ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННУЮ ЗАЯВКУ
[0001] По настоящей заявке РСТ испрашивается приоритет предварительной заявки США номер 61/903840, поданной 13 ноября 2013 года. Этот документ включен в настоящее описание посредством ссылки в полном объеме.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0002] Настоящее изобретение относится к фармацевтическим композициям и способам лечения или профилактики инфекции, вызванной гриппом, у пациентов.
УРОВЕНЬ ТЕХНИКИ
[0003] Грипп в основном передается от человека к человеку посредством крупных капель, содержащих вирус, которые образуются при кашле или чихании инфицированного человека; эти крупные капли могут затем оседать на поверхность слизистой оболочки верхних дыхательных путей чувствительных индивидуумов, которые находятся вблизи (например, в пределах 6 футов) от инфицированных людей. Передача также может произойти в результате прямого или непрямого контакта с выделениями из дыхательных путей, например, дотрагиваясь до поверхностей, зараженных вирусом гриппа, а затем касаясь глаз, носа или рта. Взрослые индивидуумы могут передавать грипп другим за 1 день до проявления симптомов и приблизительно в течение 5 дней после начала симптомов. Маленькие дети и индивиды с ослабленной иммунной системой могут быть заразными в течение 10 или более дней после появления симптомов.
[0004] Вирусы гриппа представляют собой РНК-вирусы семейства Orthomyxoviridae, который состоит из пяти родов: Вирус гриппа A, вирус гриппа B, вирус гриппа C, вирус ISA и вирус Тогото.
[0005] Род вируса гриппа А имеет один вид, вирус гриппа А. Дикие водоплавающие птицы являются природными хозяевами большого разнообразия гриппа А. Иногда вирусы передаются другим видам, что затем может вызывать катастрофические вспышки среди домашней птицы или приводить к пандемии гриппа у человека. Вирусы типа А являются наиболее вирулентными патогенами для человека среди трех типов гриппа и вызывают наиболее тяжелое заболевание. Вирус гриппа А можно подразделить на различные серотипы на основании гуморального (антительного) ответа на эти вирусы. Серотипами, которые были подтверждены у человека, расположены по количеству известных смертей человека при пандемии, являются: H1N1 (который вызвал Испанский грипп в 1918 году), H2N2 (который вызвал Азиатский грипп в 1957 году), H3N2 (который вызвал Гонконгский грипп в 1968 году), вирус H5N1 (угроза пандемии в сезон гриппа 2007-08), H7N7 (который имеет необычайную зоонозную силу), H1N2 (эндемический грипп у людей и свиней), H9N2, H7N2, H7N3 и H10N7.
[0006] Род вируса гриппа В имеет один вид, вирус гриппа В. Грипп B инфицирует практически только людей и менее распространен, чем грипп А. Как известно, единственным животным, восприимчивым к инфекции гриппом типа В, является тюлень. Скорость мутирования этого гриппа в 2-3 раза медленнее, чем типа А и, следовательно, он менее генетически разнообразен и имеет только один серотип гриппа B. В результате такого недостаточного антигенного разнообразия некоторый уровень иммунитета к гриппу B обычно приобретается в раннем возрасте. Тем не менее, грипп B мутирует в достаточной степени и стойкий иммунитет к нему невозможен. Такой пониженный уровень антигенного изменения, в сочетании с ограниченным диапазоном хозяев (ингибирующий межвидовой антигенный сдвиг), гарантирует, отсутствие пандемии гриппа В.
[0007] Вирус гриппа рода С имеет один вид, вирус гриппа С, который инфицирует людей и свиней и может привести к тяжелому заболеванию и локальным эпидемиям. При этом, грипп С встречается реже, чем другие типы и, как правило, по-видимому вызывает легкое заболевание у детей.
[0008] Вирусы гриппа A, B и C очень похожи по своей структуре. Диаметр частица вируса составляет 80-120 нм и, как правило, он имеет почти сферическую форму, хотя могут возникать нитчатые формы. Геном представляет собой не один фрагмент нуклеиновой кислоты, что необычно для вируса; вместо этого, он содержит семь или восемь частей сегментированной отрицательной смысловой РНК. Геном гриппа A кодирует 11 белков: гемагглютинин (HA), нейраминидазу (NA), нуклеопротеины (NP), M1, M2, NS1, NS2 (NEP), PA, PB1, PB1-F2 и PB2.
[0009] HA и NA являются крупными гликопротеинами снаружи вирусных частиц. НА представляет собой лектин, который опосредует связывание вируса с клетками-мишенями и проникновение вирусного генома в клетку-мишень, а NA участвует в высвобождении потомства вируса из инфицированных клеток путем расщепления сахаров, которые связывают зрелые вирусные частицы. Таким образом, эти белки были мишенями антивирусных препаратов. Кроме того, они являются антигенами, в отношении которых продуцируются антитела. Вирусы гриппа А классифицируют на подтипы, на основе гуморальных (антительных) ответов на HA и NA, на этом основано различие Н и N (см. выше), например, H5N1.
[0010] Грипп приводит к непосредственным расходам в результате потери трудоспособности и связанного с ним медицинского лечения, а также к косвенным расходам на профилактические меры. В Соединенных Штатах на борьбу с гриппом затрачивается в целом свыше $10 миллиардов в год, но по проведенным расчетом пандемия в будущем может привести к сотням миллиардов долларов на непосредственные и косвенные расходы. Профилактические расходы также велики. Правительствами по всему миру было потрачено миллиарды долларов США на подготовку и планирование борьбы с возможной пандемией птичьего гриппа H5N1, принимая во внимание расходы, связанными с приобретением лекарств и вакцин, а также на разработку учений по борьбе с бедствием и стратегий для улучшения пограничного контроля.
[0011] Современные варианты лечения гриппа включают вакцинацию и химиотерапию или химиопрофилактику противовирусными препаратами. Вакцинация против гриппа вакциной против гриппа часто рекомендуется для групп c высоким риском, таких как дети и пожилые люди, или люди, которые страдают астмой, диабетом или сердечным заболеванием. Тем не менее, можно сделать прививку и при этом заболеть гриппом. Состав вакцины меняется каждый сезон для нескольких конкретных штаммов вируса гриппа, но не может включать в себя все штаммы, активно инфицирующие людей в мире в течение этого сезона. Для получения состава и производства миллионов доз, необходимых для борьбы с сезонными эпидемиями, производителям требуется шесть месяцев; иногда, в течение этого времени появляется новый штамм или активизируется штамм, упущенный из вида, и инфицирует людей, хотя они были вакцинированы (как при гриппе Фуцзяни H3N2 в сезоне 2003-2004). Кроме того, можно заразиться непосредственно перед вакцинацией и заболеть тем самым штаммом, для защиты от которого предполагалась вакцина, так как до того, как вакцина становится эффективной, может пройти несколько недель.
[0012] Кроме того, эффективность вакцин против гриппа является переменной величиной. Из-за высокой скорости мутации вируса конкретная вакцина против гриппа, как правило, обеспечивает защиту не более чем на несколько лет. Состав вакцина на один год может оказаться неэффективным в следующем году, так как вирус гриппа быстро меняется с течением времени, и другие штаммы становятся доминирующими.
[0013] Кроме того, из-за отсутствия ферментов, исправляющих РНК, РНК-зависимая РНК-полимераза вРНК гриппа делает ошибку, вставку одного нуклеотида, примерно каждые 10 тысяч нуклеотидов, что соответствует примерной длине вРНК гриппа. Таким образом, почти каждый вновь полученный вирус гриппа обладает мутант-антигенным дрейфом. Разделение генома на восемь отдельных сегментов вРНК позволяет смешивать или пересортировывать вРНК, если клетка инфицирована более чем одной вирусной линией. Происходящее в результате быстрое изменение в генетике вирусов обеспечивает антигенные сдвиги и позволяет вирусу инфицировать новые виды хозяев и быстро преодолевать защитный иммунитет.
[0014] Противовирусные препараты также могут быть использованы для лечения гриппа, особенно эффективны ингибиторы нейраминидазы, но вирусы могут вырабатывать устойчивость к стандартным противовирусным препаратам.
[0015] Таким образом, по-прежнему существует потребность в препаратах для лечения инфекций, вызванных гриппом, таких как препараты с расширенным окном лечения и/или пониженной чувствительностью к вирусному титру.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0016] Настоящее изобретение в целом относится к фармацевтическим композициям, которые содержат соль HCl Соединения (1)•хH2O (где х равно от 0 до 3), к способам получения таких фармацевтических композиций, к способам лечения гриппа с использованием таких фармацевтических композиций, к способам снижения количества вируса гриппа с использованием таких фармацевтических композиций, а также к способам ингибирования репликации вирусов гриппа с использованием таких фармацевтических композиций. Соединение (1) представлено следующей структурной формулой
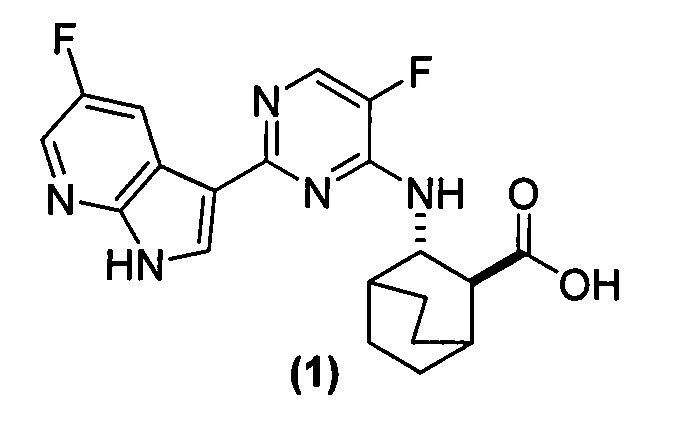 .
.
[0017] Один из вариантов осуществления настоящего изобретения относится к фармацевтической композиции, содержащей а) соль HCl Соединения (1)•хH2O, где Соединение (1) представлено структурной формулой выше, где х равно от 0 до 3; и b) один или несколько эксципиентов, содержащих наполнитель, дезинтегрирующий агент, смачивающий агент, связующее вещество, скользящее вещество, смазывающее вещество или любую их комбинацию, где соль HCl Соединения (1)•хH2O имеет концентрацию от 5 масс.% до 95 масс.% по массе композиции, и один или более наполнителей имеет концентрацию от 5 масс.% до 95 масс.% по массе композиции.
[0018] В некоторых вариантах осуществления фармацевтическая композиция по существу не содержит скользящее вещество или смачивающий агент.
[0019] В некоторых вариантах осуществления х равен от 0.5 до 3. Например, х равно 0,5.
[0020] В некоторых вариантах осуществления соль HCl Соединения (1)•хH2O имеет кристаллическую форму.
[0021] В некоторых вариантах осуществления фармацевтическая композиция дополнительно содержит от 10 масс.% до 80 масс.% наполнителя по массе фармацевтической композиции. В других вариантах осуществления наполнитель содержит микрокристаллическую целлюлозу, лактозу или любую их комбинацию.
[0022] В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция дополнительно содержит от 1 масс.% до 10 масс.% дезинтегрирующего агента по массе фармацевтической композиции. В других вариантах осуществления дезинтегрирующий агент включает кроскармеллозу, кросповидон, полипласдон, крахмал, металл крахмал гликолят или любую их комбинацию. И, в некоторых вариантах осуществления, дезинтегрирующий агент включает кроскармеллозу натрия, полиплаcдон или любую их комбинацию.
[0023] В некоторых вариантах осуществления фармацевтическая композиция содержит от 0,1 масс.% до 5 масс.% по массе связующего вещества по массе фармацевтической композиции. В других вариантах осуществления связующее вещество содержит поливинилпирролидон, крахмал, сахар, микрокристаллическую целлюлозу, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу или любую их комбинацию.
[0024] В некоторых вариантах осуществления фармацевтическая композиция содержит от 0,5 масс.% до 5 масс.% смазочного вещества по массе фармацевтической композиции. В других вариантах осуществления смазывающее вещество содержит металл стеарат, металл стеарилфумарат или любую их комбинацию. Например, смазывающее вещество содержит натрия стеарилфумарат, магния стеарат или любую их комбинацию. И в некоторых примерах, смазывающее вещество содержит натрия стеарилфумарат.
[0025] В некоторых вариантах осуществления фармацевтическая композиция содержит а) от 20 масс.% до 80 масс.% Формы А соли HCl Соединения (1)•1/2H2О по массе фармацевтической композиции; b) от 1 масс.% до 10 масс.% дезинтегрирующего агента по массе фармацевтической композиции; и с) от 20 масс.% до 80 масс.% наполнителя по массе фармацевтической композиции.
[0026] В некоторых вариантах осуществления фармацевтическая композиция содержит а) от 20 масс.% до 80 масс.% Формы А соли HCl Соединения (1)•1/2H2О по массе фармацевтической композиции; b) от 1 масс.% до 10 масс.% дезинтегрирующего агента по массе фармацевтической композиции; с) от 0,1 масс.% до 5 масс.% связующего вещества по массе фармацевтической композиции; и d) от 20 масс.% до 80 масс.% наполнителя по массе фармацевтической композиции.
[0027] В некоторых вариантах осуществления фармацевтическая композиция содержит а) от 20 масс.% до 80 масс.% Формы А соли HCl Соединения (1)•1/2H2О по массе фармацевтической композиции; b) от 1 масс.% до 10 масс.% дезинтегрирующего агента по массе фармацевтической композиции; c) от 0,1 масс.% до 5 масс.% связующего вещества по массе фармацевтической композиции; d) от 20 масс.% до 80 масс.% наполнителя по массе фармацевтической композиции; и е) от 0,5 масс.% до 5 масс.% смазочного вещества по массе композиции.
[0028] В некоторых вариантах осуществления фармацевтическая композиция содержит a) от 35 масс.% до 75 масс.% Формы А соли HCl Соединения (1)•1/2H2O по массе фармацевтической композиции; b) от 1 масс.% до 7 масс.% дезинтегрирующего агента по массе фармацевтической композиции, где дезинтегрирующий агент выбран из кроскармеллозы, кросповидона, полиплаcдона, металл крахмалгликолята, крахмала или любой их композиции; c) от 0,5 масс.% до 2 масс.% связующего вещества по массе фармацевтической композиции, где связующее вещество выбрано из поливинилпирролидона, крахмала, сахара, микрокристаллической целлюлозы, гидроксипропилметилцеллюлозы, гидроксипропилцеллюлозы или гидроксиэтилцеллюлозы или любой их композиции; d) от 25 масс.% до 50 масс.% наполнителя по массе фармацевтической композиции; где наполнитель выбран из микрокристаллической целлюлозы, лактозы, сорбита, целлюлозы, фосфата кальция, крахмала или сахара или любой их композиции; и e) 0,5 масс.% до 3 масс.% смазывающего вещества по массе композиции, где смазывающее вещество выбрано из металл стеарата, метал стеарилфумарат или любой их композиции.
[0029] В некоторых вариантах осуществления фармацевтическая композиция содержит a) от 35 масс.% до 75 масс.% Формы А соли HCl Соединения (1)•1/2H2O по массе фармацевтической композиции; b) от 3 масс.% до 7 масс.% дезинтегрирующего агента по массе фармацевтической композиции, где дезинтегрирующий агент содержит кроскармеллозу; c) от 0,5 масс.% до 2 масс.% связующего вещества по массе фармацевтической композиции, где связующее вещество содержит поливинилпирролидон; d) от 25 масс.% до 50 масс.% наполнителя по массе фармацевтической композиции; где наполнитель в данном случае содержит микрокристаллическую целлюлозу и лактозу; и e) от 0,5 масс.% до 3 масс.% смазывающего вещества по массе композиции, где смазывающее вещество содержит метал стеарилфумарат.
[0030] В некоторых вариантах осуществления фармацевтическая композиция содержит: a) от 35 масс.% до 75 масс.% Формы А соли HCl Соединения (1)•1/2H2O по массе фармацевтической композиции; b) от 3 масс.% до 7 масс.% кроскармеллозы по массе фармацевтической композиции; c) от 0,5 масс.% до 2 масс.% поливинилпирролидона по массе фармацевтической композиции; d) от 25 масс.% до 50 масс.% наполнителя по массе фармацевтической композиции; где наполнитель содержит микрокристаллическую целлюлозу и лактозу; и e) от 0,5 масс.% до 3 масс.% натрия стеарилфумарата по массе композиции.
[0031] В некоторых вариантах осуществления фармацевтическая композиция содержит a) от 35 масс.% до 65 масс.% Формы А соли HCl Соединения (1)•1/2H2O по массе фармацевтической композиции; b) от 3 масс.% до 7 масс.% кроскармеллозы натрия по массе фармацевтической композиции; c) 0,5 масс.% до 2 масс.% поливинилпирролидона со средней молекулярной массой от 3000 до 5000 по массе фармацевтической композиции; d) от 30 масс.% до 40 масс.% микрокристаллической целлюлозы по массе фармацевтической композиции; e) от 5 масс.% до 10 масс.% лактозы моногидрата по массе фармацевтической композиции; и f) по 1 масс.% до 3 масс.% натрия стеарилфумарата по массе композиции.
[0032] Еще один вариант осуществления относится к фармацевтической композиции, содержащей: а) от 1 мг/мл до 20 мг/мл Соединения (1) в воде, где Соединение (1) представлено структурной формулой выше; и от 0,01 М до 0,1 М фармацевтически приемлемого модификатора рН.
[0033] В некоторых вариантах осуществления источником Соединения (1) является соль HCl Соединения (1)•хН2O, где х равно от 0 до 3. В некоторых вариантах осуществления х равно 0,5. И, в некоторых вариантах осуществления, соль HCl Соединения (1)•xH2O образует Форму А соли HCl Соединения (1)•1/2H2О.
[0034] В некоторых вариантах осуществления модификатор рН включает NaOH, KOH, NH4OH, HCl, карбонат, бикарбонат, одноосновной фосфат, двухосновный фосфат, ацетат или любую их комбинацию.
[0035] В некоторых вариантах осуществления модификатор рН содержит агент фосфатного буфера. И в некоторых вариантах осуществления агент фосфатного буфера включает мононатрия фосфат, динатрия фосфат или любую их комбинацию.
[0036] В некоторых вариантах осуществления фармацевтическая композиция содержит от 1 масс.% до 20 масс.% комплексообразующего агента по массе фармацевтической композиции. В некоторых вариантах осуществления комплексообразующий агент содержит циклодекстрин, полисорбат, касторовое масло или любую их комбинацию. И, в некоторых вариантах осуществления комплексообразующий агент содержит циклодекстрин, содержащий альфа циклодекстрин, бета циклодекстрин, гамма циклодекстрин, гидроксипропилметилцеллюлоза-бета-циклодекстрин, сульфо-бутиловый эфир-бета-циклодекстрин, полианионный бета-циклодекстрин или любую их комбинацию; полисорбат, содержащий полиоксиэтилен (20) сорбит моноолеат; касторовое масло, содержащее полиокси 40 гидрогенизированное касторовое масло, полиокси 35 касторовое масло или любую их комбинацию; или любую их комбинацию.
[0037] В некоторых вариантах осуществления фармацевтическая композиция содержит декстрозу, маннит или любую их комбинацию.
[0038] Еще один вариант осуществления настоящего изобретения относится к способу получения фармацевтической композиции, включающему получение смеси Соединения (1), содержащей: а) от 5 масс.% до 95 масс.% соли HCl Соединения (1)•xH2O по массе фармацевтической композиции, где Соединение (1) представлено структурной формулой, приведенной выше, где x равно от 0 до 3; и b) один или более эксципиентов, содержащих наполнитель, дезинтегрирующий агент, смачивающий агент, связующее вещество, скользящее вещество, смазывающее вещество или любую их композицию, где смесь содержит от 5 масс.% до 95 масс.% одного или более эксципиентов.
[0039] В некоторых вариантах осуществления стадия получения смеси Соединения (1) включает: смешивание соли HCl Соединения (1)•хH2O и одного или нескольких внутригранулярных эксципиентов с получением гранул Соединения (1), где гранулы Соединения (1) содержат от 60 масс.% до 90 масс.% соли HCl Соединения (1)•хH2O по массе гранул и от 10 масс.% до 40 масс.% одного или нескольких эксципиентов по массе гранул; и смешивание гранул Соединения (1) с одним или несколькими внегранулярными эксципиентами дает фармацевтическую композицию, содержащую от 15 масс.% до 40 масс.% одного или нескольких внегранулярных эксципиентов по массе фармацевтической композиции.
[0040] В некоторых вариантах осуществления гранулы Соединения (1) содержат от 10 масс.% до 40 масс.% наполнителя по массе гранул, фармацевтическая композиция содержит от 15 масс.% до 40 масс.% наполнителя по массе фармацевтической композиции, или и то, и другое.
[0041] В некоторых вариантах осуществления изобретения наполнитель содержит микрокристаллическую целлюлозу, лактозу или любую их комбинацию.
[0042] В некоторых вариантах осуществления смесь Соединения (1) дополнительно содержит связующее вещество, дезинтегрирующий агент, смазывающее вещество или любую их комбинацию.
[0043] В некоторых вариантах осуществления стадия получения смеси Соединения (1) включает: смешивание i) от 70 масс.% до 85 масс.% соли HCl Соединения (1)•xH2O по массе гранул Соединения (1); и ii) одного или нескольких внутригранулярных эксципиентов, содержащих от 14 масс.% до 25 масс.% наполнителя по массе гранул и от 1 масс.% до 5 масс.% дезинтегрирующего агента по массе гранул с получением гранул Соединения (1); и смешивание гранул Соединения (1) с одним или несколькими внегранулярными эксципиентами, содержащими от 15 масс.% до 40 масс.% наполнителя по массе фармацевтической композиции, от 0,5 масс.% до 5 масс.% дезинтегрирующего агента по массе фармацевтической композиции и от 0,5 масс.% до 5 масс.% смазывающего вещества по массе фармацевтической композиции.
[0044] В некоторых вариантах осуществления стадия получения смеси Соединения (1) включает: получение раствора связующего вещества, содержащего воду и от 0,5 масс.% до 5 масс.% связующего вещества по массе гранул Соединения (1); получение внутригранулярной композиции, содержащей i) от 70 масс.% до 85 масс.% соли HCl Соединения (1)•xH2O по массе гранул Соединения (1); и ii) внутригранулярный эксципиент, который включает от 14 масс.% до 25 масс.% наполнителя по массе гранул Соединения (1) и от 1 масс.% до 5 масс.% дезинтегрирующего агента по массе гранул Соединения (1); и смешивание раствора связующего вещества и внутригранулярной композиции с получением гранул Соединения (1); и смешивание гранул Соединения (1) с одним или несколькими внегранулярными эксципиентами, содержащими от 15 масс.% до 40 масс.% наполнителя по массе фармацевтической композиции, от 0,5 масс.% до 5 масс.% дезинтегрирующего агента по массе фармацевтической композиции, и от 0,5 масс.% до 5 масс.% смазывающего вещества по массе фармацевтической композиции.
[0045] В некоторых вариантах осуществления стадия смешивания связующего раствора и предгранулярной композиции включает i) подачу внутригранулярной композиции в двухшнековый экструдер; и ii) введение связующего раствора в двухшнековый экструдер.
[0046] В некоторых вариантах осуществления связующий раствор содержит от 30 масс.% до 50 масс.% воды по массе внутригранулярной композиции.
[0047] В некоторых вариантах осуществления изобретения наполнитель содержит микрокристаллическую целлюлозу, лактозу или любую их комбинацию.
[0048] В некоторых вариантах осуществления связующее вещество содержит гидроксилпропилцеллюлозу, поливинилпирролидон или любую их комбинацию.
[0049] В некоторых вариантах осуществления дезинтегрирующий агент включает кроскармеллозу натрия, кросповидон, натрий крахмал гликолят или любую их комбинацию.
[0050] В некоторых вариантах осуществления смазывающее вещество содержит металл стеарат, металл стеарилфумарат или любую их комбинацию.
[0051] В некоторых вариантах осуществления связующее вещество содержит поливинилпирролидон со средней молекулярной массой от 3,000 до 5000; наполнитель содержит микрокристаллическую целлюлозу и моногидрат лактозы; дезинтегрирующий агент содержит натрий кроскармеллозу; и смазывающее вещество содержит натрия стеарилфумарат.
[0052] В некоторых вариантах осуществления, способ дополнительно включает прессование смеси Соединения (1) в таблетку.
[0053] Еще один вариант осуществления настоящего изобретения относится к способу получения фармацевтической композиции, включающему: смешивание a) соли HCl Соединения (1)•хH2O, где Соединение (1) представлено структурной формулой, приведенной выше, и где x равно 0-3; и от 0,01 M до 0,1 M pH модификатора, с получением смеси, содержащей от 1 мг/мл до 20 мг/мл Соединения (1) в воде.
[0054] В некоторых вариантах осуществления х равно 0,5. В некоторых вариантах осуществления соль HCl Соединения (1)•xH2O представляет собой Форму А соли HCl Соединения (1)•1/2H2О.
[0055] Другой вариант осуществления настоящего изобретения относится к способу снижения количества вируса гриппа в биологическом в образце in vitro или у индивидуума, включающему введение в образец или индивидууму эффективного количества фармацевтической композиции, такой как любая фармацевтическая композиция, описанная в настоящем документе.
[0056] Еще один вариант осуществления настоящего изобретения относится к способу ингибирования репликации вирусов гриппа в биологическом образце in vitrо или у индивидуума, включающему введение в образец или индивидууму эффективного количества фармацевтической композиции, такой как любая фармацевтическая композиция, описанная в настоящем документе.
[0057] Еще один вариант осуществления настоящего изобретения относится к способу лечения гриппа у индивидуума, включающему введение индивидууму терапевтически эффективного количества фармацевтической композиции, такой как любая фармацевтическая композиция, описанная в настоящем документе.
[0058] Некоторые из этих вариантов осуществления дополнительно включают совместное введение одного или более дополнительных терапевтических средств в образец или индивидууму. И, в некоторых вариантах осуществления настоящего изобретения дополнительные терапевтические средства включают противовирусное лекарственное средство (например, ингибитор нейраминидазы (например, озельтамивир, занамивир или любую их комбинацию), ингибитор полимеразы (например, флавипиравир) или любую их комбинацию.
[0059] В некоторых вариантах осуществления вирусами гриппа являются вирусы гриппа A.
[0060] Еще один вариант осуществления настоящего изобретения относится к режиму дозирования, включающему введение индивидууму эффективного количества фармацевтической композиции, такой как любая из описанных в настоящем документе, в дозировке от 100 мг до 1600 мг соли HCl Соединения (1)•хН2O, где х равно от 0 до 3 (например, 1/2).
КРАТКОЕ ОПИСАНИЕ ФИГУР
[0061] Фиг.1 и 2 представляют собой изображение порошковой рентгеновской дифракции (XRPD) и спектр твердотельной спектроскопии ядерно-магнитного резонанса на С13 (C13 SSNMR) Формы А соли HCl Соединения (1)•1/2H2O, соответственно.
[0062] Фиг.3 и 4 представляют собой изображение XRPD и спектр С13 SSNMR Формы F соли HCl Соединения (1)•3Н2O, соответственно.
[0063] Фиг.5 и 6 представляют собой изображение XRPD и спектр C13 SSNMR формы D соли HCl Соединения (1), соответственно.
[0064] Фиг.7А-7D представляют собой графики, показывающие растворимость формы А соли HCl Соединения (1)•1/2H2О относительно концентрации комплексообразующего агента: Tween® 80 на Фиг.7A; Кремофор® на Фиг.7B; Каптизол® на Фиг.7С; и Кавитрон® на Фиг.7D.
[0065] Фиг.8 представляет собой график, показывающий AUC выделения вируса для группы, получавшей дозу 1200 мг/600 мг, Формы А соли HCl Соединения (1)•1/2 Н2O на модели с заражением человека живым аттенуированным вирусом.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0066] Настоящее изобретение относится к фармацевтическим композициям, которые содержат соль HCl Соединения (1)•×H2O (где х равно от 0 до 3), к способам получения таких фармацевтических композиций, к способам лечения гриппа, к способам снижения количества вируса гриппа, а также к способам ингибирования репликации вируса гриппа, используя такие фармацевтические композиции.
[0067] I. ОПРЕДЕЛЕНИЯ
[0068] Используемый в настоящем описании термин "эксципиент" представляет собой неактивный ингредиент в фармацевтической композиции. Примеры эксципиентов включают наполнители или разбавители, смачивающие агенты (например, поверхностно-активные вещества), связующие вещества, скользящие вещества, смазывающие вещества, дезинтегрирующие агенты или тому подобное.
[0069] Используемый в настоящем описании термин "дезинтегрирующий агент" представляет собой эксципиент, который гидратирует фармацевтическую композицию и способствует дисперсии таблетки. Примеры дезинтегрирующих агентов включают натрий кроскармеллозу, полипласдон (то есть, поперечно-сшитый поливинилпирролидон), натрий крахмал гликолят или любую их комбинацию.
[0070] Используемый в настоящем описании термин "разбавитель" или "наполнитель" представляет собой эксципиент, который дает объем фармацевтической композиции. Примеры наполнителей включают лактозу, сорбит, целлюлозу, кальций фосфаты, различные виды крахмала, сахара (например, маннит, сахароза или тому подобное) или любую их комбинацию.
[0071] Используемый в настоящем описании термин "смачивающий агент" или "поверхностно-активное вещество» представляет собой эксципиент, который придает фармацевтическим композициям улучшенную растворимость и/или смачиваемость. Примеры смачивающих агентов включают натрий лаурилсульфат (SLS), натрий стеарилфумарат (SSF), полиоксиэтилен 20 сорбит моноолеат (например, Tween™) или любую их комбинацию.
[0072] Используемый в настоящем описании термин "связующее вещество" представляет собой эксципиент, который придает фармацевтической композиции улучшенную когезию или предел прочности при растяжении (например, твердость). Примеры связующих веществ включают двухосновный кальций фосфат, сахарозу, кукурузный (маис) крахмал, микрокристаллическую целлюлозу и модифицированную целлюлозу (например, гидроксиметилцеллюлозу).
[0073] Используемый в настоящем описании термин "скользящее вещество" представляет собой эксципиент, который придает фармацевтическим композициям улучшенные реологические свойства. Примеры скользящих веществ включают коллоидный диоксид кремния и/или тальк.
[0074] Используемый в настоящем описании термин "краситель" представляет собой эксципиент, который придает фармацевтической композиции желаемый цвет. Примеры красителей включают коммерчески доступные пигменты, такие как FD&C Blue # 1 Aluminium Lake, FD&C Blue # 2, другие цвета FD & C Blue, диоксид титана, оксид железа и/или их комбинации. Другие красители включают коммерчески доступные пигменты, такие как FD&C Green # 3.
[0075] Используемый в настоящем описании термин "смазывающее вещество" представляет собой эксципиент, который добавляют к фармацевтическим композициям, которые прессуют в таблетки. Смазывающее вещество способствует уплотнению гранул в таблетки и выбросу таблетки из фармацевтической композиции из штамповочного пресса. Примеры смазывающих веществ включают магния стеарат (стеарин), гидрогенизированное масло, натрия стеарилфумарат или любую их комбинацию.
[0076] II. ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ
[0077] Один из вариантов осуществления настоящего изобретения относится к фармацевтическим композициям солей HCl Соединения (1)•хН2О.
[0078] Соединение (1), представленное следующей структурной формулой
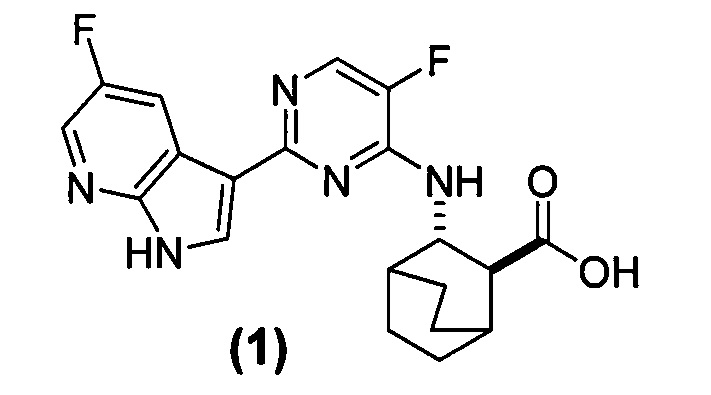
и его фармацевтически приемлемые соли могут ингибировать репликацию вируса гриппа и также описаны в публикации WO 2010/148197. В настоящем изобретении используют соли HCl Соединения (1)•хH2O, где х равно от 0 до 3, например, 0; 0,5; 1; 2 или 3 в составах фармацевтических композиций.
[0079] Соли HCl Соединения (1)•хН2О могут существовать в виде различных полиморфных форм. Как известно в данной области, полиморфизм представляет собой способность соединения кристаллизоваться в виде более одного отдельного кристаллического или "полиморфного" вида. Полиморф представляет собой твердую кристаллическую фазу соединения по меньшей мере с двумя различными относительными расположениями или полиморфными формами молекулы этого соединения в твердом состоянии. Полиморфные формы любого конкретного соединения определяются той же химической формулой или композицией, и различаются по химической структуре как кристаллические структуры двух различных химических соединений. Как правило, различные полиморфные формы могут быть охарактеризованы с помощью аналитических методов, таких как порошковая рентгеновская дифракция (XRPD) с получением профиля, термогравиметрический анализ (TGA) и дифференциальная сканирующая калориметрия (DSC), или по точке плавления или другими способами, известными в данной области. Используемый в настоящем описании термин "полиморфная форма" включает в себя сольваты и чистую полиморфную форму, которая не имеет никаких сольватов.
[0080] Используемый в настоящем описании термин "Соединение (1) " означает форму свободного основания Соединения (1). Соответственно, "соль HCl Соединения (1)" означает соль HCl свободного основания соединения. Следует отметить, что соли HCl Соединения (1) могут быть сольватированными или несольватированными, если не указано иного. Термин "соль HCl Соединения (1)•хH2O", включает в себя гидраты солей HCl Соединения (1), если х не равен нулю (например, 0,5; 1; 2 или 3), а также безводные соли HCl Соединения (1), если х равен нулю. Следует также отметить, что соли HCl Соединения (1)•хН2O могут быть кристаллическими или аморфными, если не указано иного.
[0081] В некоторых вариантах осуществления в настоящем изобретении используется соль НСl Соединения (1)•хH2O, где х равно от 0,5 до 3. В других вариантах осуществления в настоящем изобретении используется соль HCl Соединения (1)•хH2O, где х равно нулю, то есть, безводная соль HCl Соединения (1). В других вариантах осуществления в настоящем изобретении используется соль HCl Соединения (1)•1/2H2О. В других вариантах осуществления в настоящем изобретении используется соль HCl Соединения (1)•3Н2О.
[0082] В одном из вариантов осуществления в настоящем изобретении используется полиморфная форма А соли HCl Соединения (1)•1/2H2О. Эта форма является полиморфной формой соли HCl Соединения (1), которая содержит воду в качестве сольвата с половинным эквивалентом на Соединение (1). В одном из конкретных вариантов осуществления форма А соли HCl Соединения (1)•1/2H2O характеризуется тем, что имеет профиль XRPD с характеристическими пиками, измеренными в 2-тета (градусы) при 10,5±0,2, 5,2±0,2, 7,4±0,2 и 12,8±0,2. В другом конкретном варианте осуществления форма А соли HCl Соединения (1)•1/2H2O характеризуется тем, что имеет профиль XRPD с характеристическими пиками, выраженными в 2-тета (градусы) в следующих положениях, указанных в Таблице 2 раздела "Примеры". В еще одном конкретном варианте осуществления форма А соли HCl Соединения (1)•1/2H2O характеризуется тем, что имеет профиль XRPD, по существу такой же, как показано на ФИГ.1. Профили XRPD получают при комнатной температуре, используя альфа излучение Cu K. В еще одном конкретном варианте осуществления полиморфная форма А соли HCl Соединения (1)•1/2H2O характеризуется тем, что имеет пики при 29,2, 107,0, 114,0, и 150,7 (± 0,3 промиль) в спектре SSNMR на C13. В еще одном конкретном варианте осуществления форма А соли HCl Соединения (1)•1/2H2O характеризуется тем, что имеет пики SSNMR на C13, перечисленные в Таблице 3 раздела "Примеры". В еще одном конкретном варианте осуществления форма А соли HCl Соединения (1)•1/2H2O характеризуется тем, что имеет спектр твердотельного SSNMR на C13, по существу, такой же, как показано на ФИГ.2.
[0083] В еще одном варианте осуществления в настоящем изобретении используется полиморфная форма F соли HCl Соединения (1)•3Н2О. Эта форма является полиморфной формой соли HCl Соединения (1), которая содержит воду в качестве сольвата с тремя эквивалентами на Соединение (1). В одном из конкретных вариантов осуществления форма F соли HCl Соединения (1)•3Н2O характеризуется тем, что имеет профиль XRPD с характеристическими пиками, выраженными в 2-тета (градусы) при 7,1±0,2, 11,9±0,2 и 12,4±0,2. В еще одном конкретном варианте осуществления, форма F соли HCl Соединения (1)•3Н2O характеризуется тем, что содержит профиль XRPD с характеристическими пиками, выраженными в 2-тета (градусы) в следующих положениях, перечисленных в Таблице 5 раздела "Примеры". В еще одном конкретном варианте осуществления форма F соли HCl Соединения (1)•3Н2O характеризуется тем, что содержит профиль XRPD, по существу такой же, как показано на ФИГ.3. Профили XRPD получают при комнатной температуре, используя альфа излучение Cu K. В еще одном конкретном варианте осуществления полиморфная форма F соли HCl Соединения (1)•3Н2O характеризуется тем, что имеет пики при 20,7, 27,4, 104,8, 142,5, 178,6 (± 0,3 промиль) в спектре SSNMR на C13. В еще одном конкретном варианте осуществления форма F соли HCl Соединения (1)•3Н2O характеризуется тем, что имеет пики SSNMR на C13, перечисленные в Таблице 6 раздела "Примеры". В еще одном конкретном варианте осуществления форма F соли HCl Соединения (1)•3Н2O характеризуется тем, что имеет спектр SSNMR на C13, по существу такой же, как показано на ФИГ.4.
[0084] В еще одном варианте осуществления в настоящем изобретении используется полиморфная форма D соли HCl Соединения (1). Эта форма является несольватированный формой соли HCl Соединения (1). В одном из конкретных вариантов осуществления форма D соли HCl Соединения (1) характеризуется тем, что имеет профиль XRPD с характеристическими пиками, выраженными в 2-тета (градусы) при 5,8±0,2, 17,1±0,2 и 19,5±0,2. В еще одном конкретном варианте осуществления форма D соли HCl Соединения (1) характеризуется тем, что имеет профиль XRPD с характеристическими пиками, выраженными в 2-тета (градусы) в положениях, указанных в Таблице 7 раздела "Примеры". В еще одном конкретном варианте осуществления форма D соли HCl Соединения (1) характеризуется тем, что имеет профиль XRPD по существу такой же, как показано на ФИГ.5. Профили XRPD получают при комнатной температуре, используя альфа излучение Cu K. В еще одном конкретном варианте осуществления Форма D соли HCl Соединения (1) характеризуется тем, что имеет пики при 29,4, 53,4, 113,3, 135,4, 177,8 (±0,3 частей на миллион) в спектре SSNMR на C13. В еще одном конкретном варианте осуществления форма D соли HCl Соединения (1) характеризуется тем, что имеет пики SSNMR на C13, перечисленные в Таблице 8 раздела "Примеры". В еще одном конкретном варианте осуществления форма D соли HCl Соединения (1) характеризуется тем, что имеет спектр SSNMR на C13, по существу такой же, как показано на ФИГ.6.
[0085] Полиморфная Форма А соли HCl Соединения (1)•1/2H2О, Форма F соли HCl Соединения (1)•3Н2О и Форма D соли HCl Соединения (1), как описано выше, может находится в выделенной, чистой форме или в смеси в виде твердой композиции, в которую примешаны другие вещества, например, другие твердые формы (например, аморфная форма, Форма А Соединения (1), или тому подобное) Соединения (1)или любые другие вещества.
[0086] В некоторых вариантах осуществления в настоящем изобретении Форма А соли HCl Соединения (1)•1/2H2О, Форма F соли HCl Соединения (1)•3Н2О и Форма D соли HCl Соединения (1) используются в выделенной твердой форме. В других вариантах осуществления в настоящем изобретении Форма А соли HCl Соединения (1)•1/2H2О, Форма F соли HCl Соединения (1)•3Н2О и Форма D соли HCl Соединения (1) используются в чистой форме. Чистая форма означает, что, например, Форма А соли HCl Соединения (1)•1/2H2O составляет более 95% (масс./масс.), например, более 98% (масс./масс.), более чем на 99% (масс./масс.%), 99,5% (масс./масс.) или более 99,9% (масс./масс.). В некоторых вариантах осуществления Форма А соли HCl Соединения (1)•1/2H2О, Форма F соли HCl Соединения (1)•3Н2О и Форма D соли HCl Соединения (1) находятся в форме композиции или смеси полиморфной формы с одним или несколькими другими кристаллическими, сольватными, аморфными или других полиморфными формами или их комбинациями. В одном конкретном варианте осуществления композиция может содержать Форму A соли HCl Соединения (1)•1/2H2O вместе с одной или несколькими другими твердыми формами Соединения (1), такими как аморфная форма, сольваты, Форма F соли HCl Соединения (1)•3H2O и Форма D соли HCl Соединения (1), и/или другие формы или их комбинации. В другом конкретном варианте осуществления композиция может содержать Форму F соли HCl Соединения (1)•3H2O вместе с одной или несколькими другими твердыми формами Соединения (1), такими как аморфная форма, сольваты, Форма A соли HCl Соединения (1)•1/2H2O, Форма D соли HCl Соединения (1) и/или другие формы или их комбинации. Еще в одном варианте осуществления композиция может содержать Форму D соли HCl Соединения (1) вместе с одной или несколькими другими твердыми формами Соединения (1), такими как аморфная форма, сольваты, Форма A соли HCl Соединения (1)•1/2H2O, Форма F соли HCl Соединения (1)•3H2O и/или другие формы или их комбинации.
[0087] В еще одном конкретном варианте осуществления композиция может содержать от следовых количеств до 100% Формы A соли HCl Соединения (1)•1/2H2O или любое количество в указанных пределах, например, 0,1%-0,5%, 0,1%-1%, 0,1%-2%, 0,1%-5%, 0,1%-10%, 0,1%-20%, 0,1%-30%, 0,1%-40% или 0,1%-50% по массе на основании общего количества Соединения (1) в фармацевтической композиции. В еще одном конкретном варианте осуществления композиция может содержать по меньшей мере 50%, 60%, 70%, 80%, 90%, 95%, 97%, 98%, 99%, 99,5% или 99,9% по массе Формы А соли HCl Соединения (1)•1/2H2O на основании общего количества Соединения (1) в фармацевтической композиции. В еще одном конкретном варианте осуществления, композиция может содержать от следовых количеств до 100% Формы F соли HCl Соединения (1)•3H2O или любое количество в указанных пределах, например, в диапазоне 0,1%-0,5%, 0,1%-1%, 0,1%-2%, 0,1%-5%, 0,1%-10%, 0,1%-20%, 0,1%-30%, 0,1%-40% или 0,1%-50% по массе на основании общего количества Соединения (1) в фармацевтической композиции. В еще одном конкретном варианте осуществления, композиция может содержать по меньшей мере 50%, 60%, 70%, 80%, 90%, 95%, 97%, 98%, 99%, 99,5% или 99,9% по массе Формы F соли HCl Соединения (1)•3H2O на основании общего количества Соединения (1) в фармацевтической композиции. В еще одном конкретном варианте осуществления композиция может содержать от следовых количеств до 100% Формы D соли HCl Соединения (1) или любое количество в указанных пределах, например, в диапазоне 0,1%-0,5%, 0,1%-1%, 0,1%-2%, 0,1%-5%, 0,1%-10%, 0,1%-20%, 0,1%-30%, 0,1%-40% или 0,1%-50% по массе на основании общего количества Соединения (1) в фармацевтической композиции. В еще одном конкретном варианте осуществления композиция может содержать по меньшей мере 50%, 60%, 70%, 80%, 90%, 95%, 97%, 98%, 99%, 99,5% или 99,9% по массе Формы D соли HCl Соединения (1) в расчете на общее количество Соединения (1) в фармацевтической композиции.
[0088] Форма А соли HCl Соединения (1)•1/2H2O может быть получена с использованием смешивания (например, перемешивания) хлористого водорода (HCl) с Соединением (1). Соединение (1) может быть сольватированным, несольватированным, аморфным или кристаллическим. Раствор, взвесь или суспензия Соединения (1) могут быть смешаны с HCl в системе растворителей, которая включает в себя воду и один или несколько органических растворителей, где система растворителей имеет активность воды, равную или больше 0,05, и равную или меньше 0,85, то есть, 0,05-0,85. Термин "активность воды" (аw), как используется в настоящем описании, известен в данной области техники и означает меру энергетического состояния воды в системе растворителей. Активность воды определяется как давление пара жидкости, разделенное на давление чистой воды при той же температуре. В частности, активность воды определяется как 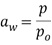 , где
, где 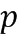 представляет собой давление пара воды в веществе, и
представляет собой давление пара воды в веществе, и 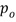 представляет собой давление пара чистой воды при той же температуре, или в случае
представляет собой давление пара чистой воды при той же температуре, или в случае 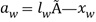 , где
, где 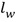 представляет собой коэффициент активности воды и
представляет собой коэффициент активности воды и 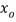 представляет собой молярную долю воды в водной фракции. Например, чистая вода имеет значение активности воды 1,0. Значения активности воды, как правило, могут быть получены либо по емкости гигрометра, либо по точке росы гигрометра. Различные виды инструментов для измерения активности воды также имеются в продаже. Альтернативно, значения активности воды смесей двух или более растворителей могут быть рассчитаны на основе количеств растворителей и известных значений активности воды растворителей.
представляет собой молярную долю воды в водной фракции. Например, чистая вода имеет значение активности воды 1,0. Значения активности воды, как правило, могут быть получены либо по емкости гигрометра, либо по точке росы гигрометра. Различные виды инструментов для измерения активности воды также имеются в продаже. Альтернативно, значения активности воды смесей двух или более растворителей могут быть рассчитаны на основе количеств растворителей и известных значений активности воды растворителей.
[0089] Пример кристаллическое Соединение (1) включает Форму А Соединения (1) (см. Примеры ниже). Эта форма является несольватированной формой соли HCl Соединения (1). В одном конкретном варианте осуществления Форма А Соединения (1) характеризуется тем, что имеет профиль XRPD с характеристическими пиками, выраженными в 2-тета (градусы) при 15,5±0,2, 18,9±0,2 и 22,0±0,2 (например, см. Таблицу 10 в разделе "Примеры"). В другом конкретном варианте осуществления Форма А Соединения (1) характеризуется тем, что имеет пики при 21,0, 28,5, 50,4, 120,8, 138,5, и 176,2 (±0,3 промилле) в спектре SSNMR на C13 (например, см. Таблицу 11 в разделе "Примеры"). Примеры сольватов Соединения (1) включают в себя сольваты 2-MeTHF, N,N-метанола, ксилола, ацетона, 2-бутанола, метилацетата, 1-пентанола, 2-пропанола, тетрагидрофурана, метилтетрагидрофурана, диметилацетамида, N,N-диметилформамида, 1,4-диоксана, 1-пентанола, 2-метил-1-пропанола, метилэтилкетона, 3-метил-1-бутанола, гептана, этилформиата, 1-бутанола, уксусной кислоты и этиленгликоля. В конкретном варианте осуществления используются сольваты 2-MeTHF (например, соединение (1)•1 (2-MeTHF)).
[0090] Системы растворителей, подходящие для получения формы А соли HCl Соединения (1)•1/2H2О могут содержать большое разнообразие комбинаций воды и органических растворителей, где активность воды системы растворителей равна или больше 0,05, и равна или меньше 0,85 (0,05-0,85). В конкретном варианте осуществления значение активности воды равно 0,4-0,6. Подходящие органические растворители включают органические растворители Класса II или Класса III, перечисленные в руководстве Международной конференции по гармонизации. Конкретные примеры подходящих органических растворителей Класса II включают хлорбензол, циклогексан, 1,2-дихлорэтен, дихлорметан (DCM), 1,2-диметоксиэтан, N,N-диметилацетамид, N,N-диметилформамид, 1,4-диоксан, 2-этоксиэтанол, формамид, гексан, 2-метоксиэтанол, метилбутилкетон, метилциклогексан, N-метилпирролидон, нитрометан, пиридин, сульфолан, тетрагидрофуран (THF), тетралин, толуол, 1,1,2-трихлорэтилен и ксилол. Конкретные примеры подходящих органических растворителей класса III включают: уксусную кислоту, ацетон, анизол, 1-бутанол, 2-бутанол, бутилацетат, трет-бутилметиловый эфир, кумол, гептан, изобутилацетат, изопропилацетат, метилацетат, 3-метил- 1-бутанол, метилэтилкетон, метилизобутилкетон, 2-метил-1-пропанол, этилацетат, этиловый эфир, этилформиат, пентан, 1-пентанол, 1-пропанол, 2-пропанол и пропилацетат. В одном из конкретных вариантов осуществления изобретения органические растворители системы растворителей выбираны из группы, состоящей из хлорбензола, циклогексана, 1,2-дихлорэтана, дихлорметана, 1,2-диметоксиэтана, гексана, 2-метоксиэтанола, метилбутилкетона, метилциклогексана, нитрометана, тетралина, ксилола, толуола, 1,1,2-трихлорэтана, ацетона, анизола, 1-бутанола, 2-бутанола, бутилацетата, трет-бутилового эфира, кумола, этанола, этилацетата, этилового эфира, этилформиата, гептана, изобутилацетата, изопропилацетата, метилацетата, 3-метил-1-бутанола, метилэтилкетона, 2-метил-1-пропанола, пентана, 1-пропанола, 1-пентанола, 2-пропанола, пропилацетата, тетрагидрофурана и метил-тетрагидрофурана. В другом конкретном варианте осуществления органические растворители системы растворителей выбраны из группы, состоящей из 2-этоксиэтанола, этиленгликоля, метанола, 2-метоксиэтанола, 1-бутанола, 2-бутанола, 3-метил-1-бутанола, 2-метил-1-пропанола, этанола, 1-пентанола, 1-пропанола, 2-пропанола, метилбутилкетона, ацетона, метилэтилкетона, метилизобутилкетона, бутилацетата, изобутилацетата, изопропилацетата, метилацетата, этилацетата, пропилацетата, пиридина, толуола и ксилола. В еще одном варианте осуществления органические растворители выбраны из группы, состоящей из ацетона, н-пропанола, изопропанола, изо-бутилацетата и уксусной кислоты. В еще одном варианте осуществления органические растворители выбраны из группы, состоящей из ацетона и изопропанола. В еще одном конкретном варианте осуществления система растворителей включает воду и ацетон. В еще одном конкретном варианте осуществления система растворителей включает воду и изопропанол.
[0091] Получение Формы А соли HCl Соединения (1)•1/2H2О может быть осуществлено при любой подходящей температуре. Как правило, получение осуществляют при температуре 5°C-75°C. В конкретном варианте осуществления получение осуществляют при температуре 15°С-75°С. В другом конкретном варианте осуществления получение осуществляют при температуре 15°С-60°С. В еще одном конкретном варианте осуществления получение осуществляют при температуре 15°С-35°С. В еще одном конкретном варианте осуществления получение осуществляют при 5°C-75°C в системе растворителей, имеющей значение активности воды 0,4-0,6. В еще одном конкретном варианте осуществления получение осуществляют при температуре 15°С-75°С в системе растворителей, имеющей значение активности воды 0,4-0,6. В еще одном конкретном варианте осуществления получение осуществляют при температуре 15°С-60°С в системе растворителей, имеющей значение активности воды 0,4-0,6. В еще одном конкретном варианте осуществления получение осуществляют при 15°С-35°С в системе растворителей, имеющей значение активности воды 0,4-0,6.
[0092] Хлористый водород может быть введен в виде раствора или газа. Одним из примеров подходящего источника хлористого водорода является раствор хлористого водорода с 30-40 массовыми процентами (например, 34 масс.% - 38 масс.%) в воде.
[0093] Форма F соли HCl Соединения (1)•3Н2O может быть получена путем смешивания HCl и Соединения (1) в системе растворителей, которая включает в себя воду или которая включает в себя воду и один или несколько органических растворителей, где система растворителей имеет активность воды, равную или больше 0,9 (≥ 0,9). Смесь может представлять собой раствор, взвесь или суспензию. Соединение (1) может быть сольватированным, несольватированным, аморфным или кристаллическим. Альтернативно, она может быть получена путем перемешивания Формы А соли HCl Соединения (1)•1/2H2O в системе растворителей, которая включает в себя воду, или которая включает воду и один или более органических растворителей, где система растворителей имеет активность воды, равную или больше 0,9. Как правило, чистая вода имеет значение активности воды 1,0. Соответственно, система растворителей, имеющая водную активность 0,9-1,0, может подходить для получения формы F соли HCl Соединения (1)•3Н2О. В конкретном варианте осуществления, смешивание или перемешивание осуществляют при температуре окружающей среды (18°C - 25°C). В еще одном конкретном варианте осуществления смешивание или перемешивание проводят при температуре 15°С-30°С. В другом конкретном варианте осуществления смешивание или перемешивание проводят при температуре 20°С-28°С (например, при 25°C). Подходящими органическими растворителями, включая конкретные примеры, для образования формы F соли HCl Соединения (1)•3Н2О, являются растворители, как описано выше для формы А соли HCl Соединения (1)•1/2H2О. В еще одном конкретном варианте осуществления система растворителей включает воду и ацетон. В еще одном конкретном варианте осуществления система растворителей включает воду и изопропанол.
[0094] Форма D соли HCl Соединения (1) может быть получена путем дегидратации Формы А соли HCl Соединения (1)•1/2H2О. Дегидратация может быть осуществлена с помощью любых подходящих средств, например, нагревом или сухой продувкой азотом, или ими вместе.
[0095] Форма А Соединения (1) может быть получена (а) путем перемешивания смеси аморфного Соединения (1) или сольвата Соединения (1) (например, сольвата 2-MeTHF Соединения (1)) в системе растворителей, которая включает в себя воду и этанол. Смесь может быть раствором или взвесью. В конкретном варианте осуществления стадию перемешивания проводят при температуре в диапазоне от 18°С до 90°С. В другом конкретном варианте осуществления стадию перемешивания (а) осуществляют при температуре кипения с обратным холодильником системы растворителей. В другом конкретном варианте осуществления система растворителей включает 5-15 масс.% воды. Примерами сольватов Соединения (1) являются сольваты, как описано выше. В конкретном варианте осуществления используются сольваты 2-MeTHF (например, Соединение (1)·1(2-MeTHF)). Более конкретно, получение дополнительно включает: (b) перемешивание аморфной формы Соединения (1) в нитрометане с образованием кристаллической затравки Формы А Соединения (1); и (с) добавление кристаллической затравки Формы А Соединения (1) к полученной смеси на стадии смешивания (а). В конкретном варианте осуществления способы дополнительно включают: (b) перемешивание аморфной формы Соединения (1) в нитрометане с образованием кристаллической затравки Формы А Соединения (1); (с) охлаждение полученной смеси на стадии смешивания (а) до температуры в диапазоне от 18°С до 60°С (например, 50-55°C или при 55°C); и (d) добавление кристаллической затравки Формы А Соединения (1) к полученной смеси стадии (с). В другом конкретном варианте осуществления способы дополнительно включает добавление воды, перед добавлением кристаллической затравки Формы А Соединения (1), к полученной смеси, которая прошла через стадию нагревания с обратным холодильником, в таком количестве, чтобы полученная система растворителей содержала воду в количестве 15-25 масс.% после добавления воды. В еще одном конкретном варианте осуществления способы дополнительно включают добавление воды к смеси, которая содержит кристаллическую затравку Формы А Соединения (1), в таком количестве, чтобы система растворителей содержала воду в количестве 35-45 масс.% после добавления воды. В еще одном конкретном варианте осуществления способы дополнительно включают охлаждение смеси, которая содержит кристаллическую затравку Формы А Соединения (1), после добавления воды, до температуры 0°C -10°C.
[0096] В одном из конкретных вариантов осуществления кристаллическая Форма А Соединения (1) может быть получена с помощью сольвата 2-MeTHF Соединения (1) в нитрометане. В одном из вариантов осуществления система растворителей для стадии нагревания с обратным холодильником, включает 5-15 масс.%, например, 10 масс.%, воды.
[0097] В одном из аспектов изобретение включает фармацевтические композиции, содержащие от 5 масс.% до 95 масс.% соли HCl Соединения (1)•хН2О по массе фармацевтической композиции, и 5 масс.% до 95 масс.% наполнителя по массе фармацевтической композиции. В одном из конкретных вариантов осуществления используется от 20 масс.% до 80 масс.% наполнителя по массе фармацевтической композиции.
[0098] Наполнители (или разбавители), как правило, включают микрокристаллическую целлюлозу (например, Avicel® PH 101), лактозы, сорбиты, целлюлозы, кальция фосфаты, различные виды крахмала, сахара (например, маннит, сахароза или тому подобное) или любую их комбинацию. Конкретные примеры наполнителей включают микрокристаллическую целлюлозу и лактозу. Конкретные примеры микрокристаллических целлюлоз включают коммерчески доступные серии Avicel®, такие как микрокристаллическая целлюлоза с размером частиц 200 меш более 70% и размером частиц 65 меш менее чем на 10% (например, Avicel® PH 101). Другими конкретными примерами микрокристаллических целлюлоз являются силикатизированные микрокристаллические целлюлозы, такие как коммерчески доступный серии Prosolv® (например, Prosolv® SMCC 50). Конкретный пример лактозы, подходящий для изобретения, включает лактозу моногидрат. Характерные количества наполнителей по отношению к общей массе фармацевтической композиции могут составлять от 5 масс.% до 95 масс.%, от 20 масс.% до 80 масс.% или от 25 масс.% до 50 масс.%.
[0099] В одном из вариантов осуществления фармацевтические композиции по настоящему изобретению дополнительно содержат от 1 масс.% до 10 масс.% дезинтегрирующего агента по массе фармацевтической композиции. В одном конкретном варианте осуществления используется от 3 масс.% до 7 масс.% дезинтегрирующего агента по массе фармацевтической композиции.
[0100] Дезинтегрирующие агенты, как правило, повышают диспергирование фармацевтических композиций. Примеры дезинтегрирующих агентов включают кроскармеллозы (например, кроскармеллоза натрия), кросповидоны, крахмал (например, кукурузный крахмал, картофельный крахмал), металл крахмал гликоляты (например, натрия крахмал гликолят) и любую их комбинацию. Конкретные примеры дезинтегрирующих агентов включают кроскармеллозу натрия (например, Ac-Di-Sol®) и натрий крахмал гликолят. Характерные количества дезинтегрирующих агентов по отношению к общей массе фармацевтической композиции может составлять от 1 масс.% до 10 масс.%, от 3 масс.% до 7 масс.% или от 1 масс.% до 5 масс.% от массы фармацевтических композиций.
[0101] В другом варианте осуществления фармацевтические композиции по изобретению дополнительно содержат от 0,1 масс.% до 5 масс.% по массе связующего вещества по массе фармацевтической композиции. В одном из конкретных вариантов осуществления используется от 0,5 масс.% до 2 масс.% по массе связующего вещества по массе фармацевтической композиции.
[0102] Связующие вещества обычно включают агенты, используемые при получении гранул активного ингредиента путем смешивания его с разбавляющими наполнителями. Характерные связующие вещества включают поливинилпирролидоны, крахмал (например, предварительно желатинизированный крахмал), сахар, микрокристаллическую целлюлозу, модифицированные целлюлозы (например, гидроксипропилметилцеллюлозы (HPMC), гидроксипропилцеллюлозы (HPC) и гидроксиэтилцеллюлозы (НЕС), а также любую их комбинацию. Конкретные примеры связующих веществ включают поливинилпирролидоны (PVP). Пример HPC включает полимер с низкой вязкостью, HPC-SL. PVP обычно характеризуется так называемой "К-величиной", которая является практической мерой вязкости полимерной композиции. PVP может быть коммерчески приобретен (например, Tokyo Chemical Industry Co., Ltd.) под товарным знаком Povidone® K12, Povidone® K17, Povidone® К25, Povidone® K30, Povidone® К60 и Povidone® K90. Конкретные примеры PVP включают растворимый высушенный распылением PVP. Более конкретный пример включает PVP со средней молекулярной массой от 3000 до 4000, такой как Povidone® К12 со средней молекулярной массой 4000. PVP может быть использован либо во влажном, либо в сухом состоянии. Характерные количества связующих веществ по отношению к общей массе фармацевтической композиции могут составлять от 0,1 масс.% до 5 масс.% или 0,5 масс.% до 2 масс.%.
[0103] В еще одном варианте осуществления фармацевтические композиции по изобретению дополнительно содержат от 0,5 масс.% до 5 масс.% смазочного вещества по массе фармацевтической композиции. В одном из конкретных вариантов осуществления используется от 0,5 масс.% до 3 масс.% или от 1 масс.% до 3 масс.% смазывающего вещества по массе фармацевтической композиции.
[0104] Смазывающие вещества, как правило, улучшают сжатие и выброс фармацевтических композиций, например, из штамповочного пресса. Характерные смазывающие вещества включают магния стеарат, стеариновую кислоту (стеарин), гидрогенизированное масло, натрия стеарилфумарат и любые их комбинации. Конкретный пример смазочных веществ включает натрия стеарилфумарат. Еще один конкретный пример смазочных веществ включает магния стеарат. Характерные количества смазочного вещества по общей массе фармацевтической композиции могут составлять от 0,5 масс.% до 5 масс.%, от 0,5 масс.% до 3 масс.% или от 1 масс.% до 3 масс.%.
[0105] В некоторых вариантах осуществления в фармацевтических композициях по изобретению может быть использован смачивающий агент. Смачивающие агенты, как правило, включают поверхностно-активные вещества, такие как неионные поверхностно-активные вещества и анионные поверхностно-активные вещества. Смачивающие агенты, подходящие для настоящего изобретения, как правило, повышают растворимость фармацевтических композиций. Примеры поверхностно-активных вещества включают натрия лаурилсульфат (SLS), полиоксиэтилированный сорбитан с жирными кислотами (например, TWEEN ТМ), эфиры сорбитана жирных кислот (например, Spans®), натрия додецилбензолсульфонат (SDBS), диоксти натрия сульфосукцинат (докузат), натриевая соль диоксихолиновой кислоты (DOSS), сорбитан моностеарат, сорбитан тристеарат, натрия N-лауроилсаркозин, олеат натрия, натрия миристат, натрия стеарат, натрия пальмитат, Gelucire 44/14, этилендиаминтетрауксусной кислоты (EDTA), витамин Е d-альфа токоферил полиэтиленгликоль 1000 сукцинат (TPGS), лецитин, MW 677-692, глутаминовая кислота мононатриевая соль моногидрат, Labrasol, PEG 8 каприловый/каприковый глицериды, Transcutol, диэтиленгликоля моноэтиловый эфир, Solutol HS-15, полиэтиленгликоль/гидроксистеарат, таурохолевая кислота, сополимеры полиоксипропилена и оксиэтилена (например, полоксамеры, также известные и коммерчески доступные как Pluronics®, например, Pluronic® L61, Pluronic® F68, Pluronic® F108 и Pluronic® F127), насыщенные полигликолизированные глицериды (Gelucirs®) и любые их комбинации. Конкретные примеры включают натрий лаурилсульфат, который представляет собой анионное поверхностно-активное вещество; и сополимеры полиоксипропилена и полиоксиэтилена, которые являются неионными поверхностно-активными веществам. Конкретные примеры сополимеров полиоксипропилена и полиоксиэтилена включают полоксамеры, такие как полоксамер с полиоксипропиленом с молекулярной массой 1,800 г/моль и с 80%-ным содержанием полиоксиэтилена (например, полоксамер 188). Характерные количества смачивающих агентов к общей массе фармацевтической композиции может составлять от 0,25 масс.% до 10 масс.% или от 1 масс.% до 5 масс.%.
[0106] Смачивающие агенты, связующие вещества, дезинтегрирующие агенты, смазывающие вещества и наполнители, подходящие для изобретения совместимы с ингредиентами фармацевтических композиций по изобретению, например, они по существу не снижают химическую стабильность.
[0107] В одном из конкретных вариантов осуществления фармацевтические композиции по настоящему изобретению содержат: а) от 20 масс.% до 80 масс.% соли HCl Соединения (1)•хН2О по массе фармацевтической композиции; b) от 1 масс.% до 10 масс.% дезинтегрирующих агентов по массе фармацевтической композиции; и с) от 20 масс.% до 80 масс.% наполнителя по массе фармацевтической композиции. В некоторых вариантах осуществления фармацевтическая композиция содержит а) от 20 масс.% до 80 масс.% Формы А соли HCl Соединения (1)•1/2H2О по массе фармацевтической композиции; b) от 1 масс.% до 10 масс.% дезинтегрирующего агента по массе фармацевтической композиции; с) от 0,1 масс.% до 5 масс.% связующего вещества по массе фармацевтической композиции; и d) от 20 масс.% до 80 масс.% наполнителя по массе фармацевтической композиции. В еще одном конкретном варианте осуществления фармацевтические композиции по изобретению содержат: a) от 20 масс.% до 80 масс.% соли HCl Соединения (1)•хH2O по массе фармацевтической композиции; b) от 1 масс.% до 10 масс.% дезинтегрирующего агента по массе фармацевтической композиции; c) от 0,1 масс.% до 5 масс.% связующего вещества по массе фармацевтической композиции; d) от 20 масс.% до 80 масс.% наполнителя по массе фармацевтической композиции; и e) от 0,5 масс.% до 5 масс.% смазывающего вещества по массе композиции. Примеры, в том числе конкретные примеры, наполнителей, дезинтегрирующих агентов, связующих веществ и смазывающих веществ являются вещества, как описано выше.
[0108] В еще одном конкретном варианте осуществления, фармацевтические композиции по изобретению содержат: a) от 35 масс.% до 75 масс.% соли HCl Соединения (1)•хH2O по массе фармацевтической композиции; b) от 1 масс.% до 7 масс.% дезинтегрирующего агента по массе фармацевтической композиции, где дезинтегрирующий агент выбран из кроскармеллозы, кросповидона, металл крахмал гликолята или крахмала или любой их композиции; c) от 0,5 масс.% до 2 масс.% связующего вещества по массе фармацевтической композиции, где связующее вещество выбрано из поливинилпирролидона, крахмала, сахара, микрокристаллической целлюлозы, гидроксипропилметилцеллюлозы, гидроксипропилцеллюлозы или гидроксиэтилцеллюлозы или любой их композиции; d) от 25 масс.% до 50 масс.% наполнителя по массе фармацевтической композиции; где наполнитель выбран из микрокристаллической целлюлозы, лактозы, сорбита, целлюлозы, кальция фосфата, крахмала или сахара или любой их композиции; и e) от 0,5 масс.% до 3 масс.% смазывающего вещества по массе композиции, где смазывающее вещество выбрано из металл стеарата и/или металл стеарилфумарата. Конкретными примерами наполнителей, дезинтегрирующих агентов, связующих веществ и смазывающих веществ являются вещества, как описано выше.
[0109] В еще одном конкретном варианте осуществления фармацевтические композиции по настоящему изобретению содержат: а) от 35 масс.% до 75 масс.% соли HCl Соединения (1)•хН2О по массе фармацевтической композиции; b) от 3 масс.% до 7 масс.% кроскармеллозы по массе фармацевтической композиции; с) от 0,5 масс.% до 2 масс.% поливинилпирролидона по массе фармацевтической композиции; d) от 25 масс.% до 50 масс.% наполнителя по массе фармацевтической композиции; где наполнитель включает микрокристаллическую целлюлозу и лактозу; и е) от 0,5 масс.% до 3 масс.% металл стеарилфумарата по массе композиции. Конкретными примерами наполнителей, дезинтегрирующих агентов, связующих веществ и смазывающих веществ являются вещества, как описано выше.
[0110] В еще одном конкретном варианте осуществления, фармацевтические композиции по изобретению содержат: a) от 35 масс.% до 75 масс.% соли HCl Соединения (1)•хH2O по массе фармацевтической композиции; b) от 3 масс.% до 7 масс.% кроскармеллозы по массе фармацевтической композиции; c) от 0,5 масс.% до 2 масс.% поливинилпирролидона по массе фармацевтической композиции; d) от 25 масс.% до 50 масс.% наполнителя по массе фармацевтической композиции; где наполнитель включает микрокристаллическую целлюлозу и лактозу; и e) от 0,5 масс.% до 3 масс.% натрия стеарилфумарата по массе композиции. Конкретными примерами наполнителей, дезинтегрирующих агентов, связующих веществ и смазывающих веществ являются вещества, как описано выше.
[0111] В еще одном конкретном варианте осуществления, фармацевтические композиции по изобретению содержат: a) от 35 масс.% до 65 масс.% соли HCl Соединения (1)•хH2O по массе фармацевтической композиции; b) от 3 масс.% до 7 масс.% кроскармеллозы натрия по массе фармацевтической композиции; c) от 0,5 масс.% до 2 масс.% поливинилпирролидона со средней молекулярной массой от 3000 до 5000 по массе фармацевтической композиции; d) от 30 масс.% до 40 масс.% микрокристаллической целлюлозы по массе фармацевтической композиции; e) от 5 масс.% до 10 масс.% лактозы моногидрата по массе фармацевтической композиции; и f) от 1 масс.% до 3 масс.% натрия стеарилфумарата по массе композиции.
[0112] В еще одном дополнительном конкретном варианте осуществления фармацевтические композиции по изобретению содержат: a) от 20 масс.% до 80 масс.% Формы А соли HCl Соединения (1)•1/2H2O по массе фармацевтической композиции; b) от 1 масс.% до 10 масс.% дезинтегрирующего агента по массе фармацевтической композиции; и c) от 20 масс.% до 80 масс.% наполнителя по массе фармацевтической композиции. В еще одном дополнительном конкретном варианте осуществления фармацевтические композиции по изобретению содержат: a) от 20 масс.% до 80 масс.% Формы А соли HCl Соединения (1)•1/2H2O по массе фармацевтической композиции; b) от 1 масс.% до 10 масс.% дезинтегрирующего агента по массе фармацевтической композиции; c) от 0,1 масс.% до 5 масс.% связующего вещества по массе фармацевтической композиции; и d) от 20 масс.% до 80 масс.% наполнителя по массе фармацевтической композиции. В еще одном дополнительном конкретном варианте осуществления фармацевтические композиции по изобретению содержат: a) от 20 масс.% до 80 масс.% Формы А соли HCl Соединения (1)•1/2H2O по массе фармацевтической композиции; b) от 1 масс.% до 10 масс.% дезинтегрирующего агента по массе фармацевтической композиции; c) от 0,1 масс.% до 5 масс.% связующего вещества по массе фармацевтической композиции; d) от 20 масс.% до 80 масс.% наполнителя по массе фармацевтической композиции; и e) от 0,5 масс.% до 5 масс.% смазывающего вещества по массе композиции. Примеры, в том числе конкретные примеры, наполнителей, дезинтегрирующих агентов, связующих веществ и смазывающих веществ являются вещества, как описано выше.
[0113] В еще одном дополнительном конкретном варианте осуществления фармацевтические композиции по изобретению содержат: a) от 35 масс.% до 75 масс.% Формы А соли HCl Соединения (1)·1/2H2O по массе фармацевтической композиции; b) от 1 масс.% до 7 масс.% дезинтегрирующего агента по массе фармацевтической композиции, где дезинтегрирующий агент выбран из кроскармеллозы, кросповидона, металл крахмалгликолята или крахмала или любой их композиции; c) от 0,5 масс.% до 2 масс.% связующего вещества по массе фармацевтической композиции, где связующее вещество выбрано из поливинилпирролидона, крахмала, сахара, микрокристаллической целлюлозы, гидроксипропилметилцеллюлозы, гидроксипропилцеллюлозы или гидроксиэтилцеллюлозы или любой их композиции; d) от 25 масс.% до 50 масс.% наполнителя по массе фармацевтической композиции; где наполнитель выбран из микрокристаллической целлюлозы, лактозы, сорбита, целлюлозы, кальция фосфата, крахмала или сахара или любой их композиции; и e) от 0,5 масс.% до 3 масс.% смазывающего вещества по массе композиции, где смазывающее вещество выбрано из металл стеарата и/или металл стеарилфумарата. Конкретными примерами наполнителей, дезинтегрирующих агентов, связующих веществ и смазывающих веществ являются вещества, как описано выше.
[0114] В еще одном дополнительном конкретном варианте осуществления фармацевтические композиции по изобретению содержат: a) от 35 масс.% до 75 масс.% Формы А соли HCl Соединения (1)·1/2H2O по массе фармацевтической композиции; b) от 3 масс.% до 7 масс.% кроскармеллозы по массе фармацевтической композиции; c) от 0,5 масс.% до 2 масс.% поливинилпирролидона по массе фармацевтической композиции; d) от 25 масс.% до 50 масс.% наполнителя по массе фармацевтической композиции; где наполнитель включает микрокристаллическую целлюлозу и лактозу; и e) от 0,5 масс.% до 3 масс.% металл стеарилфумарата по массе композиции. Конкретными примерами наполнителей, дезинтегрирующих агентов, связующих веществ и смазывающих веществ являются вещества, как описано выше.
[0115] В еще одном дополнительном конкретном варианте осуществления фармацевтические композиции по изобретению содержат: a) от 35 масс.% до 75 масс.% Формы А соли HCl Соединения (1)•1/2H2O по массе фармацевтической композиции; b) от 3 масс.% до 7 масс.% кроскармеллозы по массе фармацевтической композиции; c) от 0,5 масс.% до 2 масс.% поливинилпирролидона по массе фармацевтической композиции; d) от 25 масс.% до 50 масс.% наполнителя по массе фармацевтической композиции; где наполнитель включает микрокристаллическую целлюлозу и лактозу; и e) от 0,5 масс.% до 3 масс.% натрия стеарилфумарата по массе композиции. Конкретными примерами наполнителей, дезинтегрирующих агентов, связующих веществ и смазывающих веществ являются вещества, как описано выше.
[0116] В еще одном дополнительном конкретном варианте осуществления фармацевтические композиции по изобретению содержат: a) от 35 масс.% до 65 масс.% Формы А соли HCl Соединения (1)•1/2H2O по массе фармацевтической композиции; b) от 3 масс.% до 7 масс.% кроскармеллозы натрия по массе фармацевтической композиции; c) от 0,5 масс.% до 2 масс.% поливинилпирролидона со средней молекулярной массой от 3000 до 5000 по массе фармацевтической композиции; d) от 30 масс.% до 40 масс.% микрокристаллической целлюлозы по массе фармацевтической композиции; e) от 5 масс.% до 10 масс.% лактозы моногидрата по массе фармацевтической композиции; и f) от 1 масс.% до 3 масс.% натрия стеарилфумарата по массе композиции.
[0117] В еще одном аспекте фармацевтические композиции по изобретению представляют собой внутривенные (ВВ) составы, которые содержат Соединение (1) в воде и 0,01 M до 0,1 M фармацевтически приемлемого рН модифицирующего агента, такого как pH буферный агент. Обычно фармацевтические композиции включают: от 1 мг/мл до 20 мг/мл Соединения (1) в растворе. Чаще фармацевтические композиции включают: от 1 мг/мл до 10 мг/мл Соединения (1) или от 1 мг/мл до 5 мг/мл Соединения (1), такого как 2 мг/мл Соединения (1). В одном из вариантов осуществления в качестве источника Соединения (1) ВВ составов используется соль HCl Соединения (1)•хH2O (где x равно от 0 до 3). Не желая быть привязанным к конкретной теории соль HCl Соединения (1)•хH2O существует в виде Соединения (1) в растворе. Характерные примеры полиморфных форм солей HCl Соединения (1)•хH2O такие, как описано выше. В одном из конкретных вариантов осуществления используется Форма A, Форма D или Форма F соли HCl Соединения (1)•хH2О. В еще одном конкретном варианте осуществления используется Форма A соли HCl Соединения (1)•1/2H2О.
[0118] Характерные примеры рН модифицирующих агентов включают NaOH, KOH, NH4OH, HCl и буферные агенты. Характерные примеры буферных агентов включают карбонаты, бикарбонаты, моноосновные фосфаты, двухосновые фосфаты и ацетаты. Особый пример буферных агентов включает агенты фосфатного буфера, такие как мононатрия фосфат и динатрия фосфат. В одном из конкретных вариантов осуществления в качестве буферного агента используется смесь мононатрия фосфата и динатрия фосфата.
[0119] В одном из вариантов осуществления ВВ составы дополнительно содержат от 1 масс.% до 20 масс.% комплексообразующего агента по массе ВВ составов. Обычные комплексообразующие агенты включают циклодекстрины (например, альфа циклодекстрин, бета циклодекстрин, гамма циклодекстрин, гидроксипропил-бета-циклодекстрин, сульфо-бутилэфир-бета-циклодекстрин и полианионный бета-циклодекстрин), полисорбаты (например, Tween® 80) и касторовые масла (например, серии Cremophor®). Конкретные примеры циклодекстринов включают альфа циклодекстрин (например, Cavamax® W6), бета циклодекстрин (например, Cavamax® W7), гамма циклодекстрин (например, Cavamax® W8), гидроксипропил-бета-циклодекстрин (например, Cavasol® W7, Cavitron® W7), сульфо-бутилэфир-бета-циклодекстрин и полианионный бета-циклодекстрин (например, Captisol®). Конкретный пример полисорбата включает полиоксиэтилен (20) сорбитан монолеат (например, Tween® 80). Конкретные примеры касторовых масел включают полиокси 40 гидрированное касторовое масло (например, Cremophor® RH 40), полиокси 35 касторовое масло (например, Cremophor® EL). В одном из конкретных вариантов осуществления комплексообразующие агенты выбраны из полиокси 40 гидрированного касторового масла, полиокси 35 касторового масла, полианионного бета-циклодекстрина или гидроксипропил-бета-циклодекстрина или любой их композиции.
[0120] В некоторых вариантах осуществления ВВ составы дополнительно содержат декстрозу и/или маннит в качестве модификаторов тоничности.
[0121] В некоторых вариантах осуществления фармацевтические композиции по изобретению дополнительно содержат краситель, такой как Opadry II белый.
[0122] В некоторых вариантах осуществления фармацевтические композиции по изобретению представляют собой твердые дозированные формы, конкретно в таблетированных формах.
[0123] В еще одном аспекте настоящее изобретение охватывает способы получения фармацевтических композиций, описанных выше. В одном из вариантов осуществления способы включают получение смеси Соединения (1), которая включает: а) от 5 масс.% до 95 масс.% соли HCl Соединения (1)•хH2O (где х равно от 0 до 3) по массе фармацевтической композиции; и b) от 5 масс.% до 95 масс.% наполнителя по массе фармацевтической композиции. В еще одном варианте осуществления способы включают получение смеси Соединения (1), которая включает: a) от 20 масс.% до 80 масс.% соли HCl Соединения (1)•хH2O (где х равно от 0 до 3) по массе фармацевтической композиции; и b) от 20 масс.% до 80 масс.% наполнителя по массе фармацевтической композиции. В одном из конкретных вариантов осуществления стадия получения смеси Соединения (1) включает: получение гранул Соединения (1), смешивание i) от 60 масс.% до 90 масс.% соли HCl Соединения (1)•хH2O по массе гранул Соединения (1) и ii) внутригранулярного эксципиента, который включает от 10 масс.% до 40 масс.% наполнителя по массе гранул Соединения (1); и смешивание гранул Соединения (1) с внегранулярным эксципиентом, который включает от 15 масс.% до 40 масс.% наполнителя по массе фармацевтической композиции.
[0124] В еще одном конкретном варианте осуществления фармацевтические композиции по изобретению дополнительно включают связующее вещество, дезинтегрирующий агент и смазывающее вещество, а стадия получения смеси Соединения (1) включает: получение гранул Соединения (1), смешивание i) от 70 масс.% до 85 масс.% соли HCl Соединения (1)•хH2O по массе гранул Соединения (1) и ii) внутригранулярного эксципиента, который включает от 14 масс.% до 25 масс.% наполнителя по массе гранул Соединения (1) и от 1 масс.% до 5 масс.% дезинтегрирующего агента по массе гранул Соединения (1); и смешивание гранул Соединения (1) с внегранулярным эксципиентом, который включает от 15 масс.% до 40 масс.% наполнителя по массе фармацевтической композиции, от 0,5 масс.% до 5 масс.% дезинтегрирующего агента по массе фармацевтической композиции, и 0,5 масс.% до 5 масс.% смазывающего вещества по массе фармацевтической композиции.
[0125] В еще одном конкретном варианте осуществления, стадия получения смеси Соединения (1) включает: получение раствора связующего вещества, который включает воду и от 0,5 масс.% до 5 масс.% связующего вещества по массе гранул; получение внутригранулярной композиции с получением гранул Соединения (1), внутригранулярная композиция включает: i) от 70 масс.% до 85 масс.% соли HCl Соединения (1)•хH2O по массе гранул Соединения (1) и ii) внутригранулярный эксципиент, который включает от 14 масс.% до 25 масс.% наполнителя по массе гранул Соединения (1) и от 1 масс.% до 5 масс.% дезинтегрирующего агента по массе гранул Соединения (1); смешивание раствора связующего вещества и предгранулярной композиции с получением гранул Соединения (1); и смешивание гранул Соединения (1) с внегранулярным эксципиентом, который включает от 15 масс.% до 40 масс.% наполнителя по массе фармацевтической композиции, от 0,5 масс.% до 5 масс.% дезинтегрирующего агента по массе фармацевтической композиции, и 0,5 масс.% до 5 масс.% смазывающего вещества по массе фармацевтической композиции.
[0126] Гранулы Соединения (1) могут быть получены в любым подходящим путем, известным в данной области, таким как влажная грануляция с помощью двухшнекового экструдера или грануляция с большим усилием сдвига. В одном из вариантов осуществления влажная грануляция с помощью двухшнекового экструдера используется для получения гранул Соединения (1). В конкретном варианте осуществления стадия смешивания раствора связующего вещества и предгранулярной композиции включает: i) подачу предгранулярной композиции в двухшнековый экструдер; и ii) введение раствора связующего вещества в двухшнековый экструдер. В дополнительном конкретном варианте осуществления раствор связующего вещества включает воду в диапазоне от 30 масс.% до 50 масс.% по массе внутригранулярной композиции.
[0127] Гранулы Соединения (1) измельчают и измельченные гранулы смешивают с внегранулярной композицией, которая включает наполнитель и другие ингредиенты, если желательно (например, дезинтегрирующий агент и/или смазывающее вещество). В некоторых вариантах осуществления от 60 масс.% до 80 масс.% измельченных гранул Соединения (1) смешивают с от 10 масс.% до 30 масс.% наполнителя и, необязательно, дополнительно с от 1 масс.% до 15 масс.% дезинтегрирующего агента и/или от 0,25 масс.% до 5 масс.% смазывающего вещества по общей объединенной массе.
[0128] В случае композиций по изобретению для таблеток, способы дополнительно включат покрытие пленкой композиций для таблеток. Обычные вещества для покрытия пленкой включают один или несколько красителей, таких как Opadry II белый.
[0129] Способы получения ВВ составов, описанных выше, также предусмотрены в настоящем документе. Как правило, способы включают смешивание: a) соли HCl Соединения (1)•хH2O (где х равно 0-3); и b) от 0,01 M до 0,1 M рН модифицирующего агента с образованием от 1 мг/мл до 20 мг/мл Соединения (1) в воде. В некоторых вариантах осуществления образуется от 1 мг/мл до 10 мг/мл Соединения (1). Как описано выше для ВВ составов, другие ингредиенты, такие как комплексообразующие агенты и/или модифицирующие агенты также могут быть смешаны с солью HCl Соединения (1)•хH2O и рН модифицирующим агентом.
[0130] Примеры, включающие конкретные примеры, солей HCl Соединения (1)•хH2O, наполнителей, дезинтегрирующих агентов, связующих веществ и смазывающих веществ, рН модифицирующих агентов, комплексообразующих агентов и модифицирующих агентов, которые могут быть использованы в способах получения фармацевтических композиций представляют собой, каждый вариант и независимо друг от друга, как описано выше для фармацевтических композиций по изобретению.
[0131] Фармацевтические композиции по изобретению являются фармацевтически приемлемыми. Как используется в настоящем описании "фармацевтически приемлемый" обозначает инертность без чрезмерного ингибирования биологической активности активного(ых) соединения(ий) (например, соли HCl Соединения (1)•хH2O) и биосовместимость (например, нетоксичный, не вызывающий воспаление, неиммуногенный или лишенный других нежелательных реакций или побочных эффектов при введении индивиду).
[0132] Фармацевтические композиции по изобретению могут дополнительно включать один или несколько фармацевтически приемлемых носителей, отличных от тех, которые описаны выше. Фармацевтически приемлемые носители должны быть биосовместимы. Можно использовать стандартные методики получения фармацевтических составов.
[0133] Некоторые примеры веществ, которые могут служить фармацевтически приемлемыми носителями включают, но не ими не ограничиваяются, ионнообменные вещества, оксид алюминия, стеарат алюминия, лецитин, белки сыворотки (например, сывороточный альбумин человека), буферные веществ (например, фосфаты или глицин), смеси частичных глицеридов насыщенных жирных кислот растительного происхождения, воду, соли или электролиты (например, протамин сульфат, динатрий гидрофосфат, калия гидрофосфат, хлорид натрия или цинковые соли), коллоидный кремнезем, магния трисиликат, поливинилпирролидон, полиакрилаты, воски, блок полимеры полиэтиленполиоксипропилена, метилцеллюлозу, гидроксипропилметилцеллюлозу, жир шерсти, сахара, такие как лактоза, глюкоза и сахароза; крахмал, такой как кукурузный крахмал и картофельный крахмал; целлюлозу и ее производные, такие как натрия карбоксиметилцеллюлоза, этилцеллюлоза и целлюлоза ацетат; порошкообразный трагант; солод; желатин; тальк; наполнители, такие как масло какао и воска для суппозиториев; масла, такие как арахисовое масло, хлопковое масло; сафлоровое масло; кунжутное масло; оливковое масло; кукурузное масло и соевое масло; гликоли; такие как пропиленгликоль или полиэтиленгликоль; эфиры, такие как этилолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и алюминия гидроксид; альгиновая кислота; апирогенная вода; изотонический солевой раствор; раствор Рингера; этиловый спирт и фосфатные буферные растворы, а также другие нетоксичные совместимые смазывающие вещества, такие как натрия лаурилсульфат и магния стеарат, а также красители, агенты высвобождения, агенты покрытия, подсластители, ароматизоторы и отдушки, консерванты и антиоксиданты также могут быть представлены в композиции, в соответствии с решением составителя.
[0134] Для целей настоящего изобретения химические элементы идентифицированы в соответствии с Периодической системой Элементов, версия CAS, Handbook of Chemistry and Physics, 75-е издание. Кроме того, общие принципы органической химии описаны в "Organic Chemistry", Thomas Sorrell, University Science Books, Sausolito: 1999, и в "Marchʹs Advanced Organic Chemistry", 5-е издание, Ed.: Smith, M.B. and March, J., John Wiley & Sons, New York: 2001, полное содержание которых приведено в настоящем описании посредством ссылки
[0135] Если не указано иного, то структуры, изображенные в настоящем описании, также подразумевают все изомерные (например, энантиомерные, диастереомерные, цис-транс, конформационные и ротационные) формы структуры. Например, конфигурации R и S для каждого асимметричного центра, (Z) и (Е) изомеры двойной связи, а также (Z) и (Е) конформационные изомеры включены в настоящее изобретение, если только один из изомеров конкретно не изображен. Как понятно специалисту в данной области техники, заместитель может свободно вращаться вокруг любой связи, способной к вращению. Например, заместители, изображенные как  , также представляют
, также представляют  .
.
[0136] Следовательно, отдельные стереохимические изомеры, а также энантиомерные, диастереомерные, цис/транс, конформационные и ротационные смеси соединений по настоящему изобретению находятся в рамках настоящего изобретения.
[0137] Если не указано иное, то все таутомерные формы соединений по настоящему изобретению входят в раки настоящего изобретения.
[0138] Кроме того, если не указано иного, то структуры, изображенные в настоящем описании, также предполагают соединения, которые отличаются только наличием одного или нескольких изотопно обогащенных атомов. Например, соединения, имеющие представленные структуры, за исключением замены водорода дейтерием или тритием, или замены углерода на 13C- или 14C-обогащенный углерод, входят в рамки настоящего изобретения. Такие соединения могут быть использованы, например, в качестве аналитических инструментов или зондов в биологических анализах. Такие соединения, особенно аналоги дейтерия (D), также могут быть терапевтически эффективными.
[0139] Соединения, описанные в настоящем документе, определены своими химическими структурами и/или химическими наименованиями. В случае, если соединение ссылается как на химическую структуру, так и на химическое название, и химическая структура и химическое название противоречат друг другу, то химическая структура преобладает при идентификации соединения.
[0140] Как будет понятно специалистам в данной области, соединения по настоящему изобретению могут существовать в виде стереоизомеров (например, оптические (+ и -), геометрические (цис и транс) и конформационные изомеры (аксиальные и экваториальные). Все такие стереоизомеры входят в рамки настоящего изобретения.
[0141] Как будет понятно специалистам в данной области соединения по настоящему изобретению могут содержать хиральный центр. Соединения формулы могут, таким образом, существовать в виде двух различных оптических изомеров (то есть (+) или (-) энантиомеров). Все такие энантиомеры и их смеси, включая рацемические смеси, входят в рамки настоящего изобретения. Единичный оптический изомер или энантиомер может быть получен способом, хорошо известным в данной области, таким как хиральная ВЭЖХ, ферментативное разделение и с помощью хирального вспомогательного агента.
[0142] В одном из вариантов осуществления соединения по настоящему изобретению представлены в форме единичного энантиомера, который по меньшей мере на 95%, по меньшей мере на 97% и по меньшей мере на 99% свободен от соответствующего энантиомера.
[0143] В еще одном дополнительном варианте осуществления соединения по настоящему изобретению находятся в форме (+) энантиомера, по меньшей мере на 95% свободного от соответствующего (-) энантиомера.
[0144] В еще одном дополнительном варианте осуществления соединения по настоящему изобретению находятся в форме (+) энантиомера, по меньшей мере на 97% свободного от соответствующего (-) энантиомера.
[0145] В еще одном дополнительном варианте осуществления соединения по настоящему изобретению находятся в форме (+) энантиомера, по меньшей мере на 99% свободного от соответствующего (-) энантиомера.
[0146] В еще одном дополнительном варианте осуществления соединения по настоящему изобретению находятся в форме (-) энантиомера, по меньшей мере на 95% свободного от соответствующего (+) энантиомера.
[0147] В еще одном дополнительном варианте осуществления соединения по настоящему изобретению находятся в форме (-) энантиомера, по меньшей мере на 97% свободного от соответствующего (+) энантиомера.
[0148] В еще одном дополнительном варианте осуществления соединения по настоящему изобретению находятся в форме (-) энантиомера, по меньшей мере на 99% свободного от соответствующего (+) энантиомера.
[0149] III. ПРИМЕНЕНИЕ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ
[0150] Один из аспектов настоящего изобретения в целом относится к применению фармацевтически приемлемых композиций, описанных выше, для ингибирования репликации вирусов гриппа в биологическом образце или у пациента, для снижения количества вируса гриппа (снижение титра вируса) в биологическом образце или у пациента, а также для лечения гриппа у пациента. Здесь и далее, если не указано иного, то различные твердые формы (например, полиморфы солей HCl Соединения (1) или их фармацевтически приемлемые соли), описанные выше, также называются, как правило, соединениями.
[0151] В одном варианте осуществления настоящее изобретение в целом относится к применению соединений, описанных в настоящем документе (например, в фармацевтически приемлемых композициях) для любого из применений, указанных выше.
[0152] В еще одном варианте осуществления соединения, описанные в настоящем документе, могут быть использованы для снижения титра вируса в биологическом образце (например, инфицированной клеточной культуре) или в организме человека (например, вирусный титр в легких у пациента).
[0153] Термины "состояние, опосредованное вирусом гриппа", "инфекция, вызванная гриппом" или "грипп", как используется в настоящем документе, являются взаимозаменяемыми и обозначают заболевание, вызванное инфекцией вирусом гриппа.
[0154] Грипп является инфекционным заболеванием, которое поражает птиц и млекопитающих, вызванным вирусами гриппа. Вирусы гриппа представляют собой РНК-вирусы семейства Orthomyxoviridae, который состоит из пяти родов: Вирус гриппа A, вирус гриппа B, вирус гриппа C, вирус ISA и вирус Тогото. Вирус гриппа рода А имеет один вид, вирус гриппа А, который может быть подразделен на различные серотипы на основе гуморального (антительного) ответа на эти вирусы: H1N1, H2N2, H3N2, H5N1, H7N7, H1N2, H9N2, H7N2, H7N3 и H10N7. Дополнительные примеры вируса гриппа А включают H3N8 и H7N9. Род вируса гриппа В имеет один вид, вирус гриппа В. Грипп B почти исключительно заражает людей и встречается реже, чем грипп А. Вирус гриппа рода С имеет один вид, вирус гриппа C, который инфицирует людей и свиней и может привести к тяжелой болезни и местным эпидемиям. При этом, грипп С встречается реже, чем другие типы и, как правило, по-видимому вызывает легкое заболевание у детей.
[0155] В некоторых вариантах осуществления настоящего изобретения, грипп или вирусы гриппа связаны с вирусом гриппа А. В некоторых конкретных вариантах осуществления настоящего изобретения, вирус гриппа А представляет собой H1N1, H2N2, H3N2 или H5N1. В некоторых конкретных вариантах осуществления настоящего изобретения, вирус гриппа А представляет собой H1N1, H3N2, H3N8, H5N1 и H7N9. В некоторых конкретных вариантах осуществления настоящего изобретения вирус гриппа А представляет собой H1N1, H3N2, H3N8 и H5N1.
[0156] У людей общими симптомами гриппа являются озноб, лихорадка, фарингит, боли в мышцах, сильная головная боль, кашель, слабость и общее недомогание. В более серьезных случаях грипп вызывает пневмонию, которая может привести к летальному исходу, особенно у маленьких детей и пожилых людей. Несмотря на то, что грипп часто путают с обычной простудой, он является гораздо более тяжелым заболеванием и вызван другим типом вируса. Грипп может вызвать тошноту и рвоту, особенно у детей, но эти симптомы более характерны для не имеющего к гриппу отношения гастроэнтериту, который иногда называют "желудочный грипп" или "24-часовой грипп".
[0157] Симптомы гриппа могут начаться совершенно неожиданно через день или два после заражения. Обычно первыми симптомами являются озноб или чувство холода, но и лихорадка также характерна для ранней стадии, при которой температура тела находится в пределах от 38 до 39°С (приблизительно 100-103°F). Многие люди настолько больны, что прикованы к постели в течение нескольких дней, чувствуя недомогание и боли во всем теле, которые сильнее в спине и ногах. Симптомы гриппа могут включать в себя: ломоту в теле, особенно в суставах и горле, невероятный холод и лихорадку, усталость, головную боль, раздражение в глазах и слезотечение, покраснение глаз, кожи (особенно лица), рта, горла и носа, боли в животе (у детей с гриппом B). Симптомы гриппа являются неспецифическими, сходными со многими возбудителями ("гриппоподобные заболевания"). Как правило, лабораторные данные необходимы для того, чтобы подтвердить диагноз.
[0158] Термины "заболевание", "расстройство" и "состояние" могут быть использованы взаимозаменяемо в настоящем документе для обозначения вируса гриппа, опосредованного медицинским или патологическим состоянием.
[0159] Используемые в настоящем описании термины "индивид" и "пациент" используются взаимозаменяемо. Термины «индивид» и «пациент» относятся к животному (например, птицам, таким как цыпленок, перепел или индейка, или млекопитающим), конкретно "млекопитающее" включает не-приматов (например, корова, свинья, лошадь, овца, кролик, морская свинка, крыса, кошка, собака, мышь) и приматов (например, обезьяна, шимпанзе и человек) и, более конкретно, человек. В одном из вариантов осуществления индивидом является животное, такое как сельскохозяйственное животное (например, лошадь, корова, свиньи или овцы) или домашнее животное (например, собака, кошка, морская свинка или кролик). В предпочтительном варианте осуществления индивидом является "человек".
[0160] Термин "биологический образец", как используется в настоящем документе, включает в себя, но ими не ограничивается, клеточные культуры или их экстракты; материал биопсии, полученный из млекопитающего или его экстрактов; кровь, слюну, мочу, фекалии, сперму, слезы или другие жидкости организма или их экстракты.
[0161] Используемый в настоящем описании термин "множественность инфекции" или "MOI" является отношением инфекционных агентов (например, фага или вируса) к мишеням инфекции (например, клеткам). Например, при ссылке на группу клеток, инокулированных инфекционными вирусными частицами, множественность инфекции или MOI представляет собой отношение, определенное количеством инфекционных вирусных частиц, внесенных в лунку, деленное на число клеток-мишеней, присутствующих в этой лунке.
[0162] Используемый в настоящем описании термин "ингибирование репликации вирусов гриппа» включает как уменьшение количества вирусных репликаций (например, сокращение по меньшей мере на 10%), так и полное прекращение вирусной репликации репликации (т.е. 100% снижение репликации вируса). В некоторых вариантах осуществления репликация вирусов гриппа ингибируется по меньшей мере на 50%, по меньшей мере на 65%, по меньшей мере на 75%, по меньшей мере на 85%, по меньшей мере на 90% или по меньшей мере на 95%.
[0163] Репликация вируса гриппа может быть измерена любым подходящим способом, известным в данной области. Например, титр вируса гриппа в биологическом образце (например, в инфицированной клеточной культуре) или в организме человека (например, вирусный титр в легких пациента) может быть измерен. Более конкретно, для анализов на основе клеток, в каждом случае клетки культивируют in vitro, вирус добавляют к культуре в присутствии или в отсутствие тестируемого агента, и после соответствующего периода времени вычисляют вирус-зависимую конечную точку. Для обычных анализов могут быть использованы клетки Мадин-Дарби почек собак (MDCK) и штамм вируса гриппа, A/Puerto Rico/8/34, адаптированный для стандартной тканевой культуры. Первый тип клеточного анализа, который может быть использован по настоящему изобретению, основан на гибели инфицированных клеток-мишеней, процесс, называемый цитопатический эффект (CPE), когда вирусная инфекция вызывает истощение клеточных ресурсов и, в конечном итоге, лизис клетки. При первом типе клеточного анализа низкую фракцию клеток в лунках планшета для микротитрования инфицируют (как правило, от 1/10 до 1/1000), позволяя вирусу совершать несколько раундов репликации в течение 48-72 часов, затем количество погибших клеток измеряют, используя уменьшение количества клеточной АТФ по сравнению с неинфицированными контролями. Второй тип клеточного анализа, который может быть использован по настоящему изобретению, основан на увеличении количества вирусспецифических молекул РНК в инфицированных клетках, при этом непосредственно измеряют уровни РНК, используя метод гибридизации ДНК с разветвленной цепью (bДНК). Во втором типе клеточного анализа сначала инфицируют небольшое количество клеток в лунках планшета для микротитрования, вирус оставляют реплицироваться в инфицированных клетках и распространяться на дополнительные раунды клеток, а затем клетки лизируются и в них измеряют содержание РНК вируса. Этот анализ быстро останавливают, как правило, через 18-36 часов, при этом все клетки-мишени остаются жизнеспособными. Вирусную РНК количественно оценивают по гибридизации с конкретными олигонуклеотидными зондами, фиксированными в лунках планшета для анализа, а затем усиление сигнала путем гибридизации с дополнительными зондами, связанными с репортерным ферментом.
[0164] Используемый в настоящем документе термин "титр вируса (или титр)" является мерой концентрации вируса. При тестировании титров можно использовать серийные разведения для получения приблизительной качественной информации из аналитической процедуры, которая по своей сути дает только положительную или отрицательную оценку. Титр соответствует самому высокому фактору разбавления, который по-прежнему дает положительный результат; например, положительные показания в первые 8 последовательных двукратных разведениях переводят в титр 1:256. Конкретным примером является вирусный титр. Для определения титра, несколько было сделано несколько разведений, например, 10-1, 10-2, 10-3, 10-4, 10-5, 10-6, 10-7, 10-8 или тому подобное. Самая низкая концентрация вируса, при которой все еще происходит инфицирование клеток, является вирусным титром.
[0165] Используемые в настоящем описании термины "лечить", "лечение" и "лечащий" относятся как терапевтическим, так и к профилактическим мероприятиям. Например, терапевтические мероприятия включают устранение или снижение прогрессирования, тяжести и/или продолжительности состояния, вызванного вирусами гриппа, или устранение одного или более симптомов (в частности, одного или более заметных симптомов) состояния, опосредованного вирусами гриппа, в результате проведения одной или нескольких терапевтических процедур (например, введения одного или более терапевтических средство, таких как соединения или композиции по настоящему изобретению). В конкретных вариантах осуществления, терапевтическое мероприятие включает в себя улучшение по крайней мере одного измеримого физического параметра состояния, опосредованного вирусом гриппа. В других вариантах осуществления терапевтическое мероприятие включает ингибирование прогрессирования состояния, опосредованного вирусом гриппа, либо физически, например, стабилизация заметного симптома, физиологически, например, стабилизация физического параметра, или их обоих. В других вариантах осуществления терапевтическое мероприятие включает снижение или стабилизацию инфекций, опосредованных вирусом гриппа. Противовирусные препараты могут быть использованы в амбулаторных условиях для лечения людей, которые уже больны гриппом, для уменьшения тяжести симптомов и уменьшения количества дней, в течение которых длится болезнь.
[0166] Термин "химиотерапия" относится к применению лекарственных средств, например, низкомолекулярных препаратов (но не "вакцин") для лечения расстройства или заболевания.
[0167] Термины "профилактика" или "профилактическое применение" и "профилактическое мероприятие", как используется в настоящем описании, относятся к любой медицинской процедуре или процедуре общественного здравоохранения, целью которых является предотвращение, а не лечение или излечение болезни. Используемые в настоящем описании термины "профилактика", "предотвращать" и "профилактический" относятся к уменьшению риска приобретения или развития данного состояния или к уменьшению или ингибированию рецидива или указанного состояния у индивида, который не болен, но который был или мог быть рядом с больным человеком. Термин "химиопрофилактика" относится к применению лекарственных средств, например, низкомолекулярных препаратов (но не "вакцин") для профилактики расстройства или заболевания.
[0168] Используемое в настоящем описании профилактическое применение включает применение в ситуациях, при которых была обнаружена вспышка инфекции, для предотвращения заражения или распространения ее в местах, где большое число людей, которые подвергаются высокому риску серьезных осложнений гриппа, живут в тесном контакте друг с другом (например, в больничной палате, детском саду, тюрьме, доме престарелых и тому подобное). Профилактическое применение также включает применение среди групп населения, которые нуждаются в защите от гриппа, но которые либо не получают защиту после вакцинации (например, из-за слабой иммунной системы), или если вакцина для них недоступна, или если они не могут быть вакцинированы из-за побочных эффектов. Такое применение также включает применение в течение двух недель после вакцинации, так как в течение этого времени вакцина все еще неэффективна. Профилактическое применение также может включать лечение индивида, который не болен гриппом или который не относится к группе повышенного риска осложнений, для уменьшения вероятности заражения гриппом и передачи гриппа индивиду с высоким риском при тесном контакте с ним (например, медицинские работники, работники дома престарелых и тому подобное).
[0169] По данным CDC США "вспышка" определяется как внезапное увеличение острого фибриллярного респираторного заболевания (AFRI), происходящее в течение периода от 48 до 72 часов, в группе людей, которые находятся в непосредственной близости друг от друга (например, в одной и той же области пансиона, в одном и том же доме и тому подобное) по сравнению с нормальной фоновой скоростью или если любой индивидуум в популяции, в отношении которого был проведен тест, оказался положительным на грипп. Один случай гриппа, подтвержденный любым способом, считается вспышкой.
[0170] "Кластер" определяется как группа из трех или более случаев AFRI, происходящих в течение периода от 48 до 72 часов в группе людей, которые находятся в непосредственной близости друг от друга (например, в одном и том же пансионе, в одном и том же доме и тому подобное).
[0171] Используемый в настоящем описании термин "источник заболевания", "первичный случай" или "пациент зеро" означает первого пациента в популяционном образце эпидемиологического исследования. При использовании в целом относительно таких пациентов в эпидемиологических исследованиях, этот термин не выделяется заглавными буквами. Если этот термин используется для обозначения конкретного индивида вместо имени этого индивида в докладе о конкретном исследовании, то термин пишется заглавными буквами как Пациент Зеро. Часто ученые ищут носителя заболевания для определения того как болезнь распространяется и в каком источнике находится заболевание между вспышками. Следует обратить внимание, что носитель заболевания является первым пациентом, который указывает на наличие вспышки. Могут быть обнаружены более ранние случаи и их обозначают как первичные, вторичные, третичные и тому подобное.
[0172] В одном варианте осуществления способы по настоящему изобретению являются профилактической или "упреждающей" мерой для пациента, в частности, для человека с предрасположенностью к осложнениям в результате инфекции, вызванной вирусом гриппа. Термин "упреждающий", используемый в настоящем описании, например, для упреждающего применения, "превентивно" и тому подобное, является профилактическим применением в тех ситуациях, при которых "источник заболевания" или "вспышка" была подтверждена, для предотвращения распространение инфекции в остальной части сообщества или группы популяции.
[0173] В другом варианте осуществления способы по изобретению применяются в качестве "упреждающей" меры к членам сообщества или группы популяции, в частности к людям, с тем, чтобы предотвратить распространение инфекции.
[0174] Используемый в настоящем описании термин "эффективное количество" относится к количеству, достаточному для того, чтобы вызвать желаемый биологический ответ. В настоящем изобретении желаемым биологическим ответом является ингибирование репликации вируса гриппа, снижение количества вирусов гриппа или уменьшение или снижение тяжести заболевания, продолжительности, прогрессии или начала инфекции вирусном гриппа, предотвращение распространения инфекции вируса гриппа, предотвращение повторения, развития, начала или прогрессирования симптомов, связанных с вирусом гриппа, или повышение или улучшение профилактического или терапевтического эффекта(ов) другого терапевтического мероприятия против гриппозных инфекций. Точное количество соединения, вводимое индивиду, будет зависеть от способа введения, типа и тяжести инфекции и от особенностей индивида, таких как общее состояние, возраст, пол, масса тела и толерантность к лекарственным средствам. Специалист будет в состоянии определить соответствующие дозировки в зависимости от этих и других факторов. При совместном введении с другими противовирусными средствами, например, при совместном введении с лекарствами против гриппа, "эффективное количество" второго агента будет зависеть от типа используемого препарата. Подходящие дозировки известны для разрешенных к применению средств и могут быть скорректированы специалистом в соответствии с состоянием индивида, типом состояния(ий), в отношении которого проводится лечение, и количеством соединения, описанного в настоящем документе. В тех случаях, когда количество четко не указано, эффективное количество следует предполагать. Например, соединения, описанные в настоящем описании, могут быть введены индивиду в интервале доз приблизительно от 0,01 до 100 мг/кг массы тела/день для терапевтического или профилактического лечения.
[0175] Как правило, режимы дозирования могут быть выбраны в соответствии с различными факторами, включая заболевание, в отношении которого проводится лечение, и тяжесть заболевания; активность конкретного используемого соединения; специфическая используемая композиция; возраст, масса тела, общее состояние здоровья, пол и режим питания пациента; время введения, путь введения и скорость экскреции специфического используемого соединения; функции почек и печени индивида; и конкретное используемое соединение или его соль, продолжительность лечения; препараты, используемые в комбинации или совместно со специфическим используемым соединением, и тому подобные факторы, хорошо известные в области медицины. Опытный специалист может легко определить и выписать эффективное количество описанных в настоящем описании соединений, необходимых для лечения, предотвращения, ингибирования (полностью или частично) или остановить прогрессирование заболевания.
[0176] Доза соединений, описанных в настоящем описании, может находиться в диапазоне от 0,01 до 100 мг/кг массы тела/сутки, от 0,01 до 50 мг/кг массы тела/день, от 0,1 до 50 мг/кг массы тела/день, или от 1 до 25 мг/кг массы тела масса тела/день. Понятно, что общее суточное количество можно вводить в виде однократной дозы или можно вводить в виде многократных доз, например, два раза в день (например, каждые 12 часов), три раза в день (например, каждые 8 часов) или четыре раза в день (например, каждые 6 часов).
[0177] В некоторых вариантах осуществления дозы соединений, описанных в настоящем документе (например, Соединение (1) и его фармацевтически приемлемые соли, включая различные твердые формы (например, Форма A соли HCl Соединения (1)•1/2H2O, Форма F соли HCl Соединения (1)•3H2O, Форма D соли HCl Соединения (1)) находятся в диапазоне от 100 мг до 1600 мг, например, от 400 мг до 1600 мг или от 400 мг до 1200 мг. Каждая доза может приниматься один раз в день (QD), два раза в день (например, каждые 12 часов (BID)) или три раза в день (например, q8h (TID)). Следует отметить, что могут быть использованы любые комбинации QD, BID и TID, если желательно, например, BID на 1-й день, а затем QD после этого, или, если ударная доза применяется на 1-й день, BID на 2-й день, а затем QD.
[0178] В одном из конкретных вариантов осуществления дозы соединений, описанных в настоящем документе, составляют от 400 мг до 1600 мг, от 400 мг до 1200 мг или от 600 мг до 1200 мг один раз в день. В другом конкретном варианте осуществления дозы соединений, описанных в настоящем документе, составляют от 400 мг до 1600 мг, от 400 мг до 1200 мг или от 300 мг до 900 мг два раза в день. В еще одном конкретном варианте осуществления дозы соединений, описанных в настоящем документе, составляют от 400 мг до 1000 мг один раз в день. В еще одном конкретном варианте осуществления, дозы соединений, описанных в настоящем документе, составляют от 600 мг до 1000 мг один раз в день. В еще одном конкретном варианте осуществления дозы соединений, описанных в настоящем документе, составляют от 600 мг до 800 мг один раз в день. В еще одном конкретном варианте осуществления дозы соединений, описанных в настоящем документе, составляют от 400 мг до 800 мг два раза в день (например, от 400 мг до 800 мг каждые 12 часов). В еще одном конкретном варианте осуществления дозы соединений, описанных в настоящем документе, составляют от 400 мг до 600 мг два раза в день.
[0179] В некоторых вариантах осуществления используется схема ударных доз. В одном из конкретных вариантов осуществления ударная доза, составляющая от 400 мг до 1600 мг, используется в день 1 лечения. В еще одном конкретном варианте осуществления ударная доза, составляющая от 600 мг до 1600 мг, используется в день 1 лечения. В еще одном конкретном варианте осуществления ударная доза, составляющая от 800 мг до 1600 мг, используется в день 1 лечения. В еще одном конкретном варианте осуществления, ударная доза, составляющая от 900 мг до 1600 мг, используется в день 1 лечения. В еще одном конкретном варианте осуществления, ударная доза, составляющая от 900 мг до 1200 мг, используется в день 1 лечения. В еще одном конкретном варианте осуществления, ударная доза, составляющая 900 мг, используется в день 1 лечения. В еще одном конкретном варианте осуществления, ударная доза, составляющая 1000 мг, используется в день 1 лечения. В еще одном конкретном варианте осуществления, ударная доза, составляющая 1200 мг, используется в день 1 лечения.
[0180] В одном из конкретных вариантов осуществления в режиме доз соединений, описанных в настоящем документе, используется ударная доза, составляющая от 600 мг до 1600 мг, на день 1 и обычная доза, равная от 300 мг до 1200 мг, в течение оставшегося периода лечения. Каждую обычную дозу можно принимать один раз в день, два раза в день, или три раза в день, или в любой их комбинации. В дополнительном специфическом варианте осуществления используется ударная доза, составляющая от 900 мг до 1600 мг, например, 900 мг, 1200 мг или 1600 мг. В еще одном дополнительном специфическом варианте осуществления используется ударная доза, составляющая от 900 мг до 1200 мг, например, 900 мг или 1200 мг. В еще одном дополнительном конкретном варианте осуществления используется обычная доза, составляющая от 400 мг до 1200 мг, например, 400 мг, 600 мг или 800 мг в течение оставшегося периода лечения. В еще одном дополнительном конкретном варианте осуществления используется обычная доза, равная от 400 мг до 1000 мг, в течение оставшегося периода лечения. В еще одном дополнительном конкретном варианте осуществления используется обычная доза, равная от 400 мг до 800 мг, в течение оставшегося периода лечения. В еще одном дополнительном конкретном варианте осуществления используется обычная доза, равная от 300 мг до 900 мг, два раза в день. В еще одном дополнительном конкретном варианте осуществления используется обычная доза, равная от 600 мг до 1200 мг один раз в день. В еще одном дополнительном конкретном варианте осуществления используется обычная доза, равная 600 мг, два раза в день на день 2, а затем 600 мг один раз в день в течение оставшегося периода лечения.
[0181] Для терапевтического мероприятия соединения, описанные в настоящем документе, могут вводиться пациенту в течение, например, 48 часов (или в пределах 40 часов или менее чем в течение 2-х дней или менее чем в течение 1,5 дней или в течение 24 часов) после появления симптомов (например, заложенности носа, боли в горле, кашля, боли, усталости, головной боли, озноба и/или испарены). Альтернативно, для терапевтического мероприятия соединения, описанные в настоящем документе, могут быть введены пациенту в течение, например, 96 часов после появления симптомов. Терапевтическое мероприятие может длиться в течение любого подходящего периода, например, в течение 3-х дней, 4-х дней, 5-ти дней, 7-ми дней, 10-ти дней, 14-ти дней и тому подобное. Для профилактического мероприятия во время вспышки заболевания соединения, описанные в настоящем документе, могут быть введены пациенту в течение, например, 2-х дней до появления симптомов в источнике заболевания, а также может быть продолжено в течение любого подходящего периода, например, в течение 7-ми дней, 10-ти дней, 14-ти дней, 20-ти дней, 28-ми дней, 35-ти дней, 42-х дней и тому подобное, вплоть до всего сезона гриппа. Сезон гриппа является ежегодно повторяющимся периодом времени, который характеризуется преобладанием вспышек гриппа. Активность гриппа иногда может быть предсказана и даже определена географически. Хотя начало основной активности гриппа в каждом сезоне изменяется в зависимости от местоположения, в любом конкретном месте эти незначительные эпидемии обычно длятся 3-4 недели до пика, а еще через 3-4 недели значительно уменьшаются. Как правило, Центр по контролю и профилактике заболеваний (CDC) собирает, накапливает и анализирует информацию о круглогодичной активности гриппа в Соединенных Штатах и готовит еженедельный отчет с октября до середины мая.
[0182] В одном из вариантов осуществления, терапевтическое мероприятие длится в течение от 1-го дня до всего сезона гриппа. В одном из конкретных вариантов осуществления терапевтическое мероприятие длится в течение от 3-х дней до 14-ти дней. В еще одном конкретном варианте осуществления терапевтическое мероприятие длится в течение от 5-ти дней до 14-ти дней. В еще одном конкретном варианте осуществления терапевтическое мероприятие длится в течение от 3-х дней до 10-ти дней. В еще одном конкретном варианте осуществления терапевтическое мероприятие длится в течение от 4-х дней до 10-ти дней. В еще одном конкретном варианте осуществления, терапевтическое мероприятие длится в течение от 5-ти дней до 10-ти дней. В еще одном конкретном варианте осуществления, терапевтическое мероприятие длится в течение от 4-х дней до 7-ми дней (например, 4 дня, 5 дней, 6 дней или 7 дней). В еще одном конкретном варианте осуществления терапевтическое мероприятие длится в течение от 5-ти дней до 7-ми дней (например, 5 дней, 6 дней или 7 дней). В одном из конкретных вариантов осуществления профилактическое лечение длится вплоть до всего сезона гриппа.
[0183] В одном из конкретных вариантов осуществления соединения, описанные в настоящем документе, вводили пациенту в течение от 3-х дней до 14-ти дней (например, от 5-ти дней до 14-ти дней) с ударной дозой, составляющей от 900 мг до 1600 мг, на 1 день и с обычной дозой, равной от 300 мг до 1200 мг, в течение оставшегося периода лечения. В еще одном конкретном варианте осуществления соединения, описанные в настоящем документе, вводили пациенту в течение от 3-х дней до 14-ти дней (например, от 5-ти дней до 14-ти дней) с ударной дозой, составляющей от 900 мг до 1200 мг, на 1 день и с обычной дозой, равной от 400 мг до 1000 мг, в течение оставшегося периода лечения. В еще одном конкретном варианте осуществления, соединения, описанные в настоящем документе, вводили пациенту в течение от 3 дней до 14 дней (например, от 5-ти дней до 14-ти дней) с ударной дозой, составляющей 900 мг до 1200 мг, на 1 день и с обычной дозой, равной от 400 мг до 800 мг, в течение оставшегося периода лечения. В еще одном конкретном варианте осуществления, соединения, описанные в настоящем документе, вводили пациенту в течение от 3 дней до 14 дней (например, от 5-ти дней до 14-ти дней) с ударной дозой, составляющей 900 мг до 1200 мг, на 1 день и с обычной дозой, равной от 400 мг до 800 мг, в течение оставшегося периода лечения. Каждая доза может быть принята один раз в день, два раза в день или три раза в день или в виде любой их композиции.
[0184] В одном из конкретных вариантов осуществления соединения, описанные в настоящем документе, вводили пациенту в течение от 3-х дней до 14-ти дней с ударной дозой, составляющей от 900 мг до 1600 мг, на 1 день и с обычной дозой, равной от 600 мг до 1000 мг, один раз в день в течение оставшегося периода лечения. В еще одном конкретном варианте осуществления соединения, описанные в настоящем документе, вводили пациенту в течение от 3-х дней до 14-ти дней с ударной дозой, составляющей от 900 мг до 1200 мг, на 1 день и с обычной дозой, равной от 600 мг до 800 мг (например, 600 мг, 650 мг, 700 мг, 750 мг или 800 мг) один раз в день в течение оставшегося периода лечения. В некоторых вариантах осуществления длительность лечения составляет от 4-х дней до 10-ти дней, от 5-ти дней до 10-ти дней или от 5-ти дней до 7-ми дней.
[0185] В одном из конкретных вариантов осуществления соединения, описанные в настоящем документе, вводили пациенту в течение от 3-х дней до 14-ти дней с ударной дозой, составляющей от 900 мг до 1600 мг, на 1 день и с обычной дозой, равной от 400 мг до 800 мг, два раза в день в течение оставшегося периода лечения. В еще одном конкретном варианте осуществления соединения, описанные в настоящем документе, вводили пациенту в течение от 3-х дней до 14-ти дней с ударной дозой, составляющей от 900 мг до 1200 мг, на день 1 и с обычной дозой, равной от 400 мг до 600 мг (например, 400 мг, 450 мг, 500 мг, 550 мг или 600 мг) два раза в день в течение оставшегося периода лечения. В некоторых вариантах осуществления длительность составляет от 4-х дней до 10-ти дней, от 5-ти дней до 10-ти дней или от 5-ти дней до 7-ми дней.
[0186] В одном из конкретных вариантов осуществления соединения, описанные в настоящем документе, вводили пациенту в течение 4-х дней или 5-ти дней с ударной дозой, составляющей от 900 мг до 1200 мг (например, 900 мг или 1200 мг), на 1 день и с обычной дозой, равной от 400 мг до 600 мг (например, 400 мг или 600 мг) два раза в день в течение оставшегося периода лечения (например, от 2-х до 4-х дней, или от 2-х до 5-ти дней). В еще одном конкретном варианте осуществления соединения, описанные в настоящем документе, вводили пациенту в течение 4-х дней или 5-ти дней с ударной дозой, составляющей от 900 мг до 1200 мг (например, 900 мг или 1200 мг), на 1 день и с обычной дозой, равной от 600 мг до 800 мг (например, 600 мг или 800 мг), один раз в день в течение оставшегося периода лечения.
[0187] В настоящем изобретении могут быть использованы различные типы способов введения, и они подробно описаны ниже в разделе под названием "Способы введения".
[0188] IV. КОМБИНИРОВАННАЯ ТЕРАПИЯ
[0189] В способе или в фармацевтической композиции по изобретению эффективное количество может быть достигнуто при использовании соединения по настоящему изобретению (включая его фармацевтически приемлемую соль или сольват (например, гидрат)) отдельно или в комбинации с дополнительным подходящим терапевтическим средством, например, противовирусным средством или вакциной. Если используется "комбинированная терапия", то эффективное количество может быть достигнуто с помощью первого количества соединения по изобретению, и второго количества дополнительного подходящего терапевтического средства (например, противовирусного агента или вакцины).
[0190] В еще одном варианте осуществления настоящего изобретения как соединение по настоящему изобретению, так и дополнительное терапевтическое средство вводят в эффективном количестве (то есть, каждое из них находится в количестве, которое будет терапевтически эффективным при самостоятельном введении). В еще одном варианте осуществления как соединение по настоящему изобретению, так и дополнительное терапевтическое средство вводят в количестве, которое само по себе не обеспечивает терапевтический эффект (субтерапевтическая доза). В еще одном варианте осуществления соединение по настоящему изобретению можно вводить в эффективном количестве, а дополнительное терапевтическое средство вводят в субтерапевтической дозе. В еще одном варианте осуществления соединение по настоящему изобретению можно вводить в субтерапевтической дозе, а дополнительное терапевтическое средство, например подходящее противораковое терапевтическое средство, вводят в эффективном количестве.
[0191] Используемый в настоящем описании термины "в комбинации" или "комбинирвоанное введение" могут быть использованы взаимозаменяемо для обозначения использования более чем одного медикамента (например, одного или более профилактических и/или терапевтических средств). Использование терминов не ограничивает порядок, в котором медикамент (например, профилактическое и/или терапевтическое средство) вводят индивиду.
[0192] Совместное введение включает введение первого и второго количества соединений для совместного введения, по существу, одновременно, например, в одной фармацевтической композиции, например, в капсуле или таблетке, имеющей фиксированное соотношение первого и второго количества, или в нескольких, раздельных капсулах или таблетках для каждого из них. Кроме того, такое совместное введение также включает использование каждого соединения последовательно в любом порядке.
[0193] В одном из вариантов осуществления настоящее изобретение относится к способам комбинированной терапии для ингибирования репликации вируса гриппа в биологических образцах или в организме пациентов, или для лечения или профилактики инфекций, вызванных вирусом гриппа, у пациентов, используя соединения, описанные в настоящем документе. Соответственно, фармацевтические композиции по настоящему изобретению также включают композиции, которые содержат ингибитор репликации вируса гриппа по настоящему изобретению в комбинации с противовирусным соединением, проявляющим противовирусную активность в отношении вируса гриппа.
[0194] Способы применения соединений, описанных в настоящем документе, и композиций по изобретению также включают комбинацию химиотерапевтического средства и соединения или композиции по изобретению, или комбинацию соединения или композиции по настоящему изобретению с другим противовирусным средством и вакцинацией против вируса гриппа.
[0195] Если совместное введение включает раздельное введение первого количества соединения по изобретению и второго количества дополнительного терапевтического средства, то соединения вводят достаточно близко по времени, чтобы иметь желаемый терапевтический эффект. Например, период времени между каждым введением, которое может привести к желаемому терапевтическому эффекту, может изменяться от нескольких минут до нескольких часов, и может быть определено с учетом свойств каждого соединения, таких как сила, растворимость, биодоступность, период полувыведения из плазмы и кинетический профиль. Например, соединение по настоящему изобретению и второе лекарственное средства могут быть введены в любом порядке в течение 24-х часов друг от друга, в течение 16-ти часов друг от друга, в течение 8-ми часов друг от друга, в течение 4-х часов друг от друга, в течение 1 часа друг от друга или в течение 30 минут друг от друга.
[0196] Более конкретно, первую терапию (например, введение профилактического или терапевтического средства, такого как соединение по изобретению) можно проводить до (например, за 5 минут, 15 минут, 30 минут, 45 минут, 1 час, 2 часа, 4 часа, 6 часов, 12 часов, 24 часа, 48 часов, 72 часов, 96 часов, 1 неделю, 2 недели, 3 недели, 4 недели, 5 недель, 6 недель, 8 недель или 12 недель до), одновременно или после (например, через 5 минут, 15 минут, 30 минут, 45 минут, 1 час, 2 часа, 4 часа, 6 часов, 12 часов, 24 часа, 48 часов, 72 часов, 96 часов, 1 неделю, 2 недели, 3 недели, 4 недели, 5 недель, 6 недель, 8 недель или через 12 недель после) проведения второй терапии (например, введения профилактического или терапевтического средства, такого как противораковое средство) индивиду.
[0197] Понятно, что способ совместного введения первого количества соединения по изобретению и второго количества дополнительного терапевтического средства может привести к усиленному или синергическому терапевтическому эффекту, причем объединенный эффект больше, чем аддитивный эффект, который мог бы быть при раздельном введении первого количества соединения по изобретению, и второго количества дополнительного терапевтического средства.
[0198] Используемый в настоящем описании термин "синергический" относится к комбинации соединения по изобретению и другой терапии (например, профилактического или терапевтического средства), которая является более эффективной, чем аддитивные эффекты терапии. Синергетический эффект комбинированной терапии (например, комбинация профилактических или терапевтических средств) может позволить использовать более низкие дозы одного или нескольких медикаментов и/или реже вводить указанные медикаменты индивиду. Возможность использовать более низкие дозы медикамента (например, профилактическое или терапевтическое средство) и/или возможность реже вводить указанный медикамент может уменьшить токсичность, связанную с введением указанного медикамента индивиду без снижения эффективности указанного медикамента для профилактики, контроля или лечения заболевания. Кроме того, синергический эффект может привести к повышению эффективности агентов для профилактики, контроля или лечения заболевания. Наконец, благодаря синергическому эффекту комбинированной терапии (например, комбинация профилактических или терапевтических средств) можно избежать или уменьшить неблагоприятные или нежелательные побочные эффекты, связанные с применением любого из этих медикаментов самостоятельно.
[0199] Когда комбинированная терапия, использующая соединения по настоящему изобретению, находится в комбинации с вакциной против гриппа, то оба терапевтических средства могут быть введены так, что промежуток времени между каждым введением может быть больше (например, несколько дней, недель или месяцев).
[0200] Наличие синергического эффекта можно определить, используя подходящие способы оценки лекарственного взаимодействия. Подходящие способы включают в себя, например, сигмоидальное уравнение для вычисления максимальной эффективности (Holford, N.H.G. and Scheiner, L.B., Clin. Pharmacokinet. 6: 429-453 (1981)), уравнение аддитивности Loewe (Loewe, S. and Muischnek, H., Arch. Exp. Pathol Pharmacol. 114: 313-326 (1926)) и уравнение медианного эффекта (Chou, T.C. and Talalay, P., Adv. Enzyme Regul. 22: 27-55 (1984)). Каждое уравнение, указанное выше, может использоваться вместе с экспериментальными данными для получения соответствующего графика для упрощения оценки эффектов лекарственной комбинации. Соответствующие графики, связанные с уравнениями, указанными выше, представляют собой кривую концентрация-эффект, кривую изоболограммы и кривую показателя аддитивности, соответственно.
[0201] Конкретные примеры, которые могут быть введены совместно с соединением, описанным в настоящем документе, включают ингибиторы нейраминидазы, такие как озельтамивир (Tamiflu®) и занамивир (Rlenza®), вирусный ионный канал (белок М2), блокаторы, такие как амантадин (Symmetrel®) и ремантадин (Flumadine®) и противовирусные препараты, описанные в WO 2003/015798, включая Т-705 в стадии разработки от Toyama Chemical, Япония. (См. также Ruruta et al., Antiviral Research, 82: 95-102 (2009), "T-705 (flavipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections"). В некоторых вариантах осуществления соединения, описанные в настоящем документе, могут быть введены совместно с обычной вакциной против гриппа. В некоторых вариантах осуществления соединения, описанные в настоящем документе, могут быть введены совместно с занамивиром. В некоторых вариантах осуществления соединения, описанные в настоящем документе, могут быть введены совместно с озельтамивиром. В некоторых вариантах осуществления соединения, описанные в настоящем документе, могут быть введены совместно с флавипиравиром (Т-705). В некоторых вариантах осуществления соединения, описанные в настоящем документе, могут быть введены совместно с амантадином или римантадином. Озельтамивир может быть введен по схеме приема в соответствии с его инструкцией. В некоторых конкретных вариантах его вводят по 75 мг два раза в день или 150 мг один раз в день.
[0202] Способы введения
[0203] Соединения и их фармацевтически приемлемые композиции, описанные выше, могут быть введены человеку и другим животным перорально, ректально, парентерально, интрацистернально, интравагинально, внутрибрюшинно, местно (в виде порошков, мазей или капель), трансбуккально, в виде перорального или назального спрея, или и тому подобное, в зависимости от тяжести инфекции, подлежащей лечению.
[0204] Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Кроме активных соединений жидкие лекарственные формы могут содержать инертные разбавители, обычно используемые в уровне техники, например, такие как вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, масло зародышей пшеницы, оливковое, касторовое и кунжутное масло), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана, а также их смеси. Кроме инертных разбавителей, композиции для перорального применения могут дополнительно включать вспомогательные вещества, такие как увлажнители, эмульгаторы и суспендирующие агенты, подсластители, вкусовые добавки и ароматизирующие агенты.
[0205] Препараты для инъекций, например, стерильные инъецируемые водные или маслянистые суспензии, могут быть получены в соответствии с информацией уровня техники, используя подходящие диспергирующие или смачивающие агенты и суспендирующие агентов. Стерильный препарат для инъекций может также быть стерильным инъецируемым раствором, суспензией или эмульсией в нетоксичном разбавителе или растворителе, приемлемом для парентерального введения, например, в виде раствора в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые могут быть использованы, можно указать воду, раствор Рингера, U.S.P и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла. Для этой цели может быть использовано любое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, для получения препаратов для инъекций используются жирные кислоты, такие как олеиновая кислота.
[0206] Составы для инъекций могут быть стерилизованы, например, фильтрацией через задерживающий бактерии фильтр или введением стерилизующих агентов в форме стерильных твердых композиций, которые могут быть растворены или диспергированы в стерильной воде или в некоторых других стерильных средах для инъекций перед применением.
[0207] Для пролонгации эффекта соединения, описанного в настоящем документе, часто желательно замедлить абсорбцию соединения после подкожной или внутримышечной инъекции. Это может быть достигнуто за счет использования жидкой суспензии кристаллического или аморфного вещества с плохой растворимостью в воде. Скорость абсорбции соединения тогда зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. Альтернативно, замедление абсорбции парентерально введенного соединения достигается посредством растворения или суспендирования соединения в масляном носителе. Инъекционные депо-формы получают путем формирования микроинкапсулирующих матриц соединения в биодеградируемых полимерах, таких как полилактид-полигликолид. В зависимости от соотношения соединения и полимера, а также природы конкретного используемого полимера, скорость высвобождения соединения можно контролировать. Примеры других поддающихся биологическому разложению полимеров включают поли(ортоэфиры) и поли(ангидриды). Депо-формы составов для инъекций также получают путем включения соединения в липосомы или микроэмульсии, которые совместимы с тканями организма.
[0208] Композиции для ректального или вагинального введения представляют собой специфические суппозитории, которые могут быть получены путем смешивания описанных в настоящем документе соединений с подходящими не раздражающими эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при температуре окружающей среды, но жидкими при температуре тела, и поэтому расплавляются в прямой кишке или вагинальной полости и высвобождают активное соединение.
[0209] Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение смешивают по меньшей мере с одним инертным, фармацевтически приемлемым эксципиентом или носителем, таким как цитрат натрия или дикальций фосфат и/или а) наполнители или сухие разбавители, такие как крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, b) связующие вещества, такие как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и гуммиарабик, с) увлажнители, такие как глицерин, d) дезинтегрирующие агенты, такие как агар-агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия, е) замедляющие растворение агенты, такие как парафин, f) ускорители абсорбции, такие как четвертичные аммониевые соединения, g) смачивающие агенты, такие как, например, цетиловый спирт и глицерина моностеарат, h) абсорбенты, такие как каолин и бентонитовая глина, и i) смазочные вещества, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае капсул, таблеток и пилюль, дозированная форма может дополнительно содержать буферные агенты.
[0210] Кроме того, твердые композиции подобного типа могут использоваться в качестве наполнителей в мягких и твердых желатиновых капсулах, используя эксципиенты, такие как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и тому подобное. Твердые дозированные формы таблеток, драже, капсул, пилюль и гранул могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные в области фармацевтических препаратов. Они могут необязательно содержать замутнители и могут представлять собой композиции, которые высвобождают активный(е) ингредиент(ы) только или предпочтительно в определенной части желудочно-кишечного тракта, необязательно, в замедленной форме. Примеры инкапсулированных композиций, которые могут использоваться, включают полимерные вещества и воски. Кроме того, твердые композиции подобного типа могут использоваться в качестве наполнителей в мягких и твердых желатиновых капсулах, используя такие эксципиенты как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и тому подобное.
[0211] Кроме того, активный ингредиент может находиться в микроинкапсулированной форме с одним или несколькими вышеуказанными эксципиентами. Твердые лекарственные формы, такие как таблетки, драже, капсулы, пилюли и гранулы, могут быть получены с покрытием и оболочками, такими как кишечнорастворимые покрытия и другие виды покрытия, контролирующие высвобождение, хорошо известные в области фармацевтических препаратов. В таких твердых лекарственных формах активное соединение может быть смешано по крайней мере с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы могут также включать, как и в обычной практике, дополнительные вещества, отличные от инертных разбавителей, например, смазывающие вещества для таблетирования и другие вспомогательные агенты для таблетирования, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль, лекарственные формы могут дополнительно содержать буферные агенты. Они могут необязательно содержать замутнители и могут представлять собой композиции, которые высвобождают активный(е) ингредиент(ы) только или предпочтительно в определенной части желудочно-кишечного тракта, необязательно, в замедленной форме. Примеры инкапсулированных композиций, которые могут использоваться, включают полимерные вещества и воски.
[0212] Лекарственные формы для местного или трансдермального введения соединения, описанного в настоящем документе, включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, ингаляторы или пластыри. Активный компонент может быть смешан в стерильных условиях с фармацевтически приемлемым носителем и любыми консервантами или агентами, которые будут необходимыми. Офтальмологические составы, ушные капли и глазные капли также входят в рамки настоящего изобретения. Кроме того, настоящее изобретение предусматривает применение трансдермальных пластырей, которые имеют дополнительное преимущество, обеспечивая контролируемую доставку соединения в организм. Такие лекарственные формы могут быть получены путем растворения или диспергирования соединения в соответствующей среде. Дополнительно могут применяться ускорители абсорбции, чтобы увеличить поступление соединения через кожу. Скорость можно контролировать либо с помощью контролирующей скорость мембраны, либо диспергируя соединение в полимерной матрице или геле.
[0213] Композиции, описанные в настоящем описании, могут быть введены перорально, парентерально, путем ингаляции, местно, ректально, назально, трансбуккально, вагинально или через имплантированный резервуар. Термин "парентеральный", используемый в настоящем документе, включает, но не ограничиваясь ими, подкожную, внутривенную, внутримышечную, внутрисуставную, внутрисиновиальную, внутригрудинную, интратекальную, внутрипеченочную, внутриочаговую и внутричерепную инъекцию или инфузию. В частности, композиции вводят перорально, внутрибрюшинно или внутривенно.
[0214] Стерильными инъекционными формами композиций, описанных в настоящем документе, могут быть водная или масляная суспензия. Эти суспензии могут быть приготовлены в соответствии с методиками, известными в данной области, используя подходящие диспергирующие или смачивающие агенты и суспендирующие агенты. Стерильный инъекционный состав также может быть стерильным инъекционным раствором или суспензией в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые могут быть использованы, можно указать воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла. С этой целью может быть использовано любое нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные могут использоваться для получения препаратов для инъекций, каких как природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно их полиоксиэтилированные варианты. Эти масляные растворы или суспензии могут также содержать спиртовой разбавитель с длинной цепью или диспергирующий агент, такой как карбоксиметилцеллюлоза или подобные диспергирующие агенты, которые обычно используются при получении фармацевтически приемлемых лекарственных форм, включая эмульсии и суспензии. Другие обычно используемые поверхностно-активные вещества, такие как Tween, Span и другие эмульгирующие агенты или усилители биодоступности, которые обычно используются для получения фармацевтически приемлемых твердых, жидких или других лекарственных форм, также могут быть использованы для целей получения.
[0215] Фармацевтические композиции, описанные в настоящем описании, могут быть введены перорально в любой перорально приемлемой лекарственной форме, включая, но ими не ограничиваясь, капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения обычно используемые носители включают, но ими не ограничиваются, лактозу и кукурузный крахмал. Также обычно добавляют смазывающие вещества, такие как стеарат магния. Для перорального введения в форме капсул подходящие разбавители включают лактозу и высушенный кукурузный крахмал. Если для перорального применения требуются водные суспензии, то активный ингредиент объединяют с эмульгирующими и суспендирующими агентами. При желании также могут быть добавлены определенные подсластители, ароматизаторы или красители.
[0216] Альтернативно, фармацевтические композиции, описанные в настоящем документе, могут быть введены в форме суппозиториев для ректального введения. Они могут быть получены путем смешивания агента с подходящим нераздражающим эксципиентом, который является твердым при комнатной температуре, но жидким при ректальной температуре и, следовательно, будет плавиться в прямой кишке с высвобождением лекарственного средства. Такие вещества включают, но ими не ограничиваются, масло какао, пчелиный воск и полиэтиленгликоли.
[0217] Фармацевтические композиции, описанные в настоящем документе, также могут применяться местно, особенно, если целью лечения являются участки или органы, легкодоступные для местного нанесения, включая болезни глаз, кожи или нижней части кишечника. Подходящие составы для местного применения могут быть легко получены для каждого из этих участков или органов.
[0218] Местное применение для нижнего отдела кишечника может быть осуществлено в виде ректальных суппозиториев (см. выше) или в виде подходящего состава для клизмы. Также могут быть использованы местные трансдермальные пластыри.
[0219] Для местного применения фармацевтические композиции могут быть получены в виде подходящей мази, содержащей активное вещество, суспендированное или растворенное в одном или нескольких носителях. Носители для местного применения соединений по настоящему изобретению включают, но ими не ограничиваются, минеральное масло, жидкий вазелин, белый вазелин, пропиленгликоль, полиоксиэтилен, полиоксипропилен, эмульгирующий воск и воду. Альтернативно, фармацевтические композиции могут быть получены в виде подходящего лосьона или крема, содержащего активные компоненты, суспендированные или растворенные в одном или более фармацевтически приемлемыми носителях. Подходящие носители включают, но ими не ограничиваются, минеральное масло, сорбитан моностеарат, полисорбат 60, цетиловые эфиры воска, цетеариловый спирт, 2 октилдодеканол, бензиловый спирт и воду.
[0220] Для офтальмологического применения фармацевтические композиции могут быть получены в виде микронизированных суспензий в изотоническом стерильном физиологическом растворе с установленным рН или, в частности, в виде растворов в изотоническом стерильном физиологическом растворе с установленным рН вместе или без консерванта, такого как хлорид бензалкония. Альтернативно, для офтальмологического применения фармацевтические композиции могут быть получены в виде мази, такой как вазелин.
[0221] Фармацевтические композиции могут быть также введены с помощью назального аэрозоля или ингаляции. Такие композиции получают в соответствии с методиками, хорошо известными в области фармацевтических составов и могут быть получены в виде растворов в физиологическом растворе, используя бензиловый спирт или другие подходящие консерванты, ускорители абсорбции для повышения биодоступности, фторуглероды и/или другие обычные солюбилизирующие или диспергирующие агенты.
[0222] Соединения для применения в способах по изобретению могут быть получены в виде стандартной лекарственной формы. Термин "единичная дозированная форма" относится к физически дискретным единицам, подходящим в качестве единичных доз для индивидов, проходящих лечение, причем каждая единица содержит предварительно определенное количество активного вещества, рассчитанное на получение желаемого терапевтического эффекта, необязательно, в сочетании с подходящим фармацевтическим носителем. Стандартная лекарственная форма может быть одной суточной дозой или одной из множества суточных доз (например, от 1 до 4 или более раз в день). Если используются множество суточных доз, то стандартная лекарственная форма может быть одинаковой или различной для каждой дозы.
[0223] V. Примеры
[0224] Пример 1: Пример 1: Общие способы XRPD, измерения C13твердотельной ЯМР, DSC и ТГА
[0225] 1A: Термогравиметрический анализ (ТГА)
[0226] Термогравиметрический анализ (ТГА) проводили на приборе ТА приборе для TGA, модель Q500 Asset Tag V014840. Твердый образец помещали в ванночку для платинового образца и нагревали при 10°С/мин до 300°С от комнатной температуры.
[0227] 1B: Измерения DSC
[0228] DSC проводили на приборе Tag V015553 TA DSC Q200 Asset. Приблизительно 1-2 мг твердого образца помещали в алюминиевую герметично закрытую ванночку для DSC с рифленой крышкой с отверстием. Клетку образец, как правило, нагревают при продувке азота.
[0229] 1C: Экспериментальная SSNMR:
[0230] Спектры твердотельной ядерно-магнитной спектроскопии (SSNMR) получали на Bruker-BioSpin 400 МГц Advance III с широким спектрометром, оснащенным зондом Bruker-BioSpin 4мм HFX. Образцы были упакованы в роторы 4 мм ZrO2 (приблизительно 70 мг или меньше, в зависимости от наличия образца). Было использована скорость вращения под магическим углом (MAS), обычно равная 12.5 кГц. Температура головки зонда была установлена на 275K, чтобы свести к минимуму влияние фрикционного нагрева во время вращения. Время протонной релаксации измеряли с помощью эксперимента по восстановлению насыщения по релаксации 1H MAS T1для того, чтобы установить правильную рециркуляционную задержку в эксперименте с 13C кросс-поляризацией (СР) MAS. Восстановление насыщения в эксперименте 13C CPMAS устанавливали по крайней мере в 1,2 раза длиннее, чем измеренное время релаксации 1H T1 для максимального увеличения соотношения сигнал углерода-шум в спектре. Время контактирования CP в эксперименте13C CPMAS было установлено на 2 мс. Использовали CP протонный импульс с линейной характеристикой (от 50% до 100%). Условие Хартмана-Хана оптимизировали на внешнем эталонном образце (глицин). Спектры фтора были получены, используя установку MAS с несвязанными протонами и повторенной задержкой, установленной примерно в 5 раз от измеренного времени релаксации 19FT1. Время релаксации фтора измеряли с помощью эксперимента по восстановлению насыщения по релаксации несвязанного протона 19F MAS T1. Оба спектра углерода и фтора были получены с помощью SPINAL64 разделение использовали с напряженностью поля около 100 кГц. Химический сдвиг был противопоставлен внешнему стандарту адамантана с резонансом, направленным в область сильного поля, равным 29.5 промилле.
[0231] 1D: Подробное описание эксперимента Bruker D8 Discover XRPD.
[0232] Параметры XRPD получали при комнатной температуре в режиме отражения, используя диффрактометр Bruker D8 Discover (Asset Tag V012842), оснащенный герметизированной пробиркой с источником и с обнаружителем Hi-Star (Bruker AXS, Madison, WI). Генератор рентгеновского излучения работал при напряжении 40 кВ и токе 35 мА. Образец порошка помещали в алюминиевый держатель. Два кадра были зарегистрированы со временем экспозиции 120 с каждый. Данные затем были интегрированы в диапазоне от 4,5°С-39° 2Ө с размером шага 0,02° и объединены в один непрерывный параметр.
[0233] Пример 2: Получение соединения (1) и сольвата 2-MeTHF Соединения (1)
[0234] Соединение (1) может быть получено, как описано в WO 2010/148197. Например, аморфное свободное основание Соединения (1) получали в соответствии с WO 2010/148197, а затем с помощью обычного хирального разделения и очистки: SCF хиральная хроматография с модификатором, который включает Et2NH (который образовывал соль Et2NH Соединения (1)), а затем обработку ионообменной смолой. Альтернативно, Соединение (1) может быть получено с помощью следующих методик как сольват 2-MeTHF:
[0235] Получение Соединения 2a (2-амино-3-бром-5-фторпиридин)
[0236] К суспензии 2-амино-5-фторпиридина (6 кг, 53,6 моль) в воде (24 л) при 14°С добавляли в течение 10 минут 48% бромистоводородную кислоту (18,5 кг, 110 моль). Реакция была экзотермической и температура поднималась до 24°C. Смесь повторно охлаждали до 12°С, затем добавляли девять порций брома (9 кг, 56,3 моль) в течение 50 минут (экзотермическая реакция, поддерживают при 20°С). Смесь перемешивали при 22°С в течение ночи и контролировали с помощью 1ЯМР аликвоты с погашенной реакцией (гасят 5 каплями в смесь 1 мл 20% K2CO3, 0,3 мл 10% Na2S2О3 и 0,7 мл DCM. Органический слой упаривали и анализировали). Смесь охлаждали до 10°С, затем гасили, добавляя бисульфит натрия (560 г, 5,4 моль) в воде (2 л), и дополнительно охлаждали до 0°С. Эту смесь добавляли к холодной (-4°С) смеси DCM (18 л) и 5,4 М гидроксида натрия (35 л, 189 моль). Порцию на дне ~35 л фильтровали через пад из Целита, и затем образовывался фазовый разрыв. Водный слой повторно экстрагировали DCM (10 л). Органические слои фильтровали через пад из 3 кг магнезола, промывали DCM (8 л). Фильтрат упаривали, растирали в порошок с гексаном и фильтровали.
[0237] Несмотря на то, что анализ в ходе процесса указал на 97% завершение, этот первоначальный продукт всех четырех пробегов, как правило, содержал ~10% SM. Его объединяли и растирали в гексане (2 л на кг вещества) при 50°С, а затем охлаждали до 15°С и фильтровали с получением Соединения 2а (30,0 кг, ~95% чистота, 149 моль, 67%). Маточные растворы первоначального растирания и повторной очистки хроматографировали (20 кг силикагеля, элюент 25-50% EtOAc в гексане) с получением дополнительного соединения 2а (4,7 кг, ~99% чистота, 24,4 моль, 11%).
[0238] Получение Соединения 3a
[0239] В инертный 400-л реактор помещали Соединение 2а (27,5 кг, чистота 96%, 138 моль), Pd (PPh3)4 (1044 г, 0,90 моль) и CuI (165 г, 0,87 моль), а затем толуол (90 кг). Смесь обескислороживали тремя циклами вакуум-азот, а затем добавляли триэтиламин (19,0 кг, 188 моль). Смесь обескислороживали еще одним циклом вакуум-азот, затем добавляли ТМS-ацетилен (16,5 кг, 168 моль). Смесь нагревали до 48°С в течение 23 ч (исходное выделение тепла составило максимально 53°C), затем охлаждали до 18°С. Суспензию фильтровали через пад из Целита и промывали толуолом (80 кг). Фильтрат промывали 12% Na2HPO4 (75 л), затем фильтровали через пад из силикагеля (25 кг), промывали 1:1 гексан:MTBE (120 л). Этот фильтрат упаривали до коричневого масла, а затем растворяли в NMP для следующей стадии. Масса раствора Соединения 3а была 58 кг, ~ 50 масс.%, 138 моль, 100%. 1H ЯМР (CDCl3, 300 MHz): δ 7,90 (с, 1H); 7,33-7,27 (м, 1H); 4,92 (с, NH2), 0,28 (с, 9H) промиль.
[0240] Получение Соединения 4а
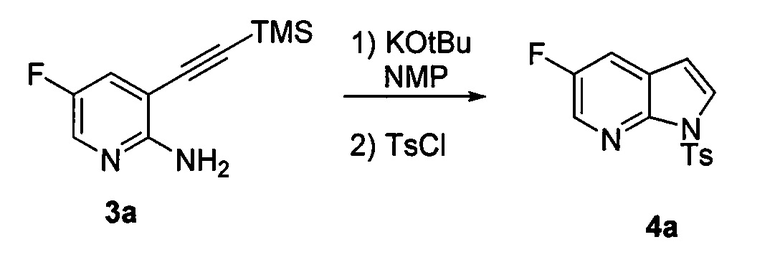
[0241] В инертный 400-л реактор помещали трет-бутоксид калия (17,5 кг, 156 моль) и NMP (45 кг). Смесь нагревали до 54°С, затем добавляли раствор Соединения 3a (29 кг, 138 моль) в NMP (38 кг) в течение 2,75 часов и промывали NMP (6 кг) (экзотермическая реакция, поддерживали при 70-77°С). Реакционную смесь перемешивали при 74°С в течение 2 часов, затем охлаждали до 30°С и добавляли раствор тозилхлорида (28,5 кг, 150 моль) в NMP (30 кг) в течение 1,5 часов и промывали с NMP (4 кг). Реакция была экзотермической и ее поддерживали на уровне 30-43°С. Реакционную смесь перемешивали в течение 1 часа при охлаждении до 20°С, затем добавляли воду (220 л) в течение 35 минут (экзотермическая реакция, поддерживали при 18-23°С). Смесь перемешивали при 20°С в течение 30 мин, затем фильтровали и промывали водой (100 л). Твердые продукты отфильтровывали с помощью DCM (250 кг), отделяли от остаточной воды, и органические вещества фильтровали через пад из магнезола (15 кг, вверху) и силикагеля (15 кг, внизу), промывая дополнительным количеством DCM (280 кг). Фильтрат концентрировали до густой взвеси (~ объем 50 л), затем добавляли MTBE (30 кг), продолжая дистилляцию при постоянном объеме (конечная температура дистиллята равна 51°С). Добавляли дополнительное количество MTBE (10 кг) и взвесь охлаждали до 15°С, фильтровали и промывали МТВЕ (40 л), получая Соединение 4а (19,13 кг, чистота 95%, 62,6 моль, 45%). Частичная концентрация фильтрата давала вторую порцию (2,55 кг, чистота 91%, 8,0 моль, 6%). 1H ЯМР (CDCl3, 300 МГц): δ 8,28-8,27 (м, 1H); 8,06-8,02 (м, 2H); 7,77 (д, J= 4,0 Гц, 1H); 7,54-7,50 (м, 1H); 7,28-7,26 (м, 2H); 6,56 (д, J= 4,0 Гц, 1H); 2,37 (с, 3H) промиль.
[0242] Получение Соединения 5a
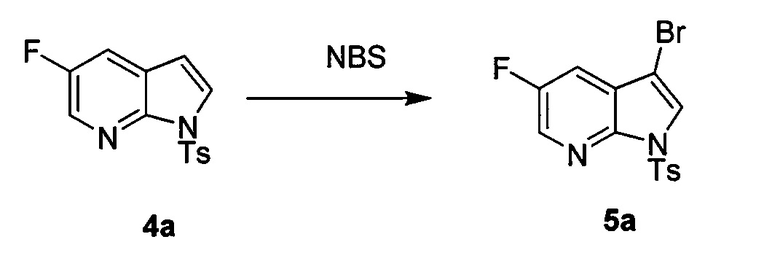
[0243] В взвесь N-бромсукцинимида (14,16 кг, 79,6 моль) в DCM (30 кг) при температуре 15°С помещали раствор Соединения 4а (19,13 кг, чистота 95%, и 2,86 кг, чистота 91%, 71,6 моль) в DCM (115 кг), промывая в DCM (20 кг). Смесь перемешивали при 25°С в течение 18 часов, а затем охлаждали до 9°С и гасили, добавляя раствор тиосульфата натрия (400 г) и 50% раствор гидроксида натрия (9,1 кг) в воде (130 л). Смесь нагревали до 20°С и слои разделяли, и органические слои промывали 12% солевым раствором (40 л). Водные слои последовательно повторно экстрагировали DCM (4х50 кг). Органические вещества объединяли и 40 л дистиллировали до азеотропной воды, затем раствор фильтровали через пад из силикагеля (15 кг, снизу) и магнезола (15 кг, верх), промывая DCM (180 кг). Фильтрат концентрировали до густой взвести (~ объем 32 л), затем добавляли гексан (15 кг). Добавляли дополнительное количество гексана (15 кг), продолжая дистилляцию при постоянном объеме (конечная температура дистиллята 52°C). Взвесь охлаждали до 16°C, фильтровали и промывали гексаном (25 кг) с получением Соединения 5a (25,6 кг, 69,3 моль 97%). 1H ЯМР (CDCl3, 300 MГц): δ 8,34-8,33 (м, 1H); 8,07 (д, J= 8,2Гц, 2H); 7,85 (с, 1H); 7,52-7,49 (м, 1H); 7,32-7,28 (м, 2H); 2,40 (с, 3H) промиль.
[0244] Получение Соединения 6а: Реакция BEFTAI
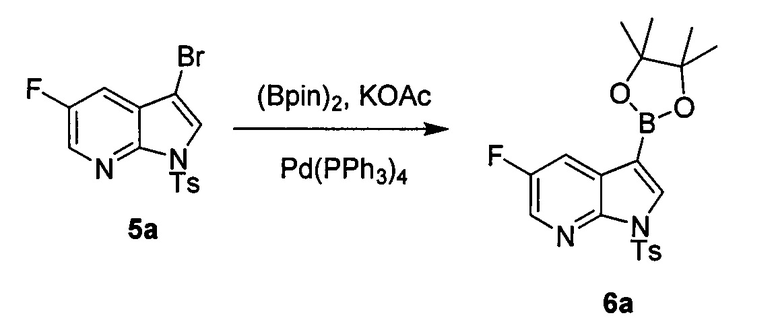
[0245] В инертный 400 л реактор помещали Соединение 5а (25,6 кг, 69,3 моль), бис (пинаколато)диборон (19 кг, 74,8 моль), ацетат калия (19 кг, 194 моль), ацетат палладия (156 г, 0,69 моль) и трифенилфосфин (564 г, 2,15 моль), а затем диоксан (172 кг), который был отдельно обескислорожен с использованием циклов вакуум-азот (х3). Смесь перемешивали и обескислороживали, используя циклы вакуум-азот (х 2), а затем нагревали до 100°C в течение 15 часов. Смесь охлаждали до 35°С, затем фильтровали, промывали 30°C THF (до 75 кг). Фильтрат упаривали и остаток растворяли в DCM (~ 90 л). Раствор перемешивали вместе с 1 кг углерода и 2 кг магнезола в течение 45 мин, затем фильтровали через пад из силикагеля (22 кг, внизу) и магнезола (10 кг, верху), промывая DCM (160 кг). Фильтрат концентрировали до густой взвести (~ объем 40 л), затем растирали в порошок при температуре 35°С и добавляли гексан (26 кг). Взвесь охлаждали до 20°С, фильтровали и промывали смесью DCM (5,3 кг) и гексана (15 кг), а затем гексаном (15 кг) и сушили в атмосфере азота на фильтре с получением Соединения 6а (23,31 кг, 56,0 моль, 81%) в виде белого твердого вещества. 1Н-ЯМР соответствует целевому продукту, ВЭЖХ 99,5%, анализ с палладием 2 промили. 1H ЯМР (CDCl3, 300 MГц): δ 8,25 (с, 1Н); 8,18 (с, 1Н); 8,09-8,02 (м, 2Н); 7,91-7,83 (м, 1Н); 7,30-7,23 (м, 2Н); 2,39 (с, 3H); 1,38 (с, 12H) промиль.
[0246] Получение Соединений 8а и 9а
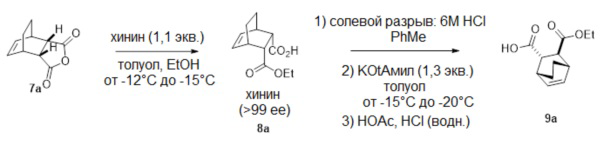
[0247] Соединение 8a: Ангидрид 7a (24,6 кг, Apex) и хинин (49,2 кг, Buchler) добавляли к реактору с последующим добавлением безводного PhMe (795,1 кг). Затем реактор охлаждали до температуры -16°С и EtOH (безводный, 41,4 кг) добавляли с такой скоростью, чтобы поддерживать внутреннюю температуру в реакторе <-12°C. Максимальная температура реакции в этом эксперименте была зарегистрирована как -16°C. Реакционную смесь затем перемешивали в течение 16 часов при -16°С. Твердый продукт сушили и оценивали с помощью 1H-ЯМР, который показал, что ангидрида не осталось. Содержимое реактора отфильтровывали. Реактор и последующий влажный осадок на фильтре промывали PhMe (безводный, 20 кг). Полученное твердое вещество помещали в центробежную сушилку при <45°C, промывая N2 в течение по крайней мере 48 часов. В этом эксперименте фактическая температура составляла 44°С, и вакуум был -30 в HG. Вещество отбирали через 2,5 дня после высушивания и ЯМР показал 3% PhMe. Еще через 8 часов образец PhMe анализировали, и он показал присутствие тех же 3% PhMe и высушивание прекращали. Масса твердого вещества белого цвета была 57,7 кг, выход 76%. 1Н-ЯМР показал соответствие со структурой и анализ Chiral SFC показал >99% ее вещества.
[0248] Соединение 9a: В реактор помещали соль хинина 8а (57,7 кг) и PhMe (250,5 кг, Aldrich класса ACS, >99,5%) и начинали перемешивать. Содержимое охлаждали до <15°С и обрабатывали 6н HCl (18 кг H2O обрабатывали 21,4 кг конц. HCl), поддерживая при этом температуру <25°С. Смесь перемешивали в течение 40 мин и визуально оценивали для того, чтобы убедиться, что никаких твердых частиц не присутствует. Перемешивание прекращали и фазам давали отстояться и фазы разделяли. Водные фазы снова экстрагировали с помощью PhMe (160 кг; количество, которое использовали, было, как правило, намного меньше, вычисл. 43 кг. Тем не менее, для эффективного перемешивания в виду минимального объема добавляли дополнительное количество PhMe. Органические фазы объединяли. Брали образцы органической фазы и прогоняли в анализе ВЭЖХ, чтобы подтвердить присутствие продукта; только для информации.
[0249] Органические фазы охлаждали до <5°C (0-5°С) и добавляли сульфат натрия (безводный, 53,1 кг) при перемешивании в течение 8 часов (в данном случае 12 часов). Содержимое реактора, содержащего органическую фазу, пропускали через фильтр, содержащий сульфат натрия (31 кг, безводный) и помещали в очищенный и высушенный реактор. Реактор промывали PhMe (57,4 кг), пропускали через фильтр в реактор емкостью 20 л. Начинали перемешивать и добавляли дополнительное количество PhMe (44 кг), и реакционную смесь охлаждали до -20°С. При этой температуре добавляли раствор PhMe трет-пентокси калия в течение 2 часов, поддерживая температуру между -15 и -22°C. Реакционную смесь поддерживали при -20°С в течение еще 30 минут перед взятием образца. Взятие образца происходило путем удаления аликвот и немедленным их гашением в 6 н. HCl. Целевое соотношение в данном случае равно 96:4 (транс:цис).
[0250] Достигнув целевого отношения в реактор помещали уксусную кислоту (2,8 кг) в течение 6 минут. Температура оставалась -20°C. Температуру затем доводили до -5°С и добавляли водный 2 н. HCl (65,7 кг воды, обработанной 15,4 кг конц. HCl). Содержимое нагревали до 5°C/-5°С, перемешивали в течение 45 мин, а затем нагревали до 20°C/-5°С при перемешивании в течение 15 минут. Перемешивание прекращали, и фазам давали отстояться. Водный слой удаляли (временное удержание). Органическую фазу промывали водой (48 кг, питьевая), перемешивали в течение 15 мин и фазам давали отстояться (по крайней мере 15 мин), и водный слой удаляли и добавляли к водному слою. В органическую фазу добавляли 1/3 буферного раствора (50 л), который был получен (7,9 кг NaH2PO4, 1,3 кг Na2HPO4 и 143,6 кг воды) и перемешивали в течение по меньшей мере 15 минут. Перемешивание прекращали и фазы оставляли разделять по крайней мере на 15 минут. Нижний слой удаляли. Другую часть буферного раствора (50 л) использовали для промывки органического слоя, как описано выше. Промывку проводили в третий раз, как описано выше.
[0251] Вакуумную дистилляцию фазы PhMe (150 л) начинали при 42°С/-13,9 фунтов на квадратный дюйм и дистиллировали до масла объемом 20 л. После значительного уменьшения объема смесь переносили в меньший сосуд для завершения дистилляции. Добавляли гептан (13,7 кг), и смесь нагревали до 40 +/-5°С в течение 30 минут, а затем содержимое охлаждали до температуры 0-5°С в течение 1,5 часов. Твердые вещества отфильтровывали и реактор промывали приблизительно 14 кг охлажденного (0-5°С) гептана. Твердые вещества сушили в вакууме перед тем как помещали в печь при температуре <40°C под домашним вакуумом (-28 фунтов на квадратный дюйм) до тех пор, пока LOD не был равен <1%. 15,3 кг, 64%, чистота согласно ВЭЖХ 96%. 1H ЯМР (400 MГц, CDCl3) δ 11,45 (ушир.с, 1H), 6,41 (т, J=7,2 Гц, 1H), 6,25 (т, J=7,2 Гц, 1H), 4,18 (м, 2H), 3,27 (м, 1H), 3,03 (м, 1H), 2,95 (м, 1H), 2,77 (м, 1H), 1,68 (м, 1H), 1,49 (м, 1H), 1,25 (т, J=7,2Hz), 1,12 (м, 1H).
[0252] Получение Соединения 10a

[0253] В трехгорлую круглодонную колбу, оснащенную механической мешалкой, датчиком температуры, обратным холодильником, капельной воронкой и входом для азота помещали Соединение 9а (145,0 г, 1 экв) и безводный толуол (Aldrich, кат. # 244511) (1408 г, 1655 мл) в атмосфере азота. Затем порциями добавляли триэтиламин (Aldrich, кат # 471283) (140 г, 193 мл, 2,14 экв) в течение 5 минут к перемешиваемому раствору, в течение чего наблюдалось выделение тепла при максимальной температуре 27°C. Начинали сбор данных с помощью ReactIR. Реакционную смесь затем нагревали до 95°С в течение 70 мин. Затем порциями добавляли дифенилфосфорилазиду (Aldrich, кат # 178756) (176,2 г, 138,0 мл, 0,99 экв) с помощью капельной воронки в течение общего времени 2,25 часа.
[0254] После завершения добавления дифенилфосфорилазиды (капельную воронку промывали небольшим количеством толуола), полученную смесь нагревали при 96°С в течение еще 50 минут. Образец реакционной смеси, разведенный в толуоле, анализировали с помощью GC/МС, который указывал на расход дифенилфосфорилазида. Затем добавляли бензиловый спирт (Aldrich, кат # 108006) (69,9 г, 67,0 мл, 1,0 экв) с помощью капельной воронки в течение 5-10 минут. Полученную смесь затем нагревали при 97°С в течение ночи (в течение примерно 19 часов). Образец реакционной смеси, разведенный в толуоле с помощью GC/МС, показал образование продукта (m/e=330). Реакционную смесь затем охлаждали до 21°С, после чего порциями добавляли воду (870 г, 870 мл) (наблюдали небольшое выделение тепла до максимальной температуры 22°С). Реакционную смесь сначала гасили, добавляя 500 г воды, и перемешивали механическим способом в течение 10 минут. Затем смесь переносили в делительную воронку, содержащую оставшиеся 370 г воды, а затем вручную перемешивали. После перемешивания и разделения фаз, органический и водный слои разделяли (отделяли водную фазу при рН~10). Органический слой затем промывали дополнительной порцией воды (870 г; 1х870 мл). Органический и водный слои разделяли (отделяли водный слой при рН~10). Собранную органическую фазу затем концентрировали досуха при пониженном давлении (водяная баня при температуре 45-50°С), получая 215 г неочищенного Соединения 10a (приблизительный объем 190 мл). 1H ЯМР и GC/МS соответствуют соединению 10А (с остаточным толуолом и бензиловым спиртом).
[0255] Получение Соединения 11а
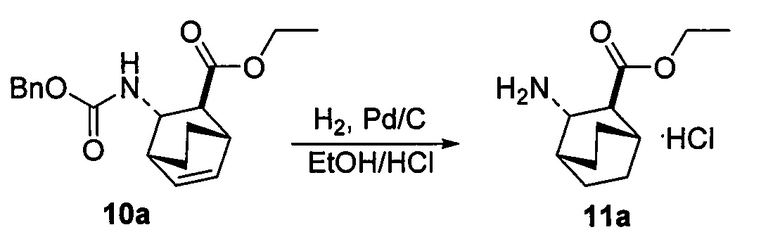
[0256] Получение HCl в этаноле: В трехгорлую круглодонную колбу, снабженную датчиком температуры, входом для азота и магнитной мешалкой, помещали этанол (1000 мл, 773 г) в атмосфере азота. Раствор перемешивали и охлаждали на бане сухой лед/ацетон до тем пор, пока внутренняя температура не достигала 12°С. Затем безводный HCl (~80 г, 2,19 моль) медленно барботировали в охлажденный раствор (наблюдали температуру от -24 до -6°С во время добавления) в течение 2 часов. После добавления раствор переносили в стеклянную колбу и оставляли нагреваться до температуры окружающей среды. Образец раствора, взятый для титрования, давал концентрацию 2,6 М. Раствор затем хранили в холодном помещении (приблизительно 5°С) в течение ночи.
[0257] Гидрирование/образование соли HCl: В стеклянную вставка в 2 галлоном автоклаве Парра помещали палладий на угле (Pd/C (Aldrich, кат # 330108), 10% в расчете на сухой вес; (влажность 50%), 13,11 г, 0,01 экв на основе Соединения 10a) в атмосфере азота, а затем увлажняли этанолом (93 г; 120 мл). Затем добавляли раствор неочищенного Соединения 10a (212 г, 1 экв) в этаноле (1246 г; 1600 мл) в стеклянную вставку (промывали небольшим объемом этанола для содействия переносу). Стеклянную вставку помещали в автоклав, а затем добавляли HCl в этаноле (получен, как описано выше, 2,6 М, 1,04 экв в расчете на Соединение 10a; 223 г; 259 мл). Автоклав герметизировали, а затем продували водородом (3х при 20 фунтах на квадратный дюйм). Затем начинали гидрирование при приложенном давлении газообразного водорода (15 фунтов на квадратный дюйм) в течение 3 часов, после чего давление было постоянным. Анализ аликвот реакционной смеси с помощью 1Н-ЯМР и GC/МС показал расход исходного материала/образование продукта. Полученную смесь затем фильтровали через пад из Целита (192 г), после чего пад из Целита промывали дополнительным количеством этанола (3х; всего во время промывки использовали 1176 г этанола). Фильтрат (зеленого цвета) затем концентрировали при пониженном давлении (на водяной бане при 45°С) до ~382 г ((~ 435 мл; 2,9 объема на основе теоретического выхода Соединения 11а. Затем к остатку добавляли изопропилацетат (1539 г, 1813 мл (12 объемов на основе теоретического выхода Соединения 11а. Полученный раствор дистиллировали в вакууме при постепенном повышении температуры.
[0258] Дистилляцию останавливали, после чего оставшийся раствор (370 г, ~365 мл общего объема, коричневатый цвет) оставляли стоять при температуре окружающей среды в течение выходных. Смесь фильтровали (для облегчения фильтрации использовали изопропилацетат) и собранные твердые продукты промывали дополнительным количеством изопропилового ацетата (2х116 мл, каждая промывка приблизительно 100 г). Затем твердое вещество сушили в вакууме при 40°C (максимальная наблюдаемая температура 42°С) в течение ночи с получением 118 г (78,1% в течение двух стадий) Соединения 11а. 1Н ЯМР вещества соответствовало структуре Соединения 11а и GC/МС показала чистоту 99%.
[0259] Получение Соединения 13а
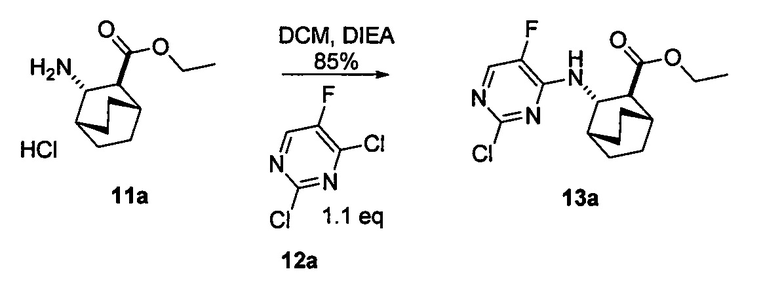
[0260] Методика A: Смесь 5-фтор-2,4-дихлорпиримидина (12a, 39,3 г, 235 ммоль, 1,1 экв) и соль амина HCl (11а, 50 г, 214 ммоль) обрабатывали с помощью СН2Cl2 (169 мл) и полученную смесь нагревали до 30°С. Полученную смесь затем медленно обрабатывали DIEA (60,8 г, 82 мл, 471 ммоль, 2,2 экв) с помощью шприцевого насоса в течение 3 часов. Пик температуры был до 32°C. Реакционную смесь перемешивали в течение 20 часов, реакционную смесь проверяли на завершение реакции с помощью ВЭЖХ и охлаждали до комнатной температуры. Полученную реакционную смесь промывали последовательно водой (211 мл, рН=8-9), 5% раствором NaHSO4 (211 мл, рН=1-2), а затем 5% водным раствором. NaCl (211 мл, рН=5-6). NaCl (211 мл, рН=5-6).
[0261] Органическую фазу затем дистиллировали при пониженном давлении до 190 мл. Помещали PhMe (422 мл) и температуру устанавливали при 70-80°C и при внутренней температуре 60-65°С до тем пор, пока объем не возвращался до 190 мл. Смесь оставляли охлаждаться приблизительно до 37°С при перемешивании - приблизительно через 10 минут начиналась кристаллизация и отмечалось повышение температуры до примерно 41°С. После уравновешивания при 37°С в суспензию помещали н-гептан (421 мл) в течение 3,5 часов, а затем охлаждали до 22°С в течение 1 часа. Смесь перемешивали в течение ночи при этой температуре перед фильтрацией. Полученное твердое вещество на фильтре промывали 10% PhMe в растворе н-гептана (2×210 мл). Затем твердое вещество сушили в печи под вакуумом при продувке N2 при 50°С в течение ночи. Полученный твердый продукт весил 62 г (выход 88%).
[0262] Методика B: В трехгорлую круглодонную колбу, оснащенную механической мешалкой, датчиком температуры, обратным холодильником, входом для азота и дополнительной капельной воронкой помещали Соединение 11а (51,2 г) и Соединение 12а (40,2 г) в атмосфере азота. Добавляли дихлорметан (173 мл, 230 г), и полученную смесь перемешивали при нагревании до внутренней температуры, равной 30°С. Затем медленно добавляли N, N-диизопропилэтиламина (85 мл, 63,09 г) с помощью капельной воронки в течение 2,5-3 часов, в течение которых наблюдалось выделение тепла до максимальной наблюдаемой температуре 33,5°С. После завершения добавления, полученный раствор перемешивали при 30-31°С в течение ночи в атмосфере азота (в течение примерно 19 часов).
[0263] Образец, 100 мкл реакционной смеси разбавляли дихлорметаном до полного объема 10 мл и раствор хорошо перемешивали. Образец разбавленной аликвоты анализировали с помощью GC/МС, что указывало на полное завершение реакции по GC/МС; при этом наблюдалось образование продукта (m/e=328)). Реакционную смесь охлаждали до 26°С и переносили в делительную воронку (при помощи дихлорметана). Затем смесь последовательно промывали водой (211 мл, 211 г, рН водный слой отделялся при ~8, небольшой слой со взвесью переносился вместе с отделением водного слоя), 5% водным раствором NaHSO4 ((получали, используя 50 г моногидрата бисульфата натрия (Aldrich кат # 233714) и 950 г воды) 211 мл, 216 г; рН при отделении водного слоя был ~ 2), а затем 5% водным раствором NaCl ((получали, используя 50 г хлорида натрия (Aldrich кат. # S9888) и 950 г воды) 211 мл, 215 г; рН водного слоя был ~4-5). Собранную органическую фазу затем концентрировали при пониженном давлении (на водяной бане при 35°С) до ~190 мл (2,7 объема на основании теоретического выхода Соединения 13а, после чего добавляли толуол (Aldrich, номер в каталоге # 179418, 422 мл, 361 г). Полученную смесь концентрировали при пониженном давлении (на водяной бане при температуре 55-65°С) до ~190 мл (2,7 объема на основании теоретического выхода Соединения 13а. Анализ образца раствора на этой стадии с помощью 1H ЯМР показал отсутствие дихлорметана. Оставшуюся смесь оставляли охлаждаться до 37°С (используя водяную баню при температуре 37°C на роторном испарителе при перемешивании). В течение этого времени наблюдалась выраженная кристаллизация. Затем смесь механически перемешивали и нагревали до температуры приблизительно 37°С (внешний источник тепла, установлен на 38°С), после чего медленно добавляли н-гептан (430 мл, 288 г, Aldrich кат. # H2198) с помощью капельной воронки в течение 3 часов. После добавления нагревание останавливали и полученную взвесь перемешивали механическим способом при охлаждении до температуры окружающей среды в течение ночи. Полученную смесь затем фильтровали и собранные твердые вещества промывали 10% толуолом в н-гептане (2×210 мл; каждую промывку получали путем смешения 21 мл (16 г) толуола и 189 мл (132 г) N- гептана). Вакуум использовали до тех пор, пока наблюдали очень мало фильтрата. Твердые вещества затем дополнительно сушили в вакууме при 50°С при продувании азотом до постоянной массы (3,5 часа) с получением 64,7 г (90%) Соединения 13а. Анализ образца твердого продукта при помощи 1Н ЯМР показал, что вещество соответствует структуре, и анализ LC показал 99,8% чистоты, используя предоставленный способ LC.
[0264] Получение Соединения 14а
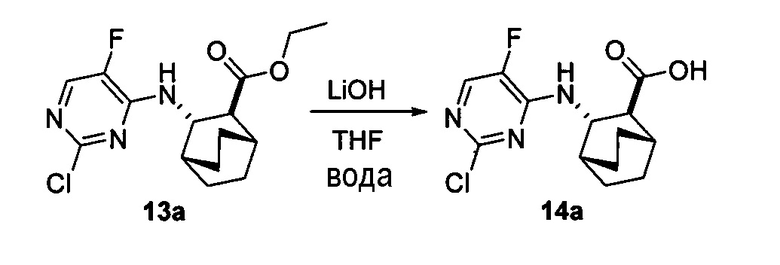
[0265] Этиловый эфир 13а (85 г, 259 ммоль) растворяли в THF (340 мл) и обрабатывали раствором LiOH (2 М, 389 мл, 778 ммоль) в течение 10 мин (температура от 21 до 24°С). Смесь нагревали до 45°С при перемешивании в течение 17 часов, после чего проверяли завершение реакции с помощью ВЭЖХ (SM не наблюдали). Реакционную смесь охлаждали до комнатной температуры и добавляли CH2Cl2 (425 мл). Затем медленно добавляли раствор лимонной кислоты (2 М, 400 мл) в течение 45 мин (при температуре до 26°C). Было отмечено, что во время добавления образовались некоторые белые твердые вещества, но они быстро растворялись при перемешивании. Реакционную смесь перемешивали в течение еще 15 мин, после чего фазы разделяли. После того, как были разделены фазы, измеряли рН водной фазы, рН=4,0. Органическую фазу промывали (перемешивание 15 мин) водой (255 мл) - фазы оставляли разделяться. Нижний слой (органический), содержащий требуемый продукт, затем хранили в холодильнике в течение ночи.
[0266] Органическую фазу концентрировали при пониженном давлении (котел устанавливали на 65°C) до 150 мл (вычисл. 1,76 объема wrt SM). IPA (510 мл) загружали и дистиллировали при пониженном давлении (85°C температурная установка холодильной машины) до 255 мл (3 объема). Уровень растворителя доводили приблизительно до 553 мл (6,5 объема) путем добавления IPA (298 мл). Затем добавляли воду (16 мл) и реакционную смесь нагревали с обратным холодильником (77°С) при энергичном перемешивании, при котором растворялись твердые вещества, осажденные на стенках сосуда. Реакционную смесь затем медленно охлаждали до 65°C (в течение 60 мин) и сохраняли все вещество в растворе (брали образец для анализа остатка в растворе). Реакционную смесь дополнительно охлаждали до 60°С, и реакционная смесь казалась слегка опалесцирующей. После перемешивания в течение 15 мин дополнительно охлаждали до 55°С. Продукт продолжал выпадать в осадок, а смесь по-прежнему была негустой и легко перемешивалась. Очень медленно (2,5-3 часа) добавляли воду (808 мл), поддерживая при этом температуру приблизительно 55°C. Затем смесь охлаждали до 22°С в течение 2 часов и оставляли перемешиваться в течение ночи. Вещество затем фильтровали и промывали смесью воды: IPA (75:25, 2x255 мл). Кислоту сушили в вакуумном сушильном шкафу при 55°С в течение ночи. Получали 69 г кислоты 14А, выход 88% твердого белого продукта. Анализ вещества с помощью ВЭЖХ - >99% чистоты.
[0267] Получение Соединения 15а: Реакция присоединения Сузуки
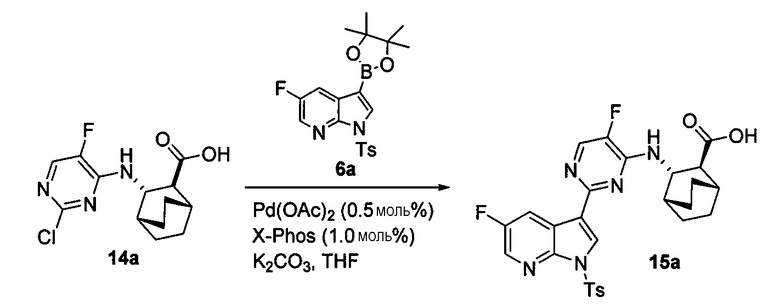
[0268] К 14а (91,4 г, 305 ммоль), 6а (158,6 г, 381 ммоль, 1,25 экв.), Pd (OAc)2 (0,34 г, 1,5 ммоль, 0,5 моль %), Х-Phos (1,45 г, 3,0 ммоль, 1,0 моль %), и К2CO3 (168,6 г, 1220 ммоль, 4 экв.) добавляли THF (731 мл, 8 объемов) и воду (29 мл, 0,32 объема). Реакционную смесь продували N2 в течение 30 мин, затем нагревали до 65-70°С и перемешивали в течение 5 часов. ВЭЖХ-анализ реакционной смеси показал 99,3% превращения. Реакционную смесь охлаждали до 22-25°С и добавляли воду. Смесь перемешивали, фазам давали разделиться и водную фазу удаляли. Раствор 18 масс.% NaCl в воде (полунасыщенный водный раствор NaCl) добавляли к органической фазе, и рН смеси доводили до 6,0-6,5 с помощью 2 н. HCl. Фазам давали разделиться и водную фазу удаляли. Органическую фазу концентрировали до минимального объема и добавляли ацетонитрил. Процесс повторяли еще один раз, и добавляли ацетонитрил до получения конечного объема 910 мл (10 объемов). Суспензию нагревали до 80-85°С в течение 6 часов, затем охлаждали до 20-25°С. Суспензию перемешивали в течение 2 часов, а затем фильтровали. Твердые вещества промывали ацетонитрилом с получением 15а (161 г, выход 89%).
[0269] Получение Соединения (1): Стадия детозилирования
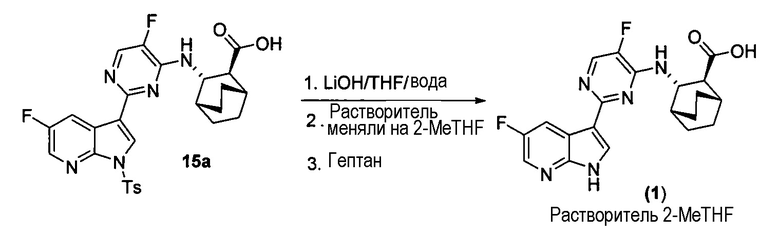
[0270] К 15а (25 г., 45.2 ммоль) добавляли THF (125 мл, 5 объемов), затем полимер МР-ТМТ (6,25 г, 25 масс.%). Смесь перемешивали при 20-25°С в течение 16 часов и фильтровали, промывая 1 объемом THF. Процесс обработки полимера и фильтрацию повторяли. Раствор THF концентрировали до 5 объемов. К смеси при 22-25°С добавляли водный раствор 2М LiOH (90,3 мл, 4 экв.). Реакционную смесь нагревали до 40-45°С и перемешивали в течение 5 часов. ВЭЖХ анализ показал 99,7% превращения. Реакционную смесь охлаждали до температуры 22-25°С и добавляли MTBE (50 мл, 2 объема). Происходило разделение фаз. Нижнюю водную фазу собирали. Водную фазу экстрагировали MTBE. Нижнюю водную фазу собирали. К водной фазе добавляли 2-MeTHF и полученную смесь перемешивали. Значение рН смеси доводили до 6,0-6,5, а нижнюю водную фазу удаляли. Органическую фазу промывали буфером с рН 6,5. Органическую фазу концентрировали до 85 мл, разбавляли 2-MeTHF (150 мл) и концентрировали до конечного объема 180 мл. Полученную взвесь нагревали до 70-75°С и перемешивали до полного растворения, затем охлаждали до 45-50°С с получением взвеси. Взвесь перемешивали в течение 1 часа, а затем добавляли гептан (180 мл). Взвесь охлаждали до температуры 20-25°С в течение 1 часа и перемешивали в течение 16 часов. Партию фильтровали, промывали гептаном твердые продукты. Твердые продукты сушили с получением сырого сольвата Соединения (1)·2-MeTHF, выход 79%.
[0271] Пример 3: Пример 3: Образование полиморфов соли HCl Соединения (1)
[0272] 3А: Получение Формы А соли HCl Соединения (1) •1/2H2О
[0273] Форма А соли HCl Соединения (1)•1/2H2O получали путем смешивания сольвата 2-метил-тетрагидрофурана (2-MeTHF) (1 эквивалент) Соединения (1) (Соединение (1)•1 (2-MeTHF)) с хлористым водородом в смеси воды и органического растворителя(ей), причем смесь из воды и органического растворителя(ей) имела активность воды 0,05-0,85. Конкретные условия реакции, используемые приведены в таблице 1 ниже.
[0274]
Условия реакции, использованные для получения Формы А соли HCl Соединения (1)•1/2H2О
(мг) 1
(2-MeTHF)
(мл)
HCl (мл)
[0275] Альтернативно, Форму А соль HCl Соединения (1)•1/2H2O также получали с помощью следующих методик: Методика A: Соединение (1)•2-MeTHF (953 г, 2,39 моль) помещали в реактор с рубашкой, 30 л, и обрабатывали IPA (15 л) и водой (0,57 л). Начинали перемешивание и реакционную смесь нагревали до 73°С для получения раствора всех компонентов, затем охлаждали до 50-55°С. При 50-55°С реакционную смесь обрабатывали свежеприготовленным раствором HCl в IPA (0,83 М, 4,34 л), медленно добавляя в течение 4 часов. Отбирали образцы реакционной смеси для проверки правильной формы с помощью XRPD. После добавления холодильную машину программировали на понижение до 0°С в течение 480 мин при перемешивании. После подтверждения формы анализом XRPD, взвесь фильтровали на двух фильтрах. Реактор промывали 3 л IPA и каждый осадок на фильтре промывали ~1,5 л IPA промывки IPA из реактора. Лепешки оставляли сохнуть на воздухе в течение ночи с помощью всасывания. Лепешки затем помещали в центробежную сушилку без нагревания в вакууме с продувкой N2 (22 в HG) в течение 24 часов. Остаточный растворитель и анализ воды показал 505 промиль IPA, 8 промиль 2-Me-THF и примерно 2,15% H2О. Вещество вынимали из печи и перемалывали до распределения с получением 805 г соли HCl Соединения (1)•1/2H2О. Методика B: Альтернативно, вместо IPA использовали ацетон, но таким же образом, как описано выше в методике А, с получением соли HCl Соединения (1)•1/2H2О.
[0276] Данные XRPD и C13SSNMR Формы А соли HCl Соединения (1)•1/2H2O показаны на Фиг.1 и 2, соответственно. Некоторые наблюдаемые пики XRPD и пики C13SSNMR суммированы в Таблицах 2 и 3, соответственно.
[0277]
Получение Формы А соли HCl Соединения (1)•1/2H2О
[0278]
Пики С13 SSNMR Формы А соли HCl Соединения (1)•1/2H2О
[0279] Было обнаружено, что полученная Форма А соли HCl Соединения (1)•1/2H2O была стабильна в следующих системах растворителей (но ими не ограничиваясь): хлорбензол, циклогексан, 1,2-дихлорэтан, дихлорметан, 1,2-диметоксиэтан, гексан, 2-метоксиэтанол, метилбутилкетона, метилциклогексан, нитрометан, тетралина, ксилол, толуол, 1,1,2-трихлорэтан, ацетон, анизол, 1-бутанол, 2-бутанол, бутилацетат, трет-бутиловый эфир, кумол, этанол, этилацетат, этиловый эфир, этилформиат, гептан, изобутиловый, изопропилацетат, метилацетат, 3-метил-1-бутанол, метилэтилкетон, 2-метил-1-пропанол, пентан, 1-пропанол, 1-пентанол, 2-пропанол, пропилацетат, тетрагидрофуран, метил-тетрагидрофуран. В частности, для тестов растворимости и стабильности Формы А соли HCl Соединения (1)•1/2H2О, образцы соединения помещали в 2 мл сосуды для ВЭЖХ с 500μл растворителя. Смесь перемешивали при температуре окружающей среды в течение 2 недель, а затем фильтровали с помощью центрифуги. Полученные твердые продукты анализировали с помощью XRPD, растворы анализировали на растворимость с помощью количественного ЯМР против стандарта гидрохинона. Результаты приведены в Таблице 4.
[0280]
Краткое представление данных формы и растворимости для Формы А соли HCl Соединения (1)
[0281] Данные термограмма (данные не показаны) были получены путем помещения образца в платиновую емкость для образцов и при нагревании при 10°С/мин в диапазоне от 300°C до комнатной температуры. Данные термограммы показали потерю массы, равную 2,1%, при от 30°до 170°С, что согласуется с теоретическим полугидратом (2,0%).
[0282] Данные DSC-термограммы были получены (данные не показаны) путем нагревания образца при 10°С/мин в диапазоне от 300°C до комнатной температуры. DSC-термограмма показала температуру дегидратации, равную 50-100°С, а затем температуру плавления/разложения, равную 200-260°С.
[0283] 3B: Получение Формы F соли HCl Соединения (1)•3Н2О
[0284] Форма F соли HCl Соединения (1)•3Н2O может быть получена путем получения взвеси Формы А соли HCl Соединения (1)•1/2H2O в изо-пропаноле и воде, или ацетоне и воде, или в воде (со значением активности воды, равной или большей 0,9).
[0285] Например, взвесь 100 мг Формы А соли HCl Соединения (1)•1/2H2О, в 5 мл изопропанол/вода или ацетон/вода при активности воды, равной 0,9, перемешивали при температуре окружающей среды в течение ночи. Супернатант удаляли и сушили под несильной струей воздуха полученное твердое вещество с получением Формы F соли HCl Соединения (1)•3Н2О.
[0286] Данные XRPD и C13SSNMR Формы F соли HCl Соединения (1)•3Н2O показаны на Фиг.3 и 4, соответственно. Некоторые наблюдаемые пики XRPD и пики C13SSNMR представлены в Таблицах 5 и 6, соответственно.
[0287]
Пики XRPD Формы F соли HCl Соединения (1)•3Н2О
[0288]
Пики С13 SSNMR Формы F соли HCl Соединения (1)•3Н2О
[0289] MDSC-термограмму получали (данные не показаны) путем нагревания образца при 2°С/мин до 350°C от -20°C и изменения на±1°C каждые 60 сек. MDSC-термограмма показала дегидратацию при ниже 150°С, плавление и перекристаллизацию при от 150°С до 200°С и деградацию при температуре выше 250°C.
[0290] Также был проведен термогравиметрический анализ (ТГА) формы. Термограмма показала потерю массы, равную 12%, при до 125°С, что близко к теоретическому тригидрату (11%). Вторая стадия потери массы ниже 200 °С была показана с помощью TGA-МС, которая представляет собой потерю HCl. Температура плавления/разложения составляет около 270-290°С.
[0291] 3C: Получение Формы D соли HCl Соединения (1)
[0292] Безводная Форма D соли HCl Соединения (1) может быть получена в целом путем дегидратации Формы А соли HCl Соединения (1)•1/2H2О. Дегидратация может быть осуществлена путем нагрева или сухой продувки азотом, или их комбинации. Например, 2 мг Формы А соли HCl Соединения (1)•1/2H2O нагревали на горячей плитке, получая желаемую безводную форму D при около 85°C.
[0293] Данные XRPD и C13 SSNMR безводной Формы D соли HCl Соединения (1) показаны на Фиг.5 и 6, соответственно. Некоторые наблюдаемые пики XRPD и пики C13SSNMR суммированы в Таблицах 7 и 8, соответственно.
[0294]
Пики XRPD для Формы D безводной соли HCl Соединения (1)
[0295]
Пики C13 SSNMR Формы D безводная соль HCl Соединения (1)
[0296] 3D: Тесты на активность воды
[0297] Конкурентное исследование взвеси Формы А соли HCl Соединения (1)•1/2H2O с затравкой Формы F соли HCl Соединения (1)•3Н2О, в воде с активностью от 0,0 до 0,8 из изопропиловый спирт/вода, показали, что форма А является наиболее стабильной формой среди Формы D безводной соли HCl Соединения (1) Формы F соли HCl Соединения (1)•3Н2О и Формы А соли HCl Соединения (1)•1/2H2O, через приблизительно 2 недели перемешивания при условиях окружающей среды. При IPA/вода с активностью 0,9, Форма А соли HCl Соединения (1)•1/2H2O превращалась в Форму F соли HCl Соединения (1)•3Н2О. Результаты этих исследований приведены в Таблице 9 ниже.
[0298]
Тесты на активность воды соли HCl Соединения (1)•1/2H2O в смесях IPA/вода
Активность (aw)
[0299] 3F: Аморфные соли HCl соединения (1)
[0300] Аморфная соль HCl Соединения (1) может быть получена путем обработки соли Me2NEt Соединения (1) (1,985 г) в воде и 2-MeTHF с 1,05 экв. NaOH, с последующей обработкой HCl для удаления амина и выхода из водного слоя (рН 2-3). Полученную взвесь концентрировали для удаления каких-либо органических соединений, а затем фильтровали. Полученное твердое вещество промывали небольшими порциями воды и сушили. Соль Me2NEt Соединения (1) получали в соответствии с WO2010/148197, а затем использовали обычное хиральное разделение и очистку: хиральная хроматография SCF с модифицирующим агентом, который включал Me2NEt (который образует соль Me2NEt Соединения (1)).
[0301] Пример 4: Образование полиморфов свободного основания Соединения (1)
[0302] 4А: Получение Формы А свободного основания Соединения (1)
[0303] Форму А свободного основания Соединения (1) (то есть, Форма А Соединения (1)) получали по следующей методике: Сырое аморфное свободное основание Соединения (1) (приблизительно 135 г) переносили в реактор объемом 4 л с рубашкой, и в реактор помещали этанол (2,67 л) и воду (0,325 л) (10% водный раствор). Смесь нагревали с обратным холодильником. Добавляли воду (300 мл) к полученной смеси на стадии 2) для получения 20% водного раствора. Полученную смесь затем охлаждали до 55°C (скорость=-1°С/мин), а затем выдерживали в течение 30 минут. Кристаллическую затравку свободного основания Формы А Соединения (1) (1,5 г, 3,756 ммоль) затем добавляли в охлажденную смесь, и полученную смесь выдерживали в течение 30 минут пока продукт осаждался. Затравка кристаллического свободного основания Формы А Соединения (1) была получена суспендированием аморфного свободного основания Соединения (1) (20 мг) в нитрометане (0,5 мл). Дополнительные затравки кристаллического свободного основания Формы А Соединения (1) получали путем получения взвеси аморфного свободного основания Соединения (1) (900 мг) в ацетонитриле (10 мл) вместе с затравками, полученными с использованием нитрометана. В смесь, содержащую затравку кристаллического свободного основания Формы А Соединения (1), медленно добавляли воду (795,0 мл) для получения 40% водного раствора. Полученную смесь медленно охлаждали до 0°C (~ -10°С/час), а затем выдерживали в течение 2 часов. Твердые вещества затем фильтровали и сушили на воздухе, а затем дополнительно сушили в сушильном шкафу при 60°С в течение 18 часов.
[0304] Альтернативно, использовали сольват 2-метил-THF свободного основания Соединения (1) вместо аморфного свободного основания Соединения (1) и аналогичными образом, как описано выше, также получали Форму А свободного основания Соединения (1).
[0305] Было обнаружено, что полученная Форма А Соединения (1) является стабильной в следующих системах растворителей (но ими не ограничиваясь): ацетонитрил, хлорбензол, хлороформ, циклогексан, 1,2-дихлорэтан, дихлорметан, 1,2-диметоксиэтан, этиленгликоль, формамид, гексан, метилбутилкетона, метилциклогексан, N-метилпирролидинон, нитрометан, тетралина, толуол, 1,1,2-трихлорэтан, уксусная кислота, анизол, 1-бутанол, бутилацетат, кумола, этилацетат, этиловый эфир, этилформиат, гептан, изобутиловый, изопропилацетат, 3-метил-1-бутанол, 2-метил-1-пропанол, пентан, пропилацетат, вода, вода-изопропанол (1:3 об./об.) и вода-ацетонитрил (1: 1 об./об.; 1:3 об./об.).
[0306] Данные XRPD и C13 SSNMR Формы А Соединения (1) представлены в таблицах 10 и 11, соответственно.
[0307]
XRPD пики формы A соединения (1)
[0308]
Пики C13 SSNMR Формы A Соединения (1)
[0309] Термогравиметрический анализ продукта, Форма А Соединения (1), был проведен (данные не показаны) на TA Instruments TGA модель Q500 путем помещения образца в платиновую емкость для образцов и последующего нагревания емкости при 10°С/мин в диапазоне от 300°C до комнатной температуры. Термограмма показала, разложение начинается приблизительно при 293°С.
[0310] ДСК-термограмма для Формы А Соединения (1) также была получена с помощью ТА Instruments DSC Q200. Образец формы нагревали при 10°С/мин до 350°С. ДСК-термограмма показала температуру плавления, равную приблизительно 278°С.
[0311] 4B: Получение гидратов свободного основания Соединения (1)
[0312] Гидратированная форма свободного основания Соединения (1) была изоморфна как формы А свободного основания Соединения (1). Форма А свободного основания Соединения (1) может свободно преобразовываться в гидратную форму, если она подвергается воздействию высокой влажности и возвращаться обратно, если влажность уменьшается. В соответствии с изменением фазы, определенной с помощью экспериментов DSC (данные не показаны), температура перехода была близка к температуре окружающей среды, и изменялась в зависимости от активности воды. Например, при температуре окружающей среды отмечалась гидратная форма, где активность воды была больше 0,6, например, 0,6-1,0.
[0313] 4C: Получение аморфного свободного основания Соединения (1)
[0314] Реакцию присоединения Сузуки проводили помещением хлоропиримидина, Соединение 13а, боронового эфира Соединения 6а, катализатора Pd (OAc)2 и лиганда (Х-Phos) в 10 объемов 2-MeTHF. Эту смесь нагревали до 65°С и добавляли 2 объема 50% водного раствора K3PO4 со скоростью, позволяющей поддерживать реакционную смесь при температуре 65°С. Обе реакции давали полное преобразование, затем смеси охлаждали до 20°С и фильтровали через целит. Водные слои удаляли, органические слои промывали 5% водным раствором NaCl, а затем концентрировали досуха с получением приблизительно 3,5 кг каждого в виде темно-зеленой пасты. Сырое масло разделяли на 4 равные части, суспендировали с 400 г SiO2 и 500 г флорисила и элюировали через 2,3 кг SiO2 на колонке с гептаном/EtOAc (фракции от 5:1 до 3: 1,2 л), объединяя все фракции, содержащие продукт. Эти фракции концентрировали досуха с получением приблизительно 2,9 кг Соединения 21а.
[0315] Соединение 21а растворяли в 10 объемах (25 л) СН3CN и обрабатывали 4 экв. HCl (4,31 л 4 н. HCl в 1,4-диоксане) при температуре 70°С в течение 15 часов. Оценка ВЭЖХ показала 100% завершение реакции и мелкую взвесь охлаждали до 20°С в течение 1 часа. Добавляли ТВМЕ (28 л, 11 об.) при 0,5 л/мин, взвесь при этом становилась густой (желеобразной) в конце добавления. Через 4-5 часов перемешивания взвесь становилась значительно менее густой. Полученные твердые вещества собирали фильтрацией отсасыванием и промывали 3х5 л TBME, с получением лепешки с низкой плотностью и сушили в потоке N2 в течение 3 дней с получением 1,71 кг (выход 86%, AUC чистота 98,9%) Соединения 22а HCl.
[0316] Раствор NaOH (55,60 мл 2М, 111,2 ммоль) добавляли к суспензии Соединения 22а· HCl (10 г, 22,23 ммоль) в 2-MeTHF (100,00 мл) при температуре 20°С. Реакционную смесь перемешивали при 60°С в течение 5 часов, а затем дополнительно при 67°C. После перемешивания в течение приблизительно 22 часов к полученной смеси добавляли 100 мл (10 об.) 2-MeTHF. Партию затем охлаждали до 0°С. К полученной смеси добавляли HCl для того, чтобы довести рН до рН 6,6, с получением сырого свободного основания Соединения (1). Сырое вещество в 60 мл (6 объемов) 2-Ме-THF нагревали до 50°С. В полученную смесь добавляли 50 мл (5 об.) н-гептана в течение 1 часа. Партию затем охлаждали до 20°С. Твердый продукт фильтровали, и твердый продукт затем очищали колоночной хроматографией (EtOAc/гептан, от 2:1 до 4:1). Данные XRPD показали аморфное свободное основание Соединения (1).
[0317] Альтернативно, наблюдали аморфное свободное основание Соединения (1) из смеси Формы А свободного основания Соединения (1) и растворителя, выбранный из 2-этоксиэтанола, 2-метоксиэтанола, трет-бутилметилового эфира, муравьиной кислоты или метилэтилкетона (например, см. Таблицу 13 ниже), которые перемешивали при температуре окружающей среды.
[0318] 4D: Получение сольвата 2-MeTHF свободного основания Соединения (1)
[0319] Соединение (1)•1(2-MeTHF) получали, как описано в примере 2 выше. Данные XRPD приведены в таблице 12.
[0320]
Пики XRPD Соединения (1)•1(2-MeTHF)
[0321] 4F: Растворимость и данных стабильности Формы А свободного основания Соединения (1) и аморфного Соединения (1) в различных системах растворителей
[0322] Растворимость и стабильность Формы А свободного основания Соединения (1) ("Форма А") и аморфного Соединение (1) ("Аморфное") в различных системах растворителей протестировали при температуре окружающей среды так образом, как описано выше, для Формы А соли HCl Соединения (1). Полученные данные приведены в Таблице 13.
[0323]
Данные растворимости и стабильности Формы А свободного основания Соединения (1) ("Форма А") и аморфного Соединения (1) ("Аморфное")
[0324] Пример 6: Составы Соединения (1)
[0325] 6A: Таблетки Соединения (1)
[0326] Композиции
[0327] Для получения таблеток использовали Форму А соли HCl Соединения (1)•1/2H2O (в дальнейшем просто Соединение (1) в Примере 6). Все эксципиенты соответствовали существующим монографиях Европейской Фармакопеи и USP/NF и были приобретены у утвержденных поставщиков.
[0328] Состав состава и размер партии для предварительной грануляции и связывающий раствор для грануляцию приведены в Таблице 14А. Размер партии связывающего раствора включал 100% избыток для калибровки насоса и заполнений линий раствора. Теоретический состав смеси сжатия также приведен в Таблице 14А. Были рассчитаны фактические величины для партии на основании выхода высушенных гранул. Состав и приблизительный размер партии суспензии для пленочного покрытия приведен в Таблице 14В и включал 100% избыток для калибровки насоса и заполнений линий суспензии. Целевое количество пленочного покрытия составило 3,0 масс.%/масс.% от массы таблетки.
[0329]
Композиции таблеток Соединения (1)
(г)
[0330]
Композиция связующего раствора
[0331]
Состав смеси для сжатия
(г)*
[0332]
Композиция для покрывающей пленки и приблизительный размер партии
(г)
[0333] Получение связующего раствора
[0334] Связующий раствор состоял из повидона и воды. Раствор получали на основе 40% водного содержимого в конечной смеси для грануляции. Таким образом, общее количество твердых веществ в растворе (повидон) составило 3,6% (масс./масс.). Избыточное количество 100% получали для заполнения линий и тому подобное. На основании визуального осмотра запуска прогона грануляции получали дополнительные исходные растворы +/- 2% (38-42%) воды в конечной смеси для грануляции. Как правило, взвешивали 87,00 г повидон К30, и 2320,00 г очищенной воды (DI) и при постоянном перемешивании добавляли повидон K30 в контейнер, содержащий DI воды. После добавления контейнер герметизировали для минимизирования испарения, и раствор перемешивали до тех пор, пока все присутствующие твердые вещества полностью не растворялись.
[0335] Схема процесса влажного гранулирования
[0336] Влажное гранулирование осуществляли с помощью методик, описанных ниже: Взвешивали избыточное (10%) количество Соединения (1), Avicel PH-101, лактозы Fastflo и кроскармеллозы натрия (см. Таблицу 14А). Их сортировали с помощью ручного сита 20 меш или конической мельницы, снабженной решетчатым ситом с размером ячеек 813 мкм при 1000 оборотах в минуту (для U5 Quadro Co-mill). Отсортированные вещества помещали в отдельные пакеты или контрейнеры. Вещества затем переносили в блендер и перемешивали в течение 15 минут, как привило, при 15 оборотах в минуту. Смешанные вещества размалывали, используя конусную мельницу U5 Quadro, оснащенную ситом с квадратными отверстиями 4 мм при 1000 оборотах в минуту. Измельченные вещества снова смешивали, повторяя стадию смешивания. Повторно смешанные вещества затем подавали в двухшнековый гранулятор. Объемную влажную смесь для гранулирования подавали в гранулятор, используя весовой питатель непрерывного действия (K-Tron или аналогичный). Полученные вещества затем гранулировали. Связующую жидкость (см. Таблицу 14а) вводили в двухшнековый гранулятор, используя перистальтический насос. Отношение скорости подачи раствора на скорость подачи порошка составляло 0,4095. Например, если скорость подачи порошка была 15,00 г/мин, то скорость подачи раствора была 0,4095*15,00=6,14 г/мин, при содержании воды 40% (в пересчете на сухую массу). Субпартии гранул собирали в сушильные лотки с заранее определенным весом. Собранные вещества равномерно разбрызгивали на лоток и сушили вещество в печи с образованием высушенных гранул. Высушенные гранулы помещали в K-Tron для непрерывного истощения подачи вещества в конусной мельнице, а затем перемалывали.
[0337] Экстрагранулярное смешивание и процесс сжатия
[0338] Экстрагранулярное смешивание и процесс сжатия проводили в соответствии с методиками, описанными ниже: Взвешивали количество экстрагранулярных наполнителей на основании состава смеси для сжатия. Взвешенные наполнители сортировали, используя U5 Comil с ситом 32C и круглый стержень лопастной мешалки при 1000 оборотах в минуту. Измельченные гранулы Соединения (1) сначала добавляли в блендер, содержащий отсортированные Avicel PH-102 и Ac-Di-Sol. Их смешивали в течение 8 минут при 16 оборотах в минуту. Стеарил натрия (SSF) сортировали через ручное сито с отверстиями 50 меш в подходящий контейнер. Часть экстрагранулированной смеси, по массе примерно в 10 раз больше количества SSF, помещали в контейнер с SSF и контейнер смешивали в течение 30 секунд перед добавлением смеси в бункерный смеситель. Все вещества затем смешивали в течение 2 минут при 16 оборотах в минуту. Конечную смесь затем прессовали в соответствии с заданными параметрами процесса сжатия таблеток.
[0339] Процесс нанесения покрытия
[0340] Пленочное покрытие наносили на сердцевины таблеток в емкости для нанесения покрытия Vector VPC 1355 в виде 15% масс./масс. Opadry II White # 33G водной суспензии. Целевое покрытие составляло 3,0% масс./масс. масса от сердцевины таблетки в приемлемом диапазоне от 2,5% до 3,5%. Для достижения этой цели, количество суспензии для покрытия, эквивалентное увеличению массы на 3,2%, распыляли, что давало 3,0% покрытия, если предположить, что эффективность покрытия составляет 95%.
[0341] Внутривенные составы (вв) Соединения (1)
[0342] Форму А соли HCl Соединения (1)•1/2H2O (в этом примере в дальнейшем просто Соединение (1)) получали в виде 2 мг/мл раствора для внутривенного (вв) введения. Состав раствора вместе с эталонным качеством и функцией каждого компонента, представлены в Таблицах 15 и 16.
[0343]
Состав раствора носителяa
(мг/50г вв раствора)
(% масс./масс.)
[0344]
Сравнение внутривенного раствора a Соединения (1)
(мг/50г вв раствора)
(% масс./масс.)
Лекарственное вещество представляло собой полугидрат соли HCl. Количество лекарственного вещества рассчитывали на основе эквивалента активного безводного свободного основания, где коэффициент преобразования из свободного основания в полугидрат соли HCl составляет 1,11.
[0345] Дополнительные фармацевтические композиции для вв введения также были получены сходным образом, как описано выше, но дополнительно содержали комплексообразующий агент, такой как Tween® 80, Cremophor®, Captisol® и Cavitron®, в 100 мМ фосфатного буфера. Данные показаны на Фиг.7А (Tween® 80), 7B (Cremophor®), 7C (Captisol®) и 7D (Cavitron®). Как показано на Фиг.7А-7D, например, композиции, имеющие приблизительно 5,0 масс.% комплексообразующего агента, дают в растворах от 5 мг/мл до 20 мг/мл Соединения (1).
[0346] Пример 7: In vivo анализ комбинации Соединения (1) вместе или без оселтамивира
[0347] Инфицированных мышей обрабатывали носителем или повышающимися уровнями доз Формы А соли HCl Соединения (1)·1/2 H2O в комбинации с клинически значимой дозой Озельтамивира, начиная через 48 часов после заражения гриппом А или за 2 часа до заражения гриппом B.
[0348] Способы: В этих исследованиях Форма А соли HCl Соединения (1), полугидрат, (в Примере 7 в дальнейшем просто Соединение (1)) была включена в состав вместе с носителем, содержащим 0,5% (масс./масс.) МС (Sigma-Aldrich, St Louis, MO) с получением гомогенной суспензии, и доза соединения была основана на соли HCl Соединения (1), полугидрат. Озельтамивир был включен в состав с дистиллированной деионизированной водой с получением гомогенной суспензии. Комбинация Соединения (1) с озельтамивиром была включена в состав с носителем, содержащим 0,5% (масс./масс.) МС. Эти комбинированные состав получали в начале каждого исследования и хранили при 4°С в течение до 10 дней при перемешивании в темноте. Все составы и носители вводили мышам с помощью перорального зонда в объеме дозы 10 мл/кг.
[0349] Самцов мышей BALB/C (5-7 недель, 17-19 г) анестезировали и заражали летальной дозой вируса гриппа A/PR/8/34 или В/Мass/3/66, адаптированного для мышей, путем интраназальной инстилляции. Восемь мышей были включены в исследуемую группу. Лечение начинали через 48 часов после заражении вирусом гриппа А или за 2 часа до заражения вирусом гриппа B. Носитель (10 мл/кг) и Соединение (1) в дозах 0,1-10 мг/кг вводили самостоятельно или в комбинации с 10 мг/кг Озельтамивиром, перорально (PO), два раза в день (BID) в течение 10 дней в исследовании гриппа А. Носитель (10 мл/кг) и Соединение (1) при дозах 1-10 мг/кг вводили самостоятельно или в комбинации с 10 мг/кг Озелтамивира, перорально (PO), два раза в день (BID) в течение 10 дней в исследовании гриппа B. Мышей взвешивали и ежедневно осматривали на наличие признаков заболевания в течение 21 дней после заражения. Кроме того, функции легких контролировали с помощью естественного WBP (Buxco, Troy, NY).
[0350] Грипп A/PR/8/34 (VR-1469) и грипп B/Mass/3/66 (VR-523) получали из АТСС (Manassas, VA). Исходные растворы получали стандартными способами, известными в данной области. Вкратце, вирус пассировали при низкой множественности инфекции в клетки Мадин-Дарби почек собак (клетки MDCK, CCL-34, ATCC), супернатант собирали через приблизительно 48 часа и центрифугировали при 650xg в течение 10 минут. Исходные растворы вируса замораживали при -80°С перед использованием. Титр вируса (TCID50/мл) рассчитывали по методу Спирмена-Каргер после серийных разведений образца вируса, инфицируя культуры MDCK в повторе, и измеряли цитопатический эффект (CPE) на основе содержания АТФ в 96 часов (CellTiter-Glo, Promega, Madison WI).
[0351] Мышей взвешивали каждый день в течение 21 дня после заражения. Данные о массе тела анализировали с помощью двухфакторного ANOVA и пост-теста Бонферрони для сравнения групп. Р-значения, равные менее 0,05, считались значимыми.
[0352] Мышей ежедневно наблюдали в течение 21 дней после заражения гриппом. Любую мышь, которая оценивалась положительной в течение четырех из последующих шести наблюдений (> 35% потеря BW, взъерошенный мех, сгорбившаяся поза, респираторный дистресс синдром, снижение подвижности или гипотермия) считали умирающей, после чего ее подвергали эвтаназии и считали это как смерть в соответствии с правилами, установленными Vertex Institutional Animal Care and Use Committee. Данные выживаемости анализированы с использованием метода Каплана-Мейера.
[0353] Мыши подвергали естественному WBP (Buxco, Troy, NY). Функцию легких выражали как длительность паузы (индекс Penh), безразмерное рассчитанное значение, которое отражает легочное сопротивление. Это значение получали на основании изменений в поддерживающем давление контейнере, которое колеблется в результате изменений дыхания животного. Бронхостеноз дыхательных путей животного влияет на поток воздуха, а следовательно, на давление в поддерживающем давление контейнере. Изменения давления отслеживали во время выдоха (PEP) и вдоха (PIP). Значения Penh вычисляли по формуле Penh=пауза х PEP/PIP, где "пауза" отражает время выдоха. Мыши акклиматизировались в камере плетизмографии в течение 15 минут, а затем собирали данные с интервалом в одну минуту, в среднем в течение 10 мин, и выражали в виде абсолютных значений Penh. Данные анализировали с помощью двухфакторного ANOVA и пост-теста Бонферрони для сравнения групп. Р-значения, равные менее 0,05, считались значимыми.
[0354] Результаты: Соединение (1) оценивали в комбинации с Озельтамивиром на способность предотвращать смертности и заболеваемость, уменьшать потери BW и предотвращать изменение или восстанавливать функцию легких на модели легочной инфекции гриппа у мышей по сравнению с эффектами Соединения (1) или Озельтамивиром, вводимыми самостоятельно. Комбинация не показала отрицательного влияния на эффективность каждого из препаратов по сравнению с каждым препаратом, вводимым самостоятельно. Кроме того, комбинация лечения показал синергию лечения гриппа А при недостаточной дозе введения каждого соединения (0.3 и 10 мг/кг Соединения (1) и Озельтамивира, соответственно) самостоятельно, тогда как комбинация увеличивала выживаемость с 0 до 100%. Соединение (1) обладает незначительной активностью против гриппа B in vivo (как ожидалось от имеющихся in vitro данных) и не влияет на эффективность Озельтамивира.
[0355] Модель гриппа A у мыши: Все контроли с носителем заболевали при исследовании на 9 или 10 день. Лечение только 1, 3 и 10 мг/кг Соединения (1) BID обеспечивало полную защиту от смерти, уменьшало потерю BW и восстанавливало функцию легких, если дозу начинали вводить через 48 часов после инфицирования по сравнению с контрольной группой с носителем (Таблица 17). Обработка 0,1 и 0,3 мг/кг Соединением (1) и 10 мг/кг Озелтамивиром, вводимыми самостоятельно, не защищало от смерти, не уменьшало потерю BW или не восстанавливало функцию легких, если лечение начинали через 48 часов после инфицирования вирусом гриппа А. Интересно, что 0,3 мг/кг Соединения (1) И Озельтамивира, вводимых вместе через 48 часа после инфицирования вирусом гриппа А, давали полную защиту от смерти, снижали потерю BW и восстанавливали функцию легких.
[0356]
Данные эффективности in vivo Соединения (1) с или без введения Озельтамивира через 48 часов после заражения гриппом A
мивир
мг/кг
(День 3)
[0357] Модель гриппа В у мыши: Все контроли с носителем заболевали при исследовании на 7 или 8 день. Введение 1, 3 или 10 мг/кг Соединения (1) самостоятельно за 2 часа до инфицирования гриппом B и затем BID в течение 10 дней не давало существенной защиты от заболеваемости, потери BW или нарушения функции легких по сравнению с контрольными группами. Озельтамивир вводили в дозе 10 мг/кг по отдельности или в сочетании с 1, 3 или 10 мг/кг Соединения (1) за 2 часа до инфицирования гриппом B, что обеспечивало полную защиту от смерти, снижало потери BW и восстанавливало функцию легких (Таблица 18).
[0358]
Данные эффективности in vivo Соединения (1) с или без введения Озельтамивира через 48 часов после заражения гриппом В
(8-й день)
(%)
(День 6/7)
[0359] Пример 8: Анализ in vivo комбинации Соединения (1) вместе с Озельтамивиром
[0360] Зараженных мышей обрабатывали носителем или повышающейся дозой Формы А соли HCl Соединения (1)•1/2H2O (в примере 8 в дальнейшем просто Соединение(1)) в комбинации с занамивиром, начиная за 24 часа до заражения вирусом гриппа 5х103 TCID50 A/PR/8/34. Суспензии для заражения гриппом A и Соединения (1) получали способом, аналогичным описанному выше в Примере 7. Зараженных мышей обрабатывали один раз IN (интраназально) занамивиром дозой 0,3 мг/кг, 1 мг/кг или 3 мг/кг за 24 часа до IN заражения 5x103 TCID50 A/PR/8/34, и Соединением (1) в дозе 0,1 мг/кг, 0,3 мг/кг или 1 мг/кг в течение 10 дней, начиная за 2 часа до заражения 5x103 TCID50 A/PR/8/34.
[0361] Результаты суммированы в Таблицах 19А и 19В ниже. Как показано в Таблице 19а ниже, комбинированная терапия Соединением (1) и занамивиром давала особые преимущества в выживании (Таблица 19а). Коэффициент эффективности, составной показатель выживания, потери массы тела и функции легких (% выживаемости/(% потери массы тела на 8-й день) * (Pehn на 6-й день)) суммированы в Таблице 19B.
[0362]
Выживаемость: Комбинированная терапия Соединением (1) и Занамивиром
1-я доза за 2 часа до инфицирования
[0363]
Коэффициент эффективности: Комбинированная терапия Соединением (1) и Занамивиром.
1-я доза за 2 часа до инфицирования
[0364] Пример 9: Профилактическая эффективности и эффективность после инфицирования Соединения (1) на модели инфекции гриппа A у мыши
[0365] Материалы и методы
[0366] Животные: Самки весом 18-20 г мышей BALB/c были получены из Jackson Laboratories (Bar Harbor, ME) для противовирусного эксперимента. Животных содержали на стандартном корме для грызунов и водопроводной воде <ad libitum>. Их помещали в карантин за 48 часов перед использованием.
[0367] Вирус: Вирус гриппа A/Калифорния/04/2009 (pndH1N1), адаптированный для мышей, получали от доктора Елены Говорковой (St. Jude Childrenʹs Research Hospital, Memphis, TN). Количество исходного вируса увеличивали в клетках MDCK, а затем титровали на летальность в мышах BALB/с. Вирус гриппа A/Victoria/3/75 (H3N2) получали из Американской коллекции типовых культур (ATCC) (Manassas, VA). Вирус пассировали семь раз из мышей адаптированным мышам, а затем один раз перевивали в клетки MDCK. Вирус дополнительно титровали на летальность в мышах BALB/с с получением подходящей смертельной дозы для заражения. Вирус грипп A/Vietnam/1203/2004 (H5N1) получали от доктора Jackie Katz из Centers for Disease Control (Atlanta, GA). Мышам вводили смертельную дозу вируса (5 MLD50, 5 PFU/мышь), которая ранее приводила к смерти в промежутке между 6 и 13 днем, 90-100% смертности на 10-й день при такой дозе.
[0368] Соединения: Озельтамивир (Тамифлю®) получали из местной аптеки. Каждая капсула Тамифлю содержит 75 мг активного компонента, озельтамивира карбоксилата после метаболизма в организме. Доза озельтамивира была основана на этом измерении. Форма А соли HCl Соединения (1) полугидрат (в Примере 9 в дальнейшем просто Соединение (1)) использовали для исследования, и доза соединения была основана на соли HCl Соединения (1) полугидрат. Оба соединения (1) и озельтамивир готовили в 0,5% растворе метилцеллюлозы (Sigma, St. Louis, MO) для перорально приема через зонд (п.о.) для введения мышам.
[0369] Ход эксперимента: Мышей анестезировали внутрибрюшинной инъекцией кетамина/ксилазина (50/5 мг/кг), и животных интраназально инфицировали 90 мкл суспензии вируса гриппа. Заражение вирусом проводили приблизительно четырьмя 50% летальными инфекционными дозами для мышей. Обработку проводили два раза в день (с интервалом 12 часов) в течение 10 дней, начиная за 2 часа до заражения вирусом или через 48 часа после заражения, как указано. Параметрами для оценки инфекции были: выживаемость, день смерти в среднем, изменения массы тела и параметров легочной инфекции (кровоизлияние в баллах, масса и титр вируса). Животных взвешивали по отдельности каждый день в течение 21 дня инфекции. Мышей, которые умерли в течение первых шести дней периода лечения, признавали умершими по другой причине, а не от инфекции вирусом гриппа, и их исключали из общего подсчета. Животных, которые умирали, учитывали.
[0370] Для оценки параметров легочной инфекции получали легкие от умерщвленных животных (первоначально 5 животных на группу были отделены для этой цели). Баллы кровоизлияния в легкие оценивали путем визуального осмотра на изменение цвета от розового до синюшного. Они возникали в областях легких, а не как постепенное изменение цвета всего легкого к более темному. Баллы кровоизлияния находились в диапазоне от 0 (нормальный) до 4 (все легкое имело синюшный цвет), и, таким образом, являются непараметрическим измерением. Легкие взвешивали и затем замораживали при -80°C. Затем размороженные легкие гомогенизировали в 1 мл клеточной культуральной среды, жидкости супернатанта центрифугировали для удаления твердых частиц, и жидкие образцы снова замораживали при -80°C. После получения 96-луночных планшетов клеток MDCK, образцы оттаивали, серийно разводили с 10-кратным увеличением разведения и титровали методом конечных точек в планшетах (1), используя 4 микролунки на разведение. Титры вируса рассчитывали как log10 50% дозы инфицированной культуры клеток на грамм легочной ткани (log10 CCID50/г).
[0371] Статистический анализ: Кривая Каплана-Меира для множественных групп сравнений анализировали с помощью лог-рангового критерия Мантеля-Кокса для определения статистической значимости. Затем парные сравнения проводили с помощью теста Гехана-Бреслоу-Вилкоксона. Относительное экспериментальное значение доводили до предельной величины порога значимости с поправкой Бонферрони на основании числа сделанных сравнений обработок. Сравнение среднего значения дня смерти и среднего балла кровоизлияния в легкие анализировали с помощью теста Крускала-Уоллиса, а затем тестом Данна множественных сравнений. Среднюю массу тела, вес легких и log10 титры вируса легких оценивали с помощью ANOVA, предполагая равную величину отклонения и нормальное распределение. После ANOVA индивидуальные значения обработки сравнивали тестом множественных сравнений Тьюки-Крамера. Анализы проводили с помощью программного обеспечения Prism® (GraphPad Software, San Diego, CA).
[0372] Результаты и обсуждения
[0373] Профилактическая доза ответ Соединения (1) исследовали на модели гриппа A у мыши. Введение дозы носителя или Соединения (1) начинали за 2 часа до инфицирования и продолжали два раза в день в течение 10 дней. Результаты представлены в Таблицах 20 и 21. Все мыши, которые получали только носитель, заболевали на 9-й день исследования и теряли, в среднем, ~ 32% массы тела (BW). Соединение (1), вводимое в дозах 1, 3 или 10 мг/кг BID, обеспечивало полную выживаемость и дозозависимое снижение потери BW. Соединение (1), вводимое в дозе 0,3 мг/кг BID, обеспечивало некоторое улучшение выживаемости (2/8 мышей), хотя у мышей была значительная потеря BW. В том же самом эксперименте мышам вводили озельтамивир в дозе 10 мг/кг BID, что клинически-эквиваленто дозе для человека (на основании AUC). Все мыши, получавшие озельтамивир, выживали и имели сходный профиль потери веса, как и мыши, которым вводили 1 мг/кг BID Соединения (1). Соединение (1) также имело эффективность на этой модели заражения вирусом гриппа A/Vietnam/1203/2004 (H5N1), если его вводили через 48 часов после инфицирования и продолжали вводить дозу BID в течение 10 дней (Таблица 22). Введение Соединения (1) в дозе 10 мг/кг давало полную защиту, как показано в Таблице 20.
[0374]
Профилактические эффекты Соединения (1) и Озельтамивира на инфекцию, вызванную гриппом A/California/04/2009 (pndH1N1) у мышей BALB/с (профилактика)
Всего
0,9e
b Усредненный день смерти мышей, которые умерли на 21 день или до этого дня,
cLog10 CCID50/г,
d Нижний предел детекции (2,6 log10),
e Незначимое по очень строгому тесту множественного сравнения Данна, но значимое от плацебо (P<0,01) по попарному двустороннему U-критерию Манна-Уитни, * P<0,05, **P<0,01, ***P<0,001, по сравнению с плацебо
[0375]
Эффекты Соединения (1) и Озельтамивира на инфекцию, вызыванную гриппом A/Victoria/3/75 (H3N2) у мышей BALB/с (профилактика)
(10 мг/кг)
b Усредненный день смерти мышей, которые умерли на 21 день или до этого дня,
cLog10 CCID50/г,
dНе значимое по очень строгому тесту множественного сравнения Данна, но значимое от плацебо (P<0,01) по попарному двустороннему U-критерию Манна-Уитни,
e См. сноску «d», но значимое от плацебо при P<0,05 уровне, **P< 0,01, *** P<0,001, по сравнению с плацебо
[0376]
Эффекты лечения (через 48 часов) Соединения (1) и Озельтамивира на инфекцию, вызванную гриппом Influenza A/Vietnam/1203/2004 (H5N1) у мышей BALB/с (профилактика)
(10 мг/кг)
(10 мг/кг)
b Усредненный день смерти мышей, которые умерли на 21 день или до этого дня,
cLog10 CCID50/г
[0377] Пример 10: Эффективность In Vitro Соединения (1) по отношению к штамму испанского гриппа
[0378] Клетки и вирусы. Клетки Madine Darby почек собак (MDCK) первоначально получали из американской коллекции культур Type (ATCC, Manassas, VA) и пассировали, используя стандартные лабораторные методики перед использованием в анализах с инфекцией. Клетки поддерживали при 37°C в среде Игла, модифицированной по методике Дульбекко (DMEM; Invitrogen, Carlsbad, CA) с добавлением 10% фетальной бычьей сыворотки (Sigma-Aldrich, St. Louis, MO), 2 мМ L-глютамина, 10 HEPES, 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина (Invitrogen). Вирус гриппа получали из ATCC, the Virus Surveillance and Diagnosis Branch of the Influenza Division of the Centers for Disease Control and Prevention (CDC; Atlanta, GA) или the Influenza Reagent Resource, Influenza Division, WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza, CDC. Для получения основных растворов вируса клетки MDCK инфицировали при низкой множественности заражения инфекции (MOI) в среде DMEM с добавлением 2 мМ L-глютамина, 10 мМ HEPES, 100 Ед/мл пенициллина, 100 мкг/мл стрептомицина и 1 мкг на мл трипсина, обработанного толилсульфонил фенилаланил хлорметил кетоном (TPCK) (USB Corp.; Santa Clara, CA). Клетки инкубировали при 37°C с 5% СО2 в течение 48 часов, после чего супернатант собирали центрифугированием при 900xg в течение 10 мин на центрифуге Beckman GS-6R. Брали аликвоты из основного раствора вируса и замораживали при -80°С.
[0379] Соединения. Свободное основание или соль HCl Соединения (1) (например, аморфная соль HCl Соединения (1), Форма A соли HCl Соединения (1), гемигидрат, аморфное свободное основание Соединения (1)) (в примере 10 в дальнешем Соединение (1)) растворяли в 100% диметил сульфоксиде (DMSO) с получением раствора с концентрацией 10 мM.
[0380] Противовирусная активность. Противовирусная активность Соединения (1) и амантадина оценивали в клетках MDCK по измерению уровеня АТФ, с помощью CellTiter-Glo (Promega; Madison, WI). Клетки MDCK помещали в черные планшеты с прозрачным дном и 384 лунками с плотностью 2x104 клеток на лунку в 50 мкл VGM. Клетки инкубировали при 37°C, 5% СО2, при насыщенной влажности для обеспечения адгезии клеток и формирования монослоя. Через 5 часов 40 мкл среды удаляли и добавляли 15 мкл вируса при MOI 0,005. Соединение добавляли как 25 мкл 10-точечного трехкратного разведения в DMEM с добавками (конечная концентрация DMSO равна 0,5%). Внутренние контроли состояли из лунок, содержащих только клетки и необработанные клетки, инфицированные вирусом. Через 72 ч инкубации в каждую лунку добавляли 20 мкл CellTiter-Glo и инкубировали при комнатной температуре в течение 10 мин. Люминесценцию измеряли с помощью читатель EnVision Multilabel (PerkinElmer; Waltham, MA). Значения EC50 (концентрация соединения, которое обеспечивает жизнеспособность 50% клеток в неинфицированном контроле) рассчитывали с помощью кривой доза соединения против данных ответа, используя метод построения кривой на 4-х параметрах, используя алгоритм Levenburg Marquardt (программное обеспечение Condoseo; Genedata, Basel, Швейцария). Тестирование in vitro hpaiH5N1 осуществляли в Southern Research Institute при предупреждении BSL-3.
[0381] Как показано в Таблице 23 ниже, Соединение (1) показало мощную активность против всех исследованных штаммов гриппа A, включая H1N1 и H3N2 ссылочные штаммы 1934-2009 годов, а также штаммы пандемии H1N1 A/California/07/2009 A/Техас/48/2009 2009 года и высоко патогенного штамма H5N1 A/VN/1203/2004 птиц. Соединение (1) было также эффективно против всех штаммов, включая штаммы, которые устойчивы к амантадину и ингибиторам нейраминидазы. Соединение показало ограниченную активностью в отношении вируса гриппа B.
[0382]
Эффективность Соединения (1) против групп штаммов гриппа
EC50 ± SD
b: устойчивость к озельтамивиру карбоксилату: Мутация NA 275Y,
с: устойчивость к озельтамивиру карбоксилату: Мутация NA 119V,
*: внешне подтвержденная фенотипическая устойчивость, последовательность данных недоступена,
[0383] Пример 11: In vitro комбинированные экспертименты с Соединением (1) и Озельтамивиром, Занамивиром или Фавипиравиром
[0384] Раствор Соединения (1) (свободное основание или соль HCl Соединения (1) как и в Примере 10) в 100% диметилсульфоксиде (DMSO) тестировали трехдневном анализе с клетками MDCK на основе CPE, которые были инфицированы A/Puerto Rico/8/34 при MOI, равном 0,01, в комбинированных экспериментах либо с ингибиторами нейраминидазы озельтамивиром и занамивиром, либо с ингибитором полимеразы T-705. Озельтамивир карбоксилат и T-705 разводили в 100% диметилсульфоксиде (DMSO); Занамивир разводили в среде Игла, модифицированного по методу Дульбекко (DMEM) с концентрацией 10 мм и хранили при -20°C. В этом исследовании использовали либо Блисс независимый способ (Macsynergy) (например, Prichard, M.N. and C. Shipman, Jr., Antiviral Res, 1990. 14(4-5): p. 181-205) или метод аддитивности Loewe/медианного эффекта (например, Chou, T.C. and P. Talalay, Adv Enzyme Regul, 1984. 22: p. 27-55). Блисс независимый способ включает тестирование комбинации различных концентраций ингибиторов в шахматном порядке, в то время как способ аддитивности Loewe включает тестирование фиксированного отношения комбинаций ингибиторов в различных разведениях фиксированного соотношения. Эксперименты также проводили, используя комбинации Соединения (1) с самим собой в качестве котроля, подтверждая аддитивность. Жизнеспособность клеток определяли с помощью CellTiter-Glo.
[0385] Результатом Блисс независимого способа были синергитические значения, равные 312 и 268, для озельтамивира карбоксилата и занамивира, соответственно; и синергитическое значение, равное 317, было получено для фавипиравира. Синергические значения больше 100 обычно считаются сильной синергией, а значения между 50 и 100 считаются умеренной синергией. Способ аддитивности Loewe показал значения CI (показатель аддитивности), равные 0,58, 0,64 и 0,89 при 50%-ном эффективном уровне для озельтамивира, занамивира и T-705, соответственно. Значения CI меньше чем 0,8 считаются сильной синергией, а значения между 0,8 и 1,0 считаются находящимися в диапазоне от аддитивных до умеренно синергетических. Вместе эти данные, как показано в Таблице 24, предлагают, что Соединение (1) является синергетическим с тестированными ингибиторами нейраминидазы и ингибитором полимеразы ингибитор.
[0386]
Вывод экспериментов in vitro синергии и антагонизма
[0387] Пример 12: Эффективность на модели инфекции гриппа А у мышей
[0388] Профилактическую дозу-ответ Соединения (1) (в аморфной форме соли HCl Соединения (1 гемигидрат) или (в этом примере - просто Соединение (1)) исследовали на модели гриппа A у мыши. Введение дозы носителя или Соединения (1) начинали за 2 часа до инфицирования и продолжали два раза в день в течение 10 дней. Все мыши, которые получали только носитель, заболевали на 9-й день исследования и теряли, в среднем, ~ 32% массы тела (BW). Соединение (1), вводимое в дозах 1, 3 или 10 мг/кг BID, обеспечивало полную выживаемость и дозозависимое снижение потери BW. Соединение (1), вводимое в дозе 0,3 мг/кг BID, обеспечивало некоторое улучшение выживаемости (2/8 мышей), хотя у мышей была значительная потеря BW. В том же самом эксперименте мышам вводили озельтамивир в дозе 10 мг/кг BID, что клинически-эквиваленто дозе для человека (на основании AUC). Все мыши, получавшие озельтамивир, выживали и имели сходный профиль потери веса как и мыши, которым вводили 1 мг/кг BID Соединения (1).
[0389] Срок, на который можно отсрочить введение Соединения (1) и при этом обеспечивать эффективность в этой модели, исследовали с помощью заражения мышей вирусом гриппа А и путем введения доз носителя, озельтамивира или Соединения (1), начиная через 24, 48, 72, 96 или 120 часа после инфицирования, продолжая вводить дозу BID в течение 10 дней (Таблица 25). Все контроли с носителем заболевали при исследовании на 8 или 9 день. Соединение (1), вводимое в дозе 1, 3 или 10 мг/кг BID, обеспечивало полную защиту от смерти и уменьшало снижение BW, если введение доз начинали через 72 часа после инфицирования по сравнению с контролем с носителем. Только введение озельтамивира в дозе 10 мг/кг обеспечивало полную защиту, если введение дозы начинали через 24 часа или ранее после инфицирования. Если начало введения соединения при этом задерживали, то Соединение (1) в дозе 3 или 10 мг/кг BID обеспечивало полное выживание через 96 часов после инфицирования и давало частичную защиту, если начало введения дозы откладывали на 120 часа после инфицирования.
[0390] Исследовали эффективность Соединения (1) в отношении уменьшения титров вируса в легких. Мышей инфицировали вирусом гриппа A и через 24 часа после этого вводили носитель, озелтамивир (10 мг/кг BID) или Соединение (1) (3, 10, 30 мг/кг BID) до извлечения легких и определения вирусной нагрузки на 6-ой день (Таблица 26). Все группы, которым вводили Соединение (1) показали надежное статистически значимое снижение титров вируса в легких по сравнению с животным, которым вводили озельтамивир и носитель.
[0391] Для получения модели PK/PD модели мышей инфицировали вирусом гриппа в течение 24 часов, а затем вводили Соединение (1) еще 24 часа. Дозы разделяли на фракции в виде одиночной дозы, в виде двух или четырех доз, вводимых каждые 12 часов или 6 часов, соответственно. Легкие извлекали, а плазму собирали для определения вирусной нагрузки легких и концентрации Соединения (1). Отдельные данные титров легких при этих схемах дозирования (q6h, q12h и q24h) переносили на кривую против отдельных значений Cmax Cmin и AUC (данные не показаны). Хотя существует четкая взаимосвязь между снижением титров в легких и Cmin, корреляция с Cmax была слабой, а корреляции с AUC еще слабее. Сильная корреляция с Cmin была тогда, когда измеренные концентрации Соединения (1) в плазме переносили на кривую против измеренных титров в легких. Половина от максимального снижения титров в легких (2-3 log) была вблизи сдвигов в сыворотке EC99 (100 нг/мл). Аналогичная корреляция была обнаружена между титром в легких и измеренными концентрациями Соединения (1) в легких (данные не показаны).
[0392]
Суммирование процента выживания и процента потери массы тела на модели гриппа А мыши
b данные того же эксперимента.
с НД, нет данных
[0393]
Суммирование титра вируса в легких и Log10 уменьшение на модели гриппа А мышей
b Титры вируса в легких определяли на 6-й день исследования.
с НД, нет данных
2 факторный ANOVA и пост-тест Бонферрони, *** P<0,001.
[0394] Пример 13: Эксперимент по проверке заражения гриппом
[0395] Ранее использовали модель инфицирования живым аттенуированным гриппом для прогнозирования эффективности препаратов против гриппа при естественной инфекции в организме челов (Calfee, D.P., Peng, A.W., Hussey, E.K., Lobo, M. and Hayden F.G. Safety and efficacy of once daily intranasal zanamivir in preventing experimental human influenza A infection. Antivir Ther. 4, 143-149 (1999); Hayden, F.G. et al. Use of the oral neuraminidase inhibitor oseltamivir in experimental human influenza. JAMA 282, 1240-1246 (1999). Проводили рандомизированное двойное слепое исследование под контролем плацебо в едином центре Формы А соли HCl Соединения (1), гемигидрат (далее в этом примере просто Соединения (1)) у здоровых добровольцев, которых инокулировали для заражения живым вирусом штамма гриппа A/Wisconsin/67/2005 (H3N2). Индивиды получали пять ежедневных доз либо плацебо (N=33), либо Соединения (1) один раз в день (QD) (в виде капсулы, состоящей из чистого Соединения (1)): 100 мг (N=16), 400 мг (N=19) или 900 мг на 1-й день, а затем 600 мг 2-й-5-й день (N=20) или 1200 мг на 1-й день, а затем 600 мг на 2-й-5-й день (N=18). Индивиды получали три раза в день тампоны в нос и три раза в день проводили запись в карточках клинических симптомов в 1-й-7-й дни и их выписывали из клиники на 8-й день, после чего их безопасность оценивали приблизительно на 28 день. Тампоны в нос исследовали на наличие вируса гриппа в культуре клеток (первичный анализ) и с помощью qRT-ПЦР (вторичный анализ).
[0396] Анализы эффективности осуществляли на наборе полного анализа (FA), определенный как все рандомизированные индивиды, которые получили по крайней мере одну дозу исследуемого препарата (Соединение (1) или плацебо) и концентрации вируса у которых были выше или равны нижнему пределу количественного определения анализа TCID50 клеточной культуры в любой момент времени в течение 48 часов после прививки, или титр ингибирования гемагглютинации которых повышался в 4 раза или более по сравнению с базовым (1-й день) в период после прививки (N=74). Набор безопасности включал всех индивидов, которые были привиты вирусом гриппа на 0-й день и которые получали по крайней мере одну дозу либо плацебо, либо Соединения (1) (N=104).
[0397] Оценка эффективности
[0398] Главным показателем в этом исследовании была демонстрация тенденции доза ответ в AUC выделения вируса между 1-м днем исследования (первый день введения дозы лекарственного средства) и 7-м днем по измерению TCID50 в анализе клеточной культуры в наборе FA. Статистически значимая тенденция доза ответ наблюдалась в медиане AUC выделения вируса в тампонах в нос (P=0,036, тест на тенденцию Джонкхиера-Терпстра). Кроме того, были проведены парные сравнения пулов плацебо и группы каждой дозой Соединения (1) для медианы AUC выделения вируса, среднего продолжения выделения и средней величины пика выделения вируса (Таблица 27). Для группы с дозой 1200/600 мг было отмечено статистически значимое снижение AUC выделения вируса (P=0,010, тест на ранговые суммы Уилкоксона), и значительные сокращения пика выделения наблюдали для группы с дозой 1200/600 мг (Фиг. 8), группы с дозой 400 мг и объединенных групп с дозой Соединения (1). Были осуществлены дополнительные анализы групп FA (данные не показаны).
[0399] Выделение гриппа через нос также количественно оценивалось qRT-ПЦР и результаты были аналогичны результатам анализа клеточной культуры. Нет никакой разницы в скорости сероконверсии между группами с дозой Соединения (1) и плацебо, как определено по 4-кратному или большему увеличению титра против гриппа относительно базовой линии до прививки, что предполагает, что доза Соединения (1), введенная через 24 часа после прививки гриппа, не влияет на скорость передачи инфекции гриппа и не прекращает последующий гуморальный иммунный ответ на инфекцию (Таблица 28A).
[0400] Индивиды записывали клинические симптомы три раза в день в дневниках. Рассчитывали AUC клинических и гриппоподобных симптомов, оцененных по баллам с 1-ого дня по 7-ой день. По сравнению с плацебо, группа с дозой 1200/600 мг Соединения (1) показала статистически значимое снижение средней продолжительности композитных клинических симптомов (P=0,001), медиана AUC гриппоподобных симптомов (P=0,040) и средняя продолжительность гриппоподобных симптомов (P<0,001) (Таблица 28B).
[0401]
Средняя AUC выделения вируса, средняя продолжительность выделения и средняя величина пика выделения вируса
17,1)
16,1)
18,0)
16,1)
8,4)
18,0)
4,63)
3,39)
НД)
4,68)
1,33)
2,43)
42,1)
41,9)
36,9)
37,1)
24,7)
41,9)
5,35)
3,39)
НД)
5,01)
0,66)
2,394)
Примечание: Статистически значимые значения P (P<0,05) выделены жирным шрифтом.
a: P=0,036 для тенденции доза ответ AUC из теста тенденции Джонкхиера-Терпстра.
b значение P рассчитывали из тест на ранговые суммы Уилкоксона.
с значение P вычисляли из ANOVA.
d значение P вычисляли из log-ранг теста.
eP=0,036 для тенденции доза ответ AUC из теста тенденции Джонкхиера-Терпстра.
fсероконверсию определяли как ≥4-кратное увеличение титра антитела против гриппа при посещения для проверки безопасности по сравнению с базовым уровнем. Значение P вычисляли с помощью точного теста Фишера.
[0402]
Медиана AUC, средняя продолжительность и средняя величина пика, композитных клинических симптомов и гриппо-подобных симптомом
23,5)
25,3)
16,0)
32,3)
5,5)
32,3)
4,73)
5,43)
4,63)
4,61)
2,24)
3,06)
17,7)
21,3)
14,0)
28,6)
4,4)
28,6)
4,73)
5,40)
4,63)
4,61)
2,24)
3,00)
Примечание: Статистически значимые значения P (P<0,05) выделены жирным шрифтом.
b значение P рассчитывали из тест на ранговые суммы Уилкоксона.
с значение P вычисляли из ANOVA.
d значение P вычисляли из log-ранг теста.
[0403] Оценка безопасности
[0404] Соединение (1) хорошо переносится, и там были не непланового вследствие соединения (1)-негативных событий, связанных с (AE) не было каких-либо серьезных побочных эффектов. Список неблагоприятных событий, происходящих у ≥10% испытуемых в любой группе лечения представлен (Таблица 29). Гриппоподобное заболевание наиболее часто приводило к неблагоприятным событиям и, как сообщалось, пропорция была приблизительно одинаковая в плацебо и в группах, получавших Соединение (1). Неблагоприятные события, которые произошли с ≥10% разницей в группах, получавших Соединение (1), и плацебо были: снижение уровня фосфора в крови (18,1%, для Соединения (1); 0% для плацебо), ринорея (Соединение (1), 4,2%; плацебо, 18,8%) и заложенность носа (1,4%, Соединение (1); 15,6%, плацебо). Кроме того, наблюдалось повышение аланинаминотрансферазы (ALT) как у плацебо, так и у реципиентов Соединения (1). Ни нарушений функции печени, ни уменьшение фосфата в сыворотке не наблюдались в исследовании с увеличением дозы впервые у человека Соединения (1) с одной дозой до 1600 мг и с несколькими дозами до 800 мг ежедневно в течение 10 дней; ранее сообщалось как о повышении ALT, так и об уменьшении фосфата сыворотки при вирусной инфекции верхних дыхательных путей.
[0405]
Список неблагоприятных событий, происходящих у ≥10% испытуемых в любой группе лечения представлен
a Одна доза в 900 мг в 1-ый день и 600 мг qd в промежутке между 2-ым днем и 5-ым днем.
b Одна доза в 1200 мг в 1-ый день и 600 мг qd в промежутке между 2-ым днем и 5-ым днем.
c Гриппо-подобное заболевание, как определено в анализе эффективности, оценивали на основе параметров, перечисленных в тексте. AE гриппоподобного заболевания определял врач.
[0406] Обсуждение
[0407] В исследовании с заражением гриппом здоровых добровольцев Соединение (1) была показана зависимость доза ответ AUC вирусного титра в тампонах в нос как в клеточных культурах TCID50, так и в qRT-ПЦР, и самая высокая оцененная доза Соединения (1) вызвала значительное снижение вирусного титра AUC, а также AUC и продолжительности симптомов гриппа. Хотя аналогичная величина улучшения выше плацебо не наблюдалась в группе со второй высокой дозой 900/600 мг (Таблица 27), эта доза не показала результаты, сходные с дозой 600 мг в отношении медианной AUC для композитных клинических симптомов и гриппо-подобных симптомов в конечных точках (Таблица 28); причины этого расхождения полностью не поняты. Хотя точная безопасность не была оценена в ходе POC, то уменьшение фосфата и увеличение ALT предполагает, что соответствующий мониторинг обоих параметров нужно будет использоваться в будущих исследованиях.
[0408] В целом, ограничения модели заражения гриппом заключаются в том, что вирус гриппа, используемый в этом исследовании, представляет собой штамм, который был специально выбран так, чтобы не дать наиболее серьезные клинические симптомы инфекции вирусом гриппа. Кроме того, количество вводимого для заражения вируса вероятно больше, чем количество гриппа при естественном заражении. Время введения дозы Соединения (1) через 24 ч после воздействия может быть невозможным сроком начала терапии в сообществе, где пациенты редко проходят диагностику или лечение до тех пор, пока у них не появляются существенные симптомы, вероятно, более чем через 24 часа после воздействия. Однако учитывая, тот факт, что при естественном заражении индивида первоначально заражаются гораздо меньшим вирусным титром временные шкалы не являются напрямую сопоставимыми.
[0409] В итоге, Соединение (1) является мощным ингибитором гриппа А PB2, который представляет особый и новый класс противовирусного средства. Свойства этого ингибитора, как описано в доклинических и клинических данных, указывают, что Соединение (1) является прекрасным кандидатом для дальнейшей оценки и имеет несколько потенциальных преимуществ перед существующими противовирусными препаратами, используемые для лечения инфекции, вызываемой гриппом.
[0410] Все ссылки, приведенные в настоящем документе, включены в качестве ссылки в полном объеме. Как используется в настоящем документе все аббревиатуры, символы и конвенции согласуются с используемыми в современной научной литературе. См., e.g., Janet S. Dodd, ed., The ACS Style Guide: A Manual for Authors and Editors, 2nd Ed., Washington, D.C.: American Chemical Society, 1997.
ДРУГИЕ ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ
[0411] Следует понимать, что хотя изобретение описано в сочетании с подробным описанием, вышеприведенное описание предназначено для иллюстрации, а не для ограничения области применения изобретения, которая определяется формулой изобретения. Другие аспекты, преимущества и изменения находятся в рамках следующей формулы изобретения.
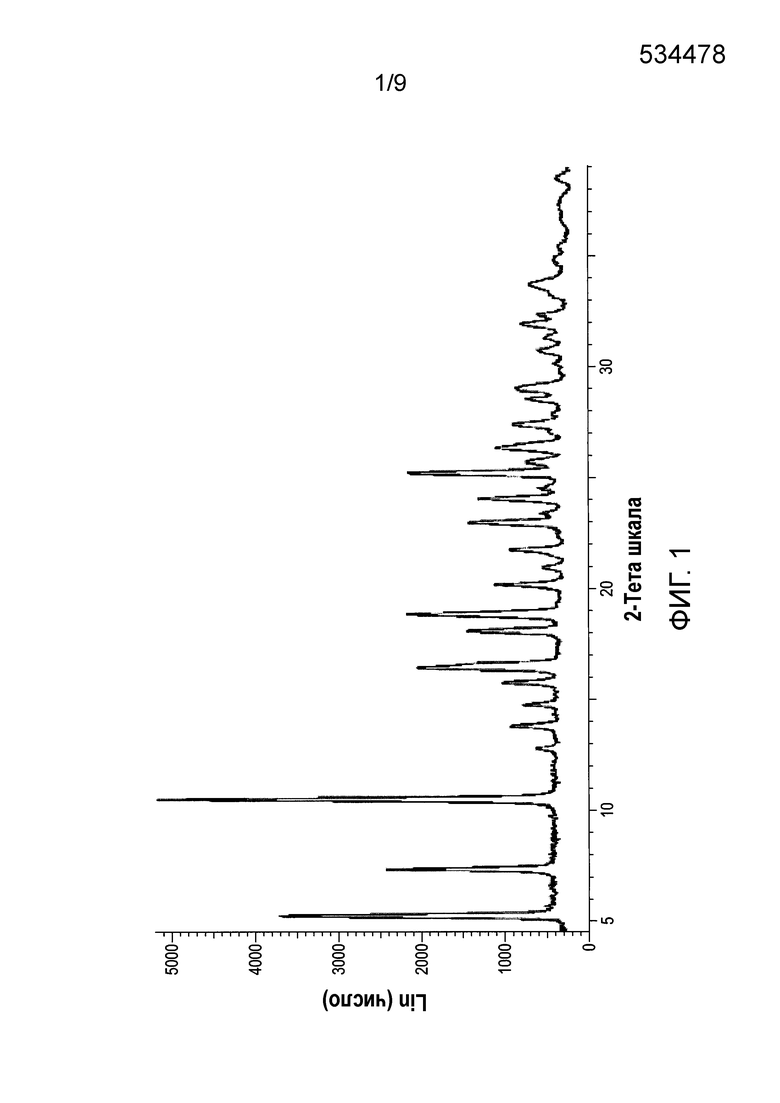
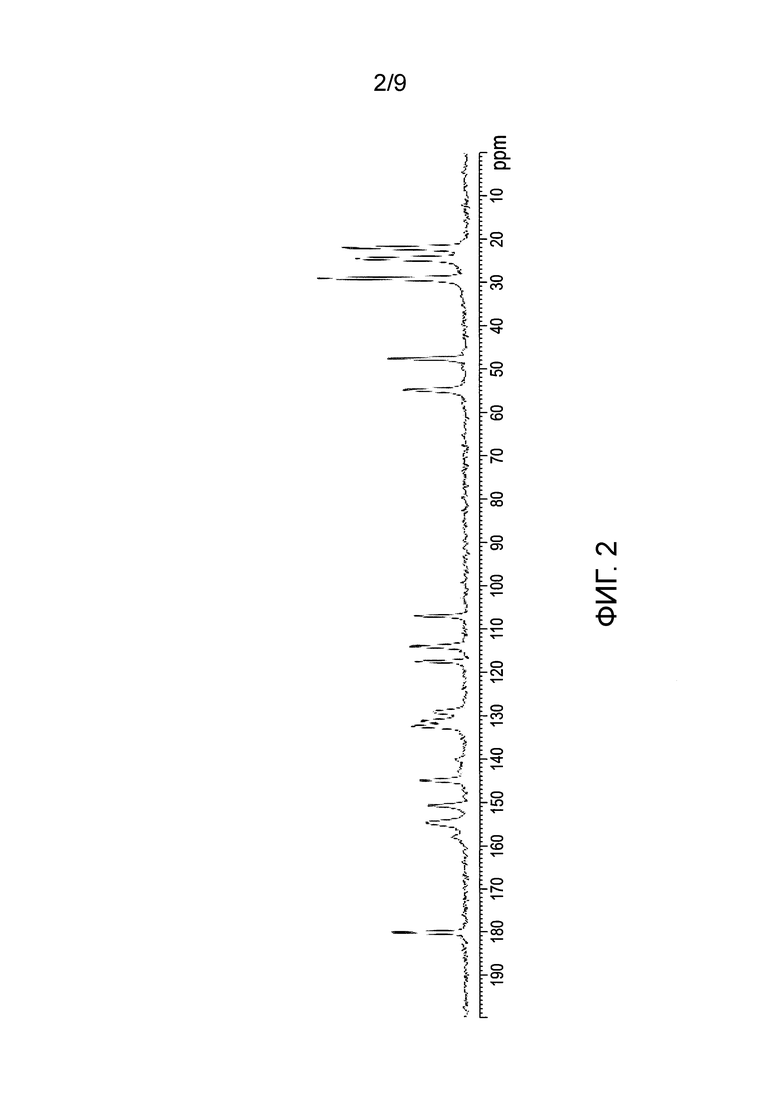
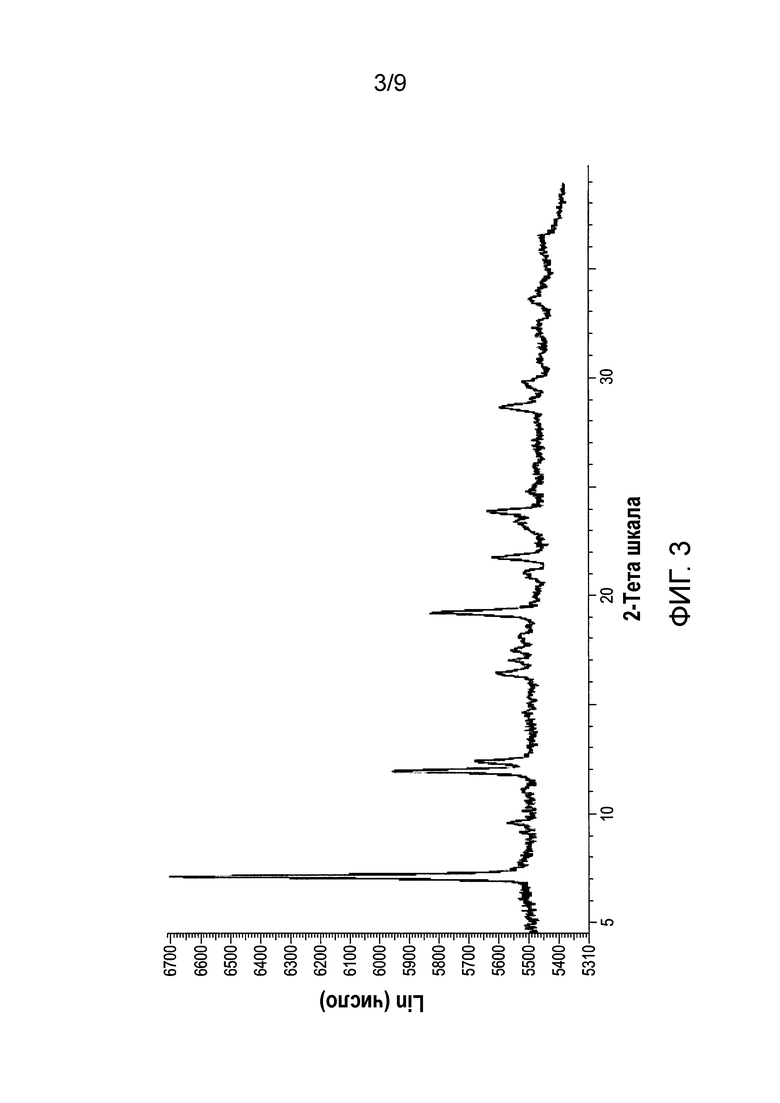
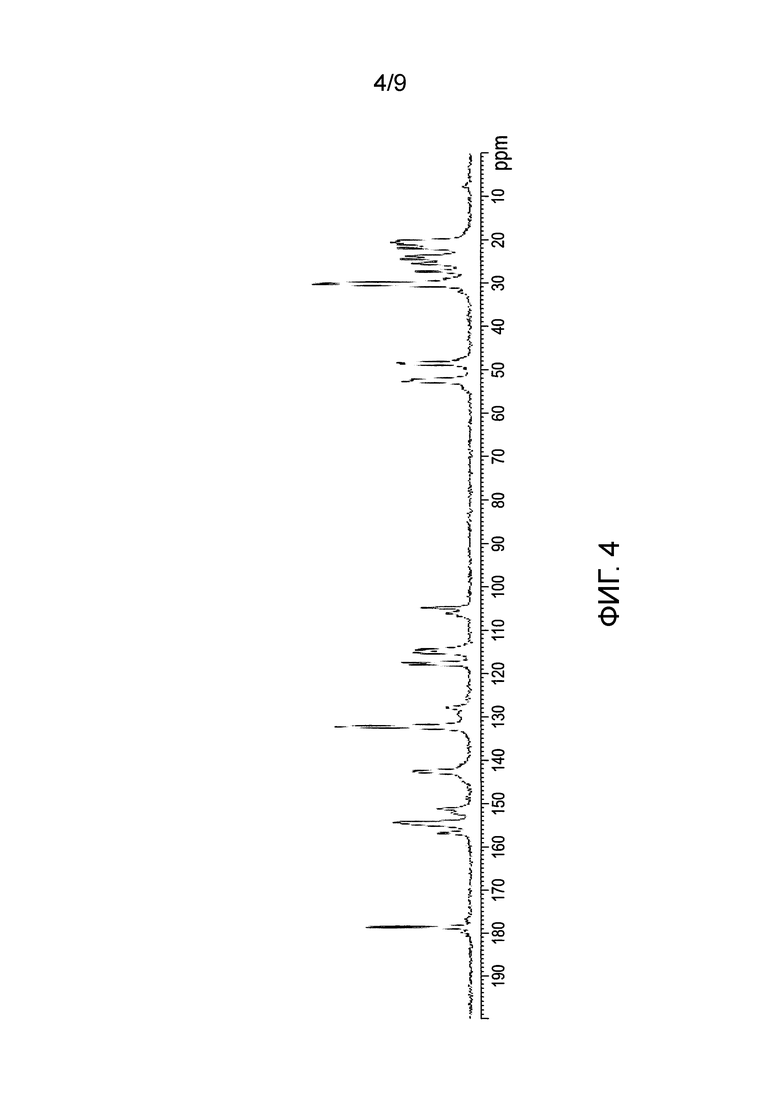
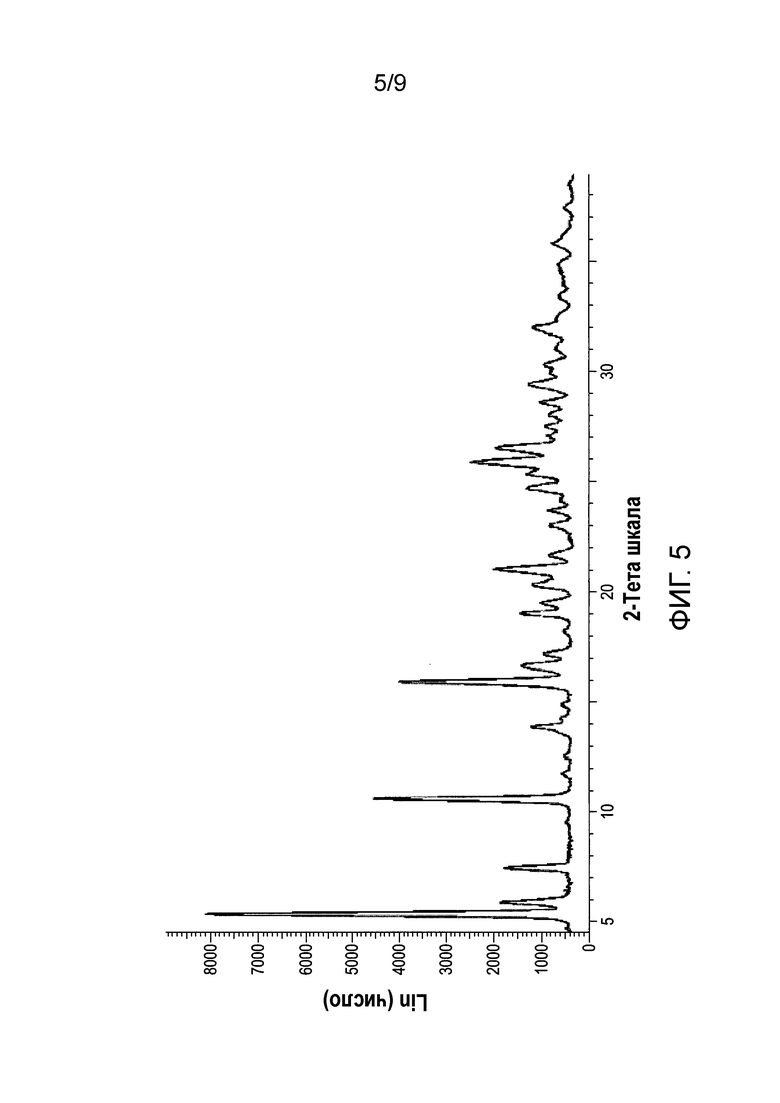
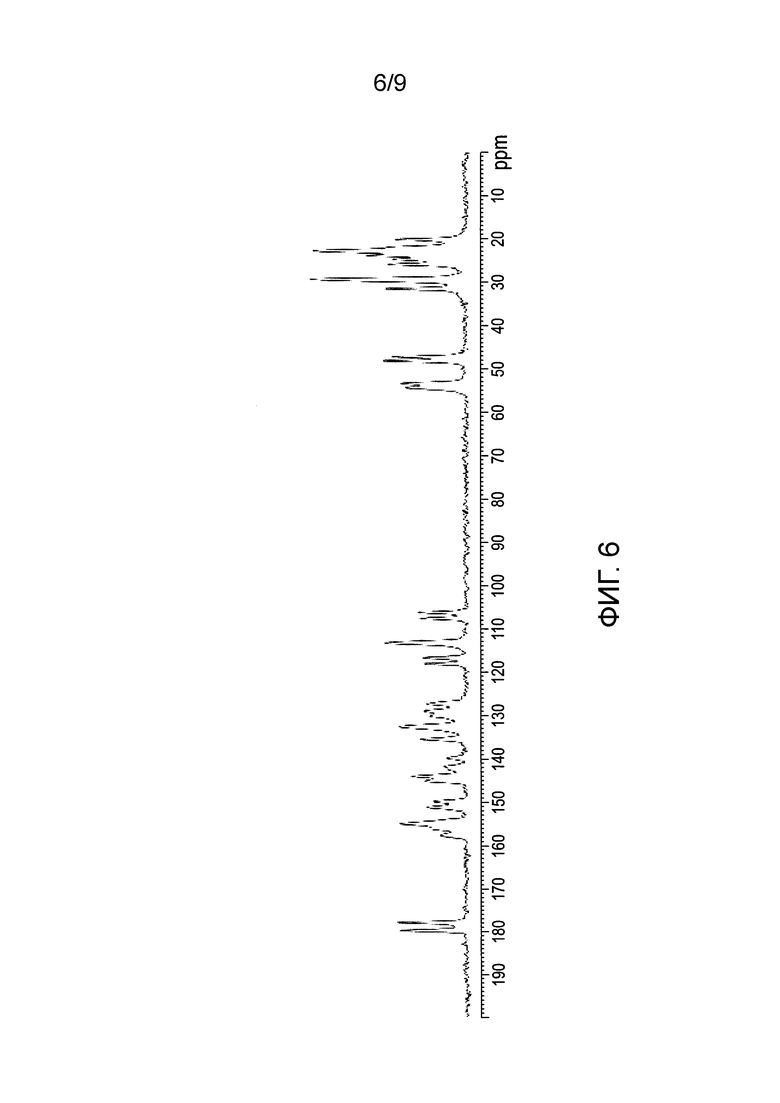
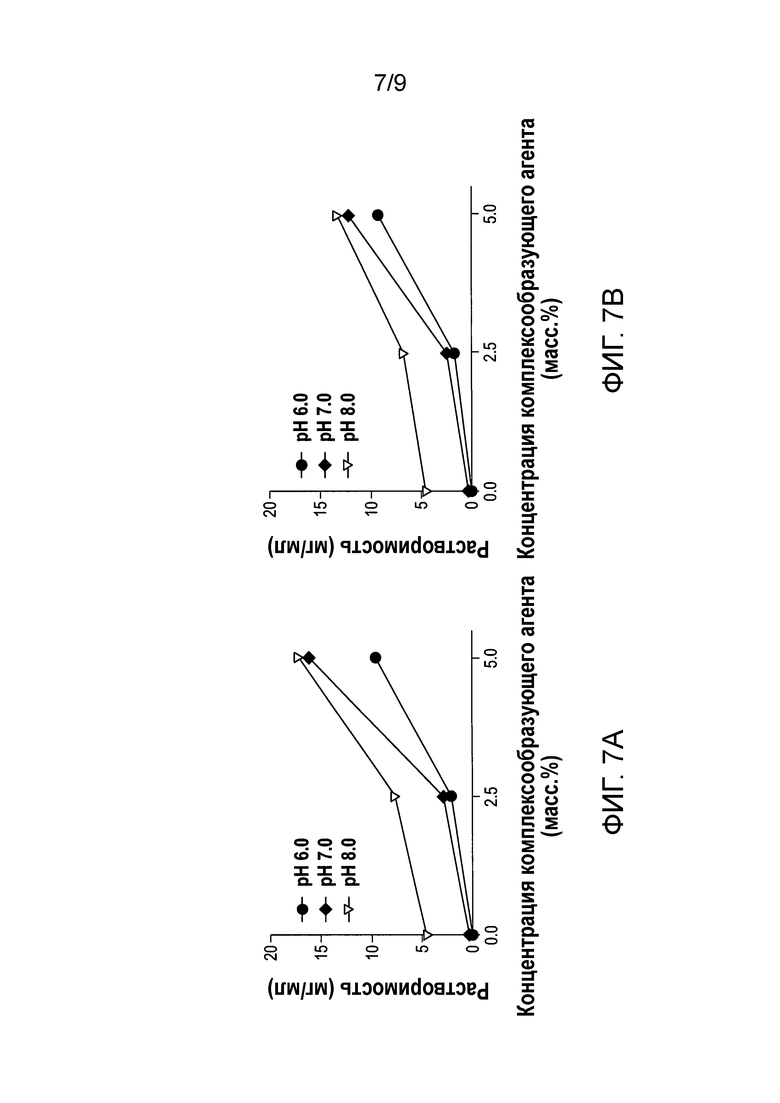
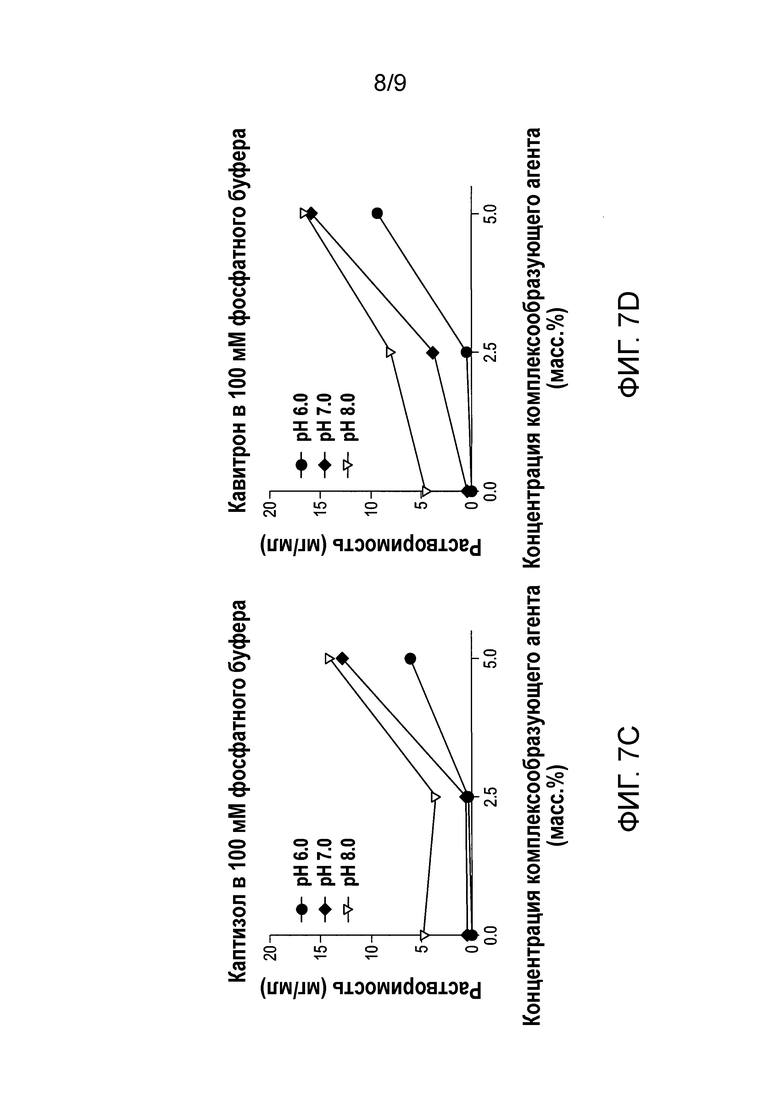
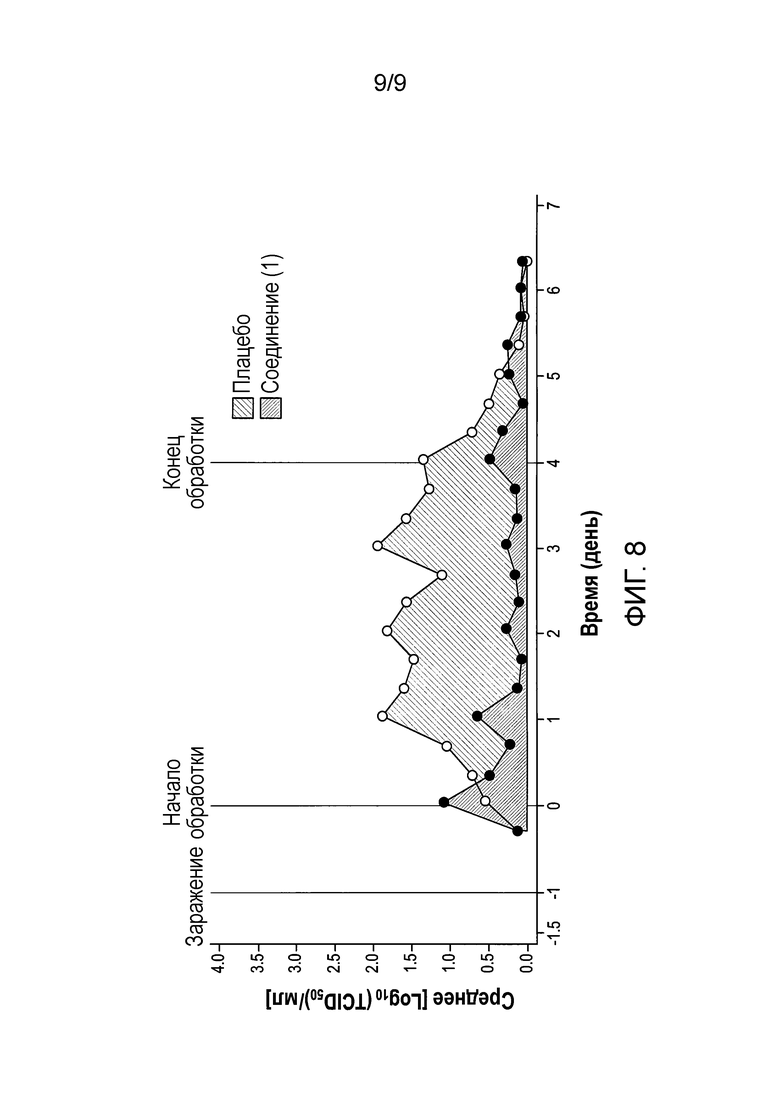
Фармацевтическая композиция предназначена для лечения гриппа А у субъекта. Композиция содержит: а) от 1 мг/мл до 20 мг/мл соединения (1) в воде и b) от 0,01 М до 0,1 М фармацевтически приемлемого модификатора рН. Источником соединения (1) является кристаллическая соль HCl соединения (1)·1/2H2O, охарактеризованная одним или более пиками, соответствующими значениям 2-тета, измеренным в градусах, 10.5±0.2, 5.2±0.2, 7.4±0.2 и 18.9±0.2 на порошковой рентгеновской дифрактограмме. Также описан способ получения фармацевтической композиции. Каждый из способов снижения количества вируса гриппа, ингибирования репликации вируса гриппа и лечения гриппа независимо использует такую фармацевтическую композицию. Композиция по изобретению характеризуется значительной эффективностью в лечении гриппа А. 5 н. и 13 з.п. ф-лы, 8 ил., 29 табл.,13 пр.
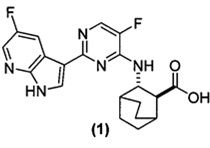
1. Фармацевтическая композиция для лечения гриппа А у субъекта, содержащая:
а) от 1 мг/мл до 20 мг/мл Соединения (1) в воде, где Соединение (1) представлено следующей структурной формулой:
 и
и
b) от 0,01 М до 0,1 М фармацевтически приемлемого модификатора рН,
где источником Соединения (1) является кристаллическая соль HCl соединения (1)⋅1/2H2O, охарактеризованная одним или более пиками, соответствующими значениям 2-тета, измеренным в градусах, 10.5 ± 0.2, 5.2 ± 0.2, 7.4 ± 0.2 и 18.9 ± 0.2 на порошковой рентгеновской дифрактограмме.
2. Фармацевтическая композиция по п. 1, где модификатор рН представляет собой pH буферный агент.
3. Фармацевтическая композиция по п. 1, где композиция содержит от 1 мг/мл до 5 мг/мл Соединения (1).
4. Фармацевтическая композиция по п. 1, дополнительно содержащая от 1 масс. % до 20 масс. % комплексообразующего агента по массе фармацевтической композиции.
5. Фармацевтическая композиция по п. 4, где комплексообразующий агент содержит циклодекстрин, полисорбат, касторовое масло или любую их комбинацию.
6. Фармацевтическая композиция по п. 5, где комплексообразующий агент содержит циклодекстрин, содержащий альфа циклодекстрин, бета циклодекстрин, гамма циклодекстрин, гидроксипропил-бета-циклодекстрин, сульфо-бутиловый эфир-бета-циклодекстрин, полианионный бета-циклодекстрин или любую их комбинацию; полисорбат, содержащий полиоксиэтилен (20) сорбит моноолеат; касторовое масло, содержащее полиокси 40 гидрогенизированное касторовое масло, полиокси 35 касторовое масло или любую их комбинацию.
7. Фармацевтическая композиция по п. 6, где комплексообразующий агент представляет собой гидроксипропил-бета-циклодекстрин.
8. Способ получения фармацевтической композиции по п. 1, включающий:
смешивание
а) кристаллической соли HCl Соединения (1)⋅1/2H2O, охарактеризованной одним или более пиками, соответствующими значениям 2-тета, измеренным в градусах, 10.5 ± 0.2, 5.2 ± 0.2, 7.4 ± 0.2 и 18.9 ± 0.2 на порошковой рентгеновской дифрактограмме, где Соединение (1) представлено следующей структурной формулой:
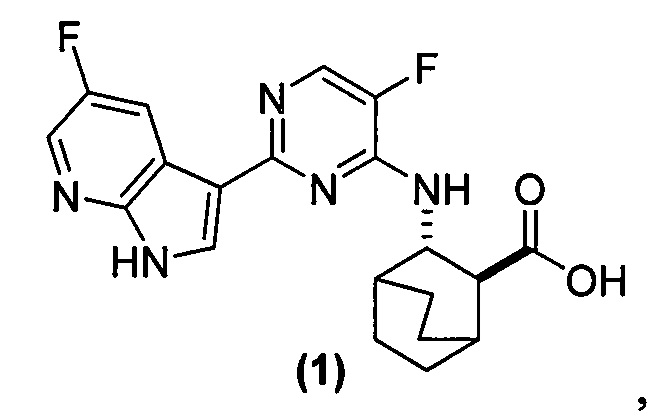
и
b) от 0,01 М до 0,1 М модификатора рН.
с получением смеси, содержащей от 1 мг/мл до 20 мг/мл Соединения (1) в растворе.
9. Способ снижения количества вируса гриппа в биологическом образце in vitro или у индивидуума, включающий введение в образец или индивидууму эффективного количества фармацевтической композиции по п. 1.
10. Способ ингибирования репликации вирусов гриппа в биологическом образце in vitro или у индивидуума, включающий введение в образец или индивидууму эффективного количества фармацевтической композиции по п. 1.
11. Способ лечения гриппа у индивидуума, включающий введение индивидууму терапевтически эффективного количества фармацевтической композиции по п. 1.
12. Способ по п. 11, дополнительно включающий совместное введение одного или более дополнительных терапевтических средств в образец или индивидууму.
13. Способ по п. 12, где дополнительные терапевтические агенты включают антивирусный препарат.
14. Способ по п. 13, где противовирусное средство включает ингибитор нейраминидазы.
15. Способ по п. 14, где ингибитор нейраминидазы включает озельтамивир, занамивир или любую их комбинацию.
16. Способ по п. 13, где противовирусное средство включает ингибитор полимеразы.
17. Способ по п. 16, где ингибитор полимеразы включает флавипиравир.
18. Способ по п. 11, где вирусами гриппа являются вирусы гриппа А.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |

