Область техники
Данное изобретение относится к химии органических соединений, фармакологии и медицине и касается способа получения N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида с чистотой выше 99,0% и не содержащего единичной примеси выше 0,1% пригодного для использования в качестве фармацевтической субстанции в производстве различных видов лекарственных форм.
Уровень техники
Получение амидов малоновой кислоты из самой кислоты или ее эфиров многократно описано в литературе. Поскольку малоновая кислота не стабильна при температурах, характерных для протекания реакции прямого амидирования карбоновых кислот, для синтеза ее амидов, как правило, используются более реакционноспособные производные, с выделением или получаемые in situ. Ниже приведены основные методы получения амидов малоновой кислоты классифицированные по типу ацилирующего агента:
Метод активированных эфиров.
Метод ацилирования аминов галогенангидридами кислот.
Метод ацилирования аминов имидазолидами карбоновых кислот
Метод ацилирования аминов эфирами кислот.
Рассмотрим детально описанные в литературе варианты реализации каждого из приведенных выше методов:
Метод активированных эфиров
Данный метод подразумевает использование специальных вспомогательных веществ, реагируя с которыми, малоновая кислота образует высокореакционноспособные интермедиаты, которые, в свою очередь, быстро и с высокими выходами способны взаимодействовать с аминами, превращаясь в целевые амиды. В качестве активирующих агентов могут быть использованы:
1) Соли (1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния)-3-оксида при комнатной температуре в среде абсолютного диметилформамида. (Tan, Guanhai; Yao, Yuchao; Gu, Yunjing; Li, Shuai; Lv, Mengjiao; Wang, Kerang; Chen, Hua; Li, Xiaoliu Bioorganic and Medicinal Chemistry Letters, 2014, vol. 24, # 13, p. 2825-2830).
2) Соли O-(бензотриазол-1-ил)-N,N,N′,N′-тетраметилурониума при комнатной температуре в среде абсолютного диметилформамида (Koval, Vasiliy S.; Arutyunyan, Albert F.; Salyanov, Victor L.; Klimova, Regina R.; Kushch, Alla A.; Rybalkina, Ekaterina Yu.; Susova, Olga Yu.; Zhuze, Alexei L. - Bioorganic and Medicinal Chemistry, 2018, vol. 26, # 9, p. 2302-2309).
3) Дициклогексилкарбодиимид при комнатной температуре в различных растворителях (Cipolla, Laura; Sgambato, Antonella; Forcella, Matilde; Fusi, Paola; Parenti, Paolo; Cardona, Francesca; Bini, Davide - Carbohydrate Research, 2014, vol. 389, # 1, p. 46-49. Wu, Hua; He, Yu-Ping; Xu, Lue; Zhang, Dong-Yang; Gong, Liu-Zhu - Angewandte Chemie - International Edition, 2014, vol. 53, # 13, p. 3466-3469, Angew. Chem., 2014, vol. 126, # 13, p. 3534-3537). Возможно так же проведение реакции с дициклогексилкарбодиимидом в присутствии бензотриазол-1-ола (Burghardt, Stephan; Hirsch, Andreas; Schade, Boris; Ludwig, Kai; Boettcher, Christoph -Angewandte Chemie - International Edition, 2005, vol. 44, # 19, p. 2976-2979). Использование бензотриазол-1-ола позволяет, в некоторых случаях, увеличить выход целевого продукта.
4) Гидрохлорид (1-(3-диметиламино)пропил)-3-этилкарбодиимида в диметил-формамиде при 25°С в присутствии основания (Goldberg, Joel; Jin, Qing; Ambroise, Yves; Satoh, Shigeki; Desharnais, Joel; Capps, Kevin; Boger, Dale L. - Journal of the American Chemical Society, 2002, vol. 124, # 4, p. 544-555.) Возможно так же проведение реакции с дициклогексилкарбодиимидом в присутствии бензотриазол-1-ола (SANOFI-AVENTIS - US2009/69368, 2009, A1). Как и в случае с дициклогексилкарбодиимидом, использование бензотриазол-1-ола позволяет, в некоторых случаях, увеличить выход целевого продукта.
5) Соли 1-метил-2-хлорпиридиния в присутствии основания в различных растворителях (Tyler; Webster - Chemical Communications, 2014, vol. 50, # 73, p. 10665-10668. Evans, Vikki; Mahon, Mary F.; Webster, Ruth L. - Tetrahedron, 2014, vol. 70, # 41, p. 7593-7597).
