Область изобретения
Настоящее изобретение относится к полиморфам и сольватам гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина (Р027), способам их получения и к содержащим их фармацевтическим композициям.
Уровень техники
Поиску новых терапевтических средств за последние годы в большой степени способствовало улучшенное понимание структуры белков и других биологических молекул, связанных с таргетными заболеваниями. Одним из важных классов указанных белков является сигма-рецептор (σ), клеточный поверхностный рецептор центральной нервной системы (ЦНС), который может иметь отношение к дисфорическим, галлюциногенным и стимулирующим сердечную деятельность эффектам опиоидов. По результатам исследований биологии и функции сигма-рецепторов были представлены свидетельства того, что лиганды сигма-рецепторов могут быть пригодными для лечения психоза и двигательных расстройств, таких как дистония и поздней дискенизии, а также двигательных расстройств, связанных с хореей Гантингтона или синдромом Туретта, и при болезни Паркинсона (Walker, J.M. et al., Pharmacological Reviews, 1990, 42, 355). Имеются сообщения о том, что известный лиганд сигма-рецептора, римказол, демонстрирует клинические эффекты при лечении психоза (Snyder, S.N., Largent, B.L. J. Neuropsychiatry 1989, 1, 7). Связывающие сайты сигма обладают предпочтительной аффинностью в отношении правовращающих изомеров определенных опиатных бензоморфанов, таких как (+)SKF 10047, (+)циклазоцин и (+)пентазоцин, а также в отношении некоторых нарколептиков, таких как галоперидол.
Сигма-рецептор имеет по меньшей мере два подтипа, которые можно дифференцировать по стереоселективным изомерам указанных фармакологически активных лекарственных средств. SKF 10047 обладает наномолярной аффинностью в отношении сайта сигма 1 (σ-1) и микромолярной аффинностью в отношении сайта сигма 2 (σ-2). Галоперидол обладает сходной аффинностью в отношении обоих подтипов. Эндогенные сигма-лиганды не известны, хотя предполагается, что прогестерон является одним из них. Возможные опосредованные сигма-сайтом лекарственные эффекты включают модуляцию функции глутаматных рецепторов, нейротрансмиттерный ответ, нейропротекцию, поведение и когнитивную способность (Quirion, R. et al., Trends Pharmacol. Sci., 1992, 13:85-86). Большинство исследований полагают, что сигма-связывающие сайты (рецепторы) представляют собой плазмалеммальные элементы каскада сигнальной трансдукции. Лекарственные средства, в отношении которых сообщалось о том, что они являются селективными сигма-лигандами, оценивали в качестве антипсихотических лекарственных средств (Hanner, M. et al., Proc. Natl. Acad. Sci., 1996, 93:8072-8077). Существование сигма-рецепторов в ЦНС, иммунной и эндокринной системах предполагает вероятность того, что это может служить в качестве связи между тремя системами.
Что касается возможного терапевтического применения агонистов или антагонистов сигма-рецепторов, предпринимаются большие усилия по поиску селективных лигандов. Так, ранее были описаны различные лиганды сигма-рецепторов. 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолин является одним из указанных многообещающих лигандов сигма-рецепторов. Указанное соединение и его синтез описаны и заявлены в WO2006/021462.
4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолин представляет собой высокоселективный антагонист рецепторов сигма-1 (σ-1). Он демонстрирует мощную анальгетическую активность при лечении и предупреждении хронической и острой боли и, особенно, нейропатической боли. Соединение имеет молекулярную массу 337,42 масс. ед. Структурная формула соединения следующая:
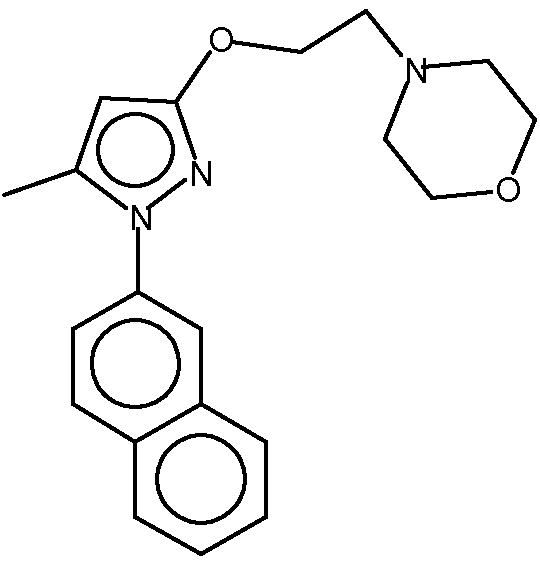
На физические свойства фармацевтического соединения в твердом состоянии могут влиять условия, при которых соединение получали в твердой форме. Физические свойства твердого состояния включают, например, текучесть размолотого твердого вещества, что влияет на простоту обращения с соединением во время получения фармацевтического продукта. Другим важным свойством твердого состояния является скорость его растворения в водной жидкости. Скорость растворения активного ингредиента в желудочном соке пациента может иметь терапевтические последствия, поскольку она представляет собой верхнюю границу скорости, с которой перорально введенный активный ингредиент может достигать крови. Твердое состояние соединения может также влиять на его растворимость, биодоступность, поведение при прессовании, стабильность или его электростатическую природу.
Полиморфизм представляет собой свойство некоторых молекул и молекулярных комплексов принимать более чем одну кристаллическую или аморфную форму в твердом состоянии. Обычно полиморфизм вызывается способностью молекулы вещества изменять свою конформацию или осуществлять различные межмолекулярные и внутримолекулярные взаимодействия, особенно водородные связи, что отражается в различных расположениях атомов в кристаллической решетке различных полиморфов. Соответственно, полиморфы представляют собой различающиеся между собой твердые вещества, имеющие одну и ту же молекулярную формулу и благоприятные и/или неблагоприятные физические свойства, отличающиеся от других форм семейства полиморфов.
Термин «сольват» относится к любой твердой форме данного соединения, в которой указанное соединение связано нековалентной связью с молекулой (молекулами) растворителя (обычно полярного растворителя).
Открытие новых кристаллических полиморфных или аморфных форм фармацевтического соединения создает возможность улучшения физических или эффективных характеристик фармацевтического продукта путем расширения ассортимента материалов, которые доступны ученому, занимающемуся составлением композиций, дизайном, например, фармацевтической лекарственной формы лекарственного средства с заданным профилем высвобождения или другими желательными характеристиками.
Таким образом, все еще существует потребность в дополнительных формах 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина для осуществления его фармацевтической разработки и реализации его потенциала и облегчения получения более совершенных композиций указанного активного фармацевтического ингредиента. В этом отношении различные морфологические формы соединения могут в широком диапазоне обладать различными свойствами, такими как, например, увеличенная термодинамическая стабильность, более высокая чистота или улучшенная биодоступность (например, улучшенное всасывание, характер растворения), а также могут становиться промежуточными соединениями для других форм или обеспечивать более совершенные композиции указанного активного фармацевтического ингредиента. Конкретные формы соединения могут также облегчить производство (например, имеющие улучшенную текучесть), обработку и хранение (например, негигроскопичные, с продолжительным сроком годности) композиций соединения или позволить использовать более низкую дозу терапевтического средства, уменьшая, таким образом, потенциальные побочные эффекты. Таким образом, важно найти подобные формы, обладающие желательными свойствами для фармацевтического применения.
