Область техники, к которой относится изобретение
Настоящее изобретение касается нового соединения, которое модулирует активность RORγ, фармацевтической композиции и применения в лечении или профилактике аутоиммунных заболеваний, воспалительных заболеваний, заболеваний обмена веществ или раковых заболеваний.
Предшествующий уровень техники
Ретиноевый орфан-рецептор гамма (RORγ) представляет собой ядерный рецептор, который связывается с ДНК и регулирует транскрипцию (НИ 1). Две изоформы RORγ, которые отличаются только по N-концу, генерируются из RORC гена; RORγ1 и RORγt (также обозначается RORγ2) (НИ 2). RORγ применяется как термин для обозначения обеих изоформ RORγ1 и RORγt.
RORγ1 экспрессируется в различных тканях, включая мышцы, почки, печень и легкие, и регулирует липогенез (НИ 3). Потеря гена RORC у мышей ускоряет дифференциацию преадипоцитов в маленькие адипопиты и защищает от инсулиновой резистентности, вызванной рационом с высоким содержанием жира. Следовательно, путем подавления функции RORγ1 можно бороться с инсулиновой резистентностью.
RORγt экспрессируется исключительно в клетках иммунной системы (НИ 4 и 5) и представляет собой главный регулятор Th17-клеточной транскрипционной сети, связанной с аутоиммунной патологией. Th17 клетки являются субпопуляцией CD4+ Т-хелперов, считающихся ключевыми драйверами воспалительного процесса при аутоиммунитете и характеризующихся выработкой превоспалительного цитокина IL-17A. Th17 клетки также экспрессируют CCR6, который участвует в миграции к сайтам воспаления, поддерживаются и расширяются под действием IL-23, при участии IL-23 рецептора (IL23R), и экспрессируют другие провоспалительные цитокины и хемокины, включая IL-17F, IL-21, IL-22, CCL20 и GM-CSF, которые совместно обеспечивают доставку других типов воспалительных клеток, особенно нейтрофилов, обуславливая патологию в поражаемой ткани. RORγt необходим для дифференциации Th17 клеток, и напрямую и опосредованно регулирует экспрессирование многих из этих провоспалительных медиаторов (НИ 6). RORγ-дефицитные мыши имеют значительно сниженное количество Th17 клеток in vivo, неспособны вырабатывать IL-17A и другие Th17-связанные цитокины ех vivo, и демонстрируют устойчивость к индуцированию различных моделей заболеваний, таких как ЕАЕ (экспериментальный аутоиммунный энцефаломиелит), дерматит, энтерит и нефрит (НИ 6, и 12-14). Таким образом, путем подавления функции RORγ можно подавлять развитие различных аутоиммунных заболеваний и воспалительных заболеваний, в которых участвуют Th17-клеточные цитокины. Кроме того, экспрессирование RORγt и, как следствие, экспрессирование Th17-клеточной транскрипционной сети наблюдалось в других типах иммунных клеток, что может также иметь значение в патогенезе заболеваний, а именно в CD8+ Т клетках, так называемых Tc17s, γδ Т клетках, естественных Т-клетках киллерах, генетически детерминированных лимфоцитах, естественных клетках-киллерах и тучных клетках (НИ 7 и 8).
Th17-клеточные цитокины и хемокины задействованы в патогенезе различных человеческих аутоиммунных и воспалительных заболеваний, включая множественный склероз, ревматоидный артрит, псориаз, псориатический артрит, анкилозирующий спондилит, кистозный фиброз, астму, хроническое обструктивное заболевание легких, эмфизему, фиброз легких, системный эритоматоз, васкулит, гранулематоз Вегенера, ревматическую полимиалгию, гигантоклеточный артериит, артериосклероз, аутоиммунный миозит, увеит, сухость глаз, воспалительную болезнь кишечника, вызванный алкоголем гепатит, неалкогольный стеатогепатит, первичный билиарный склероз, вирусный гепатит и диабет 1 типа (НИ 9-11).
Известно, что RORγt оказывает ингибирующее действие на анти-онкогенную активность Th9 клеток, подтипа Т-хелперов (НИ 15). У RORγ-дефицитных мышей усилена выработка IL-9 в Th9 клетках, и образование опухоли замедляется у мышей, которым инъецировали клетки меланомы. Поэтому полагают, что при подавлении функции RORγ можно активировать работу Th9 клеток и подавлять образование меланомы и других злокачественных опухолей.
Из изложенного выше можно ожидать, что модулятор RORγ будет обеспечивать терапевтические или профилактические преимущества при лечении заболеваний обмена веществ, таких как диабет; аутоиммунных заболеваний или воспалительных заболеваний, и меланомы и других раковых заболеваний.
Список процитированной литературы
Непатентные источники (НИ)
НИ 1: Gigure, Endocrine. Reviews. 20: 689-725, 1999
НИ 2: Jetten, Nucl. Recept. Signal. 7: е003, 2009
НИ 3: Meissburger et al., EMBO Mol. Med. 3: 637-651, 2011
НИ 4: Hirose et al., Biochem. Biophys. Res. Commun. 30: 1976-1983, 1994
НИ 5: Eberl and Littman., Science. 9: 248-251, 2004
НИ 6: Ivanov et al., Cell 126: 1121-1133, 2006
НИ 7: Sutton et al., Eur. J. Imnnmol. 42: 2221-2231, 2012
НИ 8: Hueber et al., J. Immunol., 184: 3336-3340, 2010
НИ 9: Miossec et al., Nature Reviews Drug Discovery 11: 763-776, 2012
НИ 10 Hammerich et al., Clin. Dev. Immunol. 2011: Article ID 345803, 2011
НИ 11 Ferraro et al., Diabetes 60: 2903-2913, 2011
НИ 12 Pantelyushin et al., J Clin Invest. 122: 2252-2256, 2012
НИ 13 Buonocore et al.. Nature 464: 1371-1375, 2010
НИ 14 Steinmetz et al., J. Am. Soc. Nephrol. 22: 472-483, 2011
НИ 15 Purwar et al., Nat. Med. 18: 1248-1254, 2012
Краткое описание изобретения
Техническая проблема
Целью настоящего изобретение является получение соединения, обладающего функцией подавления активности RORγ.
Решение проблемы
Авторы настоящего изобретения провели различные исследования с целью достичь указанной выше цели и, в результате, нашли новое соединение, имеющее формулу (I), или его фармацевтически приемлемую соль, соединение или его фармацевтически приемлемую соль, обладающие функцией подавления активности RORγ. Таким образом, настоящее изобретение заключается в следующем.
(1) Соединение, имеющее формулу (I), или его фармацевтически приемлемая соль:
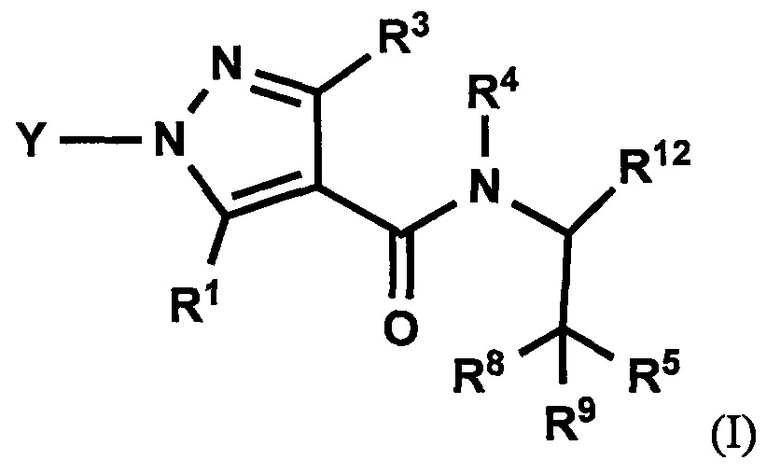
где:
R1 выбран из F, Cl, Br, C1-С6 алкильной группы, замещенной 0, 1, 2 или 3 Ra группами, и С3-C8 циклоалкильной группы, замещенной 0, 1, 2 или 3 Ra группами;
Y выбран из С4-С6 циклоалкильной группы, С6-С9 бициклоалкильной группы и С6-С9 спироалкильной группы, которые все замещены R2 группой, 0 или 1 R6 группой, и 0, 1, 2 или 3 R7 группами;
R2 выбран из -ОН, -CO2H, -SO3H, -CONH2, -SO2NH2, (C1-С6 алкокси)карбонильной группы, замещенной 0, 1, 2 или 3 Rc группами, (C1-С6 алкил)аминокарбонильной группы, замещенной 0, 1, 2 или 3 Rc группами, C1-С6 алкилсульфонильной группы, замещенной 0, 1, 2 или 3 Rc группами, C1-С6 алкиламиносульфонильной группы, замещенной 0, 1, 2 или 3 Rc группами, (гидроксикарбонил)(С1-С3 алкильной) группы, замещенной 0, 1, 2 или 3 Rc группами, (C1-С6 алкокси)карбонил(С1-С3 алкильной) группы, замещенной 0, 1, 2 или 3 Rc группами, (C1-С6 алкил)сульфонил(С1-С3 алкильной) группы, замещенной 0, 1, 2 или 3 Rc группами, и (С2-С6 алкенил)(С1-С3 алкильной) группы, замещенной 0, 1, 2 или 3 Rc группами;
R6 и R7 независимо выбраны из Н, F, -ОН, -NH2, -CN, C1-С6 алкильной группы, замещенной 0, 1, 2 или 3 Rb группами, и C1-С6 алкокси-группы, замещенной 0, 1, 2 или 3 Rb группами;
R3 выбран из Н, F, Cl, -CH3 и -CF3;
R4 выбран из C1-С6 алкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (С2-С6 алкенил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (С2-С6 алкинил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (C1-С6 алкокси)(С2-С4 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (С6-С10 арил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, (5-10-членный гетероарил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, С3-C8 циклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С3-C8 циклоалкенильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С3-C8 циклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С3-C8 циклоалкенил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, 3-8-членной гетероциклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, и (3-8-членный гетероциклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 спироалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С6-С9 спироалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 спирогетероалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, C5-C9 бициклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С5-С9 бициклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 гетеробициклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, и (С6-С9 гетеробициклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами;
R5 выбран из С6-С10 арильной группы, замещенной 0, 1, 2, 3, 4 или 5 Ri группами, 5-10-членной гетероарильной группы, замещенной 0, 1, 2, 3, или 4 Ri группами, С3-C8 циклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rj группами, С3-C8 циклоалкенильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rj группами, и 3-8-членной гетероциклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rj группами;
R8 и R9 независимо выбраны из Н, F, -ОН, -NH2, C1-С3 алкильной группы, замещенной 0, 1, 2 или 3 Rh группами, и C1-С6 алкокси-группы, замещенной 0, 1, 2 или 3 Rh группами;
или R8 и R9 вместе образуют оксо-группу или тиоксо-группу;
R12 представляет собой Н; или R4 и R12 вместе представляют собой -CRmRm-CR13R14-CRmRm- или -CR13R14-CRmRm-CRmRm-, формируя пирролидиновый цикл;
R13 выбран из Н, C1-С6 алкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, С6-С10 арильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, С6-С10 арилокси-группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, (С2-С6 алкенил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (С2-С6 алкинил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (C1-С6 алкокси)(С2-C4 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (С6-С10 арил)(C1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, (5-10-членный гетероарил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, С3-С8 циклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С3-C8 циклоалкенильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С3-C8 циклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С3-C8 циклоалкенил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, 3-8-членной гетероциклоалкильной группы, замещенной 0, 1, 2, 3 4 или 5 Rg группами, и (3-8-членный гетероциклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 спироалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С6-С9 спироалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 спирогетероалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 бициклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (C5-C9 бициклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 гетеробициклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, и (С6-С9 гетеробициклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами;
R14 независимо выбран из Н и C1-С6 алкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами; или R13 и R14 вместе формируют С3-C8 циклоалкановое кольцо, замещенное 0, 1, 2, 3, 4 или 5 Rg группами, С3-C8 циклоалкеновое кольцо, замещенное 0, 1, 2, 3, 4 или 5 Rg группами, или 3-8-членное гетероциклоалкановое кольцо, замещенное 0, 1, 2, 3, 4 или 5 Rg группами;
Rm независимо выбран из Н, F, Cl, -CH3 и -CF3;
Rg и Rj независимо выбраны из F, Cl, C1-С6 алкильной группы, -ОН, -CN, -NH2, -NO2, -CO2H, C1-С6 алкокси-группы, моно(C1-С6 алкил)амино-группы, ди(С1-С6 алкил)амино-группы, -CF3, C1-С6 алкиленовой группы, замещенной 0, 1, 2 или 3 R1 группами, С2-С6 алкениленовой группы, замещенной 0, 1, 2 или 3 R1 группами, и оксо-группы;
Rf и Ri независимо выбраны из F, Cl, Br, -ОН, -CN, -NO2, -CO2H, C1-С6 алкильной группы, замещенной 0, 1, 2 или 3 Rk группами, С2-С6 алкенильной группы, замещенной 0, 1, 2 или 3 Rk группами, С2-С6 алкинильной группы, замещенной 0, 1, 2 или 3 Rk группами, С3-C8 циклоалкильной группы, замещенной 0, 1, 2 или 3 Rk группами, C1-С6 алкокси-группы, замещенной 0, 1, 2 или 3 Rk группами, С3-C8 циклоалкилокси-группы, замещенной 0, 1, 2 или 3 Rk группами, -SH, C1-С6 алкилтио-группы, замещенной 0, 1, 2 или 3 Rk группами, С3-C8 циклоалкилтио-группы, замещенной 0, 1, 2 или 3 Rk группами, (C1-С6 алкил)карбонильной группы, замещенной 0, 1, 2 или 3 Rk группами, (C1-С6 алкокси)карбонильной группы, замещенной 0, 1, 2 или 3 Rk группами, (C1-С6 алкил)аминокарбонильной группы, замещенной 0, 1, 2 или 3 Rk группами, 3-8-членной гетероциклоалкильной группы, замещенной 0, 1, 2 или 3 Rk группами, C1-С6 алкилсульфонильной группы, замещенной 0, 1, 2 или 3 Rk группами, -NH2, моно(C1-С6 алкил)амино-группы, замещенной 0, 1, 2 или 3 Rk группами, и ди(С1-С6 алкил)амино-группы, замещенной 0, 1, 2 или 3 Rk группами; и
Ra, Rb, Rc, Re, Rh, Rk и Rl независимо выбраны из F, C1-C4 алкильной группы, -ОН, -CN, -NO2, -NH2, -CO2H, C1-С6 алкокси-группы, моно(C1-С6 алкил)амино-группы, ди(С1-С6 алкил)амино-группы, -CF3 и оксо-группы.
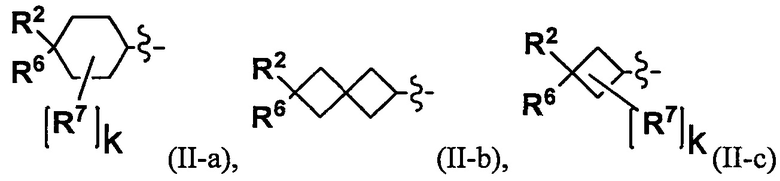
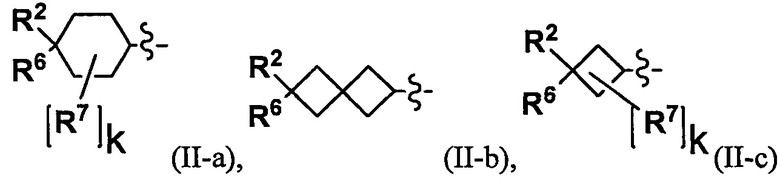
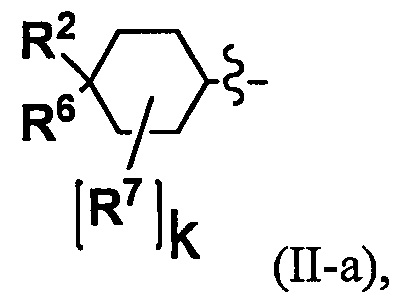
(2) Соединение по п. 1 или его фармацевтически приемлемая соль, где Y выбран из формулы (II-а), формулы (II-b), формулы (II-с) и формулы (II-d):
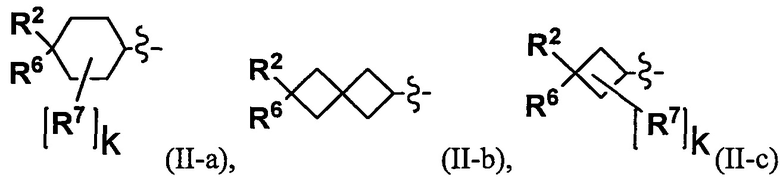 or
or 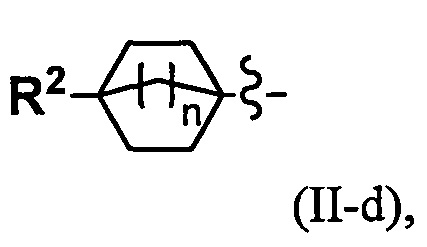
где:
k равно 0, 1 или 2;
и n равно 1, 2 или 3.
(3) Соединение по п. 2 или его фармацевтически приемлемая соль, где Y представляет собой группу, имеющую следующую формулу (II-а):
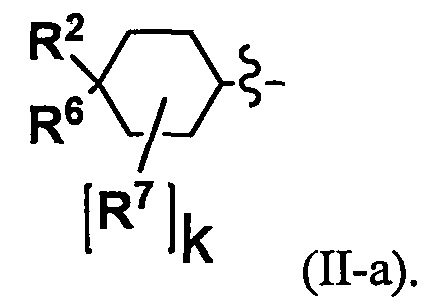
(4) Соединение по п. 2 или его фармацевтически приемлемая соль, где Y представляет собой группу, имеющую следующую формулу (II-d):
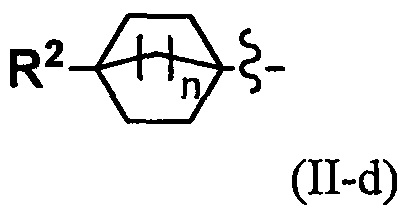
и n равно 2.
(5) Соединение по любому из пунктов 1-4 или его фармацевтически приемлемая соль, где R3 представляет собой Н.
(6) Соединение по любому из пунктов 1-5 или его фармацевтически приемлемая соль, где R2 представляет собой -СО2Н или гидроксикарбонилметильную группу, замещенную 0, 1 или 2 Rc группами.
(7) Соединение по любому из пунктов 1-6 или его фармацевтически приемлемая соль, где R12 представляет собой Н.
(8) Соединение по любому из пунктов 1-7 или его фармацевтически приемлемая соль, где R8 и R9 вместе формируют оксо-группу или оба R8 и R9 представляют собой Н.
(9) Соединение по любому из пунктов 1-8 или его фармацевтически приемлемая соль, где R1 представляет собой -CF3, -CF2H или Cl.
(10) Соединение по любому из пунктов 1-9 или его фармацевтически приемлемая соль, где R5 представляет собой С6-С10 арильную группу, замещенную 0, 1, 2, 3, 4 или 5 Ri группами, или 5-10-членную гетероарильную группу, замещенную 0, 1, 2, 3 или 4 Ri группами.
(11) Соединение по любому из пунктов 1-10 или его фармацевтически приемлемая соль, где R4 представляет собой C1-С6 алкильную группу, замещенную 0, 1, 2 или 3 Re группами, (С6-С10 арил)(C1-С3 алкильную) группу, замещенную 0, 1, 2, 3, 4 или 5 Rf группами, С3-C8 циклоалкильную группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами, (С3-C8 циклоалкил)(С1-С3 алкильную) группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 спироалкильную группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами, (С6-C9 спироалкил)(С1-С3 алкильную) группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами, C5-C9 бициклоалкильную группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами, (C6-C9 бициклоалкил)(С1-С3 алкильную) группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами, или (С6-С9 гетеробициклоалкил)(С1-С3 алкильную) группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами.
(12) Способ лечения или профилактики заболевания с применением соединения по любому из пунктов 1-11 или его фармацевтически приемлемой соли, где заболевание представляет собой множественный склероз, хронический ревматоидный артрит, анкилозирующий спондилит, системный эритоматоз, псориаз, псориатический артрит, воспалительную болезнь кишечника или астму.
(13) Фармацевтическая композиция, содержащая соединение по любому из пунктов 1-11 или его фармацевтически приемлемую соль.
Положительный эффект изобретения
В настоящем изобретении описано новое соединение, имеющее прекрасную ингибирующую активность в отношении RORγ, и способ его производства. Кроме того, соединение по настоящему изобретению или его фармацевтически приемлемая соль могут применяться в качестве терапевтического средства или средства профилактики против аутоиммунных заболеваний, воспалительных заболеваний (например, множественный склероз, хронический ревматоидный артрит, анкилозирующий спондилит, системный эритоматоз, псориаз, псориатический артрит, воспалительная болезнь кишечника и астма), заболеваний обмена веществ (особенно диабета), раковых заболеваний (особенно злокачественной меланомы) и т.п.
Описание вариантов осуществления
Далее разъясняются термины, применяющиеся независимо или в комбинации в настоящем описании. Если не указано особо, разъяснение по каждому заместителю является общим для каждого положения. Кроме того, когда какой-либо варьируемый заместитель (например, Rj и т.п.) присутствует в каких-либо соответствующих элементах конструкции (например, Rf, Ri, и т.п.), его значение является независимым в соответствующих элементах конструкции. Кроме того, комбинация заместителей и варьируемые заместители допустимы только тогда, когда такая комбинация дает химически устойчивое соединение. Когда заместитель сам замещен двумя или более группами, эти несколько групп могут быть присоединены к одному атому углерода или к разным атомам углерода, при условии формирования устойчивой структуры.
Каждая группа в соединениях, имеющих Формулу (I) по настоящему изобретению, имеет описанное ниже определение. Порядок написания в каждой группе указывает на порядок связей в Формуле (I). Например, "(С3-C8 циклоалкил)(С1-С3 алкильная) группа" в R4 представлена группой, где "C1-С3 алкильная группа" связана с атомом азота в Формуле (I), и "С3-C8 циклоалкильная группа" и "C1-С3 алкильная группа" связаны.
Кроме того, число, расположенное справа от атома углерода, означает число атомов углерода. Например, "C1-С6" означает группу, содержащую "1-6 атомов углерода". Само собой разумеется, что в настоящем изобретении различное число атомов углерода означает группу, содержащее указанное число атомов углерода. Например, "C1-С4 алкильная группа" означает алкильную группу, содержащую 1-4 атомов углерода. Для других групп определение числа атомов углерода осуществляется таким же образом.
В настоящем изобретении "C1-С6 алкильная группа" означает насыщенную линейную или разветвленную алифатическую углеводородную группу, содержащую 1-6 атомов углерода. Например, в качестве примеров можно привести метальную группу, этильную группу, н-пропильную группу, н-бутильную группу, н-пентильную группу, н-гексильную группу, изопропильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, изопентильную группу, 2-метилбутильную группу, 3-метилбутильную группу, 1-этилпропильную группу, 1,1-диметилпропильную группу, 1,2-диметилпропильную группу, неопентильную группу, 4-метилпентильную группу, 3-метилпентильную группу, 2-метилпентильную группу, 1-метилпентильную группу, 3,3-диметилбутильную группу, 2,2-диметилбутильную группу, 1,1-диметилбутильную группу, 1,2-диметилбутильную группу, 1,3-диметилбутильную группу, 2,3-диметилбутильную группу, 1-этилбутильную группу, 2-этилбутильную группу, и т.п.
В настоящем изобретении "C1-C4 алкильная группа" означает насыщенную линейную или разветвленную алифатическую углеводородную группу, содержащую 1-4 атомов углерода. Например, в качестве примеров можно привести метальную группу, этильную группу, н-пропильную группу, изопропильную группу и н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу и т.п.
В настоящем изобретении "С2-C4 алкильная группа" означает насыщенную линейную или разветвленную алифатическую углеводородную группу, содержащую 2-4 атомов углерода. Например, в качестве примеров можно привести этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу и т.п.
В настоящем изобретении "C1-С3 алкильная группа" означает насыщенную линейную или разветвленную алифатическую углеводородную группу, содержащую 1-3 атомов углерода. Например, в качестве примеров можно привести метальную группу, этильную группу, н-пропильную группу, изопропильную группу, и т.п.
В настоящем изобретении "С2-С6 алкенильная группа" означает линейную или разветвленную алифатическую углеводородную группу, содержащую 2-6 атомов углерода с ненасыщенной двойной связью. Например, в качестве примеров можно привести винильную группу, 1-пропенильную группу, 2-пропенильную группу, 2-метил-1-пропенильную группу, 2-метил-2-пропенильную группу, 2-бутен-1-ильную группу, 3-бутен-1-ильную группу, 2-пентен-1-ильную группу, 3-пентен-1-ильную группу, 4-пентен-1-ильную группу, 5-гексен-1-ильную группу, 4-гексен-1-ильную группу, 3-гексен-1-ильную группу, 2-гексен-1-ильную группу, 3-метил-2-бутен-1-ильную группу, 3-метил-3-пентен-1-ильную группу, 3-метил-2-пентен-1-ильную группу, 4-метил-3-пентен-1-ильную группу, 4-метил-2-пентен-1-ильную группу, 2-метил-2-пентен-1-ильную группу, и т.п.
В настоящем изобретении "С2-Сб алкинильная группа" означает линейную или разветвленную алифатическую углеводородную группу, содержащую 2-6 атомов углерода с ненасыщенной тройной связью. Например, в качестве примеров можно привести этинильную группу, 1-пропин-1-ильную группу, 2-пропин-1-ильную группу, 2-бутин-1-ильную группу, 3-бутин-1-ильную группу, 2-пентин-1-ильную группу, 3-пентин-1-ильную группу, 4-пентин-1-ильную группу, 5-гексин-1-ильную группу, 4-гексин-1-ильную группу, 3-гексин-1-ильную группу, 2-гексин-1-ильную группу, и т.п.
В настоящем изобретении "C1-С6 алкиленовая группа" означает двухвалентную группу, образовавшуюся при удалении атома водорода от "C1-С6 алкильной группы". Например, в качестве примеров можно привести метилен, этилен, пропилен, бутилен, пентилен, гексилен, и т.п. C1-С6 алкиленовая группа может быть связана с одним атомом углерода или с двумя разными атомами углерода с образованием цикла.
В настоящем изобретении "С2-С6 алкениленовая группа" означает двухвалентную группу, имеющую двойную связь в каком-либо положении "С2-С6 алкиленовой группы". В качестве примеров можно привести винилен, пропенилен, 1-бутенилен, 2-бутенилен, 1-пентенилен, 2-пентенилен, 1-гексенилен, 2-гексенилен, 3-гексенилен, и т.п.
В настоящем изобретении "С3-C8 циклоалкильная группа" означает циклическую алкильную группу, содержащую 3-8 атомов углерода. Например, в качестве примеров можно привести циклопропильную группу, циклобутильную группу, циклопентильную группу, циклогексильную группу, циклогептильную группу, циклооктильную группу, и т.п.
В настоящем изобретении "C4-С6 циклоалкильная группа" означает циклическую алкильную группу, содержащую 4-6 атомов углерода. Например, в качестве примеров можно привести циклобутильную группу, циклопентильную группу, циклогексильную группу, и т.п.
В настоящем изобретении "С6-С9 бициклоалкильная группа" означает бициклическую алкильную группу, содержащую 6-9 атомов углерода. Например, в качестве примеров можно привести бицикло[3.1.0]гексанильную группу, бицикло[2.2.0]гексанильную группу, бицикло[2.1.1]гексанильную группу, бицикло[3.2.0]гептанильную группу, бицикло[2.2.1]гептанильную группу, бицикло[3.1.1]гептанильную группу, бицикло[4.1.0]гептанильную группу, октагидропенталенильную группу, бицикло[2.2.2]октанильную группу, бицикло[3.2.1]октанильную группу, бицикло[4.2.0]октанильную группу, бицикло[4.1.1]октанильную группу, бицикло[5.1.0]октанильную группу, октагидро-1H-инденильную группу, бицикло[3.2.2]нонанильную группу, бицикло[3.3.1]нонанильную группу, бицикло[4.2.1]нонанильную группу, бицикло[5.2.0]нонанильную группу, и т.п.
В настоящем изобретении "C5-С9 бициклоалкильная группа" означает бициклическую алкильную группу, содержащую 5-9 атомов углерода. Например, в качестве примеров можно привести бицикло[1.1.1]пентанильную группу, бицикло[3.1.0]гексанильную группу, бицикло[2.2.0]гексанильную группу, бицикло[2.1.1]гексанильную группу, бицикло[3.2.0]гептанильную группу, бицикло[2.2.1]гептанильную группу, бицикло[3.1.1]гептанильную группу, бицикло[4.1.0]гептанильную группу, октагидропенталенильную группу, бицикло[2.2.2]октанильную группу, бицикло[3.2.1]октанильную группу, бицикло[4.2.0]октанильную группу, бицикло[4.1.1]октанильную группу, бицикло[5.1.0]октанильную группу, октагидро-1H-инденильную группу, бицикло[3.2.2]нонанильную группу, бицикло[3.3.1]нонанильную группу, бицикло[4.2.1]нонанильную группу, бицикло[5.2.0]нонанильную группу, и т.п.
В настоящем изобретении "спироалкильная группа" означает группу, состоящую из двух циклоалкильных фрагментов, которые имеют ровно один общий атом. «С6-С9 спироалкильная группа» означает спироалкильную группу, содержащую 6-9 атомов углерода. Например, в качестве примеров можно привести спиро[2.3]гексанильную группу, спиро[2.4]гептанильную группу, спиро[3.3]гептанильную группу, спиро[2.5]октанильную группу, спиро[3.4]октанильную группу, спиро[2.6]нонанильную группу, спиро[3.5]нонанильную группу, спиро[4.4]нонанильную группу, и т.п.
В настоящем изобретении "(С6-С9 спироалкил)(С1-С3 алкильная) группа" означает группу, полученную путем замещения "C1-С3 алкильной группы" " (С6-С9 спироалкильной) группой" по какому-либо положению. Например, в качестве примеров можно привести спиро[2.3]гексанил метальную группу, спиро[2.4]гептанил метальную группу, спиро[3.3]гептанил метальную группу, спиро[2.5]октанил метальную группу, спиро[3.4]октанил метальную группу, спиро[2.6]нонанил метальную группу, спиро[3.5]нонанил метальную группу, спиро[4.4]нонанил метальную группу, и т.п.
В настоящем изобретении "С3-C8 циклоалкенильная группа" означает группу, содержащую двойную связь в каком-либо положении "С3-С8 циклоалкильной группы", содержащей 3-8 атомов углерода. Например, в качестве примеров можно привести циклопропенильную группу, циклобутенильную группу, циклопентенильную группу, циклогексенильную группу, циклогептенильную группу, циклооктенильную группу и т.п.
В настоящем изобретении "(С3-C8 циклоалкил)(С1-С3 алкильная) группа" означает группу, полученную путем замещения "C1-С3 алкильной группы" "С3-C8 циклоалкильной группой" по какому-либо положению. Например, в качестве примеров можно привести циклопропилметильную группу, циклопропилэтильную группу, циклопропилпропильную группу, циклобутилметильную группу, циклобутилэтильную группу, циклопентилметильную группу, циклопентилэтильную группу, циклогексилметильную группу, циклогексилэтильную группу, циклогептилметильную группу, циклогептилэтильную группу, циклооктилметильную группу, и т.п.
В настоящем изобретении "(С3-C8 циклоалкенил)(С1-С3 алкильная) группа" означает группу, полученную путем замещения "C1-С3 алкильной группы" "С3-C8 циклоалкенильной группой" по какому-либо положению. Например, в качестве примеров можно привести циклопропенилметильную группу, циклопропенилэтильную группу, циклопропенилпропильную группу, циклобутенилметильную группу, циклобутенилэтильную группу, циклопентенилметильную группу, циклопентенилэтильную группу, циклогексенилметильную группу, циклогексенилэтильную группу, циклогептенилметильную группу, циклогептенилэтильную группу, циклооктенилметильную группу, и т.п.
В настоящем изобретении "(С2-С6 алкенил)(С1-С3 алкильная) группа" означает группу, полученную путем замещения "C1-С3 алкильной группы" "С2-С6 алкенильной группой" по какому-либо положению. Например, в качестве примеров можно привести 2-пропенильную группу, 1-метил-2-пропенильную группу, 2-метил-2-пропенильную группу, 2-бутен-1-ильную группу, 3-бутен-1-ильную группу, 2-пентен-1-ильную группу, 3-пентен-1-ильную группу, 4-пентен-1-ильную группу, 5-гексен-1-ильную группу, 4-гексен-1-ильную группу, 3-гексен-1-ильную группу, 2-гексен-1-ильную группу, 1-метил-2-бутен-1-ильную группу, 1-этил-2-бутен-1-ильную группу, 2-метил-2-бутен-1-ильную группу, 3-метил-2-бутен-1-ильную группу, 3-метил-3-пентен-1-ильную группу, 3-метил-2-пентен-1-ильную группу, 3-этил-2-пентен-1-ильную группу, 4-метил-3-пентен-1-ильную группу, 4-метил-2-пентен-1-ильную группу, 2-метил-2-пентен-1-ильную группу, и т.п.
В настоящем изобретении "(С2-С6 алкинил)(С1-С3 алкильная) группа" означает группу, полученную путем замещения "C1-С3 алкильной группы" "С2-С6 алкинильной группой" по какому-либо положению. Например, в качестве примеров можно привести 2-пропин-1-ильную группу, 1-метил-2-пропин-1-ильную группу, 1-этил-2-пропин-1-ильную группу, 2-бутин-1-ильную группу, 1-метил-2-бутин-1-ильную группу, 1-этил-2-бутин-1-ильную группу, 3-бутин-1-ильную группу, 1-метил-3-бутин-1-ильную группу, 1-этил-3-бутан-1-ильную группу, 2-пентин-1-ильную группу, 1-метил-2-пентин-1-ильную группу, 3-пентин-1-ильную группу, 1-метил-3-пентин-1-ильную группу, 4-пентин-1-ильную группу, 5-гексин-1-ильную группу, 4-гексин-1-ильную группу, 3-гексин-1-ильную группу, 2-гексин-1-ильную группу, и т.п.
В настоящем изобретении "C1-С6 алкокси-группа" означает группу, полученную путем замещения окси-группы "C1-С6 алкильной группой". Например, в качестве примеров можно привести метокси-группу, этокси-группу, н-пропокси-группу, изопропокси-группу, н-бутокси-группу, втор-бутокси-группу, 2-метилпропокси-группу, n-пентилокси-группу, изопентилокси-группу, 2-метилбутокси-группу, 1-этилпропокси-группу, 2,2-диметилпропокси-группу, н-гексилокси-группу, 4-метилпентокси-группу, 3-метилпентокси-группу, 2-метилпентокси-группу, 3,3-диметилбутокси-группу, 2,2-диметилбутокси-группу, 1,1-диметилбутокси-группу, трет-бутокси-группу и т.п.
В настоящем изобретении "(C1-С6 алкокси)(С2-C4 алкил)" означает группу, полученную путем замещения "C2-C4 алкильной группы" "C1-С6 алкокси-группой" или, другими словами, группу, полученную путем замены одного атома углерода C4-С11 алкильной группы одним атомом кислорода в каком-либо химически допустимом положении. Например, в качестве примеров можно привести метоксиэтильную группу, этоксиэтильную группу, пропилоксиэтильную группу, изопропилоксиэтильную группу, бутилоксиэтильную группу, изобутилоксиэтильную группу, втор-бутилоксиэтильную группу, трет-бутилоксиэтильную группу, изопентилоксиэтильную группу, 2-метилбутилоксиэтильную группу, 3-метилбутилоксиэтильную группу, 1-этилпропилоксиэтильную группу, 1,1-диметилпропилоксиэтильную группу, 1,2-диметилпропилоксиэтильную группу, неопентилоксиэтильную группу, гексилоксиэтильную группу, 4-метилпентилоксиэтильную группу, 3-метилпентилоксиэтильную группу, 2-метилпентилоксиэтильную группу, 1-метилпентилоксиэтильную группу, 3,3-диметилбутилоксиэтильную группу, 2,2-диметилбутилоксиэтильную группу, 1,1-диметилбутилоксиэтильную группу, 1,2-диметилбутилоксиэтильную группу, 1,3-диметилбутилоксиэтильную группу, 2,3-диметилбутилоксиэтильную группу, 1-этилбутилоксиэтильную группу, 2-этилбутилоксиэтильную группу, метоксипропильную группу, этоксипропильную группу, пропилоксипропильную группу, изопропилоксипропильную группу, бутилоксипропильную группу, изобутилоксипропильную группу, втор-бутилоксипропильную группу, трет-бутилоксипропильную группу, изопентилоксипропильную группу, 2-метилбутилоксипропильную группу, 3-метилбутилоксипропильную группу, 1-этилпропилоксипропильную группу, 1,1-диметилпропилоксипропильную группу, 1,2-диметилпропилоксипропильную группу, неопентилоксипропильную группу, гексилоксипропильную группу, 4-метилпентилоксипропильную группу, 3-метилпентилоксипропильную группу, 2-метилпентилоксипропильную группу, 1-метилпентилоксипропильную группу, 3,3-диметилбутилоксипропильную группу, 2,2-диметилбутилоксипропильную группу, 1,1-диметилбутилоксипропильную группу, 1,2-диметилбутилоксипропильную группу, 1,3-диметилбутилоксипропильную группу, 2,3-диметилбутилоксипропильную группу, 1-этилбутилоксипропильную группу, 2-этилбутилоксипропильную группу, метоксибутильную группу, этоксибутильную группу, пропилоксибутильную группу, изопропилоксибутильную группу, бутилоксибутильную группу, изобутилоксибутильную группу, втор-бутилоксибутильную группу, трет-бутилоксибутильную группу, изопентилоксибутильную группу, 2-метилбутилоксибутильную группу, 3-метилбутилоксибутильную группу, 1-этилпропилоксибутильную группу, 1,1-диметилпропилоксибутильную группу, 1,2-диметилпропилоксибутильную группу, неопентилоксибутильную группу, гексилоксибутильную группу, 4-метилпентилоксибутильную группу, 3-метилпентилоксибутильную группу, 2-метилпентилоксибутильную группу, 1-метилпентилоксибутильную группу, 3,3-диметилбутилоксибутильную группу, 2,2-диметилбутилоксибутильную группу, 1,1-диметилбутилоксибутильную группу, 1,2-диметилбутилоксибутильную группу, 1,3-диметилбутилоксибутильную группу, 2,3-диметилбутилоксибутильную группу, 1-этилбутилоксибутильную группу, 2-этилбутилоксибутильную группу, и т.п.
В настоящем изобретении "C1-С6 алкилтио-группа" означает группу, полученную путем замещения тио-группы "C1-С6 алкильной группой". Например, в качестве примеров можно привести метилтио-группу, этилтио-группу, пропилтио-группу, изопропилтио-группу, бутилтио-группу, изобутилтио-группу, втор-бутилтио-группу, трет-бутилтио-группу, пентилтио-группу, неопентилтио-группу, трет-пентилтио-группу, 2-метилбутилтио-группу, гексилтио-группу, изогексилтио-группу, и т.п.
В настоящем изобретении "С3-C8 циклоалкилтио-группа" означает группу, полученную путем замещения тио-группы "С3-C8 циклоалкильной группой". Например, в качестве примеров можно привести циклопропилтио-группу, циклобутилтио-группу, циклопентилтио-группу, циклогексилтио-группу, циклогептилтио-группу, циклооктилтио-группу, и т.п.
В настоящем изобретении "(C1-С6 алкил)карбонильная группа" означает группу, полученную путем замещения карбонильной группы "C1-С6 алкильной группой". Например, в качестве примеров можно привести ацетильную группу, пропионильную группу, бутирильную группу, изобутирильную группу, н-пентилкарбонильную группу, втор-бутилкарбонильную группу, трет-бутилкарбонильную группу, изопентилкарбонильную группу, 2-метилбутилкарбонильную группу, 3-метилбутилкарбонильную группу, 1-этилпропилкарбонильную группу, 1,1-диметилпропилкарбонильную группу, 1,2-диметилпропилкарбонильную группу, неопентилкарбонильную группу, 4-метилпентилкарбонильную группу, 3-метилпентилкарбонильную группу, 2-метилпентилкарбонильную группу, 1-метилпентилкарбонильную группу, 3,3-диметилбутилкарбонильную группу, 2,2-диметилбутилкарбонильную группу, 1,1-диметилбутилкарбонильную группу, 1,2-диметилбутилкарбонильную группу, 1,3-диметилбутилкарбонильную группу, 2,3-диметилбутилкарбонильную группу, 1-этилбутилкарбонильную группу, 2-этилбутилкарбонильную группу, н-гексилкарбонильную группу, и т.п.
В настоящем изобретении "(C1-С6 алкокси)карбонильная группа" означает группу, полученную путем замещения карбонильной группы "C1-С6 алкокси-группой". Например, в качестве примеров можно привести метоксикарбонильную группу, этоксикарбонильную группу, н-пропоксикарбонильную группу, изопропоксикарбонильную группу, н-бутоксикарбонильную группу, изобутоксикарбонильную группу, втор-бутоксикарбонильную группу, трет-бутоксикарбонильную группу, н-пентоксикарбонильную группу, изопентоксикарбонильную группу, 2-метилбутоксикарбонильную группу, 3-метилбутоксикарбонильную группу, 1-этилпропоксикарбонильную группу, 1,1-диметилпропоксикарбонильную группу, 1,2-диметилпропоксикарбонильную группу, неопентоксикарбонильную группу, 4-метилпентоксикарбонильную группу, 3-метилпентоксикарбонил, 2-метилпентоксикарбонильную группу, 1-метилпентоксикарбонильную группу, 3,3-диметилбутоксикарбонильную группу, 2,2-диметилбутоксикарбонильную группу, 1,1-диметилбутоксикарбонильную группу, 1,2-диметилбутоксикарбонильную группу, 1,3-диметилбутоксикарбонильную группу, 2,3-диметилбутоксикарбонильную группу, 1-этилбутоксикарбонильную группу, 2-этилбутоксикарбонильную группу, н-гексоксикарбонильную группу, и т.п.
В настоящем изобретении "С3-C8 циклоалкилокси-группа" означает группу, полученную путем замещения окси-группы "С3-C8 циклоалкильной группой". Например, в качестве примеров можно привести циклопропилокси-группу, циклобутилокси-группу, циклопентилокси-группу, циклогексилокси-группу, циклогептилокси-группу, циклооктилокси-группу, и т.п.
В настоящем изобретении "моно(C1-С6 алкил)амино-группа" означает группу, полученную путем замещения амино-группы "C1-С6 алкильной группой". Например, в качестве примеров можно привести метиламино-группу, этиламино-группу, пропиламино-группу, изопропиламино-группу, бутиламино-группу, изобутиламино-группу, втор-бутиламино-группу, трет-бутиламино-группу, пентиламино-группу, гексиламино-группу, и т.п.
В настоящем изобретении "ди(С1-С6 алкил)амино-группа" означает группу, полученную путем замещения амино-группы двумя одинаковыми или разными "C1-С6 алкильными группами". Например, в качестве примеров можно привести диметиламино-группу, диэтиламино-группу, дипропиламино-группу, диизопропиламино-группу, дибутиламино-группу, диизобутиламино-группу, ди(втор-бутил)амино-группу, ди(трет-бутил)амино-группу, дипентиламино-группу, дигексиламино-группу, и т.п.
В настоящем изобретении "(C1-С6 алкил)аминокарбонильная группа" означает группу, полученную путем замещения карбонильной группы "(C1-С6 алкил)амино-группой". Например, в качестве примеров можно привести метиламинокарбонильную группу, этиламинокарбонильную группу, пропиламинокарбонильную группу, изопропиламинокарбонильную группу, бутиламинокарбонильную группу, изобутиламинокарбонильную группу, втор-бутиламинокарбонильную группу, трет-бутиламинокарбонильную группу, пентиламинокарбонильную группу, гексиламинокарбонильную группу, и т.п.
В настоящем изобретении "C1-С6 алкилсульфонильная группа" означает группу, полученную путем замещения сульфонильной группы "C1-С6 алкильной группой". Например, в качестве примеров можно привести метилсульфонильную группу, этилсульфонильной группы, пропилсульфонильную группу, изопропилсульфонильную группу, бутилсульфонильную группу, изобутилсульфонильную группу, втор-бутилсульфонильную группу, трет-бутилсульфонильную группу, пентилсульфонильную группу, гексилсульфонильную группу, и т.п.
В настоящем изобретении "C1-С6 алкиламиносульфонильная группа" означает группу, полученную путем замещения сульфонильной группы "моно(C1-С6 алкил)амино-группой". Например, в качестве примеров можно привести метиламиносульфонильную группу, этиламиносульфонильную группу, пропиламиносульфонильную группу, изопропиламиносульфонильную группу, бутиламиносульфонильную группу, изобутиламиносульфонильную группу, втор-бутиламиносульфонильную группу, трет-бутиламиносульфонильную группу, пентиламиносульфонильную группу, гексиламиносульфонильную группу, и т.п.
В настоящем изобретении "(гидроксикарбонил)(С1-С3 алкильная) группа" означает группу, полученную путем замещения "C1-С3 алкильной группы" "(гидроксикарбонильной) группой" по какому-либо положению. Например, в качестве примеров можно привести гидроксикарбонилметильную группу, (1-гидроксикарбонил)этильную группу, (2-гидроксикарбонил)этильную группу, (3-гидроксикарбонил)пропильную группу, (2-гидроксикарбонил)пропильную группу, (1-гидроксикарбонил)пропильную группу, (1-гидроксикарбонил)(1-метил)этильную группу, и т.п.
В настоящем изобретении "(C1-C6 алкокси)карбонил(С1-С3 алкильная) группа" означает группу, полученную путем замещения "C1-С3 алкильной группы" "(C1-С6 алкокси)карбонильной группой" по какому-либо положению. Например, в качестве примеров можно привести метоксикарбонилметильную группу, метоксикарбонилэтильную группу, (3-метоксикарбонил)пропильную группу, (2-метоксикарбонил)пропильную группу, (1-метоксикарбонил)пропильную группу, (1-метоксикарбонил)(1-метил)этильную группу, этоксикарбонилметильную группу, этоксикарбонилэтильную группу, (3-этоксикарбонил)пропильную группу, (2-этоксикарбонил)пропильную группу, (1-этоксикарбонил)пропильную группу, (1-этоксикарбонил)(1-метил)этильную группу, и т.п.
В настоящем изобретении "(C1-С6 алкил)сульфонил(С1-С3 алкильная) группа" означает группу, полученную путем замещения "C1-С3 алкильной группы" "(C1-С6 алкил)сульфонильной группой" по какому-либо положению. Например, в качестве примеров можно привести метилсульфонилметильную группу, метилсульфонилэтильную группу, (3-метилсульфонил)пропильную группу, (2-метилсульфонил)пропильную группу, (1-метилсульфонил)пропильную группу, (1-метилсульфонил)(1-метил)этильную группу, этилсульфонилметильную группу, этилсульфонилэтильную группу, (3-этилсульфонил)пропильную группу, (2-этилсульфонил)пропильную группу, (1-этилсульфонил)пропильную группу, (1-этилсульфонил)(1-метил)этильную группу, и т.п.
В настоящем изобретении "С6-С10 арильная группа" означает ароматическую углеводородную группу, содержащую 6-10 атомов углерода. Например, в качестве примеров можно привести фенильную группу, нафтильную группу, инденильную группу, тетрагидронафтильную группу, инданильную группу, азуленильную группу, и т.п.
В настоящем изобретении "С6-С10 арилокси-группа" означает группу, полученную путем замещения окси-группы "С6-С10 арильной группой". Например, в качестве примеров можно привести фенилокси-группу, нафтилокси-группу, инденилокси-группу, тетрагидронафтилокси-группу, инданилокси-группу, азуленилокси-группу, и т.п.
В настоящем изобретении "(С6-С10 арил)(С1-С3 алкильная) группа" означает группу, полученную путем замещения "C1-С3 алкильной группы" "С6-С10 арильной группой". Например, в качестве примеров можно привести бензильную группу, фенэтильную группу, фенилпропильную группу, нафтилметильную группу, и т.п.
В настоящем изобретении "5-10-членная гетероарильная группа" означает 5-10-членную моноциклическую или бициклическую гетероциклическую группу, обладающую ароматичностью, где указанная гетероциклическая группа содержит 1-5 гетероатомов, выбранных из кислорода, серы и азота. Кроме того, в случае бициклической ароматической гетероциклической группы, если один цикл является ароматическим или ароматическим гетероциклическим, другой цикл может быть неароматическим циклом. В такой ароматической гетероциклической группе, число соответствующих гетероатомов и их комбинаций особо не ограничено, при условии, что цикл с указанным числом членов может формироваться и устойчиво существовать. В качестве примеров такой «5-10-членной гетероарильной группы» можно привести пиридильную группу, пиразильную группу, пиримидильную группу, пиридазинильную группу, фурильную группу, тиенильную группу, пиррольную группу, пиразолильную группу, 1,3-диоксаинданильную группу, изоксазолильную группу, изотиазолильную группу, бензофуранильную группу, изобензофурильную группу, бензотиенильную группу, индолильную группу, изоиндолильную группу, хроманильную группу, бензотиазолильную группу, бензоимидазолильную группу, бензоксазолильную группу, пиранильную группу, имидазолильную группу, оксазолильную группу, тиазолильную группу, триазинильную группу, триазолильную группу, фуразанильную группу, тиадиазолильную, дигидробензофурильную группу, дигидроизобензофурильную группу, дигидрохинолильную группу, дигидроизохинолильную группу, дигидробензоксазолильную группу, дигидроптеридинильную группу, бензоксазолильную группу, бензизоксазолильную группу, бензодиоксазолильную группу, хинолильную группу, изохинолильную группу, бензотриазолильную группу, птеридинильную группу, пуринильную группу, хиноксалинильную группу, хиназолинильную группу, циннолинильную группу, тетразолильную группу, и т.п.
В настоящем изобретении "(5-10-членный гетероарил)(С1-С3 алкильная) группа" означает группу, полученную путем замещения "C1-С3 алкильной группы" "5-10-членной гетероарильной группой". Например, в качестве примеров можно привести пиридилметильную группу, тиенилметильную группу, тиазолилметильную группу, бензотиазолилметильную группу, бензотиофенилметильную группу, и т.п.
В настоящем изобретении "3-8-членная гетероциклоалкильная группа" означает 3-8-членную алифатическую гетероциклическую группу, которая может быть насыщенной или частично ненасыщенной, где цикл содержит 1-4 гетероатомов, выбранных из кислорода, серы и азота. Например, в качестве примеров можно привести пиперидильную группу, тетрагидрофуранильную группу, тетрагидропиранильную группу, тетрагидротиенильную группу, морфолильную группу, и т.п.
В настоящем изобретении "(3-8-членный гетероциклоалкил)(С1-С3 алкильная) группа" означает группу, полученную путем замещения "C1-С3 алкильной группы" "3-8-членной гетероциклоалкильной группой". Например, в качестве примеров можно привести пиперидилметильную группу, тетрагидрофуранилметильную группу, тетрагидропиранилметильную группу, тетрагидротиенилметильную группу, морфолиноэтильную группу, оксетан-3-илметильную группу, и т.п.
В настоящем изобретении "спирогетероалкильная группа" означает спироалкильную группу, в которой 1-4 атома углерода заменены на 1-4 гетероатома, выбранные из кислорода, серы и азота. "С6-С9 спирогетероалкильная группа" означает спироалкильную группу, содержащую 6-9 атомов углерода. Например, в качестве примеров можно привести 4-оксаспиро[2.4]гептанильную группу, 4-оксаспиро[2.5]октанеильную группу, и т.п.
В настоящем изобретении "(C5-С9 бициклоалкил)(С1-С3 алкильная) группа" означает группу, полученную путем замещения "C1-С3 алкильной группы" "C5-С9 бициклоалкильной группой" по какому-либо положению. Например, в качестве примеров можно привести бицикло[1.1.1]пентанил метальную группу, бицикло[3.1.0]гексанил метальную группу, бицикло[3.1.0]гексанил этильную группу, бицикло[2.2.0]гексанил метальную группу, бицикло[2.2.0]гексанил этильную группу, бицикло[2.1.1]гексанил метальную группу, бицикло[2.1.1]гексанил этильную группу, бицикло[3.2.0]гептанил метальную группу, бицикло[3.2.0]гептанил этильную группу, бицикло[2.2.1]гептанил метальную группу, бицикло[2.2.1]гептанил этильную группу, бицикло[3.1.1]гептанил метальную группу, бицикло[4.1.0]гептанил метальную группу, октагидропенталенил метальную группу, бицикло[2.2.2]октанил метальную группу, бицикло[3.2.1]октанил метальную группу, бицикло[4.2.0]октанил метальную группу, бицикло[4.1.1]октанил метальную группу, бицикло[5.1.0]октанил метальную группу, октагидро-1Н-инденил метальную группу, бицикло[3.2.2]нонанил метальную группу, бицикло[3.3.1]нонанил метальную группу, бицикло[4.2.1]нонанил метальную группу, бицикло[5.2.0]нонанил метальную группу, и т.п.
В настоящем изобретении "гетеробициклоалкильная группа" означает бициклоалкильную группу, 1-4 атома углерода заменены на 1-4 гетероатома, выбранные из кислорода, серы и азота. "С6-C9 гетеробициклоалкильная группа" означает гетеробициклоалкильную группу, содержащую 6-9 атомов углерода. Например, в качестве примеров можно привести 7-оксабицикло[2.2.1]гептанильную группу и т.п.
В настоящем изобретении "(С6-С9 гетеробициклоалкил)(С1-С3 алкильная) группа" означает группу, полученную путем замещения "C1-С3 алкильной группы" "С6-С9 гетеробициклоалкильной группой" по какому-либо положению. Например, в качестве примеров можно привести 7-оксабицикло[2.2.1]гептанил метальную группу, 7-оксабицикло[2.2.1]гептанил этильную группу, и т.п.
В настоящем изобретении в "C1-С6 алкильной группе, замещенной 0, 1, 2 или 3 Ra группами", когда C1-С6 алкильная группа замещена несколькими Ra группами, каждая Ra группа может быть выбрана независимо, и C1-С6 алкильная группа может быть замещена одинаковыми Ra группами или разными Ra группами. Кроме того, другие выражения, такие как "C1-С6 алкильная группа, замещенная 0, 1, 2 или 3 Rb группами" и т.п., имеют сходные значения.
Настоящее изобретение касается соединения, имеющего формулу (I), или его фармацевтически приемлемой соли:
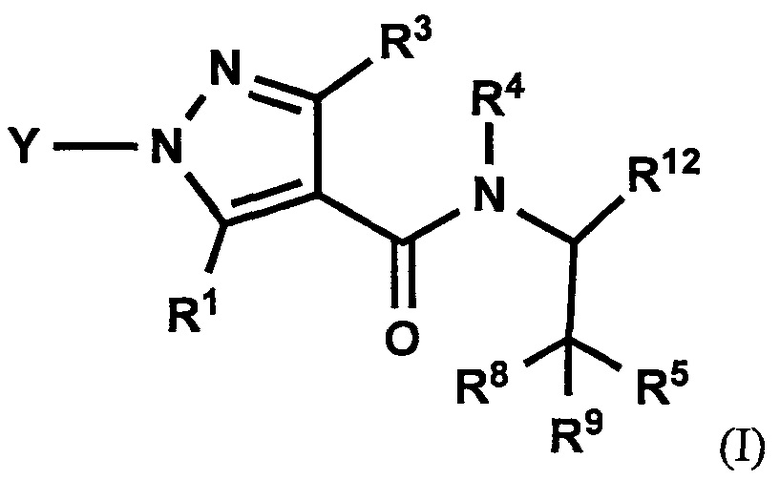
В формуле (I), R1 выбран из F, Cl, Br, C1-С6 алкильной группы, замещенной 0, 1, 2 или 3 Ra группами, и С3-C8 циклоалкильной группы, замещенной 0, 1, 2 или 3 Ra группами;
где Ra независимо выбран из F, C1-C4 алкильной группы, -ОН, -CN, -NO2, -NH2, -СО2Н, C1-С6 алкокси-группы, моно(C1-С6 алкил)амино-группы, ди(С1-С6 алкил)амино-группы, -CF3 и оксо-группы.
"C1-С6 алкильная группа, замещенная 0, 1, 2 или 3 Ra группами" в R1 предпочтительно представляет собой C1-С3 алкильную группу, замещенную 0, 1, 2 или 3 Ra группами, и более предпочтительно представляет собой трифторметильную группу или дифторметильную группу.
"С3-C8 циклоалкильная группа, замещенная 0, 1, 2 или 3 Ra группами" в R1 предпочтительно представляет собой С3-C4 циклоалкильную группу, замещенную 0, 1, 2 или 3 Ra группами, более предпочтительно представляет собой циклопропильную группу, замещенную 0, 1, 2 или 3 Ra группами.
В целом, R1 предпочтительно представляет собой Cl, C1-C4 алкильную группу, замещенную 0, 1, 2 или 3 Ra группами, или циклопропильную группу, замещенную 0, 1, 2 или 3 Ra группами, и более предпочтительно представляет собой трифторметильную группу, дифторметильную группу или Cl.
В формуле (I), Y представляет собой C4-С6 циклоалкильную группу, С6-С9 бициклоалкильную группу или С6-С9 спироалкильную группу, которые все замещены R2 группой, 0 или 1 R6 группами и 0, 1, 2 или 3 R7 группами;
где R2 выбран из -ОН, -CO2H, -SO3H, -CONH2, -SO2NH2, (C1-С6 алкокси)карбонильной группы, замещенной 0, 1, 2 или 3 Rc группами, (C1-С6 алкил)аминокарбонильной группы, замещенной 0, 1, 2 или 3 Rc группами, C1-С6 алкилсульфонильной группы, замещенной 0, 1, 2 или 3 Rc группами, C1-С6 алкиламиносульфонильной группы, замещенной 0, 1, 2 или 3 Rc группами, (гидроксикарбонил)(С1-С3 алкильной) группы, замещенной 0, 1, 2 или 3 Rc группами, (C1-С6 алкокси)карбонил(С1-С3 алкильной) группы, замещенной 0, 1, 2 или 3 Rc группами, (C1-С6 алкил)сульфонил(С1-С3 алкильной) группы, замещенной 0, 1, 2 или 3 Rc группами, и (С2-С6 алкенил)(С1-С3 алкильной) группы, замещенной 0, 1, 2 или 3 Rc группами;
R6 и R7 независимо выбраны из Н, F, -ОН, -NH2, -CN, C1-С6 алкильной группы, замещенной 0, 1, 2 или 3 Rb группами, и C1-С6 алкокси-группы, замещенной 0, 1, 2 или 3 Rb группами;
где Rb и Rc независимо выбраны из F, C1-C4 алкильной группы, -ОН, -CN, -NO2, -NH2, -CO2H, C1-С6 алкокси-группы моно(C1-С6 алкил)амино-группы, ди(С1-С6 алкил)амино-группы, -CF3 и оксо-группы;
"С4-С6 циклоалкильная группа, С6-С9 бициклоалкильная группа или С6-С9 спироалкильная группа, которые все замещены R2 группой, 0 или 1 R6 группами и 0, 1, 2 или 3 R7 группами" в Y предпочтительно представляют собой группу, имеющую следующую формулу (II-а), формулу (II-b), формулу (II-с) или формулу (II-d):
 или
или 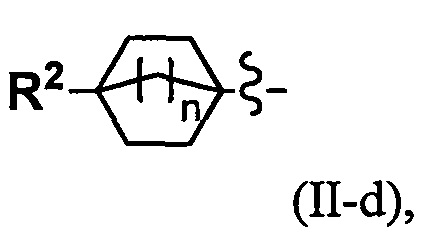
где:
k равно 0, 1 или 2;
и n равно 1, 2 или 3.
В случае группы, имеющей формулу (II-а), формулу (II-b), формулу (II-с) или формулу (II-d), Y предпочтительно представляет собой группу, имеющую формулу (II-а), формулу (II-с) или формулу (II-d); и более предпочтительно - группу, имеющую формулу (II-а) или формулу (II-d).
Переменная n предпочтительно равна 2 в группе, имеющей следующую формулу (II-d).
R2 в Y предпочтительно представляет собой -CO2H, -SO3H, -CONH2, -SO2NH2, (C1-С2 алкил)аминокарбонильную группу замещенную 0 или 1 Rc группами, C1-C2 алкилсульфонильную группу, замещенную 0 или 1 Rc группами, C1-С2 алкиламиносульфонильную группу замещенную 0 или 1 Rc группами, или (гидроксикарбонил)(С1-С3 алкильную) группу, замещенную 0, 1, 2 или 3 Rc группами, и более предпочтительно представляет собой -CO2H или гидроксикарбонилметильную группу замещенную 0, 1 или 2 Rc группами.
R6 в Y предпочтительно представляет собой Н или C1-С4 алкильную группу без Rb группы, и более предпочтительно представляет собой Н, метальную группу или этильную группу.
R7 в Y предпочтительно представляет собой Н или C1-С2 алкильную группу без Rb группы, и более предпочтительно представляет собой Н или метальную группу.
В формуле (I), R3 выбран из Н, F, Cl, -CH3 и -CF3. R3 предпочтительно представляет собой Н.
В формуле (I), R4 выбран из C1-С6 алкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (C1-С6 алкенил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (С2-С6 алкинил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (C1-С6 алкокси)(С2-C4 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (С6-С10 арил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, (5-10-членный гетероарил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, С3-C8 циклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С3-C8 циклоалкенильной группы, замещенной 0, 1, 2, 3, 4 или 5Rg группами, (С3-C8 циклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С3-C8 циклоалкенил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, 3-8-членный гетероциклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (3-8-членный гетероциклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 спироалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С6-С9 спироалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 спирогетероалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С5-С9 бициклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (C5-C9 бициклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 гетеробициклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, и (С6-С9 гетеробициклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами;
где Re независимо выбран из F, C1-C4 алкильной группы, -ОН, -CN, -NO2, -NH2, -CO2H, C1-С6 алкокси-группы, моно(C1-С6 алкил)амино-группы, ди(С1-С6 алкил)амино-группы, -CF3 и оксо-группы;
Rf независимо выбран из F, Cl, Br, -ОН, -CN, -NO2, -CO2H, C1-С6 алкильной группы, замещенной 0, 1, 2 или 3 Rk группами, С2-С6 алкенильной группы, замещенной 0, 1, 2 или 3 Rk группами, С2-С6 алкинильной группы, замещенной 0, 1, 2 или 3 Rk группами, С3-C8 циклоалкильной группы, замещенной 0, 1, 2 или 3 Rk группами, C1-С6 алкокси-группы, замещенной 0, 1, 2 или 3 Rk группами, С3-C8 циклоалкилокси-группы, замещенной 0, 1, 2 или 3 Rk группами, -SH, C1-С6 алкилтио-группы, замещенной 0, 1, 2 или 3 Rk группами, С3-C8 циклоалкилтио-группы, замещенной 0, 1, 2 или 3 Rk группами, (C1-С6 алкил)карбонильной группы, замещенной 0, 1, 2 или 3 Rk группами, (C1-С6 алкокси)карбонильной группы, замещенной 0, 1, 2 или 3 Rk группами, (C1-С6 алкил)аминокарбонильной группы, замещенной 0, 1, 2 или 3 Rk группами, 3-8-членной гетероциклоалкильной группы, замещенной 0, 1, 2 или 3 Rk группами, C1-С6 алкилсульфонильной группы, замещенной 0, 1, 2 или 3 Rk группами, -NH2, моно(C1-С6 алкил)амино-группы, замещенной 0, 1, 2 или 3 Rk группами, и ди(С1-С6 алкил)амино-группы, замещенной 0, 1, 2 или 3 Rk группами;
где Rk независимо выбран из F, C1-С4 алкильной группы, -ОН, -CN, -NO2, -NH2, -CO2H, C1-С6 алкокси-группы, моно(C1-С6 алкил)амино-группы, ди(С1-С6 алкил)амино-группы, -CF3 и оксо-группы;
Rg независимо выбран из F, Cl, C1-С6 алкильной группы, -ОН, -CN, -NH2, -NO2, -CO2H, C1-С6 алкокси-группы, моно(C1-С6 алкил)амино-группы, ди(С1-С6 алкил)амино-группы, -CF3, C1-С6 алкиленовой группы, замещенной 0, 1, 2 или 3 Rl группами, С2-С6 алкениленовой группы, замещенной 0, 1, 2 или 3 Rl группами, и оксо-группы;
где Rl независимо выбран из F, C1-C4 алкильной группы, -ОН, -CN, -NO2, -NH2, -CO2H, C1-С6 алкокси-группы, моно(C1-С6 алкил)амино-группы, ди(C1-С6 алкил)амино-группы, -CF3 и оксо-группы.
"C1-С6 алкильная группа, замещенная 0, 1, 2, 3, 4 или 5Re группами», в R4 предпочтительно представляет собой С2-С6 алкильную группу, замещенную 0, 1, 2, 3, 4 или 5Re, и более предпочтительно - трет-бутилметильную группу или 3,3,3-трифтор-2,2-диметилпропильную группу.
"(С2-С6 алкенил)(С1-С3 алкильная) группа, замещенная 0, 1, 2, 3, 4 или 5 Re группами", в R4 предпочтительно представляет собой группу, содержащую 3-6 атомов углерода в (C2-С6 алкенил)(С1-С3 алкиле), и более предпочтительно - 3-метил-2-бутен-1-ил группу.
"(С2-С6 алкинил)(С1-С3 алкильной) группа, замещенная 0, 1, 2, 3, 4 или 5Re группами", в R4 предпочтительно представляет собой группу, содержащую 4-8 атомов углерода в (С2-С6 алкинил)(С1-С3 алкиле), и более предпочтительно - 4,4-диметил-2-пентин-1-ильную группу.
"(C1-С6 алкокси)(С2-С4 алкильной) группа, замещенная 0, 1, 2, 3, 4 или 5Re группами", в R4 предпочтительно представляет собой группу, содержащую 3-7 атомов углерода в (C1-С6 алкокси)(С2-C4 алкиле), более предпочтительно - C1-C4 алкоксиэтильную группу, замещенную 0, 1, 2 или 3 алкильными группами, и еще более предпочтительно - 2,2-диметил-2-метоксиэтильную группу или 2-(трет-бутокси)этильную группу.
"(С6-С10 арил)(С1-С3 алкильной) группа, замещенная 0, 1, 2, 3, 4 или 5 Rf группами", в R4 предпочтительно представляет собой бензильную группу, замещенную 0, 1, 2, 3, 4 или 5 Rf более предпочтительно - бензильную группу, замещенную 1, 2 или 3 группами, выбранными из F и Cl, или незамещенную бензильную группу; и еще более предпочтительно представляет собой 4-фторбензильную группу, 3,5-дифторбензильную группу или 4-(трифторметил)бензильную группу.
"(5-10-членный гетероарил)(С1-С3 алкильная) группа, замещенная 0, 1, 2, 3, 4 или 5 Rf группами", в R4 предпочтительно представляет собой пиридилметильную группу, тиенилметильную группу, тиазолилметильную группу или фуранилметильную группу.
"С3-C8 циклоалкильная группа, замещенная 0, 1, 2, 3, 4 или 5 Rg группами", в R4 предпочтительно представляет собой С3-С6 циклоалкильную группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами, и более предпочтительно - 2,2-диметилциклобутильную группу или 4,4-диметилциклогексильную группу.
"(С3-C8 циклоалкил)(С1-С3 алкильная) группа, замещенная 0, 1, 2, 3, 4 или 5 Rg группами", в R4 предпочтительно представляет собой С3-С6 циклоалкилметильную группу, замещенную 0, 1, 2, 3 или 4 Rg группами; и более предпочтительно представляет собой (1-фторциклопентил)метильную группу, (3,3-диметилциклобутил)метильную группу, (1-метилциклобутил)метильную группу, (1-(трифторметил)циклобутил) метильную группу, (1-(трифторметил)циклопропил)метильную группу или (1-метилциклопропил)метильную группу.
"3-8-членная гетероциклоалкильная группа, замещенная 0, 1, 2, 3, 4 или 5 Rg группами", в R4 предпочтительно представляет собой 3-6-членную гетероциклоалкильную группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами.
" (3-8-членный гетероциклоалкил)(С1-С3 алкильная) группа, замещенная 0, 1, 2, 3, 4 или 5 Rg группами", в R4 предпочтительно представляет собой 3-6-членную гетероциклоалкилметильную группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами; более предпочтительно - тетрагидрофуранилметильную группу, замещенную 1, 2 или 3 группами, выбранными из F, C1-C4 алкильной группы и C1-С6 алкиленовой группы, замещенной 0, 1, 2 или 3 R1 группами.
"С6-С9 спироалкильная группа, замещенная 0, 1, 2, 3, 4 или 5 Rg группами", в R4 предпочтительно представляет собой C7-C8 спироалкильный цикл, замещенный 0, 1, 2, 3, 4 или 5 Rg группами; более предпочтительно - спиро[2.5]октан-1-ильную группу, спиро[3.5]нонан-1-ильную группу, спиро[3.3]гептан-1-ильную группу или спиро[3.3]гептан-2-ильную группу.
"(С6-С9 спироалкил)(С1-С3 алкильная) группа, замещенная 0, 1, 2, 3, 4 или 5 Rg группами", в R4 предпочтительно представляет собой С6-C8 спироалкилметильную группу замещенную 0, 1, 2, 3, 4 или 5 Rg группами; более предпочтительно - спиро[2.5]октан-6-илметильнуюгруппу, замещенную 0, 1, 2 или 3 Rg группами, или спиро[2.3]гексан-5-илметильную группу, замещенную 0, 1, 2 или 3 Rg группами; и еще более предпочтительно представляет собой спиро[2.5]октан-6-илметильную группу, (5-фтор-спиро[2.3]гексан)-5-илметильную группу или спиро[2.3]гексан-5-илметильную группу.
"С6-С9 спирогетероалкильная группа, замещенная 0, 1, 2, 3, 4 или 5 Rg группами", в R4 предпочтительно представляет собой C7-C8 спирогетероалкильный цикл, замещенный 0, 1, 2, 3, 4 или 5 Rg группами.
"C5-С9 бициклоалкильная группа, замещенная 0, 1, 2, 3, 4 или 5 Rg группами", в R4 предпочтительно представляет собой С6-C8 бициклоалкильное кольцо, замещенное 0, 1, 2, 3, 4 или 5 Rg группами; более предпочтительно - бицикло[3.1.0]гексан-3-ильную группу, замещенную 0, 1,2 или 3 Rg группами; и еще более предпочтительно представляет собой 6,6-диметилбицикло[3.1.0]гексан-3-ильную группу.
"(C5-С9 бициклоалкил)(С1-С3 алкильная) группа, замещенная 0, 1, 2, 3, 4 или 5 Rg группами", в R4 предпочтительно представляет собой C5-C7 бициклоалкилметильную группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами; более предпочтительно - (бицикло[1.1.1]пентан-1-ил)метильную группу, замещенную 0, 1, 2 или 3 Rg группами, или (бицикло[2.2.1]гептан-1-ил)метильную группу, замещенную 0, 1, 2 или 3 Rg группами;и еще более предпочтительно представляет собой (4-метилбицикло[2.2.1]гептан-1-ил)метильную группу или (бицикло[1.1.1]pentan-1-ил)метильную группу.
"(С6-С9 гетеробициклоалкил)(С1-С3 алкильная) группа, замещенная 0, 1, 2, 3, 4 или 5 Rg группами", в R4 предпочтительно представляет собой C6-C7 гетеробициклоалкил метильную группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами; более предпочтительно -(7-оксабицикло[2.2.1]гептан-1-ил)метильную группу, замещенную 0, 1, 2 или 3 Rg группами; и еще более предпочтительно представляет собой (4-метил-7-оксабицикло[2.2.1]гептан-1-ил)метильную группу или (7-оксабицикло[2.2.1]гептан-1-ил)метильную группу.
В формуле (I), R5 выбран из С6-С10 арильной группы, замещенной 0, 1, 2, 3, 4 или 5 Ri группами, 5-10-членной гетероарильной группы, замещенной 0, 1, 2, 3, или 4 Ri группами, С3-C8 циклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rj группами, С3-C8 циклоалкенильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rj группами, и 3-8-членной гетероциклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rj группами;
где Ri независимо выбран из F, Cl, Br, -ОН, -CN, -NO2, -CO2H, C1-С6 алкильной группы, замещенной 0, 1, 2 или 3 Rk группами, C2-С6 алкенильной группы, замещенной 0, 1, 2 или 3 Rk группами, С2-С6 алкинильной группы, замещенной 0, 1, 2 или 3 Rk группами, С3-C8 циклоалкильной группы, замещенной 0, 1, 2 или 3 Rk группами, C1-С6 алкокси-группы, замещенной 0, 1, 2 или 3 Rk группами, С3-C8 циклоалкилокси-группы, замещенной 0, 1, 2 или 3 Rk группами, -SH, C1-С6 алкилтио-группы, замещенной 0, 1, 2 или 3 Rk группами, С3-C8 циклоалкилтио-группы, замещенной 0, 1, 2 или 3 Rk группами, (C1-С6 алкил)карбонильной группы, замещенной 0, 1, 2 или 3 Rk группами, (C1-С6 алкокси)карбонильной группы, замещенной 0, 1, 2 или 3 Rk группами, (C1-С6 алкил)аминокарбонильной группы, замещенной 0, 1, 2 или 3 Rk группами, 3-8-членной гетероциклоалкильной группы, замещенной 0, 1, 2 или 3 Rk группами, C1-С6 алкилсульфонильной группы, замещенной 0, 1, 2 или 3 Rk группами, -NH2, моно(C1-С6 алкил)амино-группы, замещенной 0, 1, 2 или 3 Rk группами, и ди(C1-С6 алкил)амино-группы, замещенной 0, 1, 2 или 3 Rk группами;
Rj независимо выбран из F, Cl, C1-С6 алкильной группы, -ОН, -CN, -NH2, -NO2, -CO2H, C1-С6 алкокси-группы, моно(C1-С6 алкил)амино-группы, ди(C1-С6 алкил)амино-группы, -CF3, C1-С6 алкиленовой группы, замещенной 0, 1, 2 или 3 Rl группами, C2-С6 алкениленовой группы, замещенной 0, 1, 2 или 3 Rl группами, оксо-группы;
где в случае, когда Rj представляет собой двухвалентную группу, выбранную из C1-С6 алкиленовой группы или С2-С6 алкениленовой группы, это означает, что каждая группа формирует связи с атомами в R5; в этом случае, две связи от каждой из указанных двухвалентных групп формируются с одним или двумя разными атомами в R5;
где Rk и Rl независимо выбраны из F, C1-C4 алкильной группы, -ОН, -CN, -NO2, -NH2, -CO2H, C1-С6 алкокси-группы, моно(C1-С6 алкил)амино-группы, ди(C1-С6 алкил)амино-группы, -CF3 и оксо-группы.
"С6-С10 арильная группа, замещенная 0, 1, 2, 3, или 4 Ri группами", в R5 предпочтительно представляет собой фенильную группу, замещенную 2-4 группами, выбранными из -ОН, -NH2, Cl, F, -CN, -CF3, -OCF3, -OCF2H, метильной группы, циклопропильной группы и метокси-группы; и более предпочтительно представляет собой 2,6-дихлорфенильную группу, 2,6-дихлор-4-фторфенильную группу, 2,6-дихлор-4-метилфенильную группу, 2,4,6-трихлорфенильную группу, 2-хлор-6-фторфенильную группу или 2,6-дихлор-3-фторфенильную группу.
"5-10-членный гетероарильная группа, замещенная 0, 1, 2, 3, или 4 Ri группами", в R5 предпочтительно представляет собой пиридильную группу, замещенную 2-3 группами, выбранными из -ОН, -NH2, Cl, F, -CN, -CF3, метильной группы и метокси-группы; и более предпочтительно представляет собой 3,5-дихлорпиридин-4-ильную группу, 3-хлор-5-метоксипиридин-4-ильную группу, 3-хлор-5-фторпиридин-4-ильную группу или 2,4-дихлор-6-метилпиридин-3-ильную группу.
В целом, R5 предпочтительно представляет собой фенильную группу, опционально замещенную 2, 3 или 4 Ri группами, или 6-членную гетероарильную группу, опционально замещенную 2 или 3 Ri группами.
В формуле (I), R8 и R9 независимо выбраны из Н, F, -ОН, -NH2, C1-С3 алкильной группы, замещенной 0, 1, 2 или 3 Rh группами, и C1-С6 алкокси-группы, замещенной 0, 1, 2 или 3 Rh группами; или R8 и R9 вместе образуют оксо-группу или тиоксо-группу;
где Rh независимо выбран из F, C1-С4 алкильной группы, -ОН, -CN, -NO2, -NH2, -CO2H, C1-С6 алкокси-группы, моно(C1-С6 алкил)амино-группы, ди(C1-С6 алкил)амино-группы, -CF3 и оксо-группы.
"C1-С3 алкильная группа, замещенная 0, 1, 2 или 3 Rh группами", в R8 и R9 предпочтительно представляет собой метальную группу, замещенную 0, 1, 2 или 3 Rh группами.
"C1-С6 алкокси- группа, замещенная 0, 1, 2 или 3 Rh группами", в R8 и R9 предпочтительно представляет собой метокси-группу, замещенную 0, 1, 2 или 3 Rh группами.
В целом, R8 и R9 предпочтительно представляют собой Н, F, -ОН или оксо-группу, и более предпочтительно представляют собой Н или оксо-группу.
В формуле (I), R12 представляет собой Н; или R4 и R12 вместе представляют собой -CRmRm-CR13R14-CRmRm- или -CR13R14-CRmRm-CRmRm-, формируя пирролидиновый цикл.
R13 выбран из Н, C1-С6 алкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, С6-С10 арильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, С6-С10 арилокси-группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, (С2-С6 алкенил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (С2-С6 алкинил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (C1-С6 алкокси)(С2-C4 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (С6-С10 арил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, (5-10-членный гетероарил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, С3-C8 циклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С3-C8 циклоалкенильной группы, замещенной 0, 1, 2, 3, 4 или 5 группами, (С3-C8 циклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С3-C8 циклоалкенил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, 3-8-членной гетероциклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, и (3-8-членный гетероциклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-C9 спироалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С6-С9 спироалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 спирогетероалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 бициклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (C5-C9 бициклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С6-С9 гетеробициклоалкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, и (С6-С9 гетеробициклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами;
R14 выбран из атома водорода и C1-С6 алкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами; или R13 и R14 вместе формируют С3-C8 циклоалкановое кольцо, замещенное 0, 1, 2, 3, 4 или 5 Rg группами, С3-C8 циклоалкеновое кольцо, замещенное 0, 1, 2, 3, 4 или 5 Rg группами, или 3-8-членное гетероциклоалкановое кольцо, замещенное 0, 1, 2, 3, 4 или 5 Rg группами;
Rm независимо выбран из Н, F, Cl, -CH3 и -CF3;
где Rg выбран из F, Cl, C1-С6 алкильной группы, -ОН, -CN, -NH2, -NO2, -CO2H, C1-С6 алкокси-группы, моно(C1-С6 алкил)амино-группы, ди(C1-С6 алкил)амино-группы, -CF3, C1-С6 алкиленовой группы, замещенной 0, 1, 2 или 3 Rl группами, C2-С6 алкениленовой группы, замещенной 0, 1, 2 или 3 Rl группами, и оксо-группы;
Rf независимо выбран из F, Cl, Br, -ОН, -CN, -NO2, -CO2H, C1-С6 алкильной группы, замещенной 0, 1, 2 или 3 Rk группами, С2-С6 алкенильной группы, замещенной 0, 1, 2 или 3 Rk группами, С2-С6 алкинильной группы, замещенной 0, 1, 2 или 3 Rk группами, С3-C8 циклоалкильной группы, замещенной 0, 1, 2 или 3 Rk группами, C1-С6 алкокси-группы, замещенной 0, 1, 2 или 3 Rk группами, С3-C8 циклоалкилокси-группы, замещенной 0, 1, 2 или 3 Rk группами, -SH, C1-С6 алкилтио-группы, замещенной 0, 1, 2 или 3 Rk группами, С3-C8 циклоалкилтио-группы, замещенной 0, 1, 2 или 3 Rk группами, (C1-С6 алкил)карбонильной группы, замещенной 0, 1, 2 или 3 Rk группами, (C1-С6 алкокси)карбонильной группы, замещенной 0, 1, 2 или 3 Rk группами, (C1-С6 алкил)аминокарбонильной группы, замещенной 0, 1, 2 или 3 Rk группами, 3-8-членной гетероциклоалкильной группы, замещенной 0, 1, 2 или 3 Rk группами, C1-С6 алкилсульфонильной группы, замещенной 0, 1, 2 или 3 Rk группами, -NH2, моно(C1-С6 алкил)амино-группы, замещенной 0, 1, 2 или 3 Rk группами, и ди(C1-С6 алкил)амино-группы, замещенной 0, 1, 2 или 3 Rk группами; и
Re и Rk независимо выбраны из F, C1-C4 алкильной группы, -ОН, -CN, -NO2, -NH2, -CO2H, C1-С6 алкокси-группы, моно(C1-С6 алкил)амино-группы, ди(C1-С6 алкил)амино-группы, -CF3 и оксо-группы.
Предпочтительно, R12 представляет собой Н; или R4 и R12 вместе представляют собой -CH2-CR13R14-CH2-, формируя пирролидиновый цикл, более предпочтительно R12 представляет собой Н.
R13 предпочтительно представляет собой C1-С6 алкильную группу, С6-С10 арильную группу, С6-С10 арилокси-группу, (С6-С10 арил)(С1-С3 алкильную) группу, или С3-C8 циклоалкенильную группу.
R14 предпочтительно представляет собой Н или CH3; или R13 и R14 вместе формируют С3-C8 циклоалкановое кольцо или С3-C8 циклоалкеновое кольцо. В формуле (I), комбинация R1, R2, R3, R4, R5, R6, R7, R8, R9, R12, R13, R14 Y, n, k, Ra, Rb, Rc, Re, Rf, Rg, Rh, Ri, Rj, Rk, Rl, Rm предпочтительно является такой, где скомбинированы перечисленные выше соответствующие предпочтительные компоненты; и более предпочтительно она является такой, где скомбинированы перечисленные выше компоненты, указанные как более предпочтительные.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R1 представляет собой C1-С6 алкильную группу, замещенную 0, 1, 2 или 3 Ra группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R1 представляет собой C1 алкильную группу, замещенную 0, 1, 2 или 3 Ra группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R1 представляет собой CF3.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R2 представляет собой CO2H.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, Y выбран из формулы (II-а), формулы (II-b), формулы (II-с) и формулы (II-d):
 и
и 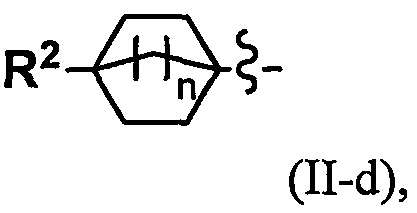
где k равно 0, 1 или 2; и n равно 1, 2 или 3.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, Y выбран из формулы (II-а) и формулы (II-d);
 или
или 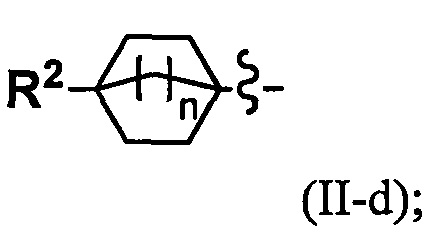
где k равно 0, 1 или 2; и n равно 1, 2 или 3.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, Y выбран из формулы (II-а) и формулы (II-d);
или
где k равно 0; и n 2.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, Y представляет собой 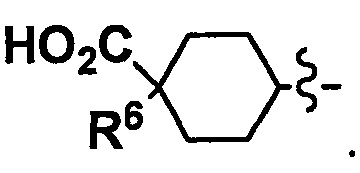
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, Y представляет собой 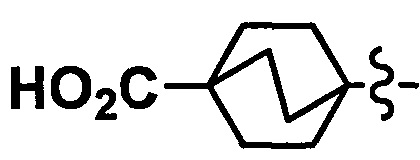 .
.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R6 выбран из F, -ОН, -NH2, -CN, C1-С6 алкильной группы, замещенной 0, 1, 2 или 3 Rb группами, и C1-С6 алкокси-группы, замещенной 0, 1, 2 или 3 Rb группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R6 представляет собой C1-С6 алкильную группу, замещенную 0, 1, 2 или 3 Rb.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R6 представляет собой CH3.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R7 независимо выбран из Н, F и C1-C6 алкильной группы, замещенной 0, 1, 2 или 3 Rb группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R7 представляет собой Н.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R2 выбран из -ОН, -CO2H, -SO3H, -CONH2 и -SO2NH2.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R3 представляет собой Н.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R4 выбран из C1-С6 алкильной группы, замещенной 0, 1, 2 или 3 Re группами, (С6-С10 арил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, С3-C8 циклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С3-C8 циклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-C9 спироалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С6-С9 спироалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, C5-С9 бициклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (C5-C9 бициклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, или (С6-С9 гетеробициклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R4 представляет собой C1-С6 алкильную группу, замещенную 0, 1, 2, 3, 4 или 5 Re группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R4 представляет собой (С6-С10 арил)(С1-С3 алкильную) группу, замещенную 0, 1, 2, 3, 4 или 5 Rf группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R4 представляет собой С3-C8 циклоалкильную группу, замещенную 0, 1, 2 или 3 Rg группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R4 представляет собой (C5-С9 бициклоалкильную) группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R4 представляет собой (С3-C8 циклоалкил)(С1-С3 алкильную) группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R4 представляет собой С6-С9 спироалкильную группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R4 представляет собой (С6-C9 спироалкил)(С1-С3 алкильную) группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R4 представляет собой (C5-C9 бициклоалкил)(С1-С3 алкильную) группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления,
R4 представляет собой (С6-С9 гетеробициклоалкил)(С1-С3 алкильную) группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R8 и R9 независимо выбраны из Н и F.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R8 и R9 вместе формируют оксо-группы.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R5 представляет собой С6-С10 арильную группу, замещенную 0, 1, 2, 3,4 или 5 Ri группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R5 представляет собой фенильную группу, замещенную 0, 1, 2, 3, 4 или 5 Ri группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R5 представляет собой 5-10-членную гетероарильную группу, замещенную 0, 1, 2, 3, или 4 Ri группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R5 представляет собой 6-членную гетероарильную группу, замещенную 0, 1, 2, 3, или 4 Ri группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R5 представляет собой пиридил, замещенный 0, 1, 2, 3, или 4 Ri группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R12 представляет собой Н.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R4 и R12 вместе представляют собой -CH2-CR13R14-CH2-, формируя пирролидиновый цикл.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R14 выбран из атома водорода и CH3. В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R13 и R14 вместе формируют С3-C8 циклоалкановое кольцо, замещенное 0, 1, 2, 3, 4 или 5 Rg группами, С3-C8 циклоалкеновое кольцо, замещенное 0, 1, 2, 3, 4 или 5 Rg группами, или 3-8-членное гетероциклоалкановое кольцо, замещенное 0, 1, 2, 3, 4 или 5 Rg группами.
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, R13 выбран из C1-С6 алкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, С6-С10 арильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, С6-С10 арилокси-группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, (С2-С6 алкенил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (С2-С6 алкинил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (C1-С6 алкокси)(С2-С4 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Re группами, (С6-С10 арил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, (5-10-членный гетероарил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rf группами, С3-С8 циклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С3-C8 циклоалкенильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С3-C8 циклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С3-C8 циклоалкенил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, 3-8-членный гетероциклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, и (3-8-членный гетероциклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-C9 спироалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (С6-С9 спироалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 спирогетероалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 бициклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (C5-C9 бициклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, С6-С9 гетеробициклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, и (С6-С9 гетеробициклоалкил)(С1-С3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами;
В другом варианте осуществления, в сочетании с любыми описанными выше или ниже вариантами осуществления, Rm представляет собой Н;
Настоящее изобретение касается также фармацевтически приемлемой соли соединения, имеющего формулу (I). Например, в настоящем изобретении описаны случаи, когда соединение, имеющее формулу (I), формирует кислотно-аддитивные соли. Кроме того, в зависимости от типа заместителя, описаны случаи, когда пиразоламидное производное образует соли с основаниями. На такие соли не накладываются какие-либо специальные ограничения, при условии, что они являются фармацевтически приемлемыми. В частности, кислотно-аддитивные соли включают соли с неорганическими кислотами, такие как гидрофторид, гидрохлорид, гидробромид, гидроиодид, фосфат, нитрат, сульфат и т.п.; органический сульфонат, такой как метансульфонат, этансульфонат, 2-гидроксиэтансульфонат, п-толуолсульфонат, бензолсульфонат, этан-1,2-дисульфонатный ион, 1,5-нафталиндисульфонатный ион, нафталин-2-сульфонатный ион и т.п.; и органический карбоксилат, такой как ацетат, трифторацетат, пропионат, оксалат, фумарат, фталат, малонат, сукцинат, глутарат, адипат, тартрат, малеат, малат, манделат, 1-гидрокси-2-нафтоат и т.п. В качестве солей с основаниями можно указать соли с неорганическими основаниями, такие как натриевые соли, калиевые соли, магниевые соли, кальциевые соли, алюминиевые соли и т.п.; и соли с органическими основаниями, такие как соли с метиламином, соли с этиламином, соли с лизином, соли с орнитином и т.п.
Раличные фармацевтически приемлемые соли соединения, имеющего формулу (I), можно получить подходящим методом, известным из предшествующего уровня техники.
Соединение, имеющее формулу (I) по настоящему изобретению, в некоторых случаях содержит изомеры. Такие изомеры включены в понятие соединения, имеющего формулу (I) по настоящему изобретению. Например, в качестве примеров можно привести изомеры в циклических и конденсированных циклических системах (Е-, Z-, цис- и трансформы), изомеры, образующиеся вследствие наличия хиральных атомов углерода (R- и S-формы, α- и β-конфигурации, энантиомеры и диастереомеры), оптически активные вещества с оптическим вращением (D-, L-, d- и l-формы), таутомеры, полярные соединения, полученные при хроматографическом разделении (высокополярное соединение и низкополярное соединение), соединения, находящиеся в равновесии, ротамеры, смеси перечисленных соединений в произвольном соотношении, рацемические смеси и т.п.
Настоящее изобретение включает также различные дейтерированные формы соединений, представленных формулой (I). Каждый атом водорода, присоединенный к атому углерода, может быть независимо заменен на атом дейтерия.
Хотя настоящее изобретение описано относительно частных аспектов или вариантов осуществления, настоящее изобретение должно пониматься на уровне современной технологии в соответствующей области, и возможны различные модификации и замены на эквиваленты без выхода за рамки сути и объема настоящего изобретения. Кроме того, в степени, допускаемой патентными законами и правилами, все публикации, патенты и заявки на патенты, процитированные в настоящем тексте, включены в настоящий текст посредством ссылки во всей их полноте, в такой же степени, как если бы каждый отдельный документ был отдельно указан как включенный в настоящий текст посредством ссылки во всей своей полноте.
Общие методы синтеза
Соединение, имеющее формулу (I) по настоящему изобретению, можно получить различными широко известными методами синтеза, учитывая особенности типов их структур или заместителей. В данном случае эффективным приемом, с точки зрения технологии производства, будет защита функциональной группы подходящей защитной группой или группой, которую можно легко превратить в функциональную группу, в процессе применения исходных веществ и промежуточных продуктов, подобранных в соответствии с целевыми функциональными группами. Такие функциональные группы включают, например, амино-группу, гидроксильную группу, карбоксильную группу и т.п. Защитные группы для них включают, например, защитные группы, описанные в книге "Protecting Groups in Organic Synthesis (третье издание, 1999)" авторов Т.W. Greene и Р.G.М. Wuts. Их можно надлежащим образом подобрать и применить в зависимости от условий реакций. В таких методах проводят реакцию введения защитной группы с последующим удалением защитной группы по необходимости или превращением в целевую группу с получением целевого соединения.
Из числа соединений, имеющих формулу (I) по настоящему изобретению, соединение (I-1) можно получить, например, следующим способом:
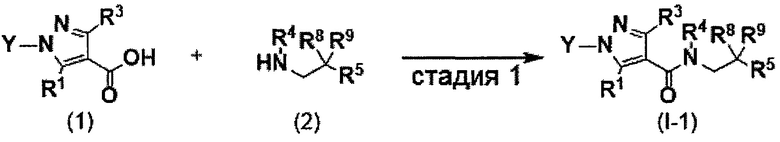
(где R8 и R9 независимо представляют собой Н; F; гидроксильную группу; амино-группу; C1-С3 алкильную группу, замещенную 0, 1, 2 или 3 Rh группами; C1-С6 алкокси-группу, замещенную 0, 1, 2 или 3 Rh группами; или R8 и R9 вместе формируют оксо-группу или тиоксо-группу. Остальные символы имеют указанные выше значения.)
(Стадия 1)
Данная стадия представляет собой способ получения соединения (I-1) реакцией соединения (1) или его реакционноспособного производного с соединением (2).
Реакционноспособное производное соединения (1) означает реакционноспособное производное по карбоксильной группе, в качестве примера можно указать хлорангидрид, ацилазид, смешанный ангидрид, симметричный ангидрид, активированный амид, активированный сложный эфир и т.п. Указанные реакционноспособные производные можно опционально подобрать в зависимости от типов применяемых карбоновых кислот.
Данную реакцию можно проводить как общую реакцию формирования амидов, описанными в литературе методами (например, Pepuchido Gousei no Kiso to Jikken by Nobuo Izumiya, etc., Maruzen, 1983, Comprehensive Organic Synthesis, Vol. 6., Pergamon Press, 1991, etc.), эквивалентными им методами или комбинацией указанных методов и общеизвестного метода. А именно, указанную реакцию можно проводить с применением конденсирующего агента, который хорошо известен квалифицированному специалисту в данной области, или методом активации сложного эфира, методом с применением смешанного ангидрида, методом с применением хлорангидрида, методом с применением карбодиимида и т.п., которые хорошо известны в данной области. Реагенты, применяющиеся в таких реакциях образования амидов, включают, например, тионил хлорид, оксалилхлорид, N,N-дициклогексилкарбодиимид, 1-метил-2-бромпиридиния иодид, N,N'-карбонилдиимидазол, дифенилфосфорил хлорид, дифенилфосфорил азид, N,N'-дисукцинимидил карбонат, N,N'-дисукцинимидил оксалат, 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид, бензотриазол-1-ил-окси-трис(пирролидинол)фосфония гексафторфосфат, 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат, 2-(5-норборнен-2,3-дикарбоксимидо)-1,1,3,3-тетраметилурония тетрафторборат, O-(N-сукцинимидил)-1,1,3,3-тетраметилурония тетрафторборат, бром-трис(пирролидино)фосфония гексафторфосфат, этил хлороформиат, изобутил хлороформиат или 2-(7-аза-1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат, и т.п. Из перечисленных предпочтительными являются, например, тионилхлорид, оксалилхлорид, 1-этил-3-(3-диметиламинопропил) карбодиимида гидрохлорид или 2-(7-аза-1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат, и т.п. В реакции образования амида можно применять основание и/или конденсирующий агент вместе с указанным выше амидообразующим агентом.
Количество применяемого конденсирующего агента строго не лимитировано, и обычно оно составляет от 0.1 эквивалентов до 100 эквивалентов на 1 эквивалент соединения (1), и предпочтительно от 0.1 эквивалентов до 10 эквивалентов.
Применяемое основание включает, например, третичный алифатический амин, такой как триметиламин, триэтиламин, N,N-диизопропилэтиламин, N-метилморфолин, N-метилпирролидин, N-метилпиперидин, N,N-диметиланилин, 1,8-диазабицикло[5.4.0]ундец-7-ен, 1,5-азабицикло[4.3.0]нон-5-ен, и т.п.; ароматические амины, такие как пиридин, 4-диметиламинопиридин, пиколин, лутидин, хинолин или изохинолин, и т.п. Более всего предпочтителен третичный алифатический амин и т.п., и особенно предпочтителен триэтиламин или N,N-диизопропилэтиламин и т.п.
Количество применяемого основания варьируется в зависимости от используемого соединения, типов растворителей и других условий реакции, однако обычно оно составляет от 0.1 эквивалентов до 100 эквивалентов на 1 эквивалент соединения (1), предпочтительно от 1 эквивалента до 5 эквивалентов.
Применяемый конденсирующий агент включает, например, N-гидроксибензотриазол гидрат, N-гидроксисукцинимид и т.п.
Количество применяемого соединения (2) варьируется в зависимости от используемого соединения, типов растворителей и других условий реакции, однако обычно оно составляет от 1 эквивалента до 10 эквивалентов на 1 эквивалент соединения (1) или его реакционноспособного производного, и предпочтительно от 1 эквивалента до 3 эквивалентов.
Реакцию обычно проводят в инертном растворителе, и примеры инертного растворителя включают тетрагидрофуран, ацетонитрил, N,N-диметилформамид, 1,4-диоксан, бензол, толуол, дихлорметан, хлороформ, тетрахлорид углерода, 1,2-дихлорэтан, пиридин и т.п., или их смеси.
Время реакции обычно составляет от 0.5 часов до 96 часов, предпочтительно от 1 часа до 24 часов.
Температура реакции в целом составляет от 0°С до температуры кипения растворителя, и предпочтительно от комнатной температуры до 80°С.
Основание, амидообразующий агент и конденсирующий агент, применяемые в данной реакции, можно применять в комбинации с одним или больше их типов.
Соединение (I-1), полученное таким образом, можно выделить и очистить методом выделения и очистки, который хорошо известен квалифицированному специалисту в данной области (например, упаривание, упаривание при пониженном давлении, кристаллизация, экстракция растворителем, переосаждение, хроматография и т.п.; в категории "общих методов синтеза", термин "способ выделения и очистки, который хорошо известен квалифицированному специалисту в данной области" имеет такой же смысл, если специально не указано иное).
Кроме того, из числа всех соединений, представленных формулой (I) по настоящему изобретению, соединения (I-2) и (I-3) можно получить, например, следующим способом:
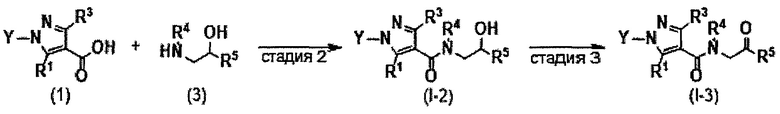
(где остальные символы имеют указанные выше значения.)
(Стадия 2)
Данная стадия представляет собой способ получения соединения (I-2) реакцией соединения (1) или его реакционноспособного производного с соединением (3).
Реакцию на данной стадии можно проводить тем же способом, как описано для стадии 1, эквивалентным ему способом или комбинацией указанных способов и общеизвестного метода.
Соединение (I-2), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
(Стадия 3)
Данная стадия представляет собой способ получения соединения (I-3) путем введения соединения (I-2) в реакцию окисления.
Данную стадию можно проводить согласно методике, хорошо известной квалифицированному специалисту в данной области. Например, окисление с помощью РСС, окисление по Сверну, окисление с помощью MnO2, окисление по Десс-Мартину, и т.п.
Например, окисление по Десс-Мартину можно проводить с применением реагента Десс-Мартина без растворителя или в растворителе, инертном к данной реакции.
Количество применяемого реагента Десс-Мартина обычно составляет от 1 эквивалента до 10 эквивалентов на 1 эквивалент соединения (I-2), предпочтительно от 1 эквивалента до 4 эквивалентов.
Реакцию на данной стадии обычно проводят в инертном растворителе. В качестве инертного растворителя можно применять, например, тетрагидрофуран, ацетонитрил, N,N-диметилформамид, диметилсульфоксид, 1,4-диоксан, бензол, толуол, дихлорметан, хлороформ, тетрахлорид углерода, 1,2-дихлорэтан и т.п.; или их смеси.
Время реакции обычно составляет от 0.5 часов до 96 часов, и предпочтительно от 1 часа до 24 часов.
Температура реакции в целом составляет от -78°С до температуры кипения растворителя, и предпочтительно от -20°С до комнатной температуры.
Соединение (I-3), полученное таким образом, можно выделить и очистить методом выделения и очистки, который хорошо известен квалифицированному специалисту в данной области.
Также, когда реагирующее вещество содержит карбоксильную группу, участвующую в реакции на первой стадии, второй стадии и третьей стадии, карбоксильную группу предпочтительно заранее защищают защитной группой, и затем защитную группу снимают по окончании реакции. Выбор такой защитной группы и условий ее удаления можно осуществить методом, описанным в ранее упомянутой книге "Protecting Groups in Organic Synthesis (третье издание, 1999)".
Кроме того, из числа соединений, представленных формулой (I) по настоящему изобретению, соединение (I-3) можно получить, например, следующим способом:
Также, из числа соединений (1), применяемых для получения соединений по настоящему изобретению, соединение (1) где R3 представляет собой Н, можно получить, например, следующим способом:
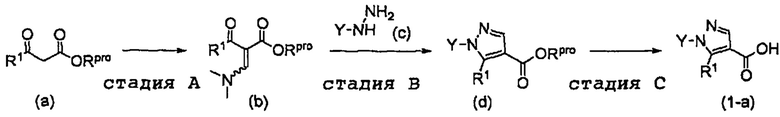
(где Rpro представляет собой защитную группу. Остальные символы имеют указанные выше значения.)
Соединение, имеющее формулу (а), можно синтезировать по методике, хорошо известной квалифицированному специалисту в данной области.
Соединение, имеющее формулу (с), можно синтезировать по методике, хорошо известной квалифицированному специалисту в данной области.
(Стадия А)
Данная стадия представляет собой способ получения соединения (b) реакцией соединения (а) с N,N-диметилформамида диметилацеталем в присутствии растворителя или без растворителя.