Однако как было показано в ходе исследований метод активированных эфиров не позволяет получать N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамид чистотой более 99% с приемлемым выходом для использования процесса в промышленности.
Метод ацилирования аминов галогенангидридами кислот
Реакцию между дихлорангидридом малоновой кислоты и аминами, как правило, проводят при охлаждении в присутствии основания (или используя в качестве основания избыток ацилируемого амина), в широком спектре растворителей. (Lei, Shan; Jin, Bo; Zhang, Qingchun; Zhang, Zhichao; Wang, Xiaofang; Peng, Rufang; Chu, Shijin - Polyhedron, 2016, vol. 119, p. 387-395. Das, Swagatika; Das, Umashankar; Sakagami, Hiroshi; Umemura, Naoki; Iwamoto, Shoko; Matsuta, Tomohiko; Kawase, Masami; (…)Gorecki, Dennis K.J.; Dimmock, Jonathan R. - European Journal of Medicinal Chemistry, 2012, vol. 51, p. 193-199). Однако, как было показано в ходе исследований, данный метод не позволяет добиться высоких выходов целевого продукта, в связи с наличием, помимо основной реакции, побочной реакции гидролиза малонилхлорида. К тому же, для промышленных синтезов нежелательно использование хлорированных производных в целях безопасности.
Метод ацилирования аминов имидазолидами карбоновых кислот
Имидазолид малоновой кислоты может быть получен из малоновой кислоты реакцией с 1,1'-карбонилдиимидазолом, в различных растворителях (Nguyen, M. V. D.; Nicolas, L.; Gaudemer, A.; Brik, M. E. - Bioorganic & Medicinal Chemistry Letters, 1998, vol. 8, # 3, p. 227-232).
Низкий выход продукта, так же, как и в случае использования метода активированных эфиров, объясняется необходимостью удалить из реакционной смеси имидазол который имеет сопоставимые значения растворимости в воде и спиртах.
Метод ацилирования аминов эфирами кислот
Данный метод подразумевает взаимодействие амина со сложными эфирами малоновой кислоты. Данные соединения относительно малореакционноспособны, но в силу своей термической стабильности, позволяют проводить реакцию при относительно высоких температурах (150-180°С). В качестве ацилирующих агентов в литературе описано использование диэтилового эфира малоновой кислоты (Devarasetty, Kiran; Tharikoppula, Giri; Sridhar, Tailor; Eppakayala, Laxminarayana; Kyasani, Mahesh; Arumugam, Premkumar; Pusuluri, Srinivas - Synthetic Communications, 2016, vol. 4 6, # 3, p. 263-274) и 2,4,6-трихлорфениловго эфира малоновой кислоты (Barbeau, Olivier R.; Cano-Soumillac, Celine; Griffin, Roger J.; Hardcastle, Ian R.; Smith, Graeme C. M.; Richardson, Caroline; Clegg, William; Harrington, Ross W.; Golding, Bernard T. - Organic and Biomolecular Chemistry, 2007, vol. 5, # 16, p. 2670-2677.).
Существование широкого разнообразия методов получения амидов малоновой кислоты связано с зависимостью селективности протекания реакции, выхода целевого продукта, а также легкость его выделения и очистки от природы ацилируемого амина.
Очевидно, что легкость очистки и выделения особенно важны в случае получения веществ, являющихся фармацевтическими субстанциями, т.к. требования к химической чистоте фармацевтических субстанций особенно высоки. По этой причине оптимизация метода синтеза является важной задачей для технологии производства фармацевтических субстанций.
Таким образом, существует потребность в разработке способа получения таких соединений, который реализуем в промышленных условиях пригоден для получения значительных количеств продукта, пригодного для использования в фармацевтике.
Как было показано на основании анализа литературных источников, для получения N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида могут быть применены различные синтетические подходы. Конкретной синтетической стратегии, обеспечивающей получения целевого продукта с высоким выходом и чистотой выше 99,0%, а так же не содержащего единичной примеси выше 0,1% в промышленных масштабах, заявителем в уровне техники не обнаружено.
Раскрытие изобретения
Задачей настоящего изобретения является разработка способа получения N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида формулы (I)

позволяющего получать целевой продукт чистотой выше 99,0% и не содержащий единичной примеси выше 0,1%, пригодный для использования в качестве фармацевтической субстанции в производстве различных видов лекарственных форм.
