ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Трансплантация является потенциальным подходом к лечению индивидуумов с аутоиммунными расстройствами, такими как сахарный диабет I типа (T1DM), но его применимость ограничена острым и хроническим иммунным отторжением трансплантированных клеток (N. Papeta et al. Transplantation 83, 174 (Jan 27, 2007); A. M. Shapiro et al. The New England journal of medicine 355, 1318 (Sep 28, 2006); J. S. Kaddis et al. JAMA 301, 1580 (Apr 15, 2009); R. P. Robertson. The New England journal of medicine 350, 694 (Feb 12, 2004); R. B. Jalili et al. Diabetes 59, 2219 (Sep, 2010); and V. Vaithilingam, The review of diabetic studies: 7, 62 (Spring, 2010)). В настоящее время с иммунным отторжением борются непрерывным подавлением системного иммунитета, т.е. подхода, который не демонстрирует достоверной долговременной эффективности, но при этом подвергает реципиента повышенным рискам инфекции и рака (A. G. Mallett, G. S. Korbutt. Tissue engineering. Part A 15, 1301 (Jun, 2009); N. Sakata et al. World journal of gastrointestinal pathophysiology 3, 19 (Feb 15, 2012); M. C. Poznansky et al. The Journal of clinical investigation 109, 1101 (Apr, 2002); and M. C. Poznansky et al. Nature medicine 6, 543 (May, 2000)). Были бы желательны альтернативные виды терапии, которые могли бы преодолеть необходимость в системном подавлении иммунитета благодаря специфическому модулированию иммунитета на ограниченном участке организма.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Полипептиды CXCL12 способны отталкивать эффекторные T-клетки, при этом мобилизуя иммуносупрессорные регуляторные T-клетки (T-супрессоры) на данном участке организма. В последнее время было установлено, что полипептиды CXCL12 способны преодолевать как острое, так и хроническое иммунное разрушение имплантированной матрицы сайт-специфичным образом, устраняя необходимость в одновременном системном подавлении иммунитета.
В одном из аспектов, настоящее изобретение относится к композициям, включающим как минимум одну клетку, инкапсулированную в матрицу, высвобождающую полипептид CXCL12 (далее по тексту иногда именуемую «элюирующей матрицей»).
В некоторых вариантах осуществления, матрица, высвобождающая полипептид CXCL12, может характеризоваться высвобождением полипептида CXCL12 со скоростью, достаточной для подавления действия эффекторных T-клеток. Например, в некоторых вариантах осуществления, матрица, высвобождающая полипептид CXCL12, может характеризоваться высвобождением полипептида CXCL12 со скоростью как минимум примерно 1,0 нг/мл/ч. В некоторых вариантах осуществления, полипептид CXCL12 может высвобождаться из матрицы, элюирующей полипептид CXCL12, со скоростью как минимум примерно 1,5 нг/мл/ч, как минимум примерно 2 нг/мл/ч, как минимум примерно 2,5 нг/мл/ч, как минимум примерно 3 нг/мл/ч, как минимум примерно 4 нг/мл/ч, как минимум примерно 5 нг/мл/ч или более. В некоторых вариантах осуществления, полипептид CXCL12 может высвобождаться со скоростью от примерно 1 нг/мл/ч до примерно 3 нг/мл/ч. В некоторых вариантах осуществления, полипептид CXCL12 может высвобождаться со скоростью примерно 1,75 нг/мл/ч. Только в качестве примера, композиция по настоящему изобретению может характеризоваться способностью отталкивать эффекторные T-клетки in vitro в камере Бойдена.
Полипептид CXCL12 может присутствовать в элюирующей матрице по настоящему изобретению в любой концентрации. В некоторых вариантах осуществления, концентрация полипептида CXCL12 может быть оптимизирована, например, для достижения желаемой скорости высвобождения и/или продолжительности высвобождения полипептида CXCL12 из элюирующей матрицы. В некоторых вариантах осуществления, полипептид CXCL12 может находиться в матрице в концентрации примерно 100 нг/мл. В некоторых вариантах осуществления, полипептид CXCL12 может находиться в матрице в концентрации от примерно 100 нг/мл до примерно 1 мкг/мл. В некоторых вариантах осуществления, концентрация полипептида CXCL12 в элюирующей матрице может сохраняться на уровне от примерно 100 нг/мл до примерно 1 мкг/мл в течение от примерно 3 месяцев до примерно 2 лет. В некоторых вариантах осуществления, концентрация полипептида CXCL12 в элюирующей матрице может сохраняться на уровне от примерно 100 нг/мл до примерно 1 мкг/мл в течение от примерно 3 месяцев до примерно 2 лет после имплантации композиции в организм субъекта. В некоторых вариантах осуществления, сохраняющаяся в матрице концентрация полипептида CXCL12 может составлять примерно 100-200 нг/мл.
Для формирования матрицы, высвобождающей полипептид CXCL12, полипептиды CXCL12 могут быть предварительно введены в элюирующую матрицу или вырабатываться in situ в элюирующей матрице. Например, в некоторых вариантах осуществления, полипептиды CXCL12 в матрице по настоящему изобретению может продуцироваться клетками, секретирующими полипептид CXCL12, или клетками, сконструированными для секреции полипептидов CXCL12. В одном из вариантов осуществления, полипептиды CXCL12 в матрице могут вырабатываться островковыми клетками, находящимися в матрице.
В некоторых вариантах осуществления, полипептиды CXCL12, находящиеся в элюирующей матрице по настоящему изобретению, могут включать аминокислотные последовательности, соответствующие виду субъекта, подвергаемого лечению. Например, в некоторых вариантах осуществления, полипептид CXCL12 может включать человеческий полипептид CXCL12.
Матрица, высвобождающая полипептид CXCL12, может характеризоваться различными структурами матрицы. Например, в некоторых вариантах осуществления, матрица, высвобождающая полипептид CXCL12, может образовывать капсулу. В некоторых вариантах осуществления, матрица, высвобождающая полипептид CXCL12, может иметь форму твердой или вспененной матрицы. В некоторых вариантах осуществления, матрица, высвобождающая полипептид CXCL12, может образовывать многокамерную или многослойную матрицу. В этих вариантах осуществления, клетка(и) и полипептид CXCL12 могут находиться в одной и той же или в разных камерах или слоях матрицы, высвобождающей полипептид CXCL12, по настоящему изобретению.
Толщина матрицы, высвобождающей полипептид CXCL12, может варьироваться, чтобы соответствовать потребностям различных применений. Только в качестве примера, толщину матрицы, высвобождающей полипептид CXCL12, можно регулировать для достижения быстрого высвобождения или медленного высвобождения полипептида CXCL12 из матрицы. В некоторых вариантах осуществления, толщина матрицы может составлять от примерно 200 до примерно 500 микронов.
Клетка(и), инкапсулированная в матрицу, высвобождающую полипептид CXCL12, может быть отобрана или получена из любого источника, любого биологического вида и/или ткани любого типа. Дополнительно или в качестве альтернативы, клетка(и), инкапсулированная в матрицу, высвобождающую полипептид CXCL12, может дифференцироваться из стволовых клеток в клетки определенного типа. В некоторых вариантах осуществления, клетка(и) инкапсулированная в матрицу, высвобождающую полипептид CXCL12, может быть аутологической. В некоторых вариантах осуществления, клетка(и), инкапсулированная в матрицу, высвобождающую полипептид CXCL12, может являться аллогенной клеткой (клетками) или ксеногенной клеткой (клетками).
В некоторых вариантах осуществления, клетка(и), инкапсулированная в матрицу, высвобождающую полипептид CXCL12, может сохранять свои функции и/или активность в течение желаемого периода времени, например, после имплантации композиции в организм субъекта. В некоторых вариантах осуществления, клетка(и) может сохранять свои функции и/или активность по крайней мере в течение примерно 1 месяца или более, в т.ч., например, как минимум примерно 2 месяцев, как минимум примерно 3 месяцев или более. В некоторых вариантах осуществления, клетка(и) может сохранять свои функции и/или активность по крайней мере в течение примерно 1 месяца или более после имплантации композиции по настоящему изобретению в организм субъекта.
В зависимости от типов клеток, инкапсулированных в матрицу, высвобождающую полипептид CXCL12, эти клетки могут осуществлять различные функции и/или проявлять различную активность. В некоторых вариантах осуществления, клетка(и) инкапсулированная в матрицу, высвобождающую полипептид CXCL12, может регулировать уровень глюкозы в крови субъекта. Например, после имплантации, клетка(и), инкапсулированная в матрицу, высвобождающую полипептид CXCL12, может выделять инсулин, реагируя на уровень глюкозы в окружающей среде. В этих вариантах осуществления, клетка(и), инкапсулированная в матрицу, высвобождающую полипептид CXCL12, может включать островковую клетку. Эта островковая клетка может являться клеткой, продуцирующей инсулин, островковой клеткой, полученной из индуцированной плюрипотентной стволовой клетки (iPS), свиной островковой клетки, человеческой островковой клетки или любых комбинаций клеток перечисленных типов.
В некоторых вариантах осуществления, клетка(и), инкапсулированная в матрицу, высвобождающую полипептид CXCL12, может быть удалена. В некоторых вариантах осуществления, эта клетка(и) может быть помещена в матрицу, высвобождающую полипептид CXCL12, in vivo.
Матрица, высвобождающая полипептид CXCL12, может включать как минимум один или несколько биосовместимых биополимеров. Эти биосовместимые полимеры могут быть биоразрушаемыми или не разрушаемыми. Биосовместимые полимеры могут являться полимерами на основе углеводов, на основе белков и/или синтетическими полимерами. В некоторых вариантах осуществления, биосовместимые полимеры можно выбирать таким образом, чтобы они были инертны в отношении инкапсулированных клеток (например, не стимулировали или не ингибировали передачу клеточных сигналов) и являлись проницаемыми для полипептида CXCL12, который должен вымываться из матрицы, и, необязательно, проницаемыми для целевой молекулы, которая должна оказывать влияние на клетки. В некоторых вариантах осуществления, биосовместимый полимер можно выбрать таким образом, чтобы средний размер пор элюирующей матрицы по настоящему изобретению отсеивал молекулы с массой превышающей примерно 130КДа.
В некоторых вариантах осуществления, матрица, высвобождающая полипептид CXCL12, может включать альгинатный гель. Этот альгинатный гель может включать маннуроновую кислоту (M) и гулуроновую кислоту (G), причем соотношение (M/G) подбирают так, чтобы добиться свойств, подходящих для индивидуальных применений. Примеры свойств альгинатного геля, которые можно оптимизировать за счет соотношения M/G, включают, не ограничиваясь этим, пороговую молекулярную массу, пористость, размер пор, прочность геля (напряжение сдвига) и/или профиль высвобождения полипептида CXCL12.
В некоторых вариантах осуществления, альгинатный гель может иметь высокое содержание маннуроновой кислоты. В некоторых вариантах осуществления, альгинатный гель может включать маннуроновую кислоту (M) и гулуроновую кислоту (G), при соотношении (M/G) равном примерно 1 или превышающем 1. В некоторых вариантах осуществления, концентрация альгинатного геля может меняться от примерно 1% масса/объем до примерно 5% масса/объем. В некоторых вариантах осуществления, концентрация алгинатного геля может составлять примерно 2% масса/объем.
В некоторых вариантах осуществления, композиция по настоящему изобретению может дополнительно включать слой клеток, которые способны экспрессировать полипептид CXCL12. В некоторых вариантах осуществления, клетки, экспрессирующие полипептид CXCL12, могут включать мезотелиальные клетки.
В некоторых вариантах осуществления, композиция по настоящему изобретению может дополнительно включать рассасывающийся слой полипептида CXCL12 поверх элюирующей матрицы.
В некоторых вариантах осуществления, композиции могут иметь такой состав, чтобы они являлись композициями, подходящими для инъекций.
В различных вариантах осуществления, композиции по настоящему изобретению могут быть имплантированы или введены с помощью инъекции в целевой участок организма субъекта для лечения заболевания или расстройства. В некоторых вариантах осуществления, композиции могут включать как минимум одну островковую клетку, инкапсулированную в матрицу, высвобождающую полипептид CXCL12. Соответственно, в некоторых вариантах осуществления, композиция, например, для лечения диабета, может быть охарактеризована как композиция, включающая аллотрансплантат или ксенотрансплантат островковых клеток, инкапсулированных в матрицу, высвобождающую полипептид CXCL12, где матрица имеет следующие характеристики: (a) толщина матрицы составляет 200-500 микронов, и концентрация полипептида CXCL12 в матрице составляет от примерно 100 нг/мл до примерно 1 мкг/мл; (b) пористость матрицы такова, что агенты, регулирующие концентрацию глюкозы в сыворотке субъекта с диабетом I типа, диффундируют через матрицу; и (c) выработка инсулина островковыми клетками основана на взаимодействии агентов с островковыми клетками, где инсулин высвобождается через матрицу со скоростью, достаточной для регулирования концентрации глюкозы в сыворотке или ее уровня в крови субъекта. Толщину матрицы, концентрацию CXCL12 и/или скорость высвобождения можно отрегулировать таким образом, чтобы подавить разрушение островковых клеток в течение периода как минимум или вплоть до примерно 4 месяцев, тем самым обеспечивая регулирование уровня глюкозы в крови субъекта в течение этого периода. В некоторых вариантах осуществления, толщину матрицы, концентрацию CXCL12 и/или скорость высвобождения можно отрегулировать таким образом, чтобы подавлять разрушение островковых клеток в течение периода примерно 6 месяцев или более.
В другом аспекте, в изобретении описаны также способы доставки островковых клеток в организм субъекта, которому это необходимо. Этот способ включает имплантацию композиций по одному или нескольким вариантам осуществления композиций, описанным в заявке, в организм субъекта, где островковые клетки регулируют уровни глюкозы в крови субъекта в течение определенного периода времени. Только в качестве примера, островковые клетки, инкапсулированные в матрицу, высвобождающую полипептид CXCL12, после имплантации в организм субъекта способны регулировать уровни глюкозы в крови субъекта в течение периода как минимум примерно 1 месяц или более, в т.ч., например, как минимум примерно 2 месяца, как минимум примерно 3 месяца, как минимум примерно 6 месяцев, как минимум примерно 9 месяцев, как минимум примерно 1 год, как минимум примерно 2 года или более.
В некоторых вариантах осуществления, островковые клетки, инкапсулированные в матрицу, высвобождающую полипептид CXCL12, способны поддерживать или восстанавливать концентрацию глюкозы в сыворотке субъекта натощак от примерно 80 мг/дл до примерно 120 мг/дл.
В некоторых вариантах осуществления, матрица, высвобождающая полипептид CXCL12, не разрушается эффекторными T-клетками или макрофагами.
В некоторых вариантах осуществления, в месте имплантации могут присутствовать регуляторные T-клетки. В некоторых вариантах осуществления, в месте имплантации могут отсутствовать эффекторные T-клетки. Только в качестве примера, присутствие регуляторных T-клеток или отсутствие эффекторных T-клеток, можно измерить с помощью цитометрии в потоке или иммуногистохимии.
В некоторых вариантах осуществления, субъекту может осуществляться неоднократная имплантация композиции, включающей как минимум одну островковую клетку, инкапсулированную с матрицу, высвобождающую полипептид CXCL12.
Еще один аспект настоящего изобретения относится к способам пополнения численности островковых клеток в организме субъекта, у которого имеется хранилище островковых клеток в форме ксенотрансплантата, при наличии такой необходимости. Этот способ включает (a) оценку времени полужизни островковых клеток, имеющихся в организме субъекта; (b) доставку островковых клеток в организм субъекта так, чтобы статистическое время полужизни островковых клеток соответствовало терапевтическому уровню, который обеспечит контроль уровней глюкозы в крови субъекта в течение определенного периода времени; и (c) повторение стадий (a) и (b), исходя из периода полужизни островковых клеток. В некоторых вариантах осуществления, время полужизни островковых клеток, имеющихся в организме субъекта, можно оценить или измерить, отслеживая изменения уровня глюкозы в крови субъекта. Например, возвращение содержания глюкозы в крови на диабетические уровни может указывать на необходимость пополнения численности островковых клеток в организме субъекта.
Кроме того, настоящее изобретение относится к композициям, включающим матрицу, высвобождающую полипептид CXCL12. Эта матрица может характеризоваться: (a) пористостью, такой что полипептид CXCL12 медленно вымывается из композиции и проникает на активный участок аутоиммунного заболевания; (b) агентом, который препятствует миграции матрицы после введения, так чтобы основная часть матрицы оставалась в месте введения и около него; где концентрация полипептида CXCL12 и скорость его вымывания выбраны таким образом, чтобы подавлять дальнейшее развитие указанного аутоиммунного заболевания. В некоторых вариантах осуществления, композиции по настоящему изобретению можно вводить инъекцией. В этих вариантах осуществления, композиции могут иметь такой состав, чтобы они были пригодными для инъекций.
Другие особенности и преимущества настоящего изобретения будут ясны из подробного описания и формулы изобретения. Таким образом, другие аспекты настоящего изобретения описаны в дальнейшей части заявки и входят в объем изобретения.
КРАТКОЕ ОПИСАНИЕ ИЛЛЮСТРАТИВНОГО МАТЕРИАЛА
Приведенное ниже по тексту подробное описание изобретения, данное в качестве примера и не претендующее на ограничение объема изобретения конкретно описанными вариантами осуществления, может быть понято при помощи приложенных чертежей, которые включены в текст заявки посредством ссылки.
Фиг.1A-1D демонстрируют, что покрытие аллогенных островков полипептидом CXCL12 с высокой концентрацией отсрочивает отторжение. Фиг.1A представляет собой кривую выживаемости, демонстрирующую долю аллотрансплантированных островков после трансплантации. Островки мышей BALB/C подвергали действию CXCL12 в концентрации примерно 100 нг/мл и примерно 1 мкг/мл или только PBS и трансплантировали под почечную капсулу диабетических мышей-реципиентов C57BL/6, обработанных STZ. Повторное наступление гипергликемии рассматривалось как показатель отторжения трансплантата, тогда как сохранение нормогликемии считалось признаком выживания аллотрансплантата. Покрытие островков примерно 1 мкг/мл, но не 100 нг/мл полипептида CXCL12 статистически достоверно увеличивало время отторжения трансплантата по сравнению с PBS контрольными образцами (p=0,012, логарифмический ранговый критерий) (12 животных на группу). Фиг.1B представляет собой группу изображений, демонстрирующих аллотрансплантаты островков. Репрезентативное окрашивание гематоксилином и эозином (H&E) участков подкапсульных трансплантатов островков продемонстрировало уменьшенную инфильтрацию мононуклеарных клеток для островков, покрытых ~1 мкг/мл CXCL12, по сравнению с непокрытым контролем (левая группа изображений). Обработка областей островковых трансплантатов, покрытых ~1 мкг/мл CXCL12 и PBS, для выявления инсулина продемонстрировала большее количество функциональных островков и более высокий уровень секреции инсулина у островков, покрытых CXCL12, по сравнению с PBS контролем (средняя группа изображений). Кроме того, флуоресцентное окрашивание продемонстрировало доказательство окрашивания CXCL12 в образцах, обработанных CXCL12, по сравнению с контрольными образцами (правая группа изображений). Совместное окрашивание CXCL12 и инсулина проявляется в виде более ярких пятен на темных участках. Фиг.1C представляет собой гистограмму, количественно отображающую число клеток CD3+, присутствующих в трансплантатах островков, по данным иммуноокрашивания CD3, и приведенные данные демонстрируют, что наблюдается статистически достоверное уменьшение инфильтрации клеток CD3+ в область трансплантата в случае трансплантатов, покрытых ~1 мкг/мл CXCL12 (p=0,001) по сравнению с непокрытыми контрольными образцами (6 животных на группу). Фиг.1D представляет собой гистограмму, количественно отображающую число клеток FoxP3+, присутствующих в трансплантатах островков, по данным окрашивания FoxP3, и приведенные данные указывают на статистически достоверное увеличение локализации клеток FoxP3+ на трансплантатах, покрытых 1 мкг/мл CXCL12 по сравнению с PBS контролем (p=0,0016)(n=6).
