ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Задачей изобретения является выявление новых соединений, которые обладают ценными свойствами, в частности таких, которые можно применять для получения лекарственных средств.
Настоящее изобретение относится к производным имидазопиразинона, которые ингибируют активность танкираз (TANKs) и поли(ADP-рибоза)полимеразы PARP-1. Таким образом, соединения согласно настоящему изобретению пригодны для лечения заболеваний, таких как злокачественное новообразование, рассеянный склероз, сердечно-сосудистые заболевания, поражения центральной нервной системы и различные формы воспаления. Настоящее изобретение также обеспечивает способы получения этих соединений, фармацевтические композиции, которые содержат эти соединения, и способы лечения заболеваний с применением фармацевтических композиций, содержащих эти соединения.
Ядерный фермент поли(ADP-рибоза)полимераза-1 (PARP-1) является представителем семейства ферментов PARP. Это растущее семейство ферментов состоит из PARPs, таких как, например: PARP-1, PARP-2, PARP-3 и Vault-PARP; и танкираз (TANKs), таких как, например: TANK-1 и TANK-2. PARP также обозначается как поли(аденозин5'-дифосфорибоза)полимераза или PARS (поли(ADP-рибоза)синтетаза).
Полагают, что TANK-1 необходима для полимеризаций связанной с митотическим веретеном поли(ADP-рибозы). Поли(ADP-рибозил)ирующая активность TANK-1 должна быть решающей для точного образования и поддержания биполярности веретена. Кроме того, полагают, что PARP активность TANK-1 необходима для нормального разделения теломер перед анафазой. Интерференция с танкиразной PARP активностью приводит к аберрантному митозу, который вызывает временную остановку клеточного цикла, возможно, вследствие активации контрольной точки веретена, с последующей клеточной гибелью. Таким образом, считают, что ингибирование танкираз имеет цитотоксическое действие на пролиферирующие опухолевые клетки (WO 2008/107478).
Ингибиторы PARP описаны у М. Rouleau и др. в Nature Reviews, том 10, 293-301 в клинических исследованиях злокачественных новообразований (таблица 2, страница 298).
В соответствии с обзором Horvath and Szabo (Drug News Perspect 20 (3), April 2007, 171-181) в самых последних исследованиях было доказано, что ингибиторы PARP усиливают гибель раковых клеток главным образом вследствие того, что они препятствуют репарации ДНК на различных уровнях. В более ранних исследованиях также было доказано, что ингибиторы PARP ингибируют ангиогенез, или путем ингибирования экспрессии фактора роста, или путем ингибирования индуцированных фактором роста клеточных пролиферативных ответов. Эти полученные данные также могут оказывать влияние на характер противораковых эффектов ингибиторов PARP in vivo.
Также в исследовании Tentori и др. (Eur. J. Cancer, 2007, 43 (14) 2124-2133) было установлено, что ингибиторы PARP аннулируют индуцированную VEGF или плацентарным фактором роста миграцию и предотвращают образование трубочкообразных сетей в клеточных системах, и повреждают ангиогенез in vivo. В исследовании также доказано, что индуцированный фактором роста ангиогенез является дефектным у PARP-1 нокаутных мышей. Результаты исследования обеспечивают подтверждение для нацеливания PARP на антиангиогенез, добавляя новые терапевтические показания к применению ингибиторов PARP в лечении злокачественного новообразования.
Хорошо известно, что дефекты в консервативных путях передачи сигналов играют ключевую роль в происхождении и поведении по существу всех злокачественных новообразований (E.A. Fearon, Cancer Cell, том 16, изд. 5, 2009, 366-368). Путь Wnt является мишенью для противораковой терапии. Ключевой особенностью пути Wnt является регулированный протеолиз (деградация) β-катенина с помощью комплекса, разрушающего β-катенин. В процесс разрушения задействованы белки, такие как WTX, АРС или Axin. Правильное разрушение β-катенина является важным для того, чтобы избежать несоответствующей активации пути Wnt, которая наблюдается при многих злокачественных новообразованиях. Танкиразы ингибируют активность Axin и, следовательно, ингибируют разрушение β-катенина. Поэтому ингибиторы танкиразы повышают разложение β-катенина. В статье журнала Nature были предложены не только важные новые сведения о белках, регулирующих передачу сигналов Wnt, но и дополнительно подтвержден подход относительно антагонизации уровней β-катенина и локализации с помощью небольших молекул (Huang и др., 2009; Nature, том 461, 614-620). Соединение XAV939 ингибирует рост DLD-1-раковых клеток. Авторами было обнаружено, что XAV9393 блокирует стимулированное посредством Wnt накопление β-катенина путем повышения уровней белков AXIN1 и AXIN2. В последующей работе авторами было установлено, что XAV939 регулирует уровни AXIN посредством ингибирования танкираз 1 и 2 (TNKS1 и TNKS2), обе из которых являются членами семейства белков поли(ADP-рибоза)полимеразы (PARP) (S.J. Hsiao and др., Biochimie 90, 2008, 83-92).
Было обнаружено, что соединения в соответствии с изобретением и их соли обладают чрезвычайно ценными фармакологическими свойствами, а также хорошей переносимостью.
В частности настоящее изобретение относится к соединениям формулы I, которые ингибируют танкиразу 1 и 2, к композициям, которые содержат эти соединения, и к способам их применения для лечения заболеваний и осложнений, индуцированных TANK.
Кроме того, соединения формулы I можно применять для выделения и исследования активности или экспрессии TANKs. К тому же, они особенно пригодны для применения в методах диагностики заболеваний, связанных с нерегулируемой или нарушенной активностью TANK.
Хозяин или пациент может принадлежать к любому из видов млекопитающих, например, к видам приматов, в частности, к людям; грызунам, включая мышей, крыс и хомяков; кроликам, лошадям, коровам, собакам, кошкам и т.д. Животные модели представляют интерес для экспериментальных исследований, обеспечивая модель для лечения болезней человека.
Восприимчивость определенной клетки к лечению соединениями в соответствии с данным изобретением может быть определена с помощью тестов in vitro. Обычно, к культуре клеток добавляют соединение в соответствии с данным изобретением в различных концентрациях в течение периода времени, который является достаточным для того, чтобы позволить активным агентам, таким, как анти IgM вызывать клеточный ответ, такой как экспрессия поверхностного маркера, как правило, от одного часа до одной недели. Исследование in vitro можно осуществлять при использовании культивируемых клеток, полученных из крови или из образца биопсии. Количество экспрессированного поверхностного маркера оценивают с помощью проточной цитометрии с применением специфических антител, которые распознают маркер.
Доза варьирует в зависимости от конкретного применяемого соединения, конкретного заболевания, состояния пациента и т.д. Обычно терапевтическая доза является достаточной для того, чтобы значительно уменьшить численность нежелательной клеточной популяции в целевой ткани наряду с поддержанием жизнеспособности пациента. Как правило, лечение продолжается до наступления значительного снижения, например, снижения, которое составляет, по меньшей мере, 50% клеточной нагрузки, и может продолжаться до по сути полного отсутствия обнаружения нежелательных клеток в организме.
ИЗВЕСТНЫЙ УРОВЕНЬ ТЕХНИКИ
Е. Wahlberg et al., Nature Biotechnology (2012), 30 (3), 283.
H. Bregman et al., Journal of Medicinal Chemistry (2013), 56 (3), 1341
Другие ингибиторы танкиразы описаны в заявках WO 2013/012723, WO 2013/010092, WO 2012/076898 и в WO 2013/008217.
В WO 201 1/089400 (CNIO, Centro Nacional de Investigaciones Oncologicas), раскрыты замещенные имидазопиразиноны, описанные как промежуточные соединения, например,
В Jpn. Kokai Tokkyo Koho (2005), JP 2005089352 A; Language: Japanese описаны:
Получение производных имидазол[1,5-а]пиразина, содержащие их фармацевтические композиции и их применения для предупреждения или лечения заболеваний, связанных с протеинтирозинкиназой (у Mukoyama, Harunobu; Nishimura, Toshihiro; Nakayama, Akiko; Kikuchi, Shinji; Komatsu, Yoshimitsu; Onoda, Hideki),
замещенные имидазопиразиноны как промежуточные соединения, например,
 .
.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Изобретение относится к соединениям формулы I
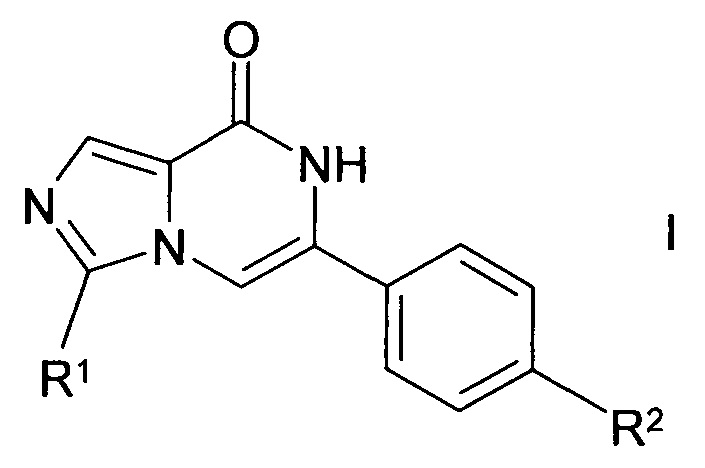
в которой
R1 означает Н или метил,
R2 означает A или Het,
А означает неразветвленный или разветвленный алкил с 1-8 атомами C, причем одна или две несмежные CH- и/или CH2-группы могут быть заменены посредством N- или О-атомов и/или причем 1-7 атомов Н могут быть заменены посредством F или Cl,
Het означает пиримидил, пиридил, пиридазинил, пиразинил, пиперидинил, пирролидинил, пиразолил, тиазолил, имидазолил, фуранил, тиофенил, пирролил, оксазолил, триазолил, оксадиазолил или тиадиазолил, каждый из которых является незамещенным или моно- или дизамещенным посредством Hal, A, CN, ОН и/или OA,
Hal означает F, Cl, Br или I,
при условии, что, если R1 означает Н, то R2 не означает 4-ОМе,
и к их фармацевтически приемлемым солям, таутомерам и стереоизомерам, включая их смеси во всех соотношениях.
Изобретение также относится к оптически активным формам (стереоизомерам), энантиомерам, рацематам, диастереомерам и гидратам и сольватам этих соединений.
Кроме того, изобретение относится к фармацевтически приемлемым производным соединений формулы I.
Термин сольваты соединений следует понимать как аддукции молекул инертного растворителя на соединениях, которые образуются вследствие силы их взаимного притяжения. Сольватами являются, например, моно- или дигидраты или алкоксиды.
Следует понимать, что изобретение также относится к сольватам солей.
Под термином фармацевтически приемлемые производные следует понимать, например, соли соединений в соответствии с изобретением, а также так называемые пролекарства соединений.
Как используют в настоящей заявке и если не указано иное, термин "пролекарство" означает производное соединения формулы I, которое можно подвергать гидролизу, окислению или иным образом реагировать в биологических условиях (in vitro или in vivo), чтобы получить активное соединение, в частности, соединение формулы I. Примеры пролекарств включают, но не ограничиваются перечисленными, производные и метаболиты соединений формулы I, которые включают биогидролизируемые фрагменты, такие как биогидролизируемые амиды, биогидролизируемые сложные эфиры, биогидролизируемые карбаматы, биогидролизируемые карбонаты, биогидролизируемые уреиды и биогидролизируемые аналоги фосфата. В некоторых вариантах осуществления пролекарства соединений с карбоксильными функциональными группами представляют собой низшие алкиловые сложные эфиры карбоновой кислоты. Сложные эфиры карбоновых кислот легко получают с помощью этерифицирования любых фрагментов карбоновых кислот, присутствующих на молекуле. Пролекарства типично могут быть получены с применением известных способов, таких как способы, описанные в Burger's Medicinal Chemistry and Drug Discovery 6-е изд. (ред. Donald J. Abraham, 2001, Wiley) и Design and Application of Prodrugs (ред. H. Bundgaard, 1985, Harwood Academic Publishers Gmfh).
Выражение "эффективное количество" означает количество лекарственного средства или фармацевтического активного компонента, которое вызывает в ткани, системе, животном или человеке биологическую или лекарственную ответную реакцию, которую стремится или желает получить, например, исследователь или лечащий врач.
Кроме того, выражение "терапевтически эффективное количество" означает то количество, которое, имеет следующие последствия по сравнению с соответствующим субъектом, который не получал этого количества:
улучшенное лечение, излечение, предотвращение или устранение заболевания, синдрома, состояния, жалобы, расстройства или побочных эффектов, а также уменьшение прогрессирования заболевания, жалобы или расстройства.
Выражение "терапевтически эффективное количество" также охватывает количества, которые эффективны для повышения нормальной физиологической функции.
Изобретение также относится к применению смесей соединений формулы I, например, смесей двух диастереомеров, например, в соотношении 1:1, 1:2, 1:3, 1:4, 1:5, 1:10, 1:100 или 1:1000.
Особенно предпочтительными являются смеси стереоизомерных соединений.
"Таутомеры" относятся к изомерным формам соединения, которые находятся в равновесии друг с другом. Концентрации изомерных форм будут зависеть от окружения, в котором находится соединение, и могут отличаться в зависимости от того, например, является ли соединение твердым веществом или находится в органическом или водном растворе.
