ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
6,6'-[[3,3',5,5'-Тетракис(1,1-Диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепин (в дальнейшем также обозначается как "соединение I") применяется в качестве лиганда гомогенных катализаторов, в частности в катализаторах родия для гидроформилирования олефинов.
Химическая структура соединения I показана на следующей формуле:
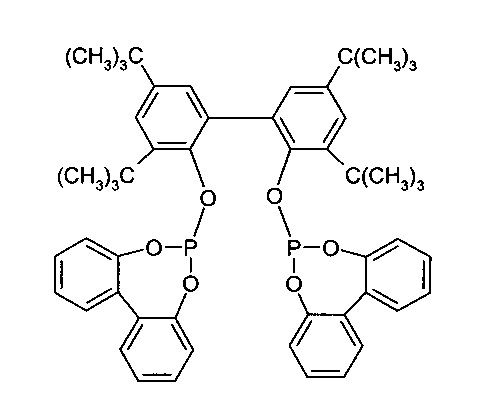
Настоящее изобретение относится к новым кристаллическим формам 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина, способам их получения и их применения.
Для потребительских свойств веществ, которые используются в промышленном масштабе, возможное существование кристаллических модификаций (также известных как кристаллические формы) или сольватов данного вещества, знание специфических свойств таких модификаций и сольватов и методов для их приготовления во многих случаях имеют решающее значение. Вещество может существовать в различных кристаллических модификациях, но также и в аморфной форме. Эти случаи называют полиморфизмом. Полиморф представляет собой твердую, кристаллическую фазу соединения, которая характеризуется определенной однородной упаковкой и расположением молекул в твердом веществе. Различные модификации одного и того же вещества проявляют различные свойства, например различаются в следующих свойствах: форма и размер кристалла, плотность, растворимость, фильтруемость, скорость растворения, устойчивость к фазовому превращению в другую модификацию, стабильность в процессе размалывания, стабильность суспензии, оптические и механические свойства, давление паров, гигроскопичность, температура плавления, устойчивость к разложению или цвет.
Органические бисфосфитные соединения типа соединения I, их получение и использование в качестве лиганда в гомогенном катализе описаны, например, в EP 0214622 A2, US 4668651, US 4748261, US 4769498, US 4885401, US 5235113, US 5391801, US 5663403, US 5728861, US 6172267, DE 10360771 A1, WO 2003/062171 и WO 2003/062251.
WO 2010/042313 описывает многостадийный процесс приготовления бисфосфита. На стадии (a) монохлорфосфит получают взаимодействием PCl3 с ароматическим диолом в суспензии в условиях реакции и в присутствии второго ароматического диола с получением смеси, содержащей монохлорфосфит, второй ароматический диол, а избыток PCl3. Суспензия содержит менее 5 мольных процентов азотистого основания, и органический растворитель выбирают из-за низкой растворимости хлористого водорода в нем. После удаления избытка PCl3, добавляют азотистое основание для осуществления конденсации монохлорфосфита со вторым ароматическим диолом с получением указанного в бисфосфита. Очистка с помощью перекристаллизации описана только в самых общих чертах.
EP 0285136 A2 описывает способ разделения вторичных органофосфитов от третичных органофосфитов, который включает (1) обработку композиции, состоящей по существу из третичных и вторичных органофосфитов, растворенных в органическом растворителе, с добавлением воды и основания для избирательного превращения вторичного органофосфита в соль и (2) отделение и выделение третичного органофосфита из указанной соли. В сравнительных примерах 8, 28 и 29 сырой третичный монофосфит (фосфит B), содержащий смесь третичного и вторичного органофосфата в виде примеси, перекристаллизовывают из ацетонитрила.
US 2003/0100787 описывает способ получения стерически затрудненных триарилфосфитов. Продукт реакции выпадает в осадок из изопропанола. Не существует никакого стимула применять соответствующий способ для получения бисфосфитных соединений.
CN 101684130 A описывает способ получения органических бисфосфитных соединений, в котором
a.) монохлорфосфит, образующий боковые крылья, растворяют в дихлорметане,
b.) ароматический диол, образующий мостиковую группу, растворяют в триэтиламине или смеси триэтиламин/дихлорметан,
c.) растворы смешивают и подвергают взаимодействию при от -40°C до +20°C,
d). Полученный раствор перемешивают при от 20 до 30°C в течение от 10 до 20 ч, и
e.) добавляют деионизированную воду к раствору, полученному на стадии d.), чтобы индуцировать разделение фаз, при котором нижняя органическая фаза содержит продукт.
Дополнительно описывается перекристаллизация полученного бисфосфита из гексана.
US 5312996 относится к процессу гидроформилирования для получения 1,6-гександиалей. В колонке 18, строка 60 и далее описывается приготовление различных лигандов, в частности соединения I (= лиганд A), с помощью реакции 1,1-бифенил-3,3'-ди-трет-бутил-5,5'-ди-трет.-бутокси-2,2-диола с хлоридитом бисфенола в качестве примера. Полученный реакционный раствор концентрируют до сиропа на роторном испарителе и добавляют ацетонитрил, чтобы осадить бисфосфитный лиганд. Полученную смесь перемешивают в течение 2 ч при комнатной температуре, фильтруют, твердые вещества промывают ацетонитрилом и сушат в вакууме. Описанная процедура не подходит для получения кристаллической несольватированной формы соединения I. Авторы настоящего изобретения обнаружили, что из соединения I и ацетонитрила при комнатной температуре получается ацетонитрил-сольват. Тем не менее, присутствие ацетонитрила в кристаллической решетке является вредным для использования в качестве лигандов для гомогенного катализа, так как ацетонитрил координирует с используемыми переходными металлами, таким образом, препятствует катализу.
A. van Rooy et al. описывают в Organometallics 1996, 15(2), 835-847 исследования гидроформилировании и характеристику объемных дифосфит-модифицированных родиевых катализаторов. Описывается получение соединения I (= лиганд 9), где продукт получают осаждением с ацетонитрилом, после перекристаллизации из смеси толуол/ацетонитрил и сушат в вакууме. Более подробные условия перекристаллизации не приводятся, и продукт не характеризуется кристаллографическими данными.
Соединения I, полученные в соответствии с такими известными способами, демонстрируют по меньшей мере один из следующих недостатков: продукт является липким, имеет тенденцию при длительном хранении в слеживанию или имеет тенденцию к образованию пыли. Такие свойства имеют негативное влияние на пригодность для использования этих соединений в промышленном масштабе, например, для изготовления катализаторов.
В настоящее время неожиданно было обнаружено, что посредством определенных процессов ранее неизвестная кристаллическая, стабильная несольватная модификация 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина, которая не демонстрирует недостатков известных твердых форм, получается с высокой степенью чистоты. Кроме того, были найдены четыре кристаллические сольвата 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина с сопоставимыми полезными свойствами.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Первая задача настоящего изобретения относится к кристаллической несольватированной форме и кристаллическому сольвату кристаллических толуол-сольвата и ацетон-сольватов 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина.
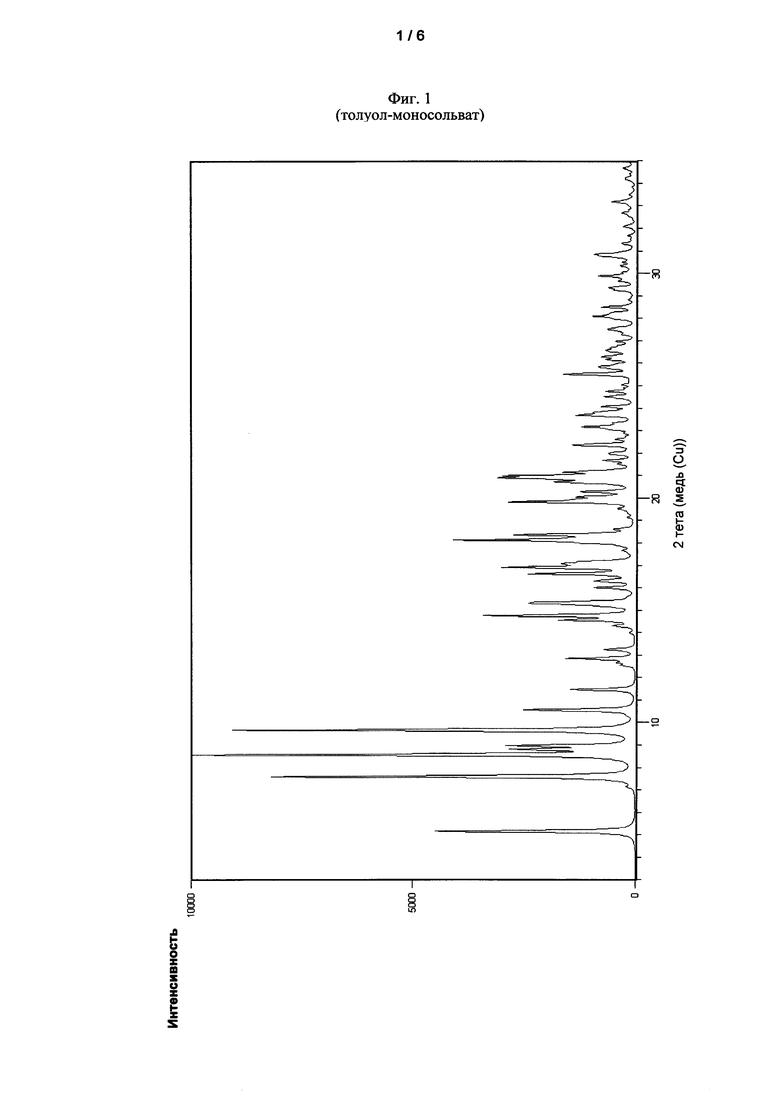
Еще одна задача настоящего изобретения относится к кристаллическому толуол-моносольвату 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина, который в рентгеновской порошковой дифрактограмме при 25°C с Cu-Kα излучением демонстрирует по меньшей мере 5 из следующих рефлексов, выраженных, как 2θ значения: 5,15°±0,20, 7,59°±0,20, 8,56°±0,20, 8,80°±0,20, 8,97°±0,20, 9,65°±0,20, 10,55°±0,20, 11,47±°0,20, 14,76°±0,20 и 15,35±0,20.
