ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] Данная заявка заявляет преимущество приоритета предварительной заявки США № 62/398713, поданной 23 сентября 2016 г., предварительной заявки США № 62/527204, поданной 30 июня 2017 г., предварительной заявки США № 62/530683, поданной 10 июля 2017 г., и предварительной заявки США № 62/539037, поданной 31 июля 2017 г., которые все в полном объеме включены в данный документ посредством ссылки.
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
[0002] Данная заявка содержит Перечень последовательностей, поданный через EFS-Web и в полном объеме включенный в данный документ посредством ссылки. Указанная копия ASCII, созданная 30 августа 2017 г., имеет название P33854-WO.SL.TXT и размер 12532 байтов.
ОБЛАСТЬ ТЕХНИКИ
[0003] Предложены варианты применения антагонистов IL-13 для лечения атопического дерматита. Также предложены способы лечения атопического дерматита и снижения тяжести атопического дерматита путем введения антагонистов IL-13.
УРОВЕНЬ ТЕХНИКИ
[0004] Атопический дерматит (АД) представляет собой хроническое рецидивирующее и временно ослабевающее воспалительное поражение кожи, поражающее все возрастные группы. Клинически АД характеризуется патологической сухостью кожи, эритематозной шелушащейся сыпью, лихенификацией, нарушением кожного барьера и интенсивным зудом (Bieber T., N Engl J Med 2008; 358: 1483-94).
[0005] Пациенты с АД несут большое бремя заболевания, что оказывает существенное негативное влияние на их качество жизни (КЖ). В одном исследовании было показано, что АД имеет больший негативный эффект на психическое здоровье пациентов, чем диабет и гипертензия (Zuberbier T, et al, J Allergy Clin Immunol 2006; 118:226-32). Пациенты с умеренным или тяжелым АД имеют высокие показатели социальной дисфункции и нарушения сна, что напрямую связано с тяжестью заболевания (Williams H, et al, J Allergy Clin Immunol 2008; 121:947-54.e15). Депрессия, тревожность и социальная дисфункция поражают не только пациентов с АД, но также отражаются на людях, осуществляющих за ними уход (Zuberbier T, et al, J Allergy Clin Immunol 2006; 118:226-32). По сравнению с псориазом, другим распространенным и изнурительным кожным заболеванием, пациенты с АД имеют более низкие показатели по шкале ролевого физического функционирования, жизненной активности, социального функционирования, ролевого эмоционального функционирования и психического здоровья (Kiebert G, et al, Int J Dermatol 2002; 41:151-8).
[0006] Интерлейкин (IL)-13 считается ключевым медиатором воспаления, связанного с Т-хелперами типа 2 (Th2), а повышенные уровни IL-13 были связаны не только с атопическим дерматитом, но и с многочисленными другими заболеваниями, включая, но не ограничиваясь этим, астму, воспалительное заболевание кишечника, идиопатический легочный фиброз (ИЛФ) и хроническую обструктивную болезнь легких (ХОБЛ) (Oh CK, et al, Eur Respir Rev 19:46-54 (2010); Fahy JV, et al, Nat Rev Immunol 15:57-65 [2015]). IL-13 вырабатывается многими типами клеток, включая клетки Th2, базофилы, эозинофилы и тучные клетки, а также эпителиальные клетки дыхательных путей и врожденные лимфоидные клетки 2 типа. IL-13 связывается с гетеродимерным рецептором IL-4Rα/IL-13Rα1, являющимся общим с IL-4, который активирует сигнальный путь STAT-6 (Hershey GK, J Allergy Clin Immunol 111(4):677-90 [2003]). Так как Th2-воспаление включает активность нескольких типов клеток в дополнение к Th2, включая врожденные лимфоидные клетки 2-го типа (ILC2), в последнее время «Th2-воспаление» называют в научной литературе «воспалением 2-го типа». Кроме клеток Th2, было обнаружено, что ILC2 являются важным источником цитокинов, таких как IL-5 и IL-13. Соответственно, такие цитокины как IL-13 и IL-5, которые раньше идентифицировали как цитокины Th2, теперь также называются в научной литературе цитокинами 2-го типа. Аналогично, болезненные состояния, связанные с такими цитокинами, включая атопический дерматит, теперь также называются заболеваниями, обусловленными 2-м типом, или заболеваниями, связанными со 2-м типом. Смотрите, например, Noonan et al, J. Allergy Clin Immunol, 132(3): 567-574 (2013); Hanania et al, Thorax 70(8): 748-56 (2015); и Cai et al, Bioanalysis 8(4): 323-332 (2016).
[0007] Эозинофильное воспаление ассоциируется с различными заболеваниями, как аллергическими, так и неаллергическими (Gonlugur (2006) Immunol. Invest. 35(1):29-45). Воспаление представляет собой восстановительный ответ живых тканей на повреждение. Для воспалительных реакций характерным является накопление лейкоцитов в поврежденной ткани вследствие выработки в самой ткани определенных химических веществ. Накопление эозинофильных лейкоцитов происходит в случае широкого ряда патологических состояний, таких как аллергические расстройства, гельминтозы и неопластические заболевания (Kudlacz et al, (2002) Inflammation 26: 111-119). Эозинофильные лейкоциты, компонент иммунной системы, являются защитными элементами слизистых оболочек. Они демонстрируют ответ не только на антигены, но также на паразитов, химические вещества и травмы.
[0008] Тканевая эозинофилия возникает при таких кожных заболеваниях, как экзема, пузырчатка, острая крапивница и токсический эпидермальный некролиз, а также при атопическом дерматите (Rzany et al, Br. J. Dermatol. 135: 6-11 (1996)). Эозинофилы накапливаются в ткани и пустых гранулярных белках при IgE-опосредованных аллергических кожных реакциях (Nielsen et al, Ann. Allergy Asthma Immunol, 85: 489-494 (2001)). Эозинофилы в комбинации с тучными клетками, вероятно, вызывают воспаление суставов (Miossec, J. Clin. Rheumatol. 3: 81-83 (1997)). Эозинофильное воспаление иногда сопровождается травмированием суставов. Эозинофилия синовиальной жидкости может быть связана с такими заболеваниями, как ревматоидный артрит, паразитарное заболевание, гиперэозинофильный синдром, болезнь Лайма и аллергические процессы, а также при гемартрозе и артрографии (Atanes et al., Scand. J. Rheumatol, 25: 183-185 (1996)). Эозинофильное воспаление также поражает кости (Yetiser et al., Int. J. Pediatr. Otorhinolaryngol, 62: 169-173 (2002)). Примеры эозинофильных мышечных заболеваний включают эозинофильный перимиозит, эозинофильный полимиозит и фокальный эозинофильный миозит (Lakhanpal et al, Semin. Arthritis Rheum., 17: 331-231 (1988)). Эозинофильные воспаления, поражающие скелетные мышцы, могут быть связаны с паразитарными инфекциями или лекарственными препаратами, или являться признаками некоторых системных расстройств гиперэозинофилии (например, идиопатического гиперэозинофильного синдрома и синдрома эозинофилии-миалгии). Эозинофилы принимают участие в воспалительном ответе на эпитопы, распознаваемые аутоиммунными антителами (Engineer et al., Cytokine, 13: 32-38 (2001)). Заболевания соединительной ткани могут приводить к нейтрофильным, эозинофильным или лимфоцитарным сосудистым воспалениям (Chen et al., J. Am. Acad. Dermatol, 35: 173-182 (1996)). Эозинофилия тканей и периферической крови может возникать при активных ревматических заболеваниях. Повышение сывороточных уровней ЭКБ при анкилозирующем спондилите, одном из видов заболевания соединительной ткани, позволяет предположить, что эозинофилы также вовлечены в процессы, лежащие в их основе (Feltelius et al., Ann. Rheum. Dis., 46: 403-407 (1987)). Гранулематоз Вегенера изредка может сопровождаться легочными узелками, плевральными выпотами и эозинофилией периферической крови (Krupsky et al., Chest, 104: 1290-1292 (1993)).
[0009] Эозинофилия периферической крови, составляющая по меньшей мере 400/мм3, может возникать в 7% случаев системного склероза, 31% случаев локализованной склеродермы и 61% случаев эозинофильного фасциита (Falanga, et al., J. Am. Acad. Dermatol, 17: 648-656 (1987)). Склеродерма приводит к воспалительному процессу, сильно напоминающему сплетения Мейсснера и Ауэрбаха, и состоит из тучных клеток и эозинофильных лейкоцитов в желудочно-кишечной системе. Эозинофильные нейротоксины могут способствовать нарушению двигательной функции желудочно-кишечного тракта, которое наблюдается при склеродерме (DeSchryver-Kecskemeti, et al. Arch. Pathol. Lab Med., 113: 394-398 (1989)).
[0010] Эозинофилы могут сопровождать локализованную (Varga, et al., Curr. Opin. Rheumatol, 9: 562-570 (1997)) или системную (Bouros et al., Am. J. Respir. Crit. Care Med., 165: 1581-1586 (2002)) пролиферацию соединительной ткани. Они могут приводить к фиброзу путем ингибирования деградации протеогликанов в фибробластах (Hernnas et al, Eur. J. Cell Biol, 59: 352-363 (1992)), а фибробласты опосредуют выживаемость эозинофилов, секретируя GM-CSF (Vancheri et al., Am. J. Respir. Cell Mol. Biol, 1: 289-214 (1989)). Эозинофилы можно обнаружить в тканях назальных (Bacherct et al., J. allergy Clin. Immunol, 107: 607-614 (2001)), бронхиальных (Arguelles, et al., Arch. Intern. Med., 143: 570-571 (1983)) и желудочно-кишечных полипов (Assarian, et al., Hum. Pathol., 16: 311-312 (1985)). Аналогично, эозинофилы могут локализоваться в воспалительных псевдоопухолях (миофибробластических опухолях). Эозинофилы часто сопутствуют воспалительным псевдоопухолям в глазной области, в случае чего состояние может напоминать ангиоэдему или аллергический риноконъюнктивит (Li et al., Ann. Allergy, 69: 101-105 (1992)).
[0011] Эозинофильное воспаление может наблюдаться при травмировании тканей (например, в результате хирургической операции или повреждения). Эозинофильное воспаление также может быть связано с сердечно-сосудистыми болезнями (например, эозинофильным миокардитом, эозинофильным коронарным артериитом, ишемической болезнью сердца, острым инфарктом миокарда, разрывом сердца). Некротические воспалительные процессы также могут включать эозинофильное воспаление (полимиозит, расслоение коронарной артерии, некротизирующие поражения при нейро-болезни Бехчета, деменцию, церебральный инфаркт).
[0012] Некоторые данные позволяют предположить, что IL-13 является ключевым патогенным компонентом при атопическом дерматите (АД). Систематически сообщается о повышенной экспрессии IL-13 при АД кожи (Hamid Q, et al, J Allergy Clin Immunol 98:225-31 [1996]; Jeong CW, et al., Clin Exp Allergy 33: 1717-24 [2003]; Tazawa T, et al., Arch Dermatol Res 295:459-64 [2004]; Neis MM, et al, J Allergy Clin Immunol 118:930-7 [2006]; Suárez-Fariñas M, et al, J Allergy Clin Immunol 132:361-70 [2013]; Choy DF, et al, J Allergy Clin Immunol.130: 1335-43 [2012]), а в некоторых сообщениях предполагается, что существует взаимосвязь между экспрессией IL-13 и тяжестью заболевания (La Grutta S, et al, Allergy 60:391-5 [2005]). Также сообщалось о повышении IL-13 в сыворотке пациентов с АД (Novak N, et al, J Invest Dermatol 2002; 119:870-5; Международная патентная заявка № PCT/US2016/022481 [публикация № WO2016149276]), а в некоторых исследованиях сообщалось о повышении количества IL-13-экспрессирующих Т-клеток в крови пациентов с АД (Akdis M, et al., J Immunol 1997; 159:4611-9; Aleksza M, et al, Br J Dermatol 2002; 147:1135-41; La Grutta S, et al., Allergy 2005; 60:391-5). Следовательно, IL-13 и его рецепторы стали терапевтическими мишенями для лечения различных заболеваний, связанный с воспалением 2 типа, включая астму, ИЛФ и АД (Corren J, et al., N Eng J Med 365: 1088-98 [2011]; Scheerens H, et al, Clin Exp Allergy 44:38-46 [2014]; Beck LA, et al, N Eng J Med 371:130-9 [2014]; Thaci D, et al, Lancet 2016; 387:40-52). Дополнительные публикации, в которых обсуждаются IL-13 при атопическом дерматите или лебрикизумаб, включают He JQ, et al, Genes Immun 2003; 4:385-89; Kim BE, et al. Clin Immunology 2008; 126, 332-7; Bhogal RK & Bona CA Int Rev Immunol 2008; 27:472-96; Kim ST, et al. J Gene Med 2009; 11 :26-37; Bieber T, et al. J Allergy Clin Immunol 2014; 133:AB404; Thaci D, et al. J Allergy Clin Immunol 2014; 133:AB192; 2. Ultsch M, et al. J Mol Biol 2013; 425:1330-1339.
[0013] Кроме того, несколько клинических исследований по изучению агентов с противовоспалительной активностью широкого спектра действия продемонстрировали снижение экспрессии IL-13, которое было связано с улучшением клинических симптомов заболевания. Например, девятнадцать взрослых пациентов с умеренным или тяжелым АД, которых лечили в течение 12 недель циклоспорином А, демонстрировали снижение экспрессии IL-13 в коже (Khattri S, et al., J Allergy Clin Immunol 2014; 133(6):1626-34), десять педиатрических пациентов, которых лечили микроэмульсией циклоспорина А, демонстрировали снижение количества Т-клеток CD3+, экспрессирующих IL-13, в крови (Bunikowski R, et al., Pediatr Allergy Immunol 2001; 12:216-23), а двадцать взрослых пациентов с умеренным или тяжелым АД, которых лечили облучением узкополосным ультрафиолетом области B, демонстрировали существенное снижение экспрессии IL-13 в коже (Tintle S, et al, J Allergy Clin Immunol 2011; 128:583-93. e1-4).
[0014] Большое число антагонистов IL-13 было описано и исследовано в клинических условиях при различных заболеваниях, связанных с воспалением 2 типа, включая астму, ХОБЛ и ИЛФ. Они включают IMA-026, IMA-638 (также называемый анрукинзумабом, INN № 910649-32-0; QAX-576); тралокинумаб (также называемый CAT-354, CAS № 1044515-88-9); и AER-001, ABT-308 (также называемый гуманизированным антителом 13C5.5). Кроме того, были разработаны некоторые антагонисты рецептора IL-4-альфа, и они являются антагонистами как IL-13, так и IL-4. Примеры антагонистов рецептора IL-4-альфа включают AMG-317, AIR-645, дупилумаб, который исследовали в клинических условиях при атопическом дерматите, а также при астме (смотрите, например, Beck LA, et al., N Eng J Med 371:130-9 [2014]), и AER-001, ловушку IL4/IL-13. Другим антагонистом IL-13 является лебрикизумаб. Лебрикизумаб представляет собой гуманизированное моноклональное антитело в виде иммуноглобулина (Ig) G4 (huIgG4) с мутацией в шарнирной области для повышения стабильности. Лебрикизумаб с высокой аффинностью специфически связывается с растворимым человеческим IL-13 и эффективно нейтрализует его функциональную активность. Лебрикизумаб ингибирует сигнализацию IL-13 через рецептор IL-4R-альфа/IL-13R-альфа1. Он блокирует связывание IL-13 с IL-4R-альфа, но не блокирует связывание IL-13 с IL-13R-альфа1 или IL-13R-альфа2. Лебрикизумаб был описан в различных публикациях и исследован в нескольких исследованиях астмы (смотрите, например, Corren et al. (2011) N Engl J Med 365: 1088-98; Scheerens et al. (2012) Am J Respir Crit Care Med 185: A3960; Jia et al. (2012) J Allergy Clin Immunol 130: 647-654 e10; Hanania et al., Thorax 2015; 70:748-756; Hanania et al, Lancet Respir Med 2016, доступная на dx(dot)doi(dot)org(slash)S2213-2600(16)30265-X, опубликованная онлайн 5 сентября 2016 г.; WO 2012/083132).
[0015] Терапевтический подход в отношении АД состоит, главным образом, в стимуляции избегания, увлажнении кожи с помощью банных процедур и применении смягчающих средств и противовоспалительной терапии, состоящей преимущественно из местных кортикостероидов (МКС). У многих пациентов лечение МКС обеспечивает некоторую степень облегчения симптомов, но не контролирует их заболевание в достаточной мере. Кроме того, применение МКС связано со многими сопутствующими заболеваниями и ограничениями, включая нагрузку на пациентов. Обзор литературы (включая PubMed, систематический обзор Nankervis, базу данных Global Resource for Eczema Trials [GREAT]) по рандомизированным, контролируемым, слепым клиническим исследованиям системного лечения иммуносупрессорами и пероральными глюкокортикоидами для лечения умеренного или тяжелого АД показывает, что применение традиционного системного лечения иммуносупрессорами часто ограничено существенными побочными явлениями; кроме того, их применение является в основном не утвержденным. Эти данные указывают на наличие существенной неудовлетворенной потребности и на важность разработки лекарственных препаратов, нацеленных на специфические пути, лежащие в основе АД, таких как недавно утвержденное анти-IL-4Rα антитело дупилумаб. Перспективой для биологической терапии при рефрактерном АД является обеспечение эффективной терапии, которая снизила бы необходимость в системной иммуносупрессивной терапии и в конечном итоге снизила бы необходимость в интенсивной МКС-терапии. Пока дупилумаб является первым и единственным биологическим препаратом, утвержденным для лечения взрослых с умеренным или тяжелым АД, его вводят в начальной дозе, составляющей 600 мг (две подкожные инъекции по 300 мг), с последующим введением 300 мг через неделю (Simpson et al., N Engl J Med. 2016; 375(24): 2335-2348).
[0016] В случае тех пациентов, которые имеют персистентное умеренное или тяжелое заболевание, не реагирующее в достаточной степени на МКС, последние руководства предлагают некоторое число вариантов терапии с поэтапным усилением (Ring J, et al, J Eur Acad Dermatol Venereol 2012; 26: 1176-93; Schneider L, et. al., J Allergy Clin Immunol 2013; 131:295-9. e1-27). Варианты с поэтапным усилением включают местные ингибиторы кальциневрина (МИК), фототерапию и иммуносупрессорные агенты, такие как пероральные кортикостероиды, циклоспорин, азатиоприн, метотрексат и микофенолат. Из них только циклоспорин утвержден для лечения умеренного или тяжелого АД (национальная лицензия во многих Европейских странах, но не в США), а его применение ограничено пациентами возрастом от 16 лет (максимум, в течение 8 недель [NEORAL®]). Хотя циклоспорин является наиболее широко изученным системным агентом, интерпретация результатов исследований и применимость в клинической практике ограничены дизайном исследований (Schmitt J, et al, J Eur Acad Dermatol Venereol 2007; 21:606-619). Исследования других иммуносупрессорных агентов, таких как азатиоприн и метотрексат, состоят, в основном, из случайных отчетов и нескольких рандомизированных контролируемых исследований (Haeck IM, et al, J Am Acad Dermatol 2011;64:1074-84; Schram ME, et al, J Allergy Clin Immunol 2011; 128:353-9). Практические назначения демонстрируют значительную характерную для каждой данной страны вариабельность, проиллюстрированную недавним исследованием Европейских дерматологов, в котором сообщается об отсутствие конкретного предпочитаемого агента: 43% применяют циклоспорин, 31% – пероральные кортикостероиды и 22% – азатиоприн (Proudfoot LE, et al, Br J Dermatol 2013; 169:901-9).
[0017] Хотя терапия с поэтапным усилением, включая системные иммуносупрессоры, применяемая для лечения пациентов с умеренным или тяжелым АД, демонстрирует данные об умеренной и хорошей эффективности, плохая переносимость вследствие побочных явлений ограничивает их длительное применение. Даже в случаях, когда циклоспорин демонстрировал значительную эффективность, приблизительно у 50% пациентов случался рецидив в течение 2 недель, а у 80% рецидив происходил в течение 6 недель после окончания терапии (Amor KT, et al, J Am Acad Dermatol 2010; 63:925-46). Продолжение применения этих агентов, несмотря на вредные побочные явления и ограничения, указывает на неудовлетворенную медицинскую потребность в более безопасных и эффективных вариантах терапии.
[0018] В попытках найти новые варианты терапии для умеренного или тяжелого АД с приемлемым профилем польза-риск было исследовано большое число биологических агентов, которые специфически нацелены на воспалительные клетки и медиаторы (Taïeb A, et al, J Dtsch Dermatol Ges 2012; 10:174-8; Guttman-Yassky E, et al, Expert Opin Biol Ther 2013; 13:549-61). Однако некоторые из этих исследований и описаний клинических случаев проводились только на небольшом числе пациентов (например, одном или двух) и продемонстрировали противоречивые данные по эффективности и/или безопасности.
[0019] Соответственно, существует потребность в новых терапевтических вариантах лечения и схемах лечения, которые будут уменьшать тяжесть симптомов атопического дерматита и максимизировать эффективность. Кроме того, существует потребность в новых терапевтических вариантах лечения и схемах лечения, которые обеспечивают улучшенный профиль безопасности с ограниченной токсичностью по сравнению с существующими вариантами лечения или обеспечивают большую переносимость или удобность для пациентов, улучшая, таким образом, соблюдение пациентами режима применения терапевтических агентов и следование схемам лечения.
[0020] Описанное в данном документе изобретение удовлетворяет некоторым из вышеописанных потребностей и обеспечивает другие преимущества.
[0021] Все ссылки, цитируемые в данном документе, включая патентные заявки и публикации, в полном объеме и в любых целях включены посредством ссылки.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[0022] Это изобретение основано, по меньшей мере частично, на удивительном и неожиданном открытии, что анти-IL-13 антагонистическое моноклональное антитело лебрикизумаб обеспечивает терапевтическое действие при введении пациентам с атопическим дерматитом с применением предложенных в данном документе схем дозирования, включая пациентов, одновременно принимающих местные кортикостероиды, согласно оценке по нескольким критериям эффективности. Соответственно, в данном документе предложены варианты применения антагонистов IL-13, включая анти-IL-13 антитела, такие как лебрикизумаб, в лечении атопического дерматита и способы лечения атопического дерматита антагонистами IL-13, включая анти-IL-13 антитела, такие как лебрикизумаб.
[0023] Соответственно, в одном аспекте предложены способы лечения атопического дерматита у пациента, включающие введение пациенту фармацевтической композиции, содержащей терапевтически эффективное количество антагониста IL-13, при этом фармацевтическая композиция снижает тяжесть заболевания у пациента, а тяжесть заболевания оценивается по критерию оценки тяжести заболевания для атопического дерматита (ADDSOM – от англ. «Atopic Dermatitis Disease Severity Outcome Measure»). В некоторых вариантах реализации изобретения атопический дерматит является умеренным или тяжелым согласно определению с помощью оценки по шкале Райка/Лангеланда (Rajka/Langeland criteria), при этом оценка по шкале Райка/Лангеланда составляет от 4,5 до 9. В некоторых вариантах реализации изобретения способ дополнительно включает введение одного или более местных кортикостероидов. В некоторых вариантах реализации изобретения один или более местных кортикостероидов вводят до введения антагониста IL-13, одновременно с введением антагониста IL-13 или после введения антагониста IL-13. В некоторых вариантах реализации изобретения один или более местных кортикостероидов выбраны из триамцинолона ацетонида, гидрокортизона и комбинации триамцинолона ацетонида и гидрокортизона. В некоторых вариантах реализации изобретения возраст пациента составляет 12 лет и более. В некоторых вариантах реализации изобретения пациент не достигает адекватного контроля при применении местных кортикостероидов. В некоторых вариантах реализации изобретения антагонист IL-13 представляет собой моноклональное анти-IL-13 антитело. В некоторых вариантах реализации изобретения анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую HVR-H1, HVR-H2 и HVR-H3, при этом соответствующие VH HVR имеют аминокислотную последовательность SEQ ID NO: 5, SEQ ID NO: 6 и SEQ ID NO: 7, и содержащее VL, содержащую HVR-L1, HVR-L2 и HVR-L3, при этом соответствующие VL HVR имеют аминокислотную последовательность SEQ ID NO: 8, SEQ ID NO: 9 и SEQ ID NO: 10. В определенных вариантах реализации изобретения анти-IL-13 антитело представляет собой IgG1, IgG2, IgG3 или IgG4. В определенных вариантах реализации изобретения анти-IL-13 антитело представляет собой человеческое, гуманизированное или химерное антитело. В некоторых вариантах реализации изобретения анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую последовательность, выбранную из SEQ ID NO: 1 и SEQ ID NO: 3, и содержащее VL, содержащую последовательность, выбранную из SEQ ID NO: 2 и SEQ ID NO: 4. В определенных вариантах реализации изобретения анти-IL-13 антитело представляет собой полноразмерное антитело или его фрагмент, которые связывают человеческий IL-13. В некоторых вариантах реализации изобретения анти-IL-13 антитело представляет собой IgG4. В одном варианте реализации изобретения анти-IL-13 антитело представляет собой лебрикизумаб. В одном варианте реализации изобретения анти-IL-13 антитело содержит тяжелую цепь, имеющую аминокислотную последовательность SEQ ID NO: 11, и легкую цепь, имеющую аминокислотную последовательность SEQ ID NO: 12. В некоторых вариантах реализации изобретения антагонист IL-13 вводят пациенту с помощью устройства для подкожного введения. В определенных таких вариантах реализации изобретения устройство для подкожного введения выбрано из предварительно наполненного шприца, одноразового шприца-ручки, микроигольного устройства, микроинфузионного устройства, безыгольного инъекционного устройства и автоинжектора. В одном варианте реализации изобретения антагонист IL-13 представляет собой лебрикизумаб, а лебрикизумаб вводят пациенту с помощью предварительно наполненного шприца. В одном варианте реализации изобретения антагонист IL-13 представляет собой лебрикизумаб, а лебрикизумаб вводят пациенту с помощью автоинжектора.
[0024] В другом аспекте фармацевтическая композиция для применения в лечении атопического дерматита содержит 125 мг или 250 мг, или 500 мг, или около 125 мг, или около 250 мг, или около 500 мг анти-IL-13 антитела. В некоторых вариантах реализации изобретения фармацевтическая композиция содержит от 110 мг до 140 мг анти-IL-13 антитела или от 120 мг до 130 мг анти-IL-13 антитела. В некоторых вариантах реализации изобретения фармацевтическая композиция содержит от 225 мг до 275 мг анти-IL-13 антитела или от 240 мг до 260 мг анти-IL-13 антитела. В некоторых вариантах реализации изобретения фармацевтическая композиция содержит от 450 мг до 550 мг анти-IL-13 антитела, или от 475 мг до 525 мг анти-IL-13 антитела, или от 490 мг до 510 мг анти-IL-13 антитела. В некоторых вариантах реализации изобретения фармацевтическая композиция содержит 125 мг, или около 125 мг, или от 110 мг до 140 мг, или от 120 мг до 130 мг анти-IL-13 антитела, и композицию вводят подкожно один раз в четыре недели. В некоторых вариантах реализации изобретения фармацевтическую композицию вводят в течение периода, составляющего 12 недель, или периода, составляющего 20 недель, или периода, составляющего 24 недели. В некоторых вариантах реализации изобретения фармацевтическая композиция содержит 250 мг, или около 250 мг, или от 225 мг до 275 мг, или от 240 мг до 260 мг анти-IL-13 антитела, и композицию вводят подкожно один раз в четыре недели или один раз в восемь недель. В некоторых вариантах реализации изобретения фармацевтическую композицию вводят в течение периода, составляющего 24 недели или более, или в течение 24 недель. В некоторых вариантах реализации атопический дерматит является умеренным или тяжелым согласно определению с помощью оценки по шкале Райка/Лангеланда, при этом оценка по шкале Райка/Лангеланда составляет от 4,5 до 9. В некоторых вариантах реализации изобретения антагонист IL-13 представляет собой моноклональное анти-IL-13 антитело. В некоторых вариантах реализации изобретения анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую HVR-H1, HVR-H2 и HVR-H3, при этом соответствующие VH HVR имеют аминокислотную последовательность SEQ ID NO: 5, SEQ ID NO: 6 и SEQ ID NO: 7, и содержащее VL, содержащую HVR-L1, HVR-L2 и HVR-L3, при этом соответствующие VL HVR имеют аминокислотную последовательность SEQ ID NO: 8, SEQ ID NO: 9 и SEQ ID NO: 10. В определенных вариантах реализации изобретения анти-IL-13 антитело представляет собой IgG1, IgG2, IgG3 или IgG4. В определенных вариантах реализации изобретения анти-IL-13 антитело представляет собой человеческое, гуманизированное или химерное антитело. В некоторых вариантах реализации изобретения анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую последовательность, выбранную из SEQ ID NO: 1 и SEQ ID NO: 3, и содержащее VL, содержащую последовательность, выбранную из SEQ ID NO: 2 и SEQ ID NO: 4. В определенных вариантах реализации изобретения анти-IL-13 антитело представляет собой полноразмерное антитело или его фрагмент, которые связывают человеческий IL-13. В некоторых вариантах реализации изобретения анти-IL-13 антитело представляет собой IgG4. В одном варианте реализации изобретения анти-IL-13 антитело представляет собой лебрикизумаб. В одном варианте реализации изобретения анти-IL-13 антитело содержит тяжелую цепь, имеющую аминокислотную последовательность SEQ ID NO: 11, и легкую цепь, имеющую аминокислотную последовательность SEQ ID NO: 12. В некоторых вариантах реализации изобретения антагонист IL-13 вводят пациенту с помощью устройства для подкожного введения. В определенных таких вариантах реализации изобретения устройство для подкожного введения выбрано из предварительно наполненного шприца, одноразового шприца-ручки, микроигольного устройства, микроинфузионного устройства, безыгольного инъекционного устройства и автоинжектора. В одном варианте реализации изобретения антагонист IL-13 представляет собой лебрикизумаб, а лебрикизумаб вводят пациенту с помощью предварительно наполненного шприца. В одном варианте реализации изобретения антагонист IL-13 представляет собой лебрикизумаб, а лебрикизумаб вводят пациенту с помощью автоинжектора. В некоторых вариантах реализации изобретения способ дополнительно включает введение одного или более местных кортикостероидов. В некоторых вариантах реализации изобретения один или более местных кортикостероидов вводят до введения антагониста IL-13, одновременно с введением антагониста IL-13 или после введения антагониста IL-13. В некоторых вариантах реализации изобретения один или более местных кортикостероидов выбраны из триамцинолона ацетонида, гидрокортизона и комбинации триамцинолона ацетонида и гидрокортизона. В некоторых вариантах реализации изобретения возраст пациента составляет 12 лет и более. В некоторых вариантах реализации изобретения пациент не достигает адекватного контроля при применении местных кортикостероидов.
[0025] В другом аспекте предложены способы лечения атопического дерматита у пациента, включающие введение пациенту фармацевтической композиции, содержащей терапевтически эффективное количество антагониста IL-13, при этом фармацевтическая композиция снижает тяжесть заболевания у пациента, а тяжесть заболевания оценивается по критерию оценки тяжести заболевания для атопического дерматита (ADDSOM), и при этом критерий оценки тяжести заболевания для атопического дерматита представляет индекс распространенности и тяжести экземы (EASI – от англ. «Eczema Area and Severity Index»), или оценку тяжести атопического дерматита (SCORAD – от англ. «Severity Scoring of Atopic Dermatitis»), или общую оценку исследователем (IGA – от англ. «Investigator Global Assessment»), или оценку результатов пациентами (PRO – от англ. «Patient Reported Outcome»). В некоторых вариантах реализации атопический дерматит является умеренным или тяжелым согласно определению с помощью оценки по шкале Райка/Лангеланда, при этом оценка по шкале Райка/Лангеланда составляет от 4,5 до 9. В некоторых вариантах реализации изобретения способ дополнительно включает введение одного или более местных кортикостероидов. В некоторых вариантах реализации изобретения один или более местных кортикостероидов вводят до введения антагониста IL-13, одновременно с введением антагониста IL-13 или после введения антагониста IL-13. В некоторых вариантах реализации изобретения один или более местных кортикостероидов выбраны из триамцинолона ацетонида, гидрокортизона и комбинации триамцинолона ацетонида и гидрокортизона. В некоторых вариантах реализации изобретения возраст пациента составляет 12 лет и более. В некоторых вариантах реализации изобретения пациент не достигает адекватного контроля при применении местных кортикостероидов. В некоторых вариантах реализации изобретения ADDSOM представляет собой EASI, а фармацевтическая композиция снижает EASI на 50%, или 75%, или 90% по сравнению с EASI, определенным до введения первой дозы фармацевтической композиции. В некоторых вариантах реализации изобретения EASI определяют через 12 недель после введения первой дозы, или через 20 недель после введения первой дозы, или через 24 недели после введения первой дозы. В некоторых вариантах реализации изобретения ADDSOM представляет собой SCORAD, а фармацевтическая композиция снижает SCORAD на 50% по сравнению со SCORAD, определенной до введения первой дозы фармацевтической композиции. В некоторых вариантах реализации изобретения ADDSOM представляет собой SCORAD, а фармацевтическая композиция снижает SCORAD на 75% по сравнению со SCORAD, определенной до введения первой дозы фармацевтической композиции. В некоторых вариантах реализации изобретения SCORAD определяют через 12 недель после введения первой дозы. В некоторых вариантах реализации изобретения ADDSOM представляет собой IGA, а фармацевтическая композиция снижает IGA до нуля или единицы. В некоторых вариантах реализации изобретения IGA определяют через 12 недель после введения первой дозы фармацевтической композиции. В некоторых вариантах реализации изобретения ADDSOM представляет собой PRO, а PRO представляет собой визуальную аналоговую шкалу (ВАШ) зуда, ВАШ бессонницы или оценку по опроснику по влиянию атопического дерматита (ADIQ – от англ. «Atopic Dermatitis Impact Questionnaire»). В некоторых вариантах реализации изобретения PRO определяют через 12 недель после введения первой дозы фармацевтической композиции. В некоторых вариантах реализации изобретения PRO представляет собой ВАШ зуда, а фармацевтическая композиция снижает ВАШ зуда на 40%-55%. В некоторых вариантах реализации изобретения PRO представляет собой ВАШ бессонницы, а фармацевтическая композиция снижает ВАШ бессонницы на 53%-61%. В некоторых вариантах реализации изобретения PRO представляет собой ADIQ, а фармацевтическая композиция снижает оценку ADIQ на 54%-65%. В некоторых вариантах реализации изобретения антагонист IL-13 представляет собой моноклональное анти-IL-13 антитело. В некоторых вариантах реализации изобретения анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую HVR-H1, HVR-H2 и HVR-H3, при этом соответствующие VH HVR имеют аминокислотную последовательность SEQ ID NO: 5, SEQ ID NO: 6 и SEQ ID NO: 7, и содержащее VL, содержащую HVR-L1, HVR-L2 и HVR-L3, при этом соответствующие VL HVR имеют аминокислотную последовательность SEQ ID NO: 8, SEQ ID NO: 9 и SEQ ID NO: 10. В определенных вариантах реализации изобретения анти-IL-13 антитело представляет собой IgG1, IgG2, IgG3 или IgG4. В определенных вариантах реализации изобретения анти-IL-13 антитело представляет собой человеческое, гуманизированное или химерное антитело. В некоторых вариантах реализации изобретения анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую последовательность, выбранную из SEQ ID NO: 1 и SEQ ID NO: 3, и содержащее VL, содержащую последовательность, выбранную из SEQ ID NO: 2 и SEQ ID NO: 4. В определенных вариантах реализации изобретения анти-IL-13 антитело представляет собой полноразмерное антитело или его фрагмент, которые связывают человеческий IL-13. В некоторых вариантах реализации изобретения анти-IL-13 антитело представляет собой IgG4. В одном варианте реализации изобретения анти-IL-13 антитело представляет собой лебрикизумаб. В одном варианте реализации изобретения анти-IL-13 антитело содержит тяжелую цепь, имеющую аминокислотную последовательность SEQ ID NO: 11, и легкую цепь, имеющую аминокислотную последовательность SEQ ID NO: 12. В некоторых вариантах реализации изобретения антагонист IL-13 вводят пациенту с помощью устройства для подкожного введения. В определенных таких вариантах реализации изобретения устройство для подкожного введения выбрано из предварительно наполненного шприца, одноразового шприца-ручки, микроигольного устройства, микроинфузионного устройства, безыгольного инъекционного устройства и автоинжектора. В одном варианте реализации изобретения антагонист IL-13 представляет собой лебрикизумаб, а лебрикизумаб вводят пациенту с помощью предварительно наполненного шприца. В одном варианте реализации изобретения антагонист IL-13 представляет собой лебрикизумаб, а лебрикизумаб вводят пациенту с помощью автоинжектора.
[0026] В другом аспекте предложены способы, включающие введение пациенту терапевтически эффективного количества антагониста IL-13, при этом терапевтически эффективное количество выбрано из 125 мг и 250 мг, а антагонист IL-13 вводят подкожно один раз в четыре недели. В некоторых вариантах реализации изобретения терапевтически эффективное количество составляет около 125 мг, или от 110 мг до 140 мг анти-IL-13 антитела, или от 120 мг до 130 мг анти-IL-13 антитела. В некоторых вариантах реализации изобретения терапевтически эффективное количество составляет около 250 мг, или от 225 мг до 275 мг анти-IL-13 антитела, или от 240 мг до 260 мг анти-IL-13 антитела. В некоторых вариантах реализации изобретения антагонист IL-13 представляет собой моноклональное анти-IL-13 антитело. В некоторых вариантах реализации изобретения анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую HVR-H1, HVR-H2 и HVR-H3, при этом соответствующие VH HVR имеют аминокислотную последовательность SEQ ID NO: 5, SEQ ID NO: 6 и SEQ ID NO: 7, и содержащее VL, содержащую HVR-L1, HVR-L2 и HVR-L3, при этом соответствующие VL HVR имеют аминокислотную последовательность SEQ ID NO: 8, SEQ ID NO: 9 и SEQ ID NO: 10. В определенных вариантах реализации изобретения анти-IL-13 антитело представляет собой IgG1, IgG2, IgG3 или IgG4. В определенных вариантах реализации изобретения анти-IL-13 антитело представляет собой человеческое, гуманизированное или химерное антитело. В некоторых вариантах реализации изобретения анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую последовательность, выбранную из SEQ ID NO: 1 и SEQ ID NO: 3, и содержащее VL, содержащую последовательность, выбранную из SEQ ID NO: 2 и SEQ ID NO: 4. В определенных вариантах реализации изобретения анти-IL-13 антитело представляет собой полноразмерное антитело или его фрагмент, которые связывают человеческий IL-13. В некоторых вариантах реализации изобретения анти-IL-13 антитело представляет собой IgG4. В одном варианте реализации изобретения анти-IL-13 антитело представляет собой лебрикизумаб. В одном варианте реализации изобретения анти-IL-13 антитело содержит тяжелую цепь, имеющую аминокислотную последовательность SEQ ID NO: 11, и легкую цепь, имеющую аминокислотную последовательность SEQ ID NO: 12. В некоторых вариантах реализации изобретения антагонист IL-13 вводят пациенту с помощью устройства для подкожного введения. В определенных таких вариантах реализации изобретения устройство для подкожного введения выбрано из предварительно наполненного шприца, одноразового шприца-ручки, микроигольного устройства, микроинфузионного устройства, безыгольного инъекционного устройства и автоинжектора. В одном варианте реализации изобретения антагонист IL-13 представляет собой лебрикизумаб, а лебрикизумаб вводят пациенту с помощью предварительно наполненного шприца. В одном варианте реализации изобретения антагонист IL-13 представляет собой лебрикизумаб, а лебрикизумаб вводят пациенту с помощью автоинжектора. В некоторых вариантах реализации атопический дерматит является умеренным или тяжелым согласно определению с помощью оценки по шкале Райка/Лангеланда, при этом оценка по шкале Райка/Лангеланда составляет от 4,5 до 9. В некоторых вариантах реализации изобретения способ дополнительно включает введение одного или более местных кортикостероидов. В некоторых вариантах реализации изобретения один или более местных кортикостероидов вводят до введения антагониста IL-13, одновременно с введением антагониста IL-13 или после введения антагониста IL-13. В некоторых вариантах реализации изобретения один или более местных кортикостероидов выбраны из триамцинолона ацетонида, гидрокортизона и комбинации триамцинолона ацетонида и гидрокортизона. В некоторых вариантах реализации изобретения возраст пациента составляет 12 лет и более. В некоторых вариантах реализации изобретения пациент не достигает адекватного контроля при применении местных кортикостероидов. В некоторых вариантах реализации изобретения терапевтически эффективное количество снижает тяжесть заболевания у пациента, а тяжесть заболевания оценивается по критерию оценки тяжести заболевания для атопического дерматита (ADDSOM). В некоторых вариантах реализации ADDSOM представляет собой индекс распространенности и тяжести экземы (EASI), или оценку тяжести атопического дерматита (SCORAD), или общую оценку исследователем (IGA), или оценку результатов пациентами (PRO). В некоторых вариантах реализации изобретения ADDSOM представляет собой EASI, а терапевтически эффективное количество снижает EASI на 50%, или 75%, или 90% по сравнению с EASI, определенным до введения первой дозы антагониста IL-13. В некоторых вариантах реализации изобретения EASI определяют через 12 недель после введения первой дозы, или через 20 недель после введения первой дозы, или через 24 недели после введения первой дозы. В некоторых вариантах реализации изобретения ADDSOM представляет собой SCORAD, а терапевтически эффективное количество снижает SCORAD на 50% по сравнению со SCORAD, определенной до введения первой дозы антагониста IL-13. В некоторых вариантах реализации изобретения ADDSOM представляет собой SCORAD, а терапевтически эффективное количество снижает SCORAD на 75% по сравнению со SCORAD, определенной до введения первой дозы антагониста IL-13. В некоторых вариантах реализации изобретения SCORAD определяют через 12 недель после введения первой дозы. В некоторых вариантах реализации изобретения ADDSOM представляет собой IGA, а терапевтически эффективное количество снижает IGA до нуля или единицы. В некоторых вариантах реализации изобретения IGA определяют через 12 недель после введения первой дозы антагониста IL-13. В некоторых вариантах реализации изобретения ADDSOM представляет собой PRO, а PRO представляет собой визуальную аналоговую шкалу (ВАШ) зуда, ВАШ бессонницы или оценку по опроснику по влиянию атопического дерматита (ADIQ). В некоторых вариантах реализации изобретения PRO определяют через 12 недель после введения первой дозы антагониста IL-13. В некоторых вариантах реализации изобретения PRO представляет собой ВАШ зуда, а терапевтически эффективное количество снижает ВАШ зуда на 40%-55%. В некоторых вариантах реализации изобретения PRO представляет собой ВАШ бессонницы, а терапевтически эффективное количество снижает ВАШ бессонницы на 53%-61%. В некоторых вариантах реализации изобретения PRO представляет собой ADIQ, а терапевтически эффективное количество снижает оценку ADIQ на 54%-65%.