Краткое описание изобретения
Авторы настоящего изобретения неожиданно обнаружили и продемонстрировали, что новые твердые формы гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина (Р027) могут обеспечивать достижение одной или более приведенных выше целей. Новые полиморфные и сольватированные формы Р027, описанные в настоящем описании, являются объективно стабильными на протяжении времени и обладают хорошими характеристиками текучести и растворения. Особенно новая и высокостабильная кристаллическая форма соединения Р027 (форма фазы I) обеспечивает выгодное получение, обработку и хранение, а также терапевтические свойства. Помимо этого, некоторые из новых твердых форм Р027 могут быть пригодными в качестве промежуточных веществ для других полезных форм, таких как кристаллическая форма фазы I Р027.
Таким образом, настоящее изобретение относится к полиморфным формам и сольватам Р027, к их применению и к нескольким способам их получения.
Гидрохлоридную соль 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина (Р027) можно получить контактированием раствора основания с хлористоводородной кислотой. Соединение Р027 имеет молекулярную массу 373,88 масс. ед., pKa 6,73 и температуру плавления 194,2°С. Соединение очень хорошо растворимо в воде и свободно растворяется в метаноле, 1н. хлористоводородной кислоте и диметилсульфоксиде. Оно плохо растворяется в этаноле, незначительно растворяется в ацетоне и практически не растворяется в этилацетате и 1н. гидроксиде натрия. Продукт демонстрирует более хороший профиль растворения и всасывания in vivo по сравнению со своим основанием.
В одном варианте осуществления настоящее изобретение относится к твердой полиморфной или сольватированной форме гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина.
Предпочтительно, указанная твердая форма выбрана из группы, состоящей из
- формы фазы I Р027, которая может быть охарактеризована, поскольку она имеет порошковую рентгеновскую дифрактограмму, демонстрирующую характерные пики в области значений угла отражения [2θ] приблизительно 5,9, 8,1, 11,3, 11,7, 14,2, 15,1, 15,8, 16,3, 16,8, 17,8, 18,1, 18,6, 19,8, 20,9, 21,9, 22,8, 23,0, 23,2, 23,6, 23,9, 24,3, 25,0, 25,1, 28,0, 28,3, 28,6, 29,0, 29,2, 30,7 и 30,9, полученные с использованием медного источника излучения (CuKa1 1,54060 Å).
- формы фазы II Р027, которая может быть охарактеризована, поскольку она имеет порошковую рентгеновскую дифрактограмму, демонстрирующую характерные пики в области значений угла отражения [2θ], как указано ниже в таблице 1:
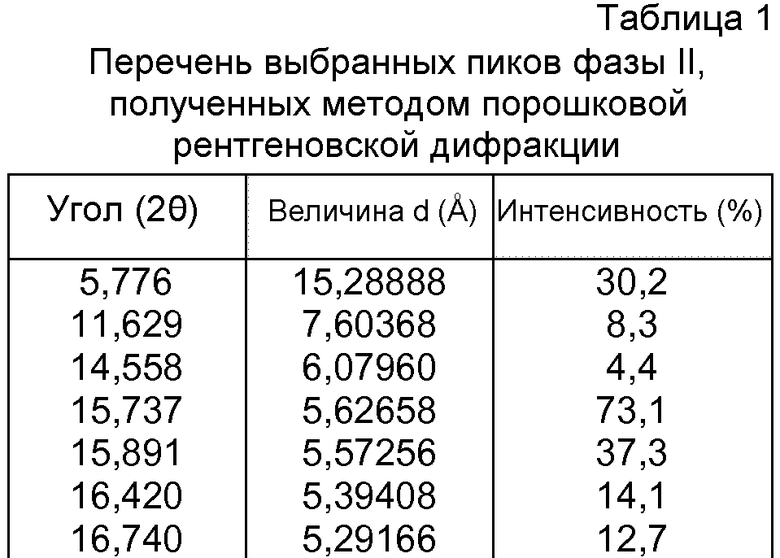
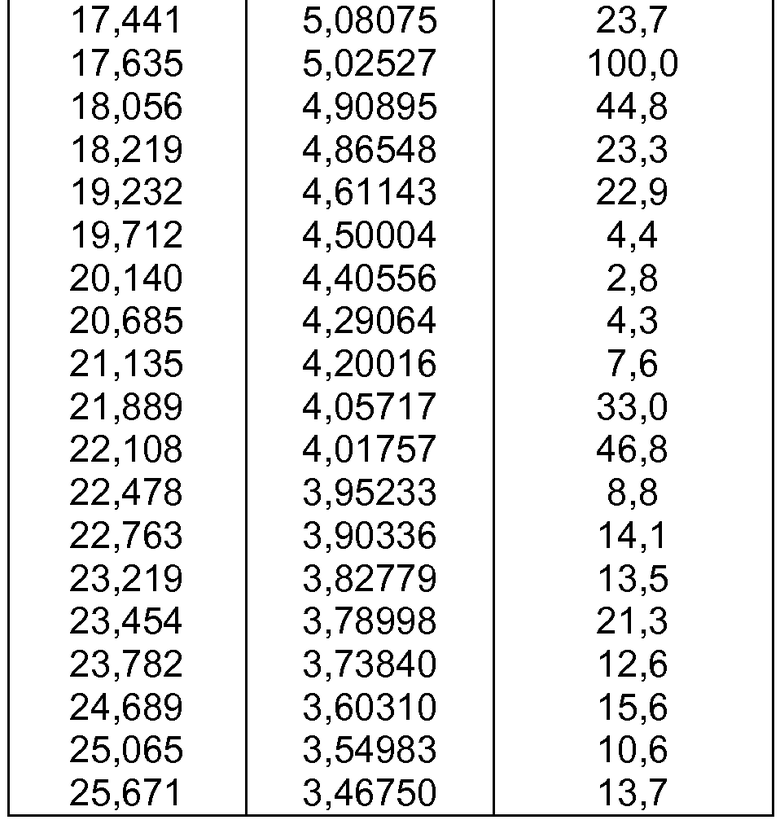
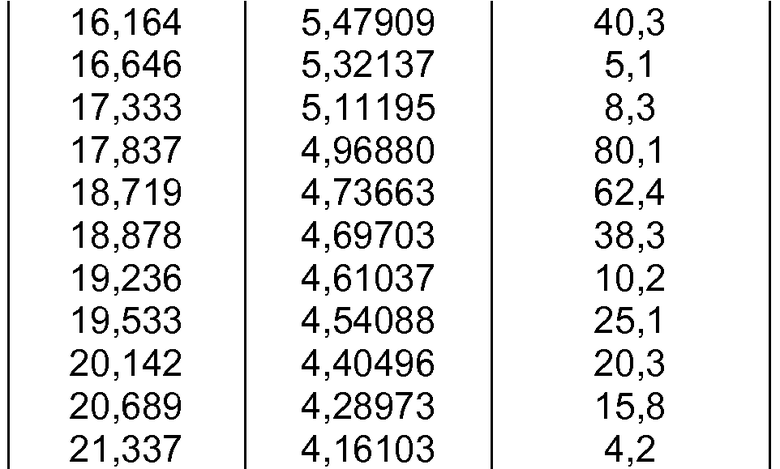
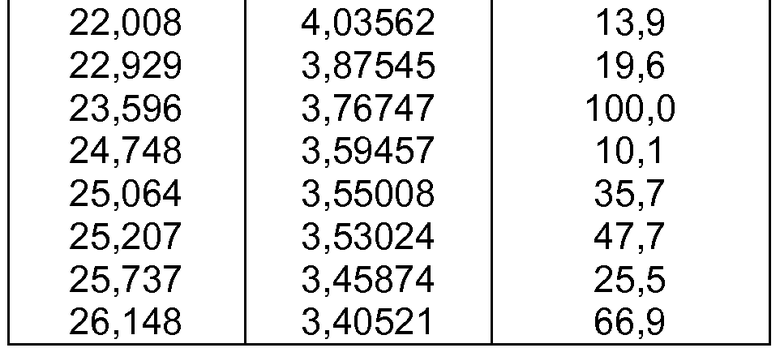
- формы фазы III Р027, которая может быть охарактеризована, поскольку она имеет порошковую рентгеновскую дифрактограмму, демонстрирующую характерные пики в области значений угла отражения [2θ], как указано ниже в таблице 2:
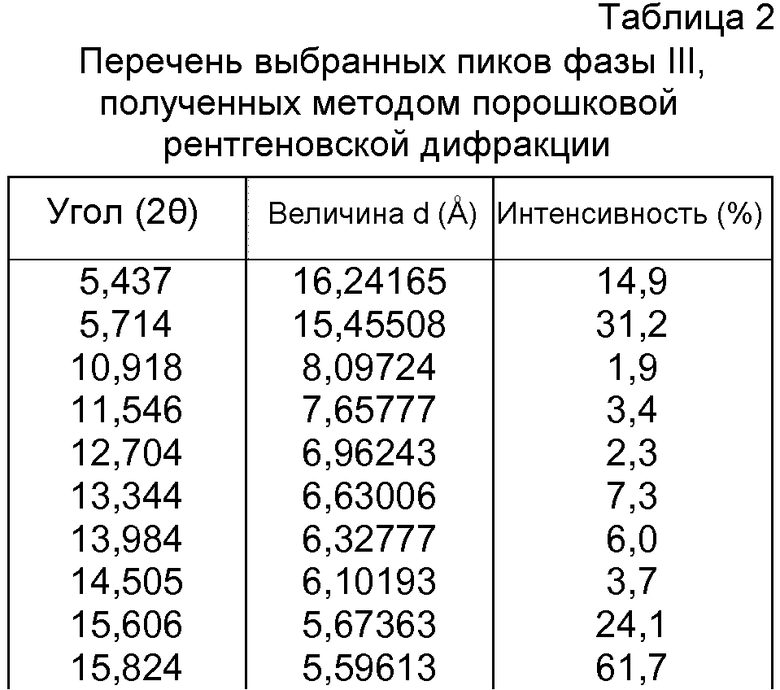
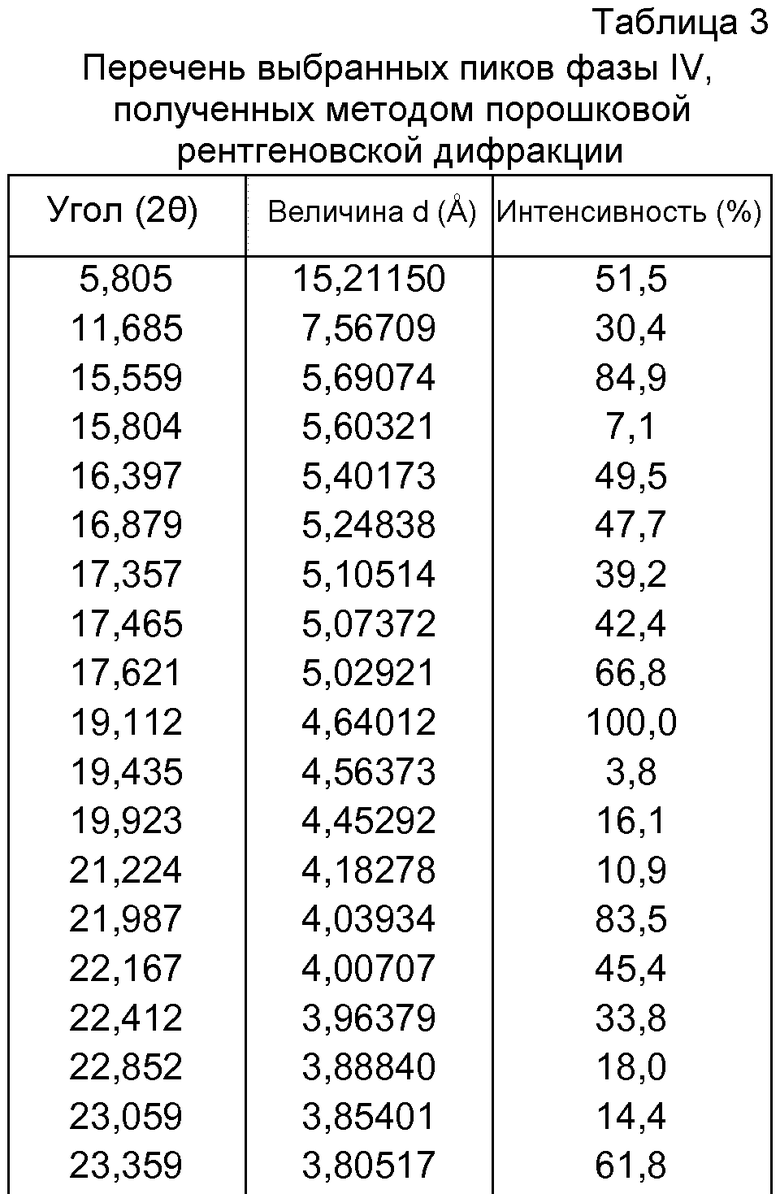
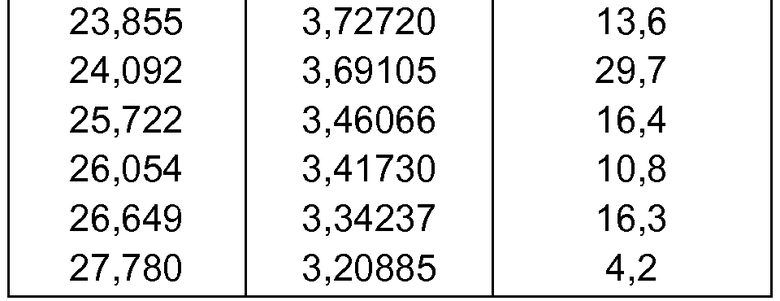
- формы фазы IV Р027, которая может быть охарактеризована, поскольку она имеет порошковую рентгеновскую дифрактограмму, демонстрирующую характерные пики в области значений угла отражения [2θ], как указано ниже в таблице 3:
- сольвата Р027 с диоксаном, который может быть охарактеризован, поскольку он имеет порошковую рентгеновскую дифрактограмму, демонстрирующую характерные пики в области значений угла отражения [2θ], как указано ниже в таблице 4:
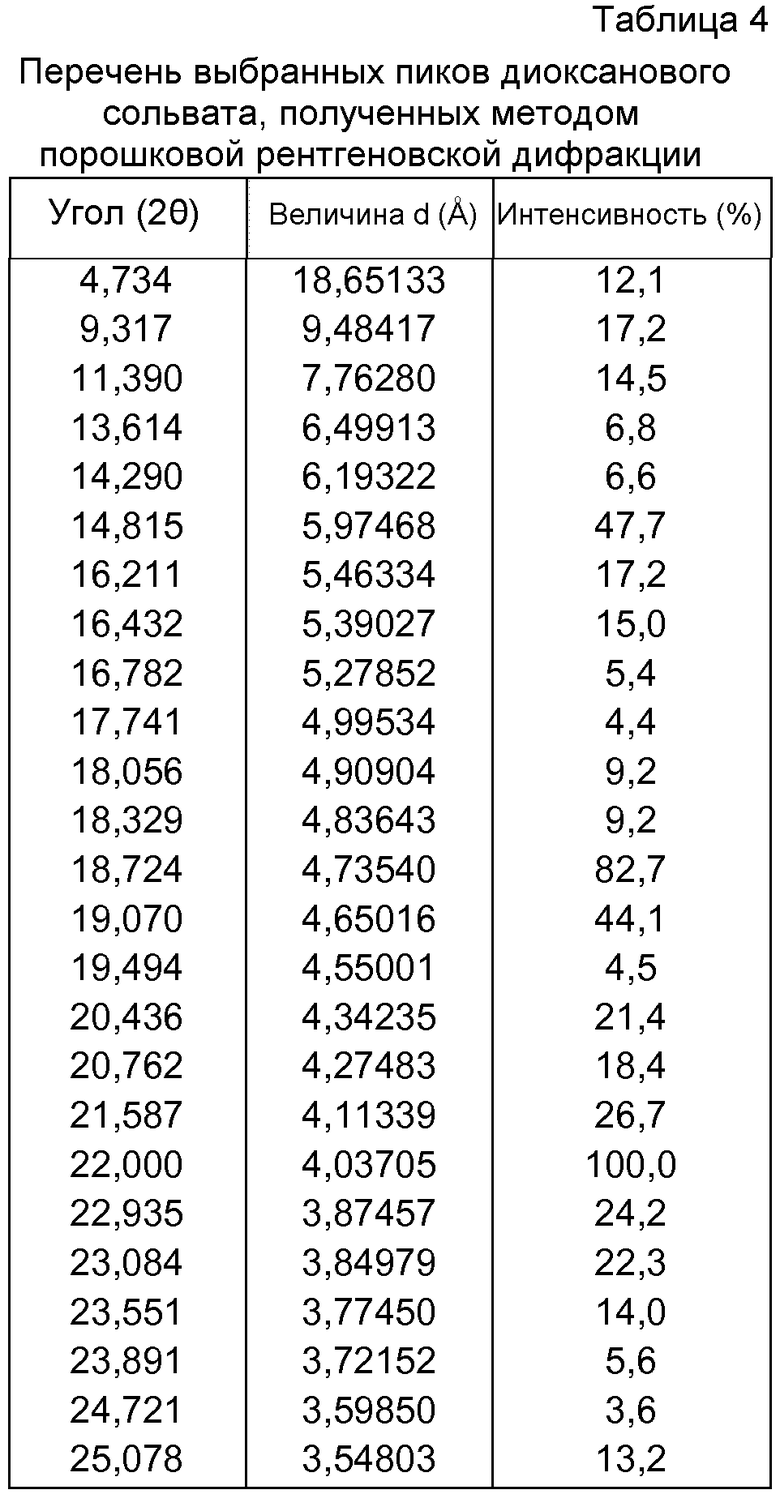
- сольвата Р027 с хлороформом, который может быть охарактеризован, поскольку он имеет порошковую рентгеновскую дифрактограмму, демонстрирующую характерные пики в области значений угла отражения [2θ], как указано ниже в таблице 5:
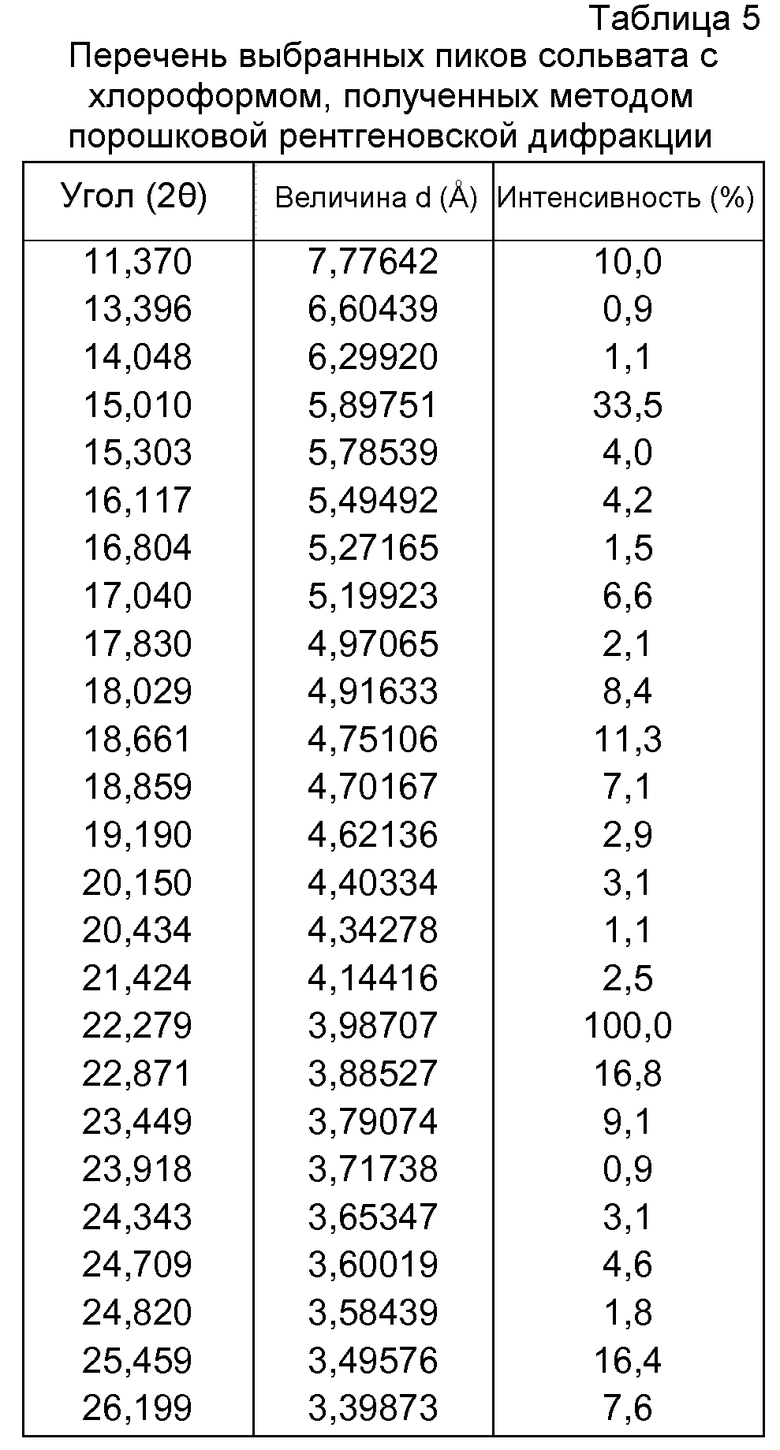
Согласно другому варианту осуществления, кристаллическая форма фазы I гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина Р027 по настоящему изобретению имеет моноклинную элементарную ячейку со следующими приблизительными измерениями:
а=29,4(3)Å
b=11,7(11)Å
с=11,0(10)Å
α=90°
β=91,3(2)°
γ=90°
Получение описанных выше полиморфных и сольватированных форм представляет дополнительные варианты осуществления настоящего изобретения.
Форма фазы I Р027 может быть получена кристаллизацией соединения Р027 в различных растворителях с использованием различных методик, таких как выпаривание растворителя при изменяющихся температурах, кристаллизация из горячих насыщенных растворов, кристаллизация добавлением антирастворителя, кристаллизация диффузией антирастворителя, кристаллизация из воды и смесей растворителей, и получение суспензий.
Форма фазы II Р027 может быть получена при полимер-индуцированной кристаллизации выпариванием растворителя.
Форма фазы III Р027 может быть получена при полимер-индуцированной кристаллизации выпариванием растворителя или кристаллизации добавлением антирастворителя.
Форма фазы IV Р027 может быть получена при полимер-индуцированной кристаллизации посредством кристаллизации добавлением антирастворителя.
Сольват Р027 с диоксаном может быть получен размалыванием с использованием капель растворителя в диоксане или кристаллизацией из горячего насыщенного раствора диоксана.
Сольват с хлороформом Р027 может быть получен при полимер-индуцированной кристаллизации выпариванием растворителя (хлороформа) или кристаллизацией из горячих насыщенных растворов хлороформа.
Другой вариант осуществления настоящего изобретения включает превращение кристаллических форм фазы II, фазы III и фазы IV, выше, в более стабильную полиморфную форму, такую как форма фазы I Р027.
Другой вариант осуществления настоящего изобретения включает превращение сольвата Р027, предпочтительно, сольвата с хлороформом, в более стабильную полиморфную форму, такую как форма фазы I.
Еще один вариант осуществления настоящего изобретения включает фармацевтические композиции, содержащие по меньшей мере одну из форм гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина, приведенных выше, особенно фазы I Р027, фазы II Р027, фазы III Р027, фазы IV Р027, сольват Р027 с хлороформом и сольват Р027 с диоксаном.
Указанные аспекты и предпочтительные варианты их осуществления также дополнительно определены в формуле изобретения.
Краткое описание фигур
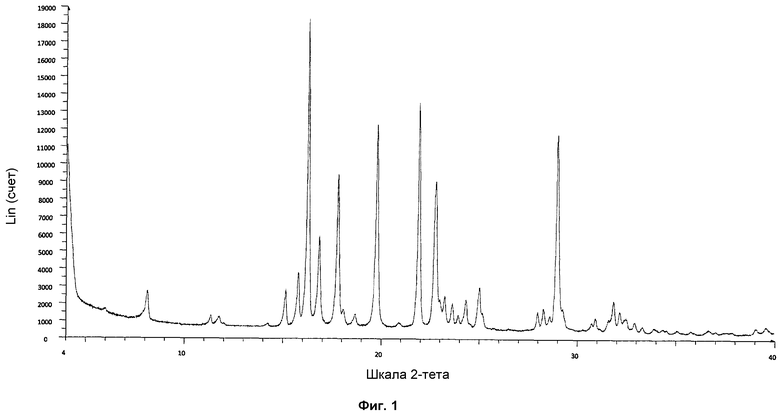
На фиг.1: показана стандартная дифрактограмма ПРД (порошковая рентгеновская дифракция).
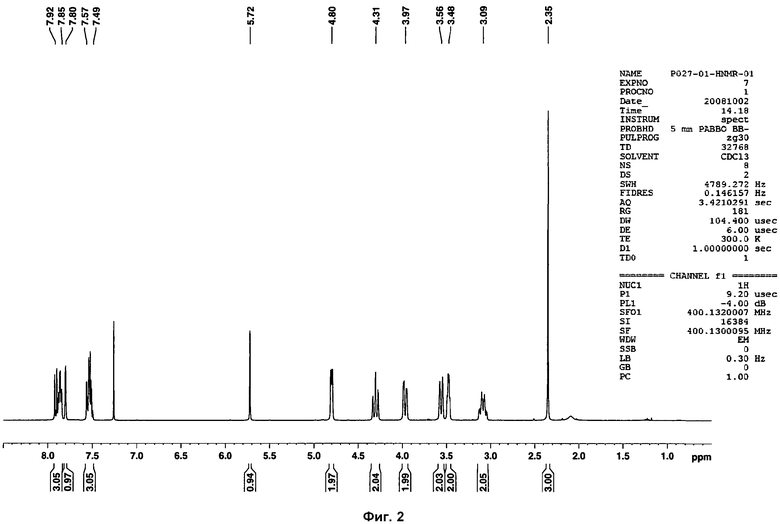
На фиг.2 показан 1Н ЯМР спектр раствора соединения Р027.
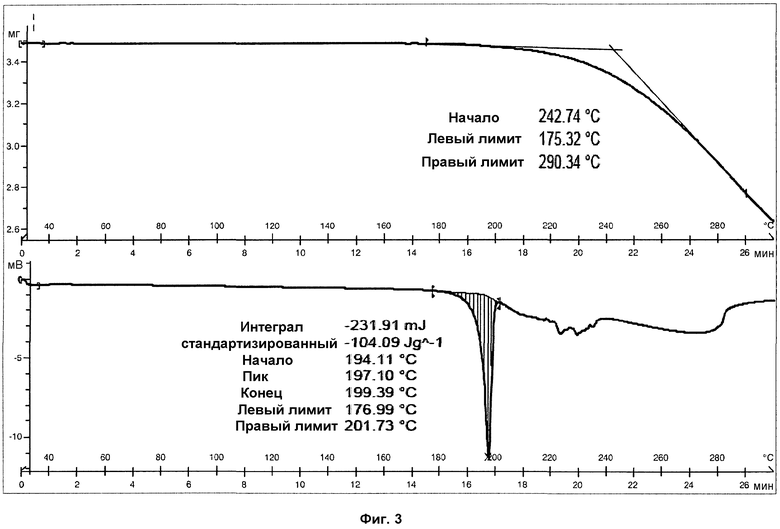
На фиг.3 показаны ДСК и ТГА анализы фазы I.
На фиг.4 показан ИК-ПФ анализ фазы I.
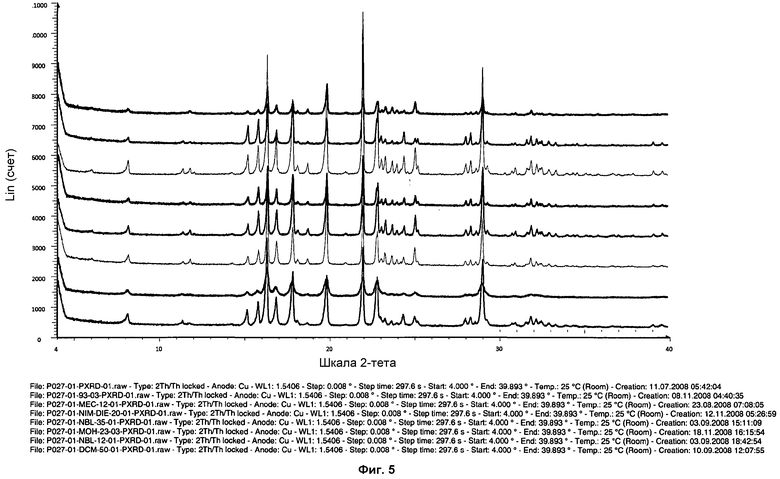
На фиг.5 показаны произвольно выбранные дифрактограммы ПРД различных твердых веществ, соответствующих фазе I, в которых можно наблюдать текстурные эффекты.