Фиг.2A-2C демонстрируют, что обработка аллогенных островков, покрытых CXCL12, низкими дозировками CsA не увеличивает выживаемость трансплантатов. На Фиг.2A показана кривая выживаемости, построенная на основании доли мышей, оставшихся не диабетическими, после трансплантации покрытых или не покрытых CXCL12 островков с обработкой или без обработки CsA. Наблюдалось достоверное уменьшение времени выживаемости трансплантата через 23 дня после трансплантации, если покрытие CXCL12 сочеталось с обработкой CsA (p=0,0245, логарифмический ранговый критерий). Фиг.2B представляет собой группу изображений, на которых показаны результаты иммуногистохимического окрашивания для выявления CD3, FoxP3 и инсулина в каждых исследованных условиях. Результаты окрашивания согласуются с данными по выживаемости, показанными на фиг.2A, и демонстрируют уменьшение инфильтрации клеток CD3+ и увеличение инфильтрации клеток FoxP3+, а также повышенную экспрессию инсулина в островках, покрытых CXCL12, по сравнению с островками, обработанными CXCL12+CsA. Фиг.2C представляет собой гистограмму, количественно отображающую число клеток CD3+ и FoxP3+, присутствующих в островковых трансплантатах, для каждого из трех исследованных условий. В трансплантатах, обработанных CXCL12+CsA, наблюдалось статистически достоверное уменьшение числа клеток FoxP3+ (p=0,0188) и увеличение числа клеток CD3+ (p=0,0002), по сравнению с трансплантатами, покрытыми только CXCL12.
На Фиг.3A-3C показано, что покрытие CXCL12 не влияет на количество C57BL/6-специфичных T-клеток CD4. Селезенки удаляли у мышей C57BL/6 через 10 дней после пересадки трансплантатов островков BALB/c и спленоциты стимулировали спленоцитами BALB/c, обработанными митомицином-C в присутствии 1 мМ BrdU. Затем клетки окрашивали для определения захвата BrdU. На Фиг.3A показано включение BrdU в спленоциты по сравнению с отрицательным окрашиванием контроля (затеменная область). На Фиг.3B показано, что отсутствовала разница с точки зрения включения BrdU в клетки у мышей, имевших островки без покрытия (линия), и островки, покрытые CXCL12 (затемненная область). Фиг.3C представляет собой гистограмму, демонстрирующую среднее число алло-специфичных T-клеток CD4 на селезенку в каждой группе мышей, и полученные результаты показывают, что покрытие островков CXCL12 не модулирует системный иммунный ответ на аллогенную ткань (n=3).
На фиг.4A-4C показаны экспериментальные данные для сингенных островков мышей NOD/LtJ, покрытых CXCL12 или обработанных PBS, трансплантированных под почечную капсулу диабетических NOD/LtJ мышей, обработанных STZ. Фиг.4A представляет собой кривую выживаемости, демонстрирующую долю мышей, которые сохранили не диабетический статус после трансплантации; имеется статистически достоверная разница между островками, обработанными CXCL12 и PBS (логарифмический ранговый критерий, p=0,017), что указывает на более продолжительное выживание и функционирование трансплантированных сингенных островков, покрытых CXCL12. Фиг.4B представляет собой гистограмму, количественно отображающую число клеток CD3+ в трансплантированных островках, и эти данные показывают, что существует статистически достоверное ослабление инфильтрации клеток CD3+ в трансплантаты островков, покрытых CXCL12 (p=0,0081). Фиг.4C представляет собой гистограмму, количественно отображающую количество клеток FoxP3+ в трансплантатах островков, и приведенные данные показывают, что наблюдается статистически достоверное увеличение локализации клеток FoxP3+ в островках, покрытых CXCL12, по сравнению с островками, обработанными PBS (p=0,0019).
На Фиг.5A-5D продемонстрированы данные для сингенных островков мышей NOD/LtJ, покрытых CXCL12 или обработанных PBS, трансплантированных под почечную капсулу мышей NOD/LtJ со спонтанным диабетом. Фиг.5A представляет собой кривую выживаемости, демонстрирующую долю мышей, которые сохранили не диабетический статус после трансплантации; статистически достоверная разница между островками, обработанными CXCL12 и PBS отсутствует (логарифмический ранговый критерий, p=0,24). Фиг.5B представляет собой группу изображений полученных в результате H&E окрашивания (левая группа изображений), демонстрирующих уменьшение инфильтрации мононуклеарных клеток в трансплантированные островки, покрытые 1 мкг/мл CXCL12 (левая группа изображений) и иммунофлуоресцентного окрашивания для выявления инсулина и CXCL12 (правая группа изображений), демонстрирующая повышение уровней обоих белков в трансплантатах, покрытых CXCL12. Фиг.5C представляет собой гистограмму, количественно отображающую число клеток CD3+ в трансплантированных островках, и приведенные данные показывают, что хотя различия в выживаемости отсутствуют, существует статистически достоверная разница в инфильтрации клеток CD3+ в трансплантаты островков, покрытые CXCL12 (p=0,0015). Фиг.5D представляет собой гистограмму, количественно отображающую количество клеток FoxP3+ в трансплантатах островков, и приведенные данные показывают, что наблюдается статистически достоверное увеличение локализации клеток FoxP3+ в островках, покрытых CXCL12, по сравнению с островками, обработанными PBS (p=0,0019).
Фиг 6A-6F демонстрируют, что включение CXCL12 в Ca-LVM альгинатные капсулы отсрочивает отторжение аллогенных и ксеногенных островков, трансплантированных в брюшную полость. Фиг.6A представляет собой график, демонстрирующий кинетику высвобождения CXCL12 из бесклеточного, поперечно-сшитого кальцием ~3,3% альгинатного инкапсулирующего материала с течением времени in vitro. Концентрация CXCL12 в несшитом альгинате натрия составляла 1 мкг/мл; значительное количество CXCL12 было потеряно в сшивающем растворе CaCl2 (n=3). Не существует различий в профилях высвобождения CXCL12 для концентраций альгината от 1,5% до 3,3% (данные не показаны). Начальная скорость высвобождения CXCL12 из капсул, включающих 1,5% альгината, в течение первых 24 часов составляла 1,75 нг/мл/ч +/-0,01 нг/мл/ч, и через четыре дня имела место стабилизация скорости высвобождения на уровне 0,18 нг/мл/ч +/-0,002 нг/мл/ч. Фиг.6B представляет собой гистограмму, демонстрирующую электростатическое взаимодействие между CXCL12 и капсулой из альгината, сшитого барием. Левая группа рисунков показывает, что в гранулах остается значительно меньшее количество CXCL12 после инкубирования с 1М NaCl по сравнению с инкубированием в отсутствии NaCl. Правая группа рисунков показывает, что достоверно большие количества CXCL12 вымываются в среду после инкубирования с 1М NaCl по сравнению с инкубированием в среде без NaCl (n=3, p*<0,05). Фиг.6C представляет собой гистограмму, отображающую активность каспазы-3, которая демонстрирует, что выключение CXCL12 статистически достоверно уменьшает активность каспазы-3 в инкапсулированных мышиных островках (p=0,0019 для ~100 нг/мл CXCL12 и p=0,00028 для ~1 мкг/мл CXCL12 относительно контроля). Мышиные островки инкапсулировали в Ca-LVM или Ca-LVM, содержащем либо ~100 нг/мл, либо ~1 мкг/мл CXCL12 (Ca-LVM-CXCL12), затем культивировали in vitro в течение 48 часов и после этого определяли активность каспазы-3. Фиг.6D представляет собой график выживаемости, показывающий долю выживших после пересадки аллотрансплантатов островков. Включение 1 мкг/мл CXCL12 в инкапсулирующий материал Ca-LVM отсрочивает отторжение аллогенных островков (n=12) для обеих групп (p=0,0237, критерий Гехана-Бреслоу-Вилкоксона). Фиг.6E представляет собой график выживаемости, демонстрирующий долю выживших после пересадки аллотрансплантатов островков. Включение ~1 мкг/мл CXCL12 также отсрочивает отторжение инкапсулированных аллогенных островков, пересаженных в организм алло-сенсибилизированных мышей NOD/LtJ (капсулы Ca-LVM, n=7; капсулы Ca-LVM-CXCL12, n=9; p=0,0066, критерий Гехана-Бреслоу-Вилкоксона). Фиг.6F представляет собой график выживаемости, демонстрирующий долю выживших после пересадки аллотрансплантатов островков. Включение 1 мкг/мл CXCL12 статистически достоверно отсрочивает отторжение инкапсулированных свиных ксеногенных островков, трансплантированных диабетическим мышам C57BL/6 (p=0,0389, логарифмический ранговый критерий). Контрольные и экспериментальные группы: капсулы Ca-LVM, капсулы Ca-LVM-10 нг/мл CXCL12, капсулы Ca-LVM-100 нг/мл CXCL12 (n=6), капсулы Ca-LVM-1 мкг/мл CXCL12 (p*<0,01)(размер группы=6) (критерий Гехана-Бреслоу-Вилкоксона).
На фиг.7A-7G показано миграционное поведение субпопуляций T-клеток в ответ на CXCL12 и связанную с ним экспрессию CXCR4. Фиг.7A и фиг.7C представляют собой гистограммы, демонстрирующие миграционную реакцию T-клеток CD3+CD8+. Фиг.7B и фиг.7D представляют собой гистограммы, демонстрирующие миграционную реакцию T-клеток CD3+CD4+CD25hi. Миграционные реакции клеток количественно определяли, как реакцию на CXCL12, островки, покрытые CXCL12 (I-CXCL12), и островки, инкапсулированные в CXCL12 (E-CXCL12). Во всех трех случаях CXCL12 применялся в концентрации ~1 мкг/мл. T-клетки CD8+ и CD4+CD25Hi подвергались незначительному хемотаксису при воздействии ~1 мкг/мл CXCL12 (M/CXCL12) и островков, покрытых или инкапсулированных CXCL12. T-клетки CD8+, но не CD4+CD25Hi, подвергались фугетаксису или химическому отталкиванию при воздействии островков, покрытых или инкапсулированных в CXCL12. Минимальные уровни как хемотаксиса, так и фугетаксиса были зарегистрированы для T-клеток CD8+ или CD4+CD25Hi при взаимодействии с островками, которые не были покрыты CXCL12 (I-Cont) или островками, инкапсулированными без добавок (E-Cont). (ns=статистически недостоверно; *p<0,05; **p<0,005, критерий Стьюдента). Для объяснения обнаруженных различий в миграционных реакциях, сравнивали экспрессию CXCR4 на T-клетках CD4+, CD8+ и регуляторных T-клетках. На Фиг.7E показан пример стратегии гейтирования для T-клеток CD8+ и регуляторных T-клеток. Фиг.7F представляет собой график, демонстрирующий среднюю интенсивность флуоресценции (MFI) при экспрессии CXCR4. Вычисляли процентную долю популяции каждых клеток, экспрессирующих CXCD4, и типовая гистограмма, представленная на фиг.7G, показывает увеличение экспрессии CXCR4 клетками Treg по сравнению с T-клетками CD4+CD25- и CD8+ (p<0,0001, критерий Стьюдента).
Фиг.8 представляет собой график, демонстрирующий кинетику удерживания CXCL12 инкапсулирующим материалом, содержащим сшитый кальцием 3,3% альгинат, в зависимости от времени in vitro. Концентрация CXCL12 в несшитом альгинате натрия составляла 1 мкг/мл (n=3). Не было зафиксировано различий в профилях высвобождения CXCL12 при концентрациях альгината от 1,5% до 3,3% (данные не показаны).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Если не указано иное, все технические и научные термины, использованные в тексте заявки, имеют те же значения, которые обычно придаются им рядовым специалистом в той области техники, к которой относится настоящее изобретение. В случае противоречия, преимущество будет иметь определение, включенное в настоящую заявку.
Термин «альгинат», известный также как «альгиновая кислота», в настоящей заявке обычно относится к углеводному полимеру (например, полисахариду), включающему как минимум два уроната сахаров.
Термин «аллогенный» означает принадлежащий особям того же вида или полученный из особей того же вида.
Термин «аллотрансплантат» относится к трансплантату клеток или ткани, полученному из особей того же вида.
Термин «ксеногенный» означает принадлежащий особям другого вида или полученный из особей другого вида.
Термин «ксенотрансплантат» относится к трансплантату клеток или ткани, полученному из особей другого вида.
Термин «эффекторная T-клетка» относится к дифференцированным T-клеткам, способным осуществлять специфичный иммунный ответ за счет высвобождения цитокинов.
Термин «регуляторная T-клетка» относится к T-клеткам, которые уменьшают или подавляют иммунный ответ B-клеток или других T-клеток на антиген.
Под «полипептидом CXCL12 или SDF-1» подразумевается белок или его фрагмент, который связывается с CXCL12-специфичными антителами и который обладает способностью вызывать хемотаксис или фугетаксис. Способность вызывать хемотаксис или фугетаксис определяют путем исследования направления миграции T-клеток (например, к интересующему агенту или от него). Смотрите, например, Poznansky et al., Nature Medicine 2000, 6:543-8.
Субъект представляет собой позвоночное животное, включая всех членов класса млекопитающих, в т.ч. человека, домашних и сельскохозяйственных животных, а также животных, содержащихся в зоопарках, спортивных и комнатных животных, например, мышей, кроликов, свиней, овец, коз, крупный рогатый скот и высших приматов.
Термины «лечить», «лечение» и т.п. в настоящем описании относятся к ослаблению или облегчению расстройства и/или связанных с ним симптомов. Следует понимать, хотя это и не исключается, что лечение расстройства или состояния не требует, чтобы расстройство, состояние или связанные с ним симптомы были полностью устранены.
В настоящей заявке термины «включает», «включающий», «содержащий», «имеющий» и т.п. могут иметь значение, приписываемое им патентным законодательством США и могут означать «содержит», «содержащий» и т.п.; аналогично, фразы «состоящий в основном из» или «состоящий в основном» имеют смысл приписываемый им в патентном законодательстве США и эти термины являются не ограничивающими, допускающими присутствие компонентов помимо перечисленных, если основные или новые характеристики описываемого предмета изобретения не подвергаются изменению из-за присутствия чего-либо кроме указанных компонентов, но при этом исключаются варианты осуществления известного уровня техники.
Другие определения будут приведены в тексте описания в связи с контекстом.
Композиции и способы по настоящему изобретению
Композиции по настоящему изобретению направлены на матрицу, высвобождающую полипептид CXCL12, в которой инкапсулирована как минимум одна клетка.
Полипептиды CXCL12 известны в технике. Смотрите, например, Poznansky et al., Nature Medicine 2000, 6:543-8. Отметим, что термины CXCL12 и SDF-1 могут являться взаимозаменяемыми. В одном из вариантов осуществления, полипептид CXCL12 имеет по крайней мере 85%, 90%, 95% или 100% идентичность аминокислотной последовательности NP 001029058 и проявляет активность хемокина или способность вызывать фугетаксис. Типовые примеры изоформ SDF1 приведены в таблице 1 (ниже по тексту):
GI:40316924
бета
GI:1352728
GI:76563933
В другом варианте осуществления, типовой полипептид CXCL12/SDF-1 имеет следующую последовательность: mnakvvvvlvlvltalclsdgkpvslsyrcpcrffeshvaranvkhlkilntpncalqivarlknnnrqvcidpklkwiqeylekalnkgrreekvgkkekigkkkrqkkrkaaqkrkn.
В еще одном варианте осуществления, полипептид CXCL12 имеет по крайней мере 85%, 90%, 95% или 100% идентичность аминокислотной последовательности изоформе дельта полипептида CXCL12 и проявляет активность хемокина или способность вызывать фугетаксис. Типовая изоформа дельта полипептида CXCL12 имеет последовательность: MNAKVVVVLVLVLTALCLSDGKPVSLSYRCPCRFFESHVARANVKHLKILNTPNCALQIVARLKNNNRQVCIDPKLKWIQEYLEKALNNLISAAPAGKRVIAGARALHPSPPRACPTARALCEIRLWPPP EWSWPSPGDV.
Матрицы, высвобождающие полипептид CXCL12, характеризуются, например, высвобождением полипептида CXCL12 со скоростью, как минимум, примерно 1,0 нг/мл/ч, например, от примерно 1,0 нг/мл/ч до примерно 3 нг/мл/ч. В конкретных вариантах осуществления, полипептид CXCL12 высвобождается со скоростью примерно 1,75 нг/мл/ч. Полипептид CXCL12 присутствует в матрице в концентрации, как минимум, примерно 100 нг/мл, например, от приблизительно 100 нг/мл до примерно 1 мкг/мл. В конкретных вариантах осуществления, полипептид CXCL12 присутствует в матрице в концентрации от примерно 100 нг/мл до примерно 1 мкг/мл в течение от примерно 3 месяцев до примерно 2 лет. Концентрации, скорости высвобождения и продолжительности высвобождения будут меняться в соответствии с выбранным типом клеток и расстройством, подвергаемым лечению, и выбор надлежащих параметров должен быть известен или очевиден специалисту в данной области. Как правило, полипептид CXCL12 высвобождается со скоростью, достаточной для отталкивания эффекторных T-клеток от конкретного участка организма. Способность матрицы, высвобождающей полипептид CXCL12, отталкивать эффекторные T-клетки можно оценить in vitro, используя анализ в камере Бойдена, как было описано ранее Poznansky et al., Journal of clinical investigation, 109, 1101 (2002).
Элюирующие матрицы могут включать биосовместимые полимеры, известные в технике, которые являются инертными по отношению к инкапсулированным клеткам (т.е. не стимулируют или не ингибируют передачу клеточных сигналов) и проницаемы для полипептида CXCL12, который должен вымываться из матрицы, а также для молекул, на которые должна реагировать матрица (например, глюкозы). Толщина матрицы составляет от примерно 200 до примерно 500 микронов, и в конкретных вариантах осуществления эта матрица образует капсулу вокруг клеток. Биосовместимые полимеры могут быть биоразрушаемыми или не биоразрушаемыми. Биосовместимый полимер может быть на углеводной основе, белковой основе и/или синтетическим, например, PLA. Биосовместимые материалы, которые подходят для применения в матрицах по настоящему изобретению, включают, не ограничиваясь перечисленными, полидиметилсилоксан (PDMS), полиглицеринсебацинат (PGS), полимолочную кислоту (PLA), поли-L-молочную кислоту (PLLA), поли-D-молочную кислоту (PDLA), полигликолид, полигликолевую кислоту (PGA), поли лактид-гликолид (PLGA), полидиоксанон, полиглюконат, сополимеры полимолочная кислота-полиэтиленоксид, модифицированную целлюлозу, коллаген, полигидроксибутират, полигидроксипропионовую кислоту, полифосфоэфир, поли(альфа-гидроксикислоту), полкапролактон, поликарбонаты, полиамиды, полиангидриды, полиаминокислоты, полиортоэфиры, полиацетали, полицианоакрилаты, разрушаемые уретаны, алифатические полиэфирполиакрилаты, полиметакрилат, ацетаты ацилзамещенной целлюлозы, не разрушаемые полиуретаны, полистиролы, поливинилхлорид, поливинилфторид, поливинилимидазол, хлорсульфонированные полиолефины, полиэтиленоксид, поливиниловый спирт, найлон силикон, поли(стирол-блок-бутадиен), полинорборнен и гидрогели. Другие подходящие полимеры можно получить согласно The Polymer Handbook, 3rd edition (Wiley, N.Y., 1989). Также могут применяться комбинации этих полимеров.
В одном из вариантов осуществления, матрица, высвобождающая полипептид CXCL12, по настоящему изобретению дополнительно включает второй слой клеток, которые экспрессируют полипептид CXCL12, например, мезотелиальных клеток. В других вариантах осуществления, внешний слой матрицы дополнительно включает рассасывающийся слой полипептида CXCL12.
В одном из вариантов осуществления, элюирующая матрица включает альгинат (например, альгиновую кислоту), и этот термин обычно относится к углеводному полимеру (например, полисахариду), включающему как минимум два уроната сахаров. Уронаты сахаров могут включать, не ограничиваясь перечисленным, соли маннуроновой кислоты (или маннуронаты), соли гулуроновой кислоты (или гулуронаты) и/или их изомеры. В некоторых вариантах осуществления альгинат может являться линейным углеводным полимером (например, полисахаридом), включающим маннуронат, гулуронат и/или их изомеры. В некоторых вариантах осуществления, альгинат может являться углеводным сополимером маннуроната, гулуроната и/или их изомеров.
