Область техники
Настоящее изобретение относится к фармацевтической композиции, содержащей 5-{4-(аминосульфонил)фенил}-2,2-диметил-4-(3-фторфенил)-3(2H)-фуранон и составу в капсулах, содержащему фармацевтическую композицию. Более конкретно, настоящее изобретение относится к фармацевтической композиции, содержащей 5-(4-(аминосульфонил)фенил}-2,2-диметил-4-(3-фторфенил)-3(2H)-фуранон, который подходит в качестве нестероидного противовоспалительного лекарственного средства вследствие его хорошей стабильности, высокой скорости растворения, улучшенной однородности, а также превосходных фармакокинетических свойств, и составу в капсулах, содержащему указанную фармацевтическую композицию.
Уровень техники
Известно, что простагландины играют важную роль в развитии воспаления. Простагландины образуются из арахидоновой кислоты под действием циклооксигеназы (далее сокращенно «СОХ»). Активность СОХ подавляют, чтобы ингибировать синтез простагландинов, в частности, PGE2, PGG2 и PGH2, что приводит к лечению воспаления.
Известны две изоформы СОХ, СОХ-1 и СОХ-2. СОХ-1 преимущественно обнаруживается в желудочно-кишечном тракте и почках, и полагают, что она поддерживает физиологические гомеостатические функции, включая целостность желудочно-кишечного тракта и функции почек. Ингибирование активности СОХ-1 может привести к угрожающим жизни токсическим эффектам, таким как язвы и кровоизлияния в желудочно-кишечном тракте. В противоположность этому, СОХ-2 индуцируется воспалительными стимулами и, как известно, ответственна за развитие воспалительного процесса.
Полагают, что ингибиторы СОХ-2 обладают широким спектром терапевтических активностей, а также противовоспалительным, обезболивающим и жаропонижающим эффектом. Например, известно, что ингибирование СОХ-2 предотвращает развитие рака, особенно рака толстой кишки [J. Clin. Invest., 99, 2254 (1997)], может применяться для лечения хронических нейродегенеративных заболеваний, таких как болезнь Альцгеймера [Neurology, 48, 626 (1997)], а также может использоваться для уменьшения объема повреждения, сопровождающего инфаркт [J. Neuroscience, 17, 2746 (1997)].
Традиционные нестероидные противовоспалительные средства (НПВС), такие как индометацин, напроксен, кетопрофен, ибупрофен, пироксикам и диклофенак ингибируют как СОХ-1, так и СОХ-2, демонстрируя токсичность для желудочно-кишечного тракта вместе с противовоспалительной эффективностью. Кроме того, такие НПВС могут демонстрировать фатальную токсичность, такую как кровотечение и язвы, возникающую в связи с ингибированием СОХ-1, что ограничивает их клиническое применение. Таким образом, селективные ингибиторы СОХ-2 могут быть использованы в качестве терапевтических агентов против воспаления и заболеваний, сопровождающихся воспалением, не оказывая токсичного эффекта на желудочно-кишечный тракт, который является общим при длительном использовании традиционных НПВС.
Недавно сообщалось, что производные 4,5-диарил-3(2H)-фуранона действуют как селективные ингибиторы СОХ-2 (патент Кореи No. 10-0495389). Когда производные фуранона используются для получения фармацевтических композиций, они должны иметь высокую скорость растворения, хорошую текучесть, оптимальное изменение массы и улучшенную однородность. Авторы настоящего изобретения обнаружили, что конкретное производное фуранона отвечает указанным требованиям. На основании этого факта авторы настоящего изобретения получили фармацевтическую композицию, содержащую производное фуранона, и состав в капсулах, содержащий фармацевтическую композицию и, наконец, реализовали настоящее изобретение.
Описание
Техническая проблема
Одной из задач настоящего изобретения является получение фармацевтической композиции, содержащей производные фуранона, с высокой скоростью растворения, хорошей текучестью, оптимальным изменением массы, а также улучшенной однородностью.
Другой задачей настоящего изобретения является создание фармацевтического состава, содержащего фармацевтическую композицию.
Техническое решение
В соответствии с одним аспектом настоящего изобретения предлагается фармацевтическая композиция, содержащая (i) соединение формулы 1:
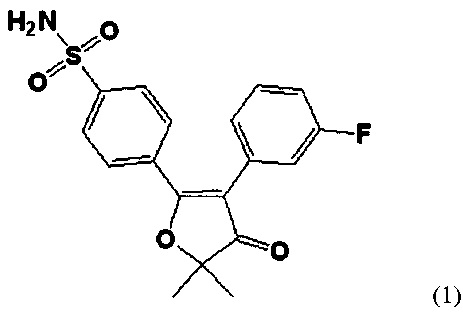
или его фармацевтически приемлемую соль, имеющее диаметр частиц 50% объема (d(0,5)) от 3 мкм до 9 мкм, (ii) фармацевтически приемлемый разбавитель, и (iii) фармацевтически приемлемое смазывающее вещество.
В соответствии с другим аспектом настоящего изобретения предлагается фармацевтический состав, содержащий фармацевтическую композицию.
Полезный эффект
Фармацевтическая композиция, содержащая 5-{4-(аминосульфонил)фенил}-2,2-диметил-4-(3-фторфенил)-3(2H)-фуранон в соответствии с настоящим изобретением, имеет преимущества хорошей стабильности, высокой скорости растворения, улучшенной однородности, а также отличные фармакокинетические свойства. Благодаря указанным преимуществам, фармацевтическая композиция согласно настоящему изобретению является эффективной для лечения воспаления или боли.
Описание чертежей
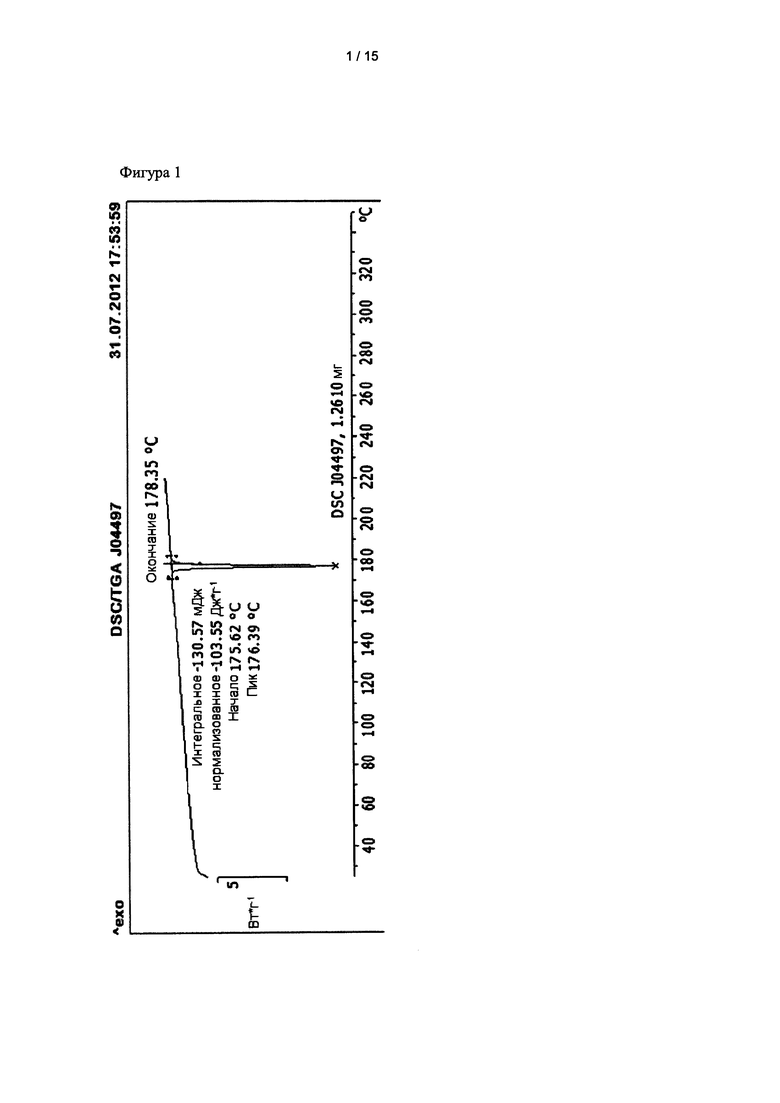
Фиг. 1 представляет собой график, показывающий результаты дифференциальной сканирующей калориметрии (ДСК) для кристаллической формы, полученной в примере получения 1.
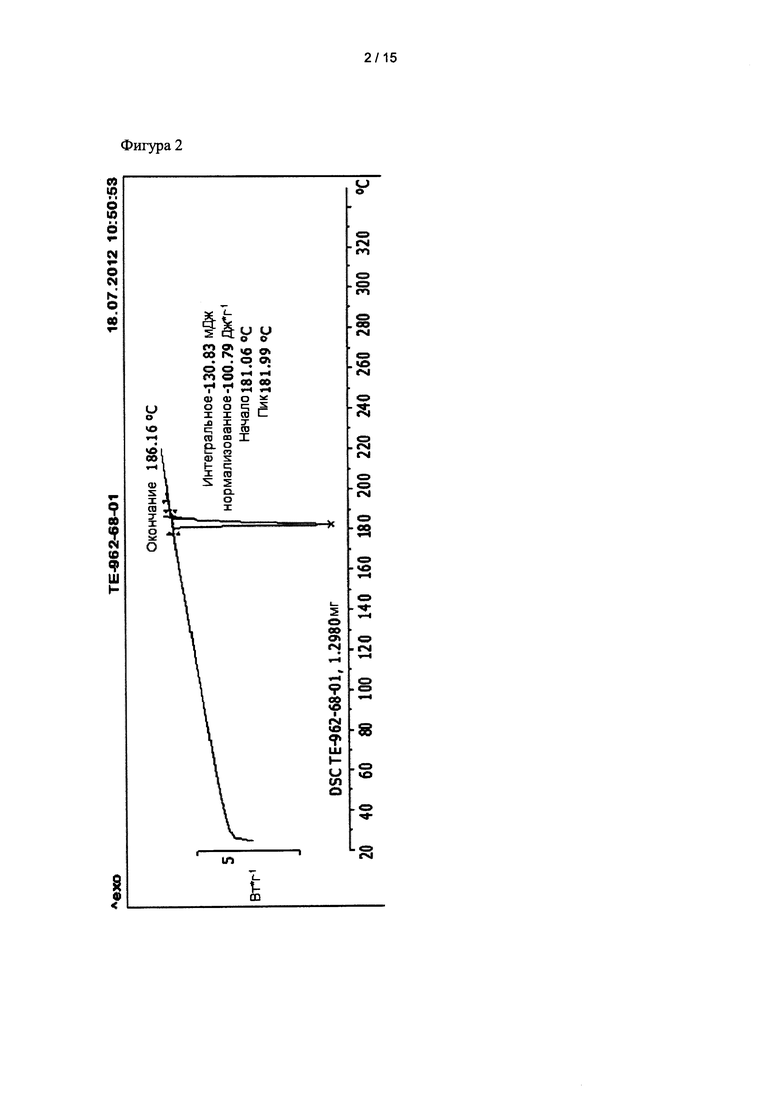
На фиг. 2 представлен график, показывающий результаты дифференциальной сканирующей калориметрии (ДСК) для кристаллической формы, полученной в примере получения 2.
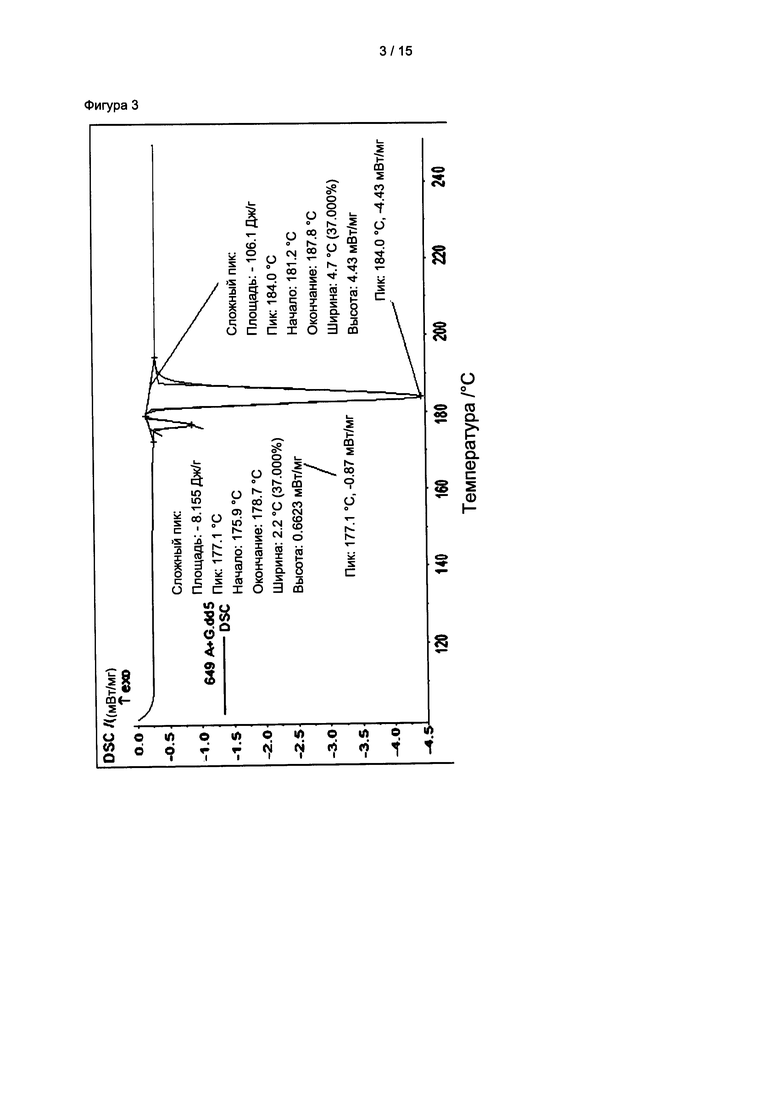
На фиг. 3 представлен график, показывающий результаты дифференциальной сканирующей калориметрии (ДСК) для кристаллической формы, полученной в примере получения 3.