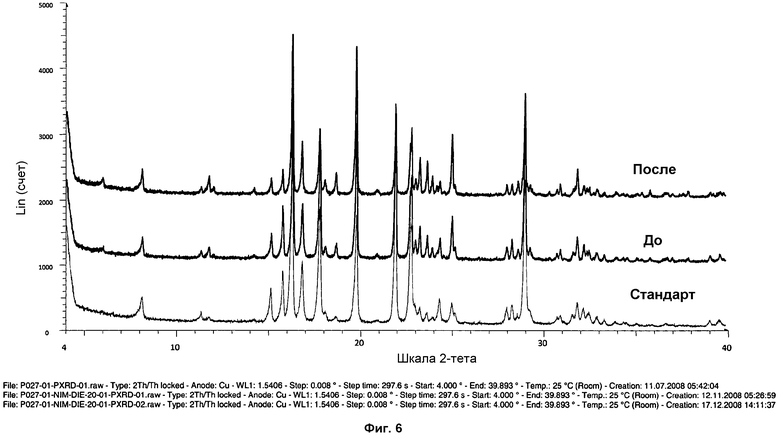
На фиг.6 показана дифрактограмма ПРД образцов фазы I до и после размалывания. Стандартная дифрактограмма ПРД фазы I показана для сравнения.
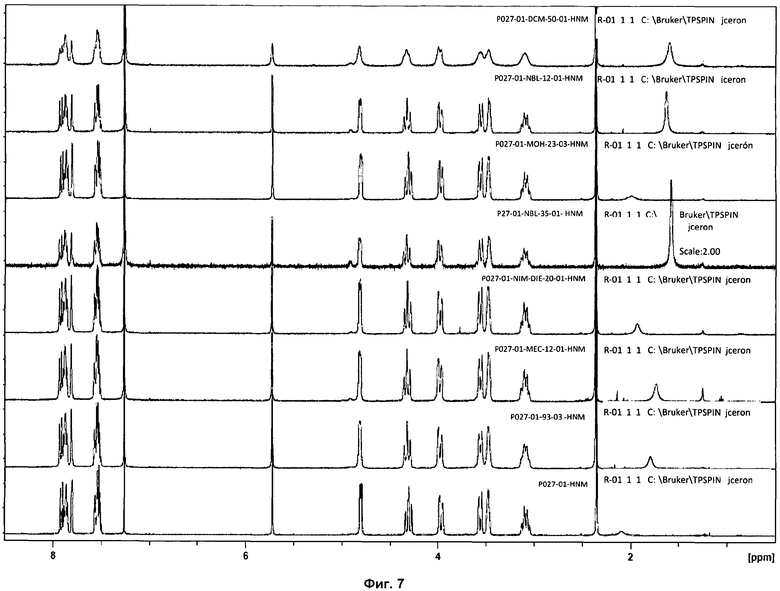
На фиг.7 показаны 1Н ЯМР спектры образцов, изображенных на фиг.6.
На фиг.8: показан ДСК анализ фазы I при скорости нагревания 5°С/мин.
На фиг.9 показан ДСК анализ фазы I при скорости нагревания 20°С/мин.
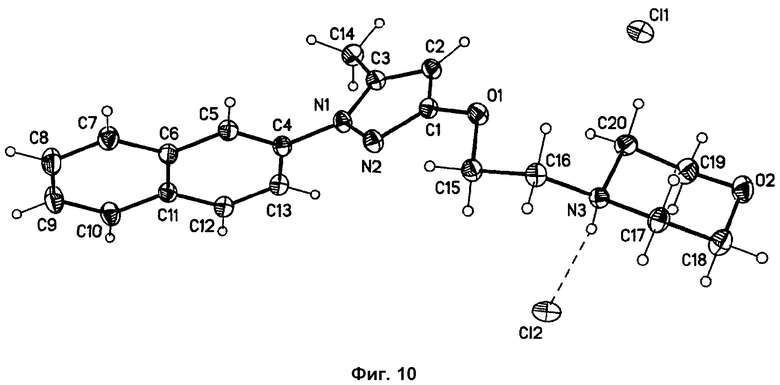
На фиг.10 представлено изображение Ortep-Plot (50%), показывающее органический катион и два независимых полуаниона хлора, содержащиеся в элементарной ячейке.
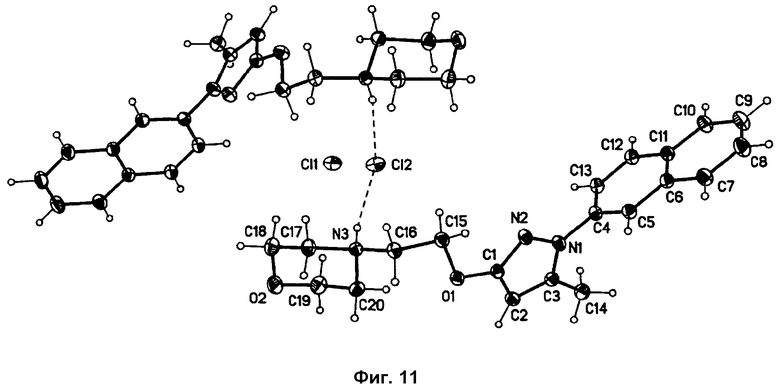
На фиг.11 представлено изображение Ortep-Plot (50%), показывающее структуру фазы I. Водородные связи отмечены прерывистой линией.
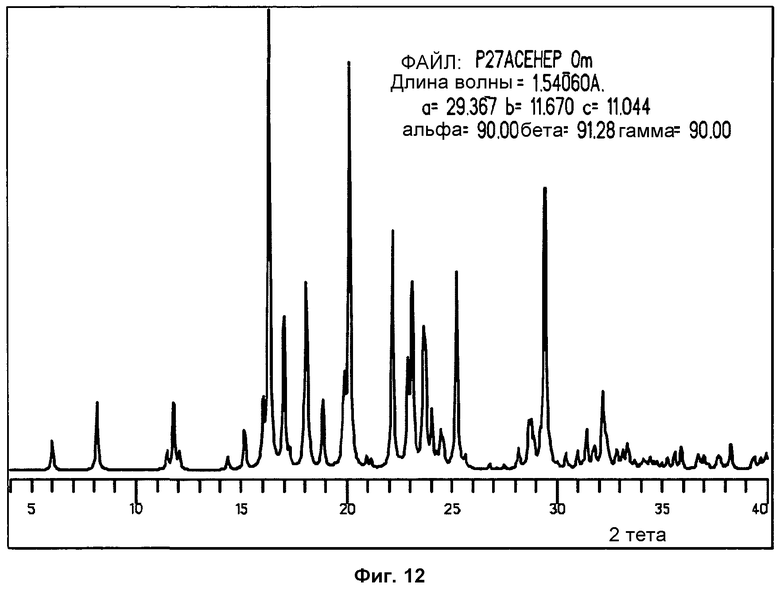
На фиг.12 показана стимулированная порошковая дифрактограмма, полученная из данных одиночного кристалла фазы I.
На фиг.13 показано сравнение стимулированной порошковой дифрактограммы, полученной из данных одиночного кристалла, и экспериментально измеренной порошковой дифрактограммы фазы I.
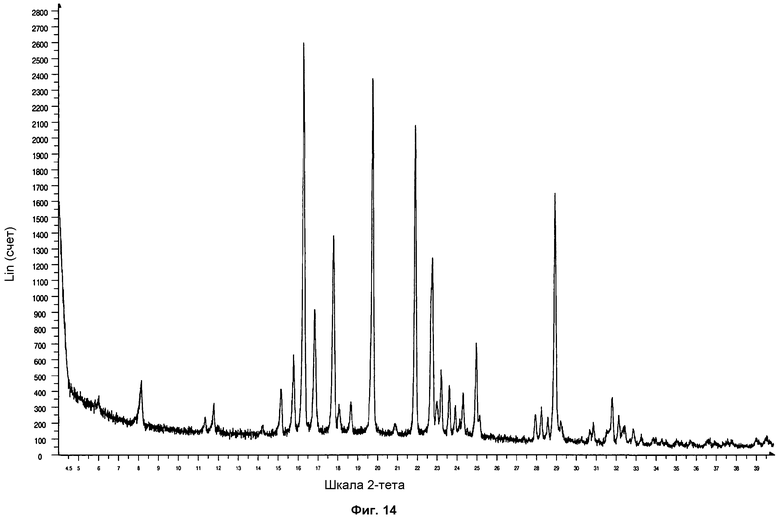
На фиг.14 показана порошковая рентгеновская дифрактограмма формы фазы I, полученной выпариванием н-бутанола при -21°С.
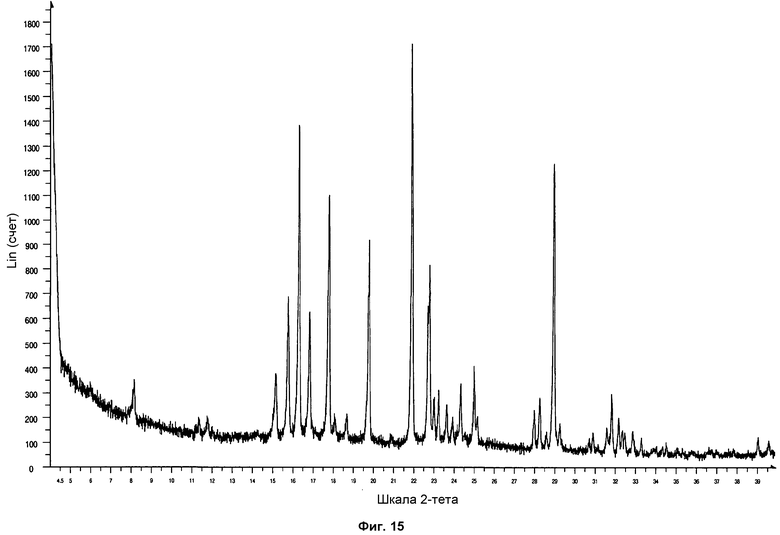
На фиг.15 показана дифрактограмма ПРД формы фазы I, полученной медленной кристаллизацией горячего насыщенного раствора соединения Р027 в метилэтилкетоне.
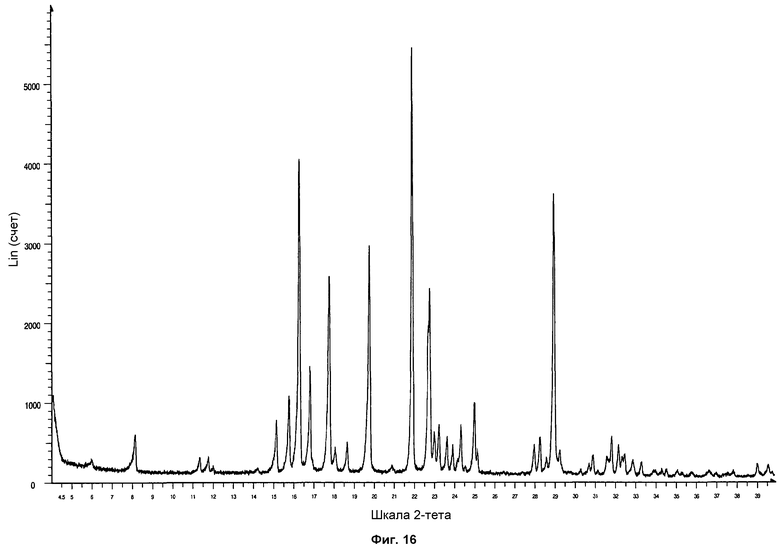
На фиг.16 показана дифрактограмма ПРД формы фазы I, полученной кристаллизацией посредством добавления раствора Р027 в метаноле в раствор н-гептана.
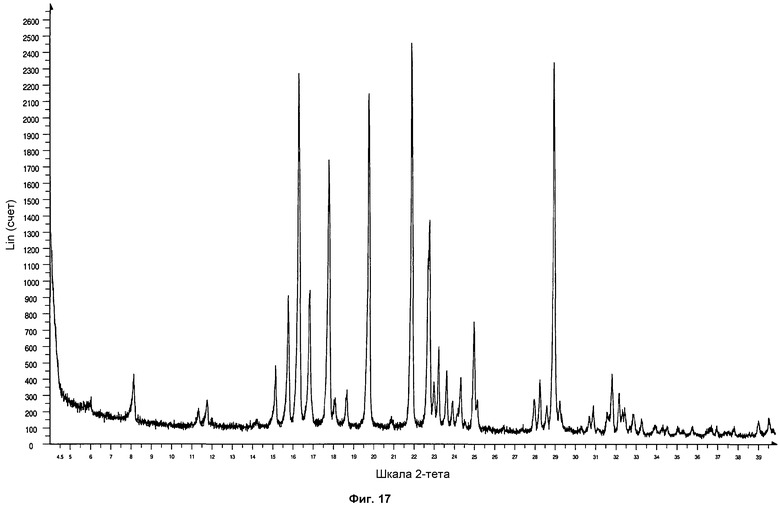
На фиг.17 показана дифрактограмма ПРД формы фазы I, полученной кристаллизацией посредством диффузии жидкость-жидкость раствора Р027 в нитрометане и растворе изопропилового эфира.
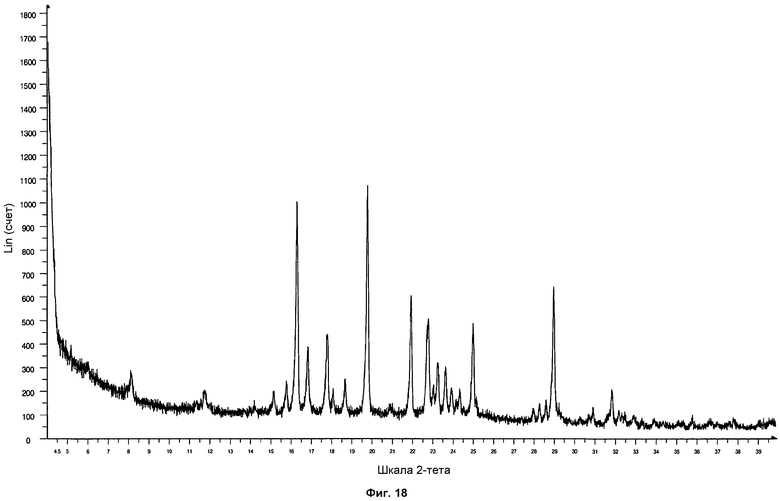
На фиг.18 показана дифрактограмма ПРД, полученная после размалывания образца формы фазы I Р027 с дихлорметаном. Дифрактограмма согласуется со стандартной дифрактограммой ПРД фазы I, демонстрируя фазовую стабильность.
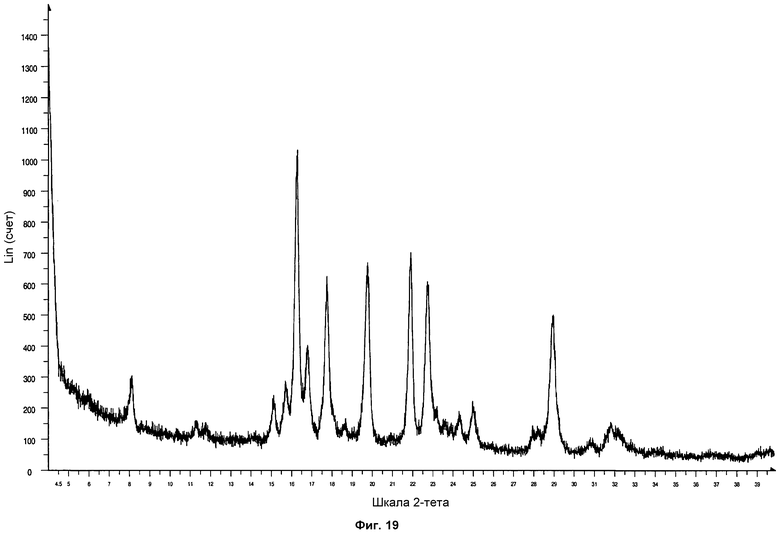
На фиг.19 показана дифрактограмма ПРД образца формы фазы I Р027 после приложения давления 30 тонн к образцу в течение 90 минут. Дифрактограмма согласуется со стандартной дифрактограммой ПРД фазы I, демонстрируя фазовую стабильность.
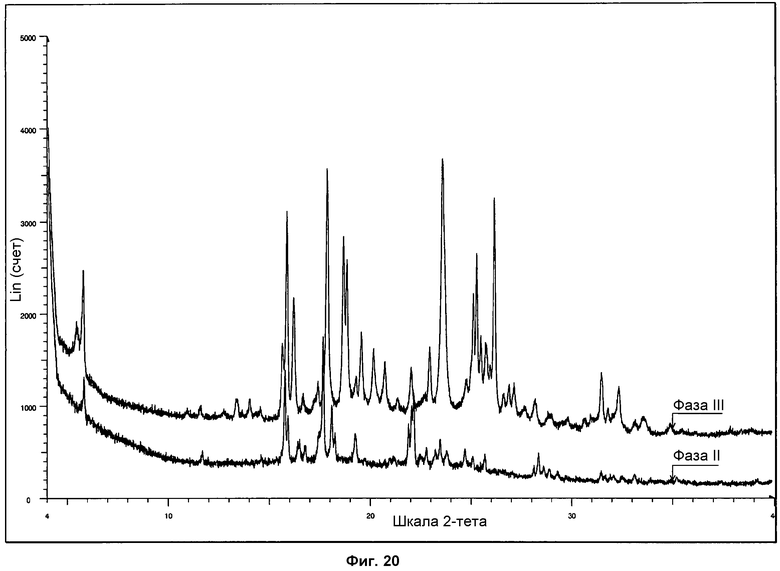
На фиг.20 представлено сравнение дифрактограмм ПРД, полученных для фазы II и фазы III.
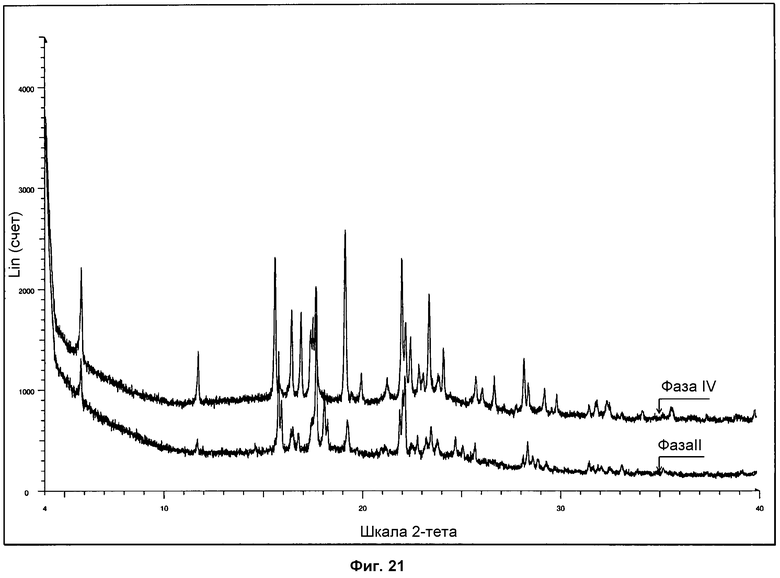
На фиг.21 представлено сравнение дифрактограмм ПРД, полученных для фазы II и фазы IV.