[0027] В другом аспекте предложены способы лечения атопического дерматита у пациента, включающие введение пациенту терапевтически эффективного количества антагониста IL-13, при этом введение включает введение по меньшей мере одной ударной дозы и введение по меньшей мере одной последующей поддерживающей дозы, и при этом по меньшей мере одну ударную дозу и каждую из по меньшей мере одной поддерживающей дозы вводят подкожно в виде постоянной дозы. В некоторых вариантах реализации атопический дерматит является умеренным или тяжелым согласно определению с помощью оценки по шкале Райка/Лангеланда, при этом оценка по шкале Райка/Лангеланда составляет от 4,5 до 9. В некоторых вариантах реализации изобретения способ дополнительно включает введение одного или более местных кортикостероидов. В некоторых вариантах реализации изобретения один или более местных кортикостероидов вводят до введения антагониста IL-13, одновременно с введением антагониста IL-13 или после введения антагониста IL-13. В некоторых вариантах реализации изобретения один или более местных кортикостероидов выбраны из триамцинолона ацетонида, гидрокортизона и комбинации триамцинолона ацетонида и гидрокортизона. В некоторых вариантах реализации изобретения возраст пациента составляет 12 лет и более. В некоторых вариантах реализации изобретения пациент не достигает адекватного контроля при применении местных кортикостероидов. В некоторых вариантах реализации изобретения антагонист IL-13 представляет собой моноклональное анти-IL-13 антитело. В некоторых вариантах реализации изобретения анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую HVR-H1, HVR-H2 и HVR-H3, при этом соответствующие VH HVR имеют аминокислотную последовательность SEQ ID NO: 5, SEQ ID NO: 6 и SEQ ID NO: 7, и содержащее VL, содержащую HVR-L1, HVR-L2 и HVR-L3, при этом соответствующие VL HVR имеют аминокислотную последовательность SEQ ID NO: 8, SEQ ID NO: 9 и SEQ ID NO: 10. В определенных вариантах реализации изобретения анти-IL-13 антитело представляет собой IgG1, IgG2, IgG3 или IgG4. В определенных вариантах реализации изобретения анти-IL-13 антитело представляет собой человеческое, гуманизированное или химерное антитело. В некоторых вариантах реализации изобретения анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую последовательность, выбранную из SEQ ID NO: 1 и SEQ ID NO: 3, и содержащее VL, содержащую последовательность, выбранную из SEQ ID NO: 2 и SEQ ID NO: 4. В определенных вариантах реализации изобретения анти-IL-13 антитело представляет собой полноразмерное антитело или его фрагмент, которые связывают человеческий IL-13. В некоторых вариантах реализации изобретения анти-IL-13 антитело представляет собой IgG4. В одном варианте реализации изобретения анти-IL-13 антитело представляет собой лебрикизумаб. В одном варианте реализации изобретения анти-IL-13 антитело содержит тяжелую цепь, имеющую аминокислотную последовательность SEQ ID NO: 11, и легкую цепь, имеющую аминокислотную последовательность SEQ ID NO: 12. В некоторых вариантах реализации изобретения ударная доза составляет 250 мг или 500 мг, а поддерживающая доза составляет 125 мг. В некоторых вариантах реализации изобретения ударная доза составляет 250 мг, а поддерживающая доза составляет 125 мг. В некоторых вариантах реализации изобретения ударная доза составляет 500 мг, а поддерживающая доза составляет 125 мг. В некоторых вариантах реализации изобретения поддерживающую дозу вводят через четыре недели после введения ударной дозы, и после этого поддерживающую дозу вводят один раз в четыре недели в течение периода лечения. В некоторых вариантах реализации изобретения ударная доза составляет 250 мг, а поддерживающая доза составляет 125 мг, при этом поддерживающую дозу вводят через четыре недели после ударной дозы, и после этого – один раз в четыре недели в течение периода лечения. В некоторых вариантах реализации изобретения ударная доза составляет 250 мг, а после введения ударной дозы следует введение второй ударной дозы через 15 суток, а поддерживающая доза составляет 125 мг. В некоторых вариантах реализации изобретения поддерживающую дозу вводят через две недели после введения второй ударной дозы, и после этого поддерживающую дозу вводят один раз в четыре недели в течение периода лечения. В некоторых вариантах реализации изобретения ударная доза составляет 250 мг, а после введения ударной дозы следует введение второй ударной дозы через 29 суток, а поддерживающая доза составляет 125 мг. В некоторых вариантах реализации изобретения поддерживающую дозу вводят через четыре недели после введения второй ударной дозы, и после этого поддерживающую дозу вводят один раз в четыре недели в течение периода лечения. В некоторых вариантах реализации изобретения ударная доза составляет 500 мг, а поддерживающая доза составляет 250 мг. В некоторых вариантах реализации изобретения поддерживающую дозу вводят через четыре недели после введения ударной дозы, и после этого поддерживающую дозу вводят один раз в четыре недели в течение периода лечения. В некоторых вариантах реализации изобретения поддерживающую дозу вводят через четыре недели после введения ударной дозы, и после этого поддерживающую дозу вводят один раз в восемь недель в течение периода лечения. В некоторых вариантах реализации изобретения ударная доза составляет 500 мг, а поддерживающая доза составляет 250 мг, при этом поддерживающую дозу вводят через четыре недели после ударной дозы, и после этого – один раз в четыре недели в течение периода лечения. В некоторых вариантах реализации изобретения период лечения составляет 24 недели или более. В некоторых вариантах реализации изобретения период лечения составляет 24 недели. В некоторых вариантах реализации изобретения терапевтически эффективное количество снижает тяжесть заболевания у пациента, а тяжесть заболевания оценивается по критерию оценки тяжести заболевания для атопического дерматита (ADDSOM). В некоторых вариантах реализации ADDSOM представляет собой индекс распространенности и тяжести экземы (EASI), или оценку тяжести атопического дерматита (SCORAD), или общую оценку исследователем (IGA), или оценку результатов пациентами (PRO). В некоторых вариантах реализации изобретения ADDSOM представляет собой EASI, а терапевтически эффективное количество снижает EASI на 50%, или 75%, или 90% по сравнению с EASI, определенным до введения первой дозы антагониста IL-13. В некоторых вариантах реализации изобретения EASI определяют через 12 недель после введения первой дозы, или через 20 недель после введения первой дозы, или через 24 недели после введения первой дозы. В некоторых вариантах реализации изобретения ADDSOM представляет собой SCORAD, а терапевтически эффективное количество снижает SCORAD на 50% по сравнению со SCORAD, определенной до введения первой дозы антагониста IL-13. В некоторых вариантах реализации изобретения ADDSOM представляет собой SCORAD, а терапевтически эффективное количество снижает SCORAD на 75% по сравнению со SCORAD, определенной до введения первой дозы антагониста IL-13. В некоторых вариантах реализации изобретения SCORAD определяют через 12 недель после введения первой дозы. В некоторых вариантах реализации изобретения ADDSOM представляет собой IGA, а терапевтически эффективное количество снижает IGA до нуля или единицы. В некоторых вариантах реализации изобретения IGA определяют через 12 недель после введения первой дозы антагониста IL-13. В некоторых вариантах реализации изобретения ADDSOM представляет собой PRO, а PRO представляет собой визуальную аналоговую шкалу (ВАШ) зуда, ВАШ бессонницы или оценку по опроснику по влиянию атопического дерматита (ADIQ – от англ. «Atopic Dermatitis Impact Questionnaire»). В некоторых вариантах реализации изобретения PRO определяют через 12 недель после введения первой дозы антагониста IL-13. В некоторых вариантах реализации изобретения PRO представляет собой ВАШ зуда, а терапевтически эффективное количество снижает ВАШ зуда на 40%-55%. В некоторых вариантах реализации изобретения PRO представляет собой ВАШ бессонницы, а терапевтически эффективное количество снижает ВАШ бессонницы на 53%-61%. В некоторых вариантах реализации изобретения PRO представляет собой ADIQ, а терапевтически эффективное количество снижает оценку ADIQ на 54%-65%. В некоторых вариантах реализации изобретения антагонист IL-13 вводят пациенту с помощью устройства для подкожного введения. В определенных таких вариантах реализации изобретения устройство для подкожного введения выбрано из предварительно наполненного шприца, одноразового шприца-ручки, микроигольного устройства, микроинфузионного устройства, безыгольного инъекционного устройства и автоинжектора. В одном варианте реализации изобретения антагонист IL-13 представляет собой лебрикизумаб, а лебрикизумаб вводят пациенту с помощью предварительно наполненного шприца. В одном варианте реализации изобретения антагонист IL-13 представляет собой лебрикизумаб, а лебрикизумаб вводят пациенту с помощью автоинжектора.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0028] На Фиг. 1 приведена схема исследования I, описанного в примере 2. Сокращения имеют следующие значения: С = сутки; Пбо = плацебо; П/К - подкожный; МКС = местный кортикостероид; Нд = неделя.
[0029] На Фиг. 2A приведена доля пациентов, достигших EASI-50 в течение 12 недель, как описано в примере 2. Пунктирная линия с незакрашенными кружками, лебрикизумаб, 250 мг, одна доза плюс местные кортикостероиды (МКС) дважды в сутки (ДС); точечно-пунктирная линия с закрашенными треугольниками, лебрикизумаб, 125 мг, одна доза плюс МКС дважды в сутки; сплошная линия с незакрашенными квадратами, лебрикизумаб, 125 мг, один раз в 4 недели (Р4Н) плюс МКС дважды в сутки; сплошная линия со значками плюс, плацебо плюс МКС дважды в сутки.
[0030] На Фиг. 2B приведена доля пациентов, достигших SCORAD-50 в течение 12 недель, как описано в примере 2. Пунктирная линия с незакрашенными кружками, лебрикизумаб, 250 мг, одна доза плюс местные кортикостероиды (МКС) дважды в сутки (ДС); точечно-пунктирная линия с закрашенными треугольниками, лебрикизумаб, 125 мг, одна доза плюс МКС дважды в сутки; сплошная линия с незакрашенными квадратами, лебрикизумаб, 125 мг, один раз в 4 недели (Р4Н) плюс МКС дважды в сутки; сплошная линия со значками плюс, плацебо плюс МКС дважды в сутки.
[0031] На Фиг. 2С приведена доля пациентов, достигших IGA 0/1 в течение 12 недель, как описано в примере 2. Пунктирная линия с незакрашенными кружками, лебрикизумаб, 250 мг, одна доза плюс местные кортикостероиды (МКС) дважды в сутки (ДС); точечно-пунктирная линия с закрашенными треугольниками, лебрикизумаб, 125 мг, одна доза плюс МКС дважды в сутки; сплошная линия с незакрашенными квадратами, лебрикизумаб, 125 мг, один раз в 4 недели (Р4Н) плюс МКС дважды в сутки; сплошная линия со значками плюс, плацебо плюс МКС дважды в сутки.
[0032] На Фиг. 3А-3С приведена доля пациентов, достигших EASI-50 (Фиг. 3A), EASI-75 (Фиг. 3B) и EASI-90 (Фиг. 3C) в течение 20 недель, в модифицированной популяции c назначенным лечением, как описано в примере 2. Пунктирная линия с незакрашенными кружками, лебрикизумаб, 250 мг, одна доза плюс местные кортикостероиды (МКС) дважды в сутки (ДС); точечно-пунктирная линия с закрашенными треугольниками, лебрикизумаб, 125 мг, одна доза плюс МКС дважды в сутки; сплошная линия с незакрашенными квадратами, лебрикизумаб, 125 мг, один раз в 4 недели (Р4Н) плюс МКС дважды в сутки; сплошная линия со значками плюс, плацебо плюс МКС дважды в сутки.
[0033] На Фиг. 4 приведено процентное изменение относительно исходного уровня EASI в течение 12 недель в модифицированной популяции c назначенным лечением, как описано в примере 2. Пунктирная линия с незакрашенными кружками, лебрикизумаб, 250 мг, одна доза плюс местные кортикостероиды (МКС) дважды в сутки (ДС); точечно-пунктирная линия с закрашенными треугольниками, лебрикизумаб, 125 мг, одна доза плюс МКС дважды в сутки; сплошная линия с незакрашенными квадратами, лебрикизумаб, 125 мг, один раз в 4 недели (Р4Н) плюс МКС дважды в сутки; сплошная линия со значками плюс, плацебо плюс МКС дважды в сутки. Приведенные экспериментальные точки представляют скорректированное среднее значение с планками стандартной погрешности. Звездочка (*) указывает, что плацебо-скорректированное изменение составляет 17,4%, P = 0,025.
[0034] На Фиг. 5 приведена продольная ФК-ФД-модель применения лебрикизумаба при атопическом дерматите, как описано в примере 3. На Фиг. 5, R(t) – оценка EASI; Kin0 – исходная константа скорости прогрессирования заболевания; kin – константа скорости прогрессирования заболевания при применении лебрикизумаба и с учетом эффекта плацебо/МКС; Edrug – лекарственный эффект лебрикизумаба на ингибирование прогрессирования заболевания; Econ – эффект плацебо/МКС (постоянный во времени); Emax – максимальный лекарственный эффект лебрикизумаба на ингибирование прогрессирования заболевания; EC50 – концентрация лебрикизумаба, которая приводит к 50% Emax; kout – константа скорости восстановления тканей; R(t=0) – исходная оценка EASI; МИВ – межиндивидуальная вариабельность; ωkout – межиндивидуальная вариабельность для kout; ωEcon – межиндивидуальная вариабельность для Econ; ρKoutXEcon – корреляция между kout и Econ; а σ – остаточная погрешность.
[0035] На Фиг. 6 приведены спрогнозированные с помощью моделирования медианные ответы EASI-75 в течение времени для схемы дозирования лебрикизумабом в группе 1 (смотрите таблицу 5), как описано в примере 3. Жирной сплошной линией представлен медианный смоделированный ответ для лечения плацебо (улучшение EASI-75 при лечении плацебо представляет вероятный вклад местных кортикостероидов в общую эффективность). Средней сплошной линией представлен медианный смоделированный ответ для схемы дозирования, включающей 125 мг лебрикизумаба, вводимых один раз в четыре недели (лебри 125 мг Р4Н). Штрихпунктирной линией представлен медианный смоделированный ответ для ударной дозы 250 мг, применяемой на 1 сутки, за которой следует поддерживающая доза 125 мг лебрикизумаба, вводимая один раз в четыре недели (лебри 250 мг ударн., 125 мг Р4Н), начиная с 4 недели. Тонкой сплошной линией представлен медианный смоделированный ответ для ударной дозы 500 мг, применяемой на 1 сутки, за которой следует поддерживающая доза 125 мг лебрикизумаба, вводимая один раз в четыре недели (лебри 500 мг ударн., 125 мг Р4Н), начиная с 4 недели. Жирной пунктирной линией представлен медианный смоделированный ответ для дозы 250 мг лебрикизумаба, вводимой на 1 и 29 сутки, за которой следует поддерживающая доза 125 мг лебрикизумаба, вводимая один раз в четыре недели (лебри 250 мг сутки 1 и 29, 125 мг Р4Н), начиная с 8 недели. Тонкой пунктирной линией представлен медианный смоделированный ответ для дозы 250 мг лебрикизумаба, вводимой на 1 и 15 сутки, за которой следует поддерживающая доза 125 мг лебрикизумаба, вводимая один раз в четыре недели (лебри 250 мг сутки 1 и 15, 125 мг Р4Н), начиная с 4 недели (29 сутки). Доверительные интервалы были удалены с графиков в целях ясности.
[0036] На Фиг. 7 приведены спрогнозированные с помощью моделирования медианные ответы EASI-75 в течение времени для схемы дозирования лебрикизумабом в группе 2 (смотрите таблицу 5), как описано в примере 3. Жирной сплошной линией представлен медианный смоделированный ответ для лечения плацебо (улучшение EASI-75 при лечении плацебо представляет вероятный вклад местных кортикостероидов в общую эффективность). Средней сплошной линией представлен медианный смоделированный ответ для схемы дозирования, включающей 125 мг лебрикизумаба, вводимые один раз в четыре недели (лебри 125 мг Р4Н). Тонкой пунктирной линией представлен медианный смоделированный ответ для схемы дозирования, включающей 250 мг лебрикизумаба, вводимые один раз в четыре недели (лебри 250 мг Р4Н). Жирной пунктирной линией представлена ударная доза 500 мг лебрикизумаба, вводимая на 1, за которой следует поддерживающая доза 250 мг лебрикизумаба, вводимая один раз в четыре недели, начиная с 4 недели (лебри 500 мг ударн., 250 мг Р4Н). Доверительные интервалы были удалены с графиков в целях ясности.
[0037] На Фиг. 8 приведены спрогнозированные с помощью моделирования медианные ответы EASI-75 в течение времени для схемы дозирования в группе 3 (смотрите таблицу 5), как описано в примере 3. Жирной сплошной линией представлен медианный смоделированный ответ для лечения плацебо (улучшение EASI-75 при лечении плацебо представляет вероятный вклад местных кортикостероидов в общую эффективность). Средней сплошной линией представлен медианный смоделированный ответ для схемы дозирования, включающей 125 мг лебрикизумаба, вводимые один раз в четыре недели (лебри 125 мг Р4Н). Тонкой пунктирной линией представлен медианный смоделированный ответ для 250 мг лебрикизумаба, вводимых один раз в восемь недель (лебри 250 мг Р8Н). Жирной пунктирной линией представлен медианный смоделированный ответ для ударной дозы 500 мг лебрикизумаба, за которой следует поддерживающая доза 250 мг лебрикизумаба, вводимая один раз в восемь недель, начиная с 4 недели (лебри 500 мг ударн., 250 мг Р8Н). Доверительные интервалы были удалены с графиков в целях ясности.
[0038] На Фиг. 9 приведены спрогнозированные с помощью моделирования медианные ответы EASI-75 в течение времени для схемы дозирования лебрикизумабом в группе 4 (смотрите таблицу 5), как описано в примере 3. Жирной сплошной линией представлен медианный смоделированный ответ для лечения плацебо (улучшение EASI-75 при лечении плацебо представляет вероятный вклад местных кортикостероидов в общую эффективность). Средней сплошной линией представлен медианный смоделированный ответ для схемы дозирования, включающей 125 мг лебрикизумаба, вводимые один раз в четыре недели (лебри 125 мг Р4Н). Жирной пунктирной линией представлен медианный смоделированный ответ для 37,5 мг лебрикизумаба, вводимых один раз в четыре недели (лебри 37,5 мг Р4Н). Тонкой пунктирной линией представлен медианный смоделированный ответ для ударной дозы 125 мг лебрикизумаба, за которой следует поддерживающая доза 37,5 мг лебрикизумаба, вводимая один раз в четыре недели, начиная с 4 недели (лебри 125 мг ударн., 37,5 мг Р4Н). Доверительные интервалы были удалены с графиков в целях ясности.
[0039] На Фиг. 10 приведена схема исследования II, описанного в примере 4. Сокращения имеют следующие значения: С = сутки; EASI = индекс распространенности и тяжести экземы; р4нд = раз в 4 недели; МКС = местные кортикостероиды; Нд = неделя.
[0040] На Фиг. 11A-11D приведена доля пациентов, достигших EASI-50 (Фиг. 11A), EASI-75 (Фиг. 11B), IGA 0/1 (Фиг. 11C) и SCORAD-50 (Фиг. 11D) на 12 неделю, как описано в примере 2, а изменение в доле пациентов, достигших EASI-50, EASI-75, IGA 0/1 и SCORAD-50, соответственно, в каждой группе исследования по сравнению с плацебо, указано точечно-пунктирной линией и стрелками вблизи каждого столбика на каждой из Фиг. 11A-11D. Сокращения имеют следующие значения: EASI = индекс распространенности и тяжести экземы; IGA = общая оценка исследователем; Р4Н (Q4W) = раз в 4 недели; SCORAD = оценка тяжести атопического дерматита; ОД = одна доза. Столбик с пятнами = лебрикизумаб, 125 мг, одна доза плюс местные кортикостероиды (МКС) дважды в сутки (ДС); заштрихованный столбика = лебрикизумаб, 250 мг, одна доза плюс МКС дважды в сутки; испещренный точками столбик = лебрикизумаб, 125 мг, один раз в 4 недели (Р4Н) плюс МКС дважды в сутки; незакрашенный столбик = плацебо плюс МКС дважды в сутки.
[0041] На Фиг. 12A-12D приведено изменение относительно исходного уровня в ВАШ зуда (Фиг. 12A), ADIQ (Фиг. 12B), ВАШ бессонницы (Фиг. 12C) и DLQI (Фиг. 12D) в течение времени, как описано в примере 2. Пунктирная линия с незакрашенными кружками, лебрикизумаб, 250 мг, одна доза плюс местные кортикостероиды (МКС) дважды в сутки (ДС); точечно-пунктирная линия с закрашенными треугольниками, лебрикизумаб, 125 мг, одна доза плюс МКС дважды в сутки; сплошная линия с незакрашенными квадратами, лебрикизумаб, 125 мг, один раз в 4 недели (Р4Н) плюс МКС дважды в сутки; сплошная линия со значками плюс, плацебо плюс МКС дважды в сутки. Сокращения имеют следующие значения: ВАШ = визуальная аналоговая шкала; ADIQ = опросник по влиянию атопического дерматита; DLQI = дерматологический индекс качества жизни.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0042] Все ссылки, цитируемые в данном документе, включая патентные заявки и публикации, в полном объеме и в любых целях включены посредством ссылки.
[0043] Если не указано иное, употребляемые в данном документе технические и научные термины имеют те же значения, которые обычно подразумеваются специалистом в области техники, к которой принадлежит это изобретение. В Singleton et al, Dictionary of Microbiology and Molecular Biology 2nd ed., J. Wiley & Sons (New York, N.Y. 1994) и March, Advanced Organic Chemistry Reactions, Mechanisms and Structure 4th ed., John Wiley & Sons (New York, N.Y. 1992) приведены общие сведения по многим терминам, употребляемым в данной заявке, известные специалисту в данной области техники.
НЕКОТОРЫЕ ОПРЕДЕЛЕНИЯ
[0044] В целях интерпретации этого описания применяются нижеприведенные термины и, в соответствующих случаях, термины, употребляемые в единственном числе, также включают множественное число и наоборот. В случае, если любое из нижеприведенных определений противоречит любому документу, включенному в данный документ посредством ссылки, приоритет имеет нижеприведенное определение.
[0045] В контексте этого описания и прилагаемой формулы изобретения форма единственного числа включает множественные отсылки, если иное четко не следует из контекста. Таким образом, например, ссылка на «белок» или «антитело» включает множество белков или антител, соответственно; ссылка на «клетку» включает смесь из клеток и т. п.
[0046] Диапазоны, приведенные в описании и прилагаемой формуле изобретения включают оба измеряемых параметра и все точки между измеряемыми параметрами. Таким образом, например, диапазон 2,0-3,0 включает 2,0, 3,0 и все точки между 2,0 и 3,0.
[0047] Термины «маркер» и «биомаркер» описаны в данном документе и употребляются взаимозаменяемо для обозначения молекул, включая ген, белок, углеводную структуру или гликолипид, метаболит, мРНК, миРНК, белок, ДНК (кДНК или геномную ДНК), число копий ДНК или эпигенетическое изменение, например, повышение, снижение или изменение метилирования ДНК (например, метилирования цитозина или метилирования CpG, не-CpG метилирования); гистоновую модификацию (например, (де)ацетилирование, (де)метелирование, (де)фосфорилирование, убиквитинирование, сумоилирование, АДФ-рибозилирование); изменение нуклеосомного расположения, экспрессию или присутствие которых в или на ткани или клетке млекопитающего можно зарегистрировать стандартными способами (или описанными в данном документе способами) и которые могут быть предикативными, диагностическими и/или прогнозными в отношении чувствительности клетки или ткани млекопитающего к схемам лечения на основании ингибирования воспалительного пути типа 2, например, ингибитором воспалительного пути типа 2, таким как анти-IL-13 антитело. Биомаркер также может представлять собой биологическую или клиническую характеристику, которую можно измерить в биологическом образце, полученном от субъекта, такую как, например, но без ограничений, число кровяных клеток.
[0048] Термин «биологический образец» включает, но не ограничивается этим, кровь, сыворотку, плазму, мононуклеарные клетки периферической крови (МКПК), мокроту, тканевую биопсию (например, образцы легких) и назальные образцы, включая назальные мазки или назальные полипы. Образец может быть получен до лечения, во время лечения или после лечения.
[0049] Термин «атопический дерматит» или «АД» обозначает хроническое рецидивирующее и временно ослабевающее воспалительное поражение кожи, характеризующееся интенсивным зудом (например, сильной чесоткой), ксерозом (например, аномальной сухостью кожи), эритематозной шелушащейся сыпью, лихенификацией, нарушением кожного барьера и чешуйчатыми и сухими экзематозными поражениями. Термин «атопический дерматит» включает, но не ограничивается этим, АД, вызванный или связанный с дисфункцией эпидермального барьера, аллергией (например, аллергией на определенную пищу, пыльцу, плесень, пыль, животных и т. д.), облучением и/или астмой. Во многих случаях хронические АД-поражения включают утолщенные участки кожи, лихенификацию и волокнистые папулы.
[0050] Предложенный в данном документе терапевтический агент включает агент, который может связываться с цитокином-мишенью, интерлейкином (IL)-13, такой как полипептид (-ы) (например, антитело, иммуноадгезин или пептитело), аптамер или малая молекула, которые могут связываться с белком, или молекула нуклеиновой кислоты, которая может связываться с молекулой нуклеиновой кислоты, кодирующей определенную в данном документе мишень (т. е. миРНК).
[0051] «Анти-IL-13-связывающий агент» относится к агенту, который связывается с человеческим IL-13. Такие связывающие агенты могут включать малую молекулу, аптамер или полипептид. Такой полипептид может включать, но не ограничивается этим, полипептид (-ы) выбранный (-ые) из группы, состоящей из иммуноадгезина, антитела, пептитела и пептида. В соответствии с одним вариантом реализации изобретения связывающий агент связывается с последовательностью человеческого IL-13 с аффинностью 1 мкМ – 1 пМ. Конкретные примеры анти-IL-13-связывающих агентов могут включать анти-IL-13 антитела, растворимый IL-13-рецептор-альфа-2, слитый с Fc человека, растворимый IL-4-рецептор-альфа, слитый с Fc человека, растворимый IL-13-рецептор-альфа, слитый с Fc человека. Типовые анти-IL-13 антитела включают, но не ограничиваются этим, IMA-026, IMA-638 (также называемый анрукинзумабом, INN № 910649-32-0; QAX-576); тралокинумаб (также называемый CAT-354, CAS № 1044515-88-9); AER-001, ABT-308 (также называемый гуманизированным антителом 13C5.5) и лебрикизумаб. В соответствии с одним вариантом реализации изобретения анти-IL-13 антитело содержит VH, содержащую последовательность, выбранную из SEQ ID NO: 1, 3 и 24, и VL, содержащую последовательность, выбранную из SEQ ID NO: 2, 4 и 25. В одном варианте реализации изобретения анти-IL-13 антитело содержит HVRH1, HVRH2, HVRH3, HVRL1, HVRL2 и HVRL3, при этом соответствующие HVR имеют аминокислотную последовательность SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, SEQ ID NO: 8, SEQ ID NO: 9 и SEQ ID NO: 10. В одном варианте реализации изобретения анти-IL-13 антитело представляет собой лебрикизумаб. В соответствии с одним вариантом реализации изобретения антитело представляет собой антитело IgG1. В соответствии с другим вариантом реализации изобретения антитело представляет собой антитело IgG4. В соответствии с одним вариантом реализации изобретения антитело IgG4 содержит мутацию S228P в константном домене. В одном варианте реализации изобретения анти-IL-13 антитело содержит мутацию Q1E в вариабельной области тяжелой цепи. В одном варианте реализации изобретения анти-IL-13 антитело содержит мутацию M4L в вариабельной области легкой цепи.
[0052] Термин «малая молекула» относится к органической молекуле, имеющей молекулярную массу от 50 дальтон до 2500 дальтон.
[0053] Термин «антитело» употребляется в самом широком смысле и, в частности, охватывает, например, моноклональные антитела, поликлональные антитела, антитела с полиэпитопной специфичностью, одноцепочечные антитела, мультиспецифические антитела, включая биспецифические антитела, и фрагменты антител, в той мере, в которой они демонстрируют желаемую антигенсвязывающую активность. Такие антитела могут быть химерными, гуманизированными, человеческими и синтетическими.
[0054] Термин «бесконтрольный» или «неконтролируемый» относится к неспособности схемы лечения минимизировать симптомы заболевания. В контексте данного документа термины «неконтролируемый» и «недостаточно контролируемый» можно употреблять взаимозаменяемо и подразумевается, что они относятся к одному состоянию. Статус контроля пациента может быть определен лечащим врачом на основании некоторого числа факторов, включая анамнез пациента, восприимчивость к лечению и уровень предписанного текущего лечения.
[0055] Термин «терапевтический агент» относится к любому агенту, который используется для лечения заболевания.
[0056] Термин «снижение дозы кортикостероида» или «СДК» означает снижение частоты и/или количества, или отказ от дальнейшего приема кортикостероида, используемого для лечения заболевания у пациента, принимающего кортикостероиды для лечения заболевания, по причине введения другого терапевтического агента. «СДК-агент» относится к терапевтическому агенту, являющемуся причиной СДК у пациента, принимающего кортикостероид.
[0057] Термин «кортикостероид» включает, но не ограничивается этим, местные кортикостероиды. Примеры местных кортикостероидов включают триамцинолона ацетонид, как правило, приготавливаемый в концентрации 0,1% в креме, и гидрокортизон, правило, приготавливаемый в концентрации 1% – 2,5% в креме. Определенные местные кортикостероиды считаются очень высокоэффективными, такие как, например, бетаметазона дипропионат, клобетазола пропионат, дифлоразондиацетат, флуоцинонид и галобетазола пропионат. Определенные местные кортикостероиды считаются высокоэффективными, такие как, например, амцинонид, дезоксиметазон, галцинонид и триамцинолона ацетонид. Определенные местные кортикостероиды считаются среднеэффективными, такие как, например, бетаметазона валерат, клокортолона пивалат, флуоцинолона ацетонид, фурандренолид, флуоцинонид, флутиказона пропионат, гидрокортизона бутират, гидрокортизона валерат, мометазона фуроат и предникарбат. Определенные местные кортикостероиды считаются низкоэффективными, такие как, например, алклометазона дипропионат, дезонид и гидрокортизон. «Ингаляционный кортикостероид» означает кортикостероид, подходящий для введения путем ингаляции. Типовыми ингаляционными кортикостероидами являются флутиказон, беклометазона дипропионата, будесонид, мометазона фуроат, циклесонид, флунизолид, триамцинолона ацетонид и любой другой кортикостероид, имеющийся на сегодняшний день или который появится в будущем. Примеры кортикостероидов, пригодных для ингаляции и смешанных с бета-2-агонистом длительного действия, включают, но не ограничиваются этим, будесонид/формотерол и флутиказон/салметерол.
[0058] Термин «ударная доза» означает дозу лекарственного препарата, которую принимают в начале курса лечения и которая выше дозы, принимаемой впоследствии, и каждой дозы, принимаемой в течение оставшегося периода лечения, которая называется «поддерживающей дозой». Как правило, ударную дозу вводят один раз или два раза. После введения ударной дозы или ударных доз вводят поддерживающую дозу, и после этого поддерживающую дозу вводят, как правило, через регулярные интервалы, в течение оставшегося курса лечения.
[0059] Термин «постоянная доза» означает, что в случае всех пациентов используют одну дозу, независимо от веса или любых индивидуальных факторов, связанных с весом или массой тела. Например, введение постоянной дозы, составляющей 100 мг антитела, означает, что каждый пациент, вне зависимости от веса, будет получать дозу в 100 мг. Иногда постоянная доза называется фиксированной дозой.
[0060] Термин «рассчитанная на вес доза» означает дозу, которую рассчитывают с учетом веса пациента. Таким образом, вводимая доза зависит от веса пациента. Например, доза 1 мг/кг антитела означает, что пациент, весящий 50 кг, будет получать дозу 50 мг, тогда как пациент, весящий 80 кг, будет получать дозу 80 мг.
[0061] «Ответ пациента» или «ответ» (и грамматические вариации этих терминов) на терапевтический агент можно оценивать, используя любые измеряемые параметры, свидетельствующие о пользе для пациента, включая, без ограничений, (1) ингибирование, в некоторой степени, прогрессирования заболевания, включая замедление и полную остановку; (2) снижение числа эпизодов и/или симптомов заболевания; (3) снижение размера поражений; (4) ингибирование (т. е. снижение, замедление или полное прекращение) инфильтрации иммунных или воспалительных клеток в прилегающие периферические органы и/или ткани; (5) ингибирование (т. е. снижение, замедление или полное прекращение) распространения заболевания; (6) снижение аутоиммунного ответа, что может, но не обязательно должно, приводить к регрессии или абляции болезненных поражений; (7) облегчение, в некоторой степени, одного или более симптомов, связанных с расстройством; и/или (8) увеличение длительности периода без признаков заболевания после лечения.
[0062] «Пациент сохраняет восприимчивость к лечению», когда восприимчивость пациента не снижается со временем в течение курса лечения. Пациент «не демонстрирует адекватный ответ», когда восприимчивость пациента снижается со временем в течение курса лечения. Например, пациент с атопическим дерматитом, чьи симптомы контролируются местными кортикостероидами (МКС) в начале лечения, но чьи симптомы не облегчаются введением МКС в более позднее время в течение курса лечения, утрачивает восприимчивость к лечению и считается не демонстрирующим адекватный ответ на МКС.
[0063] «Устройство для подкожного введения» относится к устройству, которое приспособлено или сконструировано для введения лекарственного препарата, например, терапевтического антитела, или фармацевтической готовой формы, подкожным путем. Типовые устройства для подкожного введения включают, но не ограничиваются этим, шприц, включая предварительно заполненный шприц, инъекционное устройство, инфузионный насос, шприц-ручку, безыгольное устройство и систему доставки на основе пластыря. Устройство для подкожного введения позволяет вводить определенный объем фармацевтической готовой формы, например, около 1,0 мл, около 1,25 мл, около 1,5 мл, около 1,75 мл или около 2,0 мл.
[0064] «Аффинность» относится к силе общих нековалентных взаимодействий между одним участком связывания молекулы (например, антитела) и ее партнера по связыванию (например, антигена). Если не указано иное, в контексте данного документа «аффинность связывания» относится к характерной аффинности связывания, которая отображает 1: 1 взаимодействие между представителями связывающейся пары (например, антителом и связывающим плечом антигена). Аффинность молекулы X в отношении ее партнера Y в общем случае может быть представлена константой диссоциации (Kд). Аффинность можно определять общепринятыми способами, известными в данной области техники, включая описанные в данном документе. Конкретные иллюстративные и типовые варианты реализации для определения аффинности связывания описаны ниже.
[0065] Антитело с «созревшей аффинностью» относится к антителу с одним или более изменениями в одной или более гипервариабельных областях (HVR) по сравнению с родительским антителом, которое не имеет таких изменений, при этом такие изменения приводят к улучшению аффинности антитела в отношении антигена.
[0066] Термины «антитело против мишени» и «антитело, которое связывается с мишенью» относятся к антителу, которое способно связывать мишень с достаточной аффинностью так, чтобы антитело можно было применять в качестве диагностического и/или терапевтического агента при нацеливании на мишень. В одном варианте реализации изобретения степень связывания антитела против мишени с неродственным, нецелевым белком составляет меньше чем около 10% от связывания антитела с мишенью по данным измерения, например, методом радиоиммуноанализа (РИА) или анализа biacore. В определенных вариантах реализации изобретения антитело, которое связывается с мишенью, имеет константу диссоциации (Kд) ≤ 1 мкМ, ≤ 100 нМ, ≤ 10 нМ, ≤ 1 нМ, ≤ 0,1 нМ, ≤ 0,01 нМ или < 0,001 нМ (например, 10-8 M или менее, например, от 10-8 M до 10-13 M, например, от 10-9 M до 10-13 M). В определенных вариантах реализации изобретения антитело против мишени связывается с эпитопом мишени, являющимся консервативным среди разных видов.
[0067] «Фрагмент антитела» относится к молекуле, отличной от интактного антитела, которая содержит часть интактного антитела, которая связывает антиген, с которым связывается интактное антитело. Примеры фрагментов антител включают, но не ограничиваются этим, одноцепочечный Fv, Fab, Fab', Fab'-SH, F(ab')2; диатела; линейные антитела; одноцепочечные молекулы антител (например, scFv); и мультиспецифические антитела, образованные из фрагментов антител.
[0068] «Антитело, которое связывается с тем же эпитопом», что и эталонное антитело, относится к антителу, которое блокирует связывание эталонного антитела со своим антигеном в конкурентном анализе на 50% или более и, наоборот, эталонное антитело блокирует связывание антитела со своим антигеном в конкурентном анализе на 50% или более. В данной области техники хорошо известны различные способы для проведения конкурентного анализа.
