Область техники, к которой относится изобретение
Настоящее изобретение в целом обеспечивает терапию для эффективного лечения и/или предупреждения заболеваний, связанных с клетками, экспрессирующими CLDN18.2, в частности, раковых заболеваний, таких как гастроэзофагеальный рак.
Уровень техники
Рак желудка и пищевода (гастроэзофагеальный; GE) относится к злокачественным опухолям с высокой неудовлетворенной медицинской потребностью. Рак желудка является второй ведущей причиной смерти от злокачественных новообразований во всем мире. За последние десятилетия увеличилась заболеваемость раком пищевода, совпадающая со сдвигом гистологического типа и локализацией первичной опухоли. В настоящее время аденокарцинома пищевода является более распространенной, чем сквамозно-клеточная карцинома в США и Западной Европе, при этом большинство опухолей локализуется в дистальном отделе пищевода. Показатель общей пятилетней выживаемости для GE рака составляет 20-25% независимо от агрессивности установленной стандартной терапии, связанной со значительными побочными эффектами.
Большинство пациентов в момент обращения за медицинской помощью имеют местно-распространенное или метастатическое заболевание и должны быть подвергнуты химиотерапии первой линии. Режимы лечения основаны на производных платины и фторпиримидина, главным образом в сочетании с третьим соединением (например, таксаном или антрациклинами). Однако лучшим, что можно ожидать, является средняя выживаемость без прогрессирования заболевания от 5 до 7 месяцев и средняя общая выживаемость от 9 до 11 месяцев.
Отсутствие значительной пользы от различных комбинированных режимов химиотерапии нового поколения для этих видов рака подтолкнуло к исследованию применения таргетных агентов. Недавно трастузумаб был одобрен для Her2/neu-положительных типов гастроэзофагеального рака. Однако, поскольку лишь ~20% пациентов экспрессируют мишень и подходят для данного лечения, потребность в области медицины все еще остается высокой.
Молекула в плотных контактах клаудин-18, сплайсированный вариант 2, (клаудин 18.2 (CLDN18.2)) является членом семейства клаудинов, белков плотных контактов. CLDN18.2 представляет собой трансмембранный белок с молекулярной массой 27,8 кДа, содержащий четыре трансмембранных домена с двумя малыми внеклеточными петлями.
В нормальных тканях отсутствует детектируемая экспрессия CLDN18.2 по данным RT-PCR, за исключением желудка. Иммуногистохимический анализ с использованием CLDN18.2-специфических антител показывает, что желудок является единственной положительной тканью.
CLDN18.2 является высокоселективным желудочным линейным антигеном, который экспрессируется исключительно на дифференцированных короткоживущих клетках эпителия желудка. CLDN18.2 сохраняется в процессе злокачественной трансформации и, таким образом, часто обнаруживается на поверхности клеток рака желудка человека. Кроме того, этот пан-опухолевый антиген эктопически активируется на значительных уровнях в аденокарциномах пищевода, поджелудочной железы и легких. Белок CLDN18.2 также локализуется в метастазах в лимфоузлах аденокарциномы рака желудка и в отдаленных метастазах, особенно в яичники (так называемые опухоли Крукенберга).
IMAB362 представляет собой химерное антитело IgG1, направленное против CLDN18.2, которое было разработано компанией Ganymed Pharmaceuticals AG. IMAB362 с высокой аффинностью и специфичностью распознает первый внеклеточный домен (ECD1) CLDN18.2. IMAB362 не связывается с какими-либо другими членами семейства клаудинов, включая близкородственный сплайсированный вариант 1 клаудина-18 (CLDN18.1). IMAB362 демонстрирует четыре точных независимых высокоактивных механизма действия в отношении специфичности к опухолевым клеткам и узлам. При связывании с мишенью IMAB362 опосредует уничтожение клеток посредством ADCC, CDC и индукции апоптоза, вызванного поперечным сшиванием мишени на поверхности опухолевых клеток, и прямого ингибирования пролиферации. Таким образом, IMAB362 эффективно лизирует CLDN18.2-положительные клетки, включая человеческие клеточные линии рака желудка in vitro и in vivo. Мыши, несущие CLDN18.2-положительные линии раковых клеток, имеют преимущества в выживаемости и до 40% мышей демонстрируют регрессию своих опухолей при лечении с помощью IMAB362.
Токсичность и PK/TK (фармакокинетический/токсикокинетический) профиль IMAB362 тщательно изучали на мышах и макаках-кабоедах, включая исследования по определению диапазона доз, 28-дневные исследования токсичности многократных доз на макаках-крабоедах и 3-месячное исследование токсичности многократных доз на мышах. Многократные дозы IMAB362 при внутривенном введении (i.v.) хорошо переносились мышами (самое длительное лечение путем еженедельного введения в течение 3 месяцев, самые высокие уровни доз составили 400 мг/кг) и мамаками-крабоедами (до 5 еженедельных применений вплоть до 100 мг/кг). Признаков системной или местной токсичности не отмечалось. В частности, желудочной токсичности не наблюдалось в любом исследовании токсичности. IMAB362 не вызывает активации иммунной системы и высвобождения цитокинов. Побочных эффектов на мужские и женские репродуктивные органы не отмечалось. IMAB362 не связывается с тканями, лишенными мишени. Исследования биораспределения у мышей показывают, что причина отсутствия желудочной токсичности, по-видимому, связана с компартментализацией плотных контактов в люминальном участке здорового эпителия желудка, что, как оказалось, значительно ухудшает доступность эпитопа IMAB362. Эта компартментализация утрачивается при злокачественной трансформации, делая эпитоп поддающимся воздействию IMAB362.
В настоящем документе представлены данные, демонстрирующие, что введение антитела к CLDN18.2, такого как IMAB362, пациенту-человеку с гастроэзофагеальным раком является безопасным и хорошо переносимым вплоть до дозы, составляющей, по меньшей мере, 1000 мг/м2. Кроме того, данные, представленные здесь, демонстрируют, что антитело является полностью функциональным у этих пациентов для осуществления эффектов против опухолевых клеток, и получено доказательство противоопухолевой активности.
Раскрытие изобретения
В настоящем изобретении в целом предлагается терапия для эффективного лечения и/или предупреждения заболеваний, связанных с клетками, экспрессирующими CLDN18.2, включающих раковые заболевания, такие как рак желудка, рак пищевода, рак поджелудочной железы, рак легкого, такой как немелкоклеточный рак легкого (NSCLC), рак яичника, рак толстой кишки, рак печени, рак головы и шеи, и рак желчного пузыря, а также их метастазы, в частности, метастазы рака желудка, такие как опухоли Крукенберга, перитонеальные метастазы и метастазы в лимфоузлы. Особенно предпочтительными раковыми заболеваниями является аденокарцинома желудка, пищевода, протока поджелудочной железы, желчных протоков, легкого и яичника.
В первом аспекте в настоящем изобретении предлагается способ лечения или предупреждения ракового заболевания, включающий введение пациенту антитела, обладающего способностью связываться с CLDN18.2, при этом антитело вводят таким образом, чтобы обеспечить уровень в сыворотке, по меньшей мере, 40 мкг/мл. В других вариантах осуществления антитело вводят таким образом, чтобы обеспечить уровень в сыворотке, по меньшей мере, 50 мкг/мл, по меньшей мере, 150 мкг/мл, по меньшей мере, 300 мкг/мл, по меньшей мере, 400 мкг/мл или, по меньшей мере, 500 мкг/мл. В различных вариантах осуществления антитело вводят таким образом, чтобы обеспечить уровень в сыворотке не более 800 мкг/мл, 700 мкг/мл, 600 мкг/мл, 550 мкг/мл или 500 мкг/мл. В одном варианте осуществления обеспечен уровень в сыворотке в диапазоне от 40 мкг/мл до 700 мкг/мл, предпочтительно от 40 мкг/мл до 600 мкг/мл, предпочтительно от 50 мкг/мл до 500 мкг/мл, такой как от 150 мкг/мл до 500 мкг/мл или от 300 мкг/мл до 500 мкг/мл. Под термином «уровень в сыворотке», используемом в настоящем описании, понимается концентрация рассматриваемого вещества в сыворотке крови. В одном варианте осуществления обеспечен уровень в сыворотке, который сохраняется в течение, по меньшей мере, 7 дней или, по меньшей мере, 14 дней. В одном варианте осуществления способ включает введение дозы/доз антитела, по меньшей мере, 300 мг/м2, например, по меньшей мере, 600 мг/м2 и предпочтительно вплоть до 1500 мг/м2, до 1200 мг/м2 или до 1000 мг/м2.
Во втором аспекте в настоящем изобретении предлагается способ лечения или предупреждения ракового заболевания, включающий введение пациенту антитела, обладающего способностью связываться с CLDN18.2, при этом антитело вводят в дозе, по меньшей мере, 300 мг/м2, например, по меньшей мере, 600 мг/м2 и предпочтительно вплоть до 1500 мг/м2, вплоть до 1200 мг/м2 или вплоть до 1000 мг/м2.
В третьем аспекте в настоящем изобретении предлагается способ лечения или предупреждения ракового заболевания, включающий введение пациенту антитела, обладающего способностью связываться с CLDN18.2, при этом по меньшей мере 50%, предпочтительно 60%, 70%, 80% или 90% раковых клеток пациента являются CLDN18.2-положительными, и/или по меньшей мере 40%, предпочтительно 50% или 60% раковых клеток пациента являются положительными в отношении поверхностной экспрессии CLDN18.2. В этом аспекте в настоящем изобретении также предлагается способ лечения или предупреждения ракового заболевания, при этом указанный способ включает: а. идентификацию пациента, демонстрирующего по меньшей мере 50%, предпочтительно 60%, 70%, 80% или 90% CLDN18.2-положительных раковых клеток и/или по меньшей мере 40%, предпочтительно 50% или 60% раковых клеток, которые являются положительными в отношении поверхностной экспрессии CLDN18.2; и b. введение указанному пациенту антитела, обладающего способностью связываться с CLDN18.2. В одном варианте осуществления, по меньшей мере, 95% или, по меньшей мере, 98% раковых клеток пациента являются CLDN18.2-положительными. В одном варианте осуществления по меньшей мере 70%, по меньшей мере 80% или по меньшей мере 90% раковых клеток пациента являются положительными в отношении поверхностной экспрессии CLDN18.2.
В одном варианте осуществления способа по любому из аспектов, описанных здесь, лечение ракового заболевания приводит к достижению стабильного заболевания. В одном варианте осуществления достигается стабильное заболевание, которое сохраняется в течение, по меньшей мере, 2 месяцев, по меньшей мере, 3 месяцев или, по меньшей мере, 6 месяцев.
В четвертом аспекте в настоящем изобретении предлагается способ достижения стабильного заболевания у ракового пациента, включающий введение пациенту антитела, обладающего способностью связываться с CLDN18.2. В одном варианте осуществления достигается стабильное заболевание, которое сохраняется в течение, по меньшей мере. 2 месяцев, по меньшей мере, 3 месяцев или, по меньшей мере, 6 месяцев.
В одном варианте осуществления способа по любому из аспектов, описанных здесь, антитело вводят в виде однократной дозы или в виде многократных доз.
В пятом аспекте в настоящем изобретении предлагается способ лечения или предупреждения ракового заболевания, включающий введение пациенту антитела, обладающего способностью связываться с CLDN18.2, при этом антитело вводят в виде многократных доз.
В случае введения антитела в соответствии с изобретением в виде многократных доз, указанное антитело предпочтительно вводят в виде по меньшей мере 3 доз, по меньшей мере 4 доз, по меньшей мере 5 доз, по меньшей мере 6 доз, по меньшей мере 7 доз, по меньшей мере 8 доз, по меньшей мере 9 доз или по меньшей мере 10 доз и предпочтительно до 30, 25, 20, 15 или 10 доз. Дозы антитела предпочтительно вводят с временными интервалами, составляющими по меньшей мере 7 дней, по меньшей мере 10 дней, по меньшей мере 14 дней или по меньшей мере 20 дней. Дозы антитела предпочтительно вводят с временными интервалами, составляющими от 7 до 30 дней, от 10 до 20 дней и предпочтительно около 14 дней.
В одном варианте осуществления способа по третьему, четвертому или пятому аспекту антитело вводят таким образом, чтобы обеспечить уровень в сыворотке, по меньшей мере, 40 мкг/мл. В других вариантах осуществления антитело вводят таким образом, чтобы обеспечить уровень в сыворотке, по меньшей мере, 50 мкг/мл, по меньшей мере 150 мкг/мл, по меньшей мере 300 мкг/мл, по меньшей мере 400 мкг/мл или по меньшей мере 500 мкг/мл. В различных вариантах осуществления антитело вводят таким образом, чтобы обеспечить уровень в сыворотке не более 800 мкг/мл, 700 мкг/мл, 600 мкг/мл, 550 мкг/мл или 500 мкг/мл. В одном варианте осуществления обеспечен уровень в сыворотке в диапазоне от 40 мкг/мл до 700 мкг/мл, предпочтительно от 40 мкг/мл до 600 мкг/мл, предпочтительно от 50 мкг/мл до 500 мкг/мл, например, от 150 мкг/мл до 500 мкг/мл или от 300 мкг/мл до 500 мкг/мл. В одном варианте осуществления обеспечен уровень в сыворотке, который сохраняется в течение, по меньшей мере, 7 дней или, по меньшей мере, 14 дней. В одном варианте осуществления способ включает введение дозы/доз антитела, составляющей, по меньшей мере, 300 мг/м2, а именно, по меньшей мере, 600 мг/м2 и предпочтительно до 1500 мг/м2, до 1200 мг/м2 или до 1000 мг/м2.
В одном варианте осуществления способа по любому из указанных выше аспектов способ дополнительно включает введение одного или нескольких лекарственных средств, выбранных из группы, состоящей из противорвотных средств, антиспазматических средств, парасимпатолитических средств и агентов, которые защищают слизистую оболочку желудка.
В шестом аспекте в настоящем изобретении предлагается способ лечения или предупреждения ракового заболевания, включающий введение пациенту антитела, обладающего способностью связываться с CLDN18.2, и одного или нескольких лекарственных средств, выбранных из группы, состоящей из противорвотных средств, антиспазматических средств, парасимпатолитических средств и агентов, которые защищают слизистую оболочку желудка.
В случае если способ по изобретению включает введение одного или нескольких лекарственных средств, выбранных из группы, состоящей из противорвотных средств, антиспазматических средств, парасимпатолитических средств и агентов, которые защищают слизистую оболочку желудка, способ в различных вариантах осуществления включает введение: (i) противорвотного средства и антиспазматического средства; (ii) антиспазматического средства и агента, который защищает слизистую оболочку желудка; (iii) противорвотного средства и агента, который защищает слизистую оболочку желудка; или (iv) противорвотного средства, антиспазматического средства и агента, который защищает слизистую оболочку желудка.
В одном варианте осуществления противорвотное средство вводят в качестве противорвотной профилактики до введения антитела. В одном варианте осуществления противорвотное средство вводят в качестве противорвотной интервенции одновременно и/или после введения антитела. В одном варианте осуществления противорвотное средство включает антагонист рецептора 5-НТ3 и/или антагонист рецептора нейрокинина 1 (NK1). Предпочтительно, антагонист рецептора NK1 включает апрепитант (например, эменд), и антагонист рецептора 5-НТ3 включает ондансетрон (например, зофран), гранисетрон (например, китрил, санкузо) или палоносетрон (например, алокси), или комбинацию двух или более из указанных.
В одном варианте осуществления антиспазматические средство включает бутилскополамин (бускопан).
В одном варианте осуществления агент, который защищает слизистую оболочку желудка, включает агент, который уменьшает выработку желудочной кислоты. В одном варианте осуществления агент, который защищает слизистую оболочку желудка, включает агент, выбранный из группы, состоящей из ингибиторов протонного насоса, мизопростола и омепразола. В одном варианте осуществления агент, который защищает слизистую оболочку желудка, включает комбинацию ингибитора протонного насоса и мизопростола. В одном варианте осуществления ингибитор протонного насоса включает в себя пантопразол (например, пантозол).
В одном варианте осуществления способ по изобретения включает введение пациенту антагониста рецептора NK1, такого как апрепитант (например, эменд), антагониста рецептора 5-НТ3, такого как ондансетрон (например, зофран), гранисетрон (например, китрил, санкузо) или палоносетрон (например, алокси), или комбинации двух или более из них, антиспазматического средства, такого как бутилскополамин (например, бускопан), и ингибитора протонного насоса, такого как пантопразол (например, пантозол).
В одном варианте осуществления способа по любому из указанных выше аспектов антитело вводят путем внутривенной (i.v.) инфузии. В одном варианте осуществления внутривенную инфузию проводят в течение периода времени, составляющего от 1 до 4 часов, предпочтительно около 2 часов.
В шестом аспекте в настоящем изобретении предлагается способ определения отвечаемости раковых пациентов на лечение или предупреждение ракового заболевания, включающий введение антитела, обладающего способностью связываться с CLDN18.2, при этом указанный способ включает стадию определения уровня одного или нескольких маркеров в крови у пациента, при этом один или несколько маркеров выбраны из группы, состоящей из СА 125, СА 15-3, СА 19-9, СЕА, IL-2, IL-15, IL-6, IFNγ и TNFα. В этом аспекте до и после введения антитела, обладающего способностью связываться с CLDN18.2, а именно после введения однократной дозы антитела, у пациента могут быть взяты биологические образцы, такие как кровь, для установления уровня одного или нескольких маркеров. Из одной и той же ткани может быть взято множество образцов для определения средних уровней и расчета возможных отклонений этих уровней. Уровень одного или нескольких маркеров после введения антитела сравнивают с уровнем, определенным до введения. Таким образом, эффект антитела на пациента может быть установлен по желательному изменению уровня маркера после введения антитела, обладающего способностью связываться с CLDN18.2. Если пациент демонстрирует желательное изменение уровня маркера после введения антитела, обладающего способностью связываться с CLDN18.2, то может быть инициировано лечение антителом, обладающим способностью связываться с CLDN18.2.
В одном варианте осуществления уровень определяют в крови, плазме или сыворотке.
В одном варианте осуществления один или несколько маркеров выбраны из группы, состоящей из СА 125, СА 15-3, СА 19-9, СЕА, IL-2, IL-15, IFNγ и TNFα, и снижение уровня, по меньшей мере, одного из маркеров после введения антитела указывает на то, что пациент отвечает на лечение или предупреждение ракового заболевания.
В одном варианте осуществления маркер представляет собой IL-6, и повышение уровня маркера после введения антитела указывает на то, что пациент отвечает на лечение или предупреждение ракового заболевания.
В восьмом аспекте в настоящем изобретении предлагается способ определения восприимчивости ракового пациента к лечению или предупреждению ракового заболевания, включающий введение антитела, обладающего способностью связываться с CLDN18.2, при этом указанный способ включает стадию определения процентного содержания CLDN18.2-положительных раковых клеток.
В этом варианте осуществления перед введением антитела, обладающего способностью связываться с CLDN18.2, у пациента может быть взят биологический образец, такой как образец опухоли (например, биопсия опухоли), для установления уровня CLDN18.2-положительных раковых клеток. Множество образцов может быть взято для определения среднего уровня и для расчета возможных отклонений этих уровней. В случае если пациент имеет желательный уровень CLDN18.2-положительных раковых клеток, может быть введено антитело, обладающее способностью связываться с CLDN18.2.
В одном варианте осуществления уровень, составляющий по меньшей мере 50%, предпочтительно 60%, 70%, 80% или 90%, по меньшей мере 95% или по меньшей мере 98% CLDN18.2-положительных раковых клеток, указывает на то, что пациент является восприимчивым к лечению или предупреждению ракового заболевания. В одном варианте осуществления уровень, составляющий по меньшей мере 40%, предпочтительно по меньшей мере 50%, по меньшей мере 60%, по меньшей мере 70%, по меньшей мере 80% или по меньшей мере 90% раковых клеток, которые являются положительными в отношении поверхностной экспрессии CLDN18.2, указывает на то, что пациент является восприимчивым к лечению или предупреждению ракового заболевания.
Антитело, обладающее способностью связываться с CLDN18.2, может связываться с нативными эпитопами CLDN18.2, присутствующими на поверхности живых клеток. В одном варианте осуществления антитело, обладающее способностью связываться с CLDN18.2, связывается с первой внеклеточной петлей CLDN18.2. В одном варианте осуществления антитело, обладающее способностью связываться с CLDN18.2, опосредует уничтожение клеток посредством одного или нескольких из следующих путей: лизиса, опосредованного зависимой от комплемента цитотоксичностью (CDC), лизиса, опосредованного зависимой от антител клеточной цитотоксичностью (ADCC), индукции апоптоза и ингибирования пролиферации. В одном варианте осуществления антитело, обладающее способностью связываться с CLDN18.2, представляет собой моноклональное, химерное или гуманизированное антитело, или фрагмент антитела. В одном варианте осуществления антитело, обладающее способностью связываться с CLDN18.2, представляет собой антитело, выбранное из группы, состоящей из (i) антитела, продуцированного и/или полученного из клона, депонированного под номером доступа DSM ACC2737, DSM ACC2738, DSM ACC2739, DSM ACC2740, DSM ACC2741, DSM ACC2742, DSM ACC2743, DSM ACC2745, DSM ACC2746, DSM ACC2747, DSM ACC2748, DSM ACC2808, DSM ACC2809 или DSM ACC2810; (ii) антитела, которое представляет собой химеризованную или гуманизированную форму антитела согласно (i); (iii) антитела, обладающего специфичностью антитела согласно (i); и (iv) антитела, содержащего участок, связывающий антиген, или антигенсвязывающий сайт, в частности, вариабельную область, антитела согласно (i) и предпочтительно обладающего специфичностью антитела согласно (i). В одном варианте осуществления антитело связывается с терапевтическим агентом, таким как токсин, радиоизотоп, лекарственное средство или цитотоксический агент.
В одном варианте осуществления рак является CLDN18.2-положительным. В одном варианте осуществления клетки рака экспрессируют CLDN18.2. В одном варианте осуществления экспрессия CLDN18.2 происходит на поверхности клеток. В одном варианте осуществления по меньшей мере 50%, предпочтительно 60%, 70%, 80% или 90% раковых клеток являются CLDN18.2-положительными, и/или по меньшей мере 40%, предпочтительно, по меньшей мере 50% раковых клеток являются положительными в отношении поверхностной экспрессии CLDN18.2. В одном варианте осуществления по меньшей мере 95% или по меньшей мере 98% раковых клеток являются CLDN18.2-положительными. В одном варианте осуществления по меньшей мере 60%, по меньшей мере 70%, по меньшей мере 80% или по меньшей мере 90% раковых клеток являются положительными в отношении поверхностной экспрессии CLDN18.2.
В одном варианте осуществления раковое заболевание выбрано из группы, состоящей из рака желудка, рака пищевода, рака поджелудочной железы, рака легкого, рака яичника, рака толстой кишки, рака печени, рака головы и шеи, рака желчного пузыря и их метастазов. Раковое заболевание может представлять собой опухоль Крукенберга, перитонеальные метастазы и/или метастазы в лимфоузлы. В одном варианте осуществления рак представляет собой аденокарциному, в частности, аденокарциному поздней стадии. В одном варианте осуществления рак выбран из группы, состоящей из рака желудка, рака пищевода, в частности, нижнего отдела пищевода, рака пищеводно-желудочного перехода и гастроэзофагеального рака. В особенно предпочтительном варианте осуществления рак представляет собой гастроэзофагеальный рак, такой как метастатический, рефракторный или рецидивирующий гастроэзофагеальный рак поздней стадии. Пациент может представлять собой HER2/neu-отрицательного пациента или пациента с HER2/neu-положительным статусом, но не подходящего для терапии транстузумабом. В одном варианте осуществления пациент получал ранее терапию, по меньшей мере, одним лекарственным средством, выбранным из группы, состоящей из аналогов пиримидина (например, фторурацил и/или капецитабин), соединений платины (например, цисплатин и/или оксалиплатин), эпирубицина, доцетаксела и детоксифицирующих агентов для противоопухолевого лечения (например, фолинат кальция и/или фолиниевая кислота). В одном варианте осуществления пациент имеет статус по шкале ECOG, равный от 0 до 1, и/или индекс Карновского от 70 до 100%. В особенно предпочтительном варианте осуществления пациент представляет собой пациента-человека.
В соответствии с изобретением CLDN18.2 предпочтительно имеет аминокислотную последовательность согласно SEQ ID NO: 1.
Настоящее изобретение также обеспечивает агенты, описанные здесь, такие как антитело, обладающее способностью связываться с CLDN18.2, для применения в описанных здесь способах.
Другие отличительные признаки и преимущества настоящего изобретения будут очевидны из следующего подробного описания и формулы изобретения.
Краткое описание чертежей
Фигура 1. Средняя концентрация в крови IMAB362 во время исследования.
Фигура 2. Активность ADCC мононуклеарных клеток периферической крови (РВМС) пациентов. (А) РВМС выделяли из образцов крови 6 пациентов через 7 дней (белый квадрат) или 14 дней (черные квадраты) после введения IMAB362. Скорости специфического лизиса клеток-мишеней рака желудка NUGC-4, экспрессирующих CLDN18.2, полученные после добавления 31,63 мкг/мл IMAB362 и РВМС от здорового донора или РВМС пациента (Е:Т=20:1), в течение 24 ч. (В) Зависимый от концентрации IMAB362 специфический лизис клеток NUGC-4, полученный через 24 ч после добавления РВМС различных пациентов (графики показывают средние значения ± стандартное отклонение, величину р рассчитывали с применением непарного t-критерия Стьюдента). (С) Кривые ADCC-ответа здоровых контрольных РВМС при добавлении возрастающих концентраций IMAB362. Анализы выполняли параллельно с каждым анализом ADCC с РВМС пациентов. (D) Кривая ADCC-ответа РВМС пациентов при добавлении возрастающих концентраций IMAB362 (для пациента 0202 недостаточно РВМС было получено для построения кривой). (Е) Полумаксимальные скорости уничтожения для всех пациентов и здоровых доноров рассчитывали с помощью программного обеспечения GraphPad Prism с использованием встроенного инструмента анализа нелинейной регрессии.
Фигура 3. Способность компонентов комплемента пациента индуцировать IMAB362-опосредованную CDC. Анализы CDC выполняли с CLDN18.2 и люцифераза-положительными клетками-мишенями СНО-K1. Клетки, сыворотку (20% об/об) и антитела инкубировали в течение 80 мин при 37°С. Образцы от пациентов готовили путем добавления свежеприготовленного IMAB362 при концентрации 0,5 мкг/мл в образцы сыворотки, взятые до инфузии (серые столбцы). HSC: Контрольный пул здоровой человеческой сыворотки с введенным IMAB362 при концентрации 0,3-10 мкг/мл (положительный контроль). Hi: Пул инактивированной путем нагревания человеческой сыворотки с введенным IMAB362 при концентрации 10 мкг/мл (отрицательный контроль). Номера пациентов указаны. Планка погрешностей: ± стандартное отклонение.
Фигура 4. Способность компонентов комплемента пациента взаимодействовать с введенным внутривенно (i.v.) IMAB362 во времени. Нормализованные CDC-анализы проводили путем доведения концентрации IMAB362 в каждом образце до 0,5 мкг/мл с использованием взятой до инфузии сыворотки каждого пациента (10-680-кратное разведение). (А/В) Анализы CDC выполняли, как описано для фигуры 3. (С) Каждая точка представляет измерение для одного пациента. Белый квадрат: 0,5 мкг/мл IMAB362 в человеческой сыворотке. Величины р получали с применением непарного t-критерия Стьюдента. Планка погрешностей: среднее значение ± стандартное отклонение.
Фигура 5. Кинетика цитотоксичности, индуцированной введенным внутривенно (i.v.) циркулирующим IMAB362. Клетки-мишени NUGC-4, РВМС одного здорового донора (Е:Т=40:1) и образцы сыворотки пациента (25% об/об) в качестве антитела и источника комплемента использовали в анализе общей цитотоксичности для измерения суммарной цитотоксической активности. Образцы сыворотки каждого пациента собирали через 1, 7, 14 и 28-32 дней после введения IMAB362. Пациентам вводили увеличивающиеся дозы IMAB362 (33-1000 мг/м2). Концентрация антитела, используемого в анализе, указана под каждым столбцом. HSC: Пул человеческой сыворотки с введенным свежеприготовленным IMAB362 при концентрации 200,0 мг/мл (EC80-100). PSC: Контрольная сыворотка пациента до инфузии с введенным свежеприготовленным IMAB362 при концентрации 200 мкг/мл. n.а.: данные отсутствуют.
Фигура 6. Кинетика ADCC-активности IMAB362 в инактивированной при нагревании сыворотке пациента. Анализ проводили, как описано для предыдущей фигуры, за исключением того, что в данном случае комплемент пациента был инактивирован при нагревании (56°С, 30 мин) для выделения ADCC-активности (черные и серые части столбцов) и для расчета дополнительных эффектов компонентов сыворотки (белые части столбцов).
Фигура 7. Активность CDC, индуцированная IMAB362, присутствующим в сыворотке пациента. Анализы CDC выполняли с CLDN18.2 и люцифераза-положительными клетками-мишенями СНО-K1. Их инкубировали в течение 80 мин с 20% (об/об) сыворотки пациента, полученной через 1, 7, 14 и 28-32 дня после инфузии антитела. Пациентам вводили IMAB362 при дозе от 33 до 1000 мг/м2. Концентрация антитела, используемого в каждом анализе, указана под каждым столбцом. HSC: Контрольный пул здоровой человеческой сыворотки с введенными понижающимися концентрациями IMAB362, как указано. PC: положительный контроль (сыворотка пациента до инфузии с введенным IMAB362 при концентрации 10 мкг/мл).
Фигура 8: Результаты фармакокинетического исследования многократных инфузий IMAB362 у пациентов. Средняя ± стандартное отклонение концентрация (мкг/мл) IMAB362 в сыворотке 4 пациентов, которым вводили многократные дозы, составляющие 300 мг/м2 (когорта 1, левая фигура) и до 30 пациентов (30 пациентов первая инфузия, 12 пациентов пятая инфузия), которым вводили многократные дозы, составляющие 600 мг/м2 (когорта 2 и когорта 3 вместе, правая фигура). Стрелки показывают инфузии IMAB362. Первую инфузию проводили на день 0.
Фигура 9. Выживаемость без прогрессирования заболевания пациентов в полной выборке пациентов для анализа (FAS).
Фигура 10. Выживаемость без прогрессирования заболевания пациентов в выборке пациентов, выполнивших требования протокола (РР) (n=20).
Осуществление изобретения
Хотя настоящее изобретение описано подробно далее, следует понимать, что данное изобретение не ограничивается конкретными методиками, протоколами и реагентами, описанными здесь, поскольку они могут варьироваться. Также следует понимать, что используемая здесь терминология необходима только с целью описания конкретных вариантов осуществления изобретения и не предполагает ограничивать объем настоящего изобретения, который будет ограничен только прилагаемой формулой изобретения. Если не определено иначе, все используемые здесь технические и научные термины имеют те же самые значения, которые обычно понимаются специалистами в данной области.
Далее описаны элементы настоящего изобретения. Данные элементы перечислены с определенными вариантами осуществления, однако следует понимать, что они могут быть комбинированы любым способом и в любом количестве для создания дополнительных вариантов осуществления. По-разному описанные примеры и предпочтительные варианты осуществления не должны рассматриваться как ограничивающие настоящее изобретение только конкретно описанными вариантами осуществления. Следует понимать, что данное описание поддерживает и охватывает варианты осуществления, которые объединяют конкретно описанные варианты осуществления с любым количеством раскрытых и/или предпочтительных элементов. Кроме того, любые перестановки и комбинации всех описанных элементов в данной заявке должны считаться раскрытыми описанием настоящей заявки, если контекст не указывает иначе.
Предпочтительно, используемым здесь терминам дают определения, как описано в руководстве "A multilingual glossary of biotechnological terms: (IUPAC Recommendations)", H.G.W. Leuenberger, B. Nagel, and H.  , Eds., Helvetica Chimica Acta, CH-4010 Basel, Switzerland, (1995).
, Eds., Helvetica Chimica Acta, CH-4010 Basel, Switzerland, (1995).
Если не указано иное, при осуществлении настоящего изобретения на практике будут использованы общепринятые методы, применяемые в области химии, биохимии, цитологии, иммунологии и технологии рекомбинантных ДНК, которые описаны в литературе в данной области (смотри, например, Molecular Cloning: A Laboratory Manual, 2nd Edition, J. Sambrook et al. eds., Cold Spring Harbor Laboratory Press, Cold Spring Harbor 1989).
Во всем данном описании и в приведенной ниже формуле изобретения, если контекст не предполагает иное, слово «включать» и варианты, такие как «включает» и «включающий», необходимо понимать как включение указанного элемента, целого числа или стадии, или группы элементов, целых чисел или стадий, но не как исключение какого-либо другого элемента, целого числа или стадии, или группы элементов, целых чисел или стадий, хотя в некоторых вариантах осуществления такой другой элемент, целое число или стадия, или группа элементов, целых чисел или стадий могут быть исключены, то есть объект изобретения состоит во включении указанного элемента, целого числа или стадии, или группы элементов, целых чисел или стадий. Термины в единственном числе (особенно в контексте формулы изобретения) необходимо интерпретировать как охватывающие как единственное, так и множественное число, если в настоящем документе не указано иное или явно не противоречит контексту. Перечисление диапазонов величин в данном описании используется всего лишь как способ сокращенного перечисления всех конкретных величин, попадающих в указанный диапазон. Если в настоящем документе не указано иное, каждая отдельная величина включена в описание, как если бы она была индивидуально цитирована в настоящем документе. Все способы, описанные здесь, могут быть выполнены в любой подходящей последовательности, если в настоящем документе не указано иное, или иное явно не противоречит контексту. Использование любого или всех примеров, или выражений, указывающих на примеры (например, «такой как»), представленных здесь, предназначено только для лучшего описания изобретения и не предполагает ограничения объема изобретения, если не заявлено иное. Никакие выражения в описании не должны рассматриваться как указания на какой-либо элемент, не указанный в формуле изобретения, существенный для осуществления изобретения на практике.
Некоторые документы процитированы по всему тексту данного описания. Каждый из документов, процитированных здесь (включая все патенты, патентные заявки, научные публикации, спецификации производителя, инструкции и т.п.), или выше, или ниже, тем самым включен здесь путем отсылки во всей полноте. Ничто здесь не должно толковаться как допущение того, что изобретение не обладает правом предшествовать такому раскрытию на основании предшествующего изобретения.
Термин «CLDN18» относится к клаудину-18 и включает любые варианты, в том числе сплайсированный вариант 1 клаудина-18 (клаудин 18.1 (CLDN18.1)) и сплайсированный вариант 2 клаудина-18 (клаудин 18.2 (CLDN18.2)).
Термин «CLDN18.2» предпочтительно относится к CLDN18.2 человека и, в частности, белку, содержащему, предпочтительно состоящему из аминокислотной последовательности согласно SEQ ID NO: 1 перечня последовательностей или варианта указанной аминокислотной последовательности.
Термин «CLDN18.1» предпочтительно относится к CLDN18.1 человека и, в частности, белку, содержащему, предпочтительно состоящему из аминокислотной последовательности согласно SEQ ID NO: 2 перечня последовательностей или варианта указанной аминокислотной последовательности.
Термин «вариант» в соответствии с изобретением относится, в частности, к мутантам, сплайсированным вариантам, конформациям, изоформам, аллельным вариантам, видовым вариантам и видовым гомологам, в частности таким, которые присутствуют естественным образом. Аллельный вариант относится к изменению в нормальной последовательности гена, значение которого часто остается неясным. Полное секвенирование гена часто выявляет многочисленные аллельные варианты для заданного гена. Видовой гомолог представляет собой последовательность нуклеиновой кислоты или аминокислотную последовательность с происхождением видов, отличным от видов заданной последовательности нуклеиновой кислоты или аминокислотной последовательности. Термин «вариант» охватывает любые посттрансляционно модифицированные варианты и конформационные варианты.
В соответствии с изобретением термин «CLDN18.2-положительный рак» означает рак, включающий раковые клетки, экспрессирующие CLDN18.2, предпочтительно на поверхности указанных раковых клеток.
Термин «клеточная поверхность» используется в соответствии с его обычным значением в данной области и, таким образом, включает внешнюю поверхность клетки, которая доступна для связывания белками и другими молекулами.
CLDN18.2 экспрессируется на поверхности клеток, если он расположен на поверхности указанных клеток и доступен для связывания CLDN18.2-специфическими антителами, добавленными в клетки.
В соответствии с изобретением CLDN18.2 значительно не экспрессируется в клетке, если уровень экспрессии является более низким по сравнению с экспрессией в клетках желудка или ткани желудка. Предпочтительно, уровень экспрессии составляет менее 10%, предпочтительно менее 5%, 3%, 2%, 1%, 0,5%, 0,1% или 0,05% экспрессии в клетках желудка или ткани желудка, или даже ниже. Предпочтительно, CLDN18.2 значительно не экспрессируется в клетке, если уровень экспрессии превышает уровень экспрессии в нераковой ткани, отличной от желудка, более чем в 2 раза, предпочтительно 1,5 раза, и предпочтительно не превышает уровень экспрессии в указанной нераковой ткани. Предпочтительно, CLDN18.2 значительно не экспрессируется в клетке, если уровень экспрессии ниже предела обнаружения и/или если уровень экспрессии слишком низкий для обеспечения связывания CLDN18.2-специфическими антителами, добавленными в клетки.
В соответствии с изобретением CLDN18.2 экспрессируется в клетке, если уровень экспрессии превышает уровень экспрессии в нераковой ткани, отличной от желудка, предпочтительно более чем в 2 раза, предпочтительно в 10 раз, 100 раз, 1000 раз или 10000 раз. Предпочтительно, CLDN18.2 экспрессируется в клетке, если уровень экспрессии выше предела обнаружения и/или если уровень экспрессии является достаточно высоким для обеспечения связывания CLDN18.2-специфическими антителами, добавленными в клетки. Предпочтительно, CLDN18.2, экспрессирующийся в клетке, экспрессируется или располагается на поверхности указанной клетки.
В соответствии с изобретением термин «заболевание» относится к любому патологическому состоянию, включая рак, в частности, тем формам рака, которые описаны в настоящем документе. Любая ссылка в настоящем документе на рак или конкретные формы рака также включает метастазы рака. В предпочтительном варианте осуществления заболевание, подлежащее лечению в соответствии с настоящей заявкой, включает клетки, экспрессирующие CLDN18.2.
«Заболевания, связанные с клетками, экспрессирующими CLDN18.2» или аналогичные выражения в соответствии с изобретением означают, что CLDN18.2 экспрессируется в клетках больной ткани или органа. В одном варианте осуществления экспрессия CLDN18.2 в клетках больной ткани или органа является повышенной по сравнению с состоянием в здоровой ткани или органе. Повышение относится к повышению по меньшей мере на 10%, в частности, по меньшей мере на 20%, по меньшей мере на 50%, по меньшей мере на 100%, по меньшей мере на 200%, по меньшей мере на 500%, по меньшей мере на 1000%, по меньшей мере на 10000%) или даже выше. В одном варианте осуществления экспрессия обнаруживается только в больной ткани, тогда как экспрессия в здоровой ткани является подавленной. В соответствии с изобретением заболевания, связанные с клетками, экспрессирующими CLDN18.2, включают раковые заболевания. Кроме того, в соответствии с изобретением раковые заболевания предпочтительно представляют собой такие заболевания, при которых раковые клетки экспрессируют CLDN18.2.
Используемый здесь термин «раковое заболевание» или «рак» включает заболевание, характеризующееся аномально регулируемым клеточным ростом, пролиферацией, дифференциацией, адгезией и/или миграцией. Три злокачественных свойства рака (неконтролируемый рост (деление выше нормы), инвазия (проникновение и деструкция прилегающих тканей) и иногда метастазирование (распространение в другие локализации в теле через лимфу или кровь)) дифференцируют рак от доброкачественных опухолей, которые являются самокупирующимися и не инвазируют или метастазируют.
Большинство видов рака формируют опухоли, но некоторые, такие как лейкоз, не формируют. Под «раковой клеткой» понимается аномальная клетка, которая растет путем быстрой неконтролируемой клеточной пролиферации и продолжает расти после прекращения действия стимула, который инициировал рост новообразования. Предпочтительно, «раковое заболевание» характеризуется клетками, экспрессирующими CLDN18.2, и раковая клетка экспрессирует CLDN18.2. Клетка, экспрессирующая CLDN18.2, предпочтительно представляет собой раковую клетку, предпочтительно описанных здесь видов рака.
В соответствии с изобретением термин «опухоль» или «опухолевое заболевание» относится к аномальному росту клеток (называемых неопластическими клетками, опухолегенными клетками или опухолевыми клетками), предпочтительно формирующих набухание или поражение. Под «опухолевой клеткой» понимается аномальная клетка, которая растет путем быстрой неконтролируемой клеточной пролиферации и продолжает расти после прекращения действия стимула, который инициировал рост новообразования. Опухоли демонстрируют частичное или полное отсутствие структурной организации и функционального взаимодействия с нормальной тканью и обычно образуют отчетливо выраженную массу ткани, которая может быть доброкачественной, предраковой или злокачественной.
В соответствии с изобретением опухоль предпочтительно представляет собой злокачественную опухоль. «Злокачественная опухоль» используется в качестве синонима рака.
«Аденокарцинома» представляет собой рак, который возникает из железистой ткани. Эта ткань также является частью более крупной категории ткани, известной как эпителиальная ткань. Эпителиальная ткань включает кожу, железы и ряд других тканей, которые выстилают полости и органы тела. Эмбриологически эпителий происходит из эктодермы, эндодермы и мезодермы. Для классификации в качестве аденокарциномы клетки необязательно должны являться частью железы, поскольку они обладают секреторными свойствами. Эта форма карциномы может возникать у некоторых высших млекопитающих, включая людей. В отличие от слабо дифференцированной, хорошо дифференцированная аденокарцинома напоминает железистую ткань, из которой она происходит. Путем окрашивания клеток, полученных путем биопсии, патолог определяет, является ли опухоль аденокарциномой или каким-либо другим типом рака. Аденокарцинома может возникать во многих тканях тела вследствие убиквитарной природы желез в организме. Несмотря на то, что каждая железа может не выделять одно и то же вещество, поскольку существует экзокринная функция клетки, она считается железистой и ее злокачественная форма, таким образом, называется аденокарциномой. Злокачественные аденокарциномы инвазируют многие ткани и часто метастазируют при условии наличия для этого достаточного времени. Аденокарцинома яичника является наиболее распространенным типом карциномы яичника. Она включает серозную и муцинозную аденокарциномы, светлоклеточную аденокациному и эндометриоидную аденокарциному.
Под «метастазированием» понимается распространение раковых клеток из исходного участка в другую часть тела. Образование метастазов представляет собой очень сложный процесс, который зависит от отрыва злокачественных клеток от первичной опухоли, инвазии внеклеточного матрикса, проницаемости эндотелиальных базальных мембран для проникновения в полость тела и сосуды, и затем, после переноса кровью, инфильтрации органов-мишеней. В конечном итоге, рост новой опухоли в участке-мишени зависит от ангиогенеза. Метастазирование опухоли часто происходит даже после удаления первичной опухоли, так как опухолевые клетки или компоненты могут оставаться и проявлять метастатический потенциал. В одном варианте осуществления термин «метастазирование» в соответствии с изобретением относится к «отдаленному метастазированию», которое имеет отношение к метастазированию, которое происходит в участках, отдаленных от первичной опухоли и системы региональных лимфатических узлов. В одном варианте осуществления термин «метастазирование» в соответствии с изобретением относится к метастазам в лимфоузлы. Одной конкретной формой метастазов, которая поддается лечению с использованием терапии по изобретению, являются метастазы, происходящие из рака желудка как первичного участка. В предпочтительных вариантах осуществления такие метастазы рака желудка называются опухолями Крукенберга, перитонеальными метастазами и/или метастазами в лимфоузлы.
Опухоль Крукенберга представляет собой редко встречающуюся метастатическую опухоль яичника, на долю которой приходится от 1% до 2% всех опухолей яичника. Прогноз опухоли Крукенберга все еще очень слабый, и одобренная терапии для опухолей Крукенберга отсутствует. Опухоль Крукенберга представляет собой метастатическую перстневидноклеточную аденокарциному яичника. Желудок является первичным участком в большинстве случаев опухоли Крукенберга (70%). Карцинома толстой кишки, аппендицита и молочной железы (в основном инвазивная лобулярная карцинома) являются следующим наиболее распространенными первичными очагами. Сообщалось о редких случаях опухоли Крукенберга, происходящей из карциномы желчного пузыря, желчных протоков, поджелудочной железы, тонкого кишечника, фатерова соска, шейки матки и мочевого пузыря/мочевого протока.
Женщины с опухолью Крукенберга, как правило, крайне молоды для пациентов с метастатической карциномой, так как обычно находятся в возрасте после 40 лет, в среднем в возрасте 45 лет. Этот молодой возраст распространения может быть отчасти связан с увеличенной частотой случаев перстневидноклеточных аденокарцином у молодых женщин. Общие проявляющиеся симптомы обычно относятся к вовлечению яичника, наиболее общими из которых являются боль в животе и вздутие живота (главным образом из-за обычно билатеральных и часто больших масс яичника). Остальные пациенты имеют неспецифические желудочно-кишечные симптомы или являются бессимптомными. Кроме того, опухоль Крукенберга по имеющимся данным связана с вирилизацией, возникающей в результате выработки гормонов стромой яичника. Асцит присутствует в 50% случаев и обычно выявляет злокачественные клетки.
Опухоли Крукенберга являются билатеральными в более чем 80% сообщенных случаях. Яичники обычно асимметрично увеличены с бугристым контуром. Рассеченные поверхности желтые или белые; они обычно твердые, хотя иногда кистозные. Важно, что капсулярная поверхность яичников с опухолями Крукенберга обычно гладкая и не имеет спаек или перитонеальных отложений. Следует отметить, что другие метастатические опухоли яичника имеют тенденцию быть связанными с поверхностными имплантами. Это может объяснять, почему макроскопическая морфология опухоли Крукенберга может обманчиво возникать в качестве первичной опухоли яичника. Однако билатеризм опухоли Крукенберга согласуется с ее метастатической природой.
Пациенты с опухолями Крукенберга имеют общий показатель смертности, который является значительно высоким. Большинство пациентов умирает в течение 2 лет (средняя выживаемость 14 месяцев). Некоторые исследования показывают, что прогноз является слабым, когда первичная опухоль идентифицирована после того, как выявлены метастазы в яичник, и прогноз становится хуже, если первичная опухоль остается скрытой.
Под термином «лечить» понимается введение соединения или композиции, или комбинации соединений или композиций субъекту для предупреждения или устранения заболевания, включая уменьшение размера опухоли или ряда опухолей у субъекта; купирование или замедление возникновения заболевания у субъекта; ингибирование или замедление развития нового заболевания у субъекта; уменьшение частоты или тяжести симптомов и/или повторных проявлений у субъекта, который в настоящий момент имеет, или который ранее имел заболевание; и/или продление, то есть увеличение продолжительности жизни субъекта.
В частности, термин «лечение заболевания» включает выздоравливание, сокращение продолжительности, уменьшение интенсивности, предупреждение, замедление или подавление прогрессирования или ухудшения, или предупреждение и отсрочку начала развития заболевания или его симптомов.
Термин «пациент» означает в соответствии с изобретением субъекта, подлежащего лечению, в частности субъекта, имеющего заболевание, в том числе человека, приматов нечеловеческого происхождения или других животных, в частности млекопитающих, таких как коровы, лошади, свиньи, овцы, козы, собаки, кошки или грызуны, такие как мыши и крысы. В особенно предпочтительном варианте осуществления пациентом является человек.
В соответствии с изобретением антитело, обладающее способностью связываться с CLDN18.2, можно вводить в сочетании, то есть одновременно, последовательно и/или после агента, стабилизирующего или повышающего экспрессию CLDN18.2.
Выражение «агент, стабилизирующий или повышающий экспрессию CLDN18.2» относится к агенту или комбинации агентов, поступление которых в клетки приводит к повышенной экспрессии CLDN18.2 на уровне РНК и/или белка, предпочтительно повышенной экспрессии белка CLDN18.2 на клеточной поверхности, по сравнению с ситуацией, когда клетки не обеспечены агентом или комбинацией агентов. Предпочтительно, клетка представляет собой раковую клетку, в частности, раковую клетку, экспрессирующую CLDN18.2, такую как клетка описанных здесь типов рака. Выражение «агент, стабилизирующий или повышающий экспрессию CLDN18.2» относится, в частности, к агенту или комбинации агентов, поступление которых в клетки приводит к более высокой плотности CLDN18.2 на поверхности указанных клеток по сравнению с ситуацией, когда клетки не обеспечены агентом или комбинацией агентов. «Стабилизация экспрессии CLDN18.2» включает, в частности, ситуацию, когда агент или комбинация агентов предотвращает снижение или уменьшает снижение экспрессии CLDN18.2, например, экспрессия CLDN18.2 будет снижаться без поступления агента или комбинации агентов, и поступление агента или комбинации агентов предотвращает указанное снижение или уменьшает указанное снижение экспрессии CLDN18.2. «Повышение экспрессии CLDN18.2» включает, в частности, ситуацию, когда агент или комбинация агентов повышает экспрессию CLDN18.2, например, экспрессия CLDN18.2 будет снижаться, оставаться в основном постоянной или повышаться без поступления агента или комбинации агентов, и поступление агента и комбинации агентов повышает экспрессию CLDN18.2 по сравнению с ситуацией без поступления агента или комбинации агентов, таким образом, полученная в результате экспрессия является более высокой по сравнению с ситуацией, когда экспрессия CLDN18.2 будет снижаться, оставаться в основном постоянной или повышаться без поступления агента или комбинации агентов.
В соответствии с изобретением выражение «агент, стабилизирующий или повышающий экспрессию CLDN18.2» включает химиотерапевтические агенты или комбинации химиотерапевтических агентов, таких как цитостатические агенты. Химиотерапевтические агенты могут воздействовать на клетки одним из следующих способов: (1) повреждать ДНК клеток таким образом, что они не могут больше репродуцироваться; (2) ингибировать синтез новых нитей ДНК таким образом, что репликация клеток невозможна; (3) останавливать митотические процессы клеток таким образом, что клетки не могут делиться на две клетки.
В соответствии с изобретением выражение «агент, стабилизирующий или повышающий экспрессию CLDN18.2» предпочтительно относится к агенту или комбинации агентов, таких как цитостатическое соединение или комбинация цитостатических соединений, поступление которых в клетки, в частности раковые клетки, приводит к получению клеток, которые блокированы или накапливаются в одной или нескольких фазах клеточного цикла, предпочтительно в одной или нескольких фазах клеточного цикла, отличных от фаз G1 и G0, предпочтительно отличных от фазы G1, предпочтительно в одной или нескольких из фаз G2 или S клеточного цикла, таких как G1/G2-, S/G2-, G2- или S-фаза клеточного цикла. Выражение «клетки, которые блокированы или накапливаются в одной или нескольких фазах клеточного цикла» означает, что процентное содержание клеток, которые находятся в указанной одной или нескольких фазах клеточного цикла, увеличивается. Каждая клетка для саморепликации проходит через цикл, включающий четыре фазы. В первой фазе, называемой G1, клетка готовится к репликации своих хромосом. Вторая стадия называется S, и в этой фазе происходит синтез ДНК и ДНК дуплицируется. Следующая фаза называется фазой G2, когда РНК и белок дуплицируются. Конечной стадией является стадия М, которая представляет собой стадию фактического клеточного деления. В этой конечной стадии дуплицированные ДНК и РНК расщепляются и расходятся к разным концам клетки, и клетка фактически делится на две идентичные функциональные клетки. Химиотерапевтические агенты, которые представляют собой агенты, повреждающие ДНК, обычно приводят к накоплению клеток в G1- и/или С2-фазе. Химиотерапевтические агенты, которые блокируют клеточный рост путем препятствования синтезу ДНК, такие как антиметаболиты, обычно приводят к накоплению клеток в S-фазе. Примерами этих лекарственных средств являются 6-меркаптопурин и 5-фторурацил.
В соответствии с изобретением выражение «агент, стабилизирующий или повышающий экспрессию CLDN18.2» включает антрациклины, такие как эпирубицин, соединения платины, такие как оксаплатин и цисплатин, нуклеозидные аналоги, такие как 5-фторурацил, или их пролекарства, таксаны, такие как доцетаксел, и аналоги камптотецина, такие как иринотекан и топотекан, а также комбинации лекарственных средств, например, комбинации лекарственных средств, включающие один или несколько антрациклинов, таких как эпирубицин, оксалиплатин и 5-фторурацил, например, комбинацию лекарственных средств, включающую оксалиплатин и 5-фторурацил, или другие комбинации лекарственных средств, описанные здесь.
В одном предпочтительном варианте осуществления «агент, стабилизирующий или повышающий экспрессию CLDN18.2» представляет собой «агент, индуцирующий иммуногенную гибель клеток».
В специфических случаях раковые клетки могут входить на путь летального стресса, связанный с испусканием комбинации сигналов, определенных в пространстве и во времени, который декодирован иммунной системой на активацию опухоль-специфических иммунных ответов (Zitvogel L. et al. (2010) Cell 140: 798-804). В таком сценарии раковые клетки начинают испускать сигналы, которые воспринимаются эффекторами врожденного иммунитета, такими как дендритные клетки, которые вызывают когнатный иммунный ответ, в который вовлечены CD8+ Т-клетки и передача сигнала IFN-γ, таким образом, что гибель раковых клеток может вызвать благоприятный противоопухолевый иммунный ответ. Эти сигналы включают преапоптотическую экспозицию кальретикулина (CRT), шаперона эндоплазматического ретикулума (ER), на поверхности клетки, преапоптотическую секрецию АТР и постапоптотическое высвобождение ядерного белка HMGB1. Взятые вместе, эти процессы составляют молекулярные детерминанты иммуногенной гибели клеток (ICD). Антрациклины, оксалиплатин и гамма-облучение способны индуцировать все сигналы, которые определяют ICD, тогда как, например, цисплатин, который лишен способности индуцировать транслокацию CRT из ER на поверхность погибающих клеток - процесс, требующий стресса ER - требует дополнения тапсигаргином, индуктором ER стресса.
В соответствии с изобретением выражение «агент, индуцирующий иммуногенную гибель клеток» относится к агенту или комбинации агентов, которые в случае поступления в клетки, в частности раковые клетки, способны индуцировать вхождение клеток на путь летального стресса, который в конечном итоге вызывает опухоль-специфические иммунные ответы. В частности, агент, индуцирующий иммуногенную гибель клеток, при поступлении в клетки индуцирует испускание клетками определенной в пространстве и во времени комбинации сигналов, включая, в частности, преапоптотическую экспозицию кальретикулина (CRT), шаперона эндоплазматического ретикулума (ER), на клеточной поверхности, преапоптотическую секрецию АТР и постапоптотическое высвобождение ядерного белка HMGB1.
В соответствии с данным изобретением выражение «агент, индуцирующий иммуногенную гибель клеток» включает антрациклины и оксалиплатин.
Антрациклины представляют собой класс лекарственных средств, широко применяющихся в химиотерапии рака, которые к тому же являются антибиотиками. По структуре все антрациклины имеют общую структуру 7,8,9,10-тетрагидротетрацен-5,12-хинона, состоящую из четырех колец, и обычно требуют гликозилирования в определенных участках.
Антрациклины предпочтительно действуют посредством примерно одного или нескольких из следующих механизмов: 1. Ингибирование синтеза ДНК и РНК путем интеркаляции между парами оснований нити ДНК/РНК, предотвращая, таким образом, репликацию быстро растущих раковых клеток, 2. Ингибирование фермента топоизомеразы II, предотвращая, таким образом, релаксацию сверхспиральной ДНК и, таким образом, блокируя транскрипцию и репликацию ДНК. 3. Создание опосредованных железом свободных радикалов кислорода, которые повреждают ДНК и клеточные мембраны.
В соответствии с изобретением термин «антрациклин» предпочтительно относится к агенту, предпочтительно противоопухолевому агенту, предназначенному для индукции апоптоза, предпочтительно путем ингибирования повторного лигирования ДНК топоизомеразой II.
Предпочтительно, в соответствии с изобретением термин «антрациклин», как правило, относится к классу соединений, имеющих следующую кольцевую структуру:
 ,
,
включая его аналоги и производные, фармацевтические соли, гидраты, сложные эфиры, конъюгаты и пролекарства.
Примеры антрациклинов и аналогов антрациклинов включают, но без ограничения, даунорубицин (дауномицин), доксорубицин (адриамицин), эпирубицин, идарубицин, родомицин, пирарубицин, валрубицин, N-трифтор-ацетил доксорубицин-14-валерат, аклациномицин, морфолинодоксорубицин (морфолино-DOX), цианоморфолино-доксорубицин (цианоморфолино-DOX, 2-пирролино-доксорубицин (2-PDOX), 5-иминодаунорубицин, митоксантрон и аклациномицин А (акларубицин). Митоксантрон является членом класса антрацендионовых соединений, являющихся аналогами антрациклинов, которые лишены сахарного звена антрациклинов, но сохраняют планарную полициклическую ароматическую кольцевую структуру, которая обеспечивает интеркаляцию в ДНК.
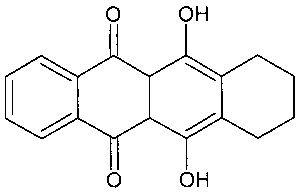
Особенно предпочтительным в качестве антрациклина в соответствии с изобретением является соединение следующей формулы:
в которой:
R1 выбран из группы, состоящей из Н и ОН, R2 выбран из группы, состоящей из Н и ОМе, R3 выбран из группы, состоящей из Н и ОН, и R4 выбран из группы, состоящей из Н и ОН.
В одном варианте осуществления R1 представляет собой Н, R2 представляет собой ОМе, R3 представляет собой Н и R4 представляет собой ОН. В другом варианте осуществления R1 представляет собой ОН, R2 представляет собой ОМе, R3 представляет собой Н, и R4 представляет собой ОН. В другом варианте осуществления R1 представляет собой ОН, R2 представляет собой ОМе, R3 представляет собой ОН и R4 представляет собой Н. В другом варианте осуществления R1 представляет собой Н, R2 представляет собой Н, R3 представляет собой Н и R4 представляет собой ОН.
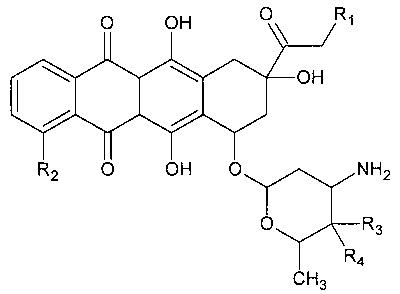
В качестве антрациклина в контексте настоящего изобретения особо рассматривается эпирубицин. Эпирубицин представляет собой антрациклиновое лекарственное средство, которое имеет следующую формулу:
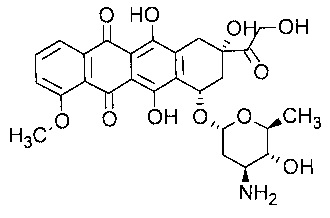
и выпускается под торговым названием Ellence в США и Pharmorubicin или Epirubicin Ebewe в других странах. В частности, термин «эпирубицин» относится к соединению (8R,10S)-10-[(2S,4S,5R,6S)-4-амино-5-гидрокси-6-метил-оксан-2-ил]окси-6,11-дигидрокси-8-(2-гидроксиацетил)-1-метокси-8-метил-9,10-дигидро-7Н-тетрацен-5,12-дион. В некоторых режимах химиотерапии эпирубицин предпочтительнее доксорубицина, наиболее популярного антрациклина, поскольку, как оказалось, вызывает меньше побочных эффектов.
В соответствии с изобретением термин «соединение платины» относится к соединениям, содержащим в своей структуре платину, такую как комплексы платины, и включает соединения, такие как цисплатин, карбоплатин и оксалиплатин.
Термин «цисплатин» или «двухвалентная платина» относится к соединению цис-диамминдихлороплатины(II) (CDDP) следующей формулы:
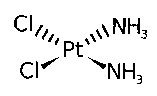 ,
,
Термин «карбоплатин» относится к соединению цис-диаммин(1,1-циклобутандикарбоксилато)платины(II) следующей формулы:
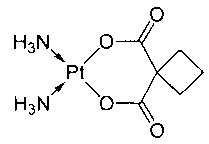 ,
,
Термин «оксалиплатин» относится к соединению, которое представляет собой соединение платины, образующее комплекс с лигандом носителя на основе диаминоциклогексана следующей формулы:
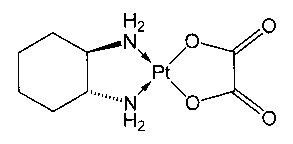
В частности, термин «оксалиплатин» относится к соединению [(1R,2R)-циклогексан-1,2-диамин](этандиоато-O,O')платина(II). Оксалиплатин, предназначенный для инъекции, также выпускается под торговым названием Eloxatine (элоксатин).
Термин «нуклеозидный аналог» относится к структурному аналогу нуклеозида, категории, которая включает аналоги пурина и аналоги пиримидина. В частности, термин «нуклеозидный аналог» относится к фторпиримидиновым производным, которые включают фторурацил и его пролекарства.
Термин «фторурацил» или «5-фторурацил» (5-FU или f5U) (присутствующие на рынке под торговыми названиями Adrucil (адруцил), Carac (карак), Efudix (Эфудикс), Efudex (эфудекс) и Fluoroplex (фтороплекс) означает соединение, которое является аналогом пиримидина следующей формулы:

В частности, термин относится к соединению 5-фтор-1Н-пиримидин-2,4-дион.
Термин «капецитабин» (Xeloda, Roche) относится к химиотерапевтическому агенту, который является про лекарством, которое превращается в 5-FU в тканях. Капецитабин, который может быть введен перорально, имеет следующую формулу:
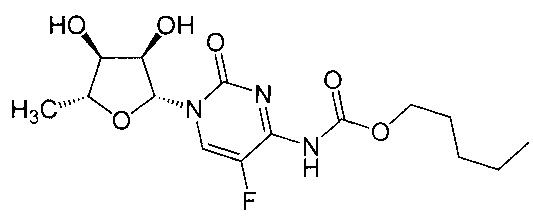
В частности, термин относится к соединению пентил [1-(3,4-дигидрокси-5-метилтетрагидрофуран-2-ил)-5-фтор-2-оксо-1Н-пиримидин-4-ил]карбамат.
Таксаны представляют собой класс дитерпеновых соединений, которые первоначально были выделены из природных источников, таких как растения рода Тис (Taxus), но некоторые были синтезированы искусственно. Основным механизмом действия лекарственных средств класса таксанов является нарушение функции микротрубочек, ингибируя, таким образом, процесс деления клеток. Таксаны включают доцетаксел (таксотер) и паклитаксел (таксол).
В соответствии с изобретением термин «доцетаксел» относится к соединению, имеющему следующую формулу:
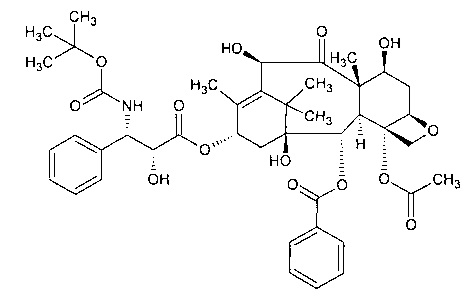
В соответствии с изобретение термин «паклитаксел» относится к соединению, имеющему следующую формулу:
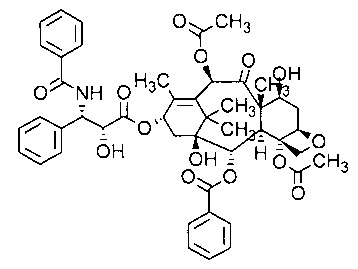
В соответствии с изобретением термин «аналог камптотецина» относится к производным соединения камптотецина (СРТ; (S)-4-этил-4-гидрокси-1Н-пирано[3',4':6,7]индолизино[1,2-b]хинолин-3,14-(4Н,12Н)-дион). Предпочтительно, термин «аналог камптотецина» относится к соединениям, имеющим следующую структуру:
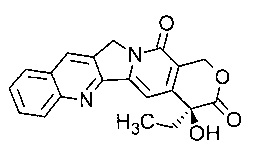
В соответствии с изобретением предпочтительными аналогами камптотецина являются ингибиторы фермента ДНК-топоизомеразы (топо I). Предпочтительными аналогами камптотецина в соответствии с изобретением являются иринотекан и топотекан.
Иринотекан представляет собой лекарственное средство, которое предотвращает ДНК от раскручивания путем ингибирования топоизомеразы I. В химических терминах иринотекан представляет собой полусинтетический аналог природного алкалоида камптотецина, имеющий следующую формулу:

В частности, термин «иринотекан» относится к соединению (S)-4,11-диэтил-3,4,12,14-тетрагидро-4-гидрокси-3,14-диоксо-1Н-пирано[3',4':6,7]-индолизино[1,2-b]хинолин-9-ил-[1,бипиперидин]-1'-карбоксилату.
Топотекан представляет собой ингибитор топоизомеразы следующей формулы:
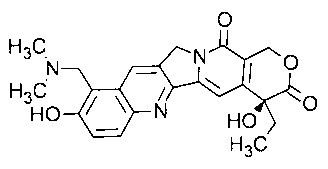
В частности, термин «топотекан» относится к соединению (S)-10-[(диметиламино)метил]-4-этил-4,9-дигидрокси-1Н-пирано[3',4':6,7]индолизино[1,2-b]хинолин-3,14(4Н,12Н)-диона моногидрохлориду.
В соответствии с изобретением агентом, стабилизирующим или повышающим экспрессию CLDN18.2, может являться химиотерапевтический агент, в частности, химиотерапевтический агент, одобренный для терапии рака, и может являться частью комбинации лекарственных средств, такой как комбинация лекарственных средств, одобренная для применения в терапии рака. Такая комбинация лекарственных средств может представлять собой комбинацию лекарственных средств, применяющуюся в химиотерапии, и может представлять собой комбинацию лекарственных средств, применяющуюся в химиотерапевтическом режиме, выбранном из группы, состоящей из химиотерапии ЕОХ, химиотерапии ECF, химиотерапии ЕСХ, химиотерапии EOF, химиотерапии FLO, химиотерапии FOLFOX, химиотерапии FOLFIRI, химиотерапии DCF и химиотерапии FLOT.
Комбинация лекарственных средств, используемая в химиотерапии ЕОХ, включает эпирубицин, оксалиплатин и капецитабин. Комбинация лекарственных средств, используемая в химиотерапии ECF, включает эпирубицин, цисплатин и 5-фторурацил. Комбинация лекарственных средств, используемая в химиотерапии ЕСХ, включает эпирубицин, цисплатин и капецитабин. Комбинация лекарственных средств, используемая в химиотерапии EOF, включает эпирубицин, оксалиплатин и 5-фторурацил.
Эпирубицин обычно вводят в дозе 50 мг/м2, цисплатин 60 мг/м2, оксалиплатин 130 мг/м2, длительную венозную инфузию 5-фторурацила проводят в дозе 200 мг/м2/день и пероральное введение капецитабина в дозе 625 мг/м2 2 раза в день в течение всего восьми 3-недельных циклов.
Комбинация лекарственных средств, используемая в химиотерапии FLO, включает 5-фторурацил, фолиниевую кислоту и оксалиплатин (обычно 24-часовая инфузия 5-фторурацила в дозе 2600 мг/м2, фолиниевая кислота в дозе 200 мг/м2 и оксалиплатин в дозе 85 мг/м2 каждые 2 недели).
FOLFOX представляет собой режим химиотерапии, состоящий из фолиниевой кислоты (лейковорина), 5-фторурацила и оксалиплатина. Рекомендуемое терапевтическое воздействие проводят каждые две недели следующим образом: День 1: оксалиплатин в дозе 85 мг/м2 путем внутривенной (IV) инфузии и лейковорин в дозе 200 мг/м2 путем внутривенной (IV) инфузии с последующим 5-FU в дозе 400 мг/м2 путем внутривенного (IV) болюсного введения, с последующим 5-FU в дозе 600 мг/м2 путем внутривенной (IV) инфузии в виде 22-часовой непрерывной инфузии; День 2: лейковорин в дозе 200 мг/м2 путем внутривенной (IV) инфузии в течение 120 минут с последующим 5-FU в дозе 400 мг/м2 путем внутривенного (IV) болюсного введения, проводимого в течение 2-4 минут, с последующим 5-FU в дозе 600 мг/м2 путем внутривенной (IV) инфузии в виде 22-часовой непрерывной инфузии.
Комбинация лекарственных средств, используемая в химиотерапии FOLFIRI, включает 5-фторурацил, лейковорин и иринотекан.
Комбинация лекарственных средств, используемая в химиотерапии DCF, включает доцетаксел, цисплатин и фторурацил.
Комбинация лекарственных средств, используемая в химиотерапии FLOT, включает доцетаксел, оксалиплатин, 5-фторурацил и фолиниевую кислоту.
Термин «фолиниевая кислота» или «лейковорин» относится к соединению, используемому в синергической комбинации с химиотерапевтическим агентом 5-фторурацилом. Фолиниевая кислота имеет следующую формулу:
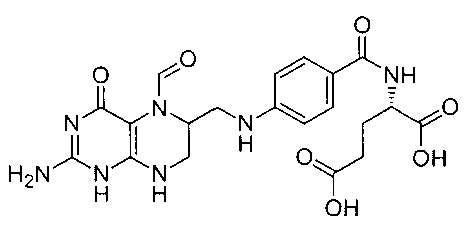
В частности, термин относится к соединению (2S)-2-{[4-[(2-амино-5-формил-4-оксо-5,6,7,8-тетрагидро-1Н-птеридин-6-ил)метиламино]бензоил]амино}пентандиовая кислота.
В соответствии с изобретением антитело, обладающее способностью связываться с CLDN18.2, можно вводить в комбинации, то есть одновременно, последовательно и/или после агента, стимулирующего γδ-Т-клетки.
γδ-Т-клетки (гамма-дельта Т-клетки) представляют небольшую субпопуляцию Т-клеток, которые содержат на своей поверхности видоизмененный Т-клеточный рецептор (TCR). Большинство Т-клеток несет Т-клеточный рецептор (TCR), состоящий из двух гликопротеиновых цепей, называемых α- и β-TCR-цепями. Напротив, в γδ-Т-клетках Т-клеточный рецептор (TCR) состоит из одной γ-цепи и одной δ-цепи. Как правило, эта группа Т-клеток является гораздо менее распространенной, чем αβ-Т-клетки. Человеческие γδ-Т-клетки играют важную роль в реакциях, контролирующих стресс, такой как инфекционные заболевания и аутоиммунитет. Предполагается, что индуцированные трансформацией изменения в опухолях также вызывают реакции, контролирующие стресс, опосредованные γδ-Т-клетками, и усиливают противоопухолевый иммунитет. Важно, что после захвата антигена активированные γδ-Т-клетки в участках поражения обеспечивают цитокины (например, INFγ, TNFα) и/или хемокины, опосредующие рекрутинг других эффекторных клеток, и демонстрируют немедленные эффекторные функции, такие как цитотоксичность (посредством пути «смерти рецептора» и цитолитических гранул) и ADCC.
Большинство γδ-Т-клеток в периферической крови экспрессируют Vγ9Vδ2-T-клеточный рецептор (TCRγδ). Vγ9Vδ2-Т-клетки являются уникальными для человека и приматов и, как полагают, играют важную роль в раннем восприятии «опасности» инвазивными агентами, так как они интенсивно размножаются во многих острых инфекциях и за несколько дней могут превысить все другие лимфоциты, например, в туберкулезе, сальмонеллезе, эрлихиозе, бруцеллезе, туляремии, листериозе, токсоплазмозе и малярии.
γδ-Т-клетки отвечают на малые непептидные фосфорилированные антигены (фосфоантигены), такие как пирофосфаты, синтезированные в бактериях, и изопентенилпирофосфат (IPP), продуцированный в клетках млекопитающих посредством мевалонатного пути. Тогда как продукция IPP в нормальных клетках является недостаточной для активации γδ-Т-клеток, дисрегуляция мевалонатного пути в клетках опухоли приводит к накоплению IPP и активации γδ-Т-клеток. Также, IPP может быть терапевтически увеличенным аминобифосфонатами, которые ингибируют фермент фарнезил-пирофосфат-синтазу мевалонатного пути (FPPS). Среди прочих, золедроновая кислота (ZA, золендронат, Zometa™, Novartis) представляет такой аминобифосфонат, который уже вводили клинически пациентам для лечения остеопороза и метастатического заболевания костей. При обработке РВМС in vitro золедроновая кислота (ZA) поглощается в основном моноцитами. Изопентенилпирофосфат (IPP) накапливается в моноцитах, которые дифференцируют в антигенпредставляющие клетки, стимулируя развитие γδ-Т-клеток. В данном контексте добавление интерлейкина-2 (IL-2) является предпочтительным в качестве фактора роста и выживаемости для активированных γδ-Т-клеток. В заключение, было описано, что некоторые алкилированные амины активируют Vγ9Vδ2-Т-клетки in vitro, однако только в миллимолярных концентрациях.
В соответствии с изобретением выражение «агент, стимулирующий γδ-Т-клетки" относится к соединениям, стимулирующим развитие γδ-Т-клеток, в частности Vγ9Vδ2-T-клеток, in vitro и/или in vivo, особенно путем индукции активации и размножения γδ-Т-клеток. Предпочтительно, выражение относится к соединениям, которые in vitro и/или in vivo повышают изопентенилпирофосфат (IPP), продуцированный в клетках млекопитающего, предпочтительно путем ингибирования фермента фарнезил-пирофосфат-синтазы (FPPS) мевалонатного пути.
Одной из конкретных групп соединений, стимулирующих γδ-Т-клетки, являются бифосфонаты, в частности, азотсодержащие бифосфонаты (N-бифосфонаты; аминобифосфонаты).
Например, бифосфонаты, подходящие для применения в данном изобретении, могут включать одно или несколько из следующих соединений, включая их аналоги и производные, фармацевтические соли, гидраты, сложные эфиры, конъюгаты и пролекарства:
[1-гидрокси-2-(1Н-имидазол-1-ил)этан-1,1-диил]бис(фосфоновая кислота), золедроновая кислота, например, золедронат;
(дихлор-фосфоно-метил)фосфоновая кислота, например, клодронат;
{1-гидрокси-3-[метил(пентил)амино]пропан-1,1-диил}бис(фосфоновая кислота), ибандроновая кислота, например, ибандронат;
(3-амино-1-гидроксипропан-1,1-диил)бис(фосфоновая кислота), памидроновая кислота, например, памидронат;
(1-гидрокси-1-фосфоно-2-пиридин-3-ил-этил)фосфоновая кислота, ризедроновая кислота, например, ризедронат;
(1-гидрокси-2-имидазо[1,2-а]пиридин-3-ил-1-фосфоноэтил)фосфоновая кислота, минодроновая кислота;
[3-(диметиламино)-1-гидроксипропан-1,1-диил]бис(фосфоновая кислота), олпадроновая кислота.
[4-амино-1-гидрокси-1-(гидрокси-оксидо-фосфорил)-бутил]фосфоновая кислота, аледроновая кислота, например, алендронат;
[(циклогептиламино)метилен]бис(фосфоновая кислота), инкадроновая кислота;
(1-гидроксиэтан-1,1-диил)бис(фосфоновая кислота), этидроновая кислота, например, этидронат; и
{[(4-хлорфенил)тио]метилен}бис(фосфоновая кислота), тилудроновая кислота.
В соответствии с изобретением золедроновая кислота (INN) или золедронат (под торговым названием Novartis торговых марок Зомета (Zometa), Зомера (Zomera), Акласта (Aclasta) и Рекласт (Reclast)) является особенно предпочтительным бифосфонатом. Зомета применяют для предупреждения переломов костей у пациентов с раком, таким как множественная миелома и рак предстательной железы. А также для лечения остеопороза. Кроме того, он может применяться для лечения опухоль-ассоциированной гиперкальцемии и быть полезным для лечения боли, вызванной метастазами в кости.
В одном особенно предпочтительном варианте осуществления агент, стимулирующий γδ-Т-клетки в соответствии с изобретением, вводят в сочетании с IL-2. Такое сочетание оказалось особенно эффективным в опосредовании размножения и активации γ9δ2-Т-клеток.
Интерлейкин-2 (IL-2) представляет собой интерлейкин, тип сигнальной молекулы цитокина в иммунной системе. Это белок, который привлекает лимфоциты и является частью естественного ответа организма на микробную инфекцию, и способен распознавать чужеродные (не свои) мишени. IL-2 опосредует свои эффекты путем связывания с IL-2-рецепторами, которые экспрессируются лимфоцитами.
IL-2, используемый в соответствии с изобретением, может быть любым IL-2, поддерживающим или обеспечивающим возможность стимуляции γδ-Т-клеток, и может быть получен из любых видов, предпочтительно человека. Il-2 может представлять собой изолированный, полученный рекомбинантно или синтетически IL-2, и может быть естественного происхождения или модифицированным IL-2.
В соответствии с изобретением термин «противорвотное средство» относится к соединению, композиции или лекарственному средству, которое является эффективным против рвоты и/или тошноты. В одном варианте осуществления противорвотное средство включает антагонист рецептора 5-НТ3 и/или антагонист рецептора нейрокинина 1 (NK1).
Антагонисты рецептора 5-НТ3 блокируют серотониновые рецепторы в центральной нервной системе и желудочно-кишечном тракте. Их примеры включают, но без ограничения: ондансетрон (зофран), который можно вводить перорально в форме таблетки, перорально в форме растворяющейся таблетки или путем инъекции; доласетрон (анземет), который можно вводить в форме таблетки или путем инъекции; гранисетрон (китрил, санкузо), который можно вводить в форме таблетки (китрил), перорального раствора (китрил), инъекции (китрил) или в форме одноразового трансдермального пластыря на плечо (санкузо); трописетрон (навобан), который можно вводить в форме пероральных капсул или инъекции; палоносетрон (алокси), который можно вводить в форме инъекции или пероральных капсул; и миртазапин (ремерон).
Антагонисты рецептора NK1 включают, но без ограничения, апрепитант (эменд).
Предпочтительной комбинацией антагониста рецептора 5-НТ3 и антагониста рецептора NK1 является комбинация ондансетрона (зофран) и апрепитанта (эменд).
Дополнительные противорвотные средства, которые могут быть использованы в соответствии с изобретением, особенно в сочетании с антагонистом рецептора 5-НТ3 и/или антагонистом рецептора NK1, включают, но без ограничения, метоклопрамид (реглан), который действует на GI тракт в качестве прокинетического средства, лоразепам, атропин, ализаприд (литикан, плитикан, суперан, вергентан) и дименгидринат (драмамин, дриминат, гравол, гравамин, вомекс, вертирозан).
В соответствии с изобретением можно вводить антиспазматическое средство (синоним: спазмолитическое средство). В соответствии с изобретением термин «антиспазматическое средство» относится к соединению, композиции или лекарственному средству, которое подавляет мышечные спазмы. Предпочтительно, антиспазматическое средство применяется для сокращения гладких мышц. Предпочтительными в соответствии с изобретением являются антиспазматические средства, которые эффективны при лечении спазматической активности в пищеварительной системе. Таким образом, предпочтительные антиспазматические средства являются эффективными в отношении ослабления желудочно-кишечных спазмов.
Антиспазматические средства включают, но без ограничения, бутилскополамин, который также известен как скополамина бутилбромид, бутилгиосцин и гиосцин бутилбромид. Он присутствует на рынке под торговым названием Buscopan (бускопан) фирмы Boehringer Ingelheim GmbH, Germany.
В соответствии с изобретением можно вводить парасимпатолитические средства. В соответствии с изобретением термин «парасимпатолитические средства» относится к соединению, композиции или лекарственному средству, которое снижает активность парасимпатической нервной системы. Парасимпатолитические средства включают, но без ограничения, атропин.
В соответствии с изобретением термин «ингибитор протонного насоса» относится к соединению, композиции или лекарственному средству, основным действием которого является значительное и продолжительное снижение выработки желудочной кислоты.
Ингибиторы протонного насоса включают производные бензимидазола и производные имидазопиридина. Примеры ингибиторов протонного насоса включают, но без ограничения, омепразол (торговые названия: Гасек (Gasec), Лосек (Losec), Прилосек (Prilosec), Зегерид (Zegerid), Оцид (Ocid), Ломак (Lomac), Омепрал (Omepral), Омез (Omez)), лансопразол (торговые названия: Превацид (Prevacid), Зотон (Zoton), Монолитум (Monolitum), Ингибитол (Inhibitol), Левант (Levant), Лупизол (Lupizole)), декслансопразол (торговые названия: Капидекс (Kapidex), Дексилант (Dexilant)), эзомепразол (торговые названия: Нексиум (Nexium), Эзотрекс (Esotrex), Эссо (esso)), пантопразол (торговые названия: Протоникс (Protonix), Сомак (Somac), Пантолок (Pantoloc), Пантозол (Pantozol), Зуркал (Zurcal), Зентро (Zentro), Пан (Pan), Контролок (Controloc), Текта (Tecta)), рабепразол (торговые названия: Ацифекс (AcipHex), Париет (Pariet), Эрраз (Erraz), Цехин (Zechin), Рабецид (Rabecid), Nzole-D, Рабелок (Rabeloc), Разо (Razo)) и илапразол (торговые названия: Илапро (Ilapro), Лупила (Lupilla), Адиза (Adiza)).
В соответствии с изобретением можно вводить другие соединения, композиции или лекарственные средства, которые оказывают защитное действие на слизистую оболочку желудка, особенно при введении нестероидного противовоспалительного лекарственного средства (NSAID).
Например, другие соединения, композиции или лекарственные средства можно вводить для предупреждения типичного побочного эффекта, образования язв желудка, вызванного NSAID, в частности, для предупреждения индуцированных NSAID язвенных болезней желудка. В одном варианте осуществления можно вводить мизопростол, который является синтетическим аналогом простагландина E1 (PGE1), который применяется для предупреждения язвенных болезней желудка, индуцированных NSAID. Мизопростол воздействует на париетальные клетки желудка, ингибируя секрецию желудочной кислоты путем опосредованного рецептором, связанного G-белком, ингибирования аденилатциклазы, что приводит к снижению уровней внутриклеточного циклического AMP и снижению активности протонного насоса на апикальной поверхности париетальной клетки.
Кроме того, доказано, что омепразол является эффективным, по меньшей мере, как мизопростол при лечении индуцированных NSAID язв, но значительно лучше переносится.
Нестероидные противовоспалительные лекарственные средства (NSAID) относятся к классу лекарственных средств, которые обеспечивают обезболивающий и жаропонижающий эффекты и в повышенных дозах противовоспалительные эффекты. Термин «нестероидные» проводит отличие этих лекарственных средств от стероидов. Наиболее известными членами этой группы лекарственных средств являются аспирин, ибупрофен и напроксен.
Одна из основных неблагоприятных реакций на лекарственное средство (ADR), связанных с NSAID, относится к желудочно-кишечным (GI) эффектам NSAID. Эти эффекты во многих случаях являются достаточно серьезными, чтобы вызвать риск прободения язвы и желудочно-кишечное кровотечение. NSAID-пациенты испытывают диспепсию, NSAID-ассоциированные неблагоприятные явления в верхнем отделе желудочно-кишечного тракта, раздражение желудочно-кишечного (GI) тракта и образование язв GI. NSAID оказывает двойное действие на GI тракт: кислые молекулы напрямую раздражают слизистую оболочку желудка, и ингибирование СОХ-1 и СОХ-2 понижает уровни защитных простагландинов. Ингибирование синтеза простагландинов в GI тракте вызывает повышение секреции желудочной кислоты, пониженную секрецию бикарбоната, пониженную секрецию слизи и пониженные трофические эффекты на слизистую оболочку эпителия. Таким образом, NSAID предпочтительно не вводят в соответствии с изобретением. Парацетамол или «ацетаминофен», который не классифицируется как NSAID, так как проявляет только слабые противовоспалительные эффекты, можно вводить в качестве обезболивающего средства в соответствии с изобретением, однако этого может быть недостаточно для управления болью и, таким образом, введение NSAID может стать необходимым, особенно для того, чтобы избежать введения опиоидов.
Обычно побочные эффекты в желудке (но необязательно кишечнике) могут быть уменьшены посредством подавления выработки кислоты, путем сопутствующего применения ингибитора протонного насоса, например, омепразола, эзомепразола; или простагландинового аналога мизопростола.
