ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННУЮ ЗАЯВКУ
Настоящая заявка имеет право согласно §119(с) раздела 35 свода законов США на приоритет предварительной заявки на патент США с № 61/428894, поданной 31 декабря 2010, и предварительной заявки на патент США с № 61/450804, поданной 9 марта 2011, которые включены сюда посредством ссылки в их полных объемах.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Аргиназа является ферментом, который катализирует зависимый от двухвалентных катионов гидролиз L-аргинина с образованием L-орнитина и мочевины. Известно, что аргиназа выполняет по меньшей мере три важные функции: (1) продукцию мочевины, (2) продукцию L-орнитина и (3) регуляцию уровней аргинина в качестве субстрата для синтаз оксида азота (также известных как NOS, эти ферменты превращают L-аргинин в цитруллин и NO).
У большинства млекопитающих существуют два изомера аргиназы: аргиназа I и аргиназа II. Аргиназа I локализуется в основном в цитоплазме печени, в то время как аргиназа II обнаруживается в митохондрии нескольких тканей, при этом более высокие концентрации обнаруживаются в почке и предстательной железе, а более низкие концентрации - в макрофагах, лактирующих молочных железах и головном мозге. Получение мочевины с помощью печеночной аргиназы является важным механизмом для экскреции азота (аммиака) в форме хорошо растворимого, нетоксичного соединения.
В тканях, в которых отсутствует полный комплект ферментов мочевинного цикла, аргиназа регулирует клеточные концентрации L-орнитина. L-орнитин является предшественником для биосинтеза полиаминов (таких как спермин и спермидин, которые играют важные роли в пролиферации и дифференциации клеток) и пролина (важного компонента коллагена, компонента фибрина и фиброзной ткани). Аргиназа также модулирует NOS-опосредованную продукцию NO посредством регуляции уровней аргинина, присутствующего в тканях. При патологических болезненных состояниях, при которых уровни внепеченочных аргиназ являются повышенными, L-аргинин потребляется более активно, что ограничивает возможность его использования в качестве субстрата для NOS. Таким образом, аргиназа и NOS, по-видимому, являются реципрокно регулируемыми. При таких болезненных состояниях особенно желательным может быть ингибирование внепеченочной аргиназы.
Избыточное количество аргиназы было связано с рядом патологических состояний человека, включая эректильную дисфункцию, атеросклероз, астму и легочную артериальную гипертензию и некоторые типы рака, такие как немелкоклеточный рак легкого, рак предстательной железы и рак поджелудочной железы. Кроме того, высокие уровни аргиназы были констатированы в моделях заболеваний человека на животных, таких как ишемия миокарда-реперфузионное повреждение, систолическая (первичная артериальная) гипертензия, атеросклероз, легочная артериальная гипертензия, эректильная дисфункция, астма и рассеянный склероз.
Пациентам с состояниями, связанными с увеличением активности аргиназы, может принести пользу лечение ингибиторами аргиназы, такими как NΩ-гидрокси-L-аргинин (L-HO-Arg), промежуточный продукт в реакции с участием синтазы NO. Однако L-OH-Arg является неселективным ингибитором, и поэтому точная роль аргиназы в патофизиологии и возможные терапевтические эффекты ингибиторов аргиназы остаются неизвестными.
Хотя желательным является несильное ингибирование печеночной аргиназы, существует подтверждение гипотезы, что мочевинный цикл является очень устойчивым. Например, Gau и его коллеги (Mol. Ther., 2009, 1:1-9) сообщили, что для спасения животного с выключенным геном аргиназы I путем терапии с использованием гена аргиназы I требуется лишь приблизительно 20% от аргиназной активности для сохранения нормальных уровней аммиака. Другими словами, до тех пор, пока аргиназная активность в печени не падает ниже 20% от нормальных уровней, мочевинный цикл может нормально функционировать и гипераммониемия не возникает. Кроме того, гетерозиготная по выключенному гену аргиназы I мышь, которая имеет лишь приблизительно 60% от нормальной активности печеночной аргиназы, имеет нормальные уровни аммиака в плазме, как сообщают Iyer и его коллеги (Mol, Cell. Biol., 2002, 22:4491-4498).
В данной области техники существует потребность в ингибиторах аргиназной активности, которые могут использоваться для лечения заболевания или нарушения у млекопитающего, причем заболевание или нарушение характеризуется либо необычно высокой аргиназной активностью, либо необычно низкими уровнями оксида азота в ткани млекопитающего. Настоящее изобретение удовлетворяет эти потребности.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение включает композицию, содержащую альфа-аминокислотное соединение, или его производное, или его соль, причем первый заместитель и второй заместитель связаны с альфа-углеродом соединения. Первый заместитель включает фрагмент, выбираемый из группы, состоящей из бороновой кислоты и N-гидроксигуанидина. Второй заместитель включает проксимальный атом азота, причем проксимальный азот является основным, причем, кроме того, проксимальный азот отделен от альфа-углерода цепью из двух, трех или четырех углеродов, при условии, что соединением не является 2-амино-6-бороно-2-(3-(пиперазин-1-ил)пропил)капроновая кислота, 2-(3-(4-ацетилпиперазин-1-ил)пропил)-2-амино-6-боронокапроновая кислота, 2-амино-6-бороно-2-(3-(4-(4-цианобензоил)пиперазин-1-ил)пропилкапроновая кислота или 2-амино-6-бороно-2-(3-(4-(3- метоксифенилкарбамоил)пиперазин-1-ил)пропил)капроновая кислота.
В одном варианте осуществления первый заместитель включает бороновую кислоту. В другом варианте осуществления первым заместителем является -(CH2)4B(OH)2 или его сложный эфир. В еще одном варианте осуществления проксимальный азот является частью первичной, вторичной или третичной аминной группы. В еще одном варианте осуществления проксимальный азот является частью гетероциклической группы. В другом варианте осуществления гетероциклическую группу выбирают из группы, состоящей из азитидина, азетидина, пирролидина, пиперидина, азепана, азокана, диазетидина, имидазолидина, пиразолидина, оксазолидина, изоксазолидина, тиазолидина, изотиазолидина, пиперазина, морфолина, их аналогов с мостиковой связью, их конденсированных комбинаций и их замещенных вариантов. В еще одном варианте осуществления второй заместитель отделен от альфа-углерода цепью из двух или трех углеродов. В другом варианте осуществления второй заместитель отделен от альфа-углерода цепью из трех углеродов. В еще одном варианте осуществления замещенная гетероциклическая группа включает по меньшей мере один заместитель, выбираемый из группы, состоящей из (C1-C6)алкила, галогена, арила и гетероарила, арил(C1-C6)алкила, гетероарил(C1-C6)алкила, -C(=O)R3, -SO2R3, -CONHR3, COOR3, OR2 и NR3R3, при условии, что если по меньшей мере один заместитель представляет собой OR2 или NR3R3, то по меньшей мере один заместитель не присоединен к тому же атому углерода, к которому присоединен атом азота гетероциклической группы; причем R2 представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил); и в каждом случае R3 независимо представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил или гетероарил(C1-C6)алкил.
В одном варианте осуществления соединением является соединение формулы (IV), или его производное, или его соль:
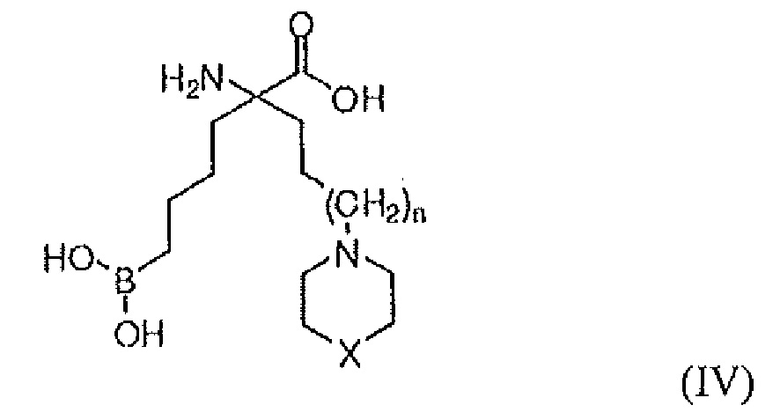
где n равно 0, 1 или 2; X представляет собой NR5, CR6R7, O, S, S(=O) или S(O)2; R7 представляет собой H, OH, OR8, CN или NR8R9; и R5, R6, R8 и R9 независимо представляют собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил); или его производное, или его соль.
В одном варианте осуществления соединение выбирают из группы, состоящей из:
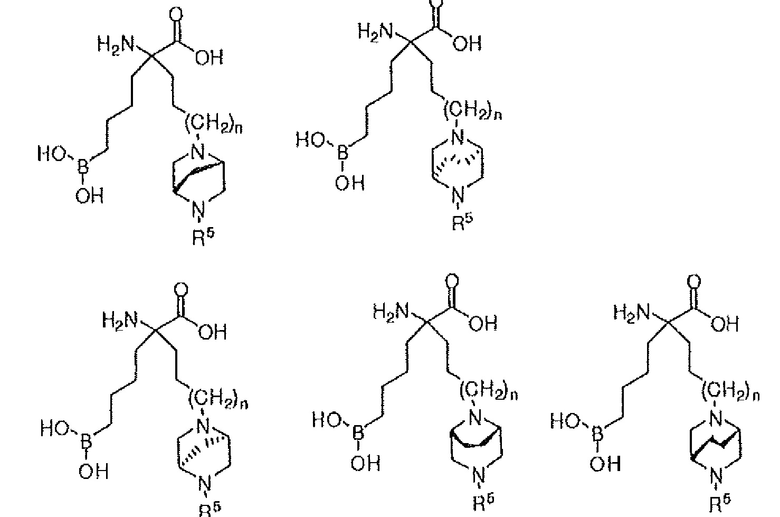
где n равно 0, 1 или 2; и R5 представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил); или их производного, или их соли.
В одном варианте осуществления соединение выбирают из группы, состоящей из:
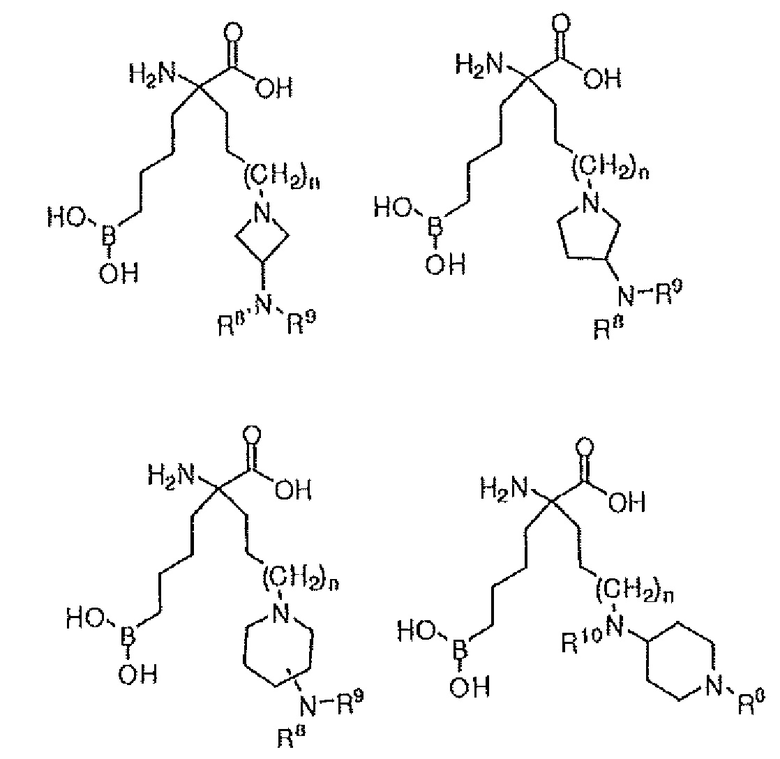
где n равно 0, 1 или 2; R8 и R9 независимо представляют собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил); и R10 представляет собой H, (C1-C6)алкил или арилалкил; или их производного, или их соли.
В одном варианте осуществления соединением является соединение формулы (V), или его производное, или его соль:
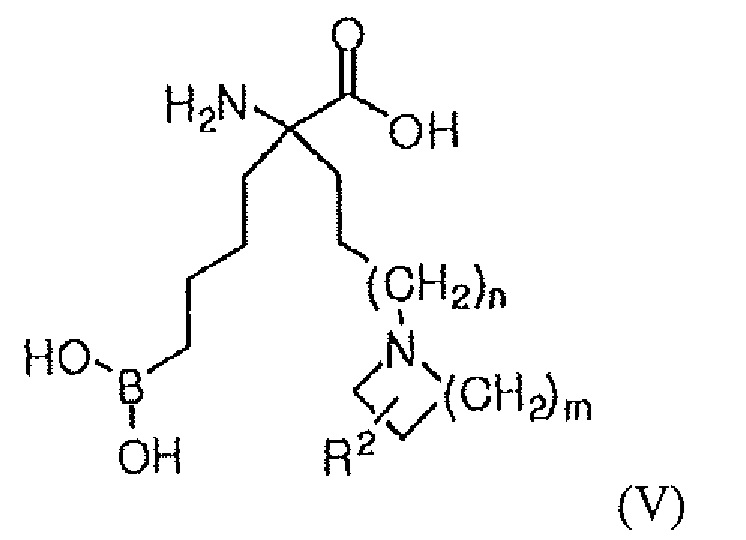
где m равно 1, 2, 3 или 4; n равно 0, 1 или 2; и R2 представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил).
В одном варианте осуществления соединением является соединение формулы (VI), или его производное, или его соль:
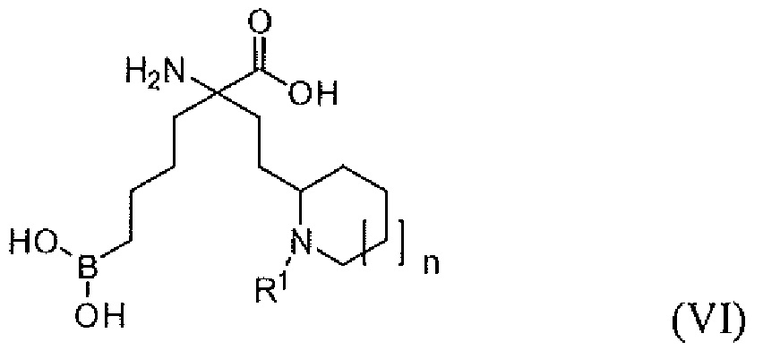
где n равно 0, 1 или 2; и R1 представляет собой H, алкил или арилалкил; и R2 представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил).
В одном варианте осуществления соединение выбирают из группы, состоящей из:
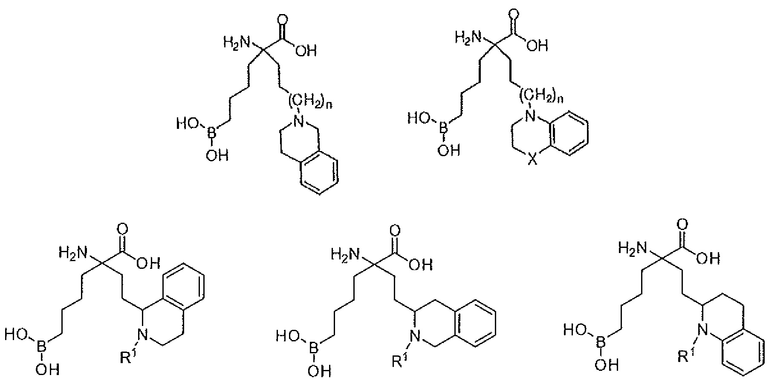
где n равно 0, 1 или 2; R1 представляет собой H, алкил или арилалкил; X представляет собой NR5, CR6R7, O, S, S(O) или S(O)2; причем, если X представляет собой CR6R7, то R7 представляет собой H, OH, OR8, CN или NR8R9; и R5, R6, R8 и R9 независимо представляют собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), - C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил); или их производного, или их соли.
В одном варианте осуществления соединение выбирают из группы, состоящей из:
где R1 и R2 независимо представляют собой H, C1-C6 алкил или арилалкил; или их производного, или их соли.
В одном варианте осуществления соединением является соединение формулы (VII), или его производное, или его соль:
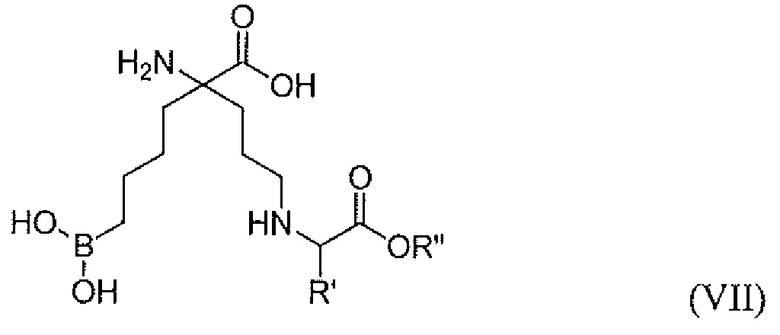
где R' представляет собой H, C1-C6 алкил, бензил, замещенный бензил, CH3SCH2CH2-, CH3S(=O)CH2CH2-, CH3S(О)2CH2CH2-, 3-индол-1H-ил-метил, HSCH2-, -CH2CH2C(=O)NH2 -CH2C(=O)NH2, CH2CH2C(=O)OH, -CH2C(=O)OH, -CH(OH)CH3, -CH2OH, -(CH2)4NH2, -(CH2)3NHC(=NH)NH2 или имидазол-4-ил-метил; R” представляет собой H или C1-C6 алкил.