Также, N,N-диметилформамида диэтилацеталь, N,N-диметилформамида диизопропилацеталь и т.п. можно применять вместо N,N-диметилформамид диметилацеталя.
Количество применяемого N,N-диметилформамида диметилацеталя обычно составляет от 1 эквивалента до 10 эквивалентов на 1 эквивалент соединения (а).
Применяющийся в данной реакции растворитель не ограничен каким-либо специальным образом при условии, что он инертен к проходящей реакции, и в частности включает, например, метанол, этанол, бензол, толуол, ксилол, тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид или их смеси.
Время реакции обычно составляет от 0.5 часов до 96 часов, и предпочтительно от 1 часа до 24 часов.
Температура реакции в целом составляет от 0°С до температуры кипения растворителя, и предпочтительно от комнатной температуры до 160°С.
Соединение (b), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки с применением способов, хорошо известных квалифицированным специалистам в данной области.
(Стадия В)
Данная стадия представляет собой способ получения соединения (d) реакцией соединения (b) с соединением, имеющим гидразиновую группу, имеющим формулу (с).
Количество применяемого соединения (с) обычно составляет от 0.5 эквивалентов до 10 эквивалентов на 1 эквивалент соединения (b), и предпочтительно от 0.7 эквивалентов до 3 эквивалентов.
На данной стадии, когда соединение (с) представляет собой соль, то необходимо применять основание для нейтрализации. Примеры таких оснований включают натрия карбонат, калия карбонат, натрия бикарбонат, калия бикарбонат, натрия ацетат, калия ацетат, натрия гидроксид, калия гидроксид, лития гидроксид, триэтиламин, N,N-диизопропилэтиламин, пиридин и т.п. Количество применяющегося основания обычно составляет от 1 эквивалента до 3 эквивалентов на 1 эквивалент соединения (с).
Применяющийся в данной реакции растворитель не ограничен каким-либо специальным образом при условии, что он инертен к проходящей реакции. В частности, его примеры включают метанол, этанол, н-пропанол, н-бутанол, изопропанол, ацетонитрил, диэтиловый эфир, тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид, дихлорметан, хлороформ, бензол, толуол, ксилол или их смеси.
Время реакции обычно составляет от 0.5 часов до 96 часов, и предпочтительно от 1 часа до 24 часов.
Температура реакции в целом составляет от 0°С до температуры кипения растворителя, и предпочтительно от комнатной температуры до 100°С.
Соединение (d), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
(Стадия С)
Данная стадия представляет собой способ получения соединения (1-а) путем удаления защитной группы Rpro с соединения (d).
Снятие защитной группы можно проводить способом, описанным в ранее упомянутой книге "Protecting Groups in Organic Synthesis (третье издание, 1999)", эквивалентным ему способом или комбинацией данных способов и общеизвестного метода. Например, когда защитная группа представляет собой бензильную группу, данную бензильную группу можно удалить методом каталитического восстановления с применением водорода и палладия как катализатора, и т.п.
Соединение (1-а), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
Кроме того, из числа соединений (2), применяющихся для получения соединения по настоящему изобретению, соединение (2-а), где R8 и R9 оба представляют собой Н, можно синтезировать, например, следующим способом:
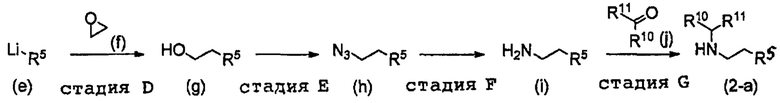
(где R10 и R11 каждый независимо представляют собой Н, группу, содержащую на один атом углерода меньше, чем углеводородная цепь в R4, или R10 и R11 вместе формируют низшую циклоалкильную или циклоалкенильную группу. Остальные символы имеют указанные выше значения.)
Соединение, имеющее формулу (f), можно синтезировать по методике, хорошо известной квалифицированному специалисту в данной области.
(Стадия D)
Данная стадия представляет собой способ получения соединения (g) реакцией литийорганического соединения (е) с этиленоксидом (f).
Количество применяемого этиленоксида (f) обычно составляет от 0.1 эквивалентов до 10 эквивалентов на 1 эквивалент соединения (е), и предпочтительно от 0.5 эквивалентов до 3 эквивалентов.
Растворитель для данной реакции не ограничен каким-либо специальным образом при условии, что он инертен к проходящей реакции, и его примеры включают тетрагидрофуран, 1,4-диоксан, диэтиловый эфир, 1,2-диметоксиэтан, н-гексан, н-гептан, дихлорметан, хлороформ, тетрахлорид углерода, 1,2-дихлорэтан, бензол, толуол, ксилол и т.п.
Время реакции обычно составляет от 0.5 часа до 48 часов, и предпочтительно от 1 часа до 24 часов.
Температура реакции в целом составляет от -78°С до температуры кипения растворителя, и предпочтительно от -78°С до комнатной температуры.
Соединение (g), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
(Стадия Е)
Данная стадия представляет собой способ получения соединения (h) реакцией соединения (g) с дифенилфосфорил азидом.
Реакцию на данной стадии можно проводить тем же способом, как описано для стадии 16, эквивалентным ему способом или комбинацией указанных способов и общеизвестного метода.
Соединение (h), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
(Стадия F)
Данная стадия представляет собой способ получения соединения (i) путем введения соединения (h) в реакцию восстановления азидной группы.
Данную стадию можно проводить по методикам, хорошо известным квалифицированному специалисту в данной области. Такие методы включают, например, метод восстановления с применением фосфина; метод каталитического восстановления с помощью водорода и палладиевого катализатора и т.п.; метод восстановления с помощью боргидрида натрия; и т.п.
Например, метод восстановления с применением фосфина можно проводить с помощью трифенилфосфина и воды в растворителе, инертном к данной реакции. В частности, его примеры включают тетрагидрофуран, ацетонитрил, N,N-диметилформамид, 1,4-диоксан, бензол, толуол, дихлорметан, хлороформ, тетрахлорид углерода, 1, 2-дихлорэтан, воду и т.п.; или их смеси.
Количество применяемого трифенилфосфина обычно составляет от 1 эквивалента до 10 эквивалентов на 1 эквивалент соединения (15), и предпочтительно от 1 до 4 эквивалентов.
Время реакции обычно составляет от 0.5 часов до 96 часов, и предпочтительно от 2 часов до 48 часов.
Температура реакции в целом составляет от 0°С до температуры кипения растворителя, и предпочтительно от комнатной температуры до температуры кипения растворителя.
Соединение (i), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
(Стадия G)
Данная стадия представляет собой способ получения соединения (2-а) реакцией соединения (i) с соединением (j) в присутствии восстановителя.
Количество применяемого на данной стадии соединения (i) обычно составляет от 0.5 эквивалентов до 10 эквивалентов на 1 эквивалент соединения (j), и предпочтительно от 0.8 эквивалентов до 4 эквивалентов.
Применяемые восстановители включают, например, боргидрид натрия, триацетоксиборгидрид натрия, цианоборгидрид натрия и т.п.
Количество применяемого восстановителя обычно составляет от 0.1 эквивалентов до 10 эквивалентов на 1 эквивалент соединения (i), и предпочтительно от 0.3 эквивалентов до 5 эквивалентов.
Применяющийся в данной реакции растворитель не ограничен каким-либо специальным образом при условии, что он инертен к проходящей реакции, и его примеры включают метанол, этанол, уксусную кислоту, тетрагидрофуран, 1,4-диоксан, дихлорметан, хлороформ, 1,2-дихлорэтан, бензол, толуол, ксилол и т.п.
Время реакции обычно составляет от 0.5 часов до 48 часов, и предпочтительно от 1 часа до 24 часов.
Температура реакции в целом составляет от 0°С до температуры кипения растворителя.
Соединение (2-а), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
Группа, имеющая следующую формулу:
(где каждый символ имеет указанное выше значение) соответствует R4.
Кроме того, из числа соединений (2), применяемых для получения соединений по настоящему изобретению, соединение (2-b) где или R8, или R9 представляет собой F, а другой из них представляет собой Н, можно синтезировать, например, следующим способом:
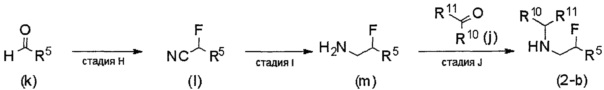
(где каждый символ имеет указанное выше значение.)
Соединение, имеющее формулу (k), можно синтезировать по методике, хорошо известной квалифицированному специалисту в данной области.
(Стадия Н)
Данная стадия представляет собой способ получения соединения (l) реакцией соединения (k) с триметилсилилцианидом в присутствии цинкового катализатора, и последующей реакцией с фторирующим агентом.
Количество применяемого триметилсилилцианида обычно составляет от 1 эквивалента до 10 эквивалентов на 1 эквивалент соединения (k), и предпочтительно от 1 эквивалента до 5 эквивалентов.
Применяемый цинковый катализатор включает, например, иодид пинка, бромид цинка и т.п.
Применяемый фторирующий агент включает, например, трифторид (N,N-диэтиламино)серы, трифторид бис(2-метоксиэтил)аминосеры, 1,1,2,2-тетрафторэтил-N,N-диметиламин и т.п.
Количество применяемого фторирующего агента обычно составляет от 1 эквивалента до 10 эквивалентов на 1 эквивалент соединения (k), и предпочтительно от 1 эквивалента до 5 эквивалентов.
Растворитель для данной реакции не ограничен каким-либо специальным образом при условии, что он инертен к проходящей реакции, и его примеры включают тетрагидрофуран, ацетонитрил, 1,4-диоксан, диэтиловый эфир, дихлорметан, хлороформ, 1,2-дихлорэтан, тетрахлорид углерода, бензол, толуол, N,N-диметилформамид и т.п.
Время реакции обычно составляет от 30 минут до 48 часов, и предпочтительно от 1 часа до 24 часов.
Температура реакции в целом составляет от 0°С до температуры кипения растворителя.
Соединение (l), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
(Стадия I)
Данная стадия представляет собой способ получения соединения (m) путем введения соединения (l) в реакцию восстановления циано-группы.
Применяемые восстановители включают, например, алюмогидрид лития, бис(2-метоксиэтокси)алюмогидрид натрия, комплекс боран-тетрагидрофуран и т.п.
Количество применяемого восстановителя обычно составляет 1 до 10 эквивалентов на 1 эквивалент соединения (l).
Растворитель для данной реакции не ограничен каким-либо специальным образом при условии, что он инертен к проходящей реакции, и его примеры включают тетрагидрофуран, 1,4-диоксан, дихлорметан, бензол, толуол, диэтиловый эфир и т.п.
Время реакции обычно составляет от 1 часа до 24 часов.
Температура реакции в целом составляет от 0°С до температуры кипения растворителя.
Соединение (m), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
(Стадия J)
Данная стадия представляет собой способ получения соединения (2-b) реакцией соединения (m) с соединением (j) в присутствии восстановителя.
Реакцию на данной стадии можно проводить тем же способом, как описано для стадии G, эквивалентным ему способом или комбинацией указанных способов и общеизвестного метода.
Соединение (2-b), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
Кроме того, из числа соединений (3), применяющихся для получения соединений по настоящему изобретению, соединение (3-Р), где или R8, или R9 представляет собой гидроксильную группу, которая защищена защитной группой, а второй из них представляет собой Н, можно синтезировать, например, следующим способом:
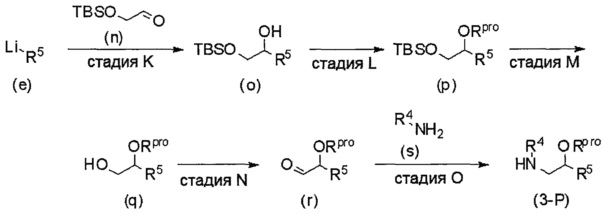
(где Rpro представляет собой защитную группу. Остальные символы имеют указанные выше значения.)
Соединение, имеющее формулу (n), можно синтезировать по методике, хорошо известной квалифицированному специалисту в данной области.
(Стадия K)
Данная стадия представляет собой способ получения соединения (о) реакцией литийорганического соединения (m) с (трет-бутилдиметилсилилокси)ацетальдегидом (n).
Реакцию на данной стадии можно проводить тем же способом, как описано для стадии D, эквивалентным ему способом или комбинацией указанных способов и общеизвестного метода.
Соединение (о), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
(Стадия L)
Данная стадия представляет собой способ введения защитной группы для гидроксильной группы соединения (о). Введение защитной группы можно проводить способом, описанным в ранее упомянутой книге "Protecting Groups in Organic Synthesis (третье издание, 1999)", эквивалентным ему способом или комбинацией указанных способов и общеизвестного метода.
Соединение (р), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
(Стадия М)
Данная стадия представляет собой способ получения соединения (q) путем удаления трет-бутилдиметилсилильной группы из соединения (р).
Снятие защитной группы можно проводить способом, описанным в ранее упомянутой книге "Protecting Groups in Organic Synthesis (третье издание, 1999)", эквивалентным ему способом или комбинацией указанных способов и общеизвестного метода, и, например, можно применять фторид тетрабутиламмония.
Соединение (q), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
(Стадия N)
Данная стадия представляет собой способ получения соединения (r) путем введения соединения (q) в реакцию окисления.
Реакцию на данной стадии можно проводить тем же способом, как описано для стадии 3, эквивалентным ему способом или комбинацией указанных способов и общеизвестного метода.
Соединение (r), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
(Стадия О)
Данная стадия представляет собой способ получения соединение (3-Р) реакцией соединения (r) с соединением (s) в присутствии восстановителя.
Реакцию на данной стадии можно проводить тем же способом, как описано для стадии г, эквивалентным ему способом или комбинацией указанных способов и общеизвестного метода.
Соединение (3-Р), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
Кроме того, из числа соединений (2), применяемых для получения соединений по настоящему изобретению, соединение (2-е), где R8 и R9 оба представляют собой F, можно синтезировать, например, следующим способом:
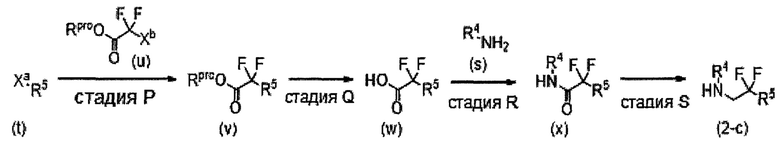
(где Xa и Xb каждый независимо представляют собой Br или I. Остальные символы имеют указанные выше значения.)
Соединение, имеющее формулу (u), можно синтезировать по методике, хорошо известной квалифицированному специалисту в данной области.
(Стадия Р)
Данная стадия представляет собой способ получения соединения (v) реакцией соединения (t) с соединением (u) в присутствии меди.
Количество применяемого соединения (t) обычно составляет от 1 эквивалента до 10 эквивалентов на 1 эквивалент соединения (u), и предпочтительно от 1 эквивалента до 3 эквивалентов.
Количество применяемой меди обычно составляет от 1 эквивалента до 10 эквивалентов на 1 эквивалент соединения (t), и предпочтительно от 1 эквивалента до 5 эквивалентов.
Применяющийся в данной реакции растворитель не ограничен каким-либо специальным образом при условии, что он инертен к проходящей реакции, и его примеры включают тетрагидрофуран, ацетонитрил, 1,4-диоксан, диметилсульфоксид, N,N-диметилформамид и т.п.
Время реакции обычно составляет от 30 минут до 48 часов.
Температура реакции в целом составляет от комнатной температуры до температуры кипения растворителя.
Соединение (v), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
(Стадия Q)
Данная стадия представляет собой способ получения соединения (w) путем удаления защитной группы Rpro из соединения (v).
Реакцию на данной стадии можно проводить тем же способом, как описано для стадии С, эквивалентным ему способом или комбинацией указанных способов и общеизвестного метода.
Соединение (w), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки с применением способов, хорошо известных квалифицированным специалистам в данной области.
(Стадия R)
Данная стадия представляет собой способ получения соединения (х) реакцией соединения (w) или его реакционноспособного производного с соединением (s).
Реакцию на данной стадии можно проводить тем же способом, как описано для стадии 1, эквивалентным ему способом или комбинацией указанных способов и общеизвестного метода.
Соединение (х), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
(Стадия S)
Данная стадия представляет собой способ получения соединения (2-c) восстановлением амидной группы соединения (х).
Реакцию на данной стадии можно проводить тем же способом, как описано для стадии I, эквивалентным ему способом или комбинацией указанных способов и общеизвестного метода.
Соединение (2-c), полученное таким образом, можно вводить в следующую стадию без выделения и очистки или после выделения и очистки методом, который хорошо известен квалифицированному специалисту в данной области.
Кроме того, соединение, имеющее формулу (I) по настоящему изобретению, может в некоторых случаях иметь таутомер и/или оптический изомер, в зависимости от типов заместителей. Однако, настоящее изобретение включает смесь таких таутомеров и изомеров, а также выделенные изомеры.
Кроме того, настоящее изобретение касается фармацевтически приемлемого пролекарства соединения, имеющего формулу (I). Термин "фармацевтически приемлемое пролекарство" означает соединение, дающее соединение, имеющее формулу (I), при сольволизе или превращении в CO2H, NH2, ОН и т.д. в физиологических условиях. Пример такой группы, которая дает пролекарство, можно найти, например, в работе Prog. Med., 5, 2157-2161 (1985), "Iyakuhin no Kaihatsu" (Hirokawa Shoten, 1990) Vol. 7., Bunshi Sekkei 163-198. В настоящем изобретении, некоторые из соединений, входящих в объем формулы (I), которые имеют группу, дающую пролекарство, могут служить пролекарством для соответствующего соединения формулы (I), которое содержит СО2Н, NH2, ОН и т.д.. Например, соединение, входящее в объем формулы (I), которое содержит алкоксикарбонильную группу, можно превратить в соответствующее производное карбоновой кислоты.
Настоящее изобретение касается также фармацевтически приемлемой соли соединения, имеющего формулу (I), и его фармацевтически приемлемого пролекарства. Такая соль включает, например, соль с галогеноводородом, таким как хлористоводородная кислота, фтористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота и т.п.; соли с неорганическими кислотами, такими как серная кислота, азотная кислота, фосфорная кислота, угольная кислота и т.п.; соли с низшими сульфокислотами, такими как метансульфокислота, этансульфокислота и т.п.; соли с арилсульфокислотами, такими как бензолсульфокислота, п-толуолсульфокислота и т.п.; соли с органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, унтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота и т.п.; и кислотно-аддитивные соли с аминокислотами, включая аспарагиновую кислоту, глутаминовую кислота и т.п. Кроме того, в зависимости от типов заместителей, соединения в настоящем изобретении могут образовывать соль с основанием. Примеры включают неорганические основания, включая такие металлы, как натрий, калий, магний, кальций, алюминий, литий и т.п.; соли с органическими основаниями, такими как метиламин, этиламин, этаноламин, гуанидин, лизин, орнитин и т.п.; и аммониевую соль и т.п.
Различные фармацевтически приемлемые соли соединения, имеющего формулу (I), можно синтезировать на основе информации, имеющейся в предшествующем уровне техники.
Соединение, имеющее формулу (I), и его фармацевтически приемлемая соль по настоящему изобретению (далее по тексту общим термином для них будет «соединение по настоящему изобретению») имеет прекрасную RORγ ингибирующую активность и может применяться как RORγ ингибитор, клинически применимый для лечения или профилактики связанных с RORγ заболеваний и симптомов. Из числа связанных с RORγ заболеваний, соединение по настоящему изобретению может применяться как терапевтическое средство или средство профилактики для, в частности, заболеваний, выбранных из аутоиммунных заболеваний и воспалительных заболеваний (например, множественный склероз, хронический ревматоидный артрит, анкилозирующий спондилит, системный эритоматоз, псориаз, псориатический артрит, воспалительная болезнь кишечника (например, болезнь Крона) и астма), болезней обмена веществ (в особенности, диабета) и раковых заболеваний (в особенности, злокачественной меланомы).
Кроме того, термин "профилактика" в настоящем изобретении означает процедуру введения фармацевтической композиции, содержащей соединение по настоящему изобретению, или ее введение пациентам, у которых еще не развилось заболевание или симптомы. Кроме того, термин "лечение" означает процедуру введения фармацевтической композиции, содержащей соединение по настоящему изобретению, или ее введение пациентам, у которых уже развилось заболевание или симптомы. Соответственно, процедура введения пациентам, у которых уже развилось заболевание или симптомы, в целях предотвращения осложнений или приступов, является одним из аспектов "лечения".
Когда соединение по настоящему изобретению применяется в качестве лекарственного средства, соединение по настоящему изобретению можно смешивать с фармацевтически приемлемым носителем (разбавителем, связующим средством, разрыхлителем, ароматизатором, отдушкой, эмульгатором, разбавителем, солюбилизатором и т.п.) и вводить в форме фармацевтической композиции или лекарственного препарата (препарата для перорального введения, инъекций и т.п.) перорально или парэнтерально. Фармацевтическую композицию можно готовить общеизвестным способом.
В настоящем описании, парэнтеральное введение включает подкожное введение, внутривенное введение, внутримышечное введение, интралеритонеальное введение, инфузионные методы и местное введение (чрескожное введение, введение в глаза, ингаляционное/бронхиальное введение, назальное введение, ректальное введение и т.п.) и т.п. Дозированные формы для перорального введения включают, например, таблетки, пилюли, гранулы, порошки, растворы, суспензии, сиропы, капсулы и т.п.
Количество соединения по настоящему изобретению, которое можно комбинировать с носителем, можно изменять в зависимости от конкретного пациента, получающего лечение, и от конкретной дозированной формы. В этой связи, конкретную дозировку для конкретного пациента определяют в зависимости от различных факторов, включая возраст, вес тела, общее состояние здоровья, пол, диету, время введения, метод введения, скорость экскреции и тяжесть заболевания во время лечения.
Количественную дозировку соединения по настоящему изобретению определяют в зависимости от возраста, веса тела, общего состояния здоровья, пола, диеты, времени введения, метода введения, скорости экскреции, тяжести заболевания у пациента, подвергающегося лечению, или с учетом других факторов. Соединение по настоящему изобретению можно вводить один или несколько раз в сутки, для взрослых в диапазоне от 0.01 мг до 1000 мг, хотя дозировка может быть различной, в зависимости от состояния пациента, веса тела, типа соединения, способам введения и т.п.
Сокращения
3-оксид гексафторфосфат
Примеры
Далее настоящее изобретение будет разъяснено на частных примерах. Однако, настоящее изобретение не ограничивается приведенными примерами.
Если не указано иное, реагенты, исходные вещества и растворители были куплены у коммерческих поставщиков (например, Aldrich, Wako Junyaku, Tokyo Kasei, Fluka, Sigma и т.п.) и использованы без дополнительной очистки.
Структуру новых выделенных соединений подтверждали масс-спектрометрией с применением одноквадрупольного прибора, оснащенного источником электроспрея, и другими подходящими аналитическими методами.
Для соединений, для которых регистрировался спектр ЯМР (300 МГц, 400 МГц или 500 МГц, MeOH-d4, ДМСО-d6, CD3CN или CDCl3), приведены значение химсдвига (δ: м.д.) и констант спин-спинового взаимодействия (J: Гц). Кроме того, перечисленные далее сокращения имеют указанные значения, соответственно: с = синглет, д = дублет, т = триплет, кв = квартет, ушир.с = уширенный синглет, м = мультиплет.
Соединения, синтезированные согласно описанным в примерах способам, были дополнительно проанализированы методом высокоэффективной жидкостной хроматографии, объединенной с масс-спектрометрией (LC/MS). В результатах масс-спектрометрии, наблюдаемое значение [М+Н]+, то есть полученное значение, показано как значение молекулярной массы соединения (М) плюс протон (Н+).
Условия проведения LCMS анализа: (UPLC/MS)
LC масс спектрометр: Waters Corporation AcquityUPLC™-SQD
Колонка: Acquity UPLC™ ВЕН С18 1.7 мкм 2.1 мм × 50 мм
УФ: PDA детектор (254 нм)
CAD: CORONA™ ULTRA детектор
Температура колонки: 40°С
ES напряжение: 3.0 кВ (капиллярное)
Напряжение на конусе: 30 В
Условия градиента:
Растворители: A: H2O/MeCN = 95/5
0.05% ТФУК
В: Н2О/MeCN = 5/95
0.05% ТФУК
Скорость потока: 0.6 мл/мин
Градиенты:
0.01-0.20 мин. Растворитель В: 2%, Растворитель А: 98%
0.20-3.0 мин, Растворитель В: от 2% до 100%, Растворитель А: от 98% до 0%
3.0-4.2 мин. Растворитель В: 100%, Растворитель А: 0%
4.2-4.21 мин, Растворитель В: от 100% до 2%, Растворитель А: от 0% до 98%
4.21-5.2 мин, Растворитель В: 2%, Растворитель А: 98%
5.2-5.5 мин. Растворитель В: 2%, Растворитель А: 98%, Скорость потока: 0.2 мл/мин
Условия проведения LCMS анализа (LC/MS метод А):
LC масс спектрометр: Agilent Technologies Corporation 1260 INFINITY™ HPLC-6130MSD
Колонка: Phenomenex Gemini™ C18 A110 3 мкм 4.6 мм × 30 мм
УФ: PDA детектор (254 нм)
Температура колонки: 40°С
Капиллярное напряжение: 3.5 кВ
Напряжение на фрагменторе: 70 В
Условия градиента:
Растворители: A: H2O/MeCN = 95/5
0.05% ТФУК
В: H2O/MeCN = 5/95
0.05% ТФУК
Скорость потока: 1.0 мл/мин
Градиенты:
0.01-0.30 мин. Растворитель В: от 2% до 10%, Растворитель А: от 98% до 90%
0.30-1.5 мин. Растворитель В: от 10% до 100%, Растворитель А: от 90% до 0%
1.5-3.5 мин. Растворитель В: 100%, Растворитель А: 0%
3.5-3.51 мин. Растворитель В: от 100% до 2%, Растворитель А: от 0% до 98%
3.51-4.5 мин. Растворитель В: 2%, Растворитель А: 98%
Условия проведения LCMS анализа (LC/MS метод В):
LC масс спектрометр: Shimadzu Corporation LCMS-2010 EV
Колонка: Shim-pack™ XR-ODII 2.0 мм × 75 мм
УФ: PDA детектор (254 нм)
Скорость потока: 0.4 мл/мин
Температура колонки: 40°С
Напряжение детектирования: 1.20 кВ
Условия градиента:
Растворители: A: H2O/MeCN = 90/5
0.1% HCO2H
В: H2O/MeCN=10/95
0.1%HCO2H
Скорость потока: 0.4 мл/мин
Градиенты:
0.01-0.50 мин, Растворитель В: 10%, Растворитель А: 90%
0.50-2.0 мин. Растворитель В: от 10% до 95%, Растворитель А: от 90% до 5%
2.0-3.8 мин. Растворитель В: 95%, Растворитель А: 5%
3.8-4.0 мин. Растворитель В: от 95% до 10%, Растворитель А: от 5% до 90%
4.0-5.0 мин. Растворитель В: 10%, Растворитель А: 90%
Справочный пример А1
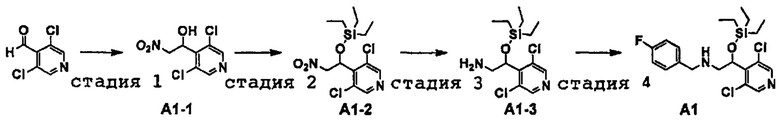
Стадия 1: 1-(3,5-дихлорпиридин-4-ил)-2-нитроэтанол (А1-1)
В раствор 3,5-дихлор-4-пиридинкарбоксальдегида (2.3 г, 13.3 ммоль) в МеОН (25 мл) добавляли нитрометан (2.2 мл, 39.9 ммоль) и метоксид натрия (861 мг, 15.9 ммоль). После добавления смесь перемешивали 1 час. Реакционную смесь гасили добавлением 2 М водного раствора HCl (7 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором поваренной соли (2 раза) и сушили над MgSO4. После удаления растворителя, остаток очищали методом колоночной хроматографии на силикагеле, получая соединение А1-1 (2.8 г, 90%) в виде белого твердого вещества.
Стадия 2: 3,5-дихлор-4-(2-нитро-1-((триэтилсилил)окси)этил)пиридин (А1-2)
В раствор соединения А1-1 (2.8 г, 11.9 ммоль) в ДМФА (15 мл) добавляли имидазол (973 мг, 14.3 ммоль) и триэтилхлорсилан (2.2 мл, 13.1 ммоль). После добавления смесь перемешивали 1 час. Реакционную смесь гасили водой и экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором поваренной соли (2 раза) и сушили над MgSO4. После удаления растворителя остаток очищали методом колоночной хроматографии на силикагеле, получая соединение А1-2 (4.1 г, 98%) в виде бесцветного масла.
Стадия 3: 2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этанамин (А1-3)
Соединение А1-2 (4.1 г, 11.6 ммоль) и никель Ренея 2800 (690 мг, в воде) в МеОН (50 мл) гидрировали в атмосфере водорода (1 атм) при комнатной температуре 8 часов. Реакционную смесь фильтровали через слой целита и промывали этилацетатом. После удаления растворителя остаток очищали методом колоночной хроматографии на силикагеле, получая соединение А1-3 (1.9 г, 50%) в виде белого твердого вещества.
Стадия 4: 2-(3,5-дихлорпиридин-4-ил)-N-(4-фторбензил)-2-((триэтилсилил)окси)этанамин (А1)
В раствор соединения А1-3 (2.8 г, 11.9 ммоль) в толуоле (6 мл) и МеОН (6 мл) добавляли 4-фторбензальдегид (360 мкл, 3.4 ммоль), и полученную смесь перемешивали при 70°С в течение 2 часов. Реакционную смесь охлаждали до 0°С, и постепенно NaBH4 добавляли. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь гасили водой и экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором поваренной соли (2 раза) и сушили над безводным Na2SO4. После удаления растворителя остаток очищали методом колоночной хроматографии на силикагеле, получая соединение А1 (1.2 г, 88%) в виде бесцветного масла.
Справочный пример А12
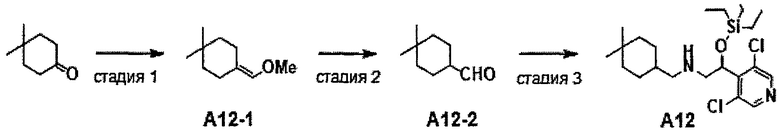
Стадия 1: 4-(метоксиметилен)-1,1-диметилциклогексан (А12-1)
н-BuLi (2.6 М раствор в гексане, 2.3 мл, 5.94 ммоль) добавляли по каплям в перемешиваемый раствор (метоксиметил)трифенилфосфония хлорида (2.04 г, 5.94 ммоль) в ТГФ (20 мл) при -78°С и перемешивали 10 минут при той же температуре и затем перемешивали 2.5 часа при комнатной температуре. Реакционную смесь охлаждали до -78°С, медленно добавляли раствор 4,4-диметилциклогексанона (500 мг, 3.96 ммоль) в ТГФ (5 мл) при -78°С. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили насыщенным водным раствором NaHCO3 (20 мл) и экстрагировали этилацетатом. Объединенные органические слои сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение А12-1 (512.2 мг, неочищенное) в виде бледно-желтого масла. Полученный неочищенный продукт использовали в следующей стадии без очистки.
Стадия 2: 4,4-диметилциклогексанкарбальдегид (А12-2)
ТФУК (2 мл) добавляли в перемешиваемый раствор соединения А12-1 (512.2 мг, неочищенное) в ДХМ (1 мл) при комнатной температуре и перемешивали 1.5 часа при той же температуре. Реакционную смесь гасили насыщенным водным раствором NaHCO3 (10 мл) и экстрагировали этилацетатом. Объединенный органический слой сушили над безводным Na2SO4, и упаривали при пониженном давлении, получая сырое соединение А12-2 в виде бледно-желтого масла. Полученный неочищенный продукт использовали в следующей стадии без очистки.
Стадия 3: 2-(3,5-дихлорпиридин-4-ил)-N-((4,4-диметилциклогексил)метил)-2-((триэтилсилил)окси)этанамин (А12)
Сырой А12-2 (52 мг) и амин А1-3 (100 мг, 311.2 ммоль) добавляли в раствор МеОН (1 мл) и толуола (1 мл) и перемешивали при 80°С 4 часа. Реакционную смесь охлаждали до комнатной температуры. МеОН (2 мл) добавляли в реакционную смесь, и добавляли в реакционную смесь NaBH4 (100 мг) при комнатной температуре. Полученную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь гасили насыщенным водным раствором NaHCO3 (10 мл) и экстрагировали этилацетатом (50 мл). Органический слой промывали насыщенным водным раствором NaHCO3 и насыщенным водным раствором поваренной соли, сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом препаративной тонкослойной хроматографии (Merck KGaA, PLC Silicagel 60 F254, 1 мм, 20×20 см с концентрирующей зоной 20×4 см, 20% EtOAc/гексан в качестве элюента), получая соединение А12 (58.6 мг, 42%) в виде бледно-желтого масла. 1Н ЯМР (CDCl3, 400 МГц): δ 8.42 (с, 2Н), 5.49 (дд, J=8.8, J=4.4 Гц, 1Н), 3.21 (дд, J=12.5, J=8.8 Гц, 1Н), 2.77 (дд, J=12.5, J=4.4 Гц, 1Н), 2.54-2.47 (м, 2Н), 1.54-1.04 (м, 9Н), 0.90-0.86 (м, 15Н), 0.62-0.49 (м, 6Н).
Справочный пример A31
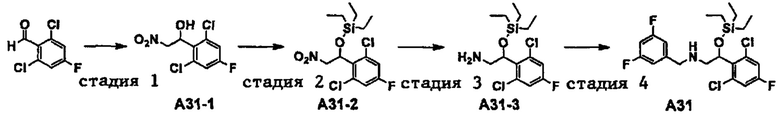
Стадия 1:1-(2,6-дихлор-4-фторфенил)-2-нитроэтанол (А31-1)
Смесь 2,6-дихлор-4-фторбензальдегида (10.0 г, 51.8 ммоль), нитрометана (2 мл) и K2CO3 (3.57 г, 25.9 ммоль) перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили водой и экстрагировали этилацетатом (2×100 мл). Объединенные органические слои промывали водой (2×50 мл) и насыщенным водным раствором поваренной соли (20 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение A31-1 (26.0 г, неочищенное) в виде желтой смолы. Полученный неочищенный продукт использовали в следующей стадии без дополнительной очистки.
Стадия 2: (1-(2,6-дихлор-4-фторфенил)-2-нитроэтокси)триэтилсилан (A31-2)
В перемешиваемый раствор соединения A31-1 (26.0 г, 102.3 ммоль) в ДМФА (100 мл) добавляли имидазол (20.9 г, 307.0 ммоль) и TES-Cl (25.7 мл, 153.5 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 1 часа. После окончания реакции, смесь гасили водой (50 мл) и экстрагировали этилацетатом (2×100 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (2×50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 0-10% EtOAc/гексан в качестве элюента), получая соединение А31-2 (32.8 г, 74%) в виде бесцветной смолы. 1H ЯМР (CDCl3, 400 МГц): δ 7.12 (с, 1Н), 7.10 (с, 1Н), 6.22 (дд, J=9.2, J=3.2 Гц, 1Н), 5.22-5.11 (м, 1Н), 4.42 (дд, J=12.2, J=3.6 Гц, 1Н), 0.84 (т, J=8.0 Гц, 9Н), 0.55-0.50 (м, 6Н).
Стадия 3: 2-(2,6-дихлор-4-фторфенил)-2-((триэтилсилил)окси)этанамин (A31-3)
В перемешиваемый раствор соединения А31-2 (15.0 г, 40.7 ммоль) в смеси EtOH/вода (60 мл, 4:1) добавляли порошок железа (22.7 г, 407.6 ммоль) и твердый NH4Cl (21.8 г, 407.6 ммоль). Полученную смесь перемешивали при 70°С 1 час. Реакционную смесь фильтровали через слой целита, промывали этилацетатом (3×150 мл) и растворитель удаляли при пониженном давлении. Остаток от упаривания суспендировали в воде (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (100 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 5% МеОН/ДХМ в качестве элюента), получая соединение A31-3 (13.0 г, 94%) в виде бесцветного масла. 1Н ЯМР (CDCl3, 400 МГц): δ 7.06 (с, 1Н), 7.04 (с, 1Н), 5.29 (дд, J=8.4, J=5.0 Гц, 1Н), 3.25 (дд, J=13.2, J=8.8 Гц, 1Н), 2.89 (дд, J=13.2, J=5.0 Гц, 1Н), 0.88 (т, J=8.0 Гц, 9Н), 0.57-0.52 (м, 6Н).
Стадия 4:2-(2,6-дихлор-4-фторфенил)-N-(3,5-дифторбензил)-2-((триэтилсилил)окси) этанамин (А31)
В перемешиваемый раствор соединения A31-3 (30.0 г, 88.7 ммоль) в МеОН (200 мл) добавляли 3,5-дифторбензальдегид (12.6 г, 88.7 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 2 часов. По окончании образования имина (мониторинг методом ТСХ), добавляли порциями твердый NaBH4 (4.9 г, 133.1 ммоль) при 0°С. Полученную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. Реакционную смесь гасили водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (2×75 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 10% EtOAc/гексан в качестве элюента), получая соединение А31 (30.0 г, 70%) в виде бесцветной смолы.
Справочный пример A35
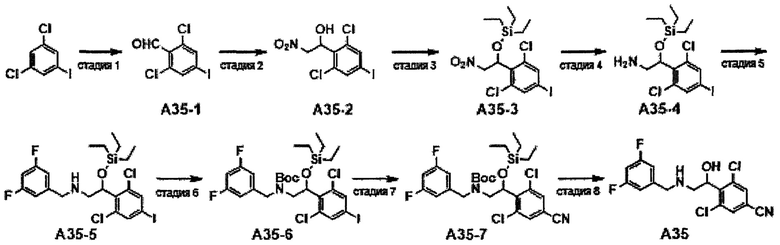
Стадия 1: 2,6-дихлор-4-иодбензальдегид (А35-1)
В перемешиваемый раствор 1,3-дихлор-5-иодбензола (4.0 г, 14.6 ммоль) в ТГФ (30 мл) добавляли по каплям LDA (2.0 М раствор в смеси ТГФ/гептан/этилбензол, 9.6 мл, 16.9 ммоль) при -78°С и перемешивали 1 час при той же температуре. Раствор ДМФА (1.7 мл, 22.0 ммоль) в ТГФ (5 мл) медленно добавляли при -78°С и перемешивали 3 часа. Реакционную смесь гасили насыщенным раствором NH4Cl (50 мл) и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным N2SO4 и упаривали при пониженном давлении. Полученный неочищенный продукт очищали методом колоночной хроматографии (силикагель, 20% EtOAc/гексан в качестве элюента), получая соединение А35-1 (1.4 г, 32%) в виде бесцветного масла.
Стадия 2: 1-(2,6-дихлор-4-иодфенил)-2-нитроэтанол (A35-2)
Соединение A35-2 (1.84 г, неочищенное) получали в виде бесцветной смолы реакцией соединения А35-1 (1.4 г, 4.8 ммоль) и K2CO3 (0.23 г, 2.0 ммоль) в CH3NO2 (10 мл) по методике, аналогичной описанной в справочном примере А1, стадия 1.
Стадия 3: (1-(2,6-дихлор-4-иодфенил)-2-нитроэтокси)триэтилсилан (А35-3)
Соединение А35-3 (2.4 г, неочищенное) получали в виде бесцветной смолы реакцией соединения А35-2 (1.84 г, 5.08 ммоль), TES-Cl (1.02 мл, 6.12 ммоль) и имидазола (1.03 г, 15.2 ммоль) в ДМФА (10 мл) по методике, аналогичной описанной в справочном примере А1, стадия 2.
Стадия 4: 2-(2,6-дихлор-4-иодфенил)-2-((триэтилсилил)окси)этанамин (A35-4)
Соединение А35-4 (2.2 г, неочищенное) получали в виде коричневого масла реакцией соединения А35-3 (2.4 г, 5.0 ммоль), Fe (2.83 г, 50.0 ммоль) и NH4Cl (2.68 г, 50.0 ммоль) в смеси EtOH/вода (4:1, 20 мл) по методике, аналогичной описанной в справочном примере А31, стадия 3.
Стадия 5: 2-(2,6-дихлор-4-иодфенил)-N-((3,5-дифторфенил)((триэтилсилил)окси)метил) этанамин (A35-5)
Соединение А35-5 (1.87 г, 67%) получали в виде бесцветной смолы реакцией соединения А35-4 (2.2 г, 5.0 ммоль), 3,5-дифторбензальдегида (0.55 мл, 5.0 ммоль) и NaBH4 (0.38 г, 10.0 ммоль) в МеОН (15 мл) по методике, аналогичной описанной в Примере A31, стадия 4.
Стадия 6: трет-бутил (2-(2,6-дихлор-4-иодфенил)-2-((триэтилсилил)окси)этил)(3,5-дифторбензил)карбамат (A35-6)
В перемешиваемый раствор соединения А35-5 (1.87 г, 3.26 ммоль) в смеси ДХМ/вода (4:1, 20 мл) добавляли NaHCO3 (0.55 г, 6.5 ммоль) и (Вос)2О (1.07 г, 4.9 ммоль) в ДХМ (8 мл) при 0°С. Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили водой (100 мл) и экстрагировали дихлорметаном (2×30 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение А35-6 (2.47 г, неочищенное) в виде бесцветной смолы.
Стадия 7: трет-бутил (2-(2,6-дихлор-4-цианофенил)-2-((триэтилсилил)окси)этил)(3,5-дифторбензил)карбамат (А35-7)
В раствор соединения А35-6 (2.0 г, 2.9 ммоль) в DMA (10 мл) в герметично закрытой пробирке добавляли Zn(CN)2 (0.7 г, 5.9 ммоль) и Pd(PPh3)4 и перемешивали 2 часа при 80°С. Реакционную смесь гасили водой (50 мл) и экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Полученный неочищенный продукт очищали методом колоночной хроматографии (силикагель, 20% EtOAc/гексан в качестве элюента), получая соединение А35-7 (1.1 г, 61%) в виде бесцветного масла.
Стадия 8: 3,5-дихлор-4-(2-((3,5-дифторбензил)амино)-1-гидроксиэтил)бензонитрил (A35)
В перемешиваемый раствор соединения А35-7 (0.2 г, 0.3 ммоль) в EtOH (10 мл) добавляли 4 М HCl (5 мл), и полученную смесь перемешивали при 80°С в течение ночи. Реакционную смесь гасили водой (50 мл), подщелачивали 10%-ным раствором NaOH до рН 9 и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Полученный неочищенный продукт очищали методом колоночной хроматографии (силикагель, 30% EtOAc/гексан в качестве элюента), получая соединение А35 (0.12 г, 99%) в виде бесцветного масла.
Справочный пример А56
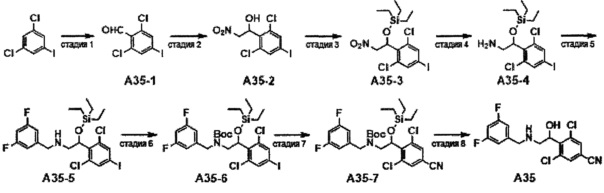
Стадия 1: 4-метилтиофен-3-карбоновая кислота (А56-1)
В перемешиваемый раствор 3-бром-4-метилтиофена (2.7 г, 15.6 ммоль) в ТГФ (35 мл) добавляли н-BuLi (1.6 М раствор в гексане, 14.6 мл, 23.3 ммоль) при -78°С по каплям в течение 15 минут, и полученную смесь перемешивали при -78°С 30 минут. Газообразный СО2 пропускали через реакционную смесь в течение 10 минут, и полученную смесь перемешивали при той же температуре 20 минут. После этого реакционную смесь нагревали до 0°С, гасили 1 М водным раствором NaOH (60 мл) и промывали этилацетатом (2×50 мл). Водный слой подкисляли до рН ~ 5 и экстрагировали дихлорметаном (2×50 мл). Объединенные органические слои промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 8% МеОН/ДХМ в качестве элюента), получая соединение А56-1 (1.5 г, 70%) в виде белого твердого вещества.
Стадия 2: 2,4-диметилтиофен-3-карбоновая кислота (А56-2)
В перемешиваемый раствор соединения А56-1 (390 мг, 2.7 ммоль) в ТГФ (4 мл) добавляли н-BuLi (1.6 М раствор в гексане, 3.8 мл, 6.0 ммоль) по каплям при -78°С в течение 10 минут. Полученную смесь перемешивали при -78°С в течение 5 минут. Добавляли по каплям раствор иодметана (0.4 мл, 6.8 ммоль) в ТГФ (1 мл), и результирующую смесь перемешивали 30 минут при -78°С. Полученную смесь оставляли нагреваться до комнатной температуры и перемешивали при той же температуре 15 часов. Реакционную смесь гасили насыщенным водным раствором NH4Cl и экстрагировали этилацетатом (2×25 мл). Объединенные органические слои промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 2% МеОН/ДХМ в качестве элюента), получая соединение А56-2 (246 мг, 57%) в виде белого твердого вещества.
Стадия 3: (2,4-диметилтиофен-3-ил)метанол (А56-3)
В перемешиваемый раствор соединения А56-2 (246 мг, 1.5 ммоль) в ТГФ (3 мл) добавляли ВН3⋅ТГФ (1 М раствор в ТГФ, 5.5 мл, 5.5 ммоль) по каплям при 0°С в течение 15 минут. Полученную смесь оставляли нагреваться до комнатной температуры и перемешивали при той же температуре 15 часов. Реакционную смесь гасили насыщенным водным раствором NaHCO3 и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (2×10 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 25% EtOAc/гексан в качестве элюента), получая соединение А56-3 (201 мг, 90%) в виде бесцветной смолы.
Стадия 4: 2,4-диметилтиофен-3-карбальдегид (А56-4)
В перемешиваемый раствор соединения А56-3 (740 мг, 5.2 ммоль) в ДХМ (18 мл) добавляли периодинан Десс-Мартина (4.6 г, 10.9 ммоль) при 0°С, и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь гасили насыщенным водным раствором Na2S2O3 и NaHCO3, и экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (2×20 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 10% EtOAc/гексан в качестве элюента), получая соединение А56-4 (275 мг, 38%) в виде желтого твердого вещества.
Стадия 5: 1-(2,4-диметилтиофен-3-ил)-2-нитроэтанол (А56-5)
Смесь соединение А56-4 (133 мг, 0.95 ммоль), нитрометана (2 мл) и K2CO3 (50 мг, 0.36 ммоль) перемешивали при комнатной температуре в течение 60 часов. Реакционную смесь гасили водой и экстрагировали этилацетатом (3×20 мл). Объединенные органические слои промывали водой (2×100 мл) и насыщенным водным раствором поваренной соли (100 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 40% EtOAc/гексан в качестве элюента), получая соединение А56-5 (80 мг, 42%) в виде желтой смолы.
Стадия 6: (1-(2,4-диметилтиофен-3-ил)-2-нитроэтокси)триэтилсилан (А56-6)
В перемешиваемый раствор соединения А56-5 (235 мг, 1.17 ммоль) в ДМФА (4 мл) добавляли имидазол (238 мг, 3.5 ммоль) и TES-Cl (0.23 мл, 1.4 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 4 часов. После окончания реакции, реакционную смесь гасили водой (50 мл) и экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (2×30 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 5% EtOAc/гексан в качестве элюента), получая соединение А56-6 (240 мг, 65%) в виде бесцветной смолы.
Стадия 7: 2-(2,4-диметилтиофен-3-ил)-2-((триэтилсилил)окси)этанамин (А56-7)
В перемешиваемый раствор соединения А56-6 (240 мг, 0.76 ммоль) в смеси EtOH/вода (10 мл, 4:1) добавляли порошкообразное железо (425 мг, 7.6 ммоль) и твердый NH4Cl (407 мг, 7.6 ммоль). Полученную смесь перемешивали при 70°С 45 минут. После окончания реакции, реакционную смесь фильтровали через слой целита и промывали МеОН (3×15 мл). Растворитель удаляли при пониженном давлении. Остаток от упаривания суспендировали в EtOAc (100 мл) и промывали водой (30 мл) и насыщенным водным раствором поваренной соли (30 мл). Органический слой сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 5% МеОН/ДХМ в качестве элюента), получая соединение А56-7 (192 мг, 88%) в виде желтой смолы.
Стадия 8: N-(3,5-дифторбензил)-2-(2,4-диметилтиофен-3-ил)-2-((триэтилсилил)окси)этанамин (А56)
В перемешиваемый раствор соединения А56-7 (192 мг, 0.67 ммоль) в МеОН (5 мл) добавляли 3,5-дифторбензальдегид (95 мг, 0.67 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 2 часов. По окончании образования имина (мониторинг методом ТСХ), добавляли порциями твердый NaBH4 (51 мг, 1.3 ммоль) при 0°С. Полученную смесь нагревали до комнатной температуры и перемешивали при той же температуре 4 часа. Реакционную смесь гасили водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (2×30 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 10% EtOAc/гексан в качестве элюента), получая соединение А56 (200 мг, 72%) в виде бесцветной смолы. 1H ЯМР (CDCl3, 300 МГц): δ 6.90-6.77 (м, 3Н), 6.71-6-60 (м, 1Н), 5.09 (дд, J=7.8, 4.2 Гц, 1Н), 3.78 (с, 2Н), 2.87 (дд, J=12.0, 7.8 Гц, 1Н), 2.71 (дд, J=12.0, 4.5 Гц, 1Н), 2.11 (д, J=0.6 Гц, 3Н), 2.06 (с, 3Н), 1.65 (ушир.с, 1Н), 0.89 (т, J=7.8 Гц, 9Н), 0.62-0.50 (м, 6Н).
Справочный пример А57
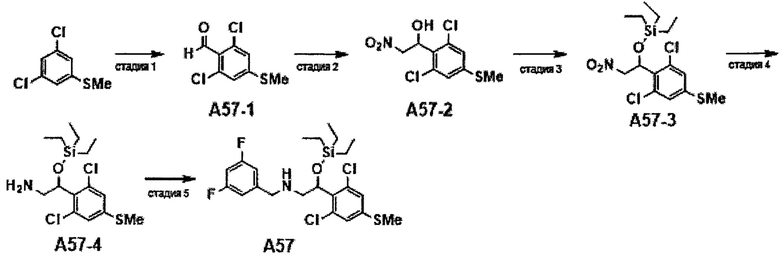
Стадия 1: 2,6-дихлор-4-(метилтио)бензальдегид (А57-1)
В перемешиваемый раствор (3,5-дихлорфенил)(метил)сульфана (1.0 г, 5.1 ммоль) в ТГФ (15 мл), добавляли по каплям н-BuLi (1.6 М раствор в ТГФ, 4.8 мл, 7.7 ммоль) при -78°С, и перемешивали 1 час при той же температуре. Раствор ДМФА (0.6 мл, 7.7 ммоль) в ТГФ (3 мл) медленно добавляли при -78°С и перемешивали 1 час. Реакционную смесь гасили насыщенным водным раствором NH4Cl (50 мл) и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали водой (30 мл), насыщенным водным раствором поваренной соли (30 мл), сушили над безводным NO2SO4 и упаривали при пониженном давлении. Полученный неочищенный продукт очищали методом колоночной хроматографии (силикагель, 20% EtOAc/гексан в качестве элюента), получая соединение А57-1 (1.4 г, 99%) в виде бесцветного масла.
Стадия 2: 1-(2,6-дихлор-4-(метилтио)фенил)-2-нитроэтанол (А57-2)
Соединение А57-2 (0.71 г, неочищенное) получали в виде бесцветной смолы реакцией соединения А57-1 (0.5 г, 2.44 ммоль) и K2CO3 (0.13 г, 0.92 ммоль) в CH3NO2 (5 мл) по методике, аналогичной описанной в справочном примере А1, стадия 1.
Стадия 3: (1-(2,6-дихлор-4-(метилтио)фенил)-2-нитроэтокси)триэтилсилан (А57-3)
Соединение А57-3 (1.0 г, неочищенное) получали в виде бесцветной смолы реакцией соединения А57-2 (0.71 г, 2.5 ммоль), TES-Cl (0.5 мл, 3.02 ммоль) и имидазола (0.51 г, 7.55 ммоль) в ДМФА (10 мл) по методике, аналогичной описанной в справочном примере А1, стадия 2.
Стадия 4: 2-(2,6-дихлор-4-(метилтио)фенил)-2-((триэтилсилил)окси)этанамин (А57-4)
Соединение А57-4 (0.98 г, неочищенное) получали в виде коричневого масла реакцией соединения А57-3 (1.0 г, 2.53 ммоль), Fe (1.42 г, 25.3 ммоль) и NH4Cl (1.34 г, 25.3 ммоль) в смеси EtOH/вода (4:1, 20 мл) по методике, аналогичной описанной в справочном примере А31, стадия 3.
Стадия 5: 2-(2,6-дихлор-4-(метилтио)фенил)-N-(3,5-дифторбензил)-2-((триэтилсилил) окси)этанамин (А57)
Соединение А57 (0.73 г, 55%) получали в виде бесцветной смолы реакцией соединения А57-4 (0.98 г, 2.69 ммоль), 3,5-дифторбензальдегида (0.29 мл, 2.69 ммоль) и NaBH4 (0.2 г, 5.36 ммоль) в МеОН (10 мл) по методике, аналогичной описанной в справочном примере А31, стадия 4. 1H ЯМР (CDCl3, 300 МГц): δ 7.10 (с, 2Н), 6.87-6.61 (м, 3Н), 5.53 (дд, J=8.6, 4.8 Гц, 1Н), 3.82 (с, 2Н), 3.23 (дд, J=12.1, 8.6 Гц, 1Н), 2.78 (дд, J=12.1, 4.8 Гц, 1Н), 2.49 (с, 3Н), 0.90-0.85 (м, 9Н), 0.58-0.50 (м, 6Н).
Справочный пример А58
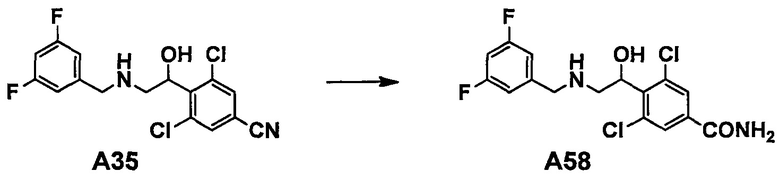
3,5-дихлор-4-(2-((3,5-дифторбензил)амино)-1-гидроксиэтил)бензамид (А58)
В перемешиваемый раствор соединения A3 5 (0.12 г, 0.29 ммоль) в смеси ТГФ/МеОН/вода (2:2:1, 5 мл) по каплям добавляли LiOH (4 М водный раствор, 0.44 мл, 1.76 ммоль) при 0°С. Полученную смесь оставляли нагреваться до комнатной температуры при продолжающемся перемешивании на 4 часа. Реакционную смесь подкисляли добавлением HCl (1 М, 6 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические слои промывали водой (10 мл), насыщенным водным раствором поваренной соли (10 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение А58 (60 мг, 47%) в виде желтого твердого вещества. LCMS (APCI): 391 (М+Н)+.
Справочный пример А59
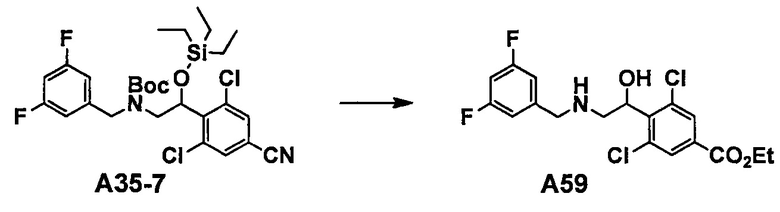
Этил 3,5-дихлор-4-(2-((3,5-дифторбензил)амино)-1-гидроксиэтил)бензоат (А59)
В перемешиваемый раствор соединения А35-7 (0.2 г, 0.3 ммоль) в EtOH (5 мл) добавляли конц. HCl (5 мл), и полученную смесь перемешивали при кипячении в течение ночи. Реакционную смесь гасили водой (50 мл), подщелачивали 10%-ным раствором NaOH до рН 9 и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Полученный неочищенный продукт очищали методом колоночной хроматографии (силикагель, 30% EtOAc/гексан в качестве элюента), получая соединение А59 (0.1 г, 92%) в виде белого твердого вещества.
Справочный пример А66
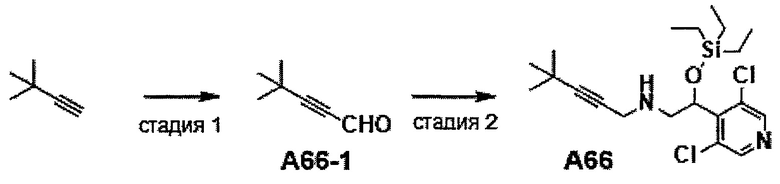
Стадия 1: 4,4-диметилпент-2-иналь (А66-1)
В перемешиваемый раствор 3,3-диметилбутан-1-ина (2.45 мл, 20 ммоль) в ТГФ (20 мл), добавляли н-BuLi (2.6 М раствор в гексане, 8.46 мл, 22 ммоль) при -78°С по каплям и перемешивали 1 час при той же температуре. Раствор ДМФА (3.85 мл, 50.0 ммоль) медленно добавляли при -78°С, и реакционную смесь оставляли нагреваться до комнатной температуры в течение ночи. Реакционную смесь гасили насыщенным раствором NH4Cl (100 мл) и экстрагировали гексаном (2×100 мл). Объединенные органические слои промывали водой (3×200 мл) и упаривали при пониженном давлении, получая соединение А66-1. Полученный неочищенный продукт использовали в следующей стадии без очистки.
Стадия 2: N-(2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)-4,4-диметилпент-2-ин-1-амин (А66)
Соединение А66 (76.1 мг, 36.6%) получали в виде бледно-желтого масла реакцией соединения А1-3 (160 мг, 0.5 ммоль), соединения А66-1 (80 мг, 0.726 ммоль), NaBH4 (120 мг) и MgSO4 (100 мг) в МеОН (6 мл) и ДХМ (3 мл) по методике, аналогичной описанной в справочном примере А31, стадия 4. 1Н ЯМР (CDCl3, 400 МГц): δ 8.43 (с, 2Н), 5.49 (дд, J=8.5, J=5.1 Гц, 1Н), 3.48 (д, J=16.4 Гц, 1Н), 3.37 (д, J=16.4 Гц, 1Н), 3.32 (дд, J=12.0, J=8.5 Гц, 1Н), 2.87 (дд, J=12.0, J=5.1 Гц, 1Н), 1.21 (с, 9Н), 0.89 (т, J=7.8 Гц, 9Н), 0.61-0.50 (м, 6Н).
Справочный пример А75
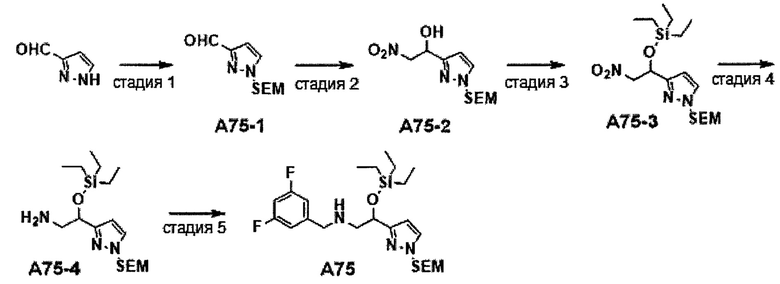
Стадия 1: 1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-3-карбальдегид (А75-1)
В перемешиваемую суспензию NaH (274 мг, 11.4 ммоль) в ДМФА (20 мл) добавляли раствор 1H-пиразол-3-карбальдегида (1.0 г, 10.4 ммоль) в ДМФА (10 мл) по каплям при 0°С, и полученную смесь перемешивали при комнатной температуре в течение 10 минут. Реакционную смесь охлаждали до 0°С и добавляли по каплям SEM-Cl (1.90 г, 11.4 ммоль). Полученную смесь нагревали до комнатной температуры и перемешивали при той же температуре 16 часов. Реакционную смесь гасили водой и экстрагировали этилацетатом (3×20 мл). Объединенные органические слои промывали водой (20 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 10% EtOAc/гексан в качестве элюента), получая соединение А75-1 (350 мг, 29%) в виде бесцветной смолы.
Стадия 2: 2-нитро-1-(1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-3-ил)этанол (А75-2)
Соединение А75-2 (428 мг, 64%) получали в виде желтой смолы реакцией соединения А75-1 (350 мг, 1.54 ммоль), CH3NO2 (1 мл) и K2CO3 (85 мг, 0.616 моль) по методике, аналогичной описанной в справочном примере А1, стадия 2.
Стадия 3: 3-(2-нитро-1-((триэтилсилил)окси)этил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол (А75-3)
Соединение А75-3 (604 мг, неочищенное) получали в виде желтой смолы реакцией соединения А75-2 (428 мг, 1.49 ммоль), TES-Cl (0.280 мл, 1.78 ммоль) и имидазола (303 мг, 4.47 ммоль) по методике, аналогичной описанной в справочном примере А1, стадия 3. Стадия 4: 2-((триэтилсилил)окси)-2-(1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-3-ил)этанамин (А75-4)
Соединение А75-4 (600 мг, неочищенное) получали в виде бесцветной смолы реакцией соединения А75-3 (604 мг, 1.51 ммоль), порошка железа (843 мг, 15.1 ммоль) и NH4Cl (806 мг, 15.1 ммоль) по методике, аналогичной описанной в справочном примере А31, стадия 3.
Стадия 5: N-(3,5-дифторбензил)-2-((триэтилсилил)окси)-2-(1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-3-ил)этанамин (А75)
Соединение А75 (40 мг, 5% за 3 стадии) получали в виде бесцветной смолы реакцией соединения А75-4 (600 мг, 1.61 ммоль), 3,5-дифторбензальдегида (206 мг, 1.45 ммоль) и NaBH4 (119 мг, 3.22 ммоль) по методике, аналогичной описанной в справочном примере А31, стадия 4. 1H ЯМР (CDCl3, 300 МГц): δ 7.59 (с, 0.7Н), 7.48 (с, 0.3Н), 6.39 (с, 1Н), 5.38-5.71 (м, 2Н), 4.91-5.08 (м, 1Н), 3.54-3.61 (м, 2Н), 2.95-3.04 (м, 2Н), 0.85-0.95 (м, 9Н), 0.59-0.62 (м, 6Н); LCMS (APCI): 499 (М+Н)+.
Справочный пример А84
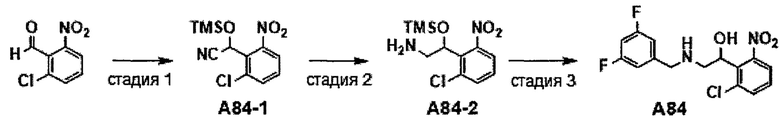
Стадия 1: 2-(2-хлор-6-нитрофенил)-2-((триметилсилил)окси)ацетонитрил (А84-1)
В перемешиваемый раствор 2-хлор-6-нитробензальдегида (1.0 г, 5.4 ммоль) в ДХМ (15 мл) добавляли TMSCN (1.0 мл, 8.1 ммоль) и NMO (0.19 г, 1.6 ммоль) при комнатной температуре и перемешивали 1 час. Реакционную смесь гасили водой (50 мл) и экстрагировали дихлорметаном (2×30 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение А84-1 (1.0 г, 67%) в виде коричневого масла.
Стадия 2: 2-(2-хлор-6-нитрофенил)-2-((триметилсилил)окси)этанамин
В перемешиваемый раствор соединения А84-1 (0.85 г, 3.0 ммоль) в ТГФ (15 мл) добавляли ВН3⋅ТГФ (1.0 М раствор в ТГФ, 17.9 мл, 17.88 ммоль) и перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь гасили МеОН и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение А84-2 (0.65 г, 75%) в виде коричневой смолы.
Стадия 3: 1-(2-хлор-6-нитрофенил)-2-((3,5-дифторбензил)амино)этанол (А84)
Соединение А84 (0.57 г, 74%) получали в виде желтого твердого вещества реакцией соединения А84-2 (0.65 г, 2.24 ммоль), 3,5-дифторбензальдегида (0.24 мл, 2.24 ммоль) и NaBH4 (0.17 г, 4.49 ммоль) в МеОН (10 мл) по методике, аналогичной описанной в справочном примере А56, стадия 8. 1Н ЯМР (CDCl3, 300 МГц): δ 7.52-7.29 (м, 3Н), 6.89-6.66 (м, 3Н), 5.22 (дд, J=10.0, 3.7 Гц, 1Н), 3.88 (с, 2Н), 3.27-3.19 (м, 1Н), 3.07 (дд, J=12.6, 3.7 Гц, 1Н); LCMS (APCI): 343 (М+Н)+.
[Справочный пример А92
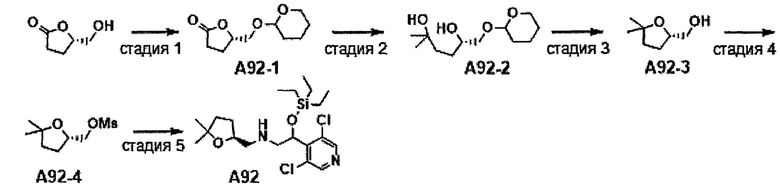
Стадия 1: (5S)-5-(((тетрагидро-2Н-пиран-2-ил)окси)метил)дигидрофуран-2(3Н)-он (А92-1)
В перемешиваемый раствор (S)-5-(гидроксиметил)дигидрофуран-2(3Н)-она (4.0 г, 34.45 ммоль) в ДХМ (20 мл) добавляли 3,4-дигидро-2Н-пиран (3.95 мл, 41.34 ммоль) и затем пиридиния п-толуолсульфонат (0.86 г, 3.44 ммоль) при комнатной температуре, и полученную смесь перемешивали 16 часов. Реакционную смесь разбавляли ДХМ (20 мл), гасили водой (40 мл) и экстрагировали дихлорметаном (2×50 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (2×20 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 50% EtOAc/гексан в качестве элюента), получая соединение А92-1 (5.85 мг, 85%) в виде бесцветной смолы.
Стадия 2: (2S)-5-метил-1-((тетрагидро-2Н-пиран-2-ил)окси)гексан-2,5-диол (А92-2)
В перемешиваемый раствор соединения А92-1 (5.85 г, 29.1 ммоль) в ТГФ (50 мл) добавляли метилмагния бромид (3.0 М в Et2O, 22.4 мл, 67.2 ммоль) по каплям при 0°С в течение 10 минут, и полученную смесь перемешивали при 0°С 4 часа. Полученную смесь оставляли нагреваться до комнатной температуры и перемешивали 15 часов. Реакционную смесь гасили насыщенным водным раствором NH4Cl и экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 90% EtOAc/гексан в качестве элюента), получая соединение А92-2 (6.09 г, 90%) в виде бесцветной смолы.
Стадия 3: (S)-(5,5-диметилтетрагидрофуран-2-ил)метанол (А92-3)
В перемешиваемый раствор соединения А92-2 (1.03 г, 4.43 ммоль) в МеОН (8 мл) добавляли моногидрат п-толуолсульфокислоты (421 мг, 2.2 ммоль) при комнатной температуре, и полученную смесь кипятили 5 час. Реакционную смесь охлаждали до комнатной температуры, гасили водой (15 мл) и экстрагировали дихлорметаном (2×25 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (20 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 35% EtOAc/гексан в качестве элюента), получая соединение А92-3 (330 мг, 57%) в виде бесцветной смолы.
Стадия 4: (S)-(5,5-диметилтетрагидрофуран-2-ил)метил метансульфонат (А92-4)
В перемешиваемый раствор соединения А92-3 (300 мг, 2.30 ммоль) в ДХМ (6 мл) добавляли Et3N (0.64 мл, 4.6 ммоль), затем метансульфонилхлорид (0.21 мл, 2.76 ммоль) при 0°С. Полученную смесь перемешивали при 0°С 30 минут. Полученную смесь оставляли нагреваться до комнатной температуры на 2 часа. Реакционную смесь гасили водой (10 мл) и экстрагировали дихлорметаном (2×20 мл). Объединенные органические слои промывали водой (20 мл), насыщенным водным раствором поваренной соли (20 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 35% EtOAc/гексан в качестве элюента), получая соединение А92-4 (310 мг, 64%) в виде бесцветной смолы.
Стадия 5: 2-(3,5-дихлорпиридин-4-ил)-N-(((S)-5,5-диметилтетрагидрофуран-2-ил)метил)-2-((триэтилсилил)окси)этанамин (А92)
Смесь соединения А92-4 (140 мг, 0.67 ммоль), соединения А1-3 (216 мг, 0.67 ммоль), Na2CO3 (710 мг, 6.7 ммоль) и изопропанола (4 мл) помещали в микроволновую пробирку. Пробирку закрывали, и полученную смесь нагревали в микроволновой печи при 120°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, гасили водой (15 мл) и экстрагировали дихлорметаном (2×25 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (20 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упариваяия очищали методом колоночной хроматографии (силикагель, 2% МеОН/ДХМ в качестве элюента), получая соединение А92 (40 мг, 14%) в виде бесцветной смолы.
Справочный пример А93
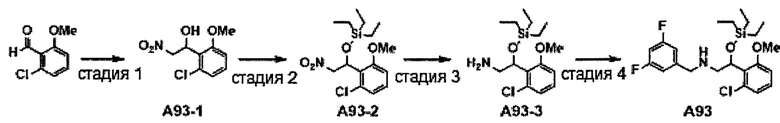
Стадия 1: 1-(2-хлор-6-метоксифенил)-2-нитроэтанол(А93-1)
Соединение А93-1 (1.35 г, неочищенное) получали в виде бесцветного масла реакцией 2-хлор-6-метоксибензальдегида (1.0 г, 5.88 ммоль) и K2CO3 (0.3 г, 2.2 ммоль) в CH3NO2 (10 мл) по методике, аналогичной описанной в справочном примере А1, стадия 1.
Стадия 2: (1-(2-хлор-6-метоксифенил)-2-нитроэтокси)триэтилсилан (А93-2)
Соединение А93-2 (2.14 г, неочищенное) получали в виде бесцветного масла реакцией соединения А93-1 (1.35 г, 5.84 ммоль), TES-Cl (1.17 мл, 7.01 ммоль) и имидазола (1.19 г, 17.53 ммоль) в ДМФА (10 мл) по методике, аналогичной описанной в справочном примере А1, стадия 2.
Стадия 3: 2-(2-хлор-6-метоксифенил)-2-((триэтилсилил)окси)этанамин (А93-3)
Соединение А93-3 (1.6 г, 84%) получали в виде бесцветного масла реакцией соединения А93-2 (2.14 г, 6.2 ммоль), Fe (3.48 г, 62.0 ммоль) и NH4Cl (3.3 г, 62.0 ммоль) в смеси EtOH/вода (4:1, 20 мл) по методике, аналогичной описанной в справочном примере А1, стадия 3.
Стадия 4: 2-(2-хлор-6-метоксифенил)-N-(3,5-дифторбензил)-2-((триэтилсилил)окси) этанамин (А93)
Соединение А93 (1.2 г, 54%) получали в виде бесцветной смолы реакцией соединения А93-3 (1.6 г, 5.16 ммоль), 3,5-дифторбензальдегида (0.56 мл, 5.16 ммоль) и NaBH4 (0.39 г, 10.2 ммоль) в МеОН (10 мл) по методике, аналогичной описанной в справочном примере А1, стадия 4. 1H ЯМР (CDCl3, 300 МГц): δ 7.13 (т, J=8.1 Гц, 1Н), 6.95-6.60 (м, 5Н), 5.58 (дд, J=8.6, 4.7 Гц, 1Н), 3.83-3.77 (м, 5Н), 3.28 (дд, J=12.0, 8.7 Гц, 1Н), 2.78 (дд, J=12.0, 4.7 Гц, 1Н), 0.87-0.82 (м, 9Н), 0.60-0.46 (м, 6Н); LCMS (APCI): 442 (М+Н)+.
Справочный пример А94
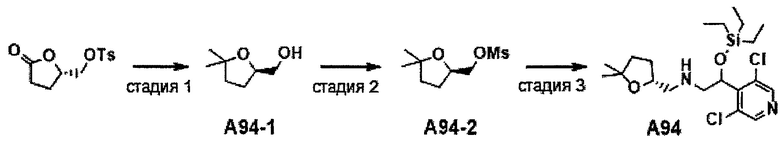
Стадия 1: (S)-(5-оксотетрагидрофуран-2-ил)метил 4-метилбензолсульфонат (А94-1)
В перемешиваемый раствор (S)-5-(гидроксиметил)дигидрофуран-2(3Н)-она (2.0 г, 17.2 ммоль) в ДХМ (20 мл) добавляли Et3N (4.8 мл, 34.44 ммоль), затем п-толуолсульфонилхлорид (3.61 г, 18.94 ммоль) при 0°С. Полученную смесь оставляли нагреваться до комнатной температуры и перемешивали при той же температуре 15 часов. Реакционную смесь гасили водой (100 мл) и экстрагировали дихлорметаном (2×50 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 50% EtOAc/гексан в качестве элюента), получая соединение А94-1 (4.06 г, 87%) в виде белого твердого вещества.
Стадия 2: (R)-(5,5-диметилтетрагидрофуран-2-ил)метанол (А94-2)
В перемешиваемый раствор соединения А94-1 (1.63 г, 6.03 ммоль) в ТГФ (20 мл) добавляли MeLi (3.0 М в диэтоксиметане, 4.4 мл, 13.26 ммоль) по каплям при -78°С в течение 10 минут, и полученную смесь перемешивали при -78°С 1 час. Полученную смесь оставляли нагреваться до комнатной температуры в течение 4 часов. Реакционную смесь гасили насыщенным водным раствором NaCl, разбавляли водой (30 мл) и экстрагировали этилацетатом (2×25 мл). Объединенные органические слои промывали водой (30 мл), насыщенным водным раствором поваренной соли (30 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 35% EtOAc/гексан в качестве элюента), получая соединение А94-2 (220 мг, 28%) в виде бесцветной смолы.
Стадия 3: (R)-(5,5-диметилтетрагидрофуран-2-ил)метил метансульфонат (А94-3)
Соединение А94-3 (351 мг, 61%) получали в виде бесцветной смолы реакцией соединения А94-2 (360 мг, 2.76 ммоль), Et3N (0.77 мл, 5.52 ммоль) и метансульфонил хлорида (0.25 мл, 3.31 ммоль) в ДХМ (5.0 мл) по методике, аналогичной описанной в справочном примере А92, стадия 4.
Стадия 4: 2-(3,5-дихлорпиридин-4-ил)-N-(((R)-5,5-диметилтетрагидрофуран-2-ил)метил)-2-((триэтилсилил)окси)этанамин (А94)
Соединение А94 (32 мг, 8%) получали в виде бесцветной смолы реакцией соединения А94-3 (200 мг, 0.96 ммоль), соединения А1-3 (247 мг, 0.77 ммоль) и Na2CO3 (508 мг, 4.8 ммоль) в изопропаноле (3.0 мл) по методике, аналогичной описанной в справочном примере А92, стадия 5.
Справочный пример А103
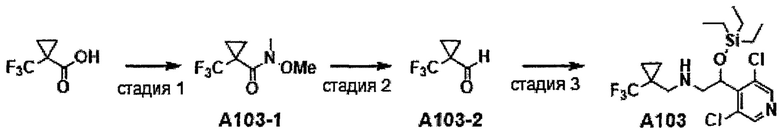
Стадия 1: N-метокси-N-метил-1-(трифторметил)циклопропанкарбоксамид (А103-1)
В смесь 1-(трифторметил)циклопропанкарбоновой кислоты (150 мг, 0.974 ммоль), 1-гидроксибензотриазола моногидрата (224 мг, 1.46 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (280 мг, 1.46 ммоль) и N,O-диметилгидроксиламина гидрохлорида (142 мг, 1.46 ммоль) в ДМФА (5 мл) добавляли DIPEA (0.50 мл, 2.92 ммоль), и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили водой (30 мл) и экстрагировали этилацетатом. Объединенный органический слой промывали водой и насыщенным водным раствором поваренной соли, сушили над MgSO4 и упаривали при пониженном давлении, получая соединение А103-1 (164 мг, 85%) в виде бледно-желтого масла.
Стадия 2: 1-(трифторметил)циклопропанкарбальдегид (А103-2)
В перемешиваемый раствор соединения А103-1 (164 мг, 0.832 ммоль) в ДХМ (2 мл) добавляли диизобутилалюминия гидрид (1 М раствор в гексане, 1.0 мл, 1.0 ммоль) при -78°С в атмосфере азота. Через 0.5 часа смесь оставляли нагреваться до 0°С и перемешивали 0.5 часа. Реакционную смесь гасили насыщенным водным раствором KHSO4 (10 мл) и экстрагировали дихлорметаном (2×4 мл). Объединенные органические слои напрямую использовали в следующей стадии без дополнительной очистки.
Стадия 3: 2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)-N-((1-(трифторметил)циклопропил)метил)этанамин (А103)
Соединение А1-3 (0.20 г, 0.622 ммоль) растворяли в ДХМ растворе, содержащем соединение А103-2. MgSO4 (0.2 г) добавляли в полученный раствор, и смесь перемешивали в течение 2 часов. Добавляли в реакционную смесь МеОН (10 мл) и NaBH4 (0.2 г), и полученную смесь перемешивали 0.5 часа. Реакционную смесь гасили водой и экстрагировали этилацетатом. Отделенный органический слой промывали насыщенным водным раствором поваренной соли, сушили над MgSO4 и упаривали при пониженном давлении. Полученный сырой продукт очищали методом колоночной хроматографии на силикагеле, элюируя 20% EtOAc в гептане, получая соединение А103 (87 мг, 32%) в виде бесцветного масла. 1H ЯМР (CDCl3, 400 МГц) δ: 8.43 (2Н, с), 5.45 (1Н, дд, J=8.5, 4.6 Гц), 3.24 (1Н, дд, J=12.2, 8.8 Гц), 2.86 (2Н, дд, J=24.4, 13.2 Гц), 2.76 (1Н, дд, J=12.2, 4.4 Гц), 0.97-0.86 (13Н, м), 0.56-0.52 (6Н, м).
Справочный пример А111
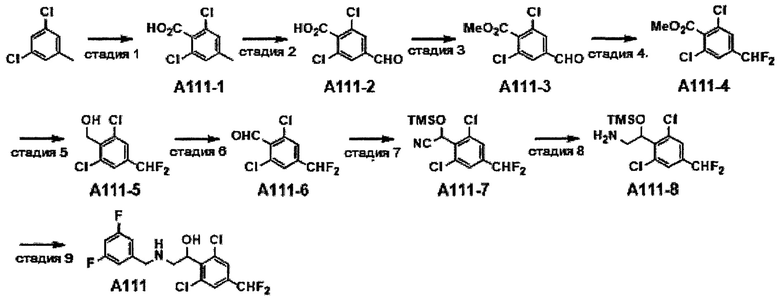
Стадия 1: 2,6-дихлор-4-метилбензойная кислота (А111-1)
В перемешиваемый раствор 1,3-дихлор-5-метилбензола (2.0 г, 12.4 ммоль) в ТГФ (20 мл) добавляли н-BuLi (2.0 М раствор в гексане, 9.3 мл, 18.6 ммоль) при -78°С по каплям в течение 10 минут, и смесь перемешивали 30 минут при -78°С. Медленно добавляли в реакционную смесь сухой лед, и полученную смесь перемешивали при той же температуре 20 минут. Затем реакционную смесь медленно нагревали до комнатной температуры, гасили 6 М раствором HCl (10 мл) и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение А111-1 (1.1 г, 44%) в виде белого твердого вещества.
Стадия 2: 2,6-дихлор-4-формилбензойная кислота (А111-2)
В перемешиваемый раствор соединения А111-1 (1.1 г, 5.3 ммоль) в ДХМ (20 мл) добавляли NBS (2.3 г, 13.4 ммоль) и дифенил оксалат (65 мг, 0.27 ммоль), после чего кипятили 40 часов. Реакционную смесь доводили до комнатной температуры и упаривали растворитель. К остатку добавляли EtOAc (10 мл), и полученный твердый осадок отфильтровывали на воронке Бюхнера. Фильтрат упаривали, и сырой продукт растворяли в EtOH (20 мл) и нагревали до 50°С. Раствор нитрата серебра (I) (1.37 г, 8.0 ммоль) в горячей воде (3 мл) добавляли в реакционную смесь по каплям и выдерживали при той же температуре 45 минут. Реакционную смесь гасили 1 М раствором HCl (10 мл), полученный твердый осадок отфильтровывали и промывали EtOH (30 мл). Фильтрат упаривали, и оставшийся водный слой экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение А111-2 (1.6 г, неочищенное) в виде коричневого масла.
Стадия 3: метил 2,6-дихлор-4-формилбензоат (А111-3)
В перемешиваемый раствор соединения А111-2 (1.1 г, 5.0 ммоль) в ДМФА (10 мл) добавляли K2CO3 (1.0 г, 7.5 ммоль) при 0°С, затем медленно добавляли MeI (0.94 мл, 15.0 ммоль), и реакционную смесь перемешивали при той же температуре 30 минут. После этого реакционную смесь гасили водой (50 мл) и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 10% EtOAc/гексан в качестве элюента), получая соединение А111-3 (0.59 г, 50%) в виде белого твердого вещества.
Стадия 4: метил 2,6-дихлор-4-(дифторметил)бензоат (А111-4)
В перемешиваемый раствор соединения А111-3 (0.36 г, 1.5 ммоль) в ДХМ (10 мл) добавляли DAST (0.37 мл, 2.8 ммоль) при -78°С по каплям, затем прикалывали МеОН, реакционную смесь перемешивали при той же температуре 15 минут и доводили до 0°С. Реакционную смесь перемешивали 30 минут при той же температуре и 16 часов при комнатной температуре. Реакционную смесь гасили насыщенным раствором NaHCO3 (20 мл) при 0°С, перемешивали 20 минут и экстрагировали дихлорметаном (2×30 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение А111-4 (0.37 г, 94%) в виде бесцветного масла.
Стадия 5: (2,6-дихлор-4-(дифторметил)фенил)метанол (А111-5)
В перемешиваемый раствор соединения А111-4 (1.44 г, 5.64 ммоль) в ТГФ (10 мл) добавляли LiA1H4 (2.0 М раствор в ТГФ, 4.23 мл, 8.46 ммоль) в ТГФ (10 мл) при -78°С по каплям в течение 15 минут, и доводили до 0°С. Реакционную смесь перемешивали 30 минут при той же температуре и 16 часов при комнатной температуре. Реакционную смесь гасили 1 М раствором HCl (20 мл) при 0°С, перемешивали 20 минут и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение А111-5 (0.59 г, 45%) в виде бесцветного масла.
Стадия 6: 2,6-дихлор-4-(дифторметил)бензальдегид (А111-6)
Соединение A111-6 (0.38 г, 65%) получали в виде бесцветного масла реакцией соединения А111-5 (0.59 г, 2.46 ммоль) и периодинана Десс-Мартина (2.1 г, 4.92 ммоль) в ДХМ (10 мл) по методике, аналогичной описанной в справочном примере А56, стадия 4.
Стадия 7: 2-(2,6-дихлор-4-(дифторметил)фенил)-2-((триметилсилил)окси)ацетонитрил (А111-7)
В перемешиваемый раствор соединения А111-6 (0.38 г, 1.6 ммоль) в ДХМ (15 мл) добавляли TMSCN (0.31 мл, 2.5 ммоль) и NMO (60 мг, 0.5 ммоль) при комнатной температуре и перемешивали 1 час. Реакционную смесь гасили водой (50 мл) и экстрагировали дихлорметаном (2×30 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение А111-7 (0.53 г, 97%) в виде желтого твердого вещества.
Стадия 8 2-(2,6-дихлор-4-(дифторметил)фенил)-2-((триметилсилил)окси)этанамин (А111-8)
В перемешиваемый раствор соединения А111-7 (0.53 г, 1.6 ммоль) в ТГФ (10 мл) добавляли ВН3⋅ТГФ (8.2 мл, 8.1 ммоль) и перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь гасили МеОН и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение А111-8 (0.5 г, неочищенное) в виде желтого масла.
Стадия 9: 1-(2,6-дихлор-4-(дифторметил)фенил)-2-((3,5-дифторбензил)амино)этанол (А111)
Соединение А111 (0.21 г, 36%) получали в виде бесцветной смолы реакцией соединения А111-8 (0.5 г, 1.52 ммоль), 3,5-дифторбензальдегида (0.16 мл, 1.52 ммоль) и NaBH4 (0.11 г, 3.0 ммоль) в МеОН (5 мл) по методике, аналогичной описанной в справочном примере А56, стадия 8. 1Н ЯМР (CDCl3, 400 МГц): δ 7.44 (с, 2Н), 6.89-6.42 (м, 4Н), 5.56-5.25 (м, 1Н), 3.87 (с, 2Н), 3.26 (дд, J=12.8, 9.6 Гц, 1Н), 2.91-2.86 (м, 1Н).
Справочный пример А112
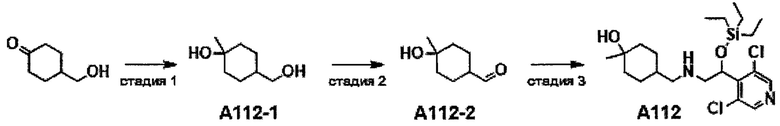
Стадия 1: 4-(гидроксиметил)-1-метилциклогексанол (А112-1)
В перемешиваемый раствор 4-(гидроксиметил)циклогексанона (1.0 г, 7.8 ммоль) в ТГФ (20 мл) добавляли метилмагния бромид (3.0 М в Et2O, 7.8 мл, 23.4 ммоль) по каплям при 0°С в течение 5 минут. Полученную смесь оставляли нагреваться до комнатной температуры и перемешивали при той же температуре в течение 2 часов. Реакционную смесь гасили насыщенным водным раствором NH4Cl и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали водой (20 мл), насыщенным водным раствором поваренной соли (20 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 80% EtOAc/гексан в качестве элюента), получая соединение А112-1 (300 мг, 27%) в виде белого твердого вещества.
Стадия 2: 4-гидрокси-4-метилциклогексанкарбальдегид (А112-2)
Соединение А112-2 (49 мг, неочищенное) получали в виде желтой пены реакцией соединения A112-1 (50 мг, 0.348 ммоль) и периодинана Десс-Мартина (206 мг, 0.48 ммоль) в ДХМ (5.0 мл) по методике, аналогичной описанной в справочном примере А56, стадия 4.
Стадия 3: 4-(((2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)амино)метил)-1-метилциклогексанол (А112)
В перемешиваемый раствор соединения А112-2 (49 мг, 0.34 ммоль) в ДХМ (15 мл) добавляли соединение А1-3 (109 мг, 0.34 ммоль), затем NaBH(OAc)3 (108 мг, 0.51 ммоль) при комнатной температуре. Полученную смесь перемешивали 4 часа при комнатной температуре. Реакционную смесь гасили насыщенным водным раствором NaHCO3 (10 мл) и экстрагировали этилацетатом (2×20 мл). Объединенный органический слой промывали водой (20 мл), насыщенным водным раствором поваренной соли (20 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 5% МеОН/ДХМ в качестве элюента), получая соединение А112 (58 мг, 37% за две стадии) в виде желтой смолы.
Справочный пример А118
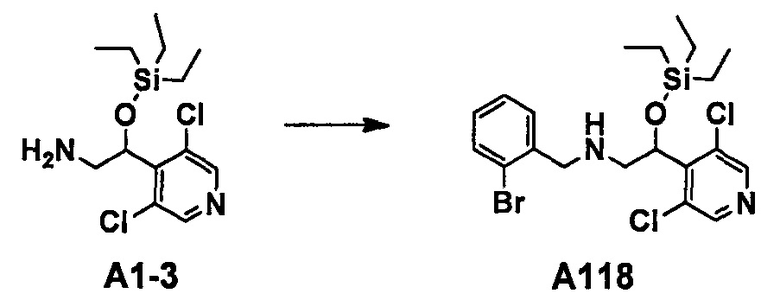
N-(2-бромбензил)-2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этанамин (А118)
Соединение A118 (1.2 г, 79%) получали в виде бесцветного масла реакцией соединения А1-3 (1.0 г, 3.16 ммоль), 2-бромбензальдегида (576 мг, 3.11 ммоль) и NaBH4 (172 мг, 4.67 ммоль) в МеОН (40 мл) по методике, аналогичной описанной в справочном примере А1, стадия 4. 1Н ЯМР (CDCl3, 400 МГц): δ 8.41 (с, 2Н), 7.53-7.51 (м, 1Н), 7.37-7.35 (м, 1Н), 7.28-7.25 (м, 1Н), 7.13-7.08 (м, 1Н), 5.55 (дд, J=8.2, 5.2 Гц, 1Н), 3.94-3.85 (м, 1Н), 3.20 (дд, J=12.1, 8.4 Гц, 1Н), 2.88 (д, J=4.8 Гц, 0.5Н), 2.86 (дд, J=12.1, 5.1 Гц, 0.5Н), 0.89-0.86 (м, 9Н), 0.58-0.51 (м, 6Н).
Справочный пример А119
Стадия 1: 1,3-дибром-2,2-диметилпропан (А119-1)
В перемешиваемый раствор трифенилфосфина (26.2 г, 0.1 моль) в CH3CN (50 мл) добавляли раствор брома (5.13 мл, 0.10 моль) в CH3CN (30 мл) по каплям при 0°С. Порциями добавляли 2,2-диметилпропан-1,3-диол (5.1 г, 0.05 моль), и реакционную смесь перемешивали при 90°С 16 часов. Растворитель удаляли при пониженном давлении. Остаток от упаривания суспендировали в МТВЕ (150 мл), и выпавший осадок отделяли фильтрованием. Фильтрат упаривали при пониженном давлении, и остаток растворяли в CH3CN и экстрагировали гексаном (3×100 мл). Объединенные гексановые экстракты упаривали при пониженном давлении, получая соединение А119-1 (6.5 г, 59%) в виде коричневого масла.
Стадия 2: дипентил 3,3-диметилциклобутан-1,1-дикарбоксилат (А119-2)
Натрий (0.98 г, 43.0 ммоль) добавляли порциями в пентанол (25 мл), и полученную смесь перемешивали при 5°С, получая прозрачный раствор. Реакционную смесь нагревали до 70°С, и затем добавляли диэтилмалонат (3.50 г, 26.0 ммоль) добавляли в течение 5 минут. Реакционную смесь нагревали до 130°С и добавляли по каплям соединение А119-1 (5.0 г, 21 ммоль) в течение 10 минут. Реакционную смесь нагревали до 130°С 4 часа. Растворитель удаляли в вакууме при 100°С. Остаток от упаривания гасили водой (100 мл) и экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты упаривали при пониженном давлении, получая соединение А119-2 (6 г, неочищенное) в виде коричневого масла. Полученный неочищенный продукт использовали в следующей стадии без очистки.
Стадия 3: 3,3-диметилциклобутан-1,1-дикарбоновая кислота (А119-3)
В раствор соединения А119-2 (6 г, неочищенное) в смеси EtOH/вода (60 мл, 2:1) добавляли раствор KOH (40%-ный водный раствор, 10 мл), и реакционную смесь перемешивали при 100°С 4 часа. После удаления растворителя при пониженном давлении, остаток суспендировали в воде (100 мл) и промывали МТВЕ. Водный слой подкисляли до рН 1 и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение А119-3 (2.5 г, неочищенное) в виде коричневой полутвердой смолы. Полученный неочищенный продукт использовали в следующей стадии без очистки.
Стадия 4: 3,3-диметилциклобутанкарбоновая кислота (А119-4)
Соединение А119-3 (2.5 г, неочищенное) нагревали в чистом виде при 200°С 2 часа, получая соединение А119-4 (900 мг, неочищенное) в виде светло-коричневой смолы.
Стадия 5: (3,3-диметилциклобутил)метанол (А119-5)
В перемешиваемую суспензию LiAlH4 (534 мг, 14.0 ммоль) в ТГФ (20 мл) добавляли раствор соединения А119-4 (900 мг, 7.0 ммоль) в ТГФ (10 мл) при 0°С, и полученную смесь перемешивали при той же температуре 3 часа. Реакционную смесь гасили водой (3 мл) и 20%-ным водным раствором NaOH (3 мл) и перемешивали при комнатной температуре в течение 10 минут. Твердый осадок отфильтровали через слой целита, и органический слой промывали водой (20 мл), насыщенным водным раствором поваренной соли (20 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 20% EtOAc/гексан в качестве элюента), получая соединение А119-5 (160 мг, 20%) в виде светло-желтого масла.
Стадия 6: 3,3-диметилциклобутанкарбальдегид (А119-6)
В перемешиваемый раствор соединения А119-5 (160 мг, 1.4 ммоль) в ДХМ (10 мл) добавляли периодинан Десс-Мартина (1.20 г, 2.8 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли ДХМ (10 мл) и гасили водным раствором Na2S2O8 (5 мл) и раствором NaHCO3 (5 мл). Органический слой промывали водой (10 мл), насыщенным водным раствором поваренной соли (10 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение А119-6 (150 мг, колич.) в виде желтого масла. Полученный неочищенный продукт использовали в следующей стадии без очистки.
Стадия 7: 2-(3,5-дихлорпиридин-4-ил)-N-((3,3-диметилциклобутил)метил)-2-((триэтил-силил)окси)этанамин (А119)
Смесь соединения А119-6 (150 мг, 1.33 ммоль) и соединения А1-3 (300 мг, 0.97 ммоль) в МеОН (10 мл) перемешивали при комнатной температуре в течение 3 часов. Порциями добавляли NaBH4 (75 мг, 1.99 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь гасили водой и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 20% EtOAc/гексан в качестве элюента), получая соединение А119 (190 мг, 36%) в виде желтой смолы. 1H ЯМР (CDCl3, 400 МГц): δ 8.42 (с, 2Н), 5.46-5.52 (м, 1Н), 3.16-3.23 (м, 1Н), 2.71-2.79 (м, 1Н), 2.53-2.69 (м, 2Н), 1.42-1.62 (м, 5Н), 1.23-1.38 (м, 6Н), 0.84-0.92 (м, 9Н), 0.49-0.58 (м, 6Н).
Справочный пример А122
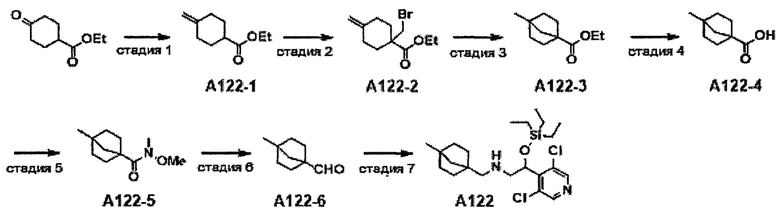
Стадия 1: этил 4-метиленциклогексанкарбоксилат (А122-1)
Лития бис(триметилсилил)амид (1.0 М раствор в ТГФ, 15 мл, 15 ммоль) добавляли по каплям в перемешиваемый раствор метилтрифенилфосфония бромида (5.36 г, 15 ммоль) в ТГФ (50 мл) при 0°С и перемешивали 40 минут при той же температуре. Раствор этил 4-оксоциклогексанкарбоксилата (2.04 г, 12 ммоль) в ТГФ (20 мл) медленно добавляли при 0°С и перемешивали 2 часа при температуре от 0°С до комнатной температуры. Реакцию гасили насыщенным водным раствором NH4Cl и экстрагировали гексаном. Отделенный органический слой сушили над MgSO4 и упаривали при пониженном давлении. Растворитель (100 мл, гексан/Et2O=5/1) добавляли в остаток и перемешивали 30 минут. Суспензию фильтровали. Фильтрат упаривали при пониженном давлении. Остаток от упаривания очищали методом хроматографии на силикагеле (5% EtOAc/гексан в качестве элюента), получая соединение А122-1 (1.478 г, 73%) в виде бесцветного масла.
Стадия 2: этил 1-(бромметил)-4-метиленциклогексанкарбоксилат (А122-2)
По каплям добавляли н-BuLi (2.6 М раствор в гексане, 2.5 мл, 6.6 ммоль) в раствор диизопропиламина (0.93 мл, 6.6 ммоль) в ТГФ (20 мл) при -78°С и перемешивали 30 минут при той же температуре. Гексаметилфосфорамид (4 мл) добавляли в реакционную смесь и перемешивали 20 минут при той же температуре. Раствор соединения А122-1 (1.01 г, 6 ммоль) в ТГФ (5 мл) добавляли и перемешивали 1 час при той же температуре. Раствор дибромметана (2.1 мл, 30 ммоль) добавляли в реакционную смесь, и полученную смесь оставляли нагреваться до комнатной температуры на 1.5 часа. Реакционную смесь разбавляли гексаном (80 мл) и AcOEt (20 мл). Отделенный органический слой промывали водой, насыщенным водным раствором NH4Cl, насыщенным водным раствором поваренной соли, сушили над MgSO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом хроматографии на силикагеле (10% EtOAc/гексан в качестве элюента), получая соединение А122-2 (1.39 г, 89%) в виде бледно-желтого масла.
Стадия 3: этил 4-метилбицикло[2.2.1]гептан-1-карбоксилат (А122-3)
В перемешиваемый раствор соединения А122-2 (783 мг, 3 ммоль) в толуоле (65 мл) добавляли трибутилолова гидрид (0.888 мл, 3.3 ммоль) и 2,2'-азобис(изобутиронитрил) (25 мг) в толуоле (20 мл), и полученную смесь перемешивали при 110°С 1 час. Реакционную смесь охлаждали и упаривали при пониженном давлении. Добавляли в остаток ДХМ (20 мл) и раствор KF (1.0 г) в воде (0.31 мл), и полученную смесь перемешивали 1 час. Реакционную смесь фильтровали через безводный Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом хроматографии на силикагеле (10% EtOAc/гексан в качестве элюента), получая соединение А122-3 (501 мг, 92%) в виде бесцветного масла.
Стадия 4: 4-метилбицикло[2.2.1]гептан-1-карбоновая кислота (А122-4)
В перемешиваемый раствор соединения А122-3 (500 мг, 2.74 ммоль) в смеси МеОН/вода (8 мл, 3:1) добавляли водный раствор LiOH (4 М, 2 мл, 8 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2.5 часов и перемешивали при 50°С еще 1.5 часа. Органический растворитель удаляли при пониженном давлении. Остаток от упаривания разбавляли водой (10 мл) и гексаном (10 мл). Водный слой подкисляли добавлением 6 М водного раствора HCl до рН 1 и экстрагировали дихлорметаном. Органические слои сушили над MgSO4 и упаривали при пониженном давлении, получая соединение А122-4 (313 мг, 74%) в виде светло-желтого твердого вещества.
Стадия 5: N-метокси-N,4-диметилбицикло[2.2.1]гептан-1-карбоксамид (А122-5)
В смесь соединения А122-4 (302 мг, 1.96 ммоль), 1-гидроксибензотриазола моногидрата (460 мг, 3 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (466 мг, 3 ммоль) и N,O-диметилгидроксиламина гидрохлорида (293 мг, 3 ммоль) в ДМФА (10 мл) добавляли DIPEA (1.03 мл, 6 ммоль), и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили водой (30 мл) и экстрагировали этилацетатом. Отделенный органический слой промывали насыщенным водным раствором NH4Cl, насыщенным водным раствором поваренной соли, сушили над MgSO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом хроматографии на силикагеле (30% EtOAc/гексан в качестве элюента), получая соединение А122-5 (271,4 мг, 70%) в виде бесцветного масла.
Стадия 6: 4-метилбицикло[2.2.1]гептан-1-карбальдегид (А122-6)
В раствор соединения А122-5 (271 мг, 1.37 ммоль) в Et2O (5 мл) добавляли суспензию LiAlH4 (52 мг, 1.37 ммоль) в Et2O (2 мл) при 0°С и перемешивали 45 минут при той же температуре. Реакционную смесь гасили насыщенным водным раствором KHSO4 (5 мл) при 0°С, перемешивали 30 минут при комнатной температуре и экстрагировали Et2O. Органический слой сушили над MgSO4 и упаривали при пониженном давлении, получая соединение А122-6 (163 мг, 86%) в виде бесцветного масла. Полученный неочищенный продукт использовали в следующей стадии без очистки.
Стадия 7: 2-(3,5-дихлорпиридин-4-ил)-N-((4-метилбицикло[2.2.1]гептан-1-ил)метил)-2-((триэтилсилил)окси)этанамин (А122)
Соединение А122 (177 мг, 80%) получали в виде бледно-желтого масла реакцией соединения А1-3 (160 мг, 0.50 ммоль), соединения А122-6 (82 мг, 0.59 ммоль), NaBH4 (120 мг) и MgSO4 (200 мг) в МеОН (4 мл) и ДХМ (3 мл) по методике, аналогичной описанной в справочном примере А31, стадия 4. 1H ЯМР (CDCl3, 400 МГц): δ 8.42 (с, 2Н), 5.50 (дд, J=8.7, J=4.6 Гц, 1Н), 3.24 (дд, J=12.6, J=8.7 Гц, 1Н), 2.78 (дд, J=12.6, J=4.6 Гц, 1Н), 2.75 (д, J=11.7 Гц, 1Н), 2.67 (д, J=11.7 Гц, 1Н), 1.54-1.32 (м, 8Н), 1.10-1.08 (м, 5Н), 0.89 (т, J=8.0 Гц, 9Н), 0.58-0.49 (м, 6Н).