В настоящей заявке термин «изомеры» относится к соединениям, имеющим одну и ту же молекулярную формулу, но различающимся по структуре. Изомеры, которые различаются только конфигурацией и/или конформацией, именуются «стереоизомерами». Термин «изомер» используется также в отношении энантиомеров. Термин «энантиомер» используется для описания одного из двух молекулярных изомеров, которые являются зеркальными отображениями друг друга, и которые невозможно совместить при наложении друг на друга. Другие термины, используемые для обозначения или ссылки на энантиомеры выключают термин «стереоизомеры» (из-за различного расположения или стереохимии у хирального центра; хотя все энантиомеры являются стереоизомерами, не все стереоизомеры являются энантиомерами) или термин «оптические изомеры» (из-за оптической активности чистых энантиомеров, которая представляет собой способность разных чистых энантиомеров вращать плоско-поляризованный свет в противоположных направлениях). Энантиомеры, как правило, имеют одинаковые физические свойства, такие как температура плавления и температура кипения, а также одинаковые спектроскопические свойства. Энантиомеры могут отличаться друг от друга с точки зрения взаимодействия с плоско-поляризованным светом и с точки зрения биологической активности. Соответственно, в некоторых вариантах осуществления, соли маннуроновой кислоты (или маннуронаты) могут включать β-D-маннуронаты. В некоторых вариантах осуществления, соли гулуроновой кислоты (или гулуронаты) могут включать α-L-гулуронаты.
В некоторых вариантах осуществления, альгинат может являться блок-полимером, включающим как минимум одну или несколько гомополимерных областей маннуроната (M-блоки), как минимум одну или несколько гомополимерных областей гулуроната (G-блоки), а также как минимум одну или несколько чередующихся структур, состоящих из маннуроната и гулуроната (MG-блоки или GM-блоки).
Процентные доли, распределение и/или длина этих блоков могут частично определять химические и/или физические свойства альгинатного геля. Например, относительно содержание G и M мономеров в альгинатных полимерах может влиять, в том числе, но не ограничиваясь этим, на размер пор, стабильность и биоразрушаемость, прочность и эластичность альгинатного геля. Не желая ограничиваться какой-либо теорией, более низкое содержание G-остатков относительно содержания M-остатков в альгинатных полимерах обычно может приводить к более биоразрушаемым гелям. Альгинатные гели с более высоким содержанием остатков G обычно могут иметь более крупный размер пор и/или более высокую прочность геля по сравнению с альгинатными гелями с более высоким содержанием остатков M, которые характеризуются меньшими размерами пор и более низкой прочностью геля. В некоторых вариантах осуществления, один или несколько альгинатных полимеров альгинатной матрицы могут включать M-блоки в количестве не менее примерно 10 масс.%, не менее примерно 20 масс.%, не менее примерно 30 масс.%, не менее примерно 40 масс.%, не менее примерно 50 масс.%, не менее примерно 60 масс.%, не менее примерно 70 масс.%, не менее примерно 80 масс.%, не менее примерно 90 масс.% или более. В некоторых вариантах осуществления, один или несколько альгинатных полимеров альгинатной матрицы могут включать G-блоки в количестве не менее примерно 10 масс.%, не менее примерно 20 масс.%, не менее примерно 30 масс.%, не менее примерно 40 масс.%, не менее примерно 50 масс.%, не менее примерно 60 масс.%, не менее примерно 70 масс.%, не менее примерно 80 масс.%, не менее примерно 90 масс.% или более. В некоторых вариантах осуществления, один или несколько альгинатных полимеров альгинатной матрицы могут включать GM- и/или MG-блоки в количестве не менее примерно 10 масс.%, не менее примерно 20 масс.%, не менее примерно 30 масс.%, не менее примерно 40 масс.%, не менее примерно 50 масс.%, не менее примерно 60 масс.%, не менее примерно 70 масс.%, не менее примерно 80 масс.%, не менее примерно 90 масс.% или более.
В некоторых вариантах осуществления, один или несколько альгинатных полимеров, входящих в альгинатную матрицу, могут иметь соотношение маннуроновой кислоты к гулуроновой кислоте (M/G) от примерно 0,01 до примерно 100, или от примерно 0,1 до примерно 50, или от примерно 0,5 до примерно 25, или от примерно 1 до примерно 20. В некоторых вариантах осуществления, один или несколько альгинатных полимеров, входящих в альгинатную матрицу, могут иметь соотношение M/G от примерно 1 до примерно 100 или от примерно 1 до примерно 50, или от примерно 1 до примерно 25, или от примерно 1 до примерно 20, или от примерно 1 до примерно 10, или от примерно 1 до примерно 5.
В некоторых вариантах осуществления, один или несколько альгинатных полимеров, входящих в альгинатную матрицу, могут иметь соотношение гулуроновой кислоты к маннуроновой кислоте (G/M) не более 1,5 или не более чем 1. Например, в некоторых вариантах осуществления, один или несколько альгинатных полимеров, входящих в альгинатную матрицу, могут иметь соотношение G/M примерно 1,5. В некоторых вариантах осуществления, один или несколько альгинатных полимеров, входящих в альгинатную матрицу, могут иметь соотношение G/M примерно 1. В некоторых вариантах осуществления, один или несколько альгинатных полимеров, входящих в альгинатную матрицу, могут иметь соотношение G/M менее, чем примерно 1,5, в т.ч., например, менее 1,4, менее 1,3, менее 1,2, менее 1,1, менее 1,0, менее 0,9, менее 0,8, менее 0,7, менее 0,6, менее 0,5, менее 0,4, менее 0,3, менее 0,2, менее 0,1, менее 0,05, менее 0,01, менее 0,0075, менее 0,005, менее 0,001 или ниже. В некоторых вариантах осуществления, один или несколько альгинатных полимеров, входящих в альгинатную матрицу, могут иметь соотношение G/M менее, чем примерно 1, в т.ч., например, менее 0,9, менее 0,8, менее 0,7, менее 0,6, менее 0,5, менее 0,4, менее 0,3, менее 0,2, менее 0,1, менее 0,05, менее 0,01, менее 0,0075, менее 0,005, менее 0,001, менее 0,0001 или ниже.
В некоторых вариантах осуществления, один или несколько альгинатных полимеров, входящих в альгинатную матрицу, могут иметь соотношение гулуроновой кислоты к маннуроновой кислоте (G/M) как минимум примерно 1,5 или более. Например, в некоторых вариантах осуществления, один или несколько альгинатных полимеров, входящих в альгинатную матрицу, могут иметь соотношение G/M примерно 1,5. В некоторых вариантах осуществления, один или несколько альгинатных полимеров, входящих в альгинатную матрицу, могут иметь соотношение G/M более, чем примерно 1,5, в т.ч., например, более 2, более 2,5, более 3, более 3,5, более 4, более 4,5, более 5, более 6, более 7, более 8, более 9, более10, более 15, более 20, более 30, более 40, более 50, более 60, более 70, более 80, более 90, более 100 или выше.
Средняя молекулярная масса альгинатных полимеров может влиять, например, на время гелеообразования, размер пор, прочность геля и/или эластичность геля. Альгинатные полимеры могут иметь среднюю молекулярную массу в диапазоне примерно от 2 КДа до 10000 КДа. Не желая ограничиваться какой-либо теорией, более низкая молекулярная масса альгинатного полимера обычно может приводить к более биоразрушаемым гелям. В некоторых вариантах осуществления, альгинатные полимеры, входящие в альгинатную матрицу, могут иметь среднюю молекулярную массу от примерно 5 КДа до примерно 10000 КДа, или от примерно 10 КДа до примерно 5000 КДа, или от примерно 25 КДа до примерно 2500 КДа, или от примерно 50 КДа до примерно 1000 КДа, или от примерно 50 КДа до примерно 500 КДа, или от примерно 50 КДа до примерно 250 КДа. В некоторых вариантах осуществления, альгинатные полимеры, входящие в альгинатную матрицу, могут иметь среднюю молекулярную массу от примерно 5 КДа до примерно 350 КДа. В некоторых вариантах осуществления, альгинатные полимеры, входящие в альгинатную матрицу, могут иметь среднюю молекулярную массу от примерно 2 КДа до примерно 100 КДа. В некоторых вариантах осуществления, альгинатные полимеры, входящие в альгинатную матрицу, имеют среднюю молекулярную массу от примерно 50 КДа до примерно 500 КДа. В некоторых вариантах осуществления, альгинатные полимеры, входящие в альгинатную матрицу, имеют среднюю молекулярную массу от примерно 50 КДа до примерно 300 КДа. В некоторых вариантах осуществления, альгинатные полимеры, входящие в альгинатную матрицу, имеют среднюю молекулярную массу от примерно 75 КДа до примерно 200 КДа. В некоторых вариантах осуществления, альгинатные полимеры, входящие в альгинатную матрицу, имеют среднюю молекулярную массу от примерно 75 КДа до примерно 150 КДа. В некоторых вариантах осуществления, альгинатные полимеры, входящие в альгинатную матрицу, имеют среднюю молекулярную массу от примерно 150 КДа до примерно 250 КДа. В некоторых вариантах осуществления, альгинатные полимеры, входящие в альгинатную матрицу, имеют среднюю молекулярную массу от примерно 100 КДа до примерно 1000 КДа.
В некоторых вариантах осуществления, альгинатные полимеры, входящие в альгинатную матрицу, могут иметь среднюю молекулярную массу менее 75 КДа или ниже. В некоторых вариантах осуществления, альгинатные полимеры, входящие в альгинатную матрицу, могут иметь среднюю молекулярную массу не менее примерно 75 КДа, не менее примерно 80 КДа, не менее примерно 90 КДа, не менее примерно 100 КДа, не менее примерно 110 КДа, не менее примерно 120 КДа, не менее примерно 130 КДа, не менее примерно 140 КДа, не менее примерно 150 КДа, не менее примерно 160 КДа, не менее примерно 170 КДа, не менее примерно 180 КДа, не менее примерно 190 КДа, не менее примерно 200 КДа, не менее примерно 250 КДа, не менее примерно 300 КДа или выше.
В одном из вариантов осуществления, альгинатные полимеры, входящие в альгинатную матрицу, имеют среднюю молекулярную массу от примерно 75 КДа до примерно 200 КДа, при соотношении гулуроновой кислоты к маннуроновой кислоте (G/M) примерно 1. В одном из вариантов осуществления, альгинатные полимеры, входящие в альгинатную матрицу, имеют среднюю молекулярную массу от примерно 75 КДа до примерно 200 КДа, при соотношении гулуроновой кислоты к маннуроновой кислоте (G/M) менее 1.
Не ограничиваясь этим, молекулярная масса может быть максимальной средней молекулярной массой (Mp), среднечисловой молекулярной массой (Mn) или среднемассовой молекулярной массой (Mw).
Альгинаты можно получать из любого источника и/или любым способом, известным в технике. В некоторых вариантах осуществления, альгинат может быть извлечен из стеблей и/или листьев морских водорослей или бурых водорослей. В некоторых вариантах осуществления альгинат можно извлекать из зеленых водорослей (Chlorophyta), бурых водорослей (Phaeophyta), красных водорослей (Rhodophyta) или любых их комбинаций. Примеры морских водорослей или бурых водорослей включают, не ограничиваясь перечисленными, различные виды Laminaria (например, но не ограничиваясь этим, Laminaria hyperborea, Laminaria digitata и Laminaria japonica), Lessonia nigrescens, Lessonia trabeculata, Durvillaea Antarctica, Ecklonia maxima, Macrocystis pyrifera, Ascophyllum nodosum, а также любые комбинации перечисленных видов.
В некоторых вариантах осуществления, альгинат может являться бактериальным альгинатом, например, полученным микробной ферментацией с применением бактерий. Примеры бактерий, которые могут применяться для получения альгината, включают, не ограничиваясь этим, Pseudomonas (например, Pseudomonas Aeruginosa) и Azotobacter (например, Azobacter Vinelandii). В некоторых вариантах осуществления, бактерии могут продуцировать полисахаридный полимер со структурой, напоминающей альгинат, например, отличающиеся наличием ацетильных групп на части гидроксилов у атомов C2 и C3.
В некоторых вариантах осуществления, альгинат может быть модифицирован. В некоторых вариантах осуществления, альгинат может быть модифицирован химически. Например, химически модифицированный альгинат может включать альгинат пропиленгликоля (PGA). В некоторых вариантах осуществления, PGA можно получить взаимодействием частично нейтрализованной альгиновой кислоты с газообразным пропиленоксидом под давлением. Пропиленоксид может взаимодействовать с альгиновой кислотой экзотермически с образованием смеси первичных/вторичных сложных эфиров.
В некоторых вариантах осуществления, альгинат может быть клинической степени чистоты, например, подходить для применения in vivo. В некоторых вариантах осуществления, альгинат может быть очищен перед применением для инкапсулирования клеток. Смотрите, например, Mallet and Korbutt, Tissue Eng Part A. 2009. 15(6):1301-1309. В некоторых вариантах осуществления, альгинат может содержать минимальные количества эндотоксинов. Например, эндотоксины могут присутствовать в альгинате в количестве не более 150 EU/грамм (EU=единица эндотоксина), не более 100 EU/грамм, не более 75 EU/грамм, не более 50 EU/грамм, не более 25 EU/грамм, не более 20 EU/грамм, не более 10 EU/грамм, не более 5 EU/грамм, не более 1 EU/грамм, не более 0,5 EU/грамм, не более 0,1 EU/грамм.
В способах по различным аспектам настоящего изобретения может применяться любой известный в технике альгинат. Примеры альгинатов, которые можно применять в композициях, описанных в настоящей заявке включают, не ограничиваясь перечисленными, альгинат натрия (натриевую соль альгиновой кислоты), альгинат калия (калиевую соль альгиновой кислоты), альгинат кальция, альгинат магния, альгинат триэтаноламина, PGA, а также любые комбинации перечисленных. В некоторых вариантах осуществления, растворимый альгинат может находиться в форме солей одновалентных катионов, включая, но не ограничиваясь этим, альгинат натрия, альгинат калия и альгинат аммония. В некоторых вариантах осуществления, альгинат может являться альгинатом кальция. В одном из вариантов осуществления, альгинат кальция может быть получен из альгината натрия, из которого удалены катионы натрия и замещены катионами кальция. Альгинаты, описанные в и/или полученные способами, описанными в международных заявках на патент №№ WO 2007/140312; WO 2006/051421; WO 2006/132661; и WO 1991/007951, а также патенте США № US 8481695, также могут применяться в композициях и способах по различным аспектам настоящего изобретения. В некоторых вариантах осуществления, коммерчески доступные альгинаты, например, полученные у FMC BioPolymer и Novamatrix, также могут входить в состав композиций и применяться в способах по различным аспектам настоящего изобретения.
Альгинат обычно образует гелевую матрицу в присутствии двухвалентных ионов и/или трехвалентных ионов. Не ограничивающие примеры двухвалентных или трехвалентных ионов, которые могут применяться для формирования альгинатных гелей, включают ионы кальция, ионы бария, ионы стронция, ионы меди, ионы цинка, ионы магния, ионы марганца, ионы кобальта, ионы свинца, ионы железа, ионы алюминия, а также любые комбинации перечисленных ионов.
В некоторых вариантах осуществления, альгинатная матрица может быть сшита поперечными ковалентными связями. Примеры агентов, позволяющих осуществлять поперечное ковалентное сшивание, которые могут применяться для сшивания альгинатов, включают, не ограничиваясь этим, карбодиимиды, оксиды аллилгалогенидов, диальдегиды, диамины и диизоцианаты.
Композиции элюирующих матриц по настоящему изобретению включают составы, подходящие для введения инъекцией, инфузией или имплантацией (подкожной, внутривенной, внутримышечной, интраперитонеальной, внутрикожной, парентеральной, ректальной и/или внутривагинальной и т.п.), игналяцией, пероральным/назальным или местным путем. Удобно получать составы в виде дозированных лекарственных форм, и такие формы можно получать любыми способами, хорошо известными в фармацевтической технике. Количество полипептида CXCL12, которое необходимо ввести в дозированную форму, будет меняться в зависимости от подвергаемого лечению реципиента, конкретной формы введения, например, инъекции или имплантации. Составы по настоящему изобретению можно получать любым способом, известным в технике для производства фармацевтических препаратов, и эти составы могут содержать подсластители, вкусоароматические агенты, красители и консерванты. В составы можно включать нетоксичные фармацевтически приемлемые эксципиенты, которые подходят для производства. Составы могут включать один или несколько разбавителей, эмульгаторов, консервантов, буферов, эксципиентов и т.д., и могут иметь такую форму, как жидкость, эмульсия, крем, лосьон, гель или пластырь, а также форму имплантата.
В матрицы, высвобождающие полипептид CXCL12 по настоящему изобретению, инкапсулирована как минимум одна клетка. Инкапсулированные клетки могут включать, не ограничиваясь перечисленным, стволовые клетки, нервные клетки, клетки гладкой или скелетной мускулатуры, миоциты, фибробласты, хондроциты, адипоциты, фибромиобласты, эктодермальные клетки, включая пластичные клетки и клетки кожи, гепатоциты, почечные клетки, клетки печени, сердечные клетки, клетки поджелудочной железы, островковые клетки, клетки, присутствующие в кишечнике, остеобласты и другие клетки, формирующие кости или хрящи, а также гематопоэтические клетки. В конкретных вариантах осуществления, указанные клетки являются клетками, продуцирующими инсулин, например, островковыми клетками (например, свиными островковыми клетками, человеческими островковыми клетками или островковыми клетками, полученными из стволовых или iPS клеток).
Элюирующие матрицы по настоящему изобретению являются пополняемыми устройствами для доставки полипептида CXCL12, которые имплантируются или другим образом вводятся в организм пациента. Например, матрица может включать входной канал в виде иглы или катетера с тем, чтобы клетки можно было вводить или удалять без извлечения матрицы из организма пациента. В качестве альтернативы, элюирующие матрицы по настоящему изобретению можно вводить субъекту (например, «сенсибилизированному субъекту») многократно, не вызывая связанного с этим иммунного отторжения.
Матрицы, высвобождающие полипептид CXCL12 по настоящему изобретению, применимы в лечении аутоиммунных заболеваний, включая, но не ограничиваясь этим, ревматоидный артрит, увеит, инсулин-зависимый сахарный диабет, гемолитические анемии, острую ревматическую лихорадку, болезнь Крона, синдром Гийена-Барре, псориаз, тиреоидит, болезнь Грейвса, миастению гравис, гломерулонефрит, аутоиммунный гепатит и системную красную волчанку.
В одном из вариантов осуществления, матрица, высвобождающая полипептид CXCL12 по настоящему изобретению, может включать островковые клетки для применения в лечении диабета. Диабет представляет собой состояние при котором в крови субъекта имеется высокий уровень сахара (глюкозы) в результате того, что организм либо не вырабатывает достаточное количество инсулина, либо клетки организма не реагируют на вырабатываемый инсулин должным образом. У здоровых людей уровни глюкозы в крови поддерживаются в узком диапазоне, главным образом за счет действия гормона инсулина. Инсулин вырабатывается бета-клетками поджелудочной железы с необходимой скоростью в ответ на концентрацию глюкозы в кровотоке, причем реакция модулируется другими факторами, в т.ч. циркулирующими питательными веществами, иннервацией островков и инкретиновыми гормонами. Инсулин поддерживает концентрацию глюкозы, ограничивая скорость высвобождения глюкозы в печени, чтобы она соответствовала скорости выведения глюкозы.
Таким образом, инсулин способствует поглощению глюкозы клетками организма для превращения ее в энергию. Если клетки организма не поглощают глюкозу, глюкоза накапливается в крови (гипергликемия), приводя к разнообразным потенциальным медицинским осложнениям. Соответственно, диабет характеризуется повышенным уровнем глюкозы в крови, приводящим ко вторичным осложнениям, таким как сердечно-сосудистые заболевания, почечная недостаточность, ретинопатия и нейропатия, если не осуществлять соответствующее регулирование уровня сахара.
Нарастание гликемии связано с двумя основными патофизиологическими процессами. Первый из них представляет собой аутоиммунную атаку на бета-клетки поджелудочной железы, вырабатывающие инсулин (диабет 1 типа), тогда как второй связан с плохим функционированием бета-клеток и нарастающей невосприимчивостью к инсулину периферических тканей (диабет 2 типа). Как и в случае диабета 1 типа, при диабете 2 типа также наблюдается гибель бета-клеток. При диабете 1 типа и часто при диабете 2 типа пациенту требуются инъекции инсулина.