В одном варианте осуществления соединением является соединение формулы (VIII), или его производное, или его соль:
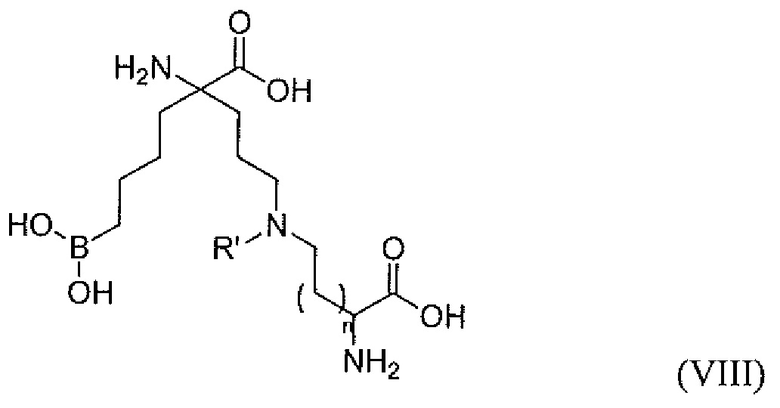
где n равно 0, 1, 2 или 3; и R' представляет собой C1-C6 алкил.
В одном варианте осуществления соединение выбирают из группы, состоящей из:
2-амино-6-бороно-2-(2-(пиперазин-1-ил)этил)капроновой кислоты;
2-амино-2-(3-(4-бензилпиперазин-1-ил)пропил)-6-боронокапроновой кислоты;
2-амино-6-бороно-2-(3-(4-(2-хлорбензил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(2-цианобензил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(2,4-дифторбензил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(2,3-дифторбензил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3,4-дихлорбензил)пиперазин-1-ил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3-(трифторметил)бензил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(4-(метилсульфонил)бензил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(4-фторбензил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3,4-дифторбензил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3,5-дифторбензил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-фенетилпиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(2-(4-(3,4-дихлорбензил)пиперазин-1-ил)этил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3,4-дихлорфенил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(2-(4-фторфенил)пиперидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(2-(пиперидин-2-ил)этил)капроновой кислоты;
2-амино-6-бороно-2-(2-(1-(3,4-дихлорбензил)пиперидин-2-ил)этил)капроновой кислоты;
2-амино-6-бороно-2-(2-(1-(3,5-дифторбензил)пиперидин-2-ил)этил)капроновой кислоты;
2-амино-6-бороно-2-(2-(1-(3,4-дифторбензил)пиперидин-2-ил)этил)капроновой кислоты;
2-амино-6-бороно-2-(2-(1-(3,4-дихлорбензил)пиперидин-3-ил)этил)капроновой кислоты;
2-амино-6-бороно-2-(3-(1-(3,4-дихлорбензил)пиперидин-2-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(1-(3,4-дифторбензил)пиперидин-2-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(1-(3,5-дифторбензил)пиперидин-2-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(3-фенилпиперидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(3-(5-фтор-1H-бензо[d]имидазол-2-ил)пиперидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3,4-дифторбензил)пиперидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(пиримидин-2-илметил)пиперидин-1-ил)пропил)капроновой кислоты;
2-(3-(3H-спиро[изобензофуран-1,4'пиперидин]-1'-ил)пропил)-2-амино-6-боронокапроновой кислоты;
2-амино-6-бороно-2-(3-(4-оксо-1-фенил-1,3,8-триазаспиро[4,5]декан-8-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(2-хлорфенил)-1H-пиразол-1-ил)пиперидин-1-ил)пропилкапроновой кислоты;
2-амино-6-бороно-2-(3-(4-(5-фенил-1,3,4-оксадиазол-2-ил)пиперидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(4-(трифторметил)фенокси)пиперидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(2-изопропилфенокси)пиперидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(4-фторфенил)пиперидин-1-ил)пропилкапроновой кислоты;
2-амино-6-бороно-2-(3-(4-(4-метоксифенил)пиперидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(4-хлорфенил)-5,6-дигидропиридин-1(2Н)-ил)пропил)капроновой кислоты;
2-амино-2-(3-(4-бензил-4-гидроксипиперидин-1-ил)пропил)-6-боронокапроновой кислоты;
2-амино-6-бороно-2-(3-(4-(4-хлорфенил)-4-гидроксипиперидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(5-(трифторметил)пиридин-2-ил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-гидроксипиперидин-1-ил)пропил)капроновой кислоты;
2-амино-2-(3-(4-((S)-2-амино-3-метилбутаноилокси)пиперидин-1-ил)пропил)-6-боронокапроновой кислоты;
2-амино-2-(3-(4-бензамидопиперидин-1-ил)пропил)-6-боронокапроновой кислоты;
2-амино-6-бороно-2-(3-(3,4-дигидроизохинолин-2(1H)-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-метил-2-фенилпиперазин-1-ил)пропил)капроновой кислоты;
2-амино-2-(3-(2-бензилпиперидин-1-ил)пропил)-6-боронокапроновой кислоты;
2-амино-6-бороно-2-(3-(2-(4-метоксифенил)пиперидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(2-(3-метоксифенил)пирролидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(2-(2-фторбензил)пирролидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(2-(2-(трифторметил)фенил)пирролидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(2-(4-фторфенил)пирролидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(2-(3-хлорфенил)пирролидин-1-ил)пропил)капроновой кислоты;
2-амино-2-(3-(2-(бифенил-4-ил)пирролидин-1-ил)пропил)-6-боронокапроновой кислоты;
2-амино-6-бороно-2-(3-(2-(3,4-дихлорфенил)пирролидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(пирролидин-1-ил)пропил)капроновой кислоты;
2-амино-2-(3-(азетидин-1-ил)пропил)-6-боронокапроновой кислоты;
2-амино-6-бороно-2-(3-(3-фенилазетидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(3-п-толилазетидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(3-(3-(3,4-дихлорфенил)уреидо)азетидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(3-(3-(4-фторфенил)уреидо)азетидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(3-(-хлор-2-фторбензамидо)азетидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-этилпиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-морфолинопропил)капроновой кислоты;
2-амино-6-бороно-2-(3-тиоморфолинопропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(тиазолидин-2-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(фенетиламино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(метил(фенетил)аминопропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(этил(нафтален-1-илметил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(метил(нафтален-1-илметил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-((циклогексилметил)(этил)амино)пропил)капроновой кислоты;
2-амино-2-(3-(бензил(этил)амино)пропил)-6-боронокапроновой кислоты;
2-амино-2-(3-(бензил(этил)амино)пропил)-6-боронокапроновой кислоты;
2-амино-6-бороно-2-(3-((4-хлорбензил)(метил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-((3,4-дихлорбензил)(метил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-((3,4-дихлорбензил)(этил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(циклогексиламино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-циклогексил(метил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(метил(тетрагидро-2H-пиран-4-ил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-циклопентил(метил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-((3-хлорбензил)(метил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(этил(тетрагидро-2H-пиран-4-ил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-((6-фторхроман-4-ил)(метил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(2-метоксиэтиламино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-((2-метоксиэтил)(метил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-((S)-1-метоксипропан-2-иламино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(диметиламино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-((2-(диметиламино)этил(метил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(2-(диметиламино)этиламино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(диэтиламино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-((1R,4R)-5-(3,4-дихлорфенилкарбамоил)-2,5-диазабицикло[2.2.2]гептан-2-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-((S)-3-метил-2-((R)-4-((3R,5S,7R,8R,9S,10S,12S,13R,14S,17R)-3,7,12-тригидрокси-10,13-диметилгексадекагидро-1H-циклопента[а]фенатрен-17-ил)пентанамидо)бутаноилокси)пиперидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(4-(пиперазин-1-ил)бутил)капроновой кислоты;
2-амино-6-бороно-2-(4-(4-(3,4-дихлорфенил)пиперазин-1-ил)бутил)капроновой кислоты;
2-амино-6-бороно-2-(4-(4-(3,4-дифторбензил)пиперидин-1-ил)бутил)капроновой кислоты;
2-амино-6-бороно-2-(4-(3,4-дигидроизохинолин-2(1H)-ил)бутил)капроновой кислоты;
2-амино-6-бороно-2-(4-(2-(4-фторфенил)пиперидин-1-ил)бутил)капроновой кислоты;
2-амино-6-бороно-2-(3-(карбоксиметиламино)пропил)капроновой кислоты;
2-амино-2-(3-(4-(бифенил-4-илметил)пиперазин-1-ил)пропил)-6-боронокапроновой кислоты;
2-амино-2-(3-(4-бензгидрилпиперазин-1-ил)пропил)-6-боронокапроновой кислоты;
2-амино-6-бороно-2-(3-(4-(4-фторбензоил)пиперидин-1-ил)пропил)капроновой кислоты;
2-(3-(3-(1H-бензо[d]имидазол-1-ил)-8-азабицикло[3.2.1]октан-8-ил)пропил)-2-амино-6-боронокапроновой кислоты;
2-амино-6-бороно-2-(3-(4-(фениламино)пиперидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3,4-дихлорбензиламино)пиперидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-((3,4-дихлорбензил)(этил)амино)пиперидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-метилпиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(3-фтор-4-фенилпиперидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(N-(3,4-дихлорбензил)октанамидо)пиперидин-1-ил)пропил)капроновой кислоты;
2-амино-3-(3-(4-бензил-4-(деканоилокси)пиперидин-1-ил)пропил)-6-боронокапроновой кислоты;
2-амино-2-(3-(3-(бензо[d]оксазол-2-ил)пиперидин-1-ил)пропил)-6-боронокапроновой кислоты;
2-амино-6-бороно-2-(3-(2-фенилпирролин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(3-(3-(3,4-дихлорфенил)уреидо)пирролидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(3-(3-(4-фторфенил)уреидо)пирролидин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(3-(3,4-дихлорфенилсульфонамидо)пирролидин-1-ил)пропил)капроновой кислоты;
2-(3-(1H-имидазол-1-ил)пропил)-2-амино-6-боронокапроновой кислоты;
2-(3-(1H-бензо[d]имидазол-1-ил)пропил)-2-амино-6-боронокапроновой кислоты;
2-амино-6-бороно-2-(3-(циклопентиламино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(тетрагидро-2Н-пиран-4-иламино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-((3R)-3-метокситетрагидро-2H-пиран-4-иламино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(метил(нафтален-2-илметил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(метил((4-метилнафтален-1-ил)метил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-((4-(3,4-дихлорфенокси)бензил)(метил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-((3',4'-дихлорбифенил-4-ил)метил)(метил)амино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-((3',4'-дихлорбифенил)-4-ил)метиламино)пропил)капроновой кислоты;
(S)-2-амино-6-бороно-2-(3-(1-карбоксилэтиламино)пропил)капроновой кислоты;
(S)-2-амино-6-бороно-2-(3-(1-карбокси-3-метилбутиламино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-((S)-1-метокси-1-оксопропан-2-иламино)пропил)капроновой кислоты;
(S)-2-амино-6-бороно-2-(3-(1-метокси-4-метил-1-оксопентан-2-иламино)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3,4-дихлорбензоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(4-метоксибензоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3-метоксибензоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(4-метилбензоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(4-фторбензоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(2-фторбензоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(2-хлорбензоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3-фторбензоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(4-(трифторметил)бензоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(4-карбамоилбензоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3,4-дихлорфенилкарбамоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(2-фторфенилкарбамоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3-фторфенилкарбамоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(4-фторфенилкарбамоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3,4-дифторфенилкарбамоил)пиперазин-1-ил)пропилкапроновой кислоты;
2-амино-6-бороно-2-(3-(4-(2,5-дифторфенилкарбамоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(2,4-дифторфенилкарбамоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(2,3-дифторфенилкарбамоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3,5-дифторфенилкарбамоил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-тозилпиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(4-фторфенилсульфонил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3-фторфенилсульфонил)пиперазин-1-ил)пропил)капроновой кислоты;
2-амино-6-бороно-2-(3-(4-(3,4-дихлорфенилсульфонил)пиперазин-1-ил)пропил)капроновой кислоты;
их соли, их производного и их смеси.
В одном варианте осуществления производным является сложноэфирное пролекарство формулы:
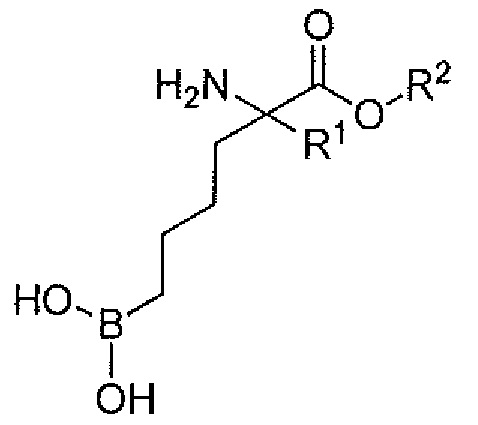
где:
R1 представляет собой второй заместитель;
R2 выбирают из группы, состоящей из C1-C6 алкила, C3-C7 циклоалкила, тетрагидрофуран-2-ил-метила, тетрагидрофуран-3-ил-метила, C3-C7 циклоалкил-метила, 2-(C3-C7 циклоалкил)этила, дигидрофуран-2(3H)-он-4-ил-метила, 2-гидроксил-этила, 2-гидроксил-2-метил-этила, фенила, 2-толила, 3-толила, 4-толила, 2-метокси-фенила, 3-метокси-фенила, 4-метокси-фенила, 2,3-дигидро-1H-инден-4-ила, 2,3-дигидро-1H-инден-5-ила, тиазол-2-ил-метила, тиазол-4-ил-метила, имидазол-2-ил-(CH2)n-, имидазол-4-ил-(CH2)n-, 2-метил-1H-бензо[d]имидазол-2-ил-(CH2)n-, R5C(=O)OCH2CH2-, R5C(=O)OCH(CH3)CH2-, R5C(=O)OCH2- или R5C(=O)OCH(CH3)-;
n равно 1, 2, 3 или 4;
R5 представляет собой H, C1-C6 алкил, C3-C7 циклоалкил, арил, гетероарил или CH(R6)NH2; и
R6 представляет собой Н, C1-C6 алкил, бензил, замещенный бензил, CH3SCH2CH2-, CH3S(=O)CH2CH2-, CH3S(О)2CH2CH2-, 3-индол-1H-ил-метил, HSCH2-, -CH2CH2C(=O)NH2, -CH2C(=O)NH2, CH2CH2C(=O)OH, -CH2C(=O)OH, -CH(OH)CH3, -CH2OH, -(CH2)4NH2, -(CH2)3NHC(=NH)NH2 или имидазол-4-ил-метил; причем бензоимидазол необязательно замещен по меньшей мере одним заместителем, выбираемым из группы, состоящей из (C1-C6)алкила, галогена и (C1-C6)алкокси.
В одном варианте осуществления производное выбирают из группы, состоящей из:
5-амино-8-(4-(3,4-дихлорбензил)пиперазин-1-ил)-5-(изопропоксикарбонил)октилбороновой кислоты;
5-амино-8-(4-(3,4-дихлорбензил)пиперазин-1-ил)-5-(изопентилоксикарбонил)октилбороновой кислоты;
5-амино-8-(4-(3,4-дихлорбензил)пиперазин-1-ил)-5-((2-(пиперидин-1-ил)этокси)карбонил)октилбороновой кислоты;
5-амино-8-(4-(3,4-дихлорбензил)пиперазин-1-ил)-5-((2-морфолиноэтокси)карбонил)октилбороновой кислоты;
5-амино-5-(метоксикарбонил)-8-(4-(4-метилсульфонил)бензил)пиперазин-1-ил)октилбороновой кислоты;
5-амино-5-(этоксикарбонил)-8-(4-(4-(метилсульфонил)бензил)пиперазин-1-ил)октилбороновой кислоты;
5-амино-8-(4-(4-(метилсульфонил)бензил)пиперазин-1-ил)-5-(пропоксикарбонил)октилбороновой кислоты;
5-амино-5-(изопропоксикарбонил)-8-(4-(4-(метилсульфонил)бензил)пиперазин-1-ил)октилбороновой кислоты;
5-амино-5-(изобутоксикарбонил)-8-(4-(4-(метилсульфонил)бензил)пиперазин-1-ил)октилбороновой кислоты;
5-амино-5-(изопентилоксикарбонил)-8-(4-(4-(метилсульфонил)бензил)пиперазин-1-ил)октилбороновой кислоты;
5-амино-8-(4-(4-(метилсульфонил)бензил)пиперазин-1-ил)-5-((пентан-3-илокси)карбонил)октилбороновой кислоты;
5-амино-5-((3-метилбутан-2-илокси)карбонил)-8-(4-(4-(метилсульфонил)бензилпиперазин-1-ил)октилбороновой кислоты;
5-амино-5-((2-метоксиэтокси)карбонил)-8-(4-(4-(метилсульфонил)бензил)пиперазин-1-ил)октилбороновой кислоты;
5-амино-5-((2-гидроксиэтокси)карбонил)-8-(4-(4-(метилсульфонил)бензил)пиперазин-1-ил)октилбороновой кислоты;
5-амино-8-(4-(4-(метилсульфонил)бензил)пиперазин-1-ил-5-((2-морфолиноэтокси)карбонил)октилбороновой кислоты;
5-амино-8-(4-(3,4-дихлорфенил)пиперазин-1-ил-5-(метоксикарбонил)октилбороновой кислоты;
их соли и их смеси.