Изобретение относится к соединениям формулы I и их солям, и к способу получения соединений формулы I и их фармацевтически применимых солей, сольватов, таутомеров и стереоизомеров, которые отличаются тем, что
соединение формулы II
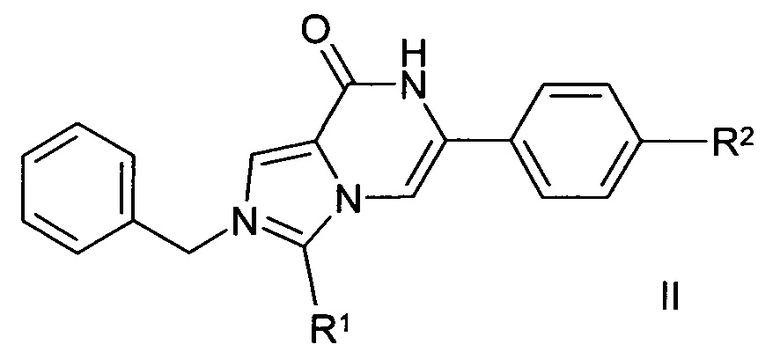
в которой R1 и R2 имеют значения, указанные в п. 1,
дебензилируют,
и/или
основание или кислоту формулы I превращают в одну из его(ее) солей.
Указанные выше и ниже радикалы R1 и R2 имеют значения, указанные для формулы I, если определенно не указано иное.
А означает алкил, который является неразветвленным (линейным) или разветвленным, и содержит 2, 3, 4, 5, 6, 7 или 8 атомов С. Предпочтительно А означает этил, пропил, изопропил, бутил, изобутил, втор-бутил или трет-бутил, кроме того также пентил, 1-, 2- или 3-метилбутил, 1,1-, 1,2- или 2,2-диметилпропил, 1-этилпропил, гексил, 1-, 2-, 3- или 4-метилпентил, 1,1-, 1,2-, 1,3-, 2,2-, 2,3- или 3,3-диметилбутил, 1- или 2-этилбутил, 1-этил-1-метилпропил, 1-этил-2-метилпропил, 1,1,2- или 1,2,2-триметилпропил, кроме того предпочтительно, например, трифторметил.
А особенно предпочтительно означает алкил, имеющий 2, 3, 4, 5 или 6 атомов C, предпочтительно этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, гексил, трифторметил, пентафторэтил или 1,1,1-трифторэтил.
Сверх того, А предпочтительно означает CH2OCH3, CH2CH2ОН или CH2CH2OCH3.
А предпочтительно также означает неразветвленный или разветвленный алкил с 1-8 атомами C, причем одна или две несмежные CH2-группы могут быть заменены посредством O-атомов и/или причем 1-3 Н-атомами могут быть заменены посредством F.
Het предпочтительно означает пиримидил, пиридил, пиридазинил, пиразинил, пиперидинил, пирролидинил, пиразолил, тиазолил, имидазолил, фуранил, тиофенил, пирролил, оксазолил, триазолил, оксадиазолил или тиадиазолил, каждый из которых является незамещенным или монозамещенным посредством А.
По всему описанию изобретения, все радикалы, которые встречаются более одного раза, могут быть одинаковыми или разными, т.е. независимыми друг от друга.
Соединения формулы I могут иметь один или несколько хиральных центров и поэтому могут встречаться в разных стереоизомерных формах. Формула I охватывает все эти формы.
Соответственно, изобретение в частности относится к соединениям формулы I, в которой по меньшей мере один из указанных радикалов имеет одно из предпочтительных значений, указанных выше. Некоторые предпочтительные группы соединений могут быть изображены с помощью следующих подформул от Iа до Id, которые соответствуют формуле I и в которых радикалы, не определенные более подробно, имеют значения, указанные для формулы I, но в которых
в Iа A означает неразветвленный или разветвленный алкил с 1-8 атомами C, причем одна или две несмежные CH2-группы могут быть заменены посредством O-атомов и/или причем 1-3 атомов Н могут быть заменены посредством F;
в Ib Het означает пиримидил, пиридил, пиридазинил, пиразинил, пиперидинил, пирролидинил, пиразолил, тиазолил, имидазолил, фуранил, тиофенил, пирролил, оксазолил, триазолил, оксадиазолил или тиадиазолил, каждый из которых является незамещенным или монозамещенным посредством A;
в Ic R1 означает Н или метил,
R2 означает A или Het,
А означает неразветвленный или разветвленный алкил с 1-8 атомами C, причем одна или две несмежные CH2-группы могут быть заменены посредством О-атомов и/или причем 1-3 атомов Н могут быть заменены посредством F,
Het означает пиримидил, пиридил, пиридазинил, пиразинил, пиперидинил, пирролидинил, пиразолил, тиазолил, имидазолил, фуранил, тиофенил, пирролил, оксазолил, триазолил, оксадиазолил или тиадиазолил, каждый из которых является незамещенным или монозамещенным посредством A;
и к их фармацевтически приемлемым солям, таутомерам и стереоизомерам, включая их смеси во всех соотношениях.
Кроме того, соединения формулы I, а также исходные вещества для их получения могут быть получены при помощи по сути известных способов, как описано в литературных источниках (например, в стандартных работах, таких как Houben-Weyl, Methoden der organischen Chemie [Методы органической химии], Georg-Thieme-Verlag, Штутгарт), точнее, в условиях реакций, которые известны и являются пригодными для указанных реакций. Также при этом можно применять разнообразные модификации, которые по сути известны, но о которых подробно не упоминается в данной заявке.
В целом, исходные соединения формулы II являются известными. Тем не менее, если они являются новыми, то их можно получить с помощью по сути, известных способов.
Соединения формулы I предпочтительно могут быть получены путем дебензилирования соединения формулы II.
Как правило, реакцию осуществляют в присутствии имидазола.
В зависимости от применяемых условий, время реакции находится в интервале от нескольких минут до 14 дней, температура реакции находится в пределах между приблизительно 60 и 250°, обычно между 80° и 200°, в частности между приблизительно 100° и приблизительно 180°.
Предпочтительно, реакцию проводят при отсутствии растворителя.
Фармацевтические соли и другие формы
Указанные соединения в соответствии с изобретением можно применять в своей конечной несолевой форме. С другой стороны, настоящее изобретение также охватывает применение таких соединений в форме их фармацевтически приемлемых солей, которые могут быть получены с помощью разнообразных органических и неорганических кислот и оснований в соответствии со способами, хорошо известными в данной области техники. Фармацевтически приемлемые солевые формы соединений формулы I получают, главным образом, при использовании традиционных способов. В случае, если соединение формулы I содержит карбоксильную группу, то его приемлемая соль может быть образована с помощью реакции соединения с приемлемым основанием для получения соответствующей соли присоединения основания. Такими основаниями являются, например, гидроксиды щелочных металлов, включая гидроксид калия, гидроксид натрия и гидроксид лития; гидроксиды щелочноземельных металлов, такие, как гидроксид бария и гидроксид кальция; алкоксиды щелочных металлов, например, этилат калия и пропилат натрия; а также различные органические основания, такие, как пиперидин, диэтаноламин и N-метилглутамин. Сюда также включены соли алюминия соединений формулы I. В случае некоторых соединений формулы I соли присоединения кислоты могут быть образованы путем обработки указанных соединений фармацевтически приемлемыми органическими и неорганическими кислотами, например, гидрогалогенидами, такими, как хлористый водород, бромистый водород или йодистый водород; другими минеральными кислотами, и их соответствующими солями такими, как, сульфат, нитрат или фосфат и т.п., и алкил- и моноарилсульфонатами, такими, как этансульфонат, толуолсульфонат и бензолсульфонат, и другими органическими кислотами и их соответствующими солями, такими, как ацетат, трифторацетат, тартрат, малеат, сукцинат, цитрат, бензоат, салицилат, аскорбат и т.п.
Таким образом, фармацевтически приемлемые соли присоединения кислоты соединений формулы I включают следующие, но не ограничиваются только ними: ацетат, адипат, альгинат, аргинат, аспартат, бензоат, бензолсульфонат (безилат), бисульфат, бисульфит, бромид, бутират, камфорат, камфорсульфонат, каприлат, хлорид, хлорбензоат, цитрат, циклопентанпропионат, диглюконат, дигидрофосфат, динитробензоат, додецилсульфат, этансульфонат, фумарат, формиат, галактерат (из муциновой кислоты), галактуронат, глюкогептаноат, глюконат, глутамат, глицерофосфат, гемисукцинат, гемисульфат, гептаноат, гексаноат, гиппурат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, йодид, изетионат, изобутират, лактат, лактобионат, малат, малеат, малонат, манделат, метафосфат, метансульфонат, метилбензоат, моногидрофосфат, 2-нафталинсульфонат, никотинат, нитрат, оксалат, олеат, пальмоат, пектинат, персульфат, фенилацетат, 3-фенилпропионат, фосфат, фосфонат, фталат.
Кроме того, основные соли соединений в соответствии с изобретением включают, но не ограничиваются только ними, соли алюминия, аммония, кальция, меди, железа(III), железа(II), лития, магния, марганца(III), марганца(II), калия, натрия и цинка. Среди перечисленных выше солей предпочтение отдают солям аммония; солям щелочных металлов-натрия и калия; и солям щелочноземельных металлов-кальция и магния. Соли соединений формулы I, которые получают из фармацевтически приемлемых органических нетоксических оснований, включают, но не ограничиваются только ними, соли первичных, вторичных и третичных аминов, замещенных аминов, также включая природные замещенные амины, циклические амины и основные ионообменные смолы, например, аргинин, бетаин, кофеин, хлорпрокаин, холин, N,N'-дибензилэтилендиамин (бензатин), дициклогексиламин, диэтаноламин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лидокаин, лизин, меглюмин, N-метил-D-глюкамин, морфолин, пиперазин, пиперидин, полиаминные смолы, прокаин, пурины, теобромин, триэтаноламин, триэтиламин, триметиламин, трипропиламин и трис-(гидроксиметил)метиламин (трометамин).
Соединения в соответствии с настоящим изобретением, которые включают основные азотсодержащие группы, могут быть кватернизированы с помощью таких агентов, как (C1-C4)-алкилгалогениды, например, метил-, этил-, изопропил- и трет-бутилхлорид, бромид и йодид; ди-(C1-C4)-алкилсульфаты, например, диметил-, диэтил- и диамилсульфат; (С10-С18)-алкилгалогениды, например, децил-, додецил-, лаурил-, миристил- и стеарилхлорид, бромид и йодид; и арил-(C1-C4)-алкилгалогениды, например, бензилхлорид и фенэтилбромид. Указанные соли позволяют получать как растворимые в воде, так и растворимые в масле соединения в соответствии с изобретением.
Предпочтительные указанные выше фармацевтические соли включают, но не ограничиваются только ними, ацетат, трифторацетат, безилат, цитрат, фумарат, глюконат, гемисукцинат, гиппурат, гидрохлорид, гидробромид, изетионат, манделат, меглюмин, нитрат, олеат, фосфонат, пивалат, фосфат натрия, стеарат, сульфат, сульфосалицилат, тартрат, тиомалат, тозилат и трометамин.
Особое предпочтение отдают гидрохлориду, дигидрохлориду, гидробромиду, малеату, мезилату, фосфату, сульфату и сукцинату.
Соли присоединения кислот основных соединений формулы I получают путем введения в контакт формы свободных оснований с достаточным количеством целевой кислоты с получением соли традиционным способом. Свободное основание можно регенерировать путем введения в контакт солевой формы с основанием и выделения свободного основания традиционным способом. Формы свободного основания в некоторой степени отличаются от своих соответствующих солевых форм своими определенными физическими свойствами, такими, как растворимость в полярных растворителях; тем не менее, для целей настоящего изобретения во всем остальном соли являются эквивалентными своим соответствующим формам свободных оснований.
Как уже было указано, фармацевтически приемлемые соли присоединения основания соединений формулы I образуют с металлами или аминами, такими, как щелочные металлы и щелочноземельные металлы или органические амины. Предпочтительные металлы представляют собой натрий, калий, магний и кальций. Предпочтительные органические амины представляют собой N,N'-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, этилендиамин, N-метил-D-глюкамин и прокаин.
Соли присоединения основания кислых соединений в соответствии с изобретением получают путем введения в контакт формы свободной кислоты с достаточным количеством целевого основания для получения соли традиционным способом. Форма свободной кислоты может быть регенерирована путем введения в контакт солевой формы с кислотой и выделения формы свободной кислоты известным способом. Формы свободной кислоты в некоторой степени отличаются от своих соответствующих солевых форм определенными физическими свойствами, такими, как растворимость в полярных растворителях, тем не менее, для целей настоящего изобретения во всем остальном соли являются эквивалентными своим соответствующим формам свободных кислот.
Если соединение в соответствии с изобретением содержит более, чем одну группу, которая способна к образованию фармацевтически приемлемых солей этого типа, то изобретение также охватывает составные соли. Примеры типичных составных солевых форм включают, но не ограничиваются только ними, битартрат, диацетат, дифумарат, димеглюмин, дифосфат, динатрий и тригидрохлорид.
В свете описанного выше можно понять, что выражение "фармацевтически приемлемая соль" в контексте данной заявки предназначено для обозначения активного компонента, который включает соединение формулы I в форме одной из его солей, особенно в том случае, если указанная солевая форма обеспечивает указанному активному компоненту улучшенные фармакокинетические свойства по сравнению со свободной формой указанного активного компонента или любой другой солевой формой указанного активного компонента, которые использовались ранее. Фармацевтически приемлемая солевая форма активного компонента изначально может также придавать указанному активному компоненту желаемое фармакокинетическое свойство, которым он ранее не обладал, а также может даже оказывать положительное влияние на фармакодинамику данного активного компонента в отношении его терапевтической активности в организме.