[0069] «Акцепторная человеческая каркасная область» в контексте данного документа представляет собой каркасную область, содержащую аминокислотную последовательность каркасной области вариабельного домена легкой цепи (VL) или каркасной области вариабельного домена тяжелой цепи (VH), полученные из каркасной области человеческого иммуноглобулина или человеческой консенсусной каркасной области, определенных ниже. Акцепторная человеческая каркасная область, «полученная из» каркасной области человеческого иммуноглобулина или человеческой консенсусной каркасной области, содержит такую же аминокислотную последовательность, что и они, или может содержать изменения в аминокислотной последовательности. В некоторых вариантах реализации изобретения число аминокислотных изменений составляет 10 или менее, 9 или менее, 8 или менее, 7 или менее, 6 или менее, 5 или менее, 4 или менее, 3 или менее или 2 или менее. В некоторых вариантах реализации изобретения акцепторная человеческая каркасная область VL идентична по последовательности с последовательностью каркасной области человеческого иммуноглобулина или последовательностью человеческой консенсусной каркасной области VL.
[0070] Термин «химерное антитело» относится к антителу, в котором часть тяжелой и/или легкой цепи получена из конкретного источника или от конкретного вида, тогда как оставшаяся часть тяжелой и/или легкой цепи получена из другого источника или от другого вида.
[0071] «Класс» антитела относится к типу константного домена или константной области тяжелой цепи. Существует пять основных классов антител: IgA, IgD, IgE, IgG и IgM, а некоторые из них можно дополнительно разделить на подклассы (изотипы), например, IgG1, IgG2, IgG3, IgG4, IgA1 и IgA2. Константные домены тяжелой цепи, которые соответствуют разным классам иммуноглобулинов, называются α, δ, ε, γ и μ, соответственно.
[0072] «Эффекторные функции» относятся к тем видам биологической активности, которые обусловлены Fc-областью антитела, которая варьируется в зависимости от изотипа антитела. Примеры эффекторных функций антитела включают: связывание C1q и комплементзависимую цитотоксичность (КЗЦ); связывание Fc-рецептора; антителозависимую клеточноопосредованную цитотоксичность (АЗКЦ); фагоцитоз; понижающую регуляцию рецепторов клеточной поверхности (например, B-клеточных рецепторов); активацию B-клеток.
[0073] «Эффективное количество» агента, например, фармацевтической готовой формы, относится к количеству, эффективному, в необходимых дозировках и в течение необходимого периода времени, для достижения желаемого терапевтического или профилактического результата.
[0074] В данном документе термин «Fc-область» используется для определения С-концевой области тяжелой цепи иммуноглобулина, которая содержит по меньшей мере часть константной области. Этот термин включает Fc-области нативной последовательности и вариантные Fc-области. В одном варианте реализации изобретения Fc-область тяжелой цепи человеческого IgG простирается от Cys226 или от Pro230 до карбокси-конца тяжелой цепи. При этом С-концевой лизин (Lys447) Fc-области может присутствовать или отсутствовать. Если в данном документе не указано иное, нумерация аминокислотных остатков в Fc-области или константной области соответствует системе нумерации EU, также называемой индексом EU, описанной в Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD, 1991.
[0075] «Каркасная область» или «FR» относится к остаткам вариабельного домена, отличным от остатков гипервариабельной области (HVR). FR вариабельного домена в общем случае состоит из четырех FR-доменов: FR1, FR2, FR3 и FR4. Соответственно, последовательности HVR и FR в общем случае расположены в следующей последовательности в VH (или VL): FR1-H1(L1)-FR2-H2(L2)-FR3-H3(L3)-FR4.
[0076] Термины «полноразмерное антитело», «интактное антитело» и «цельное антитело» взаимозаменяемо употребляются для обозначения антитела, имеющего структуру, по существу аналогичную структуре нативного антитела, или имеющего тяжелые цепи, которые содержат Fc-область по определению данного документа.
[0077] Термины «клетка-хозяин», «линия клеток-хозяев» и «культура клеток-хозяев» употребляются взаимозаменяемо и относятся к клеткам, в которые была внесена экзогенная нуклеиновая кислота, включая потомство таких клеток. Клетки-хозяева включают «трансформантов» и «трансформированные клетки», которые включают первичные трансформированные клетки и полученное от них потомство вне зависимости от числа пассажей. Потомство может не быть полностью идентичным по содержанию нуклеиновой кислоты с родительской клеткой, но может содержать мутации. Мутантное потомство, которое имеет такую же функцию или биологическую активность, что были получены в процессе скрининга или отбора в исходной трансформированной клетке, включено в данный документ.
[0078] «Человеческое антитело» представляет собой антитело, которое имеет аминокислотную последовательность, соответствующую последовательности антитела, вырабатываемого человеком или человеческой клеткой, или полученного из нечеловеческого источника, в котором используется репертуар человеческих антител или других последовательностей, кодирующих человеческие антитела. Из этого определения человеческого антитела, в частности, исключены гуманизированные антитела, содержащие нечеловеческие антигенсвязывающие остатки.
[0079] «Человеческая консенсусная каркасная область» представляет собой каркасную область, которая представляет наиболее часто встречающиеся аминокислотные остатки в выборке каркасных последовательностей VL или VH человеческого иммуноглобулина. В общем случае выборка последовательностей VL или VH человеческого иммуноглобулина принадлежит подгруппе последовательностей вариабельных доменов. В общем случае подгруппа последовательностей представляет подгруппу, описанную в Kabat et al., Sequences of Proteins of Immunological Interest, Fifth Edition, NIH Publication 91-3242, Bethesda MD (1991), vols. 1-3. В одном варианте реализации изобретения в случае VL подгруппа представляет подгруппу каппа I, описанную в Kabat et al., выше. В одном варианте реализации изобретения в случае VH подгруппа представляет подгруппу III, описанную в Kabat et al., выше.
[0080] «Гуманизированное» антитело относится к химерному антителу, содержащему аминокислотные остатки из нечеловеческих HVR и аминокислотные остатки из человеческих FR. В определенных вариантах реализации изобретения гуманизированное антитело содержит практически все из по меньшей мере одного и, как правило, двух, вариабельных доменов, в которых все или практически все HVR (например, CDR) соответствуют областям нечеловеческого антитела, а все или практически все FR соответствуют областям человеческого антитела. Гуманизированное антитело, необязательно, может содержать по меньшей мере часть константной области антитела, полученную из человеческого антитела. «Гуманизированная форма» антитела, например, нечеловеческого антитела, относится к антителу, которое подверглось гуманизации.
[0081] Термин «гипервариабельная область» или «HVR» относится к каждой из областей вариабельного домена антитела, которые являются гипервариабельными по последовательности и/или образуют структурно определенные петли («гипервариабельные петли»). В общем случае нативные четырехцепочечные антитела содержат шесть HVR; три в VH (H1, H2, H3) и три в VL (L1, L2, L3). HVR в общем случае содержат аминокислотные остатки из гипервариабельных петель и/или из «определяющих комплементарность областей» (CDR), при этом последние, как правило, имеют наивысшую вариабельность последовательности и/или вовлечены в распознавание антигена. В контексте данного документа область HVR содержит любое число остатков, расположенных в пределах позиций 24-36 (для HVRL1), 46-56 (для HVRL2), 89-97 (для HVRL3), 26-35B (для HVRH1), 47-65 (для HVRH2) и 93-102 (для HVRH3).
[0082] «Индивид», или «пациент», или «субъект» является млекопитающим. Млекопитающие включают, но не ограничиваются этим, одомашненных животных (например, коров, овец, кошек, собак и лошадей), приматов (например, людей и отличных от людей приматов, таких как обезьяны), кроликов и грызунов (например, мышей и крыс). В определенных вариантах реализации изобретения индивид, или пациент, или субъект является человеком. В некоторых вариантах реализации изобретения «индивид», или «пациент», или «субъект» в контексте данного документа представляет любого субъекта-человека, подлежащего лечению, который демонстрирует или демонстрировал один или более признаков, симптомов или других индикаторов астмы или респираторного патологического состояния. Предполагается, что в качестве субъекта могут быть включены любые субъекты, включенные в клинические исследования, не демонстрирующие никаких признаков заболевания, или субъекты, включенные в эпидемиологические исследования, или субъекты, которые когда-либо выступали контрольными субъектами. Субъект мог ранее проходить лечение антагонистом IL-13 или другим лекарственным препаратом или не проходить такое лечение. Субъект может быть наивным в отношении антагониста IL-13 на момент начала лечения согласно данному документу, т. е. субъект мог ранее не проходить лечение, например, антагонистом IL-13, в момент включения в исследование (т. е. в установленный момент времени до введения первой дозы антагониста IL-13 в способе лечения согласно данному документу, например, день проведения скрининга субъекта перед началом лечения). Такие «наивные» субъекты в общем случае считаются кандидатами для лечения таким (-ими) лекарственным (-ыми) препаратом (-ами).
[0083] «Педиатрический» индивид, или пациент, или субъект представляет собой человека с момента рождения до 18 лет (или в возрасте 0-18 лет). В некоторых вариантах реализации изобретения возраст педиатрического индивида, или пациента, или субъекта составляет от 2 до 6, от 2 до 17, от 6 до 11, от 6 до 18, от 6 до 17, от 8 до 17, от 12 до 17 или от 12 до 18 лет.
[0084] «Выделенное» антитело представляет собой антитело, которое было отделено от компонентов его естественного окружения. В некоторых вариантах реализации изобретения антитело очищено до более чем 95% или 99% чистоты согласно определению, например, электрофоретическим (например, ДСН-ПААГ, изоэлектрическое фокусирование (ИЭФ), капиллярный электрофорез) или хроматографическим (например, ионообменная или обращенно-фазовая ВЭЖХ) методом. Обзор методов оценки чистоты антитела смотрите, например, в Flatman et al, J. Chromatogr. B 848:79-87 (2007).
[0085] «Выделенная» нуклеиновая кислота относится к молекуле нуклеиновой кислоты, которая была отделена от компонентов ее естественного окружения. Выделенная нуклеиновая кислота включает молекулу нуклеиновой кислоты, содержащуюся в клетках, которые обычно содержат молекулу нуклеиновой кислоты, но молекула нуклеиновой кислоты находится вне хромосомы или в хромосомной локации, отличной от ее естественной хромосомной локации.
[0086] «Выделенная молекула нуклеиновой кислоты, кодирующая антитело против мишени» относится к одной или более молекулам нуклеиновых кислоты, кодирующих тяжелую и легкую цепи антитела (или их фрагменты), включая такие молекулы нуклеиновых кислот в одном векторе или отдельных векторах, и такие молекулы нуклеиновых кислот, присутствующие в одной или более локациях в клетке-хозяине.
[0087] Термин «моноклональное антитело» относится к антителу, полученному из популяции практически гомогенных антител, т. е. отдельные антитела, составляющие популяцию являются идентичными и/или связывают один эпитоп, за исключением возможных вариантных антител, например, содержащих мутации природного происхождения или возникших во время получения препарата моноклональных антител, причем такие варианты в общем случае присутствуют в незначительных количествах. В отличие от препаратов поликлональных антител, которые, как правило, содержат разные антитела, направленные против разных детерминант (эпитопов), каждое моноклональное антитело препарата моноклональных антител направлено против одной детерминанты на антигене. Таким образом, модификатор «моноклональное» указывает на характеристику антитела, как полученного из практически гомогенной популяции антител, и не должен восприниматься как требующий получения антитела каким-либо конкретным способом. Например, моноклональные антитела для применения в соответствии с предложенными в данном документе способами, можно получать разными методами, включая, но не ограничиваясь этим, гибридомный метод, методы рекомбинантных ДНК, методы фагового дисплея и методы с применением трансгенных животных, содержащих все или часть локусов человеческого иммуноглобулина, при этом такие методы и другие типовые методы получения моноклональных антител описаны в данном документе.
[0088] «Оголенное антитело» относится к антителу, которое не конъюгировано с гетерологичным компонентом (например, цитотоксическим компонентом) или радиоактивной меткой. Оголенное антитело может присутствовать в фармацевтической готовой форме.
[0089] «Нативные антитела» относятся к молекулам иммуноглобулина природного происхождения с разными структурами. Например, нативные антитела IgG представляют собой гетеротетрамерные гликопротеины массой около 150000 дальтон, состоящие из двух идентичных легких цепей и двух идентичных тяжелых цепей, которые связаны дисульфидными связями. От N- до C-конца каждая тяжелая цепь содержит вариабельную область (VH), также называемую вариабельным тяжелым доменом или вариабельным доменом тяжелой цепи, за которой следуют три константных домена (CH1, CH2 и CH3). Аналогично, от N- до C-конца каждая легкая цепь содержит вариабельную область (VL), также называемую вариабельным легким доменом или вариабельным доменом легкой цепи, за которой следует константный домен легкой цепи (CL). Легкая цепь антитела может относиться к одному из двух типов, называемых каппа (κ) и лямбда (λ) на основании аминокислотной последовательности ее константного домена.
[0090] Термин «вкладыш упаковки» используется для обозначения инструкций, обычно включенных в коммерческие упаковки терапевтических продуктов, которые содержат информацию по показаниям, применению, дозировке, введению, комбинированной терапии, противопоказаниям и/или предупреждения относительно применения таких терапевтических продуктов. Термин «вкладыш упаковки» также используется для обозначения инструкций, обычно включенных в коммерческие упаковки диагностических продуктов, которые содержат информацию по предполагаемому применению, принципу проведения испытания, приготовлению и обращению с реагентами, сбору и подготовке образцов, калибровке анализа и процедуре анализа, клиническим характеристикам и данным о точности, таким как чувствительность и специфичность анализа.
[0091] «Процент (%) идентичности аминокислотной последовательности» по отношению к эталонной полипептидной последовательности определяется как процент аминокислотных остатков в кандидатной последовательности, которые идентичны с аминокислотными остатками в эталонной полипептидной последовательности, после выравнивания последовательностей и внесения гэпов, в случае необходимости, для достижения максимального процента идентичности последовательности, и не учитывая любые консервативные замены как часть идентичности последовательности. Выравнивание в целях определения процента идентичности аминокислотной последовательности можно проводить различными путями, которые известны в данной области техники, например, используя общедоступное компьютерное программное обеспечение, такое как программы BLAST, BLAST-2, ALIGN или Megalign (DNASTAR). Специалисты в данной области техники могут определить соответствующие параметры для выравнивания последовательностей, включая любые алгоритмы, необходимые для достижения максимального выравнивания вдоль всей длины сравниваемых последовательностей. При этом в целях данного документа значения % идентичности аминокислотной последовательности получают, используя компьютерную программу для сравнения последовательностей ALIGN-2. Авторские права на компьютерную программу для сравнения последовательностей ALIGN-2 принадлежат Genentech, Inc., а исходный программный код был подан вместе с документацией пользователя в Бюро регистрации авторских прав США, Вашингтон, округ Колумбия, 20559, где он был зарегистрирован под номером регистрации авторского права США № TXU510087. Программа ALIGN-2 находится в общем доступе от Genentech, Inc., Южный Сан-Франциско, Калифорния, или может быть скомпилирована из исходного программного кода. Программу ALIGN-2 необходимо компилировать для использования в операционной системе UNIX, включая цифровую UNIX V4.0D. Все параметры для сравнения последовательностей установлены в программе ALIGN-2 и не варьируются.
[0092] В ситуациях, когда ALIGN-2 применяют для сравнения аминокислотных последовательностей, % идентичности аминокислотной последовательности заданной аминокислотной последовательности А с или против заданной аминокислотной последовательности B (что можно, в альтернативном варианте, перефразировать как заданная аминокислотная последовательность А, которая имеет или содержит определенный % идентичности аминокислотной последовательности с или против заданной аминокислотной последовательности B) рассчитывают следующим образом:
[0093] 100 умножить на отношение X/Y, где X – число аминокислотных остатков, которые были оценены как идентичные совпадения программой выравнивания последовательностей ALIGN-2 при выравнивании этой программой A и B, и где Y – общее число аминокислотных остатков в B. Следует понимать, что если длина аминокислотной последовательности А не равна длине аминокислотной последовательности B, % идентичности аминокислотной последовательности A относительно B не будет равен % идентичности аминокислотной последовательности B относительно A. Если специально не указано иное, все значения % идентичности аминокислотной последовательности, используемые в данном документе, получены, как описано в предыдущем параграфе, с помощью компьютерной программы ALIGN-2.
[0094] Термин «фармацевтическая готовая форма» или «фармацевтическая композиция» относится к препарату, находящемуся в такой форме, чтобы обеспечивать эффективность биологической активности содержащегося в нем активного ингредиента, и который не содержит дополнительных компонентов, являющихся неприемлемо токсичными для субъекта, которому будут вводить готовую форму.
[0095] «Фармацевтически приемлемый носитель» относится к ингредиенту в фармацевтической готовой форме, отличному от активного ингредиента, который является нетоксичным для субъекта. Фармацевтически приемлемые носители включают, но не ограничиваются этим, буфер, вспомогательное вещество, стабилизатор или консервант.
[0096] Термин «мишень» относится к любой нативной молекуле из любого источника, относящегося к позвоночным, включая млекопитающих, таких как приматы (например, люди), и грызунов (например, мышей и крыс), если не указано иное. Этот термин включает «полноразмерную» не подвергнутую процессингу мишень, а также любую форму мишени, получаемую в результате процессинга в клетке. Этот термин также включает варианты мишеней природного происхождения, например, сплайс-варианты или аллельные варианты.
[0097] Термин «лечение» (и его грамматические вариации, такие как «лечить») относится к клиническому вмешательству в попытке изменения естественного течения заболевания у проходящего лечение индивида, и может проводиться как для профилактики, так и во время клинической патологии. Желаемые эффекты лечения включают, но не ограничиваются этим, предотвращение появления или повторного появления заболевания, облегчение симптомов, уменьшение любых прямых или непрямых патологических последствий заболевания, предотвращение метастазирования, снижение скорости прогрессирования заболевания, уменьшение интенсивности или ослабление болезненного состояния и ремиссию или улучшение прогноза. В некоторых вариантах реализации изобретения антитела применяют для замедления развития заболевания или для замедления прогрессирования заболевания.
[0098] Термин «вариабельная область» или «вариабельный домен» относится к домену тяжелой или легкой цепи антитела, который вовлечен в связывание антитела с антигеном. Вариабельные домены тяжелой цепи и легкой цепи (VH и VL соответственно) нативного антитела в общем случае имеют аналогичные структуры, при этом каждый домен содержит четыре консервативные каркасные области (FR) и три гипервариабельные области (HVR). (Смотрите, например, Kindt et al. Kuby Immunology, 6th ed., W.H. Freeman and Co., страница 91 (2007).) Одного домена VH или VL может быть достаточно для обеспечения антигенсвязывающей специфичности. Кроме того, антитела, которые связывают конкретный антиген, можно выделять, используя домен VH или VL из антитела, которое связывает этот антиген, для проведения скрининга библиотеки комплементарных доменов VL или VH соответственно. Смотрите, например, Portolano et al, J. Immunol. 150: 880-887 (1993); Clarkson et al., Nature 352:624-628 (1991).
[0099] Термин «вектор» относится к молекуле нуклеиновой кислоты, способной увеличивать число копий другой нуклеиновой кислоты, с которой она связана. Этот термин включает вектор в виде способной к саморепликации структуры нуклеиновой кислоты, а также вектор, внедренный в геном клетки-хозяина, в которую он был внесен. Определенные векторы способны управлять экспрессией нуклеиновых кислот, с которыми они функционально связаны. Такие векторы называются в данном документе «экспрессионными векторами».
[00100] Термин «обострение» у пациента с диагнозом атопического дерматита означает определяемое повышение степени тяжести поражений в течение периода, составляющего по меньшей мере 3 суток, при непрерывном лечении, соответствующее клинически значимому повышению тяжести заболевания (по оценке лечащего врача и/или самого пациента), при котором требуется усиление терапии (смотрите, например, Darsow et al, J Eur Acad Dermatol Venereol 2010; 24:317-28).
[00101] Атопический дерматит можно диагностировать, используя «критерии Hanifin/Rajka». Диагностические критерии Hanifin/Rajka описаны в Acta Derm Venereol (Stockh) 1980; Suppl 92:44-7. Чтобы поставить диагноз атопического дерматите необходимо, чтобы у пациента присутствовали по меньшей мере 3 «основных признака» и 3 или более второстепенных признаков, перечисленных ниже. Основные признаки включают зуд, типичную морфологию и распределение, такое как искривленная лихенификация или линейность, хронический или хронически-рецидивирующий дерматит и персональный или семейный анамнез атопии, например, астмы, аллергического ринита, атопического дерматита. Второстепенные признаки включают ксероз, ихтиоз, ладонную гиперлинейность или волосяной кератоз, среднюю (типа 1) реактивность при кожной пробе, повышение сывороточного IgE, ранний возраст начала, расположенность к кожным инфекциям (в особенности Staph. aureus и Herpes simplex), нарушение клеточноопосредованного иммунитета, расположенность к неспецифическому дерматиту кистей и стоп, экзему сосков, хейлит, рецидивирующий конъюнктивит, складку Денни-Моргана, кератоконус, переднюю субкапсулярную катаракту, потемнение в глазах, бледность лица/эритему лица, себорейную экзему, передние складки на шее и чесотку при потении. Дополнительные второстепенные критерии включают непереносимость шерсти и липидных растворителей, перифолликулярную акцентуацию, непереносимость пищи, влияние окружающих или эмоциональных факторов на течение заболевания и белый дермографизм/медленное обесцвечивание эритемы.
[00102] Тяжесть атопического дерматита можно определить по «шкале Райка/Лангеланда», как описано в Rajka G and Langeland T, Acta Derm Venereol (Stockh) 1989; 144 (Suppl): 13-4. Оценочным категориям тяжести заболевания присваивается оценка от 1 до 3: I) величина пораженной площади тела, II) течение заболевания, например, более или менее 3 месяцев на протяжении одного года или непрерывное течение заболевания, и III) интенсивность в диапазоне от слабой чесотки до сильной чесотки, обычно беспокоящей во время ночного сна. Возможны оценки 1,5 или 2,5. Общую тяжесть заболевания определяют по сумме отдельных оценок по этим трем оценочным категориям заболевания, а тяжесть определяют по сумме этих оценок, при этом легкое заболевание определяется общей оценкой 3-4, умеренное – оценкой 4,5-7,5, а тяжелое – общей оценкой 8-9.
[00103] Термин «критерий оценки тяжести заболевания для атопического дерматита» или «ADDSOM» означает определение некоторых признаков, симптомов, характеристик или параметров, которые связаны с атопическим дерматитом и которые могут быть количественно или качественно оценены. Типовые критерии ADDSOM включают, но не ограничиваются этим, «индекс распространенности и тяжести экземы» (EASI), «оценку тяжести атопического дерматита» (SCORAD), «общую оценку исследователем» (IGA), «общую оценку признаков исследователем» (IGSA), оценку атопического дерматита Райка/Лангеланда и оценку результатов пациентами, включая, но не ограничиваясь этим, визуальную аналоговую шкалу зуда (аспект тяжести заболевания, оцениваемый как составляющая SCORAD), визуальную аналоговую шкалу бессонницы (аспект тяжести заболевания, оцениваемый как составляющая SCORAD), дневник симптомов атопического дерматита (ADSD), опросник по влиянию атопического дерматита (ADIQ), дерматологический индекс качества жизни (DLQI) (Finlay and Khan, Clin Exper Dermatol 1994; 19:210) и 5-D шкалу чесотки (Elman et al., Br J Dermatol 2010; 162(3):587-593).
[00104] «Индекс распространенности и тяжести экземы» или «EASI» представляет собой утвержденный показатель, используемый в клинических условиях для оценки тяжести и степени АД, Hanifin et al., Exp Dermatol 2001; 10: 11-18. Клиническим врачом или другим специалистом-медиком оцениваются четыре отдельных участка тела: голова и шея (ГиШ), верхние конечности (ВК; включая внешние подмышечные впадины и ладони), торс (включая внутренние подмышечные впадины и пах), и нижние конечности (НК; включая ягодицы и стопы). Для каждого участка тела оценивают площадь пораженной поверхности тела (ППТ) и приписывают оценку от 0 до 6 (или, необязательно, 0-7, где 0 соответствует отсутствию сыпи) для расчета процента пораженной ППТ (0% – 100%); каждый участок отдельно оценивают для определения средней степени тяжести (0-3: отсутствие, легкая, умеренная, тяжелая), при этом допускаются половинные значения, в отношении каждого из четырех клинических признаков: эритема, уплотнение/появление папул, экскориация и лихенификация. Каждому участку тела приписывается средняя оценка от 0 до 12; общая оценка приписывается участку тела на основании суммы отдельных оценок по клиническим признакам (максимум = 12) х оценку по пораженной площади (максимум = 6) х индекс участка тела (ГиШ – 0,1, ВК – 0,2, торс – 0,3, НК – 0,4). Общая оценка (0-72) приписывается на основании суммы общих оценок для каждого из четырех участков тела.
[00105] «Общая оценка исследователем» или «IGA» представляет собой показатель оценки, используемый в клинических условиях для определения тяжести АД и клинического ответа на лечение на основании пятиточечной шкалы. Оценка 0 (чистая кожа) означает отсутствие воспалительный признаков атопического дерматита, а оценка 1 (почти чистая кожа) означает, что присутствует только видимая эритема и только видимое появление папул и уплотнение. Оценки 2, 3 или 4 (легкое, умеренное, тяжелое заболевание) основаны на тяжести эритемы, появлении папул и уплотнения, выделении экссудата и образовании корки. В «общей оценке признаков исследователем» или «IGSA» используется градация оценки поражений в диапазоне от чистой кожи до тяжелого заболевания на основании оценки эритемы, отека, лихенифицированных бляшек или папул и экскориации. Кроме того, градация оценки поражений может быть повышена или понижена на основании степени и расположения кожных поражений.
[00106] «Оценка тяжести атопического дерматита» или «SCORAD» представляет собой клиническую оценку тяжести АД, разработанную Европейской специальной группой по атопическому дерматиту (консенсусный доклад, Dermatology 1993; 186:23-31). Оцениваются три аспекта тяжести заболевания: (i) величина площади тела, пораженной воспалением, с оценкой 0-100, обозначаемой «А» в общей оценке, (ii) интенсивность шести клинических признаков: эритемы, отека/образования папул, выделения экссудата/образования корки, экскориации, лихенификации и сухости, каждому из которых приписывается оценка 0-3 на основании тяжести (отсутствие заболевания, легкое, умеренное, тяжелое заболевание) с получением общей оценки в диапазоне 0-18, обозначаемой «B» в общей оценке, и (iii) два субъективных показателя, в которых используется оценка пациентами: визуальная аналоговая шкала зуда (в диапазоне от 0 [отсутствие чесотки] до 10 [наихудшая чесотка, которую можно представить]) и визуальная аналоговая шкала бессонницы (в диапазоне от 0 [отсутствие бессонницы] до 10 [наихудшая бессонница, которую можно представить]), каждая в виде среднего за последние 3 суток или ночей, обозначаемая «С» в общей оценке. Общую оценку (0-103) определяют по формуле: A/5+(7B/2)+C.
[00107] Термин «оценка результатов пациентами» или «PRO» обозначает утвержденный опросник или инструмент, используемый в клинической практике или в клинических исследованиях для оценки качества жизни с точки зрения пациента в связи с симптомами заболевания атопическим дерматитом у пациента, которые могут включать эмоциональное и функциональное воздействие заболевания. PRO представляет собой отчет о состоянии здоровья пациента, исходящий непосредственно от пациента, без интерпретации реакции пациента клиническим врачом или кем-либо еще. Результат может быть определен в абсолютных терминах (например, тяжесть симптома, признака или состояния заболевания) или в виде изменения относительно предыдущих показателей. В клинических исследованиях инструмент PRO можно использовать для определения эффекта медицинского вмешательства на один или более показателей (т. е. определяемых показателей, таких как симптом или ряд симптомов, эффект на конкретную функцию или ряд функций, или ряд симптомов или функций, являющихся показательными для определения тяжести состояния здоровья). Типовые инструменты PRO, используемые для оценки АД включают, но не ограничиваются этим, визуальную аналоговую шкалу зуда (аспект тяжести заболевания, оцениваемый как составляющая SCORAD), визуальную аналоговую шкалу бессонницы (аспект тяжести заболевания, оцениваемый как составляющая SCORAD), дневник симптомов атопического дерматита (ADSD), опросник по влиянию атопического дерматита (ADIQ), дерматологический индекс качества жизни (DLQI) (Finlay and Khan, Clin Exper Dermatol 1994; 19:210) и 5-D шкалу чесотки (Elman et al., Br J Dermatol 2010; 162(3):587-593).
КОМПОЗИЦИИ И СПОСОБЫ
[00108] Это изобретение основано, по меньшей мере частично, на удивительном и неожиданном открытии, что анти-IL-13 антагонистическое моноклональное антитело лебрикизумаб обеспечивает терапевтическое действие при введении пациентам с атопическим дерматитом с применением предложенных в данном документе схем дозирования, включая пациентов, одновременно принимающих местные кортикостероиды, согласно оценке по нескольким критериям эффективности результатов. Соответственно, в данном документе предложены способы лечения атопического дерматита антагонистами IL-13, включая анти-IL-13 антитела, такие как лебрикизумаб.
[00109] Лебрикизумаб представляет собой гуманизированное, моноклональное антитело IgG4, которое связывает IL-13 с высокой аффинностью и блокирует сигнализацию через активный гетеродимер IL-4R-альфа/IL-13R-альфа1. Лебрикизумаб клинически исследовали при астме в различных дозах, а результаты были представлены в литературе и обобщены ниже.
[00110] В исследовании MILLY взрослым со слабо контролируемой, несмотря на лечение ингаляционными кортикостероидами, астмой вводили 250 мг лебрикизумаба или плацебо один раз в четыре недели всего в течение шести месяцев. Corren et al, N Engl J Med 2011; 365:1088-98. Пациенты, которых лечили лебрикизумабом, в частности, пациенты в подгруппе с высоким уровнем биомаркера (высоким уровнем сывороточного периостина), демонстрировали большее улучшение легочной функции согласно определению FEV1, чем пациенты в группе плацебо. Corren et al, выше.
[00111] Исследование MOLLY представляло собой исследование с подбором дозы, в котором пациенты с астмой, не принимавшие ингаляционные кортикостероиды, получали 125 мг лебрикизумаба, или 250 мг лебрикизумаба, или 500 мг лебрикизумаба, или плацебо один раз в четыре недели всего в течение 12 недель. Noonan et al, J Allergy Clin Immunol 2013; 132:567-74. Хотя среднее относительное изменение в FEV1 было численно выше во всех группах с разными дозами лебрикизумаба по сравнению с группой плацебо, эта разница не была ни статистически ни клинически значимой. Noonan et al, выше. Кроме того, исследование MOLLY не смогло продемонстрировать зависимость доза-ответ. Noonan et al, выше. Одним из выводов из результатов является то, что блокирования одного цитокина, IL-13, в популяции пациентов с астмой в исследовании MOLLY, было недостаточно для улучшения легочной функции, определяемой по FEV1, по сравнению с существующими вариантами лечения. Noonan et al, выше.
[00112] В исследованиях LUTE и VERSE три разных дозы лебрикизумаба, 37,5 мг, 125 мг и 250 мг, вводимых один раз в четыре недели, исследовали на пациентах с умеренной и тяжелой астмой, не контролируемой приемом ингаляционных кортикостероидов. Hanania et al., Thorax 2015; 70:748-756. Лечение лебрикизумабом снижало уровень астматических обострений, которые были наиболее выраженными у пациентов с повышенным уровнем биомаркера (высоким уровнем сывороточного периостина) (все дозы: 60% снижение), чем у пациентов с низким уровнем биомаркера (низким уровнем сывороточного периостина) (все дозы: 5% снижение); однако отсутствуют четкие данные по зависимости доза-ответ. Hanania et al., 2015, выше. После лечения лебрикизумабом также улучшалась легочная функция, при этом наибольшее увеличение FEV1 наблюдалось у пациентов с высоким уровнем периостина по сравнению с пациентами с низким уровнем периостина. Hanania et al., 2015, выше.
[00113] Как описано в данном документе, пациенты как с АД, так и с астмой имели повышенные уровни биомаркеров, связанных с биологией IL-13, и опосредованную Th-2 гиперчувствительность. Соответственно, посчитали, что уровни доз лебрикизумаба, приводящие к улучшению клинических показателей при астме, также могли бы демонстрировать биологическую активность у пациентов с АД. Таким образом, исходные дозы, предложенные для исследования на пациентах с АД, приведенные в описании клинического исследования I и клинического исследования II в примерах были основаны на опубликованных клинических данных, полученных в случае астмы, как описано выше.
[00114] После инициации клинического исследования I и клинического исследования II, описанных в данном документе, были проанализированы результаты клинических исследований фазы 3 лебрикизумаба при астме (LAVOLTA I и LAVOLTA II) и опубликованы онлайн 5 сентября 2016 г. в Hanania et al., Lancet Respir Med 2016, доступные на dx(dot)doi(dot)org(slash)S2213-2600(16)30265-X. LAVOLTA I и II представляли собой исследования фазы 3 с повторяющимся дизайном для оценки эффективности и безопасности лебрикизумаба у пациентов с неконтролируемой, несмотря на прием ингаляционных кортикостероидов и по меньшей мере одного второго контролирующего препарата, астмой. Эти исследования фазы 3 были разработаны с целью получения разрешения на маркетинг от органов здравоохранения по всему миру при условии демонстрации статистически значимой эффективности в обоих исследованиях и приемлемого профиля безопасности.
[00115] В LAVOLTA I и II пациентов лечили 37,5 мг лебрикизумаба, или 125 мг лебрикизумаба, или плацебо, которые вводили один раз в четыре недели в течение 52 недель. Основным измеряемым параметром эффективности было снижение уровня обострения заболевания. Пациентов стратифицировали в соответствии с высоким уровнем биомаркера (высоким сывороточным уровнем периостина или высоким числом эозинофилов в крови) или низким уровнем биомаркера (низким сывороточным уровнем периостина или низким числом эозинофилов в крови). Как сообщалось в Hanania et al, 2016, выше, результаты по эффективности в обоих исследованиях не согласовались между собой. Лебрикизумаб значительно снижал уровень обострения у пациентов с высоким уровнем биомаркера в LAVOLTA I, но не в LAVOLTA II. Hanania et al., 2016, выше. Оба исследования не смогли продемонстрировать четкой зависимости доза-ответ, хотя результаты по фармакокинетике и фармакодинамике согласовались с результатами исследований фазы II, описанных выше, что указывает на аналогичное воздействие лекарственного препарата и ингибирование пути IL-13. Hanania et al., 2016, выше.
[00116] Соответственно, хотя схемы дозирования лебрикизумаба, исследуемые в описанных в данном документе клиническом исследовании I и клиническом исследовании II атопического дерматита, были основаны на клиническом опыте для астмы, не было уверенности в том, что лебрикизумаб может обеспечивать клинически значимый результат у пациентов с атопическим дерматитом при применении схем дозирования, описанных для клинического исследования I и клинического исследования II. Несогласующиеся результаты для лебрикизумаба у пациентов с астмой и отсутствие четкой зависимости доза-ответ среди нескольких исследований, в общих чертах описанных выше, включая результаты, приведенные для исследований LAVOLTA I и II фазы 3, обуславливают такую неуверенность в отношении терапевтической пользы лебрикизумаба у пациентов с атопическим дерматитом. Перед появлением описанного в данном документе изобретения нельзя было предсказать, сможет ли лебрикизумаб обеспечивать клинически значимый результат у пациентов с атопическим дерматитом при применении схем дозирования, описанных в данном документе.
Типовые антитела
Анти-IL-13 антитела
[00117] В одном аспекте в изобретении предложены выделенные антитела, которые связываются с человеческим IL-13.
[00118] Типовые анти-IL-13 антитела известны и включают, например, но не ограничиваются этим, лебрикизумаб, IMA-026, IMA-638 (также называемый анрукинзумабом, INN № 910649-32-0; QAX-576); тралокинумаб (также называемый CAT-354, CAS № 1044515-88-9); AER-001, ABT-308 (также называемый гуманизированным антителом 13C5.5). Примеры таких анти-IL-13 антител и других ингибиторов IL-13 описаны, например, в WO 2005/062967, WO2008/086395, WO2006/085938, US 7615213, US 7501121, WO2007/036745, WO2010/073119, WO2007/045477, WO 2014/165771. В одном варианте реализации изобретения анти-IL-13 антитело представляет собой гуманизированное антитело IgG4. В одном варианте реализации изобретения анти-IL-13 антитело представляет собой лебрикизумаб. В одном варианте реализации изобретения анти-IL-13 антитело содержит три HVR тяжелой цепи, HVR-H1 (SEQ ID NO: 5), HVR-H2 (SEQ ID NO: 6) и HVR-H3 (SEQ ID NO: 7). В одном варианте реализации изобретения анти-IL-13 антитело содержит три HVR легкой цепи, HVR-L1 (SEQ ID NO: 8), HVR-L2 (SEQ ID NO: 9) и HVR-L3 (SEQ ID NO: 10). В одном варианте реализации изобретения анти-IL-13 антитело содержит три HVR тяжелой цепи и три HVR легкой цепи, HVR-H1 (SEQ ID NO: 5), HVR-H2 (SEQ ID NO: 6), HVR-H3 (SEQ ID NO: 7), HVR-L1 (SEQ ID NO: 8), HVR-L2 (SEQ ID NO: 9) и HVR-L3 (SEQ ID NO: 10). В одном варианте реализации изобретения анти-IL-13 антитело содержит вариабельную область тяжелой цепи, VH, имеющую аминокислотную последовательность, выбранную из SEQ ID NO: 1, 3 и 24. В одном варианте реализации изобретения анти-IL-13 антитело содержит вариабельную область легкой цепи, VL, имеющую аминокислотную последовательность, выбранную из SEQ ID NO: 2, 4 и 25. В одном варианте реализации изобретения анти-IL-13 антитело содержит вариабельную область тяжелой цепи, VH, имеющую аминокислотную последовательность, выбранную из SEQ ID NO: 1, 3 и 24, и вариабельную область легкой цепи, VL, имеющую аминокислотную последовательность, выбранную из SEQ ID NO: 2, 4 и 25.
[00119] В другом варианте реализации изобретения антитело содержит последовательности вариабельной области SEQ ID NO: 1 и SEQ ID NO: 2. В другом варианте реализации изобретения антитело содержит последовательности вариабельной области SEQ ID NO: 1 и SEQ ID NO: 4. В другом варианте реализации изобретения антитело содержит последовательности вариабельной области SEQ ID NO: 1 и SEQ ID NO: 25. В другом варианте реализации изобретения антитело содержит последовательности вариабельной области SEQ ID NO: 3 и SEQ ID NO: 2. В другом варианте реализации изобретения антитело содержит последовательности вариабельной области SEQ ID NO: 3 и SEQ ID NO: 4. В другом варианте реализации изобретения антитело содержит последовательности вариабельной области SEQ ID NO: 3 и SEQ ID NO: 25. В другом варианте реализации изобретения антитело содержит последовательности вариабельной области SEQ ID NO: 24 и SEQ ID NO: 2. В другом варианте реализации изобретения антитело содержит последовательности вариабельной области SEQ ID NO: 24 и SEQ ID NO:4. В другом варианте реализации изобретения антитело содержит последовательности вариабельной области SEQ ID NO: 24 и SEQ ID NO: 25.
[00120] В любом из вышеприведенных вариантов реализации изобретения анти-IL-13 антитело может быть гуманизированным. В одном варианте реализации изобретения анти-IL-13 антитело содержит HVR, как в любом из вышеприведенных вариантов реализации изобретения, и дополнительно содержит акцепторную человеческую каркасную область, например, каркасную область человеческого иммуноглобулина или человеческую консенсусную каркасную область.