В одном варианте осуществления композиция дополнительно включает по меньшей мере один фармацевтически приемлемый носитель.
В одном варианте осуществления композиция дополнительно включает ингибитор, выбираемый из группы, состоящей из ингибитора фосфодиэстеразы-1 (PDE1), ингибитора фосфодиэстеразы-2 (PDE2), ингибитора фосфодиэстеразы-3 (PDE3), ингибитора фосфодиэстеразы-4 (PDE4), ингибитора фосфодиэстеразы-5 (PDE5), неспецифического ингибитора PDE, который ингибирует по меньшей мере два фермента, выбираемых из группы, состоящей из PDE1, PDE2, PDE3, PDE4 и PDE5, и их комбинации.
В одном варианте осуществления композиция дополнительно включает по меньшей мере один фармацевтически приемлемый носитель и ингибитор, выбираемый из группы, состоящей из ингибитора фосфодиэстеразы-1 (PDE1), ингибитора фосфодиэстеразы-2 (PDE2), ингибитора фосфодиэстеразы-3 (PDE3), ингибитора фосфодиэстеразы-4 (PDE4), ингибитора фосфодиэстеразы-5 (PDE5), неспецифического ингибитора PDE, который ингибирует по меньшей мере два фермента, выбираемых из группы, состоящей из PDE1, PDE2, PDE3, PDE4 и PDE5, и их комбинации.
В одном варианте осуществления соединение включает способный формировать изображение фрагмент, выбираемый из группы, состоящей из флуоресцентной метки, испускающего гамма-лучи радиоизотопа, испускающего позитроны радиоизотопа, контрастного агента для получения изображения методом магнитного резонанса, рентгеноконтрастного агента и контрастного агента для эхографии.
Настоящее изобретение также включает способ ингибирования аргиназы у млекопитающего. Способ включает введение млекопитающему эффективного количества препарата, содержащего альфа-аминокислотное соединение, или его производное, или его соль, причем первый заместитель и второй заместитель связаны с альфа-углеродом соединения. Первый заместитель включает фрагмент, выбираемый из группы, состоящей из бороновой кислоты и N-гидроксигуанидина. Второй заместитель включает проксимальный атом азота, причем проксимальный азот является основным, причем, кроме того, проксимальный азот отделен от альфа-углерода цепью из двух, трех или четырех углеродов, при условии, что соединением не является 2-амино-6-бороно-2-(3-(пиперазин-1-ил)пропил)капроновая кислота, 2-(3-(4-ацетилпиперазин-1-ил)пропил)-2-амино-6-боронокапроновая кислота, 2-амино-6-бороно-2-(3-(4-(4-цианобензоил)пиперазин-1-ил)пропилкапроновая кислота или 2-амино-6-бороно-2-(3-(4-(3-метоксифенилкарбамоил)пиперазин-1-ил)пропил)капроновая кислота.
В одном варианте осуществления аргиназой является аргиназа дрожжей, бактерии, паразита или млекопитающего. В другом варианте осуществления аргиназой млекопитающего является аргиназа типа I человека или аргиназа типа II человека. В еще одном варианте осуществления препарат вводят млекопитающему по меньшей мере одним путем, выбираемым из группы, состоящей из перорального, назального, легочного, чрескожного, интраназального, офтальмологического, ректального и парентерального введения, причем парентеральное введение включает подкожное, внутривенное, интрауретральное или внутримышечное введение.
Настоящее изобретение дополнительно включает способ лечения нарушения или заболевания у млекопитающего. Способ включает введение млекопитающему терапевтически эффективного количества препарата, содержащего по меньшей мере один фармацевтически приемлемый носитель и альфа-аминокислотное соединение, или его производное, или его соль, причем первый заместитель и второй заместитель связаны с альфа-углеродом соединения. Первый заместитель включает фрагмент, выбираемый из группы, состоящей из бороновой кислоты и N-гидроксигуанидина. Второй заместитель включает проксимальный атом азота, причем проксимальный азот является основным, причем, кроме того, проксимальный азот отделен от альфа-углерода цепью из двух, трех или четырех углеродов, при условии, что соединением не является 2-амино-6-бороно-2-(3-(пиперазин-1-ил)пропил)капроновая кислота, 2-(3-(4-ацетилпиперазин-1-ил)пропил)-2-амино-6-боронокапроновая кислота, 2-амино-6-бороно-2-(3-(4-(4-цианобензоил)пиперазин-1-ил)пропилкапроновая кислота или 2-амино-6-бороно-2-(3-(4-(3- метоксифенилкарбамоил)пиперазин-1-ил)пропил)капроновая кислота.
В одном варианте осуществления нарушение или заболевание характеризуется необычно высокой аргиназной активностью или необычно низкой активностью синтазы оксида азота в ткани млекопитающего.
В одном варианте осуществления нарушение или заболевание выбирают из группы, состоящей из состояния, связанного с ишемически-реперфузионным повреждением, идиопатическим фиброзом легких, легочной артериальной гипертензией, сильной коронарной вазодилатацией, астмой, острым респираторным дистресс-синдромом, хроническим обструктивным заболеванием легких (COPD), бронхолегочной дисплазией, гипоксической дыхательной недостаточностью, муковисцидозом, субарахноидальным кровоизлиянием, тромбозом, микробной инфекцией, раком, заживлением ран, консервацией крови, гипертрофией сердца, заболеванием желудочно-кишечного тракта, воспалительным заболеванием легких, нарушением полового возбуждения, сердечно-сосудистым нарушением, заболеванием, вызванным патогенным микроорганизмом, иммунологическим нарушением, раком, преждевременными родами, болезнью Рейно, псориазом, ревматоидным артритом и болезнью Пейрони, причем состояние, связанное с ишемически-реперфузионным повреждением, включает ишемию миокарда-реперфузионное повреждение, трансплантацию органа, острую почечную недостаточность или вазоокклюзивные кризисы при серповидноклеточной анемии.
В одном варианте осуществления препарат вводят млекопитающему по меньшей мере одним путем, выбираемым из группы, состоящей из перорального, назального, легочного, чрескожного, интраназального, офтальмологического, ректального и парентерального введения, причем парентеральное введение включает подкожное, внутривенное, интрауретральное или внутримышечное введение.
Настоящее изобретение также включает способ диагностирования сверхэкспрессии аргиназы у млекопитающего. Способ включает стадии введения млекопитающему диагностически эффективного количества препарата, содержащего альфа-аминокислотное соединение, его производное или его фармацевтически приемлемую соль, и получение изображения млекопитающего. Первый заместитель и второй заместитель связаны с альфа-углеродом соединения. Первый заместитель включает фрагмент, выбираемый из группы, состоящей из бороновой кислоты и N-гидроксигуанидина. Второй заместитель включает проксимальный атом азота, причем проксимальный азот является основным, причем, кроме того, проксимальный азот отделен от альфа-углерода цепью из двух, трех или четырех углеродов, при условии, что соединением не является 2-амино-6-бороно-2-(3-(пиперазин-1-ил)пропил)капроновая кислота, 2-(3-(4-ацетилпиперазин-1-ил)пропил)-2-амино-6-боронокапроновая кислота, 2-амино-6-бороно-2-(3-(4-(4-цианобензоил)пиперазин-1-ил)пропилкапроновая кислота или 2-амино-6-бороно-2-(3-(4-(3- метоксифенилкарбамоил)пиперазин-1-ил)пропил)капроновая кислота. Соединение включает формирующий изображение заместитель, который создает возможность для in vivo визуализации соединения.
В одном варианте осуществления сверхэкспрессия аргиназы связана с астмой, раком, бактериальной инфекцией или их комбинациями. В другом варианте осуществления формирующий изображение заместитель выбирают из группы, состоящей из флуоресцентной метки, испускающего гамма-лучи радиоизотопа, испускающего позитроны радиоизотопа, контрастного агента для получения изображения методом магнитного резонанса, рентгеноконтрастного агента и контрастного агента для эхографии.
Настоящее изобретение дополнительно включает способ расслабления гладкой мышцы или усиления расслабления гладкой мышцы у млекопитающего. Способ включает введение млекопитающему терапевтически эффективного количества препарата, содержащего по меньшей мере один фармацевтически приемлемый носитель и альфа-аминокислотное соединение, или его производное, или его соль, причем первый заместитель и второй заместитель связаны с альфа-углеродом соединения. Первый заместитель включает фрагмент, выбираемый из группы, состоящей из бороновой кислоты и N-гидроксигуанидина. Второй заместитель включает проксимальный атом азота, причем проксимальный азот является основным, причем, кроме того, проксимальный азот отделен от альфа-углерода цепью из двух, трех или четырех углеродов, при условии, что соединением не является 2-амино-6-бороно-2-(3-(пиперазин-1-ил)пропил)капроновая кислота, 2-(3-(4-ацетилпиперазин-1-ил)пропил)-2-амино-6-боронокапроновая кислота, 2-амино-6-бороно-2-(3-(4-(4-цианобензоил)пиперазин-1-ил)пропилкапроновая кислота или 2-амино-6-бороно-2-(3-(4-(3-метоксифенилкарбамоил)пиперазин-1-ил)пропил)капроновая кислота.
В одном варианте осуществления гладкую мышцу выбирают из группы, состоящей из гладкой мышцы желудочно-кишечного тракта, гладкой мышцы анального сфинктера, мышцы пищеводного сфинктера, пещеристого тела, сфинктера Одди, артериальной гладкой мышцы, сердечной гладкой мышцы, легочной гладкой мышцы, почечной гладкой мышцы, маточной гладкой мышцы, влагалищной гладкой мышцы, гладкой мышцы шейки матки, плацентарной гладкой мышцы, гладкой мышцы глаза и их комбинации. В другом варианте осуществления препарат вводят млекопитающему по меньшей мере одним путем, выбираемым из группы, состоящей из перорального, назального, легочного, чрескожного, интраназального, офтальмологического, ректального и парентерального введения, причем парентеральное введение включает подкожное, внутривенное, интрауретральное или внутримышечное введение.
Настоящее изобретение также включает способ обеспечения освобождения от иммуносупрессии у млекопитающего. Способ включает введение млекопитающему терапевтически эффективного количества препарата, содержащего по меньшей мере один фармацевтически приемлемый носитель и альфа-аминокислотное соединение, или его производное, или его соль, причем первый заместитель и второй заместитель связаны с альфа-углеродом соединения. Первый заместитель включает фрагмент, выбираемый из группы, состоящей из бороновой кислоты и N-гидроксигуанидина. Второй заместитель включает проксимальный атом азота, причем проксимальный азот является основным, причем, кроме того, проксимальный азот отделен от альфа-углерода цепью из двух, трех или четырех углеродов, при условии, что соединением не является 2-амино-6-бороно-2-(3-(пиперазин-1-ил)пропил)капроновая кислота, 2-(3-(4-ацетилпиперазин-1-ил)пропил)-2-амино-6-боронокапроновая кислота, 2-амино-6-бороно-2-(3-(4-(4-цианобензоил)пиперазин-1-ил)пропилкапроновая кислота или 2-амино-6-бороно-2-(3-(4-(3-метоксифенилкарбамоил)пиперазин-1-ил)пропил)капроновая кислота.
В одном варианте осуществления млекопитающее страдает заболеванием или состоянием, выбираемым из группы, состоящей из хронического инфекционного заболевания, бактериальной инфекции, паразитарной инфекции, травмы, лепры, туберкулеза, трансплантации печени, рака и их комбинации. В другом варианте осуществления препарат вводят млекопитающему по меньшей мере одним путем, выбираемым из группы, состоящей из перорального, назального, легочного, чрескожного, интраназального, офтальмологического, ректального и парентерального введения, причем парентеральное введение включает подкожное, внутривенное, интрауретральное или внутримышечное введение.
Способ дополнительно включает способ ингибирования продукции орнитина у млекопитающего. Способ включает введение млекопитающему терапевтически эффективного количества препарата, содержащего по меньшей мере один фармацевтически приемлемый носитель и альфа-аминокислотное соединение, или его производное, или его соль, причем первый заместитель и второй заместитель связаны с альфа-углеродом соединения. Первый заместитель включает фрагмент, выбираемый из группы, состоящей из бороновой кислоты и N-гидроксигуанидина. Второй заместитель включает проксимальный атом азота, причем проксимальный азот является основным, причем, кроме того, проксимальный азот отделен от альфа-углерода цепью из двух, трех или четырех углеродов, при условии, что соединением не является 2-амино-6-бороно-2-(3-(пиперазин-1-ил)пропил)капроновая кислота, 2-(3-(4-ацетилпиперазин-1-ил)пропил)-2-амино-6-боронокапроновая кислота, 2-амино-6-бороно-2-(3-(4-(4-цианобензоил)пиперазин-1-ил)пропилкапроновая кислота или 2-амино-6-бороно-2-(3-(4-(3- метоксифенилкарбамоил)пиперазин-1-ил)пропил)капроновая кислота.
В одном варианте осуществления млекопитающее страдает заболеванием или состоянием, выбираемым из группы, состоящей из рака и фиброза. В другом варианте осуществления препарат вводят млекопитающему по меньшей мере одним путем, выбираемым из группы, состоящей из перорального, назального, легочного, чрескожного, интраназального, офтальмологического, ректального и парентерального введения, причем парентеральное введение включает подкожное, внутривенное, интрауретральное или внутримышечное введение.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
С целью иллюстрации настоящего изобретения некоторые варианты осуществления настоящего изобретения представлены на чертежах. Однако настоящее изобретение не ограничивается определенными компоновками и средствами вариантов осуществления, представленных на чертежах.
Фиг.1 является схемой, иллюстрирующей жидкофазный синтез выбранных исходных материалов.
Фиг.2 является схемой, иллюстрирующей синтез выбранных исходных материалов.
Фиг.3 является схемой, иллюстрирующей неограничивающий пример жидкофазного синтеза соединения настоящего изобретения.
Фиг.4 является схемой, иллюстрирующей синтез выбранных исходных материалов.
Фиг.5 является схемой, иллюстрирующей снятие защитных групп с аминокислот.
Фиг.6 является схемой, иллюстрирующей синтез соединения настоящего изобретения.
Фиг.7 является схемой, иллюстрирующей альтернативный синтез соединения настоящего изобретения.
Фиг.8 является схемой, иллюстрирующей альтернативный синтез соединения настоящего изобретения.
Фиг.9 является схемой, иллюстрирующей манипулирование защитными группами, использованное при синтезе соединения настоящего изобретения.
Фиг.10 является схемой, иллюстрирующей некоторые реакции снятия защитных групп, использованные для образования соединения настоящего изобретения.
Фиг.11 является схемой, иллюстрирующей некоторые хиральные промежуточные продукты, использованные для образования соединения 1.
Фиг.12 является схемой, иллюстрирующей некоторые способы разделения основных рацемических промежуточных продуктов и хиральный синтез соединения 1.
Фиг.13 является схемой, иллюстрирующей приготовление некоторых являющихся пролекарствами сложноэфирных промежуточных продуктов.
Фиг.14 является схемой, иллюстрирующей некоторые способы снятия всех защитных групп с являющихся пролекарствами сложных эфиров.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение включает ингибиторы ферментов, композиции, включающие такие ингибиторы, и их применения. В неограничивающем аспекте настоящее изобретение включает ингибиторы аргиназы, композиции, содержащие такие ингибиторы аргиназы, и способы диагностирования и/или лечения состояний, характеризующихся необычно высокой аргиназной активностью или необычно низкими уровнями оксида азота, используя композиции настоящего изобретения.
Определения
Используемые выше и на протяжении всего описания следующие термины, если не указано иное, как следует понимать, имеют следующие значения.
Используемый в описании термин «приблизительно», когда относится к измеряемой величине, такой как количество, протяженность по времени и т.п., как подразумевается, охватывает вариации, составляющие ±20%, предпочтительно ±10%, более предпочтительно ±5%, даже более предпочтительно ±1% и даже еще более предпочтительно ±0,1% от указанной величины, поскольку такие вариации являются адекватными для выполнения описываемых способов и композиций.
Используемый в описании термин «ABH» относится к 2(S)-амино-6-боронокапроновой кислоте. Используемый в описании термин «BEC» относится к S-(2-бороноэтил)-L-цистеину.