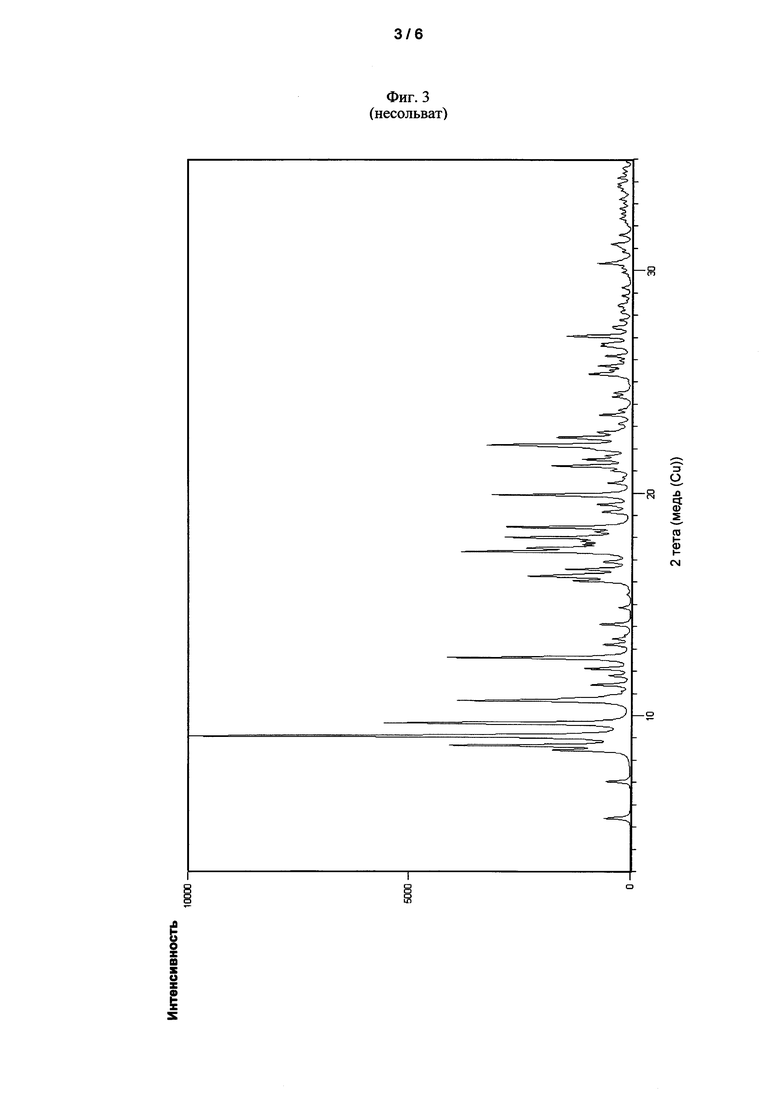
Еще одна задача настоящего изобретения относится к несольватированной кристаллической форме 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина, которая в рентгеновской порошковой дифрактограмме при 25°C с Cu-Kα излучением демонстрирует по меньшей мере 5 из следующих рефлексов, выраженных, как 2θ значения: 5,39°±0,20, 7,04±0,20, 8,44°±0,20, 8,65°±0,20, 9,08°±0,20, 9,66°±0,20, 10,66°±0,20, 12,60±°0,20, 16,25°±0,20 и 17,36°±0,20.
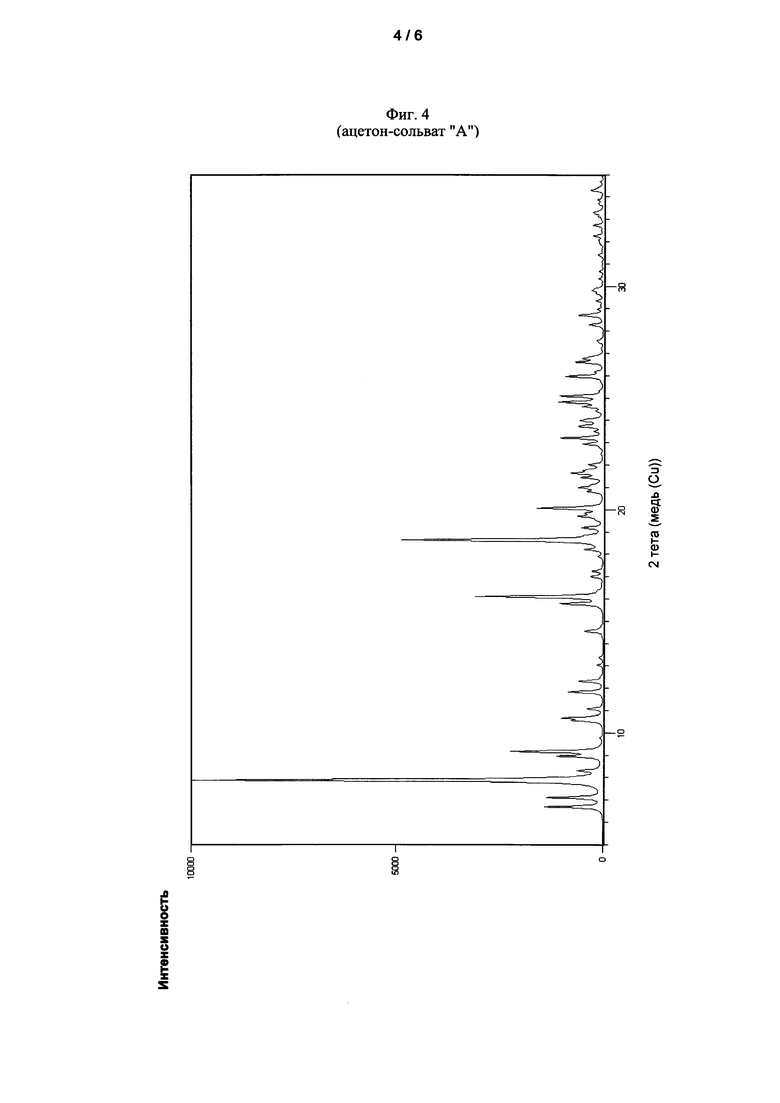
Еще одна задача настоящего изобретения относится к кристаллическому ацетон-сольвату "A" 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2,-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепину, который в рентгеновской порошковой дифрактограмме при 25°C с Cu-Kα-излучением демонстрирует по меньшей мере 5 из следующих рефлексов, выраженных, как 2θ значения: 6,67°±0,20, 7,11°±0,20, 7,87°±0,20, 8,31°±0,20, 8,96°±0,20, 9,17°±0,20 10,68°±0,20, 15,78°±0,20, 16,10°±0,20 и 18,63°±0,20.
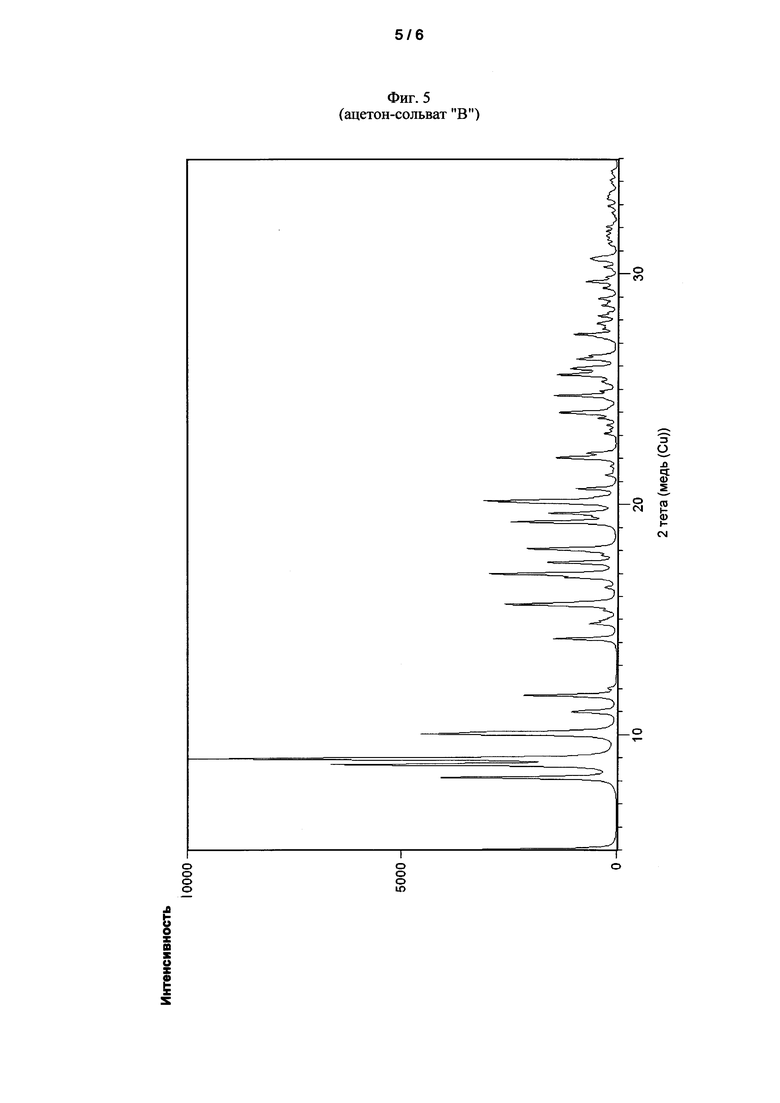
Еще одна задача настоящего изобретения относится к кристаллическому ацетон-сольвату «B» 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина, который в рентгеновской порошковой дифрактограмме при 25°C с Cu-Kα излучением демонстрирует по меньшей мере 5 из следующих рефлексов, выраженных, как 2θ значения: 8,13±0,2, 8,70±0,2°, 8,95±0,2°, 10,02±0,2°, 10,98±0,2°, 11,71±0,2°, 14,16±0,2°, 15,65±0,2°, 16,98±0,2° и 0±18,08±0,2°.
