Изобретение относится к медицине и молекулярной биологии, а именно к фтизиатрии, и может быть использовано в лабораторной диагностике туберкулеза легких.
Возбудителем туберкулеза легких являются различные виды микобактерий из группы Mycobacterium tuberculosis complex. По данным ВОЗ туберкулез остается одной из самых больших угроз здравоохранению в мире: в 2014 году 9,6 миллиона человек заболели туберкулезом и 1,5 миллиона человек умерли от этой болезни [1]. Туберкулез может поражать как легкие, так и другие органы и ткани человека: глаза, кости, кожу, мочеполовую систему, кишечник [2].
Обнаружение микобактерий туберкулезного комплекса в биологических материалах является одним из основных диагностических критериев во фтизиатрии [3]. В настоящее время традиционные микробиологические методы выявления возбудителя по своей эффективности уже не удовлетворяют клиницистов. Быстрый метод бактериоскопии обладает низкой чувствительностью: для обнаружения микобактерий туберкулеза (МВТ) необходимо, чтобы 1 мл материала содержал не менее 100 тыс. микробных клеток. Люминесцентная микроскопия увеличивает чувствительность бактериоскопии на 10-30%. Чувствительность бактериологического посева значительно выше - 20-100 МВТ, однако исследование занимает длительное время - один-два месяца. В связи с этим МВТ в нашей стране выявляют в среднем лишь у 50-60% больных активным туберкулезом. Определение устойчивости микобактерий классическими бактериологическими методами занимает 1-2 мес, которые можно считать «потерянными» для больного [4]. В настоящее время разработан ряд методов, позволяющих идентифицировать микобактерию и диагностировать ее лекарственную устойчивость микобактерий с использованием различных молекулярно-генетических подходов, например ПЦР, гибридизация ДНК, секвенирование [1]. Методы идентификации и выявления лекарственной резистентности, основанные на анализе ДНК микобактерий, выполняются в течение нескольких часов, показывают достаточную специфичность и чувствительность. Таким образом, существует насущная необходимость уменьшения их цены, упрощения их выполнения, увеличения их надежности и адаптации к клинико-диагностическим лабораториям фтизиатрических учреждений.
Важным этапом ПЦР диагностики туберкулеза является выделение ДНК изолятов из клинического образца. Для выделения используются: легочные и внелегочные образцы, стул, кровь, моча, сыворотка, бронхоальвеолярный лаваж. Особую трудность представляет собой выделение ДНК Mycobacterium tuberculosis из ткани легкого, причиной этому является несколько факторов: в основном тест-системы подразумевают добавление лизирующего буфера без гомогенизации, состав лизирующего буфера не известен, сама микобактерия имеет плотную «воскоподобную» оболочку, что требует тщательного подбора состава самого буфера.
Целью данной работы являлось создание методики для выделения ДНК изолятов Mycobacterium tuberculosis ткани легкого (операционный материал), а именно подбор состава деконтаминирующего и лизирующего буфера для выделения ДНК из тканей легкого (операционный материал) с использованием гомогенизатора в специальных пробирках, содержащих стеклянные и керамические шарики.
Аналогом предлагаемого способа является метод выделения ДНК с использованием коммерческого лизирующего буфера «DNAzol® Reagent» (Thermo Fisher Scientific, США), имеющего в своем составе гуанидин [6]. Далее следует преципитация спиртом и растворение полученной ДНК в растворе 8 мМ NaOH. Время выделения ДНК при использовании данного набора незначительно, эффективность выделения геномной ДНК варьирует в зависимости от типа образца: клеток, цельной крови или ткани, состав лизирующего буфера является «коммерческой тайной».
Наиболее близким к заявленному способу – прототипом - является способ выделения ДНК, предложенный Holmes D.S. и Bonner J. [5]. В данной работе лизирующий буфер имеет следующий состав: 7М Urea, 100 мМ Tris HCl рН 8.0, 10 мМ EDTA рН 8.0, 350 мМ NaCl, 2% SDS. Образец ткани подвергается гомогенизации при добавлении лизирующего буфера, далее следует фенол-хлороформная экстракция с незначительным добавлением изоамилового спирта (фенол: хлороформ: изоамиловый спирт (ФХИ)=25:24:1), и дальнейшая преципитация спиртом. Данный метод обладает как преимуществами, так и недостатками. К достоинствам метода нужно причислить относительную дешевизну и быстроту выделения ДНК, к недостаткам стоит отнести меньшее количество ДНК, нежели при использовании Протеиназы К, а также наличие стадии фенол-хлороформной экстракции, что требует наличия вытяжного шкафа.