«Заболеванием» является состояние здоровья животного, в котором животное не может сохранять гомеостаз, и в случае которого, если заболевание не уменьшать, то здоровье животного продолжает ухудшаться. В отличие от этого, «нарушением» у животного является состояние здоровья, в котором животное способно сохранять гомеостаз, но в котором состояние здоровья животного является менее благоприятным, чем то, которое было бы в отсутствие нарушения. Оставленное без лечения нарушение не вызывает обязательно дальнейшее ухудшение состояния здоровья животного.
Используемый в описании термин «лечение» относится к профилактическому, радикальному и паллиативному лечению заболевания или недуга, особенно у являющегося млекопитающим пациента, нуждающегося в таком лечении, предпочтительно у являющегося человеком пациента.
Используемый в описании термин «введение» относится к акту дачи или предоставления композиции или соединения пациенту самим пациентом или тем, кто ухаживает за больным, например, медицинским специалистом или т.п., включая акт проглатывания пациентом или нанесения на пациента или т.п., при котором композиция или соединение может проявлять свои действия.
Под термином «эффективное количество», используемым в описании, подразумевается количество ингибитора, которое является достаточным для предупреждения, ослабления или устранения симптомов или состояния, которые вызывают беспокойство. Квалифицированному специалисту будет понятно, что это количество варьирует и может быть без труда определено на основе ряда факторов, таких как заболевание или состояние, подвергаемое лечению, возраст и состояние здоровья и физическое состояние животного, подвергаемого лечению, тяжесть заболевания, конкретное вводимое соединение и т.п. Как правило, доза будет устанавливаться на уровне между 0,01 мг/кг и 250 мг/кг. Настоящее изобретение не ограничивается каким-либо конкретным способом введения.
Используемый в описании термин «фармацевтически приемлемые» относится к таким соединениям, материалам, композициям или лекарственным формам, которые, по результатам тщательной медицинской оценки, подходят для контактирования с тканями людей и животных без чрезмерной токсичности, раздражения, аллергической реакции или других проблемных ситуаций в соответствии с надлежащим соотношением пользы и риска.
Используемый в описании термин «фармацевтически приемлемые соли» относится к производным описываемых соединений, причем исходное соединение модифицируют, готовя его соли, образуемые из кислот или оснований, включая соли присоединения кислот и соли присоединения оснований. Примеры фармацевтически приемлемых солей включают, но не ограничиваются ими, образуемые из минеральных или органических кислот соли основных остатков, таких как амины; образуемые из щелочей или органические соли кислотных остатков, таких как карбоновые кислоты; и т.п.
Термин «соли присоединения кислот» относится к соответствующему производному в виде соли исходного соединения, которое было получено посредством присоединения кислоты. Фармацевтически приемлемые соли включают традиционные соли или четвертичные аммонийные соли исходного соединения, образуемые, например, из неорганических или органических кислот. Например, такие традиционные соли включают, но не ограничиваются ими, те, которые получают из неорганических кислот, таких как хлористоводородная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная кислота и т.п.; и соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, памовая, малеиновая, адипиновая, альгиновая, аспарагиновая, гидромалеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая кислота, бензолсульфокислота, толуолсульфокислота, метансульфокислота, 2-нафталинсульфокислота, этандисульфокислота, щавелевая, изэтионовая, глюкогептановая, глицерофосфорная, гептановая, капроновая, хлористоводородная, бромистоводородная, йодистоводородная, 2-нафталинсульфокислота, пектиновая, фосфорная, серная, 3-фенилпропионовая, пикриновая, триметилуксусная, тиоциановая, п-толуолсульфокислота, масляная, камфорная, камфорсульфоновая, диглюконовая, циклопентанпропионовая, бисерная, додецилсерная кислота, этансульфокислота и ундекановая кислота и т.п.
Термин «соль присоединения основания» относится к соответствующему производному в виде соли исходного соединения, которое было получено посредством присоединения основания. Также содержащие основный азот группы могут быть подвергнуты кватернизации с использованием таких агентов, как (низший алкил)галогениды, таких как метил-, этил-, пропил- и бутилхлориды, бромиды и йодиды; диалкилсульфаты, такие как диметил-, диэтил-, дибутил- и диамилсульфаты, длинноцепочечные алкилгалогениды, такие как децил-, лаурил-, миристил- и стеарилхлориды, бромиды и йодиды, арилалкилгалогениды, такие как бензил- и фенетилбромиды, и другие. Фармацевтически приемлемые соли включают традиционные соли или четвертичные аммонийные соли исходного соединения, образуемые, например, из неорганических или органических оснований. Например, такие традиционные соли включают, но не ограничиваются ими, те, которые получают из неорганических оснований, таких как гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид магния и гидроксид аммония, и соли, полученные из органических аминов, таких как метиламин, этиламин, изопропиламин, пиперидин, пиперазин, пирролидин, этаноламин, морфолин, диазепин, этилендиамин, пиридин, хинолин, хинуклидин и т.п.
Используемый в описании термин «фармацевтически приемлемый носитель» означает фармацевтически приемлемый материал, композицию или носитель, такой как жидкий или твердый наполнитель, стабилизатор, диспергирующий агент, суспендирующий агент, разбавитель, эксципиент, загуститель, растворитель или материал для инкапсулирования, вовлеченный в перенос или транспортировку соединения, применимого в настоящем изобретении, внутри пациента или к пациенту таким образом, чтобы оно могло выполнять предназначенную ему функцию. Обычно такие конструкции переносятся или транспортируются от одного органа, или части тела, к другому органу, или части тела. Каждый носитель должен быть «приемлемым» в том смысле, что он является совместимым с другими ингредиентами препарата, включая соединение, применимое в настоящем изобретении, и не вредным для пациента. Некоторые примеры материалов, которые могут служить в качестве фармацевтически приемлемых носителей, включают сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлозу и ее производные, такие как натриевая соль карбоксиметилцеллюлозы, этилцеллюлоза и ацетат целлюлозы; порошкообразный трагакант; солод; желатин; тальк; эксципиенты, такие как масло какао и парафины для суппозиториев; масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат, агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; поверхностно-активные вещества; альгиновую кислоту; апирогенную воду; изотонический солевой раствор; раствор Рингера; этиловый спирт; фосфатные буферные растворы; и другие нетоксичные совместимые вещества, используемые в фармацевтических препаратах. Используемый в описании термин «фармацевтически приемлемый носитель» также включает любые и всякие покрытия, антибактериальные и противогрибковые средства, и замедляющие абсорбцию средства и т.п., которые совместимы с активностью соединения, применимого в настоящем изобретении, и являются физиологически приемлемыми для пациента. В композиции также могут быть включены дополнительные активные соединения. «Фармацевтически приемлемый носитель» может дополнительно включать фармацевтически приемлемую соль соединения, применимого в настоящем изобретении. Другие дополнительные ингредиенты, которые могут быть включены в фармацевтические композиции, используемые для осуществления на практике настоящего изобретения, известны в данной области техники и описаны, например, в публикации Remington's Pharmaceutical Sciences (Genaro, Ed., Mack Publishing Co,, 1985, Easton, PA), которая включена в описание посредством ссылки.
За исключением случаев, когда какая-либо среда или агент несовместим(а) с активным соединением, предусматривается ее (его) применение в композициях. В композиции могут также быть включены дополнительные активные соединения.
Используемый в описании термин, «единица дозирования» относится к физически дискретным единицам, подходящим в качестве однократных доз для конкретного пациента, подвергаемого лечению. Каждая единица может содержать заранее заданное количество активного соединения(й), рассчитанное так, чтобы вызвать желаемый терапевтический эффект(ы), в сочетании с необходимым фармацевтическим носителем. Спецификация для форм единиц дозирования настоящего изобретения может быть обусловлена (a) уникальными характеристиками активного соединения(й) и конкретным терапевтическим эффектом(ами), который должен быть достигнут, и (b) ограничениями, связанными с областью составления смеси такого активного соединения(й).
Используемый в описании термин «пациент» относится к животному, включая млекопитающее, предпочтительно к человеку.
«Аминокислотой», если не указано иное, является альфа-аминокислота, содержащая альфа-углерод (также известный как α-углерод, Cα или αC), аминогруппу, присоединенную к α-углероду, карбоксильную группу, присоединенную к α-углероду, и по меньшей мере одну группу в виде боковой цепи, присоединенную к α-углероду. Эта по меньшей мере одна боковая цепь будет охватывать те, которые обнаруживаются в двадцати природных аминокислотах, такие как боковые цепи в глицине, аланине, лизине или глутаминовой кислоте, а также боковые цепи, которые не обнаруживаются в известных аминокислотах (неприродные аминокислоты).
Используемый в описании термин «основный азот» относится к атому азота, который имеет неподеленную пару электронов, имеющуюся в распоряжении для связывания с протоном и образования аммоний-йона. Основный азот может быть частью первичного, вторичного или третичного амина. Кроме того, основный азот может быть частью гетероарильного кольца, когда неподеленная пара электронов атома N может связываться с протоном. Основный азот может образовывать водородную связь в качестве акцептора или образовывать соляные мостики с кислотными группами на белках, такими как боковая цепь глутаминовой или аспарагиновой кислоты.
«Инструктивный материал», как этот термин используется в описании, включает публикацию, запись, диаграмму или любое другое средство выражения, которое может быть использовано для сообщения о применимости нуклеиновой кислоты, пептида и/или соединения настоящего изобретения в наборе для осуществления ослабления или лечения различных заболеваний или нарушений, перечисленных в описании. Необязательно, или альтернативно, инструктивный материал может описывать один или более способов ослабления заболеваний или нарушений в клетке или ткани животного. Инструктивный материал набора может, например, быть прикреплен к контейнеру, который содержит нуклеиновую кислоту, пептид и/или соединение настоящего изобретения, или пересылаться вместе с контейнером, который содержит нуклеиновую кислоту, пептид и/или соединение. Альтернативно, инструктивный материал может пересылаться отдельно от контейнера в расчете на то, что получатель будет использовать инструктивный материал и соединение совместно.
Как используется в определениях в описании, Ra в каждом случае независимо представляет собой H, OH, алкил (необязательно замещенный одним или более R4), алкокси (необязательно замещенный одним или более R4), галоген, трифторметил, алканоилокси (необязательно замещенный одним или более R4), метилендиокси, бензилокси (необязательно замещенный одним или более R4), фенилокси (необязательно замещенный одним или более R4), нафтилокси (необязательно замещенный одним или более R4), нитро, трифторметокси, нитрил, алкенил (необязательно замещенный одним или более R4), алкинил, сульфоксид, сульфонил, сульфонамидо, арил (необязательно замещенный одним или более R4), гетероарил (необязательно замещенный одним или более R4), арилоил (необязательно замещенный одним или более R4), гетероарилоил (необязательно замещенный одним или более R4), гетероарилокси (необязательно замещенный одним или более R4), гетероарилметилокси (необязательно замещенный одним или более R4), алканоил, алкоксикарбонил, алкиламинокарбонил или амино.
Как используется в описании, в представленных определениях R4 в каждом случае независимо представляет собой (C1-C20)алкил, (C2-C20)алкенил, (C1-C20)алкинил, галоген, нитрил, нитро, (C5-C50)арил, (C3-C50)гетероарил, содержащий по меньшей мере один гетероатом, выбираемый из N, О и S; (C5-C50)арил(C1-C20)алкил, гетероарил(C1-C20)алкил, (C5-C50)арилокси(C1-C20)алкил, гетероарилокси(C1-C20)алкил, (C5-C50)ариламино(C1-C20)алкил, гетероариламино(C1-C20)алкил, амино(C1-C20)алкил, -Rx-C(=O)-Ry, -Rx-О-Rz или -L-Y.
«Алкил», как используется в описании, относится к алифатической углеводородной цепи из 1 до приблизительно 20 атомов углерода, предпочтительно от 1 до 10 атомов углерода, более предпочтительно от 1 до 6 атомов углерода и даже более предпочтительно от 1 до 4 атомов углерода и включает неразветвленные и разветвленные цепи, такие как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил и изогексил. Низший алкил относится к алкилу, содержащему от 1 до 4 атомов углерода. Алкильные группы могут быть необязательно замещены одним или более Ra, определенных в описании.
«Алкиленил», как используется в описании, относится к двухвалентному аналогу «алкила», определенному в описании (например, метиленилу, этиленилу, пропиленилу и т.д.). Алкиленильные группы могут быть необязательно замещены одним или более Ra, определенных в описании.
«Алкенил» или «олефиновая группа», как используется в описании, относится к алкильной группе из по меньшей мере двух атомов углерода, имеющей одну или более двойных связей, где алкил является группой, определенной выше. Алкенильные группы могут быть необязательно замещены одним или более Ra, определенных в описании.
«Гидрокси(C1-C20)алкил», как используется в описании, относится к алкильной группе, определенной выше, замещенной по меньшей мере одной гидроксигруппой.
«Гидрокси(C2-C20)алкенил», как используется в описании, относится к алкенильной группе, определенной выше, замещенной по меньшей мере одной гидроксигруппой.
«Алкинил», как используется в описании, относится к алкильной группе из по меньшей мере двух атомов углерода, имеющей одну или более тройных связей, где алкил является группой, определенной выше. Алкинильные группы могут быть необязательно замещены одним или более Ra, определенных в описании.
«Арил», как используется в описании, относится к необязательно замещенной, моно-, ди-, три- и другой полициклической ароматической кольцевой системе, содержащей от приблизительно 5 до приблизительно 50 атомов углерода (и все комбинации и субкомбинации диапазонов и конкретных чисел атомов углерода в нем), при этом предпочтительным является от приблизительно 6 до приблизительно 10 углеродов. Неограничивающие примеры включают, например, фенил, нафтил, антраценил и фенантренил. Арильные группы могут быть необязательно замещены одним или более Ra, определенных в описании.
«Гетероарил», как используется в описании, относится к необязательно замещенной, моно-, ди-, три- и другой полициклической ароматической кольцевой системе, которая включает по меньшей мере один и предпочтительно от 1 до приблизительно 4 членов колец, являющихся гетероатомами - серой, кислородом или азотом. Гетероарильные группы могут содержать, например, от приблизительно 3 до приблизительно 50 атомов углерода (и все комбинации и субкомбинации диапазонов и конкретных чисел атомов углерода в нем), при этом предпочтительным является от приблизительно 6 до приблизительно 10 углеродов. Неограничивающие примеры гетероарильных групп включают, например, пирролил, фурил, пиридил, 1,2,4-тиадиазолил, пиримидил, тиенил, изотиазолил, имидазолил, тетразолил, пиразинил, пиримидил, хинолил, изохинолил, тиофенил, бензотиенил, изобензофурил, пиразолил, индолил, пуринил, карбазолил, бензимидазолил и изоксазолил. Гетероарильные группы могут быть необязательно замещены одним или более Ra, определенных в описании.
«(C5-C50)Арил(C1-C20)алкил», как используется в описании, относится к группе R-R'-, где R представляет собой арильную группу, а R' представляет собой алкиленил, определенный выше.
«Гетероарил(C1-C20)алкил», как используется в описании, относится к группе R-R'-, где R представляет собой гетероарильную группу, а R' представляет собой алкиленил, определенный выше.
«(C5-C50)арилокси(C1-C20)алкил», как используется в описании, относится к группе R-O-R'-, где R представляет собой арильную группу, а R' представляет собой алкиленил, определенный выше.
«Гетероарилокси(C1-C20)алкил», как используется в описании, относится к группе R-O-R'-, где R представляет собой гетероарильную группу, а R' представляет собой алкиленил, определенный выше.
«(C5-C50)ариламино(C1-C20)алкил», как используется в описании, относится к группе R-NH-R'-, где R представляет собой арильную группу, а R' представляет собой алкиленил, определенный выше.
«Гетероарилоксиамино(C1-C20)алкил», как используется в описании, относится к группе R-NH-R'-, где R представляет собой гетероарильную группу, а R' представляет собой алкиленил, определенный выше.
«Амино(C1-C20)алкил», как используется в описании, относится к группе N(R”)-R'-, где R” представляет собой водород или (C1-C6)алкильную группу, а R' представляет собой алкиленил, определенный выше.
«Циклоалкил», как используется в описании, относится к необязательно замещенной алкильной группе, содержащей одно или более колец в своих структурах, содержащих от 3 до приблизительно 20 атомов углерода (и все комбинации и субкомбинации диапазонов и конкретных чисел атомов углерода в нем), при этом предпочтительным является от 3 до приблизительно 10 атомов углерода. Полициклические структуры могут быть с мостиковыми связями или конденсированными кольцевыми структурами. Группы включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил, циклооктил, 2-[4-изопропил-1-метил-7-окса-бицикло[2.2.1]гептанил], 2-[1,2,3,4-тетрагидро-нафталенил] и адамантил.
«Гетероциклоалкил», как используется в описании, относится к необязательно замещенной циклоалкильной группе, содержащей одно или более колец в своих структурах, содержащих от 2 до приблизительно 20 атомов углерода (и все комбинации и субкомбинации диапазонов и конкретных чисел атомов углерода в нем), при этом предпочтительным является от 2 до приблизительно 10 атомов углерода, помимо по меньшей мере одного гетероатома, независимо выбираемого из группы, состоящей из N, O и S. Полициклические структуры могут быть с мостиковыми связями или конденсированными кольцевыми структурами. Группы включают, но не ограничиваются ими, азиридинил, пирролидинил, пирролидино, пиперидинил, пиперидино, пиперазинил, пиперазино, морфолинил, морфолино, тиоморфолинил, тиоморфолино, тетрагидрофуранил, тетрагидротиофуранил, тетрагидропиранил и пиранил.