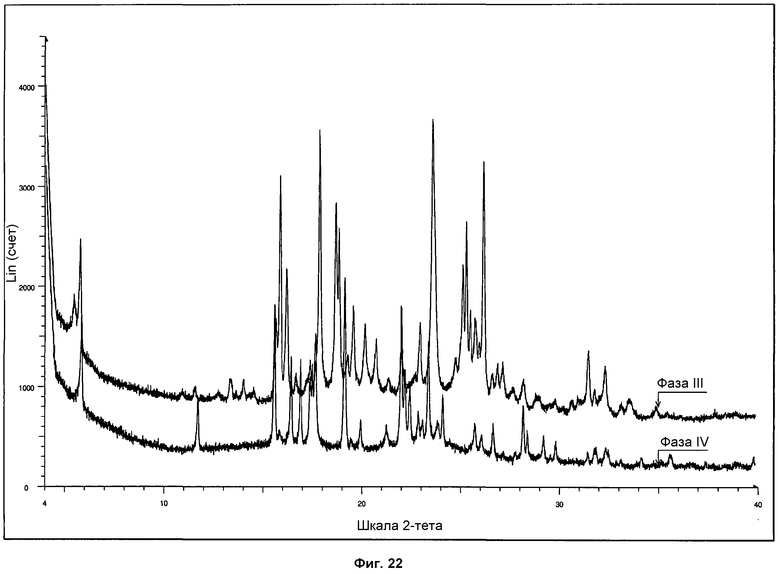
На фиг.22 представлено сравнение дифрактограмм ПРД, полученных для фазы III и фазы IV.
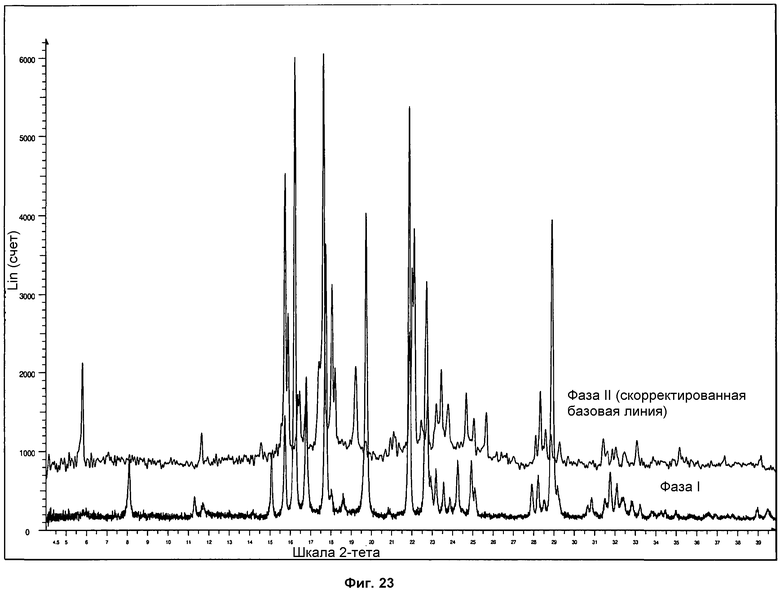
На фиг.23 представлено сравнение дифрактограмм ПРД, полученных для фазы I и фазы II.
На фиг.24 показана стандартная дифрактограмма ПРД фазы II.
На фиг.25 показан 1Н ЯМР спектр фазы II.
На фиг.26 показаны ДСК и ТГА анализы фазы II.
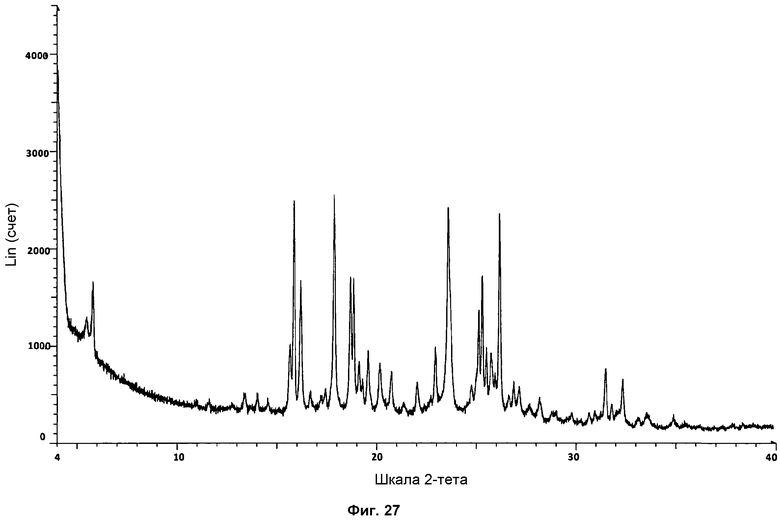
На фиг.27 показана стандартная дифрактограмма ПРД фазы III.
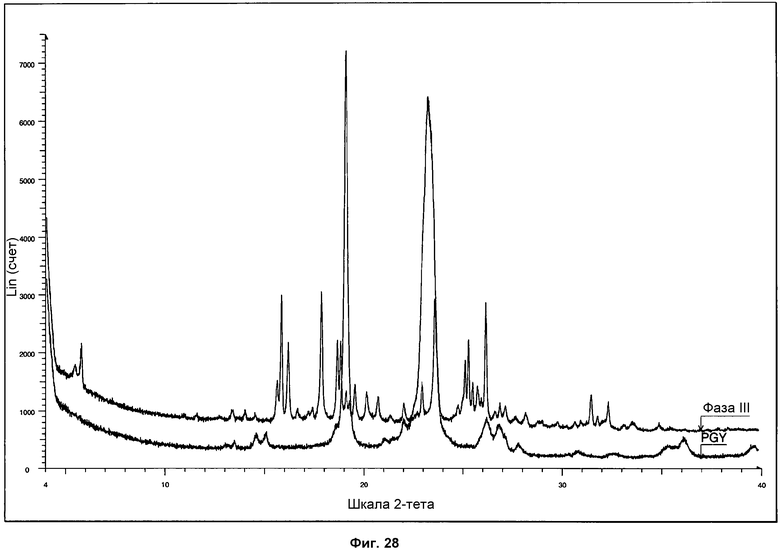
На фиг.28 показано сравнение дифрактограмм ПРД, полученных для полиэтиленгликоля и фазы III.
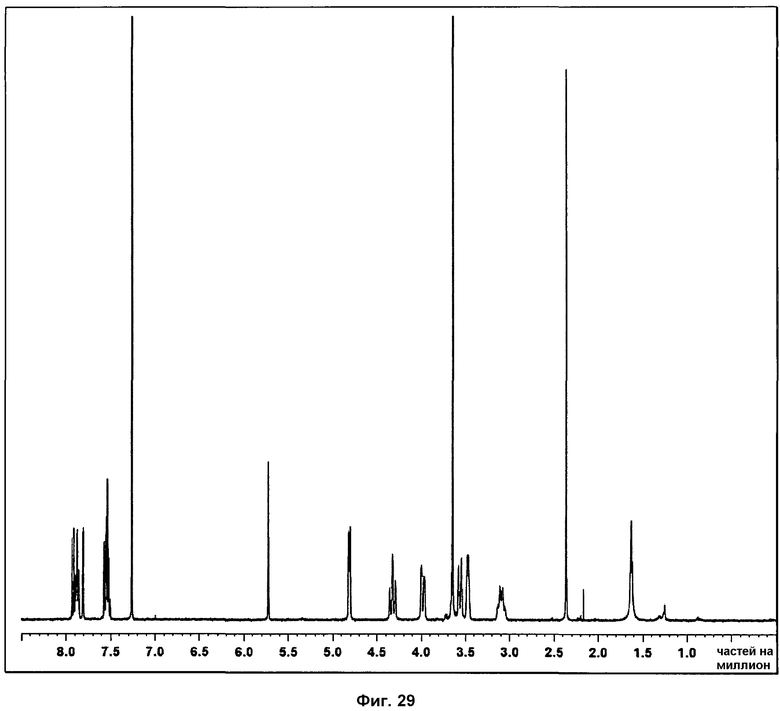
На фиг.29 показан 1Н ЯМР спектр фазы III.
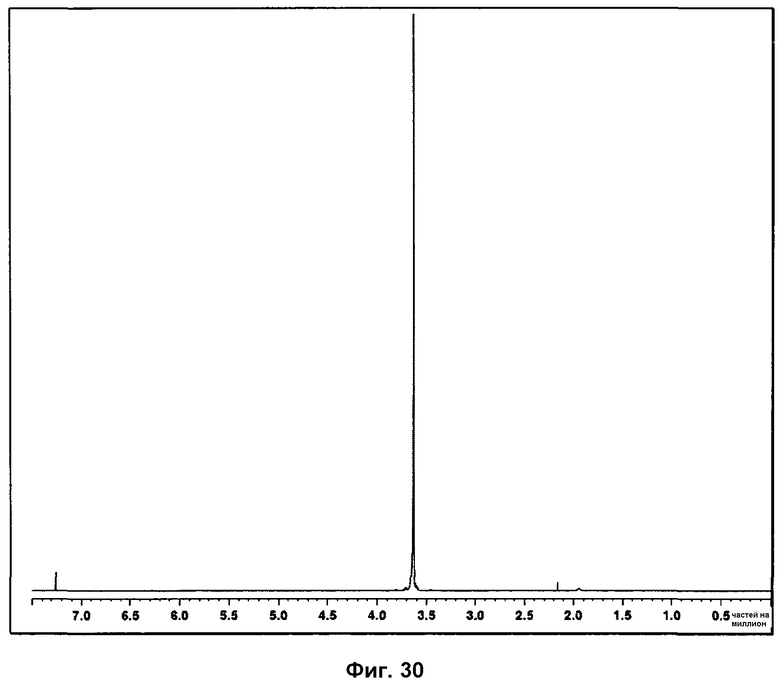
На фиг.30 показан 1Н ЯМР спектр полиэтиленгликоля.
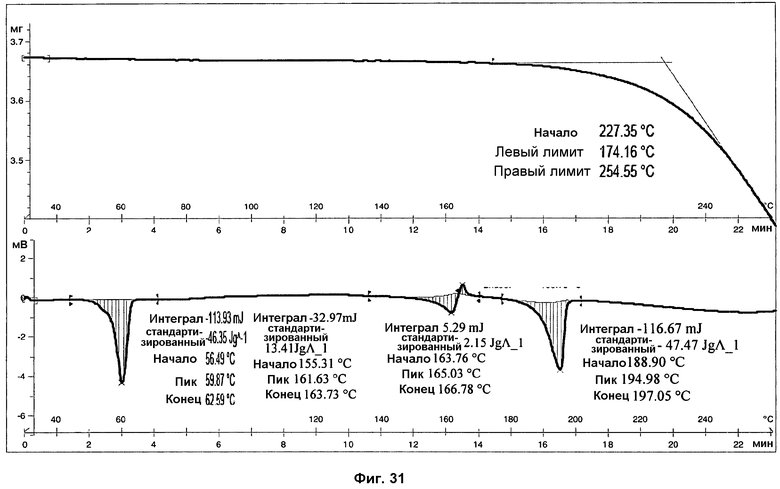
На фиг.31 показаны ДСК и ТГА анализы фазы III.
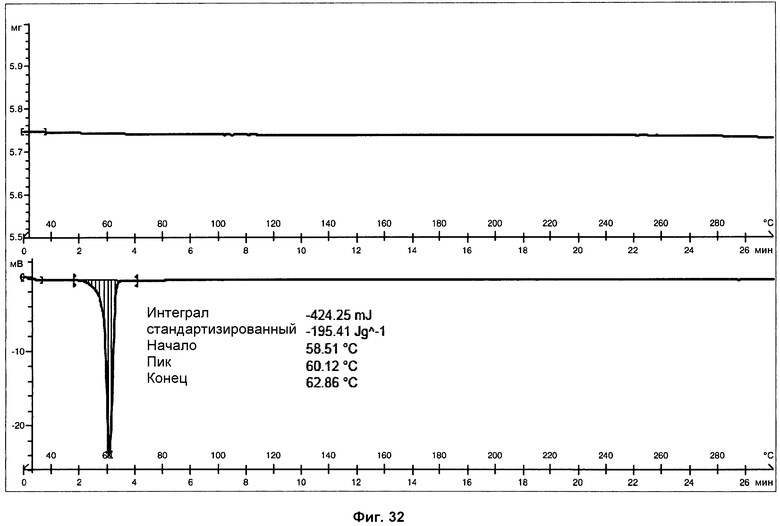
На фиг.32 показаны ДСК и ТГА анализы полиэтиленгликоля.
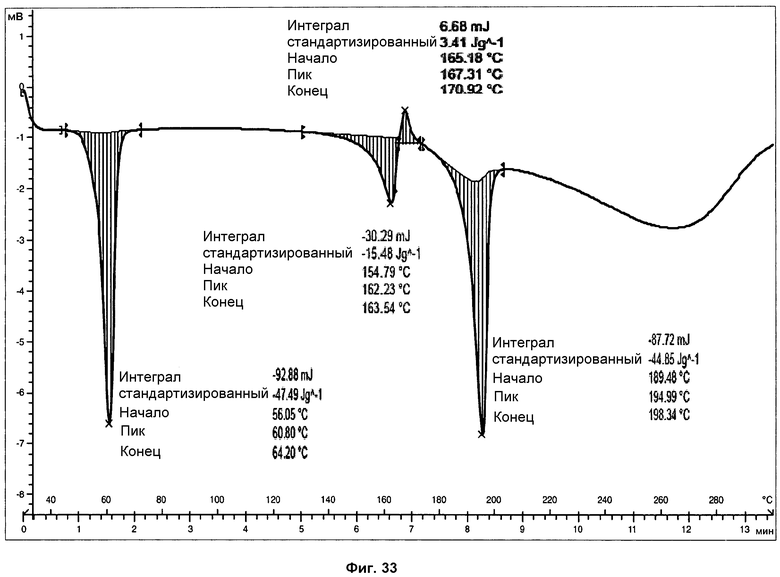
На фиг.33 показаны ДСК анализы фазы III при скорости нагревания 20°С/мин.
На фиг.34 показаны ДСК анализы фазы III при скорости нагревания 30°С/мин.
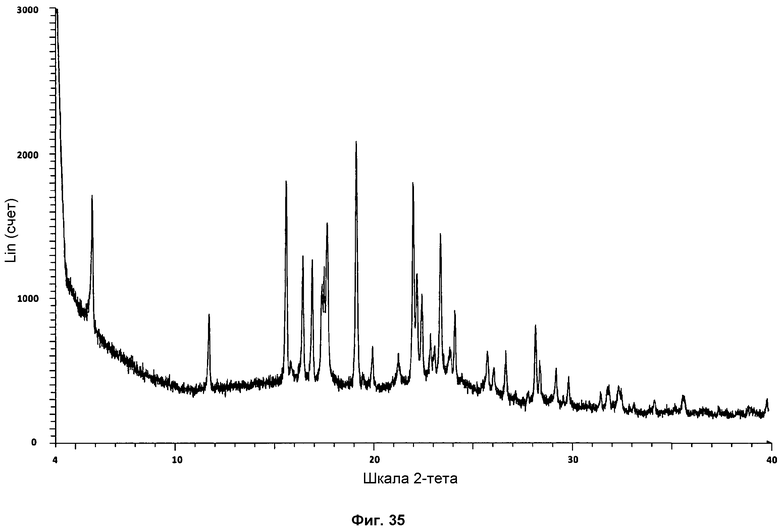
На фиг.35 показана стандартная дифрактограмма ПРД фазы IV.
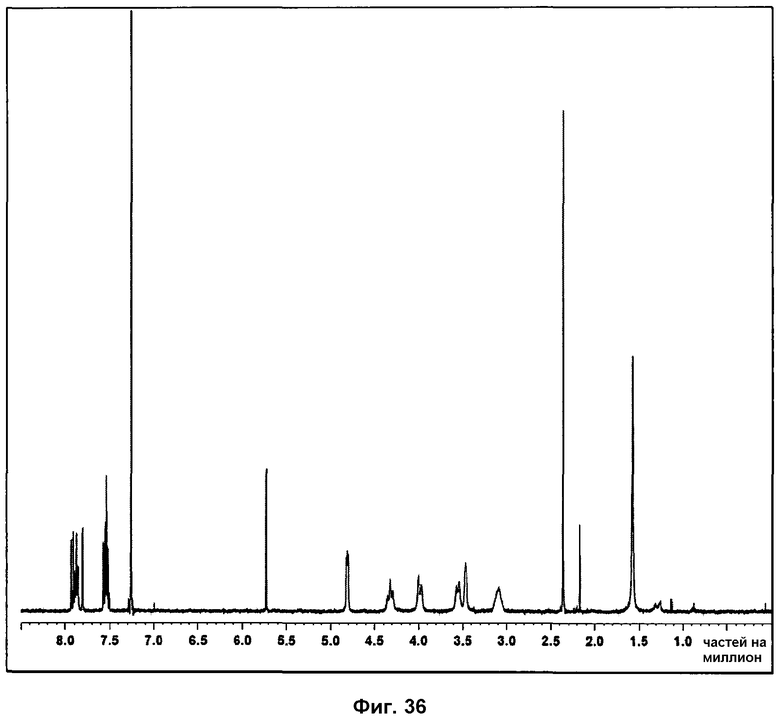
На фиг.36 показан 1Н ЯМР спектр фазы IV.