Задачей предлагаемого изобретения является увеличение эффективности выделения ДНК из ткани легкого, быстроты выделения. Особое внимание уделялось наличию доступных реактивов для российских клинических лабораторий.
Способ основан на использовании лизирующего, деконтаминирующего буфера, следующего состава: 5М GuSCN, 100 мМ TRIS HCl рН 8.0, 10 мМ EDTA рН 8.0, 100 мМ NaCl, 0.5% SDS; а также на использовании специальных пробирок, содержащих стеклянные, керамические шарики, соударение которых с образцом в процессе осцилляции ротора гомогенизатора MagNA (Roche, Швейцария) позволяет быстро и эффективно выделить ДНК, использовать реактивы, коммерчески доступные для клинических лабораторий.
Вначале нами был подобран лизирующий, деконтаминирующий буфер с учетом «воскоподобной» стенки микобактерий, богатой миколовыми кислотами. В качестве хаотропного агента был выбран гуанидин изотиоцианат (5М GuSCN), как более активный в ряду «активности» (эффективности) при экстракции белков из мембран: CCl3COO->SCN->гуанидин>ClO4->Br->NO3->мочевина [7]. При этом мочевина обладает большим солюбилизирующим действием при 0°C, чем при 25°C. Додецил сульфат натрия относится к анионным поверхностно-активным веществам, обладает бактерицидным действием, разрушая стенки грамотрицательных бактерий, используется для отделения белков от нуклеиновых кислот, солюбилизирует белки. Мы снизили процент SDS в лизирующем буфере до 0.5% для уменьшения новообразования при гомогенизации. В качестве хелатирующего агента была использована этилендиаминтетрауксусная кислота (EDTA), предотвращающая воздействие на ДНК клеточных нуклеаз, поскольку связывает необходимые для их работы катионы Mg2+ [8]. Для образования осадка нуклеиновых кислот при преципитации спиртом мы добавляли 100 мМ NaCl.
Ткань легкого (операционный материал) может содержать в себе эпителий, кровеносные сосуды, эластичные волокна, соединительную ткань, потому необходима тщательная гомогенизация образца. В качестве гомогенизатора мы использовали MagNA Lyser (Roche, Швейцария), который в автоматическом режиме гомогенизирует образцы и разрушает клетки, облегчая процесс получения супернатанта, используемого для последующего выделения и очистки нуклеиновых кислот (http://roche-applied-science.ru/producte/vyibor-produkta/proba/pribor-magna-lyser/). В прибор помещаются специальные пробирки («Вектор-Бест», Новосибирск, РФ), содержащие керамические и стеклянные шарики, исследуемый материал и лизирующий буфер. В процессе осцилляции ротора происходит соударение шариков с тканью легкого (операционным материалом) и, как следствие, гомогенизация образца и разрушение клеток. Гомогенизация проходит в специальной герметично закрытой пробирке, благодаря чему предотвращает контакт пользователя с инфицированным материалом, что немаловажно при работе с микроорганизмами 3-4 групп патогенности.
Метод выделения ДНК из тканей легкого заключается в следующем:
1. Поместить в специальные пробирки, содержащие керамические шарики, ткань легкого (операционный материал) массой около 0,25 г; пробирки предварительно подписать нестираемым перманентным маркером.
2. Добавить в пробирки 500 мкл лизирующего буфера (5М GuSCN, 100 мМ TRIS HCl рН 8.0, 10 мМ EDTA рН 8.0, 100 мМ NaCl, 0.5% SDS);
3. Поместить пробирки в гомогенизатор MagNA (Roche, Швейцария).
4. Установить следующие параметры для гомогенизации: 7000 об/мин, 60 секунд. Включить гомогенизатор.
5. Центрифугировать пробирки на скорости 6000 об/мин 5 минут.
6. Перенести супернатант в чистые, подписанные перманентным маркером 1,5 мл пробирки типа «Eppendorf».
7. Провести преципитацию изопропиловым спиртом из расчета 0.8 V на образец (примерно, 400 мкл), аккуратно перемешать, поставить на 20 минут на - 18°C, в морозилку.
