Перекрестные ссылки на родственные заявки
В настоящей заявке заявлен приоритет предварительной патентной заявки US 61/767761, поданной 21 февраля 2013, которая включена в данное описание изобретения посредством ссылки во всей своей полноте.
Область изобретения
Настоящее изобретение относится к свободному основанию 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она, обладающему улучшенными физико-химическими свойствами. Изобретение также относится к фармацевтическим композициям и лекарственным формам, содержащим свободное основание, и к способам изготовления и применения таких соединений, композиций и лекарственных форм в лечении клеточно-пролиферативных заболеваний, таких как рак.
Предшествующий уровень техники
Соединение 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]пиримидин-7-он (также упоминаемое в данном описании изобретения как "соединение 1"), может быть представлено структурой:

и также известно как палбоциклиб или PD-0332991. Соединение 1 является сильным и селективным ингибитором CDK4 и CDK6.
Соединение 1, и его фармацевтически приемлемые соли раскрыты в международной публикации WO 2003/062236 и в патентах US 6936612, 7208489 и 7456168, где описано получение соединения 1 в виде его гидрохлоридной соли. В международной публикации WO 2005/005426 и в патентах US 7345171 и 7863278 описано получение свободного основания и различных солей присоединения моно- и дикислоты соединения 1, включая полиморфные формы изэтионатной соли. Способ получения соединения 1 в виде моно-изэтионатной соли описан в международной публикации WO 2008/032157 и в патенте US 7781583. Содержания каждой из вышеупомянутых ссылок включены в данное описание изобретения посредством ссылки во всей их полноте.
Хотя соединение 1 является сильным и селективным ингибитором CDK4/CDK6, его применение в качестве свободного основания представляло затруднения для фармацевтической разработки. Свободное основание, полученное посредством традиционных процедур разрушения солей, например, как в Примере 4 WO 2005/005426, было очень склонным к статической электризации и образовывало небольшие первичные частицы, которые агломерировались в большие твердые агломераты, которые было трудно диспергировать посредством просеивания, и которые были непригодны для дальнейшей обработки. В настоящем изобретении предлагается свободное основание соединения 1, имеющее более крупный размер первичных частиц, которое демонстрирует улучшенные физико-химические свойства и технологичность.
Краткое изложение сущности изобретения
Свободное основание соединения 1, 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]пиримидин-7-он, может существовать в одной или более полиморфных формах, включая Форму А и Форму В, где Форма А является более устойчивой кристаллической формой. Свободное основание может быть безводным или может содержать варьирующиеся количества воды или одного или более растворителей.
В настоящем изобретении предлагается кристаллическое свободное основание соединения 1, имеющее более крупный размер первичных частиц, значительно уменьшенную удельную площадь поверхности и более низкие показатели поверхностной энергии, чем свободное основание, полученное посредством традиционных методов разрушения солей, описанных в данной области техники. Свободное основание соединения 1 с большим размером частиц, раскрытое в данном описании изобретения можно отличить посредством ряда способов.
Полиморфные и твердые формы по изобретению могут быть распознаны посредством порошковой рентгеновской дифрактометрии (PXRD), ЯМР твердого тела (ssNMR), дифференциальной сканирующей калориметрии (DSC), вибрационной спектроскопии (например, ИК(инфракрасной) и Рамановской спектроскопии), микроскопии в поляризованном свете (PLM), сканирующей электронной микроскопии (SEM), оптической микроскопии с нагревательным столиком, электронной кристаллографии, рентгеновской дифрактометрии монокристаллов, количественного анализа, анализа размера частиц (PSA) (например размера частиц, распределения частиц по размерам (PSD) и формы частиц), анализа удельной площади поверхности (SSA), анализа поверхностной энергии (например, обращенной газовой хроматографией, или IGC), посредством исследований растворимости и исследований растворения, или посредством комбинации этих методов.
В одном аспекте изобретения предлагается кристаллическое свободное основание 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она, имеющее удельную поверхность менее или равную 2 м2/г. В некоторых воплощениях свободное основание имеет удельную поверхность менее или равную 1 м2/г.
В предпочтительных воплощениях кристаллическое свободное основание соединения 1 представляет собой полиморфную Форму А свободного основания. В таких воплощениях кристаллическое свободное основание имеет PXRD-дифрактограмму, содержащую пик при угле дифракции (2θ) 10,1±0,2. В других таких воплощениях кристаллическое свободное основание имеет PXRD-дифрактограмму, содержащую пики при углах дифракции (2θ) 8,0±0,2 и 10,1±0,2. В других воплощениях кристаллическое свободное основание имеет PXRD-дифрактограмму, содержащую пики при углах дифракции (2θ) 8,0±0,2, 10,1±0,2 и 11,5±0,2. В других воплощениях кристаллическое свободное основание имеет PXRD-дифрактограмму, содержащую пики при углах дифракции (2θ) 8,0±0,2, 10,1±0,2, 10,3±0,2 и 11,5±0,2. В других воплощениях кристаллическое свободное основание имеет PXRD-дифрактограмму, содержащую пики при углах дифракции (2θ), по существу, таких же, как показано на Фиг. 1.
В некоторых воплощениях кристаллическое свободное основание соединения 1 (Форма А) имеет спектр 13С ЯМР твердого тела (ssNMR), содержащий следующие резонансные значения (млн-1): 12,5 млн-1±0,2 млн-1. В других воплощениях кристаллическое свободное основание имеет спектр 13С ЯМР твердого тела, содержащий следующие резонансные значения (млн-1): 12,5 млн-1 и 112,4 млн-1±0,2 млн-1. В других воплощениях кристаллическое свободное основание имеет спектр 13С ЯМР твердого тела, содержащий следующие резонансные значения (млн-1): 12,5 млн-1 и 112,4 млн-1 и 143,2 млн-1±0,2 млн-1.
В некоторых воплощениях, описанных здесь, свободное основание соединения 1 по изобретению распознается посредством анализа размера частиц. В некоторых таких воплощениях кристаллическое свободное основание имеет размер первичных частиц от примерно 5 мкм до примерно 150 мкм, предпочтительно от примерно 10 мкм до примерно 100 мкм, или более предпочтительно от примерно 15 мкм до примерно 80 мкм. В других таких воплощениях кристаллическое свободное основание имеет распределение первичных частиц по размерам, характеризующееся: (1) величиной D10 от примерно 5 мкм до примерно 10 мкм; (2) величиной D50 от примерно 10 мкм до примерно 45 мкм; или (3) величиной D90 от примерно 30 мкм до примерно 125 мкм; или комбинацией (1), (2) и (3). В дополнительных воплощениях кристаллическое свободное основание имеет соотношение (D90-D10)/D50 распределения первичных частиц по размерам от примерно 2 до примерно 3. В дополнительных воплощениях кристаллическое свободное основание имеет средний объемный диаметр (D[4,3]) от примерно 15 мкм до примерно 125 мкм.
В некоторых воплощениях кристаллическое свободное основание соединения 1 является безводным. В других воплощениях кристаллическое свободное основание соединения 1 представляет собой сольват, в частности гидрат.
В другом аспекте изобретения предлагается фармацевтическая композиция, содержащая кристаллическое свободное основание соединения 1, имеющее большой размер первичных частиц, по изобретению, и фармацевтически приемлемый носитель, разбавитель или эксципиент. Часто фармацевтическая композиция содержит полиморфную Форму А свободного основания.
В изобретении дополнительно предлагается капсула, содержащая такую фармацевтическую композицию по изобретению. В некоторых таких воплощениях капсула содержит от 0,1 до 200 мг и, предпочтительно, от 25 до 150 мг свободного основания соединения 1 (предпочтительно в виде полиморфной Формы А), имеющего большой размер первичных частиц, как описано в данном описании изобретения.
В еще одном аспекте изобретения предложен способ лечения рака у млекопитающего, предпочтительно человека, включающий введение млекопитающему терапевтически эффективного количества фармацевтической композиции по изобретению. Способ лечения может дополнительно включать введение соединения 1 в комбинации с одним или более дополнительными терапевтическими агентами.
В других аспектах изобретения предлагаются способы получения свободного основания соединения 1 с большим размером первичных частиц, как описано в данном описании изобретения. Один способ включает растворение частиц свободного основания соединения 1 с небольшим размером в смеси первого растворителя и второго растворителя и нагревание до достижения растворения, охлаждение до подходящей температуры, введение затравочных кристаллов свободного основания соединения 1 (Формы А) с последующей кристаллизацией с получением свободного основания соединения 1 с большим размером частиц. Используемые в этом способе частицы свободного основания с небольшим размером могут быть выделены в традиционном способе разрушения солей, например путем кислотного гидролиза промежуточного винилового эфира с получением соли присоединения кислоты, с последующим подщелачиванием, как описано в Примере 5. Другой способ включает кислотный гидролиз промежуточного винилового эфира в смеси воды и первого растворителя, который может требовать нагревания для достижения растворения, добавление второго растворителя и подщелачивание с получением второй смеси, содержащей свободное основание, образованное in situ, нагревание, если требуется, для достижения растворения и отгонки воды, и получение затравочных кристаллов свободного основания соединения 1 (Формы А) при подходящей температуре, с последующей кристаллизацией с получением свободного основания соединения 1, имеющего большой размер первичных частиц. В изобретении дополнительно предлагается свободное основание соединения 1, полученное посредством этих способов, имеющее свойства, описанные в данном описании изобретения.
В каждом из вышеупомянутых способов первый растворитель представляет собой спирт, и второй растворитель представляет собой ароматический растворитель. Подходящие спирты включают, но без ограничения ими, относительно высококипящие спирты, такие как н-бутанол, трет-бутанол, н-пропанол, пентанол, 1,4-бутандиол или пропиленгликоль, и тому подобные. Подходящие ароматические растворители включают, но без ограничения ими, анизол, мезитилен, метта-ксилол, хлорбензол, пиридин, и тому подобные. Для увеличения выхода, эти способы могут включать нагревание или охлаждение до температур выше или ниже комнатной температуры. Часто реакционные смеси могут быть нагреты до температур, в интервале от примерно 30°С до примерно 150°С, и чаще от примерно 50°С до примерно 120°С, для достижения растворения. Во время кристаллизации может быть желательным охлаждение реакционной смеси до температуры, которая равна или ниже комнатной температуры, например от примерно 0°С до примерно 30°С, предпочтительно до примерно 5°С, до примерно 10°С, примерно 15°С или примерно 20°С.
Эти и другие аспекты и воплощения дополнительно описаны в подробном описании, предложенном здесь. Каждое из воплощений, описанных в данном описании изобретения, можно комбинировать с любым другим воплощением, описанным в данном описании изобретения, не противоречащим воплощению, с которым его комбинируют.
Краткое описание графических материалов
На Фиг. 1 показана PXRD-дифрактограмма полиморфной Формы А свободного основания соединения 1.
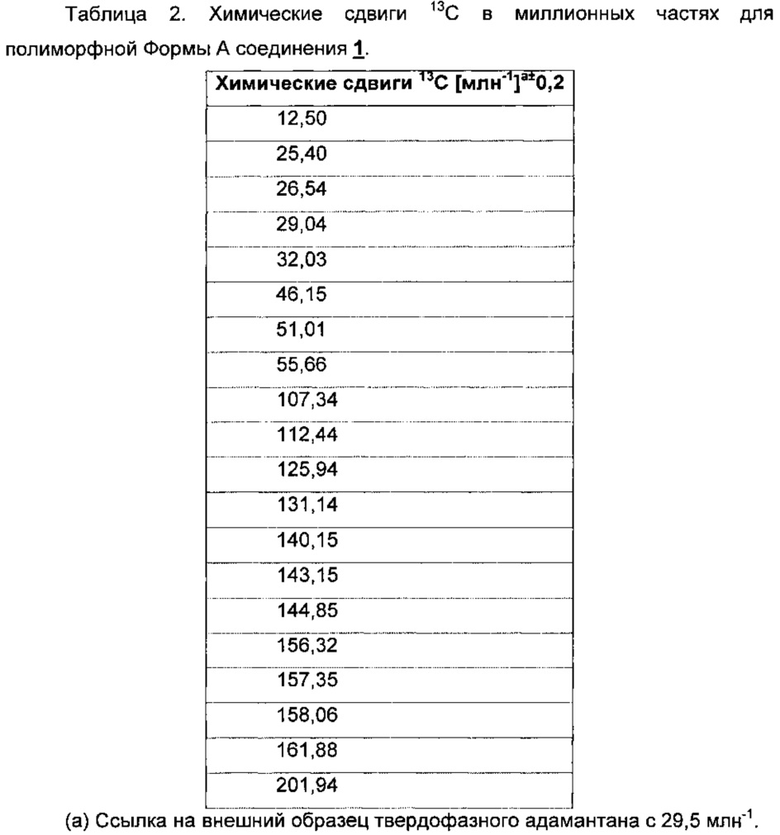
На Фиг. 2 показан спектр углеродной CPMAS (ЯМР с кросс-поляризацией и вращением образца под магическим углом) свободного основания соединения 1, полиморфной Формы А. Пики, отмеченные звездочками, представляют собой боковые полосы при вращении.
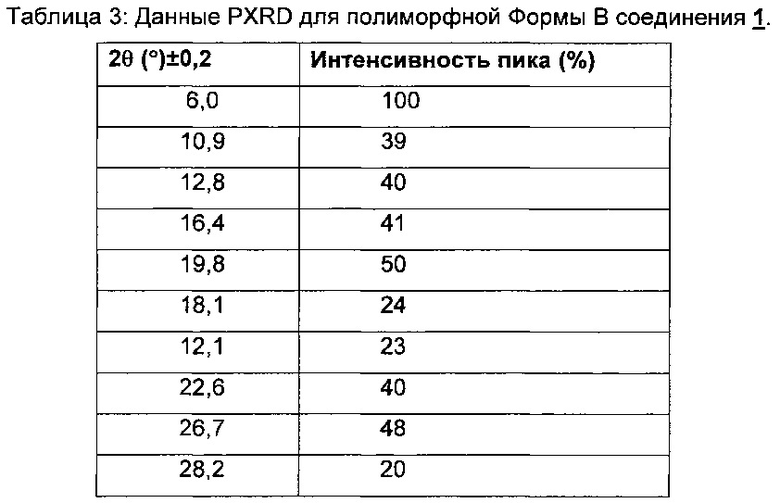
На Фиг. 3 показана PXRD-дифрактограмма свободного основания соединения 1, полиморфной Формы В.
На Фиг. 4 показан спектр углеродной CPMAS свободного основания соединения 1, полиморфной Формы В. Пики, отмеченные звездочками, представляют собой боковые полосы при вращении.
На Фиг. 5 показано полученное при помощи сканирующей электронной микроскопии (200х увеличение) изображение свободного основания соединения 1 полиморфной Формы A, API (активный фармацевтический ингредиент), перекристаллизованного из смеси 40% н-BuOH/анизол.
На Фиг. 6 показано полученное при помощи сканирующей электронной микроскопии (1500х увеличение) изображение свободного основания соединения 1, полиморфной Формы A, API, выделенного стандартным способом получения свободного основания.
На Фиг. 7 показано распределение размеров частиц свободного основания соединения 1, полиморфной Формы A, API, перекристаллизованного из смеси 40% н-BuOH/анизол.
На Фиг. 8 показано распределение по размерам частиц свободного основания соединения 1, полиморфной Формы A, API, выделенного стандартным способом получения свободного основания.
На Фиг. 9 показано полученное при помощи микроскопии в поляризованном свете (PLM) (200х увеличение) изображение свободного основания соединения 1, полиморфной Формы A, API, перекристаллизованного из смеси 40% н-BuOH/анизол.
Подробное описание изобретения
Настоящее изобретение можно понять более легко посредством ссылки на следующее подробное описание изобретения и Примеры, включенные в него. Следует понимать, что терминология, использованная в данном описании изобретения, предназначено только для целей описания конкретных воплощений и не предназначена для ограничения. Кроме того, следует понимать, что если в данном описании изобретения не определено конкретно, используемой здесь терминологии следует придавать ее традиционное значение, которое известно в релевантном уровне техники.
При использовании в данном описании изобретения форма единственного числа "a", "an" и "the" включает существительные во множественном числе, если не указано иное. Например, "а" заместитель включает один или более заместителей.
При использовании в данном описании изобретения термин "примерно" означает в пределах статистически значимого диапазона величины, такой как определенный диапазон концентрации, период времени, молекулярная масса, размер частиц, температура или рН. Такой диапазон может находиться в пределах порядка величины, обычно в пределах 20%, более типично в пределах 10%, и еще более типично в пределах 5% от указанной величины или диапазона. Иногда такой диапазон может находиться в пределах типичной погрешности эксперимента в стандартных способах, используемых для измерения и/или определения заданной величины или диапазона. Допустимое отклонение, охватываемое термином "примерно", будет зависеть от конкретной исследуемой системы и легко может быть оценено специалистом в данной области техники. Всякий раз, когда в данной заявке описан диапазон, каждое целое число в пределах диапазона также рассматривается как воплощение изобретения.
При использовании в данном описании изобретения, если не указано иное, термин "аномальный рост клеток" относится к росту клеток, который не зависит от нормальных регуляторных механизмов (например, к потере контактного ингибирования). "Аномальные клеточно-пролиферативные заболевания" представляют собой заболевания, характеризующиеся аномальным ростом клеток, например рак.
Термин "рак" включает как солидные опухоли, так и гематологические злокачественные новообразования. Раковые заболевания включают, но без ограничения ими, рак молочной железы, рак яичника, рак шейки матки, рак эндометрия, рак предстательной железы, рак яичек, рак поджелудочной железы, рак пищевода, рак головы и шеи, рак желудка, рак мочевого пузыря, рак легкого (например, аденокарциному, NSCLC (немелкоклеточный рак легкого) и SCLC (мелкоклеточный рак легкого)), рак кости (например, остеосаркому), рак толстой кишки, рак прямой кишки, рак щитовидной железы, раковые заболевания головного мозга и центральной нервной системы, глиобластому, нейробластому, нейроэндокринный рак, рабдоидный рак, кератоакантому, плоскоклеточный рак, семиному, меланому, саркому (например, липосаркому), рак мочевого пузыря, рак печени (например, печеночно-клеточный рак), рак почек (например, гипернефрому), миелоидные расстройства (например, AML (острый миелобластный лейкоз), CML (хронический миелолейкоз), миелодипластический синдром и промиелоцитарный лейкоз) и лимфоидные расстройства (например, лейкоз, множественную миелому, мантийноклеточную лимфому, ALL (острый лимфоидный лейкоз), CLL (хронический лимфоидный лейкоз), В-клеточную лимфому, Т-клеточную лимфому, лимфому Ходжкина, неходжкинскую лимфому, лимфому волосистых клеток).
Выражение "фармацевтически приемлемый" относится к веществам, которые в рамках обоснованного медицинского суждения подходят для применения в контакте с тканями пациентов без чрезмерной токсичности, раздражения, аллергической реакции и тому подобного, соответствуют разумному соотношению польза/риск и эффективны при использовании их по назначению.
Термин "млекопитающее" при использовании в данном описании изобретения, может представлять собой человека или другое млекопитающее (например, собаку, кошку, кролика, крысу, мышь, лошадь, обезьяну, другого примата низшего класса и так далее). Предпочтительно млекопитающее представляет собой человека.
При использовании в данном описании изобретения, если не указано иное, термин "лечение" означает реверсию, облегчение, замедление развития, или предупреждение расстройства или состояния, к которому применяется такой термин, или одного или более симптомов такого расстройства или состояния. Термин "лечение", при использовании в данном описании изобретения, если не указано иное, относится к акту лечения, где "лечение" определено непосредственно выше.
При использовании в данном описании изобретения "эффективное" количество относится к количеству соединения, агента, вещества, препарата или композиции, которое достаточно, чтобы привести к снижению серьезности симптомов заболевания, увеличению частоты и продолжительности периодов без симптомов заболевания, или к предупреждению ухудшения или нетрудоспособности из-за болезненного недуга. Это количество может быть представлено в виде однократной дозы или согласно режиму многократного приема, одного или в комбинации с другими соединениями, агентами или веществами. Специалист в данной области техники может определить такие количества на основе таких факторов, как величина пациента, серьезность симптомов у пациента, и конкретная композиция или выбранный путь введения.
