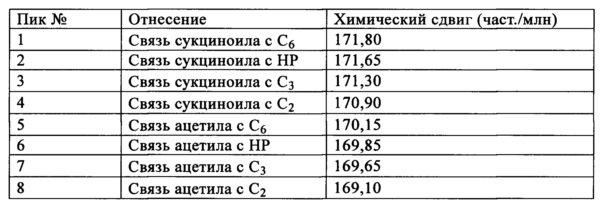
Для определения площадей пиков пики разлагали на компоненты и оцененные площади нормировали на количества ацетила и сукциноила в положении СЗ, которые определяли по описанным ниже методикам.
Определение содержаний свободных кислот и ацетильных и сукциноильных групп - Статья НФ (национальный фармакологический справочник) USP 34-NF 29 Hypromellose Acetate Succinate.
Содержания свободных кислот и ацетильных и сукциноильных групп определяли по методикам, описанным в национальном фармакологическом справочнике Фармакопеи Соединенных Штатов Америки 2011: статья НФ USP 34-NF 29 Hypromellose Acetate Succinate.
1. Предельное количество свободных уксусной и янтарной кислот
Раствор фосфорной кислоты - В мерную колбу объемом 50 мл переносили 1,0 мл 1,25 М раствора фосфорной кислоты и доводили до метки водой.
0,02 М Фосфатный буфер - 5,44 г Дигидрофосфата калия растворяли в 2 л воды.
Растворитель - 0,02 М Фосфатный буфер с добавлением 1 н. раствора гидроксида натрия для обеспечения значения рН, равного 7,5.
Исходный раствор уксусной кислоты - В мерную колбу с притертой пробкой объемом 100 мл добавляли примерно 20 мл воды. Колбу помещали на весы и определяли массу тары. В колбу переносили 2,0 мл ледяной уксусной кислоты и определяли массу добавленной уксусной кислоты. В колбу добавляли воду до метки. 6,0 мл Этого раствора переносили в мерную колбу объемом 100 мл и доводили до метки водой.
Исходный раствор янтарной кислоты - Примерно 130 мг янтарной кислоты добавляли в мерную колбу объемом 100 мл. Добавляли примерно 50 мл воды и содержимое взбалтывали до полного растворения янтарной кислоты. В колбу добавляли воду до метки.
Подвижная фаза - Значение рН 0,02 М фосфатного буфера доводили до равного 2,8 путем проводимого по каплям добавления 6 М раствора фосфорной кислоты и пропускали через полиамидный фильтр с размером пор, равным 0,22 мкм.
Раствор стандарта - В мерную колбу объемом 25 мл переносили 4,0 мл исходного раствора уксусной кислоты. В эту же колбу переносили 4,0 мл исходного раствора янтарной кислоты, доводили до метки подвижной фазой и перемешивали. Раствор готовили дважды.
Исследуемый раствор - 4,0 мл Растворителя переносили в стеклянный сосуд, содержащий примерно 102 мг ГПМЦ-АС, и содержимое перемешивали в течение примерно 2 ч. Затем в тот же сосуд переносили 4,0 мл раствора фосфорной кислоты для обеспечения значения рН исследуемого раствора, равного примерно 3 или менее. Сосуд несколько раз переворачивали для обеспечения полного перемешивания, центрифугировали и прозрачную надосадочную жидкость использовали в виде исследуемого раствора.
Хроматографическая система (USP 34 Chromatography <621>) - Жидкостной хроматограф был снабжен детектором, работающим при 215 нм, и колонкой 4,6 мм×15 см, которая содержала насадочный материал L1, 5 мкм (а именно, Restek UltraAqueous С18, 5 мкм, 150×4,6 мм, Cat. # 9178565-700). Температуру колонки поддерживали равной примерно 30°C. Скорость потока составляла примерно 1 мл/мин и длительность эксперимента составляла примерно 15 мин. Анализировали раствор стандарта и определяли площади пиков, как указано в методике: эффективность колонки, определенная по площади пика янтарной кислоты, составляла не менее 8000 теоретических тарелок; коэффициент асимметрии этого пика составлял от 0,9 до 1,5; и относительное S, стандартное отклонение, для проводимых 6-ти повторных инжектирований составляло не более 2,0% для каждого пика. Дважды анализировали раствор стандарта и определяли площади пиков, как указано в методике. После каждой последовательности экспериментов колонку промывали сначала смесью примерно 50% воды и примерно 50% ацетонитрила в течение примерно 60 мин и затем 100% метанолом в течение примерно 60 мин. Колонку хранили в 100% метаноле.
Процедура - Раствор стандарта и исследуемый раствор, обладающие равными объемами (10 мкл), по отдельности инжектировали в хроматограф, получали хроматограммы и определяли площади пиков, соответствующих уксусной и янтарной кислотам.
Выраженное в процентах количество свободной уксусной кислоты, содержащееся во фрагменте ГПМЦ-АС, рассчитывали по формуле:
0,0768(WA/W)(rUA/rSA),
где WA обозначает выраженную в мг массу ледяной уксусной кислоты, использующейся для приготовления исходного раствора уксусной кислоты; W обозначает выраженную в мг массу ГПМЦ-АС, использующегося для приготовления исследуемого раствора; и rUA и rSA обозначают площади пиков уксусной кислоты, полученные для исследуемого раствора и раствора стандарта соответственно.
Выраженное в процентах количество свободной янтарной кислоты, Sfree, содержащееся во фрагменте ГПМЦ-АС, рассчитывали по формуле:
l,28(WS/W)(rUS/rSS),
где WS обозначает выраженную в мг массу янтарной кислоты, использующейся для приготовления исходного раствора янтарной кислоты; rUS и rSS обозначают площади пиков янтарной кислоты, полученные для исследуемого раствора и раствора стандарта соответственно; и W является таким, как определено выше.
2. Содержание ацетильных и сукциноильных групп
Раствор фосфорной кислоты, исходный раствор уксусной кислоты, исходный раствор янтарной кислоты, подвижная фаза, раствор стандарта и хроматографическая система - Использовали такие, как указано в исследовании для определения предельного количества свободных уксусной и янтарной кислот.
Исследуемый раствор - 4,0 мл 1,0 н. Раствора гидроксида натрия переносили в стеклянный сосуд, содержащий примерно 12,4 мг ГПМЦ-АС. Раствор перемешивали в течение 4 ч. Затем в тот же сосуд добавляли 4,0 мл 1,25 М раствора фосфорной кислоты для обеспечения значения рН раствора, равного примерно 3 или менее. Сосуд, содержащий раствор исследуемого образца, несколько раз переворачивали для обеспечения полного перемешивания и пропускали через фильтр с размером пор, равным 0,22 мкм. Прозрачный фильтрат использовали в виде использовали в виде исследуемого раствора.
Процедура - Раствор стандарта и исследуемый раствор, обладающие равными объемами (10 мкл), по отдельности инжектировали в хроматограф, получали хроматограммы и определяли площади пиков, соответствующих уксусной и янтарной кислотам.
Выраженное в процентах количество уксусной кислоты, А, содержащееся во фрагменте ГПМЦ-АС, рассчитывали по формуле:
0,0768(WA/WU)(rUA/rSA),
где WA обозначает выраженную в мг массу уксусной кислоты, использующейся для приготовления исходного раствора уксусной кислоты; WU обозначает выраженную в мг массу ГПМЦ-АС, использующегося для приготовления исследуемого раствора; и rUA и rSA обозначают площади пиков уксусной кислоты, полученные для исследуемого раствора и раствора стандарта соответственно.
Выраженное в процентах количество ацетильных групп (-СОСН3) рассчитывали во фрагменте ацетат-сукцината гидроксипропилметилцеллюлозы по формуле:
0,717(A-Afree),
где Afree обозначает выраженное в процентах количество свободной уксусной кислоты, полученное в исследовании для определения предельного количества свободных уксусной и янтарной кислот; и А является таким, как определено выше.
Выраженное в процентах количество янтарной кислоты, содержащееся во фрагменте ГПМЦ-АС, рассчитывали по формуле:
1,28(WS/WU)(rUS/rSS),
где WS обозначает выраженную в мг массу янтарной кислоты, использующейся для приготовления исходного раствора янтарной кислоты; WU является таким, как определено выше; и rUS и rSS обозначают площади пиков янтарной кислоты, полученные для исследуемого раствора и раствора стандарта соответственно.
Выраженное в процентах количество сукциноильных групп (-СОС2Н4СООН) рассчитывали во фрагменте ГПМЦ-АС по формуле:
0,856(S-Sfree)
где S является таким, как определено выше; и Sfree обозначает выраженное в процентах количество свободной янтарной кислоты, полученное в исследовании для определения предельного количества свободных уксусной и янтарной кислот.
Определение содержаний гидроксипропоксигрупп и метоксигрупп - Статья ФСША USP 34-NF 29 Hypromellose.
Содержания гидроксипропоксигрупп и метоксигрупп определяли по методикам, описанным в национальном фармакологическом справочнике Фармакопеи Соединенных Штатов Америки, 2011: статья ФСША USP 34-NF 29 Hypromellose.
1. Процедура
Прибор - В качестве реакционного сосуда использовали герметизированный сосуд для сыворотки объемом 5 мл, высотой 50 мм, внешним диаметром, равным 20 мм, и внутренним диаметром горлышка, равным 13 мм. Сосуд был снабжен герметичной крышкой мембранного типа, состоящей из бутильного каучука с лицевой поверхностью из политетрафторэтилена, и воздухонепроницаемым уплотнением с алюминиевым зажимом или любой уплотняющей системой, обеспечивающей достаточную воздухонепроницаемость. Использовали нагревательное устройство, включающее нагревательный модуль, который содержал алюминиевый блок квадратной формы с отверстиями диаметром 20 мм и глубиной 32 мм, в которые помещали реакционные сосуды. Нагревательный модуль также был снабжен магнитной мешалкой, обеспечивающей перемешивание содержимого сосуда, или использовали вибрационный шюттльаппарат, обеспечивающий качательные движения, примерно 100 качаний/мин.
Йодистоводородная кислота - Использовали реагент, обладающий типичной концентрацией HI, равной примерно 57%.
Раствор внутреннего стандарта - 30 мг/мл н-Октана в о-ксилоле.
Раствор стандарта - 2,0 мл Йодистоводородной кислоты и 2,0 мл раствора внутреннего стандарта добавляли в подходящий сосуд для сыворотки, содержащий примерно 60 и 100 мг адипиновой кислоты. Сосуд надежно закрывали подходящей мембранной пробкой и взвешивали. Через мембрану шприцем добавляли примерно 15 мл и 22 мл изопропилйодида. Сосуд повторно взвешивали и по разнице рассчитывали массу добавленного изопропилйодида. Добавляли 45 мл метилйодида и повторно взвешивали, по разнице рассчитывали массу добавленного изопропилйодида. Реакционный сосуд тщательно встряхивали и слоям давали разделиться. Верхний слой использовали в качестве раствора стандарта.
Раствор образца - 0,065 г Высушенного ГПМЦ-АС переносили в толстостенный реакционный сосуд объемом 5 мл, снабженный герметичной крышкой мембранного типа. В сосуд пипеткой добавляли примерно 60 и 100 мг адипиновой кислоты и 2,0 мл раствора внутреннего стандарта. К смеси пипеткой добавляли 2,0 мл йодистоводородной кислоты, сосуд сразу плотно закрывали и взвешивали. Содержимое сосуда непрерывно перемешивали с использованием магнитной мешалки, которой был снабжен нагревательный модуль, или с помощью вибрационного шюттльаппарата. Содержимое нагревали и выдерживали при температуре, равной примерно 130±2°C, в течение примерно 60 мин. Если не было возможности использовать вибрационный шюттльаппарат или магнитную мешалку, то сосуд тщательно встряхивали вручную в течение первых 30 мин нагревания с интервалами в 5 мин. Сосуду давали охладиться, и его взвешивали.
2. Хроматографическая система
Режим - ГХ (газовая хроматография)
Детектор - теплопроводности или водородный пламенный ионизационный детектор
Колонка - стеклянная колонка 3-4 мм × 1,8-3 м, наполненная 20% жидкой фазой G28 на подложке S1C 100-120 меш, которая не силанизирована.
Температура колонки - 100°C
Газ-носитель - в случае детектора теплопроводности использовали гелий; в случае водородного пламенного ионизационного детектора использовали гелий или азот.
Скорость потока - в случае раствора стандарта скорость потока регулировали таким образом, что время удерживания внутреннего стандарта составляло примерно 10 мин.