[00121] В другом аспекте анти-IL-13 антитело содержит последовательность вариабельного домена тяжелой цепи (VH), имеющую по меньшей мере 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% или 100% идентичности последовательности с аминокислотной последовательностью SEQ ID NO: 1. В определенных вариантах реализации изобретения последовательность VH, имеющая по меньшей мере 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% или 99% идентичности, содержит замены (например, консервативные замены), вставки или делеции относительно эталонной последовательности, но анти-IL-13 антитело, содержащее такую последовательность, сохраняет способность связываться с человеческим IL-13. В определенных вариантах реализации изобретения была проведены замена, вставка и/или делеция всего 1-10 аминокислот в SEQ ID NO: 1. В определенных вариантах реализации изобретения замены, вставки или делеции находятся в областях за пределами HVR (т. е. в FR). Необязательно, анти-IL-13 антитело содержит последовательность VH в SEQ ID NO: 1, включая посттрансляционные модификации этой последовательности. Необязательно, анти-IL-13 антитело содержит последовательность VH в SEQ ID NO: 3, включая посттрансляционные модификации этой последовательности. Необязательно, анти-IL-13 антитело содержит последовательность VH в SEQ ID NO: 24, включая посттрансляционные модификации этой последовательности.
[00122] В другом аспекте предложено анти-IL-13 антитело, содержащее последовательность вариабельного домена легкой цепи (VL), имеющую по меньшей мере 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% или 100% идентичности последовательности с аминокислотной последовательностью SEQ ID NO: 2. В определенных вариантах реализации изобретения последовательность VL, имеющая по меньшей мере 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% или 99% идентичности, содержит замены (например, консервативные замены), вставки или делеции относительно эталонной последовательности, но анти-IL-13 антитело, содержащее такую последовательность, сохраняет способность связываться с человеческим IL-13. В определенных вариантах реализации изобретения была проведены замена, вставка и/или делеция всего 1-10 аминокислот в SEQ ID NO: 2. В определенных вариантах реализации изобретения замены, вставки или делеции находятся в областях за пределами HVR (т. е. в FR). Необязательно, анти-IL-13 антитело содержит последовательность VL в SEQ ID NO: 2, включая посттрансляционные модификации этой последовательности. Необязательно, анти-IL-13 антитело содержит последовательность VL в SEQ ID NO: 4, включая посттрансляционные модификации этой последовательности. Необязательно, анти-IL-13 антитело содержит последовательность VL в SEQ ID NO: 25, включая посттрансляционные модификации этой последовательности.
[00123] В другом аспекте предложено анти-IL-13 антитело, содержащее VH согласно любому из вышеприведенных вариантов реализации изобретения и VL согласно любому из вышеприведенных вариантов реализации изобретения.
[00124] В дополнительном аспекте в изобретении предложено антитело, которое связывается с тем же эпитопом, что и предложенное в данном документе анти-IL-13 антитело. Например, в определенных вариантах реализации изобретения предложено антитело, которое связывается с тем же эпитопом или может конкурентно ингибироваться анти-IL-13 антителом, содержащим последовательность VH с SEQ ID NO: 1 и последовательность VL с SEQ ID NO: 2.
[00125] В дополнительном аспекте изобретения анти-IL-13 антитело в соответствии с любым из вышеприведенных вариантов реализации может представлять собой моноклональное антитело, включая химерное, гуманизированное или человеческое антитело. В одном варианте реализации изобретения анти-IL-13 антитело представляет собой фрагмент антитела, например, Fv, Fab, Fab', scFv, диатело или фрагмент F(ab')2. В другом варианте реализации изобретения антитело представляет собой полноразмерное антитело, например, интактное антитело IgG1 или IgG4 или антитело другого класса или изотипа, описанного в данном документе. В соответствии с другим вариантом реализации изобретения антитело представляет собой биспецифическое антитело. В одном варианте реализации изобретения биспецифическое антитело содержит HVR или содержит области VH и VL, описанные выше.
[00126] В одном варианте реализации изобретения анти-IL-13 антитело содержит три HVR тяжелой цепи, HVR-H1 (SEQ ID NO: 13), HVR-H2 (SEQ ID NO: 14) и HVR-H3 (SEQ ID NO: 15). В одном варианте реализации изобретения анти-IL-13 антитело содержит три HVR легкой цепи, HVR-L1 (SEQ ID NO: 16), HVR-L2 (SEQ ID NO: 17) и HVR-L3 (SEQ ID NO: 18). В одном варианте реализации изобретения анти-IL-13 антитело содержит три HVR тяжелой цепи и три HVR легкой цепи, HVR-H1 (SEQ ID NO: 13), HVR-H2 (SEQ ID NO: 14), HVR-H3 (SEQ ID NO: 15), HVR-L1 (SEQ ID NO: 16), HVR-L2 (SEQ ID NO: 17) и HVR-L3 (SEQ ID NO: 18). В одном варианте реализации изобретения анти-IL-13 антитело содержит вариабельную область тяжелой цепи, VH, имеющую аминокислотную последовательность SEQ ID NO: 12. В одном варианте реализации изобретения анти-IL-13 антитело содержит вариабельную область легкой цепи, VL, имеющую аминокислотную последовательность SEQ ID NO: 11. В одном варианте реализации изобретения анти-IL-13 антитело содержит вариабельную область тяжелой цепи, VH, имеющую аминокислотную последовательность SEQ ID NO: 12, и вариабельную область легкой цепи, VL, имеющую аминокислотную последовательность SEQ ID NO: 11.
[00127] В любом из вышеприведенных вариантов реализации изобретения анти-IL-13 антитело может быть гуманизированным. В одном варианте реализации изобретения анти-IL-13 антитело содержит HVR, как в любом из вышеприведенных вариантов реализации изобретения, и дополнительно содержит акцепторную человеческую каркасную область, например, каркасную область человеческого иммуноглобулина или человеческую консенсусную каркасную область.
[00128] В другом аспекте анти-IL-13 антитело содержит последовательность вариабельного домена тяжелой цепи (VH), имеющую по меньшей мере 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% или 100% идентичности последовательности с аминокислотной последовательностью SEQ ID NO: 12. В определенных вариантах реализации изобретения последовательность VH, имеющая по меньшей мере 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% или 99% идентичности последовательности, содержит замены (например, консервативные замены), вставки или делеции относительно эталонной последовательности, но анти-IL-13 антитело, содержащее такую последовательность, сохраняет способность связываться с человеческим IL-13. В определенных вариантах реализации изобретения была проведены замена, вставка и/или делеция всего 1-10 аминокислот в SEQ ID NO: 12. В определенных вариантах реализации изобретения замены, вставки или делеции находятся в областях за пределами HVR (т. е. в FR). Необязательно, анти-IL-13 антитело содержит последовательность VH в SEQ ID NO: 12, включая посттрансляционные модификации этой последовательности.
[00129] В другом аспекте предложено анти-IL-13 антитело, содержащее последовательность вариабельного домена легкой цепи (VL), имеющую по меньшей мере 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% или 100% идентичности последовательности с аминокислотной последовательностью SEQ ID NO: 11. В определенных вариантах реализации изобретения последовательность VL, имеющая по меньшей мере 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% или 99% идентичности, содержит замены (например, консервативные замены), вставки или делеции относительно эталонной последовательности, но анти-IL-13 антитело, содержащее такую последовательность, сохраняет способность связываться с человеческим IL-13. В определенных вариантах реализации изобретения была проведены замена, вставка и/или делеция всего 1-10 аминокислот в SEQ ID NO: 11. В определенных вариантах реализации изобретения замены, вставки или делеции находятся в областях за пределами HVR (т. е. в FR). Необязательно, анти-IL-13 антитело содержит последовательность VL в SEQ ID NO: 11, включая посттрансляционные модификации этой последовательности.
[00130] В другом аспекте предложено анти-IL-13 антитело, содержащее VH согласно любому из вышеприведенных вариантов реализации изобретения и VL согласно любому из вышеприведенных вариантов реализации изобретения.
[00131] В дополнительном аспекте в изобретении предложено антитело, которое связывается с тем же эпитопом, что и предложенное в данном документе анти-IL-13 антитело. Например, в определенных вариантах реализации изобретения предложено антитело, которое связывается с тем же эпитопом или может конкурентно ингибироваться анти-IL-13 антителом, содержащим последовательность VH SEQ ID NO: 12 и последовательность VL с SEQ ID NO: 11.
[00132] В дополнительном аспекте изобретения анти-IL-13 антитело в соответствии с любым из вышеприведенных вариантов реализации может представлять собой моноклональное антитело, включая химерное, гуманизированное или человеческое антитело. В одном варианте реализации изобретения анти-IL-13 антитело представляет собой фрагмент антитела, например, Fv, Fab, Fab', scFv, диатело или фрагмент F(ab')2. В другом варианте реализации изобретения антитело представляет собой полноразмерное антитело, например, интактное антитело IgG1 или IgG4 или антитело другого класса или изотипа, описанного в данном документе. В соответствии с другим вариантом реализации изобретения антитело представляет собой биспецифическое антитело. В одном варианте реализации изобретения биспецифическое антитело содержит HVR или содержит области VH и VL, описанные выше.
[00133] В дополнительном аспекте анти-IL-13 антитело в соответствии с любым из вышеприведенных вариантов реализации изобретения может обладать любыми характеристиками, по отдельности или в комбинации, описанными ниже в разделах 1-7:
1. Аффинность антител
[00134] В определенных вариантах реализации изобретения предложенное в данном документе антитело имеет константу диссоциации (Kд) ≤ 1 мкМ, ≤ 100 нМ, ≤ 10 нМ, ≤ 1 нМ, ≤ 0,1 нМ, ≤ 0,01 нМ или < 0,001 нМ (например, 10-8 M или менее, например, от 10-8 M до 10-13 M, например, от 10-9 M до 10-13 M).
[00135] В одном варианте реализации изобретения Kд измеряют с помощью анализа связывания радиоактивно меченого антигена (РИА), проводимого с Fab-версией представляющего интерес антитела и его антигеном, как описано ниже. Аффинность связывания Fab в отношении антигена в растворе измеряют, уравновешивая Fab минимальной концентрацией (125I)-меченого антигена в присутствии ряда титров немеченого антигена, затем проводя захват связанного антигена на покрытом анти-Fab антителом планшете (смотрите, например, Chen et al., J. Mol. Biol. 293:865-881(1999)). Чтобы установить условия для анализа, многолуночные планшеты MICROTITER® (Thermo Scientific) покрывают в течение ночи 5 мкг/мл захватывающего анти-Fab антитела (Cappel Labs) в 50 мМ карбонате натрия (pH 9,6), а после этого блокируют 2% (масс./об.) бычьего сывороточного альбумина в ФСБ в течение двух-пяти часов при комнатной температуре (приблизительно 23°C). В неадсорбирующем планшете (Nunc #269620) смешивают 100 пМ или 26 пМ [125I]-антигена с серийными разведениями представляющего интерес Fab (например, в соответствии с оценкой анти-VEGF антитела, Fab-12, в Presta et al, Cancer Res. 57:4593-4599 (1997)). Представляющий интерес Fab инкубируют в течение ночи; при этом инкубацию можно продолжать в течение более длительного периода (например, около 65 часов), чтобы гарантировать достижение равновесия. После этого смеси переносят в планшет для захвата для инкубации при комнатной температуре (например, в течение одного часа). Затем раствор удаляют, а планшет промывают восемь раз 0,1% полисорбатом 20 (ТВИН-20®) в ФСБ. Когда планшеты высыхают, добавляют 150 мкл/лунку сцинтиллятора (MICROSCINT-20 TM; Packard) и считывают планшеты на гамма-счетчике TOPCOUNT TM (Packard) в течение десяти минут. Концентрации каждого Fab, которые дают меньше или 20% максимального связывания, отбирают для использования в конкурентном анализе связывания.
[00136] В соответствии с другим вариантом реализации изобретения Kд измеряют с помощью анализа методом поверхностного плазмонного резонанса, используя BIACORE®-2000 или BIACORE ®-3000 (BIAcore, Inc., Piscataway, NJ) при 25°C с иммобилизованным на чипе CM5 антигеном при ~10 единицах ответа (ЕО). Вкратце, биосенсорные чипы на основе карбоксиметилированного декстрана (CM5, BIACORE, Inc.) активируют N-этил-N'- (3-диметиламинопропил)-карбодиимида гидрохлоридом (EDC) и N-гидроксисукцинимидом (NHS) в соответствии с инструкциями поставщика. Антиген разводят 10 мМ ацетата натрия, pH 4,8, до 5 мкг/мл (~0,2 мкМ) перед вводом при скорости потока 5 мкл/минуту до достижения приблизительно 10 единиц ответа (ЕО) сопряженного белка. После ввода антигена вводят 1 М этаноламина, чтобы блокировать непрореагировавшие группы. Для кинетических измерений двукратные серийные разведения Fab (от 0,78 нМ до 500 нМ) вводят в ФСБ с 0,05% полисорбатом 20 (ТВИН-20TM), поверхностно-активное вещество (ФСБТ), при 25°C при скорости потока приблизительно 25 мкл/мин. Скорость ассоциации (kасс.) и скорость диссоциации (kдисс.) рассчитывают, используя простую взаимно-однозначную модель связывания Ленгмюра (оценочное программное обеспечение BIACORE®, версия 3.2), путем одновременной аппроксимации сенсограмм ассоциации и диссоциации. Равновесную константу диссоциации (Kд) рассчитывают как соотношение kдисс./kасс.. Смотрите, например, Chen et al, J. Mol. Biol. 293:865-881 (1999). Если скорость ассоциации превышает 106 М-1 с-1 по данным вышеприведенного метода поверхностного плазмонного резонанса, тогда скорость ассоциации можно определить, используя методику гашения флуоресценции, в которой измеряется повышение или снижение интенсивности испускания флуоресценции (возбуждение = 295 нм; испускание = 340 нм, 16 нм полоса пропускания) при 25oC 20 нМ комплекса антиген-антитело (в Fab форме) в ФСБ, pH 7,2, в присутствии возрастающих концентраций антигена с помощью спектрометра, такого как спектрофотометр с возможностью остановки потока (Aviv Instruments) или спектрофотометр серии 8000 SLM-AMINCO TM (ThermoSpectronic) с кюветой с перемешиванием.
2. Фрагменты антител
[00137] В определенных вариантах реализации изобретения предложенное в данном документе антитело представляет собой фрагмент антитела. Фрагменты антител включают, но не ограничиваются этим, фрагменты Fab, Fab', Fab'-SH, F(ab')2, Fv и scFv и другие фрагменты, описанные ниже. Обзор некоторых фрагментов антител смотрите в Hudson et al. Nat. Med. 9: 129-134 (2003). Обзор фрагментов scFv смотрите, например, в Pluckthün, в The Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg and Moore eds., (Springer-Verlag, New York), pp. 269-315 (1994); также смотрите WO 93/16185; и патенты США № 5571894 и 5587458. Описание фрагментов Fab и F(ab')2, содержащих эпитоп связывания рецептора реутилизации и имеющих повышенное in vivo время полужизни, смотрите в патенте США № 5869046.
[00138] Диатела представляют собой фрагменты антител с двумя антигенсвязывающими участками, которые могут быть бивалентными или биспецифическими. Смотрите, например, EP 404097; WO 1993/01161; Hudson et al, Nat. Med. 9: 129-134 (2003); и Hollinger et al, Proc. Natl. Acad. Sci. USA 90: 6444-6448 (1993). Триатела и тетратела также описаны в Hudson et al., Nat. Med. 9: 129-134 (2003).
[00139] Однодоменные антитела представляют собой фрагменты антител, содержащие весь или часть вариабельного домена тяжелой цепи или весь или часть вариабельного домена легкой цепи антитела. В определенных вариантах реализации изобретения однодоменное антитело представляет собой человеческое однодоменное антитело (Domantis, Inc., Waltham, MA; смотрите, например, патент США № 6248516 B1).
[00140] Фрагменты антител могут быть получены различными способами, включая, но не ограничиваясь этим, протеолитическое расщепление интактного антитела, а также выработку рекомбинантными клетками-хозяевами (например, E. coli или фага), как описано в данном документе.
3. Химерные и гуманизированные антитела
[00141] В определенных вариантах реализации изобретения предложенное в данном документе антитело представляет собой химерное антитело. Некоторые химерные антитела описаны, например, в патенте США № 4816567; и Morrison et al, Proc. Natl. Acad. Sci. USA, 81:6851-6855 (1984)). В одном примере химерное антитело содержит нечеловеческую вариабельную область (например, вариабельную область, полученную от мыши, крысы, хомяка, кролика или отличного от человека примата, такого как обезьяна) и человеческую константную область. В дополнительно примере химерное антитело представляет собой антитело с «переключенным классом», класс или подкласс которого был изменен относительно класса родительского антитела. Химерные антитела включают их антигенсвязывающие фрагменты.
[00142] В определенных вариантах реализации изобретения химерное антитело представляет собой гуманизированное антитело. Как правило, нечеловеческое антитело гуманизируют, чтобы снизить его иммуногенность для человека, в то же время сохраняя специфичность и аффинность родительского нечеловеческого антитела. В общем случае гуманизированное антитело содержит один или более вариабельных доменов, в которых HVR, например, CDR (или их части) получены из нечеловеческого антитела, а FR (или их части) получены из последовательностей человеческого антитела. Гуманизированное антитело, необязательно, также может содержать по меньшей мере часть человеческой константной области. В некоторых вариантах реализации изобретения некоторые остатки FR в гуманизированном антителе замещены соответствующими остатками из нечеловеческого антитела (например, антитела, из которого получены остатки HVR), например, для сохранения или улучшения специфичности или аффинности антитела.
[00143] Обзор гуманизированных антител и способов их получения приведен, например, в Almagro and Fransson, Front. Biosci. 13: 1619-1633 (2008), а дополнительное описание приведено, например, в Riechmann et al, Nature 332:323-329 (1988); Queen et al, Proc. Nat'l Acad. Sci. USA 86: 10029-10033 (1989); патентах США № 5821337, 7527791, 6982321 и 7087409; Kashmiri et al, Methods 36:25-34 (2005) (где описано прививание SDR (a-CDR)); Padlan, Mol. Immunol. 28:489-498 (1991) (где описано «изменение поверхности»); Dall'Acqua et al., Methods 36:43-60 (2005) (где описана «перетасовка FR»); и Osbourn et al, Methods 36:61-68 (2005) и Klimka et al, Br. J. Cancer, 83:252-260 (2000) (где описан подход «направляемого отбора» в отношении перетасовки FR).
[00144] Человеческие каркасные области, которые можно использовать для гуманизации, включают, но не ограничиваются этим: каркасные области, выбранные методом «наилучшего соответствия» (смотрите, например, Sims et al. J. Immunol. 151 :2296 (1993)); каркасные области, полученные из консенсусной последовательности человеческих антител конкретной подгруппы вариабельных областей легкой или тяжелой цепи (смотрите, например, Carter et al. Proc. Natl. Acad. Sci. USA, 89:4285 (1992); и Presta et al. J. Immunol, 151 :2623 (1993)); человеческие зрелые (прошедшие соматическую мутацию) каркасные области или каркасные области человеческой зародышевой линии (смотрите, например, Almagro and Fransson, Front. Biosci. 13: 1619-1633 (2008)); и каркасные области, полученные при скрининге библиотек FR (смотрите, например, Baca et al., J. Biol. Chem. 272: 10678-10684 (1997) и Rosok et al., J. Biol. Chem. 271:22611-22618 (1996)).
4. Человеческие антитела
[00145] В определенных вариантах реализации изобретения предложенное в данном документе антитело представляет собой человеческое антитело. Человеческие антитела можно получать, используя различные методики, известные в данной области техники. Человеческие антитела в целом описаны в van Dijk and van de Winkel, Curr. Opin. Pharmacol. 5: 368-74 (2001) и Lonberg, Curr. Opin. Immunol. 20:450-459 (2008).
[00146] Человеческие антитела можно получать путем введения иммуногена трансгенному животному, которое было модифицировано так, чтобы вырабатывать интактные человеческие антитела или интактные антитела с человеческими вариабельными областями в ответ на антигенную стимуляцию. Такие животные, как правило, содержат все или часть локусов человеческого иммуноглобулина, которые замещают локусы эндогенного иммуноглобулина или которые присутствуют внехромосомно или случайным образом интегрированы в хромосомы животного. У таких трансгенных мышей локусы эндогенного иммуноглобулина в общем случае инактивированы. Обзор способов получения человеческих антител от трансгенных животных смотрите в Lonberg, Nat. Biotech. 23: 1117-1125 (2005). Также смотрите, например, патенты США № 6075181 и 6150584, где описывается технология XENOMOUSETM; патент США № 5770429, где описывается технология HUMAB®; патент США № 7041870, где описывается технология K-M MOUSE®, и публикацию патентной заявки США № US 2007/0061900, где описывается технология VELOCIMOUSE®). Человеческие вариабельные области из интактных антител, вырабатываемых такими животными, можно дополнительно модифицировать, например, комбинируя с разными человеческими константными областями.
[00147] Человеческие антитела также можно получать способами на основе гибридом. Были описаны линии клеток миеломы человека и гетеромиеломы мышей и человека для получения человеческих моноклональных антител. (Смотрите, например, Kozbor J. Immunol, 133: 3001 (1984); Brodeur et al, Monoclonal Antibody Production Techniques and Applications, pp. 51-63 (Marcel Dekker, Inc., New York, 1987); и Boemer et al, J. Immunol, 147: 86 (1991).) Человеческие антитела, созданные с помощью технологии человеческой B-клеточной гибридомы, также описаны в Li et al, Proc. Natl. Acad. Sci. USA, 103:3557-3562 (2006). Дополнительные способы включают описанные, например, в патенте США № 7189826 (где описано получение моноклональных человеческих антител IgM из линий клеток гибридомы) и Ni, Xiandai Mianyixue, 26(4): 265-268 (2006) (где описаны человек-человеческие гибридомы). Технология человеческой гибридомы (технология триомы) также описана в Vollmers and Brandlein, Histology and Histopathology, 20(3):927-937 (2005) и Vollmers and Brandlein, Methods and Findings in Experimental and Clinical Pharmacology, 27(3): 185-91 (2005).
[00148] Человеческие антитела также можно создавать, выделяя последовательности вариабельного домена Fv-клона, выбранные из человеческих библиотек фагового дисплея. Такие последовательности вариабельного домена затем можно комбинировать с любым необходимым человеческим константным доменом. Методики отбора человеческих антител из библиотек антител описаны ниже.
5. Полученные из библиотек антитела
[00149] Антитела согласно изобретению могут быть выделены путем скрининга комбинаторных библиотек для получения антител с необходимой активностью или активностями. Например, в данной области техники известно много способов создания библиотек фагового дисплея и скрининга таких библиотек в отношении антител, обладающих необходимыми характеристиками связывания. Обзор таких способов приведен, например, в Hoogenboom et al. in Methods in Molecular Biology 178: 1-37 (O'Brien et al, ed., Human Press, Totowa, NJ, 2001), и дополнительно они описаны, например, в McCafferty et al, Nature 348:552-554; Clackson et al, Nature 352: 624-628 (1991); Marks et al, J. Mol. Biol. 222: 581-597 (1992); Marks and Bradbury, in Methods in Molecular Biology 248: 161-175 (Lo, ed., Human Press, Totowa, NJ, 2003); Sidhu et al., J. Mol. Biol. 338(2): 299-310 (2004); Lee et al, J. Mol. Biol. 340(5): 1073-1093 (2004); Fellouse, Proc. Natl. Acad. Sci. USA 101(34): 12467-12472 (2004); и Lee et al., J. Immunol. Methods 284(1-2): 119-132(2004).
[00150] В определенных способах с применением фагового дисплея репертуары генов VH и VL отдельно клонируют посредством полимеразной цепной реакции (ПЦР) и случайным образом рекомбинируют в фаговых библиотеках, после чего можно проводить их скрининг в отношении антигенсвязывающего фага, как описано в Winter et al, Ann. Rev. Immunol, 12: 433-455 (1994). Фаг, как правило, отображает фрагменты антител в виде одноцепочечных фрагментов Fv (scFv) или в виде фрагментов Fab. Библиотеки из иммунизированных источников обеспечивают высокоаффинные антитела к иммуногену без необходимости создания гибридом. В альтернативном варианте можно клонировать наивный репертуар (например, от человека), чтобы обеспечить единый источник антител к широкому диапазону несобственных, а также собственных антигенов, без какой-либо иммунизации, как описано в Griffiths et al, EMBO J, 12: 725-734 (1993). И наконец, наивные библиотеки также можно получать синтетическим образом, клонируя неперестроенные V-генные сегменты из стволовых клеток и используя ПЦР-праймеры, содержащие случайную последовательность, для кодирования высоковариабельных областей CDR3 и для осуществления перестройки in vitro, как описано в Hoogenboom and Winter, J. Mol. Biol., 227: 381-388 (1992). Патентные публикации, описывающие фаговые библиотеки человеческих антител, включают, например: патент США № 5750373 и патентные публикации США № 2005/0079574, 2005/0119455, 2005/0266000, 2007/0117126, 2007/0160598, 2007/0237764, 2007/0292936 и 2009/0002360.
[00151] Антитела или фрагменты антител, выделенные из библиотек человеческих антител, считаются в данном документе человеческими антителами или фрагментами человеческих антител.
6. Мультиспецифические антитела
[00152] В определенных вариантах реализации изобретения предложенное в данном документе антитело представляет собой мультиспецифическое антитело, например, биспецифическое антитело. Мультиспецифические антитела представляют собой моноклональные антитела, имеющие специфичность связывания в отношении по меньшей мере двух разных участков. В определенных вариантах реализации изобретения одна специфичность связывания относится к IL-13, а другая – к любому другому антигену. В определенных вариантах реализации изобретения биспецифические антитела могут связываться с двумя разными эпитопами IL-13. Биспецифические антитела также можно использовать для локализации цитотоксических агентов в клетках. Биспецифические антитела можно получать в виде полноразмерных антител или фрагментов антител.
[00153] Методики получения мультиспецифических антител включают, но не ограничиваются этим, рекомбинантную коэкспрессию двух пар тяжелая цепь – легкая цепь иммуноглобулина, имеющих разную специфичность (смотрите Milstein and Cuello, Nature 305: 537 (1983)), WO 93/08829 и Traunecker et al., EMBO J. 10: 3655 (1991)), и конструирование методом «выступ-во-впадину» (смотрите, например, патент США № 5731168). Мультиспецифические антитела также можно получать путем создания с эффекта электростатического взаимодействия для получения Fc-гетеродимерных молекул антител (WO 2009/089004A1); перекрестного сшивания двух или более антител или фрагментов (смотрите, например, патент США № 4676980 и Brennan et al, Science, 229: 81 (1985)); использования лейциновых молний для получения биспецифических антител (смотрите, например, Kostelny et al., J. Immunol., 148(5): 1547-1553 (1992)); использования технологии «диател» для получения биспецифических фрагментов антител (смотрите, например, Hollinger et al, Proc. Natl. Acad. Sci. USA, 90:6444-6448 (1993)); и использования димеров одноцепочечных Fv (sFv) (смотрите, например, Gruber et al., J. Immunol, 152:5368 (1994)); и получения триспецифических антител, как описано, например, в Tutt et al. J. Immunol. 147: 60 (1991).
[00154] Сконструированные антитела с тремя или более функциональными антигенсвязывающими участками, включая «антитела-осьминоги», также включены в данный документ (смотрите, например US 2006/0025576A1).
[00155] В данном документе антитело или фрагмент антитела также включают «FAb двойного действия» или «DAF», содержащий антигенсвязывающий участок, который связывается с IL-13, а также с другим, отличным антигеном (смотрите, например, US 2008/0069820).
7. Варианты антител
[00156] В определенных вариантах реализации изобретения предусмотрены варианты аминокислотных последовательностей предложенных в данном документе антител. Например, может существовать необходимость в улучшении аффинности связывания и/или других биологических свойств антитела. Варианты аминокислотных последовательностей антитела можно получать путем внесения соответствующих модификаций в нуклеотидную последовательность, кодирующую антитело, или путем пептидного синтеза. Такие модификации включают, например, делеции, и/или вставки, и/или замены остатков в пределах аминокислотных последовательностей антитела. Можно осуществлять любую комбинацию делеции, вставки и замены для получения конечной конструкции при условии, что конечная конструкция обладает необходимыми характеристиками, например, связывает антиген.
Варианты, содержащие замены, вставки и делеции
[00157] В определенных вариантах реализации изобретения предложены варианты антител, имеющие одну или более аминокислотных замен. Представляющие интерес для заместительного мутагенеза участки включают HVR и FR. Консервативные замены приведены в таблице 1 под заголовком «консервативные замены». Более существенные изменения приведены в таблице 1 под заголовком «типовые замены» и дополнительно описаны ниже в связи с классами аминокислотных боковых цепей. Аминокислотные замены можно вносить в представляющее интерес антитело и проводить скрининг продуктов в отношении необходимой активности, например, сохранения/улучшения связывания антигена, снижения иммуногенности или улучшения АЗКЦ или КЗЦ.
Таблица 1
остаток
замены
замены
[00158] Аминокислоты можно сгруппировать в соответствии с общими свойствами боковых цепей:
(1) гидрофобные: Норлейцин, Met, Ala, Val, Leu, Ile;
(2) нейтральные гидрофильные: Cys, Ser, Thr, Asn, Gln;
(3) кислые: Asp, Glu;
(4) основные: His, Lys, Arg;
(5) остатки, влияющие на ориентацию цепи: Gly, Pro;
(6) ароматические: Trp, Tyr, Phe.
[00159] Неконсервативные замены включают замену представителя одного из этих классов представителем другого класса.
[00160] Один из типов заместительного варианта включает замещение одного или более остатков гипервариабельной области родительского антитела (например, гуманизированного или человеческого антитела). В общем случае получаемые в результате варианты, отобранные для дополнительного исследования, будут иметь модификации (например, улучшения) в определенных биологических свойствах (например, повышение аффинности, снижение иммуногенности) по сравнению с родительским антителом и/или будут по существу сохранять определенные биологические свойства родительского антитела. Типовым заместительным вариантом является антитело с созревшей аффинностью, которое удобно получать, например, используя методики достижения созревания аффинности на основе фагового дисплея, такие как описаны в данном документе. Вкратце, проводят мутацию одного или более участков HVR и скрининг вариантных антител, отображаемых фагом, в отношении конкретной биологической активности (например, аффинности связывания).
[00161] Изменения (например, замены) можно осуществлять в HVR, например, для улучшения аффинности антитела. Такие изменения можно осуществлять в «горячих точках» HVR, т. е. остатках, кодируемых кодонами, которые подвергаются мутации с высокой частотой во время процесса соматического созревания (смотрите, например, Chowdhury, Methods Mol. Biol. 207: 179-196 (2008)), и/или SDR (a-CDR), исследуя получаемые в результате вариантные VH или VL в отношении аффинности связывания. Созревание аффинности путем конструирования и повторного отбора из вторичных библиотек было описано, например, в Hoogenboom et al. in Methods in Molecular Biology 178: 1-37 (O'Brien et al., ed., Human Press, Totowa, NJ, (2001).) В некоторых вариантах реализации созревания аффинности разнообразие вносят в выбранные для созревания гены вариабельной области различными способами (например, ПЦР с внесением ошибок, перетасовки цепей или олигонуклеотид-направленного мутагенеза). Затем создают вторичную библиотеку. Затем проводят скрининг библиотеки, чтобы идентифицировать любые варианты антител с необходимой аффинностью. Другой способ внесения разнообразия включает HVR-направленные подходы, в которых рандомизируют несколько остатков HVR (например, 4-6 остатков за раз). В частности, можно идентифицировать остатки HVR, вовлеченные в связывание антигена, например, используя аланин-сканирующий мутагенез или моделирование. В частности, часто мишенями являются CDR-H3 и CDR-L3.
[00162] В определенных вариантах реализации изобретения замены, вставки или делеции могут находиться в пределах одной или более HVR до той меры, пока такие изменения существенно не снижают способность антитела связывать антиген. Например, в HVR можно осуществлять консервативные изменения (например, приведенные в данном документе консервативные замены), которые существенно не снижают аффинность связывания. Такие изменения могут находиться за пределами «горячих точек» HVR или SDR. В определенных вариантах реализации вариантных последовательностей VH и VL, приведенных выше, каждая HVR является неизменной или содержит не более одной, двух или трех аминокислотных замен.
[00163] Эффективный способ идентификации остатков или областей антитела, которые могут быть мишенями для мутагенеза, называется «аланин-сканирующим мутагенезом» и описан в Cunningham and Wells (1989) Science, 244: 1081-1085. В этом способе идентифицируют остаток или группу целевых остатков (например, заряженных остатков таких как arg, asp, his, lys и glu) и замещают нейтральной или отрицательно заряженной аминокислотой (например, аланином или полиаланином) для определения, будет ли это влиять на взаимодействие антитела с антигеном. Дополнительные замены можно вносить в аминокислотные локации, демонстрирующие функциональную чувствительность к исходным заменам. В альтернативном или дополнительном варианте определяют кристаллическую структуру комплекса антиген-антитело для идентификации точек контакта между антителом и антигеном. Такие контактные остатки и соседние с ними остатки могут служить мишенями или удаляться как кандидаты на замену. Можно проводить скрининг вариантов для определения, имеют ли они необходимые свойства.
[00164] Аминокислотные вставки включают амино- и/или карбокси-концевые слияния с диапазоном длины от одного остатка до полипептидов, содержащих сто или более остатков, а также вставки одного или некоторого количества аминокислотных остатков внутри последовательности. Примеры концевых вставок включают антитело с N-концевым остатком метионила. Другие содержащие вставки варианты молекулы антитела включают слияние N- или C-конца антитела с ферментом (например, для ADEPT) или полипептидом, который повышает сывороточное время полужизни антитела.
Гликозилированные варианты
[00165] В определенных вариантах реализации изобретения предложенное в данном документе антитело изменяют для увеличения или уменьшения степени гликозилирования антитела. Добавление или удаление участков гликозилирования в антителе удобно осуществлять, изменяя аминокислотную последовательность так, чтобы создать или удалить один или более участков гликозилирования.
[00166] Если антитело содержит Fc-область, можно изменять присоединенный к ней углевод. Нативные антитела, вырабатываемые клетками млекопитающих, как правило, содержат разветвленный, биантеннарный олигосахарид, который в общем случае присоединен N-связью к Asn297 CH2-домена Fc-области. Смотрите, например, Wright et al. TIBTECH 15:26-32 (1997). Олигосахарид может содержать различные углеводы, например, маннозу, N-ацетилглюкозамин (GlcNAc), галактозу и сиаловую кислоту, а также фукозу, присоединенную к GlcNAc в «стволовой» части биантеннарной олигосахаридной структуры. В некоторых вариантах реализации изобретения модификации олигосахарида в антителе согласно изобретению можно проводить для создания вариантов антител с определенными улучшенными свойствами.
[00167] В одном варианте реализации изобретения предложены варианты антител, имеющие углеводную структуру, в которой отсутствует фукоза, присоединенная (прямым или непрямым образом) к Fc-области. Например, количество фукозы в таком антителе может составлять от 1% до 80%, от 1% до 65%, от 5% до 65% или от 20% до 40%. Количество фукозы определяют, рассчитывая отношение среднего количества фукозы в сахарной цепи в Asn297 к сумме всех гликоструктур, присоединенных к Asn 297 (например, комплексных, гибридных структур и структур с высоким содержанием маннозы), определяемой методом масс-спектроскопии MALDI-TOF, как описано, например, в WO 2008/077546. Asn297 относится к остатку аспарагина, расположенному приблизительно в позиции 297 в Fc-области (нумерация Eu остатков Fc-области); при этом Asn297 также может располагаться приблизительно на ± 3 выше или ниже позиции 297, т. е. между позициями 294 и 300, вследствие незначительных вариаций последовательностей в антителах. Такие фукозилированные варианты имеют улучшенную функцию АЗКЦ. Смотрите, например, патентные публикации США № US 2003/0157108 (Presta, L.); US 2004/0093621 (Kyowa Hakko Kogyo Co., Ltd). Примеры публикаций, относящихся к «дефукозилированным» или «фукозо-дефицитным» вариантам антител, включают: US 2003/0157108; WO 2000/61739; WO 2001/29246; US 2003/0115614; US 2002/0164328; US 2004/0093621; US 2004/0132140; US 2004/0110704; US 2004/0110282; US 2004/0109865; WO 2003/085119; WO 2003/084570; WO 2005/035586; WO 2005/035778; WO2005/053742; WO2002/031140; Okazaki et al. J. Mol. Biol. 336: 1239-1249 (2004); Yamane-Ohnuki et al. Biotech. Bioeng. 87: 614 (2004). Примеры линий клеток, способных вырабатывать дефукозилированные антитела, включают клетки Lec13 CHO с дефицитом белкового фукозилирования (Ripka et al. Arch. Biochem. Biophys. 249:533-545 (1986); патентная заявка США № US 2003/0157108 A1, Presta, L; и WO 2004/056312 A1, Adams et al., в особенности пример 11), и линии клеток с нокаутом, такие как клетки CHO с нокаутом гена альфа-1,6-фукозилтрансферазы, FUT8 (смотрите, например, Yamane-Ohnuki et al. Biotech. Bioeng. 87: 614 (2004); Kanda, Y. et al., Biotechnol. Bioeng., 94(4):680-688 (2006); и WO2003/085107).
[00168] Дополнительно предложены варианты антител с разделенными надвое олигосахаридами, например, в которых биантеннарный олигосахарид, присоединенный к Fc-области антитела, разделен надвое GlcNAc. Такие варианты антител могут иметь сниженное фукозилирование и/или улучшенную функцию АЗКЦ. Примеры таких вариантов антител описаны, например, в WO 2003/011878 (Jean-Mairet et al.); патенте США № 6602684 (Umana et al.); и US 2005/0123546 (Umana et al.). Также предложены варианты антител с по меньшей мере одним остатком галактозы в олигосахариде, присоединенном к Fc-области. Такие варианты антител могут иметь улучшенную функцию КЗЦ. Такие варианты антител описаны, например, в WO 1997/30087 (Patel et al.); WO 1998/58964 (Raju, S.); и WO 1999/22764 (Raju, S.).
Варианты Fc-области
[00169] В определенных вариантах реализации изобретения одна или более аминокислотных модификаций могут быть внесены в Fc-область предложенного в данном документе антитела, что позволяет создавать варианты Fc-области. Вариант Fc-области может содержать человеческую последовательность Fc-области (например, Fc-области человеческого IgG1, IgG2, IgG3 или IgG4), содержащую аминокислотную модификацию (например, замену) в одной или более аминокислотных позициях.
[00170] В определенных вариантах реализации в изобретении предусмотрен вариант антитела, который обладает некоторыми, но не всеми эффекторными функциями, которые делают его желаемым кандидатом для применений, в которых важным является время полужизни антитела in vivo, но при этом определенные эффекторные функции (такие как комплементзависимая цитотоксичность и АЗКЦ) являются необязательными или вредными. Анализ in vitro и/или in vivo цитотоксичности можно проводить для подтверждения снижения/прекращения активности КЗЦ и/или АЗКЦ. Например, анализ связывания Fc-рецептора (FcR) можно проводить, чтобы гарантировать, что у антитела отсутствует способность связывать FcγR (и следовательно, отсутствует активность АЗКЦ), но при этом оно сохраняет способность связывать FcRn. Основные клетки, опосредующие АЗКЦ, NK-клетки, экспрессируют только FcγRIII, тогда как моноциты экспрессируют FcγRI, FcγRII и FcγRIII. Данные по экспрессии FcR обобщены в таблице 3 на странице 464 в Ravetch and Kinet, Annu. Rev. Immunol. 9:457-492 (1991). Неограничивающие примеры in vitro анализов для оценки активности АЗКЦ представляющей интерес молекулы описаны в патенте США № 5500362 (смотрите, например, Hellstrom, I. et al. Proc. Nat'l Acad. Sci. USA 83:7059-7063 (1986)) и Hellstrom, I et al, Proc. Nat'l Acad. Sci. USA 82: 1499-1502 (1985); 5821337 (смотрите Bruggemann, M. et al., J. Exp. Med. 166: 1351-1361 (1987)). В альтернативном варианте можно применять методы нерадиоактивного анализа (смотрите, например, нерадиоактивный анализ цитотоксичности ACTI™ для проточной цитометрии (CellTechnology, Inc. Mountain View, CA; и нерадиоактивный анализ цитотоксичности CytoTox 96® (Promega, Madison, WI). Применяемые в таких видах анализа эффекторные клетки включают мононуклеарные клетки периферической крови (МКПК) и естественные клетки-киллеры (NK). В альтернативном или дополнительном варианте активность АЗКЦ представляющей интерес молекулы можно оценивать in vivo, например, в животной модели, как описано в Clynes et al. Proc. Nat'l Acad. Sci. USA 95:652-656 (1998). Также можно проводить анализ связывания C1q для подтверждения того, что антитело не способно связывать C1q и, следовательно, у него отсутствует активность КЗЦ. Смотрите, например, ИФА связывания C1q и C3c в WO 2006/029879 и WO 2005/100402. Чтобы оценить активацию комплемента, можно проводить анализ КЗЦ (смотрите, например, Gazzano-Santoro et al., J. Immunol. Methods 202: 163 (1996); Cragg, M.S. et al., Blood 101: 1045-1052 (2003); и Cragg, M.S. and M.J. Glennie, Blood 103:2738-2743 (2004)). Определение связывания FcRn и in vivo выведения/времени полужизни также можно проводить, используя известные в данной области техники способы (смотрите, например, Petkova, S.B. et al., Int'l. Immunol. 18(12): 1759-1769 (2006)).