"Стандартная лекарственная форма" при использовании в данном описании изобретения относится к физически обособленной единице композиции по изобретению, подходящей для пациента, подлежащего лечению. Понятно, однако, что общее суточное применение композиций по настоящему изобретению будет определяться лечащим врачом в рамках обоснованного медицинского суждения. Конкретный эффективный уровень дозы для любого конкретного пациента будет зависеть от ряда факторов, включающих расстройство, подлежащее лечению, и серьезность расстройства; конкретную применяемую композицию; возраст, массу тела, общее состояние здоровья, пол и питание пациента; время введения, продолжительность лечения; лекарственные средства и/или дополнительные виды терапии, применяемые в комбинации или совмещаемые с композициями по изобретению, и подобных факторов хорошо известных в медицинских навыках.
При использовании в данном описании изобретения термин "по существу такой же" со ссылкой на положения пиков дифракции рентгеновских лучей означает, что во внимание приняты типичное положение пика и колебания интенсивности. Например, специалисту в данной области техники будет ясно, что положения пиков (20) будут показывать некоторую изменчивость в разных аппаратах, обычно вплоть до 0,2° или 0,1°. Кроме того, специалисту в данной области техники будет ясно, что относительные интенсивности пиков будут показывать изменчивость в различных аппаратах, а также изменчивость, обусловленную степенью кристалличности, предпочтительной ориентацией, подготовленной поверхностью образца и другими факторами, известными специалисту в данной области техники, и их следует воспринимать только как качественные показатели.
Термин "сольват" при использовании в данном описании изобретения относится к кристаллической форме вещества, которая содержит растворитель. Термин "гидрат" относится к сольвату, где растворитель представляет собой воду.
Термин "введение затравки" при использовании в данном описании изобретения означает добавление кристаллов в систему кристаллизации для инициирования или усиления образования центров кристаллизации или действия в качестве субстрата для дополнительной кристаллизации.
При использовании в данном описании изобретения термин "API" или "активный фармацевтический ингредиент" относится к свободному основанию 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она.
При использовании в данном описании изобретения термин "первичные частицы" относится к отдельным кристаллам API.
При использовании в данном описании изобретения термин "агломераты" относится к прочно связанным кристаллам API, которые трудно раздробить на первичные частицы во время обработки и анализа размеров частиц.
В настоящем изобретении предлагается свободное основание соединения 1, имеющее более крупный размер первичных частиц, значительно уменьшенную удельную площадь поверхности и более низкие показатели поверхностной энергии, чем свободное основание, полученное посредством традиционных способов разрушения солей. Для удобства свободное основание соединения 1, предложенное в изобретении, иногда может упоминаться в данном описании изобретения как свободное основание с "большим размером (первичных) частиц". В этом состоит отличие от свободного основания соединения 1, полученного посредством традиционных способов разрушения солей, которое иногда называется свободным основанием с "небольшим размером (первичных) частиц". Специалисту в данной области техники понятно, что ссылка на "небольшой размер частиц" в данном случае относится к размеру частиц отдельных кристаллов API, и склонность "небольших" частиц образовывать большие агломераты не принимается во внимание.
В некоторых воплощениях изобретения, описанных здесь, кристаллическое свободное основание соединения 1 отличается удельной площадью поверхности (SSA). Таким образом, в одном аспекте изобретения предлагается кристаллическое свободное основание 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она, имеющее удельную площадь поверхности (SSA) меньше или равную 2 м2/г. В некоторых воплощениях свободное основание имеет удельную площадь поверхности (SSA) меньше или равную 1 м2/г. В других воплощениях свободное основание соединения 1 имеет SSA≤0,9 м2/г, ≤0,8 м2/г или ≤0,7 м2/г. В других воплощениях свободное основание соединения 1 имеет SSA от 0,2 м2/г до 2 м2/г, от 0,5 м2/г до 1,5 м2/г, или от 0,5 м2/г до 1 м2/г.
В некоторых воплощениях, описанных здесь, кристаллическое свободное основание соединения 1 отличается дисперсионной поверхностной энергией. Таким образом, в одном аспекте изобретения предлагается кристаллическое свободное основание 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она, имеющее дисперсионную поверхностную энергию меньше или равную 60 мДж/м2. В некоторых воплощениях свободное основание имеет дисперсионную поверхностную энергию ≤55 мДж/м2, ≤50 мДж/м2, ≤45 мДж/м2 или ≤40 мДж/м2. В дополнительных воплощениях свободное основание соединения 1 имеет дисперсионную поверхностную энергию от 20 мДж/м2 до 60 мДж/м2, от 25 мДж/м2 до 50 мДж/м2 или от 30 мДж/м2 до 50 мДж/м2.
В предпочтительных воплощениях кристаллическое свободное основание соединения 1 представляет собой полиморфную Форму А свободного основания. В некоторых таких воплощениях кристаллическая форма имеет PXRD-дифрактограмму, содержащую пик с углом дифракции (2θ) 10,1±0,2. В других таких воплощениях кристаллическая форма имеет PXRD-дифрактограмму, содержащую пики с углами дифракции (2θ) 8,0±0,2 и 10,1±0,2. В других воплощениях кристаллическая форма имеет PXRD-дифрактограмму, содержащую пики с углами дифракции (2θ) 8,0±0,2, 10,1±0,2 и 11,5±0,2. В других воплощениях кристаллическая форма имеет PXRD-дифрактограмму, содержащую пики с углами дифракции (2θ) 8,0±0,2, 10,1±0,2, 10,3±0,2 и 11,5±0,2. В других воплощениях кристаллическая форма имеет PXRD-дифрактограмму, содержащую пики с углами дифракции (2θ) 5,1±0,2, 8,0±0,2, 10,1±0,2 и 11,5±0,2. В других воплощениях кристаллическая форма имеет PXRD-дифрактограмму, содержащую пики с углами дифракции (2θ) 8,0±0,2, 10,1±0,2, 11,5±0,2 и 19,7±0,2. В других воплощениях кристаллическая форма имеет PXRD-дифрактограмму, содержащую пики с углами дифракции (2θ) 8,0±0,2, 10,1±0,2, 11,5±0,2, и 22,5±0,2. В других воплощениях кристаллическая форма имеет PXRD-дифрактограмму, содержащую пики с углами дифракции (2θ), по существу такими же, как показано на Фиг. 1.
В некоторых воплощениях кристаллическое свободное основание соединения 1 (Форма А) имеет спектр 13С ЯМР твердого тела, содержащий следующие резонансные значения (млн-1): 12,5 млн-1±0,2 млн-1. В других воплощениях кристаллическая форма имеет спектр 13С ЯМР твердого тела, содержащий следующие резонансные значения (млн-1): 12,5 млн-1 и 112,4 млн-1±0,2 млн-1. В других воплощениях кристаллическая форма имеет спектр 13С ЯМР твердого тела, содержащий следующие резонансные значения (млн-1): или 12,5 млн-1, 112,4 млн-1 и 143,2 млн-1±0,2 млн-1.
В некоторых воплощениях, описанных здесь, кристаллическое свободное основание соединения 1 можно отличить посредством анализа размера частиц. В некоторых таких воплощениях свободное основание имеет размер первичных частиц от примерно 5 мкм до примерно 150 мкм, предпочтительно от примерно 10 мкм до примерно 100 мкм, и более предпочтительно от примерно 15 мкм до примерно 80 мкм.
В других таких воплощениях свободное основание имеет распределение первичных частиц по размерам, характеризующееся: (1) величиной D10 от примерно 5 мкм до примерно 10 мкм; (2) величиной D50 от примерно 10 мкм до примерно 45 мкм; или (3) величиной D90 от примерно 30 мкм до примерно 125 мкм; или комбинацией (1), (2) и (3). В дополнительных воплощениях свободное основание имеет соотношение (D90-D10)/D50 в распределении первичных частиц по размерам от примерно 2 до примерно 3. В других воплощениях свободное основание имеет средний объемный диаметр (D[4,3]) от примерно 15 мкм до примерно 125 мкм.
В одном аспекте изобретения предлагается кристаллическое свободное основание 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]пиримидин-7-он, имеющее размер первичных частиц больше примерно 5 мкм. В некоторых воплощениях свободное основание имеет размер первичных частиц больше примерно 7,5 мкм. В других воплощениях свободное основание имеет размер первичных частиц больше примерно 10 мкм. В других таких воплощениях свободное основание имеет размер первичных частиц больше примерно 12,5 мкм. В других таких воплощениях свободное основание имеет размер первичных частиц больше примерно 15 мкм.
В другом аспекте изобретения предлагается кристаллическое свободное основание 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-он, имеющее размер первичных частиц от примерно 5 мкм до примерно 200 мкм. В некоторых воплощениях это свободное основание имеет размер первичных частиц: от примерно 5 мкм до примерно 175 мкм; от примерно 5 мкм до примерно 150 мкм; от примерно 5 мкм до примерно 125 мкм; от примерно 5 мкм до примерно 100 мкм; от примерно 5 мкм до примерно 75 мкм; от примерно 10 мкм до примерно 200 мкм; от примерно 10 мкм до примерно 175 мкм; от примерно 10 мкм до примерно 150 мкм; от примерно 10 мкм до примерно 125 мкм; от примерно 10 мкм до примерно 100 мкм; от примерно 10 мкм до примерно 75 мкм; от примерно 15 мкм до примерно 200 мкм; от примерно 15 мкм до примерно 175 мкм; от примерно 15 мкм до примерно 150 мкм; от примерно 15 мкм до примерно 125 мкм; от примерно 15 мкм до примерно 100 мкм; или от примерно 15 мкм до примерно 75 мкм.
В еще одном аспекте изобретения предлагается кристаллическое свободное основание 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1 -ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-он с распределением первичных частиц по размерам, имеющим по меньшей мере одно из:
а) величину D10 от примерно 5 мкм до примерно 10 мкм;
б) величину D50 от примерно 10 мкм до примерно 45 мкм; и
в) величину D90 от примерно 30 мкм до примерно 125 мкм.
В некоторых таких воплощениях свободное основание имеет величину D10 от примерно 5 мкм до примерно 10 мкм. В других таких воплощениях свободное основание имеет величину D90 от примерно 30 мкм до примерно 125 мкм. В других таких воплощениях свободное основание имеет величину D50 от примерно 10 мкм до примерно 45 мкм. В некоторых таких воплощениях свободное основание имеет D10 величину от примерно 5 мкм до примерно 10 мкм и величину D90 от примерно 30 мкм до примерно 125 мкм. В других воплощениях свободное основание имеет величину D10 от примерно 5 мкм до примерно 10 мкм, величину D90 от примерно 30 мкм до примерно 125 мкм и величину D50 от примерно 10 мкм до примерно 45 мкм.
В другом аспекте изобретения предлагается кристаллическое свободное основание 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2- иламино)-8H-пиридо[2,3-d]пиримидин-7-он с распределением первичных частиц по размерам, имеющим по меньшей мере одно из:
г) величину D10 от примерно 5 мкм до примерно 10 мкм;
д) величину D50 от примерно 10 мкм до примерно 25 мкм; и
е) величину D90 от примерно 30 мкм до примерно 75 мкм.
В некоторых таких воплощениях свободное основание имеет величину D10 от примерно 5 мкм до примерно 10 мкм. В других таких воплощениях свободное основание имеет величину D90 от примерно 30 мкм до примерно 75 мкм. В других таких воплощениях свободное основание имеет величину D50 от примерно 10 мкм до примерно 25 мкм. В некоторых таких воплощениях свободное основание имеет величину D10 от примерно 5 мкм до примерно 10 мкм и величину D90 от примерно 30 мкм до примерно 75 мкм. В других воплощениях свободное основание имеет величину D10 от примерно 5 мкм до примерно 10 мкм, величину D90 от примерно 30 мкм до примерно 755 мкм и величину D50 от примерно 10 мкм до примерно 25 мкм.
В других воплощениях свободное основание имеет распределение первичных частиц по размерам с величиной D10 от примерно 5 мкм до примерно 7,5 мкм; от примерно 5 мкм до примерно 10 мкм; от примерно 5 мкм до примерно 12,5 мкм; или от примерно 5 мкм до примерно 15 мкм.
В других воплощениях свободное основание имеет распределение первичных частиц по размерам с величиной D50: от примерно 10 мкм до примерно 50 мкм; от примерно 10 мкм до примерно 45 мкм; от примерно 10 мкм до примерно 40 мкм; от примерно 10 мкм до примерно 35 мкм; от примерно 10 мкм до примерно 30 мкм; от примерно 10 мкм до примерно 25 мкм; или от примерно 10 мкм до примерно 20 мкм.
В других воплощениях свободное основание имеет распределение первичных частиц по размерам с величиной D90: от примерно 30 мкм до примерно 175 мкм; от примерно 30 мкм до примерно 160 мкм; от примерно 30 мкм до примерно 150 мкм; от примерно 30 мкм до примерно 140 мкм; от примерно 30 мкм до примерно 130 мкм; от примерно 30 мкм до примерно 125 мкм; от примерно 30 мкм до примерно 120 мкм; от примерно 30 мкм до примерно 115 мкм; от примерно 30 мкм до примерно 110 мкм; от примерно 30 мкм до примерно 100 мкм; от примерно 30 мкм до примерно 75 мкм; от примерно 30 мкм до примерно 70 мкм; от примерно 30 мкм до примерно 65 мкм; от примерно 30 мкм до примерно 60 мкм; от примерно 30 мкм до примерно 55 мкм; от примерно 30 мкм до примерно 50 мкм; или от примерно 30 мкм до примерно 45 мкм.
Каждое из вышеупомянутых значений для воплощений D10 можно комбинировать с любым значением D50 и/или значением D90, не противоречащим ему. Каждое из вышеупомянутых значений для воплощений D50 можно комбинировать с любым значением D10 и/или значением D90, не противоречащей ему. Каждое из вышеупомянутых значений для воплощений D90 можно комбинировать с любым значением D10 и/или значением D50, не противоречащим ему.
В еще одном аспекте изобретения предлагается кристаллическое свободное основание 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]пиримидин-7-он, имеющее соотношение (D90-D10)/D50 в распределении первичных частиц по размерам от примерно 2 до примерно 3. В некоторых таких воплощениях свободное основание имеет размер первичных частиц от примерно 5 мкм до примерно 150 мкм.
В некоторых воплощениях этого аспекта свободное основание имеет соотношение (D90-D10)/D50 в распределении первичных частиц по размеру: от примерно 2 до примерно 2,75; от примерно 2 до примерно 2,5; от примерно 2 до примерно 2,25. В других воплощениях соотношение равно примерно 2,0, примерно 2,1, примерно 2,2, примерно 2,3, примерно 2,4, примерно 2,5, примерно 2,6, примерно 2,7, примерно 2,8, примерно 2, или примерно 3,0.
В еще одном аспекте изобретения предлагается кристаллическое свободное основание 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-он, имеющее средний объемный диаметр (D[4,3]) от примерно 15 мкм до примерно 125 мкм. В некоторых воплощениях свободное основание имеет D[4,3] от примерно 50 мкм до примерно 100 мкм. В других воплощениях свободное основание имеет D[4,3] от примерно 15 мкм до примерно 30 мкм.
В других воплощениях свободное основание имеет D[4,3]: от примерно 15 мкм до примерно 100 мкм; от примерно 15 мкм до примерно 90 мкм; от примерно 15 мкм до примерно 80 мкм; от примерно 15 мкм до примерно 70 мкм; от примерно 15 мкм до примерно 60 мкм; от примерно 15 мкм до примерно 50 мкм; от примерно 15 мкм до примерно 40 мкм; от примерно 25 мкм до примерно 120 мкм; от примерно 25 мкм до примерно 100 мкм; от примерно 25 мкм до примерно 90 мкм; от примерно 25 мкм до примерно 80 мкм; от примерно 25 мкм до примерно 70 мкм; от примерно 25 мкм до примерно 60 мкм; от примерно 25 мкм до примерно 50 мкм; от примерно 25 мкм до примерно 40 мкм; примерно 25 мкм; примерно 30 мкм; примерно 35 мкм; примерно 40 мкм; примерно 45 мкм; примерно 50 мкм; примерно 55 мкм; примерно 60 мкм; примерно 65 мкм; примерно 70 мкм; примерно 75 мкм; до примерно 80 мкм; примерно 90 мкм; примерно 100 мкм; примерно 105 мкм; примерно 110 мкм; примерно 115 мкм; или примерно 120 мкм.
В еще одном аспекте изобретения предлагается фармацевтическая композиция, содержащая свободное основание по изобретению и фармацевтически приемлемый носитель, разбавитель или эксципиент. В изобретении, кроме того, предлагается капсула, содержащая такую фармацевтическую композицию по изобретению.
В некоторых воплощениях капсула содержит от 0,1 до 200 мг полиморфной Формы А 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-с0пиримидин-7-она. В других воплощениях капсула содержит от 25 до 150 мг полиморфной Формы А 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она. В других воплощениях капсула содержит от 50 до 150 мг полиморфной Формы А 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]пиримидин-7-она. В других воплощениях капсула содержит от 50 до 100 мг полиморфной Формы А 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она. В других воплощениях капсула содержит от 75 до 150 мг полиморфной Формы А 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она.
В еще одном аспекте изобретения предлагается способ лечения рака у млекопитающего, в том числе человека, включающий введение млекопитающему терапевтически эффективного количества фармацевтической композиции по изобретению. В некоторых таких воплощениях фармацевтическую композицию вводят в капсуле. Капсула может содержать от 0,1 до 200 мг полиморфной Формы А свободного основания 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d] пиримидин-7-она. В других воплощениях капсула может содержать от 25 до 150 мг полиморфной Формы А свободного основания 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она. В других воплощениях капсула может содержать от 50 до 150 мг полиморфной Формы А свободного основания 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она.
Методы характеристики кристаллического свободного основания соединения 1 по изобретению включают, но без ограничения ими, порошковую рентгеновскую дифрактометрию (PXRD), ЯМР твердого тела (ssNMR), дифференциальную сканирующую калориметрию (ДСК), вибрационную спектроскопию (например, ИК и Рамановскую спектроскопию), микроскопию в поляризованном свете (PLM), сканирующую электронную микроскопию (SEM), оптическую микроскопию с нагревательным столиком, электронную кристаллографию, рентгеновскую дифрактометрию монокристаллов, количественный анализ, анализ размеров частиц (PSA) (например размера частиц, распределения частиц по размерам (PSD) и формы частиц), анализ удельной поверхности (SSA), анализ поверхностной энергии (например, обращенная газовая хроматография или IGC), посредством исследований растворимости и исследований растворения, или посредством комбинации этих методов.
В других аспектах изобретения предлагаются способы получения свободного основания соединения 1, имеющего большой размер первичных частиц, как описано в данном описании изобретения. Один способ включает растворение частиц свободного основания соединения 1 небольшого размера в смеси первого растворителя и второго растворителя и нагревание до достижения растворения, охлаждение до подходящей температуры, обеспечение затравки кристаллов свободного основания соединения 1 (Форма А) с последующей кристаллизацией с получением частиц свободного основания соединения 1 большого размера. Частицы свободного основания с небольшим размером, используемые в этом способе, могут быть выделены традиционным способом разрушения солей, например, посредством кислотного гидролиза промежуточного винилового эфира с получением соли присоединения кислоты с последующим подщелачиванием, как описано в Примере 5.
В одном воплощении изобретения предлагается способ получения частиц свободного основания 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]пиримидин-7-она (Формы А) большого размера, включающий: (а) суспендирование свободного основания 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она в смеси первого растворителя и второго растворителя и нагревание до достижения растворения; (б) охлаждение до подходящей температуры и обеспечение затравки кристаллов свободного основания 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]пиримидин-7-она (Формы А); (в) постепенное охлаждение смеси для достижения кристаллизации; и (г) выделение свободного основания 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-s]пиримидин-7-она (Формы А), имеющего большой размер частиц.
В еще одном воплощении в изобретении предлагается способ получения частиц свободного основания 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она (Формы А) большого размера, включающий: (а) суспендирование свободного основания 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]пиримидин-7-она в смеси н-бутанола и анизола и нагревание до примерно 95-100°С до достижения растворения; (б) охлаждение до примерно 80°С и обеспечение затравки кристаллов свободного основания (Формы А) 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]пиримидин-7-она; (в) поддержание смеси примерно при 80°С в течение примерно 3 часов и затем постепенное охлаждение до примерно 10°С с достижением кристаллизации; и (г) фильтрование для выделения свободного основания (Формы А) 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она, имеющего большой размер частиц.