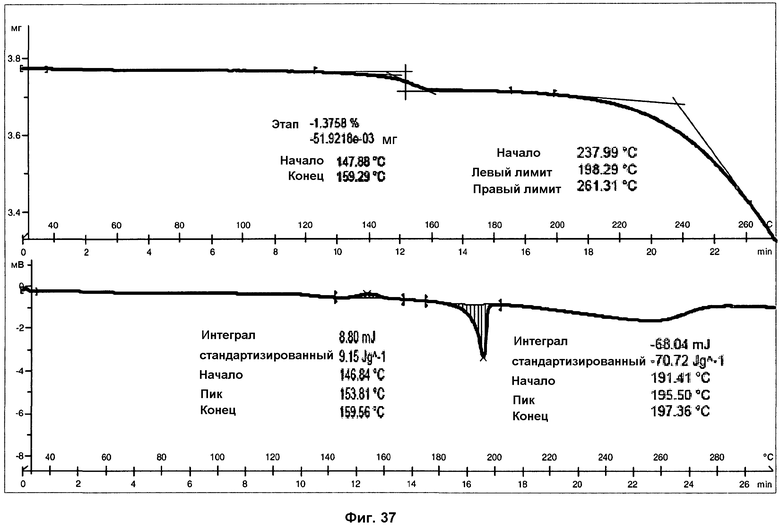
На фиг.37 показаны ДСК и ТГА анализы фазы IV.
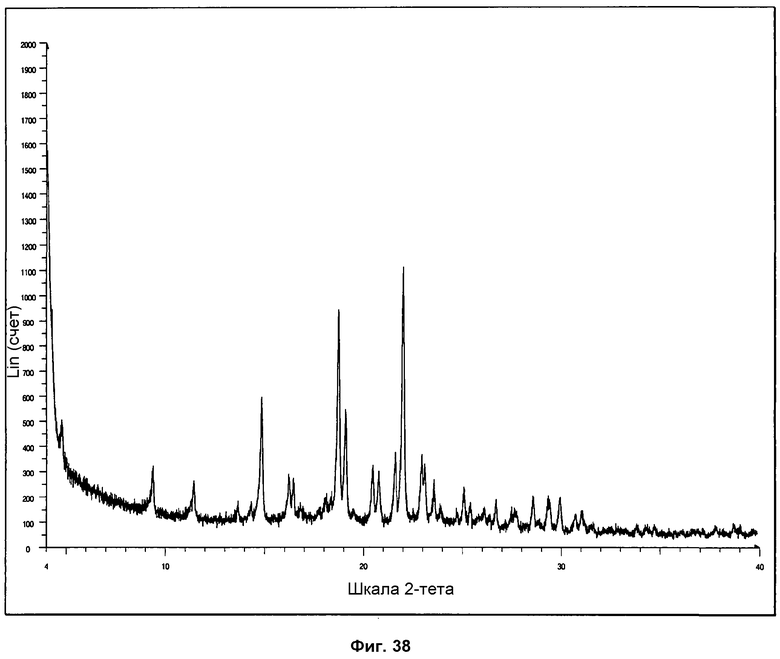
На фиг.38 показана стандартная дифрактограмма ПРД сольвата с диоксаном.
На фиг.39 показан 1Н ЯМР спектр сольвата с диоксаном.
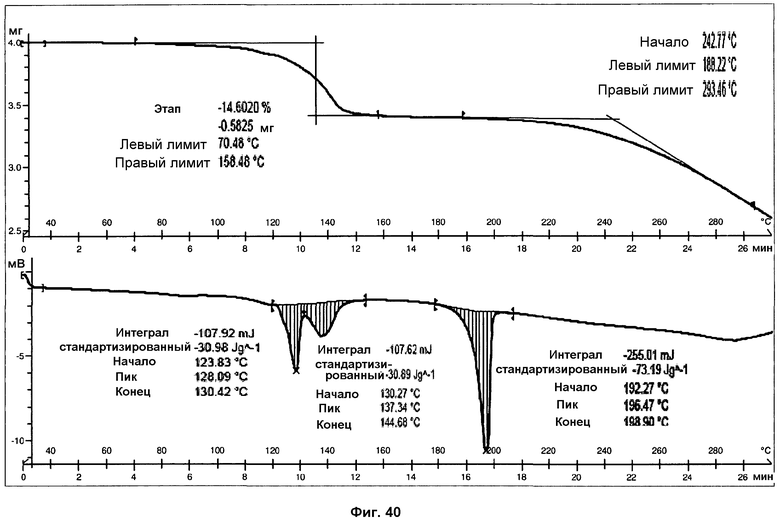
На фиг.40 показаны ДСК и ТГА анализы сольвата с диоксаном.
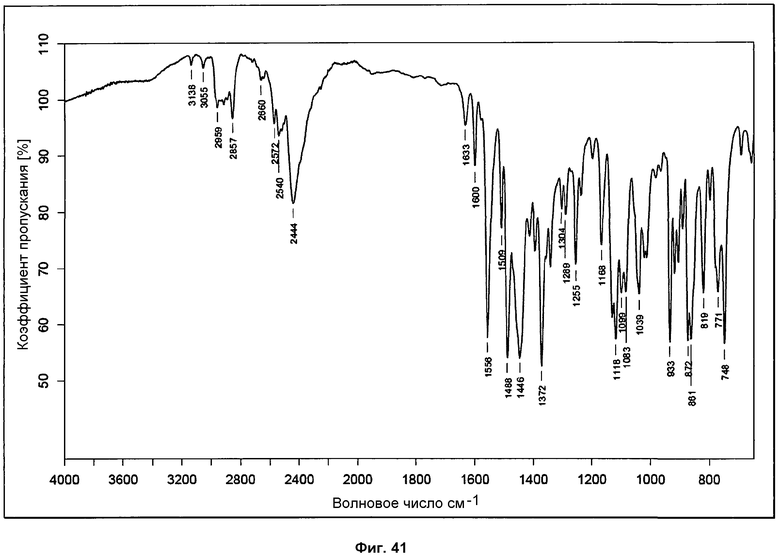
На фиг.41 показан ИК-ПФ анализ сольвата с диоксаном.
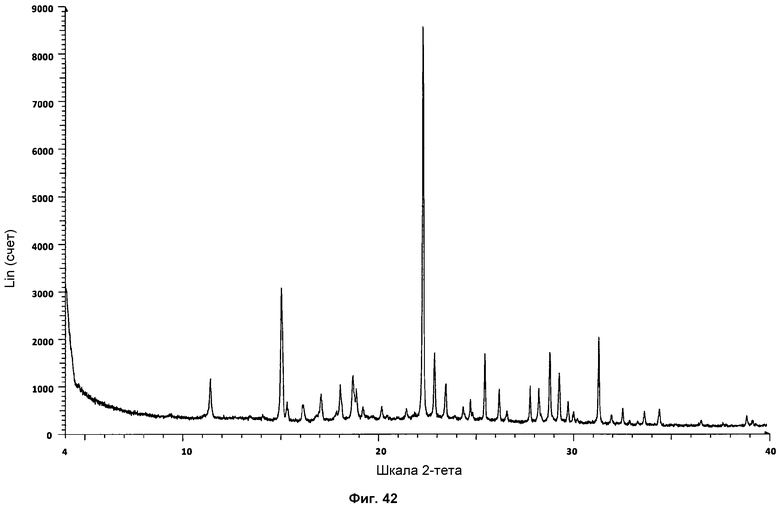
На фиг.42 показана стандартная дифрактограмма ПРД сольвата с хлороформом.
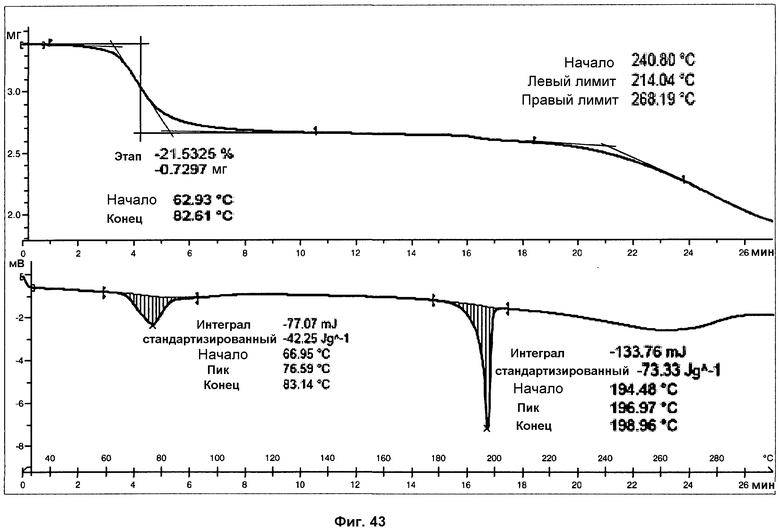
На фиг.43 показаны ДСК и ТГА анализы сольвата с хлороформом.
Подробное описание изобретения
Авторы обнаружили новые твердые формы гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина (Р027), которые обеспечивают выгодное получение, обработку, хранение и терапевтические свойства. Указанные соединения имеют преимущества благодаря тому факту, что они являются твердыми веществами, что упрощает выделению, очистку и обработку. Помимо этого, форма фазы I указанного соединения является высокостабильной и может быть помещен в композиции и введена, обеспечивая стабильные композиции и хорошие фармакологические свойства. Помимо этого, новые формы Р027 можно использовать для получения других форм, таких как кристаллическая форма фазы I Р027.
Используемый в настоящем описании термин «приблизительно» означает незначительное изменение описываемой величины, предпочтительно, в пределах 10 процентов описываемой величины. Тем не менее, термин «приблизительно» может означать более высокую толерантность изменения, зависящую, например, от используемой экспериментальной методики. Указанные изменения описываемой величины понятны специалистам в данной области и находятся в пределах контекста настоящего изобретения. Кроме того, для обеспечения более лаконичного описания некоторые из количественных выражений, приведенных в настоящем описании, не квалифицируются термином «приблизительно». Понятно, что, используется ли термин «приблизительно» недвусмысленно или нет, любое количество, приведенное в настоящем описании, относится к истинной данной величине, а также относится к приближению к указанной данной величине, которая будет корректно выведена специалистом в данной области, включая эквиваленты и приближения, зависящие от условий эксперимента и/или измерения для указанной данной величины.
Используемый в настоящем описании термин «комнатная температура» или его сокращение «кт» означает температуру от 20 до 25°С.
Характеристики новых форм Р027, описанных в настоящем описании, изучали с использованием порошковой рентгеновской дифракции (ПРД), дифференциальной сканирующей калориметрии (ДСК), термогравиметрического анализа и инфракрасной спектроскопии с преобразованием Фурье (ИК-ПФ). Настоящее изобретение в одном аспекте относится к самим новым твердым формам Р027, не зависимо от методики, используемой для изучения их характеристик. Таким образом, методики и результаты, представленные в настоящем описании, не предназначены для ограничения настоящего изобретения, а служат для его описания. Специалист в данной области сможет, с использованием руководства и результатов, описанных в настоящем описании, сравнивать и характеризовать, используя доступные ему методики, различные полиморфы и сольваты соединения гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина (Р027).
Получение твердых образцов соединения Р027 осуществляли в ряду из 40 растворителей (таблица 6). Растворители выбирали в соответствии с предыдущим опытом, чтобы охватить обширный ряд свойств.
Для того чтобы планировать скрининг кристаллизации, определяли растворимость Р027 при комнатной температуре в ряде растворителей таблицы 6 с использованием следующей методики (таблица 7): 10 мг образца суспендировали при комнатной температуре в 0,2 мл соответствующего растворителя и осуществляли последовательные добавления (сначала 0,2 мл и в конце 0,5 мл) растворителя до полного растворения твердого вещества или максимум до 8 мл. После каждого добавления растворителя суспензию интенсивно перемешивали в течение 10-15 минут и изучали визуально на предмет полного растворения твердого вещества. Пределы растворимости представлены в таблице 7.
2 Твердое вещество растворяли при 80°С. Раствор оставляли при комнатной температуре, и твердого вещества не наблюдали.
Растворители, в которых Р027 не растворялся, использовали в качестве антирастворителей (например, растворители с растворимостью <1,2 мг/мл). Например, н-гептан (НЕР), метил-трет-бутиловый эфир (МТЕ) и диизопропиловый эфир (DIE) использовали в качестве антирастворителей. Остальные растворители использовали в качестве растворяющих растворителей при различных изученных стратегиях кристаллизации.
Для того чтобы охватить наиболее широкие возможные пределы кристаллизации, применяли несколько методик кристаллизации с использованием растворителей, описанных в таблице 6. Использовали методики, ориентированные на получение термодинамически стабильной фазы, а также методики, направленные на получение кинетически благоприятных фаз. Помимо этого, изучали опосредованные растворителем и не использующие растворителя методики кристаллизации. Перечень методик кристаллизации, которые использовались в настоящем изобретении, следующий:
- выпаривание растворителя с использованием двух скоростей при комнатной температуре,
- выпаривание растворителя при различных температурах: -21,4 и 60°С,
- кристаллизация из горячих насыщенных растворов с использованием двух скоростей охлаждения,
- кристаллизация, нацеленная на получение гидратов,
- кристаллизация добавлением антирастворителя,
- кристаллизация диффузией антирастворителя,
- эксперименты с размалыванием,
- эксперименты с давлением,
- эксперименты с взвесями (суспензиями).
Помимо стандартных методик кристаллизации, использовали новую методологию с участием полимеров для инициации кристаллизации новых твердых веществ. Как описано в литературе, использование полимеров может благоприятствовать образованию новых кристаллических фаз (M. Lang et al., J. Am. Chem. Soc., 2002, 124, 14834; C. Price et al., J. Am. Chem. Soc., 2005, 127, 5512). Кроме того, присутствие полимеров может поддерживать образование одиночных кристаллов большего размера и стабилизировать образование сольватов. Серии полимеров (см. таблицу 8) добавляли в каталитических количествах к раствору Р027 и кристаллизовали с использованием следующих методик:
- выпаривание растворителя при комнатной температуре,
- кристаллизация из горячих насыщенных растворов,
- кристаллизация добавлением антирастворителя,
- эксперименты с размалыванием.
Используемый в настоящем описании термин «каталитическое количество» означает субстехиометрическое количество полимера по отношению к соединению Р027; предпочтительно, менее 25% масс. от количества (масс.) соединения Р027. В конкретном варианте осуществления настоящего изобретения «каталитические количества» составляют менее 20% масс. от соединения Р027. В более конкретном варианте осуществления настоящего изобретения «каталитические количества» составляют менее 10% масс. от соединения Р027.
Все твердые вещества, полученные с использованием различных методик кристаллизации, характеризовали с помощью ПРД и классифицировали, согласно полученным различным дифрактограмм ПРД. Дополнительные проводившиеся анализы также принимали во внимание для классификации твердых веществ (см. экспериментальный раздел).
Следующие формы Р027 были идентифицированы и охарактеризованы среди полученных твердых веществ: форма фазы I Р027, форма фазы II Р027, форма фазы III Р027, форма фазы IV Р027, сольват Р027 с диоксаном и сольват Р027 с хлороформом.
В одном варианте осуществления настоящего изобретения форму фазы I Р027 получают растворением соединения Р027 в подходящем растворителе, и затем выпариванием растворителя с получением кристаллической формы фазы I. Согласно одному варианту указанного способа, соединение Р027 растворяют при температуре приблизительно от комнатной температуры до 120°С. В другом варианте указанного способа растворитель выпаривают при температуре приблизительно от -21°С до 60°С. В еще одном варианте указанного способа раствор Р027 оставляют медленно охлаждаться. В еще одном варианте указанного способа раствор Р027 охлаждают быстро.
В другом варианте осуществления настоящего изобретения форму фазы I Р027 получают смешиванием раствора Р027 и антирастворителя. В варианте указанного способа раствор Р027 добавляют к антирастворителю. В другом варианте указанного способа антирастворитель добавляют к раствору Р027. В еще одном варианте указанного способа раствор Р027 и антирастворитель смешивают при температуре приблизительно от комнатной температуры до 90°С.
В еще одном варианте осуществления настоящего изобретения форму фазы I Р027 получают объединением раствора Р027 и антирастворителя через диффузию. В варианте указанного способа диффузия представляет собой диффузию жидкость-жидкость. В другом варианте указанного способа диффузия представляет собой диффузию газ-жидкость.
В еще одном варианте осуществления настоящего изобретения форму фазы I Р027 собирают из смесей Р027, воды и растворителей.
В еще одном варианте осуществления настоящего изобретения форму фазы I Р027 получают из суспензий, содержащих соединение Р027. В варианте указанного способа температуру суспензии поддерживают в пределах приблизительно от комнатной температуры до 80°С.
В еще одном варианте осуществления настоящего изобретения раствор хлористоводородной кислоты и 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина смешивают с получением соединения Р027. Предпочтительно, антирастворитель добавляют к смеси для инициации кристаллизации соединения Р027.
Различные варианты осуществления настоящего изобретения могут потребовать дополнительных стадий, таких как центрифугирование, для дальнейшего выделения формы фазы I Р027.
Форму фазы II, форму фазы III и форму фазы IV Р027 можно получить с использованием полимер-индуцированной кристаллизации, выпариванием растворителя или кристаллизацией добавлением антирастворителя. Таким образом, другой вариант осуществления настоящего изобретения относится к способу получения полиморфных форм гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина, включающему:
а) растворение гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина в подходящем растворителе или смеси растворителей в присутствии каталитических количеств полимера, и
b) выпаривание растворителя или растворителей или добавление антирастворителя.
В предпочтительном варианте осуществления настоящего изобретения форму фазы II Р027 получают выпариванием раствора Р027 в воде в присутствии каталитических количеств поли(винилового спирта).
В другом предпочтительном варианте осуществления настоящего изобретения форму фазы III Р027 получают выпариванием раствора Р027 в воде или в ацетоне в присутствии каталитических количеств поли(этиленгликоля). Форму фазы III Р027 можно также удобно получить добавлением диизопропилового эфира в качестве антирастворителя к раствору Р027 в воде в присутствии каталитических количеств поли(этиленгликоля).
В другом предпочтительном варианте осуществления настоящего изобретения форму фазы IV Р027 получают с использованием хлороформа в качестве растворителя, диизопропилового эфира в качестве антирастворителя и следующих полимеров: поливинилпирролидона (PVP), поли(акриловой кислоты) (РАА), полипропилена (PPL), поли(стирол-со-дивинилбензола) (PSV), поли(тетрафторэтилена) (PTF), поли(винилового спирта) (PVH), полиакриламида (PAD) и поли(метилметакрилата) (РММ).