[00171] Антитела со сниженной эффекторной функцией включают антитела с заменой одного или более остатков Fc-области 238, 265, 269, 270, 297, 327 и 329 (патент США № 6737056). Такие Fc-мутанты включают Fc-мутантов с заменами в двух или более аминокислотных позициях 265, 269, 270, 297 и 327, включая так называемый Fc-мутант «DANA» с заменой остатков 265 и 297 на аланин (патент США № 7332581).
[00172] Описаны некоторые варианты антител с улучшенным или сниженным связыванием с FcR. (Смотрите, например, патент США № 6737056; WO 2004/056312, и Shields et al., J. Biol. Chem. 9(2): 6591-6604 (2001).)
[00173] В определенных вариантах реализации изобретения вариант антитела содержит Fc-область с одной или более аминокислотными заменами, которые улучшают АЗКЦ, например, заменами в позициях 298, 333 и/или 334 Fc-области (нумерация остатков EU).
[00174] В некоторых вариантах реализации изобретения осуществляют изменения в Fc-области, которые приводят к изменению (т. е. как улучшению, так и снижению) связывания C1q и/или комплементзависимой цитотоксичности (КЗЦ), например, как описано в патенте США № 6194551, WO 99/51642 и Idusogie et al. J. Immunol. 164: 4178-4184 (2000).
[00175] Антитела с повышенным временем полужизни и улучшенным связыванием с неонатальным Fc-рецептором (FcRn), который отвечает за перенос материнских IgG в плод (Guyer et al., J. Immunol. 117:587 (1976) и Kim et al., J. Immunol. 24:249 (1994)), описаны в US2005/0014934A1 (Hinton et al.). Эти антитела содержат Fc-область с одной или более заменами, которые улучшают связывание Fc-области с FcRn. Такие Fc-варианты включают варианты с заменами в одном или более остатках Fc-области: 238, 256, 265, 272, 286, 303, 305, 307, 311, 312, 317, 340, 356, 360, 362, 376, 378, 380, 382, 413, 424 или 434, например, заменой остатки Fc-области 434 (патент США № 7371826).
[00176] Также смотрите Duncan & Winter, Nature 322:738-40 (1988); патент США № 5648260; патент США № 5624821; и WO 94/29351 в отношении других примеров вариантов Fc-области.
Цистеин-сконструированные варианты антител
[00177] В определенных вариантах реализации изобретения может существовать необходимость создания цистеин-сконструированных антител, например, «THIOMAB», в которых один или более остатков антитела замещены остатками цистеина. В конкретных вариантах реализации изобретения замещенные остатки находятся в доступных участках антитела. При замене этих остатков цистеином реактивные тиольные группы оказываются в доступных участках антитела, что можно использовать для конъюгации антитела с другими соединениями, такими как лекарственные соединения или линкерно-лекарственные соединения, для создания иммуноконъюгата, как дополнительно описано в данном документе. В определенных вариантах реализации изобретения любой один или более из следующих остатков могут быть замещены цистеином: V205 (нумерация Kabat) легкой цепи; A118 (нумерация EU) тяжелой цепи; и S400 (нумерация EU) Fc-области тяжелой цепи. Цистеин-сконструированные антитела можно создавать, как описано в патенте США № 7521541.
Производные антител
[00178] В определенных вариантах реализации изобретения предложенное в данном документе антитело может быть дополнительно модифицировано так, чтобы оно содержало дополнительные небелковые соединения, известные в данной области техники и легко доступные. Соединения, подходящие для дериватизации антитела, включают, но не ограничиваются этим, водорастворимые полимеры. Неограничивающие примеры водорастворимых полимеров включают, но не ограничиваются этим, полиэтиленгликоль (ПЭГ), сополимеры этиленгликоля/пропиленгликоля, карбоксиметилцеллюлозу, декстран, поливиниловый спирт, поливинилпирролидон, поли-1,3-диоксолан, поли-1,3,6-триоксан, сополимер этилена/малеинового ангидрида, полиаминокислоты (как гомополимеры, так и случайные сополимеры) и декстран или поли(н-винилпирролидон)полиэтиленгликоль, гомополимеры пропиленгликоля, сополимеры полипропиленоксида/этиленоксида, полиоксиэтилированные полиолы (например, глицерин), поливиниловый спирт и их смеси. Пропиональдегид полиэтиленгликоля может иметь преимущества в производстве благодаря своей стабильности в воде. Полимер может иметь любую молекулярную массу и может быть разветвленным или неразветвленным. Число полимеров, присоединенных к антителу, может варьироваться, и в случае присоединения более одного полимера, они могут представлять собой одинаковые или разные молекулы. В общем случае число и/или тип полимеров, используемых для дериватизации, можно определить на основании факторов, включающих, но не ограничивающихся этим, конкретные свойства или функции антитела, подлежащие улучшению, то, будет ли производное антитела применяться в терапии в определенных условиях, и т. д.
[00179] В другом варианте реализации изобретения предложены конъюгаты антитела и небелкового соединения, которые могут избирательным образом нагреваться при воздействии излучения. В одном варианте реализации изобретения небелковое соединение представляет собой углеродную нанотрубку (Kam et al., Proc. Natl. Acad. Sci. USA 102: 11600-11605 (2005)). Излучение может иметь любую длину волны и включает, но не ограничивается этим, длины волн, которые не вредят обычным клеткам, но которые вызывают нагревание небелкового соединения до температуры, при которой погибают клетки, находящиеся вблизи комплекса антитело-небелковое соединение.
Рекомбинантные способы и композиции
[00180] Антитела можно получать, используя рекомбинантные способы и композиции, например, как описано в патенте США № 4816567. В одном варианте реализации изобретения предложена выделенная нуклеиновая кислота, кодирующая описанное в данном документе антитело. Такая нуклеиновая кислота может кодировать аминокислотную последовательность, составляющую VL, и/или аминокислотную последовательность, составляющую VH антитела (например, легкую и/или тяжелую цепи антитела). В дополнительном варианте реализации изобретения предложены один или более векторов (например, экспрессионных векторов), содержащих такую нуклеиновую кислоту. В дополнительном варианте реализации изобретения предложена клетка-хозяин, содержащая такую нуклеиновую кислоту. В одном таком варианте реализации изобретения клетка-хозяин содержит (например, была трансформирована с помощью): (1) вектор, содержащий нуклеиновую кислоту, которая кодирует аминокислотную последовательность, составляющую VL антитела, и аминокислотную последовательность, составляющую VH антитела, или (2) первый вектор, содержащий нуклеиновую кислоту, которая кодирует аминокислотную последовательность, составляющую VL антитела, и второй вектор, содержащий нуклеиновую кислоту, которая кодирует аминокислотную последовательность, составляющую VH антитела. В одном варианте реализации изобретения клетка-хозяин является эукариотической, например, клеткой яичника китайского хомяка (CHO) или лимфоидной клеткой (например, клеткой Y0, NS0, Sp20). В одном варианте реализации изобретения предложен способ получения антитела, включающий культивирование клетки-хозяина, содержащей нуклеиновую кислоту, кодирующую антитело, как указано выше, в условиях, подходящих для экспрессии антитела, и, необязательно, выделение антитела из клетки-хозяина (или содержащей клетки-хозяев культуральной среды).
[00181] Для рекомбинантного получения антитела нуклеиновую кислоту, кодирующую антитело, например, описанное выше, выделяют и вставляют в один или более векторов для дополнительного клонирования и/или экспрессии в клетке-хозяине. Выделение и секвенирование такой нуклеиновой кислоты легко можно провести, используя традиционные процедуры (например, используя олигонуклеотидные зонды, способные специфически связываться с генами, кодирующими тяжелую и легкую цепи антитела).
[00182] Подходящие клетки-хозяева для клонирования или экспрессии кодирующих антитело векторов включают описанные в данном документе клетки прокариот или эукариот. Например, антитела можно получать в бактериях, в частности, когда отсутствует необходимость в гликозилировании и эффекторной функции Fc. Информацию об экспрессии фрагментов антител и полипептидов в бактериях смотрите, например, в патентах США № 5648237, 5789199 и 5840523. (Также смотрите Charlton, Methods in Molecular Biology, Vol. 248 (B.K.C. Lo, ed., Humana Press, Totowa, NJ, 2003), pp. 245-254, где описана экспрессия фрагментов антител в E. coli.) После экспрессии антитело можно выделять из пасты бактериальных клеток в растворимую фракцию и дополнительно очищать.
[00183] Кроме прокариот, эукариотические микробы, такие как нитевидные фаги или дрожжи, являются подходящими хозяевами для клонирования или экспрессии кодирующих антитело векторов, включая грибы и штаммы дрожжей, чьи пути гликозилирования были «гуманизированы», что приводит к получению антитела с частично или полностью человеческим профилем гликозилирования. Смотрите Gerngross, Nat. Biotech. 22: 1409-1414 (2004) и Li et al., Nat. Biotech. 24:210-215 (2006).
[00184] Подходящие клетки-хозяева для экспрессии гликозилированного антитела также получают из многоклеточных организмов (беспозвоночных и позвоночных). Примеры клеток беспозвоночных включают клетки растений и насекомых. Были определены многочисленные бакуловирусные штаммы, которые можно использовать в сочетании с клетками насекомых, в частности, для трансфекции клеток Spodoptera frugiperda.
[00185] Также в качестве хозяев можно использовать культуры растительных клеток. Смотрите, например, патенты США № 5959177, 6040498, 6420548, 7125978 и 6417429 (где описана технология PLANTIBODIESTM для получения антител в трансгенных растениях).
[00186] Также в качестве хозяев можно использовать клетки позвоночных. Например, можно использовать линии клеток млекопитающих, адаптированных для роста в суспензии. Другими примерами применимых линий клеток млекопитающих являются линия клеток почки обезьяны CV1, трансформированная SV40 (COS-7); линия клеток эмбриональной почки человека (293 или клетки 293, описанные, например, в Graham et al., J. Gen Virol. 36:59 (1977)); клетки почки новорожденного хомяка (BHK); клетки Сертоли мышей (клетки TM4, описанные, например, в Mather, Biol. Reprod. 23:243-251 (1980)); клетки почки обезьяны (CV1); клетки почки африканской зеленой мартышки (VERO-76); клетки карциномы шейки матки человека (HELA); клетки почки собаки (MDCK); клетки печени серой крысы (BRL 3A); клетки легких человека (W138); клетки печени человека (Hep G2); клетки опухоли молочной железы мышей (MMT 060562); клетки TRI, описанные, например, в Mather et al., Annals N.Y. Acad. Sci. 383:44-68 (1982); клетки MRC 5; и клетки FS4. Другие применимые линии клеток-хозяев млекопитающих включают клетки яичника китайского хомяка (CHO), включая клетки DHFR-CHO (Urlaub et al., Proc. Natl. Acad. Sci. USA 77:4216 (1980)); и линии клеток миеломы, такие как Y0, NS0 и Sp2/0. Обзор некоторых линий клеток-хозяев млекопитающих, подходящих для получения антител, смотрите, например, в Yazaki and Wu, Methods in Molecular Biology, Vol. 248 (B.K.C. Lo, ed., Humana Press, Totowa, NJ), pp. 255-268 (2003).
Фармацевтические готовые формы
[00187] Фармацевтические готовые формы, также называемые в данном документе фармацевтическими композициями, анти-IL-13 антитела, описанного в данном документе, можно готовить, смешивая такое антитело или молекулу, имеющие необходимую степень чистоты, с одним или более необязательными фармацевтически приемлемыми носителями (Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980)), в лиофилизированной форме или в виде водных растворов. Фармацевтически приемлемые носители в общем случае являются нетоксичными для реципиентов в применяемых дозировках и концентрациях и включают, но не ограничиваются этим: буферы, например, фосфатный, цитратный и на основе других органических кислот; антиоксиданты, включая аскорбиновую кислоту и метионин; консерванты (такие как октадецилдиметилбензиламмония хлорид; гексаметония хлорид; бензалкония хлорид; бензетония хлорид; фенол, бутил или бензиловый спирт; алкилпарабены, такие как метил- или пропилпарабен; катехол; резорцинол; циклогексанол; 3-пентанол; и м-крезол); низкомолекулярные (менее чем около 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатирующие агенты, такие как ЭДТА; сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как натрий; комплексы металлов (например, комплексы Zn-белок); и/или неионные поверхностно-активные вещества, такие как полиэтиленгликоль (ПЭГ). Типовые фармацевтически приемлемые носители согласно данному документу включают агенты для диспергирования лекарственных препаратов в интерстициальном пространстве, такие как растворимые нейтрально-активные гликопротеины на основе гиалуронидазы (sHASEGP), например, человеческие растворимые гликопротеины гиалуронидазы PH-20, такие как rHuPH20 (HYLENEX®, Baxter International, Inc.). Некоторые типовые sHASEGP и способы их применения, включая rHuPH20, описаны в патентных публикациях США № 2005/0260186 и 2006/0104968. В одном аспекте sHASEGP комбинируют с одной или более другими глюкозаминогликаназами, такими как хондроитиназы.
[00188] Типовые лиофилизированные готовые формы антител описаны в патенте США № 6267958. Водные готовые формы антител включают описанные в патенте США № 6171586 и WO2006/044908, причем последние готовые формы содержат гистидин-ацетатный буфер.
[00189] Готовые формы согласно данному документу также могут содержать более одного активного ингредиента в случае необходимости для конкретного подлежащего лечению показания, предпочтительно ингредиенты с взаимодополняющей активностью, которые не оказывают друг на друга негативного влияния. Например, может существовать необходимость дополнительно обеспечить контроллер с ингибитором пути Th2. Такие активные ингредиенты присутствуют в комбинации в количествах, эффективных для предполагаемой цели.
[00190] Активные ингредиенты могут быть заключены в микрокапсулах, приготовленных, например, путем коацервации или межфазной полимеризации, например, гидроксиметилцеллюлозные или желатиновые микрокапсулы и поли-(метилметакрилатные) микрокапсулы, соответственно, в коллоидных системах доставки лекарственных препаратов (например, липосомы, альбуминовые микросферы, микроэмульсии, наночастицы и нанокапсулы) или в макроэмульсиях. Такие методики описаны в Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980).
[00191] Можно готовить препараты с продленным высвобождением. Подходящие примеры препаратов с продленным высвобождением включают полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие антитело, при этом матрицы имеют вид формованных изделий, например, пленок или микрокапсул.
[00192] Готовые формы для применения для in vivo введения в общем случае являются стерильными. Стерильность можно без труда обеспечивать, например, путем фильтрации через стерильные фильтровальные мембраны.
Терапевтические способы и композиции
[00193] В определенных вариантах реализации изобретения предложены способы лечения атопического дерматита, включающие введение терапевтически эффективного количества лебрикизумаба пациенту с атопическим дерматитом, при этом лечение приводит к снижению тяжести заболевания согласно определению или оценке по критерию оценки тяжести заболевания для атопического дерматита (ADDSOM). В некоторых вариантах реализации изобретения атопический дерматит является умеренным или тяжелым согласно определению с помощью оценки по шкале Райка/Лангеланда. Оценка по шкале Райка/Лангеланда от 4,5 до 9, как правило, считается соответствующей атопическому дерматиту от умеренного до тяжелого. В некоторых вариантах реализации изобретения способы дополнительно включают введение одного или более местных кортикостероидов. Местные кортикостероиды можно вводить до (т. е. перед) введения антагониста IL-13, одновременно с введением антагониста IL-13 или после введения антагониста IL-13. Примеры местных кортикостероидов включают, но не ограничиваются этим, триамцинолона ацетонид, гидрокортизон и комбинацию триамцинолона ацетонида и гидрокортизона. Триамцинолона ацетонид, как правило, готовят в концентрации 0,1% в креме, а гидрокортизон, правило, готовят в концентрации 1% – 2,5% в креме. Определенные местные кортикостероиды считаются очень эффективными, такие как, например, бетаметазона дипропионат, клобетазола пропионат, дифлоразондиацетат, флуоцинонид и галобетазола пропионат. Определенные местные кортикостероиды считаются высокоэффективными, такие как, например, амцинонид, дезоксиметазон, галцинонид и триамцинолона ацетонид. Определенные местные кортикостероиды считаются среднеэффективными, такие как, например, бетаметазона валерат, клокортолона пивалат, флуоцинолона ацетонид, фурандренолид, флуоцинонид, флутиказона пропионат, гидрокортизона бутират, гидрокортизона валерат, мометазона фуроат и предникарбат. Определенные местные кортикостероиды считаются низкоэффективными, такие как, например, алклометазона дипропионат, дезонид и гидрокортизон. МКС можно применять к пораженным участкам один раз в сутки, два раза в сутки, три раза в сутки или по мере необходимости. В некоторых вариантах реализации изобретения пациент не достигает адекватного контроля при применении местных кортикостероидов.
[00194] В данной области техники известны различные ADDSOM, которые описаны в данном документе в дополнение к новому опроснику по влиянию атопического дерматита (ADIQ). Типовые ADDSOM включают, но не ограничиваются этим, «индекс распространенности и тяжести экземы» (EASI), «оценку тяжести атопического дерматита» (SCORAD), «общую оценку исследователем» (IGA), «общую оценку признаков исследователем» (IGSA), оценку атопического дерматита Райка/Лангеланда и оценку результатов пациентами, включая, но не ограничиваясь этим, визуальную аналоговую шкалу зуда (аспект тяжести заболевания, оцениваемый как составляющая SCORAD), визуальную аналоговую шкалу бессонницы (аспект тяжести заболевания, оцениваемый как составляющая SCORAD), дневник симптомов атопического дерматита (ADSD), опросник по влиянию атопического дерматита (ADIQ), дерматологический индекс качества жизни (DLQI) (Finlay and Khan, Clin Exper Dermatol 1994; 19:210) и 5-D шкалу чесотки (Elman et al., Br J Dermatol 2010; 162(3):587-593).
[00195] Предложенный в данном документе терапевтический агент можно вводить любыми подходящими путями, включая парентеральный, подкожный, внутрибрюшинный, внутрилегочный и интраназальный. Парентеральные инфузии включают внутримышечное, внутривенное, внутриартериальное, внутрибрюшинное или подкожное введение. В определенных вариантах реализации изобретения дозирование осуществляют с помощью инъекций, например, внутривенных или подкожных инъекций. В другом варианте реализации изобретения терапевтический агент вводят, используя шприц (например, предварительно наполненный или нет) или автоинжектор. В определенных вариантах реализации изобретения терапевтический агент применяют местным образом.
[00196] В определенных вариантах реализации изобретения анти-IL13 антитело согласно изобретению вводят, используя, например, устройство для самостоятельного проведения инъекции, автоинжектор или другое устройство, сконструированное для самостоятельного введения. В определенных вариантах реализации изобретения анти-IL13 антитело согласно изобретению вводят, используя устройство для подкожного введения. В данной области техники известны и коммерчески доступны различные устройства самостоятельного проведения инъекции и устройства для подкожного введения, включая автоинжектор. Типовые устройства включают, но не ограничиваются этим, предварительно наполненные шприцы (такие как BD HYPAK SCF®, READYFILL™ и STERIFILL SCF™ от Becton Dickinson; предварительно наполненные шприцы из сополимера CLEARSHOT™ от Baxter; и предварительно наполненные шприцы Daikyo Seiko CRYSTAL ZENITH®, доступные от West Pharmaceutical Services); одноразовые шприцы-ручки, такие как BD Pen от Becton Dickinson; ультраострые и микроигольные устройства (такие как INJECT-EASE™ и микроинфузионные устройства от Becton Dickinson; и H-PATCH™, доступные от Valeritas), а также безыгольные устройства для инъекций (такие как BIOJECTOR® и IJECT®, доступные от Bioject; и SOF-SERTER® и устройства на основе пластырей, доступные от Medtronic). В определенных вариантах реализации изобретения анти-IL13 антитело согласно изобретению вводят, используя автоинжектор, состоящий из различных компонентов, например, как описано в патентных публикациях США №№ 2014/0114247, 2014/0148763 и 2013/0131590; патентах США №№ 7597685, 7896850 и 8617109, которые все включены посредством ссылки. Предусмотрена возможность совместных готовых форм или совместного введения с помощью таких устройств самостоятельного проведения инъекции или устройств для подкожного введения анти-IL13 антитела с по меньшей мере вторым терапевтическим соединением.
[00197] В случае предотвращения или лечения заболевания подходящая дозировка антитела согласно изобретению (при отдельном применении или в комбинации в одним или более другими дополнительными терапевтическими агентами) будет зависеть от типа заболевания, подлежащего лечению, типа антитела, тяжести и течения заболевания, от того, вводят ли антитело в превентивных или терапевтических целях, предварительной терапии, анамнеза пациента и его ответа на антитело, а также от решения лечащего врача. Антитело удобно вводить пациенту за один раз или в течение некоторого количества приемов. В определенных вариантах реализации изобретения антитело согласно изобретению вводят в виде постоянной дозы (а не дозы на основе массы), составляющей 125 мг, или 250 мг, или около 125 мг, или около 250 мг, или постоянной дозы от 110 мг до 140 мг, или постоянной дозы от 120 мг до 130 мг, или постоянной дозы от 225 мг до 275 мг, или постоянной дозы от 240 мг до 260 мг. В определенных вариантах реализации изобретения дозу вводят путем подкожной инъекции один раз в четыре недели или один раз в восемь недель в течение некоторого периода времени. В определенных вариантах реализации изобретения период времени составляет 12 недель, или 20 недель, или 24 недели, или 9 месяцев, или один год, два года, пять лет, десять лет, 15 лет, 20 лет, или в течение всей жизни пациента. Прогресс этой терапии легко отслеживать с помощью стандартных методик и анализов. В определенных вариантах реализации изобретения пациент имеет умеренный или тяжелый атопический дерматит и принимает МКС, в случае необходимости, во время лечения антителом согласно изобретению. В определенных вариантах реализации изобретения МКС применяют раз в сутки или два раза в сутки, или три раза в сутки или более. В определенных вариантах реализации изобретения пациент способен снижать употребление МКС в течение времени введения антитела согласно изобретению. Такое снижение употребления МКС называется «снижением дозы» или «снижением потребности в МКС». Период времени, соответствующий сниженному употреблению, может составлять одну, две, три или четыре недели, или два, три, четыре, пять или шесть месяцев.
[00198] Антагонисты IL-13 согласно изобретению, в соответствии с некоторыми вариантами его реализации, включают введение в комбинации с одним или более дополнительными терапевтическими агентами. Термин «в комбинации с» означает, что дополнительный (-ые) терапевтический (-ие) агент (-ы) вводят до, после или одновременно с фармацевтической композицией, содержащей антагонист IL-13. Термин «в комбинации с» также включает последовательное или одновременное введение антагониста IL-13 и второго терапевтического агента.
[00199] Например, в случае введения «до» фармацевтической композиции, содержащей антагонист IL-13, дополнительный терапевтический агент можно вводить за около 72 часа, около 60 часов, около 48 часов, около 36 часов, около 24 часов, около 12 часов, около 10 часов, около 8 часов, около 6 часов, около 4 часа, около 2 часа, около 1 часа, около 30 минут, около 15 минут или около 10 минут до введения фармацевтической композиции, содержащей антагонист IL-13. Например, в случае введения «после» фармацевтической композиции, содержащей антагонист IL-13, дополнительный терапевтический агент можно вводить через около 10 минут, около 15 минут, около 30 минут, около 1 часа, около 2 часа, около 4 часа, около 6 часов, около 8 часов, около 10 часов, около 12 часов, около 24 часа, около 36 часов, около 48 часов, около 60 часов или около 72 часа после введения фармацевтической композиции, содержащей антагонист IL-13. Введение «одновременно» с фармацевтической композицией, содержащей антагонист IL-13, означает, что дополнительный терапевтический агент вводят субъекту в отдельной дозированной форме в течение менее чем 5 минут (до, после или в одно время) относительно введения фармацевтической композиции, содержащей антагонист IL-13, или вводят субъекту в одной комбинированной дозированной форме, содержащей как дополнительный терапевтический агент, так и антагонист IL-13.
[00200] Дополнительный терапевтический агент может представлять собой, например, другой антагонист IL-13, антагонист IL-4R, антагонист IL-1, антагонист IL-6, антагонист IL-6R, антагонист TNF, антагонист IL-8, антагонист IL-9, антагонист IL-17, антагонист IL-5, антагонист IgE, антагонист CD48, антагонист IL-31, антагонист тимусного стромального лимфопоэтина (TSLP), антибиотики на основе интерферона-гамма (IFN.gamma.), местные кортикостероиды, такролимус, пимекролимус, микофенолат, циклоспорин, азатиоприн, метотрексат, кромолин-натрий, ингибиторы протеиназы или их комбинации. В определенных вариантах реализации изобретения фармацевтическую композицию, содержащую антагонист IL-13, вводят субъекту в сочетании с пероральным иммуносупрессантом, местным ингибитором кальциневрина, таким как, например, пимекролимус или такролимус, или пероральным кортикостероидом, таким как, например, преднизолон. В определенных вариантах реализации изобретения фармацевтическую композицию, содержащую антагонист IL-13, вводят субъекту в сочетании с не фармацевтической терапией, такой как терапия ультрафиолетовым (УФ) светом.
[00201] В определенных вариантах реализации изобретения антитело согласно изобретению вводят один или более раз в виде постоянной (т. е. не зависящей от массы) ударной дозы с последующим введением, один или более раз, в виде постоянной поддерживающей дозы. В определенных вариантах реализации изобретения ударная доза составляет 250 мг или 500 мг, а поддерживающая доза составляет 125 мг. В определенных вариантах реализации изобретения вводят две ударные дозы по 250 мг с интервалом в 15 суток или 29 суток. В определенных вариантах реализации изобретения поддерживающую дозу вводят через две недели после последней ударной дозы или через четыре недели после последней ударной дозы, и после этого – каждые четыре недели. В некоторых вариантах реализации изобретения ударная доза составляет 500 мг, а поддерживающая доза составляет 250 мг. В некоторых вариантах реализации изобретения поддерживающую дозу в 250 мг вводят через четыре недели после ударной дозы, и после этого – каждые четыре недели или каждые восемь недель.
Изделия
[00202] В дополнительном аспекте изобретения предложено изделие, содержащее материалы, применимые для лечения, предотвращения и/или диагностики описанных выше расстройств. Изделие содержит контейнер и этикетку или упаковочный вкладыш, находящиеся на контейнере или прилагающиеся к нему. Подходящие контейнеры включают, например, бутылки, флаконы, шприцы, пакеты для В/В растворов, автоинжекторы и т. д. Контейнеры могут быть выполнены из разных материалов, таких как стекло или пластик. Контейнер содержит композицию, которая находится сама по себе или в комбинации с другой композицией, эффективную для лечения, предотвращения и/или диагностирования патологического состояния, и может иметь стерильное отверстие для доступа (например, контейнер может представлять собой пакет для внутривенного раствора или флакон с пробкой, прокалываемой гиподермической иглой для инъекций). По меньшей мере один активный агент в композиции представляет собой антитело согласно изобретению. На этикетке или упаковочном вкладыше указано, что композицию используют для лечения данного патологического состояния. Кроме того, изделие может содержать (а) первый контейнер с находящейся в нем композицией, при этом композиция содержит антитело согласно изобретению; и (b) второй контейнер с находящейся в нем композицией, при этом композиция содержит дополнительный терапевтический агент. В этом варианте реализации изобретения изделие может дополнительно содержать упаковочный вкладыш, на котором указано, что композицию можно использовать для лечения конкретного патологического состояния. В альтернативном или дополнительном варианте изделие может дополнительно содержать второй (или третий) контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (БВДИ), фосфатно-солевой буфер, раствор Рингера и раствор декстрозы. Он может дополнительно содержать материалы, необходимые с коммерческой и пользовательской точки зрения, включая другие буферы, разбавители, фильтры, иглы, шприцы и автоинжекторы.
ПРИМЕРЫ
ПРИМЕР 1 – Анти-IL-13 антитела
[00203] Анти-IL-13 антитела, включая моноклональные, гуманизированные анти-IL-13 антитела, были описаны ранее (смотрите, например, WO2005062967). Одним моноклональным гуманизированным анти-IL-13 антителом, описанным в данном документе, является лебрикизумаб, антитело IgG4, также смотрите таблицу с перечнем последовательностей. Лебрикизумаб применяли в описанных ниже исследованиях, приготовленный в концентрации 125 мг/мл в фармацевтически приемлемой композиции, поставляемой в предварительно наполненном шприце.
ПРИМЕР 2 – Клиническое исследование I
[00204] Клиническое исследование I представляло собой рандомизированное, двойное слепое, плацебо-контролируемое исследование фазы II для оценки безопасности и эффективности лебрикизумаба у пациентов (возрастом 18-75 лет) с персистентным умеренным или тяжелым атопическим дерматитом, который адекватно не контролируется местными кортикостероидами (МКС). В этом исследовании лебрикизумаб исследовали в комбинации с МКС. Схема исследования приведена на Фиг. 1.
[00205] Скрининг. Пациенты, подлежащие включению в исследование должны были иметь умеренный или тяжелый АД, присутствующий в течение по меньшей мере 1 года, и должны были соответствовать всем критериям включения. Оценку пациентов проводили во время скрининга для того, чтобы оценить пригодность для включения на этап вводного периода, и снова в конце вводного периода для того, чтобы оценить пригодность для включения на этап периода лечения и рандомизации в отношении исследуемого препарата.
[00206] Критерии включения были следующими: возраст от 18 до 75 лет; АД, диагностированный по критериям Hanifin/Rajka, присутствующий в течение по меньшей мере 1 года, на момент скрининга; АД от умеренного до серьезного по градации в соответствии со шкалой Райка/Лангеланда на момент скрининга; анамнез отсутствия адекватного ответа на длящуюся ≥ 1 месяца (в течение 3 месяцев перед скрининговым визитом) схему лечения с по меньшей мере ежесуточным применением МКС и регулярным применением смягчающих средств для лечения АД; оценка EASI ≥ 14 на момент скрининга и в конце вводного периода (визит 3), оценка IGA ≥ 3 (5-точечная шкала) на момент скрининга и в конце вводного периода (визит 3); распространение АД на ≥ 10% площади поверхности тела (ППТ) на момент скрининга; и оценка ВАШ зуда (определяемая как часть SCORAD) ≥ 3 на момент скрининга.
[00207] Критерии исключения были следующими: применение в прошлом и/или настоящем любой терапии на основе анти-IL-13 или анти-IL-4/IL-13, включая лебрикизумаб; применение исследуемого агента в течение 4 недель перед скринингом или в течение 5 периодов полужизни исследуемого агента, в зависимости от того, какой из периодов больше; анамнез тяжелой аллергической реакции или анафилактической реакции на биологический агент или подтвержденная гиперчувствительность к любому компоненту инъекции лебрикизумаба; гиперчувствительность к МКС или к любым другим ингредиентам, содержащимся в МКС-продукте, применяемом в исследовании; применение каких-либо дополняющих, альтернативных или гомеопатических лекарственных средств, включая, но не ограничиваясь этим, фитотерапию, традиционные или нетрадиционные растительные лекарственные средства, незаменимые жирные кислоты или акупунктуру, в течение 7 суток перед вводным периодом или необходимость в таких лекарственных средствах во время лечения; масса тела < 40 кг или индекс массы тела > 38 кг/м2; признаки других кожных патологий, включая, но не ограничиваясь этим, Т-клеточную лимфому или аллергический контактный дерматит; признаки или текущее лечение (включая лечение местными антибиотиками) активной кожной инфекции на момент скрининга (Сутки -15); определенные инфекции; анамнез недавних (< 1 года) паразитарных инфекций, в особенности вызванных нематодами (например, Ascaris, Ancylostoma), платигельминтами (например, Schistosoma), или анамнез листериозных инфекций; активная форма туберкулеза, требующая лечения, в течение 12 месяцев перед визитом 1; признаки острого или хронического гепатита или подтвержденный цирроз печени; подтвержденный иммунодефицит, включая инфекцию ВИЧ; применение МИК (местного ингибитора кальциневрина) во время скрининга; посещение солярия в течение 4 недель перед скрининговым визитом; иммунотерапия аллергенами в течение 3 месяцев от скрининга; получение живой аттенуированной вакцины в течение 4 недель перед исходным визитом (сутки 1); запланированная хирургическая операция во время исследования; клинически значимая аномалия при исследовании ЭКГ или лабораторных исследованиях (гематология, биохимический анализ сыворотки и анализ мочи); повышение АСТ, АЛТ или общего билирубина ≥ 2,0 x относительно верхнего предела нормы (ВПН) во время скрининга; подтвержденное текущее злокачественное образование или текущее исследование в отношении потенциального злокачественного образования, включая базально- или плоскоклеточную карциному кожи или карциному in situ; анамнез злокачественного образования в течение 5 лет перед скринингом, за исключением должным образом вылеченной карциномы in situ шейки матки, немеланомной карциномы кожи; рака матки стадии I; и другие клинически значимые заболевания, неконтролируемые несмотря на лечение.
[00208] Вводный период. После скринингового визита подходящие пациенты проходили 2-недельный вводный период (сутки от -14 до -1), во время которого инициировали установленную протоколом схему применения местной терапии. На первые сутки вводного периода (сутки -14) пациенты получали МКС для применения в течение всего вводного периода. Пациентам было необходимо применять следующую схему местной терапии ежесуточно: смягчающее средство ко всем ксеротическим участкам кожи по меньшей мере один раз в сутки; крем с содержанием МКС, состоящий из МКС средней эффективности (0,1% триамцинолона ацетонид), два раза в сутки, только в местах активных кожных поражений. В случае поражений лица или опрелых участков можно было использовать МКС низкой эффективности (крем 2,5% гидрокортизона) вместо 0,1% триамцинолона ацетонида с разрешения исследователя.
[00209] В конце вводного периода проводили вторую оценку пригодности для рандомизации в отношении исследуемого лекарственного препарата (лебрикизумаб или плацебо), а пациенты должны были продолжать демонстрировать умеренный или тяжелый АД, определяемый по оценке EASI ≥ 14 и оценке IGA ≥ 3.
[00210] Период лечения. В конце вводного периода рандомизировали пациентов, которые: 1) демонстрировали соблюдение установленной протоколом схемы приема МКС и 2) которые продолжали отвечать описанным выше критериям пригодности. Планировалось рандомизировать приблизительно 200 пациентов (1:1:1:1) в одну из следующих групп лечения (смотрите Фиг. 1): (1) Группа 1: лебрикизумаб, 250 мг, вводимый подкожно (П/К), одна доза (сутки 1) с последующими 2 дозами плацебо (на неделю 4 и на неделю 8), всего в количестве 3 доз + крем МКС; (2) Группа 2: лебрикизумаб, 125 мг П/К, одна доза (сутки 1) с последующими 2 дозами плацебо (на неделю 4 и на неделю 8), всего в количестве 3 доз + крем МКС; (3) Группа 3: лебрикизумаб, 125 мг П/К, раз в 4 недели (Р4Н), всего в количестве 3 доз + крем МКС; и (4) Группа 4: плацебо П/К, Р4Н, всего в количестве 3 доз + крем МКС. Три активные дозы были включены, чтобы получить характеристики как взаимосвязи воздействие – ответ, так и требований к частоте дозирования.
[00211] Пациенты, рандомизированные в исследовании по этим четырем группам лечения, получали исследуемый лекарственный препарат (лебрикизумаб или плацебо П/К) в комбинации с МКС. В течение 12-недельного периода лечения эти пациенты продолжали применять смягчающее средство по меньшей мере один раз в сутки и крем 0,1% триамцинолона ацетонида, как описано для вводного периода (два раза в сутки ко всем активным поражениям кожи на теле). В случае поражений лица или опрелых участков можно было использовать МКС низкой эффективности (крем 2,5% гидрокортизона) вместо 0,1% триамцинолона ацетонида с разрешения исследователя. Пациенты делали записи о своем применении местных кремов и отвечали на относящиеся к PRO вопросы, используя заполняемый вручную электронный дневник eDiary в продолжении всего периода лечения. При каждом визите в ходе исследования пациенты должны были приносить свои контейнеры с МКС в исследовательский центр для взвешивания.
[00212] Период последующего наблюдения в целях безопасности. Всех пациентов, которые завершили лечение согласно исследованию во время плацебо-контролируемого периода лечения (недели 1-12), наблюдали в целях безопасности в течение дополнительных 8 недель (недели 13-20). Во время этого периода последующего наблюдения в целях безопасности пациенты больше не должны были применять местную схему лечения, предписанную во время вводного периода и периода лечения. Вместо этого крем 0,1% триамцинолона ацетонида и/или крем 2,5% гидрокортизона можно было применять на усмотрение пациента и исследователя. При каждом визите в ходе исследования пациенты должны были приносить свои контейнеры с МКС в исследовательский центр для взвешивания. Пациентов поощряли к продолжению применения смягчающих средств к ксеротическим участкам кожи по меньшей мере один раз в сутки. Допускалось повышение дозы в терапии АД в любое время в течение исследования, если, по мнению исследователя, это было клинически показано.
[00213] В любое время в течение этого исследования не допускалось применение местных ингибиторов кальциневрина (МИК). Пациентам, которые применяли МИК во время скрининга, разрешалось участвовать в исследовании, если они соглашались прекратить применение МИК на время исследования, и если, по мнению исследователя, было безопасно прекратить применение МИК и использовать вместо этого предписанный протоколом крем МКС.
[00214] Обоснование выбора дозы и схемы применения лебрикизумаба. Доза лебрикизумаба для АД была основана на имеющихся клинических данных по программе для астмы и аналогии в биологии IL-13 и ожидаемых ФК-характеристиках лебрикизумаба в двух популяциях пациентов. Пациенты как с АД, так и с астмой имели повышенные уровни биомаркеров, связанных с биологией IL-13, и опосредованную Th-2 гиперчувствительность. В частности, IL-13 повышен в легких пациентов с астмой и играет патогенную роль при астме, тогда как недавно появились соответствующие данные о повышенной экспрессии IL-13 в пораженной АД коже.
[00215] Кроме того, учитывая, что основным механизмом выведения лебрикизумаба является неспецифический эндоцитоз и катаболизм с минимальным вкладом со стороны опосредованного мишенью выведения, ожидается, что ФК-характеристики лебрикизумаба будут аналогичными у пациентов с астмой и АД. Хотя в литературе имеется ограниченная информация относительно распределения моноклональных антител по различным тканям, доступные данные по животным моделям позволяют предположить аналогичные коэффициенты распределения в плазме и тканях для моноклональных антител в тканях легких и кожи (Shah DK et al., mAbs 2013; 5(2):297-305). Соответственно, уровни доз лебрикизумаба, приводящие к улучшению клинических показателей при астме, также могут демонстрировать биологическую активность у пациентов с АД.
