Настоящее изобретение относится к химической технологии производства катализаторов, в частности катализаторов гидрокрекинга и гидроочистки тяжелых остатков нефти, вязкой и высоковязкой нефти.
Остаточные и вновь открываемые месторождения нефти включают большое количество трудноизвлекаемых запасов. Это так называемые вязкие и высоковязкие нефти. Основными недостатками данного типа нефтей являются высокое содержание гетероатомных соединений азота и серы, тяжелых металлов, смолистых и асфальтеновых компонентов, высокая плотность и вязкость. Данные факторы приводят к сложностям при добыче и транспортировке нефти. Это определяет необходимость разработки новых способов снижения вязкости нефти непосредственно на месторождениях. При этом разработка новых технологий позволит включить в переработку тяжелые остатки нефти на нефтеперерабатывающих заводах с увеличением выхода целевых продуктов.
В настоящий момент времени для переработки тяжелых остатков нефти, вязкой и высоковязкой нефти наибольший интерес приобретают процессы гидропереработки. Несмотря на необходимость наличия значительных количеств водорода и капиталовложений данные процессы позволяют существенно увеличить выход светлых дистиллятов и, соответственно, целевых продуктов.
Известен катализатор (Патент Российская федерация №2414297 «Катализатор на носителе нерегулярной, несферической формы и способ гидроконверсии тяжелых нефтяных фракций», МПК B01J 21/04, B01J 23/40, B01J 23/24, B01J 23/74, B01J 35/02, B01J 35/10, C10G 47/10, C10G 45/04, приоритет от 14.12.2006, опубликован 20.03.2011) для гидрообработки и/или гидроконверсии тяжелых углеводородов. Этот катализатор включает носитель на основе оксида алюминия, по меньшей мере, один каталитический металл или соединение каталитического металла группы VIB и/или VIII, пористая структура которого состоит из множества соприкасающихся агломератов, образованных каждый множеством иглообразных пластинок. Пластинки каждого агломерата, как правило, ориентированы радиально одни по отношению к другим и по отношению к центру агломерата. Вышеуказанный носитель имеет нерегулярную и несферическую форму и находится в большинстве своем в виде фрагментов, получаемых путем дробления шариков из оксида алюминия. Носитель получают согласно способу, включающему следующие стадии: а) гранулирование, исходя из порошка активного оксида алюминия, обладающего плохо кристаллизованной и/или аморфной структурой, с целью получения агломератов в виде шариков; b) созревание во влажной атмосфере при температуре в диапазоне от 60°С до 100°С, затем высушивание вышеуказанных шариков; с) просеивание для рекуперации фракции вышеуказанных шариков; d) дробление вышеуказанной фракции; е) прокаливание, по меньшей мере, части вышеуказанной дробленой фракции при температуре в диапазоне от 250°С до 900°С; f) кислотное импрегнирование и гидротермальная обработка при температуре в диапазоне от 80°С до 250°С; g) высушивание, затем прокаливание при температуре в диапазоне от 500°С до 1100°С. Технический результат - описанный катализатор проявляет улучшенные рабочие характеристики при его использовании для гидроконверсии/гидрообработки тяжелых углеводородов. Недостатком данного катализатора является многостадийная процедура его приготовления.
Разработан катализатор гидрокрекинга углеводородного сырья (Патент Российская федерация №2607908 «Способ приготовления катализатора гидрокрекинга углеводородного сырья», МПК - B01J 37/02, B01J 38/62, B01J 20/16, B01J 23/883, C10G 47/12, приоритет от 05.11.2015, опубликован 11.01.2017). Катализатор включает в свой состав никель, молибден, алюминий и кремний. Способ включает приготовление гранулированного носителя, содержащего оксид алюминия и 50-80 масс % аморфного алюмосиликата с массовым отношением Si/Al от 0,6 до 0,85, нанесение на полученный гранулированный носитель биметаллических комплексных соединений [Ni(H2O)x(L)y]2[Mo4O11(C6H5O7)2], где L - частично депротонированная форма лимонной кислоты С6Н5О7; х=0 или 2; у=0 или 1, сушку катализатора при температуре 100-250°С. Компоненты в получаемом катализаторе содержатся в следующих концентрациях, масс %: [Ni(H2O)x(L)y]2[Mo4O11(C6H5O7)2] - 13,1-23,3; аморфный алюмосиликат - 40,0-61,3; γ-Al2O3 - остальное, что соответствует содержанию в прокаленном при 550°С катализаторе, масс %: MoO3 - 7,0-13,0; NiO - 1,8-3,4; аморфный алюмосиликат - 43,1-66,9; γ-Al2O3 - остальное. Технический результат - использование в гидрокрекинге катализатора, приготовленного указанным способом, обеспечивает получение дизельной фракции с высоким выходом. Недостатком данного катализатора является низкая активность.
Наиболее близким аналогом выбран катализатор по патенту Российской Федерации №2506997 «Катализатор переработки тяжелых нефтяных фракций», МПК B01J 23/74, B01J 23/24, B01J 35/10, В82В 1/00, C10G 45/00, приоритет от 27.08.2012, опубликован 20.02.2014. В катализаторе активный компонент, выбираемый из соединений никеля, или кобальта, или молибдена, или вольфрама или любой их комбинации, нанесен на неорганический пористый носитель, состоящий из оксида алюминия, диоксидов кремния, титана или циркония, алюмосиликатов или железосиликатов, или любой их комбинации, отличающийся тем, что указанный катализатор содержит макропоры, образующие регулярную пространственную структуру макропор, причем доля макропор размером более 50 нм составляет не менее 30% в общем удельном объеме пор указанного катализатора. Данный катализатор обладает недостаточной активностью, невысоким значением массообмена за счет невысоких диффузионных характеристик, низким значением межрегенерационного периода работы.
Задачей предлагаемого изобретения является увеличение активности катализатора, длительности его межрегенерационного периода работы и улучшение массообмена. повышение эффективности гидрокрекинга и гидроочистки.
Техническим результатом является улучшение диффузионных характеристик катализатора, исключение диффузных ограничений транспорта исходных соединений и продуктов реакции и блокировки пор углеродистыми отложениями. Другим техническим результатом является высокая скорость гидрокрекинга и гидроочистки молекул тяжелых остатков нефти, вязкой и высоковязкой нефти, тяжелого и сверхтяжелого нефтяного сырья с высоким выходом компонентов масел и низкой скоростью формирования углеводородных отложений и углеводородов газовой фазы. Это обеспечит решение поставленной задачи.
Поставленная задача решается и технический результат реализуется разработкой катализатора гидрокрекинга и гидроочистки тяжелых остатков нефти, вязкой и высоковязкой нефти, тяжелого и сверхтяжелого нефтяного сырья, который содержит неорганический пористый носитель, активный компонент, выбираемый из соединений никеля, кобальта, молибдена, вольфрама или любой их комбинации, нанесенный на носитель.
Отличием изобретения от прототипа является следующее:
- в качестве носителя взят высокопористый ячеистый носитель из металла, выбранного из ряда: никель, хром, медь, железо, титан, алюминий в индивидуальной форме или в комбинациях друг с другом, или из оксида алюминия, или из оксида железа, или в комбинации друг с другом;
- носитель характеризуется пористостью не менее 85%, средним размером пор (ячеек) 0,5-6,0 мм;
- на высокопористом ячеистом носителе закреплен слой вторичного носителя, выбранного из ряда: цеолит, оксид алюминия, оксид железа, оксид кремния, оксид титана, оксид циркония, алюмосиликат, железосиликат, глина или любая их комбинация;
- вторичный носитель характеризуется толщиной от 10 мкм до 2000 мкм, удельной поверхностью не менее 20 м2/г, объемом пор от 0,1 до 1,0 см3/г, в котором поры диаметром более 5 нм составляют не менее 50% общего объема пор;
- вторичный носитель характеризуется наличием кислотных центров Бренстеда и Льюиса, при этом согласно данным температурно-программируемой десорбции аммиака количество средних и сильных кислотных центров Бренстеда и Льюиса с температурными диапазонами десорбции аммиака 250-350°С и более 350°С составляет 1-1500 и 1-1500 мкмоль/г соответственно, а соотношение средних и сильных кислотных центров Бренстеда и Льюиса составляет 1-10:1-5;
- вторичный носитель в общем составе катализатора составляет не менее 5 масс %;
- активный компонент закреплен на вторичном носителе, при следующем соотношении компонентов, масс %:
При указанном наборе ограничительных и отличительных признаков:
- высокопористый ячеистый носитель, характеризующийся пористостью не менее 85% и средним размером пор (ячеек) 0,5-6,0 мм, обеспечивает высокий массо- и теплоперенос, низкое гидравлическое сопротивление;
- наличие вторичного носителя толщиной от 10 мкм до 2000 мкм, с удельной поверхностью не менее 20 м2/г, объемом пор от 0,1 до 1,0 см3/г, в котором поры диаметром более 5 нм составляют не менее 50% общего объема пор, с содержанием не менее 5 масс % обеспечивает оптимальное распределение активного компонента, возможность увеличения его общего количества до 40% масс, с возможностью формирования его мелких активных частиц, отсутствие диффузионных ограничений доступа реакционных молекул к активным центрам, что приводит к увеличению активности катализатора и длительности межрегенерационного периода работы;
- наличие средних и сильных кислотных центров Бренстеда и Льюиса с температурными диапазонами десорбции аммиака 250-350°С и более 350°С в количестве 1-1500 и 1-1500 мкмоль/г соответственно, при соотношении средних и сильных кислотных центров Бренстеда и Льюиса 1-10:1-5 обеспечивает высокую скорость крекинга молекул тяжелых остатков нефти, вязкой и высоковязкой нефти с высоким выходом компонентов масел и низкой скоростью формирования углеводородных отложений и углеводородов газовой фазы.
В результате использования катализатора на основе высокопористого носителя с тонким слоем вторичного носителя в соответствии с данным изобретением, процесс гидрокрекинга и гидроочистки тяжелых остатков нефти, вязкой и высоковязкой нефти осуществляется более эффективно, что отражается в значительном снижении вязкости, увеличении выхода целевых углеводородов, входящих в состав масел и фракций с температурой кипения ниже 350°С, удаления серы, снижении протекания побочных процессов формирования легких углеводородов, которые входят в газовую фазу.
Известен катализатор селективного гидрирования ацетиленовых и диеновых углеводородов в С2-С5+-углеводородных фракциях с использованием основного и вторичного носителя (патент РФ №2547258). Его использование также обеспечивает улучшение тепло- и массообмена и диффузионных характеристик, но другого технологического процесса - селективного гидрирования. В отличие от этого известного решения в предложенном катализаторе вторичный носитель характеризуется наличием средних и сильных кислотных центров Бренстеда и Льюиса с температурными диапазонами десорбции аммиака 250-350°С и более 350°С в количестве 1-1500 и 1-1500 мкмоль/г соответственно, при соотношении средних и сильных кислотных центров Бренстеда и Льюиса 1-10:1-5 для обеспечения высокой скорости гидрокрекинга молекул вязкой нефти, тяжелого и сверхтяжелого нефтяного сырья с высоким выходом компонентов масел и низкой скорости формирования углеводородных отложений и углеводородов газовой фазы, при этом как сам признак, так и полученный результат за счет его использования из уровня техники не известны. Вторичный носитель при этом может быть выбран из более широкого перечня соединений.
Катализатор получали следующим образом.
Высокопористый носитель из металла получали методом электрохимического осаждения металла на металлизированную подложку ретикулированной сетчато-ячеистой пенополиуретановой матрицы или шликерным способом.
Электрохимическое осаждение металла на металлизированную подложку ретикулированной сетчато-ячеистой пенополиуретановой матрицы осуществляли в три стадии: нанесение электропроводного слоя на пенополиуретановую матрицу, электрохимическое осаждение металлического покрытия на металлизированную пенополиуретановую матрицу и удаление органической пенополиуретановой матрицы путем прокалки при температуре 300-1900°С в течение 2-8 часов.
Нанесение металла шликерным способом осуществляли методом дублирования ретикулированной сетчато-ячеистой пенополиуретановой матрицы путем нанесения металлических порошков на пенополиуретановую матрицу с последующим циклическим воздействием (сжатие-растяжение). Далее избыток шликера удаляли отжатием образца до заданной массы. Затем последовательно проводили сушку пропитанных заготовок при температуре 100-120°С в течение 1-4 часов и обжиг при температуре 1200-1900°С в течение 2-8 часов.
В качестве исходного металла высокопористого носителя использовали никель, хром, медь, железо, титан, алюминий в индивидуальной форме или в комбинациях друг с другом.
Высокопористый носитель из оксида алюминия, из оксида железа или в комбинации друг с другом получали шликерным способом, который заключался в пропитывании растворами соединений алюминия ретикулированной сетчато-ячеистой пенополиуретановой матрицы путем циклического воздействия (сжатие-растяжение). Далее избыток шликера удаляли отжатием образца до заданной массы. Затем последовательно проводили сушку пропитанных заготовок при температуре 100-120°С в течение 1-4 часов и обжиг при температуре 1200-1700°С в течение 2-8 часов. При этом формировали различные модификации оксида алюминия (γ-Al2O3, β-Al2O3, η-Al2O3, δ-Al2O3, θ-Al2O3, α-Al2O3), оксида железа в индивидуальной форме или в комбинациях с друг с другом.
В качестве исходного соединения для получения высокопористого носителя из оксида алюминия использовали различные соли, гидроксиды, оксиды алюминия в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения для получения высокопористого носителя из оксида железа использовали различные соли, гидроксиды, оксиды железа в индивидуальной форме или в комбинациях друг с другом.
При этом при получении высокопористого носителя из металла, оксида алюминия или оксида железа шликерным способом для увеличения седиментационной устойчивости и прочности адгезии при закрепления шликера на поверхности ретикулированной сетчато-ячеистой пенополиуретановой матрицы в водный раствор шликера возможно добавление раствора поверхностно активного вещества.
Для формирования вторичного слоя из цеолита, оксида алюминия, оксида кремния, оксида титана, оксида циркония, алюмосиликата, железосиликата, оксида железа, глины, или любой их комбинации поверхность высокопористого носителя из металла, оксида алюминия или оксида железа пропитывали соединениями данных элементов. Для этого образец высокопористого носителя помещали в камеру смесителя. Затем дозировали водные или органические растворы исходных соединений элементов. Пропитку высокопористого носителя растворами соединений элементов осуществляли в течение 0,1-2 часов при температуре 20-80°С. После пропитки высокопористый носитель сушили в течение 0,5-2 часов при температуре 30-250°С. Для формирования вторичного слоя из цеолита, оксида алюминия, оксида кремния, оксида титана, оксида циркония, алюмосиликата, железосиликата, оксида железа, глины, или любой их комбинации толщиной с общей массовой долей не менее 5 мас. % от общей массы катализатора пропитку высокопористого носителя из металла или оксида алюминия соединениями элементов проводили один или более раз с сушкой и/или прокалкой после каждой пропитки. При этом при получении вторичного слоя из цеолита, оксида алюминия, оксида кремния, оксида титана, оксида циркония, алюмосиликата, железосиликата, оксида железа, глины, или любой их комбинации на поверхности высокопористого ячеистого материала для увеличения седиментационной устойчивости и прочности адгезии при закрепления соединения алюминия на поверхности высокопористого носителя из металла и оксида алюминия в водный раствор соединения элемента возможно добавление раствора поверхностно активного вещества. Далее для перевода нанесенного соединения элемента в оксидную форму проводили прокаливание высокопористого ячеистого носителя с соединением элемента. Носитель с нанесенным соединением элемента прокаливали при температурах от 250°С до 1200°С в течение 2-8 часов для формирования различных модификаций цеолита, оксида алюминия, оксида кремния, оксида титана, оксида циркония, алюмосиликата, железосиликата, оксида железа, глины, или любой их комбинации в индивидуальной форме или в комбинациях с друг с другом. Вторичный слой из оксида алюминия характеризуется толщиной от 10 мкм до 2000 мкм, удельной поверхностью не менее 20 м2/г, объемом пор от 0,1 до 1,0 см3/г, порами диаметром более 5 нм, которые составляют не менее 50% общего объема пор. Вторичный носитель оксид алюминия в общем составе катализатора составляет не менее 5% масс. Использование слоя вторичного оксида алюминия с толщиной не менее 10 мкм и более 2000 мкм и общим количеством не менее 5 масс % обусловлено необходимостью формирования определенного количества активной фазы для превращения углеводородов вязкой нефти, тяжелых и сверхтяжелых остатков нефти.
Введение активного компонента, выбранного из соединений никеля, кобальта, молибдена, вольфрама или любой их комбинации осуществляли на стадии закрепления вторичного носителя на высокопористом ячеистом носителе и/или методом пропитки высокопористого ячеистого носителя с закрепленным вторичным носителем. После нанесения соединения активного компонента катализатор сушили при температурах от 50°С до 250°С в течение 1-2 часов и прокаливали при температурах от 250°С до 800°С в течение 2-8 часов.
В качестве исходного соединения никеля использовали гидроксид никеля, нитрат никеля, сульфат никеля, фосфат никеля, хлорид никеля, карбонат никеля, оксалат никеля, ацетат никеля, ацетилацетонат никеля, металлоорганические комплексные соединения никеля в индивидуальной форме или в комбинациях друг с другом.
В качестве источника исходного соединения кобальта использовали оксид кобальта (II), оксид кобальта (II, III), оксид кобальта (III), гидроксид кобальта, фторид кобальта (II), бромид кобальта (II), хлорид кобальта (II), хлорид кобальта (III), йодид кобальта (II), нитрат кобальта (II), карбонат кобальта (II), оксалат кобальта (II), ацетат кобальта (II), ацетилацетонат кобальта (II), ацетилацетонат кобальта (III), хромат кобальта (II), комплексные соединения с кобальтсодержащим анионом (например, дисульфатокобальтиат калия, гексанитрокобальтиат натрия и т.п.), в индивидуальной форме или в комбинациях друг с другом.
В качестве источника исходного соединения молибдена использовали оксид молибдена (III), оксид молибдена (IV), оксид молибдена (V), оксид молибдена (VI), гидроксид молибдена (III), бромид молибдена (II), бромид молибдена (III), бромид молибдена (IV), хлорид молибдена (II), хлорид молибдена (III), хлорид молибдена (IV), хлорид молибдена (V), сульфид молибдена (II), сульфид молибдена (III), сульфид молибдена (IV), фосфат молибдена, молибденовая кислота, надмолибденовая кислота, парамолибдат аммония, оксалат молибдена (VI), ацетат молибдена (II), молибден (VI) диоксид бис(ацетилацетонат), металлоорганические комплексные соединения молибдена в индивидуальной форме или в комбинациях друг с другом.
В качестве источника исходного соединения вольфрама использовали, оксид вольфрама (IV), оксид вольфрама (VI), фторид вольфрама (VI), бромид вольфрама (II), бромид вольфрама (V), бромид вольфрама (VI), хлорид вольфрама (II), хлорид вольфрама (IV), хлорид вольфрама (V), хлорид вольфрама (VI), вольфрамовая кислота, оксалат вольфрама (V), металлоорганические комплексные соединения вольфрама в индивидуальной форме или в комбинациях друг с другом.
Катализатор также может содержать дополнительно модифицирующий элемент. Модифицирующий элемент представляет собой, по крайней мере, один из металлов, выбранных из группы: К, Na, Mg, Zn, Cu, Re, Bi, Mn, Ba, Sn, B, Ce, Cs, Fe, Cr, Ti, Zr, Si, редкоземельные элементы, или любую их комбинацию. При необходимости введение модифицирующего элемента осуществляли: на стадии синтеза высокопористого ячеистого материла и/или нанесением методом пропитки высокопористого ячеистого носителя и/или на стадии закрепления вторичного носителя на высокопористом ячеистом носителе и/или методом пропитки высокопористого ячеистого носителя с закрепленным вторичным носителем.
После нанесения соединения модифицирующего элемента катализатор сушили при температурах от 50°С до 250°С в течение 1-2 часов и прокаливали при температурах от 250°С до 800°С в течение 2-8 часов.
В качестве исходного соединения калия использовали гидроксид калия, нитрат калия, бромид калия, сернистокислый калий, сульфат калия, сернокислый калий, карбонат калия, фосфат калия, хлорид калия, оксалат калия, металлоорганические комплексные соединения калия в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения натрия использовали гидроксид натрия, нитрат натрия, бромид натрия, гидросернистокислый натрий, сернистокислый натрий, сульфат натрия, карбонат натрия, фосфат натрия, хлорид натрия, оксалат натрия, металлоорганические комплексные соединения натрия в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения магния использовали гидроксид магния, нитрат магния, нитрит магния, карбонат магния, фосфат магния, ацетат магния, оксалат магния, металлоорганические комплексные соединения магния в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения цинка использовали гидроксид цинка, сульфат цинка, нитрат цинка, фосфат цинка, хлорид цинка, карбонат цинка, хромат цинка, дихромат цинка, хромит цинка, оксалат цинка, ацетат цинка, ацетилацетонат цинка, металлоорганические комплексные соединения цинка в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения меди использовали сульфат меди, гидроксид меди, нитрат меди, нитрит меди, карбонат меди, хромит меди, хромат меди, дихромат меди, ацетат меди, ацетилацетонат меди, комплексы меди с многоатомными спиртами (например, глицерат меди и т.п.) или их смеси, металлоорганические комплексные соединения меди в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения рения использовали рениевую кислоту, хлорид рения, бромид рения, сульфид рения, перренат аммония, оксид рения (IV), оксид рения (VII), металлоорганические комплексные соединения рения в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения висмута использовали нитрат висмута, гидроксид висмута, сульфид висмута, дихромат висмута, гидроксид висмута, йодид висмута, оксикарбонат висмута, оксихлорид висмута, металлоорганические комплексные соединения висмута в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения марганца использовали нитрат марганца (II), хлорид марганца (II), бромид марганца (II), сульфат марганца (II), карбонат марганца (II), перманганат марганца, металлоорганические комплексные соединения марганца (II) в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения бария использовали гидроксид бария, нитрат бария, бромид бария, йодид бария, гидросернистокислый барий, карбонат бария, сульфат бария, формиат бария, металлоорганические комплексные соединения бария в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения олова использовали оксид олова (II), оксид олова (IV), гидроксид олова (II), сульфат олова (II), сульфат олова (IV), хлорид олова (II), хлорид олова (IV), сульфид олова (II), сульфид олова (IV), гексахлоростаннат (IV) водорода, гексагидроксостаннат (IV) натрия, металлоорганические комплексные соединения олова в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения бора использовали оксид бора, тригидроксид бора, борную кислоту, фторид бора, бромид бора, йодид бора, сульфид бора, борофтористоводородную кислоту, метаборат натрия, тетраборат натрия, тетрафтороборат аммония, тетрафтороборат калия, тетрагидридоборат лития, тетрагидридоборат натрия, тетрагидроксоборат натрия, трифтороамминбор, тетрафтороборат натрия, металлоорганические комплексные соединения бора в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения церия использовали оксид церия (III), оксид церия (IV), нитрат церия (III), сульфат церия (III), сульфат церия (IV), сульфид церия (III), карбонат церия (III), фосфат церия (III), оксалат церия (III), ацетат церия (III), ацетилацетонат церия (III), металлоорганические комплексные соединения церия в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения цезия использовали оксид цезия, фторид цезия, хлорид цезия, бромид цезия, йодид цезия, сульфид цезия, нитрат цезия, сульфат цезия, оксалат цезия, ацетат цезия, ацетилацетонат цезия, металлоорганические комплексные соединения цезия в индивидуальной форме или в комбинациях друг с другом.
В качестве источника исходного соединения железа использовали оксид железа (III), оксид железа (II, III), гидроксид железа (III), нитрат железа (III), сульфат железа (II), сульфат кобальта (III), фосфат железа (II), фосфат железа (III), фторид железа (II), фторид железа (III), бромид железа (II), бромид железа (III), йодид железа (II), хлорид железа (II), хлорид железа (III), карбонат железа (II), оксалат железа (II), оксалат железа (III), ацетат железа (II), ацетилацетонат железа (III), в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения титана использовали нитрат хрома, сульфат хрома, фосфат хрома, оксалат хрома, хромовую кислоту, хромат калия, металлоорганические комплексные соединения хрома в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения титана использовали оксид титана (III), оксид титана (IV), нитрат титана (II), фторид титана (III), фторид титана (IV), хлорид титана (II), хлорид титана (III), хлорид титана (IV), бромид титана (IV), йодид титана (IV), сульфат титана (III), пирофосфат титана, оксалат титана (III), титановую кислоту, металлоорганические комплексные соединения титана в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения циркония использовали гидроксид циркония, оксид циркония, нитрат циркония, фторид циркония, хлорид циркония, бромид циркония, йодид циркония, сульфат циркония, оксалат циркония, металлоорганические комплексные соединения циркония в индивидуальной форме или в комбинациях друг с другом.
В качестве исходного соединения кремния использовали оксид кремния, гидроксид кремния, кремневая кислота, кремневольфрамовая кислота, кремнифтористоводородная кислота, кремнийорганические соединения, комплексные соединения кремния в индивидуальной форме или в комбинациях друг с другом.
В качестве растворителей для нанесения исходных соединений модифицирующего элемента использовали дистиллированную воду, водные растворы уксусной, серной, азотной, хлорной и др. кислот, диметиламин, триметиламин, пропиламин, бутиламин, толуол, бензол, ацетонитрил, хлористый метилен, аммиачные растворы, хлороформ, ацетон, гидрогксиламин, органические эфиры, спирты или их смеси и др. растворители.
Величина удельной поверхности определялась методом низкотемпературной адсорбции азота (БЭТ). Расчеты порометрического объема и распределения объемов пор по диаметрам вторичного оксида алюминия осуществлялся по десорбционной ветви изотермы по стандартной процедуре Баррета-Джойнера-Хайленду (С. Грег, К. Синг. Адсорбция, удельная поверхность, пористость. М., Мир, 1984, 306 с.). Сущность метода определения параметров пористой системы катализатора изложена в методике ASTMD 3663-99 «Стандартный метод исследования площади поверхности и объема пор катализаторов и носителей катализаторов».
Фазовый состав носителя и катализатора определялся методом дифракции рентгеновских лучей. Съемку рентгенограмм проводили с использованием длинноволнового излучения CuKα и графитового монохроматора на дифрагирующем пучке. Диапазон записи углов в шкале 2θ составляет от 5 до 95 град.
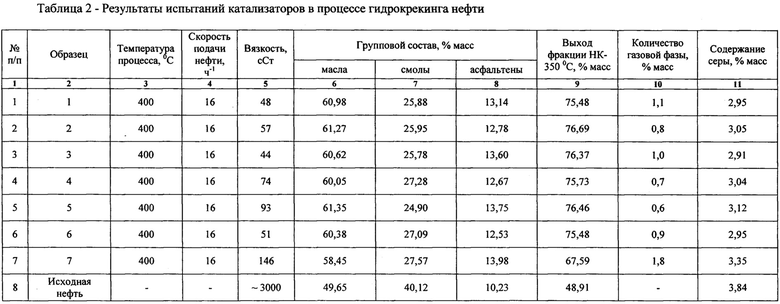
Испытания катализаторов в процессе гидрокрекинга и гидроочистки вязкости нефти осуществляется в адиабатическом реакторе с стационарным слоем катализатора при температуре 350-415°С, давлении 40 кгс/см2, скорости подачи вязкой нефти 14 ч-1. В реактор дополнительно подавали водород исходя из объемного соотношении водород: сырье 50-100 нл/л. Сырьем во всех экспериментах являлась нефть с кинематической вязкостью, равной 3000 сСт (таблица 2). Измерения вязкости исходной и обработанной нефти проводили на приборе Brookfield DV2T.
Групповой состав исходной и обработанной нефти исследовали методом жидкостно-адсорбционной хроматографии. Исходная нефть содержит: масла (алканы, цикланы и моноароматические арены) в количестве до 49,65 масс %; смолы (бензольные смолы (полиароматические углеводороды) и спирто-бензольные (кислые) смолы) - до 40,12 масс %; и асфальтены - до 10,23 масс % (таблица 2).
Элементный анализ исходной и обработанной нефти проводили методом рентгенофлюоресцентного анализа. Исходная нефть содержит до 3,84% масс серы (таблица 2).
Термогравиметрический анализ образцов нефти для определения фракционного состава был выполнен с помощью прибора NETZSCH TG 209 F1 Libra в интервале температур 30-800°С в корундовых тиглях при скорости нагревания 10 К мин-1; перед началом режима нагревания все образцы термостатировались при 30°С в течение 5 минут. Поток реакционного газа (азот) был установлен на уровне 30 мл мин-1, защитного (азот) 20 мл мин-1. Образец каждой из нефтей был измерен один раз; масса навески колебалась в пределах 4-7 мг.
Термопрограммированную десорбцию аммиака для определения концентрации кислотных центров Бренстеда и Льюиса проводили при помощи анализатора AutoChem 2950 HP. Для этого образец в количестве 0,5 г загружали в кварцевый реактор с последующей дегазацией в электрической печи прибора при 550°С, скорость нагрева составляет 10°С/мин. Скорость подачи газа носителя (Не) 10 мл/мин. Носитель насыщали смесью 10% NH3 в Не при комнатной температуре в течение часа. После чего образец продували аргоном при 100°С для удаления физически сорбированного аммиака. Спектры десорбции записывали в температурном диапазоне от 25-700°С. Погрешность определения составляет ± 10%.
Показатели тепло- и массопереноса оценивали в ходе испытаний катализаторов в реакции гидрокрекинга и гидроочистки вязкой нефти. Суть метода заключалась в измерении максимальной разницы показателей температуры вдоль оси поперечного сечения реактора, расположенного в центре катализаторного слоя. Диаметр сечения реактора составлял 30 мм.
Показатель эффективности диффузии оценивался по расчетному значению  - модуль Тиле. При <0,5 процесс идет в кинетической области, и степень использования поверхности близка к единице, диффузионные ограничения отсутствуют. При >2,5 процесс протекает в области внутренней диффузии, и используется лишь ограниченная приповерхностным слоем зерна часть катализатора, отмечаются диффузионные ограничения.
- модуль Тиле. При <0,5 процесс идет в кинетической области, и степень использования поверхности близка к единице, диффузионные ограничения отсутствуют. При >2,5 процесс протекает в области внутренней диффузии, и используется лишь ограниченная приповерхностным слоем зерна часть катализатора, отмечаются диффузионные ограничения.
В Таблице 1 приведены исходные данные катализаторов для каталитических испытаний в процессе гидрокрекинга вязкой нефти, в таблице 2 - результаты испытаний данных катализаторов. Пример 7 в Таблицах 1 и 2 характеризует данные прототипа. Строка 8 Таблицы 2 содержит показатели исходной нефти, приведенные для сравнения.
В таблице 2 количество газовой фазы характеризует скорость протекания побочных процессов формирования легких углеводородов
Примеры конкретного осуществления изобретения описаны ниже.
Пример 1. Катализатор готовят последовательной пропиткой высокопористого ячеистого никелевого носителя с вторичным слоем оксида алюминия δ-модификации растворами ацетата цинка и парамолибдата аммония. Высокопористый ячеистый никелевый носитель характеризуется пористостью 87%, средним диаметром пор 1,2 мм. Вторичный носитель из оксида алюминия δ-модификации и оксида железа обладает величиной удельной поверхности 164 м2/г, объемом пор 0,58 см3/г, мезопорами диаметром более 5 нм, которые вместе составляют 63% общего объема пор. Толщина слоя оксида алюминия δ-модификации составляет 45 мкм. Вторичный носитель характеризуется наличием кислотных центров Бренстеда и Льюиса, при этом согласно данным температурно-программируемой десорбции аммиака количество средних и сильных кислотных центров Бренстеда и Льюиса с температурными диапазонами десорбции аммиака 250-350°С и более 350°С составляет 174 и 79 мкмоль/г соответственно, а соотношение средних и сильных кислотных центров Бренстеда и Льюиса составляет 2,2:1,0.
Высокопористый ячеистый никелевый носитель с вторичным слоем из оксида алюминия δ-модификации и оксида железа в количестве 81,1 г помещают в камеру смесителя, дозируют 200 мл водного раствора, содержащего 25,05 г ацетата цинка. Пропитку осуществляют в течение 3 часов при температуре пропиточного раствора 80°С. После пропитки высокопористого ячеистого никелевого носителя с вторичным слоем оксида алюминия δ-модификации водным раствором ацетата цинка удаляют растворитель при температуре 90°С в течение 2 часов. Затем высокопористый ячеистый никелевый носитель с вторичным слоем из оксида алюминия δ-модификации и оксида железа, пропитанный ацетатом цинка, сушат при температуре 60°С в течение 2 часов и прокаливают для перевода цинка в окисное состояние при температуре 500°С в течение 4 часов.
Далее высокопористый ячеистый никелевый носитель с вторичным слоем из оксида алюминия δ-модификации и оксида железа с оксидом цинка в количестве 100 г помещают в камеру смесителя, дозируют 450 мл водного раствора, содержащего 139,44 г парамолибдата аммония. Пропитку осуществляют в течение 2 часов при температуре пропиточного раствора 80°С. После пропитки носителя парамолибдатом аммония удаляют растворитель при температуре 110°С в течение 3 часов. Затем носитель, пропитанный парамолибдатом аммония, сушат при температуре 250°С в течение 1 часа, прокаливают при температуре 500°С в течение 2 часов и восстанавливают в атмосфере водорода при температуре 150°С в течение 2 часов.
Готовый катализатор характеризуется перепадом температуры по диагонали сечения катализаторного слоя 0,7°С, модулем Тиля - 0,12.
Применяемый катализатор имеет следующий состав:
Zn - 7,4% масс;
Mo - 11,5% масс;
δ-Al2O3+Fe2O3 - 7,3% масс;
Высокопористый ячеистый никелевый носитель - 73,8% масс.
Результаты каталитических испытаний данного образца представлены в Таблице 2.
Пример 2. Катализатор готовят последовательной пропиткой высокопористого ячеистого железного носителя с вторичным слоем оксида алюминия δ-модификации и оксида титана водными растворами нитрата цезия, нитрата никеля и нитрата кобальта. Высокопористый ячеистый железный носитель характеризуется пористостью 85%, средним диаметром пор 0,5 мм. Вторичный носитель оксид алюминия δ-модификации и оксид титана обладает величиной удельной поверхности 225 м2/г, объемом пор 0,1 см3/г, мезопорами диаметром более 5 нм, которые вместе составляют 50% общего объема пор. Толщина слоя оксида алюминия δ-модификации и оксида титана составляет 10 мкм. Вторичный носитель характеризуется наличием кислотных центров Бренстеда и Льюиса, при этом согласно данным температурно-программируемой десорбции аммиака количество средних и сильных кислотных центров Бренстеда и Льюиса с температурными диапазонами десорбции аммиака 250-350°С и более 350°С составляет 15 и 3 мкмоль/г соответственно, а соотношение средних и сильных кислотных центров Бренстеда и Льюиса составляет 5,0:1,0.
Высокопористый ячеистый железный носитель с вторичным слоем оксида алюминия δ-модификации и оксида титана в количестве 100 г помещают в камеру смесителя, дозируют 300 мл водного раствора, содержащего 124 г нитрата церия. Пропитку осуществляют в течение 3 часов при температуре пропиточного раствора 60°С. После пропитки высокопористого ячеистого железного носителя с вторичным слоем оксида алюминия δ-модификации и оксида титана водным раствором нитрата церия удаляют растворитель при температуре 90°С в течение 2 часов. Затем высокопористый ячеистый железный носитель с вторичным слоем оксида алюминия δ-модификации и оксида титана, пропитанный нитратом церия, сушат при температуре 110°С в течение 1 часа и прокаливают при температуре 600°С в течение 8 часов для перевода нитрата церия в оксид церия.
Далее высокопористый ячеистый железный носитель с вторичным слоем оксида алюминия δ-модификации и оксида титана в количестве 100 г помещают в камеру смесителя, дозируют 300 мл водного раствора, содержащего 49,32 г нитрата никеля и 41,43 г нитрата кобальта. Пропитку осуществляют в течение 3 часов при температуре пропиточного раствора 70°С. После пропитки носителя нитратом никеля и нитратом кобальта удаляют растворитель при температуре 150°С в течение 2 часов. Затем носитель, пропитанный нитратом никеля и нитратом кобальта, сушат при температуре 200°С в течение 2 часов, прокаливают при температуре 800°С в течение 4 часов и восстанавливают в атмосфере водорода при температуре 200°С в течение 2 часов.
Готовый катализатор характеризуется перепадом температуры по диагонали сечения катализаторного слоя 0,5°С, модулем Тиля - 0,24.
Применяемый катализатор имеет следующий состав:
Се - 40,0% масс;
Ni - 10,0% масс;
Со - 8,4% масс;
δ-Al2O3+TiO2 - 9,8% масс;
Высокопористый ячеистый железный носитель - 31,8% масс.
Результаты каталитических испытаний данного образца представлены в Таблице 2.
Пример 3. Катализатор готовят последовательной пропиткой высокопористого ячеистого алюминиевого носителя с вторичным слоем алюмосиликата водными растворами ацетата магния и нитрата никеля. Высокопористый ячеистый алюминиевый носитель характеризуется пористостью 94% с средним диаметром пор 2,4 мм. Вторичный носитель алюмосиликат обладает величиной удельной поверхности 40 м2/г, объемом пор 0,83 см3/г, мезопорами диаметром более 5 нм, которые вместе составляют 87% общего объема пор. Толщина слоя вторичного носителя алюмосиликата составляет 263 мкм. Вторичный носитель характеризуется наличием кислотных центров Бренстеда и Льюиса, при этом согласно данным температурно-программируемой десорбции аммиака количество средних и сильных кислотных центров Бренстеда и Льюиса с температурными диапазонами десорбции аммиака 250-350°С и более 350°С составляет 1 и 1 мкмоль/г соответственно, а соотношение средних и сильных кислотных центров Бренстеда и Льюиса составляет 1,0:1,0.
Высокопористый ячеистый алюминиевый носитель с вторичным слоем алюмосиликата в количестве 100 г помещают в камеру смесителя, дозируют 100 мл водного раствора, содержащего 0,45 г ацетата магния. Пропитку осуществляют в течение 3 часов при температуре пропиточного раствора 70°С. После пропитки высокопористого ячеистого алюминиевого носителя с вторичным слоем алюмосиликата водным раствором ацетата магния удаляют растворитель при температуре 90°С в течение 1 часа. Затем высокопористый ячеистый алюминиевый носитель с вторичным слоем алюмосиликата, сушат при температуре 50°С в течение 2 часов и прокаливают при температуре 250°С в течение 5 часов для перевода исходного ацетата магния в оксид магния.
Далее высокопористый ячеистый алюминиевый носитель с вторичным слоем алюмосиликата в количестве 100 г помещают в камеру смесителя, дозируют 200 мл водного раствора, содержащего 84,84 г нитрата никеля. Пропитку осуществляют в течение 2 часов при температуре пропиточного раствора 60°С. После пропитки носителя нитратом никеля удаляют растворитель при температуре 110°С в течение 1 часа. Затем носитель, пропитанный нитратом никеля, сушат при температуре 150°С в течение 2 часа, прокаливают при температуре 600°С в течение 8 часов и восстанавливают в атмосфере водорода при температуре 400°С в течение 3 часов.
Готовый катализатор характеризуется перепадом температуры по диагонали сечения катализаторного слоя 0,4°С, модулем Тиля - 0,07.
Применяемый катализатор имеет следующий состав:
Mg - 0,05% масс;
Ni - 40,0% масс;
Al2O3+SiO2 - 5,0% масс;
Высокопористый ячеистый алюминиевый носитель - 54,95% масс.
Результаты каталитических испытаний данного образца представлены в Таблице 2.
Пример 4. Катализатор готовят последовательной пропиткой высокопористого ячеистого носителя оксида алюминия α-модификации с вторичным слоем оксида алюминия γ-модификации и оксида циркония водными растворами нитрата меди, парамолибдата аммония и вольфрамовой кислотой. Высокопористый ячеистый носитель оксида алюминия α-модификации характеризуется пористостью 90% с средним диаметром пор 3,7 мм. Вторичный носитель оксид алюминия γ-модификации и оксид циркония обладает величиной удельной поверхности 20 м2/г, объемом пор 1,0 см3/г, мезопорами диаметром более 5 нм, которые вместе составляют 94% общего объема пор. Толщина слоя вторичного носителя составляет 523 мкм. Вторичный носитель характеризуется наличием кислотных центров Бренстеда и Льюиса, при этом согласно данным температурно-программируемой десорбции аммиака количество средних и сильных кислотных центров Бренстеда и Льюиса с температурными диапазонами десорбции аммиака 250-350°С и более 350°С составляет 1500 и 238 мкмоль/г соответственно, а соотношение средних и сильных кислотных центров Бренстеда и Льюиса составляет 6,3:1,0.
Высокопористый ячеистый носитель оксид алюминия α-модификации с вторичным слоем оксида алюминия γ-модификации и оксида циркония в количестве 100 г помещают в камеру смесителя, дозируют 300 мл водного раствора, содержащего 53,65 г нитрата меди. Пропитку осуществляют в течение 3 часов при температуре пропиточного раствора 60°С. После пропитки высокопористого ячеистого носителя оксида алюминия α-модификации с вторичным слоем оксида алюминия γ-модификации и оксида циркония водным раствором нитрата меди удаляют растворитель при температуре 90°С в течение 2 часов. Затем высокопористый ячеистый носитель оксид алюминия α-модификации с вторичным слоем оксида алюминия γ-модификации и оксида циркония, пропитанный нитратом меди, сушат при температуре 250°С в течение 2 часов и прокаливают при температуре 800°С в течение 5 часов для перевода нитрата меди в оксид меди.
Далее высокопористый ячеистый носитель оксид алюминия α-модификации с вторичным слоем оксида алюминия γ-модификации и оксида циркония в количестве 100 г помещают в камеру смесителя, дозируют 400 мл водного раствора, содержащего 145,5 г парамолибдата аммония и 10,87 г вольфрамовой кислоты. Пропитку осуществляют в течение 1 часа при температуре пропиточного раствора 50°С. После пропитки носителя парамолибдатом аммония и вольфрамовой кислотой удаляют растворитель при температуре 120°С в течение 2 часов. Затем носитель, пропитанный парамолибдатом аммония и вольфрамовой кислотой, сушат при температуре 120°С в течение 6 часов, прокаливают при температуре 600°С в течение 3 часов и восстанавливают в атмосфере водорода при температуре 300°С в течение 4 часов.
Готовый катализатор характеризуется перепадом температуры по диагонали сечения катализаторного слоя 0,5°С, модулем Тиля - 0,07.
Применяемый катализатор имеет следующий состав:
Cu - 11,6% масс;
Мо- 12,0% масс;
W - 8,0% масс;
γ-Al2O3+ZrO2 - 10,3% масс;
Высокопористый ячеистый носитель оксид алюминия α-модификации - 58,1% масс.
Результаты каталитических испытаний данного образца представлены в Таблице 2.
Пример 5. Катализатор готовят пропиткой высокопористого ячеистого носителя оксида алюминия α-модификации с вторичным слоем оксида алюминия δ-модификации и цеолита ZSM-5 водным раствором нитрата кобальта. Высокопористый ячеистый носитель оксида алюминия α-модификации характеризуется пористостью 91% с средним диаметром пор 4,1 мм. Вторичный носитель оксид алюминия δ-модификации и цеолит ZSM-5 обладает величиной удельной поверхности 146 м2/г, объемом пор 0,58 см3/г, мезопорами диаметром более 5 нм, которые вместе составляют 73% общего объема пор. Толщина слоя вторичного слоя оксида алюминия δ-модификации и цеолита ZSM-5 составляет 2000 мкм. Вторичный носитель характеризуется наличием кислотных центров Бренстеда и Льюиса, при этом согласно данным температурно-программируемой десорбции аммиака количество средних и сильных кислотных центров Бренстеда и Льюиса с температурными диапазонами десорбции аммиака 250-350°С и более 350°С составляет 300 и 1500 мкмоль/г соответственно, а соотношение средних и сильных кислотных центров Бренстеда и Льюиса составляет 1,0:5,0.
Высокопористый ячеистый носитель оксида алюминия α-модификации с вторичным слоем оксида алюминия δ-модификации и цеолита ZSM-5 в количестве 100 г помещают в камеру смесителя, дозируют 300 мл водного раствора, содержащего 0,25 г нитрата кобальта. Пропитку осуществляют в течение 3 часов при температуре пропиточного раствора 70°С. После пропитки высокопористого ячеистого носителя оксида алюминия α-модификации с вторичным слоем оксида алюминия δ-модификации и цеолита ZSM-5 водным раствором нитрата кобальта удаляют растворитель при температуре 90°С в течение 2 часов. Затем высокопористый ячеистый носитель оксид алюминия α-модификации с вторичным слоем оксида алюминия δ-модификации и цеолита ZSM-5, пропитанный нитратом кобальта, сушат при температуре 50°С в течение 2 часов, прокаливают при температуре 500°С в течение 6 часов и восстанавливают в атмосфере водорода при температуре 400°С в течение 4 часов.
Готовый катализатор характеризуется перепадом температуры по диагонали сечения катализаторного слоя 0,3°С, модулем Тиля - 0,15.
Катализатор имеет следующий состав:
Со - 0,05% масс;
δ-Al2O3+ZSM-5 - 12,6% масс;
Высокопористый ячеистый оксид алюминия α-модификации - 87,35% масс.
Результаты каталитических испытаний данного образца представлены в Таблице 2.
Пример 6. Катализатор готовят пропиткой высокопористого ячеистого носителя оксида алюминия α-модификации с вторичным слоем оксида кремния и железосиликата водным раствором нитрата никеля и нитрата кобальта. Высокопористый ячеистый оксид алюминия α-модификации характеризуется пористостью 94% с средним диаметром пор 6,0 мм. Вторичный носитель оксид кремния и железосиликат обладает величиной удельной поверхности 85 м2/г, объемом пор 0,73 см3/г, мезопорами диаметром более 5 нм, которые вместе составляют 85% общего объема пор. Толщина слоя оксида кремния и железосиликата составляет 734 мкм. Вторичный носитель характеризуется наличием кислотных центров Бренстеда и Льюиса, при этом согласно данным температурно-программируемой десорбции аммиака количество средних и сильных кислотных центров Бренстеда и Льюиса с температурными диапазонами десорбции аммиака 250-350°С и более 350°С составляет 35 и 3,5 мкмоль/г соответственно, а соотношение средних и сильных кислотных центров Бренстеда и Льюиса составляет 10,0:1,0.
Высокопористый ячеистый носитель оксида алюминия α-модификации с вторичным слоем оксида кремния и железосиликата в количестве 100 г помещают в камеру смесителя, дозируют 300 мл водного раствора, содержащего 25,15 г нитрата никеля и 18,74 г нитрата кобальта. Пропитку осуществляют в течение 2 часов при температуре пропиточного раствора 60°С. После пропитки высокопористого ячеистого носителя оксида алюминия α-модификации с вторичным слоем оксида кремния и железосиликата водным раствором нитрата никеля и нитрата кобальта удаляют растворитель при температуре 90°С в течение 3 часов. Затем высокопористый ячеистый носитель оксид алюминия α-модификации с вторичным слоем оксида кремния и железосиликата, пропитанный нитратом никеля и нитратом кобальта, сушат при температуре 110°С в течение 2 часов, прокаливают при температуре 250°С в течение 6 часов и восстанавливают в атмосфере водорода при температуре 500°С в течение 2 часов.
Готовый катализатор характеризуется перепадом температуры по диагонали сечения катализаторного слоя 0,6°С, модулем Тиля - 0,13.
Катализатор имеет следующий состав:
Ni - 5,1% масс;
Со - 3,8% масс;
SiO2+FeSiO3 - 40,0% масс;
Высокопористый ячеистый оксид алюминия α-модификации - 51,1% масс.
Результаты каталитических испытаний данного образца представлены в Таблице 2.
Как видно из приведенных примеров, предлагаемый катализатор превосходит по активности и селективности в реакциях гидрокрекинга вязкой нефти катализатор сравнения (пример 7). Это обусловлено использованием высокопористого ячеистого носителя с вторичным слоем, что приводит к увеличению массо- и теплообмена, к исключению диффузионных ограничений транспорта исходных соединений и продуктов реакции и блокировки пор углеродистыми отложениями. Это в свою очередь приводит к более высокой скорости переработки макромолекул углеводородов исходной нефти и более длительному межрегенерациооному периоду работы катализатора. Также отмечается большее уменьшение количества серы в обработанной нефти.
Более высокая активность образцов катализатора гидрокрекинга данного изобретения по сравнению с катализатором прототипа обусловлена наличием тонкого слоя вторичного носителя. Именно наличие вторичного носителя толщиной от 10 мкм до 2000 мкм, удельной поверхностью не менее 20 м2/г, объемом пор от 0,1 до 1,0 см3/г, в котором поры диаметром более 5 нм составляют более 50% общего объема пор, обеспечивает оптимальное распределение активных компонентов на поверхности катализатора с высоким количеством активных центров, доступных для реагирующих молекул. Это в свою очередь приводит к увеличению активности катализатора в реакциях гидрокрекинга углеводородов тяжелых остатков нефти, вязкой и высоковязкой нефти.
Наличие средних и сильных кислотных центров Бренстеда и Льюиса с температурными диапазонами десорбции аммиака 250-350°С и более 350°С в количестве 1-1500 и 1-1500 мкмоль/г соответственно, при соотношении средних и сильных кислотных центров Бренстеда и Льюиса 1-10:1-5, обеспечивает высокую скорость гидрокрекинга молекул вязкой нефти, тяжелого и сверхтяжелого нефтяного сырья с высоким выходом компонентов масел и низкой скоростью формирования углеводородных отложений и углеводородов газовой фазы.

Примечание: ВПЯМ - высокопористый ячеистый материал;
ΔТ - градиент температур по сечению катализаторного слоя, °С;
- модуль Тиле;
образец №7 - прототип имеет следующий состав, % масс:
Al2O3/SiO2 - 72;
Со - 8,7;
Мо - 19,3,
сформован в виде экструдатов диаметром 2,5 мм.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИЗАТОР КРЕКИНГА ТЯЖЕЛЫХ ОСТАТКОВ НЕФТИ, ВЯЗКОЙ И ВЫСОКОВЯЗКОЙ НЕФТИ | 2019 |
|
RU2691650C1 |
| КАТАЛИЗАТОР СЕЛЕКТИВНОГО ГИДРИРОВАНИЯ АЦЕТИЛЕНОВЫХ И ДИЕНОВЫХ УГЛЕВОДОРОДОВ В С-С-УГЛЕВОДОРОДНЫХ ФРАКЦИЯХ | 2014 |
|
RU2547258C1 |
| КАТАЛИЗАТОР ДЕГИДРИРОВАНИЯ C-C ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ | 2019 |
|
RU2705808C1 |
| Способ получения микросферического катализатора дегидрирования парафиновых C-C углеводородов | 2016 |
|
RU2620815C1 |
| Способ получения микросферического катализатора дегидрирования парафиновых C-C углеводородов | 2016 |
|
RU2626323C1 |
| НОСИТЕЛЬ ДЛЯ КАТАЛИЗАТОРА ГИДРООЧИСТКИ ДИЗЕЛЬНЫХ ФРАКЦИЙ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2023 |
|
RU2811917C1 |
| Состав и способ приготовления катализатора гидрирования диолефинов | 2019 |
|
RU2714138C1 |
| КАТАЛИЗАТОР ДЛЯ СЕЛЕКТИВНОГО ГИДРИРОВАНИЯ ДИЕНОВЫХ УГЛЕВОДОРОДОВ | 2006 |
|
RU2292952C1 |
| Катализатор защитного слоя для реакторов гидрогенизационной переработки нефтяного сырья и способ его получения | 2021 |
|
RU2761528C1 |
| КАТАЛИЗАТОР ПЕРЕРАБОТКИ ТЯЖЕЛЫХ НЕФТЯНЫХ ФРАКЦИЙ | 2012 |
|
RU2506997C1 |
Изобретение относится к технологии производства катализаторов гидрокрекинга и гидроочистки тяжелых остатков нефти, вязкой и высоковязкой нефти. Заявленный катализатор содержит высокопористый ячеистый носитель, выполненный из металла: никель, хром, медь, железо, титан, алюминий в индивидуальной форме или в комбинациях друг с другом, или из оксида алюминия, или оксида железа, или в комбинации друг с другом, активный компонент, выбранный из ряда: никель, хром, медь, железо, титан, алюминий, оксид алюминия оксид железа с пористостью не менее 85%, средним размером пор (ячеек) 0,5-6,0 мм, на высокопористом ячеистом носителе закреплен слой вторичного носителя, выбранного из ряда: цеолит, оксид алюминия, оксид железа, оксид кремния, оксид титана, оксид циркония, алюмосиликат, железосиликат, глина или любая их комбинация, вторичный носитель характеризуется толщиной от 10 до 2000 мкм, удельной поверхностью не менее 20 м2/г, объемом пор от 0,1 до 1,0 см3/г, в котором поры диаметром более 5 нм составляют не менее 50% общего объема пор, наличием кислотных центров Бренстеда и Льюиса, при этом согласно данным температурно-программируемой десорбции аммиака количество средних и сильных кислотных центров Бренстеда и Льюиса с температурными диапазонами десорбции аммиака 250-350°С и более 350°С составляет 1-1500 и 1-1500 мкмоль/г соответственно, а соотношение средних и сильных кислотных центров Бренстеда и Льюиса составляет 1-10:1-5, вторичный носитель в общем составе катализатора составляет не менее 5 мас.%, активный компонент закреплен на вторичном носителе при следующем соотношении компонентов, мас.%: вторичный носитель 5,0-40,0, активный компонент 0,05-40,0, модифицирующий элемент 0-40,0, высокопористый ячеистый материал - остальное. Технический результат заключается в увеличении активности катализатора, длительности его межрегенерационного периода работы и улучшении массообмена, повышении эффективности гидрокрекинга и гидроочистки. 2 табл., 7 пр.
Катализатор гидрокрекинга и гидроочистки тяжелых остатков нефти, вязкой и высоковязкой нефти, который содержит неорганический пористый носитель, активный компонент, выбираемый из соединений никеля, кобальта, молибдена, вольфрама или любой их комбинации, нанесенный на носитель, отличающийся тем, что в качестве носителя взят высокопористый ячеистый носитель из металла, выбранного из ряда: никель, хром, медь, железо, титан, алюминий в индивидуальной форме или в комбинациях друг с другом, или из оксида алюминия, или оксида железа, или в комбинации друг с другом, высокопористый ячеистый носитель характеризуется пористостью не менее 85%, средним размером пор (ячеек) 0,5-6,0 мм, на высокопористом ячеистом носителе закреплен слой вторичного носителя, выбранного из ряда: цеолит, оксид алюминия, оксид железа, оксид кремния, оксид титана, оксид циркония, алюмосиликат, железосиликат, глина или любой их комбинации, вторичный носитель характеризуется толщиной от 10 до 2000 мкм, удельной поверхностью не менее 20 м2/г, объемом пор от 0,1 до 1,0 см3/г, в котором поры диаметром более 5 нм составляют не менее 50% общего объема пор, вторичный носитель характеризуется наличием кислотных центров Бренстеда и Льюиса, при этом согласно данным температурно-программируемой десорбции аммиака количество средних и сильных кислотных центров Бренстеда и Льюиса с температурными диапазонами десорбции аммиака 250-350°С и более 350°С составляет 1-1500 и 1-1500 мкмоль/г соответственно, а соотношение средних и сильных кислотных центров Бренстеда и Льюиса составляет 1-10:1-5, вторичный носитель в общем составе катализатора составляет не менее 5 мас.%, активный компонент закреплен на вторичном носителе, при следующем соотношении компонентов, мас.%:
| КАТАЛИЗАТОР ПЕРЕРАБОТКИ ТЯЖЕЛЫХ НЕФТЯНЫХ ФРАКЦИЙ | 2012 |
|
RU2506997C1 |
| Способ приготовления катализатора гидрокрекинга углеводородного сырья | 2015 |
|
RU2607908C1 |
| US 6187710 B1, 13.02.2001 | |||
| WO 2007126419 A2, 08.11.2007 | |||
| КАТАЛИЗАТОР, СПОСОБ ПОЛУЧЕНИЯ НОСИТЕЛЯ, СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА И ПРОЦЕСС ГИДРООБЕССЕРИВАНИЯ ДИЗЕЛЬНЫХ ФРАКЦИЙ | 2006 |
|
RU2311959C1 |