Справочный пример А124
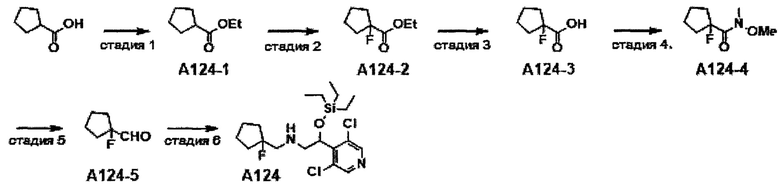
Стадия 1: этил циклопентанкарбоксилат (А124-1)
В раствор циклопентанкарбоксилата (1.14 г, 10 ммоль) в EtOH (5 мл) добавляли H2SO4 (0.1 мл) при комнатной температуре. Полученную смесь оставляли нагреваться до 80°С и перемешивали при той же температуре 3.5 часа. Реакционную смесь охлаждали до комнатной температуры и выливали в насыщенный водный раствор NaHCO3 (40 мл). Полученную смесь перемешивали при комнатной температуре в течение 30 минут и экстрагировали этилацетатом. Органический слой сушили над MgSO4 и упаривали при пониженном давлении, получая соединение А124-1 (1.01 г, 71%) в виде бледно-желтого масла. Полученный неочищенный продукт использовали в следующей стадии без очистки.
Стадия 2: этил 1-фторциклопентанкарбоксилат (А124-2)
Добавляли по каплям н-BuLi (2.6 М раствор в гексане, 4.0 мл, 10.5 ммоль) в раствор диизопропиламина (1.55 мл, 11 ммоль) в ТГФ (40 мл) при -78°С и перемешивали 30 минут при той же температуре. Добавляли раствор соединения А124-1 (1.00 г, 7 ммоль) в ТГФ (10 мл), и полученную смесь перемешивали 50 минут при той же температуре. Реакционную смесь оставляли нагреваться до 0°С на 1 час. Добавляли раствор N-фтор-N-(фенилсульфонил)бензолсульфонамида (3.47 г, 10 ммоль) в ТГФ (10 мл), и полученную смесь перемешивали 1 час при той же температуре. Реакционную смесь оставляли нагреваться до комнатной температуры в течение ночи. Реакцию гасили насыщенным водным раствором NH4Cl и экстрагировали этилацетатом. Отделенный органический слой упаривали при пониженном давлении. Остаток от упаривания очищали методом хроматографии на силикагеле (10% EtOAc/гексан в качестве элюента), получая соединение А124-2 (911 мг, 81%) в виде желтого масла.
Стадия 3: 1-фторциклопентанкарбоновая кислота (А124-3)
В перемешиваемый раствор соединения А124-2 (910 мг, 5.68 ммоль) в смеси EtOH/ТГФ/вода (7 мл, 4:2:1) добавляли водный раствор LiOH (4 М, 3 мл, 12 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2.5 часов. Органический растворитель удаляли при пониженном давлении. Остаток от упаривания подкисляли добавлением 2 М водного раствора HCl до рН 1 и экстрагировали этилацетатом. Органический слой сушили над MgSO4 и упаривали при пониженном давлении, получая соединение А124-3 (709 мг, 95%) в виде коричневого масла. Полученный неочищенный продукт использовали в следующей стадии без очистки.
Стадия 4: 1-фтор-N-метокси-N-метилциклопентанкарбоксамид (А124-4)
В смесь соединения А124-3 (709 мг, 5.37 ммоль), 1-гидроксибензотриазола (986 мг, 6.44 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (1.0 г, 6.44 ммоль) и N,O-диметилгидроксиламина гидрохлорида (628 мг, 6.44 ммоль) в ДМФА (10 мл) добавляли триэтиламин (1.12 мл, 8.05 ммоль), и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили 2 М водным раствором HCl (30 мл) и экстрагировали этилацетатом. Отделенный органический слой промывали водой, насыщенным водным раствором поваренной соли, сушили над MgSO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом хроматографии на силикагеле (20% EtOAc/гексан в качестве элюента), получая соединение А124-4 (543 мг, 58%) в виде желтого масла.
Стадия 5: 1-фторциклопентанкарбальдегид (А124-5)
В раствор соединения А124-4 (140 мг, 0.8 ммоль) в Et2O (20 мл) добавляли LiAlH4 (33 мг, 0.88 ммоль) при 0°С и перемешивали 5 часов при той же температуре. Реакционную смесь гасили насыщенным водным раствором KHSO4 (5 мл) при 0°С и экстрагировали Et2O. Объединенный органический слой сушили над MgSO4 и упаривали при пониженном давлении, получая соединение А124-5. Полученный неочищенный продукт использовали в следующей стадии без очистки.
Стадия 6: 2-(3,5-дихлорпиридин-4-ил)-N-((1-фторциклопентил)метил)-2-((триэтил-силил)окси)этанамин (А124)
Соединение А124 (207 мг, 68%) получали реакцией соединения А1-3 (233 мг, 0.73 ммоль), соединения А124-5 (93 мг, 0.8 ммоль), NaBH(OAc)3 (231 мг, 1.09 ммоль), MgSO4 (93 мг) и АсОН (0.042 мл, 0.73 ммоль) в ДХМ (2 мл) по методике, аналогичной описанной в справочном примере А31, стадия 4. 1H ЯМР (CDCl3, 400 МГц): δ 8.43 (с, 2Н), 5.49 (дд, J=8.5, J=4.5 Гц, 1Н), 3.26 (дд, J=12.6, J=8.5 Гц, 1Н), 2.87 (д, J=21.0 Гц, 2Н), 2.83 (дд, J=12.6, J=4.5 Гц, 1Н), 1.93-1.60 (м, 8Н), 0.88 (т, J=7.8 Гц, 9Н), 0.60-0.49 (м, 6Н).
Справочный пример А141

Стадия 1: 3-метиленциклобутанкарбоновая кислота (А141-1)
В перемешиваемый раствор KOH (10 г, 178 ммоль) в воде (15 мл) и EtOH (15 мл) добавляли 3-метиленциклобутанкарбонитрил (3.92 г, 42 ммоль) при комнатной температуре в течение 10 минут. Полученную смесь оставляли нагреваться до 90°С и перемешивали при той же температуре 3.5 часа. Реакционную смесь упаривали при пониженном давлении. Остаток от упаривания растворяли в воде (10 мл) при 0°С. Полученную смесь подкисляли добавлением 6 М водного раствора HCl до рН 1 и экстрагировали дихлорметаном. Органический слой сушили над MgSO4 и упаривали при пониженном давлении, получая соединение А141-1 (4.65 г, 98%) в виде бесцветного масла. Полученный продукт использовали в следующей стадии без дополнительной очистки.
Стадия 2: метил 3-метиленциклобутанкарбоксилат (А141-2)
Триметилсилилдиазометан (2.0 М раствор в гексане, 25 мл, 50 ммоль) добавляли в перемешиваемый раствор соединения А141-1 (4.64 г, 41.4 ммоль) в ДХМ (25 мл) и МеОН (5 мл) по каплям при 0°С в течение 5 минут. Полученную смесь оставляли нагреваться до комнатной температуры и перемешивали при той же температуре 30 минут. Реакционную смесь гасили добавлением АсОН (0.45 мл) и упаривали при пониженном давлении. Остаток от упаривания очищали методом хроматографии на силикагеле (20% ДХМ/гексан в качестве элюента), получая соединение А141-2 (3.8 г, 73%) в виде бесцветного масла.
Стадия 3: метил спиро[2.3]гексан-5-карбоксилат (А141-3)
В раствор диэтилцинка (1.0 М раствор в гексане, 46 мл, 46 ммоль) в ДХМ (200 мл) добавляли раствор ТФУК (3.54 мл, 46 ммоль) в ДХМ (50 мл) по каплям при 0°С в течение 30 минут. Добавляли по каплям раствор дииодметана (3.7 мл, 46 ммоль) в ДХМ (50 мл) при 0°С в течение 45 минут. Полученную смесь перемешивали при той же температуре 1 час. Раствор соединения А141-2 (2.52 г, 20 ммоль) в ДХМ (30 мл) добавляли в реакционную смесь. Полученную смесь оставляли нагреваться до комнатной температуры в течение ночи. Реакционную смесь гасили насыщенным водным раствором NH4Cl (200 мл) и экстрагировали дихлорметаном. Отделенный органический слой сушили над MgSO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом хроматографии на силикагеле (20% EtOAc/гексан в качестве элюента), получая соединение А141-3 (1.77 г, 63%) в виде бесцветного масла.
Стадия 4: спиро[2.3]гексан-5-карбоновая кислота (А141-4)
В перемешиваемый раствор LiOH (4 М в воде, 10 мл, 40 ммоль) в воде (10 мл) и МеОН (20 мл) добавляли соединение А141-3 (1.76 г, 12.6 ммоль) при комнатной температуре. Полученную смесь перемешивали при комнатной температуре в течение 40 минут. Реакционную смесь упаривали при пониженном давлении до объема раствора примерно 20 мл. Полученный раствор подкисляли добавлением 6 М водного раствора HCl до рН 1 и экстрагировали дихлорметаном. Органический слой сушили над MgSO4 и упаривали при пониженном давлении, получая соединение А141-4 (1.51 г, 95%) в виде бесцветного масла. Полученный продукт использовали в следующей стадии без дополнительной очистки.
Стадия 5: N-метокси-N-метилспиро[2.3]гексан-5-карбоксамид (А141-5)
В смесь соединения А141-4 (1.51 мг, 12.0 ммоль), 1-гидроксибензотриазола моногидрата (2.30 г, 15 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (2.33 г, 15 ммоль) и N,O-диметилгидроксиламина гидрохлорида (1.46 г, 15 ммоль) в ДМФА (20 мл) добавляли DIPEA (3.43 мл, 20 ммоль), и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили водой и экстрагировали гексаном и EtOAc. Отделенный органический слой промывали 1 М водным раствором HCl (100 мл), водой, насыщенным водным раствором Na2CO3 (2×100 мл), насыщенным водным раствором поваренной соли, сушили над MgSO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом хроматографии на силикагеле (75% EtOAc/гексан в качестве элюента), получая соединение А141-5 (1.72 г, 84%) в виде бесцветного масла.
Стадия 6: спиро[2.3]гексан-5-карбальдегид (А141-6)
В раствор соединения А141-5 (677 мг, 4 ммоль) в Et2O (15 мл) добавляли суспензию LiAlH4 (152 мг, 4 ммоль) в Et2O (5 мл) при 0°С в течение 5 минут и перемешивали 2 часа при той же температуре. Реакционную смесь гасили насыщенным водным раствором KHSO4 (10 мл) при 0°С и экстрагировали Et2O. Объединенный органический слой сушили над MgSO4 и упаривали при пониженном давлении, получая соединение А141-6 (351 мг, 80%) в виде бесцветного масла. Полученный неочищенный продукт использовали в следующей стадии без очистки.
Стадия 7: 2-(2,4,6-трихлорфенил)-N-(спиро[2.3]гексан-5-илметил)-2-((триэтилсилил)окси) этанамин (А141)
Соединение А141 (123 мг, 39%) получали в виде бледно-желтого масла реакцией 2-(2,4,6-трихлорфенил)-2-((триэтилсилил)окси)этанамина (248 мг, 0.7 ммоль), соединения А141-6 (100 мг, 0.91 ммоль), NaBH4 (212 мг) и MgSO4 (100 мг) в МеОН (1.4 мл) и ТГФ (3.5 мл) по методике, аналогичной описанной в справочном примере А31, стадия 4. 1H ЯМР (CDCl3, 400 МГц): δ 7.29 (с, 2Н), 5.53 (дд, J=9.0, J=4.6 Гц, 1Н), 3.26 (дд, J=12.2, J=8.8 Гц, 1Н), 2.85-2.71 (м, 3Н), 2.62-2.51 (м, 1Н), 2.17-2.10 (м, 2Н), 1.86-1.81 (м, 2Н), 0.87 (т, J=7.8 Гц, 9Н), 0.57-0.50 (м, 6Н), 0.43-0.33 (м, 4Н).
Справочный пример А194
1-(2,6-дихлор-3-фторфенил)-2-(((1-(трифторметил)циклопропил)метил)амино)этанол
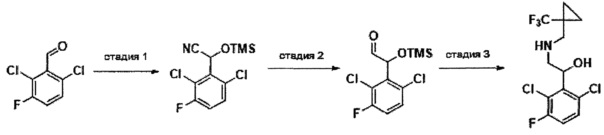
Стадия 1: 2-(2,6-дихлор-3-фторфенил)-2-((триметилсилил)окси)ацетонитрил
В круглодонную колбу объемом 200 мл помещали раствор 2,6-дихлор-3-фторбензальдегида (2.29 г, 11.87 ммоль), ДХМ (23 мл), TMSCN (1.9 мл, 14.24 ммоль) и добавляли иодид цинка (0.379 г, 1.187 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 4 часов. Затем реакционную смесь промывали водой (2×20 мл) и насыщенным водным раствором поваренной соли. Органический слой упаривали при пониженном давлении. Полученный сырой продукт очищали методом колоночной хроматографии (силикагель, элюент: 0%-30% EtOAc/гептан), получая 2-(2,6-дихлор-3-фторфенил)-2-((триметилсилил)окси)ацетонитрил (1.435 г, 4.91 ммоль, 41.4% выход) в виде бесцветного масла. 1Н ЯМР (400 МГц, CDCl3) δ 7.32-7.42 (м, 1Н); 7.12-7.24 (м, 1Н); 6.17-6.30 (м, 1Н); 0.12-0.33 (м, 9Н).
Стадия 2: 2-(2,6-дихлор-3-фторфенил)-2-((триметилсилил)окси)ацетальдегид
В круглодонную колбу объемом 100 мл помещали 2-(2,6-дихлор-3-фторфенил)-2-((триметилсилил)окси)ацетонитрил (0.50 г, 1.711 ммоль) и ДХМ (9 мл). Реакционную смесь продували азотом и охлаждали до -64°С. В атмосфере азота добавляли по каплям диизобутилалюминий гидрид, 1.0 М раствор в гексане (2.6 мл, 2.6 ммоль). Полученную смесь перемешивали при -64°С. Через 2 часа реакцию гасили. Поддерживая температуру <-65°С, осторожно добавляли по каплям МеОН (1.4 мл, 34.2 ммоль) в реакционную смесь, затем насыщенный раствор сегнетовой соли (5 мл). Полученную смесь оставляли нагреваться до комнатной температуры и перемешивали 30 минут. Добавляли воду и ДХМ, и водный слой экстрагировали дихлорметаном. Объединенный органический слой промывали насыщенным водным раствором поваренной соли, сушили над безводным MgSO4 и упаривали, получая 2-(2,6-дихлор-3-фторфенил)-2-((триметилсилил)окси) ацетальдегид в виде бесцветного масла (0.517 г, неочищенное).
Стадия 3: 1-(2,6-дихлор-3-фторфенил)-2-(((1-(трифторметил)циклопропил)метил)амино)этанол
В раствор сырого 2-(2,6-дихлор-3-фторфенил)-2-((триметилсилил)окси) ацетальдегида (0.258 г, 0.874 ммоль) в MeCN (9 мл) добавляли (1-(трифторметил) циклопропил)метанамин (0.122 г, 0.874 ммоль), затем АсОН (0.050 мл, 0.874 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли NaBH(ОАс)3 (0.370 г, 1.748 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 23 часов. Потом ее гасили насыщенным водным раствором NaHCO3 и перемешивали 30 минут. Экстрагировали дихлорметаном (2×5 мл). Объединенный органический слой промывали насыщенным водным раствором поваренной соли, сушили над безводным MgSO4 и упаривали при пониженном давлении, получая желтое масло. Полученное желтое масло растворяли в 2 мл ТГФ. Затем добавляли TBAF, 1.0 М раствор в ТГФ (0.874 мл, 0.874 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 минут. Ее гасили насыщенным водным раствором NaHCO3 и экстрагировали дихлорметаном. Объединенный органический слой сушили над безводным MgSO4 и упаривали при пониженном давлении. Полученный сырой продукт очищали методом колоночной хроматографии (силикагель, элюент: 0%-50% EtOAc/гептан), получая 1-(2,6-дихлор-3-фторфенил)-2-(((1-(трифторметил) циклопропил)метил)амино)этанол (116 мг, 0.335 ммоль, 38.3% выход) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7.25-7.31 (м, 1Н), 7.06 (дд, J=8.9, 8.0 Гц, 1Н), 5.45 (дд, J=9.7, 4.5 Гц, 1Н), 3.45 (ушир.с, 1Н), 3.28 (дд, J=12.6, 9.8 Гц, 1Н), 2.89-2.92 (м, 3Н), 0.99-1.04 (м, 2Н), 0.69-0.76 (м, 2Н); LCMS: 346.0 [М+Н]+.
Справочный пример А224
2-(2,6-дихлор-4-фторфенил)-N-((1-метилциклопропил)метил)-2-((триэтилсилил)окси)этанамин

Смесь 1-метилциклопропанкарбальдегида (31.6 мг, 0.375 ммоль) и 2-(2,6-дихлор-4-фторфенил)-2-((триэтилсилил)окси)этанамина (127 мг, 0.375 ммоль) в МеОН (1.9 мл) перемешивали при комнатной температуре в течение 3 часов. Добавляли порциями NaBH4 (14.20 мг, 0.375 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 40 минут. Полученную смесь упаривали и очищали методом препаративной ТСХ, элюируя 5% МеОН/ДХМ, получая 2-(2,6-дихлор-4-фторфенил)-N-((1-метилциклопропил)метил)-2-((триэтилсилил)окси)этанамин (111 мг, 0.273 ммоль, 72.8% выход). 1H ЯМР (500 МГц, CDCl3) δ 7.29 (с, 1Н), 7.06-7.10 (м, 2Н), 5.56 (ушир.с, 1Н), 3.33 (т, J=10.51 Гц, 1Н), 2.81 (д, J=9.17 Гц, 1Н), 2.60-2.67 (м, 1Н), 2.44 (д, J=11.86 Гц, 1Н), 1.46-1.59 (м, 1Н), 1.13 (с, 3Н), 0.85-0.96 (м, 9Н), 0.50-0.63 (м, 6Н), 0.36 (ушир.с, 2Н), 0.30 (ушир.с, 2Н); LCMS (ESI) m/z 406.0 (М+Н)+.
Справочный пример А258
2-(2,6-дихлорфенил)-N-((1-метилциклопропил)метил)-2-((триэтилсилил)окси)этанамин
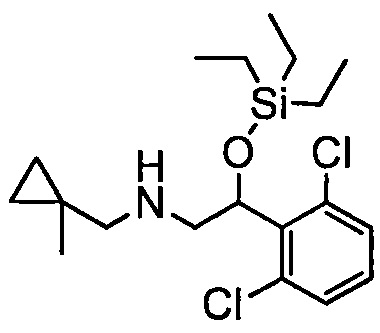
В смесь 1-метилциклопропанкарбальдегида (32.8 мг, 0.390 ммоль) в ДХМ (2.0 мл) добавляли 2-(2,6-дихлорфенил)-2-((триэтилсилил)окси)этанамин (125 мг, 0.390 ммоль), затем NaBH(ОАс)3 (124 мг, 0.585 ммоль). Через 45 минут реакцию гасили насыщенным водным раствором NaHCO3. Слои разделяли. Водный слой экстрагировали дихлорметаном. Объединенные органические слои упаривали, затем очищали методом препаративной ТСХ, элюируя 5% МеОН/ДХМ, получая 2-(2,6-дихлорфенил)-N-((1-метилциклопропил)метил)-2-((триэтилсилил)окси)этанамин (95 мг, 0.245 ммоль, 62.7% выход). 1Н ЯМР (500 МГц, CDCl3) δ 7.28-7.30 (м, 2Н), 7.12-7.16 (м, 1Н), 5.65 (ушир.с, 1Н), 3.37-3.44 (м, 1Н), 2.82-2.90 (м, 1Н), 2.69 (ушир.с, 1Н), 2.48 (д, J=11.86 Гц, 1Н), 1.60 (ушир.с, 1Н), 1.15 (с, 3Н), 0.87-0.92 (м, 9Н), 0.51-0.63 (м, 6Н), 0.28-0.44 (м, 4Н); LCMS (ESI) m/z 388.3 (М+Н)+.
Справочный пример А259
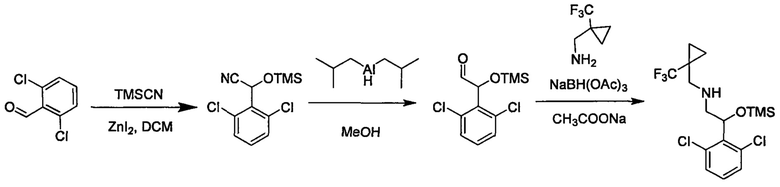
Стадия 1: 2-(2,6-дихлорфенил)-2-((триметилсилил)окси)ацетонитрил
В круглодонную колбу объемом 100 мл помещали раствор 2,6-дихлорбензальдегида (5.08 г, 29.0 ммоль) и TMSCN (4.64 мл, 34.8 ммоль) в ДХМ (60 мл). Добавляли иодид цинка (0.926 г, 2.90 ммоль), и полученную смесь перемешивали при комнатной температуре 3 часа. Реакционную смесь разбавляли ДХМ (200 мл). Органический слой промывали водой (2×20 мл) и насыщенным водным раствором поваренной соли (20 мл), органический слой фильтровали через целит и упаривали. Остаток от упаривания очищали методом флэш-хроматографии на 100 г Biotage SNAP картридже, используя 0-40% EtOAc в гептане, получая 2-(2,6-дихлорфенил)-2-((триметилсилил)окси)ацетонитрил (3.01 г, 38%).
Стадия 2: 2-(2,6-дихлорфенил)-2-((триметилсилил)окси)ацетальдегид.
В раствор 2-(2,6-дихлорфенил)-2-((триметилсилил)окси)ацетонитрила (1.372 г, 5.00 ммоль) в ДХМ (23.16 мл) добавляли 1.0 М раствор диизобутилалюминия гидрида в гексане (7.50 мл, 7.50 ммоль) при -78°С по каплям в течение 20 минут. Реакцию осторожно гасили сначала добавлением МеОН (1 мл, 24.97 ммоль) и затем 1.5 М раствора сегнетовой соли (5.00 мл, 7.50 ммоль). Колбу вынимали из бани, оставляли нагреваться до комнатной температуры и экстрагировали этилацетатом (20 мл). Органический слой отделяли и промывали насыщенным водным раствором поваренной соли, фильтровали через слой целита и упаривали, получая 2-(2,6-дихлорфенил)-2-((триметилсилил)окси) ацетальдегид (1.34 г, 97%) в виде белого твердого вещества.
Стадия 3: 2-(2,6-дихлорфенил)-N-((1-(трифторметил)циклопропил)метил)-2-((триметил-силил)окси)этанамин.
В раствор сырого 2-(2,6-дихлорфенил)-2-((триметилсилил)окси)ацетальдегида (0.35 г, 1.263 ммоль) в ДХМ (6.31 мл) добавляли (1-(трифторметил)циклопропил)метанамин (0.176 г, 1.263 ммоль) и NaBH(OAc)3 (0.374 мл, 2.53 ммоль) и перемешивали 2 часа при комнатной температуре. Реакцию гасили насыщенным водным раствором NH4Cl и разбавляли добавлением ДХМ (50 мл). Органический слой пропускали через разделитель фаз и упаривали, получая 2-(2,6-дихлорфенил)-N-((1-(трифторметил)циклопропил)метил)-2-((триметилсилил)окси)этанамин (0.378 г, 70%) в виде светло-желтого масла. Его использовали в следующей стадии без дополнительной очистки.
Справочный пример А260
2-(2,6-дихлорфенил)-N-((1-метилциклобутил)метил)-2-((триэтилсилил)окси)этанамин
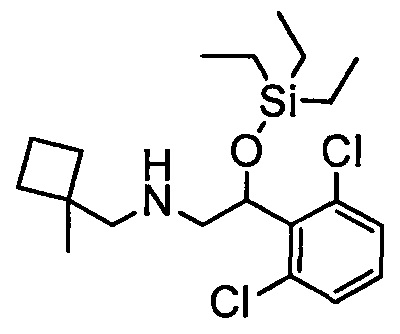
В смесь 1-метилциклобутанкарбальдегида (38.3 мг, 0.390 ммоль) в ДХМ (2.0 мл) добавляли 2-(2,6-дихлорфенил)-2-((триэтилсилил)окси)этанамин (125 мг, 0.390 ммоль), затем NaBH(ОАс)3 (124 мг, 0.585 ммоль). Через 45 минут реакцию гасили насыщенным водным раствором NaHCO3. Слои разделяли. Водный слой экстрагировали дихлорметаном. Объединенные органические слои упаривали и очищали методом препаративной ТСХ, элюируя 5% МеОН/ДХМ, получая 2-(2,6-дихлорфенил)-N-((1-метилциклобутил)метил)-2-((триэтилсилил)окси)этанамин (89 мг, 0.221 ммоль, 56.7% выход). 1Н ЯМР (500 МГц, CDCl3) δ 7.18-7.22 (м, 2Н), 6.98-7.10 (м, 1Н), 5.57 (ушир.с, 1Н), 3.30 (т, J=10.70 Гц, 1Н), 2.74 (ушир.с, 1Н), 2.60 (ушир.с, 1Н), 2.51 (д, J=10.03 Гц, 1Н), 1.68-1.89 (м, 4Н), 1.60 (ушир.с, 2Н), 1.47 (ушир.с, 1Н), 1.03-1.14 (м, 3Н), 0.76-0.84 (м, 9Н), 0.41-0.54 (м, 6Н); LCMS (ESI) m/z 402.4 (М+Н)+.
Справочный пример А262
2-(2,6-дихлорфенил)-N-((5-фторспиро[2.3]гексан-5-ил)метил)-2-((триэтилсилил)окси)этанамин
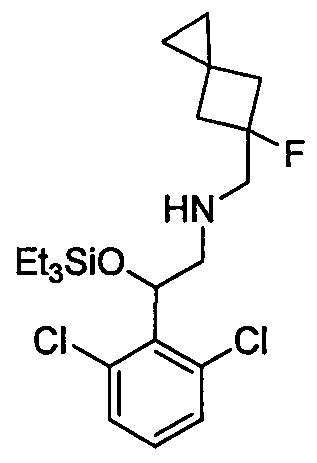
Спиро[2.3]гексан-5-карбальдегид (300 мг, 2.72 ммоль) и N-этил-N-изопропилпропан-2-амин (546 мкл, 3.13 ммоль) смешивали в MeCN (5 мл) и добавляли по каплям триметилсилил трифторметансульфонат (517 мкл, 2.86 ммоль). Полученный раствор перемешивали 30 минут и добавляли Selectfluor (1061 мг, 3.00 ммоль) в MeCN (5 мл). Полученный раствор перемешивали и обрабатывали ультразвуком еще 30 минут. Добавляли 2-(2,6-дихлорфенил)-2-((триэтилсилил)окси)этанамин (785 мг, 2.451 ммоль) и АсОН (187 мкл, 3.27 ммоль). Полученный раствор перемешивали 30 минут и добавляли NaBH(OAc)3 (1154 мг, 5.45 ммоль), раствор перемешивали еще 2 часа. Полученный раствор гасили насыщенным водным раствором NaHCO3, водный слой экстрагировали этилацетатом, объединенные органические слои промывали насыщенным водным раствором поваренной соли и сушили над безводным Na2SO4, фильтровали и упаривали. Полученный продукт очищали колоночной хроматографией на силикагеле (40 г колонка) с применением 0-100% EtOAc в гептане, получая 2-(2,6-дихлорфенил)-N-((5-фторспиро[2.3]гексан-5-ил)метил)-2-((триэтилсилил)окси)этанамин (300 мг, 0.694 ммоль, 25.5% выход). MS m/z=432 [М+Н]+.
Справочный пример А267
2-(2,6-дихлорфенил)-N-(спиро[2.5]октан-6-илметил)-2-((триэтилсилил)окси)этанамин
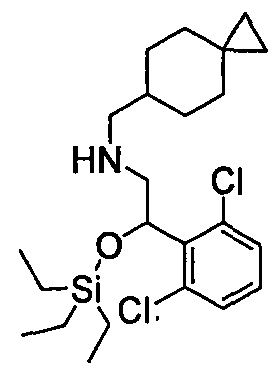
В раствор f 2-(2,6-дихлорфенил)-2-((триэтилсилил)окси)этанамина (248 мг, 0.774 ммоль) в ДХМ (2581 мкл) добавляли спиро[2.5]октане-6-карбальдегид (107 мг, 0.774 ммоль), АсОН (35.5 мкл, 0.619 ммоль) и NaBH(ОАс)3 (246 мг, 1.161 ммоль). Мутную смесь перемешивали при комнатной температуре в течение ночи. Полученную смесь гасили добавлением 0.5 М раствора NaOH, и смесь перемешивали при комнатной температуре 30 минут. Наблюдалось выделение газа. Слои разделяли. Органический слой сушили над Na2SO4 и упаривали. Остаток от упаривания очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 100% EtOAc в гексане, получая 2-(2,6-дихлорфенил)-N-(спиро[2.5]октан-6-илметил)-2-((триэтилсилил)окси) этанамин.
Справочный пример А275
N-(2-(3-хлорхинолин-4-ил)-2-((триэтилсилил)окси)этил)-2,2-диметилпропан-1-амин
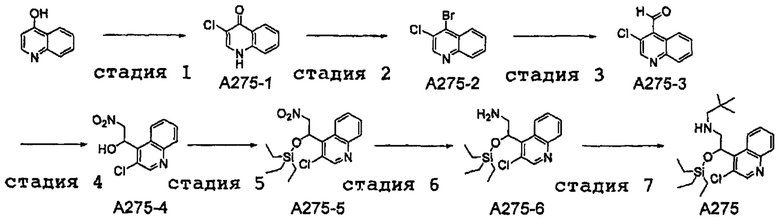
Стадия 1: 3-хлорхинолин-4(1Н)-он (А275-1)
В смесь 4-гидроксихинолина (5.33 г, 36.7 ммоль) в АсОН (184 мл) добавляли N-хлорсукцинимид (6.37 г, 47.7 ммоль), и полученную желтую гомогенную смесь перемешивали и нагревали до 60°С. Через 3 часа смесь охлаждали до комнатной температуры и упаривали в вакууме. Добавляли насыщенный водный раствор NaHCO3 (300 мл) до тех пор, пока рН не достиг ~8.5. Выпавший твердый осадок отделяли фильтрованием, промывали водой (300 мл) и сушили в высоком вакууме, получая 3-хлорхинолин-4(1Н)-он (А275-1) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 12.28 (1Н, ушир.с), 8.40 (1Н, д, J=6.5 Гц), 8.15 (1Н, дд, J=8.2, 1.4 Гц), 7.65-7.73 (1Н, м), 7.58-7.63 (1Н, м), 7.39 (1Н, ддд, J=8.1, 6.9, 1.2 Гц); LCMS (ESI) m/z 180.1 (M+H)+.
Стадия 2: 4-бром-3-хлорхинолин (А275-2)
В охлажденную суспензию 3-хлорхинолин-4(1Н)-она (А275-1) (5.15 г, 28.7 ммоль) в ДМФА (43.4 мл) при 0°С добавляли трибромид фосфора (2.77 мл, 29.5 ммоль) по каплям в течение 3 минут, и затем смесь становилась оранжевой и гомогенной. Через 4 минуты образовывался желтый осадок, и полученную желтую гетерогенную смесь дополнительно перемешивали при 0°С еще 15 минут. Через 15 минут охлаждающую баню удаляли, и желтую гетерогенную смесь перемешивали при комнатной температуре. Через 15 часов, смесь выливали в ледяную воду (300 мл) и перемешивали при 0°С 20 минут. Полученную смесь затем нейтрализовывали добавлением 2 М раствора NaOH (50 мл) до рН>9 (рН бумага). Выпавший осадок отделяли фильтрованием, промывали полученный осадок водой (400 мл) и сушили в высоком вакууме, получая 4-бром-3-хлорхинолин (А275-2) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8.96 (1Н, с), 8.20 (1Н, дц, J=8.2, 1.6 Гц), 8.12 (1Н, дд, J=8.3, 0.9 Гц), 7.81-7.93 (2Н, м); LCMS (ESI) m/z 242.0 [М+Н (79Br)]+ и 243.9 [М+Н (81Br)]+.
Стадия 3: 3-хлорхинолин-4-карбальдегид (А275-3)
В колбу помещали 4-бром-3-хлорхинолин (А275-2) (1.00 г, 4.12 ммоль) и ТГФ (16.5 мл) в атмосфере азота, и раствор охлаждали до -78°С. В охлажденную смесь добавляли н-бутиллитий (2.5 М раствор в гексан, 1.65 мл, 4.12 ммоль), и полученную смесь перемешивали при -78°С в течение 1 часа. В смесь по каплям добавляли ДМФА (1.60 мл, 20.6 ммоль), и оставляли нагреваться до комнатной температуры. Через 4 часа смесь гасили насыщенным водным раствором NH4Cl (20 мл). Полученную смесь разделяли между водой (50 мл) и EtOAc (50 мл). Водный слой экстрагировали этилацетатом (1×50 мл). Органический экстракт сушили над MgSO4. Полученный раствор фильтровали и упаривали в вакууме получая сырой продукт в виде коричневого сиропа. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом хроматографии на преупакованной REDISEP™ колонке с силикагелем (80 г), элюируя в градиенте от 0% до 20% EtOAc в гексане, и сушили в высоком вакууме, получая 3-хлорхинолин-4-карбальдегид (А275-3) в виде коричневого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 10.74 (1Н, с), 9.10 (1Н, с), 8.68-8.73 (1Н, м), 8.15 (1Н, дд, J=8.5, 0,9 Гц), 7.79-7.92 (2Н, м); LCMS (ESI) m/z 192.1 (M+H)+.
Стадия 4: 1-(3-хлорхинолин-4-ил)-2-нитроэтанол (А275-4)
В прозрачный коричневый раствор 3-хлорхинолин-4-карбальдегида (А275-3) (0.362 г, 1.89 ммоль) в ТГФ (1.9 мл) при комнатной температуре добавляли карбонат калия (0.078 г, 0.566 ммоль) и нитрометан (1.420 мл, 26.4 ммоль). Коричневую гомогенную смесь перемешивали при комнатной температуре. Через 4 часа реакционную смесь гасили водой (50 мл) и экстрагировали этилацетатом (2×50 мл). Органический экстракт промывали насыщенным водным раствором NaCl (1×50 мл) и сушили над Na2SO4. Полученный раствор фильтровали, упаривали в вакууме и сушили в высоком вакууме, получая 1-(3-хлорхинолин-4-ил)-2-нитроэтанол (А275-4) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8.88-8.93 (1Н, м), 8.73 (1Н, дд, J=8.6, 0.8 Гц), 8.08 (1Н, дд, J=84, 1.0 Гц), 7.82 (1Н, ддд, J=84, 6.9, 1.5 Гц), 7.72 (1Н, ддд, J=8.5, 6.9, 1.4 Гц), 6.91 (1Н, дд, J=4.5, 1.0 Гц), 6.26 (1Н, ддд, J=10.0, 4.6, 3.6 Гц), 5.03-5.12 (1Н, м), 4.94-5.01 (1Н, м); LC-MS (ESI) m/z 253.1 (М+Н)+.
Стадия 5: 3-хлор-4-(2-нитро-1-((триэтилсилил)окси)этил)хинолин (А275-5)
В прозрачный коричневый раствор 1-(3-хлорхинолин-4-ил)-2-нитроэтанола (А275-4) (0.423 г, 1.68 ммоль) в ДМФА (4.19 мл) при комнатной температуре добавляли имидазол (0.342 г, 5.03 ммоль) и триэтилсилил хлорид (0.341 мл, 2.01 ммоль). Полученную смесь перемешивали при комнатной температуре. Через 2 часа смесь гасили водой (50 мл) и экстрагировали этилацетатом (2×50 мл). Органический экстракт промывали 1 М раствором LiCl (1×50 мл) и насыщенным водным раствором поваренной соли (1×50 мл), и сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде желтого сиропа. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом хроматографии на преупакованной REDISEP™ колонке с силикагелем (40 г), элюируя в градиенте от 0% до 10% EtOAc в гексане, и сушили в высоком вакууме, получая 3-хлор-4-(2-нитро-1-((триэтилсилил)окси)этил)хинолин (А275-5). 1H ЯМР (400 МГц, ДМСО-d6) δ 8.95 (1Н, с), 8.67 (1Н, д, J=7.4 Гц), 8.10 (1Н, дд, J=8А, 0.8 Гц), 7.84 (1Н, тд, J=7.6, 1.4 Гц), 7.71-7.79 (1Н, м), 6.38 (1Н, дд, J=9.8, 2.5 Гц), 5.14-5.23 (1Н, м), 5.03-5.11 (1Н, м), 0.65-0.74 (9Н, м), 0.32-0.51 (6Н, м); LCMS (ESI) m/z 367.1 (M+H)+.
Стадия 6: 2-(3-хлорхинолин-4-ил)-2-((триэтилсилил)окси)этанамин (А275-6)
В прозрачный желтый раствор 3-хлор-4-(2-нитро-1-((триэтилсилил)окси)этил) хинолина (0.511 г, 1.39 ммоль) в EtOH (7.96 мл) и воде (1.99 мл) при комнатной температуре добавляли порошок железа (0.778 г, 13.9 ммоль) и хлорид аммония (0.745 г, 13.9 ммоль). Темно-коричневую результирующую смесь перемешивали и нагревали до 60°С. Через 4 часа смесь охлаждали до комнатной температуры и фильтровали через слой силикагеля, промывали осадок на фильтре метанолом (3×30 мл). Объединенные фильтраты упаривали в вакууме. Остаток от упаривания разделяли между EtOAc (100 мл) и водой (50 мл). Полученную смесь (рН -4.0) промывали насыщенным водным раствором NaHCO3 (1×50 мл), водой (1×50 мл) и насыщенным водным раствором поваренной соли (1×50 мл), сушили над безводным Na2SO4, упаривали в вакууме, и сушили в высоком вакууме получая 2-(3-хлорхинолин-4-ил)-2-((триэтилсилил)окси)этанамин (А275-6) в виде желтого сиропа. 1H ЯМР (400 МГц, ДМСО-d6) δ 8.84 (1Н, с), 8.72 (1Н, д, J=8.2 Гц), 8.04 (1Н, дд, J=84, 1.0 Гц), 7.76 (1Н, ддд, J=8А, 6.9, 1.4 Гц), 7.64 (1Н, ддд, J=8.5, 7.0, 1.3 Гц), 5.52 (1Н, дд, J=7.6, 5.5 Гц), 3.16 (1Н, дд, J=13.0, 7.9 Гц), 2.88 (1Н, дд, J=13.0, 5.4 Гц), 1.74 (1Н, ушир.с), 0.71-0.80 (1Н, м), 0.71-0.80 (9Н, м), 0.37-0.57 (6Н, м); LCMS (ESI) m/z 337.1 (M+H)+.
Стадия 7: N-(2-(3-хлорхинолин-4-ил)-2-((триэтилсилил)окси)этил)-2,2-диметилпропан-1-амин (А275)
В прозрачный желтый раствор 2-(3-хлорхинолин-4-ил)-2-((триэтилсилил)окси) этанамина (А275-6) (0.217 г, 0.644 ммоль) в ДХМ (2.15 мл) добавляли триметилацетальдегид (0.077 мл, 0.71 ммоль), АсОН (0.045 мл, 0.77 ммоль) и NaBH(OAc)3 (0.205 г, 0.966 ммоль). Желтую гомогенную смесь перемешивали при комнатной температуре. Через 2 часа смесь гасили водой (20 мл) и нейтрализовывали 0.5 М раствором NaOH (10 мл) до рН ~9.0. Реакционную смесь экстрагировали дихлорметаном (2×50 мл). Органический экстракт промывали насыщенным водным раствором NaCl (1×50 мл) и сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде желтого сиропа. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом хроматографии на преупакованной REDISEP™ колонке с силикагелем (40 г), элюируя в градиенте от 0% до 20% EtOAc в гексане, и сушили в высоком вакууме, получая N-(2-(3-хлорхинолин-4-ил)-2-((триэтилсилил)окси)этил)-2,2-диметилпропан-1-амин (А275) в виде бесцветного сиропа. 1H ЯМР (400 МГц, ДМСО-d6) δ 8.84 (1Н, с), 8.75 (1Н, д, J=7.2 Гц), 8.04 (1Н, дд, J=84, 1.0 Гц), 7.77 (1Н, ддд, J=84, 6.9, 1.4 Гц), 7.61-7.68 (1Н, м), 5.70 (1Н, дд, J=7.7, 5.0 Гц), 3.26 (1Н, дд, J=12.6, 8.1 Гц), 2.85 (1Н, дд, J=12.6, 5.0 Гц), 2.23-2.39 (2Н, м), 1.72 (1Н, ушир.с), 0.81 (9Н, с), 0.72-0.79 (9Н, м), 0.36-0.56 (6Н, м); LCMS (ESI) m/z 407.1 (M+H)+.
Справочный пример А281
2-(3,5-дихлорпиридин-4-ил)-N-((5-метилтетрагидрофуран-2-ил)метил)-2-((триэтилсилил)окси)этанамин
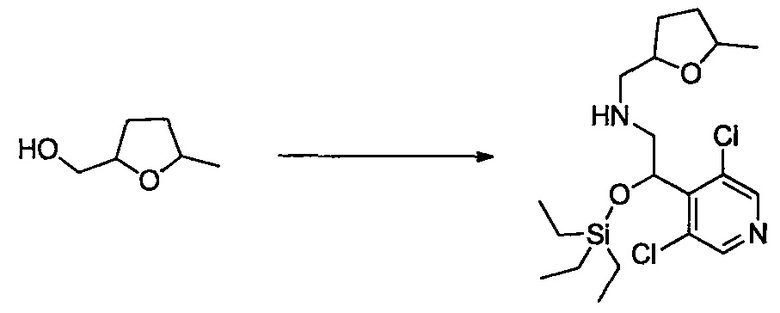
В прозрачный раствор 5-метилтетрагидрофуран-2-метанола в ДХМ добавляли периодинан Десс-Мартина (1.2 экв.). Полученную смесь перемешивали при комнатной температуре в течение ночи. Неочищенную смесь напрямую добавляли в раствор 2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этанамин (1 экв.) в ДХМ, затем добавляли АсОН (1.2 экв.) и NaBH(ОАс)3 (1.5 экв.). Реакционную смесь перемешивали при комнатной температуре. Через 2 часа смесь гасили насыщенным водным раствором Na2S2O3 и насыщенным NaHCO3. Реакционную смесь экстрагировали дихлорметаном. Органический экстракт сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 25% EtOAc в гептане, получая 2-(3,5-дихлорпиридин-4-ил)-N-((5-метилтетрагидрофуран-2-ил)метил)-2-((триэтилсилил)окси)этанамин (А281) в виде светло-желтого сиропа. 1H ЯМР (300 МГц, ДМСО-d6) δ 8.58 (2Н, с), 5.34-5.46 (1Н, м), 3.71-3.90 (2Н, м), 3.10 (1Н, дт, J=12.5, 8.1 Гц), 2.90 (1Н, тд, J=12.1, 6.0 Гц), 2.52-2.67 (2Н, м), 1.71-2.07 (3Н, м), 1.47-1.64 (1Н, м), 1.19-1.38 (1Н, м), 1.11 (3Н, т, J=6.3 Гц), 0.77-0.89 (9Н, м), 0.40-0.62 (6Н, м); LCMS (ESI) m/z 419.1 (М+Н)+.
Справочный пример А294
N-(2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)-3,3,3-трифтор-2,2-диметилпропан-1-амин

Стадия 01: 3,3,3-трифтор-N-метокси-N,2,2-триметилпропанамид (А294-1)
В прозрачный раствор 3,3,3-трифтор-2,2-диметилпропионовой кислоты (5.000 г, 32.0 ммоль) в MeCN (22.88 мл) добавляли триэтиламин (9.82 мл, 70.5 ммоль), затем HATU (12.79 г, 33.6 ммоль), и полученную смесь перемешивали при комнатной температуре. Через 15 мин, в полученную темную прозрачную жидкость добавляли N,O-диметилгидроксиламина гидрохлорид (3.44 г, 35.2 ммоль), и полученную смесь перемешивали при комнатной температуре. Через 18 часов реакционную смесь разбавили EtOAc (100 мл) и промывали 1 н. раствором HCl (2×100 мл) и насыщенным раствором NaCl (5×100 мл) и сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде оранжевого твердого вещества. Полученное оранжевое твердое вещество адсорбировали на слое силикагеля и очищали методом хроматографии на силикагеле, элюируя в градиенте от 0% до 25% EtOAc в гептане, получая 3,3,3-трифтор-N-метокси-N,2,2-триметилпропанамид (5.0503 г, 25.4 ммоль, 79% выход) в виде желтой жидкости. 1Н ЯМР (300 МГц, CDCl3) δ 3.71 (3Н, с), 3.22 (3Н, с), 1.51 (6Н, д, J=0.7 Гц); LCMS (ESI) m/z 200.1 (М+Н)+.
Стадия 2: 3,3,3-трифтор-2,2-диметилпропаналь
В трехгорлую круглодонную колбу объемом 250 мл, оснащенную S-образной насадкой для подачи азота и для термопары, добавляли литийалюминий гидрид, 1 М раствор в Et2O (25.3 мл, 25.3 ммоль) при 0°С. В охлажденную смесь добавляли раствор 3,3,3-трифтор-N-метокси-N,2,2-триметилпропанамида (А294-1) (5.0325 г, 25.3 ммоль) в Et2O (47.7 мл) по каплям в течение 35 минут при 0°С. После окончания добавления, реакционную смесь дополнительно перемешивали при 0°С. Через 2 часа смесь осторожно гасили при 0°С водой (0.96 мл), NaOH (15%, 0.96 мл) и водой (2.88 мл), и полученную смесь интенсивно перемешивали 40 минут. Реакционную смесь разбавляли Et2O (50 мл), добавляли Na2SO4 и затем фильтровали через слой силикагеля, промывали Et2O (100 мл). Фильтрат упаривали в вакууме, получая 3,3,3-трифтор-2,2-диметилпропаналь (А294-2) (3.2304 г, 23.06 ммоль, 91% выход) в виде желтой жидкости. 1Н ЯМР (400 МГц, CDCl3) δ 9.69 (1Н, д, J=7.4 Гц), 1.31 (6Н, с).
Стадия 3: N-(2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)-3,3,3-трифтор-2,2-диметилпропан-1-амин (А294)
В желтую прозрачную смесь 2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этанамина (3.57 г, 11.11 ммоль) в ДХМ (37.0 мл) добавляли 3,3,3-трифтор-2,2-диметилпропаналь (11.11 ммоль) в ДХМ, затем АсОН (0.770 мл, 13.33 ммоль) и NaBH(OAc)3 (3.53 г, 16.67 ммоль). Полученную желтую гетерогенную смесь перемешивали при комнатной температуре. Через 8 часов смесь гасили насыщенным водным раствором NaHCO3 (100 мл). Реакционную смесь экстрагировали дихлорметаном (2×100 мл). Органический экстракт сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде оранжевого сиропа. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 20% EtOAc в гептане, получая N-(2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)-3,3,3-трифтор-2,2-диметилпропан-1-амин (А294) (3.4393 г, 7.72 ммоль, 69.5% выход) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 8.44 (2Н, с), 5.48 (1Н, дд, J=7.7, 4.4 Гц), 3.27 (1Н, дд, J=12.3, 8.4 Гц), 2.58-2.83 (3Н, м), 1.25-1.44 (1Н, м), 1.10 (6Н, с), 0.85-0.94 (9Н, м), 0.47-0.64 (6Н, м); LCMS (ESI) m/z 445.1 (M+H)+.
Приведенные далее вторичные амины получали по методике, аналогичной описанной выше в справочных примерах:
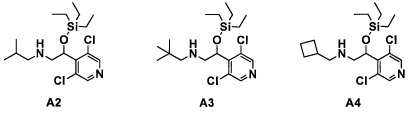
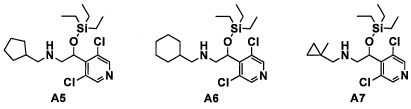
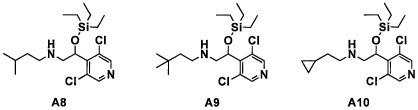
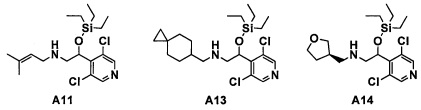
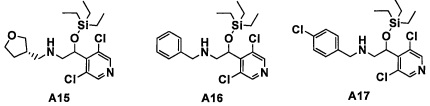
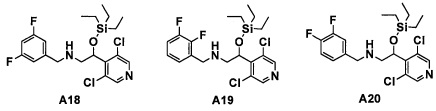
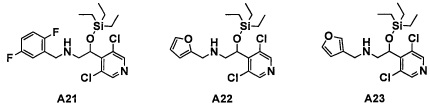
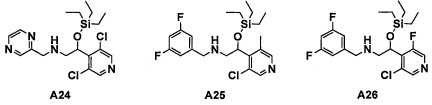
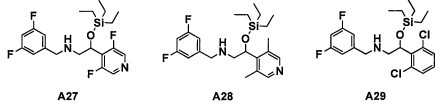
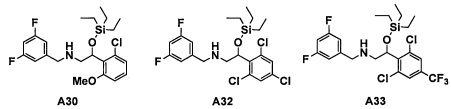
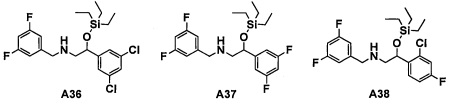
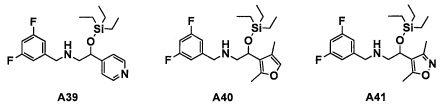
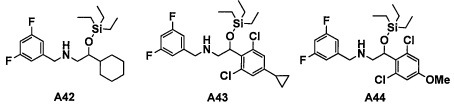
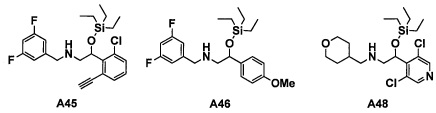
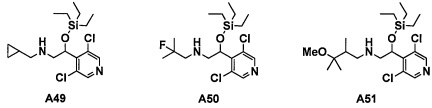
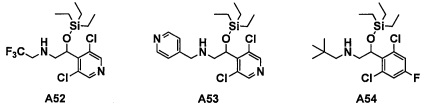
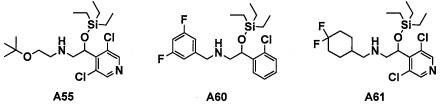
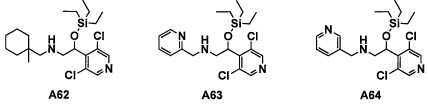
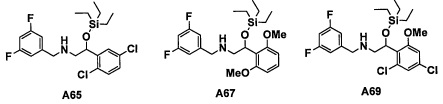
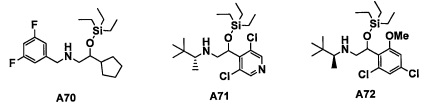
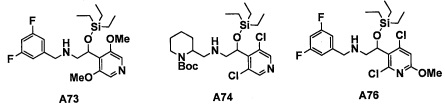
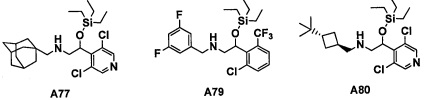
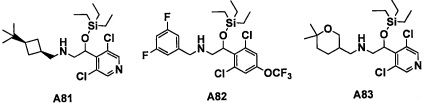
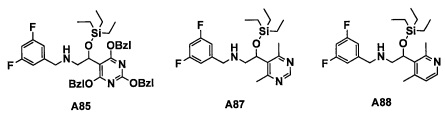
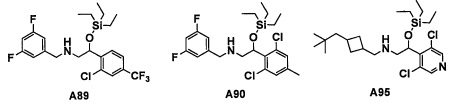
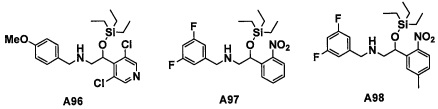
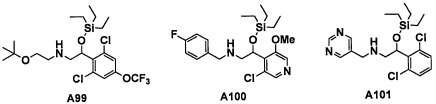
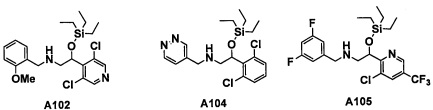
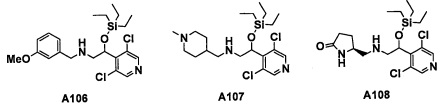
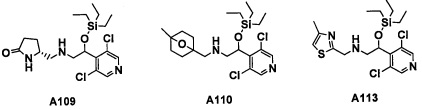
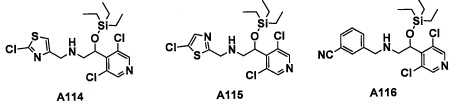
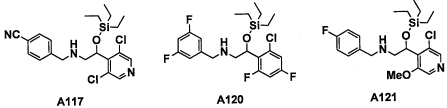
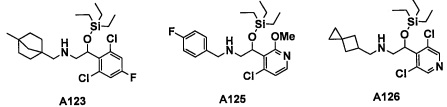
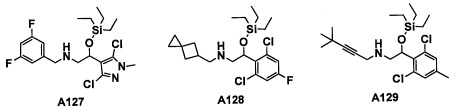
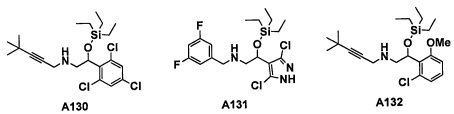
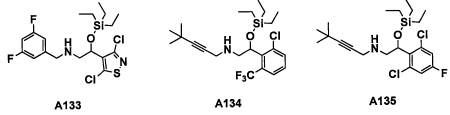
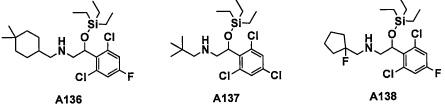
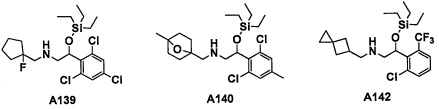
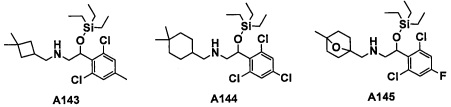
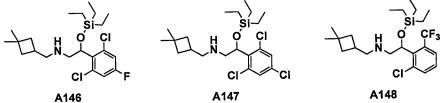
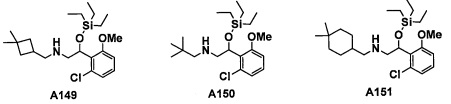
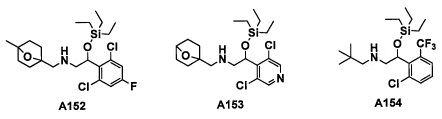
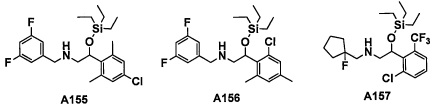
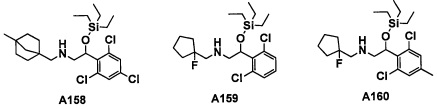
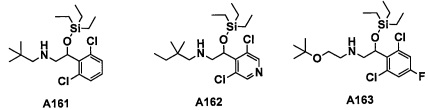
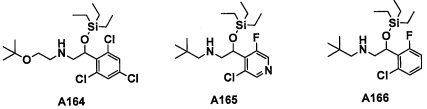
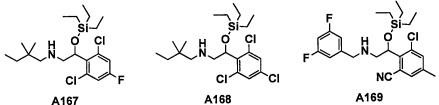
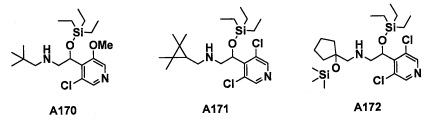
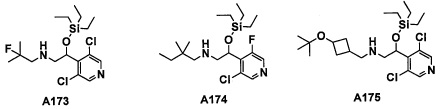
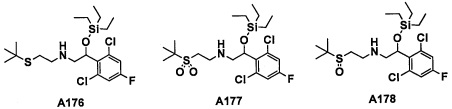
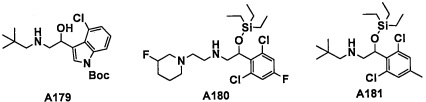
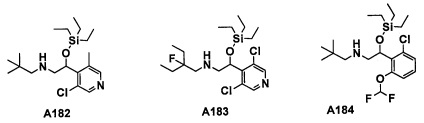
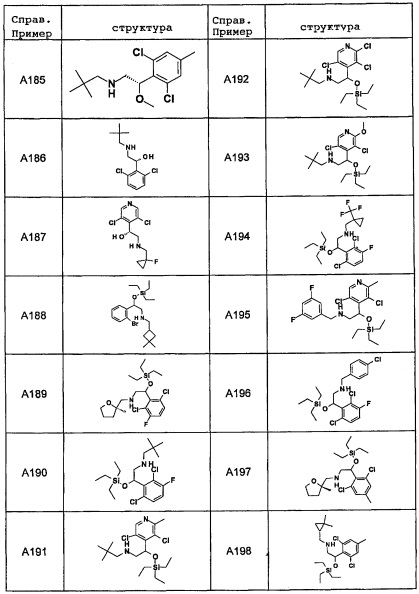
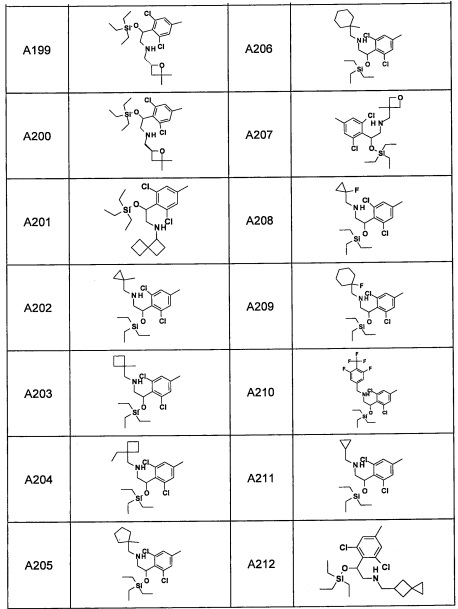
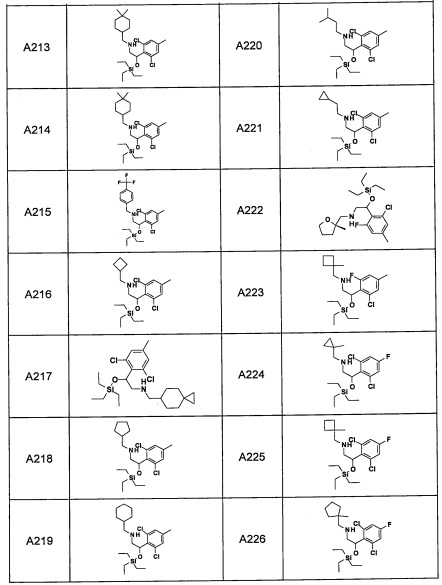
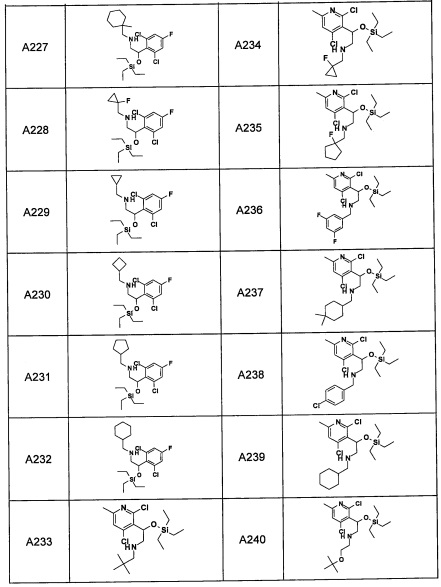
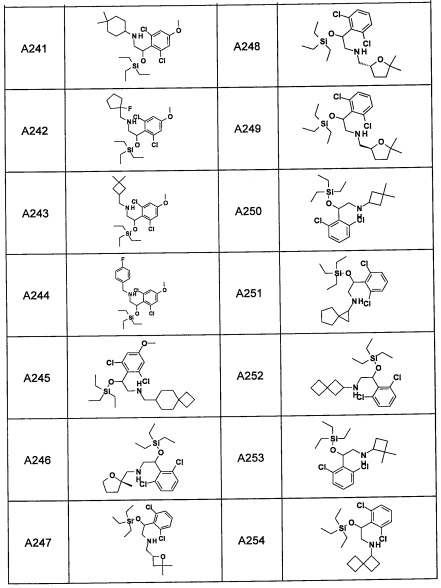
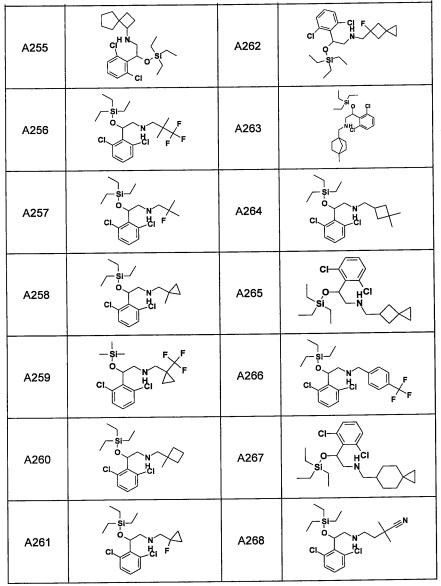
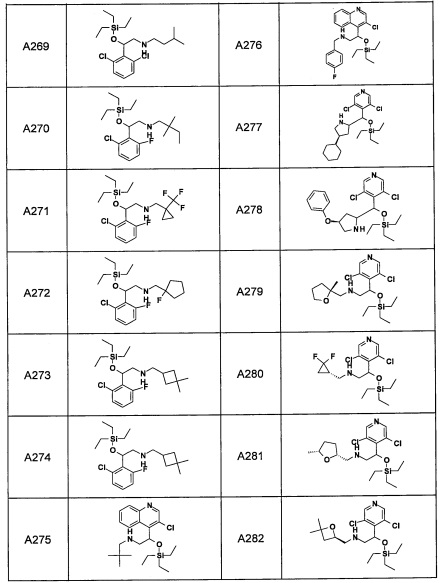
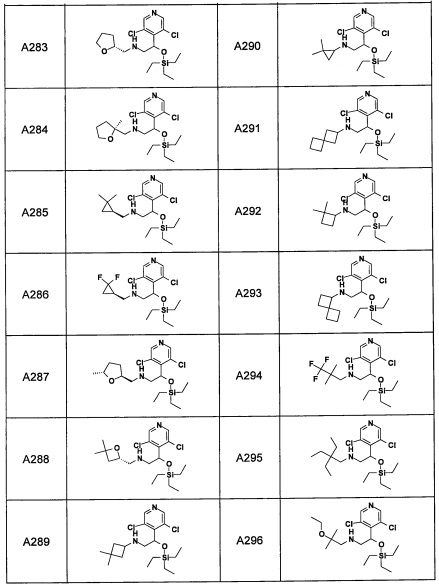
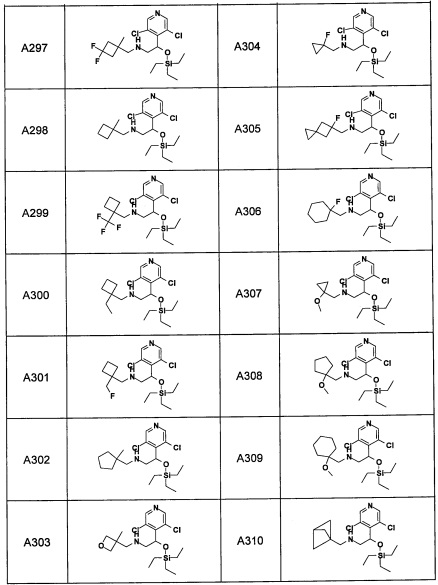
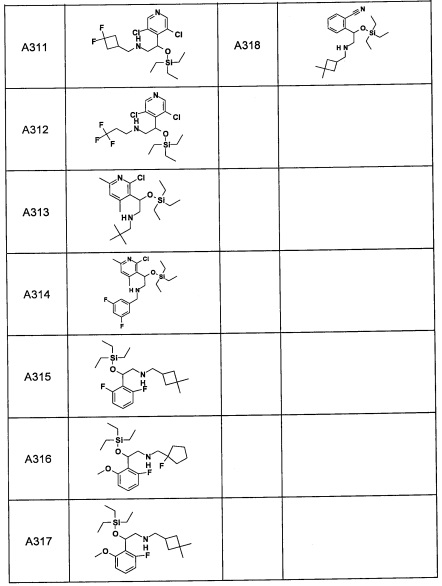
Справочный пример В1
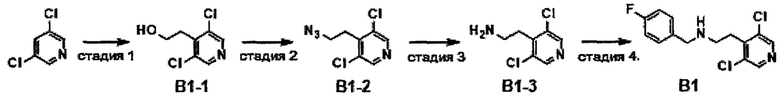
Стадия 1: 2-(3,5-дихлорпиридин-4-ил)этанол (В1-1)
В раствор 3,5-дихлорпиридина (4.0 г, 27.0 ммоль) в ТГФ (70 мл) добавляли LDA (1.8 М раствор в смеси ТГФ/гептан/этилбензол, 22.0 мл, 39.6 ммоль) при -78°С, и полученную смесь перемешивали при той же температуре 2 часа, и затем добавляли этиленоксид (1.2 М раствор в ТГФ, 25 мл, 30.0 ммоль). Реакционную смесь оставляли постепенно нагреваться до комнатной температуры и перемешивали в течение 1 часа при комнатной температуре. Реакционную смесь гасили насыщенным водным раствором NH4Cl и экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором поваренной соли (2 раза) и сушили над MgSO4. После удаления растворителя остаток очищали методом колоночной хроматографии на силикагеле, получая соединение В 1-1 (3.1 г, 60%) в виде желтого твердого вещества.
Стадия 2: 4-(2-азидоэтил)-3,5-дихлорпиридин (В1-2)
В раствор соединения В1-1 (3.1 г, 16.2 ммоль) в ТГФ (60 мл) добавляли DIAD (6.3 мл, 32.0 ммоль), трифенилфосфин (8.52 г, 32.5 ммоль) и DPPA (6.98 мл, 32.5 ммоль) при 0°С. Реакционную смесь оставляли постепенно нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 4.5 часов. Реакционную смесь гасили водой и экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором поваренной соли (х 2) и сушили над MgSO4. После удаления растворителя, остаток очищали методом колоночной хроматографии на силикагеле, получая соединение В1-2 (2.4 г, 68%) в виде желтого масла.
Стадия 3: 2-(3,5-дихлорпиридин-4-ил)этанамин (В1-3)
В раствор соединения В1-2 (2.4 г, 11.1 ммоль) в ТГФ (25 мл) добавляли трифенилфосфин (2.9 г, 22.1 ммоль) при 0°С. Полученную смесь перемешивали при комнатной температуре в течение 2 часов и затем добавляли воду (2.5 мл). Реакционную смесь оставляли постепенно нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 22 часов. Реакционную смесь гасили 2 М водным раствором HCl (10 мл) и разбавляли добавлением EtOAc. Водный слой промывали EtOAc 3 раза и затем подщелачивали 2М водным раствором NaOH до рН 12. Водный слой экстрагировали этилацетатом, промывали насыщенным водным раствором поваренной соли (х 2) и сушили над MgSO4. Сушка раствора в высоком вакууме дала соединение В1-3 (1.9 г, 90%) в виде белого твердого вещества.
Стадия 4: 2-(3,5-дихлорпиридин-4-ил)-N-(4-фторбензил)этанамин (В1)
В раствор соединения В1-3 (2.9 г, 15.2 ммоль) в МеОН (30 мл) добавляли 4-фторбензальдегид (1.89 г, 15.2 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь охлаждали до 0°С и постепенно добавляли NaBH4 (1.16 г, 30.4 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь гасили водой и экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором поваренной соли (2 раза) и сушили над MgSO4. После удаления растворителя остаток очищали методом колоночной хроматографии на силикагеле, получая соединение В1 (3.4 г, 75%) в виде светло-желтого твердого вещества.
Справочный пример В2
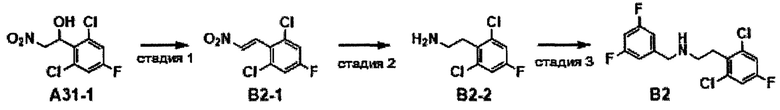
Стадия 1: 1,3-дихлор-5-фтор-2-(2-нитровинил)бензол(В2-1)
В перемешиваемый раствор соединения А31-1 (1.3 г, 5.1 ммоль) в диоксане (10 мл) добавляли 6 М HCl (20 мл) при комнатной температуре, и полученную смесь перемешивали при кипячении в течение ночи. Реакционную смесь нейтрализовывали 10%-ным раствором NaOH и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 10% EtOAc/гексан в качестве элюента), получая соединение В2-1 (0.22 г, 18%) в виде бесцветного масла.
Стадия 2: 2-(2,6-дихлор-4-фторфенил)этанамин (В2-2)
В перемешиваемый раствор LiBH4 (3.0 М, 4.2 мл, 12.5 ммоль) в ТГФ (5 мл) добавляли TMS-Cl (3.2 мл, 25.2 ммоль) по каплям при комнатной температуре, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Пропускали газообразный N2 через реакционную смесь в течение 5 минут для удаления остатков образующегося триметилсилана. Раствор соединения В2-1 (0.22 г, 3.1 ммоль) в ТГФ (2 мл) добавляли по каплям в полученную смесь при комнатной температуре и кипятили 1 час. Реакционную смесь охлаждали до 0°С и осторожно гасили метанолом (10 мл). Растворитель упаривали при пониженном давлении, и остаток разделяли между 20%-ным раствором KOH (10 мл) и ДХМ (20 мл). Органический слой сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 20% EtOAc/гексан в качестве элюента), получая соединение В2-2 (0.21 г, 99%) в виде бесцветного масла.
Стадия 3: 2-(2,6-дихлор-4-фторфенил)-N-(3,5-дифторбензил)этанамин (В2)
Соединение В2 (0.21 г, 69%) получали в виде бесцветной смолы реакцией соединения В2-2 (0.19 г, 0.91 ммоль), 3,5-дифторбензальдегида (0.1 мл, 0.91 ммоль) и NaBH4 (70 мг, 1.8 ммоль) в МеОН (5 мл) по методике, аналогичной описанной в справочном примере В1, стадия 4. 1H ЯМР (CDCl3, 300 МГц): δ 7.13-7.05 (м, 2Н), 6.87-6.59 (м, 3Н), 3.83 (с, 2Н), 3.12-2.80 (м, 4Н).
Справочный пример В3
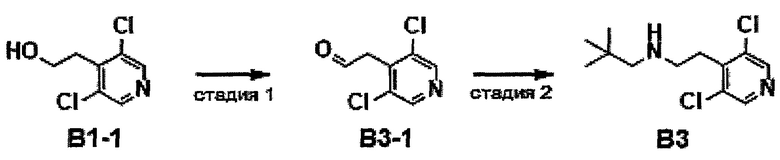
Стадия 1: 2-(3,5-дихлорпиридин-4-ил)ацетальдегид (В3-1)
Соединение В1-1 (1.0 г, 5.21 ммоль) растворяли в ДХМ (26.0 мл) и добавляли периодинан Десс-Мартина (2.43 г, 5.73 ммоль). Полученный раствор перемешивали 1 час. Реакционную смесь гасили добавлением 50 мл 5%-ного раствора Na2S2O3, органический слой промывали насыщенным водным раствором NaHCO3, сушили над безводным Na2SO4 и упаривали. Полученный продукт очищали методом колоночной хроматографии на силикагеле (40 г колонка) с применением 0-100% EtOAc в гептане, получая соединение В3-1 (750 мг, 3.95 ммоль, 76% выход). LC/MS (ESI+) m/z=189.9 (М+Н)+.
Стадия 2: N-(2-(3,5-дихлорпиридин-4-ил)этил)-2,2-диметилпропан-1-амин (В3)
Соединение В3-1 (0.65 г, 3.42 ммоль) растворяли в ДХМ (17 мл) в инертной атмосфере, затем добавляли 2,2-диметилпропан-1-амин (0.605 мл, 5.13 ммоль), затем ледяную АсОН (0.198 мл, 3.42 ммоль). Полученный раствор перемешивали 15 минут и затем добавляли NaBH(ОАс)3 (1.450 г, 6.84 ммоль). Полученный раствор гасили 15 мл насыщенного раствора NaHCO3 и перемешивали 45 минут. Органический слой отделяли и упаривали. Полученный продукт очищали колоночной хроматографией на силикагеле (40 г колонка) с применением 0-100% EtOAc в гептане, получая соединение В3 (775 мг, 2.97 ммоль, 87% выход). LC/MS (ESI+) m/z=261.0 (М+Н)+.
Справочный пример В13
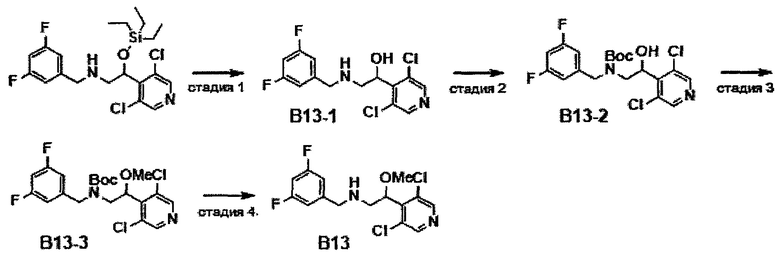
Стадия 1: 1-(3,5-дихлорпиридин-4-ил)-2-((3,5-дифторбензил)амино)этанол (В13-1)
В перемешиваемый раствор 2-(3,5-дихлорпиридин-4-ил)-N-(3,5-дифторбензил)-2-((триэтилсилил)окси)этанамина (0.2 г, 0.44 ммоль) в ТГФ (5 мл) по каплям добавляли TBAF (1.0 М раствор в ТГФ, 0.9 мл, 0.88 ммоль) при 0°С, и полученную смесь оставляли нагреваться с 0°С до комнатной температуры при перемешивании в течение 2 часов. Реакционную смесь гасили насыщенным водным раствором MH4Cl и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали водой (20 мл), насыщенным водным раствором поваренной соли (20 мл) и сушили над безводным Na2SO4. Растворитель упаривали при пониженном давлении, получая соединение В13-1 (0.2 г, неочищенное) в виде коричневой смолы.
Стадия 2: трет-бутил (2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(3,5-дифторбензил)карбамат(В13-2)
В перемешиваемый раствор соединения В13-1 (0.2 г, 0.6 ммоль) в смеси ДХМ/вода (4:1, 5 мл) добавляли NaHCO3 (0.1 г, 1.2 ммоль) и (Вос)2O (0.19 г, 0.9 ммоль) в ДХМ (2 мл) при 0°С. Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили водой (50 мл) и экстрагировали дихлорметаном (2×30 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 20% EtOAc/гексан в качестве элюента), получая соединение В13-2 (0.17 г, 65%) в виде бесцветного масла.
Стадия 3: трет-бутил (2-(3,5-дихлорпиридин-4-ил)-2-метоксиэтил)(3,5-дифторбензил)карбамат(В13-3)
В перемешиваемый раствор соединения В13-2 (0.1 г, 0.2 ммоль) в ТГФ (5 мл) добавляли NaH (14 мг, 0.5 ммоль), затем по каплям добавляли MeI (44 мкл, 0.7 ммоль) при 0°С. Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили водой (50 мл) и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали водой (30 мл), насыщенным водным раствором поваренной соли (30 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 10% EtOAc/гексан в качестве элюента), получая соединение В13-3 (0.11 г, 99%) в виде бесцветного масла.
Стадия 4: трет-бутил 2-(3,5-дихлорпиридин-4-ил)-N-(3,5-дифторбензил)-2-метоксиэтанамин (В13)
В перемешиваемый раствор соединения В13-3 (0.28 г, 0.6 ммоль) в диоксане (5 мл) добавляли 4 М раствор HCl (в диоксане, 1.9 мл, 7.4 ммоль) при комнатной температуре, и полученную смесь перемешивали в течение ночи. Растворитель упаривали при пониженном давлении, получая соединение В13-3 (0.1 г, 48%) в виде белого твердого вещества. 1H ЯМР (CDCl3, 300 МГц): δ 8.45 (с, 2Н), 6.90-6.63 (м, 3Н), 5.14 (дд, J=8.9, 4.1 Гц, 1Н), 3.89-3.77 (м, 2Н), 3.30-3.23 (м, 4Н), 2.78 (дд, J=12.6, 4.1 Гц, 1Н).
Справочный пример В15
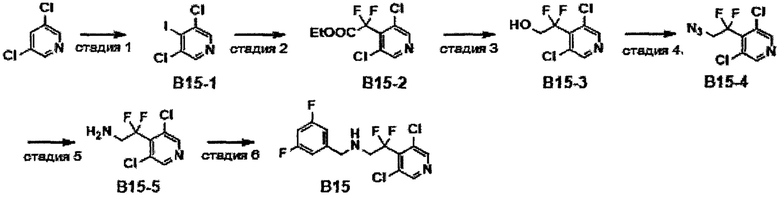
Стадия 1: 3,5-дихлор-4-иодпиридин (В15-1)
В перемешиваемый раствор 3,5-дихлорпиридина (3.0 г, 20.4 ммоль) в ТГФ (15 мл) добавляли LDA (2.0 М раствор в смеси ТГФ/гептан/этилбензол, 12.14 мл, 24.4 ммоль) по каплям при 0°С, и полученную смесь перемешивали при той же температуре 1 час. Раствор иода (2.7 г, 21.4 ммоль) в ТГФ (10 мл) по каплям добавляли в полученную смесь. По окончании добавления, смесь перемешивали при той же температуре 1 час. Реакционную смесь гасили водой (40 мл) и экстрагировали этилацетатом (4×50 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение В15-1 (3.2 г, 57%) в виде желтой смолы.
Стадия 2: этил-2-(3,5-дихлорпиридин-4-ил)-2,2-дифторацетат (В15-2)
Смесь соединения В15-1 (530 мг, 0.83 ммоль), этил 2-бром-2,2-дифторацетата (0.12 мл, 1.38 ммоль) и Cu (800 мг, 12.5 ммоль) в ДМСО (10 мл) нагревали при 55°С 16 часов. Реакционную смесь охлаждали до комнатной температуры, гасили насыщенным раствором NH4Cl (100 мл) и экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 30% EtOAc/гексан в качестве элюента), получая соединение В 15-2 (315 мг, 60%) в виде желтовато-коричневой смолы.
Стадия 3: 2-(3,5-дихлорпиридин-4-ил)-2,2-дифторэтанол (В15-3)
В перемешиваемый раствор соединения В15-2 (315 мг, 1.16 ммоль) в EtOH (10 мл) порциями добавляли твердый NaBH4 (16.2 мг, 1.74 ммоль) при 0°С. Полученную смесь нагревали до комнатной температуры и перемешивали при той же температуре в течение 2 часов. Реакционную смесь гасили водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (2×30 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 55% EtOAc/гексан в качестве элюента), получая соединение В15-3 (180 мг, 44%) в виде бесцветной смолы.
Стадия 4: 4-(2-азидо-1,1-дифторэтил)-3,5-дихлорпиридин (В15-4)
В перемешиваемый раствор соединения В15-3 (140 мг, 0.72 ммоль) в ТГФ (5 мл) добавляли DIAD (0.31 мл, 1.60 ммоль), DPPA (0.34 мл, 1.60 ммоль) и PPh3 (420 мг, 1.60 ммоль) при 0°С. Полученную смесь нагревали до комнатной температуры и перемешивали при той же температуре 16 часов. Реакционную смесь гасили водой (10 мл) и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (20 мл), сушили над безводным Na2SO4, и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 20% EtOAc/гексан в качестве элюента), получая соединение В15-4 (80 мг, 55%) в виде желтой смолы.
Стадия 5: 2-(3,5-дихлорпиридин-4-ил)-2,2-дифторэтанамин (В15-5)
В перемешиваемый раствор соединения В15-4 (80 мг, 0.31 ммоль) в EtOAc (2 мл) добавляли (СН3)3Р (0.47 мл, 0.47 ммоль) и H2O (0.5 мл). Полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли EtOAc (10 мл) и промывали водой (10 мл). Органический слой промывали насыщенным водным раствором поваренной соли (10 мл), сушили над безводным Na2SO4, и упаривали при пониженном давлении, получая соединение В15-5 (60 мг) в виде желтой смолы. Сырой остаток использовали в следующей стадии без очистки.
Стадия 6: 2-(3,5-дихлорпиридин-4-ил)-N-(3,5-дифторбензил)-2,2-дифторэтанамин (В15)
Смесь соединения В15-5 (113 мг, 0.49 ммоль), 3,5-дифторбензальдегида (70 мг, 0.49 ммоль) и NaBH(ОАс)3 (316 мг, 1.49 ммоль) в ДХМ перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь гасили водой (20 мл) и экстрагировали дихлорметаном (2×25 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 20% EtOAc/гексан в качестве элюента), получая соединение В15 (66 мг, 38%) в виде белого твердого вещества. 1H ЯМР (CDCl3, 300 МГц): δ 8.54-8.53 (м, 2Н), 6.73-6.66 (м, 3Н), 3.86 (с, 2Н), 3.36-3.45 (т, J=28.7 Гц, 2Н); LCMS (APCI): 353 (М+Н)+.
Справочный пример В19
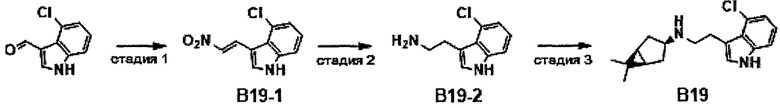
Стадия 1: (Е)-4-хлор-3-(2-нитровинил)-1Н-индоле (В19-1)
Смесь 4-хлориндол-3-карбальдегида (314 мг, 1.75 ммоль) и ацетата аммония (404 мг, 5.25 ммоль) в нитрометане (6 мл) перемешивали при 100°С 20 минут. Реакционную смесь охлаждали, разбавляли водой и экстрагировали этилацетатом (2×40 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли, сушили над MgSO4 и упаривали при пониженном давлении. Полученный сырой продукт очищали методом колоночной хроматографии на силикагеле (50-100% EtOAc/гептан), получая соединение В19-1 (224 мг, 58%) в виде оранжевого твердого вещества.
Стадия 2: 2-(4-хлор-1Н-индол-3-ил)этанамин (В19-2)
Раствор соединения В19-1 (1.46 г, 6.56 ммоль) в ТГФ (25 мл) добавляли в перемешиваемую суспензию литийалюминий гидрида (995 мг, 26.2 ммоль) в ТГФ (50 мл) при комнатной температуре. Полученную смесь кипятили в течение 2 часов и оставляли охлаждаться до комнатной температуры. Реакцию гасили добавлением воды по каплям (1.3 мл), затем 15%-ного водного раствора NaOH (1.3 мл), затем снова воды (3.25 мл). После интенсивного перемешивания в течение 14 часов, смесь фильтровали через целит и упаривали фильтрат. Остаток от упаривания растворяли в EtOAc и затем экстрагировали 2 н. водным раствором HCl (2×20 мл). Объединенные водные слои подщелачивали добавлением 5н. водного раствора NaOH и экстрагировали этилацетатом (2×40 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли, сушили над MgSO4, фильтровали и упаривали при пониженном давлении, получая соединение В19-2 (1.02 г, 80%) в виде темно-красного сиропа.
Стадия 3: (1R,3r,5S)-N-(2-(4-хлор-1Н-индол-3-ил)этил)-6,6-диметилбицикло[3.1.0]гексан-3-амин (В19)
Соединение В19 (22 мг, 14%) получали реакцией соединения В19-2 (100 мг, 0.514 ммоль), соединения С22-5 (128 мг, 1.03 ммоль), NaBH(ОАс)3 (326 мг, 1.54 ммоль) и АсОН (0.108 мл, 2.05 ммоль) в ДХМ (2 мл) по методике, аналогичной описанной в справочном примере А31, стадия 4. 1H ЯМР (CDCl3, 400 МГц) δ: 8.09 (1Н, ушир.с), 7.26-7.22 (1Н, м), 7.07-7.05 (3Н, м), 3.57-3.47 (1Н, м), 3.13 (2Н, т, J=7.3 Гц), 2.89 (2Н, т, J=7.3 Гц), 2.17-2.10 (2Н, м), 1.03-0.93 (10Н, м).
Справочный пример В50
3,5-дихлор-4-(((2R)-4-изопропилпирролидин-2-ил)метил)пиридин (В50)
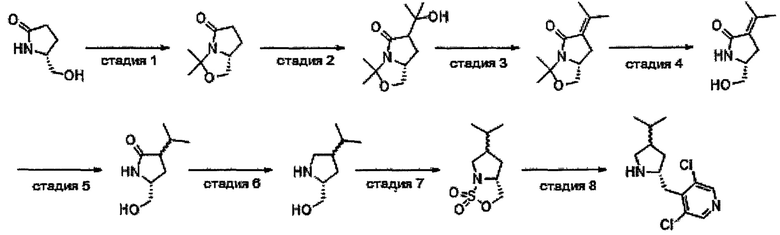
Стадия 1: (R)-3,3-диметилтетрагидропирроло[1,2-с]оксазол-5(3)-он
Реакционную колбу снабжали насадкой Дина-Старка, затем 2,2-диметоксипропан (17.09 мл, 139 ммоль) добавляли в перемешиваемую смесь (R)-(-)-5-(гидроксиметил)-2-пирролидинона (5.353 г, 46.5 ммоль) и п-толуолсульфокислоты моногидрата (0.126 г, 0.662 ммоль) в толуоле (100 мл). Реакционную смесь кипятили в течение 1.5 часов и оставляли перемешиваться при комнатной температуре в течение ночи. Растворитель упаривали, получая (R)-3,3-диметилтетрагидропирроло[1,2-с]оксазол-5(3H)-он (7.22 г, 100% выход) в виде светло-желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 4.18 (тт, J=8.8, 6.2 Гц, 1Н), 4.00 (дд, J=8.1, 5.8 Гц, 1Н), 3.40 (т, J=8.6 Гц, 1Н), 2.69 (ддд, J=16.4, 12.1, 8.6 Гц, 1Н), 2.33 (дд, J=16.3, 9.1 Гц, 1Н), 2.02-2.11 (м, 1Н), 1.73 (тт, J=12.1, 8.9 Гц, 1Н), 1.53 (с, 3Н), 1.33 (с, 3Н). m/z (ESI, +ve) 156 (M+H).
Стадия 2: (7aR)-6-(2-гидроксипропан-2-ил)-3,3-диметилтетрагидропирроло[1,2-с]оксазол-5(3H)-он
Раствор (R)-3,3-диметилтетрагидропирроло[1,2-с]оксазол-5(3H)-она (6.68 г, 43.0 ммоль) в ТГФ (100 мл) охлаждали до -78°С, добавляли диизопропиламид лития, 2.0 М раствор в смеси ТГФ/гептан/этилбензол (43.0 мл, 86 ммоль) и перемешивали при -78°С 1 час. В результирующую смесь добавляли ацетон (99.8%, безводный, acroseal) (6.32 мл, 86 ммоль) при -78°С и затем оставляли смесь нагреваться до комнатной температуры на 16 часов. Реакцию гасили насыщенным водным раствором NH4Cl и экстрагировали этилацетатом (2×200 мл). Объединенные экстракты промывали насыщенным водным раствором поваренной соли, сушили над Na2SO4, фильтровали и упаривали, получая (7aR)-6-(2-гидроксипропан-2-ил)-3,3-диметилтетрагидропирроло[1,2-с]оксазол-5(3H)-он (6.088 г, 28.5 ммоль, 66.3% выход) в виде желтого масла. 1H ЯМР (400 МГц, ДМСО-d6) δ 4.50 (с, 1Н), 4.07-4.18 (м, 1Н), 3.98 (дд, J=8.0, 5.7 Гц, 1Н), 3.32-3.35 (м, 1Н), 2.50-2.56 (м, 1Н), 2.22 (ддд, J=13.4, 7.2, 2.0 Гц, 1Н), 1.83 (ддд, J=13.3, 10.4, 7.6 Гц, 1Н), 1.54 (с, 3Н), 1.32 (с, 3Н), 1.21 (с, 3Н), 1.14 (с, 3Н). m/z (ESI, +ve) 214 (М+Н).
Стадия 3: (R)-3,3-диметил-6-(пропан-2-илиден)тетрагидропирроло[1,2-с]оксазол-5(3H)-он
В раствор (7aR)-6-(2-гидроксипропан-2-ил)-3,3-диметилтетрагидропирроло-[1,2-с]оксазол-5(3H)-она (5.06 г, 23.73 ммоль) в ДХМ (50 мл) при комнатной температуре добавляли метансульфонил хлорид (2.75 мл, 35.6 ммоль), затем триэтиламин (16.50 мл, 119 ммоль) и затем нагревали при 55°С в течение 1 часа. В результирующую смесь добавляли еще метансульфонил хлорид (2.75 мл, 35.6 ммоль) и нагревали еще 1 час. Реакционную смесь оставляли охлаждаться до комнатной температуры, гасили водой (50 мл) и экстрагировали дихлорметаном (2×100 мл). Объединенные экстракты промывали насыщенным водным раствором поваренной соли, сушили над MgSO4, фильтровали и упаривали, получая сырой (R)-3,3-диметил-6-(пропан-2-илиден)тетрагидропирроло[1,2-с]оксазол-5(3H)-он в виде коричневого масла, который использовали в следующей стадии без дополнительной очистки, m/z (ESI, +ve) 196 (М+Н).
Стадия 4: (R)-5-(гидроксиметил)-3-(пропан-2-илиден)пирролидин-2-он
В раствор (R)-3,3-диметил-6-(пропан-2-илиден)тетрагидропирроло[1,2-с]оксазол-5(3H)-она (4.63 г, 23.73 ммоль) в МеОН (50 мл) при комнатной температуре добавляли n-толуолсульфокислоты моногидрат (0.451 г, 2.373 ммоль) и затем нагревали при 60°С 45 минут. Растворитель упаривали, сырой продукт адсорбировали на слое силикагеля и очищали хроматографически на преупакованной REDISEP™ колонке с силикагелем (80 г), элюируя в градиенте от 0% до 10% МеОН в ДХМ, получая (R)-5-(гидроксиметил)-3-(пропан-2-илиден)пирролидин-2-он (2.223 г, 14.32 ммоль, 60.4% выход) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 6.60 (ушир.с, 1Н), 3.74 (тд, J=8.0, 3.9 Гц, 1Н), 3.67 (дд, J=11.1, 3.6 Гц, 1Н), 3.44 (дд, J=11.1, 7.3 Гц, 1Н), 2.75-2.86 (м, 1Н), 2.81 (дд, J=16.5, 8.7 Гц, 1Н), 2.33-2.43 (м, 1Н), 2.23 (с, 3Н), 1.77 (с, 3Н). m/z (ESI, +ve) 156 (М+Н).
Стадия 5: (5R)-5-(гидроксиметил)-3-изопропилпирролидин-2-он
Смесь (R)-5-(гидроксиметил)-3-(пропан-2-илиден)пирролидин-2-она (2.223 г, 14.32 ммоль) и оксида платины (iv) (0.325 г, 1.432 ммоль) в EtOAc (40 мл)/МеОН (4 мл) при комнатной температуре перемешивали в реакторе для проведения реакций под давлением Н2 (от 28 фунт/кв.дюйм до 2 фунт/кв.дюйм) в течение ночи. Результирующую смесь фильтровали через слой целита, промывали этилацетатом и упаривали, получая (5R)-5-(гидроксиметил)-3-изопропилпирролидин-2-он (2.251 г, 14.32 ммоль, 90% выход) в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 6.56-6.71 (м, 1Н), 3.64-3.80 (м, 2Н), 3.37-3.53 (м, 1Н), 2.48 (тд, J=9.9, 4.5 Гц, 2Н), 2.14-2.27 (м, 1Н), 1.97-2.13 (м, 1Н), 1.50 (ддд, J=12.7, 10.7, 8.3 Гц, 1Н), 0.98 (д, J=6.8 Гц, 3Н), 0.86 (д, J=6.8 Гц, 3Н). m/z (ESI, +ve) 158 (M+H).
Стадия 6: ((2R)-4-изопропилпирролидин-2-ил)метанол
В раствор (5R)-5-(гидроксиметил)-3-изопропилпирролидин-2-она (2.251 г, 14.32 ммоль) в ТГФ (25 мл) медленно по каплям добавляли литийалюминий гидрид, 1.0 М раствор в ТГФ (20.05 мл, 20.05 ммоль) при комнатной температуре. Результирующую смесь затем кипятили при 75°С в течение 2 часов. Добавляли еще литийалюминий гидрид, 1.0 М раствор в ТГФ (20.05 мл, 20.05 ммоль), и полученную смесь кипятили в течение ночи. Через 18 часов реакционную смесь оставляли охлаждаться до 0°С. Реакцию гасили насыщенным водным раствором сегнетовой соли. Реакционную смесь интенсивно перемешивали в течение 1 часа и разделяли слои. Водный слой экстрагировали этилацетатом дважды, органические слои объединяли, промывали насыщенным водным раствором поваренной соли, сушили над MgSO4, фильтровали и упаривали в вакууме, получая ((2R)-4-изопропилпирролидин-2-ил)метанол (1.645 г, 11.49 ммоль, 80% выход) в виде светло-желтого масла. Полученный сырой продукт использовали в следующей стадии без дополнительной очистки, m/z (ESI, +ve) 144 (М+Н).
Стадия 7: (3aR)-5-изопропилтетрагидро-3Н-пирроло[1,2-с][1,2,3]оксатиазол 1,1-диоксид
Раствор ((2R)-4-изопропилпирролидин-2-ил)метанола (1.639 г, 11.44 ммоль) и триэтиламина (3.18 мл, 22.89 ммоль) в ДХМ (100 мл) охлаждали до -78°С. В полученную смесь по каплям добавляли сульфурил хлорид, 1.0 М раствор в ДХМ (13.73 мл, 13.73 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры в течение ночи. Реакционную смесь упаривали на слое силикагеля и очищали на хроматографе ISCO на преупакованной REDISEP™ колонке с силикагелем (40 г), элюируя в градиенте от 0% до 10% МеОН (с 2 М NH3) в ДХМ, получая (3aR)-5-изопропилтетрагидро-3Н-пирроло[1,2-с][1,2,3]оксатиазол 1,1-диоксид (211.9 мг, 1.032 ммоль, 9% выход) в виде светло-желтого масла, m/z (ESI, +ve) 206 (М+Н).
Стадия 8: 3,5-дихлор-4-(((2R)-4-изопропилпирролидин-2-ил)метил)пиридин
В раствор 3,5-дихлорпиридина (228 мг, 1.542 ммоль) в ТГФ (2.6 мл) при -78°С по каплям добавляли диизопропиламид лития, 2.0 М раствор в смеси гептан/ТГФ/этилбензол (0.976 мл, 1.953 ммоль). После перемешивания в течение 45 минут, добавляли по каплям раствор (3aR)-5-изопропилтетрагидро-3Н-пирроло[1,2-с][1,2,3]оксатиазол 1,1-диоксида (211 мг, 1.028 ммоль) в ТГФ (3.0 мл) при -78°С. Результирующую смесь оставляли нагреваться до комнатной температуры и затем перемешивали 3 часа. После упаривания растворителя, к полученному коричневому твердому веществу добавляли 2 н. раствор HCl (3 мл) и EtOH (3 мл) и нагревали до 80°С в течение 2 часов. Реакционную смесь упаривали для удаления EtOH. В результирующую смесь добавляли лед и подщелачивали 2н. раствором NaOH до рН~10 и экстрагировали этилацетатом (2×10 мл). Экстракты сушили, упаривали и очищали на хроматографе ISCO на преупакованной REDISEP™ колонке с силикагелем (12 г), элюируя в градиенте от 0% до 5% МеОН (с 2 М NH3) в ДХМ, получая 3,5-дихлор-4-(((2R)-4-изопропилпирролидин-2-ил)метил)пиридин (102 мг, 0.373 ммоль, 36.3% выход) в виде оранжевого масла. 1H ЯМР (400 МГц, ДМСО-d6) δ 8.56 (с, 2Н), 3.35-3.51 (м, 1Н), 2.84-3.08 (м, 3Н), 2.35-2.44 (м, 1Н), 1.80-1.93 (м, 1Н), 1.55-1.69 (м, 1Н), 1.31-1.49 (м, 2Н), 1.02-1.17 (м, 1Н), 0.85 (т, J=6.7 Гц, 6Н). m/z (ESI, +ve) 273 (M+H).
Справочный пример В52
(R)-3,5-дихлор-4-((4,4-диаллилпирролидин-2-ил)метил)пиридин
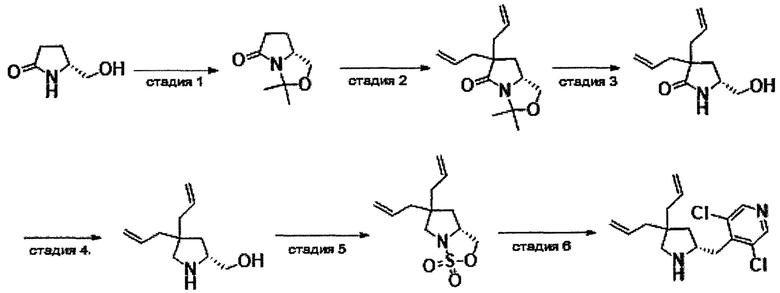
Стадия 1: (R)-3,3-диметилтетрагидропирроло[1,2-с]оксазол-5(3Н)-он
В перемешиваемую суспензию (R)-(-)-5-(гидроксиметил)-2-пирролидинона (2.20 г, 19.11 ммоль) и п-толуолсульфокислоты (0.018 г, 0.096 ммоль) в толуоле (54.6 мл) добавляли 2,2-диметоксипропан (7.02 мл, 57.3 ммоль), и реакционную смесь кипятили в течение 2 часов. Реакционную колбу снабжали насадкой Дина-Старка, затем добавляли 2,2-диметоксипропан (7.02 мл, 57.3 ммоль), и реакционную смесь кипятили в течение ночи. Растворитель упаривали, получая (R)-3,3-диметилтетрагидропирроло[1,2-с]оксазол-5(3Н)-он (3.04 г, 19.59 ммоль, 103% выход) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 4.27 (тт, J=6.01, 9.00 Гц, 1Н), 4.09 (дд, J=5.65, 8.24 Гц, 1Н), 3.43-3.50 (м, 1Н), 2.81 (ддд, J=5.53, 12.19, 16.65 Гц, 1Н), 2.55 (ддд, J=1.01, 9.15, 16.64 Гц, 1Н), 2.13-2.23 (м, 1Н), 1.72-1.80 (м, 1Н), 1.66-1.72 (м, 3Н), 1.48 (с, 3Н).
Стадия 2: (R)-6,6-диаллил-3,3-диметилтетрагидропирроло[1,2-с]оксазол-5(3Н)-он
Раствор (R)-3,3-диметилтетрагидропирроло[1,2-с]оксазол-5(3Н)-она (2.55 г, 16.43 ммоль) в ТГФ (54.8 мл) охлаждали до -78°С, добавляли раствор диизопропиламида лития (14.79 мл, 29.6 ммоль). Полученный раствор перемешивали при этой температуре в течение 1 часа, после чего добавляли аллилбромид (2.133 мл, 24.65 ммоль). Реакционную смесь нагревали до комнатной температуры (1 час), затем охлаждали до -78°С, после чего добавляли диизопропиламид лития (14.79 мл, 29.6 ммоль). Полученную смесь перемешивали при -78°С в течение 1 часа. после чего добавляли аллилбромид (2.133 мл, 24.65 ммоль). Полученную смесь медленно нагревали до комнатной температуры и перемешивали в течение ночи. Реакцию гасили насыщенным водным раствором NH4Cl и экстрагировали этилацетатом. Объединенные экстракты промывали насыщенным водным раствором поваренной соли, сушили и упаривали. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом хроматографии на преупакованной REDISEP™ колонке с силикагелем (80 г), элюируя в градиенте от 0% до 25% EtOAc в гексане, получая (R)-6,6-диаллил-3,3-диметилтетрагидропирроло[1,2-с]оксазол-5(3Н)-он (3.31 г, 14.07 ммоль, 86% выход) в виде светло-желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 5.66-5.90 (м, 2Н), 5.06-5.19 (м, 4Н), 4.01-4.11 (м, 2Н), 3.29-3.38 (м, 1Н), 2.32-2.48 (м, 2Н), 2.20-2.29 (м, 1Н), 2.12 (дд, J=5.97, 13.79 Гц, 1Н), 1.86-1.98 (м, 1Н), 1.73-1.84 (м, 1Н), 1.65 (с, 3Н), 1.46 (с, 3Н).
Стадия 3: (R)-3,3-диаллил-5-(гидроксиметил)пирролидин-2-он
В раствор (R)-6,6-диаллил-3,3-диметилтетрагидропирроло[1,2-с]оксазол-5(3Н)-она (0.75 г, 3.19 ммоль) в МеОН (12 мл) добавляли п-толуолсульфокислоты моногидрат (0.061 г, 0.319 ммоль). Результирующую смесь нагревали до кипения в течение 2 часов. ТСХ показала полную конверсию. Растворитель упаривали, и неочищенное вещество адсорбировали на слое силикагеля и очищали методом хроматографии на преупакованной REDISEP™ колонке с силикагелем (12 г), элюируя в градиенте от 0% до 6% МеОН в ДХМ, получая (R)-3,3-диаллил-5-(гидроксиметил)пирролидин-2-он (0.62 г, 3.18 ммоль, 100% выход) в виде белого масла. 1Н ЯМР (400 МГц, CDCl3) δ 6.68 (ушир.с, 1Н), 5.67-5.86 (м, 2Н), 5.06-5.20 (м, 4Н), 3.62-3.74 (м, 2Н), 3.36-3.45 (м, 1Н), 2.37 (ддд, J=6.45, 11.86, 13.15 Гц, 2Н), 2.19 (ддд, J=4.79, 8.40, 13.45 Гц, 2Н), 1.99 (дд, J=7.72, 13.37 Гц, 1Н), 1.69 (дд, J=7.44,13.40 Гц, 1Н).