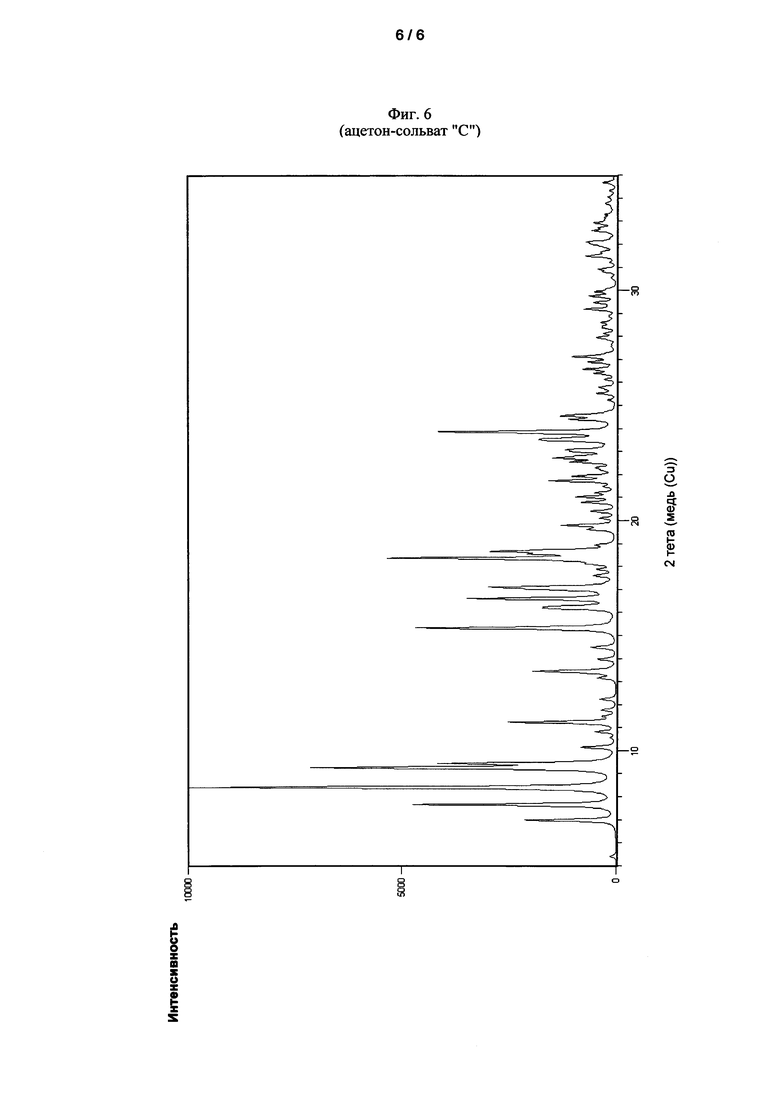
Еще одна задача настоящего изобретения относится к кристаллическому ацетон-сольвату "C" 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина, который в рентгеновской порошковой дифрактограмме при 25°C с Cu-Kα излучением демонстрирует по меньшей мере 5 из следующих рефлексов, выраженных, как 2θ значения: 5,40°±0,20, 6,97°±0,20, 7,64°±0,20, 8,39°±0,20, 9,24°±0,20, 9,44°±0,20, 11,23°±0,20, 13,46°±0,20, 15,32°±0,20 и 18,35°±0,20.
Дополнительными задачами настоящего изобретения являются способы получения кристаллических форм соединения I.
Еще одной задачей настоящего изобретения является применение кристаллической формы соединения I, как определено выше и далее, для получения катализатора на основе переходного металла для гидроформилирования, гидроцианирования или гидрирования.
Еще одной задачей настоящего изобретения является способ получения катализатора на основе переходного металла, в котором кристаллическая форма соединения I, как определено выше и далее, обеспечивается и приводится в контакт с соединением или комплексом переходного металла в инертном растворителе.
Еще одной задачей настоящего изобретения является катализатор на основе переходного металла, который можно получить способом, в котором кристаллическая форма соединения I, как определено выше и далее, обеспечивается и приводится в контакт с соединением или комплексом переходного металла в инертном растворителе.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Кристаллические формы соединения I в соответствии с настоящим изобретением имеют следующие преимущества:
- низкая липкость,
- меньшая склонность к слеживанию,
- меньшая склонность пылеобразованию,
- высокая объемная плотность,
- высокая чистота, что позволяет использовать в качестве лигандов в процессах промышленного масштаба.
В частности, кристаллические формы в соответствии с настоящим изобретением более просты в обращении, чем другие известные твердые формы 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина, так как они получены в виде дискретных кристаллов или кристаллитов или кристаллических агломератов.
Целью настоящего изобретения также является композиция, содержащая по меньшей мере 50 мас. % в расчете на общую массу композиции по меньшей мере одну кристаллическую форму соединения I в соответствии с настоящим изобретением. Дополнительными соединениями композиции могут быть кристаллические формы соединения I, отличные от кристаллических форм по изобретению, соединение I в аморфной форме, и соединения, отличные от соединения I. Предпочтительно композиция содержит по меньшей мере 75 мас. %, более предпочтительно по меньшей мере 85 мас. %, в особенности по меньшей мере 90 мас. %, особенно по меньшей мере 95 мас. % в расчете на общую массу композиции, по меньшей мере одной кристаллической формы соединения I в соответствии с настоящим изобретением.
Композиция по меньшей мере одной кристаллической формы соединения I в соответствии с настоящим изобретением содержит предпочтительно по меньшей мере 75 мас. %, более предпочтительно по меньшей мере 85 мас. %, в особенности по меньшей мере 90 мас. %, особенно по меньшей мере 95 мас. %, более предпочтительно по меньшей мере 98 мас. %, например, по меньшей мере 99 мас. % по меньшей мере одной кристаллической формы соединения I в соответствии с настоящим изобретением, на основе общего содержания соединения I.
Еще одна задача настоящего изобретения относится к композиции соединения I, которая содержит по меньшей мере два (т.е. 2, 3, 4 или 5) кристаллические формы, выбранные из
- толуол-моносольвата, как определено в настоящем описании,
- несольватированной кристаллической формы, как определено в настоящем описании,
- ацетон-сольвата "A", как определено в настоящем описании,
- ацетон-сольвата "B", как определено в настоящем описании,
- ацетон-сольвата "C", как определено в настоящем описании.
В принципе синтез 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина (соединение I), используемого в качестве исходного материала для получения кристаллического сольвата и несольватных форм по изобретению, может быть осуществлено известными способами. Подходящими способами являются способы для синтеза органических дифосфитов, которые описаны, например, в EP 0214622 A2, US 4668651, US 4748261, US 4769498, US 4885401, US 5235113, US 5391801, US 5663403, US 5728861, US 6172267, DE 10360771 A1, WO 2003/062171 и WO 2003/062251. Раскрытия этих документов включены в настоящее описание посредством ссылки.
В приемлемом варианте выполнения соединение I получают с помощью процесса, включающего следующие стадии:
a) взаимодействие первого ароматического диола формулы (A1)
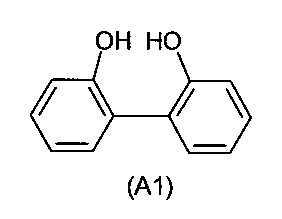
с PCl3 с получением монохлорфосфита (A2)
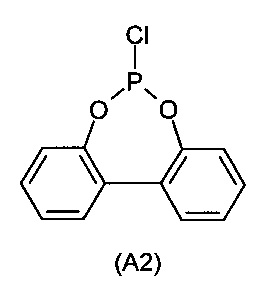
b) взаимодействие монохлорфосфита (A2) со вторым ароматическим диолом (A3)
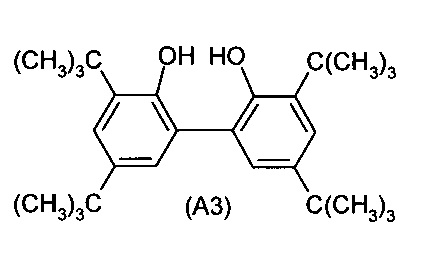
с получением соединения I.
Существуют несколько методов для удаления галогеноводородов, образующихся в реакции конденсации. Такие способы известны специалистам в данной области техники и описаны, в частности, в вышеупомянутых документах. Одной из возможностей является добавление по меньшей мере стехиометрического количества основания. Типичными основаниями, используемыми для удаления галогеноводородов, являются азотистые основания.
В предпочтительном варианте выполнения соединение I получают по способу, описанному в WO 2003/062171 и WO 2003/062251 (например, в соответствии с примером 17). Идея этих документов включена в настоящее описание посредством ссылки. В соответствии с этим методом галогеноводороды, образованные в по меньшей мере одной из реакций конденсации, отделяют от реакционной смеси с помощью вспомогательного основания. Указанное основание образует соль с галогеноводородами, которая является жидкой при температуре, при которой ценное вещество существенно не разлагается в процессе разделения и, где соль вспомогательного основания и ценное вещество или раствор ценного вещества образуют две несмешивающиеся фаз жидкости.
Кристаллический толуол-моносольват соединения I
Кристаллический толуол-моносольват соединения I может быть идентифицирован с помощью рентгеновской порошковой дифрактометрии на основании его дифракционной диаграммы. Таким образом, рентгеновская порошковая дифрактограмма, записанная при 25°C с использованием Cu-Kα излучения (1,54178 Å), демонстрирует по меньшей мере 5, часто по меньшей мере 6, в особенности по меньшей мере 7, и особенно все рефлексы, указанные в следующей таблице 1, как значения 20, и как межплоскостные расстояния d:
Исследования монокристаллов толуол-моносольвата соединения I показывают, что основная кристаллическая структура является ромбической. Элементарная ячейка имеет пространственную группу Pbca. Характерные данные по кристаллической структуре толуол-моносольвата соединения I (определенной при -173°C) приведены в таблице 2.
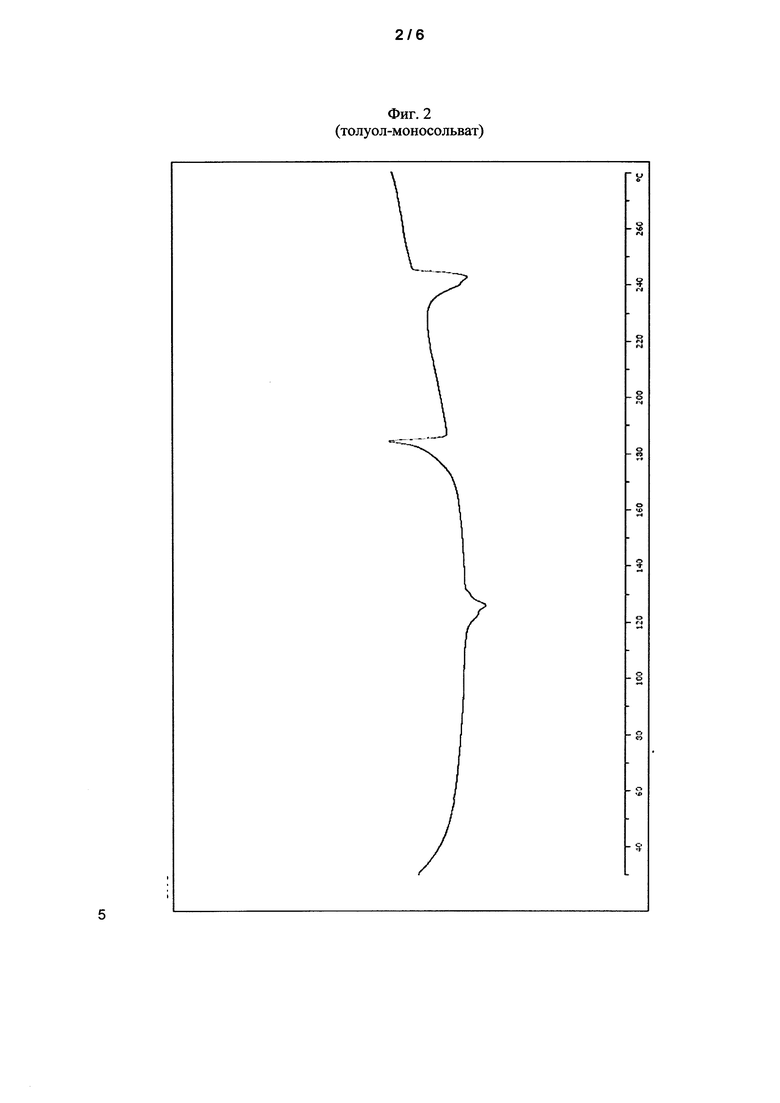
Толуол-моносольват соединения I показывает характеристические пики в дифференциальной сканирующей калориметрии (ДСК) (см. Фиг. 2). При 126°C происходит десольватация, и кристаллическая форма превращается в аморфную форму. При 184°C наблюдается вторичная кристаллизация в стабильную, несольватную форму. Дополнительный пик при 243°C может быть связан с плавлением несольватной формы.