Изотопы
Кроме того, также предусматривают, что соединение формулы I включает его изотопно-меченые формы. Изотопно-меченная форма соединения формулы I является идентичной указанному соединению, за исключением того факта, что один или несколько атомов указанного соединения были замещены атомом или атомами, которые имеют атомную массу или атомное число, отличающееся от атомной массы или атомного числа указанного атома, который обычно встречается в природе. Примеры изотопов, которые являются легкодоступными коммерчески и которые могут быть введены в соединение формулы I хорошо известными способами, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, например, 2Н, 3Н, 13С, 14С, 15N, 18O, 17O, 31Р, 35Р, 35S, 18F и соответственно 36CI. Соединение формулы I, его пролекарственная форма или фармацевтически приемлемая соль, которые содержат один или несколько указанных выше изотопов и/или другие изотопы других атомов также входят в объем притязаний настоящего изобретения. Меченое изотопом соединение формулы I можно применять в ряде способов. Например, меченное изотопами соединение формулы I, например, в которое введен радиоактивный изотоп, такой, как 3Н или 14С, являются пригодным для анализов распределения лекарственного средства и/или субстрата в тканях. Эти радиоактивные изотопы, например, тритий (3Н) и углерод-14 (14С), являются особенно предпочтительными вследствие простоты получения и превосходной способности к выявлению. Введение более тяжелых изотопов, например, дейтерия (2Н), в соединение формулы I, имеет терапевтические преимущества, благодаря большей метаболической стабильности указанного соединения, меченого изотопами. Большая метаболическая стабильность проявляется непосредственно в повышении времени полураспада in vivo или снижении требуемой дозы, что в большинстве условий будет представлять предпочтительный вариант осуществления настоящего изобретения. Как правило, меченое изотопом соединение формулы I может быть получено путем осуществления методик, раскрытых в схемах синтеза и в относящемся к ним описании, в разделе, касающемся примеров и способов получения, описанных в настоящей заявке, путем замены немеченого изотопами реагента его соответствующим легко доступным реагентом, меченым изотопом.
Дейтерий (2Н) также может быть введен в соединение формулы I с целью изменения окислительного метаболизма соединения путем первичного кинетического изотопного эффекта. Первичный кинетический изотопный эффект представляет собой изменение скорости химической реакции, которое происходит по причине замещения изотопного ядра, что, в свою очередь, вызывается изменением энергий основного состояния, что необходимо для образования ковалентной связи после указанного изотопного замещения. Замещение тяжелым изотопом обычно приводит к снижению энергии основного состояния для химической связи, вызывая, таким образом, уменьшение скорости скорость-лимитирующей стадии разрушения связи. Когда происходит разрушение связи в или вблизи участка седлообразной конфигурации вдоль координаты реакции образования нескольких продуктов, коэффициент распределения продуктов может существенно изменяться. Для объяснения: в случае, если дейтерий связывается с атомом углерода в положении, в котором не происходит обмен, различия скорости kM/kD=2-7 являются типичными. Если такое отличие в скорости с успехом применяется к соединению формулы I, чувствительному к окислению, то профиль этого соединения in vivo может быть изменен радикально и приводить к улучшению фармакокинетических свойств.
В процессе обнаружения и совершенствования терапевтических средств специалист в данной области техники пытается оптимизировать фармакокинетические параметры наряду с сохранением желательных свойств in vitro. Рационально предположить, что многие соединения со слабыми фармакокинетическими профилями чувствительны к окислительному метаболизму. Доступные в настоящее время анализы in vitro микросом печени обеспечивают ценную информацию о ходе окислительного метаболизма такого типа, что, в свою очередь, позволяет получить рациональную модель меченных дейтерием соединений формулы I с улучшенной стабильностью вплоть до резистентности к такому окислительному метаболизму. Таким образом, получают значительные улучшения фармакокинетических профилей соединений формулы I, что может быть выражено количественно в величинах увеличения периода полураспада in vivo (t/2), в концентрации при максимальном терапевтическом эффекте (Cmax), площадью под кривой ответа на определенную дозу (AUC) и F; в величинах уменьшения клиренса, дозы и материальных затрат.
Приведенное далее предназначено для иллюстрации указанного выше: соединение формулы I, которое имеет многочисленные потенциальные сайты для окислительного метаболизма, например, атомы водорода бензила и атомы водорода, соединенные с атомом азота, получают как серии аналогов, в которых различные комбинации атомов водорода заменяются атомами дейтерия так, что некоторые, большинство или все из указанных атомов водорода заменяются на атомы дейтерия. Определения периода полураспада обеспечивают благоприятное и точное определение степени улучшения резистентности к окислительному метаболизму. Таким образом определяют, что период полураспада исходного соединения может быть продлен вплоть до 100% как результат такого замещения водорода дейтерием.
Замещение водорода дейтерием в соединении формулы I можно также применять для достижения благоприятного изменения спектра метаболитов исходного соединения с целью уменьшения или устранения нежелательных токсических метаболитов. Например, когда токсический метаболит возникает при окислительном расщеплении углерод-водородной связи (С-Н), то с достаточной вероятностью предполагается, что меченый дейтерием аналог значительно уменьшит или устранит выработку нежелательного метаболита, даже в случае, когда отдельное окисление не является лимитирующей скорость стадией. Дополнительная информация, относящаяся к уровню техники в отношении замещения водорода дейтерием, может быть найдена, например, в работе Hanzlik и др., J. Org. Chem. 55, 3992-3997, 1990; Reider и др., J. Org. Chem. 52, 3326-3334, 1987; Foster, Adv. Drug Res. 14, 1-40, 1985; Gillette и др., Biochemistry 33 (10) 2927-2937, 1994; и Jarman и др. Carcinogenesis 16 (4), 683-688, 1993.
Кроме того, изобретение относится к лекарственным средствам, содержащим по меньшей мере одно соединение формулы I и/или его фармацевтически приемлемые производные, сольваты и стереоизомеры, включая их смеси во всех соотношениях, и необязательно наполнители и/или вспомогательные вещества.
Фармацевтические составы можно вводить в виде дозированных единиц, которые содержат заранее установленное количество активного компонента на дозированную единицу. Такая единица может включать, например, от 0,5 мг до 1 г, предпочтительно от 1 мг до 700 мг, более предпочтительно от 5 мг до 100 мг, соединения в соответствии с изобретением, в зависимости от состояния, которое лечат лечению, способа введения, а также возраста, веса тела и состояния пациента, или фармацевтические составы можно вводить в виде дозированных единиц, которые содержат заранее установленное количество активного компонента на дозированную единицу. Предпочтительными составами дозированных единиц являются те, которые содержат суточную дозу или часть суточной дозы, как указано выше, или соответствующую ей порцию активного компонента. Фармацевтические составы этого типа также могут быть получены способом, который хорошо известен в области фармацевтики.
Фармацевтические составы могут быть приспособлены для введения при помощи любого приемлемого способа, например, путем перорального (включая буккальное или подъязычное), ректального, назального, местного (включая буккальное, подъязычное или трансдермальное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное или внутрикожное) введения. Такие составы могут быть приготовлены с помощью любого способа, известного в области фармацевтики, например, путем объединения активного компонента с наполнителем(ями) или вспомогательным(ыми) веществом(ами).
Фармацевтические составы, предназначенные для перорального введения, можно вводить в виде отдельных единиц, таких как, например, капсулы или таблетки; порошки или гранулы; растворы или суспензии в водных или неводных жидкостях; съедобных пен или пенящихся пищевых продуктов; или жидких эмульсий масло-в-воде или жидких эмульсий вода-в-масле.
Так, например, в случае перорального введения в виде таблетки или капсулы, активный компонент может быть объединен с пероральным, нетоксичным и фармацевтически приемлемым инертным наполнителем, таким как, например, этанол, глицерин, вода и т.п. Порошки получают путем измельчения соединения до необходимого малого размера и смешивания его с фармацевтическим наполнителем, измельченным аналогичным способом, таким как, например, пищевой углевод, такой как, например, крахмал или маннит. Равным образом могут присутствовать ароматизатор, консервант, диспергирующее вещество и краситель.
Капсулы получают путем приготовления порошковой смеси, как описано выше, и заполнения нею формованных желатиновых капсул. Перед заполнением капсул к порошковой смеси можно добавлять придающие скользкость и смазывающие вещества, такие как, например, высокодисперсная кремниевая кислота, тальк, стеарат магния, стеарат кальция или полиэтиленгликоль в твердой форме. Для улучшения доступности лекарственного средства, заключенного в капсулу, также можно добавлять дезинтегрирующее вещество или солюбилизатор, такой как, например, агар-агар, карбонат кальция или карбонат натрия.
Кроме того, при желании или необходимости в смесь также можно добавлять пригодные связующие вещества, смазывающие вещества, дезинтеграторы, а также красители. Пригодные связующие включают крахмал, желатин, природные сахара, такие как, например, глюкоза или бета-лактоза, подсластители, полученные из кукурузы, природные и синтетические смолы, такие как, например, аравийская камедь, трагакантовая камедь или альгинат натрия, карбоксиметилцеллюлоза, полиэтиленгликоль, воски и т.п. Смазывающие вещества, которые можно применять в таких дозированных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п. Дезинтеграторы включают, но не ограничиваются только ними, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и т.п. Лекарственные средства в виде таблеток получают, например, путем приготовления порошковой смеси, гранулирования или сухого прессования смеси, добавления смазывающего вещества и дезинтегратора и прессования полученной смеси в таблетки. Порошковую смесь готовят путем смешивания соединения, измельченного приемлемым образом, с разбавителем или основанием, как описано выше, и необязательно со связующим веществом, таким как, например, карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, замедлителем растворения, таким как, например, парафин, усилителем поглощения, таким как, например, четвертичная соль, и/или абсорбентом, таким как, например, бентонит, каолин или фосфат дикальция. Порошковую смесь можно гранулировать путем смачивания связующим веществом, таким как, например, сироп, крахмальная паста, слизь акации или растворы целлюлозы или полимерных веществ и прессования ее через сито. В качестве альтернативы грануляции, порошковую смесь можно пропускать через таблетировочную машину, получая куски неправильной формы, которые распадаются, образуя гранулы. Гранулы можно замасливать путем добавления стеариновой кислоты, стеарата, талька или минерального масла для предотвращения слипания в таблетировочной литейной форме. После этого смазанную смесь спрессовывают, получая таблетки. Соединения в соответствии с изобретением также можно объединять с сыпучим инертным наполнителем и затем подвергать прямому прессованию, получая таблетки без осуществления стадий грануляции или сухого прессования. Таблетки также можно покрывать прозрачным или светонепроницаемым защитным слоем, состоящим из шеллакового герметизирующего слоя, слоя сахара или полимерного вещества и глянцевого слоя воска. К этим покрытиям также можно добавлять красители для возможности различения между разными дозированными единицами.
Жидкости для перорального введения, такие как, например, раствор, сиропы и эликсиры, могут быть приготовлены в виде дозированных единиц таким образом, чтобы они содержали заранее установленное количество соединения. Сиропы могут быть получены путем растворения соединения в водном растворе с подходящим ароматизатором, тогда как эликсиры готовят с применением нетоксичного спиртового наполнителя. Суспензии могут быть приготовлены путем диспергирования соединения в нетоксичном наполнителе. Также можно добавлять солюбилизаторы и эмульгаторы, такие как, например, этоксилированные изостеариловые спирты и полиоксиэтиленовые эфиры сорбита, консерванты, ароматические добавки, такие как, например, масло перечной мяты, или натуральные заменители сахара или сахарин, или другие искусственные подсластители и т.п.
При желании составы для перорального введения в виде дозированных единиц могут быть инкапсулированы в микрокапсулы. Также состав может быть приготовлен таким образом, чтобы пролонгировать или замедлить высвобождение, например, путем применения покрытий или введения вещества в виде частиц в полимеры, воск и т.п.
Соединения формулы I и их фармацевтические приемлемые соли, таутомеры и стереоизомеры также можно вводить в виде липосомных систем доставки, таких как, например, небольшие однослойные пузырьки, большие однослойные пузырьки и многослойные пузырьки. Липосомы могут быть образованы с помощью различных фосфолипидов, таких как, например, холестерин, стеариламин или фосфатидилхолины.
Соединения формулы I и их соли, таутомеры и стереоизомеры также могут быть доставлены путем применения моноклональных антител в качестве индивидуальных носителей, к которым присоединены молекулы соединения. Соединения также могут быть соединены с растворимыми полимерами в качестве нацеливающих носителей лекарственных средств. Такими полимерами могут являться поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидофенол, полигидроксиэтиласпартамидофенол или полиэтиленоксидполилизин, замещенный пальмитоиловыми радикалами. Кроме того, соединения можно связывать с биоразлагаемыми полимерами, которые пригодны для обеспечения контролируемого высвобождения лекарственного средства, например полимолочной кислотой, поли-эпсилон-капролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидроксипиранами, полицианоакрилатами и перекрестно-сшитыми или амфипатическими блок-сополимерами гидрогелей.
Фармацевтические составы, предназначенные для трансдермального введения, можно вводить в виде независимых пластырей для пролонгированного, тесного контакта с эпидермисом реципиента. Таким образом, например, активный компонент может быть доставлен из пластыря путем ионофореза, как в общих чертах описано в Pharmaceutical Research, 3 (6), 318 (1986).
Фармацевтические соединения, предназначенные для местного введения, могут быть приготовлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел.
Для лечения глаз или других наружных тканей, например рта и кожи, предпочтительно применяются составы в виде мази или крема для локального нанесения. Для приготовления состава в виде мази, активный компонент может применяться с парафиновым или смешиваемым с водой мазевым основанием. Альтернативно, для получения крема активный компонент может быть приготовлен с основой для крема типа масло-в-воде или основой вода-в-масле.
Фармацевтические составы, предназначенные для местного применения для глаз, включают глазные капли, в которых активный компонент растворен или суспендирован в приемлемом носителе, предпочтительно в водном растворителе.
Фармацевтические составы, предназначенные для местного введения в полость рта, включают лепешки, пастилки и жидкости для полоскания рта.
Фармацевтические составы, предназначенные для ректального введения, можно вводить в виде суппозиториев или клизм.