Стадия 4: (R)-(4,4-диаллилпирролидин-2-ил)метанол
Раствор (R)-3,3-диаллил-5-(гидроксиметил)пирролидин-2-она (0.43 г, 2.202 ммоль) в ТГФ (5.51 мл) охлаждали до 0°С, добавляли литийалюминий гидрид, 1.0 М раствор в ТГФ (2.86 мл, 2.86 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Добавляли еще литийалюминий гидрид, 1.0 М раствор в ТГФ (2.86 мл, 2.86 ммоль) и кипятили в течение 6 часов. Добавляли еще литийалюминий гидрид, 1.0 М раствор в ТГФ (2.86 мл, 2.86 ммоль), и полученную смесь кипятили в течение ночи. Реакционную смесь охлаждали до 0°С, после чего медленно добавляли водный раствор сегнетовой соли. Полученную суспензию экстрагировали этилацетатом (10 мл). Объединенные экстракты промывали насыщенным водным раствором поваренной соли, сушили и упаривали, получая (R)-(4,4-диаллилпирролидин-2-ил)метанол (0.34 г, 1.876 ммоль, 85% выход) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 5.72-5.88 (м, 2Н), 5.00-5.17 (м, 4Н), 3.49-3.59 (м, 1Н), 3.30-3.46 (м, 2Н), 2.79 (д, J=11.30 Гц, 1Н), 2.67 (д, J=11.35 Гц, 1Н), 2.08-2.19 (м, 4Н), 1.72 (дд, J=6.97,13.04 Гц, 1Н), 1.22-1.39 (м, 1Н).
Стадия 5: (R)-5,5-диаллилтетрагидро-3Н-пирроло[1,2-с][1,2,3]оксатиазол 1,1-диоксид
Раствор триэтиламина (2.460 мл, 17.65 ммоль) и (R)-(4,4-диаллилпирролидин-2-ил)метанола (1.60 г, 8.83 ммоль) в ДХМ (44.1 мл) охлаждали до -78°С. В полученную смесь добавляли сульфурил хлорид (0.859 мл, 10.59 ммоль) в ДХМ (44 мл) по каплям в течение 1 часа. Реакционную смесь выдерживали при этой температуре 3 часа, затем оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Полученную смесь промывали 1 н. водным раствором HCl (30 мл × 2), насыщенным водным раствором поваренной соли (30 мл), сушили, фильтровали и упаривали. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом хроматографии на преупакованной REDISEP™ колонке с силикагелем (40 г), элюируя в градиенте от 0% до 30% EtOAc в гексане, получая (R)-5,5-диаллилтетрагидро-3Н-пирроло[1,2-с][1,2,3]оксатиазол 1,1-диоксид (0.66 г, 2.71 ммоль, 30.7% выход) в виде светло-желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 5.71-5.86 (м, 2Н), 5.10-5.20 (м, 4Н), 4.57 (дд, J=6.63, 8.76 Гц, 1Н), 4.24-4.36 (м, 1Н), 4.19 (дд, J=4.66, 8.76 Гц, 1Н), 3.21-3.32 (м, 2Н), 2.19-2.29 (м, 4Н), 2.03-2.18 (м, 1Н), 1.57-1.63 (м, 1Н).
Стадия 6: (R)-3,5-дихлор-4-((4,4-диаллилпирролидин-2-ил)метил)пиридин
В раствор 3,5-дихлорпиридина (1.069 г, 7.22 ммоль) в ТГФ (12.04 мл) при -78°С по каплям добавляли диизопропиламид лития, 2.0 М раствор в смеси гептан/ТГФ/этилбензол (4.57 мл, 9.15 ммоль). После перемешивания в течение 1 часа, по каплям добавляли раствор (R)-5,5-диаллилтетрагидро-3Н-пирроло[1,2-с][1,2,3]оксатиазол 1,1-диоксида (1.172 г, 4.82 ммоль) в ТГФ (10 мл) при -78°С, и полученную смесь оставляли нагреваться до комнатной температуры при перемешивании на 6 часов. После упаривания растворителя, в полученную бежевую пену добавляли горячий 2 н. раствор HCl (12 мл) и EtOH (12 мл) на ночь. Полученную смесь охлаждали до комнатной температуры и подщелачивали 1 н. раствором NaOH и экстрагировали этилацетатом. Экстракты сушили, упаривали и очищали методом хроматографии на преупакованной REDISEP™ колонке с силикагелем (40 г), элюируя в градиенте от 1% до 6% МеОН в ДХМ, получая (R)-3,5-дихлор-4-((4,4-диаллилпирролидин-2-ил)метил)пиридин (0.70 г, 2.249 ммоль, 46.7% выход) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 8.46 (с, 2Н), 5.66-5.86 (м, 2Н), 5.03-5.18 (м, 4Н), 3.59-3.72 (м, 1Н), 3.25 (д, J=7.15 Гц, 1Н), 2.97 (д, J=11.51 Гц, 1Н), 2.82 (д, J=11.51 Гц, 1Н), 2.10-2.28 (м, 4Н), 1.78 (дд, J=13.06, 6.95 Гц, 1Н), 1.51-1.61 (м, 1Н); LCMS (ESI) m/z 311.0 (M+H)+.
Справочный пример В53
(R)-3-((3,5-дихлорпиридин-4-ил)метил)-2-азаспиро[4.4]нон-7-ен
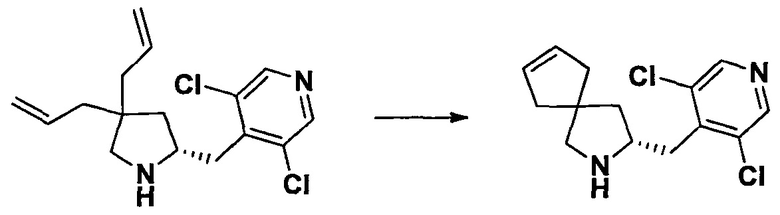
Смесь (R)-3,5-дихлор-4-((4,4-диаллилпирролидин-2-ил)метил)пиридина (3.1 г, 9.96 ммоль) и катализатора Граббса 2-го поколения (1.691 г, 1.992 ммоль) в ДХМ (996 мл). Полученную смесь перемешивали при 40°С 20 часов. Полученную смесь упаривали и адсорбировали на слой силикагеля и очищали методом хроматографии на колонке Biotage (100 г), элюируя в градиенте от 1% до 50% 1М NH3/MeOH в ДХМ, получая (R)-3-((3,5-дихлорпиридин-4-ил)метил)-2-азаспиро[4.4]нон-7-ен (1.0 г, 3.53 ммоль, 35.5% выход) в виде темно-коричневого масла. 1H ЯМР (400 МГц, CDCl3) δ 8.45 (с, 2Н), 5.61-5.72 (м, 2Н), 3.69-3.82 (м, 1Н), 3.25 (ушир.с, 2Н), 3.05 (д, J=10.47 Гц, 1Н), 2.89-2.97 (м, 1Н), 2.47 (ушир.с, 2Н), 2.23-2.37 (м, 2Н), 1.93 (дд, J=6.84, 12.59 Гц, 1Н), 1.69-1.82 (м, 1Н); LCMS (ESI) m/z 283.0 (M+H)+.
Справочный пример В54
(R)-3-((3,5-дихлорпиридин-4-ил)метил)-2-азаспиро[4.4]нон-7-ен
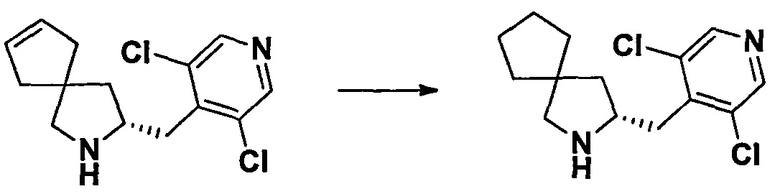
Смесь (R)-3-((3,5-дихлорпиридин-4-ил)метил)-2-азаспиро[4.4]нон-7-ена (0.090 г, 0.318 ммоль) и 10 вес. % Pd на активированном угле (0.034 г, 0.032 ммоль) в EtOAc (4 мл) перемешивали под шариком с водородом при комнатной температуре 3 часа. Исходное вещество превращалось в целевой продукт с моно-хлор продуктом (~ 4:1). Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом хроматографии на преупакованной REDISEP™ колонке с силикагелем (12 г), элюируя в градиенте от 5% до 50% 1М NH3/МеОН в ДХМ, получая (R)-3-((3,5-дихлорпиридин-4-ил)метил)-2-азаспиро[4.4]нонан (0.053 г, 0.186 ммоль, 58.5% выход) в виде коричневого масла. 1Н ЯМР (400 МГц, CDCl3) δ 8.42-8.50 (м, 2Н), 3.63-3.83 (м, 1Н), 3.28 (ушир.с, 2Н), 3.02 (д, J=10.37 Гц, 1Н), 2.87 (ушир.с, 1Н), 1.73-1.83 (м, 1Н), 1.54-1.72 (м, 9Н), 1.42-1.53 (м, 1Н); LCMS (ESI) m/z 285.0 (М+Н)+.
Приведенные далее вторичные амины получали по методике, аналогичной описанной выше в справочных примерах:
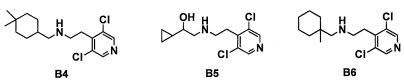
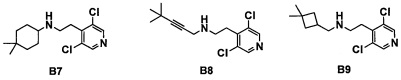
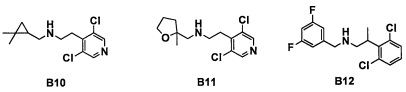
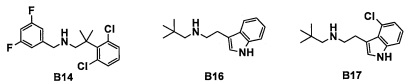
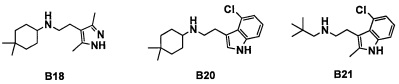
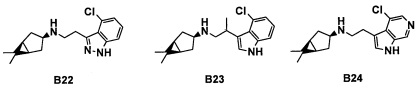
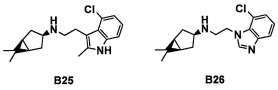
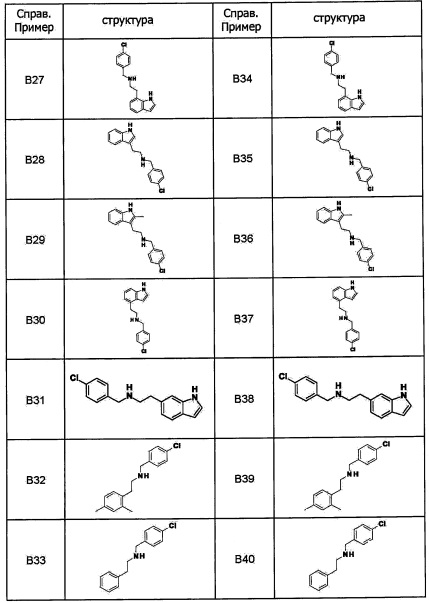
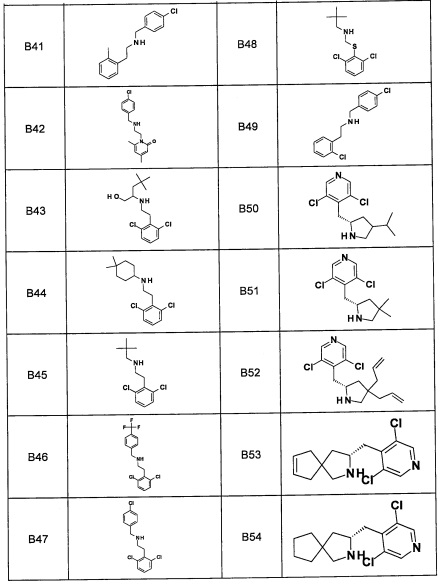
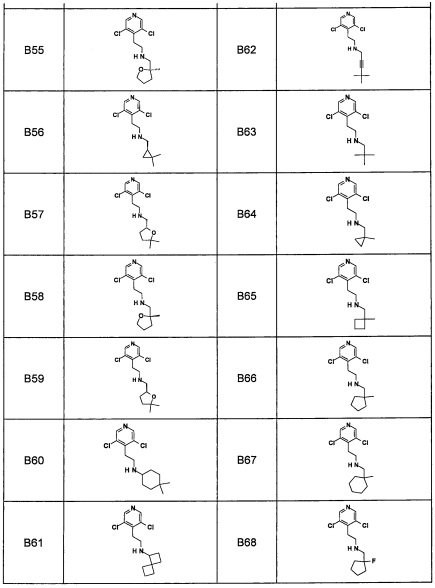
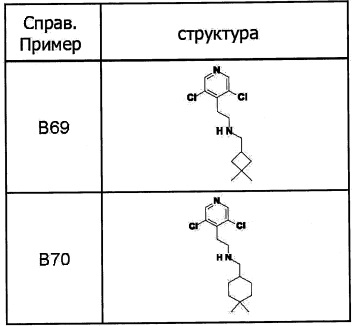
Справочный пример С1
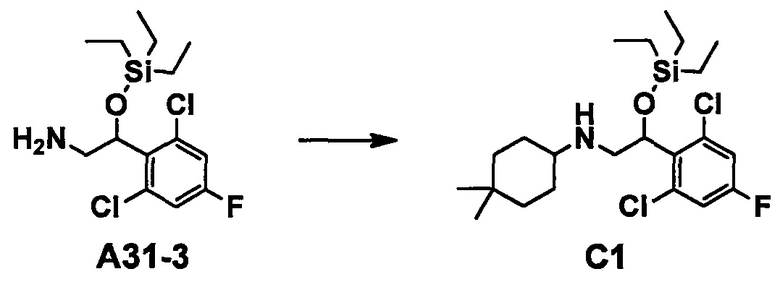
N-(2-(2,6-дихлор-4-фторфенил)-2-((триэтилсилил)окси)этил)-4,4-диметилциклогексанамин (С1)
В перемешиваемый раствор соединения A31-3 (107 мг, 0.32 ммоль) в ДХМ (2 мл) добавляли 4,4-диметилциклогексанон (40 мг, 0.32 ммоль), NaBH(ОАс)3 (83 мг, 0.38 ммоль) и АсОН (101 мг, 0.47 ммоль). Результирующую смесь перемешивали при комнатной температуре в течение 17 часов, затем гасили 0.5 М водным раствором NaOH (10 мл) и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (10 мл), сушили над MgSO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии на силикагеле (элюент: от 5% до 30% EtOAc/гексан), получая соединение С1 (122 мг, 86%) в виде бесцветного сиропа.
Справочный пример С22
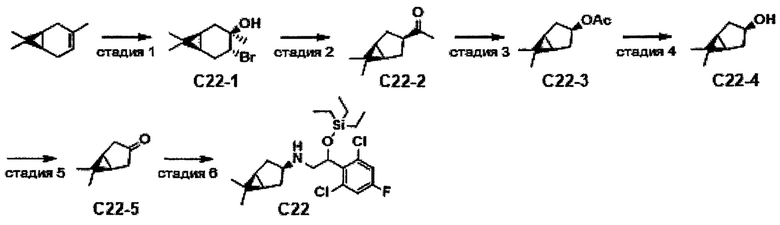
Стадия 1: (1S,3R,4R,6R)-4-бром-3,7,7-триметилбицикло[4.1.0]гептан-3-ол (С22-1)
Суспензию (+)-3-карена (4.09 г, 30 ммоль), СаСО3 (3.90 г, 39 ммоль) и NBS (6.94 г, 39 ммоль) в воде (15 мл) и 1,4-диоксане (30 мл) перемешивали при комнатной температуре в течение 1 часа. Полученную смесь разбавляли водой (75 мл) и экстрагировали Et2O (100 мл). Органический слой промывали водой (3×50 мл), насыщенным водным раствором Na2S2O3 (50 мл), сушили над MgSO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом хроматографии на силикагеле (10% EtOAc/гексан в качестве элюента), получая соединение С22-1 (4.53 г, 65%) в виде белого твердого вещества.
Стадия 2: 1-((1R,3R,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)этанон (С22-2)
В раствор соединения С22-1 (4.53 г, 19.4 ммоль) в воде (9 мл) и 1,4-диоксане (127 мл) добавляли оксид серебра (I) (12.16 г, 52.5 ммоль) и перемешивали при комнатной температуре в течение 22 часов. Полученную смесь фильтровали через слой целита, и фильтрат упаривали при пониженном давлении. Остаток от упаривания разбавляли водой и экстрагировали Et2O. Органический слой промывали водой, сушили над MgSO4 и упаривали при пониженном давлении, получая соединение С22-2 (2.86 г, 99%) в виде бледно-желтого масла. Полученный неочищенный продукт использовали в следующей стадии без очистки.
Стадия 3: (1R,3R,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил ацетат (С22-3)
В раствор соединения С22-2 (2.86 г, 18.8 ммоль) в ДХМ (57 мл) добавляли м-хлорпероксибензойную кислоту (6.02 г, 24.4 ммоль) при 0°С и перемешивали при комнатной температуре в течение 15 часов. Реакционную смесь гасили 0.2 М водным раствором NaOH и экстрагировали дихлорметаном (80 мл и 2×50 мл). Объединенные органические слои промывали насыщенным водным раствором NaHCO3, водой и насыщенным водным раствором поваренной соли, сушили над MgSO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом хроматографии на силикагеле (10% EtOAc/гексан в качестве элюента), получая соединение С22-3 (2.35 г, 74%) в виде бесцветной смолы.
Стадия 4: (1R,3R,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ол (С22-4)
В раствор соединения С22-3 (2.35 г, 14.0 ммоль) в смеси EtOH/вода (63 мл, 2:1) добавляли водный раствор LiOH (4 М, 21 мл, 84 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2.5 часов. Полученную смесь разбавляли водой и экстрагировали этилацетатом (2×80 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли, сушили над MgSO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом хроматографии на силикагеле (35% EtOAc/гексан в качестве элюента), получая соединение С22-4 (1.54 г, 88%) в виде бесцветного масла.
Стадия 5: (1R,5S)-6,6-диметилбицикло[3.1.0]гексан-3-он (С22-5)
Соединение С22-4 (240 мг, 1.9 ммоль) растворяли в ДХМ (5 мл) и добавляли периодинан Десс-Мартина (968 мг, 2.28 ммоль). Реакционную смесь перемешивали 3 часа. Реакционную смесь гасили 5%-ным раствором Na2S2O3 и экстрагировали Et2O (30 мл). Органический слой промывали насыщенным водным раствором NaHCO3 дважды, сушили над MgSO4 и упаривали при пониженном давлении, получая соединение С22-5 (261 мг, колич.) в виде бесцветной смолы. Полученный неочищенный продукт использовали в следующей стадии без очистки.
Стадия 6: (1R,3R,5S)-N-(2-(2,6-дихлор-4-фторфенил)-2-((триэтилсилил)окси)этил)-6,6-диметилбицикло[3.1,0]гексан-3-амин (С22)
Соединение С22 (75 мг, 74%) получали реакцией соединения A31-3 (77 мг, 0.228 ммоль), соединения С22-5 (31 мг, 0.250 ммоль), NaBH(ОАс)3 (72 мг, 0.341 ммоль) и АсОН (0.013 мл, 0.228 ммоль) в ДХМ (2 мл) по методике, аналогичной описанной в справочном примере А31, стадия 4. 1H ЯМР (CDCl3, 400 МГц): δ 7.04 (д, J=8.3 Гц, 2Н), 5.48 (дц, J=9.2, J=4.5 Гц, 1Н), 3.60-3.51 (м, 1Н), 3.19 (дд, J=12.2, J=9.2 Гц, 1Н), 2.65 (дд, J=12.2, J=4.5 Гц, 1Н), 2.17-2.07 (м, 2Н), 1.06-0.97 (м, 10Н), 0.87 (т, J=8.0 Гц, 9Н), 0.58-0.47 (м, 6Н).
Справочный пример С45
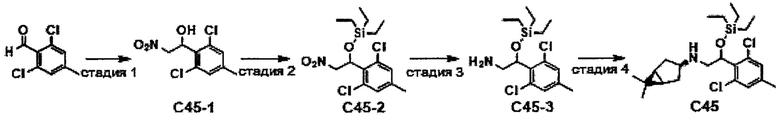
Стадия 1: 1-(2,6-дихлор-4-метилфенил)-2-нитроэтанол(С45-1)
Соединение С45-1 (1.25 г, 96%) получали в виде бесцветной смолы реакцией 2,6-дихлор-4-метилбензальдегида (1.0 г, 5.3 ммоль) и K2CO3 (0.28 г, 2.0 ммоль) в CH3NO2 (10 мл) по методике, аналогичной описанной в Примере А1, стадия 2.
Стадия 2: (1-(2,6-дихлор-4-метилфенил)-2-нитроэтокси)триэтилсилан (С45-2)
Соединение С45-2 (1.8 г, неочищенное) получали в виде бесцветной смолы реакцией соединения С45-1 (1.25 г, 1.0 ммоль), TES-CL (1.0 мл, 1.2 ммоль) и имидазола (1.2 г, 3.0 ммоль) в ДМФА (10 мл) по методике, аналогичной описанной в справочном примере А1, стадия 3.
Стадия 3: 2-(2,6-дихлор-4-метилфенил)-2-((триэтилсилил)окси)этанамин (С45-3)
Соединение С45-3 (1.56 г, 94%) получали в виде коричневого масла реакцией соединения С45-2 (1.8 г, 4.9 ммоль), Fe (2.76 г, 49.3 ммоль) и NH4Cl (2.62 г, 49.3 ммоль) в смеси EtOH/вода (4:1, 20 мл) по методике, аналогичной описанной в справочном примере А31, стадия 3.
Стадия 4: (1R,3R,5S)-N-(2-(2,6-дихлор-4-метилфенил)-2-((триэтилсилил)окси)этил)-6,6-диметилбицикло[3.1.0]гексан-3-амин (С45)
Соединение С45 (75 мг, 44%) получали реакцией С45-3 (130 мг, 0.389 ммоль), кетона С22-5 (49 мг, 0.394 ммоль), NaBH(ОАс)3 (125 мг, 0.590 ммоль) и АсОН (0.023 мл, 0.402 ммоль) в ДХМ (3 мл) по методике, аналогичной описанной в справочном примере А31, стадия 4. 1H ЯМР (CDCl3, 400 МГц) δ: 7.07 (2Н, с), 5.49 (1Н, дд, J=9.3, 4.4 Гц), 3.61-3.52 (1Н, м), 3.20 (1Н, дд, J=12.2, 9.3 Гц), 2.64 (1Н, дд, J=12.2, 4.4 Гц), 2.27 (3Н, с), 2.17-2.08 (2Н, м), 1.08-0.97 (10Н, м), 0.86 (9Н, т, J=7.8 Гц), 0.56-0.49 (6Н, м).
Справочный пример С46
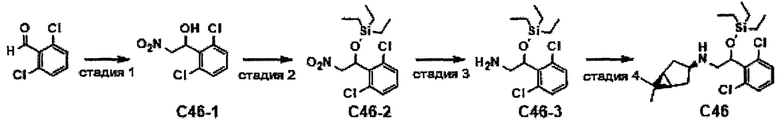
Стадия 1: 1-(2,6-дихлорфенил)-2-нитроэтанол (С46-1)
Соединение С46-1 (0.67 г, неочищенное) получали в виде желтой смолы реакцией 2,6-дихлорбензальдегида (0.5 г, 2.85 ммоль) и K2CO3 (0.15 г, 1.08 ммоль) в CH3NO2 (10 мл) по методике, аналогичной описанной в справочном примере А1, стадия 2.
Стадия 2: (1-(2,6-дихлорфенил)-2-нитроэтокси)триэтилсилан (С46-2)
Соединение С46-2 (0.95 г, 52%) получали в виде бесцветного масла реакцией соединения С46-1 (0.67 г, 2.83 ммоль), TES-CL (0.57 мл, 3.4 ммоль) и имидазола (0.58 г, 8.5 ммоль) в ДМФА (10 мл) по методике, аналогичной описанной в справочном примере А1, стадия 3.
Стадия 3: 2-(2,6-дихлорфенил)-2-((триэтилсилил)окси)этанамин (С46-3)
Соединение С46-3 (0.86 г, неочищенное) получали в виде бесцветного масла реакцией соединения С46-2 (0.95 г, 2.84 ммоль), Fe (1.59 г, 28.4 ммоль) и NH4Cl (1.51 г, 28.4 ммоль) в смеси EtOH/вода (4:1, 20 мл) по методике, аналогичной описанной в справочном примере А31, стадия 3.
Стадия 4: (1R,3R,5S)-N-(2-(2,6-дихлорфенил)-2-((триэтилсилил)окси)этил)-6,6-диметил-бицикло[3.1.0]гексан-3-амин (С46)
Соединение С46 (94 мг, 78%) получали реакцией соединения С46-3 (90 мг, 0.281 ммоль), кетона С22-5 (42 мг, 0.337 ммоль), NaBH(ОАс)3 (89 мг, 0.421 ммоль) и АсОН (0.016 мл, 0.281 ммоль) в ДХМ (2 мл) по методике, аналогичной описанной в справочном примере А31, стадия 4. 1H ЯМР (CDCl3, 400 МГц) δ: 7.30-7.26 (2Н, м), 7.09 (1Н, т, J=7.8 Гц), 5.53 (1Н, дд, J=9.3, 4.4 Гц), 3.62-3.53 (1Н, м), 3.23 (1Н, дд, J=12.2, 9.3 Гц), 2.66 (1Н, дд, J=12.2, 4.4 Гц), 2.17-2.10 (2Н, м), 1.04-0.99 (8Н, м), 0.90-0.84 (11Н, м), 0.60-0.45 (6Н, м).
Справочный пример С80
(1R,3R,5S)-N-(2-(2,6-дихлор-3-фторфенил)-2-((триэтилсилил)окси)этил)-6,6-диметилбицикло[3.1.0]гексан-3-амин
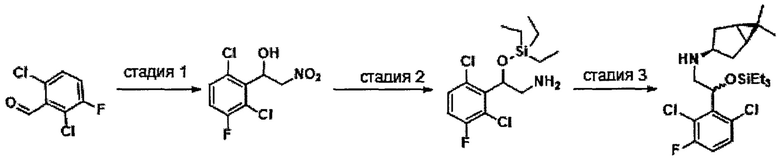
Стадия 1: 1-(2,6-дихлор-3-фторфенил)-2-нитроэтанол
В 3-горлой круглодонной колбе объемом 100 мл, свежеизмельченный карбонат калия (0.486 г, 3.51 ммоль) добавляли в раствор 2,6-дихлор-3-фторбензальдегида (2.26 г, 11.71 ммоль) в ТГФ (12 мл) при комнатной температуре. Затем добавляли нитрометан (8.88 мл, 164 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Полученную смесь гасили водой (15 мл) и экстрагировали этилацетатом (3×15 мл). Объединенный органический слой промывали насыщенным водным раствором поваренной соли, сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая 1-(2,6-дихлор-3-фторфенил)-2-нитроэтанол (2.97 г, 11.69 ммоль, 100% выход) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7.36 (дд, J=8.9,4.8 Гц, 1Н), 7.17 (дд, J=8.9, 7.8 Гц, 1Н), 6.27 (м, 1Н), 5.19 (дд, J=13.3, 10.1 Гц, 1Н), 4.57 (дд, J=13.3, 3.4 Гц, 1Н), 3.20 (ушир.с, 1Н).
Стадия 2: 2-(2,6-дихлор-3-фторфенил)-2-((триэтилсилил)окси)этанамин
В 3-горлую круглодонную колбу объемом 100 мл помещали (1-(2,6-дихлор-3-фторфенил)-2-нитроэтокси)триэтилсилан (3.64 г, 9.88 ммоль) в EtOH (16 мл) и воде (4 мл) при комнатной температуре, затем добавляли железо (5.52 г, 99 ммоль) и хлорид аммония (5.29 г, 99 ммоль). Колбу продували азотом и нагревали до 60°С в атмосфере азота 3 часа. Полученную смесь охлаждали до комнатной температуры, разбавляли добавлением 40 мл МеОН, обрабатывали ультразвуком 10 минут. Затем раствор декантировали через слой целита. Этот процесс повторяли три раза. Фильтрат упаривали до ~30 мл и разбавляли EtOAc (120 мл). Твердый осадок отфильтровывали и отбрасывали. Фильтрат упаривали при пониженном давлении. Остаток разбавляли 50 мл EtOAc, промывали водой, насыщенным водным раствором поваренной соли, сушили над безводным MgSO4 и упаривали, получая 2-(2,6-дихлор-3-фторфенил)-2-((триэтилсилил)окси)этанамина гидрохлорид в виде не совсем белого твердого вещества. Полученную HCl соль разбавляли 50 мл ДХМ. Суспензию подщелачивали насыщенным водным раствором NaHCO3 (рН=9). Органический слой отделяли, промывали насыщенным водным раствором поваренной соли, сушили над безводным MgSO4 и упаривали, получая 2-(2,6-дихлор-3-фторфенил)-2-((триэтилсилил)окси)этанамин (2.73 г, 8.07 ммоль, 82% выход) в виде коричневого масла. 1H ЯМР (400 МГц, CDCl3) δ 7.23-7.29 (м, 1Н), 6.99-7.06 (м, 1Н), 5.35 (дд, J=8.6, 4.9 Гц, 1Н), 3.29 (дд, J=13.1, 8.7 Гц, 1Н), 2.92 (дд, J=13.2, 4.9 Гц, 1Н), 0.83-0.93 (м, 9Н), 0.46-0.61 (м, 6Н); LCMS: 338.2 [М+Н]+.
Стадия 3: (1R,3R,5S)-N-(2-(2,6-дихлор-3-фторфенил)-2-((триэтилсилил)окси)этил)-6,6-диметилбицикло[3.1.0]гексан-3-амин
(1R,5S)-6,6-диметилбицикло[3.1.0]гексан-3-он (0.181 г, 1.457 ммоль) и 2-(2,6-дихлор-3-фторфенил)-2-((триэтилсилил)окси)этанамин (0.493 г, 1.457 ммоль) объединяли в безводном EtOH (7 мл) в атмосфере азота при комнатной температуре и добавляли тетраизопропоксититан (0.86 мл, 2.91 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем добавляли NaBH4 (0.083 г, 2.186 ммоль). Через 2 часа реакционный раствор гасили насыщенным водным раствором хлорида аммония (3 мл) и подщелачивали насыщенным водным раствором NaHCO3. EtOH затем удаляли при пониженном давлении, и раствор разбавляли водой EtOAc. Добавляли целит, и раствор интенсивно перемешивали 15 минут. Полученный раствор фильтровали через слой целита. Водный слой экстрагировали этилацетатом, и объединенные органические слои промывали насыщенным водным раствором поваренной соли и сушили над безводным Na2SO4, фильтровали и упаривали, получая желтое масло. Полученный сырой продукт очищали методом колоночной хроматографии (силикагель, элюент: от 0% до 10% EtOAc/гептан), получая (1R,3r,5S)-N-(2-(2,6-дихлор-3-фторфенил)-2-((триэтилсилил)окси)этил)-6,6-диметилбицикло[3.1.0]гексан-3-амин (414 мг, 0.927 ммоль, 63.6% выход) в виде бесцветного масла.
1Н ЯМР (400 МГц, CDCl3) δ 7.24 (дд, J=8.9, 4.9 Гц, 1Н), 6.98-7.04 (м, 1Н), 5.54 (ушир.с, 1Н), 3.59 (r, J=5.8 Гц, 1Н), 3.18-3.31 (м, 1Н), 2.71 (д, J=12.3 Гц, 1Н), 2.15 (д, J=5.1 Гц, 1Н), 1.22-1.34 (м, 4Н), 1.06 (д, J=5.8 Гц, 2Н), 0.99 (д, J=5.0 Гц, 6Н), 0.84-0.93 (м, 9Н), 0.47-0.59 (м, 6Н); LCMS: 446.2 [М+Н]+.
Приведенные далее вторичные амины получали по методике, аналогичной описанной выше в справочных примерах:
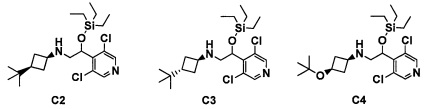
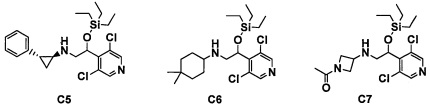
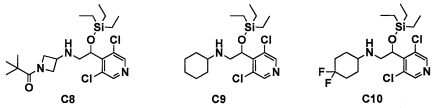
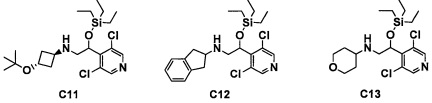
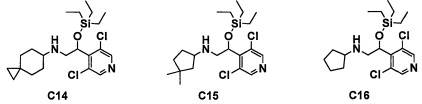
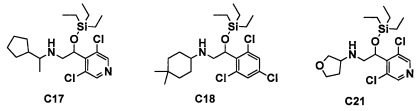
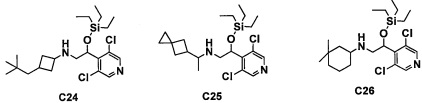
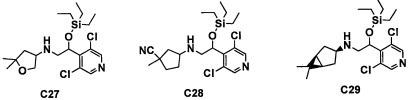
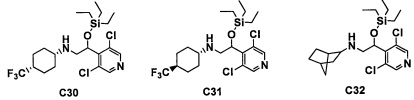
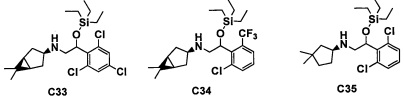
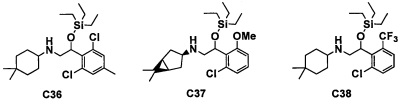
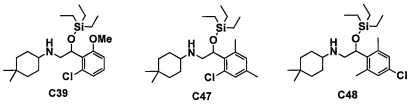
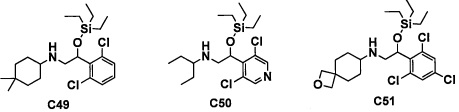
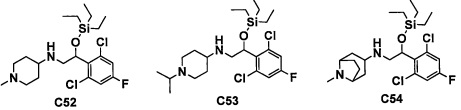

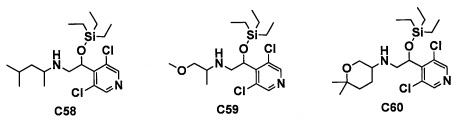
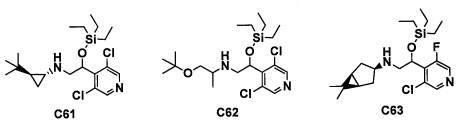
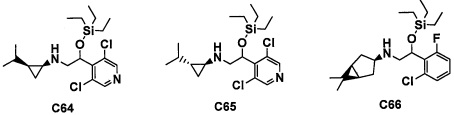
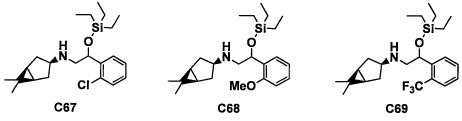
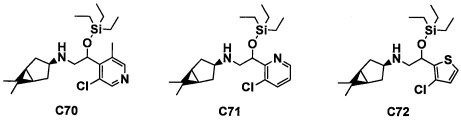
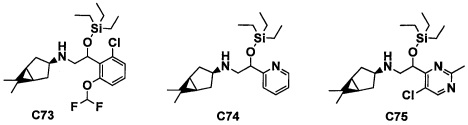
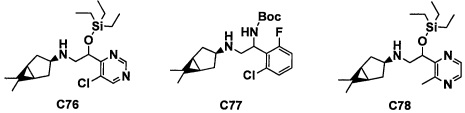
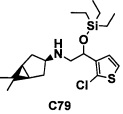
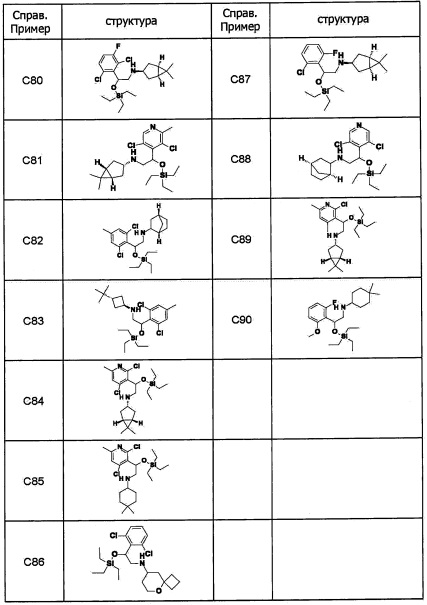
Справочный пример D1
Стадия 1: трет-бутил 2-(транс-4-(этоксикарбонил)циклогексил)гидразинкарбоксилат (D1-1)
В раствор этилового эфира 4-циклогексанонкарбоновой кислоты (5.0 г, 29.0 ммоль) и трет-бутил карбазата (3.9 г, 29.4 ммоль) в дихлорметане (250 мл) и АсОН (4 мл) постепенно добавляли NaBH(ОАс)3 (18.7 г, 88.0 ммоль) при 0°С. После добавления смесь перемешивали при той же температуре 3 часа, затем оставляли нагреваться до комнатной температуры и перемешивали 20 часов. Реакционную смесь выливали в насыщенный водный раствор Na2CO3 и экстрагировали дихлорметаном. ДХМ экстракты промывали насыщенным водным раствором поваренной соли (2 раза) и сушили над MgSO4. После удаления растворителя, остаток очищали методом колоночной хроматографии на силикагеле, получая соединение D1-1 (3.0 г, 36%) в виде белого твердого вещества.
Стадия 2: этил транс-4-гидразинилциклогексанкарбоксилат гидрохлорид (D1-2)
В раствор соединения D1-1 в EtOH (25 мл) добавляли 4 М раствор HCl (в ТГФ, 25 мл, 100 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Сушка раствора в высоком вакууме дала соединение D1-2 (2.8 г, колич.) в виде белого твердого вещества.
Стадия 3: бензил 4,4,4-трифтор-3-оксобутаноат (D1-3)
В раствор этил 4,4,4-трифтор-3-оксобутаноата (17.0 г, 92.3 ммоль) в толуоле (80 мл) добавляли бензиловый спирт (11.4 мл, 109.6 ммоль). Полученную смесь перемешивали при 120°С с насадкой Дина-Старка 5 часов, и затем реакционную смесь охлаждали до 0°С. Сушка раствора в высоком вакууме дала соединение D1-3 (21.2 г, колич.) в виде бесцветного масла, которое использовали в следующей стадии без дополнительной очистки.
Стадия 4: бензил 2-((диметиламино)метилен)-4,4,4-трифтор-3-оксобутаноат (D1-4)
В раствор соединения D1-3 (21.2 г, 92.3 ммоль) и АсОН (10.6 мл, 184.7 ммоль) в ТГФ (100 мл) по каплям добавляли N,N-диметилформамида диизопропилацеталь (38.6 мл, 184.7 ммоль) в течение 25 минут, и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь выливали в насыщенный водный раствор NaHCO3 и экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором поваренной соли (2 раза) и сушили над MgSO4. После удаления растворителя остаток очищали методом колоночной хроматографии на силикагеле, получая соединение D1-4 (17.1 г, 91%) в виде желтого масла.
Стадия 5: бензил 1-(транс-4-(этоксикарбонил)циклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат (D1-5)
В раствор соединения D1-2 (2.8 г, 10.5 ммоль) в EtOH (50 мл) добавляли DIPEA (3.2 мл, 12.6 ммоль) и соединение D1-4 (3.3 г, 11.0 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 1.5 часов. Реакцию гасили насыщенным водным раствором поваренной соли и экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором поваренной соли (х 2) и сушили над MgSO4. После удаления растворителя остаток очищали методом колоночной хроматографии на силикагеле, получая соединение D1-5 (3.5 г, 78%) в виде бесцветного масла.
Стадия 6: 1-(транс-4-(этоксикарбонил)циклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота (D1)
Соединение D1-5 (3.5 г, 8.2 ммоль) и 10% Pd на угле (300 мг) в EtOAc (40 мл) гидрировали в атмосфере водорода (1 атм) при комнатной температуре 25 часов. Реакционную смесь фильтровали через слой целита и промывали этилацетатом. Сушка раствора в высоком вакууме дала соединение D1 (2.6 г, 95%) в виде белого твердого вещества.
Справочный пример D2
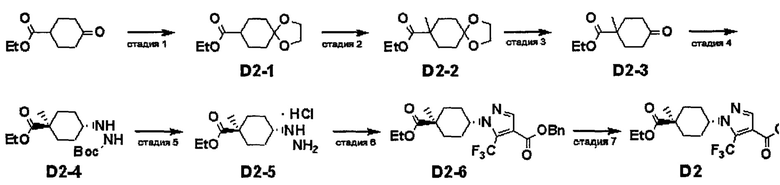
Стадия 1: этил-1,4-диоксаспиро[4.5]декан-8-карбоксилат (D2-1)
Смесь этил-4-оксоциклогексанкарбоксилата (10 г, 58.75 ммоль), этиленгликоля (4.97 мл, 88.13 ммоль) и п-TsOH (кат.) в толуоле (80 мл) кипятили в течение 16 часов в колбе, оснащенной насадкой Дина-Старка. После окончания реакции, смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении, получая соединение D2-1 (9.6 г, неочищенное) в виде коричневого масла. Полученный неочищенный продукт использовали в следующей стадии без дополнительной очистки. 1H ЯМР (CDCl3, 400 МГц): δ 4.15-4.09 (м, 2Н), 3.95 (с, 4Н), 2.36-2.03 (м, 1Н), 1.97-1.91 (м, 2Н), 1.85-1.75 (м, 4Н), 1.66-1.52 (м, 2Н), 1.26-1.27 (м, 3Н).
Стадия 2: этил-8-метил-1,4-диоксаспиро[4.5]декан-8-карбоксилат (D2-2)
В перемешиваемый раствор соединения D2-1 (5.1 г, 23.83 ммоль) в ТГФ (15 мл) добавляли LDA (2.0 М раствор в смеси ТГФ/гептан/этилбензол, 17.8 мл, 35.74 ммоль) по каплям при -78°С в течение 15 минут. Полученную смесь перемешивали при -78°С 30 минут. Раствор иодметана (2.23 мл, 35.74 ммоль) в ТГФ (1 мл) добавляли в полученную смесь по каплям, и результирующий раствор перемешивали 30 минут при -78°С. Полученную смесь оставляли нагреваться до комнатной температуры и перемешивали 16 часов. Реакцию гасили насыщенным водным раствором NH4Cl и экстрагировали этилацетатом (2×25 мл). Объединенные органические слои промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 2% EtOAc/гексан в качестве элюента), получая соединение D2-2 (2.7 г, 50%) в виде бесцветного масла. 1Н ЯМР (CDCl3, 400 МГц): δ 4.14 (кв, J=7.2 Гц, 2Н), 3.93 (с, 4Н), 2.15-2.10 (м, 2Н), 1.65-1.60 (м, 4Н), 1.54-1.49 (м, 2Н), 1.25 (т, J=7.2 Гц, 3Н), 1.18 (с, 3Н).
Стадия 3: этил-1-метил-4-оксоциклогексанкарбоксилат (D2-3)
В раствор соединения D2-2 (8.4 г, 36.84 ммоль) в ацетоне (100 мл) по каплям добавляли HCl (3 М раствор в воде, 50 мл) при комнатной температуре, и результирующую смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь гасили водой (100 мл) и экстрагировали этилацетатом (2×25 мл). Объединенные органические слои промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая соединение D2-3 (6.3 g) в виде светло-желтого масла. Полученный неочищенный продукт использовали в следующей стадии без дополнительной очистки. 1Н ЯМР (CDCl3, 400 МГц): δ 4.22 (кв, J=7.0 Гц, 2Н), 2.47-2.38 (м, 4Н), 2.34-2.30 (м, 2Н), 1.72-1.64 (м, 2Н), 1.31-1.29 (м, 6Н).
Стадия 4: трет-бутил-2-(транс-4-(этоксикарбонил)-4-метилциклогексил) гидразин-карбоксилат (D2-4)
В смесь соединения D2-3 (30 г, 163.0 ммоль) и трет-бутилгидразин карбоксилата (21.5 г, 163.0 ммоль) в изопропаноле (200 мл) добавляли АсОН (каталитическое количество), и полученную смесь перемешивали при комнатной температуре в течение 2 часов. По окончании формирования имина (мониторинг методом ТСХ), смесь охлаждали до 0°С, и порциями добавляли твердый NaBH3CN (30.7 г, 489.1 ммоль). Значение рН реакционной смеси доводили до 5-6 с помощью АсОН, и продолжали перемешивание в течение 3 часов при комнатной температуре. Полученную смесь гасили водой (100 мл) и экстрагировали этилацетатом (2×200 мл). Объединенные органические слои промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 30% EtOAc/гексан) (Примечание: полярное пятно соответствовало транс-изомеру), получая соединение D2-4 (12.0 г, 34%) в виде белого твердого вещества.
Стадия 5: этил транс-4-гидразинил-1-метилциклогексанкарбоксилат гидрохлорид (D2-5)
В раствор соединения D2-4 (36.0 г, 120.0 ммоль) в EtOH (100 мл) добавляли HCl (4 М раствор в 1,4-диоксане, 350 мл) по каплям при 0°С, и результирующую смесь перемешивали при комнатной температуре в течение 18 часов. Растворитель удаляли при пониженном давлении, и остаток растирали в Et2O, получая соединение D2-5 (31.0 г, 95%) в виде белого твердого вещества. Полученный неочищенный продукт использовали в следующей стадии без дополнительной очистки. 1Н ЯМР (CDCl3, 400 МГц): δ 7.24-7.00 (ушир.с, 4Н), 4.13 (кв, J=7.2 Гц, 2Н), 3.44 (ушир.с, 1Н), 2.08-2.05 (м, 2Н), 1.97-1.90 (м, 2Н), 1.81-1.80 (м, 4Н), 1.30-1.26 (м, 6Н).
Стадия 6: бензил-1-(транс-4-(этоксикарбонил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат (D2-6)
В раствор соединения D2-5 (31.0 г, 113.9 ммоль) в EtOH (150 мл) добавляли DIPEA (39.4 мл, 227.9 ммоль) по каплям, и полученную смесь перемешивали при комнатной температуре в течение 5 минут. Добавляли по каплям раствор соединения D1-4 (37.7 г, 125.3 ммоль) в EtOH (10 мл), и результирующую смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь гасили водой (200 мл) и экстрагировали этилацетатом (2×200 мл). Объединенный органический слой промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 15% EtOAc/гексан в качестве элюента), получая соединение D2-6 (20.0 г, 40%) в виде коричневой смолы. 1Н ЯМР (CDCl3 400 МГц): δ 7.94 (с, 1Н), 7,40-7.35 (м, 5Н), 5.30 (с, 2Н), 4.36 (м, 1Н), 4.15 (кв, J=7.2 Гц, 2Н), 2.24-2.19 (м, 2Н), 1.88-1.87 (м, 6Н), 1.3 (с, 3Н), 1.26 (т, J=7.2 Гц, 3Н).
Стадия 7: транс-4-(этоксикарбонил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота (D2)
Смесь соединения D2-6 (20.0 г, 45.6 ммоль) и 5% Pd на угле (10.0 г, 50% по весу) в МеОН (200 мл) перемешивали в атмосфере водорода (1 атм) 4 часа. Полученную смесь фильтровали через слой целита, промывали этилацетатом (3×100 мл) и упаривали при пониженном давлении. Остаток от упаривания растирали в смеси 10% EtOAc/гексан (2×25 мл), получая соединение D2 (13.0 г, 82%) в виде белого твердого вещества. 1H ЯМР (CDCl3, 300 МГц): δ 8.03 (с, 1Н), 4.42-4.41 (м, 1Н), 4.15 (кв, J=7.2 Гц, 2Н), 2.25-2.21 (м, 2Н), 1.92-1.88 (м, 6Н), 1.35 (с, 3Н), 1.27 (т, J=7.0 Гц, 3Н).
Справочный пример D19
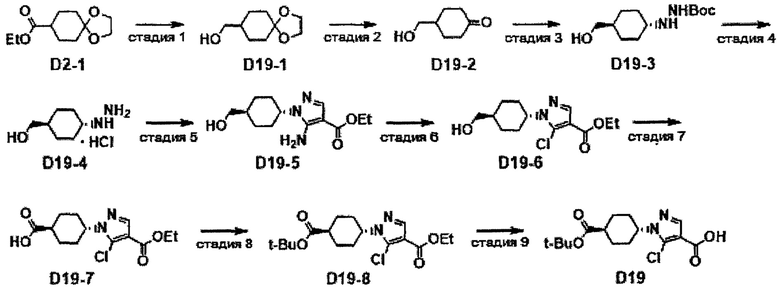
Стадия 1: 1,4-диоксаспиро[4.5]декан-8-илметанол (D19-1)
В суспензию LiAlH4 (5.69 г, 150 ммоль) в ТГФ (100 мл) по каплям добавляли раствор соединения D2-1 (21.4 г, 100 ммоль) в ТГФ (100 мл) при 0°С, и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь охлаждали до 0°С, гасили водой (7 мл) и 6 М раствором NaOH (7 мл) и перемешивали при комнатной температуре в течение 20 минут. В полученную смесь добавляли Na2SO4 (10 г), фильтровали через слой целита и промывали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (100 мл), водой (100 мл) и упаривали при пониженном давлении, получая соединение D19-1 (17.0 г, колич.) в виде бесцветного масла. Полученный неочищенный продукт использовали в следующей стадии без очистки.
Стадия 2: 4-(гидроксиметил)циклогексанон (D19-2)
В перемешиваемый раствор соединения D19-1 (17.0 г, 9.88 ммоль) в ацетоне (100 мл) добавляли водный раствор HCl (2 М, 38 мл), и полученную смесь перемешивали при комнатной температуре в течение 18 часов. Растворитель удаляли при пониженном давлении, затем разбавляли водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали водой, сушили над Na2SO4 и упаривали при пониженном давлении, получая соединение D19-2 (7.5 г, 51%) в виде бесцветной смолы.
Стадия 3: трет-бутил 2-(транс-4-(гидроксиметил)циклогексил)гидразинкарбоксилат (D19-3)
Смесь соединения D19-2 (2.0 г, 15.5 ммоль) и Вос-гидразина (2.26 г, 17 ммоль) в изопропаноле (20 мл) перемешивали при комнатной температуре в течение 16 часов. Добавляли Na(CN)BH3 (2.92 г, 45.6 ммоль) и АсОН (1 мл, кат.), и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь гасили водой (50 мл) и экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали водой, сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 50% EtOAc/гексан в качестве элюента), получая соединение D19-3 (820 мг, 22%) в виде белого полукристаллического вещества.
Стадия 4: (транс-4-гидразинилциклогексил)метанол гидрохлорид (D19-4)
В перемешиваемую смесь соединения D19-3 (1.8 г, 7.3 ммоль) в диоксане (40 мл) добавляли HCl (20 мл, 73 ммоль, 4 М раствор в диоксане), и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель удаляли при пониженном давлении, сушили на высоковакуумном насосе, получая соединение D19-4 (1.7 г, неочищенное) в виде не совсем белого твердого вещества.
Стадия 5: этил-5-амино-1-(транс-4-(гидроксиметил)циклогексил)-1Н-пиразол-4-карбоксилат (D19-5)
В раствор соединения D19-4 (720 мг, 3.31 ммоль) в EtOH (20 мл) добавляли этил-2-циано-3-этоксиакрилат (448 мг, 2.65 ммоль) и NaOAc (571 мг, 6.96 ммоль), и полученную смесь перемешивали при 70°С 18 часов. Растворитель удаляли при пониженном давлении, остаток суспендировали в воде (20 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические слои промывали водой, сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом обращенно-фазной колоночной хроматографии (С18 силикагель, 30% CH3CN/вода в качестве элюента), получая соединение D19-5 (320 мг, 37%) в виде красновато-коричневого твердого вещества.
Стадия 6: этил-5-хлор-1-(транс-4-(гидроксиметил)циклогексил)-1Н-пиразол-4-карбоксилат (D19-6)
В суспензию CuCl (103 мг, 1.04 ммоль) в CH3CN (5 мл) по каплям добавляли трет-бутилнитрит (0.134 мл, 1.125 ммоль) при 0°С. Раствор соединения D19-5 (200 мг, 0.749 ммоль) в CH3CN (4 мл) добавляли по каплям в полученную смесь при 0°С, и перемешивали при той же температуре 5 минут. Полученную смесь перемешивали при комнатной температуре в течение 30 минут и при 70°С 30 минут. Реакционную смесь гасили водой (10 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические слои промывали водой, сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 40% EtOAc/гексан в качестве элюента), получая соединение D19-6 (68 мг, 31%) в виде коричневого полукристаллического вещества.
Стадия 7: транс-4-(5-хлор-4-(этоксикарбонил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (D 19-7)
В суспензию H5IO6 (159 мг, 0.698 ммоль) в CH3CN добавляли CrO3 (0.6 мг, 0.0061 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Полученную смесь охлаждали до 0°С и добавляли по каплям раствор соединения D19-6 (100 мг, 0.349 ммоль). Реакционную смесь перемешивали при той же температуре 30 минут. Органический растворитель удаляли при пониженном давлении, остаток суспендировали в воде (10 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические слои промывали водой, сушили над Na2SO4 и упаривали при пониженном давлении, получая соединение D19-7 (105 мг, колич.) в виде не совсем белого твердого вещества.
Стадия 8: этил-1-(транс-4-(трет-бутоксикарбонил)циклогексил)-5-хлор-1Н-пиразол-4-карбоксилат (D19-8)
В смесь соединения D19-7 (105 мг, 0.35 ммоль) и Вос-ангидрида (152 мг, 0.70 ммоль) в т-BuOH (5 мл) добавляли DMAP (13 мг, 0.105 ммоль), и полученную смесь перемешивали при 35°С 16 часов. Реакционную смесь гасили водой (10 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические слои промывали водой, сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом обращенно-фазной колоночной хроматографии (С18 силикагель, 90% CH3CN/вода в качестве элюента), получая соединение D19-8 (70 мг, 56%) в виде бесцветной смолы.
Стадия 9: 1-(транс-4-(трет-бутоксикарбонил)циклогексил)-5-хлор-1Н-пиразол-4-карбоновая кислота (D19)
В перемешиваемый раствор соединения D19-8 (70 мг, 0.233 ммоль) в смеси ТГФ/МеОН (4 мл, 1:1) добавляли раствор LiOH (44 мг, 1.86 ммоль) в воде (1 мл). Полученную смесь перемешивали при комнатной температуре в течение 4 часов. Органический растворитель удаляли при пониженном давлении. Остаток от упаривания разбавляли водой (5 мл), подкисляли 20%-ным водным раствором KHSO4 до рН 4 и экстрагировали этилацетатом (3×10 мл), получая соединение D19 (62 мг, 90%) в виде белого твердого вещества. 1H ЯМР (CDCl3, 300 МГц): δ 8.01 (с, 1Н), 4.29-4.37 (м, 1Н), 2.25-2.43 (м, 1Н), 2.10-2.19 (м, 2Н), 1.99-2.09 (м, 4Н), 1.52-1.65 (м, 2Н), 1.45 (с, 9Н).
Справочный пример D20
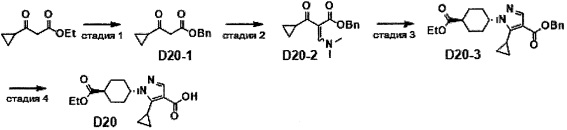
Стадия 1: бензил 3-циклопропил-3-оксопропаноат (D20-1)
Смесь этил 3-циклопропил-3-оксопропаноата (5.0 г, 32.0 ммоль), бензилового спирта (8.2 мл, 80.0 ммоль) и LiOCl (680 мг, 6.4 ммоль) в толуоле (50 мл) кипятили в течение 48 часов в колбе, снабженной насадкой Дина-Старка. Реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении, получая соединение D20-1 (5.2 г, неочищенное) в виде коричневого масла.
Стадия 2: бензил 2-(циклопропанкарбонил)-3-(диметиламино)акрилат (D20-2)
Смесь соединения D20-1 (1.0 г, 4.58 ммоль) и диметилформамида диметилацеталя (0.61 мл, 4.58 ммоль) в 1,4-диоксане (25 мл) перемешивали при 100°С 13 часов. Реакционную смесь гасили водой (20 мл) и экстрагировали этилацетатом (2×25 мл). Объединенные органические слои промывали водой (25 мл), насыщенным водным раствором поваренной соли (25 мл), сушили над Na2SO4 и упаривали при пониженном давлении, получая соединение D20-2 (1.2 г, неочищенное) в виде желтовато-коричневой смолы.
Стадия 3: бензил 5-циклопропил-1-(транс-4-(этоксикарбонил)циклогексил)-1Н-пиразол-4-карбоксилат (D20-3)
В раствор соединения D1-2 (809 мг, 2.67 ммоль) в EtOH (20 мл) по каплям добавляли DIPEA (0.45 мл, 2.61 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 5 минут, затем добавляли по каплям раствор соединения D20-2 (600 мг, 2.18 ммоль) в EtOH (5 мл), и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь гасили водой (200 мл) и экстрагировали этилацетатом (2×25 мл). Объединенные органические слои промывали водой (25 мл), насыщенным водным раствором поваренной соли (25 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 20% EtOAc/гексан в качестве элюента), получая соединение D20-3 (425 мг, не чистый) в виде желтой смолы.
Стадия 4: 5-циклопропил-1-(транс-4-(этоксикарбонил)циклогексил)-1Н-пиразол-4-карбоновая кислота (D20)
В перемешиваемый раствор соединения D20-3 (425 мг, 1.07 ммоль) в смеси ТГФ/МеОН (20 мл, 1:1) добавляли 10% Pd на угле (80 мг, 20% по весу), и полученную смесь перемешивали в атмосфере водорода (1 атм) в течение 2 часов. Полученную смесь фильтровали через слой целита и промывали этилацетатом (3×50 мл). Фильтрат упаривали при пониженном давлении. Остаток от упаривания растирали в смеси 10% EtOAc/гексан (2×20 мл), получая соединение D20 (200 мг, неочищенное) в виде белого твердого вещества.
Справочный пример D22
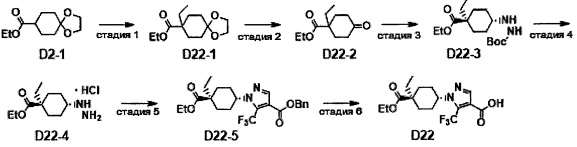
Стадия 1: этил 8-этил-1,4-диоксаспиро[4.5]декан-8-карбоксилат (D22-1)
В перемешиваемый раствор соединения D2-1 (2.1 г, 9.80 ммоль) в ТГФ (24 мл) по каплям добавляли LDA (2.0 М раствор в смеси ТГФ/гептан/этилбензол, 7.3 мл, 14.7 ммоль) при -78°С в течение 5 минут. Полученную смесь перемешивали при -78°С 15 минут, после чего добавляли EtBr (1.09 мл, 14.7 ммоль). Реакционную смесь перемешивали при -78°С 1 час. Полученную смесь в виде желтовато-коричневой смолы оставляли нагреваться до комнатной температуры и перемешивали при той же температуре 1 час. Реакционную смесь гасили насыщенным водным раствором NH4Cl и экстрагировали этилацетатом (2×25 мл). Объединенные органические слои промывали водой (20 мл), насыщенным водным раствором поваренной соли (20 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 20% EtOAc/гексан в качестве элюента), получая соединение D22-1 (2.07 г, 87%) в виде бесцветной смолы.
Стадия 2: этил 1-этил-4-оксоциклогексанкарбоксилат (D22-2)
В перемешиваемый раствор соединения D22-1 (2.07 г, 8.54 ммоль) в ацетоне (60 мл) добавляли водный раствор HCl (2 М, 40 мл) при комнатной температуре. Полученную смесь перемешивали при той же температуре 16 часов. Ацетон удаляли при пониженном давлении. Остаток от упаривания подщелачивали водным раствором NaHCO3 и экстрагировали дихлорметаном (2×30 мл). Объединенные органические слои промывали водой (20 мл), насыщенным водным раствором поваренной соли (20 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 20% EtOAc/гексан в качестве элюента), получая соединение D22-2 (1.85 г, 99%) в виде бесцветной смолы.
Стадия 3: трет-бутил 2-(транс-4-(этоксикарбонил)-4-этилциклогексил)гидразинкарбоксилат (D22-3)
Соединение D22-3 (1.57 г, 53%) получали в виде белого твердого вещества реакцией соединения D22-2 (1.87 г, 9.43 ммоль), трет-бутил гидразинкарбоксилата (1.24 г, 9.4 ммоль), АсОН (кат.) и NaBH3CN (1.78 г, 28.29 ммоль) в изопропаноле (20 мл) по методике, аналогичной описанной в справочном примере D2, стадия 4.
Стадия 4: этил транс-1-этил-4-гидразинилциклогексанкарбоксилат гидрохлорид (D22-4)
Соединение D22-4 (1.36 г, 100%) получали в виде белого твердого вещества реакцией соединения D22-3 (1.50 г, 4.78 ммоль) и HCl (4 М раствор в 1,4-диоксане, 8.3 мл, 33.4 ммоль) по методике, аналогичной описанной в справочном примере D2, стадия 5.
Стадия 5: бензил 1-(транс-4-(этоксикарбонил)-4-этилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат (D22-5)
Соединение D22-5 (820 мг, 86%) получали в виде бесцветной смолы реакцией соединения D22-4 (600 мг, 2.1 ммоль), соединения D1-4 (669 мг, 2.2 ммоль) и DIPEA (0.43 мл, 2.52 ммоль) в EtOH (12 мл) по методике, аналогичной описанной в справочном примере D2, стадия 6.
Стадия 6: 1-(транс-4-(этоксикарбонил)-4-этилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота (D22)
Соединение D22 (285 мг, 98%) получали в виде белого твердого вещества реакцией соединения D22-5 (363 мг, 0.80 ммоль), 5% Pd на угле (85 мг, 30% по весу) и Н2 (1 атм) в МеОН (6 мл) по методике, аналогичной описанной в справочном примере D2, стадия 7.
Справочный пример D26
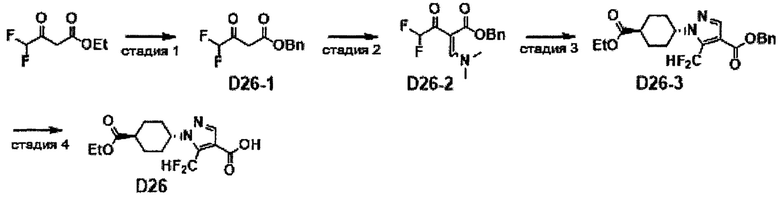
Стадия 1: бензил-4,4-дифтор-3-оксобутаноат (D26-1)
Соединение D26-1 (7.5 мг, неочищенное) получали в виде желтого масла реакцией этил-4,4-дифтор-3-оксобутаноата (5 г, 0.12 ммоль) и BnOH (3.25 г, 30.0 ммоль) в толуоле (50 мл) по методике, аналогичной описанной в справочном примере D1, стадия 3.
Стадия 2: бензил-2-((диметиламино)метилен)-4,4-дифтор-3-оксобутаноат (D26-2)
Соединение D26-2 (5.8 г, неочищенное) получали в виде желтого масла реакцией соединения D26-1 (5.3 г, 23.2 ммоль), диметилформамида диметилацеталя (6.2 мл, 46.4 ммоль) и АсОН (2.05 мл, 46.4 ммоль) в ТГФ (50 мл) по методике, аналогичной описанной в справочном примере D1, стадия 4.
Стадия 3: бензил-5-(дифторметил)-транс-4-(этоксикарбонил)циклогексил)-1Н-пиразол-4-карбоксилат (D26-3)
Соединение D26-3 (520 мг, 16%) получали в виде светло-желтого твердого вещества реакцией соединения D26-2 (1.50 г, 5.28 ммоль), соединения D1-2 (1.6 г, 5.28 ммоль) и DIPEA (1.8 мл, 10.5 ммоль) в EtOH (30 мл) по методике, аналогичной описанной в справочном примере D1, стадия 5.
Стадия 4: 5-(дифторметил)-транс-4-(этоксикарбонил)циклогексил)-1Н-пиразол-4-карбоновая кислота (D26)
Соединение D26 (255 мг, 63%) получали в виде белого твердого вещества реакцией соединения D26-3 (520 мг, 1.28 ммоль) и 5% Pd на угле (70 мг, 30% по весу) в EtOH (30 мл) по методике, аналогичной описанной в справочном примере D1, стадия 6.
LCMS (APCI): 317 (М+Н)+.
Справочный пример D27
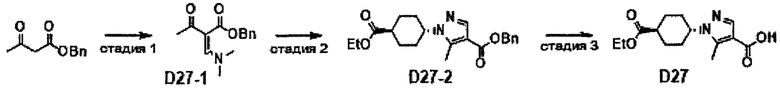
Стадия 1: бензил 2-((диметиламино)метилен)-3-оксобутаноат (D27-1)
В перемешиваемый бензил 3-оксобутаноат (1.1 г, 5.7 ммоль) добавляли по каплям диметилформамида диметилацеталь (1 мл, 7.4 ммоль) при комнатной температуре. Полученную смесь перемешивали 16 часов при комнатной температуре. Реакционную смесь упаривали при пониженном давлении, и остаток подвергали азеотропной отгонке с толуолом (3×10 мл), получая соединение D27-1 в виде коричневого масла (1.4 г, колич.).
Стадия 2: бензил 1-((транс-4-(этоксикарбонил)циклогексил)-5-метил-1Н-пиразол-4-карбоксилат (D27-2)
В раствор соединения D1-2 (1.12 г, 4.3 ммоль) в EtOH (10 мл) по каплям добавляли DIPEA (1.2 мл, 6.7 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 5 минут. Добавляли по каплям раствор соединения D27-1 (0.97 г, 3.94 ммоль) в EtOH (5 мл), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили водой (20 мл) и экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 20% EtOAc/гексан в качестве элюента), получая соединение D27-2 (0.78 г, 54%) в виде белого твердого вещества.
Стадия 3: 1-((транс-4-(этоксикарбонил)циклогексил)-5-метил-1Н-пиразол-4-карбоновая кислота (D27)
В перемешиваемый раствор соединения D27-2 (0.78 г, 2.1 ммоль) в МеОН (10 мл) добавляли 5% Pd на угле (0.19 г, 25% по весу), и полученную смесь перемешивали в атмосфере водорода (1 атм) в течение 2 часов. Полученную смесь фильтровали через слой целита и промывали МеОН (3×20 мл). Фильтрат промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания растирали в 5% EtOAc/гексан (20 мл), получая соединение D27 (0.5 г, 85%) в виде белого твердого вещества. 1Н ЯМР (300 МГц, ДМСО-d6): δ 1.19 (т, J=7.2 Гц), 1.56 (м, 2Н), 1.88 (м, 4Н), 2.00 (м, 2Н), 2.35 (м, 1Н), 2.50 (с, 3Н), 4.07 (кв, J=7.2 Гц, 2Н), 4.20 (м, 1Н), 7.72 (с, 1Н), 12.10 (с, 1Н).
Справочный пример D28
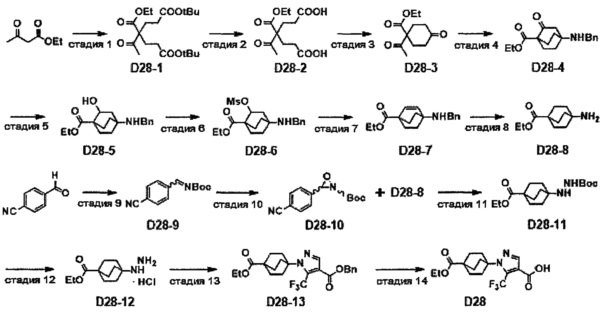
Стадия 1: 1,5-ди-трет-бутил 3-этил 3-ацетилпентан-1,3,5-трикарбоксилат (D28-1)
В перемешиваемый раствор этил 3-оксобутаноата (45 г, 345 ммоль) и Triton-B (40 вес. %, раствор в воде, 1.08 мг, 6.90 ммоль) в трет-BuOH (54 мл) по каплям добавляли трет-бутил акрилат (100.72 г, 691 ммоль) в течение 30 минут в атмосфере азота. Полученный раствор перемешивали при комнатной температуре в течение 24 часов. Реакционную смесь разделяли между водой (200 мл) и EtOAc (200 мл). Водный слой промывали EtOAc (2×50 мл). Объединенные органические слои промывали водой (200 мл), насыщенным водным раствором поваренной соли (200 мл), сушили над Na2SO4 и упаривали при пониженном давлении, получая соединение D28-1 (140 г, колич.) в виде бледно-желтого масла. 1H ЯМР (CDCl3, 400 МГц): δ 4.20 (кв, J=7.2 Гц, 2Н), 2.24-2.09 (м, 8Н), 1.58 (с, 3Н), 1.43 (с, 18Н), 1.31 (т, J=7.2 Гц, 3Н).
Стадия 2: 4-ацетил-4-(этоксикарбонил)гептандиовая кислота (D28-2)
В перемешиваемый раствор соединения D28-1 (140 г, 326 ммоль) в ДХМ (350 мл) добавляли ТФУК (350 мл) в ДХМ (350 мл) при 0°С, и полученную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и остаток упаривали совместно с толуолом (3×200 мл), получая соединение D28-2 (85 г, колич.) в виде не совсем белого твердого вещества.
Стадия 3: этил-1-ацетил-4-оксоциклогексанкарбоксилат (D28-3)
В перемешиваемую суспензию соединения D28-2 (85 г, 310 ммоль) в уксусном ангидриде (255 мл) добавляли пиридин (27 мл), и полученную смесь перемешивали при 145°С в течение 2 часов. Растворитель удаляли при пониженном давлении, остаток суспендировали в воде (200 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии на силикагеле (14% EtOAc/гексан в качестве элюента), получая соединение D28-3 (11 г, 17%) в виде коричневой смолы. 1Н ЯМР (CDCl3, 400 МГц): δ 4.28 (кв, J=7.2 Гц, 2Н), 2.44-2.42 (м, 6Н), 2.23-2.20 (м, 5Н), 1.31 (т, J=7.2 Гц, 3Н).
Стадия 4: этил 4-(бензиламино)-2-оксобицикло[2.2.2]октан-1-карбоксилат (D28-4)
В перемешиваемую смесь соединения D28-3 (25.0 г, 117 моль) и бензиламина (38.6 мл, 353 моль) в толуоле (250 мл) добавляли п-TsOH (0.22 г, 1.17 ммоль), и полученную смесь кипятили в течение 8 часов в колбе, снабженной насадкой Дина-Старка. Реакционную смесь охлаждали до комнатной температуры. Добавляли в реакционную смесь HCl (3 М, 250 мл), и результирующую смесь перемешивали 30 минут. Полученную смесь нейтрализовывали 6 М водным раствором NaOH до рН 7. Реакционную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии на силикагеле (50% EtOAc/гексан в качестве элюента), получая соединение D28-4 (30 г, 85%) в виде не совсем белого твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 7.40-7.21 (м, 5Н), 6.44-6.32 (м, 2Н), 4.20 (кв, J=7.2 Гц, 2Н), 3.74 (с, 1Н), 2.45 (с, 2Н), 2.30-2.20 (м, 2Н), 2.10-1.95 (м, 2Н), 1.89-1.75 (м, 4Н), 1.27 (т, J=6.8 Гц, 3Н).
Стадия 5: этил-4-(бензиламино)-2-гидроксибицикло[2.2.2]октан-1-карбоксилат (D28-5)
В перемешиваемый раствор соединения D28-4 (30.0 г, 99.0 ммоль) в EtOH (300 мл) порциями добавляли твердый NaBH4 (5.64 г, 148 ммоль) при 0°С. Результирующую смесь перемешивали при комнатной температуре в течение 30 минут. Полученную смесь гасили водой (100 мл) и экстрагировали этилацетатом (3×200 мл). Объединенные органические слои промывали водой (150 мл), насыщенным водным раствором поваренной соли (150 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии на силикагеле (80% EtOAc/гексан в качестве элюента), получая соединение D28-5 (14 г, 46%) в виде белого твердого вещества.
Стадия 6: этил-4-(бензиламино)-2-((метилсульфонил)окси)бицикло[2.2.2]октан-1-карбоксилат (D28-6)
В перемешиваемый раствор соединения D28-5 (14.0 г, 46.0 ммоль) и Et3N (12.8 мл, 57.5 ммоль) в смеси ТГФ/толуол (125 мл, 1:4) добавляли MsCl (4.47 мл, 57.5 ммоль) при 0°С, и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь гасили водой (100 мл) и экстрагировали толуолом (50 мл). Органический слой отделяли, сушили над Na2SO4 и упаривали при пониженном давлении, получая соединение D28-6 (14 г, неочищенное). Полученный неочищенный продукт использовали в следующей стадии без дополнительной очистки.
Стадия 7: этил-4-(бензиламино)бицикло[2.2.2]окт-2-ен-1-карбоксилат (D28-7)
В перемешиваемый раствор соединения D28-6 (17.6 г, 46.3 моль) и NaI (1.38 г, 9.25 ммоль) в толуоле (170 мл) добавляли DBU (34.65 мл, 231 ммоль) и DMA (50 мл), и результирующую смесь перемешивали при 120°С 43 часа. Реакционную смесь гасили водой (100 мл) и экстрагировали этилацетатом (2×100 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над безводным Na2SO4, и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 50% EtOAc/гексан в качестве элюента), получая соединение D28-7 (8 г, 61%, за две стадии) в виде не совсем белого твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 7.36-7.32 (м, 5Н), 6.44 (д, J=8.8 Гц, 1Н), 6.32 (д, J=8.8 Гц, 1Н), 4.19 (кв, J=7.2 Гц, 2Н), 3.86 (с, 2Н), 2.04-1.97 (м, 2Н), 1.65-1.50 (м, 6Н), 1.28 (т, J=7.2 Гц, 3Н).
Стадия 8: этил-4-аминобицикло[2.2.2]октан-1-карбоксилат (D28-8)
В перемешиваемый раствор соединения D28-7 (8.0 г, 28.0 ммоль) в МеОН (80 мл) добавляли 10% Pd на угле (1.6 г, 20% по весу), и результирующую смесь перемешивали 5 часов в атмосфере водорода (1 атм). Реакционную смесь фильтровали через слой целита и промывали МеОН (2×30 мл). Фильтрат упаривали при пониженном давлении, получая соединение D28-8 (5.2 г, 94%) в виде бесцветной смолы. 1Н ЯМР (CDCl3, 400 МГц): δ 4.00 (кв, J=7.2 Гц, 2Н), 1.88-1.84 (м, 4Н), 1.56-1.55 (м, 8Н), 1.15 (т, J=7.2 Гц, 3Н).
Стадия 9: трет-бутил 4-цианобензилиденкарбамат (D28-9)
Смесь 4-формилбензонитрила (12.0 г, 9.16 моль) и трет-бутил (трифенилфосфоранилиден)карбамата (36.3 г, 9.61 моль) в толуоле (60 мл) кипятили в течение 18 часов. Выпавший твердый осадок отфильтровывали. Фильтрат упаривали при пониженном давлении, получая соединение D28-9 (13 г, неочищенное) в виде бесцветной смолы.
Стадия 10: трет-бутил 3-(4-цианофенил)-1,2-оксазиридин-2-карбоксилат (D28-10, смесь цис- и транс- изомеров)
В перемешиваемый раствор соединения D28-9 (13 г, 1.67 ммоль) в CHCl3 (220 мл) добавляли охлажденный раствор K2CO3 (50 г) в воде (400 мл) при 0°С, и полученную смесь интенсивно перемешивали. Добавляли охлажденный раствор Oxone (80 г) в воде (800 мл), и результирующую смесь перемешивали при 0°С 50 минут. Реакционную смесь подвергали десяти таким циклам обработки. Объединенный органический слой отделяли, промывали водой (200 мл), насыщенным водным раствором поваренной соли (200 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом обращенно-фазной колоночной хроматографии (С18 силикагель, 45-50% CH3CN/вода в качестве элюента), получая соединение D28-10 (1.3 г, 14% за две стадии) в виде белого твердого вещества. 1H ЯМР (CDCl3, 400 МГц, смесь цис- и транс-): δ 7.73-7.58 (м, 6.5Н), 5.29 (с, 0.3Н), 5.06 (с, 1Н), 1.57 (с, 3Н), 1.55 (с, 9Н).
Стадия 11: трет-бутил 2-(4-(этоксикарбонил)бицикло[2.2.2]октан-1-ил)гидразин-карбоксилат (D28-11)
Смесь соединения D28-8 (0.8 г, 4.04 ммоль) и соединения D28-10 (1.03 г, 4.24 ммоль) в ДХМ (20 мл) перемешивали 3 часа при 0°С. Реакционную смесь гасили водой (10 мл) и экстрагировали дихлорметаном (2×10 мл). Объединенные органические слои промывали водой (10 мл), насыщенным водным раствором поваренной соли (10 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 10% EtOAc/гексан в качестве элюента), получая соединение D28-11 (0.6 г, 50%) в виде белого твердого вещества.
Стадия 12: этил-4-гидразинилбицикло [2.2.2]октан-1-карбоксилата гидрохлорид (D28-12)
Смесь соединения D28-11 (0.6 г, 1.92 ммоль) и 4 М раствора HCl в диоксане (4.80 мл, 19.2 ммоль) перемешивали при комнатной температуре в течение 18 часов. Растворитель удаляли при пониженном давлении. Остаток от упаривания дважды упаривали совместно с гексаном, получая соединение D28-12 (0.58 г, неочищенное) в виде белого твердого вещества.
Стадия 13: бензил 1-(4-(этоксикарбонил)бицикло[2.2.2]октан-1-ил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат (D28-13)
В перемешиваемую смесь соединения D28-12 (0.58 г, 2.04 ммоль) и DIPEA (0.69 мл, 4.08 ммоль) в EtOH (10 мл) добавляли раствор соединения D1-4 (0.64 г, 2.15 ммоль) в EtOH (10 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили водой (20 мл) и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали водой (20 мл), насыщенным водным раствором поваренной соли (20 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 10% EtOAc/гексан в качестве элюента), получая соединение D28-13 (0.2 г, 21%) в виде светло-желтой смолы. 1Н ЯМР (CDCl3 400 МГц): δ 7.81-7.80 (с, 1Н), 7.39-7.25 (м, 5Н), 5.29 (с, 2Н), 4.11 (кв, J=7.2 Гц, 2Н), 2.27-2.23 (м, 6Н), 2.02-1.99 (м, 6Н), 1.25 (т, J=7.2 Гц, 3Н).
Стадия 14: 1-(4-(этоксикарбонил)бицикло[2.2.2]октан-1-ил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота (D28)
В перемешиваемый раствор соединения D28-13 (0.2 г, 0.44 ммоль) в МеОН добавляли 10% Pd на угле (40 мг, 30% по весу), и результирующую смесь перемешивали в атмосфере водорода (1 атм) 5 часов. Реакционную смесь фильтровали через слой целита, промывали МеОН (3×30 мл). Фильтрат упаривали при пониженном давлении. Остаток от упаривания растирали в гексане (2×10 мл) и твердое вещество отфильтровывали, получая соединение D28 (0.15 г, 93%) в виде белого твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 7.90 (с, 1Н), 4.14 (кв, J=7.2 Гц, 2Н), 2.30-2.26 (м, 6Н), 2.04-2.00 (м, 6Н), 1.25 (т, J=7.2 Гц, 3Н).
Справочный пример D30
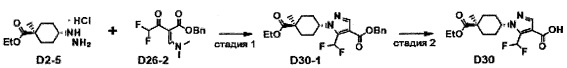
Стадия 1: бензил-5-(дифторметил)-транс-4-(этоксикарбонил)-4-метилциклогексил)-1Н-пиразол-4-карбоксилат (D30-1)
Соединение D30-1 (1.91 г, 50%) получали в виде светло-желтого твердого вещества реакцией соединения D26-2 (2.7 г, 9.55 ммоль), соединения D2-5 (2.6 г, 9.55 ммоль) и DIPEA (3.3 мл, 19.1 ммоль) в EtOH (50 мл) по методике, аналогичной описанной в справочном примере D1, стадия 5. 1Н ЯМР (CDCl3, 400 МГц): δ 7.94 (с, 1Н), 7.40-7.35 (м, 6Н), 5.30 (с, 2Н), 4.36 (м, 1Н), 4.15 (кв, J=7.2 Гц, 2Н), 2.24-2.19 (м, 2Н), 1.88-1.87 (м, 6Н), 1.3 (с, 3Н), 1.26 (т, J=7.2 Гц, 3Н).
Стадия 2: 1-транс-4-(этоксикарбонил)циклогексил-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота (D30)
Соединение D30 (1.19 г, 79%) получали в виде белого твердого вещества реакцией соединения D30-1 (1.91 г, 4.54 ммоль) и 5% Pd на угле (200 мг, 10% по весу) в EtOH (30 мл) по методике, аналогичной описанной в справочном примере D1, стадия 6. 1Н ЯМР (CDCl3, 300 МГц): δ 8.03 (с, 1Н), 7.51 (т, J=51.6 Гц), 4.4-4.42 (м, 1Н), 4.15 (кв, J=7.2 Гц, 2Н), 2.2-2.25 (м, 2Н), 1.88-1.92 (м, 6Н), 1.35 (с, 3Н), 1.27 (т, J=7.0 Гц, 3Н).
Справочный пример D33
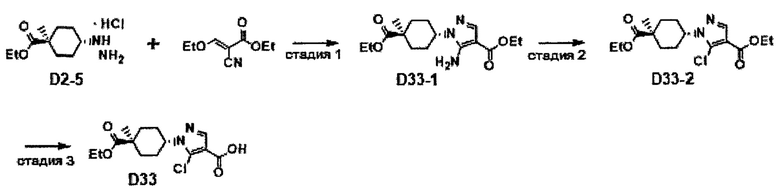
Стадия 1: Этил 5-амино-1-(транс-4-(этоксикарбонил)-4-метилциклогексил)-1Н-пиразол-4-карбоксилат (D33-1)
В раствор этил 2-циано-3-этоксиакрилата (19 г, 70 ммоль) и соединения D2-5 (11.96 г, 70 ммоль) в EtOH (100 мл) добавляли ацетат натрия (11.54 г, 140 ммоль), и полученную смесь кипятили в течение 6 часов. Реакционную смесь гасили водой и экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором поваренной соли, сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 30% EtOAc/гексан в качестве элюента), получая соединение D33-1 (16 г, 45%) в виде желтого твердого вещества.
Стадия 2: этил 5-хлор-1-(транс-4-(этоксикарбонил)-4-метилциклогексил)-1Н-пиразол-4-карбоксилат (D33-2)
В перемешиваемую смесь хлорида меди (I) (0.77 г, 7.8 ммоль) в CH3CN (10 мл) при 0°С добавляли трет-бутилнитрит (0.92 мл, 7.8 ммоль). Раствор соединения D33-1 (1.26 г, 3.9 ммоль) в CH3CN (10 мл) добавляли по каплям в полученную смесь при той же температуре. Реакционную смесь нагревали до комнатной температуры и перемешивали при той же температуре в течение 1 часа и при 60°С еще 1 час. Реакционную смесь гасили 6 М раствором HCl (10 мл) при 0°С и экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 30% EtOAc/гексан в качестве элюента), получая соединение D33-2 (0.3 г, 37%) в виде бесцветной смолы.
Стадия 3: 5-хлор-1-(транс-4-(этоксикарбонил)-4-метилциклогексил)-1Н-пиразол-4-карбоновая кислота (D33)
В раствор соединения D33-2 (0.6 г, 1.75 ммоль) в EtOH (10 мл) по каплям добавляли 1н. раствор NaOH при комнатной температуре. Полученную смесь перемешивали 45 минут. Значение рН реакционной смеси доводили до 3 и экстрагировали этилацетатом (2×200 мл). Объединенные органические слои промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом обращенно-фазной колоночной хроматографии (С18 силикагель, 80% CH3CN/вода в качестве элюента), получая соединение D33 (0.4 г, 55%) в виде не совсем белого твердого вещества.
Справочный пример D41
1-((1S,3R,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота
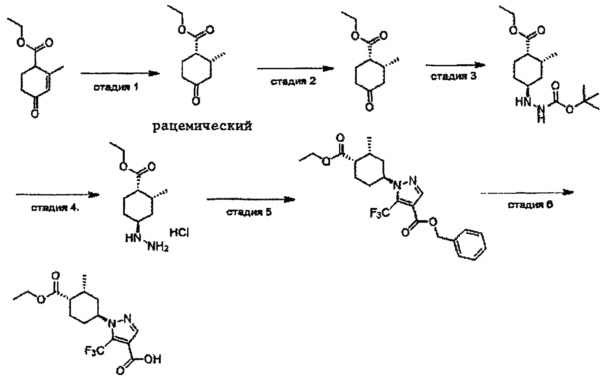
Стадия 1: (1S,2R)-этил 2-метил-4-оксоциклогексанкарбоксилат (рацемический)
В колбу аппарата Парра добавляли 10% палладий на угле (wet degussa тип) (4.47 г, 4.20 ммоль) в EtOH (378 мл). Затем добавляли в реакционную смесь этил 2-метил-4-оксо-2-циклогексен-1-карбоксилат (23.65 мл, 140 ммоль) и 5н. раствор хлористоводородной кислоты (1.679 мл, 8.40 ммоль). Колбу откачивали и затем заполняли водородом (50 фунт/кв.дюйм). Полученную смесь оставляли перемешиваться в условиях гидрирования на 30 минут. Прохождение реакции отслеживали методами LC/MS и ТСХ (50% EtOAc/гексан; окрашивание перманганатом калия), которые показали окончание реакции. Полученную смесь фильтровали через слой целита и осадок на фильтре промывали EtOH. Полученную смесь упаривали в вакууме. Полученный сырой продукт очищали хроматографией на колонке Interchim (15 микрон) с силикагелем (220 г), элюируя в градиенте 0-50% EtOAc в гексане, получая (1S,2R)-этил 2-метил-4-оксоциклогексанкарбоксилат (18.277 г, 99 ммоль, 70.9% выход) (рацемический) в виде светло-желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 4.19 (дтт, 2Н), 2.85 (тд, J=4.25, 8.31 Гц, 1Н), 2.43-2.58 (м, 4Н), 2.31 (ддд, J=6.06, 8.75, 14.72 Гц, 1Н), 2.01-2.21 (м, 2Н), 1.29 (т, J=7.14 Гц, 3Н), 0.98 (д, J=6.85 Гц, 3Н); LCMS (ESI) m/z 185.0 (М+Н)+.
Стадия 2: (1S,2R)-этил 2-метил-4-оксоциклогексанкарбоксилат (хиральный)
(1S,2R)-этил 2-метил-4-оксоциклогексанкарбоксилат (рацемический) разделяли на хиральный пик 1 и хиральный пик 2 нормальнофазной ВЭЖХ; Varian Cardinals SD1 нормальнофазная система (10×50 см; AS колонка 20 микрон). Метод: 10% EtOH в гептане, скорость потока: 400 мл/мин. Детектирование: 220 нм, 300 нм. Данный метод очистки дал пик 1 (1S,2R)-этил 2-метил-4-оксоциклогексанкарбоксилат (>98% ее) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 4.19 (ддквинт, 2Н), 2.85 (тд, J=4.25, 8.31 Гц, 1Н), 2.43-2.58 (м, 4Н), 2.31 (ддд, J=6.16, 8.66, 14.72 Гц, 1Н), 2.01-2.21 (м, 2Н), 1.24-1.32 (м, 3Н), 0.98 (д, J=6.85 Гц, 3Н); LCMS (ESI) m/z 185.0 (М+Н)+. Пик 2 (1R,2S)-этил 2-метил-4-оксоциклогексанкарбоксилат (>95% ее) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 4.19 (ддквинт, 2Н), 2.85 (тд, J=4.13, 8.36 Гц, 1Н), 2.43-2.58 (м, 4Н), 2.31 (ддд, J=6.16, 8.66, 14.72 Гц, 1Н), 2.01-2.21 (м, 2Н), 1.29 (т, J=7.14 Гц, 3Н), 0.98 (д, J=6.85 Гц, 3Н); LCMS (ESI) m/z 185.0 (М+Н)+.
Стадия 3: трет-бутил 2-((1S,3R,4S)-4-(этоксикарбонил)-3-метилциклогексил) гидразинкарбоксилат
В 3-горлую круглодонную колбу объемом 500 мл помешали (1S,2R)-этил 2-метил-4-оксоциклогексанкарбоксилат (10.00 г, 54.3 ммоль) в хлороформе (201 мл). Затем добавляли в реакционную смесь ледяную АсОН (3.13 мл, 54.3 ммоль) и трет-бутил карбазат (7.89 г, 59.7 ммоль). Колюу помещали в заранее нагретую баню (30°С) и оставляли перемешиваться на 10 минут. Затем медленно добавляли в реакционную смесь NaBH(ОАс)3 (34.5 г, 163 ммоль) небольшими порциями. Баню удаляли по завершении добавления и смесь оставляли перемешиваться в инертной атмосфере 16 часов. Прохождение реакции отслеживали методами LC/MS и ТСХ (30% EtOAc/ДХМ; окрашивание нингидрином), которые показали окончание реакции. Полученную смесь нейтрализовывали медленным добавлением в реакционную смесь насыщенного водного раствора NaHCO3. После нейтрализации слои разделяли, и водный слой экстрагировали дихлорметаном (3х). Объединенные органические экстракты сушили над Na2SO4, фильтровали и упаривали в вакууме. Неочищенный образец анализировали методом ТСХ (30% EtOAc/гексан; окрашивание нингидрином; Пик 1: Rf=0.46 & Пик 2: Rf=0.38). Неочищенное вещество делили на две порции и очищали методом хроматографии на колонке Interchim (25 микрон) с силикагелем (300 г) *(Применяли две 300-граммовые колонки), элюируя в градиенте 0-30% EtOAc в гексане, получая трет-бутил 2-((1R,3R,4S)-4-(этоксикарбонил)-3-метилциклогексил) гидразинкарбоксилат (8.512 г, 28.3 ммоль, 52.2% выход) (Пик 1; цис) 1H ЯМР (400 МГц, CDCl3) δ 6.03-6.28 (м, 1Н), 4.07-4.16 (м, 2Н), 3.59-3.90 (м, 1Н), 2.76-2.97 (м, 1Н), 2.55 (д, J=2.74 Гц, 1Н), 2.01 (дд, J=3.03, 13.40 Гц, 1Н), 1.59-1.77 (м, 3Н), 1.49-1.56 (м, 2Н), 1.46 (с, 10Н), 1.19-1.31 (м, 3Н), 1.02 (д, J=7.04 Гц, 3Н); LCMS (ESI) m/z 301.1 (М+Н)+, и трет-бутил 2-((1S,3R,4S)-4-(этоксикарбонил)-3-метилциклогексил) гидразинкарбоксилат (5.089 г, 16.94 ммоль, 31.2% выход) (Пик 2; транс) 1Н ЯМР (400 МГц, ДМСО-d6) δ 7.89-8.27 (м, 1Н), 5.75 (с, 1Н), 4.08-4.19 (м, 1Н), 2.74-2.93 (м, 1Н), 2.21-2.46 (м, 2Н), 1.99 (с, 1Н), 1.66 (д, J=3.91 Гц, 3Н), 1.38 (с, 9Н), 1.14-1.26 (м, 5Н), 0.79 (д, J=7.04 Гц, 3Н); LCMS (ESI) m/z 301.1 (М+Н)+.
Стадия 4: (1S,2R,4S)-этил 4-гидразинил-2-метилциклогексанкарбоксилат гидрохлорид
В круглодонную колбу объемом 250 мл добавляли трет-бутил 2-((1S,3R,4S)-4-(этоксикарбонил)-3-метилциклогексил)гидразинкарбоксилат (5.089 г, 16.94 ммоль) в EtOH (56.5 мл). Затем добавляли в реакционную смесь хлороводород, 4.0 М раствор в 1,4-диоксане (72.0 мл, 288 ммоль). Полученную смесь оставляли перемешиваться в инертной атмосфере в течение ночи. Прохождение реакции отслеживали методом ТСХ (30% EtOAc в гексане; окрашивание нингидрином), который показал окончание реакции. Полученную смесь упаривали в вакууме. Остаток от упаривания разбавляли гексаном и упаривали в вакууме. Получали (1S,2R,4S)-этил 4-гидразинил-2-метилциклогексанкарбоксилат гидрохлорид (4.60 г) в виде белого твердого вещества. Полученное вещество использовали в следующей стадии синтеза без дополнительной очистки. LCMS (ESI) m/z 201.2(M+H)+.
Стадия 5: бензил 1-((1S,3R,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат
В круглодонную колбу объемом 250 мл помещали (1S,2R,4S)-этил 4-гидразинил-2-метилциклогексанкарбоксилата гидрохлорид (4.00 г, 16.90 ммоль) и DIPEA (4.43 мл, 25.3 ммоль) в EtOH (84 мл). Затем в реакционную смесь по каплям добавляли раствор (Z)-бензил 2-((диметиламино)метилен)-4,4,4-трифтор-3-оксобутаноата (5.09 г, 16.90 ммоль) в EtOH (84 мл). Результирующую реакционную смесь оставляли перемешиваться в инертной атмосфере при комнатной температуре в течение ночи. Прохождение реакции отслеживали методами LC/MS и ТСХ (30% EtOAc/гексан), которые показали наличие главным образом целевого продукта LCMS (ESI) m/z 461.2 (М+Na)+, без остатков исходного вещества. Реакционную смесь упаривали в вакууме. Полученный сырой продукт очищали хроматографией на колонке Interchim (25 микрон) с силикагелем (200 г), элюируя в градиенте 0-30% EtOAc в гексане, получая бензил 1-((1S,3R,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат (5.631 г, 12.84 ммоль, 76% выход) в виде светло-желтого масла. 1H ЯМР (400 МГц, ДМСО-d6) δ 8.14 (с, 1Н), 7.32-7.45 (м, 5Н), 5.30 (с, 2Н), 4.55-4.65 (м, 1Н), 4.02-4.15 (м, 2Н), 2.65 (тд, J=4.50, 11.54 Гц, 1Н), 2.13 (дт, J=4.50,12.42 Гц, 1Н), 1.95-2.04 (м, 2Н), 1.73 (д, J=4.89 Гц, 3Н), 1.16-1.23 (м, 3Н), 0.92 (д, J=7.04 Гц, 3Н); LCMS (ESI) m/z 461.2 (M+Na)+.
Стадия 6: 1-((1S,3R,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота
*(Гидрирование проводили в портативном аппарате)
В виалу для проведения реакции под давлением помещали палладий 10 вес. % (по сухому остатку) на активированном угле, влажный (1.367 г, 1.284 ммоль) в токе N2 (газ). Затем добавляли раствор бензил 1-((1S,3R,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилата (5.631 г, 12.84 ммоль) в 1:1 смеси EtOH (32.1 мл)/EtOAc (32.1 мл). Атмосферу над реакционной смесью продували газообразным водородом (3х). Реакционную смесь интенсивно перемешивали в условиях гидрирования (35 фунт/кв.дюйм) 2.5 часа. Прохождение реакции отслеживали методом LC/MS, который показал окончание реакции LCMS (ESI) m/z 371.2 (M+Na)+. Полученную смесь фильтровали через слой целита, и фильтрат упаривали в вакууме. Остаток от упаривания разбавляли гексаном и интенсивно размешивали. Осадок отделяли фильтрованием и промывали гексаном. Получали 1-((1S,3R,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновую кислоту (3.810 г, 10.94 ммоль, 85% выход) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7.91-8.21 (м, 1Н), 4.47-4.69 (м, 1Н), 4.01-4.16 (м, 2Н), 2.56-2.70 (м, 1Н), 2.12 (дт, J=4.21, 12.37 Гц, 1Н), 1.93-2.06 (м, 2Н), 1.71-1.90 (м, 3Н), 1.19 (т, J=7.04 Гц, 3Н), 0.92 (д, J=7.04 Гц, 3Н); LCMS (ESI) m/z 371.2(M+Na)+.
Справочный пример D43
1-((1R,3R,4R)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота в смеси с 1-((1S,3S,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновой кислотой (1:1) (D43)

Стадия 1: трет-бутил 2-((1R,3R,4R)-4-(этоксикарбонил)-3-метилциклогексил)гидразинкарбоксилат в смеси с трет-бутил 2-((1S,3S,4S)-4-(этоксикарбонил)-3-метилциклогексил)гидразинкарбоксилатом (1:1) (D43-1)
В гомогенную рацемическую смесь (1R,2R)-этил 2-метил-4-оксоциклогексанкарбоксилата и (1S,2S)-этил 2-метил-4-оксоциклогексанкарбоксилата (1:1) (1.600 г, 8.68 ммоль) добавляли трет-бутил карбазат (1.263 г, 9.55 ммоль), АсОН (1.038 мл, 17.98 ммоль) и NaBH(ОАс)3 (6.00 г, 28.3 ммоль). Светло-желтую гетерогенную смесь перемешивали при комнатной температуре. Через 24 часа LCMS (ESI) и ТСХ показали завершение реакции, наличие двух пиков 323.1 (M+Na).
[ТСХ]: (30% EtOAc в гексане, окрашивание фосфомолибденовой кислотой в EtOH)
Rf реагента = 0.47, Rf 1,4-цис-целевого продукта = 0.42, Rf 1,4-транс-целевого продукта = 0.25. Реакционную смесь выливали в насыщенный водный раствор NaHCO3 (150 мл). Реакционную смесь экстрагировали дихлорметаном (2×100 мл). Органический экстракт сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде бесцветного масла. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 25% EtOAc в гексане, получая две фракции:
Первая фракция - более высокое пятно (1,4-цис): (Rf=0.42 для 30% EtOAc в гексане) трет-бутил 2-((1R,3S,4S)-4-(этоксикарбонил)-3-метилциклогексил)гидразинкарбоксилат с трет-бутил 2-((1S,3R,4R)-4-(этоксикарбонил)-3-метилциклогексил)гидразинкарбоксилатом (1:1) (1.4418 г, 4.80 ммоль, 55.3% выход) в виде светло-желтого сиропа: 1H ЯМР (300 МГц, CDCl3) δ 6.05 (1Н, ушир.с), 4.14 (2H, кв, J=7.1 Гц), 3.25 (1Н, ушир.с), 1.12-2.22 (21Н, м), 0.88 (3Н, д, J=6.6 Гц); LCMS (ESI) m/z 301.1 (M+H)+ и m/z 323.1 (M+Na)+.
Вторая фракция - более низкое пятно (1,4-транс): Целевой продукт (Rf=0.25 при 30% EtOAc в гексане) трет-бутил 2-((1R,3R,4R)-4-(этоксикарбонил)-3-метилциклогексил)гидразинкарбоксилат с трет-бутил 2-((1S,3S,4S)-4-(этоксикарбонил)-3-метилциклогексил)гидразинкарбоксилатом (1:1) (D43-1) (0.5467 г, 1.820 ммоль, 20.96% выход) в виде не совсем белого сиропообразного твердого вещества. 1H ЯМР (300 МГц, CDCl3) δ 6.05 (1Н, ушир.с), 4.06-4.23 (2Н, м), 2.81-2.99 (1Н, м), 1.65-2.07 (5Н, м), 1.39-1.56 (10Н, м), 1.20-1.31 (4Н, м), 0.99-1.16 (1Н, м), 0.79-0.96 (4Н, м); LCMS (ESI) m/z 323.1 (M+Na)+.
[Примечание]: Вторую фракцию использовали в Стадии 2.
Стадия 2: (1R,2R,4R)-этил 4-гидразинил-2-метилциклогексанкарбоксилат и (1S,2S,4S)-этил 4-гидразинил-2-метилциклогексанкарбоксилат (1:1) дигидрохлорид (D43-2)
В смесь трет-бутил 2-((1R,3R,4R)-4-(этоксикарбонил)-3-метилциклогексил)гидразинкарбоксилата и трет-бутил 2-((1S,3S,4S)-4-(этоксикарбонил)-3-метилциклогексил)гидразинкарбоксилата (1:1) (D42-1) (0.5245 г, 1.746 ммоль) в EtOH (4.37 мл) добавляли хлороводород, 4 М раствор в 1,4-диоксане (4.37 мл, 17.46 ммоль). Прозрачную светло-желтую смесь перемешивали при комнатной температуре. Через 42 часа (белая гетерогенная смесь), LC-MS (ESI) показал завершение реакции, сформировался целевой продукт (m/z 201.2 (М+1)). Полученную смесь упаривали в вакууме, получая (1R,2R,4R)-этил 4-гидразинил-2-метилциклогексанкарбоксилат и (1S,2S,4S)-этил 4-гидразинил-2-метилциклогексанкарбоксилат (1:1) дигидрохлорид (D43-2) в виде светло-желтого твердого вещества. 1Н ЯМР (300 МГц, ДМСО-d6) δ 4.07 (2Н, кв, J=7.0 Гц), 2.88-3.05 (1Н, м), 2.04 (2Н, т, J=11.6 Гц), 1.80-1.96 (2Н, м), 1.52-1.73 (1Н, м), 1.12-1.46 (5Н, м), 0.78-1.08 (4Н, м); LCMS (ESI) m/z 201.2 (M+H)+.