Инжектируемое количество - 1-2 мл
3. Анализ
Образцы - верхний слой раствора стандарта и раствор образца
Выраженное в процентах количество -ОСН3 во фрагменте ГПМЦ-АС рассчитывали, как:
Результат = 21,864×(RUa/RSa)×(WSa/WU)
RUa = отношение площади пика метилйодида к площади пика н-октана, полученное для раствора образца
RSa = отношение площади пика метилйодида к площади пика н-октана, полученное для раствора стандарта
WSa = масса метилйодида в растворе стандарта (мг)
WU = масса ГПМЦ-АС в растворе образца в пересчете на массу в сухом состоянии (мг)
Выраженное в процентах количество -ОС3Н6ОН во фрагменте ГПМЦ-АС рассчитывали, как:
Результат = 44,17×(RUb/RSb)×(WSb/WU)
RUb = отношение площади пика изопропилйодида к площади пика н-октана, полученное для раствора образца
RSb = отношение площади пика изопропилйодида к площади пика н-октана, полученное для раствора стандарта
WSb = масса изопропилйодида в растворе стандарта (мг)
WU = масса ГПМЦ-АС в растворе образца в пересчете на массу в сухом состоянии (мг)
Определение молекулярно-массового (ММ) распределения с помощью эксклюзионной хроматографии (ЭКХ)
Молекулярная масса является суммой атомных масс атомов, содержащихся в молекуле. При использовании в настоящем изобретении применительно к полимерам термины "молекулярная масса", "средняя молекулярная масса" и "кажущаяся молекулярная масса" означает среднее арифметическое значение молекулярной массы отдельных макромолекул, определенной с помощью эксклюзионной хроматографии (ЭКХ).
Средние значения относительной молекулярной массы рассчитывали путем сравнения данных, полученных с помощью аналитической ЭКХ, со значениям для стандартов поли(этиленгликоль/этиленоксид) (ПЭГ/ПЭО), обладающих узким молекулярно-массовым распределением.
1. Установка для хроматографии
Все содержащиеся в установке модули Waters выпускаются фирмой Waters Corporation, 34 Maple Street, Milford, MA 01757, USA. Установку заменили аналогичной, выпускающейся другой фирмой (фирмами).
Система подачи растворителя - Waters М515
Автоматический пробоотборник - Waters М717
Детектор дифференциального показателя преломления (ДПП) - Waters М2414 для относительной ЭКХ**
Группа (группы) колонок - см. подробное описание в приведенном ниже разделе "условия проведения анализа"
Программное обеспечение - Waters Empower 2
* Диапазон ПП (показатель преломления) - от 1,00 до 1,75 ЕПП (единицы показателя преломления)
Диапазон измерений - 7×10-7 ЕПП
Смещение - 2×10-7 ЕПП
2. Условия проведения анализа с помощью ЭКХ
Подвижная фаза - 55% 0,1 М ацетат лития/45% этанол
Скорость потока - 0,8 мл/мин
Колонки - TSKgel quard (6 мм×40 мм) + 2 колонки Linear TSK GMPWx1; 13 мкм; 300 мм × 7,8 мм (TOSOH Bioscience LLC, 3604 Horizon Drive, Suite 100, King of Prussia, PA 19406, USA)
Температура колонки - 35°C
Температура детектора ДПП (дифференциальный показатель преломления) - 35°C
Калибровка - стандарты ПЭО/ПЭГ, обладающие узким молекулярно-массовым распределением (PSS-USA, Inc. Amherst Fields Research Park, 160 Old Farm Road, Amherst, MA 01002)
Концентрация образца - обычно 1 мг/мл (если не указано иное) Инжектируемый объем - 200 мкл
Определение температуры стеклования
Температурой стеклования, Tg, является температура, при которой аморфное твердое вещество, такое как стекло или полимер, становится хрупким или жестким при охлаждении, или мягким или пластичным при нагревании. Tg можно определить, например, путем дифференциальной сканирующей калориметрии (ДСК). С помощью ДСК измеряют разницу между количеством тепла, необходимого для повышения температуры образца и эталона, как функцию температуры. Во время фазового перехода, такого как переход из аморфного состояния в кристаллическое состояние, изменяется количество необходимого тепла. В случае твердого вещества, которое в основном не содержит кристаллических компонентов, наличие одной температуры стеклования означает, что твердое вещество представляет собой молекулярную дисперсию.
Определение температуры стеклования (Tg) проводили с помощью ДСК (дифференциальный сканирующий калориметр) Q2000, выпускающегося фирмой ТА Instruments. Примерно 5 мг образца помещали в стандартный алюминиевый тигель. Исследование нагревание-охлаждение-нагревание при линейном изменение температуры проводили в температурном диапазоне, составляющем примерно от -20 до 190°C, и при скорости нагревания и охлаждения, равной примерно 20°C/мин. Температуру стеклования определяли из результатов, полученных при втором нагревании, и ее определяли на основании ширины на половине высоты кривой для теплового потока.
Измерение вязкости
4,3 г Гидроксида натрия растворяли в не содержащей диоксид углерода воде и получали 1000 мл раствора гидроксида натрия. Раствор гидроксида натрия добавляли к 2,00 г предварительно высушенной ГПМЦ-АС и получали 100,0 г. В сосуд вставляли пробку и содержимое растворяли путем непрерывного встряхивания в течение примерно 30 мин. Температуру раствора устанавливали равной 20±0,1°C. Вязкость определяли с помощью вискозиметра Уббелоде или эквивалентного вискозиметра в соответствии с руководством ФСША 34 NF29 - Procedure for Cellulose Derivative under Viscosity <911>, полное содержание которого явно включено в настоящее изобретение в качестве ссылки. Эти измерения также можно провести с использованием автоматического вискозиметра Cannon Mini-PV-x.
Синтез ГПМЦ-АС
Сравнительный пример 1 - Синтез ГПМЦ-АС при 115°C (методика А)
В высушенный в сушильном шкафу стеклянный реактор объемом 1 л, снабженный патрубком для ввода и вывода азота, холодильником и верхней механической мешалкой, добавляли уксусную кислоту. Медленно добавляли ГПМЦ (Benecel™ Е5, выпускающийся фирмой Ashland, Inc.), перемешивая смесь при 200 об/мин. Добавляли уксусный ангидрид и смесь перемешивали при температуре окружающей среды в течение примерно 30 мин. Последовательно добавляли янтарный ангидрид и ацетат натрия и смесь перемешивали при 300 об/мин, нагревая до 115°C. После установления внутренней температуры, равной 115°C, перемешивание продолжали в течение примерно 3 ч. Затем температуру понижали. Реакционную смесь охлаждали до температуры окружающей среды и смешивали с водой и осаждалось почти белое твердое вещество. Осадок промывали дополнительным количеством воды и сушили в сушильном устройстве с псевдоожиженным слоем при 65°C. В таблице 1 приведены разные образцы ГПМЦ-АС, полученные по методике А с использованием разных количеств реагентов.
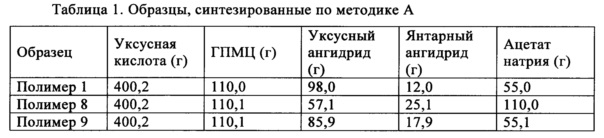
Пример 1А - Синтез ГПМЦ-АС (методика В)
В высушенный в сушильном шкафу стеклянный реактор объемом 1 л, снабженный патрубком для ввода и вывода азота, холодильником и верхней механической мешалкой, добавляли уксусную кислоту. Медленно добавляли ГПМЦ, перемешивая смесь при 200 об/мин. Последовательно добавляли уксусный ангидрид и ацетат натрия и смесь нагревали при 115°C при перемешивании при 300 об/мин. После установления внутренней температуры, равной 115°C, перемешивание продолжали в течение примерно 30 мин (начальное время выдерживания). Добавляли янтарный ангидрид и смесь перемешивали при примерно 115°C в течение примерно 2,5 ч (время выдерживания после второго добавления). Реакционную смесь охлаждали до температуры окружающей среды и смешивали с водой и осаждалось почти белое твердое вещество. Осадок промывали водой и сушили в сушильном устройстве с псевдоожиженным слоем при 65°C. В таблице 2А приведены полимеры 2-4 и полимер 10, полученные с использованием разных количеств реагентов.
Пример 1В - Синтез ГПМЦ-АС (методика В1)
В высушенный в сушильном шкафу стеклянный реактор объемом 1 л, снабженный патрубком для ввода и вывода азота, холодильником и верхней механической мешалкой, добавляли уксусную кислоту. Медленно добавляли ГПМЦ, перемешивая смесь при 200 об/мин. Добавляли уксусный ангидрид и ацетат натрия и смесь нагревали при 115°C при перемешивании при 300 об/мин. После установления внутренней температуры, равной 115°C, перемешивание продолжали в течение примерно 2 ч в случае полимера 11 и в течение примерно 1,0 ч в случае полимера 12, приведенных в таблице 2А (начальное время выдерживания). Добавляли янтарный ангидрид и смесь перемешивали при примерно 115°C в течение примерно 2,5 ч (время выдерживания после второго добавления). Затем медленно добавляли воду, поддерживая температуру реакционной смеси, равной от 45 до 85°C, до завершения добавления воды. Реакционную смесь охлаждали до температуры окружающей среды и смешивали с водой и осаждалось почти белое твердое вещество. Осадок промывали дополнительным количеством воды и сушили в сушильном устройстве с псевдоожиженным слоем при 65°C. В таблице 2А приведены полученные полимеры 11-12.
Пример 1С - Синтез ГПМЦ-АС (методика В2)
В высушенный в сушильном шкафу стеклянный реактор объемом 1 л, снабженный патрубком для ввода и вывода азота, холодильником и верхней механической мешалкой, добавляли уксусную кислоту. Медленно добавляли ГПМЦ, перемешивая смесь при 200 об/мин. Добавляли уксусный ангидрид и ацетат натрия и смесь нагревали при 85°C. После установления внутренней температуры, равной 85°C, перемешивание продолжали в течение примерно 1 ч (начальное время выдерживания). Добавляли янтарный ангидрид и смесь нагревали при 85°C в течение примерно 1,5 ч и затем в течение примерно 30 мин нагревали до 115°C и выдерживали при 115°C в течение примерно 2,5 ч (время выдерживания после второго добавления составляло всего 4,5 ч). Реакционную смесь охлаждали до температуры окружающей среды и смешивали с водой и осаждалось почти белое твердое вещество. Осадок промывали водой и сушили в сушильном устройстве с псевдоожиженным слоем при 65°C. В таблице 2А приведен полученный полимер 13.

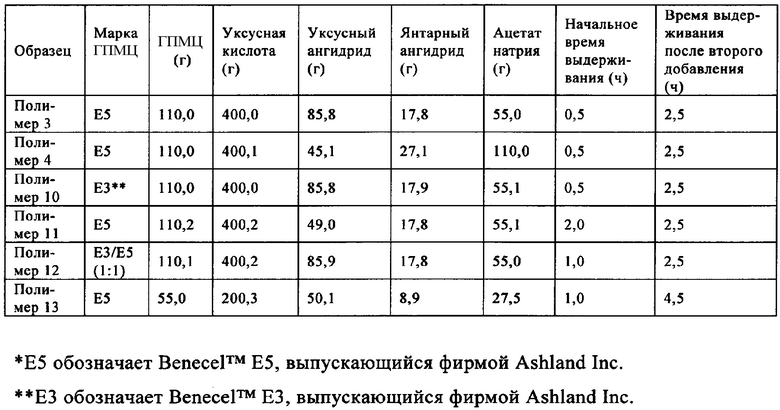
Пример 1D - Синтез ГПМЦ-АС (методика В3)
В высушенный в сушильном шкафу стеклянный реактор объемом 1 л, снабженный патрубком для ввода и вывода азота, холодильником и верхней механической мешалкой, добавляли уксусную кислоту. Медленно добавляли ГПМЦ, перемешивая смесь при 200 об/мин. Добавляли уксусный ангидрид и ацетат натрия и смесь перемешивали при 300 об/мин и при температуре окружающей среды в течение периода времени (время выдерживания после первого добавления). Затем смесь нагревали при 85°C. После установления внутренней температуры, равной 85°C, перемешивание продолжали в течение периода времени (начальное время выдерживания). Добавляли янтарный ангидрид и смесь перемешивали при примерно 85°C в течение периода времени (время выдерживания после второго добавления). Затем медленно добавляли воду, поддерживая температуру реакционной смеси, равной от 45 до 85°C, до завершения добавления воды. Реакционную смесь охлаждали до температуры окружающей среды и смешивали с водой и осаждалось почти белое твердое вещество. Осадок промывали водой и сушили в сушильном устройстве с псевдоожиженным слоем при 65°C. В таблице 2В приведены полимеры 14-18, полученные с использованием разных количеств реагентов.