Фармацевтические составы, предназначенные для назального введения, в которых носитель представляет собой твердое вещество, включают крупнозернистый порошок, имеющий размер частичек, например, в пределах 20-500 микрон, который вводят путем вдыхания, то есть путем быстрого вдоха через нос из контейнера, содержащего порошок, который придерживают возле носа. Пригодные составы для введения в виде назального спрея или носовых капель с жидкостью в качестве носителя включают растворы активного вещества в воде или в масле.
Фармацевтические составы, предназначенные для введения путем ингаляции, включают тонкоизмельченные частички в виде тонкого порошка или аэрозоля, которые могут быть получены с помощью различных диспергирующих устройств под давлением при помощи аэрозолей, распылителей или инсуффляторов.
Фармацевтические составы, предназначенные для вагинального введения, можно вводить в виде маточных колец, тампонов, кремов, гелей, паст, пен или составов для распыления.
Фармацевтические составы, предназначенные для парентерального введения, включают водные или неводные стерильные растворы для инъекций, содержащие антиоксиданты, буферы, бактериостатические вещества и растворенные вещества, с помощью которых состав поддерживается изотоническим по отношению к крови реципиента, которого лечат; и водные или неводные стерильные суспензии, которые могут содержать суспензионную среду и загустители. Составы могут быть введены с помощью емкостей для однократного или многократного введения, например, запечатанных ампул и флаконов, и их можно хранить в лиофилизированном состоянии, при этом непосредственно перед введением необходимо только добавить стерильную жидкость-носитель, например, воду для инъекций. Растворы и суспензии для инъекций, приготовленные согласно рецептуре, могут быть приготовлены из стерильных порошков, гранул и таблеток.
Само собой разумеется, что дополнительно к предпочтительным описанным выше составляющим, составы также могут содержать другие вещества, которые используют в данной области техники для конкретных типов составов; например, составы, пригодные для перорального введения, могут содержать ароматизаторы.
Терапевтически эффективное количество соединения формулы I зависит от многих факторов, включая, например, возраст и вес животного, определенное состояние, которое необходимо лечить, и его тяжесть, природу лекарственного средства и способ введения, и в конечном счете оно может быть определено лечащим врачом или ветеринаром. Тем не менее, эффективное количество соединения в соответствии с изобретением, как правило, находится в пределах от 0,1 до 100 мг/кг веса тела реципиента (млекопитающего) в сутки и предпочтительно обычно находится в пределах от 1 до 10 мг/кг веса тела в сутки. Таким образом, действующее суточное количество для взрослого млекопитающего весом 70 кг обычно может составлять от 70 до 700 мг, причем это количество может вводиться в виде отдельной дозы один раз в сутки или обычно в виде циклов частичных доз (таких как, например, два, три, четыре, пять или шесть раз) в сутки, так что общая суточная доза является такой же самой. Эффективное количество его соли, сольвата или физиологически функционального производного может быть определено в виде доли эффективного количества соединения в соответствии с изобретением как таковое. Также можно предположить, что аналогичные дозы пригодны для лечения других состояний, описанных выше.
Комбинированное лечение этого типа можно осуществлять с помощью одновременного, последовательного или раздельного дозирования индивидуальных компонентов лечения. В комбинированных продуктах такого типа применяются соединения в соответствии с изобретением.
Кроме того, изобретение относится к лекарственным средствам, содержащим по меньшей мере одно соединение формулы I и/или его фармацевтически приемлемые соли, таутомеры и стереоизомеры, включая их смеси во всех соотношениях, и по меньшей мере один дополнительный активный компонент лекарственного средства.
Изобретение также относится к комплекту (набору), состоящему из отдельных упаковок
(а) эффективного количества соединения формулы I и/или его фармацевтически приемлемых солей, таутомеров и стереоизомеров, включая их смеси во всех соотношениях,
и
(б) эффективного количества дополнительного активного компонента лекарственного средства.
Комплект содержит в себе пригодные емкости, такие как коробки, отдельные бутылочки, пакеты или ампулы. Комплект может включать, например, отдельные ампулы, каждая из которых содержит эффективное количество соединения формулы I и/или его фармацевтически приемлемых солей, таутомеров и стереоизомеров, включая их смеси во всех соотношениях,
и эффективное количество дополнительного активного компонента лекарственного средства в растворенной или лиофилизированной форме.
Термин "лечение", используемый в настоящей заявке, обозначает облегчение, полностью или частично, симптомов, связанных с нарушением или заболеванием, или замедление, или остановку дальнейшего прогрессирования или ухудшения этих симптомов, или предотвращение или профилактику заболевания или нарушения у субъекта, имеющего риск развития такого заболевания или нарушения.
Термин "эффективное количество" применительно к соединению формулы (I) может обозначать количество, способное облегчать, полностью или частично, симптомы, связанные с нарушением или заболеванием, или замедлять или останавливать дальнейшее прогрессирование или ухудшение этих симптомов, или предотвращать или обеспечивать профилактику заболевания или нарушения у субъекта, имеющего заболевание или с риском развития заболевания, описанного в настоящей заявке, такого как воспалительные состояния, иммунологические состояния, злокачественные новообразования или метаболические состояния.
В одном варианте осуществления эффективное количество соединения формулы (I) представляет собой количество, которое ингибирует танкиразу в клетке, например, in vitro или in vivo. В некоторых вариантах осуществления, эффективное количество соединения формулы (I) ингибирует танкиразу в клетке на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% или 99%, по сравнению с активностью танкиразы в необработанной клетке. Эффективное количество соединения формулы (I), например, в фармацевтической композиции, может находиться на уровне, который превышает желаемый эффект; например, от приблизительно 0,005 мг/кг веса тела субъекта до приблизительно 10 мг/кг веса тела субъекта в единичной лекарственной форме как для перорального, так и для парентерального введения.
ПРИМЕНЕНИЕ
Соединения согласно настоящему изобретению пригодны в качестве фармацевтически активных компонентов для млекопитающих, в особенности для людей, для лечения злокачественного новообразования, рассеянного склероза, сердечно-сосудистых заболеваний, поражений центральной нервной системы и различных форм воспаления.
Настоящее изобретение охватывает применение соединений формулы I и/или их физиологически приемлемых солей, таутомеров и стереоизомеров для приготовления лекарственного средства для лечения или предотвращения злокачественного новообразования, рассеянного склероза, сердечно-сосудистых заболеваний, поражений центральной нервной системы и различных форм воспаления.
Примеры воспалительных заболеваний включают ревматоидный артрит, псориаз, контактный дерматит, отсроченная реакция гиперчувствительности и другие.
Также включено применение соединений формулы I и/или их физиологически приемлемых солей, таутомеров и стереоизомеров для приготовления лекарственного средства для лечения или предотвращения заболевания, индуцированного танкиразой, или состояния, индуцированного танкиразой у млекопитающего, при котором в этом способе терапевтически эффективное количество соединения в соответствии с изобретением вводят больному млекопитающему, нуждающемуся в таком лечении. Терапевтическое количество изменяется в зависимости от конкретного заболевания и легко может быть определено специалистом в данной области техники.
Выражение "заболевания или состояния, вызванные танкиразой" относится к патологическим состояниям, которые зависят от активности одной или нескольких танкираз. Заболевания, связанные с активностью танкиразы, включают злокачественное новообразование, рассеянный склероз, сердечнососудистые заболевания, поражения центральной нервной системы и различные формы воспаления.
Настоящее изобретение, в особенности, относится к соединениям формулы I и к их фармацевтически приемлемым солям, таутомерам и стереоизомерам, включая их смеси во всех соотношениях,
для применения в лечении заболеваний, при которых имеет значение ингибирование, регуляция и/или модуляция ингибирования танкиразы.
В особенности, настоящее изобретение относится к соединениям формулы I и к их фармацевтически приемлемым солям, таутомерам и стереоизомерам, включая их смеси во всех соотношениях, для применения для ингибирования танкиразы.
В особенности настоящее изобретение относится к соединениям формулы I и к их фармацевтически приемлемым солям, таутомерам и стереоизомерам, включая их смеси во всех соотношениях, для применения в лечении злокачественного новообразования, рассеянного склероза, сердечно-сосудистых заболеваний, поражений центральной нервной системы и различных форм воспаления.
Настоящее изобретение, в частности, относится к способам лечения или предотвращения злокачественного новообразования, рассеянного склероза, сердечно-сосудистых заболеваний, поражений центральной нервной системы и различных форм воспаления, которые включают введение субъекту, который в этом нуждается, эффективного количества соединения формулы I или его фармацевтически приемлемой соли, таутомера, стереоизомера или сольвата.
Характерные злокачественные новообразования, для лечения или предотвращения которых являются полезными соединения формулы I, включают, но не ограничиваются таковыми, рак головы, шеи, глаз, рта, горла, пищевода, бронхов, гортани, глотки, груди, костей, легких, ободочной кишки, прямой кишки, желудка, предстательной железы, мочевого пузыря, матки, шейки матки, молочной железы, яичников, яичек или других репродуктивных органов, кожи, щитовидной железы, крови, лимфатических узлов, почек, печени, поджелудочной железы, головного мозга, центральной нервной системы, солидных опухолей и злокачественного перерождения крови.
Характерные сердечно-сосудистые заболевания, для лечения или предотвращения которых пригодны соединения формулы I, включают, но не ограничиваются таковыми, рестеноз, атеросклероз и его последствия, такие как инсульт, инфаркт миокарда, ишемические повреждения сердца, легких, кишечника, почек, печени, поджелудочной железы, селезенки или мозга.
Настоящее изобретение относится к способу лечения пролиферативного, аутоиммунного, противовоспалительного или инфекционного заболевания или нарушения, который включает введение субъекту, который в этом нуждается, терапевтически эффективного количества соединения формулы I.
Предпочтительно, настоящее изобретение относится к способу, где заболевание представляет собой злокачественное новообразование.
Особенно предпочтительно настоящее изобретение относится к способу, где заболевание представляет собой злокачественное новообразование, причем введение является одновременным, последовательным или чередующимся с введением по меньшей мере одного другого активного лекарственного средства.
Описанные соединения формулы I можно вводить в комбинации с другими известными лекарственными средствами, включая противораковые средства. Как применяют в настоящем изобретении, термин "противораковое средство" относится к любому средству, которое вводят пациенту со злокачественным новообразованием для лечения рака.
Определенное выше противораковое лечение можно применять в виде монотерапии или дополнительно к соединениям формулы I, описанным в настоящей заявке, могут включать в себя стандартное оперативное вмешательство или лучевую терапию или лекарственную терапию. Такая лекарственная терапия, например, химиотерапия или таргетная терапия, может включать одно или несколько, но предпочтительно одно из следующих противоопухолевых средств:
Алкилирующие средства
такие как алтретамин, бендамустин, бусульфан, кармустин, хлорамбуцил, хлорметин, циклофосфамид, дакарбазин, ифосфамид, импросульфан тозилат, ломустин, мельфалан, митобронитол, митолактол, нимустин, ранимустин, темозоломид, тиотепа, треосульфан, мехлороэтамин, карбоквон;
апазиквон, фотемустин, глуфосфамид, палифосфамид, пипоброман, трофосфамид, урамустин, ТН-3024, VAL-0834;
Соединения платины
такие как карбоплатин, цисплатин, эптаплатин, мириплатина гидрат, оксалиплатин, лобаплатин, недаплатин, пикоплатин, сатраплатин;
лобаплатин, недаплатин, пикоплатин, сатраплатин;
Средства, изменяющие ДНК
такие как амрубицин, бисантрен, децитабин, митоксантрон, прокарбазин, трабектедин, клофарабин;
амсакрин, бросталлицин, пиксантрон, ларомустин1,3;
Ингибиторы топоизомеразы
такие как этопозид, иринотекан, разоксан, собузоксан, тенипозид, топотекан,
амонафид, белотекан, эллиптиния ацетат, ворелоксин;
Модификаторы микротрубочек
такие как кабазитаксел, доцетаксел, эрибулин, иксабепилон, паклитаксел, винбластин, винкристин, винорелбин, виндезин, винфлунин,
фосбретабулин, тезетаксел;
Антиметаболиты
такие как аспарагиназа3, азацитидин, левофолинат кальция, капецитабин, кладрибин, цитарабин, эноцитабин, флоксуридин, флударабин, фторурацил, гемцитабин, меркаптопурин, метотрексат, неларабин, пеметрексед, пралатрексат, азатиоприн, тиогуанин, кармофур; доксифлуридин, элацитарабин, ралтитрексед, сапацитабин, тегафур2,3, триметрексат;
Противораковые антибиотики
такие как блеомицин, дактиномицин, доксорубицин, эпирубицин, идарубицин, левамизол, милтефозин, митомицин С, ромидепсин, стрептозоцин, валрубицин, зиностатин, зорубицин, даунуробицин, пликамицин; акларубицин, пепломицин, пирарубицин;
Гормоны/антагонисты
такие как абареликс, абиратерон, бикалутамид, бусерелин, калустерон, хлортрианизен, дегареликс, дексаметазон, эстрадиол, флуокортолон флуоксиместерон, флутамид, фулвестрант, гозерелин, гистрелин, лейпрорелин, мегестрол, митотан, нафарелин, нандролон, нилутамид, октреотид, преднизолон, ралоксифен, тамоксифен, тиротропин альфа, торемифен, трилостан, трипторелин, диэтилстильбэстрол;
аколбифен, даназол, деслорелин, эпитиостанол, ортеронел, энзалутамид1,3;
Ингибиторы ароматазы
такие как аминоглутетимид, анастрозол, эксеместан, фадрозол, летрозол, тестолактон;
форместан;
Низкомолекулярные ингибиторы киназ
такие как кризотиниб, дазатиниб, эрлотиниб, иматиниб, лапатиниб, нилотиниб, пазопаниб, регорафениб, руксолитиниб, сорафениб, санитиниб, вандетаниб, вемурафениб, босутиниб, гефитиниб, акситиниб;
афатиниб, алисертиб, дабрафениб, дакомитиниб, динациклиб, довитиниб, энзастаурин, нинтеданиб, ленватиниб, линифаниб, линситиниб, маситиниб, мидостаурин, мотесаниб, нератиниб, орантиниб, перифосин, понатиниб, радотиниб, ригосертиб, типифарниб, тивантиниб, тивозаниб, траметиниб, пимазертиб, бриваниб аланинат, седираниб, апатиниб4, кабозантиниб S-малат1,3, ибрутиниб1,3, икотиниб4, бупарлисиб2, ципатиниб4, кобиметиниб1,3, иделалисиб1,3, федратиниб1, XL-6474;
Фотосенсибилизаторы
такие как метоксален3;
порфимер натрия, талапорфин, темопорфин;
Антитела
такие как алемтузумаб, бесилесомаб, брентуксимаб ведотин, цетуксимаб, деносумаб, ипилимумаб, офатумумаб, панитумумаб, ритуксимаб, тозитумомаб,
трастузумаб, бевацизумаб, пертузумаб2,3;
катумаксомаб, элотузумаб, эпратузумаб, фарлетузумаб, могамулизумаб, нецитумумаб, нимотузумаб, обинутузумаб, окаратузумаб, ореговомаб, рамуцирумаб, рилотумумаб, силтуксимаб, тоцилизумаб, залутумумаб, занолимумаб, матузумаб, далотузумаб1,2,3, онартузумаб1,3, ракотумомаб1, табалумаб1,3, EMD-5257974, ниволумаб1,3;
Цитокины
такие как альдеслейкин, интерферон альфа2, интерферон альфа2а, интерферон альфа2b2.3;
целмолейкин, тазонермин, тецелейкин, опрелвекин1,3, рекомбинантный интерферон бета-1а4;
Конъюгаты лекарственных препаратов
такие как денилейкин дифтитокс, ибритумомаб тиуксетан, йобенгуан 1123, преднимустин, трастузумаб эмтансин, эстрамустин, гемтузумаб, озогамицин, афлиберцепт;
цинтредекин бесудотокс, эдотреотид, инотузумаб озогамицин, наптумомаб эстафенатокс, опортузумаб монатокс, технеций (99mТс) арцитумомаб1,3,
винтафолид1,3;
Вакцины
такие как сипулейцел3, витеспен3, эмепепимут-S3, онкоВАКС4, риндопепимут3, троВакс4, MGN-16014, MGN-17034;
Прочие препараты
алитретиноин, бексаротен, бортезомиб, эверолимус, ибандроновая кислота, имиквимод, леналидомид, лентинан, метирозин, мифамуртид, памидроновая кислота, пэгаспаргаза, пентостатин, сипулейцел3, сизофиран, тамибаротен, темсиролимус, талидомид, третиноин, висмодегиб, золедроновая кислота, вориностат;
целекоксиб, циленгитид, энтиностат, этанидазол, ганетеспиб, идроноксил, инипариб, иксазомиб, лонидамин, ниморазол, панобиностат, перетиноин, плитидепсин, помалидомид, прокодазол, ридафоролимус, тасквинимод, телотристат, тимальфазин, тирапазамин, тоседостат, трабедерсен, убенимекс, вальсподар, гендицин4,
пицибанил4, реолизин4, гидрохлорид ретаспимицина1,3, требананиб2,3, вирулизин4, карфилзомиб1,3, эндостатин4, иммукотел4, белиностат3, MGN-17034;
1 Предл. МНН (предложенное Международное Непатентованное Название)
2 Рекоменд. МНН (рекомендованное Международное Непатентованное Название)
3 Название в США (наименование препарата по Справочнику национальных непатентованных названий США)
4 МНН отсутствует.