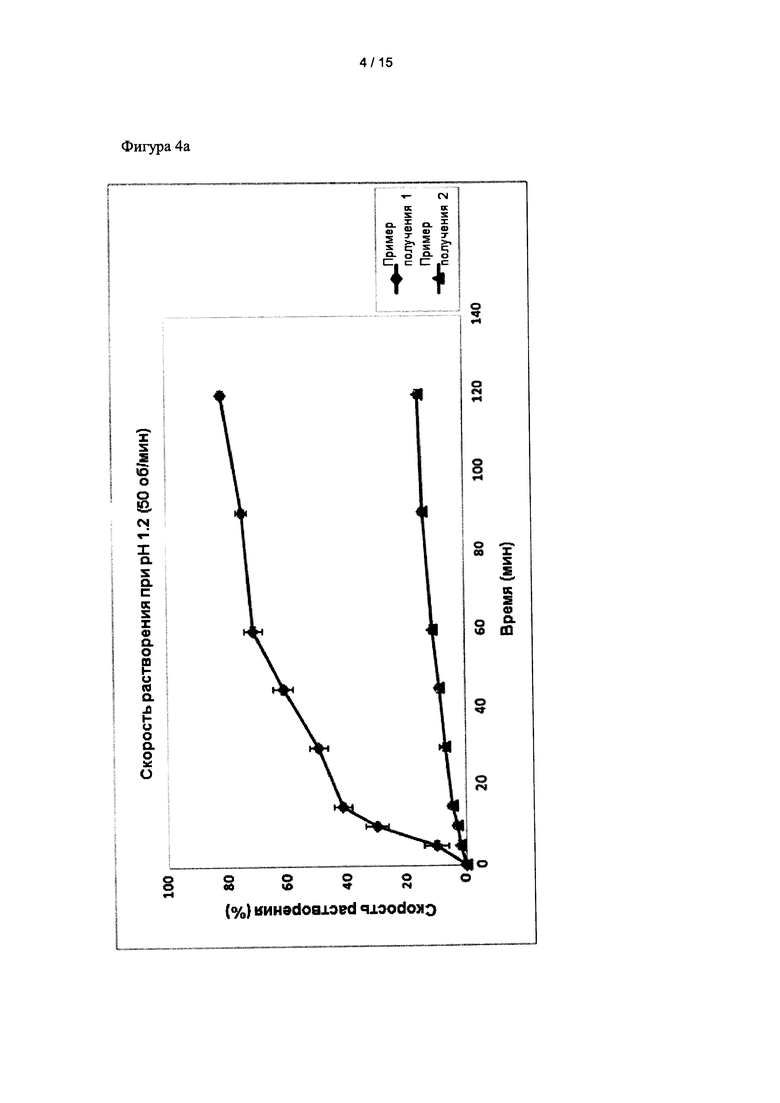
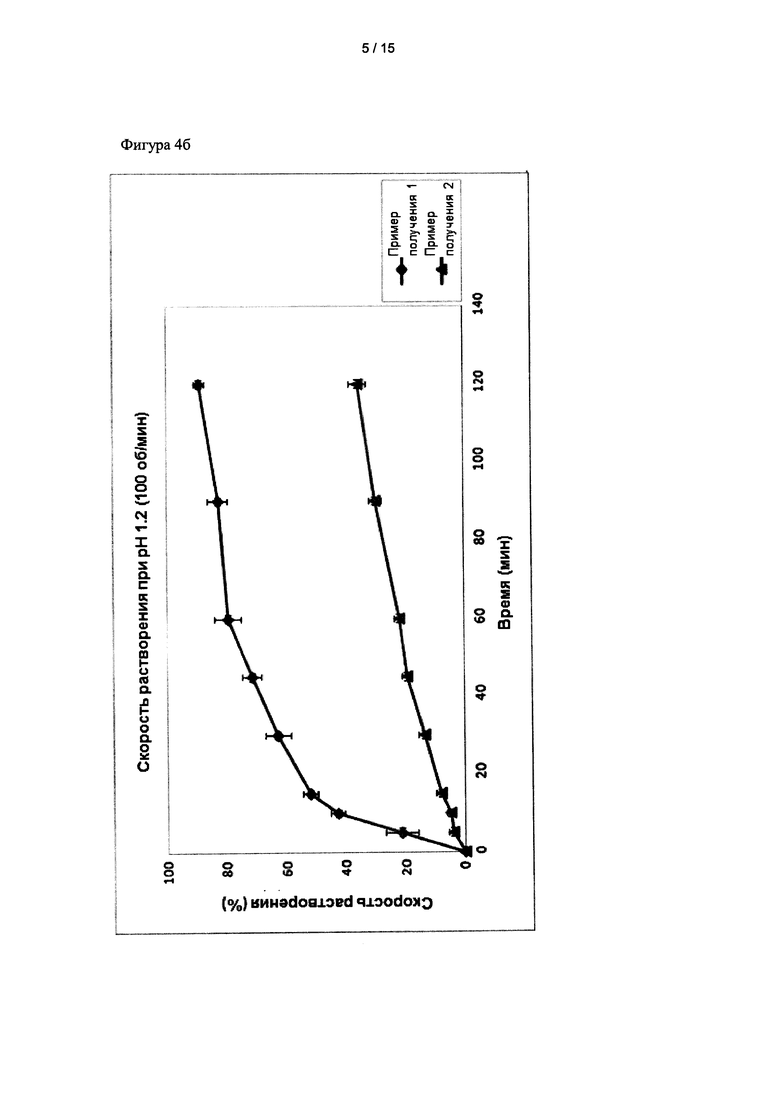
Фиг. 4а и 4б показывают графики скорости растворения кристаллических форм, полученных в примерах получения 1 и 2, при различных скоростях перемешивания - 50 оборотов в минуту (фиг. 4а) и 100 оборотов в минуту (фиг. 4б).
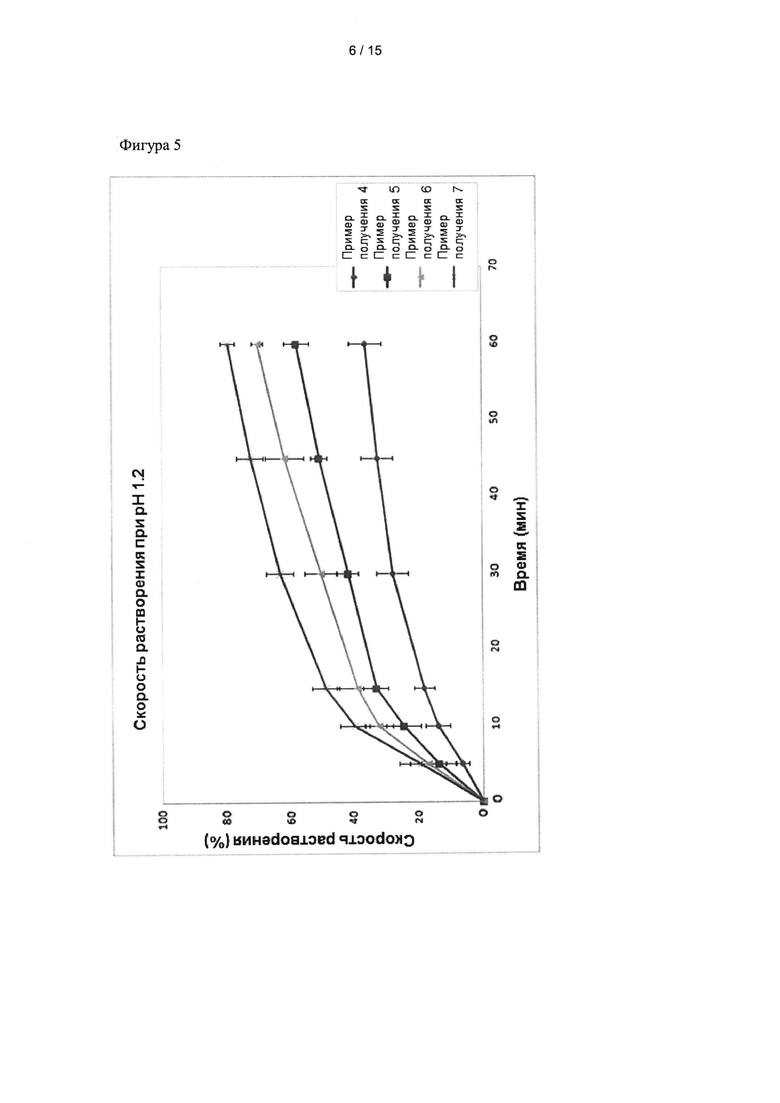
Фиг. 5 представляет собой график, показывающий скорость растворения смесей кристаллических форм, приготовленных в примерах получения с 1 по 7.
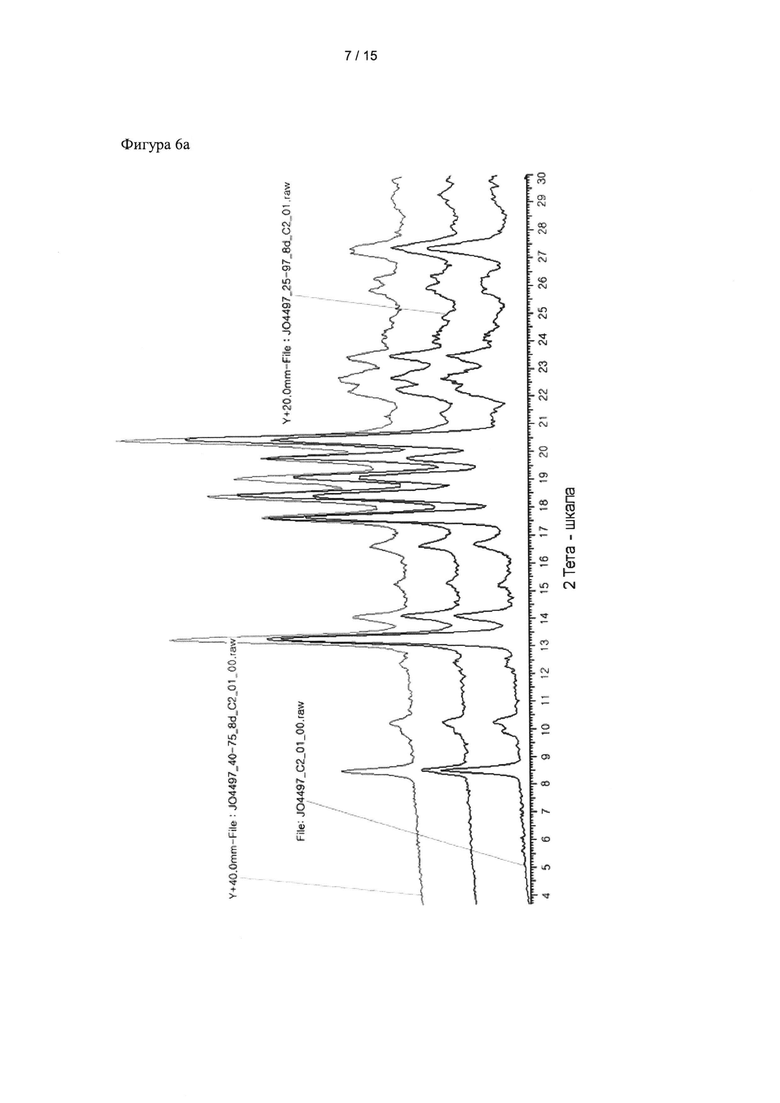
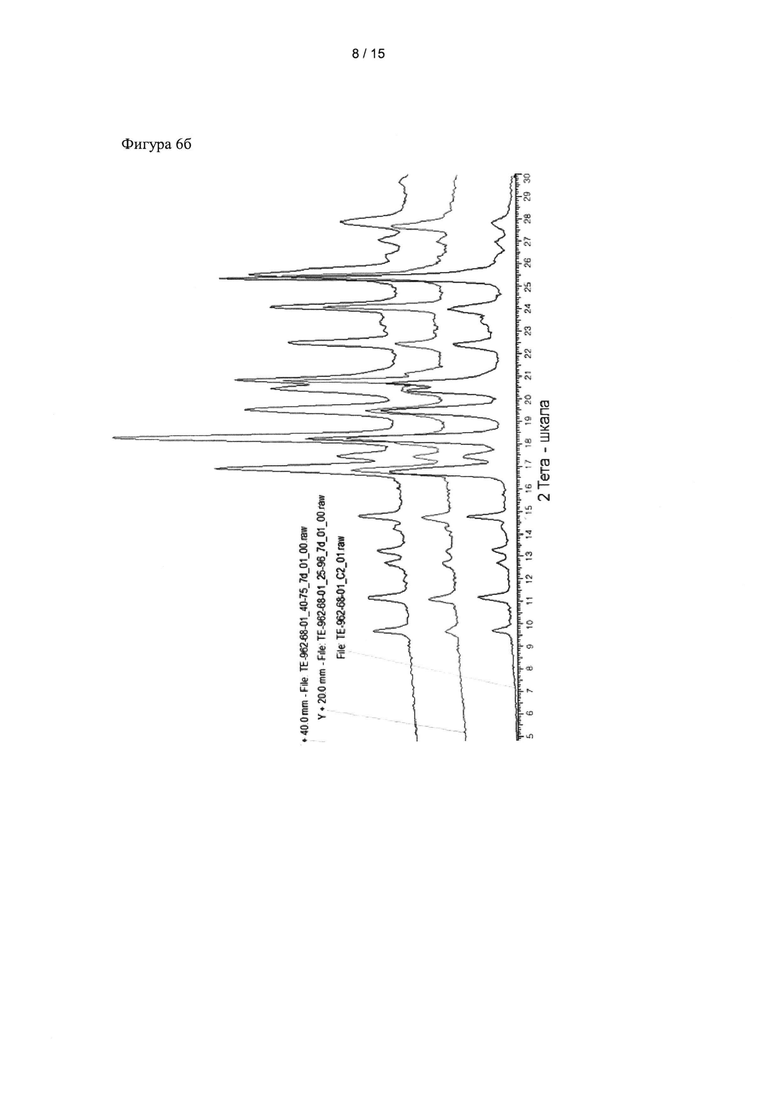
Фиг. 6а и 6б показывают результаты рентгеноструктурного анализа для кристаллических форм, полученных в примерах получения 1 (фиг. 6а) и 2 (фиг. 6б), после хранения в различных условиях.