Сахарный диабет 1 типа обычно характеризуется утратой продуцирующих инсулин бета-клеток, находящихся в островках Лангерганса поджелудочной железы, что приводит к дефициту инсулина. Диабет этого типа может быть далее подразделен на иммунно-опосредованный или идиопатический. Большинство случаев диабета 1 типа имеют иммунно-опосредованную природу, при которой утрата бета-клеток происходит вследствие иммунной атаки, опосредованной бета-клетками. Сахарный диабет 2 типа характеризуется дисфункцией бета-клеток в комбинации с невосприимчивостью к инсулину. Считается, что в нарушение реакции тканей организма на инсулин вовлечены рецепторы инсулина. Как и в случае диабета 1 типа, недостаточная масса бета-клеток также является патогенным фактором у многих пациентов с диабетом 2 типа. На ранних стадиях диабета 2 типа гипергликемию можно обратить, применяя ряд мер и медикаментов, которые улучшают секрецию инсулина и уменьшают выработку глюкозы в печени. По мере прогрессирования заболевания, возникает нарушение секреции инсулина, и у некоторых пациентов может возникнуть необходимость в терапевтическом замещении инсулина.
Регуляторные T-клетки являются субпопуляцией T-клеток CD4+, вырабатываемых тимусом, которые, как хорошо известно, играют значительную роль в поддержании иммунологической толерантности. Регуляторные T-клетки играют активную роль в модулировании иммунитета и подавляют аллоиммунные реакции отторжения трансплантата (C. A. Piccirillo. Cytokine 43, 395 (Sep, 2008); G. Xia et al. Translational research: the journal of laboratory and clinical medicine 153, 60 (Feb, 2009); K. J. Wood. Transplantation proceedings 43, 2135 (Jul-Aug, 2011); and G. Feng et al. Transplantation 86, 578 (Aug 27, 2008)). Регуляторные T-клетки предотвращают аутоиммунный диабет у мышей и подавляют аутореактивное разрушение трансплантированных островков (M. J. Richer et al. PloS one 7, e31153 (2012) and D. R. Tonkin et al. Immunol 181, 4516 (Oct 1, 2008)). Трансплантация островков является потенциальным подходом к лечению диабета, однако в более ранних исследованиях трансплантации островков подавление системного иммунитета не позволяло добиться долговременного регулирования уровней глюкозы в крови из-за иммунно-опосредованного отторжения трансплантированных островков. Включение полипептида CXCL12 в матрицу, инкапсулирующую трансплантированные островки, обеспечивает как физический, так и биологический барьер против клеточно-опосредованного и гуморального иммунного воздействия на островки. Полипептид CXCL12 отталкивает эффекторные T-клетки и мобилизует иммуносупрессорные регуляторные T-клетки, за счет чего уменьшает или устраняет необходимость системного подавления иммунитета. Соответственно, в одном из вариантов осуществления, матрица, высвобождающая полипептид CXCL12 по настоящему изобретению, применима для регенерации, замещения или замены (частичной или полной) как минимум части поджелудочной железы у пациента с недостаточным количеством клеток поджелудочной железы, в частности бета-клеток, без одновременного подавления иммунитета. Любой пациент, у которого поджелудочная железа не вырабатывает достаточное количество инсулина или фактически не вырабатывает инсулин, может получить пользу от такой терапии. Недостаточная выработка инсулина включает выработку пониженных количеств инсулина по сравнению с нормальным (здоровым) субъектом, а также включает субъектов, которые вырабатывают инсулин в количестве, сравнимом с нормальным (здоровым) субъектом, но которым требуются более высокие уровни инсулина, например, из-за невосприимчивости к инсулину, избыточного потребления пищи, ожирения в тяжелой форме и т.п.
Матрицы, высвобождающие полипептид CXCL12 по настоящему изобретению, селективно мобилизуют регуляторные T-клетки и за счет этого пролонгируют выживание имплантированной матрицы и обеспечивают защиту от иммунного разрушения даже у сенсибилизированного хозяина. Соответственно, матрицы, высвобождающие полипептид CXCL12 по настоящему изобретению, при желании могут быть восстановлены и введены повторно без отторжения иммунной системой. Кроме того, матрицы, высвобождающие полипептид CXCL12 по настоящему изобретению, обеспечивают долговременное выживание островков и непрерывную выработку полипептида CXCL12 инкапсулированными островками в течение по крайней мере от примерно 1 месяца до примерно 2 лет. В течение этого времени, концентрация глюкозы в крови субъекта натощак сохраняется на уровне от примерно 80 мг/мл до примерно 120 мг/дл. Соответственно, матрица, высвобождающая полипептид CXCL12 по настоящему изобретению, особенно применима при лечении диабета.
В конкретном варианте осуществления, матрица, высвобождающая полипептид CXCL12, предназначенная для применения в лечении диабета, включает от примерно 1,5 до примерно 2% масса/объем сшитого кальцием альгината с высоким содержанием маннуроновой кислоты, от примерно 100 нг/мл до примерно 1 мкг/мл полипептида CXCL12 и как минимум одну островковую клетку, где полипептид CXCL12 высвобождается со скоростью от примерно 1,0 нг/мл/ч до примерно 3 нг/мл/ч. Типовая методика инкапсулирования начинается с получения островковых клеток, путем смешивания островков донора с 2 мл профильтрованной смеси дитизон/PBS. 80 мг альгината-LVM (Pronova UP LVM Sodium Alginate) смешивают с 5 мл 300 мОсмо NaCl до растворения. От примерно 800 до примерно 1000 островков центрифугируют в cRPM в течение 3 минут при 300 об/мин × G, ресуспендируют в DMEDM и вновь центрифугируют при 300 об/мин × G в течение 3 минут. Для смешивания 0,75 мл альгината с островками можно использовать 1 мл шприц. Альгинат и островки можно загрузить в 60-мл шприц, используя иглу 18g. Заполненный шприц помещают в инкапсулятор с установленным на максимум напряжением (например, 1,21 КВ), частотой, установленной на 1500 и амплитудой, установленной на максимум. После инкапсулирования перемешивают капсулы в течение примерно 5 минут в 300 мОсмо CaCl2, фильтруют капсулы в свежий стакан, промывают и культивируют с DMEM.
Настоящее изобретение дополнительно описано с привлечением расположенных ниже по тексту иллюстративных не ограничивающих примеров, которые обеспечивают лучшее понимание настоящего изобретения и многих из его преимуществ.
ПРИМЕРЫ
Приведенные ниже примеры иллюстрируют некоторые варианты осуществления и аспекты настоящего изобретения. Специалисту в соответствующей области техники должно быть очевидно, что различные модификации, дополнения, замены и т.п. могут быть осуществлены без изменения сути и выхода за пределы объема изобретения, и такие модификации и варианты охвачены объемом изобретения, который определен приведенной ниже формулой изобретения. Следующие далее примеры никоим образом не ограничивают изобретение.
Трансплантация островков представляет собой потенциально эффективный подход к лечению диабета 1 типа. Однако трансплантация островков обычно требует системного подавления иммунитета для борьбы с иммунно-опосредованным отторжением трансплантированных островков и имеются ограниченные источники человеческих островков. Приведенные ниже примеры показывают, что хемокин, а именно CXCL12, может отталкивать эффекторные T-клетки при этом мобилизуя иммуносупрессорные регуляторные T-клетки (Tregs) на соответствующем участке организма, и покрытие или инкапсулирование донорных островков CXCL12 может вызывать местную изоляцию от действия иммунной системы и защищать алло- или ксенотрансплантаты без системного подавления иммунитета. В приведенных ниже примерах трансплантацию островков осуществляли в мышиных моделей инсулин-зависимого диабета. Покрытие островков CXCL12 или микроинкапсулирование островков с помощью альгината, включающего этот хемокин, т.е. CXCL12, приводило к увеличению срока выживания и функционирования алло- и ксено- островков, а также к селективному увеличению инфильтрации Treg. Эти данные, как показано ниже, указывают, что применение CXCL12 в качестве покрытия или компонента альгинатного инкапсулирующего материала вызывает локальную изоляцию алло- и ксено-трансплантатов островков, при этом отменяя необходимость системного подавления иммунитета.
Пример 1: Непосредственное покрытие аллогенных островков полипептидом CXCL12
В этом примере предполагалось определить, может ли покрытие аллогенных островков полипептидами CXCL12 перед трансплантацией привести к увеличению срока выживания и функционирования островков, а также к аккумулированию регуляторных T-клеток (Tregs) в месте нахождения трансплантата. Капсула с островками обычно содержит фибронектин и, не ограничиваясь какой-либо конкретной теорией, CXCL12 способен стабильно связываться и вымываться из этого матричного белка (15). Соответственно, островки, извлеченные из мышей BALB/C, подергали действию буферного раствора (например, PBS) или покрывали CXCL12 в концентрации примерно 100 нг/мл или примерно 1 мкг/мл и трансплантировали под капсулу левой почки диабетических мышей C57BL/6, обработанных стрептозоцином (STZ). Мышей умерщвляли в тот момент, когда они возвращались к диабетическому состоянию, которое определяли по двум последовательно зарегистрированным уровням глюкозы >250 мг/дл. Аллогенные трансплантаты островков, покрытые CXCL12 в концентрации ~1 мкг/мл позволяли поддерживать мышей-реципиентов в не диабетическом состоянии в течение достоверно более продолжительного времени по сравнению с аллогенными островками, на которые действовали только PBS (p=0,012; логарифмический ранговый критерий Каплана-Майера) (Фиг.1A). Островки, покрытые 100 нг/мл полипептида CXCL12, подвергались отторжению со скоростью, аналогичной скорости отторжения островков, обработанных PBS (p=0,31). Инфильтрация мононуклеарных клеток в трансплантаты уменьшалась в случае покрытия CXCL12 в концентрации примерно 1 мкг/мл по сравнению с PBS контролем (Фиг.1B). Экспрессия инсулина и наличие CXCL12 также показано для аллотрансплантатов, покрытых CXCL12, по сравнению с трансплантатами, обработанными PBS (фиг.1B). Количественно определяли инфильтрацию T-клеток CD3 и FoxP3 в трансплантаты. Инфильтрация T-клеток CD3+ в трансплантаты островков достоверно сокращалась при покрытии CXCL12 по сравнению с аллотрансплантатами, обработанными PBS, по данным иммуногистохимии (Фиг.1C, p=0,001). Далее, покрытие аллотрансплантатов островков CXCL12 приводило к достоверному росту инфильтрации T-клеток FoxP3+ в трансплантат, покрытый CXCL12, и вокруг него, по сравнению с аллотрансплантатами, обработанными PBS (Фиг.1D, p=0,0016).
Пример 2: Одновременное применение покрытия CXCL12 и низких доз циклоспорина A
В следующем эксперименте предполагалось определить, может ли сопутствующее использование системного подавления иммунитета, например, в форме введения низкой дозы циклоспорина A (CsA) (например, 2 мг/кг), улучшить выживаемость аллотрансплантата островков, покрытого CXCL12. Хотя в этих экспериментах было показано, что покрытие аллотрансплантата CXCL12 само по себе продлевает выживаемость островков, по сравнению с контролем, обработанным PBS(p=0,0245), покрытие CXCL12 в комбинации с введением низких доз CsA не приводит к более продолжительному выживанию островков, причем комбинация покрытия CXCL12 и обработки CsA могла привести к уменьшению выживаемости островков по сравнению только с покрытием CXCL12 (p=0,046) (Фиг.2A). Достоверная разница между выживаемостью островков и между контрольными и экспериментальными группами была показана через 23 дня после трансплантации. Окрашивание трансплантатов островков выявило сильную инфильтрацию T-клеток СВ3+ у животных с CXCL12 покрытием, дополнительно получавших CsA, по сравнению с животными, у которых было только покрытие CXCL12 (p=0,0002), или только получавших CsA (p=0,0027) (Фиг.2B-2C). Добавление CsA в низких дозировках животным с трансплантатами островков, покрытыми CXCL12, приводило также к достоверному уменьшению инфильтрации клеток FoxP3 в трансплантат, по сравнению с островками, покрытыми только CXCL12 (p=0,0188) (Фиг.2C). Ранее обсуждалось, что CsA ингибирует определенные элементы сигнального пути CXCL12 и, например, опосредованную хемокинами миграцию клеток (16). Данные настоящего примера показывают, что одновременное применение покрытия CXCL12 и циклоспорина A в низких дозировках не приводит к дополнительному усилению защиты аллотрансплантатов островков от действия иммунной системы.
Поскольку островки, покрытые CXCL12, продемонстрировали более продолжительное выживание по сравнению с островками, обработанными PBS, на следующем этапе планировалось определить, является ли причиной этого явления воздействие хемокина на клеточно-опосредованный иммунный ответ против островков. Для выяснения уровня аллореактивности, осуществляли реакцию смешанной культуры лимфоцитов (MLR), отобранных у мышей через 21 день после введения животным трансплантатов островков покрытых или не покрытых CXCL12. Между двумя этими группами не было обнаружено различий, и это свидетельствует о том, что CXCL12 создает нишу в организме, в которой может замедляться отторжение, опосредованное T-клетками, но это не влияет на развитие иммунной реакции на островки (Фиг. 3A-3C). Таким образом, не желая ограничиваться конкретной теорией, в некоторых вариантах осуществления, покрытие CXCL12 может вызывать локальную изоляцию от действия иммунной системы, а не предотвращать возникновение системного анти-аллогенного клеточно-опосредованного ответа.
Пример 3: Трансплантация сингенных островков, покрытых CXCL12, преддиабетическим и диабетическим мышиным моделям.
Для определения может ли покрытие CXCL12 способствовать ослаблению или предотвращению отторжения при трансплантации сингенных островков, использовали диабетических мышей NOD/LtJ. В этой модели, сингенные островки не диабетических мышей NOD/LtJ трансплантировали диабетическим мышам NOD/LtJ, обработанным STZ. Покрытие сингенных островков CXCL12 приводило к достоверно более продолжительному периоду нормогликемии по сравнению с островками, обработанными PBS (Фиг.4A) (p=0,017). Гистопатологические и иммуногистохимические исследования продемонстрировали сильную инфильтрацию мононуклеарных клеток в островки, обработанные PBS, но не в островки, покрытые CXCL12 (изображения не показаны). Т.е. окрашивание H&E продемонстрировало уменьшение инфильтрации мононуклеарных клеток в трансплантаты островков, покрытые ~1 мкг/мл CXCL12, и иммунофлуоресцентное окрашивание на инсулин и CXCL12 продемонстрировало увеличение уровней обоих белков в трансплантатах, покрытых CXCL12. Окрашивание на CXCL12 и инсулин в этих экспериментах продемонстрировало здоровые вырабатывающие инсулин и CXCL12-положительные островки, по сравнению с трансплантатами, обработанными PBS. Покрытие сингенных островков CXCL12 также сократило инфильтрацию T-клеток CD3+ в островки донора по сравнению с сингенными островками NOD/LtJ, обработанными PBS (Фиг.4B, p=0,0081). Покрытие сингенных островков NOD/LtJ CXCL12 приводило также к достоверному увеличению числа клеток FoxP3+ в трансплантатах островков по сравнению с контролем, обработанным PBS (p=0,0019) (Фиг.4C). Хотя покрытие сингенных островков CXCL12 не уменьшало скорость повторного появления диабета у мышей NOD/LtJ со спонтанным диабетом (Фиг.5A) (p=0,24), окрашивание гематоксилином и эозином и на инсулин/CXCL12 продемонстрировало ослабление инфильтрации мононуклеарных клеток и увеличение экспрессии инсулина в островках, покрытых CXCL12, по сравнению с контролем (Фиг.5B-5C). Кроме того, наблюдалось соответствующее уменьшение инфильтрации T-клеток CD3+ и увеличение инфильтрации клеток FoxP3+ в трансплантаты, покрытые CXCL12 по сравнению с PBS-контрольными образцами (Фиг.5D). Подводя итог, покрытие сингенных островков CXCL12 может предупредить отторжение в преддиабетической модели, но необязательно в диабетической модели. Это отсутствие способности покрытия CXCL12 стимулировать выживание в ситуации сингенного трансплантата можно отнести на счет, например, неэффективности блокирования развивающегося гуморального иммунного ответа против островков CXCL12 покрытием трансплантированных островков.
Пример 4: Применение альгинатного инкапсулирующего материала (островковые клетки, инкапсулированные в альгинате), включающего CXCL12 у сенсибилизированных и не сенсибилизированных реципиентов
Выше обсуждалось, что преждевременная экспрессия аутоантител против инсулина коррелирует с прогрессированием диабета и, вероятно, чаще всего является признаком инсулита (воспаления островков) (17-19). Как показано в примере 3, покрытие островков CXCL12 для трансплантации является более эффективным в качестве меры в ситуации заранее сформировавшихся антител против островков. В этом примере предполагалось определить, способно ли включение CXCL12 в альгинатные микрокапсулы защитить трансплантированные островки за счет обеспечения как физического, так и биологического барьера для клеточно-опосредованной и гуморальной иммунной реакции против островков.
В одном из вариантов осуществления, инкапсулирующая матрица включала 2% сшитого кальцием альгината низкой вязкости с высоким содержанием маннуроновой кислоты (Ca-LVM) в который был включен полипептид CXCL12 (далее по тексту этот материал именуется ʺCa-LVM-CXCL12ʺ). На Фиг.6A показано, что применение инкапсулирующего материала Ca-LVM-CXCL12 приводит к пролонгированному высвобождению CXCL12 in vitro со скоростью примерно 1,75 +/- 0,4 нг/мл/ч после первоначального быстрого высвобождения в течение первых 3 часов. Кроме того наблюдалось пролонгированное удерживание CXCL12 в альгинатной матрице, которое приводило к сохранению остаточной концентрации CXCL12 в капсулах от 100 до 200 нг/мл через 22 дня in vitro инкубирования капсул, не содержащих клеток (данные не показаны). Не желая ограничиваться какой-либо теорией, пролонгированное удерживание CXCL12 в матрице, вероятно, происходит в результате электростатических взаимодействий между положительно заряженным хемокином (pI=9) и отрицательно заряженным альгинатом (pI=2) (20). Это предположение подтверждается тем фактом, что CXCL12 эффективно вымывался в среду в результате инкубирования с 1М NaCl по сравнению со средой, не содержащей NaCl. Аналогично CXCL12 удерживался в капсуле при инкубировании в отсутствии NaCl, но извлекался из капсулы в 1М NaCl (Фиг.6B). Поскольку CXCL12, как было показано, является фактором выживания островков, оценивали также влияние включения CXCL12 в инкапсулирующий материал Ca-LVM на выживаемость островков. Представленные в заявке данные показывают, что включение CXCL12 в инкапсулирующий материал статистически достоверно снижало уровень активности каспазы-3 в инкапсулированных островках, который определяли как минимум через 48 часов культивирования in vitro по сравнению с немодифицированным альгинатом Ca-LVM (100 нг/мл CXCL12; p=0,0019) (1 мкг/мл; CXCL12; p=0,00028) (Фиг.6c).
Исследовали трансплантацию аллогенных островков в инкапсулирующем материале Ca-LVM-CXCL12 мышам NOD/LtJ со спонтанным диабетом без системного подавления иммунитета. Если аллогенные островки трансплантировали в брюшную полость диабетических мышей NOD/LtJ, включение CXCL12 в альгинат Ca-LVM достоверно продлевало функционирование островков по сравнению с немодифицированным альгинатом Ca-LVM (среднее число дней после трансплантации в не диабетическом состоянии Ca-LVM-CXCL12=136; Ca-LVM=62) (p=0,048) (Фиг.6D). Гистопатологические исследования капсул Ca-LVM-CXCL12, извлеченных через 12 недель после трансплантации, показало отсутствие изменений морфологии островков в отличие от капсул из немодифицированного Ca-LVM, в которых островки подверглись некрозу или дегенерации (данные не показаны). С помощью фазово-контрастной микроскопии и H&E окрашивания, было установлено, что островки, инкапсулированные в Ca-LVM-CXCL12, оказались живыми и неповрежденными через 6 недель после трансплантации, в противоположность подвергшихся некрозу островкам, инкапсулированным в немодифицированный Ca-LVM. Соответственно, включение CXCL12 улучшает жизнеспособность островков и ослабляет некроз через шесть недель после трансплантации. Затем аллогенные островки мышей C57BL/6 трансплантировали мышам NOD/LtJ со спонтанным диабетом, которым ранее пересаживали трансплантат кожи мышей C57/B6, подвергшийся отторжению. Аллогенные островки, инкапсулированные в Ca-LVM-CXCL12, выживали и сохраняли нормогликемическое состояние реципиентов достоверно дольше, чем островки, инкапсулированные в Ca-LVM без добавки (Фиг.6E). Это показывает, что включение CXCL12 в инкапсулирующий материал защищает островки в организме мыши-реципиента от ответа иммунной памяти.