Следующие сокращения соответственно относятся к приведенным ниже определениям:
водн. (водный), ч (час), г (грамм), л (литр), мг (миллиграмм), МГц (мегагерц), мин. (минута), мм (миллиметр), ммоль (миллимоль), мМ (миллимолярный), т.пл. (температура плавления), экв. (эквивалент), мл (миллилитр), мкл (микролитр), ACN (ацетонитрил), АсОН (уксусная кислота), CDCl3 (дейтерированный хлороформ), CD3OD (дейтерированный метанол), CH3CN (ацетонитрил), c-hex (циклогексан), DCC (дициклогексил карбодиимид), ДХМ (дихлорметан), DIC (диизопропил карбодиимид), DIEA (диизопропилэтил-амин), ДМФА (диметилформамид), ДМСО (диметилсульфоксид), ДМСО-d6 (дейтерированный диметилсульфоксид), EDC (1-(3-диметиламинопропил)-3-этилкарбодиимид), ЭРИ (электрораспылительная ионизация), EtOAc (этилацетат), Et2O (простой диэтиловый эфир), EtOH (этанол), HATU гексафторфосфат (диметиламино-([1,2,3]триазоло[4,5-b]пиридин-3-илокси)-метилен]-диметиламмония), ВЭЖХ (высокоэффективная жидкостная хроматография), i-PrOH (2-пропанол), K2CO3 (карбонат калия), ЖХ (жидкостная хроматография), МеОН (метанол), MgSO4 (сульфат магния), МС (масс-спектрометрия), МТВЕ (метил-трет-бутиловый эфир), NaHCO3 (бикарбонат натрия), NaBH4 (борогидрид натрия), NMM (N-метилморфолин), ЯМР (ядерный магнитный резонанс), РуВОР (гексафторфосфат бензотриазол-1-илокси-трис-пирролидинофосфония), КТ (комнатная температура), Rt (время удержания), SPE (твердофазная экстракция), TBTU (тетрафторборат 2-(1-Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония), TEA (триэтиламин), ТФУ (трифторуксусная кислота), ТГФ (тетрагидрофуран), ТСХ (тонкослойная хроматография), УФ (ультрафиолет).
Описание анализов in vitro
Сокращения:
GST = глутатион-S-трансфераза
FRET = резонансный перенос энергии флуоресценции
HTRF® = (гомогенная флуоресценция с временным разрешением)
HEPES = буфер 4-(2-гидроксиэтил)-1-пиперазин этансульфоновая кислота
DTT = дитиотреитол
BSA = бычий сывороточный альбумин
CHAPS = детергент;
CHAPS = 3-[(3-холамидопропил)диметиламмонио]-1-пропансульфонат
Стрептавидин-XLent® представляет собой высококачественный конъюгат стрептавидин-XL665, для которого были оптимизированы условия сочетания для получения конъюгата с улучшенными характеристиками для некоторых анализов, в частности тех, для которых требуется высокая чувствительность.
Тестирование биохимической активности танкираз 1 и 2: Анализ аутопарсилирования
Анализ аутопарсилирования осуществляют в две стадии: ферментативная реакция, в которой GST-меченая танкираза-1, соотв. танкираза-2 переносит биотинилированную ADP-рибозу на себя из биотинилированного NAD в качестве ко-субстрата, и реакция обнаружения, где анализируют FRET с временным разрешением между меченным криптатом анти-GST, который связан с GST меткой фермента и Xlent®)-меченным стрептавидином, связанным с биотин-парсилированным остатком. Аутопарсилирующую активность определяют непосредственно через повышение HTRF сигнала.
Анализ аутопарсилирования осуществляют в формате анализа HTRF® на 384 лунки (Cisbio, Codolet, France) в микротитрационных планшетах низкого объема Greiner nb на 384 лунки и применяют для экрана с высокой пропускной способностью. 250 нМ GST-меченную танкиразу-1 (1023-1327 а/к), соответственно приблизительно 250 нМ GST-меченную танкиразу-2 (873-1166 а/к) и 5 мкМ bio-NAD (Biolog, Life science Inst., Бремен, Германия) в качестве ко-субстрата инкубируют в общем объеме 5 мкл (50 мМ HEPES, 4 мМ Mg-хлорид, 0.05% Pluronic F-68, 1.4 мМ DTT, 0.5% ДМСО, pH 7.7) в отсутствии или в присутствии тестируемого соединения (10 концентраций разведения) в течение 90 мин при 30°С.Реакцию останавливают путем добавления 1 мкл 50 мМ раствора EDTA. Добавляют 2 мкл раствора для обнаружения (1.6 мкМ SA-Xlent® (Cisbio, Кодоле, Франция), 7.4 нМ анти-GST-K® (Eu-меченное анти-GST, Cisbio, Кодоле, Франция) в 50 мМ HEPES, 800 мМ KF, 0.1% BSA, 20 мМ EDTA, 0.1% CHAPS, pH 7.0). После инкубирования в течение 1 ч. при комнатной температуре HTRF измеряют с помощью многорежимного считывающего устройства Envision (Perkin Elmer LAS Germany GmbH) при длине волны возбуждения 340 нм (лазерный режим) и длинах волн эмиссии 615 нм и 665 нм. Определяют соотношение сигналов эмиссии. Полное используемое значение представляет реакцию без ингибитора. Используемое значение фармакологического нуля представляет собой XAV-939 (Tocris) в конечной концентрации 5 мкМ. Ингибирующие значения (IC50) определяют, используя или программу Symyx Assay Explorer®, или Condosseo® от GeneData.
Измерение клеточного ингибирования танкиразы
Поскольку было описано, что танкиразы модулируют клеточный уровень Axin2 (Huang и др., 2009; Nature), то повышение уровня Axin2 используют в качестве считываемых данных для определения клеточного ингибирования танкираз в исследовании на основе Luminex.
Клетки клеточной линии карциномы ободочной кишки DLD1 высевают в планшеты на 96 лунок плотностью 1.5×104 клеток на лунку. На следующий день, клетки обрабатывают серийными разведениями исследуемого соединения за семь стадий в виде трех повторов с конечной концентрацией ДМСО 0.3%. Через 24 часа, клетки лизируют в лизирующем буфере (20 мМ Tris/HCl pH 8.0, 150 мМ NaCl, 1% NP40, 10% глицерин), и лизаты очищают путем центрифугирования через фильтровальный планшет на 96 лунок (0.65 мкм). Белок Axin2 выделяют из клеточных лизатов путем инкубирования с моноклональным анти-Axin2 антителом (R&D Systems #МАВ6078), которое связывается с флуоресцентными карбоксишариками. После этого, связанный Axin2 специфически обнаруживают с помощью поликлонального анти- Axin2 антитела (Cell Signaling #2151) и пригодного РЕ-флуоресцентного вторичного антитела. Количество выделенного Axin2 белка определяют с помощью прибора Luminex200 (Luminex Corporation) в соответствии с инструкциями производителя путем подсчитывания 100 событий на лунку. Ингибирование танкиразы исследуемыми соединениями приводит к более высоким уровням Axin2, которые прямо коррелируют с повышением обнаруживаемой флуоресценции. В качестве контролей клетки обрабатывают только растворителем (нейтральный контроль) и сравнительным ингибитором танкиразы IWR-2 (3Е-06 М), который служит в качестве контроля для максимального повышения Axin2. Для анализа, полученные данные нормализуют по отношению к необработанному контролю с растворителем и подгоняют для определения значений ЕС50, с применением программного обеспечения Assay Explorer (Accelrys).
Описание исследования PARP1
Тестирование биохимической активности PARP-1: Исследование аутопарсилирования
Анализ аутопарсилирования осуществляют в две стадии: ферментативная реакция, в которой His-меченная Parp-1 переносит биотинилированную ADP-рибозу/ADP-рибозу на себя с биотинилированного NAD/NAD в качестве ко-субстрата, и реакция обнаружения, где анализируют FRET с временным разрешением между меченным криптатом анти-His антителом, связанным с His меткой фермента и Xlent®-меченным стрептавидином, связанным с биотин-парсилированным остатком. Аутопарсилирующую активность определяют непосредственно путем повышения HTRF сигнала.
Анализ аутопарсилирования осуществляют в формате исследования на 384 лунки HTRF® (Cisbio, Codolet, France) в микротитрационных планшетах низкого объема на 384 лунки Greiner nb. 35 нМ His-меченную Parp-1 (человеческая, рекомбинантная, Enzo Life Sciences GmbH, Лёррах, Германия), и смесь 125 нМ bio-NAD (Biolog, Life science Inst., Бремен, Германия) и 800 нМ NAD в качестве ко-субстрата инкубируют в общем объеме 6 мкл (100 мМ Tris/HCl, 4 мМ Mg-хлорид, 0,01% IGEPAL® СА630, 1 мМ DTT, 0,5% ДМСО, pH 8, 13 нг/мкл активированной ДНК (BPS Bioscience, San Diego, US)) при отсутствии или в присутствии исследуемого соединения (10 концентраций разведения) в течение 150 мин при 23°С. Реакцию останавливают путем добавления 4 мкл раствора стоп/обнаружение (70 нМ SA-Xlent® (Cisbio, Codolet, France), 2.5 нМ анти-His-K® (Eu-меченное анти-His, Cisbio, Codolet, France) в 50 мМ HEPES, 400 мМ KF, 0.1% BSA, 20 мМ EDTA, pH 7.0). После инкубирования в течение 1 ч при комнатной температуре HTRF измеряют с помощью многорежимного считывающего устройства Envision (Perkin Elmer LAS Germany GmbH) при длине волны возбуждения 340 нм (лазерный режим) и длинах волн эмиссии 615 нм и 665 нм. Определяют соотношение сигналов эмиссии. Полное используемое значение представляет реакцию без ингибитора. Используемое значение фармакологического нуля представляет собой Olaparib (LClabs, Woburn, US) в конечной концентрации 1 мкМ. Определяют ингибирующие значения (IC50), используя либо программу Symyx Assay Explorer®, либо Condosseo® от GeneData.