Сольват с диоксаном Р027 можно получить в эксперименте размалыванием с использованием капель растворителя в диоксане или посредством кристаллизации из горячего насыщенного раствора диоксана. Сольват с хлороформом Р027 можно получить с использованием полимер-индуцированных кристаллизаций посредством выпаривания растворителя (хлороформа) или кристаллизацией из горячих насыщенных растворов хлороформа.
Таким образом, другой вариант осуществления настоящего изобретения относится к способу получения сольватированных форм гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина, включающему по меньшей мере одну из трех альтернатив i)-iii):
i) размалывание с использованием капель растворителя, включающее:
а) загрузку гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина вместе с каталитическим количеством подходящего растворителя в контейнер шаровой мельницы; и
b) размалывание;
ii) кристаллизацию из горячего насыщенного раствора подходящего растворителя; или
iii) полимер-индуцированную кристаллизацию, включающую:
а) растворение гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина в подходящем растворителе в присутствии каталитическим количеством полимера, и
b) выпаривание растворителя или кристаллизацию в горячем насыщенном растворе растворителя.
В предпочтительном варианте осуществления настоящего изобретения сольват с диоксаном Р027 получают:
а) загрузкой гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина вместе с каталитическим количеством диоксана в контейнер шаровой мельницы; и
b) размалыванием; или
ii) кристаллизацией из горячего насыщенного раствора диоксана.
В предпочтительном варианте осуществления настоящего изобретения сольват с хлороформом Р027 получают:
а) растворением гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина в хлороформе в присутствии каталитического количества полимера, выбранного из группы, состоящей из поли(этиленгликоля), поливинилпирролидона, поли(акриловой кислоты), нейлона 6/6, полипропилена, поли(тетрафторэтилена), поли(винилацетата), поли(винилового спирта), полиакриламида и полисульфона; и
b) выпариванием хлороформа или кристаллизацией в горячем насыщенном растворе хлороформа.
Другой вариант осуществления настоящего изобретения включает применение кристаллических форм фазы II, фазы III и фазы IV Р027 для получения более стабильной полиморфной формы фазы I Р027. В одном варианте осуществления настоящего изобретения превращение осуществляют нагреванием кристаллических форм фазы II, фазы III и фазы IV в полиморфную форму фазы I.
В анализах ДСК фаз II, III и IV наблюдались широкие экзотермические пики, которые соответствуют переходу твердое вещество-твердое вещество. Переход твердое вещество-твердое вещество (перекристаллизация) фазы II в фазу I наблюдался при 145°С. Переход твердое вещество-твердое вещество (перекристаллизация) фазы III в фазу I наблюдался при 150-170°С. Переход твердое вещество-твердое вещество (перекристаллизация) фазы IV в фазу I наблюдался при 147°С.
Таким образом, другой вариант осуществления настоящего изобретения относится к получению формы фазы I Р027, включающему стадию нагревания кристаллических форм фазы II, фазы III и фазы IV Р027 при температуре приблизительно от 140°С до 170°С.
Другой вариант осуществления настоящего изобретения включает превращение сольвата Р027, предпочтительно, сольвата с хлороформом, в более стабильную полиморфную форму, такую как форма фазы I. После сушки сольвата с диоксаном в течение 4 часов при 60°С, 80°С и 100°С наблюдалось превращение в фазу I. Полученные твердые вещества характеризовали с использованием ПРД.
Еще один вариант осуществления настоящего изобретения включает фармацевтические композиции, содержащие по меньшей мере одну из форм гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина, особенно фазы I Р027, фазы II Р027, фазы III Р027, фазы IV Р027, сольват Р027 с хлороформом и сольват Р027 с диоксаном.
С учетом описания изобретения в общих чертах, его легче будет понять с помощью следующих примеров, которые представлены в качестве иллюстрации и не предназначены для ограничения настоящего изобретения.
Примеры
Оборудование для изучения характеристик твердых форм гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина
а) Порошковая рентгеновская дифракция (ПРД)
Приблизительно 20 мг необработанных образцов подготавливали на стандартных держателях с использованием двух пленок из полиацетата.
Порошковые дифрактограммы получали на системе порошковой дифракции D8 Advance Series 2Theta/Theta с использованием CuKα излучения в геометрии прохождения (длина волны: 1,54060). Система была оборудована подсчитывающим единичные фотоны PSD VANTEC-1, германиевым монохроматором, с устройством автоматической смены предметного столика на девяносто позиций, фиксированными прорезями дивергенции и радиальным соллером. Использованные программы: сбор данных с использованием DIFFRAC плюс XRD Commander V.2.5.1 и оценка с использованием EVA V.12.0.
b) Протонный ядерно-магнитный резонанс (1Н ЯМР)
Анализы протонного ядерно-магнитного резонанса записывали в дейтерированном хлороформе (CDCl3) с использованием ЯМР спектрометра Bruker Avance 400 Ultrashield, оборудованным z-градиентным 5 мм ВВО (Broad Observe) детектором с АТМ и автоматическим пробоотборником BACS-120. Спектры получали, растворяя 2-10 мг образца в 0,6 мл дейтерированного растворителя.
с) Дифференциальная сканирующая калориметрия (ДСК)
Стандартные анализы ДСК записывали на Mettler Toledo DSC822e. Образцы 1-2 мг отвешивали в алюминиевые тигели 40 мкл с крышками с точечным отверстием и нагревали в атмосфере азота (50 мл/мин) от 30 до 300°С со скоростью 10°С/мин. Сбор и оценку данных осуществляли с использованием компьютерной программы STARe.
d) Термогравиметрический анализ (ТГА)
Термогравиметрические анализы записывали на Mettler Toledo SDTA851e. Образцы 3-4 мг отвешивали (используя микровесы MX5, Mettler) в открытые алюминиевые тигели 40 мкл с крышками с точечным отверстием и нагревали со скоростью 10°С/мин от 30 до 500°С в атмосфере азота (80 мл/мин). Сбор и оценку данных осуществляли с использованием компьютерной программы STARe.
е) Инфракрасная спектроскопия с преобразованием Фурье (ИК-ПФ)
Спектры ИК-ПФ записывали с использованием Bruker Tensor 27, оборудованного ATR системой однократного отражения с решеткой с золотым покрытием MKII, источником средней инфракрасной области спектра в качестве источника возбуждения и детектором DTGS. Спектры получали в 32 сканированиях с разрешением 4 см-1. Для осуществления анализа подготовки образцов не требовалось.
f) Рентгеновский дифракционный анализ единичных кристаллов (SCXRD)
Измеряемые кристаллы выбирали с использованием цейссовского стереомикроскопа в поляризованном свете и подготавливали в инертных условиях, погруженными в перфторполиэфир, как в защитное масло, для манипуляций. Определение структуры кристаллов осуществляли с помощью дифрактометра Bruker-Nonius, оборудованного двумерным детектором APPEX 2 4K CCD, вращающимся анодом с MoKα излучением, зеркалами Montel в качестве монохроматора и низкотемпературным устройством Kryoflex (Т=100 К). Омега- и фи-сканограммы сбора данных. Использованные программы: сбор данных Apex2 V. 1.0-22 (Bruker-Nonius 2004), предварительная обработка данных Saint+Version 6.22 (Bruker-Nonius) и поправка на поглощение SADABS V. 2.10 (2003). Решение о структуре кристаллов достигалось с использованием прямых методов, как в SHELXTL Version 6.10 (Sheldrick, Universtitat Gottingen (Германия), 2000), и визуализировалось с использованием программы ХР. Недостающие атомы впоследствии локализовали с помощью дифференциального синтеза Фурье и добавляли к перечню атомов. Уточнение методом наименьших квадратов на F0 2 с использованием всех измеренных интенсивностей осуществляли с помощью программы SHELXTL Version 6.10 (Sheldrick, Universtitat Gottingen (Германия), 2000). Все атомы. Не являющиеся водородом, были уточнены, включая параметры анизотропной перестановки.
Исходный синтез соединения Р027
Гидрохлорид 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина получали, следуя следующим протоколам:
1) 50,8 литра 6н. раствора хлористоводородной кислоты/пропан-2-ола добавляли к раствору 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина (85 кг) в этаноле (290 л) при Т>35°С. Затем 213 литров метил-трет-бутилового эфира добавляли к суспензии. Смесь охлаждали позднее до 0-5°С. Полученное твердое вещество выделяли центрифугированием, с получением 90 кг соединения Р027.
2) 27 мл 6н. раствора хлористоводородной кислоты/пропан-2-ола добавляли к раствору 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина (44,5 г) в этаноле (120 мл) при Т>35°С. Затем суспензию охлаждали до 0-5°С. Полученное твердое вещество выделяли фильтрованием, с получением 47 г соединения Р027.
Пример 1
Получение и определение характеристик кристаллической формы фазы I гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина
Пример 1.1: Выпаривание растворителя с двумя скоростями при комнатной температуре
От 10 до 20 мг соединения Р027 растворяли в минимальном количестве соответствующих растворителей при комнатной температуре (кт), 60°С и 80°С. Полученные растворы оставляли быстро выпариваться в открытых флаконах или медленно, в закрытых пробирках, проткнутых иглой, при комнатной температуре (см. таблицы 9 и 10). Указанные растворы, которые не до конца выпарились спустя 3 месяца, оставляли выпариваться при комнатной температуре в открытых флаконах. Полученные твердые образцы анализировали посредством ПРД. Образцы продемонстрировали дифрактограмму, соответствующую стандартной ПРД фазы I.
Пример 1.2: Выпаривание растворителя при различных температурах
От 10 до 20 мг соединения Р027 растворяли в минимальном количестве соответствующих растворителей при комнатной температуре (кт), 60°С или 80°С. Полученные растворы оставляли выпариваться в открытых флаконах при трех различных температурах: 60°С, 4°С и -21°С (см. таблицы 11, 12 и 13). Указанные растворы, которые не до конца выпарились спустя 3 месяца, оставляли выпариваться при комнатной температуре в открытых флаконах. Полученные твердые образцы анализировали посредством ПРД. Образцы продемонстрировали дифрактограмму, соответствующую стандартной дифрактограмме ПРД фазы I. На фиг.14 проиллюстрирована дифрактограмма ПРД формы фазы I, полученной выпариванием раствора н-бутанола при -21°С, согласно настоящему протоколу.
Пример 1.3: Кристаллизация из горячих насыщенных растворов
От 10 до 30 мг соединения Р027 растворяли в минимальном количестве соответствующего растворителя при высокой температуре для получения насыщенных растворов. Затем растворы охлаждали двумя различными способами:
1) Медленное охлаждение при комнатной температуре (медленная кристаллизация) [см. таблицу 14].
2) Быстрое охлаждение погружением в ледяную баню (быстрая кристаллизация) [см. таблицу 15].
После охлаждения до комнатной температуры, полученные твердые вещества отделяли фильтрованием или центрифугированием. Если твердые вещества не образовывались, раствор выдерживали при 4°С несколько дней на первой стадии. Все твердые вещества, которые образовывались на указанной стадии, выделяли из раствора. Если твердые вещества не образовывались во время первой стадии, раствор выдерживали при -21°С еще несколько дней. Все твердые вещества, которые образовывались на указанной второй стадии, выделяли из раствора. Растворы, которые не кристаллизовались во время второй стадии, оставляли выпариваться досуха при комнатной температуре. Твердое вещество отфильтровывали в нескольких экспериментах, когда кристаллизация наблюдалась до полного выпаривания.
Полученные твердые образцы анализировали посредством ПРД. Образцы показали дифрактограмму, соответствующую стандартной дифрактограмме ПРД фазы I. На фиг.15 проиллюстрирована дифрактограмма формы фазы I, полученной медленной кристаллизацией горячего насыщенного раствора Р027 в метилэтилкетоне.
Пример 1.4: Мелкомасштабная кристаллизация посредством добавления антирастворителя
От 10 до 20 мг соединения Р027 растворяли в минимальном количестве соответствующего растворяющего агента при высокой температуре или при комнатной температуре. Диизопропиловый эфир (DIE) или н-гептан (НЕР) использовали в качестве антирастворителей. Следовали следующим протоколам:
1) Антирастворитель добавляли по каплям к раствору Р027 при интенсивном перемешивании при комнатной температуре или при высокой температуре (см. таблицы 16 и 17).
2) Раствор Р027 добавляли по каплям к 4 мл антирастворителя при интенсивном перемешивании при комнатной температуре или при высокой температуре (см. таблицы 18 и 19).
Полученные после смешивания растворяющего агента и антирастворителя твердые вещества отделяли фильтрованием или центрифугированием. Если твердые вещества не образовывались, раствор выдерживали при 4°С несколько дней на первой стадии. Все твердые вещества, которые образовывались на указанной стадии, выделяли из раствора. Если твердые вещества не образовывались во время первой стадии, раствор выдерживали при -21°С еще несколько дней. Все твердые вещества, которые образовывались на указанной второй стадии, выделяли из раствора. Растворы, которые не кристаллизовались во время второй стадии, оставляли выпариваться досуха при комнатной температуре. Твердое вещество отфильтровывали в нескольких экспериментах, когда кристаллизация наблюдалась до полного выпаривания.
Полученные твердые образцы анализировали посредством ПРД. Образцы показали дифрактограмму, соответствующую стандартной дифрактограмме ПРД фазы I. На фиг.16 проиллюстрирована дифрактограмма ПРД формы фазы I, полученной кристаллизацией добавлением раствора Р027 в метаноле к раствору н-гептана.
Пример 1.5: Крупномасштабная кристаллизация посредством добавления антирастворителя
133 литра метил-трет-бутилового эфира добавляли к раствору соединения Р027 (45 кг) в этаноле (265 л) при Т>35°С. Затем суспензию охлаждали до 0-5°С. Полученное твердое вещество выделяли центрифугированием, с получением 40,2 кг гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина.
Пример 1.6: Кристаллизация диффузией антирастворителя
От 10 до 50 мг соединения Р027 растворяли в минимальном количестве соответствующих растворителей при высокой температуре или при комнатной температуре. Использовали различные растворяющие агенты. Следовали следующим протоколам:
1) Диффузия жидкость-жидкость. Антирастворитель осторожно добавляли на раствор Р027 с образованием двух отдельных фаз. Твердое вещество кристаллизовалось благодаря диффузии фаз (см. таблицу 20).
2) Диффузия газ-жидкость. Первый контейнер с раствором Р027 вставляли во второй, большего размера, принимающий контейнер, содержавший антирастворитель. Диффузия газа антирастворителя над раствором Р027 инициировала кристаллизацию фазы I (см. таблицу 21).
Полученные твердые образцы анализировали посредством ПРД. Образцы показали дифрактограмму, соответствующую стандартной дифрактограмме ПРД фазы I. На фиг.17 проиллюстрирована дифрактограмма ПРД формы фазы I, полученной кристаллизацией диффузией жидкость-жидкость изопропилового эфира в раствор Р027 в нитрометане.
Пример 1.7: Кристаллизация из смесей воды и растворителя
От 10 до 20 мг соединения Р027 растворяли в минимальном количестве соответствующего растворителя, насыщенного водой. Растворители смешивали в различных соотношениях с водой, в соответствии с их смешиваемостью (см. таблицу 22).
Растворы оставляли кристаллизоваться при комнатной температуре в закрытой пробирке на две недели. Если твердые вещества не образовывались, раствор выдерживали при 4°С несколько дней. Все твердые вещества, которые образовывались на указанной стадии, выделяли из раствора. Если твердые вещества не образовывались во время первой стадии, раствор оставляли выпариваться досуха при комнатной температуре.
Полученные твердые образцы анализировали посредством ПРД. Образцы показали дифрактограмму, соответствующую стандартной дифрактограмме ПРД фазы I.
Пример 1.8: Размалывание
Приблизительно 40 мг фазы I Р027 помещали в контейнер шаровой мельницы вместе с каталитическими количествами соответствующего растворителя (три капли). Фазу I Р027 и растворитель размалывали с максимальной частотой 30 сек-1 в течение 30 минут (см. таблицу 23).
Полученные твердые образцы анализировали посредством ПРД. Образцы показали дифрактограмму, соответствующую стандартной дифрактограмме ПРД фазы I, что показывает, что фаза I Р027 является стабильной после размалывания. На фиг.18 проиллюстрирована дифрактограмма формы фазы I, полученной размалыванием Р027 с дихлорметаном.
Пример 1.9: Давление
Таблетки фазы I Р027 получали на гидравлическом прессе при трех различных значениях давления (5, 7,5 и 10 тонн) в течение различных периодов времени (5, 30 и 90 минут) [см. таблицу 24].
Полученные твердые образцы анализировали посредством ПРД. Образцы показали дифрактограмму, соответствующую стандартной дифрактограмме ПРД фазы I, что показывает, что фаза I Р027 является стабильной под давлением. На фиг.19 проиллюстрирована дифрактограмма формы фазы I, полученной приложением давления 30 тонн к Р027 в течение 90 минут.
Пример 1.10: Получение суспензий
От 30 до 400 мг соединения Р027 перемешивали в 4 мл соответствующего растворителя в течение i) 48 часов при комнатной температуре или (ii) 24 часов при 80°С (см. таблицу 25).
Все суспензии фильтровали. Полученные твердые образцы анализировали посредством ПРД. Образцы показали дифрактограмму, соответствующую стандартной дифрактограмме ПРД фазы I.
Характеризация кристаллической формы фазы I гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина
Форма фазы I Р027 показывает дифрактограмму ПРД, имеющую характерные пики в области значений угла отражения [2θ] приблизительно 5,9, 8,1, 11,3, 11,7, 14,2, 15,1, 15,8, 16,3, 16,8, 17,8, 18,1, 18,6, 19,8, 20,9, 21,9, 22,8, 23,0, 23,2, 23,6, 23,9, 24,3, 25,0, 25,1, 28,0, 28,3, 28,6, 29,0, 29,2, 30,7 и 30,9, полученными с использованием медного источника излучения (CuKa1 1,54060 Å).