Получение кристаллического толуол-моносольвата 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина в соответствии с настоящим изобретением осуществляют посредством кристаллизации из толуольного раствора.
Еще одной задачей настоящего изобретения является способ получения кристаллического толуол-моносольвата соединения I, как описано выше, включающий:
i) получение раствора соединения I в толуоле, который является пересыщенным при температуре 30°C, и
ii) обеспечение соединения I для кристаллизации при температуре не более 30°C.
Предпочтительно, на стадии i) получают насыщенный раствор соединения I при температуре по меньшей мере 80°C и атмосферном давлении. Более предпочтительно, на стадии i) получают насыщенный раствор соединения I при температуре кипения с обратным холодильником и давлении окружающей среды. Понятно, что этот раствор станет пересыщенный соединением I при более низких температурах, пока не начнется кристаллизации соединения I.
Для приготовления раствора посредством растворения соединения I по существу могут быть использованы любые известные формы соединения I. Часто будет использоваться аморфный 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепин или смесь различных кристаллических модификаций или смесь аморфного и кристаллического 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо [d,f][1,3,2]-диоксафосфепина.
В соответствии с особым вариантом выполнения раствор соединения I может быть также получен посредством химической реакции, которая приводит к реакционной смеси, которая содержит соединение I, в случае необходимости после удаления реагентов и/или побочных продуктов. В настоящем описании эта методика может быть использована для осуществления реакции в толуоле в качестве органического растворителя или посредством переноса реакционного продукта в толуол в качестве растворителя известными способами.
Решающее значение для формирования кристаллического толуол-моносольвата соединения I имеет то, что кристаллизацию на стадии ii) осуществляют при температуре не более 30°C.
В первом предпочтительном варианте выполнения изобретения кристаллизацию на стадии ii) осуществляют посредством охлаждения раствора соединения I в толуоле до температуры не более чем 30°C, где раствор имеет концентрацию соединения I, которая является пересыщенной при температуре не более чем 30°C. Образование кристаллов при более высоких температурах может быть предотвращено, например, посредством выбора достаточно высокой скорости охлаждения и/или избегания присутствия затравочных кристаллов. Если затравочные кристаллы кристаллического толуол-моносольвата соединения I применяются, они добавляются при температуре не более чем 30°C.
В другом предпочтительном варианте выполнения изобретения кристаллизацию на стадии ii) осуществляют посредством добавления раствора соединения I в толуоле, имеющего температуру, по меньшей мере около 65°C, предпочтительно по меньшей мере 80°C, в сосуд, содержащий метанол, имеющий температуру не более чем 30°C, и в котором во время добавления и кристаллизации температура растворителя-смеси в сосуде поддерживается на уровне не более чем 30°C. Температура смеси растворителей в сосуде может поддерживаться при значении не более чем 30°C, например, по меньшей мере одним из следующих способов:
- с использованием метанола с достаточно низкой начальной температурой,
- с помощью достаточно большого количества метанола,
- охлаждением сосуда во время добавления и кристаллизации.
Кристаллизация кристаллического толуол-моносольвата 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина при необходимости может быть стимулирована или ускорена посредством затравочных кристаллов кристаллического толуол-моносольвата соединения I. Затравочные кристаллы толуол-моносольвата соединения I обычно добавляли перед кристаллизацией.
Если затравочные кристаллы использовали для кристаллизации, их количество предпочтительно составляло от 0,001 до 10 мас. %, более предпочтительно от 0,005 до 5 мас. %, в особенности от 0,01 до 1 мас. % и особенно от 0,05 до 0,5 мас. %, считая на растворенное соединение I.
Отделение кристаллического толуол-моносольвата соединения I от продукта кристаллизации, то есть отделение толуол-моносольвата от маточного раствора достигался с помощью обычных методик для отделения твердых компонентов от жидкостей, например, фильтрацией, центрифугированием или декантацией. В подходящем варианте выполнения выделенное твердое вещество промывали предпочтительно толуолом. Промывание обычно осуществляли при температурах ниже 30°C, часто ниже 25°C и, в частности, ниже 20°C для того, чтобы обеспечить потери ценного вещества как можно на более низком уровне. Далее, кристаллический толуол-моносольват соединения I может быть высушен и затем подаваться для дальнейшей обработки.
Содержание кристаллического толуол-моносольвата соединения I, в расчете на общее количество соединения I, как правило, составляет по меньшей мере 90% и часто по меньшей мере 95% и особенно по меньшей мере 97%.
Несольватированная кристаллическая форма соединения I.
Неожиданно было обнаружено, что в то время как при достаточно низкой температуре можно получить вышеуказанный толуол-сольват соединения I, при более высокой температуре может быть выделена стабильная несольватированная кристаллическая форма соединения I.
Несольватированная кристаллическая форма соединения I может быть идентифицирована с помощью рентгеновской порошковой дифрактометрии на основании ее дифракционной диаграммы. Таким образом, рентгеновская порошковая дифрактограмма, записанная при 25°C с использованием Cu-Kα излучения (1,54178 Å), демонстрирует по меньшей мере 5, часто по меньшей мере 6, в особенности по меньшей мере 7, и особенно все рефлексы, указанные в следующей таблице 3 как 2θ-значения и как межплоскостные расстояния d:
Исследования монокристаллов несольватированной формы соединения I показывают, что основная кристаллическая структура является моноклинной. Элементарная ячейка имеет пространственную группу P 21/c. Характерные данные по кристаллической структуре несольватированной формы соединения I (определенной при -173°C) приведены в таблице 4.
Несольват соединения I показывает в ДСК пик, отнесенный к плавящемуся при 243°C.
Еще одной задачей настоящего изобретения является способ получения несольватированной кристаллической формы соединения I, в котором 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепину дают возможность кристаллизоваться при температуре выше 65°C или, в котором 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепин суспендируют в растворителе и взвесь перемешивают в суспензии при температуре по меньшей мере, 65°C.
В предпочтительном варианте выполнения суспендированное вещество перемешивают в суспензии при температуре по меньшей мере 75°C, более предпочтительно по меньшей мере 85°C, особенно при температуре выше точки кипения толуола при давлении окружающей среды.
Предпочтительно растворитель выбирают из алкилбензолов, арилалкилэфиров, хлорбензола и их смесей. Предпочтительно, растворитель выбирают из растворителей и смесей растворителей, имеющих точку кипения по меньшей мере 100°C при 1013 мбар.
Подходящими растворителями являются, например, толуол, ди-н-бутиловый эфир и их смеси. Особенно предпочтительный растворитель содержит или состоит из толуола.
Предпочтительно время существования суспензии, по меньшей мере 10 минут, более предпочтительно по меньшей мере 30 минут, в особенности по меньшей мере 1 час.
Предпочтительно суспензию отделяют от маточного раствора при температуре по меньшей мере 65°C, более предпочтительно по меньшей мере 75°C, в особенности по меньшей мере 85°C.
Выделение суспендированных твердых частиц, т.е. кристаллической несольватированной формы соединения I из маточного раствора может быть осуществлено с помощью обычных методик для отделения твердых компонентов от жидкостей, например, фильтрацией, центрифугированием или декантацией. В подходящем варианте выполнения выделенное твердое вещество промывали, предпочтительно толуолом. Промывание обычно осуществляли при температуре по меньшей мере 65°C. Далее кристаллическая несольватированная форма соединения I может быть высушена и затем подаваться для дальнейшей обработки.
Кристаллический ацетон-сольват «A» соединения I
Кристаллический ацетон-сольват «A» соединения I может быть идентифицирован с помощью рентгеновской порошковой дифрактометрии на основании его дифракционной диаграммы. Таким образом, рентгеновская порошковая дифрактометрия, записанная при 25°C с использованием Cu-Kα излучения (1,54178 Å) демонстрирует по меньшей мере 5, часто по меньшей мере 6, в особенности по меньшей мере 7, и особенно все рефлексы, указанные в следующей таблице 5, как 2θ значения, и как межплоскостные расстояния d:
Исследования монокристаллов ацетон-сольвата «A» соединения I показывало, что основная кристаллическая структура моноклинная. Элементарная ячейка имеет пространственную группу P 21/n. Характеристические данные по кристаллической структуре ацетон-сольвата "А" соединения I (определенные при -173°C) суммированы в таблице 6.
Еще одной задачей настоящего изобретения является способ получения кристаллического ацетон-сольвата «А» соединения I, как определено выше, включающий:
I) приготовление суспензии соединения I в ацетоне,
II) нагревание суспензии, чтобы перевести соединение I в раствор, и
III) охлаждение раствора, полученного на стадии II) и осуществление кристаллизации.
Предпочтительно, на стадии I) суспензия соединения I в ацетоне получают при температуре от 10 до 30°C.
Предпочтительно на стадии II) суспензию нагревают до температуры по меньшей мере 50°C, предпочтительно до температуры кипения с обратным холодильником. Нагревание предпочтительно осуществляют при давлении окружающей среды. Ацетон имеет температуру кипения 56°C при атмосферном давлении (101,3 кПа).
Предпочтительно, на стадии III) раствор, полученный на стадии II), охлаждают до температуры от температуры кипения (около 56°) до температуру окружающей среды (около 20°C).
Для приготовления раствора соединения I по существу любая известная форма соединения I может быть использована. Часто будет использоваться аморфный 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепин или смесь различных кристаллических модификаций или смесь аморфного и кристаллического 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина.
Выделение кристаллического ацетон-сольвата A соединения I из маточного раствора, достигается с помощью обычных методик для отделения твердых компонентов от жидкостей, например, фильтрацией, центрифугированием или декантацией. В подходящем варианте выполнения выделенный ацетон-сольват будет промываться, предпочтительно ацетоном. Промывание обычно осуществляют при температурах ниже 30°C, часто ниже 25°C и, в особенности, ниже 20°C для того, чтобы обеспечить потери ценного вещества как можно более низком уровне.