Пример 1F - Синтез ГПМЦ-АС (методика В4)
В высушенный в сушильном шкафу стеклянный реактор объемом 1 л, снабженный патрубком для ввода и вывода азота, холодильником и верхней механической мешалкой, добавляли уксусную кислоту. Медленно добавляли ГПМЦ (Benecel™ Е3), перемешивая смесь при 200 об/мин. Добавляли ацетат натрия и смесь нагревали при 85°C при перемешивании при 300 об/мин. После установления внутренней температуры, равной 85°C, добавляли уксусный ангидрид и перемешивание продолжали в течение периода времени (начальное время выдерживания). Добавляли янтарный ангидрид и смесь перемешивали при примерно 85°C в течение периода времени (время выдерживания после второго добавления). Затем медленно добавляли воду, поддерживая температуру реакционной смеси, равной от 45 до 85°C, до завершения добавления воды. Реакционную смесь охлаждали до температуры окружающей среды и смешивали с водой и осаждалось почти белое твердое вещество. Осадок промывали водой и сушили в сушильном устройстве с псевдоожиженным слоем при 65°C. В таблице 2 В приведен полученный полимер 19.
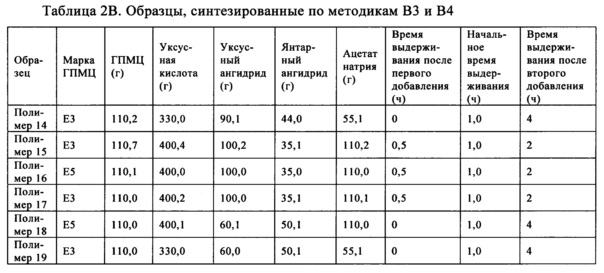
Пример 2А - Синтез ГПМЦ-АС (методика С)
В высушенный в сушильном шкафу стеклянный реактор объемом 1 л, снабженный патрубком для ввода и вывода азота, холодильником и верхней механической мешалкой, добавляли уксусную кислоту. Медленно добавляли ГПМЦ (Benecel™ Е5), перемешивая смесь при 200 об/мин. Последовательно добавляли янтарный ангидрид и ацетат натрия и смесь нагревали при 115°C при перемешивании при 300 об/мин. После установления внутренней температуры, равной 115°C, перемешивание продолжали в течение примерно 30 мин. Добавляли уксусный ангидрид и смесь перемешивали примерно при 115°C в течение примерно 2,5 ч. Реакционную смесь охлаждали до температуры окружающей среды и смешивали с водой и осаждалось почти белое твердое вещество. Осадок перемешивали и промывали водой и сушили в сушильном устройстве с псевдоожиженным слоем при 65°C. В таблице 3А приведены полимеры 5-7 и полимеры 20-22, полученные с использованием разных количеств реагентов.
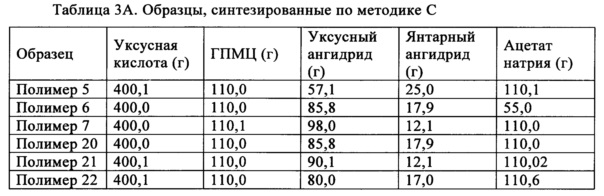
Пример 2В - Синтез ГПМЦ-АС (методика С1)
В высушенный в сушильном шкафу стеклянный реактор объемом 1 л, снабженный патрубком для ввода и вывода азота, холодильником и верхней механической мешалкой, добавляли уксусную кислоту. Медленно добавляли ГПМЦ, перемешивая смесь при 200 об/мин. Добавляли ацетат натрия и смесь нагревали при 85°C при перемешивании при 300 об/мин. После установления внутренней температуры, равной 85°C, добавляли янтарный ангидрид и перемешивание продолжали в течение периода времени (время выдерживания после первого добавления). Добавляли уксусный ангидрид и смесь перемешивали примерно при 85°C в течение периода времени (время выдерживания после второго добавления). Затем медленно добавляли воду, поддерживая температуру реакционной смеси, равной от 45 до 85°C, до завершения добавления воды. Реакционную смесь охлаждали до температуры окружающей среды и смешивали с водой и осаждалось почти белое твердое вещество. Осадок промывали водой и сушили в сушильном устройстве с псевдоожиженным слоем при 65°C. В таблице 3В приведены полимеры 23-26, полученные с использованием разных количеств реагентов.
Пример 2С - Синтез ГПМЦ-АС (методика С2)
В высушенный в сушильном шкафу стеклянный реактор объемом 1 л, снабженный патрубком для ввода и вывода азота, холодильником и верхней механической мешалкой, добавляли уксусную кислоту. Медленно добавляли ГПМЦ, перемешивая смесь при 200 об/мин. Добавляли ацетат натрия и смесь нагревали при 85°C при перемешивании при 300 об/мин. После установления внутренней температуры, равной 85°C, добавляли янтарный ангидрид и перемешивание продолжали в течение периода времени (время выдерживания после первого добавления). Добавляли уксусный ангидрид (66,5 г) и смесь перемешивали при примерно 85°C в течение периода времени (время выдерживания после второго добавления). Добавляли вторую порцию уксусного ангидрида (60,5 г) и смесь перемешивали при 85°C в течение примерно 2,5 ч. Затем медленно добавляли воду, поддерживая температуру реакционной смеси, равной от 45 до 85°C, до завершения добавления воды. Реакционную смесь охлаждали до температуры окружающей среды и смешивали с водой и осаждалось почти белое твердое вещество. Осадок перемешивали и промывали водой и сушили в сушильном устройстве с псевдоожиженным слоем при 65°C. В таблице 3В приведен полученный полимер 27.
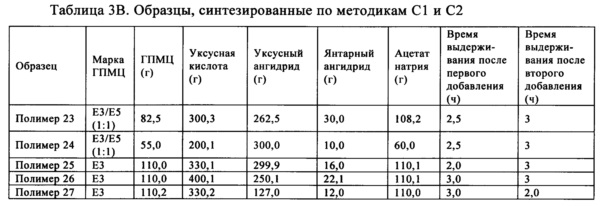
Характеризация образцов ГПМЦ-АС
В таблице 4 представлены характеризующие данные для образцов. Для сравнения приведены данные для имеющихся в продаже образцов, выпускающихся фирмой Shin-Etsu, соответствующих названиям Shin-Etsu AQOAT LF, Shin-Etsu AQOAT MF и Shin-Etsu AQOAT HF. "F" обозначает марку тонкодисперсного порошка, обладающего средним размером частиц, равным примерно 5 мкм.
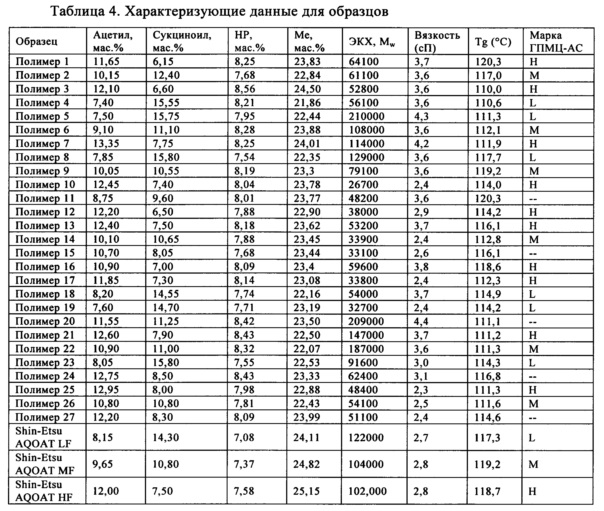
В таблице 5 приведено распределение ацетильных и сукциноильных заместителей, находящихся в положениях CHP-ОН, С2-ОН, С3-ОН и C6-ОН для ЗБГ, которое определяли путем анализа с помощью 13С ЯМР. Полученное на основании данных, приведенных в таблице 5, выраженное в процентах содержание ацетила и сукциноила в положениях гидроксигрупп С6 и С3, приведено в таблице 5А.
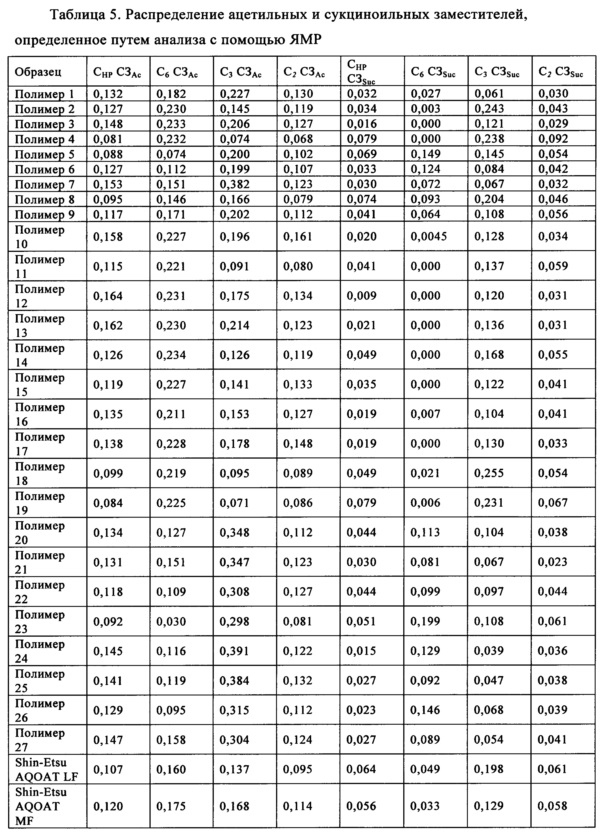
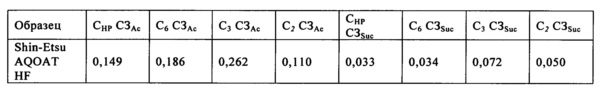
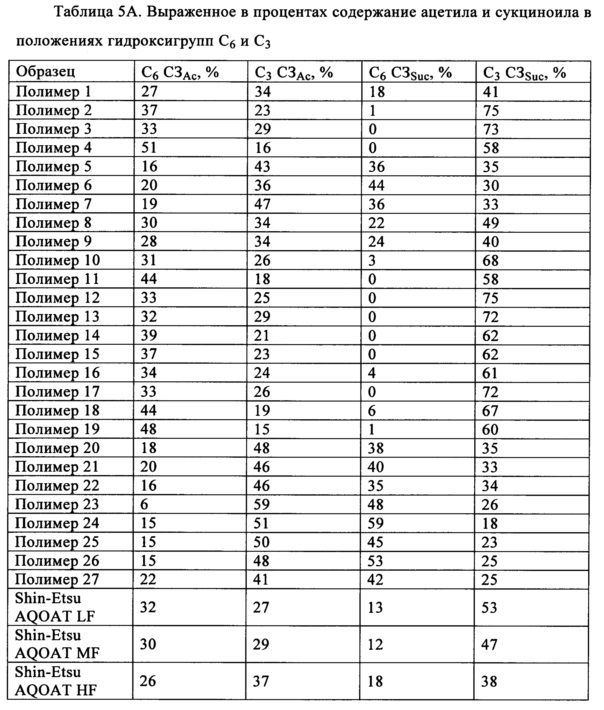
Измерение индекса расплава ГПМЦ-АС
Индекс текучести расплава ГПМЦ-АС измеряли с использованием Tinius Olsen Thermodyne 5208. Примерно 5 г порошка ГПМЦ-АС помещали в пуансон и уплотняли, избегая попадания воздуха. В пуансон вставляли поршень. Температуру пуансона повышали до равной примерно 100°C и в течение примерно 5 мин давали установиться равновесию. Затем к поршню прикладывали нагрузку, составляющую 5 кг, и экструдат, если его получали, собирали в течение 6 мин. С поршня снимали нагрузку, составляющую 5 кг. Затем температуру пуансона повышали на 10°C и в течение примерно 5 мин давали установиться равновесию. Такую же процедуру повторяли для сбора экструдата при повышенной температуре до тех пор, пока в пуансоне совсем не оставалось ГПМЦ-АС. Регистрировали время экструзии и количества экструдата. Индекс текучести расплава рассчитывали, как (количество граммов полимера)/(время течения, равное 10 мин). В таблице 6 приведены результаты.