Другой способ включает кислотный гидролиз промежуточного винилового эфира в смеси воды и первого растворителя, что может требовать нагревания для достижения растворения, добавление второго растворителя и подщелачивание с получением второй смеси, содержащей свободное основание, образованное in situ, нагревание, если требуется, для достижения растворения и для отгонки воды, охлаждение до подходящей температуры, обеспечение затравки кристаллов свободного основания (Формы А) соединения 1 с последующей кристаллизацией с получением свободного основания соединения 1, имеющего большой размер первичных частиц.
В одном воплощении изобретения предлагается способ получения частиц свободного основания 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1 -ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она (Формы А) большого размера, включающий: (а) суспендирование трет-бутилового эфира 4-{6-[6-(1-бутоксил-винил)-8-циклопентил-5-метил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-иламино]-пиридин-3-ил}-пиперазин-1-карбоновой кислоты в смеси воды и первого растворителя и нагревание для достижения растворения; (б) добавление кислоты и взаимодействие с получением соли присоединения кислоты к 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]-пиримидин-7-ону in situ; (в) добавление второго растворителя и водного основания до достижения рН больше или равного 10; (г) отделение органического слоя и нагревание для отгонки воды; (д) охлаждение до подходящей температуры и обеспечение кристаллов затравки свободного основания (Формы А) 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она; (е) постепенное охлаждение смеси до достижения кристаллизации; и (ж) выделение свободного основания (Формы А) 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она, имеющего большой размер частиц.
В другом воплощении изобретения предлагается способ получения частиц свободного основания (Формы А) 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она большого размера, включающий: (а) суспендирование трет-бутилового эфира 4-{6-[6-(1-бутоксил-винил)-8-циклопентил-5-метил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-иламино]-пиридин-3-ил}-пиперазин-1-карбоновой кислоты в смеси воды и н-бутанола и нагревание до примерно 70°С для достижения растворения; (б) добавление концентрированной HCl и нагревание примерно при 70°С в течение 4-6 часов; (в) добавление анизола и водного NaOH с получением двухфазной смеси, имеющей рН выше 10; (г) разделение слоев и нагревание органического слоя до примерно 120°С для отгонки воды; (д) охлаждение до примерно 80°С и обеспечение кристаллов затравки свободного основания (Формы А) 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]пиримидин-7-она; (е) поддержание смеси примерно при 80°С в течение примерно 3 часов и затем постепенное охлаждение до примерно 10°С для достижения кристаллизации; и (ж) фильтрование с выделением свободного основания 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она (Формы А), имеющего большой размер частиц.
В некоторых воплощениях каждого из вышеупомянутых способов способ обеспечивает получение свободного основания соединения 1, имеющего удельную поверхность меньше или равную 2 м2/г. В других воплощениях каждого из вышеупомянутых способов способ обеспечивает получение свободного основания соединения 1, имеющего удельную поверхность меньше или равную 1 м2/г. В других воплощениях каждого из вышеупомянутых способов способ обеспечивает получение свободного основания соединения 1, имеющего размер первичных частиц от примерно 5 мкм до примерно 150 мкм, предпочтительно от примерно 10 мкм до примерно 100 мкм и более предпочтительно от примерно 15 мкм до примерно 80 мкм. В других воплощениях каждого из вышеупомянутых способов способ обеспечивает получение свободного основания соединения 1, имеющего распределение первичных частиц по размерам, характеризующееся: (1) величиной D10 от примерно 5 мкм до примерно 10 мкм; (2) величиной D90 от примерно 30 мкм до примерно 125 мкм; или (3) величиной D50 от примерно 10 мкм до примерно 45 мкм; или комбинацией (1), (2) и (3). В других воплощениях каждого из вышеупомянутых способов способ обеспечивает получение свободного основания соединения 1, имеющего соотношение (D90-D10)/D50 в распределении первичных частиц по размерам от примерно 2 до примерно 3. В других воплощениях каждого из вышеупомянутых способов способ обеспечивает получение свободного основания соединения 1, имеющего средний объемный диаметр (D[4,3]) от примерно 15 мкм до примерно 125 мкм.
В другом аспекте изобретения предлагается свободное основание соединения 1, как описано в данном описании изобретения, полученное в соответствии с одним из этих способов. В некоторых воплощениях изобретения предлагается кристаллическое свободное основание 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8Н-пиридо[2,3-d]пиримидин-7-она (Форма А), полученное в соответствии с любым из способов, описанных в данном описании изобретения. В некоторых таких воплощениях свободное основание, полученное способами, описанными здесь, может быть охарактеризовано при помощи его SSA, PSA или поверхностной энергии, или посредством комбинации этих способов, самих по себе или в дополнительной комбинации с PXRD или ssNMR. В некоторых таких воплощениях кристаллическое свободное основание имеет остаточное содержание растворителя от 0,05 до 0,25% масс, анизола и/или от 0,05 до 0,25% масс, н-бутанола. В других таких воплощениях кристаллическое свободное основание имеет остаточное содержание растворителя менее или равное 0,5% масс. анизола и менее или равное 0,5% масс. н-бутанола и предпочтительно менее или равное 0,25% масс. анизола и менее или равное 0,25% масс. н-бутанола.
В каждом из указанных выше способов первый растворитель представляет собой спирт, и второй растворитель представляет собой ароматический растворитель. Подходящие спирты включают, но без ограничения ими, относительно высококипящие спирты, такие как н-бутанол, трет-бутанол, н-пропанол, пентанол, 1,4-бутандиол или пропиленгликоль и подобные. Подходящие ароматические растворители включают, но без ограничения, анизол, мезитилен, мета-ксилол, хлорбензол, пиридин и подобные.
В некоторых таких воплощениях смесь растворителей содержит 10% спирта, 15% спирта, 20% спирта, 25% спирта, 30% спирта, 35% спирта, 40% спирта, 45% спирта, 50% спирта, 60% спирта, 70% спирта или более 70% спирта, где остальной частью является ароматический растворитель. В других таких воплощениях смесь растворителей содержит 90% ароматического соединения, 85% ароматического соединения, 80% ароматического соединения, 75% ароматического соединения, 70% ароматического соединения, 65% ароматического соединения, 60% ароматического соединения, 55% ароматического соединения, 50% ароматические, 40% ароматические, 30% ароматические, или менее 30% ароматического соединения, где остальной частью является спиртовой растворитель.
В одном предпочтительном воплощении первый растворитель представляет собой н-бутанол. В другом предпочтительном воплощении второй растворитель представляет собой анизол. В особенно предпочтительном воплощении первый растворитель представляет собой н-бутанол и второй растворитель представляет собой анизол. В некоторых таких воплощениях смесь растворителей содержит 10% н-бутанол/анизол, 15% н-бутанол/анизол, 20% н-бутанол/анизол, 25% н-бутанол/анизол, 30% н-бутанол/анизол, 35% н-бутанол/анизол, 40% н-бутанол/анизол, 45% н-бутанол/анизол, 50% н-бутанол/анизол, 60% н-бутанол/анизол, 70% н-бутанол/анизол или более 70% н-бутанол/анизол. В некоторых предпочтительных воплощениях смесь растворителей содержит от примерно 20 до примерно 50% н-бутанол/анизол. В особенно предпочтительном воплощении смесь растворителей содержит примерно 40% н-бутанол/анизол.
Для улучшения выходов способы могут включать нагревание или охлаждение до температур выше или ниже комнатной температуры. Часто для достижения растворения реакционные смеси могут быть нагреты до температур в диапазоне от примерно 30°С до примерно 150°С и еще чаще от примерно 50°С до примерно 120°С. Во время кристаллизации может быть желательно охлаждать реакционную смесь до температуры, которая равна или ниже комнатной температуры, например от примерно 0°С до примерно 30°С, предпочтительно до примерно 5°С, примерно 10°С, примерно 15°С или примерно 20°С.
В дополнительных воплощениях свободное основание соединения 1 представляет собой полиморфную Форму А, имеющую порошковую рентгеновскую дифрактограмму, содержащую пик с углом дифракции (2θ) 10,1±0,2. В других воплощениях кристаллическая форма имеет порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ) 10,1±0,2 и 22,5±0,2. В других воплощениях этого аспекта кристаллическая форма имеет порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ) 5,1±0,2, 10,1±0,2 и 22,5±0,2. В других воплощениях кристаллическая форма имеет порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ) 5,1±0,2, 10,1±0,2, 19,7±0,2 и 22,5±0,2. В других воплощениях кристаллическая форма имеет порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ) 5,1±0,2, 10,1±0,2, 17,1±0,2, 19,7±0,2 и 22,5±0,2. В дополнительных воплощениях кристаллическая форма имеет порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ) 5,1±0,2, 10,1±0,2, 11,5±0,2, 17,1±0,2, 19,7±0,2 и 22,5±0,2. В других воплощениях кристаллическая форма имеет порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ) 5,1±0,2, 10,1±0,2, 11,5±0,2, 17,1±0,2, 18,7±0,2, 19,7±0,2 и 22,5±0,2. В некоторых воплощениях этого аспекта кристаллическая форма имеет дифрактограмму дифракции рентгеновских лучей на порошке (PXRD), содержащую пики с углами дифракции (2θ) по существу такими же, как показано на Фиг. 1.
Дифрактограмма рентгеновских лучей на порошке (PXRD) полиморфной Формы А свободного основания показана на Фиг. 1 и соответствующие данные представлены в Таблице 1.
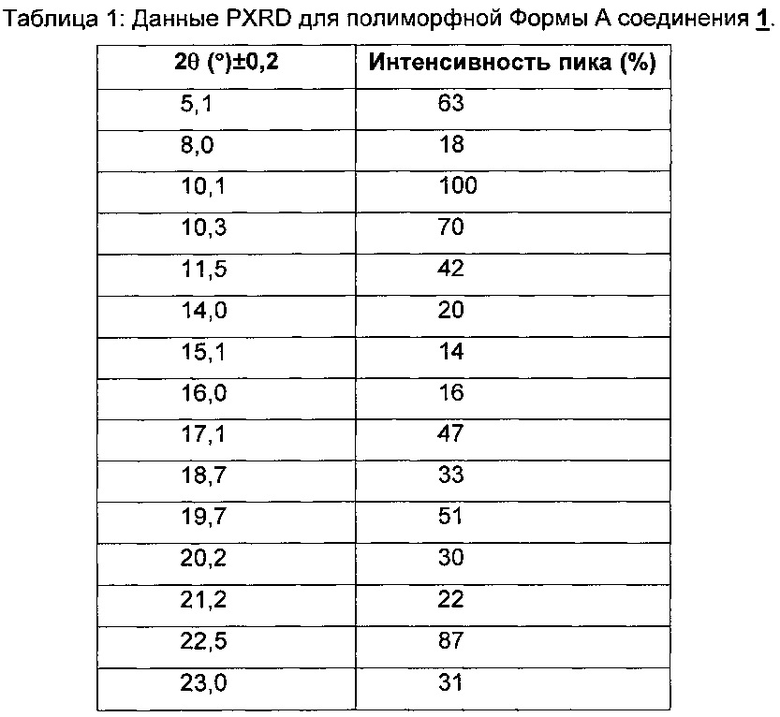
Ядерный магнитный резонанс твердого тела (ssNMR) для кристаллического свободного основания (Формы А) соединения 1 показан на Фиг. 2 и соответствующие данные представлены в Таблице 2.
В другом аспекте изобретения предлагается кристаллическое свободное основание соединения 1, которое представляет собой полиморфную Форму В свободного основания соединения 1. В некоторых воплощениях этого аспекта кристаллическая форма имеет порошковую рентгеновскую дифрактограмму, содержащую пик с углом дифракции (2θ) 6,0±0,2. В других воплощениях этого аспекта кристаллическая форма имеет порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ) 6,0±0,2 и 19,8±0,2. В других воплощениях этого аспекта кристаллическая форма имеет порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ) 6,0±0,2, 19,8±0,2 и 26,7±0,2. В других воплощениях кристаллическая форма имеет порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ) 6,0±0,2, 16,4±0,2, 19,8±0,2 и 26,7±0,2. В других воплощениях кристаллическая форма имеет порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ) 6,0±0,2, 12,8±0,2, 16,4±0,2, 19,8±0,2 и 26,7±0,2. В дополнительных воплощениях кристаллическая форма имеет порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ) 6,0±0,2, 12,8±0,2, 16,4±0,2, 19,8±0,2, 22,6±0,2 и 26,7±0,2. В других воплощениях кристаллическая форма имеет порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ) 6,0±0,2, 10,9±0,2, 12,8±0,2, 16,4±0,2, 19,8±0,2, 22,6±0,2 и 26,7±0,2. В некоторых воплощениях этого аспекта кристаллическая форма имеет PXRD-дифрактограмму, содержащую пики с углами дифракции (2θ) по существу такими же, как показано на Фиг. 3. Дифрактограмма дифракции рентгеновских лучей на порошке (PXRD) для полиморфной Формы В свободного основания показана на Фиг. 3 и соответствующие данные представлены в Таблице 3.
Ядерный магнитный резонанс твердого тела (ssNMR) для кристаллического свободного основания (Формы В) 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]пиримидин-7-она показан на Фиг. 4, соответствующие данные представлены в табличной форме в Таблице 4.

Для каждого измерения дифракции рентгеновских лучей на порошке образец свободного основания помещали в кювету, расположенную на плоской поверхности держателя, и предметное стекло использовали для выравнивания поверхности образца. Держатель, который содержит образец, помещали в дифрактометр, и источник рентгеновских лучей облучал образец, сначала с небольшим углом относительно плоскости поверхности держателя. Затем рентгеновские лучи постепенно перемещали по дуге, что последовательно увеличивало угол между падающим лучом и плоскостью поверхности держателя. На каждом шаге сканирования сцинтилляционный счетчик определял количество дифрагированного излучения, которое регистрировали в виде функции от 2θ(°). Программное обеспечение инструмента отображает результаты сканирования дифрагированного излучения в виде зависимости интенсивности от 2θ(°).
В Таблицах 1 и 3 приведены значительные пики PXRD (то есть те, которые имеют отношение высоты пика к шуму более 3,5) для свободного основания соединения 1, имеющего полиморфную Форму А или Форму В соответственно. Представленный перечень характеристических пиков представляет собой не единственный возможный перечень характеристических пиков. Специалисты обычной квалификации в области идентификации полиморфов могут выбрать другие совокупности характеристических пиков, которые также будут отличать один полиморф от другого.
Различия в дифрактограммах PXRD отдельных измерений одного и того же полиморфа могут возникать по многим причинам. Источники ошибок включают отклонения в приготовлении образца (например, в высоте образца), инструментальные погрешности, погрешности калибровки и ошибки оператора (включая ошибки в определении положений пиков). Предпочтительная ориентация, то есть отсутствие случайной ориентации кристаллов в образце для PXRD, может приводить к значительным различиям относительных высот пиков. Погрешности калибровки и отклонения в высоте образца часто приводят к сдвигу всех пиков дифрактограммы в одном и том же направлении и на одну и ту же величину. Небольшие различия в высоте образца на плоском держателе могут приводить к большим смещениям положений пиков PXRD. Систематическое исследование, показывающее, что разница в высоте образца 1 мм может приводить к сдвигам пиков вплоть до 1° 2θ, смотри в Chen et al., J. Pharmaceutical and Biomedical analysis (2001) 26:63.
Во многих случаях сдвиги пиков в дифрактограммах, появляющиеся в результате систематической ошибки, могут быть устранены при помощи введения поправочного коэффициента для сдвига (например, путем применением поправочного коэффициента ко всем значениям положений пиков) или посредством повторной калибровки дифрактометра. Обычно одни и те же способы можно использовать для корректировки различий между дифрактометрами, для того чтобы можно было привести в соответствие полученные положения пиков PXRD от двух разных инструментов. Кроме того, когда эти способы применяют к измерениям PXRD от одинаковых или разных дифрактометров, положения пиков для конкретного полиморфа обычно совпадают в пределах примерно ±0,2° 2θ.
Раскрытые соединения включают все фармацевтически приемлемые изотопные варианты. Изотопный вариант представляет собой соединение, в котором по меньшей мере один атом замещен атомом, имеющим такой же атомный номер, но атомную массу, отличающуюся от атомной массы, обычно встречающейся в природе. Полезные изотопы включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора. Типичные изотопы, таким образом, включают, без ограничения ими, 2Н, 3Н, 13С, 14С, 15N, 17O, 18O, 32Р, 35S, 18F и 36Cl.
Замещение изотопами в раскрытых соединениях, такими как дейтерий, то есть 2Н, может обеспечить некоторые терапевтические преимущества, вытекающие из большей метаболической стабильности, например увеличенный период полувыведения in vivo или потребность в уменьшенных дозах и, следовательно, может быть более полезным в некоторых обстоятельствах. Кроме того, некоторые изотопные варианты, например, включающие радиоактивный изотоп, являются полезными в исследованиях распределения лекарственного средства и/или субстрата в тканях. Радиоактивные изотопы тритий, то есть 3Н, и углерод-14, то есть 14С, являются особенно полезными для этой цели, вследствие простоты их включения и готовых средств обнаружения.
Изотопные варианты раскрытых соединений обычно могут быть приготовлены обычными способами, известными специалисту в данной области техники, или способами, аналогичными способам, описанным в сопутствующих Примерах, с использованием подходящих изотопных вариантов подходящих реагентов. Фармацевтически приемлемые сольваты раскрытых соединений включают сольваты, в которых растворитель при кристаллизации может быть изотопно замещенным, например D2O, d6-ацетон, d6-DMSO.
Эксперименты по растворимости
В US 7345171 сообщалось, что свободное основание соединения 1, полученное традиционным способом разрушения солей, имело плохую растворимость в воде (9 мкг/мл) при рН 7,9 и демонстрировало низкую биодоступность в исследованиях на животных. Сообщалось, что свободное основание находится в своей наиболее устойчивой кристаллической фазе согласно экспериментам с суспензией (то есть в Форме А). На Фиг. 17 в US 7345171 представлены изотермы адсорбции/десорбции воды для свободного основания Формы А. Как отмечалось ранее, это вещество соответствует частицам свободного основания соединения 1 небольшого размера, описанным в данном описании изобретения.
Свободное основание соединения 1 (Форма А) имеет высокую склонность прилипать к отверстиям в процессе изготовления частиц лекарственного средства. Так как прилипание к отверстиям связано с площадью поверхности API, контроль над размером частиц API является чрезвычайно важным для минимизации прилипания во время изготовления лекарственного продукта. В дополнение к проблемам с прилипанием к отверстиям, было обнаружено, что свободное основание соединения 1, выделенное непосредственно из стандартного способа разрушения солей имеет высокую склонность к статической электризации и образует большие (примерно 500 микрон) твердые агломераты, которые не диспергировались при просеивании. Свободное основание API с аналогичными плохими физическими свойствами получали путем получения свободного основания существующей изэтионатной соли API или путем нейтрализации образованной in situ соли на конечной стадии синтеза API. В любом способе получали небольшие первичные частицы API из-за быстрой кристаллизации, вызванной резким изменением растворимости при регулировании рН. Во всех случаях свободное основание выделяли в виде более устойчивого полиморфа Формы А.
На Фиг. 6 показано полученное при помощи сканирующей электронной микроскопии (SEM) изображение типичных небольших первичных частиц, образованных посредством получения свободного основания и экспериментов с нейтрализацией, описанных выше. Измерение распределения частиц по размерам для партии соединения 1 (Форма А), полученной посредством этого способа выделения свободного основания, представлен на Фиг. 8. Второй способ распределения частиц по размерам был вызван присутствием больших агломератов, которые также видны на изображении SEM на Фиг. 6. Попытки модифицировать процесс получения свободного основания не были успешными в улучшении физических свойств полученного API. Так как способ получения свободного основания приводил к выделению API с плохими физическими свойствами, была осуществлена работа по нахождению способа перекристаллизации, который мог бы улучшить физические свойства API.