«Галоген», как используется в описании, относится к хлору, брому, фтору и йоду.
«Алкокси», как используется в описании, относится к группе R-O-, где R представляет собой алкильную группу из 1-6 атомов углерода.
«Алкоксикарбонил», как используется в описании, относится к группе R-О-C(=O)-, где R представляет собой алкильную группу из 1-6 атомов углерода.
«Алканоил», как используется в описании, относится к группе R-C(=O)-, где R представляет собой алкильную группу из 1-6 атомов углерода.
«Алканоилокси», как используется в описании, относится к группе R-C(=O)-О-, где R представляет собой алкильную группу из 1-6 атомов углерода.
«Алкиламинокарбонил», как используется в описании, относится к группе R-NH-C(=O)-, где R представляет собой алкильную группу из 1-6 атомов углерода.
«Алкилкарбониламино», как используется в описании, относится к группе R-C(=O)-NH, где R представляет собой алкильную группу из 1-6 атомов углерода.
«Гетероарилметил», как используется в описании, относится к группе R-CH2-, где R представляет собой гетероарильную группу, определенную выше.
«Гетероарилметилокси», как используется в описании, относится к группе R-CH2-O-, где R представляет собой гетероарильную группу, определенную выше.
«Гетероарилокси», как используется в описании, относится к группе R-O-, где R представляет собой гетероарильную группу, определенную выше.
«Гетероарилметилокси», как используется в описании, относится к группе R-CH2-O-, где R представляет собой гетероарильную группу, определенную выше.
«Гетероцикл» или «гетероциклил», как используется в описании, относится к стабильному (5-7)-членному моноциклическому или бициклическому, или (7-10)-членному бициклическому гетероциклическому кольцу, или его радикалу, которое является насыщенным, частично ненасыщенным или ненасыщенным (ароматическим), и которое содержит атомы углерода и от 1 до 4 гетероатомов, независимо выбираемых из группы, состоящей из N, O и S, и включает любую бициклическую группу, в которой любое из определенных выше гетероциклических колец конденсировано с бензольным кольцом. Являющиеся азотом и серой гетероатомы могут быть необязательно окисленными. Гетероциклическое кольцо может быть присоединено к своей боковой группе у любого гетероатома или атома углерода, который приводит к стабильной структуре. Гетероциклические кольца, описываемые в описании, могут быть замещены у атома углерода или атома азота, если полученное соединение является стабильным. Если специально отмечено, атом азота в гетероцикле может быть необязательно кватернизован. Предпочтительным является то, что когда общее число атомов S и О в гетероцикле превышает один, то эти гетероатомы не примыкают друг к другу. Предпочтительно, когда число атомов S и О в гетероцикле составляет не более одного. Примеры гетероциклов включают, но не ограничиваются ими, 1H-индазол, 2-пирролидонил, 2H,6H-1,5,2-дитиазинил, 2H-пирролил, 3H-индолил, 4-пиперидонил, 4aH-карбазол, 4H-хинолизинил, 6H-1,2,5-тиадиазинил, акридинил, азоцинил, бензимидазолил, бензофуранил, бензотиофуранил, бензотиофенил, бензоксазолил, бензтиазолил, бензтриазолил, бензтетразолил, безизоксазолил, бензизотиазолил, бензимидазалонил, карбазолил, 4H-карбазолил, α-, β- или γ-карболинил, хроманил, хроменил, циннолинил, декагидрохинолинил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуран, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1Н-индазолил, индоленил, индолинил, индолизинил, индолил, изобензофуранил, изохроманил, изоиндазолил, изоиндолинил, изоиндолил, изохинолинил, изотиазолил, изоксазолил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, оксазолидинилпиримидинил, фенантридинил, фенантролинил, феноксазинил, феназинил, фенотиазинил, феноксатиинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, птеридинил, пиперидонил, 4-пиперидонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазол, пиридоимидазол, пиридотиазол, пиридинил, пиридил, пиримидинил, пирролидинил, пирролинил, пирролил, хиназолинил, хинолинил, 4H-хинолизинил, хиноксалинил, хинуклидинил, карболинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, 6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, тиенил, тиенотиазолил, тиенооксазолил, тиеноимидазолил, тиофенил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил, ксантенил. Предпочтительные гетероциклы включают, но не ограничиваются ими, пиридинил, фуранил, тиенил, пирролил, пиразолил, имидазолил, индолил, бензимидазолил, 1H-индазолил, оксазолидинил, бензотриазолил, бензизоксазолил, оксиндолил, бензоксазолинил или изатинил. Также включены конденсированные кольцевые соединения или спиросоединения, содержащие, например, приведенные выше гетероциклы.
«Сульфоксид», как используется в описании, относится к соединению или фрагменту, содержащему группу -S(=O)-.
«Сульфонамидо», как используется в описании, относится к фрагменту, содержащему группу -S(О)2NH-.
«Сульфонил», как используется в описании, относится к фрагменту, содержащему группу -S(О)2-.
«Алифатическая связь», как используется в описании, относится к любой двухвалентной алкиленильной группе (например, метиленилу, этиленилу, пропиленилу и т.д.), включая группы, имеющие общую формулу -(CH2)m-, где m является целым числом от 1 до 6.
«Ароматическая связь», как используется в описании, относится к любой двухвалентной арильной группе, такой как группа -(C6H4)-.
«Остаток, способный формировать изображение фрагмента», или просто «способный формировать изображение фрагмент», как используется в описании, относится к любому фрагменту, обычно известному в данной области техники и, в частности, определенному в описании, который содержит одну или более групп, которые можно выявить или непосредственно, или опосредованно в in vivo или in vitro диагностическом способе визуализации, и включает, например, один или более фрагментов, которые испускают или могут быть причиной испускания обнаруживаемого излучения (например, в результате радиоактивного распада, возбуждения флуоресценции, активации спинового резонанса и т.д.), группы, которые оказывают влияние на локальные электромагнитные поля (например, парамагнитные, суперпарамагнитные, ферримагнитные или ферримагнитные разновидности), группы, которые поглощают или рассеивают энергию излучения (например, хромофоры, частицы (в том числе содержащие газ или жидкость везикулы), тяжелые элементы и их соединения и т.д.), и группы, которые порождают выявляемое вещество (например, генераторы газовых микропузырьков). Примеры способных формировать изображение фрагментов можно выбрать из группы, состоящей из испускающих гамма-лучи радиоизотопов, испускающих позитроны радиоизотопов, контрастных агентов для получения изображения методом магнитного резонанса (например, хелатных комплексов гадолиния), рентгеноконтрастных агентов (например, йодированных рентгеноконтрастных ароматических веществ) или контрастного агента для эхографии (например, липосом, содержащих эхогенное соединение).
На протяжении всего этого описания различные аспекты настоящего изобретения могут быть представлены в формате диапазона. Следует понимать, что описание в формате диапазона представлено исключительно для удобства и краткости и не должно рассматриваться как жесткое ограничение объема настоящего изобретения. Соответственно, описание диапазона, как следует рассматривать, содержит конкретно выявляемые все возможные поддиапазоны, а также отдельные численные значения внутри этого диапазона. Например, описание такого диапазона, как от 1 до 6, как следует рассматривать, содержит конкретно выявляемые поддиапазоны, такие как от 1 до 3, от 1 до 4, от 1 до 5, от 2 до 4, от 2 до 6, от 3 до 6 и т.д., а также отдельные численные значения внутри этого диапазона, например, 1, 2, 2,7, 3, 4, 5, 5,3 и 6. Это применимо вне зависимости от ширины диапазона.
Описание
Настоящее изобретение включает селективный ингибитор аргиназы. В одном варианте осуществления соединение настоящего изобретения включает альфа-аминокислоту, или ее производное, с первым заместителем и вторым заместителем у альфа-углерода (Cα). Первый заместитель включает фрагмент, выбираемый из группы, состоящей из бороновой кислоты и N-гидроксигуанидина. Другим заместителем является фрагмент, включающий проксимальный азот, причем проксимальный азот является основным и отделен от альфа-углерода линкером из двух-четырех углеродов.
В неограничивающем аспекте присутствие проксимального азота, который является основным, связанным через C2-C4 цепь с альфа-углеродом, придает неожиданные свойства соединению настоящего изобретения. В неограничивающем примере соединения настоящего изобретения могут ингибировать обе аргиназы (аргиназу I и аргиназу II) нечувствительным к pH образом. Это является значительным усовершенствованием по сравнению с известными ингибиторами типа бороновой кислоты, такими как ABH (2(S)-амино-6-боронокапроновая кислота) и BEC (S-(2-бороноэтил)-L-цистеин).
Физиологический pH организма человека (т.е. pH жидкости в субклеточных или клеточных компонентах, тканях или органах) составляет от 7,35 до 7,45. Хотя pH в некоторых клеточных микросредах (например, субклеточных компартментах, таких как митохондрии) может быть более основным (с pH вплоть до 8,5), большая часть нормальной физиологии, как полагают, происходит при физиологическом pH (см. Abad et al., 2004, J. Biol. Chem. 279:11521-11529). Однако оптимальная каталитическая активность аргиназы I или II имеет место при очень основном pH, составляющим 9,5. Большинство in vitro исследований для определения активности фермента - аргиназы выполняют при рН для оптимальной каталитической активности (т.е. pH 9,5), хотя это значение pH не является особенно физиологически релевантным. Для более физиологически релевантного количественного определения ингибирования аргиназы исследование фермента следует проводить при pH 7,5 или вблизи этого значения, но ингибиторная активность известных ингибиторов типа бороновой кислоты (таких как ABH и BEC) намного ниже при pH 7,5, чем при pH 9,5 (Colleluori & Ash, 2001, Biochem. 40, 9356-9362).
В неограничивающем варианте осуществления соединения настоящего изобретения не проявляют уменьшение ингибиторной в отношении аргиназы активности при изменении pH с 9,5 до 7,5. В другом неограничивающем варианте осуществления ингибиторная в отношении аргиназы активность соединений настоящего изобретения выше при pH<9,5, чем при pH 9,5. Например, когда экспериментальный pH уменьшали с 9,5 до 7,5, в примере 8 демонстрируется увеличение активности ингибирования аргиназы I человека, и не выявлено изменение в большой степени активности ингибирования аргиназы II человека. На основе своих увеличенных значений ингибиторной активности при физиологическом pH (по сравнению с известными ингибиторами типа бороновой кислоты) соединения настоящего изобретения оказывают неожиданно увеличенные биологические эффекты на аргиназу in vivo. Таким образом, соединения настоящего изобретения могут более эффективно модулировать или контролировать заболевания, характеризующиеся необычно высокой аргиназной активностью или необычно низкими уровнями оксида азота, чем известные ингибиторы типа бороновой кислоты.
В неограничивающем варианте осуществления ингибиторы настоящего изобретения селективно ингибируют аргиназу I по сравнению с аргиназой II при значении pH, которое меньше 9,5 (т.е. pIC50 для аргиназы I больше pIC50 для аргиназы II при значении pH, которое меньше 9,5). В другом неограничивающем варианте осуществления фрагмент, содержащий проксимальный азот, выбирают так, что соединение настоящего изобретения более селективно ингибирует аргиназу I по сравнению с аргиназой II (т.е. pIC50 для аргиназы I больше pIC50 для аргиназы II). В другом неограничивающем примере, если проксимальный азот в соединении настоящего изобретения является частью гетероциклической группы, включающей амидную, мочевинную или сульфонамидную функциональную группу, селективность соединения по отношению к аргиназе I по сравнению с аргиназой II значительно больше, чем в случае соединения настоящего изобретения, в котором отсутствует такая структурная особенность.
Соединения настоящего изобретения
В одном аспекте настоящее изобретение включает соединение, включающее альфа-аминокислоту с двумя заместителями при альфа-углероде. Первый заместитель включает фрагмент, выбираемый из группы, состоящей из бороновой кислоты и N-гидроксигуанидина. Второй заместитель включает фрагмент, содержащий проксимальный азот, причем проксимальный азот является основным и связан с альфа-углеродом (Cα) с помощью цепи из двух-четырех углеродов (C2-C4 цепи).
В одном варианте осуществления соединением не является 2-амино-6-бороно-2-(3-(пиперазин-1-ил)пропил)капроновая кислота, 2-(3-(4-ацетилпиперазин-1-ил)пропил)-2-амино-6-боронокапроновая кислота, 2-амино-6-бороно-2-(3-(4-(4-цианобензоил)пиперазин-1-ил)пропилкапроновая кислота или 2-амино-6-бороно-2-(3-(4-(3-метоксифенилкарбамоил)пиперазин-1-ил)пропилкапроновая кислота.
В неограничивающем варианте осуществления первый заместитель способен взаимодействовать с аргиназными остатками. Первый заместитель включает, например, бороновую кислоту, N-гидроксигуанидин и другие известные эквиваленты/заместители. В одном варианте осуществления первым заместителем является бороновая кислота. В настоящем изобретении предусматриваются бороновые кислоты, имеющие остовы различных длин. В другом варианте осуществления первый заместитель включает бороновую кислоту, связанную с альфа-углеродом через н-бутильную группу.
В неограничивающем варианте осуществления второй заместитель способен устанавливать связывающие взаимодействия в поверхностном углублении активного центра и области по обе стороны от поверхностных углублений активного центра аргиназы паразита, бактериальной аргиназы, аргиназы I или аргиназы II. Если этот заместитель может образовывать сильные взаимодействия с являющимся мишенью белком - аргиназой, то можно идентифицировать соединения с увеличенной активностью или селективностью по сравнению с соединениями, описанными в известном уровне техники. Второй заместитель включает углеродную цепь и проксимальный азот. В одном варианте осуществления проксимальный азот присоединен к альфа-углероду через цепь из двух-четырех углеродов. В другом варианте осуществления проксимальный азот присоединен к альфа-углероду через цепь из трех углеродов. Еще в одном варианте осуществления проксимальный азот является основным. Еще в одном варианте осуществления проксимальный азот является частью первичной, вторичной или третичной аминной группы.
В неограничивающем аспекте, если длина углеродной цепи составляет менее 4 атомов углерода, наличие проксимального азота в качестве части гетероциклической группы облегчает синтез соединения и уменьшает степень или вероятность циклизации проксимального азота с альфа-карбоновой кислотой. В одном варианте осуществления углеродная цепь не является разветвленной. В другом варианте осуществления углеродная цепь замещена по меньшей мере одной C1-C4 алкильной группой. В еще одном варианте осуществления углеродная цепь является частью гетероциклической группы, содержащей проксимальный азот.
В одном варианте осуществления настоящее изобретение включает соединение формулы (I) или (II), или его соль:
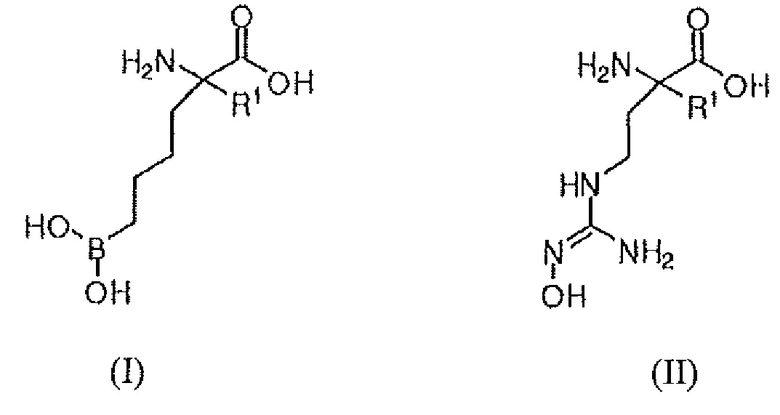
где:
R1 представляет собой (C2-4)-N(Z)Y;
C2-4 представляет собой цепь из двух, трех или четырех углеродов;
N является проксимальным азотом;
Z представляет собой H, алкил, циклоалкил, гетероциклоалкил, арил, гетероарил, арилалкил или алкоксиалкил;
Y представляет собой Н, алкил, циклоалкил, гетероциклоалкил, арил, гетероарил, арилалкил или алкоксиалкил;
где, если Z и Y являются циклическими, то Z и Y могут быть дискретными или связанными с образованием групп с мостиковой связью и конденсированных групп; и
где Z и Y могут быть дискретными или связанными вместе с N с образованием общей гетероциклоалкильной группы, которая может содержать один или более гетероатомов и может быть необязательно замещена алкилом, амино, галогеном, алкиламино, диалкиламино, арилом, гетероарилом, амидом, арилсульфонилом, алкилсульфонилом, сульфонамидом, арилкарбонилом, алкилкарбонилом, мочевиной, циано, гидрокси, алкокси, аралкокси, арилокси, аминокарбокси, арилалкилом или гетероарилалкилом.
Подходящие гетероциклические группы, предусматриваемые в настоящем изобретении, включают, например, азиридин, азетидин, пирролидин, пиперидин, азепан, азокан, диазетидин, имидазолидин, пиразолидин, оксазолидин, изоксазолидин, тиазолидин, изотиазолидин, пиперазин, морфолин, их аналоги с мостиковой связью, их конденсированные комбинации и их замещенные варианты.