Содержание кристаллического ацетон-сольвата A соединения I в расчете на общее количество соединения I, как правило составляет по меньшей мере 90% и часто по меньшей мере 95% и особенно по меньшей мере 97%.
Кристаллический ацетон-сольват "B" соединения I
Кристаллический ацетон-сольват «В» соединения I может быть идентифицирован с помощью рентгеновской порошковой дифрактометрии на основании его дифракционной диаграммы. Таким образом, рентгеновская порошковая дифрактограмма, записанная при 25°C с использованием Cu-Kα излучения (1,54178 Å), демонстрирует по меньшей мере 5, часто по меньшей мере 6, в особенности по меньшей мере 7, и особенно все рефлексы, указанные в следующей таблице 7, как 2θ значения, и как межплоскостные расстояния d:
Исследования монокристаллов ацетон-сольвата "B" соединения I показывают, что основная кристаллическая структура является моноклинной. Элементарная ячейка имеет пространственную группу P 21/c. Характерные данные по кристаллической структуре ацетон-сольвата «В» соединения I (определенные при -173°C) приведены в таблице 8.
Еще одной задачей настоящего изобретения является способ получения кристаллического ацетон-сольвата "B" соединения I, как определено выше, включающий:
I) обеспечение раствора соединения I в ацетоне,
II) что позволяет части растворителя раствора, обеспеченного на стадии I, испарится и, таким образом, индуцировать кристаллизацию.
Предпочтительно, на стадии I) раствор соединения I в ацетоне обеспечивается при температуре от 40 до 20°C. В частности, раствор соединения I в ацетоне обеспечивается при температуре окружающей среды.
Раствор соединения I может, например, быть получен посредством растворения соединения I в ацетоне.
Предпочтительно на стадии II) раствор охлаждают до температуры не более 15°C, предпочтительно самое большее 10°C.
Выделение кристаллического ацетон-сольвата «B» соединения I из маточного раствора достигается с помощью обычных методик для отделения твердых компонентов от жидкостей, например, фильтрацией, центрифугированием или декантацией. В подходящем варианте выполнения выделенное твердое вещество промывают, предпочтительно ацетоном. Промывание обычно осуществляют при температурах ниже 30°C, часто ниже 25°C и, в особенности ниже 20°C, для того чтобы обеспечить потери ценного вещества на как можно более низком уровне.
Содержание кристаллического ацетон-сольвата «B» соединения I, считая на общее количество соединения I, составляет, как правило, по меньшей мере 90% и часто по меньшей мере 95% и особенно по меньшей мере 97%.
Кристаллический ацетон-сольват "C" соединения I
Кристаллический ацетон-сольват "C" соединения I может быть идентифицирован с помощью рентгеновской порошковой дифрактометрии на основании его дифракционной диаграммы. Таким образом, рентгеновская порошковая дифрактограмма, записанная при 25°C с использованием Cu-Kα излучения (1,54178 Å), демонстрирует по меньшей мере 5, часто по меньшей мере 6, в особенности по меньшей мере 7 и особенно все рефлексы, указанные в следующей таблице 9, как 2θ значения, и как межплоскостные расстояния d:
Исследования монокристаллов ацетон-сольвата «C» соединения I показывают, что основная кристаллическая структура является триклинной. Элементарная ячейка имеет пространственную группу P-1. Характерные данные по кристаллической структуре ацетон-сольвата "C" соединения I (определенной при -173°C) приведены в таблице 8.
Еще одной задачей настоящего изобретения является способ получения кристаллического ацетон-сольвата "C" соединения I, как определено выше, включающий:
I) приготовление при температуре от около 15 до 25°C насыщенного раствора соединения I в ацетоне,
II) нагревание раствора, полученного на стадии I), до температуры, которая на от около 15 до 25°C выше чем температура на стадии I),
III) что позволяет нагретому раствору, полученному на стадии II), охладиться до температуры, которая на от около 5 до 15°C ниже температуры стадии I),
где стадии от I) до III) повторяются, по меньшей мере в 10 раз.
Предпочтительно, на стадии I) насыщенный раствор соединения I в ацетоне получают при температуре окружающей среды.
Предпочтительно, на стадии II) раствор нагревают до температуры, которая на около 20°C выше чем температура на стадии I).
Предпочтительно, на стадии III) раствор оставляли охлаждаться до температуры, которая от около 5 до 15°C ниже температуры стадии I).
Предпочтительно, стадии от I) до III) повторяются от 10 до 15 раз, более предпочтительно 10 раз.
Выделение кристаллического ацетон-сольвата "C" соединения I из маточного раствора достигается с помощью обычных методик для отделения твердых компонентов из жидкостей, например, фильтрацией, центрифугированием или декантацией. В подходящем варианте выполнения выделенный ацетон-сольват "C" промывают, предпочтительно ацетоном. Промывание обычно осуществляют при температурах ниже 30°C, часто ниже 25°C и, в особенности ниже 20°C для того, чтобы обеспечить потери ценного вещества на как можно более низком уровне.
Содержание кристаллического ацетон-сольвата "C" соединения I, в расчете на общее количество соединения I составляет, как правило, по меньшей мере 90% и часто по меньшей мере 95% и особенно по меньшей мере 97%.
Кристаллические сольватные и не сольватные формы 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина в соответствии с настоящим изобретением особенно пригодны в качестве лигандов катализатора на основе переходного металла для гидроформилирования, гидроцианирования или гидрирования.
Как упоминалось ранее, кристаллические формы соединения I в соответствии с настоящим изобретением, в том числе сольваты соединения I, и их композиции, содержащие основное количество этих кристаллических форм, имеют полезные свойства, в частности, следующие свойства:
- низкую липкость,
- низкую склонность к слеживанию,
- низкую склонность к образованию пыли,
- высокую объемную плотность и
- высокую чистоту соединения I.
Неожиданно было также обнаружено, что кристаллические формы по настоящему изобретению, содержащие растворитель в кристаллической решетке, могут быть использованы в качестве лигандов для катализаторов на основе переходных металлов без отрицательного влияния растворителя на формирование гомогенных катализаторов и/или каталитической реакции.
Кристаллические формы по изобретению также характеризуется хорошей текучестью.
Низкая склонность к слеживанию приводит к тому, что кристаллические формы по настоящему изобретению могут храниться даже в течение длительных периодов времени до их использования. Предпочтительно, механическое разрушение перед использованием во многих случаях не является необходимым.
Кристаллические формы по изобретению позволяет легче регулировать, например, процедуры взвешивания, фасовки и дозирования, в которых минимизируется образование пыли, которая может представлять опасность для здоровья при вдыхании или попадании на кожу или в глаза.
Как результат этих свойств, кристаллические формы по изобретению в частности подходят для изготовления катализаторов на основе переходных металлов.
В отличие от ацетонитрила, содержащегося в ацетонитрил-сольватах соединения I, толуол и ацетон, содержащиеся в толуол- или ацетон-сольватах соединения I в условиях гомогенного катализа не взаимодействуют с каталитически активными центрами металла катализаторов на основе переходных металлов, полученных из переходного металла и соединения I и, следовательно, не мешают катализу на основе переходного металла, например, за счет снижения каталитической активности или селективности.
Еще одной задачей настоящего изобретения является применение кристаллической формы соединения I, как определено выше, для получения катализатора на основе переходного металла для гидроформилирования, гидроцианирования или гидрирования.
Еще одной задачей настоящего изобретения является способ получения катализатора на основе переходного металла, в котором кристаллическая форма соединения I, как определено выше, обеспечивается и приводится в контакт с соединением или комплексом переходного металла в инертном растворителе.
Еще одной задачей настоящего изобретения является катализатор на основе переходного металла, который можно получить способом, в котором кристаллическая форма соединения I, как определено выше и далее, обеспечивается и приводится в контакт с соединением или комплексом переходного металла в инертном растворителе.
Следующие утверждения в равной степени относятся катализаторам по изобретению, а также к способу их получения и их применения.
Предпочтительно, чтобы катализатор по изобретению получали бы из 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина, который состоит из по меньшей мере на 50 мас. %, более предпочтительно по меньшей мере на 75 мас. %, в особенности, по меньшей мере на 90 мас. %, по меньшей мере одной кристаллической формы, выбранной из сольватных и несольватных форм 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина в соответствии с настоящим изобретением, и их смесей.
В первом варианте выполнения катализатор содержит кристаллический толуол-моносольват соединения I в качестве лиганда. Кроме того, предпочтительно катализатор содержит по меньшей мере 50 мас. %, более предпочтительно по меньшей мере 75 мас. %, в особенности по меньшей мере 90 мас. % кристаллического толуол-моносольвата соединения I в расчете на общую массу соединения I, используемого в качестве лиганда.
Во втором варианте выполнения катализатор содержит кристаллическую несольватированную форму соединения I в качестве лиганда. Кроме того, предпочтительно катализатор содержит по меньшей мере 50 мас. %, более предпочтительно по меньшей мере 75 мас. %, в особенности по меньшей мере 90 мас. % кристаллической несольватированной формы соединения I в расчете на общую массу соединения I, используемого в качестве лиганда.
В третьем варианте выполнения катализатор содержит кристаллический ацетон-сольват "A" соединения I в качестве лиганда. Кроме того, предпочтительно катализатор содержит по меньшей мере 50 мас. %, более предпочтительно по меньшей мере 75 мас. %, в особенности по меньшей мере 90 мас. % кристаллического ацетон-сольвата "A" соединения I в расчете на общую массу соединения I, используемого в качестве лиганда.
В четвертом варианте выполнения катализатор содержит кристаллический ацетон-сольват "B" соединения I в качестве лиганда. Кроме того, предпочтительно катализатор содержит, по меньшей мере 50 мас. %, более предпочтительно по меньшей мере 75 мас. %, в особенности по меньшей мере 90 мас. % кристаллического ацетон-сольвата "B" соединения I в расчете на общую массу соединения I, применяемого в качестве лиганда.
В пятом варианте выполнения катализатор содержит кристаллический ацетон-сольват "B" соединения I в качестве лиганда. Кроме того, предпочтительно катализатор содержит по меньшей мере 50 мас. %, более предпочтительно по меньшей мере 75 мас. %, в особенности по меньшей мере 90 мас. % кристаллического ацетон-сольвата "C" соединения I в расчете на общую массу соединение I, используемого в качестве лиганда.
В общем, концентрация металла в реакционной среде лежит в диапазоне от около 1 до 10000 частей на миллион. Молярное отношение лиганда к переходному металлу, обычно находится в интервале от около 0,5:1 до 1000:1, предпочтительно от 1:1 до 500:1.