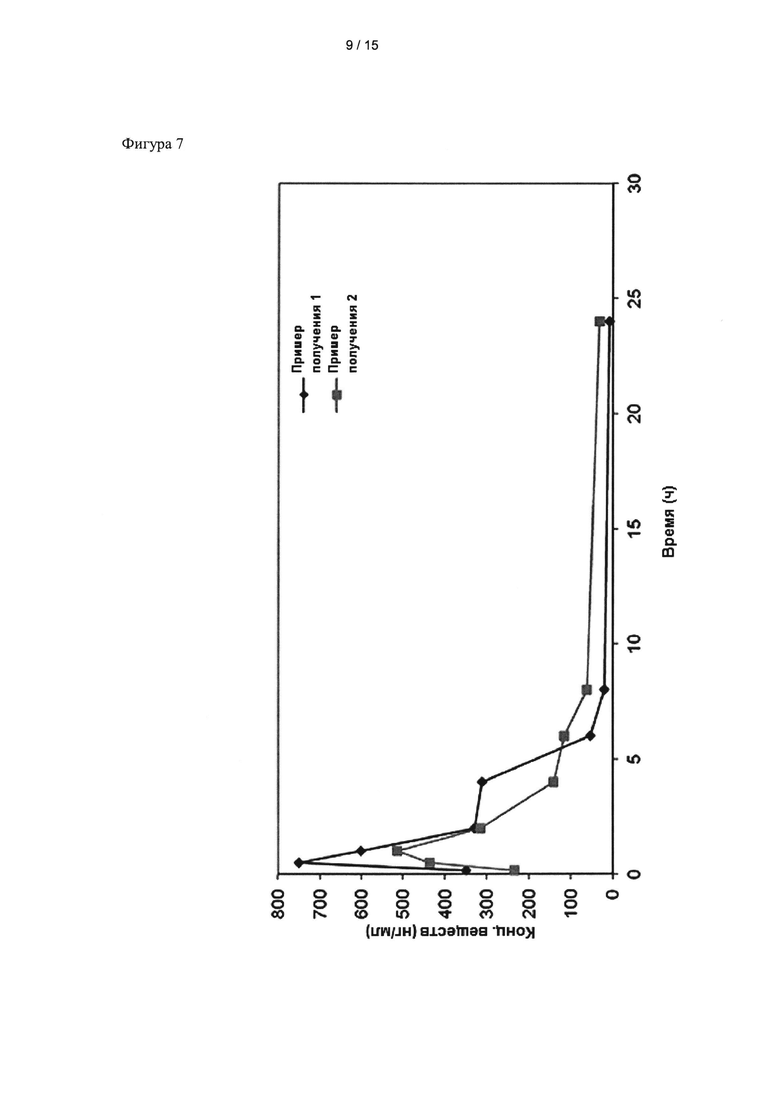
Фиг. 7 представляет собой график, показывающий фармакокинетические свойства кристаллических форм, приготовленных в примерах получения 1 и 2, на крысах.
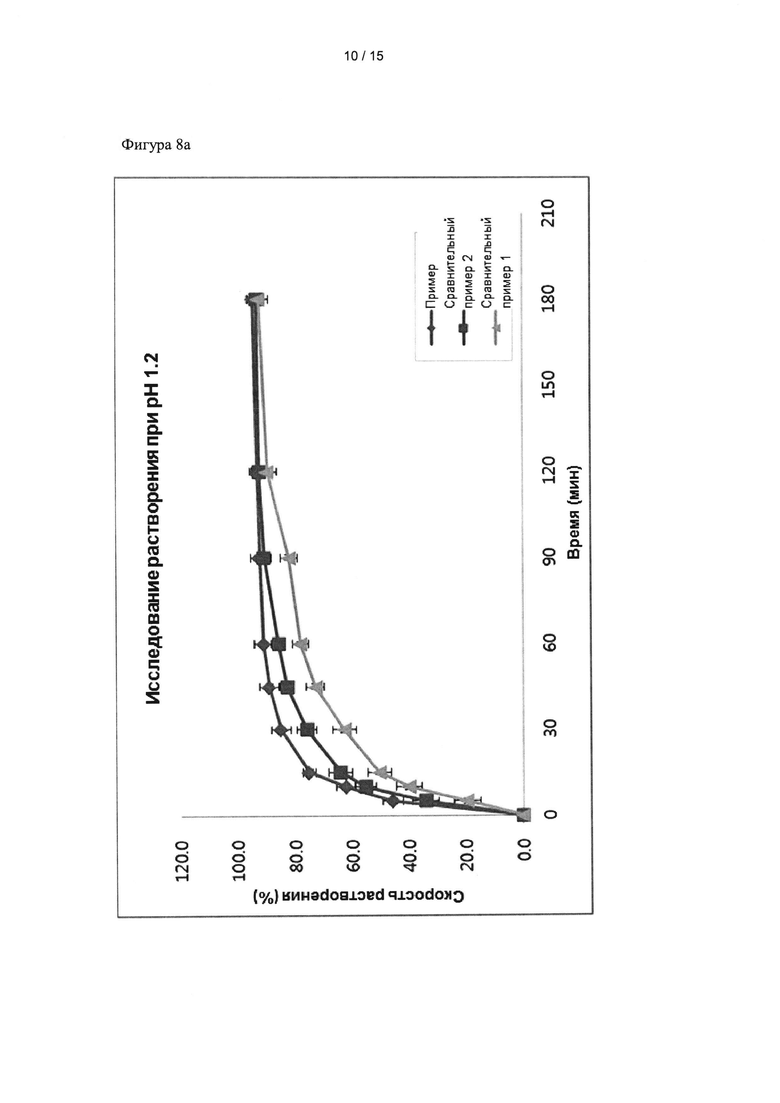
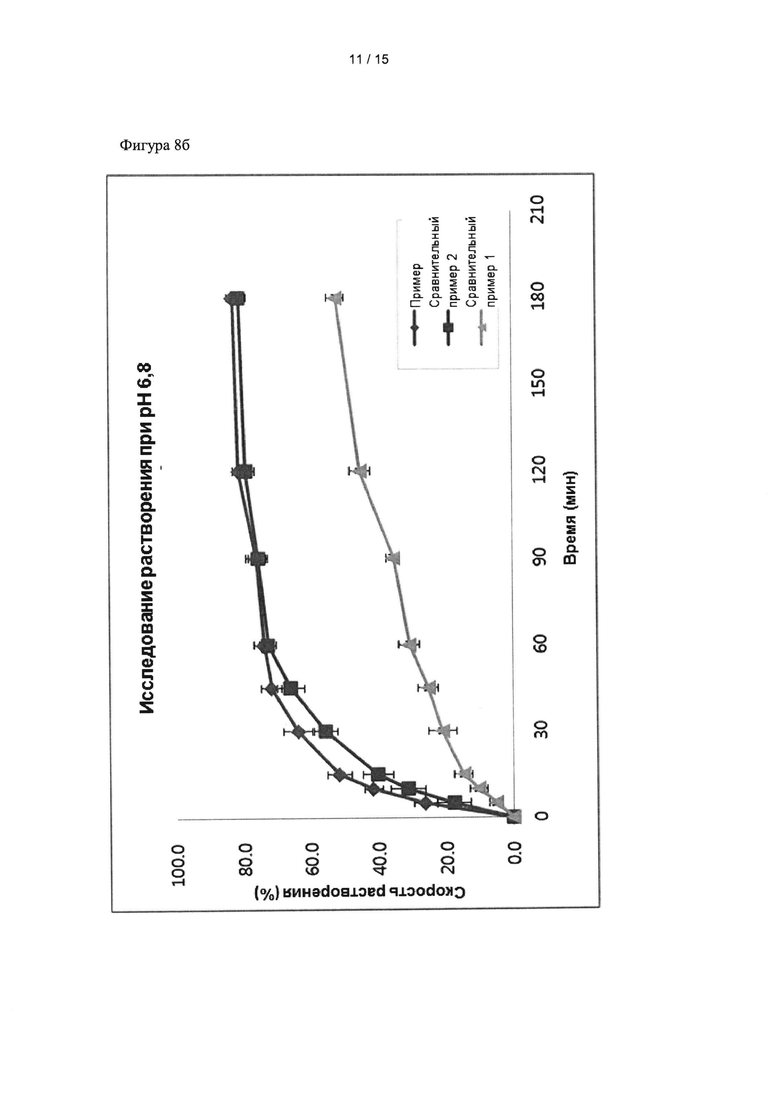
Фиг. 8а и 8б показывают графики скорости растворения кристаллических форм примера 1 и сравнительных примеров 1 и 2 для различных растворов.