Стадия 3: бензил 1-((1R,3R,4R)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат и бензил 1-((1S,3S,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат (1:1) (D43-3)
В смесь (1R,2R,4R)-этил 4-гидразинил-2-метилциклогексанкарбоксилата и (1S,2S,4S)-этил 4-гидразинил-2-метилциклогексанкарбоксилата (1:1) дигидрохлорида (D42-2) (0.413 г, 1.745 ммоль) в EtOH (13.42 мл) добавляли DIPEA (0.669 мл, 3.84 ммоль), затем раствор (2)-бензил 2-((диметиламино)метилен)-4,4,4-трифтор-3-оксобутаноата (0.526 г, 1.745 ммоль) в EtOH (5 мл). Прозрачную коричневую смесь перемешивали при комнатной температуре. Через 15 часов, LC-MS (ESI) показал завершение реакции, сформировался целевой продукт (m/z 439.1 (М+1)). Реакционную смесь упаривали в вакууме. Остаток от упаривания разбавляли водой (50 мл) и экстрагировали этилацетатом (2×100 мл). Органический экстракт промывали насыщенным водным раствором NaCl (1×100 мл) и сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде коричневого сиропа. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 10% EtOAc в гексане, получая бензил 1-((1R,3R,4R)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат и бензил 1-((1S,3S,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат (1:1) (D43-3) (0.4258 г, 0.971 ммоль, 55.7% выход) в виде желтого сиропа: 1H ЯМР (300 МГц, ДМСО-d6) δ 8.06-8.17 (1Н, м), 7.29-7.50 (5Н, м), 5.29 (2Н, с), 4.42-4.60 (1Н, м), 4.10 (2Н, кв, J=7.1 Гц), 1.48-2.13 (8Н, м), 1.19 (3Н, т, J=7.1 Гц), 0.89 (3Н, д, J=6.0 Гц); LCMS (ESI) m/z 439.1 (М+Н)+.
Стадия 4: 1-((1R,3R,4R)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота и 1-((1S,3S,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота (1:1) (D43)
В виалу для проведения реакции под давлением помещали палладий 10 вес. % на активированном угле (0.103 г, 0.097 ммоль) в токе азота. Затем добавляли в виалу раствор бензил 1-((1R,3R,4R)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилата и бензил 1-((1S,3S,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилата (1:1) (D43-3) (0.4258 г, 0.971 ммоль) в 1:1 смеси EtOH (2.428 мл)/EtOAc (2.428 мл). Атмосферу над реакционной смесью продували газообразным водородом (3 раза). Реакционную смесь интенсивно перемешивали в условиях гидрирования (33 фунт/кв.дюйм) при 21°С. Через 3 часа LCMS (ESI) показал завершение реакции. Реакционную смесь продували азотом 30 минут. Полученную смесь фильтровали через слой целита, и осадок на фильтре промывали EtOAc. Фильтрат упаривали в вакууме, получая 1-((1R,3R,4R)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновую кислоту и 1-((1S,3S,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновую кислоту (1:1) (D43) (0.3224 г, 0.926 ммоль, 95% выход) в виде светло-желтого твердого вещества:
1H ЯМР (300 МГц, ДМСО-d6) δ 13.14 (1Н, ушир.с), 8.01 (1Н, с), 4.40-4.59 (1Н, м), 4.10 (2Н, кв, J=7.0 Гц), 1.48-2.16 (8Н, м), 1.20 (3Н, т, J=7.1 Гц), 0.89 (3Н, д, J=6.0 Гц); LCMS (ESI) m/z 349.1 (M+H)+.
Справочный пример D48
1-((3aS,5R,7aS)-3а-(метоксикарбонил)октагидро-1Н-инден-5-ил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота
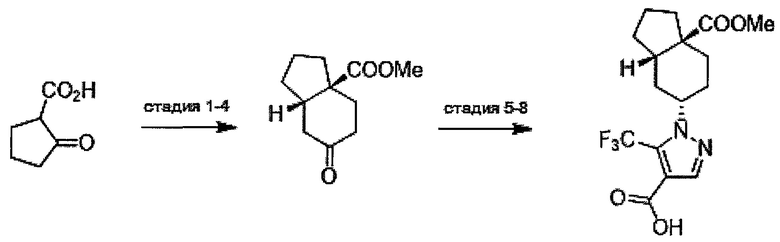
Стадия 1: метил 2-оксо-1-(3-оксобутил)циклопентанкарбоксилат
Раствор метил 2-оксоциклопентапекарбоксилата (2.000 мл, 14.07 ммоль), метил винил кетона (1.381 мл, 16.88 ммоль) и триэтиламина (2.94 мл, 21.10 ммоль) в толуоле (20 мл) нагревали до 40°С в течение 24 часов. Реакционную смесь доводили до комнатной температуры, разбавляли добавлением EtOAc, промывали насыщенным водным раствором NH4Cl, сушили над Na2SO4, фильтровали, упаривали и хроматографировали на силикагеле с применением 0-50% гептана/EtOAc, получая бесцветное масло метил 2-оксо-1-(3-оксобутил)циклопентанкарбоксилата (2.0 г, 9.42 ммоль, 67.0% выход).
Стадия 2: метил 5-(пирролидин-1-ил)-2,6,7,7а-тетрагидро-1Н-инден-7а-карбоксилат
Раствор метил 2-оксо-1-(3-оксобутил)циклопентанкарбоксилата (2.0 г, 9.42 ммоль, 67.0% выход) и пирролидина (2.354 мл, 28.1 ммоль) в сухом толуоле (25 мл) нагревали до кипения в атмосфере азота с насадкой Дина-Старка 16 часов. Реакцию доводили до конца и упаривали. Остаток от упаривания растворяли в EtOAc, промывали водой, насыщенным водным раствором поваренной соли, сушили над Na2SO4, фильтровали и упаривали, получая зеленоватое масло, представляющее собой метил 5-(пирролидин-1-ил)-2,6,7,7а-тетрагидро-1Н-инден-7а-карбоксилат (3.3 г, 13.34 ммоль, 95% выход), который использовали без очистки.
Стадия 3: метил 6-оксо-2,3,3а,4,5,6-гексагидро-1Н-инден-3а-карбоксилат
Неочищенный енамин со Стадии 2 растворяли в толуоле (20 мл), добавляли раствор натрия ацетата (1.360 мл, 25.3 ммоль) в смеси АсОН/вода (4/4 мл), и результирующую смесь нагревали до кипения в атмосфере азота в течение 2 часов. Реакцию доводили до конца, смесь разбавляли добавлением EtOAc, промывали водой, насыщенным раствором NH4Cl, насыщенным раствором NaHCO3, насыщенным водным раствором поваренной соли, сушили над Na2SO4, фильтровали, упаривали и хроматографировали на силикагеле с применением смеси 0-30% гептан/EtOAc, получая метил 6-оксо-2,3,3а,4,5,6-гексагидро-1Н-инден-3а-карбоксилат (1.32 г, 6.80 ммоль, 48.3% выход) в виде ярко-желтого масла. MS m/z=195.2 [M+H]+.
Стадия 4: (3aS,7aR)-метил 6-оксооктагидро-1H-инден-3а-карбоксилат
В перемешиваемый раствор метил 6-оксо-2,3,3а,4,5,6-гексагидро-1Н-инден-3а-карбоксилата (1.32 г, 6.80 ммоль) в EtOH (30 мл) добавляли палладий на угле 10 вес. %(по сухому остатку) влажный, degussa type e101 ne/w (0.120 мл, 6.80 ммоль), и результирующую смесь гидрировали в приборе для гидрирования 3 часа. Полученную смесь фильтровали через целит, упаривали и хроматографировали на силикагеле с применнеием смеси 0-25% гептан/гексан, получая (3aS,7aR)-метил 6-оксооктагидро-1H-инден-3а-карбоксилат (0.278 г, 1.417 ммоль, 20.84% выход) и (3aS,7aS)-метил 6-оксооктагидро-1Н-инден-3а-карбоксилат (0.394 г, 2.008 ммоль, 29.5% выход) в виде бесцветного масла. MS m/z=181.2 [M+H]+.
Стадии 5-8. 1-((3aS,5R,7aS)-3а-(метоксикарбонил)октагидро-1Н-инден-5-ил)-5-(трифторметил)-1Н-пиразол-4-карбоновую кислоту получали из (3aS,7aR)-метил 6-оксооктагидро-1Н-инден-3а-карбоксилата по методикам, аналогичным описанным в примере D22. MS m/z=361.2 [М+Н]+.
[Пример D55]
транс-1-(4-(Этоксикарбонил)-3,3-диметилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота (рацемическая смесь)
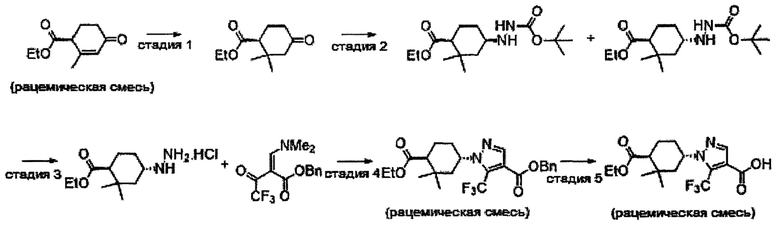
Стадия 1:
Этил 2,2-диметил-4-оксоциклогексанкарбоксилат (рацемическая смесь)
Метиллитий (170 мл 1.6 М раствор в Et2O, 260 ммоль) добавляли в перемешиваемую смесь иодида меди (I) (25 г, 130 ммоль) и Et2O (130 мл), при -40°С в атмосфере азота. После перемешивания в течение 10 минут при -40°С, добавляли этил 2-метил-4-оксо-2-циклогексен-1-карбоксилат (12 г, 66 ммоль). После перемешивания в течение 30 минут при -40°С, реакционную смесь оставляли нагреваться до -20°С. После перемешивания в течение 90 минут при -20°С, последовательно добавляли насыщенный водный раствор хлорида аммония и EtOAc, смесь разделяли между дополнительным количеством насыщенного водного раствора хлорида аммония и EtOAc, слои разделяли, органическую часть последовательно промывали насыщенным водным раствором хлорида аммония (2×) и насыщенным водным раствором поваренной соли, сушили (Na2SO4), фильтровали, и фильтрат упаривали. Остаток от упаривания растворяли в ДХМ, силикагель (39 г) добавляли в раствор, и летучие компоненты удаляли при пониженном давлении. Остаток от упаривания очищали колоночной хроматографией на силикагеле (элюирование в градиенте; от 19:1 до 9:1 гексан-EtOAc), получая этил 2,2-диметил-4-оксоциклогексанкарбоксилат (8.9 г, 68% выход; рацемическая смесь) в виде прозрачного желтого масла.
Стадия 2:
трет-Бутил транс-2-4-(этоксикарбонил)-3,3-диметилциклогексил)гидразинкарбоксилат (рацемическая смесь)
NaBH(ОАс)3 (29 г, 140 ммоль) добавляли в перемешиваемый раствор этил 2,2-диметил-4-оксоциклогексанкарбоксилата (8.9 г, 45 ммоль, со Стадии 1; рацемическое вещество), трет-бутил карбазата (6.5 г, 49 ммоль), ледяной АсОН (7.8 мл, 140 ммоль) и ТГФ (90 мл). После перемешивания в течение 26 часов, реакционную смесь добавляли в насыщенный водный раствор NaHCO3, смесь перемешивали 60 минут, разделяли между EtOAc и дополнительным количеством насыщенного водного раствора NaHCO3, слои разделяли, органическую часть промывали последовательно насыщенным водным раствором NaHCO3 и насыщенным водным раствором поваренной соли, сушили (Na2SO4), фильтровали, и фильтрат упаривали при пониженном давлении. Остаток от упаривания растворяли в ДХМ, добавляли в раствор силикагель (42 г), и летучие компоненты удаляли при пониженном давлении. Остаток от упаривания очищали колоночной хроматографией на силикагеле (элюирование в градиенте; от 9:1 до 4:1 гексан-EtOAc), и выделенное вещество, содержащее целевой продукт, снова очищали методом флэш-хроматографии на силикагеле (5:1 гексан-EtOAc), получая трет-бутил транс-2-4-(этоксикарбонил)-3,3-диметилциклогексил)гидразинкарбоксилат (0.79 г, 5.6% выход; рацемическая смесь) в виде прозрачного бесцветного масла.
Стадия 3:
Этил транс-4-гидразинил-2,2-диметилциклогексанкарбоксилат гидрохлорид (рацемическая смесь)
Хлороводород (3.1 мл 4.0 М раствора в 1,4-диоксане, 13 ммоль) добавляли в перемешиваемый раствор трет-бутил транс-2-4-(этоксикарбонил)-3,3-диметилциклогексил)гидразинкарбоксилата (0.79 г, 2.5 ммоль, со Стадии 2; рацемическое вещество) и EtOH (5.0 мл), и затем реакционную смесь нагревали до 60°С. После перемешивания в течение 3 часов при 60°С, реакционную смесь оставляли охлаждаться до комнатной температуры и затем упаривали при пониженном давлении, получая этил транс-4-гидразинил-2,2-диметилциклогексанкарбоксилата гидрохлорид (0.63 г, 100% выход; рацемическая смесь) в виде не совсем белого твердого вещества.
Стадия 4:
Бензил транс-1-4-(этоксикарбонил)-3,3-диметилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат (рацемическая смесь)
Раствор (Z)-бензил 2-((диметиламино)метилен)-4,4,4-трифтор-3-оксобутаноата (0.76 г, 2.5 ммоль) и EtOH (2.4 мл) добавляли в перемешиваемый раствор этил транс-4-гидразинил-2,2-диметилциклогексанкарбоксилата гидрохлорида (0.63 г, 2.5 ммоль, со Стадии 3; рацемическая смесь), DIPEA (0.96 мл, 5.5 ммоль) и EtOH (6.0 мл). После перемешивания в течение 20 часов, реакционную смесь упаривали при пониженном давлении, остаток разделяли между EtOAc и насыщенным водным раствором NaHCO3, слои разделяли, органическую часть промывали последовательно насыщенным водным раствором NaHCO3 и насыщенным водным раствором поваренной соли, сушили (Na2SO4), фильтровали, и фильтрат упаривали при пониженном давлении. Остаток от упаривания растворяли в ДХМ, силикагель (5.0 г) добавляли в раствор, и летучие компоненты удаляли при пониженном давлении. Остаток от упаривания очищали колоночной хроматографией на силикагеле (19:1 гексан-EtOAc), получая бензил транс-1-4-(этоксикарбонил)-3,3-диметилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат (0.76 г, 67% выход; рацемическая смесь) в виде прозрачного бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 7.94 (с, 1Н), 7.46-7.29 (м, 5Н), 5.30 (с, 2Н), 4.67-4.52 (м, 1Н), 4.25-4.05 (м, 2Н), 2.35-2.23 (м, 1Н), 2.12-1.84 (м, 5Н), 1.69 (дд, J=3.2, 12.8 Гц, 1Н), 1.27 (т, J=7.1 Гц, 3Н), 1.09 (с, 3Н), 1.07 (с, 3Н). LCMS (ESI): 453.0 (М+Н)+.
Стадия 5:
транс-1-(4-(Этоксикарбонил)-3,3-диметилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота (рацемическая смесь)
Перемешиваемую смесь бензил транс-1-4-(этоксикарбонил)-3,3-диметилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилата (0.76 г, 1.7 ммоль, со Стадии 4; рацемическая смесь), палладия (0) (10 вес. % по сухому веществу, влажный) на активированном угле (0.18 г, 0.17 ммоль), EtOAc (4.2 мл) и EtOH (4.2 мл) обрабатывали газообразным водородом (33 фунт/кв.дюйм). После перемешивания в течение 2 часов, реакционную смесь фильтровали и фильтрат упаривали при пониженном давлении, получая транс-1-(4-(этоксикарбонил)-3,3-диметилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновую кислоту (0.59 г, 97% выход; рацемическая смесь) в виде бесцветного твердого вещества. LCMS (ESI): 363.0 (М+Н)+.
Справочный пример D60
1-(((+/-)-цис)-2-аллилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота в виде рацемата (D60)
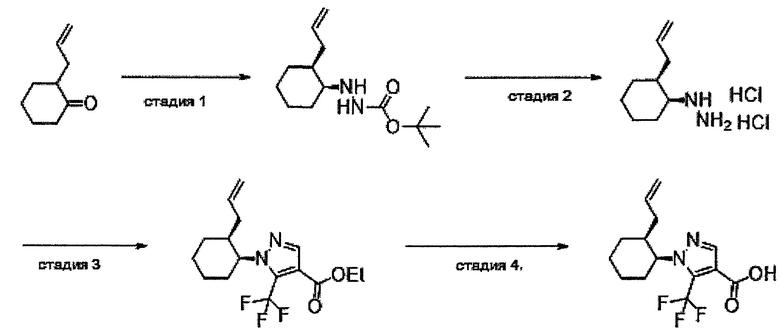
Стадия 1: трет-бутил 2-(((+/-)цис)-2-аллилциклогексил)гидразинкарбоксилат в виде рацемата
В раствор трет-бутил карбазата (0.966 г, 7.31 ммоль), 2-аллилциклогексанона (1.00 г, 7.24 ммоль) и АсОН (1.00 мл, 17.47 ммоль) при 0°С добавляли NaBH(ОАс)3 (4.60 г, 21.71 ммоль), и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь медленно добавляли в насыщенный водный раствор Na2CO3. Слои разделяли, и водный слой дважды экстрагировали дихлорметаном. Органические слои объединяли, промывали насыщенным водным раствором поваренной соли, сушили над Na2SO4, декантировали и упаривали в вакууме, получая бесцветный сироп. ЯМР показал присутствие ~0.16:1 смеси изомеров. Полученный сироп очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 50% EtOAc в гексане. Пик, элюирующийся первым, собирали и упаривали в вакууме, получая трет-бутил 2-(((+/-)цис)-2-аллилциклогексил)гидразинкарбоксилат в виде рацемата.
Стадия 2: (((+/-)цис)-2-аллилциклогексил)гидразина дигидрохлорид в виде рацемата
4 М раствор HCl в диоксане (11.79 мл, 47.2 ммоль) добавляли в раствор трет-бутил 2-(((+/-)цис)-2-аллилциклогексил)гидразинкарбоксилата в виде рацемата (1.20 г, 4.72 ммоль) в EtOH (11.79 мл), и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь упаривали в вакууме, получая (((+/-)цис)-2-аллилциклогексил)гидразина дигидрохлорид в виде рацемического белого твердого вещества.
Стадия 3: этил 1-(((+/-)цис)-2-аллилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат в виде рацемата
Раствор (Z)-этил 2-((диметиламино)метилен)-4,4,4-трифтор-3-оксобутаноата (1.073 г, 4.49 ммоль) в EtOH (11 мл) медленно добавляли в раствор (((+/-)цис)-2-аллилциклогексил)гидразина дигидрохлорида в виде рацемата (1.07 г, 4.71 ммоль) и DIPEA (1.724 мл, 9.87 ммоль) в EtOH (22.43 мл) при комнатной температуре. Через 6 часов, реакционную смесь упаривали в вакууме, разбавляли водой и дважды экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным водным раствором поваренной соли, сушили над Na2SO4, декантировали и упаривали в вакууме, получая оранжевое масло. Полученную смесь очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 35% EtOAc в гексане, получая этил 1-(((+/-)цис)-2-аллилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат в виде рацемата в виде бледно-желтого масла.
Стадия 4: 1-(((+/-)-цис)-2-аллилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота в виде рацемата
Раствор лития гидроксида гидрата (1.265 г, 30.2 ммоль) в воде добавляли в раствор этил 1-(((+/-)цис)-2-аллилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилата в виде рацемата (0.996 г, 3.02 ммоль) в ТГФ и МеОН, и полученную смесь перемешивали при комнатной температуре в течение ночи. Полученную смесь упаривали в вакууме. Полученный мутный раствор разбавляли водой, получая прозрачный раствор. Значение рН доводили до 1 добавлением 1 М раствора HCl, и полученную смесь интенсивно перемешивали 30 минут. Выпавший осадок отделяли фильтрованием под вакуумом, получая 1-(((+/-)-цис)-2-аллилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновую кислоту в виде рацемата (D60) в виде белого твердого вещества.
Справочный пример D68 (цис и транс)
1-((1R,4R)-4-(2-этокси-2-оксоэтил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота и 1-((1S,4S)-4-(2-этокси-2-оксоэтил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота
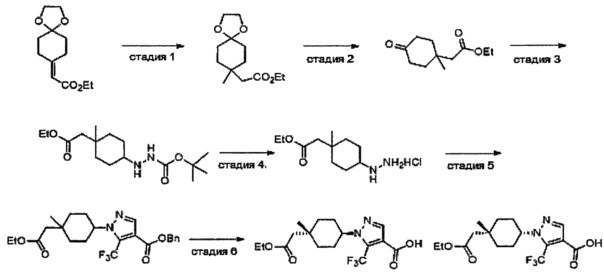
Стадия 1: этил 2-(8-метил-1,4-диоксаспиро[4.5]декан-8-ил)ацетат
В раствор CuI (5.8 г, 30 ммоль, 3.24 экв.) в Et2O (100 мл) в атмосфере N2 при 0°С по каплям добавляли 3.0 М раствор MeLi (21.3 мл, 64 ммоль, 6.8 экв.) в диметоксиэтане. Полученный раствор перемешивали при 0°С 10 минут и эфирный растворитель удаляли из реакционной смеси в вакууме (120 торр) при 0°С. Затем добавляли в остаток ДХМ (100 мл), и реакционную смесь охлаждали до -78°С. Добавляли TMSCl (4.4 мл, 35 ммоль, 3.7 экв.), затем этил 2-(1,4-диоксаспиро[4.5]декан-8-илиден)ацетат (JW Pharmlab, Levittown, PA; 2.127 г, 9.4 ммоль) в ДХМ (10 мл). Реакционную смесь перемешивали в течение ночи и гасили водным раствором NH4Cl. Полученную черную суспензию фильтровали через целит, органический слой отделяли, промывали, сушили и очищали методом хроматографии на силикагеле (EtOAc/гексан, до 15%) на 80-граммовой колонке, получая этил 2-(8-метил-1,4-диоксаспиро[4.5]декан-8-ил)ацетат (1.6 г, 6.60 ммоль, 70.2% выход) в виде бесцветной жидкости: 1H ЯМР (500 МГц, CDCl3) δ 1.07 (с, 3Н), 1.19-1.33 (м, 3Н), 1.49-1.67 (м, 8Н), 2.27 (с, 2Н), 3.94 (с, 4Н), 4.09-4.16 (м, 2Н).
Стадия 2: этил 2-(1-метил-4-оксоциклогексил)ацетат
Воду (0.5 мл) добавляли в перемешиваемый раствор этил 2-(8-метил-1,4-диоксаспиро[4.5]декан-8-ил)ацетата (1.6 г, 6.60 ммоль) и муравьиной кислоты (10 мл) при комнатной температуре. Анализ реакционной смеси методом LCMS показал исчезновение исходного вещества и формирование целевого продукта. Реакционную смесь упаривали при пониженном давлении, и остаток разделяли между EtOAc и насыщенным водным раствором поваренной соли, слои разделяли, органическую часть промывали насыщенным водным раствором поваренной соли (2х), сушили (Na2SO4), фильтровали, и фильтрат упаривали при пониженном давлении, получая бледно-желтую жидкость - этил 2-(1-метил-4-оксоциклогексил)ацетат (1.6 г, 8.07 ммоль, 86% выход): 1H ЯМР (500 МГц, CDCl3) δ 1.22-1.31 (м, 6Н), 1.77-1.91 (м, 4Н), 2.39-2.43 (м, 6Н), 4.12-4.23 (м, 2Н).
Стадия 3: трет-бутил 2-(4-(2-этокси-2-оксоэтил)-4-метилциклогексил) гидразин-карбоксилат
Этил 2-(1-метил-4-оксоциклогексил)ацетат (1.5 г, 7.57 ммоль) и трет-бутил карбазат (1.100 г, 8.32 ммоль) растворяли в хлороформе (30 мл) и добавляли АсОН (1.0 мл) и NaBH(ОАс)3 (5.65 г) при охлаждении на ледяной бане. Полученную смесь постепенно нагревали обратно до комнатной температуры, и полученную смесь перемешивали 4 часа. Реакционную смесь выливали в насыщенный водный раствор NaHCO3, и полученную смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (гексан : EtOAc, 100%-35%), получая трет-бутил 2-(4-(2-этокси-2-оксоэтил)-4-метилциклогексил)гидразинкарбоксилат (1.72 г, 5.47 ммоль, 72.3% выход) в виде смеси изомеров (бесцветное масло). LCMS=315.4 (М+Н)+.
Стадия 4: этил 2-(4-гидразинил-1-метилциклогексил)ацетата гидрохлорид
В трет-бутил 2-(4-(2-этокси-2-оксоэтил)-4-метилциклогексил)гидразинкарбоксилат (1.7 г, 5.41 ммоль) в EtOH (5 мл) добавляли HCl (4 М раствор в 1,4-диоксане, 10 мл) по каплям при 0°С. Полученную смесь перемешивали при комнатной температуре в течение 4 часов и упаривали, получая белое твердое вещество, использовавшееся без дополнительной очистки в следующей стадии.
Стадия 5: бензил 1-(4-(2-этокси-2-оксоэтил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат
Раствор (Z)-бензил 2-((диметиламино)метилен)-4,4,4-трифтор-3-оксобутаноата (2.018 г, 6.70 ммоль) в EtOH (20 мл) добавляли по каплям в раствор этил 2-(4-гидразинил-1-метилциклогексил)ацетата гидрохлорида (1.6 г, 6,38 ммоль) и DIPEA (2.452 мл, 14.04 ммоль) в EtOH (31.9 мл) при комнатной температуре. Реакционную смесь оставляли перемешиваться в течение ночи. Растворитель удаляли, и оставшееся масло очищали на колонке 40 г REDISEP™ Gold SiO2, элюируя смесью 0-25% EtOAc/гексан, применяя метод разделения Gold. Фракции, содержащие целевой продукт, объединяли и упаривали в вакууме, получая бензил 1-(4-(2-этокси-2-оксоэтил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат (2.12 г, 4.69 ммоль, 73.4% выход) в виде смеси изомеров (бесцветный сироп). LCMS=453.4 (М+Н)+.
Стадия 6: 1-((1R,4R)-4-(2-этокси-2-оксоэтил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота и 1-((1S,4S)-4-(2-этокси-2-оксоэтил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота
Бензил 1-(4-(2-этокси-2-оксоэтил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксилат (2.1 г, 4.64 ммоль) растворяли в EtOH (10 мл) и EtOAc (10 мл) и добавляли Pd/C (10%, 210 мг) в колбу для проведения реакций под давлением, в атмосфере N2. Реакционный сосуд снабжали системой управления давлением, к одному отводу подключали к вакууму, а второй к баллону с водородом. Устанавливали давление 20 фунт/кв.дюйм, и реакционную систему дважды вакуумировали и заполняли водородом. Затем клапаны закрывали, и реакционную смесь перемешивали в течение 2 часов. Давление составляло 5 фунт/кв.дюйм, и LCMS подтвердил окончание реакции. Фильтрование через целит и удаление растворителей дало масло (1.5 г). Полученное вещество разделяли методом препаративной СЖХ: 150×50 мм AD-H колонка с 18 мл/мин МеОН (20 мМ NH3) + 162 г/мин СО2, 10% со-растворитель при 180 г/мин. Темп. = 29°С, Даление на выходе = 100 бар. Длина волны = 230 нм. Вкалывали 0.5 мл образца 1500 мг, растворенного в 20 мл смеси 1:1 МеОН : ДХМ; с = 75 мг/мл и 37.5 мг за один вкол. Время цикла 11 минут, время анализа 15 минут, получая Пик 1: белая твердая 1-((1R,4R)-4-(2-этокси-2-оксоэтил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота (600 мг, 1.656 ммоль, 35.7% выход): 1H ЯМР (500 МГц, CD2Cl2) δ 1.14 (с, 3Н), 1.23-1.28 (м, 3Н), 1.46-1.58 (м, 2Н), 1.67-1.77 (м, 2Н), 1.79-1.87 (м, 2Н), 2.16-2.28 (м, 4Н), 4.08-4.14 (м, 2Н), 4.32 (тт, J=11.7, 4.1 Гц, 1Н), 6.76 (ушир.с, 1Н), 7.94 (с, 1Н). LCMS=363.3 (М+Н)+; Пик 2: 1-((1S,4S)-4-(2-этокси-2-оксоэтил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновая кислота (700 мг, 1.932 ммоль, 41.6% выход): 1Н ЯМР (500 МГц, CDCl3) δ 1.09 (с, 3Н), 1.25-1.28 (м, 3Н), 1.33-1.43 (м, 2Н), 1.82-1.88 (м, 4Н), 2.17-2.32 (м, 2Н), 2.47 (с, 2Н), 4.12-4.17 (м, 2Н), 4.35 (тт, J=11.7, 3.9 Гц, 1Н), 6.72 (ушир.с, 1Н), 7.98 (с, 1Н). LCMS=363.4 (М+Н)+.
Перечисленные ниже пиразолкарбоновые кислоты получали по методикам, аналогичным описанным выше для сравнительных примеров.
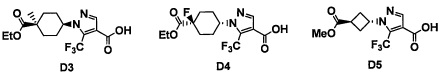
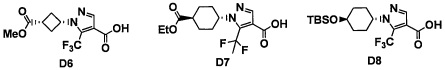
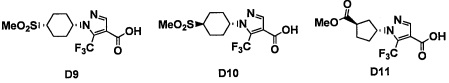
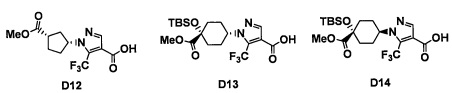
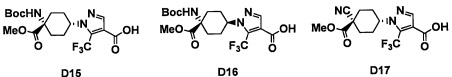
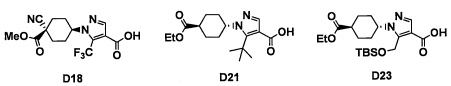
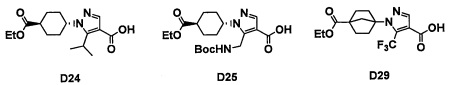
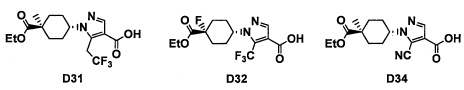
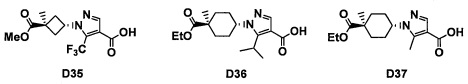
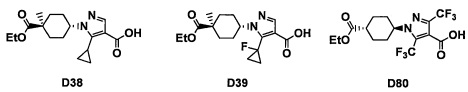
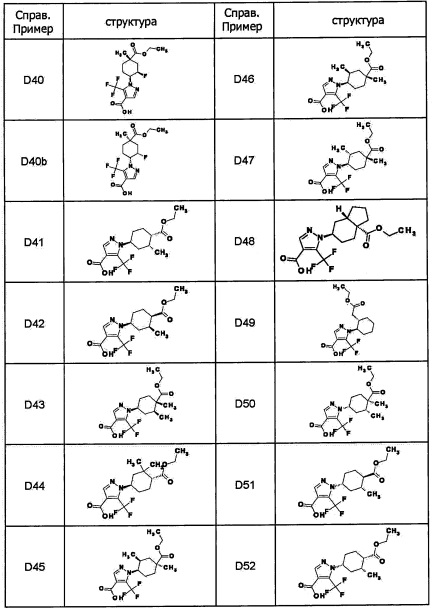
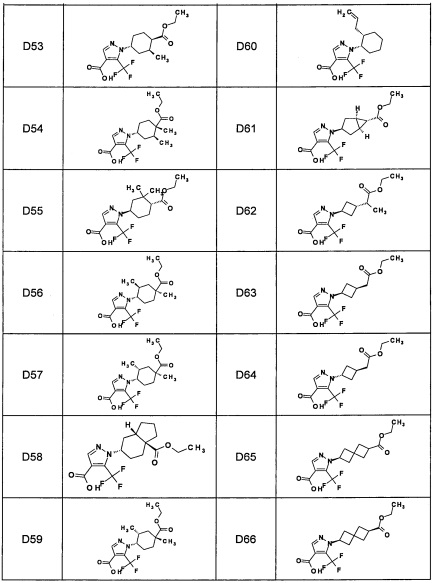
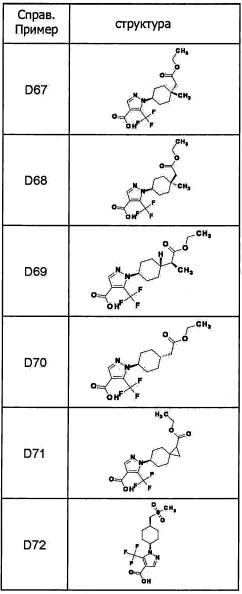
Пример 1
транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(4-фторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота

Стадия 1: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-(триэтилсилилоху)этил)(4-фторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (1-1)
В смесь кислоты D1 (6.22 г, 18.6 ммоль) и амина А1 (8.67 г, 20.4 ммоль) в ДМФА (100 мл) добавляли HATU (8.48 г, 22.3 ммоль) и DIPEA (4.74 мл, 27.9 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь гасили водой (200 мл) и экстрагировали этилацетатом (2×100 мл). Объединенные органические слои промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над Na2SO4 и упаривали при пониженном давлении, получая соединение 1-1 (15 г, неочищенное) в виде коричневой смолы.
Стадия 2: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(4-фторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (1-2)
В перемешиваемый раствор соединения 1-1 (15 г, 20.2 ммоль) в ТГФ (20 мл) добавляли TBAF (1.0 М раствор в ТГФ, 40.4 мл, 40.4 ммоль) по каплям при 0°С, и полученную смесь оставляли нагреваться с 0°С до комнатной температуры при перемешивании в течение 2 час. Реакционную смесь гасили насыщенным водным раствором NH4Cl (100 мл) и экстрагировали этилацетатом (2×150 мл). Объединенные органические слои промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, элюент: 70% EtOAc/гексан), получая соединение 1-2 (9.9 г, 84% за две стадии) в виде желто-коричневой смолы. 1H ЯМР (CDCl3): присутствуют ротамеры δ 8.42 и 8.38 (2Н, 2х с); 7.57 и 7.53 (1Н, 2х с); 7.41-7.35 и 7.14-7.09 (4Н, 2х м); 5.61-5.45 (1Н, м); 5.10-4.50 (3Н, м); 4.25-3.90 (4Н, м); 3.31-3.15 (1Н, м); 2.23-2.16 (6Н, м); 1.65-1.51 (2Н, м); 1.28-1.23 (3Н, м); LCMS: 631 (М+Н)+.
Стадия 3: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(4-фторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (1-3)
В перемешиваемый раствор соединения 1-2 (9.9 г, 15.6 ммоль) в ДХМ (120 мл) порциями добавляли периодинан Десс-Мартина (21.9 г, 21.9 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь гасили NaHCO3 (50 мл, насыщенный водный раствор) и Na2S2O3 (50 мл, насыщенный водный раствор), затем экстрагировали дихлорметаном (2×150 мл). Объединенные органические слои промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, элюент: 10% EtOAc/гексан), получая соединение 1-3 (9.12 г, 92%) в виде белого твердого вещества. 1H ЯМР (CDCl3): присутствуют ротамеры δ 8.74 и 8.67 (2Н, 2х с); 7.85 и 7.79 (1Н, 2х с); 7.30-7.26 (1Н, м); 7.41-7.37 и 7.22-7.15 (3Н, 2х м); 4.73-4.51 (4Н, м); 4.27-4.21 (1Н, м); 4.07 (2Н, кв, J=7.2 Гц); 2.50-2.48 (1Н, м); 2.06-1.93 (6Н, м); 1.59-1.54 (2Н, м); 1.18 (3Н, т, J=6.9 Гц); LCMS: 629 (М+Н)+.
Стадия 4: транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(4-фторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (1)
В перемешиваемый раствор соединения 1-3 (9.12 г, 14.5 ммоль) в смеси ТГФ/вода/EtOH (77 мл, 7:1:7) по каплям добавляли LiOH (4.0 М водный раствор, 4.45 мл, 57.9 ммоль) при 0°С. Полученную смесь оставляли нагреваться до комнатной температуры при продолжающемся перемешивании на 4 часа. Реакционную смесь подкисляли добавлением HCl (1 М, 60 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали водой (100 мл), насыщенным водным раствором поваренной соли (100 мл), сушили над Na2SO4 и упаривали при пониженном давлении, получая соединение из Примера 1 (8.0 г, 94%) в виде белого твердого вещества. 1H ЯМР (CDCl3): присутствуют ротамеры δ 8.53 и 8.47 (2Н, 2х с); 7.69 и 7.60 (1Н, 2х с); 7.31-7.28 (1Н, м); 7.16-7.12 (1Н, м); 7.06-7.02 (2Н, м); 4.83 и 4.65 (2Н, 2х с); 4.61 и 4.30 (2Н, 2х с), 4.27-4.21 (1Н, м); 2.78 (1Н, м); 2.44-2.40 (2Н, м); 2.26-2.15 (2Н, м); 1.96-1.86 (2Н, м); 1.74-1.67 (2Н, м); LCMS (ESI): 601.2 (М+Н)+.
Пример 2
транс-4-(4-((2-(2,6-дихлор-4-фторфенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
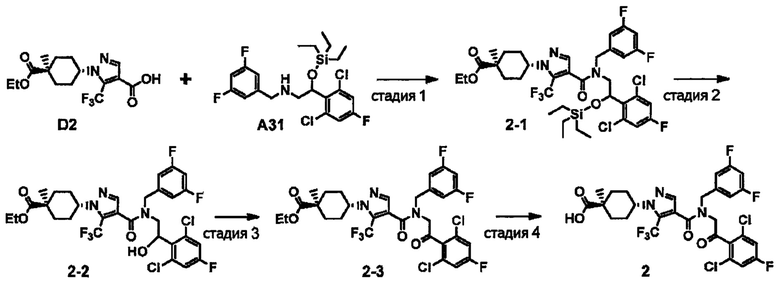
Стадия 1: этил транс-4-(4-((2-(2,6-дихлор-4-фторфенил)-2-((триэтилсилил)окси)этил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (2-1)
В раствор кислоты D2 (12.5 г, 35.9 ммоль) и (COCl)2 (4.62 мл, 39.51 ммоль) в ДХМ (150 мл) добавляли ДМФА (каталитическое количество), и результирующую смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь упаривали при пониженном давлении и сушили в высоком вакууме. Остаток от упаривания растворяли в ДХМ (10 мл) и добавляли по каплям в смесь амина А31 (18.3 г, 39.5 ммоль) и Et3N (10.0 мл, 71.8 ммоль) в ДХМ (150 мл) при 0°С. После окончания реакции (мониторинг методом ТСХ), смесь гасили водой (50 мл) и экстрагировали дихлорметаном (2×100 мл). Объединенный органический слой промывали насыщенным водным раствором поваренной соли (20 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 0-10% EtOAc/гексан в качестве элюента), получая соединение 2-1 (27.0 г, 91%) в виде бесцветной смолы. 1Н ЯМР (CDCl3): присутствуют ротамеры δ 7.54 и 7.47 (1Н, 2х с); 7.02-6.98 (2Н, м); 6.87-6.86 и 6.56-6.54 (2Н, 2х м); 6.73-6.71 (1Н, м); 5.90-5.88 и 5.50-5.47 (1Н, 2х м); 4.99-4.29 (2Н, м); 4.18-4.12 и 3.30-3.26 (4Н, 2х м); 3.87-3.81 (1Н, м); 2.21-2.16 (2Н, м); 1.89-1.88 (6Н, м); 1.35-1.24 (6Н, м); 0.91-0.84 (9Н, м); 0.58-0.48 (6Н, м).
Стадия 2: этил транс-4-(4-((2-(2,6-дихлор-4-фторфенил)-2-гидроксиэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (2-2)
Соединение 2-2 получали по методике, аналогичной описанной в Примере 1, стадия 2.
Стадия 3: этил транс-4-(4-((2-(2,6-дихлор-4-фторфенил)-2-оксоэтил)(3,5-дифторбензил) карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (2-3)
Соединение 2-3 получали по методике, аналогичной описанной в Примере 1, стадия 3.
Стадия 4: транс-4-(4-((2-(2,6-дихлор-4-фторфенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота (2)
Соединение из Примера 2 получали по методике, аналогичной описанной в Примере 1, стадия 4. 1H ЯМР (CDCl3): присутствуют ротамеры δ 8.55 и 8.49 (2Н, 2х с); 7.66 и 7.62 (1Н, 2х с); 6.85-6.69 (3Н, м); 4.83 и 4.70 (2Н, 2х с); 4.62 и 4.34 (2Н, 2х с); 4.29-4.21 (1Н, м); 2.25-2.17 (2Н, м); 1.94-1.88 (6Н, м); 1.41 и 1.40 (3Н, 2х с) LCMS (ESI): 650.2 (М+Н)+.
Пример 3
транс-4-(4-((3,5-дифторбензил)(2-(2,4-диметилтиофен-3-ил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
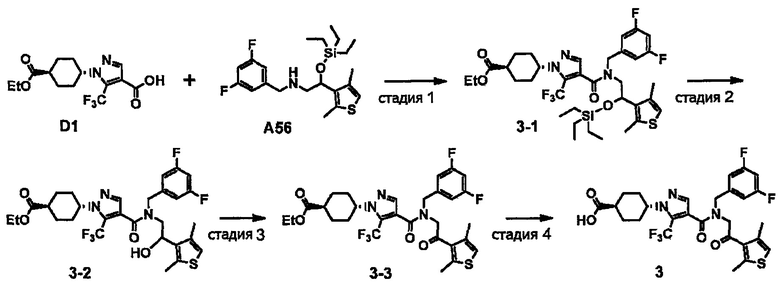
Стадия 1 и 2: этил транс-4-(4-((3,5-дифторбензил)(2-(2,4-диметилтиофен-3-ил)-2-гидроксиэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (3-2)
В смесь кислоты D1 (162 мг, 0.48 ммоль) и амина А56 (200 мг, 0.48 ммоль) в ДМФА (4 мл) добавляли DIPEA (0.12 мл, 0.72 ммоль) и HATU (221 мг, 0.58 ммоль) при комнатной температуре и перемешивали при той же температуре 4 часа. Реакционную смесь гасили водой (50 мл) и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (20 мл), сушили над Na2SO4 и упаривали при пониженном давлении, получая желтый остаток.
В перемешиваемый раствор полученного желтого остатка от упаривания по каплям добавляли TBAF (1 М раствор в ТГФ, 0.96 мл, 0.96 ммоль) при комнатной температуре. Полученную смесь перемешивали при той же температуре 1 час. Реакционную смесь гасили насыщенным водным раствором NaHCO3 и экстрагировали этилацетатом (2×20 мл). Органические слои промывали насыщенным водным раствором поваренной соли (2×10 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 10% EtOAc/гексан в качестве элюента), получая соединение 3-2 (290 мг, 97%) в виде бесцветной смолы.
Стадия 3: этил транс-4-(4-((3,5-дифторбензил)(2-(2,4-диметилтиофен-3-ил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (3-3)
В перемешиваемый раствор соединения 3-2 (290 мг, 0.47 ммоль) в ДХМ (8 мл) добавляли периодинан Десс-Мартина (401 мг, 0.94 ммоль) при 0°С, и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь гасили насыщенным водным раствором Na2S2O3 и NaHCO3, и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (2×10 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 30% EtOAc/гексан в качестве элюента), получая соединение 3-3 (220 мг, 78%) в виде бесцветной смолы.
Стадия 4: транс-4-(4-((3,5-дифторбензил)(2-(2,4-диметилтиофен-3-ил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (3)
В раствор соединения 3-3 (220 мг, 0.37 ммоль) в EtOH (1 мл), ТГФ (1 мл) и Н2О (0.2 мл) по каплям добавляли LiOH (4 М водный раствор, 0.55 мл, 2.2 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили добавлением по каплям 1 М водного раствора HCl (рН доводили до 4.0) и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (2×10 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом обращенно-фазной колоночной хроматографии (С18 силикагель, 56% вода/CH3CN в качестве элюента), получая соединение из Примера 3 (56 мг, 26%) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6): присутствуют ротамеры δ 7.63 и 7.50 (1Н, 2х с); 7.14 и 7.09 (1Н, 2х с); 6.83-6.81 (1Н, м); 6.77-6.68 (2Н, м); 4.78 и 4.69 (2Н, 2х с); 4.59 и 4.28 (2Н, 2х с); 4.27-4.18 (1Н, м); 2.49-2.38 (4Н, м); 2.25-2.18 (5Н, м); 2.10-1.97 (4Н, м); 1.70-1.57 (2Н, м); LCMS (APCI): 584 (М+Н)+.
Пример 4
транс-4-(4-((2-(2,6-дихлор-4-(метилсульфонил)фенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
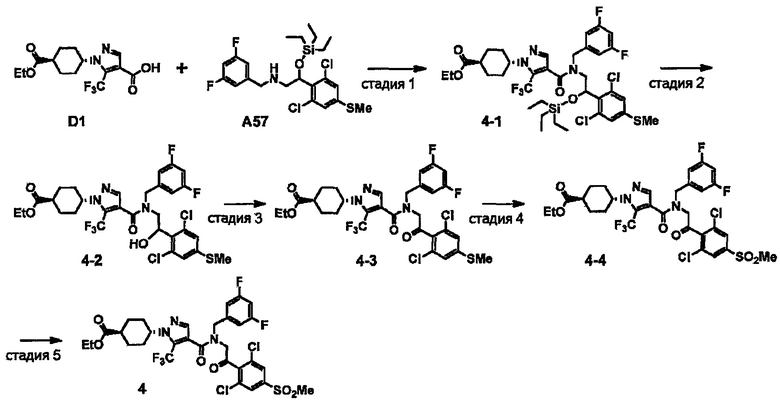
Стадия 1: этил транс-4-(4-((2-(2,6-дихлор-4-(метилтио)фенил)-2-((триэтилсилил)окси)этил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (4-1)
Соединение 4-1 (0.44 г, неочищенное) получали в виде коричневой смолы реакцией амина А57 (0.26 г, 0.52 ммоль), кислоты D1 (0.17 г, 0.52 ммоль), HATU (0.24 г, 0.63 ммоль) и DEPEA (0.13 мл, 0.79 ммоль) в ДМФА (5 мл) по методике, аналогичной описанной в Примере 1.
Стадия 2: этил транс-4-(4-((2-(2,6-дихлор-4-(метилтио)фенил)-2-гидроксиэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (4-2)
Соединение 4-2 (0.38 г, 91%) получали в виде коричневой смолы реакцией соединения 4-1 (0.44 г, 0.59 ммоль) и TBAF (1.0 М раствор в ТГФ, 0.31 мл, 1.19 ммоль) в ТГФ (10 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(4-((2-(2,6-дихлор-4-(метилтио)фенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (4-3)
Соединение 4-3 (0.1 г, 26%) получали в виде бесцветной смолы реакцией соединения 4-2 (0.38 г, 0.61 ммоль) и периодинана Десс-Мартина (0.52 г, 1.22 ммоль) в ДХМ (10 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: этил транс-4-(4-((2-(2,6-дихлор-4-(метилсульфонил)фенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (4-4)
В перемешиваемый раствор соединения 4-3 (0.1 г, 0.1 ммоль) в ДХМ (5 мл) добавляли м-СРВА (84 мг, 0.48 ммоль) при комнатной температуре. Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили водой (30 мл) и экстрагировали дихлорметаном (2×20 мл). Объединенные органические слои промывали 10%-ным раствором NaOH (20 мл), водой (30 мл), насыщенным водным раствором поваренной соли (30 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 20% EtOAc/гексан в качестве элюента), получая соединение 4-4 (0.17 г, 65%) в виде бесцветного масла.
Стадия 5: транс-4-(4-((2-(2,6-дихлор-4-(метилсульфонил)фенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (4)
Соединение из Примера 4 (50 мг, 52%) получали в виде белого твердого вещества реакцией соединения 4-4 (0.1 г, 0.13 ммоль) и LiOH (20 мг, 0.82 ммоль) в смеси ТГФ/МеОН/вода (2:2:1, 5 мл) по методике, аналогичной описанной в Примере 1. 1H ЯМР (ДМСО-d6): присутствуют ротамеры δ 12.21 (1Н, ушир.с); 8.10 и 8.03 (2Н, 2х с); 7.88 и 7.86 (1Н, 2х с); 7.20-7.14 (1Н, м); 7.11-7.08 и 6.95-6.92 (2Н, 2х м); 4.85 и 4.73 (2Н, 2х с); 4.69 и 4.57 (2Н, 2х с); 4.28-4.17 (1Н, м); 3.37 и 3.32 (3Н, 2х с); 2.35-2.29 (1Н, м); 2.07-2.02 (2Н, м); 1.98-1.90 (4Н, м); 1.60-1.49 (2Н, м); LCMS (APCI): 696 (М+Н)+.
Пример 5
N-(2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)-N-(3,5-дифторбензил)-1-(транс-4-(гидроксикарбамоил)циклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксамид
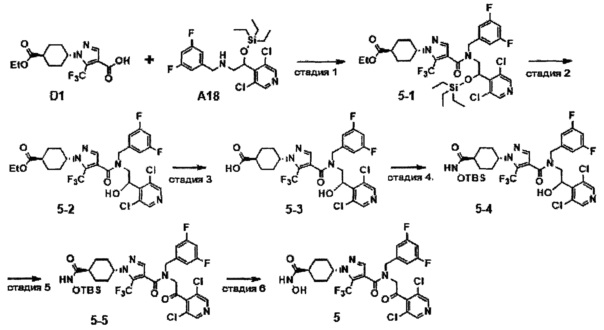
Стадия 1: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (5-1)
Соединение 5-1 (633 мг, неочищенное) получали в виде коричневой смолы реакцией кислоты D1, амина А18 (400 мг, 0.89 ммоль), HATU (408 мг, 1.07 ммоль) и DIPEA (0.23 мл, 1.34 ммоль) в ДМФА (6.0 мл) по методике, аналогичной описанной в Примере 1.
Стадия 2: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (5-2)
Соединение 5-2 (410 мг, 71%) получали в виде желтого твердого вещества реакцией соединения 5-1 (633 мг, 0.83 ммоль) и TBAF (1 М раствор в ТГФ, 1.65 мл, 1.65 ммоль) в ТГФ (3.0 мл) по методике, аналогичной описанной в Примере 1. LCMS: 649 (М+Н)+.
Стадия 3: транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (5-3)
Соединение 5-3 (185 мг, 86%) получали в виде белого твердого вещества реакцией соединения 5-2 (224 мг, 0.34 ммоль) и LiOH⋅H2O (87 мг, 2.06 ммоль) в ТГФ (3.0 мл), EtOH (2.0 мл) и воде (2.0 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: 1-(транс-4-(((трет-бутилдиметилсилил)окси)карбамоил)циклогексил)-N-(2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)-N-(3,5-дифторбензил)-5-(трифторметил)-1Н-пиразол-4-карбоксамид (5-4)
Соединение 5-4 (173 мг, 84%) получали в виде белого твердого вещества реакцией соединения 5-3 (170 мг, 0.27 ммоль), O-(трет-бутилдиметилсилил)гидроксиламина (41 мг, 0.27 ммоль), HATU (124 мг, 0.32 ммоль) и DIPEA (0.07 мл, 0.41 ммоль) в ДМФА (3.0 мл) по методике, аналогичной описанной в Примере 1. LCMS: 750 (М+Н)+.
Стадия 5: 1-(транс-4-(((трет-бутилдиметилсилил)окси)карбамоил)циклогексил)-N-(2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)-N-(3,5-дифторбензил)-5-(трифторметил)-1Н-пиразол-4-карбоксамид (5-5)
Соединение 5-5 (100 мг, 58%) получали в виде бесцветной смолы реакцией соединения 5-4 (173 мг, 0.23 ммоль) и периодинана Десс-Мартина (117 мг, 0.27 ммоль) в ДХМ (20.0 мл) по методике, аналогичной описанной в Примере 1. LCMS: 748 (М+Н)+.
Стадия 6: N-(2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)-N-(3,5-дифторбензил)-1-(транс-4-(гидроксикарбамоил)циклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксамид (5)
В перемешиваемый раствор соединения 5-5 (100 мг, 0.13 ммоль) в ТГФ (8 мл) по каплям добавляли TBAF (1 М раствор в ТГФ, 0.20 мл, 0.20 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь гасили МеОН (2 мл) и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (2×10 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 7% МеОН/ДХМ в качестве элюента), получая соединение из Примера 5 (19 мг, 22%) в виде белого твердого вещества. 1H ЯМР (CDCl3): присутствуют ротамеры δ 8.54 и 8.48 (2Н, 2х с); 7.64 и 7.60 (1Н, 2х с); 6.84-6.68 (3Н, м); 4.82-4.25 (5Н, м); 2.23-2.04 (7Н, м); 1.83-1.73 (2Н, м); LCMS (APCI): 634 (М+Н)+.
Пример 6
N-(2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)-N-(3,5-дифторбензил)-1-(транс-4-(метоксикарбамоил)циклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксамид
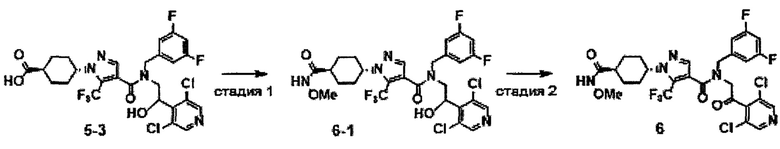
Стадия 1: N-(2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)-N-(3,5-дифторбензил)-1-(транс-4-(метоксикарбамоил)циклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксамид (6-1)
В смесь соединения 5-3 (75 мг, 0.12 ммоль) и 0-метилгидроксиламин гидрохлорида (10 мг, 0.12 ммоль) в ДМФА (3 мл) добавляли HATU (55 мг, 0.14 ммоль) и DIPEA (0.05 мл, 0.30 ммоль), и смесь перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь гасили водой и экстрагировали этилацетатом. Объединенные органические слои промывали водой, насыщенным водным раствором поваренной соли, сушили над Na2SO4 и упаривали при пониженном давлении, получая сырое соединение 6-1 (65 мг, 82%) в виде белой пены. LCMS: 650 (М+Н)+.
Стадия 2: N-(2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)-N-(3,5-дифторбензил)-1-(транс-4-(метоксикарбамоил)циклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоксамид (6)
Соединение из Примера 6 (15 мг, 23%) получали в виде белого твердого вещества реакцией соединения 6-1 (65 мг, 0.099 ммоль) и периодинана Десс-Мартина (85 мг, 0.19 ммоль) в ДХМ (5.0 мл) по методике, аналогичной описанной в Примере 1. 1H ЯМР (CDCl3): присутствуют ротамеры δ 8.54 и 8.48 (2Н, 2х с); 8.07 (1Н, ушир.с); 7.64 и 7.60 (1Н, 2х с); 6.84-6.68 (3Н, м); 4.82-4.25 (5Н, м); 3.81 и 3.78 (3Н, 2х с); 2.10-2.01 (7Н, м); 1.84-1.75 (2Н, м); LCMS (APCI): 648 (М+Н)+.
Пример 7
транс-4-(4-((3,5-дифторбензил)(2-(2-гидрокси-6-метоксифенил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
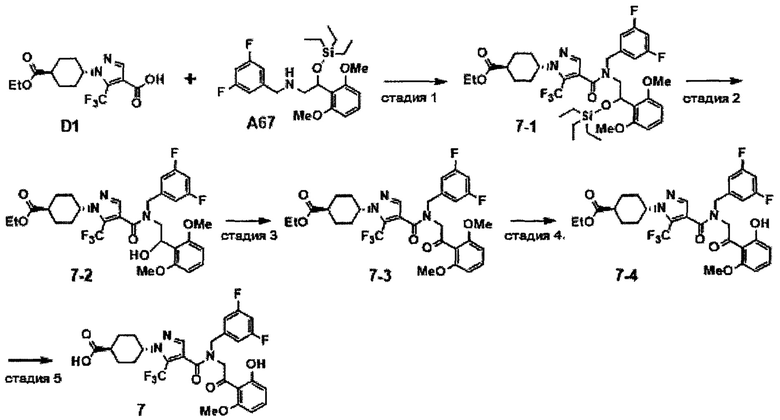
Стадия 1: этил транс-4-(4-((3,5-дифторбензил)(2-(2,6-диметоксифенил)-2-((триэтилсилил)окси)этил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (7-1)
Соединение 7-1 (0.23 г, неочищенное) получали в виде коричневой смолы реакцией амина А67 (0.13 г, 0.3 ммоль), кислоты D1 (0.1 г, 0.3 ммоль), HATU (0.13 г, 0.35 ммоль) и DIPEA (76 мкл, 0.44 ммоль) в ДМФА (5 мл) по методике, аналогичной описанной в Примере 1.
Стадия 2: этил транс-4-(4-((3,5-дифторбензил)(2-(2,6-диметоксифенил)-2-гидроксиэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (7-2)
Соединение 7-2 (0.22 г, неочищенное) получали в виде коричневой смолы реакцией соединения 7-1 (0.23 г, 0.3 ммоль) и TBAF (1.0 М раствор в ТГФ, 0.61 мл, 0.6 ммоль) в ТГФ (5 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(4-((3,5-дифторбензил)(2-(2,6-диметоксифенил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (7-3)
Соединение 7-3 (0.16 г, 73%) получали в виде бесцветной смолы реакцией соединения 7-2 (0.22 г, 0.34 ммоль) и периодинана Десс-Мартина (0.29 г, 0.69 ммоль) в ДХМ (10 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: этил транс-4-(4-((3,5-дифторбензил)(2-(2-гидрокси-6-метоксифенил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (7-4)
В перемешиваемый раствор соединения 7-3 (50 мг, 0.07 ммоль) в ДХМ (5 мл) добавляли BBr3 (1.0 М раствор в ДХМ, 1.5 мл, 1.4 ммоль) при комнатной температуре, и полученную смесь перемешивали 16 часов. Растворитель упаривали при пониженном давлении, и полученный остаток очищали методом колоночной хроматографии (силикагель, 30% EtOAc/гексан в качестве элюента), получая соединение 7-4 (32 мг, 65%) в виде коричневой смолы.
Стадия 5: транс-4-(4-((3,5-дифторбензил)(2-(2-гидрокси-6-метоксифенил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (7)
Соединение из Примера 7 (15 мг, 50%) получали в виде белого твердого вещества реакцией соединения 7-4 (32 мг, 0.05 ммоль) и LiOH (6.2 мг, 0.25 ммоль) в смеси ТГТФ/МеОН/вода (2:2:1, 5 мл) по методике, аналогичной описанной в Примере 1. 1H ЯМР (ДМСО-d6): присутствуют ротамеры δ 11.81 (1Н, ушир.с); 10.89 (1Н, ушир.с); 7.76 и 7.64 (1Н, 2х с); 7.42-7.23 (1Н, м); 7.18-6.86 (3Н, м); 6.61-6.46 (2Н, м); 4.82-4.51 (4Н, м); 4.25-4.13 (1Н, м); 3.84 и 3.65 (3Н, 2х с); 2.28-2.21 (1Н, м); 2.03-1.89 (6Н, м); 1.55-1.44 (2Н, м); LCMS (APCI): 596 (М+Н)+.
Пример 8
транс-4-(4-((3,5-дифторбензил)(2-оксо-2-(1Н-пиразол-3-ил)этил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
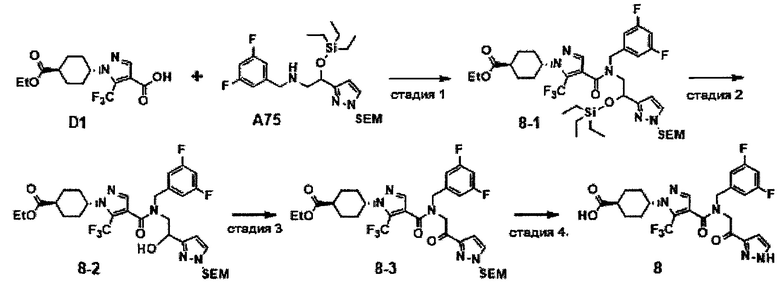
Стадия 1: этил транс-4-(4-((3,5-дифторбензил)(2-((триэтилсилил)окси)-2-(1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-3-ил)этил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (8-1)
Соединение 8-1 (34 мг, не чистое) получали в виде бесцветной смолы реакцией амина А75 (40 мг, 0.080 ммоль), кислоты D1 (26 мг, 0.080 ммоль), HATU (36.4 мг, 0.096 ммоль) и DIPEA (0.020 мл, 0.120 ммоль) в ДМФА (5 мл) по методике, аналогичной описанной в Примере 1.
Стадия 2: этил транс-4-(4-((3,5-дифторбензил)(2-гидрокси-2-(1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-3-ил)этил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (8-2)
Соединение 8-2 (25 мг, неочищенное) получали в виде бесцветной смолы реакцией соединения 8-1 (34 мг, 0.048 ммоль) и TBAF (1 М раствор в ТГФ, 0.10 мл, 0.10 ммоль) в ТГФ (3 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(4-((3,5-дифторбензил)(2-оксо-2-(1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-3-ил)этил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (8-3)
Соединение 8-3 (40 мг, 50%) получали в виде не совсем белого твердого вещества реакцией соединения 8-2 (80 мг, 0.114 ммоль) и периодинана Десс-Мартина (97 мг, 0.228 ммоль) в ДХМ (5 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: транс-4-(4-((3,5-дифторбензил)(2-оксо-2-(1Н-пиразол-3-ил)этил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (8)
В перемешиваемый раствор соединения 8-3 (75 мг, 0.107 ммоль) в 1,4-диоксане (2 мл) добавляли HCl (12 М, 0.5 мл). Полученную смесь перемешивали при 80°С в течение 2 часов. Растворитель удаляли при пониженном давлении. Остаток от упаривания растворяли в 1,4-диоксане (2 мл) и добавляли NH4OH (0.5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли при пониженном давлении, и остаток очищали методом обращенно-фазной колоночной хроматографии (С18 силикагель, 70% CH3CN/вода в качестве элюента), получая соединение из Примера 8 (10 мг, 16%) в виде белого твердого вещества. 1H ЯМР (CD3OD): присутствуют ротамеры δ 7.74-7.50 (2Н, м); 6.98-6.95 (1Н, м); 6.87-6.78 (3Н, м); 5.00 и 4.78 (2Н, 2х с); 4.74 и 4.64 (2Н, 2х с); 4.28-4.21 (1Н, м); 2.36-2.28 (1Н, м); 2.17-2.09 (2Н, м); 2.02-1.93 (4Н, м); 1.62-1.55 (2Н, м); LCMS (APCI): 540 (М+Н)+.
Пример 9
4-(4-((3,5-дифторбензил)(2-(2,6-дигидроксифенил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
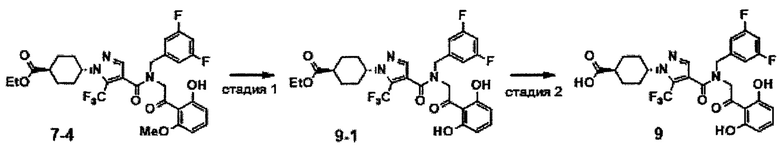
Стадия 1: этил транс-4-(4-((3,5-дифторбензил)(2-(2-гидрокси-6-метоксифенил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (9-1)
В раствор соединения 7-4 (50 мг, 0.082 ммоль) в дихлорэтане (3 мл) добавляли BBr3 (0.822 мл, 0.822 ммоль, 1 М раствор в ДХМ) по каплям, и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь упаривали при пониженном давлении, получая соединение 9-1 в виде коричневого масла (50 мг, колич.).
Стадия 2: 4-(4-((3,5-дифторбензил)(2-(2,6-дигидроксифенил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (9)
В соединение 9-1 (50 мг, 0.082 ммоль) добавляли избыток BBr3 (1 М раствор в ДХМ) по каплям, и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь упаривали при пониженном давлении. Остаток от упаривания очищали методом обращенно-фазной колоночной хроматографии (С18 силикагель, 55% вода/CH3CN в качестве элюента), получая соединение из Примера 9 (3 мг, 6%) в виде белого твердого вещества. 1H ЯМР (ДМСО-d6): присутствуют ротамеры δ 11.91 (1Н, ушир.с); 7.79 и 7.61 (1Н, 2x с); 7.28-6.88 (4H, м); 6.36-6.02 (2Н, м); 4.89-4.13 (5Н, м); 2.29-2.22 (1Н, м); 2.05-1.83 (6Н, м); 1.55-1.46 (2Н, м); LCMS (ESI): 582 (М+Н)+.
Пример 10
транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2,2-дифторэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
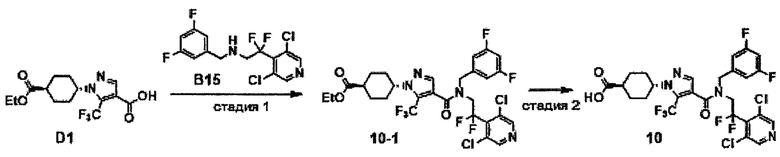
Стадия 1: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2,2-дифторэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (10-1)
В смесь кислоты D1 (56.7 мг, 0.16 ммоль) и амина В15 (60 мг, 0.016 ммоль) в пиридине (4 мл) добавляли POCl3 (0.02 мл, 0.25 ммоль) по каплям при 0°С и перемешивали при той же температуре 1 час. Реакционную смесь гасили насыщенным водным раствором KHPO4 (5 мл) и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (20 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 15% EtOAc/гексан в качестве элюента), получая соединение 10-1 (25 мг, 22%) в виде светло-желтого твердого вещества.
Стадия 2: транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2,2-дифторэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (10)
Соединение из Примера 10 (11 мг, 46%) получали в виде белого твердого вещества реакцией соединения 10-1 (25 мг, 0.37 ммоль) и LiOH (27 мг, 0.11 ммоль) в EtOH (0.5 мл), ТГФ (0.5 мл) и H2O (0.2 мл) по методике, аналогичной описанной в Примере 1. 1H ЯМР (ДМСО-d6): присутствуют ротамеры δ 8.75 и 8.69 (2Н, 2х с); 7.91 и 7.74 (1Н, 2х с); 7.21-6.75 (3Н, м); 4.86 и 4.76 (2Н, 2х с); 4.63-4.00 (3Н, м); 2.34-2.23 (1Н, м); 2.09-1.77 (6Н, м); 1.61-1.44 (2Н, м); LCMS (APCI): 641 (М+Н)+.
Пример 11
транс-4-(4-((2-(2-амино-6-хлорфенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
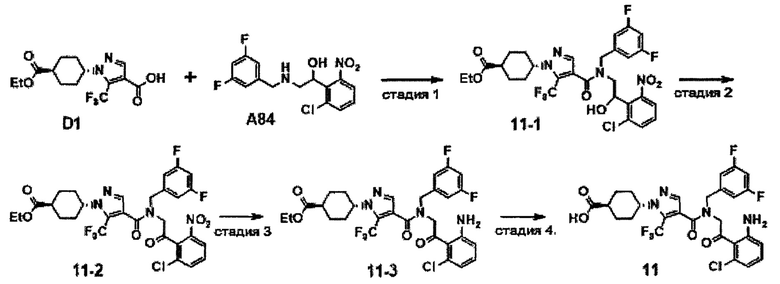
Стадия 1: этил транс-4-(4-((2-(2-хлор-6-нитрофенил)-2-гидроксиэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (11-1)
Соединение 11-1 (0.40 г, неочищенное) получали в виде бледно-желтой смолы реакцией амина А84 (0.2 г, 0.58 ммоль), кислоты D1 (0.19 г, 0.58 ммоль), HATU (0.26 г, 0.7 ммоль) и DIPEA (0.14 мл, 0.87 ммоль) в ДМФА (10 мл) по методике, аналогичной описанной в Примере 1.
Стадия 2: этил транс-4-(4-((2-(2-хлор-6-нитрофенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (11-2)
Соединение 11-2 (0.28 г, 70%) получали в виде бесцветной смолы реакцией соединения 11-1 (0.40 г, 0.6 ммоль) и периодинана Десс-Мартина (0.51 г, 1.2 ммоль) в ДХМ (10 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(4-((2-(2-амино-6-хлорфенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (11-3)
Соединение 11-3 (70 мг, 74%) получали в виде желтой смолы реакцией соединения 11-2 (0.1 г, 0.15 ммоль), Fe (85 мг, 1.52 ммоль) и NH4Cl (81 мг, 1.52 ммоль) в смеси EtOH/вода (4:1, 5 мл) по методике, аналогичной описанной в справочном примере А56, стадия 7.
Стадия 4: транс-4-(4-((2-(2-амино-6-хлорфенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (11)
Соединение из Примера 11 (8 мг, 33%) получали в виде желтого твердого вещества реакцией соединения 11-3 (20 мг, 0.03 ммоль) и LiOH (3.8 мг, 0.16 ммоль) в смеси ТГФ/МеОН/вода (2:2:1, 5 мл) по методике, аналогичной описанной в Примере 1. 1H ЯМР (ДМСО-d6): присутствуют ротамеры δ 12.17 (1Н, ушир.с); 7.79 и 7.79 (1Н, 2x с); 7.17-6.93 (4H, м); 6.70-6.54 (2Н, м); 5.80 (1Н, ушир.с); 5.53 (1Н, ушир.с); 4.77-4.59 (4Н, м); 4.23-4.16 (1Н, м); 2.29-2.22 (1Н, м); 2.06-1.88 (6Н, м); 1.57-1.46 (2Н, м); LCMS (APCI): 599 (М+Н)+.
Пример 12
транс-4-(4-((3,5-дифторбензил)(2-оксо-2-(2,4,6-тригидроксипиримидин-5-ил)этил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
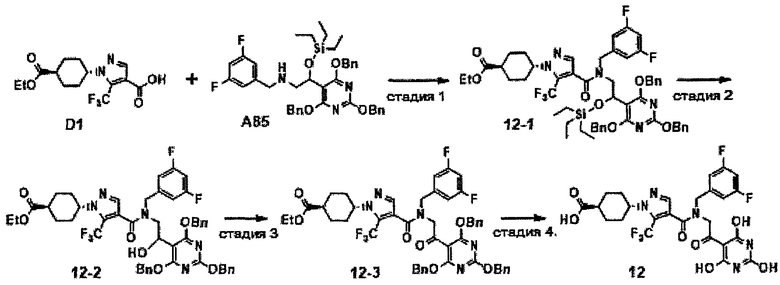
Стадия 1: этил транс-4-(4-((3,5-дифторбензил)(2-((триэтилсилил)окси)-2-(2,4,6-трис(бензилокси)пиримидин-5-ил)этил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (12-1)
Соединение 12-1 получали (0.45 г, неочищенное) в виде коричневой смолы реакцией амина А85 (0.3 г, 0.4 ммоль), кислоты D1 (0.14 г, 0.4 ммоль), HATU (0.19 г, 0.5 ммоль) и DIPEA (0.11 мл, 0.6 ммоль) в ДМФА (5 мл) по методике, аналогичной описанной в Примере 1.
Стадия 2: этил транс-4-(4-((3,5-дифторбензил)(2-гидрокси-2-(2,4,6-трис(бензилокси)пиримидин-5-ил)этил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (12-2)
Соединение 12-2 получали (0.31 г, 79%) в виде коричневой смолы реакцией соединения 12-1 (0.45 г, 0.4 ммоль) и TBAF (1 М раствор в ТГФ, 0.9 мл, 0.8 ммоль) в ТГФ (10 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(4-((3,5-дифторбензил)(2-оксо-2-(2,4,6-трис(бензилокси)пиримидин-5-ил)этил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (12-3)
Соединение 12-3 получали (0.31 г, колич.) в виде бесцветной смолы реакцией соединения 12-2 (0.31 г, 0.3 ммоль) и периодинана Десс-Мартина (0.29 г, 0.7 ммоль) в ДХМ (10 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: транс-4-(4-((3,5-дифторбензил)(2-оксо-2-(2,4,6-тригидроксипиримидин-5-ил)этил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (пример 12)
В перемешиваемый раствор соединения 12-3 (0.1 г, 0.1 ммоль) в диоксане (5 мл) добавляли 6 М HCl (5 мл) при комнатной температуре, и полученную смесь перемешивали при 80°С в течение 2 часов. Добавляли в реакционную смесь 10%-ный раствор NaOH до рН 5 и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом обращенно-фазной колоночной хроматографии (С18 силикагель, 75% вода/CH3CN в качестве элюента), получая соединение из Примера 12 (8 мг, 12%) в виде белого твердого вещества. 1H ЯМР (ДМСО-d6): присутствуют ротамеры δ 7.78 и 7.72 (1Н, 2х с); 7.18-6.89 (3Н, м); 4.92-4.56 (4Н, м); 4.22-4.15 (1Н, м); 2.33-2.25 (1Н, м); 2.05-1.87 (6Н, м); 1.57-1.46 (2Н, м); LCMS (APCI): 600 (М+Н)+.
Пример 13
транс-4-(4-((2-(2-ацетамидо-6-хлорфенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
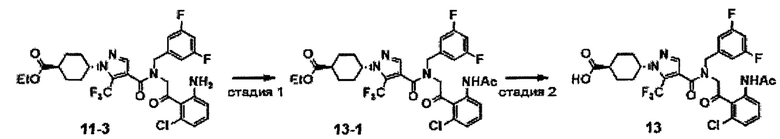
Стадия 1: этил транс-4-(4-((2-(2-ацетамидо-6-хлорфенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (13-1)
В перемешиваемый раствор соединения 11-3 (45 мг, 0.07 ммоль) в 1:1 смеси пиридина и ДХМ (5 мл) добавляли CH3COCl (6 мкл, 0.08 ммоль) при 0°С и перемешивали в течение 2 часов. Реакционную смесь гасили водой (20 мл) и экстрагировали дихлорметаном (2×10 мл). Объединенные органические слои промывали водой (50 мл), насыщенным водным раствором поваренной соли (50 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 20% EtOAc/гексан в качестве элюента), получая соединение 13-1 (50 мг, колич.) в виде желтого твердого вещества.
Стадия 2: транс-4-(4-((2-(2-ацетамидо-6-хлорфенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (13)
Соединение из Примера 13 (25 мг, 52%) получали в виде белого твердого вещества реакцией соединения 13-1 (50 мг, 0.07 ммоль) и LiOH (9 мг, 0.37 ммоль) в смеси ТГФ/МеОН/вода (2:2:1, 5 мл) по методике, аналогичной описанной в Примере 1. 1Н ЯМР (ДМСО-d6): присутствуют ротамеры δ 10.10 и 9.74 (1Н, 2х с), 7.80 и 7.78 (1Н, 2х с); 7.59-6.91 (6Н, м); 4.82-4.61 (4Н, м); 4.26-4.15 (1Н, м); 2.34-2.24 (1Н, м); 2.07-1.89 (9Н, м); 1.58-1.47 (2Н, м); LCMS (APCI): 641 (М+Н)+.
Пример 14
транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(((S)-5,5-диметилТГФ-2-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
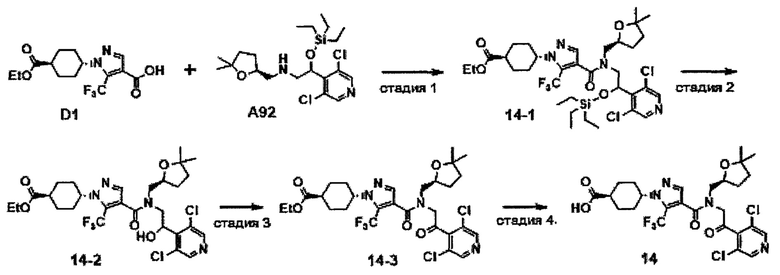
Стадия 1: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)(((S)-5,5-диметилТГФ-2-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (14-1)
Соединение 14-1 (42 мг, 61%) получали в виде желтой пены реакцией кислоты D1 (31 мг, 0.09 ммоль), амина А92 (40 мг, 0.09 ммоль), HATU (42 мг, 0.11 ммоль) и DIPEA (0.024 мл, 0.138 ммоль) в ДМФА (3.0 мл) по методике, аналогичной описанной в Примере 1. LCMS (APCI): 749 (М+Н)+.
Стадия 2: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(((S)-5,5-диметилТГФ-2-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (14-2)
Соединение 14-2 (22 мг, 62%) получали в виде бесцветной смолы реакцией соединения 14-1 (42 мг, 0.056 ммоль) и TBAF (1 М раствор в ТГФ, 0.11 мл, 0.11 ммоль) в ТГФ (3.0 мл) по методике, аналогичной описанной в Примере 1.
LCMS (APCI): 635 (М+Н)+.
Стадия 3: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(((S)-5,5-диметилТГФ-2-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (14-3)
Соединение 14-3 (15 мг, 70%) получали в виде бесцветной смолы реакцией соединения 14-2 (22 мг, 0.034 ммоль) и периодинана Десс-Мартина (29 мг, 0.069 ммоль) в ДХМ (4.0 мл) по методике, аналогичной описанной в Примере 1.
LCMS (APCI): 633 (М+Н)+.
Стадия 4: транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(((S)-5,5-диметилТГФ-2-ил) метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (14)
Соединение из Примера 14 (7.5 мг, 54%) получали в виде белого твердого вещества реакцией соединения 14-3 (15 мг, неочищенное) и LiOH⋅H2O (8 мг, 0.19 ммоль) в смеси МеОН/ТГФ/Н2O (4 мл, 1:1:0.5) по методике, аналогичной описанной в Примере 1. 1H ЯМР (ДМСО-d6): присутствуют ротамеры δ 12.16 (1Н, ушир.с), 8.79 и 8.72 (2Н, 2х с); 7.84 и 7.69 (1Н, 2х с); 5.01-4.76 (2Н, м); 4.30-3.81 (3Н, м); 2.33-2.26 (1Н, м); 2.06-1.45 (13Н, м); 1.19-1.04 (6Н, м); LCMS (APCI): 605 (М+Н)+.
Пример 15
транс-4-(4-((2-(2-хлор-6-гидроксифенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота

Стадия 1: этил транс-4-(4-((2-(2-хлор-6-метоксифенил)-2-((триэтилсилил)окси)этил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (15-1)
Соединение 15-1 (0.41 г, неочищенное) получали в виде коричневой смолы реакцией амина А93 (0.23 г, 0.52 ммоль), кислоты D1 (0.17 г, 0.52 ммоль), HATU (0.23 г, 0.62 ммоль) и DIPEA (0.133 мл, 0.78 ммоль) в ДМФА (10 мл) по методике, аналогичной описанной в Примере 1.
Стадия 2: этил транс-4-(4-((2-(2-хлор-6-метоксифенил)-2-гидроксиэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1H-пиразол-1-ил)циклогексанкарбоксилат (15-2)
Соединение 15-2 (0.36 г, неочищенное) получали в виде желтой смолы реакцией соединения 15-1 (0.41 г, 0.54 ммоль) и TBAF (1.0 М раствор в ТГФ, 1.1 мл, 1.08 ммоль) в ТГФ (10 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(4-((2-(2-хлор-6-метоксифенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (15-3)
Соединение 15-3 (0.25 г, 70%) получали в виде белого твердого вещества реакцией соединения 15-2 (0.36 г, 0.56 ммоль) и периодинана Десс-Мартина (0.47 г, 1.12 ммоль) в ДХМ (10 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: этил транс-4-(4-((2-(2-хлор-6-гидроксифенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (15-4)
В перемешиваемый раствор соединения 15-3 (0.25 г, 0.39 ммоль) в ДХМ (10 мл) добавляли BBr3 (1.0 М раствор в ДХМ, 3.9 мл, 3.9 ммоль) при комнатной температуре, и полученную смесь перемешивали 16 часов. Растворитель упаривали при пониженном давлении, и полученный остаток очищали методом колоночной хроматографии (силикагель, 30% EtOAc/гексан в качестве элюента), получая соединение 15-4 (85 мг, 35%) в виде желтого масла.
Стадия 5: транс-4-(4-((2-(2-хлор-6-гидроксифенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (15)
Соединение из Примера 15 (55 мг, 68%) получали в виде белого твердого вещества реакцией соединения 15-4 (85 мг, 0.13 ммоль) и LiOH (16.3 мг, 0.67 ммоль) в смеси ТГФ/МеОН/вода (2:2:1, 5 мл) по методике, аналогичной описанной в Примере 1.
1Н ЯМР (ДМСО-d6): присутствуют ротамеры δ 7.76 и 7.74 (1Н, 2х с); 7.28-6.78 (6Н, м); 4.75-4.52 (4Н, м); 4.26-4.15 (1Н, м); 2.33-2.26 (1Н, м); 2.07-1.90 (6Н, м); 1.58-1.47 (2Н, м); LCMS (APCI): 600 (М+Н)+.
Пример 16
транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(((R)-5,5-диметилТГФ-2-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
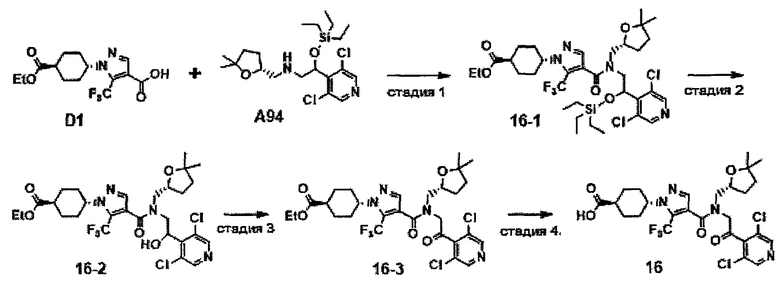
Стадия 1: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)(((R)-5,5-диметилТГФ-2-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил) циклогексанкарбоксилат (16-1)
Соединение 16-1 (29 мг, 52%) получали в виде бесцветной смолы реакцией кислоты D1 (25 мг, 0.074 ммоль), амина А94 (32 мг, 0.074 ммоль), HATU (34 мг, 0.088 ммоль) и DIPEA (0.019 мл, 0.11 ммоль) в ДМФА (3.0 мл) по методике, аналогичной описанной в Примере 1. LCMS (APCI): 749 (М+Н)+.
Стадия 2: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(((R)-5,5-диметилТГФ-2-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (16-2)
Соединение 16-2 (25 мг, неочищенное) получали в виде бесцветной смолы реакцией соединения 16-1 (29 мг, 0.038 ммоль) и TBAF (1 М раствор в ТГФ, 0.076 мл, 0.076 ммоль) в ТГФ (3.0 мл) по методике, аналогичной описанной в Примере 1. LCMS (APCI): 635 (М+Н)+.
Стадия 3: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(((R)-5,5-диметилТГФ-2-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (16-3)
Соединение 16-3 (23 мг, неочищенное) получали в виде бесцветной смолы реакцией соединения 16-2 (25 мг, неочищенное) и периодинана Десс-Мартина (33 мг, 0.078 ммоль) в ДХМ (4.0 мл) по методике, аналогичной описанной в Примере 1. LCMS (APCI): 633 (M+H)+.
Стадия 4: транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(((R)-5,5-диметилТГФ-2-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (16)
Соединение из Примера 16 (8.5 мг, 39%) получали в виде белого твердого вещества реакцией соединения 16-3 (23 мг, неочищенное) и LiOH⋅H2O (12 мг, 0.29 ммоль) в смеси МеОН/ТГФ/H2O (4 мл, 1:1:0.5) по методике, аналогичной описанной в Примере 1. 1Н ЯМР (ДМСО-d6): присутствуют ротамеры δ 12.19 (1Н, ушир.с), 8.79 и 8.72 (2Н, 2х с); 7.84 и 7.69 (1Н, 2х с); 5.01-4.76 (2Н, м); 4.27-3.80 (3Н, м); 2.33-2.26 (1Н, м); 2.06-1.45 (13Н, м); 1.19-1.04 (6Н, м); LCMS (APCI): 605 (М+Н)+.
Пример 17
транс-4-(5-циклопропил-4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
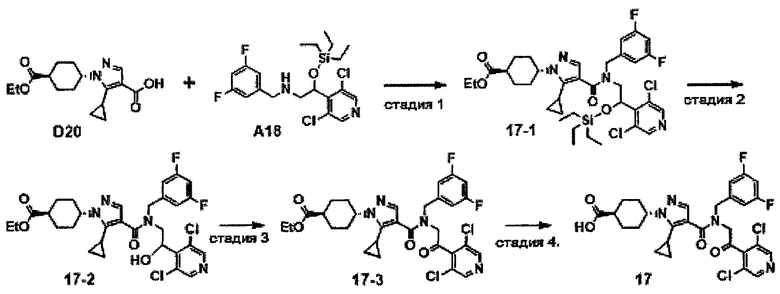
Стадия 1: этил транс-4-(5-циклопропил-4-((2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)(3,5-дифторбензил)карбамоил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (17-1)
Соединение 17-1 (130 мг, неочищенное) получали в виде бесцветной смолы реакцией кислоты D20 (200 мг, 0.56 ммоль), амина А18 (233 мг, 0.52 ммоль), HATU (296 мг, 0.78 ммоль) и DIPEA (0.165 мл, 0.97 ммоль) в ДМФА (10 мл) по методике, аналогичной описанной в Примере 1.
Стадия 2: этил транс-4-(5-циклопропил-4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(3,5-дифторбензил)карбамоил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (17-2)
Соединение 17-2 (80 мг, неочищенное) получали в виде бесцветной смолы реакцией соединения 17-1 (80 мг, 0.128 ммоль) и TBAF (1 М раствор в ТГФ, 0.190 мл, 0.190 ммоль) в ТГФ (4 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(5-циклопропил-4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (17-3)
Соединение 17-3 (60 мг, 75%) получали в виде не совсем белого твердого вещества реакцией соединения 17-2 (80 мг, 0.128 ммоль) и периодинана Десс-Мартина (110 мг, 0.250 ммоль) в ДХМ (10 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: транс-4-(5-циклопропил-4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (17)
Соединение из Примера 17 (8 мг, 12%) получали в виде белого твердого вещества реакцией соединения 17-3 (70 мг, 0.113 ммоль) и LiOH (8.4 мг, 0.330 ммоль) в смеси EtOH/ТГФ/вода (5 мл, 2:2:1) по методике, аналогичной описанной в Примере 1. 1H ЯМР (ДМСО-d6): присутствуют ротамеры δ 8.76 и 8.69 (2Н, 2х с); 7.46 и 7.35 (1Н, 2х с); 7.18-7.12 и 6.95-6.91 (3Н, м); 4.76-4.62 (4Н, м); 4.44-4.36 (1Н, м); 2.26-2.17 (1Н, м); 2.03-2.00 (2Н, м); 1.87-1.83 (5Н, м); 1.57-1.47 (2Н, м); 0.95-0.90 (2Н, м); 0.68-0.64 (2Н, м); LCMS (APCI): 591 (М+Н)+.
Пример 18
транс-4-(4-((2-(2,6-дихлор-4-(дифторметил)фенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
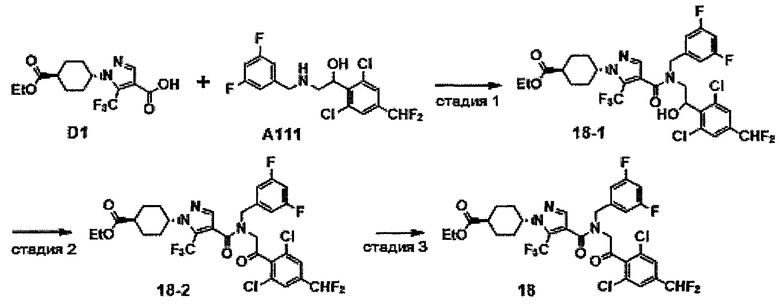
Стадия 1: этил транс-4-(4-((2-(2,6-дихлор-4-(дифторметил)фенил)-2-гидроксиэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1H-пиразол-1-ил)циклогексанкарбоксилат (18-1)
Соединение 18-1 (0.18 г, неочищенное) получали в виде коричневой смолы реакцией амина А111 (0.10 г, 0.26 ммоль), кислоты D1 (87 мг, 0.26 ммоль), HATU (0.12 г, 0.31 ммоль) и DIPEA (67 мкл, 0.39 ммоль) в ДМФА (5 мл) по методике, аналогичной описанной в Примере 1.
Стадия 2: этил транс-4-(4-((2-(2,6-дихлор-4-(дифторметил)фенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (18-2)
Соединение 18-2 (0.12 г, 67%) получали в виде бесцветной смолы реакцией соединения 18-1 (0.18 г, 0.26 ммоль) и периодинана Десс-Мартина (0.22 г, 0.52 ммоль) в ДХМ (5 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: транс-4-(4-((2-(2,6-дихлор-4-(дифторметил)фенил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (18)
Соединение из Примера 18 (15 мг, 26%) получали в виде белого твердого вещества реакцией соединения 18-2 (60 мг, 0.08 ммоль) и LiOH (11 мг, 0.43 ммоль) в смеси ТГФ/МеОН/вода (2:2:1, 5 мл) по методике, аналогичной описанной в Примере 1. 1H ЯМР (ДМСО-d6): присутствуют ротамеры δ 12.20 (1Н, ушир.с); 7.87-7.75 (3Н, м); 7.21-6.88 (4Н, м); 4.84 и 4.72 (2Н, 2х с); 4.68 и 4.57 (2Н, 2х с); 4.28-4.17 (1Н, м); 2.34-2.27 (1Н, м); 2.07-1.90 (6Н, м); 1.59-1.49 (2Н, м); LCMS (APCI): 668 (М+Н)+.
Пример 19
транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)((4-гидрокси-4-метилциклогексил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
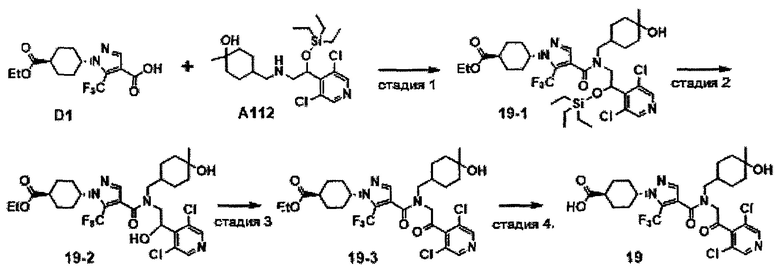
Стадия 1: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)((4-гидрокси-4-метилциклогексил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (19-1)
Соединение 19-1 (72 мг, неочищенное) получали в виде бесцветной смолы реакцией кислоты D1 (44 мг, 0.129 ммоль), амина А112 (58 мг, 0.129 ммоль), HATU (59 мг, 0.155 ммоль) и DIPEA (0.034 мл, 0.194 ммоль) в ДМФА (4.0 мл) по методике, аналогичной описанной в Примере 1.
Стадия 2: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)((4-гидрокси-4-метилциклогексил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (19-2)
Соединение 19-2 (46 мг, 55% за две стадии) получали в виде бесцветной смолы реакцией соединения 19-1 (72 мг, неочищенное) и TBAF (1 М раствор в ТГФ, 0.18 мл, 0.18 ммоль) в ТГФ (4.0 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)((4-гидрокси-4-метилциклогексил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (19-3)
Соединение 19-3 (45 мг, неочищенное) получали в виде бесцветной смолы реакцией соединения 19-2 (46 мг, 0.071 ммоль) и периодинана Десс-Мартина (60 мг, 0.14 ммоль) в ДХМ (5.0 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)((4-гидрокси-4-метилциклогексил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (19)
Соединение из Примера 19 (16 мг, 38%) получали в виде белого твердого вещества реакцией соединения 19-3 (45 мг, 0.069 ммоль) и LiOH⋅H2O (18 мг, 0.41 ммоль) в смеси EtOH/ТГФ/Н2О (4 мл, 1:1:0.5) по методике, аналогичной описанной в Примере 1.
1Н ЯМР (ДМСО-d6): присутствуют ротамеры δ 12.24 (1Н, ушир.с); 8.80 и 8.73 (2Н, 2х с); 7.80 и 7.70 (1Н, 2х с); 4.83 и 4.67 (2Н, 2х с); 4.28-4.18 (1Н, м); 3.93 и 3.87 (1Н, 2х с); 3.49-3.47 и 3.18-3.11 (1Н, 2х м); 2.33-2.25 (1Н, м); 2.06-1.90 (6Н, м); 1.58-1.01 (14Н, м); LCMS (APCI): 617 (М-Н)+.
Пример 20
транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(дифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
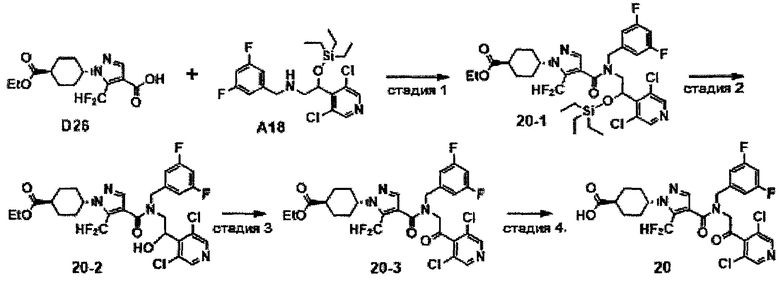
Стадия 1: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)(3,5-дифторбензил)карбамоил)-5-(дифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (20-1)
Соединение 20-1 (130 мг, 55%) получали в виде белого твердого вещества реакцией кислоты D26 (100 мг, 0.31 ммоль), амина А18 (105 мг, 0.31 ммоль), (COCl)2 (0.03 мл, 0.37 ммоль), Et3N (0.08 мл, 0.63 ммоль) и ДМФА (кат.) в ДХМ по методике, аналогичной описанной в Примере 2.
Стадия 2: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(3,5-дифторбензил)карбамоил)-5-(дифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (20-2)
Соединение 20-2 (101 мг, 91%) получали в виде не совсем белого твердого вещества реакцией соединения 20-1 (130 мг, 0.54 ммоль) и TBAF (1.0 М раствор в ТГФ, 0.24 мл, 0.24 ммоль) в ТГФ (4 мл) по методике, аналогичной описанной в Примере 2.
Стадия 3: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(дифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (20-3)
Соединение 20-3 (30 мг, 33%) получали в виде не совсем белого твердого вещества реакцией соединения 20-2 (101 мг, 0.15 ммоль) и периодинана Десс-Мартина (101 мг, 0.23 ммоль) в ДХМ (8 мл) по методике, аналогичной описанной в Примере 2.
Стадия 4: транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(дифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (20)
В раствор соединения 20-3 (106 мг, 0.17 ммоль) в диоксане (6 мл) добавляли 6 М раствор HCl (6 мл) и нагревали при 80°С 16 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли H2O и экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали насыщенным водным раствором поваренной соли (2×10 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом обращенно-фазной колоночной хроматографии (С18 силикагель, 75% CH3CN /вода в качестве элюента), получая соединение из Примера 20 (36 мг, 62%) в виде белого твердого вещества. 1H ЯМР (ДМСО-d6): присутствуют ротамеры δ 8.76 и 8.69 (2Н, 2х с); 7.77 и 7.67 (1Н, 2х с); 7.45-7.00 (4Н, м); 4.91-4.70 (4Н, м); 4.36-4.29 (1Н, м); 2.31-2.24 (1Н, м); 2.06-2.02 (2Н, м); 1.94-1.89 (4Н, м); 1.56-1.45 (2Н, м); LCMS (ESI): 599 (М+Н)+.
Пример 21
транс-4-(4-((2-(2-амино-4-хлорпиридин-3-ил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
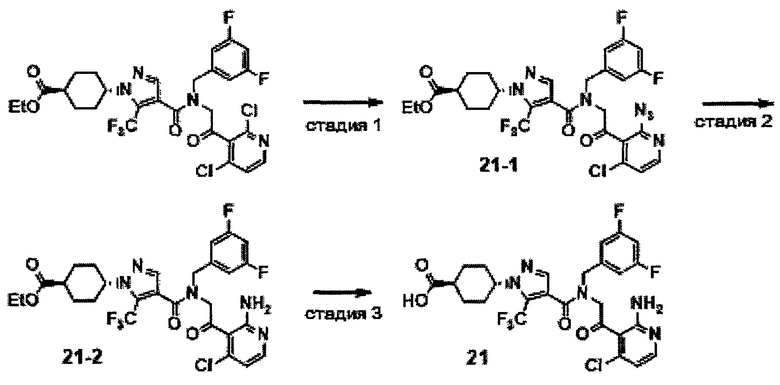
Стадия 1: этил транс-4-(4-((2-(2-азидо-4-хлорпиридин-3-ил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (21-1)
В перемешиваемый раствор этил транс-4-(4-((2-(2,4-дихлорпиридин-3-ил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилата (100 мг, 0.15 ммоль) в ДМФА (5 мл) добавляли NaN3 (50 мг, 0.7 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь гасили водой (20 мл) и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали водой (50 мл) и насыщенным водным раствором поваренной соли (50 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 30% EtOAc/гексан в качестве элюента), получая соединение 21-1 (60 мг, 59%) в виде коричневого масла.
Стадия 2: этил транс-4-(4-((2-(2-амино-4-хлорпиридин-3-ил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (21-2)
В перемешиваемый раствор соединения 21-1 (60 мг, 0.09 ммоль) в ТГФ (5 мл) добавляли Me3P (1.0 М раствор в ТГФ, 0.18 мл, 0.18 ммоль) при 0°С, и полученную смесь перемешивали при комнатной температуре в течение 2 часов. H2O (0.06 мл) добавляли в реакционную смесь при 0°С, и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель удаляли при пониженном давлении, и остаток очищали методом колоночной хроматографии (силикагель, 30% EtOAc/гексан в качестве элюента), получая соединение 21-2 (40 мг, 69%) в виде желтого масла.
Стадия 3: транс-4-(4-((2-(2-амино-4-хлорпиридин-3-ил)-2-оксоэтил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (21)
В перемешиваемый раствор соединения 21-2 (40 мг, 0.06 ммоль) в ТГФ/МеОН (4 мл, 1:1) добавляли раствор LiOH (7.7 мг, 0.3 ммоль) в воде (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли водой (10 мл), подкисляли 0.5 М HCl (до рН 5) и экстрагировали этилацетатом (3×10 мл). Объединенные органические слои промывали водой (10 мл), насыщенным водным раствором поваренной соли (10 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом обращенно-фазной колоночной хроматографии (С18 силикагель, 70% CH3CN/вода в качестве элюента), получая соединение из Примера 21 (25 мг, 66%) в виде белого твердого вещества. 1H ЯМР (ДМСО-d6): присутствуют ротамеры δ 12.14 (1Н, ушир.с); 8.75 и 8.46 (1Н, 2x с); 7.91 (2H, ушир.с); 7.73 и 7.49 (1Н, 2x с); 7.16-7.01 (2H, м); 6.89-6.85 (1Н, м); 6.78 и 6.73 (1Н, 2x с); 5.02 и 4.83 (2H, 2x с); 4.65 и 4.55 (2H, 2x с); 4.20-4.12 (1Н, м); 2.33-2.21 (1Н, м); 2.04-1.79 (6Н, м); 1.56-1.43 (2H, м); LCMS (APCI): 600 (М+Н)+.
Пример 22
транс-4-(4-((2-цианобензил)(2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
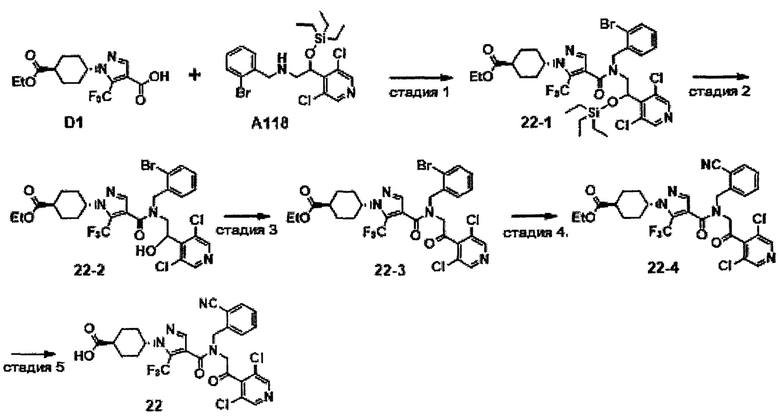
Стадия 1: этил транс-4-(4-((2-бромбензил)(2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (22-1)
Соединение 22-1 (310 мг, неочищенное) получали в виде желтого масла реакцией амина А118 (200 мг, 0.40 ммоль), кислоты D1 (136 мг, 0.40 ммоль), HATU (232 мг, 0.61 ммоль) и DIPEA (0.14 мл, 0.81 ммоль) в ДМФА (5 мл) по методике, аналогичной описанной в Примере 1.
Стадия 2: этил транс-4-(4-((2-бромбензил)(2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (22-2)
Соединение 22-2 (175 мг, 73%) получали в виде не совсем белого твердого вещества реакцией соединения 22-1 (310 мг, 0.38 ммоль) и TBAF (1.0 М раствор в ТГФ, 0.57 мл, 0.57 ммоль) в ТГФ (4 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(4-((2-бромбензил)(2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (22-3)
Соединение 22-3 (150 мг, 86%) получали в виде не совсем белого твердого вещества реакцией соединения 22-2 (175 мг, 0.25 ммоль) и периодинана Десс-Мартина (161 мг, 0.37 ммоль) в ДХМ (10 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: этил транс-4-(4-((2-цианобензил)(2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (22-4)
Смесь соединения 22-3 (100 мг, 0.14 ммоль) и Zn(CN)2 (34 мг, 0.28 ммоль) в DMA (8 мл) продували аргоном 10 минут. Добавляли Pd(PPh3)4 (33.4 мг, 0.02 ммоль) и полученную смесь нагревали при 100°С 3 часа. Реакционную смесь охлаждали до комнатной температуры, гасили водой (10 мл) и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали водой (10 мл) и насыщенным водным раствором поваренной соли (10 мл), сушили над Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (силикагель, 30% EtOAc/гексан), получая соединение 22-4 (27 мг, 29%) в виде не совсем белого твердого вещества.
Стадия 5: транс-4-(4-((2-цианобензил)(2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (22)
Соединение из Примера 22 (15 мг, 39%) получали в виде белого твердого вещества реакцией соединения 22-4 (40 мг, 0.06 ммоль) и LiOH (4.3 мг, 0.03 ммоль) в EtOH (2 мл), ТГФ (2 мл) и H2O (1 мл) по методике, аналогичной описанной в Примере 1. 1H ЯМР (ДМСО-d6): присутствуют ротамеры δ 8.56-8.50 (2Н, м); 7.91 (2Н, ушир.с); 7.94 (1Н, с); 7.54-7.42 и 7.17 (3Н, м и с); 7.08 (1Н, с); 5.62-5.58 и 4.64-4.59 (1Н, 2х м); 4.33-4.17 (1Н, м); 2.39-2.33 (1Н, м); 2.10-1.93 (7Н, м); LCMS (APCI): 608 (М+Н)+.