После этого предполагалось определить, можно ли защитить ксеногенные островки альгинатным инкапсулирующим материалом, включающим CXCL12. Ксеногенные свиные островки инкапсулировали в Ca-LVM или Ca-LVM с CXCL12 в концентрации примерно 10 нг/мл, примерно 100 нг/мл или примерно 1 мкг/мл, и трансплантировали в брюшную полость диабетических мышей C57BL/6. Свиные островки, инкапсулированные в Ca-LVM, содержащий CXCL12 в концентрации примерно 1 мкг/мл, поддерживали нормогликемию у мыши-реципиента в течение достоверно более продолжительного периода времени, чем в случае островков, инкапсулированных в Ca-LVM или Ca-LVM-CXCL12 (~10 нг/мл) (Фиг.6F).
Пример 5: Влияние покрытия или инкапсулирования CXCL12 на T-клетки CD8+ и клетки Treg in vitro
Для определения того, включает ли механизм, посредством которого покрытие CXCL12 или введение этого полипептида в инкапсулирующий материал, поддерживает иммунную изоляцию ксено- или алло-трансплантата островков, селективное отталкивание T-клеток CD8+ и притяжение Treg клеток CD4+ к месту нахождения трансплантата, была исследована экспрессия CXCR4 на субпопуляциях T-клеток и миграция этих субпопуляций у мышей NOD/LtJ с помощью поточной цитометрии и анализа миграции клеток, соответственно. Исследовали реакцию T-клеток, изолированных из мышей NOD/LtJ, на среду, рекомбинантный CXCL12, островки, покрытые CXCL12 или островки, инкапсулированные в Ca-LVM-CXCL12 с применением анализа в камере Бойдена. Верхние отсеки загружали очищенными T-клетками CD3+CD8+ (Фиг.7A и 7C) или Treg клетками CD4+CD25hi+ (Фиг.7B и 7D) для каждого условия исследования. Верхние и нижние лунки заполняли средой, CXCL12 (~1 мкг/мл), островками, покрытыми CXCL12 (~1 мкг/мл), или инкапсулированными в Ca-LVM-CXCL12 (~1 мкг/мл). Как T-клетки CD3+CD8+, так и Treg клетки CD4+CD25Hi подвергались хемотаксису в ответ на CXCL12, островки покрытые CXCL12, или островки, инкапсулированные в Ca-LVM-CXCL12. Однако значительно более сильная фугетаксическая реакция была зарегистрирована для клеток CD8+, которые инкубировали с CXCL12, островками покрытыми CXCL12, или островками, инкапсулированными в Ca-LVM-CXCL12, по сравнению с клетками Treg. В условиях эксперимента не было зарегистрировано обнаружимых уровней фугетаксиса клеток Treg.
Затем предполагалось определить, может ли различающаяся миграционная реакция на CXCL12 T-клеток CD8+ и Treg клеток CD4+ быть частично обусловлена различной экспрессией хемокин-распознающего рецептора, т.е. CXCR4, на двух этих субпопуляциях T-клеток. T-клетки CD8+ из селезенки мышей NOD/LtJ экспрессируют значительно более низкие уровни CXCR4, чем Treg клетки CD4+CD25HiFoxP3+ (p<0,005) (Фиг.7E и 7F). Эти данные показывают, что покрытие CXCL12 или включение CXCL12 в инкапсулирующий материал, окружающий аллотрансплантаты островков, может привести к преимущественной мобилизации клеток Treg в трансплантате, при отталкивании T-клеток CD8+ в данной модели трансплантата.
Обсуждение
Данные, представленные в примерах 1-5, показывают, что покрытие островков CXCL12 может вести к отсрочиванию отторжения островков при трансплантации аллогенных и сингенных островков диабетическим мышам NOD/LtJ, обработанным STZ. Конкретно, было установлено, что инкапсулирующий материал на основе Ca-LVM, включающий CXCL12, защищает аллогенные островки от конституционального гуморального и клеточно-опосредованного иммунного ответа при трансплантации в организм диабетических и сенсибилизированных мышей NOD/LtJ.
Модели трансплантатов в примерах 1-5 показывают, что покрытие или инкапсулирование островков CXCL12 приводит к накоплению клеток Treg в месте нахождения трансплантата.
Соответственно, не ограничиваясь какой-либо теорией, как в опухолях, так и в трансплантатах удерживание/накопление клеток Treg может быть связано с появлением иммуносупрессорного микроокружения. Представленные в настоящей заявке данные показывают, что покрытие или инкапсулирование аллогенных или ксеногенных островков CXCL12 может вести к отсрочиванию отторжения островков и сопутствующему увеличению времени функционирования островков, за счет развития локальной изоляции от действия иммунной системы. Обнаружение того, что включение CXCL12 в альгинатный инкапсулирующий материал клинической степени чистоты, окружающий трансплантированные аллогенные и ксеногенные островки, делает возможным более продолжительное функционирование островков и их защиту от разрушения иммунной системой сенсибилизированного организма-хозяина без системного подавления иммунитета, является неожиданным открытием, которое можно реализовать в клинической практике.
Типовые материалы и методики, использованные в примерах 1-5
Животные и инициирование диабета. Использовали самок мышей BALB/c (H2d) в возрасте 6 недель, самок мышей C57BL/6 (H2b) в возрасте 6 недель и самок мышей NOD/LtJ (H2g7) в возрасте 6-8 недель. Животных можно приобрести, например, у Jackson Laboratory (Bar Harbor, ME). Все процедуры с животными выполняли, следуя положениям Службы Общественного Здравоохранения по уходу за лабораторными животными, и эти процедуры были одобрены подкомитетом по уходу за исследуемыми животными при Массачусетском многопрофильном госпитале (Massachusetts General Hospital). Гипергликемия самопроизвольно возникала у мышей NOD/LtJ в возрасте 18-20 недель, и ее инициировали у 4- и 6-недельных мышей NOD/LtJ и 6-недельных мышей C57BL/6 интраперитонеальной инъекцией 200 мг/кг стрептозотоцина (Sigma-Aldrich, St.Louis, MO). Мышей, у которых три раза подряд регистрировали уровень глюкозы в крови более 250 мг/дл, считали гипергликемическими.
Извлечение островков поджелудочной железы, покрытие CXCL12 и включение хемокина в альгинатный инкапсулирующий материал. Первичные островки извлекали из мыши-донора, как описано в Papeta et al., Transplantation 83, 174 (2007). Например, островки извлекали у самок мышей BALB/c в возрасте 6 недель, самок мышей C57BL/6 в возрасте 6 недель или самок мышей NOD/LtJ в возрасте 4 недель. В поджелудочные железы через общий желчевыносящий проток вливали Liberase TL (например, ~83 мкг/мл) (Roche Diagnostics, Indianapolis, IN) и выдерживали в течение ~20 минут при ~37°C. Островки очищали на градиенте плотности полисахароза/глюкоза (Mediatech, Manassas, VA) и отбирали под микроскопом. Полученные островки культивировали в RPMI 1640 (Mediatech с добавкой 10% сыворотки телят, 1% пенициллина/стрептомицина и 1% L-глутамина) в течение как минимум 2 дней или более, чтобы дать им восстановиться. Минимум 450 островков, которые предполагалось трансплантировать через почечную капсулу, инкубировали перед трансплантацией при ~37°C в течение ~3 часов в DPBS (Mediatech) с или без ~100 нг/мл или ~1 мкг/мл CXCL12 (PeproTech Inc, Rocky Hill, NJ). Отдельно перед инкапсулированием вводили CXCL12 в жидкий Na-LVM альгинат (ультра чистый LVM, Novamatrix) в концентрации ~1 мкг/мл.
Получение и трансплантация капсул из Ca-LVM альгината. Примерно 1000 островков C57BL/6 смешивали с ~0,75 мл ~1,5% альгината (ультра чистый LVM, Novamatrix, Drammen, Norway)) в ~300 мОсмо растворе NaCl с или без ~1 мкг/мл CXCL12. Затем смесь пропускали через шприцевый инкапсулятор (Inotech Research Encapsulator, IE-50R) используя 300 мкм насадку с напряжением 1,21 КВ и вибрирующую на частоте 1500 Гц. Осуществляли поперечное сшивание альгината, например, в 300 мОсмо (примерно 118 мМ) растворе CaCl2 в течение примерно 5 минут, фильтровали и промывали DMEM (Mediatech) для удаления избытка кальция.
Удерживание и высвобождение CXCL12 из альгинатных капсул и анализ каспазы-3. Перед формированием капсул, 3,6% LVM смешивали с ~10 мкг/мл концентрированным раствором CXCL12 (Peprotech, Rocky Hill, NJ), получая ~3,3% LVM с содержанием CXCL12 ~1 мг/мл. Капсулы из LVM, сшитого кальцием, формировали с помощью электростатического генератора капель при напряжении 5 КВ, используя насадку 0,5 мм и скорость потока 30 мл/ч. Бесклеточные капсулы, содержащие CXCL12 инкубировали в обработанных многолуночных планшетах для нетканевых культур в среде DMEM (Sigma Aldrich, St.Louis, MO) с 1,6 г/л альбумина бычьей сыворотки (Sigma Aldrich). Соотношение между капсулами и средой в ходе эксперимента поддерживали на уровне примерно 1:2 объем/объем. В любой момент времени отбирали ~0,1 мл образец капсул и солюбилизировали его, используя 110 мМ раствор цитрата натрия. Кроме того, в любой момент времени удаляли всю среду и добавляли свежую среду. Среду и солюбилизированные образцы капсул анализировали на содержание CXCL12 с помощью ELISA (R&D Systems, Minneapolis, MN). Для определения влияния инкапсулирования в комбинации с CXCL12 на апоптоз островков, мышиные островки инкапсулировали как описано в заявке, в альгинатный инкапсулирующий материал, содержащий или не содержащий ~1 мкг/мл CXCL12. В качестве контроля, островки можно было также подвергнуть действию стрептозотоцина - известного инициатора апоптоза островков. Например, островки можно культивировать в этих условиях in vitro в течение 48 часов и определять активность каспазы-3 в инкапсулированных островках с помощью анализа на основе ELISA (R and D systems, Minneapolis, MN).
Высаливание CXCL12 из альгинатных капсул. Формировали капсулы, содержащие CXCL12, как описано выше со следующей модификацией; капсулы сшивали с использованием 20 мМ хлорида кальция для предотвращения их растворения в растворе хлорида натрия высокой концентрации. Капсулы инкубировали в ~1М NaCl c ~0,1% BSA в течение ~6 часов, после чего раствор собирали и шарики солюбилизировали, используя 200 мМ раствор динатриевой соли этилендиаминтетрауксусной кислоты (ED2SS), pH 9,0. Раствор и солюбилизированные образцы капсул анализировали на содержание CXCL12 с помощью ELISA, как описано выше. Контрольные капсулы инкубировали в растворе CaCl2, не содержащем NaCl, и определяли удержанный и высвободившийся из капсул CXCL12.
Модели трансплантации островков. Островки, подвергшиеся различной обработке, трансплантировали под капсулу левой почки мышей-реципиентов или инкапсулировали в Ca-LVM альгинат и трансплантировали через брюшную полость. Для трансплантации использовали четыре различные типовые модели: (1) островки BALB/C трансплантировали под почечную капсулу STZ-инициированных диабетических мышей C57BL/6; (2) островки преддиабетических (ранее сенсибилизированных) мышей NOD/LtJ трансплантировали под почечную капсулу мышей NOD/LtJ со спонтанным диабетом; (3) островки преддиабетических (ранее сенсибилизированных) мышей NOD/LtJ трансплантировали под почечную капсулу диабетических (ранее сенсибилизированных, с STZ-инициированным диабетом) мышей NOD/LtJ в возрасте 6 недель; и (4) островки мышей C57BL/6 инкапсулировали в альгинат Ca-LVM и трансплантировали в брюшную полость мышей NOD/LtJ в возрасте 6 недель с диабетом, инициированным STZ.
Мониторинг гликемического статуса реципиентов. Определяли уровень глюкозы в крови в хвостовой вене реципиентов как минимум два раза в неделю и по полученным данным судили о функционировании трансплантатов островков. Отторжение трансплантатов определяли по возврату гипергликемии (который характеризовался, например, по трем последовательным результатам определения глюкозы более 250 мг/дл). После отторжения трансплантатов мышей умерщвляли.
Иммуногистохимия и иммунофлуоресцентное окрашивание. Почки, с находящимися в них подкапсульными трансплантатами островков, фиксировали в 4% формальдегиде, заливали в парафин и готовили препараты, получая 5 мкм срезы трансплантатов. Часть срезов окрашивали гематоксилином и эозином (H&E). Также осуществляли иммуногистохимию, используя первичные антитела против CD3 (Dako, Denmark), FoxP3 (eBioscience, CA) и инсулина (Dako, Denmark). В пару к каждому первичному антителу подбирали подходящее вторичное антитело, и в качестве субстрата для окрашивания использовали DAB (3,3'-диаминобензидин). Окрашивание ядер (в синий цвет) осуществляли с помощью гемалюма Майера для реализации способа окрашивания комплекса авидин-биотин IHC. Изображения регистрировали при помощи камеры на микроскопе Zeiss Axio Observer Z1 при увеличении 10×, причем производили ручной подсчет срезов островков для определения инфильтрации T-клеток CD3+ и/или FoxP3+. Количества клеток, положительных для каждого маркера, регистрировали в 5 случайно выбранных полях зрения при высоком увеличении вблизи места нахождения трансплантата. Иммунофлуоресцентное окрашивание также проводили с использованием флуоресцентных первичных антител против инсулина (антитела морской свинки против инсулина, 1:800, Invitrogen) и CXCL12 (кроличьи против мышиного CXCL12, 1:200, Cell Sciences). Срезы промывали, инкубировали с первичными антителами в течение ночи при 4°C и затем инкубировали с подходящими вторичными антителами в течение одного часа при комнатной температуре (козий IgG против морской свинки [Dylight 488, 1:200, Jackson Immunoresearch] и козий IgG против кролика [Dylight 549, 1:200, Jackson Immunoresearch]). После обработки вторичными антителами, ткани промывали, проводили контр-окрашивание To-Pro-3 (1:5000, Invitrogen) и готовили препараты для микроскопического исследования. Цифровые изображения иммунофлуоресцентных препаратов получали с применением конфокальной микроскопии (LSM 5 Pascal, Carl Zeiss).
Обработка циклоспорином A. Диабетическим мышам C57BL/6 трансплантировали 450 островков BALB/C, покрытых PBS или CXCL12 (~ 1 мкг/мл). Реципиентов делили на три экспериментальные группы, которые получали (A) инъекцию циклоспорина A (CsA, Sigma) для реципиентов островков, покрытых PBS; (B) инъекцию CsA для реципиентов островков, покрытых CXCL12; и (C) инъекцию PBS для реципиентов островков, покрытых CXCL12. Для групп A и B, CsA готовили растворением в этаноле и вводили подкожной инъекцией в дозировке ~2 мг/кг в день трансплантации и затем ежедневно до отторжения трансплантата.
Реакция смешанной культуры лимфоцитов. Мышей C57BL/6 (n=3/группу) для отбора иммунореактивных клеток распределяли в одну из трех групп: с трансплантатами островков, покрытыми CXCL12, с не покрытыми трансплантатами островков, или без какого-либо вмешательства. Через 10 дней после осуществления указанных выше операций, селезенки животных подвергали механическому разрушению и фильтровали, красные кровяные клетки подвергали лизису, используя буфер M-lyse (R&D Systems). Аллогенные стимулирующие клетки из селезенок необработанных мышей BALB/C получали аналогичным образом и инкубировали с 20 мкг/мл митомицином C (Sigma Aldrich) в течение 1 часа при 37°C, несколько раз промывали средой и культивировали совместно с иммунореактивными клетками в соотношении 1:1 при 37°C в атмосфере 5% CO2 в течение трех дней. За 18 часов до завершения стимулирования, клетки возбуждали 1 мМ раствором бромдезоксиуридина (BrdU, BD Biosciences). После 72 часов культивирования клетки промывали и затем окрашивали антителами против мышиных CD3 (APC-Cy7), CD8(PerCP) и CD4(PE-Cy7) (BD Biosciences). Клетки пермеабилизировали с помощью Cytofix/Cytoperm (BD Biosciences), подвергали расщеплению действием DNase (Sigma Aldrich) и окрашивали PE-конъюгированным антителом против BrdU (BD Biosciences). Выполняли проточную цитометрию на приборе LSRII (BD Biosciences) и анализировали данные в программе FlowJo (Tree Star) для определения числа T-клеток на селезенку, которые подверглись стимулированию и захватили BrdU, за вычетом числа нестимулированных клеток на селезенку, которые захватили BrdU.
Исследование миграции клеток. Проводили анализ миграции клеток двух типов. Во-первых определяли миграцию клеток с использованием камеры Бойдена (96-луночный формат, поры 3 мкм; ChemoTx System, Neuro Probe Inc, Gaithersburg, MA), как было описано ранее Poznansky et al., Journal of clinical investigation, 109, 1101 (2002). Например, ~30 мкл RPMI 1640 с добавкой 0,5% FBS вместе с 50 контрольными островками или островками, покрытыми 1 мкг/мл CXCL12 (PeproTech Inc, Rocky Hill, NJ) помещали в верхние и нижние отсеки. 7000 клеток T-reg или T-клеток CD8, изолированных с помощью наборов для выделения клеток CD4, CD25 и/или CD8 MACS, соответственно (Miltenyi Biotec, Auburn, CA) помещали в верхние отсеки камеры Бойдена. После трехчасового инкубирования при 37°C и 5% CO2, клетки с верхней поверхности мембраны удаляли, и мигрировавшие клетки в нижнем отсеке подсчитывали три раза. В отдельном исследовании, определяли также миграцию в инкапсулированные островки при включенном или не включенном в состав капсул CXCL12, используя систему Transwell (24-луночный формат, поры 3 мкм, Corning). Вкратце, 500 мкл RPMI 1640 с добавкой 0,5% FBS, либо только среду, либо с добавлением 10 контрольных капсул, либо с добавлением капсул, содержащих 1 мкг/мл CXCL12, помещали в нижний отсек. 100 мкл среды, либо в чистом виде, либо с различными капсулами, помещали в верхний отсек вместе с 105 клеток желаемой популяции. Клетки инкубировали в течение 3 часов при 37°C и 5% CO2, и проводили три независимых подсчета количества мигрировавших клеток. В обоих экспериментах вычисляли коэффициент миграции, как соотношение числом клеток, подсчитанным в присутствии CXCL12 и в контрольных образцах в присутствии только среды.
Количественное определение экспрессии CXCR4 на субпопуляциях T-клеток. У необработанных мышей NOD/LtJ собирали спленоциты и красные кровяные клетки подвергали лизису с использованием буфера Mouse Erythrocyte Lysing Buffer (R&D Systems). Затем клетки окрашивали для выявления поверхностных маркеров CD3ε, CD4, CD8α, CD25 и CXCR4 (CD184) (CD3ε: клон 145-2C11; CD4: клон RM 4-5; CD8α: клон 53-6.7; CD25: клон PC61; CXCR4: клон 2B11, BD Biosciences), фиксировали пермеабилизировали с помощью Cytofix/Cytoperm (BD Biosciences) согласно инструкции производителя и окрашивали для выявления внутриклеточного маркера FoxP3 (клон MF23, BD Biosciences). Выполняли проточную цитометрию на приборе 4 Laser LSR II (BD Biosciences). Данные анализировали с использованием программы FlowJo (Tree Star) и осуществляли гейтирование (отбор данных только по интересующей популяции клеток) с использованием стратегии флуоресценция-минус-один (fluorescence-minus-one).
Статистический анализ. Выживаемость трансплантатов островков в различных группах сравнивали с помощью способа Каплана-Майера, и данные по выживаемости анализировали с помощью программы статистического анализа GraphPadPrism 5. Числовые переменные сравнивали с использованием t-критерия Стьюдента. Значение p менее 0,05 считалось статистически достоверным.
Из приведенного выше описания должно быть очевидно, что в описанное изобретение можно вносить изменения и модификации, чтобы привести его в соответствие с различными применениями и условиями. Такие варианты осуществления также охвачены приведенной ниже формулой изобретения. Упоминание перечная элементов в любом определении переменной величины в тексте заявки, включает определения этой переменной, как любого отдельного элемента или комбинации (или подкомбинации) перечисленных элементов. Упоминание в тексте заявки какого-либо варианта осуществления включает этот вариант осуществления в качестве отдельного варианта осуществления или в комбинации с любым другим вариантом осуществления или его частями.