Заместители на гетероциклических группах могут включать одну или более из следующих групп: (C1-C6)алкил, галоген, арил и гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)R3, -SO2R3, -CONHR3, COOR3, OR2 и NR3R3, при условии, что если по меньшей мере одним заместителем является OR2 или NR3R3, то по меньшей мере один заместитель не присоединен к тому же атому углерода, к которому присоединен атом азота гетероциклической группы.
R2 представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил).
R3 в каждом случае независимо представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил или гетероарил(C1-C6)алкил.
В одном подварианте осуществления соединением формулы (I) не является 2-амино-6-бороно-2-(3-(пиперазин-1-ил)пропил)капроновая кислота, 2-(3-(4-ацетилпиперазин-1-ил)пропил)-2-амино-6-боронокапроновая кислота, 2-амино-6-бороно-2-(3-(4-(4-цианобензоил)пиперазин-1-ил)пропилкапроновая кислота или 2-амино-6-бороно-2-(3-(4-(3-метоксифенилкарбамоил)пиперазин-1-ил)пропилкапроновая кислота.
В одном варианте осуществления настоящее изобретение включает соединение формулы (III) или его соль:
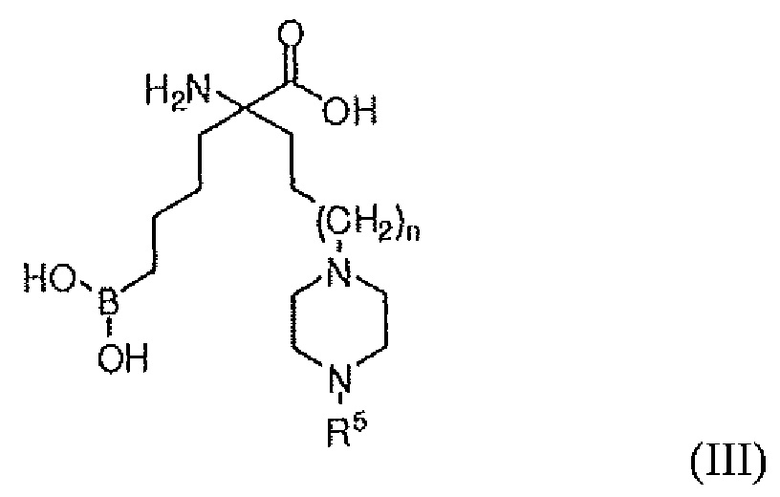
где:
n равно 0, 1 или 2; и
R5 представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил).
В одном подварианте осуществления R5 представляет собой фенил, бензил, ацетамид, бензамид или замещенный бензамид.
В одном варианте осуществления настоящее изобретение включает соединение формулы (IV) или его соль:
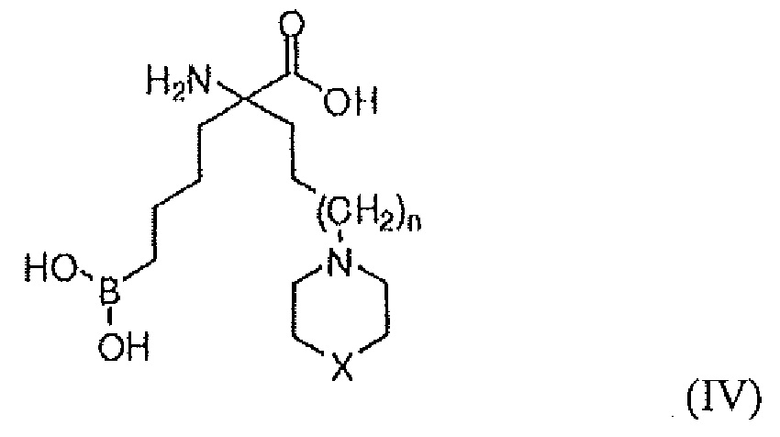
где:
n равно 0, 1 или 2;
X представляет собой NR5, CR6R7, O, S, S(=O) или S(O)2;
R7 представляет собой H, OH, OR8, CN или NR8R9; и
R5, R6, R8 и R9 независимо представляют собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил).
В одном подварианте осуществления X представляет собой NR5. В другом варианте осуществления R5 представляет собой фенил, бензил, ацетамид, бензамид или замещенный бензамид.
В одном варианте осуществления гетероциклическая группа является группой с мостиковой связью. Неограничивающими примерами соединений настоящего изобретения с гетероциклическими группами с мостиковой связью являются следующие соединения:
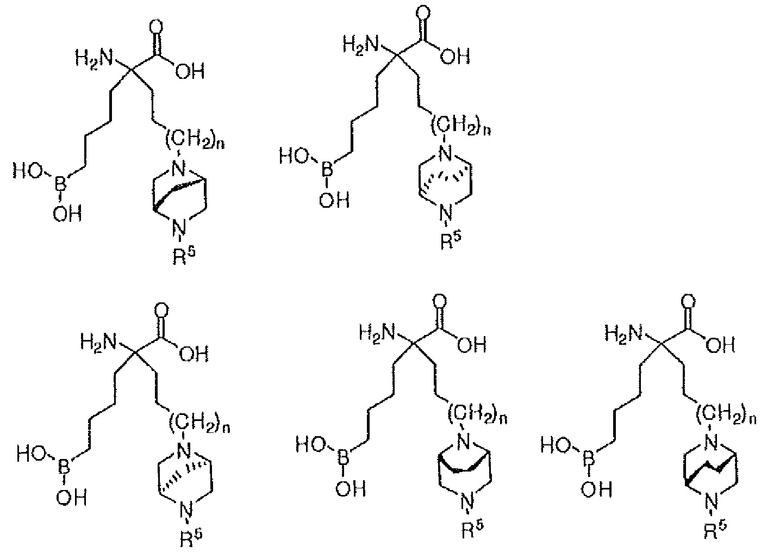
где:
n равно 0, 1 или 2; и
R5 представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил).
В одном варианте осуществления гетероциклическая группа замещена аминогруппой или содержит аминогруппу. Неограничивающие примеры таких соединений продемонстрированы ниже:
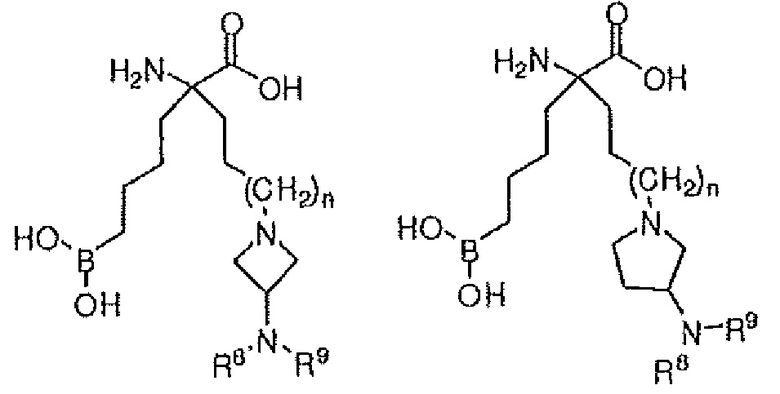
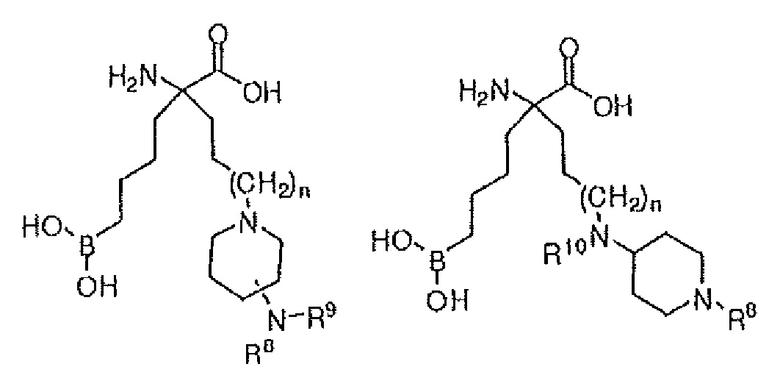
где:
n равно 0, 1 или 2;
R8 и R9 независимо представляют собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил); и
R10 представляет собой H, (C1-C6)алкил или арилалкил.
В одном варианте осуществления настоящее изобретение включает соединение формулы (V) или его соль:
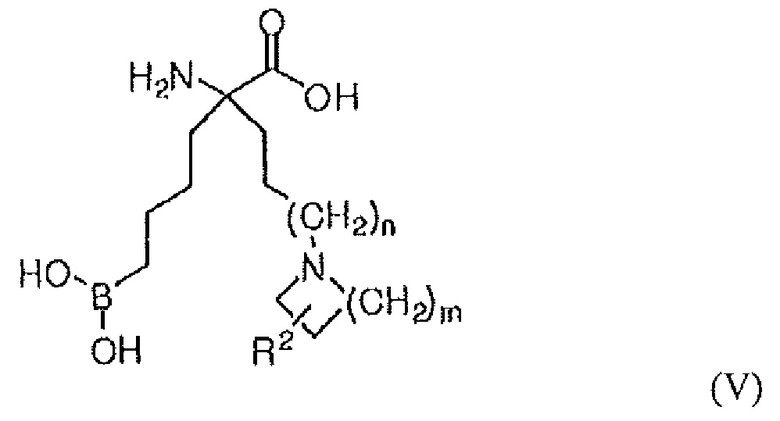
где:
m равно 1, 2, 3 или 4;
n равно 0, 1 или 2; и
R2 представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил).
Заместители на гетероциклических группах могут включать одну или более из следующих групп: (C1-C6)алкил, галоген, арил и гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)R3, -SO2R3, -CONHR3, COOR3, OR2 и NR3R3, при условии, что если по меньшей мере одним заместителем является OR2 или NR3R3, то по меньшей мере один заместитель не присоединен к тому же атому углерода, к которому присоединен атом азота гетероциклической группы.
R2 представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил).
R3 в каждом случае независимо представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил или гетероарил(C1-C6)алкил.
В одном варианте осуществления настоящее изобретение включает соединение формулы (VI) или его соль:
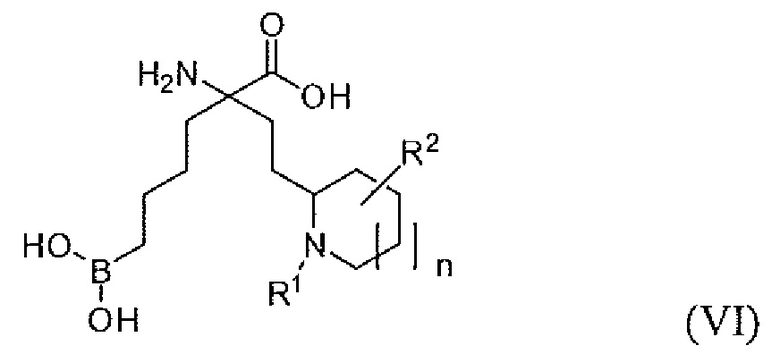
где:
n равно 0, 1 или 2; и
R1 представляет собой H, алкил или арилалкил; и
R2 представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил).
В одном варианте осуществления гетероциклическая группа конденсирована с другой циклической группой. Неограничивающие примеры такого варианта осуществления продемонстрированы ниже:
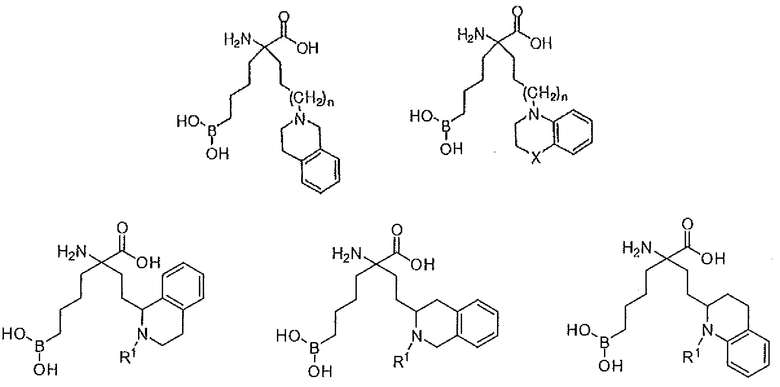
где:
n равно 0, 1 или 2;
R1 представляет собой H, алкил или арилалкил;
X представляет собой NR5, CR6R7, O, S, S(O) или S(O)2;
причем, если X представляет собой CR6R7, то R7 представляет собой H, OH, OR8, CN или NR8R9; и
R5, R6, R8 и R9 независимо представляют собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил).
Арильное кольцо может быть необязательно замещено одной или более из следующих групп: H, (C1-C6)алкил, арил, галоген, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил).
Заместители на гетероциклических группах могут включать одну или более из следующих групп: (C1-C6)алкил, галоген, арил и гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)R3, -SO2R3, -CONHR3, COOR3, OR2 и NR3R3, при условии, что если по меньшей мере одним заместителем является OR2 или NR3R3, то по меньшей мере один заместитель не присоединен к тому же атому углерода, к которому присоединен атом азота гетероциклической группы.
R2 представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил).
R3 в каждом случае независимо представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил или гетероарил(C1-C6)алкил.
В одном варианте осуществления проксимальный атом азота является частью анилиновой и пиридиновой группы. Неограничивающие примеры такого варианта осуществления продемонстрированы ниже:
где R1 и R2 независимо представляют собой H, C1-C6 алкил или арилалкил. В одном варианте осуществления соединением не является 2-амино-6-(бороно-2-(пиридин-3-илметил)капроновая кислота.
Арильное или гетероарильное кольцо может быть необязательно замещено одной или более из следующих групп: H, (C1-C6)алкил, арил, галоген, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил).
Заместители на гетероциклических группах могут включать одну или более из следующих групп: (C1-C6)алкил, галоген, арил и гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)R3, -SO2R3, -CONHR3, COOR3, OR2 и NR3R3, при условии, что если по меньшей мере одним заместителем является OR2 или NR3R3, то по меньшей мере один заместитель не присоединен к тому же атому углерода, к которому присоединен атом азота гетероциклической группы.
R2 представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил, гетероарил(C1-C6)алкил, -C(=O)(C1-C6)алкил, -C(=O)(арил), -C(=O)(гетероарил), -SO2(C1-C6)алкил, -SO2(арил), -SO2(гетероарил), -CONH(C1-C6)алкил, -CONH(арил) или -CONH(гетероарил).
R3 в каждом случае независимо представляет собой H, (C1-C6)алкил, арил, гетероарил, арил(C1-C6)алкил или гетероарил(C1-C6)алкил.
В одном варианте осуществления настоящее изобретение включает соединение формулы (VII) или его соль:
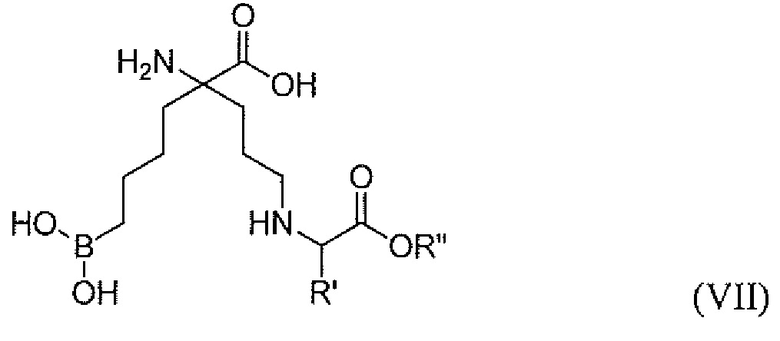
где:
R' представляет собой H, C1-C6 алкил, бензил, замещенный бензил, CH3SCH2CH2-, CH3S(=O)CH2CH2-, CH3S(О)2CH2CH2-, 3-индол-1H-ил-метил, HSCH2-, -CH2CH2C(=O)NH2 -CH2C(=O)NH2, CH2CH2C(=O)OH, -CH2C(=O)OH, -CH(OH)CH3, -CH2OH, -(CH2)4NH2, -(CH2)3NHC(=NH)NH2 или имидазол-4-ил-метил;
R” представляет собой H или C1-C6 алкил.
В одном варианте осуществления настоящее изобретение включает соединение формулы (VIII) или его соль:
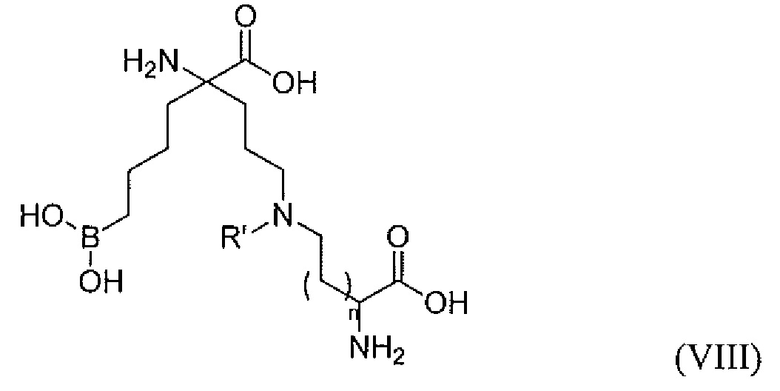
где:
n равно 0, 1, 2 или 3;
R' представляет собой Н или C1-C6 алкил.