Ранние скрининговые эксперименты с кристаллизацией свободного основания соединения 1 завершились идентификацией системы растворителей, которая позволяет выделять частицы с улучшенными физическими свойствами. При помощи комбинации скрининга растворимости и мелкомасштабных исследований перекристаллизации были изучены многочисленные потенциальные системы растворителей.
Мелкомасштабные исследования кристаллизации
Выполняли ряд мелкомасштабных экспериментов по кристаллизации для идентификации потенциальной системы растворителей для перекристаллизации, а также для оценки влияния растворителя на форму выделенных первичных частиц свободного основания. Исходную совокупность из 14 скрининговых исследований выполняли в 10 мг масштабе, используя герметично закрытые флаконы и внешний источник тепла для нагревания образцов 50 мг/мл вплоть до температуры дефлегмации. При помощи визуального наблюдения идентифицировали образцы, которые переходили в раствор, и для характеристики полученных частиц использовали фотомикроскопию. Результаты этих исходных скрининговых экспериментов в отношении кристаллизации представлены в Таблице 5.
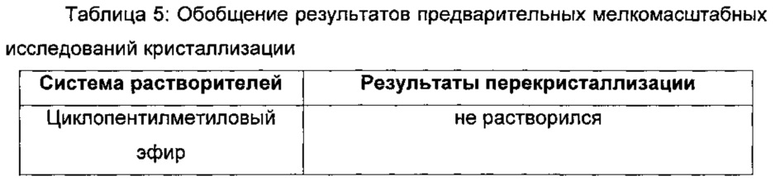
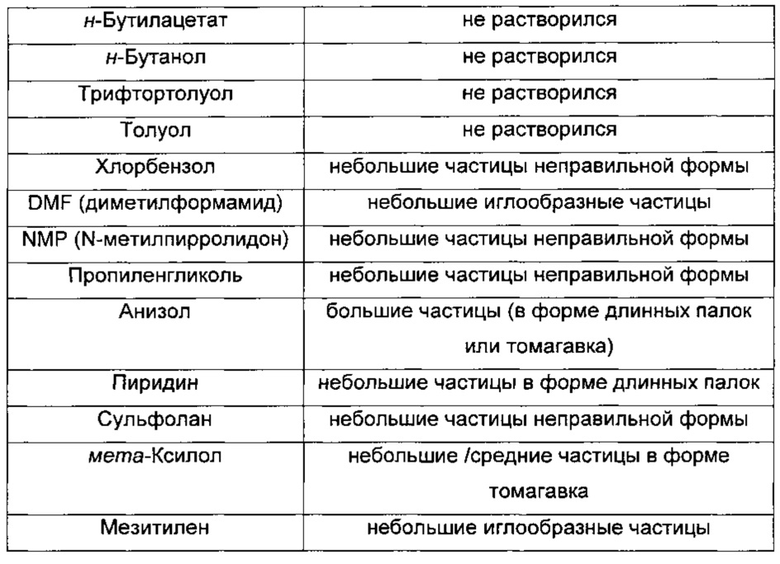
На основании этих мелкомасштабных исследований кристаллизации, дополнительные исследования кристаллизации и растворимости сфокусировались на анизоле, так как полученные частицы были большими и так как анизол является растворителем III класса согласно ICH (Международная конференция по гармонизации). В этом скрининговом исследовании в качестве потенциальных систем растворителей на основе полученных частиц также были идентифицированы пиридин, мета-ксилол и мезитилен, хотя ни один из этих растворителей не внесен в список III класса согласно ICH.
Для перекристаллизации твердого вещества также использовали следующие растворители: изопропанол, изобутанол, этанол, этилацетат, толуол, тетрагидрофуран и диоксан. Каждый из этих растворителей образовывал полиморфную Форму А кристаллического твердого вещества соединения 1, которая была такой же, как исходная кристаллическая форма, полученная из дихлорметана.
Исследования растворимости
Параллельно с начальными мелкомасштабных исследованиями кристаллизации проводили ряд исследований растворимости со свободным основанием соединения 1 для идентификации возможной системы перекристаллизации. В начальном скрининговом исследовании растворимости при комнатной температуре подвергали скринингу всего 23 растворителя. Это исследование показало, что свободное основание соединения 1 имеет низкую растворимость в ряде органических растворителей, за исключением только метиленхлорида, который показывает растворимость более 1 мг/мл (3,0 мг/мл). Затем проводили целевые исследования растворимости при более высоких температурах. В последующем исследовании изучали группу из 16 систем растворителей с постоянной концентрацией 25 мг/мл и измеряли температуру растворения, применяя метод изучения кинетической растворимость вплоть до максимальной температуры 110°С.
Синергический характер растворимости, предсказанные посредством модели растворимости COSMOtherm соединения 1, использовали для выбора двухкомпонентных и трехкомпонентных систем растворителей, включенных в это скрининговое исследование. Результаты этих исследований приведены в Таблице 6. В экспериментах, обозначенных в таблице как "более 110°С", соединение 1 не растворялось в растворителе при нагревании до 110°С, что указывает на то, что растворимость в этом растворителе составляет менее 25 мг/мл при 110°С.


Последующее исследование посредством UPLC/MS насыщенного раствора из экспериментов №3 и №11 в Таблице 6 показало присутствие ранее не замеченного пика примеси, означающее, что в этих экспериментах происходило разрушение.
Хотя смеси пропиленгликоль/н-BuOH/анизол показали улучшенную растворимость по сравнению со смесями н-BuOH/анизол, первую систему растворителей не рассматривали из-за возможных проблем, связанных с работой с пропиленгликолем, из-за его высокой вязкости и точки кипения, которые могут вызывать проблемы при масштабировании.
На основе этих скрининговых исследований смесь 40% н-бутанола и анизола выбрали в качестве системы растворителей при кристаллизации для дальнейшей работы из-за относительно высокой растворимости, химической стабильности API и свойств частиц перекристаллизованного соединения 1 API. Эту систему растворителей использовали в последующем производстве для получения первичных частиц API большего размера, которые имели уменьшенное прилипание, не были склонны к статической электризации и не содержали агломератов.
Используя эту смесь растворителей, соединение 1 растворяли при помощи 40 мл/г растворителя (концентрация 25 мг/мл) при нагревании до 95-100°С, и затем кристаллизовали с использованием регулируемого профиля охлаждения и введения затравки для индукции образования центров кристаллизации. Фиг. 9 представляет собой PLM-изображение партии соединения 1 в лабораторном масштабе, перекристаллизованного с использованием данного метода перекристаллизации, в то время как на Фиг. 7 показано распределение частиц по размерам для трех партий перекристаллизованного API. Этот способ перекристаллизации приводит к выделению частиц API соединения 1 с большим размером первичных частиц, что приводит к снижению тенденции к прилипанию в процессе изготовления лекарственного продукта. Такое перекристаллизованное соединение 1 API не образует агломератов и также имеет такое положительное качество, как отсутствие склонности к статической электризации.
Посредством комбинации скрининговых исследований растворимости и мелкомасштабных перекристаллизации изучили большое количество возможных систем растворителей для перекристаллизации свободного основания соединения 1. На основе результатов этих скрининговых исследований была выбрана смесь 40% н-бутанол/анизол в качестве предпочтительной системы растворителей для кристаллизации на основании относительно высокой растворимости, химической стабильности API и свойств частиц перекристаллизованного соединения 1. Более крупный размер частиц и улучшенные свойства частиц API, выделенных при помощи этого способа перекристаллизации, облегчали разработку способа изготовления лекарственного продукт из свободного основания соединения 1.
Оценка размера частиц
Размер частиц перекристаллизованных веществ оценивали, используя методы лазерной дифракции. Лазерная дифракция признана стандартами и руководящими органами, включая ISO (Международная организация по стандартизации) и ASTM (Американское общество испытания материалов) и широко используется для определения распределений частиц по размерам. При проведении оценки образец пропускают через лазерный луч, что приводит к рассеиванию лазерного света в некотором диапазоне углов. Детекторы, помещенные в фиксированных углах, измеряют интенсивность рассеянного света в этом положении. Затем используют математическую модель (теория Фраунгофера или Ми) для создания распределения частиц по размерам.
Размер частиц анализировали, используя метод лазерной дифракции (или малоугловое светорассеяние), посредством диспергирования сухого порошка образца сжатым воздухом. Конкретно, распределение частиц по размерам анализировали, используя систему Sympatec HELOS RODOS, оснащенную механизмом подачи сухого порошка Vibri. Образец порошка диспергировали при помощи давления рассеивания 0,5 бар (5×104 Па). В некоторых случаях использовали микродозирующее устройство Aspiros и образец порошка диспергировали при помощи давления рассеивания 0,2 бар (2×104 Па). Подбирали подходящие линзы, чтобы охватить диапазон размеров частиц каждого образца.
При определениях размера частиц медианное значение определяют как значение, при котором половина группы находится выше этой точки, и половина находится ниже этой точки. В распределении частиц по размерам медиана называется D50. D50 представляет собой размер в микронах, который разделяет распределение на две половины, одна из которых выше и вторая ниже этого диаметра. Выражение Dv50 или D[v,0.5] иногда используют для медианы распределения по объему.
Мода представляет собой пик распределения по частоте. Распределение частиц может содержать более одной моды, например, когда частицы существуют в виде первичных частиц и агломератов.
В качестве значения ширины распределения иногда используют диапазон и его определяют как соотношение (D[v,0.9]-D[v,0.1])/D[v,0.5] или (D90-D10)/D50.
Ширина распределения также может быть охарактеризована путем указания одного, двух или предпочтительно трех значений на оси X, обычно некоторой комбинации D10, D50 и D90. Медиана, D50, была определена выше, как такой диаметр, где половина группы лежит ниже этой величины. Аналогично, 90 процентов распределения лежит ниже D90, и 10 процентов группы лежит ниже D10.
Термин D[4,3] относится к среднему значению объема или среднему значению массового момента. Результаты лазерной дифракции приведены по объему и среднее значение по объему можно использовать для определения центральной точки распределения. Величина D[4,3] чувствительна к присутствию больших частиц в распределении.
Композиция
Настоящее изобретение также относится к фармацевтическим композициям, содержащим свободное основание полиморфной Формы А соединения 1, описанное в данном описании изобретения. Фармацевтические композиции по настоящему изобретению могут, например, находиться в форме, подходящей для перорального введения, в виде таблетки, капсулы, драже, порошка, препаратов с замедленным высвобождением, раствора, суспензии, для парентеральной инъекции в виде стерильного раствора, суспензии или эмульсии, для местного введения в виде мази или крема, или для введения в прямую кишку в виде суппозитория. Фармацевтическая композиция может быть представлена в стандартных лекарственных формах, подходящих для однократного введения точных дозировок. Фармацевтическая композиция включает обычный фармацевтический носитель или эксципиент и соединение согласно изобретению в качестве активного ингредиента. Кроме того, она может включать другие медицинские или фармацевтические агенты, носители, вспомогательные лекарственные средства и так далее.
Подходящие фармацевтические носители включают инертные разбавители или наполнители, воду и различные органические растворители. Фармацевтические композиции могут, если требуется, содержать дополнительные ингредиенты, такие как ароматизаторы, связывающие вещества, эксципиенты и подобные. Таким образом, для перорального введения таблетки, содержащие различные эксципиенты, такие как лимонная кислота, можно использовать вместе с различными разрыхлителями, такими как крахмал, альгиновая кислота и некоторые комплексные силикаты, и со связывающими агентами, такими как сахароза, желатин и гуммиарабик. Кроме того, смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк, часто являются полезными для таблетирования. Твердые композиции аналогичного типа также могут применяться в мягких и твердых заполненных желатиновых капсулах. Предпочтительные вещества включают лактозу или молочный сахар и высокомолекулярные полиэтиленгликоли. Если для перорального введения желательны водные суспензии или эликсиры, активное соединение в них можно комбинировать с различными подсластителями или ароматизаторами, красящими веществами или красителями и, если требуется, с эмульгаторами или суспендирующими агентами, вместе с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин или их комбинации.
Способы получения различных фармацевтических композиций с определенным количеством активного соединения известны или очевидны специалисту в данной области техники. Например, смотри Remington's Pharmaceutical Sciences. Mack Publishing Company, Easter, Pa., 15th Edition (1975).
Раскрытое соединение может быть введено одно или в комбинации с другими лекарственными средствами и обычно будет введено в виде композиции вместе с одним или более фармацевтически приемлемыми эксципиентами. Термин "эксципиент" описывает любой ингредиент, отличный от соединения 1 и его солей. Выбор эксципиента в большой степени будет зависеть от конкретного пути введения.
Раскрытые соединения могут быть введены перорально. Пероральное введение может включать проглатывание, так что соединение поступает в желудочно-кишечный тракт, или можно использовать буккальное или сублингвальное введение, посредством которого соединение поступает в кровоток непосредственно изо рта.
Композиции, подходящие для перорального введения, включают твердые композиции, такие как таблетки, капсулы, содержащие частицы, жидкости или порошки, пастилки (включая заполненные жидкостью), жевательные таблетки, мульти- и наночастицы, гели, твердый раствор, липосому, пленки (включая мукоадгезивные), овули, спреи и жидкие композиции. Жидкие композиции включают суспензии, растворы, сиропы и эликсиры. Таких композиции могут применяться в качестве наполнителей в мягких или твердых капсулах и обычно содержат носитель, например воду, EtOH, полиэтиленгликоль, пропиленгликоль, метил целлюлозу или подходящее масло, и один или более эмульгаторов и/или суспендирующих агентов. Жидкие композиции также могут быть получены восстановлением из твердых композиций, например из саше.
Раскрытые соединения также можно использовать в быстрорастворяющихся лекарственных формах, быстрораспадающихся лекарственных формах, таких как описанные в Liang and Chen, Expert Opinion in Therapeutic Patents (2001) 11 (6):981-986.
Для лекарственных форм в виде таблеток, в зависимости от дозы, лекарственное средство может составлять от 1% масс. до 80% масс. лекарственной формы, обычно от 5% масс. до 60% масс. лекарственной формы. В дополнение к лекарственному средству таблетки обычно содержат разрыхлитель. Примеры разрыхлителей включают натрия крахмала гликолят, натрия карбоксиметилцеллюлозу, кальция карбоксиметилцеллюлозу, кроскармеллозу натрия, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, замещенную низшим алкилом гидроксипропилцеллюлозу, крахмал, клейстеризованный крахмал и альгинат натрия. Обычно разрыхлитель составляет от 1% масс. до 25% масс., предпочтительно от 5% масс. до 20% масс. лекарственной формы.
Связывающие вещества обычно используют для придания когезивных свойств таблеточной композиции. Подходящие связывающие вещества включают микрокристаллическую целлюлозу, желатин, сахара, полиэтиленгликоль, натуральные и синтетические камеди, поливинилпирролидон, клейстеризованный крахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. Таблетки также могут содержать разбавители, такие как лактоза (моногидрат, высушенный распылением моногидрат, безводная и подобные), маннит, ксилит, декстрозу, сахарозу, сорбит, микрокристаллическую целлюлозу, крахмал и двухзамещенный дигидрофосфат кальция.
Таблетки возможно могут включать также поверхностно-активные агенты, такие как натрия лаурилсульфат и полисорбат 80, и скользящие вещества, такие как диоксид кремния и тальк. Поверхностно-активные агенты, если присутствуют, могут составлять от 0,2% масс., до 5% масс., таблетки, и скользящие вещества могут составлять от 0,2% масс., до 1% масс., таблетки.
Таблетки также обычно содержат смазывающие вещества, такие как стеарат магния, стеарат кальция, стеарат цинка, натрия стеарилфумарат и смеси стеарата магния с натрия лаурилсульфатом. Смазывающие вещества обычно составляют от 0,25% масс, до 10% масс., предпочтительно от 0,5% масс., до 3% масс., таблетки. Другие ингредиенты могут включать консерванты, антиоксиданты, ароматизаторы и красители.
Таблеточные смеси можно непосредственно прессовать с образованием таблеток. Таблеточные смеси или части смесей, альтернативно, можно подвергать влажному, сухому гранулированию или гранулированию расплавов, замораживанию расплавов, или их можно экструдировать перед таблетированием. Конечная композиция может содержать один или более слоев и может быть покрытой или непокрытой оболочкой. Типичные таблетки содержат вплоть до примерно 80% лекарственного средства, от примерно 10% масс., до примерно 90% масс., связывающего вещества, от примерно 0% масс., до примерно 85% масс., разбавителя, от примерно 2% масс., до примерно 10% масс., разрыхлителя и от примерно 0,25% масс., до примерно 10% масс., смазывающего вещества. Дополнительные подробности, касающиеся композиции таблеток смотри в Н. Lieberman и L. Lachman, Pharmaceutical Dosage forms: Tablets, Vol. 1 (1980).
Твердые композиции для перорального введения могут быть приготовлены в виде препарата с немедленным и/или модифицированным высвобождением. Композиции с модифицированным высвобождением включают отложенное, замедленное, прерывистое, контролируемое, направленное и программируемое высвобождение. Общее описание подходящих композиций с модифицированным высвобождением смотри в патенте US 6106864. Подробности других полезных методов высвобождения, таких как высокоэнергетические дисперсии и осмотические и покрытые оболочкой частицы, смотри в Verma et al, Pharmaceutical Technology On-line (2001) 25(2):1-14. Обсуждение применения жевательной резинки для достижения контролируемого высвобождения смотри в WO 00/35298.
Раскрытые соединения также могут быть введены непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие способы парентерального введения включают внутривенное, внутриартериальное, внутрибрюшинное, внутриоболочечное, внутрижелудочковое, внутриуретральное, внутригрудинное, внутричерепное, внутримышечное и подкожное введение. Подходящие устройства для парентерального введения включают игольные (включая микроигольные) инжекторы, безыгольные инжекторы и инфузионные способы.
Парентеральные композиции обычно представляют собой водные растворы, которые могут содержать эксципиенты, такие как соли, углеводы и буферные агенты (предпочтительно до рН от 3 до 9), но для некоторых применений они могут быть более подходящим образом приготовлены в виде стерильного неводного раствора или в виде сухой формы для использования вместе с подходящим носителем, таким как стерильная апирогенная вода. Получение парентеральных композиций в стерильных условиях, например, посредством лиофилизации, может быть легко выполнено с использованием стандартных фармацевтических методов, хорошо известных специалисту в данной области техники. Типичные формы для парентерального введения включают растворы или суспензии активных соединений в стерильных водных растворах, например водные растворы пропиленгликоля или декстрозы. Такие лекарственные формы могут быть соответствующим образом забуферены, если это требуется.
Растворимость раскрытых соединений, используемых в получении парентеральных растворов, может быть увеличена путем использования подходящих способов приготовления препаратов, таких как включение агентов, усиливающих растворимость. Композиции для парентерального введения могут быть приготовлены в виде препарата с немедленным и/или модифицированным высвобождением, как описано выше. Таким образом, раскрытые соединения могут быть приготовлены в виде препарата в более твердой форме для введения в виде имплантированного депо, обеспечивающего длительное высвобождение активного соединения.
Соединения по изобретению также могут быть введены местно на кожу или слизистую оболочку, либо кожно, либо чрескожно. Обычные композиции для этой цели включают гели, гидрогели, лосьоны, растворы, крема, мази, присыпки, повязки, пенки, пленки, кожные пластыри, прокладки, импланты, губки, волокна, бандажи и микроэмульсии. Также можно использовать липосомы. Обычные носители включают спирт, воду, минеральное масло, вазелиновое масло, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Местные композиции также могут включать вещества, способствующие проникновению. Смотри, например, Finnin and Morgan, J Pharm Sci (1999) 88(10):955-958.
Другие способы местного введения включают доставку посредством ионтофореза, электропорации, фонофореза, сонофореза и безыгольной (например, POWDERJECT) или микроигольной инъекции. Композиции для местного введения могут быть приготовлены в виде препарата с немедленным и/или модифицированным высвобождением, как описано выше.