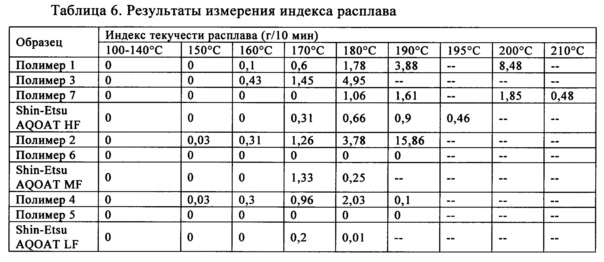
Исследование предупреждения зародышеобразования
Исследование предупреждения зародышеобразования проводили по методике перенасыщения, как это описано выше. В емкости объемом 2 унции 26,3 мг ГПМЦ-АС добавляли к 26,3 мл фосфатного буферного раствора FaSSIF, рН=6,5. Смесь или встряхивали в помещенном в водяную баню орбитальном встряхивающем устройстве при 200 встряхиваний/мин при 37°C в течение 5 ч, или выдерживали при комнатной температуре, равной примерно 25°C, в течение ночи и нагревали в течение 1 ч до 37°C перед добавлением перенасыщенного раствора нифедипина. Перенасыщенный раствор нифедипина получали путем добавления 1,5 г нифедипина (растворяемое вещество) к 30 г метанола и использования обработки ультразвуком для растворения растворяемого вещества. Затем 0,79 г перенасыщенного раствора нифедипина в течение примерно 30 с по каплям добавляли в емкость объемом 2 унции, содержащую ГПМЦ-АС и FaSSIF. Емкость объемом 2 унции непрерывно встряхивали при 200 встряхиваний/мин. В разные моменты времени отбирали образцы объемом 1 мл и их центрифугировали в мини-центрифуге (Minispin Plus®, выпускающаяся фирмой Eppendorf) при 14500 об/мин в течение 4 мин. Затем 0,1 мл надосадочной жидкости, полученной после центрифугирования, добавляли в сосуды из оранжевого стекла для ВЭЖХ и разбавляли с помощью 1 мл метанола. Концентрацию нифедипина определяли с помощью ВЭЖХ. Использовали колонку Restek Ultra Aqueous C18 при 40°C и подвижную фазу вода/ацетонитрил 70/30 в изократическом режиме при УФ-детектировании при 235 нм. Аликвоты образцов (2 мкл) инжектировали в колонку и элюировали при 0,2 мл/мин. Концентрацию нифедипина в каждом сосуде для ВЭЖХ пересчитывали в концентрацию нифедипина в соответствующей емкости объемом 2 унции с учетом коэффициента разведения и выражали в мг/мл. Экспериментальные результаты приведены в таблице 7.
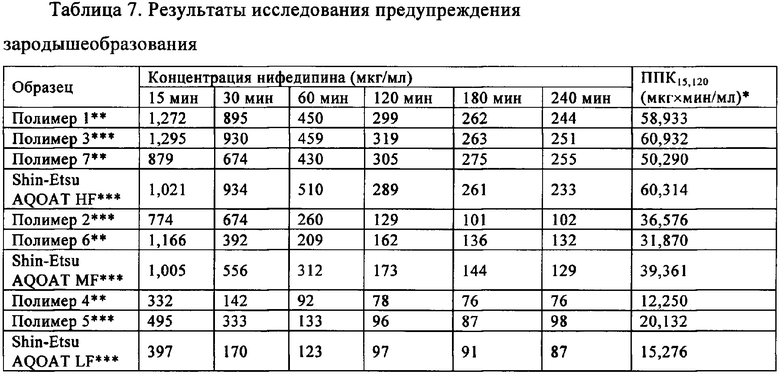
*: ППК15,120 обозначает площадь под кривой в промежутке времени от 15 до 120 мин
**: Образцы HPMCAS смешивали с FaSSIF, рН 6,5, и выдерживали при комнатной температуре в течение ночи и нагревали в течение 1 ч до 37°C перед добавлением перенасыщенного раствора нифедипина.
***: Перед проведением исследования перенасыщения смеси HPMCAS и FaSSIF, рН 6,5, встряхивали на водяной бане при 37°C в течение 5 ч.
Исследование твердости таблетки ГПМЦ-АС
Порошок ГПМЦ-АС просеивали через сито 40 меш. В случае марок ГПМЦ-АС - гранул Shin-Etsu AQOAT до просеивания полимеры размалывали с помощью CuisinArt в течение 30 с. Затем просеянные полимеры перемешивали с 1% раствором стеариновой кислоты в смесителе Turbula в течение 30 с и прессовали с помощью пресса Beta Press в плоские со скошенной кромкой (ПССК) таблетки диаметром 3/8 дюйма и массой 280 мг. Прочность таблетки при раздавливании определяли с помощью прибора для определения прочности на раздавливание, выпускающегося фирмой Key International Inc. Все исследования повторяли 5 раз.
Насыпную плотность/плотность утряски гранул измеряли с использованием мерного цилиндра объемом 10 мл. Полимер взвешивали, затем помещали в цилиндр и регистрировали объем до и после утряски. Количество встряхиваний равнялось 100. Каждый эксперимент повторяли трижды.
Размер гранул определяли с помощью лазерного дифракционного анализатора размера сухих частиц Sympatec Helo. Результаты приведены в таблице 8.
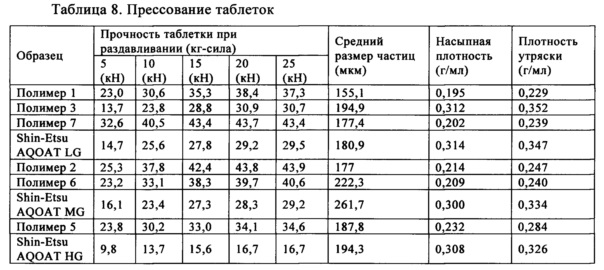
Индекс текучести расплава смеси ГПМЦ-АС и нифедипина
Индекс текучести расплава смеси ГПМЦ-АС и нифедипина измеряли с использованием Tinius Olsen Thermodyne 5208. До проведения измерения ГПМЦ-АС (примерно 75 мас. %) и нифедипин (примерно 25 мас. %) перемешивали в смесителе Turbula в течение примерно 5 мин. Примерно 5 г смеси помещали в пуансон и уплотняли, избегая попадания воздуха. В пуансон вставляли поршень. Температуру пуансона повышали до равной примерно 100°C и в течение примерно 5 мин давали установиться равновесию. К поршню прикладывали нагрузку, составляющую 5 кг, и экструдат, если его получали, собирали в течение 6 мин. С поршня снимали нагрузку, составляющую 5 кг. Затем температуру пуансона повышали на 10°C и в течение примерно 5 мин давали установиться равновесию. Такую же процедуру повторяли для сбора экструдата при повышенной температуре до тех пор, пока в пуансоне совсем не оставалось смеси. Регистрировали время экструзии и количества экструдата. Индекс текучести расплава рассчитывали, как (количество граммов полимера)/(время течения, равное 10 мин). В таблице 9 приведены результаты.
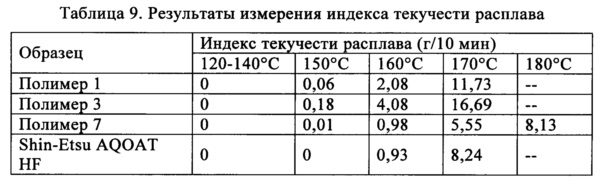
Высушенная распылительной сушкой дисперсия (РСД) ГПМЦ-АС
Растворы для распылительной сушки получали путем растворения 5% (мас./мас.) твердых веществ (ГПМЦ-АС и лекарственное средство) в смеси растворителей дихлорметан : метанол состава 2:1 (мас./мас.) (10 г твердых веществ +190 г растворителя). Отношение количества лекарственного средства к количеству ГПМЦ-АС было разным и зависело от АФИ. Для получения содержания лекарственного средства, составляющего 50%, использовали образцы, содержащие 5 г АФИ и 5 г ГПМЦ-АС. Для получения содержания лекарственного средства, составляющего 60%, использовали образцы, содержащие 6 г АФИ и 4 г ГПМЦ-АС. Распылительную сушку проводили с помощью устройства для распылительной сушки GEA SD Micro™ (выпускается фирмой GEA Process Engineering A/S, Soeborg, Denmark). Загружаемый материал распыляли с использованием двухжидкостного сопла Шлика размером 0,5 мм, при обеспечении температуры на входе, равной 85°C, скорости потока технологического газа, равной 25 кг/ч, давления распыляющего газа, равного 0,5 бар, и скорости потока распыляющего газа, равной 1,5 кг/ч. Скорость подачи жидкой загрузки регулировали для обеспечения температуры на выходе, равной 55°C. После распылительной сушки высушенные распылительной сушкой дисперсии (высушенные распылительной сушкой образцы) сушили в вакууме при 40°C при давлении, пониженном на 25 дюймов рт. ст., в течение 24 ч.
Чтобы продемонстрировать превосходную растворимость дисперсий, предлагаемых в настоящем изобретении (и предлагаемых в нем решениях), по сравнению с дисперсиями, в которых используют продукты фирмы Shin-Etsu, и с обычными препаратами, не содержащими полимеры ГПМЦ-АС, исследовали кинетику растворимости высушенных распылительной сушкой образцов. Растворение проводили с использованием μDISS Profiler™ (выпускается фирмой Pion Inc., MA, USA). Каждый высушенный распылительной сушкой порошок отвешивали таким образом, что в каждый сосуд добавляли объем, эквивалентный типичной суточной дозе лекарственного средства, и его корректировали с учетом содержания лекарственного средства в образцах. Например, в случае фелодипина целевая масса АФИ равнялась 9,5 мг в 20 мл FaSSIF. Отвешивали высушенный распылительной сушкой образец, содержащий 9,5 мг АФИ-фелодипина. В случае эзетимиба отвешивали высушенный распылительной сушкой образец, содержащий 10 мг АФИ. В сосудах к 20 мл FaSSIF добавляли отвешенные высушенные распылительной сушкой образцы и затем нагревали при 37°C. Сосуды выдерживали при постоянной скорости перемешивания, равной 300 об/мин. Исследование растворения проводили in situ с помощью волоконно-оптических датчиков в разные моменты времени и эти результаты анализировали при соответствующей длине волны и получали концентрацию растворенного АФИ. На основании полученных in situ результатов измерений рассчитывали разные параметры растворимости
Результаты исследования солюбилизации высушенной распылительной сушкой дисперсии, содержащей фелодипин при содержании лекарственного средства, составляющем 50%, приведены в таблице 10. Результаты исследования солюбилизации высушенной распылительной сушкой дисперсии, содержащей эзетимиб при содержании лекарственного средства, составляющем 60 мас. %, приведены в таблице 11.
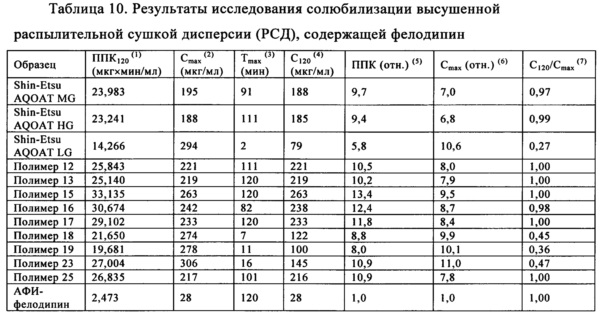
(1) ППК120 = площадь под кривой (ППК) для концентрации АФИ в течение первых 120 мин исследования
(2) Cmax = максимальная концентрация АФИ в растворе в течение первых 120 мин
(3) Tmax = время, при котором обеспечивалась Cmax
(4) С120 = концентрация АФИ при времени =120 мин
(5) ППК (отн.) = отношение ППК120 дисперсии полимер/лекарственное средство к ППК120 только АФИ
(6) Cmax (отн.) = отношение Cmax дисперсии полимер/лекарственное средство к Cmax только АФИ
(7) C120/Cmax = отношение С120 к Cmax для дисперсии полимер/лекарственное средство или только АФИ
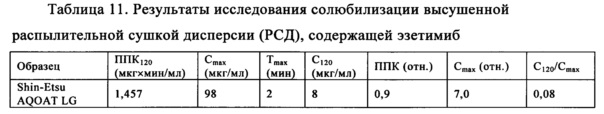
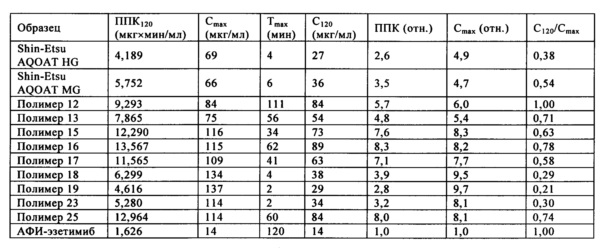
Динамические реологические исследования ГПМЦ-АС
Динамическое реологическое исследования характеристик полимеров ГПМЦ-АС проводили с использованием реометра AR G2, снабженного системой регулирования температуры - камерой для испытаний на воздействие окружающей среды (КИОС) (выпускается фирмой ТА Instruments, New Castle, DE, USA). Для испытания использовали изготовленные из нержавеющей стали параллельные пластины диаметром 25 мм. Проводили эксперименты двух типов - испытание при постоянной температуре с разверткой по частоте и динамическое испытание с линейным изменением температуры. Исследования проводили в атмосфере азота.