Соединения настоящего изобретения содержат хиральные центры, обеспечивая различные стереоизомерные формы, такие как диастереомерные смеси, энантиомерные смеси, а также оптические изомеры. Отдельные оптические изомеры можно получить непосредственно посредством асимметрического или стереоспецифического синтеза или посредством обычного хирального разделения оптических изомеров из энантиомерной смеси.
Некоторые из соединений настоящего изобретения могут содержать хиральные центры сверх Cα, и такие соединения могут существовать в форме стереоизомеров (т.е. энантиомеров или диастереоизомеров). Настоящее изобретение включает все такие стереоизомеры и любые их смеси, включая рацемические смеси. Рацемические смеси стереоизомеров, а также по существу чистые стереоизомеры, находятся в пределах объема настоящего изобретения. Используемый в описании термин «по существу чистые» относится к тому, что по меньшей мере приблизительно 90 мол.%, более предпочтительно по меньшей мере приблизительно 95 мол.%, даже более предпочтительно по меньшей мере приблизительно 98 мол.% и даже более предпочтительно по меньшей мере приблизительно 98 мол.% желаемого стереоизомера присутствует относительно других возможных стереоизомеров. Предпочтительные энантиомеры можно выделить из рацемических смесей любым способом, известным квалифицированным в данной области техники специалистам, включая высокоэффективную жидкостную хроматографию (ВЭЖХ) и образование хиральных солей и их кристаллизацию, или получить способами, описанными в описании. См., например, Jacques et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen, S.H. et al., 1977, Tetrahedron, 33:2725; Eliel, E.L., Stereochemistry of Carbon Compounds, (McGraw-Hill, NY, 1962); Wilen, S.H, Tables of Resolving Agents and Optical Resolutions, p. 268 (E.L. Eliel, Ed., University of Notre Dame Press, Notre Dame, IN 1972).
В некоторых предпочтительных вариантах осуществления соединения представляют собой L-стереоизомерные формы, аналогично соединениям, продемонстрированным ниже:
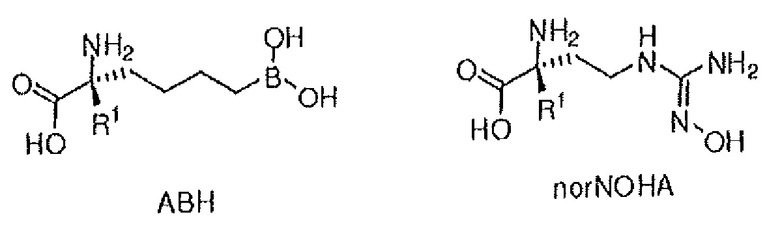
В неограничивающем аспекте в результате структурных и функциональных исследований, описанных в описании, предположили, что «L»-стереохимия каждой аминокислоты (определенной выше) способствует прочному связыванию в активном центре фермента; «D»-стереоизомеры, по-видимому, не связываются настолько же прочно или, по-видимому, являются менее эффективными. Альтернативно констатировали, что предпочтительная стереохимия аналогична стереоспецифической замене (R)-водорода в ABH на R1. В зависимости от относительного приоритета двух заместителей при альфа-углероде соединение может иметь или R-, или S-стереохимию. Обычно ингибиторные активности R- и S-стереоизомеров могут отличаться, и поэтому предпочтительной стереохимией является форма, которая более эффективно позволяет молекуле функционировать в качестве ингибитора аргиназы. В связи с этим, в одном неограничивающем аспекте в результате структурных и функциональных исследований предположили, что определенная выше стереохимия αC является предпочтительной для прочного связывания в активном центре фермента; тогда как стереоизомеры с противоположной конфигурацией не связываются настолько же прочно или являются менее эффективными.
Альтернативно, стереоизомеры можно определить в случаях, когда ProS водород глицина, представленного ниже, заменен на X1, который вписывается в активный центр фермента. Второй заместитель, R1, замещает ProR водород глицина. В соответствии с правилами Кана-Ингольда-Прелога обозначение R или S для стереоизомеров зависит от иерархии, основанной на атомах, соединенных с хиральным углеродом. В случае предпочтительных соединений настоящего изобретения, в которых ProS H заменен на -(CH2)4B, а ProR H заменен функциональными группами -(CH2)2-4N-, стереохимией в αC является R. В случае предпочтительных соединений настоящего изобретения, в которых ProS H заменен функциональной группой norNOHA, а ProR H заменен функциональной группой -(CH2)2N-, стереохимией в αC является R. Однако, если ProR H заменен функциональными группами -(CH2)3-4N-, стереохимией в αC является S.
Настоящее изобретение включает пролекарства аминокислоты настоящего изобретения или пролекарства одного из связывающих фрагментов (таких как бороновая кислота или N-гидроксигуанидин, например). «Пролекарство», как используется в описании, означает соединение, которое превращается in vivo метаболическим образом (например, в результате гидролиза) в соединение настоящего изобретения. Различные формы пролекарств известны в данной области техники, например, как обсуждается в Bundgaard, (ed.), Design of Prodrugs, Elsevier (1985); Widder et al. (ed.), Methods in Enzymology, vol. 4, Academic Press (1985); Krogsgaard-Larsen et al. (ed). "Design and Application of Prodrugs," Textbook of Drug Design and Development, Chapter 5, 113-191 (1991), Bundgaard et al., 1992, J. Drug Deliv. Rev. 8:1-38, Bundgaard, 1988, J. Pharm. Sci. 77:285 и далее; и Higuchi and Stella (eds.), Prodrugs as Novel Drug Delivery Systems, American Chemical Society (1975). В неограничивающем варианте осуществления в качестве пролекарств получают сложные эфиры альфа-карбоновой кислоты для увеличения пероральной биодоступности, в соответствии с чем сложный эфир является стабильным в желудке и желудочно-кишечном тракте, оптимально переносится через выстилку желудочно-кишечного тракта в кровоток, а затем превращается под действием повсеместных эстераз в крови и/или в печени во фрагмент в виде карбоновой кислоты. Аналогичный принцип, основанный на образовании сложного эфира, можно было бы воплотить в отношении фрагмента в виде бороновой кислоты. Более того, можно использовать принцип двойного сложного эфира для пролекарства в отношении обоих фрагментов в виде карбоновой кислоты и бороновой кислоты.
Неограничивающие примеры сложноэфирных пролекарств, предусмотренных в настоящем изобретении, представлены ниже:
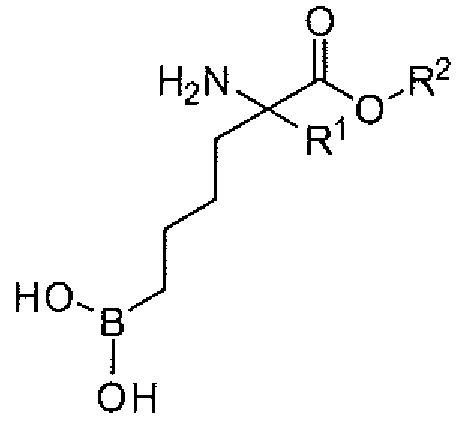
где:
R1 можно определить как любое соединение, описанное в описании в любом другом месте;
R2 выбирают из группы, состоящей из C1-C6 алкила, C3-C7 циклоалкила, тетрагидрофуран-2-ил-метила, тетрагидрофуран-3-ил-метила, C3-C7 циклоалкил-метила, 2-(C3-C7 циклоалкил)-этила, дигидрофуран-2(3H)-он-4-ил-метила, 2-гидроксил-этила, 2-гидроксил-2-метил-этила, фенила, 2-толила, 3-толила, 4-толила, 2-метокси-фенила, 3-метокси-фенила, 4-метокси-фенила, 2,3-дигидро-1H-инден-4-ила, 2,3-дигидро-1H-инден-5-ила, тиазол-2-ил-метила, тиазол-4-ил-метила, имидазол-2-ил-(CH2)n-, имидазол-4-ил-(CH2)n-, 2-метил-1H-бензо[d]имидазол-2-ил-(CH2)n-, R5C(=O)OCH2CH2-, R5C(=O)OCH(CH3)CH2-, R5C(=O)OCH2- или R5C(=O)OCH(CH3)-;
n равно 1, 2, 3 или 4;
R5 представляет собой H, C1-C6 алкил, C3-C7 циклоалкил, арил, гетероарил или CH(R6)NH2; и
R6 представляет собой Н, C1-C6 алкил, бензил, замещенный бензил, CH3SCH2CH2-, CH3S(=O)CH2CH2-, CH3S(О)2CH2CH2-, 3-индол-1H-ил-метил, HSCH2-, -CH2CH2C(=O)NH2, -CH2C(=O)NH2, CH2CH2C(=O)OH, -CH2C(=O)OH, -CH(OH)CH3, -CH2OH, -(CH2)4NH2, -(CH2)3NHC(=NH)NH2 или имидазол-4-ил-метил.
Бензоимидазол может быть необязательно замещен по меньшей мере одним заместителем, выбираемым из группы, состоящей из (C1-C6)алкила, галогена и (C1-C6)алкокси.
Кроме того, аминокислота настоящего изобретения может существовать в несольватированной, а также сольватированной форме, с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п. Как правило, сольватированные формы считаются эквивалентными несольватированным формам для цели настоящего изобретения.
Синтез соединений настоящего изобретения
Соединения настоящего изобретения могут быть получены рядом способов, хорошо известных квалифицированным в данной области техники специалистам. Соединения можно синтезировать, например, с использованием способов, описанных ниже, или вариаций на их основе, как это понятно квалифицированному специалисту. Используемые при получении соединений настоящего изобретения реагенты могут быть или получены коммерческим путем, или могут быть получены с помощью стандартных процедур, описанных в литературе. Все процессы, описанные в связи с настоящим изобретением, как предусматривается, осуществляются на практике в любом масштабе, включая миллиграммовый, граммовый, многограммовый, килограммовый, многокилограммовый или коммерческий промышленный масштаб. Квалифицированному в данной области техники специалисту будет понятно, что синтез таких вторых замещающих групп в α-положении аминокислоты является трудным процессом синтеза, судя по отсутствию коммерчески доступных α,α-двузамещенных аминокислот. См., например, Vogt et al., 2007, Org, Biomol, Chem. 5:406-30.
На фиг.1-14 проиллюстрированы некоторые общие механизмы для получения соединений настоящего изобретения. На фиг.1 схематически проиллюстрирован жидкофазный синтез применимых исходных материалов 4a (R=метил), 4b (R=этил) и 4c (R=трет-бутил). Как проиллюстрировано на фиг.1, осуществляют реакцию гидрохлоридной соли алкилового эфира глицина 2a-c с бензофенонимином (O'Donnell, et al., 1982, J. Org. Chem. 47:2663) при комнатной температуре с получением соответствующего кетимин-защищенного эфира глицина 4a-c. Это трансиминирование особенно применимо для трет-бутилового эфира 4c. Альтернативно, можно осуществить реакцию алкиловых эфиров α-бромуксусной кислоты 3a-c с бензофеноном в кипящем ацетонитриле, как показано во второй реакции, приводящей к 4a-c (O'Donnell, 2004, Acc. Chem. Res. 37:506).
На фиг.2 проиллюстрирован синтез дополнительных выбранных исходных материалов. Как проиллюстрировано на фиг.2, соединение 7 получают, осуществляя реакцию замещенных или незамещенных пиперазинов (6) с 3-бром-1-пропанолом (5) в присутствии основания, такого как карбонат калия, при повышенной температуре в течение нескольких часов. Когда R представляет собой алкил или арилалкил, этот исходный продукт можно получить, осуществляя реакцию 8 с альдегидом 9 в условиях восстановительного аминирования с получением 10. При выделении продукта 7 или 10, их можно превратить в алкилйодид (11), осуществляя реакцию с йодом в присутствии имидазола и трифенилфосфина (Garegg & Samuelsson, 1979, J. Chem. Soc. Chem. Commun. 978). Альтернативно, связанный со смолой трифенилфосфин можно также использовать для образования соединения 11, которое можно использовать на стадиях алкилирования в виде R'-X в соответствии со способами, описанными в описании (Classon et, al., 1988, J. Org, Chem. 53:6126).
На фиг.3 проиллюстрирован приводимый в качестве примера жидкофазный синтез соединения 1 настоящего изобретения, в котором боковая цепь R1 содержит по-разному замещенные пиперазины. Осуществляют реакцию соединения 4c с LiHMDS при низкой температуре, как описано в публикации Reddy et al., 2007, Org. Biomol. Chem. 5:889, а затем добавляют один эквивалент 11 и обеспечивают возможность протекания реакции при комнатной температуре в течение нескольких часов с получением соединения 12. Вторую боковую цепь можно ввести путем обработки промежуточного соединения 12 сильным основанием, таким как n-BuLi, LDA, или предпочтительным основанием KHMDS, в безводных условиях при низкой температуре, а затем добавления одного эквивалента кротилбромида с получением соединения 13. Гидроборирование кротильной боковой цепи для получения боковой цепи в виде боронатного сложного эфира осуществляют путем обработки пинаколбораном в присутствии иридиевого катализатора (подобно процедуре, описанной в публикации Yamamoto et al., 2004, Tetrahedron 60:10695) с получением соединения 14.
Когда R=Boc в соединении 14, дополнительные фрагменты можно ввести с использованием схемы синтеза, представленной на фиг.4. Соединение 14 обрабатывают 25% ТФУК/DCM в безводных условиях в атмосфере аргона в течение от 30 мин до одного часа, что приводит к соединению 15. Можно осуществить реакцию этого промежуточного продукта в безводном ДМФА с избыточным количеством карбоната калия и алкил- или арилалкилгалогенидом с получением соединения 16. Кроме того, 15 можно подвергнуть ацилированию либо с использованием хлорангидрида, получая соединение 17, либо с использованием изоцианата, получая мочевину 18.
С соединений 14, 15, 16, 17 и 18 можно снять все защитные группы с получением конечных продуктов 1, как представлено на фиг.5. В зависимости от стабильности группы R в соединении 14-18, это соединение можно обработать 4 н. HCl при 70°C в течение нескольких часов или с использованием двустадийной процедуры, в которой трет-бутиловый сложный эфир удаляют, сначала используя 50-100% ТФУК в DCM, в течение 1-2 ч, а затем осуществляя реакцию этого промежуточного продукта с избыточным количеством фенилбороновой кислоты в 1 н. водном растворе HCl и диэтиловом эфире в течение нескольких часов.
Соединения данного изобретения 1 можно также синтезировать, как представлено на фиг.6 и 7. Кетаминную защитную группу в соединении 14 можно отщепить путем обработки 1 н. HCl в ТГФ при 0°C или комнатной температуре в течение от 15 мин до 1 ч, а затем этот α-амин вновь защитить, проводя реакцию с бензилхлорформиатом, с получением соединения 19. Boc-группу в этом промежуточном продукте можно удалить путем обработки 25% ТФУК/DCM в течение 30 мин с получением соединения 20, которое можно подвергнуть ацилированию, используя сульфонилхлориды для получения сульфонамидов 21, хлорангидриды для получения амидов 22 или изоцианаты для получения мочевины 23.
Представленный на фиг.7 синтез начинается с соединения 4c, которое обрабатывают LiHMDS при низкой температуре, как описано в публикации Reddy et al, 2007, Org. Biomol. Chem. 5:889, а затем добавляют 1 эквивалент 3-бромпропокси-трет-бутилдиметилсилана и обеспечивают возможность протекания реакции при комнатной температуре в течение нескольких часов с получением соединения 24. Вторую боковую цепь можно ввести путем обработки промежуточного соединения 24 сильным основанием, таким как n-BuLi, LDA, или предпочтительным основанием KHMDS, в безводных условиях при низкой температуре, а затем добавления 1 эквивалента кротилбромида с получением соединения 25. Кетаминную защитную группу в 25 удаляют избирательно путем обработки 1 н. гидрохлоридом гидроксиламина в ТГФ (1:2) в течение нескольких часов, а полученный α-амин 26 защищают путем обработки бензилхлорформиатом в щелочных условиях с получением 27. Гидроборирование кротильной боковой цепи для получения боковой цепи в виде боронатного сложного эфира осуществляют путем обработки пинаколбораном в присутствии иридиевого катализатора, подобно процедуре, описанной в публикации Yamamoto et al., 2004, Tetrahedron 60:10695, с получением соединения 28. Удаления защитной группы TBS и функционализации гидроксильной группы достигают путем обработки 28 слабым водным раствором кислоты в органическом растворе или TBAF с получением промежуточного соединения 29, с последующим осуществлением реакции с йодом в присутствии трифенилфосфина и имидазола с получением 30. Можно осуществить реакцию различных нуклеофильных аминов с соединением 30 в щелочных условиях в ДМФА с получением 31. Эти нуклеофильные амины могут включать реагенты, приводящие к образованию соединений 20-23, а также множество других реагентов, приводящих к образованию соединения 1 настоящего изобретения.