Термин «антиген» относится к агенту, такому как белок или пептид, содержащему эпитоп, против которого направлен иммунный ответ и/или должен быть направлен. В предпочтительном варианте осуществления антиген представляет собой опухоль-ассоциированный антиген, такой как CLDN18.2, то есть компонент раковых клеток, который может быть получен из цитоплазмы, клеточной поверхности и клеточного ядра, в частности такие антигены, которые продуцируются, предпочтительно в больших количествах, внутриклеточно или в качестве поверхностных антигенов на раковых клетках.
В контексте настоящего изобретения термин «опухоль-ассоциированный антиген» предпочтительно относится к белкам, которые в нормальных условиях специфически экспрессируются в ограниченном числе тканей и/или органов, или на специфических стадиях развития, и экспрессируются или аберрантно экспрессируются в одной или более опухолях или раковых тканях. В контексте настоящего изобретения опухоль-ассоциированный антиген предпочтительно связан с клеточной поверхностью раковой клетки и предпочтительно не экспрессируется или только редко экспрессируется в нормальных тканях.
Термин «эпитоп» относится к антигенной детерминанте в молекуле, то есть к части в молекуле, которая распознается иммунной системой, например, которая распознается антителом. Например, эпитопы представляют собой дискретные трехмерные участки на антигене, которые распознаются иммунной системой. Эпитопы обычно состоят из химически активных групп, расположенных на поверхности молекул, таких как аминокислотные или сахарные боковые цепи, и обычно обладают специфическими трехмерными структурными характеристиками, а также специфическими характеристиками зарядов. Конформационные и неконформационные эпитопы различают по тому, что в присутствии денатурирующих растворителей исчезает связывание с первыми, но не со вторыми эпитопами. Эпитоп белка, такого как CLDN18.2, предпочтительно содержит непрерывную и прерывистую часть указанного белка и содержит предпочтительно от 5 до 100, предпочтительно от 5 до 50, более предпочтительно от 8 до 30, наиболее предпочтительно от 10 до 25 аминокислот в длину, например, эпитоп может иметь предпочтительно 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 или 25 аминокислот в длину.
Термин «антитело» относится к гликопротеину, включающему, по меньшей мере, две тяжелые (Н) цепи и две легкие (L) цепи, связанные между собой дисульфидными связями, и включает любую молекулу, содержащую его антигенсвязывающий участок. Термин «антитело» включает моноклональные антитела и фрагменты или производные антител, включая, без ограничения, антитела человека, гуманизированные антитела, химерные антитела, одноцепочечные антитела, например, scFv и антиген-связывающие фрагменты антитела, такие как фрагменты Fab и Fab' и также включает все рекомбинантные формы антител, например, антител, экспрессирующихся в прокариотах, негликолизированные антитела и любые антиген-связывающие фрагменты антител и производные, описанные здесь. Каждая тяжелая цепь состоит из вариабельной области тяжелой цепи (обозначенной здесь как VH) и константной области тяжелой цепи. Каждая легкая цепь состоит из вариабельной области легкой цепи (обозначенной здесь как VL) и константной области легкой цепи. Области VH и VL могут дополнительно подразделяться на участки гипервариабельности, называемые участками, определяющими комплементарность (CDR), перемежающиеся более консервативными участками, называемыми каркасными участками (FR). Каждая VH и VL состоит из трех CDR и четырех FR, расположенных в направлении от амино-конца к карбокси-концу в следующем порядке: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. Вариабельные области тяжелой и легкой цепей содержат связывающий домен, который взаимодействует с антигеном. Константные области антител могут опосредовать связывание иммуноглобулина с тканями-хозяевами или факторами, включающими различные клетки иммунной системы (например, эффекторные клетки) и первый компонент (C1q) классической системы комплемента.
Антитела, описанные в настоящем документе, могут представлять собой человеческие антитела. Термин «человеческое антитело», используемый здесь, включает антитело, имеющее вариабельную и константную области от родительских последовательностей иммуноглобулинов человека. Человеческие антитела, описанные здесь, могут включать аминокислотные остатки, которые не кодируются родительскими последовательностями иммуноглобулинов человека (например, вследствие мутации, введенных с помощью случайного или сайт-направленного мутагенеза in vitro или с помощью соматической мутации in vivo).
Термин «гуманизированное антитело» относится к молекуле, имеющей в своем составе участок связывания антигена, который в значительной степени происходит из иммуноглобулина тех видов, которые не относятся к человеку, где оставшаяся структура молекулы иммуноглобулина базируется на структуре и/или последовательности иммуноглобулина человека. Участок, связывающий антиген, может включать полные вариабельные области, объединенные с константными областями, или только участки, определяющие комплементарность (CDR), привитые на подходящие каркасные участки в вариабельных областях. Участки, связывающие антиген, могут быть дикого типа или модифицированными с помощью одной или нескольких аминокислотных замен, например, модифицированными таким образом, чтобы более точно походить на иммуноглобулин человека. Некоторые формы гуманизированных антител сохраняют все последовательности CDR (например, гуманизированное мышиное антитело, которое содержит все шесть CDR из мышиного антитела). Другие формы имеют один или несколько CDR, которые отличаются от исходного антитела.
Термин «химерное антитело» относится к таким антителам, в которых одна часть каждой аминокислотной последовательности тяжелых и легких цепей гомологична соответствующим последовательностям в антителах, происходящих из определенных видов или принадлежащим определенному классу, тогда как оставшийся сегмент цепи гомологичен соответствующим последовательностям у других видов. Обычно вариабельная область как в легких, так и в тяжелых цепях имитирует вариабельные области антител, полученных из одного вида млекопитающих, тогда как константные участки гомологичны последовательностям антител, полученным из другого вида. Одним несомненным преимуществом таких химерных форм является то, что вариабельную область можно удобным образом получить из известных в настоящее время источников, применяя легко доступные В-клетки или гибридомы из организмов-хозяев, которые не являются человеком, в комбинации с константными областями, полученными, например, из препаратов клеток человека. В то время как вариабельная область имеет преимущество, состоящее в легкости получения и в том, что специфичность не зависит от источника получения, константная область, источником которой является человек, менее вероятно вызывает иммунный ответ у субъекта-человека при введении антител, чем это делала бы константная область, полученная из источника, не являющегося человеком. Однако определение не ограничено этим специфическим примером.
Термины «антигенсвязывающий участок» антитела (или просто «связывающий участок») или «антигенсвязывающий фрагмент» антитела (или просто «связывающий фрагмент»), или аналогичные термины относятся к одному или нескольким фрагментам антитела, которые сохраняют способность специфически связываться с антигеном. Было показано, что антигенсвязывающая функция антитела может осуществляться фрагментами полноразмерного антитела. Примеры связывающих фрагментов, охватывающие термин «антигенсвязывающий участок» антитела, включают (i) Fab-фрагменты, моновалентные фрагменты, состоящие из доменов VL, VH, CL и СН; (ii) F(ab')2-фрагменты, бивалентные фрагменты, включающие два Fab-фрагмента, связанные дисульфидным мостиком в шарнирном участке; (iii) Fd-фрагменты, состоящие из доменов VH и СН; (iv) Fv-фрагменты, состоящие из доменов VL и VH одного плеча антитела, (v) dAb-фрагменты (Ward et al., (1989) Nature 341: 544-546), которые состоят из VH-домена; (vi) изолированные участки, определяющие комплементарность (CDR), и (vii) комбинации двух или более изолированных CDR, которые могут быть необязательно присоединены с помощью синтетического линкера. Кроме того, хотя два домена Fv-фрагмента, VL и VH, кодируются разными генами, они могут быть соединены с применением рекомбинантных методов с помощью синтетического линкера, который дает им возможность стать единой белковой цепью, в которой пара участков VL и VH образует моновалентные молекулы (известные как одноцепочечный Fv (scFv); смотри, например, Bird et al. (1988) Science 242: 423-426; и Huston et al. (1988) Proc. Natl. Acad. Sci. USA 85: 5879-5883). Такие одноцепочечные антитела также включены в термин «антигенсвязывающий участок» антитела. Еще одним примером являются слитые белки связывающего домена иммуноглобулина, включающие (i) полипептид связывающего домена, который слит с полипептидом шарнирного участка иммуноглобулина, (ii) константную область СН2 тяжелой цепи иммуноглобулина, слитую с шарнирным участком; и (iii) константную область СН3 тяжелой цепи иммуноглобулина, слитую с константной областью СН2. Полипептид связывающего домена может быть вариабельной областью тяжелой цепи или вариабельной областью легкой цепи. Слитые белки связывающего домена иммуноглобулина, кроме того, раскрыты в заявках US 2003/0118592 и US 2003/0133939. Эти фрагменты антитела получают с помощью общепринятых методик, известных специалистам в данной области, и проводят скрининг фрагментов на применение так же, как в случае интактных антител.
Термин «биспецифическая молекула» включает любой агент, например, белок, пептид или комплекс белка или пептида, который обладает двумя различными специфичностями связывания. Например, молекула может связываться или взаимодействовать с (а) антигеном клеточной поверхности, и (b) Fc-рецептором на поверхности эффекторной клетки. Термин «мультиспецифичная молекула» или «гетероспецифичная молекула» включает любой агент, например, белок, пептид или комплекса белка или пептида, который обладает более чем двумя различными специфичностями связывания. Например, молекула может связываться или взаимодействовать с (а) антигеном клеточной поверхности, (b) Fc-рецептором на поверхности эффекторной клетки, и (с) по меньшей мере, с еще одним компонентом. Соответственно, изобретение включает, но без ограничения, биспецифичные, триспецифичные и другие мультиспецифичные молекулы, действие которых направлено на CLDN18.2 и другие мишени, такие как Fc-рецепторы на эффекторных клетках. Термин «биспецифичные антитела» также включает диатела. Диатела представляют собой бивалентные, биспецифичные антитела, в которых VH- и VL-домены экспрессируются на одной полипептидной цепи, но с использованием линкера, который является слишком коротким, чтобы позволить спаривание двух доменов на одной и той же цепи, что заставляет домены спариваться с комплементарными доменами другой цепи и создавать два антигенсвязывающих сайта (смотри, например, Holliger, P., et al. (1993) Proc. Natl. Acad. Sci. USA 90: 6444-6448; Poljak, R.J., et al. (1994) Structure 2: 1121-1123).
Антитело может быть конъюгировано с терапевтическим фрагментом или агентом, таким как цитотоксин, лекарственным средством (например, иммуносупрессантом) или радиоизотопом. Цитотоксин или цитотоксический агент включает любой агент, который является пагубным для клеток и, в частности, убивает их. Примеры включают таксол, цитохалазин В, грамицидин D, этидия бромид, эметин, митомицин, этопозид, тенопозид, винкристин, винбластин, колхицин, доксорубицин, даунорубицин, дигидрокси антрацин дион, митоксантрон, митрамицин, актиномицин D, 1-дегидротестостерон, глюкокортикоиды, прокаин, тетракаин, лидокаин, пропранолол и пиромицин и их аналоги или гомологи. Подходящие терапевтические агенты для формирования конъюгатов антитела включают, но без ограничения, антиметаболиты (например, метотрексат, 6-меркаптопурин, 6-тиогуанин, цитарабин, флударабин, 5-фторурацил декарбазин), алкилирующие агенты (например, мехлорэтамин, тиоепа хлорамбуцил, мелфалан, кармустин (BSNU) и ломустин (CCNU), циклофосфамид, бусульфан, дибромманнитол, стрептозотоцин, митомицин С и цис-дихлордиамин палладия (II) (DDP) цисплатин), антрациклины (например, даунорубицин (исходное название дауномицин) и доксорубицин), антибиотики (например, дактиномицин (исходное название актиномицин), блеомицин, митрамицин и антрамицин (АМС) и антимитотические агенты (например, винкристин и винбластин). В предпочтительном варианте осуществления терапевтический агент представляет собой цитотоксический агент или радиотоксический агент. В другом варианте осуществления терапевтический агент представляет собой иммуносупрессант. В еще другом варианте осуществления терапевтический агент представляет собой GM-CSF. В предпочтительном варианте осуществления терапевтический агент представляет собой доксорубицин, цисплатин, блеомицин, сульфат, кармустин, хлорамбуцил, циклофосфамид или рицин А.
Антитела также могут быть конъюгированы с радиоизотопом, например, иодом-131, иттрий-90 или индий-111 для генерации цитотоксических радиофармацевтических средств.
Конъюгаты антител изобретения могут быть использованы для модификации заданного биологического ответа и фрагмент лекарственного средства не считается ограниченным классическими химическими терапевтическими агентами. Например, фрагмент лекарственного средства может представлять собой белок или полипептид, обладающий желательной биологической активностью. Такие белки могут включать, например, ферментативно активный токсин или его активный фрагмент, такой как абрин, рицин А, экзотоксин псевдонома или токсин дифтерии; белок, такой как фактор некроза опухоли или интерферон-γ; или модификаторы биологического ответа, такие как, например, лимфокины, интерлейкин-1 ("IL-1"), интерлейкин-2 ("IL-2"), интерлейкин-6 ("IL-6"), гранулоцитарный макрофагальный колониестимулирущий фактор ("GM-CSF"), гранулоцитарный колониестимулирующий фактор ("G-CSF"), или другие факторы роста.
Методики для конъюгирования такой терапевтической группы с антителами хорошо известны, смотри, например, Arnon et al., "Monoclonal Antibodies For Immunotargeting Of Drugs In Cancer Therapy", in Monoclonal Antibodies And Cancer Therapy, Reisfeld et al. (eds.), pp. 243-56 (Alan R. Liss, Inc. 1985); Hellstrom et al., "Antibodies For Drug Delivery", in Controlled Drug Delivery (2nd Ed.), Robinson et al. (eds.), pp. 623-53 (Marcel Dekker, Inc. 1987); Thorpe, "Antibody Carriers Of Cytotoxic Agents In Cancer Therapy: A Review", in Monoclonal Antibodies '84: Biological And Clinical Applications, Pincheraet al. (eds.), pp. 475-506 (1985); "Analysis, Results, And Future Prospective Of The Therapeutic Use Of Radiolabeled Antibody In Cancer Therapy", in Monoclonal Antibodies For Cancer Detection And Therapy, Baldwin et al. (eds.), pp. 303-16 (Academic Press 1985), и Thorpe et al., "The Preparation And Cytotoxic Properties Of Antibody-Toxin Conjugates", Immunol. Rev., 62: 119-58 (1982).
Используемый здесь термин антитело «происходит» из специфической родительской последовательности, если антитело получают из системы с помощью иммунизации животного или с помощью скрининга библиотеки генов иммуноглобулинов, и если отобранное антитело по своей аминокислотной последовательности, по меньшей мере, на 90%, более предпочтительно, по меньшей мере, на 95%, еще более предпочтительно, по меньшей мере, на 96%, 97%, 98% или 99% идентично аминокислотной последовательности, кодируемой родительским геном иммуноглобулина. Обычно антитело, происходящее из специфической родительской последовательности, будет отличаться не более чем по 10 аминокислотам, более предпочтительно, не более чем по 5, или еще более предпочтительно, не более чем по 4, 3, 2 или 1 аминокислоте от аминокислотной последовательности, кодируемой родительским геном иммуноглобулина.
Используемый здесь термин «гетероантитела» относится к двум или более антителам, их производным или антигенсвязывающим участками, связанным вместе, по меньшей мере, два из которых обладают различными специфичностями. Эти различные специфичности включают специфичность связывания с Fc-рецептором на эффекторной клетке и специфичность связывания с антигеном или эпитопом на клетке-мишени, например, на опухолевой клетке.
Антитела, описанные здесь, могут представлять собой моноклональные антитела. Термин «моноклональное антитело», используемый здесь, относится к препарату молекул антитела одного единственного молекулярного состава. Моноклональное антитело демонстрирует один и тот же сайт специфического связывания и аффинность. В одном варианте осуществления моноклональные антитела продуцируют с помощью гибридомы, которая включает В-клетку, полученную из животного, которое не является человеком, например, из мыши, слитую или иммортализованную клетку.
Антитела, описанные здесь, могут представлять собой рекомбинантные антитела. Термин «рекомбинантное антитело», используемый здесь, включает все антитела, которые получают, экспрессируют, создают или выделяют с помощью рекомбинантных способов, такие как (а) антитела, выделенные из животного (например, мыши), которое является трансгенным или трансхромосомным в отношении генов иммуноглобулинов или из гибридом, полученных из него, (b) антитела, выделенные из клетки-хозяина, трансформированной для экспрессии антитела, например, из трансфектомы, (с) антитела, выделенные из библиотеки рекомбинантных, комбинаторных антител, и (d) антитела, полученные, экспрессированные, созданные или выделенные с помощью других способов, которые включают сплайсинг последовательностей иммуноглобулиновых генов, приводящий к другим последовательностям ДНК.
Антитела, описанные здесь, могут быть получены из разных видов, включая, но без ограничения, мышь, крысу, кролика, морскую свинку и человека.
Антитела, описанные в настоящем документе, включают поликлональные и моноклональные антитела, и включают IgA-антитела, такие как IgA1 или IgA2, IgG1, IgG2, IgG3, IgG4, IgE, IgM и IgD. В различных вариантах осуществления антитело представляет собой IgG1-антитело, более предпочтительно IgG1-антитело, изотипы IgG1-каппа или IgG1-лямбда (то есть IgG1, κ, λ), IgG2a-антитело (например, IgG2a, κ, λ), IgG2b-антитело (например, IgG2b, κ, λ), IgG3-антитело (например, IgG3, κ, λ) или IgG4-антитело (например, IgG4, κ, λ).
Термин «трансфектома», используемый здесь, включает рекомбинантные эукариотические клетки-хозяева, экспрессирующие антитело, такие как СНО-клетки, NS/0-клетки, HEK293-клетки, HEK293T-клетки, клетки растений или грибы, включая клетки дрожжей.
Используемый здесь термин «гетерологичное антитело» определен в отношении трансгенного организма, продуцирующего такое антитело. Этот термин относится к антителу, имеющему аминокислотную последовательность или последовательность кодирующей нуклеиновой кислоты, соответствующей той последовательности, которая обнаружена в организме, в состав которого не входит трансгенный организм, и которая обычно происходит из других видов, не являющихся трансгенным организмом.
Используемое здесь «гетерогибридное антитело» относится к антителу, имеющему легкие и тяжелые цепи различного организменного происхождения. Например, антитело, имеющее тяжелую цепь иммуноглобулина человека, связанную с легкой цепью иммуноглобулина мыши, является гетерогибридным антителом.
Изобретение включает все антитела и производные антител, описанные выше, которые в целях изобретения охвачены термином «антитело». Термин «производные антитела» относятся к любой модифицированной форме антитела, например, конъюгату антитела и другого агента или антитела, или фрагмента антитела.
Описанные здесь антитела предпочтительно являются выделенными. Используемое здесь «выделенное антитело» относится к антителу, которое в значительной степени свободно от других антител, обладающих другой антигенной специфичностью (например, выделенное антитело, которое специфически связывается с CLDN18.2, в значительной степени свободно от антител, которые специфически связывают антигены, которые не являются CLDN18.2). Выделенное антитело, которое специфически связывается с эпитопом, изоформой или вариантом CLDN18.2 человека, может, однако, обладать кросс-реактивностью по отношению к другим родственным антигенам, например, из других видов (например, видовым гомологам CLDN18.2). Кроме того, выделенное антитело может быть в значительной степени свободно от другого клеточного материала и/или химических реагентов. В одном варианте осуществления комбинация «выделенных» моноклональных антител относится к антителам, имеющим различную специфичность, которые объединены в ясно определенную композицию или смесь.
Термин «связывание» в соответствии с изобретением предпочтительно относится к специфическому связыванию.
В соответствии с настоящим изобретением антитело способно к связыванию с предварительно определенной мишенью, если оно обладает значительной аффинностью в отношении указанной предварительно определенной мишени и связывается с указанной предварительно определенной мишенью в стандартных анализах. «Аффинность» или «аффинность связывания» часто измеряется равновесной константой диссоциации (KD). Предпочтительно термин «значительная аффинность» относится к связыванию с предварительно определенной мишенью с константой диссоциации (KD), равной 10-5 М или ниже, 10-6 М или ниже, 10-7 М или ниже, 10-8 М или ниже, 10-9 М или ниже, 10-10 М или ниже, 10-11 М или ниже, или 10-12 М или ниже.
Антитело (по существу) не способно к связыванию с мишенью, если оно не обладает значительной аффинностью в отношении указанной мишени и не связывается в значительной степени, в частности, не связывается обнаруживаемым образом с указанной мишенью в стандартных анализах. Предпочтительно, антитело не связывается с указанной мишенью обнаруживаемым образом в случае присутствия при концентрации до 2, предпочтительно 10, более предпочтительно 20, в частности 50 или 100 мкг/мл или выше. Предпочтительно, антитело не обладает значительной аффинностью в отношении мишени, если оно связывается с указанной мишенью с KD, которая, по меньшей мере, в 10-раз, 100-раз, 103-раз, 104-раз, 105-раз или 106-раз выше, чем KD для связывания с предварительно установленной мишенью, с которой антитело способно связываться. Например, если KD для связывания антитела с мишенью, с которой антитело способно связываться, равно 10-7 М, KD для связывания с мишенью, в отношении которой антитело не обладает значительной аффинностью, будет равно, по меньшей мере, 10-6 М, 10-5 М, 10-4 М, 10-3 М, 10-2 М или 10-1 М.
Антитело является специфическим в отношении предварительно определенной мишени, если оно способно к связыванию с указанной предварительно определенной мишенью, при этом оно не способно к связыванию с другими мишенями, то есть не обладает значительной аффинностью в отношении других мишеней и значительно не связывается с другими мишенями в стандартных анализах. В соответствии с изобретением антитело является специфическим в отношении CLDN18.2, если оно способно связываться с CLDN18.2, но (по существу) не способно связываться с другими мишенями. Предпочтительно, антитело является специфическим в отношении CLDN18.2, если аффинность и связывание с такими другими мишенями значительно не превышают аффинность или связывание с CLDN18.2-неродственными белками, такими как бычий сывороточный альбумин (BSA), казеин, человеческий сывороточных альбумин (HSA) или неклаудиновые трансмембранные белки, такие как молекулы МНС или рецептор трансферрина, или любой другой указанный полипептид. Предпочтительно, антитело является специфическим в отношении предварительно определенной мишени, если оно связывается с указанной мишенью с KD, которая, по меньшей мере, в 10-раз, 100-раз, 103-раз, 104-раз, 105-раз или 106-раз ниже, чем KD для связывания с мишенью, в отношении которой оно является специфическим. Например, если KD для связывания антитела с мишенью, в отношении которой оно является специфическим, равно 10-7 М, KD для связывания с мишенью, в отношении которой оно не является специфическим, будет равно, по меньшей мере, 10-6 М, 10-5 М, 10-4 М, 10-3 М, 10-2 М или 10-1 М.
Связывание антитела с мишенью может быть определено экспериментально с использованием любого подходящего способа; смотри, например, Berzofsky et al., "Antibody-Antigen Interactions" In Fundamental Immunology, Paul, W.E., Ed., Raven Press New York, N Y (1984), Kuby, Janis Immunology, W.H. Freeman and Company New York, N Y (1992), и описанные здесь способы. Аффинности могут быть легко определены с использованием обычных методов, таких как равновесный диализ, с использованием прибора BIAcore 2000, с использованием общих процедур, указанных производителем; с помощью радиоммуноанализа с использованием радиомеченого антигена мишени; или с помощью другого способа, известного специалисту в данной области. Данные аффинности могут быть подвергнуты анализу, например, с помощью способа Scatchard et al., Ann N.Y. Acad. ScL, 51: 660 (1949). Измеренная аффинность взаимодействия конкретное антитело-антиген может изменяться при измерении в различных условиях, например, концентрации соли, рН. Таким образом, измерения аффинности и других параметров связывания антигена, например, KD, IC50, проведены с помощью стандартизированных растворов антитела и антигена, и стандартизированного буфера.
Используемый здесь «изотип» относится к классу антител (например, IgM или IgG1), который кодируется генами константной области тяжелых цепей.
Используемый здесь «переключение изотипов» относится к явлению, с помощью которого класс или изотип антитела изменяется с одного класса Ig на один из других классов Ig.
Термин «встречающийся в природе», используемый в настоящем документе в отношении объекта, относится к тому факту, что объект может быть обнаружен в природе. Например, полипептидная или полинуклеотидная последовательность, которая существует в организме (включая вирусы), которую можно выделить из природного источника и которая не была специально модифицирована человеком в лаборатории, является встречающейся в природе последовательностью.
Термин «перегруппированная», используемый здесь, относится к конфигурации тяжелой цепи или легкой цепи иммуноглобулинового локуса, в котором сегмент V непосредственно примыкает к сегменту D-J или J-сегменту в конформации, кодирующей практически полный VH- или VL-домен, соответственно. Перегруппированный локус гена иммуноглобулина (антитела) можно идентифицировать путем сравнения с родительской ДНК; перегруппированный локус будет иметь, по меньшей мере, один рекомбинированный элемент гептамерной/нонамерной гомологии.
Термин «неперегруппированная» или «родительская конфигурация», используемый здесь со ссылкой на V-сегмент, относится к конфигурации, в которой V-сегмент не является рекомбинированным таким образом, что непосредственно примыкает к сегменту D или J.
В соответствии с изобретением антитело, обладающее способностью связываться с CLDN18.2, представляет собой антитело, способное связываться с эпитопом CLDN18.2, предпочтительно эпитопом, локализованным в пределах внеклеточночных доменов CLDN18.2, в частности, первого внеклеточного домена, предпочтительно в аминокислотных положениях с 29 по 78 CLDN18.2. В конкретных вариантах осуществления антитело, обладающее способностью связываться с CLDN18.2, представляет собой антитело, способное связываться с (i) эпитопом CLDN18.2, который не присутствует у CLDN18.2, предпочтительно с SEQ ID NO: 3, 4 и 5, (ii) эпитопом, локализованным в петле CLDN18.2, предпочтительно с SEQ ID NO: 8, (iii) эпитопом, локализованным в петле 2 CLDN18.2, предпочтительно с SEQ ID NO: 10, (iv) эпитопом, локализованным в петле D3 CLDN18.2, предпочтительно с SEQ ID NO: 11, (v) эпитопом, который включает петлю 1 CLDN18.2 и петлю D3 CLDN18.2, или (vi) негликолизированным эпитопом, локализованным в петле D3 CLDN18.2, предпочтительно с SEQ ID NO: 9.
В соответствии с изобретением антитело, обладающее способностью связываться с CLDN18.2, предпочтительно представляет собой антитело, обладающее способностью связываться с CLDN18.2, но не с CLDN18.1. Предпочтительно, антитело, обладающее способностью связываться с CLDN18.2, является специфическим в отношении CLDN18.2. Предпочтительно, антитело, обладающее способностью связываться с CLDN18.2, предпочтительно представляет собой антитело, обладающее способностью связываться с CLDN18.2, экспрессирующимся на клеточной поверхности. В особенно предпочтительных вариантах осуществления антитело, обладающее способностью связываться с CLDN18.2, связывается с нативными эпитопами CLDN18.2, присутствующими на поверхности живых клеток. Предпочтительно, антитело, обладающее способностью связываться с CLDN18.2, связывается с одним или несколькими пептидами, выбранными из группы, состоящей из SEQ ID NO: 1, 3-11, 44, 46 и 48-50. Предпочтительно, антитело, обладающее способностью связываться с CLDN18.2, является специфическим в отношении указанных выше белков, пептидов или иммуногенных фрагментов или их производных. Антитело, обладающее способностью связываться с CLDN18.2, может быть получено с помощью способа, включающего стадию иммунизации животного белком или пептидом, содержащим аминокислотную последовательность, выбранную из группы, состоящей из SEQ ID NO: 1, 3-11, 44, 46 и 48-50 или нуклеиновой кислоты или клетки-хозяина, экспрессирующей указанный белок или пептид. Предпочтительно, антитело связывается с раковыми клетками, в частности, клетками типов рака, указанных выше, и, предпочтительно, не связывается значительно с нераковыми клетками.
Предпочтительно, связывание антитела, обладающего способностью связываться с CLDN18.2, с клетками, экспрессирующими CLDN18.2, индуцирует или опосредует уничтожение клеток, экспрессирующих CLDN18.2. Клетки, экспрессирующие CLDN18.2, представляют собой раковые клетки и, в частности, выбраны из группы, состоящей из опухолегенных клеток рака желудка, пищевода, поджелудочной железы, легких, яичника, толстой кишки, печени, головы и шеи, и желчного пузыря. Предпочтительно, антитело индуцирует или опосредует уничтожение клеток путем индукции одного или более из лизиса, опосредованного зависимой от комплемента цитотоксичностью (CDC), лизиса, опосредованного зависимой от антитела клеточной цитотоксичностью (ADCC), апоптоза и ингибирования пролиферации клеток, экспрессирующих CLDN18.2. Предпочтительно, ADCC-опосредованный лизис клеток происходит в присутствии эффекторных клеток, которые в конкретных вариантах осуществления выбраны из группы, состоящей из моноцитов, мононуклеарных клеток, NK-клеток и PMN. Ингибирование пролиферации клеток может быть измерено in vitro путем определения пролиферации клеток в анализе с использованием бромдеоксиуридина (5-бром-2-деоксиуридина, BrdU). BrdU представляет собой синтетический нуклеозид, который является аналогом тимидина и может быть включен во вновь синтезированную ДНК реплицирующих клеток (во время S-фазы клеточного цикла), заменяя тимидин во время репликации ДНК. Обнаружение включенного химического соединения с использованием, например, антител, специфических в отношении BrdU, указывает на клетки, которые активно реплицировали их ДНК.
В предпочтительных вариантах осуществления антитела, описанные здесь, могут быть охарактеризованы одним или нескольким из следующих свойств:
a) специфичность в отношении CLDN18.2;
b) аффинность связывания с CLDN18.2, равная примерно 100 нМ или меньше, предпочтительно, примерно 5-10 нМ или меньше, и, более предпочтительно, примерно 1-3 нМ или меньше;
c) способность индуцировать или опосредовать CDC на CLDN18.2-положительных клетках;
d) способность индуцировать или опосредовать ADCC на CLDN18.2-положительных клетках;
e) способность ингибировать рост CLDN18.2-положительных клеток;
f) способность индуцировать апоптоз CLDN18.2-положительных клеток.
В особенно предпочтительном варианте осуществления антитело, обладающее способностью связываться с CLDN18.2, продуцируется гибридомой, депонированной в DSMZ (Mascheroder Weg 1b, 31824 Braunschweig, Germany; new address: Inhoffenstr. 7B, 31824 Braunschweig, Germany) и имеющей следующее назначение и номер доступа:
a. 182-D1106-055, номер доступа DSM ACC2737, депонировано 19 октября 2005 года
b. 182-D1106-056, номер доступа DSM ACC2738, депонировано 19 октября 2005 года
c. 182-D1106-057, номер доступа DSM ACC2739, депонировано 19 октября 2005 года
d. 182-D1106-058, номер доступа DSM ACC2740, депонировано 19 октября 2005 года
e. 182-D1106-059, номер доступа DSM ACC2741, депонировано 19 октября 2005 года
f. 182-D1106-062, номер доступа DSM ACC2742, депонировано 19 октября 2005 года,
g. 182-D1106-067, номер доступа DSM ACC2743, депонировано 19 октября 2005 года
h. 182-D758-035, номер доступа DSM ACC2745, депонировано 17 ноября 2005 года
i. 182-D758-036, номер доступа DSM ACC2746, депонировано 17 ноября 2005 года
j. 182-D758-040, номер доступа DSM ACC2747, депонировано 17 ноября 2005 года
k. 182-D1106-061, номер доступа DSM ACC2748, депонировано 17 ноября 2005 года
l. 182-D1106-279, номер доступа DSM ACC2808, депонировано 26 октября 2006 года
m. 182-D1106-294, номер доступа DSM ACC2809, депонировано 26 октября 2006 года
n. 182-D1106-362, номер доступа DSM ACC2810, депонировано 26 октября 2006 года.
Предпочтительные антитела в соответствии с изобретением представляют собой антитела, продуцируемые или получаемые из описанных выше гибридом, то есть 37G11 в случае 182-D1106-055, 37Н8 в случае 182-D1106-056, 38G5 в случае 182-D1106-057, 38Н3 в случае 182-D1106-058, 39F11 в случае 182-01106-059, 43А11 в случае 182-D1106-062, 61С2 в случае 182-D1106-067, 26В5 в случае 182-D758-035, 26D12 в случае 182-D758-036, 28D10 в случае 182-D758-040, 42Е12 в случае 182-D1106-061, 125Е1 в случае 182-D1106-279, 163Е12 в случае 182-D1106-294, и 175D10 в случае 182-D1106-362; и их химеризованные и гуманизированные формы.
Предпочтительные химеризованные антитела и их последовательности показаны в следующей таблице.
В предпочтительных вариантах осуществления антитела, в частности, химеризованные формы антител в соответствии с изобретением включают антитела, включающие константную область (СН) тяжелых цепей, включающую аминокислотную последовательность константной области тяжелых цепей иммуноглобулина человека, такую как аминокислотная последовательность, представленная SEQ ID NO: 13 или ее фрагментом. В следующих предпочтительных вариантах осуществления антитела, в частности, химерные формы антител в соответствии с изобретением включают антитела, включающие константную область (CL) легких цепей, включающую аминокислотную последовательность константной области легких цепей иммуноглобулина человека, такую как аминокислотная последовательность, представленная SEQ ID NO: 12 или ее фрагментом. В особенно предпочтительном варианте осуществления антитела, в частности, химерные формы антител в соответствии с изобретением включают антитела, которые включают СН, включающую аминокислотную последовательность СН человека, такую как аминокислотная последовательность, представленная SEQ ID NO: 13 или ее фрагментом, и которые включают CL, включающую аминокислотную последовательность CL человека, такую как аминокислотная последовательность, представленная SEQ ID NO: 12 или ее фрагментом.
В одном варианте осуществления антитело, обладающее способностью связываться с CLDN18.2, представляет собой химерное мышиное/человеческое IgG1 моноклональное антитело, содержащее каппа, мышиную вариабельную легкую цепь, человеческую константную область каппа легкой цепи аллотипа Km(3), мышиную вариабельную область тяжелой цепи, человеческую константную область IgG1 аллотип G1m(3).
В специфических предпочтительных вариантах осуществления химерные формы антител включают антитела, включающие тяжелую цепь, включающую аминокислотную последовательность, выбранную из группы, состоящей из SEQ ID NO: 14, 15, 16, 17, 18, 19 и ее фрагмента, и/или включающую легкую цепь, включающую аминокислотную последовательность, выбранную из группы, состоящей из SEQ ID NO: 20, 21, 22, 23, 24, 25, 26, 27, 28 или ее фрагмента.
В специфических предпочтительных вариантах осуществления химерные формы антител включают антитела, включающие комбинацию тяжелых цепей и легких цепей, выбранных из следующих возможных вариантов от (i) до (ix), приведенных ниже:
(i) тяжелая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 14 или ее фрагментом, и легкая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 21 или ее фрагментом;
(ii) тяжелая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 15 или ее фрагментом, и легкая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 20 или ее фрагментом;
(iii) тяжелая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 16 или ее фрагментом, и легкая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 22 или ее фрагментом;
(iv) тяжелая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 18 или ее фрагментом, и легкая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 25 или ее фрагментом;
(v) тяжелая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 17 или ее фрагментом, и легкая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 24 или ее фрагментом;
(vi) тяжелая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 19 или ее фрагментом, и легкая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 23 или ее фрагментом;
(vii) тяжелая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 19 или ее фрагментом, и легкая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 26 или ее фрагментом;
(viii) тяжелая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 19 или ее фрагментом, и легкая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 27 или ее фрагментом;
(ix) тяжелая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 19 или ее фрагментом, и легкая цепь включает аминокислотную последовательность, представленную SEQ ID NO: 28 или ее фрагментом.
Антитело в соответствии с (v) является особенно предпочтительным.
Термин «фрагмент» или «фрагмент аминокислотной последовательности», используемый выше, относится к части последовательности антитела, например, к последовательности, которая представлена последовательностью антитела, укороченной на N- и/или С-конце, которая, если заменяет указанную последовательность антитела, то сохраняет связывание указанного антитела с CLDN18.2 и предпочтительно функции указанного антитела, как описано здесь, например, CDC-опосредованный лизис или ADCC-опосредованный лизис. Предпочтительно, фрагмент аминокислотной последовательности включает, по меньшей мере, 80%, предпочтительно, по меньшей мере, 90%, 95%, 96%, 97%, 98% или 99% аминокислотных остатков указанной аминокислотной последовательности. Фрагмент аминокислотной последовательности, выбранный из группы, состоящей из SEQ ID NOs: 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27 и 28, предпочтительно относится к указанной последовательности, с N-конца которой удалены 17, 18, 19, 20, 21, 22 или 23 аминокислоты.
В предпочтительном варианте осуществления антитело, обладающее способностью связываться с CLDN18.2, включает вариабельную область тяжелых цепей (VH), включающую аминокислотную последовательность, выбранную из группы, состоящей из SEQ ID NO: 29, 30, 31, 32, 33, 34 и ее фрагмента.
В предпочтительном варианте осуществления антитело, обладающее способностью связываться с CLDN18.2, включает вариабельную область легких цепей (VL), включающую аминокислотную последовательность, выбранную из группы, состоящей из SEQ ID NO: 35, 36, 37, 38, 39, 40, 41, 42, 43 и ее фрагмента.
В специфических предпочтительных вариантах осуществления антитело, обладающее способностью связываться с CLDN18.2, включает комбинацию вариабельной области тяжелых цепей (VH) и вариабельную область легких цепей (VL), выбранную из возможных вариантов от (i) до (ix), приведенных ниже:
(i) VH включает аминокислотную последовательность, представленную SEQ ID NO: 29 или ее фрагментом, и VL включает аминокислотную последовательность, представленную SEQ ID NO: 36 или ее фрагментом;
(ii) VH включает аминокислотную последовательность, представленную SEQ ID NO: 30 или ее фрагментом, и VL включает аминокислотную последовательность, представленную SEQ ID NO: 35 или ее фрагментом;
(iii) VH включает аминокислотную последовательность, представленную SEQ ID NO: 31 или ее фрагментом, и VL включает аминокислотную последовательность, представленную SEQ ID NO: 37 или ее фрагментом;
(iv) VH включает аминокислотную последовательность, представленную SEQ ID NO: 33 или ее фрагментом, и VL включает аминокислотную последовательность, представленную SEQ ID NO: 40 или ее фрагментом;
(v) VH включает аминокислотную последовательность, представленную SEQ ID NO: 32 или ее фрагментом, и VL включает аминокислотную последовательность, представленную SEQ ID NO: 39 или ее фрагментом;
(vi) VH включает аминокислотную последовательность, представленную SEQ ID NO: 34 или ее фрагментом, и VL включает аминокислотную последовательность, представленную SEQ ID NO: 38 или ее фрагментом;
(vii) VH включает аминокислотную последовательность, представленную SEQ ID NO: 34 или ее фрагментом, и VL включает аминокислотную последовательность, представленную SEQ ID NO: 41 или ее фрагментом;
(viii) VH включает аминокислотную последовательность, представленную SEQ ID NO: 34 или ее фрагментом, и VL включает аминокислотную последовательность, представленную SEQ ID NO: 42 или ее фрагментом;
(ix) VH включает аминокислотную последовательность, представленную SEQ ID NO: 34 или ее фрагментом, и VL включает аминокислотную последовательность, представленную SEQ ID NO: 43 или ее фрагментом.
Антитело в соответствии с (v) является особенно предпочтительным.
В предпочтительном варианте осуществления антитело, обладающее способностью связываться с CLDN18.2, включает VH, включающую набор участков, определяющих комплементарность, CDR1, CDR2 и CDR3, выбранных из следующих вариантов от (i) до (vi):
(i) CDR1: положения 45-52 SEQ ID NO: 14, CDR2: положения 70-77 SEQ ID NO: 14, CDR3: положения 116-125 SEQ ID NO: 14;
(ii) CDR1: положения 45-52 SEQ ID NO: 15, CDR2: положения 70-77 SEQ ID NO: 15, CDR3: положения 116-126 SEQ ID NO: 15;
(iii) CDR1: положения 45-52 SEQ ID NO: 16, CDR2: положения 70-77 SEQ ID NO: 16, CDR3: положения 116-124 SEQ ID NO: 16;
(iv) CDR1: положения 45-52 SEQ ID NO: 17, CDR2: положения 70-77 SEQ ID NO: 17, CDR3: положения 116-126 SEQ ID NO: 17;
(v) CDR1: положения 44-51 SEQ ID NO: 18, CDR2: положения 69-76 SEQ ID NO: 18, CDR3: положения 115-125 SEQ ID NO: 18; и
(vi) CDR1: положения 45-53 SEQ ID NO: 19, CDR2: положения 71-78 SEQ ID NO: 19, CDR3: положения 117-128 SEQ ID NO: 19.
В предпочтительном варианте осуществления антитело, обладающее способностью связываться с CLDN18.2, включает VL, включающую набор участков, определяющих комплементарность, CDR1, CDR2 и CDR3, выбранных из следующих вариантов от (i) до (ix):
(i) CDR1: положения 47-58 SEQ ID NO: 20, CDR2: положения 76-78 SEQ ID NO: 20, CDR3: положения 115-123 SEQ ID NO: 20;
(ii) CDR1: положения 49-53 SEQ ID NO: 21, CDR2: положения 71-73 SEQ ID NO: 21, CDR3: положения 110-118 SEQ ID NO: 21;
(iii) CDR1: положения 47-52 SEQ ID NO: 22, CDR2: положения 70-72 SEQ ID NO: 22, CDR3: положения 109-117 SEQ ID NO: 22;
(iv) CDR1: положения 47-58 SEQ ID NO: 23, CDR2: положения 76-78 SEQ ID NO: 23, CDR3: положения 115-123 SEQ ID NO: 23;
(v) CDR1: положения 47-58 SEQ ID NO: 24, CDR2: положения 76-78 SEQ ID NO: 24, CDR3: положения 115-123 SEQ ID NO: 24;
(vi) CDR1: положения 47-58 SEQ ID NO: 25, CDR2: положения 76-78 SEQ ID NO: 25, CDR3: положения 115-122 SEQ ID NO: 25;
(vii) CDR1: положения 47-58 SEQ ID NO: 26, CDR2: положения 76-78 SEQ ID NO: 26, CDR3: положения 115-123 SEQ ID NO: 26;
(viii) CDR1: положения 47-58 SEQ ID NO: 27, CDR2: положения 76-78 SEQ ID NO: 27, CDR3: положения 115-123 SEQ ID NO: 27; и
(ix) CDR1: положения 47-52 SEQ ID NO: 28, CDR2: положения 70-72 SEQ ID NO: 28, CDR3: положения 109-117 SEQ ID NO: 28.