Эксперимент при постоянной температуре с разверткой по частоте проводили при 170°C. Образец порошка ГПМЦ-АС помещали на пластину для испытаний с помощью держателя для загрузки образца. Образец приводили в равновесие при 170°C в течение примерно 2 мин, затем пластины для испытаний устанавливали так, что между ними оставался зазор (т.е. 1 мм). Эксперимент при постоянной температуре с разверткой по частоте проводили со сбором данных при 5 частотах в каждой декаде в диапазоне от 0,1 до 600 рад/с.
Динамическое испытание с линейным изменением температуры проводили при температурах, находящихся в диапазоне от 150 до 200°C. Образец порошка ГПМЦ-АС помещали на пластину для испытаний при 170°C с помощью держателя для загрузки образца. Образец приводили в равновесие при 170°C в течение примерно 2 мин, затем пластины для испытаний устанавливали так, что между ними оставался зазор (т.е. 1 мм). После загрузки температуру понижали до 150°C при обеспечении постоянного осевого усилия, равного 0+/-0,2 Н. Затем образец приводили в равновесие при 150°C в течение 5 мин, затем проводили испытание. Измерения проводили в диапазоне линейного изменения температуры, составляющего от 150 до 200°C, при скорости нагрева, равной 2°C/мин. Испытания проводили при частоте, установленной равной 6,28 рад/с (т.е. 1 Гц) и деформации, находящейся в диапазоне линейной вязкоупругости для каждого образца.
Результаты исследования реологических характеристик представлены в виде разных параметров, которые можно легко преобразовать друг в друга. С использованием исходных данных зависимости динамического модуля упругости G’(ω) и модуля потерь G"(ω) от угловой частоты (ω) применяют следующие уравнения (J.D. Ferry, "Viscoelastic Properties of Polymers", John Wiley & Sons, (1980) 3rd Edition):
Динамические модули: G*(ω)=(G'(ω)2+G"(ω)2)0,5
Тангенс угла сдвига фаз: δ=G"(ω)/G'(ω)
Динамическая вязкость: Eta*(ω)=G*(ω)/ω
На фиг. 1, 3 и 5 приведены результаты испытаний с разверткой по частоте для образцов ГПМЦ-АС марок Н, М и L, полученных по различным методикам синтеза, в сравнении с результатами для продуктов фирмы Shin-Etsu марок Н, М и L соответственно. На фиг. 2, 4 и 6 приведены результаты испытаний с линейным изменением температуры для этих образцов в сравнении с результатами для соответствующих продуктов фирмы Shin-Etsu соответственно. В случае образцов марки Н вязкость расплава полимера 3 является более низкой, тогда как вязкость расплава полимера 7 является более высокой, чем вязкости расплавов продуктов фирмы Shin-Etsu. Вязкости расплавов полимера 1 и продукта фирмы Shin-Etsu являются сходными. В случае образцов марки М вязкость расплава полимера 2 является более низкой, тогда как вязкость расплава полимера 6 является более высокой, чем вязкости расплавов продуктов фирмы Shin-Etsu. Вязкости расплавов полимера 9 и продукта фирмы Shin-Etsu являются сходными. В случае образцов марки L вязкость расплава полимера 4 является более низкой, тогда как вязкости расплавов полимеров 5 и 8 являются более высокими, чем вязкости расплавов продуктов фирмы Shin-Etsu.