Раскрытые соединения также могут быть введены интраназально или посредством ингаляции обычно в форме сухого порошка (либо отдельно, в виде смеси, например в сухой смеси с лактозой, либо в виде частиц со смешанными компонентами, например смешанными с фосфолипидами) из ингалятора для сухого порошка или в виде аэрозольного спрея из находящегося под давлением контейнера, помпы, спрея, распылителя (предпочтительно распылителя с использованием электрогидродинамики для получения мелкодисперного тумана), или небулайзера, с применением или без применения подходящего пропеллента, такого как дихлорфторметан. Находящийся под давлением контейнер, помпа, спрей, распылитель или небулайзер содержит раствор или суспензию, которые содержат активное соединение, агент для диспергирования, солюбилизации или продления высвобождения активного соединения (например EtOH или водный EtOH), один или более растворителей, которые служат в качестве пропеллента, и, возможно, поверхностно-активное вещество, такое как сорбитантриолеат или олигомолочная кислота.
Перед использованием в сухой порошковой композиции или в суспензионной композиции лекарственный продукт микронизируют до размера, подходящего для доставки путем ингаляции (обычно менее 5 микрон). Это может быть достигнуто посредством любого подходящего способа измельчения, например посредством размола на спиральной струйной мельнице, размола на струйной мельнице с кипящим слоем, обработки сверхкритической жидкости с образованием наночастиц, гомогенизации под высоким давлением или сушки распылением.
Капсулы, блистеры и картриджи (изготовленные, например, из желатина или гидроксипопилметилцеллюлозы) для использования в ингаляторе или инсуффляторе могут быть приготовлены в виде препарата, который содержит порошковую смесь активного соединения, подходящей порошковой основы, такой как лактоза или крахмал, и модификатор характеристик, такой как L-лейцин, маннит или стеарат магния. Лактоза может быть безводной или, предпочтительно, моногидратированной. Другие подходящие эксципиенты включают декстран, глюкозу, мальтозу, сорбит, ксилит, фруктозу, сахарозу и трегалозу.
Подходящая композиция в форме раствора для использования в распылителе с использованием электрогидродинамики с получением мелкодисперного тумана может содержать от 1 мкг до 20 мг соединения по изобретению на одно нажатие и объем, высвобождаемый при одном нажатии, может варьироваться от 1 мкл до 100 мкл. Типичная композиция может содержать соединение 1, пропиленгликоль, стерильную воду, EtOH и NaCl. Альтернативные растворители, которые можно использовать вместо пропиленгликоля, включают глицерин и полиэтиленгликоль.
Композиции для ингаляционного/интраназального введения могут быть изготовлены в виде препаратов для немедленного и/или модифицированного высвобождения с использованием, например, сополимера DL-молочной и гликолевой кислот (PGLA). Подходящие ароматизаторы, такие как ментол и левоментол, или подсластители, такие как сахарин или сахаринат натрия, могут быть добавлены в композиции, предназначенные для ингаляционного/интраназального введения.
В случае ингаляторов для сухого порошка и аэрозолей единица дозирования регулируется посредством клапана, который доставляет отмеренное количество. Устройства в соответствии с изобретением обычно скомпонованы для введения отмеренной дозы или "пуфа", содержащего от 100 до 1000 мкг активного фармацевтического ингредиента. Полная суточная доза, которая может быть введена в однократной дозе или, чаще, в виде разделенных доз в течение суток, обычно находится в диапазоне от 100 мкг до 10 мг.
Активные соединения могут быть введены ректально или вагинально, например, в форме суппозитория, пессария или клизмы. Кокосовое масло представляет собой традиционную основу суппозитория, но при необходимости можно использовать различные альтернативы. Композиции для ректального/вагинального введения могут быть изготовлены в виде препарата для немедленного и/или модифицированного высвобождения, как описано выше.
Раскрытые соединения также могут быть введены непосредственно в глаз или ухо, обычно в форме капель микронизированной суспензии или раствора в изотоническом, стерильном солевом растворе с отрегулированным рН. Другие композиции, подходящие для глазного и ушного введения, включают мази, биоразлагаемые (например, абсорбируемые гелевые губки, коллаген) и небиоразлагаемые (например силиконовые) импланты, прокладки, линзы и системы микрочастиц или везикул, такие как ниосомы или липосомы. Полимер, такой как поперечно-сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер (например, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза) или гетерополисахаридный полимер (например геллановая камедь) может быть включен вместе с консервантом, таким как бензалкония хлорид. Такие композиции также могут быть доставлены при помощи ионтофореза. Композиции для глазного/ушного введения могут быть изготовлены в виде препарата для немедленного и/или модифицированного высвобождения, как описано выше.
Раскрытые соединения можно комбинировать с растворимыми макромолекулярными соединениями, такими как циклодекстрин или полиэтиленгликоль-содержащие полимеры, для улучшения их растворимости, скорости растворения, маскирования вкуса, биодоступности и/или стабильности. Например, обнаружено, что комплексы лекарственное средство-циклодекстрин обычно полезны для большинства лекарственных форм и путей введения. Можно использовать как комплексы включения, так и комплексы без включения. В качестве альтернативы прямому комплексообразованию с лекарственным средством, циклодекстрин можно использовать в качестве вспомогательной добавки, то есть в качестве носителя, разбавителя или солюбилизатора. Для этих целей обычно используют альфа-, бета- и гамма-циклодекстрины. Смотри, например, международные заявки на патент WO 91/11172, WO 94/02518 и WO 98/55148.
Терапевтически эффективная доза соединения 1 варьируется от примерно 0,01 мг/кг до примерно 100 мг/кг массы тела в сутки. Обычные дозы для взрослых составляют от примерно 0,1 мг до примерно 3000 мг в сутки. Количество активного компонента в стандартной дозе препарата можно варьировать или регулировать от примерно 0,1 мг до примерно 500 мг, предпочтительно от примерно 0,6 мг до 100 мг, согласно конкретному применению и эффективности активного компонента. Композиция также может, если требуется, содержать другие совместимые терапевтические агенты. Пациенту, нуждающемуся в лечении, вводят дозу от примерно 0,6 до примерно 500 мг в сутки, либо однократно, либо многократными дозами в течение 24-часового периода. Такое лечение можно повторять последовательными интервалами столько, сколько необходимо.
Расстройства или состояния, вызванные аномальной пролиферацией клеток, включают рак и пролиферацию гладких мышц сосудов, ассоциированную с атеросклерозом, послеоперационный стеноз и рестеноз сосудов, и эндометриоз. Аутоиммунные заболевания включают псориаз, воспалительный ревматоидный артрит, волчанку, диабет 1 типа, диабетическую нефропатию, рассеянный склероз, гломерулонефрит и отторжение органа-трансплантата, включая болезнь «трансплантат против хозяина».
В одном воплощении настоящего изобретения предлагается способ лечения аномального роста клеток у млекопитающего, включая человека, нуждающегося в таком лечении, включающий введение указанному млекопитающему терапевтически эффективного количества кристаллического свободного основания соединения 1 по изобретению, описанного здесь. В часто встречающихся воплощениях свободное основание представляет собой полиморфную Форму А.
В другом воплощении аномальный рост клеток представляет собой рак, включая как солидные опухоли, так и гематологические злокачественные заболевания. В некоторых таких воплощениях рак выбран из рака молочной железы, рака яичника, рака шейки матки, рака эндометрия, рака предстательной железы, рака яичек, рака поджелудочной железы, рака пищевода, рака головы и шеи, рака желудка, рака мочевого пузыря, рака легкого (например, аденокарциномы, NSCLC и SCLC), рака кости (например, остеосаркомы), рака толстой кишки, рака прямой кишки, рака щитовидной железы, раковых заболеваний головного мозга и центральной нервной системы, глиобластомы, нейробластомы, нейроэндокринного рака, рабдоидного рака, кератоакантомы, плоскоклеточного рака, семиномы, меланомы, саркомы (например липосаркомы), рака мочевого пузыря, рака печени (например печеночно-клеточного рака), рака почек (например гипернефромы), миелоидных расстройств (например AML, CML, миелодипластического синдрома и промиелоцитарного лейкоза) и лимфоидных расстройств (например лейкоза, множественной миеломы, мантийноклеточной лимфомы, ALL, CLL, В-клеточной лимфомы, Т-клеточной лимфомы, лимфомы Ходжкина, неходжкинской лимфомы, лейкоза волосатых клеток).
Общая схема синтеза
Примеры и получения, предложенные ниже, дополнительно иллюстрируют и поясняют конкретные аспекты воплощений изобретения. Следует понимать, что объем настоящего изобретения не ограничен никоим образом объемом следующих примеров.
Примеры
Общие методы и вещества
Порошковая рентгеновская дифракция (PXRD)
Данные PXRD собирали согласно следующему протоколу. Образец (2 мг) помещали на предметное стекло с нулевым фоновым излучением. Затем образец помещали в Discover D8 (Bruker AXS Instruments), оснащенный детектором GADDS. В системе использовали медный источник рентгеновских лучей, поддерживаемый при 40 кВ и 40 мА с получением эмиссии CUα1 с длиной волны 1,5406 ангстрем. Данные собирали в диапазоне от 4 до 40° 2θ с шагом сканирования 0,02° с шагом временем 60,1 секунд. Пики дифракции обычно измерены с погрешностью ±0,2 градуса (2θ).
Измерительные приборы и способ ssNMR
Данные ssNMR собирали согласно следующему протоколу. Спектры собирали на датчике 4 мм и 7 мм BL CPMAS Bruker-Biospin, установленном в широкопросветном ЯМР-спектрометре Bruker-Biospin Avance III 500 МГц. Роторы 4 мм ориентировали под магическим углом и вращали при 15,0 кГц. Роторы 7 мм ориентировали под магическим углом и вращали при 7,0 кГц. Все спектры получали в условиях окружающей среды (температуру не контролировали).
Спектры 13С твердого тела собирали с использованием эксперимента с кросс-поляризацией и протонной развязкой при вращении образца под магическим углом (CPMAS). Пики резонансов регистрировали в миллионных долях (млн-1) ±0,2 млн-1.
Дифференциальная сканирующая калориметрия (ДСК)
Измерения ДСК выполняют, используя Q1000 Thermal Analysis Instruments. Образец помещают в герметично закрытую алюминиевую кювету с микроотверстием. Типичная масса образца составляет 1,6 мг. Образец уравновешивали при 25°С и затем линейно повышали температуру до 250°С со скоростью сканирования 10°С/мин. Сухой азот использовали в качестве газа для продувки.
Измерение удельной площади поверхности (SSA) по Брунауэру-Эмметту-Теллеру (BET)
SSA-измерения собирали согласно следующему протоколу. Для определения удельной площади поверхности сухого порошка активного фармацевтического ингредиента использовали образование монослоя молекул газа на поверхности кристалла. Изготавливали не содержащий влаги и атмосферных паров образец посредством применения нагревания и продувания газообразным азотом. Затем температуру образца уменьшали до температуры жидкого азота для адсорбции газа-адсорбата (азота). Данные о количестве адсорбированного газа и давлении использовали для создания графика изотермы адсорбции. Затем данные преобразовывали в величину удельной площади поверхности, используя математический алгоритм, основанный на так называемой теории Брунауэра, Эмметта и Теллера (BET) (смотри, например, J. Am. Chem. Soc., 1938, 60:309). Удельную площадь поверхности измеряли, используя статический многоточечный или одноточечный способ адсорбции газа, который полностью описан в ISO 9277:2010 и в экспериментальной части ниже.
Измерение поверхностной энергии посредством обращенно-фазовой газовой хроматографии (IGC)
Измерения поверхностной энергии с применением IGC собирали согласно следующему протоколу. Достаточное количество образца помещали в силанизированную стеклянную колонку с порошковой массой, надежно закрепленной в колонке посредством пробок из стекловаты, вставленных в оба конца. Колонку кондиционировали путем пропускания потока сухого азота через порошковую массу в течение времени, достаточного для удаления любых абсорбатов с поверхности. Измерения выполняли посредством ввода ряда образцов паров алканов (нонана, октана, гептана и гексана) в поток газа-носителя в концентрациях, достаточно низких чтобы допускать бесконечное разбавление паров алкана в потоке азота, и регистрации времени, которое требовалось для элюирования каждого пара через колонку. График зависимости времени удерживания (скорректированного с учетом «мертвого объема» - пустот между частицами в заполненной колонке) как функции от площади поперечного сечения и поверхностного натяжения используемых молекул образцов паров алканов приводил к линии с наклоном, показывающим поверхностную энергию исследуемого твердого порошка.
Примеры синтеза
Пример 1. Получение трет-бутилового эфира 4-(6-амино-пиридин-3-ил)пиперазин-1-карбоновой кислоты
Стадия А. Получение трет-бутилового эфира 4-(6-нитро-пиридин-3-ил)-пиперазин-1-карбоновой кислоты
В сосуд добавляли 5-бром-2-нитропиридин (10,0 г, 1,0 экв.) вместе с DMSO (25 мл, 2,5 об.). Добавляли N-Boc-пиперазин (13,8 г, 1,5 экв.), затем триэтиламин (7,5 г, 1,5 экв.) и LiCl (2,1 г, 1,0 экв.). Смесь нагревали до 60-65°С в течение минимум 12 часов.
Воду (5 мл, 0,5 об.) медленно добавляли в сосуд при 60-65°С. Смесь выдерживали при 60-65°С в течение одного часа, затем охлаждали до комнатной температуры. Суспензию выдерживали при 20-25°С в течение 1 часа и затем фильтровали на бумажном фильтре Whatman™ №2. Осадок промывали водой (50 мл, 5 об.). Неочищенные твердые вещества собирали и переносили обратно в чистый сосуд.
Воду (100 мл, 10 об.) добавляли в сосуд, содержащий твердые вещества, и смесь нагревали до 35-40°С в течение 2 часов, затем фильтровали, пока он был теплым, на бумажном фильтре Whatman™ №2. Твердые вещества промывали водой (40 мл, 4 об.) и оставляли сушиться в течение ночи в вакуумном сушильном шкафу при 50-55°С. Трет-бутиловый эфир 4-(6-нитро-пиридин-3-ил)-пиперазин-1-карбоновой кислоты выделяли в виде желтого твердого вещества (собрано 14,1 г; выход ~93%).
Стадия В. Получение трет-бутилового эфира 4-(6-амино-пиридин-3-ил)-пиперазин-1-карбоновой кислоты
В сосуд добавляли трет-бутиловый эфир 4-(6-нитро-пиридин-3-ил)-пиперазин-1-карбоновой кислоты (12,0 г, 1,0 экв.) вместе с этилацетатом (48 мл, 4,0 об.). В суспензию добавляли 5% Pd на активированном угле в виде водной суспензии с влажностью 50% (480 мг, 4% масс/масс.) и сосуд продували три раза азотом. Сосуд продували три раза водородом и затем повышали давление водорода до 50 фунтов на квадратный дюйм (3,45×105 Па). Смесь нагревали до 42-47°С и оставляли перемешиваться до прекращения поглощения водорода (по меньшей мере 8 часов).
Смесь продуктов фильтровали и промывали этилацетатом (2×1,5 мл). Объединенные фильтраты концентрировали при пониженном давлении до объема 6 мл (2 об.). В раствор добавляли н-гептан (54 мл, 4,5 об.) и смесь дистиллировали при пониженном давлении до объема 6 мл (2 об.). В раствор добавляли н-гептан (54 мл, 4,5 об.). Полученную густую суспензию охлаждали до 20-25°С и оставляли перемешиваться в течение 2 часов. Суспензию фильтровали и осадок на фильтре промывали н-гептаном (36 мл, 3 об.). Твердые вещества оставляли сушиться в течение ночи в вакуумном сушильном шкафу при 50-55°С. Трет-бутиловый эфир 4-(6-амино-пиридин-3-ил)-пиперазин-1-карбоновой кислоты выделяли в виде бледно-оранжевого твердого вещества (собрано 10,4 г; выход ~96%). 1Н ЯМР (500 МГц, DMSO-d6): δ 7,62 (dd, J=2,99, 0,60 Гц, 1Н), 7.17 (dd, J=8,85, 2,99 Гц, 1Н), 6.40 (dd, J=8,85, 0,60 Гц, 1Н), 5.45 (bs, 2Н), 3.43 (m, 2Н), 2.85 (m, 2Н), 1.41 (s, 9Н); 13С ЯМР (125 МГц, DMSO-d6): δ 154,8, 153,8, 138,7, 136,8, 125,9, 108,3, 78,9, 50,5, 43,8, 43,0, 28,0; МСВР (масс-спектроскопия высокого разрешения): Рассчитано для C14H23N4O2 (М+Н)+: 279,18155. Обнаружено: 279,18173.
Пример 2. Получение 6-бром-2-хлор-8-циклопентил-5-метил-8H-пиридо[2,3-d]пиримидин-7-она
Стадия А. Получение 5-бром-2-хлор-6-циклопентиламино-пиримидина
В сосуд добавляли абсолютный этанол (3000 мл, 3,0 об.), затем 5-бром-2,4-дихлорпиримидин (ММ (молекулярная масса) 227,87; 1000 г, 1,0 экв.). Добавляли триэтиламин (612 мл, 1,0 экв.) и затем медленно добавляли циклопентиламин (ММ 85,15; 520 мл, 1,2 экв.) в течение 2 часов для обеспечения умеренного количества выделившегося тепла. При необходимости, после завершения добавления циклопентиламина реакционную в смесь вносили затравку 5-бром-2-хлор-6-циклопентиламино-пиримидина (5 г, 0,5% масс), чтобы индуцировать кристаллизацию. Реакционную смесь перемешивали при 25°С в течение 2 часов.
Воду (2500 мл, 2,5 об.) добавляли в сосуд при 20-25°С со скоростью 30 мл/мин. Смесь охлаждали до 8-12°С со скоростью 2°С/мин. Суспензию выдерживали при 8-12°С в течение 1 часа и затем фильтровали на бумажном фильтре Whatman™ №2. Осадок промывали н-гептаном (2000 мл). Осадок ресуспендировали с н-гептаном на фильтре-влагоотделителе (2000 мл). Вещество сушили в течение ночи в вакуумном сушильном шкафу при 50-55°С с получением 5-бром-2-хлор-6-циклопентиламино-пиримидина (1020 г; 84%) в виде белого твердого вещества.
Стадия В. Получение 2-хлор-8-циклопентил-5-метил-8Н-пиридо[2,3-d]пиримидин-7-она
В сосуд добавляли 5-бром-2-хлор-6-циклопентиламино-пиримидин (10,0 г, 1,0 экв.) вместе с N-метилпирролидоном (NMP) (50 мл, 5,0 об.) при температуре окружающей среды. К реакционной смеси добавляли кротоновую кислоту (4,7 г, 1,5 экв.) и триэтиламин (20,2 мл, 4,0 экв.). Сосуд дегазировали и продували три раза азотом. В дегазированную реакционную смесь добавляли Pd(OAc)2 (0,25 г, 0,03 экв.). Сосуд дегазировали и продували три раза азотом, используя такой же способ, как на стадии 3. Смесь нагревали до 65°С и оставляли перемешиваться до тех пор, пока исходное вещество не было поглощено (по меньшей мере 6 часов).
Уксусный ангидрид (6,8 мл, 2,0 экв.) добавляли в реакционную смесь. Реакционную смесь оставляли взаимодействовать при 65°С до тех пор, пока исходное вещество не было поглощено (обычно 1-2 часа).
Реакционную смесь охлаждали до 20°С и добавляли Н2О (100 мл, 10 об.) для растворения солей триэтиламин⋅HBr и осаждения 2-хлор-8-циклопентил-5-метил-8Н-пиридо[2,3-d]пиримидин-7-она. Вещество гранулировали при 20°С в течение 1 часа. Твердые вещества фильтровали и промывали Н2О (20 мл, 2,0 об.) и смесью 4:1 изопропанол/H2O (50 мл, 5,0 об.). Неочищенный продукт сушили в вакууме при 55-70°С с получением 2-хлор-8-циклопентил-5-метил-8Н-пиридо[2,3-d]пиримидин-7-она (7,8 г; 81%) в виде твердого вещества, имеющего цвет от желто-коричневого до серого.
