ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННУЮ ЗАЯВКУ
[0001] По данной заявке PCT испрашивается приоритет на основании предварительной заявки США № 61/903893, зарегистрированной 13 ноября 2013. Данный документ включен в настоящую заявку во всей своей полноте путем ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0002] Настоящее изобретение относится к способам и промежуточным соединениям для получения соединений, подходящих для применения в качестве ингибиторов репликации вируса гриппа.
УРОВЕНЬ ТЕХНИКИ
[0003] Грипп распространяется по всему миру сезонными эпидемиями, приводя к гибели сотен тысяч людей ежегодно и миллионов в пандемические годы. Например, в 20-м столетии произошли три пандемии гриппа, которые погубили десятки миллионов людей, причем каждая из данных пандемий была вызвана появлением нового штамм вируса в человеческой популяции. Часто данные новые штаммы являются результатом распространения существующего вируса гриппа от других видов животных на людей.
[0004] Грипп передается от человека человеку главным образом через крупные нагруженные вирусом капельки, которые образуются, когда зараженные люди кашляют или чихают; данные крупные капельки могут впоследствии оседать на поверхностях слизистой верхних дыхательных путей восприимчивых индивидуумов, которые находятся вблизи (например, в пределах 6 футов) от зараженных людей. Передача может происходить также через прямой контакт или непрямой контакт с секретами органов дыхания, такой как прикосновение к поверхностям, зараженным вирусом гриппа, а затем прикосновение к глазам, носу или рту. Взрослые могут распространять грипп, передавая его другим людям в период в диапазоне от 1 суток до появления симптомов до приблизительно 5 суток после возникновения симптомов. Маленькие дети и люди с ослабленной иммунной системой могут быть заразными на протяжении 10 или более суток после появления симптомов.
[0005] Вирусы гриппа представляют собой РНК-вирусы семейства Orthomyxoviridae, которое включает в себя пять родов: вирус Influenza A, вирус Influenza B, вирус Influenza C, вирус ISA и вирус Thogoto (Тогото).
[0006] Род вируса Influenza A имеет один вид, вирус гриппа A. Дикие водоплавающие птицы являются естественными хозяевами разнообразных типов гриппа A. Эпизодически вирусы передаются другим видам и тогда могут вызывать опустошающие вспышки среди домашней птицы или вызывать пандемии в человеческой популяции. Среди трех типов гриппа вирусы типа A представляют собой наиболее вирулентные патогены человека и вызывают наиболее тяжелое заболевание. Вирус гриппа A можно подразделить на различные серотипы, основываясь на гуморальном ответе на данные вирусы. Серотипами, которые были подтверждены в человеческой популяции, приведенными в порядке числа известных пандемических смертей среди людей, являются: H1N1 (который вызвал испанский грипп в 1918), H2N2 (который вызвал азиатский грипп в 1957), H3N2 (который вызвал гонконгский грипп в 1968), H5N1 (угроза пандемии в сезон гриппа в 2007-2008), H7N7 (который обладает необычным зоонозным потенциалом), H1N2 (эндемичен у людей и свиней), H9N2, H7N2, H7N3 и H10N7.
[0007] Род вируса Influenza B имеет один вид, вирус гриппа B. Гриппом B заражаются почти исключительно люди, и он менее распространен, чем грипп A. Единственным другим животным, для которого известна восприимчивость к инфекции гриппа B, является тюлень. Данный тип гриппа мутирует со скоростью в 2-3 раза медленнее, чем тип A и, следовательно, его генетическое разнообразие выражено в меньшей степени, так что он имеет только один серотип гриппа B. Как результат такого отсутствия антигенного разнообразия иммунитет к гриппу B обычно приобретается в раннем возрасте. Однако грипп B мутирует в достаточной степени для того, чтобы длительный иммунитет был невозможен. Такая сниженная скорость антигенного изменения в сочетании с ограниченным кругом хозяев (препятствующим межвидовой антигенной изменчивости) обеспечивает невозможность возникновения пандемий гриппа B.
[0008] Род вируса Influenza C имеет один вид, вирус гриппа C, который инфицирует людей и свиней и может вызывать тяжелое заболевание и локальные эпидемии. Однако вирус C менее распространен, чем другие типы и, кажется, обычно вызывает легкое заболевание у детей.
[0009] Вирусы A, B и C весьма похожи по структуре. Вирусная частица имеет диаметр 80-120 нанометров и обычно является в общем сферической, хотя могут встречаться нитевидные формы. Необычно для вируса то, что его геном представляет собой не единственную единицу нуклеиновой кислоты, но вместо этого он содержит семь или восемь единиц сегментированной отрицательно-смысловой РНК. Геном гриппа A кодирует 11 белков: гемагглютинин (HA), нейраминидазу (NA), нуклеопротеин (NP), M1, M2, NS1, NS2(NEP), PA, PB1, PB1-F2 и PB2.
[0010] HA и NA представляют собой крупные гликопротеины на внешней стороне вирусных частиц. HA представляет собой лектин, который опосредует связывание вируса с клетками-мишенями и проникновение вирусного генома в клетку-мишень, тогда как NA вовлечен в высвобождение дочерних вирусов из инфицированных клеток, расщепляя сахара, которые связывают зрелые вирусные частицы. Таким образом, данные белки представляли собой мишени для противовирусных лекарственных средств. Более того, они представляют собой антигены, к которым могут вырабатываться антитела. Вирусы гриппа A классифицируют на подтипы, принимая за основу гуморальный ответ на HA и NA, что дает базу для разграничения по H и N (смотри выше), как, например, в H5N1.
[0011] Грипп создает прямые издержки из-за снижения производительности труда и связанного с ним медицинского лечения, а также непрямые издержки, обусловленные профилактическими мерами. В Соединенных Штатах грипп ответственен за суммарные издержки в более чем 10 миллиардов долларов в год, при том, что согласно произведенным оценкам будущая пандемия могла бы вызвать прямые и непрямые издержки в сотни миллиардов долларов. Издержки на профилактику также высоки. По всему миру правительства израсходовали миллиарды долларов США на подготовку к потенциальной пандемии птичьего гриппа H5N1 и на планирование действий на случай ее возникновения, причем издержки были связаны с покупкой лекарственных средств и вакцин, а также разработкой тренировочных мероприятий на случай катастрофического развития событий и стратегий для улучшенного пограничного контроля.
[0012] Современные варианты лечения гриппа включают в себя вакцинацию и химиотерапию или химиопрофилактику противовирусными препаратами. Вакцинация от гриппа противогриппозной вакциной часто рекомендуется для групп высокого риска, таких как дети и пожилые люди, или в случае людей, которые страдают астмой, диабетом или заболеванием сердца. Однако существует возможность заболевания гриппом, даже будучи вакцинированным. Рецептуру вакцины изменяют каждый сезон для нескольких конкретных штаммов гриппа, но она просто не может включать в себя все штаммы, активно инфицирующие людей по всему миру в данный сезон. Производителям может потребоваться шесть месяцев для того, чтобы разработать рецептуру и произвести миллионы доз, требующихся для того, чтобы справиться с сезонными эпидемиями; эпизодически новый или не получивший достаточного внимания штамм становится заметным в указанный период времени и инфицирует людей, хотя они были вакцинированы (как в случае вспышки гриппа H3N2 в провинции Фуцзянь (Fujian flu) в сезон гриппа в 2003-2004). Непосредственно перед вакцинацией также возможно инфицирование и развитие заболевания из-за того же самого штамма, от которого должна защитить вакцина, поскольку для того, чтобы вакцина достигла эффективности может потребоваться несколько недель.
[0013] Более того, эффективность данных вакцин от гриппа непостоянна. Из-за высокой скорости мутации вируса конкретная вакцина от гриппа обычно обеспечивает защиту на срок не более нескольких лет. Вакцина, составленная для одного года, может быть неэффективной в следующем году, поскольку вирус гриппа с течением времени быстро изменяется, и начинают доминировать другие штаммы.
[0014] Также, из-за отсутствия РНК-корректирующих ферментов РНК-зависимая РНК-полимераза гриппа вРНК делает одну ошибку однонуклеотидной вставки приблизительно на каждые 10 тысяч нуклеотидов, что представляет собой приблизительную длину вРНК гриппа. Следовательно, почти каждый вновь произведенный вирус гриппа представляет собой мутантный антигенный дрейф. Разделение генома на восемь отдельных сегментов вРНК делает возможным смешение или рекомбинацию нескольких вРНК, если одна клетка инфицирована более чем одной вирусной линией. Являющееся результатом этого быстрое изменение в генетике вируса приводит к антигенным сдвигам и дает возможность вирусу инфицировать новые виды-хозяева и быстро преодолевать защитный иммунитет.
[0015] Противовирусные лекарственные средства также могут быть использованы для лечения гриппа, причем ингибиторы нейраминидазы являются особенно эффективными, но вирусы могут развивать устойчивость к стандартным противовирусным лекарственным средствам.
[0016] Таким образом, все еще существует потребность в лекарственных средствах для лечения инфекций, вызванных вирусами гриппа, таких как, например, лекарственные средства с расширенным терапевтическим окном и/или сниженным порогом чувствительности к титру вируса. Более того, существует потребность в способах получения таких лекарственных средств эффективным образом.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0017] Настоящее изобретение в общем относится к способам получения соединения (1) или его фармацевтически приемлемой соли и к способам получения некоторых промежуточных соединений, служащих для этого:
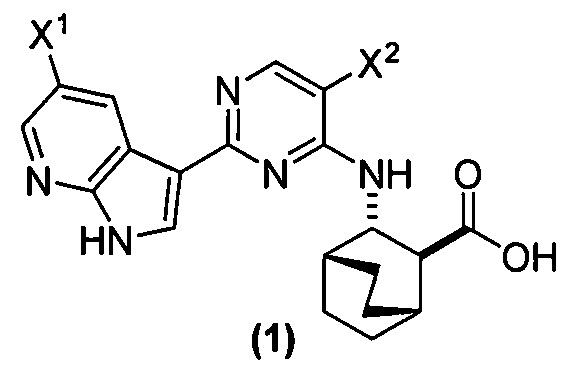 , где X1 и X2, каждый, независимо представляют собой -F или -Cl.
, где X1 и X2, каждый, независимо представляют собой -F или -Cl.
[0018] В одном варианте осуществления изобретение относится к способу получения соединения (1) или его фармацевтически приемлемой соли. Способ включает в себя
(a) взаимодействие соединения (X)  или его фармацевтически приемлемой соли с соединением (Y)
или его фармацевтически приемлемой соли с соединением (Y) 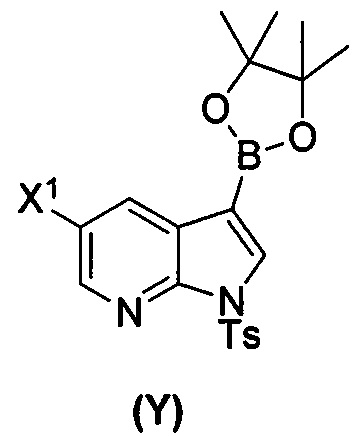 в присутствии палладиевого катализатора и основания с образованием соединения (Z)
в присутствии палладиевого катализатора и основания с образованием соединения (Z) 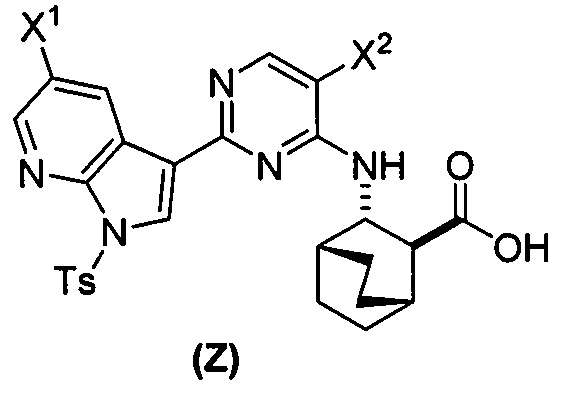 или его фармацевтически приемлемой соли; и
или его фармацевтически приемлемой соли; и
(b) удаление защитной группы Ts (тозил) из соединения (Z) или его фармацевтически приемлемой соли с образованием соединения (1) или его фармацевтически приемлемой соли.
[0019] В некоторых вариантах осуществления палладиевый катализатор образуется на месте (in situ). В некоторых вариантах осуществления данный палладиевый катализатор представляет собой комплекс палладий-Xphos, в котором XPhos представляет собой 2-дициклогексилфосфино-2’,4’,6’-триизопропилбифенил. В других вариантах осуществления комплекс палладий-Xphos получают на месте (in situ) смешением источника Pd(0) или Pd(II) с XPhos. Также, в некоторых вариантах осуществления источник Pd(0) или Pd(II) включает в себя Pd2(dba)3, Pd(OAc)2, PdCl2 или их любое сочетание, где dba представляет собой дибензилиденацетон, а OAc представляет собой ацетат. Например, комплекс палладий-Xphos получают на месте (in situ) смешением Pd(OAc)2 и XPhos.
[0020] В других вариантах осуществления основание включает в себя фосфатное основание или карбонатное основание. Например, фосфатное основание или карбонатное основание выбирают из Na2CO3, K2CO3, K3PO4 или Na3PO4.
[0021] В некоторых вариантах осуществления реакцию соединения (X) с соединением (Y), дающую соединение (Z), как предусмотрено выше на стадии (a), осуществляют в системе растворителя, содержащей воду и органический растворитель, выбранный из 2-метил-ТГФ или ТГФ или любого их сочетания.
[0022] В других вариантах осуществления удаление защитного тозила (Ts) из соединения (Z), как предусмотрено выше на стадии (b), включает в себя обработку соединения (Z) или его фармацевтически приемлемой соли неорганическим гидроксидом, включающим в себя LiOH, NaOH, KOH или любое их сочетание.
[0023] Некоторые варианты осуществления дополнительно включают в себя
(c) взаимодействие соединения (F) 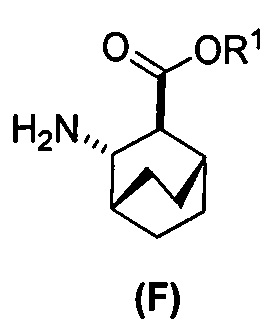 или его фармацевтически приемлемой соли с соединением (G)
или его фармацевтически приемлемой соли с соединением (G) 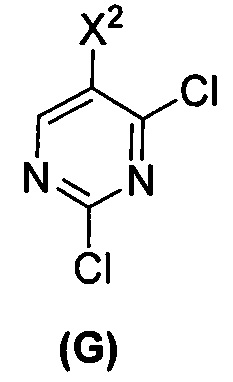 с образованием соединения (H)
с образованием соединения (H) 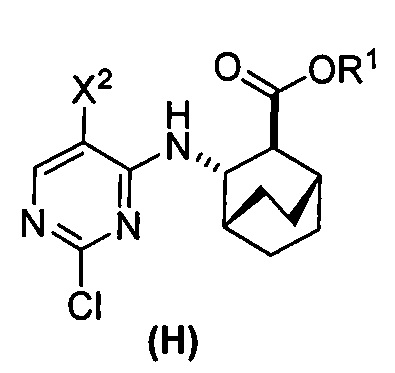 или его фармацевтически приемлемой соли, где R1 представляет собой C1-4-алкил; и
или его фармацевтически приемлемой соли, где R1 представляет собой C1-4-алкил; и
(d) гидролиз соединения (H) или его фармацевтически приемлемой соли с образованием соединения (X) 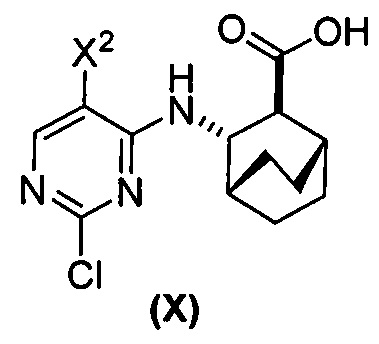 или его фармацевтически приемлемой соли.
или его фармацевтически приемлемой соли.
[0024] Некоторые варианты осуществления дополнительно включают в себя
(e) взаимодействие соединения (C) 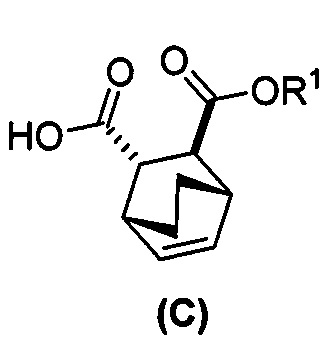 или его фармацевтически приемлемой соли с дифенилфосфорилазидом и с бензиловым спиртом с образованием соединения (D)
или его фармацевтически приемлемой соли с дифенилфосфорилазидом и с бензиловым спиртом с образованием соединения (D) 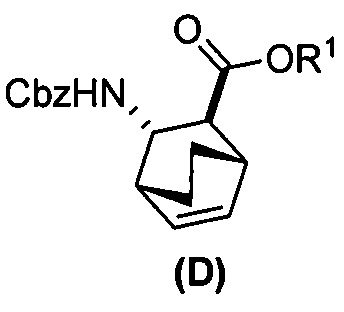 или его фармацевтически приемлемой соли, где Cbz представляет собой карбоксибензил; и
или его фармацевтически приемлемой соли, где Cbz представляет собой карбоксибензил; и
(f) взаимодействие соединения (D) или его фармацевтически приемлемой соли с H2 в присутствии Pd-катализатора на угле с образованием соединения (F) 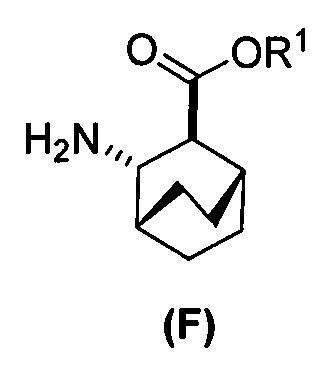 или его фармацевтически приемлемой соли.
или его фармацевтически приемлемой соли.
[0025] Некоторые варианты осуществления дополнительно включают в себя
(g) взаимодействие соединения (A) 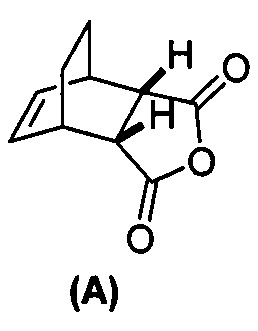 с хинином
с хинином  и R1-OH с образованием аддукта хинина и соединения (C-1)
и R1-OH с образованием аддукта хинина и соединения (C-1) 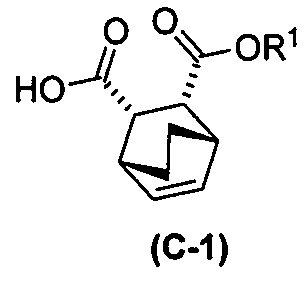 , где R1 представляет собой C1-4-алкил;
, где R1 представляет собой C1-4-алкил;
(h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl с образованием соединения (C-1) или его фармацевтически приемлемой соли; и
(i) эпимеризацию соединения (C-1) или его фармацевтически приемлемой соли с образованием соединения (C) 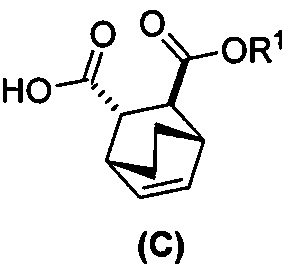 или его фармацевтически приемлемой соли.
или его фармацевтически приемлемой соли.
[0026] В некоторых вариантах осуществления стадия эпимеризации vii) включает в себя обработку соединения (C-1) C1-6-алкоксидом. В некоторых вариантах осуществления C1-6-алкоксид включает в себя трет-бутоксид, трет-амилат или любое их сочетание. В других вариантах осуществления R1 представляет собой этил.
[0027] Некоторые варианты осуществления дополнительно включают в себя гидрирование соединения (S) 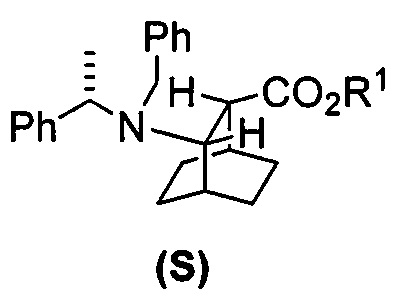 или его фармацевтически приемлемой соли, где Ph представляет собой фенил, в присутствии палладиевого катализатора с образованием соединения (F) или его фармацевтически приемлемой соли, где палладиевый катализатор включает в себя Pd(0) на угле (Pd(0)/C), Pd(OH)2 на угле или любое их сочетание.
или его фармацевтически приемлемой соли, где Ph представляет собой фенил, в присутствии палладиевого катализатора с образованием соединения (F) или его фармацевтически приемлемой соли, где палладиевый катализатор включает в себя Pd(0) на угле (Pd(0)/C), Pd(OH)2 на угле или любое их сочетание.
[0028] Некоторые варианты осуществления дополнительно включают в себя взаимодействие соединения (R) 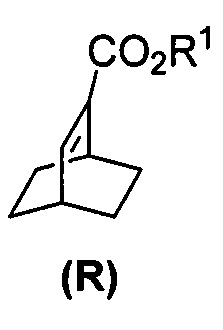 с S-(-)-N-бензил-альфа-метилбензиламинолитием с образованием соединения (S)
с S-(-)-N-бензил-альфа-метилбензиламинолитием с образованием соединения (S) 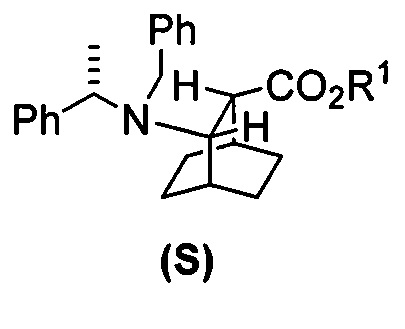 или его фармацевтически приемлемой соли.
или его фармацевтически приемлемой соли.
[0029] Некоторые варианты осуществления дополнительно включают в себя
(j) взаимодействие 1,3-циклогексадиена с CH≡CHC(O)OR1 в присутствии алюминиевого катализатора с образованием соединения (Q) 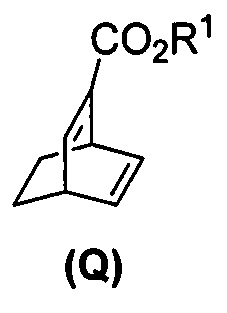 , где R1 представляет собой C1-4-алкил, и
, где R1 представляет собой C1-4-алкил, и
(k) гидрирование соединения (Q) с образованием соединения (R) 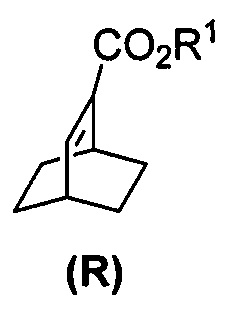 .
.
[0030] В некоторых вариантах осуществления R1 представляет собой этил.
[0031] В некоторых вариантах осуществления алюминиевый катализатор включает в себя EtAlCl2, Et2AlCl, смесь AlCl3 и триоктилалюминия или любое их сочетание.
[0032] В некоторых вариантах осуществления гидрирование соединения (R) включает в себя взаимодействие соединения (R) с H2 в присутствии Rh(I)-катализатора или отравленного Pd(0)-катализатора.
[0033] В некоторых вариантах осуществления Rh(I)-катализатор включает в себя (PPh3)3RhCl, смесь (PPh3)3RhCl и этилропиолата или любое их сочетание, где Ph представляет собой фенил.
[0034] В некоторых вариантах осуществления отравленный Pd(0)-катализатор включает в себя отравленный свинцом Pd(0)-катализатор на CaCO3 (Pd(Pb)/CaCO3).
[0035] Некоторые варианты осуществления дополнительно включают в себя взаимодействие соединения (O) 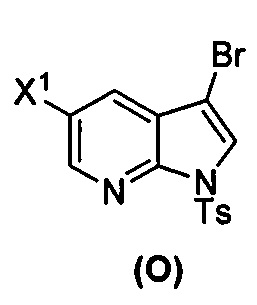 с бис(пинаколато)дибором в присутствии палладиевого катализатора, содержащего фосфиновый лиганд, с образованием соединения (Y)
с бис(пинаколато)дибором в присутствии палладиевого катализатора, содержащего фосфиновый лиганд, с образованием соединения (Y) 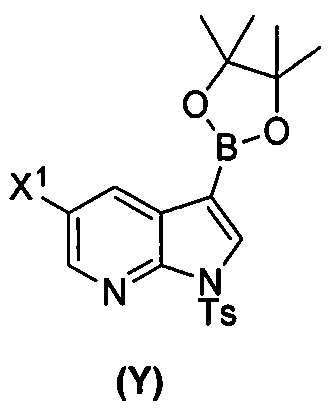 .
.
[0036] В некоторых вариантах осуществления палладиевый катализатор, содержащий фосфиновый лиганд, представляет собой Pd(Ph3P)4.
[0037] Некоторые варианты осуществления дополнительно включают в себя обработку соединения (N) 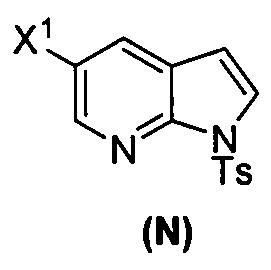 бромирующим агентом, включающим в себя Br2, N-бромсукцинимид, 1,3-дибром-5,5-диметилгидантоин или любое их сочетание, с образованием соединения (O).
бромирующим агентом, включающим в себя Br2, N-бромсукцинимид, 1,3-дибром-5,5-диметилгидантоин или любое их сочетание, с образованием соединения (O).
[0038] Некоторые варианты осуществления дополнительно включают в себя
(1) взаимодействие соединения (J)  или его фармацевтически приемлемой соли с иодирующим агентом или бромирующим агентом с образованием соединения (K)
или его фармацевтически приемлемой соли с иодирующим агентом или бромирующим агентом с образованием соединения (K) 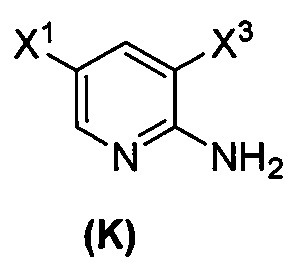 или его фармацевтически приемлемой соли, где X3 представляет собой Br или I;
или его фармацевтически приемлемой соли, где X3 представляет собой Br или I;
(m) взаимодействие соединения (K) или его фармацевтически приемлемой соли с триметилсилилацетиленом с образованием соединения (L) 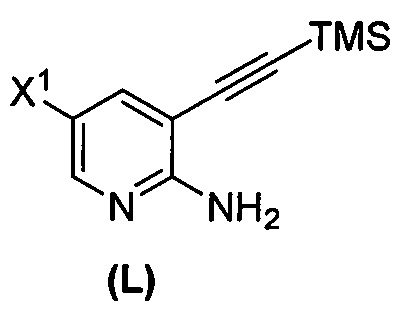 или его фармацевтически приемлемой соли, где TMS представляет собой триметилсилил;
или его фармацевтически приемлемой соли, где TMS представляет собой триметилсилил;
(n) взаимодействие соединения (L) или его фармацевтически приемлемой соли с C1-6-алкоксидным основанием с образованием соединения (P) 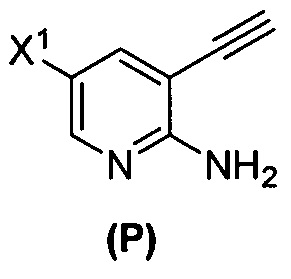 или его фармацевтически приемлемой соли;
или его фармацевтически приемлемой соли;
(o) взаимодействие соединения (P) с трет-бутоксидом калия, трет-амилатом калия или любым их сочетанием с образованием соединения (M) 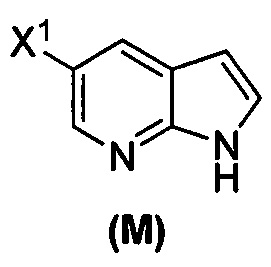 или его фармацевтически приемлемой соли; и
или его фармацевтически приемлемой соли; и
(p) тозилирование соединения (M) или его фармацевтически приемлемой соли с образованием соединения (N) 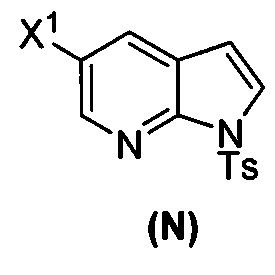 .
.
[0039] В некоторых вариантах осуществления C1-6-алкоксидное основание включает в себя трет-амилат калия, трет-бутоксид калия, метоксид калия, трет-амилат натрия, трет-бутоксид натрия, метоксид натрия или любые их сочетания.
[0040] В некоторых вариантах осуществления реакцию соединения (K) или его фармацевтически приемлемой соли с триметилсилилацетиленом осуществляют в присутствии палладиевого катализатора, включающего в себя Pd(Ph3P)4, Pd(PPh3)2Cl2, Pd(dppf)2Cl2 или любое их сочетание, катализатора в виде галогенида меди (I) или любого их сочетания.
[0041] В некоторых вариантах осуществления реакцию соединения (K) или его фармацевтически приемлемой соли с триметилсилилацетиленом осуществляют в присутствии CuI, Pd(Ph3P)4, Pd(PPh3)2Cl2, Pd(dppf)2Cl2 или любого их сочетания.
[0042] В некоторых вариантах осуществления стадию тозилирования xiv) осуществляют путем проведения реакции соединения (M) или его фармацевтически приемлемой соли с TsCl.
[0043] В некоторых вариантах осуществления соединение (J) или его фармацевтически приемлемую соль вводят в реакцию с иодирующим агентом, включающим в себя I2, ICl, N-иодсукцинимид, и где X3 представляет собой I. В других вариантах осуществления иодирующий агент представляет собой I2.
[0044] В некоторых вариантах осуществления соединение (J) или его фармацевтически приемлемую соль вводят в реакцию с бромирующим агентом, включающим в себя Br2, N-бромсукцинимид, 1,3-дибром-5,5-диметилгидантоин или любое их сочетание, и где X3 представляет собой Br. В других вариантах осуществления бромирующий агент представляет собой Br2.
[0045] Некоторые варианты осуществления дополнительно включают в себя
(q) взаимодействие соединения (K) 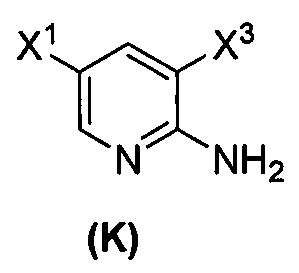 или его фармацевтически приемлемой соли с ацетальдегидом в присутствии палладиевого катализатора, включающего в себя смесь бис(дибензилиденацетон)палладия и третичного фосфинового лиганда, PR3, где R представляет собой C1-6-алкил или C5-6-циклоалкил, с образованием соединения (M)
или его фармацевтически приемлемой соли с ацетальдегидом в присутствии палладиевого катализатора, включающего в себя смесь бис(дибензилиденацетон)палладия и третичного фосфинового лиганда, PR3, где R представляет собой C1-6-алкил или C5-6-циклоалкил, с образованием соединения (M) 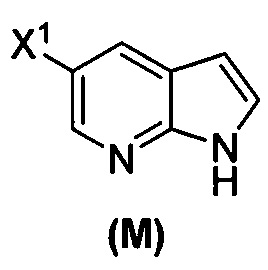 или его фармацевтически приемлемой соли, где X3 представляет собой Br или I; и
или его фармацевтически приемлемой соли, где X3 представляет собой Br или I; и
(p) тозилирование соединения (M) или его фармацевтически приемлемой соли с образованием соединения (N) 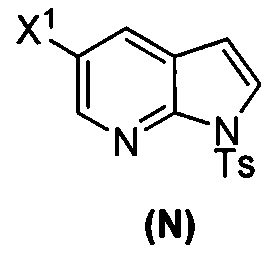 .
.
[0046] В некоторых вариантах осуществления третичный фосфиновый лиганд, PR3, включает в себя P(tBu)3, PCy3, P(i-Pr)3, P(Bu3), PEt3, PMe3 или любое их сочетание. Например, третичный фосфиновый лиганд включает в себя P(tBu)3.
[0047] Некоторые варианты осуществления дополнительно включают в себя после стадии (b) удаления защиты обработку соединения (1) HCl в системе растворителя, содержащей воду и один или более органических растворителей, с образованием соли соединения (1) с HCl, где органический растворитель выбирают из ацетонитрила, хлорбензола, хлороформа, циклогексана, 1,2-дихлорэтена, дихлорметана, 1,2-диметоксиэтана, N,N-диметилацетамида, N,N-диметилформамида, 1,4-диоксана, 2-этоксиэтанола, этиленгликоля, формамида, гексана, метанола, 2-метоксиэтанола, метилбутилкетона, метилциклогексана, N-метилпирролидона, нитрометана, пиридина, сульфолана, тетрагидрофурана (ТГФ), тетралина, толуола, 1,1,2-трихлорэтена, ксилола, уксусной кислоты, ацетона, анизола, 1-бутанола, 2-бутанола, бутилацетата, трет-бутилметилового простого эфира, кумола, гептана, изобутилацетата, изопропилацетата, метилацетата, 3-метил-1-бутанола, метилэтилкетона, метилизобутилкетона, 2-метил-1-пропанола, диметилсульфоксида, этанола, этилацетата, этилового простого эфира, этилформиата, муравьиной кислоты, пентана, 1-пентанола, 1-пропанола, 2-пропанола, пропилацетата или любого их сочетания. В некоторых вариантах осуществления органические растворители системы растворителя выбирают из группы, состоящей из 2-этоксиэтанола, этиленгликоля, метанола, 2-метоксиэтанола, 1-бутанола, 2-бутанола, 3-метил-1-бутанола, 2-метил-1-пропанола, этанола, 1-пентанола, 1-пропанола, 2-пропанола, метилбутилкетона, ацетона, метилэтилкетона, метилизобутилкетона, бутилацетата, изобутилацетата, изопропилацетата, метилацетата, этилацетата, пропилацетата, пиридина, толуола и ксилола.
[0048] В некоторых вариантах осуществления система растворителя включает в себя воду и ацетон или воду и изопропанол.
[0049] В другом варианте осуществления изобретение относится к способу получения соединения (1) или его фармацевтически приемлемой соли, где способ включает в себя:
(g) взаимодействие соединения (A) 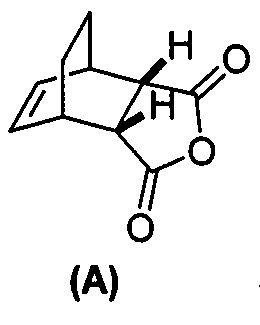 с хинином
с хинином 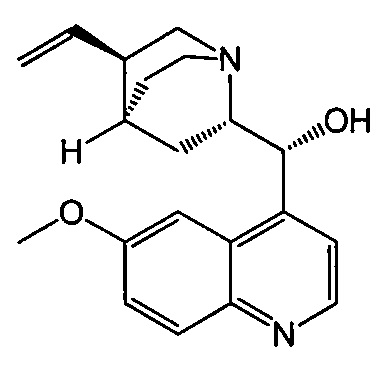 и этиловым спиртом с образованием аддукта хинина и соединения (C-1)
и этиловым спиртом с образованием аддукта хинина и соединения (C-1) 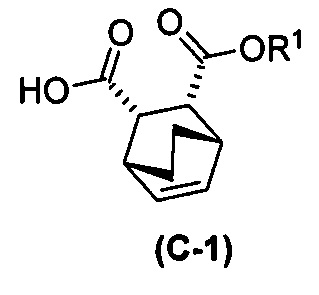 , где R1 представляет собой этил;
, где R1 представляет собой этил;
(h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl с образованием соединения (C-1) или его фармацевтически приемлемой соли;
(i) эпимеризацию соединения (C-1) в соединение (C) 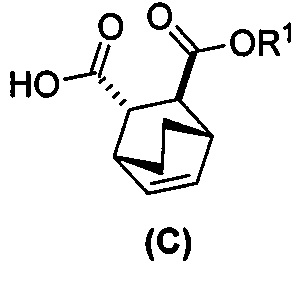 или его фармацевтически приемлемую соль;
или его фармацевтически приемлемую соль;
(e) взаимодействие соединения (C) или его фармацевтически приемлемой соли с дифенилфосфорилазидом и бензиловым спиртом с образованием соединения (D) 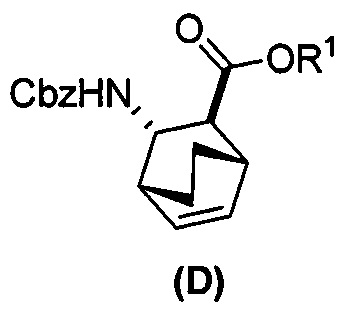 , где Cbz представляет собой карбоксибензил;
, где Cbz представляет собой карбоксибензил;
(f) взаимодействие соединения (D) или его фармацевтически приемлемой соли с H2 в присутствии Pd-катализатора на угле (Pd(0)/C) с образованием соединения (F) 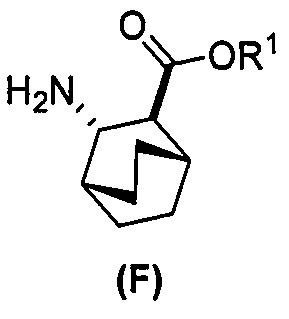 или его фармацевтически приемлемой соли;
или его фармацевтически приемлемой соли;
(c) взаимодействие соединения (F) или его фармацевтически приемлемой соли с соединением (G) 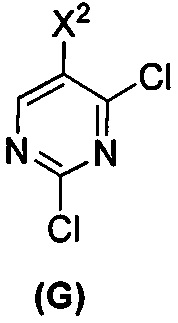 с образованием соединения (H)
с образованием соединения (H) 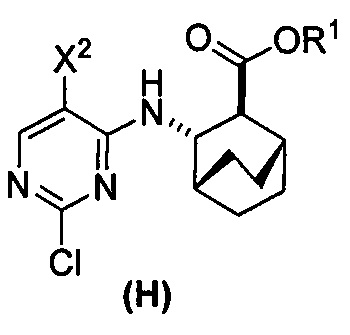 или его фармацевтически приемлемой соли;
или его фармацевтически приемлемой соли;
(d) гидролиз соединения (H) или его фармацевтически приемлемой соли с образованием соединения (X) 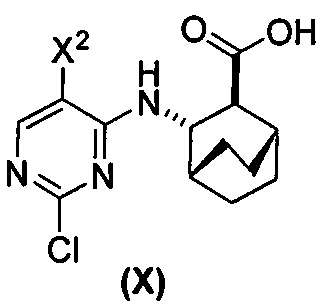 или его фармацевтически приемлемой соли;
или его фармацевтически приемлемой соли;
(a) взаимодействие соединения (X) или его фармацевтически приемлемой соли с соединением (Y)  в присутствии палладиевого катализатора с образованием соединения (Z)
в присутствии палладиевого катализатора с образованием соединения (Z) 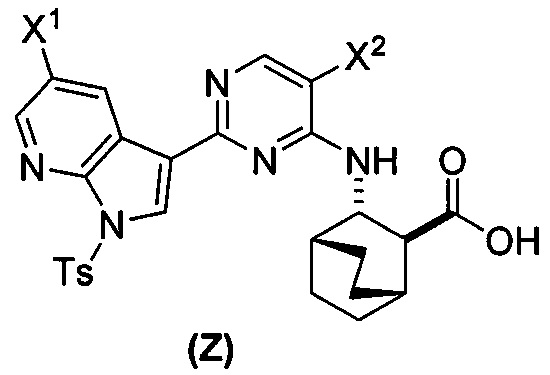 или его фармацевтически приемлемой соли; и
или его фармацевтически приемлемой соли; и
(b) удаление защитной группы Ts из соединения (Z) или его фармацевтически приемлемой соли с образованием соединения (1) или его фармацевтически приемлемой соли, где X1 и X2 независимо представляют собой -F или -Cl; и каждый R1 независимо представляет собой этил.
[0050] В другом варианте осуществления изобретение относится к способу получения соединения (1) или его фармацевтически приемлемой соли, где способ включает в себя:
(q) взаимодействие соединения (K)  или его фармацевтически приемлемой соли с ацетальдегидом в присутствии палладиевого катализатора с образованием соединения (M)
или его фармацевтически приемлемой соли с ацетальдегидом в присутствии палладиевого катализатора с образованием соединения (M) 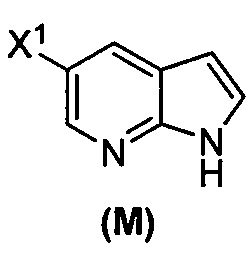 или его фармацевтически приемлемой соли;
или его фармацевтически приемлемой соли;
(p) тозилирование соединения (M) или его фармацевтически приемлемой соли с образованием соединения (N) 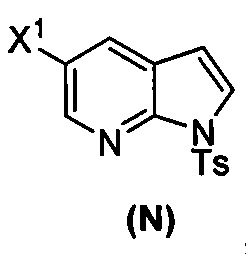 ;
;
(s) бромирование соединения (N) с образованием соединения (O) 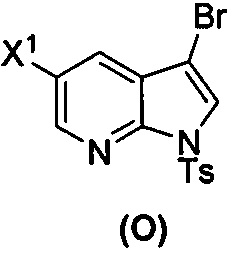 ;
;
(t) взаимодействие соединения (O) с бис(пинаколато)дибором в присутствии палладиевого катализатора с образованием соединения (Y) 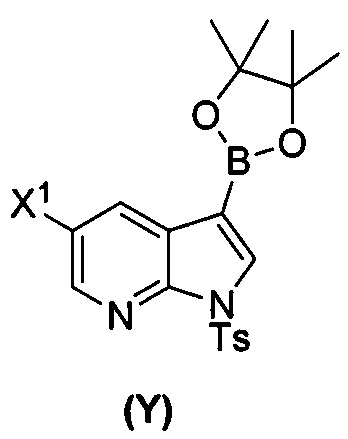 ;
;
(a) взаимодействие соединения (X) 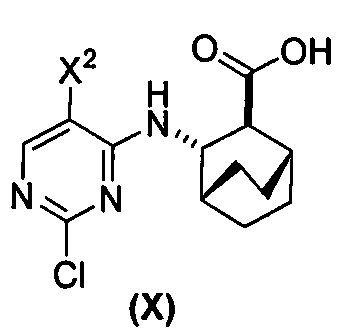 или его фармацевтически приемлемой соли с соединением (Y) в присутствии палладиевого катализатора с образованием соединения (Z)
или его фармацевтически приемлемой соли с соединением (Y) в присутствии палладиевого катализатора с образованием соединения (Z) 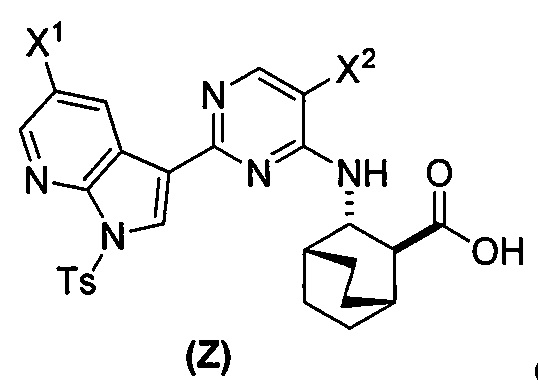 или его фармацевтически приемлемой соли; и
или его фармацевтически приемлемой соли; и
(b) удаление защитной группы Ts из соединения (Z) или его фармацевтически приемлемой соли с образованием соединения (1) или его фармацевтически приемлемой соли. X1 и X2 независимо представляют собой -F или -Cl; X3 представляет собой -Br; и каждый R1 независимо представляет собой этил.
[0051] В другом варианте осуществления изобретение относится к способу получения соединения (2):
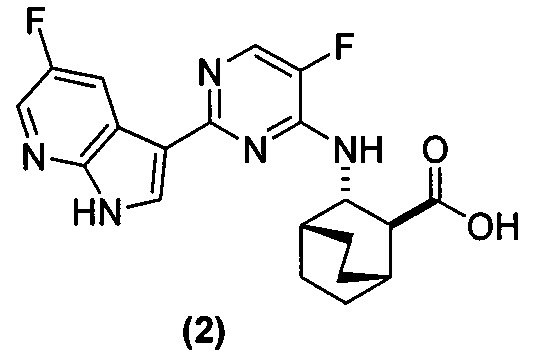 или его фармацевтически приемлемой соли, где способ включает в себя:
или его фармацевтически приемлемой соли, где способ включает в себя:
(g) взаимодействие соединения (A) 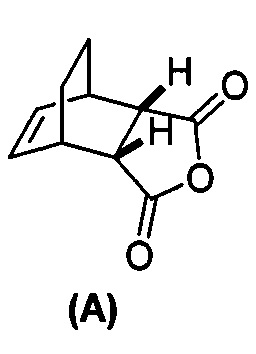 с хинином
с хинином 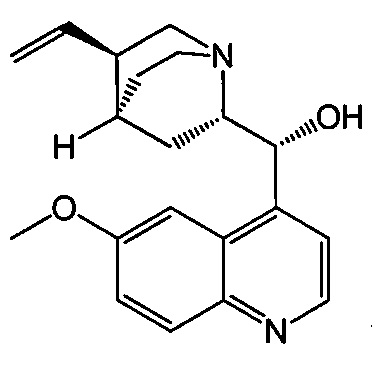 и этиловым спиртом с образованием аддукта хинина и соединения (C-1)
и этиловым спиртом с образованием аддукта хинина и соединения (C-1) 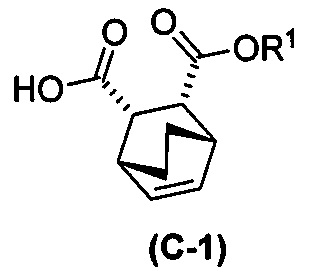 ;
;
(h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl с образованием соединения (C-1);
(i-1) взаимодействие соединения (C-1) с C1-6-алкоксидом, выбранным из трет-бутоксида или трет-амилата с образованием соединения (C) 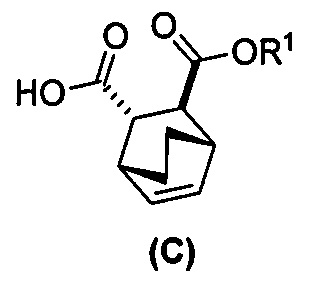 или его фармацевтически приемлемой соли;
или его фармацевтически приемлемой соли;
(e) взаимодействие соединения (C) с дифенилфосфорилазидом, а затем с бензиловым спиртом с образованием соединения (D) 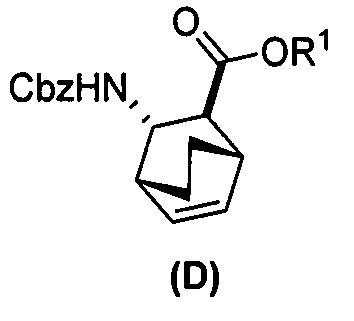 ;
;
(f) взаимодействие соединения (D) или его фармацевтически приемлемой соли с H2 в присутствии Pd-катализатора на угле (Pd(0)/C) с образованием соли соединения (F) 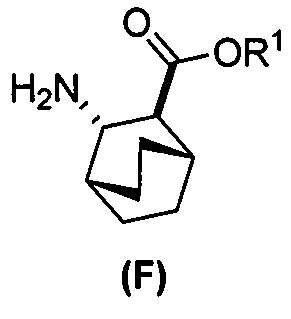 с HCl;
с HCl;
(r) взаимодействие соли соединения (F) с HCl с соединением (G) 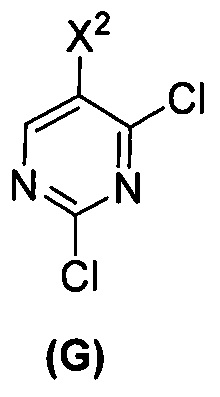 с образованием соединения (H)
с образованием соединения (H) 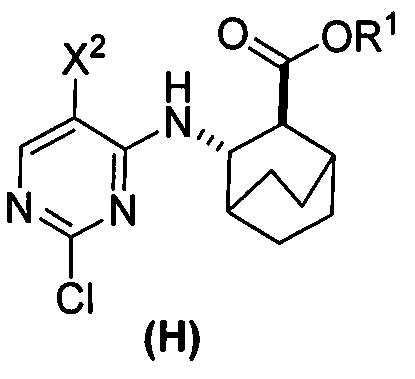 ;
;
(h-1) гидролиз соединения (H) с образованием соединения (X-2) 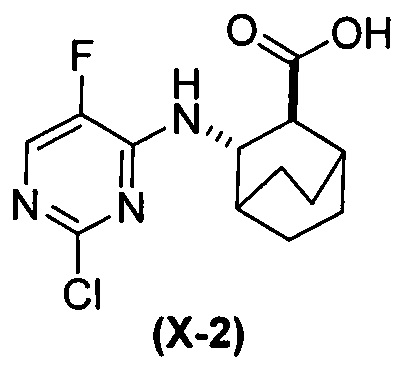 ;
;
(l) иодирование или бромирование соединения (J) 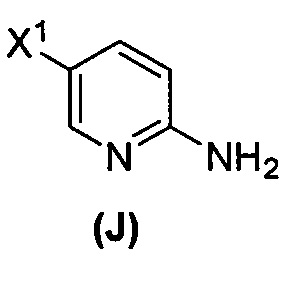 с образованием соединения (K)
с образованием соединения (K) 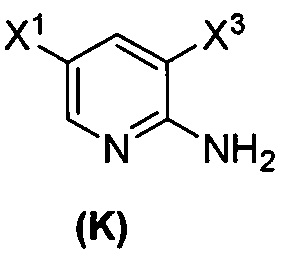 ;
;
(q-1) взаимодействие соединения (K) с триметилсилилацетиленом с образованием соединения (L) 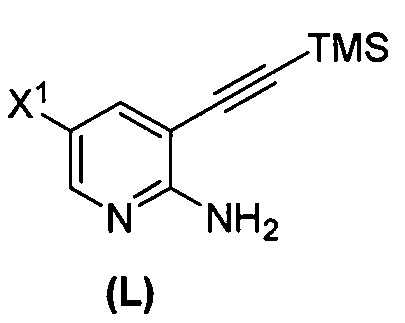 ;
;
(j) взаимодействие соединения (L) с C1-6-алкоксидом с образованием соединения (M)  ;
;
(k) тозилирование соединения (M) с образованием соединения (N) 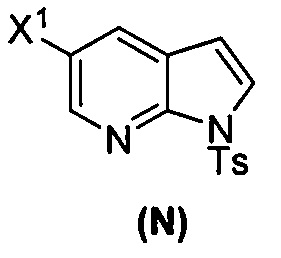 ;
;
(s) бромирование соединения (N) с образованием соединения (O) 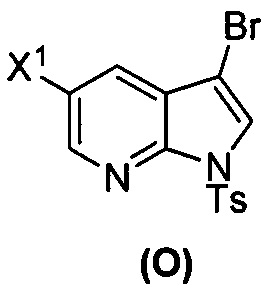 ;
;
(t) взаимодействие соединения (O) с бис(пинаколато)дибором в присутствии Pd(Ph3P)4 с образованием соединения (Y-2) 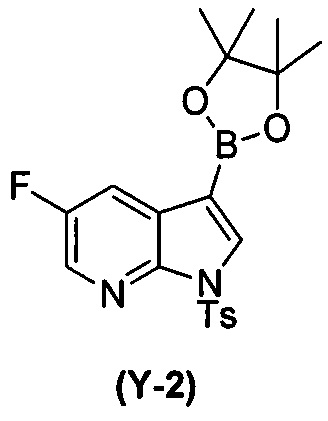 ;
;
(a) взаимодействие соединения (X-2) с соединением (Y-2) в присутствии комплекса палладий-XPhos и фосфатного или карбонатного основания, выбранного из K2CO3 или K3PO4, с образованием соединения (Z-2): 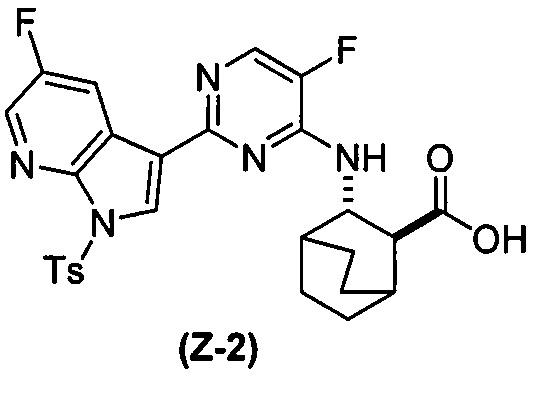 или его фармацевтически приемлемой соли; и
или его фармацевтически приемлемой соли; и
(b) удаление защитной группы Ts из соединения (Z-2) или его фармацевтически приемлемой соли с образованием соединения (2) или его фармацевтически приемлемой соли; и где: Cbz представляет собой каробксибензил; XPhos представляет собой 2-дициклогексилфосфино-2’,4’,6’-триизопропилбифенил; каждый R1 независимо представляет собой этил; каждый X1 независимо представляет собой F; каждый X2 независимо представляет собой F; и каждый X3 независимо представляет собой Br или I.
[0052] В другом варианте осуществления изобретение относится к способу получения соединения (C) или его фармацевтически приемлемой соли, который включает в себя:
(g) взаимодействиевзаимодействие соединения (A) 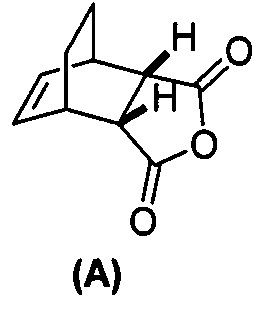 с хинином
с хинином 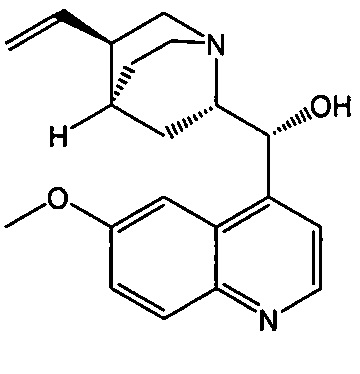 и этиловым спиртом с образованием аддукта хинина и соединения (C-1)
и этиловым спиртом с образованием аддукта хинина и соединения (C-1) 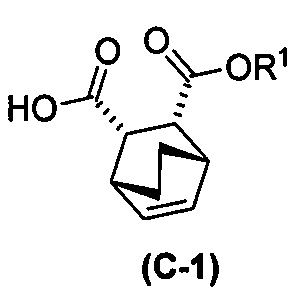 , где R1 представляет собой этил;
, где R1 представляет собой этил;
(h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl с образованием соединения (C-1);
(i) эпимеризацию соединения (C-1) в соединение (C) 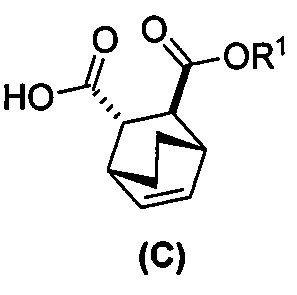 или его фармацевтически приемлемую соль.
или его фармацевтически приемлемую соль.
[0053] В другом варианте осуществления изобретение относится к способу получения соединения(N), который включает в себя:
(q) взаимодействие соединения (K) 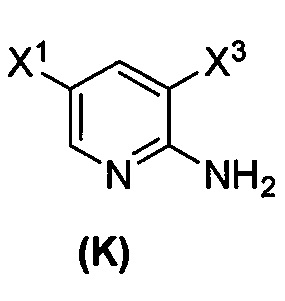 или его фармацевтически приемлемой соли с ацетальдегидом в присутствии палладиевого катализатора с образованием соединения (M)
или его фармацевтически приемлемой соли с ацетальдегидом в присутствии палладиевого катализатора с образованием соединения (M) 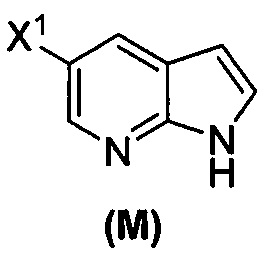 или его фармацевтически приемлемой соли, где X1 представляет собой F или Cl, а X3 представляет собой Br; и
или его фармацевтически приемлемой соли, где X1 представляет собой F или Cl, а X3 представляет собой Br; и
(p) тозилирование соединения (M) или его фармацевтически приемлемой соли с образованием соединения (N): 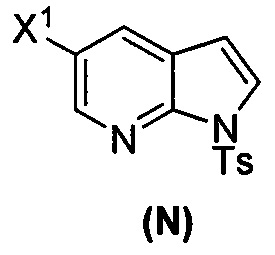 .
.
ОПИСАНИЕ ЧЕРТЕЖА
[0054] На Фиг. 1 представлена диаграмма, показывающая AUC вирусовыделения для дозовой группы с введением 1200 мг/600 мг формы A соли с HCl соединения (1)•1/2H2O в человеческой модели введения провокационной пробы живого, ослабленного гриппа.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0055] I. ОБЩЕПРИНЯТЫЕ СОКРАЩЕНИЯ
[0056] II. ПОЛУЧЕНИЕ СОЕДИНЕНИЙ
[0057] Следует отметить, что описанные здесь стадии могут быть осуществлены в любом хронологическом порядке независимо от того, какой буквой обозначена стадия. Например, стадия (a) может предшествовать стадии (g), стадии (e), стадии (f) иди стадии (s) или следовать за ними.
[0058] Соединение (1)
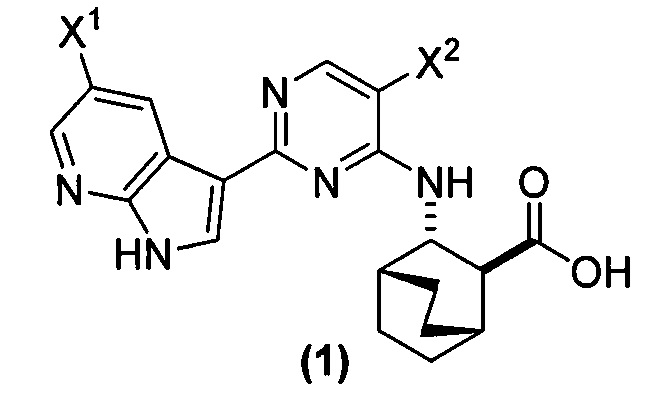 , соединение (2) (где X1 и X2 соединения (1), оба, представляют собой -F) и их фармацевтически приемлемые соли представляют собой ингибиторы репликации вирусов гриппа и могут быть использованы для лечения гриппа у пациента, как описано в WO 2010/148197. В одном конкретном варианте осуществления X1 представляет собой -F и X2 представляет собой -F. В другом конкретном варианте осуществления X1 представляет собой -Cl и X2 представляет собой -F. В другом конкретном варианте осуществления X1 представляет собой -Cl и X2 представляет собой -Cl. В другом конкретном варианте осуществления X1 представляет собой -F и X2 представляет собой -Cl.
, соединение (2) (где X1 и X2 соединения (1), оба, представляют собой -F) и их фармацевтически приемлемые соли представляют собой ингибиторы репликации вирусов гриппа и могут быть использованы для лечения гриппа у пациента, как описано в WO 2010/148197. В одном конкретном варианте осуществления X1 представляет собой -F и X2 представляет собой -F. В другом конкретном варианте осуществления X1 представляет собой -Cl и X2 представляет собой -F. В другом конкретном варианте осуществления X1 представляет собой -Cl и X2 представляет собой -Cl. В другом конкретном варианте осуществления X1 представляет собой -F и X2 представляет собой -Cl.
[0059] В одном варианте осуществления соединения (1) и (2) и их фармацевтически приемлемые соли могут быть получены, как показано на схеме 1: (a) проведением реакции соединения (X): 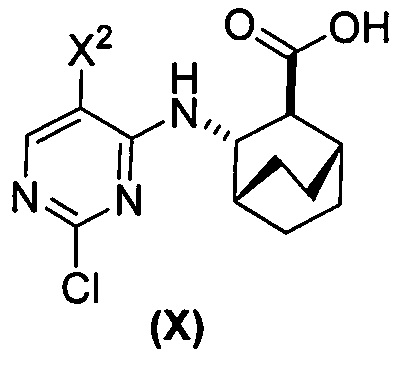 или его фармацевтически приемлемой соли с соединением (Y):
или его фармацевтически приемлемой соли с соединением (Y): 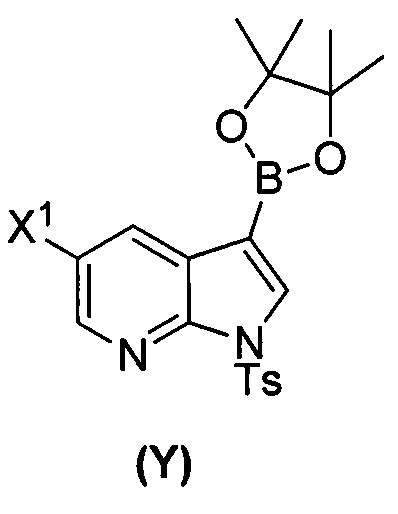 в присутствии комплекса палладий-XPhos и фосфатного или карбонатного основания с образованием соединения (Z) или его фармацевтически приемлемой соли:
в присутствии комплекса палладий-XPhos и фосфатного или карбонатного основания с образованием соединения (Z) или его фармацевтически приемлемой соли: 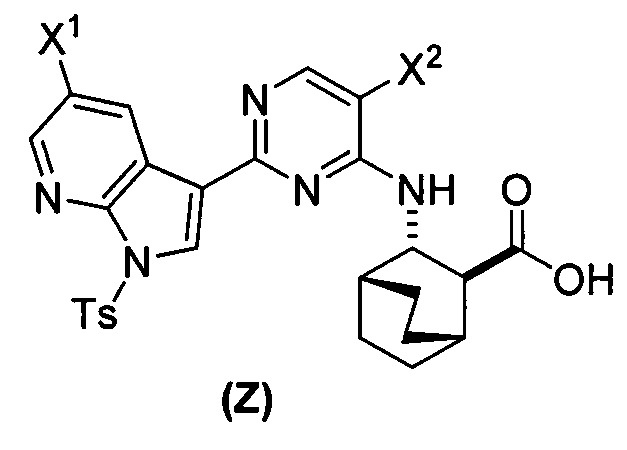 ; и (b) удалением защитной тозильной (Ts) группы из соединения (Z) или его фармацевтически приемлемой соли. X1 представляет собой -F или -Cl; Ts представляет собой тозил; и XPhos представляет собой 2-дициклогексилфосфино-2’,4’,6’-триизопропилбифенил (
; и (b) удалением защитной тозильной (Ts) группы из соединения (Z) или его фармацевтически приемлемой соли. X1 представляет собой -F или -Cl; Ts представляет собой тозил; и XPhos представляет собой 2-дициклогексилфосфино-2’,4’,6’-триизопропилбифенил (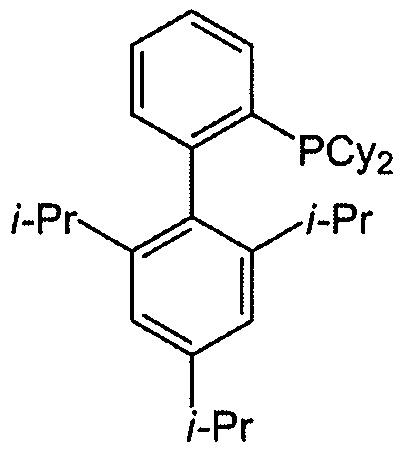 , где Cy представляет собой циклогексил и i-Pr представляет собой изопропил).
, где Cy представляет собой циклогексил и i-Pr представляет собой изопропил).
[0060] Комплекс палладий-XPhos может быть использован как предварительно полученный реагент или, альтернативно, может быть получен на месте (in situ). В одном конкретном варианте осуществления комплекс палладий-XPhos получают смешением источника Pd(0) или Pd(II) с XPhos. Типичные примеры источников Pd(0) или Pd(II) включают Pd2(dba)3, Pd(OAc)2 и PdCl2, где dba представляет собой дибензилиденацетон, а OAc представляет собой ацетат. В одном конкретном варианте осуществления комплекс палладий-XPhos получают на месте (in situ), смешивая Pd(OAc)2 и XPhos.
[0061] Реакцию соединения (X) или его фармацевтически приемлемой соли и соединения (Y) осуществляют в присутствии фосфатного или карбонатного основания. Типичные примеры фосфатных или карбонатных оснований включают Na2CO3, K2CO3, K3PO4 и Na3PO4. В одном конкретном варианте осуществления основание включает K2CO3 или K3PO4. В другом конкретном варианте осуществления основание включает K2CO3. В другом варианте осуществления основание включает K3PO4.
[0062] Реакция соединения (X) или его фармацевтически приемлемой соли и соединения (Y) может быть осуществлена в любой подходящей системе растворителя. В одном конкретном варианте осуществления реакцию осуществляют в системе растворителя, которая включает воду и органический растворитель, выбранный из 2-Me-ТГФ или ТГФ или их сочетания. В другом конкретном варианте осуществления реакцию осуществляют в системе растворителя, которая включает воду и ТГФ. В другом конкретном варианте осуществления реакцию осуществляют в системе растворителя, которая включает воду и 2-Me-ТГФ.
[0063] Стадия (b) удаления защиты может быть осуществлена в любых подходящих условиях, известных в уровне техники для удаления защитной тозильной группы. В одном конкретном варианте осуществления на стадии удаления защиты используют обработку соединения (Z) или его фармацевтически приемлемой соли неорганическим гидроксидом. Типичные примеры подходящих неорганических гидроксидов включают LiOH, NaOH и KOH. В одном конкретном варианте осуществления используют LiOH. В другом конкретном варианте осуществления на стадии (b) удаления защиты используют LiOH в системе растворителя, которая включает ТГФ.
[0064] Схема 1:
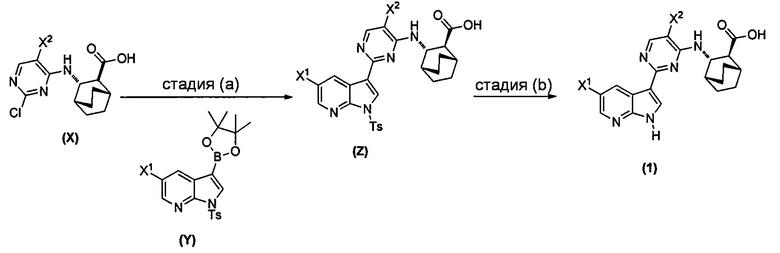
[0065] В другом варианте осуществления соединение (2) и его фармацевтически приемлемые соли могут быть получены, как показано на схеме 1-A: (a) проведением реакции соединения (X-2): 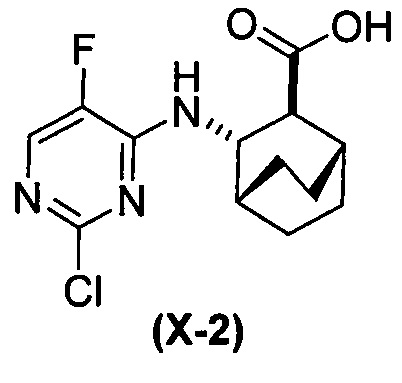 или его фармацевтически приемлемой соли с соединением (Y-2):
или его фармацевтически приемлемой соли с соединением (Y-2): 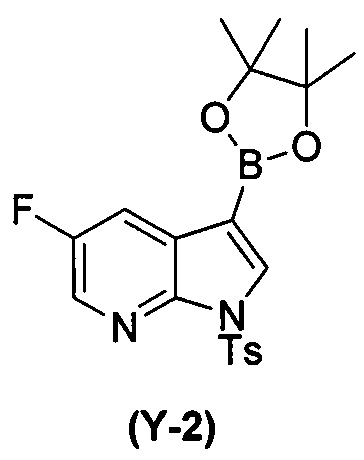 в присутствии комплекса палладий-XPhos и фосфатного или карбонатного основания с образованием соединения (Z-2) или его фармацевтически приемлемой соли:
в присутствии комплекса палладий-XPhos и фосфатного или карбонатного основания с образованием соединения (Z-2) или его фармацевтически приемлемой соли: 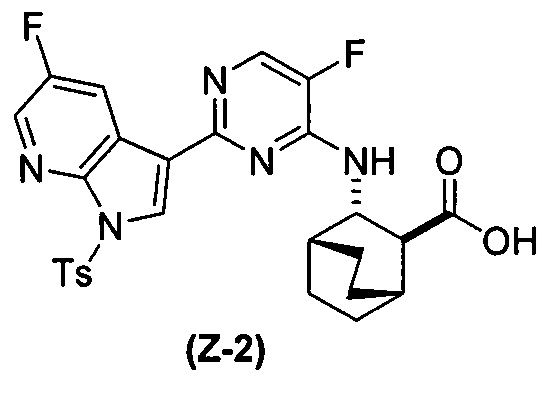 ; и (b) удалением защитной тозильной (Ts) группы из соединения (Z-2) или его фармацевтически приемлемой соли. Подходящие условия реакции для каждой из стадий (a) и (b) и их конкретные примеры, включая фосфатное или карбонатное основание, аналогичны таковым, описанным выше применительно к схеме 1.
; и (b) удалением защитной тозильной (Ts) группы из соединения (Z-2) или его фармацевтически приемлемой соли. Подходящие условия реакции для каждой из стадий (a) и (b) и их конкретные примеры, включая фосфатное или карбонатное основание, аналогичны таковым, описанным выше применительно к схеме 1.
[0066] Схема 1-A:
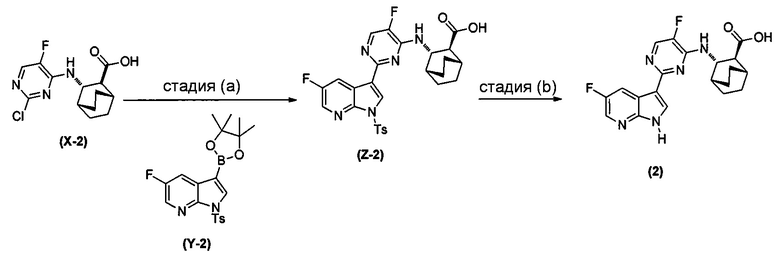
[0067] В другом варианте осуществления на стадии (a) схем 1 и 1-A (например, в реакциях между соединениями (X) и (Y) и между соединениями (X-2) и (Y-2)) можно дополнительно использовать поглотитель Pd (например, смолу или уголь) после реакций, но перед стадией (b) удаления защитной группы Ts, чтобы удалить или уменьшить количества всякого остаточного Pd-катализатора. Типичный пример подходящих поглотителей Pd включает смолу из полистирола с присоединенным тримеркаптотриазином (например, смолу MP-TMP).
[0068] В предлагаемых в изобретении способах получения соединений (1) и (2) и их фармацевтически приемлемых солей, показанных на схемах 1 и 1-A, используют соединения (X) и (Z), каждое из которых имеет свободную группу карбоновой кислоты. Без ограничения какой-либо конкретной теорией, следует отметить, что использование соединений (X) и (Z) вместо соответствующих им сложных эфиров может давать энантиомерно чистые соединения (1) и (2) и их фармацевтически приемлемые соли более удобным и эффективным образом, поскольку соединения (X) и (Z) обычно представляют собой твердые вещества, тогда как соответствующие им сложные эфиры представляют собой масла. Маслянистый материал обычно сложнее очистить по сравнению с твердым материалом, что может ухудшать общий выход и/или энантиомерную чистоту конечного продукта, особенно при крупномасштабном получении, таком как коммерческое производство соединений (1) и (2) или их фармацевтически приемлемых солей. В частности, соответствующие сложные эфиры соединений (X) и (Z) обычно подвергаются эпимеризации (например, в ходе реакции Сузуки соединения (X) с получением соединения (Z) и в ходе удаления тозильной группы Ts из соединения (Z)), что может ухудшать общий выход и/или энантиомерную чистоту конечного продукта.
[0069] В другом варианте осуществления в способах по изобретению используют получение соединений (X) и их фармацевтически приемлемых солей, показанное ниже на схеме 2. Получение включает: (c) взаимодействие соединения (F) или его фармацевтически приемлемой соли: 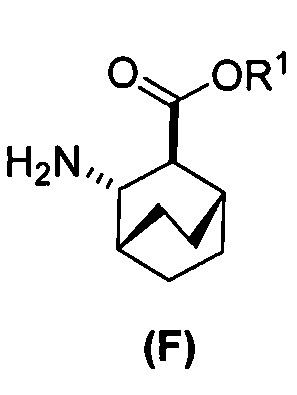 с соединением (G):
с соединением (G): 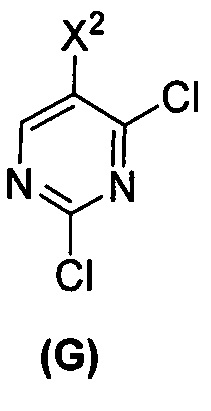 с образованием соединения (H):
с образованием соединения (H): 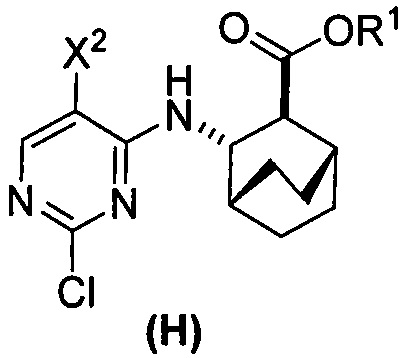 или его фармацевтически приемлемой соли; и (d) гидролиз соединения (H) или его фармацевтически приемлемой соли с образованием соединения (X):
или его фармацевтически приемлемой соли; и (d) гидролиз соединения (H) или его фармацевтически приемлемой соли с образованием соединения (X): 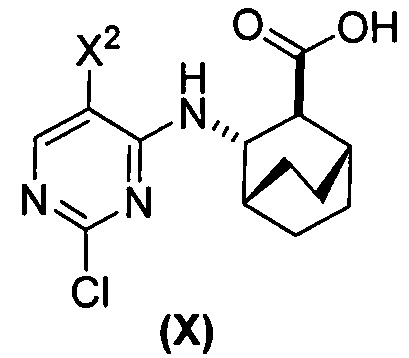 или его фармацевтически приемлемой соли. R1 представляет собой C1-4-алкил, такой как этил или метил. В одном конкретном варианте осуществления, R1 представляет собой этил. В другом конкретном варианте осуществления, R1 представляет собой метил.
или его фармацевтически приемлемой соли. R1 представляет собой C1-4-алкил, такой как этил или метил. В одном конкретном варианте осуществления, R1 представляет собой этил. В другом конкретном варианте осуществления, R1 представляет собой метил.
[0070] Схема 2:
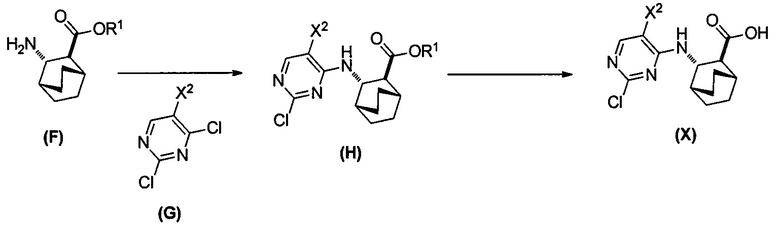
[0071] Сочетание друг с другом соединения (G) и соединения (F) или его фармацевтически приемлемой соли (стадия c) и гидролиз соединения (H) или его фармацевтически приемлемой соли (стадия d) может быть осуществлено в любых подходящих условиях, известных в данной области. В одном конкретном варианте осуществления сочетание друг с другом соединения (F) или его фармацевтически приемлемой соли и соединения (G) осуществляют при температуре в диапазоне от 15°C до 40°C (например, от 25°C до 35°C) в присутствии основания, такого как аминное основание. Типичные примеры таких аминных оснований включают N,N-диизопропилэтиламин, триэтиламин, N,N-диэтилметиламин и так далее. В другом конкретном варианте осуществления гидролиз соединения (H) или его фармацевтически приемлемой соли осуществляют в присутствии основания, такого как неорганическое основание. Типичные примеры таких неорганических оснований включают LiOH, NaOH, KOH и так далее. В одном дополнительном конкретном варианте осуществления гидролиз осуществляют, обрабатывая соединение (H) или его фармацевтически приемлемую соль LiOH при температуре в диапазоне от 20°C до 50°C, таком как 35°C до 50°C (например, 45°C).
[0072] В другом варианте осуществления в способах изобретения используют получение соединения (F) или его фармацевтически приемлемой соли, показанное ниже на схеме 3.
[0073] Схема 3:
 .
.
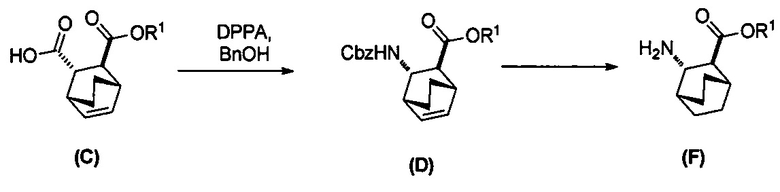
[0074] Получение соединения (F) или его фармацевтически приемлемой соли включает: (e) взаимодействие соединения (C): 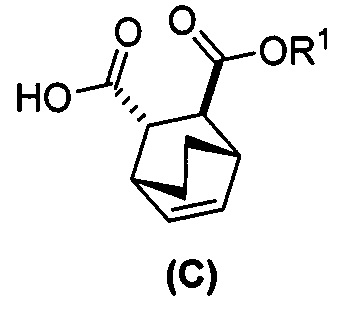 или его фармацевтически приемлемой соли с дифенилфосфорилазидом (DPPA), а затем с бензиловым спиртом с образованием соединения (D):
или его фармацевтически приемлемой соли с дифенилфосфорилазидом (DPPA), а затем с бензиловым спиртом с образованием соединения (D):  или его фармацевтически приемлемой соли; (f) взаимодействие соединения (D) или его фармацевтически приемлемой соли с H2 в присутствии Pd-катализатора на угле (Pd(0)/C) с образованием соединения (F)
или его фармацевтически приемлемой соли; (f) взаимодействие соединения (D) или его фармацевтически приемлемой соли с H2 в присутствии Pd-катализатора на угле (Pd(0)/C) с образованием соединения (F) 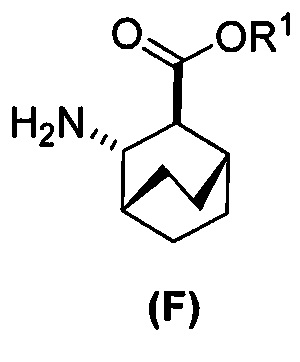 или его фармацевтически приемлемой соли. Cbz представляет собой карбоксибензил.
или его фармацевтически приемлемой соли. Cbz представляет собой карбоксибензил.
[0075] Реакция соединения (C) или его фармацевтически приемлемой соли с DPPA и с бензиловым спиртом с образованием соединения (D) или его фармацевтически приемлемой соли может быть осуществлена в любых условиях, подходящих для перегруппировки Курциуса. Без ограничения какой-либо конкретной теорией, следует отметить, что в ходе перегруппировки Курциуса в реакции DPPA с соединением (C) или его фармацевтически приемлемой солью образуется ацилнитреновое промежуточное соединение, которое затем перегруппировывается в изоцианатное промежуточное соединение. Изоцианатное промежуточное соединение впоследствии реагирует с бензиловым спиртом, давая Cbz-защищенный амин в соединении (D) или его фармацевтически приемлемой соли. В одном конкретном варианте осуществления перегруппировку Курциуса осуществляют при температуре в диапазоне от 60°C до 100°C, таком как от 90°C до 100°C или от 95°C до 100°C. В одном конкретном варианте осуществления реакцию осуществляют в присутствии основания, такого как аминное основание. Типичные примеры таких аминных оснований включают триэтиламин, N,N-диизопропилэтиламин, N,N-диэтилметиламин и так далее. В другом конкретном варианте осуществления перегруппировку Курциуса осуществляют, используя проточный аппарат, при повышенной температуре, такой как от 90°C до 110°C.
[0076] Гидрирование двойной связи с удалением защитной группы Cbz из соединения (D) или его фармацевтически приемлемой соли может быть осуществлено в любых подходящих условиях, известных в уровне техники для общего гидрирования двойной связи и удаления защитной группы Cbz. В одном конкретном варианте осуществления соединение (D) или его фармацевтически приемлемая соль реагирует с H2 в присутствии Pd(0)-катализатора на угле (Pd/C), такого как 10% (по массе на сухую основу) Pd/C. В другом конкретном варианте осуществления продукт реакции соединения (D) или его фармацевтически приемлемой соли с H2 далее обрабатывают HCl в этаноле с образованием соли соединения (F) с HCl.
[0077] В другом варианте осуществления в способах изобретения используют получение соединения (C) или его фармацевтически приемлемой соли, показанное ниже на схеме 4.
[0078] Схема 4:
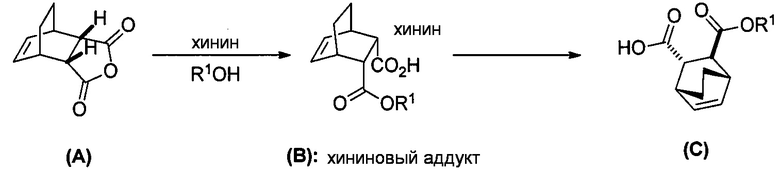 .
.
[0079] Получение соединения (C) или его фармацевтически приемлемой соли включает: (g) взаимодействие соединения (A): 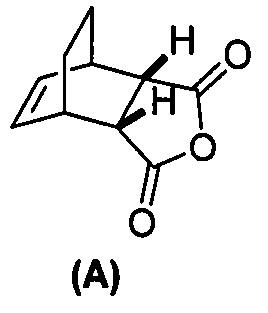 с хинином:
с хинином: 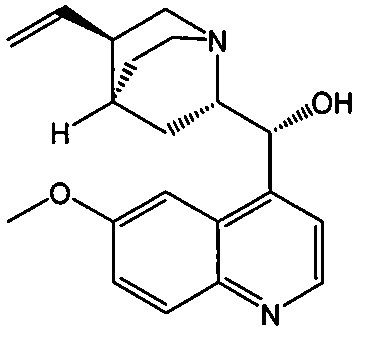 и R1OH с образованием аддукта хинина и соединения (C-1):
и R1OH с образованием аддукта хинина и соединения (C-1): 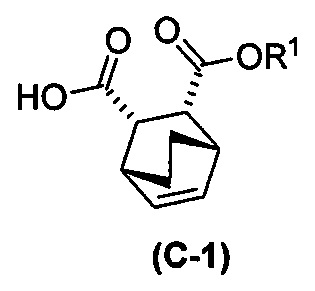 или его фармацевтически приемлемой соли; (h) разрушение аддукта путем обработки аддукта HCl с образованием соединения (C-1) или его фармацевтически приемлемой соли, где R1 представляет собой C1-4-алкил (например, метил, этил, пропил, изопропил или бутил); и эпимеризацию соединения (C-1) или его фармацевтически приемлемой соли в соединение (C) или его фармацевтически приемлемую соль. В одном конкретном варианте осуществления R1 представляет собой этил. Эпимеризация соединения (C-1) или его фармацевтически приемлемой соли может быть проведена, применяя любые подходящие условия, известные в уровне техники. Типично ее осуществляют, обрабатывая соединение основанием, таким как алкоксид. В одном конкретном варианте осуществления применяют C1-6-алкоксид (например, C1-6-алкоксид щелочного металла (например, натрия или калия) или щелочноземельного металла (например, кальция или магния)). В другом конкретном варианте осуществления применяют трет-бутоксид (например, трет-бутоксид калия) или трет-амилат (например, трет-амилат калия).
или его фармацевтически приемлемой соли; (h) разрушение аддукта путем обработки аддукта HCl с образованием соединения (C-1) или его фармацевтически приемлемой соли, где R1 представляет собой C1-4-алкил (например, метил, этил, пропил, изопропил или бутил); и эпимеризацию соединения (C-1) или его фармацевтически приемлемой соли в соединение (C) или его фармацевтически приемлемую соль. В одном конкретном варианте осуществления R1 представляет собой этил. Эпимеризация соединения (C-1) или его фармацевтически приемлемой соли может быть проведена, применяя любые подходящие условия, известные в уровне техники. Типично ее осуществляют, обрабатывая соединение основанием, таким как алкоксид. В одном конкретном варианте осуществления применяют C1-6-алкоксид (например, C1-6-алкоксид щелочного металла (например, натрия или калия) или щелочноземельного металла (например, кальция или магния)). В другом конкретном варианте осуществления применяют трет-бутоксид (например, трет-бутоксид калия) или трет-амилат (например, трет-амилат калия).
[0080] В одном конкретном варианте осуществления в способах изобретения используют получение соединения (X) или его фармацевтически приемлемой соли, как показано ниже на схеме 5.
[0081] Схема 5:
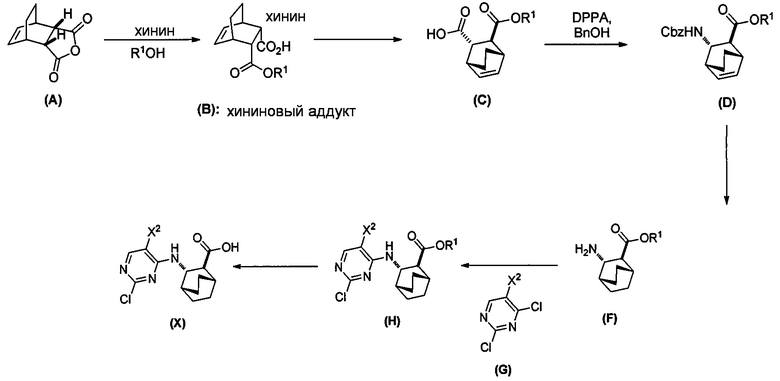
[0082] Подходящие условия реакции для каждой из стадий на схеме 5 и конкретные их примеры аналогичны таковым, описанным выше.
[0083] В другом варианте осуществления в способах изобретения используют получение соединения (F) или его фармацевтически приемлемой соли. В одном конкретном варианте осуществления получают соль соединения (F) с HCl (например, смотри схему 6). Получение включает проведение гидрогенолиза соединения (S): 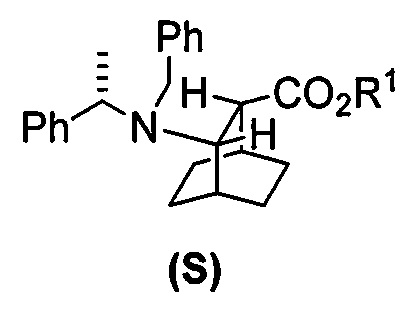 или его фармацевтически приемлемой соли, где Ph представляет собой фенил, в присутствии палладиевого катализатора с образованием соединения (F) или его фармацевтически приемлемой соли, где Ph представляет собой фенил. Типично гидрогенолиз относится к химической реакции, при которой одинарная связь углерод-углерод или углерод-гетероатом (например, N, O или S) расщепляется или подвергается “лизису” под действием водорода. Без ограничения какой-либо конкретной теорией, следует отметить, что в ходе гидрогенолиза соединения (S) или его фармацевтически приемлемой соли расщепляются связи углерод-азот -N(CH2Ph)(CH(CH3)Ph)) соединения (S). Типично, гидрогенолиз проводят каталитически, используя газообразный водород. Подходящие примеры палладиевых катализаторов для стадии гидрогенолиза включают Pd(0) на угле (Pd/C), Pd(OH)2 на угле (Pd(OH)2/C) и их сочетание.
или его фармацевтически приемлемой соли, где Ph представляет собой фенил, в присутствии палладиевого катализатора с образованием соединения (F) или его фармацевтически приемлемой соли, где Ph представляет собой фенил. Типично гидрогенолиз относится к химической реакции, при которой одинарная связь углерод-углерод или углерод-гетероатом (например, N, O или S) расщепляется или подвергается “лизису” под действием водорода. Без ограничения какой-либо конкретной теорией, следует отметить, что в ходе гидрогенолиза соединения (S) или его фармацевтически приемлемой соли расщепляются связи углерод-азот -N(CH2Ph)(CH(CH3)Ph)) соединения (S). Типично, гидрогенолиз проводят каталитически, используя газообразный водород. Подходящие примеры палладиевых катализаторов для стадии гидрогенолиза включают Pd(0) на угле (Pd/C), Pd(OH)2 на угле (Pd(OH)2/C) и их сочетание.
[0084] В одном конкретном варианте осуществления стадию гидрогенолиза осуществляют в присутствии HCl (например, 37,7 масс. % в воде) и она дает соль соединения (F) с HCl.
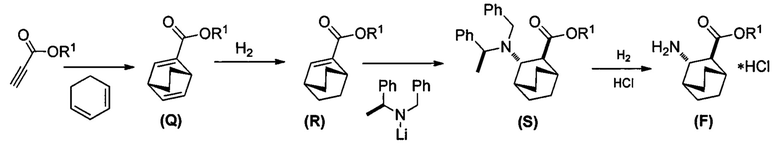
[0085] Схема 6:
[0086] В другом варианте осуществления в способах изобретения используют получение соединения (S) или его фармацевтически приемлемой соли. Получение включает: взаимодействие соединения (R): 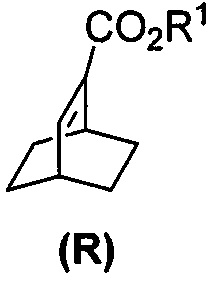 с S-(-)-N-бензил-альфа-метилбензиламинолитием с образованием соединения (S):
с S-(-)-N-бензил-альфа-метилбензиламинолитием с образованием соединения (S): 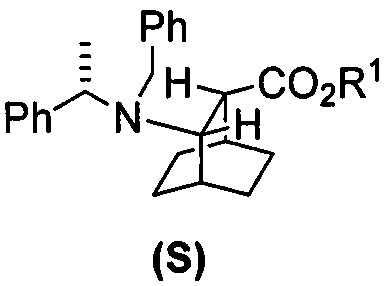 или его фармацевтически приемлемой соли. Данная реакция может быть осуществлена в любых подходящих условиях проведения реакции, известных в уровне техники. В одном конкретном варианте осуществления S-(-)-N-бензил-альфа-метилбензиламинолитий получают на месте (in situ), смешивая S-(-)-N-бензил-альфа-метилбензиламин с алкиллитием, таким как н-бутиллитий.
или его фармацевтически приемлемой соли. Данная реакция может быть осуществлена в любых подходящих условиях проведения реакции, известных в уровне техники. В одном конкретном варианте осуществления S-(-)-N-бензил-альфа-метилбензиламинолитий получают на месте (in situ), смешивая S-(-)-N-бензил-альфа-метилбензиламин с алкиллитием, таким как н-бутиллитий.
[0087] В другом варианте осуществления в способах изобретения используют получение соединения (R). Получение включает:(j) взаимодействие 1,3-циклогексадиена с CH≡CHC(O)OR1 в присутствии алюминиевого катализатора с образованием соединения (Q): 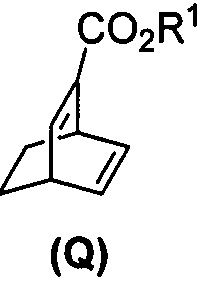 ; и (k) гидрирование соединения (Q) с образованием соединения (R):
; и (k) гидрирование соединения (Q) с образованием соединения (R):  . R1 представляет собой C1-4-алкил. В конкретном варианте осуществления R1 представляет собой этил. В другом конкретном варианте осуществления R1 представляет собой метил.
. R1 представляет собой C1-4-алкил. В конкретном варианте осуществления R1 представляет собой этил. В другом конкретном варианте осуществления R1 представляет собой метил.
[0088] Стадия (j) из предыдущего абзаца представляет собой реакцию Дильса-Альдера между 1,3-циклогексадиеном и CH≡CHC(O)OR1. Для осуществления стадии (j) можно использовать любые подходящие алюминиевые катализаторы реакции Дильса-Альдера, известные в уровне техники. Подходящие примеры алюминиевых катализаторов включают EtAlCl2 (Et=этил), Et2AlCl и смесь AlCl3 и триоктилалюминия. Стадия (k) гидрирования из предыдущего абзаца может быть осуществлена в любых подходящих условиях, известных в уровне техники для общего гидрирования. В одном конкретном варианте осуществления стадия (k) включает взаимодействие соединения (R) с H2 в присутствии Rh(I)-катализатора или отравленного Pd(0)-катализатора. Подходящие примеры Rh(I)-катализаторов включают (PPh3)3RhCl и смесь (PPh3)3RhCl и этилпропиолата, где Ph представляет собой фенил. Отравленный Pd(0)-катализатор относится к Pd(0)-катализатору, в котором другое соединение химически связано с его активными участками на поверхности для регулирования реакционной способности Pd. Отравленные участки больше не могут ускорять реакцию, для катализа которой предназначен катализатор. В общем, отравление катализаторов может быть использовано для улучшения селективности реакций. Подходящий пример отравленного Pd(0)-катализатора включает отравленный свинцом Pd(0)-катализатор на CaCO3 (Pd(Pb)/CaCO3).
[0089] В другом варианте осуществления в способах изобретения используют получение соединения (Y): 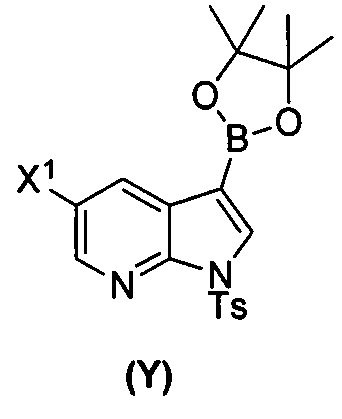 . Получение соединения (Y) включает : взаимодействие соединения (O):
. Получение соединения (Y) включает : взаимодействие соединения (O): 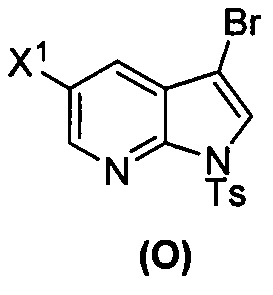 с бис(пинаколато)дибором в присутствии палладиевого катализатора с образованием соединения (Y). Смотри, например, схему 7:
с бис(пинаколато)дибором в присутствии палладиевого катализатора с образованием соединения (Y). Смотри, например, схему 7:
[0090] Схема 7:
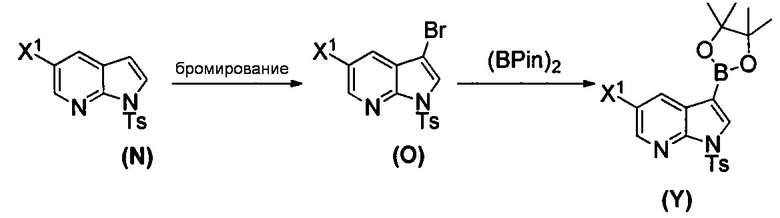
[0091] Типичный пример подходящего палладиевого катализатора включает Pd(Ph3P)4.
[0092] В другом варианте осуществления в способах изобретения используют взаимодействие соединения (O) с бис(пинаколато)дибором в присутствии палладиевого катализатора для получения соединения (Y) и дополнительно используют получение соединения (O), как показано выше на схеме 7. Получение соединения (O) включает бромирование соединения (N): 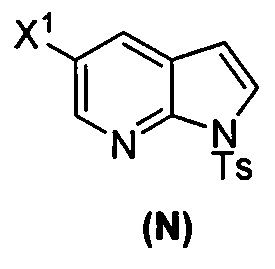 бромирующим агентом. Типичные примеры бромирующих агентов включают Br2, NBS и DBDMH, где NBS представляет собой N-бромсукцинимид, а DBDMH представляет собой 1,3-дибром-5,5-диметилгидантоин. В одном конкретном варианте осуществления бромирующий агент включает Br2 или NBS. В другом конкретном варианте осуществления бромирующий агент включает NBS. В другом конкретном варианте осуществления бромирующий агент включает Br2.
бромирующим агентом. Типичные примеры бромирующих агентов включают Br2, NBS и DBDMH, где NBS представляет собой N-бромсукцинимид, а DBDMH представляет собой 1,3-дибром-5,5-диметилгидантоин. В одном конкретном варианте осуществления бромирующий агент включает Br2 или NBS. В другом конкретном варианте осуществления бромирующий агент включает NBS. В другом конкретном варианте осуществления бромирующий агент включает Br2.
[0093] В другом варианте осуществления в способах изобретения используют получение соединения (N), как изображено на схеме 8.
[0094] Схема 8:
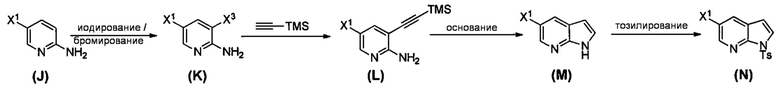
[0095] Получение соединения (N) включает: (l) взаимодействие соединения (J): 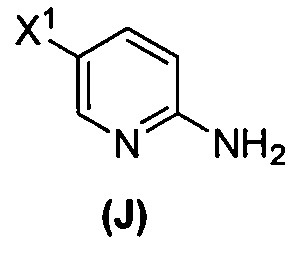 или его фармацевтически приемлемой соли с иодирующим агентом или бромирующим агентом с образованием соединения (K):
или его фармацевтически приемлемой соли с иодирующим агентом или бромирующим агентом с образованием соединения (K): 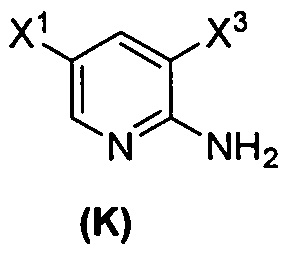 или его фармацевтически приемлемой соли; (m) взаимодействие соединения (K) или его фармацевтически приемлемой соли с триметилсилилацетиленом с образованием соединения (L):
или его фармацевтически приемлемой соли; (m) взаимодействие соединения (K) или его фармацевтически приемлемой соли с триметилсилилацетиленом с образованием соединения (L): 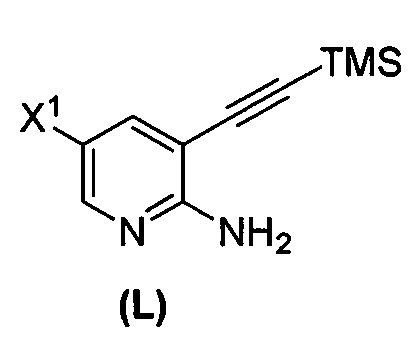 или его фармацевтически приемлемой соли; (n) взаимодействие соединения (L) или его фармацевтически приемлемой соли с C1-6-алкоксидным основанием с образованием соединения (M):
или его фармацевтически приемлемой соли; (n) взаимодействие соединения (L) или его фармацевтически приемлемой соли с C1-6-алкоксидным основанием с образованием соединения (M): 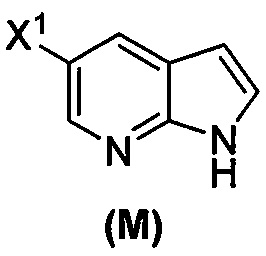 или его фармацевтически приемлемой соли; и (p) тозилирование соединения (M) или его фармацевтически приемлемой соли с образованием соединения (N):
или его фармацевтически приемлемой соли; и (p) тозилирование соединения (M) или его фармацевтически приемлемой соли с образованием соединения (N): 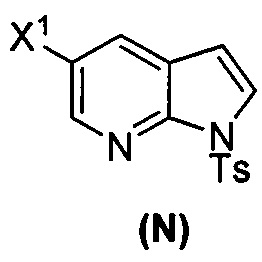 , где X1 представляет собой -F или -Cl; X3 представляет собой -Br или -I; TMS представляет собой триметилсилил; и Ts представляет собой тозил.
, где X1 представляет собой -F или -Cl; X3 представляет собой -Br или -I; TMS представляет собой триметилсилил; и Ts представляет собой тозил.
[0096] Иодирование или бромирование, упомянутое на схеме 8, может быть произведено, используя подходящие условия и реагенты, известные в уровне техники. Типичные примеры бромирующих агентов включают Br2, NBS и DBDMH, где NBS представляет собой N-бромсукцинимид, а DBDMH представляет собой 1,3-дибром-5,5-диметилгидантоин. Типичные примеры иодирующих агентов включают I2, ICl и NIS, где NIS представляет собой N-иодсукцинимид. В одном конкретном варианте осуществления используют бромирование. В другом конкретном варианте осуществления бромирование используют, применяя Br2. В другом конкретном варианте осуществления бромирование используют, применяя NBS. В другом конкретном варианте осуществления используют иодирование. В другом конкретном варианте осуществления иодирование используют, применяя I2.
[0097] Реакция соединения (K) или его фармацевтически приемлемой соли с триметилсилилацетиленом может быть осуществлена в любых подходящих условиях, известных в уровне техники для проведения сочетания по Соногашире между арилгалогенидами и триметилсилилацетиленом. Типично, реакцию осуществляют в присутствии палладиевого катализатора и/или катализатора в виде галогенида меди (I). Типичный пример галогенидов меди (I) включает CuI. Типичные примеры палладиевых катализаторов включают Pd(Ph3P)4 (Ph=фенил), Pd(PPh3)2Cl2, Pd(dppf)2Cl2 dppf=1,1'-бис(дифенилфосфино)ферроцен), Pd(acac)2 (acac=ацетилацетонат), PdCl2(PCy3)2 (Cy=циклогексил), Pd2(dba)3 (dba = дибензилиденацетон) и любое их сочетание. В одном конкретном варианте осуществления реакцию осуществляют в присутствии палладиевого катализатора и/или катализатора в виде галогенида меди (I). В другом конкретном варианте осуществления реакцию осуществляют в присутствии аминного основания (например, C1-4-алкиламина, такого как триэтиламин, N,N-диизопропилэтиламин, N,N-диэтилметиламин) и палладиевого катализатора и/или катализатора в виде галогенида меди (I). В другом конкретном варианте осуществления реакцию осуществляют в присутствии CuI и Pd-катализатора, выбранного из Pd(Ph3P)4, Pd(PPh3)2Cl2, Pd(dppf)2Cl2 или любого их сочетания. В другом конкретном варианте осуществления реакцию осуществляют в присутствии C1-4-алкиламина (например, триэтиламина) и CuI и Pd-катализатора, выбранного из Pd(Ph3P)4, Pd(PPh3)2Cl2, Pd(dppf)2Cl2 или любого их сочетания.
[0098] Стадия (n) реакции соединения (L) или его фармацевтически приемлемой соли с C1-6-алкоксидным основанием может быть также проведена, используя подходящие условия и реагенты, известные в уровне техники. Типичные примеры C1-6-алкоксидов описаны выше. Конкретные примеры включают трет-амилат, трет-бутоксид и метоксид (такие как трет-амилат калия, трет-бутоксид калия и метоксид натрия) и любые их сочетания. В одном конкретном варианте осуществления используют трет-бутоксид калия. В другом конкретном варианте осуществления стадия (n) включает взаимодействие соединения (L) или его фармацевтически приемлемой соли с метоксидом натрия с образованием соединения (P): 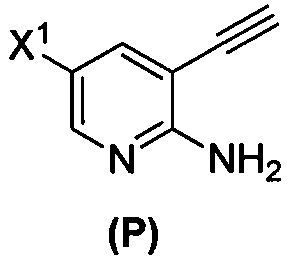 или его фармацевтически приемлемой соли, которое впоследствии вводят в реакцию с трет-бутоксидом калия и/или трет-амилатом калия с образованием соединения (M):
или его фармацевтически приемлемой соли, которое впоследствии вводят в реакцию с трет-бутоксидом калия и/или трет-амилатом калия с образованием соединения (M): 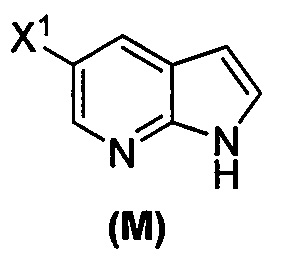 или его фармацевтически приемлемой соли.
или его фармацевтически приемлемой соли.
[0099] Стадия (p) тозилирования может быть осуществлена в любых подходящих условиях, известных в уровне техник для тозилирования. В одном конкретном варианте осуществления стадию тозилирования осуществляют путем проведения реакции соединения (M) или его фармацевтически приемлемой соли с TsCl.
[0100] В другом варианте осуществления в изобретении используют получение соединения (N), как показано на схеме 9.
[0101] Схема 9:
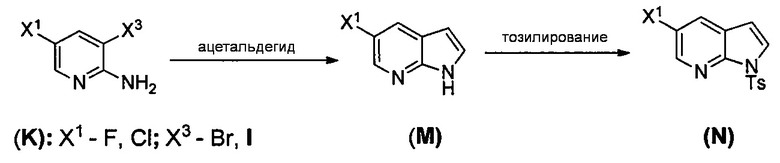
[0102] Получение включает:(q) взаимодействие соединения (K): 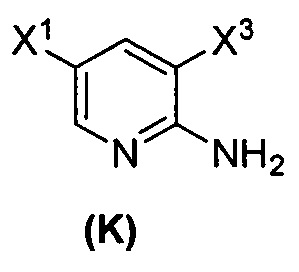 или его фармацевтически приемлемой соли с ацетальдегидом в присутствии палладиевого катализатора с образованием соединения (M):
или его фармацевтически приемлемой соли с ацетальдегидом в присутствии палладиевого катализатора с образованием соединения (M): 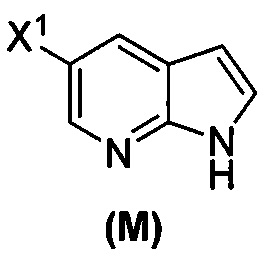 или его фармацевтически приемлемой соли, где X3 представляет собой -Br или -I; и (p) тозилирование соединения (M) или его фармацевтически приемлемой соли с образованием соединения (N):
или его фармацевтически приемлемой соли, где X3 представляет собой -Br или -I; и (p) тозилирование соединения (M) или его фармацевтически приемлемой соли с образованием соединения (N): 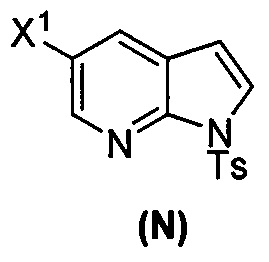 .
.
[0103] Типичные примеры палладиевых катализаторов для реакции соединения (K) или его фармацевтически приемлемой соли с ацетальдегидом включают смеси бис(дибензилиденацетон)палладия и третичного фосфинового лиганда, PR3, где R представляет собой C1-6-алкил или C5-6-циклоалкил. Типичные примеры третичных фосфиновых лигандов, PR3, включают P(tBu)3, PCy3, P(i-Pr)3, P(Bu3), PEt3, PMe3 или их смесь. В одном конкретном варианте осуществления используют P(tBu)3.
[0104] В одном конкретном варианте осуществления получение соединения (N), как показано на схеме 9, дополнительно включает обработку реакционной смеси, содержащей соединение (K) или его фармацевтически приемлемую соль и ацетальдегид, карбонатным основанием, таким как Na2CO3, перед стадией тозилирования с образованием соединения (N).
[0105] Стадия (p) тозилирования может быть осуществлена в любых подходящих условиях, известных в уровне техники для тозилирования. В одном конкретном варианте осуществления стадию тозилирования осуществляют путем проведения реакции соединения (M) или его фармацевтически приемлемой соли с TsCl.
[0106] В другом варианте осуществления в способах изобретения используют получение соединения (Y), как показано на схеме. Примеры и условия для каждой стадии на схеме 10 представляют собой независимо таковые, как описаны выше.
[0107] Схема 10:

[0108] В другом варианте осуществления способы изобретения служат для получения соединения (1) или его фармацевтически приемлемой соли, где способы включают:(g) взаимодействие соединения (A): 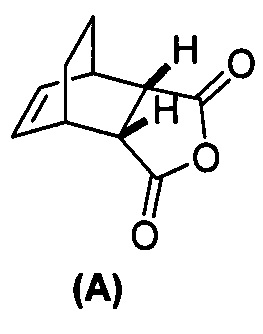 с хинином:
с хинином:  и этиловым спиртом с образованием аддукта хинина и соединения (C-1):
и этиловым спиртом с образованием аддукта хинина и соединения (C-1): 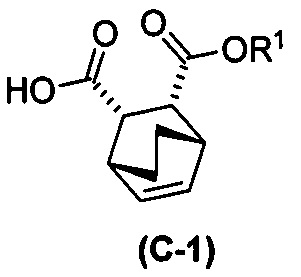 ; (h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl с образованием соединения (C-1) или его фармацевтически приемлемой соли; (i) эпимеризацию соединения (C-1) или его фармацевтически приемлемой соли в соединение (C):
; (h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl с образованием соединения (C-1) или его фармацевтически приемлемой соли; (i) эпимеризацию соединения (C-1) или его фармацевтически приемлемой соли в соединение (C): 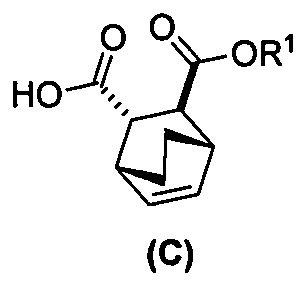 или его фармацевтически приемлемую соль; (e) взаимодействие соединения (C) или его фармацевтически приемлемой соли с дифенилфосфорилазидом (DPPA) и с бензиловым спиртом с образованием соединения (D):
или его фармацевтически приемлемую соль; (e) взаимодействие соединения (C) или его фармацевтически приемлемой соли с дифенилфосфорилазидом (DPPA) и с бензиловым спиртом с образованием соединения (D): 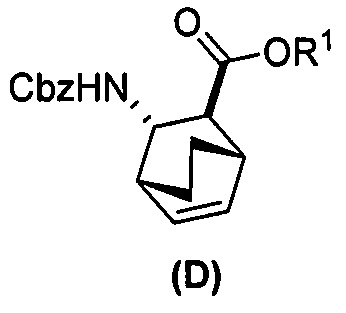 , где Cbz представляет собой карбоксибензил; (f) взаимодействие соединения (D) или его фармацевтически приемлемой соли с H2 в присутствии Pd-катализатора на угле (Pd(0)/C) с образованием соединения (F)
, где Cbz представляет собой карбоксибензил; (f) взаимодействие соединения (D) или его фармацевтически приемлемой соли с H2 в присутствии Pd-катализатора на угле (Pd(0)/C) с образованием соединения (F) 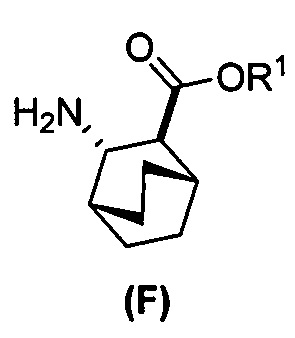 или его фармацевтически приемлемой соли; (c) взаимодействие соединения (F) или его фармацевтически приемлемой соли с соединением (G):
или его фармацевтически приемлемой соли; (c) взаимодействие соединения (F) или его фармацевтически приемлемой соли с соединением (G): 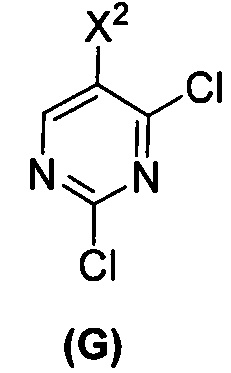 с образованием соединения (H):
с образованием соединения (H):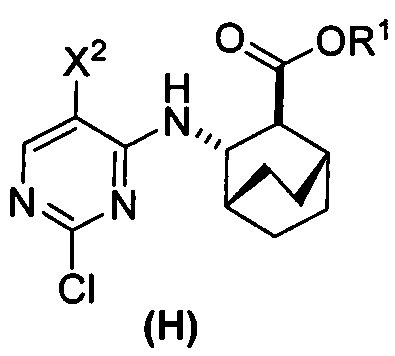 или его фармацевтически приемлемой соли; (d) гидролиз соединения (H) или его фармацевтически приемлемой соли с образованием соединения (X);
или его фармацевтически приемлемой соли; (d) гидролиз соединения (H) или его фармацевтически приемлемой соли с образованием соединения (X); 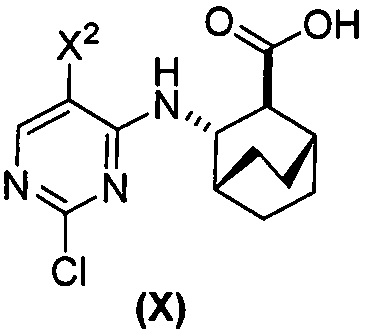 или его фармацевтически приемлемой соли; (a) взаимодействие соединения (X) или его фармацевтически приемлемой соли с соединением (Y):
или его фармацевтически приемлемой соли; (a) взаимодействие соединения (X) или его фармацевтически приемлемой соли с соединением (Y): 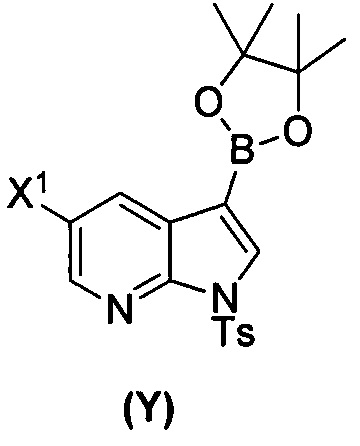 в присутствии палладиевого катализатора с образованием соединения (Z):
в присутствии палладиевого катализатора с образованием соединения (Z): 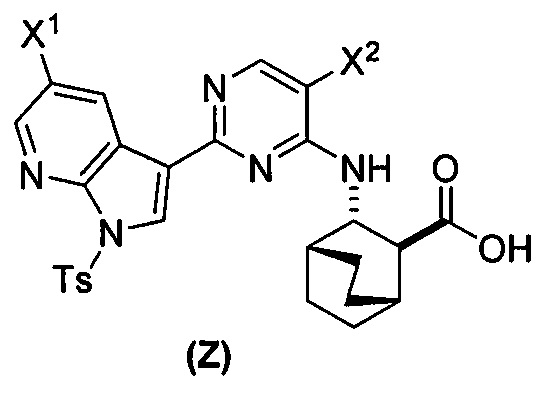 или его фармацевтически приемлемой соли; и (b) удаление защитной группы Ts из соединения (Z) или его фармацевтически приемлемой соли с образованием соединения (1) или его фармацевтически приемлемой соли. Каждый из X1 и X2 независимо представляет собой -F или -Cl; и R1 представляет собой этил. Подходящие условия и реагенты, включая конкретные для каждой стадии, представляют собой таковые, как описано выше применительно к схемам 1-10. В одном конкретном варианте осуществления стадию h) реакции соединения (X) или его фармацевтически приемлемой соли с соединением (Y) осуществляют в присутствии комплекса палладий-XPhos и фосфатного или карбонатного основания. Конкретные примеры фосфатных и карбонатных оснований представляют собой таковые, как описано выше. В другом конкретном варианте осуществления стадию h) реакции соединения (X) или его фармацевтически приемлемой соли с соединением (Y) осуществляют в системе растворителя, которая включает воду и органический растворитель, выбранный из 2-метил-ТГФ или ТГФ или их сочетания. В другом конкретном варианте осуществления стадия (i) удаления защиты включает обработку соединения (Z) или его фармацевтически приемлемой соли неорганическим гидроксидом, выбранным из группы, состоящей из LiOH, NaOH и KOH. В другом конкретном варианте осуществления стадия (i) удаления защиты включает обработку соединения (Z) или его фармацевтически приемлемой соли LiOH в системе растворителя, которая включает ТГФ. В другом конкретном варианте осуществления стадия (d) гидрирования соединения (D) включает взаимодействие соединения (D) с H2 в присутствии Pd-катализатора на угле (Pd/C). В другом варианте осуществления эпимеризацию соединения (C-1) или его фармацевтически приемлемой соли проводят, обрабатывая соединение C1-6-алкоксидом. Конкретные примеры C1-6-алкоксида представляют собой таковые, как описано выше.
или его фармацевтически приемлемой соли; и (b) удаление защитной группы Ts из соединения (Z) или его фармацевтически приемлемой соли с образованием соединения (1) или его фармацевтически приемлемой соли. Каждый из X1 и X2 независимо представляет собой -F или -Cl; и R1 представляет собой этил. Подходящие условия и реагенты, включая конкретные для каждой стадии, представляют собой таковые, как описано выше применительно к схемам 1-10. В одном конкретном варианте осуществления стадию h) реакции соединения (X) или его фармацевтически приемлемой соли с соединением (Y) осуществляют в присутствии комплекса палладий-XPhos и фосфатного или карбонатного основания. Конкретные примеры фосфатных и карбонатных оснований представляют собой таковые, как описано выше. В другом конкретном варианте осуществления стадию h) реакции соединения (X) или его фармацевтически приемлемой соли с соединением (Y) осуществляют в системе растворителя, которая включает воду и органический растворитель, выбранный из 2-метил-ТГФ или ТГФ или их сочетания. В другом конкретном варианте осуществления стадия (i) удаления защиты включает обработку соединения (Z) или его фармацевтически приемлемой соли неорганическим гидроксидом, выбранным из группы, состоящей из LiOH, NaOH и KOH. В другом конкретном варианте осуществления стадия (i) удаления защиты включает обработку соединения (Z) или его фармацевтически приемлемой соли LiOH в системе растворителя, которая включает ТГФ. В другом конкретном варианте осуществления стадия (d) гидрирования соединения (D) включает взаимодействие соединения (D) с H2 в присутствии Pd-катализатора на угле (Pd/C). В другом варианте осуществления эпимеризацию соединения (C-1) или его фармацевтически приемлемой соли проводят, обрабатывая соединение C1-6-алкоксидом. Конкретные примеры C1-6-алкоксида представляют собой таковые, как описано выше.
[0109] В другом конкретном варианте осуществления способы изобретения служат для получения соединения (2) или его фармацевтически приемлемой соли, где способы включают стадии (a) по (i) из предыдущего абзаца, где каждый из X1 и X2 независимо представляет собой -F, а R1 представляет собой этил. Подходящие условия реакций для каждой из стадий и конкретные их примеры представляют собой таковые, как описано выше для получения соединения (1) или его фармацевтически приемлемой соли.
[0110] В другом варианте осуществления способы изобретения служат для получения соединения (1) или его фармацевтически приемлемой соли. Способы включают: (q) взаимодействие соединения (K) или его фармацевтически приемлемой соли: 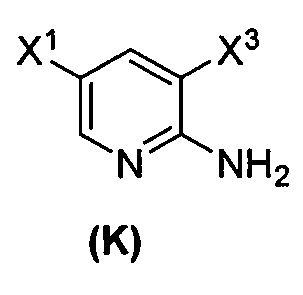 с ацетальдегидом в присутствии палладиевого катализатора с формированием соединения (M):
с ацетальдегидом в присутствии палладиевого катализатора с формированием соединения (M): 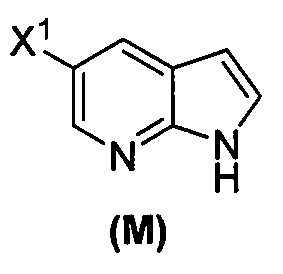 или его фармацевтически приемлемой соли; (p) тозилирование соединения (M) или его фармацевтически приемлемой соли с образованием соединения (N):
или его фармацевтически приемлемой соли; (p) тозилирование соединения (M) или его фармацевтически приемлемой соли с образованием соединения (N): 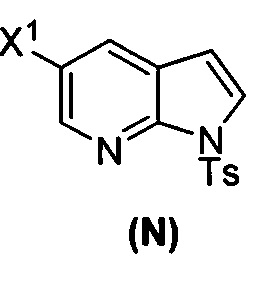 ; (s) бромирование соединения (N) с образованием соединения (O):
; (s) бромирование соединения (N) с образованием соединения (O): 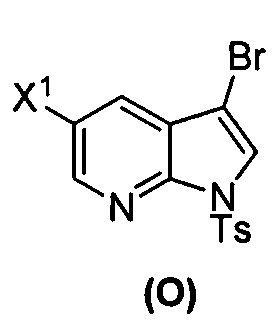 ; (t) взаимодействие соединения (O) с бис(пинаколато)дибором в присутствии палладиевого катализатора с образованием соединения (Y):
; (t) взаимодействие соединения (O) с бис(пинаколато)дибором в присутствии палладиевого катализатора с образованием соединения (Y): 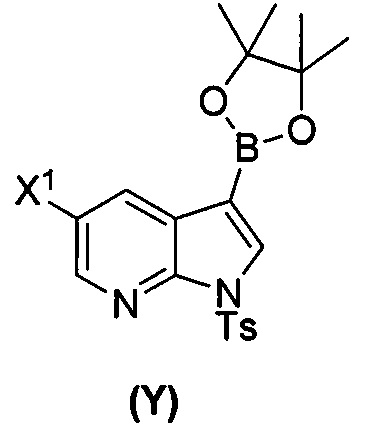 ; (a) взаимодействие соединения (X):
; (a) взаимодействие соединения (X): 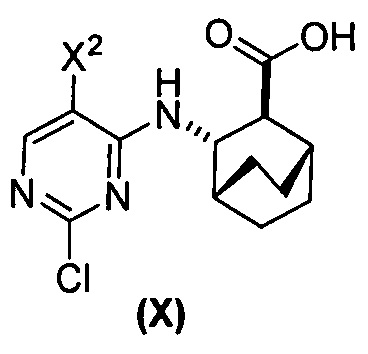 или его фармацевтически приемлемой соли с соединением (Y) в присутствии палладиевого катализатора с образованием соединения (Z):
или его фармацевтически приемлемой соли с соединением (Y) в присутствии палладиевого катализатора с образованием соединения (Z): 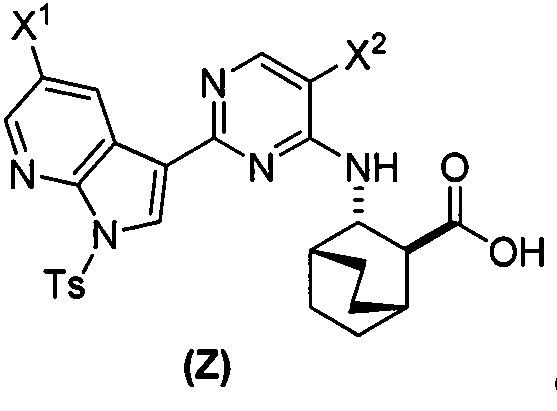 или его фармацевтически приемлемой соли; и удаление (b) защитной группы Ts из соединения (Z) или его фармацевтически приемлемой соли с образованием соединения (1) или его фармацевтически приемлемой соли. Каждый из X1 и X2 независимо представляет собой -F или -Cl; и X3 представляет собой -Br. Подходящие условия и реагенты, включая конкретные для каждой стадии, представляют собой таковые, как описано выше применительно к схемам 1-10. В одном конкретном варианте осуществления стадию (e) реакции соединения (X) с соединением (Y) осуществляют в присутствии комплекса палладий-XPhos и фосфатного или карбонатного основания. Конкретные примеры фосфатных и карбонатных оснований представляют собой таковые, как описано выше. В другом конкретном варианте осуществления стадию (e) реакции соединения (X) или его фармацевтически приемлемой соли с соединением (Y) осуществляют в системе растворителя, которая включает воду и органический растворитель, выбранный из 2-метил-ТГФ или ТГФ или их сочетания. В другом конкретном варианте осуществления стадия (f) удаления защиты включает обработку соединения (Z) или его фармацевтически приемлемой соли неорганическим гидроксидом, выбранным из группы, состоящей из LiOH, NaOH и KOH. В другом конкретном варианте осуществления стадия (f) удаления защиты включает обработку соединения (Z) или его фармацевтически приемлемой соли LiOH в системе растворителя, которая включает ТГФ. В другом конкретном варианте осуществления палладиевый катализатор стадии (a) реакции соединения (K) или его фармацевтически приемлемой соли с ацетальдегидом включает бис(дибензилиденацетон)палладий и третичный фосфиновый лиганд, PR3, где R представляет собой C1-6-алкил или C5-6-циклоалкил. В другом конкретном варианте осуществления третичный фосфиновый лиганд включает P(tBu)3.
или его фармацевтически приемлемой соли; и удаление (b) защитной группы Ts из соединения (Z) или его фармацевтически приемлемой соли с образованием соединения (1) или его фармацевтически приемлемой соли. Каждый из X1 и X2 независимо представляет собой -F или -Cl; и X3 представляет собой -Br. Подходящие условия и реагенты, включая конкретные для каждой стадии, представляют собой таковые, как описано выше применительно к схемам 1-10. В одном конкретном варианте осуществления стадию (e) реакции соединения (X) с соединением (Y) осуществляют в присутствии комплекса палладий-XPhos и фосфатного или карбонатного основания. Конкретные примеры фосфатных и карбонатных оснований представляют собой таковые, как описано выше. В другом конкретном варианте осуществления стадию (e) реакции соединения (X) или его фармацевтически приемлемой соли с соединением (Y) осуществляют в системе растворителя, которая включает воду и органический растворитель, выбранный из 2-метил-ТГФ или ТГФ или их сочетания. В другом конкретном варианте осуществления стадия (f) удаления защиты включает обработку соединения (Z) или его фармацевтически приемлемой соли неорганическим гидроксидом, выбранным из группы, состоящей из LiOH, NaOH и KOH. В другом конкретном варианте осуществления стадия (f) удаления защиты включает обработку соединения (Z) или его фармацевтически приемлемой соли LiOH в системе растворителя, которая включает ТГФ. В другом конкретном варианте осуществления палладиевый катализатор стадии (a) реакции соединения (K) или его фармацевтически приемлемой соли с ацетальдегидом включает бис(дибензилиденацетон)палладий и третичный фосфиновый лиганд, PR3, где R представляет собой C1-6-алкил или C5-6-циклоалкил. В другом конкретном варианте осуществления третичный фосфиновый лиганд включает P(tBu)3.
[0111] В другом конкретном варианте осуществления способы изобретения служат для получения соединения (2) или его фармацевтически приемлемой соли, где способы включают стадии (a) по (f) из предыдущего абзаца, где каждый из X1 и X2 независимо представляет собой -F. Подходящие условия реакций для каждой из стадий и конкретные их примеры представляют собой таковые, как описано выше для получения соединения (1) или его фармацевтически приемлемой соли.
[0112] В другом варианте осуществления способы изобретения служат для получения соединения (2) или его фармацевтически приемлемой соли. Способы включают:(g) взаимодействие соединения (A): 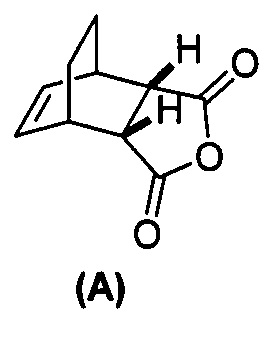 с хинином:
с хинином:  и этиловым спиртом с образованием аддукта хинина и соединения (C-1):
и этиловым спиртом с образованием аддукта хинина и соединения (C-1): 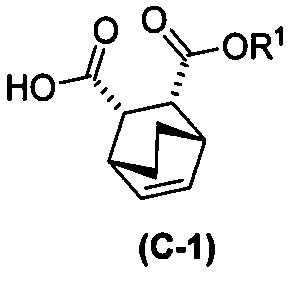 ; (h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl с образованием соединения (C-1) или его фармацевтически приемлемой соли; (i) эпимеризацию соединения (C-1) в соединение (C):
; (h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl с образованием соединения (C-1) или его фармацевтически приемлемой соли; (i) эпимеризацию соединения (C-1) в соединение (C): 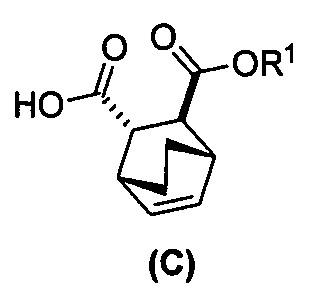 или его фармацевтически приемлемую соль; (e) взаимодействие соединения (C) с дифенилфосфорилазидом и затем с бензиловым спиртом с образованием соединения (D):
или его фармацевтически приемлемую соль; (e) взаимодействие соединения (C) с дифенилфосфорилазидом и затем с бензиловым спиртом с образованием соединения (D): 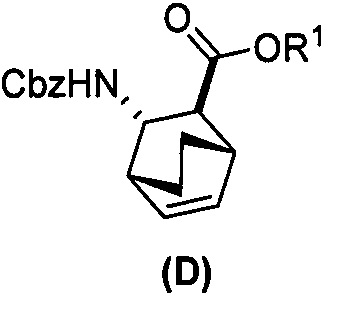 ; (f) взаимодействие соединения (D) или его фармацевтически приемлемой соли с H2 в присутствии Pd-катализатора на угле (Pd(0)/C) с образованием соли соединения (F) с HCl; (r) взаимодействие соли соединения (F) с HCl с соединением (G):
; (f) взаимодействие соединения (D) или его фармацевтически приемлемой соли с H2 в присутствии Pd-катализатора на угле (Pd(0)/C) с образованием соли соединения (F) с HCl; (r) взаимодействие соли соединения (F) с HCl с соединением (G): 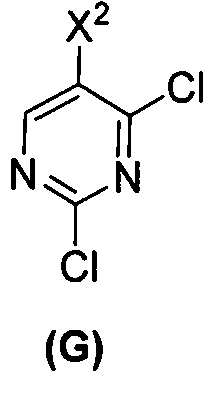 с образованием соединения (H):
с образованием соединения (H): 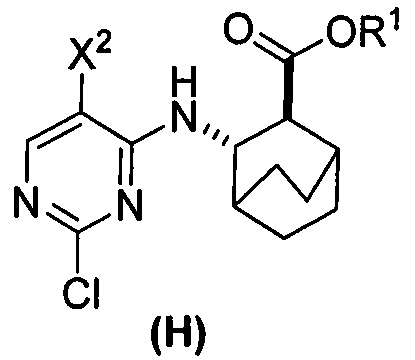 ; (h-1) гидролиз соединения (H) с образованием соединения (X-2):
; (h-1) гидролиз соединения (H) с образованием соединения (X-2): 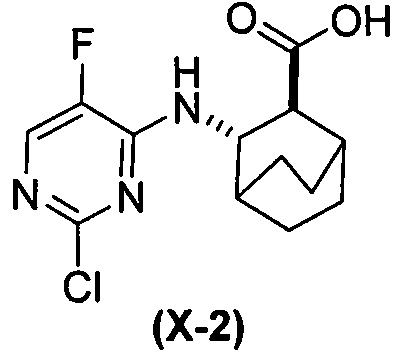 ; (l) иодирование или бромирование соединения (J):
; (l) иодирование или бромирование соединения (J):  с образованием соединения (K):
с образованием соединения (K): 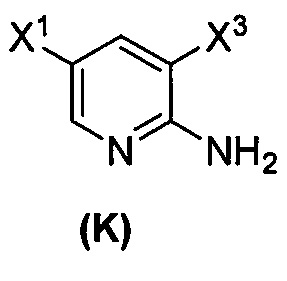 ; (q-1) взаимодействие соединения (K) с триметилсилилацетиленом с образованием соединения (L):
; (q-1) взаимодействие соединения (K) с триметилсилилацетиленом с образованием соединения (L): 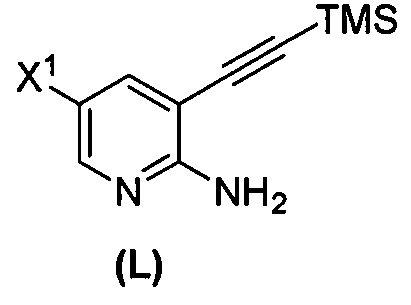 ; (j) взаимодействие соединения (L) с C1-6-алкоксидом с образованием соединения (M):
; (j) взаимодействие соединения (L) с C1-6-алкоксидом с образованием соединения (M): 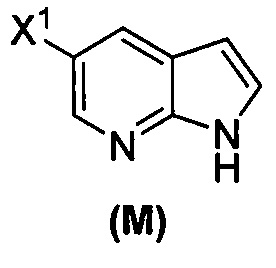 ; (k) тозилирование соединения (M) с образованием соединения (N):
; (k) тозилирование соединения (M) с образованием соединения (N):  ; (s) бромирование соединения (N) с образованием соединения (O):
; (s) бромирование соединения (N) с образованием соединения (O): 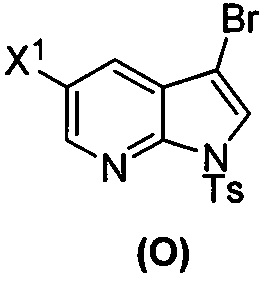 ; (t) взаимодействие соединения (O) с бис(пинаколато)дибором в присутствии Pd(Ph3P)4 с образованием соединения (Y-2):
; (t) взаимодействие соединения (O) с бис(пинаколато)дибором в присутствии Pd(Ph3P)4 с образованием соединения (Y-2): 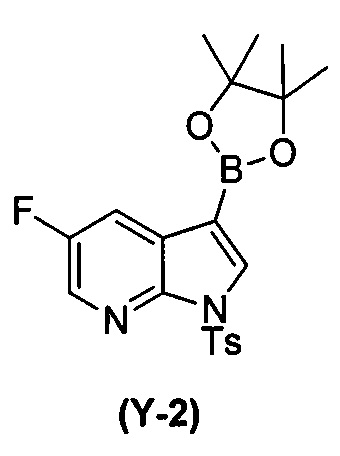 ; (n) взаимодействие соединения (X-2) с соединением (Y-2) в присутствии комплекса палладий-XPhos и фосфатного или карбонатного основания, выбранного из K2CO3 или K3PO4, с образованием соединения (Z-2):
; (n) взаимодействие соединения (X-2) с соединением (Y-2) в присутствии комплекса палладий-XPhos и фосфатного или карбонатного основания, выбранного из K2CO3 или K3PO4, с образованием соединения (Z-2): 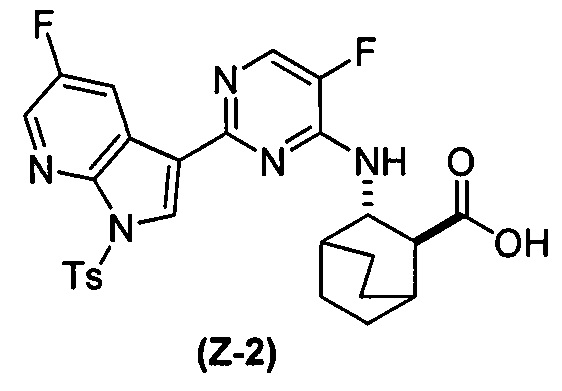 или его фармацевтически приемлемой соли; и удаление (o) защитной группы Ts из соединения (Z-2) или его фармацевтически приемлемой соли с образованием соединения (2) или его фармацевтически приемлемой соли. Каждый R1 независимо представляет собой этил; каждый X1 независимо представляет собой -F; каждый X2 независимо представляет собой -F; и каждый X3 независимо представляет собой -Br или -I. Подходящие условия и реагенты, включая конкретные для каждой стадии, представляют собой таковые, как описано выше применительно к схемам 1-10.
или его фармацевтически приемлемой соли; и удаление (o) защитной группы Ts из соединения (Z-2) или его фармацевтически приемлемой соли с образованием соединения (2) или его фармацевтически приемлемой соли. Каждый R1 независимо представляет собой этил; каждый X1 независимо представляет собой -F; каждый X2 независимо представляет собой -F; и каждый X3 независимо представляет собой -Br или -I. Подходящие условия и реагенты, включая конкретные для каждой стадии, представляют собой таковые, как описано выше применительно к схемам 1-10.
[0113] В другом варианте осуществления изобретение относится к способам получения соединения (C) или его фармацевтически приемлемой соли. Способы включают:(g) взаимодействие соединения (A): 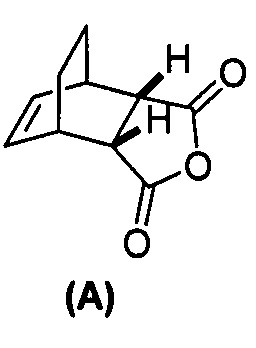 с хинином:
с хинином:  и этиловым спиртом с образованием аддукта хинина и соединения (C-1):
и этиловым спиртом с образованием аддукта хинина и соединения (C-1): 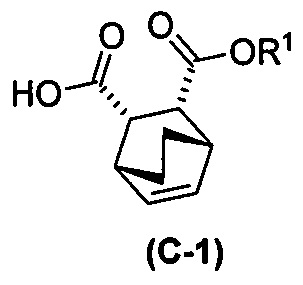 ; (h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl с образованием соединения (C-1) или его фармацевтически приемлемой соли; и (i) эпимеризацию соединения (C-1) в соединение (C):
; (h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl с образованием соединения (C-1) или его фармацевтически приемлемой соли; и (i) эпимеризацию соединения (C-1) в соединение (C): 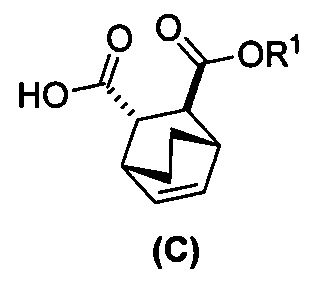 или его фармацевтически приемлемую соль. Без ограничения какой-либо конкретной теорией, следует отметить, что аддукт хинина и соединения (C), имеющего этил в качестве R1, осаждается из реакционной смеси, образованной хинином и соединением (A), что может давать соединение (C) в энантиомерно чистой форме с энантиомерной чистотой более 99%.
или его фармацевтически приемлемую соль. Без ограничения какой-либо конкретной теорией, следует отметить, что аддукт хинина и соединения (C), имеющего этил в качестве R1, осаждается из реакционной смеси, образованной хинином и соединением (A), что может давать соединение (C) в энантиомерно чистой форме с энантиомерной чистотой более 99%.
[0114] В другом варианте осуществления изобретение относится к способам получения соединения (N). Способы включают : (q) взаимодействие соединения (K): 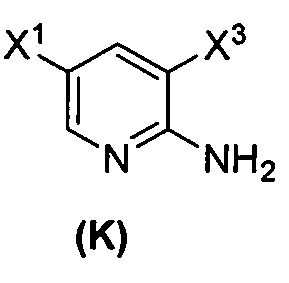 или его фармацевтически приемлемой соли с ацетальдегидом в присутствии палладиевого катализатора с образованием соединения (M):
или его фармацевтически приемлемой соли с ацетальдегидом в присутствии палладиевого катализатора с образованием соединения (M): 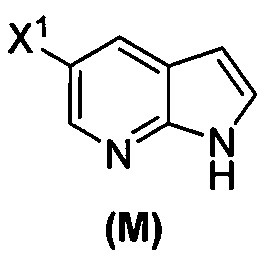 или его фармацевтически приемлемой соли, где X1 представляет собой -F или -Cl, и X3 представляет собой -Br или -I; и (p) тозилирование соединения (M) или его фармацевтически приемлемой соли с образованием соединения (N):
или его фармацевтически приемлемой соли, где X1 представляет собой -F или -Cl, и X3 представляет собой -Br или -I; и (p) тозилирование соединения (M) или его фармацевтически приемлемой соли с образованием соединения (N): 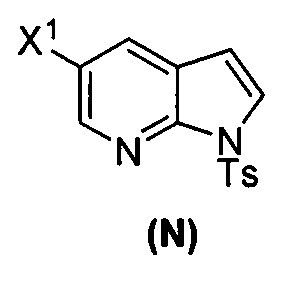 . В одном конкретном варианте осуществления X1 представляет собой -F. В другом конкретном варианте осуществления X3 представляет собой -Br. В другом конкретном варианте осуществления X1 представляет собой -F и X3 представляет собой -Br. Без ограничения какой-либо конкретной теорией, следует отметить, что получение соединения (N) согласно схеме 9 имеет несколько преимуществ перед таковым согласно схеме 8 (соединения (K) → соединение (N)) в том, что получение соединения (N) согласно схеме 9 обычно дает лучшие выходы и в целом требует меньшего фильтрования. Оно также является экономичным при крупномасштабных реакциях, таких как реакции коммерческого масштаба, из-за относительно меньшей стоимости ацетальдегида по сравнению с таковой триметилсилилацетилена.
. В одном конкретном варианте осуществления X1 представляет собой -F. В другом конкретном варианте осуществления X3 представляет собой -Br. В другом конкретном варианте осуществления X1 представляет собой -F и X3 представляет собой -Br. Без ограничения какой-либо конкретной теорией, следует отметить, что получение соединения (N) согласно схеме 9 имеет несколько преимуществ перед таковым согласно схеме 8 (соединения (K) → соединение (N)) в том, что получение соединения (N) согласно схеме 9 обычно дает лучшие выходы и в целом требует меньшего фильтрования. Оно также является экономичным при крупномасштабных реакциях, таких как реакции коммерческого масштаба, из-за относительно меньшей стоимости ацетальдегида по сравнению с таковой триметилсилилацетилена.
[0115] В другом варианте осуществления в способах изобретения дополнительно используют обработку соединения (1) после стадии (b) удаления защиты, показанной выше на схеме 1, с помощью HCl с образованием соли соединения (1) с HCl. В одном конкретном варианте осуществления обработку HCl осуществляют в системе растворителя, которая включает воду и один или более органических растворителей, с образованием соли соединения (1) с HCl, где органические растворители независимо выбирают из органических растворителей класса II, выбираемых из группы, состоящей из: хлорбензола, циклогексана, 1,2-дихлорэтена, дихлорметана, 1,2-диметоксиэтана, N,N-диметилацетамида, N,N-диметилформамида, 1,4-диоксана, 2-этоксиэтанола, формамида, гексана, 2-метоксиэтанола, метилбутилкетона, метилциклогексана, N-метилпирролидона, нитрометана, пиридина, сульфолана, тетрагидрофурана (ТГФ), тетралина, толуола, 1,1,2-трихлорэтена и ксилола, или органических растворителей класса III, выбираемых из группы, состоящей из: уксусной кислоты, ацетона, анизола, 1-бутанола, 2-бутанола, бутилацетата, трет-бутилметилового простого эфира, кумола, гептана, изобутилацетата, изопропилацетата, метилацетата, 3-метил-1-бутанола, метилэтилкетона, метилизобутилкетона, 2-метил-1-пропанола, этилацетата, этилового простого эфира, этилформиата, пентана, 1-пентанола, 1-пропанола, 2-пропанола и пропилацетата. В другом конкретном варианте осуществления органические растворители системы растворителя выбирают из группы, состоящей из: хлорбензола, циклогексана, 1,2-дихлорэтана, дихлорметана, 1,2-диметоксиэтана, формамида, гексана, 2-метоксиэтанола, метилбутилкетона, метилциклогексана, нитрометана, тетралина, ксилола, толуола, 1,1,2-трихлорэтана, ацетона, анизола, 1-бутанола, 2-бутанола, бутилацетата, т-бутилметилового простого эфира, кумола, этанола, этилацетата, этилового простого эфира, этилформиата, гептана, изобутилацетата, изопропилацетата, метилацетата, 3-метил-1-бутанола, метилэтилкетона, 2-метил-1-пропанола, пентана, 1-пропанола, 1-пентанола, 2-пропанола, пропилацетата, тетрагидрофурана и метилтетрагидрофурана. В другом конкретном варианте осуществления органические растворители системы растворителя выбирают из группы, состоящей из: 2-этоксиэтанола, этиленгликоля, метанола, 2-метоксиэтанола, 1-бутанола, 2-бутанола, 3-метил-1-бутанола, 2-метил-1-пропанола, этанола, 1-пентанола, 1-пропанола, 2-пропанола, метилбутилкетона, ацетона, метилэтилкетона, метилизобутилкетона, бутилацетата, изобутилацетата, изопропилацетата, метилацетата, этилацетата, пропилацетата, пиридина, толуола и ксилола. В другом конкретном варианте осуществления система растворителя включает воду и ацетон или воду и изопропанол.
[0116] В другом варианте осуществления в способах изобретения дополнительно используют обработку соединения (2) после стадии (b) удаления защиты, показанной выше на схеме 1-A, с помощью HCl с образованием соли соединения (2) с HCl. подходящие системы растворителя, включая конкретные примеры, представляют собой таковые, как описано выше для соединения (1).
[0117] Конкретные иллюстративные условия, подходящие для каждой стадии схем 1-10, которые - каждое и независимо - могут быть использованы в способах изобретения, описаны ниже в разделе с описанием примеров.
[0118] Вышеописанные способы изобретения могут быть использованы для получения конкретных кристаллических форм соединения (1) или его фармацевтически приемлемых солей. Например, соединение (1) может существовать в виде различных полиморфных форм или образовывать различные полиморфные формы. Как известно в уровне техники, полиморфизм представляет собой способность соединения кристаллизоваться в виде более чем одной различающихся кристаллических или “полиморфных” разновидностей. Полиморф представляет твердую кристаллическую фазу соединения с по меньшей мере двумя разными расположениями или полиморфными формами молекулы такого соединения в твердом состоянии. Полиморфные формы любого данного соединения определяются той же самой химической формулой или композицией и различаются по химической структуре в той же степени, как кристаллические структуры двух разных химических соединений. В общем, разные полиморфы могут быть охарактеризованы аналитическими методами, такими как картина рентгеновской порошковой дифракции (XRPD), термогравиметрический анализ (ТГА) и дифференциальная сканирующая калориметрия (ДСК), или температурой плавления полиморфа, или же другой методикой, известной в уровне техники. Использованный здесь термин “полиморфная форма” включает сольваты и чистую полиморфную форму, которая не имеет каких-либо сольватов. Необходимо отметить, что соединение (1) и соли соединения (1) могут быть сольватированными или несольватированными, если иное не указано. Также, необходимо отметить, что соединение Compound (1) и соли соединения (1) могут быть кристаллическими или аморфными, если иное не указано.
[0119] Примером твердых форм соединения (1) или его фармацевтически приемлемых солей является полиморфная форма A соли соединения (1) с HCl на 1/2H2O. Данная форма представляет собой полиморфную форму соли соединения (1) с HCl, которая включает воду в качестве сольвата в количестве половины эквивалента на соединение (1). В одном конкретном варианте осуществления форма A соли соединения (1) с HCl на 1/2H2O характеризуется как имеющая картину XRPD с характеристичными пиками, измеренными в углах 2-тета (градусы), при 10,5±0,2, 5,2±0,2, 7,4±0,2 и 12,8±0,2. В другом конкретном варианте осуществления форма A соли соединения (1) с HCl на 1/2H2O характеризуется как имеющая картину XRPD с характеристичными пиками, измеренными в углах 2-тета (градусы), в следующих положениях, представленных в Таблице 3A описания примеров. Картины XRPD получают при комнатной температуре, используя CuK-альфа излучение. В другом конкретном варианте осуществления полиморфная форма A соли соединения (1) с HCl на 1/2H2O характеризуется как имеющая пики в спектре твердотельного ЯМР 13C (C13 SSNMR) при 29,2, 107,0, 114,0 и 150,7 (±0,3 м.д.). В другом конкретном варианте осуществления форма A соли соединения (1) с HCl на 1/2H2O характеризуется как имеющая C13-SSNMR-пики, представленные в Таблице 3B описания примеров.
[0120] Другим примером твердых форм соединения (1) или его фармацевтически приемлемых солей является полиморфная форма F соли соединения (1) с HCl на 3H2O. Данная форма представляет собой полиморфную форму соли соединения (1) с HCl, которая включает воду в качестве сольвата в количестве трех эквивалентов на соединение (1). В одном конкретном варианте осуществления форма F соли соединения (1) с HCl на 3H2O характеризуется как имеющая картину XRPD с характеристичными пиками, измеренными в углах 2-тета (градусы), при 7,1±0,2, 11,9±0,2 и 12,4±0,2. В другом конкретном варианте осуществления форма F соли соединения (1) с HCl на 3H2O характеризуется как имеющая картину XRPD с характеристичными пиками, выраженными в углах 2-тета (градусы), в следующих положениях, представленных в Таблице 5 описания примеров. Картины XRPD получают при комнатной температуре, используя CuK-альфа излучение. В другом конкретном варианте осуществления полиморфная форма F соли соединения (1) с HCl на 3H2O характеризуется как имеющая пики в спектре твердотельного ЯМР 13C (C13 SSNMR) при 20,7, 27,4, 104,8, 142,5, 178,6 (±0,3 м.д.). В другом конкретном варианте осуществления форма F соли соединения (1) с HCl на 3H2O характеризуется как имеющая C13-SSNMR-пики, представленные в Таблице 6 описания примеров.
[0121] Другим примером твердых форм соединения (1) или его фармацевтически приемлемых солей является полиморфная форма D соли соединения (1) с HCl. Данная форма представляет собой несольватированную форму соли соединения (1) с HCl. В одном конкретном варианте осуществления форма D соли соединения (1) с HCl характеризуется как имеющая картину XRPD с характеристичными пиками, измеренными в углах 2-тета (градусы), при 5,8±0,2, 17,1±0,2 и 19,5±0,2. В другом конкретном варианте осуществления форма D соли соединения (1) с HCl характеризуется как имеющая картину XRPD с характеристичными пиками, измеренными в углах 2-тета (градусы), в положениях, представленных в Таблице 7 описания примеров. Картины XRPD получают при комнатной температуре, используя CuK-альфа излучение. В другом конкретном варианте осуществления форма D соли соединения (1) с HCl характеризуется как имеющая пики в спектре твердотельного ЯМР 13C (C13 SSNMR) при 29,4, 53,4, 113,3, 135,4, 177,8 (±0,3 м.д.). В другом конкретном варианте осуществления форма D соли соединения (1) с HCl характеризуется как имеющая C13-SSNMR-пики, представленные в Таблице 8 описания примеров.
[0122] Другим примером твердых форм соединения (1) или его фармацевтически приемлемых солей является полиморфная форма A соединения (1). Данная форма представляет собой несольватированную форму свободного основания соединения (1). В одном конкретном варианте осуществления форма A соединения (1) характеризуется как имеющая картину XRPD с характеристичными пиками, измеренными в углах 2-тета (градусы), при 15,5±0,2, 18,9±0,2 и 22,0±0,2. В другом конкретном варианте осуществления форма A соединения (1) характеризуется как имеющая картину XRPD с характеристичными пиками, измеренными в углах 2-тета (градусы), в положениях, представленных в Таблице 10 описания примеров. Картины XRPD получают при комнатной температуре, используя CuK-альфа излучение. В другом конкретном варианте осуществления форма A соединения (1) характеризуется как имеющая пики в спектре твердотельного ЯМР 13C (C13 SSNMR) при 21,0, 28,5, 50,4, 120,8, 138,5 и 176,2 (±0,3 м.д.). В другом конкретном варианте осуществления форма A соединения (1) характеризуется как имеющая C13-SSNMR-пики, представленные в Таблице 11 описания примеров.
[0123] Другим примером твердых форм соединения (1) или его фармацевтически приемлемых солей является полиморфная форма A тозилатной соли соединения (1). Данная форма представляет собой несольватированную форму тозилатной соли соединения (1). В одном конкретном варианте осуществления форма A тозилатной соли соединения (1) характеризуется как имеющая картину XRPD с характеристичными пиками, измеренными в углах 2-тета (градусы), в следующих положениях, представленных в Таблице 14 описания примеров. Картины XRPD получают при комнатной температуре, используя CuK-альфа излучение.
[0124] Другим примером твердых форм соединения (1) или его фармацевтически приемлемых солей являются сольваты соединения (1) с 2-Me-ТГФ. В одном конкретном варианте осуществления сольваты включают 0,5-1,5 эквивалента 2-Me-ТГФ на соединение (1), как, например, 1 эквивалент 2-Me-ТГФ на соединение (1). В другом конкретном варианте осуществления сольваты включают 1 эквивалент 2-Me-ТГФ и характеризуются тем, что имеют определенные пики XRPD, представленные в Таблице 12 описания примеров.
[0125] Форма A соли соединения (1) с HCl на 1/2H2O может быть получена, используя смешение (например, перемешивание) хлорида водорода (HCl) с соединением (1). Соединение (1) может быть сольватированным, несольватированным, аморфным или кристаллическим. Раствор, взвесь или суспензия соединения (1) может быть смешана с HCl в системе растворителя, которая включает воду и один или более органических растворителей, где система растворителя имеет активность воды, равную или превышающую 0,05 и равную или меньшую 0,85, то есть 0,05-0,85. Термин “активность воды” (aw) использован здесь в известном в уровне техники смысле и означает меру энергетического состоянии воды в системе растворителя. Ее определяют как давление пара жидкости, поделенное на давление пара чистой воды при той же температуре. Конкретно, ее определяют как 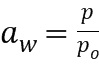 , где
, где  представляет собой давление пара воды в веществе, а
представляет собой давление пара воды в веществе, а  представляет собой давление пара чистой воды при той же температуре, или как
представляет собой давление пара чистой воды при той же температуре, или как 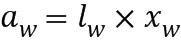 , где
, где 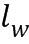 представляет собой коэффициент активности воды, а
представляет собой коэффициент активности воды, а 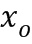 представляет собой мольную долю воды в водной фракции. Например, чистая вода имеет значение активности, равное 1,0. Значения активности воды обычно можно получить с помощью либо емкостного гигрометра, либо гигрометра, измеряющего точку росы. Коммерчески доступны также различные типы приборов для измерения активности воды. Альтернативно, значения активности воды в смеси двух или более растворителей можно рассчитать, основываясь на количествах растворителей и известных значениях активности воды в растворителях.
представляет собой мольную долю воды в водной фракции. Например, чистая вода имеет значение активности, равное 1,0. Значения активности воды обычно можно получить с помощью либо емкостного гигрометра, либо гигрометра, измеряющего точку росы. Коммерчески доступны также различные типы приборов для измерения активности воды. Альтернативно, значения активности воды в смеси двух или более растворителей можно рассчитать, основываясь на количествах растворителей и известных значениях активности воды в растворителях.
[0126] Пример кристаллического соединения (1) включает форму A соединения (1). Примеры сольватов соединения (1) включают сольваты 2-Me-ТГФ, метанола, ксилола, ацетона, 2-бутанола, метилацетата, 1-пентанола, 2-пропанола, тетрагидрофурана, метилтетрагидрофурана, диметилацетамида, N,N-диметилформамида, 1,4-диоксана, 1-пентанола, 2-метил-1-пропанола, метилэтилкетона, 3-метил-1-бутанола, гептана, этилформиата, 1-бутанола, уксусной кислоты и этиленгликоля. В одном конкретном варианте осуществления используют сольваты 2-Me-ТГФ (например, соединение (1)·1(2-Me-ТГФ)).
[0127] Системы растворителя, подходящие для получения формы A соли соединения (1) с HCl на 1/2H2O, могут состоять из разнообразных сочетаний воды и органических растворителей, где активность воды в системах растворителя равна или превышает 0,05 и равна или меньше 0,85 (0,05-0,85). В одном конкретном варианте осуществления значение активности воды составляет 0,4-0,6. Подходящие органические растворители включают органические растворители класса II или класса III, список которых приведен в руководстве Международной конференции по гармонизации (International Conference on Harmonization Guidelines). Конкретные примеры подходящих органических растворителей класса II включают хлорбензол, циклогексан, 1,2-дихлорэтен, дихлорметан, 1,2-диметоксиэтан, N,N-диметилацетамид, N,N-диметилформамид, 1,4-диоксан, 2-этоксиэтанол, формамид, гексан, 2-метоксиэтанол, метилбутилкетон, метилциклогексан, N-метилпирролидон, нитрометан, пиридин, сульфолан, тетрагидрофуран (ТГФ), тетралин, толуол, 1,1,2-трихлорэтен и ксилол. Конкретные примеры подходящих органических растворителей класса III включают : уксусную кислоту, ацетон, анизол, 1-бутанол, 2-бутанол, бутилацетат, трет-бутилметиловый простой эфир, кумол, гептан, изобутилацетат, изопропилацетат, метилацетат, 3-метил-1-бутанол, метилэтилкетон, метилизобутилкетон, 2-метил-1-пропанол, этилацетат, этиловый простой эфир, этилформиат, пентан, 1-пентанол, 1-пропанол, 2-пропанол и пропилацетат. В одном конкретном варианте осуществления органические растворители системы растворителя выбирают из группы, состоящей из хлорбензола, циклогексана, 1,2-дихлорэтана, дихлорметана, 1,2-диметоксиэтана, формамида, гексана, 2-метоксиэтанола, метилбутилкетона, метилциклогексана, нитрометана, тетралина, ксилола, толуола, 1,1,2-трихлорэтана, ацетона, анизола, 1-бутанола, 2-бутанола, бутилацетата, т-бутилметилового простого эфира, кумола, этанола, этилацетата, этилового простого эфира, этилформиата, гептана, изобутилацетата, изопропилацетата, метилацетата, 3-метил-1-бутанола, метилэтилкетона, 2-метил-1-пропанола, пентана, 1-пропанола, 1-пентанола, 2-пропанола, пропилацетата, тетрагидрофурана и метилтетрагидрофурана. В другом конкретном варианте осуществления органические растворители системы растворителя выбирают из группы, состоящей из 2-этоксиэтанола, этиленгликоля, метанола, 2-метоксиэтанола, 1-бутанола, 2-бутанола, 3-метил-1-бутанола, 2-метил-1-пропанола, этанола, 1-пентанола, 1-пропанола, 2-пропанола, метилбутилкетона, ацетона, метилэтилкетона, метилизобутилкетона, бутилацетата, изобутилацетата, изопропилацетата, метилацетата, этилацетата, пропилацетата, пиридина, толуола и ксилола. В другом варианте осуществления органические растворители выбирают из группы, состоящей из ацетона, н-пропанола, изопропанола, изобутилацетата и уксусной кислоты. В другом варианте осуществления органические растворители выбирают из группы, состоящей из ацетона и изопропанола. В другом конкретном варианте осуществления система растворителя включает воду и ацетон. В другом конкретном варианте осуществления система растворителя включает воду и изопропанол.
[0128] Получение формы A соли соединения (1) с HCl на 1/2H2O может быть осуществлено при любой подходящей температуре. Типично получение осуществляют при температуре от 5°C до 75°C. В одном конкретном варианте осуществления получение осуществляют при температуре от 15°C до 75°C. В другом конкретном варианте осуществления получение осуществляют при температуре от 15°C до 60°C. В другом конкретном варианте осуществления получение осуществляют при температуре от 15°C до 35°C. В другом конкретном варианте осуществления получение осуществляют при 5-75°C в системе растворителя, имеющей значение активности воды 0,4-0,6. В другом конкретном варианте осуществления получение осуществляют при температуре от 15°C до 75°C в системе растворителя, имеющей значение активности воды 0,4-0,6. В другом конкретном варианте осуществления получение осуществляют при температуре от 15°C до 60°C в системе растворителя, имеющей значение активности воды 0,4-0,6. В другом конкретном варианте осуществления получение осуществляют при 15-35°C в системе растворителя, имеющей значение активности воды 0,4-0,6.
[0129] Хлорид водорода может быть введен как раствор или газ. Одним примером подходящего источника хлорида водорода является раствор хлорида водорода с содержанием 30-40 массовых процентов (например, 34 масс. % - 38 масс. %) в воде.
[0130] Форма F соли соединения (1) с HCl на 3H2O может быть получена смешением HCl и соединения (1) в системе растворителя, которая включает воду или которая включает воду и один или более органических растворителей, где система растворителя имеет активность воды, равную или превышающую 0,9 (≥0,9). Смесь может представлять собой раствор, взвесь или суспензию. Соединение (1) может быть сольватированным, несольватированным, аморфным или кристаллическим. Альтернативно, она может быть получена перемешиванием формы A соли соединения (1) с HCl на 1/2H2O в системе растворителя, которая включает воду или которая включает воду и один или более органических растворителей, где система растворителя имеет активность воды, равную или превышающую 0,9. Типично, чистая вода имеет значение активности воды, равное 1,0. Соответственно, система растворителя, имеющая активность воды 0,9-1,0, может являться подходящей для получения формы F соли соединения (1) с HCl на 3H2O. В одном конкретном варианте осуществления смешение или перемешивание осуществляют при окружающей температуре (18-25°C). В другом конкретном варианте осуществления смешение или перемешивание осуществляют при температуре от 15°C до 30°C. В другом конкретном варианте осуществления смешение или перемешивание осуществляют при температуре от 20°C до 28°C (например, 25°C). Подходящие органические растворители, включая конкретные примеры, для формирования формы F соли соединения (1) с HCl на 3H2O представляют собой таковые, как описано выше для формы A соли соединения (1) с HCl на 1/2H2O. В другом конкретном варианте осуществления система растворителя включает воду и ацетон. В другом конкретном варианте осуществления система растворителя включает воду и изопропанол.
[0131] Форма D соли соединения (1) с HCl может быть получена дегидратацией формы A соли соединения (1) с HCl на 1/2H2O. Дегидратация может быть выполнена любым подходящим способом, таким как нагрев или продувка сухим азотом или ими обоими.
[0132] Форма A соединения (1) может быть получена (a) перемешиванием смеси аморфного соединения (1) или сольвата соединения (1) (такого как сольват соединения (1) с 2-Me-ТГФ) в системе растворителя, которая включает воду и этанол. Смесь может представлять собой раствор или взвесь. В одном конкретном варианте осуществления стадию перемешивания осуществляют при температуре в диапазоне от 18°C до 90°C. В другом конкретном варианте осуществления стадию (a) перемешивания осуществляют при температуре кипения системы растворителя. В другом конкретном варианте осуществления система растворителя включает воду в количестве 5-15 масс. %. Примеры сольватов соединения (1) представляют собой таковые, описанные выше. В одном конкретном варианте осуществления используют сольваты 2-Me-ТГФ (например, соединение (1)·1(2-Me-ТГФ)).
[0133] В другом варианте осуществления способы получения формы A соединения (1) дополнительно включают: (b) перемешивание аморфной формы соединения (1) в нитрометане с образованием кристаллической затравки формы A соединения (1); и (c) добавление кристаллической затравки формы A соединения (1) к смеси, получаемой в результате стадии (a) смешения. В одном конкретном варианте осуществления способы дополнительно включают : (b) перемешивание аморфной формы соединения (1) в нитрометане с образованием кристаллической затравки формы A соединения (1); (c) охлаждение смеси, полученной на стадии (a) смешения, до температуры в диапазоне от 18°C до 60°C (например, 50-55°C или 55°C); и (d) добавление кристаллической затравки формы A соединения (1) к смеси, полученной на стадии (c). В другом конкретном варианте осуществления способы дополнительно включают перед добавлением кристаллической затравки формы A соединения (1) добавление воды к полученной смеси, которая подверглась стадии кипячения с обратным холодильником, в таком количестве, чтобы полученная система растворителя включала воду в количестве 15-25 масс. % после добавления воды. В другом конкретном варианте осуществления способы дополнительно включают добавление воды к смеси, которая включает кристаллическую затравку формы A соединения (1), в таком количестве, чтобы полученная система растворителя включала воду в количестве 35-45 масс. % после добавления воды. В другом конкретном варианте осуществления способы дополнительно включают охлаждение смеси, которая включает кристаллическую затравку формы A соединения (1), после добавления воды, до температуры от 0°C до 10°C.
[0134] В одном конкретном варианте осуществления кристаллическая затравка формы A соединения (1) может быть получена из сольвата 2-Me-ТГФ соединения (1) в нитрометане. В одном варианте осуществления система растворителя для стадии кипячения с обратным холодильником включает воду в количестве 5-15 масс .%, как, например, 10 масс. %.
[0135] Форма A тозилатной соли соединения (1) может быть получена перемешиванием смеси аморфного соединения (1) или сольвата соединения (1) (такого, как сольват соединения (1) с 2-Me-ТГФ), п-толуолсульфоновой кислоты и системы растворителя, которая включает ацетонитрил. В одном конкретном варианте осуществления стадию смешения или перемешивания осуществляют при окружающей температуре. В другом конкретном варианте осуществления стадию смешения или перемешивания осуществляют при температуре от 15°C до 30°C. В другом конкретном варианте осуществления стадию смешения или перемешивания осуществляют при температуре от 20°C до 30°C (например, 25°C). Подходящие примеры сольватов соединения (1), включая конкретные примеры, представляют собой таковые, как описано выше применительно к получению формы A соединения (1).
[0136] В другом варианте осуществления изобретение охватывает аморфные формы соединения (1) и его фармацевтически приемлемых солей, такие как аморфная соль соединения (1) с HCl и аморфное соединение (1). В другом варианте осуществления изобретение также охватывает форму B гидрата соединения (1). Форма B гидрата соединения (1) изоморфна форме A соединения (1), показывая те же пики XRPD, что и форма A соединения (1), но она образуется в присутствии воды, например, в системе, имеющей активность воды более 0,6, такую как 0,6-1,0, при окружающей температуре.
[0137] Настоящее изобретение охватывает описанные выше полиморфные формы соединения (1) в выделенной в индивидуальном состоянии, чистой форме или в виде смеси как твердой композиции в случае смешения с другими материалами, например, другими формами (то есть с аморфной формой, формой A соединения (1) и так далее) соединения (I), или любыми другими материалами.
[0138] В одном аспекте настоящее изобретение предоставляет полиморфные формы, такие как форма A соли соединения (1)·с HCl на 1/2 H2O, форма F соли соединения (1)·с HCl на 3 H2O, форма D соли соединения (1)·с HCl, форма A соединения (1), форма B гидрата соединения (1) и форма A тозилатной соли соединения (1), в выделенной в индивидуальном состоянии твердой форме. В другом аспекте настоящее изобретение предоставляет аморфную форму соединения (1) и его фармацевтически приемлемых солей, такую как аморфная соль соединения (1) с HCl и аморфное соединение (1), в выделенной в индивидуальном состоянии твердой форме.
[0139] В дополнительном аспекте настоящее изобретение предоставляет полиморфные формы, такие как форма A соли соединения (1)·с HCl на 1/2 H2O, форма F соли соединения (1)·с HCl на 3 H2O, форма D соли соединения (1)·с HCl, форма A соединения (1), форма B гидрата соединения (1) и форма A тозилатной соли соединения (1), в чистой форме. Чистая форма означает, что конкретная полиморфная форма имеет содержание более 95% (масс./масс.), например, более 98% (масс./масс.), более 99% (масс./масс.), более 99,5% (масс./масс.) или более 99,9% (масс./масс.). В другом дополнительном аспекте предоставлены аморфные формы соединения (1) или его фармацевтически приемлемых солей в чистой форме. Чистая форма означает, что аморфная форма имеет содержание более 95% (масс./масс.), например, более 98% (масс./масс.), более 99% (масс./масс.), более 99,5% (масс./масс.) или более 99,9% (масс./масс.).
[0140] Конкретнее, настоящее изобретение предоставляет каждую из упомянутых полиморфных форм в форме композиции или смеси полиморфной формы с одной или более другими кристаллическими, сольватными, аморфными или другими полиморфными формами или их сочетаний. Например, в одном варианте осуществления композиция содержит форму A соли соединения (1) с HCl на 1/2 H2O наряду с одной или более другими полиморфными формами соединения (1), такими как аморфная форма, сольваты, форма D соли соединения (1) с HCl, форма F соли соединения (1) с HCl на 3 H2O, форма A соединения (1) и/или другие формы или их сочетания. Аналогично в другом варианте осуществления композиция содержит форму F соли соединения (1) с HCl на 3 H2O наряду с одной или более другими полиморфными формами соединения (1), такими как аморфная форма, сольваты, форма A соли соединения (1) с HCl на 1/2 H2O, форма D соли соединения (1) с HCl, форма A содинения (1) и/или другие формы или их сочетания. Аналогично в другом варианте осуществления композиция содержит форму D соли соединения (1) с HCl наряду с одной или более другими полиморфными формами соединения (1), такими как аморфная форма, сольваты, форма A соли соединения (1) с HCl на 1/2 H2O, форма F соли соединения (1) с HCl на 3 H2O, форма A соединения (1) и/или другие формы или их сочетания. В другом варианте осуществления композиция содержит форму A соединения (1) наряду с одной или более другими полиморфными формами соединения (1), такими как аморфная форма, гидраты, сольваты и/или другие формы или их сочетания. В другом варианте осуществления композиция содержит форму A тозилатной соли соединения (1) наряду с одной или более другими полиморфными формами соединения (1), такими как аморфная форма, гидраты, сольваты и/или другие формы или их сочетания. Конкретнее, композиция сожжет содержать от следовых количеств вплоть до 100% конкретной полиморфной формы или любое количество между данными крайними значениями, например, 0,1%-0,5%, 0,1%-1%, 0,1%-2%, 0,1%-5%, 0,1%-10%, 0,1%-20%, 0,1%-30%, 0,1%-40% или 0,1%-50% по массе в расчете на суммарное количество соединения (1) в композиции. Альтернативно, композиция может содержать по меньшей мере 50%, 60%, 70%, 80%, 90%, 95%, 97%, 98%, 99%, 99,5% или 99,9% по массе конкретной полиморфной формы в расчете на суммарное количество соединения (1) в композиции.
[0141] Описанные здесь соединения определяются здесь своими химическими структурами и/или химическими наименованиями. Когда соединение описывается как химической структурой, так и химическим наименованием, а химическая структура и химическое наименование не соответствуют друг другу, химическая структура является определяющей в идентификации соединения.
[0142] Специалистам в данной области будет понятно, что в способах настоящего изобретения некоторые функциональные группы, такие как гидроксильные или аминогруппы в исходных реагентах или промежуточных соединениях могут нуждаться в защите защитными группами. Таким образом, получение описанных выше соединений может включать на различных стадиях введение и удаление одной или более защитных групп. Защита функциональных групп и снятие защиты с функциональных групп описаны в монографии “Protective Groups in Organic Chemistry” под редакцией J. W. F. McOmie, Plenum Press (1973) и в монографии “Protective Groups in Organic Synthesis”, 3-е издание, T. W. Greene и P. G. M. Wuts, Wiley Interscience, и в монографии “Protecting Groups”, 3-е издание, P. J. Kocienski, Thieme (2005).
[0143] В контексте данного изобретения химические элементы определены в соответствии с Периодической таблицей элементов, версия CAS, Handbook of Chemistry and Physics, 75-е издание. Кроме того, общие принципы органической химии описаны в “Organic Chemistry”, Thomas Sorrell, University Science Books, Sausolito: 1999, и в “March’s Advanced Organic Chemistry”, 5-е издание, под редакцией Smith, M.B. и March, J., John Wiley & Sons, New York: 2001, полное содержание которых включено в настоящую заявку путем ссылки.
[0144] Термин “алифатический” или “алифатическая группа”, использованный в настоящей заявке, означает прямоцепочечную (то есть неразветвленную) или разветвленную углеводородную цепь, которая является полностью насыщенной или которая содержит один или более фрагментов ненасыщенности, но является неароматической. Если иное не указано, алифатические группы содержат 1-10 алифатических атомов углерода. В некоторых вариантах осуществления алифатические группы содержат 1-6 алифатических атомов углерода, а в других вариантах осуществления алифатические группы содержат 1-4 алифатических атома углерода. Алифатические группы могут быть линейными или разветвленными, замещенными или незамещенными алкильными, алкенильными или алкинильными группами. Конкретные примеры включают следующие, но не ограничены ими: метил, этил, изопропил, н-пропил, втор-бутил, винил, н-бутенил, этинил и трет-бутил и ацетилен.
[0145] Термин “циклоалифатический” (или “карбоцикл” или “карбоциклил”, или “карбоциклический”) относится к неароматической, только углеродсодержащей, кольцевой системе, которая может являться насыщенной или содержит один или более фрагментов ненасыщенности, имея от трех до четырнадцати кольцевых атомов углерода. В некоторых вариантах осуществления число атомов углерода составляет от 3 до 10. В других вариантах осуществления число атомов углерода составляет от 4 до 7. В других вариантах осуществления число атомов углерода составляет 5 или 6. Данный термин включает моноциклические, бициклические или полициклические, конденсированные, спиро- или мостиковые карбоциклические кольцевые системы. Данный термин также включает полициклические кольцевые системы, в которых карбоциклическое кольцо может быть “конденсировано” с одним или более неароматическими карбоциклическими или гетероциклическими кольцами или одним или более ароматическими кольцами или с сочетанием названных, где радикал или точка присоединения находится на карбоциклическом кольце. “Конденсированные” бициклические кольцевые системы содержат два кольца, которые делят между собой два смежных кольцевых атома. Мостиковая бициклическая группа содержит два кольца, которые делят между собой три или четыре смежных кольцевых атома. Бициклические кольцевые спиро-системы делят между собой один кольцевой атом. Примеры циклоалифатических групп включают следующие, но не ограничены ими: циклоалкильные и циклоалкенильные группы. Конкретные примеры включают следующие, но не ограничены ими: циклогексил, циклопропенил и циклобутил.
[0146] Использованный здесь термин “алкил” означает насыщенный углеводород с прямой или разветвленной цепью. В некоторых вариантах осуществления “алкил” представляет собой C1-C6-алкил или C1-C4-алкил. Использованный здесь термин “циклоалкил” означает насыщенный углеводород с циклической цепью. В некоторых вариантах осуществления “циклоалкил” представляет собой C3-C8-алкил или C5-C6-алкил. В контексте настоящей заявки каждый из “алкила” или “циклоалкила” необязательно может являться замещенным, как изложено ниже.
[0147] Подходящие заместители при насыщенном углероде алкильной, алифатической, циклоалкильной или циклоалифатической группы выбирают из группы, состоящей из: галогена; -R°; -OR°; -SR°; 1,2-метилендиокси; 1,2-этилендиокси; фенила (Ph), необязательно замещенного R°; -O(Ph), необязательно замещенного R°; -(CH2)1-2(Ph), необязательно замещенного R°; -CH=CH(Ph), необязательно замещенного -R°; -NO2; -CN; -N(R°)2; -NR°C(O)R°; -NR°C(S)R°; -NR°C(O)N(R°)2; -NR°C(S)N(R°)2; -NR°CO2R°; -NR°NR°C(O)R°; -NR°NR°C(O)N(R°)2; -NR°NR°CO2R°; -C(O)C(O)R°; -C(O)CH2C(O)R°; -CO2R°; -C(O)R°; -C(S)R°; -C(O)N(R°)2; -C(S)N(R°)2; -OC(O)N(R°)2; -OC(O)R°; -C(O)N(OR°)R°; -C(NOR°)R°; -S(O)2R°; -S(O)3R°; -SO2N(R°)2; -S(O)R°; -NR°SO2N(R°)2; -NR°SO2R°; -N(OR°)R°; -C(=NH)-N(R°)2; или -(CH2)0-2NHC(O)R°; где каждый независимый случай R° выбирают из водорода, необязательно замещенного C1-6-алифатического, незамещенного 5-6-членного гетероарильного или гетероциклического кольца, фенила, -O(Ph) или -CH2(Ph), либо два независимых случая R°, располагаясь при одном заместителе или при разных заместителях, совместно с атомом(ами), к которому(ым) присоединена каждая группа R°, образуют 5-8-членное гетероциклическое, карбоциклическое арильное или гетероарильное кольцо или 3-8-членное циклоалкильное кольцо, где указанное гетероарильное или гетероциклическое кольцо имеет 1-3 гетероатома, независимо выбранные из азота, кислорода или серы. Необязательные заместители при алифатической группе R° выбирают из NH2, NH(C1-4-алифатический радикал), N(C1-4-алифатический радикал)2, галогена, C1-4-алифатического радикала, OH, O(C1-4-алифатический радикал), NO2, CN, CO2H, CO2(C1-4-алифатический радикал), O(гало-C1-4-алифатический радикал) или гало-C1-4-алифатического радикала, CHO, N(CO)(C1-4-алифатический радикал), C(O)N(C1-4-алифатический радикал), где каждая из вышеупомянутых C1-4-алифатических групп в R° является незамещенной. Дополнительные заместители включают : =O, =S, =NNHR*, =NN(R*)2, =NNHC(O)R*, =NNHCO2(C1-4-алкил), =NNHSO2(C1-4-алкил) или =NR*, где каждый R* независимо выбирают из водорода или необязательно замещенного C1-6-алифатического радикала. Необязательные заместители при алифатической группе R* выбирают из NH2, NH(C1-4-алифатический радикал), N(C1-4-алифатический радикал)2, галогена, C1-4-алифатического радикала, OH, O(C1-4-алифатический радикал), NO2, CN, CO2H, CO2(C1-4-алифатический радикал), O(гало-C1-4-алифатический радикал) или гало(C1-4- алифатический радикал), где каждая из вышеупомянутых C1-4-алифатических групп в R* является незамещенной.
[0148] Использованный здесь термин “алкокси” относится к алкильной группе, как определено ранее, присоединенной к молекуле через атом кислорода (“алкокси”, например, -O-алкил).
[0149] Использованные здесь термины “галоген”, “гало” и “гал” означают F, Cl, Br или I.
[0150] Термины “галоалкил”, “галоалкенил”, “галоалифатический” и “галоалкокси” означают алкил, алкенил, алифатический радикал или алкокси, соответственно замещенные одним или более атомами галогена. Данные термины включают перфторированные алкильные группы, такие как -CF3 и -CF2CF3.
[0151] В контексте настоящей заявки “цикло”, “циклический”, “циклическая группа” или “циклический фрагмент” включают моно-, би- и трициклические кольцевые системы, включая циклоалифатический радикал, гетероциклоалифатический радикал, карбоциклический арил или гетероарил, каждый из которых был определен выше.
[0152] В контексте настоящей заявки “бициклическая кольцевая система” включает 8-12-членные структуры (например, 9-, 10- или 11-членные), которые образуют два кольца, где два кольца имеют по меньшей мере один общий атом (например, 2 общих атома). Бициклические кольцевые системы включают бициклоалифатические радикалы (например, бициклоалкил или бициклоалкенил), бициклогетероалифатические радикалы, бициклические карбоциклические арилы и бициклические гетероарилы.
[0153] В контексте настоящей заявки “мостиковая бициклическая кольцевая система” относится к бициклической гетероциклоалифатической кольцевой системе или бициклической циклоалифатической кольцевой системе, в которой кольца соединены мостиком. Примеры мостиковых бициклических кольцевых систем включают следующие, но не ограничены ими: адамантанил, ноборнанил, бицикло[3.2.1]октил, бицикло[2.2.2]октил, бицикло[3.3.1]нонил, бицикло[3.2.3]нонил, 2-окса-бицикло[2.2.2]октил, 1-аза-бицикло[2.2.2]октил, 3-аза-бицикло[3.2.1]октил и 2,6-диокса-трицикло[3.3.1.03,7]нонил. Мостиковая бициклическая кольцевая система необязательно может быть замещена одним или более заместителями, такими как алкил (включая карбоксиалкил, гидроксиалкил и галоалкил, такой как трифторметил), алкенил, алкинил, циклоалкил, (циклоалкил)алкил, гетероциклоалкил, (гетероциклоалкил)алкил, карбоциклический арил, гетероарил, алкокси, циклоалкилокси, гетероциклоалкилокси, (карбоциклический арил)окси, гетероарилокси, аралкилокси, гетероаралкилокси, ароил, гетероароил, нитро, карбокси, алкоксикарбонил, алкилкарбонилокси, аминокарбонил, алкилкарбониламино, циклоалкилкарбониламино, (циклоалкилалкил)карбониламино, (карбоциклический арил)карбониламино, аралкилкарбониламино, (гетероциклоалкил)карбониламино, (гетероциклоалкилалкил)карбониламино, гетероарилкарбониламино, гетероаралкилкарбониламино, циано, гало, гидрокси, ацил, меркапто, алкилсульфанил, сульфокси, мочевино, тиомочевино, сульфамоил, сульфамид, оксо или карбамоил.
[0154] В контексте настоящей заявки “мостик” относится к связи или атому или неразветвленной цепи атомов, соединяющим две разные части молекулы. Два атома, которые соединены мостиком (обычно, но не всегда два третичных атома углерода), обозначают как “узлы мостика”.
[0155] Использованный здесь термин “спиро” относится к кольцевым системам, имеющим один атом (обычно четвертичный углерод) в качестве единственного общего атома для двух колец.
[0156] Термин “кольцевой атом” означает атом, такой как C, N, O или S, который находится в кольце ароматической группы, циклоалкильной группы или неароматического гетероциклического кольца.
[0157] “Замещаемый кольцевой атом” в ароматической группе представляет собой кольцевой атом углерода или азота, связанный с атомом водорода. Водород необязательно может быть замещен подходящей замещающей группой. Таким образом, термин “замещаемый кольцевой атом” не включает кольцевые атомы азота или углерода, которые являются общими в случае двух конденсированных колец. Кроме того, “замещаемый кольцевой атом” не включает кольцевые атомы углерода или азота, когда из структуры следует, что они уже присоединены к фрагменту, отличному от водорода.
[0158] Термин “гетероатом” означает один или более из кислорода, серы, азота, фосфора или кремния (включая любую окисленную форму азота, серы, фосфора или кремния; кватернизованную форму любого основного азота, или замещаемый азот гетероциклического кольца, например, N (как в 3,4-дигидро-2H-пирролиле), NH (как в пирролидиниле) или NR+ (как в
N-замещенном пирролидиниле)).
[0159] Набор заместителей и сочетаний заместителей, предусматриваемый данным изобретением, включает таковые, которые приводят к образованию стабильных или химически реализуемых соединений. Использованный здесь термин “стабильный” относится к соединениям, которые по существу не изменяются, когда они подвергаются воздействию условий, обеспечивающих возможность их получения, обнаружения и, в частности, их извлечения, очистки и применения для одной или более раскрытых здесь целей. В некоторых вариантах осуществления стабильное соединение или химически реализуемое соединение представляет собой соединение, которое по существу не изменяется при выдерживании при температуре 40°C или менее в отсутствие влаги или других химически активных условий в течение по меньшей мере недели. Рассматриваются только те варианты выбора и сочетания заместителей, которые приводят к стабильной структуре. Такие варианты выбора и сочетания будут ясны специалистам обычной квалификации в данной области и могут быть выявлены без излишнего экспериментирования.
[0160] Использованные здесь термины “защитная группа” и “защищающая группа” являются взаимозаменяемыми и относятся к агенту, используемому для временного блокирования одной или более желаемых функциональных групп в соединении, имеющем несколько реакционноспособных участков. В некоторых вариантах осуществления защитная группа имеет одно или более, или в особенности все из следующих характеристик: а) селективно присоединяется к функциональной группе с хорошим выходом, давая защищенный субстрат, который b) стабилен по отношению к реакциям, протекающим на одном или более других реакционноспособных центрах; и c) является селективно удаляемой с хорошим выходом посредством реагентов, которые не затрагивают регенерированную, лишенную защиты функциональную группу. Как было бы понятно специалисту в данной области, в некоторых случаях реагенты не затрагивают другие реакционноспособные группы в соединении. В других случаях реагенты также могут реагировать с другими реакционноспособными группами в соединении. Примеры защитных групп подробно описаны в руководстве Greene, T. W., Wuts, P. G, “Protective Groups in Organic Synthesis”, третье издание, John Wiley & Sons, Нью-Йорк: 1999 (и в других изданиях данной книги), полное содержание которого включено в настоящую заявку путем ссылки. Использованный здесь термин “защищающая азот группа” относится к агенту, используемому для временного блокирования одного или более желаемых азотных реакционноспособных центров в многофункциональном соединении. Предпочтительные защищающие азот группы также обладают характеристиками, приведенными выше в качестве примера, и некоторые иллюстративные защищающие азот группы также подробно описаны в главе 7 руководства Greene, T.W., Wuts, P.G. “Protective Groups in Organic Synthesis”, третье издание, John Wiley & Sons, Нью-Йорк: 1999, полное содержание которого включено в настоящую заявку путем ссылки.
[0161] Использованный здесь термин “вытесняемый фрагмент” или “уходящая группа” относится к группе, которая связана с алифатической или ароматической группой, как определено здесь, и подвергается вытеснению при нуклеофильной атаке нуклеофилом.
[0162] Если иное не указано, подразумевается, что изображенные здесь структуры также включают все изомерные (например, энантиомерные, диастереомерные, цис-транс, конформационные и ротационные) формы структуры. Например, R- и S-конфигурации для каждого асимметричного центра, (Z)- и (E)-изомеры по двойной связи и конформационные (Z)- и (E)-изомеры включены в данное изобретение, если только лишь один из изомеров не изображен конкретным образом. Как было бы понятно специалисту в данной области, заместитель может свободно вращаться вокруг любых способных к вращению связей. Например, заместитель, изображенный как
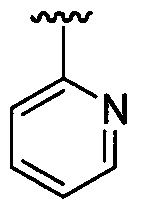 , также представляет
, также представляет
 .
.
[0163] Следовательно, в объем данного изобретения входят единственные стереохимические изомеры, а также энантиомерные, диастереомерные, цис-/транс-, конформационные и ротационные смеси настоящих соединений.
[0164] Если иное не указано, все таутомерные формы соединений изобретения входят в объем изобретения.
[0165] Дополнительно, если иное не указано, также подразумевается, что изображенные здесь структуры включают соединения, которые отличаются только присутствием одного или более изотопно обогащенных атомов. Например, соединения, имеющие настоящие структуры, за исключением замещения водорода дейтерием или тритием или замещения углерода 13C- или 14C-обогащенным углеродом, входят в объем данного изобретения. Такие соединения полезны для использования, например, в качестве аналитических средств или зондов в биологических исследованиях. Такие соединения, в особенности дейтерированные аналоги, также могут быть полезны с терапевтической точки зрения.
[0166] Описанные здесь соединения определяются своими химическими структурами и/или химическими наименованиями. Когда соединение описывается как химической структурой, так и химическим наименованием, а химическая структура и химическое наименование не соответствуют друг другу, химическая структура является определяющей в идентификации соединения.
[0167] Специалистам в данной области будет понятно, что соединения согласно настоящему изобретению могут содержать хиральный центр. Соединения определенной формулы могут, таким образом, существовать в форме двух разных оптических изомеров (то есть (+)- или (-)-энантиомеров). Все такие энантиомеры и их смеси, включая рацемические смеси, включены в объем изобретения. Единственный оптический изомер или энантиомер может быть получен способом, хорошо известным в уровне техники, таким как хиральная ВЭЖХ, ферментативное расщепление и использование хирального вспомогательного реагента.
[0168] В одном варианте осуществления соединения согласно настоящему изобретению предоставлены в форме единственного энантиомера, свободного по меньшей мере на 95%, по меньшей мере на 97% и по меньшей мере на 99% от соответствующего ему энантиомера.
[0169] В дополнительном варианте осуществления соединения согласно настоящему изобретению находятся в форме (+)-энантиомера, свободного по меньшей мере на 95% от соответствующего (-)-энантиомера.
[0170] В дополнительном варианте осуществления соединения согласно настоящему изобретению находятся в форме (+)-энантиомера, свободного по меньшей мере на 97% от соответствующего (-)-энантиомера.
[0171] В дополнительном варианте осуществления соединения согласно настоящему изобретению находятся в форме (+)-энантиомера, свободного по меньшей мере на 99% от соответствующего (-)-энантиомера.
[0172] В дополнительном варианте осуществления соединения согласно настоящему изобретению находятся в форме (-)-энантиомера, свободного по меньшей мере на 95% от соответствующего (+)-энантиомера.
[0173] В дополнительном варианте осуществления соединения согласно настоящему изобретению находятся в форме (-)-энантиомера, свободного по меньшей мере на 97% от соответствующего (+)-энантиомера.
[0174] В дополнительном варианте осуществления соединения согласно настоящему изобретению находятся в форме (-)-энантиомера, свободного по меньшей мере на 99% от соответствующего (+)-энантиомера.
[0175] III. ПРИМЕНЕНИЕ СОЕДИНЕНИЙ
[0176] Раскрытые здесь соединения могут быть использованы для ингибирования репликации вирусов гриппа в биологическом образце или у пациента, для уменьшения количества вирусов гриппа (уменьшения вирусного титра) в биологическом образце или у пациента и для лечения гриппа у пациента. В одном варианте осуществления настоящее изобретение в общем относится к применению раскрытых здесь соединений (например, в фармацевтически приемлемых композициях) для любых из описанных выше вариантов использования.
[0177] В другом варианте осуществления раскрытые здесь соединения могут быть использованы для уменьшения вирусного титра в биологическом образце (например, в инфицированной клеточной культуре) или у людей (например, легочного вирусного титра у пациента).
[0178] Использованные здесь термины “состояние, опосредованное вирусом гриппа”, “обусловленная гриппом инфекция” или “грипп” использованы взаимозаменяемо для обозначения заболевания, вызванного инфекцией вирусом гриппа.
[0179] Грипп представляет собой инфекционное заболевание, которое поражает птиц и млекопитающих, вызываемое вирусами гриппа. Вирусы гриппа представляют собой РНК-вирусы семейства Orthomyxoviridae, которое включает пять родов: вирус Influenza A, вирус Influenza B, вирус Influenza C, вирус ISA и вирус Thogoto (Тогото). Род вируса Influenza A имеет один вид, вирус гриппа A, который может быть подразделен на различные серотипы, основываясь на гуморальном ответе на данные вирусы: H1N1, H2N2, H3N2, H5N1, H7N7, H1N2, H9N2, H7N2, H7N3 и H10N7. Дополнительные примеры вируса гриппа A включают H3N8 и H7N9. Род вируса Influenza B имеет один вид, вирус гриппа B. Грипп B почти исключительно инфицирует людей и является менее распространенным, чем грипп A. Род вируса Influenza C имеет один вид, вирус гриппа C, который инфицирует людей и свиней и может вызывать тяжелое заболевание и локальные эпидемии. Однако вирус гриппа C менее распространен, чем другие типы и, кажется, обычно вызывает легкое заболевание у детей.
[0180] В некоторых вариантах осуществления изобретения грипп или вирусы гриппа относятся к вирусам Influenza A или B. В некоторых вариантах осуществления изобретения грипп или вирусы гриппа относятся к вирусу Influenza A. В некоторых конкретных вариантах осуществления изобретения вирус Influenza A представляет собой H1N1, H2N2, H3N2 или H5N1. В некоторых конкретных вариантах осуществления изобретения вирус Influenza A представляет собой H1N1, H3N2, H3N8, H5N1 и H7N9. В некоторых конкретных вариантах осуществления изобретения вирус Influenza A представляет собой H1N1, H3N2, H3N8 и H5N1.
[0181] У людей обычными симптомами гриппа являются озноб, лихорадка, фарингит, боль в мышцах, сильная головная боль, кашель, слабость и общее недомогание. В более серьезных случаях грипп вызывает пневмонию, которая может быть смертельной, особенно у маленьких детей и пожилых. Хотя его часто путают с простудой, грипп является намного более тяжелым заболеванием и вызывается другим типом вируса. Грипп может вызывать тошноту и рвоту, в особенности у детей, но данные симптомы более характерны для не связанного с ним гастроэнтерита, который иногда называют “желудочным гриппом” или “24-часовым гриппом”.
[0182] Симптомы гриппа могут появляться совершенно неожиданно по прошествии от одних до двух суток после инфицирования. Обычно первыми симптомами являются озноб или ощущение познабливания, но лихорадка также является обычной на ранней стадии инфицирования, причем температуры тела находятся в диапазоне 38-39°C (приблизительно 100-103°F). Многие люди настолько сильно заболевают, что становятся прикованными к постели на несколько суток, испытывая боли и ломоту по всему телу, которые сильнее всего выражены в спине и ногах. Симптомы гриппа могут включать : боли в теле, в особенности в суставах и горле, сильные озноб и лихорадку, слабость, головную боль, раздраженные и слезящиеся глаза, покрасневшие глаза, кожу (в особенности лица), ротовую полость, горло и нос, боль в животе (у детей с гриппом B). Симптомы гриппа являются неспецифичными, перекрываясь со многими патогенами (“гриппоподобное заболевание”). Обычно для того, чтобы подтвердить диагноз, необходимы лабораторные данные.
[0183] Термины “заболевание”, “нарушение” и “состояние” могут быть использованы здесь взаимозаменяемо для указания на медицинское или патологическое состояние, опосредованное вирусом гриппа.
[0184] Использованные здесь термины “субъект” и “пациент” использованы взаимозаменяемо. Термины “субъект” и “пациент” относятся к животному (например, птице, такой как курица, перепел или индейка, или млекопитающему), в частности “млекопитающему” включающему непримата (например, корову, свинью, лошадь, овцу, кролика, морскую свинку, крысу, кошку, собаку и мышь) и примата (например, обезьяну, шимпанзе и человека), и конкретнее - человека. В одном варианте осуществления субъект представляет собой не являющееся человеком животное, такое как сельскохозяйственное животное (например, лошадь, корову, свинью или овцу), или домашнее животное (например, собаку, кошку, морскую свинку или кролика). В предпочтительном варианте осуществления субъект представляет собой “человека”.
[0185] Использованный здесь термин “биологический образец” включает без ограничения клеточные культуры или их экстракты; биопсийный материал, полученный от млекопитающего, или его экстракты; кровь, слюну, мочу, фекалии, сперму, слезы или другие жидкости организма или их экстракты.
[0186] В контексте настоящей заявки “множественность заражения” или “MOI” представляет собой отношение числа of инфекционных агентов (например, фага или вируса) к числу мишеней инфекции (например, клеток). Например, при указании на группу клеток, инокулированных инфекционными вирусными частицами, множественность заражения или MOI представляет собой отношение, определяемое числом инфекционных вирусных частиц, осевших в лунке планшета, поделенное на число клеток-мишеней, присутствующих в этой лунке.
[0187] Использованный здесь термин “ингибирование репликации вирусов гриппа” включает как уменьшение степени репликации вирусов (например, уменьшение по меньшей мере на 10%), так и полный арест репликации вирусов (то есть 100% уменьшение степени репликации вирусов). В некоторых вариантах осуществления репликация вирусов гриппа ингибируется по меньшей мере на 50%, по меньшей мере на 65%, по меньшей мере на 75%, по меньшей мере на 85%, по меньшей мере на 90% или по меньшей мере на 95%.
[0188] Репликация вирусов гриппа может быть измерена любым подходящим способом, известным в уровне техники. Например, может быть измерен титр вирусов гриппа в биологическом образце (например, инфицированной клеточной культуре) или у людей (например, легочный вирусный титр у пациента). Конкретнее, при исследованиях на клетках в каждом случае клетки культивируют in vitro, вирус вносят в культуру в присутствии или отсутствии испытываемого агента и по прошествии подходящего промежутка времени оценивают вирусозависимую конечную точку. В типичных исследованиях можно использовать клетки Мадин-Дарби почек собак (MDCK) и стандартный штамм гриппа A/Puerto Rico/8/34, адаптированный к тканевой культуре. В основе первого типа исследования клеток, который может быть использован в изобретении, лежит гибель инфицированных клеток-мишеней, то есть процесс, называемый цитопатическим эффектом (CPE), где вирусная инфекция вызывает истощение ресурсов клетки и в конечном итоге лизис клетки. В первом типе исследования клеток в лунках микротитрационного планшета инфицируют малую долю клеток (типично от 1/10 до 1/1000), вирусу позволяют пройти через несколько циклов репликации в течение 48-72 часов, затем измеряют величину гибели клеток по уменьшению содержания клеточного АТФ по сравнению с неинфицированными контролями. В основе второго типа исследования клеток, который может быть использован в изобретении, лежит умножение вирусспецифичных молекул РНК в инфицированных клетках, причем уровни РНК непосредственно измеряют, используя метод гибридазиии с ДНК, имеющей разветвленную цепь (bDNA). Во втором типе исследования клеток, малое число клеток первоначально инфицируют в лунках микротитрационного планшета, вирусу позволяют реплицироваться в инфицированных клетках и распространиться на другие клетки, затем клетки подвергают лизису и измеряют содержание вирусной РНК. Данное исследование останавливают рано, обычно спустя 18-36 часов, когда все клктки-мишени еще живы. Вирусную РНК количественно определяют путем гибридизации с особыми олигонуклиотидными зондами, прикрепленными к лункам аналитического планшета, затем амплификации сигнала путем гибридизации с дополнительными зондами, связанными с репортерным ферментом.
[0189] В контексте настоящей заявки “вирусный титр” представляет собой меру концентрации вирусов. При исследовании тира может применяться серийное разведение, чтобы получить апроксимированную количественную информацию из аналитической методики, которая по своей природе дает лишь оценку положительно или отрицательно. Титр соответсвует наивысшему фактору разведения, который еще дает положительный отсчет; например, положительные отсчеты в первых 8 серийных двукратных разведениях дают титр 1:256. Конкретным примером является вирусный титр. Для определения тира будут приготавливаться несколько разведений, таких как 10-1, 10-2, 10-3, 10-4, 10-5, 10-6, 10-7, 10-8. Наименьшая концентрация вируса, которая еще приводит к инфицированию клеток, представляет собой вирусный титр.
[0190] Использованные здесь термины “лечить”, “лечение” и “осуществлять лечение” относятся как к вариантам терапевтического, так и профилактического лечения. Например, варианты терапевтического лечения включают уменьшение или ослабление прогрессирования, тяжести и/или продолжительности состояний, опосредованных вирусами гриппа, или ослабление одного или более симптомов (а именно, одного или более явных симптомов) состояний, опосредованных вирусами гриппа, что является результатом введения одного или более терапевтических средств (например, одного или более терапевтических агентов, таких как соединение или композиция изобретения). В конкретных вариантах осуществления терапевтическое лечение включает ослабление по меньшей мере одного измеряемого физического параметра состояния, опосредованного вирусами гриппа. В других вариантах осуществления терапевтическое лечение включает торможение прогрессирования состояния, опосредованного вирусами гриппа, либо физически, например путем стабилизации явного симптома, физиологически, например путем стабилизации физического параметра, либо обоими путями. В других вариантах осуществления терапевтическое лечение включает уменьшение или стабилизацию инфекций, опосредованных вирусами гриппа. Противовирусные лекарственные средства могут быть использованы в амбулаторных условиях для лечения людей, которые уже болеют гриппом, чтобы уменьшить тяжесть симптомов и уменьшить число дней, когда они больны.
[0191] Термин “химиотерапия” относится к применению лекарственных средств, например лекарственных средств в виде малых молекул (а не “вакцин”) для лечения нарушения или заболевания.
[0192] Использованные здесь термины “профилактика” или “профилактическое применение” и “профилактическое лечение” относятся к любой медицинской или здравоохранительной процедуре, целью которой является предотвращение, а не лечение или вылечивание заболевания. Использованные здесь термины “предотвращать”, “предотвращение” и “осуществление предотвращения” относятся к уменьшению риска приобретения или развития данного состояния или к уменьшению или торможению рецидива указанного состояния у субъекта, который не болен, но который находился или может находиться вблизи индивидуума, имеющего данное заболевание. Термин “химиопрофилактика” относится к применению лекарственных средств, например лекарственных средств в виде малых молекул (а не “вакцин”) для предотвращения нарушения или заболевания.
[0193] В контексте данной заявки профилактическое применение включает применение в ситуациях, в которых была зарегистрирована вспышка, чтобы предотвратить контактную передачу или распространение инфекции в местах, где в близком контакте друг с другом пребывает много людей, которые имеют высокий риск серьезных осложнений гриппа (например, в больничной палате, центр дневной помощи, тюрьма, центр сестринского ухода и так далее). Оно также включает применение среди групп населения, которым необходима защита от гриппа, но которые либо не приобретают защиту после вакцинации (например, из-за слабой иммунной системы), либо когда вакцина им недоступна, либо когда они не могут получить вакцину по причине побочных эффектов. Оно также включает применение на протяжении двух недель после вакцинации, поскольку в течение этого времени вакцина еще неэффективна. Профилактическое применение может также включать осуществление лечения индивидуума, который не болен гриппом или который не рассматривается, как имеющий высокий риск осложнений, чтобы уменьшить вероятность инфицирования гриппом и передачи его индивидууму с таким высоким риском, который находится в близком контакте с ним (например, работники системы здравоохранения, работники центров сестринского ухода и так далее).
[0194] Согласно Центру по контролю за заболеваниями США (US CDC), “вспышка” гриппа определяется как внезапное возрастание случаев острого лихорадочного респираторного заболевания (AFRI), наблюдающееся в пределах периода 48-72 часов в группе людей, которые находятся в непосредственной близости друг к другу (например, в одной и той же зоне учреждения для проживания нуждающихся в помощи людей, в одном и том же домохозяйстве и так далее), относительно нормальной фоновой величины, или когда любой субъект в популяции дает положительный результат при исследовании на грипп. Один случай гриппа, подтвержденного любым методом испытания, рассматривается как вспышка.
[0195] “Кластер” определяется как группа трех или более случаев AFRI, наблюдающихся в пределах периода 48-72 часов в группе людей, которые находятся в непосредственной близости друг к другу (например, в одной и той же зоне учреждения для проживания нуждающихся в помощи людей, в одном и том же домохозяйстве и так далее).
[0196] В контексте настоящей заявки “индекс-случай”, “первичный случай” или “нулевой пациент” является первоначальным пациентом в популяционной выборке эпидемиологического исследования. При использовании в общем смысле для указания на таких пациентов в эпидемиологических исследованиях данный термин не записывают с прописной буквы. Когда данный термин используют для указания конкретного индивидуума вместо имени такого индивидуума в отчете о конкретном исследовании, термин записывают с прописной буквы в форме Нулевой пациент. Часто исследователи осуществляют поиск индекс-случая для того, чтобы определить, как распространяется заболевание и в каких резервуарах сохраняется заболевание между вспышками. Следует отметить, что индекс-случай является первым пациентом, который указывает на существование вспышки. Могут быть обнаружены более ранние случаи и их обозначают как первичный, вторичный, третичный и так далее.
[0197] В одном варианте осуществления способы изобретения представляют собой превентивную или “упреждающую” меру для пациента, в частности человека, имеющего предрасположенность к осложнениям, являющимся результатом инфекции вирусом гриппа. Термин “упреждающий”, использованный здесь, например, в упреждающем применении, “с упреждающей целью” и так далее, относится к профилактическому применению в ситуациях, в которых был подтвержден “индекс-случай” или была подтверждена “вспышка”, чтобы предотвратить распространение инфекции в остальном сообществе или популяционной группе.
[0198] В другом варианте осуществления способы изобретения применяют в качестве “упреждающей” меры для членов сообщества или популяционной группы, в частности людей, чтобы предотвратить распространение инфекции.
[0199] В контексте настоящей заявки “эффективное количество” относится к количеству, достаточному, чтобы добиться желаемого биологического ответа. В настоящем изобретении желаемый биологический ответ заключается в том, чтобы ингибировать репликацию вируса гриппа, уменьшить количество вирусов гриппа или уменьшить или ослабить тяжесть, длительность, прогрессирование или манифестацию обусловленной вирусами гриппа инфекции, предотвратить развитие обусловленной вирусами гриппа инфекции, предотвратить рецидив, развитие, манифестацию или прогрессирование симптомов, связанных с обусловленной вирусами гриппа инфекцией, либо повысить или улучшить профилактический или терапевтический эффект(ы) другой терапии, используемой против гриппозных инфекций. Точное количество соединения, введенное субъекту, будет зависеть от способа введения, типа и тяжести инфекции и характеристик субъекта, таких как общее состояние здоровья, возраст, пол, масса тела и переносимость лекарственных средств. Специалист в данной области сможет определить надлежащие дозировки в зависимости от данных и других факторов. В случае совместного введения с другими противовирусными агентами, например совместного введения с противогриппозным лекарственным средством, “эффективное количество” второго агента будет зависеть от типа используемого лекарственного средства. Для одобренных агентов известны подходящие дозировки, и они могут быть подобраны специалистом в данной области в соответствии с состоянием пациента, типом состояния(ий), подвергаемых лечению, и количеством описанного здесь соединения, которое используют. В случаях, когда количество явно не указано, для эффективного количества следует предложить предположительное значение. Например, раскрытые здесь соединения могут быть введены субъекту в дозировках в диапазоне приблизительно от 0,01 до 100 мг/кг массы тела/сутки для терапевтического или профилактического лечения.
[0200] В общем, режимы дозирования могут быть выбраны в соответствии с разнообразными факторами, включая подвергаемое лечению нарушение и тяжесть нарушения; активность конкретного применяемого соединения; конкретную применяемую композицию; возраст, массу тела, общее состояние здоровья, пол и диету пациента; время введения, путь введения и скорость экскреции конкретного применяемого соединения; почечную и печеночную функцию субъекта; и конкретное применяемое соединение или его соль, продолжительность лечения; лекарственные средства, используемые в сочетании или совместно с конкретным применяемым соединением, и подобные им факторы, хорошо известные в медицинских областях. Специалист в данной области может легко определить и предписать эффективное количество описанных здесь соединений, требуемое для лечения, предотвращения, торможения (полностью или частично) или остановки прогрессирования заболевания.
[0201] Дозировки описанных здесь соединений могут находиться в диапазоне от 0,01 до 100 мг/кг массы тела/сутки, от 0,01 до 50 мг/кг массы тела/сутки, от 0,1 до 50 мг/кг массы тела/сутки или от 1 до 25 мг/кг массы тела/сутки. Понятно, что суммарное суточное количество может быть введено в единственной дозе или может быть введено в нескольких дозировках, таких как дважды в сутки (например, каждые 12 часов), три раза в сутки (например, каждые 8 часов) или четыре раза в сутки (например, каждые 6 часов).
[0202] В некоторых вариантах осуществления дозировки описанных здесь соединений (например, соединения (1) и его фармацевтически приемлемых солей, включая различные твердые формы) находятся в диапазоне от 100 мг до 1600 мг, таком как от 400 мг до 1600 мг или от 400 мг до 1200 мг. Каждая доза может быть принята один раз в сутки (QD), дважды в сутки (например, каждые 12 часов (BID)) или три раза в сутки (например, q8h (TID)). Следует отметить, что по желанию могут быть использованы любые сочетания QD, BID и TID, такие как BID в 1 сутки, после чего следует QD.
[0203] В одном конкретном варианте осуществления дозировки описанных здесь соединений составляют от 400 мг до 1600 мг, от 400 мг до 1200 мг или от 600 мг до 1200 мг один раз в сутки. В другом конкретном варианте осуществления дозировки описанных здесь соединений составляют от 400 мг до 1600 мг, от 400 мг до 1200 мг или от 300 мг до 900 мг дважды в сутки. В другом конкретном варианте осуществления дозировки описанных здесь соединений составляют от 400 мг до to 1000 мг один раз в сутки. В другом конкретном варианте осуществления дозировки описанных здесь соединений составляют от 600 мг до 1000 мг один раз в сутки. В другом конкретном варианте осуществления дозировки описанных здесь соединений составляют от 600 мг до 800 мг один раз в сутки. В другом конкретном варианте осуществления дозировки описанных здесь соединений составляют от 400 мг до 800 мг дважды в сутки (например, от 400 мг до 800 мг каждые 12 часов). В другом конкретном варианте осуществления дозировки описанных здесь соединений составляют от 400 мг до 600 мг дважды в сутки.
[0204] В некоторых вариантах осуществления используют режим введения ударной дозы. В одном конкретном варианте осуществления в 1 сутки лечения используют ударную дозу от 400 мг до 1600 мг. В другом конкретном варианте осуществления в 1 сутки лечения используют ударную дозу от 600 мг до 1600 мг. В другом конкретном варианте осуществления в 1 сутки лечения используют ударную дозу от 800 мг до 1600 мг. В другом конкретном варианте осуществления в 1 сутки лечения используют ударную дозу от 900 мг до 1600 мг. В другом конкретном варианте осуществления в 1 сутки лечения используют ударную дозу от 900 мг до 1200 мг. В другом конкретном варианте осуществления в 1 сутки лечения используют ударную дозу в 900 мг. В другом конкретном варианте осуществления в 1 сутки лечения используют ударную дозу в 1000 мг. В другом конкретном варианте осуществления в 1 сутки лечения используют ударную дозу в 1200 мг.
[0205] В одном конкретном варианте осуществления в режиме дозирования описанных здесь соединений используется ударная дозировка от 600 мг до 1600 мг в 1 сутки при регулярной дозировке от 300 мг до 1200 мг в течение остального срока лечения. Каждая регулярная доза может быть принята один раз в сутки, дважды в сутки или три раза в сутки или в виде любого сочетания указанных приемов. В дополнительном конкретном варианте осуществления используется ударная дозировка от 900 мг до 1600 мг, такая как 900 мг, 1200 мг или 1600 мг. В другом дополнительном конкретном варианте осуществления используется ударная дозировка от 900 мг до 1200 мг, такая как 900 мг или 1200 мг. В другом дополнительном конкретном варианте осуществления в течение остального срока лечения используется регулярная дозировка от 400 мг до 1200 мг, такая как 400 мг, 600 мг или 800 мг. В другом дополнительном конкретном варианте осуществления в течение остального срока лечения используется регулярная дозировка от 400 мг до 1000 мг. В другом дополнительном конкретном варианте осуществления в течение остального срока лечения используется регулярная дозировка от 400 мг до 800 мг. В другом дополнительном конкретном варианте осуществления дважды в сутки используется регулярная дозировка от 300 мг до 900 мг. В другом дополнительном конкретном варианте осуществления один раз в сутки используется регулярная дозировка от 600 мг до 1200 мг. В другом дополнительном конкретном варианте осуществления на 2 сутки используется регулярная дозировка в 600 мг два раза в сутки с последующими 600 мг один раз в сутки в течение остального срока лечения.
[0206] Для проведения терапевтического лечения описанные здесь соединения могут быть введены пациенту в пределах, например, 48 часов (или в пределах 40 часов или менее, чем через 2 дня, или менее, чем через 1,5 дня или в пределах 24 часов) после появления симптомов (например, заложенности носа, боли в горле, кашля, ломоты, слабости, головных болей и озноба/потения). Альтернативно, для проведения терапевтического лечения описанные здесь соединения могут быть введены пациенту в пределах, например, 96 часов после появления симптомов. Терапевтическое лечение может длиться в течение любого подходящего срока, например, в течение 3 суток, 4 суток, 5 суток, 7 суток, 10 суток, 14 суток и так далее. Для проведения профилактического лечения при вспышке в группе компактно проживающего населения описанные здесь соединения могут быть введены пациенту в индекс-случае в пределах, например, 2 суток после появления симптомов, и такое лечение может быть продолжено в течение любого подходящего срока, например, в течение 7 суток, 10 суток, 14 суток, 20 суток, 28 суток, 35 суток, 42 суток и так далее вплоть до продолжительности всего сезона гриппа. Сезон гриппа представляет собой ежегодно повторяющийся период времени, характеризующийся широким распространением вспышек гриппа. Активность гриппа иногда может быть предсказана и даже географически отслежена. Хотя начало основной активности гриппа в каждом сезоне изменяется в зависимости от местоположения, в любом конкретном местоположении данные небольшие эпидемии обычно достигают своего максимума за 3-4 недели и значительно уменьшаются в течение следующих 3-4 недель. Обычно Центр по контролю за заболеваниями (CDC) круглогодично собирает, компилирует и анализирует информацию об активности гриппа в Соединенных Штатах и подготавливает еженедельный отчет с октября по середину мая.
[0207] В одном варианте осуществления терапевтическое лечение длится в течение от 1 дня до продолжительности всего сезона гриппа. В одном конкретном варианте осуществления терапевтическое лечение длится в течение от 3 суток до 14 суток. В другом конкретном варианте осуществления терапевтическое лечение длится в течение от 5 суток до 14 суток. В другом конкретном варианте осуществления терапевтическое лечение длится в течение от 3 суток до 10 суток. В другом конкретном варианте осуществления терапевтическое лечение длится в течение от 4 суток до 10 суток. В другом конкретном варианте осуществления терапевтическое лечение длится в течение от 5 суток до 10 суток. В другом конкретном варианте осуществления терапевтическое лечение длится в течение от 4 суток до 7 суток (например, 4 суток, 5 суток, 6 суток или 7 суток). В другом конкретном варианте осуществления терапевтическое лечение длится в течение от 5 суток до 7 суток (например, 5 суток, 6 суток или 7 суток). В одном конкретном варианте осуществления профилактическое лечение длится вплоть до продолжительности всего сезона гриппа.
[0208] В одном конкретном варианте осуществления описанные здесь соединения вводят пациенту в течение от 3 суток до 14 суток (например, от 5 суток до 14 суток) с ударной дозировкой от 900 мг до 1600 мг в 1 сутки и с регулярной дозировкой от 300 мг до 1200 мг в течение остального срока лечения. В другом конкретном варианте осуществления описанные здесь соединения вводят пациенту в течение от 3 суток до 14 суток (например, от 5 суток до 14 суток) с ударной дозировкой от 900 мг до 1200 мг в 1 сутки и с регулярной дозировкой от 400 мг до 1000 мг в течение остального срока лечения. В другом конкретном варианте осуществления описанные здесь соединения вводят пациенту в течение от 3 суток до 14 суток (например, от 5 суток до 14 суток) с ударной дозировкой от 900 мг до 1200 мг в 1 сутки и с регулярной дозировкой от 400 мг до 800 мг в течение остального срока лечения. В другом конкретном варианте осуществления описанные здесь соединения вводят пациенту в течение от 3 суток до 14 суток (например, от 5 суток до 14 суток) с ударной дозировкой от 900 мг до 1200 мг в 1 сутки и с регулярной дозировкой от 400 мг до 800 мг в течение остального срока лечения. Каждая доза может быть принята один раз в сутки, дважды в сутки или три раза в сутки или в виде любого сочетания указанных приемов.
[0209] В одном конкретном варианте осуществления описанные здесь соединения вводят пациенту в течение от 3 суток до 14 суток с ударной дозировкой от 900 мг до 1600 мг в 1 сутки и с регулярной дозировкой от 600 мг до 1000 мг один раз в сутки в течение остального срока лечения. В другом конкретном варианте осуществления описанные здесь соединения вводят пациенту в течение от 3 суток до 14 суток с ударной дозировкой от 900 мг до 1200 мг в 1 сутки и с регулярной дозировкой от 600 мг до 800 мг (например, 600 мг, 650 мг, 700 мг, 750 мг или 800 мг) один раз в сутки в течение остального срока лечения. В некоторых вариантах осуществления срок лечения составляет от 4 суток до 10 суток, от 5 суток до 10 суток или от 5 суток до 7 суток.
[0210] В одном конкретном варианте осуществления описанные здесь соединения вводят пациенту в течение от 3 суток до 14 суток с ударной дозировкой от 900 мг до 1600 мг в 1 сутки и с регулярной дозировкой от 400 мг до 800 мг дважды в сутки в течение остального срока лечения. В другом конкретном варианте осуществления описанные здесь соединения вводят пациенту в течение от 3 суток до 14 суток с ударной дозировкой от 900 мг до 1200 мг в 1 сутки и с регулярной дозировкой от 400 мг до 600 мг (например, 400 мг, 450 мг, 500 мг, 550 мг или 600 мг) дважды в сутки в течение остального срока лечения. В некоторых вариантах осуществления срок составляет от 4 суток до 10 суток, от 5 суток до 10 суток или от 5 суток до 7 суток.
[0211] В одном конкретном варианте осуществления описанные здесь соединения вводят пациенту в течение 4 суток или 5 суток с ударной дозировкой от 900 мг до 1200 мг (например, 900 мг или 1200 мг) в 1 сутки и с регулярной дозировкой от 400 мг до 600 мг (например, 400 мг или 600 мг) дважды в сутки в течение остального срока лечения (например, со 2 по 4 дни или со 2 по 5 дни). В другом конкретном варианте осуществления описанные здесь соединения вводят пациенту в течение 4 суток или 5 суток с ударной дозировкой от 900 мг до 1200 мг (например, 900 мг или 1200 мг) в 1 сутки и с регулярной дозировкой от 600 мг до 800 мг (например, 600 мг или 800 мг) один раз в сутки в течение остального срока лечения.
[0212] В изобретении можно использовать различные типы способов введения, и они подробно описаны ниже в разделе, озаглавленном “Способы введения”.
[0213] IV. КОМБИНИРОВАННАЯ ТЕРАПИЯ
[0214] Эффективное количество может быть обеспечено в способе или в фармацевтической композиции изобретения, используя только соединение изобретения (включая фармацевтически приемлемую соль или сольват (например, гидрат)) или в сочетании с дополнительным терапевтическим агентом, например, противовирусным агентом или вакциной. Когда используют “комбинированную терапию”, эффективное количество может быть обеспечено, используя первое количество соединения изобретения и второе количество дополнительного подходящего терапевтического агента (например, противовирусного агента или вакцины).
[0215] В другом варианте осуществления данного изобретения соединение изобретения и дополнительный терапевтический агент, каждые, вводят в эффективном количестве (то есть каждые в количестве, которое было бы терапевтически эффективным, если бы они были введены единственными). В другом варианте осуществления соединение изобретения и дополнительный терапевтический агент, каждые, вводят в количестве, которое само по себе не обеспечивает терапевтического эффекта (субтерапевтическая доза). В другом варианте осуществления соединение изобретения может быть введено в эффективном количестве, тогда как дополнительный терапевтический агент вводят в субтерапевтической дозе. В другом варианте осуществления соединение изобретения может быть введено в субтерапевтической дозе, тогда как дополнительный терапевтический агент, например, подходящий агент для терапии рака вводят в эффективном количестве.
[0216] Использованные здесь термины “в сочетании” или “совместное введение” могут быть использованы взаимозаменяемо для указания на использование более чем одного терапевтического средства (например, одного или более профилактических и/или терапевтических агентов). Использование данных терминов не накладывает ограничений на порядок, в котором терапевтические средства (например, профилактические и/или терапевтические агенты) вводят субъекту.
[0217] Совместное введение охватывает введение первого и второго количеств соединений, используемых при совместном введении, по существу одновременно, как, например, в единственной фармацевтической композиции, например, капсуле или таблетке, содержащей фиксированное соотношение первого и второго количеств, или в нескольких, отдельных капсулах или таблетках для каждого количества. Кроме того, такое совместное введение также охватывает использование каждого соединения последовательно в любом порядке.
[0218] В одном варианте осуществления настоящее изобретение относится к способам комбинированной терапии для ингибирования репликации вирусов гриппа в биологических образцах или у пациентов или для лечения или предотвращения инфекций, обусловленных вирусами гриппа, у пациентов, используя описанные здесь соединения. Соответственно, фармацевтические композиции изобретения также включают таковые, содержащие ингибитор репликации вирусов гриппа данного изобретения в сочетании с противовирусным соединением, проявляющим активность против вирусов гриппа.
[0219] Способы применения описанных здесь соединений и композиций изобретения также включают себя сочетание химиотерапии с соединением или композицией изобретения или с сочетанием соединения или композиции данного изобретения с другим противовирусным агентом и вакцинацией противогриппозной вакциной.
[0220] Когда совместное введение включает раздельное введение первого количества соединения изобретения и второго количества дополнительного терапевтического агента, соединения вводят достаточно близко друг к другу по времени, чтобы получить желаемый терапевтический эффект. Например, период времени между каждым введением, который может приводить к желаемому терапевтическому эффекту, может изменяться в диапазоне от минут до часов и может быть определен, принимая во внимание свойства каждого соединения, такие как активность, растворимость, биодоступность, время полувыведения из плазмы и кинетический профиль. Например, соединение изобретения и второй терапевтический агент могут быть введены в любом порядке в течение 24 часов друг за другом, в течение 16 часов друг за другом, в течение 8 часов друг за другом, в течение 4 часов друг за другом, в течение 1 часа друг за другом или в течение 30 минут друг за другом.
[0221] Конкретнее, первое терапевтическое средство (например, профилактический или терапевтический агент, такой как соединение изобретения) может быть введено субъекту перед (например, за 5 минут до, за 15 минут до, за 30 минут до, за 45 минут до, за 1 час до, за 2 часа до, за 4 часа до, за 6 часов до, за 12 часов до, за 24 часа до, за 48 часов до, за 72 часа до, за 96 часов до, за 1 недель до, за 2 недели до, за 3 недели до, за 4 недели до, за 5 недель до, за 6 недель до, за 8 недель до или за 12 недель до), одновременно с или после (например, спустя 5 минут, 15 минут, 30 минут, 45 минут, 1 час, 2 часа, 4 часа, 6 часов, 12 часов, 24 часа, 48 часов, 72 часа, 96 часов, 1 недель, 2 недели, 3 недели, 4 недели, 5 недель, 6 недель, 8 недель или 12 недель) введения второго терапевтического средства (например, профилактического или терапевтического агента, такого как противораковый агент).
[0222] Понятно, что способ совместного введения первого количества соединения изобретения и второго количества дополнительного терапевтического агента может приводить к усиленному или синергетическому терапевтическому эффекту, где объединенный эффект больше, чем аддитивный эффект, который появлялся бы в результате раздельного введения первого количества соединения изобретения и второго количества дополнительного терапевтического агента.
[0223] Использованный здесь термин “синергетический” относится к сочетанию соединения изобретения и другого терапевтического средства (например, профилактического или терапевтического агента), которое более эффективно, чем аддитивные эффекты терапевтических средств. Синергетический эффект сочетания терапевтических средств (например, сочетания профилактических или терапевтических агентов) может допускать использование меньших дозировок одного или более терапевтических средств и/или менее частое введение указанных терапевтических средств субъекту. Возможность использовать меньшие дозировки терапевтического средства (например, профилактического или терапевтического агента) и/или вводить указанное терапевтическое средство менее часто может приводить к снижению токсичности, связанной с введением указанного терапевтического средства субъекту без уменьшения эффективности указанного терапевтического средства в предотвращении, сдерживании или лечении нарушения. Кроме того, синергетический эффект может приводить к повышению эффективности агентов в плане предотвращения, сдерживания или лечения нарушения. Наконец, синергетический эффект сочетания терапевтических средств (например, сочетания профилактических или терапевтических агентов) может устранить или уменьшить вредные или нежелательные побочные эффекты, связанные с использованием каждого терапевтического средства по отдельности.
[0224] Когда комбинированная терапия, использующая соединения настоящего изобретения, сочетается с противогриппозной вакциной, оба терапевтических агента могут быть введены таким образом, что период времени между каждым введением может быть дольше (например, составляя дни, недели или месяцы).
[0225] Наличие синергетического эффекта может быть определено, используя подходящие способы оценки взаимодействия лекарственных средств. Подходящие способы включают, например, сигмоидальное уравнение для расчета максимальной эффективности Emax (Holford, N.H.G. and Scheiner, L.B., Clin. Pharmacokinet. 6: 429-453 (1981)), уравнение аддитивности Loewe (Loewe, S. and Muischnek, H., Arch. Exp. Pathol Pharmacol. 114: 313-326 (1926)) и уравнение медианного эффекта (Chou, T.C. and Talalay, P., Adv. Enzyme Regul. 22: 27-55 (1984)). Каждое вышеупомянутое уравнение может быть применено к экспериментальным данным для построения соответствующей диаграммы, способствующей оценке эффектов сочетания лекарственных средств. Соответствующие диаграммы, относящиеся к вышеупомянутым уравнениям, представляют собой соответственно кривую типа “концентрация-эффект”, кривую изоболограммы и кривую показателя аддитивности.
[0226] Конкретные примеры, которые могут быть совместно введены с описанным здесь соединением, включают ингибиторы нейраминидазы, такие как осельтамивир (Tamiflu®) и занамивир (Rlenza®), блокаторы ионных каналов вируса (белка M2), такие как амантадин (Symmetrel®) и римантадин (Flumadine®), и противовирусные лекарственные средства, описанные в WO 2003/015798, включая средство T-705, разрабатываемое Toyama Chemical, Япония. (Смотри также Ruruta et al., Antiviral Research, 82: 95-102 (2009), “T-705 (flavipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections”). В некоторых вариантах осуществления описанные здесь соединения могут быть совместно введены с традиционной противогриппозной вакциной. В некоторых вариантах осуществления описанные здесь соединения могут быть совместно введены с занамивиром. В некоторых вариантах осуществления описанные здесь соединения могут быть совместно введены с осельтамивиром. В некоторых вариантах осуществления описанные здесь соединения могут быть совместно введены с T-705. В некоторых вариантах осуществления описанные здесь соединения могут быть совместно введены с амантадином или римантадином. Осельтамивир может быть введен в режиме дозировки, указанном в описании к нему. В некоторых конкретных вариантах осуществления его вводят в дозе 75 мг дважды в сутки или 150 мг один раз в сутки.
[0227] V. ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
[0228] Из описанных здесь соединений могут быть составлены фармацевтические композиции, которые дополнительно содержат фармацевтически приемлемые носитель, разбавитель, вспомогательное средство или наполнитель. В одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей соединение изобретения, описанное выше, и фармацевтически приемлемые носитель, разбавитель, вспомогательное средство или наполнитель. В одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей эффективное количество соединения настоящего изобретения или его фармацевтически приемлемую соль и фармацевтически приемлемые носитель, разбавитель, вспомогательное средство или наполнитель. Фармацевтически приемлемые носители включают, например, фармацевтические разбавители, формообразующие или носители, надлежащим образом выбираемые с учетом предполагаемой формы введения и согласующиеся с общепринятыми фармацевтическими практиками.
[0229] “Эффективное количество” включает “терапевтически эффективное количество” и “профилактически эффективное количество”. Термин “терапевтически эффективное количество” относится к количеству, эффективному в лечении и/или облегчении инфекции, обусловленной вирусами гриппа, у пациента, инфицированного гриппом. Термин “профилактически эффективное количество” относится к количеству, эффективному в предотвращении и/или существенном уменьшении вероятности или масштаба вспышки инфекции, обусловленной вирусами гриппа. Конкретные примеры эффективных количеств описаны выше в разделе, озаглавленном “Применение соединений”.
[0230] Фармацевтически приемлемый носитель может содержать инертные ингредиенты, которые не подавляют в негативной степени биологическую активность соединений. Фармацевтически приемлемые носители должны быть биосовместимыми, например, нетоксичными, не вызывающими воспаление, неиммуногенными или лишенными других нежелательных реакций или побочных эффектов при введении субъекту. Могут быть использованы стандартные методики составления фармацевтических рецептур.
[0231] В контексте данной заявки фармацевтически приемлемые носитель, вспомогательное средство или наполнитель включают всякие и любые растворители, разбавители или другой жидкий наполнитель, вспомогательные средства диспергирования или суспендирования, поверхностно-активные агенты, изотонические агенты, загустители или эмульгаторы, консерванты, твердые связующие, смазывающие агенты и тому подобное, подходящие для конкретной желаемой дозировочной формы. В руководстве Remington's Pharmaceutical Sciences, издание шестнадцатое, E. W. Martin (Mack Publishing Co., Easton, Pa., 1980), раскрыты различные носители, используемые в составлении фармацевтически приемлемых композиций, и известные методики их приготовления. За исключением ситуации, когда любая общепринятая среда-носитель несовместима с описанными здесь соединениями, такими как создание любого нежелательного биологического эффекта или иного вредного взаимодействия с любым(и) другим(и) компонентом(ами) фармацевтически приемлемой композиции, применение такой среды-носителя предусматривается входящим в объем данного изобретения. Использованная здесь фраза “побочные эффекты” охватывает нежелательные и вредные эффекты терапевтического средства (например, профилактического или терапевтического агента). Побочные эффекты всегда являются нежелательными, но нежелательные эффекты необязательно являются вредными. Вредный эффект от терапевтического средства (например, профилактического или терапевтического агента) мог бы являться пагубным или связанным с неудобством или опасным. Побочные эффекты включают следующие, но не ограничены ими: лихорадку, озноб, вялость, желудочно-кишечную токсичность (включая желудочные и кишечные изъязвления и эрозии), тошноту, рвоту, нейротоксичность, нефротоксичность, почечную токсичность (включая такие состояния, как папиллярный некроз и хронический интерстициальный нефрит), гепатотоксичность (включая повышенные уровни печеночных ферментов в сыворотке), миелотоксичность (включая лейкопению, миелосупрессию, тромбоцитопению и анемию), сухость во рту, металлический вкус, удлинение беременности, слабость, сонливость, боль (включая боль в мышцах, боль в костях и головную боль), облысение, астению, головокружение, экстрапирамидальные симптомы, акатизию, сердечнососудистые нарушения и сексуальную дисфункцию.
[0232] Некоторые примеры материалов, которые могут служить в качестве фармацевтически приемлемых носителей, включают следующие, но не ограничены ими: ионообменники, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки (такие как человеческий сывороточный альбумин), буферные вещества (такие как твин 80, фосфаты, глицин, сорбиновую кислоту или сорбат калия), смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты (такие как сульфат протамина, двунатриевый гидрофосфат, гидрофосфат калия, хлорид натрия или соли цинка), коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, полиакрилаты, воски, блок-полимеры полиэтилена-полиоксипропилена, метилцеллюлозу, гидроксипропилметилцеллюлозу, ланолин, сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлозу и ее производные, такие как натриевая карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; порошковый трагакант; солод; желатин; тальк; формообразующие, такие как масло какао и воски для суппозиториев; масла, такие как арахисовое масло, хлопковое масло; сафлоровое масло; кунжутное масло; оливковое масло; кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль или полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид алюминия; альгиновую кислоту; апирогенную воду; изотонический солевой раствор; раствор Рингера; этиловый спирт и фосфатные буферные растворы, а также другие нетоксичные совместимые смазывающие агенты, такие как лаурилсульфат натрия и стеарат магния, наряду с которыми по усмотрению составителя в композиции могут также присутствовать красящие агенты, высвобождающие агенты, покрывающие агенты, подсластители, вкусовые агенты и ароматизирующие агенты, консерванты и антиоксиданты.
[0233] VI. СПОСОБЫ ВВЕДЕНИЯ
[0234] Описанные выше соединения и фармацевтически приемлемые композиции могут быть введены людям и другим животным перорально, ректально, парентерально, интрацистернально, интравагинально, интраперитонеально, местно (в порошках, мазях или каплях), букально в виде перорального или назального спрея, либо способами, подобными упомянутым, в зависимости от тяжести подвергаемой лечению инфекции.
[0235] Жидкие дозировочные формы для перорального введения включают следующие, но не ограничены ими: фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным соединениям жидкие дозировочные формы могут содержать инертные разбавители, обычно используемые в данной области, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, из проростков, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана, и смеси указанных разбавителей. Кроме инертных разбавителей пероральные композиции могут также включать вспомогательные средства, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, подсластители, вкусовые агенты и ароматизирующие агенты.
[0236] Рецептуры инъекционных препаратов, например, стерильных инъекционных водных или масляных суспензий, могут быть составлены согласно известному уровню техники, используя подходящие диспергирующие или смачивающие агенты и суспендирующие агенты. Стерильный инъекционный препарат может также представлять собой стерильный инъекционный раствор, суспензию или эмульсию в нетоксичном парентерально приемлемом разбавителе или растворителе, например, раствор в 1,3-бутандиоле. Среди приемлемых наполнителей и растворителей, которые могут быть использованы, можно упомянуть воду, раствор Рингера, соответствующий Фармакопеи США и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды традиционно используют стерильные, нелетучие масла. Для этой цели можно использовать любое нераздражающее нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, при приготовлении инъекционных форм используют жирные кислоты, такие как олеиновая кислота.
[0237] Инъекционные рецептуры могут быть стерилизованы, например, путем фильтрации через удерживающий бактерии фильтр, или, путем включения стерилизующих агентов, в форме стерильных твердых композиций, которые могут быть растворены или диспергированы в стерильной воде или другой стерильной инъекционной среде перед использованием.
[0238] Чтобы пролонгировать эффект описанного здесь соединения, часто желательно замедлять абсорбцию соединения, введенного подкожной или внутримышечной инъекцией. Это может быть достигнуто путем использования жидкой суспензии кристаллического или аморфного материала, обладающего плохой растворимостью в воде. Тогда скорость абсорбции соединения зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. Альтернативно, замедленная абсорбция парентерально введенной формы соединения достигается путем растворения или суспендирования соединения в масляном наполнителе. Инъекционные депо-формы готовят путем формирования микроинкапсулированных матриц соединения в биоразлагаемых полимерах, таких как полилактид-полигликолид. В зависимости от соотношения количеств соединения и полимера и природы конкретного используемого полимера сожно контролировать скорость высвобождения соединения. Примеры других биоразлагаемых полимеров включают поли(ортоэфиры) и поли(ангидриды). Инъекционные депо-рецептуры также готовят путем включения соединения в липосомы или микроэмульсии, которые совместимы с тканями организма.
[0239] Конкретные примеры композиций для ректального или вагинального введения представляют собой суппозитории, которые могут быть приготовлены путем смешения описанных здесь соединений с подходящими нераздражающими формообразующими или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при температуре окружающей среды, но жидкими при температуре тела, и, следовательно, расплавляются в прямой кишке или вагинальной полости и высвобождают активное соединение.
[0240] Твердые дозировочные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых дозировочных формах активное соединение смешивают по меньшей мере с одним инертным, фармацевтически приемлемым формообразующим или носителем, таким как цитрат натрия или фосфат дикальция, и/или a) наполнителями или сухими разбавителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и салициловая кислота, b) связующими, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и гуммиарабик, c) увлажнителями, такими как глицерин, d) разрыхлителями, такими как агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты и карбонат натрия, e) замедлителями растворения, такими как парафин, f) ускорителями абсорбции, такими как четвертичные аммониевые соединения, g) смачивающими агентами, такими как, например, цетиловый спирт и моностеарат глицерина, h) абсорбентами, такими как каолиновая и бентонитовая глина, и i) смазывающими агентами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае капсул, таблеток и пилюль дозировочная форма может также содержать буферные агенты.
[0241] Твердые композиции подобного типа могут быть также использованы в качестве наполнителей в мягких и твердых желатиновых капсулах, используя такие формообразующие, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и тому подобное. Твердые дозировочные формы таблеток, драже, капсул, пилюль и гранул могут быть приготовлены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные в области изготовления фармацевтических композиций. Они могут необязательно содержать опалесцирующие агенты и могут быть также образованы композицией, из которой высвобождается(ются) активный(е) ингредиент(ы) только или предпочтительно в определенной части кишечника необязательно в замедленном режиме. Примеры внедряющих композиций, которые могут быть использованы, включают полимерные вещества и воски. Твердые композиции подобного типа могут быть также использованы в качестве наполнителей в мягких и твердых желатиновых капсулах, используя такие формообразующие, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и тому подобное.
[0242] Активные соединения могут также находиться в микроинкапсулированной форме с одним или более формообразующими, как отмечено выше. Твердые дозировочные формы таблеток, драже, капсул, пилюль и гранул могут быть приготовлены с покрытиями и оболочками, такими как энтеросолюбильные покрытия, покрытия для контролируемого высвобождения и другие покрытия, хорошо известные в области изготовления фармацевтических композиций. В таких твердых дозировочных формах активное соединение может быть смешано по меньшей с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие дозировочные формы могут также содержать, как это обычно принято на практике, дополнительные вещества, отличные от инертных разбавителей, например, смазывающие агенты для изготовления таблеток и другие вспомогательные средства таблетирования, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль дозировочные формы могут также содержать буферные агенты. Они могут необязательно содержать опалесцирующие агенты и могут быть также образованы композицией, из которой высвобождается(ются) активный(е) ингредиент(ы) только или предпочтительно в определенной части кишечника необязательно в замедленном режиме. Примеры внедряющих композиций, которые могут быть использованы, включают полимерные вещества и воски.
[0243] Дозировочные формы для местного или трансдермального введения описанного здесь соединения включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, лекарственные формы для ингаляции или пластыри. Активный компонент смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами или буферами, если это требуется. Подразумевается, что глазная рецептура, ушные капли и глазные капли также входят в объем данного изобретения. Кроме того, настоящее изобретение охватывает использование трансдермальных пластырей, которые обладают дополнительным преимуществом, обеспечивая контроируемую доставку соединения в организм. Такие дозировочные формы могут быть изготовлены растворением или диспергированием соединения в подходящей среде. Чтобы увеличить продвижение соединения через кожу, также могут быть использованы усилители абсорбции. Скорость можно контролировать либо предусматривая мембрану, контролирующую скорость высвобождения, либо диспергируя соединение в полимерной матрице или геле.
[0244] Описанные здесь композиции могут быть введены перорально, парентерально, спреем для ингаляций, местно, ректально, назально, букально, вагинально или посредством имплантированного резервуара. Использованный здесь термин “парентерально” включает, но не ограничен ими: подкожную, внутривенную, внутримышечную, внутрисуставную, интрасиновиальную, внутригрудинную, интратекальную, внутрипеченочную, внутриочаговую и интракраниальную методики инъекции или инфузии. А именно, композиции вводят перорально, интраперитонеально или внутривенно.
[0245] Стерильные инъекционные формы описанных здесь композиций могут представлять собой водную или масляную суспензию. Рецептуры данных суспензий могут быть составлены согласно методикам, известным в данной области, используя подходящие диспергирующие или смачивающие агенты и суспендирующие агенты. Стерильный инъекционный препарат также может представлять собой стерильный инъекционный раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, например, раствор в 1,3-бутандиоле. Среди приемлемых наполнителей и растворителей, которые могут быть использованы, можно упомянуть воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды традиционно используют стерильные, нелетучие масла. Для этой цели можно использовать любое нераздражающее нелетучее масло, включая синтетические моно- или диглицериды. При приготовлении инъекционных форм используют жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, так как представляют собой природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, в особенности в их полиоксиэтилированных вариантах. Такие масляные растворы или суспензии могут также содержать разбавитель или диспергирующий агент в виде спирта с длинной цепью, такой как карбоксиметилцеллюлоза или похожие диспергирующие агенты, которые обычно используются при составлении рецептур фармацевтически приемлемых дозировочных форм, включая эмульсии и суспензии. В целях составления рецептур могут быть также использованы другие обычно применяемые поверхностно-активные вещества, такие как твины, спаны и другие эмульгирующие агенты или усилители биодоступности, которые обычно используются при производстве фармацевтически приемлемых твердых, жидких или других дозировочных форм.
[0246] Описанные здесь фармацевтические композиции могут быть введены перорально в любой перорально приемлемой дозировочной форме, включая следующие, но не ограниченные ими: капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения обычно используемые носители включают следующие, но не ограничены ими: лактозу и кукурузный крахмал. Типично добавляют также смазывающие агенты, такие как стеарат магния. Для перорального применения в форме капсул подходящие для использования разбавители включают лактозу и высушенный кукурузный крахмал. Кода для перорального применения требуются водные суспензии, активный ингредиент объединяют с эмульгирующими и суспендирующими агентами. Если того желают, также могут быть добавлены некоторые подсластители, вкусовые или красящие агенты.
[0247] Альтернативно, описанные здесь фармацевтические композиции могут быть введены в форме суппозиториев для ректального введения. Они могут быть приготовлены путем смешения агента с подходящим нераздражающим формообразующим, которое является твердым при комнатной температуре, но жидким при ректальной температуре, и, следовательно, будет расплавляться в прямой кишке, высвобождая лекарственное средство. Такие материалы включают следующие, но не ограничены ими: масло какао, пчелиный воск и полиэтиленгликоли.
[0248] Описанные здесь фармацевтические композиции могут быть также введены местно, в особенности когда цель лечения включает области или органы, легко доступные для местного нанесения, включая заболевания глаз, кожи или нижних отделов желудочно-кишечного тракта. Подходящие рецептуры для местного применения легко получаются для каждых из данных областей или каждого из данных органов.
[0249] Местное нанесение в случае нижних отделов желудочно-кишечного тракта может быть осуществлено рецептурой ректального суппозитория (смотри выше) или подходящей рецептурой для введения клизмой. Также можно использовать местные трансдермальные пластыри.
[0250] Для местного нанесения рецептуры фармацевтических композиций могут быть составлены в виде подходящей мази, содержащей активный компонент, суспендированный или растворенный в одном или более носителях. Носители для местного нанесения соединений данного изобретения включают следующие, но не ограничены ими: минеральное масло, жидкий вазелин, белый вазелин, пропиленгликоль, полиоксиэтиленовое, полиоксипропиленовое соединение, эмульгирующийся воск и воду. Альтернативно, рецептуры фармацевтических композиций могут быть составлены в виде подходящего лосьона или крема, содержащего активные компоненты, суспендированные или растворенные в одном или более фармацевтически приемлемых носителях. Подходящие носители включают следующие, но не ограничены ими: минеральное масло, моностеарат сорбитана, полисорбат 60, цетиловые сложноэфирные воски, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду.
[0251] Для глазного применения рецептуры фармацевтических композиций могут быть составлены в виде микросуспензий в изотоническом, стерильном солевом растворе с отрегулированным значением pH или, конкретнее, в виде раствором в изотоническом, стерильном солевом растворе с отрегулированным значением pH, содержащем консервант, такой как хлорид бензилалкония, либо без него. Альтернативно, для глазного применения рецептуры фармацевтических композиций могут быть составлены в виде мази, такой как мазь на основе вазелина.
[0252] Фармацевтические композиции могут быть также введены назальным аэрозолем или ингаляцией. Такие композиции получают согласно методикам, хорошо известным в области составления фармацевтических композиций, и они могут быть получены в виде растворов в солевом растворе, используя бензиловый спирт или другие подходящие консерванты, промоторы абсорбции для повышения биодоступности, фторуглероды и/или другие общепринятые солюбилизирующие или диспергирующие агенты.
[0253] На основе соединений может быть составлена единичная дозировочная форма. Термин “единичная дозировочная форма” относится к физически дискретным единицам, подходящим в качестве единичной дозировки для субъектов, которым проводится лечение, причем каждая единица содержит заданное количество активного материала, рассчитанное на то, чтобы производить желаемый терапевтический эффект, необязательно в сочетании с подходящим фармацевтическим носителем. Единичная дозировочная форма может быть предназначена для введения единственной дневной дозы или одной или нескольких дневных доз (например, от 1 до 4 или более раз в сутки). Когда используют несколько дневных доз, единичная дозировочная форма может быть одинаковой или разной для каждой дозы.
[0254] VII. ПРИМЕРЫ
[0255] Пример 1: Получение 2-амино-3-бром-5-фторпиридина (соединение 2a)
[0256] Методика A: К взвеси 2-амино-5-фторпиридина (6 кг, 53,6 моль) в воде (24 л) при 14°C прибавляли в течение 10 минут 48% бромистоводородной кислоты (18,5 кг, 110 моль). Реакция была экзотермической и температура поднималась до 24°C. Смесь повторно охлаждали до 12°C, затем прибавляли бром (9 кг, 56,3 моль) девятью порциями в течение 50 минут (экзотермическая реакция, температуру поддерживали при 20°C). Смесь перемешивали при 22°C в течение ночи и отслеживали с помощью 1H-ЯМР погашенной аликвоты (гасили, прибавляя 5 капель в смесь 1 мл 20% K2CO3, 0,3 мл 10% Na2S2O3 и 0,7 мл DCM. Органический слой упаривали и исследовали). Смесь охлаждали до 10°C, затем гасили путем прибавления бисульфита натрия (560 г, 5,4 моль) в доле (2 л) и дополнительно охлаждали до 0°C. Данную смесь прибавляли к охлажденной (-4°C) смеси DCM (18 л) и 5,4 M гидроксида натрия (35 л, 189 моль). Нижние ~35 л фильтровали через слой целлита, а затем осуществляли разделение фаз. Водный слой повторно экстрагировали DCM (10 л). Органические компоненты фильтровали через слой из 3 кг магнезола, промывая его DCM (8 л). Фильтрат упаривали, затирали с гексаном и отфильтровывали.
[0257] Несмотря на то, что проводимое в ходе реакции исследование, указывало на 97% завершение процесса, данный исходный продукт из всех четырех загрузок обычно содержал ~10% исходного материала (SM). Данные продукты объединяли и затирали в гексане (2 л на кг материала) при 50°C, затем охлаждали до 15°C и фильтровали, что давало соединение 2a (30,0 кг, чистота ~95%, 149 моль, 67%). Маточные растворы от первоначального затирания и повторной очистки хроматографировали (20 кг силикагеля, элюент 25-50% EtOAc в гексане), что давало дополнительную порцию соединения 2a (4,7 кг, чистота ~99%, 24,4 моль, 11%).
[0258] Методика B: В альтернативном варианте бромирование осуществляли, используя HOAc вместо HBr. В одном конкретном примере аминопиридин (952 г, 8,49 моль) растворяли в HOAc (7 л) и обрабатывали NaOAc (1,04 кг, 12,7 моль) с последующим прибавлением по каплям Br2 (используя охлаждаемую льдом капельную воронку для охлаждения реакции). После прибавления Br2 реакции позволяли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь выливали в воду и создавали основную среду, прибавляя 6 н. NaOH. Реакционную смесь экстрагировали EtOAc. Значительное количество твердого вещества не растворялось в органической или водной фазе. Смесь целиком фильтровали и фазы разделялись. Органический слой сушили (MgSO4), фильтровали через слой SiO2, элюируя с помощью EtOAc. Фильтрат упаривали, что давало коричневое твердое вещество, 889 г.
[0259] Методика C: В альтернативном варианте бромирование осуществляли, используя H2SO4. В одном конкретном примере к 93% серной кислоте (12,5 кг, 119 моль) в воде (26 л) в реакторе на 50 л прибавляли 2-амино-5-фторпиридин (6,5 кг, 58 моль). Температуру регулировали до 30°C, затем десятью порциями в течение трех часов прибавляли бром (10 кг, 63 моль). Смесь перемешивали при 45°C в течение 18 часов, затем при 50°C в течение 5 часов. Смесь охлаждали до 15°C для обработки в реакторе на 400 л.
[0260] Четыре вышеописанных реакции (4×6,5 кг) объединяли и гасили в смеси 50% гидроксида натрия (110 кг, 1375 моль) и тиосульфата натрия (1,8 кг, 11,4 моль) в воде (100 л) при -3°C в течение одного часа. Температуру регулировали до 32°C, и взвесь фильтровали и промывали водой (80 л), что давало влажный от воды сырой продукт (62 кг). Вторую серию из трех реакций (3×6,5 кг SM) проводили похожим образом, что давало влажный от воды сырой продукт (41 кг). Сырые продукты (103 кг) растворяли (некоторое количество нерастворимых веществ) в толуоле (280 кг) при 25-30°C. Прибавляли рассол (20 кг), но разделение фаз было невозможно из-за наличия твердых веществ. Смесь фильтровали через слой целита, промывая толуолом, и тогда слои разделялись. Органические материалы концентрировали до объема 347 л для азеотропной отгонки остаточной воды в целях использования для получения соединения 3a. Аликвоту использовали для определения концентрации продукта, находя, что она составляла 181 г на литр раствора. Выход равен 62,8 кг. Дополнительные 600 г выделяли экстракцией водного/рассольного слоя этилацетатом (10 л) и последующим фильтрованием через слой магнезола, упариванием и затиранием с гексаном. Суммарный выход составляет 82%.
[0261] Получение соединения 3a
[0262] Методика A: В инертный реактор на 400 л загружали 2a (27,5 кг, чистота 96%, 138 моль), Pd(PPh3)4 (1044 г, 0,90 моль) и CuI (165 г, 0,87 моль), после чего прибавляли толуол (90 кг). Смесь освобождали от кислорода с помощью трех циклов откачивания вакуумом-заполнения азотом, затем прибавляли триэтиламин (19,0 кг, 188 моль). Реакционную смесь освобождали от кислорода с помощью еще одного цикла откачивания вакуумом-заполнения азотом, затем прибавляли TMS-ацетилен (16,5 кг, 168 моль). Смесь нагревали до 48°C в течение 23 часов (начальная экзотермия повышала температуру максимум до 53°C), затем охлаждали до 18°C. Взвесь фильтровали через слой целлита и промывали его толуолом (80 кг). Фильтрат промывали 12% Na2HPO4 (75 л), затем фильтровали через слой силикагеля (25 кг), промывая смесью 1:1 гексан:MTBE (120 л). Данный фильтрат упаривали с получением коричневого масла, а затем его растворяли NMP для проведения следующей стадии. Масса раствора соединения 3a составляет 58 кг, ~50 масс. %, 138 моль, 100%. 1H-ЯМР (CDCl3, 300 МГц): δ 7,90 (с, 1H); 7,33-7,27 (м, 1H); 4,92 (с, NH2), 0,28 (с, 9H) м.д.
[0263] Методика B: 2-Амино-3-бром-5-фторпиридин ( 2a : 10,7 г, 56 ммоль) обрабатывали CuI (1,72 г, 9,03 ммоль), Pd (dppf)Cl2 (2,87 г, 3,92 ммоль), TMS-ацетиленом (8,25 г, 11,8 мл, 84 ммоль), ТГФ (200 мл) и Et3N (190 мл) и нагревали, кипятя с обратным холодильником в течение ночи. О завершении реакции судили по результатам ТСХ и выливали реакционную смесь в воду (200 мл). Фазы разделялись, и фазы экстрагировали EtOAc (3 × 200 мл). Органические фазы объединяли и сушили (MgSO4), фильтровали, а фильтрат концентрировали в вакууме, что давало масло, которое затвердевало в вакууме. Твердое вещество растворяли в CH2Cl2 и пропускали раствор через слой SiO2, элюируя с помощью CH2Cl2, что давало желтое твердое вещество, 11,7 г, выход 93%.
[0264] Получение соединения 4a
[0265] В инертный реактор на 400 л загружали т-бутоксид калия (17,5 кг, 156 моль) и NMP (45 кг). Смесь нагревали до 54°C, затем в течение 2,75 часов прибавляли раствор соединения 3a (29 кг, 138 моль) в NMP (38 кг) и смывали его NMP (6 кг) (экзотермическая реакция, температуру поддерживали при 70-77°C). Реакционную смесь перемешивали при 74°C в течение 2 часов, затем охлаждали до 30°C и в течение 1,5 часа прибавляли раствор тозилхлорида (28,5 кг, 150 моль) в NMP (30 кг) и смывали его NMP (4 кг). Реакция была экзотермической и ее температуру поддерживали при 30-43°C. Реакционную смесь перемешивали в течение 1 часа, охлаждая до 20°C, затем в течение 35 минут прибавляли воду (220 л) (экзотермическая реакция, температуру поддерживали при 18-23°C). Смесь перемешивали при 20°C в течение 30 минут, затем фильтровали и промывали водой (100 л). Твердые вещества растворяли на фильтре в DCM (250 кг), проводили отделение от остаточной воды и органический материал фильтровали через слой магнезола (15 кг, верх) и силикагеля (15 кг, низ), смывая дополнительным количеством DCM (280 кг). Фильтрат концентрировали до вязкой взвеси (объемом ~50 л), затем прибавляли MTBE (30 кг), продолжая отгонку, поддерживая постоянный объем (конечная температура дистиллята составляла 51°C). Прибавляли дополнительное количество MTBE (10 кг) и охлаждали взвесь до 15°C, фильтровали и промывали с помощью MTBE (40 л), что давало соединение 4a (19,13 кг, чистота 95%, 62,6 моль, 45%). Частичное концентрирование фильтрата давала вторую порцию продукта (2,55 кг, чистота 91%, 8,0 моль, 6%). 1H-ЯМР (CDCl3, 300 МГц): δ 8,28-8,27 (м, 1H); 8,06-8,02 (м, 2H); 7,77 (д, J = 4,0 Гц, 1H); 7,54-7,50 (м, 1H); 7,28-7,26 (м, 2H); 6,56 (д, J = 4,0 Гц, 1H); 2,37 (с, 3H) м.д.
[0266] Получение соединения 5a
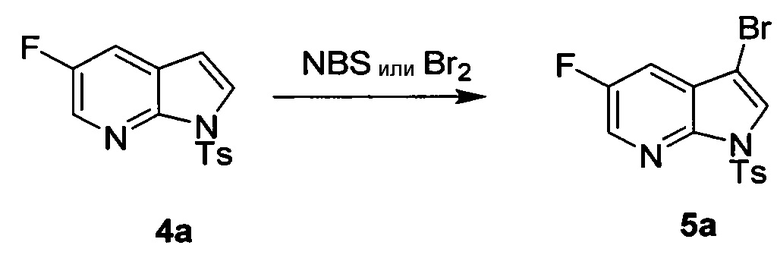
[0267] Методика A: К взвеси N-бромсукцинимида (14,16 кг, 79,6 моль) в DCM (30 кг) при 15°C добавляли раствор соединения 4a (19,13 кг, чистота 95%, и 2,86 кг, чистота 91%, 71,6 моль) в DCM (115 кг), смывая его с помощью DCM (20 кг). Смесь перемешивали при 25°C в течение 18 часов, а затем охлаждали до 9°C и гасили добавлением раствора тиосульфата натрия (400 г) и 50% гидроксида натрия (9,1 кг) в воде (130 л). Смесь нагревали до 20°C и разделяли слои, и органические материалы промывали 12% рассолом (40 л). Водные слои последовательно повторно экстрагировали DCM (4 × 50 кг). Органические материалы объединяли и отгоняли 40 л, чтобы азеотропно удалить воду, затем раствор фильтровали через слой силикагеля (15 кг, низ) и магнезола (15 кг, верх), смывая с помощью DCM (180 кг). Фильтрат концентрировали в густую взвесь (объемом ~32 л), затем добавляли гексан (15 кг). Прибавляли дополнительное количество гексана (15 кг), продолжая отгонку, поддерживая постоянный объем (конечная температура дистиллята составляла 52°C). Взвесь охлаждали до 16°C, фильтровали и промывали гексаном (25 кг), что давало соединение 5a (25,6 кг, 69,3 моль, 97%). 1H-ЯМР (CDCl3, 300 МГц): δ 8,34-8,33 (м, 1H); 8,07 (д J = 8,2 Гц, 2H); 7,85 (с, 1H); 7,52-7,49 (м, 1H); 7,32-7,28 (м, 2H); 2,40 (с, 3H) м.д.
[0268] Методика B: Раствор Br2 (115 мл, 1,15 экв.) в CH2Cl2 (1 л) прибавляли по каплям к раствору соединения 4a (566 г, 1,95 моль) в CH2Cl2 (4 л) в течение 90 минут. В ходе прибавления температура повышалась от 16 до 23°C и реакционную смесь охлаждали с помощью бани изо льда и соли до 10°C. После завершения прибавления температура достигала 12°C. Суспензию (в ходе прибавления образовывалось оранжевое твердое вещество) перемешивали в течение 30 минут. Реакционную смесь перемешивали при комнатной температуре (RT) в течение ночи. Осторожно в течение 5-10 минут прибавляли насыщенный водный раствор NaHCO3 (4 л). Реакционную смесь интенсивно перемешивали в течение 1 часа и позволяли разделиться слоям. Полученный раствор фильтровали через фильтр. Органический слой промывали насыщенным водным раствором NaHCO3 (2 л) и рассолом (2 × 1 л), сушили над Na2SO4 и пропускали через силикагель (2 кг), элюируя с помощью CH2Cl2 (всего ~10 л). Растворители (~20 л) удаляли при пониженном давлении, что давало соединение 5a (580 г) в виде белого твердого вещества. Продукт повторно растворяли в CH2Cl2 (2,5 л) и фильтровали через другой фильтр, имеющий силикагель (2 кг), элюируя с помощью CH2Cl2. После удаления растворителей при пониженном давлении соединение 5a (568 г, выход 79%) получали в виде беловатого твердого вещества. После тестовой реакции для следующей стадии оставшийся материал промывали смесью гептанов (2×) и сушили, что давало лучшие результаты на следующей стадии. 1H-ЯМР (CDCl3, 300 МГц): δ 8,34-8,33 (м, 1H); 8,07 (д, J = 8,2 Гц, 2H); 7,85 (с, 1H); 7,52-7,49 (м, 1H); 7,32-7,28 (м, 2H); 2,40 (с, 3H) м.д.
[0269] Получение соединения 6a: реакция BEFTAI
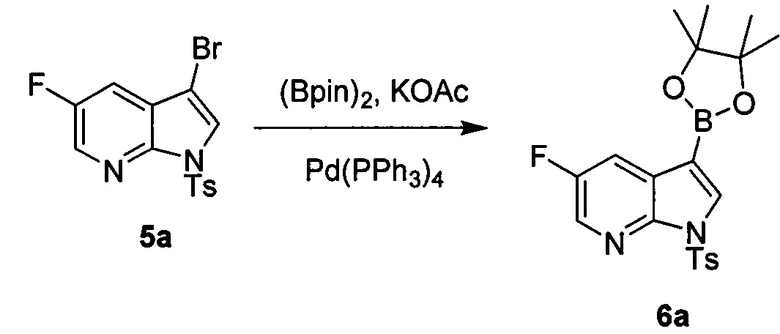
[0270] В инертный реактор на 400 л загружали соединение 5a (25,6 кг, 69,3 моль), бис(пинаколато)дибор (19 кг, 74,8 моль), ацетат калия (19 кг, 194 моль), ацетат палладия (156 г, 0,69 моль) и трифенилфосфин (564 г, 2,15 моль) с последующим добавлением диоксана (172 кг), который отдельно освобождали от кислорода, используя циклы откачивания вакуумом-заполнения азотом (×3). Смесь перемешивали и освобождали от кислорода, используя циклы откачивания вакуумом-заполнения азотом (×2), затем нагревали до 100°C в течение 15 часов. Смесь охлаждали до 35°C, затем фильтровали, промывая нагретым до 30°C ТГФ (75 кг). Фильтрат упаривали, а остаток растворяли в DCM (~90 л). Раствор перемешивали с 1 кг угля и 2 кг магнезола в течение 45 минут, затем фильтровали через слой силикагеля (22 кг, низ) и магнезола (10 кг, верх), промывая с помощью DCM (160 кг). Фильтрат концентрировали в густую взвесь (объемом ~40 л), затем затирали при 35°C и прибавляли гексан (26 кг). Взвесь охлаждали до 20°C, фильтровали и промывали смесью DCM (5,3 кг) и гексана (15 кг), затем гексаном (15 кг) и сушили под азотом на фильтре, что давало соединение 6a (23,31 кг, 56,0 моль, 81%) в виде белого твердого вещества. 1H-ЯМР соответствует желаемому продукту, чистота по ВЭЖХ 99,5%, анализ на палладий – содержание 2 м.д. 1H-ЯМР (CDCl3, 300 МГц): δ 8,25 (с, 1H); 8,18 (с, 1H); 8,09-8,02 (м, 2H); 7,91-7,83 (м, 1H); 7,30-7,23 (м, 2H); 2,39 (с, 3H); 1,38 (с, 12H) м.д.
[0271] Получение соединений 8a и 9a
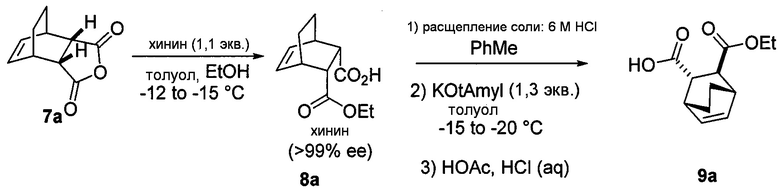
[0272] Соединение 8a: Ангидрид 7a (24,6 кг, Apex) и хинин (49,2 кг, Buchler) вводили в реактор, за чем следовало прибавление безводного PhMe (795,1 кг). Затем реактор охлаждали до -16°C и прибавляли EtOH (безводный, 41,4 кг) с такой скоростью, чтобы поддерживать температуру внутри реактора < -12°C. Максимальная зарегистрированная температура реакции для данного эксперимента составляла -16°C. Затем реакционную смесь перемешивали в течение 16 ч при -16°C. Отбирали и фильтровали образец. Твердое вещество сушили и анализировали посредством 1H-ЯМР, данные которого показывали, что ангидрида не оставалось. Содержимое реактора фильтровали. Реактор и полученный из него влажный фильтрованный остаток промывали PhMe (безводный, 20 кг). Полученное твердое вещество помещали в сушилку с поддонами и выдерживали в ней при < 45°C, продувая N2 в течение по меньшей мере 48 ч. В данном эксперименте фактическая температура составляла 44°C, а вакуум составлял -30 дюймов Hg (-762 мм рт. ст.). Спустя 2,5 дня сушки отбирали образец материала, и данные ЯМР показывали присутствие 3% PhMe. После дополнительной сушки в течение 8 ч, анализ на количество PhMe показывал присутствие тех же 3% PhMe и сушку прекращали. Масса белого твердого вещества составляла 57,7 кг, выход 76%. 1H-ЯМР соответствовал структуре, а анализ методом хиральной сверхкритической флюидной хроматографии (SFC) показывал, что материал имеет чистоту >99% de.
[0273] Соединение 9a: В реактор загружали хининовую соль 8a (57,7 кг) и PhMe (250,5 кг, Aldrich, чистота по ACS >99,5%) и включали мешалку. Содержимое охлаждали до <15°C и обрабатывали 6 N HCl (18 кг H2O обрабатывали 21,4 кг конц. HCl), поддерживая температуру <25°C. Смесь перемешивали в течение 40 мин и визуально осматривали, проверяя на отсутствие твердых веществ. Перемешивание прекращали и позволяли фазам осесть и разделяли фазы. Водные фазы снова экстрагировали PhMe (160 кг; обычно использованное количество было значительно меньше, по расчету 43 кг. Однако из-за минимального объема для эффективного перемешивания прибавляли дополнительное количество PhMe). Органические фазы объединяли. Отбирают образец органической фазы и проводят анализ ВЭЖХ, чтобы убедиться в присутствии продукта; испытание проводят только для получения информации.
[0274] Органические фазы охлаждали до <5°C (0-5°C) и прибавляли к ним сульфат натрия (безводный, 53,1 кг), перемешивая в течение 8 ч (в данном случае 12 ч). Содержимое реактора, содержащего органическую фазу, пропускали через фильтр, содержащий сульфат натрия (31 кг, безводный) и подавали в чистый и высушенный реактор. Реактор промывали PhMe (57,4 кг), пропускали через фильтр в реактор 201. Включали мешалку и прибавляли дополнительное количество PhMe (44 кг), и реакционную смесь охлаждали до -20°C. При данной температуре в течение 2 ч прибавляли раствор трет-пентоксида калия в PhMe, поддерживая температуру между -15°C и -22°C. Реакционную смесь выдерживали при приблизительно -20°C в течение дополнительных 30 мин перед отбором образца. Получение образца осуществлялось путем отбора аликвоты с немедленным гашением в 6 N HCl.
[0275] После достижения целевого соотношения (96:4 (транс:цис)) в реактор в течение 6 мин загружали уксусную кислоту (2,8 кг). Температура оставалась при -20°C. Затем температуру устанавливали равной -5°C и прибавляли водный раствор 2 N HCl (65,7 кг воды обрабатывали 15,4 кг конц. HCl). Содержимое нагревали до 5°C +/- 5°C, перемешивали в течение 45 мин перед нагреванием до 20°C +/- 5°C при перемешивании в течение 15 мин. Мешалку останавливали и позволяли осесть фазам. Водный слой удаляли. Органическую фазу промывали водой (48 кг, питьевая), перемешивали в течение 15 мин и позволяли осесть фазам (по меньшей мере 15 мин), и водный слой удаляли и прибавляли к водному слою. Буферный раствор в количестве 1/3 (50 л), который был получен из 7,9 кг NaH2PO4, 1,3 кг Na2HPO4 и 143,6 кг воды, прибавляли к органической фазе и перемешивали в течение по меньшей мере 15 мин. Перемешивание прекращали и фазам позволяли разделиться в течение по меньшей мере 15 мин. Нижний слой отбрасывали. Другую часть буферного раствора (50 л) использовали для промывки органического слоя, как описано ранее. Промывку проводили третий раз, как описано выше.
[0276] Вакуумную перегонку фазы PhMe (150 л) начинали при 42°C /-13,9 фунт/кв. дюйм изб. (-0,946 атм) и проводили до получения масла объемом приблизительно 20 л. После существенного уменьшения объема смесь переносили в меньший сосуд для завершения перегонки. Прибавляли смесь гептанов (13,7 кг) и смесь нагревали до 40 +/- 5°C в течение 30 мин, затем содержимое охлаждали до 0-5°C в течение 1,5 ч. Твердые вещества отфильтровывали и реактор промывали приблизительно 14 кг охлажденной (0-5°C) смеси гептанов. Твердым веществам позволяли высохнуть в вакууме перед их помещением в печь при <40°C, откачиваемую бытовым вакуумом (-28 фунт/кв. дюйм изб.) (-1,905 атм), до значения потери при сушке (LOD) <1%. Получали 15,3 кг, 64%, чистота по ВЭЖХ 96%. 1H-ЯМР (400 МГц, CDCl3): δ 11,45 (уш. s, 1H), 6,41 (т, J=7,2 Гц, 1H), 6,25 (т, J=7,2 Гц, 1H), 4,18 (м, 2H), 3,27 (м, 1H), 3,03 (м, 1H), 2,95 (м, 1H), 2,77 (м, 1H), 1,68 (м, 1H), 1,49 (м, 1H), 1,25 (т, J=7,2 Гц), 1,12 (м, 1H).
[0277] Получение соединения 10a
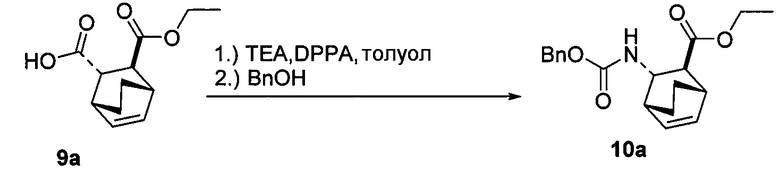
[0278] В трехгорлую колбу, оборудованную механической мешалкой, температурным зондом, обратным холодильником, капельной воронкой и впуском азота, в атмосфере азота загружали соединение 9a (145,0 г, 1 эквив.) и безводный толуол (Aldrich, каталожный номер 244511) (1408 г, 1655 мл). Затем порциями в течение 5 минут к перемешиваемому раствору прибавляли триэтиламин (Aldrich, каталожный номер 471283) (140 г, 193 мл, 2,14 эквив.), в ходе чего наблюдался экзотермический эффект до максимальной температуры 27°C. Начинали сбор данных с помощью ReactIR. Затем реакционную смесь нагревали до 95°C в течение 70 минут. Затем из капельной воронки порциями в течение суммарного времени 2,25 часов прибавляли дифенилфосфорилазид (Aldrich, каталожный номер 178756) (176,2 г; 138,0 мл, 0,99 эквив.).
[0279] После завершения прибавления дифенилфосфорилазида (капельную воронку промывали малым количеством толуола) полученную смесь нагревали при 96°C в течение дополнительных 50 минут. Образец реакционной смеси, разведенной в толуоле, анализировали посредством анализа GC/MS (газовая хроматография/масс-спектрометрия), который указывал на израсходование дифенилфосфорилазида. Затем из капельной воронки в течение 5-10 минут прибавляли бензиловый спирт (Aldrich, каталожный номер 108006) (69,9 г, 67,0 мл, 1,0 эквив.). Затем полученную смесь нагревали при 97°C в течение ночи (приблизительно в течение 19 часов). Образец реакционной смеси, разведенной в толуоле, при анализе GC/MS указывал на образование продукта (m/e =330). Затем реакционную смесь охлаждали до 21°C, после чего порциями прибавляли воду (870 г, 870 мл) (наблюдалась слабая экзотермия с максимальной температурой 22°C). Реакционную смесь сначала гасили прибавлением 500 г воды и механически перемешивали в течение 10 минут. Затем смесь переносили в делительную воронку, содержащую остальные 370 г воды, а затем перемешивали вручную. После перемешивания и разделения фаз, органический и водный слои разделяли (водный имел pH ~10). Затем органический слой промывали дополнительной порцией воды (870 г; 1 × 870 мл). Органический и водный слои разделяли (водный имел pH ~10). Затем собранную органическую фазу концентрировали досуха при пониженном давлении (водяная баня при 45-50°C), что давало 215 г сырого соединения 10a (приблизительный объем 190 мл). 1H-ЯМР и GC/MS соответствовали соединению 10a (присутствовали остатки толуола и бензилового спирта).
[0280] Получение соединения 11a
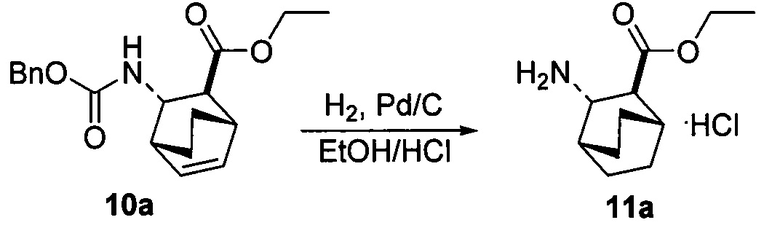
[0281] a.) Получение HCl в этаноле: В трехгорлую колбу, оборудованную температурным зондом, впуском азота и магнитной мешалкой, в атмосфере азота загружали этанол (1000 мл, 773 г). Жидкость перемешивали и охлаждали в бане с сухим льдом/ацетоном до тех пор, пока температура внутри не достигла -12°C. Затем в охлажденный раствор медленно в течение 2 часов барботировали безводный HCl (~80 г, 2,19 моль) (в ходе прибавления наблюдалась температура от -24°C до -6°C). После прибавления раствор переносили в стеклянную бутыль и позволяли ему нагреться до температуры окружающей среды. Образец раствора подвергали титрованию, дававшему значение концентрации 2,6 M. Затем раствор выдерживали в течение ночи в холодном помещении (при приблизительно 5°C).
[0282] b.) Гидрирование/образование соли с HCl: В атмосфере азота в стеклянную вставку автоклава Parr на 2 галлона (7,57 л) загружали палладий на угле (Pd/C (Aldrich, каталожный номер 330108), 10% на сухую массу; (50% на влажную), 13,11 г, 0,01 эквив. относительно соединения 10a), а затем увлажняли этанолом (93 г; 120 мл). Затем в стеклянную вставку прибавляли раствор сырого соединения 10a (212 г, 1 экв.) в этаноле (1246 г; 1600 мл) (небольшая обмывка этанолом для содействия переносу). Стеклянную вставку помещали в автоклав, после чего прибавляли HCl в этаноле (получен, как описано выше; 2,6 M; 1,04 эквив. относительно соединения 10a; 223 г; 259 мл). Автоклав герметично закрывали, а затем продували водородом (3× при 20 фунт/кв. дюйм (1,4 атм)). Затем начинали гидрирование при подаваемом давлении газообразного водорода (15 фунт/кв. дюйм (1 атм)) в течение 3 часов, во время чего давление водорода было постоянным. Анализ аликвоты реакционной смеси посредством 1H-ЯМР и GC/MS указывал на израсходование исходного материала/образование продукта. Затем полученную смесь фильтровали через слой целита (192 г), после чего слой целита промывали дополнительным этанолом (3×; всего в ходе промывок использовали 1176 г этанола). Затем фильтрат (зеленого цвета) концентрировали при пониженном давлении (водяная баня при 45°C) до ~382 г (~435 мл; 2,9 объема в расчете на теоретический выход соединения 11a). Затем к остатку прибавляли изопропилацетат (1539 г; 1813 мл (12 объемов в расчете на теоретический выход соединения 11a). Полученный раствор подвергали перегонке в вакууме при постепенном увеличении температуры.
[0283] Перегонку прекращали, после чего оставшийся раствор (370 г, суммарный объем ~365 мл; коричневатого цвета) оставляли стоять при температуре окружающей среды на протяжении выходных. Смесь фильтровали (для содействия фильтрованию использовали изопропилацетат) и собранное твердое вещество промывали дополнительным изопропилацетатом (2 × 116 мл; каждая промывка производилась приблизительно 100 г). Затем твердое вещество сушили в вакууме при 40°C (максимальная наблюдаемая температура 42°C) в течение ночи, что давало 118 г (78,1% на две стадии) соединения 11a. 1H-ЯМР материала согласовывался со структурой соединения 11a, а GC/MS указывали на чистоту 99%.
[0284] Получение соединения 13a
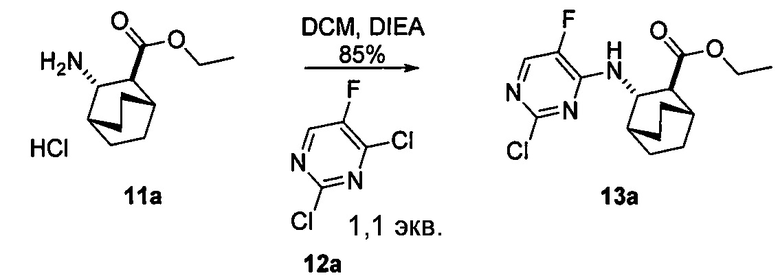
[0285] Методика A: Смесь 5-фтор-2,4-дихлорпиримидина (12a, 39,3 г, 235 ммоль, 1,1 эквив.) и соли амина с HCl (11a, 50 г, 214 ммоль) обрабатывали CH2Cl2 (169 мл), и данную смесь нагревали до 30°C. Затем смесь медленно обрабатывали в течение 3 ч DIEA (60,8 г, 82 мл, 471 ммоль, 2,2 эквив.), используя шприцевой насос. Максимальная температура достигала 32°C. Реакционную смесь перемешивали в течение 20 ч, о завершении реакции судили по данным ВЭЖХ и охлаждали реакционную смесь до комнатной температуры. Полученную реакционную смесь последовательно промывали водой, (211 мл, pH=8-9), 5% NaHSO4 (211 мл, pH=1-2), затем 5% водным NaCl (211 мл, pH=5-6).
[0286] Затем органическую фазу отгоняли при пониженном давлении до 190 мл. Загружали PhMe (422 мл) и устанавливали температуру равной 70-80°C, а внутреннюю температуру равной 60-65°C, продолжая отгонку до уменьшения объема обратно до 190 мл. При перемешивании смеси позволяли охладиться приблизительно до 37°C и спустя приблизительно 10 мин началась кристаллизация и наблюдалось увеличение температуры приблизительно до 41°C. После достижения равновесия при 37°C в суспензию вводили н-гептан (421 мл) в течение 3,5 ч с последующим охлаждением до 22°C в течение 1 ч. Смесь оставляли перемешиваться в течение ночи при указанной температуре перед фильтрованием. Полученное на фильтре твердое вещество промывали 10% раствором PhMe в н-гептане (2×210 мл). Затем твердое вещество сушили в течении ночи в печи в вакууме при продувке N2 при 50°C. Полученное твердое вещество весило 62 г (выход 88%).
[0287] Методика B: В атмосфере азота в трехгорлую колбу, оборудованную механической мешалкой, температурным зондом, обратным холодильником, впуском азота и капельной воронкой, загружали соединение 11a (51,2 г) и соединение 12a (40,2 г). Прибавляли дихлорметан (173 мл, 230 г) и перемешивали полученную смесь, нагревая до температуры внутри, равной 30°C. Затем из капельной воронки медленно в течении 2,5-3 часов прибавляли N,N-диизопропилэтиламин (85 мл, 63,09 г), во время чего наблюдалась экзотермия с максимальной наблюденной температурой 33,5°C. После завершения прибавления полученный в результате раствор перемешивали в атмосфере азота при 30-31°C в течение ночи (приблизительно в течение 19 часов).
[0288] Образец реакционной смеси объемом 100 мкл разводили дихлорметаном до суммарного объема 10 мл и хорошо перемешивали раствор. Образец разведенной аликвоты анализировали посредством анализа GC/MS, который указывал, что реакция завершилась согласно данным GC/MS; наблюдалось образование продукта (m/e = 328). Реакционную смесь охлаждали до 26°C и переносили в делительную воронку (с помощью дихлорметана). Затем смесь последовательно промывали водой (211 мл, 211 г; значение pH водной фазы составляло ~8; небольшой слой шубы переносили с водной фазой), 5% водным NaHSO4 ((получен, используя 50 г моногидрата бисульфата натрия (Aldrich, каталожный номер 233714) и 950 г воды), 211 мл, 216 г; значение pH водной фазы составляло ~2), а затем 5% водным NaCl ((получен, используя 50 г хлорида натрия (Aldrich, каталожный номер S9888) и 950 г воды), 211 мл, 215 г; значение pH водной фазы составляло ~4-5). Затем собранную органическую фазу концентрировали при пониженном давлении (водная баня при 35°C) до ~190 мл (2,7 объема в расчете на теоретический выход соединения 13a), после чего прибавляли толуол (Aldrich, каталожный номер 179418, 422 мл, 361 г). Полученную смесь концентрировали при пониженном давлении (водяная баня при 55-65°C) до ~190 мл (2.7 объема в расчете на теоретический выход соединения 13a). Анализ образца раствора на данной стадии посредством 1H-ЯМР указывал на отсутствие дихлорметана. Полученной смеси позволяли охладиться до 37°C (используя водяную баню при 37°C на роторном испарителе при перемешивании). В это время наблюдалась выраженная кристаллизация. Затем смесь механически перемешивали и нагревали приблизительно до 37°C (внешний источник тепла, отрегулированный на 38°C), после чего в течение 3 часов из капельной воронки медленно прибавляли н-гептан (430 мл, 288 г; Aldrich, каталожный номер H2198). После прибавления нагрев прекращали и полученную взвесь механически перемешивали в течение ночи, охлаждая до температуры окружающей среды. Затем полученную смесь фильтровали, а отделенное твердое вещество промывали 10% толуола в н-гептане (2×210 мл; каждый раствор для промывки готовили, смешивая 21 мл (16 г) толуола и 189 мл (132 г) н-гептана). Вакуум использовали до тех пор, пока не наблюдалось присутствие весьма малого количества фильтрата. Затем твердое вещество дополнительно сушили в вакууме при 50°C в потоке азота до постоянной массы (3,5 часа), что давало 64,7 г (90%) соединения 13a. Анализ образца твердого вещества посредством 1H-ЯМР показывал, что материал соответствует структуре, а анализ жидкостной хроматографией (LC) указывал на чистоту 99,8% при использовании предусмотренного метода LC.
[0289] Получение соединения 14a
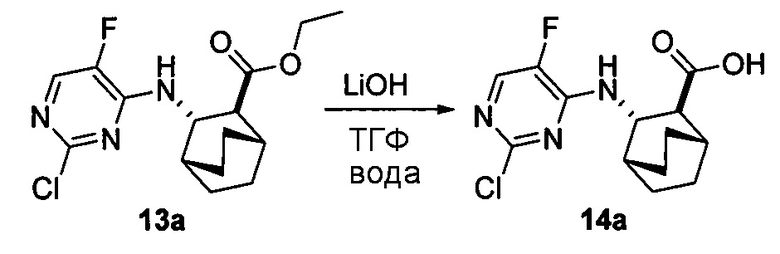
[0290] Этиловый сложный эфир 13a (85 г, 259 ммоль) растворяли в ТГФ (340 мл) и обрабатывали раствором LiOH (2 M, 389 мл, 778 ммоль) в течение 10 мин (температура от 21°C до 24°C). Смесь нагревали до 45°C при перемешивании в течение 17 ч, где по достижении указанного времени согласно данным ВЭЖХ судили о завершении реакции (исходный материал (SM) не наблюдался). Реакционную смесь охлаждали до комнатной температуры и прибавляли CH2Cl2 (425 мл). Затем медленно в течение 45 мин (температура вплоть до 26°C) прибавляли раствор лимонной кислоты (2 M, 400 мл). Отмечали, что в ходе прибавления образовывалось некоторое количество белого вещества, но оно быстро растворялось при перемешивании. Реакционную смесь перемешивали в течение дополнительных 15 мин перед тем, как фазам дали разделиться. После того, как фазы разделились, измерение pH водной фазы показало, что она имела pH=4,0. Органическую фазу промывали (перемешивание в течение 15 мин) водой (255 мл) и давали фазам разделиться. Затем нижний слой (органический), содержащий желаемый продукт, выдерживали в холодильнике в течение ночи.
[0291] Органическую фазу концентрировали при пониженном давлении (температуру нагревательной емкости устанавливали на 65°C) до приблизительно 150 мл (примерно 1,76 объема относительно исходного материала (SM)). Загружали IPA (510 мл) и перегоняли при пониженном давлении (температура холодильника установлена на 85°C) до 255 мл (3 объема). Количество растворителя доводили приблизительно до 553 мл (6,5 объемов), добавляя IPA (298 мл). Затем прибавляли воду (16 мл) и нагревали реакционную смесь до кипения (77°C) при хорошем перемешивании, что приводило к растворению твердого вещества, осевшего на стенках сосуда. Затем реакционную смесь медленно охлаждали до 65°C (в течение 60 мин) и выдерживали при этой температуре, причем весь материал еще находился в растворе (отбирали образец для анализа на остаточный растворитель). Реакционную смесь дополнительно охлаждали до 60°C, и реакционная смесь становилась слегка мутной. После перемешивания в течение 15 мин дополнительно охлаждали до 55°C. Хотя осаждается больше продукта, смесь все еще остается жидкой и легко промешивается. Весьма медленно (2,5-3 ч) прибавляли воду (808 мл), поддерживая температуру около 55°C. Затем смесь охлаждали до 22°C в течение 2 ч и позволяли ей перемешиваться в течение ночи. Затем материал фильтровали и промывали смесью воды:IPA (75:25, 2×255 мл). Кислоту сушили в вакуумной печи в течение ночи при 55°C. Получали 69 г кислоты 14a, выход белого твердого вещества 88%. Согласно анализу ВЭЖХ материал имел чистоту >99%.
[0292] Получение соединения 15a: сочетание по Сузуки
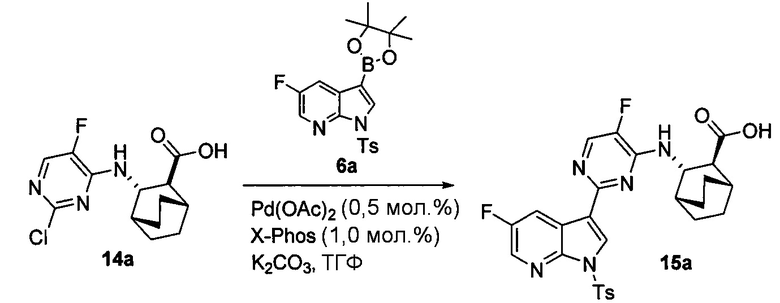
[0293] К соединению 14a (91,4 г, 305 ммоль), соединению 6a (158,6 г, 381 ммоль, 1,25 эквив.), Pd(OAc)2 (0,34 г, 1,5 ммоль, 0,5 мол. %), X-Phos (1,45 г, 3,0 ммоль, 1,0 мол. %) и K2CO3 (168,6 г, 1220 ммоль, 4 эквив.) прибавляли ТГФ (731 мл, 8 объемов) и воду (29 мл, 0,32 объема). В течение 30 мин в реакционную смесь барботировали N2, затем ее нагревали до 65-70°C и перемешивали в течение 5 ч. Анализ ВЭЖХ реакционной смеси показывал конверсию 99,3%. Реакционную смесь охлаждали до 22-25°C и прибавляли воду. Смесь перемешивали, фазам позволяли разделиться, и водную фазу декантировали. К органической фазе прибавляли раствор 18 масс. % NaCl в воде (полунасыщенный водный раствор NaCl) и используя 2 N HCl регулировали pH смеси до 6,0-6,5. Фазам позволяли разделиться и водную фазу декантировали. Органическую фазу концентрировали до минимального объема и прибавляли ацетонитрил. Данный процесс повторяли еще один раз и прибавляли ацетонитрил, чтобы довести до конечного объема, равного 910 мл (10 объемов). Взвесь нагревали до 80-85°C в течение 6 ч, затем охлаждали до 20-25°C. Перемешивали взвесь в течение 2 ч, затем фильтровали. Твердое вещество промывали ацетонитрилом, что давало соединение 15a (161 г, выход 89%).
[0294] Получение соединения (2): Стадия удаления тозильной группы
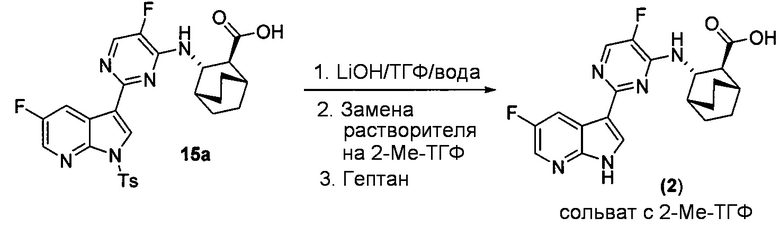
[0295] К соединению 15a (25 г, 45,2 ммоль) прибавляли ТГФ (125 мл, 5 объемов), затем смолу MP-TMT (6,25 г, 25 масс. %). Смесь перемешивали при 20-25°C в течение 16 ч и фильтровали, промывая 1 объемом ТГФ. Процесс обработки смолой и фильтрования повторяли. Раствор в ТГФ концентрировали до 5 объемов. При 22-25°C к смеси прибавляли водный раствор 2 M LiOH (90,3 мл, 4 эквив.). Реакционную смесь нагревали до 40-45°C и перемешивали в течение 5 ч. Анализ ВЭЖХ показывал конверсию 99,7%. Реакционную смесь охлаждали до 22-25°C и прибавляли MTBE (50 мл, 2 объема). Происходило разделение фаз. Нижнюю водную фазу собирали. Водную фазу экстрагировали MTBE. Нижнюю водную фазу собирали. К водной фазе прибавляли 2-Me-ТГФ и перемешивали смесь. pH смеси регулировали до 6,0-6,5 и декантировали нижнюю водную фазу. Органическую фазу промывали буфером с pH 6,5. Органическую фазу концентрировали до 85 мл, разбавляли 2-Me-THF (150 мл) и концентрировали до конечного объема 180 мл. Полученную взвесь нагревали до 70-75°C и перемешивали до полного растворения, затем охлаждали до 45-50°C, что давало взвесь. Взвесь перемешивали в течение 1 ч, затем прибавляли гептан (180 мл). Взвесь охлаждали до 20-25°C в течение 1 ч и перемешивали в течение 16 ч. Массу фильтровали, промывали твердое вещество гептаном. Твердое вещество сушили, что давало сырой сольват соединения (2)·с 2-Me-ТГФ, выход 79%.
[0296] Получение гемигидрата соли соединения (2) с HCl: Образование соли
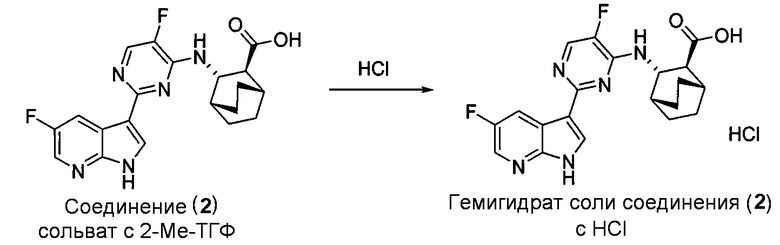
[0297] Методика A: Соединение (2)⋅2-Me-ТГФ (953 г, 2,39 моль) помещали в реактор с рубашкой объемом 30 л и обрабатывали IPA (15 л) и водой (0,57 л). Запускали мешалку и реакционную смесь нагревали до 73°C, переводя все в раствор, затем охлаждали до 50-55°C. При 50-55°C реакционную смесь обрабатывали свежеприготовленным HCl в IPA (0,83 M, 4,34 л), медленно добавляя в течение 4 ч. Из реакции отбирали образец для проверки на нужную форму методом XRPD. После прибавления холодильник программировали на линейное охлаждение до 0°C в течение 480 мин при перемешивании. После подтверждения формы посредством анализа XRPD взвесь фильтровали на двух фильтрах. Реактор промывали 3 л IPA и каждую отфильтрованную массу промывали ~1,5 л IPA, представляющего собой IPA от промывки реактора. Отфильтрованным массам давали возможность высохнуть на воздухе в течение ночи, применяя отсос. Затем отфильтрованные массы помещали в сушилку с поддонами, выдерживая в ней в вакууме с продувкой N2 (22 дюйма Hg (560 мм рт. ст.).) в течение 24 ч, не используя нагрев. Анализ на остатки растворителя и воды показал присутствие 505 м.д. IPA, 8 м.д. 2-Me-ТГФ и приблизительно 2,15% H2O. Материал извлекали из печи и перемалывали для удаления комков, что давало 805 г соли соединения (2) с HCl на 1/2H2O.
[0298] Методика B: В альтернативном варианте вместо IPA использовали ацетон, но аналогично описанному выше для методики A, получая соль соединения (2) с HCl на 1/2H2O.
[0299] Пример 2: Альтернативные варианты получения некоторых соединений и условия реакции Сузуки
[0300] A. Получение соединения 3a примера 1
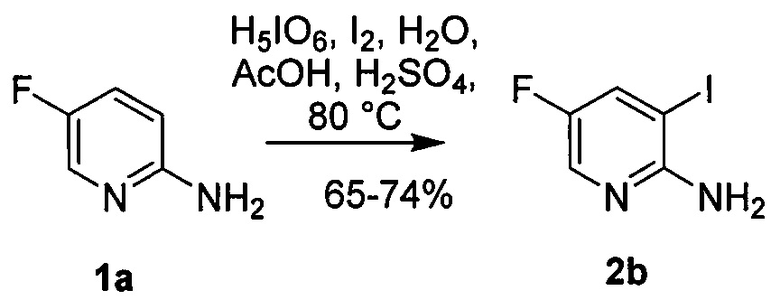
[0301] Стадия 1: 5-Фтор-3-иодпиридин-2-амин (2b)
[0302] H2SO4 (120 мл) прибавляли по каплям в течение 5 минут к раствору 2-амино-5-фторпиридина (1 кг, 8,9 моль) в AcOH (4 л) и H2O (1 л). Прибавляли периодную кислоту (H5IO6; 450 г, 1,97 моль, 0,22 экв.) и I2 (1 кг, 3,94 моль, 0,44 экв.) и перемешивали реакционную смесь в течение ночи при 82°C (внутри). Анализ образца (разведение H2O, подщелачивание 30% NaOH, экстрагирование EtOAc, концентрирование) показал присутствие в нем 13-15% исходного материала. Прибавляли дополнительное количество H5IO6 (80 г) и I2 (180 г) и продолжали перемешивание при 80°C в течение ночи. Внешний нагрев удаляли и реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь выливали на воду со льдом (8 л), подщелачивали 33% водным NaOH (необходимо ~6,5 л) и перемешивали в течение 2 ч. Осажденный продукт отделяли фильтрованием и промывали горячей H2O (8×3 л). Промытый продукт фильтрования оставляли на ночь, после чего продукт промывали смесью гептанов (3×). Продукт сушили в печи при 45°C на протяжении выходных. Соединение 2b (1390 г, выход 65%) получали в виде черного твердого вещества. К гептановому слою прибавляли H2O и оставляли на выходные. Темный водный слой отделяли от светло-желтого органического слоя, который концентрировали досуха. Таким образом получали дополнительную порцию соединения 2b (95 г, суммарный выход 70%) в виде желтого твердого вещества. 1H-ЯМР (ДМСО-d6, 300 МГц): δ 7,95-7,88 (м, 2H) м.д.. 1H-ЯМР (CDCl3, 300 МГц): δ 7,95-7,90 (м, 1H); 7,68-7,62 (м, 1H); 4,85 (с, NH2) м.д.
[0303] Стадия 2: 5-Фтор-3-((триметилсилил)этинил)пиридин-2-амин (3a)
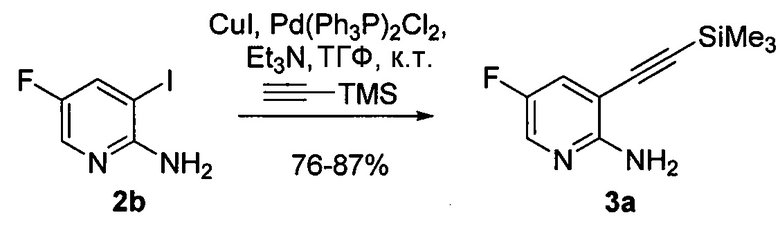
[0304] Раствор соединения 2b (790 г, 3,3 моль) в ТГФ (2,9 л) дегазировали (3×), используя циклы N2(г)/вакуум. Продувку с помощью N2 (г) начинали вслед за прибавлением CuI (6,32 г, 0,01 экв.), PdCl2(PPh3)2 (23,4 г, 0,01 экв.) и Et3N (1,4 л, 3 экв.). Продувку продолжали в течение 10 минут и реакционную смесь еще раз дегазировали, после чего в течение 40-45 минут следовало прибавление по каплям триметилсилилацетилена (605 мл, 1,3 экв.). В ходе прибавления экзотермическая реакция самопроизвольно не начиналась и реакционную смесь нагревали до ~45°C. Внешний нагрев удаляли. В это время начиналась экзотермическая реакция и температура достигала ~66°C (через 40 минут прибавление завершали). Реакционной смеси давали перемешиваться в течение дополнительных 2 ч, после чего температура снижалась до 26°C. Анализ образца (фильтрование через целит, концентрирование) показал полную конверсию, и реакционную смесь разводили EtOAc (3 л). Раствор фильтровали через силикагель (2 кг), элюируя с помощью EtOAc (в сумме 9 л). Растворители удаляли при пониженном давлении, что давало соединение 3a (642 г, выход 93%) в виде темного масла. 1H-ЯМР (CDCl3, 300 МГц): δ 7,90 (с, 1H); 7,33-7,27 (м, 1H); 4,92 (с, NH2), 0,28 (с, 9H) м.д.
[0305] B. Получение соединения 4a примера 1
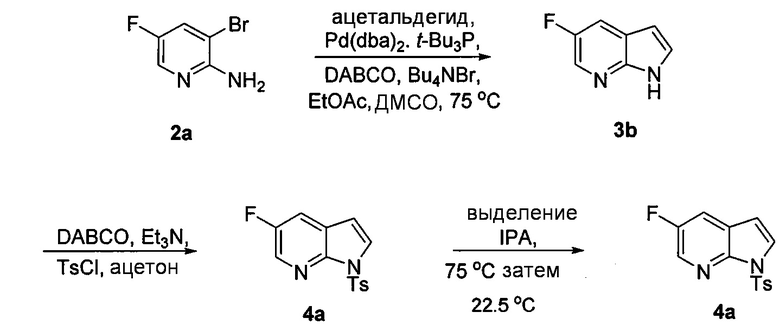
[0306] Стадия 1: 5-Фтор-1H-пирроло[2,3-b]пиридин (3b)

[0307] В колбу для работы под давлением на 500 мл, продутую азотом, загружали 3-бром-5-фторпиридин-2-амин (соединение 2a) (20 г, 104,7 ммоль, 1 эквив.), DABCO (17,6 г, 157,0 ммоль, 1,5 эквив.) и бромид тетрабутиламмония (3,38 г, 10,5 ммоль, 0,1 эквив.). В колбу загружали диметилсульфоксид (безводный, 40 мл) и этилацетат (безводный, 120 мл) и в течение 30 мин через полученную смесь барботировали азот. Загружали бис(дибензилиденацетон)палладий (0) (3,01 г, 5,24 ммоль, 0,05 эквив.), 10% масс./масс. раствор три-трет-бутилфосфина в гексане (21,2 г, 10,47 ммоль, 0,1 эквив.) и ацетальдегид (5,08 г, 115,2 ммоль, 1,1 эквив.) и герметично закрывали колбу. Смесь перемешивали в течение 1 ч при комнатной температуре, затем нагревали на масляной бане при 76,5°C в течение 5 ч. Массу охлаждали и отбирали образец для анализа ВЭЖХ. После того, как наблюдали полную конверсию в соединение 3b (обычно конверсию 100% спустя 5 ч), массу гасили водой (40 мл). Проводили обратную экстракцию водной фазы этилацетатом (40 мл) и объединенные органические материалы фильтровали через слой целита, чтобы удалить мелкодисперсное твердое вещество. Целит промывали этилацетатом (40 мл), и полученный раствор сырого продукта обрабатывали 5% Na2CO3 (60 мл) и при перемешивании барботировали через него азот в течение 30 мин. Полученную органическую фазу промывали водой (60 мл) и концентрировали при <30°C до 43 мл. Определенный с использованием стандартов ВЭЖХ выход в растворе составлял 13,1 г (92%) в расчете на 3-бром-5-фторпиридин-2-амин.
[0308] Стадия 2: 5-Фтор-1-тозил-1H-пирроло[2,3-b]пиридин (4a)
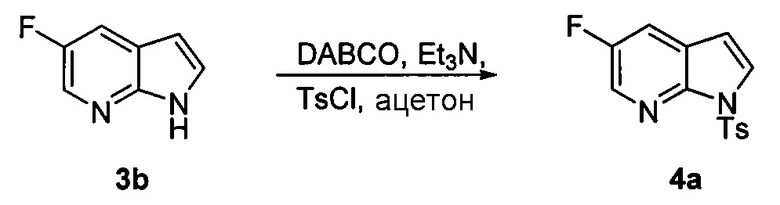
[0309] К раствору сырого соединения 3b (считая на 14,26 г (100% со стадии 1), 1 эквив.) в этилацетате со стадии 1 прибавляли ацетон (71,2 мл), DABCO (5,8 г, 52,35 ммоль, 0,5 эквив.) и триэтиламин (29,4 мл, 209,4 ммоль, 2 эквив.), и реакционную колбу продували азотом. В отдельной колбе готовили раствор тозилхлорида (29,9 г, 157,0 ммоль, 1,5 эквив.) в ацетоне (35,6 мл). Раствор тозилхлорида прибавляли к раствору соединения 3b при комнатной температуре в течение 30 мин at room temperature. Через 4 часа реакционную смесь анализировали посредством ВЭЖХ на % конверсии. Когда количество оставшегося соединения 3b составляло <0,2% AUC (обычно через 4 ч), реакцию гасили водой (10 мл), перемешивали в течение 30 мин и прибавляли HCl (80 мл) и дихлорметан (144 мл). Массу перемешивали в течение 30 мин. Водную фазу экстрагировали дихлорметаном (43 мл), и объединенные органические материалы промывали 5% NaCl (72 мл). Выход соединения 4a в растворе на данной стадии составлял 27,0 г (97%) в расчете на стандарты ВЭЖХ.
[0310] Стадия 3: Выделение 5-фтор-1-тозил-1H-пирроло[2,3-b]пиридина (4a)
[0311] Раствор сырого соединения 4a со стадии 2 концентрировали в вакууме до 57 мл, добавляли к нему 2-пропанол (184 мл) и концентрировали до 120,8 мл. Полученную смесь нагревали до 83,6°C. После перемешивания при указанной температуре в течение 1 ч смесь охлаждали до 22,5°C в течение 2 ч и выдерживали при указанной температуре в течение 20 ч. Затем взвесь фильтровали и промывали реактор и отделенное твердое вещество смесью вода/2-пропанол 25/75 (2×80 мл). Материал сушили в течение 7 ч в вакууме в токе азота при 56°C. Соединение 4a выделяли с выходом 81% (24,5 г) в расчете на 3-бром-5-фторпиридин-2-амин (3 стадии, включая выделение), оно имело чистоту AUC 98,4%. 1H-ЯМР (CDCl3, 300 МГц): δ 8,28-8,27 (м, 1H); 8,06-8,02 (м, 2H); 7,77 (д, J = 4,0 Гц, 1H); 7,54-7,50 (м, 1H); 7,28-7,26 (м, 2H); 6,56 (д, J = 4,0 Гц, 1H); 2,37 (с, 3H) м.д.
[0312] C. Получение соединения 4a примера 1
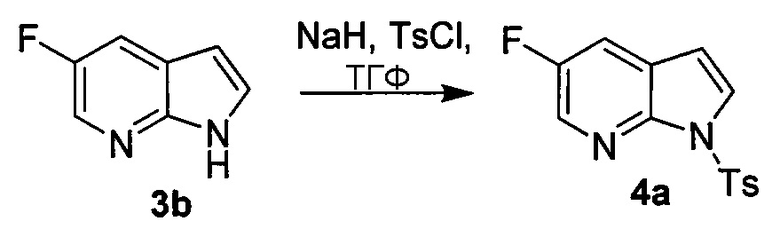
[0313] Способ A: Соединение 3b (280 г, 2 моль) растворяли в ТГФ (6 л) и раствор охлаждали в ледяной бане до <10°C. В течение 30 минут порциями прибавляли дисперсию 60% NaH (95 г, 57 г NaH, 1,15 экв.) в минеральном масле. Температуру поддерживали в диапазоне 5-10°C. Реакционную смесь перемешивали в течение 40 минут. В течение 40 минут по каплям прибавляли раствор пара-толуолсульфонилхлорида (408 г, 1,04 экв.) в ТГФ (суммарный объем раствора 2,5 л). Внешнее охлаждение удаляли и перемешивали реакционную смесь в течение 70 минут, по прошествии которых температура достигала 8°C. Анализ ЯМР образца (разведение EtOAc, промывка насыщенным NaHCO3 и концентрирование) показал, что реакция завершилась. Реакционную смесь гасили насыщенным водным раствором NaHCO3 (2 л) и разводили EtOAc (8 л). Слои разделяли и органический слой делили на 2 части. Каждую часть промывали насыщенным водным раствором NaHCO3 (2×1,5 л) и рассолом (2×1 л). Части объединяли, сушили над Na2SO4 и фильтровали через силикагель (2 кг), элюируя с помощью EtOAc (всего ~10 л). Растворители (~28 л) удаляли при пониженном давлении, а полученное в результате твердое вещество (628 г) переносили на фильтр и промывали смесью гептанов (2×). Примечание: первый гептановый промывочный раствор имел оранжево-красный цвет, а второй гептановый промывочный раствор был почти бесцветным. После сушки получали чистое соединение 4a (566 г, чистота 94,6%) в виде желто-коричневого твердого вещества. 1H-ЯМР (CDCl3, 300 МГц): δ 8,28-8,27 (м, 1H); 8,06-8,02 (м, 2H); 7,77 (д, J=4,0 Гц, 1H); 7,54-7,50 (м, 1H); 7,28-7,26 (м, 2H); 6,56 (д, J=4,0 Гц, 1H); 2,37 (с, 3H) м.д.
[0314] Способ B:
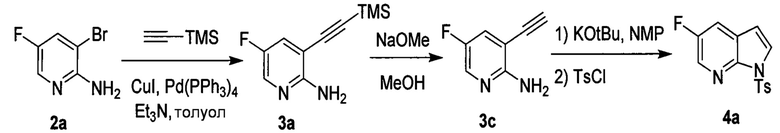
[0315] Стадия 1: Получение соединения 3c
[0316] В инертный реактор на 400 л, содержащий толуоловый раствор соединения 2a (185 л, ~33,5 кг, 175 моль), загружали Pd(PPh3)4 (1215 г, 1,05 моль) и CuI (200 г, 1,05 моль). Смесь освобождали от кислорода с помощью двух циклов откачивания вакуумом-заполнения азотом, затем прибавляли триэтиламин (23 кг, 227 моль). Смесь освобождали от кислорода с помощью еще одного цикла откачивания вакуумом-заполнения азотом, затем прибавляли TMS-ацетилен (19 кг, 193 моль). Смесь нагревали до 50°C в течение 22 часов, затем до 54°C в течение дополнительных 9 часов и охлаждали до 25°C в течение ночи. Одной порцией прибавляли 25 масс. % метоксида натрия в метаноле (41,6 кг, 193 моль) и смесь перемешивали при 25°C в течение 40 минут. Смесь охлаждали до 20°C, затем одной порцией прибавляли уксусную кислоту (2 л, 35 моль) и перемешивали в течение 1 часа. Взвесь фильтровали через слой целита, промывая его толуолом (30 кг) и сохраняли фильтрат для выделения продукта. Аналогичным образом проводили второй эксперимент, исходя из ~29,8 кг толуолового раствора соединения 2a (156 моль).
[0317] Фильтраты из обоих экспериментов концентрировали до объема ~220 л (чтобы испарить метанол) и разводили свежим толуолом до объема ~290 л. Данный раствор фильтровали через слой магнезола (20 кг), промывая его MTBE (240 л). Фильтрат упаривали с получением густой взвеси (объемом ~75 л), затем при ~35°C прибавляли гексан (65 кг). Взвесь охлаждали до 20°C. При попытке фильтрования твердое вещество не извлекалось из реактора, но жидкость извлекалась легко. После отведения жидкости твердое вещество в емкости промывали гексаном (93 кг). Твердое вещество снова оставалось в емкости, а промывная жидкость извлекалась. Твердое вещество сушили в емкости с помощью вакуума (чистота ~95%, 249 моль, выход 75%).
[0318] Фильтрат концентрировали и остаток распределяли между DCM (25 л) и 2 M HCl (30 л). Верхний водный слой промывали DCM (5 л). Органические слои последовательно повторно экстрагировали с помощью 2 M HCl (6 л). Водные слои перемешивали с DCM (25 л) и pH регулировали до ~8 прибавлением K3PO4 (1 кг), затем 8 M NaOH (8,3 л). Слои разделяли (продукт находится в органической фазе), и водный слой повторно экстрагировали DCM (5 л). Органические слои фильтровали через слой магнезола (3 кг), промывая его DCM (7 л). Фильтрат концентрировали досуха, затирали с гексаном (4 л) при 45°C, охлаждали до 20°C, фильтровали и промывали гексаном, что давало дополнительное количество для стадии 2 (8,25 кг, чистота ~95%, 58 моль, 17%) в виде оранжево-коричневого твердого вещества.
[0319] Стадия 2: Получение соединения 4a
[0320] В инертный реактор на 400 загружали NMP (80 кг) и т-бутоксид калия (40 кг, 357 моль). Смесь нагревали до 59°C, затем в течение 2 часов прибавляли раствор для стадии 2 (44 кг, чистота ~95%, 307 моль) в NMP (80 кг) и промывали с помощью NMP (10 кг) (экзотермия, поддерживали 70-83°C). Реакционную смесь перемешивали при 75°C в течение 1 часа. Анализ ЯМР показал, что образец, погашенный в DCM/NaHCO3, не содержал остатков исходного материала. Образец, погашенный избытком TsCl, а затем обработанный DCM/NaHCO3, показал остаток ~4% N-H. Смесь охлаждали до 48°C и прибавляли дополнительное количество т-бутоксида калия (2 кг, 18 моль). Смесь дополнительно охлаждали до 37°C и в течение 1,5 часа прибавляли раствор тозилхлорида (62 кг, 326 моль) в NMP (60 кг) и промывали с помощью NMP (4 кг) (экзотермия, поддерживали 30-45°C). Реакционную смесь перемешивали в течение 1 часа, охлаждая до 20°C.
[0321] Анализ образца, погашенного в DCM/NaHCO3, показал ~9% остатка N-H. Прибавляли дополнительное количество тозилхлорида (3 кг, 16 моль) и в течение ночи перемешивали смесь при 20°C, затем переносили в инертный реактор на 800 л, промывая с помощью NMP (10 кг). В течение 2,5 часов прибавляли воду при 5°C (500 л) (экзотермия, поддерживали 17-23°C). Смесь перемешивали при 17°C в течение 30 минут. При попытке фильтрования смесь не извлекалась бы из реактора. После предоставления возможности осесть жидкость отсасывали сверху (через фильтр). Твердое вещество в емкости смачивали водой (100 л, не перемешивалось бы), затем жидкость снова отсасывали сверху. Данную промывку повторяли с дополнительными 100 л (оставляя стоять на ночь), а затем с 200 л воды (оставляя стоять на 7 часов). На выходные в реакторе создавали медленный ток азота, проходящий через твердое вещество, и выходящий из донного вентиля (через фильтр). Полученное твердое вещество растворяли в DCM (400 кг) и отделяли от остаточной воды. Водную фазу повторно экстрагировали DCM (50 кг). Объединенные органические материалы подвергали перегонке, чтобы удалить ~30 л растворителя (и азеотропно удалить остаточную воду), а затем фильтровали через слой магнезола (30 кг), а затем слой силикагеля (50 кг), промывая дополнительным количеством DCM (~600 кг). Фильтрат концентрировали в густую взвесь (объемом ~110 л), затем порциями прибавляли MTBE (65 кг), продолжая отгонку до конечной температуры паров 50°C (конечный объем 145 л). Взвесь охлаждали до 15°C, фильтровали и промывали с помощью MTBE (65 кг), что давало продукт (43,46 кг, 150 моль, 49%) в виде бледно-оранжевого твердого вещества. Частичное концентрирование фильтрата давало вторую порцию (2,65 кг, чистота ~93%, 8,5 моль, 3%). Данный фильтрат концентрировали досуха, затем распределяли между DCM (60 л) и 2,2 M NaOH (35 л). Органический слой промывали водой (2×30 л), затем рассолом (20 л) и фильтровали через слой силикагеля (35 кг), элюируя с помощью DCM. Фильтрат концентрировали и остаток затирали с MTBE (20 л) и фильтровали, что давало третью порцию (3,72 кг, 12,8 моль, 4%).
[0323] D. Получение соединения 11a примера 1
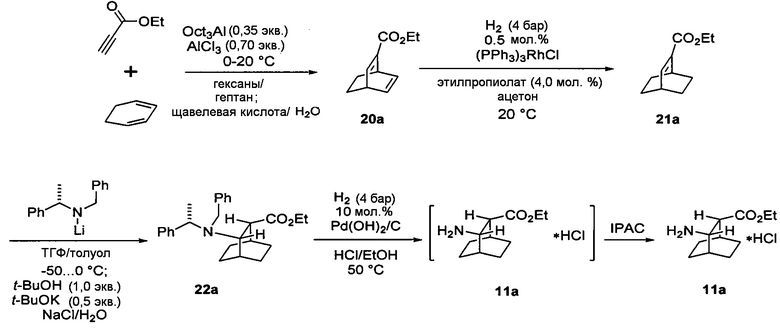
[0324] Получение соединения 20a: реакция Дильса-Альдера
[0325] AlCl3 (380,6 г, 2,85 моль, 0,7 эквив.) загружали в реактор на 10 л с донным дренажом и с рубашкой от Chemglass, который продували N2, после чего следовало прибавление гептана (1,6 л, 4 объема). Смесь охлаждали до 0°C. В течение 40 мин через капельную воронку прибавляли триоктилалюминий (2,99 л, 1,43 моль, 0,35 эквив., 25 масс. % в смеси гексанов). Бледно-зеленой легкой взвеси позволяли перемешиваться в течение 1 ч. В течение 1 ч прибавляли этилпропиолат (400 г, 413 мл, 1,0 эквив.). Температура в конце прибавления составляла 6,0°C. В течение 3 ч прибавляли 1,3-циклогексадиен (425 г, 494 мл, 1,3 эквив.). Реакцию оставляли перемешиваться в течение 16 ч. Внешний вид реакционной смеси изменялся с легкой взвеси оранжевого цвета на гомогенный оранжевый раствор. Реакцию охлаждали до 0°C. В атмосфере N2 в реактор на 30 л с донным дренажом и с рубашкой от Chemglass помещали 9% раствор щавелевой кислоты в воде и охлаждали до 0°C. Реакционную смесь переносили из реактора на 10 л в реактор для гашения на 30 л порциями в течение 1 ч. Реактор на 10 л промывали гептаном (800 мл, 2 объема), и раствор от промывки переносили в реактор для гашения на 30 л. Погашенную реакционную смесь нагревали до 22,5°C, перемешивая. Перемешивание прекращали и фазам давали возможность разделиться. Нижнюю водную фазу отводили. В реактор для гашения на 30 л загружали воду (800 мл, 2 объема) и перемешивали смесь в течение 30 мин. Перемешивание прекращали и фазам давали возможность разделиться. Нижнюю водную фазу отводили, а верхнюю органическую фазу концентрировали на роторном испарителе (температура бани составляла 40-50°C) вместе с раствором, полученном в другом эксперименте в тех же условиях и в том же масштабе. Масса концентрированного материала составляла 1771 г, и было обнаружено, что он содержит 83% продукта (остаток приходился на остаточный октан). Выход рассчитан на 101%. Чистота по ВЭЖХ составляла 99,39% AUC. 1H-ЯМР (400 МГц, CDCl3) δ 7,27 (дд, J=6,5, 1,8 Гц, 1H), 6,37 (ддд, J=7,5, 6,2, 1,5 Гц, 1H), 6,26 (ддд, J=7,3, 5,9, 1,5 Гц, 1H), 4,23-4,13 (м, 3H), 3,78-3,70 (м, 1H), 1,40-1,19 (м, 7H).
[0326] Получение соединения 21a
[0327] Катализатор Уилкинсона Rh(PPh3)3Cl (22,97 г, 25 ммоль, 0,005 эквив.) прибавляли к сырому соединению 20a (1068 г, 83 масс. %, 4,97 моль, 1 эквив.). Суспензию переносили в гидрогенизатор на 3 л от Buchi, температура рубашки которого была установлена на 20°C. Бутыль, которая содержала исходный материал, промывали ацетоном (885 мл, 1 объем) и смывы переносили в гидрогенизатор. Прибавляли этилпропиолат (19,49 г, 20,2 мл, 200 ммоль, 0,04 эквив.). Реакционную смесь перемешивали при давлении газообразного водорода 4 бар при 20°C в течение 17 ч. Наблюдалась конверсия 99,6% AUC в желаемое соединение 21a. Сырое соединение 20a в количестве 697 г подвергали реакции в тех же условиях и достигали практически такой же конверсии. Оба сырых раствора соединения 21a концентрировали на роторном испарителе при температуре в бане 40-50°C. Получали 1913 г сырого продукта. Анализ по массам с использованием очищенного стандарта давал по расчету значение активного содержания 70,9%: 1357 г активного продукта, выход 92% на две стадии. 1H-ЯМР (400 МГц, CDCl3) δ 7,34 (дд, J=6,9, 1,7 Гц, 1H), 4,20 (кв, J=7,1 Гц, 2H), 3,17 (м, 1H), 2,69-2,73 (м, 1H), 1,54-1,60 (м, 4H), 1,31 (т, J = 7,1 Гц, 3H), 1,23-1,27 (м, 4H).
[0328] Получение соединения 22a
[0329] S-(-)-N-Бензил-α-метилбензиламин (585 г, 579 мл, 2,77 моль, 1.1 эквив.) загружали в круглодонную колбу на 22 л, после чего прибавляли безводный ТГФ (5,1 л, 11 объемов). Начинали перемешивание и охлаждали колбу в бане с сухим льдом/ацетоном до 0°C. В течение 50 мин прибавляли н-BuLi (1,1 л, 2,77 моль, 1,1 эквив., 2,5 M раствор в смеси гексанов). Реакционную смесь перемешивали в течение 15 мин. Реакционную смесь охлаждали до -42,2°C в бане с сухим льдом/ацетоном в течение 10 мин. Соединение (R) (640 г, 70,9 масс. %, 454 г активного вещества, 2,52 моль, 1,0 эквив.) в толуоле (640 мл, 1,4 объема) прибавляли в течение 30 мин, поддерживая температуру внутри от -45°C до -40°C. Реакционную смесь перемешивали в течение 1 ч. В течение 20 мин прибавляли раствор t-BuOH (186 г, 240 мл, 2,52 моль, 1,0 эквив.) в безводном ТГФ (95 мл, 0,2 объема), зачем следовало прибавление раствора t-BuOK (1,26 л, 1,26 моль, 0,5 эквив., 1,0 M в ТГФ), который прибавляли в течение 20 мин, поддерживая температуру в ходе всего прибавления внутри от -45°C до -40°C. Реакционную смесь доводили до комнатной температуры и оставляли перемешиваться на 18 ч. Реакционную смесь охлаждали до 0°C и прибавляли раствор хлорида натрия (160 г) в воде (3,0 л). Смесь нагревали до 20°C и позволяли фазам разделиться. Нижний водный слой отводили. При перемешивании в реактор загружали воду (3 л). Температура повышалась от 21°C до 26°C. Смесь перемешивали в течение 30 мин, затем перемешивание прекращали и в течение 30 мин позволяли фазам осесть. Собирали верхнюю органическую фазу. Полученный в данном эксперименте раствор упаривали на роторном испарителе вместе с растворами двух других проведенных ранее экспериментов, которые осуществляли в тех же условиях и в похожем масштабе. Суммарная масса сырого продукта (желтое масло) составляла 3630 г. Материал использовали на следующей стадии. 1H-ЯМР (400 МГц, CDCl3) δ 7,49-7,41 (м, 2H), 7,41-7,14 (м, 8H), 4,27-4,18 (м, 1H), 4,09-3,93 (м, 2H), 3,89-3,81 (м, 1H), 3,48 (м, 2H), 2,52-2,45 (м, 1H), 2,08-1,95 (м, 2H), 1,76 (м, 1H), 1,70-1,21 (м, 7H), 1,44 (д, J=6,8 Гц, 3H), 1,19 (кв, J=7,2 Гц, 3H).
[0330] Получение соединения 11a
[0331] Катализатор Перлмана (Pd(OH)2/C, 20 масс. % на подложке, 50% воды, всего 500 г, 50 г активного компонента, 0,6 моль 0,095 эквив.) загружали в гидрогенизатор на 20 л от Buchi, температура рубашки которого была установлена на 20°C. Прибавляли соединение 22a (фактическая масса 1815 г, предполагаемая масса активного вещества 1468 г, 3,75 моль, 1,0 эквив.), после чего прибавляли EtOH (7,5 л, 5,1 объема в расчете на предполагаемую активную загрузку). В течение 20 мин прибавляли конц. HCl (37,7 масс. % в воде, 305 мл, 363 г, 137 г активного вещества, 3,75 моль, 1,0 эквив.). Значение pH реакционной среды измеряли бумагой для определения pH, и регистрировали, что оно составляло 1. Реакционную смесь гидрировали при давлении H2 1 бар и нагревали до 50°C. Как только достигалось значение 50°C, давление H2 в реакторе дополнительно увеличивали до 4 бар. Реакцию выдерживали в вышеописанных условиях в течение 96 ч. Катализатор отфильтровывали на целите, гидрогенизатор промывали EtOH (2 л) и использовали промывной раствор также для промывки слоя целита, и соединяли его с основным фильтратом. Этанольный раствор сырого продукта, соединения 11a, концентрировали на роторном испарителе вместе с продуктом предшествующего эксперимента, который был получен по существу в тех же условиях и том же масштабе. Получали густую пасту. В отгоночную колбу роторного испарителя, которая содержала продукт в предшествующей стадии, прибавляли изо-пропилацетат (2 л). Затем отгоночную колбу вращали при атмосферном давлении в N2 в течение 30 мин, чтобы суспендировать твердое вещество. Растворитель отгоняли в вакууме. Прибавляли другую порцию изо-пропилацетата (2 л) и отгоночную колбу вращали при атмосферном давлении в N2, чтобы суспендировать твердое вещество. Взвесь переносили в реактор с рубашкой на 30 л от Chemglass. Отгоночную колбу промывали изо-пропилацетатом (2 л) и прибавляли в реактор промывочный раствор. Прибавляли изо-пропилацетат (12 л, всего 16 л, 5,5 объемов в расчете на предполагаемое количество активного соединения 11a). Желтую суспензию перемешивали в течение 20 ч при комнатной температуре 24,4°C. Взвесь фильтровали на фарфоровом фильтре с фильтровальной бумагой. Реактор промывали изо-пропилацетатом (4 л) и использовали промывочный раствор для промывки отфильтрованной массы. Получали белую, плотную отфильтрованную массу. Ее оставляли на несколько часов под втягивающим воздействием вакуума. Массу сушили в вакуумной печи при 40°C в токе N2 в течение 20 ч. Количество полученного продукта составляло 924 г (3,95 моль, выход 48% в расчете на этилпропиолат). Чистота (GC) составляла 98,31% AUC. 1H-ЯМР (400 МГц, MeOH-d4) δ 4,28-4,14 (м, 2H), 3,87-3,79 (м, 1H), 2,63-2,56 (м, 1H), 2,07 (дд, J=5,4, 2,7 Гц, 1H), 1,95-1,86 (м, 1H), 1,86-1,72 (м, 2H), 1,73-1,53 (м, 4H), 1,54-1,37 (м, 2H), 1,28 (т, J=7,1 Гц, 3H). 13C-ЯМР (101 МГц, MeOH-d4) δ 174,03, 62,37, 52,00, 48,89, 29,58, 28,96, 25,94, 25,05, 21,48, 19,28, 14,56.
[0332] F. Скриниг условий реакции Сузуки для получения соединения (Z-2)
[0333] Осуществляли тринадцать экспериментов по получению соединения (Z-2) массой 1 г, используя для этого различные каталитические системы, включая применение сочетания Pd(OAc)2/X-Phos в целях сопоставления. Оказалось, что сочетание Pd(OAc)2/X-Phos давало превосходные результаты в сравнении с другими катализаторами.
[0334] В реакционный флакон объемом 40 мл, снабженный накручивающейся крышкой-септой, загружали соединение (X-2) (1,0 г, 3,3 ммоль, 1,0 эквив.), соединение (Y-2) (1,7 г, 4,2 ммоль, 1,25 эквив.), катализатор (0,25 мол. %) и карбонат калия (1,8 г, 13,3 ммоль, 4,0 экв.), за чем следовало добавление ТГФ (8 мл, 8 объемов). После начала перемешивания густую взвесь дегазировали с помощью 3 циклов откачивания вакуумом/заполнения азотом и нагревали до 60-65°C. Как только достигалась желаемая температура, в течение периода 15 минут прибавляли дегазированную воду (0,32 мл, 0,3 объема). После завершения прибавления воды реакционной смеси давали перемешиваться при установленной температуре и в различные временные точки отбирали образцы для анализа ВЭЖХ (Таблица 1).
[0335] Таблица 1: Скрининг катализаторов для получения соединения (Z-2)
[0336] Пример 3: Формирование полиморфов солей соединения (1) с HCl
[0337] 3A: Получение формы A соли соединения (1) с HCl на 1/2H2O
[0338] Форму A соли соединения (1) с HCl на 1/2H2O получали смешением 2-метилтетрагидрофуранового (2-Me-ТГФ) сольвата (1 эквивалент) соединения (1) (соединение (1)⋅1 (2-Me-ТГФ)) с хлоридом водорода в смеси воды и органического(их) растворителя(ей), где смесь воды и органического(их) растворителя(ей) имела активность воды 0,05-0,85. Конкретные использованные условия реакций приведены ниже в Таблице 2.
[0339] Таблица 2: Условия реакций, использованные для получения формы A соли соединения (1) с HCl на 1/2H2O
(2-Me-ТГФ)
*две стадии: iPrOH/AcOH, а затем повторное суспендирование в смеси ацетон/вода
[0340] Альтернативно, форму A соли соединения (1) с на 1/2H2O также получали по следующим методикам: Методика A: Соединение (1)⋅2-Me-ТГФ (953 г, 2,39 моль) помещали в реактор с рубашкой на 30 л и обрабатывали IPA (15 л) и водой (0,57 л). Запускали мешалку и нагревали реакционную смесь до 73°C, что перевести все содержимое в раствор, затем охлаждали до 50-55°C. При 50-55°C реакционную смесь обрабатывали свежеприготовленным раствором HCl в IPA (0,83 M, 4,34 л) путем медленного прибавления в течение 4 ч. Из реакции отбирали образец для проверки методом XRPD на присутствие нужной формы. После прибавления холодильник программировали на линейное охлаждение до 0°C в течение 480 мин при перемешивании. После подтверждения формы посредством анализа XRPD взвесь фильтровали на двух фильтрах. Реактор промывали 3 л IPA и каждую отфильтрованную массу промывали ~1,5 л IPA, представляющего собой IPA от промывки реактора. Отфильтрованным массам давали возможность высохнуть на воздухе в течение ночи, применяя отсос. Затем отфильтрованные массы помещали в сушилку с поддонами, выдерживая в ней в вакууме с продувкой N2 (22 дюйма Hg (560 мм рт. ст.).) в течение 24 ч, не используя нагрев. Анализ на остатки растворителя и воды показал присутствие 505 м.д. IPA, 8 м.д. 2-Me-ТГФ и приблизительно 2,15% H2O. Материал извлекали из печи и перемалывали для удаления комков, что давало 805 г соли соединения (2) с HCl на 1/2H2O. Методика B: В альтернативном варианте вместо IPA использовали ацетон, но аналогично описанному выше для методики A, получая соль соединения (1) с HCl на 1/2H2O.
[0341] Некоторые наблюденные пики XRPD и пики 13C-SSNMR приведены в Таблицах 3A и 3B соответственно.
[0342] Таблица 3A: Пики XRPD для формы A соли соединения (1) с HCl на 1/2H2O
[0343] Таблица 3B: Пики 13C-SSNMR для формы A соли соединения (1) с HCl на 1/2H2O
[± 3 м.д.]
[0344] Было обнаружено, что полученная форма A соли соединения (1) с HCl на 1/2H2O является стабильной в следующих системах растворителя (но без ограничения ими): хлорбензол, циклогексан, 1,2-дихлорэтан, дихлорметан, 1,2-диметоксиэтан, гексан, 2-метоксиэтанол, метилбутилкетон, метилциклогексан, нитрометан, тетралин, ксилол, толуол, 1,1,2-трихлорэтан, ацетон, анизол, 1-бутанол, 2-бутанол, бутилацетат, трет-бутилметиловый простой эфир, кумол, этанол, этилацетат, этиловый простой эфир, этилформиат, гептан, изобутилацетат, изопропилацетат, метилацетат, 3-метил-1-бутанол, метилэтилкетон, 2-метил-1-пропанол, пентан, 1-пропанол, 1-пентанол, 2-пропанол, пропилацетат, тетрагидрофуран, метилтетрагидрофуран. А именно, для испытаний на растворимость и стабильность формы A соли соединения (1) с HCl на 1/2H2O образцы соединения загружали во флаконы для ВЭЖХ на 2 мл с 500 мкл растворителя. Смесь перемешивали при температуре окружающей среды в течение 2 недель, а затем фильтровали посредством центрифуги. Полученные твердые вещества анализировали посредством XRPD, растворы анализировали на растворимость посредством количественного ЯМР относительно гидрохинонового стандарта. Результаты сведены в Таблице 4.
[0345] Таблица 4: Сводка данных по растворимости формы A соли соединения (1) с HCl и формам, образующимся в растворителях
[0346] Данные термограммы (данные не показаны) получали, помещая образец в платиновый тигель для образцов и нагревая со скоростью 10°C/мин от комнатной температуры до 300°C. Данные термограммы демонстрировали потерю массы, составляющую 2,1% в диапазоне от 30°C до 170°C, что согласовывалось с теоретическим значением для гемигидрата (2,0%).
[0347] Данные термограммы ДСК (данные не показаны) получали, нагревая образец со скоростью 10°C/мин от комнатной температуры до 300°C. Термограмма ДСК показывала температуру начала дегидратации при 50-100°C, после чего следовала температура начала плавления/разложения при 200-260°C.
[0348] 3B: Получение формы F соли соединения (1) с HCl на 3H2O
[0349] Форма F соли соединения (1) с HCl на 3H2O может быть получена путем суспендирования формы A соли соединения (1) с HCl на 1/2H2O в изопропаноле и воде, либо ацетоне и воде, либо воде (где значение активности воды равно, либо превышает 0,9).
[0350] Например, в течение ночи при температуре окружающей среды перемешивали взвесь 100 мг формы A соли соединения (1) с HCl на 1/2H2O в 5 мл смеси изопропанол/вода или ацетон/вода с активностью воды 0,9. Декантация надосадочной жидкости и осторожная сушка сухим воздухом полученного твердого материала давала форму F соли соединения (1) с HCl на 3H2O.
[0351] Некоторые наблюденные пики XRPD и пики 13C-SSNMR приведены в Таблицах 5 и 6 соответственно.
[0352] Таблица 5: Пики XRPD для формы F соли соединения (1) с HCl на 3H2O
[± 3 м.д.]
[0354] Термограмму MDSC (данные не показаны) получали, нагревая образец со скоростью 2°C/мин от -20°C до 350°C и с модуляцией при ±1°C каждые 60 сек. Термограмма MDSC показывала дегидратацию ниже 150°C, плавление и перекристаллизацию между 150°C и 200°C и разложение выше 250°C.
[0355] Также проводили термогравиметрический анализ (ТГА) данной формы. Термограмма показала потерю массы, составляющую 12%, вплоть до 125°C, что было близко к теоретическому значению для тригидрата (11%). По данным ТГА-МС потеря массы на второй ступени ниже 200°C представляла собой потерю HCl. Начало плавления/разложения находилось при 270-290°C.
[0356] 3C: Получение формы D соли соединения ( 1 ) с HCl
[0357] Безводную форму D соли соединения ( 1 ) с HCl можно, в общем, получить дегидратацией формы A соли соединения ( 1 ) с HCl на 1/2H2O. Дегидратация могла бы быть произведена путем нагрева или продувки с использованием сухота азота, либо применяя сочетание двух данных подходов. Например, на горячей плитке нагревали 2 мг формы A соли соединения ( 1 ) с HCl на 1/2H2O, получая желаемую безводную форму D приблизительно при 85°C.
[0358] Некоторые наблюденные пики XRPD и пики 13C-SSNMR приведены в Таблицах 7 и 8 соответственно.
[0359] Таблица 7: Пики XRPD для формы D безводной соли соединения (1) с HCl
[0360] Таблица 8: Пики 13C-SSNMR для формы D безводной соли соединения (1) с HCl
[± 3 м.д.]
[0361] 3D: Испытания по определению влияния активности воды
[0362] Конкурентное исследование в среде взвеси для формы A соли соединения (1) с HCl на 1/2H2O, содержащей зерна формы F соли соединения (1) с HCl на 3H2O, при значениях активности воды от 0,0 до 0,8 в смеси изопропиловый спирт/вода показало, что после перемешивания приблизительно в течение 2 недель в условиях окружающей среды форма A является наиболее стабильной формой среди формы D безводной соли соединения (1) с HCl, формы F соли соединения (1) с HCl на 3H2O формы A соли соединения (1) с HCl на 1/2H2O. При активности 0,9 в смеси IPA/вода форма A соли соединения (1) с HCl на 1/2H2O превращалась в форму F соли соединения (1) с HCl на 3H2O. Результаты данных исследований сведены в приведенной ниже Таблице 9.
[0363] Таблица 9: Испытания по влиянию активности воды на соль соединения (1) с HCl на 1/2H2O в смесях IPA/вода
[0364] 3F: Аморфная соль соединения (1) с HCl
[0365] Аморфную соль соединения ( 1 ) с HCl можно было бы сформировать обработкой соли соединения ( 1 ) с Me2NEt (1,985 г) в воде и 2-Me-ТГФ с помощью 1,05 экв. NaOH с последующей обработкой с помощью HCl для удаления амина и извлечения из водного слоя (pH 2-3). Полученную взвесь концентрировали, чтобы удалить любые органические материалы, а затем фильтровали. Полученное твердое вещество промывали небольшими количествами воды и сушили. Соль соединения (1) с Me2NEt получали согласно WO 2010/148197 с последующими общепринятыми хиральным разделением и очисткой: хиральная хроматография SFC с модификатором, который включал Me2NEt (что давало соль соединения (1) с Me2NEt).
[0366] Пример 4: Формирование полиморфов свободного основания соединения (1)
[0367] 4A: Получение формы A свободного основания соединения ( 1 )
[0368] Форму A свободного основания соединения ( 1 ) получали по следующей методике: Сырое аморфное свободное основание соединения ( 1 ) (приблизительно 135 г) переносили в реактор с рубашкой на 4 л и загружали в реактор этанол (2,67 л) и воду (0,325 л) (раствор с 10% воды). Смесь нагревали до кипения. К полученной смеси стадии 2) прибавляли воду (300 мл), чтобы получить раствор с 20% воды. Затем полученную смесь охлаждали до 55°C (со скоростью -1°C/мин) и впоследствии выдерживали в течение 30 минут. Затем в охлажденную смесь вносили кристаллическую затравку формы A свободного основания соединения (1) (1,5 г, 3,756 ммоль) и полученную смесь выдерживали в течение 30 минут, во время чего осаждался продукт. Затравку формы A кристаллического свободного основания соединения (1) получали суспендированием аморфного свободного основания соединения (1) (20 мг) в нитрометане (0,5 мл). Дополнительные затравочные материалы формы A кристаллического свободного основания соединения (1) получали суспендированием аморфного свободного основания соединения (1) (900 мг) в ацетонитриле (10 мл) с затравкой, полученной с использованием нитрометана. В смесь, содержащую затравку формы A кристаллического свободного основания соединения (1) медленно прибавляли воду (795,0 мл), чтобы получить раствор с 40% воды. Полученную смесь медленно охлаждали до 0°C (~ -10°C/час) и впоследствии выдерживали в течение 2 часов. Затем твердый материал фильтровали и сушили на воздухе, а затем дополнительно сушили в печи при 60°C в течение 18 часов.
[0369] В альтернативном варианте вместо аморфного свободного основания соединения ( 1 ) использовали сольват свободного основания ( 1 ) с 2-метил-ТГФ, и форму A свободного основания соединения ( 1 ) также получали способом, аналогичным описанному выше.
[0370] Было обнаружено, что полученная форма A соединения (1) является стабильной в следующих системах растворителя (но без ограничения ими): ацетонитрил, хлорбензол, хлороформ, циклогексан, 1,2-дихлорэтан, дихлорметан, 1,2-диметоксиэтан, этиленгликоль, формамид, гексан, метилбутилкетон, метилциклогексан, N-метилпирролидинон, нитрометан, тетралин, толуол, 1,1,2-трихлорэтан, уксусная кислота, анизол, 1-бутанол, бутилацетат, кумол, этилацетат, этиловый простой эфир, этилформиат, гептан, изобутилацетат, изопропилацетат, 3-метил-1-бутанол, 2-метил-1-пропанол, пентан, пропилацетат, вода, вода-изопропанол (1:3 объем/объем) и вода-ацетонитрил (1:1 объем/объем; 1:3 объем/объем).
[0371] Некоторые наблюденные пики XRPD и пики 13C-SSNMR приведены в Таблицах 10 и 11 соответственно.
[0372] Таблица 10: Пики XRPD для формы A соединения ( 1 )
[0373] Таблица 11: Пики 13C-SSNMR для формы A соединения (1)
[± 3 м.д.]
[0374] Термогравиметрический анализ продукта, формы A соединения (1), проводили (данные здесь не показаны) на приборе от TA Instruments TGA, модель Q500, помещая образец продукта в платиновый тигель для образцов и впоследствии нагревая тигель со скоростью 10°C/мин от комнатной температуры до 300°C. Термограмма демонстрировала начало разложения около 293°C.
[0375] Используя прибор TA Instruments DSC Q200A, также получали термограмму ДСК для формы A соединения (1). Образец формы нагревали со скоростью 10°C/мин до 350°C. Термограмма ДСК показывала, что температура плавления находилась около 278°C.
[0376] 4B: Получение формы B гидратов свободного основания соединения ( 1 )
[0377] Гидратированная форма свободного основания соединения (1) была изоморфна форме A свободного основания соединения (1): форма A свободного основания соединения (1) могла легко превращаться в гидратированную форму B, когда она подвергалась воздействию высокой влажности, и возвращаться в исходное состояние, когда влажность уменьшали. В соответствии с данными о фазовых переходах, определенными используя эксперименты ДСК (данные не показаны), температура перехода была близка к температуре окружающей среды и изменялась в зависимости от активности воды. Например, при температуре окружающей среды, гидратная форма наблюдалась, когда активность воды превышала 0,6, составляя, например, 0,6-1,0.
[0378] 4C: Получение аморфного свободного основания соединения (1)
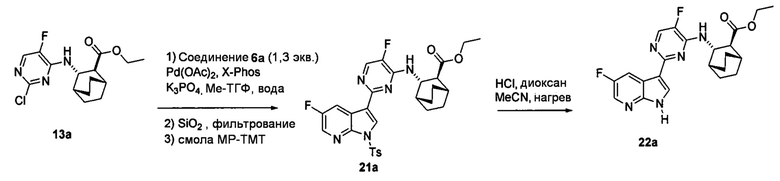
[0379] Сочетание по Сузуки проводили беря хлорпиримидин, то есть соединение 13a, сложный эфир бороновой кислоты, то есть соединение 6a, Pd(OAc)2 в качестве катализатора и лиганд (X-Phos) в 10 объемах 2-Me-ТГФ. Данную смесь нагревали до 65°C и прибавляли 2 объема 50% водного раствора K3PO4 с такой скоростью, чтобы поддерживать температуру реакционной смеси при 65°C. Обе реакции достигали полной конверсии, после чего их охлаждали до 20°C и фильтровали через целит. Водные слои отделяли и отбрасывали, органические слои промывали 5% водным раствором NaCl, а затем концентрировали досуха, что давало приблизительно 3,5 кг темно-зеленой пасты в каждом эксперименте. Сырое масло делили на 4 равные порции, суспендировали с 400 г SiO2 и 500 г Florisil и элюировали через колонку с 2,3 кг SiO2 смесью гептан/EtOAc (от 5:1 до 3:1, фракции объемом 2 л), объединяя все фракции, содержащие продукт. Данные фракции концентрировали досуха, что давало приблизительно 2,9 кг соединения 21a.
[0380] Соединение 21a растворяли в 10 объемах (25 л) CH3CN и обрабатывали 4 экв. HCl (4,31 л 4 н. HCl в 1,4-диоксане) при 70°C в течение 15 ч. О завершении реакции на 100% судили по ВЭЖХ и охлаждали жидкую взвесь до 20°C в течение 1 ч. Прибавляли TBME (28 л, 11 объемов) со скоростью 0,5 л/мин, причем в конце прибавления взвесь становилась очень густой (желеподобной). После 4-5 ч перемешивания взвесь становилась более жидкой. Полученное твердое вещество отделяли фильтрованием с отсосом и промывали TBME 3 × 5 л, что давало фильтрованную массу низкой плотности и сушили в токе N2 в течение 3 дней, что давало 1,71 кг (выход 86%, чистота AUC 98,9%) соединения 22a⋅HCl.
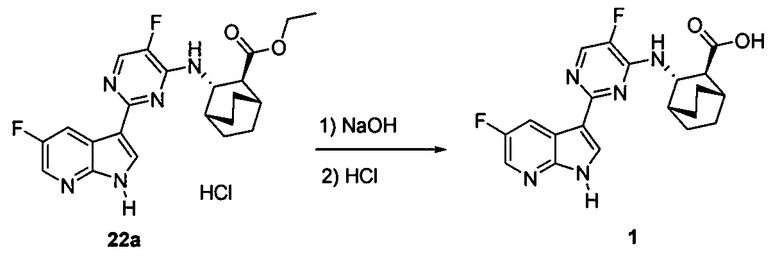
[0381] Раствор NaOH (55,60 мл с концентрацией 2 M, 111,2 ммоль) прибавляли к суспензии соединения 22a⋅HCl (10 г, 22,23 ммоль) в 2-Me-ТГФ (100,00 мл) при 20°C. Реакционную смесь перемешивали при 60°C в течение 5 ч, а затем дополнительно при 67°C. После перемешивания приблизительно в течение 22 часов к полученной смеси прибавляли 100 мл (10 объемов) 2-Me-ТГФ. Затем массу охлаждали до 0°C. К полученной смеси прибавляли HCl, чтобы отрегулировать pH до значения pH 6,6, получая сырое свободное основание соединения (1). Сырой материал нагревали до 50°C в 60 мл (6 объемов) 2-Me-ТГФ. В течение 1 часа в полученную смесь добавляли 50 мл (5 объемов) н-гептана. Затем массу охлаждали до 20°C. Твердый продукт фильтровали и дополнительно очищали колоночной хроматографией (EtOAc/гептан от 2:1 до 4:1). Данные XRPD для него указывали на аморфное свободное основание (1).
[0382] В альтернативном варианте образование аморфного свободного основания соединения (1) наблюдалось из смеси формы A свободного основания соединения (1) и растворителя, выбранного из 2-этоксиэтанола, 2-метоксиэтанола, трет-бутилметилового простого эфира, муравьиной кислоты или метилэтилкетона (например, смотри ниже Таблицу 13), которую перемешивали при температуре окружающей среды.
[0383] 4D: Получение сольвата свободного основания соединения (1) с 2-Me-ТГФ
[0384] Соединение (1)⋅1(2-Me-ТГФ) получали, как описано выше в примере 2. Некоторые наблюденные пики XRPD для соединения приведены в Таблице 12.
[0386] 4F: Данные по растворимости и стабильности для формы A свободного основания соединения (1) и аморфного соединения (1) в различных системах растворителя
[0387] Растворимость и стабильность формы A свободного основания соединения (1) (“форма A”) и аморфного соединения (1) (“аморфное”) в различных системах растворителя испытывали при температуре окружающей среды аналогично тому, как описано выше для испытаний, проведенных для формы A соли соединения (1) с HCl. Полученные данные сведены в Таблице 13.
[0388] Таблица 13: Данные по растворимости и стабильности формы A свободного основания соединения (1) (“форма A”) и аморфного соединения (1) (“аморфное”)
[0389] Пример 5: Получение формы A тозилатной соли соединения (1)
[0390] Форму A тозилатной соли соединения (1) получали суспендированием аморфного свободного основания соединения (1) (500 мг) и п-толуолсульфоновой кислоты в ацетонитриле (20 мл). Образцы перемешивали в течение ночи. Некоторые наблюденные пики XRPD для соединения приведены в Таблице 14.
[0391] В альтернативном варианте, чтобы получить форму A тозилата соединения (1) способом, аналогичным описанному выше, вместо аморфного свободного основания соединения (1) можно было использовать сольват свободного основания (1) с 2-метил-ТГФ.
[0392] Таблица 14: Пики XRPD для формы A тозилатной соли соединения (1)
[0393] Пример 6: Рецептуры соединения (1)
[0394] A. Таблетки соединения (1)
[0395] Композиции
[0396] Для формирования таблеток использовали форму A соли соединения (1) с HCl на 1/2H2O (здесь далее упоминаемую просто как соединение (1) в контексте примера 6). Все формообразующие отвечали требованиям текущих статей Европейской фармакопеи European и Фармакопеи США USP/NF и приобретались у одобренных поставщиков.
[0397] Композиция рецептуры и размер загрузки для смеси перед гранулированием и связующий раствор для гранулирования приведены в Таблице 15A. Размер загрузки связующего раствора включал 100% избыток для настройки насоса и заливки линий подачи раствора. Теоретическая прессованная композиция смеси также приведена в Таблице 15B. Фактические количества для загрузки рассчитывали, основываясь на выходе высушенных гранул. Композиция и приблизительный размер загрузки суспензии для нанесения пленочного покрытия приведены в Таблице 15B и включали 100% избыток для настройки насоса и заливки линий подачи суспензии. Целевое количество пленочного покрытия составляло 3,0% масс./масс. массы таблетки.
[0398] Таблица 15A: Композиции таблеток соединения (1)
[0399] Таблица 15B: Композиция суспензии для нанесения пленочного покрытия и приблизительный размер загрузки
[0400] Приготовление связующего раствора
[0401] Связующий раствор состоял из повидона и воды. Раствор готовили, чтобы получить в окончательном материале гранулирования содержание воды, составляющее 40%. Таким образом, суммарное количество твердых веществ в растворе (повидон) составляло 3,6% (масс./масс.). Избыточное количество, равное 100%, готовили для заливки линий и так далее. Основываясь на визуальном наблюдении за началом процесса гранулирования, для окончательного материала гранулировании готовили дополнительные стоковые растворы с содержанием воды 38-42% +/- 2%. В типичном случае отвешивали 87,00 г повидона K30 и 2320,00 г очищенной (деионизированной, DI) воды, и при постоянном перемешивании прибавляли повидон K30 в контейнер, содержащий DI-воду. После прибавления контейнер герметично закрывали, чтобы свести к минимуму испарение, а раствор перемешивали до полного растворения всего присутствующего твердого вещества.
[0402] Технологическая схема влажного гранулирования
[0403] Влажное гранулирование осуществляли по нижеописанным методикам: Отвешивали избыточное (10%) количество соединения (1), Avicel PH-101, лактозу Fastflo и кросскармеллозу натрия (смотри Таблицу 15A). Их просеивали, используя ручное сито 20 меш или конусную мельницу, оборудованную решетчатым ситом для просеивания с отверстиями 813 мкм, при скорости вращения 1000 об/мин (для мельницы U5 Quadro Co-mill). Просеянные материалы помещали в индивидуальные мешки или контейнеры. Затем материалы переносили в смеситель и перемешивали в течение 15 минут типично со скоростью 15 об/мин. Смешанные материалы перемалывали, используя конусную мельницу U5 Quadro, оборудованную ситом с квадратными отверстиями 4 мм, при 1000 об/мин. Молотые материалы снова перемешивали, повторяя стадию смешения. Затем повторно смешанные материалы подавали в двухшнековый гранулятор. Массу влажного материала для гранулирования подавали в гранулятор, используя дозирующий питатель непрерывного действия (K-tron или подобный ему). Затем полученные материалы гранулировали. Связующий раствор (смотри Таблицу 15A) впрыскивали в двухшнековый гранулятор, используя перистальтический насос. Отношение скорости подачи раствора к скорости подачи порошка составляло 0,4095. Например, если скорость подачи порошка составляла 15,00 г/мин, скорость подачи раствора составляла 0,4095*15,00=6,14 г/мин при содержании воды 40% (в расчете на сухую массу). Гранулированные предварительные партии собирали в предварительно тарированные сушильные поддоны. Собранные материалы равномерно распределяли по поддону, и сушили материал в печи, формируя высушенные гранулы. Высушенные гранулы помещали в аппарат K-tron для их непрерывной подачи в конусную мельницу и впоследствии перемалывали.
[0404] Внегранульное смешение и процесс прессования
[0405] Внегранульное смешение и процесс прессования осуществляли по нижеописанным методикам: Отвешивали количество внегранульных формообразующих, основываясь на композиции прессованной смеси. Отвешенные формообразующие просеивали, используя U5 Comil с ситом 32C и лопасть с круглым стержнем, при 1000 об/мин. Сначала в смеситель, содержащий просеянные Avicel PH-102 и Ac-Di-Sol, вносили молотые гранулы соединения (1). Компоненты смешивали в течение 8 минут при 16 об/мин. В подходящий контейнер просеивали стеарил натрия (SSF) через ручное сито 50 меш. Часть внегранульной смеси, масса которой была приблизительно равна 10-кратной массе количества SSF, помещали в контейнер с SSF и смешивали в течение 30 секунд перед внесением смеси в бункерный смеситель. Затем все материалы перемешивали в течение 2 минут при 16 об/мин. Затем конечную смесь прессовали в соответствии с предписанными параметрами процесса прессования в таблетки.
[0406] Процесс нанесения пленочного покрытия
[0407] Пленочное покрытие наносили на сердцевины таблеток в дражировочном аппарате Vector VPC 1355 в виде водной суспензии 15% масс./масс. Opadry II white 33G. Целевое количество покрытия составляло 3,0% масс./масс. от массы сердцевины таблетки с приемлемым диапазоном отклонения от 2,5% до 3,5%. Для осуществления нанесения распыляли количество кроющей суспензии, эквивалентное приросту массы на 3,2%, что давало 3,0% покрытия, если принять эффективность нанесения покрытия равной 95%.
[0408] Внутривенные (IV) рецептуры соединения (1)
[0409] Форму A соли соединения (1) с HCl на 1/2H2O (здесь далее упоминаемую просто как соединение (1) в контексте примера 6) предоставляли в виде раствора с концентрацией 2 мг/мл для внутривенного (IV) введения. Композиция раствора наряду с указанием качества и функцией каждого компонента представлены в Таблицах 16A и 16B.
[0410] Таблица 16A: Композиция наполнителя для раствораa
a pH раствора регулировали NaOH или HCl
a pH раствора регулировали NaOH или HCl. Плотность раствора составляет 1,000 г/см3.
b Лекарственная субстанция представляла собой гемигидрат соли с HCl. Количество лекарственной субстанции рассчитывали, основываясь на эквиваленте активного безводного свободного основания, где фактор перевода из свободного основания в гемигидрат соли с HCl равен 1,11.
[0412] Пример 7: Исследование In Vivo сочетания соединения (1) с осельтамивиром или без него
[0413] Инфицированным мышам вводили наполнитель или возрастающие уровни доз формы A соли соединения (1) с HCl на 1/2H2O в сочетании с клинически обусловленной дозой осельтамивира, начиная спустя 48 часов после провокации гриппа A или за 2 часа до провокации гриппа B.
[0414] Методы: В данных исследованиях из формы A гемигидрата соли соединения (1) с HCl (здесь далее упоминаемой просто как соединение (1) в контексте примера 7) составляли рецептуру в наполнителе, содержащем 0,5% (масс./об.) MC (Sigma-Aldrich, Сент-Луис, Миссури), что давало гомогенную суспензию, и при расчете дозы соединения учитывали, что использовался гемигидрат соли соединения (1) с HCl. Рецептуру с осельтамивиром составляли в дистиллированной деионизированной воде, что давало гомогенную суспензию. Из сочетания соединения (1) с осельтамивиром составляли рецептуру в наполнителе, содержащем 0,5% (масс./об.) MC. Комбинированные рецептуры готовили в начале каждого исследования и хранили при 4°C в течение не более 10 суток в темноте, перемешивая. Все рецептуры и наполнители вводили мышам через пероральный зонд в дозировочном объеме 10 мл/кг.
[0415] Самцов мышей Balb/c (5-7 недель, 17-19 грамм) анестезировали и инокулировали летальной дозой адаптированного к мышам вируса гриппа A/PR/8/34 или B/Mass/3/66 путем интраназальной инстилляции. В каждую исследуемую группу входило восемь мышей. Лечение начинали через 48 часов после инокуляции гриппом A или за 2 часа до инокуляции гриппом B. Наполнитель (10 мл/кг) и соединение (1) в дозах 0,1-10 мг/кг вводили в виде монотерапии или в сочетании с 10 мг/кг осельтамивира, перорально (PO) дважды в сутки (BID) в течение 10 суток в случае исследования с гриппом A. Наполнитель (10 мл/кг) и соединение (1) в дозах 1-10 мг/кг вводили в виде монотерапии или в сочетании с 10 мг/кг осельтамивира, перорально (PO) дважды в сутки (BID) в течение 10 суток в случае исследования с гриппом B. На протяжении 21 дня после инфицирования мышей ежедневно взвешивали и обследовали для выявления признаков заболевания. Дополнительно отслеживали функцию легких, используя неограниченную плетизмографию всего тела (WBP) (Buxco, Трой, Нью-Йорк).
[0416] Штаммы гриппа A/PR/8/34 (VR-1469) и гриппа B/Mass/3/66 (VR-523) получали от ATCC (Манассас, Виргиния). Вирусный исходный материал готовили стандартными методами, известными в данной области. Вкратце, вирус подвергали пассажу с низкой множественностью инфекции в клетках Мадин-Дарби почек собак (клетки MDCK cells, CCL-34, ATCC), надосадочную жидкость отбирали приблизительно спустя 48 часов и центрифугировали при 650×g в течение 10 минут. Вирусный исходный материал замораживали при -80°C до момента использования. Вирусные титры (TCID50/мл) рассчитывали методом Spearman-Karger (Спирмана-Каргера) после серийного разведения образца вируса, инфицирования реплицирующихся культур MDCK и измерения цитопатического эффекта (CPE), основываясь на содержании АТФ в момент 96 часов (CellTiter-Glo, Promega, Мэдисон, Висконсин).
[0417] На протяжении 21 дня после инфицирования мышей ежедневно взвешивали. Данные по массе тела анализировали для сравнения групп, используя двухфакторный анализ ANOVA и тест Бонферрони. Значения, для которых P меньшие 0,05, рассматривали как значимые.
[0418] На протяжении 21 дня после инфицирования мышей ежедневно осматривали. Любую мышь, которая набирала положительные баллы по четырем из следующих шести признаков (потеря массы тела (BW) >35%, взъерошенная шерсть, сгорбленная поза, затруднение дыхания, сниженная подвижность или гипотермия), считали умирающей, затем подвергали эвтаназии и засчитывали как смерть согласно рекомендациям, разработанным Vertex Institutional Animal Care and Use Committee. Данные по выживаемости анализировали, используя метод Каплана-Мейера (Kaplan Meier).
[0419] Мышей подвергали неограниченной плетизмографии всего тела (WBP) (Buxco, Трой, Нью-Йорк). Функцию легких выражают как увеличенную паузу (Penh), безразмерное расчетное значение, которое отражает легочное сопротивление. Данное значение получают из изменений давления в камере с животным, которое изменяется вследствие изменений характера дыхания животного. Сужение бронхов в дыхательных путях животного будет влиять на поток воздуха и, следовательно, давление в камере с животным. Изменения давления отслеживают при выдохе (PEP) и вдохе (PIP). Значения Penh рассчитывали по формуле Penh = пауза × PEP/PIP, где “пауза” отражает длительность выдоха. Мышей акклиматизировали в камере плетизмографа в течение 15 минут, затем собирали данные с интервалами в одну минуту, усредняли по 10 минутам и выражали как абсолютные значения Penh. Данные анализировали для сравнения групп, используя двухфакторный анализ ANOVA и тест Бонферрони. Значения, для которых P меньшие 0,05, рассматривали как значимые.
[0420] Результаты: Соединение (1) оценивали в сочетании с осельтамивиром на способность соединения предотвращать смертность и заболеваемость, потерю массы тела и предотвращать ухудшение функции легких и/или восстанавливать функцию легких в мышиной модели гриппозной легочной инфекции в сравнении монотерапией соединением (1) или осельтамивиром. Сочетание не показало отрицательного воздействия на эффективность каждого из лекарственных средств в сравнении с каждым лекарственным средством, введенным в одиночку. Кроме того, комбинированное лечение показало синергизм при лечении гриппа A, поскольку недостаточная доза для каждого соединения при применении в одиночку (0,3 и 10 мг/кг для соединения (1) и осельтамивира соответственно) при комбинированном использовании увеличивала выживаемость от 0 до 100 процентов. Соединение (1) обладает малой активностью по отношению к гриппу B in vivo (как ожидалось из доступных данных исследований in vitro) и не уменьшает эффективность осельтамивира.
[0421] Мышиная модель гриппа A: Все леченные наполнителем контроли пали жертвой заболевания к 9 или 10 дню. Лечение в режиме BID (дважды в сутки) в дозах 1, 3 и 10 мг/кг лишь соединением (1) обеспечивало полную защиту от смерти, уменьшало потерю массы тела (BW) и восстанавливало функцию легких, когда введение доз начиналось через 48 часов после инфицирования, в сравнении с контролями, получавшими наполнитель (Таблица 17). Лечение соединением (1) в дозе 0,1 и 0,3 мг/кг и осельтамивиром в дозе 10 мг/кг в режиме монотерапии не защищало от смерти, не уменьшало потерю BW или не восстанавливало функцию легких, когда лечение начиналось через 48 часов после инфицирования гриппом A. Интересно, что 0,3 мг/кг соединения (1) и осельтамивир, введенные совместно через 48 часов после инфицирования гриппом A, обеспечивали полную защиту от смерти, уменьшали потерю BW и восстанавливали функцию легких.
[0422] Таблица 17: Данные по эффективности in vivo соединения (1) с осельтамивиром или без него при введении через 48 часов после инфицирования гриппом A
(3 сутки)
(3 сутки)
[0423] Мышиная модель гриппа B: Все леченные наполнителем контроли пали жертвой заболевания к 7 или 8 дню. Введение только соединения (1) в дозах 1, 3 или 10 мг/кг за 2 ч до инфицирования гриппом B и продолженное в режиме BID в течение 10 суток не обеспечивало значимой защиты от заболеваемости, потери BW или потери функции легких в сравнении с контролями. Осельтамивир, введенный в дозе 10 мг/кг в виде монотерапии или в сочетании с 1, 3 или 10 мг/кг соединения (1) за 2 ч до инфицирования гриппом B, обеспечивал полную защиту от смерти, уменьшал потерю BW и восстанавливал функцию легких (Таблица 18).
[0424] Таблица 18: Данные по эффективности in vivo соединения (1) с осельтамивиром или без него при введении за 2 ч до инфицирования гриппом B
[0425] Пример 8: Исследование In Vivo сочетания соединения (1) с занамивиром
[0426] Инфицированным мышам вводили наполнитель или возрастающие уровни доз формы A соли соединения (1) с HCl на 1/2H2O (здесь далее упоминаемой просто как соединение (1) в контексте примера 8) в сочетании с занамивиром, начиная за 24 часа до провокации гриппа A дозой 5×103 TCID50 штамма A/PR/8/34. Провокацию гриппа A и приготовление суспензии соединения (1) осуществляли аналогично тому, как описано выше в примере 7. Зараженным мышам осуществляли единственное интраназальное (IN) введение занамивира в дозе 0,3 мг/кг, 1 мг/кг или 3 мг/кг за 24 часа перед провокацией IN дозой 5×103 TCID50 штамма A/PR/8/34 и соединения (1) в дозе 0,1 мг/кг, 0,3 мг/кг или 1 мг/кг в режиме BID в течение 10 суток за 2 часа до провокации дозой 5x103 TCID50 штамма A/PR/8/34.
[0427] Результаты сведены в приведенных ниже Таблицах 19A и 19B. Как видно из Таблицы 19A ниже, комбинированная терапия соединением (1) и занамивиром обеспечивала дополнительный полезный эффект выживания (Таблица 19A). Коэффициент эффективности, комбинированная мера выживаемости, потери массы тела и функции легких (% выживаемости/(% потери массы тела на 8 сутки)*(Penh на 6 сутки)) приведен в Таблице 19B.
[0428] Таблица 19A: Показатель выживаемости: комбинированная терапия соединением (1) с занамивиром
1ая доза за 2 ч до инфицирования
[0429] Таблица 19B: Коэффициент эффективности: комбинированная терапия соединением (1) с занамивиром
1ая доза за 2 ч до инфицирования
[0430] Пример 9: Профилактическая и постинфекционная эффективность соединения (1) в мышиной модели инфекции гриппа A
[0431] Материалы и методы
[0432] Животные: Самок мышей BALB/c массой 18-20 г получали от Jackson Laboratories (Бар Харбор, Мэн), и их использовали в эксперименте на противовирусную активность. Животных держали на стандартной диете для грызунов и поили водопроводной водой ad libitum. За 48 часов до использования их подвергали карантину.
[0433] Вирус: Адаптированный к мышам вирус гриппа A/California/04/2009 (pndH1N1) получали от доктора Елены Говорковой (Elena Govorkova) (St. Jude Children’s Research Hospital, Мемфис, Теннесси). Исходный вирусный материал амплифицировали в клетках MDCK с последующим титрованием по летальности для мышей BALB/c. Вирус гриппа A/Victoria/3/75 (H3N2) получали от American Type Culture Collection (Манассас, Виргиния). Вирус подвергали пассажу семь раз на мышах, чтобы адаптировать его к мышам, с последующим одним пассажем в клетках MDCK. Далее вирус титровали по летальности для мышей BALB/c, чтобы получить надлежащую летальную провокационную дозу. Вирус гриппа A/Vietnam/1203/2004 (H5N1) получали от доктора Jackie Katz, Centers for Disease Control (Атланта, Джорджия). Мышам вводили летальную дозу вируса (5 MLD50, 5 PFU/мышь), которая ранее приводила к смерти между 6-13 днями при смертности 90-100% на 10 суток при такой дозе.
[0434] Соединения: Осельтамивир (как Tamiflu®) получали из местной аптеки. Каждая капсула Tamiflu содержит пролекарство, которое при метаболизме в организме дает 75 мг активного компонента, то есть карбоксилата осельтамивира. Доза осельтамивира основывалась на данном измерении. В исследовании использовали форму A гемигидрата соли соединения (1) с HCl (здесь далее упоминаемую просто как соединение (1) в контексте примера 9), и доза соединения основывалась на факте использования гемигидрата соли соединения (1) с HCl. Как для соединения (1), так и осельтамивира препарат готовили в 0,5% метилцеллюлозы (Sigma, Сент-Луис, Миссури) для перорального введения мышам через зонд (p.o.).
[0435] Дизайн эксперимента: Мышей анестезировали интраперитонеальной инъекцией кетамина/ксилазина (50/5 мг/кг), и инфицировали животных интраназально 90 мкл суспензии вируса гриппа. Провокационная доза вируса была приблизительно равна четырем инфекционным дозам, вызывающим 50% летальность у мышей. Лечение проводили дважды в сутки (с интервалами 12 часов) в течение 10 суток, начиная за 2 часа до вирусной провокации или спустя 48 часов после провокации в соответствии с показаниями. Параметрами для оценки инфекции являлись выживаемость, средний срок гибели, изменения массы тела и параметры легочной инфекции (геморрагический балл, масса и вирусный титр). Животных взвешивали индивидуально через сутки вплоть до 21 дня инфекции. Мыши, которые погибли в первые шесть суток периода лечения, рассматривались как погибшие от причин, отличных от инфекции вируса гриппа, и были исключены из итогового подсчета.
[0436] Для оценки параметров легочной инфекции извлекали легкие забитых животных (изначально 5 животных из группы резервировали для данной цели). Балл легочной геморрагии оценивали визуальным осмотром для определения изменений цвета с розового на темно-фиолетовый. Данное явление наблюдается в легких участками, а не в виде постепенного изменения цвета всего легкого на более темный. Геморрагические баллы изменялись в диапазоне от 0 (норма) до 4 (все легкое обнаруживает темно-фиолетовый цвет), и, таким образом, данный показатель является непараметрическим измерением. Легкие взвешивали, а затем замораживали при -80°C. Позже размороженные легкие гомогенизировали в 1 мл среды для клеточных культур, надосадочные жидкости центрифугировали, чтобы удалить материал в форме взвешенных частиц, и жидкие образцы повторно замораживали при -80°C. После подготовки 96-луночных планшетов с клетками MDCK образцы размораживали, серийно разводили с 10-кратными шагами разведения и титровали методом конечной точки в планшетах (1), используя 4 микролунки на разведение. Вирусные титры рассчитывали как log10 50% инфицирующих клеточную культуру доз на грамм легочной ткани (log10 CCID50/г).
[0437] Статистический анализ: Для определения статистической значимости посредством логарифмического рангового критерия Мантеля-Кокса анализировали графики Каплана-Мейера для сравнений множества групп. Впоследствии посредством критерия Гехана-Бреслоу-Уилкоксона проводили парные сравнения. Относительную экспериментальную значимость регулировали к скорректированному порогу значимости по Бонферрони на основе числа произведенных сравнений вариантов лечения. Сравнения среднего срока гибели и среднего балла геморрагии легких анализировали посредством критерия Краскала-Уоллиса с последующим применением критерия Данна для множественных сравнений. Средние массы тела, массы легких и log10 вирусных титров в легких оценивали методом ANOVA в предположении равных дисперсий и нормального распределения. После проведения оценки методом ANOVA, значения индивидуальных вариантов лечения сравнивали посредством критерия Тьюки-Крамера для множественных сравнений. Анализы проводили, используя программное обеспечение Prism® (GraphPad Software, Сан-Диего, Калифорния).
[0438] Обсуждение результатов
[0439] Ответ на профилактические дозы соединения (1) исследовали в мышиной модели гриппа A. Введение доз наполнителя или соединения (1) начинали за 2 ч до инфицирования и продолжали дважды в сутки на протяжении 10 суток. Результаты сведены в Таблицах 20 и 21. Все мыши, которые получали только наполнитель, пали жертвой инфекции к 9 дню исследования и теряли в среднем ~32% своей массы тела (BW). Соединение (1), введенное в режиме BID в дозе 1, 3 или 10 мг/кг, обеспечивало полную выживаемость и дозозависимое снижение потери BW. Соединение (1), введенное в режиме BID в дозе 0,3 мг/кг, обеспечивало некоторый полезный эффект выживаемости (2/8 мышей), хотя мыши имели значительную потерю BW. В этом же эксперименте мышам вводили дозу осельтамивира в режиме BID 10 мг/кг, клинически эквивалентную человеческой дозе (основанной на AUC). Все мыши, получавшие введение осельтамивира, выжили с профилем потери массы, схожим с таковым мышей, которым вводили соединение (1) в режиме 1 мг/кг BID.
[0440] Соединение (1) по прежнему проявляло эффективность в данной модели, спровоцированной вирусом гриппа A/Vietnam/1203/2004 (H5N1), когда его вводили с момента 48 часов после инфицирования в непрерывном режиме BID на протяжении 10 суток (Таблица 22). Введение соединения (1) в дозе 10 мг/кг обеспечивало полную защиту, как показано в Таблице 20.
[0441] Таблица 20: Профилактические эффекты соединения (1) и осельтамивира в отношении инфекции вируса гриппа A/California/04/2009 (pndH1N1) у мышей BALB/c (профилактика)
(6 сутки)
(10 мг/кг)
b Средний срок гибели мышей, которые погибали на 21 сутки или раньше.
c Log10 CCID50/г.
d Ниже предела определения (2,6 log10).
e Не является значимым при применении очень строгого критерия Данна для множественных сравнений, но является значимым относительно плацебо (P<0,01) при применении парного двустороннего U-критерия Манна-Уитни. * P<0,05, ** P<0,01, *** P<0,001, сравнение с плацебо.
[0442] Таблица 21: Эффекты соединения (1) и осельтамивира в отношении инфекции вируса гриппа A/Victoria/3/75 (H3N2) у мышей BALB/c (профилактика)
(6 сутки)
b Средний срок гибели мышей, которые погибали на 21 сутки или раньше.
c Log10 CCID50/г.
d Не является значимым при применении очень строгого критерия Данна для множественных сравнений, но является значимым относительно плацебо (P<0,01) при применении парного двустороннего U-критерия Манна-Уитни.
e То же, что в сноске “d”, но является значимым относительно плацебо на уровне P<0,05. ** P<0,01, *** P<0,001, сравнение с плацебо.
[0443] Таблица 22: Лечебные эффекты (через 48 ч) соединения (1) и осельтамивира в отношении инфекции вируса гриппа A/Vietnam/1203/2004 (H5N1) у мышей BALB/c
(6 сутки)
(10 мг/кг)
b Средний срок гибели мышей, которые погибали на 21 сутки или раньше.
c Log10 CCID50/г.
[0444] Пример 10: Эффективность In Vitro соединения (1) в отношении панели штаммов гриппа
[0445] Клетки и вирусы. Изначально клетки Мадин-Дарби почек собак (MDCK) получали от American Type Culture Collection (ATCC, Манассас, Виргиния), и перед их использованием в исследованиях инфекции проводили пассаж, используя стандартные лабораторные методики. Клетки выдерживали при 37°C в модифицированной по Дульбекко среде Игла (DMEM; Invitrogen, Карлсбад, Калифорния), содержащей 10% эмбриональной бычьей сыворотки (Sigma-Aldrich, Сент-Луис, Миссури), 2 мM L-глутамина, 10 мM HEPES, 100 ед/мл пенициллина и 100 мкг/мл стрептомицина (Invitrogen). Вирусы гриппа получали от ATCC, Virus Surveillance and Diagnosis Branch of the Influenza Division of the Centers for Disease Control and Prevention (CDC; Атланта, Джорджия) или от Influenza Reagent Resource, Influenza Division, WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza, CDC. Для получения вирусного исходного материала клетки MDCK инфицировали с низкой множественностью инфекции (MOI) в среде DMEM, содержащей 2 мM L-глутамина, 10 мM HEPES, 100 ед/мл пенициллина, 100 мкг/мл стрептомицина и 1 мкг на мл трипсина, обработанного толилсульфонилфенилаланилхлорметилкетоном (TPCK) (USB Corp.; Санта-Клара, Калифорния). Клетки инкубировали при 37°C в атмосфере 5% CO2 в течение периода времени 48 ч, по прошествии которого надосадочную жидкость отделяли с помощью центрифугирования при 900×g в течение 10 мин, используя центрифугу Beckman GS-6R. Вирусный исходный материал аликвотировали и замораживали при -80°C.
[0446] Соединения. Свободное основание или соль с HCl соединения (1) (например, аморфную соль соединения (1) с HCl, форму A гемигидрата соли соединения (1) с HCl, аморфное свободное основание соединения (1)) (здесь далее упоминаемые просто как соединение (1) в контексте примера 10) растворяли в 100% диметилсульфоксиде (ДМСО) для получения раствора с концентрацией 10 мM.
[0447] Противовирусная активность. Противовирусную активность соединения (1) и амантадина оценивали на клетках MDCK, измеряя уровни АТФ, используя CellTiter-Glo (Promega; Мэдисон, Висконсин). Клетки MDCK высеивали в черные, имеющие прозрачное дно, 384-луночные планшеты с плотностью 2×104 клеток на лунку в 50 мкл VGM. Клетки инкубировали при 37°C в атмосфере 5% CO2 с насыщенной влажностью, позволяя клеткам сцепиться друг с другом и образовать монослой. Через 5 ч удаляли 40 мкл среды и прибавляли 15 мкл вирусного материала с MOI=0,005. Соединение прибавляли в объеме 25 мкл десятиточечного трехкратного разведения в среде DMEM, содержащей добавки (конечная концентрация ДМСО составляла 0,5%). Внутренние контроли состояли из лунок, содержащих только клетки и необработанные клетки, инфицированные вирусом. После 72 ч инкубирования в каждую лунку прибавляли 20 мкл CellTiter-Glo и инкубировали при комнатной температуре в течение 10 мин. Люминесценцию измеряли, используя ридер EnVision Multilabel (PerkinElmer; Уолтем, Массачусетс). Значения EC50 (концентрация соединения, которая обеспечивает 50% жизнеспособность клеток относительно неинфицированного контроля) рассчитывали, строя зависимости между дозой соединения и данными по ответу, используя 4-параметрический метод подгонки кривой, применяя алгоритм Левенберга—Марквардта (программное обеспечение Condoseo; Genedata, Базель, Швейцария). In vitro исследование hpaiH5N1 проводили в Southern Research Institute, соблюдая меры предосторожности в рамках 3-го уровня безопасности (BSL-3).
[0448] Как показано в нижеприведенной Таблице 23, соединение (1) демонстрировало сильную активность в отношении всех исследованных штаммов гриппа A, включая референтные штаммы H1N1 и H3N2 с 1934 по 2009, а также штаммы H1N1 пандемии 2009 A/California/07/2009, A/Texas/48/2009 и высокопатогенный штамм птичьего гриппа H5N1 A/VN/1203/2004. Соединение (1) было равным образом эффективно в отношении всех штаммов, включая таковые, которые устойчивы к амантадину и ингибиторам нейраминидазы. Оно показывало ограниченную активность в отношении вируса гриппа B.
[0449] Таблица 23: Эффективность соединения (1) в отношении панели штаммов гриппа
EC50±SD
(1) (нМ)
b: устойчивость к карбоксилату осельтамивира: мутация NA 275Y.
c: устойчивость к карбоксилату осельтамивира: мутация NA 119V.
*: внешне подтвержденная фенотипическая устойчивость, данные о последовательности недоступны.
[0450] Пример 11: Эксперименты In Vitro с использованием сочетания соединения (1) и осельтамивира, занамивира или фавипиравира
[0451] Раствор соединения (1) (свободное основание или соль с HCl соединения (1), как в примере 10) в 100% диметилсульфоксида (ДМСО) исследовали в трехдневном анализе на клетках MDCK, основанном на цитопатическом эффекте (CPE), где клетки были инфицированы штаммом A/Puerto Rico/8/34 при MOI = 0,01, в экспериментах с использованием сочетаний либо с ингибиторами нейраминидазы, карбоксилатом осельтамивира и занамивиром, либо с ингибитором полимеразы, T-705. Карбоксилат осельтамивира T-705 растворяли в 100% диметилсульфоксида (ДМСО); занамивир растворяли в модифицированной по Дульбекко среде Игла (DMEM) в концентрации 10 мM и хранили при -20°C. В исследовании использовали либо метод независимости Блисса (Bliss) (Macsynergy) (например, Prichard, M.N. and C. Shipman, Jr., Antiviral Res, 1990. 14(4-5): стр. 181-205), либо метод аддитивности Леве (Loewe)/медианного эффекта (например, Chou, T.C. and P. Talalay, Adv Enzyme Regul, 1984. 22: стр. 27-55). Метод независимости Блисса включает исследование сочетаний различных концентраций ингибиторов в шахматном порядке, тогда как метод независимости Леве включает исследование сочетания фиксированного соотношения ингибиторов при различных разведениях фиксированного соотношения. Проводили также эксперименты с использованием сочетаний соединения (1) с самим собой в качестве контроля, подтверждая аддитивность. Жизнеспособность клеток определяли, используя CellTiter-Glo.
[0452] Метод независимости Блисса показал объемы синергии, составляющие 312 и 268 для карбоксилата осельтамивира и занамивира соответственно; а для фавипиравира был получен объем синергии, составляющий 317. Объемы синергии, превышающие 100, обычно рассматриваются как соответствующие сильной синергии, а объемы между 50 и 100 рассматриваются как соответствующие умеренной синергии. Способ аддитивности Леве дал значения C.I. (показатель аддитивности) 0,58, 0,64 и 0,89 при 50% уровне эффекта для осельтамивира, занамивира T-705 соответственно. Значения C.I. меньшие 0,8 рассматриваются как соответствующие сильной синергии, тогда как значения между 0,8 и 1,0 рассматриваются как соответствующие аддитивности вплоть до легкой синергии. Эти данные, сопоставленные вместе, как показано в Таблице 24, указывают на то, что соединение (1) проявляет синергетический эффект с исследованными ингибиторами нейраминидазы и ингибитором полимеразы.
[0453] Таблица 24: Сводка результатов экспериментов по определению in vitro синергии и антагонизма
[0454] Пример 12: Эффективность в мышиной модели инфекции гриппа A
[0455] Ответ на профилактические дозы соединения (1) (в аморфной форме или форме A гемигидрата соли соединения (1) с HCl (здесь далее в данном примере упоминаемых просто как соединение (1)) исследовали в мышиной модели гриппа A. Введение доз наполнителя или соединения (1) начинали за 2 ч до инфицирования и продолжали дважды в сутки на протяжении 10 суток. Все мыши, которые получали только наполнитель, пали жертвой инфекции к 9 дню исследования и теряли в среднем ~32% своей массы тела (BW). Соединение (1), введенное в режиме BID в дозе 1, 3 или 10 мг/кг, обеспечивало полную выживаемость и дозозависимое снижение потери BW. Соединение (1), введенное в режиме BID в дозе 0,3 мг/кг, обеспечивало некоторый полезный эффект выживаемости (2/8 мышей), хотя мыши имели значительную потерю BW. В этом же эксперименте мышам вводили дозу осельтамивира в режиме BID 10 мг/кг, клинически эквивалентную человеческой дозе (основанной на AUC). Все мыши, получавшие введение осельтамивира, выжили с профилем потери массы, схожим с таковым мышей, которым вводили соединение (1) в режиме 1 мг/кг BID.
[0456] Степень, в которой введение соединения (1) могло бы быть отложено и все еще обеспечивало бы полезный эффект в данной модели, исследовали, провоцируя у мышей инфекцию вирусом гриппа A и вводя им наполнитель, осельтамивир или соединение (1), начиная введения в моменты времени 24, 48, 72, 96 или 120 ч после инфицирования, продолжая вводить дозу в режиме BID в течение 10 суток (Таблица 25). Все контроли, получавшие наполнитель, пали жертвой заболевания к 8 или 9 дню исследования. По сравнению с контролями, получавшими наполнитель, соединение (1), введенное в режиме BID в дозе 1, 3 или 10 мг/кг, обеспечивало полную защиту от смерти и снижало потерю BW, когда введение доз начинали в период вплоть до 72 ч после инфицирования. Введение лишь дозы 10 мг/кг осельтамивира в режиме BID обеспечивало полную защиту, когда введение дозы начинали в пределах 24 ч или менее после инфицирования. Когда начало введения соединения откладывали еще больше, соединение (1) в дозах 3 или 10 мг/кг в режиме BID обеспечивало полную выживаемость в случае введения в момент 96 ч после инфицирования и частичную защиту, когда начало введения доз откладывали на 120 ч после инфицирования.
[0457] Исследовали эффективность соединения (1) по снижению вирусных тиров в легких. Мышей инфицировали гриппом A и через 24 ч вводили им наполнитель, осельтамивир (10 мг/кг, BID) или соединение (1) (3, 10, 30 мг/кг, BID) вплоть до извлечения легких и определения вирусной нагрузки на 6 сутки (Таблица 26). Все группы с введением соединения (1) показали выраженные, статистически значимые снижения вирусных тиров в легких по сравнению с животными, получавшими введения осельтамивира и наполнителя.
[0458] Чтобы установить фармакокинетическую/фармакодинамическую (PK/PD) модель, мышей инфицировали вирусом гриппа в течение 24 ч, а затем вводили соединение (1) в течение дополнительных 24 ч. Дозы фракционировали на единственную дозу, две или четыре дозы, вводимые каждые 12 ч или 6 ч соответственно. Легкие и плазму собирали для определения вирусных нагрузок в легких и концентраций соединения (1). Индивидуальные данные по титру в легких из данных режимов введения доз (q6h, q12h и q24h) откладывали на графике в зависимости от индивидуальных значений Cmax, Cmin или AUC (данные не показаны). Хотя имелась явная корреляция между снижением легочного титра и Cmin, имелась малая корреляция с Cmax и лишь слабая корреляция с AUC. Имелась сильная корреляция с Cmin, когда измеренные концентрации соединения (1) в плазме откладывали на графике от измеренных легочных титров. Полумаксимальное снижение легочных титров (2-3 log) происходит вблизи концентрации EC99 (100 нг/мл), учитывающей влияние сывороточных белков. Похожую корреляцию обнаруживали между легочным титром и измеренными концентрациями соединения (1) в легких (данные не показаны).
[0459] Таблица 25: Сводка данных по проценту выживаемости и проценту потери массы тела в мышиной модели гриппа A
(мг/кг; BID)
b Данные из того же эксперимента.
c ND – не определено
[0460] Таблица 26: Сводка данных по вирусному титру в легких и Log10 уменьшения в мышиной модели гриппа A
(Log10 TCID50)b
(Log10 TCID50)b
b Вирусные титры в легких определяли на 6 сутки исследования.
c ND – не определено.
2-факторный анализ ANOVA c тестом Бонферрони, ***P<0,001.
[0461] Пример 13: Экспериментальная проверка концепции с провокацией гриппа
[0462] Модель провокации живым, ослабленным вирусом гриппа использовали ранее для предсказания эффективности антигриппозных противовирусных препаратов при естественной инфекции у людей (Calfee, D.P., Peng, A.W., Hussey, E.K., Lobo, M. & Hayden F.G. Safety and efficacy of once daily intranasal zanamivir in preventing experimental human influenza A infection. Antivir Ther. 4 , 143-149 (1999); Hayden, F.G. et al. Use of the oral neuraminidase inhibitor oseltamivir in experimental human influenza. JAMA 282 , 1240-1246 (1999)). Рандомизированное, двойное слепое, плацебо-контролируемое, одноцентровое исследование формы A гемигидрата соли с HCl соединения ( 1 ) (здесь и далее в данном примере упоминаемой просто как соединение ( 1 )) проводили на здоровых добровольцах, инокулированных живым провокационным штаммом вируса гриппа A/Wisconsin/67/2005 (H3N2). Субъекты получали пять дневных доз либо плацебо (N=33), либо соединения ( 1 ) один раз в сутки (QD) (в капсульной форме, состоящей из чистого соединения ( 1 )): 100 мг (N=16), 400 мг (N=19) или 900 мг в 1 сутки с последующими 600 мг на 2-5 дни (N=20) или 1200 мг в 1 сутки с последующими 600 мг на 2-5 дни (N=18). Трижды в сутки у субъектов брали мазки из носа, и в бальных картах регистрировались трижды в сутки клинические симптомы на протяжении 1-7 суток, и выписывали из учреждения на 8 сутки, продолжая наблюдать их в целях безопасности приблизительно по 28 сутки. Мазки из носа исследовали на вирус гриппа в клеточной культуре (первичный анализ) и посредством qRT-PCR (вторичный анализ).
[0463] Анализы эффективности проводили на полной выборке (FA), определяемой как все рандомизированные субъекты, которые получали по меньшей мере одну дозу исследованного лекарственного средства (соединение ( 1 ) или плацебо) и у которых концентрации вирусов были выше или равны нижнему пределу количественного определения в анализе по определению TCID50 в клеточной культуре в любой временной точке в пределах 48 ч после инокуляции, или у которых титр торможения гемагглютинации возрастал в 4 раза или более относительно базовой линии (1 сутки) в период после инокуляции (N=74). Выборка для оценки безопасности включала всех субъектов, которые были инокулированы гриппом в сутки 0 и которые получали по меньшей мере одну дозу либо плацебо, либо соединения ( 1 ) (N=104).
[0464] Оценка эффективности
[0465] Первичной мерой в данном исследовании являлась демонстрация тренда дозозависимого эффекта в выделении вируса по AUC между 1 сутками исследования (первые сутки введения дозы лекарственного средства) и 7 сутками исследования, для чего на полной FA-выборке измеряли TCID50 в анализе на клеточной культуре. Статистически значимый тренд дозозависимого эффекта наблюдался в медианном выделении вируса по AUC в мазках из носа (P=0,036, тренд-критерий Джонкхиера-Терпстра (Jonckheere-Terpstra)). Кроме того, проводили парные сравнения между объединенной группой плацебо и каждой группой, получавшей дозу соединения ( 1 ), для определения медианного выделения вируса по AUC, медианной продолжительности выделения и средней величины пикового выделения вируса (Таблица 27). Статистически значимое уменьшение в выделении вируса по AUC наблюдали для группы с дозой 1200/600 мг (P=0,010, критерий суммы рангов Уилкоксона), а значимые снижения пикового выделения наблюдали для группы с дозой 1200/600 мг (Фиг. 1), группы с дозой 400 мг объединенных групп, получавших дозу соединения ( 1 ). Проводили дополнительные анализы для FA-группы (не показаны).
[0466] Выделение вируса из носа также количественно определяли посредством qRT-PCR и результаты были схожи с таковыми, наблюдавшимися в случае клеточной культуры. Отсутствовала разница в степенях сероконверсии между группами с введениями доз соединения ( 1 ) и плацебо, как определено по 4-кратному или более увеличению противогриппозного титра относительно базовой линии перед инокуляцией, что указывает на то, что соединение ( 1 ), введенное через 24 ч после инокуляции гриппа, не влияло на степень зараженности инфекцией гриппа и не устраняло последующий гуморальный иммунный ответ на инфекцию (Таблица 28A).
[0467] Субъекты регистрировали клинические симптомы три раза в сутки в дневниках. Рассчитывали AUC для баллов клинических и гриппоподобных симптомов с 1 суток по 7 сутки. В сравнении с плацебо группа, получавшая дозу соединения ( 1 ) 1200/600 мг, показала статистически значимое уменьшение медианной продолжительности совокупных клинических симптомов (P=0,001), медианного значения AUC для гриппоподобных симптомов (P=0,040) и медианной продолжительности гриппоподобных симптомов (P<0,001) (Таблица 28B).
[0468] Таблица 28A: Медианное выделение вируса по AUC, медианная продолжительность выделения и средняя величина пикового выделения вируса
17,1)
16,1)
18,0)
16,1)
8,4)
18,0)
[сутки]
4,63)
3,39)
NA)
4,68)
1,33)
2,43)
42,1)
41,9)
36,9)
37,1)
24,7)
41,9)
[сутки]
5,35)
3,39)
NA)
5,01)
0,66)
2,394)
AUC: площадь под кривой значения от времени; CI: доверительный интервал; NA: не применимо; qRT-PCR: количественная полимеразная цепная реакция с обратной транскриптазой; SD: стандартное отклонение; TCID50: доза заражения 50% культурной ткани.
Примечание: Статистически значимые значения P (P<0,05) выделены жирным шрифтом.
a P=0,036 для тренда дозозависимого эффекта AUC по тренд-критерию Джонкхиера-Терпстра.
b Значение P рассчитано по критерию суммы рангов Уилкоксона.
c Значение P рассчитано по ANOVA.
d Значение P рассчитано по критерию суммы рангов.
e P = 0,031 для тренда дозозависимого эффекта AUC по тренд-критерию Джонкхиера-Терпстра.
f Сероконверсию определяли как ≥4-кратное увеличение титра противогриппозных антител при визите последующего наблюдения в сравнении с базовой линией. Значение P рассчитывали, используя точный критерий Фишера.
[0469] Таблица 28B: Медианное значение AUC, медианная продолжительность и средняя величина пика для совокупного клинического симптома и гриппоподобного симптома
23,5)
25,3)
16,0)
32,3)
5,5)
32,3)
[сутки]
4,73)
5,43)
4,63)
4,61)
2,24)
3,06)
17,7)
21,3)
14,0)
28,6)
4,4)
28,6)
[сутки]
4,73)
5,40)
4,63)
4,61)
2,24)
3,00)
AUC: площадь под кривой значения от времени; CI: доверительный интервал; NA: не применимо.
Примечание: Статистически значимые значения P (P<0,05) выделены жирным шрифтом.
b Значение P рассчитано по критерию суммы рангов Уилкоксона.
c Значение P рассчитано по ANOVA.
d Значение P рассчитано по критерию суммы рангов.
[0470] Оценка безопасности
[0471] Соединение (1) хорошо переносилось и случаи прекращения исследования из-за связанных с соединением (1) неблагоприятных явлений (AE) отсутствовали, отсутствовали также какие-либо серьезные неблагоприятные явления. Список неблагоприятных явлений, встречающихся у ≥10% субъектов в любой получавшей лечение группе, представлен в Таблице 29. Гриппоподобное недомогание являлось наиболее часто регистрируемым неблагоприятным явлением, и жалобы на него выражала приблизительно равная доля субъектов в группах, получавших плацебо и соединение (1). Неблагоприятные явления, которые наблюдались с разницей во встречаемости ≥10% между группами, получавшими соединение (1), и реципиентами плацебо представляли собой: пониженный уровень фосфора в крови (18,1%, соединение (1); 0%, плацебо), ринорею (соединение (1), 4,2%; 18,8%, плацебо), и заложенность носа (1,4%, соединение (1); 15,6% плацебо). Кроме того, как у реципиентов плацебо, так и соединения (1) наблюдались повышения уровня аланинаминотрансферазы (ALT). Ни нарушения функции печени, ни снижения уровня сывороточного фосфата не наблюдались в проводимом впервые на человеке исследовании с увеличением дозы соединения (1) при использовании единственных доз вплоть до 1600 мг и нескольких доз вплоть до 800 мг ежесуточно в течение 10 суток; как о повышениях уровня ALT, так и о снижениях уровня сывороточного фосфата сообщалось ранее в случае вирусных инфекций верхних дыхательных путей.
[0472] Таблица 29: Список неблагоприятных явлений, встречающихся у ≥10% субъектов в любой получавшей лечение группе
Примечания: Субъект с несколькими событиями подсчитывался один раз, как имеющий AE. Субъекты могут попадать в несколько категорий.
a Единственная ударная доза 900 мг в 1 сутки и 600 мг qd во 2-5 сутки.
b Единственная ударная доза 1200 мг в сутки 1 и 600 мг qd во 2-5 сутки.
c Гриппоподобное недомогание, определяемое в анализе эффективности, оценивали, основываясь на параметрах, перечисленных в тексте. Факт AE гриппоподобного недомогания устанавливался врачом.
[0473] Обсуждение
[0474] В провокационном исследовании гриппа на здоровых добровольцах соединение (1) продемонстрировало тренд дозозависимого эффекта в вирусном титре по in AUC в мазках из носа при определении как посредством анализа на TCID50 в клеточной культуре, так и посредством qRT-PCR, наиболее высокая оцененная доза соединения (1) вызывала значительное снижение вирусного титра по AUC, а также в AUC и продолжительности симптомов гриппа. Хотя схожая величина улучшения по сравнению с плацебо не наблюдалась в группе со второй наиболее высокой дозой, 900/600 мг (Таблица 27), данная доза действительно демонстрировала схожие результаты с группой, получавшей дозу 1200/600 мг, в плане медианного значения AUC для конечных точек совокупного клинического симптома и гриппоподобного симптома (Таблица 28); причины данного расхождения полностью не ясны. Хотя в исследовании проверки концепции (исследование POC) определенные проблемы в плане безопасности не встречались, наблюденные снижения уровня фосфата и повышения уровня ALT указывают на то, что в будущих исследованиях будет необходимо использование надлежащего мониторинга.
[0475] В общем, ограничения модели провокации гриппа состоят в том, что использованный в данном исследовании вирус гриппа представляет собой штамм, который был специально выбран, чтобы не вызывать наиболее тяжелые клинические симптомы инфекции вируса гриппа. Кроме того, введенный вирусный инокулят, вероятно, превышает инокулят при естественном воздействии гриппа. Время введения соединения (1) через 24 ч после заражения не может представлять собой реалистичные временные рамки для начала терапии в амбулаторных условиях, в которых пациенты не часто обращаются за диагностикой или лечением до тех пор, пока у них не разовьются выраженные симптомы, на что потребуется, вероятно, более 24 ч после заражения. Однако, учитывая, что естественно зараженные субъекты изначально инокулируются значительно меньшим вирусным титром, масштабы времени нельзя непосредственно несопоставимы.
[0476] Подводя итог, можно отметить, что соединение (1) является эффективным PB2-ингибитором гриппа A, который представляет отличающийся от ранее известных и новый класс противовирусных агентов. Свойства данного ингибитора, описанные с привлечением как преклинических, так и клинических данных, указывают на то, что соединение (1) является перспективным кандидатом для дальнейшей оценки, обладая несколькими потенциальными преимуществами над современными противовирусными агентами, используемыми для лечения инфекции гриппа.
[0477] Все приведенные здесь ссылки включены в настоящую заявку во всей своей полноте путем ссылки. Все использованные здесь аббревиатуры, символы и условные обозначения согласуются с таковыми, используемыми в современной научной литературе. Смотри, например, Janet S. Dodd, ed., The ACS Style Guide: A Manual for Authors and Editors, 2nd Ed., Washington, D.C.: American Chemical Society, 1997.
ДРУГИЕ ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ
[0478] Следует понимать, что хотя изобретение описано во взаимосвязи со своим подробным описанием, вышеприведенное описание предназначено иллюстрировать и не ограничивать объем изобретения, который определяется объемом прилагаемой формулы изобретения. Другие аспекты, преимущества и модификации входят в объем нижеследующей формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ ПОЛИМЕРАЗЫ | 2011 |
|
RU2718690C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ ПОЛИМЕРАЗЫ | 2011 |
|
RU2599013C2 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИРУСОВ ГРИППА | 2014 |
|
RU2700415C1 |
| СОСТАВЫ СОЕДИНЕНИЙ АЗАИНДОЛА | 2014 |
|
RU2685730C1 |
| Композиции и способы для лечения вирусных инфекций | 2019 |
|
RU2816717C2 |
| Способы лечения гриппа | 2018 |
|
RU2769317C2 |
| СТАБИЛИЗАТОРЫ ТУЧНЫХ КЛЕТОК ЛЕЧЕНИЯ ГИПЕРЦИТОКИНЕМИИ И ВИРУСНОЙ ИНФЕКЦИИ | 2017 |
|
RU2760682C2 |
| ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ ДЛЯ ЛЕЧЕНИЯ ГРИППА, ХАРАКТЕРИЗУЮЩИЙСЯ ТЕМ, ЧТО В НЕМ ОБЪЕДИНЕНЫ ИНГИБИТОР КЭП-ЗАВИСИМОЙ ЭНДОНУКЛЕАЗЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО ПРОТИВ ГРИППА | 2016 |
|
RU2745071C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЗАМЕЩЕННЫЕ ПОЛИЦИКЛИЧЕСКИЕ ПИРИДОНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРОЛЕКАРСТВО | 2017 |
|
RU2727962C1 |
| ПОЛИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ КАРБАМОИЛПИРИДОНА, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2770096C1 |
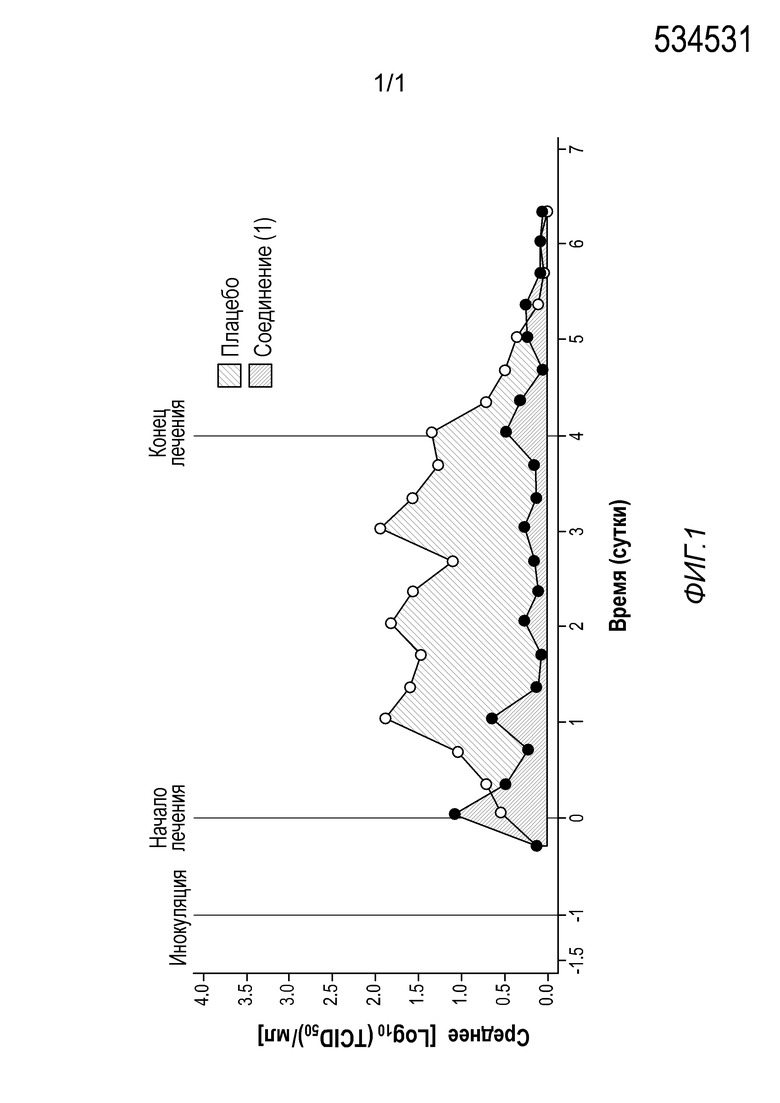
Изобретение относится к вариантам улучшенного способа получения соединения формулы (1) или его фармацевтически приемлемой соли. Соединение формулы (1) используется в качестве ингибитора репликации вируса гриппа. Способ получения соединения (1), имеющего структурную формулу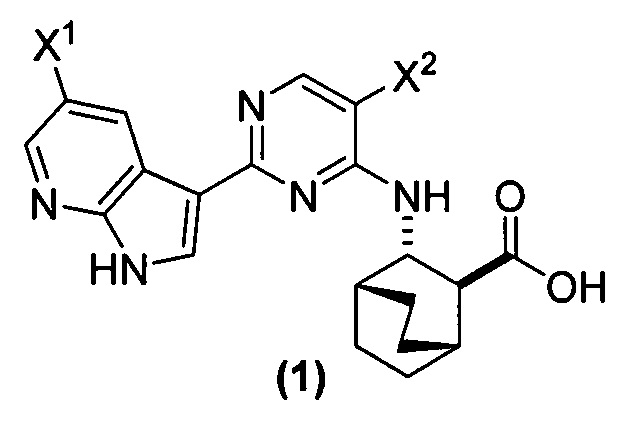 ,
,
включает a) взаимодействие соединения (X) или его фармацевтически приемлемой соли с соединением (Y)
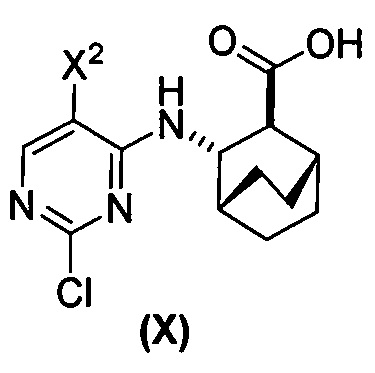
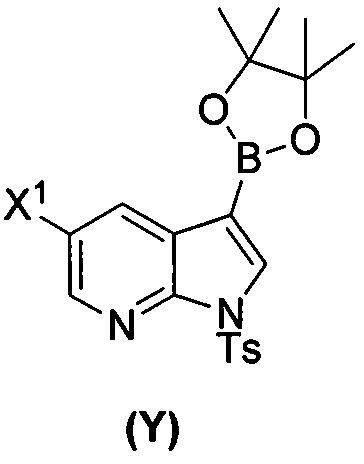
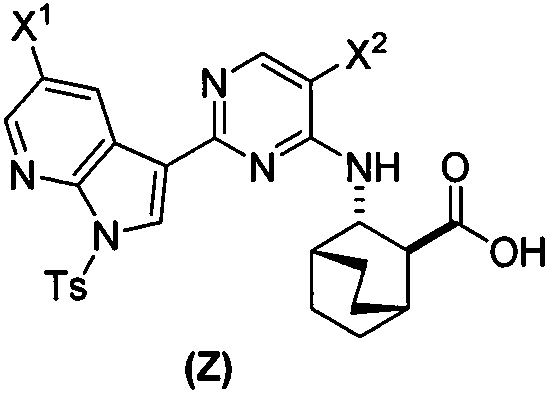 в присутствии палладиевого катализатора и основания с образованием соединения (Z) или его фармацевтически приемлемой соли; и (b) удаление защитной группы Ts из соединения (Z) или его фармацевтически приемлемой соли с образованием соединения (1) или его фармацевтически приемлемой соли. При этом каждый из X1 и X2 независимо представляет собой -F или -Cl; Ts представляет собой тозил; палладиевый катализатор содержит комплекс палладий-XPhos, в котором XPhos представляет собой 2-дициклогексилфосфино-2’,4’,6’-триизопропилбифенил; основание представляет собой фосфатное основание или карбонатное основание, и взаимодействие на стадии (а) проводят в системе растворителей, содержащей воду и органический растворитель, выбранный из 2-метил-тетрагидрофурана или тетрагидрофурана (ТГФ), или любого их сочетания. Комплекс палладий-XPhos может быть получен in situ путем смешения Pd(OAc)2 и XPhos. Основание представляет собой фосфатное основание или карбонатное основание, выбранное из Na2CO3, K2CO3, K3PO4 или Na3PO4.На стадии (b) соединение (Z) или его фармацевтически приемлемую соль обрабатывают неорганическим гидроксидом, включающим LiOH, NaOH, KOH или любое их сочетание в системе растворителя, которая содержит THF. Либо способ получения соединения (I) включает дополнительно стадии (g) взаимодействие соединения (A)
в присутствии палладиевого катализатора и основания с образованием соединения (Z) или его фармацевтически приемлемой соли; и (b) удаление защитной группы Ts из соединения (Z) или его фармацевтически приемлемой соли с образованием соединения (1) или его фармацевтически приемлемой соли. При этом каждый из X1 и X2 независимо представляет собой -F или -Cl; Ts представляет собой тозил; палладиевый катализатор содержит комплекс палладий-XPhos, в котором XPhos представляет собой 2-дициклогексилфосфино-2’,4’,6’-триизопропилбифенил; основание представляет собой фосфатное основание или карбонатное основание, и взаимодействие на стадии (а) проводят в системе растворителей, содержащей воду и органический растворитель, выбранный из 2-метил-тетрагидрофурана или тетрагидрофурана (ТГФ), или любого их сочетания. Комплекс палладий-XPhos может быть получен in situ путем смешения Pd(OAc)2 и XPhos. Основание представляет собой фосфатное основание или карбонатное основание, выбранное из Na2CO3, K2CO3, K3PO4 или Na3PO4.На стадии (b) соединение (Z) или его фармацевтически приемлемую соль обрабатывают неорганическим гидроксидом, включающим LiOH, NaOH, KOH или любое их сочетание в системе растворителя, которая содержит THF. Либо способ получения соединения (I) включает дополнительно стадии (g) взаимодействие соединения (A)
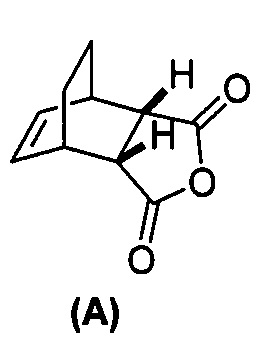 с хинином
с хинином  и этиловым спиртом при температуре от -12°C до -16°C с образованием аддукта хинина и соединения (C-1), h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl при температуре менее чем 25°C с образованием соединения (C-1) или его фармацевтически приемлемой соли; (i) эпимеризацию соединения (C-1) или его фармацевтически приемлемой соли с образованием соединения (C) или его фармацевтически приемлемой соли,где эпимеризации включает обработку соединения (C-1) C1-6-алкоксидом щелочного металла при температуре от -15°C до -22°C; (e) взаимодействие соединения (C) или его фармацевтически приемлемой соли с дифенилфосфорилазидом и бензиловым спиртом с образованием соединения (D), где Cbz представляет собой карбоксибензил;(f) взаимодействие соединения (D) или его фармацевтически приемлемой соли с H2 в присутствии Pd-катализатора на угле (Pd(0)/C) с образованием соединения (F)
и этиловым спиртом при температуре от -12°C до -16°C с образованием аддукта хинина и соединения (C-1), h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl при температуре менее чем 25°C с образованием соединения (C-1) или его фармацевтически приемлемой соли; (i) эпимеризацию соединения (C-1) или его фармацевтически приемлемой соли с образованием соединения (C) или его фармацевтически приемлемой соли,где эпимеризации включает обработку соединения (C-1) C1-6-алкоксидом щелочного металла при температуре от -15°C до -22°C; (e) взаимодействие соединения (C) или его фармацевтически приемлемой соли с дифенилфосфорилазидом и бензиловым спиртом с образованием соединения (D), где Cbz представляет собой карбоксибензил;(f) взаимодействие соединения (D) или его фармацевтически приемлемой соли с H2 в присутствии Pd-катализатора на угле (Pd(0)/C) с образованием соединения (F) 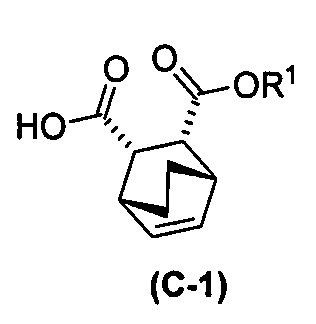
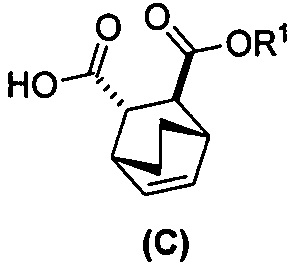

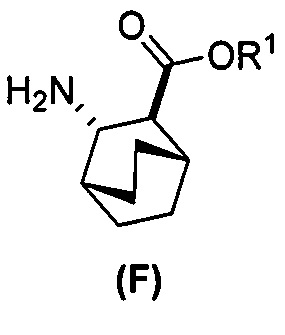
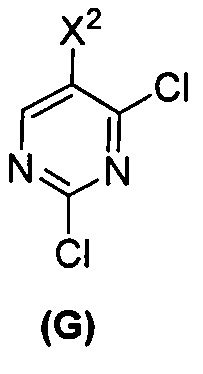
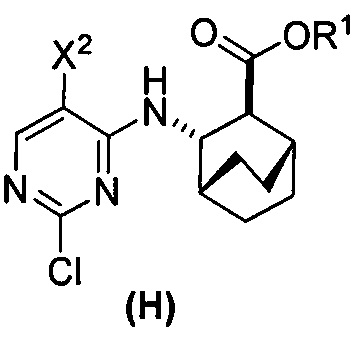
или его фармацевтически приемлемой соли;(c) взаимодействие соединения (F) или его фармацевтически приемлемой соли с соединением (G) с образованием соединения (H) или его фармацевтически приемлемой соли;(d) гидролиз соединения (H) или его фармацевтически приемлемой соли с образованием соединения (X) или его фармацевтически приемлемой соли. Далее осуществляют стадий а)- (b) для получения соединении (1) как указано выше. Изобретение также включает способ получения соединения(1) из соединения 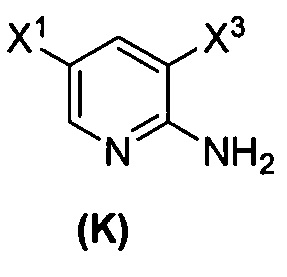 и, а также способ получения соединения (С) и способ получения соединения (2), соответствующего структурной формуле (1), где каждый Х1 и Х2 означает F. Способ позволяет получить соединение (1) со степенью превращения 99,9% в течение 1,25 часа, что значительно ускоряет известные ранее способы при осуществлении способа с другими катализаторами. 5н. и 52 з.п. ф-лы, 29 табл., 13 пр.
и, а также способ получения соединения (С) и способ получения соединения (2), соответствующего структурной формуле (1), где каждый Х1 и Х2 означает F. Способ позволяет получить соединение (1) со степенью превращения 99,9% в течение 1,25 часа, что значительно ускоряет известные ранее способы при осуществлении способа с другими катализаторами. 5н. и 52 з.п. ф-лы, 29 табл., 13 пр.
1. Способ получения соединения (1) или его фармацевтически приемлемой соли, где соединение (1) представлено следующей структурной формулой:
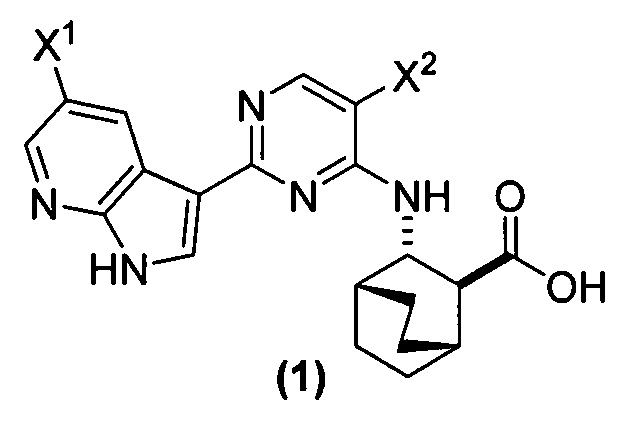 ,
,
включающий:
(a) взаимодействие соединения (X) 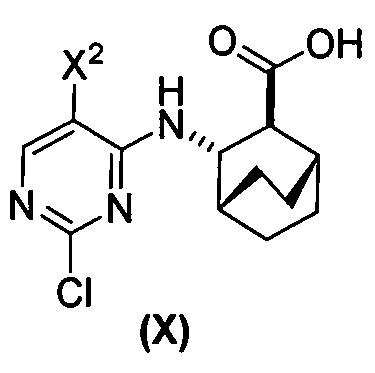 или его фармацевтически приемлемой соли с соединением (Y)
или его фармацевтически приемлемой соли с соединением (Y) 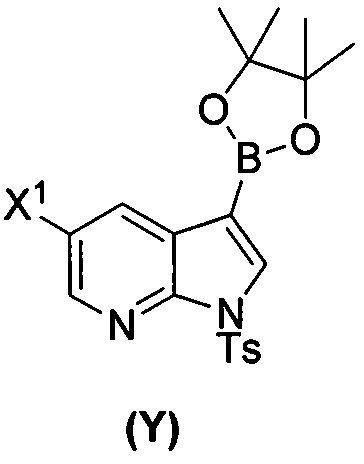 в присутствии палладиевого катализатора и основания с образованием соединения (Z)
в присутствии палладиевого катализатора и основания с образованием соединения (Z) 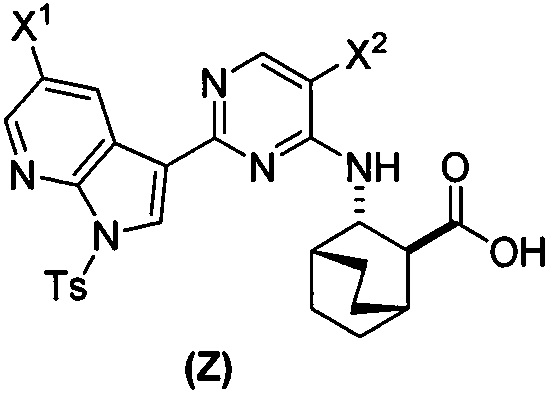 или его фармацевтически приемлемой соли; и
или его фармацевтически приемлемой соли; и
(b) удаление защитной группы Ts из соединения (Z) или его фармацевтически приемлемой соли с образованием соединения (1) или его фармацевтически приемлемой соли;
где:
каждый из X1 и X2 независимо представляет собой -F или -Cl;
Ts представляет собой тозил;
палладиевый катализатор содержит комплекс палладий-XPhos, в котором XPhos представляет собой 2-дициклогексилфосфино-2’,4’,6’-триизопропилбифенил;
основание представляет собой фосфатное основание или карбонатное основание, и
взаимодействие на стадии (а) проводят в системе растворителей, содержащей воду и органический растворитель, выбранный из 2-метил-тетрагидрофурана или тетрагидрофурана (ТГФ), или любого их сочетания.
2. Способ по п. 1, где комплекс палладий-XPhos получают in situ путем смешения Pd(OAc)2 и XPhos.
3. Способ по п.1, где каждый из X1 и X2 представляет собой -F.
4. Способ по п.1, где X1 представляет собой -Cl и X2 представляет собой -F.
5. Способ по любому из пп. 1-4, где основание представляет собой фосфатное основание или карбонатное основание, выбранное из Na2CO3, K2CO3, K3PO4 или Na3PO4.
6. Способ по п. 5, где основание выбирают из K2CO3 или K3PO4.
7. Способ по п. 1, где стадия (b) включает обработку соединения (Z) или его фармацевтически приемлемой соли неорганическим гидроксидом, включающим LiOH, NaOH, KOH или любое их сочетание.
8. Способ по п. 7, в котором стадия (b) включает обработку соединения (Z) или его фармацевтически приемлемой соли LiOH в системе растворителя, которая содержит THF.
9. Способ по п. 1, дополнительно включающий:
(c) взаимодействие соединения (F) 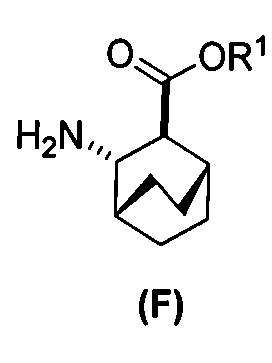 или его фармацевтически приемлемой соли с соединением (G)
или его фармацевтически приемлемой соли с соединением (G) 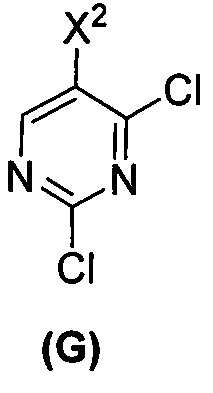 с образованием соединения (H)
с образованием соединения (H) 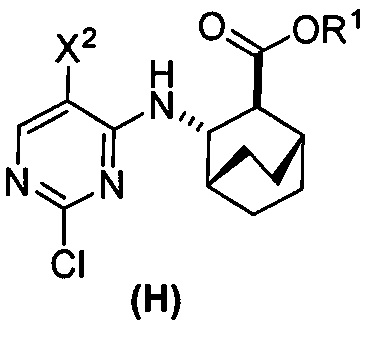 или его фармацевтически приемлемой соли, где R1 представляет собой C1-4-алкил; и
или его фармацевтически приемлемой соли, где R1 представляет собой C1-4-алкил; и
(d) гидролиз соединения (H) или его фармацевтически приемлемой соли с образованием соединения (X) 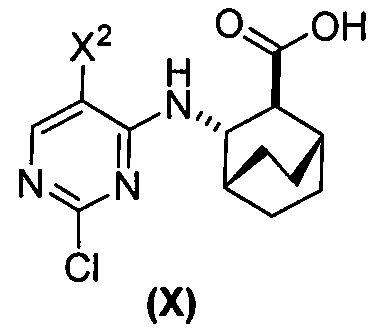 или его фармацевтически приемлемой соли.
или его фармацевтически приемлемой соли.
10. Способ по п. 9, дополнительно включающий:
(e) взаимодействие соединения (C) 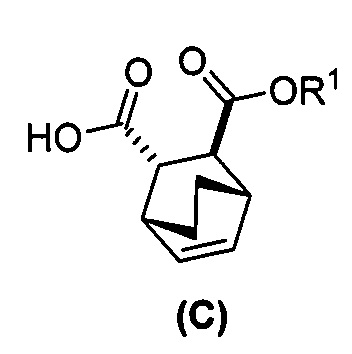 или его фармацевтически приемлемой соли с дифенилфосфорилазидом и с бензиловым спиртом с образованием соединения (D)
или его фармацевтически приемлемой соли с дифенилфосфорилазидом и с бензиловым спиртом с образованием соединения (D) 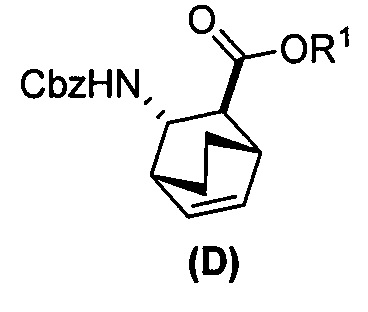 или его фармацевтически приемлемой соли, где Cbz представляет собой карбоксибензил; и
или его фармацевтически приемлемой соли, где Cbz представляет собой карбоксибензил; и
(f) взаимодействие соединения (D) или его фармацевтически приемлемой соли с H2 в присутствии Pd-катализатора на угле с образованием соединения (F) 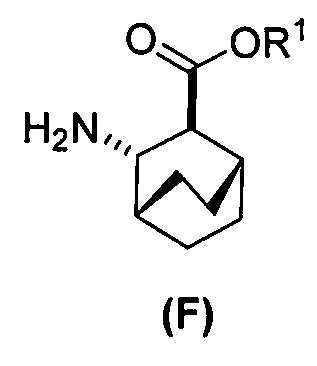 или его фармацевтически приемлемой соли.
или его фармацевтически приемлемой соли.
11. Способ по п. 10, дополнительно включающий:
(g) взаимодействие соединения (A) 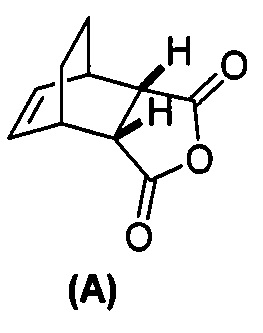 с хинином
с хинином 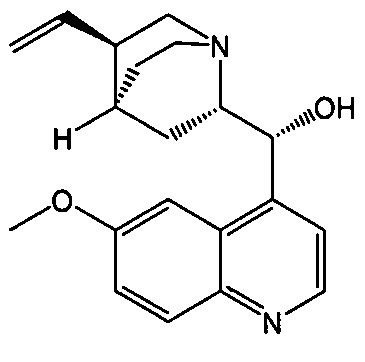
и R1-OH при температуре от -12°C до -16°C с образованием аддукта хинина и соединения (C-1) 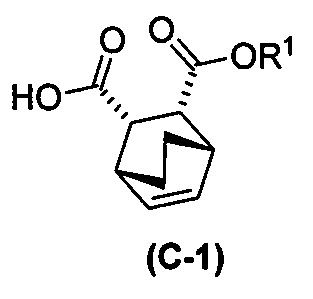 , где R1 представляет собой C1-4-алкил;
, где R1 представляет собой C1-4-алкил;
(h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl при температуре менее чем 25°C с образованием соединения (C-1) или его фармацевтически приемлемой соли; и
(i) эпимеризацию соединения (C-1) или его фармацевтически приемлемой соли с образованием соединения (C) 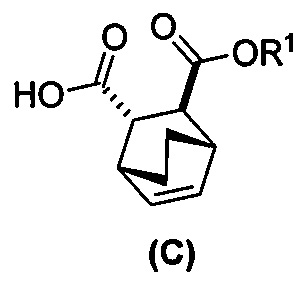 или его фармацевтически приемлемой соли,
или его фармацевтически приемлемой соли,
где эпимеризация на стадии (i) включает обработку соединения (C-1) C1-6-алкоксидом щелочного металла при температуре от -15°C до -22°C.
12. Способ по п. 11, где C1-6-алкоксид щелочного металла включает трет-бутоксид, трет-амилат или любое их сочетание.
13. Способ по п. 11, где R1 представляет собой этил.
14. Способ по п. 9, дополнительно включающий гидрирование соединения (S) 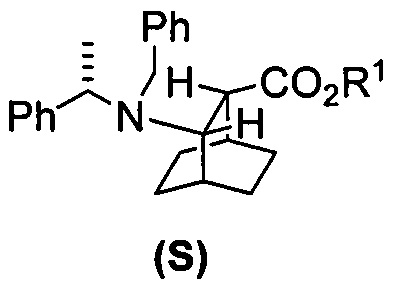 или его фармацевтически приемлемой соли, где Ph представляет собой фенил, в присутствии палладиевого катализатора с образованием соединения (F) или его фармацевтически приемлемой соли, где палладиевый катализатор представляет собой Pd(0) на угле (Pd(0)/C), Pd(OH)2 на угле или любое их сочетание.
или его фармацевтически приемлемой соли, где Ph представляет собой фенил, в присутствии палладиевого катализатора с образованием соединения (F) или его фармацевтически приемлемой соли, где палладиевый катализатор представляет собой Pd(0) на угле (Pd(0)/C), Pd(OH)2 на угле или любое их сочетание.
15. Способ по п. 14, дополнительно включающий взаимодействие соединения (R)  с S-(-)-N-бензил-альфа-метилбензиламинолитием с образованием соединения (S)
с S-(-)-N-бензил-альфа-метилбензиламинолитием с образованием соединения (S) 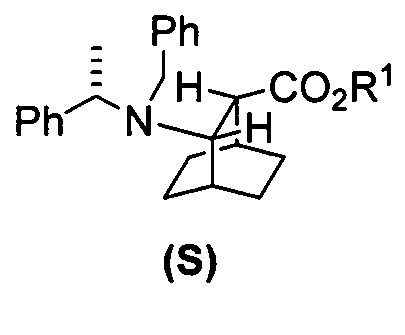 или его фармацевтически приемлемой соли.
или его фармацевтически приемлемой соли.
16. Способ по п. 15, дополнительно включающий:
(j) взаимодействие 1,3-циклогексадиена с CH≡CHC(O)OR1 в присутствии алюминиевого катализатора с образованием соединения (Q) 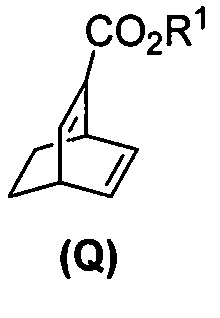 , где R1 представляет собой C1-4-алкил, и
, где R1 представляет собой C1-4-алкил, и
(k) гидрирование соединения (Q) с образованием соединения (R) 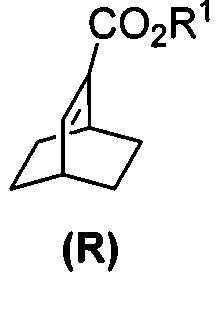 ,
,
где гидрирование соединения (Q) на стадии (k) представляет собой взаимодействие соединения (Q) с H2 в присутствии Rh(I)-катализатора или отравленного Pd(0)-катализатора.
17. Способ по п. 16, где R1 представляет собой этил.
18. Способ по п. 16, где алюминиевый катализатор представляет собой EtAlCl2, Et2AlCl, смесь AlCl3 и триоктилалюминия или любое их сочетание.
19. Способ по п. 16, где Rh(I)-катализатор представляет собой (PPh3)3RhCl, смесь (PPh3)3RhCl и этилропиолата или любое их сочетание, где Ph представляет собой фенил.
20. Способ по п. 16, где отравленный Pd(0)-катализатор представляет собой отравленный свинцом Pd(0)-катализатор на CaCO3 (Pd(Pb)/CaCO3).
21. Способ по п. 1, дополнительно включающий взаимодействие соединения (O)  с бис(пинаколато)дибором в присутствии палладиевого катализатора, содержащего фосфиновый лиганд, с образованием соединения (Y)
с бис(пинаколато)дибором в присутствии палладиевого катализатора, содержащего фосфиновый лиганд, с образованием соединения (Y) 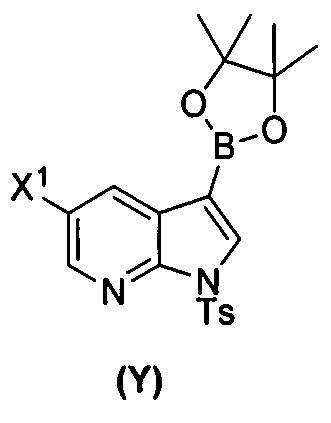 .
.
22. Способ по п. 21, где палладиевый катализатор, содержащий фосфиновый лиганд, представляет собой Pd(Ph3P)4.
23. Способ по п. 21, дополнительно включающий обработку соединения (N) 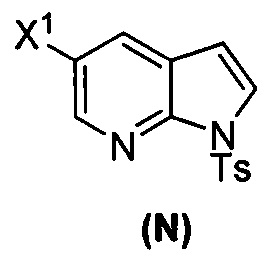 бромирующим агентом, представляющим собой Br2, N-бромсукцинимид, 1,3-дибром-5,5-диметилгидантоин или любое их сочетание, с образованием соединения (O).
бромирующим агентом, представляющим собой Br2, N-бромсукцинимид, 1,3-дибром-5,5-диметилгидантоин или любое их сочетание, с образованием соединения (O).
24. Способ по любому из пп. 21-23, дополнительно включающий:
l) взаимодействие соединения (J) 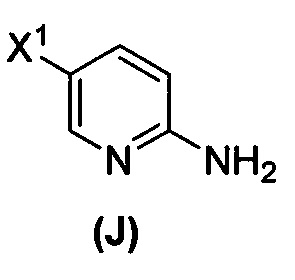 или его фармацевтически приемлемой соли с иодирующим агентом или бромирующим агентом с образованием соединения (K)
или его фармацевтически приемлемой соли с иодирующим агентом или бромирующим агентом с образованием соединения (K) 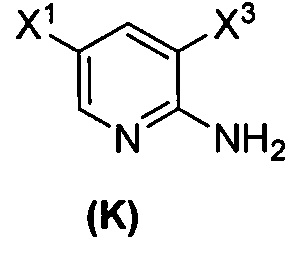 или его фармацевтически приемлемой соли, где X3 представляет собой Br или I;
или его фармацевтически приемлемой соли, где X3 представляет собой Br или I;
(m) взаимодействие соединения (K) или его фармацевтически приемлемой соли с триметилсилилацетиленом с образованием соединения (L) 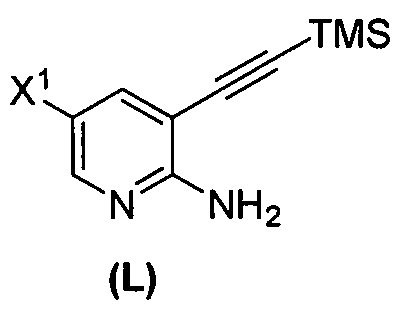 или его фармацевтически приемлемой соли, где TMS представляет собой триметилсилил;
или его фармацевтически приемлемой соли, где TMS представляет собой триметилсилил;
(n) взаимодействие соединения (L) или его фармацевтически приемлемой соли с C1-6-алкоксидным основанием с образованием соединения (P) 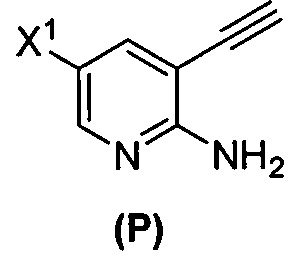 или его фармацевтически приемлемой соли;
или его фармацевтически приемлемой соли;
(o) взаимодействие соединения (P) с трет-бутоксидом калия, трет-амилатом калия или любым их сочетанием с образованием соединения (M) 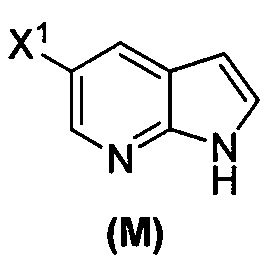 или его фармацевтически приемлемой соли; и
или его фармацевтически приемлемой соли; и
(p) тозилирование соединения (M) или его фармацевтически приемлемой соли с образованием соединения (N) 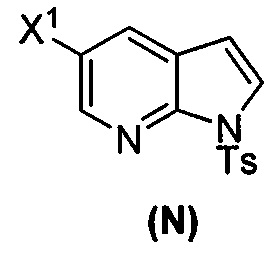 .
.
25. Способ по п. 24, где C1-6-алкоксидное основание включает трет-амилат калия, трет-бутоксид калия, метоксид калия, трет-амилат натрия, трет-бутоксид натрия, метоксид натрия или любые их сочетания.
26. Способ по п. 24, где реакцию соединения (K) или его фармацевтически приемлемой соли с триметилсилилацетиленом осуществляют в присутствии палладиевого катализатора, представляющего собой Pd(Ph3P)4, Pd(PPh3)2Cl2, Pd(dppf)2Cl2 или любое их сочетание, катализатора, представляющего собой галогенид меди (I), или любого их сочетания.
27. Способ по п. 26, где взаимодействие соединения (K) или его фармацевтически приемлемой соли с триметилсилилацетиленом осуществляют в присутствии CuI, Pd(Ph3P)4, Pd(PPh3)2Cl2, Pd(dppf)2Cl2 или любого их сочетания.
28. Способ по п. 24, где стадию тозилирования(p) осуществляют путем проведения реакции соединения (M) или его фармацевтически приемлемой соли с TsCl.
29. Способ по п. 24, где соединение (J) или его фармацевтически приемлемую соль вводят в реакцию с иодирующим агентом, представляющим собой I2, ICl, N-иодсукцинимид, и где X3 представляет собой I.
30. Способ по п. 29, где иодирующий агент представляет собой I2.
31. Способ по п. 24, где соединение (J) или его фармацевтически приемлемую соль вводят в реакцию с бромирующим агентом, представляющим собой Br2, N-бромсукцинимид, 1,3-дибром-5,5-диметилгидантоин или любое их сочетание, и где X3 представляет собой Br.
32. Способ по п. 31, где бромирующий агент представляет собой is Br2.
33. Способ по п. 23, дополнительно включающий:
(q) взаимодействие соединения (K) 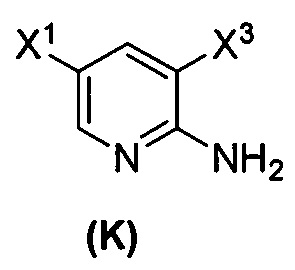 или его фармацевтически приемлемой соли с ацетальдегидом в присутствии палладиевого катализатора, представляющего собой смесь бис(дибензилиденацетон)палладия и третичного фосфинового лиганда, PR3, где R представляет собой C1-6-алкил или C5-6-циклоалкил, с образованием соединения (M)
или его фармацевтически приемлемой соли с ацетальдегидом в присутствии палладиевого катализатора, представляющего собой смесь бис(дибензилиденацетон)палладия и третичного фосфинового лиганда, PR3, где R представляет собой C1-6-алкил или C5-6-циклоалкил, с образованием соединения (M) 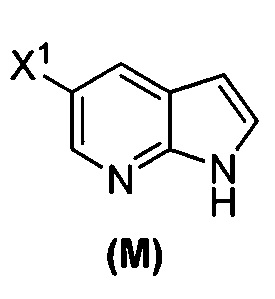 или его фармацевтически приемлемой соли, где X3 представляет собой Br или I; и
или его фармацевтически приемлемой соли, где X3 представляет собой Br или I; и
(p) тозилирование соединения (M) или его фармацевтически приемлемой соли с образованием соединения (N) 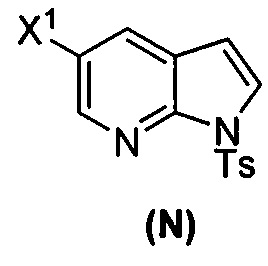 .
.
34. Способ по п. 33, где третичный фосфиновый лиганд, PR3, представляет собой P(tBu)3, PCy3, P(i-Pr)3, P(Bu3), PEt3, PMe3 или любое их сочетание.
35. Способ по п. 34, третичный фосфиновый лиганд представляет собой P(tBu)3.
36. Способ по п. 1, дополнительно включающий проведение после стадии (b) удаления защиты стадии обработки соединения (1) HCl в системе растворителя, содержащей воду и один или более органических растворителей, с образованием соли соединения (1) с HCl, где органический растворитель выбирают из ацетонитрила, хлорбензола, хлороформа, циклогексана, 1,2-дихлорэтена, дихлорметана, 1,2-диметоксиэтана, N,N-диметилацетамида, N,N-диметилформамида, 1,4-диоксана, 2-этоксиэтанола, этиленгликоля, формамида, гексана, метанола, 2-метоксиэтанола, метилбутилкетона, метилциклогексана, N-метилпирролидона, нитрометана, пиридина, сульфолана, тетрагидрофурана (ТГФ), тетралина, толуола, 1,1,2-трихлорэтена, ксилола, уксусной кислоты, ацетона, анизола, 1-бутанола, 2-бутанола, бутилацетата, трет-бутилметилового простого эфира, кумола, гептана, изобутилацетата, изопропилацетата, метилацетата, 3-метил-1-бутанола, метилэтилкетона, метилизобутилкетона, 2-метил-1-пропанола, диметилсульфоксида, этанола, этилацетата, этилового простого эфира, этилформиата, муравьиной кислоты, пентана, 1-пентанола, изопропанол, 1-пропанола, 2-пропанола, пропилацетата или любого их сочетания.
37. Способ по п. 36, где органические растворители системы растворителя выбирают из группы, состоящей из 2-этоксиэтанола, этиленгликоля, метанола, 2-метоксиэтанола, 1-бутанола, 2-бутанола, 3-метил-1-бутанола, 2-метил-1-пропанола, этанола, 1-пентанола, 1-пропанола, 2-пропанола, метилбутилкетона, ацетона, метилэтилкетона, метилизобутилкетона, изопропанол, бутилацетата, изобутилацетата, изопропилацетата, метилацетата, этилацетата, пропилацетата, пиридина, толуола и ксилола.
38. Способ по п. 37, где система растворителя включает воду и ацетон или воду и изопропанол.
39. Способ получения соединения (1) или его фармацевтически приемлемой соли, где соединение (1) представлено следующей структурной формулой:
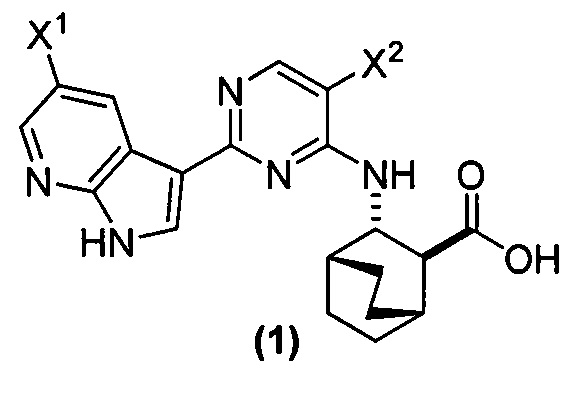 ,
,
включающий:
(g) взаимодействие соединения (A) 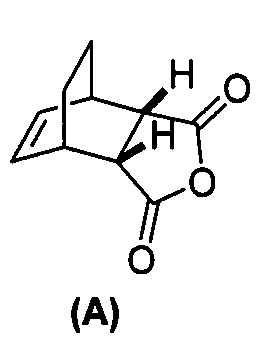 с хинином
с хинином 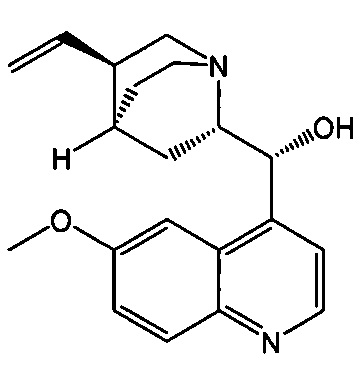 и этиловым спиртом при температуре от -12°C до -16°C с образованием аддукта хинина и соединения (C-1)
и этиловым спиртом при температуре от -12°C до -16°C с образованием аддукта хинина и соединения (C-1) 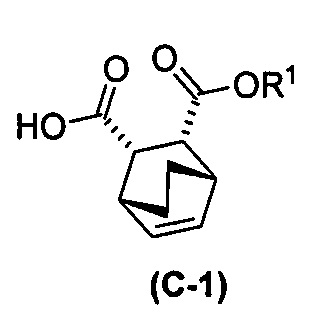 ;
;
(h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl при температуре менее чем 25°C с образованием соединения (C-1) или его фармацевтически приемлемой соли;
(i) эпимеризацию соединения (C-1) или его фармацевтически приемлемой соли с образованием соединения (C) 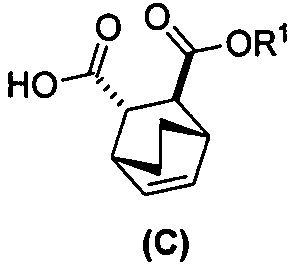 или его фармацевтически приемлемой соли,
или его фармацевтически приемлемой соли,
где эпимеризации включает обработку соединения (C-1) C1-6-алкоксидом щелочного металла при температуре от -15°C до
-22°C;
(e) взаимодействие соединения (C) или его фармацевтически приемлемой соли с дифенилфосфорилазидом и бензиловым спиртом с образованием соединения (D) 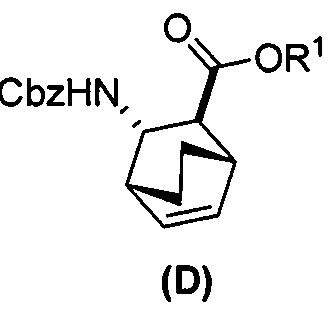 , где Cbz представляет собой карбоксибензил;
, где Cbz представляет собой карбоксибензил;
(f) взаимодействие соединения (D) или его фармацевтически приемлемой соли с H2 в присутствии Pd-катализатора на угле (Pd(0)/C) с образованием соединения (F) 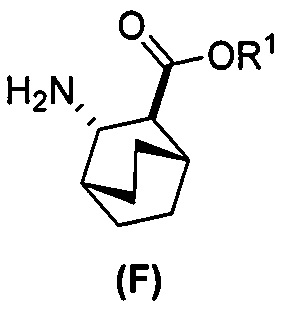 или его фармацевтически приемлемой соли;
или его фармацевтически приемлемой соли;
(c) взаимодействие соединения (F) или его фармацевтически приемлемой соли с соединением (G) 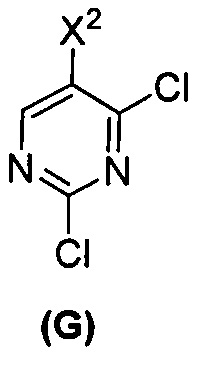 с образованием соединения (H)
с образованием соединения (H) 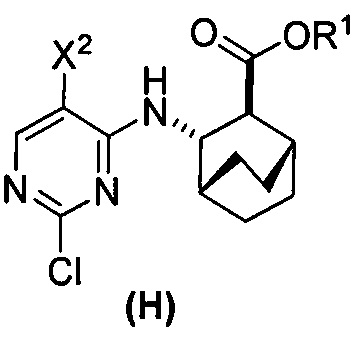 или его фармацевтически приемлемой соли;
или его фармацевтически приемлемой соли;
(d) гидролиз соединения (H) или его фармацевтически приемлемой соли с образованием соединения (X) 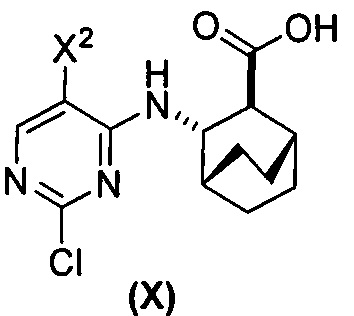 или его фармацевтически приемлемой соли;
или его фармацевтически приемлемой соли;
(a) взаимодействие соединения (X) или его фармацевтически приемлемой соли с соединением (Y) 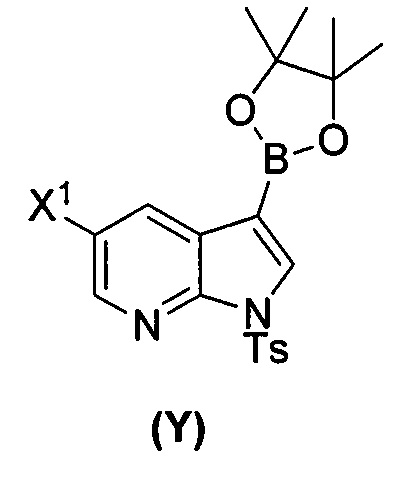 в присутствии палладиевого катализатора и фосфатного или карбонатного основания в системе растворителей, содержащей воду и органический растворитель, выбранный из из 2-метил-ТГФ или ТГФ, или любого их сочетания с образованием соединения (Z)
в присутствии палладиевого катализатора и фосфатного или карбонатного основания в системе растворителей, содержащей воду и органический растворитель, выбранный из из 2-метил-ТГФ или ТГФ, или любого их сочетания с образованием соединения (Z) 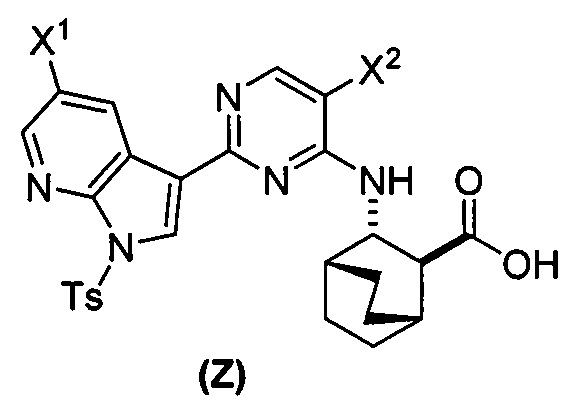 или его фармацевтически приемлемой соли,
или его фармацевтически приемлемой соли,
где палладиевый катализатор содержит комплекс палладий-Xphos, где XPhos представляет собой 2-дициклогексилфосфино-2’,4’,6’-триизопропилбифенил; и
(b) удаление защитной группы Ts из соединения (Z) или его фармацевтически приемлемой соли с образованием соединения (1) или его фармацевтически приемлемой соли; и
где: X1 и X2 независимо представляют собой -F или -Cl;
Ts представляет собой тозил;
и каждый R1 независимо представляет собой этил.
40. Способ по п. 39, где карбонатное основание включает Na2CO3, K2CO3 или их сочетание и фосфатное основание включает K3PO4, Na3PO4 или их сочетание.
41. Способ по п. 40, где карбонатное основание включает K2CO3 и фосфатное основание включает K3PO4.
42. Способ по п. 39, где основание включает K2CO3.
43. Способ по п. 39, где стадия (b) удаления защиты включает обработку соединения (Z) или его фармацевтически приемлемой соли неорганическим гидроксидом, включающим LiOH, NaOH, KOH или любое их сочетание.
44. Способ по п. 39, где каждый из X1 и X2 представляет собой -F.
45. Способ по п. 39, где X1 представляет собой -Cl и X2 представляет собой -F.
46. Способ по п. 39, где C1-6-алкоксид щелочного металла включает трет-бутоксид,трет-амилат или любое их сочетание.
47. Способ получения соединения (1) или его фармацевтически приемлемой соли, где соединение (1) представлено следующей структурной формулой:
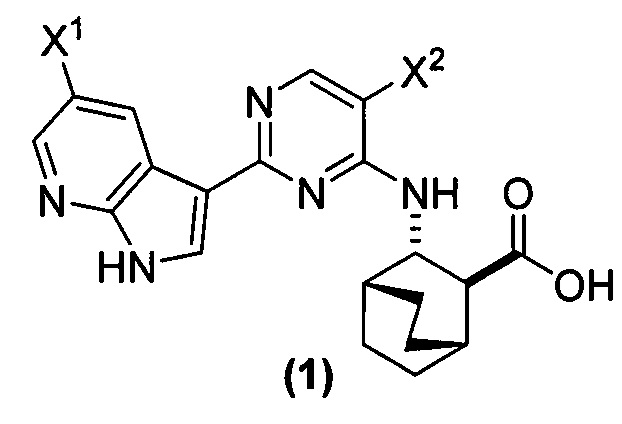 ,
,
включающий:
(q) взаимодействие соединения (K) 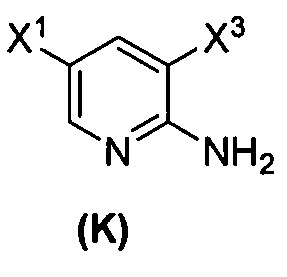 или его фармацевтически приемлемой соли с ацетальдегидом в присутствии первого палладиевого катализатора с образованием соединения (M)
или его фармацевтически приемлемой соли с ацетальдегидом в присутствии первого палладиевого катализатора с образованием соединения (M) 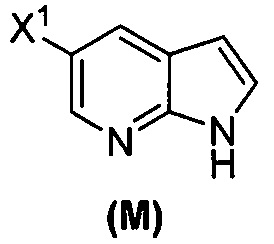 или его фармацевтически приемлемой соли;
или его фармацевтически приемлемой соли;
(p) тозилирование соединения (M) или его фармацевтически приемлемой соли с образованием соединения (N) 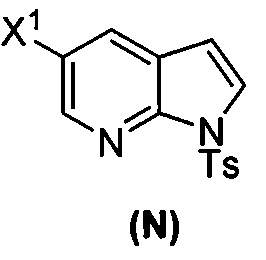 ;
;
(s) бромирование соединения (N) с образованием соединения (O) 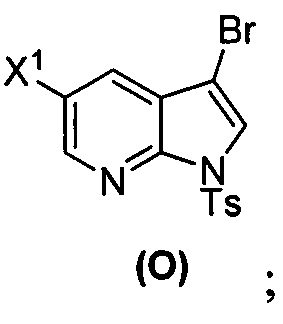
(t) взаимодействие соединения (O) с бис(пинаколато)дибором в присутствии палладиевого катализатора с образованием соединения (Y) 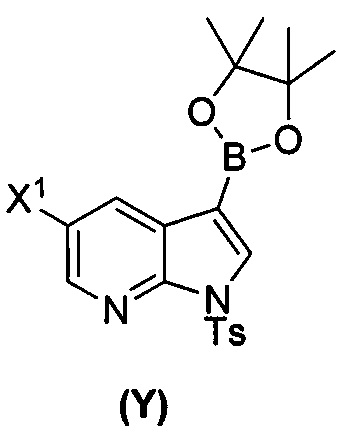 ;
;
(a) взаимодействие соединения (X)  или его фармацевтически приемлемой соли с соединением (Y) в присутствии второго палладиевого катализатора с образованием соединения (Z)
или его фармацевтически приемлемой соли с соединением (Y) в присутствии второго палладиевого катализатора с образованием соединения (Z) 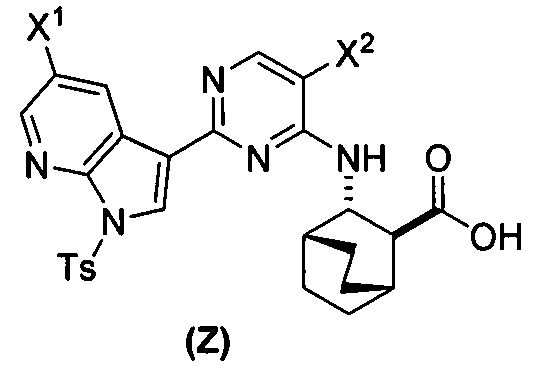 или его фармацевтически приемлемой соли; и
или его фармацевтически приемлемой соли; и
(b) удаление защитной группы Ts из соединения (Z) или его фармацевтически приемлемой соли с образованием соединения (1) или его фармацевтически приемлемой соли, где X1 и X2 независимо представляют собой -F или -Cl; X3 представляет собой -Br; и Ts представляет собой тозил.
48. Способ по п. 47, где второй палладиевый катализатор стадии (а) содержит комплекс палладий-Xphos и фосфатное основание или карбонатное основание, где XPhos представляет собой 2-дициклогексилфосфино-2’,4’,6’-триизопропилбифенил.
49. Способ по п. 48, где фосфатное основание представляет собой K3PO4 и карбонатное основание представляет собой K2CO3.
50. Способ по п. 47, где стадия (b) удаления защиты представляет собой обработку соединения (Z) или его фармацевтически приемлемой соли неорганическим гидроксидом, включающим LiOH, NaOH, KOH или любое их сочетание.
51. Способ по любому из пп. 47-50, где первый палладиевый катализатор стадии (q) содержит смесь бис(дибензилиденацетон)палладия и третичного фосфинового лиганда, PR3, где R представляет собой C1-6-алкил или C5-6-циклоалкил.
52. Способ по п. 51, где третичный фосфиновый лиганд включает P(tBu)3.
53. Способ по п. 47, где каждый из X1 и X2 представляет собой -F.
54. Способ по п. 47 , где X1 представляет собой -Cl и X2 представляет собой -F.
55. Способ получения соединения (2) или его фармацевтически приемлемой соли, где соединение (2) представлено следующей структурной формулой:
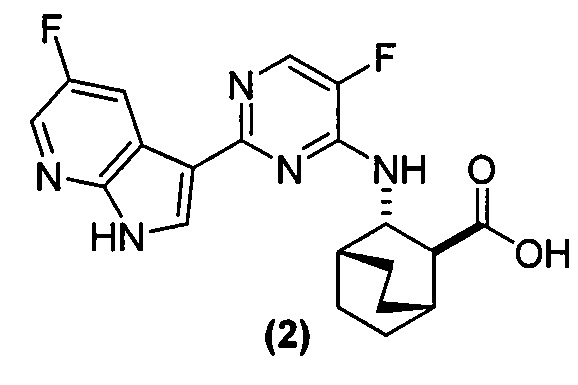 ,
,
включающий:
(g) взаимодействие соединения (A) 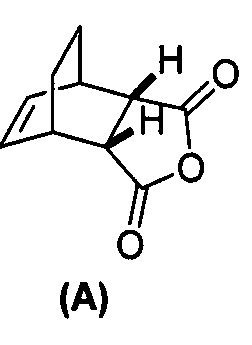 с хинином
с хинином  и этиловым спиртом при температуре от -12°C до -16°C с образованием аддукта хинина и соединения (C-1)
и этиловым спиртом при температуре от -12°C до -16°C с образованием аддукта хинина и соединения (C-1) 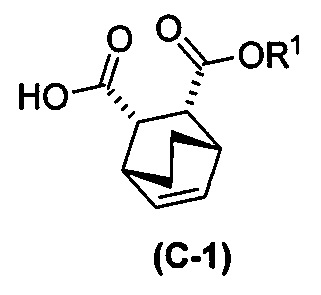 ;
;
(h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl при температуре менее чем 25°C с образованием соединения (C-1) или его фармацевтически приемлемой соли;
(i) эпимеризация соединения (C-1) или его фармацевтически приемлемой соли, где эпимеризация представляет собой обработку соединения (C-1) C1-6-алкоксидом щелочного металла, выбранным из трет-бутоксида щелочного металла или трет-амилата щелочного металла при температуре от -15°C до -22°C с образованием соединения (C) 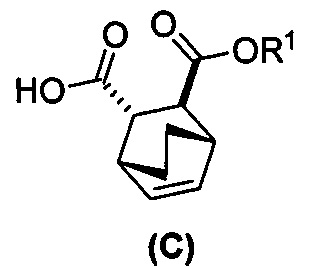 или его фармацевтически приемлемой соли;
или его фармацевтически приемлемой соли;
(e) взаимодействие соединения (C) с дифенилфосфорилазидом, а затем с бензиловым спиртом с образованием соединения (D) 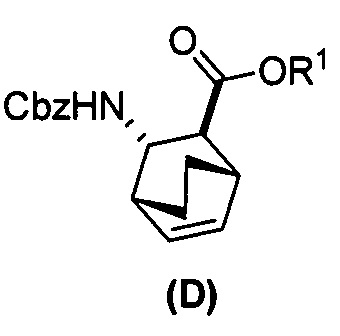 ;
;
(f) взаимодействие соединения (D) или его фармацевтически приемлемой соли с H2 в присутствии Pd-катализатора на угле (Pd(0)/C) с образованием соли соединения (F) 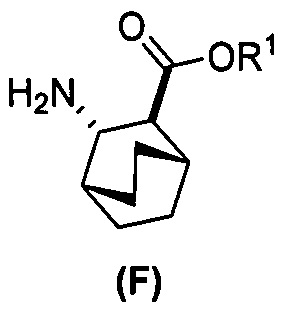 с HCl;
с HCl;
(r) взаимодействие соли соединения (F) с HCl с соединением (G) 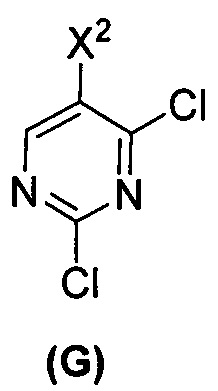 с образованием соединения (H)
с образованием соединения (H) 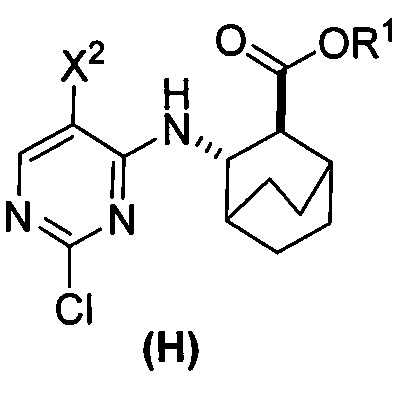 ;
;
(h-1) гидролиз соединения (H) с образованием соединения (X-2) 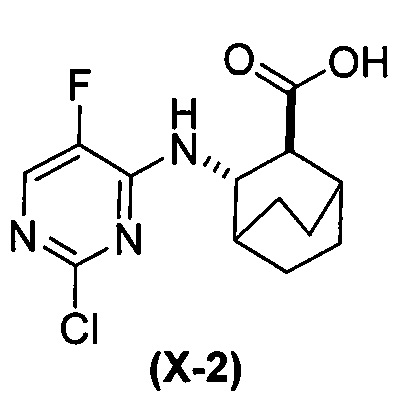 ;
;
(l) иодирование или бромирование соединения (J) 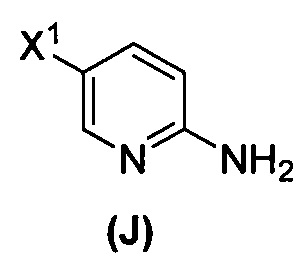 с образованием соединения (K)
с образованием соединения (K) 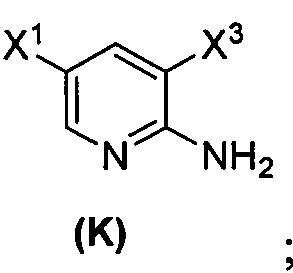 ;
;
(q-1) взаимодействие соединения (K) с триметилсилилацетиленом с образованием соединения (L) 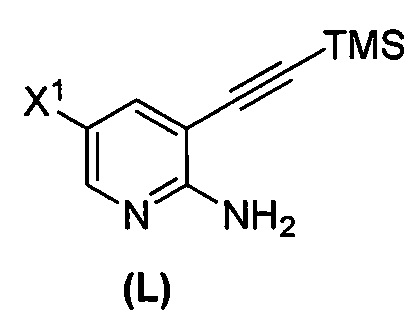 ;
;
(j) взаимодействие соединения (L) с C1-6-алкоксидом с образованием соединения (M) 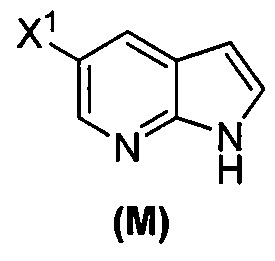 ;
;
(k) тозилирование соединения (M) с образованием соединения (N) 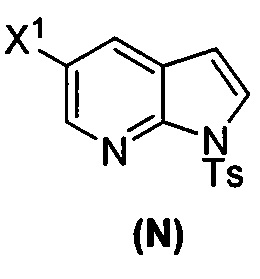 ;
;
(s) бромирование соединения (N) с образованием соединения (O) 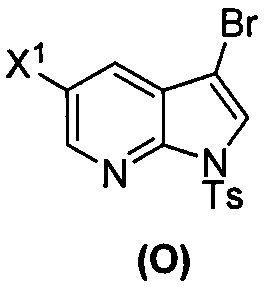 ;
;
(t) взаимодействие соединения (O) с бис(пинаколато)дибором в присутствии Pd(Ph3P)4 с образованием соединения (Y-2)  ;
;
(n) взаимодействие соединения (X-2) с соединением (Y-2) в присутствии комплекса палладий-XPhos и фосфатного или карбонатного основания, выбранного из K2CO3 или K3PO4, с образованием соединения (Z-2): 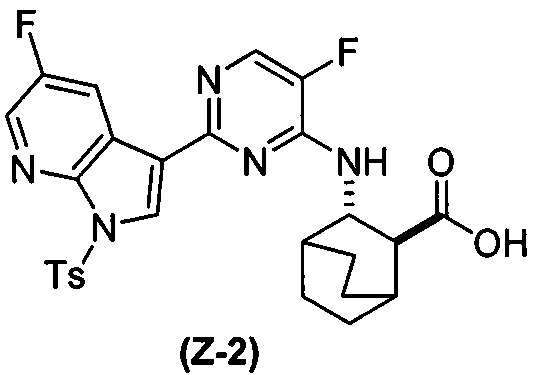 или его фармацевтически приемлемой соли; и
или его фармацевтически приемлемой соли; и
(o) удаление защитной группы Ts из соединения (Z-2) или его фармацевтически приемлемой соли с образованием соединения (2) или его фармацевтически приемлемой соли; и где:
Cbz представляет собой каробксибензил;
XPhos представляет собой 2-дициклогексилфосфино-2’,4’,6’-триизопропилбифенил;
Ts представляет собой тозил;
каждый R1 независимо представляет собой этил;
каждый X1 независимо представляет собой F;
каждый X2 независимо представляет собой F; и
каждый X3 независимо представляет собой Br или I.
56. Способ получения соединения (C) или его фармацевтически приемлемой соли, включающий:
(g) взаимодействие соединения (A) 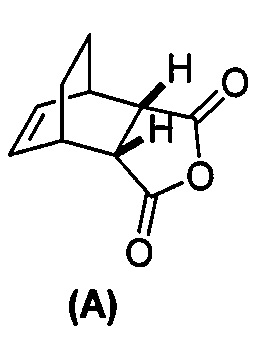 с хинином
с хинином 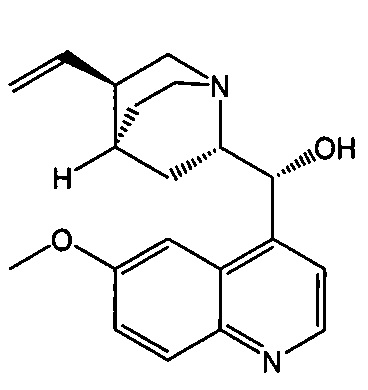 и этиловым спиртом при температуре от -12°C до -16°C с образованием аддукта хинина и соединения (C-1)
и этиловым спиртом при температуре от -12°C до -16°C с образованием аддукта хинина и соединения (C-1) 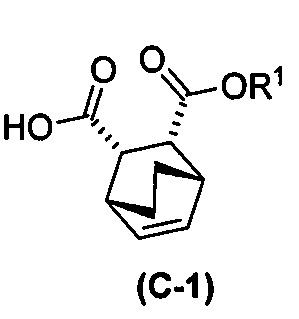 , где R1 представляет собой этил;
, где R1 представляет собой этил;
(h) разрушение аддукта хинина и соединения (C-1) путем обработки аддукта HCl при температуре менее чем 25°C с образованием соединения (C-1) или его фармацевтически приемлемой соли; и
(i) эпимеризацию соединения (C-1) или его фармацевтически приемлемой соли с образованием соединения (C) 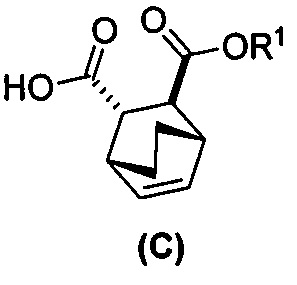 или его фармацевтически приемлемой соли, где эпимеризация на стадии (i) представляет собой обработку соединения (C-1) C1-6-алкоксидом щелочного металла при температуре от -15°C до -22°C.
или его фармацевтически приемлемой соли, где эпимеризация на стадии (i) представляет собой обработку соединения (C-1) C1-6-алкоксидом щелочного металла при температуре от -15°C до -22°C.
57. Способ по п. 56, где C1-6-алкоксид щелочного металла представляет собой трет-бутоксид, трет-амилат или любое их сочетание.
| WO 2010148197 А1, 23.12.2010 | |||
| US 4349562 A1, 14.09.1982 | |||
| Conception Nemecek et al., Design of Potent IGF1-R Inhibitors Related to Bis-azaindoles, Chem Biol Drug Des, 2010; 76:100-106 | |||
| JAESCHKE G | |||
| et al., Highly Enantioselective Opening of Cyclic meso-Anhydrides to Isopropyl Hemiesters with TI-TADDOLates: AN Alternative Hidrolitic Enzimes, Journal of Organic Chemistry,1998, vol.63, pages 1190-1197 | |||
| Zhengren Xu et al., Palladium-Catalyzed Indole and Azaindole Synthesis by Direct Annulation of Electron-Poor o-Chloroanilines and o-Chloroaminopyridines with Aldehydes, Synthesis, 2008, no.24, p.3981-3987 | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Способ прикрепления барашков к рогулькам мокрых ватеров | 1922 |
|
SU174A1 |
| АЗАИНДОЛЫ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ JAK И ДРУГИХ ПРОТЕИНКИНАЗ | 2005 |
|
RU2403252C2 |
| KHASELEV N | |||
| et al., The Role of the C-C double bond in alcohol elimination from NH+ ions of unsaturated bicyclyc esters upon chemical ionisation, Journal of mass spectrometry, vol.30, no.11, pp.1533-1538. | |||