В предпочтительном варианте осуществления антитело, обладающее способностью связываться с CLDN18.2, включает комбинацию VH и VL, каждая из которых включает набор участков, определяющих комплементарность, CDR1, CDR2 и CDR3, выбранных из следующих вариантов от (i) до (ix):
(i) VH: CDR1: положения 45-52 SEQ ID NO: 14, CDR2: положения 70-77 SEQ ID NO: 14, CDR3: положения 116-125 SEQ ID NO: 14, VL: CDR1: положения 49-53 SEQ ID NO: 21, CDR2: положения 71-73 SEQ ID NO: 21, CDR3: положения 110-118 SEQ ID NO: 21;
(ii) VH: CDR1: положения 45-52 SEQ ID NO: 15, CDR2: положения 70-77 SEQ ID NO: 15, CDR3: положения 116-126 SEQ ID NO: 15, VL: CDR1: положения 47-58 SEQ ID NO: 20, CDR2: положения 76-78 SEQ ID NO: 20, CDR3: положения 115-123 SEQ ID NO: 20;
(iii) VH: CDR1: положения 45-52 SEQ ID NO: 16, CDR2: положения 70-77 SEQ ID NO: 16, CDR3: положения 116-124 SEQ ID NO: 16, VL: CDR1: положения 47-52 SEQ ID NO: 22, CDR2: положения 70-72 SEQ ID NO: 22, CDR3: положения 109-117 SEQ ID NO: 22;
(iv) VH: CDR1: положения 44-51 SEQ ID NO: 18, CDR2: положения 69-76 SEQ ID NO: 18, CDR3: положения 115-125 SEQ ID NO: 18, VL: CDR1: положения 47-58 SEQ ID NO: 25, CDR2: положения 76-78 SEQ ID NO: 25, CDR3: положения 115-122 SEQ ID NO: 25;
(v) VH: CDR1: положения 45-52 SEQ ID NO: 17, CDR2: положения 70-77 SEQ ID NO: 17, CDR3: положения 116-126 SEQ ID NO: 17, VL: CDR1: положения 47-58 SEQ ID NO: 24, CDR2: положения 76-78 SEQ ID NO: 24, CDR3: положения 115-123 SEQ ID NO: 24;
(vi) VH: CDR1: положения 45-53 SEQ ID NO: 19, CDR2: положения 71-78 SEQ ID NO: 19, CDR3: положения 117-128 SEQ ID NO: 19, VL: CDR1: положения 47-58 SEQ ID NO: 23, CDR2: положения 76-78 SEQ ID NO: 23, CDR3: положения 115-123 SEQ ID NO: 23;
(vii) VH: CDR1: положения 45-53 SEQ ID NO: 19, CDR2: положения 71-78 SEQ ID NO: 19, CDR3: положения 117-128 SEQ ID NO: 19, VL: CDR1: положения 47-58 SEQ ID NO: 26, CDR2: положения 76-78 SEQ ID NO: 26, CDR3: положения 115-123 SEQ ID NO: 26;
(viii) VH: CDR1: положения 45-53 SEQ ID NO: 19, CDR2: положения 71-78 SEQ ID NO: 19, CDR3: положения 117-128 SEQ ID NO: 19, VL: CDR1: положения 47-58 SEQ ID NO: 27, CDR2: положения 76-78 SEQ ID NO: 27, CDR3: положения 115-123 SEQ ID NO: 27, и
(ix) VH: CDR1: положения 45-53 of SEQ ID NO: 19, CDR2: положения 71-78 of SEQ ID NO: 19, CDR3: положения 117-128 SEQ ID NO: 19, VL: CDR1: положения 47-52 SEQ ID NO: 28, CDR2: положения 70-72 SEQ ID NO: 28, CDR3: положения 109-117 SEQ ID NO: 28.
В следующих предпочтительных вариантах осуществления антитело, обладающее способностью связываться с CLDN18.2, предпочтительно включает один или несколько участков, определяющих комплементарность, (CDR), предпочтительно, по меньшей мере, CDR3-вариабельный участок, вариабельных областей тяжелых цепей (VH) и/или вариабельных областей легких цепей (VL) моноклонального антитела против CLDN18.2, предпочтительно моноклонального антитела против CLDN18.2, описанного здесь, и предпочтительно включает один или несколько участков, определяющих комплементарность, (CDR), предпочтительно, по меньшей мере, CDR3-вариабельный участок, вариабельных областей тяжелых цепей (VH) и вариабельных областей легких цепей (VL), описанные в настоящем документе. В одном варианте осуществления один или несколько участков, определяющих комплементарность, (CDR) выбраны из набора описанных здесь участков, определяющих комплементарность, CDR1, CDR2 и CDR3. В особенно предпочтительном варианте осуществления антитело, обладающее способностью связываться с CLDN18.2, предпочтительно включает участки, определяющие комплементарность, CDR1, CDR2 и CDR3 вариабельной области тяжелых цепей (VH) и/или вариабельной области легких цепей (VL) моноклонального антитела против CLDN18.2, предпочтительно моноклонального антитела против CLDN18.2, описанного здесь, и предпочтительно включает участки, определяющие комплементарность, CDR1, CDR2 и CDR3 вариабельных областей тяжелых цепей (VH) и/или вариабельных областей легких цепей (VL), описанных здесь.
В одном варианте осуществления антитело, включающее один или более CDR, набор CDR или комбинацию наборов CDR, как описано здесь, включает указанные CDR вместе с их промежуточными каркасными участками. Предпочтительно, эта часть также будет включать, по меньшей мере, около 50% любого из первого и четвертого каркасных участков или обоих вместе, при этом 50% являются 50% С-концевой части первого каркасного участка и 50% N-концевой части четвертого каркасного участка. Конструирование антитела, осуществленное с помощью методик рекомбинантных ДНК, может приводить к введению остатков на N- или С-конце в вариабельных областях, кодируемых линкерами, введенными для того, чтобы облегчить клонирование или другие стадии обработки, включающие введение линкеров для присоединения вариабельных областей настоящего изобретения к дополнительным белковым последовательностям, включающим тяжелые цепи иммуноглобулинов, другие вариабельные домены (например, при получении димерных антител) или белковые метки.
В одном варианте осуществления антитело, включающее один или более CDR, набор CDR или комбинацию наборов CDR, как описано здесь, включает указанные CDR в каркасном участке антитела человека.
Ссылка в тексте на антитело, включающее в их тяжелой цепи специфическую цепь или специфический участок или последовательность, предпочтительно относится к случаю, когда все тяжелые цепи указанного антитела включают указанную специфическую цепь, участок или последовательность. Это распространяется и на легкую цепь антитела.
Термин «нуклеиновая кислота», используемый здесь, включает ДНК и РНК. Нуклеиновая кислота может быть одноцепочечной или двухцепочечной, но предпочтительно представляет собой двухцепочечную ДНК.
В соответствии с изобретением термин «экспрессия» применяют в его общем значении, и он включает образование ДНК или РНК и белка/пептида. Указанный термин также включает частичную экспрессию нуклеиновых кислот. Кроме того, экспрессия может проходить временно или стабильно.
Идея изобретения, представленная в настоящем документе, в отношении последовательностей специфических аминокислот, например, тех, которые показаны в перечисленных последовательностях, сформулирована таким образом, что также касается и вариантов указанных специфических последовательностей, которые в результате являются функциональным эквивалентом указанных специфических последовательностей, например, аминокислотных последовательностей, проявляющих свойства, идентичные или похожие на свойства специфических аминокислотных последовательностей. Одно важное свойство состоит в сохранении связывания антитела с его мишенью или в обеспечении эффекторных функций антитела. Предпочтительно, чтобы последовательность, которая является вариантом по отношению к специфической последовательности, в случае, если она заменяет специфическую последовательность в антителе, сохраняла связывание указанного антитела с CLDN18.2 и предпочтительно функции указанного антитела, как описано здесь, например, лизис, опосредованный CDC или ADCC.
Специалистам в данной области следует понимать, в частности, что последовательности CDR, гипервариабельные и вариабельные области можно модифицировать без утраты способности к связыванию с CLDN18.2. Например, CDR-участки будут или идентичны, или высоко гомологичны участкам, описанным в настоящем документе. Под «высокогомологичными» участками подразумевается, что в CDR могут быть произведены от 1 до 5, предпочтительно от 1 до 4, например, от 1 до 3 или 1, или 2 замены. Кроме того, гипервариабельные и вариабельные области могут быть модифицированы таким образом, что они будут демонстрировать значительную гомологию с областями антител, особым образом раскрытыми в настоящем документе.
Для целей настоящего изобретения «варианты» аминокислотной последовательности включают варианты аминокислотных вставок, варианты аминокислотных добавлений, варианты аминокислотных делеций и/или варианты аминокислотных замен. Варианты аминокислотных делеций, которые включают делецию на N-конце и/или С-конце белка, также называются N-концевыми и/или С-концевыми укороченными вариантами.
Варианты аминокислотных вставок включают вставки одной или двух, или более аминокислот в конкретную аминокислотную последовательность. В случае вариантов аминокислотной последовательности, имеющей вставку, один или более аминокислотных остатков являются вставленными в конкретный участок в аминокислотной последовательности, хотя возможна также случайная вставка по данным соответствующего скрининга конечного продукта.
Варианты аминокислотного добавления включают амино- и/или карбокси-концевые слияния одной или более аминокислот, например 1, 2, 3, 5, 10, 20, 30, 50 или более аминокислот.
Варианты аминокилотной делеций характеризуются удалением одной или более аминокислот из последовательности, например, удалением 1, 2, 3, 5, 10, 20, 30, 50 или более аминокислот. Делеций могут быть произведены в любом положении белка.
Варианты аминокислотного замещения характеризуются, по меньшей мере, одним остатком в последовательности, которая удалена, и другим остатком, который вставлен на ее место. Предпочтение отдается модификациям в положениях в аминокислотной последовательности, которые не являются консервативными между гомологичным белками или пептидами, и/или замене аминокислот другими аминокислотами, обладающими сходными свойствами. Предпочтительно, аминокислотные изменения в вариантах белков являются консервативными аминокислотными изменениями, то есть замещениями сходно заряженных или незаряженных аминокислот. Консервативное аминокислотное изменение включает замещение одной из семейства аминокислот, которая относится к их боковым цепям. Аминокислоты естественного происхождения обычно делятся на четыре семейства: кислотные (аспартат, глутамат), основные (лизин, аргинин, гистидин), неполярные (аланин, валин, лейцин, изолейцин, пролин, фенилаланин, метионин, триптофан), и незаряженные полярные (глицин, аспарагин, глутамат, цистеин, серии, треонин, тирозин) аминокислоты. Фенилаланин, триптофан и тирозин иногда классифицируются вместе как ароматические аминокислоты.
Предпочтительно степень сходства, предпочтительно идентичности между заданной аминокислотной последовательностью и аминокислотной последовательностью, которая является вариантом указанной заданной аминокислотной последовательности, будет составлять, по меньшей мере, 60%, 65%, 70%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% или 99%. Степень сходства или идентичности представлена предпочтительно для участка аминокислоты, который составляет по меньшей мере примерно 10%, по меньшей мере примерно 20%, по меньшей мере примерно 30%, по меньшей мере примерно 40%, по меньшей мере примерно 50%, по меньшей мере примерно 60%, по меньшей мере примерно 70%, по меньшей мере примерно 80%, по меньшей мере примерно 90% или по меньшей мере примерно 100% от общей длины референсной аминокислотной последовательности. Например, если референсная аминокислотная последовательность состоит из 200 аминокислот, степень сходства или идентичности представлена для по меньшей мере 20, по меньшей мере 40, по меньшей мере 60, по меньшей мере 80, по меньшей мере 100, по меньшей мере 120, по меньшей мере 140, по меньшей мере 160, по меньшей мере 180 или по меньшей мере 200 аминокислот, предпочтительно непрерывных аминокислот. В предпочтительных вариантах осуществления степень сходства или идентичности представлена для полной длины референсной аминокислотной последовательности. Выравнивание для определения сходства последовательности, предпочтительно идентичности последовательности может быть сделано с помощью известных в данной области инструментов, предпочтительно с использованием наилучшего выравнивания последовательности, например, с использованием Align, с использованием стандартных наборов, предпочтительно EMBOSS::needle, Matrix: Blosum62, Gap Open 10.0, Gap Extend 0.5.
«Сходство последовательностей» показывает процент аминокислот, которые или идентичны, или которые представлены консервативными аминокислотными заменами. «Идентичность последовательностей» между двумя аминокислотным последовательностями показывает процент аминокислот, которые идентичны в последовательностях.
Термин «процент идентичности» предназначен для обозначения процента аминокислотных остатков, которые идентичны между двумя последовательностями, подлежащими сравнению, полученный после наилучшего выравнивания, этот процент является только статистическим и разницу между двумя последовательностями распределяют случайным образом и по всей их длине. Сравнение последовательностей между двумя аминокислотными последовательностями обычно проводят путем сравнения этих последовательностей после проведения их оптимального выравнивания, при этом указанное сравнение проводят с помощью сегмента или «окна сравнения» для идентификации и сравнения локальных участков сходства последовательностей. Оптимальное выравнивание последовательностей для сравнения можно получить не только вручную, но и с помощью алгоритма локальной гомологии согласно Smith and Waterman, 1981, Ads App. Math. 2, 482, с помощью алгоритма локальной гомологии согласно Neddleman and Wunsch, 1970, J. Mol. Biol. 48, 443, с помощью метода поиска сходства согласно Pearson and Lipman, 1988, Proc. Natl Acad. Sci. USA 85, 2444 или с помощью компьютерных программ, в которых используют алгоритмы (GAP, BESTFIT, FASTA, BLAST Р, BLAST N и TFASTA в пакете программного обеспечения Wisconsin Genetics Software Package, Genetics Computer Group, 575 Science Drive, Madison, Wis.).
Процент идентичности рассчитывают с помощью определения количества идентичных положений в двух сравниваемых последовательностях, делят это количество на количество сравниваемых положений и умножают полученный результат на 100, чтобы найти процент идентичности между этими двумя последовательностями.
Термин «трансгенное животное» относится к животному, имеющему геном, включающий один или несколько трансгенов, предпочтительно трансгенов тяжелых и/или легких цепей, или трансхромосомы (интегрированные или не интегрированные в природную геномную ДНК животного), и которое предпочтительно способно экспрессировать трансгены. Например, трансгенная мышь может обладать трансгеном легкой цепи иммуноглобулина человека и как трансгеном тяжелой цепи иммуноглобулина человека, так и трансхромосомой тяжелой цепи иммуноглобулина человека, так что мышь продуцирует человеческие анти-CLDN18.2 антитела, если ее иммунизируют антигеном CLDN18.2 и/или клетками, экспрессирующими CLDN18.2. Трансген тяжелых цепей иммуноглобулина человека может быть интегрирован в хромосомальную ДНК мыши, как и в случае трансгенных мышей, например, мышей HuMAb, таких как мыши НСо7 или НСо12, или трансген тяжелых цепей иммуноглобулина человека может сохраняться вне хромосом, как в случае трансхромосомных мышей (например, КМ), описанных в патенте WO 02/43478. Такие трансгенные и трансхромосомальные мыши могут быть способны к продуцированию множественных изотипов моноклональных антител человека к CLDN18.2 (например, IgG, IgA и/или IgE) после V-D-J-рекомбинации и переключения изотипов.
«Сокращение», «уменьшение» или «ингибирование», используемый здесь, означает общее уменьшение или способность вызывать общее уменьшение, предпочтительно на 5% или более, 10% или более, 20% или более, более предпочтительно 50% или более, и наиболее предпочтительно 75% или более, уровня, например, уровня экспрессии или уровня пролиферации клеток.
Термины, такие как «увеличение» или «усиление» предпочтительно относятся к увеличению или усилению, по меньшей мере на 10%, предпочтительно по меньшей мере на 20%, предпочтительно по меньшей мере на 30%, более предпочтительно по меньшей мере на 40%, более предпочтительно по меньшей мере на 50%, даже более предпочтительно по меньшей мере на 80%, и наиболее предпочтительно по меньшей мере на 100%, по меньшей мере на 200%, по меньшей мере на 500%, по меньшей мере на 1000%, по меньшей мере на 10000% или даже более.
Механизм действия mAb
Хотя ниже представлен анализ механизмов, которые лежат в основе терапевтической эффективности антител изобретения, его ни в коей мере не следует рассматривать как ограничение изобретения.
Антитела, описанные в настоящем документе, предпочтительно взаимодействуют с компонентами иммунной системы, предпочтительно посредством ADCC и CDC. Антитела изобретения могут быть использованы для целенаправленной доставки различных агентов (например, радиоизотопов, лекарственных средств или токсинов) с целью непосредственного уничтожения опухолевых клеток и могут быть использованы синергически с обычными химиотерапевтическими средствами, воздействующими на опухоль через дополнительные механизмы действия, которые могут включать противоопухолевые иммунные ответы, которые могут быть опасными из-за цитотоксических побочных эффектов химиотерапевтических средств на Т-лимфоциты. Однако антитела, описанные здесь, могут также проявлять эффект просто путем связывания с CLDN18.2 на клеточной поверхности, таким образом, например, блокируя пролиферацию клеток.
Зависимая от антител клеточно-опосредованная цитотоксичность
ADCC описывает способность эффекторных клеток, в частности лимфоцитов, для которых предпочтительно необходимо, чтобы клетка-мишень была помечена антителом, вызывать лизис клеток, как описано в настоящем документе.
ADCC предпочтительно происходит, если антитела связываются с антигенами на опухолевых клетках и Fc-домены антитела контактируют с Fc-рецепторами (FcR) на поверхности иммунных эффекторных клеток. Установлено несколько семейств Fc-рецепторов, и специфические популяции клеток обычно экспрессируют определенные Fc-рецепторы. ADCC можно рассматривать как механизм прямой индукции непосредственного разрушения опухоли в различной степени, которая приводит к презентации антигенов и индукции направленных на опухоль Т-клеточных ответов. Предпочтительно, индукция ADCC in vivo будет приводить к направленным на опухоль Т-клеточным ответам и образованию антител в организме хозяина.
Комплемент-зависимая цитотоксичность
CDC представляет собой другой способ лизиса клеток, который может контролироваться антителами. IgM представляет собой наиболее эффективный изотип для активации комплемента. Оба иммуноглобулина IgG1 и IgG3 также высокоэффективны при направлении CDC по классическому пути активации комплемента. Предпочтительно, в этом каскаде образование комплексов антиген-антитело приводит к демаскированию множественных участков связывания C1q в непосредственной близости от CH2-доменов участвующих молекул антител, таких как молекулы IgG (C1q представляет собой один из трех субкомпонентов комплемента С1). Предпочтительно эти демаскированные участки связывания C1q превращают ранее низкоаффинное взаимодействие C1q-IgG во взаимодействие с высокой авидностью, которое запускает каскад событий, включающих серию белков комплемента и приводит к протеолитическому высвобождению хемотаксических/активирующих веществ С3а и С5а эффектных клеток. Предпочтительно, комплементный каскад заканчивается образованием мембраноатакующего комплекса, который создает поры в клеточной мембране, облегчающие свободное перемещение воды и растворенных веществ внутрь клетки и из клетки.
Антитела изобретения можно продуцировать с помощью разнообразных методов, включающих общепринятые методы создания моноклональных антител, например, стандартную методику гибридизации соматических клеток по Kohler and Milstein, Nature 256: 495 (1975). Хотя процедуры гибридизации соматических клеток являются предпочтительными, в принципе, можно применять и другие способы получения моноклональных антител, например, вирусную или онкогенную трансформацию В-лимфоцитов или методы фагового дисплея с использованием библиотеки генов антител.
Предпочтительная животная модель для получения гибридом, которые секретируют моноклональные антитела представляет собой систему с использованием мышей. Получение гибридом у мышей является очень надежной процедурой. Протоколы иммунизации и способы выделения иммунизированных спленоцитов, предназначенных для слияния, известны в данной области. Партнеры по слиянию (например, клетки мышиной миеломы) и способы слияния также известны.
Другими предпочтительные животные системы для получения гибридом, которые секретируют моноклональные антитела, представляют собой системы с использованием крыс и кроликов (например, описанные в работе Spieker-Polet et al., Proc. Natl. Acad. Sci. U.S.A. 92: 9348 (1995), смотри также работу Rossi et al., Am. J. Clin. Pathol. 124: 295 (2005)).
В еще одном предпочтительном варианте осуществления человеческие моноклональные антитела можно создать с помощью трансгенных или трансхромосомных мышей, включающих в большей степени компоненты иммунной системы человека, а не мышей. Такие трансгенные и трансхромосомные мыши включают мышей, известных как мыши HuMAb и КМ, соответственно, и обобщенно называются здесь как «трансгенные мыши». Образование антител человека в таких трансгенных мышах можно проводить, как было подробно описано для CD20 в WO2004 035607.
Еще одной стратегией для образования моноклональных антител является непосредственное выделение генов, кодирующих антитела, из лимфоцитов, продуцирующих антитела определенной специфичности, например, смотри работу Babcock et al., 1996; A novel strategy for generating monoclonal antibodies from single, isolated lymphocytes producing antibodies of defined specificities. Более подробную информацию о конструировании рекомбинантных антител смотри также в работах Welschof и Kraus, Recombinant antibodes for cancer therapy ISBN-0-89603-918-8 и Benny K.C. Lo Antibody Engineering ISBN 1-58829-092-1.
Чтобы вызвать образование антител, мышей можно иммунизировать конъюгированными с носителем пептидами из последовательности антигена, то есть последовательности, против которой направлены антитела, обогащенным препаратом рекомбинантно экспрессированного антигена или их фрагментами и/или клетками, экспрессирующими антиген, как было описано. Альтернативно, мышей можно иммунизировать ДНК, кодирующей антиген, или ее фрагментами. В случае, если иммунизации с использованием очищенного или обогащенного препарата антигена не дает результата, чтобы вызвать иммунный ответ, мышей также можно иммунизировать клетками, экспрессирующими антиген, например, клеточной линией.
За иммунным ответом можно следить путем ведения протокола иммунизации, образцы плазмы и сыворотки отбирают из хвостовой вены или с помощью ретроорбитального кровотечения. Клетки мышей с достаточно высоким титром иммуноглобулина могут быть использованы для слияния. Мышей за 3 дня перед умерщвлением и удалением селезенки можно подвергнуть бустерной иммунизации путем внутрибрюшинной или внутривенной инъекции антиген-экспрессирующих клеток, чтобы увеличить размер гибридом, секретирующих специфическое антитело.
Для создания гибридом, продуцирующих моноклональные антитела, клетки спленоцитов и лимфатических узлов иммунизированных мышей можно выделить и провести слияние с подходящей линией иммортализованных клеток, таких как клеточная линия мышиной миеломы. Затем можно протестировать способность полученных гибридом продуцировать антиген-специфические антитела. Затем с помощью метода ELISA в отдельных лунках можно определить наличие гибридом, секретирующих антитела. С помощью иммунофлуоресценции и FACS-анализа, используя экспрессирующие антиген клетки, можно идентифицировать антитела, специфичные к антигену. Гибридомы, секретирующие антитела, можно снова высеять, снова отсортировать и, если они все еще позитивны в отношении моноклональных антител, субклонировать с помощью метода предельных разведений. Стабильные субклоны затем можно культивировать in vitro, чтобы вызвать образование антитела в тканевой культуральной среде для определения их характеристик.
Антитела также можно получать в трансфекторе клеток хозяина, используя, например, комбинацию методов рекомбинантных ДНК и способов трансфекции генов, которые хорошо известны в данной области (Morrison, S. (1985) Science 229: 1202).
Например, в одном варианте осуществления ген(ы), представляющий интерес например, гены антитела, можно лигировать в экспрессионный вектор, такой как эукариотическая экспрессионная плазмида, например, та, которую применяют в системе экспрессии гена GS, раскрытой в WO 87/04462, WO 89/01036 и ЕР 338 841, или другие экспрессионные системы, хорошо известные в данной области. Очищенную плазмиду с генами клонированного антитела можно вводить в эукариотические клетки-хозяева, такие как СНО-клетки, NS/0-клетки, HEK293T-клетки или HEK293-клетки, или, альтернативно, другие эукариотические клетки, подобные растительным клеткам, клеткам грибов или дрожжей. Способ, применяемый для введения этих генов, может быть одним из методов, описанных в данной области, например, таких как электропорация, применение липофектина, липофектамина или др. После введения этих генов антител в клетки-хозяева, клетки, экспрессирующие антитело, могут быть идентифицированы и отобраны. Эти клетки представляют собой трансфектомы, которые затем могут быть размножены до экспрессионного уровня и выращены в укрупненном масштабе для получения антител. Рекомбинатные антитела могут быть выделены и очищены из культуральных супернатантов и/или клеток.
Альтернативно, клонированные гены антител могут быть экспрессированы в других экспрессионных системах, включающих прокариотические клетки, такие как микроорганизмы, например, Е. Coli. Кроме того, антитела могут быть получены в трансгенных животных, которые не являются человеком, например, в составе овечьего или кроличьего молока или в составе куриных яиц, или в трансгенных растениях; смотри, например, работы Verma, R., et al. (1998) J. Immunol. Meth. 216: 165-181; Pollock, et al. (1999) J. Immunol. Meth. 231: 147-157; и Fischer, R., et al. (1999) Biol. Chem. 380: 825-839.
Химеризация
Мышиные моноклональные антитела можно применять у людей в качестве лечебных антител, если их пометить токсинами или радиоактивными изотопами. Немеченые мышиные антитела при повторном применении высоко иммуногенны для человека, и это приводит к снижению терапевтического эффекта. Основная иммуногенность опосредована константными областями тяжелых цепей. Иммуногенность мышиных антител для человека может быть снижена или полностью устранена, если соответствующие антитела являются химеризованным или гуманизированными. Химерные антитела представляют собой антитела, различные компоненты которых происходят из различных видов животных, например, это антитела, имеющие вариабельную область мышиного антитела и константную область иммуноглобулина человека. Химеризация антител достигается путем присоединения вариабельных областей тяжелой и легкой цепи мышиных антител к константной области тяжелой и легкой цепи антител человека (например, как это описано в работе Kraus et al., in Methods in Molecular Biology series, Recombinant antibodies for cancer therapy ISBN-0-89603-918-8). В предпочтительном варианте осуществления химерные антитела создают путем присоединения константной области легкой каппа-цепи иммуноглобулина человека к вариабельной области легких цепей иммуноглобулина мыши. В также предпочтительном варианте осуществления химерные антитела можно создать путем присоединения константной области легкой лямбда-цепи иммуноглобулина человека к вариабельной области легких цепей иммуноглобулина мыши. Предпочтительными константными областями тяжелых цепей для создания химерных антител являются IgG1, IgG3 и IgG4. Другими предпочтительными константными областями тяжелых цепей для создания химерных антител являются IgG2, IgA, IgD и IgM.
Гуманизация
Антитела взаимодействуют с антигенами-мишенями преимущественно с помощью аминокислотных остатков, которые расположены в шести участках, определяющих комплементарность, (CDR) тяжелых и легких цепей. Поэтому аминокислотные последовательности участков CDR среди индивидуальных антител различаются сильнее, чем последовательности, расположенные вне CDR. Так как последовательности CDR отвечают за большую часть взаимодействий антитело-антиген, возможно экспрессировать рекомбинантные антитела, которые имитируют свойства специфических встречающихся в природе антител, с помощью конструирования экспрессионных векторов, включающих последовательности CDR из специфического встречающегося в природе антитела, пересаженные на каркасные последовательности другого антитела с другими свойствами (смотри, например, работы Riechmann, L. et al. (1998) Nature 332: 323-327; Jones, P. et al. (1986) Nature 321: 522-525; и Queen, C. et al. (1989) Proc. Natl. Acad. Sci. U.S.A. 86: 10029-10033). Такие каркасные последовательности могут быть получены из общедоступных баз данных ДНК, которые включают генетические последовательности генов родительского антитела. Эти родительские последовательности будут отличаться от последовательностей гена зрелого антитела, так как они не будут включать полностью укомплектованные изменяемые гены, которые образуются путем соединения V(D)J-воссоединения в процессе созревания В-клеток. Родительские последовательности также будут отличаться от последовательностей высокоаффинных вторичных антител по всей индивидуальной вариабельной области.
Способность антитела связываться с антигеном можно определить с помощью стандартных анализов связывания (например, ELISA, вестерн-блоттинг, иммунофлуоресценция и проточно-цитометрический анализ).
Чтобы очистить антитела, отобранные гибридомы могут быть выращены в двухлитровых вращающихся колбах, предназначенных для очистки моноклональных антител. Альтернативно, антитела могут быть получены в биореакторах диализного типа. Супернатанты можно фильтровать и, при необходимости, концентрировать перед аффинной хроматографией на протеин G-сефарозе или протеин А-сефарозе. Чтобы гарантировать чистоту, элюированные IgG можно контролировать с помощью гель-электрофореза и высокоэффективной жидкостной хроматографии. Буферный раствор можно поменять на PBS, и концентрацию можно определить по оптическому поглощению при 280 нм, используя коэффициент экстинкции, равный 1,43. Моноклональные антитела можно распределить по аликвотам и хранить при -80°С.
Чтобы определить, связываются ли отобранные моноклональные антитела со специфическими эпитопами, можно применять сайт-направленный мутагенез или множественный сайт-направленный мутагенез.
Чтобы определить изотип антител, можно проводить анализы изотипов ELISA с различными коммерческими наборами (например, Zymed, Roche Diagnostics). Лунки микротитровальных планшетов могут быть покрыты антителами к Ig мыши. После блокировки планшетов проводят реакцию с моноклональными антителами или очищенными контролями изотипов при комнатной температуре в течение двух часов. Затем содержимое лунок может взаимодействовать с мышиными IgG1-, IgG2a-, IgG2b- или IgG3-, IgA- или IgM-специфическими образцами, конъюгированными с пероксидазой. После промывания в планшетах при добавлении субстрата ABTS (1 мг/мл) развивается окрашивание, которое анализируют при 405-650 нм. Альтернативно, может быть использован набор IsoStrip Mouse Monoclonal Antibody Isotyping Kit (Roche, Cat. No. 1493027) в соответствии с инструкцией фирмы-производителя.
Чтобы продемонстрировать присутствие антител в сыворотке крови иммунизированных мышей или связывание моноклональных антител с живыми клетками, экспрессирующими антиген, может быть использован проточно-цитометрический анализ. Клеточные линии, экспрессирующие антиген естественным образом или после трансфекции, и отрицательные контроли, лишенные экспрессии антигена (выращенные в стандартных условиях роста), можно смешивать с различными концентрациями моноклональных антител в супернатантах гибридомы или в PBS, содержащем 1% эмбриональную бычью сыворотку (FBS), и инкубировать при 4°С в течение 30 мин. После промывания АРС- или Alexa647-меченое антитело к IgG может связываться с моноклональным антителом, связавшим антиген, в тех же самых условиях окрашивания первичных антител. Образцы можно проанализировать с помощью проточной цитометрии с помощью прибора FACS, используя свойства светового и бокового рассеяния, чтобы пропускать единичные живые клетки. Чтобы отличить антиген-специфические моноклональные антитела от неспецифически связывающих компонентов в единичном измерении, можно применять метод ко-трансфекции. Клетки, временно трансфицированные плазмидами, кодирующими антиген, и флуоресцентным маркером, можно окрашивать, как описано выше. Трансфицированные клетки можно обнаружить в другом флуоресцентном канале, а не там, где детектируют клетки, окрашенные антителами. Так как большинство трансфицированных клеток экспрессируют оба трансгена, антиген-специфические моноклональные антитела предпочтительно связываются с клетками, экспрессирующими флуоресцентный маркер, тогда как неспецифические антитела связываются на сравнимом уровне с нетрансфицированными клетками. В дополнение или вместо проточной цитометрии может быть использован альтернативный анализ с использованием флуоресцентной микроскопии. Клетки могут быть окрашены точно так, как описано выше, и исследованы с помощью флуоресцентной микроскопии.
Чтобы продемонстрировать присутствие антител в сыворотке крови иммунизированных мышей или связывание моноклональных антител с живыми клетками, экспрессирующими антиген, можно применять иммунофлуоресцентную микроскопию. Например, клеточные линии, экспрессирующие антиген естественным образом или после трансфекции, и отрицательные контроли, лишенные экспрессии антигена, выращивают на предметных стеклах с лунками в стандартных условиях роста в среде DMEM/F12, дополненной 10%-ной фетальной телячьей сывороткой (FCS), 2 мМ L-глутамином, 100 МЕд/мл пенициллина и 100 мкг/мл стрептомицина. Клетки затем можно фиксировать метанолом или параформальдегидом или можно оставлять необработанными. Затем можно провести реакцию клеток с моноклональными антителами против антигена в течение 30 мин при 25°С. После промывания в тех же самых условиях можно провести реакцию клеток с Alexa555-мечеными вторичными антителами к IgG мыши (Molecular Probes). Затем клетки можно исследовать с помощью флуоресцентной микроскопии.
Можно получить клеточные экстракты из клеток, экспрессирующих антиген, и подходящие отрицательные контроли и подвергнуть их электрофорезу в полиакриламидном геле в присутствии додецилсульфата натрия (SDS). После электрофореза разделенные антигены переносят на нитроцеллюлозные мембраны, блокируют и исследуют с помощью тестируемых моноклональных антител. Связывание IgG можно обнаружить с помощью антител к IgG мыши, конъюгированных с пероксидазой, и развитию окраски в присутствии субстрата усиленной хемилюминесценции (ECL).
Антитела можно дополнительно протестировать на реактивность по отношению к антигену с помощью иммуногистохимического анализа, который хорошо известен специалистам в данной области, например, используя фиксированные параформальдегидом или ацетоном криосрезы или срезы тканей, залитые парафином, приготовленные из образцов нераковых тканей или раковых тканей, полученных от пациентов во время обычных хирургических операций или полученных от мышей, несущих ксенотрансплантатные опухоли, привитые с помощью клеточных линий, экспрессирующих антиген спонтанно или после трансфекции. Для иммуноокрашивания антитела, реактивные по отношению к антигену, можно инкубировать с козьими антителами к мышиным иммуноглобулинами или с козьими антителами к кроличьим иммуноглобулинам, конъюгированным с пероксидазой хрена (DAKO) в соответствии с инструкциями фирмы-производителя.
Можно тестировать способность антител опосредовать фагоцитоз и лизис клеток, экспрессирующих CLDN18.2. Тестирование активности моноклональных антител in vitro обеспечит начальный скрининг перед тестированием в моделях in vivo.
Зависимая от антител клеточно-опосредованная цитотоксичность (ADCC):
Вкратце, полиморфно-ядерные клетки (PMN), NK-клетки, моноциты, мононуклеарные клетки или другие эффекторные клетки от здоровых доноров можно очистить путем центрифугирования в градиенте плотности Ficol-Hypaque с последующим лизисом примесных эритроцитов. Промытые эффекторные клетки можно суспендировать в RPMI, дополненной 10%-ной инактивированной нагреванием фетальной телячьей сывороткой или, альтернативно, 5%-ной инактивированной нагреванием сывороткой человека, и смешать с 51Cr-мечеными клетками-мишенями, экспрессирующими CLDN18.2, при различном соотношении эффекторных клеток и клеток-мишеней. Альтернативно, клетки-мишени можно пометить лигандом, усиливающим флуоресценцию (BATDA). Сильно флуоресцирующий хелат европия с усиливающим лигандом, который высвобождается мертвыми клетками, можно измерять с помощью флуориметра. В другом альтернативном способе можно применять трансфекцию клеток-мишеней люциферазой. Добавленный Люцифер желтый может затем быть окислен только жизнеспособными клетками. Затем можно добавить очищенные антитела к CLDN18.2 класса IgG в различных концентрациях. Иррелевантные IgG человека могут быть использованы в качестве отрицательного контроля. Анализы можно проводить в течение от 4 до 20 часов при 37°С, в зависимости от типа применяемых эффекторных клеток. Образцы можно анализировать в отношении цитолиза с помощью измерения высвобождения 51Cr или по присутствию хелата EuTDA в культуральном супернатанте. Альтернативно, люминесценция, вызванная окислением Люцифера желтого, может быть показателем жизнеспособных клеток.
Моноклональные антитела к CLDN18.2 также можно тестировать в различных комбинациях, чтобы определить, усиливается ли цитолиз множественными моноклональными антителами.
Зависимая от комплемента цитотоксичность (CDC)
С помощью разнообразных известных методов можно протестировать способность моноклональных антитела к CLDN18.2 опосредовать CDC. Например, сыворотка крови как источник комплемента может быть получена из крови способом, известным специалистам в данной области. Чтобы определить CDC-активность mAb, можно применять различные методы. Например, может быть измерено высвобождение 51Cr или повышенный уровень проницаемости мембраны можно оценивать с помощью анализа проницаемости клеток для пропидия иодида (PI). Вкратце, клетки-мишени можно промыть и инкубировать их в количестве 5×105/мл с различными концентрациями mAb в течение 10-30 мин при комнатной температуре или при 37°С. Затем можно добавить сыворотку или плазму крови в конечной концентрации 20% (об/об) и инкубировать клетки при 37°С в течение 20-30 мин. Все клетки из каждого образца можно добавить к раствору PI в пробирку для FACS. Затем смесь может быть немедленно проанализирована с помощью проточно-цитометрического анализа с использованием прибора FACSArray.
В альтернативном анализе индукцию CDC можно определить на адгезивных клетках. В одном варианте осуществления этого анализа клетки засевают в микротитровальные планшеты для тканевых культур с плоским дном за 24 ч до анализа при плотности 3×104/лунку. На следующий день среду роста удаляют и клетки инкубируют в трех повторностях с антителами. Контрольные клетки инкубируют со средой роста или средой роста, содержащей 0,2%-ный сапонин для определения базального уровня лизиса и максимального лизиса, соответственно. После инкубации в течение 20 мин при комнатной температуре удаляют супернатант и к клеткам добавляют 20%-ную (об/об) плазму или сыворотку крови человека в DMEM (предварительно прогретую до 37°С) и инкубируют еще 20 мин при 37°С. Все клетки из каждого образца можно добавлять к раствору пропидия иодида (10 мкг/мл). Затем супернатанты заменяют PBS, содержащим 2,5 мкг/мл этидий бромида и при возбуждении светом при 520 нм измеряют флуоресценцию при 600 нм с помощью прибора Tecan Safire. Процент специфического лизиса рассчитывают по следующей формуле: % специфического лизиса = (флуоресценция образца - фоновая флуоресценция) / (флуоресценция при максимальном лизисе - фоновая флуоресценция) × 100.
Индукция апоптоза и ингибирование пролиферации клеток моноклональными антителами:
Чтобы протестировать способность вызывать апоптоз, моноклональные антитела к CLDN18.2 можно, например, инкубировать с CLDN18.2-положительными опухолевыми клетками, например, с SNU-16, DAN-G, KATO-III или CLDN18.2-трансфицированными опухолевыми клетками при 37°С в течение примерно 20 часов. Клетки можно собрать, промыть в аннексин-V-связывающем буфере (BD biosciences) и инкубировать с аннексином V, конъюгированным с FITC или АРС (BD biosciences) в течение 15 мин в условиях темноты. Все клетки из каждого образца могут быть добавлены к раствору PI (10 мкг/мл в PBS) в пробирку для FACS и немедленно проанализированы с помощью проточной цитометрии (как описано выше). Альтернативно, общее ингибирование клеточной пролиферации моноклональными антителами можно определить с помощью коммерчески доступных наборов. Набор DELFIA Cell Proliferation Kit (Perkin-Elmer, Cat. No. AD0200) представляет собой неизотопный иммуноанализ, основанный на измерении включения 5-бром-2'-деоксиуридина (BrdU) во время синтеза ДНК пролиферирующими клетками в микропланшетах. Включенный BrdU обнаруживают с помощью моноклонального антитела, меченого европием. Для обеспечения возможности обнаружения антител клетки фиксируют, а ДНК денатурируют с помощью раствора для фиксации. Несвязавшееся антитело удаляют промыванием и добавляют индуктор DELFIA, чтобы обеспечить диссоциацию ионов европия из меченого антитела в раствор, где они образуют интенсивно флуоресцирующие хелаты с компонентами индуктора DELFIA. Измеряемая флуоресценция, использующая при определении флуорометрию с разрешением во времени, пропорциональна уровню синтеза ДНК в клетке каждой лунки.
Доклинические исследования
Моноклональные антитела, которые связываются с CLDN18.2, также могут быть протестированы в модели in vivo (например, у иммунодефицитных мышей, несущих ксенотрансплантатные опухоли, привитые с помощью клеточных линий, экспрессирующих CLDN18.2, например, линий DAN-G, SNU-16 или KATO-III, или после трансфекции, например, HEK293) для определения их эффективности в отношении контроля роста CLDN18.2-экспрессирующих опухолевых клеток.
Исследования in vivo после ксенотрансплантации CLDN18.2-экспрессирующих опухолевых клеток в мышей или других животных с ослабленным иммунитетом можно проводить с использованием антител изобретения. Антитела можно вводить мышам, у которых нет опухолей, а затем с помощью инъекции вводить опухолевые клетки для измерения влияния антитела на предотвращение образования опухолей или симптомов, связанных с опухолями. Антитела можно вводить несущим опухоль мышам для определения терапевтической эффективности соответствующих антител в отношении уменьшения роста опухоли, метастазирования или симптомов, связанных с опухолями. Применение антител можно сочетать с применением других веществ, таких как цитостатические лекарственные средства, ингибиторы фактора роста, блокаторы клеточного цикла, ингибиторы ангиогенеза или другие антитела для определения синергической эффективности и потенциальной токсичности комбинаций. Для анализа токсичных побочных эффектов, опосредованных антителами, животным можно ввести антитела или контрольные реагенты и тщательно исследовать симптомы, которые возможно связаны с терапией антителами к CLDN18.2. Возможные побочные эффекты при применении антител к CLDN18.2 in vivo включают токсичность в CLDN18.2-экспрессирующих тканях, включая желудок. Антитела, распознающие CLDN18.2 у человека и у других видов, например у мышей, особенно полезны для прогнозирования потенциальных побочных эффектов, опосредованных применением моноклональных антител к CLDN18.2 у человека.
Картирование эпитопов, распознаваемых антителами, можно выполнять, как подробно описано в изданиях "Epitope Mapping Protocols (Methods in Molecular Biology) by Glenn E. Morris ISBN-089603-375-9 и "Epitope Mapping: A Practical Approach" Practical Approach Series, 248 by Olwyn M.R. Westwood, Frank C. Hay.
Соединения и агенты, описанные здесь, можно вводить в форме любой подходящей фармацевтической композиции.
Фармацевтические композиции обычно представлены в виде стандартной лекарственной формы и могут быть приготовлены известным способом per se. Например, фармацевтическая композиция может быть представлена в форме раствора или суспензии.
Фармацевтическая композиция может содержать соли, буферные вещества, консерванты, носители, разбавители и/или вспомогательные вещества, все из которых предпочтительно являются фармацевтически приемлемыми. Термин «фармацевтически приемлемый» относится к нетоксичности материала, который не влияет на действие активного компонента фармацевтической композиции.
Соли, которые не являются фармацевтически приемлемыми, могут быть использованы для приготовления фармацевтически приемлемых солей и включены в изобретение. Фармацевтически приемлемые соли этого типа включают неограничивающим образом такие соли, которые получены из следующих кислот: соляная, бромистоводородная, серная, азотная, фосфорная, малеиновая, уксусная, салициловая, лимонная, муравьиная, малоновая, янтарная и т.п. Фармацевтически приемлемые соли могут быть также приготовлены в виде солей щелочных металлов или солей щелочноземельных металлов, таких как соли натрия, соли калия или соли кальция.
Подходящие буферные вещества для использования в фармацевтической композиции включают уксусную кислоту в виде соли, лимонную кислоту в виде соли, борную кислоту в виде соли и фосфорную кислоту в виде соли.
Подходящие консерванты для использования в фармацевтической композиции включают бензалкония хлорид, хлорбутанол, парабен и тимеросал.
Лекарственная форма для инъекции может содержать фармацевтически приемлемое вспомогательное вещество, такое как лактат Рингера.
Термин «носитель» относится к органическому или неорганическому компоненту естественной или синтетической природы, в котором содержится активный компонент для содействия, усиления или обеспечения возможности применения. В соответствии с изобретением термин «носитель» также включает один или несколько совместимых твердых или жидких наполнителей, разбавителей или инкапсулирующих веществ, которые являются подходящими для введения пациенту.
Возможные вещества носителя для парентерального введения представляют собой, например, стерильную воду, раствор Рингреа, лактат Рингреа, стерильный раствор хлорида натрия, полиалкиленгликоли, гидрированные нафталины и, в частности, биосовместимые лактидные полимеры, сополимеры лактид/гликолид или сополимеры полиоксиэтилен/полиоксипропилен.
Термин «вспомогательное вещество» при использовании в настоящем документе означает все вещества, которые могут присутствовать в фармацевтической композиции и которые не являются активными ингредиентами, такие как, например, носители, связующие вещества, скользящие вещества, загустители, поверхностно-активные агенты, консерванты, эмульгаторы, буферы, вкусовые агенты или красители.
Агенты и композиции, описанные здесь, можно вводить любым общепринятым путем, таким как парентеральное введение, включая инъекцию или инфузию. Предпочтительным является парентеральное введение, например, внутривенное, внутриартериальное, подкожное, внутрикожное или внутримышечное.
Композиции, подходящие для парентерального введения, обычно содержат стерильный водный или неводный препарат активного соединения, который предпочтительно является изотоничным крови реципиента. Примерами совместимых носителей и растворителей являются раствор Рингера и изотоничный раствор хлорида натрия. Кроме того, обычно стерильные фиксированные масла используются в качестве раствора или суспензионной среды.
Агенты и композиции, описанные здесь, вводят в эффективных количествах. «Эффективное количество» относится к количеству, которое достигает желательной реакции или желательного эффекта отдельно или вместе с дополнительными дозами. В случае лечения конкретного заболевания или конкретного состояния желательная реакция предпочтительно относится к ингибированию течения заболевания. Это включает замедление развития заболевания и, в частности, прерывание или обратное развитие заболевания. Желательной реакцией при лечении заболевания или состояния может также являться задержка начала или предупреждение начала развития указанного заболевания или указанного состояния.
Эффективное количество агента или композиции, описанной здесь, будет зависеть от состояния, подлежащего лечению, тяжести заболевания, индивидуальных параметров пациента, включая возраст, физиологическое состояние, размер и вес, продолжительность лечения, тип сопутствующей терапии (в случае наличия), определенный способ введения и аналогичные факторы. Соответственно, вводимые дозы агентов, описанных здесь, могут зависеть от таких различных параметров. В случае, если реакция у пациента недостаточна при введении начальной дозы, можно применять более высокие дозы (или эффективно более высокие дозы, достигаемые иным, более локализованным путем введения).
Агенты и композиции, описанные здесь, можно вводить пациентам, например, in vivo, для лечения или предупреждения различных нарушений, таких как описанные здесь. Предпочтительные пациенты включают пациентов-людей, имеющих нарушения, которые могут быть скорректированы или интенсивность которых может быть уменьшена путем введения описанных здесь агентов и композиций. Это включает нарушения, в которые вовлечены клетки, характеризующиеся измененной картиной экспрессии CLDN18.2.
Например, в одном варианте осуществления описанные здесь антитела можно применять для лечения пациента, имеющего раковое заболевание, например, описанное здесь раковое заболевание, характеризующееся присутствием раковых клеток, экспрессирующих CLDN18.2.
Фармацевтические композиции и способы лечения, описанные в соответствии с изобретением, можно также применять для иммунизации или вакцинации для предупреждения описанного здесь заболевания.
Настоящее изобретение далее проиллюстрировано следующими примерами, которые не должны рассматриваться как ограничивающие объем изобретения.
Примеры
Пример 1. Клиническое, впервые проводимое на человеке, с однократным введением, многоцентровое открытое исследование фазы I с эскалацией дозы путем внутривенной (i.v.) инфузии, оценивающее безопасность и переносимость IMAB362 у госпитализированных пациентов с гастроэзофагеальным раком поздней стадии
Клиническое, впервые проводимое на человеке, с однократным введением, многоцентровое открытое исследование фазы I с эскалацией дозы путем внутривенной (i.v.) инфузии у человека с использованием IMAB362 выполняли для определения максимальной переносимой или подходящей однократной дозы (MTD) IMAB362, изучения безопасности, переносимости и профиля неблагоприятных явлений, вызванных IMAB362, определения фармакокинетического профиля однократных возрастающих доз IMAB362, определения иммуногенности применения однократной дозы IMAB362, и определения потенциальной противоопухолевой активности IMAB362 у пациентов с гастроэзофагеальным (GE) раком поздней стадии.
Данное исследование было разработано как впервые проводимое на человеке, многоцентровое, нерандомизированное, с эскалацией однократной дозы у разных пациентов, открытое клиническое исследование фазы I с использованием однократной внутривенной инфузии IMAB362 и периодом последующего наблюдения с отсутствием лечения в течение 4 недель.
Для включения в исследование пациенты должны удовлетворять следующим критериям включения:
- метастатическое, рефракторное или рецидивирующее заболевание гастроэзофагеальным раком поздней стадии, подтвержденным гистологией;
- экспрессия CLDN18.2, подтвержденная иммуногистохимией или наличие образца ткани опухоли, подходящей для определения экспрессии CLDN18.2;
- предшествующая стандартная химиотерапия, включающая фторпиримидин, соединение платины и/или эпирубицин, и - в случае, если клинически приемлемо -доцетаксел;
- по меньшей мере 1 измеряемый участок заболевания в соответствии с критерием RECIST (компьютерная томография (СТ) или магнитно-резонансная томография (MRT) не ранее, чем за 6 недель до включения в исследование);
- возраст 18 лет или старше;
- письменное информированное согласие после получения информации об исследовании;
- общее состояние (performance status, PS) по шкале ECOG 0-1 балла или по шкале Карновского 70-100%;
- ожидаемая продолжительность жизни >3 месяцев;
- количество тромбоцитов ≥100000/мм3;
- гемоглобин ≥10 г/дл;
- INR <1,5;
- билирубин в норме;
- AST и ALT <2,5 раза выше верхней границы нормы (upper limit of normal, ULN) (в 5 раз выше ULN в случае наличия метастазов в печени);
- креатинин <1,5 × ULN;
- для женщин с репродуктивным потенциалом (последняя менструация менее чем 2 года назад до включения в исследование): Отрицательный тест на наличие беременности (β-HCG) на начало исследования и использование двух высокоэффективных методов контрацепции в течение 8 недель после инфузии исследуемого лекарственного средства;
- пациенты мужского пола должны использовать общепринятые методы контрацепции в течение 8 недель после инфузии исследуемого лекарственного средства.
Пациенты, удовлетворяющие одному или нескольким из следующих критериев, не подлежат включению в исследование:
- беременность или кормление грудью;
- предшествующая аллергическая реакция или непереносимость моноклонального антитела, включая гуманизированные и химерные антитела;
- предшествующее включение в настоящее исследование;
- менее 3 недель после предшествующей противоопухолевой химиотерапии или лучевой терапии;
- другие экспериментальные препараты или устройства параллельно или в течение 4 недель до данного исследования;
- другие сопутствующие противоопухолевые средства или терапии;
- история положительного теста на антитело к вирусу иммунодефицита человека (ВИЧ);
- известный гепатит;
- неконтролируемое или тяжелое заболевание, включая, но без ограничения, любое из следующих:
- продолжающаяся или активная инфекция, требующая парентеральных антибиотиков;
- симптоматическая застойная сердечная недостаточность;
- нестабильная стенокардия;
- неконтролируемая гипертония;
- клинически значимая сердечная аритмия;
- инфаркт миокарда в течение последних 6 месяцев;
- желудочное кровотечение в течение последних четырех недель;
- симптоматическая пептическая язва;
- клинические симптомы церебральных метастазов или подтвержденных результатами лабораторных исследований метастазов;
- психиатрическое заболевание или социальные ситуации, которые будут препятствовать приверженности к лечению;
- сопутствующее введение антикоагулянтных средств с антагонистами витамина К (например, кумадин);
- сопутствующее введение терапевтических доз гепарина (профилактические дозы допускаются).
Из общего количества 29 пациентов 15 пациентов получали исследуемое лекарственное средство и были распределены в одну из дозовых когорт (33, 100, 300, 600 или 1000 мг IMAB362/м2). Эти пациенты составили выборку для оценки безопасности (safety population, SP). Так как потенциально дозолимитирующих токсичностей не возникало ни в одной из этих дозовых групп, тестирования дополнительных пациентов для подтверждения возможных дозолимитирующих токсичностей не требовалось. Таким образом, исследуемое лекарственное средство получали не более 3 пациентов в каждой дозовой когорте, то есть всего 15 пациентов.
Распределение пациентов по когортам с различными дозами IMAB362 представлено в таблице 1 ниже.
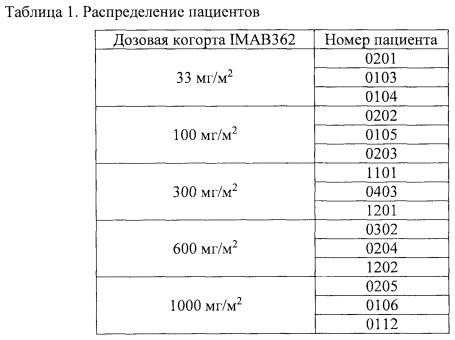
Ни один из пациентов преждевременно не прекратил исследование, то есть все пациенты завершили исследование в соответствии с протоколом.
А. ОЦЕНКА БЕЗОПАСНОСТИ
Было обнаружено, что IMAB362 является безопасным и хорошо переносимым.
Только 25 нежелательных явлений (adverse events, АЕ), которые возникли у 8 пациентов, были оценены как связанные с проводимой терапией. Связанные с проводимой терапией нежелательные явления (АЕ) были сходными между дозовыми группами. Более половины этих нежелательных явлений (АЕ) представляли собой нарушения со стороны желудочно-кишечного тракта (в основном тошнота, рвота). Только одно из связанных с проводимой терапией нежелательных явлений (АЕ) было оценено как тяжелое (рвота), тогда как все другие были легкими или умеренными. Все связанные с проводимой терапией нежелательные явления (АЕ) прекращались, за исключением одного случая дисгевзии (степень 1 согласно СТС (легкая)) с неизвестным исходом и случая повышенной активности GGT (степень 2 согласно СТС (умеренная)), которая не восстановилась до исходного уровня.
Дозолимитирующей токсичности (DLT), определенной как нежелательное явление (АЕ), связанное с проводимой терапией, возникающей во время или в течение четырех недель после инфузии исследуемого лекарственного средства и представляющей токсичность 3 степени (за исключением тошноты) или токсичность 4 или 5 степени (согласно СТС версия 3.0), не наблюдалось в любой из дозовых групп. Таким образом, в настоящем исследовании была определена максимальная переносимая или подходящая однократная доза (MTD) IMAB362, составляющая 1000 мг/м2.
Серьезного нежелательного явления (SAE) и предполагаемой непредвиденной серьезной нежелательной реакции (SUSAR) в настоящем исследовании не возникало.
Только 7 пациентов имели, по меньшей мере, один лабораторный показатель, выходящий за рамки нормальных значений, оцененный как степень 3 (тяжелая). Зависимости доза-эффект и четкой связи с исследуемым лекарственным средством не наблюдалось. Лабораторных показателей степени 4 согласно СТС (угрожающие жизни) или 5 (смерть) не отмечалось.
В заключение, значимых различий в профиле АЕ и других параметров безопасности между дозовыми группами не наблюдалось. В целом, наблюдалось, что IMAB362, вводимый в виде однократной дозы, является безопасным и хорошо переносимым с тошнотой и рвотой, которые являются наиболее распространенными связанным с проводимой терапией нежелательными явлениями.
В. ОЦЕНКА ФАРМАКОКИНЕТИКИ И ИММУНОГЕННОСТИ
Для определения концентрации лекарственного средства уровни IMAB362 в сыворотке крови всех пациентов измеряли непосредственно перед инфузией исследуемого препарата, в конце инфузии, через 3, 8, 12 и 24 часа после завершения инфузии, и на дни 3 (V3), 5 (V4), 8 (V5), 15 (V6) и 29 (V7).
Общий обзор уровней IMAB362 в сыворотке крови в течение исследования для каждого пациента представлен в таблице 2. По неизвестным причинам для одного пациента (номер 1201) в дозовой группе 300 мг/м2 низкий уровень IMAB362 в сыворотке крови (12,633 мкг/мл) был измерен еще до инфузии исследуемого лекарственного средства (V2, день 0).
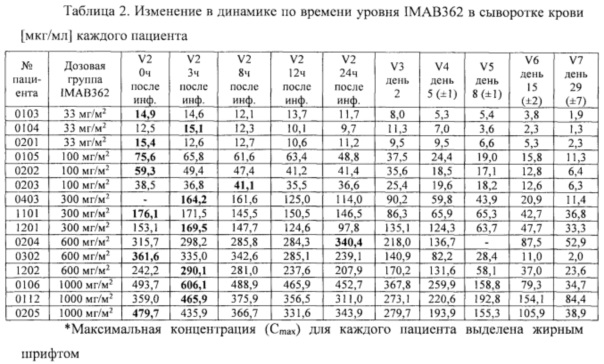
Наблюдаемые средние максимальные концентрации (Cmax) на дозовую группу показаны в таблице 3. Возрастающие средние значения Cmax соответствует возрастающим дозам IMAB362, вводимым путем инфузии.
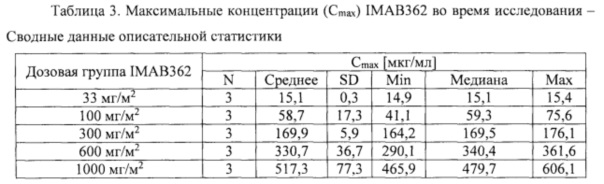
В графическом виде средние концентрации IMAB362 в крови во время исследования представлены на фигуре 1.
Самые высокие уровни IMAB362 были измерены в период времени от непосредственного завершения инфузии до 8 часов после ее завершения. Через 3 часа после завершения инфузии средняя концентрация IMAB362 составила 14,1 мкг/мл в группе 33 мг/м2, 50,7 мкг/мл в группе 100 мг/м2, 164,2 мкг/мл в группе 300 мг/м2, 307,8 мкг/мл в группе 600 мг/м2 и 502,6 мкг/мл в группе 1000 мг/м2.
Фармакокинетика IMAB362 является дозозависимой. Наиболее высокие уровни доз наблюдались в течение первых 8 часов после 2-часовой инфузии. Средний период полувыведения IMAB362 составил в целом 8,5 дней, варьируя от около 5 до около 12 дней в различных дозовых группах.
В результате исследований механизма действия in vitro было определено, что при концентрации IMAB362, составляющей 50 мкг/мл, можно ожидать четкого осуществления эффектов против опухолевых клеток посредством ADCC, CDC и ингибирования пролиферации, и что значения ЕС50 ADCC и CDC, которые рассматриваются в качестве основных механизмов действия, покрываются даже половиной этого уровня концентрации. Исходя из этого, для более точной оценки в исследованиях многократных доз IMAB362 были установлены уровни, составляющие 300 мг/м2 и 600 мг/м2. Пациенты, которые получали IMAB362 в дозе 300 мг/м2 и 600 мг/м2, были очевидным образом выше этих уровней на день 8 (V5) и близки к 50 мкг/мл на день 15 (V6).
Признаки наличия антител к лекарственному препарату у пациентов после этой однократной дозы IMAB362 отсутствовали.
С. ОЦЕНКА ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТИ
Первичным критерием для оценки потенциальной противоопухолевой активности являлся статус опухоли в соответствии с классификацией RECIST (версия 1.0) в период со 2 по 5 неделю после инфузии IMAB362 (V6/V7). Так как все пациенты завершили исследование согласно протоколу, оценки были сделаны исключительно на V7, то есть в период с 4 по 5 неделю после инфузии лекарственного средства. Всех пациентов оценивали с помощью СТ.
Три пациента не имели измеряемого заболевания (пациенты 1101 и 1201 не имели целевого опухолевого очага, для пациента 0302 соответствующие данные отсутствовали), но были включены в популяцию для анализа противоопухолевой активности, так как это не являлось официальной оценкой эффективности.
В целом, ни для одного из пациентов ответ не был оценен как полный или частичный. Стабильное заболевание наблюдалось у одного из 15 пациентов в дозовой группе 600 мг/м2. Несмотря на то, что у подвергнутых лечению пациентов процентное содержание опухолевых клеток, окрашенных положительно по CLDN18.2, находилось в диапазоне от 1% до 80% (до 50% опухолевых клеток с окрашиванием мембраны), 90% или более опухолевых клеток этого пациента были окрашены положительно по CLDN18.2 с большой долей опухолевых клеток, демонстрирующих окрашивание мембраны. Два пациента из дозовой группы 300 мг/м2 также не показали прогрессирования, и поскольку они не имели целевого опухолевого очага, то не могли быть подвергнуты оценке на объективный ответ опухоли и оценивались как non-CR, non-PD. Продолжительность SD составила около 2 месяцев. Продолжительность non-CR, non-PD составила около двух месяцев и 6 недель, соответственно.
Обзор общего ответа пациента представлен в таблице 4.
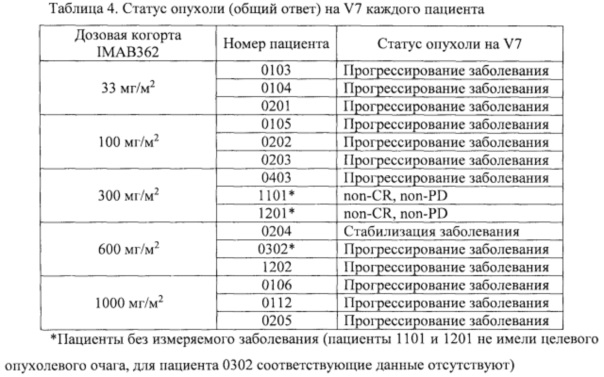
Различные параметры, содействующие оценке статуса опухоли (общего ответа) описаны далее.
В таблице 5В представлены данные, касающиеся изменения суммы самых длинных диаметров (целевого опухолевого очага), статуса дополнительных опухолевых очагов после терапии IMAB362, возникновения новых очагов, обзор результатов оценки после терапии IMAB362 (оценка проведена на V7).
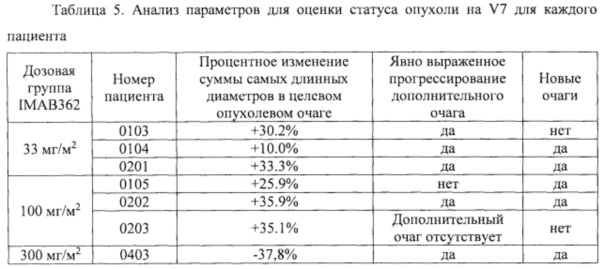
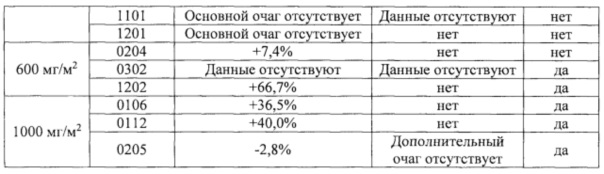
Процентное изменение суммы самых длинных диаметров целевого опухолевого очага с V1 по V7 не показало какого-либо заметного различия для разных вводимых доз.
Для дополнительных очагов опухолевого поражения четко выраженное прогрессирование (с V1 по V7) отмечалось чаще у пациентов при более низких уровнях доз, но не при уровнях доз 600 мг/м2 и 1000 мг/м2.
У одного пациента в группе 300 мг/м2 (0403) четко выраженное прогрессирование наблюдалось в дополнительном опухолевом очаге и уменьшение самого длинного диаметра в одном лимфатическом узле целевого опухолевого очага.
В отношении новых очагов не наблюдалось преобладания какой-либо одной из дозовых групп.
В случае пациентов 0302 (дозовая группа 600 мг/м2) и 0205 (дозовая группа 1000 мг/м2) возникновение новых опухолевых очагов являлось причиной оценки общего ответа как прогрессирующего заболевания.
Для оценки статуса дополнительных опухолевых очагов согласно RECIST, уровень опухолевых антигенов СА 125, СА 15-3, СА 19-9 и СЕА в сыворотке крови определяли в центральной лаборатории на V2 (день 1, перед инфузией), V6 и V7.
Обзор содержания опухолевых маркеров в сыворотке крови для 3 пациентов с общим ответом, по меньшей мере, стабильного заболевания представлен в таблице 6.
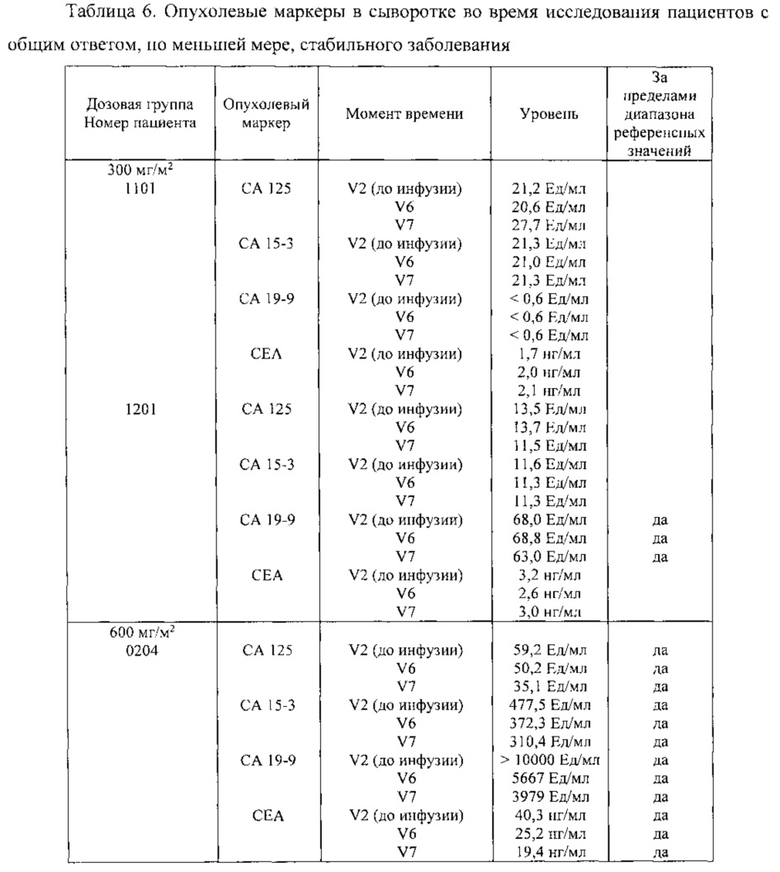
Из 3 пациентов со стабильным заболеванием или Non-CR/Non-PD в соответствии с визуализацией два пациента имели стабильные уровни опухолевых маркеров в период наблюдения. Один пациент (0204) показал значительное снижение всех 4 опухолевых маркеров после лечения. Большинство пациентов с прогрессированием заболевания, напротив, испытывали повышение уровней опухолевых маркеров.
Статус опухоли (в соответствии с классификацией RECIST) в период с 4 по 5 неделю после инфузии IMAB362 (V6/V7) сравнивали со статусом на момент начала исследования. В целом, ни для одного из пациентов ответ не был оценен как полный или частичный. Один из 15 пациентов (в дозовой группе 600 мг/м2) показал стабильное заболевание в конце исследования. Два пациента в дозовой группе 300 мг/м2 с не поддающимся измерению заболеванием показали non-CR/non-PD. Наряду с этим, уровни опухолевых маркеров у этих трех пациентов оставались стабильными (2 пациента) или даже значительно снизились (1 пациент). Большинство пациентов с прогрессированием заболевания показали увеличение уровней опухолевых маркеров в динамике по времени.
В отношении параметров, содействующих оценке статуса опухоли (общий ответ), уменьшение в одном очаге наблюдалось в дозовой группе 300 мг/м2. При скрининге (V1) 13 из 15 пациентов имели всего 32 дополнительных опухолевых очага. После терапии IMAB362 (оцененной на V7) четко выраженное прогрессирование дополнительного опухолевого очага отмечалось всего для 5 пациентов, 3 в дозовой группе 33 мг/м2, 1 в группе 100 мг/м2 и 1 в дозовой группе 300 мг/м2. Ни для одного из этих 5 пациентов общий ответ не был оценен как прогрессирование заболевания только по причине прогрессирования их дополнительных опухолевых очагов. В течение периода исследования было зафиксировано всего 17 новых очагов, равномерно распределенных по дозовым группам. В случае 2 пациентов (в дозовых группах 600 мг/м2 и 1000 мг/м2) возникновение новых очагов являлось причиной оценки общего ответа как прогрессирования заболевания.
Более того, дополнительные данные, полученные у отобранных пациентов, показали, что компоненты сыворотки и РВМС пациентов являются полностью функциональными и активными в отношении опосредования основных механизмов действия IMAB362 CDC и ADCC, соответственно.
В заключение, признаки противоопухолевой активности (стабильное заболевание, снижение опухолевых маркеров) наблюдались в дозовых группах 300 мг/м2 и 600 мг/м2. Вследствие малого объема выборки дозовых групп трудно сделать выводы относительно тенденций, касающихся эффективности.
С. ОБЩИЕ ВЫВОДЫ
Данное исследование разработано как впервые проводимое на человеке, многоцентровое, нерандомизированное, с эскалацией однократной дозы у разных пациентов, открытое клиническое исследование фазы I с однократной внутривенной инфузией IMAB362 и 4-недельным периодом наблюдения с отсутствием лечения.
Всего 15 пациентов получали исследуемый препарат и были распределены в одну из дозовых когорт (33, 100, 300, 600 или 1000 мг IMAB362/м2). Дозовые группы могут рассматриваться как сравнимые. Значимых несоответствий, касающихся демографических данных и исходных характеристик, не наблюдалось.
В отношении основной цели исследования, ни в одной из дозовых групп дозолимитирующей токсичности (DLT) не наблюдалось. Таким образом, подходящая однократная доза IMAB362 в настоящем исследовании составила 1000 мг/м2. IMAB362 был безопасным и хорошо переносимым, при этом тошнота и рвота являлись наиболее распространенными связанными с проводимой терапией неблагоприятными явлениями.
Было обнаружено, что профиль АЕ и частота возникновения АЕ сходны в различных дозовых группах. Значительных различий между дозовыми группами не наблюдалось у ряда отдельных пациентов с клинически значимыми ухудшениями по любому из гематологических, биохимических или коагуляционных параметров.
В отношении потенциальной противоопухолевой активности IMAB362 в соответствии с критериями RECIST полный или частичный ответ не наблюдался ни для одного из пациентов. Один из 15 пациентов (в дозовой группе 600 мг/м2) показал стабильное заболевание в конце исследования. Два пациента в дозовой группе 300 мг/м2 с не поддающимся измерению заболеванием показали non-CR/non-PD. Из этих 3 пациентов со стабильным заболеванием в соответствии с визуализацией два пациента имели стабильные уровни опухолевых маркеров в период наблюдения. Один пациент показал значительное снижение всех 4 опухолевых маркеров после лечения.
Данное и фармакокинетические исследования, показывающие, что целевые уровни IMAB362 в сыворотке достигаются при уровнях доз, составляющих 600 мг/м2, подтверждают, что эта доза подлежит дополнительному исследованию.
Кроме того, дополнительные данные подтверждают, что иммунные эффекторы пациента являются полностью функциональными и активными в отношении опосредования главных механизмов действия IMAB362 CDC и ADCC, соответственно.
Пример 2. Активность лекарственного средства
Цели анализов in vitro, выполняемых для этой фазы I клинического исследования, включали анализ (i) способности эффекторных клеток, присутствующих в крови пациента, индуцировать IMAB362-зависимую ADCC; (ii) способность системы комплемента пациента индуцировать IMAB362-зависимую CDC; и (iii) изменение способности IMAB362 индуцировать ADCC и CDC после введения пациентам.
Различные типы анализов выполняли для исследования цитолитической активности, индуцированной IMAB362 после введения пациентам. Анализы выполняли с использованием сыворотки пациентов или РВМС пациентов, выделенных из образцов крови (таблица 7). Для сравнения и подтверждения функциональности анализов CDC и ADCC с использованием сыворотки, пул сывороток человека (полученных от здоровых субъектов-людей), в котором стерильно разведен свежеприготовленный IMAB362, был параллельно включен в каждый анализ. Для тестирования функциональности анализа ADCC с использованием РВМС, клетки крови, изолированные от здорового донора, использовали в качестве положительного контроля в том же самом анализе для каждого пациента.
А. МАТЕРИАЛЫ И МЕТОДЫ
Для различных in vitro анализов образцы сыворотки пациентов собирали до инфузии IMAB362 и через 1, 7, 14 и 28-32 дней после введения антитела IMAB362 (таблица 7). Их использовали в качестве источника антитела IMAB362 и комплемента в CDC, или в качестве источника антитела в анализе ADCC сыворотки. Сыворотку пациентов до инфузии использовали в качестве отрицательного контроля «нет IMAB362» и для разведения образцов сыворотки пациентов для доведения концентрации IMAB362 до 0,5 мкг/мл. Свежеприготовленные образцы крови собирали через 14 дней после инфузии (через 7 дней для пациента 0203) и использовали в качестве источника эффекторных клеток для анализа ADCC.
Образцы крови собирали у пациентов (таблица 7), сыворотку собирали и разделяли на аликвоты, которые немедленно помещали на хранение при -80°С. Анализы всех этих образцов выполняли в одном отдельном эксперименте после сбора всех 24 образцов сыворотки. Для ADCC свежеприготовленные образцы крови (15 мл Na2EDTA) использовали для выделения РВМС и на следующий день выполняли анализы ADCC.
Способность РВМС пациентов индуцировать ADCC в сочетании с IMAB362 тестировали ex vivo путем использования свежеприготовленных антикоагулированных с помощью 15 мл Na2-EDTA образцов крови, полученных от пациентов через 14 дней (через 7 дней для пациента 0203) после введения IMAB362. После получения образцов крови РВМС выделяли путем центрифугирования в градиенте плотности фиколла. РВМС культивировали в течение 24 ч, и на следующий день выполняли анализ ADCC с трансфицированными люциферазой CLDN18.2-положительными клетками рака желудка человека NUGC4 в качестве мишеней в сочетании с различными концентрациями экзогенно добавленного IMAB362. РВМС добавляли в соотношении Е:Т, равном 20:1, и пробы инкубировали в течение 24 ч при 37°С, 5% СО2. РВМС, полученные от здорового донора, тестировали в таких же условиях параллельно с анализом валидности пробы (положительная контрольная проба). Эти исходные РВМС хранили в жидком N2, и для каждого анализа ADCC с РВМС пациентов отбирали аликвоту из этого исходного раствора РВМС и анализировали параллельно.
Использовали материалы со следующими характеристиками:
- CLDN18.2-положительные клетки-мишени: временно трансфицированные люциферазой клетки рака желудка NUGC4-10cH11E10;
- Положительные контрольные эффекторные клетки: РВМС, полученные от здорового донора (замороженный в N2-исходный раствор, партия номер: 276-SMS-09-00706, 4e7c/vial, MNZ, 08.07.07.SJA);
- Функциональное контрольное антитело: IMAB362 в серийных разведениях (0,4 нг/мл - 126,5 мкг/мл);
- Отрицательное контрольное антитело: контроль изотипа (ритуксимаб, 126,5 мкг/мл).
Способность компонентов сыворотки пациента индуцировать комплемент-зависимую цитотоксичность (CDC) в сочетании с IMAB362 анализировали ex vivo в динамике по времени. Образцы сыворотки собирали и хранили при -80°С, и образцы от всех пациентов анализировали параллельно в этом же самом эксперименте. Дополнительно к сыворотке до инфузии, в которую было экзогенно добавлено фиксированное количество 0,5 мкг/мл IMAB362 (представляющее концентрацию in vitro ЕС50), также тестировали образцы, собранные через 1, 7, 14 и 28-32 дней после введения IMAB362, в которых концентрацию циркулирующего IMAB362 доводили до 0,5 мкг/мл (CDC с нормализацией). Конечная концентрация в сыворотке в каждой пробе была доведена до 20%. Трансфицированные люциферазой клетки СНО-K1, стабильно трансфицированные CLDN18.2, использовали в качестве мишеней. Для сравнения тестировали пул сывороток здоровых доноров-людей с введенным IMAB362.
Использовали материалы со следующими характеристиками:
- CLDN18.2-положительные клетки-мишени: Стабильно трансфицированные клетки СНО-K1 р740 luci #2А5.
- Аналитический положительный контроль: серийные разведения IMAB362 (1:3,16), приготовленные в пуле сывороток человека от здоровых доноров, до конечных концентраций в диапазоне от 31,6 нг/мл до 10,0 мкг/мл.
- Функциональное контрольное антитело: IMAB362, доведенный до конечной аналитической концентрации 0,5 мкг/мл в каждом образце сыворотки до инфузии.
- Аналитическое отрицательное контрольное антитело: Контролирующее изотип антитело, разведенное в пуле сывороток человека (ритуксимаб).
Кинетику общей цитотоксичности, опосредованной циркуляцией IMAB362 у человека, а также его способность индуцировать ADCC и CDC анализировали в анализе «одна пробирка».
Сыворотку от каждого пациента собирали через 7, 14 и 28-32 дней после внутривенного введения IMAB362 и, таким образом, включая факторы комплемента пациента плюс циркулирующий IMAB362 тестировали в этом анализе. Сыворотку в каждом анализе доводили до конечной концентрации 25% (об/об). РВМС здоровых контролей добавляли в качестве эффекторных клеток, тогда как клетки NUGC-4 служили в качестве клеток-мишеней в соотношении Е:Т, равном 40:1.
В дополнительных условиях сыворотка была инактивирована путем нагревания, разрушая активность комплемента. Этот второй анализ, таким образом, отражает исключительно активность ADCC, индуцированную IMAB362, присутствующим в сыворотке пациента.
Во время фазы I исследования образцы сыворотки собирали и хранили при -80°С. Все образцы, полученные от пациентов, анализировали параллельно в этом же самом эксперименте.
Использовали материал со следующими характеристиками:
- CLDN18.2-положительные клетки-мишени: стабильно трансфицированные люциферазой клетки рака желудка NUGC-4 10СН11 luci eGFP #2.
- Эффекторные клетки: РВМС от здорового донора (свежеприготовленная лейкоцитарная пленка).
- Функциональное контрольное антитело: серийные разведения IMAB362 (0,26 нг/мл - 200,0 мкг/мл), введенные в пул сывороток человека.
- Положительный контрольный образец: Образец сыворотки пациента до инфузии с введенным IMAB362 (200,0 мкг/мл) (представляющей EC80-100 для IMAB362 в этом анализе).
- Аналитическое отрицательное контрольное антитело: Контролирующее изотип антитело в пуле сывороток человека (ритуксимаб).
Способность IMAB362 вступать во взаимодействие и активировать комплемент, присутствующий в сыворотке пациента, и индуцировать комплемент-зависимую цитотоксичность (CDC) после продолжительной циркуляции в крови пациента анализировали ex vivo через 1, 7, 14 и 28-32 дней после введения IMAB362. Анализ выполняли путем непосредственного использования образцов сыворотки пациентов в анализе (CDC без нормализации). В качестве положительного контроля использовали сыворотку до инфузии, в которую было экзогенно добавлено фиксированное количество 10 мкг/мл IMAB362 (представляющее концентрацию in vitro ЕС90-100). Конечную концентрацию в сыворотке в каждом образце доводили до 20%. В качестве мишеней использовали трансфицированные люциферазой клетки СНО-K1, стабильно трансфицированные CLDN18.2. Для сравнения тестировали пул сывороток здоровых доноров-людей с введенным IMAB362.
Во время фазы I исследования образцы сыворотки собирали и хранили при -80°С. Все образцы от пациентов анализировали параллельно в этом же самом эксперименте.
Использовали материал со следующими характеристиками:
- CLDN18.2-положительные клетки-мишени: Стабильно трансфицированные клетки СНО-K1 р740 luci #2А5.
- Функциональное контрольное антитело: Серийные разведения IMAB362 (1:3,16), приготовленные в пуле сывороток человека от здоровых доноров, до конечной концентрации в диапазоне от 31,6 нг/мл до 10,0 мкг/мл.
- Положительный контрольный образец: Образцы сыворотки пациента до инфузии, каждый с введенным IMAB362 (10,0 мкг/мл) {in vitro концентрация CDC-EC90-100).
- Аналитическое отрицательное контрольное антитело: Контролирующее изотип антитело, разведенное в пуле сывороток человека.
В. РЕЗУЛЬТАТЫ
Способность РВМС пациентов опосредовать ADCC
Для анализа способности иммунных клеток пациентов лизировать опухолевые клетки, экспрессирующие CLDN18.2, клетки рака желудка NUGC-4, эндогенно экспрессирующие CLDN18.2, инкубировали с увеличивающимися концентрациями IMAB362 и РВМС пациентов. Пробы с РВМС от здорового донора включены в качестве функционального контроля.
РВМС пациентов показали зависимые от дозы IMAB362 скорости лизиса с максимальной величиной от 27% до 77% при концентрации ~30 мкг/мл. Это значительно не отличается (t-критерий Стьюдента для одной выборки) от максимальных скоростей лизиса, составляющих от 14% до 56%, полученных с использованием здоровых контрольных РВМС, тестированных в таких же анализах (фигура 2). Активность ADCC была наиболее значительной для пациента 0204.
Эти данные показывают, что РВМС пациентов с раком желудка не хуже в отношении индукции ADCC человеческих CLDN18.2-положительных клеток рака желудка в сочетании с IMAB362 по сравнению с РВМС, полученным от здоровых доноров.
Способность системы комплемента пациентов индуцировать CDC
Тестировали способность комплемента пациента взаимодействовать с IMAB362, присутствующим в сыворотке, и индуцировать CDC. В образцы сыворотки до инфузии вводили 0,5 мкг/мл свежеприготовленного IMAB362, и активность CDC сравнивали с такой же концентрацией антитела, введенного в пул сывороток человека. Образцы сыворотки/антитела инкубировали с клетками СНО-K1 р740 luci #2А5, и лизис определяли через 80 мин путем измерения активности люциферазы.
Все пациенты были способны индуцировать значительную CDC в пределах 80 мин (фигура 3). Для 5 из 6 пациентов наблюдались максимальные скорости лизиса в диапазоне от 50% до 71%. Это сравнимо с данными, полученными при параллельном тестировании с пулом сывороток от здоровых контролей (64,5%). Примечательно, что пациент 0204 показал самую высокую активность CDC со свежеприготовленным IMAB362 (93,9%).
Способность растворимых эффекторов в сыворотке пациентов индуцировать уничтожение клеток с помощью введенного внутривенно циркулирующего IMAB362
Далее, способность сыворотки пациентов взаимодействовать с внутривенно введенным IMAB362 в течение интервала времени его циркуляции у пациента изучали путем тестирования образцов сыворотки, собранных в различные моменты времени после введения IMAB362 в анализах CDC на CLDN18.2-положительных клетках-мишенях СНО-K1. Образцы сыворотки являлись источником для специфических для пациентов растворимых эффекторов, включая комплемент, а также для IMAB362. Концентрации IMAB362 в образцах сыворотки определяли с помощью ELISA (vivoScience) (таблица 8) и доводили до конечной концентрации IMAB362, равной 0,5 мкг/мл (медиана ЕС50 IMAB362), с использованием соответствующей сыворотки каждого пациента до инфузии в качестве разбавителей. Так как концентрации IMAB362 различаются в зависимости от вводимой дозы и времени отбора крови, фактор разведения для образцов значительно различался между пациентами, изменяясь от 4,6-кратного до 688-кратного. Пул сывороток от здоровых доноров (HSC) использовали в качестве контроля (фигура 4).
По сравнению с положительным контролем (сыворотка до инфузии соответствующего пациента + свежеприготовленный IMAB362) активность по уничтожению клеток сохранялась в течение первых 24 ч, тогда как цитолитическая активность образцов сыворотки, собранных неделей позже, снижалась, прогрессируя дальше в последующие недели (фигура 4). Даже в этом случае значительная цитотоксичность осуществлялась сывороткой пациентов даже через 2 недели после введения IMAB362. Утрата активности CDC через 28-32 дня является значительной и наиболее выраженной у пациентов, получавших низкие дозы IMAB362 (фигура 4). Оказалось, что у пациентов, получавших высокие дозы (0204; 600 мг/м2 и 0205; 1000 мг/м2), активность CDC сохранялась лучше в течение исследуемого периода. На основе имеющихся в наличии данных можно сделать заключение о том, что лежащий в основе этого ухудшения механизм до сих пор остается неясным.
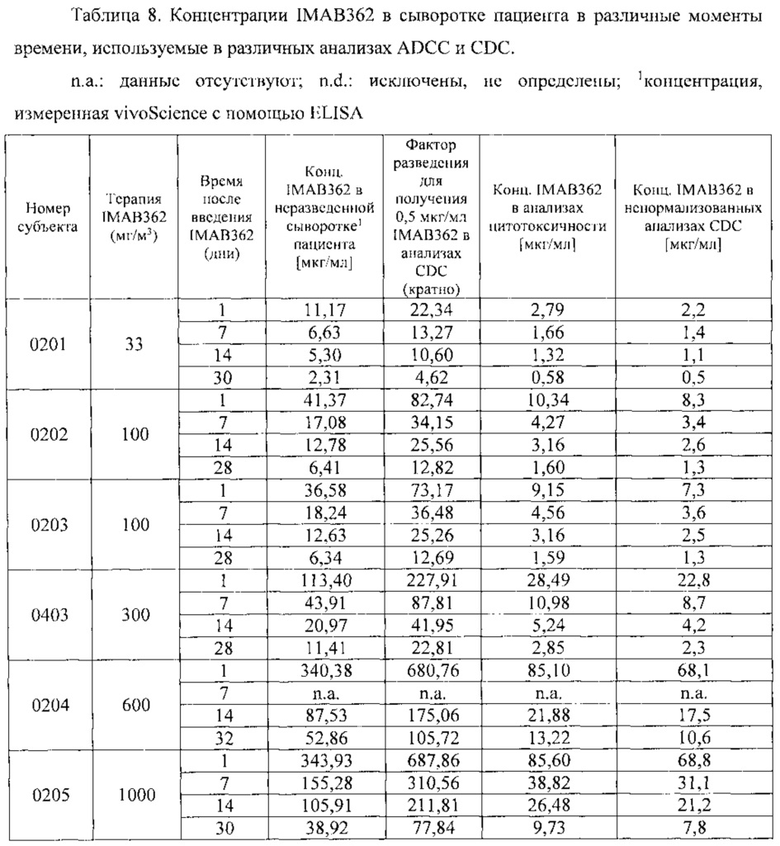
Эффект компонентов сыворотки на индуцированную IMAB362 цитотоксичность
ADCC-активность mAB может быть нарушена в присутствии человеческой сыворотки. Изучали эффект сыворотки пациента на активность ADCC. С этой целью сыворотку каждого пациента собирали через 7, 14 и 28-32 дней после введения IMAB362 и, таким образом, использовали представляющие факторы комплемента пациента плюс циркулирующий введенный внутривенно IMAB362. Все образцы сыворотки пациентов разводили до конечной концентрации сыворотки 25% (об/об) и рассчитывали оставшуюся концентрацию IMAB362 в каждом аналитическом образце пациента (таблица 8). В этом ADCC-анализе РВМС от одного здорового донора использовали в качестве эффекторов, и клетки NUGC-4 использовали в качестве клеток-мишеней (соотношение Е:Т=40:1). Все анализы для всех пациентов выполняли параллельно в одном эксперименте с использование одинаковых условий, клеток-мишеней и донорных РВМС для обеспечения сопоставимости. В качестве контроля функционального анализа в пул сывороток здоровых людей вводили IMAB362 (200,0 мкг/мл). В качестве дополнительного положительного контроля в индивидуальные образцы сыворотки пациентов до инфузии вводили 200,0 мкг/мл IMAB362 (представляющую in vitro EC80-100 для IMAB362 в этой системе).
Во всех анализах наблюдалось, что антитела IMAB362, присутствующие в сыворотке пациента после введения, являются высоко активными и индуцируют цитотоксичность (фигура 5). Биологическая активность IMAB362 сохранялась в течение 28-32 дней после введения со специфическим уничтожением все еще выше 48% во всех дозовых группах. Общие различия между дозовыми группами были удивительным образом умеренными, предполагая эффект насыщения. У пациентов, получавших лечение более низкими дозами (33-200 мг/м2) умеренное уменьшение специфического уничтожения от 77,7-87,4% до 48,3-66,8% наблюдалось в течение времени, коррелирующего с уменьшением концентраций антитела в сыворотке (фигура 5 верхняя группа). Наиболее высокая активность, стабильно сохраняющаяся в течение времени, наблюдалась у пациентов, получавших лечение IMAB362 в дозе 600 или 1000 мг/м2 (фигура 5 нижняя группа/панель).
Данный анализ повторяли с образцами сыворотки, в которых факторы комплемента были инактивированы путем их инкубации при 56°С в течение 30 мин. Цитотоксичность в случае инактивированных с помощью нагревания образцов сыворотки пациентов была ниже по сравнению с цитотоксичностью, полученной в случае необработанных образцов сыворотки во всех случаях. Сходные уменьшения также наблюдались с инактивированным с помощью нагревания пулом от здоровых доноров (HSC, фигура 6).
В целом, эти данные указывают на то, что сыворотка пациентов не ингибирует ADCC способности растворимых компонентов сыворотки, но вместо этого способствует общей цитолитической активности, индуцированной IMAB362.
Кинетика IMAB362-опосредованной CDC в сыворотке пациентов
Для определения кинетики CDC-способности IMAB362 в сыворотке от пациентов различных дозовых групп образцы сыворотки собирали через 1, 7, 14 и 28 дней после введения IMAB362.
Снова, эта сыворотка служит в качестве источника для комплемента, а также для IMAB362. Конечные концентрации в сыворотке доводили до конечного объема 20% (об/об). Конечные концентрации IMAB362 в каждом образце CDC-анализа приведены в таблице 7. В качестве положительного контроля в образцы от пациента до инфузии вводили свежеприготовленное антитело IMAB362 до конечной концентрации 10 мкг/мл (in vitro ЕС95 IMAB362 в этой системе CDC-анализа). Кроме того, для функционального контроля CDC-анализа готовили серийные разведения IMAB362 (0,032-10 мкг/мл) в пуле сывороток человека. Стандартизированную пробу с клетками СНО-K1, стабильно трансфицированными CLDN18.2 и люциферазой, использовали в качестве клеток-мишеней. Все образцы сыворотки отбирали и тестировали параллельно в одном и том же эксперименте.
CDC-активность хорошо коррелирует с концентрацией антитела в каждом образце сыворотки (фигура 7). Наиболее важно, данные предполагают, что CDC-опосредованная цитотоксическая активность сохраняется в течение 4 недель. В частности, пациенты высоких дозовых групп не показали снижения CDC-активности в течение этого времени.
Краткие выводы и заключения
Оказалось, что у пациентов с GEC не нарушена способность индуцировать ADCC и CDC CLDN18.2-экспрессирующих клеток-мишеней в сочетании с IMAB362. Примечательно, что максимальный лизис, наблюдаемый путем ADCC и CDC, а также ЕС50, измеренная для ADCC, были самыми высокими для пациента 0204, который имел наиболее значительный клинический и сывороточный ответ на опухолевый антиген.
Анализы ex vivo CDC с циркулирующим IMAB362 в различные моменты времени после его введения показали, что еще 2 недели после введения имеется достаточно активная циркуляция IMAB362 у пациентов для индукции значительных ADCC и CDC.
CDC-активность пациентов в сочетании с циркулирующим IMAB362 снижается с течением времени по неизвестным до сих пор причинам.
Пример 3. Цитокины
Уровни цитокинов в сыворотке могут служить в качестве индикаторов иммунного статуса пациента. В этом клиническом исследовании целью анализа цитокинов в первую очередь являлось содействие контролю безопасности. Цитокины рассматривались в этом дополнительном анализе с точки зрения определения возможных кандидатов в биомаркеры.
Уровень цитокинов определяли на день 1 до инфузии IMAB362 и на дни 3 и 5 цикла лечения. Исследуемые цитокины включали провоспалительные (IL-1, IL-6, IL-12, IFNγ, TNFα) и противовоспалительные (IL-4, IL-10) цитокины, а также цитокины, необходимые для роста и функции Т-клеток (IL-2), и пролиферации NK-клеток (IL-2, IL-15).
Цитокины анализировали с помощью ELISA и проточной цитометрией (Interlab). Цитокины анализировали в соответствии с Interlab SOP-MU-IMM.M.0144. 05 «Flow Cytomix Cytokin- Check IL4, IL6, IL13, TNF-alpha, IFN-gamma, MCP-1, IL10 IL2, IL1-β, IL12p70, IL8, IL17A, IL23» и SOP-MU-IMM.M.0151.02 «Humanes Interleukin 15».
Уровни цитокинов IL-1, IL-2, IL-4, IL-6, IL-10, IL-12, IL-15, IFNγ и TNFα в сыворотке анализировали для 14 из 15 пациентов (таблица 9). Для пациента 0403 уровни цитокинов не были определены (300 мг/м2). Только значения уровней цитокинов в сыворотке, которые были выше диапазона референсных значений, анализировали на изменение во времени. Значения референсного диапазона были определены Interlab (смотри CSR GM-IMAB-001).
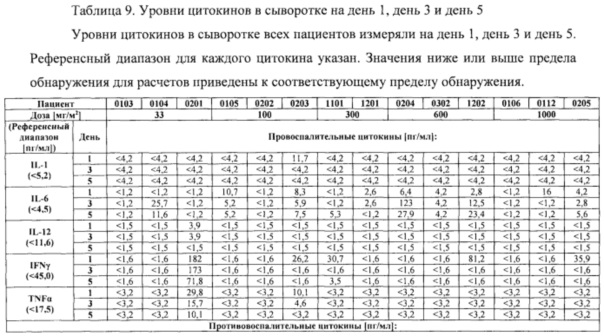
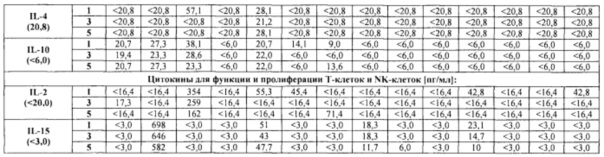
Уровни провоспалительных цитокинов (IL-1, IL-6, IL-12, IFNγ, TNFα) были выше соответствующих референсных диапазонов у 9 из 14 пациентов (0104, 0105, 0201, 0203, 0204, 0112, 1202, 0112, 0205). Уровни IFNγ оценивали у двух пациентов (0201, 1202). Уровень TNFα оценивали у одного из этих двух пациентов (0201). У обоих пациентов уровни IFNγ и TNFα были повышенными до введения IMAB362 и пониженными в последующие дни. Уровни IL-6 были повышенными у восьми пациентов (0104, 0105, 0203, 1101, 0204, 0112, 1202, 0205). Отсутствует ясная картина изменений уровня IL-6 в отношении введения IMAB362 и зависимости доза-эффект. Уровни IL-6 у пациента 0204 (600 мг/м2 IMAB362) не были повышенными до введения, но значительно повысились через 2 дня после инфузии, картина, не проявленная ни одним другим пациентом. Уровни IL-1 и IL-12 оставались в пределах соответствующего референсного диапазона для всех пациентов.
Уровни противовоспалительных цитокинов (IL-4, IL-10) были выше соответствующего референсного диапазона у 6 из 14 пациентов (0103, 0104, 0201, 0202, 0203, 1101). Уровни IL-10 были повышенными у шести пациентов (0103, 0104, 0201, 0202, 0203, 1101), уровни IL-4 были повышенными у двух из этих пациентов (0201, 0202). Колебания уровней противовоспалительных цитокинов не показывают четкой картины в отношении введения IMAB362 и зависимости доза-эффект.
Уровни цитокинов IL-2 в IL-15 для функции и пролиферации Т-клеток и NK-клеток были выше соответствующего референсного диапазона у 9 из 14 пациентов (0104, 0201, 0202, 0203, 1101, 1201, 0204, 1202, 0205). Уровни IL-2 были выше референсного диапазона у шести пациентов (0201, 0202, 0203, 1101, 1202, 0205). Уровни IL-15 были выше референсного диапазона у пяти пациентов (0104, 0202, 1201, 0204, 1202). Семь из девяти пациентов (0104, 0201, 0202, 0203, 1201, 1202, 0205) с повышенными уровнями IL-2/IL-15 до введения продемонстрировали снижение уровня цитокинов в последующие дни: уровни IL-2/IL-15 были выше соответствующего референсного диапазона до введения IMAB362 и снижались на второй и четвертый день после введения IMAB362. Наиболее значительное снижение в этой группе наблюдалось для концентраций IL-2 в сыворотке. У всех пяти пациентов (0201, 0202, 0203, 1202, 0205) с повышенными уровнями IL-2 до инфузии снижение до менее чем 50% соответствующих уровней до инфузии наблюдалось на четвертый день после введения. Это снижение может наблюдаться также у одного пациента (0201) со значительно повышенным уровнем IL-2 (354 пг/мл) до введения 33 мг/м2 IMAB362.
Другой профиль концентрации IL-2 был продемонстрирован пациентом 1101 (300 мг/м2 IMAB362) с уровнями IL-2 в референсном диапазоне до инфузии и через 2 дня, но повышенной концентрацией IL-2 на четвертый день после вливания.
Уровень IL-15 был пониженным на четвертый день после введения у всех четырех пациентов (0104, 0202, 1201, 1202) с повышенными уровнями IL-15 до инфузии. Этот профиль концентрации имеет значительное сходство с наблюдаемым профилем концентрации IL-2, хотя относительное снижение уровня не является выраженным.
Зависимость доза-эффект не может быть установлена для любого из анализируемых цитокинов.
Резюмируя вышесказанное, анализы уровней у пациентов до лечения показали, что уровни IL-6, IL-10, IL-2, IL-15 являются повышенными у значительной доли пациентов с гастроэзофагеальным заболеванием поздней стадии. Напротив, ни один или только отдельные пациенты имели повышенные уровни IL-1, IL-12, IL-4, IFNγ и TNFα.
Анализ изменения уровней цитокинов в течение первых 5 дней после лечения IMAB362 привел к следующим результатам. Было обнаружено, что у всех пяти пациентов с повышенными уровнями IL-2 эти уровни значительно снизились, при этом четверо из пяти пациентов достигли нормальных референсных значений. Аналогичным образом, у всех четырех пациентов с повышенными уровнями IL-15 наблюдалось умеренное снижение после введения IMAB362. Снижение повышенных уровней после лечения также наблюдалось для отдельных пациентов с повышенными уровнями IFNγ и TNFα, соответственно. Напротив, уровень IL-6 повысился после введения IMAB362, при этом четверо пациентов до лечения и 7 из 14 пациентов на день 5 после лечения показали IL-6 выше референсных уровней.
Пример 4. Международное, многоцентровое, открытое исследование фазы IIа с использованием многократных доз, оценивающее эффективность и безопасность многократных доз IMAB362 у пациентов с аденокарциномой желудка или нижнего отдела пищевода поздней стадии
Международное, многоцентровое, открытое исследование фазы IIа с использованием многократных доз проводили для изучения эффективности и безопасности многократных доз IMAB362 у пациентов с аденокарциномой желудка или нижнего отдела пищевода поздней стадии. Главной целью данного исследования являлось исследование скорости ремиссии (CR, PR) в соответствии с RECIST. Дополнительными целями данного исследования являлись: частота и тяжесть нежелательных явлений согласно СТСАЕ v3.0 и переносимость многократных доз IMAB362; время выживания без прогрессирования (PFS): время от начала первой инфузии до даты первого наблюдаемого прогрессирования заболевания или смерти по любой причине (независимо от того, что возникает в первую очередь); иммуногенность по данным анализа человеческих антихимерных антител; качество жизни; частота клинической эффективности (CR, PR и SD в соответствии с RECIST); и фармакокинетика IMAB362 по уровням в сыворотке.
Пациенты подвергались скринингу на определение присутствия CLDN18.2-мишени для IMAB362 в их опухолях. Статус CLDN18.2 определяли с помощью иммуногистохимии с помощью антитела к клаудину-18 в соответствии со стандартизированным протоколом. В исследование были включены пациенты с опухолями, по меньшей мере, 50% клеток которых были окрашены с интенсивностью, по меньшей мере, 2+ (двойная интенсивность). Критерии включения и исключения проверяли во время скринингового визита (V1). Пациентов набирали из университетских госпиталей, специализирующихся на лечении гастроэзофагеального рака.
Пациенты должны удовлетворять всем из следующих критериев включения:
Метастатическое, рефракторное или рецидивирующее заболевание аденокарциномы желудка или нижнего отдела пищевода поздней стадии, подтвержденное гистологией,
- Экспрессия CLDN18.2, подтвержденная иммуногистохимией залитого в парафин образца опухолевой ткани, по меньшей мере, в 50% опухолевых клеток с интенсивностью окрашивания, по меньшей мере, 2+ (по шкале от 0 до 3+),
- По меньшей мере, один поддающийся измерению участок заболевания в соответствии с критерием RECIST (проведения компьютерной томографии (СТ) или магнитно-резонансной томографии (MRI) не ранее, чем за 2 недели до визита 2),
- Возраст ≥18 лет,
- Письменное информированное согласие,
- Общее состояние здоровья по шкале ECOG (PS) 0-1 или индекс Карновского 70-100%,
- Ожидаемая продолжительность жизни >3 месяцев,
- Количество тромбоцитов ≥100000/мм3,
- Гемоглобин ≥10 г/дл,
- Билирубин в норме,
- AST и ALT <2,5 раз превышает верхний предел нормы (ULN) (в 5 раз превышает ULN в случае присутствия метастазов в печени),
- Креатинин <1,5 × ULN,
- Для женщин с репродуктивным потенциалом (последняя менструация менее чем за 2 года до включения): Отрицательный тест на наличие беременности (β-HCG) на момент начала исследования и использование двух высокоэффективных способов контрацепции во время фазы лечения и в течение 8 недель после последней инфузии исследуемого лекарственного средства,
- Пациенты мужского пола, у которых сексуальными партнерами были женщины с репродуктивным потенциалом, должны использовать одобренный способ контрацепции во время фазы лечения и в течение 8 недель после последней инфузии исследуемого лекарственного средства.
Пациенты, удовлетворяющие любому одному или нескольким из следующих критериев исключения, не допускаются к включению в исследование:
- Беременность или кормление грудью
- Предшествующая тяжелая аллергическая реакция или непереносимость моноклонального антитела, включая гуманизированные или химерные антитела
- Менее 3 недель после предшествующей химио- или лучевой терапии
- Другие экспериментальные препараты или устройства одновременно или в пределах 4 недель до данного исследования
- Другие сопутствующие противораковые терапии (не по показаниям данного исследования)
- Известная ВИЧ-инфекция или известный активный гепатит (А, В, С)
- Сопутствующая антикоагуляция с антагонистами витамина K (например, кумадин, маркумар)
- Терапевтические дозы гепарина (профилактические дозы допускаются)
- Неконтролируемое заболевание, включая, но без ограничения любое из следующих:
- Продолжающаяся или активная инфекция, требующая парентеральных антибиотиков
- Сердечная недостаточность с застойными явлениями
- Нестабильная стенокардия
- Неконтролируемая гипертония
- Клинически значимая сердечная аритмия
- Инфаркт миокарда в течение последних 6 месяцев
- Кровоточащая язва желудка в течение последних четырех недель
- Симптоматическая пептическая язва
- Клинические симптомы метастазов в головном мозге
- Психиатрическое заболевание или социальные ситуации, которые препятствуют приверженности лечению
Все пациенты всех когорт получали повторные дозы IMAB362 каждые две недели во время визитов 2, 5, 6, 7 и 8 (5 применений). Процедуры с эскалацией дозы охватывали следующие когорты с двумя различными дозам (антитело/площадь поверхности тела) IMAB362:
Раствор антитела вводили в виде 2 ч внутривенной инфузии каждые две недели. Важно, чтобы продолжительность инфузии составляла не менее 2 часов. Для инфузии следует использовать инфузионную систему (например, Infusomat® fmS) для контроля времени инфузии. Для применения лекарственного средства следует использовать набор для инфузии, поставляемый вместе с исследуемым лекарственным средством, которое было протестировано изготовителем на совместимость. Инфузию лекарственного средства следует проводить утром. Квалифицированный врач должен быть доступен во время инфузии и в течение 24 часов после инфузии.
Тридцать семь пациентов получали, по меньшей мере, одну терапию. К сожалению, для 3 из них в базе данных имеется неполная документация, поэтому в популяцию пациентов, получивших хотя бы одну дозу исследуемого препарата (all patients treated set, APT set), будут включены 34 пациента и использованы для анализа безопасности. Четыре, 6 и 24 пациента распределяли соответственно в когорту 1 с 300 мг/м2 IMAB362, когорту 2 с 600 мг/м2 IMAB362 и когорту 3 с 600 мг/м2 IMAB362.
Во время фазы лечения один пациент из когорты 1, три пациента из когорты 2 и 12 пациентов из когорты 3 прервали исследования до получения 5 инфузий IMAB362 и завершили визит 9 (включая вторую визуализацию опухоли) через две недели после пятой инфузии. Эти пациенты были заменены.
Два пациента в когорте 2 не имели измеряемого заболевания на момент начала исследования и были исключены из анализов эффективности. Возникали незначительные отклонения от протокола, такие как оценка опухоли на исходном уровне на >14 дней раньше, чем Визит 2 (n=3; 8,8%), гемоглобин <10 г/дл (n=5; 14,7%), отклоняющиеся от нормы показатели билирубина (n=3, 8,8%), ALT или AST >2.5 ULN (>5 ULN в случае метастазов в печени) (n=2; 5,9%), показатель для креатинина >1,5 ULN (n=1; 2,9%) и увеличенный промежуток времени (>15 дней) между периодом скрининга и началом лечения (n=2; 5,9%), но не приводили к исключению из любого анализа. Один пациент имел инфаркт миокарда в течение последних 6 месяцев. Отказ от исполнения был предоставлен.
Так как в когорте 2 и 3 пациенты получали одинаковую дозу, составляющую 600 мг/м2, было решено анализировать этих пациентов как одну группу. Все пациенты (n=34) АРТ-популяции принадлежали к белой (кавказоидной) расе. Средний возраст составил 62 года (45-65 лет) в дозовой группе 300 мг/м2 и 61 год (42-77 лет) в дозовой группе 600 мг/м2.
Обзор локализации рака и результат гистопатологической оценки показан в Таблице 10. Средний период времени между первым диагнозом и скрининговым визитом для этой группы составил 16 месяцев (минимум 2,7 / максимум 56). Статус экспрессии HER2/neu был в основном неизвестным для пациентов, за исключением 5 пациентов, получавших лечение 600 мг/м2. Один из этих 5 пациентов был НЕK2/neu-положительным.
Система классификации степени онкологических заболеваний (TNM) была установлена для рака желудка (n=16) и пищевода или гастроэзофагеального соединения (n=19). В АРТ-популяции 25% пациентов, представленных с первичными опухолями желудка, были классифицированы с Т1 или 2, 31% представлены с Т3, 25% с Т4 первичными опухолями и для 19% это было неизвестно. Шестьдесят девять (69) % пациентов в АРТ-популяции имели, по меньшей мере, один или два инфильтрованных лимфатических узла, обозначенные согласно классификации N1, и 56% пациентов, страдали периферическими метастазами (M1) на момент постановки диагноза. Шестьдесят девять (69%) пациентов с раком пищевода или гастоэзофагельного соединения были диагностированы с ≥ Т3. По меньшей мере об одном или двух инфильтрованных лимфатических узлах (N1) сообщалось для 84% пациентов. Кроме того, 84% пациентов имели периферические метастазы.


На основе системно-органных классов (System Organ Class, SOC) Медицинского словаря регуляторной деятельности MedDRA наиболее частыми клинически значимыми перенесенными заболеваниями были хирургические процедуры у 25 пациентов (73,5%), химиотерапия у 30 пациентов (88,2%) и облучение у 7 пациентов (79,4%). В большинстве случаев хирургическое вмешательство состояло в хирургическом удалении органов (таком как гастроэктомия (72%), резекция пищевода (16%), иссечение лимфатических узлов (32%), холецистэктомия (20%)). Все пациенты, за исключение четырех, имели, по меньшей мере, одну предшествующую терапию их заболевания согласно исследованию. На основе WHO DD АТС наиболее часто используемыми лекарственными средствами являлись аналоги пиримидина (фторурацил и/или капецитабин), соединения платины (цисплатин и/или оксалиплатин) и детоксифирующие агенты для противоопухолевого лечения (фолинат кальция и/или фолиниевая кислота). Другие предшествующие терапии (прекращающееся самое позднее в день инфузии) также были документально зафиксированы.
Всего 30 из 34 пациентов (88,2%) имели, по меньшей мере, одно сопутствующее заболевания, то есть заболевание, которое продолжалось на день инфузии исследуемого препарата. На основе MedDRA SOC наиболее распространенными диагнозами являлись «нарушения со стороны желудочно-кишечного тракта» у 19 пациентов (56%), «общие нарушение» у 12 пациентов (35%), «нарушения со стороны обмена веществ и питания» у 10 пациентов (29%) и «нарушения со стороны скелетно-мышечной и соединительной ткани» у 8 пациентов (23,5%). Сопутствующими терапиями в основном являлись лекарственные средства против кислотозависимых заболеваний (17 пациентов; 50%), болеутоляющие средства (12 пациентов; 35,3%) и лекарственные препараты для заболеваний желудочно-кишечного тракта (10 пациентов; 29,4%).
А. ОЦЕНКА БЕЗОПАСНОСТИ
Так как инъекции исследуемого препарата вводились исследователями в исследовательских центрах и пациенты должны были оставаться в госпитале для наблюдения в течение 24 часов и вплоть до 72 часов, общая приверженность лечению в соответствии с протоколом исследования была гарантирована. Распределение включенных в исследование субъектов по дозовым когортам выполнялось, как указано в протоколе исследования (под контролем DSMB). Продолжительность исследования, определенная как время от даты скрининговой части 1 визита до последнего дня исследования изменялась от минимум 18 дней до максимум 355 дней. Средняя продолжительность исследования составила 106 дней. 16 пациентов прервали исследование преждевременно перед целевым визитом 9.
Пациенты во всех дозовых группах получали в среднем от 4,5 до 5 инфузий IMAB362. Средняя продолжительность одной инфузии IMAB362 в АРТ-популяции составила 125 минут. Имелся один пациент с продолжительностью инфузии менее 120 минут, указанной в протоколе. Этот пациент остановил инфузию по причине рвоты и преждевременно прервал исследование.
Анализ безопасности проводили для АРТ-популяции, включающей 34 пациента, которые получали, по меньшей мере, одну дозу 300 мг/м2 (n=4) или 600 мг/м2 (n=30). Нежелательные явления в количестве, равном двести сорок одно (241), по описанию терапевта были кодированы в соответствии со словарем MedDRA и переведены в предпочтительные термины. Нежелательные явления в соответствии с предпочтительными терминами подсчитывали только один раз для каждого пациента (также, если такое же нежелательное явление возникало более одного раза для этого пациента во время исследования). Самый высокий балл согласно NCI-CTC, возникающий у каждого пациента, записывали. Тридцать два (32, 94%) пациента имели, по меньшей мере, одно нежелательное явление (независимо от взаимосвязи) во время исследования. Для 2 пациентов не было зафиксировано нежелательных явлений. Всего 6 пациентов (18%) не испытывали нежелательное явления, возможно связанного с лекарственным средством. Сто четыре (104) связанных с лекарственным средством нежелательных явления с помощью предпочтительных терминов сообщались для 28 пациентов. Восемь (8) возможно связанных с лекарственным средством серьезных нежелательных явления было зафиксировано для 4 пациентов. Количество пациентов в дозовой группе с более низкой дозой (300 мг/м2) были слишком малым для обеспечения тщательного сравнения между обеими дозовыми группами. Частота случаев пациентов с нежелательными явлениями, связанными с лекарственными препаратом, в когорте 300 мг/м2 и группе 600 мг/м2 (когорта 2 и 3) составила 75 и 83%, соответственно.
Всего наиболее часто сообщаемыми нежелательными явлениями (АЕ) согласно системно-органным классам (SOC) являлись «нарушения со стороны желудочно-кишечного тракта» (27/34 пациентов, 79.4%), «общие расстройства и нарушения в месте введения» (26/34 пациентов, 76.5%). На основе MedDRA РТ наиболее часто сообщаемыми нежелательными явлениями (АЕ) являлись «тошнота» (57 явлений у 18 пациентов), «рвота» (52 явлений у 16 пациентов) и «утомляемость» (20 явлений у 14 пациентов). Всего только 192 из зафиксированных нежелательных явлений (АЕ) оценивалось исследователями как связанные с исследуемым лекарственным средством. Эти связанные с проводимым лечением нежелательные явления (АЕ) были классифицированы в 104 различных предпочтительных термина и наблюдались у 28/34 пациентов.
Большинство связанных с проводимым лечением нежелательных явлений были от легких до умеренных. Имелось 8 (23,5%) пациентов с умеренными, связанными с лекарственным средством нежелательными явлениями, возникающими во время лечения, и 12 (35,3%) пациентов с тяжелыми, связанными с лекарственным средством нежелательными явлениями, возникающими во время лечения.
Связанные с лекарственным средством нежелательные явления (АЕ) сильной интенсивности отмечались у 2 пациентов в дозовой группе 300 мг/м2, рвота и у одного пациента сопутствующая тошнота. В дозовой группе 600 мг/м2 10 пациентов испытывали тяжелые нежелательные явления, связанные с лекарственным средством, 6 пациентов с рвотой, из которых 3 пациента дополнительно испытывали тошноту, один пациент с гиперчувствительностью (аллергической реакцией), один пациент с гиперсекрецией слюны, один пациент с обезвоживанием, и один пациент с гипоальбуминемией. У последних двух пациентов также отмечалась тошнота и рвота. Два пациента страдали связанной с лекарственным средством гиперчувствительностью (аллергической реакцией) во время инфузии лекарственного средства, одна из которых была классифицирована как умеренная или одна как тяжелая. Оба пациента восстановились после прекращения инфузии.
Во всех отмеченных нежелательных явлениях, возникающих в ходе лечения, действие исследуемого лекарственного средства для 12/34 пациентов было неизбежно связано с нежелательным явлением. В 7 (21%) случаях нежелательное явление приводило к длительному прерыванию исследования. Основное нежелательное явление было связано с лекарственным средством у 3 (гиперчувствительность (аллергическая реакция) (n=2), рвота и боль в животе) и не связано с лекарственным средством у других четырех пациентов (ухудшение общего состояния здоровья (n=3), пневмония). У одного пациента доза была уменьшена, и у другого пациента введение дозы было отложено на 4 дня по причине серьезной рвоты с тошнотой. У трех пациентов инфузия была прервана/продлена. Двадцать семь пациентов (79%) получали сопутствующую терапию по причине нежелательного явления. Одиннадцать пациентов были госпитализированы.
У 13 пациентов было зафиксировано 31 серьезное нежелательное явление (SAE). Один пациент умер во время фазы второго скрининга исследования. Двенадцать пациентов имели другие серьезные нежелательные явления, которые были связаны с исследуемым лекарственным средством у четырех пациентов. Рвота, тошнота и связанные нежелательные явления, такие как желудочно-кишечное (GI) кровотечение и эксикоз, были оценены исследователями как связанные с исследуемым препаратом. В настоящем исследовании было отмечено 4 SAR и 2 SUSAR (рвота и рвота с GI кровотечением). Конечным исходом была смерть в семи случаях. Ни один из смертельных исходов не был классифицирован исследователями как связанный с исследуемым препаратом.
Один пациент был мужского пола кавказоидной расы в возрасте 45 лет с хорошим общим состоянием здоровья (статус состояния здоровья согласно ECOG стадии 1, индекс Карновского 80%) со статусом диеты для уменьшения веса тела (BMI 19.3).
Пациент получал инфузии 300 мг/м2 IMAB362 каждые две недели, 04 ноября, 22 ноября и третью 6 декабря 2011 года. До исследования пациент уже страдал тошнотой и рвотой 1 степени. 7 ноября 2010 года была диагностирована рвота 3 степени. Так как она оценивалась как серьезная, пациент был госпитализирован. Когда рвота изменилась на 1 степень 17 ноября 2010 года и в итоге полностью прекратилась, пациент в тот же день был выписан из госпиталя. Перед второй и третьей инфузией IMAB362 пациент получал лечение сильными премедикационными лекарственными средствами (ализаприд, апрепитант, метоклопрамид, димегидринат) в качестве профилактики тошноты и рвоты, таким образом, он не страдал снова тошнотой и рвотой. Исследователь оценил рвоту как связанную с исследуемым лекарственным средством. Отчет был получен спонсором 19 января 2011 года и серьезное нежелательное явление (SAE) оценивалось как неожидаемое, но связанное с исследуемым лекарственным средством и, таким образом, указанное как SUSAR.
Один пациент был мужского рода кавказоидной расы в возрасте 77 лет. Он находился в очень хорошем общем состоянии (общее состояние здоровья согласно ECOG: стадия 0, индекс Карновского: 100%) с нормальным статусом диеты (BMI 24) при скрининге. Перед исследованием пациент уже страдал тошнотой и поэтому получал лечение по мере необходимости метоклопрамидом. Пациент получил только однократно 600 мг/м2 IMAB362 9 ноября 2011 года, так как исследование было прервано по причине смерти. Плевральный выпот в левом легком был диагностирован с помощью рентгенографии перед инфузией и описан как SAE. На следующее утро начался гематемезис. После введения пантопразола и 8 мг ондансетрона, внутривенно, рвота уменьшилась, и гематемезис прекратился в тот же день. Рвота уменьшилась с 3 степени до 2 степени и окончательно прекратилась 12 ноября 2011 года, так что пациент был выписан из госпиталя 13 ноября 2011 года. Исследователи оценивали событие как связанное с исследуемым лекарственным средством. Отчет быть получен спонсором 10 ноября 2011 года, и событие расценивалось как неожидаемое, но связанное с исследуемым лекарственным средством и поэтому было указано как SUSAR. Общее состояние пациента ухудшилось, у него развилась почечная недостаточность и, к сожалению, он умер 6 декабря 2011 года.
Один пациент был мужского рода кавказоидной расы в возрасте 42 лет в очень хорошем общем состоянии (общее состояние здоровья согласно ECOG: стадия 0, индекс Карновского: 100%) со статусом нормального питания (BMI 26). Пациент получил две инфузии 600 мг/м2 IMAB362. 20 марта 2012 года пациент получил первое введение исследуемого лекарственного средства. Так как он страдал тошнотой и серьезной рвотой, скорость инфузии была уменьшена через 35 минут после начала инфузии. Симптомы лечили с помощью 40 мг пантопразола и 3 мг гранисетрона, и 2 ампул внутривенно бутилскополамина, и 80 мг внутривенно апрепитанта. Это серьезное нежелательное явление привело к продолжительной госпитализации. Исследователь оценил данное явление как связанное с исследуемым лекарственным средством. Отчет о SAE был получен спонсором 21 марта 2012 года, и событие было оценено как ожидаемое и связанное с исследуемым лекарственным средством. Через несколько дней, 24 марта 2012 года пациент был госпитализирован снова по причине серьезного обезвоживания, которое было вызвано тошнотой и рвотой. Кроме того, пациент страдал от боли в пищеводе. Он получил 1 г внутривенно метамизола, бупренорфиновый пластырь и инфузии против обезвоживания. 30 марта 2012 года симптомы ослабли и пациент регидратировал. Исследователь оценил данное событие как не связанное с исследуемым лекарственным средством. Отчет о SAE был получен спонсором 26 марта 2012 года, и событие оценивалось как неожидаемое и не связанное с исследуемым лекарственным средством. 3 апреля 2012 года пациент получил вторую инфузию, которая снова вызвала нежелательные явления в виде тошноты и рвоты. Пациент получил 30 капель перорально метоклопраминда и 1 ампулу внутривенно дименгидраната. Так как симптомы ухудшились 5 апреля 2012 года, они были оценены как тяжелые. Кроме того, пациента мучила дисфагия, и поэтому сильно сократился прием пищи. 15 апреля 2012 года симптомы прошли. Исследователь оценил данное событие как связанное с исследуемым лекарственным средством. Отчет о SAE был получен спонсором 19 апреля 1012 года, и событие оценивалось как ожидаемое и связанное с исследуемым лекарственным средством.
Один пациент был мужского рода кавказоидной расы в возрасте 73 лет в хорошем общем состоянии (общее состояние здоровья согласно ECOG: стадия 1, индекс Карновского: 90%) со статусом нормального питания (BMI 26). С 8 ноября 2011 года по 3 января 2012 года пациент получал пять запланированных введений исследуемого лекарственного средства IMAB362 в дозе 600 мг/м2 каждые две недели. 8 ноября 2011 года пациент получил первое введение IMAB362. Во время и после инфузии он страдал тошнотой и рвотой. Симптомы стали тяжелыми 9 ноября 2011 года. После лечения метоклопрамидом симптомы прошли днем позже. Исследователь оценил событие как связанное с исследуемым лекарственным средством. Отчет о SAE был получен спонсором 10 ноября 2011 года, и событие оценивалось как ожидаемое и связанное с исследуемым лекарственным средством. 6 декабря 2011 года была проведена третья инфузия. Пациент страдал легкой рвотой и умеренной тошнотой, получал лечение клемастином, ранитидином и ондансетроном. Рвота продолжалась один день. Тошнота продолжалась в течение 7 дней. Исследование прервали 16 января 2012 года вследствие прогрессирования заболевания. Визит контрольного наблюдения не проводили.
В заключение было обнаружено, что IMAB362 является безопасным и хорошо переносимым у популяции тяжелых, ранее проходивших лечение пациентов с аденокарциномой желудка, пищевода или гастроэзофагеального соединения поздней стадии. Всего наиболее часто сообщаемыми нежелательными явлениями (АЕ) согласно системно-органным классам (SOC) являлись «нарушения со стороны желудочно-кишечного тракта» (27/34 пациентов, 79.4%), «общие расстройства и нарушения в месте введения» (26/34 пациентов, 76.5%).
На основе MedDRA РТ наиболее часто сообщаемыми нежелательными явлениями (АЕ) являлись «тошнота» (57 явлений у 18 пациентов), «рвота» (52 явления у 16 пациентов) и «утомляемость» (20 явлений у 14 пациентов).
Всего 192 из зафиксированных нежелательных явлений (АЕ) оценивались исследователями как связанные с исследуемым препаратом. Эти связанные с проводимым лечением нежелательные явления (АЕ) наблюдались у 28 из 34 пациентов. Восемьдесят три (83) процента этих связанных АЕ представляли собой нарушения со стороны желудочно-кишечного тракта (68%, 130 АЕ), зафиксированные у 25 пациентов и общие нарушения (15%, 29 АЕ), зафиксированные у 16 пациентов.
На основе MedDRA РТ наиболее связанные с проводимым лечением нежелательные явления были от легких до умеренных с тошнотой (50%), рвотой (47%), утомляемостью (27%), болью в животе (15%), периферическим отеком (15%), сниженным аппетитом (12%) и диареей (12%), возникающей у более 10% пациентов.
Два пациента страдали связанной с проводимым лечением гиперчувствительностью (аллергической реакцией) во время инфузии лекарственного средства, одна из которых была классифицирована как умеренная и одна как тяжелая. Оба пациента восстановились после прекращения инфузии.
Отклоняющихся от нормы связанных с исследуемым лекарственным средством лабораторных величин СТС 4 степени (угрожающих жизни) или 5 (смерть) не отмечалось.
Имелось 12 (35,3%) пациентов с возникающими во время лечения нежелательными явлениями. Связанные с лекарственным средством нежелательные явления (АЕ) сильной интенсивности отмечались у 2 пациентов в дозовой группе 300 мг/м2, рвота и у одного пациента сопутствующая тошнота. В дозовой группе 600 мг/м2 10 пациентов испытывали тяжелые связанные с лекарственным средством нежелательные явления, 6 пациентов с рвотой, из которых 3 пациента испытывали дополнительно тошноту, один пациент с гиперчувствительностью (аллергическая реакция), один пациент с гиперсекрецией слюны, один пациент с обезвоживанием и один пациент с гипоальбуминемией. У последних двух пациентов также отмечалась рвота и тошнота.
На момент анализа 13 пациентов восстановились от всех связанных с лекарственным средством нежелательных явлений, 2 пациента находились на стадии восстановления, 11 пациентов не восстановились, по меньшей мере, от одного нежелательного явления (АЕ) и для 2 статус был неизвестен. Из 11 пациентов, у которых, по меньшей мере, одно связанное с лекарственным средством нежелательное явление не прекратилось, 9 имели нарушения со стороны желудочно-кишечного тракта (4 тошнота, 2 рвота).
У 13 пациентов было зафиксировано 31 нежелательное явление (АЕ), в том числе 7 смертельных исходов. Один пациент умер во время фазы скрининга, то есть до начала инфузии исследуемого лекарственного средства, и был, таким образом, классифицирован как явление, связанное со скринингом (screening event). У четырех пациентов возникшие в ходе лечения нежелательные явления (SAE) со стороны желудочно-кишечного тракта, такие как рвота (n=4), тошнота (n=2), эксикоз (n=1) и желудочно-кишечное (GI) кровотечение (n=1) оценивались как связанные с лечением. Один из этих пациентов с рвотой получал 300 мг/м2, остальные трое получали 600 мг/м2 IMAB362. Трое из этих четырех пациентов восстановились, за исключением одного, который умер по причине почечной недостаточности, не связанной с проводимым лечением.
Частота возникновения нежелательных явлений, связанных с лекарственным средством, была сравнимой между дозовыми группами 300 мг/м2 и 600 мг/м2, составляющая 75% и 83% пациентов, соответственно. Частота и тяжесть тошноты, рвоты и утомляемости также были сравнимы между обеими дозовыми группами. Не существовало четкой взаимосвязи между дозой и частотой/тяжестью нежелательных явлений.
Профиль нежелательных явлений с большинством нежелательных явлений (АЕ), отмечаемых со стороны желудочно-кишечного тракта, согласуется с основным заболеванием, а также профилем экспрессии CLDN18.2. Предполагается, что тошнота и рвота являются целевым эффектом (on-target effect), так как CLDN18.2 также экспрессируется на клетках эпителия желудка (в плотных контактах).
В целом, наблюдалось, что IMAB362, вводимый в виде многократных доз, составляющих 300 мг/м2 и 600 мг/м2, является безопасным и хорошо переносимым, с рвотой и тошнотой, которые являются наиболее распространенными связанными с проводимым лечением нежелательными явлениями.
В. ОЦЕНКА ФАРМАКОКИНЕТИКИ И ИММУНОГЕННОСТИ
Предварительные данные, касающиеся концентрации лекарственного средства для применения многократных доз IMAB362 имеются для четырех пациентов из первой и 34 пациентов из второй и третьей когорт, которые получали 300 мг/м2 и 600 мг/м2 IMAB362, соответственно.
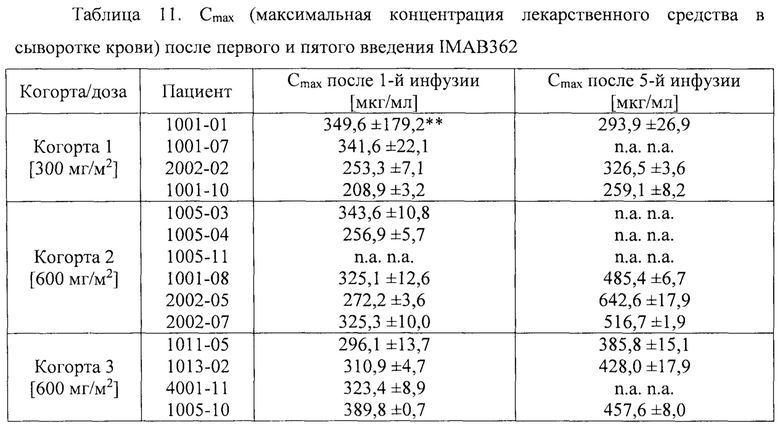
Образцы крови собирали перед каждой инфузией. После первой инфузии дополнительные образцы собирали в конце инфузии и через 1, 1.5, 2, 3, 4, 6, 12, 24 часа, 3 и 6 дней после завершения инфузии. После последней инфузии образцы собирали в конце инфузии и через 1, 1,5, 2 часа и 14 дней, а также через 4-8 недель после завершения последней инфузии. Анализируемого вещества не было обнаружено в образцах до введения доз от пациентов, распределенных в когорту 1-3.
После первой инфузии IMAB362 значения Cmax находились в интервале от 208,9 мкг/мл до 349,6 мкг/мл для первой когорты. Для второй и третьей когорт, взятые вместе, значения Cmax находились в интервале от 269,1 мкг/мл до 575,1 мкг/мл после первого применения.
В образцах сыворотки, взятых в последующие моменты времени (от V3 до V5), наблюдалось зависимое от времени снижение концентрации IMAB362 (фигура 8). Во время визита 5 до второй инфузии минимальные уровни в сыворотке в диапазоне от 11,3 мкг/мл до 36,8 мкг/мл (среднее значение 22,5±10,5 мкг/мл) были определены для когорты 1, и в диапазоне от 17,0 мкг/мл до 100,2 мкг/мл (среднее значение 54,5±29,0 мкг/мл) для взятых вместе когорт 2 и 3.
Во время визита 8 (день 57) до пятой инфузии минимальные уровни в сыворотке в диапазоне от 32,4 мкг/мл до 67,1 мкг/мл (среднее значение 46,1±18,5 мкг/мл) были определены для когорты 1, и в диапазоне от 28,3 мкг/мл до 301,6 мкг/мл (среднее значение 147,2±93,1 мкг/мл) для когорт 2 и 3 (таблица 12).
После инфузии во время визита 8, значения Cmax находились в интервале от 259,1 мкг/мл до 326,5 мкг/мл для первой когорты, и в интервале от 278,1 мкг/мл до 642,6 мкг/мл для когорт 2 и 3 (таблица 11).
Для когорты 1 средние значения Cmax были определены через 90 мин после первой инфузии IMAB362 (270,6±63,9 мкг/мл) и через 90 мин после пятой инфузии (279,2±27,7). Для взятых вместе когорт 2 и 3 средние значения Cmax были определены в конце первой инфузии IMAB362 (340,8±80,2 мкг/мл) и через 60 мин после пятой инфузии (443,3±97,7) (таблица 12).
Таким образом, измеренные уровни IMAB362 в сыворотке крови показали, что у пациентов, получавших лечение в дозе 300 мг/м2, концентрация IMAB362 в сыворотке крови опускалась ниже желательного уровня, составляющего 50-100 мкг/мл в период между 2-х недельными циклами. При дозе 600 мг/м2, напротив, у большинства пациентов уровни IMAB362 в сыворотке крови составили выше 50 мкг/мл даже через 2 недели после первого применения. Через 7-29 дней (в среднем 15 дней) после 5-го введения уровень дозы составил выше 50 мкг/мл (среднее значение 151,3±90,1 мкг/мл).
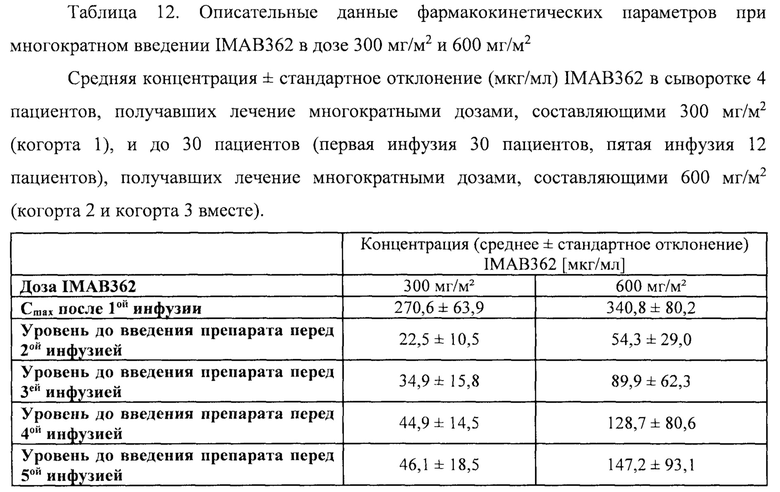

Умеренное накопление IMAB362 наблюдалось от цикла к циклу. Коэффициент накопления изменялся в диапазоне от 1,03-кратного до 3,52-кратного по отношению к первому значению до введения дозы перед второй инфузией (среднее значение 2,04).
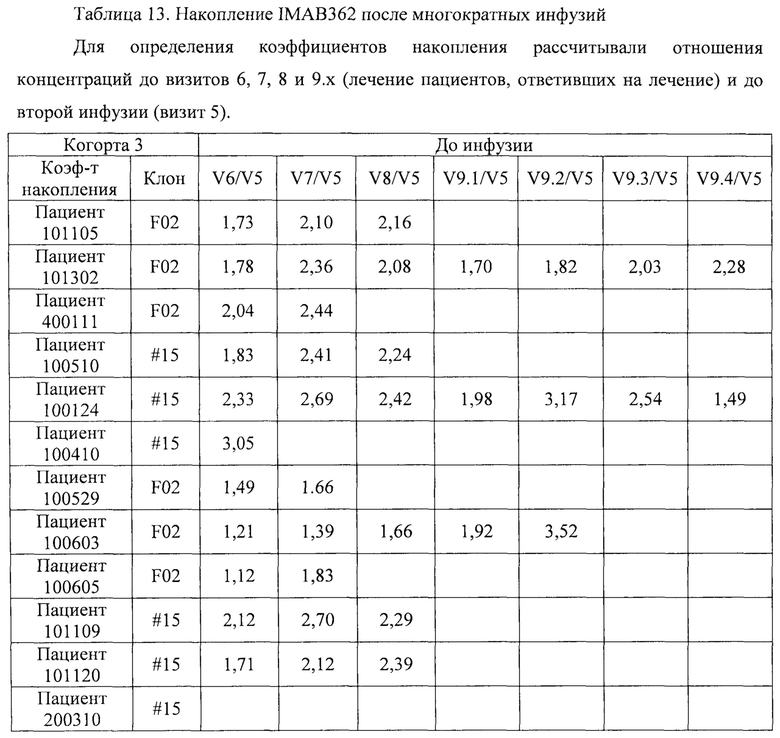
В заключение, было обнаружено, что фармакокинетика IMAB362 является дозозависимой.
После первой инфузии IMAB362 значения Cmax находились в интервале от 208,9 мкг/мл до 349,6 мкг/мл для первой когорты. Для второй и третьей когорт, взятых вместе, значения Cmax находились в интервале от 269,1 мкг/мл до 575,1 мкг/мл после первого применения.
В образцах сыворотки, взятых в последующие моменты времени (от V3 до V5), наблюдалось зависимое от времени снижение концентрации IMAB362. Во время визита 5 до второй инфузии минимальные уровни в сыворотке в диапазоне от 11,3 мкг/мл до 36.8 мкг/мл (среднее значение 22,5±10,5 мкг/мл) были определены для когорты 1, и в диапазоне от 17,0 мкг/мл до 100,2 мкг/мл (среднее значение 54,5±29,0 мкг/мл) для когорт 2 и 3, взятых вместе.
Во время визита 8 (день 57) до пятой инфузии минимальные уровни в сыворотке в диапазоне от 32,4 мкг/мл до 67,1 мкг/мл (среднее значение 46,1±18,5 мкг/мл) были определены для когорты 1, и в диапазоне от 28,3 мкг/мл до 301,6 мкг/мл (среднее значение 147,2±93,1 мкг/мл) для когорт 2 и 3.
После 5-ой инфузии во время визита 8 значения Cmax находились в интервале от 259,1 мкг/мл до 326,5 мкг/мл для первой когорты, и в интервале от 278,1 мкг/мл до 642,6 мкг/мл для когорт 2 и 3.
Для когорты 1 средние значения Cmax были определены через 90 мин после первой инфузии IMAB362 (270,6±63,9 мкг/мл) и через 90 мин после пятой инфузии (279,2±27,7). Для взятых вместе когорт 2 и 3 средние значения Cmax были определены в конце первой инфузии IMAB362 (340,8±80,2 мкг/мл) и через 60 мин после пятой инфузии (443,3±97,7).
Таким образом, измеренные уровни IMAB362 в сыворотке крови показывают, что у пациентов, получавших лечение в дозе 300 мг/м2, концентрация IMAB362 в сыворотке крови опускалась ниже желательного уровня, составляющего 50-100 мкг/мл в период между 2-х недельными циклами. Напротив, при дозе 600 мг/м2 у большинства пациентов уровни IMAB362 в сыворотке крови составили выше 50 мкг/мл даже через 2 недели после первого применения. Через 7-29 дней (среднее число дней равно 15) после 5-го введения уровень дозы составил выше 50 мкг/мл (среднее значение 151,3±90,1 мкг/мл).
С. ОЦЕНКА ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТИ
Полная выборка пациентов для анализа (Full analysis set, FAS):
Включены все субъекты, которые получали исследуемый препарат, по меньшей мере, однократно, и для которых имеются данные по эффективности лечения.
На момент анализа в исследование было включено 50 пациентов при дозе 600 мг/м2. Девять из них были включены недавно и дополнительные данные на текущий момент отсутствовали при причине их недавнего включения. Для десяти пациентов не проводилась вторая визуализация опухоли и эти пациенты, таким образом, не были включены в FAS-выборку. FAS-выборка включает 31 пациента.
Средний возраст составил 57 лет в диапазоне от 35 до 77 лет. Пациенты FAS-выборки имели средний индекс Карновского, равный 90% (в диапазоне 70-100%). Большинство (81%) пациентов ранее получали терапию в виде, по меньшей мере, одного химиотерапевтического режима. Шесть (6) пациентов ранее не получали химиотерапевтического режима.

Медиана числа предшествующих химиотерапевтических режимов составила 2,0 (в интервале от 0 до 5). Химиотерапевтические режимы для гастроэзофагеального рака в основном состоят из различных комбинаций производного 5-FU, соединения платины, таксанов, эпирубицина, иринотекана, трастузумаба для HER2/neu-положительных пациентов и других экспериментальных агентов. В FAS-выборке 81% пациентов получали, по меньшей мере, однократно 5-FU или капецитабин, и 74% получали лечение соединением платины, по меньшей мере, однократно перед включением в исследование. Шесть (6, 19%) пациентов ранее получали лечение трастузумабом или другими экспериментальными агентами. Шесть (6, 19%) пациентов также получали лучевую терапию до начала исследования.
Вследствие поздней стадии заболевания пациенты имели медиану метастатических участков, равную 2,0 (в интервале от 1,0 до 4,0). Наиболее значительными являются лимфатические узлы (19 pts, 61%); печень (13 pts, 42%); асциты (8 pts, 26%) и брюшная полость (7 pts, 23%).
Общая частота контроля заболевания составила 39% (таблица 15). Четыре пациента имели частичный ответ и 8 пациентов имели стабилизацию заболевания. Первую повторную оценку для этих пациентов проводили в период между 8 и 11 неделями после первой инфузии, за исключением двух пациентов, для которых первая повторная оценка опухоли была проведена через 6 недель, соответственно.

У 6 пациентов из 12 с контролем клинического течения заболевания, по меньшей мере, один опухолевый маркер (СЕА; СА19-9; СА125; СА15-3), который был повышенным на момент начала исследования, снизился на 35-76% за период исследования. У трех пациентов все опухолевые маркеры были ниже предельного значения и для одного пациента результаты относительно опухолевого маркера отсутствовали.
Примечательно, что у 4 пациентов с прогрессированием заболевания в качестве лучшего ответа также имелось снижение опухолевого маркера на 29-54% в период исследования.
Частичные ответы были достигнуты через 2,3 месяца лечения (два пациента), 6,5 месяцев (один пациент) и 4,8 месяцев (один пациент) соответственно. Для одного пациента был подтвержден частичный ответ (PR), который продолжался еще в течение 4,4 месяцев, что привело к выживаемости без прогрессирования заболевания (PFS), составляющей 9,2 месяцев для этого пациента. Для других трех пациентов подтверждения были сделаны через 6 (один пациент) и 12 недель (два пациента), соответственно. Более подробная информация представлена в таблице 16.
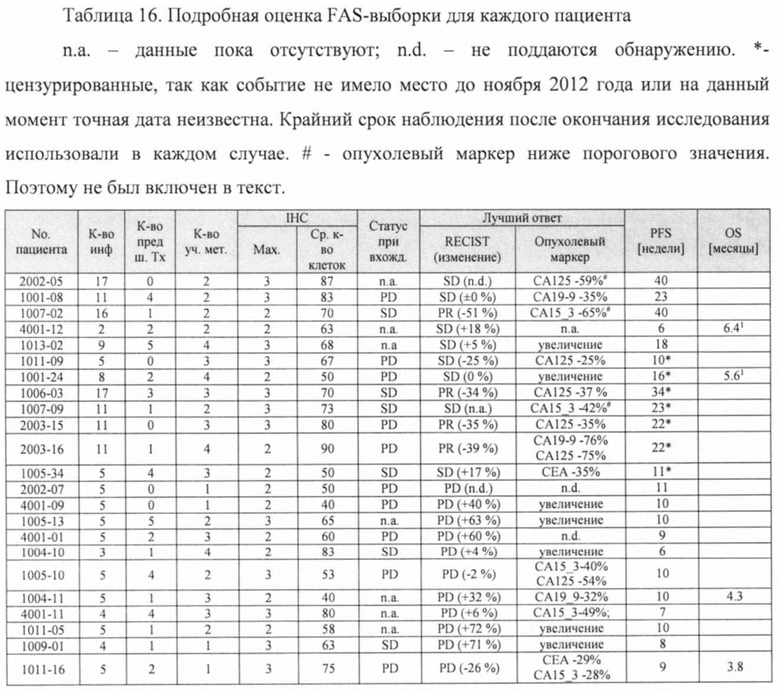

Средняя выживаемости без прогрессирования заболевания для пациентов в FAS-выборке составила 10 недель (минимум 4 недели; максимум 40 недель). По причине ограниченной доступности событий медиана выживаемости без прогрессирования заболевания для пациентов с клинической пользой (PR+SD), показанная на фигуре 9, имеет предельную величину. Пациенты без клинической пользы (PD) имели медиану выживаемости без прогрессирования заболевания, составляющую 9 недель (минимум 4 недели; максимум 11 недель) (фигура 9).
Различия отсутствовали между пациентами с клинической пользой (PR или SD в качестве лучшего ответа) или прогрессированием заболевания (PD в качестве лучшего ответа) в отношении возраста (средний возраст 56 лет против 59 лет), отсутствия предшествующего химиотерапевтического режима (в среднем 1,9 против 2,1), индекса Карновского (в среднем 89% против 88%). Только количество метастатических участков было меньше в группе пациентов, ответивших на лечение, со средним значением 1,9 по сравнению с 2,3 в группе пациентов, не ответивших на лечение. Различие не является статистически значимым.
Интенсивность (средняя и максимальная) IHC-окрашивания была сходной между пациентами с клинической пользой и прогрессированием заболевания. Количество окрашенных клеток различалось между обеими группами. Максимальное количество окрашенных клеток и среднее количество окрашенных клеток было выше у пациентов с клинической пользой со средним значением 77% против 67% и 71% против 60%, соответственно.
Различия также наблюдались в отношении локализации метастаз. У пациентов с клинической пользой частота плеврального выпота (25% против 5%), перитонеального карциноматоза (42% против 11%) и асцитов (42% против 16%) была выше по сравнению с пациентами с прогрессированием заболевания в качестве лучшего ответа. Присутствие метастазов в печени было значительно ниже (17% против 58%) у пациентов с клинической пользой.
Выборка пациентов, выполнивших требования протокола (РР):
РР-популяция включала всех пациентов, которые завершили раздел лечения (до визита 9) без какого-либо существенного отклонения от протокола.
Из 31 пациента в FAS-выборке два пациента имели существенное нарушение протокола (целевой опухолевый очаг отсутствует) и девять пациентов не завершили протокол исследования до визита 9 и, таким образом, получили меньше 5 требуемых инфузий IMAB362. РР-выборка включает 20 пациентов.
Средняя возраста составляла 60 лет в диапазоне от 35 до 77 лет. Пациенты РР-выборки имели средний индекс Карновского 90% (в диапазоне 70-100%). Большинство (80%) пациентов ранее получали лечение в виде, по меньшей мере, одного химиотерапевтического режима. Четыре (4) пациента не получали ранее химиотерапевтического режима лечения.

Средняя числа предшествующих химиотерапевтических режимов составила 2,0 (в диапазоне от 0 до 5). Химиотерапевтические режимы для гастроэзофагеального рака в основном состоят из различных комбинаций производного 5-FU, соединения платины, таксанов, эпирубицина, иринотекана, трастузумаба для HER2/neu-положительных пациентов и других экспериментальных препаратов. В РР-выборке 80% пациентов получали, по меньшей мере, однократно 5-FU или капецитабин и 75% получали лечение соединением платины, по меньшей мере, однократно до включения в исследование. Пять (5, 25%) пациентов получали ранее терапию трастузумабом или другими экспериментальными агентами. Более подробная информация представлена в таблице 17. Пять (5, 25%) пациентов получали лучевую терапию до начала исследования.
Вследствие поздней стадии заболевания пациенты имели медиану метастатических участков, равную 3,0 (в интервале от 1,0 до 4,0). Наиболее значимыми были лимфатические узлы (13 pts, 65%); печень (9 pts, 45%); асциты (6 pts, 30%) и легкое (5 pts, 25%).
Общая частота контроля заболевания составила 50%. Четыре пациента имели подтвержденный частичный ответ и 6 стабилизацию заболевания. Первую повторную оценку для этих пациентов проводили в период между 8 и 11 неделями после первой инфузии, за исключением одного пациента, для которого первую повторную оценку опухоли проводили через 6 недель, соответственно (таблица 18).

У 6 пациентов из 10 с контролем развития клинического заболевания, по меньшей мере, один опухолевый маркер (СЕА; СА19-9; СА125; СА15-3), который был повышен на момент начала исследования, снизился на 35-76% за период исследования. У двух пациентов все опухолевые маркеры были ниже предельного значения и для одного пациента результаты относительно опухолевого маркера отсутствовали.
Примечательно, что 3 пациента с прогрессированием заболевания в качестве лучшего ответа также имели снижение опухолевого маркера на 29-54% за период исследования.
Частичные ответы были достигнуты через 2,3 месяца лечения (два пациента), 6,5 месяцев (один пациент) и 4,8 месяцев (один пациент), соответственно. Для одного пациента был подтвержден частичный ответ (PR), который продолжался еще в течение 4,4 месяцев, что привело к PFS 9,2 месяцев для этого пациента. Для остальных трех пациентов подтверждения были сделаны через 6 (один пациент) и 12 недель (два пациента), соответственно. Более подробная информация представлена в таблице 19.
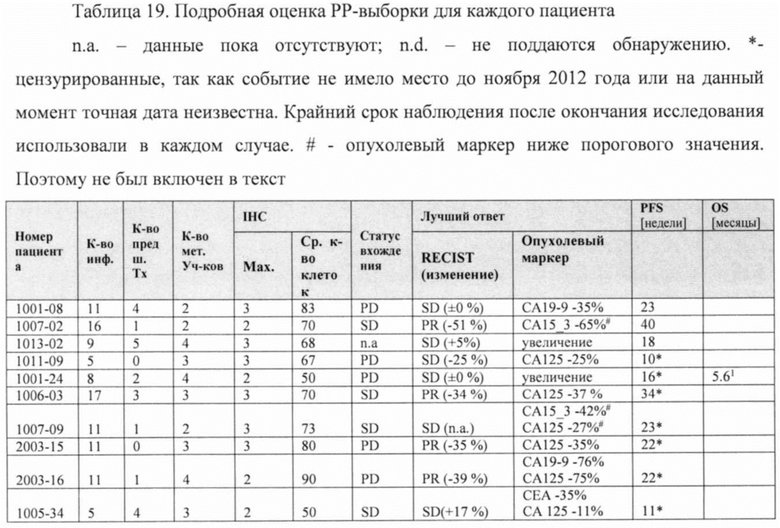

Средняя выживаемости без прогрессирования заболевания для пациентов в РР-выборке составила 18 недель (минимум 9 недель; максимум 40 недель). Вследствие ограниченной доступности событий медиана выживаемости без прогрессирования заболевания для пациентов с клинической пользой (PR+SD), показанная на фигуре 10, имеет предельную величину. Пациенты без клинической пользы (PD) имели медиану выживаемости без прогрессирования заболевания, составляющую 10 недель (минимум 9 недель; максимум 10 недель) (фигура 10).
Различий между пациентами с клинической пользой (PR или SD в качестве лучшего ответа) или прогрессированием заболевания (PD в качестве лучшего ответа) отсутствовали в отношении возраста (средний возраст 57 против 62 лет), предшествующих химиотерапевтических режимов (в среднем 2,1 против 2,0), индекса Карновского (в среднем 88 против 88%). Только количество метастатических участков было выше у пациента с клинической пользой со средним количеством 3,0 по сравнению с 2,2 у пациента без клинической пользы. Различие не является статистически значимым.
Интенсивность (средняя и максимальная) IHC-окрашивания была сходной между пациентами с клинической пользой и прогрессированием заболевания. Количество окрашенных клеток различалось между обеими группами. Максимальное количество окрашенных клеток и среднее количество окрашенных клеток было выше у пациентов с клинической пользой с максимальным показателем 76% против 66% и средним показателем 70% против 66%, соответственно.
Различия также наблюдались в отношении локализации метастазов. У пациентов с клинической пользой частота плеврального выпота (30% против 10%), перитонеального карциноматоза (40% против 0%) и асцитов (40% против 20%) были выше по сравнению с пациентами с прогрессированием заболевания в качестве лучшего ответа. Присутствие метастазов в печени было значительно ниже (20% против 70%) у пациентов с клинической пользой.
В заключение, статус опухоли (в соответствии с RECIST) через 2 недели после 5-й инфузии IMAB362 (V9) сравнивали со статусом, определенным на момент начала исследования. Для 31 пациента (FAS) проводилось, по меньшей мере, одно стадирование после начала исследования. В исследование были включены пациенты с заболеванием на конечной стадии с медианой предшествующих химиотерапий 2,0 и метастатических участков 2,0.
Подтвержденный частичный ответ был установлен у четырех пациентов, что привело к частоте общего ответа, составляющей 13%. У троих из них на текущий момент ответ продолжался, поэтому продолжительность ответа не может быть рассчитана. Кроме того, восемь пациентов имели стабилизацию заболевания, что привело к частоте общего ответа, составляющей 39%. На момент проведения анализа выживаемость без прогрессирования заболевания для этих 12 пациентов с клинической пользой находилась в пределах от 6 до 40 недель. Медиана не может быть рассчитана, так как событие не было записано для 7 из этих пациентов на текущий момент. У 9 пациентов с клинической пользой, по меньшей мере, один опухолевый маркер был повышен на момент начала исследования и снизился на -35-76% у 6 из них одновременно. Примечательно, что 4 пациента с прогрессированием заболевания в качестве лучшего ответа также имели снижение на -29-54%, по меньшей мере, одного из повышенных опухолевых маркеров за период исследования. Общая медиана выживаемости без прогрессирования заболевания составила 10 недель в интервале от 4 до ~40 недель.
У пациентов с клинической пользой (4 PR + 8 SD = 39%) частота возникновения перитонеального карциноматоза, плеврального выпота и асцитов была выше, и частота возникновения метастазов в печени была ниже, чем у пациентов без клинической пользы. С другой стороны, один пациент с подтвержденным частичным ответом имел частые метастазы в печени.
На основании текущего набора данных можно предположить, что пациенты с клинической пользой имели более высокое число клеток с IHC-положительным окрашиванием.
Кроме того, дополнительные данные, собранные у выбранных пациентов, показали, что компоненты сыворотки крови пациентов и РВМС пациентов являются полностью функциональными и активными в отношении опосредования основных механизмов действия IMAB362 CDC и ADCC, соответственно.
В заключение, наблюдается противоопухолевая активность (частичный ответ, стабильное заболевание, снижение опухолевого маркера), и IMAB362 заслуживает дальнейшего исследования.
D. ОБЩИЕ ВЫВОДЫ
Данное исследование разработано как фаза IIa мультицентрового, нерандомизированного, с эскалацией многократных доз у разных пациентов, открытого клинического исследования с использованием 3 когорт. Для данного клинического исследования отбирались пациенты, которые являлись рефракторными к стандартному лечению или для которых одобренная терапия отсутствовала.
Для этого промежуточного отчета 34 пациента подлежали оценке в анализе на безопасность (АРТ-выборка), из которых 4 были включены в когорту 1 (300 мг/м2), 6 в когорту 2 (600 мг/м2) и 20 в когорту 3 (600 мг/м2). IMAB362, вводимый по схеме многократного дозирования, являлся безопасным и хорошо переносимым у получавших ранее интенсивное лечение пациентов с гастроэзофагеальным раком, с тошнотой и рвотой, которые являлись наиболее распространенными, связанными с проводимым лечением, нежелательными явлениями. Большинство нежелательных явлений были от легких до умеренных. Имелось только два пациента с аллергическими реакциями, одна умеренной степени, одна тяжелой. Степень 4 и степень 5 нежелательных явлений отсутствовали (включая лабораторные параметры) на этой фазе IIa исследования и предыдущей фазе I исследования. То, что IMAB352 до сих пор не вызывал нежелательных явлений 4 степени, является удивительным, так как большинство зарегистрированных моноклональных антител ассоциированы с угрожающими жизни степенями 4 и 5 нежелательных явлений. Показание к применению бевацизумаба для лечения метастатического рака молочной железы было аннулировано FDA в ноябре 2011 года после первоначального предварительного одобрения в 2008 году. Бевацизумаб не продлевал жизнь и вызывал серьезное высокое кровяное давление и кровоизлияние с перфорацией стенки кишечника и перфорацией носовой перегородки. Цетуксимаб вызывает подобные угревой сыпи высыпания и инфузионные реакции 3-4 степени, анафилаксию и остановку сердца, необходимость антигистаминной профилактики дифенгидрамином перед лечением. Трастузумаб все еще широко применяется, хотя вызывает симптоматическую сердечную дисфункцию у 2-7% пациентов, о чем известно в течение более 10 лет.
Первичным критерием для оценки потенциальной противоопухолевой активности являлся статус опухоли в соответствии с RECIST. Имелись пациенты в количестве 31 человека, которые имели, по меньшей мере, одну такую оценку после включения в исследование и, таким образом, были включены в FAS. Четыре PR и 8 SD у 31 (RR 13%, DCR 39%) прошедшего ранее интенсивное лечение пациента очень хорошо согласуются с результатами ответов, полученными в других исследованиях фазы II с использованием утвержденной таргетной монотерапии в качестве терапии вторичного заболевания или заболевания на поздней стадии.
Цетуксимаб, антагонист EGFR, достигал частоты объективного ответа (Response Rate, RR), составляющей 3% (с дополнительными 7% SD) на поздней стадии гастроэзофагеального рака (GEC) (большинство пациентов имели 2 или более метастатических участков и предшествующие терапии), измеренной через 8 недель на фазе II исследования с участием тридцати пациентов. Во втором исследовании с участием 55 пациентов на поздней стадии цетуксимаб привел к 5% RR и дополнительно 11% SD, измеренным через 8 недель. Сходные показатели частоты ответа были достигнуты у EGFR-положительных рефракторных пациентов с метастатическим колоректальным раком (mCRC), для терапии которого позднее был одобрен цетуксимаб.
Сунитиниб и эрлотиниб тестировали у пациентов с гастроэзофагеальным раком (GEC) поздней стадии в различных исследованиях фазы II с участием всего 150 пациентов. Частота контроля заболевания (DCR) через 6-8 недель варьировала между 16 и 39%, и частота объективного ответа находилась в диапазоне от 3 до 7%, соответственно.
Частота объективных ответов на фазе II для трастузумаба в качестве дополнительной терапии рака молочной железы составила 11% с дополнительными 9% SD ≥6 месяцев. Для эрлонитиба в случае рака легкого, подвергнутого ранее лечению, отмечалась частота объективного ответа, составляющая 9%. Сорафениб достигал частоты объективного ответа (RR) от 2% до 18% в двух исследованиях фазы II в случае рака почки, и для темсиролимуса частота объективного ответа (RR) составила 7% при исследовании рака почки. Позже эти соединения таргетной терапии были дополнительно разработаны в комбинации с химиотерапией и стали зарегистрированными для этих применений.
IMAB362 представляет собой безопасное и эффективное антитело. Как предполагалось исходя из превосходной тканевой специфичности целевой поверхностной молекулы и связывания антитела с высокой точностью, экспериментальное лекарственное средство хорошо переносится по сравнению с другой присутствующей на рынке таргетной терапией. Кроме того, у нескольких пациентов наблюдались признаки клинической активности, сравнимой или превосходящей результаты фазы II исследований других, уже присутствующих на рынке таргетных терапий.
Пример 5. Тошнота/рвота, вызванная IMAB362
Наблюдалось, что IMAB362 вызывает тошноту/рвоту вплоть до степени 3 NCI-СТС. Симптоматику можно описать следующим образом: (i) не зависит от дозы, (ii) острое начало, в основном в течение первых минут инфузии, может продолжаться после завершения инфузии, (iii) начинается со спазмов в эпигастральной области, гиперсаливации, (iv) рвота может начинаться без предшествующих проявлений, (v) редко возникает у пациентов с тотальной гастрэктомией, (vi) реакция при первой инфузии является показателем увеличения симптомов от цикла к циклу.
Тот факт, что эти нежелательные реакции редко возникают у пациентов, которые были подвергнуты тотальной гастрэктомии, дает основание предполагать, что лежащий в основе механизм является целевым эффектом. Рвота, вызванная IMAB362, возникает чаще, чем тошнота и ее возникновение часто отмечается без предшествующей тошноты. Начало может быть как острым, так и отсроченным. Предполагается, что незначительные количества IMAB362 связываются с ограниченно доступным эпитопом плотных контактов. Это приводит к локальному разрыву плотных контактов и просачиванию желудочной кислоты в подслизистый слой. Возникшие в результате реакции тканей и спазмы инициируют каскад тошноты/рвоты.
Таким образом, рекомендуемыми мерами противодействия являются эффективная противорвотная профилактика и защита слизистой оболочки желудка.
Например, пациенты должны получать противорвотную профилактику до начала приема препарата. Для профилактических и лечебных мероприятий рекомендована комбинация рецептора NK-1 (например, апрепитант/эменд) и блокатора 5-НТ3-рецептора (например, ондансетрон/зофран), которая может быть расширена дополнительными соединениями. Прием противорвотного препарата предпочтительно осуществляют в течение, по меньшей мере, первых трех дней каждого цикла. Может быть рассмотрено профилактическое введение бутилскополамина/бускопана незадолго до каждой инфузии IMAB362.
Любая мера для защиты слизистой оболочки может уменьшить симптомы со стороны желудка. В этом отношении можно применять ингибиторы протонного насоса и/или мизопростол, которые можно вводить, например, на дни 1-2 или 3 каждого цикла. Нестероидные противовоспалительные лекарственные средства (NSAID) применять не следует, но применение ацетаминофена допускается. Если ацетаминофен является не эффективным в управлении болью, можно применять NSAID, если для управления болью требуется избежать терапии опиоидами. Пациенты, получающие NSAID, предпочтительно получают лечение ингибиторами протонного насоса и/или мизопростолом.
Таким образом, противорвотную профилактику и защиту слизистой оболочки желудка можно начинать незадолго до инфузии IMAB362. Например, можно вводить следующую комбинацию, при этом внутривенное введение является предпочтительным:
• NK-1 RA: например, апрепитант/эменд (150 мг IV)
• 5-НТ3 RA: например, палоносетрон (0,25 мг IV), ондансетрон/зофран (8 мг IV), гранисетрон (3 мг IV)
• бутилскополамин/бускопан
• Ингибитор протонного насоса: пантопразол/пантозол
Необязательно, можно также вводить метоклопрамид/МСР, лоразепам и/или атропин.
IMAB362 представляет собой антитело, в основе которого лежат иммунологические механизмы действия, которые могут быть нарушены иммуносупрессорными соединениями. По этой причине в противорвотной профилактике следует избегать применения стероидов и применять их только в случае, если другие соединения оказались неэффективными.
Кроме того, необходимо внимательно относиться к применению IMAB362. Например, рекомендуется тщательный контроль в течение первых 15-30 мин. При необходимости скорость инфузии должна быть снижена (например, до 4 ч вместо 2 ч) и должны быть включены перерывы в инфузии.
Прием противорвотных лекарственных средств, а также защиту слизистой оболочки желудка можно продолжать, например, до дня 3 каждого цикла.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ РАКА, ВКЛЮЧАЮЩАЯ АНТИТЕЛА ПРОТИВ КЛАУДИНА 18.2 | 2014 |
|
RU2832015C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ С ИСПОЛЬЗОВАНИЕМ АНТИТЕЛ ПРОТИВ КЛАУДИНА 18.2 ДЛЯ ЛЕЧЕНИЯ РАКА | 2014 |
|
RU2699549C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ С ИСПОЛЬЗОВАНИЕМ АНТИТЕЛ К КЛАУДИНУ 18.2 ДЛЯ ЛЕЧЕНИЯ РАКА | 2013 |
|
RU2662066C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ С ИСПОЛЬЗОВАНИЕМ АНТИТЕЛ К КЛАУДИНУ 18.2 ДЛЯ ЛЕЧЕНИЯ РАКА | 2013 |
|
RU2665321C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ С ИСПОЛЬЗОВАНИЕМ АНТИТЕЛ К КЛАУДИНУ 18.2 ДЛЯ ЛЕЧЕНИЯ РАКА | 2013 |
|
RU2789478C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ С ИСПОЛЬЗОВАНИЕМ АНТИТЕЛ ПРОТИВ КЛАУДИНА 18.2 ДЛЯ ЛЕЧЕНИЯ РАКА | 2014 |
|
RU2792932C2 |
| КОНЪЮГАТЫ ЛЕКАРСТВЕННЫХ СРЕДСТВ, СОДЕРЖАЩИЕ АНТИТЕЛА ПРОТИВ КЛАУДИНА 18.2 | 2016 |
|
RU2841168C2 |
| НОВЫЕ АНТИ-CLDN18.2 АНТИТЕЛА | 2020 |
|
RU2830887C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ПРОГНОЗИРОВАНИЯ ТЕРАПЕВТИЧЕСКОЙ ЭФФЕКТИВНОСТИ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ И ПРОГНОЗА ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2016 |
|
RU2785291C2 |
| АНТИТЕЛА ПРОТИВ КЛАУДИНА 18.2 ДЛЯ ДИАГНОСТИКИ РАКА | 2013 |
|
RU2661772C2 |






































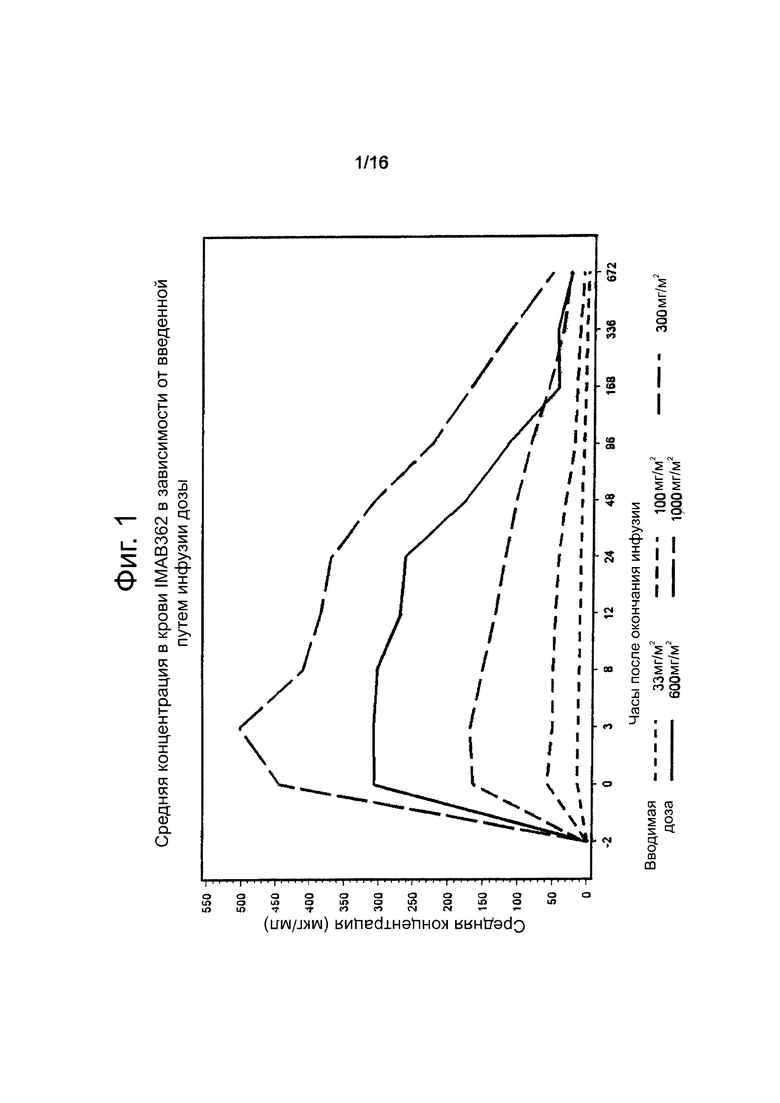
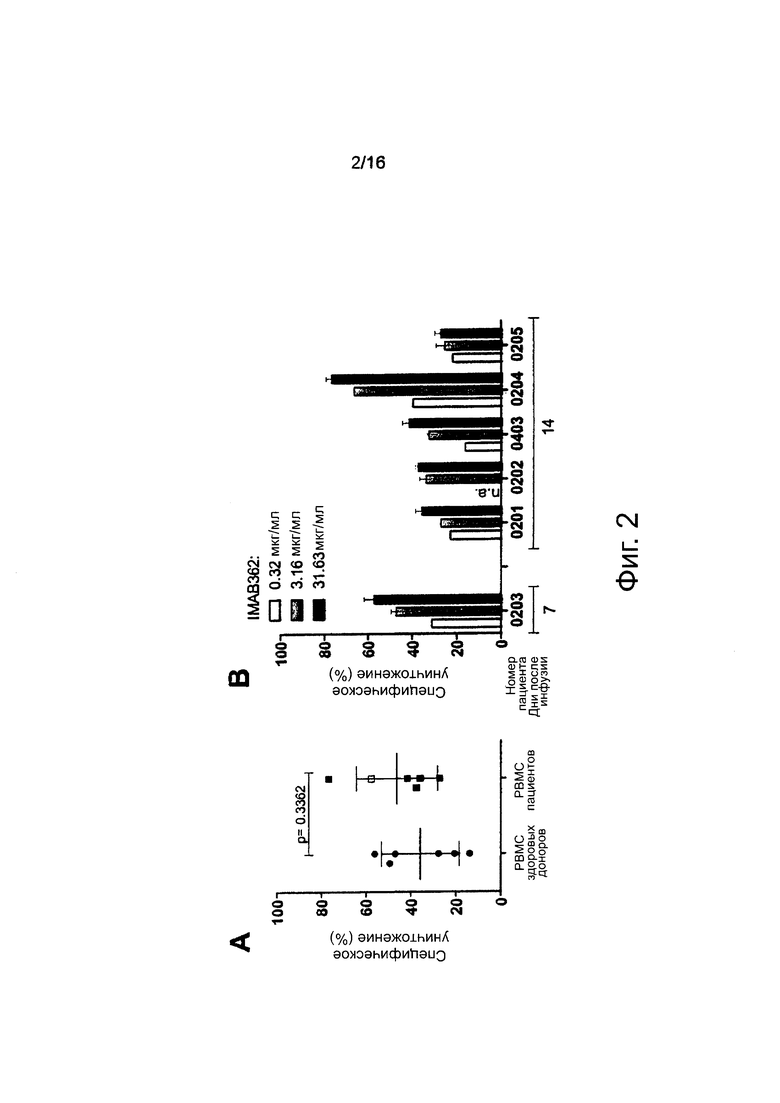
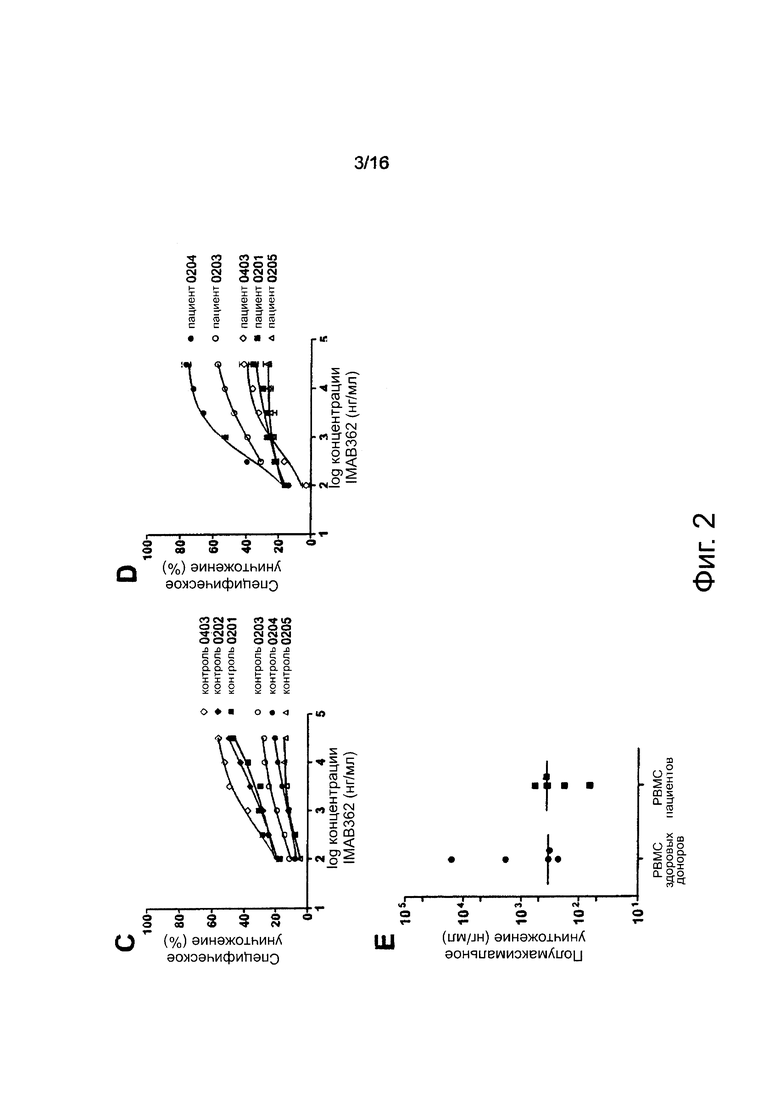
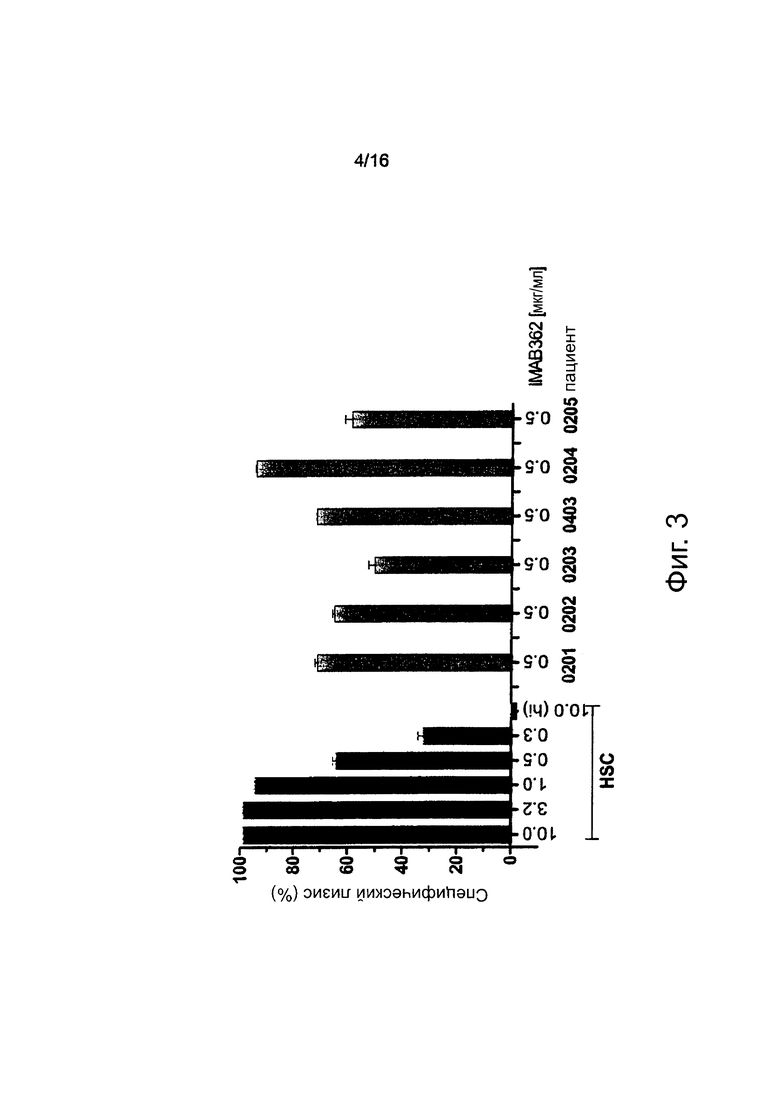
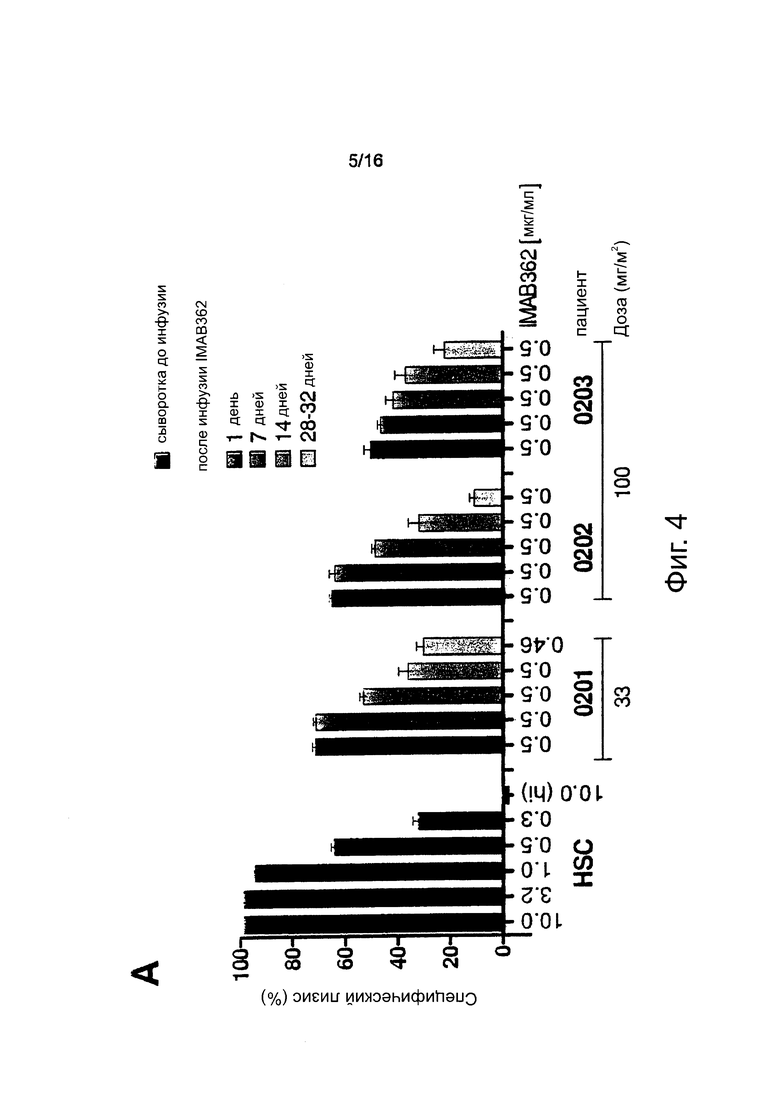
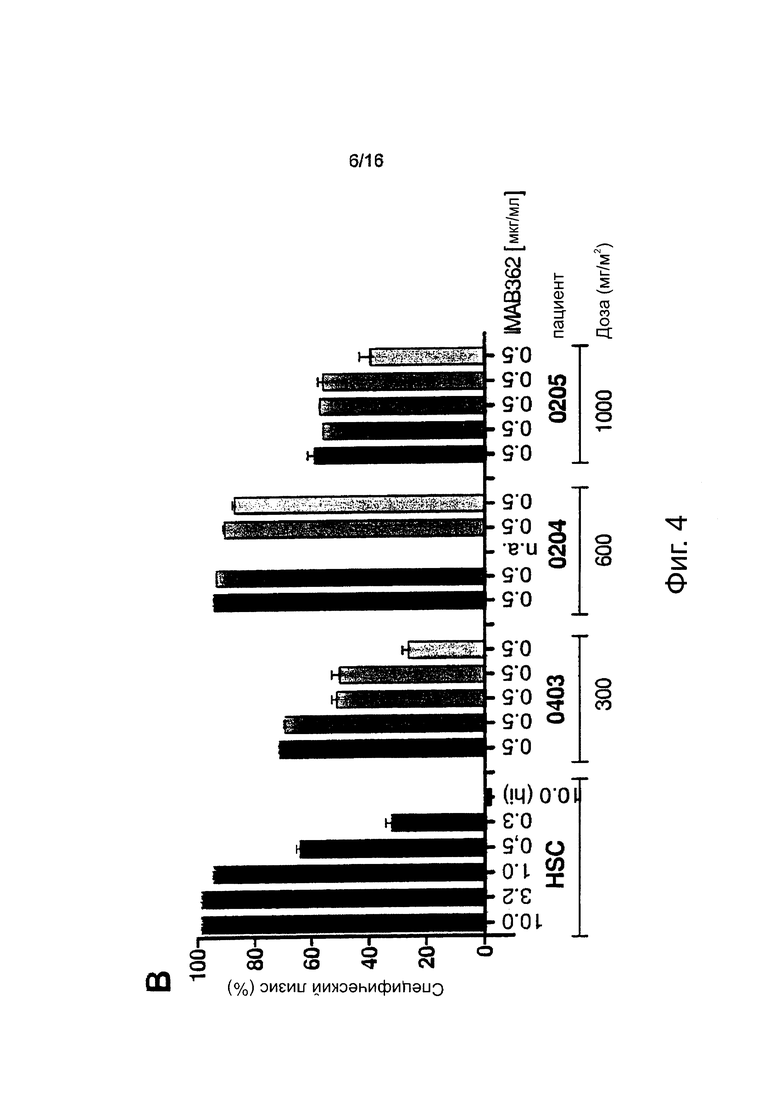
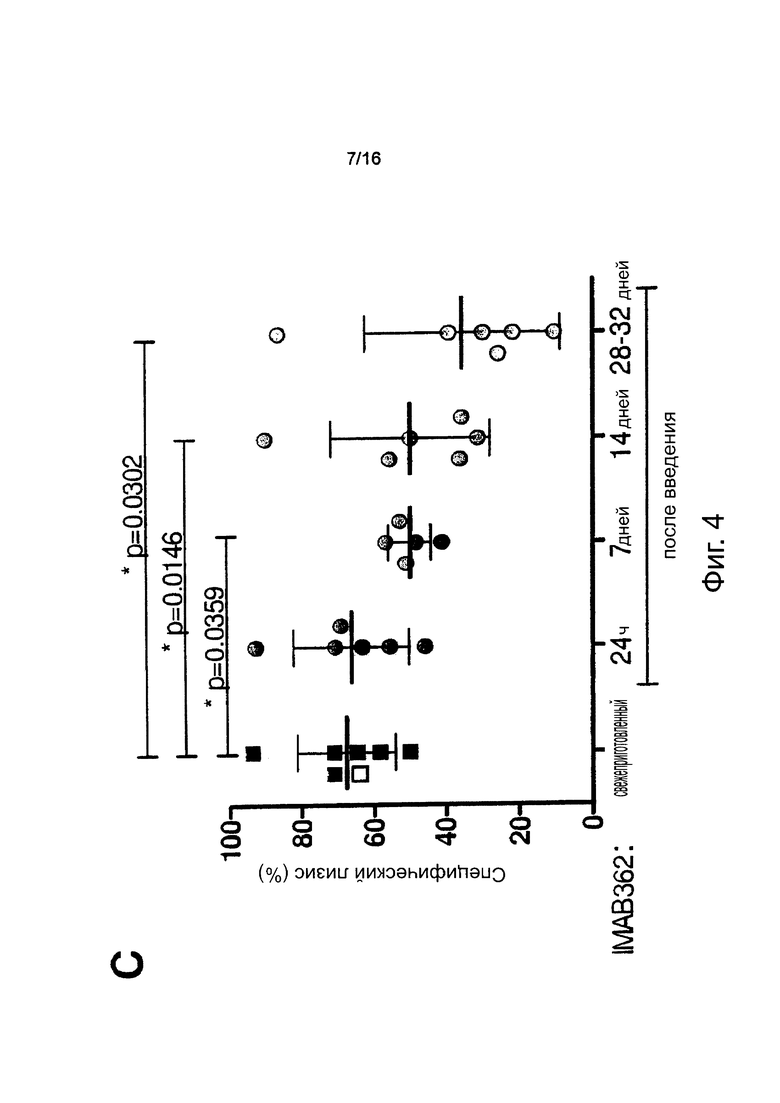
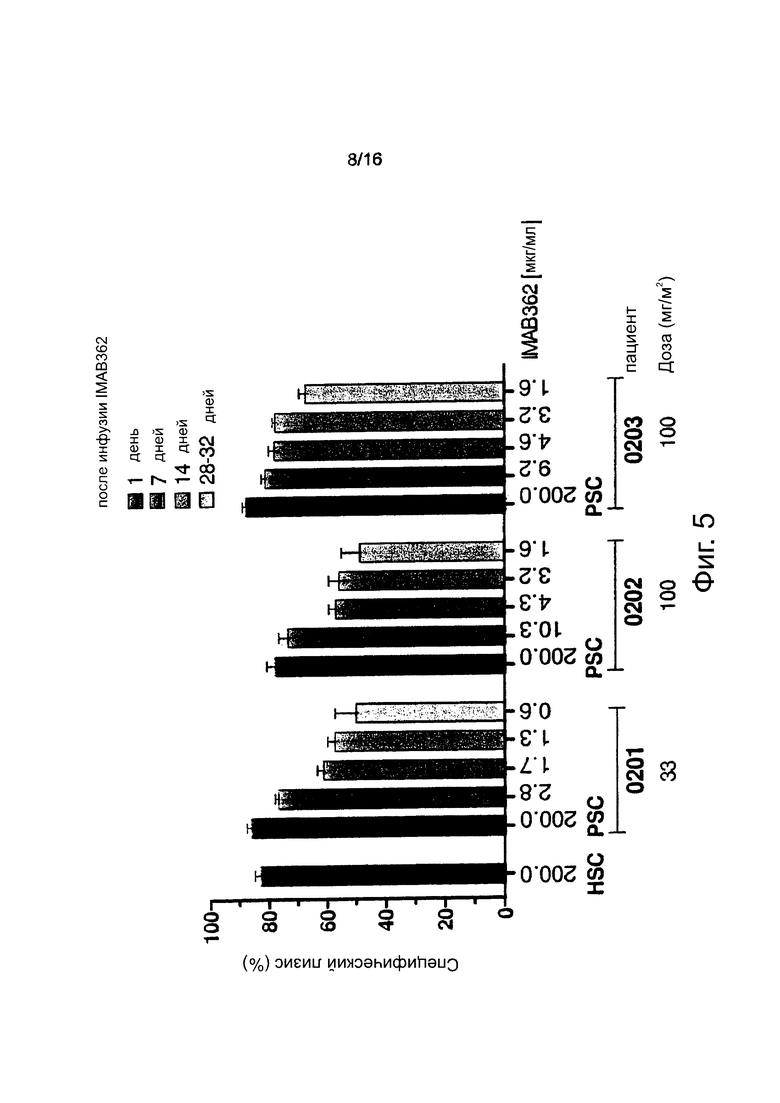
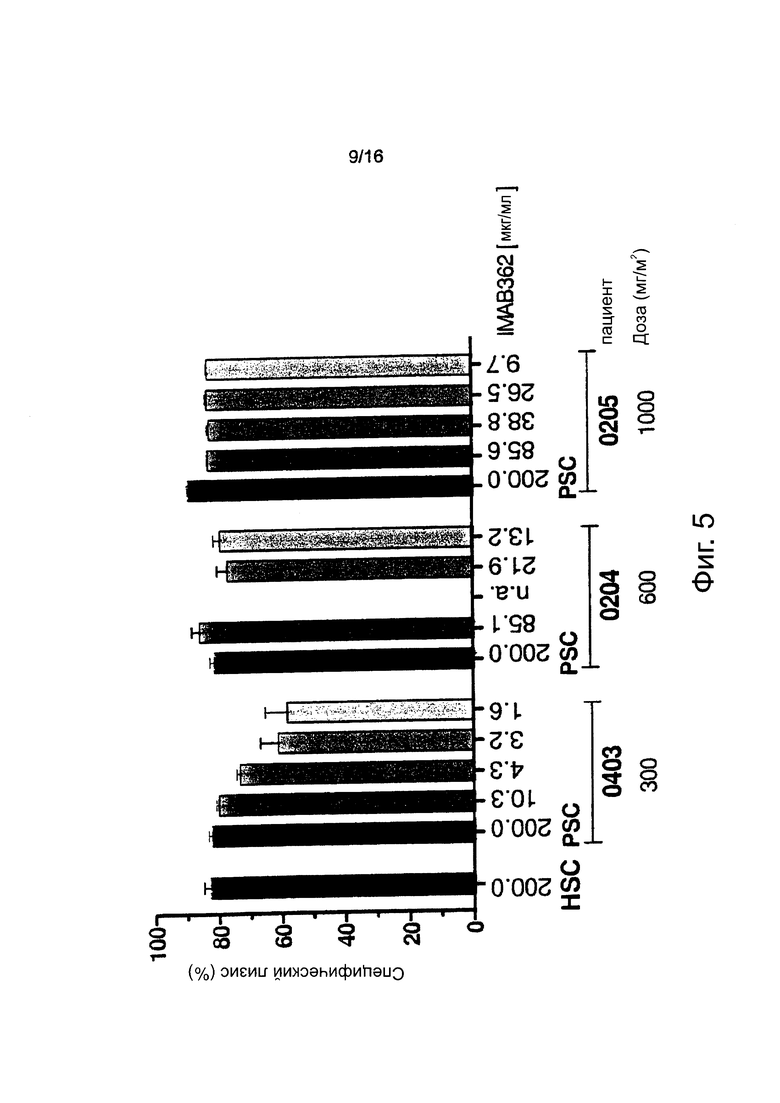
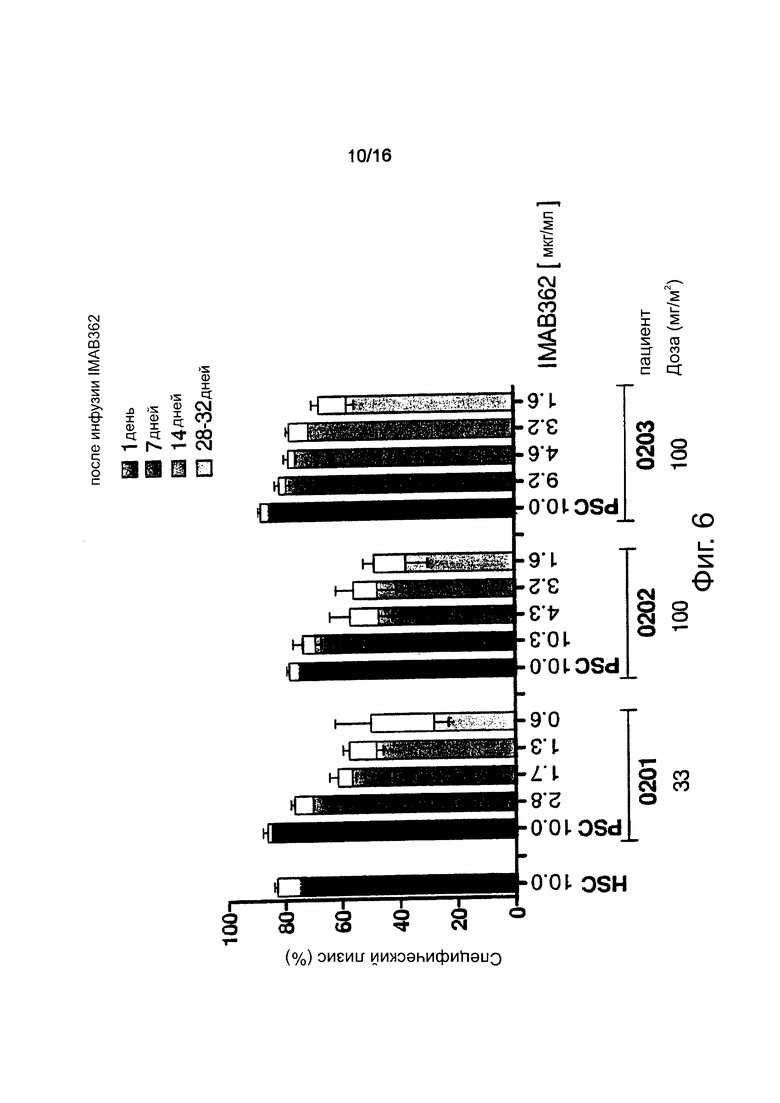
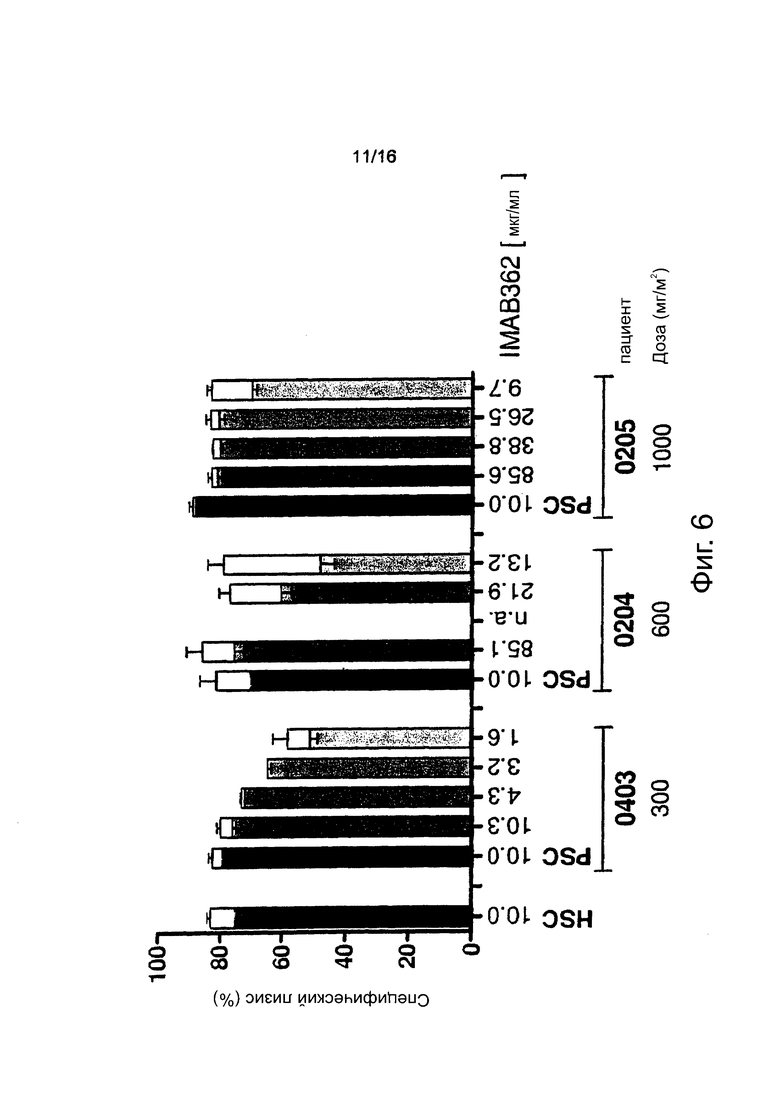
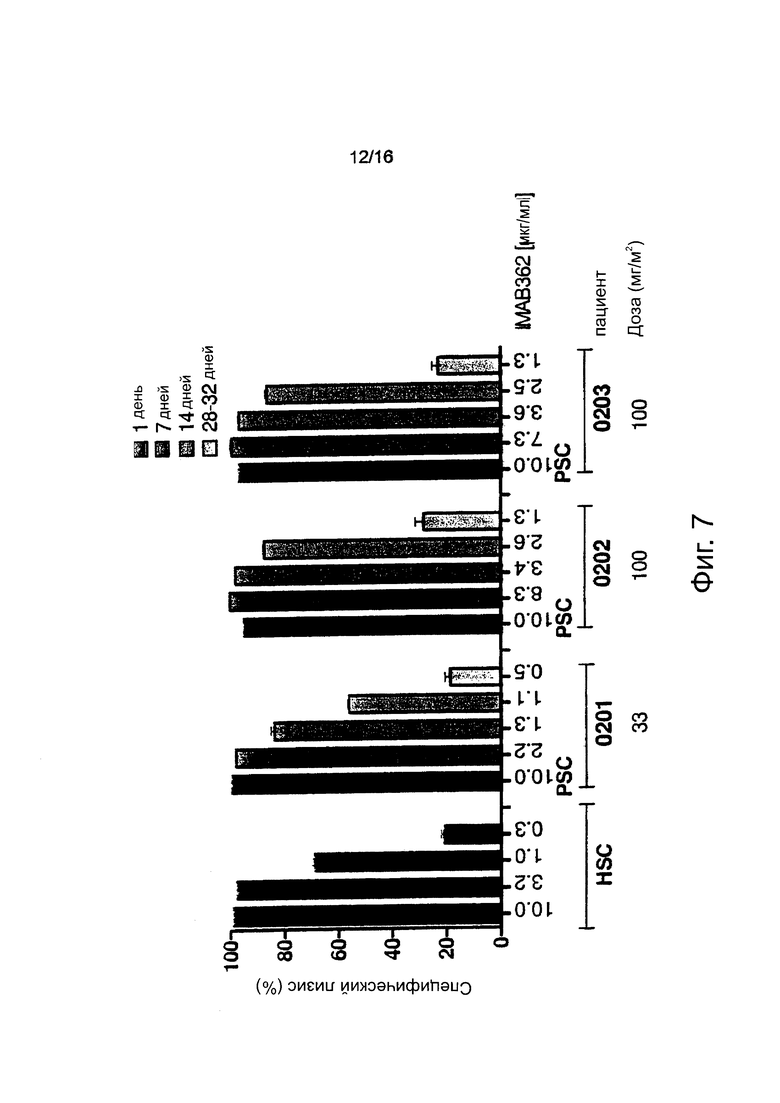
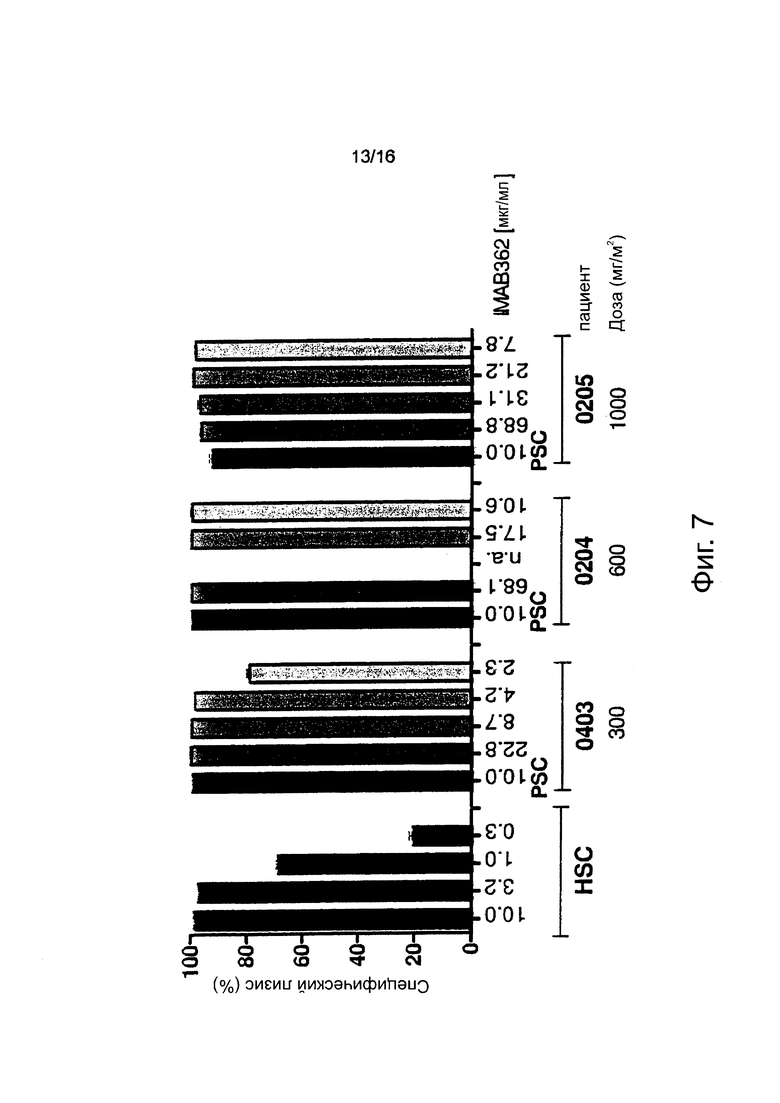
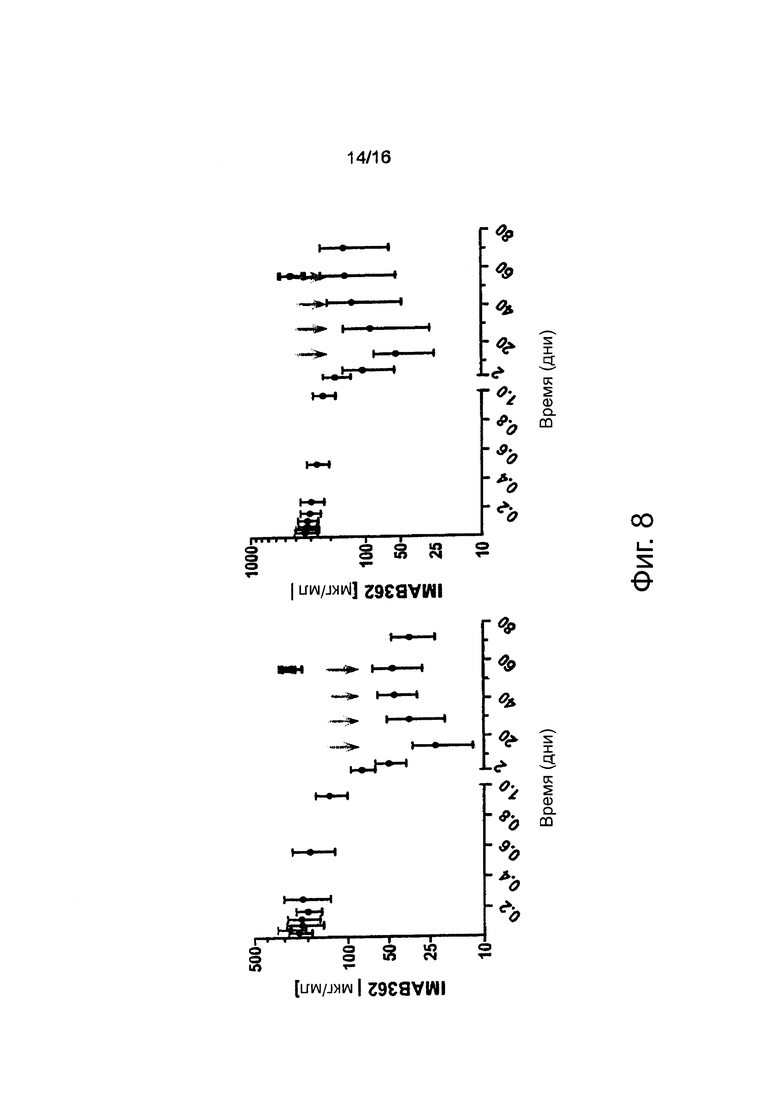

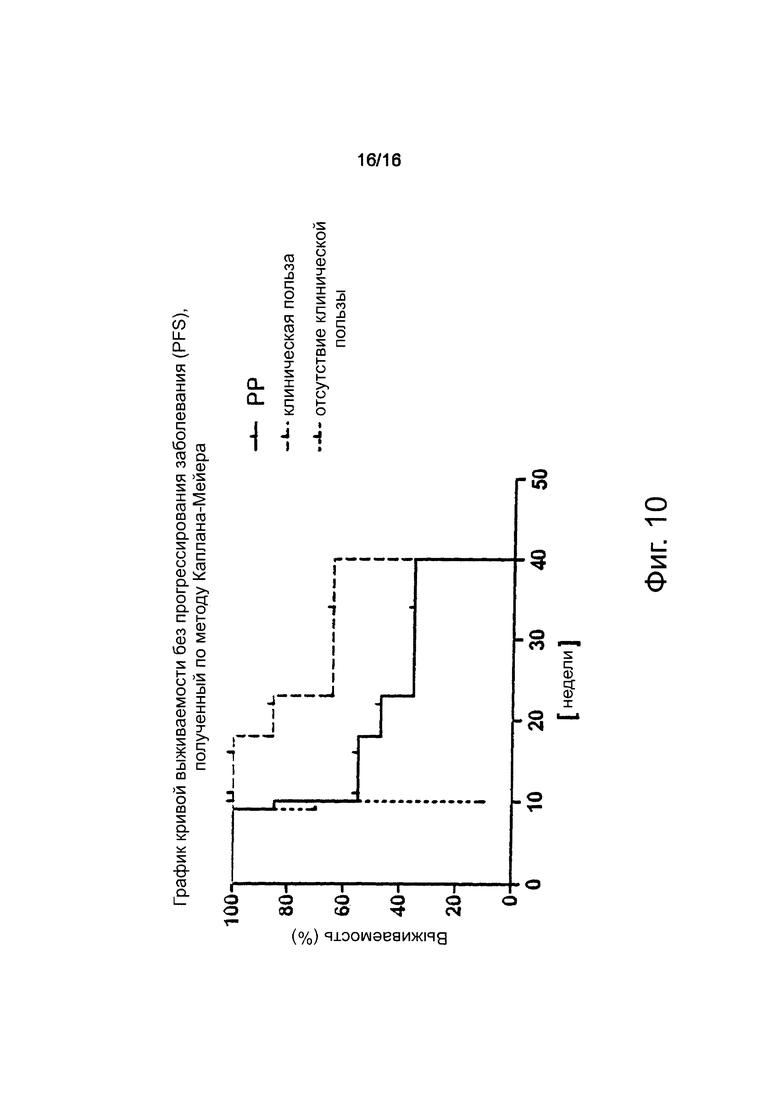
Группа изобретений относится к медицине и предназначена для лечения рака. Пациенту вводят антитело, обладающее способностью связываться с CLDN18.2, причем антитело вводят таким образом, чтобы обеспечить уровни в сыворотке по меньшей мере 40 мкг/мл. Также описан вариант способа, в котором указанное антитело вводят в дозе по меньшей мере 300 мг/м2. Группа изобретений позволяет расширить арсенал терапевтических средств. 2 н. и 25 з.п. ф-лы, 10 ил., 19 табл., 5 пр.
1. Способ лечения или предупреждения ракового заболевания, включающий введение пациенту антитела, обладающего способностью связываться с CLDN18.2, отличающийся тем, что антитело вводят таким образом, чтобы обеспечить уровни в сыворотке по меньшей мере 40 мкг/мл.
2. Способ по п. 1, в котором уровень в сыворотке обеспечивают в диапазоне от 40 до 700 мкг/мл.
3. Способ по п. 1 или 2, в котором уровень в сыворотке обеспечивают в течение периода, составляющего по меньшей мере 7 дней.
4. Способ по п. 1 или 2, который включает введение дозы антитела, составляющей по меньшей мере 300 мг/м2.
5. Способ по п. 1 или 2, в котором лечение ракового заболевания приводит к достижению стабильного заболевания.
6. Способ по п. 5, в котором стабильное заболевание достигается на период, составляющий по меньшей мере 2 месяца.
7. Способ по п. 1 или 2, в котором антитело вводят в виде однократной дозы или многократных доз.
8. Способ по п. 1 или 2, дополнительно включающий введение одного или нескольких препаратов, выбранных из группы, состоящей из противорвотных средств, антиспазматических средств, парасимпатолитических веществ и агентов, которые защищают слизистую оболочку желудка.
9. Способ по п. 8, который включает введение пациенту антагониста рецептора нейрокинина 1 (NK), такого как апрепитант (например, эменд), антагониста рецептора 5-НТ3, такого как ондансетрон (например, зофран), гранисетрон (например, китрил, санкузо) или палоносетрон (например, алокси), или комбинации двух или более из них, антиспазматического средства, такого как бутилскополамин (например, бускопан), и ингибитора протонного насоса, такого как пантопразол (например, пантозол).
10. Способ по п. 1 или 2, в котором антитело вводят путем внутривенной инфузии.
11. Способ по п. 10, в котором внутривенную инфузию проводят в течение периода времени, составляющего от 1 до 4 часов.
12. Способ по п. 1 или 2, в котором антитело опосредует уничтожение клеток посредством одного или нескольких путей, выбранных из лизиса, опосредованного комплементзависимой цитотоксичностью (CDC), лизиса, опосредованного зависимой от антитела клеточной цитотоксичностью (ADCC), индукции апоптоза и ингибирования пролиферации.
13. Способ по п. 1 или 2, в котором антитело представляет собой антитело, выбранное из группы, состоящей из (i) антитела, продуцированного или полученного из клона, депонированного под номером доступа DSM ACC2737, DSM ACC2738, DSM ACC2739, DSM ACC2740, DSM ACC2741, DSM ACC2742, DSM ACC2743, DSM ACC2745, DSM ACC2746, DSM ACC2747, DSM ACC2748, DSM ACC2808, DSM ACC2809 или DSM ACC2810, (ii) антитела, которое является химеризованной или гуманизированной формой антитела согласно (i), (iii) антитела, обладающего специфичностью антитела согласно (i), и (iv) антитела, содержащего антигенсвязывающий участок или антигенсвязывающий сайт, в частности вариабельную область антитела согласно (i), и предпочтительно обладающего специфичностью антитела согласно (i).
14. Способ по п. 1 или 2, где рак представляет собой гастроэзофагеальный рак.
15. Способ по п. 1 или 2, где пациентом является человек.
16. Способ лечения или предупреждения ракового заболевания, включающий введение пациенту антитела, обладающего способностью связываться с CLDN18.2, отличающийся тем, что указанное антитело вводят в дозе по меньшей мере 300 мг/м2.
17. Способ по п. 16, в котором лечение ракового заболевания приводит к достижению стабильного заболевания.
18. Способ по п. 17, в котором стабильное заболевание достигается на период, составляющий по меньшей мере 2 месяца.
19. Способ по п. 16, в котором антитело вводят в виде однократной дозы или многократных доз.
20. Способ по п. 16, дополнительно включающий введение одного или нескольких препаратов, выбранных из группы, состоящей из противорвотных средств, антиспазматических средств, парасимпатолитических веществ и агентов, которые защищают слизистую оболочку желудка.
21. Способ по п. 20, который включает введение пациенту антагониста рецептора нейрокинина 1 (NK), такого как апрепитант (например, эменд), антагониста рецептора 5-НТ3, такого как ондансетрон (например, зофран), гранисетрон (например, китрил, санкузо) или палоносетрон (например, алокси), или комбинации двух или более из них, антиспазматического средства, такого как бутилскополамин (например, бускопан), и ингибитора протонного насоса, такого как пантопразол (например, пантозол).
22. Способ по п. 16, в котором антитело вводят путем внутривенной инфузии.
23. Способ по п. 22, в котором внутривенную инфузию проводят в течение периода времени, составляющего от 1 до 4 часов.
24. Способ по п. 16, в котором антитело опосредует уничтожение клеток посредством одного или нескольких путей, выбранных из лизиса, опосредованного комплементзависимой цитотоксичностью (CDC), лизиса, опосредованного зависимой от антитела клеточной цитотоксичностью (ADCC), индукции апоптоза и ингибирования пролиферации.
25. Способ по п. 16, в котором антитело представляет собой антитело, выбранное из группы, состоящей из (i) антитела, продуцированного или полученного из клона, депонированного под номером доступа DSM ACC2737, DSM ACC2738, DSM ACC2739, DSM ACC2740, DSM ACC2741, DSM ACC2742, DSM ACC2743, DSM ACC2745, DSM ACC2746, DSM ACC2747, DSM ACC2748, DSM ACC2808, DSM ACC2809 или DSM ACC2810, (ii) антитела, которое является химеризованной или гуманизированной формой антитела согласно (i), (iii) антитела, обладающего специфичностью антитела согласно (i), и (iv) антитела, содержащего антигенсвязывающий участок или антигенсвязывающий сайт, в частности вариабельную область антитела согласно (i), и предпочтительно обладающего специфичностью антитела согласно (i).
26. Способ по п. 16, где рак представляет собой гастроэзофагеальный рак.
27. Способ по п. 16, где пациентом является человек.
| WO2007059997 A1, 31.05.2007 | |||
| WO2007090670 A1, 16.08.2007 | |||
| SAHIN U et al., Claudin-18 Splice Variant 2 Is a Pan-Cancer Target Suitable for Therapeutic Antibody Development Clin Cancer Res | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| СУШИЛКА С ВРАЩАЮЩИМСЯ ГОРИЗОНТАЛЬНЫМ ДНОМ И НАСАЖЕННЫМИ НА ВАЛУ ЛОПАСТЯМИ ДЛЯ ПЕРЕМЕШИВАНИЯ | 1926 |
|
SU7624A1 |
| PMID:19047087. | |||