Стадия С. Получение 6-бром-2-хлор-8-циклопентил-5-метил-8Н-пиридо[2,3-d]пиримидин-7-она
В эмалированный сосуд добавляли 2-хлор-8-циклопентил-5-метил-8Н-пиридо[2,3-d]пиримидин-7-он (9,35 г, 1,0 экв.) вместе с ацетонитрилом (65 мл, 7,0 об). Добавляли N-бромсукцинимид (9,67 г, 1,5 экв.) и щавелевую кислоту (0,65 г, 0,2 экв.). Реакционную смесь нагревали до 60±5°С. Реакционную смесь перемешивали при 60°С до поглощения исходного вещества (по меньшей мере 6 часов). Суспензию охлаждали до 20°С и добавляли Н2О (9 мл, 1 об.). В суспензию добавляли раствор бисульфита натрия (3,88 г, 1,0 экв.) в H2O (38 мл, 4 об.). Суспензию гранулировали в течение 1 часа, затем фильтровали непосредственно на бумажном фильтре Whatman №2. Реакционный сосуд промывали водой (19 мл, 2 об.), затем смесью 7:3 метанол/ацетонитрил (28 мл, 3 об.), и промывные воды переносили на осадок на фильтре. Продукт сушили в вакуумном сушильном шкафу при 50-55°С. 6-Бром-2-хлор-8-циклопентил-5-метил-8H-пиридо[2,3-d]пиримидин-7-он (10,52 г; 87%) выделяли в виде бледно-желтого твердого вещества.
Продукт дополнительно очищали посредством перекристаллизации из толуола и н-гептанов. Толуол (60 мл, 6 об.) и 6-бром-2-хлор-8-циклопентил-5-метил-8Н-пиридо[2,3-d]пиримидин-7-он (10,00 г, 1 экв.) добавляли в реакционный сосуд и нагревали до 80°С. Теплую реакционную смесь фильтровали через подходящий картридж, чтобы обеспечить удаление нерастворимого Pd и других нерастворимых примесей. Фильтрующий картридж промывал 80°С толуолом (5 мл, 0,5 об.). Суспензию охлаждали до 25°С со скоростью 1°С/мин. н-Гептан (70 мл, 7 об.) добавляли в реакционную суспензию со скоростью 1 мл/мин. Суспензию дополнительно охлаждали до 0°С со скоростью 1°С/мин. Суспензию гранулировали при 0°С в течение по меньшей мере 1 часа.
Суспензию фильтровали непосредственно на бумажном фильтре Whatman №2. н-Гептан (30 мл, 3 об.) загружали в реакционный сосуд и промывную воду переносили на осадок на фильтре и продукт сушили в вакуумном сушильном шкафу при 50-55°С. 6-Бром-2-хлор-8-циклопентил-5-метил-8Н-пиридо[2,3-d]пиримидин-7-он (8,73 г, 87%) выделяли в виде твердого вещества кремового цвета. 1Н ЯМР (500 МГц, DMSO-d6): δ 9.20 (s, 1Н), 5.82 (m, 1Н), 2.65 (s, 3Н), 2.11 (m, 2Н), 2.04 (m, 2Н), 1.86 (m, 2Н), 1.64 (m, 2Н); 13С ЯМР (125 МГц, DMSO-d6): δ 158,2, 158,2, 157,6, 154,1, 144,0, 120,9, 113,0, 54,4, 28,3, 25,7, 18,3; МСВР: Рассчитано для C13H14N3O1Br1Cl1 (М+Н)+: 342,00033, Обнаружено: 342,00037.
Пример 3. Получение трет-бутилового эфира 4-{6-[6-бром-8-циклопентил-5-метил-7-оксо-7,8-дигидро-пиридо[2,3-d]пиримидин-2-иламино]пиридин-3-ил)-пиперазин-1-карбоновой кислоты
В продутый азотом сухой реактор загружали тетрагидрофуран (900 мл, 15 мл/г). Температуру партии устанавливали на 20°С и начинали взбалтывание со скоростью 250 об/мин. В реактор загружали трет-бутиловый эфир 4-(6-амино-пиридин-3-ил)-пиперазин-1-карбоновой кислоты (63,4 г, 0,2278 моль, 1,3 экв.) и смесь выдерживали при 20°С в течение 30 мин до растворения исходного вещества. В реактор загружали хлористый изопропилмагний (93,9 г, 0,193 моль, 1-ая загрузка 1,1 экв.) (2,0 М в THF, 1,1 экв.) при помощи насоса в течение 30 мин. Партию поддерживали при 20°С в течение 40 мин. В реактор загружали 6-бром-2-хлор-8-циклопентил-5-метил-8Н-пиридо[2,3-d]пиримидин-7-он (60,1 г, 0,1755 моль, 1 экс), весь сразу, и промывали при помощи THF (50 мл промывка). Дополнительную загрузку хлористого изопропилмагния (93,9 г, 0,193 моль, 1,1 экв. - 2-ая загрузка (2,0 М в THF, 1,1 экв.) добавляли при помощи насоса в течение 30 мин. Партию выдерживали при 20°С в течение 90 мин и затем нагревали от 20°С до 60°С.
После взаимодействия смесь THF (2,86 об.) и НОАс (1 экв.) использовали для гашения реакции. Затем в партию вносили затравку 0,5% масс/масс. трет-бутилового эфира 4-{6-[6-бром-8-циклопентил-5-метил-7-оксо-7,8-дигидро-пиридо[2,3-d]пиримидин-2-иламино]-пиридин-3-ил}-пиперазин-1-карбоновой кислоты и загружали смесь THF (1,14 об.) и НОАс (0,4 экв.) для завершения осаждения. После охлаждения до 20°С порцию фильтровали, промывали ацетоном (4 об.), водой (6 об.) и ацетоном (4 об.).
Влажный осадок сушили в вакууме при 65°С до постоянной массы с получением трет-бутилового эфира 4-{6-[6-бром-8-циклопентил-5-метил-7-оксо-7,8-дигидро-пиридо[2,3-d]пиримидин-2-иламино]-пиридин-3-ил}-пиперазин-1-карбоновой кислоты с выходом 93%. 1Н ЯМР (600 МГц, THF-d8): δ 9.36 (s, 1Н), 8.87 (s, 1H), 8.22 (d, J=8,8 Гц, 1H), 8.04 (d, J=2,9 Гц, 1H), 7.39 (dd, J=8,8, 2,9 Гц, 1H), 6.10 (m, 1H), 3.55 (уширенный, 4Н), 3.09 (уширенный, 4Н), 2.60 (s, 3Н), 2.30 (m, 2Н), 2.09 (m, 2Н), 1.85 (m, 2Н), 1.66 (m, 2Н), 1.46 (s, 9Н); 13С ЯМР (150 МГц, THF-d6): δ 159,5, 158,9, 157,7, 156,0, 155,0, 147,2, 144,62, 144,56, 138,0, 126,7, 117,6, 114,2, 108,4, 79,9, 55,5, 50,6, 44,7, 29,0, 28,7, 26,9, 18,1; МСВР: Рассчитано для C27H35N7O3Br1 (М+Н)+: 584,19797, Обнаружено: 584,19811.
Пример 4. Получение трет-бутилового эфира 4-{6-[6-(1-бутоксил-винил)-8-циклопентил-5-метил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-иламино]-пиридин-3-ил}-пиперазин-1-карбоновой кислоты
В продутый азотом сухой реактор загружали 1-бутанол (60 мл, 6 мл/г) и трет-бутиловый эфир 4-{6-[6-бром-8-циклопентил-5-метил-7-оксо-7,8-дигидро-пиридо[2,3-d]пиримидин-2-иламино]-пиридин-3-ил}-пиперазин-1-карбоновой кислоты (10 г, 0,017 моль) и добавляли бутил виниловый эфир (5,1 г, 0,051 моль, 3,0 экв.). Добавляли диизопропилэтиламин (5,3 г, 0,041 моль, 2,4 экв.) и смесь барботировали азотом через барботажную трубку в течение 30 минут. Добавляли ацетат палладия (0,16 г, 0,00068 моль, 0,0400 экв.) и бис(2-дифенилфосфинофенил)эфир (0,45 г, 0,00082 моль, 0,04800 экв.). Смесь нагревали до 95°С в течение 30 минут и партию перемешивали при 95°С в течение 2 часов. Смесь охлаждали до 80°С и отбирали пробы для наблюдения за окончанием реакции. После окончания добавляли воду (15 мл, 1,5 мл/г) и 1-бутанол (30 мл, 3 мл/г).
Раствор фильтровали через фильтр 0,45 микрон для удаления осажденного палладия. Добавляли воду (35 мл, 3,5 мл/г), затем 1,2 диаминопропан (6,3 г, 0,085 моль, 5,0 экв.). Смесь перемешивали при 70°С в течение по меньшей мере 30 минут. Перемешивание останавливали и смесь оставляли отстаиваться в течение 15 минут. Нижнюю, водную фазу отделяли и смесь охлаждали до 60°С в течение 30 минут. В смесь вносили затравку трет-бутилового эфира 4-{6-[6-(1-бутоксил-винил)-8-циклопентил-5-метил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-иламино]пиридин-3-ил}-пиперазин-1-карбоновой кислоты (Форма С) (50 мг, 0,005 г/г) и смесь выдерживали при 60°С в течение 90 минут.
После наблюдения кристаллизация смесь охлаждали до 50°С в течение одного часа и выдерживали при 50°С в течение трех часов. Смесь охлаждали до 30°С в течение трех часов и выдерживали при 30°С в течение двух часов, затем охлаждали до 20°С в течение четырех часов и выдерживали при 20°С в течение четырех часов. Суспензию фильтровали и промывали 1-бутанолом (10 мл, 1 мл/г). Осадок на фильтре продували и в смесь загружали 1-бутанол (10 мл, 1 мл/г) и суспензию перемешивали при 20°С в течение 1 часа. Осадок на фильтре продували. Смесь промывали метил-трет-бутиловым эфиром (20 мл, 2 мл/г) и осадок полностью освобождали от жидкости, используя продолжительные периоды продувания (2 часа или более). Осадок сушили при 70°С. Выход составляет 75-80%. 1Н ЯМР (600 МГц, DMSO-d6): δ 10.0 (s, 1Н), 8.87 (s, 1Н), 8.07 (d, J=2,9 Гц, 1Н), 7.91 (d, J=9,0 Гц, 1Н), 7.48 (dd, J=9,0, 2,9 Гц, 1Н), 5.83 (m, 1Н), 4.47 (d, J=1,6 Гц, 1Н), 4.05 (d, J=1,6 Гц, 1Н), 3.77 (t, J=6,4 Гц, 2Н), 3.48 (уширенный, 4Н), 3.11 (уширенный, 4Н), 2.37 (s, 3Н), 2.22 (m, 2Н), 1.89 (m, 2Н), 1.75 (m, 2Н), 1.61 (m, 2Н), 1.58 (m, 2Н), 1.43 (s, 9Н), 1.38 (m, 2Н), 0.90 (t, J=7,39 Гц, 3Н); 13С ЯМР (150 МГц, DMSO-d6): δ 160,9, 158,2, 157,3, 155,2, 154,6, 153,7, 145,0, 143,0, 142,6, 136,0, 125,8, 125,5, 114,6, 106,6, 87,8, 78,9, 66,8, 52,8, 48,5, 43,4, 42,5, 30,3, 28,0, 27,4, 25,1, 18,8, 14,4, 13,6; МСВР: Рассчитано для C33H46N7O4 (М+Н)+: 604,36058, обнаружено: 604,36049.
Промежуточный бутоксил-виниловый эфир может быть выделен в одной из нескольких полиморфных форм. Форму А выделяли в виде кинетического продукта при отсутствии затравки, в то время как Форму В выделяли в нескольких случаях, но наблюдали редко. Наиболее устойчивую кристаллическую форму бутоксил-винилового эфира, Форму С, получали путем внесения в реакционную смесь затравки кристаллов Формы С. Любую из этих полиморфных форм можно использовать в получении свободного основания соединения 1, но полиморфная Форма С бутоксил-винилового эфира является предпочтительной из-за легкой фильтруемости.
Данные PXRD для полиморфных Форм А, В и С промежуточного бутоксил-винилового эфира представлены в Таблицах 7, 8 и 9 соответственно.

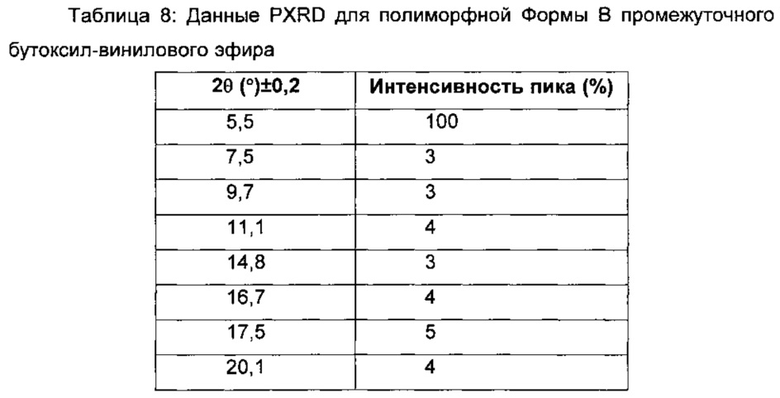
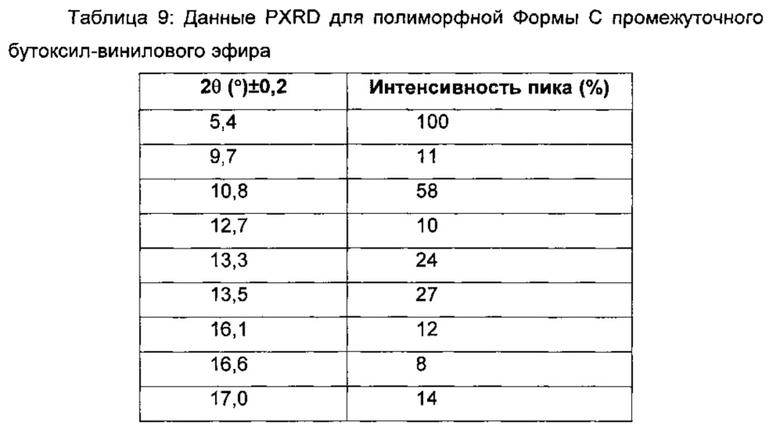
Пример 5. Получение свободного основания соединения 1 с небольшим размером частиц посредством способа разрушения солей
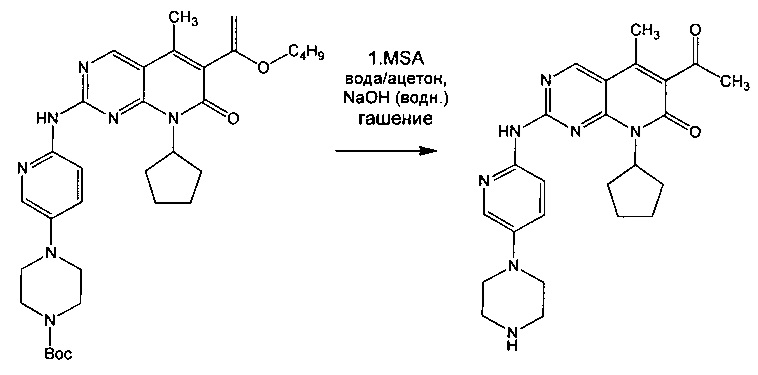
В реактор добавляли трет-бутиловый эфир 4-{6-[6-(1-бутоксил-винил)-8-циклопентил-5-метил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-иламино]-пиридин-3-ил}-пиперазин-1-карбоновой кислоты (2,70 кг, 4,47 мол, 1,0 экв.), затем смесь воды (27,00 л, 10 л/кг) и ацетона (13,50 л, 5 л/кг). Желтую суспензию нагревали до температуры от 50°С до 55°С. Раствор метансульфоновой кислоты (2,15 кг, 22,36 мол, 5,0 экв.), разбавленной водой (5,40 л, 2 л/кг исходного вещества) и ацетоном (5,40 л, 2 л/кг исходного вещества) добавляли в реактор в течение примерно 10 минут. Реакционную смесь выдерживали при температуре от 45°С до 55°С в течение по меньшей мере 12 часов. Во время реакции получали прозрачный желтый раствор.
Реакционную смесь охлаждали до 35°С и смесь 5% масс, раствора гидроксида натрия добавляли партиями в реактор для повышения рН реакционной смеси до значения более 9. Реактор охлаждали до температуры от 20°С до 25°С, гранулировали и фильтровали. Осадок промывали водой, затем ацетоном и сушили в вакууме.
Посредством этого способа образовывалось свободное основание соединения 1 с первичными частицами небольшого размера, которое было эквивалентно веществу, полученному при обработке гидрохлоридной соли соединения 1 водным NaOH в Примере 4 в WO 2005/005426.
В дополнение к типичной методике, предложенной выше (соответствующей Эксперименту S в Таблице 10), подвергали скринингу ряд кислот и водных систем растворителей для определения влияния на взаимодействие и последующее гашение и выделение свободного основания соединения 1. Скрининговые эксперименты в лабораторном масштабе проводили для определения условий взаимодействия для превращения промежуточного винилового эфира в свободное основание соединения 1. Результаты этих экспериментов по скринингу реакционных смесей представлены в Таблице 10, и они указывают на универсальность способа.

Пример 6. Преобразование свободного основания с небольшим размером частиц в свободное основание соединения 1 с большим размером частиц

В реактор добавляли свободное основание соединения 1 (20 г, 44,69 ммоль, 1,0 экв.), полученное согласно Примеру 5, затем 1-бутанол (320 мл, 16 мл/г) и анизол (480 мл, 24 мл/г). Желтую суспензию нагревали до температуры от 95°С до 100°С для достижения растворения. Реактор охлаждали до 80°С. Для индуцирования кристаллизации в раствор, находящийся в реакторе, загружали суспензию затравки, содержащую затравочные кристаллы (0,1 г, 0,2 ммоль, 0,005 экв.) свободного основания (Форма А) соединения 1, суспендированные в 1-бутаноле (5 мл, 0,25 мл/г исходного вещества). Полученную суспензию перемешивали при 80°С в течение 3 часов. Суспензию охлаждали до 10°С со скоростью 0,2°С/мин в течение 350 минут, гранулировали и фильтровали. Осадок промывали анизолом, затем гептаном и сушили в вакууме.
Этим способом получали кристаллы свободного основания соединения 1 с большим размером первичных частиц, которые были эквивалентными свободному основанию, полученному с использованием однореакторного способа, описанного в Примере 7 ниже.
Пример 7. Однореакторный способ получения свободного основания соединения 1 с большим размером частиц
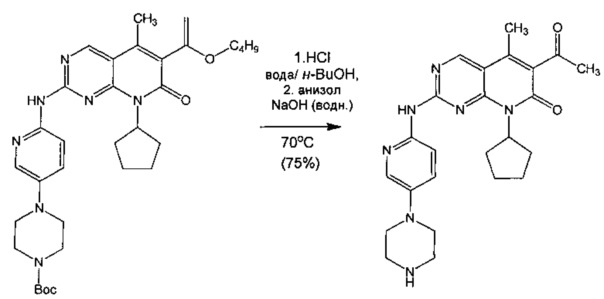
В реактор добавляли воду (200 мл, 10 мл/г) и трет-бутиловый эфир 4-{6-[6-(1-бутоксил-винил)-8-циклопентил-5-метил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-иламино]-пиридин-3-ил}-пиперазин-1-карбоновой кислоты (20 г, 33,1 ммоль, 1,0 экв.), затем 1-BuOH (232 мл, 11,6 мл/г) для смывания любых твердых веществ в реактор. Желтую суспензию нагревали до 70°С. Образовывалась двухфазная жидкая смесь. Концентрированный раствор HCl (16,3 г, 165,5 ммоль, 5,0 экв.) добавляли в реактор в течение примерно 10 минут. Реакционную смесь выдерживали при 70°С в течение 4-6 часов. Через 3 часа получали прозрачный желтый двухфазный раствор.
В реакционную смесь добавляли анизол (356 мл, 17,8 мл/г). Поддерживая смесь при 70°С, в реактор добавляли раствор водного NaOH (17,2 г, 172,1 ммоль, 5,2 экв.) (40% масс, раствор) в течение 20 минут для повышения рН реакционной смеси до более 10. Двухфазную смесь перемешивали в течение 30 минут после окончания добавления NaOH.