На фиг. 7, 9 и 11 приведены результаты испытаний с разверткой по частоте для образцов ГПМЦ-АС марки Н, обладающих высокой молекулярной массой и низкой молекулярной массой, и для образцов ГПМЦ-АС марки L в сравнении с результатами для соответствующих продуктов фирмы Shin-Etsu марок Н и L соответственно. На фиг. 8, 10 и 12 приведены результаты испытаний с линейным изменением температуры для этих образцов в сравнении с результатами для соответствующих продуктов фирмы Shin-Etsu соответственно. В случае образцов марки Н, обладающих высокой молекулярной массой, вязкость расплава продукта фирмы Shin-Etsu является немного более высокой, чем вязкости расплавов полимера 13 и полимера 16. В случае образцов марки Н, обладающих низкой молекулярной массой, вязкость расплава продукта фирмы Shin-Etsu является более высокой, чем вязкости расплавов полимера 12 и полимера 17. В случае образцов марки L вязкость расплава продукта фирмы Shin-Etsu является более высокой, чем вязкость расплава полимера 19, но сходна с вязкостью расплава полимера 18.
На фиг. 13-16 приведены зависимости динамического модуля упругости G' и модуля потерь G" от температуры для полимера 1, полимера 3, полимера 7 и Shin-Etsu AQOAT HF соответственно. Для полимера 1 и полимера 3 наблюдается температура точки пересечения, при которой G' равен G". Температура точки пересечения для полимера 1 выше температуры точки пересечения для полимера 3. Для полимера 7 и продукта фирмы Shin-Etsu не наблюдается точка пересечения, хотя в случае продукта фирмы Shin-Etsu при увеличении температуры G' приближается к G".
Также установлено, что при включении ацетильных и сукциноильных групп в полимер ГПМЦ происходит разное увеличение среднемассовой молекулярной массы полимеров ГПМЦ-АС, полученных по разным методикам синтеза. Обычно выраженное в процентах увеличение среднемассовой молекулярной массы ГПМЦ-АС, полученного по методике А, составляет более 60%. Выраженное в процентах увеличение среднемассовой молекулярной массы ГПМЦ-АС, полученного по методике В, составляет менее 60%, тогда как выраженное в процентах увеличение среднемассовой молекулярной массы ГПМЦ-АС, полученного по методике С, составляет более 100%.
Разумеется, невозможно описать все приемлемые комбинации компонентов или методологий для изложения раскрытой информации, но специалист с общей подготовкой в данной области техники может понять, что возможны многочисленные другие комбинации и перестановки раскрытой информации. Поэтому подразумевается, что раскрытая информаций включает все такие изменения, модификации и варианты, которые входят в сущность и объем прилагаемой формулы изобретения.