Описание ELISA исследования TNKS1 и TNKS2
Тестирование биохимической активности TNK 1 и 2: определение активность с помощью ELISA (анализ аутопарсилирования)
Для анализа аутопарсилирующей активности TNKS 1 и 2 осуществляют ELISA: На первой стадии GST меченую TNKS захватывают на покрытом глутатионом планшете. После этого осуществляют исследование активности с биотинилированным NAD при отсутствии /в присутствии соединений. В процессе ферментативной реакции GST меченая TNKS переносит биотинилированную ADP-рибозу на себя с биотинилированного NAD в качестве ко-субстрата. Для обнаружения добавляют конъюгат стрептавидин-HRP, который связывается с биотинилированной TNKS, и таким образом захватывается на планшеты. Определяют количество биотинилированной соотв. аутопарсилированной TNKS с люминесцентным субстратом для HRP. Уровень сигнала люминесценции прямо коррелируется с количеством аутопарсилированной TNKS и, следовательно, с активностью TNKS.
Определение активности с помощью ELISA осуществляют в микротитрационных планшетах на 384 лунки, покрытых глутатионом (планшеты для захвата Express, покрытые глутатионом, Biocat, Гейдельберг, Германия). Планшеты предварительно уравновешивают с помощью PBS. Затем планшеты инкубируют с 50 мкл 20 нг/лунку GST-меченной Tnks-1 (1023-1327 а/к, собственного производства), соответственно GST-меченной Tnks-2 (873-1166 а/к, собственного производства) в буфере для исследования (50 мМ HEPES, 4 мМ Mg-хлорид, 0.05% Pluronic F-68, 2 мМ DTT, pH 7.7) в течение ночи при 4°С. Планшеты промывают 3 раза с помощью PBS-Tween-20. Лунки блокируют путем инкубирования при комнатной температуре в течение 20 минут с 50 мкл блокирующего буфера (PBS, 0.05% Tween-20, 0.5% BSA). После этого планшеты промывают 3 раза с помощью PBS-Tween-20. Ферментативную реакцию осуществляют в 50 мкл реакционного раствора (50 мМ HEPES, 4 мМ Mg-хлорид, 0.05% Pluronic F-68, 1.4 мМ DTT, 0.5% ДМСО, pH 7.7) с 10 мкМ bio-NAD (Biolog, Life science Inst., Бремен, Германия) в качестве ко-субстрата при отсутствии или присутствии тестируемого соединения (10 концентраций разведения) в течение 1 часа при 30°С. Реакцию останавливают путем промывания 3 раза PBS-Tween-20. Для обнаружения добавляют 50 мкл 20 нг/мкл стрептавидин, HRP конъюгата (MoBiTec, Геттинген, Германия) в PBS/0.05%Tween-20/0.01%BSA и планшеты инкубируют в течение 30 минут при комнатной температуре. После трехразового промывания PBS-Tween-20 добавляют 50 мкл SuperSignal ELISA Femto Maximum чувствительного субстратного раствора (ThermoFisherScientific (Pierce), Бонн, Германия). После инкубирования в течение 1 минуты при комнатной температуре, измеряют люминесцентные сигналы с помощью многорежимного считывающего устройства Envision (Perkin Elmer LAS Germany GmbH) при 700 нм. Полное используемое значение представляет реакцию без ингибитора. Используемое значение фармакологического нуля представляет собой XAV-939 (Tocris) в конечной концентрации 5 мкМ. Определяют ингибирующие значения (IC50), используя либо программу Symyx Assay Explorer®, либо Condosseo® от GeneData.
Все приведенные выше и ниже температуры указаны в градусах Цельсия °С. В последующих примерах "обычная обработка" обозначает: при необходимости добавляют воду, pH устанавливают, при необходимости, до значений от 2 до 10, в зависимости от строения конечного продукта, смесь экстрагируют этилацетатом или дихлорметаном, фазы разделяют, органическую фазу высушивают над сульфатом натрия и упаривают, и остаток очищают при помощи хроматографии на силикагеле и/или кристаллизации.
1Н ЯМР записывали на спектрометре Bruker 400 или 500 МГц, используя остаточный сигнал дейтерированного растворителя в качестве внутреннего стандарта. Химические сдвиги (5) представлены в виде част, на млн. относительно остаточного сигнала растворителя (δ=2.49 част, на млн. для 1Н ЯМР в ДМСО-d6). Данные 1Н ЯМР представлены следующим образом: химический сдвиг (мультиплетность, константы взаимодействия и число атомов водорода). Мультиплетность сокращали следующим образом: s (синглет), d (дублет), t (триплет), q (квартет), m (мультиплет), br (широкий).
Условия ВЭЖХ/МС (А)
колонка: Chromolith SpeedROD RP-18e, 50×4.6 мм2
градиент: А:B = от 96:4 до 0:100 за 3.4 мин
скорость потока: 2.40 мл/мин
элюент A: вода + 0.05% муравьиная кислота
Элюент B: ацетонитрил + 0.04% муравьиная кислота
длина волны: 220 нм
масс-спектроскопия: позитивный режим
Пример 1
Синтез 6-n-толил-7H-имидазо[1,5-а]пиразин-8-она ("Al")
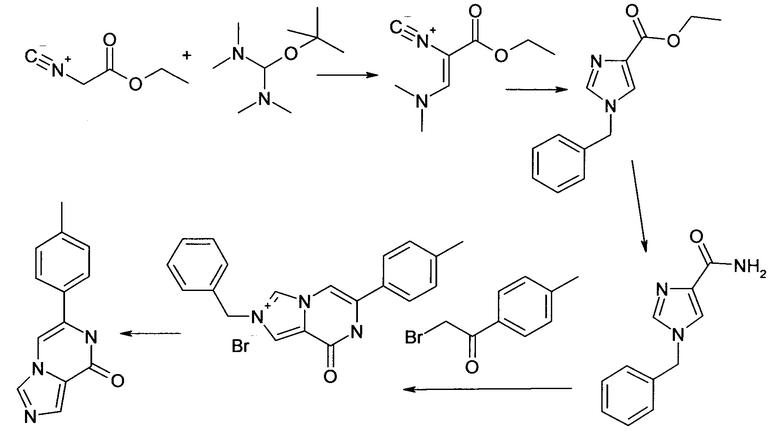
1.1 Этиловый эфир (Z)-3-диметиламино-2-изоциано-акриловой кислоты
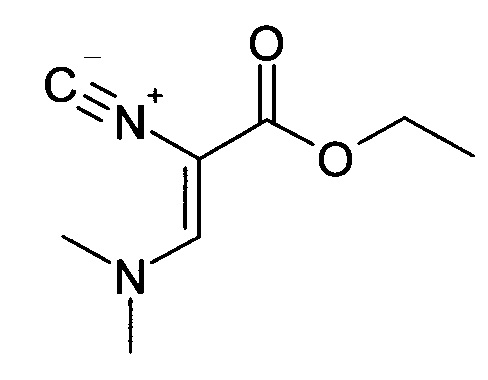
К этилизоцианоацетату (3.22 мл, 29.44 ммоль) трет-бутокси-бис(диметиламино)метан (12.16 мл, 58.88 ммоль) добавляли и смесь перемешивали в течение 14 ч. под аргоном при комнатной температуре. Реакционную смесь выпаривали досуха и остаток (5.6 г (98%), масло коричневого цвета) использовали в следующей стадии без дополнительной очистки; ВЭЖХ-МС: Ву=1.64; [М+Н] 169.
1.2 Этиловый эфир 1-бензил-1H-имидазол-4-карбоновой кислоты
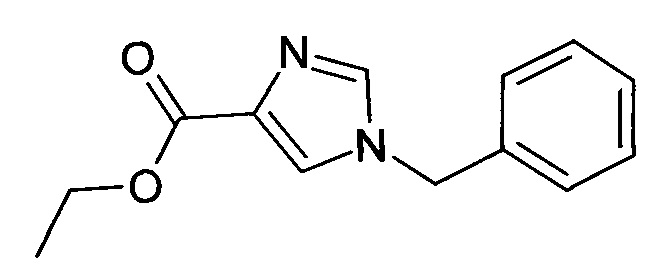
К этиловому эфиру (Z)-3-диметиламино-2-изоциано-акриловой кислоты (5.6 г, 28.97 ммоль) добавляли бензиламин (3.48 мл, 31.86 ммоль) и реакционную смесь перемешивали при 70°C в течение 14 ч. Реакционную смесь концентрировали, и остаток очищали посредством флэш-хроматографии (элюент: ДХМ-метанол). Целевые фракции объединяли и выпаривали досуха с получением 3.93 г (57%), масло коричневого цвета; ВЭЖХ-МС: Ву=1.72; [М+Н] 231.
1.3 Амид 1-бензил-1H-имидазол-4-карбоновой кислоты
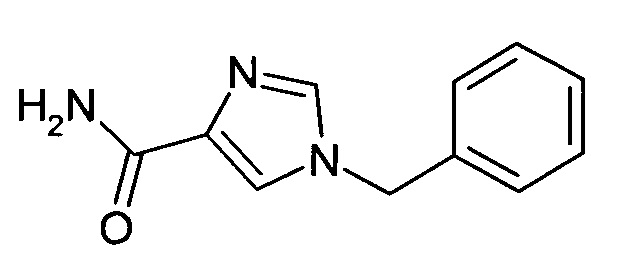
Этиловый эфир 1-бензил-1H-имидазол-4-карбоновой кислоты (3.93 г, 17.07 ммоль) и хлорид аммония (0.27 г, 5.12 ммоль) обрабатывали раствором гидроксида аммония (32%, 45 мл) и перемешивали в автоклаве при 105°C в течение 14 ч. Реакционную смесь охлаждали до комнатной температуры, осадок отфильтровывали, промывали водой и сушили при 50°C в вакууме; выход: 2.13 г (61%); ВЭЖХ-МС: Ву=1.30; [М+Н] 202.
1.4 Бромид 2-бензил-8-оксо-6-n-толил-7,8-дигидро-имидазо[1,5-а]пиразин-2-ия
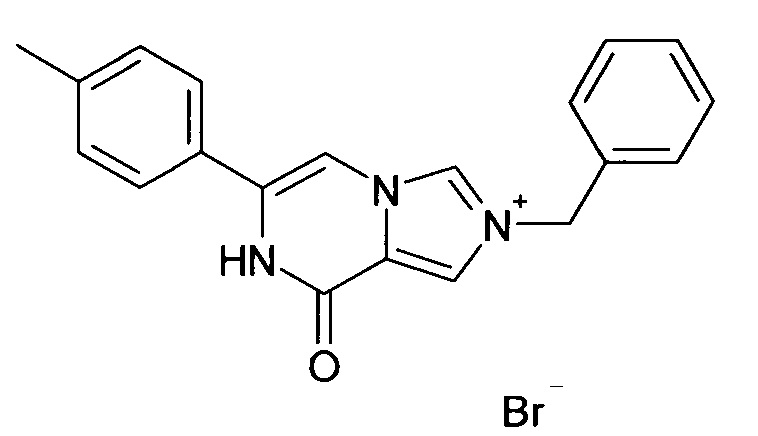
Амид 1-бензил-1Н-имидазол-4-карбоновой кислоты (500 мг, 2.49 ммоль) и 2-бром-1-n-толил-этанон (635.3 мг, 2,98 ммоль) растворяли в ДМФА/CH3CN -3/7 (10 мл) и перемешивали при 90°C в течение 14 ч. Реакционную смесь охлаждали до комнатной температуры, осадок отфильтровывали, промывали диэтиловым эфиром и сушили; выход: 452 мг (46%); ВЭЖХ-МС: By=1.43; [М] 316.
1.5 6-n-Толил-7H-имидазо[1,5-а]пиразин-8-он ("А1")
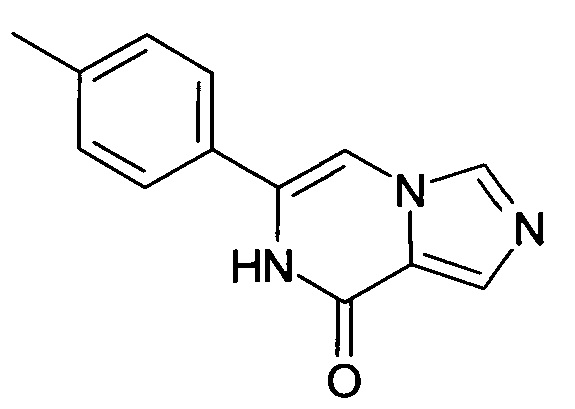
Смесь бромида 2-бензил-8-оксо-6-n-толил-7,8-дигидро-имидазо[1,5-а]пиразин-2-ия (451.0 мг, 1.138 ммоль) и имидазола (3.87 г, 56.90 ммоль) нагревали под атмосферой азота до 175°C и перемешивали в течение 5 ч. Реакционную смесь охлаждали и выливали на лед. Осадок фильтровали отсасыванием, промывали диэтиловым эфиром и сушили; выход: 141 мг (55%); ВЭЖХ-МС: Ву=1.50; [М+Н] 226;
1Н ЯМР (500 МГц, ДМСО-d6) δ=10.93 (s, 1Н), 8.25 (d, J=0.9 Hz, 1Н), 7.77 (s, 1H), 7.74 (s, 1H), 7.59-7.54 (m, 2H), 7.31-7.26 (m, 2H), 2.36 (s, 3H).
Аналогичным образом получали следующие примеры:
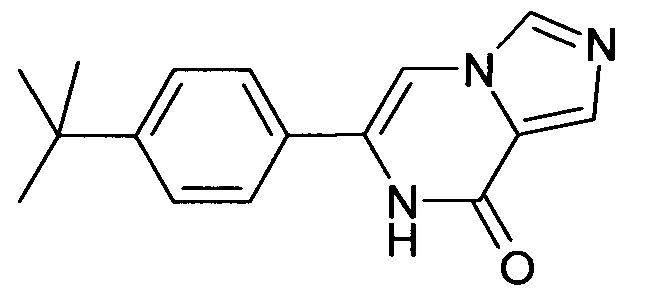
6-(4-трет-Бутил-фенил)-7H-имидазо[1,5-а]пиразин-8-он ("А2")
Выход: 13.5 мг (11%); ВЭЖХ-МС: Ву=1.97; [М+Н] 268;
1Н ЯМР (400 МГц, ДМСO-d6) δ=10.92 (s, 1Н), 8.27 (s, 1H), 7.78 (s, 1Н), 7.75 (s, 1H), 7.64-7.58 (m, 2Н), 7.53-7.47 (m, 2Н), 1.31 (s, 9Н).