Фазы разделяли и органическую фазу дважды промывали водой. Затем партию нагревали до 80°С и без крупинок фильтровали в сосуд для кристаллизации, промывая фильтр бутанолом. Затем партию дистиллировали для удаления воды и доводили температуру до 120°С. Затем партию охлаждали до 80°С и вводили затравку в виде затравочной суспензии, содержащей затравочные кристаллы (0,015 г, 0,033 ммоль, 0,1% масс. в расчете на соединение 1) свободного основания (Форма А) соединения 1, и 1-BuOH (10 мл, 0,5 мл/г). Затем партию охлаждали до 30°С со скоростью 0,2°С/мин и затем выдерживал в течение трех циклов, где температуру понижали каждый раз на 10°С. В конечном цикле партию охлаждали до 10°С, гранулировали и фильтровали. Осадок дважды промывали гептаном и сушили в вакууме. После сушки подтвердили, что образец представляет собой монокристаллическую полиморфную Форму А.
1Н ЯМР (600 МГц, DMSO-d6/TFA): δ 10.41 (s, 0.75Н), 9.03 (s, 0.25Н), 8.98 (s, 2Н), 8.12 (d, J=3,0 Гц, 1H), 7.90 (d, J=9,1 Гц, 1H), 7.63 (dd, J=9,1, 3,0 Гц, 1H), 5.84 (m, 1Н), 3.40 (уширенный, 4Н), 3.29 (уширенный, 4Н), 2.43 (s, 3Н), 2.33 (s, 3Н), 2.21 (m, 2Н), 1.91 (m, 2Н), 1.79 (m, 2Н), 1.59 (m, 2Н); 13С ЯМР (150 МГц, DMSO-d6/TFA): δ 202,4, 160,7, 154,8, 158,3, 158,0, 144,9, 142,3, 142,0, 134,6, 129,7, 126,7, 115,3, 107,0, 53,0, 45,6, 42,6, 31,3, 27,6, 25,2, 13,7; МСВР: Рассчитано для C24H30N7O2 (М+Н)+: 448,24555, обнаружено: 448,24540.
Ниже представлены сравнительные данные PSA, SSA и поверхностной энергии для композиций свободного основания соединения 1 с небольшим размером первичных частиц и с большим размером первичных частиц. Во всех случаях свободное основание выделяли в виде полиморфной Формы А.
Порошковая рентгеновская дифракция (PXRD)
Экспериментальная часть
Анализ порошковой дифракции проводили, используя дифрактометр Bruker D8, оснащенный Си источником излучения, с фиксированными размерами щелей (расходимость = 1,0 мм, антирассеивающая щель = 0,6 мм и приемная щель = 0,6 мм) и сцинтилляционным детектором-счетчиком. Данные собирали на гониометре Theta-Theta при длине волны Cu Кα1=1,54056 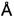 от 3,0 до 40,0 градусов 2-тэта, используя величину шага 0,040 градусов и время шага 2,0 секунд. Напряжение и силу тока в рентгеновской трубке устанавливали равными 40 кВ и 40 мА соответственно. Образцы подготавливали путем размещения на никелевом диске (Gasser & Sons, Inc. Commack, NY) и вращали во время сбора данных. Данные собирали и анализировали, используя программу Bruker DIFFRAC Plus (Version 2.6). Файлы (.raw) с данными PXRD не обрабатывали до поиска пика. Обычно использовали пороговую величину 1 и величину ширины 0,3 для предварительной оценки пиков. Результат автоматических оценок проверяли визуально, чтобы убедиться в его достоверности, и при необходимости вручную выполняли корректировки. Кроме того, пики оценивали вручную в спектрах, если это было целесообразно.
от 3,0 до 40,0 градусов 2-тэта, используя величину шага 0,040 градусов и время шага 2,0 секунд. Напряжение и силу тока в рентгеновской трубке устанавливали равными 40 кВ и 40 мА соответственно. Образцы подготавливали путем размещения на никелевом диске (Gasser & Sons, Inc. Commack, NY) и вращали во время сбора данных. Данные собирали и анализировали, используя программу Bruker DIFFRAC Plus (Version 2.6). Файлы (.raw) с данными PXRD не обрабатывали до поиска пика. Обычно использовали пороговую величину 1 и величину ширины 0,3 для предварительной оценки пиков. Результат автоматических оценок проверяли визуально, чтобы убедиться в его достоверности, и при необходимости вручную выполняли корректировки. Кроме того, пики оценивали вручную в спектрах, если это было целесообразно.
ssNMR Экспериментальная часть
Углеродные спектры Формы А получали на 4 мм роторе за 2048 сканирований при задержке повторного цикла 25 секунд и 2 миллисекундах кросс-поляризации. Во время сбора данных производили отщепление протонов при 100 кГц. Углеродные спектры Формы В получали на 4 мм роторе за 2048 сканирований, за 128 сканирований собирали при задержке повторного цикла 4,5 секунд с 2 миллисекундами кросс-поляризации. Во время сбора данных производили отщепление протонов при 70 кГц и полное подавление боковых полос при вращении (TOSS).
Способ измерения
Примерно 80 мг образца помещали в 4 мм ротор из ZrO2. Спектры собирали при температуре и давлении окружающей среды на 4 мм датчике CPMAS Bruker-Biospin, установленном в широкопросветном ЯМР-спектрометре Bruker-Biospin Avance III 500 МГц (частота 1Н). Наполненный ротор ориентировали под магическим углом и вращали при 15,0 кГц. Спектры 13С твердого тела собирали с использованием эксперимента кросс-поляризации и вращения под магическим углом (CPMAS) с фазомодулированным отщеплением протона. Время контакта при кросс-поляризации устанавливали равным 2,0 мс. Во время сбора данных для отщепления протона использовали поле примерно 100 кГц. Углеродный спектр Формы А соединения 1 получали за 512 сканирований при задержке повторного цикла 25 секунд. Спектр показан на Фиг. 2 и данные представлены в Таблице 2. Углеродный спектр Формы В соединения 1 получали за 2048 сканирований при задержке повторного цикла 4,5 секунды. Углеродные спектры привязывали, используя в качестве внешнего стандарта кристаллический адамантан и устанавливая его резонанс в сторону сильного поля равным 29,5 млн-1. Спектр показан на Фиг. 4 и данные представлены в Таблице 4.
Анализ размера частиц
Размер частиц анализировали, используя метод лазерной дифракции (или малоугольное рассеивания света), посредством диспергирования сухого порошка образца при помощи сжатого воздуха. Конкретно, распределение частиц по размерам анализировали, используя систему Sympatec HELOS RODOS, оснащенную дозатором сухого порошка Vibri. Образец порошка диспергировали при давлении рассеивания 0,5 бар (5×104 Па). В некоторых случаях применяли микродозирующее устройство Aspiros и образец порошка диспергировали при давлении рассеивания 0,2 бар (2×104 Па). Выбирали подходящие объективы, чтобы охватить диапазон размеров частиц каждого образца.
Результаты
Сравнительные данные для четырех партий API представлены в Таблице 11 ниже с применением устройств либо Vibri, либо Aspiros для диспергирования образца. Партия №4 имела D90 примерно 75 мкм, в то время как обе партии №№1 и 2 имели D90 примерно 45 мкм. Данные о размере частиц, полученные посредством лазерной дифракции, подтверждают наблюдения SEM для этих партий.
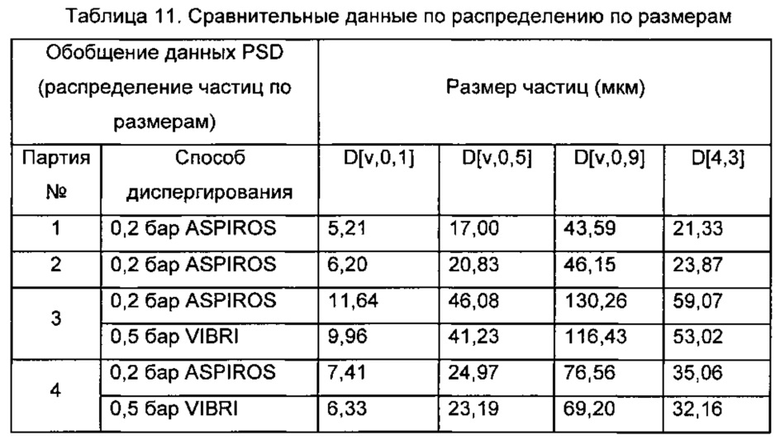
Сканирующая электронная микроскопия (SEM)
Сканирующую электронную микроскопию выполняли в стандартных условиях. На Фиг. 5 представлено полученное посредством SEM (200x увеличение) изображение Формы А свободного основания соединения 1, перекристаллизованной из смеси 40% BuOH/анизол. На Фиг. 6 представлено полученное посредством SEM (1500х увеличение) изображение Формы А свободного основания соединения 1, выделенной посредством стандартного способа получения свободного основания
Анализ прилипания
Пуансон MASS (Проверка адгезии вещества на прилипание) был разработан собственными силами для количественной оценки склонности к прилипанию таблеточных композиций посредством взвешивания количества прилипшего порошка на съемном наконечнике пуансона после ряда сжатий. Это испытание дает возможность разработчикам композиций объективно и быстро оценить риска прилипания к пуансону во время разработки лекарственного продукта и устранить прилипание, наблюдаемое во время изготовления таблеток для клиник.
Для приготовления образца для испытания на MASS пуансоне, 10 г API разбавляли в слегка смазанной стандартной смеси (10% API, 89,75% Avicel РН102 и 0,25% стеарата магния) и смешивали в бутылке (500 мл бутылка из желтого стекла) за 500 вращений. Массу порошка, прилипшего к съемному наконечнику пуансона (1/2" круглый плоскоцилиндрический), оценивали, используя микровесы периодически вплоть до 100 сжатий ~250 мгВ таблеток в целевой твердой фракции 0,85.
Профиль MASS пуансона свободного основания соединения 1, смешанного в стандартной смеси, продемонстрировал положительный ответ. Фотографии наконечников пуансона в конце выполнения сжатий подтвердили, что порошок прилипал к наконечникам (данные не показаны). Для информации, контрольный образец стандартной смеси не является липким и будет иметь менее 10 мкг прилипшего порошка. Обнаружено, что исследуемый способ причисляет склонность к прилипанию новых серий API к сериям известных веществ.
Измерение удельной площади поверхности (SSA) (BET (метод Брунауэра - Эммета - Теллера) с азотом)
Аппаратура
Удельную площадь поверхности (SSA) (BET с азотом) измеряли, используя анализатора удельной площади поверхности Micromeritics TriStar II 3020 вместе с устройством Micromeritics SmartPrep (Micromeritics U.K. Ltd., Ste 2, The Stables Hexton Manor, Hexton, Hertfordshire SG5 3JH, England). Образцы подвергали анализу адсорбции азота согласно BET для определения удельной площади поверхности образцов.
Установки
Версия программного обеспечения: TriStar II Confirm (1.03 или эквивалент)
Адсорбат: Азот
Пробирка для образца: 3/8" мм плоскодонная ячейка со стеклянными палочками наполнителя
Масса образцов *: Приблизительно ¾ полной ячейки
Приготовление образца: SmartPrep (дегазирование в потоке с использованием азота)
Условия выхода газа: 16 часов при 25°С в потоке газа (нарастание со скоростью 10°С/мин)
Изотермический кожух: использовали
Изотермические точки сбора: 11 точек BET в диапазоне 0,05-0,30 Р/Ро
Изотермический диапазон анализа данных: 7 точек BET в диапазоне 0,05-0,20 Р/Ро
Испытание на герметичность: 120 с
Свободное пространство: измеряли
Время откачивания: 1 ч
Продолжительность тестирования после дегазирования: 180 с
Интервал уравновешивания: 10 с
Таймаут уравновешивания: 600 с
*Масса образца варьируется в соответствии с размером частиц испытываемого образца. Для образцов, где размер частиц был относительно небольшим, примерно 0,50 г вещества требовалось для заполнения баллона ячейки на ¾, а когда размер частиц образца являлся относительно большим, требовалось 0,75 г вещества для заполнения баллона ячейки на ¾.
Вычисления и отчет
Удельную площадь поверхности регистрировали в диапазоне 0,05-0,20 Р/Ро, используя 7 точек BET в определении с тремя повторами. Определяли массу образца, удельную площадь поверхности, константу BET (величину 'С') и коэффициент корреляции для каждого повтора.
Результаты
В Таблице 12 представлены BET-N2 SSA для четырех партий API свободного основания соединения 1, где одна содержит первичные частицы API небольшого размера, полученные посредством традиционного способа разрушения солей (партия 5), и три партии содержат частицы API большого размера, полученные согласно настоящему изобретению. Партия 5 содержала свободное основание соединения 1, имеющее небольшие первичные частицы и большие агломераты, которые были очень склонны к статической электризации и прилипанию. Партия 6 была получена с применением циклического изменения температуры и имела типичное распределение частиц по размерам (PSD) свободного основания соединения 1 с большим размером частиц, с VMD (объемный срединный диаметр) примерно 17 мкм. Партия 7 показывала PSD, аналогичный партии 6. Партия 8 представляет собой типичную ICH партию частиц свободного основания соединения 1 большого размера, также полученного посредством циклического изменения температуры. Те же самые партии использовали в определениях поверхностной энергии ниже.
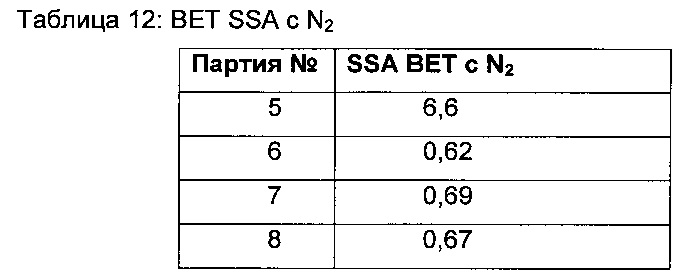
Измерение поверхностной энергии посредством обращенно-фазовой газовой хроматографии (IGC)
Достаточное количество образца помещали в силанизированную стеклянную колонку с порошковой массой, надежно закрепленной в колонке посредством пробок из стекловаты, вставленных в оба конца Колонку уравновешивали путем пропускания потока сухого азота через порошковую массу в течение времени, достаточного для удаления любых адсорбатов с поверхности. Измерения выполняли посредством ввода ряда образцов паров алканов (нонана, октана, гептана и гексана) в поток газа-носителя в достаточно низких концентрациях, чтобы допускать неограниченное разбавление паров алкана в потоке азота, и регистрации времени, необходимого для элюирования каждого пара через колонку. Графическое изображение времени удерживания (скорректированное с учетом «мертвого объема», пространства между частицами в заполненной колонке) как функции от площади поперечного сечения и поверхностного натяжения используемых тест-молекул паров алканов давало линию с наклоном, показывающим поверхностную энергию исследуемого порошка твердого вещества
Результаты
В Таблице 13 представлены данные о дисперсионной поверхностной энергии (мДж/м2), полученные для четырех партий свободного основания соединения 1, то есть партий 5-8, описанных выше в отношении данных о SSA. Партия 5 представляет собой свободное основание с частицами небольшого размера, и партии 6-8 включают свободное основание API с частицами большого размера.