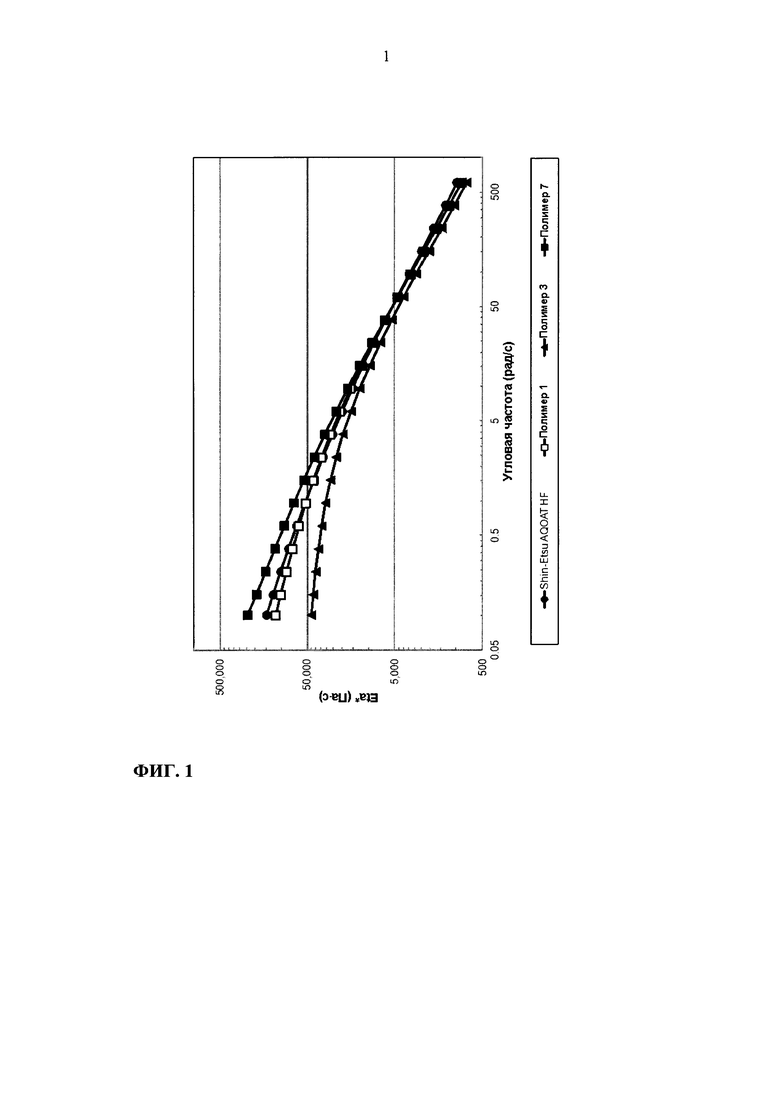

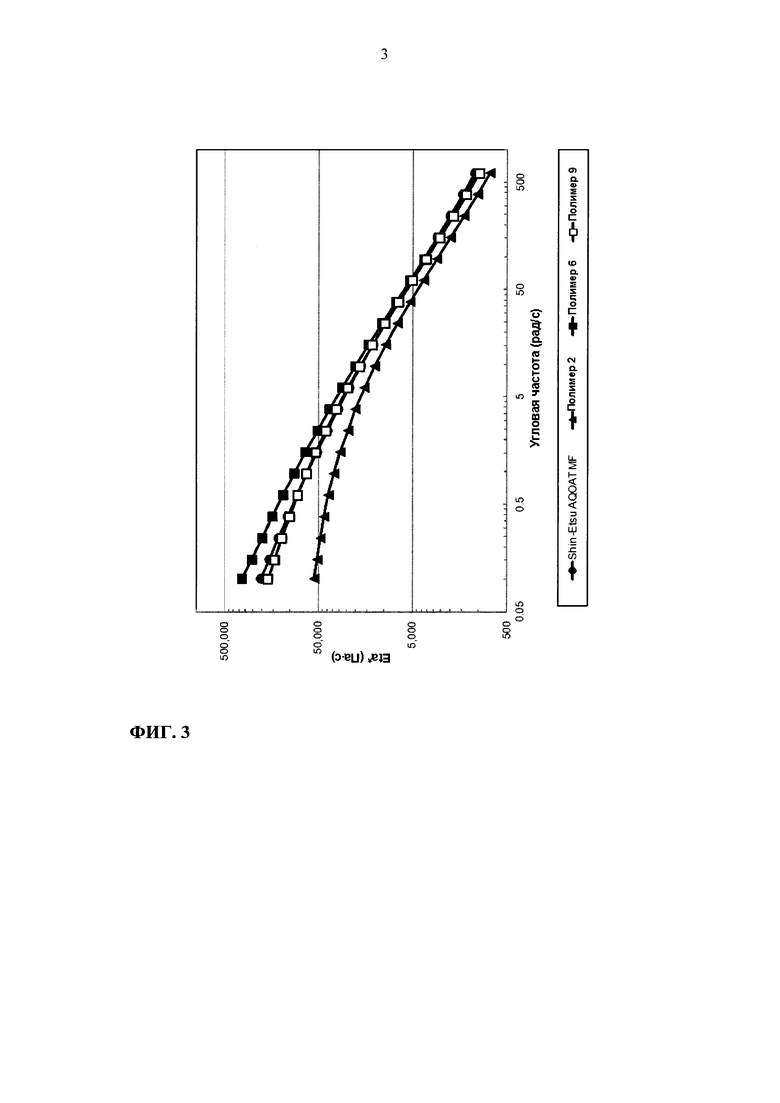
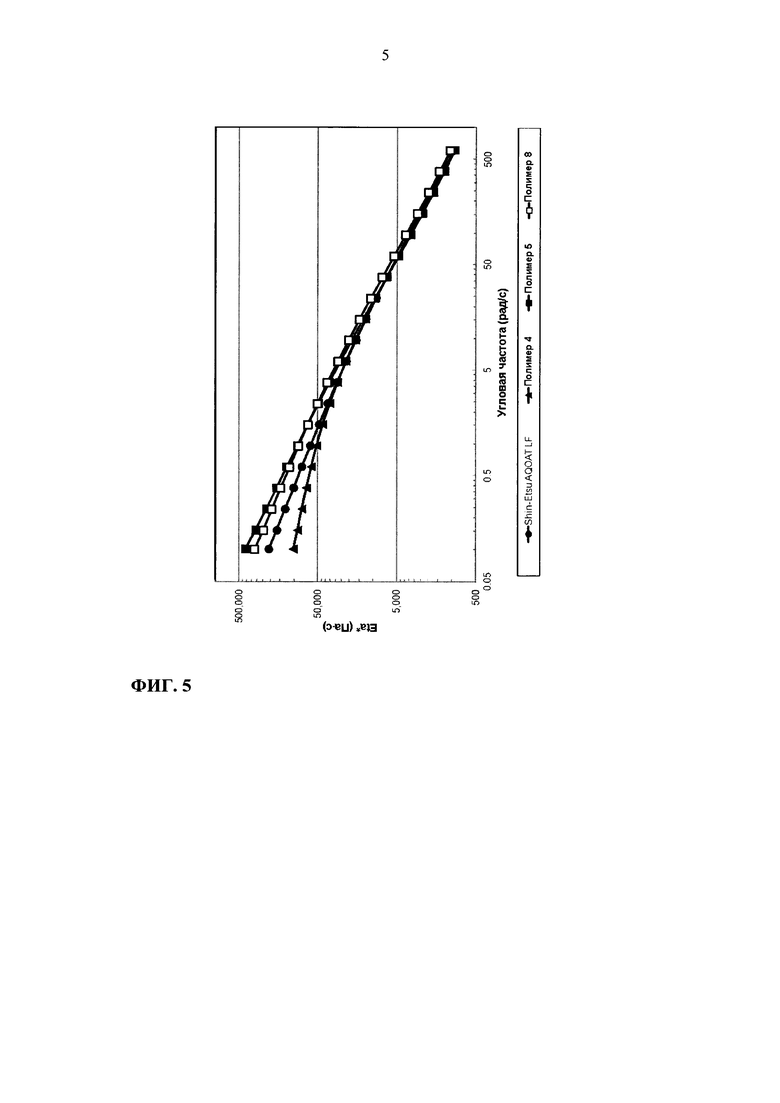
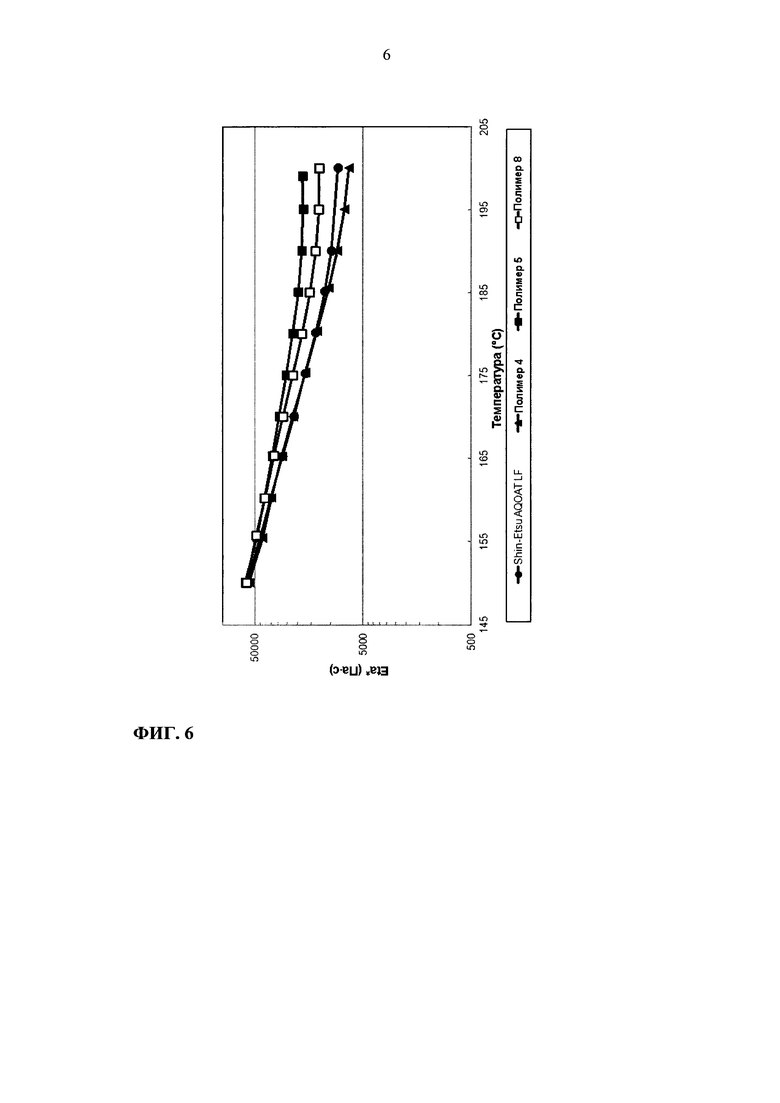
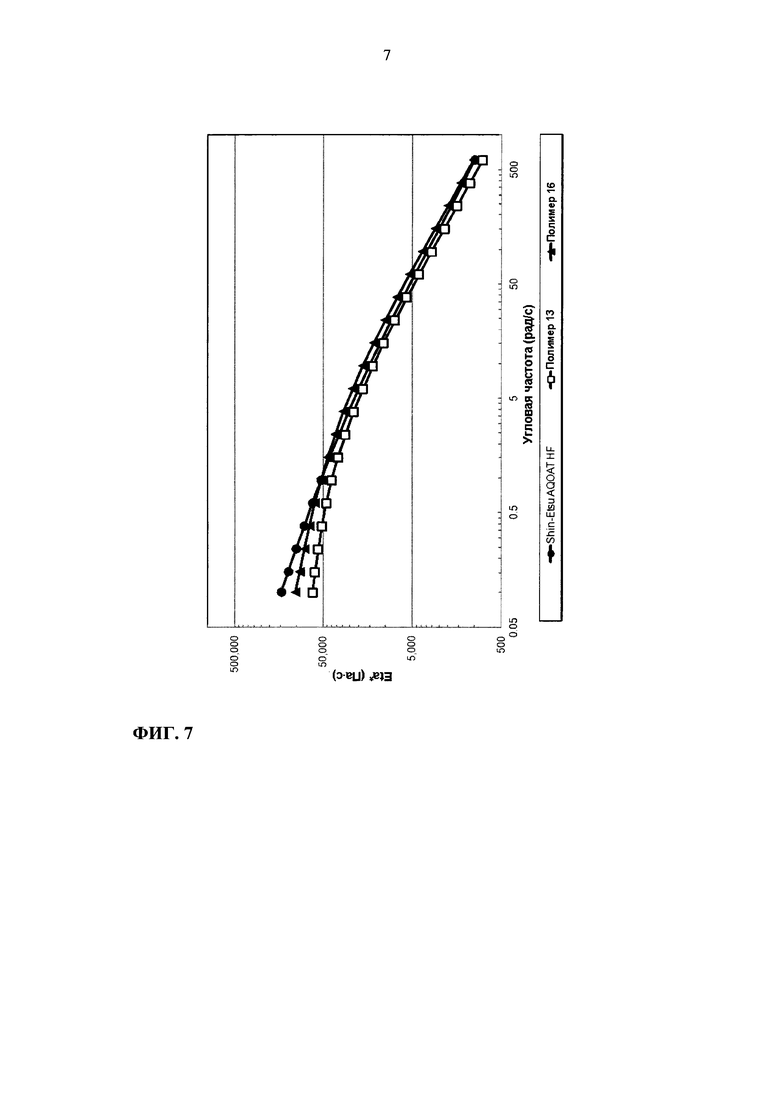
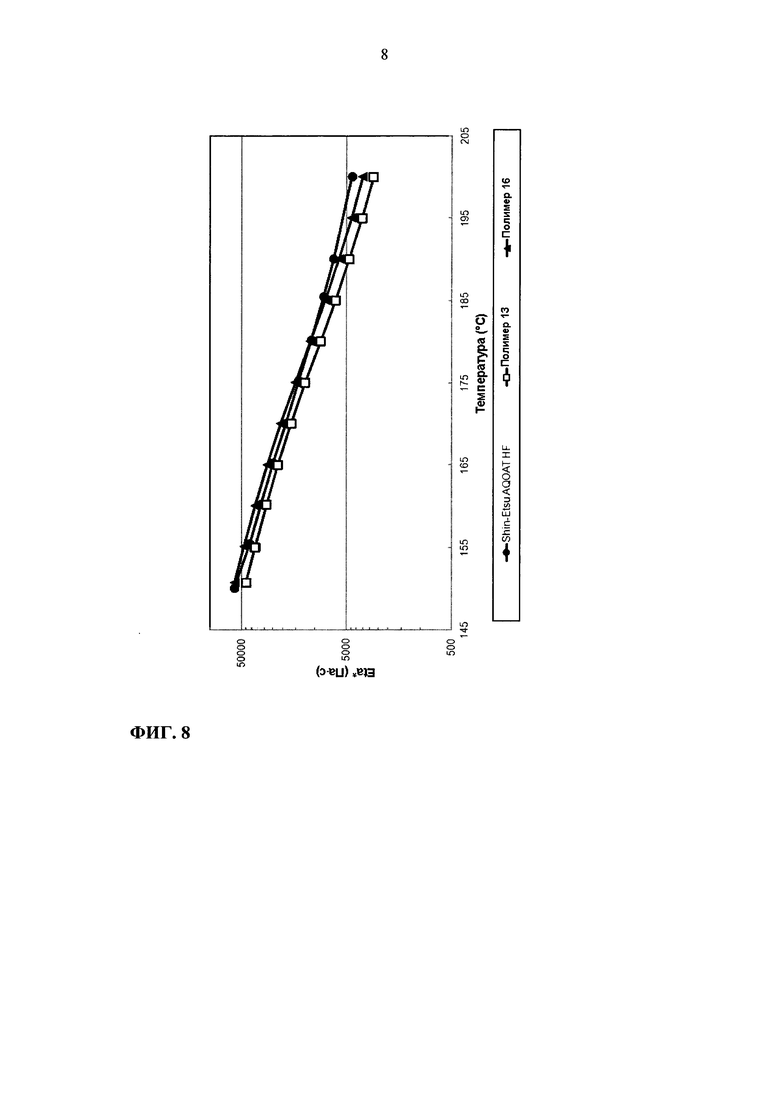
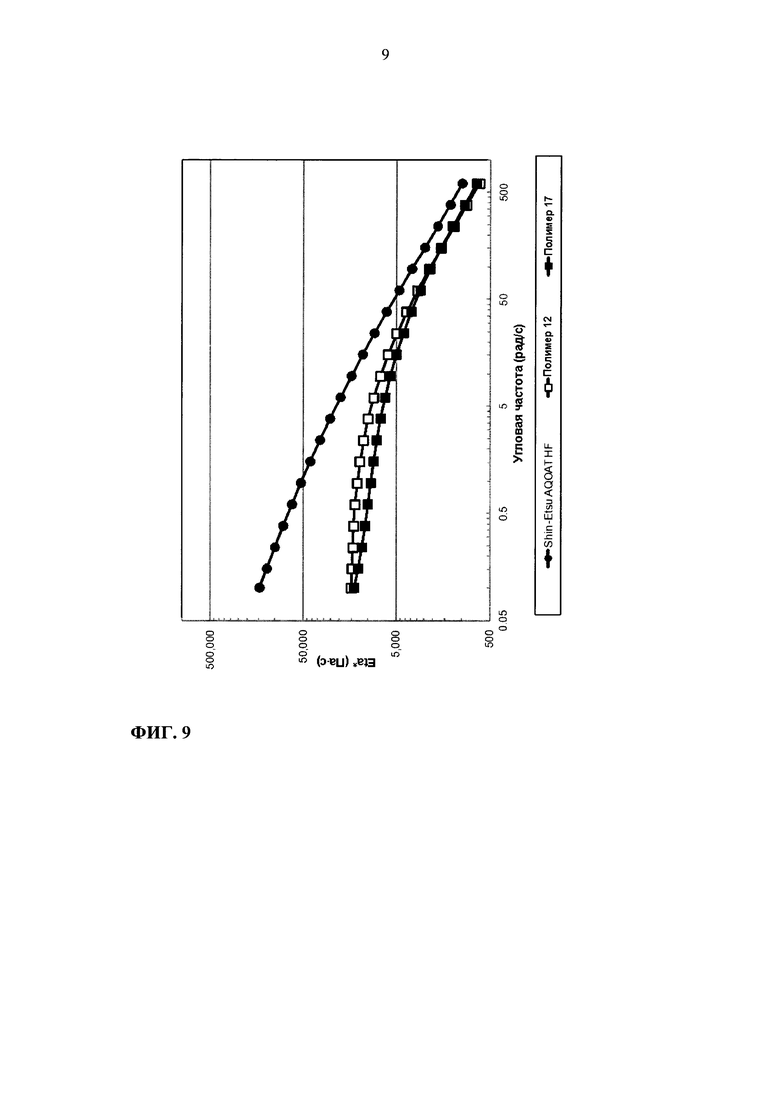
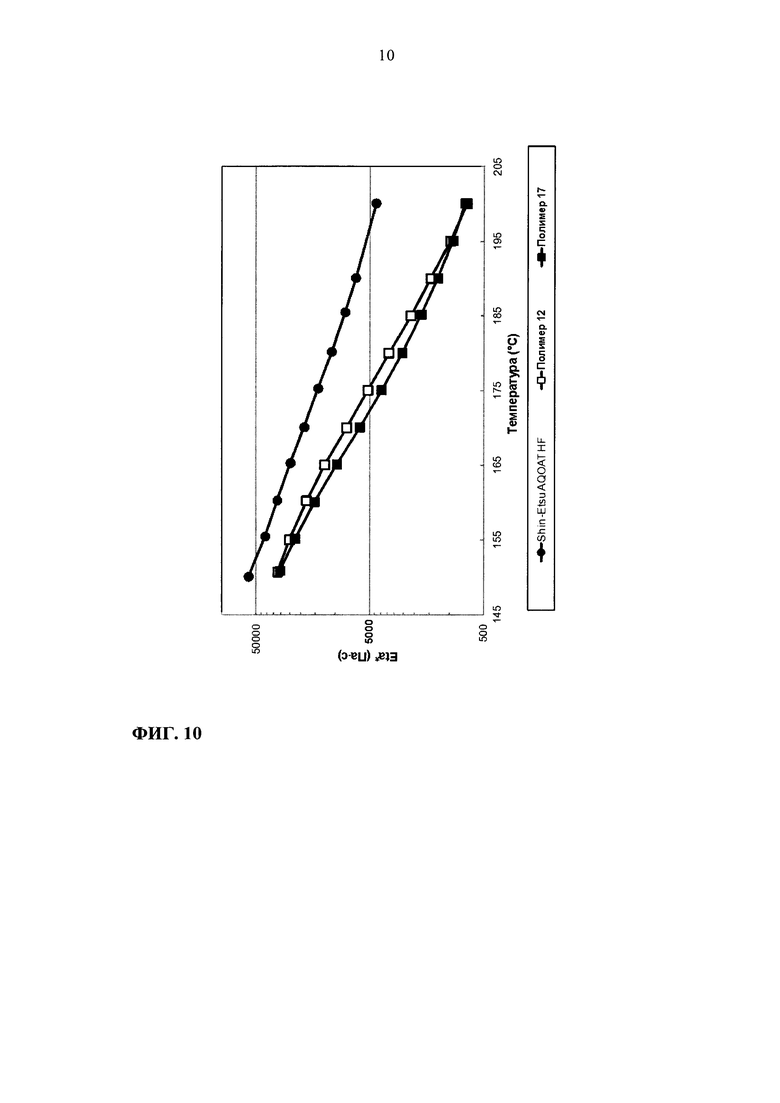
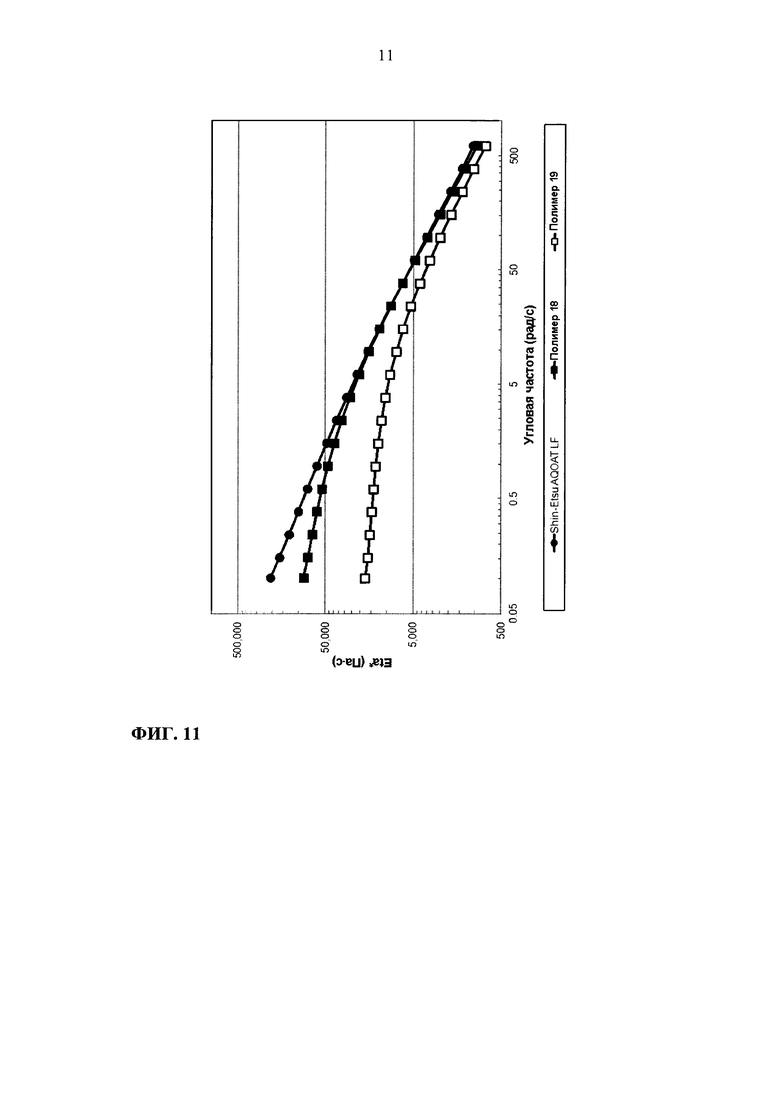
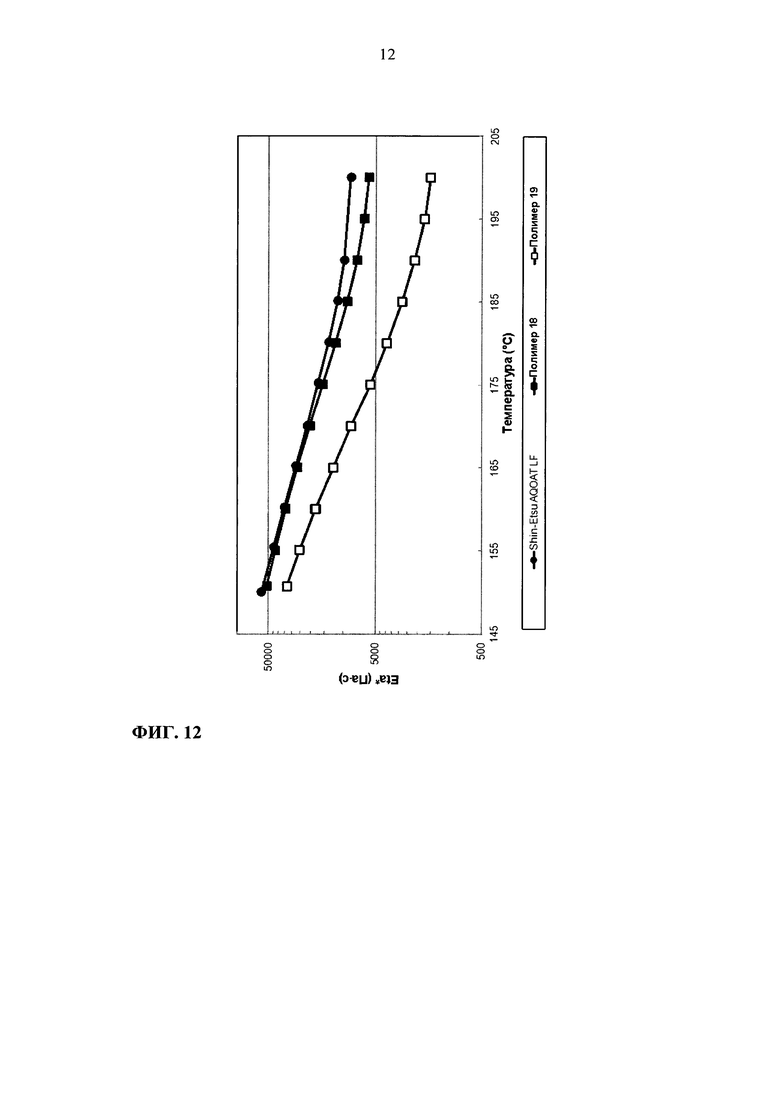
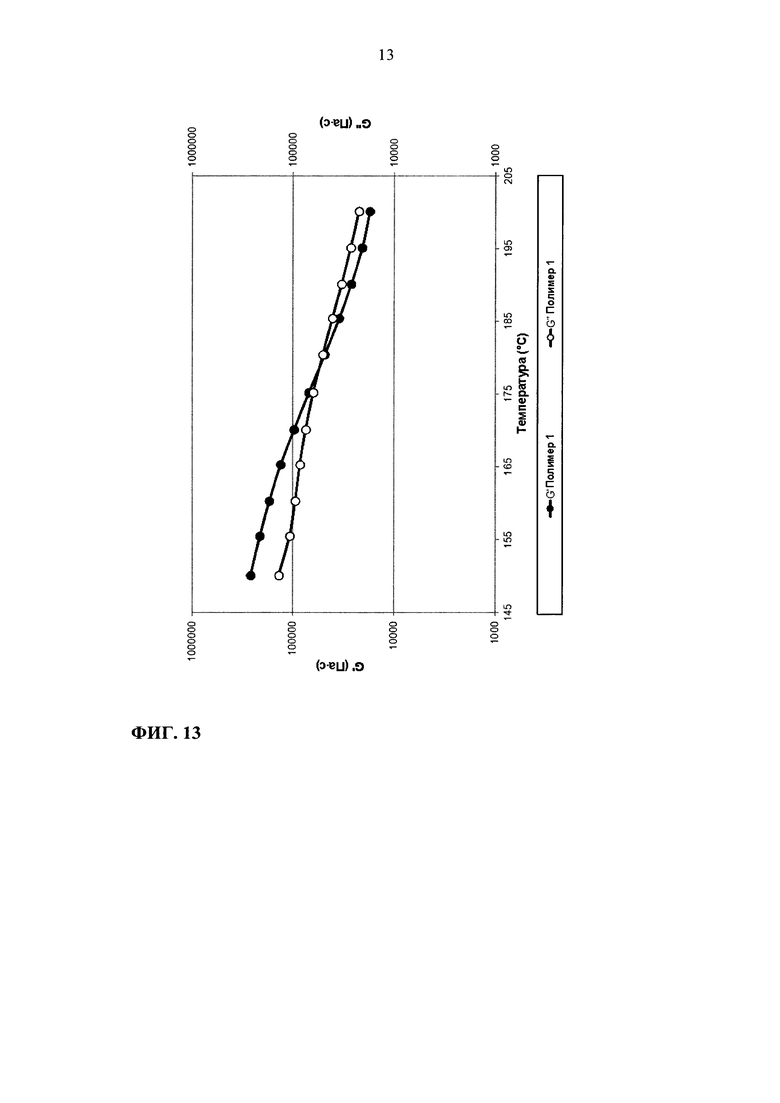
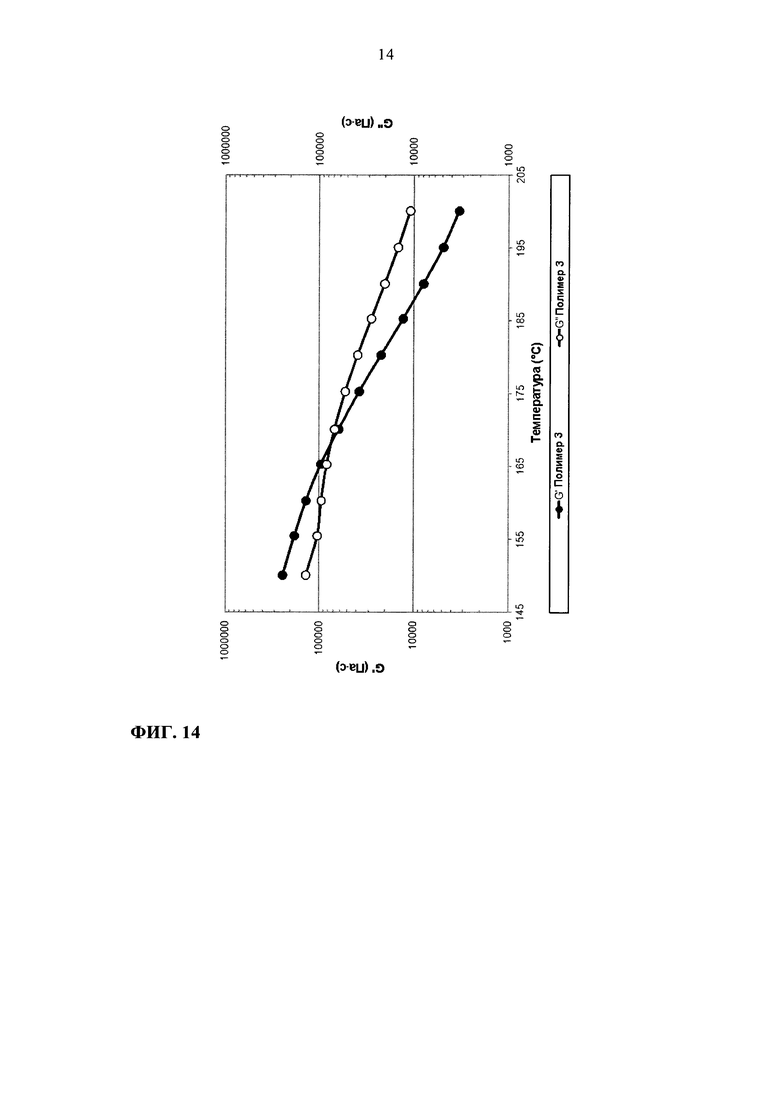
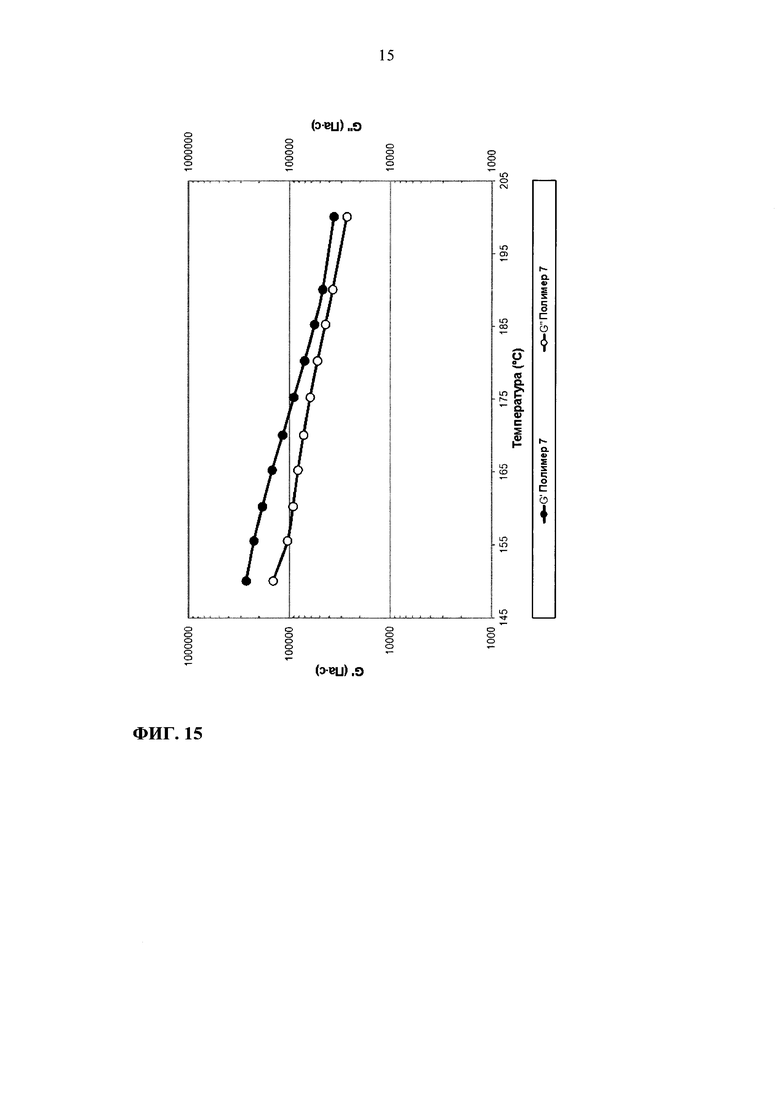
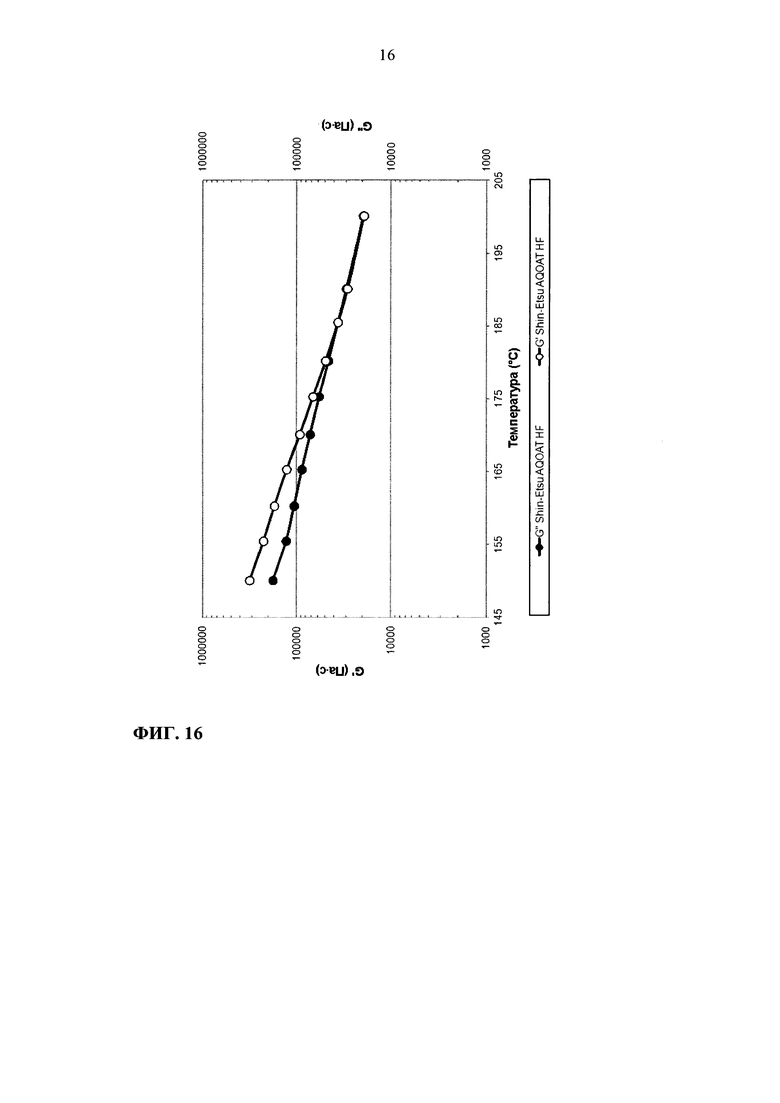
Изобретение относится к полимеру, предназначенному для повышения эффективности лекарственного средства и улучшения обрабатываемости, включающему ацетат-сукцинат гидроксипропилметилцеллюлозы (ГПМЦ-АС), в котором выраженная в процентах полная степень замещения (СЗ) сукциноилом в положении C6-ОН меньше или равна 6% (С6 СЗSuc, % ≤ 6%) и в положении С3-ОН составляет от 58 до 84% (58% < С3 СЗSuc, % < 84%), и выраженная в процентах полная СЗ ацетилом в положении C6-ОН составляет от 33 до 51% (33% < С6 СЗAc, % < 51%) и в положении С3-ОН составляет от 16 до 20% (16% < С3 СЗАс, % < 20%). Изобретение относится к композиции, предназначенной для повышения эффективности и улучшения обрабатываемости лекарственного средства, которая содержит лекарственное средство и укахзанный полимер, а также способу получения ацетат-сукцината гидроксипропилметилцеллюлозы (ГПМЦ-АС), обладающего выраженной в процентах полной степенью замещения (СЗ) сукциноилом, в положении C6-ОН меньше или равной 6% (С6 СЗSuc, % ≤ 6%) и в положении С3-ОН составляющей от 58 до 84% (58% < С3 СЗSuc, % < 84%), и выраженной в процентах полной СЗ ацетилом, в положении C6-ОН составляющей от 33 до 51% (33% < С6 СЗAc, % < 51%) и в положении С3-ОН составляющей от 16 до 20% (16% < С3 СЗАс, % < 20%), включающему стадии (а) введения в реакцию уксусного ангидрида и ацетата натрия с гидроксипропилметилцеллюлозой при температуре, находящейся в диапазоне от примерно 85 до примерно 115°C, с получением промежуточного продукта; (b) поддерживания температуры в течение периода времени от 30 мин до 2,5 ч и (с) введения в реакцию янтарного ангидрида с промежуточным продуктом при температуре в течение периода времени от 2,5 до 23,5 ч с получением ГПМЦ-АС. 3 н. и 7 з.п. ф-лы, 16 ил., 11 табл.
1. Полимер, предназначенный для повышения эффективности лекарственного средства и улучшения обрабатываемости, включающий ацетат-сукцинат гидроксипропилметилцеллюлозы (ГПМЦ-АС), в котором выраженная в процентах полная степень замещения (СЗ) сукциноилом в положении C6-ОН меньше или равна 6% (С6 СЗSuc, % ≤ 6%) и в положении С3-ОН составляет от 58 до 84% (58% < С3 СЗSuc, % < 84%), и выраженная в процентах полная СЗ ацетилом в положении C6-ОН составляет от 33 до 51% (33% < С6 СЗAc, % < 51%) и в положении С3-ОН составляет от 16 до 20% (16% < С3 СЗАс, % < 20%).