[00216] Как описано выше, в этом исследовании фазы II исследовали три разных схемы дозирования: (1) доза 125 мг Р4Н (Группа 3) была включена в качестве потенциально эффективной дозы. 125 мг Р4Н представлял схему с наибольшей дозой (т. е. наибольшим общим воздействием), которую исследовали в базовых исследованиях фазы III применения лебрикизумаба взрослыми пациентами с астмой (LAVOLTA I и II), проводимых на момент начала исследования. Учитывая ожидаемую аналогию в роли IL-13 и фармакокинетике между астмой и АД, предположили, что эта доза могла бы быть эффективной для пациентов с АД; (2) однократная 250 мг доза (Группа 1) была предложена в качестве альтернативной потенциально эффективной дозы. Ожидалось, что эта доза будет приводить к меньшему общему воздействию, чем 125 мг Р4Н в течение первых 12 недель. Установив определение первичного измеряемого параметра на 12 неделе, исследовали однократную 250 мг дозу, чтобы выяснить потенциал поквартального дозирования; и (3) однократная 125 мг доза (Группа 2) была предложена в качестве частично эффективной дозы. Учитывая, что для этой дозы ожидалось, что общее воздействие будет в 2-3 раза меньше, чем при схемах с применением 125 мг Р4Н и однократной дозы 250 мг, данные по всем трем группам дозирования можно использовать, чтобы получить характеристики взаимосвязи воздействие-ответ для лебрикизумаба у пациентов с АД.
[00217] Однако, как описано выше, результаты исследований LAVOLTA I и II не были согласующимися. Hanania et al., Lancet Respir Med 2016, доступная на dx(dot)doi(dot)org(slash)S2213-2600(16)30265-X, опубликованная 5 сентября 2016 г. Оба исследования не смогли продемонстрировать четкой зависимости доза-ответ, хотя результаты по фармакокинетике и фармакодинамике согласовались с результатами исследований фазы II, описанных выше, что указывает на аналогичное воздействие лекарственного препарата и ингибирование пути IL-13. Hanania et al., 2016, выше. Соответственно, не было уверенности в том, что лебрикизумаб будет обеспечивать клинически значимое улучшение показателей у пациентов с атопическим дерматитом при применении описанных в данном документе схем дозирования.
[00218] Первичный измеряемый параметр в исследовании фазы II определяли на 12 неделе. Ожидалось, что средняя сывороточная концентрация лебрикизумаба на 12 неделе будет составлять более 90% стационарного значения для пациентов, получающих 125 мг Р4Н. Кроме того, в случае дозы 125 мг Р4Н прогнозируемая остаточная концентрация на 12 неделе будет находиться в рамках диапазона, в котором лебрикизумаб демонстрировал биологическую активность в клинических исследованиях астмы фазы II. В случае однократной дозы 250 мг прогнозируемое среднее воздействие в течение 12 недель превышает воздействие при применении схемы 37,5 мг Р4Н, которая также демонстрировала биологическую активность в клинических исследованиях астмы фазы II; Hanania et al, Thorax 2015; 70:748-756.
[00219] Обоснование для контрольной группы. Пациенты в 4-ой группе лечения получали плацебо П/К + крем МКС и, следовательно, служили в качестве активной сравнительной группы. Эту группу лечения использовали в качестве контроля для П/К применения лебрикизумаба для того, чтобы точно оценить эффективность и эффект безопасности лебрикизумаба в качестве вспомогательной терапии с МКС.
[00220] Активная сравнительная МКС-группа была включена, так как применение МКС является стандартом лечения в клинической практике и продолжается, когда начинается усиливающаяся терапия (например, системными агентами) (руководства Европейской академии дерматовенерологии [EADV] 2009; Darsow et al, J Eur Acad Dermatol Venereol 2010; 24:317-28). Кроме того, прекращение применения МКС у пациентов с персистентным умеренным или тяжелым заболеванием может привести к существенному ухудшению симптомов АД и бремени заболевания.
[00221] Критерии результатов эффективности. Первичным критерием результатов эффективности был процент пациентов, достигших EASI-50 (50% снижения оценки EASI по сравнению с исходным уровнем) на 12 неделе. Вторичные критерии результатов эффективности в этом исследовании были следующими: (i) процентное и абсолютное изменение относительно исходного уровня оценки EASI на 12 неделе; (ii) процент пациентов, достигших 75% снижения относительно исходного уровня оценки EASI (EASI-75) на 12 неделе; (iii) процент пациентов, достигших оценки IGA 0 или 1 на 12 неделе; (iv) процент пациентов со снижением на ≥ 2 пунктов относительно исходного уровня в IGA на 12 неделе; (v) абсолютное изменение относительно исходного уровня в IGA на 12 неделе; (vi) процент пациентов, достигших оценки IGSA 0 или 1 на 12 неделе; (vii) процент пациентов со снижением на ≥ 2 пунктов относительно исходного уровня в IGSA на 12 неделе; (viii) абсолютное изменение относительно исходного уровня в IGSA на 12 неделе; (ix) процентное и абсолютное изменение относительно исходного уровня в SCORAD на 12 неделе; (x) процент пациентов со снижением на 50% или 75% относительно исходного уровня в SCORAD-50/75 на 12 неделе; (xi) процент пациентов, достигших EASI-50 на 12 неделе и сохраняющих EASI-50 на 16 и 20 неделе; (xii) процент пациентов, достигших оценки IGA 0 или 1 на 12 неделе и сохраняющих оценку IGA 0 или 1 на 16 и 20 неделе; (xiii) процент пациентов, достигших оценки IGSA 0 или 1 на 12 неделе и сохраняющих оценку IGSA 0 или 1 на 16 и 20 неделе; (xiv) процент пациентов, достигших SCORAD-50 на 12 неделе и сохраняющих SCORAD-50 на 16 и 20 неделе; (xv) процентное изменение относительно исходного уровня в общем % пораженной площади поверхности тела (ППТ) на 12 неделе; (xvi) абсолютное и процентное изменение относительно исходного уровня в зуде, определяемое по ВАШ зуда (оцениваемое в качестве части SCORAD) на 12 неделе; (xvii) абсолютное и процентное изменение относительно исходного уровня в зуде, определяемое по 5-D шкале чесотки на 12 неделе; (xviii) общее употребление (в граммах) МКС относительно исходного уровня к 12 неделе; (xvix) общее употребление (в граммах) МКС от 12 недели до окончания исследования или раннего прекращения исследования; (xx) число случаев обострения заболевания относительно исходного уровня к 12 неделе; (xxi) изменение в симптомах АД относительно исходного уровня к 12 неделе, оцениваемое по ADSD; (xxii) изменение в связанных со здоровьем пунктах опросника по КЖ относительно исходного уровня к 12 неделе, оцениваемое по ADIQ; и (xxiii) изменение в связанных со здоровьем пунктах опросника по КЖ относительно исходного уровня к 12 неделе, оцениваемое по DLQI.
[00222] Критерии результатов безопасности. Критерии результатов безопасности в этом исследовании были следующими: (i) частота и тяжесть нежелательных явлений, возникших в ходе лечения; (ii) количество человеческих антител против терапевтического средства (АПТС) на исходном уровне и во время исследования; (iii) частота и тяжесть инфекций кожи и других систем органов во время исследования; клиническое определение инфекции кожи было следующим: диагноз инфекции кожи ставили на основании клинической оценки исследователя, включая, но не ограничиваясь этим, наличие корок цвета меда, серозные выделения, пустулы или боль в месте сыпи, которые могут быть связаны с системными характеристиками, включая повышение температуры или обострение заболевания АД (по определению выше); и (iv) количество случаев возвращения заболевания после прекращения приема исследуемого лекарственного препарата, оцениваемое исследователем; клиническое определение возвращения заболевания было следующим: существенное ухудшение тяжести заболевания после прекращения терапии до уровня тяжести, большего, чем перед началом терапии (Hijnen et al, J Eur Acad Dermatol Venereol 2007; 21(1) 85-9).
[00223] Опросник по влиянию атопического дерматита (ADIQ). Мы разработали для АД специальный связанный со здоровьем инструмент оценки качества жизни для пациентов возрастом от 12 лет и старше, следуя руководству по оценке результатов пациентами (PRO) Управления США по санитарному надзору за качеством пищевых продуктов и медикаментов (доступному на www(dot)fda(dot)gov(slash)downloads(slash)Drugs(slash)Guidances(slash)UCM193282(dot)pdf. В частности, был сделан обзор литературы для оценки информации по АД PRO. Со взрослыми возрастом 18-75 лет (n = 15) и подростками возрастом 12-17 лет (n = 15) с умеренным или тяжелым АД были проведены собеседования для определения концепции (ОК) для определения признаков и симптомов АД, которые являются важными для пациентов. Для качественного анализа записей собеседований использовали теоретически обоснованный подход. Провели совещание по составлению пунктов (ССП) для просмотра данных и разработки предварительных вопросов ADIQ на основании полученных в ОК-собеседованиях данных от пациентов.
[00224] Черновой вариант ADIQ, который тестировали в когнитивных собеседованиях с 34 взрослыми пациентами и пациентами-подростками, подтвердившими ясность и релевантность концепции, включенной в ADIQ, приведен ниже в таблице 2. Психометрическая валидация не включена.
Таблица 2. Опросник по влиянию атопического дерматита (ADIQ).
1 = Слегка подавлен
2 = Несколько подавлен
3 = Довольно сильно подавлен
4 = Сильно подавлен
1 = Слегка беспокоен
2 = Несколько беспокоен
3 = Довольно сильно беспокоен
4 = Сильно беспокоен
1 = Слегка раздражен
2 = Несколько раздражен
3 = Довольно сильно раздражен
4 = Сильно раздражен
1 = Слегка разочарован
2 = Несколько разочарован
3 = Довольно сильно разочарован
4 = Сильно разочарован
1 = Слегка смущался
2 = Несколько смущался
3 = Довольно сильно смущался
4 = Сильно смущался
1 = Слегка стеснялся
2 = Несколько стеснялся
3 = Довольно сильно стеснялся
4 = Сильно стеснялся
1 = Слегка
2 = Несколько
3 = Довольно сильно
4 = Сильно
1 = Слегка
2 = Несколько
3 = Довольно сильно
4 = Сильно
1 = Слегка утомлен
2 = Несколько утомлен
3 = Довольно сильно утомлен
4 = Сильно утомлен
1 = Слегка
2 = Несколько
3 = Довольно сильно
4 = Сильно
1 = Слегка
2 = Несколько
3 = Довольно сильно
4 = Сильно
1 = Редко
2 = Некоторое время
3 = Большую часть времени
4 = Все время
1 = Редко
2 = Некоторое время
3 = Большую часть времени
4 = Все время
1 = Редко
2 = Некоторое время
3 = Большую часть времени
4 = Все время
1 = Слегка
2 = Несколько
3 = Довольно сильно
4 = Сильно
1 = Слегка
2 = Несколько
3 = Довольно сильно
4 = Сильно
1 = Редко
2 = Некоторое время
3 = Большую часть времени
4 = Все время
Результаты
[00225] В этом исследовании проводили лечение 209 пациентов. На 12 неделе большая часть пациентов, которых лечили лебрикизумабом (125 мг каждые четыре недели) плюс МКС, достигали EASI-50 (Фиг. 2A и Фиг. 3A), EASI-75 (Фиг. 3B), EASI-90 (Фиг. 3C) и SCORAD-50 (Фиг. 2B) по сравнению с плацебо. Кроме того, на 12 неделе процент пациентов, которые достигли EASI-50, первичного критерия эффективности, был большим для всех групп лечения лебрикизумабом по сравнению с плацебо (62,3%), но статистическая значимость достигалась только в группе с многократными дозами (82,4% [p = 0,026] для группы 125 мг Р4Н), как показано на Фиг. 11A. В целом, доля пациентов, достигших ответа EASI-75, вторичного критерия эффективности, была большей для всех групп лечения разными дозами лебрикизумаба, но была значимо большей только в группе 125 мг Р4Н (54,9% [p = 0,036]) по сравнению с плацебо (34,0%) на 12 неделе, как показано на Фиг. 11B. Процент пациентов, которые достигли IGA 0/1 на 12 неделе, был большим для всех групп лечения лебрикизумабом по сравнению с плацебо, но не достигал статистической значимости. Однако, как и в случае других критериев, эти данные позволяют предположить наличие взаимосвязи доза-ответ для IGA (Фиг. 11C), а группа 125 мг Р4Н демонстрировала дальнейшее улучшение в последние недели периода лечения (Фиг. 2C). В отношении SCORAD-50 больше пациентов в группе с дозой лебрикизумаба 125 мг Р4Н (51,0% [p = 0,018]) и группах с ОД 250 мг (47,2% [p = 0,030]) достигали этого измеряемого параметра на 12 неделе по сравнению с плацебо (26,4%; Фиг. 11D). Все группы лечения лебрикизумабом демонстрировали улучшение в отношении пораженной ППТ (таблица 3). Наибольшее снижение пораженной ППТ на 12 неделе наблюдалось в группе лечения лебрикизумабом 125 мг Р4Н (57,7% снижения), хотя в группе плацебо также наблюдалось существенное улучшение (47,4%). Плацебо-скорректированная эффективность для ППТ не была статистически значимой (p = 0,38).
[00226] Лечение лебрикизумабом приводило к улучшению по ВАШ зуда (таблица 3 и таблица 7). Улучшение по ВАШ зуда в течение 2-недельного вводного периода с применением МКС было количественно большим, чем улучшение критериев тяжести заболевания. Наблюдалась высокая степень соблюдения лечения с применением МКС во всех группах лечения, при этом применение МКС в среднем составляло 86,8% (125 мг ОД), 86,7% (250 мг ОД), 91,9% (125 мг Р4Н) и 88,2% (плацебо) суток, начиная с исходного уровня и до 12 недели.
[00227] Также наблюдали большее улучшение по ВАШ бессонницы, а также тенденцию к улучшению в IGA 0/1, ВАШ зуда и результатах АД-специфического связанного со здоровьем опросника по качеству жизни (QoL-КЖ), в частности, опросника по влиянию атопического дерматита (ADIQ), у пациентов, которых лечили 125 мг лебрикизумаба раз в 4 недели плюс МКС, по сравнению с плацебо на 12 неделе (таблица 3, таблица 7, Фиг. 12A, Фиг. 12B и Фиг. 12C). Также наблюдалось дозозависимое улучшение в оценке DLQI, при этом численно большее снижение относительно исходного уровня было у всех групп лечения лебрикизумабом по сравнению с группами плацебо (таблица 3, таблица 7 и Фиг. 12D).
[00228] Также оценивали фармакокинетические параметры следующим образом. Образцы сыворотки для анализа фармакокинетики лебрикизумаба получали на 1 сутки (до приема дозы) и на неделе 1, 4 (до приема дозы), 6, 8 (до приема дозы), 12, 16 и 20 для всех схем дозирования. Сывороточные концентрации лебрикизумаба обобщали по типу лечения и визиту, используя описательную статистику для пациентов, которые соблюдали одну из схем лечения лебрикизумабом. Представленные ФК-параметры включают Cмакс на 1 неделе, Cмин на 4, 8 и 12 неделях и элиминационное время полужизни. Средние ФК-параметры и соответствующие стандартные отклонения для каждой схемы дозирования лебрикизумабом приведены в таблице 8. Результаты показывают, что фармакокинетика лебрикизумаба у пациентов с АД согласовалась с результатами по фармакокинетике лебрикизумаба у взрослых с астмой, представленными в ранее опубликованных литературных источниках (смотрите, например, Hanania et al, Thorax 70(8): 748-756 (2015)), где представлены линейные и пропорциональные дозе характеристики со временем полужизни 19-22 суток.
[00229] Уровень нежелательных явлений в целом был аналогичным среди групп лечения (66,7% для всех групп лечения лебрикизумабом и 66,0% для плацебо), и в основном они были слабыми или умеренными по тяжести (таблица 9). В частности, у трех (2%) пациентов в группе лечения лебрикизумабом (все дозы вместе) и у одного (2%) пациента в группе плацебо наблюдали НЯ (кожную инфекцию в группе 125 мг П/К, тревожность и миопатию в группе 125 мг Р4Н, и атопический дерматит в группе плацебо), которые привели к исключению из исследования. У одного (1%) пациента в группе лечения лебрикизумабом наблюдали НЯ (желудочно-кишечная вирусная инфекция), которое привело к прерыванию применения дозы. Представляющие интерес случаи смерти, случаи анафилаксии, злокачественных образований или определенных протоколом паразитарных или направленных внутриклеточных инфекций отсутствовали. Реакции в месте инъекции возникали не часто (1,3% для всех групп лечения лебрикизумабом и 1,9% для плацебо); все явления были несерьезными, длились в среднем 1-3 суток и не приводили к приостановке лечения.
[00230] Инфекции герпеса возникали не часто и только среди пациентов, которых лечили лебрикизумабом (n = 6 [3,8%]; простой герпес у n = 4 [2,6%] и опоясывающий герпес у n = 2 [1,3%]); все явления были несерьезными и не приводили к приостановке лечения. Кроме того, не было случаев герпетической экземы. В группах, которые лечили лебрикизумабом (все дозы вместе), было 14 (9%) пациентов с инфекциями кожи и девять (17%) пациентов в группе плацебо. Связанные с эозинофилами НЯ также наблюдались не часто и возникли только у пяти пациентов, которых лечили лебрикизумабом (3,2%; 3 случая «эозинофилии» и 2 случая «повышения числа эфозинофилов»); все явления были несерьезными, слабыми или умеренными по интенсивности и не приводили к прерыванию лечения. Максимальное число эозинофилов у этих пяти пациентов находилось в диапазоне от 0,91 до 3,2 x 109/л и из них три соответствовали эозинофилии 2 степени (1501-5000 клеток/мм3). У этих пяти пациентов не наблюдалось связанных с указанными явлениями клинических симптомов. Кроме того, наблюдаемое повышение согласовалось с тем, что наблюдали в предыдущих исследованиях лебрикизумаба (Hanania et al, Thorax 2015:70(8):748-756; Hanania et al, Lancet Respir Med 2016; 4(10): 781-796). Мы провели оценку конъюнктивита, учитывая наблюдаемое ранее несоответствие в биологических исследованиях в этой группе заболеваний (Thaci et al., Lancet 2016: 387 (10013): 40-52). Всего 15 (9,6%) пациентов в объединенной группе лечения лебрикизумабом и четыре пациента (7,5%) в группе плацебо имели НЯ конъюнктивита; все явления были несерьезными и не приводили к приостановке лечения.
Таблица 3. Ключевые результаты по эффективности на 12 неделе.
ADIQ = опросник по влиянию атопического дерматита; ППТ = площадь поверхности тела; ДИ = доверительный интервал; DLQI = дерматологический индекс качества жизни; EASI = индекс распространенности и тяжести экземы; IGA = общая оценка исследователем; SCORAD = оценка тяжести атопического дерматита; СП = стандартная погрешность; ВАШ = визуальная аналоговая шкала.
Таблица 7. Процентное изменение в EASI, SCORAD, IGA, ВАШ зуда и проценте пораженной ППТ от скрининга до исходного уровня (вводный период).
(n = 52)
(n = 53)
(n = 51)
(n = 53)
(n = 209)
ППТ = площадь поверхности тела; EASI = индекс распространенности и тяжести экземы; IGA = общая оценка исследователем; Р4Н = раз в 4 недели; ОД = одна доза; SCORAD = оценка тяжести атопического дерматита; ОД = одна доза; СП = стандартная погрешность; ВАШ = визуальная аналоговая шкала.
Таблица 8. Средние фармакокинетические параметры лебрикизумаба после однократного или многократного подкожного введения.
Cмакс, н1 = максимальная концентрация лебрикизумаба на 1 неделе, Cмакс, н4 = наблюдаемая минимальная концентрация на 4 неделе, Cмакс, н8 = наблюдаемая минимальная концентрация на 8 неделе, Cмакс, н12 = наблюдаемая минимальная концентрация на 12 неделе, t1/2 = элиминационное время полужизни.
Таблица 9. Обзор ключевой информации по безопасности, недели 0-20.
(n = 54)
(n = 52)
(n = 50)
(n = 156)
(n = 53)
Миопатия
НЯ = нежелательное явление; Р4Н = раз в четыре недели; СНЯ = серьезное нежелательное явление; ОД = одна доза. * Заслепленные данные пересматривали для того, чтобы установить случаи анафилаксии по критериям Сэмпсона. † Инфекции, связанные с исследуемым препаратом оценивались исследователем.
[00231] Также мы наблюдали выраженную зависимость доза-ответ для данных, представленных на Фиг. 2A. Одна доза 250 мг была численно лучше в ранние моменты времени по сравнению с одной дозой 125 мг. Кроме того, данные, представленные на Фиг. 2A, позволяют предположить, что доза 125 мг, вводимая раз в четыре недели, не достигает плато эффективности на 12 неделе. Эти данные позволяют предположить потенциальное преимущество более высокой дозы (например, 250 мг Р4Н) или ударной дозы.
[00232] Кроме того, на 20 неделе в модифицированной популяции с назначенным лечением большая часть пациентов, которых лечили 125 мг лебрикизумаба раз в 4 недели плюс МКС, достигли EASI-50 (Фиг. 3A), EASI-75 (Фиг. 3B) и EASI-90 (Фиг. 3C) по сравнению с плацебо. Также смотрите таблицу 9. Также у пациентов, которых лечили лебрикизумабом, наблюдали большее процентное снижение относительно исходного уровня в EASI в течение 12 недель по сравнению с плацебо (Фиг. 4). Следует отметить, что пациенты, которые получали 125 мг лебрикизумаба раз в четыре недели в течение 12 недель, демонстрировали 70,5% изменение (снижение) в EASI по сравнению с 53,1% изменением (снижением) в группе плацебо. Эта 17,4% разница между лечением лебрикизумабом и плацебо была статистически значимой, P = 0,025.
Таблица 9. Обобщенные ключевые результаты по эффективности на 20 неделе.
ППТ = площадь поверхности тела; EASI = индекс распространенности и тяжести экземы; IGA = общая оценка исследователем; SCORAD = оценка тяжести атопического дерматита; ОД = одна доза; СП = стандартная погрешность; ВАШ = визуальная аналоговая шкала.
[00233] Также мы оценивали симптомы пациентов и связанное со здоровьем качество их жизни по визуальной аналоговой шкале (ВАШ) зуда SCORAD, ВАШ бессонницы и ADIQ. Улучшение относительно исходного уровня было продемонстрировано в ВАШ зуда, ВАШ бессонницы и ADIQ (данные приведены в таблице 3). Изменения со времени скрининга также изучали с помощью апостериорного анализа. Пациенты, которых лечили 125 мг лебрикизумаба, вводимого раз в четыре недели, демонстрировали снижение ВАШ зуда 55,2% (p = 0,03 по сравнению с плацебо) со времени скрининга на 12 неделе, тогда как плацебо приводило к снижению 39,3% со времени скрининга. Также наблюдали улучшение в ВАШ бессонницы со времени скрининга на 12 неделе со снижением 61,5% (p = 0,04 по сравнению с плацебо) при лечении лебрикизумабом и снижение 39,3% при применении плацебо. Наблюдали снижение 64,7% (p = 0,02 по сравнению с плацебо) в ADIQ со времени скрининга при лечении лебрикизумабом и снижение 41,8% при применении плацебо. Разница в этих измеряемых параметрах в сравнении с плацебо для групп, в которых лебрикизумаб применяли только в виде одной дозы (125 мг и 250 мг), в большинстве случаев была статистически значимой.
[00234] В целом, эти результаты демонстрируют, что нацеливание на IL-13 при умеренном или тяжелом АД обеспечивает клинически значимое плацебо-скорректированное улучшение в случае многих показателей тяжести, в общем случае дозозависимым образом. Ежемесячное дозирование существенно увеличило часть пациентов, достигающих EASI-50, EASI-75 и SCORAD-50, с тенденцией к увеличению количества пациентов, достигающих IGA 0/1, и улучшению в ВАШ зуда. Эти улучшения были видны на фоне интенсивного применения МКС, с которым связаны существенные ответы в группе плацебо. Уровень нежелательных явления в общем случае был аналогичным среди групп лечения, и в большинстве случаев они были слабыми или умеренными по тяжести.
[00235] Кроме того, результаты показывают, что лебрикизумаб обеспечивает пользу лечения на фоне полноценного применения МКС у пациентов с умеренным или тяжелым АД, которые не демонстрировали адекватный ответ на соответствующее стандарту лечение МКС и имели исходные характеристики, которые относили эту популяцию пациентов к соответствующему большей степени тяжести концу спектра. Исследование достигло своего первичного измеряемого параметра – процента пациентов с EASI-50 на 12 неделе – со статистически значимым числом пациентов в группе лечения 125 мг лебрикизумаба Р4Н, достигшими этого уровня снижения в исходной оценке EASI. Кроме того, кривые ответа, имеющие наклон вверх, в течение последних недель периода лечения, позволяют предположить, что плато ответа могло не быть достигнуто к 12 неделе для группы лечения 125 мг лебрикизумаба Р4Н, а большая продолжительность лечения может приводить к улучшению эффективности. Также наблюдалась зависимость доза-ответ, причем доза лебрикизумаба 125 мг Р4Н демонстрировала численно большие уровни ответа для большинства измеряемых параметров, в частности, EASI, а доза лебрикизумаба 125 мг ОД демонстрировала наименьшие клинические результаты на 12 неделе.
[00236] Следует отметить, что группа с ОД лебрикизумаба 250 мг демонстрировала численно большие ответы в более ранние моменты времени для некоторых показателей, что позволяет предположить потенциальную пользу применения более высоких доз или ударной дозы. Для всех основных измеряемых параметров эффективности наблюдалось непрерывное улучшение в течение курса 12-недельного периода лечения. Разница во многих вторичных измеряемых параметрах эффективности на 12 неделе была статистически значимой при нескорректированном 5% уровне значимости для пациентов в группе лечения 125 мг лебрикизумаба Р4Н по сравнению с пациентами в группе плацебо. Они включали процент пациентов с EASI-75, процент пациентов с SCORAD-50, процентное изменение относительно исходного уровня в оценке EASI и процентное изменение относительно исходного уровня в SCORAD. Улучшение в IGA и ВАШ зуда было численно большим, чем при применении плацебо. Зуд считается основным фактором снижения КЖ пациентов с умеренным или тяжелым АД, а наблюдаемое численно большее улучшение КЖ (по определению DLQI и ADIQ) и сна при дозировании 125 мг лебрикизумаба Р4Н, вероятно, связано с улучшением в отношении зуда. В общем случае эффект лебрикизумаба также сохранялся во время 8-недельного периода последующего наблюдения для некоторых показателей, включая ответы EASI, IGA (0/1) и SCORAD, до 20 недели.
[00237] Учитывая зависимость доза-ответ, наблюдаемую для многих измеряемых параметров, и тенденцию к улучшению эффективности при увеличении дозы и длительности лечения, резонно предположить, что дополнительное увеличение дозы (в виде ударной дозы или более высокой дозы, например, 250 мг Р4Н) и/или продолжительности лечения может привести к улучшению эффективности. Кроме того, мы наблюдали разницу в зависимости доза-ответ между FEV1 у пациентов с астмой (Hanania et al, Thorax 2015; 70(8):748-756) и измеряемыми параметрами EASI/IGA у пациентов с АД, что позволяет предположить, что при АД действительно могут требоваться более высокие дозы лебрикизумаба для достижения плато ответа. Это согласовалось бы с более высокой нагрузкой IL-13 у пациентов с атопическим дерматитом по сравнению с пациентами с астмой.
[00238] По протоколу этого исследования от пациентов требовалось соблюдение применения МКС дважды в сутки во время 2-недельного вводного периода для того, чтобы они были пригодными для рандомизации. Кроме того, пациенты были пригодными для рандомизации только в том случае, если они демонстрировали достаточную тяжесть АД после этого вводного периода. Хотя пациенты, включенные в это исследования, не демонстрировали адекватный контроль с помощью МКС, включение этого периода с применением МКС приводило к существенному улучшению заболевания. Это улучшение было продемонстрировано снижением исходных оценок тяжести АД, в особенности показателей, связанных с чесоткой, вероятно, приводя к тому, что демонстрация дополнительного улучшения выше исходного уровня является более сложной.
[00239] Во время периода лечения протокол требовал продолжения применения МКС дважды в сутки, а пациенты были обеспечены ежедневными напоминаниями в электронных дневниках; пациенты в этом исследовании применяли МКС с высоким уровнем соблюдения режима, составляющим 88%, во всех группах лечения. Хотя ежесуточное применение МКС, как правило, рекомендуется для острых поражений, а не в виде длительного и бесконечного применения, цель этого исследования фазы 2 для подтверждения концепции состояла в выяснении потенциальной эффективности лебрикизумаба в дополнение к интенсивному применению МКС (Eichenfield et al, J Am Acad Dermatol. 2014; 71(1): 116-132), а не в оценке умеренного применения МКС. Рассматривалось менее частое применение МКС, так как эта схема МКС может не отражать долгосрочное лечение или клиническую практику. Однако эта схема согласуется с утвержденным применением МКС, и существовали регуляторные вопросы относительно применения не по утвержденным показаниям с меньшей частотой, чем предусмотрена для утвержденного применения продукта. В то же время, не возникало вопросов относительно того, что альтернативный дизайн, который не разрешает фоновое применение МКС, такой как монотерапия лебрикизумабом, в значительной мере приводил бы к выбыванию пациентов и/или неудачному результату лечения пациентов в контрольной группе. Действительно, исследования вариантов биологической терапии в виде монотерапии приводили к уровню выбывания/неудачного результата лечения пациентов порядка приблизительно 50% в контрольной группе (Simpson et al, N Engl J Med. 2016; 375(24):2335-2348) в противоположность уровню выбывания 13% для группы плацебо в этом исследовании. Интенсивное применение МКС во время 12-недельного периода лечения, вероятно, объясняет значительный уровень ответа, наблюдаемый в группе плацебо. Было показано, что длительное и частое применение МКС приводит в прогрессирующему улучшению в АД, но большинство руководств предлагают ограничение ежедневного применения для избежания НЯ. Смотрите, например, Brunner et al., J Allergy Clin Immunol. 2016; 138(1): 169-178; Schneider et al, J Allergy Clin Immunol. 2013; 131(2):295-9.e1-27; и Hanifin et al, J Am Acad Dermatol. 2004; 50(3):391-404. Кроме того, значительный уровень ответа мог быть в группе плацебо, поэтому не наблюдалось большей лечебной разницы между группами лечения плацебо и лебрикизумабом. Несмотря на относительно высокую эффективность длительного и частого применения МКС, наблюдалось значительное улучшение, в частности, в признаках АД (EASI) и общих оценках (SCORAD), при добавлении лечения лебрикизумабом.
[00240] Дупилумаб, анти-IL-4Rα моноклональное антитело демонстрировало эффективность у пациентов с умеренным или тяжелым АД и недавно было утверждено для применения при АД. Считается, что IL-4Rα является важной рецепторной субъединицей для сигнализации как IL-4, так и IL-13. Исследования дупилумаба у пациентов с АД обеспечивают понимание потенциала блокирования IL-13 для лечения АД с оговоркой, что не была установлена относительная важность IL-4 по сравнению с IL-13 при АД. IL-13 и IL-4 имеют сходные биологические и эффекторные функции; например, оба играют роль в развитии Т-хелперов 2, выработке IgE, привлечении эозинофилов, нарушении целостности эпителиального барьера посредством понижающей регуляции противомикробных пептидов, фиброзе и снижении экспрессии филаггрина. Смотрите, например, Paternoster et al, Nat Genet. 2011; 44(2):187-192; Kagami et al., Clin Exp Immunol. 2005; 141(3):459-466. Вследствие такой высокой степени сходства в биологии, блокирование одного IL-13 потенциально могло бы обеспечить сравнимое улучшение в АД с комбинированным блокированием IL-13 и IL-4, с более специфическим направленным действием. Кроме того, нацеливание на растворимый цитокин, такой как IL-13, может обеспечить преимущество нелинейного ФК-профиля с получаемым в результате улучшением в частоте дозирования. Это частично объясняет способность дозировать лебрикизумаб Р4Н и может позволить осуществлять менее частое дозирование во время поддерживающего периода. В противоположность этому, нацеливание на рецептор связано с опосредованным мишенью выведением лекарственного препарата, что может приводить к быстрому снижению концентрации после прекращения или прерывания приема лекарственного препарата, как в случае с дупилумабом, который дозируют Р2Н (Kovalenko et al, CPT Pharmacometrics Syst Pharmacol. (2016) Nov; 5(11):617-624).
[00241] Результаты этого исследования позволяют предположить, что IL-13-опосредованные пути сигнализации играют важную роль в патогенезе АД, а блокирование этого цитокина может приводить к значительному улучшению клинических результатов. Пациенты с умеренным или тяжелым АД демонстрировали улучшения при лечении лебрикизумабом, даже в случае однократных доз и интенсивного применения МКС. Дополнительные исследования большей продолжительности, с более частым дозированием или применением более высоких доз и в большей популяции, с разными фоновыми схемами (или в их отсутствие) помогут выяснить роль нацеливания только на IL-13 при АД.
ПРИМЕР 3 – Альтернативные схемы дозирования
[00242] Чтобы оценить возможность разработки альтернативных схем дозирования лебрикизумабом и прогнозирования их эффективности, мы разработали новую, основанную на механизме продольную ФК-ФД модель, описанную ниже. Эту модель использовали для моделирования прогнозированной эффективности, например, оценки EASI-75, различных альтернативных схем дозирования, например, включая, но не ограничиваясь этим, различные ударные дозы с применением впоследствии различных поддерживающих доз, например, постоянных доз лебрикизумаба, вводимых через регулярные интервалы времени, или различных меньших постоянных доз или больших постоянных доз, или увеличенные интервалы лечения лебрикизумабом.
[00243] Разработка модели началась с исходных данных по фармакокинетике и эффективности, которые были получены во время проведения клинического исследования I фазы II, описанного выше. Данные по фармакокинетике лебрикизумаба у пациентов с атопическим дерматитом согласовались с прогнозами, сделанными по популяционной ФК-модели применения лебрикизумаба, которая была разработана на основе концентраций лекарственного препарата, полученных для пациентов с астмой и здоровых волонтеров (данные не показаны). Это позволило использовать популяционную ФК-модель применения лебрикизумаба для облегчения разработки ФК-ФД модели при атопическом дерматите, описанной ниже.
[00244] ФК-ФД модель применения лебрикизумаба при атопическом дерматите была разработана на основании оценок EASI за период вплоть до 12 недели из клинического исследования I и ранее установленной популяционной ФК-модели применения лебрикизумаба. Эффектом ФД-ответа было снижение оценки EASI. В эту модель также были включены эффекты плацебо и/или введения МКС. Ковариатный анализ позволяет предположить, что исходная оценка EASI была существенной ковариатой для Kout, которая представляет собой константу скорости восстановления ткани, и Econ, которая представляет собой эффект плацебо/МКС. Конечная модель приведена на Фиг. 5, а параметры модели приведены в таблице 4.
Таблица 4. Параметры модели применения лебрикизумаба при атопическом дерматите
Доверительные интервалы представляют 2,5-ый и 97,5-ый процентили параметров, оцениваемых для набора из 1000 образцов с повторениями; объяснение символов смотрите в описании Фиг. 5.
[00245] Эта модель включает некоторые предположения, такие как предположения, что зависимость доза-ответ является аналогичной для всех концентраций лекарственного препарата, при этом мы обнаружили, что она адекватно описывает изменение исходной оценки EASI, наблюдаемое в исследовании 1. Кроме того, модель могла очень хорошо прогнозировать ответы EASI-50, EASI-75 и EASI-90 вплоть до 12 недели в исследовании 1, несмотря на то, что не была разработана для этой цели. Этот результат обеспечивает дополнительное подтверждение модели и повышение нашей уверенности в ее точности и корректности.
[00246] Мы использовали конечную модель, чтобы смоделировать некоторое число схем дозирования, а затем мы прогнозировали оценки EASI в различное время в течение лечения. Смоделированные схемы дозирования приведены ниже в таблице 5. Все варианты моделирования проводили в течение 24-недельного периода времени. Схему дозирования 125 мг с применением раз в четыре недели использовали в качестве эталонного параметра для сравнения всех моделируемых результатов. Как показано в таблице 5, смоделированные схемы дозирования были в целом категоризированы по четырем группам: Схемой для Группы 1 были варианты применения ударной дозы и поддерживающей дозы, схемой для Группы 2 были варианты применения 250 мг, вводимых один раз в четыре недели, схемой для Группы 3 были варианты применения 250 мг, вводимых один раз в восемь недель, а схемой для Группы 4 были варианты применения с меньшей дозой: 37,5 мг, вводимой один раз в четыре недели.
Таблица 5. Схемы дозирования, смоделированные с помощью модели применения лебрикизумаба при АД
[00247] Моделирование для пациентов, достигших со временем EASI-75, при применении каждой их схем дозирования в Группе 1 показано на Фиг. 6. ФК-ФД модель смогла спрогнозировать все варианты применения ударной дозы, приводящие к небольшим улучшениям в ответе EASI-75 по сравнению со схемой с применением только 125 мг один раз в четыре недели. Это улучшение было наиболее очевидным в средней части кривой (например, 56-84 сутки) и было наиболее размытым для поздних моментов времени (140-168 сутки).
[00248] Моделирование для пациентов, достигших со временем EASI-75, при применении каждой их схем дозирования в Группе 2 показано на Фиг. 7. ФК-ФД модель смогла спрогнозировать, что схемы на основе 250 мг один раз в четыре недели начинают демонстрировать отличие (т. е. улучшение в EASI-75) от схемы 125 мг один раз в четыре недели в средней части кривой, и это отличие сохраняется в более поздние моменты времени.
[00249] Моделирование для пациентов, достигших со временем EASI-75, при применении каждой их схем дозирования в Группе 3 показано на Фиг. 8. ФК-ФД модель смогла спрогнозировать, что схемы, включающие 250 мг, вводимых один раз в восемь недель, имеют сравнимую эффективность со схемой, включающей 125 мг, вводимых один раз в четыре недели, с небольшим улучшением в незанятые недели (например, на 12 неделе и 20 неделе) и небольшим ухудшением результата за неделю до приема следующей дозы (например, на 8 неделе и 16 неделе).
[00250] Моделирование для пациентов, достигших со временем EASI-75, при применении каждой их схем дозирования в Группе 4 показано на Фиг. 9. ФК-ФД модель смогла спрогнозировать, что схемы, включающие 37,5 мг, вводимых один раз в четыре недели, были менее эффективными, чем схемы дозирования, включающие 125 мг, вводимых один раз в четыре недели.
[00251] В целом, ФК-ФД модель применения лебрикизумаба при атопическом дерматите смогла спрогнозировать, что все схемы дозирования, включающие введение лебрикизумаба «один раз в четыре недели», обеспечивают эквивалентные или лучшие оценки EASI-75 по сравнению с оценками EASI-75, получаемыми при введении 125 мг один раз в четыре недели (доза, клинически исследованная в описанном выше клиническом исследовании I фазы II), за исключением схем дозирования, которые включали дозу лебрикизумаба 37,5 мг, вводимую один раз в четыре недели. Это моделирование показало, что 37,5 мг лебрикизумаба, вводимые один раз в четыре недели, по прогнозам будут менее эффективными согласно оценкам EASI-75, чем любая из других моделируемых схем дозирования.
[00252] ФК-ФД модель применения лебрикизумаба при атопическом дерматите также смогла спрогнозировать, что наилучший результат EASI-75 получают при введении 250 мг один раз в четыре недели (с ударной дозой или без нее) при длительности лечения 16-24 недели. Модель также смогла спрогнозировать, что включение ударной дозы улучшает результат EASI-75 в более ранние моменты времени (например, на 8 или 12 недели), но влияние этого улучшения по сравнению со схемами дозирования с отсутствием ударной дозы снижается со временем (например, на 20 или 24 недели). Таким образом, включение ударной дозы может обеспечить улучшение результатов лечения пациентов на ранних сроках курса лечения. И наконец, модель смогла спрогнозировать, что введение 250 мг лебрикизумаба один раз в восемь недель приводит к улучшению EASI-75, аналогичному результатам EASI-75, спрогнозированным для введения 125 мг лебрикизумаба один раз в четыре недели. Общее улучшение в EASI-75 для схемы «250 мг Р8Н» согласно прогнозам должно быть несколько выше в течение недель, когда не вводится доза 250 мг (например, на 4 неделе), и немного ниже, чем «схема 125 мг Р4Н» непосредственно перед введением дозы 250 мг (например, на 8 неделю). Таким образом, описанные здесь результаты моделирования демонстрируют, что согласно прогнозам некоторое число схем дозирования лебрикизумаба будут обеспечивать улучшение терапевтических результатов у пациентов с атопическим дерматитом.