6-(4-Трифторметил-фенил)-7H-имидазо[1,5-а]пиразин-8-он ("A3")
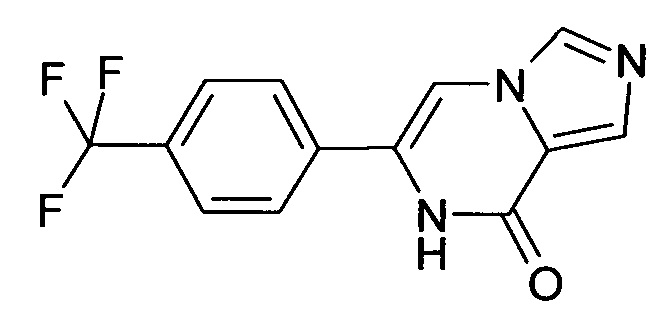
Выход: 39.5 мг (20%); ВЭЖХ-МС: Ву=1.75; [М+Н] 280; 1Н ЯМР (400 МГц, ДМСО-d6) δ=11.15 (s, 1Н), 8.33 (s, 1Н), 7.96-7.80 (m, 6Н).
Пример 4
Синтез 6-[4-(1-метил-1Н-пиразол-4-ил)-фенил]-7H-имидазо[1,5-а]пиразин-8-она ("А4")
4.1 6-(4-Бром-фенил)-7H-имидазо[1,5-а]пиразин-8-он
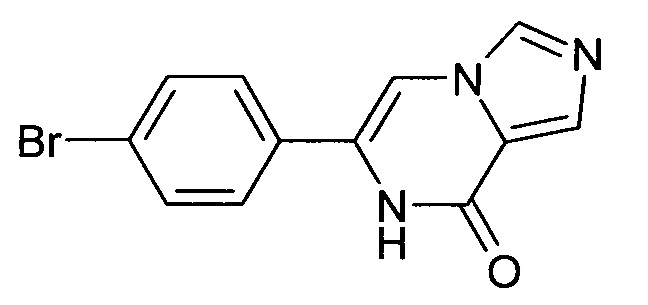
Выход: 1.6 г (98%); ВЭЖХ-МС: Ву=1.65; [М+Н] 291.
4.2 6-[4-(1-Метил-1Н-пиразол-4-ил)-фенил]-7H-имидазо[1,5-а]пиразин-8-он
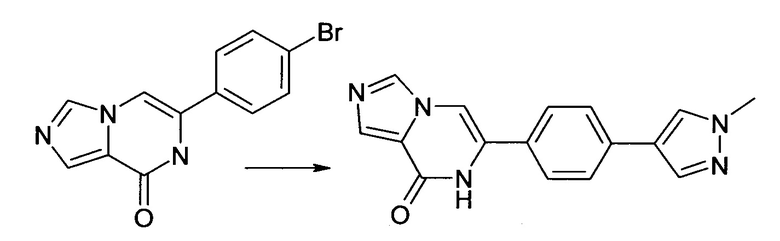
6-(4-Бром-фенил)-7H-имидазо[1,5-а]пиразин-8-он (пример 4, 150.0 мг, 0.517 ммоль), 1-метил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-пиразол (118,3 мг, 0,569 ммоль) и гидрокарбонат натрия (52.1 мг, 0,620 ммоль) суспендировали в ДМФА/вода - 2/1 (1.5 мл), продували азотом и нагревали до 40°С. Добавляли хлорид бис(трифенилфосфин)-палладия(II) (10.5 мг, 0,015 ммоль) и реакционную смесь нагревали до 80°C в течение 14 ч. Температуру увеличивали до 90°C и реакционную смесь перемешивали в течение дополнительных 5 ч. Реакционную смесь разбавляли с водой. Полученный осадок фильтровали отсасыванием и очищали посредством флэш-хроматографии; выход: 17 мг (11%); ВЭЖХ-МС: Ву=1.40; [М+Н] 292;
1Н ЯМР (400 МГц, ДМСО-d6) δ=10.82 (brs, 1Н), 8.21(s, 2Н), 7.93 (s, 1Н), 7.80 (s, 1Н), 7.73-7.61 (m, 5Н), 3.87 (s, 3Н).
Пример 5
Синтез 6-(4-гидроксиметил-фенил)-7Н-имидазо[1,5-а]пиразин-8-она ("А5")
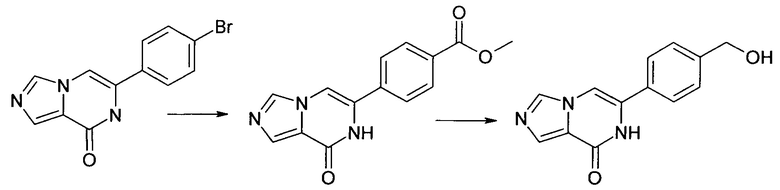
5.1 Метиловый эфир 4-(8-оксо-7,8-дигидро-имидазо[1,5-а]пиразин-6-ил)-бензойной кислоты
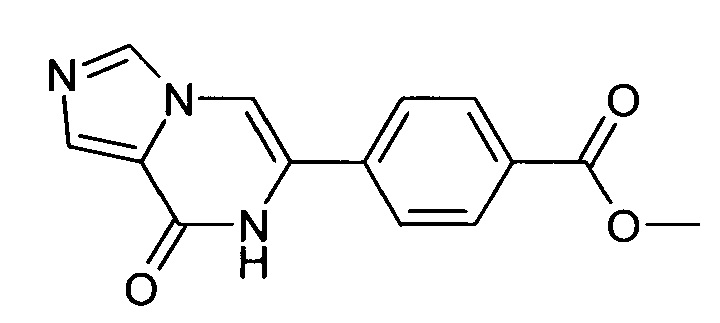
В автоклаве, раствор 6-(4-бром-фенил)-7H-имидазо[1,5-а]пиразин-8-она (500 мг, 1.723 ммоль) и триэтиламина (260 мг, 2.569 ммоль) в метанол/толуол - 1/1 (16 мл) продували азотом. Добавляли (1,1'-бис(дифенилфосфино)-ферроцен)дихлорпалладий(II), дихлорметан (43 мг, 0.053 ммоль) и 1,1-бис-(дифенилфосфино)-ферроцен (39 мг, 0.070 ммоль). Затем автоклав наполняли оксидом углерода и нагревали до 100°С. Автоклав выдерживали при этой температуре в течение 6 часов под давлением оксида углерода в 2-4 бар. Давление в автоклаве сбрасывали до атмосферного. Выпавшее в осадок твердое вещество отфильтровывали отсасыванием, промывали метанолом и высушивали в вакууме; выход: 314 мг (68%), ВЭЖХ-МС: By=1.47; [М+Н] 270.
5.2 6-(4-Гидроксиметил-фенил)-7Н-имидазо[1,5-а]пиразин-8-он
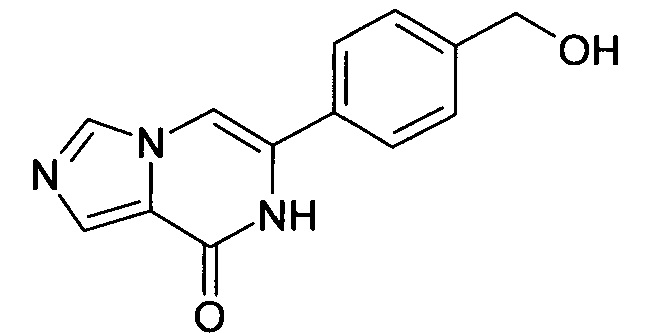
Метиловый эфир 4-(8-оксо-7,8-дигидро-имидазо[1,5-а]пиразин-6-ил)-бензойной кислоты (100 мг, 0.371 ммоль) суспендировали в ТГФ (5 мл) и обрабатывали гидридом лития и алюминия (2,0 моль в ТГФ, 0.371 мл, 0.743 ммоль) и перемешивали при температуре окружающей среды в течение 14 ч. Реакционную смесь гасили небольшим количеством метанола, подкисленного посредством 1 мл 1 N хлористоводородной кислоты и фильтровали. Фильтрат экстрагировали три раза дихлорметаном, объединенные органические слои промывали водой и рассолом, высушивали над сульфатом натрия, фильтровали и выпаривали досуха. Водный слой выпаривали досуха. Объединенные остатки очищали посредством хроматографии; выход: 55 мг (61%); ВЭЖХ-МС: Ву=1.05; [М+Н] 242;
1Н ЯМР (400 МГц, ДМСО-d6) 5=10.96 (s, 1Н), 8.27 (s, 1Н), 7.79-7.74 (m, 2Н), 7.66-7.61 (m, 2Н), 7.45-7.37 (m, 2Н), 5.25 (t, J=5.7 Hz, 1Н), 4.55 (d, J=5.7 Hz, 2H).
Пример 6
Синтез 6-[4-(1-гидрокси-1-метил-этил)-фенил]-7Н-имидазо[1,5-а]пиразин-8-она ("А6")
Метиловый эфир 4-(8-оксо-7,8-дигидро-имидазо[1,5-а]пиразин-6-ил)-бензойной кислоты (70 мг, 0.252 ммоль) суспендировали в ТГФ (4 мл), добавляли хлорид церия (68.4 мг, 0.277 ммоль) и смесь перемешивали в течение 1 ч. при температуре окружающей среды. Добавляли хлорид метилмагния (3 М в ТГФ, 0.39 мл; 1,059 ммоль) и реакционную смесь перемешивали в течение 1 ч. при температуре окружающей среды. Реакционную смесь гасили посредством 5% лимонной кислоты. Образованный осадок отфильтровывали и фильтрат концентрировали. Образованный осадок отфильтровывали отсасыванием и высушивали; выход: 50 мг (74%); ВЭЖХ-МС: Ву=1.30; [М+Н] 270; 1Н ЯМР (400 МГц, ДМСO-d6) δ=10.92 (s, 1Н), 8.31 (s, 1Н), 7.82 (s, 1Н), 7.76 (s, 1Н), 7.64-7.51 (m, 4Н), 5.14 (s, 1Н), 1.44 (s, 6Н).
Пример 7
Синтез 6-(4-трет-бутил-фенил)-3-метил-7H-имидазо[1,5-а]пиразин-8-она ("А7")
7.1 Этиловый эфир 2-метил-1Н-имидазол-4-карбоновой кислоты
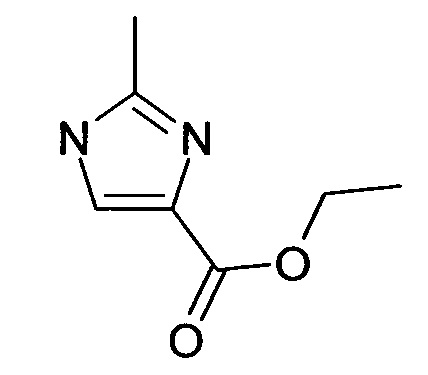
2-Метил-1Н-имидазол-4-карбоновой кислоты (1.43 g; 11.299 ммоль) растворяли в этаноле (90.0 мл). Добавляли HCl в диоксане (4 М, 7.50 мл) и смесь нагревали при 90°C в течение 18 ч. Раствор концентрировали. Осадок распределяли между этилацетатом и насыщенным раствором NaHCO3. Водный слой экстрагировали два раза этилацетатом. Объединенные органические слои промывали рассолом и высушивали над Na2SO4, фильтровали и выпаривали досуха; выход: 1.43 г (82%).
7.2 Этиловый эфир 1-бензил-2-метил-1Н-имидазол-4-карбоновой кислоты
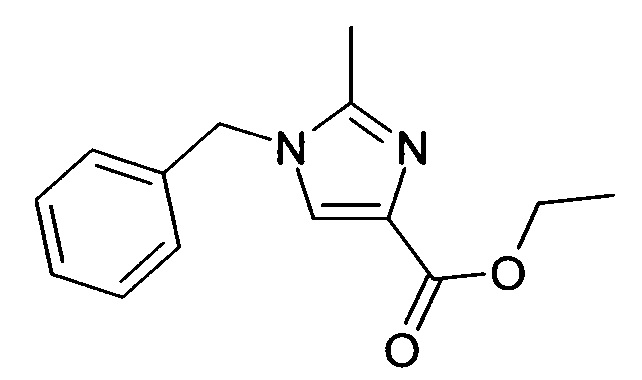
К раствору этилового эфира 2-метил-1Н-имидазол-4-карбоновой кислоты (1.43 г; 9.276 ммоль) в ацетонитриле (80 мл) добавляли карбонат цезия (6.04 г; 18.551 ммоль). По каплям добавляли бензилбромид (1.59 г; 9.276 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 14 ч. Реакционную смесь концентрировали в вакууме, разбавляли с водой и экстрагировали два раза этилацетатом. Объединенные органические слои высушивали над сульфатом натрия, фильтровали и выпаривали досуха. Маслянистый остаток использовали в следующей стадии без какой-либо дополнительной очистки; выход: 940 мг (29%).