Все публикации и патентные заявки, указанные в данном описании изобретения, и все ссылки, указанные в нем, включены в данное описание изобретения посредством ссылки, как если бы каждая отдельная публикация, или патентная заявка, или ссылка были конкретно и индивидуально указаны как включенные посредством ссылки. Хотя вышеупомянутое изобретение описано подробно посредством иллюстрации и примера для целей ясности понимания, специалисту в данной области техники будет очевидно в свете идей данного изобретения, что в изобретении могут быть осуществлены некоторые изменения и модификации без отклонения от существа или объема прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИЗЕТИОНАТНАЯ СОЛЬ СЕЛЕКТИВНОГО ИНГИБИТОРА CDK4 | 2004 |
|
RU2317296C2 |
| НОВЫЕ КРИСТАЛЛИЧЕСКИЕ ФОРМЫ НАТРИЕВОЙ СОЛИ(4-{ 4-[5-(6-ТРИФТОРМЕТИЛ-ПИРИДИН-3-ИЛАМИНО) ПИРИДИН-2-ИЛ] ФЕНИЛ} ЦИКЛОГЕКСИЛ) УКСУСНОЙ КИСЛОТЫ | 2011 |
|
RU2612556C2 |
| КРИСТАЛЛИЧЕСКИЙ ИНГИБИТОР FGFR4 И ЕГО ПРИМЕНЕНИЕ | 2016 |
|
RU2763328C2 |
| ПОЛИМОРФНЫЕ И АМОРФНЫЕ ФОРМЫ {2-ФТОР-5-[3-((Е)-2-ПИРИДИН-2-ИЛВИНИЛ)-1Н-ИНДАЗОЛ-6-ИЛАМИНО]ФЕНИЛ}АМИДА 2,5-ДИМЕТИЛ-2Н-ПИРАЗОЛ-3-КАРБОНОВОЙ КИСЛОТЫ | 2005 |
|
RU2324692C1 |
| ПОЛИМОРФЫ С-MET/HGFR ИНГИБИТОРА | 2006 |
|
RU2387650C2 |
| АНТИПРОЛИФЕРАТИВНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ PAH | 2020 |
|
RU2786588C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ И СОЛЬВАТЫ АВТ-263 ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С БЕЛКОМ BCL-2 | 2010 |
|
RU2551376C2 |
| СОЛИ И КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНДУЦИРУЮЩЕГО АПОПТОЗ АГЕНТА | 2011 |
|
RU2628560C2 |
| ГИДРОХЛОРИД (1S,2S,4R)-4-{ 4-[(1S)-2,3-ДИГИДРО-1Н-ИНДЕН-1-ИЛАМИНО]-7Н-ПИРРОЛО [2,3-d]ПИРИМИДИН-7-ИЛ} -2-ГИДРОКСИЦИКЛОПЕНТИЛ)МЕТИЛ СУЛЬФАМАТА | 2010 |
|
RU2562245C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-[5-(3,5-ДИФТОРБЕНЗИЛ)-1Н-ИНДАЗОЛ-3-ИЛ]-4-(4-МЕТИЛПИПЕРАЗИН-1-ИЛ)-2-(ТЕТРАГИДРОПИРАН-4-ИЛАМИНО)БЕНЗАМИДА | 2013 |
|
RU2602071C2 |
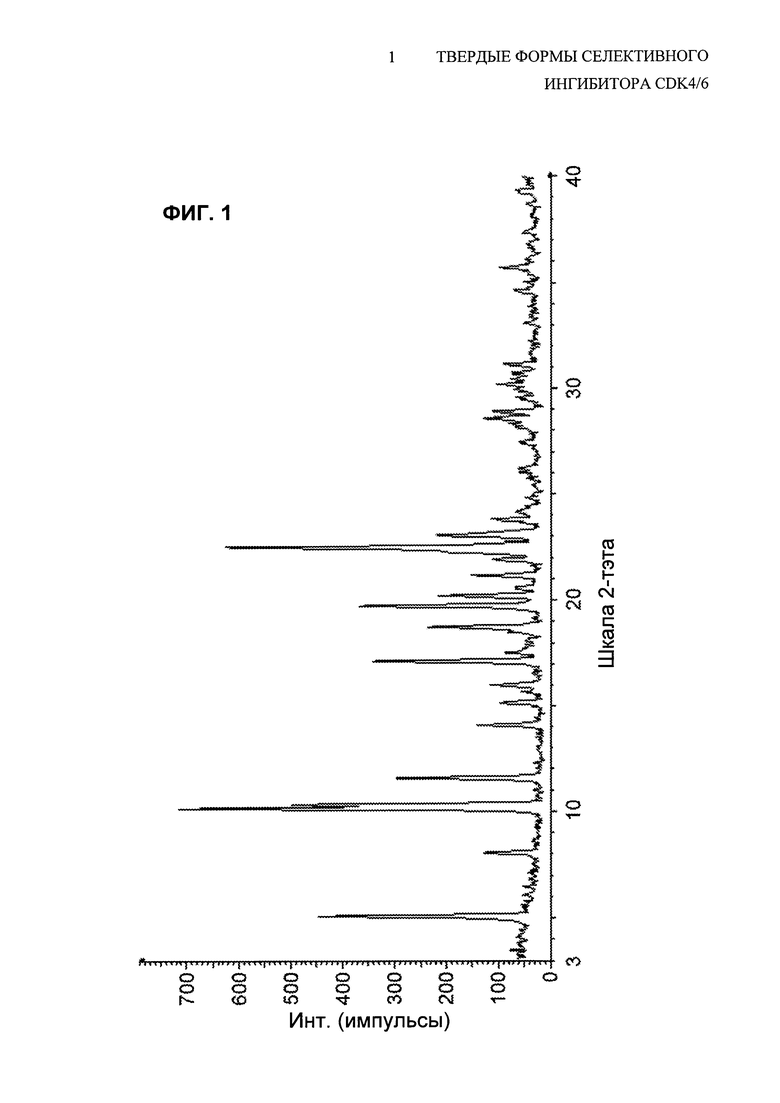
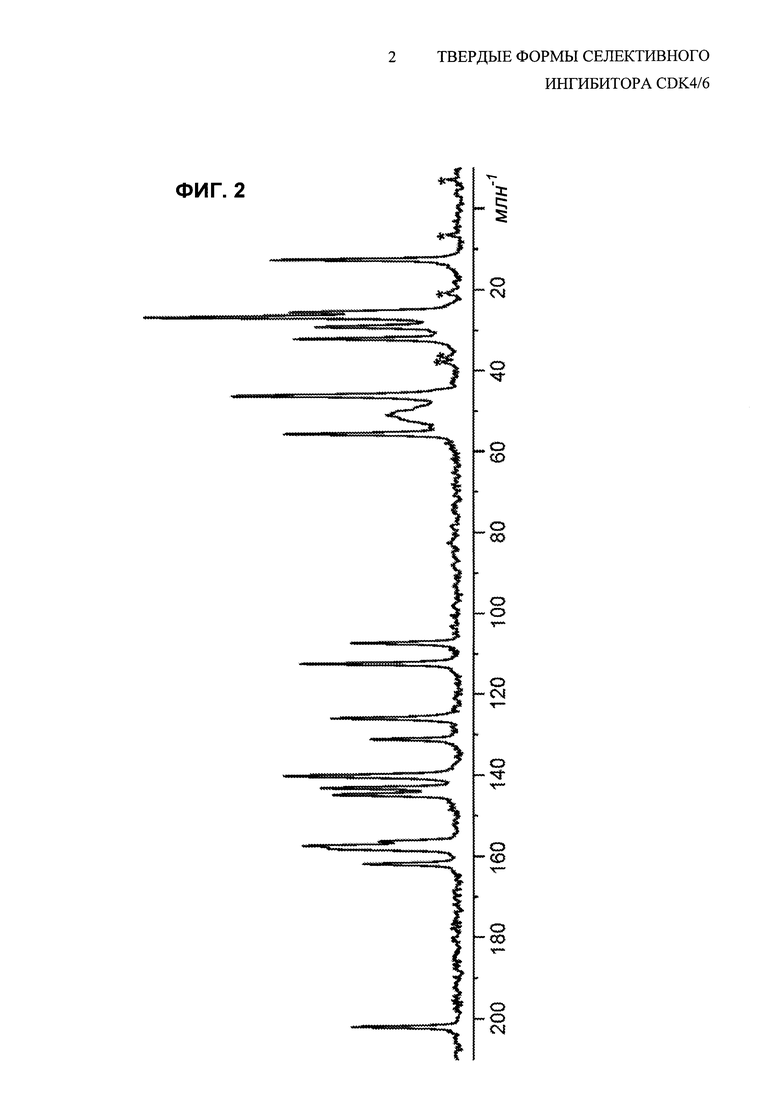

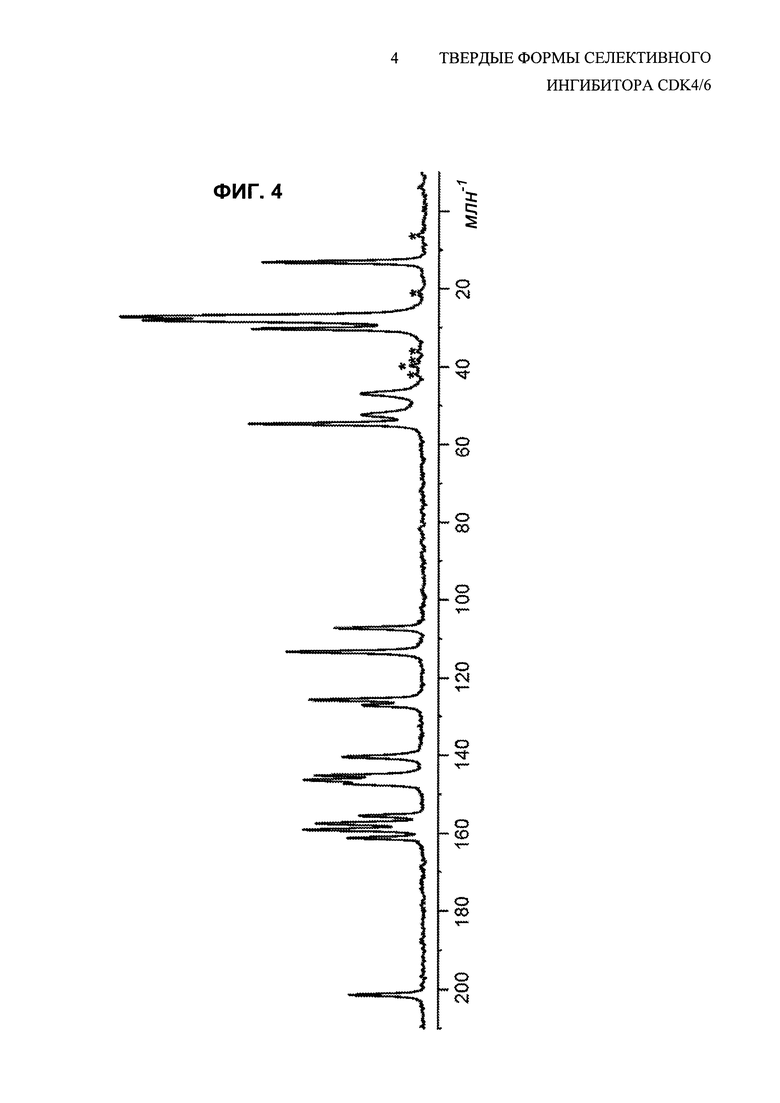
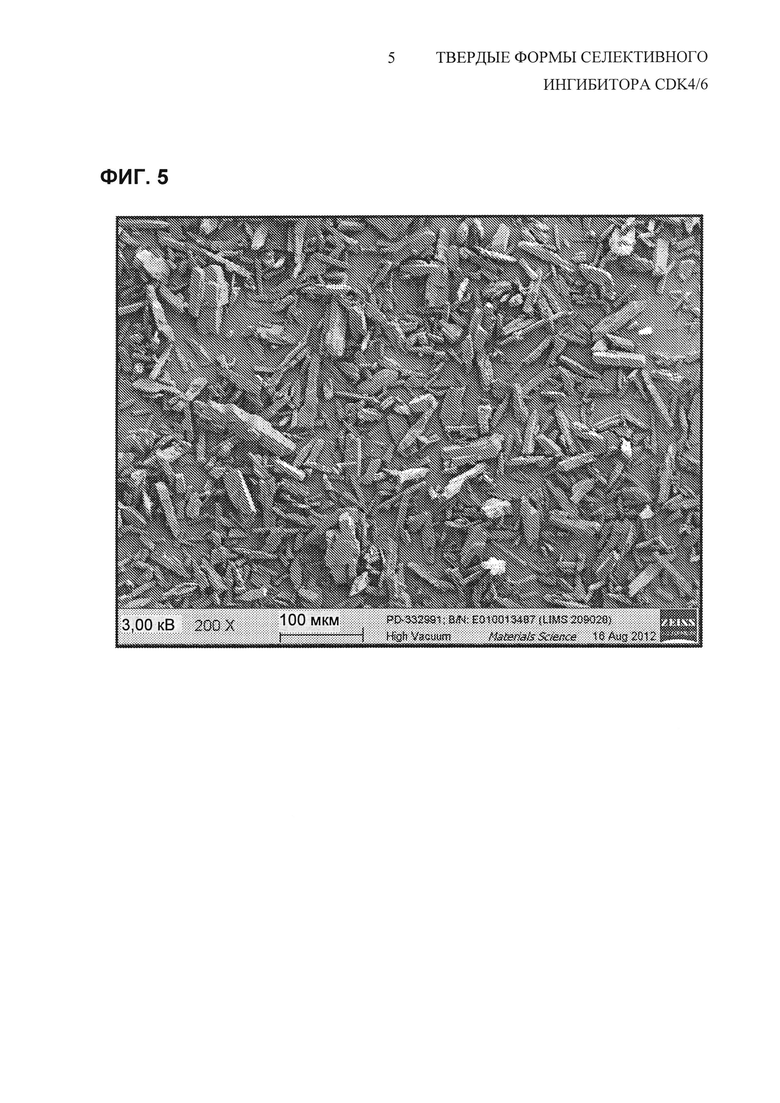

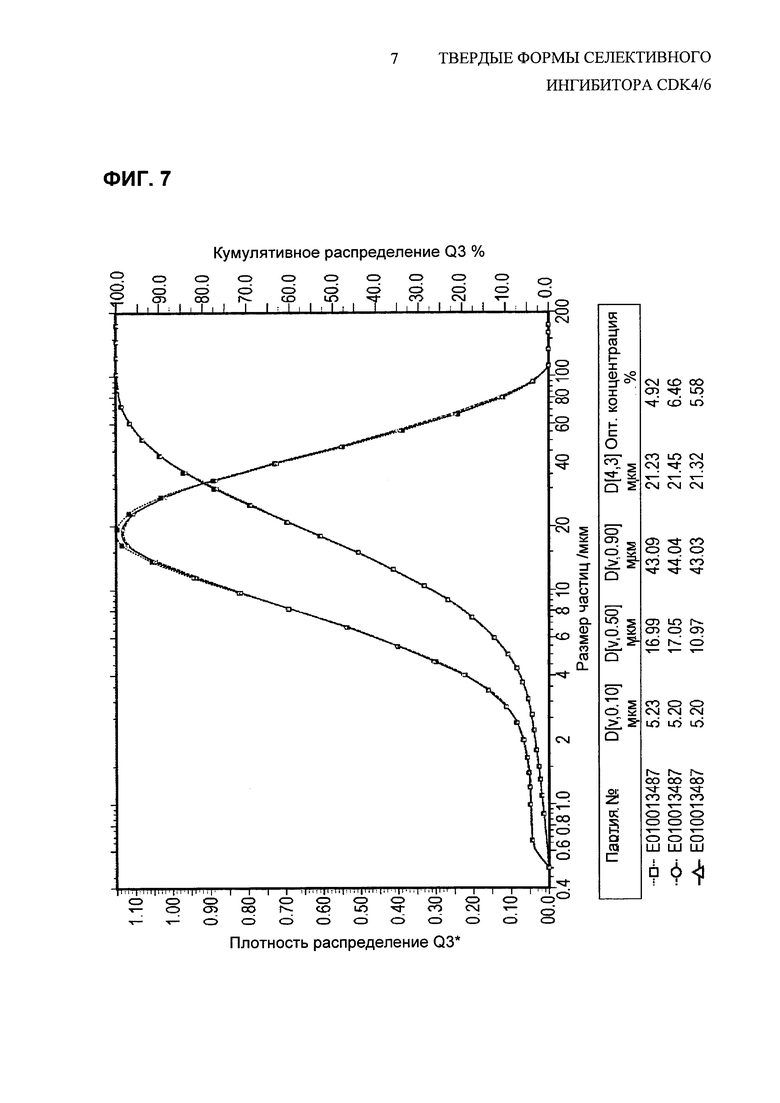
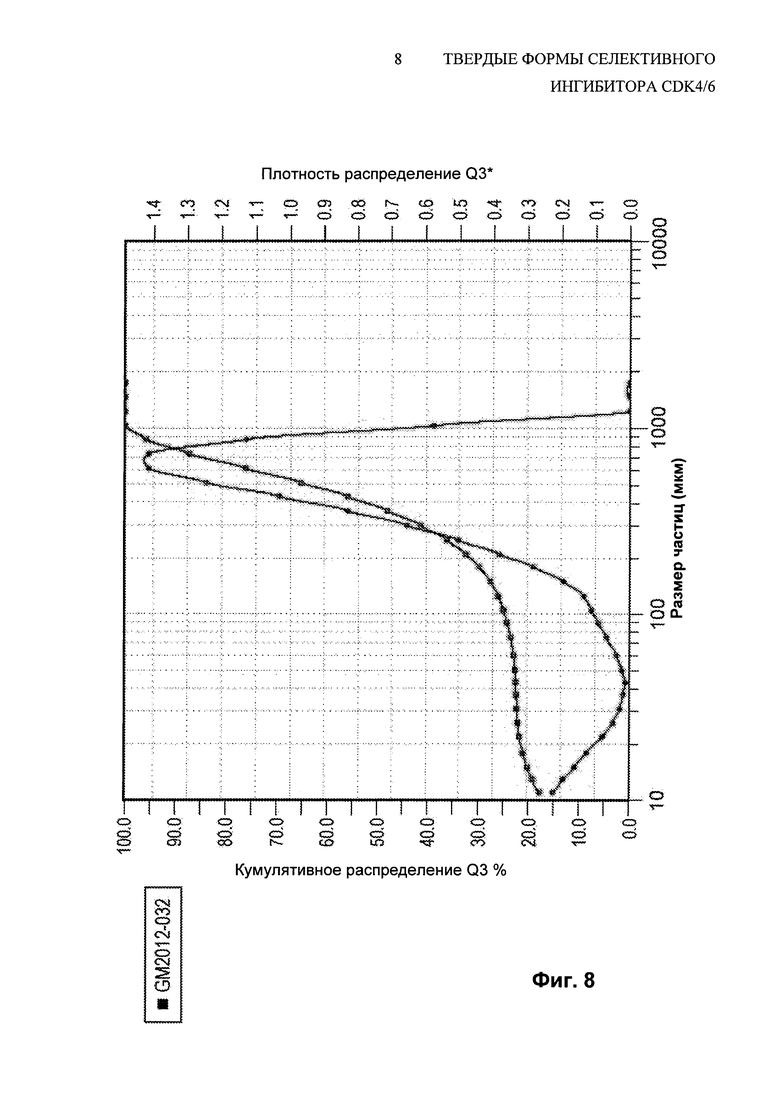
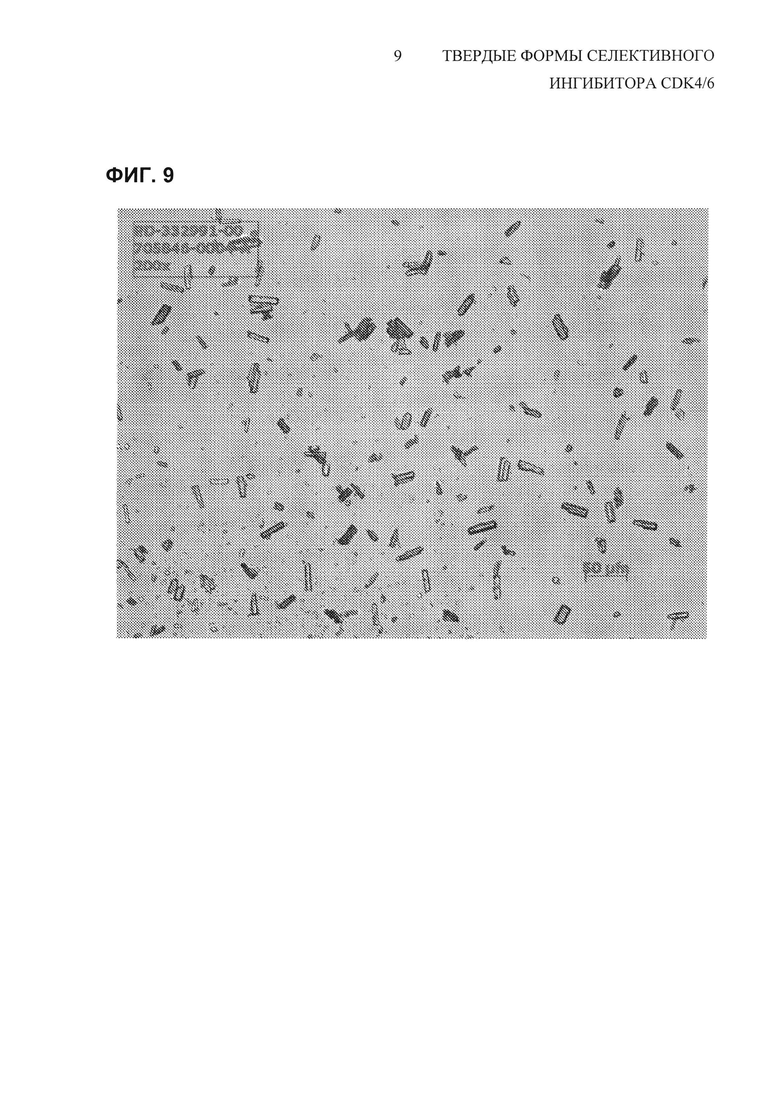
Изобретение относится к кристаллическому свободному основанию 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]пиримидин-7-она, представляющему собой полиморфную Форму А, имеющему удельную площадь поверхности менее или равную 2 м2/г и порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ) 8,0±0,2, 10,1±0,2 и 11,5±0,2, а также к фармацевтическим композициям на его основе. Технический результат: получено свободное основание, имеющее более крупный размер частиц и улучшенные физико-химические свойства, которое может применяться в лечении клеточно-пролиферативных заболеваний, таких как рак. 2 н. и 11 з.п. ф-лы, 9 ил., 13 табл., 7 пр.
1. Кристаллическое свободное основание 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]пиримидин-7-она, представляющее собой полиморфную Форму А, имеющее удельную площадь поверхности менее или равную 2 м2/г и порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ) 8,0±0,2, 10,1±0,2 и 11,5±0,2.
2. Свободное основание по п. 1, имеющее удельную площадь поверхности менее или равную 1 м2/г.
3. Свободное основание по п. 1, имеющее порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ) 8,0±0,2, 10,1±0,2, 10,3±0,2 и 11,5±0,2.
4. Свободное основание по п. 1, имеющее порошковую рентгеновскую дифрактограмму, содержащую пики с углами дифракции (2θ), по существу, такими, как показано на Фиг. 1.
5. Свободное основание по п. 1, имеющее спектр 13С ЯМР твердого тела, содержащий следующие резонансные значения (млн-1): 12,5 млн-1 ±0,2 млн-1.
6. Свободное основание по п. 5, имеющее спектр 13С ЯМР твердого тела, содержащий следующие резонансные значения (млн-1): 12,5 млн-1 и 112,4 млн-1 ±0,2 млн-1.
7. Свободное основание по п. 5, имеющее спектр 13С ЯМР твердого тела, содержащий следующие резонансные значения (млн-1): 12,5 млн-1, 112,4 млн-1 и 143,2 млн-1 ±0,2 млн-1.
8. Свободное основание по п. 1, имеющее размер первичных частиц от примерно 5 до примерно 150 мкм.
9. Свободное основание по п. 1, имеющее распределение первичных частиц по размерам, характеризующееся (1) величиной D10 от примерно 5 до примерно 10 мкм; (2) величиной D90 от примерно 30 до примерно 125 мкм; или (3) величиной D50 от примерно 10 до примерно 45 мкм; или комбинацией (1), (2) и (3).
10. Свободное основание по любому из пп. 1-9, имеющее соотношение (D90-D10)/D50 в распределении первичных частиц по размерам от примерно 2 до примерно 3.
11. Фармацевтическая композиция для лечения рака, содержащая свободное основание по любому из пп. 1-10 и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или эксципиент.
12. Свободное основание по п. 1, имеющее распределение первичных частиц по размерам, характеризующееся величиной D90 от 30 мкм ±20% до 65 мкм ±20%.
13. Свободное основание по п. 1, имеющее средний объемный диаметр D[4,3] от 15 мкм ±20% до 40 мкм ±20%.
| ИЗЕТИОНАТНАЯ СОЛЬ СЕЛЕКТИВНОГО ИНГИБИТОРА CDK4 | 2004 |
|
RU2317296C2 |
| УСТРОЙСТВО АКТИВНОГО КОНТРОЛЯ НА ВНУТРИШЛНФОВАЛЬНОМ СТАНКЕ | 0 |
|
SU170741A1 |
| WO03062236 A1, 31.07.2003. | |||