ПРИМЕР 4 – Клиническое исследование II
[00253] Клинические исследования II представляло собой рандомизированное, открытое исследование фазы II для оценки безопасности и эффективности монотерапии лебрикизумабом у взрослых пациентов (возрастом 18-75 лет) с персистентным умеренным или тяжелым АД, который адекватно не контролируется МКС. Схема исследования II приведена на Фиг. 10.
[00254] Скрининг. Пациенты, подходящие для включения в исследование, должны были соответствовать всем описанным ниже критериям пригодности.
[00255] Критерии включения были следующими: возраст от 18 до 75 лет; АД, диагностированный по критериям Hanifin/Rajka, присутствующий в течение по меньшей мере 1 года на момент скрининга; АД от умеренного до серьезного по градации в соответствии со шкалой Райка/Лангеланда на момент скрининга; анамнез отсутствия адекватного ответа на длящуюся ≥ 1 месяца (в течение 3 месяцев перед скрининговым визитом) схему лечения с по меньшей мере ежесуточным применением МКС и регулярным применением смягчающих средств для лечения АД; оценка EASI ≥ 14 на момент скрининга и в конце вводного периода (визит 3), оценка IGA ≥ 3 (5-точечная шкала) на момент скрининга и в конце вводного периода (визит 3); распространение АД на ≥ 10% площади поверхности тела (ППТ) на момент скрининга; и оценка ВАШ зуда (определяемая как часть SCORAD) ≥ 3 на момент скрининга; соблюдение установленной протоколом схемы применения МКС в течение 2-недельного вводного периода (по меньшей мере 10 из 14 суток) на момент начала периода лечения (1 сутки).
[00256] Критерии исключения были следующими: применение в прошлом и/или настоящем любой терапии на основе анти-IL-13 или анти-IL-4/IL-13, включая лебрикизумаб; применение исследуемого агента в течение 4 недель перед скринингом или в течение 5 периодов полужизни исследуемого агента, в зависимости от того, какой из периодов больше; анамнез тяжелой аллергической реакции или анафилактической реакции на биологический агент или подтвержденная гиперчувствительность к любому компоненту инъекции лебрикизумаба; гиперчувствительность к МКС или к любым другим ингредиентам, содержащимся в МКС-продукте, применяемом в исследовании; применение каких-либо дополняющих, альтернативных или гомеопатических лекарственных средств, включая, но не ограничиваясь этим, фитотерапию, традиционные или нетрадиционные растительные лекарственные средства, незаменимые жирные кислоты или акупунктуру, в течение 7 суток перед вводным периодом или необходимость в таких лекарственных средствах во время исследования; масса тела < 40 кг или индекс массы тела > 38 кг/м2; признаки других кожных патологий, включая, но не ограничиваясь этим, Т-клеточную лимфому или аллергический контактный дерматит; признаки или текущее лечение (включая лечение местными антибиотиками) активной кожной инфекции на момент скрининга (Сутки -15); определенные инфекции; анамнез недавних или активных (в течение 6 месяцев) паразитарных инфекций, в особенности вызванных нематодами (например, Ascaris, Ancylostoma), платигельминтами (например, Schistosoma), или анамнез листериозных инфекций; активная форма туберкулеза, требующая лечения, в течение 12 месяцев перед визитом 1; признаки острого или хронического гепатита или подтвержденный цирроз печени; подтвержденный иммунодефицит, включая инфекцию ВИЧ; применение МИК (местного ингибитора кальциневрина) во время скрининга; посещение солярия в течение 4 недель перед скрининговым визитом; иммунотерапия аллергенами в течение 3 месяцев от скрининга; получение живой аттенуированной вакцины в течение 4 недель перед исходным визитом (сутки 1); запланированная хирургическая операция во время исследования; клинически значимая аномалия при исследовании ЭКГ или лабораторных исследованиях (гематология, биохимический анализ сыворотки и анализ мочи); повышение АСТ, АЛТ или общего билирубина ≥ 2,0 x относительно верхнего предела нормы (ВПН) во время скрининга; подтвержденное текущее злокачественное образование или текущее исследование в отношении потенциального злокачественного образования, включая базально- или плоскоклеточную карциному кожи или карциному in situ; анамнез злокачественного образования в течение 5 лет перед скринингом, за исключением должным образом вылеченной карциномы in situ шейки матки, немеланомной карциномы кожи; рака матки стадии I; и другие клинически значимые заболевания, неконтролируемые несмотря на лечение.
[00257] Вводный период. После скрининга подходящие пациенты проходили 2-недельный вводный период (сутки от -14 до -1). После вводного периода пациенты прекращали применять используемые до исследования МКС (и другие показанные при АД средства) и начинали соблюдать установленную протоколом схему местной терапии, описанную ниже. На первые сутки вводного периода (сутки -14) пациенты получали МКС для применения в течение всего вводного периода. В частности, пациенты применяли крем 0,1% триамцинолона ацетонида для нанесения на поверхность тела и крем 2,5% гидрокортизона для нанесения на лицо и опрелости. Согласно инструкциям пациенты должны были наносить смягчающее средство на все ксеротические кожные поверхности по меньшей мере один раз в сутки и наносить крем на активные кожные поражения дважды в сутки и не наносить крем на непораженные участки.
[00258] В конце вводного периода проводили оценку тяжести заболевания, а пациенты, которые продолжали демонстрировать АД от умеренного до тяжелого согласно EASI и IGA (смотрите критерии включения, выше), подходили для рандомизации в отношении исследуемого лекарственного препарата (лебрикизумаб) или только МКС.
[00259] Период лечения (недели 1-12). В конце вводного периода проводили рандомизацию пациентов, которые соблюдали установленную протоколом схему применения МКС и продолжали удовлетворять критериям пригодности (смотрите критерии включения, выше). Всего провели рандомизацию (1:1) приблизительно 50 пациентов в одну из следующих групп лечения (см. Фиг. 10). Группа 1: 125 мг лебрикизумаба, вводимые путем подкожной (П/К) инъекции раз в 4 недели (Р4Н), всего в количестве 3 доз; и Группа 2: Один МКС-крем. Пациенты, рандомизированные в Группу 1, не придерживались установленной протоколом схемы применения МКС, а пациенты, рандомизированные в Группу 2, продолжали применять МКС, используя такую же схему применения МКС, что и во время вводного периода (два раза в сутки только к пораженной коже).
[00260] Период последующего наблюдения (недели 13-20). Всех пациентов, которые завершили лечение согласно исследованию во время периода лечения (недели 1-12), наблюдали в целях безопасности в течение дополнительных 8 недель (недели 13-20). Во время этого периода последующего наблюдения в целях безопасности пациенты в Группе 2 (только МКС) больше не должны были придерживаться установленной протоколом схемы применения МКС, предписанной во время вводного периода и периода лечения. Вместо этого, можно было наносить крем 0,1% триамцинолона ацетонида и/или крем 2,5% гидрокортизона только на активные кожные поражения на усмотрение пациента и исследователя. Аналогично, пациенты в Группе 1 лечения (монотерапия лебрикизумабом) могли наносить крем 0,1% триамцинолона ацетонида и/или крем 2,5% гидрокортизона только на активные кожные поражения на усмотрение пациента и исследователя. Всех пациентов поощряли к продолжению применения смягчающих средств к ксеротическим участкам кожи по меньшей мере один раз в сутки.
[00261] В любое время в течение этого исследования не допускалось применение местных ингибиторов кальциневрина (МИК). Пациентам, которые применяли МИК во время скрининга, разрешалось участвовать в исследовании, если они соглашались прекратить применение МИК на время исследования, и если, по мнению исследователя, было безопасно прекратить применение МИК и использовать вместо этого предписанный протоколом крем МКС.
[00262] Обоснование выбора дозы и схемы применения лебрикизумаба. Обоснование дозы и схемы применения лебрикизумаба в исследовании II было аналогичным обоснованию, описанному выше для исследования I. В этом исследовании фазы II изучали схему, включающую одну дозу лебрикизумаба (125 мг Р4Н [Группа 1]). Схема, включающая дозу 125 мг Р4Н, была включена в качестве потенциально эффективной дозы и была схемой с наибольшей дозой, которую исследовали в базовых исследованиях лебрикизумаба фазы III (LAVOLTA I и II) при астме у взрослых. Учитывая ожидаемую аналогию в роли IL-13 и фармакокинетике между астмой и АД, предположили, что эта доза могла бы быть эффективной для пациентов с АД.
[00263] Однако, как описано выше, результаты исследований LAVOLTA I и II не были согласующимися. Hanania et al., Lancet Respir Med 2016, доступная на dx(dot)doi(dot)org(slash)S2213-2600(16)30265-X, опубликованная 5 сентября 2016 г. Оба исследования не смогли продемонстрировать четкой зависимости доза-ответ, хотя результаты по фармакокинетике и фармакодинамике согласовались с результатами исследований фазы II, описанных выше, что указывает на аналогичное воздействие лекарственного препарата и ингибирование пути IL-13. Hanania et al., 2016, выше. Соответственно, не было уверенности в том, что лебрикизумаб будет обеспечивать клинически значимое улучшение показателей у пациентов с атопическим дерматитом при применении описанных в данном документе схем дозирования.
[00264] Первичный измеряемый параметр в исследовании фазы II определяли на 12 неделе. Ожидалось, что средняя сывороточная концентрация лебрикизумаба на 12 неделе будет составлять более 90% стационарного значения для пациентов, получающих 125 мг Р4Н. Кроме того, в случае дозы 125 мг Р4Н прогнозируемая остаточная концентрация на 12 неделе будет находиться в рамках диапазона, в котором лебрикизумаб демонстрировал биологическую активность в клинических исследованиях астмы фазы II.
[00265] Обоснование для контрольной группы. Пациенты в 2-ой группе лечения получали только крем МКС и, следовательно, служили в качестве активной сравнительной группы. Эту группу лечения использовали в качестве контроля для П/К применения лебрикизумаба для того, чтобы точно оценить безопасность и эффективность лебрикизумаба в качестве монотерапии по сравнению с МКС.
[00266] Активная сравнительная МКС-группа была включена, так как применение МКС является стандартом лечения в клинической практике (руководства Европейской академии дерматовенерологии [EADV] 2009; Darsow et al, J Eur Acad Dermatol Venereol 2010; 24:317-28; American Academy of Dermatology 2014 guidelines; Eichenfield et al, J Am Acad Dermatol 2014:71(1): 116-32).
[00267] Критерии результатов эффективности. Поисковые критерии результатов эффективности монотерапии лебрикизумабом по сравнению с применением одних МКС в этом исследовании были следующими, по порядку: (i) процент пациентов с 50% или 75% (EASI 50/75) снижением относительно исходного уровня EASI на 12 неделе (EASI-50 и EASI-75 были определены как 50% и 75% снижение, соответственно, в оценке EASI на 12 неделе по сравнению с исходным уровнем); (ii) процентное и абсолютное изменение относительно исходного уровня в оценке EASI на 12 неделе; (iii) процент пациентов, достигших оценки IGA 0 или 1 на 12 неделе; (iv) процент пациентов со снижением ≥ 2 пунктов относительно исходного уровня в IGA на 12 неделе; (v) абсолютное изменение относительно исходного уровня в IGA на 12 неделе; (vi) процент пациентов, достигших оценки IGSA 0 или 1 на 12 неделе; (vii) процент пациентов со снижением ≥ 2 пунктов относительно исходного уровня в IGSA на 12 неделе; (viii) абсолютное изменение относительно исходного уровня в IGSA на 12 неделе; (ix) процентное и абсолютное изменение относительно исходного уровня в SCORAD на 12 неделе; (x) процент пациентов с 50% или 75% снижением (SCORAD 50/75) относительно исходного уровня в оценке SCORAD на 12 неделе; (xi) процентное изменение относительно исходного уровня в общей пораженной площади поверхности тела (ППТ) на 12 неделе; (xii) абсолютное и процентное изменение относительно исходного уровня в зуде по ВАШ зуда (оцениваемое в качестве части SCORAD) на 12 неделе; (xiii) число обострений заболевания от исходного уровня до 12 недели; и (xiv) процент пациентов, которые получали не установленные протоколом МКС до 12 недели.
[00268] Критерии для результатов безопасности. Критерии результатов безопасности в этом исследовании были следующими: (i) частота нежелательных явлений, возникших в ходе лечения, на 12 неделю при применении лебрикизумаба в виде монотерапии по сравнению с применением только МКС; (ii) количество человеческих антител против терапевтического средства (АПТС) на исходном уровне и во время исследования; (iii) частота и тяжесть инфекций кожи и других систем органов во время исследования; клиническое определение инфекции кожи было следующим: диагноз инфекции кожи ставили на основании клинической оценки исследователя, включая, но не ограничиваясь этим, наличие корок цвета меда, серозные выделения, пустулы или боль в месте сыпи, которые могут быть связаны с системными характеристиками, включая повышение температуры или обострение заболевания АД (по определению выше); (iv) количество случаев возвращения заболевания после прекращения приема исследуемого лекарственного препарата, оцениваемое исследователем; клиническое определение возвращения заболевания было следующим: существенное ухудшение тяжести заболевания после прекращения терапии до уровня тяжести, большего, чем перед началом терапии (Hijnen et al, J Eur Acad Dermatol Venereol 2007; 21(1) 85-9); (v) частота реакций в месте инъекции от исходного уровня до 12 недели; и (vi) частота и тяжесть нежелательных явлений, возникших в ходе лечения, включая представляющие особый интерес нежелательные явления, от исходного уровня до 12 недели.
[00269] Результаты. Ключевые результаты по эффективности для клинического исследования II обобщены в таблице 6, ниже. Данные показывают, что монотерапия лебрикизумабом в течение 12-недельного периода исследования была эффективной. Результаты EASI-75 и EASI-90 были аналогичными для групп лечения лебрикизумабом и МКС, тогда как результаты IGA, SCORAD и оценки зуда были несколько ниже в группе лебрикизумаба, чем в группе МКС.
Таблица 6. Ключевые результаты по эффективности на 12 неделе.
(n = 27)
(n = 28)
(n = 27)
(n = 28)
* у пациентов с исходной ВАШ зуда ≥ 3.
Аминокислотные последовательности анти-IL13 антитела (лебрикизумаба)
[00270] В таблице ниже приведены аминокислотные последовательности областей CDR-H1, CDR-H2, CDR-H3, CDR-L1, CDR-L2 и CDR-L3 лебрикизумаба наряду с последовательностями VH, VL, последовательностями тяжелой цепи и последовательностями легкой цепи. Как указано в нижеприведенной таблице последовательностей и описано выше, VH и тяжелая цепь могут содержать N-концевой глутамин, и тяжелая цепь также может содержать С-концевой лизин. Как хорошо известно в данной области техники, N-концевые остатки глутамина могут образовывать пироглутамат, а С-концевые остатки лизина могут быть удалены во время процесса производства.
ТАБЛИЦА ПОСЛЕДОВАТЕЛЬНОСТЕЙ
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЛЕЧЕНИЯ КОЖНОЙ ИНФЕКЦИИ ПУТЕМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2015 |
|
RU2704999C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ТЯЖЕЛОГО АТОПИЧЕСКОГО ДЕРМАТИТА ПУТЕМ ВВЕДЕНИЯ ИНГИБИТОРА IL-4R | 2017 |
|
RU2759630C2 |
| СПОСОБЫ ЛЕЧЕНИЯ АТОПИЧЕСКОГО ДЕРМАТИТА ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2020 |
|
RU2828020C2 |
| СПОСОБЫ ДИАГНОСТИКИ И ЛЕЧЕНИЯ, СВЯЗАННЫЕ С ИНГИБИРОВАНИЕМ TH2 | 2011 |
|
RU2578468C2 |
| СПОСОБЫ ДИАГНОСТИКИ И ЛЕЧЕНИЯ, СВЯЗАННЫЕ С ИНГИБИРОВАНИЕМ ТН2 | 2011 |
|
RU2737245C2 |
| СПОСОБЫ ПОВЫШЕНИЯ ЭФФЕКТИВНОСТИ ВАКЦИНЫ ПУТЕМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2017 |
|
RU2753869C2 |
| СПОСОБЫ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ АЛЛЕРГИИ ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2017 |
|
RU2777328C2 |
| СПОСОБ ЛЕЧЕНИЯ АТОПИЧЕСКОГО ДЕРМАТИТА ПОСРЕДСТВОМ ВВЕДЕНИЯ ИНГИБИТОРА ИЛ-4R | 2019 |
|
RU2801204C2 |
| СПОСОБЫ ДИАГНОСТИКИ И ЛЕЧЕНИЯ, СВЯЗАННЫЕ С ИНГИБИРОВАНИЕМ TH2 | 2020 |
|
RU2832013C2 |
| ЛЕЧЕНИЕ АЛЛЕРГИЧЕСКИХ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ IgE | 2018 |
|
RU2800765C2 |
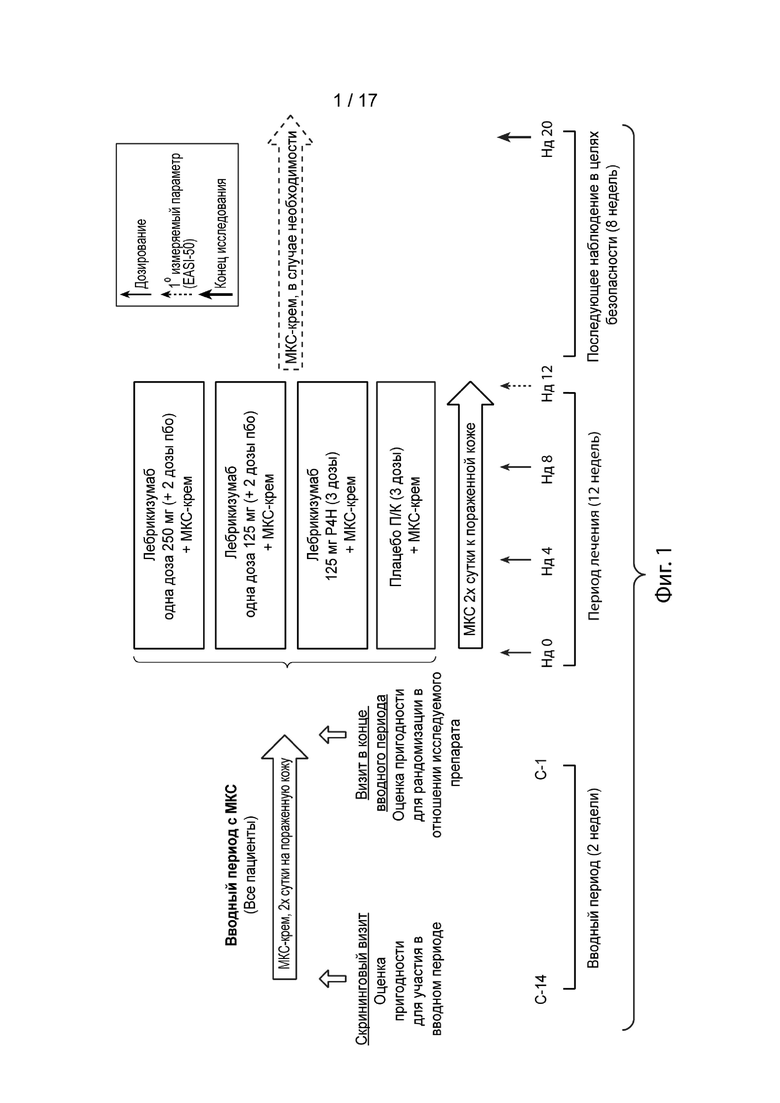
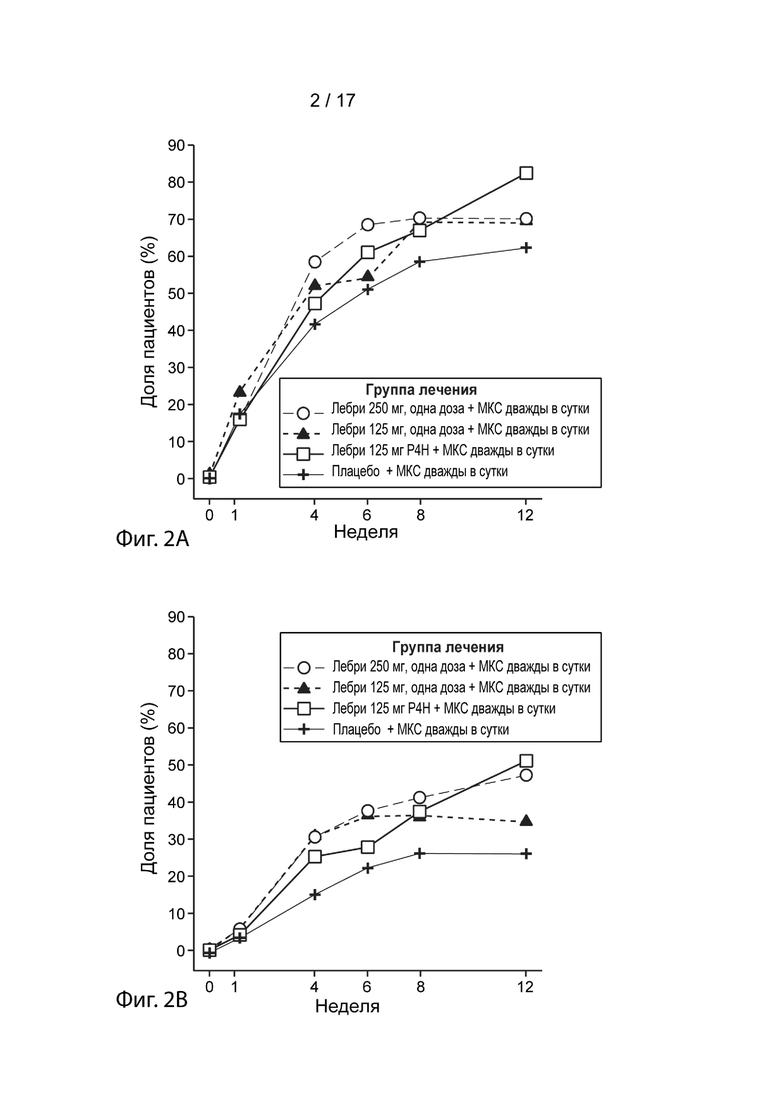
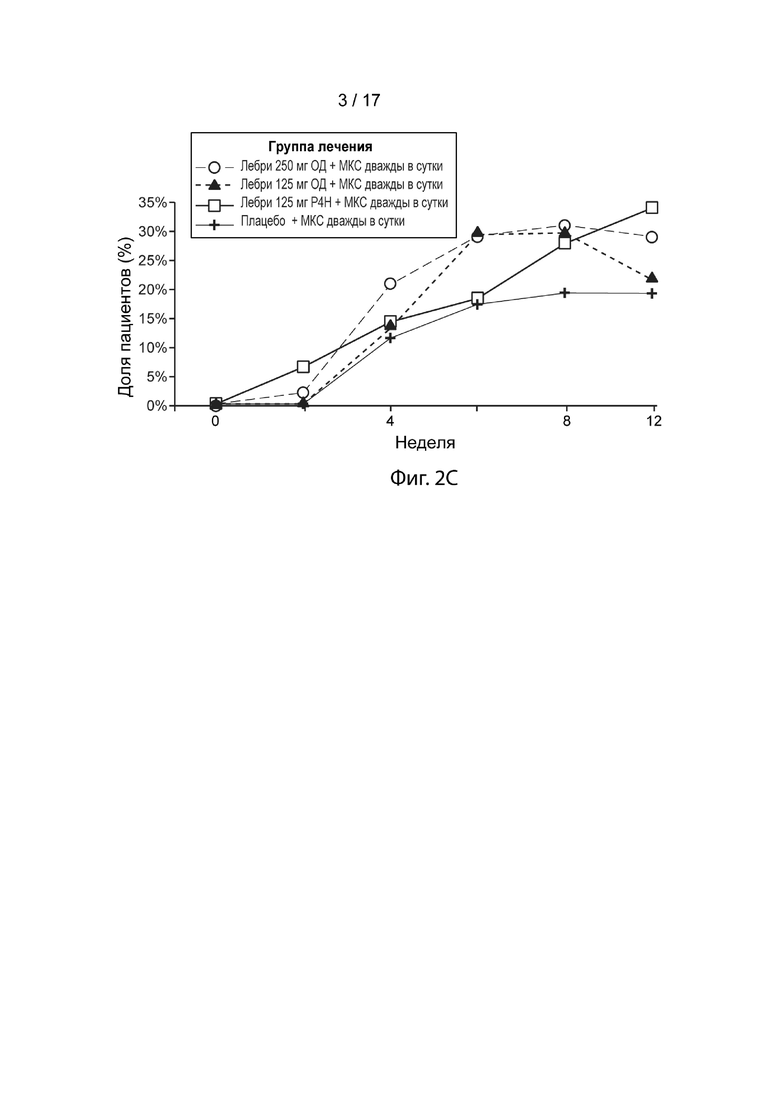
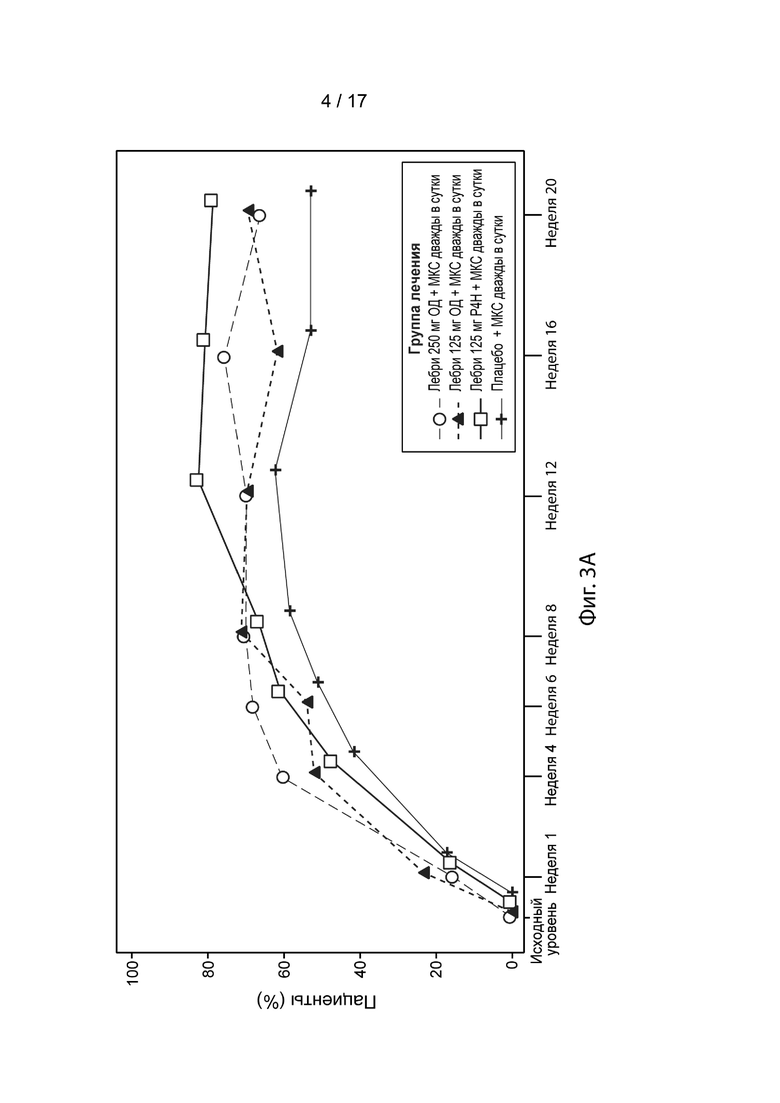
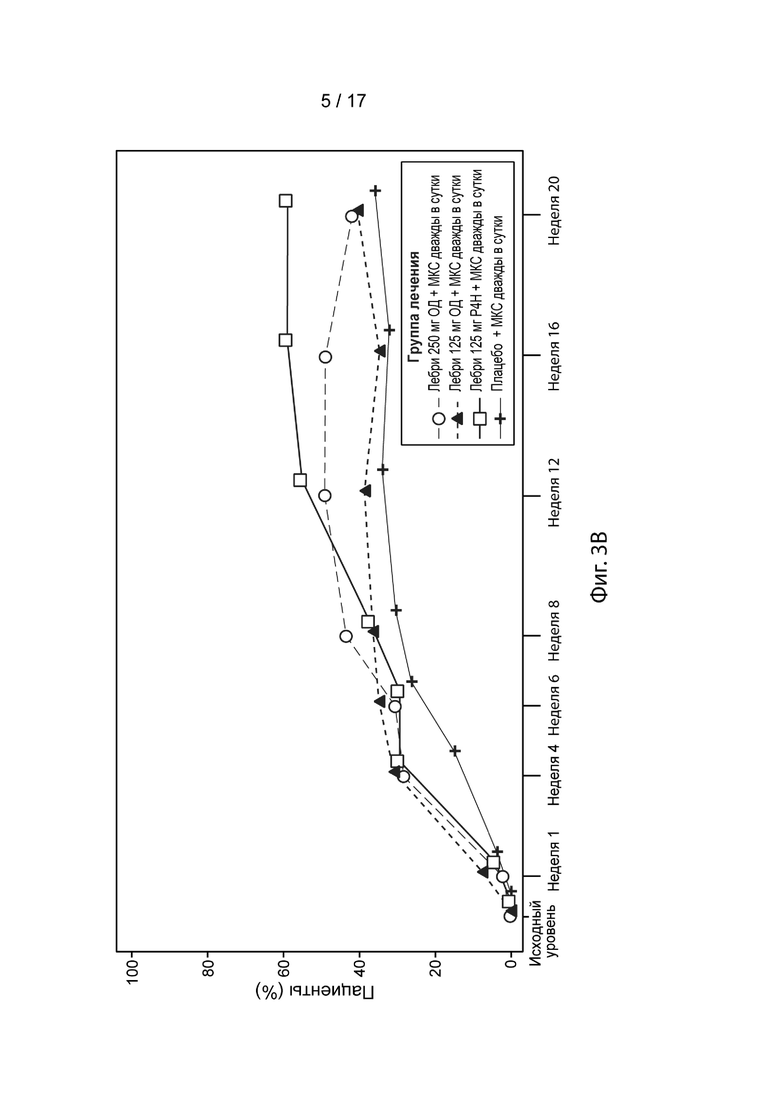
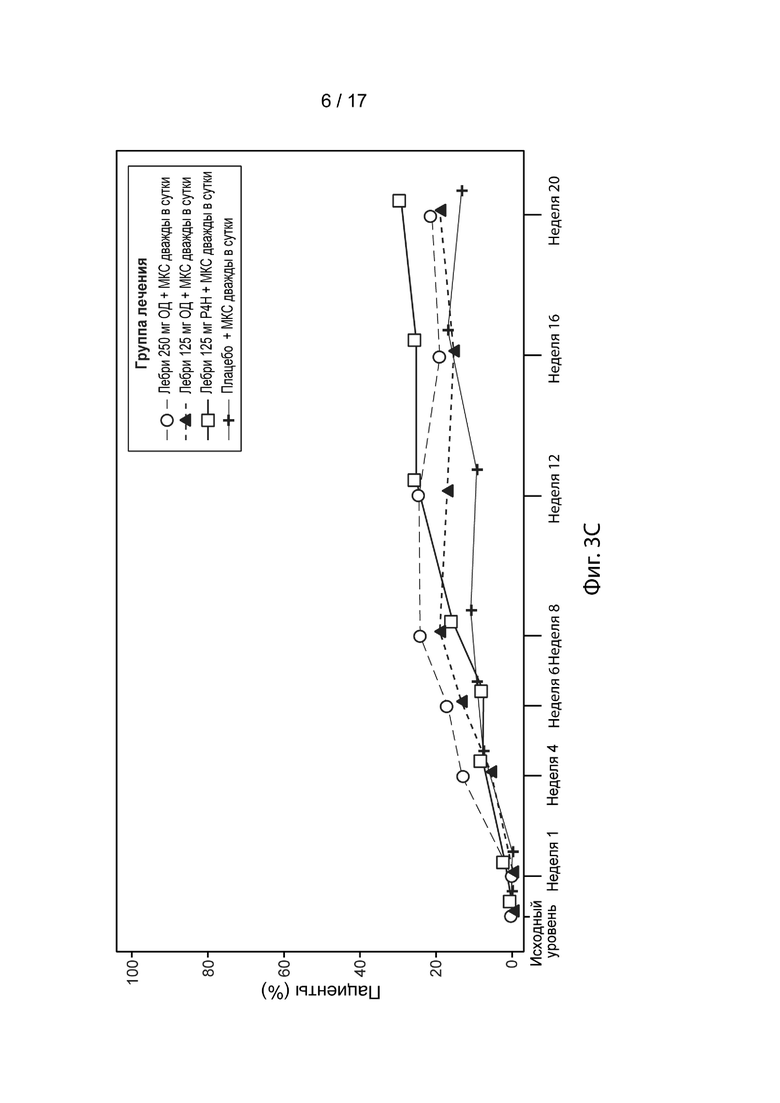
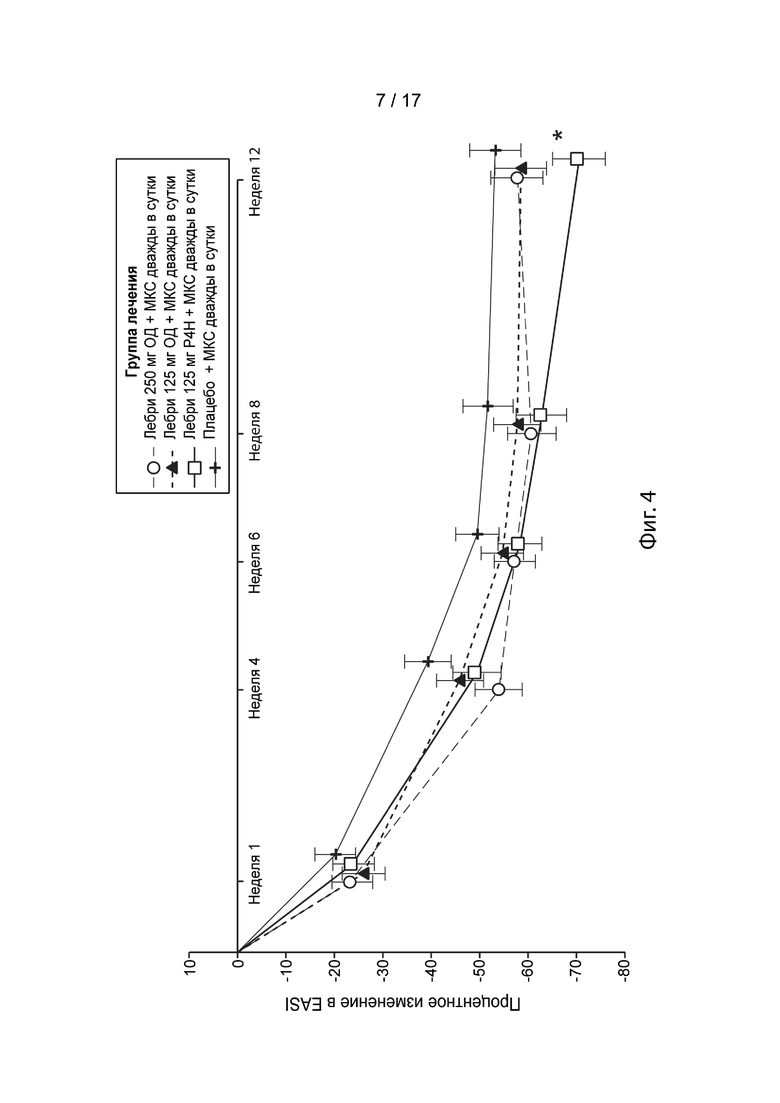
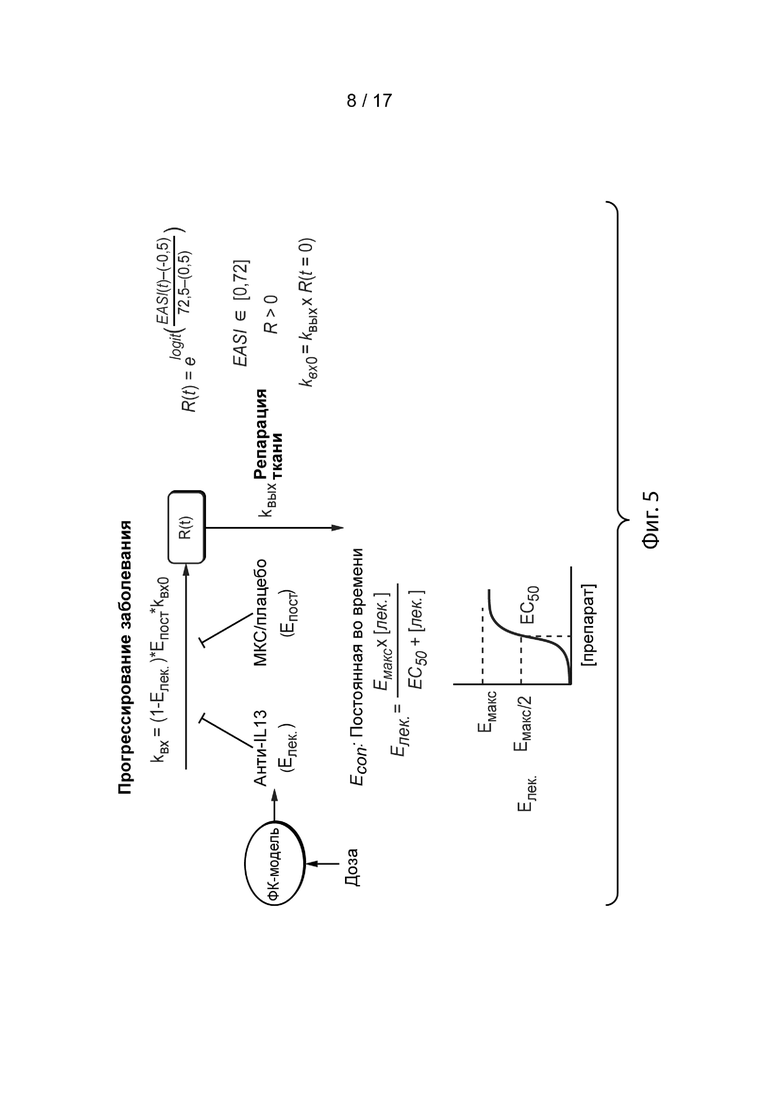
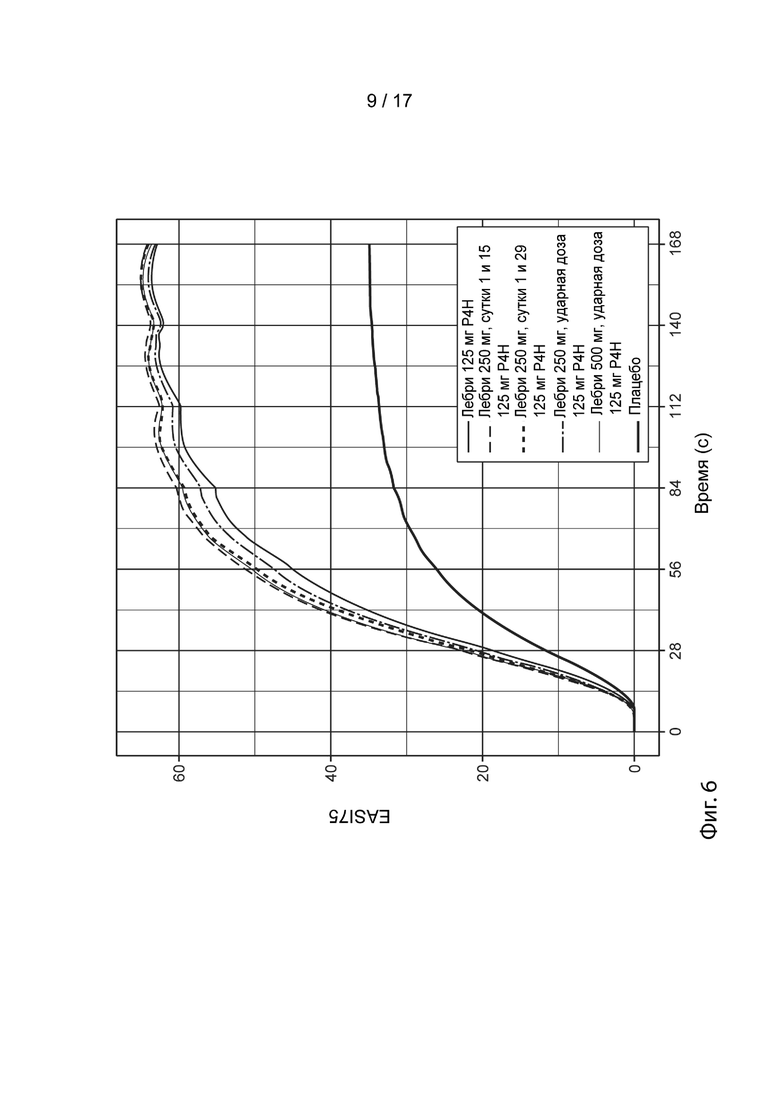
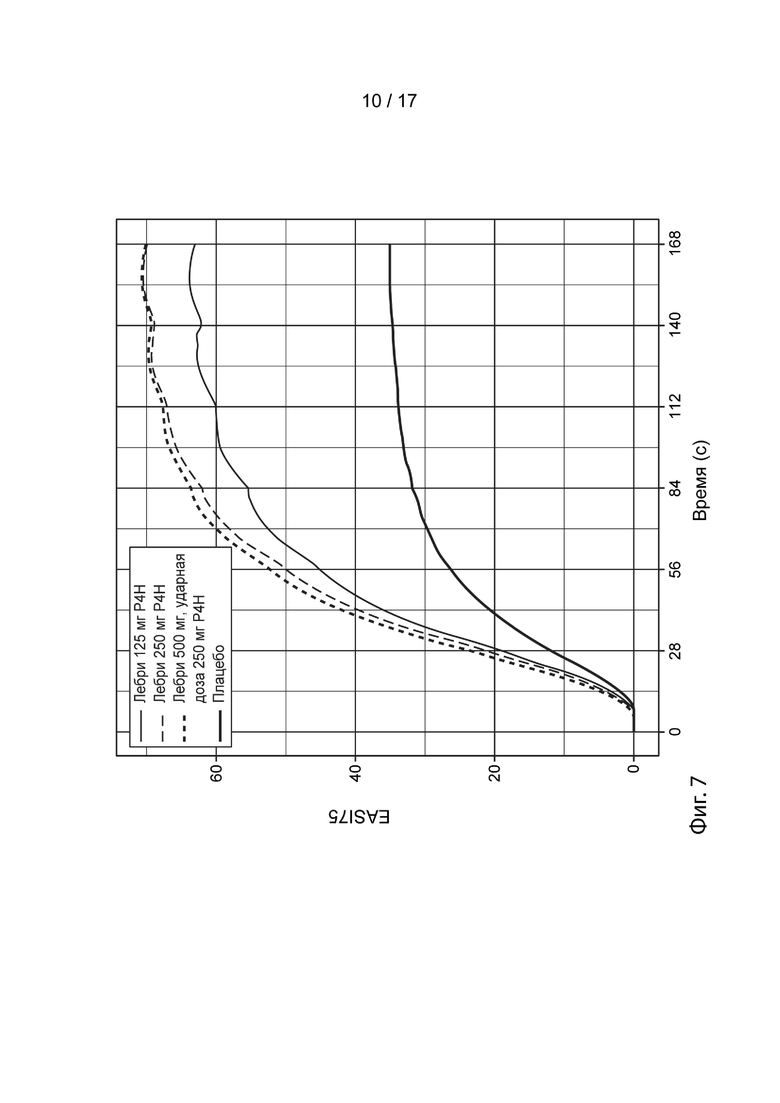
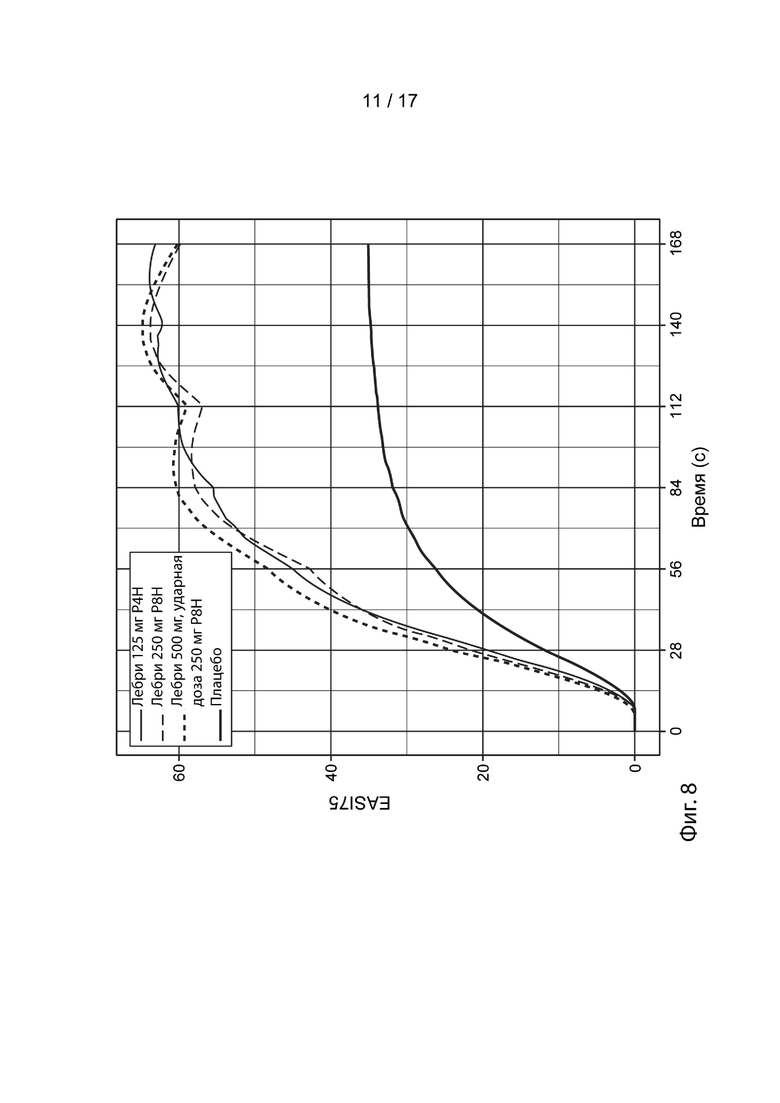
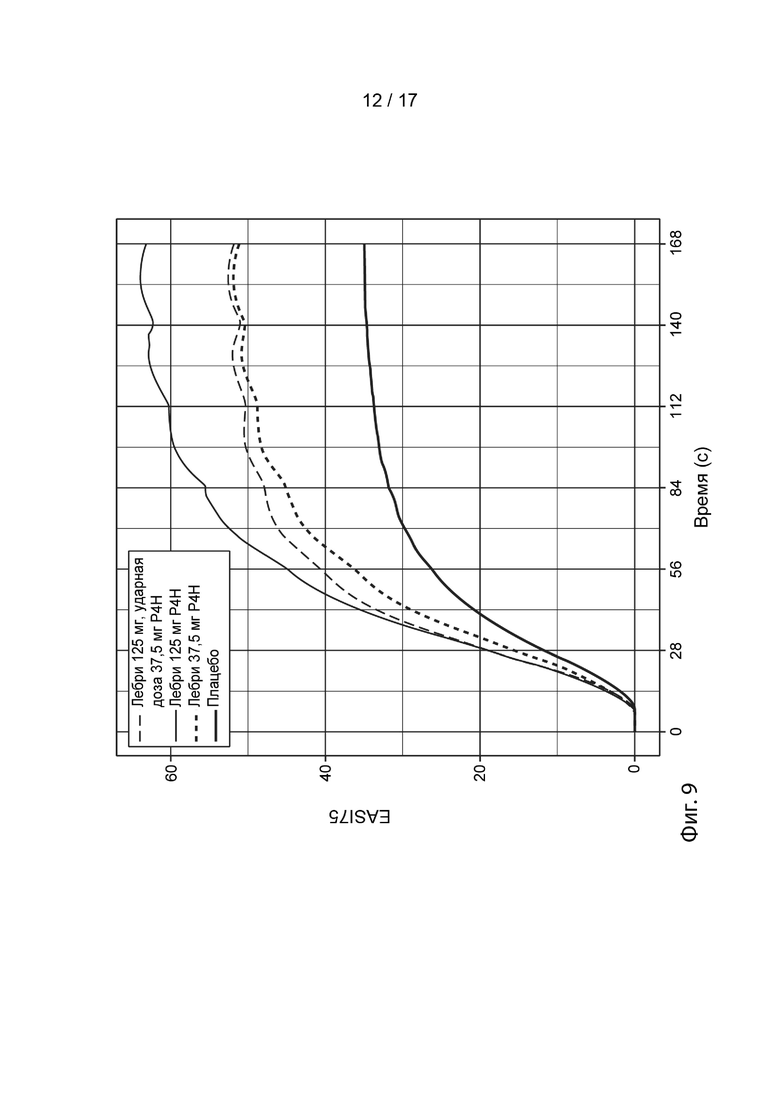
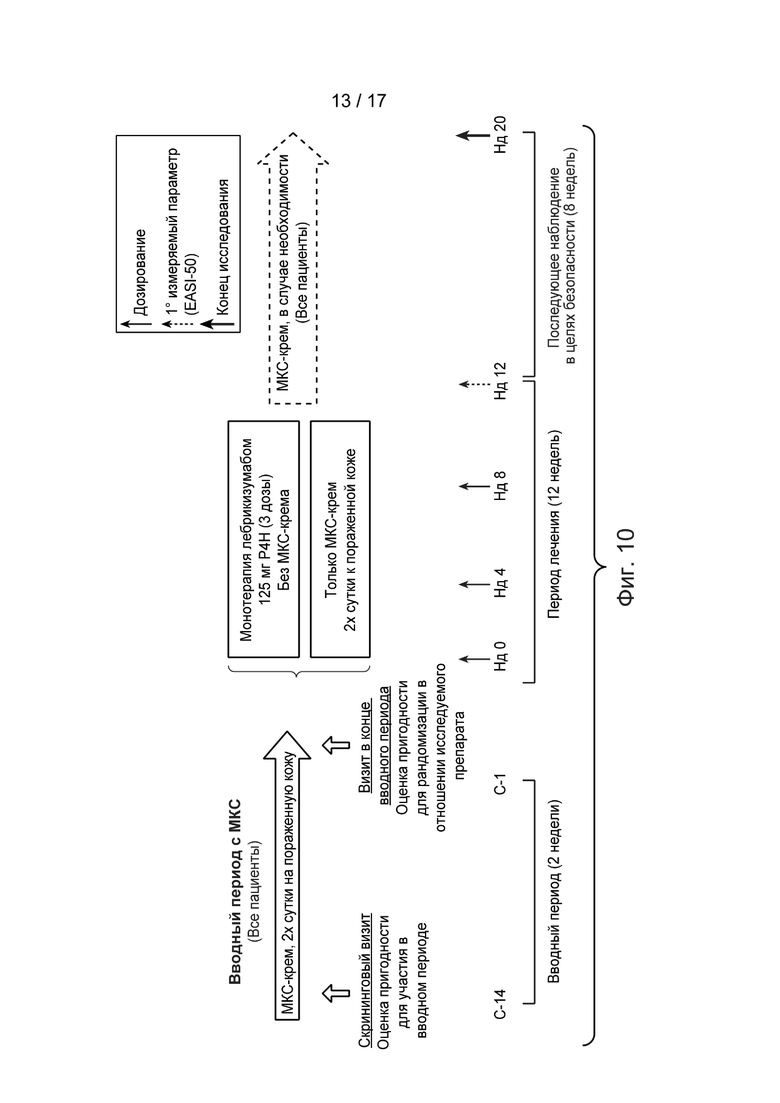
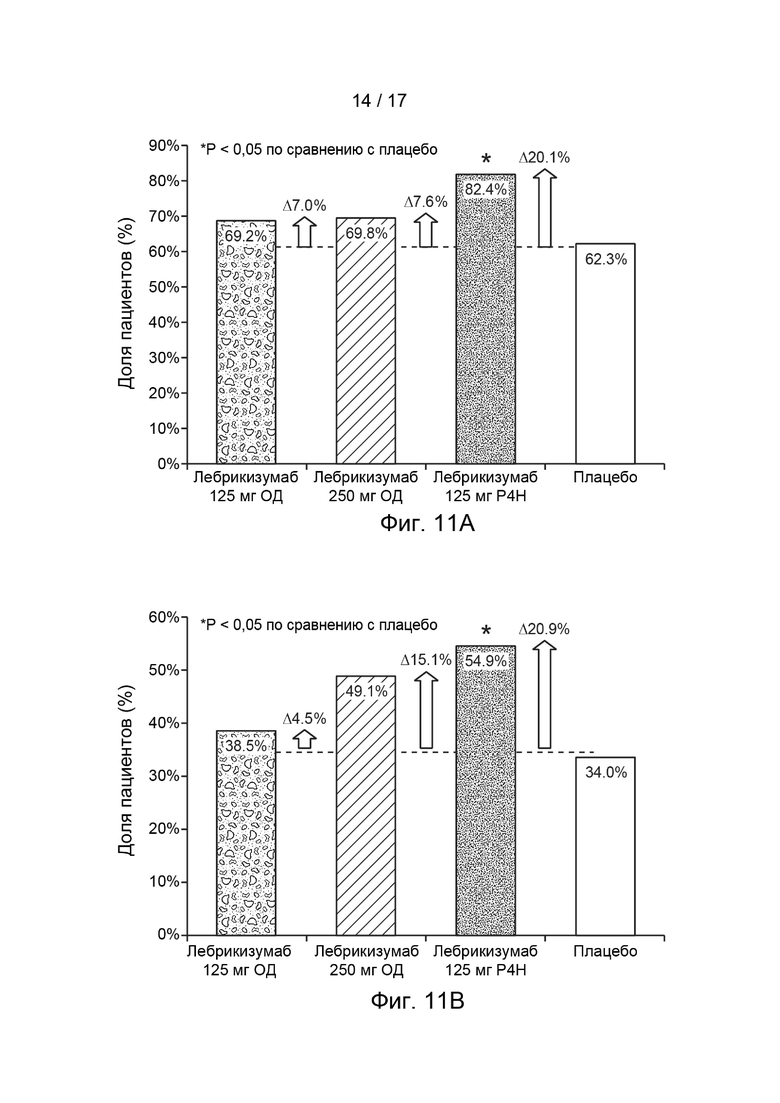