Специалист в данной области техники будет выбирать переходный металл в зависимости от реакции, которую необходимо катализировать. Переходный металл предпочтительно представляет собой металл из групп 8, 9 или 10 периодической таблицы элементов, предпочтительно из металлов групп 9 и 10 (т.е. Co, Ni, Rh, Pd, Ir, Pt).
В дополнение к описанным выше лигандам, катализаторы по изобретению могут иметь по меньшей мере один лиганд, который предпочтительно выбирают из: карбоксилатов, ацетилацетонатов, арилсульфонатов и алкилсульфонатов, гидридов, CO, олефинов, диенов, циклоолефинов, таких как циклооктадиен и норборнадиен, нитрилы, ароматические соединения и гетероароматические соединения, простые эфиры, и монодентатные, бидентатные и полидентатные фосфорамидиты и фосфитные лиганды. Главным образом, дополнительные лиганды выбирают из гидрида, CO и олефинов (т.е. видов, которые способны образовывать активный катализатор для реакции гидроформилирования).
Катализаторы по изобретению (или приготовленные по способу настоящего изобретения, или полученные с использованием кристаллической формы соединения I в соответствии с настоящим изобретением) особенно пригодны в качестве катализатора для реакции гидроформилирования. Дополнительным объектом настоящего изобретения является способ гидроформилировани соединения, которое содержат по меньшей мере одну ненасыщенную этиленовую двойную связь посредством реакции с монооксидом углерода и водородом в присутствии катализатора, содержащего по меньшей мере один комплекс металла, выбранного из кобальта или родия. Особое предпочтение отдается использованию родия.
В предпочтительном варианте выполнения катализаторы гидроформилирования получают in situ, в реакторе, используемом для реакции гидроформилирования. Однако, катализаторы согласно изобретению, если желательно, также могут быть получены отдельно и выделены обычными способами. Для получения катализаторов в соответствии с настоящим изобретением in situ, например, по меньшей мере одна кристаллическая форма соединения I согласно изобретению, соединение или комплекс переходным металлом, при необходимости, по меньшей мере с еще одним дополнительным лигандом и активатором может взаимодействовать в инертном растворителе в условиях гидроформилирования.
Подходящими соединениями или комплексами родия для приготовления катализаторов гидроформилирования являются, например, соли родия(II) и родия(III), такие как карбоксилат родия(II) или родия(III), ацетат родия(II) и родия(III) и т.д. Также подходящими являются комплексы родия, такие как бис(карбонил)ацетилацетонат, ацетилацетонато бисэтиленродий(I), ацетилацетонато циклооктадиенил родия(I), ацетилацетонато норборнадиенил родия(I), ацетилацетонато карбонила трифенилфосфин родия(I) и т.д.
Подходящими соединениями кобальта для получения катализаторов гидроформилирования являются, например, кобальт(II) сульфат, кобальт(II) карбонат, их аминные или гидратные комплексы, карбоксилаты кобальта, такие как ацетат кобальта, этилгексаноат кобальта, нафтаноат кобальта, а также комплекс капроата кобальта. Карбонильные комплексы кобальта, такие как дикобальт октакарбонил, тетракобальт додекакарбонил и гексакобальт гексадекакарбонил могут быть также использованы в настоящем изобретении.
Соединения кобальта или родия, которые были упомянуты и дополнительно подходящие соединения, в принципе известны и адекватно описаны в литературе, либо могут быть получены специалистами в данной области техники, таким же образом, как уже известные соединения.
При гидроформилировании и/или при разработке катализатора могут быть предприняты ряд мер для повышения каталитической активности и/или предотвращения разложения катализатора. Такие способы описаны, в частности, в патентных документах EP 0590613, EP 0865418, EP 0874796, EP 0874797, EP 0876321, EP 0876322, EP 0904259, EP 1019352, EP 1019353. Раскрытия этих документов включено в настоящее описание посредством ссылки.
Растворителями предпочтительно являются альдегиды, которые образуются при гидроформилировании специфических олефинов, а также их высококипящие последующие продукты реакции, например, продукты альдольной конденсации. Растворители, которые подобным образом являются пригодными, представляют собой ароматические углеводороды, такие как толуол и ксилолы, углеводороды или смеси углеводородов, а также для разбавления вышеуказанных альдегидов и последующих продуктов альдегидов. Дополнительными растворителями являются сложные эфиры алифатических карбоновых кислот с алканолами, например Texanol™ и сложные эфиры ароматических карбоновых кислот, например, C8-C13-диалкилфталаты.
Что касается получения и применения катализаторов гидроформилирования и их применения в гомогенном катализе можно сослаться, например, на EP 0214622 A2, US 4668651, US 4748261, US 4769498, US 4885401, US 5235113, US 5391801, US 5663403, US 5728861, US 6172267, DE 10360771 A1, WO 2003/062171 и WO 2003/062251.
Полезными субстратами для процесса гидроформилирования в соответствии с настоящим изобретением, в принципе, являются все соединения, которые содержат одну или несколько этиленовых ненасыщенных двойных связей. Они включают, например, олефины, такие как ос-олефины, внутренние олефины с неразветвленной цепью и разветвленные, циклические олефины и олефины с заместителями, которые являются, по существу инертным в условиях гидроформилирования. Предпочтительными являются олефиновое сырье, содержащее олефины, имеющие от 2 до 12, в особенности от 3 до 8 атомов углерода.
Подходящими α-олефинами являются, например, этилен, пропилен, 1-бутен, 1-пентен, 1-гексен, 1-гептен, 1-октен, 1-нонен, 1-децен, 1-ундецен, 1-додецен и т.д. Предпочтительными разветвленными внутренними олефинами, являются С4-С20-олефины, такие как 2-метил-2-бутен, 2-метил-2-пентен, 3-метил-2-пентен, смеси разветвленных внутренних гептенов, смеси разветвленных внутренних октенов, смеси разветвленных внутренних ноненов, смеси разветвленных внутренних деценов, смеси разветвленных внутренних ундеценов, смеси разветвленных внутренних додеценов и т.д. Кроме того, олефинами, пригодными для гидроформилирования, являются С5-С8-циклоалкены, такие как циклопентен, циклогексен, циклогептен, циклооктен и их производные, например, их С1-С20-алкильные производные, имеющие от 1 до 5 алкильных заместителей. Дополнительные олефины, пригодные для гидроформилирования представляют собой винилароматические углеводороды, такие как стирол, α-метилстирол, 4-изобутилстирол и т.д. Другими олефинами, пригодными для гидроформилирования, являются сложные эфиры и амиды α,β-этиленненасыщенных монокарбоновых и/или дикарбоновых кислот, например, метил-3-пентеноат, метил-4-пентеноат, метил олеат, метилакрилат, метилметакрилат, ненасыщенные нитрилы, такие как 3-пентеннитрил, 4-пентеннитрил, акрилонитрил, простые виниловые эфиры, такие как винил метиловый эфир, винил этиловый эфир, винил пропиловый эфир и т.д., С1-С20-алкенолы, С1-С20-алкендиолы и алкадиенолы, такие как 2,7-октадиен-1-ол. Кроме того, пригодными субстратами являются диены или полиены, имеющие изолированные или сопряженные двойные связи. К ним относятся, например, 1,3-бутадиен, 1,4-пентадиен, 1,5-гексадиен, 1,6-гептадиен, 1,7-октадиен, винилциклогексен, дициклопентадиен, 1,5,9-циклооктатриен а также гомополимеры и сополимеры бутадиена.
В предпочтительном варианте выполнения использовали промышленно доступную смесь олефинов в реакции гидроформилирования. Подходят, например олефиновые смеси, которые образуются в результате крекинга углеводородов при переработке нефти, например, при каталитическом крекинге, таком как крекинг на флюидизированном катализаторе (FCC), термический крекинг или гидрокрекинг с последующим дегидрированием.
Предпочтительной смесью промышленных олефинов является С3-фракция. Пропиленовое сырье, которое пригодно в качестве исходного материала для способа по настоящему изобретению, может содержать часть пропана в дополнение к пропилену. Оно содержит, например, от 0,5 до 40 мас. %, предпочтительно от 2 до 30 мас. %, в особенности от 3 до 10 мас. % пропана. Предпочтительным примером является "химически чистый пропилен", который содержит от 3 до 10 мас. % пропана. Его получают, например, посредством реакции бензино-лигроиновой фракции или природного газа в установке парового крекинга и последующим выделением продукта реакции посредством перегонки. Еще одним примером подходящего пропиленового сырья является "пропилен квалификации ректификованный", который имеет содержание пропана, от 20 до 30%.
Еще одной предпочтительной смесью промышленных олефинов является С4-фракция. С4-Фракции могут быть получены, например, посредством жидкофазного каталитического крекинга или парофазного крекинга газойля или парового крекинга бензино-лигроиновой фракции. В зависимости от состава С4-фракции, делается различие между общей С4-фракцией (сырой С4-фракцией), рафинатом I, полученным после удаления 1,3-бутадиена, и рафинатом II, полученным после удаления изобутена. Особенно предпочтительным является рафинат II.
Еще одной задачей настоящего изобретения является применение катализатора переходного металла, содержащего в качестве лиганда кристаллическую форму соединения I, для гидроцианирования.
Катализаторы, используемые для гидроцианирования, также содержат комплексы переходного металла VIII группы, в частности, никеля, рутения, родия, палладия, платины, предпочтительно никеля, палладия или платины и особенно предпочтительно никеля. Получение комплексов металлов может быть осуществлено, как описано выше. То же самое относится к приготовлению in-situ катализаторов гидроцианирования по изобретению. Способы гидроцианирования описаны в J. March, Advanced Organic Chemistry, 4th edition, pp. 811-812, который включен в данное описание посредством ссылки.
Что касается получения и применения катализаторов гидроцианирования, и их применения в гомогенном катализе, можно сослаться, например, на US 6127567.
Еще одной задачей настоящего изобретения является применение катализатора на основе переходного металла, содержащего в качестве лиганда кристаллическую форму соединения I, для гидрирования.
Катализаторы в соответствии с настоящим изобретением, используемые для гидрирования, предпочтительно содержат, по меньшей мере, один металл из группы 9 или 10 группы Периодической таблицы элементов, то есть металл, выбранный из группы Rh, Ir, Ni, Co, Pd и Pt.
Количество катализатора, которое будет использоваться, зависит среди прочего от соответствующего каталитически активного металла и от формы, в которой он используется, и может быть определено для каждого конкретного случая специалистом в данной области техники. Так, например, Ni- или Co-содержащей катализатор гидрогенизации используется в количестве предпочтительно от 0,1 до 70 мас. %, особенно предпочтительно от 0,5 до 20 мас. % и в особенности от 1 до 10 мас. % в расчете на массу соединения, подлежащего гидрированию. Количество указанного катализатора основывается на количестве активного металла, то есть на каталитически активном соединении катализатора. Когда используются благородные металлы, содержащие, например, родий, рутений, платину или палладий, они используются в количествах, которые приблизительно в 10 раз меньше.
Гидрирование предпочтительно проводится при температуре в интервале от 0 до 250°C, особенно предпочтительно в диапазоне от 20 до 200°C и, в особенности, в диапазоне от 50 до 150°C.
Реакционное давление в реакции гидрогенизации предпочтительно находится в диапазоне от 1 до 300 бар, особенно предпочтительно в диапазоне от 50 до 250 бар, в особенности в диапазоне от 150 до 230 бар.
Как реакционное давление, так и температура реакции зависят, помимо прочего, от активности и количества используемого катализатора гидрогенизации, и может быть определено в каждом конкретном случае специалистом в данной области техники.
Гидрогенизацию можно проводить в подходящем растворителе или в объеме (без растворителя). Подходящими растворителями являются такие, которые являются инертными в условиях реакции, т.е. которые не вступает в реакцию ни с исходным материалом ни с продуктом, так и сами по себе не изменяются, и могут быть отделены без проблем от полученных изоалканов. Подходящие растворители включают, например, ациклические и циклические простые эфиры, например, диэтиловый эфир, метил-трет-бутиловый эфир, тетрагидрофуран или 1,4-диоксан, и спирты, в частности C1-C3-алканолы, такие как метанол, этанол н пропанол или изопропанол. Смеси вышеуказанных растворителей также пригодны.
Водород необходимый для гидрогенизации может быть использован либо в чистом виде или в виде газовых смесей, содержащих водород. Однако, последний не должен содержать мешающих количеств каталитических ядов, таких как сера-содержащих соединений или CO. Примерами подходящих газовых смесей, содержащих водород, являются смеси из реформинг-процесса. Тем не менее, предпочтение отдается использованию водорода в чистом виде.
Гидрогенизацию можно проводить непрерывно или периодически.
Гидрогенизацию обычно проводят в начальной стадии посредством загрузки соединения, которое должно быть гидрогенизировано, в случае необходимости в растворителе. Этот реакционный раствор затем предпочтительно смешивают с катализатором гидрогенизации прежде, чем вводят водород. В зависимости от используемого катализатора гидрогенизации, гидрогенизацию осуществляют при повышенной температуре и/или давлении выше атмосферного. Когда реакцию проводят под давлением, можно использовать обычные сосуды под давлением, известные из известного уровня техники, например, автоклавы, автоклавы с помешиванием и реакторы давления. Если гидрогенизацию осуществляется под давлением водорода не выше атмосферного, возможны обычные реакционные аппараты настоящего уровня техники, которые подходят для атмосферном давления. Примерами являются обычные аппараты с мешалкой, которые предпочтительно оборудованы испарительным охлаждением, подходящими мешалками, приспособлениями для подачи, если соответствующие элементы теплообменника и материалы облицовки внутренней поверхности являются инертными. В случае непрерывной реакции, гидрогенизацию можно проводить при атмосферном давлении в реакционных сосудах, трубчатых реакторах, реакторах с неподвижным слоем катализатора и т.п., которые являются обычными для этой цели.
Следующие чертежи и примеры служат для иллюстрации изобретения и не должны рассматриваться как ограничивающие.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1 показывает рентгеновскую порошковую дифрактограмму кристаллического толуол-моносольвата 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина (соединение I). Рентгеновская порошковая дифрактограмма была записана при условиях, изложенных ниже.
Фиг. 2 показывает ДСК кристаллического толуол-моносольвата соединения I.
Фиг. 3 показывает рентгеновскую порошковую дифрактограмму несольватированной кристаллической формы соединения I. Рентгеновская порошковая дифрактограмма была записана при условиях, изложенных ниже.
Фиг. 4 показывает рентгеновскую порошковую дифрактограмму кристаллического ацетон-сольвата "A" соединения I. Рентгеновская порошковая дифрактограмма была записана при условиях, изложенных ниже.
Фиг. 5 показывает рентгеновскую порошковую дифрактограмму кристаллического ацетон-сольвата "B" соединения I. Рентгеновская порошковая дифрактограмма была записана при условиях, изложенных ниже.
Фиг. 6 показывает рентгеновскую порошковую дифрактограмму кристаллического ацетон-сольвата "C" соединения I. Рентгеновская порошковая дифрактограмма была записана при условиях, изложенных ниже.
ПРИМЕРЫ
Рентгеновские порошковые дифрактограммы были записаны с помощью дифрактометра Panalytical X'Pert Pro (изготовитель: Panalytical) в геометрии отражения в диапазоне от 2θ=3° до 35° с шагом 0,0167°, с использованием Cu-Kα излучения (1,54178 Å) при 25°C. Записанные значения 2θ были использованы для расчета установленных межплоскостных расстояний d. Интенсивность пиков (ось y: интенсивность линий) строится в зависимости от 2θ угла (ось x в градусах 2θ).
Рентгеновские порошковые дифрактограммы были записаны с помощью дифрактометра Panalytical X'Pert Pro (изготовитель: Panalytical) в геометрии отражения в диапазоне от 2θ=3° до 35° с шагом 0,0167°, с использованием Cu-Kα излучения (1,54178 Å) при 25°C. Записанные значения 2θ были использованы для расчета установленных межплоскостных расстояний d. Интенсивность пиков (ось y: интенсивность линий) строится в зависимости от 20 угла (ось x в градусах 2θ).
Данные дифракции рентгеновских лучей на монокристалле собирали при 100 K на детекторе Bruker AXS SMART 6000 CCD с использованием Cu-Kα излучения, либо вращающимся анодом или микроисточником, оба оснащенные многослойными зеркалами. Структуры были решены с помощью методов двойного пространственного циклирования и уточнены значения F2 с использованием пакета программ SHELX TL (Bruker AXS, 2003). Применяли мульти-скан поправку для систематических ошибок с использованием SADABS (G.М. Sheldrick, University of Gottingen, 2010).
ДСК (дифференциальная сканирующая калориметрия) проводили на модуле Mettler Toledo DSC 822e. Образец помещали в обжимные, но вентилируемые алюминиевые кюветы (размер образца составлял 10 мг). Термические свойства анализировали в диапазоне от 30 до 280°C с использованием скорости нагрева 5°C/мин и скорости потока азота при 150 мл/в течение всего эксперимента.
Значения точек плавления и полиморфных переходов подтверждали с помощью Mettler Hot Stage в сочетании с оптическим микроскопом.
Пример 1:
Получение толуол-моносольвата соединения I.
В перемешиваемой трехгорлой колбе с холодильником и впускным патрубком для азота суспендировали 30 г 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина в 50 мл толуола при температуре окружающей среды и нагревали в масляной бане в течение 3 ч до температуры кипения растворителя (температура масляной ванны: 120°C). Полученный насыщенный раствор фильтровали горячим, и фильтрат затем охлаждали до температуры около 25°C. После выдерживания в течение ночи образовавшиеся кристаллы отфильтровывали и сушили в вакууме при 30°C в течение 13 ч.
Продукт кристаллизации демонстрировал рентгеновскую порошковую дифрактограмму, показанную на Фиг. 1, и ДСК, показанную на Фиг. 2. После десольватации (пик при 126°C) получали аморфную форму. При нагревании аморфная форма кристаллизуется в стабильную несольватированную форму (пик повторной кристаллизации при 184°C), которая плавится при 243°C.
Пример 2:
Получение несольватированной формы соединения I.
В трехгорлой колбе с мешалкой и с холодильником и впускным патрубком для азота суспендировали 50,41 г 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина в 30,67 г толуола при температуре окружающей среды и нагревали в масляной бане в течение 3 ч при температуре кипения (температура масляной бани: 120°C). Горячую смесь, содержащую кристаллы, пропускали через вакуум-фильтр, который нагревали до приблизительно 80°C. Полученные кристаллы затем охлаждали до температуры окружающей среды и сушили в вакууме при 30°C в течение 20 ч.
Продукт кристаллизации демонстрировал рентгеновскую порошковую дифрактограмму, показанную на Фиг. 3.
Пример 3:
Получение ацетон-сольвата "A" соединения I.
В 500 мл трехгорлую колбу с мешалкой и с холодильником, охладителем и впускным патрубком для аргона азота суспендировали 20 г 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина в 300 г ацетона при температуре окружающей среды и нагревали на масляной бане в течение 3 ч при температуре кипения с обратным холодильником (температура масляной бани: 70°C). Полученный раствор фильтровали в горячем состоянии, и фильтрат затем охлаждали до температуры окружающей среды. После перемешивания в течение ночи, закристаллизованный продукт отделяли, сушили в вакууме (0,15 мбар) при 30°C в течение 2 ч и анализировали.
Продукт кристаллизации демонстрировал рентгеновскую порошковую дифрактограмму, показанную на Фиг. 4.
Ячейка: P 21/n, Z=4, Z'=1; =12,755(2) Å, b=26,444(5) Å, c=14,490(3) Å; α=90,00, β=103,655(8) γ=90,00; V=4749,24 Å3, R-фактор=6,6%.
Пример 4:
Получение ацетон-сольвата «B» соединения I.
Флакон загружали 0,5 мл ацетона и около 2 мг 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина. Прозрачный раствор медленно упаривали при 5°C в холодильнике в течение нескольких дней. Через нескольких дней наблюдалось образование кристаллов в растворе. Затем подходящий кристалл выделяли под микроскопом и подвергали идентификации рентгеновской структуры.
Ячейка: моноклинная P 21/c, Z=4, Z'=1; a=19.597(6) Å, b=11,849(4) Å, c=21,936(7) Å; α=90,00, β=115,865°(14), γ=90,00; V=4583(3) Å3, R-фактор 5,4%.
Продукт кристаллизации демонстрировал рентгеновскую порошковую дифрактограмму, показанную на Фиг. 5.
Пример 5:
Получение ацетон-сольвата «C» соединения I.
Во флаконе 500 мг 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепин растворяли в 20 мл ацетона. Флакон с прозрачным раствором герметизировали и подвергали циклам нагревания/охлаждения от 10°C до 40°C 10 раз. После 10 циклов наблюдали образование кристаллов в растворе. Затем под микроскопом выделяли подходящий кристалл и подвергали идентификации рентгеновской структуры.
Ячейка: триклинная P-1, Z=2, Z'=1; a=11,568(3) Å, b=12,738(3) Å, c=16,419(4) Å; α=95,632 (10), β=90,723 (9), γ=92,197 (10); V=2405,6 (10) Å3, R-фактор=3,6%.
Продукт кристаллизации демонстрировал рентгеновскую порошковую дифрактограмму, показанную на Фиг. 6.