Под единичной примесью в данном изобретении подразумевается любое индивидуальное химическое соединение, которое может образовываться в ходе осуществления процесса синтеза по изобретению.
Техническим результатом данного изобретения является предоставление способа, который позволяет получать N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамид формулы (I) чистотой выше 99,0 мас.%, указанный продукт не содержит единичной примеси более 0,1 мас.%, указанный продукт пригоден для использования в качестве фармацевтической субстанции для производства различных видов лекарственных форм.
Указанный технический результат достигается путем нагревания диметилового эфира малоновой кислоты (2) с гистамином (1) в бутаноле при температуре 90-110°С.
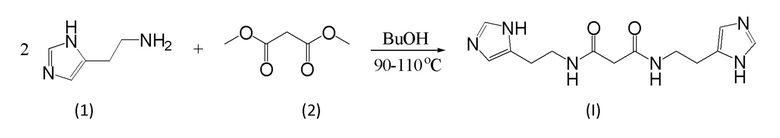
Схема получения N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида может дополнительно включать стадию очистки путем кристаллизации из воды или органического растворителя.
Соединение (N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамид формулы (I) и способ его получения путем взаимодействия диэтилового эфира малоновой кислоты и гистамина, описаны в патенте на изобретение RU 2 665 688. Воспроизведение описанной в нем методики позволяет получать образец N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида с чистотой менее 90%.
Попытки осуществить синтез N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида путем нагревания стехиометрических соотношений диэтилмалоната и гистамина до 170°С приводили к значительному осмолению реакционной смеси и образованию по данным ВЭЖХ значительного количества (до 38%) побочных продуктов.
Неожиданно было обнаружено, что замена диэтилмалоната на диметилмалонат, позволила снизить температуру проведения реакции, добиться высокой конверсии. Кроме того, использование в разработанной методике растворителя (бутанола) позволило сделать методику масштабируемой для получения соединения формулы (I) в промышленных количествах. В результате проведенной оптимизации, удалось увеличить общий выход целевого соединения с 44% до 60%, при этом продукт по данным ВЭЖХ не содержит каких-либо примесей.
Не желая связывать себя теорией, Заявитель предполагает, что найденная им комбинация используемых исходных соединений и растворителя позволяет эффективно провести реакцию в более мягких условиях и, как следствие, замедлить скорость протекания побочных реакций, в том числе процесс образования продуктов термической деградации (осмоления) а также приводит к отсутствию локальных перегревов (они также способствуют осмолению, что затрудняет извлечение реакционной массы из реактора).
Краткое описание фигур
Фиг. 1. Кривые ВЭЖХ анализа образца N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида, полученного взаимодействием диметилмалоната и гистамина в бутаноле при 100°С и перекристаллизации из этанола с обработкой горячего раствора активированным углем.
Фиг. 2. Кривые ВЭЖХ анализа образца N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида, полученного взаимодействием диметилмалоната и гистамина в бутаноле при 100°С и перекристаллизации из метанола с обработкой горячего раствора активированным углем.
Фиг. 3. Кривые ВЭЖХ анализа образца N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида полученного взаимодействием диметилмалоната и гистамина в бутаноле при 100°С и перекристаллизации из воды с обработкой горячего раствора активированным углем.