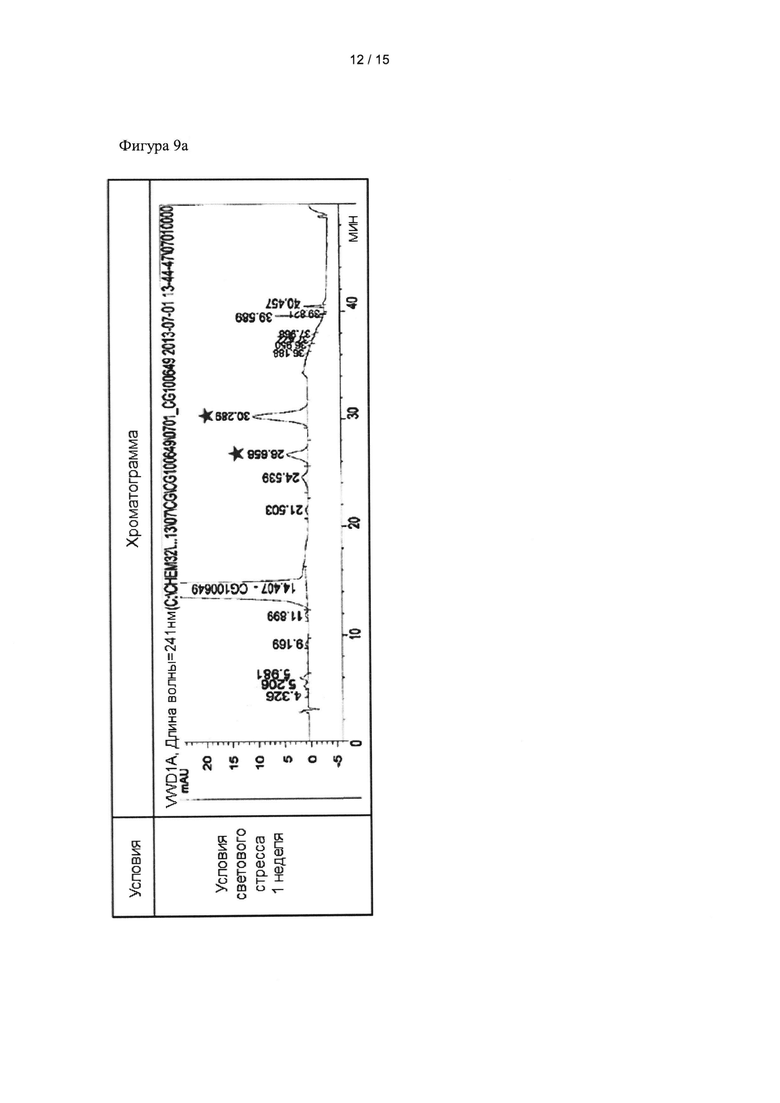
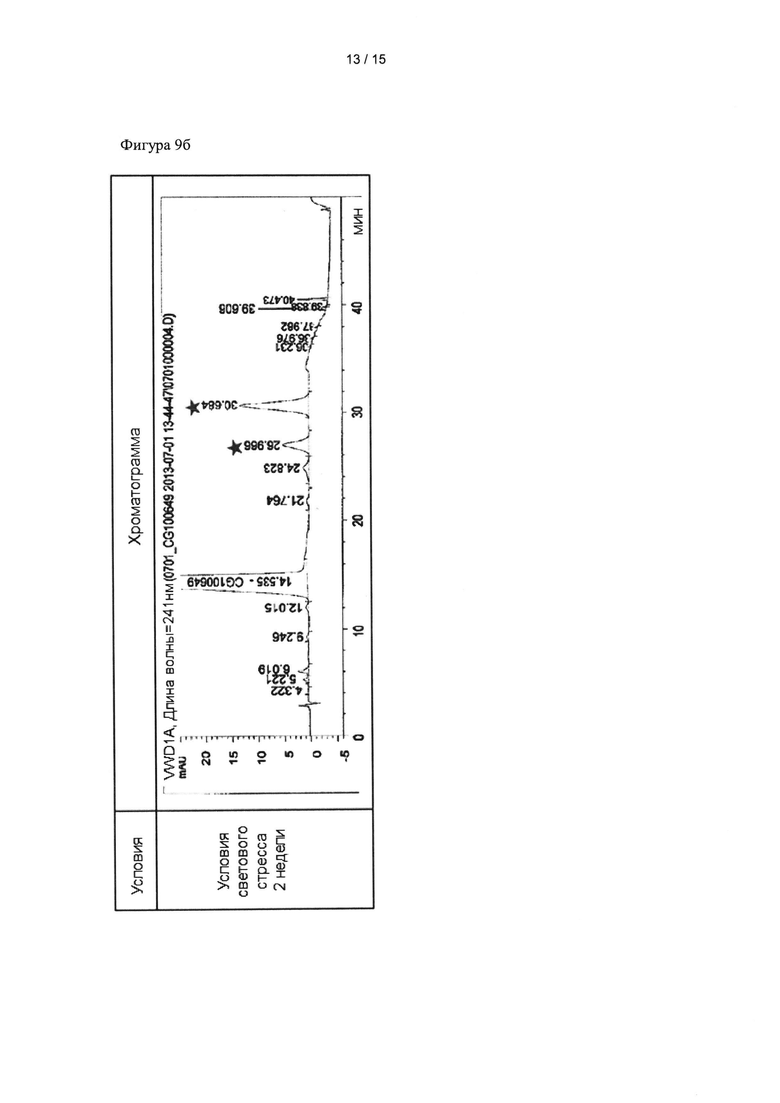
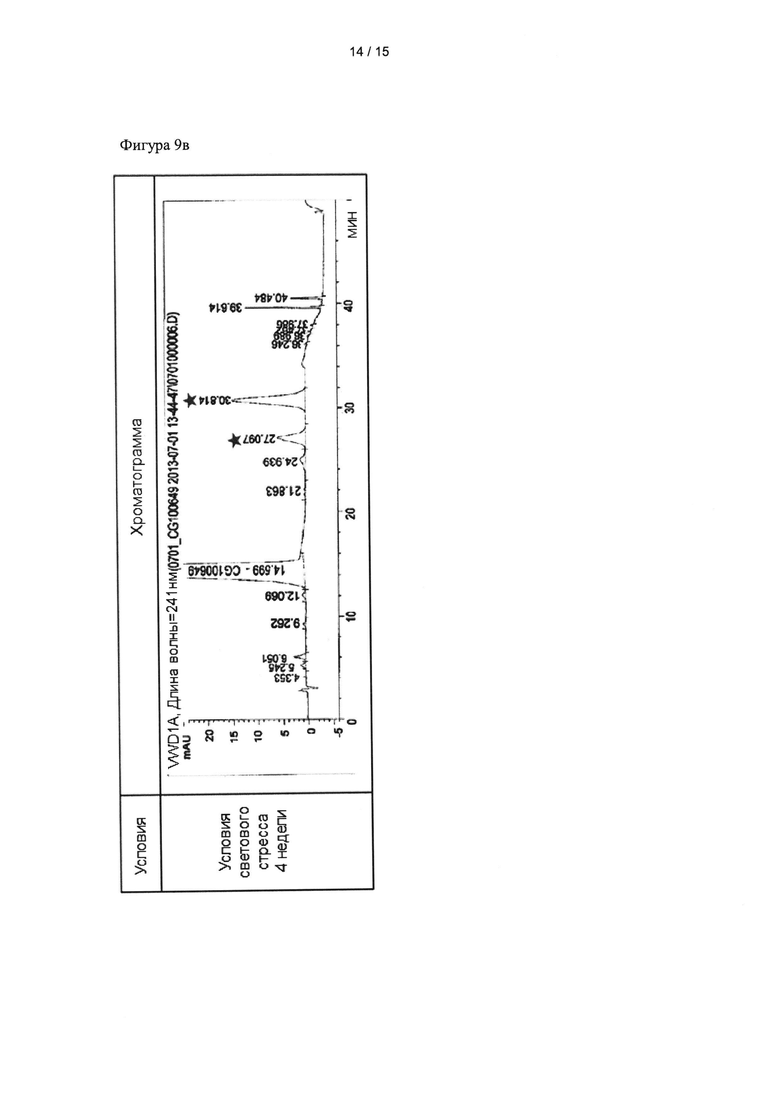
Фиг. 9а-9в представляют собой хроматограммы композиций, содержащих кристаллическую форму из примера 1, полученные с помощью ВЭЖХ после хранения в условиях светового стресса; пики, отмеченные  , указывают, что соответствующие вещества, образовавшиеся в условиях светового стресса, превысили соответствующие контрольные значения, определенные способами для родственных веществ.
, указывают, что соответствующие вещества, образовавшиеся в условиях светового стресса, превысили соответствующие контрольные значения, определенные способами для родственных веществ.
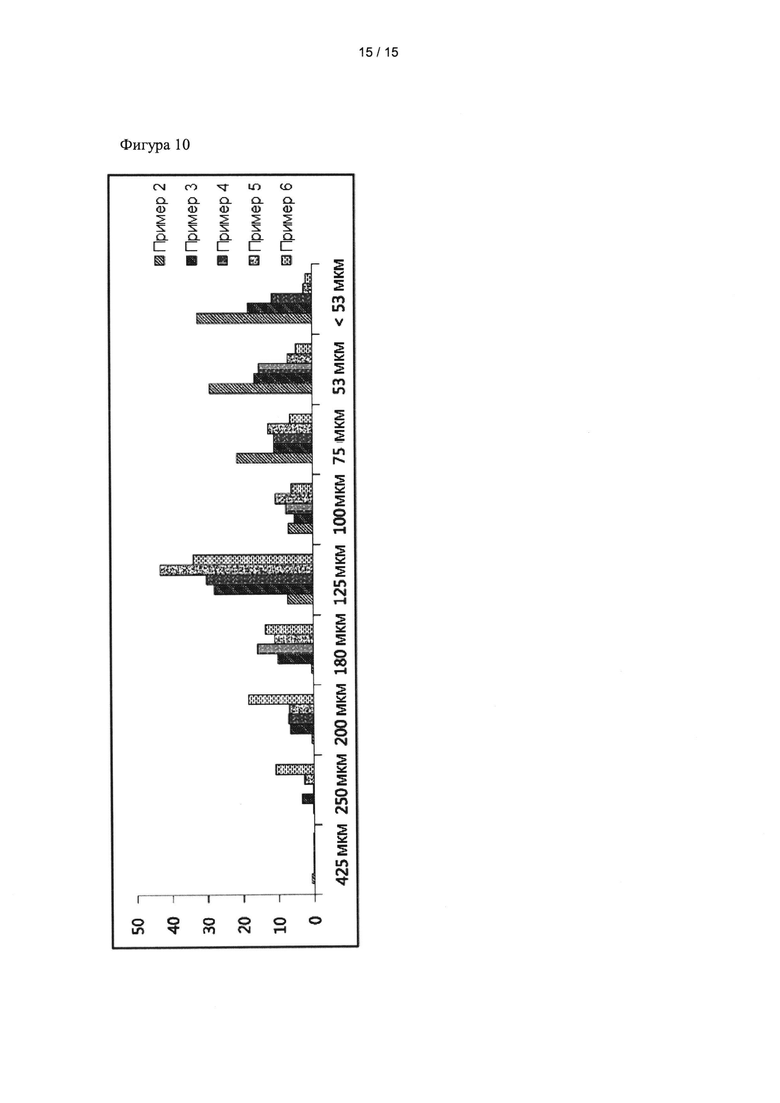
Фиг. 10 представляет собой график, показывающий распределение частиц по размеру в составах, полученных в примерах со 2 по 6.
Подробное описание изобретения
Далее настоящее изобретение будет описано подробно.
Настоящее изобретение относится к фармацевтической композиции, содержащей (i) соединение формулы 1:
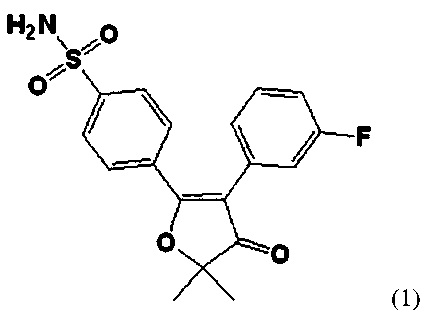
или его фармацевтически приемлемую соль, имеющее диаметр частиц 50% объема (d(0,5)) от 3 мкм до 9 мкм, (ii) фармацевтически приемлемый разбавитель, и (iii) фармацевтически приемлемое смазывающее вещество.
Соединение формулы 1 используют в качестве активного ингредиента в фармацевтической композиции согласно настоящему изобретению. Соединение формулы 1 представляет собой селективный ингибитор СОХ-2, химическое название 5-{4-(аминосульфонил)фенил}-2,2-диметил-4-(3-фторфенил)-3(2H)-фуранон. Известно, что Соединение формулы 1 имеет сниженную токсичность для желудочно-кишечного тракта и эффективно в отношении воспалительных заболеваний, ассоциированных с воспалением заболеваний, боли, солидных опухолей, заболеваний, ассоциированных с ангиогенезом, болезни Альцгеймера, атак, конвульсий, инсультов и эпилепсии по сравнению с традиционными НПВС (см. Корейский патент N 10-0495389).
Соединение формулы 1 характеризуется тем, что диаметр частиц 50% объема (d(0,5)) составляет от 3 мкм до 9 мкм и, необязательно, диаметр частиц 90% объема (d(0,9)) составляет от 10 мкм до 100 мкм. Диаметр частиц 50% объема (d(0,5)) означает такой диаметр частиц, когда, если частицы вводят от меньшего диаметра к большему, кумулятивная частота объемного распределения достигает 50% от общего объема, диаметр частиц 90% объема (d(0,9)) означает, такой диаметр частиц, когда, если частицы вводят от меньшего диаметра к большему, кумулятивная частота объемного распределения достигает 90% от общего объема. Соединение формулы 1 используют в количестве от 0,5 до 20% по массе в расчете на общую массу фармацевтической композиции. Благодаря использованию меньшего количества, диаметра частиц 50% объема (d(0,5)) от 3 мкм до 9 мкм, и, возможно, диаметра частиц 90% объема (d(0,9)) от 10 мкм до 100 мкм, проще обеспечить равномерность содержания соединения формулы 1 при измельчении с использованием разбавителя, и этим достигается дальнейшее улучшение скорости растворения соединения формулы 1 (фиг. 8а и 8б). d(0,5) может составлять от 3 мкм до 8 мкм в некоторых вариантах реализации, от 4 мкм до 9 мкм в еще одном варианте реализации, и от 4 мкм до 8 мкм в дополнительных вариантах реализации. d(0,9) составлять от 10 мкм до 80 мкм в некоторых вариантах реализации, от 10 мкм до 50 мкм в еще одном варианте реализации, и от 10 мкм до 20 мкм в дополнительных вариантах реализации.
Соединение формулы 1 может существовать в кристаллической форме А, кристаллической форме G или в виде их смеси.
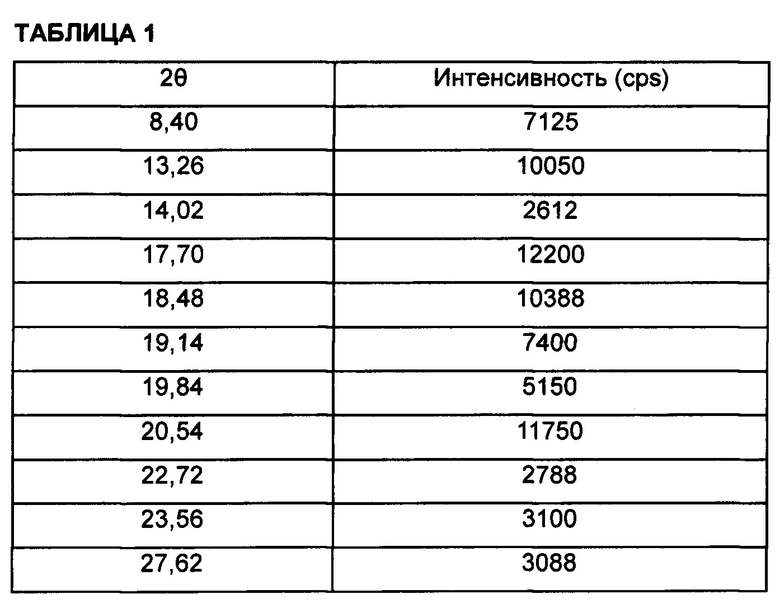
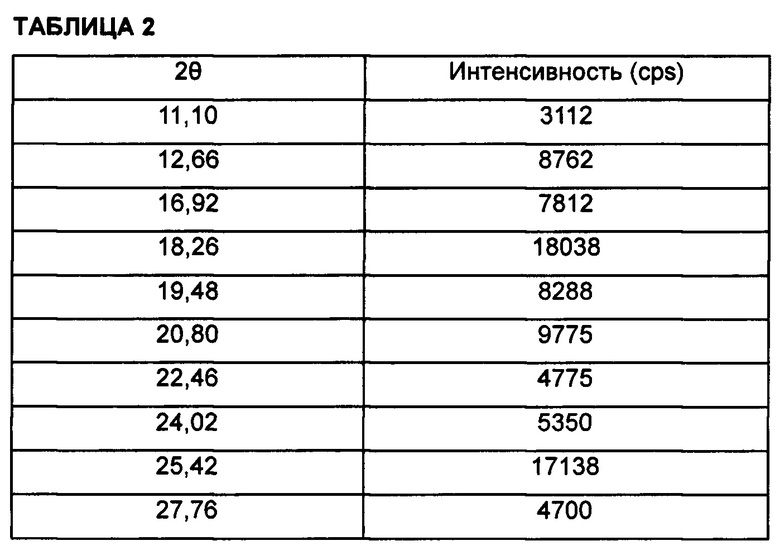
По результатам экспериментов, проведенных авторами настоящего изобретения, кристаллическая форма А имеет результаты рентгеноструктурного анализа, представленные в таблице 1, и дифференциальной сканирующей калориметрии (ДСК), показанной на фиг. 1. Кристаллическая форма G имеет результаты рентгеноструктурного анализа, представленные в таблице 2, и дифференциальной сканирующей калориметрии (ДСК), показанной на фиг. 2.
Авторы настоящего изобретения получили кристаллические формы от В до F перекристаллизацией кристаллической формы А из подходящих растворителей, таких как трет-бутилметиловый эфир, изопропиловый спирт, метиловый спирт, этиловый спирт и ацетонитрил. Тем не менее, кристаллические формы от В до F, как правило, возвращаются к кристаллической форме А при хранении при 40°С и 75% относительной влажности в течение 4-х дней. В противоположность этому, кристаллические формы А и G были весьма стабильны. В частности, когда частицы в кристаллической форме А присутствуют в большем количестве, в частности, кристаллическая форма А присутствует в количестве 50% по массе в расчете на общую массу кристаллических форм, была получена более высокая скорость растворения. Соответственно, предпочтительно, чтобы соединение формулы 1 содержало по меньшей мере 50% по массе кристаллической формы А в расчете на общую массу соединения.
Состояния кристаллических форм А и G остаются стабильными в течение длительного хранения и при ускоренных условиях хранения.
Соединение формулы 1 может быть использовано в количестве от 0,5 до 20% по массе, предпочтительно, 1% по массе в расчете на общую массу фармацевтической композиции.
Соединение формулы 1 может существовать в форме фармацевтически приемлемой соли.
Фармацевтическая композиция согласно настоящему изобретению содержит фармацевтически приемлемый разбавитель и фармацевтически приемлемое смазывающее вещество в дополнение к активному ингредиенту.
Разбавитель может быть использован в количестве от 75 до 99% по массе в расчете на общую массу фармацевтической композиции. В качестве разбавителя могут быть упомянуты, например, силикатированная микрокристаллическая целлюлоза (например, силикатированная микрокристаллическая целлюлоза 50 или 90), микрокристаллическая целлюлоза, целлюлоза, лактоза или их комбинация (например, Cellactose® 80). Использование силикатированной микрокристаллической целлюлозы является предпочтительным.
Смазывающее вещество может быть использован в количестве от 0,1 до 5% по массе, предпочтительно, 1% по массе в расчете на общую массу фармацевтической композиции. В качестве смазывающего вещества могут быть упомянуты, например, тальк или стеариновая кислота. Использование талька является предпочтительным.
Фармацевтическая композиция согласно настоящему изобретению может дополнительно содержать одну или несколько фармацевтически приемлемых добавок, обычно используемых в фармацевтической области, в дополнение к разбавителю и смазывающему веществу.
Фармацевтическая композиция может быть использована для профилактики или лечения воспалительных заболеваний, ассоциированных с воспалением заболеваний, боли, солидных опухолей, заболеваний, ассоциированных с ангиогенезом, болезни Альцгеймера, атак, конвульсий, инсульта или эпилепсии. Фармацевтическая композиция предпочтительно используется для профилактики или лечения воспалительных заболеваний, ассоциированных с воспалением заболеваний или боли.
Фармацевтическая композиция согласно настоящему изобретению может быть переработана в различные фармацевтические составы.
Составы могут быть в виде таблеток, порошков, гранул, капсул, суспензий, ингаляционных спреев и растворов для инъекций. Предпочтительно, эти составы являются капсулами, более предпочтительно, твердыми капсулами.
Фармацевтическую композицию согласно настоящему изобретению можно вводить различными путями, включая, но не ограничиваясь ими, перорально, внутривенно, подкожно и путем местного применения.
Фармацевтическую композицию согласно настоящему изобретению можно вводить в суточной дозе от 0,1 до 100 мг/кг массы тела пациента. Суточная доза может варьироваться в зависимости от показаний, условий или состояния пациента. Фармацевтическая композиция согласно настоящему изобретению может быть введена в соответствии с различными схемами введения, например, один, два и три раза в день, но не ограничивается этими схемами.
Настоящее изобретение будет описано подробно со ссылкой на следующие примеры, в том числе тестовые примеры. Тем не менее, эти примеры приводятся только для иллюстрации и не предназначены для ограничения объема настоящего изобретения.
Пример получения 1: получение 5-{4-(аминосульфонил)фенил}-2,2-диметил-4-(3-фторфенил)-3(2H)-фуранона и характеристика его кристаллической формы (кристаллическая форма А)
5-{4-(аминосульфонил)фенил}-2,2-диметил-4-(3-фторфенил)-3(2H)-фуранон получали в соответствии с методикой, описанной в примере 4 патента Кореи №10-0495389.
В частности, 4-бром-2,2-диметил-5-4-(аминосульфонил)фенил-3(2H)-фуранон (170 мг) растворяли в 30 мл толуола и 10 мл этанола. Раствор перемешивали. К полученному раствору по каплям добавляли 25 мг тетракис(трифенилфосфин) палладия (0), 10 мл насыщенного водного раствора бикарбоната натрия и 100 мг 3-фторбензолборной кислоты. После перемешивания при 90°С в течение 12 ч растворители удаляли из реакционного раствора при пониженном давлении и остаток экстрагировали водой и дихлорметаном. Органический слой концентрировали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии (гексан/этилацетат), получая 120 мг 5-{4-(аминосульфонил)фенил}-2,2-диметил-4-(3-фторфенил)-3(2H)-фуранона в виде твердого вещества.
(1) Рентгеноструктурный анализ (XRD)
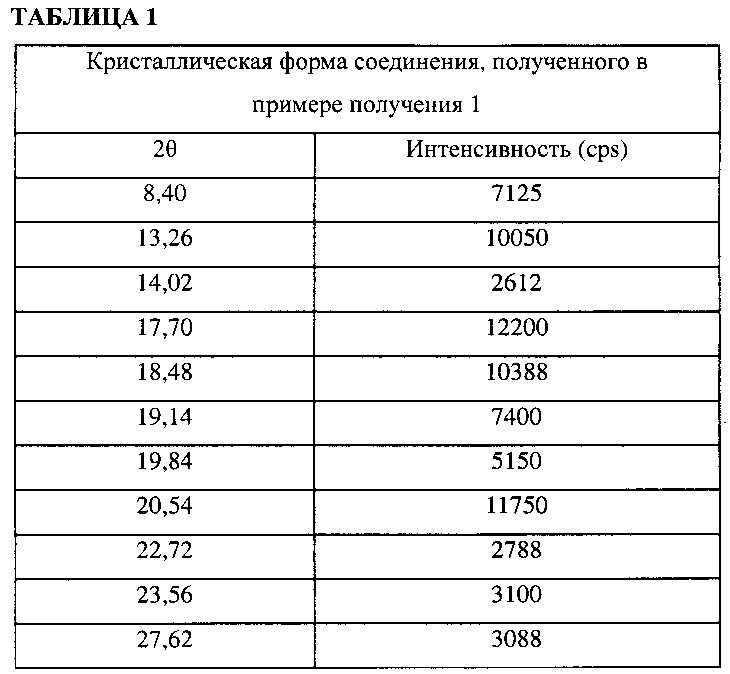
После того как соединение, полученное в примере получения 1, кристаллизовали общим методом кристаллизации, его кристаллическую форму охарактеризовывали с помощью рентгеноструктурного анализа (XRD). Рентгеноструктурный анализ проводили с использованием рентгеновского дифрактометра высокого разрешения Ultima III (Rigaku, Япония) с Cu излучением.
Экспериментальные результаты приведены в таблице 1.
(2) Дифференциальная сканирующая калориметрия (ДСК)
Кристаллическую форму соединения, полученного в примере получения 1, анализировали с помощью дифференциальной сканирующей калориметрии (ДСК). Анализ ДСК проводили с использованием DSC 823е (Mettler Toledo, Швейцария). Около 1-2,3 мг образца кристаллической формы помещали на алюминиевый поддон и нагревали со скоростью 10°С/мин от 25°С до 220°С. Данные анализировали с использованием STARe v9.20 (Proteus®).
Экспериментальные результаты показаны на рис. 1.
Кристаллическую форму соединения, полученного в примере получения 1, с результатами анализов XRD и ДСК, назвали «кристаллическая форма А».
Пример получения 2: получение 5-{4-(аминосульфонил)фенил}-2,2-диметил-4-(3-фторфенил)-3(2H)-фуранона и характеристика его кристаллической формы (кристаллическая форма G)
Кристаллическую форму соединения, полученного в примере получения 1, изменили с использованием прибора ДСК (Q2000, ТА Instruments, Великобритания или DSC 823е, Mettler Toledo, Швейцария). В частности, 5 мг образца кристаллической формы А помещали на алюминиевый поддон и подвергали циклу нагрев-поддержание температуры-охлаждение в приборе ТА для получения новой кристаллической формы. Цикл состоял из пяти этапов: нагрев со скоростью 10°С/мин от 25°С до 180°С (стадия 1); поддержание температуры 180°С в течение 5 мин (стадия 2); охлаждение со скоростью 10°С/мин от 180°С до 25°С (стадия 3); поддержание температуры 25°С в течение 1 мин (стадия 4); и нагревание со скоростью 10°С/мин от 25°С до 170°С (этап 5). На протяжении всего получения кристаллической формы соединения поддерживали продувку азотом со скоростью 50 мл/мин.
(1) Рентгеноструктурный анализ (XRD)
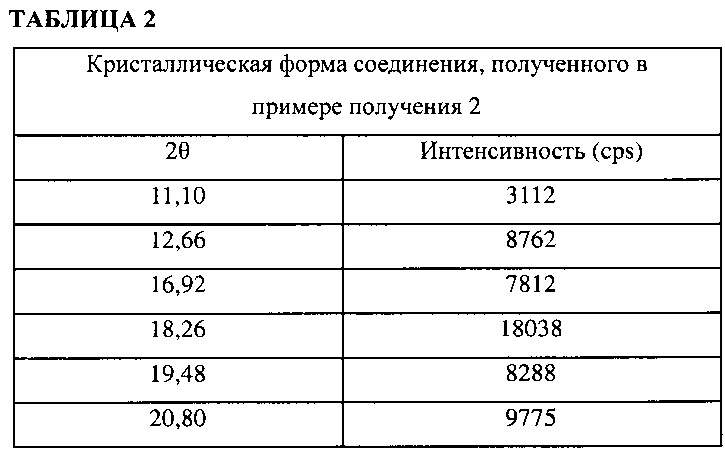
Кристаллическую форму соединения, полученного в примере получения 2, характеризовали с помощью рентгеноструктурного анализа (XRD). Рентгеноструктурный анализ проводили с использованием рентгеновского дифрактометра высокого разрешения Ultima III (Rigaku, Япония) с Cu излучением.
Экспериментальные результаты приведены в таблице 2.
(2) Дифференциальная сканирующая калориметрия (ДСК)
Кристаллическую форму соединения, полученного в примере получения 2, анализировали с помощью дифференциальной сканирующей калориметрии (ДСК). Анализ ДСК проводили с использованием DSC 823е (Mettler Toledo, Швейцария). Около 1-2,3 мг образца кристаллической формы помещали на алюминиевый поддон и нагревали со скоростью 10°С/мин от 25°С до 220°С. Данные анализировали с использованием STARe v9.20 (Proteus®).
Экспериментальные результаты показаны на фиг. 2.
Результаты анализов XRD и ДСК подтверждают, что кристаллическая форма соединения, полученного в примере получения 2, довольно сильно отличается от кристаллической формы А соединения, полученного в примере получения 1. Кристаллическую форму соединения, полученного в примере получения 2 с данными результатами анализов XRD и ДСК назвали «кристаллическая форма G».
Пример получения 3: получение и характеристика смеси кристаллических форм (кристаллическая форма А + кристаллическая форма G) 5-{4-(аминосульфонил)фенил}-2,2-диметил-4-(3-фторфенил)-3(2H)-фуранона
Кристаллические формы примеров получения 1 и 2 смешивали в массовом соотношении 50:50 с получением смеси. Смесь исследовали на предмет, сохранились ли характеристики кристаллических форм.
Смесь кристаллических форм А и G анализировали с помощью дифференциальной сканирующей калориметрии (ДСК). Анализ ДСК проводили с использованием DSC 200 F3 Maia® (NETZSCH). Около 1-5 мг образца смеси помещали на алюминиевый поддон и нагревали со скоростью 20°С/мин от 25 до 100°С и при скорости 10°С/мин от 100 до 250°С. Данные анализировали с помощью STARe v9.20 (Proteus®).
Экспериментальные результаты показаны на фиг. 3.
Как показано на фиг. 3, ДСК график смеси кристаллических форм, приготовленной в примере получения 3, показывает эндотермические пики, соответствующие кристаллическим формам примеров получения 1 и 2. Эти результаты показывают, что кристаллические формы А и G сохраняют свои характеристики даже при смешивании.
Тестовый пример 1: Анализ скорости растворения различных кристаллических форм
В этом примере изучали скорости растворения кристаллических форм соединения формулы 1. В частности, кристаллической формой А соединения из примера получения 1 и кристаллической формой G соединения из примера получения 2 наполняли твердые капсулы, а затем растворяли 900 мл раствора с рН 1,2 при различных скоростях перемешивания - 50 и 100 оборотов в минуту и температуре 37±0,5°С в течение 2 часов. Растворенные частицы анализировали в следующих условиях ВЭЖХ:
<Условия ВЭЖХ>
Колонка: Hypurity С18, 250×4,6 мм, 5 мкм или эквивалентная колонка
Детектор: УФ-абсорбционный спектрометр (измеряли при 325 нм)
Объем впрыска: 100 мкл
Скорость потока: 1,5 мл/мин
Температура колонки: 30°С
Подвижная фаза: А-ацетонитрил, В-вода, А : В=60:40, об/об%
Время анализа: 5 мин
Экспериментальные результаты, полученные при 50 оборотах в минуту и 100 оборотах в минуту, показаны на фиг. 4а и 4б соответственно.
Как видно на фиг. 4а и 4б, кристаллическая форма А показала более высокую скорость растворения, чем кристаллическая форма G при двух разных скоростях перемешивания. Эти результаты показывают, что кристаллические формы соединения формулы 1 показывают различные скорости растворения и большая доля кристаллической формы А был бы полезной для достижения желаемой скорости растворения. Более высокие скорости растворения композиций, содержащих большие доли кристаллической формы А, подтвердили в примерах получения 4-7.
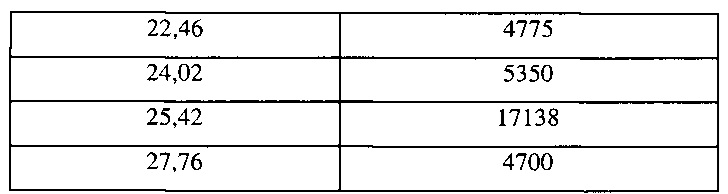
Примеры получения 4-7: получение смесей частиц, содержащих кристаллические формы в различных соотношениях
Кристаллическую форму А примера получения 1 и кристаллическую форму G примера получения 2 смешивали в соотношениях, показанных в таблице 3. Исследовали скорости растворения смесей.
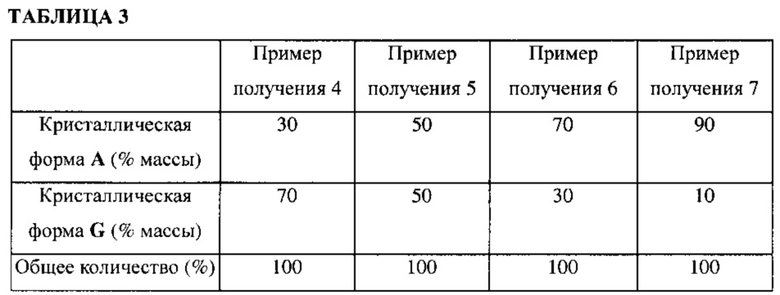
Тестовый пример 2: Анализ скорости растворения смесей, содержащих кристаллические формы в различных соотношениях
В этом примере исследовали скорости растворения частиц смесей кристаллических форм А и G в различных соотношениях. В частности, 2 мг каждой из смесей, приготовленных в примерах получения 4-7, засыпали в твердые капсулы и затем растворяли в 900 мл раствора с рН 1,2 при скорости перемешивания 100 оборотов в минуту и при температуре 37±0,5°С в течение 2 часов. Растворенные частицы анализировали при тех же условиях ВЭЖХ, как описано в тестовом примере 1. Результаты эксперимента представлены на фиг. 5.
Как видно на фиг. 5, скорость растворения возрастает с увеличением доли кристаллической формы А.
Тестовый пример 3: Анализ стабильности кристаллических форм
Кристаллические формы соединения формулы 1 оценивали на стабильность при хранении. Соединениями с кристаллической формой А примера получения 1 и кристаллической формой G примера получения 2 заполнили различные твердые капсулы и хранили в жестких условиях повышенной влажности (25°С/97% относительной влажности) и ускоренных условиях хранения (40°С/75% относительной влажности) в течение 7 дней. Рентгеноструктурный анализ проводили в соответствии со способом, описанным в примерах 1 и 2.
Результаты анализа представлены на фиг. 6а и 6б.
Как видно на фиг. 6а и 6б, состояния кристаллической формы А соединения формулы 1 поддерживались стабильными в жестких условиях повышенной влажности и ускоренных условиях хранения.
Тестовый пример 4: Анализ фармакокинетических свойств кристаллических форм
Фармакокинетические свойства различных кристаллических форм соединения формулы 1 проанализировали in vivo. Около 5 мг соединения с кристаллической формой А примера получения 1 и кристаллической формой G примера получения 2 суспендировали в 10 мл 0,5% раствора метилцеллюлозы для получения композиции для перорального применения. 6-недельных самцов крыс SD (Orient Bio. Inc., Корея) разделили на две группы. Около 3 мл (10 мл/кг) пероральной композиции однократно вводили перорально каждой крысе и отбирали образцы крови из крыс с предварительно определенными интервалами, равными 0,167, 0,5, 1,0, 2,0, 4,0, 6,0, 8,0, и 24,0 ч. Образцы крови использовали для анализа фармакокинетических параметров кристаллической формы.
Фармакокинетические параметры кристаллических форм анализировали с использованием Waters Quattro premier ХЕ 2795 Alliance НТ (Waters) при следующих условиях: скорость потока = 0,25 мл/мин, температура колонки = 40°С, вводимый объем = 7 мкл, и подвижная фаза = А: 1 мМ ацетата аммония и 0,1% уксусной кислоты (35%), B : ACN (65%). Линейность обеспечивали 8 различными концентрациями стандартных растворов.
Пероральные составы, в том числе соединения примера получения 1, и пероральные составы, в том числе соединения примера получения 2, вводили различным крысам. График уровня соединений в крови представлен на фиг. 7. Cmax (нг/мл), Tmax (ч), и AUC (ч * нг/мл) рассчитывали из графика, значения приведены в таблице 4.
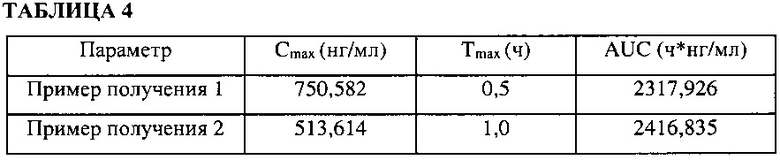
Как показано на фиг. 7, кристаллическая форма А примера получения 1 показала более высокую скорость растворения in vivo, чем кристаллическая форма G примера получения 2. Как можно видеть из результатов, приведенных в таблице 4, кристаллическая форма А примера получения 1 имела более высокие значения Cmax и Tmax, чем кристаллическая форма G примера получения 2, что свидетельствует о большей эффективности кристаллической формы А примера получения 1.
Пример 1 и сравнительные примеры 1 и 2: Получение кристаллической формы с частицами различного размера
Для того чтобы сравнить характеристики соединения формулы 1 в качестве лекарственного средства в зависимости от размера частиц, кристаллические формы переработали в частицы различного диаметра по следующим методикам.
<Сравнительный пример 1>
Кристаллическая форма, полученная в примере получения 1, называется «Сравнительный пример 1».
<Сравнительный пример 2>
Кристаллическую форму сравнительного примера 1 получили путем измельчения с использованием мельницы (струйная мельница, JE POWDER) при следующих условиях: шнековый питатель = 7 оборотов в минуту, мешалка = 7 оборотов в минуту, давление эжектора = 5,0 кг/см2, а рабочее давление = 3,5 кг/см2 Измельченную кристаллическую форму называли «Сравнительный пример 2».
<Пример 1>
Кристаллическую форму сравнительного примера 1 получили путем измельчения с использованием мельницы (струйная мельница, JE POWDER) при следующих условиях: шнековый питатель = 7 оборотов в минуту, мешалка = 7 оборотов в минуту, давление эжектора = 5,0 кг/см2, а рабочее давление = 3,5 кг/см2. Измельченную кристаллическую форму назвали «Пример 1».
Тестовый пример 5: Анализ размеров частиц и измерение скорости растворения кристаллической формы с частицами различного размера
<5-1> Анализ размеров частиц
Размеры частиц кристаллической формы сравнительных примеров 1 и 2 и в примере 1 анализировали с помощью лазерного анализатора размера частиц на основе дифракции (Mastersizer 2000®, Malvern). Каждый образец помещали в сухой модуль (Scirocco 2000®, Malvern) при давлении 2 бар, затем измеряли диаметр частиц 50% объема (d(0,5)) и диаметр частиц 90% объема (D(0,9)). Экспериментальные результаты приведены в таблице 5.
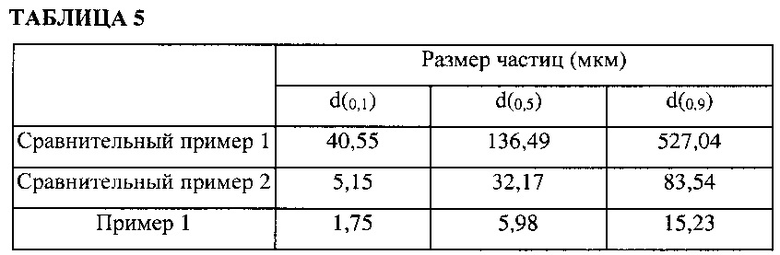
Как видно из результатов в таблице 5, частицы из сравнительного примера 1 имели диаметр частиц 50% объема (d(0,5)) 136,49 мкм и диаметр частиц 90% объема (d(0,9)) 527,04 мкм, частицы из сравнительного примера 2 имели диаметр частиц 50% объема (d(0,5)) 32,17 мкм и диаметр частиц 90% объема (d(0,9)) 83,54 мкм, а частицы из примера 1 имели диаметр частиц 50% объема (d(0,5)) 5,98 мкм и диаметр частиц 90% объема (d(0,9)) от 15,23 мкм. Исходя из этих результатов, подтверждено, что кристаллические формы сравнительных примеров 1 и 2 и примера 1 имели различные распределения частиц по размерам.
<5-2> Анализ скоростей растворения кристаллической формы с различными размерами частиц
В этом примере исследовали скорости растворения кристаллической формы с частицами различного размера. Частицами сравнительных примеров 1 и 2 и примера 1 заполняли различные твердые желатиновые капсулы (2 мг на капсулу), а затем растворяли в 900 мл раствора с рН 1,2 и 900 мл раствора с рН 6,8 при скорости перемешивания 100 оборотов в минуту и температуре 37±0,5°С в течение 3 часов. Растворенные частицы анализировали в тех же условиях ВЭЖХ, как описано в тестовом примере 1.
Результаты показаны на фиг. 8а и 8б.
Как видно из фиг. 8а и 8б, частицы из примера 1, имеющие диаметр частиц 50% объема (d(0,5)) 3-9 мкм и диаметр частиц 90% объема (d(0,9)) 10-50 мкм показали более высокие скорости растворения, чем частицы сравнительных примеров 1 и 2, чей диаметр частиц 50% объема и диаметр частиц 90% объема был вне диапазонов распределения частиц по размерам примера 1. Эти результаты показывают, что более высокая скорость растворения может быть достигнута, когда диаметр частиц 50% объема (d(0,5)) и диаметр частиц 90% объема (d(0,9)) кристаллической формы соединения формулы 1 соответствует диапазонам 3-9 мкм и 10-50 мкм соответственно.
Тестовый пример 6: Анализ устойчивости кристаллической формы соединения формулы 1
<6-1> Термостабильность
Кристаллической формой А примера 1 наполняли твердые капсулы (2 мг на капсулу), упакованные с РТР, и хранили в течение 72 ч в жестких температурных условиях, показанных в таблице 7. Во время хранения наблюдали появление кристаллической формы А, время удерживания основного пика, количества (в %) родственных веществ и содержание действующего вещества. Время удерживания основного пика, количество родственных веществ и содержание действующего вещества анализировали с помощью высокоэффективной жидкостной хроматографии в следующих условиях. Результаты приведены в таблице 6.
<ВЭЖХ условия для анализа родственных веществ>
Колонка: Hypurity С18, 250×4,6 мм, 5 м или эквивалентная колонка
Детектор: УФ-абсорбционный спектрометр (измерение при 241 нм)
Объем впрыска: 20 мкл
Скорость потока: 1,0 мл / мин
Температура колонки: 30°С
Подвижная фаза: А - ацетонитрил, В - 0,1% об/об трифторуксусной кислоты (ТФУ) в воде
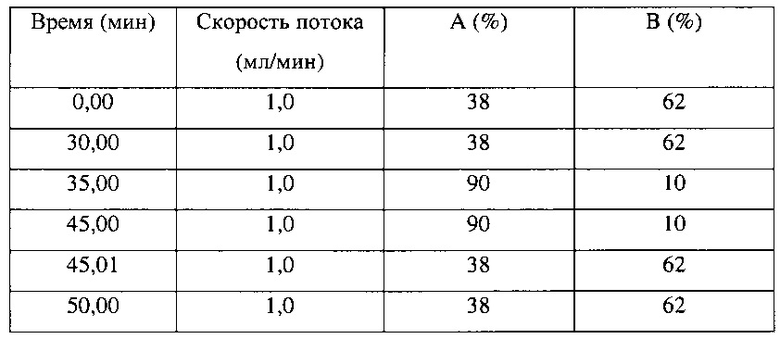
<ВЭЖХ условия для анализа содержания соединений>
Колонка: Hypurity С18, 250×4,6 мм, 5 м или эквивалентная колонка
Детектор: УФ-абсорбционный спектрометр (измерение при 325 нм)
Объем впрыска: 20 мкл
Скорость потока: 1,5 мл/мин Температура колонки: 30°С
Подвижная фаза: А - ацетонитрил, В - вода, А : В=60:40, об/об%
Время анализа: 5 мин
Разбавитель: вода : ацетонитрил =50:50, об/об%
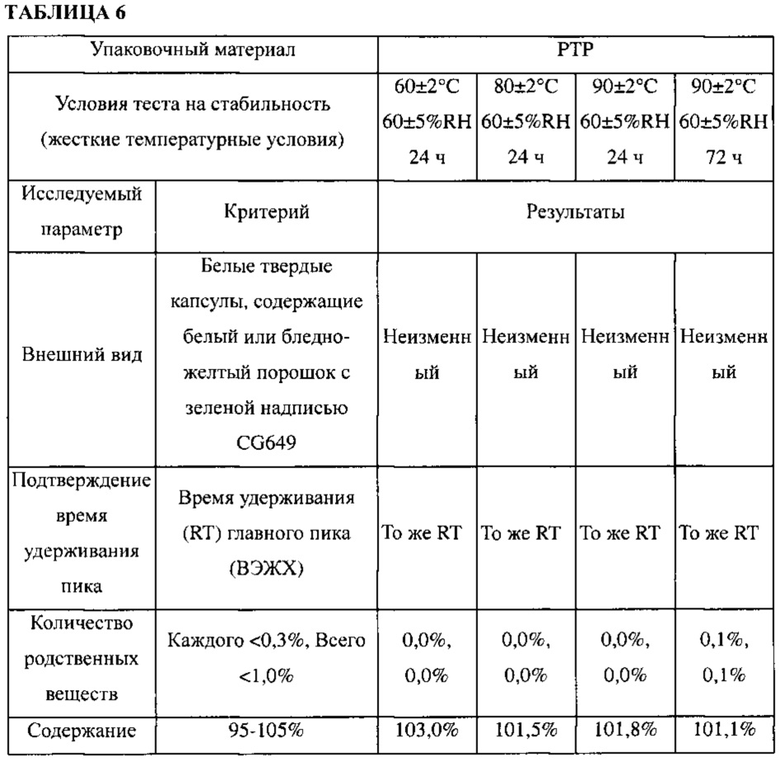
Как видно из результатов, приведенных в таблице 6, внешний вид кристаллической формы А остался неизменным, и при жестких температурных условиях не наблюдали значительного уменьшения содержания кристаллической формы А или значительного увеличения количества родственных веществ. Эти результаты демонстрируют высокую стабильность кристаллической формы А при температурных условиях.
<6-2> Стабильность при различной влажности
Кристаллическую форму А соединения примера 1 оценивали на стабильность при различной влажности таким же образом, как и в тестовом примере <6-1>. Кристаллическую форму А помещали в твердые капсулы (2 мг на капсулу) и хранили в жестких условиях повышенной влажности, показанных в таблице 7. Затем анализировали внешний вид кристаллической формы А, время удерживания основного пика, количество (%) родственных веществ и содержание соединения. Результаты приведены в таблице 7.
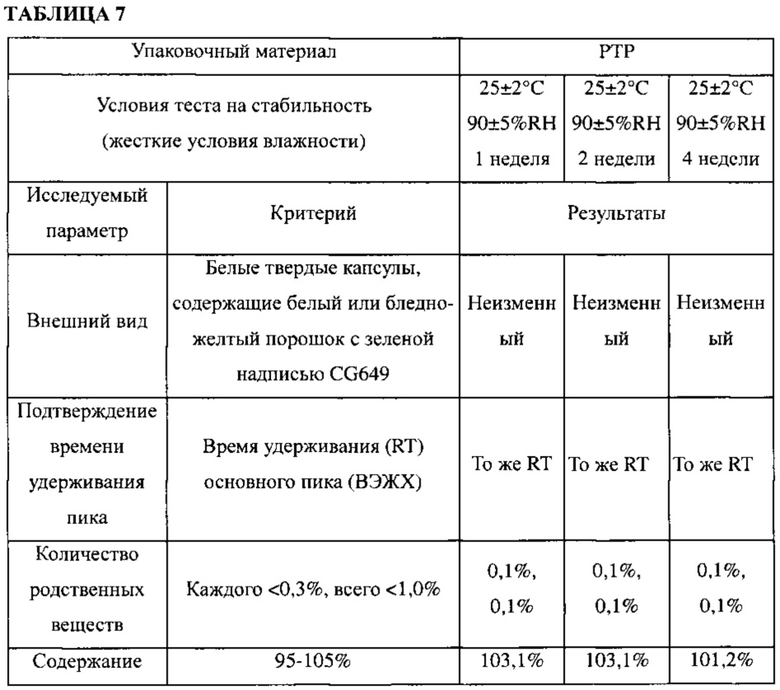
Как видно из результатов, приведенных в таблице 7, внешний вид кристаллической формы А остался неизменным, и не происходит значительного уменьшения содержания кристаллической формы А или значительного увеличения количества родственных веществ в жестких условиях повышенной влажности. Эти результаты демонстрируют высокую стабильность кристаллической формы А в условиях высокой влажности.
<6-3> Стабильность на свету
Кристаллическую форму А примера 1 оценивали на устойчивость на свету таким же образом, как и в тестовом примере <6-1>. Кристаллическую форму А помещали в твердые капсулы (2 мг на капсулу) и хранили в условиях светового стресса, показанных в таблице 8. После этого анализировали внешний вид кристаллической формы А, время удерживания основного пика, количество (%) родственных веществ и содержание соединения. Результаты представлены в таблице 8 и фиг. 9а-9в.
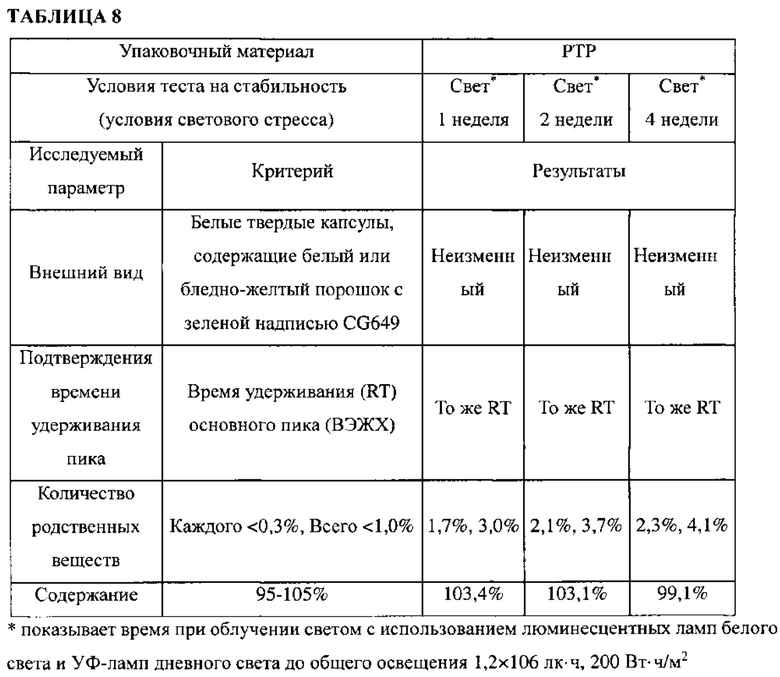
Как видно из таблицы 8 и фиг. 9а-9в, кристаллическая форма А образовывала родственные вещества, превышающие критерии, в течение 7 дней при хранении в условиях светового стресса. Эти результаты приводят к выводу, что исходные материалы должны храниться и составы должны храниться и производиться в темное время суток или в условиях, защищенных от воздействия сильного света. Легкие условия экранирования необходимы в реальных процессах.
Примеры 2-11: Получение состава в капсулах, содержащего кристаллическую форму А
Для того чтобы найти оптимальные фармацевтические добавки, пригодные для кристаллической формы А, для получения составов в капсулах использовали разбавители и смазывающие вещества, приведенные в таблицах 9 и 10.
Индекс Карра каждого состава в капсулах измеряли с помощью метода Карра, используя тестер насыпной плотности (ERWEKA, SVM 101) и угол естественного откоса каждого состава в капсулах определяли с помощью метода фиксированной воронки, например, капельным методом.