Пример 23
транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)((3,3-диметилциклобутил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота
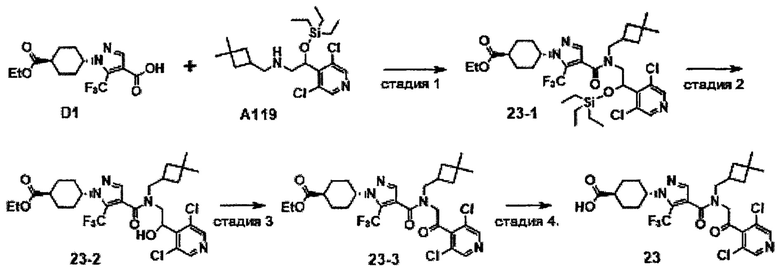
Стадия 1: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)((3,3-диметилциклобутил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил) циклогексанкарбоксилат (23-1)
Соединение 23-1 (180 мг, неочищенное) получали в виде желтой смолы реакцией амина А119 (190 мг, 0.455 ммоль), кислоты D1 (167 мг, 0.50 ммоль), оксалилхлорида (0.086 мл, 1.0 ммоль), Et3N (0.10 мл, 0.68 ммоль) и ДМФА (кат.) в ДХМ (10 мл) по методике, аналогичной описанной в Примере 2.
Стадия 2: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)((3,3-диметилциклобутил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (23-2)
Соединение 23-2 (60 мг, 20%, за 2 стадии) получали в виде бесцветной смолы реакцией соединения 23-1 (180 мг, 0.245 ммоль) и TBAF (0.5 мл, 0.5 ммоль, 1 М раствор в ТГФ) в ТГФ (3 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)((3,3-диметил-циклобутил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоксилат (23-3)
Соединение 23-3 (60 мг, колич.) получали в виде белого твердого вещества реакцией соединения 23-2 (60 мг, 0.096 ммоль) и периодинана Десс-Мартина (83 мг, 0.193 ммоль) в ДХМ (10 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)((3,3-диметилциклобутил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)циклогексанкарбоновая кислота (23)
Соединение из Примера 23 (30 мг, 52%) получали в виде не совсем белого твердого вещества реакцией соединения 23-3 (60 мг, 0.097 ммоль) и LiOH (19 мг, 0.048 ммоль) в смеси ТГФ/вода/МеОН (5 мл, 2:2:1) по методике, аналогичной описанной в Примере 1. 1H ЯМР (CD3OD): присутствуют ротамеры δ 8.65 и 8.58 (2Н, 2х с); 7.74 и 7.57 (1Н, 2х с); 4.81 и 4.63 (2Н, 2х с); 4.36-4.24 (1Н, м); 3.62 и 3.42 (2Н, 2х д, J=7.5 Гц); 2.67-2.34 (2Н, м); 2.21-2.02 (6Н, м); 1.92-1.81 (2Н, м); 1.70-1.56 (3Н, м); 1.37-1.29 (1Н, м); 1.15-0.93 (6Н, м); LCMS (APCI): 591 (М+Н)+.
Пример 186
транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)-(2,2-диметилпропил)карбамоил)-5-(трифторметил)пиразол-1-ил)-1-метилциклогексан-1-карбоновая кислота
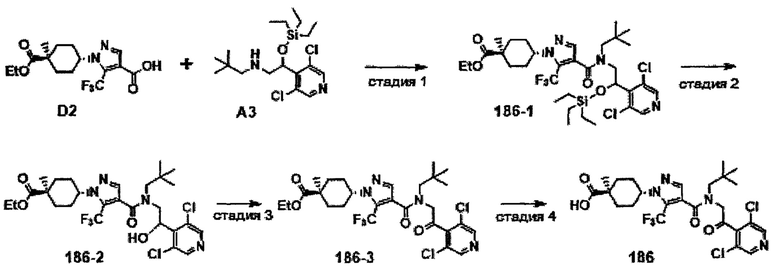
Стадия 1: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексан карбоксилат (186-1)
Соединение 186-1 получали реакцией амина A3 (60 мг, 0.153 ммоль), кислоты D2 (56 мг, 0.161 ммоль), оксалилхлорида (0.028 мл, 0.322 ммоль), 1н. раствора NaOH (0.92 мл, 0.920 ммоль) и ДМФА (кат.) в ДХМ (1 мл) по методике, аналогичной описанной в Примере 2.
Стадия 2: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (186-2)
Соединение 186-2 (89 мг, 96% за 2 стадии) получали в виде бесцветного сиропа реакцией соединения 186-1 (неочищ.) и TBAF (0.169 мл, 0.169 ммоль, 1 М раствор в ТГФ) в ТГФ (1 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (186-3)
Соединение 186-3 (69 мг, 78%) получали в виде белого твердого вещества реакцией соединения 186-2 (89 мг, 0.147 ммоль) и периодинана Десс-Мартина (87 мг, 0.205 ммоль) в ДХМ (2 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)-(2,2-диметилпропил)карбамоил)-5-(трифторметил)пиразол-1-ил)-1-метилциклогексан-1-карбоновая кислота (186)
Соединение из Примера 186 (51 мг, 77%) получали в виде белого твердого вещества реакцией соединения 186-3 (69 мг, 0.114 ммоль) и 4 н. раствора LiOH (0.228 мл, 0.912 ммоль) в ТГФ/вода/МеОН (1 мл, 2:1:2) по методике, аналогичной описанной в Примере 1. 1Н ЯМР (CDCl3): присутствуют ротамеры δ 8.57 и 8.50 (2Н, 2х с); 7.71 и 7.57 (1Н, 2х с); 4.87 и 4.53 (2Н, 2х с); 4.25-4.18 (1Н, м); 3.43-3.35 (2Н, м); 2.25-2.15 (2Н, м); 1.95-1.85 (6Н, м); 1.42 и 1.40 (3Н, 2х с); 1.01 и 0.85 (9Н, 2х с); LCMS (ESI): 577.2 (М+Н)+.
Пример 233
4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)-(2,2-диметилпропил)карбамоил)-5-(трифторметил)пиразол-1-ил)бицикло[2.2.2]октан-1-карбоновая кислота
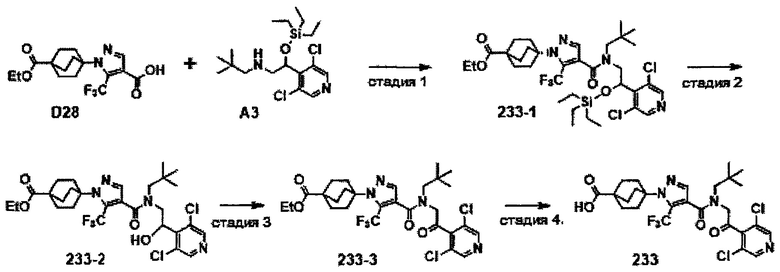
Стадия 1: этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)бицикло[2.2.2]октан-1-карбоксилат (233-1)
Соединение 233-1 получали реакцией амина A3 (58 мг, 0.148 ммоль), кислоты D28 (50 мг, 0.139 ммоль), оксалилхлорида (0.024 мл, 0.278 ммоль), 1 н. раствора NaOH (0.833 мл, 0.833 ммоль) и ДМФА (кат.) в ДХМ (1 мл) по методике, аналогичной описанной в Примере 2.
Стадия 2: этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)бицикло[2.2.2]октан-1-карбоксилат (233-2)
Соединение 233-2 (67 мг, 78% за 2 стадии) получали в виде бесцветного сиропа реакцией соединения 233-1 (неочищ.) и TBAF (0.148 мл, 0.148 ммоль, 1 М раствор в ТГФ) в ТГФ (1 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)бицикло[2.2.2]октан-1-карбоксилат (233-3)
Соединение 233-3 (56 мг, 84%) получали в виде белого твердого вещества реакцией соединения 233-2 (67 мг, 0.108 ммоль) и периодинана Десс-Мартина (64 мг, 0.151 ммоль) в ДХМ (2 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)-(2,2-диметилпропил)карбамоил)-5-(трифторметил)пиразол-1-ил)бицикло[2.2.2]октан-1-карбоновая кислота (233)
Соединение из Примера 233 (27 мг, 51%) получали в виде белого твердого вещества реакцией соединения 233-3 (56 мг, 0.091 ммоль) и 4 н. раствора LiOH (0.091 мл, 0.363 ммоль) в смеси ТГФ/вода/МеОН (0.7 мл, 3:1:3) по методике, аналогичной описанной в Примере 1. 1H ЯМР (CDCl3): присутствуют ротамеры δ 8.57 и 8.50 (2Н, 2х с); 7.64 и 7.49 (1Н, 2х с); 4.84 и 4.50 (2Н, 2х с); 3.61-3.26 (2Н, м); 2.32-2.22 (6Н, м); 2.08-2.04 (6Н, м); 1.01 и 0.85 (9Н, 2х с); LCMS (ESI): 589.2 (М+Н)+.
Пример 276
транс-4-(5-хлор-4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)-(2,2-диметилпропил)карбамоил)пиразол-1-ил)-1-метилциклогексан-1-карбоновая кислота
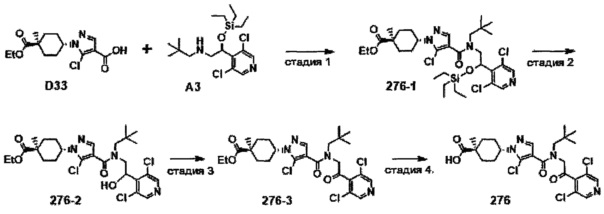
Стадия 1: этил транс-4-(5-хлор-4-((2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)(неопентил)карбамоил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (276-1)
Соединение 276-1 получали реакцией амина A3 (124 мг, 0.31 ммоль), кислоты D33 (100 мг, 0.31 ммоль), оксалилхлорида (0.082 мл, 0.95 ммоль), Et3N (0.088 мл, 0.66 ммоль) и ДМФА (кат.) в ДХМ (5 мл) по методике, аналогичной описанной в Примере 2.
Стадия 2: этил транс-4-(5-хлор-4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(неопентил)карбамоил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (276-2)
Соединение 276-2 (160 мг, 88%, за 2 стадии) получали в виде белого твердого вещества реакцией соединения 276-1 (неочищ.) и TBAF (0.5 мл, 0.5 ммоль, 1 М раствор в ТГФ) в ТГФ (2 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(5-хлор-4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(неопентил)карбамоил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (276-3)
Соединение 276-3 (130 мг, 81%) получали в виде не совсем белого твердого вещества реакцией соединения 276-2 (160 мг, 0.279 ммоль) и периодинана Десс-Мартина (233 мг, 0.56 ммоль) в ДХМ (10 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: транс-4-(5-хлор-4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)-(2,2-диметил-пропил)карбамоил)пиразол-1-ил)-1-метилциклогексан-1-карбоновая кислота (276)
Соединение из Примера 276 (60 мг, 47%) получали в виде белого твердого вещества реакцией соединения 276-3 (130 мг, 0.224 ммоль) и LiOH (26.9 мг, 1.123 ммоль) в смеси ТГФ/EtOH/вода (11 мл, 5:5:1) по методике, аналогичной описанной в Примере 1. 1Н ЯМР (ДМСО-d6): присутствуют ротамеры δ 12.25 (1Н, ушир.с); 8.77 и 8.72 (2Н, 2х с); 7.79 и 7.73 (1Н, 2х с); 4.84 и 4.77 (2Н, 2х с); 4.36-4.28 (1Н, м); 2.02-1.72 (8Н, м); 1.21 (3Н, с); 0.95 и 0.74 (9Н, 2х с); LCMS (APCI): 545 (М+Н)+.
Пример 277
транс-4-(4-((2-(2,6-дихлор-4-метилфенил)-2-оксоэтил)((1r,3r,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
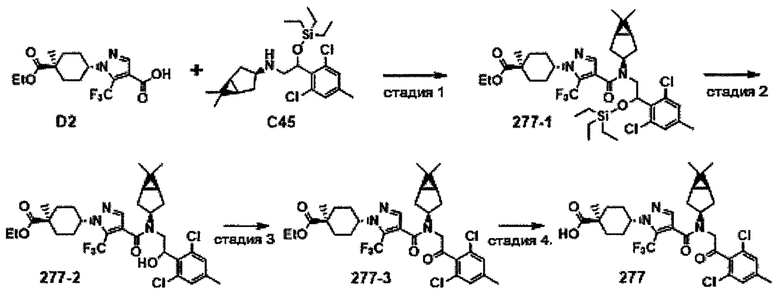
Стадия 1: этил транс-4-(4-((2-(2,6-дихлор-4-метилфенил)-2-((триэтилсилил)окси)этил)((1R,3r,5S)-6,б-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (277-1)
Соединение 277-1 получали реакцией амина С45 (74 мг, 0.167 ммоль), кислоты D2 (55 мг, 0.158 ммоль), HATU (72 мг, 0.190 ммоль) и DIPEA (0.055 мл, 0.320 ммоль) в ДМФА (2 мл) по методике, аналогичной описанной в Примере 1.
Стадия 2: этил транс-4-(4-((2-(2,6-дихлор-4-метилфенил)-2-гидроксиэтил)((1R,3r,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (277-2)
Соединение 277-2 (85 мг, 82% за 2 стадии) получали в виде желтого полутвердого вещества реакцией соединения 277-1 (неочищ.) и TBAF (0.3 мл, 0.3 ммоль, 1 М раствор в ТГФ) в ТГФ (2 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(4-((2-(2,6-дихлор-4-метилфенил)-2-оксоэтил)((1R,3r,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1H-пиразол-1-ил)-1-метилциклогексанкарбоксилат (277-3)
Соединение 277-3 (80 мг, 95%) получали в виде бледно-желтого полутвердого вещества реакцией соединения 277-2 (85 мг, 0.129 ммоль) и периодинана Десс-Мартина (66 мг, 0.155 ммоль) в ДХМ (2 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: транс-4-(4-((2-(2,6-дихлор-4-метилфенил)-2-((триэтилсилил)окси)этил)((1R,3r,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (277)
Соединение из Примера 277 (58 мг, 75%) получали в виде белого твердого вещества реакцией соединения 277-3 (80 мг, 0.122 ммоль) и 4 н. раствора LiOH (0.31 мл, 1.22 ммоль) в ТГФ/вода/МеОН (1.5 мл, 2:1:2) по методике, аналогичной описанной в Примере 1. 1Н ЯМР (CDCl3): присутствуют ротамеры δ 7.69 и 7.54 (1Н, 2х с); 7.16 и 7.08 (2Н, 2х с); 5.10-5.00 и 4.36-4.14 (2Н, 2х м); 4.59 и 4.39 (2Н, 2х с); 2.35 и 2.31 (3Н, 2х с); 2.31-1.85 (10Н, м); 1.42 и 1.39 (3Н, 2х с); 1.39-1.23 (2Н, м); 1.09-0.95 (8Н, м); LCMS (ESI): 628.3 (М+Н)+.
Пример 278
транс-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1R,3r,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
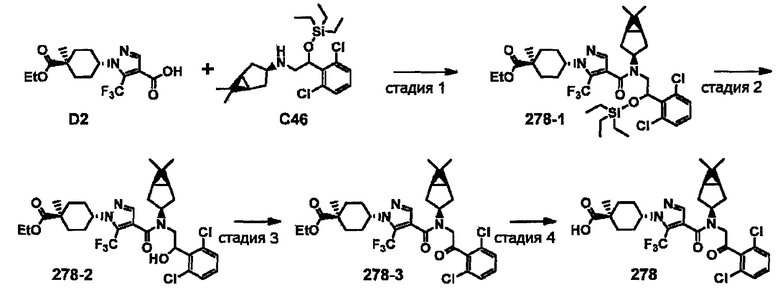
Стадия 1: этил транс-4-(4-((2-(2,6-дихлорфенил)-2-((триэтилсилил)окси)этил)((1R,3r,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (278-1)
Соединение 278-1 получали реакцией амина С46 (94 мг, 0.219 ммоль), кислоты D2 (76 мг, 0.219 ммоль), оксалилхлорида (0.038 мл, 0.438 ммоль), DIPEA (0.114 мл, 0.657 ммоль) и ДМФА (кат.) в ДХМ (1 мл) по методике, аналогичной описанной в Примере 2.
Стадия 2: этил транс-4-(4-((2-(2,6-дихлорфенил)-2-гидроксиэтил)((1R,3r,5S)-6,6-диметилбицикло [3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (278-2)
Соединение 278-2 (102 мг, 72% за 2 стадии) получали в виде бесцветного масла реакцией соединения 278-1 (неочищ.) и TBAF (0.22 мл, 0.22 ммоль, 1 М раствор в ТГФ) в ТГФ (1 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1R,3r,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (278-3)
Соединение 278-3 (84 мг, 83%) получали в виде белого твердого вещества реакцией соединения 278-2 (102 мг, 0.158 ммоль) и периодинана Десс-Мартина (100 мг, 0.237 ммоль) в ДХМ (1 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: транс-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1R,3r,5S)-6,6-диметил-бицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1H-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота (278)
Соединение из Примера 278 (35 мг, 44%) получали в виде белого твердого вещества реакцией соединения 278-3 (84 мг, 0.131 ммоль) и 4 н. раствора LiOH (0.33 мл, 1.31 ммоль) в смеси ТГФ/вода/МеОН (1.5 мл, 2:1:2) по методике, аналогичной описанной в Примере 1. 1H ЯМР (CDCl3): присутствуют ротамеры δ 7.70 и 7.55 (1Н, 2х с); 7.36-7.26 (3Н, м); 5.10-5.00 и 4.37-4.15 (2Н, 2х м); 4.61 и 4.42 (2Н, 2х с); 2.31-1.87 (10Н, м); 1.42 и 1.39 (3Н, 2х с); 1.39-1.23 (2Н, м); 1.09-0.95 (8Н, м); LCMS (ESI): 614.2 (М+Н)+.
Пример 330
транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)-((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)пиразол-1-ил)-1-метилциклогексан-1-карбоновая кислота
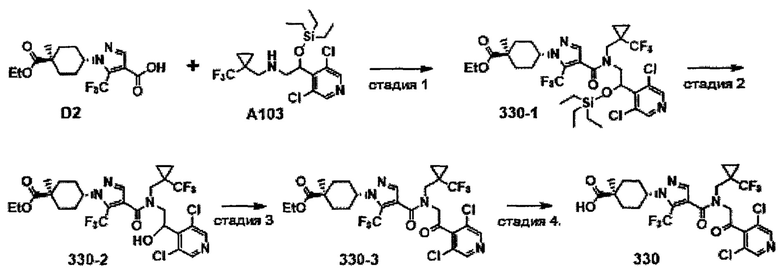
Стадия 1: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (330-1)
Соединение 330-1 получали реакцией амина А103 (44 мг, 0.098 ммоль), кислоты D2 (34 мг, 0.098 ммоль), оксалилхлорида (0.017 мл, 0.196 ммоль), 1н. раствора NaOH (0.49 мл, 0.491 ммоль) и ДМФА (кат.) в ДХМ (1 мл) по методике, аналогичной описанной в Примере 2.
Стадия 2: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (330-2)
Соединение 330-2 (37 мг, 58% за 2 стадии) получали в виде бесцветного масла реакцией соединения 330-1 (неочищ.) и TBAF (0.098 мл, 0.098 ммоль, 1 М раствор в ТГФ) в ТГФ (1 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (330-3)
Соединение 330-3 (31 мг, 83%) получали в виде бесцветного масла реакцией 330-2 (37 мг, 0.056 ммоль) и периодинана Десс-Мартина (36 мг, 0.085 ммоль) в ДХМ (1 мл) по методике, аналогичной описанной в Примере 1.
Стадия 4: транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)-((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)пиразол-1-ил)-1-метилциклогексан-1-карбоновая кислота (330)
Соединение из Примера 330 (17 мг, 58%) получали в виде белого твердого вещества реакцией соединения 330-3 (31 мг, 0.047 ммоль) и 4 н. раствора LiOH (0.12 мл, 0.467 ммоль) в ТГФ/вода/МеОН (0.75 мл, 2:1:2) по методике, аналогичной описанной в Примере 1. 1Н ЯМР (CDCl3): присутствуют ротамеры δ 8.59 и 8.52 (2Н, 2х с); 7.65 и 7.54 (1Н, 2х с); 4.95 и 4.58 (2Н, 2х с); 4.28-4.20 (1Н, м); 3.84 и 3.74 (2Н, 2х с); 2.26-2.16 (2Н, м); 1.95-1.85 (6Н, м); 1.42 и 1.41 (3Н, 2х с); 1.12-1.06 (4Н, м); LCMS (ESI): 629.2 (М+Н)+.
Пример 343
транс-4-(4-((2-(4-хлор-1Н-индол-3-ил)этил)((1R,3r,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1H-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
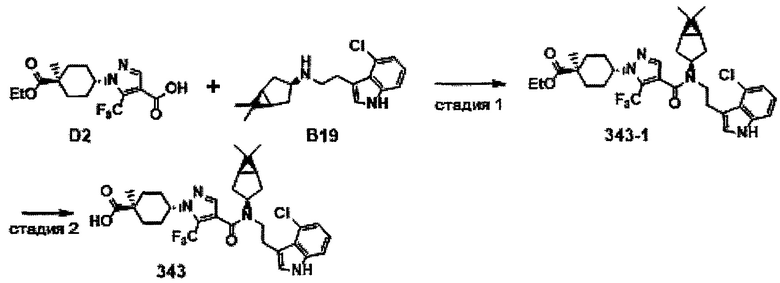
Стадия 1: этил транс-4-(4-((2-(4-хлор-1Н-индол-3-ил)этил)((1R,3r,5S)-6,6-диметил-бицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (343-1)
Соединение 343-1 (42 мг, 91%) получали в виде желтой смолы реакцией амина В19 (22 мг, 0.073 ммоль), кислоты D2 (25 мг, 0.073 ммоль), HATU (33 мг, 0.087 ммоль) и DIPEA (0.037 мл, 0.218 ммоль) в ДМФА (1 мл) по методике, аналогичной описанной в Примере 1.
Стадия 2: транс-4-(4-((2-(4-хлор-1Н-индол-3-ил)этил)((1R,3r,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота (343)
Соединение из Примера 343 (31 мг, 78%) получали в виде белого твердого вещества реакцией соединения 343-1 (42 мг, 0.066 ммоль) и 4 н. раствора LiOH (0.066 мл, 0.265 ммоль) в ТГФ/вода/МеОН (0.5 мл, 2:1:2) по методике, аналогичной описанной в Примере 1. 1H ЯМР (CDCl3): присутствуют ротамеры δ 8.20 (1Н, ушир.с); 7.53-6.90 (5Н, м); 4.68-4.61 и 4.29-4.02 (2Н, 2х м); 3.64-3.61 и 3.53-3.49 (2Н, 2х м); 3.35-3.32 и 3.04-3.00 (2Н, 2х м); 2.28-1.86 (8Н, м); 1.70-1.60 (2Н, м); 1.41 и 1.39 (3Н, 2х с); 1.28-0.85 (10Н, м); LCMS (ESI): 606.0 (М+Н)+.
Пример 361 и 362
транс-4-(4-(((2R)-2-(2,6-дихлор-4-фторфенил)-2-гидроксиэтил)-(4,4-диметилциклогексил)карбамоил)-5-(трифторметил)пиразол-1-ил)-1-метилциклогексан-1-карбоновая кислота или транс-4-(4-(((2S)-2-(2,6-дихлор-4-фторфенил)-2-гидроксиэтил)-(4,4-диметилциклогексил)карбамоил)-5-(трифторметил)пиразол-1-ил)-1-метилциклогексан-1-карбоновая кислота
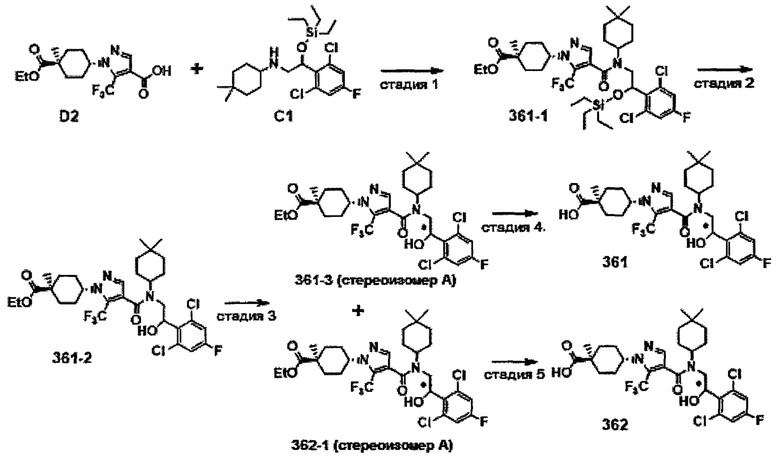
Стадия 1: этил транс-4-(4-((2-(2,6-дихлор-4-фторфенил)-2-((триэтилсилил)окси)этил)(4,4-диметилциклогексил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (361-1)
Соединение 361-1 получали реакцией амина С1 (89 мг, 0.199 ммоль), кислоты D2 (60 мг, 0.172 ммоль), оксалилхлорида (0.044 мл, 0.517 ммоль), DIPEA (0.090 мл, 0.517 ммоль) и ДМФА (кат.) в ДХМ (2 мл) по методике, аналогичной описанной в Примере 2.
Стадия 2: этил транс-4-(4-((2-(2,6-дихлор-4-метилфенил)-2-гидроксиэтил)(4,4-диметилциклогексил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (377-2)
Соединение 361-2 (100 мг, 88% за 2 стадии) получали реакцией 361-1 (неочищ.) и TBAF (0.26 мл, 0.258 ммоль, 1 М раствор в ТГФ) в ТГФ (2 мл) по методике, аналогичной описанной в Примере 1.
Стадия 3: этил транс-4-(4-(((S)-2-(2,6-дихлор-4-метилфенил)-2-гидроксиэтил)(4,4-диметилциклогексил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат и этил транс-4-(4-(((R)-2-(2,6-дихлор-4-метилфенил)-2-гидроксиэтил)(4,4-диметилциклогексил)карбамоил)-5-(трифторметил)-1H-пиразол-1-ил)-1-метилциклогексанкарбоксилат (361-3 и 362-1)
Соединение 361-2 (100 мг, 0.151 ммоль) очищали методом хиральной ВЭЖХ (250×20 мм DAICEL CHIRALPAK™ IA 5 мкм колонка с 16 мл/мин н-гексан/IPA (96/4)), получая 361-3 (49 мг, 49%) в качестве изомера, элюирующегося первым, и соединение 362-1 (48 мг, 48%) в качестве изомера, элюирующегося вторым.
Стадия 4: транс-4-(4-(((2R)-2-(2,6-дихлор-4-фторфенил)-2-гидроксиэтил)-(4,4-диметил-циклогексил)карбамоил)-5-(трифторметил)пиразол-1-ил)-1-метилциклогексан-1-карбоновая кислота или транс-4-(4-(((2S)-2-(2,6-дихлор-4-фторфенил)-2-гидроксиэтил)-(4,4-диметилциклогексил)карбамоил)-5-(трифторметил)пиразол-1-ил)-1-метилциклогексан-1-карбоновая кислота (361)
Соединение из Примера 361 (37 мг, 79%) получали в виде белого твердого вещества реакцией соединения 361-3 (49 мг, 0.074 ммоль) и 4 н. раствора LiOH (0.25 мл, 1.00 ммоль) в смеси EtOH/вода (1.5 мл, 2:1) по методике, аналогичной описанной в Примере 1. LCMS (ESI): 636.3 (М+Н)+.
Стадия 5: транс-4-(4-(((2R)-2-(2,6-дихлор-4-фторфенил)-2-гидроксиэтил)-(4,4-диметил-циклогексил)карбамоил)-5-(трифторметил)пиразол-1-ил)-1-метилциклогексан-1-карбоновая кислота или транс-4-(4-(((2S)-2-(2,6-дихлор-4-фторфенил)-2-гидроксиэтил)-(4,4-диметилциклогексил)карбамоил)-5-(трифторметил)пиразол-1-ил)-1-метилциклогексан-1-карбоновая кислота (362)
Соединение из Примера 362 (39 мг, 85%) получали в виде белого твердого вещества реакцией соединения 362-1 (48 мг, 0.073 ммоль) и 4 н. раствора LiOH (0.25 мл, 1.00 ммоль) в смеси EtOH/вода (1.5 мл, 2:1) по методике, аналогичной описанной в Примере 1. 1H ЯМР (CDCl3): присутствуют ротамеры δ 7.57 и 7.56 (1Н, 2х с); 7.11 и 7.05 (2Н, 2х д, J=7.8 Гц); 5.62-5.47 (1Н, м); 4.86 (1Н, ушир.с); 4.70-4.64 и 4.09-4.02 (1Н, 2х м); 4.30-4.20 (1Н, м); 3.46-3.26 (2Н, м); 2.31-2.18 (2Н, м); 1.99-1.66 (8Н, м); 1.47-1.26 (7Н, м); 1.12-0.86 (8Н, м); LCMS (ESI): 636.0 (M+H)+.
Пример 569
транс-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)(спиро[25]окт-6-илметил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
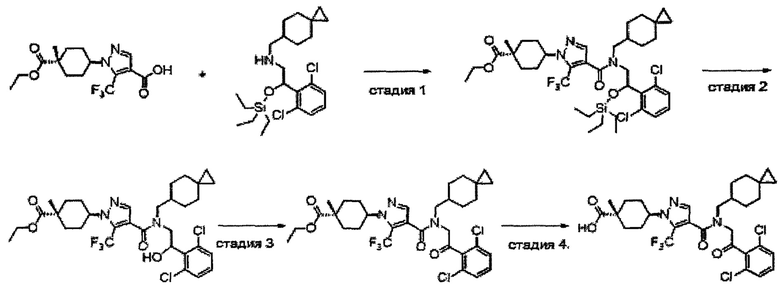
Стадия 1 и Стадия 2: этил транс-4-(4-((2-(2,6-дихлорфенил)-2-гидроксиэтил)(спиро[2.5] октан-6-илметил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
В раствор 1-((транс)-4-(этоксикарбонил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновой кислоты (0.157 г, 0.452 ммоль) и оксалилхлорида (0.049 мл, 0.565 ммоль) в ДХМ (4.52 мл) добавляли ДМФА (1 каплю), и полученную смесь перемешивали при комнатной температуре. Через 1 час реакционную смесь упаривали в вакууме. В остаток добавляли раствор 2-(2,6-дихлорфенил)-N-(спиро[2.5]октан-6-илметил)-2-((триэтилсилил)окси)этанамина (0.200 г, 0.452 ммоль) в ТГФ (4.5 мл), затем DEPEA (0.158 мл, 0.904 ммоль), и полученную смесь перемешивали при комнатной температуре. Через 17 часов в реакционную смесь добавляли раствор TBAF, 1.0 М раствор в ТГФ (0.904 мл, 0.904 ммоль), и полученную смесь перемешивали при комнатной температуре. Через 7 часов реакционную смесь разбавляли водой (50 мл) и насыщенным водным раствором поваренной соли (50 мл). Реакционную смесь экстрагировали этилацетатом (2×50 мл). Органический экстракт промывали насыщенным водным раствором NaCl (1×50 мл) и сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде светло-желтого сиропа. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 50% EtOAc в гептане, получая этил транс-4-(4-((2-(2,6-дихлорфенил)-2-гидроксиэтил)(спиро[2.5]октан-6-илметил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (0.0815 г, 0.124 ммоль, 27.4% выход) в виде бесцветной смолы.
Стадия 3: этил транс-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)(спиро[2.5]октан-6-илметил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
В раствор этил транс-4-(4-((2-(2,6-дихлорфенил)-2-гидроксиэтил)(спиро[2.5]октан-6-илметил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексан карбоксилата (0.0815 г, 0.124 ммоль) в ДХМ (1.238 мл) добавляли периодинан Десс-Мартина (0.079 г, 0.186 ммоль), и полученную смесь перемешивали при комнатной температуре. Через 6 часов смесь гасили насыщенным водным раствором Na2S2CO3 (50 мл) и насыщенным водным раствором NaHCO3 (50 мл). Реакционную смесь экстрагировали дихлорметаном (2×100 мл). Органический экстракт сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде белого твердого вещества. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 35% EtOAc в гептане, получая этил транс-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)(спиро[2.5]октан-6-илметил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (0.0685 г, 0.104 ммоль, 84% выход) в виде бесцветного сиропа.
Стадия 4: транс-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)(спиро[25]окт-6-илметил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
В раствор этил транс-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)(спиро[2.5]октан-6-илметил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексан карбоксилата (0.0685 г, 0.104 ммоль) в ТГФ (0.750 мл) добавляли раствор лития гидроксида гидрата (0.044 г, 1.043 ммоль) в воде (0.500 мл), и полученную смесь перемешивали и нагревали до 50°С в течение ночи. ТГФ и МеОН удаляли в вакууме, и оставшийся мутный раствор разбавляли водой (3 мл), получая прозрачный раствор. 1 М HCl добавляли для доведения рН до 1. Полученную смесь перемешивали 30 минут, после чего осадок отделяли фитрованием под вакуумом, получая белое твердое вещество. Полученное вещество очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 5% МеОН в ДХМ, получая Пример 569 (0.0457 г, 0.073 ммоль, 69.7% выход) в виде белого твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 7.40-7.79 (4Н, м), 5.30-5.54 (1Н, м), 4.87 (1Н, с), 4.64 (1Н, с), 4.22-4.36 (1Н, м), 3.50 (2Н, д, J=7.2 Гц), 2.12-2.30 (2Н, м), 1.54-1.98 (10Н, м), 1.27-1.41 (5Н, м), 0.78-1.05 (3Н, м), 0.07-0.37 (4Н, м), (присутствуют ротамеры); LCMS (ESI) m/z 628.2 (М+Н)+.
Пример 688
транс-4-(4-(((2R)-2-((3,5-дихлор-4-пиридинил)метил)-4,4-диметил-1-пирролидинил)карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
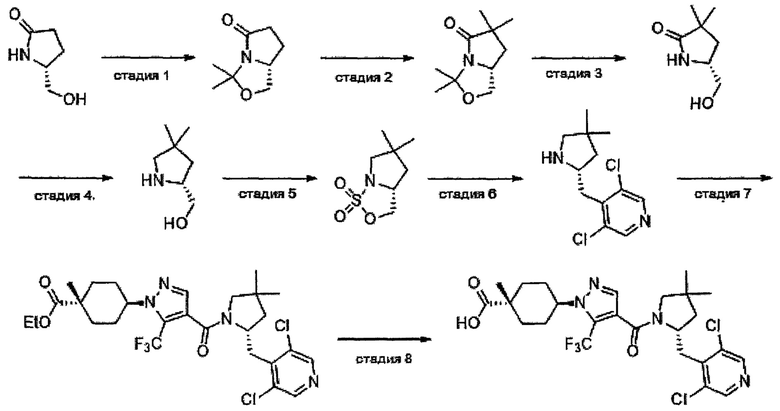
Стадия 1: Получено согласно заявке на патент WO 2013004290A1. В перемешиваемую суспензию (R)-(-)-5-(гидроксиметил)-2-пирролидинона (Sigma Aldrich Chemical Company, St. Louis, МО, 5.36 г, 46.5 ммоль) и п-толуолсульфокислоты (44 мг, 0.233 ммоль) в толуоле (100 мл) добавляли 2,2-диметоксипропан (17.1 мл, 140 ммоль), и реакционную смесь кипятили в течение 1.5 часов. Реакционную колбу снабжали насадкой Дина-Старка, затем добавляли еще 2,2-диметоксипропан (17.1 мл, 140 ммоль), и реакционную смесь кипятили в течение 36 часов. Растворитель упаривали, получая (R)-3,3-диметилтетрагидропирроло[1,2-с]оксазол-5(3Н)-он в виде оранжевого воскообразного твердого вещества. MS (ESI) 156.1 [М+Н]+. Полученный сырой продукт использовали в следующей стадии без дополнительной очистки.
Стадия 2: Получено согласно заявке на патент WO 2013004290A1. В раствор (R)-3,3-диметилтетрагидропирроло[1,2-с]оксазол-5(3Н)-она (3.50 г, 22.55 ммоль) в ТГФ (75 мл), охлажденный до -78°С, добавляли раствор лития диизопропиламида (2.0М раствор в смеси гептан/ТГФ/этилбензол, 20.30 мл, 40.6 ммоль). Полученный раствор перемешивали при этой температуре в течение 1 часа, после чего добавляли иодметан (2.12 мл, 33.8 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа, затем охлаждали до -78°С, после чего добавляли лития диизопропиламид (2.0 М раствор в смеси гептан/ТГФ/этилбензол, 20.30 мл, 40.6 ммоль). Полученную смесь перемешивали при -78°С в течение 1 часа, после чего добавляли еще иодметан (2.12 мл, 33.8 ммоль). Полученную смесь медленно нагревали до комнатной температуры и перемешивали в течение ночи (16 часов). Реакцию гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (2×75 мл). Объединенные органические экстракты промывали насыщенным водным раствором поваренной соли, сушили над MgSO4, фильтровали и упаривали, получая сырой (R)-3,3,6,6 тетраметилтетрагидропирроло[1,2-с]оксазол-5(3Н)-он в виде оранжевой смолы. MS (ESI) 184.1 [М+Н]+.
Стадия 3: Получено согласно заявке на патент WO 2013004290. В раствор (R)-3,3,6,6-тетраметилтетрагидропирроло[1,2-с]оксазол-5(3Н)-она (4.13 г, 22.54 ммоль) в МеОН (90 мл) добавляли п-толуолсульфокислоты моногидрат (0.429 г, 2.254 ммоль). Результирующую смесь кипятили в течение 2 часов. Растворитель удаляли при пониженном давлении (роторный испаритель), неочищенное вещество адсорбировали на слое силикагеля и очищали методом хроматографии на ISCO Combiflash™ RF (40 г Grace Reverlis колонка, применяя градиент 0-20% МеОН в ДХМ), получая (R)-5-(гидроксиметил)-3,3-диметилпирролидин-2-он (2.91 г, 20.31 ммоль, 90% выход) в виде белого полутвердого вещества. MS (ESI) 144.1 [М+Н]+.
Стадия 4: Получено согласно патенту США 20070032433 А1. В раствор (20-5-(гидроксиметил)-3,3-диметилпирролидин-2-она (2.91 г, 20.30 ммоль) в ТГФ (50.8 мл) охлажденный до 0°С, добавляли литийалюминий гидрид (2.0 М раствор в ТГФ, 12.18 мл, 24.36 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи (16 часов). Добавляли еще литийалюминий гидрид (2.0 М раствор в ТГФ, 12.18 мл, 24.36 ммоль), и раствор кипятили в течение 6 часов. Реакционную смесь охлаждали и добавляли еще литийалюминий гидрид (2.0 М раствор в ТГФ, 12.18 мл, 24.36 ммоль), и полученную смесь кипятили в течение ночи. Реакционную смесь охлаждали до 0°С в ледяной бане перед добавлением воды (3.67 мл), затем 15%-ного водного раствора NaOH (3.67 мл) и воды (10.9 мл). Затем интенсивно перемешивали при комнатной температуре в течение 1 часа и фильтровали через среднеплотный стеклянный фильтр с ватой и целитом, промывая EtOAc. Полученный раствор затем упаривали, получая сырой (R)-(4,4-диметилпирролидин-2-ил)метанол (2.29 г, 17.73 ммоль, 87% выход) в виде желтого вязкого масла. MS (ESI) 130.1 [М+Н]+.
Стадия 5: Раствор триэтиламина (4.94 мл, 35.4 ммоль) и (R)-(4,4-диметилпирролидин-2-ил)метанола (2.29 г, 17.72 ммоль) в ДХМ (89 мл) охлаждали до -78°С. В полученную смесь добавляли сульфурил хлорид (1.0 М раствор в ДХМ, 21.27 мл, 21.27 ммоль) в течение 15 секунд. Реакционную смесь выдерживали при этой температуре ~ 3 часа, оставляли нагреваться до комнатной температуры и перемешивали в течение ночи (16 час). Полученную смесь промывали водным 1 н. раствором HCl (30 мл × 2), насыщенным водным раствором поваренной соли (40 мл), сушили над MgSO4, фильтровали и упаривали, получая сырой продукт в виде коричнево-оранжевого масла, кристаллизующегося при стоянии. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом хроматографии на ISCO Combiflash™ RF (40 г колонка Grace Reverlis, применяя градиент 0-60% EtOAc в гептане), получая (R)-5,5-диметилтетрагидро-3Н-пирроло[1,2-с][1,2,3]оксатиазол 1,1-диоксид (708 мг, 3.70 ммоль, 21% выход) в виде белого кристаллического вещества. MS (ESI) 192.1 [М+Н]+.
Стадия 6: В раствор 3,5-дихлорпиридина (796 мг, 5.38 ммоль) в ТГФ (9.0 мл) при -78°С по каплям добавляли лития диизопропиламид (2.0 М раствор в смеси гептан/ТГФ/ этилбензол, 3.41 мл, 6.82 ммоль). После перемешивания в течение 1 часа при этой температуре, добавляли по каплям раствор (R)-5,5-диметилтетрагидро-3Н-пирроло[1,2-с][1,2,3]оксатиазол 1,1-диоксида (686 мг, 3.59 ммоль) в ТГФ (9.0 мл) при -78°С, и полученную смесь оставляли нагреваться до комнатной температуры на 3 часа, после чего перемешивали при комнатной температуре в течение 4 часов. После упаривания растворителя, к слученной бежевойпене добавляли горячий (80°С) 2н. раствор HCl (8 мл) и EtOH (8 мл) на ночь. Реакционную смесь упаривали при пониженном давлении (роторный испаритель), в полученную смесь добавляли лед, подщелачивали 5н. раствором NaOH (8 мл) и экстрагировали этилацетатом (2×75 мл). Органические экстракты сушили, упаривали и очищали методом хроматографии на ISCO Combiflash™ RF (25 г колонка Thomson SingleStep, применяя градиент 0-10% МеОН в ДХМ), получая (R)-3,5-дихлор-4-((4,4-диметилпирролидин-2-ил)метил)пиридин (748 мг, 2.89 ммоль, 80% выход) в виде оранжевого масла. MS (ESI) 259.1,261.0 [М+Н]+.
Стадия 7: В 1-((1R,4R)-4-(этоксикарбонил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновую кислоту (445 мг, 1.28 ммоль) добавляли ДХМ (8 мл) и три капли ДМФА, охлаждали до 0°С в ледяной бане и добавляли оксалилхлорид (0.16 мл, 1.82 ммоль) медленно по каплям. Реакционную смесь вынимали из ледяной бани и оставляли перемешиваться при комнатной температуре на 1.5 часа. Летучие компоненты удаляли при пониженном давлении (роторный испаритель), к полученному сырому ацилхлориду добавляли ДХМ (10 мл), охлаждали в ледяной бане и медленно добавляли (R)-3,5-дихлор-4-((4,4-диметилпирролидин-2-ил)метил)пиридин (315 мг, 1.22 ммоль) (в ДХМ 5 мл) по каплям, затем DIPEA (0.64 мл, 3.65 ммоль). Полученный раствор вынимали из ледяной бани, оставляли нагреваться до комнатной температуры и перемешивали 1 час. Растворитель упаривали, и неочищенное вещество адсорбировали на слое силикагеля и очищали методом хроматографии на ISCO Combiflash™ RF (40 г колонка Thomson SingleStep, применяя градиент 0-40% EtOAc в гептане), получая (1R,4R)-этил 4-(4-((R)-2-((3,5-дихлорпиридин-4-ил)метил)-4,4-диметилпирролидин-1-карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (489 мг, 0.83 ммоль, 68% выход) в виде светло-желтого аморфного твердого вещества после сушки в вакуумной печи в течение 48 часов при 40°С. MS (ESI) 589.3/591.2 [М+Н]+.
Стадия 8: В смесь (1R,4R)-этил 4-(4-((R)-2-((3,5-дихлорпиридин-4-ил)метил)-4,4-диметилпирролидин-1-карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилата (464 мг, 0.787 ммоль) в ТГФ (3.9 мл) и МеОН (3.9 мл) добавляли лития гидроксид моногидрат (1.0 М водный раствор, 3.9 мл, 3.94 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи (16 час). Органику удаляли при пониженном давлении (роторный испаритель), и водный раствор подкисляли добавлением 1 н. раствором HCl, что приводило к образованию осадка. Полученную смесь экстрагировали этилацетатом (2×40 мл). Объединенные экстракты промывали насыщенным водным раствором поваренной соли, сушили над безводным MgSO4, фильтровали и упаривали. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом хроматографии на ISCO Combiflash™ RF (40 г колонка Thomson SingleStep, применяя градиент 0-8% МеОН в ДХМ), получая (1R,4R)-4-(4-((R)-2-((3,5-дихлорпиридин-4-ил)метил)-4,4-диметилпирролидин-1-карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновую кислоту (258 мг, 0.46 ммоль, 59% выход) в виде белой аморфной пены. MS (ESI) 561.0, 563.1 [М+Н]+.
Пример 692
транс-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
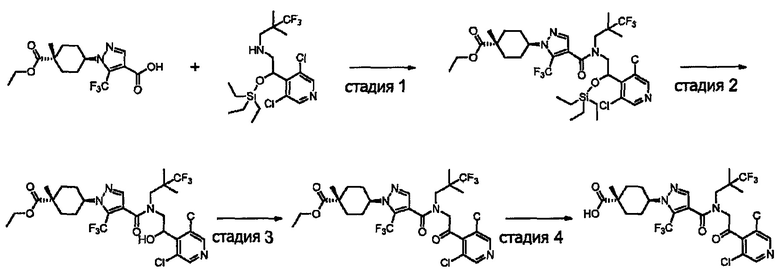
Стадия 1 и Стадия 2: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
В немного мутный раствор 1-((1R,4R)-4-(этоксикарбонил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновой кислоты (1.0102 г, 2.90 ммоль) в ДХМ (29.0 мл) добавляли оксалилхлорид (0.307 мл, 3.63 ммоль), затем ДМФА (1 каплю), и светло-желтую немного мутную реакционную смесь перемешивали при комнатной температуре. Через 3 часа смесь упаривали в вакууме, получая этил транс-4-(4-(хлоркарбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат в виде светло-желтого сиропа. В остаток добавляли раствор N-(2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил) окси)этил)-3,3,3-трифтор-2,2-диметилпропан-1-амина (1.292 г, 2.90 ммоль) в ТГФ (29.0 мл), затем DIPEA (2.021 мл, 11.60 ммоль), и полученную смесь перемешивали при комнатной температуре. Через 19 часов LCMS (ESI) показал, что образовался этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат: LCMS (ESI) m/z 775.1 (М+Н)+.
В реакционную смесь добавляли раствор TBAF, 1.0 М раствор в ТГФ (11.60 мл, 11.60 ммоль), и полученную смесь перемешивали при комнатной температуре. Через 30 минут LC-MS (ESI) показал завершение реакции. Реакционную смесь разбавляли водой (100 мл) и насыщенным водным раствором поваренной соли (100 мл). Реакционную смесь экстрагировали этилацетатом (2×100 мл). Органический экстракт промывали насыщенным водным раствором NaCl (1×100 мл) и сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде светло-желтого сиропа. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 50% EtOAc в гептане, получая этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (1.6938 г, 2.56 ммоль, 88% выход) в виде белого резинообразного твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8.46-8.63 (2Н, м), 7.71-7.83 (1Н, м), 6.11 (1Н, д, J=4.1 Гц), 5.19-5.33 (1Н, м), 4.27 (1Н, т, J=11.0 Гц), 4.09 (2Н, кв, J=7.2 Гц), 3.39-3.97 (4Н, м), 2.02-2.19 (2Н, м), 1.66-1.97 (6Н, м), 1.14-1.30 (12Н, м), В ЯМР содержится несколько наборов пиков из-за наличия диастереомеров и ротамеров; LCMS (ESI) m/z 661.1 (М+Н)+.
Стадия 3: этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
В прозрачный раствор этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилата (1.6826 г, 2.54 ммоль) в ДХМ (25.4 мл) добавляли периодинан Десс-Мартина (1.618 г, 3.82 ммоль). Полученную белую мутную смесь перемешивали при комнатной температуре. Через 1 час смесь гасили насыщенным водным раствором Na2S2O3 (50 мл) и насыщенным водным раствором NaHCO3 (50 мл). Реакционную смесь экстрагировали дихлорметаном (2×100 мл). Органический экстракт сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде белого твердого вещества. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 30% EtOAc в гептане, получая этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (1.5825 г, 2.400 ммоль, 94% выход) в виде белого резинообразного твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8.45-8.64 (2Н, м), 7.51-7.78 (1Н, м), 4.52 (2Н, с), 4.09-4.30 (3Н, м), 3.70 (2Н, ушир.с), 2.12-2.32 (2Н, м), 1.79-2.00 (6Н, м), 1.02-1.46 (12Н, м), присутствуют ротамеры; LCMS (ESI) m/z 659.0 (М+Н)+.
Стадия 4: транс-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
В прозрачную смесь этил транс-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилата (1.5739 г, 2.387 ммоль) в ТГФ (9.55 мл), EtOH (9.55 мл) и воде (4.77 мл) добавляли 2 М раствор LiOH в воде (11.93 мл, 23.87 ммоль). После добавления 2 М раствора LiOH, белая гетерогенная смесь становилась желтой мутной смесью. Полученную желтую мутную смесь перемешивали и нагревали до 60°С. Через 15 часов реакционную смесь упаривали в вакууме для удаления ТГФ и EtOH. Результирующий водный раствор разбавляли водой (30 мл). Значение рН раствора доводили до ~3.0 1 н. раствором HCl, и выпавший осадок отделяли фильтрованием под вакуумом, промывали водой и лиофилизовывали в течение ночи, получая Пример 692 (1.3955 г, 2.210 ммоль, 93% выход) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12.27 (1Н, ушир.с), 8.58-8.83 (2Н, м), 7.75-8.02 (1Н, м), 4.68-5.43 (2Н, м), 4.26 (1Н, т, J=11.0 Гц), 3.46-3.90 (2Н, м), 1.97-2.17 (2Н, м), 1.69-1.92 (6Н, м), 1.00-1.39 (9Н, м), присутствуют ротамеры; LCMS (ESI) m/z 631.0 (М+Н)+.
Пример 713
(1R,4R)-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((5-фторспиро[2.3]гексан-5-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
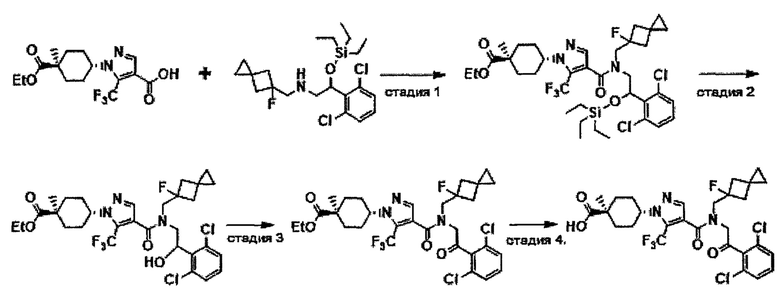
Стадия 1: этил транс-4-(4-((2-(2,6-дихлор-4-фторфенил)-2-((триэтилсилил)окси)этил)(3,5-дифторбензил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
1-((1R,4R)-4-(этоксикарбонил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновую кислоту (3.48 г, 9.99 ммоль) растворяли в ДХМ (30 мл) и добавляли тионил хлорид (0.875 мл, 11.99 ммоль), затем 1 каплю ДМФА. Реакционную смесь кипятили 2.5 часа. Растворители удаляли в вакууме, и остаток помещали в морозильник в течение ночи. Затвердевшее вещество затем сушили в вакууме в течение 1 часа, получая (1S,4S)-этил 4-(4-(хлоркарбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат. 2-(2,6-Дихлорфенил)-N-((5-фторспиро[2.3]гексан-5-ил)метил)-2-((триэтилсилил)окси)этанамин (150 мг, 0.347 ммоль) растворяли в 2 мл ДХМ и добавляли (1S,4S)-этил 4-(4-(хлоркарбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (127 мг, 0.347 ммоль), растворенный в 2 мл ДХМ, затем добавляли триэтиламин (242 мкл, 1.734 ммоль). Полученный раствор перемешивали в течение 1 часа и упаривали, получая сырой (1R,4R)-этил 4-(4-((2-(2,6-дихлорфенил)-2-((триэтилсилил)окси)этил)((5-фторспиро[2.3]гексан-5-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (265 мг, 0.347 ммоль, 100% выход).
Стадия 2: (1R,4R)-этил 4-(4-((2-(2,6-дихлорфенил)-2-гидроксиэтил)((5-фторспиро[2.3]гексан-5-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
В перемешиваемый раствор (1R,4R)-этил 4-(4-((2-(2,6-дихлорфенил)-2-((триэтилсилил)окси)этил)((5-фторспиро[2.3]гексан-5-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилата (265 мг, 0.347 ммоль) в 2 мл ТГФ добавляли TBAF (695 мкл, 0.695 ммоль), и полученную смесь перемешивали 1 час. Реакционную смесь гасили насыщенным водным раствором NH4Cl и экстрагировали этилацетатом. Объединенные органические слои промывали водой и насыщенным водным раствором поваренной соли, сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая сырой (1R,4R)-этил 4-(4-((2-(2,6-дихлорфенил)-2-гидроксиэтил)((5-фторспиро[2.3]гексан-5-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (225 мг, 0.347 ммоль, 100% выход). MS w/z=648[M+H]+.
Стадия 3: (1R,4R)-этил 4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((5-фторспиро[2.3]гексан-5-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
(1R,4R)-этил 4-(4-((2-(2,6-дихлорфенил)-2-гидроксиэтил)((5-фторспиро[2.3]гексан-5-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексан карбоксилат (225 мг, 0.347 ммоль) растворяли в 10 мл ДХМ и добавляли периодинан Десс-Мартина (184 мг, 0.434 ммоль). Полученный раствор перемешивали 1 час. Полученный раствор гасили 5%-ным раствором Na2S2O3, промывали насыщенным NaHCO3, сушили над Na2SO4 и упаривали. Полученный продукт очищали колоночной хроматографией на силикагеле (40 г колонка) с 0-100% EtOAc в гептане, получая (1R,4R)-этил 4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((5-фторспиро[2.3]гексан-5-ил)метил)карбамоил)-5-(трифторметил)-1H-пиразол-1-ил)-1-метилциклогексанкарбоксилат (140 мг, 0.217 ммоль, 62.4% выход). MS m/z=646 [М+Н]+.
Стадия 4: (1R,4R)-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((5-фторспиро[2.3]гексан-5-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
(1R,4R)-этил 4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((5-фторспиро[2.3]гексан-5-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексан карбоксилат (140 мг, 0.217 ммоль) и гидроксид лития (100 мг, 4.18 ммоль) смешивали в 5 мл МеОН, 5 мл ТГФ и 2 мл воды. Полученный раствор нагревали до 50°С на 3 часа. Полученный раствор подкисляли 6 н. раствором HCl и разбавляли водой. Полученный продукт экстрагировали этилацетатом, сушили над Na2SO4, фильтровали и упаривали, получая (1R,4R)-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((5-фторспиро[2.3]гексан-5-ил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновую кислоту (115 мг, 0.186 ммоль, 86% выход). 1H ЯМР (400 МГц, CD3OD, смесь ротамеров) δ 7.88 (с, 0.2Н) 7.66 (с, 0.8Н) 7.40-7.50 (м, 3Н) 4.11 (м, 3Н) 2.43-2.61 (м, 2Н) 2.15-2.32 (м, 3Н) 1.81-2.05 (м, 7Н) 1.17-1.43 (м, 5Н) 0.42-0.70 (м, 4Н) LC/MS (ESI+) m/z=618 (М+Н)+.
Пример 716
транс-4-(4-((2-(2,6-дихлор-4-фторфенил)-2-оксоэтил)((1-метилциклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
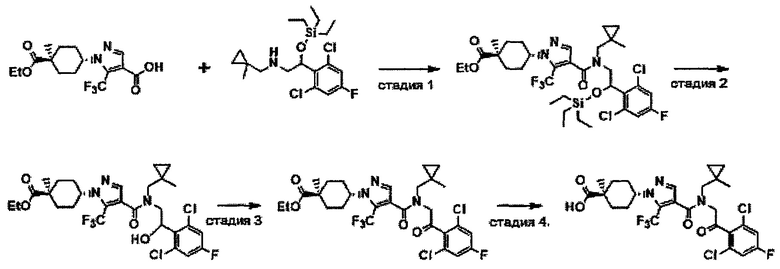
Стадия 1: этил транс-4-(4-((2-(2,6-дихлор-4-фторфенил)-2-((триэтилсилил)окси)этил)((1-метилциклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
Данное соединение получали по методике, аналогичной описанной в примере 1, стадия 1, без хроматографической очистки. LCMS (ESI) m/z 735.8 (М+Н)+.
Стадия 2: этил транс-4-(4-((2-(2,6-дихлор-4-фторфенил)-2-гидроксиэтил)((1-метилцикло-пропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
Данное соединение получали по методике, аналогичной описанной в примере 1, стадия 2, без хроматографической очистки. LCMS (ESI) m/z 623.9 (М+Н)+.
Стадия 3: этил транс-4-(4-((2-(2,6-дихлор-4-фторфенил)-2-оксоэтил)((1-метилцикло-пропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
Данное соединение получали по методике, аналогичной описанной в примере 1, стадия 3. 1H ЯМР (500 МГц, CDCl3): присутствуют ротамеры δ 7.65 и 7.58 (2 х с, 1Н), 7.16 и 7.15 (2 х с, 1Н), 7.09 и 7.08 (2 х с, 1Н), 4.98 (с, 1Н), 4.60 (с, 1Н), 4.22-4.33 (м, 1Н), 4.15-4.22 (м, 2Н), 3.36 (с, 1Н), 2.15-2.31 (м, 2Н), 1.84-1.99 (м, 6Н), 1.39 и 1.37 (2 х с, 3Н), 1.30 (тд, J=7.09, 2.32 Гц, 3Н), 1.12 и 0.98 (2 х с, 3Н), 0.48-0.56 (м, 1Н), 0.34-0.43 (м, 3Н); LCMS (ESI) m/z 619.8 (М+Н)+.
Стадия 4: транс-4-(4-((2-(2,6-дихлор-4-фторфенил)-2-оксоэтил)((1-метилциклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
Данное соединение получали в виде белого твердого вещества по методикам, аналогичным описанным в примере 1, стадия 4. 1H ЯМР (500 МГц, ДМСО-d6): присутствуют ротамеры δ 12.29 (с, 1Н), 7.52-7.82 (м, 3Н), 5.17 и 4.87 (2 х с, 1Н), 4.69 (с, 1Н), 4.18-4.32 (м, 1Н), 3.52 и 3.38 (2 х с, 1Н), 3.32 и 3.22 (2 х с, 1Н), 2.00-2.16 (м, 2Н), 1.73-1.91 (м, 6Н), 1.24 и 1.08 (2 х с, 3Н), 1.05 и 0.91 (2 х с, 3Н), 0.44-0.57 (м, 1Н), 0.23-0.38 (м, 3Н); LCMS (ESI) m/z 592.1 (М+Н)+.
Пример 729
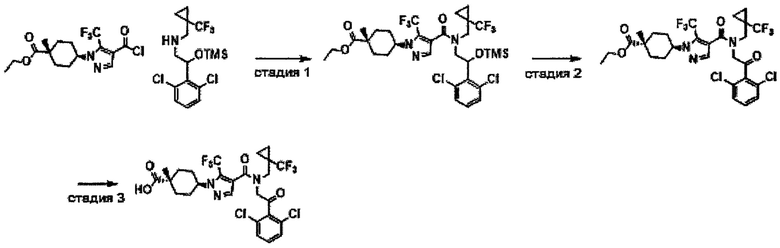
Стадия 1: (1R,4R)-этил 4-(4-((2-(2,6-дихлорфенил)-2-((триметилсилил)окси)этил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат.
В раствор 2-(2,6-дихлорфенил)-N-((1-(трифторметил)циклопропил)метил)-2-((триметилсилил)окси)этанамина (0.15 г, 0.375 ммоль) в ДХМ (3 мл) добавляли этил 4-(4-(хлоркарбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (0.137 г, 0.375 ммоль), затем триэтиламин (0.104 мл, 0.749 ммоль) и перемешивали при комнатной температуре 15 минут. Реакционную смесь загружали на 25-граммовую колонку (MPLC) и элюировали смесью гексан : EtOAc (0-50%), получая (1R,4R)-этил 4-(4-((2-(2,6-дихлорфенил)-2-((триметилсилил)окси)этил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (0.177 г, 0.242 ммоль, 65%) в виде прозрачного масла.
Стадия 2: (1R,4R)-этил 4-(4-((2-(2,6-дихлорфенил)-2-гидроксиэтил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
В раствор (1R,4R)-этил 4-(4-((2-(2,6-дихлорфенил)-2-((триметилсилил)окси)этил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилата (0.177 г, 0.242 ммоль) в 2-Ме-ТГФ (0.808 мл) добавляли тетр-н-бутиламмония фторид (0.291 мл, 0.291 ммоль). Полученную смесь перемешивали при комнатной температуре 1 час. Реакционную смесь гасили насыщенным водным раствором NH4Cl (1 мл) и разбавляли добавлением EtOAc (50 мл) и воды (20 мл). Органический слой упаривали при пониженном давлении, получая (1R,4R)-этил 4-(4-((2-(2,6-дихлорфенил)-2-гидроксиэтил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат в виде не совсем белого твердого вещества. Его растворяли в ДХМ (3 мл), добавляли периодинан Десс-Мартина (0.134 г, 0.315 ммоль), и реакционную смесь перемешивали при комнатной температуре 16 часов. Затем в смесь добавляли Na2S2O3 (5 мл), затем насыщенный раствор NaHCO3 (2 мл) и ДХМ (20 мл) и перемешивали 15 минут. Органический слой пропускали через разделитель фаз и упаривали. Сырой продукт очищали методом MPLC (25 г колонка) и элюировали смесью гексан : EtOAc (10-40%), получая (1R,4R)-этил 4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (0.13 г, 82%) в виде твердого белого аморфного вещества.
Стадия 3: (1R,4R)-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1-(трифторметил)циклопропил) метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота.
В раствор (1R,4R)-этил 4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилата (0.13 г, 0.198 ммоль) в 2Ме-ТГФ (0.660 мл), МеОН (0.660 мл) и воде (0.660 мл) добавляли гидроксид лития (0.047 г, 1.980 ммоль) и перемешивали при 40°С 1 час. Реакционную смесь подкисляли добавлением 2 н. раствор HCl до рН 2 и экстрагировали этилацетатом (2×30 мл). Органический слой сушили над безводным Na2SO4, фильтровали и упаривали, получая (1R,4R)-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновую кислоту (0.1 г, 75%) в виде твердого белого аморфного вещества. 1Н ЯМР δ (ДМСО-d6): присутствуют ротамеры 12.22 (1Н, ушир.с); 9.79 (1Н, 2х с); 7.69 и 7.67 (1Н, 2х с); 7.55 и 7.54 (1Н, 2х с); 7.46 и 7.44 (1Н, 2х с); 5.19 (1Н, м); 4.30-4.20 (2Н, м); 3.78 (2Н, м); 2.28-2.20 (2Н, м); 2.18-1.98 (3Н, м); 1.88-1.47 (8Н, м); 1.24 и 1.23 (3Н, 2х с) LCMS (ESI): 628.0 (М+Н)+.
Пример 759
транс-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1-метилциклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота

Стадия 1: транс-этил 4-(4-((2-(2,6-дихлорфенил)-2-гидроксиэтил)((1-метилциклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
В раствор 1-(транс-4-(этоксикарбонил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновой кислоты (2.88 г, 8.27 ммоль) в ДХМ добавляли тионил хлорид (0.663 мл, 9.10 ммоль), затем 1 каплю ДМФА. На колбу затем помещали обратный холодильник, и полученную смесь перемешивали 4 часа при 40°С, затем перемешивали в течение ночи при комнатной температуре. Растворители удаляли в вакууме, и остаток сушили в вакууме, получая транс-этил 4-(4-(хлоркарбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (2.87 г, 95% выход), который использовали без дополнительной очистки. В раствор 2-(2,6-дихлорфенил)-N-((1-метилциклопропил)метил)-2-((триэтилсилил)окси)этанамина (95 мг, 0.245 ммоль) в ДХМ (1.2 мл) добавляли DIPEA (85 мкл, 0.489 ммоль) и транс-этил 4-(4-(хлоркарбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (90 мг, 0.245 ммоль). Через 45 минут добавляли TBAF (1 М раствор в ТГФ) (905 мкл, 0.905 ммоль). Через 2 часа добавляли в реакционную смесь 1 М водный раствор HCl. Органический слой отделяли, и водный слой экстрагировали дихлорметаном. Объединенные органические слои промывали насыщенным водным раствором NaHCO3, сушили над Na2SO4 и упаривали, получая сырой транс-этил 4-(4-((2-(2,6-дихлорфенил)-2-гидроксиэтил)((1-метилциклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (206 мг), который использовали в следующей стадии без очистки.
Стадия 2: транс-этил 4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1-метилциклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
В раствор транс-этил 4-(4-((2-(2,6-дихлорфенил)-2-гидроксиэтил)((1-метилциклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилата (206 мг, 0.341 ммоль) в ДХМ (3.5 мл) добавляли периодинан Десс-Мартина (217 мг, 0.511 ммоль). Через 40 минут добавляли 1 М водный раствор Na2S2O3 и насыщенный раствором NaHCO3. Полученную смесь перемешивали в течение 1 часа, органический слой отделяли, водный слой экстрагировали дихлорметаном. Объединенные органические слои упаривали. Остаток от упаривания очищали методом препаративной ТСХ, элюируя 30% EtOAc/гексан и получая транс-этил 4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1-метилциклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (100 мг, 0.166 ммоль, 48.7% выход).
Стадия 3: транс-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1-метилциклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
В смесь транс-этил 4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1-метилциклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилата (100 мг, 0.166 ммоль) в МеОН (1.5 мл), ТГФ (1.5 мл) и воде (1 мл) добавляли лития гидроксид моногидрат (69 мг, 1.66 ммоль). Полученную смесь нагревали при 50°С 90 минут. Большую часть МеОН и ТГФ удаляли в вакууме. Полученную смесь доводили до рН 1 1 М водным раствором HCl. Полученную смесь перемешивали 15 минут, выпавший осадок отфильтровывали, промывали водой и сушили в вакууме, получая транс-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1-метилциклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновую кислоту (82 мг, 0.143 ммоль, 86% выход). 1H ЯМР (400 МГц, ДМСО-d6): смесь ротамеров и кето-енольных таутомеров δ 12.24 (ушир.с, 1Н), 9.59 (с, 0.2Н), 7.80 (с, 0.2Н), 7.73 (с, 0.55Н), 7.72 (с, 0.25Н), 7.32-7.63 (м, 3Н), 5.15 (с, 0.2Н), 4.88 (ушир.с, 0.5Н), 4.70 (ушир.с, 1.1Н), 4.15-4.35 (м, 1Н), 3.52 (с, 0.4Н), 3.32 (с, 1.1Н), 3.22 (с, 0.5Н), 1.99-2.16 (м, 2Н), 1.70-1.93 (м, 6Н), 1.21-1.28 (м, 3Н), 0.88-1.11 (м, 3Н), 0.45-0.58 (м, 2Н), 0.23-0.35 (м, 2Н). LCMS (APCI): 574.3 (М+Н)+.
Пример 760
транс-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1-метилциклобутил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
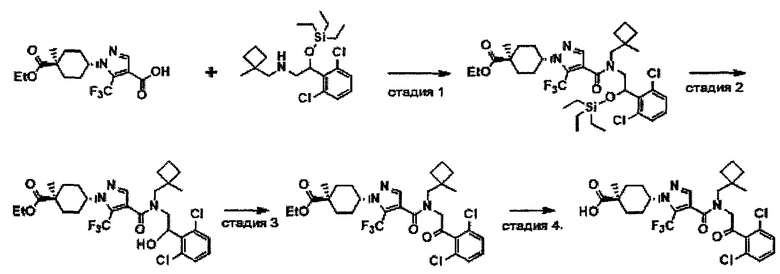
Стадия 1: этил транс-4-(4-((2-(2,6-дихлорфенил)-2-((триэтилсилил)окси)этил)((1-метилциклобутил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
Данное соединение получали по методике, аналогичной описанной в примере 1, стадия 1, без хроматографической очистки.
Стадия 2: этил транс-4-(4-((2-(2,6-дихлорфенил)-2-гидроксиэтил)((1-метилциклобутил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
Данное соединение получали по методике, аналогичной описанной в примере 1, стадия 2, без хроматографической очистки. LCMS (ESI) m/z 618.3 (М+Н)+.
Стадия 3: этил транс-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1-метилциклобутил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
Данное соединение получали по методике, аналогичной описанной в примере 1, стадия 3. 1H ЯМР (500 МГц, ДМСО-d6): присутствуют ротамеры δ 7.44-7.77 (м, 4Н), 4.63 (2 х с, 2Н), 4.26 (м, 1Н), 4.09 (кв, J=7.13 Гц, 2Н), 3.48 (ушир.с, 2Н), 1.98-2.13 (м, 4Н), 1.82-1.96 (м, 3Н), 1.74-1.82 (м, 5Н), 1.58-1.67 (м, 2Н), 1.14-1.28 (м, 9Н); LCMS (ESI) m/z 616.3 (М+Н)+.
Стадия 4: транс-4-(4-((2-(2,6-дихлорфенил)-2-оксоэтил)((1-метилциклобутил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
Данное соединение получали в виде белого твердого вещества по методике, аналогичной описанной в примере 1, стадия 4. 1H ЯМР (500 МГц, ДМСО-d6): присутствуют ротамеры δ 12.18 (ушир.с, 1Н), 9.58 (д, J=7.10 Гц, 1Н), 7.66 (с, 1Н), 7.27-7.48 (м, 3Н), 5.05 (д, J=111 Гц, 1Н), 4.09-4.22 (м, 1Н), 3.55-3.61 (м, 1Н), 1.91-2.05 (м, 4Н), 1.67-1.85 (м, 8Н), 1.44-1.61 (м, 2Н), 1.16 (с, 3Н), 1.10 (с, 3Н); LCMS (ESI) m/z 588.3 (М+Н)+.
Пример 785
(1S,4S)-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2,2-диметилциклогексанкарбоновая кислота
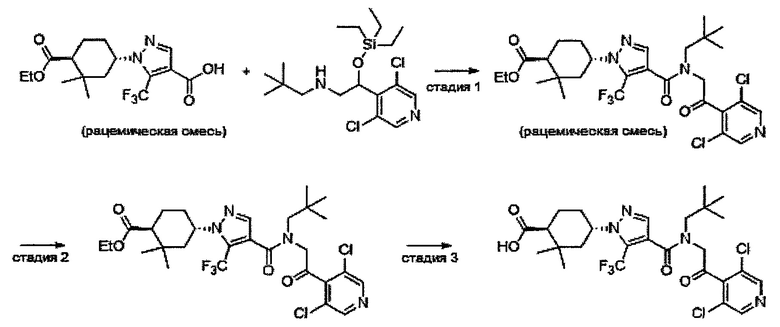
Стадия 1: Этил танс-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2,2-диметилциклогексанкарбоксилат (рацемическая смесь).
Оксалилхлорид (64 мкл, 0.72 ммоль) и ДМФА (1 каплю) последовательно добавляли в перемешиваемый раствор транс-1-(4-(этоксикарбонил)-3,3-диметилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновой кислоты (0.20 г, 0.55 ммоль; рацемическая смесь) в ДХМ (5.5 мл). После перемешивания в течение 2 часов реакционную смесь упаривали при пониженном давлении. Остаток от упаривания растворяли в ТГФ (4.5 мл) и добавляли раствор N-(2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)-2,2-диметилпропан-1-амина (0.22 г, 0.55 ммоль) в ТГФ (1.0 мл), затем DIPEA (0.29 мл, 1.7 ммоль). После перемешивания в течение 30 минут, добавляли TBAF (1.7 мл 1.0 М раствора в ТГФ, 1.7 ммоль). После перемешивания в течение 1 часа, реакционную смесь разделяли между EtOAc и насыщенным водным раствором NaHCO3, слои разделяли, органическую часть промывали последовательно насыщенным водным раствором NaHCO3 (2×) и насыщенным водным раствором поваренной соли, сушили (Na2SO4), фильтровали, и фильтрат упаривали при пониженном давлении. Остаток от упаривания растворяли в ДХМ (5.5 мл), и в полученный раствор добавляли периодинан Десс-Мартина (0.26 г, 0,60 ммоль). После перемешивания в течение 10 минут, реакционную смесь упаривали при пониженном давлении, остаток разделяли между ТГФ-EtOAc (1:1 об/об) и насыщенным водным раствором NaHCO3, слои разделяли, органическую часть промывали последовательно насыщенным водным раствором NaHCO3 и насыщенным водным раствором поваренной соли, сушили (Na2SO4), фильтровали, и фильтрат упаривали при пониженном давлении. Остаток от упаривания растворяли в ДХМ, силикагель (1.0 г) добавляли в раствор, и летучие компоненты удаляли при пониженном давлении. Остаток от упаривания очищали колоночной хроматографией на силикагеле (элюирование в градиенте; от 9:1 до 4:1 гексан-EtOAc), получая этил транс-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2,2-диметилциклогексанкарбоксилат (0.27 г, 80% общий выход; рацемическая смесь) в виде бесцветного твердого вещества.
Стадия 2: Этил (1S,4S)-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(2,2-диметилпропил) карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2,2-диметилциклогексанкарбоксилат.
Этил транс-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2,2-диметилциклогексанкарбоксилат (0.22 г, со Стадии 1; рацемическая смесь) разделяли с применением препаративной высокоэффективной жидкостной хроматографии (CHERALPAK™ AD-H колонка от Chiral Technologies, Inc., West Chester, PA (250 мм × 30 мм, 5 мкм колонка), элюируя смесью гептан/EtOH (90:10 об/об) при скорости потока 50 мл/мин), получая два продукта с энантиомерным избытком более 97%.
Пик 1: Этил (1R,4R)-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2,2-диметилциклогексанкарбоксилат (0.10 г) в виде бесцветного твердого вещества. Пик 2: Этил (1S,4S)-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2,2-диметилциклогексанкарбоксилат (0.098 г) в виде бесцветного твердого вещества.
Стадия 3:
(1S,4S)-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2,2-диметилциклогексанкарбоновая кислота
NaOH (1.6 мл 1.0 М водного раствора, 1.6 ммоль) добавляли в перемешиваемый раствор этил (1S,4S)-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(2,2-диметилпропил) карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2,2-диметилциклогексанкарбоксилата (0.098 г, 0.16 ммоль, со Стадии 2) в ТГФ (1.6 мл) и EtOH (1.6 мл), и затем реакционную смесь нагревали до 60°С. После перемешивания в течение 40 часов, реакционную смесь оставляли охлаждаться до комнатной температуры и затем упаривали при пониженном давлении. Остаток от упаривания растворяли водой (10 мл), добавляли в раствор концентрированную соляную кислоту (10 капель), результирующую гетерогенную смесь фильтровали, осадок на фильтре промывали водой, растворяли в Et2O, раствор фильтровали, и фильтрат упаривали при пониженном давлении. Остаток от упаривания растворяли в ДХМ, раствор фильтровали, и фильтрат упаривали при пониженном давлении, получая (1S,4S)-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2,2-диметилциклогексанкарбоновую кислоту (0.082 г, 88% выход) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3) основной ротамер/таутомер (протон карбоновой кислоты не наблюдается) δ 8.50 (с, 2Н), 7.55 (с, 1Н), 4.61-4.35 (м, 3Н), 3.70-3.16 (м, 2Н), 2.47-2.31 (м, 1Н), 2.16-1.86 (м, 5Н), 1.81-1.57 (м, 1Н), 1.17 (ушир.с, 3Н), 1.10 (ушир.с, 3Н), 1.01 (ушир.с, 9Н); LCMS (ESI): 591.0 (M+H)+.
Пример 754: получали из рацемического этилового эфира со Стадии 1 примера 785.
Пример 784: получали из (1R,4R)-этилового эфира со Стадии 2 примера 785.
Пример 807: получали аналогично примеру 754.
Пример 791
(транс-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексил)уксусная кислота
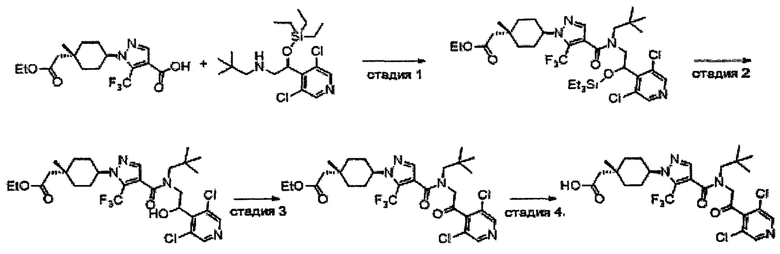
Стадии 1 и 2: 2-((1R,4R)-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексил)ацетат
Стадии 1 и 2 проводили аналогично Примеру 1, получая этил 2-((1R,4R)-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексил)ацетат.
Стадия 3: этил 2-((1R,4R)-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексил)ацетат
В раствор этил 2-((1R,4R)-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексил)ацетата (134.7 мг, 0.217 ммоль) в ДХМ (2 мл) добавляли периодинан Десс-Мартина (129 мг, 0.303 ммоль). Результирующую смесь перемешивали при комнатной температуре 30 минут. Реакционную смесь гасили NaHCO3 (5 мл, насыщенный водный раствор) и Na2S2O3 (5 мл, насыщенный водный раствор), затем экстрагировали CH2Cl2 (2×15 мл). Объединенные органические слои промывали водой (10 мл), насыщенным водным раствором поваренной соли (10 мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии (24 г Gold, 0% -50% EtOAc/Гексан), получая белое твердое вещество, представляющее собой чистый этил 2-((1R,4R)-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексил)ацетат (85.5 мг, 0.138 ммоль, 63.7% выход). LCMS=618 (М+Н)+.
Стадия 4: (транс-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексил)уксусная кислота.
В раствор этил 2-((1R,4R)-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексил)ацетата (85.5 мг, 0.138 ммоль) в ТГФ (2 мл)/EtOH (0.500 мл) добавляли LiOH, 1 M водный раствор (0.552 мл, 0.552 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель частично удаляли. Водный раствор подкисляли до рН 2. Выпавший осадок фильтровали, промывали водой и оставляли сушиться на воздухе, получая белое твердое вещество, представляющее собой чистую 2-((1R,4R)-4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексил)уксусную кислоту (82 мг, 0.139 ммоль, 100% выход) в виде смеси таутомеров. 1H ЯМР (500 МГц, ДМСО-d6) δ 0.90-1.01 (м, 9Н) 1.05-1.14 (м, 3Н) 1.46-1.59 (м, 2Н) 1.61-1.83 (м, 4Н) 2.00-2.21 (м, 2Н) 3.49 (с, 2Н) 4.05-4.23 (м, 1Н) 5.34 (с, 1Н) 7.75 (с, 1Н) 8.62 (с, 2Н) 9.88 (с, 1Н). LCMS=590.0 (М+Н)+.
Пример 795
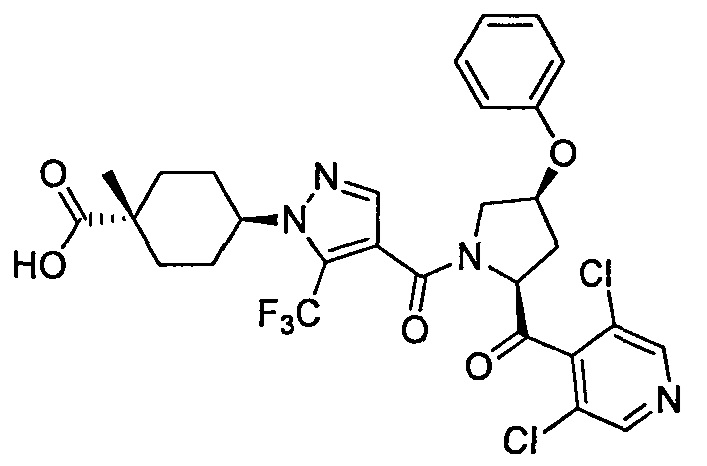
транс-4-(4-(((2S,4S)-2-((3,5-дихлор-4-пиридинил)карбонил)-4-фенокси-1-пирролидинил)карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота.
Указанное в заголовке соединение получали согласно примеру 822, используя (2S,4S)-Вос-4-фенокси-пирролидин-2-карбоновую кислоту (Chem Impex Int'l. Wood Dale, IL, 2.07 г, 6.74 ммоль). Полученную смесь эпимеров разделяли методом препаративной СФХ в описанных далее условиях. Стадия 1: препаративная СФХ ОХ-Н (5 мкм, 21 мм × 25 см), Органический модификатор: 15% МеОН. F=70 мл/мин, Т=40°С, BPR=100 бар, 220 нм. Р=151 бар. Весь образец (605 мг) растворяли в МеОН (10 мл) ~60 мг/мл, вкалывали 0.5 мл.
Стадия 2: Препаративная СФХ: Переочистка Пика 2. ОХ-Н (5 мкм, 21 мм × 25 см), Органический модификатор: 25% МеОН. F=70 мл/мин, Т=40°С, BPR=100 бар, 220 нм. Р=165 бар. Весь образец растворяли в МеОН (10 мл), ~60 мг/мл), вкалывали 1.0 мл.
Стадия 3: Препаративная СФХ: Переочистка Пика 1. ОХ-Н (5 мкм, 21 мм × 25 см)
Органический модификатор: 25% МеОН. F=70 мл/мин, Т=40°С, BPR=100 бар, 220 нм. Р=165 бар. Весь образец растворяли в МеОН (10 мл), вкалывали 1.0 мл. MS (ESI) 639.0, 641.0 [М+Н]+. Примечание: данный эпимер представлял собой пик, элюирующийся вторым в описанных выше условиях разделения.
Пример 796
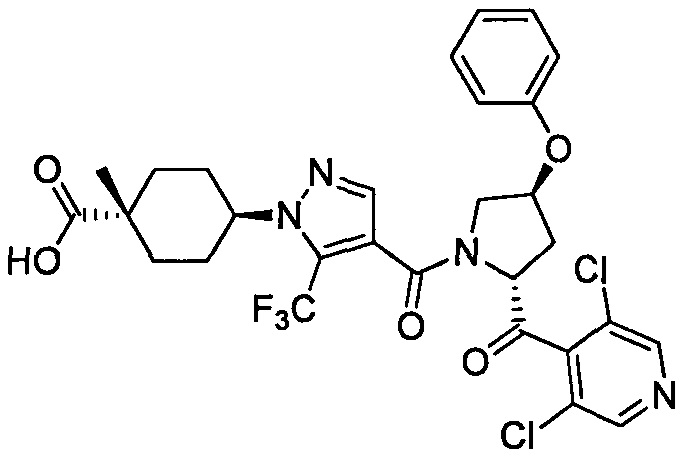
транс-4-(4-(((2R,4S)-2-((3,5-дихлор-4-пиридинил)карбонил)-4-фенокси-1-пирролидинил)карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота.
Указанное в заголовке соединение выделяли (107 мг, 0.17 ммоль, 11% выход) в виде светло-желтого аморфного твердого вещества после препаративного СФХ разделения смеси эпимеров (по положению С2 в пирролидине) из Примера 712. MS (ESI) 639.0, 641.0 [М+H]+. Примечание: данный эпимер представлял собой пик, элюирующийся третьим в описанных выше условиях разделения для Примера 795.
Пример 797
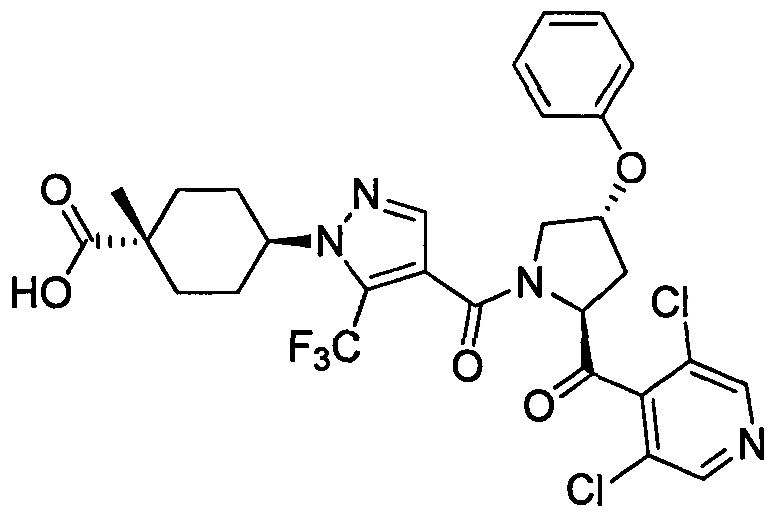
транс-4-(4-(((2S,4R)-2-((3,5-дихлор-4-пиридинил)карбонил)-4-фенокси-1-пирролидинил)карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота.
Указанное в заголовке соединение выделяли (6.7 мг, 10.48 мкмоль, 0.7% выход) в виде светло-желтого аморфного твердого вещества после препаративного СФХ разделения смеси эпимеров (по положению С2 в пирролидине) из Примера 795. MS (ESI) 639.0, 641.0 [М+Н]+. Примечание: данный эпимер представлял собой пик, элюирующийся первым в описанных выше условиях разделения для Примера 795.
Пример 798
(1R,4R)-4-(4-((2-(2,6-дихлор-3-фторфенил)-2-оксоэтил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
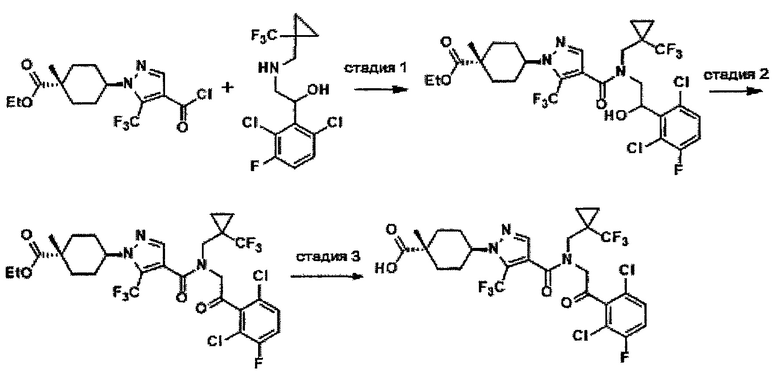
Стадия 1: (1R,4R)-этил 4-(4-((2-(2,6-дихлор-3-фторфенил)-2-гадроксиэтил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
В раствор 1-(2,6-дихлор-3-фторфенил)-2-(((1-(трифторметил)циклопропил)метил)амино)этанола (116 мг, 0.335 ммоль) и (1R,4R)-этил 4-(4-(хлоркарбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилата (147 мг, 0.402 ммоль) в ДХМ (2.3 мл) добавляли DIPEA (117 мкл, 0.670 ммоль). Реакционную смесь перемешивали при комнатной температуре. Через 1.5 часа реакционную смесь гасили насыщенным водным раствором NaHCO3 и экстрагировали дихлорметаном. Объединенные органические слои промывали водой, насыщенным водным раствором поваренной соли, сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая светло-желтое масло. Полученный сырой продукт очищали методом колоночной хроматографии (силикагель, элюент: 10% - 70% EtOAc/гептан), получая (1R,4R)-этил 4-(4-((2-(2,6-дихлор-3-фторфенил)-2-гидроксиэтил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (150 мг, 0.222 ммоль, 66.2% выход) в виде белого твердого вещества. LCMS: 675.9 (М+Н)+.
Стадия 2: (1R,4R)-этил 4-(4-((2-(2,6-дихлор-3-фторфенил)-2-оксоэтил)((1-(трифторметил) циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
Смесь (1R,4R)-этил 4-(4-((2-(2,6-дихлор-3-фторфенил)-2-гидроксиэтил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилата (150 мг, 0.222 ммоль), TEMPO (3.46 мг, 0.022 ммоль), ДХМ (2.2 мл) и 1 М водного раствора NaHCO3 (554 мкл, 0.554 ммоль) перемешивали при 0°С. Затем медленно добавляли гипохлорит натрия, 5.65-6% (1.5 мл, 1.1 ммоль). Через 1 час реакцию гасили насыщенным водным раствором Na2S2O3 при 0°С и экстрагировали дихлорметаном (10 мл). Органический слой сушили над безводным MgSO4, и упаривали при пониженном давлении, получая бесцветный остаток. Полученный сырой продукт очищали методом колоночной хроматографии (силикагель, элюент: 0%-40% EtOAc/гептан), получая (1R,4R)-этил 4-(4-((2-(2,6-дихлор-3-фторфенил)-2-оксоэтил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (113 мг, 0.168 ммоль, 76% выход) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7.54 (с, 1Н), 7.25-7.30 (м, 1Н), 7.15-7.21 (м, 1Н), 4.57 (с, 2Н), 4.12-4.20 (м, 3Н), 3.86 и 3.75 (2Н, 2 х с,), 2.12-2.29 (м, 2Н), 1.81-1.97 (м, 6Н), 1.34-1.39 (м, 3Н), 1.25-1.31 (м, 3Н), 1.07 (д, J=6А Гц, 4Н); LCMS: 674.1 [M+H]+.
Стадия 3: (1R,4R)-4-(4-((2-(2,6-дихлор-3-фторфенил)-2-оксоэтил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
В смесь (1R,4R)-этил 4-(4-((2-(2,6-дихлор-3-фторфенил)-2-оксоэтил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилата (113 мг, 0.168 ммоль) в МеОН (0.4 мл) и ТГФ (0.4 мл) (1:1 соотношение) добавляли 2н. водный раствор NaOH (0.42 мкл, 0.838 ммоль). Реакционную смесь нагревали при 50°С в течение 2 часов. Смесь упаривали, охлаждали до 0°С и подкисляли 1 н. водным раствором HCl. Белый твердый осадок отделяли, промывали водой и сушили при пониженном давлении, получая (1R,4R)-4-(4-((2-(2,6-дихлор-3-фторфенил)-2-оксоэтил)((1-(трифторметил)циклопропил)метил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновую кислоту (86 мг, 0.133 ммоль, 79% выход). 1H ЯМР (400 МГц, ДМСО-d6) δ 12.26 (ушир.с, 1Н), 7.68 (с, 1Н), 7.62 (д, J=6.5 Гц, 2Н), 4.88 и 4.71 (2Н, 2х с), 4.27 (м, 1Н), 3.77 и 3.67 (2Н, 2х м), 1.98-2.16 (м, 2Н), 1.69-1.90 (м, 6Н), 1.20-1.27 (м, 3Н), 1.01 (ушир.с, 4Н); LCMS: 645.9 [M+H]+.
Пример 813
(1S,2S,4S)-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоновая кислота
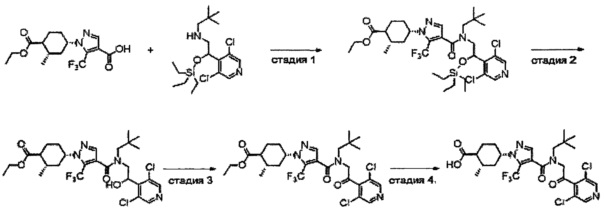
Стадия 1 и Стадия 2: (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат и его (1R,2R,4R)-изомер
В светло-желтый прозрачный раствор 1-((1S,3S,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновой кислоты и ее (1R,3R,4R)-изомера (0.3128 г, 0.898 ммоль) в ДХМ (8.98 мл) добавляли оксалилхлорид (0.095 мл, 1.123 ммоль), затем ДМФА (1 каплю), и полученную светло-желтую прозрачную реакционную смесь перемешивали при комнатной температуре. Через 2 часа смесь упаривали в вакууме, получая (1S,2S,4S)-этил 4-(4-(хлоркарбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат и его (1R,2R,4R)-изомер в виде коричневого сиропообразного вещества. В этот остаток добавляли раствор N-(2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)-2,2-диметилпропан-1-амина (0.352 г, 0.898 ммоль) в ТГФ (8.98 мл), затем DIPEA (0.626 мл, 3.59 ммоль). Полученную коричневую гетерогенную смесь перемешивали при комнатной температуре. Через 3 часа LC-M8 (ESI) показал, что образовался интермедиат (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат и его (1R,2R,4R)-изомер: LCMS (ESI) m/z 721.1 (М+Н)+.
В реакционную смесь добавляли раствор TBAF, 1.0 М раствор в ТГФ (3.59 мл, 3.59 ммоль). Через 1 час реакционную смесь разбавляли водой (30 мл) и насыщенным водным раствором поваренной соли (30 мл). Реакционную смесь экстрагировали этилацетатом (2×50 мл). Органический экстракт промывали насыщенным водным раствором NaCl (1×100 мл) и сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде орнажевого сиропа. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 50% EtOAc в гексане, получая (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат и его (1R,2R,4R)-изомер (0.4742 г, 0.781 ммоль, 87% выход) в виде не совсем белого сиропообразного твердого вещества: 1H ЯМР (300 МГц, ДМСО-d6) δ 8.44-8.63 (2Н, м), 7.57-7.71 (1Н, м), 6.01 (1Н, д, J=4А Гц), 5.28 (1Н, дт, J=9.0,4.5 Гц), 4.32 (1Н, д, J=7.2 Гц), 4.11 (2Н, кв, J=7.1 Гц), 3.85 (1Н, дд, J=14.6, 9.2 Гц), 3.53 (2Н, д, J=13.0 Гц), 3.32-3.41 (1Н, м), 1.49-2.16 (8Н, м), 1.21 (3Н, т, J=7.1 Гц), 0.63-0.98 (12Н, м), (несколько наборов пиков из-за наличия диастереомеров и ротамеров); LCMS (ESI) m/z 607.1 (М+Н)+.
Стадия 3: (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат
В светло-желтый прозрачный раствор (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилата и его (1R,2R,4R)-изомера (0.4702 г, 0.774 ммоль) в ДХМ (12.90 мл) добавляли периодинан Десс-Мартина (0.492 г, 1.161 ммоль). Результируюущую белую мутную смесь перемешивали при комнатной температуре. Через 2 часа смесь гасили насыщенным водным раствором NaHCO3 (30 мл) и насыщенным водным раствором Na2S2O3 (30 мл). Реакционную смесь экстрагировали дихлорметаном (2×50 мл). Органический экстракт сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде бесцветного сиропа. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 50% EtOAc в гексане получая (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат и его (1R,2R,4R)-изомер (0.437 г, 0.722 ммоль, 93% выход): 1Н ЯМР (400 МГц, CDCl3) δ 8.46-8.61 (2Н, м), 7.51-7.74 (1Н, м), 4.49-4.92 (2Н, м), 4.25-4.39 (1Н, м), 4.18 (2Н, кв, J=7.1 Гц), 3.29-3.61 (2Н, м), 1.63-2.18 (8Н, м), 1.29 (3Н, т, J=7.1 Гц), 0.81-1.06 (12Н, м), присутствуют ротамеры; LCMS (ESI) m/z 605.0 (М+Н)+.
Данную рацемическую смесь разделяли методом СФХ, получая две фракции:
Стереохимию каждой фракции определяли экспортно.
Первый пик на SFC IA колонке: (1R,2R,4R)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат (0.1588 г, 0.262 ммоль, 33.9% выход) в виде белого твердого вещества: 1H ЯМР (300 МГц, ДМСО-d6) δ 8.68-8.87 (2Н, м), 7.71-7.89 (1Н, м), 4.65-4.92 (2Н, м), 4.33 (1Н, ушир.с), 4.10 (2Н, кв, J=7.0 Гц), 3.24-3.30 (2Н, м), 1.49-2.17 (8Н, м), 1.20 (3Н, т, J=7.1 Гц), 0.73-1.00 (12Н, м); LCMS (ESI) m/z 605.0 (М+Н)+.
Второй пик на SFC IA колонке: (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат (0.1526 г, 0.252 ммоль, 32.6% выход) в виде белого твердого вещества: 1H ЯМР (300 МГц, ДМСО-d6) δ 8.70-8.87 (2Н, м), 7.72-7.89 (1Н, м), 4.64-4.93 (2Н, м), 4.34 (1Н, д, J=5.1 Гц), 4.10 (2Н, кв, J=7.0 Гц), 3.27 (2Н, ушир.с), 1.50-2.18 (8Н, м), 1.20 (3Н, т, J=7.1 Гц), 0.72-1.01 (12Н, м); LCMS (ESI) m/z 605.0 (М+Н)+.
Стадия 4: (1S,2S,4S)-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоновая кислота.
В смесь рацемической смеси (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилата (0.1245 г, 0.206 ммоль), ТГФ (1.645 мл), EtOH (1.645 мл) и воды (0.822 мл) добавляли 2 М раствор LiOH в воде (1.028 мл, 2.056 ммоль). Полученную белую гомогенную смесь перемешивали и нагревали при 60°С. Через 17 часов реакционную смесь упаривали в вакууме для удаления ТГФ и EtOH. Результирующий водный раствор разбавляли водой (10 мл). Значение рН раствора доводили до ~3.0 добавлением 2 н. раствора HCl, выпавший осадок отделяли фильтрованием под вакуумом, и лиофилизовывали в течение ночи, получая Пример 813 (0.0939 г, 0.163 ммоль, 79% выход) в виде белого твердого вещества. 1H ЯМР (300 МГц, ДМСО-d6) δ 12.19 (1Н, ушир.с), 8.57-9.91 (2Н, м), 7.72-7.88 (1Н, м), 4.65-5.39 (2Н, м), 4.32 (1Н, д, J=4.5 Гц), 3.22-3.53 (2Н, м), 1.46-2.10 (8Н, м), 0.72-1.03 (12Н, м), присутствуют ротамеры; LC-MS (ESI) m/z 577.1 (M+H)+.
Экспертно приписывали стереохимическую конфигурацию (1S,2S,4S).
Пример 822

транс-4-(4-(((2R,4S)-4-циклогексил-2-((3,5-дихлор-4-пиридинил)карбонил)-1-пирролидинил)карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
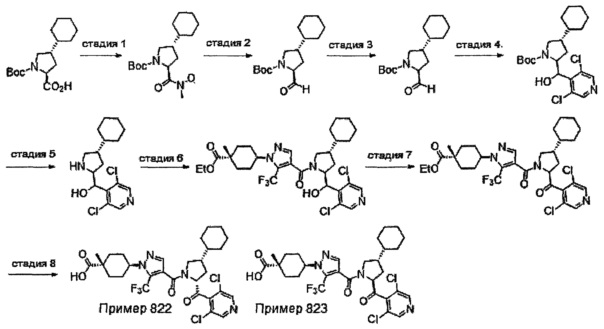
Стадия 1: К (2S,4S)-Вос-4-циклогексил-пирролидин-2-карбоновой кислоте (Chem Impex Int'l, Wood Dale, IL, 997 мг, 3.35 ммоль) добавляли ДХМ (25 мл), затем 1,1'-карбонилдиимидазол (598 мг, 3,69 ммоль). Полученный раствор оставляли перемешиваться при комнатной температуре на 1.5 часа, затем в реакционную смесь добавляли N,О-диметилгидроксиламина гидрохлорид (360 мг, 3.69 ммоль) и оставляли перемешиваться на выходные при комнатной температуре. Реакционную смесь разбавляли EtOAc (50 мл), промывали насыщенным раствором NaHCO3 (30 мл) и насыщенным водным раствором поваренной соли (30 мл), сушили над MgSO4, фильтровали и упаривали, получая сырой (2S,4S)-трет-бутил 4-циклогексил-2-(метокси(метил)карбамоил)пирролидин-1-карбоксилат (1.14 г, 3.35 ммоль, 99% выход) в виде прозрачного бесцветного вязкого масла. MS (ESI) 363.2 [М+Na]+. Полученный сырой продукт использовали в следующей стадии без дополнительной очистки.
Стадия 2: (2S,4S)-трет-бутил 4-циклогексил-2-(метокси(метил)карбамоил)пирролидин-1-карбоксилат (1.14 г, 3.35 ммоль) добавляли в ТГФ (20 мл), охлаждали до 0°С в ледяной бане и затем добавляли литийалюминий гидрид (1.0 М раствор в ТГФ, 3.35 мл, 3.35 ммоль) медленно по каплям в течение 3 минут. Полученный раствор перемешивали при 0°С 45 минут. Реакционную смесь гасили раствором натрия калия тратрата, перемешивали при комнатной температуре в течение 20 минут, затем экстрагировали этилацетатом (3×50 мл), промывали насыщенным водным раствором поваренной соли и сушили над MgSO4, фильтровали и упаривали, получая сырой (2S,4S)-трет-бугил 4-циклогексил-2-формилпирролидин-1-карбоксилат в виде прозрачного вязкого масла. MS (ESI) 304.1 [М+Na]+. Полученный сырой продукт использовали в следующей стадии без дополнительной очистки.
Стадия 3: (2S,4S-трет-бутил 4-циклогексил-2-формилпирролидин-1-карбоксилат (943 мг, 3.35 ммоль) добавляли в ТГФ (20 мл) и DBU (1.0 мл, 6.70 ммоль) и оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь упаривали досуха на роторном испарителе, добавляли ДХМ и насыщенный раствор NH4Cl и экстрагировали, промывали насыщенным водным раствором поваренной соли, сушили над MgSO4, фильтровали и упаривали, получая смесь сырого (2S,4S)-трет-бутил 4-циклогексил-2-формилпирролидин-1-карбоксилата и (2R,4S)-трет-бутил 4-циклогексил-2-формилпирролидин-1-карбоксилата (470 мг, 1.67 ммоль, 99% выход) в виде прозрачного бесцветного вязкого масла. MS (ESI) 304.1 [М+Na]+.