СПИСОК ЛИТЕРАТУРЫ
Все патенты, заявки на патенты и публикации, упомянутые в настоящем описании включены в заявку посредством ссылки в той же степени, как если бы для каждого отдельного патента или публикации было конкретно и индивидуально указано, что он включен в заявку посредством ссылки. Включение посредством ссылки в настоящую заявку относится, не ограничиваясь этим, к следующим источникам:
1. N. Papeta et al., Long-term survival of transplanted allogeneic cells engineered to express a T cell chemorepellent. Transplantation 83, 174 (Jan 27, 2007).
2. A. M. Shapiro et al., International trial of the Edmonton protocol for islet transplantation. The New England journal of medicine 355, 1318 (Sep 28, 2006).
3. J. S. Kaddis et al., Human pancreatic islets and diabetes research. JAMA: the journal of the American Medical Association 301, 1580 (Apr 15, 2009).
4. R. P. Robertson, Islet transplantation as a treatment for diabetes - a work in progress. The New England journal of medicine 350, 694 (Feb 12, 2004).
5. R. B. Jalili et al., Local expression of indoleamine 2,3 dioxygenase in syngeneic fibroblasts significantly prolongs survival of an engineered three-dimensional islet allograft. Diabetes 59, 2219 (Sep, 2010).
6. V. Vaithilingam, J. Oberholzer, G. J. Guillemin, B. E. Tuch, The humanized NOD/SCID mouse as a preclinical model to study the fate of encapsulated human islets. The review of diabetic studies: RDS 7, 62 (Spring, 2010).
7. A. G. Mallett, G. S. Korbutt, Alginate modification improves long-term survival and function of transplanted encapsulated islets. Tissue engineering. Part A 15, 1301 (Jun, 2009).
8. N. Sakata et al., Encapsulated islets transplantation: Past, present and future. World journal of gastrointestinal pathophysiology 3, 19 (Feb 15, 2012).
9. M. C. Poznansky et al., Thymocyte emigration is mediated by active movement away from stroma-derived factors. The Journal of clinical investigation 109, 1101 (Apr, 2002).
10. M. C. Poznansky et al., Active movement of T cells away from a chemokine. Nature medicine 6, 543 (May, 2000).
11. M. Khattar et al., Novel sphingosine-1-phosphate receptor modulator KRP203 combined with locally delivered regulatory T cells induces permanent acceptance of pancreatic islet allografts. Transplantation 95, 919 (Apr 15, 2013).
12. F. Vianello, I. T. Olszak, M. C. Poznansky, Fugetaxis: active movement of leukocytes away from a chemokinetic agent. J Mol Med (Berl) 83, 752 (Oct, 2005).
13. E. Righi et al., CXCL12/CXCR4 blockade induces multimodal antitumor effects that prolong survival in an immunocompetent mouse model of ovarian cancer. Cancer research 71, 5522 (Aug 15, 2011).
14. Z. Liu, J. F. Habener, Stromal cell-derived factor-1 promotes survival of pancreatic beta cells by the stabilisation of beta-catenin and activation of transcription factor 7-like 2 (TCF7L2). Diabetologia 52, 1589 (Aug, 2009).
15. A. J. Pelletier et al., Presentation of chemokine SDF-1 alpha by fibronectin mediates directed migration of T cells. Blood 96, 2682 (Oct 15, 2000).
16. A. Datta et al., Differential effects of immunosuppressive drugs on T-cell motility. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 6, 2871 (Dec, 2006).
17. L. Yu et al., Early expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence for early determination of subsequent diabetes. Proceedings of the National Academy of Sciences of the United States of America 97, 1701 (Feb 15, 2000).
18. G. S. Eisenbarth, Insulin autoimmunity: immunogenetics/immunopathogenesis of type 1A diabetes. Annals of the New York Academy of Sciences 1005, 109 (Nov, 2003).
19. E. Bonifacio et al., International Workshop on Lessons From Animal Models for Human Type 1 Diabetes: identification of insulin but not glutamic acid decarboxylase or IA-2 as specific autoantigens of humoral autoimmunity in nonobese diabetic mice. Diabetes 50, 2451 (Nov, 2001).
20. Y. Wang, D. J. Irvine, Engineering chemoattractant gradients using chemokine-releasing polysaccharide microspheres. Biomaterials 32, 4903 (Jul, 2011).
21. T. Yano, Z. Liu, J. Donovan, M. K. Thomas, J. F. Habener, Stromal cell derived factor-1 (SDF-1)/CXCL12 attenuates diabetes in mice and promotes pancreatic beta-cell survival by activation of the prosurvival kinase Akt. Diabetes 56, 2946 (Dec, 2007).
22. Q. Ma et al., Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proceedings of the National Academy of Sciences of the United States of America 95, 9448 (Aug 4, 1998).
23. T. Netelenbos et al., Proteoglycans guide SDF-1-induced migration of hematopoietic progenitor cells. Journal of leukocyte biology 72, 353 (Aug, 2002).
24. C. A. Piccirillo, Regulatory T cells in health and disease. Cytokine 43, 395 (Sep, 2008).
25. G. Xia, M. Shah, X. Luo, Prevention of allograft rejection by amplification of Foxp3(+)CD4(+)CD25(+) regulatory T cells. Translational research: the journal of laboratory and clinical medicine 153, 60 (Feb, 2009).
26. K. J. Wood, Regulatory T cells in transplantation. Transplantation proceedings 43, 2135 (Jul-Aug, 2011).
27. G. Feng, K. J. Wood, A. Bushell, Interferon-gamma conditioning ex vivo generates CD25+CD62L+Foxp3+ regulatory T cells that prevent allograft rejection: potential avenues for cellular therapy. Transplantation 86, 578 (Aug 27, 2008).
28. M. J. Richer, D. J. Lavallee, I. Shanina, M. S. Horwitz, Immunomodulation of antigen presenting cells promotes natural regulatory T cells that prevent autoimmune diabetes in NOD mice. PloS one 7, e31153 (2012).
29. D. R. Tonkin, J. He, G. Barbour, K. Haskins, Regulatory T cells prevent transfer of type 1 diabetes in NOD mice only when their antigen is present in vivo. J Immunol 181, 4516 (Oct 1, 2008).
30. K. Hire, D. K. Ngo, K. M. Stewart-Maynard, B. Hering, P. Bansal-Pakala, FoxP3+, and not CD25+, T cells increase post-transplant in islet allotransplant recipients following anti-CD25+ rATG immunotherapy. Cellular immunology 274, 83 (2012).
31. Q. Shi, J. R. Lees, D. W. Scott, D. L. Farber, S. T. Bartlett, Endogenous expansion of regulatory T cells leads to long-term islet graft survival in diabetic NOD mice. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 12, 1124 (May, 2012).
32. R. S. Francis et al., Induction of transplantation tolerance converts potential effector T cells into graft-protective regulatory T cells. European journal of immunology 41, 726 (Mar, 2011).