2. Композиция, предназначенная для повышения эффективности и улучшения обрабатываемости лекарственного средства, содержащая лекарственное средство и полимер, где полимером является ацетат-сукцинат гидроксипропилметилцеллюлозы (ГПМЦ-АС), в котором выраженная в процентах полная степень замещения (СЗ) сукциноилом в положении C6-ОН меньше или равна 6% (С6 С3Suc, % < 6%) и в положении С3-ОН составляет от 58 до 84% (58% < С3 С3Suc, % < 84%), и выраженная в процентах полная СЗ ацетилом в положении C6-ОН составляет от 33 до 51% (33% < С6 СЗАс, % < 51%) и в положении С3-ОН составляет от 16 до 20% (16% < С3 СЗАс, % < 20%).
3. Композиция по п. 2, в которой лекарственное средство является обладающим низкой растворимостью лекарственным средством.
4. Композиция по п. 3, где композиция представляет собой твердую аморфную дисперсию, содержащую обладающее низкой растворимостью лекарственное средство и полимер.
5. Композиция по п. 3, где композиция представляет собой физическую смесь обладающего низкой растворимостью лекарственного средства и полимера.
6. Способ получения ацетат-сукцината гидроксипропилметилцеллюлозы (ГПМЦ-АС), обладающего выраженной в процентах полной степенью замещения (СЗ) сукциноилом, в положении C6-ОН меньше или равной 6% (С6 СЗSuc, % < 6%) и в положении С3-ОН составляющей от 58 до 84% (58% < С3 С3Suc, % < 84%), и выраженной в процентах полной СЗ ацетилом в, положении C6-ОН составляющей от 33 до 51% (33% < С6 СЗAc, % < 51%) и в положении С3-ОН составляющей от 16 до 20% (16% < С3 СЗAc, % < 20%), включающий стадии:
(а) введения в реакцию уксусного ангидрида и ацетата натрия с гидроксипропилметилцеллюлозой при температуре, находящейся в диапазоне от примерно 85 до примерно 115°C, с получением промежуточного продукта;
(b) поддерживания температуры в течение периода времени от 30 мин до 2,5 ч; и
(с) введения в реакцию янтарного ангидрида с промежуточным продуктом при температуре в течение периода времени от 2,5 до 23,5 ч с получением ГПМЦ-АС.
7. Способ по п. 6, в котором на стадии (с) время меняют от 2,5 до 15,5 ч.
8. Способ по п. 7, в котором на стадии (с) время меняют от 2,5 до 5,5 ч.
9. Способ по п. 6, в котором температура находится в диапазоне от 95 до 115°C.
10. Способ по п. 6, в котором температура находится в диапазоне от 95 до 110°C.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| ВРАЩАЮЩАЯСЯ ПЕЧЬ | 0 |
|
SU219426A1 |
| US 5776501A, 07.07.1998 | |||
| СТАБИЛЬНЫЕ ПЕРОРАЛЬНЫЕ СУСПЕНЗИИ НЕДИГИДРАТНОГО АЗИТРОМИЦИНА | 2004 |
|
RU2325149C2 |