Стадия 4: Лития диизопропиламид (2.0 М раствор в смеси гептан/ТГФ/этилбензол, 3.51 мл, 7.02 ммоль) добавляли к 3,5-дихлорпиридину (820 мг, 5.54 ммоль), рстворенному в ТГФ (15 мл), охлажденному до -78°С, и перемешивали при этой температуре 1 час. Добавляли раствор (2S,4S)-Трет-бутил 4-циклогексил-2-формилпирролидин-1-карбоксилата (1.04 г, 3.70 ммоль) и его эпимера по С2 положению пирролидина в ТГФ (11 мл), удаляли из охлаждающей бани, оставляли нагреваться до комнатной температуры и перемешивали в течение 2 часов. Полученный раствор гасили насыщенным раствором хлорида аммония, водный слой экстрагировали этилацетатом (2×50 мл), органический слой промывали насыщенным водным раствором поваренной соли (30 мл) и сушили над безводным сульфатом магния, фильтровали и упаривали. Полученный неочищенный продукт очищали на ISCO Combiflash™ RF (40 г Grace Reverlis колонка, применяя градиент 0-80% EtOAc в гептане), получая (2S,4S)-трет-бутил 4-циклогексил-2-((S)-(3,5-дихлорпиридин-4-ил)(гидрокси)метил)пирролидин-1-карбоксилат (1.20 г, 2.79 ммоль, 76% выход) в виде смеси эпимеров. MS (ESI) 451.1,453.1 [М+Na]+.
Стадия 5: (2S,4S)-трет-бутил 4-циклогексил-2-((S)-(3,5-дихлорпиридин-4-ил)(гидрокси)метил)пирролидин-1-карбоксилат (1.20 г, 2.79 ммоль) и его эпимер по С2 положению пирролидина добавляли в ДХМ (10 мл) и ТФУК (7 мл, 91 ммоль) и оставляли перемешиваться при комнатной температуре на 1.5 часа. Реакционную смесь упаривали на роторном испарителе, и сырой остаток очищали на ISCO Combiflash™ RF (40 г Grace Reveleris колонка, применяя градиент 0-20% 2М NH3/МеОН в ДХМ), получая (S)-((2S,4S)-4-циклогексилпирролидин-2-ил)(3,5-дихлорпиридин-4-ил)метанол 2,2,2-трифторацетат (755 мг, 1.705 ммоль, 61% выход) вместе с его эпимером по С2 положению пирролидина в виде светло-коричневой пены. MS (ESI) 329.0, 331.1 [М+Н]+.
Стадия 6: Оксалилхлорид (0.22 мл, 2.55 ммоль) добавляли в раствор 1-((1R,4R)-4-(этоксикарбонил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновой кислоты (593 мг, 1.70 ммоль) в ДХМ (10.0 мл), затем добавляли 2 капли ДМФА, при охлаждении на ледяной бане. Полученный раствор затем вынимали из охлаждающей бани и оставляли перемешиваться при комнатной температуре на 1 час. Реакционную смесь упаривали досуха на роторном испарителе, и в сырой остаток добавляли ДХМ (10.0 мл) и охлаждали до 0°С. В перемешиваемый раствор затем добавляли (S)-((2S,4S)-4-циклогексилпирролидин-2-ил)(3,5-дихлорпиридин-4-ил)метанол 2,2,2-трифторацетат (755 мг, 1.70 ммоль) и DIPEA (0.89 мл, 5.11 ммоль) в ДХМ (10 мл) и оставляли нагреваться до комнатной температуры и перемешивали 1 час. Реакционную смесь упаривали досуха при пониженном давлении (роторный испаритель), и сырой остаток очищали на ISCO Combiflash™ RF (40 г Grace Reveleris колонка, применяя градиент 0-100% EtOAc в гептане), получая (1S,4R)-этил 4-(4-((2S,4S)-4-циклогексил-2-((S)-(3,5-дихлорпиридин-4-ил)(гидрокси)метил)пирролидин-1-карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (640 мг, 0.970 ммоль, 57% выход) вместе с его эпимером по С2 положению пирролидина в виде светло-коричневой пены. MS (ESI) 659.2, 661.1 [М+H]+.
Стадия 7: К периодинану Десс-Мартина (823 мг, 1.94 ммоль) и (1R,4R)-этил 4-(4-((2R,4S)-4-циклогексил-2-((R)-(3,5-дихлорпиридин-4-ил)(гидрокси)метил)пирролидин-1-карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилату (640 мг, 0.97 ммоль) в виде смеси с его эпимером по С2 положению пирролидина добавляли ДХМ (10 мл) и оставляли перемешиваться при комнатной температуре на 3 часа. В реакционную смесь добавляли насыщенный раствор NaHCO3 и твердый метабисульфит натрия. Реакционную смесь затем экстрагировали дихлорметаном (2×75 мл), сушили над MgSO4, фильтровали и упаривали, получая сырой продукт в виде светло-оранжевой пены. Этот остаток очищали на ISCO Combiflash™ RF (25 г Grace Reverlis колонка, применяя градиент 0-70% EtOAc в гептане), получая (1R,4R)-этил 4-(4-((2R,4S)-4-циклогексил-2-(3,5-дихлоризоникотиноил)пирролидин-1-карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (605 мг, 95%) вместе с его эпимером по С2 положению пирролидина в виде светло-желтой пены. MS (ESI) 657.0, 659.0 [М+Н]+.
Стадия 8: К (1S,4R)-этил 4-(4-((2S,4S)-4-циклогексил-2-(3,5-дихлоризоникотиноил)пирролидин-1-карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилату (454 мг, 0.69 ммоль) и его эпимеру по С2 положению пирролидина в ТГФ (3.5 мл) и МеОН (3.5 мл) добавляли лития гидроксид моногидрат (1.0 М раствор, 3.5 мл, 3.45 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи (16 час), органику удаляли при пониженном давлении (роторный испаритель), и результирующий водный раствор подкисляли добавлением 1 н. раствора HCl, что вызывало выпаление твердого осадка. Полученную смесь экстрагировали этилацетатом (2×40 мл). Объединенные экстракты промывали насыщенным водным раствором поваренной соли, сушили над безводным MgSO4, фильтровали и упаривали. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом хроматографии на ISCO Combiflash™ RF (25 г Thomson SingleStep колонка, применяя градиент 0-100% [10% МеОН в ДХМ] в ДХМ), получая смесь двух продуктов, эпимерных по С2 положению пирролидина. Полученное вещество разделяли методом препаративной СФХ в следующих условиях: ОХ колонка (SN=2121, 5 мкм, 21 мм × 25 см, 50/50/50 р=172), Органический модификатор: 25% МеОН с 20 мМ NH3. F=70 мл/мин, Т=40°С, BPR=100 бар, 220 нм. Р=165 бар, весь образец (416 мг) растворяли в 8 мл МеОН, ~ 52 мг/мл), вкалывали 1.0 мл, получая транс-4-(4-(((2R,4S)-4-циклогексил-2-((3,5-дихлор-4-пиридинил)карбонил)-1-пирролидинил)карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновую кислоту (55.3 мг, 0.088 ммоль, 13% выход) в виде светло-желтого аморфного твердого вещества. MS (ESI) 629.1, 631.1 [М+Н]+. Примечание: данный эпимер представлял собой пик, элюирующийся первым в описанных выше условиях разделения.
Пример 823
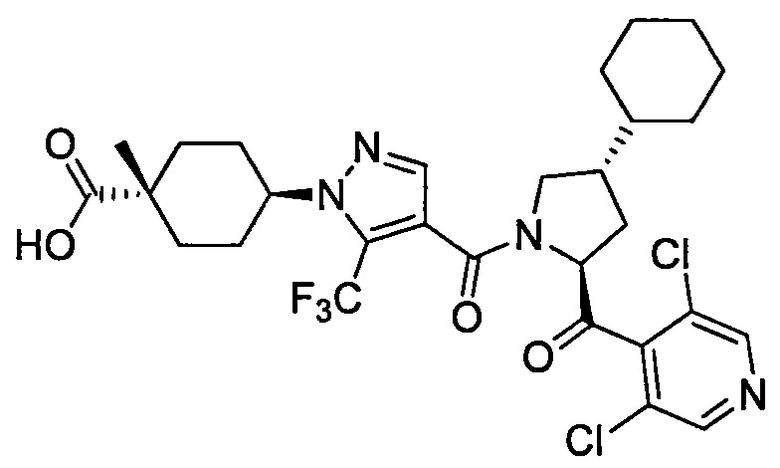
транс-4-(4-(((2S,4S)-4-циклогексил-2-((3,5-дихлор-4-пиридинил)карбонил)-1-пирролидинил)карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота.
Указанное в заголовке соединение выделяли (291 мг, 0.46 ммоль, 67% выход) в виде светло-желтого аморфного твердого вещества после препаративного СФХ разделения смеси эпимеров (по положению С2 в пирролидине) из Примера 739. MS (ESI) 629.1, 631.1 [М+Н]+. Примечание: данный эпимер представлял собой пик, элюирующийся вторым в описанных выше условиях разделения.
Пример 827
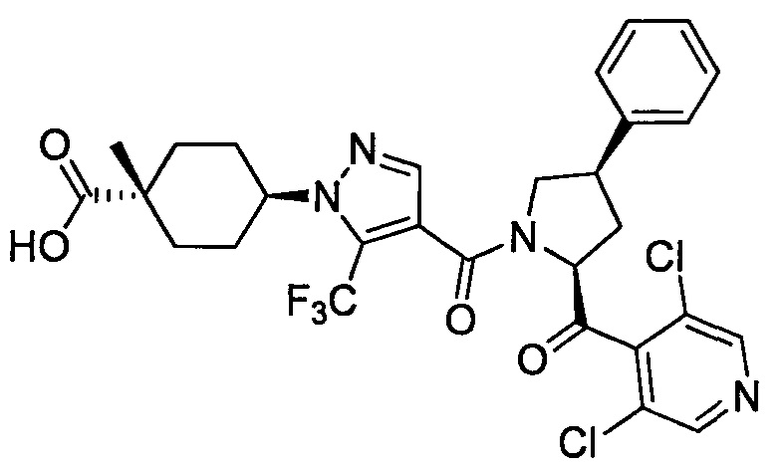
транс-4-(4-(((2S,4R)-2-((3,5-дихлор-4-пиридинил)карбонил)-4-фенил-1-пирролидинил)карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
Указанное в заголовке соединение получали согласно Примеру 822 с применением (2S,4R)-1-(трет-бутоксикарбонил)-4-фенилпирролидин-2-карбоновой кислоты (Frontier Scientific, Newark, DE, 1.00 г, 3.43 ммоль) и выделяли (63.7 мг, 0.10 ммоль, 18% выход) в виде белого аморфного твердого вещества. Полученную смесь эпимеров разделяли с помощью препаративной СФХ в следующих условиях. Колонка: CHIRALPAK™ AZ-H (Reversed) (250×21 мм, 5 мкм), Подвижная фаза: 82:18 (А:В), А: Жидкий CO2, В: EtOH. Скорость потока: 70 мл/мин. Температура колонки/печи: 40°С, давление на входе 186-193 бар. SN: 403121. MS (ESI) 623.0, 625.0 [M+Н]+. Примечание: данный эпимер представлял собой пик, элюирующийся вторым в описанных выше условиях разделения.
Пример 828
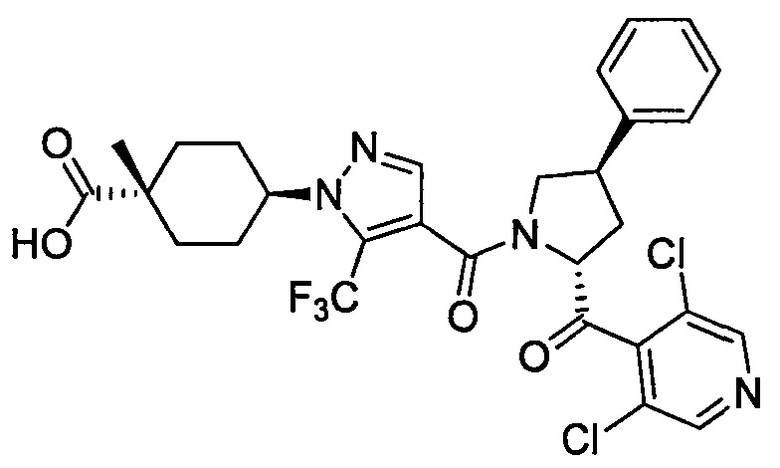
транс-4-(4-(((2R,4S)-2-((3,5-дихлор-4-пиридинил)карбонил)-4-фенил-1-пирролидинил)карбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота.
Указанное в заголовке соединение выделяли (135 мг, 0.217 ммоль, 39% выход) в виде белой пены после препаративного СФХ разделения смеси эпимеров (по положению С2 в пирролидине) из Примера 827. MS (ESI) 623.0, 625.0 [M+Н]+. Примечание: данный эпимер представлял собой пик, элюирующийся третьим в описанных выше условиях разделения для Примера 827.
Пример 845
(1S,2R,4S)-4-(4-((2-(2,6-дихлор-3-фторфенил)-2-оксоэтил)((1R,3r,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоновая кислота.
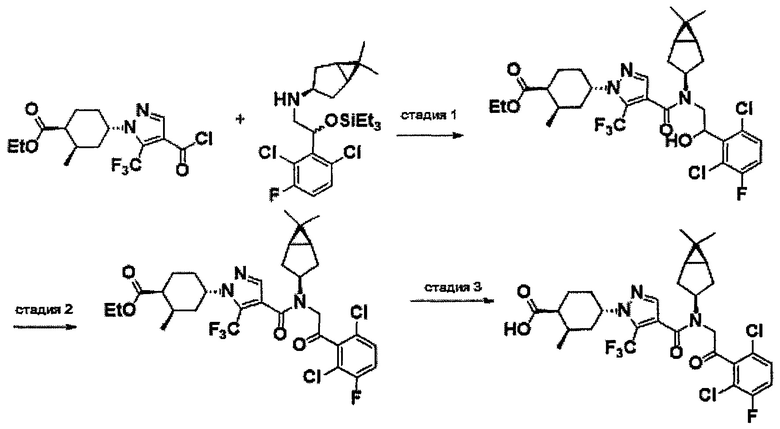
Стадия 1: (1S,2R,4S)-этил 4-(4-((2-(2,6-дихлор-3-фторфенил)-2-гидроксиэтил)((1R,3r,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат.
В раствор (1R,3R,5S)-N-(2-(2,6-дихлор-3-фторфенил)-2-((триэтилсилил)окси)этил)-6,6-диметилбицикло[3.1.0]гексан-3-амина (97 мг, 0.217 ммоль) и (1S,2R,4S)-этил 4-(4-(хлоркарбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилата (96 мг, 0.261 ммоль) в ДХМ (0.8 мл) добавляли DIPEA (76 мкл, 0.434 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили насыщенным водным раствором NaHCO3 и экстрагировали дихлорметаном (3×10 мл). Органический слой объединяли, сушили над безводным MgSO4, фильтровали и упаривали, получая продукт в виде желтого остатка. Остаток от упаривания растворяли в ТГФ (0.75 мл), затем добавляли 1.0 М раствор TBAF в ТГФ (434 мкл, 0.434 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 0.5 часа. Его гасили насыщенным водным раствором NaHCO3 и экстрагировали дихлорметаном. Объединенный органический слой промывали водой, насыщенным водным раствором поваренной соли, сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая светло-желтое масло. Полученный сырой продукт очищали методом колоночной хроматографии (силикагель, элюент: 0%-40% EtOAc/гептан), получая (1S,2R,4S)-этил 4-(4-((2-(2,6-дихлор-3-фторфенил)-2-гидроксиэтил)((1R,3R,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат (96 мг, 0.145 ммоль, 66.7% выход) в виде белого твердого вещества. LCMS: 662.1 [М+Н]+.
Стадия 2: (1S,2R,4S)-этил 4-(4-((2-(2,6-дихлор-3-фторфенил)-2-оксоэтил)((1R,3R,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат
(1S,2R,4S)-этил 4-(4-((2-(2,6-дихлор-3-фторфенил)-2-гидроксиэтил)((1R,3r,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат (96 мг, 0.145 ммоль) растворяли в ДХМ (3 мл) и добавляли периодинан Десс-Мартина (77 мг, 0.181 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь гасили 5%-ным раствором Na2S2O3, промывали насыщенным раствором NaHCO3, сушили над безводным Na2SO4 и упаривали. Полученный неочищенный продукт очищали методом колоночной хроматографии (силикагель, элюент: 0-40% EtOAc/гептан), получая (1S,2R,4S)-этил 4-(4-((2-(2,6-дихлор-3-фторфенил)-2-оксоэтил)((1R,3R,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат (90 мг, 0.136 ммоль, 94% выход) в виде вязкого белого масла. LCMS 660.0 [М+Н]+.
Стадия 3: (1S,2R,4S)-4-(4-((2-(2,6-дихлор-3-фторфенил)-2-оксоэтил)((1R,3R,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоновая кислота.
В смесь (1S,2R,4S)-этил 4-(4-((2-(2,6-дихлор-3-фторфенил)-2-оксоэтил)((1R,3R,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилата (90 мг, 0.136 ммоль), МеОН (0.34 мл) и ТГФ (0.34 мл) (1:1 соотношение) добавляли 2н. водный раствор NaOH (0.34 мл, 0.68 ммоль). Реакционную смесь нагревали при 50°С 3 часа. Смесь упаривали, охлаждали до 0°С и подкисляли 1н. водным раствором HCl. Белый твердый осадок отделяли, промывали водой и сушили при пониженном давлении, получая (1S,2R,4S)-4-(4-((2-(2,6-дихлор-3-фторфенил)-2-оксоэтил)((1R,3R,5S)-6,6-диметилбицикло[3.1.0]гексан-3-ил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоновую кислоту (58 мг, 0.092 ммоль, 67.3% выход). 1Н ЯМР (500 МГц, ДМСО-d6) δ 12.18 (ушир.с, 1Н), 7.87-7.56 (м, 2Н), 7.40-7.51 (м, 1Н), 4.96-5.10 (м, 1Н), 4.53-4.77 (м, 1Н), 4.11-4.52 (м, 2Н), 2.54-2.64 (м, 1Н), 1.66-2.24 (м, 10Н), 1.35-1.59 (м, 2Н), 1.19-1.29 (м, 1Н), 0.82-1.14 (м, 15Н); LCMS: 632.2 [М+Н]+.
Пример 853
транс-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)((1S,4S)-7-оксабицикло[221]гепт-1-илметил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
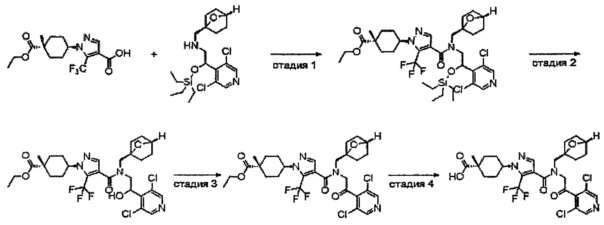
Стадия 1 и Стадия 2: этил транс-4-(4-(((1S,4S)-7-оксабицикло[2.2.1]гептан-1-илметил)(2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат.
В прозрачный раствор 1-((1R,4R)-4-(этоксикарбонил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновой кислоты (0.121 г, 0.348 ммоль) в ДХМ (3.48 мл) добавляли оксалилхлорид (0.037 мл, 0.435 ммоль), затем ДМФА (1 каплю), и полученную прозрачную реакционную смесь перемешивали при комнатной температуре. Через 5 часов смесь упаривали в вакууме, получая (1R,4R)-этил 4-(4-(хлоркарбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат в виде светло-желтого сиропа. В остаток от упаривания добавляли раствор N-((1S,4S)-7-оксабицикло[2.2.1]гептан-1-илметил)-2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этанамина (0.150 г, 0.348 ммоль) в ТГФ (3.48 мл), затем DIPEA (0.242 мл, 1.391 ммоль). Полученную желтую гетерогенную смесь перемешивали при комнатной температуре. Через 13 часов LCMS показал образовано промежуточного этилтранс-4-(4-(((1S,4S)-7-оксабицикло[2.2.1]гептан-1-илметил)(2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилата: LCMS (ESI) m/z 761.2 (М+Н)+. В реакционную смесь добавляли раствор TBAF, 1.0 М раствор в ТГФ (1.391 мл, 1.391 ммоль). Через 4 часа реакционную смесь разбавляли водой (30 мл) и насыщенным водным раствором поваренной соли (30 мл). Реакционную смесь экстрагировали этилацетатом (2×50 мл). Органический экстракт промывали насыщенным водным раствором NaCl (1×100 мл) и сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде светло-желтого сиропа. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 50% EtOAc в гептане, получая этил транс-4-(4-(((1S,4S)-7-оксабицикло[2.2.1]гептан-1-илметил)(2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (0.1083 г, 0.167 ммоль, 48.1% выход) в виде бесцветного сиропообразного твердого вещества:
1H ЯМР (400 МГц, ДМСО-d6) δ 8.43-8.59 (2Н, м), 7.49-7.84 (1Н, м), 5.95-6.04 (1Н, м), 5.32-5.63 (1Н, м), 3.51-4.54 (8Н, м), 1.11-2.22 (22Н, м), (присутствуют диастереомеры и ротамеры); LCMS (ESI) m/z 647.2 (М+Н)+.
Стадия 3: этил транс-4-(4-(((1S,4S)-7-оксабицикло[2.2.1]гептан-1-илметил)(2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат
В прозрачный раствор этил транс-4-(4-(((1S,4S)-7-оксабицикло[2.2.1]гептан-1-илметил)(2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилата (0.1015 г, 0.157 ммоль) в ДХМ (2.61 мл) добавляли периодинан Десс-Мартина (0.100 г, 0.235 ммоль). Результирующую мутную реакционную смесь перемешивали при комнатной температуре. Через 2 часа смесь гасили насыщенным водным раствором Na2S2O3 (30 мл) и насыщенным водным раствором NaHCO3 (30 мл). Реакционную смесь экстрагировали дихлорметаном (2×50 мл). Органический экстракт сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде светло-желтого сиропа. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 50% EtOAc в гептане, получая этил транс-4-(4-(((1S,4S)-7-оксабицикло[2.2.1]гептан-1-илметил)(2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилат (0.0841 г, 0.130 ммоль, 83% выход) в виде бесцветного сиропа. 1H ЯМР (400 МГц, CDCl3) δ 8.43-8.60 (2Н, м), 7.51-7.68 (1Н, м), 3.71-5.12 (8Н, м), 1.21-2.36 (22Н, м), присутствуют ротамеры; LC-MS (ESI) m/z 645.0 (М+Н)+.
Стадия 4: транс-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)((1S,4S)-7-оксабицикло[221]гепт-1-илметил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота
В прозрачный раствор этил транс-4-(4-(((1S,4S)-7-оксабицикло[2.2.1]гептан-1-илметил)(2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоксилата (0.0786 г, 0.122 ммоль) в ТГФ (0.974 мл), EtOH (0.974 мл) и воде (0.487 мл) добавляли 2 М раствор LiOH в воде (0.609 мл, 1.218 ммоль). Полученную желтую гомогенную смесь перемешивали и нагревали до 60°С. Через 10 часов реакционную смесь упаривали в вакууме для удаления ТГФ и EtOH. Результирующий водный раствор разбавляли водой (10 мл). Значение рН раствора доводили до ~3.0 добавлением 2 н. раствора HCl, и выпавший осадок отделяли фильтрованием под вакуумом, промывали водой и лиофилизовывали в течение ночи, получая Пример 853 в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 12.25 (1Н, ушир.с), 8.57-8.84 (2Н, м), 7.69-7.86 (1Н, м), 4.77-5.00 (2Н, м), 4.40-4.53 (1Н, м), 4.25 (1Н, т, J=11.3 Гц), 3.64-4.06 (2Н, м), 1.10-2.20 (19Н, м), присутствуют ротамеры; LCMS (ESI) m/z 617.0 (М+Н)+.
Пример 872
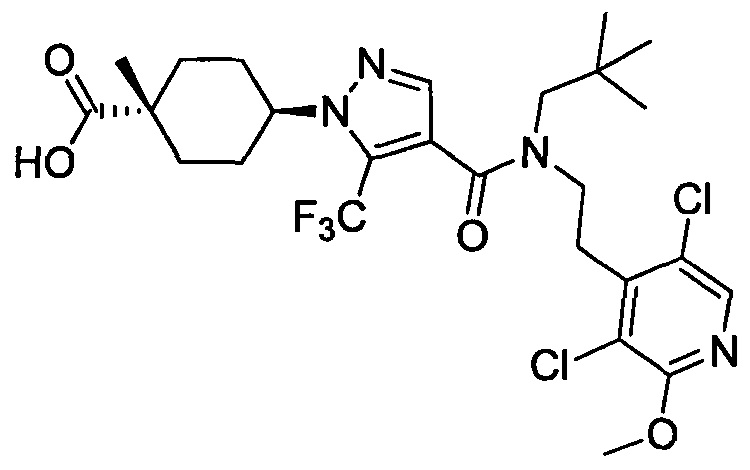
транс-4-(4-((2-(3,5-дихлор-2-метокси-4-пиридинил)этил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота.
Указанное в заголовке соединение получали из N-(2-(3,5-дихлор-2-метоксипиридин-4-ил)этил)-2,2-диметилпропан-1-амина и 1-((1R,4R)-4-(этоксикарбонил)-4-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновой кислоты по методикам, аналогичным описанным в Примере 545. MS (ESI) 593.2, 595.1 [М+H]+.
Пример 879
(1S,2S,4S)-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоновая кислота
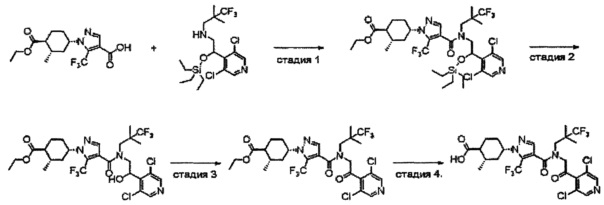
Стадия 1 и Стадия 2: (1R,2R,4R)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат с (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилатом (1:1)
В немного мутную смесь 1-((1R,3R,4R)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновой кислоты в смеси с 1-((1S,3S,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновой кислотой (1:1) (0.300 г, 0.431 ммоль) в ДХМ (17.23 мл) добавляли оксалилхлорид (0.091 мл, 1.077 ммоль), затем ДМФА (1 каплю), и результирующую светло-желтую немного мутную реакционную смесь перемешивали при комнатной температуре. Через 1.5 часа смесь упаривали в вакууме, получая (1R,2R,4R)-этил 4-(4-(хлоркарбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат с (1S,2S,4S)-этил 4-(4-(хлоркарбонил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилатом (1:1) в виде светло-желтого сиропа.
В полученный желтый сироп добавляли раствор N-(2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)-3,3,3-трифтор-2,2-диметилпропан-1-амина (0.384 г, 0.862 ммоль) в ТГФ (17.23 мл), затем DIPEA (0.600 мл, 3.45 ммоль). Полученную желтую гомогенную смесь перемешивали при комнатной температуре. Через 4 часов LCMS (ESI) показал формирование промежуточного (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-((триэтилсилил)окси)этил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилата, содержащего его (1R,2R,4R) изомер: LCMS (ESI) m/z 775.1 (М+Н)+.
В реакционную смесь добавляли раствор TBAF, 1.0 М раствор в ТГФ (3.45 мл, 3.45 ммоль), и полученную желтую гомогенную смесь перемешивали при комнатной температуре. Через 20 минут реакционную смесь разбавляли водой (50 мл) и насыщенным водным раствором поваренной соли (50 мл). Реакционную смесь экстрагировали этилацетатом (2×50 мл). Органический экстракт промывали насыщенным водным раствором NaCl (1×100 мл) и сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде светло-желтого сиропа. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 50% EtOAc в гептане, получая (1R,2R,4R)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат с (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилатом (1:1) (0.4648 г, 0.351 ммоль, 82% выход) в виде бесцветного сиропа: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8.46-8.63 (2Н, м), 7.70-7.82 (1Н, м), 6.11 (1Н, д, J=3.3 Гц), 5.20-5.32 (1Н, м), 4.34 (1Н, д, J=8.0 Гц), 4.06-4.16 (2Н, м), 3.43-3.97 (4Н, м), 1.53-2.15 (8Н, м), 0.82-1.31 (12Н, м), (диастереомеры и ротамеры); LCMS (ESI) m/z 661.1 (М+Н)+.
Стадия 3: (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат
В прозрачный раствор (1R,2R,4R)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилата с (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-гидроксиэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилатом (1:1) (0.458 г, 0.346 ммоль) в ДХМ (11.54 мл) добавляли периодинан Десс-Мартина (0.441 г, 1.039 ммоль). Результирующую белую мутную смесь перемешивали при комнатной температуре. Через 14 часов смесь гасили насыщенным водным раствором Na2S2O3 (50 мл) и насыщенным водным раствором NaHCO3 (50 мл). Реакционную смесь экстрагировали дихлорметаном (2×50 мл). Органический экстракт сушили над Na2SO4. Полученный раствор фильтровали и упаривали в вакууме, получая сырой продукт в виде белого твердого вещества. Полученный сырой продукт адсорбировали на слое силикагеля и очищали методом колоночной хроматографии на силикагеле, элюируя в градиенте от 0% до 50% EtOAc в гексане, получая (1R,2R,4R)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат с (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилатом (1:1) (0.3795 г, 0.288 ммоль, 83% выход) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8.70-8.85 (2Н, м), 7.73-7.99 (1Н, м), 4.69-4.93 (2Н, м), 4.35 (1Н, д, J=3.7 Гц), 4.10 (2Н, кв, J=7.0 Гц), 3.51-3.87 (2Н, м), 1.51-2.15 (8Н, м), 0.87-1.23 (12Н, м), присутствуют ротамеры; LCMS (ESI) m/z 659.0 (М+Н)+.
Полученную рацемическую смесь разделяли методом СФХ, получая две фракции, где стереохимию каждой фракции определяли экспертно. Первый пик на SFC IA колонке: (1R,2R,4R)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат (0.1371 г, 0.208 ммоль, 43.4% выход) в виде белого твердого вещества: 1Н ЯМР (400 МГц, CDCl3) δ 8.47-8.63 (2Н, м), 7.51-7.77 (1Н, м), 4.52 (2Н, с), 4.26-4.39 (1Н, м), 4.18 (2Н, кв, J=7.1 Гц), 3.70 (2Н, ушир.с), 1.62-2.17 (8Н, м), 1.29 (3Н, т, J=7.1 Гц), 1.24 (6Н, с), 1.00 (3Н, д, J=6.1 Гц), присутствуют ротамеры; LCMS (ESI) m/z 659.0 (М+Н)+.
Второй пик на SFC IA колонке: (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат (0.1447 г, 0.219 ммоль, 45.8% выход) в виде белого порошка: 1H ЯМР (400 МГц, CDCl3) δ 8.44-8.64 (2Н, м), 7.51-7.77 (1Н, м), 4.52 (2Н, с), 4.25-4.38 (1Н, м), 4.18 (2Н, кв, J=7.1 Гц), 3.57-3.98 (2Н, м), 1.63-2.14 (8Н, м), 1.29 (3Н, т, J=7.1 Гц), 1.24 (6Н, с), 1.00 (3Н, д, J=6.1 Гц), присутствуют ротамеры; LCMS (ESI) m/z 659.0 (М+Н)+.
Стадия 4: (1S,2S,4S)-4-(4-((2-(3,5-дихлор-4-пиридинил)-2-оксоэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоновая кислота
В прозрачный раствор (1S,2S,4S)-этил 4-(4-((2-(3,5-дихлорпиридин-4-ил)-2-оксоэтил)(3,3,3-трифтор-2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилата (0.1345 г, 0.204 ммоль) в ТГФ (1.632 мл), EtOH (1.632 мл) и воде (0.816 мл) добавляли 2 М раствор LiOH в воде (1.020 мл, 2.040 ммоль). Полученную светло-желтую немного мутную смесь перемешивали и нагревали до 60°С. Через 4 часа реакционную смесь упаривали в вакууме для удаления ТГФ и EtOH. Результирующий водный раствор разбавляли водой (10 мл). Значение рН раствора доводили до ~3.0 добавлением 1 н. раствора HCl, и выпавший осадок отделяли фильтрованием под вакуумом, промывали водой и лиофилизовывали в течение ночи, получая Пример 879 (0.1151 г, 0.182 ммоль, 89% выход) в виде белого твердого вещества: 1H ЯМР (400 МГц, ДМСО-d6) δ 12.18 (1Н, ушир.с), 8.59-8.86 (2Н, м), 7.73-8.02 (1Н, м), 4.65-5.49 (2Н, м), 4.33 (1Н, д, J=8.4 Гц), 3.44-3.94 (2Н, м), 1.48-2.10 (8Н, м), 0.85-1.36 (9Н, м), присутствуют ротамеры; LCMS (ESI) m/z 631.1 (М+Н)+. Экспертно приписывали стереохимическую конфигурацию (1S,2S,4S).
Пример 885
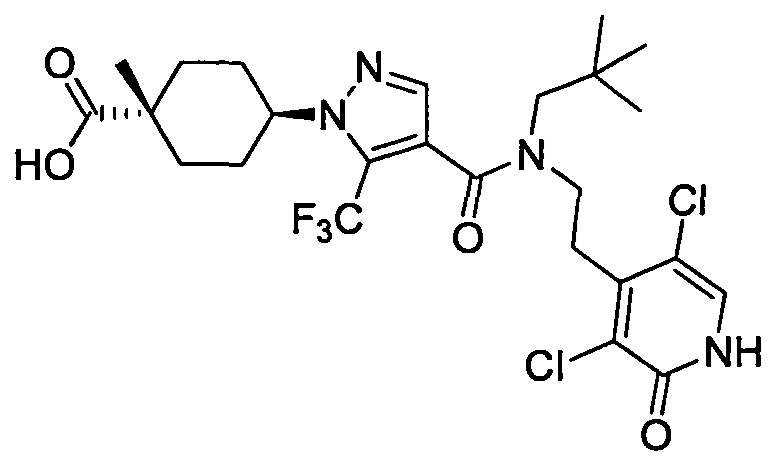
тиранс-4-(4-((2-(3,5-дихлор-2-оксо-1,2-дигидро-4-пиридинил)этил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-1-метилциклогексанкарбоновая кислота.
Указанное в заголовке соединение получали по методике, аналогичной описанной в Примере 886, и выделяли (36.7 мг, 0.063 ммоль, 54% выход) в виде белого аморфного твердого вещества. MS (ESI) 579.0, 581.0 [М+Н]+.
Пример 886
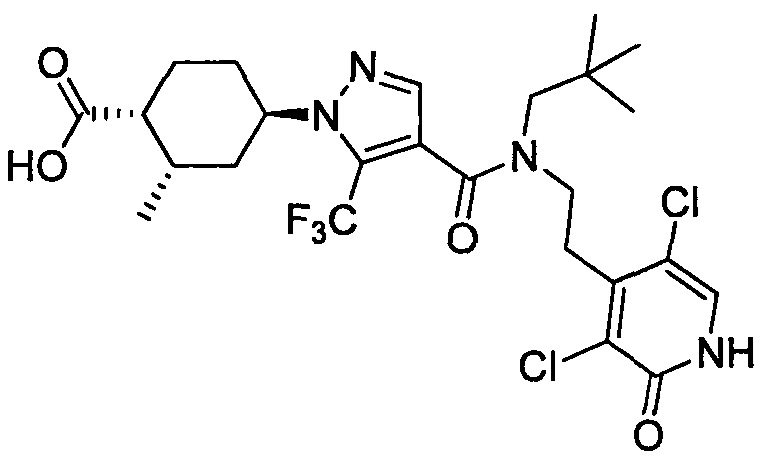
(1S,2R,4S)-4-(4-((2-(3,5-дихлор-2-оксо-1,2-дигидро-4-пиридинил)этил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоновая кислота
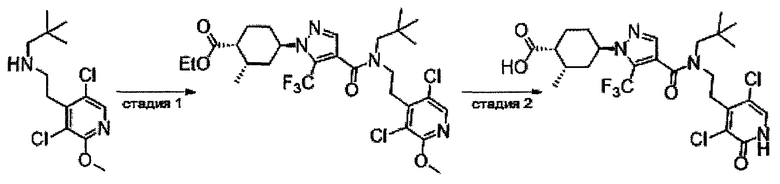