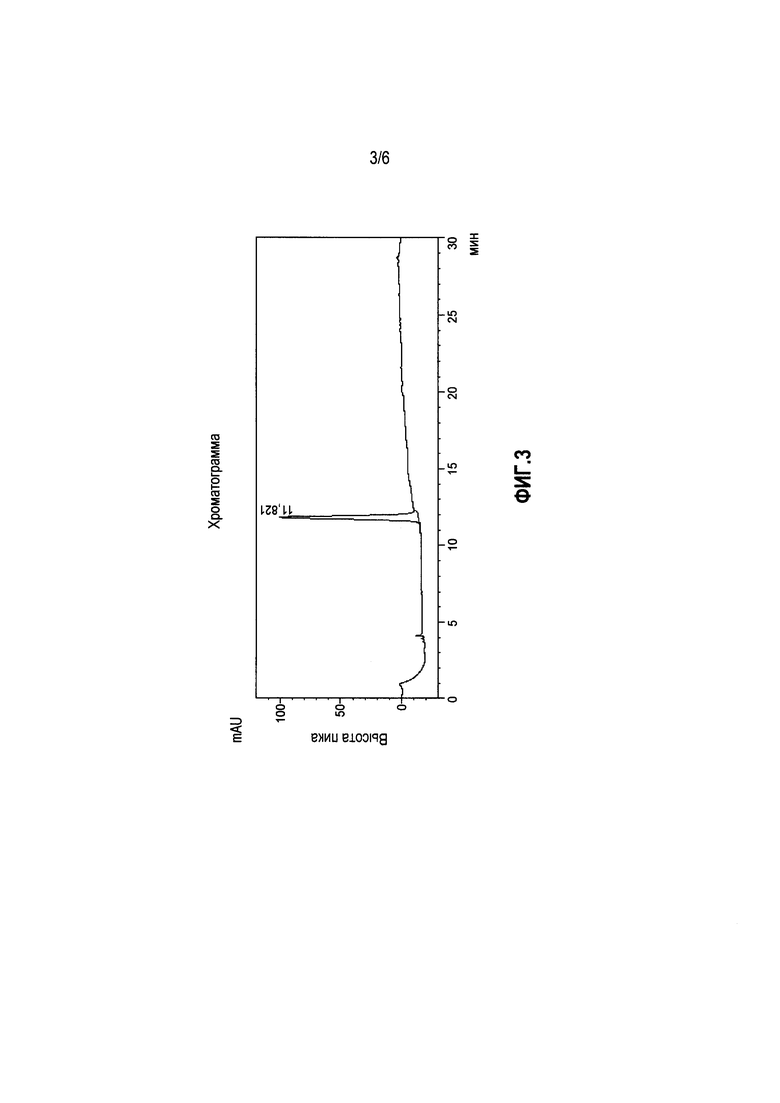
Фиг. 4. Спектр ядерного магнитного резонанса 1H (Bruker DRX500,13, 500,13 МГц, ДМСО-d6) образца N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида.
Фиг. 5. Спектр ядерного магнитного резонанса 13С (Bruker DRX500,13 125,76 МГц, ДМСО-d6) образца N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида.
Фиг. 6. Масс-спектр высокого разрешения образца N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида.
Подробное раскрытие изобретения
Для решения задачи получения N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида формулы (I) чистотой выше 99,0% и не содержащего единичной примеси выше 0,1%, исходя из литературных данных, был осуществлен синтез данного соединения согласно всем описанным методам с целью определения выхода и оценки методов выделения и очистки N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида и произведено сравнение со способом по изобретению.
Анализ образцов методом ВЭЖХ осуществляли на жидкостном хроматографе с автосэмплером, УФ-детектором, оснащенным колонкой Gemini C18, 4.6×250 мм, 5μм.
Анализ образцов методом ЯМР осуществляли на приборе Bruker DRX с рабочей частотой 500,13, 500,13 МГц, в ДМСО-d6.
Анализ образцов методом масс-спектрометрии осуществляли на приборе Bruker micrOTOF-Q с ионизацией электрораспылением.
Пример 1. (сравнительный) Синтез N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида методом активированных эфиров
Реакцию между гистамином и малоновой кислотой в присутствии гексафторфосфата (1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния)-3-оксида (метод а1), тетрафторбората O-(бензтриазол-1-ил)-N,N,N′,N′-тетраметилурониума (метод а2), гидрохлоридом (1-(3-диметиламино)пропил)-3-этилкарбодиимида (метод а4), и гидройодидом 1-метил-2-хлорпиридиния (метод а5) проводили в среде диметилформамида, являющегося полярным, апротонным растворителем, способным хорошо растворять гистамин и являющимся наиболее используемым растворителем в данном типе превращений. Реакции проводили при комнатной температуре по следующей процедуре:
к раствору 22,2 г (0,2 моль) гистамина в 120 мл безводного, дегазированного диметилформамида прибавляли при перемешивании 10,4 г (0,1 моль) высушенной в вакууме малоновой кислоты, 35 мл (~0,25 моль) безводного триэтиламина и 0,22 моль активирующего агента. Реакционную смесь перемешивали 4 часа, растворители удаляли в вакууме, остаток суспендировали в 100 мл холодной воды, осадок отфильтровывали и промывали водой до исчезновения по данным ВЭЖХ сопутствующих продуктов реакции.
Несмотря на высокую конверсию (по данным ВЭЖХ) исходных веществ, общий выход N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида в качестве продукта оказался чрезвычайно низким. Это связано с тем, что сопутствующие продукты реакции, образующиеся из активирующего агента, хоть и имеют более высокую, по равнению с N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамидом растворимость в воде, однако объемы воды, требуемые для их полного удаления (до уровня менее 5%) оказались достаточно велики, что привело к значительным потерям целевого вещества, а плохая растворимость целевого продукта в несмешивающихся с водой растворителях (дихлорметан, бензол, гексан) не позволяла выделять продукт экстракцией из водной фазы.
Также была осуществлена реакция образования амида (целевого продукта формулы (I)) под действием дициклогексилкарбодиимида, т.к. известно, что образующаяся из него в результате синтеза дициклогексилмочевина обладает низкой растворимостью в воде. Реакцию проводили в тетрагидрофуране и диметилформамиде без добавления дополнительных оснований (например, триэтиламина, диизопропилэтиламина). В результате с высоким выходом был получен N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамид, который однако, даже после двукратного горячего фильтрования водного раствора содержал примесь дициклогексилмочевины. Полученные результаты экспериментальных исследований получения N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида методом активированных эфиров приведены в Таблице 1.
Таблица 1
метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния)-3-оксида
(метод а1)
(метод а2)
(метод а4)
(метод а5)
Как видно из полученных данных, метод активированных эфиров не позволяет получать N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамид чистотой более 99% с приемлемым выходом.
Пример 2 (сравнительный) Синтез N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида ацилированием гистамина малоилхлоридом
Первоначально реакцию ацилирования гистамина малоилхлоридом проводили с концентрированным водным раствором гистамина (~20% (масс.)) при охлаждении. Данный эксперимент был осуществлен в двух вариантах: в первом было использовано стехиометрическое соотношение реагентов, а во втором двукратный избыток гистамина. В обоих случаях, даже на количествах вещества, типично используемых в масштабах лаборатории, происходило быстрое загустевание реакционной смеси делающее невозможным ее дальнейшее перемешивание, и как следствие, обеспечение адекватного массо- и теплообмена.
Следствием этого стала низкая конверсия гистамина и образование целого ряда трудноотделимых побочных продуктов.
Попытка проведения реакции в разбавленным раствором (~5% (масс.)), показала, что в данном случае в качестве основной реакции, протекает гидролиз малонилхлорида, а не ацилирование гистамина.
Попытка использовать в качестве среды для ацилирования диметилформамида хоть и позволила обеспечить высокие конверсию гистамина и выход продукта, приводила к формированию ряда трудноудаляемых примесей, в частности N-формилгистамина и бисдиметиламида малоновой кислоты. Образование данных примесей, по-видимому, связано с относительно высокой реакционной способностью диметилформамида по отношению к сильным нуклеофилам. Несмотря на низкую температуру проведения реакции (-5-(-)10°С) и интенсивное перемешивание, локальные перегревы приводили к протеканию данных побочных реакций.
Таким образом выделение продукта, полученного по данному методу, осложнялось образованием трудноотделяемых, из-за близких значений растворимости, примесей. Так, перекристаллизация образца N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида, полученного данным способом из воды или этанола, практически не повлияла на содержание примесей в образце.
Принимая во внимание необходимость дальнейшего использования гетерогенных ионообменных носителей для выделения свободного основания N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида, данный подход так же был признан неудовлетворительным. Полученные результаты экспериментальных исследований получения N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида взаимодействием малонилхлорида с гистамином приведены в Таблице 2.
Таблица 2
Пример 3 (сравнительный) Ацилирование гистамина бисимидазолидом малоновый кислоты
Имидазолид малоновой кислоты был получен из малоновой кислоты и карбонилдиимидазола in situ и не выделялся.
Ацилирование проводилось по следующей методике: к раствору 10,4 г (0,1 моль) малоновой кислоты в 200 мл безводного тетрагидрофурана прибавили 34,0 г (0,21 моль) карбонилдимидазола. Реакционную смесь перемешивали 30 мин. при комнатной температуре, а затем еще 2 часа при 50°С. Реакционную смесь охлаждали и прибавляли 22,2 (0,2 моль) гистамина и перемешивали 18 часов. Растворитель удаляли в вакууме, к остатку прибавляли 100 мл холодной воды, осадок отфильтровывали и промывали холодной водой 3×50 мл. Выход N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида 13,6 г (47%).
Низкий выход продукта, так же как и в случае использования метода активированных эфиров, можно объяснить необходимостью удалить из реакционной смеси имидазол который имеет сопоставимые значения растворимости в воде и спиртах.
Так же, в результате анализа ВЭЖХ было найдено, что в качестве побочного продукта в данной реакции образуется бис(2-этил-4(5)-имидазолил)мочевина.
Совокупность полученных данных свидетельствует о нецелесообразности применения данного метода для решения задачи настоящего изобретения.
Пример 4. Ацилирование гистамина различными сложными эфирами малоновой кислоты
Как указано выше, был известен способ получение соединения формулы (I) путем взаимодействия диэтилового эфира малоновой кислоты с гистамином в отсутствии растворителя.
Заявителем было изучено взаимодействие с гистамином дифенилового и диметилового эфиров малоновой кислоты. Реакцию проводили в различных растворителях. Результаты экспериментальных исследований получения N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида путем взаимодействия сложных эфиров малоновой кислоты с гистамином приведены в таблице 3.
Таблица 3
Таблица 3 - Продолжение
(сравнительный)
Как видно из приведенных данных, наиболее перспективным представляется проведение реакции дифенил- и диметилмалоната с гистамином в бутаноле при температуре около 100°С. Данные условия обеспечивают высокую конверсию гистамина и приводят к образованию N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида, не содержащего значительного количества примесей. С целью решения задачи данного изобретения была изучена возможность дальнейшей очистки образцов N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида полученных из дифенил- и диметилмалоната с гистамином в бутаноле при 100°С. Результаты экспериментальных исследований по очистке N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида представлены в таблице 4.
Таблица 4
малонамид полученный реакцией гистамина с диметил-малонатом
малонамид полученный реакцией гистамина с дифенил-малонатом
Данные ВЭЖХ, подтверждающие чистоту продуктов по опытному методу 13 (таб. 4), приведены на фигурах 1, 2 и 3, соответственно.
Также для опытного образца, полученного методом 13 (таб. 4), очищенного путем кристаллизации из воды с обработкой горячего раствора активированным углем были получены спектры ЯМР:
1H (Bruker DRX500,13, 500,13 МГц, ДМСО-d6) образца N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида. (1H NMR (500 MHz, DMSO-d6) δ: 11.82 (s, 1H, NHимидазол,), 8.09 (t, J=5.6 Hz, 1H, CONH), 7.51 (s, 1H, H2), 6.79 (s, 1H, H5), 3.28 (q, J=7.2 Hz, 2H, Hα), 3.01 (s, 2H, COCH2CO), 2.63 (t, J=7.2 Hz, 2H, Hβ).
13С (Bruker DRX500,13 125,76 МГц, ДМСО-d6) образца N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида: δ: 166.9 (CO), 134.7 (C5), 134.3 (C3), 116.9 (C2), 43.5 (CH2), 39.0 (Cα), 26.8 (Cβ).
Указанные спектры 1H и 13C образца приведены на фиг.4 и фиг.5., соответственно а также на фиг.6 приведен масс-спектр высокого разрешения.
Данные спектрального анализа образца подтверждают высокую чистоту исследуемого опытного образца.
В ходе создания настоящего изобретения найдено, что реакция гистамина с диметилмалонатом в бутаноле при температуре предпочтительно до 100°С, в сочетании с кристаллизацией продукта из этанола, метанола или воды с обработкой горячего раствора активированным углем позволяет получать N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамид чистотой выше 99,0% и не содержащий единичной примеси выше 0,1% (см. фиг. 1), пригодный для использования в качестве фармацевтической субстанции в производстве различных видов лекарственных форм.
Резюмируя приведенные экспериментальные данные, авторы отмечают, что поскольку диметилмалонат, в отличии от дифенилмалоната, является в настоящий момент более коммерчески доступным реагентом, и образцы, полученные с его использованием, являются более высокочистыми, чем образцы, полученные из дифенилмалоната, применение диметилмалоната для ацилирования гистамина представляется наиболее целесообразным.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА N,N'-БИС-[2-(1Н-ИМИДАЗОЛ-4-ИЛ)ЭТИЛ]ПРОПАНДИАМИДА И ЕЕ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2022 |
|
RU2823162C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ N,N'-БИС[2-(1Н-ИМИДАЗОЛ-4-ИЛ)ЭТИЛ]МАЛОНАМИДА | 2022 |
|
RU2818073C2 |
| Способ получения 4,4'-(пропандиамидо)дибензоата натрия | 2016 |
|
RU2624729C1 |
| ЖИДКАЯ ЛЕКАРСТВЕННАЯ ФОРМА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ РИНИТА | 2022 |
|
RU2808034C2 |
| N-АЦИЛЬНЫЕ ПРОИЗВОДНЫЕ БИОГЕННЫХ АМИНОВ - МОДУЛЯТОРЫ ПЕРЕКИСНОГО ОКИСЛЕНИЯ ЛИПИДОВ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2093520C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3, 4-БИС(3-АМИНОФУРАЗАН-4-ИЛ)-ФУРАЗАНА И ЕГО N, N'-ДИАЦИЛЬНЫХ ПРОИЗВОДНЫХ | 2012 |
|
RU2489428C1 |
| ПСЕВДОДИПЕПТИДЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СПОСОБЫ ЛЕЧЕНИЯ | 1994 |
|
RU2166510C2 |
| КРИСТАЛЛИЧЕСКИЙ ИНГИБИТОР АХАТ | 2006 |
|
RU2394020C2 |
| СПОСОБ ПОЛУЧЕНИЯ 6-МЕТИЛ-2-(4-МЕТИЛФЕНИЛ)-ИМИДАЗОЛО[1,2-А]-ПИРИДИН-3-(N, N-ДИМЕТИЛАЦЕТАМИДА), ЭФИРЫ, КРИСТАЛЛИЧЕСКИЕ ЭФИРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ | 2000 |
|
RU2254334C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАРНОЗИНА | 1990 |
|
RU2030422C1 |




Способ получения N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида формулы (I) реакцией диметилмалоната с гистамином в полярном органическом растворителе при температуре 85-120°С, включающий стадию кристаллизации из воды и/или полярного органического растворителя. Способ позволяет получать продукт чистотой от 99,0 мас.% и пригоден для получения субстанции в фармацевтической промышленности. 5 з.п. ф-лы, 6 ил., 4 пр., 4 табл.

1. Способ получения N,N'-бис[2-(1Н-имидазол-4-ил)этил]малонамида формулы

реакцией диметилмалоната с гистамином в полярном органическом растворителе при температуре 85-120°С, включающий стадию кристаллизации из воды и/или полярного органического растворителя.
2. Способ по п.1, включающий проведение реакции в растворе, содержащем 3-30 мас.% гистамина.
3. Способ по п.1 или 2, включающий проведение реакции в бутаноле, пропаноле, изопропаноле, этиленгликоле, диоксане, или комбинации указанных растворителей, или их смесях с водой.
4. Способ по п.1, включающий использование на стадии кристаллизации воды, метанола, этанола, н-пропанола, изопропанола, этиленгликоля и других спиртов, или их смеси с водой и/или друг с другом.
5. Способ по любому из пп.1-4, дополнительно включающий внесение затравки в раствор для кристаллизации продукта формулы (I).
6. Способ по любому из пп.1-5, включающий следущие стадии:
к 15% (по массе) раствору гистамина в бутаноле, или изопропаноле, или этаноле, необязательно в виде смеси друг с другом, при этом смесь необязательно содержит воду в количестве до 10 мас.%, прибавляют диметилмалонат в количестве 0,4-0,55 моль-экв.,
полученную смесь выдерживают при температуре от 90 до 110°С до достижения содержания гистамина в реакционной смеси менее 3% (мас.) в течение 4-8 часов, охлаждают, осадок отфильтровывают, растворяют в горячей воде, добавляют 1-10% (мас.) активированного угля, перемешивают, горячий раствор фильтруют и оставляют для кристаллизации.
| WO 2014168523 A1, 16.10.2014 | |||
| АСПАРТИЛЬНЫЕ ПРОИЗВОДНЫЕ ГИСТАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ ФЕРМЕНТОВ АНТИОКСИДАНТНОЙ ЗАЩИТЫ | 2005 |
|
RU2287524C1 |