Различия в значениях интенсивности пиков дифрактограммы ПРД можно было наблюдать в зависимости от методики кристаллизации или используемого для кристаллизации растворителя (см. фиг.5). Выраженные различия в значениях интенсивности пиков могли иметь место вследствие предпочтительных ориентаций, текстурных эффектов кристаллов, и они не являются показателем присутствия различных кристаллических фаз. Неидеальные кристаллические фазы определяются расположением пиков, а не значениями интенсивности пиков. Различия в значениях интенсивности пиков могли иметь место вследствие различных конфигураций измерительных устройств (трансмиссия или отражение) или текстурных эффектов, связанных с предпочтительными ориентациями кристаллов.
Для того чтобы проверить, имели ли место различия в значениях интенсивности пиков вследствие текстурных эффектов, некоторые выбранные образцы осторожно размалывали в агатовой ступке и измеряли. После гомогенизации образцов текстурные эффекты становились менее выраженными или исчезали (см. фиг.6).
Кроме того, несколько образцов фазы I анализировали посредством 1Н ЯМР, с целью проверить стабильность соли. Химические сдвиги и интеграции сигналов 1Н ЯМР совпадали для всех образцов, и признаков утраты HCl или разложения образцов не наблюдалось (см. фиг.7).
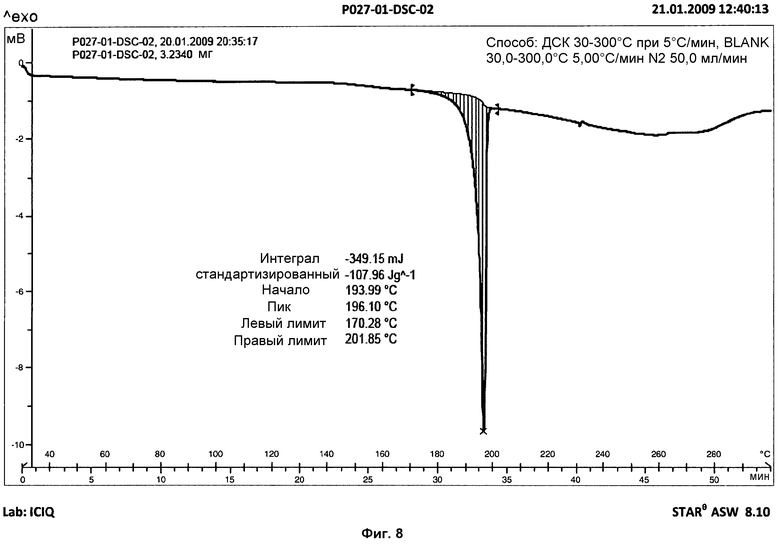
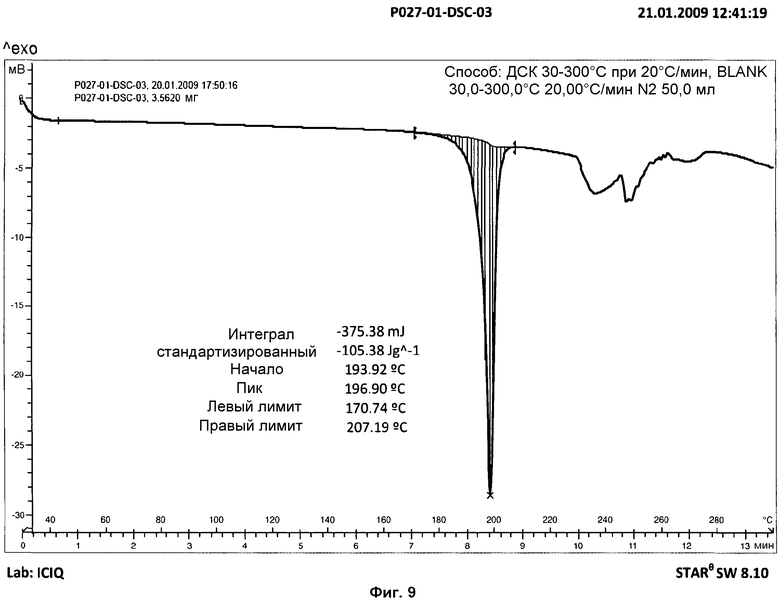
Анализ ДСК образцов фазы I осуществляли при скорости нагревания 10°С/мин. Анализ представил острый эндотермический пик, который не возвращался к базовой линии, с началом на 194°С и энтальпией 103 Дж/г, соответствующий плавлению с последующим разложением продукта (см. фиг.3). В дополнительных анализах ДСК того же образца, осуществлявшихся при скорости нагревания 5°С/мин и 20°С/мин, наблюдали, что температура начала эндотермического пика не изменяется в зависимости от скорости нагревания (см. фиг.8 и 9).
В ТГА образца фазы I наблюдалась потеря массы вследствие разложения образца при температурах выше 195°С (см. фиг.3). Потеря массы не наблюдалась при температурах ниже 195°С, что указывает на отсутствие растворителя. Температура начала потери массы при ТГА совпадала с температурой плавления, подтверждая, что образец разлагается при плавлении.
В спектре ИК-ПФ фазы I Р027 присутствуют интенсивные пики приблизительно на 2965, 2609, 1632, 1600, 1559, 1508, 1490, 1439, 1376, 1301, 1257, 1242, 1169, 1129, 1103, 1042, 1010, 932, 914, 862, 828 и 753 см-1 (см. фиг.4).
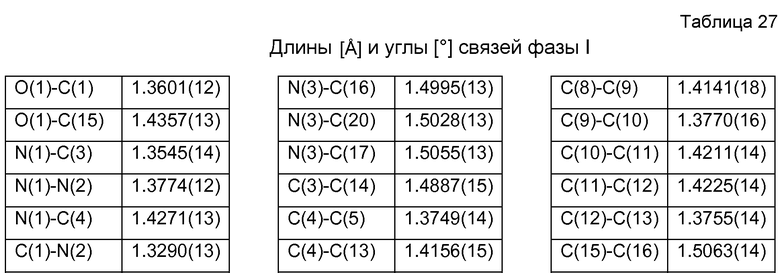
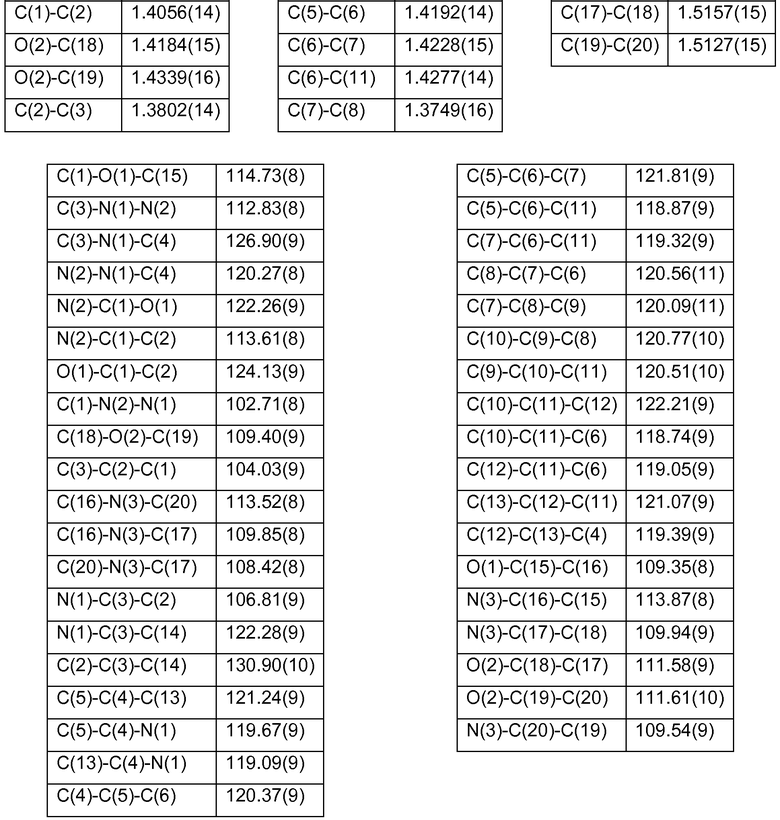
Структурное определение кристаллической формы фазы I гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина посредством рентгеновского дифракционного анализа единичных кристаллов
Идентичность и структуру кристаллов фазы I Р027 исследовали с использованием рентгеновского дифракционного анализа единичных кристаллов. Подходящие кристаллы получали медленной диффузией н-гептана в концентрированный раствор продукта в ацетоне. Поскольку отобранные кристаллы в большинстве своем были сдвоены, малый фрагмент пластины (0,30×0,30×0,07 мм3) отделяли микроскальпелем и использовали для рентгеновского определения структуры единичного кристалла. В таблице 26 показаны использовавшиеся условия измерения, константы ячейки и результаты, полученные рентгеновским структурным дифракционным анализом единичных кристаллов. В таблице 27 приведены выбранные расстояния и углы между связями фазы I для рентгеновского определения структуры, осуществлявшегося при 100 К.
Условия измерения, константы ячейки и результаты
b=11,6704(11) Å, β=91,284(2)
с=11,0(10) Å, γ=90°
-17<=l<=19
Форма фазы I кристаллизуется в центросимметричном пространстве С2/с с одной катионной молекулой и двумя независимыми полуанионами хлора в элементарной ячейке (см. фиг.10). Каждая катионная молекула делит два аниона хлора с соседними катионными молекулами. Один из общих атомов хлора связан с положительно заряженными N-H-группами двух соседних катионных молекул, образуя две водородные связи (Cl1...N3-дистанция: 3,13 Å) [см. фиг.10 и 11]. Второй общий атом хлора располагается в межмолекулярном пространстве, обеспечивая только слабые взаимодействия с окружающими молекулами (кратчайшей дистанцией является дистанция С12...С17: 3,56 Å).
Порошковая дифрактограмма, модулированная из данных по единичным кристаллам, показывает хорошее соответствие экспериментально измеренной стандартной порошковой дифрактограмме фазы I. Совмещение подтверждает чистоту фазы. Небольшие вариации в позициях пиков происходят вследствие разницы температур, при которых измерялись сравниваемые порошковые дифрактограммы (модулированные при -173°С и экспериментально измеренные при комнатной температуре). На фиг.12 и 13 показана модулированная порошковая дифрактограмма фазы I и ее сравнение с экспериментально измеренной дифрактограммой, соответственно.
Пример 2
Получение и определение характеристик кристаллической формы фазы II гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина
При первоначальном скрининге смесь фазы I и фазы получали выпариванием растворителя в нескольких растворителях (метаноле, воде, смеси диизопропилового эфира и воды, смеси нитрометана, диоксана и воды, и смеси гептана и воды). Указанную новую фазу II можно воспроизвести в чистом виде в ходе скрининга, осуществляемого с использованием полимеров путем выпаривания раствора Р027 в воде и в присутствии каталитических количеств поли(винилового спирта).
Кристаллизация формы фазы II выпариванием растворителя при комнатной температуре: Образец гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина (20-25 мг) растворяли в минимальном количестве воды (0,7 мл) при комнатной температуре и добавляли небольшое количество поли(винилового спирта) (2-3 мг) в соответствующий раствор. Полученный раствор или суспензии оставляли выпариваться на две недели в открытых флаконах при комнатной температуре.
Сравнение дифрактограммы ПРД фазы I и фазы II показано на фиг.23. Можно видеть, что фаза II, полученная с использованием поли(винилового спирта), является чистой, и на дифрактограмме не выявляются пики фазы I.
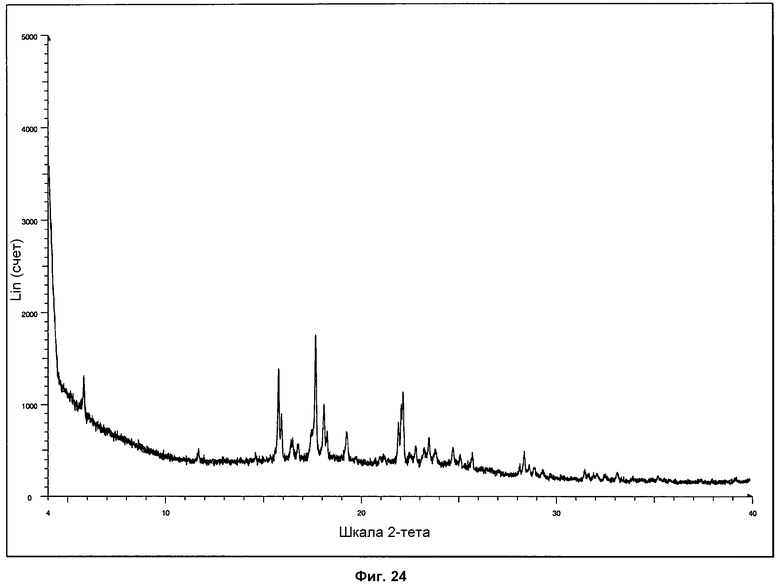
Стандартная дифрактограмма ПРД для фазы II показана на фиг.24.
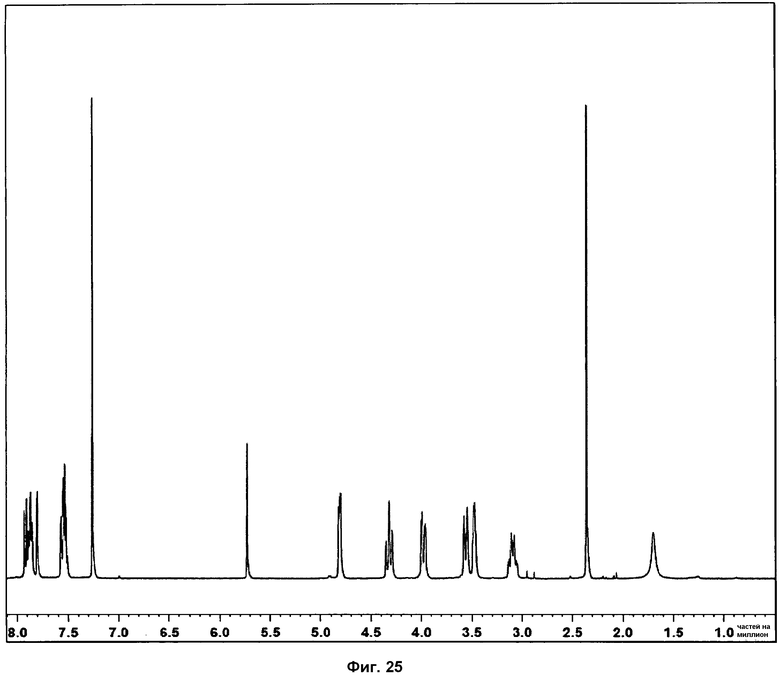
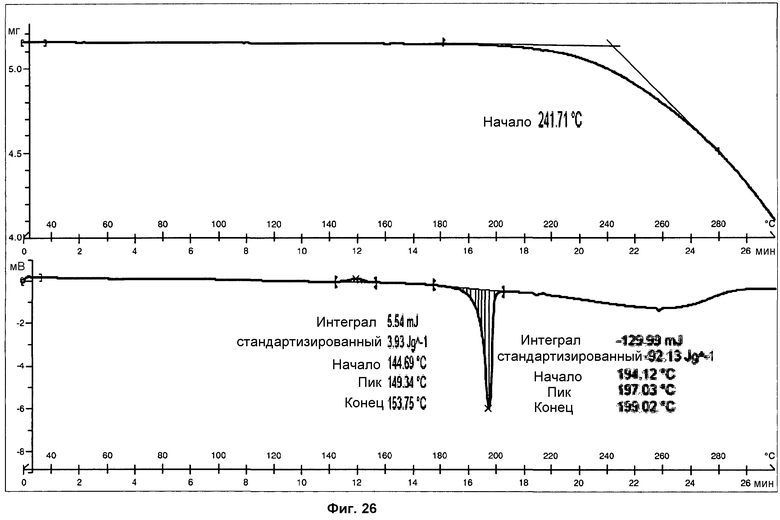
Определение характеристик посредством 1Н ЯМР, ДСК и ТГА показано на фиг.25 и 26.
Спектр 1Н ЯМР, полученный из смеси фаз I и II, является идентичным спектру, полученному для фазы I, указывая на то, что фаза II не является продуктом разложения. Спектры, полученные для фазы I и фазы II, сравниваются на фиг.25. Разницы в сдвигах релевантных атомов водорода не наблюдается.
Анализ ДСК образцов фазы II, осуществленный при скорости нагревания 10°С/мин, показал слабый широкий экзотермический пик, с началом при 145°С и энтальпией 4 Дж/г, и острый эндотермический пик, с началом при 194°С и энтальпией 92 Дж/г, соответствующий плавлению с последующим разложением продукта (см. фиг.26). Малый экзотермический пик при 145°С предполагает, что фаза II должная быть метастабильной фазой, монотропически родственной фазе I. Таким образом, ДСК действительно показывает переход твердое вещество-твердое вещество, фазы II в фазу I, с последующим слиянием фазы I.
В анализе ТГ образца фазы II (фиг.26) наблюдалась потеря массы вследствие разложения образца при температурах выше 195°С. Температура начала потери массы при ТГА совпадает с температурой плавления, подтверждая, что образец разлагается при плавлении. Потери массы не наблюдалось при температурах ниже 180°С, что указывает на отсутствие растворителя. ТГ анализ твердого вещества, содержащего фазу II, является идентичным анализу, полученному для фазы I.
Пример 3
Получение и определение характеристик кристаллической формы фазы III гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина
Форму фазы III получали посредством полимер-индуцированной кристаллизации. Указанное твердое вещество получали в четырех экспериментах, всегда в присутствии поли(этиленгликоля). В трех случаях его получали выпариванием воды или ацетона, и в одном случае его получали добавлением диизопропилового эфира в качестве антирастворителя к раствору в воде.
Кристаллизация формы фазы выпариванием растворителя при комнатной температуре: Образец гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина (20-25 мг) растворяли в минимальном количестве воды (0,7 мл) или ацетона (5,7 мл) при комнатной температуре и добавляли небольшое количество поли(этиленгликоля) (2-3 мг) в соответствующий раствор. Полученный раствор или суспензии оставляли выпариваться на две недели в открытых флаконах при комнатной температуре.
Кристаллизация формы фазы добавлением антирастворителя: Образец гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина (20-25 мг) вместе с поли(этиленгликолем) (3-4 мг) растворяли в минимальном количестве воды при комнатной температуре и добавляли диизопропиловый эфир (10 мл) при интенсивном перемешивании. Конечную суспензию оставляли выпариваться.