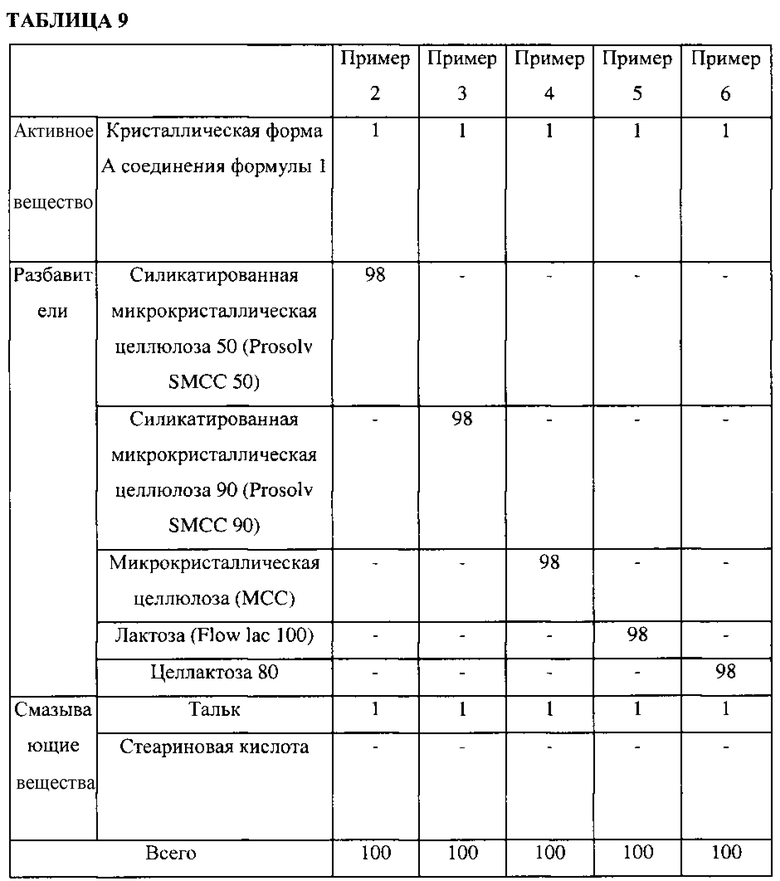

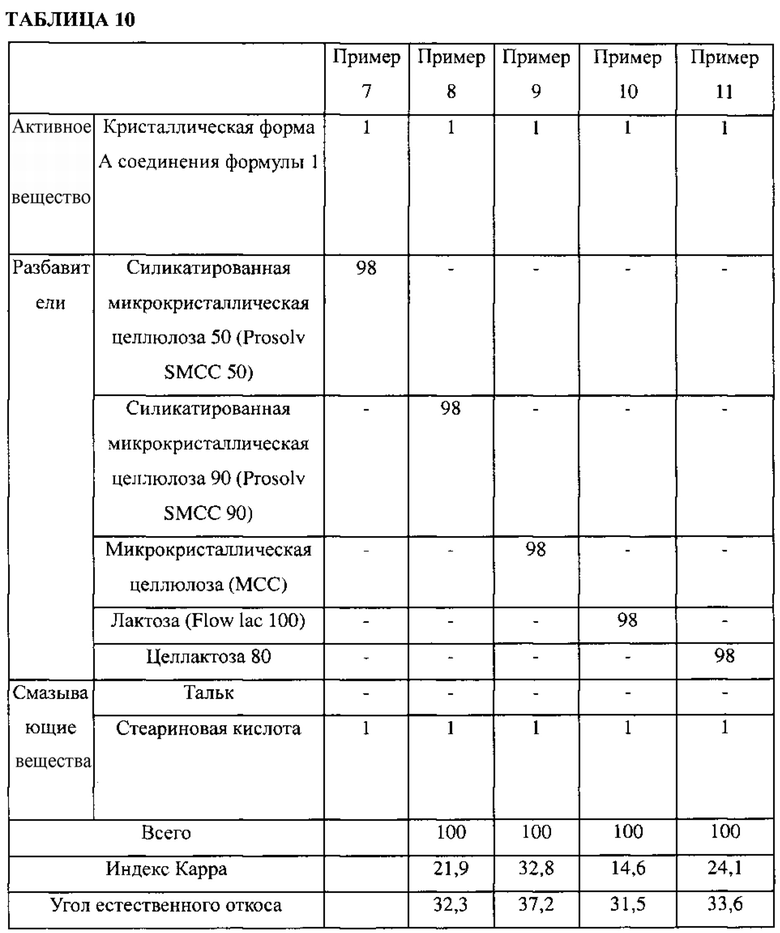
Как видно из результатов в таблицах 9 и 10, составы в капсулах, содержащие силикатированную микрокристаллическую целлюлозу 50, силикатированную микрокристаллическую целлюлозу 90, микрокристаллическую целлюлозу, лактозу или целлактозу 80 в качестве разбавителя и тальк или стеариновую кислоту в качестве смазывающего вещества (примеры 2 и 11) имели углы естественного откоса в диапазоне от 30 до 40°, а индекс Карра в диапазоне от 21 до 30%. В пределах этих диапазонов обеспечивается хорошая текучесть порошков, таким образом, композиции пригодны для заполнения капсул. Тем не менее, составы в капсулах примеров от 7 до 11, изготовленные с использованием стеариновой кислоты в качестве смазывающего вещества, имели значительно более высокое содержание воды, несмотря на те же экспериментальные условия, что и в примерах 2-6. Таким образом, можно сделать вывод о том, что составы в капсулах примеров от 7 до 11 трудно производить в очень влажной среде или сезоне года, и, следовательно, использование талька в качестве смазывающего вещества было бы более желательным.
Тестовый пример 7: Измерение распределений частиц по размерам
Распределение размера частиц в составах, полученных в примерах 2-6, измерили с использованием 40-, 60-, 70-, 80-, 120-, 140-, 200-, и 270-меш стандартных сит в соответствии с методом ситовой классификации (метод II), описанным в стандартных методах исследования размеров частиц корейской фармакопеи. Результаты показаны на фиг. 10.
Как показано на фиг. 10, распределение частиц по размеру сильно различалось в зависимости от вида используемого разбавителя и смазывающего вещества.
В частности, состав примера 2, полученный с использованием силикатированной микрокристаллической целлюлозы 50 в качестве разбавителя, показал равномерное распределение размера частиц в диапазоне диаметров частиц менее 125 мкм, что указывает на высокую однородность смешивания. Силикатированная микрокристаллическая целлюлоза 50 будет более подходящей для использования в композиции согласно настоящему изобретению вследствие ее высокой текучести, улучшенных смазывающих эффектов и легкости смешивания по сравнению с другими разбавителями.
Тестовый пример 8: Анализ однородности состава
Составы в капсулах, полученные в примерах 2-6, исследовали на однородность в соответствии со способом исследования однородности, описанным в стандартных способах испытаний однородности рецептуры в корейской фармакопее. Из каждого состава в капсулах взяли по шесть образцов. Измеряли содержание основного вещества в образцах для определения среднего содержания, стандартного отклонения и оценочного значения (AV). Экспериментальные результаты приведены в таблице 11.
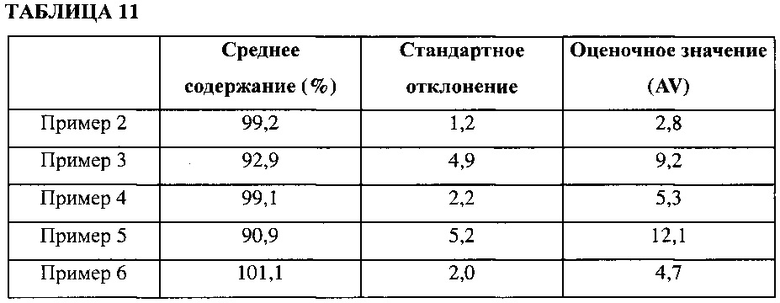
Как видно из результатов, приведенных в таблице 11, обнаружили композиции, имеющие хорошую однородность. В частности, состав примера 2 имел наименьшее оценочное значение (AV), что указывает на лучшую однородность.
Настоящее изобретение относится к противовоспалительной фармацевтической композиции, содержащей 5-{4-(аминосульфонил)фенил}-2,2-диметил-4-(3-фторфенил)-3(2Н)-фуранон в кристаллической форме А, кристаллической форме G или в виде смеси указанных кристаллических форм, фармацевтически приемлемый разбавитель и фармацевтически приемлемое смазывающее вещество. Указанное активное вещество имеет диаметр частиц 50% объема (d(0,5)) от 3 до 9 мкм. Разбавитель выбран из группы, состоящей из силикатированной микрокристаллической целлюлозы, микрокристаллической целлюлозы, целлюлозы, лактозы, а также их комбинации, и смазывающее вещество представляет собой тальк или стеариновую кислоту. Фармацевтическая композиция согласно настоящему изобретению имеет преимущества вследствие хорошей стабильности, высокой скорости растворения, улучшенной однородности, а также удовлетворительных фармакокинетических свойств. Благодаря этим преимуществам фармацевтическая композиция согласно настоящему изобретению является эффективной для лечения воспаления или боли. 2 н. и 6 з.п. ф-лы, 10 ил., 11 табл., 11 пр.
1. Противовоспалительная фармацевтическая композиция, содержащая (I) соединение формулы 1
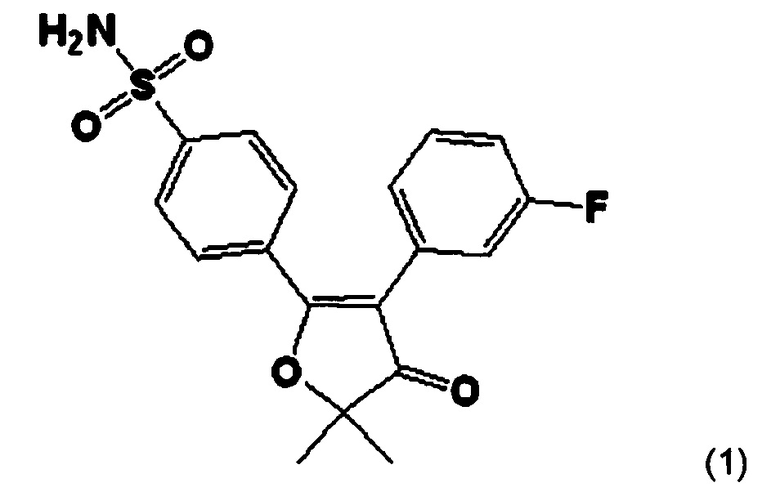
или его фармацевтически приемлемую соль, имеющее диаметр частиц 50% объема (d(0,5)) от 3 до 9 мкм, (II) фармацевтически приемлемый разбавитель и (III) фармацевтически приемлемое смазывающее вещество,
где соединение формулы 1 находится в кристаллической форме А, кристаллической форме G или в виде смеси указанных кристаллических форм,
при этом кристаллическая форма А характеризуется результатами рентгеноструктурного анализа, полученными с помощью Cu-излучения, представленными в таблице 1, и результатами дифференциальной сканирующей калориметрии (ДСК), полученными при скорости нагревания 10°С/мин от 25 до 220°С, где полученный профиль ДСК имеет пик в интервале от 175,62 до 178,35°С
при этом кристаллическая форма G характеризуется результатами рентгеноструктурного анализа, полученными с помощью Cu-излучения, представленными в таблице 2, и результатами дифференциальной сканирующей калориметрии (ДСК), полученными при скорости нагревания 10°С/мин от 25 до 220°С, где полученный профиль ДСК имеет пик в интервале от 181,06 до 186,16°С
при этом разбавитель выбран из группы, состоящей из силикатированной микрокристаллической целлюлозы, микрокристаллической целлюлозы, целлюлозы, лактозы, а также их комбинации, и смазывающее вещество представляет собой тальк или стеариновую кислоту.
2. Противовоспалительная фармацевтическая композиция по п. 1, отличающаяся тем, что соединение формулы 1 имеет диаметр частиц 90% объема (d(0,9)) от 10 до 100 мкм.
3. Противовоспалительная фармацевтическая композиция по любому из пп. 1, 2, отличающаяся тем, что соединение формулы 1 существует в виде смеси кристаллической формы А и кристаллической формы G.
4. Противовоспалительная фармацевтическая композиция по любому из пп. 1, 2, отличающаяся тем, что соединение формулы 1 содержит по меньшей мере 50% по массе кристаллической формы А.
5. Противовоспалительная фармацевтическая композиция по любому из пп. 1, 2, отличающаяся тем, что разбавитель представляет собой силикатированную микрокристаллическую целлюлозу.
6. Противовоспалительная фармацевтическая композиция по любому из пп. 1, 2, отличающаяся тем, что смазывающее вещество представляет собой тальк.
7. Противовоспалительная фармацевтическая композиция по любому из пп. 1, 2, отличающаяся тем, что фармацевтическая композиция содержит 1% по массе соединения формулы 1, 98% по массе разбавителя и 1% по массе смазывающего вещества.
8. Противовоспалительный фармацевтический состав в капсулах, содержащий фармацевтическую композицию по любому из пп. 1-7.
| WO 00/61571 A1, 19.10.2000 | |||
| EA 004432 B1, 29.04.2004 | |||
| KR 20130078147 A, 10.07.2013 | |||
| Огнетушитель | 1925 |
|
SU2205A1 |
| KR 20100096512 A, 02.09.2010 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| US 5536752 A, 16.07.1996 | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |