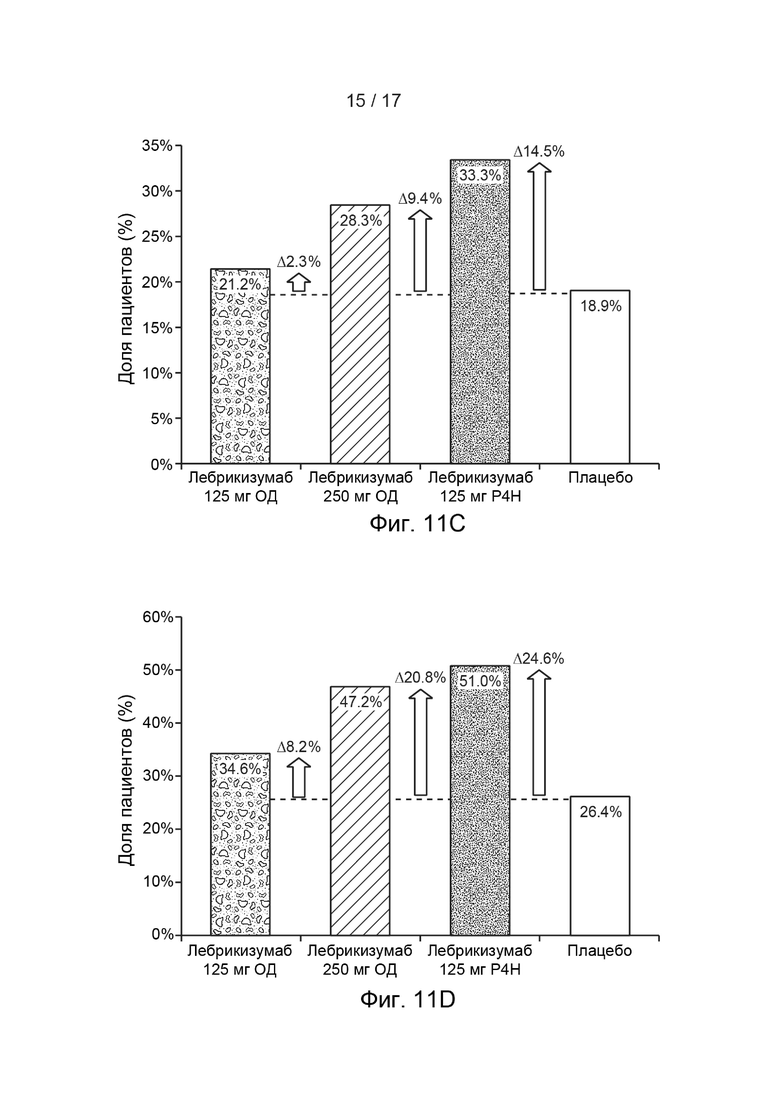
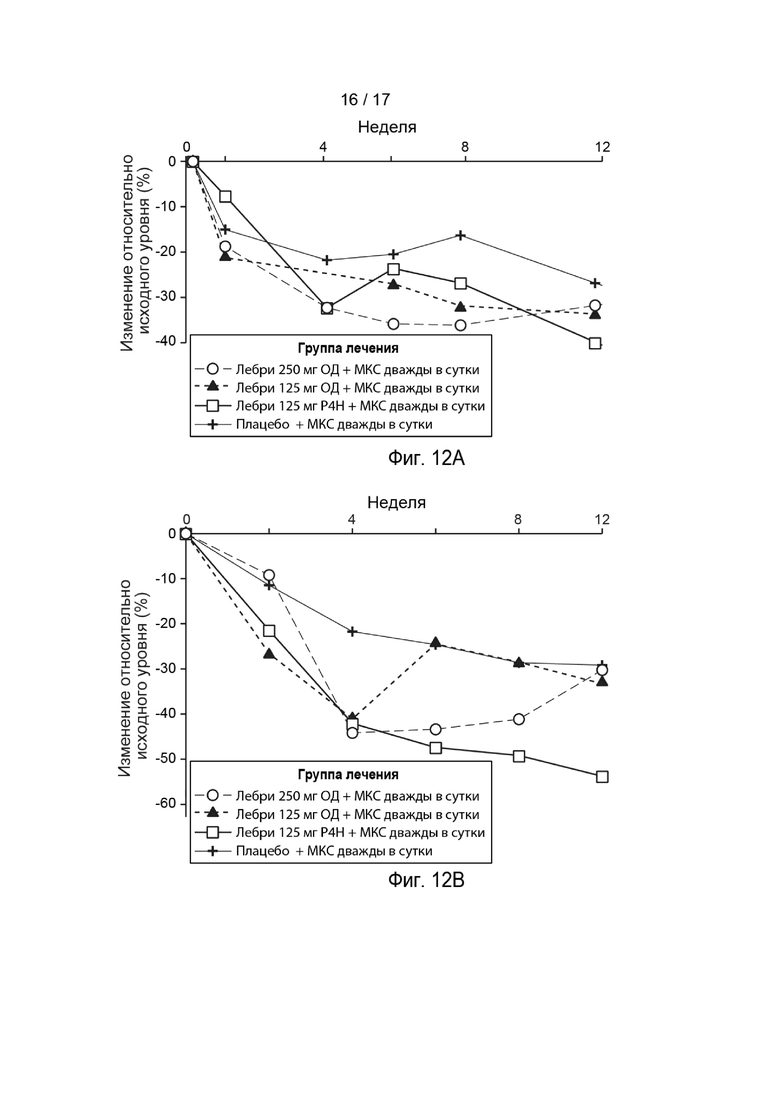
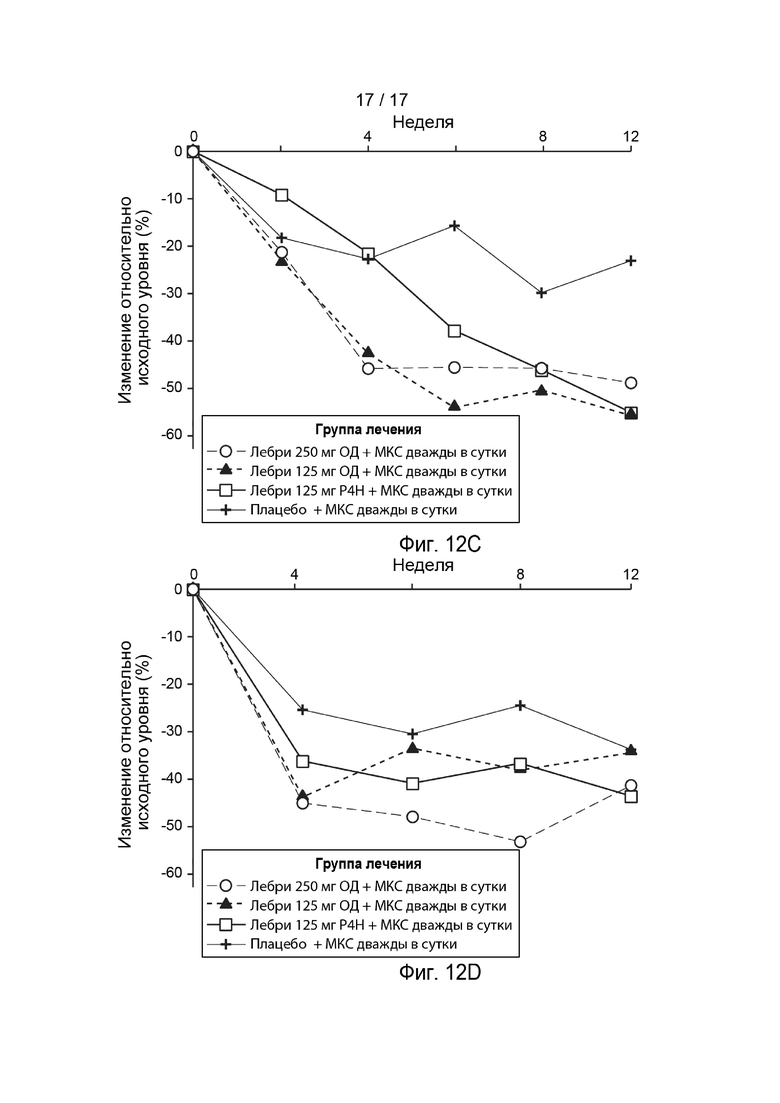
Группа изобретений относится к области медицины, а именно к дерматологии и иммунологии, и предназначена для лечения атопического дерматита у пациента. Для лечения атопического дерматита у пациента осуществляют введение пациенту фармацевтической композиции, содержащей 125 мг, или 250 мг, или 500 мг анти-IL-13 антитела. Фармацевтическая композиция снижает тяжесть заболевания у пациента, которая оценивается по критерию оценки тяжести заболевания для атопического дерматита (Atopic Dermatitis Disease Severity Outcome Measure). Анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую HVRH1, HVR-H2 и HVR-H3, при этом соответствующие VH HVR имеют аминокислотную последовательность SEQ ID NO: 5, SEQ ID NO: 6 и SEQ ID NO: 7, и содержащее VL, содержащую HVR-L1, HVR-L2 и HVR-L3, при этом соответствующие VL HVR имеют аминокислотную последовательность SEQ ID NO: 8, SEQ ID NO: 9 и SEQ ID NO: 10. В другом воплощении осуществляют введение пациенту 125 мг или 250 мг анти-IL-13 антитела подкожно один раз в четыре недели или один раз в восемь недель. Также представлен способ лечения, включающий введение анти-IL-13 антитела в одной ударной дозе, которая составляет 250 мг или 500 мг, и одной последующей поддерживающей дозе, которая составляет 125 мг или 250 мг, при этом каждую из ударной дозы и поддерживающей дозы вводят подкожно в виде постоянной дозы. Использование группы изобретений позволяет уменьшить тяжесть симптомов атопического дерматита и максимизировать эффективность лечения. 3 н. и 73 з.п. ф-лы, 12 ил., 9 табл., 4 пр.
1. Способ лечения атопического дерматита у пациента, включающий введение пациенту фармацевтической композиции, содержащей 125 мг, или 250 мг, или 500 мг анти-IL-13 антитела, при этом фармацевтическая композиция снижает тяжесть заболевания у пациента, где тяжесть заболевания оценивается по критерию оценки тяжести заболевания для атопического дерматита (Atopic Dermatitis Disease Severity Outcome Measure) и где анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую HVRH1, HVR-H2 и HVR-H3, при этом соответствующие VH HVR имеют аминокислотную последовательность SEQ ID NO: 5, SEQ ID NO: 6 и SEQ ID NO: 7, и содержащее VL, содержащую HVR-L1, HVR-L2 и HVR-L3, при этом соответствующие VL HVR имеют аминокислотную последовательность SEQ ID NO: 8, SEQ ID NO: 9 и SEQ ID NO: 10.
2. Способ по п. 1, отличающийся тем, что анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую последовательность, выбранную из SEQ ID NO: 1 и SEQ ID NO: 3, и содержащее VL, содержащую последовательность, выбранную из SEQ ID NO: 2 и SEQ ID NO: 4.
3. Способ по п. 1, отличающийся тем, что анти-IL-13 антитело содержит тяжелую цепь, имеющую аминокислотную последовательность SEQ ID NO: 11, и легкую цепь, имеющую аминокислотную последовательность SEQ ID NO: 12.
4. Способ по любому из пп. 1-3, отличающийся тем, что фармацевтическая композиция содержит 125 мг анти-IL-13 антитела и при этом композицию вводят подкожно один раз в четыре недели.
5. Способ по любому из пп. 1-3, отличающийся тем, что фармацевтическая композиция содержит 250 мг анти-IL-13 антитела и при этом композицию вводят подкожно один раз в четыре недели.
6. Способ по любому из пп. 1-3, отличающийся тем, что фармацевтическая композиция содержит 250 мг анти-IL-13 антитела и при этом композицию вводят подкожно один раз в восемь недель.
7. Способ по любому из пп. 4-6, отличающийся тем, что фармацевтическую композицию вводят в течение периода, составляющего 24 недели или более.
8. Способ по любому из пп. 1-7, отличающийся тем, что критерий оценки тяжести заболевания для атопического дерматита представляет индекс распространенности и тяжести экземы (EASI – от англ. «Eczema Area and Severity Index»).
9. Способ по п. 8, отличающийся тем, что фармацевтическая композиция снижает EASI на 50%, или 75%, или 90% по сравнению с EASI, определенным до введения первой дозы фармацевтической композиции.
10. Способ по п. 9, отличающийся тем, что EASI определяют через 12 недель после введения первой дозы, или через 20 недель после введения первой дозы, или через 24 недели после введения первой дозы.
11. Способ по любому из пп. 1-7, отличающийся тем, что критерий оценки тяжести заболевания для атопического дерматита представляет оценку тяжести атопического дерматита (SCORAD – от англ. «Severity Scoring of Atopic Dermatitis»).
12. Способ по п. 11, отличающийся тем, что фармацевтическая композиция снижает SCORAD на 50% или 75% по сравнению со SCORAD, определенной до введения первой дозы фармацевтической композиции.
13. Способ по п. 12, отличающийся тем, что SCORAD определяют через 12 недель после введения первой дозы.
14. Способ по любому из пп. 1-7, отличающийся тем, что критерий оценки тяжести заболевания для атопического дерматита представляет общую оценку исследователем (IGA – от англ. «Investigator Global Assessment»).
15. Способ по п. 14, отличающийся тем, что фармацевтическая композиция снижает IGA до нуля или единицы.
16. Способ по п. 15, отличающийся тем, что IGA определяют через 12 недель после введения первой дозы фармацевтической композиции.
17. Способ по любому из пп. 1-7, отличающийся тем, что критерий оценки тяжести заболевания для атопического дерматита представляет оценку результатов пациентами (PRO – от англ. «Patient Reported Outcome»), и причем PRO представляет собой визуальную аналоговую шкалу (ВАШ) зуда, ВАШ бессонницы или оценку по опроснику по влиянию атопического дерматита (ADIQ – от англ. «Atopic Dermatitis Impact Questionnaire»).
18. Способ по п. 17, отличающийся тем, что PRO определяют через 12 недель после введения первой дозы фармацевтической композиции.
19. Способ по п. 17 или 18, отличающийся тем, что фармацевтическая композиция снижает ВАШ зуда на 40%-55%.
20. Способ по п. 17 или 18, отличающийся тем, что фармацевтическая композиция снижает ВАШ бессонницы на 53%-61%.
21. Способ по п. 17 или 18, отличающийся тем, что фармацевтическая композиция снижает оценку ADIQ на 54%-65%.
22. Способ лечения атопического дерматита у пациента, включающий введение пациенту 125 мг или 250 мг анти-IL-13 антитела, где анти-IL-13 антитело вводят подкожно один раз в четыре недели или один раз в восемь недель и где анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую HVR-H1, HVR-H2 и HVR-H3, при этом соответствующие VH HVR имеют аминокислотную последовательность SEQ ID NO: 5, SEQ ID NO: 6 и SEQ ID NO: 7, и содержащее VL, содержащую HVR-L1, HVR-L2 и HVR-L3, при этом соответствующие VL HVR имеют аминокислотную последовательность SEQ ID NO: 8, SEQ ID NO: 9 и SEQ ID NO: 10.
23. Способ по п. 22, отличающийся тем, что анти-IL-13 антитело вводят в количестве 125 мг один раз в четыре недели.
24. Способ по п. 22, отличающийся тем, что анти-IL-13 антитело вводят в количестве 250 мг один раз в четыре недели.
25. Способ по п. 22, отличающийся тем, что анти-IL-13 антитело вводят в количестве 250 мг один раз в восемь недель.
26. Способ по любому из пп. 22-25, отличающийся тем, что анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую последовательность, выбранную из SEQ ID NO: 1 и SEQ ID NO: 3, и содержащее VL, содержащую последовательность, выбранную из SEQ ID NO: 2 и SEQ ID NO: 4.
27. Способ по любому из пп. 22-25, отличающийся тем, что анти-IL-13 антитело содержит тяжелую цепь, имеющую аминокислотную последовательность SEQ ID NO: 11, и легкую цепь, имеющую аминокислотную последовательность SEQ ID NO: 12.
28. Способ по любому из пп. 22-27, отличающийся тем, что терапевтически эффективное количество снижает тяжесть заболевания у пациента, а тяжесть заболевания оценивается по критерию оценки тяжести заболевания для атопического дерматита (ADDSOM).
29. Способ по п. 28, отличающийся тем, что критерий оценки тяжести заболевания для атопического дерматита представляет индекс распространенности и тяжести экземы (EASI).
30. Способ по п. 29, отличающийся тем, что терапевтически эффективное количество снижает EASI на 50%, или 75%, или 90% по сравнению с EASI, определенным до введения первой дозы анти-IL-13 антитела.
31. Способ по п. 30, отличающийся тем, что EASI определяют через 12 недель после введения первой дозы, или через 20 недель после введения первой дозы, или через 24 недели после введения первой дозы.
32. Способ по п. 28, отличающийся тем, что критерий оценки тяжести заболевания для атопического дерматита представляет оценку тяжести атопического дерматита (SCORAD).
33. Способ по п. 32, отличающийся тем, что терапевтически эффективное количество снижает SCORAD на 50% или 75 % по сравнению со SCORAD, определенной до введения первой дозы анти-IL-13 антитела.
34. Способ по п. 32, отличающийся тем, что SCORAD определяют через 12 недель после введения первой дозы.
35. Способ по п. 28, отличающийся тем, что критерий оценки тяжести заболевания для атопического дерматита представляет общую оценку исследователем (IGA).
36. Способ по п. 35, отличающийся тем, что терапевтически эффективное количество снижает IGA до нуля или единицы.
37. Способ по п. 36, отличающийся тем, что IGA определяют через 12 недель после введения первой дозы анти-IL-13 антитела.
38. Способ по п. 28, отличающийся тем, что критерий оценки тяжести заболевания для атопического дерматита представляет оценку результатов пациентами (PRO), и где PRO представляет собой визуальную аналоговую шкалу (ВАШ) зуда, ВАШ бессонницы или оценку по опроснику по влиянию атопического дерматита (ADIQ).
39. Способ по п. 38, отличающийся тем, что PRO определяют через 12 недель после введения первой дозы анти-IL-13 антитела.
40. Способ по п. 38 или 39, отличающийся тем, что терапевтически эффективное количество снижает ВАШ зуда на 40%-55%.
41. Способ по п. 38 или 39, отличающийся тем, что терапевтически эффективное количество снижает ВАШ бессонницы на 53%-61%.
42. Способ по п. 38 или 39, отличающийся тем, что терапевтически эффективное количество снижает ADIQ на 54%-65%.
43. Способ лечения атопического дерматита у пациента, включающий введение пациенту терапевтически эффективного количества анти-IL-13 антитела, при этом анти-IL-13 антитело вводят пациенту в одной ударной дозе, которая составляет 250 мг или 500 мг, и одной последующей поддерживающей дозе, которая составляет 125 мг или 250 мг, при этом каждую из ударной дозы и поддерживающей дозы вводят подкожно в виде постоянной дозы, и где анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую HVR-H1, HVR-H2 и HVR-H3, при этом соответствующие VH HVR имеют аминокислотную последовательность SEQ ID NO: 5, SEQ ID NO: 6 и SEQ ID NO: 7, и содержащее VL, содержащую HVR-L1, HVR-L2 и HVR-L3, при этом соответствующие VL HVR имеют аминокислотную последовательность SEQ ID NO: 8, SEQ ID NO: 9 и SEQ ID NO: 10.
44. Способ по п. 43, отличающийся тем, что анти-IL-13 антитело представляет собой антитело, содержащее VH, содержащую последовательность, выбранную из SEQ ID NO: 1 и SEQ ID NO: 3, и содержащее VL, содержащую последовательность, выбранную из SEQ ID NO: 2 и SEQ ID NO: 4.
45. Способ по п. 43, отличающийся тем, что анти-IL-13 антитело содержит тяжелую цепь, имеющую аминокислотную последовательность SEQ ID NO: 11, и легкую цепь, имеющую аминокислотную последовательность SEQ ID NO: 12.
46. Способ по п. 43, отличающийся тем, что ударная доза составляет 250 мг, а поддерживающая доза составляет 125 мг.
47. Способ по п. 43, отличающийся тем, что ударная доза составляет 250 мг, при этом после введения ударной дозы следует введение второй ударной дозы через 15 суток.
48. Способ по п. 47, отличающийся тем, что поддерживающую дозу вводят через две недели после введения второй ударной дозы.
49. Способ по п. 43, отличающийся тем, что ударная доза составляет 250 мг, при этом после введения ударной дозы следует введение второй ударной дозы через 29 суток.
50. Способ по п. 49, отличающийся тем, что поддерживающую дозу вводят через четыре недели после введения второй ударной дозы, и причем поддерживающую дозу вводят один раз в четыре недели после этого в течение периода лечения.
51. Способ по п. 43, отличающийся тем, что ударная доза составляет 500 мг, а поддерживающая доза составляет 250 мг.
52. Способ по п. 51, отличающийся тем, что поддерживающую дозу вводят через четыре недели после введения ударной дозы, и причем поддерживающую дозу вводят один раз в четыре недели после этого в течение периода лечения.
53. Способ по любому из пп. 50 или 52, отличающийся тем, что период лечения составляет 24 недели или более.
54. Способ по любому из пп. 43-53, отличающийся тем, что анти-IL-13 антитело снижает тяжесть заболевания у пациента, а тяжесть заболевания оценивается по критерию оценки тяжести заболевания для атопического дерматита (ADDSOM).
55. Способ по п. 54, отличающийся тем, что критерий оценки тяжести заболевания для атопического дерматита представляет индекс распространенности и тяжести экземы (EASI).
56. Способ по п. 55, отличающийся тем, что анти-IL-13 антитело снижает EASI на 50%, или 75%, или 90% по сравнению с EASI, определенным до введения первой дозы анти-IL-13 антитела.
57. Способ по п. 56, отличающийся тем, что EASI определяют через 12 недель после введения первой дозы, или через 20 недель после введения первой дозы, или через 24 недели после введения первой дозы.
58. Способ по п. 54, отличающийся тем, что критерий оценки тяжести заболевания для атопического дерматита представляет оценку тяжести атопического дерматита (SCORAD).
59. Способ по п. 58, отличающийся тем, что анти-IL-13 антитело снижает SCORAD на 50% или 75% по сравнению со SCORAD, определенной до введения первой дозы анти-IL-13 антитела.
60. Способ по п. 59, отличающийся тем, что SCORAD определяют через 12 недель после введения первой дозы.
61. Способ по п. 54, отличающийся тем, что критерий оценки тяжести заболевания для атопического дерматита представляет общую оценку исследователем (IGA).
62. Способ по п. 61, отличающийся тем, что анти-IL-13 антитело снижает IGA до нуля или единицы.
63. Способ по п. 62, отличающийся тем, что IGA определяют через 12 недель после введения первой дозы анти-IL-13 антитела.
64. Способ по п. 54, отличающийся тем, что критерий оценки тяжести заболевания для атопического дерматита представляет оценку результатов пациентами (PRO), и где PRO представляет собой визуальную аналоговую шкалу (ВАШ) зуда, ВАШ бессонницы или оценку по опроснику по влиянию атопического дерматита (ADIQ).
65. Способ по п. 64, отличающийся тем, что PRO определяют через 12 недель после введения первой дозы анти-IL-13 антитела.
66. Способ по п. 64 или 65, отличающийся тем, что анти-IL-13 антитело снижает ВАШ зуда на 40%-55%.
67. Способ по п. 64 или 65, отличающийся тем, что анти-IL-13 антитело снижает ВАШ бессонницы на 53%-61%.
68. Способ по п. 64 или 65, отличающийся тем, что анти-IL-13 антитело снижает ADIQ на 54%-65%.
69. Способ по любому из пп. 1-68, отличающийся тем, что атопический дерматит является умеренным или тяжелым согласно определению с помощью оценки по шкале Райка/Лангеланда, при этом оценка по шкале Райка/Лангеланда составляет от 4,5 до 9.
70. Способ по любому из пп. 1-69, отличающийся тем, что анти-IL-13 антитело вводят пациенту с помощью устройства для подкожного введения.
71. Способ по п. 70, отличающийся тем, что устройство для подкожного введения выбрано из предварительно наполненного шприца, одноразового шприца-ручки, микроигольного устройства, микроинфузионного устройства, безыгольного инъекционного устройства и автоинжектора.
72. Способ по любому из пп. 1-71, отличающийся тем, что способ дополнительно включает введение одного или более местных кортикостероидов.
73. Способ по п. 72, отличающийся тем, что один или более местных кортикостероидов вводят до введения анти-IL-13 антитела, одновременно с введением анти-IL-13 антитела или после введения анти-IL-13 антитела.
74. Способ по п. 73, отличающийся тем, что один или более местных кортикостероидов выбраны из триамцинолона ацетонида, гидрокортизона и комбинации триамцинолона ацетонида и гидрокортизона.
75. Способ по любому из пп. 1-74, отличающийся тем, что возраст пациента составляет 12 лет и более.
76. Способ по любому из пп. 1-75, отличающийся тем, что пациент не достигает адекватного контроля при применении местных кортикостероидов.
| HOWELL MD et al | |||
| Past, present, and future for biologic intervention in atopic dermatitis | |||
| Allergy, 2015, 70(8): 887-96, c,887 кол.2, c.891 кол.1-2, табл.1 | |||
| A Study of Lebrikizumab in Participants With Persistent Moderate to Severe Atopic Dermatitis | |||
| СПОСОБ ПРОИЗВОДСТВА КОНСЕРВОВ "СУП ИЗ БАРАНИНЫ И ТЫКВЫ" СПЕЦИАЛЬНОГО НАЗНАЧЕНИЯ (ВАРИАНТЫ) | 2007 |
|
RU2340234C1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