Определение характеристик фазы III осуществляли посредством 1Н ЯМР, ДСК и ТГА. Репрезентативная дифрактограмма ПРД для фазы III показана на фиг.27. Путем сравнения дифрактограммы фазы III с дифрактограммой поли(этиленгликоля) можно ясно различить два сильнейших характерных сигнала полимера на 19,1° и 23,2° при 2θ (см. сравнение на фиг.28). Пик на 19,1° при 2θ можно наблюдать в виде слабого сигнала, и широкий пик на 23,2° при 2θ также можно наблюдать, слегка сдвинутым к 23,6° при 2θ на дифрактограмме фазы III.
Определение характеристик посредством 1Н ЯМР, ДСК и ТГА показано на фиг.29 и 31.
На спектре 1Н ЯМР фазы III присутствие характерных сигналов Р027 указывает на то, что образец не разложился. Помимо этого, во всех измеренных спектрах, наблюдался характерный пик, соответствующий поли(этиленгликолю), указывая на то, что фаза II всегда смешана с указанным полимером. Спектр 1Н ЯМР поли(этиленгликоля) представлен на фиг.30.
Анализ ДСК образцов фазы III (см. фиг.31), осуществленный при скорости нагревания 10°С/мин, представляет первый острый эндотермический пик, с началом при 56°С и энтальпией 46 Дж/г, соответствующий плавлению поли(этиленгликоля). ДСК чистого поли(этиленгликоля) показана на фиг.32. В пределах от 150 до 175°С ДСК показывает двойной пик, первый эндотермический, а затем экзотермический, соответствующий, вероятно, плавлению фазы III, перекрывающемуся с перекристаллизацией в фазу I. И наконец, можно наблюдать эндотермический пик, с началом при 190°С и энтальпией 47 Дж/г, соответствующий плавлению с последующим разложением фазы I. Помимо этого, анализы ДСК того же самого образца, осуществленные при скорости нагревания 20°С/мин (фиг.33), и 30°С/мин (фиг.34), показали, что температура начала эндотермических пиков не изменяется со скоростью нагревания. Это указывает на то, что эндотермические пики соответствуют температурам плавления.
В анализе ТГ фазы III (фиг.31) наблюдалась потеря массы вследствие разложения образца при температурах выше 180°С. Потери массы не наблюдалось при температурах ниже 180°С, что указывает на отсутствие растворителя. Температура начала потери массы при ТГА совпадает с температурой плавления, подтверждая, что образец разлагается при плавлении.
Пример 4
Получение и определение характеристик кристаллической формы фазы IV гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина
Форму фазы IV получали только посредством полимер-индуцированной кристаллизации. Указанную фазу получали в экспериментах с использованием хлороформа в качестве растворителя и диизопропилового эфира в качестве антирастворителя. Твердое вещество фазы IV получали с использованием следующих полимеров: поливинилпирролидона (PVP), поли(акриловой кислоты) (РАА), полипропилена (PPL), поли(стирол-со-дивинилбензола) (PSV), поли(тетрафторэтилена) (PTF), поли(винилового спирта) (PVH), полиакриламида (PAD) и поли(метилметакрилата (РММ). Полимеры PVP, PAA, PSV, PVH, PAD и РММ являются аморфными, и полимеры PPL и PTF являются кристаллическими. Только в образце фазы IV, полученном с кристаллическим PTF, можно было выявить слабый пик полимеров в дифрактограмме ПРД.
Кристаллизация формы фазы добавлением антирастворителя: Образец гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина (20-25 мг) вместе с 3-4 мг соответствующего полимера (поливинилпирролидона, поли(акриловой кислоты), полипропилена, поли(стирол-со-дивинилбензола), поли(тетрафторэтилена), поли(винилового спирта), полиакриламида, поли(метилметакрилата)) растворяли в минимальном количестве хлороформа при комнатной температуре и добавляли диизопропиловый эфир (2 мл) при интенсивном перемешивании. Конечное твердое вещество отделяли центрифугированием.
Определение характеристик фазы IV осуществляли посредством ПРД, 1Н ЯМР, ДСК и ТГА.
Репрезентативная дифрактограмма ПРД для фазы IV показана на фиг.35.
Определение характеристик посредством 1Н ЯМР, ДСК и ТГА показано на фиг.36 и 37.
На спектре 1Н ЯМР фазы IV (см. фиг.36) присутствие характерных сигналов Р027 указывает на то, что образец не разложился. Не было выявлено сигналов, соответствующих полимерам.
Анализ ДСК фазы IV (см. фиг.37), осуществленный при скорости нагревания 10°С/мин, представляет широкий экзотермический пик, с началом при 147°С и энтальпией 9 Дж/г, соответствующий, вероятно, переходу твердое вещество-твердое фазы IV в фазу I. И наконец, можно наблюдать эндотермический пик, с началом при 191°С и энтальпией 71 Дж/г, соответствующий плавлению с последующим разложением фазы I.
В анализе ТГ фазы IV (фиг.37) можно наблюдать небольшую потерю массы, соответствующую 1,4% образца, при температурах от 120°С до 170°С. Разложение образца наблюдалась при температурах выше 190°С. Потеря массы, вероятно, соответствует небольшим количествам воды или дихлорметана, которые утрачиваются в процессе перехода. Температура начала более высокой потери массы при ТГА совпадает с температурой плавления, подтверждая, что образец разлагается при плавлении.
Пример 5
Получение и определение характеристик сольвата гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина с диоксаном
Новую кристаллическую сольватированную фазу, называемую сольватом с диоксаном, получали в эксперименте размалыванием с использованием капель растворителя в диоксане или посредством кристаллизации из горячего насыщенного раствора в диоксане. Сольват с диоксаном кристаллизуется в форме небольших липких кристаллитов. Репрезентативная дифрактограмма ПРД для сольвата показана на фиг.38. Определение характеристик посредством 1Н ЯМР, ДСК, ТГА и ИК-ПФ показано на фиг.39-41.
Эксперимент с размалыванием: 50 мг соединения вместе с каталитическими количествами диоксана (три капли) размалывали в шаровой мельнице при 30 сек-1 в течение 30 минут. Для экспериментов с размалыванием использовали шаровую мельницу Retsch MM400.
Кристаллизация из горячего насыщенного раствора: 0,5 г гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина растворяли в диоксане (80 мл) при 80°С. Полученный раствор охлаждали до 40°С, и твердое вещество начинало кристаллизоваться. Полученную суспензию выдерживали при 40°С в течение 2 часов при осторожном перемешивании, охлаждали до комнатной температуры и выдерживали при указанной температуре в течение 2 часов при осторожном перемешивании. Конечное твердое вещество отфильтровывали.
Анализ ДСК сольвата с диоксаном при скорости нагревания 10°С/мин представляет два перекрывающихся эндотермических пика, с началом при 124°С и 130°С, вероятно, в силу утраты диоксана, и третий эндотермический пик, с началом при 192°С и энтальпией 73 Дж/г, соответствующий плавлению с последующим разложением продукта (фиг.40).
В анализе ТГ сольвата с диоксаном (фиг.40) можно наблюдать потерю массы 14,6% в силу утраты диоксана (теоретическое содержание диоксана для моносольвата с диоксаном составляет 19%), при температурах от 100°С до 160°С. Разложение образца наблюдалось при температурах выше 190°С. Температура начала потери массы в силу разложения при ТГА совпадает с эндотермическим пиком при ДСК, подтверждая, что образец разлагается при плавлении. В 1Н ЯМР спектре можно наблюдать характерный сигнал диоксана, что подтверждает присутствие указанного растворителя (см. фиг.39).
Спектр ИК-ПФ, характерный для сольвата с диоксаном, представлен на фиг.41 и представляет интенсивные пики на 3138, 3055, 2959, 2857, 2660, 2572, 2540, 2444, 1633, 1600, 1556, 1509, 1488, 1446, 1372, 1304, 1289, 1255, 1168, 1118, 1099, 1083, 1039, 933, 872, 861, 819, 771 и 748 см-1.
Постепенное увеличение количества сольвата с диоксаном осуществляли, начиная с 50, 100 и 500 мг соединения. Полученные результаты в каждом случае собраны в таблице 28.
Твердые вещества, полученные при первоначальном скрининге и при постепенном увеличении количества до 100 и 500 мг, давали одну и ту же кристаллическую фазу. При масштабе 50 мг твердое вещество получали в экспериментах размалывания с использованием капель растворителя, в диоксане. При масштабах 100 мг и 500 мг твердое вещество кристаллизовалось во время охлаждения до комнатной температуры горячего насыщенного раствора в диоксане.
Пример 6
Получение и определение характеристик сольвата гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина с хлороформом
Новую кристаллическую сольватированную фазу, названную сольватом с хлороформом, получали в ходе полимер-индуцированных кристаллизаций. Сольват с хлороформом Р027 получали выпариванием хлороформа или кристаллизацией горячих насыщенных хлороформенных растворов с использованием следующих полимеров: поли(этиленгликоля) (PGY), поливинилпирролидона (PVP), поли(акриловой кислоты) (PAA), нейлона 6/6 (NYL), полипропилена (PPL), поли(тетрафторэтилена) (PTF), поли(винилацетата) (PVA), поли(винилового спирта) (PVH), полиакриламида (PAD) и полисульфона (PLS). Полимеры PGY, PPL и PTF являются кристаллическими, и остальные - аморфными. В дифрактограммах ПРД сигналов кристаллических полимеров не наблюдалось. Сольват с хлороформом в большинстве случаев кристаллизуется в форме больших кристаллов, которые, возможно, стабилизируются в присутствии полимеров. Репрезентативная дифрактограмма ПРД для сольвата показана на фиг.42. Определение характеристик посредством ДСК и ТГА показано на фиг.43.
Кристаллизация сольвата с хлороформом выпариванием растворителя: Образец гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина (20-25 мг) растворяли в 0,6 мл хлороформа и добавляли 3-4 мг соответствующего полимера (поли(этиленгликоля), поливинилпирролидона, поли(акриловой кислоты), нейлона 6/6, полипропилена, поли(тетрафторэтилена), поли(винилацетата), поли(винилового спирта), полиакриламида, полисульфона). Суспензию оставляли выпариваться. Спустя 24 часа полученное твердое вещество анализировали посредством ПРД, ДСК и ТГА.
Анализ ДСК сольвата с хлороформом при скорости нагревания 10°С/мин представляет широкий эндотермический пик, с началом при 67°С и энтальпией 42 Дж/г, в силу утраты хлороформа, и второй острый эндотермический пик, с началом при 194°С и энтальпией 73 Дж/г, соответствующий плавлению с последующим разложением фазы I (фиг.43).
В анализе ТГ сольвата с хлороформом (фиг.43) можно наблюдать потерю массы 21,5% в силу утраты хлороформа (теоретическое содержание хлороформа для моносольвата с хлороформом составляет 22,6%), при температурах от 50°С до 120°С. Разложение образца наблюдалась при температурах выше 190°С.




Изобретение относится к полиморфам и сольватам гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина (Р027), способам их получения и к содержащим их фармацевтическим композициям. 12 н. и 5 з.п. ф-лы, 43 ил., 28 табл., 6 пр.
1. Твердая полиморфная или сольватированная форма гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина, выбранная из группы, состоящей из:
a) полиморфной формы фазы I, характеризующейся порошковой рентгеновской дифрактограммой, демонстрирующей характерные пики в области значений угла отражения [2θ в градусах] приблизительно 5,9, 8,1, 11,3, 11,7, 14,2, 15,1, 15,8, 16,3, 16,8, 17,8, 18,1, 18,6, 19,8, 20,9, 21,9, 22,8, 23,0, 23,2, 23,6, 23,9, 24,3, 25,0, 25,1, 28,0, 28,3, 28,6, 29,0, 29,2, 30,7 и 30,9;
b) полиморфной формы фазы II, характеризующейся порошковой рентгеновской дифрактограммой, демонстрирующей характерные пики в области значений угла отражения [2θ в градусах] приблизительно 5,776, 11,629, 14,558, 15,737, 15,891, 16,420, 16,740, 17,441, 17,635, 18,056, 18,219, 19,232, 19,712, 20,140, 20,685, 21,135, 21,889, 22,108, 22,478, 22,763, 23,219, 23,454, 23,782, 24,689, 25,065 и 25,671;
c) полиморфной формы фазы III, характеризующейся порошковой рентгеновской дифрактограммой, демонстрирующей характерные пики в области значений угла отражения [2θ в градусах] приблизительно 5,437, 5,714, 10,918, 11,546, 12,704, 13,344, 13,984, 14,505, 15,606, 15,824, 16,164, 16,646, 17,333, 17,837, 18,719, 18,878, 19,236, 19,533, 20,142, 20,689, 21,337, 22,008, 22,929, 23,596, 24,748, 25,064, 25,207, 25,737 и 26,148;
d) полиморфной формы фазы IV, характеризующейся порошковой рентгеновской дифрактограммой, демонстрирующей характерные пики в области значений угла отражения [2θ в градусах] приблизительно 5,805, 11,685, 15,559, 15,804, 16,397, 16,879, 17,357, 17,465, 17,621, 19,112, 19,435, 19,923, 21,224, 21,987, 22,167, 22,412, 22,852, 23,059, 23,359, 23,855, 24,092, 25,722, 26,054, 26,649 и 27,780;
e) сольвата с диоксаном, характеризующегося порошковой рентгеновской дифрактограммой, демонстрирующей характерные пики в области значений угла отражения [2θ в градусах] приблизительно 4,734, 9,317, 11,390, 13,614, 14,290, 14,815, 16,211, 16,432, 16,782, 17,741, 18,056, 18,329, 18,724, 19,070, 19,494, 20,436, 20,762, 21,587, 22,000, 22,935, 23,084, 23,551, 23,891, 24,721 и 25,078; и
f) сольвата с хлороформом, характеризующегося порошковой рентгеновской дифрактограммой, демонстрирующей характерные пики в области значений угла отражения [2θ в градусах] приблизительно 11,370, 13,396, 14,048, 15,010, 15,303, 16,117, 16,804, 17,040, 17,830, 18,029, 18,661, 18,859, 19,190, 20,150, 20,434, 21,424, 22,279, 22,871, 23,449, 23,918, 24,343, 24,709, 24,820, 25,459 и 26,199;
где указанные значения получены с использованием излучения меди (CuKa1 1,54060 Ǻ).
2. Способ получения полиморфной формы фазы I по п. 1, включающий:
a) растворение гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина в подходящем растворителе, и
b) выпаривание растворителя.
3. Способ по п. 2, в котором гидрохлорид 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина растворяют при температуре от комнатной температуры до 120°С и/или растворитель выпаривают при температуре от -21°С до 60°С.
4. Способ получения полиморфной формы фазы I по п. 1, в котором раствор, включающий гидрохлорид 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина, и подходящий антирастворитель смешивают.
5. Способ по п. 4, в котором смешивание осуществляют при температуре от комнатной температуры до 90°С.
6. Способ по п. 4, в котором смешивание осуществляют посредством диффузии жидкость-жидкость или диффузии газ-жидкость.
7. Способ по п. 2, в котором воду добавляют к раствору.
8. Способ получения полиморфной формы фазы I по п. 1, в котором получают суспензию, включающую гидрохлорид 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина.
9. Способ по п. 8, в котором суспензию поддерживают при температуре от комнатной температуры до 80°С.
10. Способ получения полиморфной формы фазы II по п. 1, включающий:
a) растворение гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина в воде в присутствии каталитических количеств поли(винилового спирта), и
b) выпаривание воды.
11. Способ получения полиморфной формы фазы III по п. 1, включающий:
а) растворение гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина в воде или ацетоне в присутствии каталитических количеств поли(этиленгликоля), и
b) выпаривание воды или ацетона;
или включающий:
a) растворение гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина в воде в присутствии каталитических количеств поли(этиленгликоля), и
b) добавление диизопропилового спирта в качестве антирастворителя.
12. Способ получения полиморфной формы фазы IV по п. 1, включающий:
a) растворение гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина в хлороформе в присутствии каталитических количеств полимера, выбранного из группы, состоящей из поливинилпирролидона, поли(акриловой кислоты), полипропилена, поли(стирол-со-дивинилбензола), поли(тетрафторэтилена), поли(винилового спирта), полиакриламида и поли(метилметакрилата), и
b) добавление диизопропилового эфира в качестве антирастворителя.
13. Способ получения сольвата с диоксаном по п. 1, включающий способ, выбранный из:
а) размалывания с использованием капель растворителя, включающего:
- загрузку гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина вместе с каталитическими количествами диоксана в контейнер шаровой мельницы; и
- размалывание; и
b) кристаллизации из горячего насыщенного раствора диоксана.
14. Способ получения сольвата с хлороформом по п. 1, включающий:
a) растворение гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина в хлороформе в присутствии каталитических количеств полимера, выбранного из группы, состоящей из поли(этиленгликоля), поливинилпирролидона, поли(акриловой кислоты), нейлона 6/6, полипропилена, поли(тетрафторэтилена), поли(винилацетата), поли(винилового спирта), полиакриламида и полисульфона; и
b) выпаривание хлороформа или кристаллизацию в горячем насыщенном растворе хлороформа.
15. Применение полиморфной формы фазы II, полиморфной формы фазы III, полиморфной формы фазы IV, сольвата с диоксаном или сольвата с хлороформом по п. 1 для получения формы фазы I гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина.
16. Способ получения фазы I гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1Н-пиразол-3-ил]окси]этил]морфолина, включающий стадию нагревания кристаллических форм фазы II, фазы III и/или фазы IV указанного соединения при температуре от 140°С до 170°С.
17. Фармацевтическая композиция, обладающая активностью антагониста рецептора сигма-1 (σ-1), содержащая эффективное количество твердой формы по п. 1.
| WO 2006021462,A1, 02.03.2006 | |||
| УСТРОЙСТВО ДЛЯ ЗАПИРАНИЯ ВЫПУСКНОГО ОТВЕРСТИЯ КОНВЕРТЕРА | 1995 |
|
RU2113501C1 |
| WO 2007098953,A1,07.09.2007 | |||
| EP 0001829875,A1, 05.09.2007 | |||
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ СОСТОЯНИЙ БЕСПОКОЙСТВА, ПРОИЗВОДНЫЕ ПИПЕРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ СОСТОЯНИЙ БЕСПОКОЙСТВА ИЛИ ЭПИЛЕПСИИ | 1992 |
|
RU2142952C1 |