Стадия 1: Оксалилхлорид (2.0М раствор в ДХМ, 0.52 мл, 1.03 ммоль) добавляли в раствор 1-((1S,3R,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновой кислоты (239 мг, 0.687 ммоль) в ДХМ (2.0 мл), затем добавляли 1 каплю ДМФА при охлаждении на ледяной бане. Полученный раствор вынимали из ледяной бани и оставляли перемешиваться при комнатной температуре на 1 час. Реакционную смесь упаривали досуха при пониженном давлении (роторный испаритель), в полученный сырой остаток добавляли ДХМ (2.0 мл) и охлаждали до 0°С. В перемешиваемый раствор затем добавляли N-(2-(3,5-дихлор-2-метоксипиридин-4-ил)этил)-2,2-диметилпропан-1-амин (200 мг, 0.687 ммоль) в ДХМ (2 мл), затем добавляли DIPEA (0.36 мл, 2.06 ммоль) и оставляли нагреваться до комнатной температуры и перемешивали в течение ночи (16 часов). Реакционную смесь упаривали досуха при пониженном давлении (роторный испаритель), и сырой остаток очищали на ISCO Combiflash™ RF (25 г Grace Reveleris колонка, применяя градиент 0-50% EtOAc в гептане), получая (1S,2R,4S)-этил 4-(4-((2-(3,5-дихлор-2-метоксипиридин-4-ил)этил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат (350 мг, 0.56 ммоль, 82% выход) в виде белого кристаллического вещества. MS (ESI) 621.2, 623.2 [М+Н]+.
Стадия 2: В (1S,2R,4S)-этил 4-(4-((2-(3,5-дихлор-2-метоксипиридин-4-ил)этил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоксилат (109 мг, 0.175 ммоль) добавляли водный раствор соляной кислоты (5.0 н., 3.00 мл, 15.00 ммоль) и хлороводород (4.0 н. раствор в 1,4-диоксане, 3.00 мл, 12.00 ммоль), оснащали колбу обратным холодильником и нагревали при 120°С 3 часа. Реакционную смесь упаривали досуха при пониженном давлении (роторный испаритель), и сырой остаток очищали на Gilson (Gemini™ Phenomenex; 30×150 мм, 5 мкм, применяя градиент 10-95% 0.1% ТФУК/CH3CN в 0.1%ТФУК/вода), упаривали в течение ночи, получая (1S,2R,4S)-4-(4-((2-(3,5-дихлор-2-оксо-1,2-дигидропиридин-4-ил)этил)(неопентил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоновую кислоту (57 мг, 0.098 ммоль, 56% выход) в виде белого аморфного твердого вещества. MS (ESI) 579.0, 581.2 [М+H]+.
Пример 887
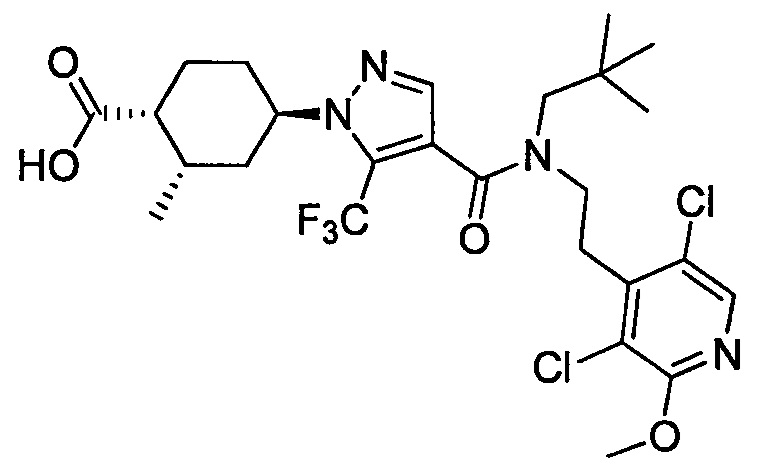
(1S,2R,4S)-4-(4-((2-(3,5-дихлор-2-метокси-4-пиридинил)этил)(2,2-диметилпропил)карбамоил)-5-(трифторметил)-1Н-пиразол-1-ил)-2-метилциклогексанкарбоновая кислота
Указанное в заголовке соединение получали из N-(2-(3,5-дихлор-2-метоксипиридин-4-ил)этил)-2,2-диметилпропан-1-амина и 1-((1S,3R,4S)-4-(этоксикарбонил)-3-метилциклогексил)-5-(трифторметил)-1Н-пиразол-4-карбоновой кислоты по методикам, аналогичным описанным в Примере 872. MS (ESI) 593.2, 595.1 [М+H]+.
Приведенные далее примеры синтезировали согласно методикам, аналогичным описанным выше.
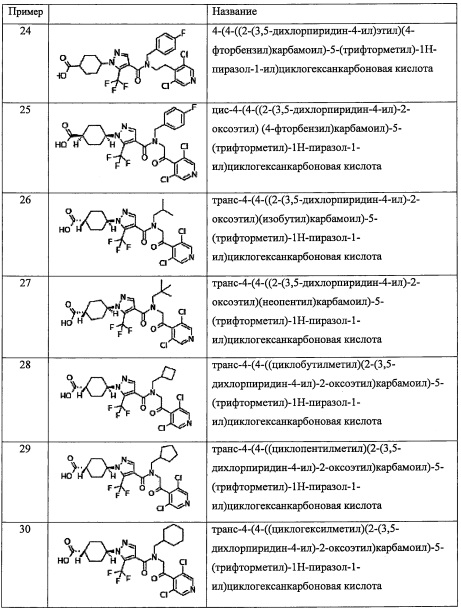
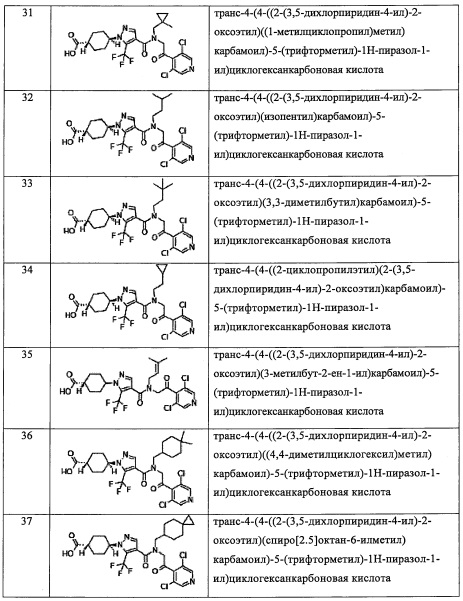
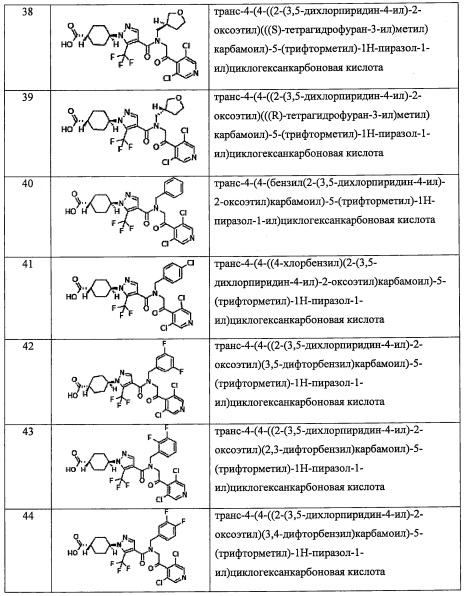
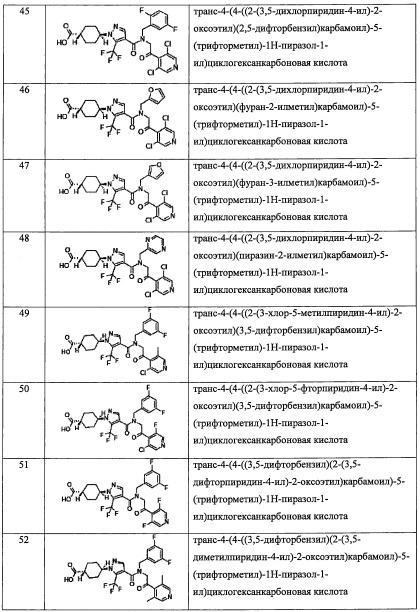
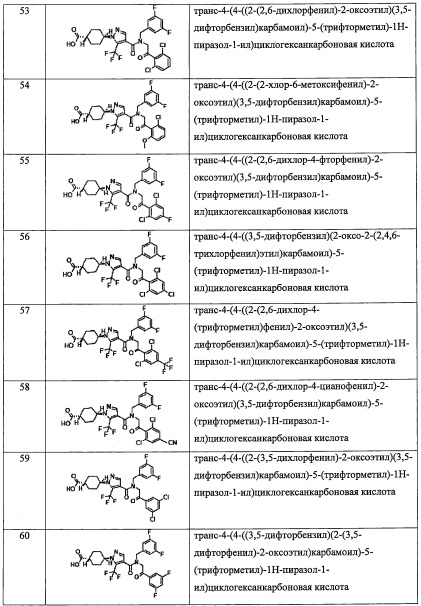
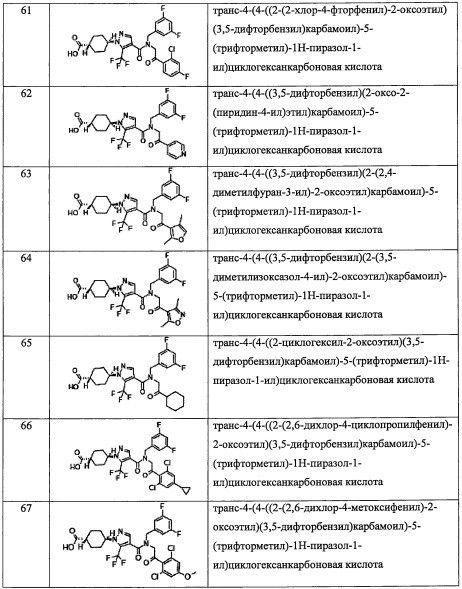
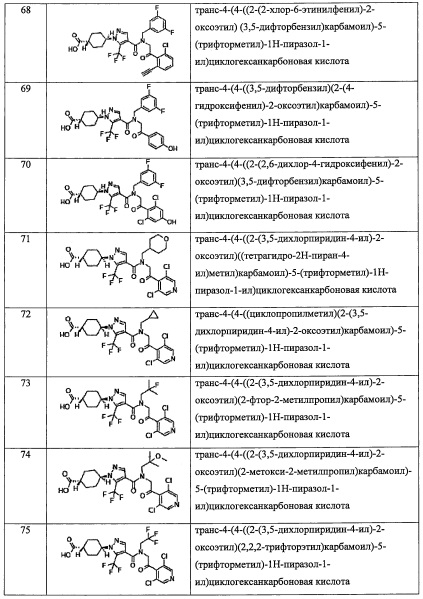
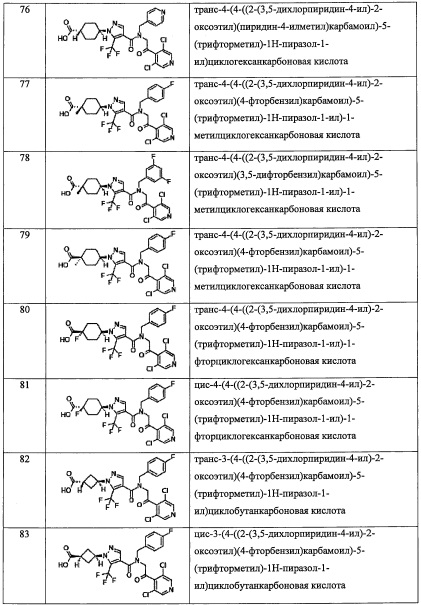
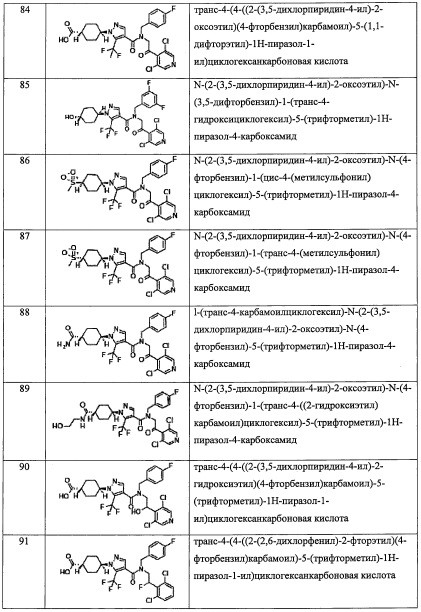
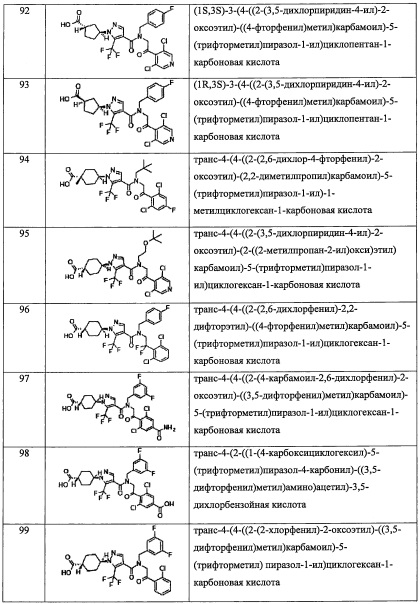
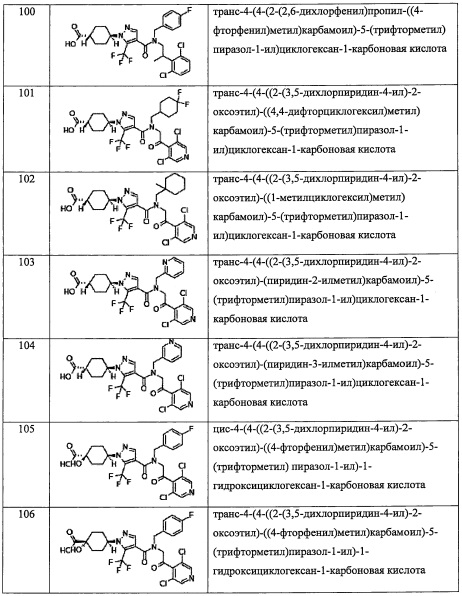
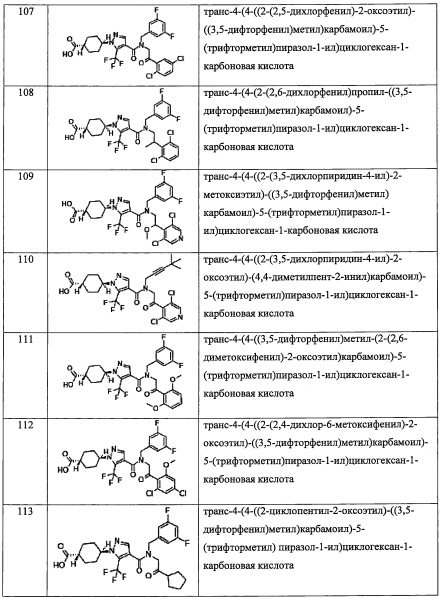
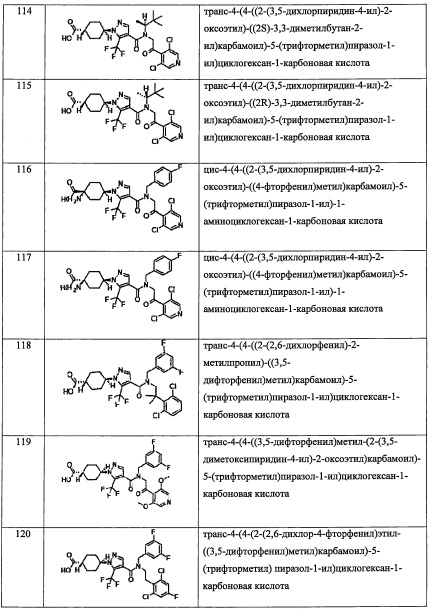
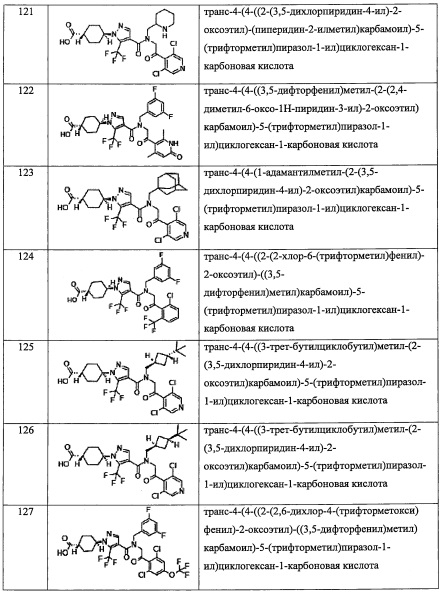
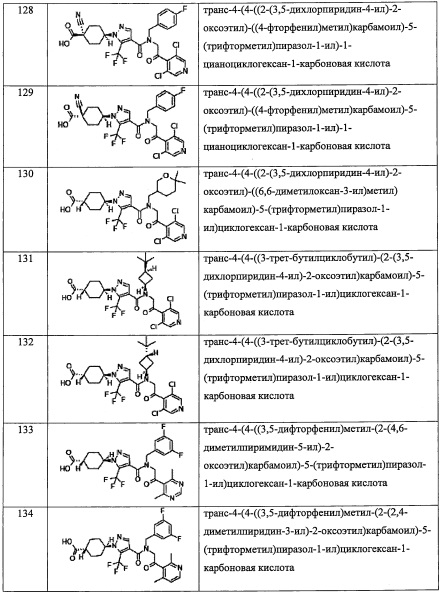
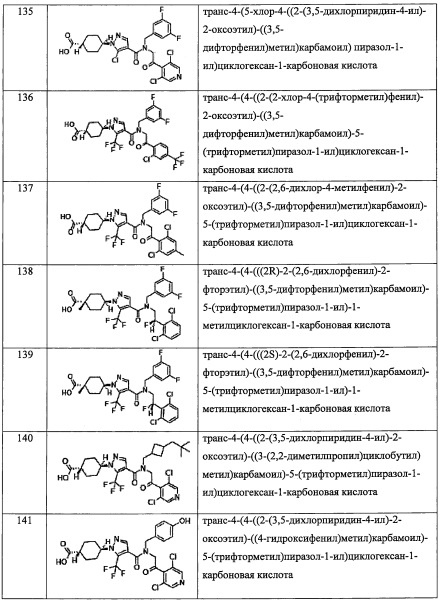
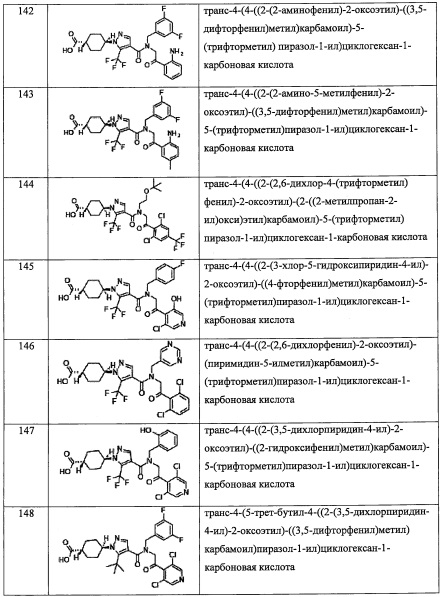
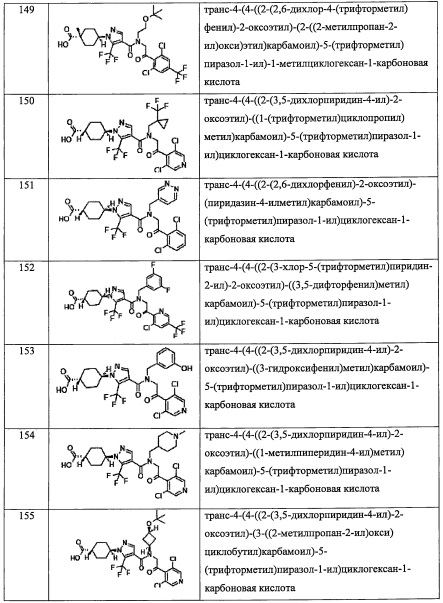
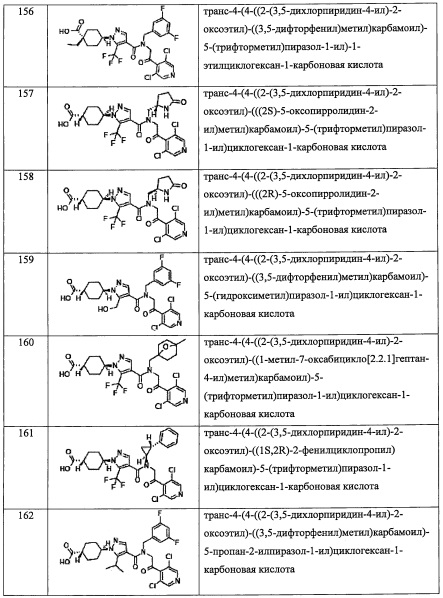
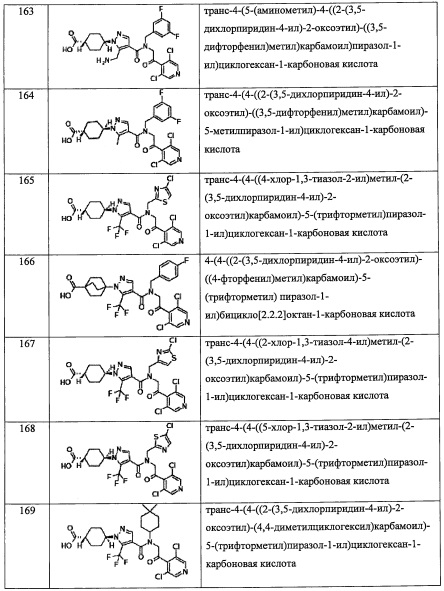
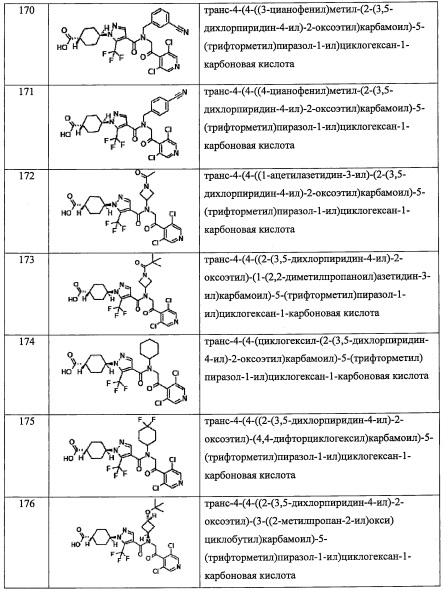
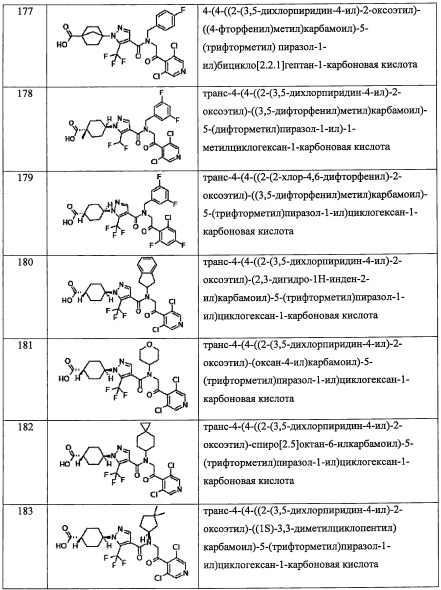
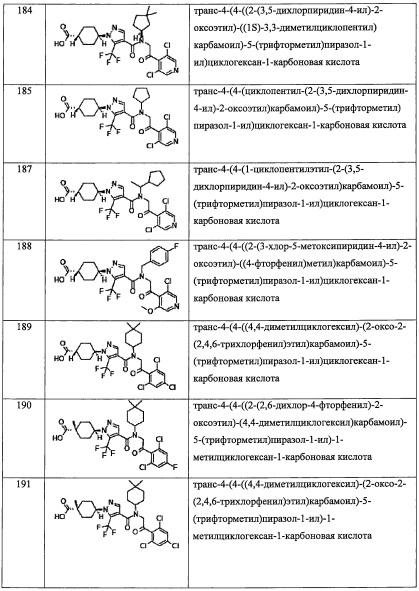
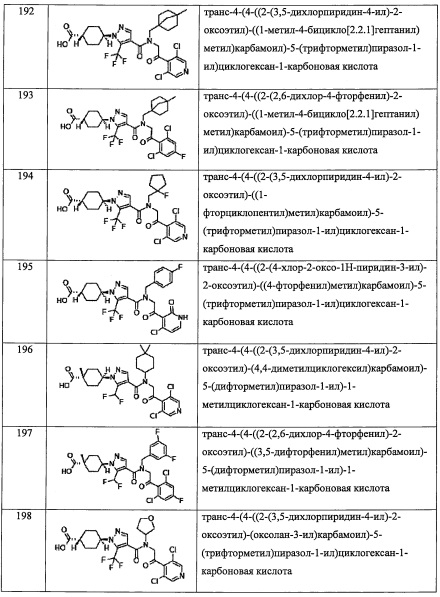
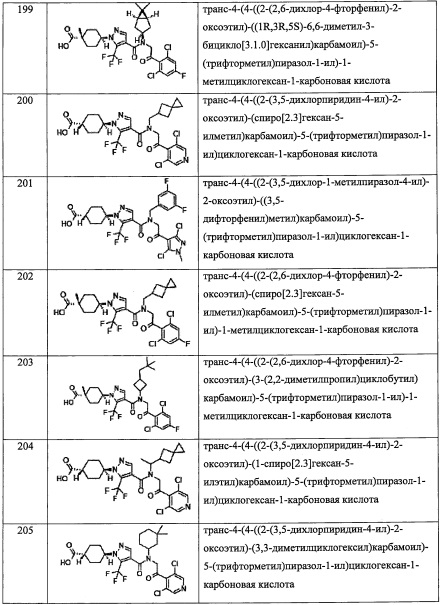
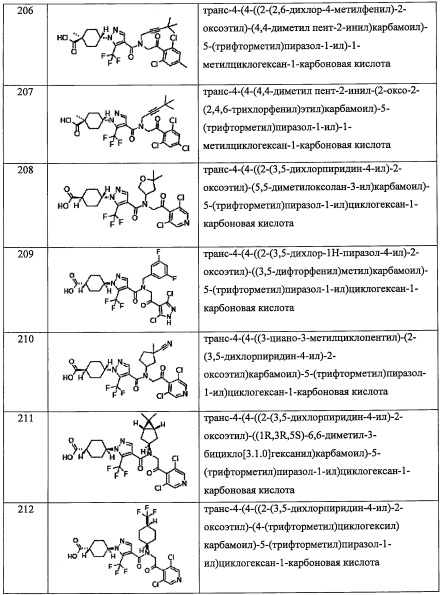
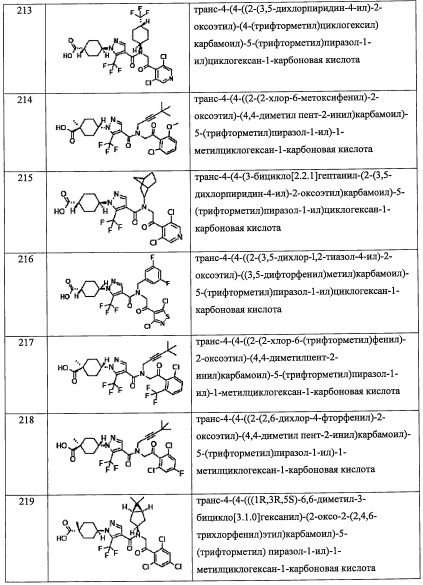
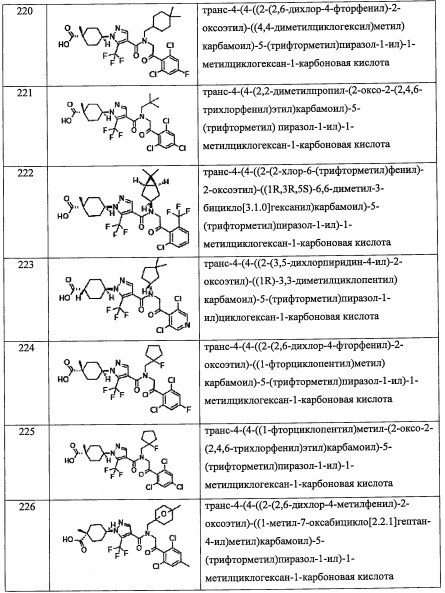
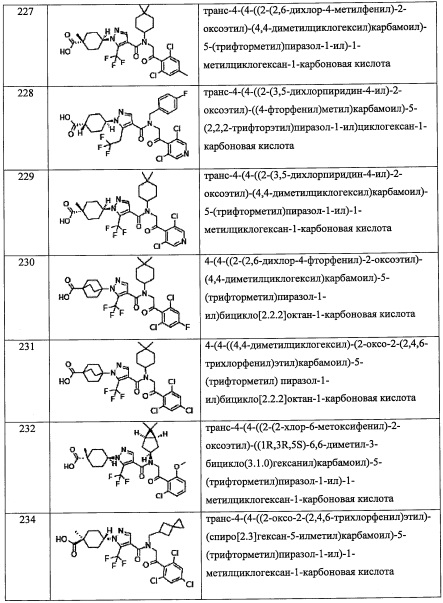
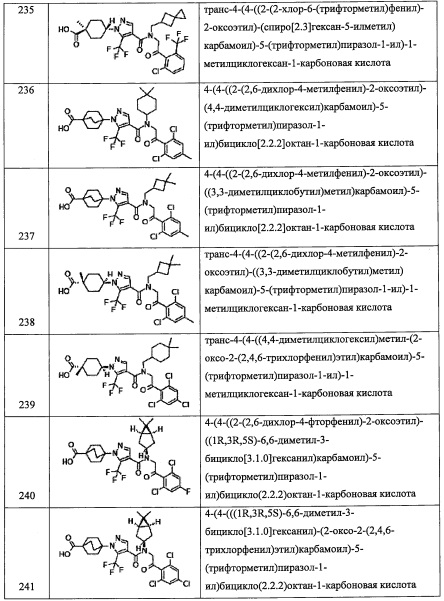
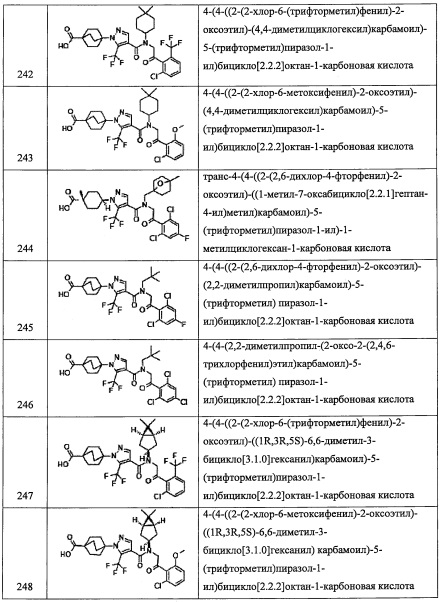
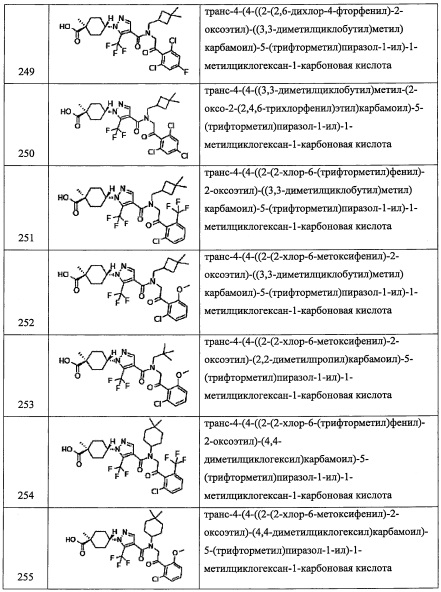
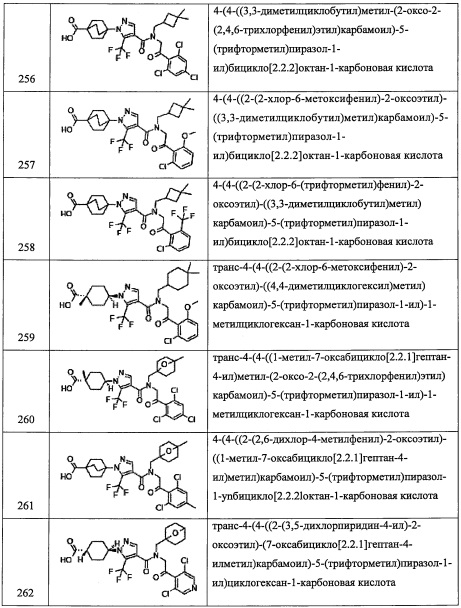
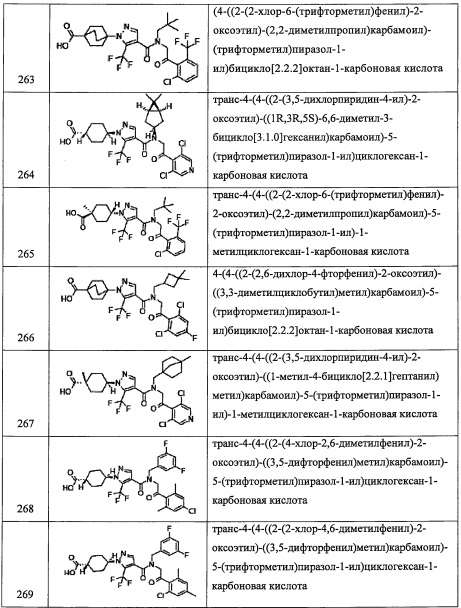
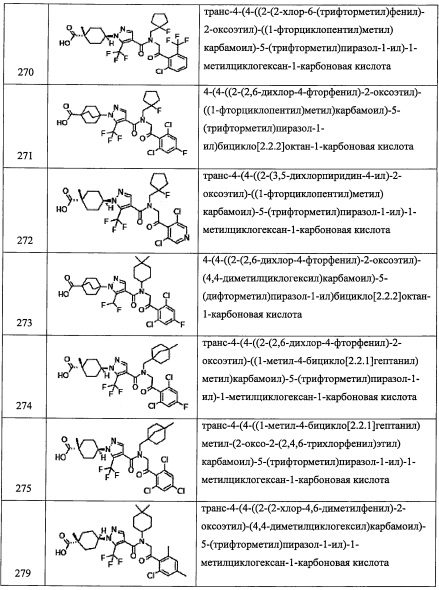
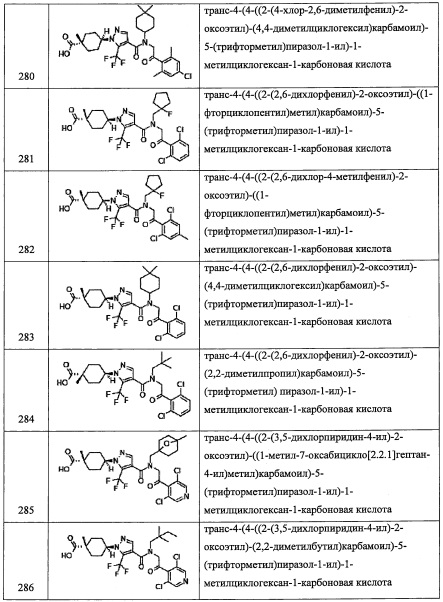
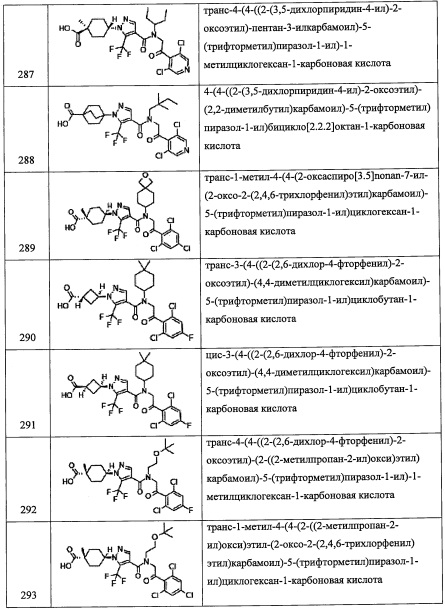
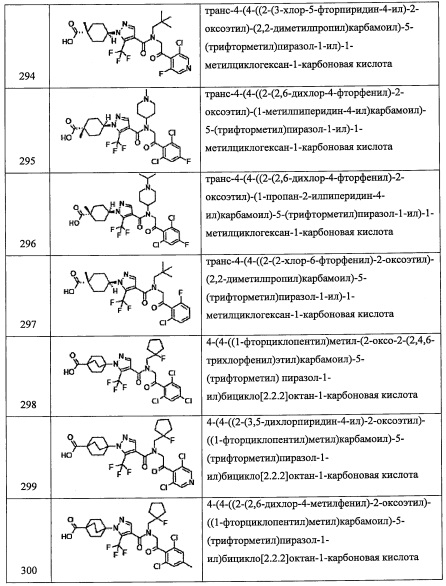

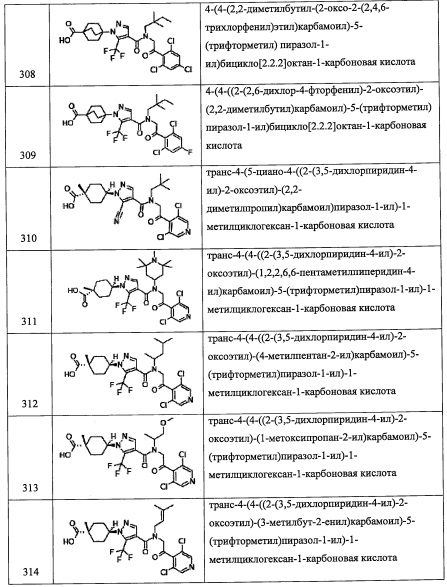
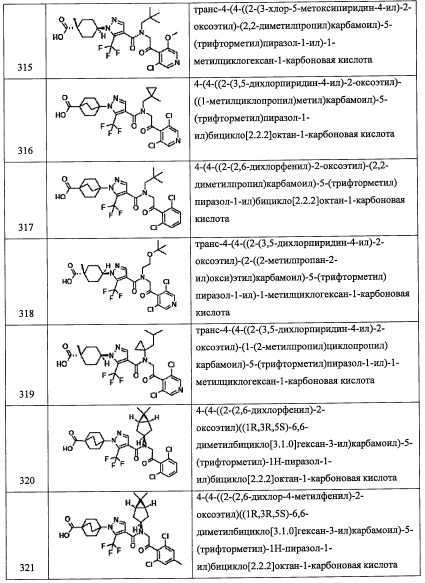
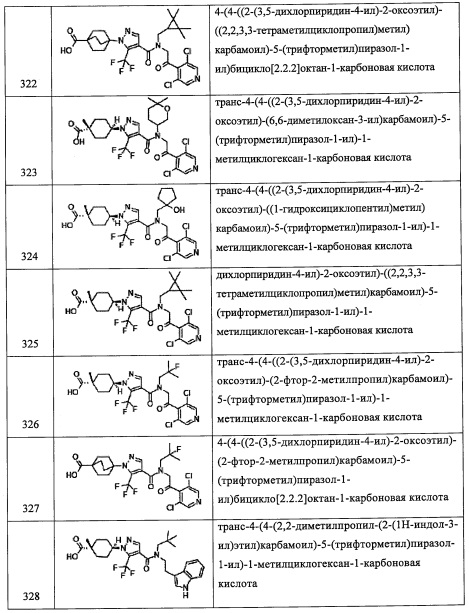
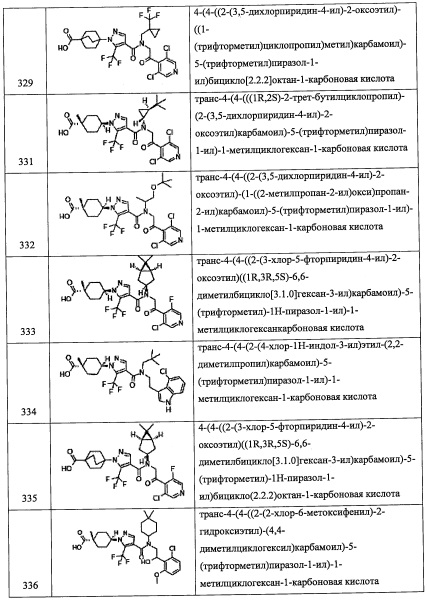
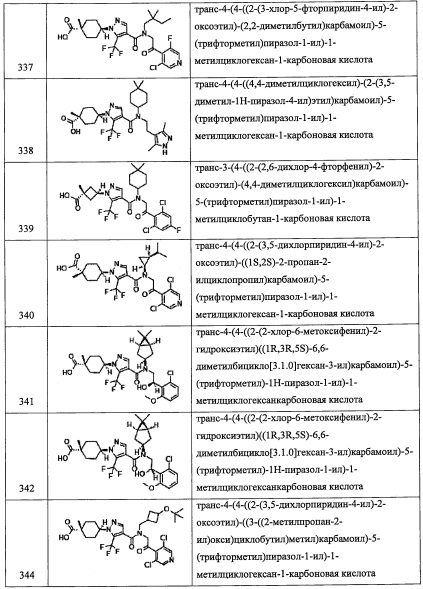
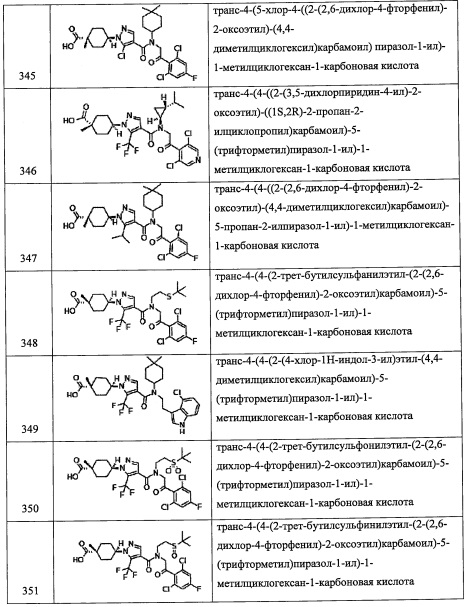
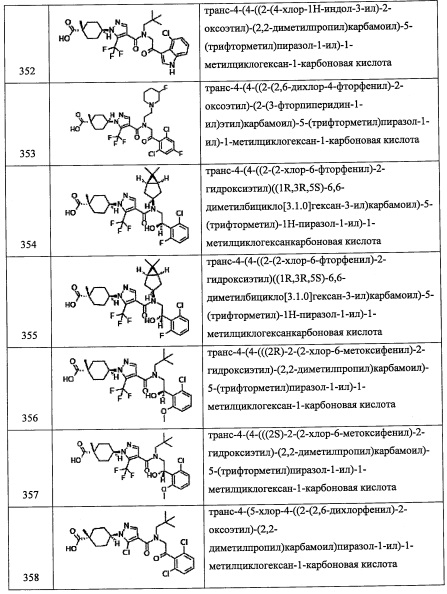
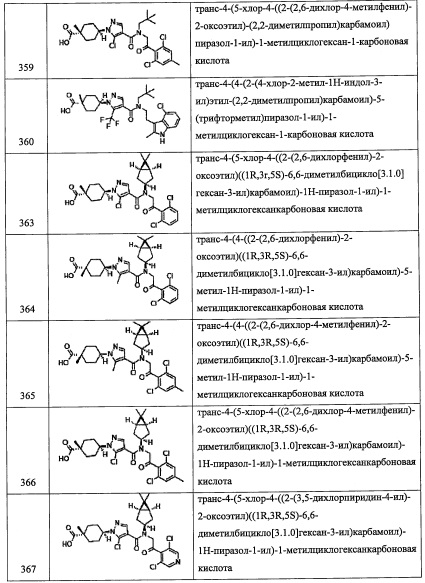
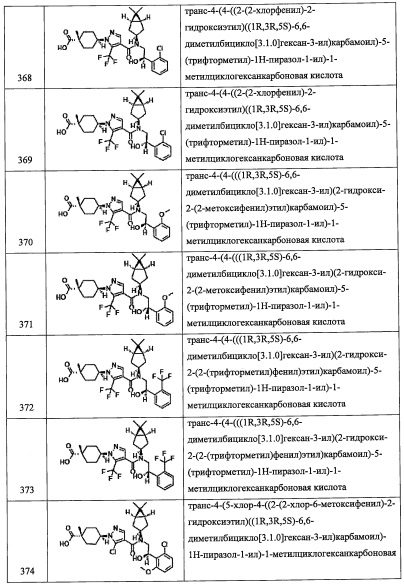
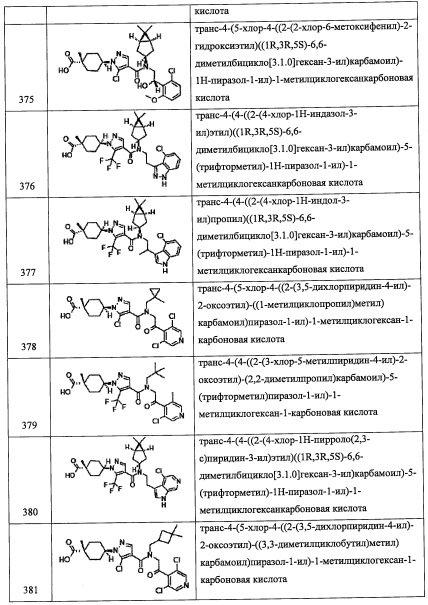
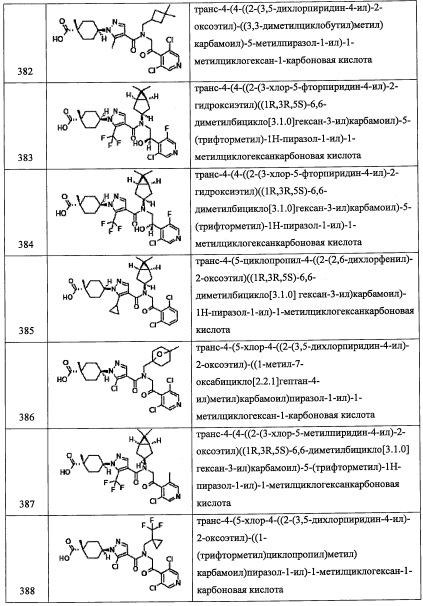
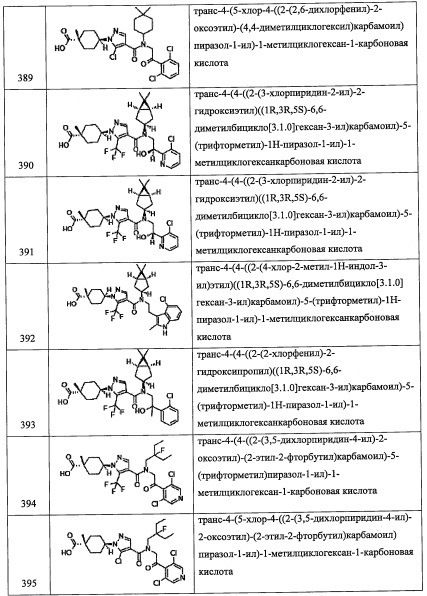
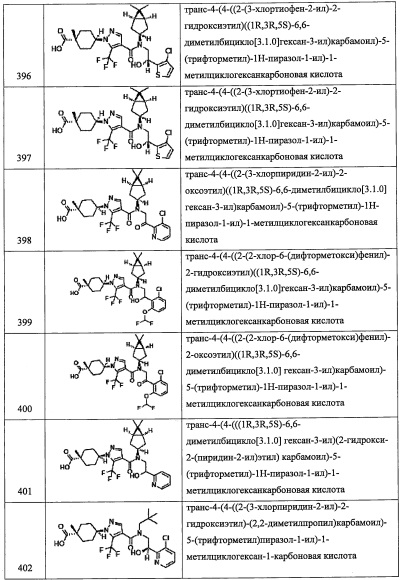
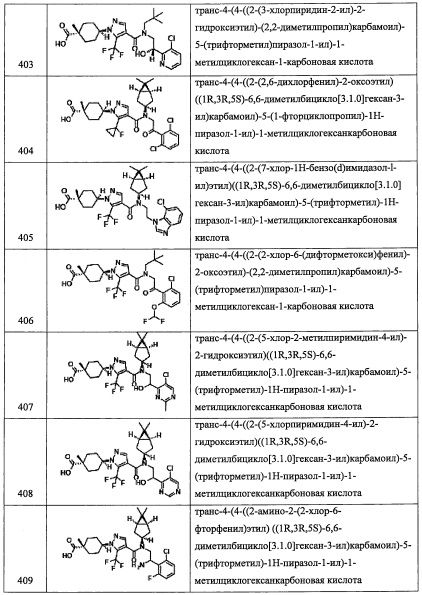
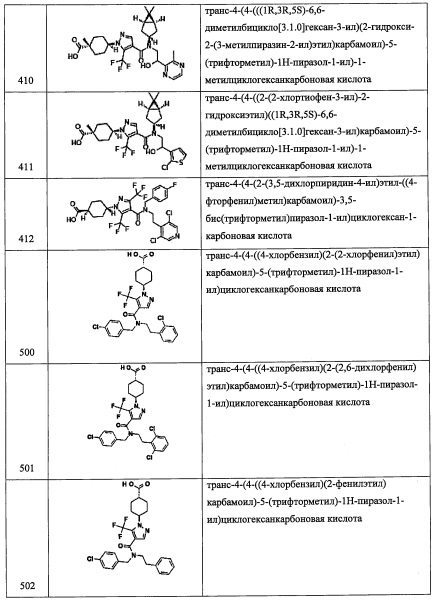
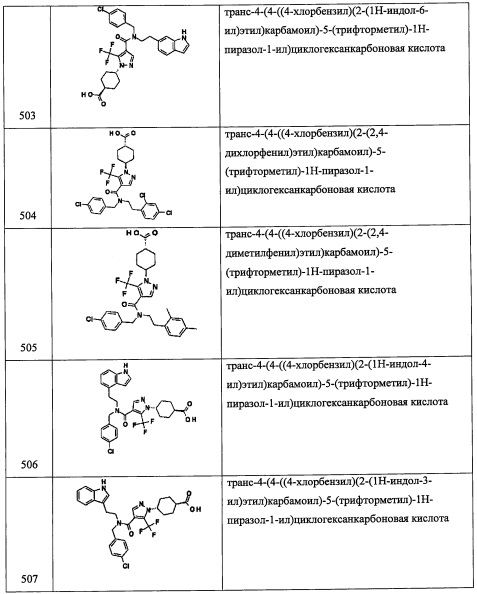
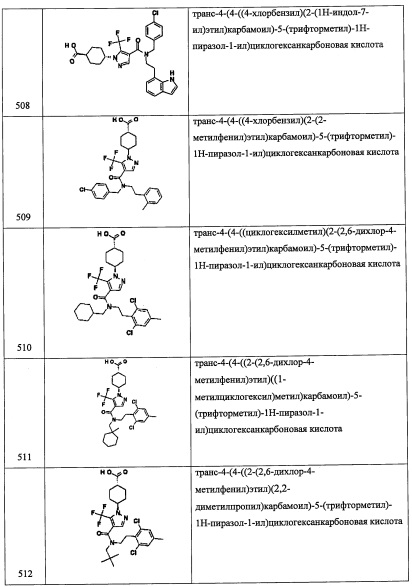
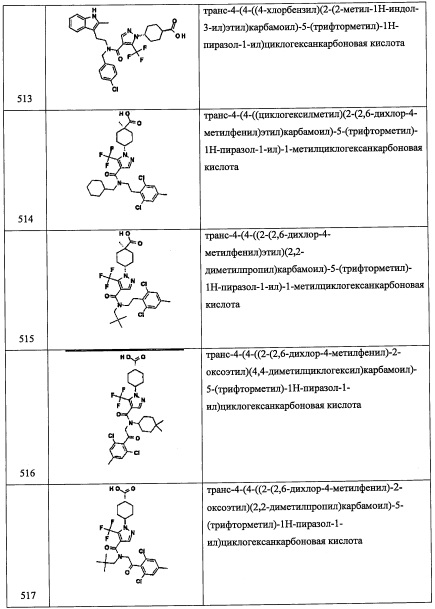
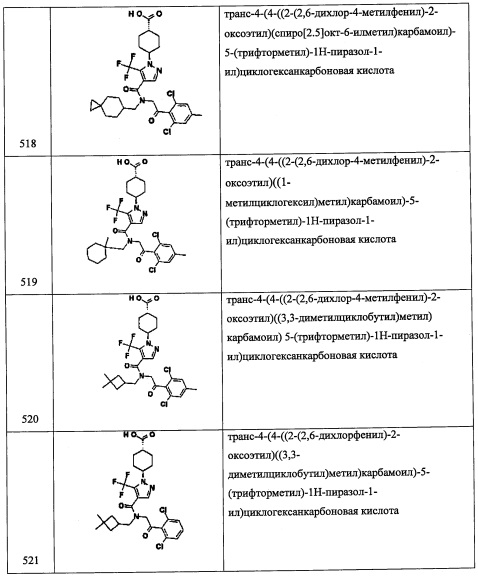
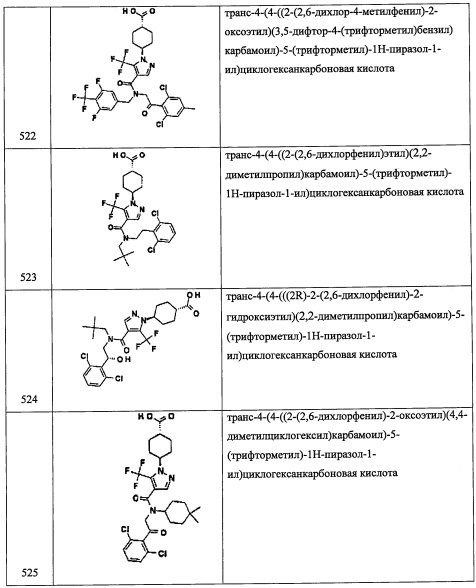
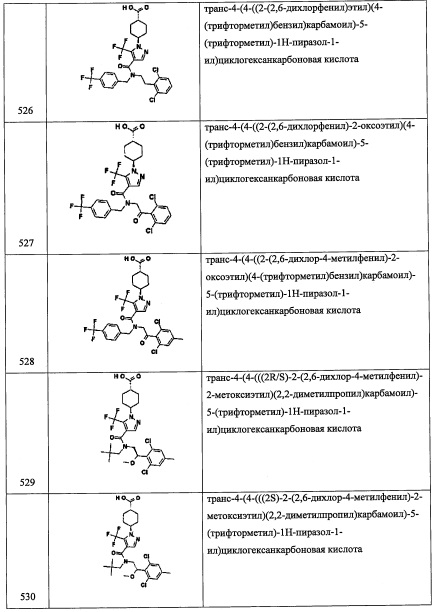
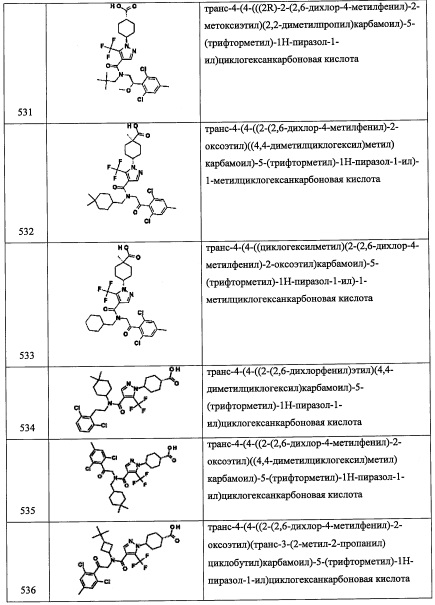
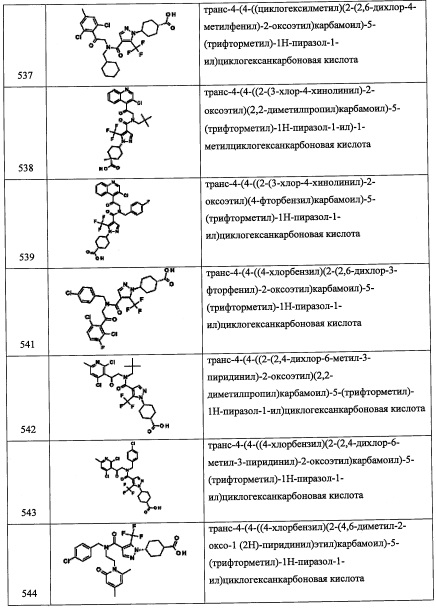
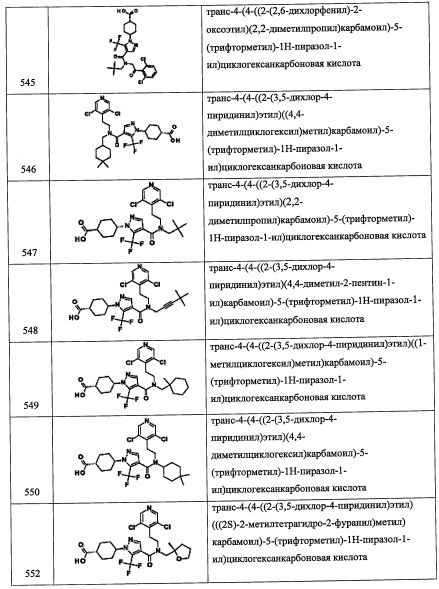
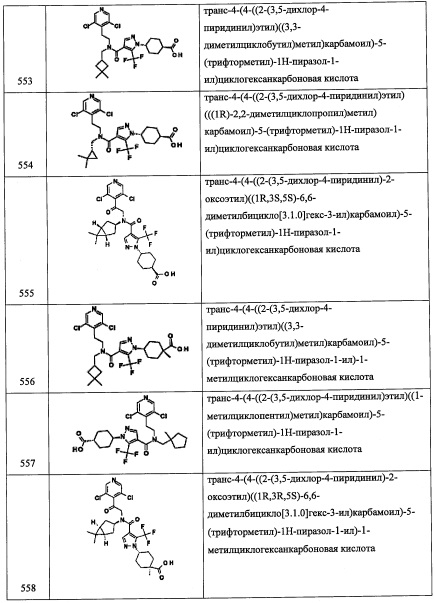
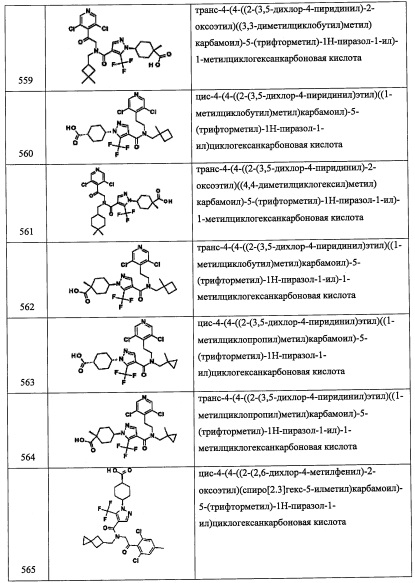
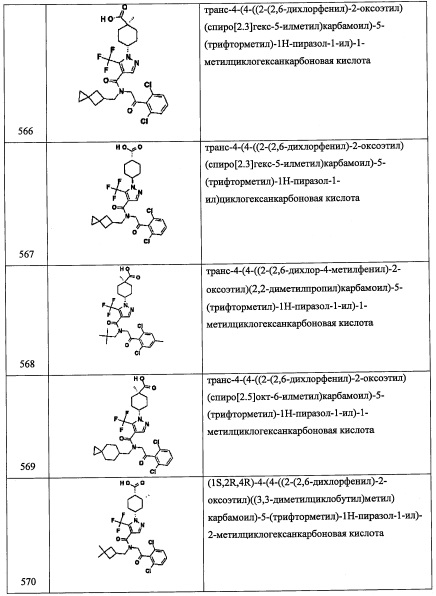
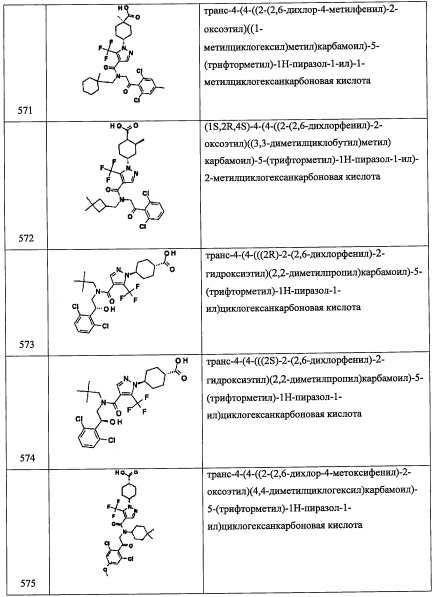
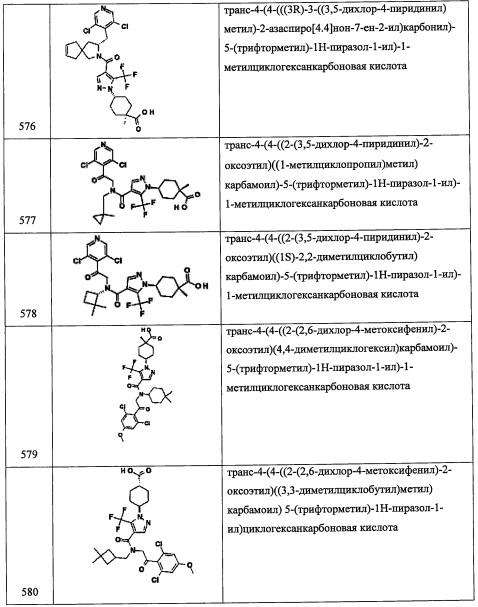
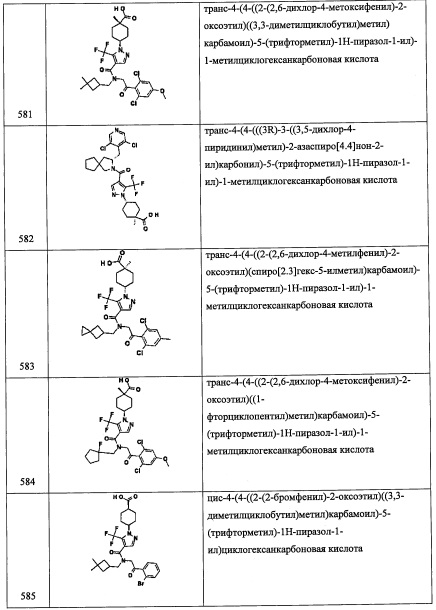
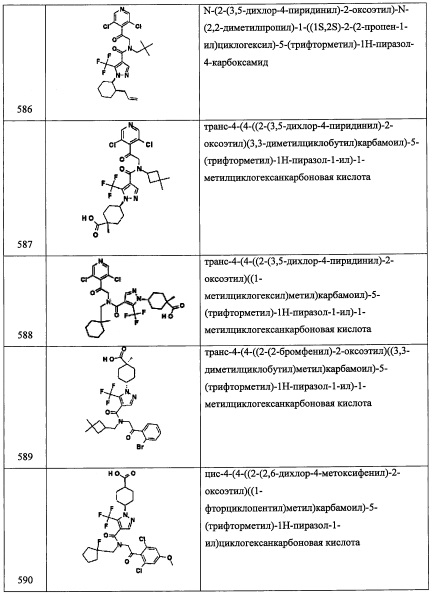
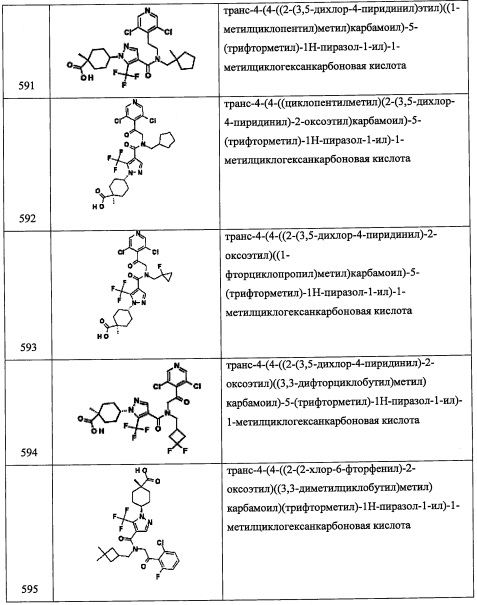
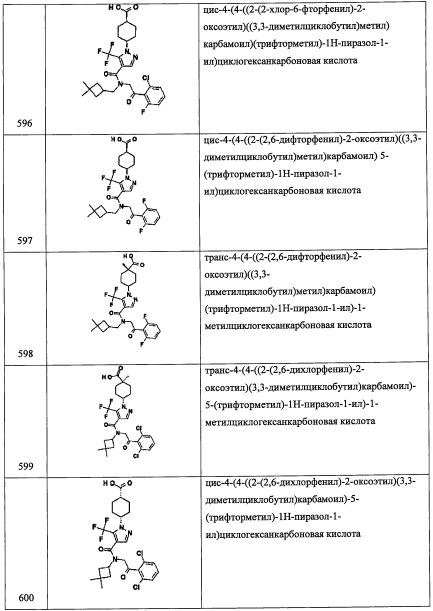
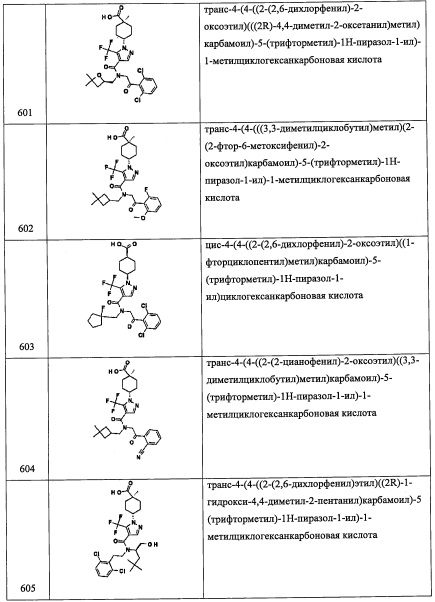
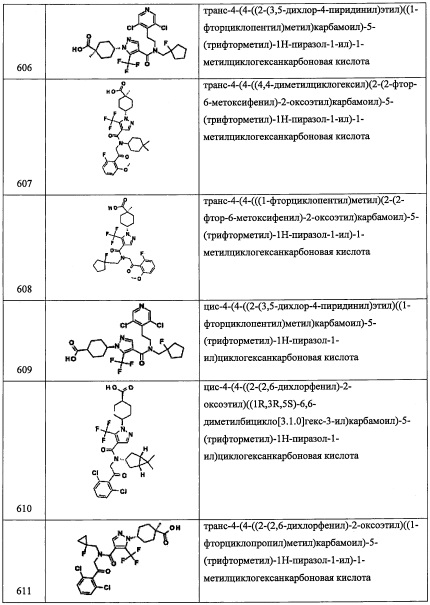
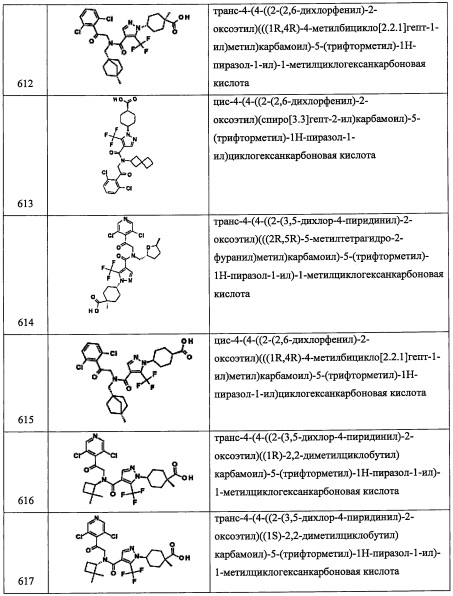
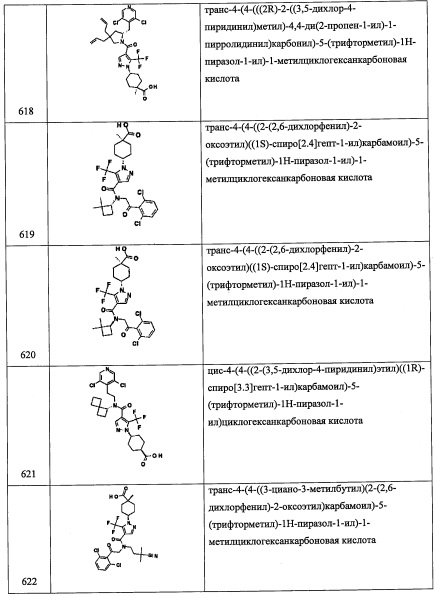
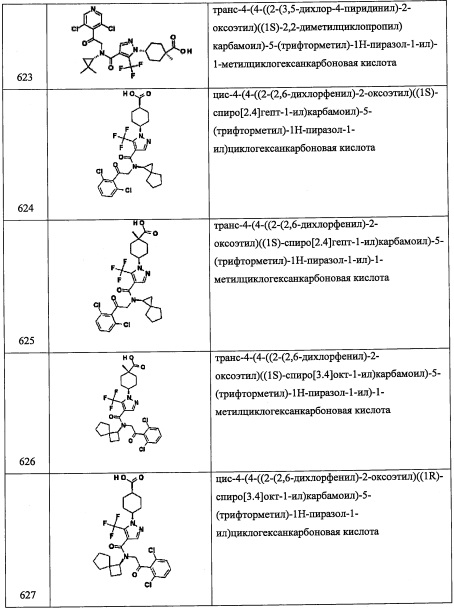
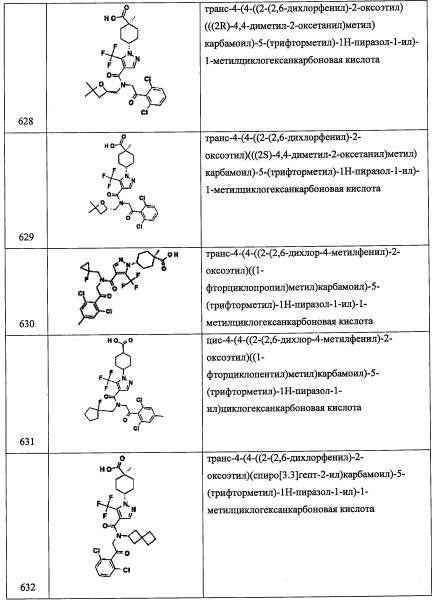
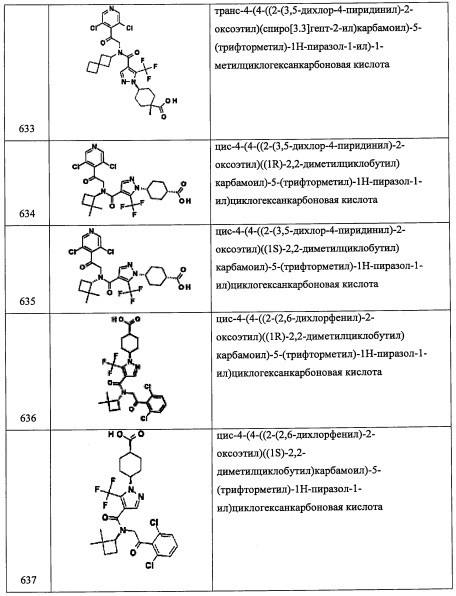
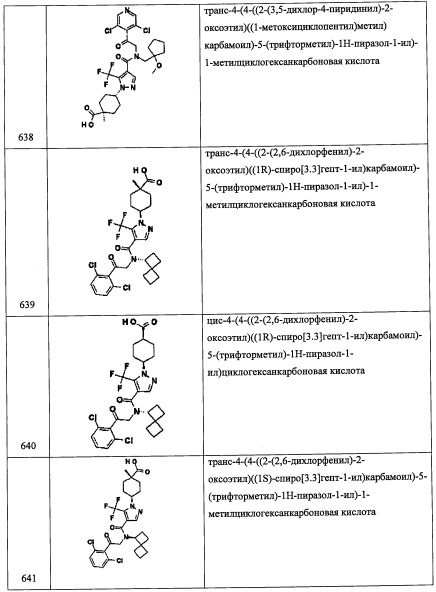
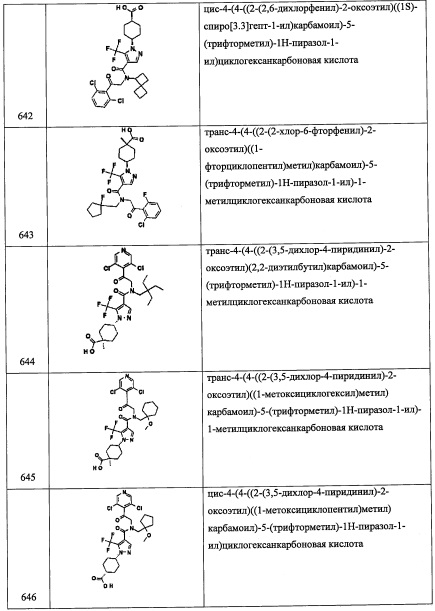
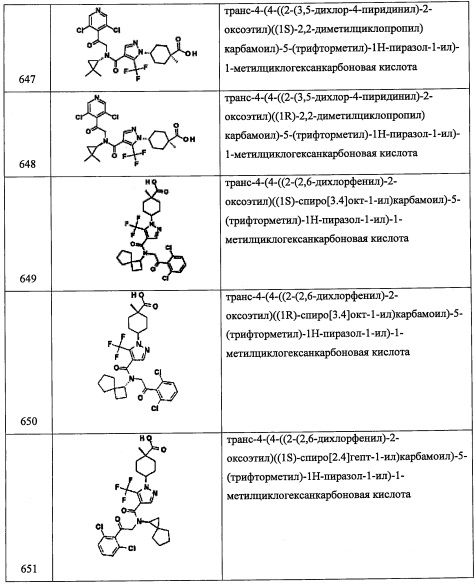
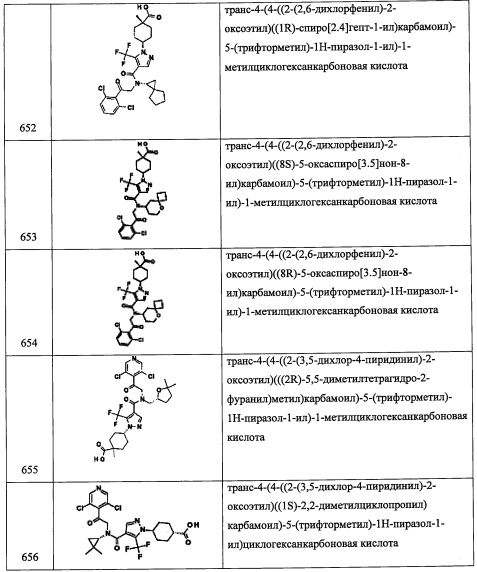
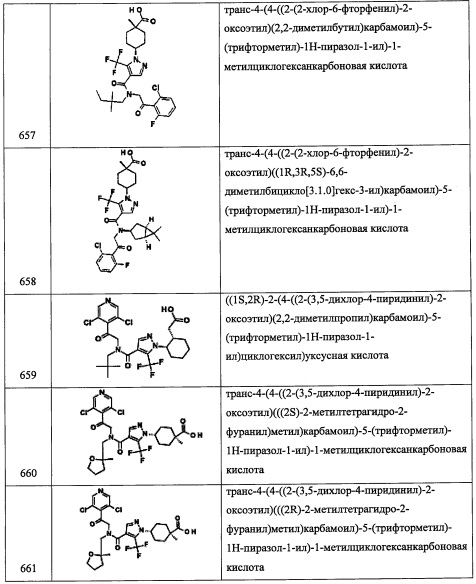
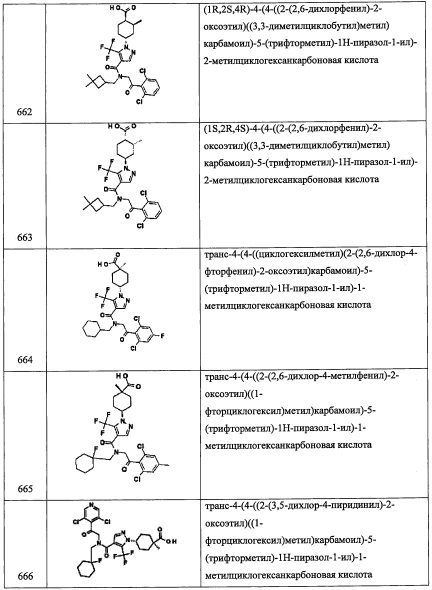
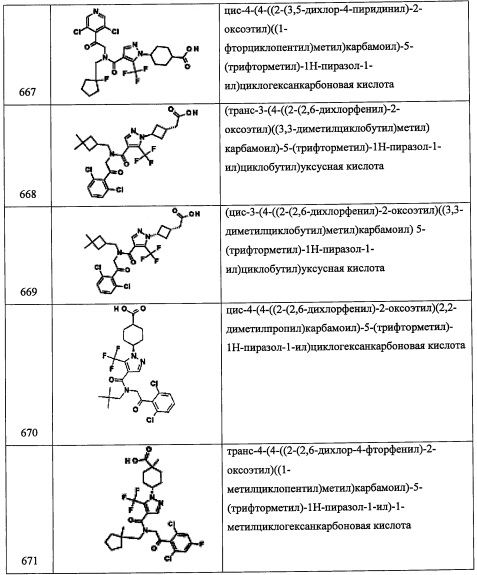
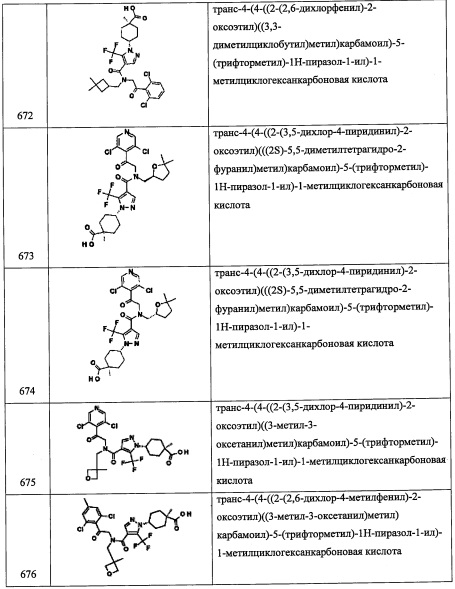
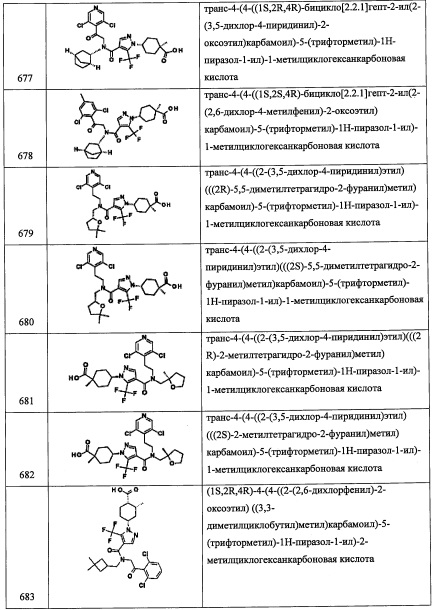
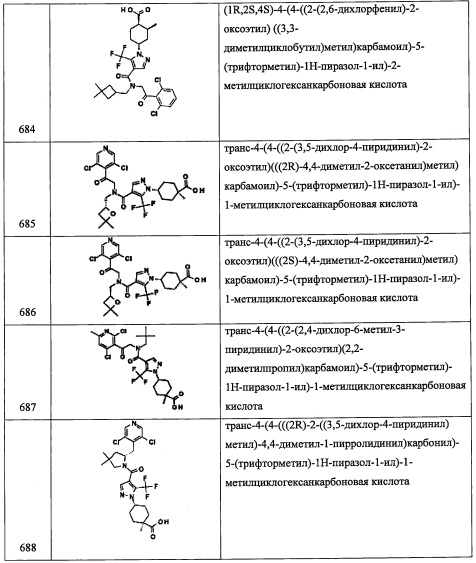
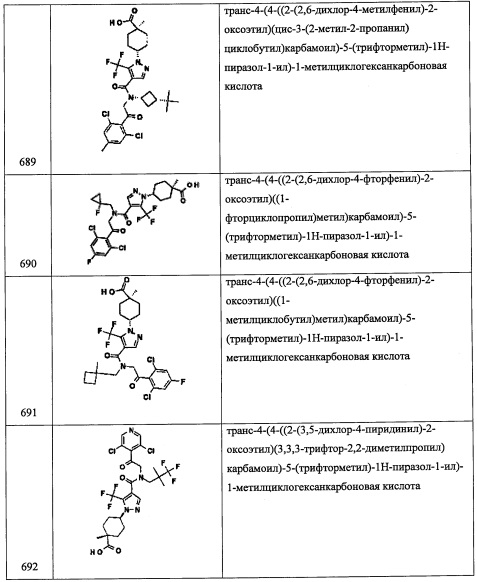
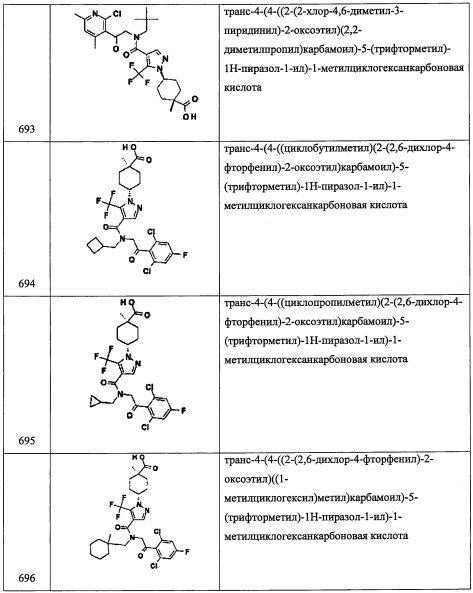
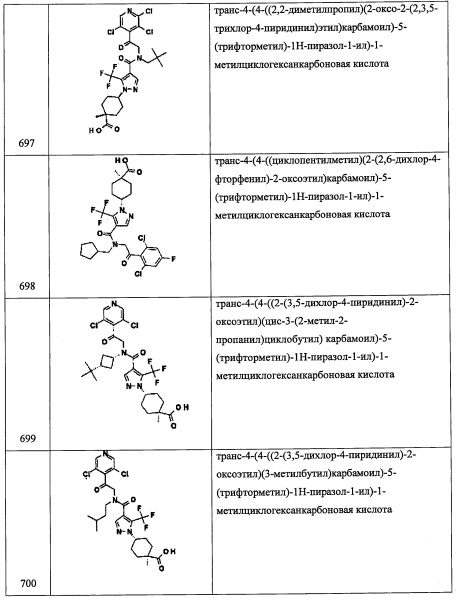
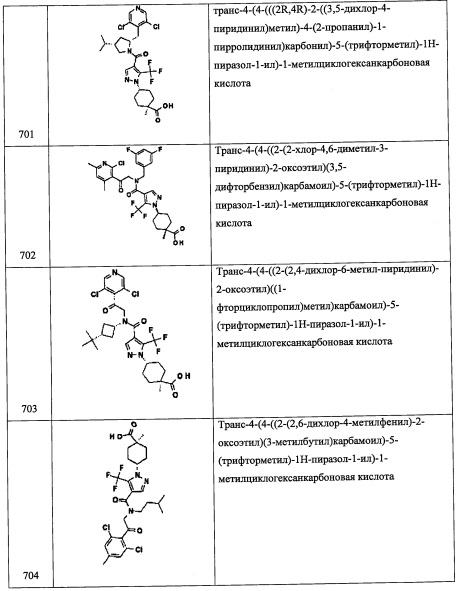
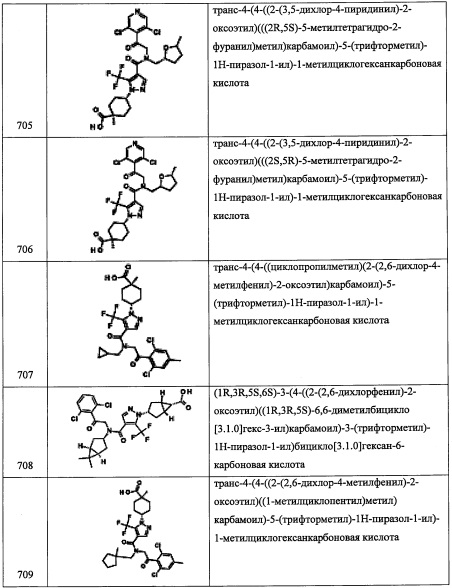
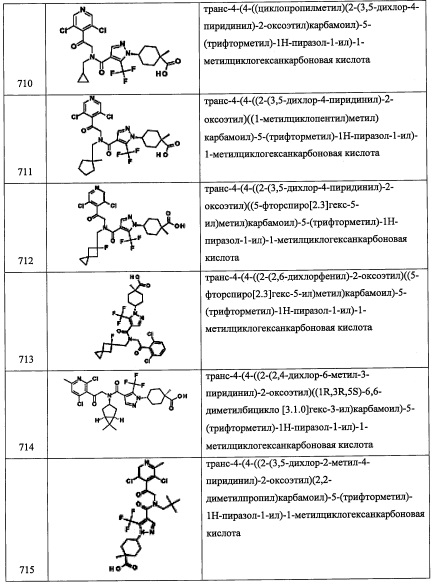
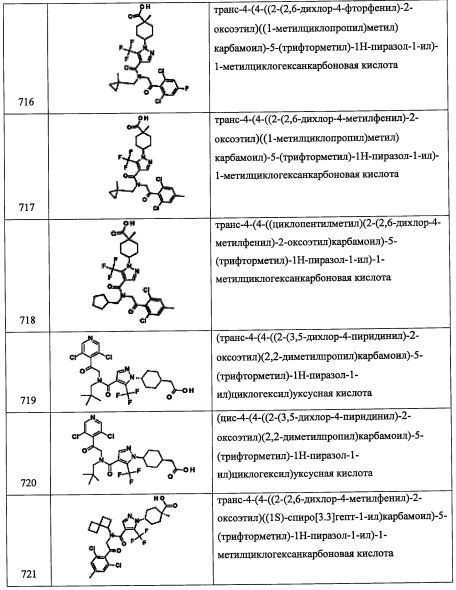
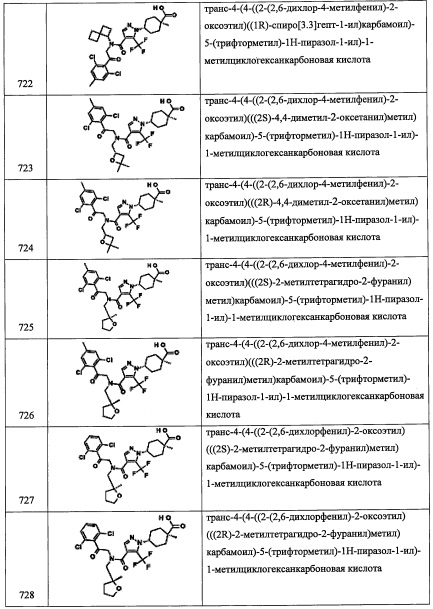
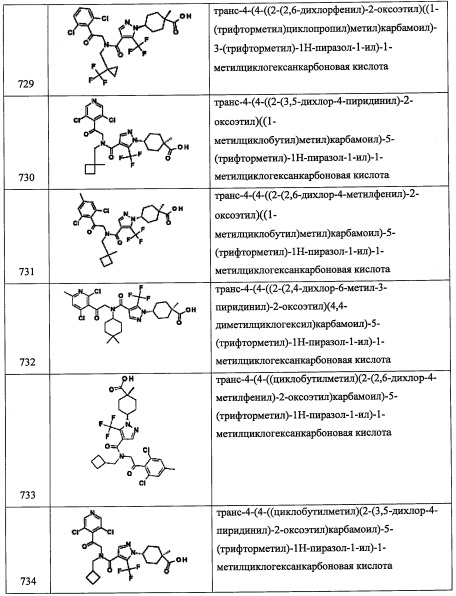
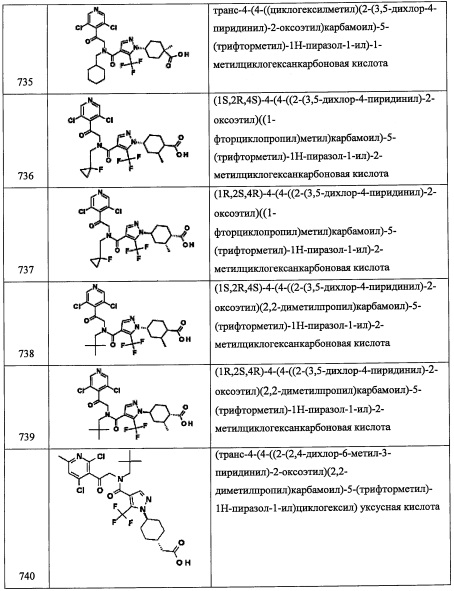
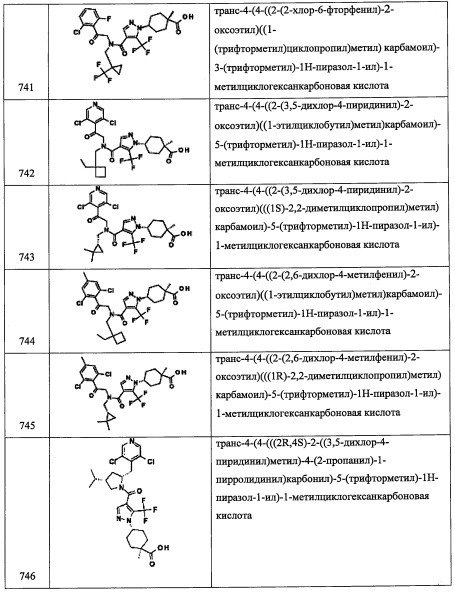
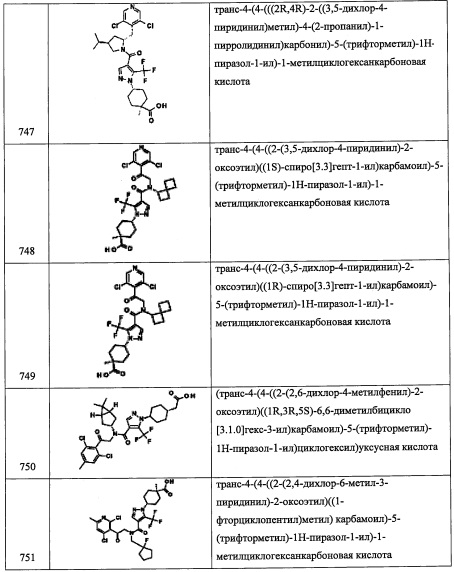
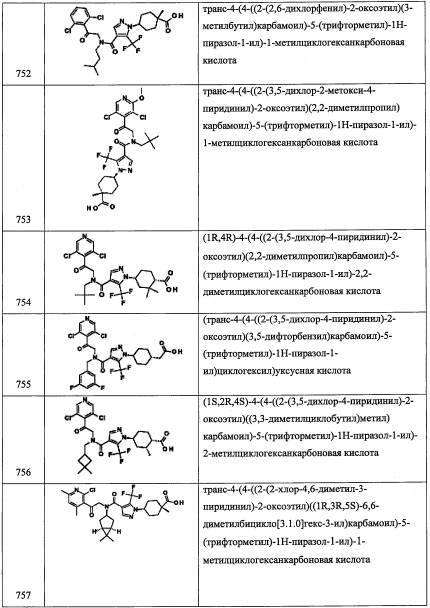
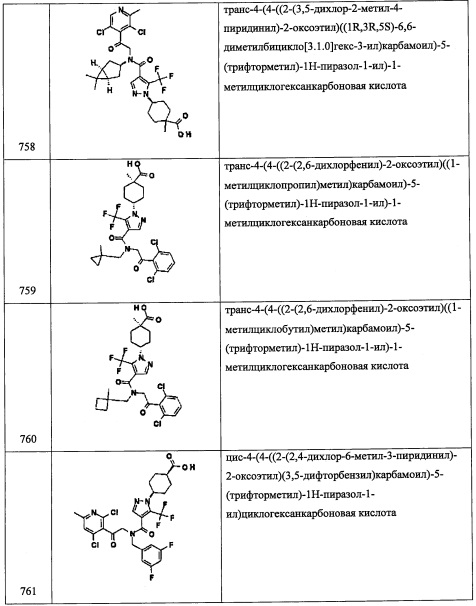
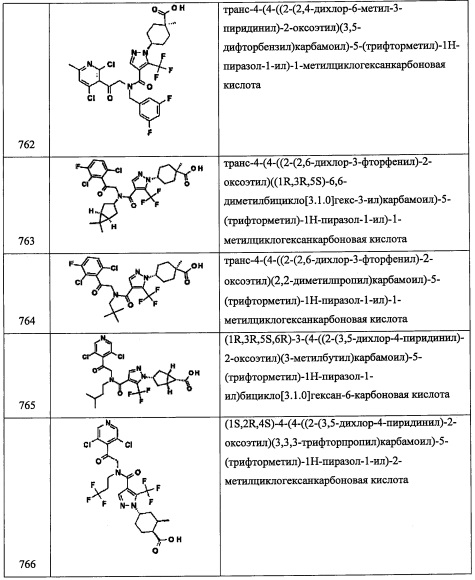
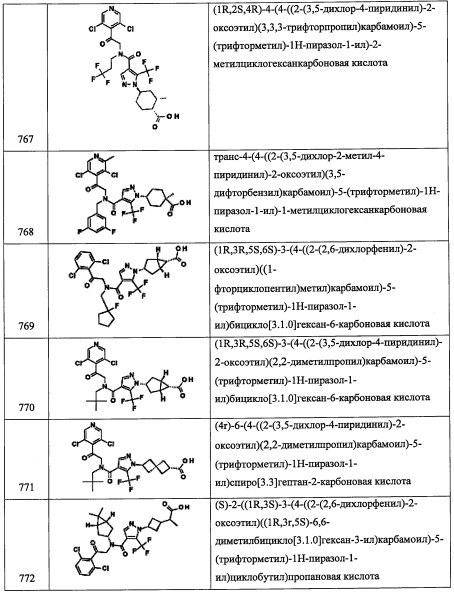
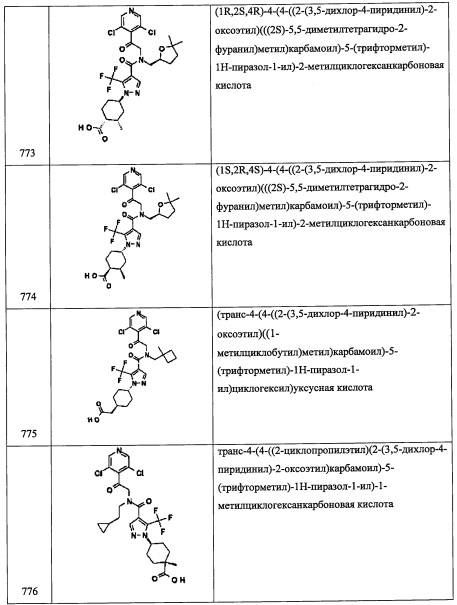
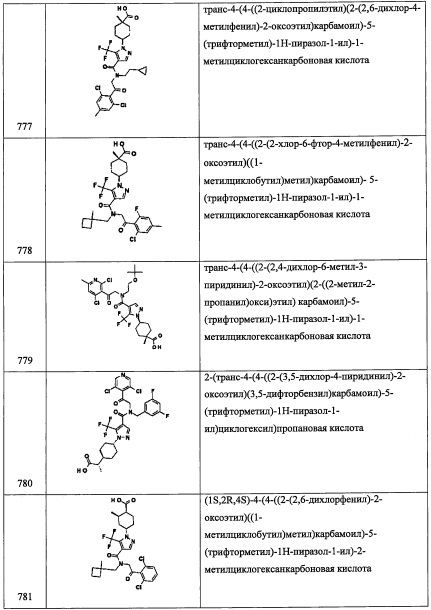
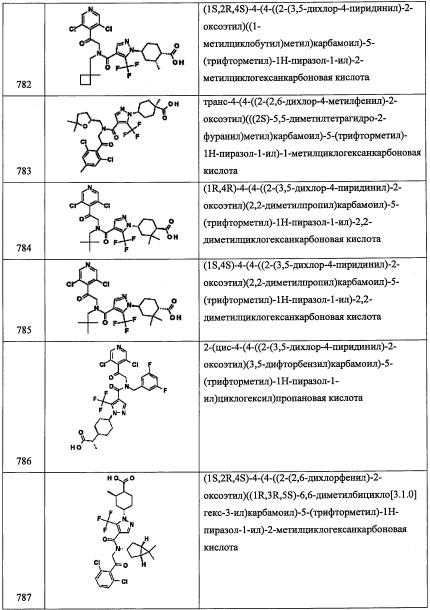
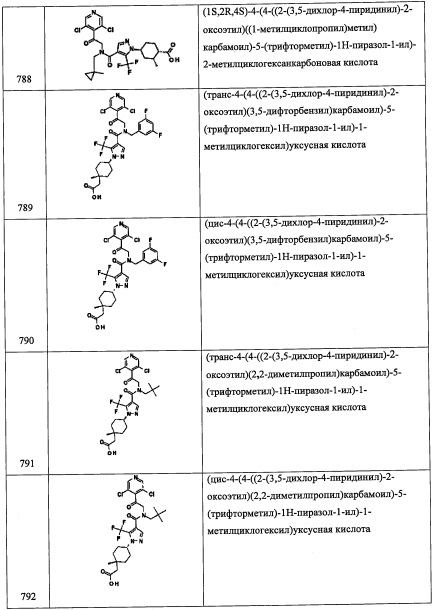
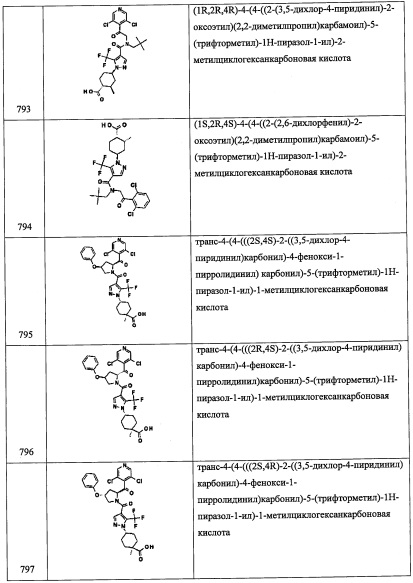
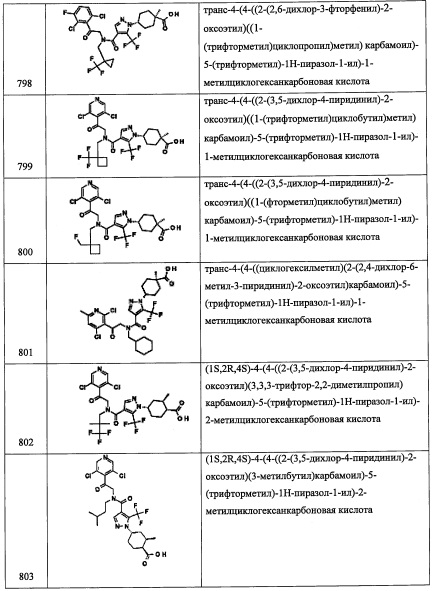
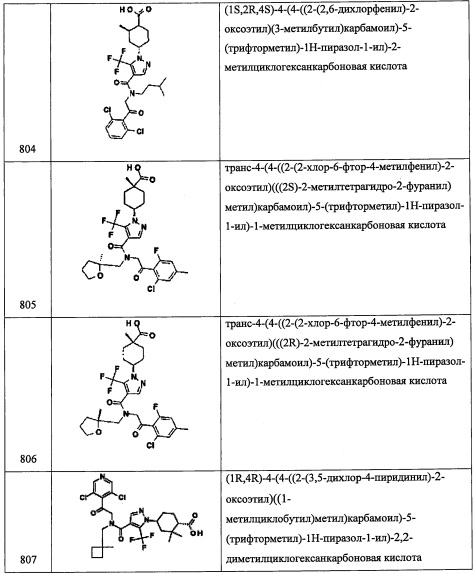
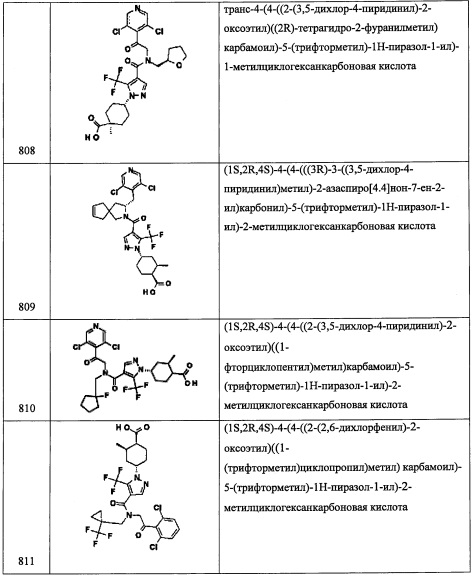
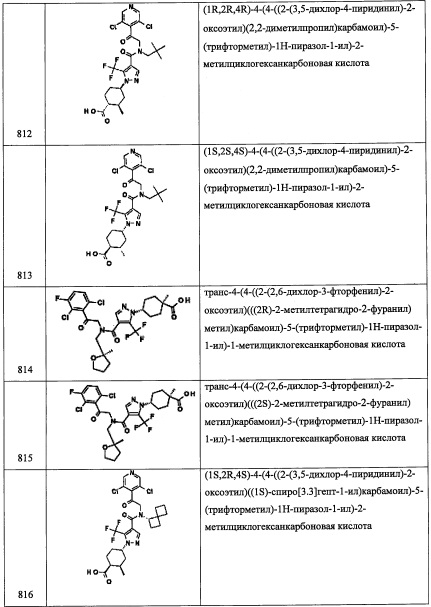
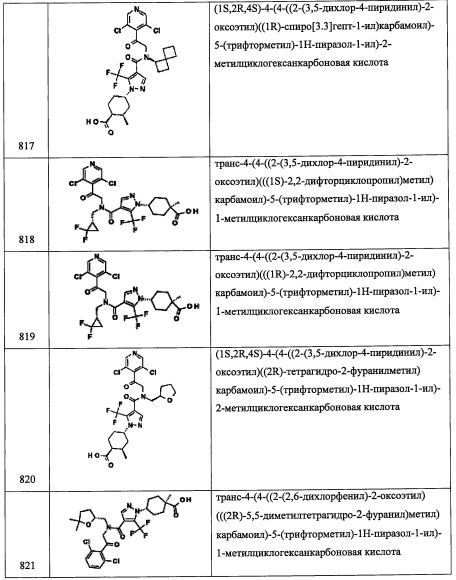
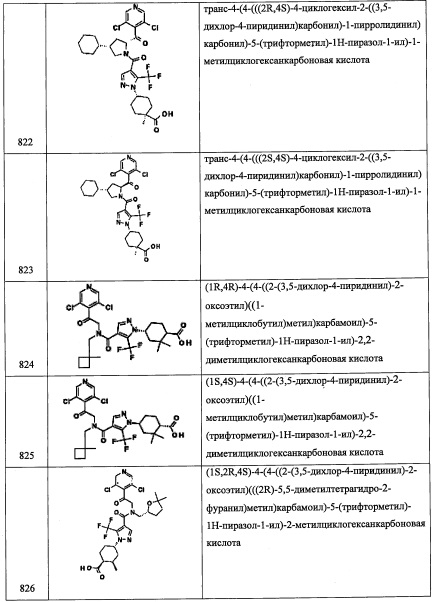
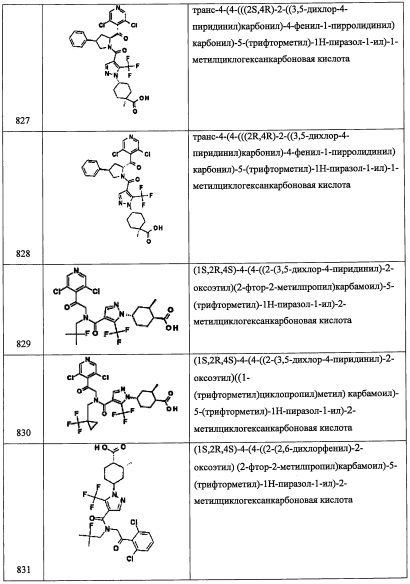
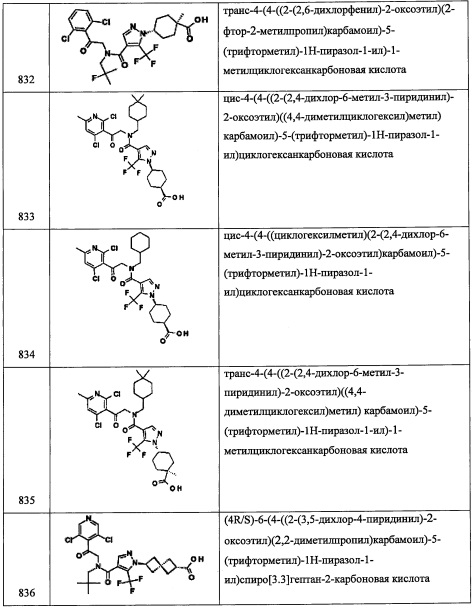
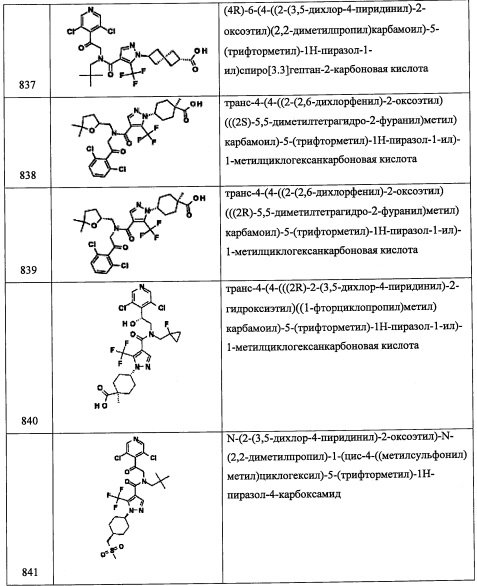
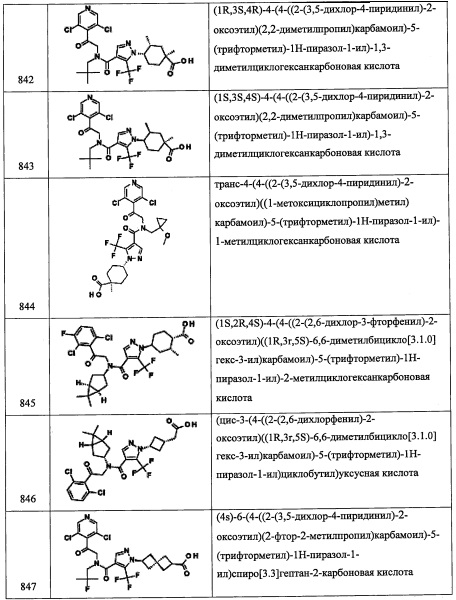
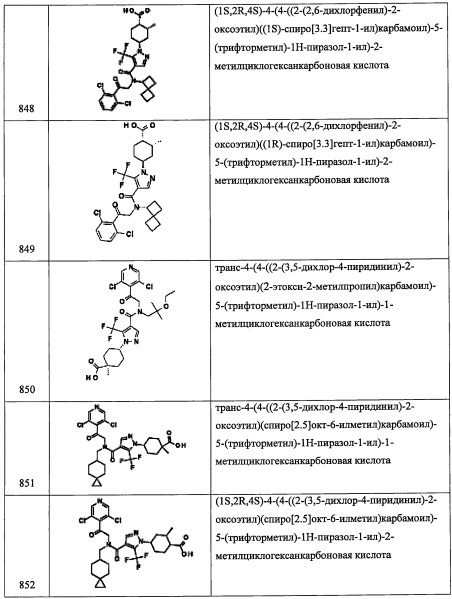
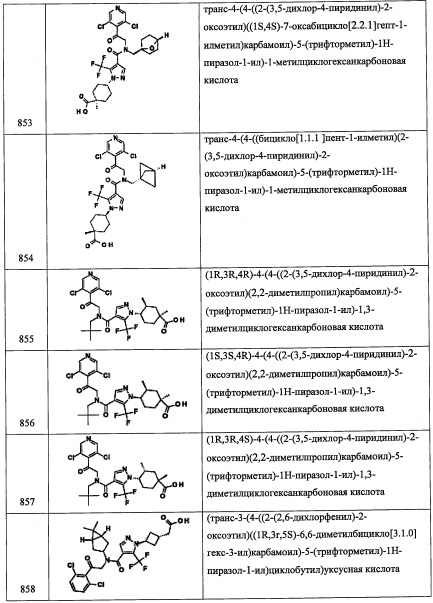
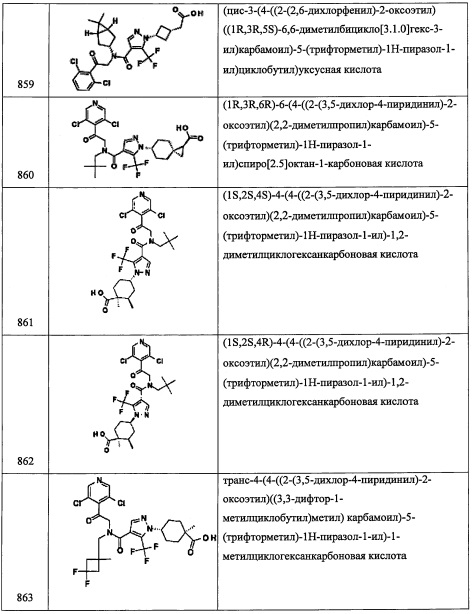
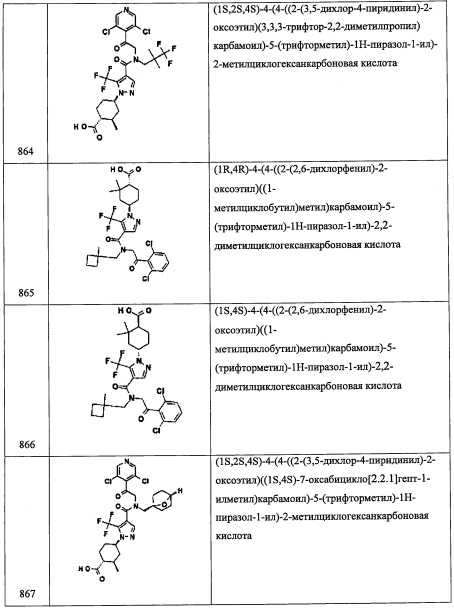
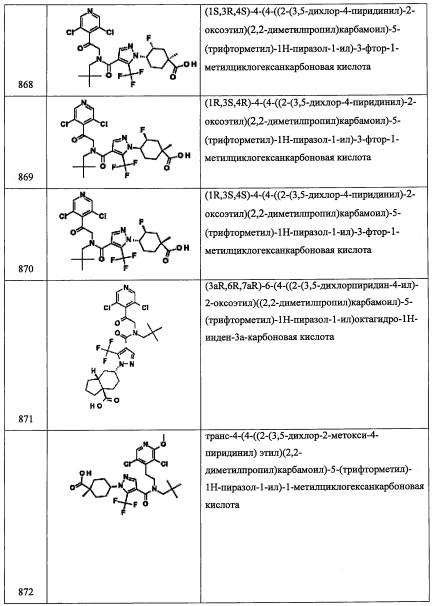

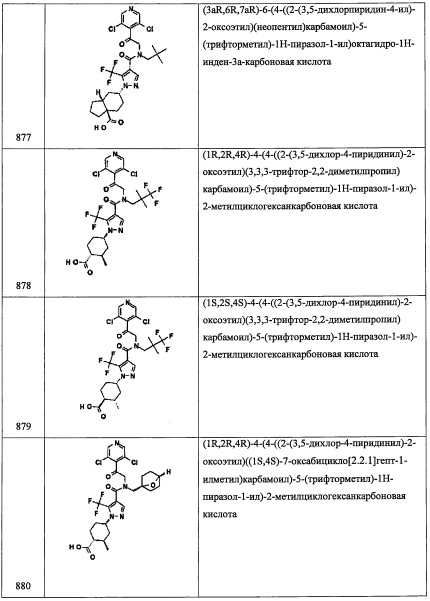
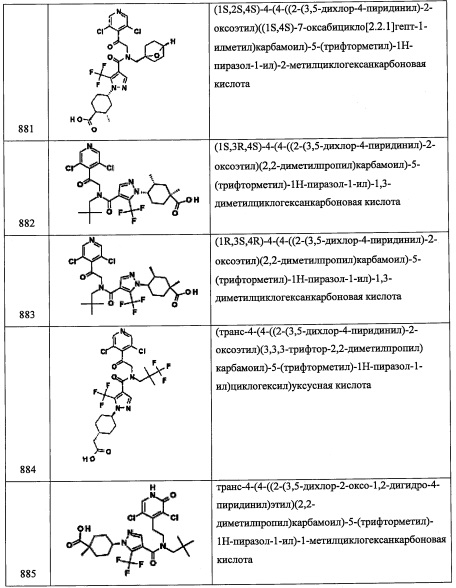
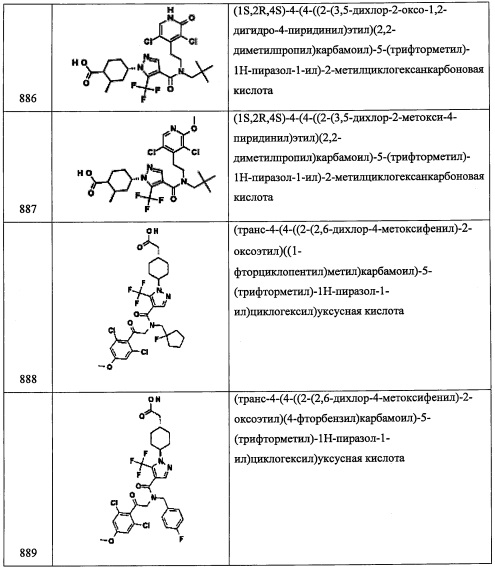
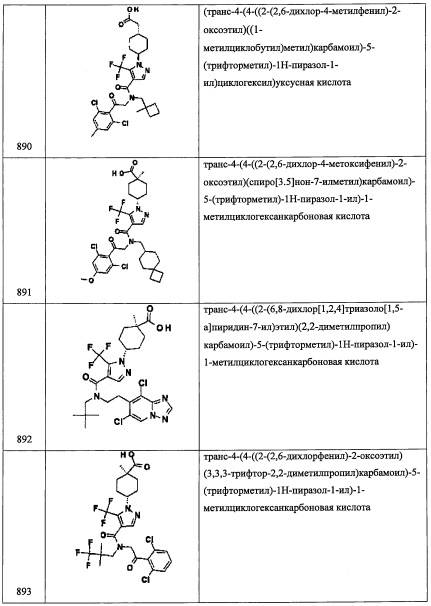
Результаты анализа перечисленных выше примеров методом ВЭЖХ-МС (LC/MS) представлены далее в таблицах.
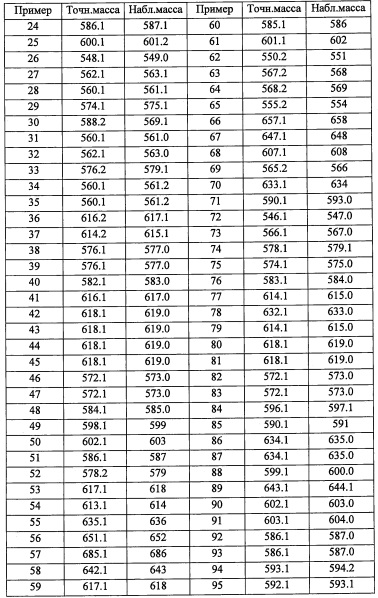
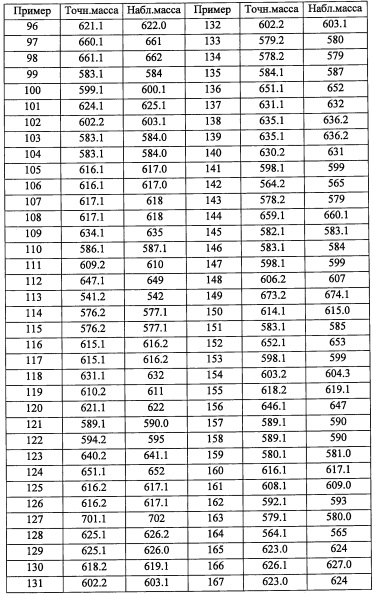
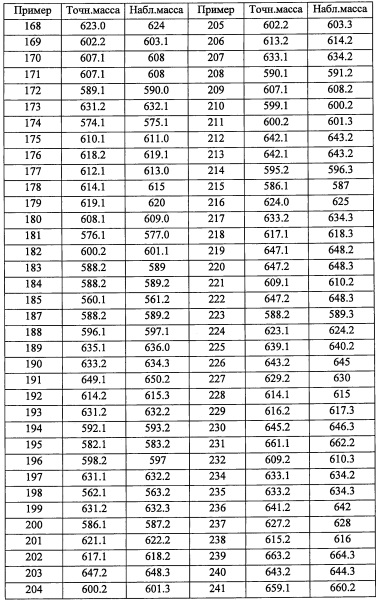
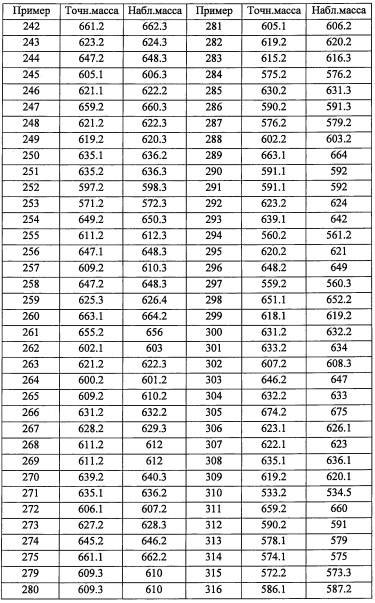
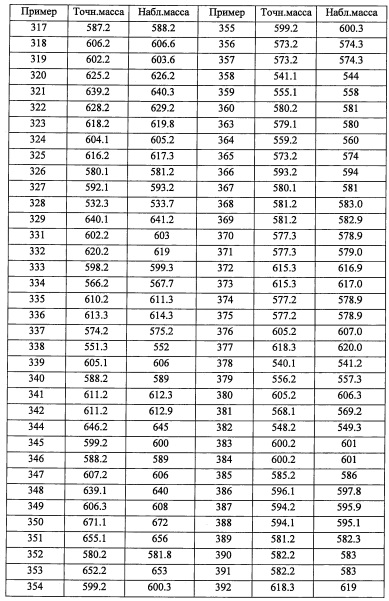
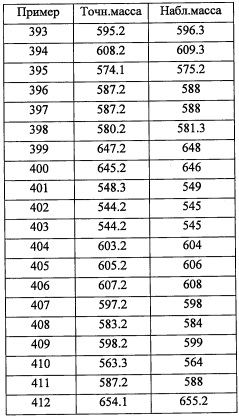
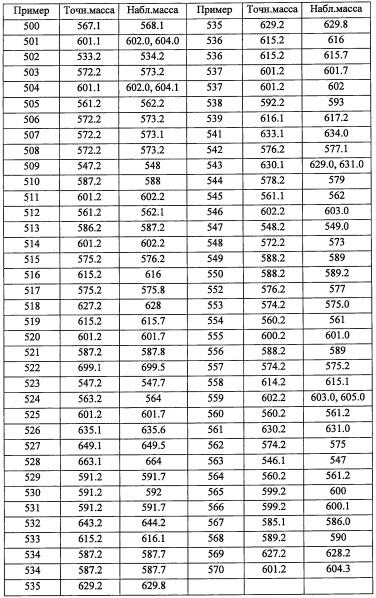
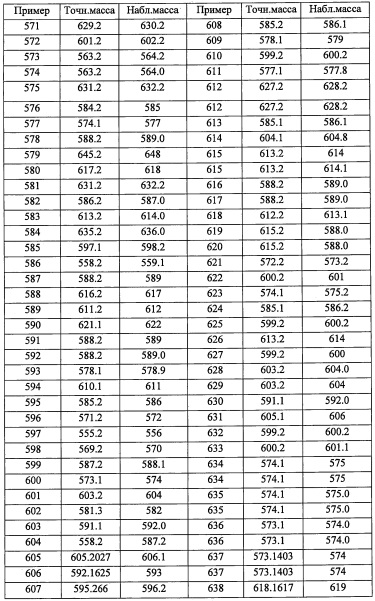
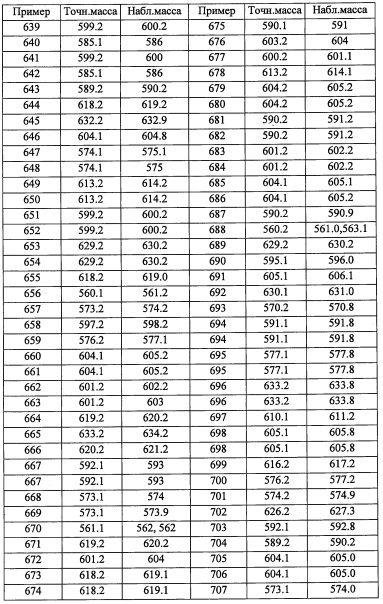
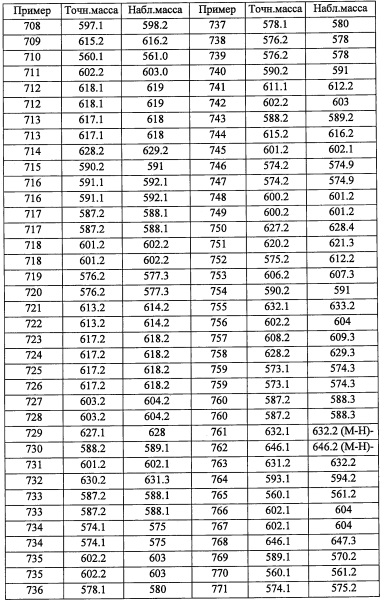
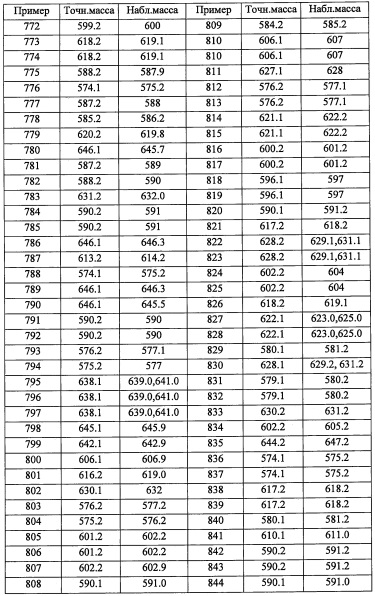
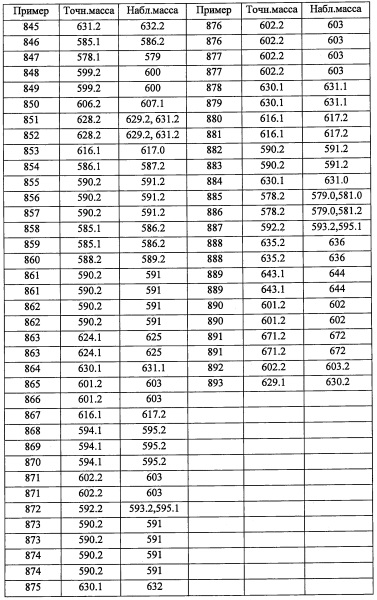
Для примеров, результаты спектроскопии приведены в таблицах ниже.
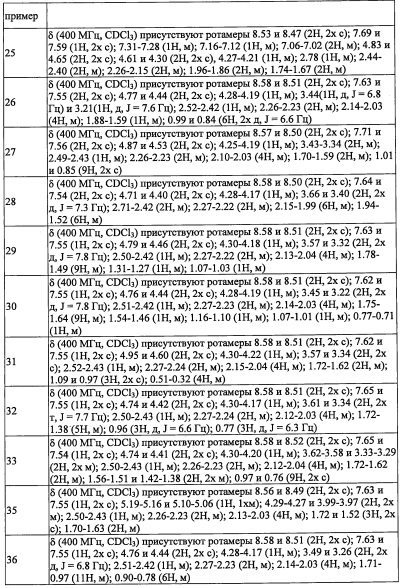
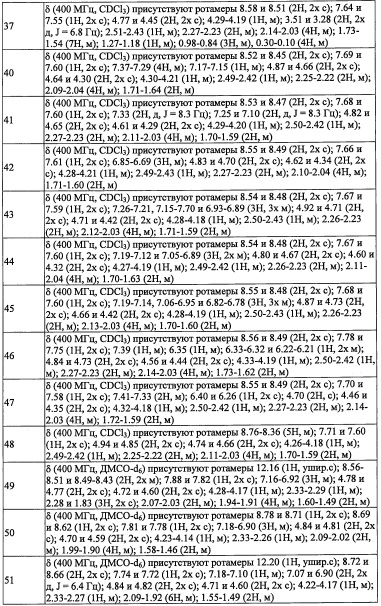
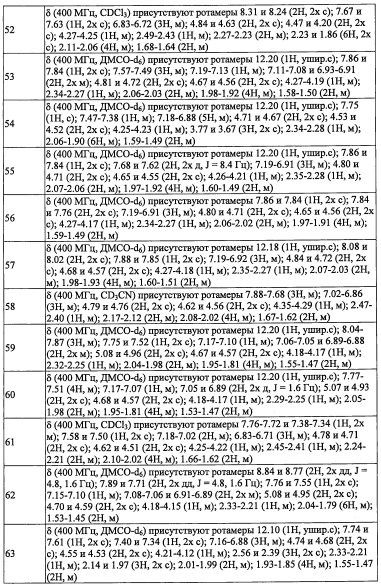
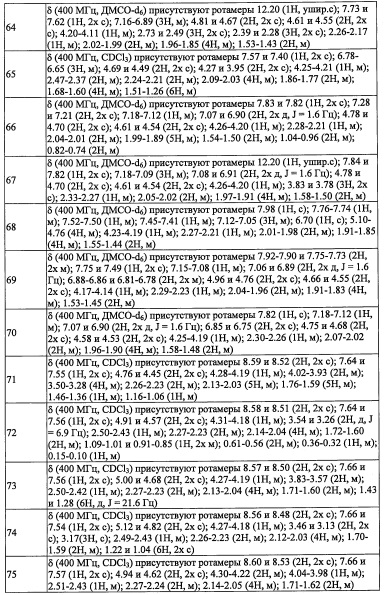
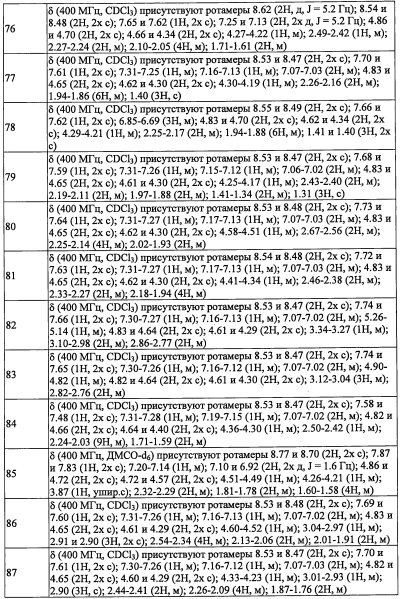
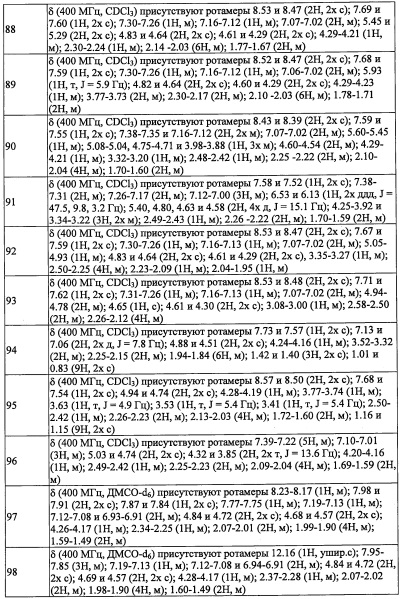
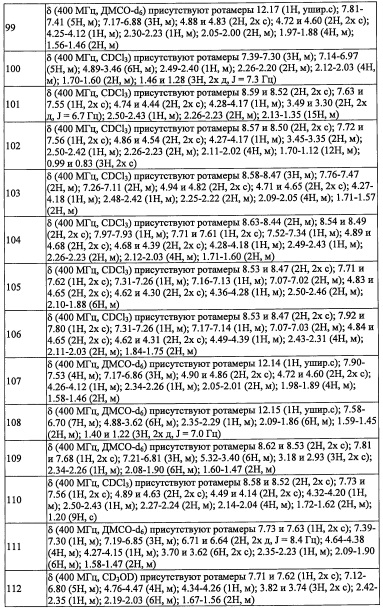
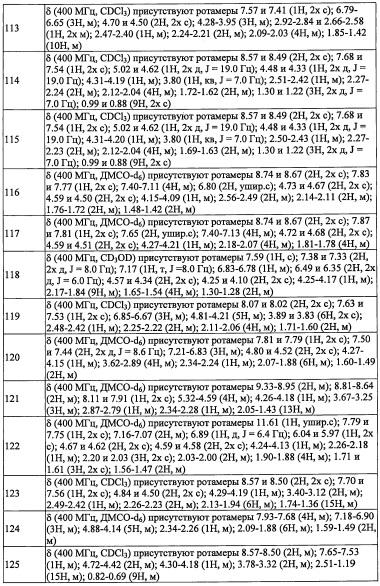
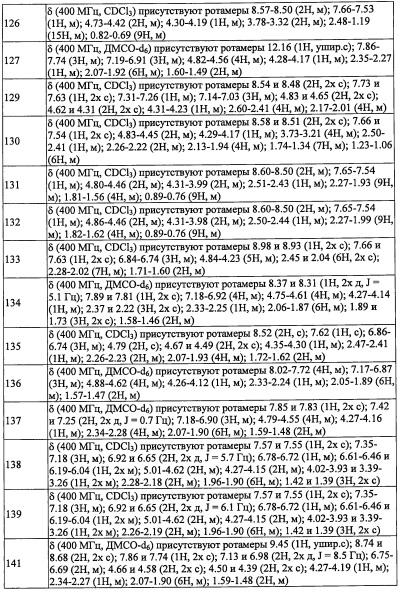
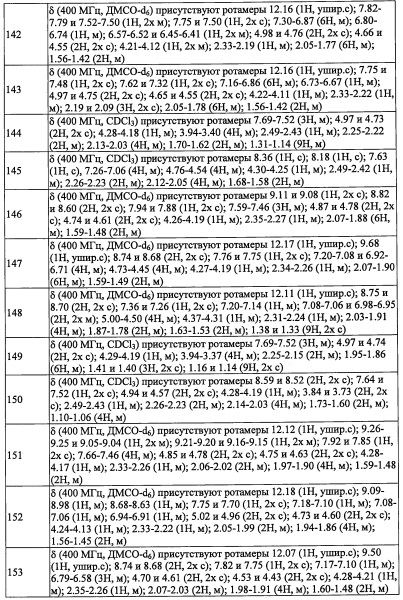
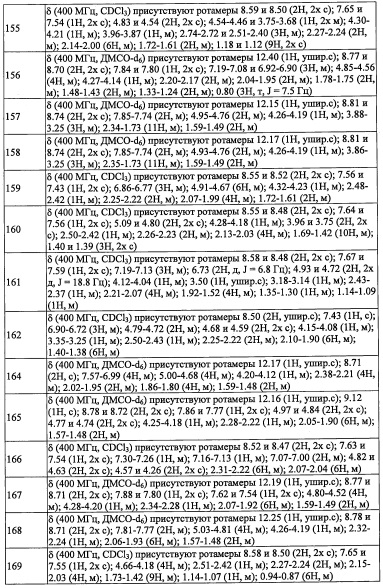
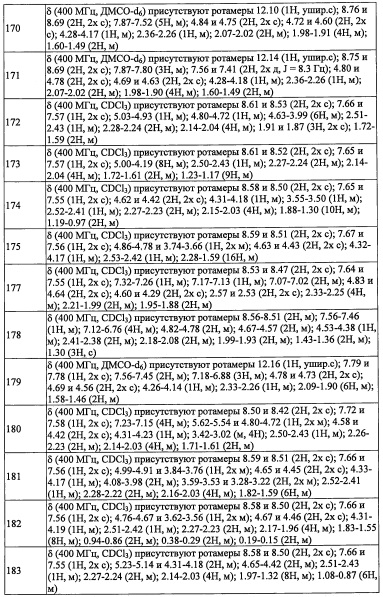
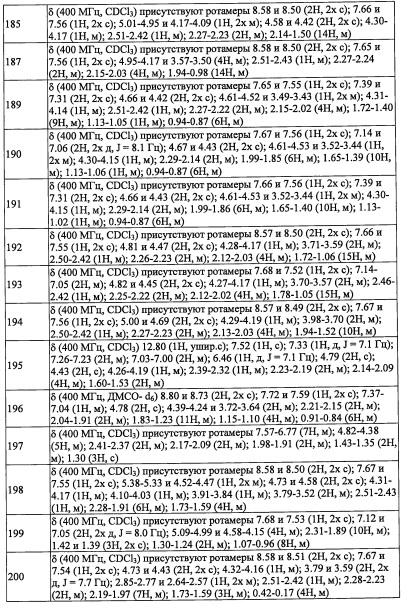
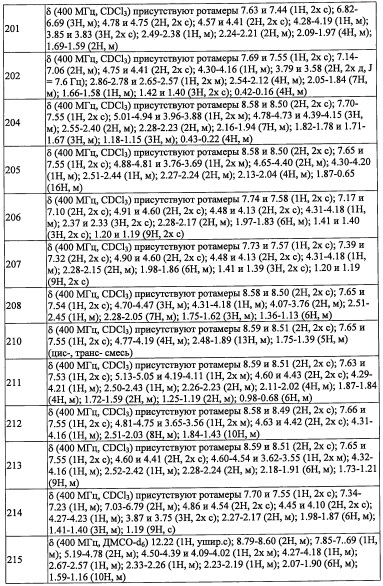
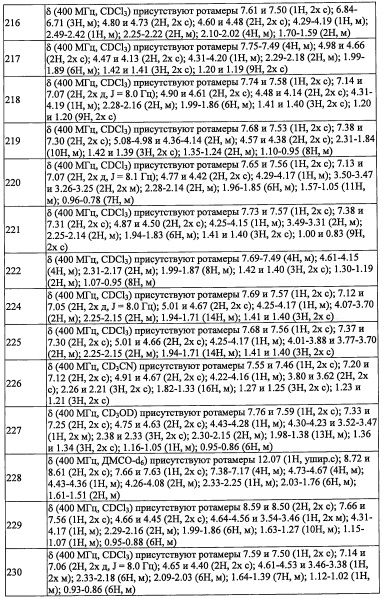
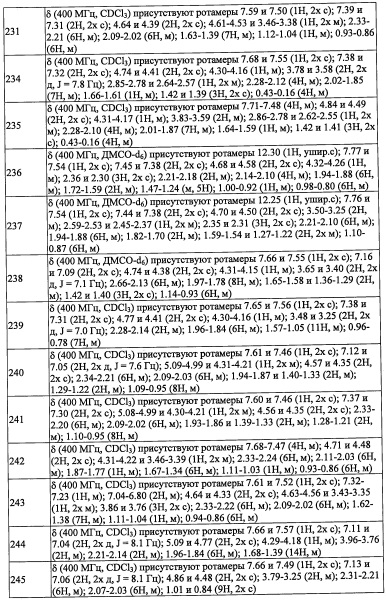
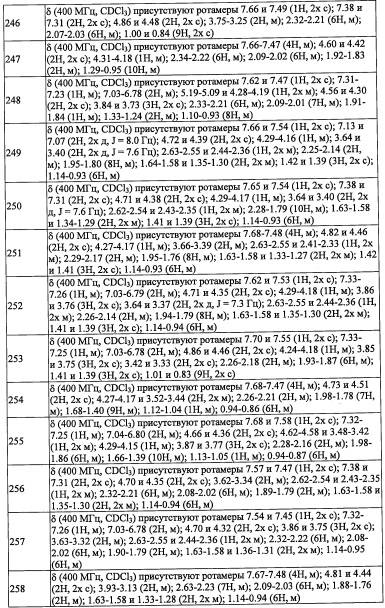
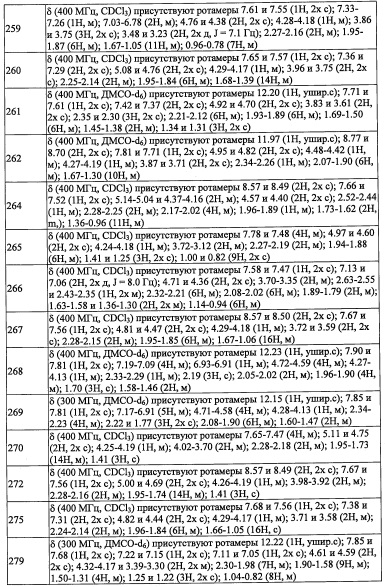
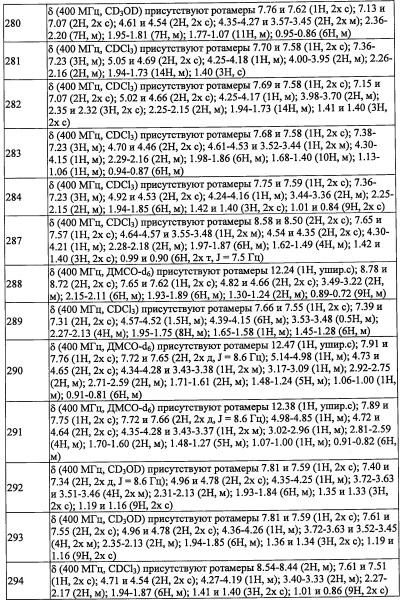
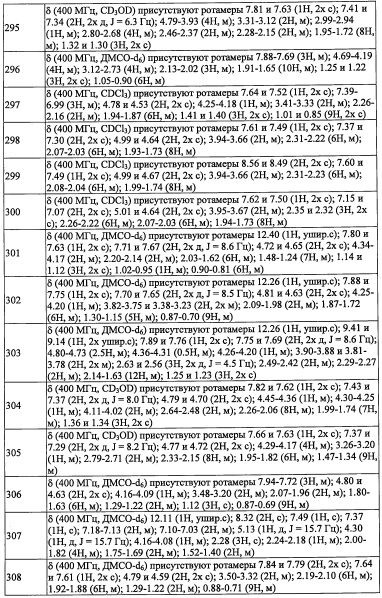
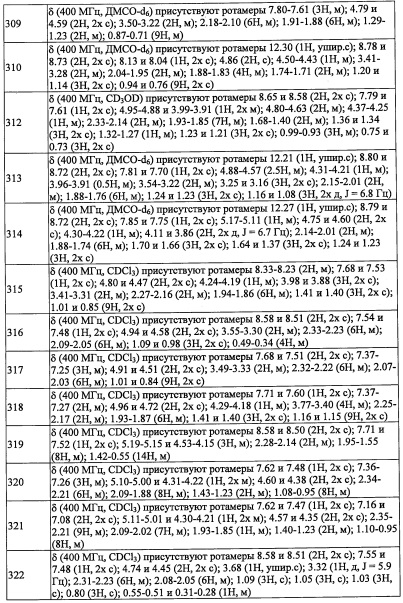
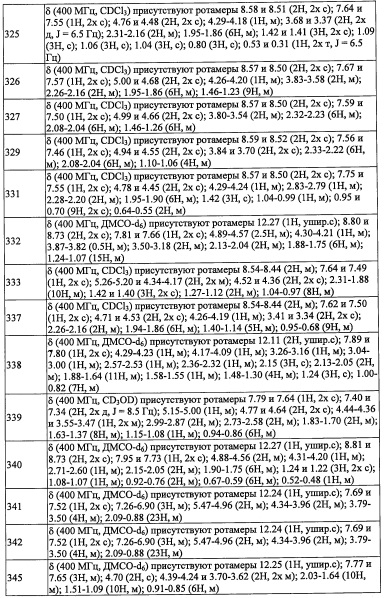
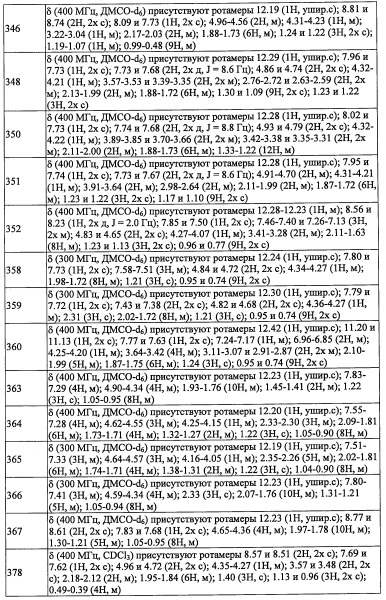
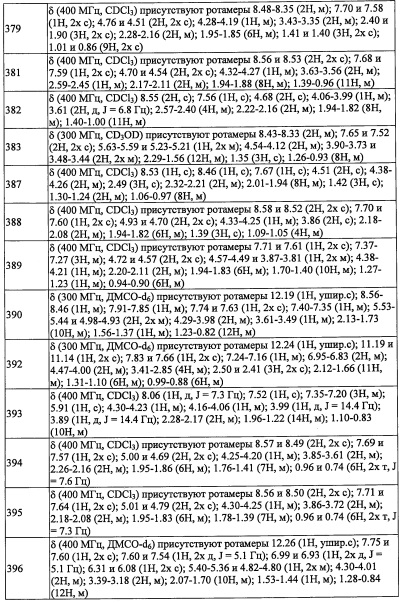
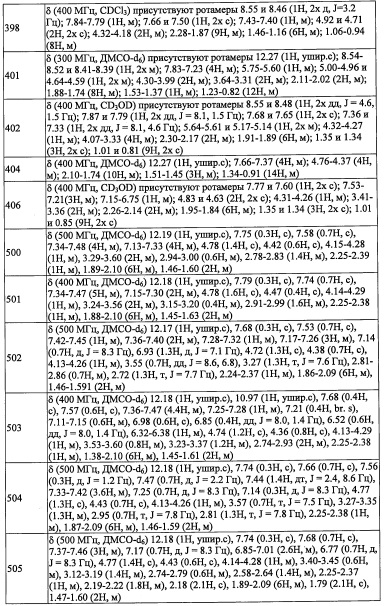
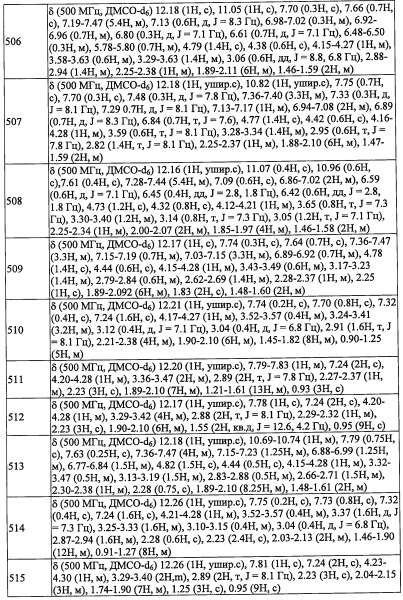
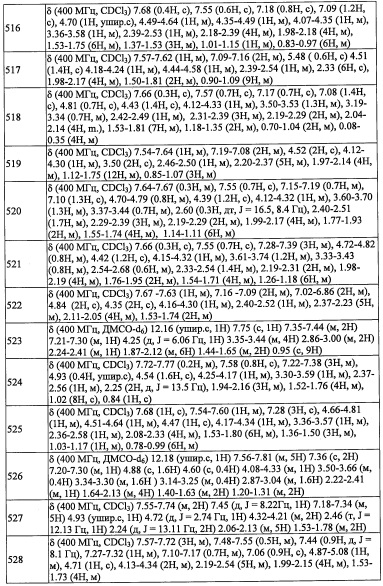
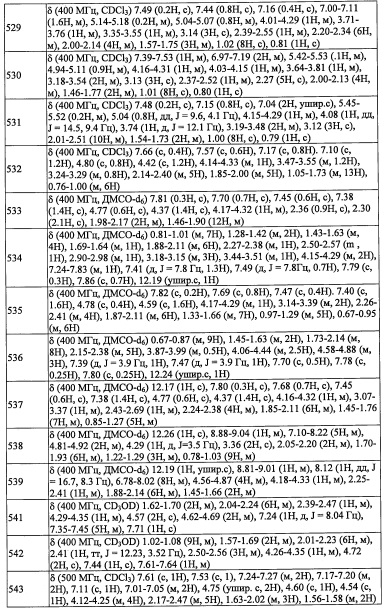

Пример 900
Анализ RORγ с репортерным геном
Применяли анализ с репортерным геном люциферазы для оценки подавления транскрипционной активности RORγ.
Экспрессирующий RORγ вектор получали введением лиганд-связывающего домена человеческого RORγ (аминокислота 247-497 в Genbank Accession NO. NM_001001523), соединенного с ДНК-связывающим доменом фактора транскрипции дрожжей GAL4, в экспрессирующий вектор рМ (Clontech). Полученный экспрессирующий вектор pM-RORγ использовали в экспериментах с трансфекцией, вместе с pGL4 люцифераза репортерной плазмидой (Promega), содержащей пять копий UAS GAL4 сайта узнавания, и pRL-CMV плазмидой (Promega), содержащей конститутивный CMV активатор и ген люциферазы Renilla.
Для приготовления смеси трансфекционного реагента/ДНК, 1 мкг pM-RORγ, 1 мкг pGL4 5xUAS, 625 пг pRL-CMV и 6.25 мкл FuGENE™ HD трансфекционного реагента (Promega) смешивали в 0.25 мл OPTI-MEM™ (Life technologies) при комнатной температуре. Одновременно готовили смесь отрицательного контроля и ДНК, используя 1 мкг рМ пустого вектора вместо pM-RORγ плазмиды. После 15 минут инкубирования, 0.25 мл смеси трансфекционного реагента/ДНК добавляли к 1000000 HEK293T клеток (АТСС) в 5 мл OPTI-MEM™, содержащей 10% очищенной на активированном угле фетальной бычьей сыворотки.
Трансфицированные клетки высевали в 384-луночный планшет (10 мкл/лунка) и добавляли в лунки 7.5 нл испытуемого соединения в 8 концентрациях от 3.5 нМ до 10.5 мкМ. Соединения растворяли в 100% ДМСО, и финальная концентрация ДМСО в данном анализе составляла 0.075%.
После 24 часов инкубирования при 37°С, 5% CO2 в инкубаторе для клеточных культур, применяли систему Dual-Glo™ Luciferase Assay System для детектирования активности согласно инструкциям от производителя (Promega, Cat. No.: E2920).
Строили график по полученным данным и вычисляли значения pIC50 с применением программы XLfit (ID Business Solutions Ltd.). Полученные результаты показаны ниже в таблицах.
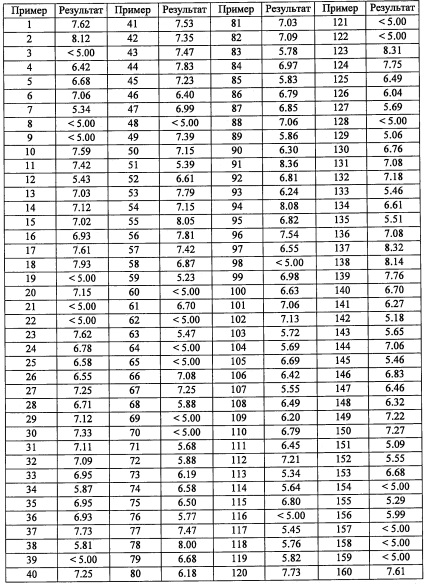
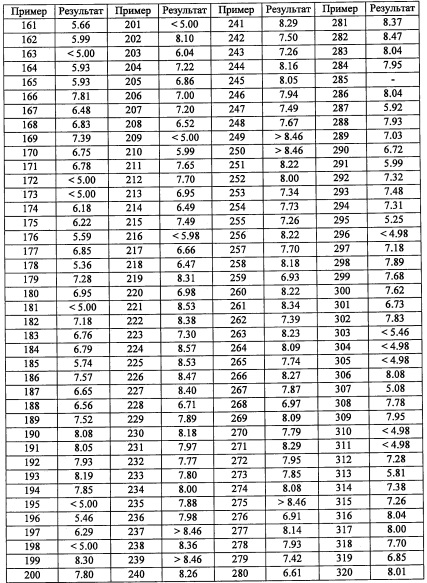
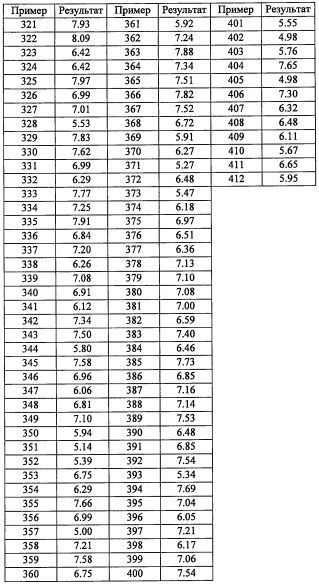
Пример 901
Alphascreen™ анализ рекрутинга RORγ коактивирующего пептида
Alphascreen™ представляет собой амплифицированный гомогенный люминисцентный анализ сближения на бусинах, который может применяться для оценки воздействия соединения на межбелковые взаимодействия. Когда биологические взаимодействия приводят к плотному сближению донорных и акцепторных бусин, реакционноспособный кислород, генерируемый при лазерном возбуждении донорных бусин, инициирует каскад люминисценции/флюоресценции в акцепторных бусинах, который приводит к сильно амплифицированному сигналу, который можно количественно оценить как свет в диапазоне 520-620 нм. Когда донорные и акцепторные бусины не находятся вблизи друг к другу, количество реакционноспособного кислорода падает, и генерируется только очень слабый фоновый сигнал.
Был разработан in vitro анализ с применением Alphascreen™ технологии для оценки подавления RORγ связывания с коактиватором GRIP1. Взаимодействие между ядерными рецепторами (ЯР) и белками-коактиваторами представляет собой ключевую стадию в передаче сигнала от рецептора к транскрипционному механизму, и его можно замерить in vitro, используя только лиганд-связывающий домен ядерного рецептора и пептид, содержащий участок связывания белка-коактиватора LXXLL с ядерным рецептором.
Для RORγ конструкции, применяемой в анализе рекрутинга коактиватора, нуклеотиды, соответствующие лиганд-связывающему домену (LBD) человеческого RORγ дикого типа (аминокислоты 262-518 из Genbank Accession No. NM_005060.3), клонировали в рЕТ24 экспрессирующий вектор (Novagen), ниже N-терминальной 6xHis и Flag-маркерной последовательностей. Рекомбинантный 6xHis:Flag-меченый человеческий RORγ-LBD белок экспрессировали в E. coli (BL-21) и очищали с помощью аффинной хроматографии на Ni Sepharose колонке, с последующей анионообменной хроматографией.
Готовили 4х смесь для анализа 6xHis:Flag-меченый человеческий RORγ-LBD с лигандом-агонистом 7-β-гидроксихолестерином в буфере для анализа (50 мМ HEPES рН 7.4, BSA 0.05%, 150 мМ NaCl, 5 мМ MgCl2, 1 мМ DTT, 0.01% Tween-20). Также готовили для контрольных лунок 4х смесь, содержащую только 6xHis:Flag-меченый человеческий RORγ-LBD.
Готовили 4х стоковый раствор биотинилированного белка-коактиватора, содержащего LXXLL участок из GRIP1 (Биотин-PKKKQNALLRYLLDKDDTKDI) в буфере для анализа.
Готовили 4х детектирующую смесь никель-хелатных Alphascreen™ акцепторных бусин (PerkinElmer) и стрептавидиновых Alphascreen™ донорных бусин (PerkinElmer) в буфере для анализа.
Тестируемые соединения распределяли в предозированный 384-луночный материнский планшет, серийно разбавляя 1 к 2, в 22 колонки, в 100%-ном ДМСО, в 40х финальной концентрации для теста, с наивысшей концентрации 4 мМ. ДМСО, не содержащий соединения, помещали в контрольные колонки. Соединения с помощью робота распределяли непосредственно в планшеты для анализа, содержащие буфер для анализа в 4х финальной концентрации для теста.
После добавления соединения, добавляли смесь для анализа, содержащую 6xHis:Flag-меченый человеческий RORγ-LBD плюс 7-β-гидроксихолестерин, биотинилированный белок-коактиватор и детектирующую смесь. Финальными условиями анализа были: 5 нМ 6xHis:Flag-меченый человеческий RORγ-LBD, 30 нМ 7-β-гидроксихолестерин, 50 нМ биотинилированный белок-коактиватор, 2.5 мкг/мл никелевых акцепторных бусин и 10 мкг/мл стрептавидиновых донорных бусин. Финальная концентрация ДМСО в анализе составляла 2.5%.
После инкубирования в течение ночи при комнатной температуре, планшеты считывали на планшет-ридере Envision™ (PerkinElmer).
Строили график по полученным данным и вычисляли значения pIC50 с применением программы Genedata Screener™ (Genedata). Полученные результаты показаны ниже в таблицах.
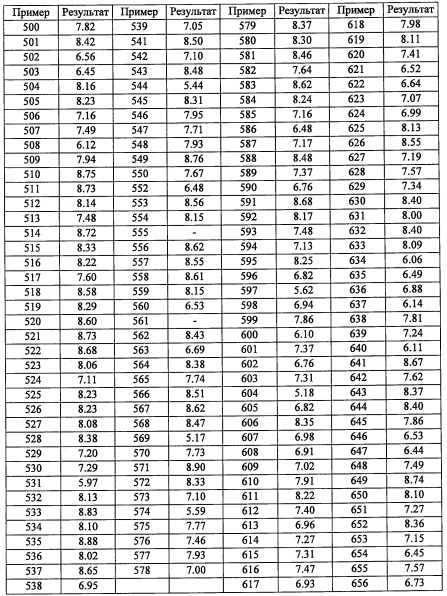
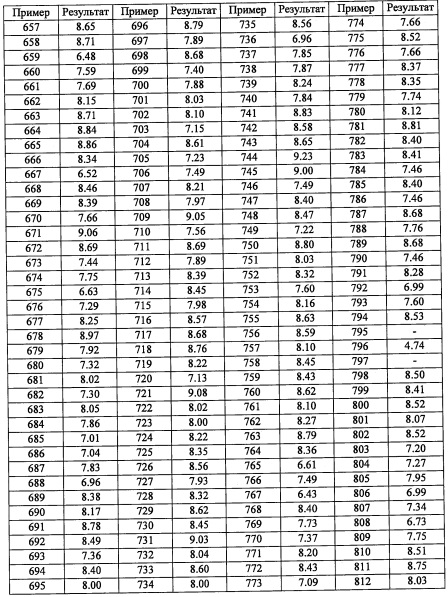
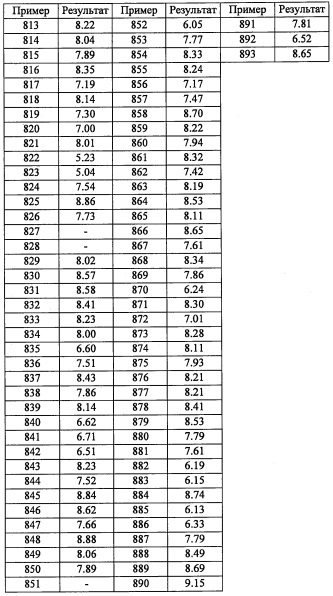
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 4-ЗАМЕЩЕННОЙ ФЕНОКСИФЕНИЛУКСУСНОЙ КИСЛОТЫ | 2007 |
|
RU2463292C2 |
| N-ацил-N'-(пиридин-2-ил) карбамиды и их аналоги, проявляющие противораковую и антипролиферативную активность | 2014 |
|
RU2769607C2 |
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2739356C2 |
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2795512C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛОПИРИМИДИНЫ И ЗАМЕЩЕННЫЕ ПУРИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ УБИКВИТИН-СПЕЦИФИЧЕСКОЙ ПРОЦЕССИРУЮЩЕЙ ПРОТЕАЗЫ 1 (USP1) | 2019 |
|
RU2833222C2 |
| ПРОИЗВОДНЫЕ БЕНЗОЙНОЙ КИСЛОТЫ КАК МОДУЛЯТОРЫ PPARα И PPARλ | 2003 |
|
RU2339613C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2015 |
|
RU2691389C2 |
| Производные 1-фенил-2-пиридинилалкиловых спиртов в качестве ингибиторов фосфодиэстеразы | 2013 |
|
RU2655170C2 |
| НОВЫЕ ГИДРОКСИСЛОЖНОЭФИРНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2734418C2 |
| НОВЫЕ ИНГИБИТОРЫ S-НИТРОЗОГЛУТАТИОНРЕДУКТАЗЫ | 2011 |
|
RU2585763C2 |
Настоящее изобретение касается нового соединения, формулы (I) или его фармацевтически приемлемой соли, которое обладает способностью подавлять активность RORγ. В формуле (I) радикалы и символы имеют значения, указанные в формуле изобретения. Настоящее изобретение касается также фармацевтической композиции, содержащей указанное соединение, и способа лечения или профилактики заболевания с применением данного соединения, где указанное заболевание представляет собой множественный склероз, хронический ревматоидный артрит, анкилозирующий спондилит, системный эритоматоз, псориаз, псориатический артрит, воспалительную болезнь кишечника или астму. 3 н. и 9 з.п. ф-лы, 8 табл., 901 пр.
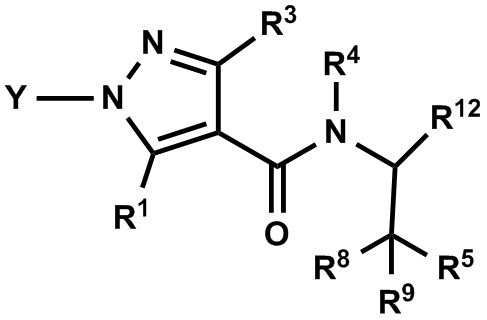 (I)
(I)
1. Соединение, имеющее формулу (I), или его фармацевтически приемлемая соль:
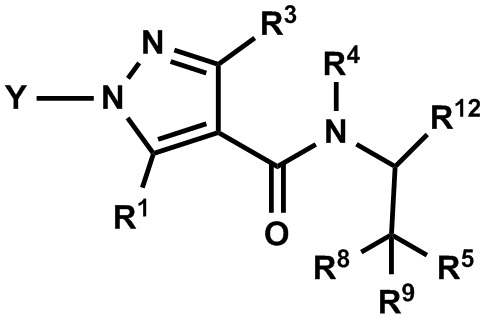 (I)
(I)
где:
R1 выбран из F, Cl, Br, C1 - C6 алкильной группы, замещенной 0, 1, 2 или 3 Ra группами, которые представляют собой F, -NH2 или -OH, и C3 - C8 циклоалкильной группы, замещенной 0 или 1 атомом F;
Y выбран из C4 - C6 циклоалкильной группы, C6 - C9 бициклоалкильной группы и C6 - C9 спироалкильной группы, которые все замещены R2 группой, 0 или 1 R6 группой и 0 или 1 R7 группой;
R2 выбран из -OH, -CO2H, -CONH2, необязательно замещенного ОН или C1 - C6 алкокси группой, (C1 - C6 алкокси)карбонильной группы, (C1 - C6 алкил)аминокарбонильной группы, необязательно замещенной ОН, C1 - C6 алкилсульфонильной группы, (гидроксикарбонил)(C1 - C3 алкильной) группы, (C1 - C6 алкокси)карбонил(C1 - C3 алкильной) группы, (C1 - C6 алкил)сульфонил(C1 - C3 алкильной) группы и (C2 - C6 алкенил)(C1 - C3 алкильной) группы;
R6 и R7 независимо выбраны из H, F, -OH, -NH2, -CN и C1 - C6 алкильной группы;
R3 представляет собой Н;
R4 выбран из C1 - C6 алкильной группы, замещенной 0, 1, 2 или 3 Re группами, (C2 - C6 алкенил)(C1 - C3 алкильной) группы, (C2 - C6 алкинил)(C1 - C3 алкильной) группы, (C1 - C6 алкокси)(C2 - C4 алкильной) группы, (C6 - C10 арил)(C1 - C3 алкильной) группы, замещенной 0, 1, 2 или 3 Rf группами, (5-10-членный гетероарил)(C1 - C3 алкильной) группы, замещенной 0, 1, 2 или 3 Rf группами, C3 - C8 циклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, C3 - C8 циклоалкенильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, (C3 - C8 циклоалкил)(C1 - C3 алкильной) группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, 3-8-членной гетероциклоалкильной группы, замещенной 0, 1, 2, 3, 4 или 5 Rg группами, и (3-8-членный гетероциклоалкил)(C1 - C3 алкильной) группы, замещенной 0, 1, 2 или 3 Rg группами, C6 - C9 спироалкильной группы, замещенной 0, 1, 2 или 3 Rg группами, (C6 - C9 спироалкил)(C1 - C3 алкильной) группы, замещенной 0, 1, 2 или 3 Rg группами, C6 - C9 спирогетероалкильной группы, замещенной 0, 1, 2 или 3 Rg группами, C5 - C9 бициклоалкильной группы, замещенной 0, 1, 2 или 3 Rg группами, (C5 - C9 бициклоалкил)(C1 - C3 алкильной) группы, замещенной 0, 1, 2 или 3 Rg группами, C6 - C9 гетеробициклоалкильной группы, замещенной 0, 1, 2 или 3 Rg группами, и (C6 - C9 гетеробициклоалкил)(C1 - C3 алкильной) группы, замещенной 0, 1, 2 или 3 Rg группами;
R5 выбран из C6 - C10 арильной группы, замещенной 0, 1, 2 или 3 Ri группами, 5-10-членной гетероарильной группы, замещенной 0, 1, 2 или 3 Ri группами, C3 - C8 циклоалкильной группы;
R8 и R9 независимо выбраны из Н, F, -OH, -NH2, C1 - C3 алкильной группы и C1 - C6 алкокси-группы; или R8 и R9 вместе образуют оксо-группу;
R12 представляет собой H; или R4 и R12 вместе представляют собой -CRmRm-CR13R14-CRmRm-, формируя пирролидиновый цикл;
R13 выбран из H, C1 - C6 алкильной группы, фенильной группы, фенокси-группы и C3 - C8 циклоалкильной группы;
R14 независимо выбран из H и C1 - C6 алкильной группы; или R13 и R14 вместе формируют C3 - C8 циклоалкановое кольцо или C3 - C8 циклоалкеновое кольцо;
Rm представляет собой Н;
Rg независимо выбран из F, Cl, C1 - C6 алкильной группы, -OH, -CN, -NH2, C1 - C6 алкокси-группы, -CF3, C1 - C6 алкиленовой группы и оксо-группы;
Rf и Ri независимо выбраны из F, Cl, Br, -OH, -CN, -CO2H, C1 - C6 алкильной группы, замещенной 0, 1, 2 или 3 атомами F, C2 - C6 алкенильной группы, C2 - C6 алкинильной группы, C3 - C8 циклоалкильной группы, C1 - C6 алкокси-группы, замещенной 0, 1, 2 или 3 атомами F, -SH, C1 - C6 алкилтио-группы, (C1 - C6 алкил)карбонильной группы, (C1 - C6 алкокси)карбонильной группы, аминокарбонильной группы, C1 - C6 алкилсульфонильной группы, -NH2, моно(C1 - C6 алкил)амино-группы и ди(C1 - C6 алкил)амино-группы; и
Re независимо выбран из F, C1 - C4 алкильной группы, -OH и -CN.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где Y выбран из формулы (II-a), формулы (II-b), формулы (II-c) и формулы (II-d):
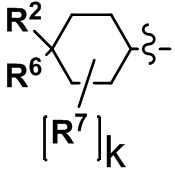 (II-a),
(II-a), 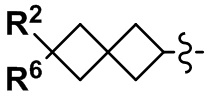 (II-b),
(II-b), 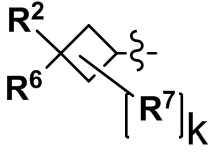 (II-c) или
(II-c) или 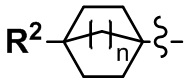 (II-d),
(II-d),
где:
k равно 0 или 1;
и n равно 1, 2 или 3.
3. Соединение по п. 2 или его фармацевтически приемлемая соль, где Y представляет собой группу, имеющую следующую формулу (II-a):
(II-a).
4. Соединение по п. 2 или его фармацевтически приемлемая соль, где Y представляет собой группу, имеющую следующую формулу (II-d):
(II-d)
и n равно 2.
5. Соединение по любому из пп. 1–4 или его фармацевтически приемлемая соль, где R2 представляет собой -CO2H или гидроксикарбонилметильную группу.
6. Соединение по любому из пп. 1–5 или его фармацевтически приемлемая соль, где R12 представляет собой H.
7. Соединение по любому из пп. 1–6 или его фармацевтически приемлемая соль, где R8 и R9 вместе формируют оксо-группу, или R8 и R9 оба представляют собой H.
8. Соединение по любому из пп. 1–7 или его фармацевтически приемлемая соль, где R1 представляет собой -CF3, -CF2H или Cl.
9. Соединение по любому из пп. 1–8 или его фармацевтически приемлемая соль, где R5 представляет собой C6 - C10 арильную группу, замещенную 0, 1, 2 или 3 Ri группами, или 5-10-членную гетероарильную группу, замещенную 0, 1, 2 или 3 Ri группами.
10. Соединение по любому из пп. 1–9 или его фармацевтически приемлемая соль, где R4 представляет собой C1 - C6 алкильную группу, замещенную 0, 1, 2 или 3 Re группами, (C6 - C10 арил)(C1 - C3 алкильную) группу, замещенную 0, 1, 2 или 3 Rf группами, C3 - C8 циклоалкильную группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами, (C3 - C8 циклоалкил)(C1 - C3 алкильную) группу, замещенную 0, 1, 2, 3, 4 или 5 Rg группами, C6 - C9 спироалкильную группу, замещенную 0, 1, 2 или 3 Rg группами, (C6 - C9 спироалкил)(C1 - C3 алкильную) группу, замещенную 0, 1, 2 или 3 Rg группами, C5 - C9 бициклоалкильную группу, замещенную 0, 1, 2 или 3 Rg группами, (C5 - C9 бициклоалкил)(C1 - C3 алкильную) группу, замещенную 0, 1, 2 или 3 Rg группами, или (C6 - C9 гетеробициклоалкил)(C1 - C3 алкильную) группу, замещенную 0, 1, 2 или 3 Rg группами.
11. Способ лечения или профилактики заболевания с применением соединения по любому из пп. 1–10 или его фармацевтически приемлемой соли, где указанное заболевание представляет собой множественный склероз, хронический ревматоидный артрит, анкилозирующий спондилит, системный эритоматоз, псориаз, псориатический артрит, воспалительную болезнь кишечника или астму.
12. Фармацевтическая композиция, обладающая RORγ ингибирующей активностью, содержащая соединение по любому из пп. 1–10 или его фармацевтически приемлемую соль.
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА (СО)ПОЛИМЕРИЗАЦИИ БУТАДИЕНА | 2010 |
|
RU2432365C1 |