Пример 6:
Получение толуол-моносольвата соединения I в промышленном масштабе.
В стеклянный реактор на 2 л с рубашкой загружали в атмосфере инертного газа 6-хлор-дибензо[d,f][1,3,2]-диоксафосфепин (445,6 г 90 мас. % раствора в толуоле, 1,60 моль) и этот раствор нагревали до 85°C. Между тем, в 2 л колбу Эрленмейера с магнитной мешалкой, загружали 1-метилимидазол (141,0 г, 1,60 моль) и толуол (791,5 г). 3,3',5,5'-Тетра(1,1-диметилэтил)-1,1'-дифенил-2,2-диол (320,5 г, 0,78 моль) добавляли к смеси при перемешивании, в результате чего образовывался почти бесцветный раствор. Этот раствор добавляли через капельную воронку в атмосфере инертного газа в течение 80 минут в стеклянный реактор. Реакционную смесь коричневого цвета, которая образуется, выдерживали при 80°C в течение еще 50 минут. Затем смесь нагревали до 90°C, и после перемешивания в течение еще десяти минут мешалку останавливали. Образовывались две фазы, и давали возможность разделиться в течение 70 минут. Нижний слой (гидрохлорид 1-метилимидазола) удаляли через нижний клапан, получая 182,7 г вязкой жидкости, которая легко затвердевает (т.пл. составляет приблизительно 80°C). Верхнюю фазу нагревали с обратным холодильником (115°C) и перемешивали в течение еще трех часов. В то же время, 4 л стеклянный реактор с рубашкой и с мешалкой помещали под 2-литровым реактором таким образом, чтобы нижнее выходное отверстие из 2-литрового реактора могло быть присоединено через теплоизолированную тефлоновую трубку через горловину 4-литрового реактора. В 4 л реактор загружали в атмосфере инертного газа метанол (2000 мл), который охлаждали до 20°C. Затем мешалку устанавливали на 355 оборотов в минуту, и сливали толуольный раствор лиганда из 2 л реактора в течение 70 минут через тефлоновую трубку в метанол таким образом, чтобы поток, выходящий из тефлоновой трубки, не контактировал ни со стенкой 4 л реактора, ни по осью или лопастями мешалки. Продукт осаждался мгновенно виде белого твердого вещества, и после завершения добавления раствора лиганда полученную в результате суспензию продукта выдерживали при перемешивании при 20°C в течение еще одного часа. Продукт отфильтровывали и 4 л реактор промывали метанолом (1000 мл). Метанольные промывные воды переносили на фильтровальный осадок, который повторно суспендировали и снова фильтровали. После трех дополнительных промываний метанолом (1000 мл каждой порции), осадок на фильтре сушили под вакуумом, и полученный продукт сушили в течение ночи при 70°C/10 мбар с получением 605,3 г бесцветного, свободно текучего порошка. Продуктом, полученным непосредственно после фильтрации, был толуол-моносольват 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина.
В зависимости от жесткости условий сушки толуол-моносольват может быть превращен в несольватированную форму I. Толуол-моносольват и смеси этих двух форм I с различным составом в зависимости от условий сушки протекают свободно и не склонны слеживаться при длительном хранении.
Хлорид (ионная хроматография) 2 мг/кг,
азот (ASTM D 5762-02) 4 мг/кг.
Настоящее изобретение относится к кристаллической форме соединения (I) 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина, которая может быть использована в химической промышленности. Предложена кристаллическая форма, выбранная из: кристаллического толуол-моносольвата, который на рентгеновской порошковой дифрактограмме при 25°C с Cu-Kα излучением демонстрирует 2θ значения: 5,15°±0,20, 7,59±0,20°, 8,56±0,20°, 8,80±0,20°, 8,97±0,20°, 9,65±0,20°, 10,55±0,20°, 11,47±0,20°, 14,76±0,20° и 15,35±0,20° или несольватированной кристаллической формы, которая на рентгеновской порошковой дифрактограмме при 25°C с Cu-Kα излучением демонстрирует 2θ значения: 5,39±0,20°, 7,04±0,20°, 8,44±0,20°, 8,65±0,20°, 9,08±0,20°, 9,66±0,20°, 10,66±0,20°, 12,60±0,20°, 16,25±0,20° и 17,36±0,20°. Предложены композиции на основе указанной формы, способы получения указанной кристаллической формы и ее применение для получения катализаторов. Указанную кристаллическую форму получают путем кристаллизации из пересыщенного раствора соединения I в толуоле при температуре не более 30°C или путем взбалтывания суспензии соединения I в растворителе, содержащем толуол, при температуре выше температуры кипения толуола. Предложены новые кристаллические формы ценного вещества с низкой липкостью, пониженной склонностью к слеживанию, низким пылеобразованием, а также эффективный способ их получения, композиции на их основе и их использование для получения ценных катализаторов на основе переходных металлов. 6 н. и 7 з.п ф-лы, 8 пр., 6 ил., 10 табл.
1. Кристаллическая форма 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина (соединение I), выбранная из:
кристаллического толуол-моносольвата, который на рентгеновской порошковой дифрактограмме при 25°C с Cu-Kα излучением демонстрирует по меньшей мере 5 из следующих рефлексов, выраженных как 2θ значения: 5,15°±0,20, 7,59±0,20°, 8,56±0,20°, 8,80±0,20°, 8,97±0,20°, 9,65±0,20°, 10,55±0,20°, 11,47±0,20°, 14,76±0,20° и 15,35±0,20° и
несольватированной кристаллической формы, которая на рентгеновской порошковой дифрактограмме при 25°C с Cu-Kα излучением демонстрирует по меньшей мере 5 из следующих рефлексов, выраженных как 2θ значения: 5,39±0,20°, 7,04±0,20°, 8,44±0,20°, 8,65±0,20°, 9,08±0,20°, 9,66±0,20°, 10,66±0,20°, 12,60±0,20°, 16,25±0,20° и 17,36±0,20°.
2. Кристаллическая несольватированная форма по п. 1, демонстрирующая в дифференциальной сканирующей калориметрии пик, отнесенный к плавлению при 243°C.
3. Композиция соединения I для получения катализатора на основе переходного металла для гидроформилирования, гидроцианирования или гидрогенизации, причем композиция содержит по меньшей мере две кристаллические формы, выбранные из
- толуол-моносольвата, как определено в п. 1,
- несольватированной кристаллической формы, как определено в п. 1 или 2,
- ацетон-сольвата "А" 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина, который на рентгеновской порошковой дифрактограмме при 25°C с Cu-Kα излучением демонстрирует по меньшей мере 5 из следующих рефлексов, выраженных как 2θ значения: 6,67±0,20°, 7,11±0,20°, 7,87±0,20°, 8,31±0,20°, 8,96±0,20°, 9,17±0,20°, 10,68±0,20°, 15,78±0,20°, 16,10±0,20° и 18,63±0,20°, - ацетон-сольвата "В", 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина, который на рентгеновской порошковой дифрактограмме при 25°C с Cu-Kα излучением демонстрирует по меньшей мере 5 из следующих рефлексов, выраженных как 2θ значения: 8,13±0,2°, 8,70±0,2°, 8,95±0,2°, 10,02±0,2°, 10,98±0,2°, 11,71±0,2°, 14,16±0,2°, 15,65±0,2°, 16,98±0,2° и 18,08±0,2°,
- ацетон-сольвата "С", 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина, который на рентгеновской порошковой дифрактограмме при 25°C с Cu-Kα излучением демонстрирует по меньшей мере 5 из следующих рефлексов, выраженных как 2θ значения: 5,40±0,20°, 6,97±0,20°, 7,64±0,20°, 8,39±0,20°, 9,24±0,20°, 9,44±0,20°, 11,23±0,20°, 13,46±0,20°, 15,32±0,20° и 18,35±0,20°.
4. Способ получения кристаллического толуол-моносольвата соединения I, как определено в п. 1, включающий:
i) получение раствора соединения I в толуоле, который пересыщен при температуре 30°C, и
ii) обеспечение кристаллизации соединения I при температуре не более 30°C.
5. Способ по п. 4, в котором на стадии i) получают насыщенный раствор соединения I при температуре по меньшей мере 80°C, предпочтительно при температуре кипения с обратным холодильником.
6. Способ по п. 4 или 5, где кристаллизацию на стадии ii) проводят посредством охлаждения раствора соединения I в толуоле до температуры не более 30°C, где раствор имеет концентрацию соединения I, которая является пересыщенной при температуре не более чем 30°C.
7. Способ по п. 4 или 5, в котором кристаллизацию на стадии ii) проводят посредством добавления раствора соединения I в толуоле, имеющего температуру по меньшей мере около 65°C, в сосуд, содержащий метанол, имеющий температуру не более чем 30°C, и в котором во время добавления и кристаллизации температуру смеси растворителей в сосуде поддерживают на уровне не более чем 30°C.
8. Способ получения несольватированной кристаллической формы соединения I, как определено в п. 1 или 2, в котором обеспечивают кристаллизацию 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепина при температуре выше 65°C или в котором 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепин суспендируют в растворителе, который содержит или состоит из толуола, и суспендированное вещество взбалтывают в суспензии при температуре выше температуры кипения толуола, при давлении окружающей среды.
9. Способ по п. 8, в котором время существования суспензии составляет по меньшей мере 10 минут, предпочтительно по меньшей мере 30 минут, в особенности по меньшей мере 1 час.
10. Способ по любому из пп. 8 или 9, в котором суспензию отделяют от маточного раствора при температуре по меньшей мере 65°C.
11. Применение кристаллической формы соединения I, как определено в п. 1 или 2, для получения катализатора на основе переходного металла для гидроформилирования, гидроцианирования или гидрогенизации.
12. Способ получения катализатора на основе переходного металла, в котором предоставляют кристаллическую форму соединения I, как определено в п. 1 или 2, и приводят в контакт с соединением или комплексом переходного металла в инертном растворителе, причем переходный металл выбирают из группы металлов групп 8, 9 или 10 периодической таблицы элементов.
13. Способ по п. 12, в котором переходный металл выбирают из родия, кобальта или никеля.
| Annemiek Van Rooy et al | |||
| Organometallics, 1996, vol | |||
| Прибор для нагревания перетягиваемых бандажей подвижного состава | 1917 |
|
SU15A1 |
| JP 9077713 A, 25.03.1997 | |||
| CN 101684130 A, 31.03.2010 | |||
| Линия для автоматической расфасовки термопластичных материалов | 1977 |
|
SU730574A1 |
| Устройство для подачи огнегасящей жидкости | 1975 |
|
SU577042A1 |
| WO2010042313 A1, 15.04.2010 | |||
| Горелка для сожигания нефти | 1922 |
|
SU1834A1 |