8. Центрифугировать при 13000 об/мин в течение 10 минут.
9. Промыть 500 мкл холодного 75% этилового спирта, после чего центрифугировать 5 минут на 13000 об/мин.
10. Сушить на 37°С в течение 30 мин при раскрытых крышках до исчезновения запаха спирта.
9. Добавить в пробирки 400 мкл 10 mM Tris-HCl, рН 8.0.
10. Поставить в шейкер на 65°С в течение 15 мин при 400 об/мин.
В плане инфекционного контроля все манипуляции проводили с учетом требований СП 1.3.2322-08 «Безопасность работы с микроорганизмами 3-4 групп патогенности (опасности) и возбудителями паразитарных болезней», МУ 1.3.1888-04 «Организация работы при исследованиях методом ПЦР материала, инфицированного патогенными биологическим агентами III-IV групп патогенности». Работу с патогенными биологическими агентами III-IV групп патогенности проводили в боксе биологической безопасности II-го класса защиты.
Концентрацию ДНК определяли спектрофотометрически по оптической плотности раствора при длине волны 260 нм в сравнении со стандартным раствором (10 mM Tris-HCl, рН 8.0). Концентрация образцов в среднем составляла 116.031 нг/мкл, при этом отношение А260/А280 в среднем составляло 1.83.
Все выделенные таким способом ДНК были использованы в качестве матриц при постановке ПЦР в режиме «реального времени». В качестве целевого фрагмента был использован фрагмент гена домашнего хозяйства (AIR12776.1), кодирующего ДНК геликазу. Размер целевого фрагмента составлял 91 н.о. При выборе олигонуклеотидных праймеров для ПЦР учитывали следующие требования: (1) олигонуклеотидные праймеры должны фланкировать последовательность AIR12776.1; (2) зонд должен быть комплементарен выбранной последовательности; (3) пары праймеров не должны образовывать праймер-димеры и неспецифичные продукты ПЦР. В результате выбрана пара праймеров и последовательность зонда, обеспечивающие наиболее специфичную и эффективную амплификацию (Таблица 1).

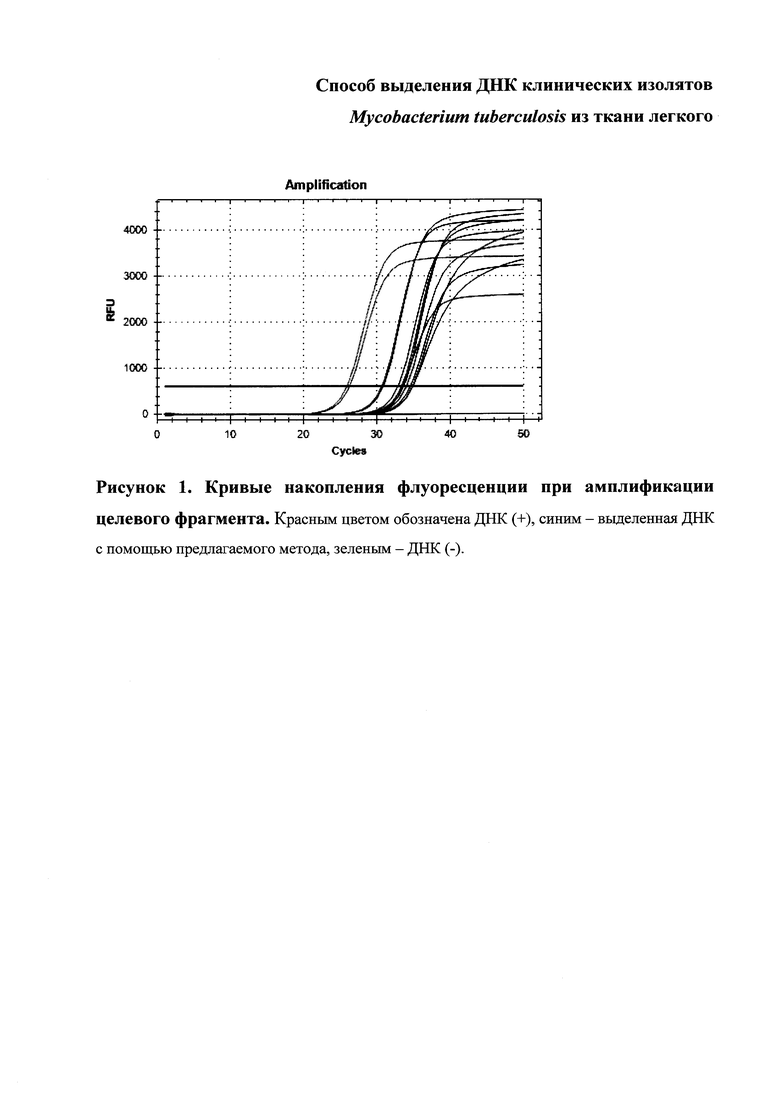
ПЦР в режиме «реального времени» с использованием технологии TaqMan проводили в объеме 20 мкл, содержащем 67 мМ трис-HCl (рН 8,9), 16 мМ сульфат аммония, 2,5 мМ MgCl2, 0,01% Твин 20, 0,2 мМ дНТФ, 0,3 мкМ растворы олигонуклеотидных праймеров, 0,1 мкМ раствор зонда, 5 нг геномной ДНК и 1 ЕД/акт Taq-полимеразы («Fermentas», США). Реакцию проводили на амплификаторе CFX96 («Bio-Rad», США) с начальной денатурацией при 96°C в течение 3 мин, далее 49 циклов с денатурацией при 96°C в течение 8 с, отжигом праймеров при 60°C в течение 40 с и последующем съемом флуоресценции на канале HEX (фильтр возбуждения /детекции - 535nm/555nm). В качестве положительного контроля использовали заведомо положительный образец ДНК Mycobacterium tuberculosis (ДНК (+)), в качестве отрицательного контроля использовали воду MillyQ (ДНК (-)). Результат амплификации целевого фрагмента оценивали по графику накопления флуоресценции (рис. 1).
Таким образом, предлагаемый способ, заключающийся в одновременном действии химических и физических факторов, а именно с применением лизирующего, деконтаминирующего буфера следующего состава: 5М GuSCN, 100 мМ TRIS НСl рН 8.0, 10 мМ EDTA рН 8.0, 100 мМ NaCl, 0.5% SDS, и с применением пробирок, содержащих керамические и стеклянные шарики, обладает высокой эффективностью выделения ДНК (в среднем 116,031 нг/мкл), качеством выделения (А260/А280 - 1.83), быстротой (процесс выделения ДНК занимает около 2-х часов).
Список литературы
[1] "Report global tuberculosis," 2015.
[2] C.S. Restrepo, R. Katre, and A. Mumbower, "Imaging Manifestations of Thoracic Tuberculosis," Radiol. Clin. North Am., vol. 54, no. 3, pp.453-473, 2016.
[3] У.Г. Бочкарев, "Актуальные вопросы генодиагностики туберкулеза," Иммунопатология, аллергология, инфектология, vol. 1, pp.92-96, 2000.
[4] D. Laurenzo and S.A. Mousa, "Mechanisms of drug resistance in Mycobacterium tuberculosis and current status of rapid molecular diagnostic testing.," Acta Trop., vol. 119, no. 1, pp.5-10, Jul. 2011.
[5] D.S. Holmes and J. Bonner, "Preparation, molecular weight, base composition, and secondary structure of giant nuclear ribonucleic acid.," Biochemistry, vol. 12, no. 12, pp.2330-8, Jun. 1973.
[6] E.M. Santos, J.F.R. Paula, P.M.C. Motta, M.B. Heinemann, R.C. Leite, J.P.A. Haddad, H.L. Del Puerto, and J.K.P. Reis, "Comparison of three methods of DNA extraction from peripheral blood mononuclear cells and lung fragments of equines.," Genet. Mol. Res., vol. 9, no. 3, pp.1591-8, 2010.
[7] К.Д.P. Досон, Д. Эллиот, У. Эллиот, Справочник биохимика. Издательство "Мир," 1991.