33. D. Chen et al., CD4+ CD25+ regulatory T-cells inhibit the islet innate immune response and promote islet engraftment. Diabetes 55, 1011 (Apr, 2006).
34. N. Marek et al., Coating human pancreatic islets with CD4(+)CD25(high)CD127(-) regulatory T cells as a novel approach for the local immunoprotection. Annals of surgery 254, 512 (Sep, 2011).
35. E. S. Yolcu et al., Pancreatic islets engineered with SA-FasL protein establish robust localized tolerance by inducing regulatory T cells in mice. J Immunol 187, 5901 (Dec 1, 2011).
36. F. Jaafar et al., Correlation of CXCL12 expression and FoxP3+ cell infiltration with human papillomavirus infection and clinicopathological progression of cervical cancer. The American journal of pathology 175, 1525 (Oct, 2009).
37. L. Zou et al., Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer research 64, 8451 (Nov 15, 2004).
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ приготовления микрокапсул с островками Лангерганса и микрокапсула по предложенному способу | 2023 |
|
RU2822875C1 |
| НОВОЕ ПРИМЕНЕНИЕ АГОНИСТОВ ПЕЧЕНОЧНОГО РЕЦЕПТОРА Х | 2006 |
|
RU2417078C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИОЛОГИЧЕСКОЙ ТКАНЕПОДОБНОЙ СТРУКТУРЫ | 2020 |
|
RU2815583C2 |
| СПОСОБ МИКРОИНКАПСУЛИРОВАНИЯ КЛЕТОК СЕРТОЛИ, ПОЛУЧАЕМЫЕ МИКРОКАПСУЛЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ ПРЕДОТВРАЩЕНИЯ И ЛЕЧЕНИЯ САХАРНОГО ДИАБЕТА 1 ТИПА | 2009 |
|
RU2598736C2 |
| Способ создания клеточных трансплантатов для лечения сахарного диабета 1-го типа | 2020 |
|
RU2765913C1 |
| ДИФФЕРЕНЦИРОВКА СТРОМАЛЬНЫХ КЛЕТОК, ПОЛУЧЕННЫХ ИЗ ЖИРОВОЙ ТКАНИ, В ЭНДОКРИННЫЕ КЛЕТКИ ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ И ИХ ИСПОЛЬЗОВАНИЕ | 2002 |
|
RU2351648C2 |
| ДИФФЕРЕНЦИРОВКА IN VITRO ПЛЮРИПОТЕНТНЫХ СТВОЛОВЫХ КЛЕТОК В ПАНКРЕАТИЧЕСКИЕ ЭНДОДЕРМАЛЬНЫЕ КЛЕТКИ (ПЭК) И ЭНДОКРИННЫЕ КЛЕТКИ | 2020 |
|
RU2764465C1 |
| ДИФФЕРЕНЦИРОВКА IN VITRO ПЛЮРИПОТЕНТНЫХ СТВОЛОВЫХ КЛЕТОК В ПАНКРЕАТИЧЕСКИЕ ЭНДОДЕРМАЛЬНЫЕ КЛЕТКИ (ПЭК) И ЭНДОКРИННЫЕ КЛЕТКИ | 2014 |
|
RU2734657C2 |
| ДИФФЕРЕНЦИРОВКА IN VITRO ПЛЮРИПОТЕНТНЫХ СТВОЛОВЫХ КЛЕТОК В ПАНКРЕАТИЧЕСКИЕ ЭНДОДЕРМАЛЬНЫЕ КЛЕТКИ (ПЭК) И ЭНДОКРИННЫЕ КЛЕТКИ | 2014 |
|
RU2670064C2 |
| САМОЖЕЛИРУЮЩИЕСЯ АЛЬГИНАТНЫЕ СИСТЕМЫ И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2393867C2 |
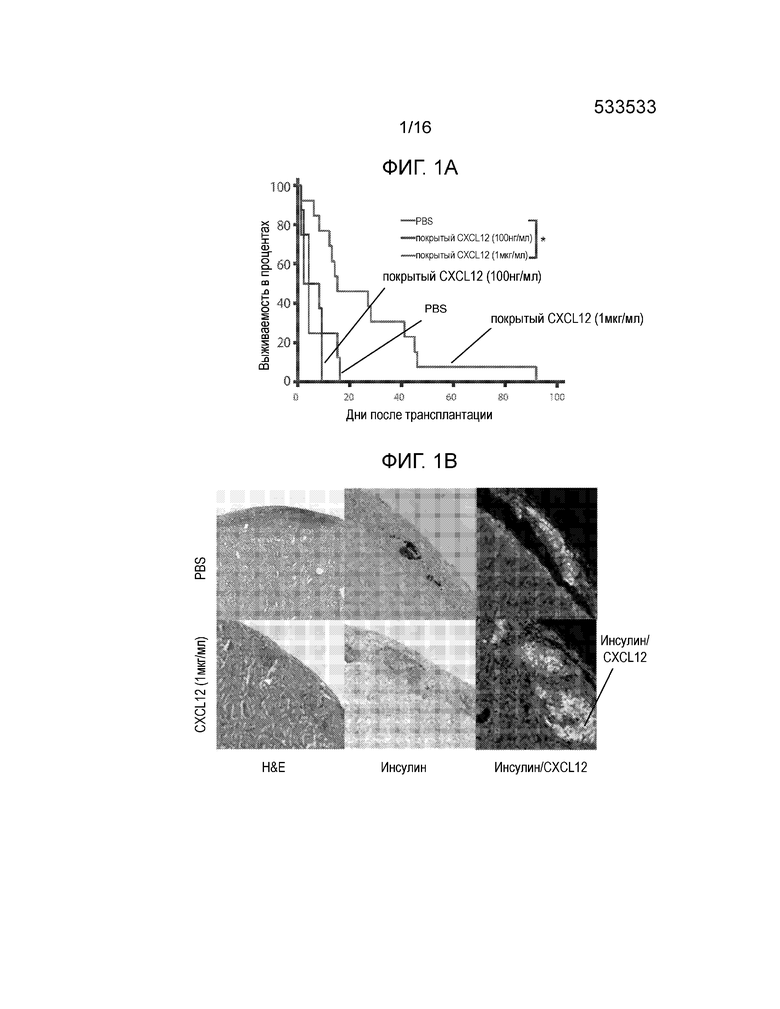
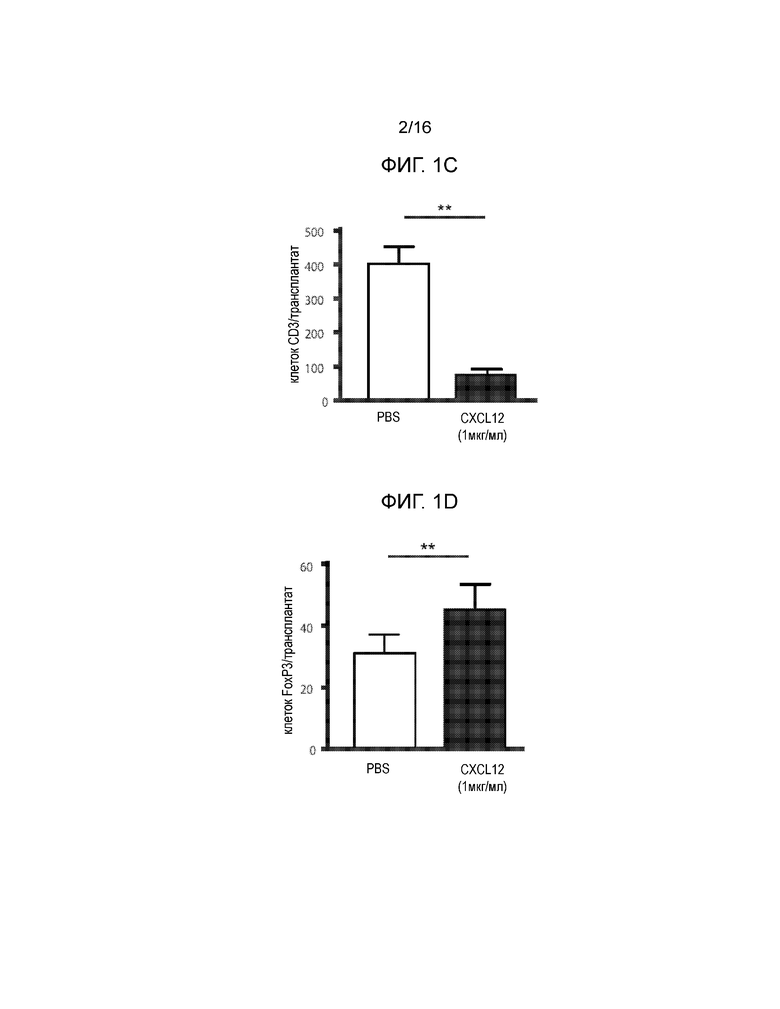
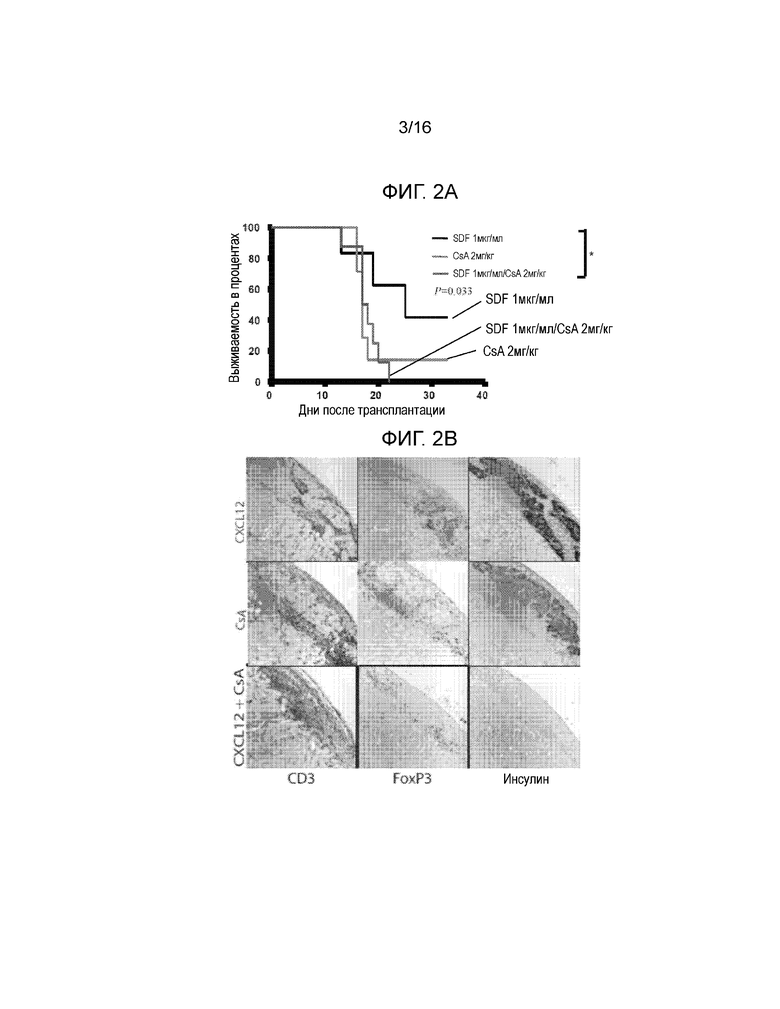
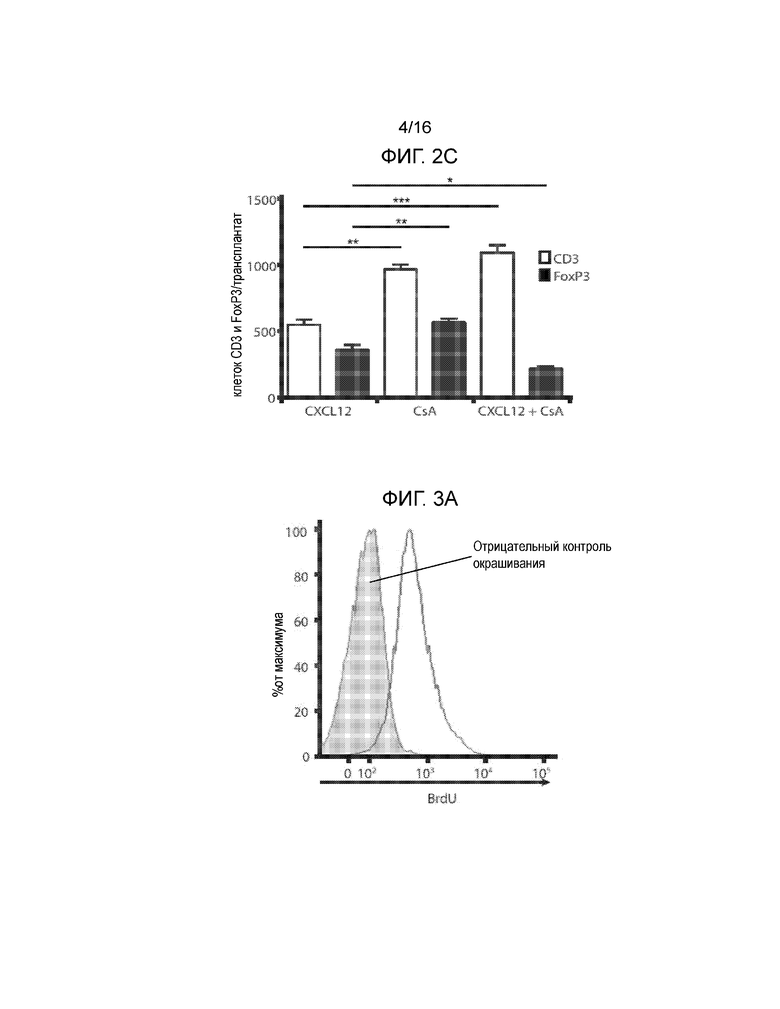
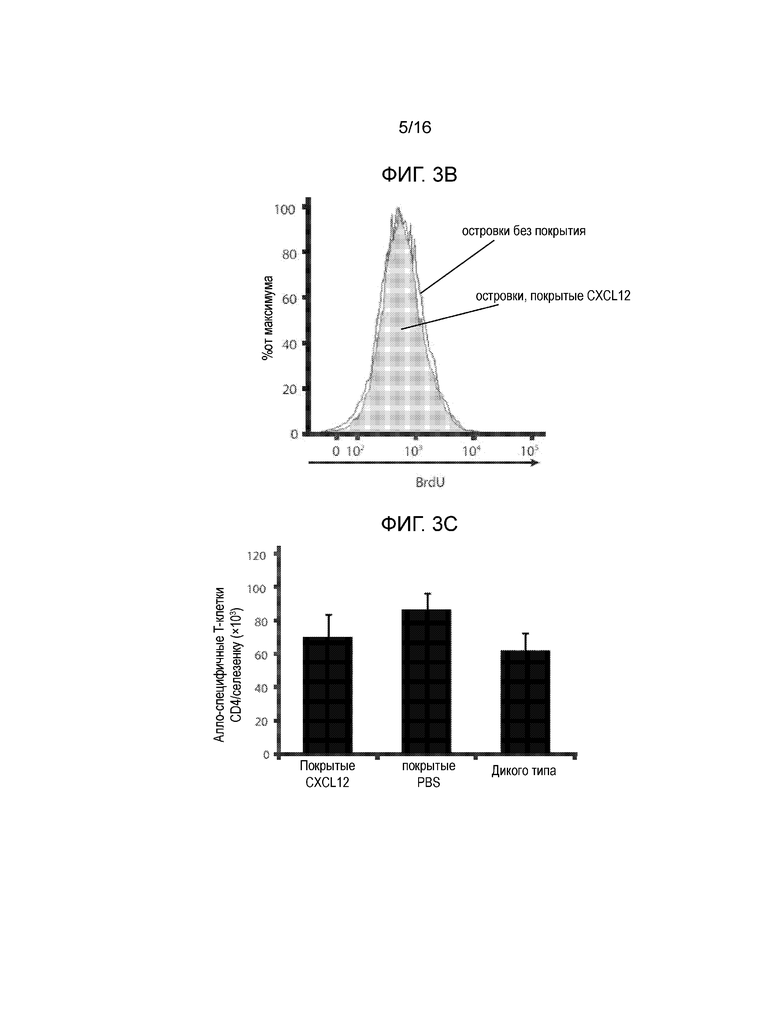
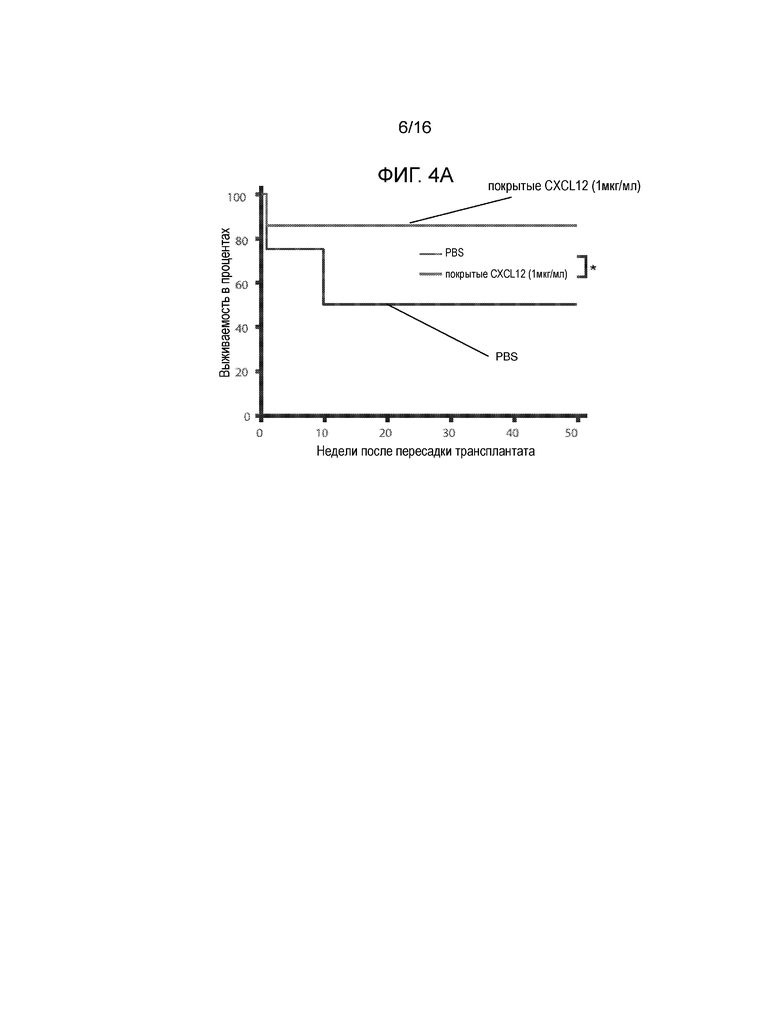
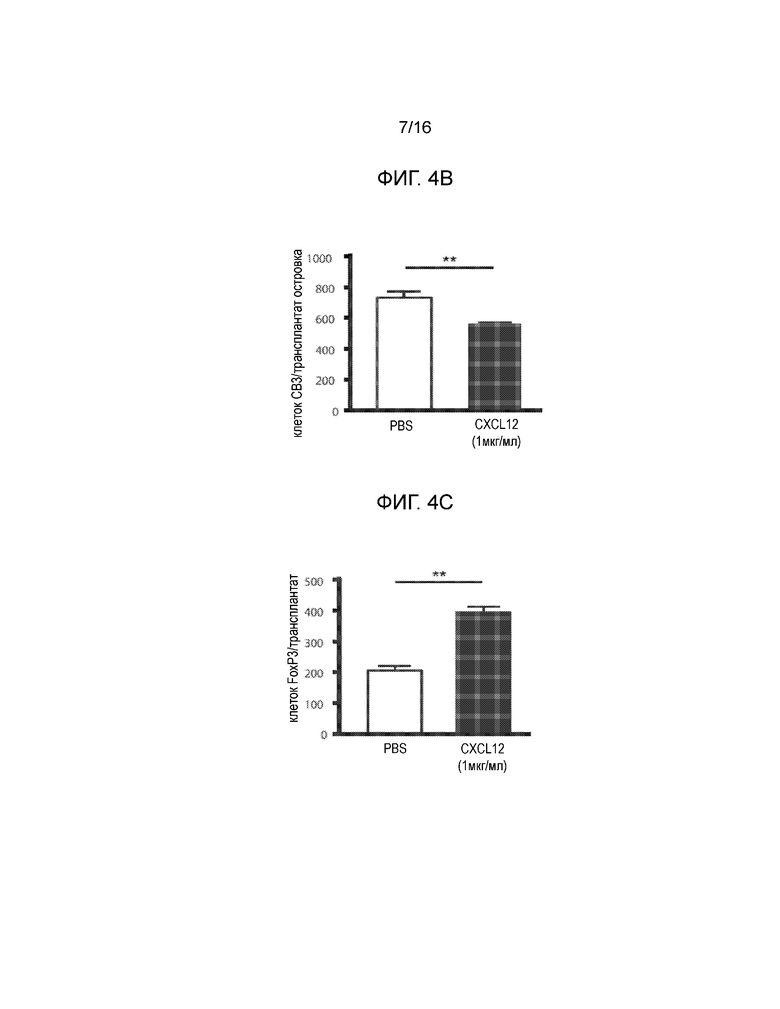
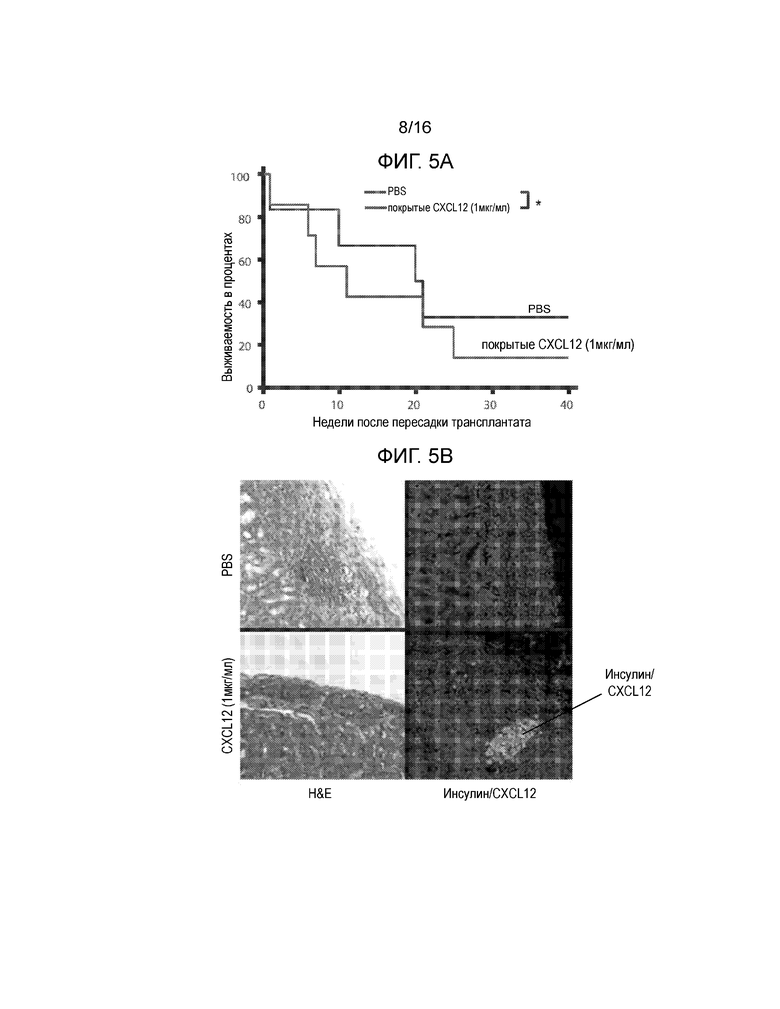
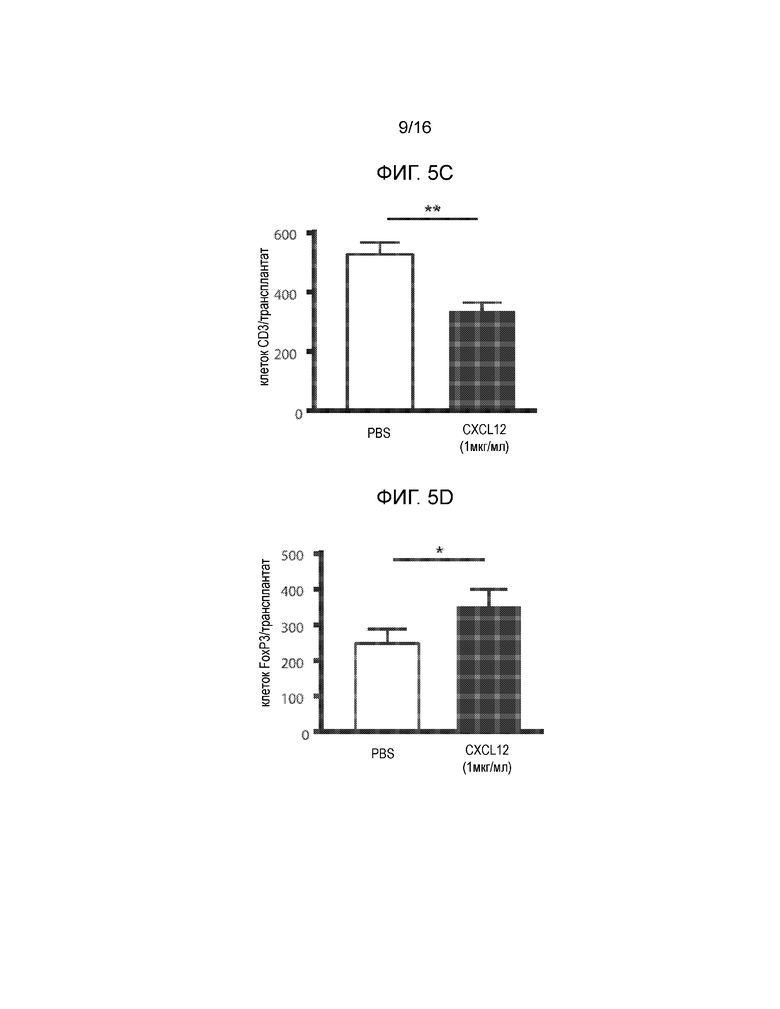
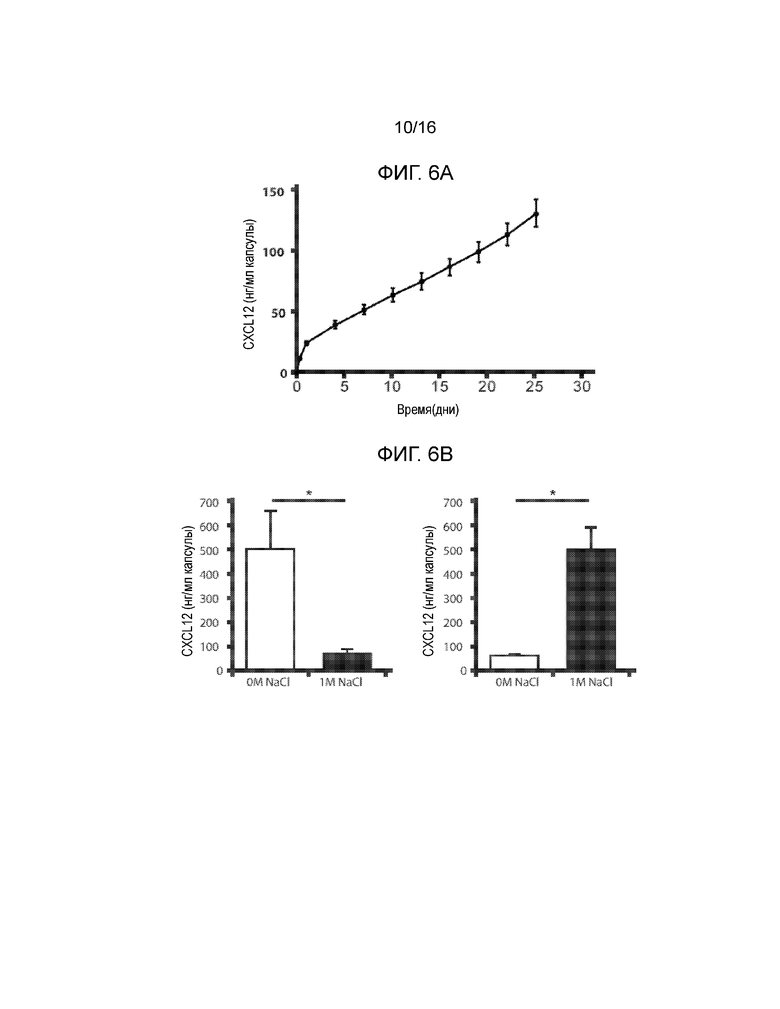
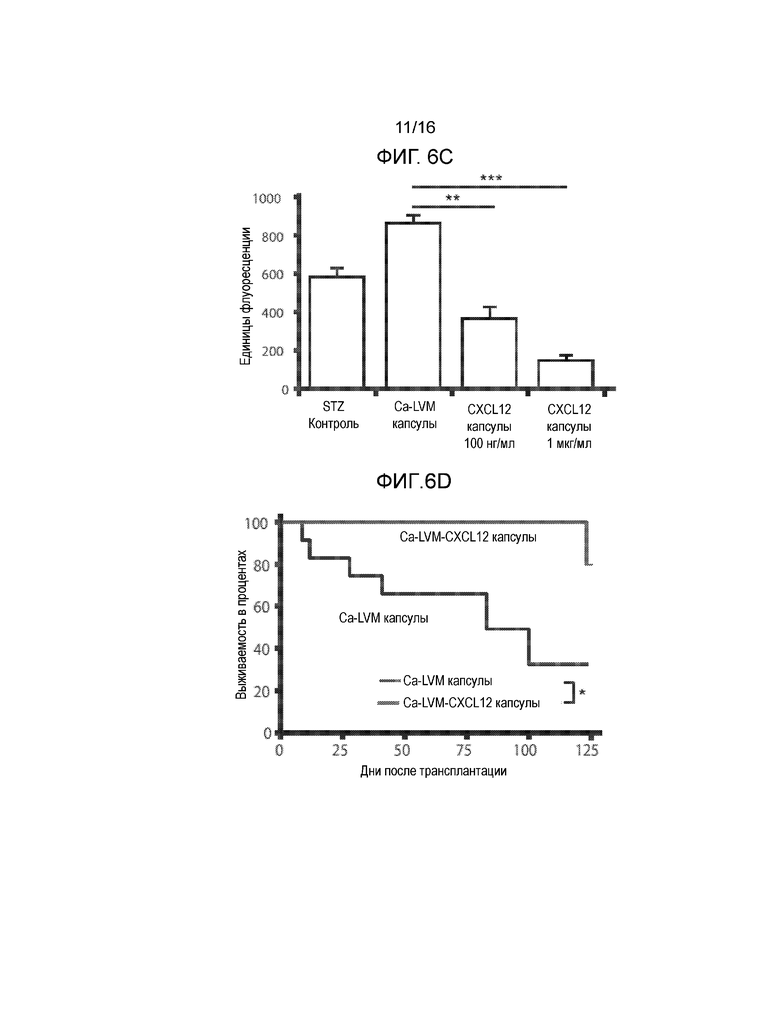
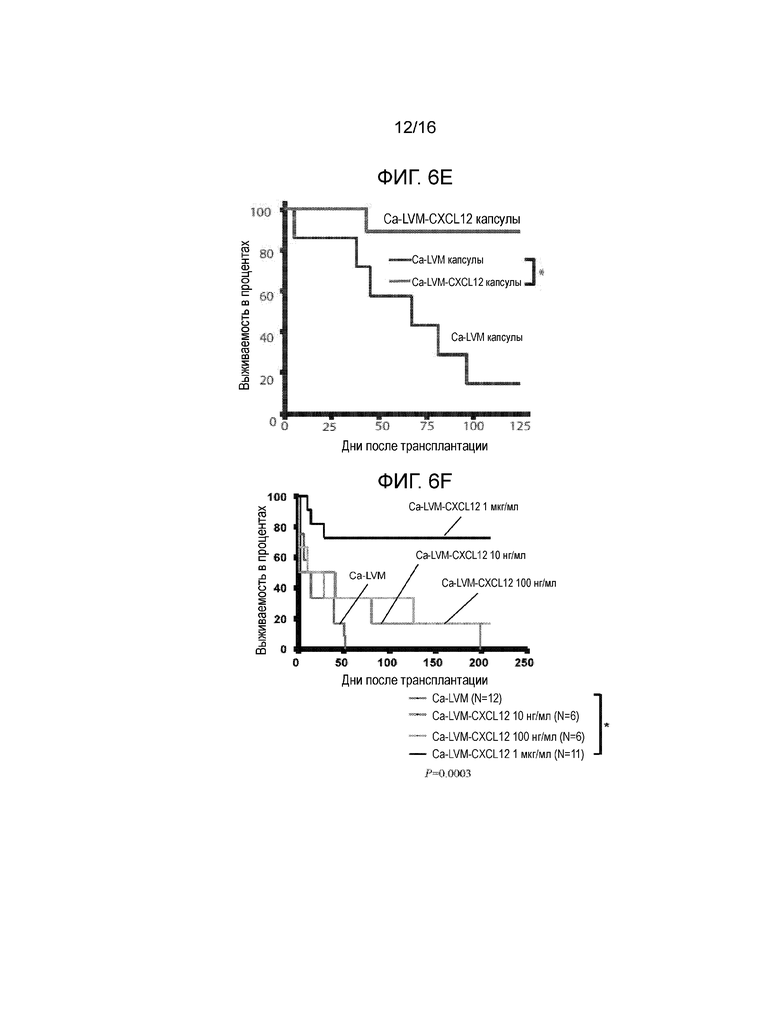
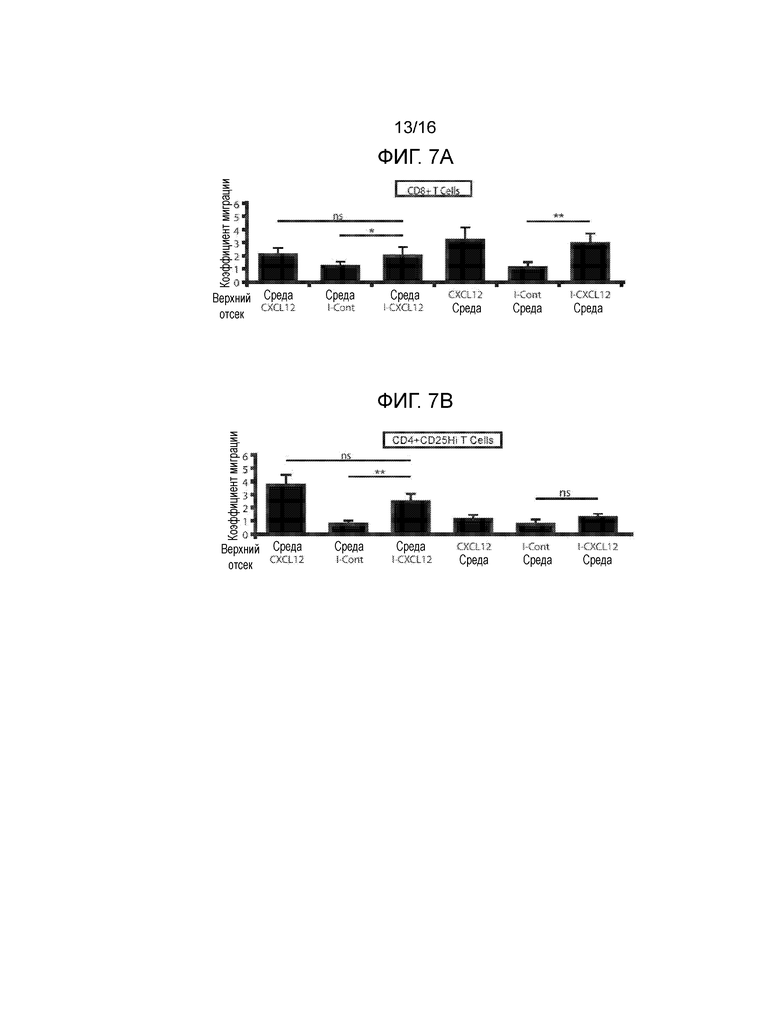
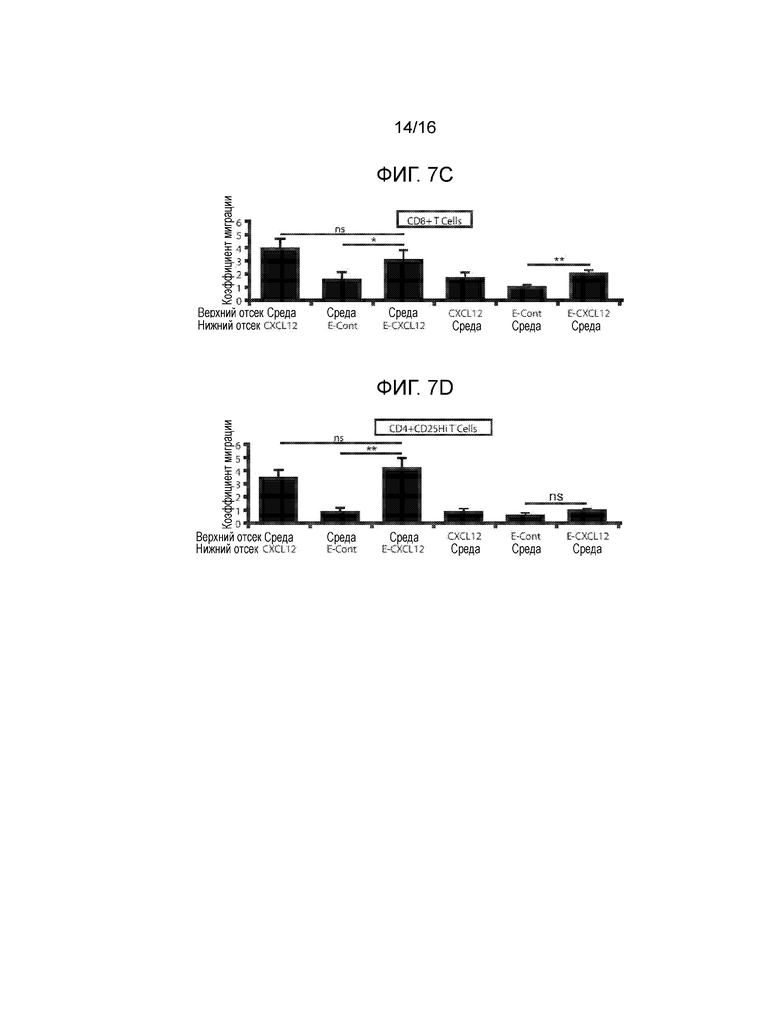
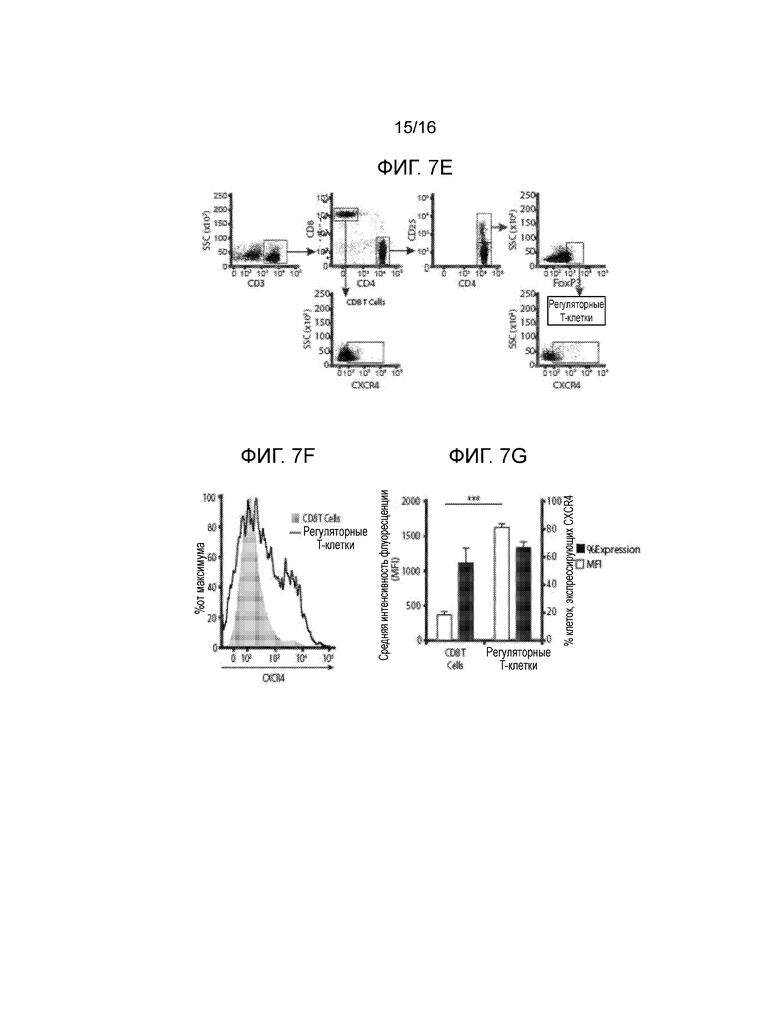
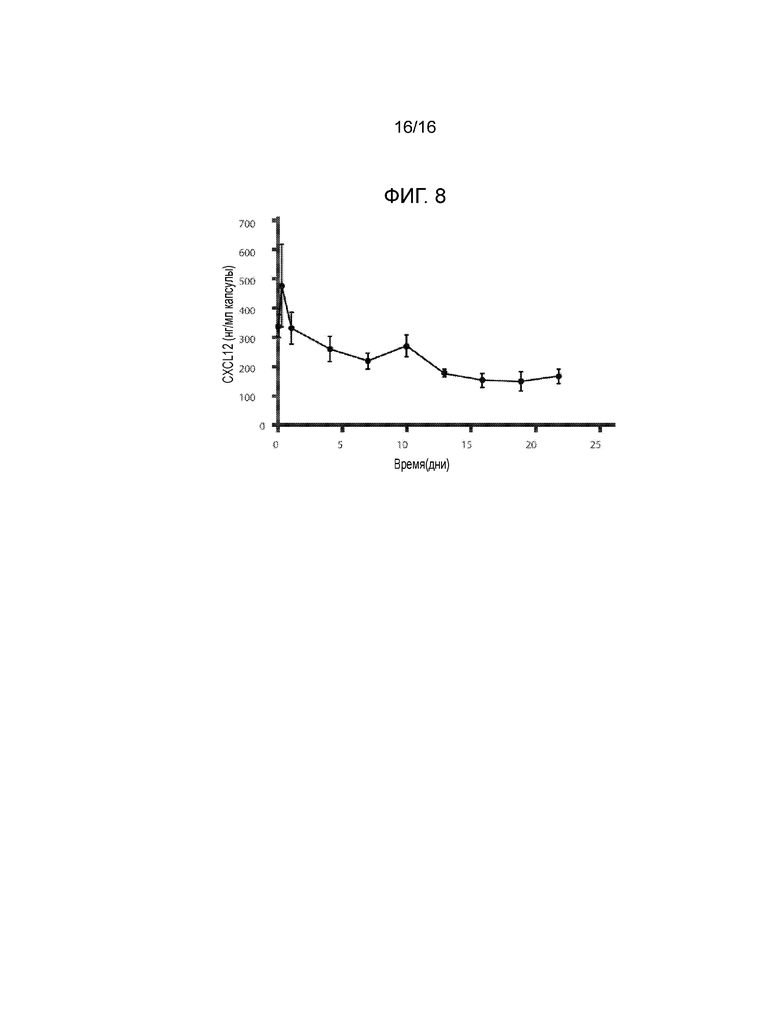
Группа изобретений относится к области медицины, а именно к иммунологии, и предназначена для лечения аутоиммунных расстройств. Элюирующая матрица, пригодная для имплантации в организм субъекта, включает как минимум одну клетку, экспрессирующую белок. Клетка инкапсулирована в пористую полимерную матрицу, высвобождающую CXCL12-полипептид. Скорость высвобождения CXCL12-полипептида из матрицы составляет как минимум 1,0 нг/мл/ч, чтобы отталкивать эффекторные T-клетки, окружающие матрицу в течение одного месяца после имплантации. Также элюирующая матрица характеризуется a) толщиной матрицы 200-500 мкм и концентрацией CXCL12-полипептида в матрице от 100 нг/мл до 1 мкг/мл; b) такой пористостью, что агенты, регулирующие концентрацию глюкозы в сыворотке субъекта с диабетом I типа, диффундируют через матрицу; c) выработкой инсулина указанными островковыми клетками, где инсулин высвобождается через матрицу со скоростью, достаточной для регулирования концентрации глюкозы в крови указанного субъекта. Использование группы изобретений повышает эффективность лечения аутоиммунных заболеваний благодаря специфическому модулированию иммунитета на ограниченном участке организма. 2 н. и 41 з.п. ф-лы, 8 ил., 1 табл., 5 пр.
1.Элюирующая матрица, пригодная для имплантации в организм субъекта, включающая как минимум одну клетку, экспрессирующую белок,
где клетка инкапсулирована в пористую полимерную матрицу, высвобождающую полипептид CXCL12, которая проницаема для белка, и, кроме того,
где скорость высвобождения полипептида CXCL12 из матрицы составляет как минимум 1,0 нг/мл/ч для того, чтобы отталкивать эффекторные T-клетки, окружающие матрицу в течение по крайней мере одного месяца после имплантации.
2. Элюирующая матрица по п. 1, где скорость высвобождения полипептида CXCL12 составляет от 1,0 нг/мл/ч до 3,0 нг/мл/ч.
3. Элюирующая матрица по п. 1 или 2, где скорость высвобождения полипептида CXCL12 составляет 1,75 нг/мл/ч.
4. Элюирующая композиция по любому из пп. 1-3, где полипептид CXCL12 присутствует в матрице в концентрации как минимум 100 нг/мл.
5. Элюирующая матрица по любому из пп. 1-4, где полипептид CXCL12 присутствует в матрице в концентрации от 100 нг/мл до 1 мкг/мл.
6. Элюирующая матрица по любому из пп. 1-5, где матрица образует капсулу.
7. Элюирующая матрица по любому из пп. 1-6, где толщина матрицы составляет от 200 до 500 мкм.
8. Элюирующая матрица по любому из пп. 1-7, где упомянутая как минимум одна клетка является аллогенной клеткой или ксеногенной клеткой в организме субъекта.
9. Элюирующая матрица по любому из пп. 1-8, где упомянутая как минимум одна клетка сохраняет активность в течение по крайней мере 1 месяца.
10. Элюирующая матрица по п. 9, где активность представляет собой регулирование уровня глюкозы в крови субъекта.
11. Элюирующая матрица по любому из пп. 1-10, где как минимум одна клетка представляет собой островковую клетку.
12. Элюирующая матрица по п. 11, где островковая клетка является клеткой, продуцирующей инсулин островковой клеткой, полученной из iPS клетки, свиной островковой клеткой или человеческой островковой клеткой.
13. Элюирующая матрица по любому из пп. 1-9, где как минимум одна клетка представляет собой миоцит, фибробласт, хондроцит, адипоцит, фибромиобласт, эктодермальную клетку, почечную клетку, клетку печени, клетку поджелудочной железы, клетку кишечника, остеобласт, гематопоэтическую клетку, нервную клетку, клетку гладкой мускулатуры, клетку скелетной мускулатуры, сердечную клетку, эпителиальную клетку, гепатоцит или стволовую клетку.