7.3 Амид 1-бензил-2-метил-1Н-имидазол-4-карбоновой кислоты
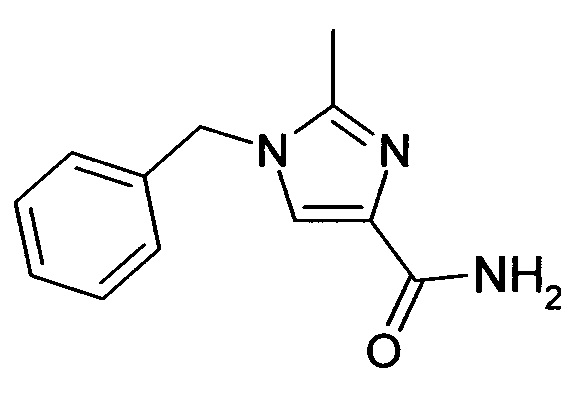
Этиловый эфир 1-бензил-2-метил-1Н-имидазол-4-карбоновой кислоты (300.0 мг, 1.228 ммоль) и хлорид аммония (20.0 мг, 0.368 ммоль) нагревали в автоклаве в растворе гидроксида аммония (32%, 20 мл) при 105°C в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры и выпаривали досуха. Осадок использовали в следующей стадии без какой-либо дополнительной очистки; выход: 132 мг (50%).
7.4 6-(4-трет-Бутил-фенил)-3-метил-7H-имидазо[1,5-а]пиразин-8-он
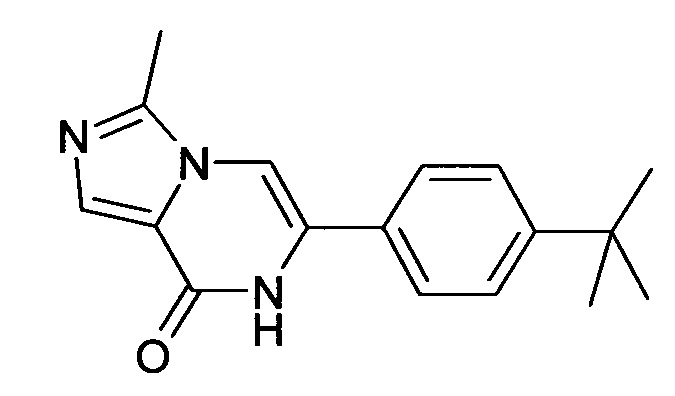
Амид 1-бензил-2-метил-1Н-имидазол-4-карбоновой кислоты (69.0 мг; 0.321 ммоль) и 1-(4-трет-бутил-фенил)-2-хлор-этанон (81.0 мг; 0.385 ммоль) растворяли в ДМФА (1 мл) и ацетонитриле (3 мл) и нагревали в микроволновой установке (СЕМ) при 160°C в течение 6 ч. Реакционную смесь концентрировали, разбавляли с водой и экстрагировали 3 раза этилацетатом. Объединенные органические слои промывали водой и рассолом, высушивали сульфатом натрия, фильтровали и выпаривали досуха. Маслянистый остаток очищали посредством хроматографии; выход: 11.0 мг (12%); ВЭЖХ-МС: By=1.85; [М+Н] 282;
1Н ЯМР (500 МГц, ДМСО-d6) δ=10.82 (s, 1Н), 7.69-7.63 (m, 3Н), 7.53-7.46 (m, 3Н), 2.57 (s, 3Н), 1.32 (s, 9Н).
Пример 8
Синтез 6-[4-(1-гидрокси-1-метил-этил)-фенил]-3-метил-7Н-имидазо[1,5-а]пиразин-она ("А8")
8.1 6-(4-Бром-фенил)-3-метил-7Н-имидазо[1,5-а]пиразин-8-он
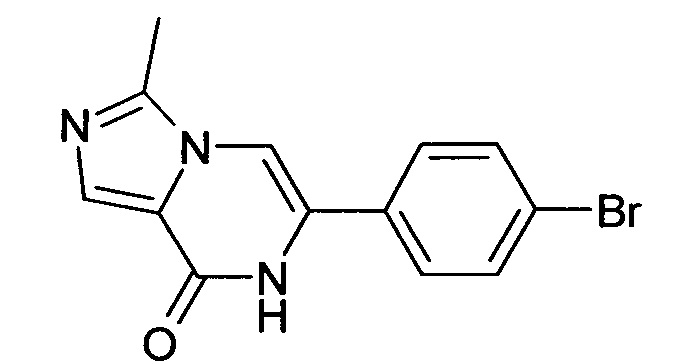
Получение, как описано для примера 7 (стадия 7.4); выход: 83.0 мг (33%);
ВЭЖ-МС: Ву=1.57; [М+Н] 304-307.
8.2 Метиловый эфир 4-(3-метил-8-оксо-7,8-дигидро-имидазо[1,5-а]пиразин-6-ил)-бензойной кислоты
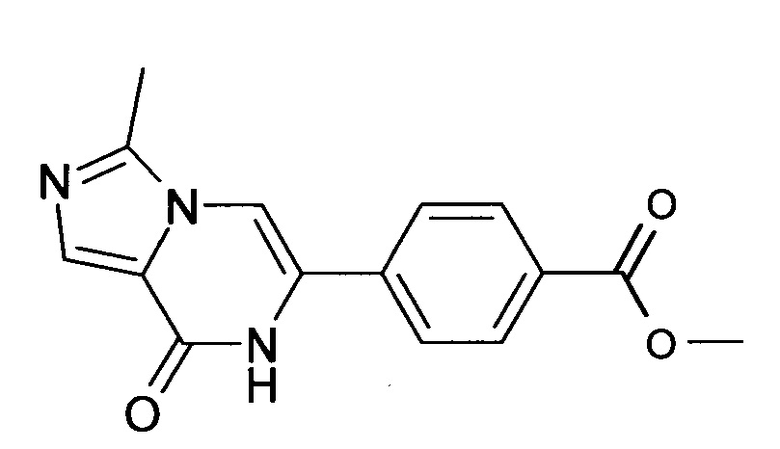
Получение, как описано для примера 5 (стадия 5.1); выход: 37.0 мг (49%); ВЭЖХ-МС: Ву=1.40; [М+Н] 284.
8.3 6-[4-(1-Гидрокси-1-метил-этил)-фенил]-3-метил-7Н-имидазо[1,5-а]пиразин-он ("А8")
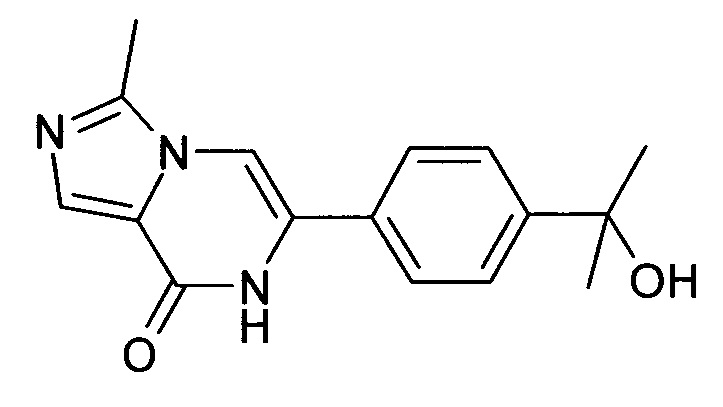
Получение, как описано для примера 6; выход: 15.0 мг (40%); ВЭЖХ-МС: Ву=1.24; [М+Н] 284; 1Н ЯМР (400 МГц, ДМСО-d6) δ=10.82 (s, 1Н), 7.72-7.62 (m, 3Н), 7.60-7.46 (m, 3Н), 5.07 (s, 1Н), 2.57 (s, 3Н), 1.45 (s, 6Н).
Фармакологические данные
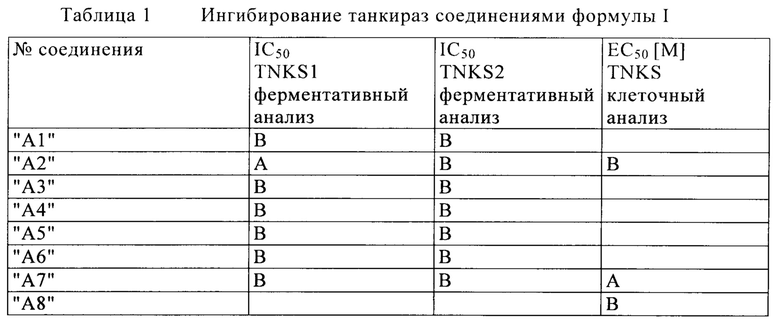
IC50: <0.3 мкМ = A 0.3-3 мкМ = В 3-50 мкМ=С
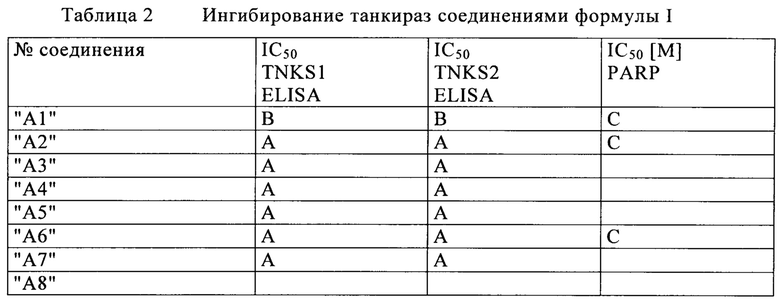
IC50: <0.3 мкМ = А 0.3-3 мкМ = В 3-50 мкМ = С
Нижеследующие примеры относятся к лекарственным средствам:
Пример A: Флаконы для инъекций
pH раствора 100 г активного компонента формулы I и 5 г гидрофосфата динатрия в 3 л бидистиллированной воды устанавливают до 6.5, с применением 2 н. соляной кислоту, стерильно фильтруют, переносят во флаконы для инъекций, лиофилизируют в стерильных условиях и запечатывают в стерильных условиях. Каждый флакон для инъекций содержит 5 мг активного компонента.
Пример Б: Суппозитории
Смесь 20 г активного компонента формулы I расплавляют со 100 г соевого лецитина и 1400 г масла какао, разливают в пресс-формы и дают остыть. Каждый суппозиторий содержит 20 мг активного компонента.
Пример B: Раствор
Раствор приготавливают из 1 г активного компонента формулы I, 9.38 г NaH2Po4⋅2Н2O, 28.48 г Na2HPO4⋅12H2O и 0.1 г хлорида бензалкония в 940 мл бидистиллированной воды. pH раствора устанавливают до 6.8, объем раствора доводят до 1 л и стерилизуют путем облучения. Этот раствор можно применять в форме глазных капель.
Пример Г: Мазь
500 мг активного компонента формулы I смешивают с 99.5 г вазелина в асептических условиях.
Пример Д: Таблетки
Смесь 1 кг активного компонента формулы I, 4 кг лактозы, 1,2 кг картофельного крахмала, 0,2 кг талька и 0,1 кг стеарата магния спрессовывают для получения таблеток обычным способом таким образом, чтобы каждая таблетка содержала 10 мг активного компонента.
Пример Е: Драже
Таблетки спрессовывают аналогично примеру Д, и затем покрывают обычным способом покрытием из сахарозы, картофельного крахмала, талька, трагаканта и красителя.
Пример Ж: Капсулы
2 кг активного компонента формулы I помещают в твердые желатиновые капсулы обычным способом таким образом, чтобы каждая капсула содержала 20 мг активного компонента.
Пример З: Ампулы
Раствор 1 кг активного компонента формулы I в 60 л бидистиллированной воды стерилизуют фильтрацией, переносят в ампулы, лиофилизируют в стерильных условиях и запечатывают в стерильных условиях. Каждая ампула содержит 10 мг активного компонента.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФТАЛАЗИНОВЫЕ ПРОИЗВОДНЫЕ | 2014 |
|
RU2663623C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2013 |
|
RU2650107C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНМОЧЕВИНЫ | 2014 |
|
RU2666894C2 |
| ПРОИЗВОДНЫЕ (АЗА-)ИЗОХИНОЛИНОНА | 2013 |
|
RU2654216C2 |
| ОКСОХИНАЗОЛИНИЛБУТАНАМИДНЫЕ ПРОИЗВОДНЫЕ | 2014 |
|
RU2669393C2 |
| ИМИДАЗОЛОНИЛХИНОЛИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ АТМ | 2016 |
|
RU2743343C2 |
| ПРОИЗВОДНОЕ ФТАЛАЗИНОНКЕТОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2011 |
|
RU2564527C2 |
| АРИЛХИНАЗОЛИНЫ | 2014 |
|
RU2701193C2 |
| БИЦИКЛИЧЕСКИЕ АГОНИСТЫ СТИМУЛЯТОРА ГЕНОВ ИНТЕРФЕРОНА STING | 2020 |
|
RU2800072C1 |
| ИНГИБИТОРЫ MAGL НА ОСНОВЕ ПИРАЗОЛА | 2018 |
|
RU2789157C2 |
Изобретение относится к новым соединениям, выбранным из группы 
, а также к лекарственным средствам на их основе. Технический результат: получены новые соединения, обладающие ингибирующей активностью в отношении танкираз (TANK). 2 н.п. ф-лы, 2 табл., 8 пр.
1. Соединение, выбранное из группы, включающей:

и его фармацевтически приемлемые соли, сольваты, таутомеры и стереоизомеры, включая их смеси во всех соотношениях.
2. Лекарственные средства, обладающие ингибирующей активностью в отношении танкираз (TANK), содержащие по меньшей мере одно соединение по п. 1 и/или его фармацевтически приемлемые соли, сольваты, таутомеры и стереоизомеры, включая их смеси во всех соотношениях в эффективном количестве, и необязательно фармацевтически приемлемый носитель, наполнитель или среду для лекарственного средства.
| WO 2013143663 A1, 03.10.2013 | |||
| Mukaiyama H et al, Synthesis ans c-Src inhibitory activity of imidazol[1,5-a]pyrazine derivatives as agent for treatment of acute ischemic stroke, Bioorganic & Medicinal Chemistry,15, 2, стр | |||
| Запорный к лапан для тушения горящих нефтяных фонтанов | 1914 |
|
SU868A1 |
| JP 2005089352 A, 07.04.2005 | |||
| RU 2010125703 A, 10.02.2012 | |||
| WO 2011089400 A1, 28.07.2011. | |||