[8] В.А. Беликов, Молекулярная биология. Практическое руководство. Издательский центр "Наука," 2013.
| название | год | авторы | номер документа |
|---|---|---|---|
| Универсальный способ выделения ДНК и лизирующая смесь для его осуществления | 2022 |
|
RU2807254C1 |
| Способ выделения ДНК из растений, пригодный для постановки ПЦР | 2017 |
|
RU2672378C1 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ОБНАРУЖЕНИЯ И ИДЕНТИФИКАЦИИ ПОСЛЕДОВАТЕЛЬНОСТЕЙ НУКЛЕИНОВЫХ КИСЛОТ В БИОЛОГИЧЕСКИХ ОБРАЗЦАХ | 2012 |
|
RU2595421C2 |
| СПОСОБ ДИАГНОСТИКИ ТУБЕРКУЛЕЗА | 1999 |
|
RU2163022C1 |
| СПОСОБ ВЫДЕЛЕНИЯ ТОТАЛЬНОЙ ДНК БАКТЕРИЙ ИЗ ОБРАЗЦОВ ПОЧВЫ, СПОСОБ ОЦЕНКИ БАКТЕРИАЛЬНОГО СОСТАВА ПОЧВ ПОСРЕДСТВОМ МЕТАГЕНОМНОГО СЕКВЕНИРОВАНИЯ И НАБОРЫ ДЛЯ ОСУЩЕСТВЛЕНИЯ СПОСОБОВ | 2024 |
|
RU2829656C1 |
| СПОСОБ ВЫДЕЛЕНИЯ ДНК ИЗ КОСТНОГО МАТЕРИАЛА | 2019 |
|
RU2724506C1 |
| СПОСОБ ОБНАРУЖЕНИЯ МИКОБАКТЕРИЙ ТУБЕРКУЛЁЗА ГЕНЕТИЧЕСКОГО КЛАСТЕРА Beijing B0/W148 | 2013 |
|
RU2551764C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ТОЧЕЧНЫХ НУКЛЕОТИДНЫХ ЗАМЕН В ДНК МИКОБАКТЕРИЙ, СПОСОБ ДИАГНОСТИКИ УСТОЙЧИВОСТИ МИКОБАКТЕРИЙ К РИФАМПИЦИНУ, БИОЧИП ДЛЯ ОСУЩЕСТВЛЕНИЯ ЭТИХ СПОСОБОВ | 2000 |
|
RU2175015C1 |
| СПОСОБ ИДЕНТИФИКАЦИИ МИКОБАКТЕРИЙ С ПОМОЩЬЮ ПОЛИМЕРАЗНОЙ ЦЕПНОЙ РЕАКЦИИ | 2010 |
|
RU2455364C2 |
| СПОСОБ ВЫДЕЛЕНИЯ И ОЧИСТКИ ДЕЗОКСИРИБОНУКЛЕИНОВЫХ КИСЛОТ | 2008 |
|
RU2400537C2 |
Изобретение относится к области медицины и молекулярной биологии, а именно к фтизиатрии, и может быть использовано в лабораторной диагностике туберкулеза легких. Способ выделения ДНК клинических изолятов Mycobacterium tuberculosis из ткани легкого заключается в добавлении к образцу ткани легкого, находящемуся в пробирке, содержащей стеклянные шарики, деконтаминирующего лизирующего буфера состава 5 М GuSCN, 100 мМ TRIS HCl рН 8.0, 10 мМ EDTA рН 8.0, 100 мМ NaCl, 0,5% SDS, с последующими центрифугированием и осаждением спиртом. Изобретение обеспечивает высокую эффективность выделения ДНК, характеризующуюся концентрацией образца в среднем 116,031 нг/мкл, высокое качество выделения ДНК, характеризующееся отношением А260/А280, равным 1,83, и быстроту процесса выделения ДНК. 1 ил., 1 табл.
Способ выделения ДНК клинических изолятов Mycobacterium tuberculosis из ткани легкого путем центрифугирования и последующим осаждением спиртом, отличающийся тем, что к образцу, находящемуся в пробирке, содержащей стеклянные шарики, добавляют деконтаминирующий лизирующий буфер следующего состава: 5 М GuSCN, 100 мМ TRIS HCl рН 8.0, 10 мМ EDTA рН 8.0, 100 мМ NaCl, 0,5% SDS.
| US 5130423 A, 14.07.1992 | |||
| US 2010184210 A1, 22.07.2010 | |||
| WO 2006088895 A2, 24.08.2006 | |||
| WO 9534574 A1, 21.12.1995 | |||
| D.S | |||
| HOLMES, et al | |||
| Preparation, Molecular Weight, Base Composition, and Secondary Structure of Giant Nuclear Ribonucleic Acid / Biochemistry, 1973, V.12, pp.2330-2338. |