14. Элюирующая матрица по любому из пп. 1-13, где матрица включает альгинатный гель.
15. Элюирующая матрица по п. 14, где альгинатный гель имеет концентрацию 2% масса/объем.
16. Элюирующая матрица по п. 14 или 15, где альгинатная матрица является поперечно-сшитой ковалентными связями.
17. Элюирующая матрица по любому из пп. 14-16, где матрица содержит от 1,5 до 2% масса/объем сшитого кальцием альгината с высоким содержанием маннуроновой кислоты.
18. Элюирующая матрица по любому из пп. 14-17, где альгинатные полимеры, входящие в состав альгинатной матрицы, имеют среднюю молекулярную массу менее 75 кДа.
19. Элюирующая матрица по любому из пп. 14-17, где альгинатные полимеры, входящие в состав альгинатной матрицы, имеют среднюю молекулярную массу от 75 кДа до 200 кДа.
20. Элюирующая матрица по любому из пп. 14-19, где в состав альгинатной матрицы входят маннуроновая кислота и гулуроновоя кислота.
21. Элюирующая матрица по п. 20, где альгинатная матрица включает маннуроновую кислоту и гулуроновую кислоту в соотношении от 1:100 до 100:1.
22. Элюирующая матрица по п. 20, где альгинатная матрица включает гулуроновую кислоту и маннуроновую кислоту в соотношении не более чем 3:2.
23. Элюирующая матрица по любому из пп. 1-22, где полипептид CXCL12 является человеческим полипептидом CXCL12.
24. Элюирующая матрица по п. 23, где человеческий полипептид CXCL12 имеет по крайней мере 85%-ную идентичность аминокислотной последовательности изоформе гамма человеческого полипептида CXCL12 или изоформе дельта.
25. Элюирующая матрица по п. 24, где человеческий полипептид CXCL12 имеет по крайней мере 95%-ную идентичность аминокислотной последовательности изоформе гамма человеческого полипептида CXCL12 или изоформе дельта.
26. Элюирующая матрица по любому из пп. 1-25, где концентрация полипептида CXCL12 в матрице поддерживается на уровне от 100 нг/мл до 1 мкг/мл в течение промежутка времени от 3 месяцев до 2 лет.
27. Элюирующая матрица по п. 26, где концентрация полипептида CXCL12, поддерживаемая в матрице, составляет 100-200 нг/мл.
28. Элюирующая матрица по любому из пп. 1-27, где полипептид CXCL12 в матрице вырабатывается островковыми клетками, находящимися в матрице.
29. Элюирующая матрица по любому из пп. 1-28, где средний размер пор матрицы не позволяет проникать молекулам, масса которых превышает 130 кД.
30. Элюирующая матрица по любому из пп 1-29, дополнительно включающая слой клеток, которые экспрессируют полипептид CXCL12.
31. Элюирующая матрица по п. 30, где упомянутые клетки являются мезотелиальными клетками.
32. Элюирующая матрица по любому из пп. 1-31, дополнительно включающая рассасывающийся слой полипептида CXCL12 поверх матрицы.
33. Элюирующая матрица по любому из пп. 1-32, где упомянутая как минимум одна клетка введена в матрицу in vivo.
34. Элюирующая матрица по любому из пп. 1-32, где элюирующая матрица характеризуется способностью отталкивать эффекторные T-клетки in vitro в камере Бойдена.
35. Элюирующая матрица по любому из пп. 1-34, доставляющая островковые клетки в организм субъекта, которому это необходимо, где как минимум одна клетка является островковой клеткой и островковые клетки регулируют уровни глюкозы в крови субъекта в течение определенного периода времени.
36. Элюирующая матрица по п. 35, где указанный период времени составляет как минимум 1 месяц.
37. Элюирующая матрица по п. 35 или 36, где островковые клетки поддерживают концентрации глюкозы в сыворотке субъекта натощак на уровне от 80 мг/дл до 120 мг/дл.
38. Элюирующая матрица по любому из пп. 35-37, которая предназначена для многократных имплантаций субъекту.
39. Элюирующая матрица по любому из пп. 35-38, где матрица не разрушается эффекторными T-клетками или макрофагами.
40. Элюирующая матрица по любому из пп. 35-39, которая предназначена для имплантации в место, где присутствуют регуляторные T-клетки.
41. Элюирующая матрица по любому из пп. 35-40, которая предназначена для имплантации в место, где отсутствуют эффекторные T-клетки.
42. Элюирующая матрица, пригодная для имплантации в организм субъекта, включающая аллотрансплантат или ксенотрансплантат островковых клеток, инкапсулированный в пористую полимерную матрицу, высвобождающую полипептид CXCL12, где указанная матрица характеризуется:
a) толщиной матрицы 200-500 мкм, и концентрацией полипептида CXCL12 в матрице от 100 нг/мл до 1 мкг/мл;
b) такой пористостью, что агенты, регулирующие концентрацию глюкозы в сыворотке субъекта с диабетом I типа, диффундируют через матрицу;
c) выработкой инсулина указанными островковыми клетками вследствие взаимодействия указанных агентов с указанными островковыми клетками, где инсулин высвобождается через матрицу со скоростью, достаточной для регулирования концентрации глюкозы в крови указанного субъекта,
где скорость высвобождения полипептида CXCL12 из матрицы составляет как минимум 1,0 нг/мл/ч для того, чтобы отталкивать эффекторные T-клетки, окружающие матрицу в течение по крайней мере одного месяца после имплантации, и
где толщину матрицы, концентрацию полипептида CXCL12 и скорость высвобождения выбирают таким образом, чтобы подавить разрушение указанных островковых клеток на период как минимум 4 месяца, тем самым обеспечивая регулирование уровней глюкозы в крови указанного субъекта в течение указанного периода.
43. Элюирующая матрица по п. 42, где указанный период составляет примерно 6 месяцев.
| US 2011045077 A1, 24.02.2011 | |||
| WANG Y et al | |||
| Engineering chemoattractant gradients using chemokine-releasing polysaccharide microspheres | |||
| Biomaterials | |||
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Способ регулирования регенерации в кристадинных приемниках | 1926 |
|
SU4904A1 |
| MOON H et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| J Korean Med Sci., 2012, 27(1), p.28 | |||
| GOMBOTZ WR et al | |||
| Protein release from alginate matrices | |||
| Advanced Drug Delivery Reviews, 2012, 64, p.197-198 | |||
| GIBLY RF et al | |||
| Advancing islet transplantation: from engraftment to the immune response | |||
| Diabetologia | |||
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| SAKATA N et al | |||
| Encapsulated islets transplantation: Past, present and future | |||
| World J Gastrointest Pathophysiol., 2012, 3(1), p.19-26. | |||