Соединения 1 настоящего изобретения, а также дополнительные примеры, можно также синтезировать в соответствии с фиг.8 и 9. Эти схемы синтеза создают возможность для улучшенных методик снятия защитных групп, как описано в описании в любом другом месте. α-Амин промежуточного продукта 26 можно защитить с помощью Boc-группы, как представлено на фиг.8, путем его обработки Boc ангидридом в присутствии основания, такого как DIEA в ТГФ, получая промежуточный продукт 32. Гидроборирование кротильной боковой цепи осуществляют путем обработки пинаколбораном в присутствии иридиевого катализатора (подобно процедуре, описанной в публикации Yamamoto et al., 2004, Tetrahedron 60:10695) с получением соединения боронатного сложного эфира 33. Удаления защитной группы TBS и функционализации гидроксильной группы достигают путем обработки 33 слабым водным раствором кислоты в органическом растворе или TBAF, с последующим осуществлением реакции с йодом в присутствии трифенилфосфина и имидазола с получением 34. Можно осуществить реакцию различных нуклеофильных аминов с соединением 34 в щелочных условиях в ДМФА с получением 31. Эти нуклеофильные амины могут включать реагенты, приводящие к образованию соединения 1 настоящего изобретения. Промежуточный продукт 32 можно также получить исходя из промежуточного продукта 25, используя две различные стратегии (фиг.9), в случае которых кетимин сначала удаляют с помощью гидролиза в слабой кислоте, который сберегает TBS эфир, и этот продукт затем обрабатывают Boc ангидридом, как описано выше. Альтернативно, промежуточный продукт 25 можно обработать кислотой для удаления и кетимина, и TBS эфира. Этот продукт вновь защищают с использованием двустадийного процесса, который включает обработку Boc ангидридом с последующей защитой гидроксильной группы с помощью TBS (используя TBSCl в DCM и DMAP в качестве основания).
На фиг.10 представлены схемы снятия всех защитных групп с соединений 20-23, 31 и 35, приводящие к получению соединения 1 настоящего изобретения. Соединения 20-23, 31 подвергают гидрогенолизу в присутствии палладиевого катализатора на угольном носителе в кислотных условиях с использованием водорода при атмосферном давлении в течение нескольких часов для удаления защитной группы Cbz. Этот промежуточный продукт затем обрабатывают 75-100% ТФУК в DCM в течение 1-2 ч с последующим осуществлением реакции с избыточным количеством фенилбороновой кислоты в смеси 1 н. водного раствора HCl и диэтилового эфира, получая соединение 1. С соединения 35 снимают все защитные группы с использованием двустадийного процесса, который включает удаление Boc и трет-бутилового эфира путем его обработки 50-100% ТФУК в течение нескольких часов, с последующим осуществлением реакции продукта с избыточным количеством фенилбороновой кислоты в смеси 1 н. водного раствора HCl и диэтилового эфира, получая соединение 1. Альтернативно, соединение 35 можно обработать 4 н. водным раствором HCl при 70-90°C в течение нескольких часов, что приводит к соединению 1. Выбор схем снятия защитных групп, проиллюстрированных на фиг.10, зависит от стабильности фрагментов NR'R” к соответствующим условиям реакций.
Как будет легко понять, присутствующие функциональные группы могут содержать защитные группы в ходе синтеза. Защитные группы известны по сути как химические функциональные группы, которые можно избирательно присоединить к функциональным группам, таким как гидроксильные группы и карбоксильные группы, или снять с них. Эти группы присутствуют в химическом соединении для придания такой функциональной группе инертности в отношении условий химических реакций, которым подвергают соединение. Любая из множества защитных групп может быть использована в настоящем изобретении. Защитные группы, которые могут использоваться в соответствии с настоящим изобретением, могут быть описаны в публикации Greene, T.W. and Wuts, P.G.M., Protective Groups in Organic Synthesis 2d. Ed., Wiley & Sons, 1991.
Соединения этого изобретения содержат хиральные центры, обеспечивая различные стереоизомерные формы, такие как диастереомерные смеси, энантиомерные смеси, а также оптические изомеры. Отдельные оптические изомеры могут быть получены непосредственно посредством асимметрического или стереоспецифического синтеза или посредством обычного хирального разделения оптических изомеров из энантиомерной смеси.
На фиг.11 продемонстрированы некоторые промежуточные продукты из фиг.7-9, которые могут быть использованы в качестве энантиомерных промежуточных продуктов для получения хирального соединения 1. Квалифицированным в данной области техники специалистам хорошо известно, что химические процессы, представленные на фиг.7-10, могут быть использованы в случае либо рацемической смеси, либо отдельного энантиомера с получением схожих результатов. Таким образом, процедуры с 36a или 37a будут приводить к одному энантиомеру желаемого соединения 1. Кроме того, 36b или 37b можно было бы подвергнуть модификации схожим образом, что приводит к получению соединения 1 с противоположной конфигурацией. В неограничивающем варианте осуществления, на основе хиральности активного энантиомера ABH и того, каким образом эти молекулы связываются с аргиназой, предполагают, что продукт 1, образованный из энантиомеров 36a или 37a, будет активным в качестве ингибитора аргиназы. Альтернативно, хиральные соединения 1 можно получить посредством специфического асимметрического синтеза или методов хирального разделения.
На фиг.12 продемонстрированы методики получения хиральных промежуточных продуктов на фиг.11. Например, как продемонстрировано на схеме A на фиг.12, способы ВЭЖХ или SFC с использованием аналитических и препаративных колонок, наполненных известными хиральными набивочными материалами, такими как ChiralPak AD-H, могли бы использоваться для разделения рацемической смеси 29, приводящего к получению хирального 36a и его энантиомера 36b. Как продемонстрировано на схеме B на фиг.12, аминоэфир 26 в виде рацемической смеси можно обработать энантиомерной солью, такой как (+)-камфорсульфокислота, с получением диастереомерных солей. Эти соли можно подвергнуть кристаллизации по отдельности, и после того как аминоэфир освободится от кристаллической соли, получают чистый энантиомер (37a).
Дополнительные хиральные кислоты можно было бы использовать для получения кристаллических диастереомерных солей. Эти хиральные кислоты будут включать (+) и (-) винную кислоту, (+) и (-) дибензоилвинную кислоту, (+) и (-) яблочную кислоту, (+) и (-) миндальную кислоту, (+) и (-) камфорсульфокислоту, (+) и (-) N-Boc-фенилаланин и (+) и (-) N-Boc-валин, среди прочих. В одном варианте осуществления оптимальная диастереомерная соль характеризуется контролируемой кинетикой кристаллизации и может быть выделена с хиральной чистотой воспроизводимо высокой степени.
На схеме C на фиг.12 представлен способ синтеза, описанный в литературе, для получения α,α-двузамещенных аминокислот с известной хиральностью (Ooi et al., 2000, J. Am. Chem. Soc, 122:5338, и Jiang et. al., 2008, Org. Proc. Res. Dev. 12:1164). Соединение 38 обрабатывают в условиях фазового перехода последовательно 3-бромпропокси-TBS эфиром, а затем кротилбромидом в присутствии хирального катализатора (40) с получением 39. Защитную группу для α-амина можно затем подвергнуть гидролизу, приводящему к желаемому хиральному промежуточному продукту 37.
С целью увеличения пероральной биодоступности соединения 1 этого изобретения можно было бы рассматривать различные стратегии получения пролекарств. В одном варианте осуществления α-карбоновую кислоту 1 превращают в сложный эфир. Этот сложный эфир является стабильным в желудочно-кишечном тракте, оптимально переносится через выстилку желудочно-кишечного тракта в кровоток, а затем превращается во фрагмент в виде карбоновой кислоты под действием повсеместных эстераз в крови или других тканях. На фиг.13 и 14 продемонстрированы некоторые способы превращения защищенных промежуточных продуктов 1 в различные сложные эфиры и затем снятия защитных групп с α-амина и бороновой кислоты с получением сложных эфиров 42.
Соединение 41 можно получить из соединения 35 (которое продемонстрировано на фиг.8 и 10) путем обработки 50-100% ТФУК/DCM при комнатной температуре в течение 1-6 ч. Как продемонстрировано на схеме A на фиг.13, соединение 41 можно впоследствии обработать спиртовым раствором HCl (используя спирт, такой как, но без ограничения, метанол) при повышенной температуре в течение нескольких часов, что приводит к образованию соединения 42. Схожие результаты можно получить путем обработки 41 тионилхлоридом в спирте при кипячении с обратным холодильником в течение 12-16 ч.
Соединения 43, 44, 45 и 46 можно получить на основе соединения 41 путем повторной защиты амина с помощью такого реагента, как бензилхлорформиат или 9-флуоренилхлорформиат. Альтернативно, эти защитные группы могут быть введены на более ранней стадии синтеза, как продемонстрировано на фиг.7 для стратегии защиты Cbz. Как продемонстрировано на схеме B на фиг.13, соединения 43-46 можно преобразовать в сложные эфиры путем обработки галогенированным реагентом, таким как этилйодид, в умеренно щелочных условиях в ДМФА или ацетонитриле с получением соответствующего продукта 47-50. Альтернативно, как продемонстрировано на схеме C на фиг.13, соединения 43-46 можно преобразовать в сложные эфиры путем обработки спиртом в присутствии DCC с получением соответствующего продукта 47-50. Альтернативно, как продемонстрировано на схеме D на фиг.13, соединения 43-46 можно преобразовать в сложные эфиры путем обработки оксалилхлоридом и последующего спиртового гидролиза с получением соответствующего продукта 47-50.
Как продемонстрировано на схеме A на фиг.14, соединение 42 можно получить путем снятия защитных групп с соединений 47, 48 и 50, представленных на фиг.10. Альтернативно, как продемонстрировано на схеме B на фиг.14, соединение 42 можно получить путем снятия защитных групп с соединения 49 двустадийным способом, сначала используя пиперазин или пиперидин в ДМФА для снятия Fmoc-группы, а затем проводя переэтерификацию продукта с использованием фенилбороновой кислоты для удаления пинакола из бороновой кислоты. Соединение 42 можно очистить методом ВЭЖХ с обращенной фазой, используя градиенты ацетонитрил/вода.
Применения соединений настоящего изобретения
В неограничивающем аспекте соединения настоящего изобретения обладают достаточной ингибиторной в отношении аргиназы активностью при значениях pH, которые меньше 9,5. В отличие от них, прототипичный ингибитор аргиназы типа бороновой кислоты, ABH, демонстрирует уменьшение ингибиторной активности в отношении обеих аргиназ при pH 7,5 по сравнению с pH 9,5. По-видимому, это является общим свойством ингибиторов типа бороновой кислоты, которые не содержат проксимальный азот, находящийся на соответствующем расстоянии в боковой цепи-заместителе при α-углероде аминокислоты.
Для иллюстрации важности проксимального азота соединения, которые не содержат проксимальный азот, такие как сравнительный пример 1 (продемонстрированный в таблице 1), сравнивали с соединениями, которые содержат проксимальный азот, такими как сравнительный пример 4. Как продемонстрировано в таблице 2, сравнительный пример 1 обладает более низкими значениями активности при pH 7,5, чем при pH 9,5, в отношении обоих типов аргиназ, в то время как сравнительный пример 4 демонстрирует увеличенную активность в отношении аргиназы I человека. Следует принять во внимание, что сравнительный пример 4 отличается от сравнительного примера 1 только заменой проксимального азота атомом азота. Аналогично, сравнение примера 8 и сравнительного примера 2 (таблица 1) и примера 11 и сравнительного примера 3 (таблица 1), которые также отличаются только присутствием проксимального азота, выявляет схожий эффект pH. В частности, пиперазины (примеры 8 и 11), содержащие проксимальный азот, демонстрируют увеличенные активности в отношении по меньшей мере одной из аргиназ при pH 7,5 по сравнению с pH 9,5, в то время как пиперидины (сравнительные примеры 2 и 3) демонстрируют уменьшенные значения активности при pH 7,5 по сравнению с pH 9,5. Примеры 17 и 28 являются аналогами, которые содержат проксимальный азот, наряду с тем, что дистальный азот пиперазина был заменен атомом углерода. Таким образом, присутствие проксимального азота на предпочтительном расстоянии в боковой цепи-заместителе при α-углероде аминокислоты связано с этим «эффектом pH».
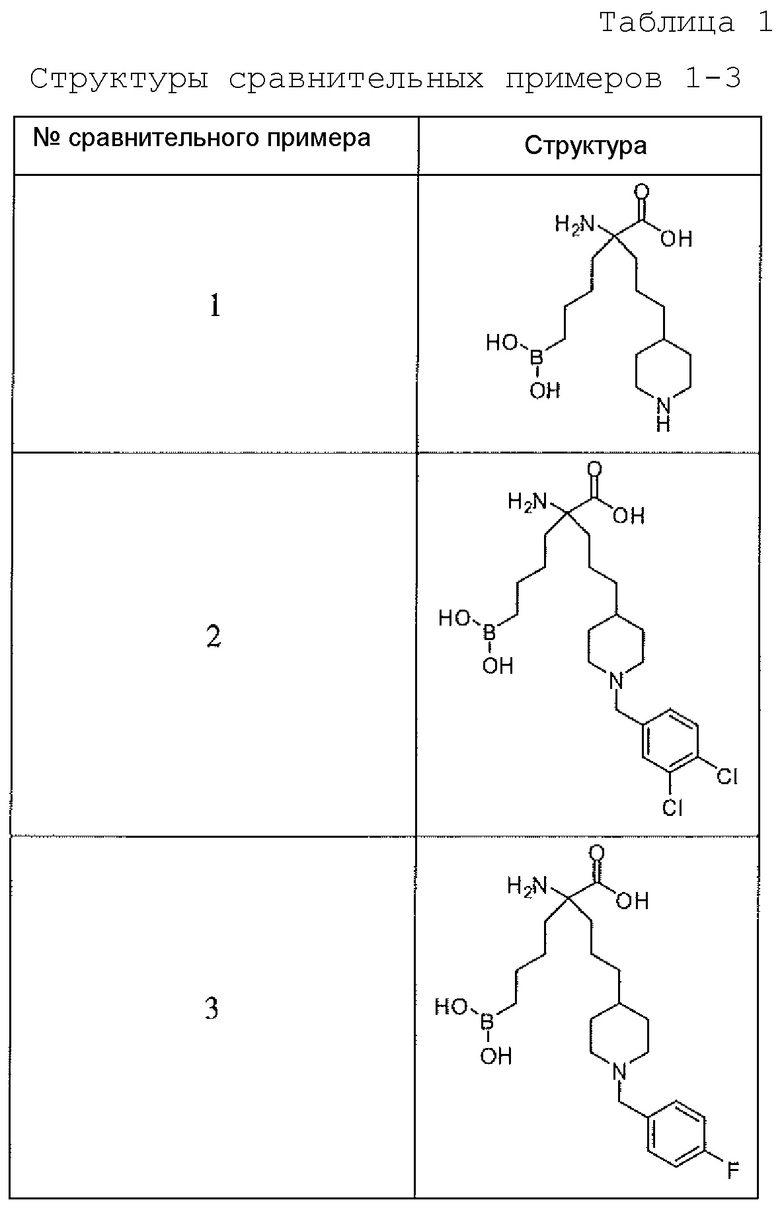
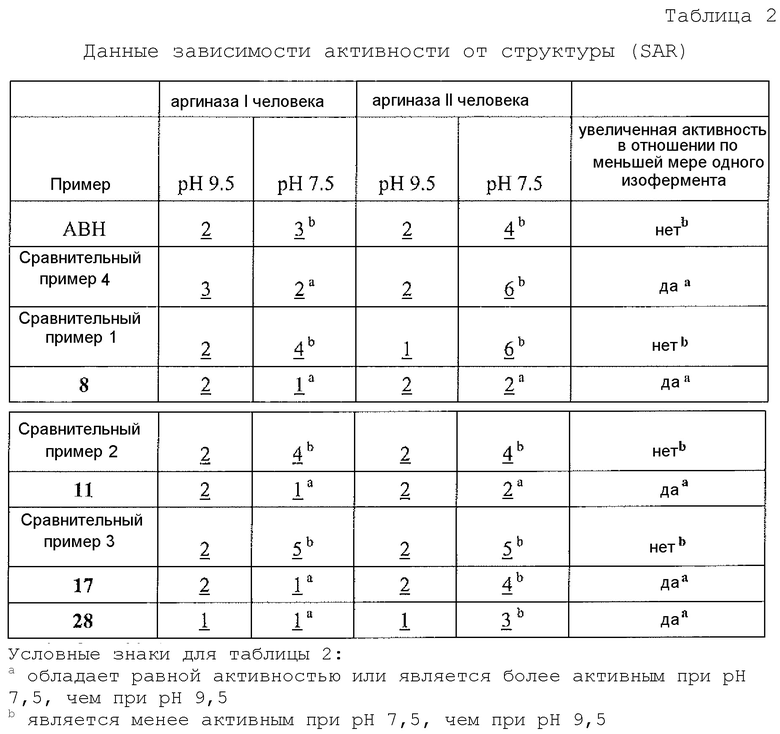
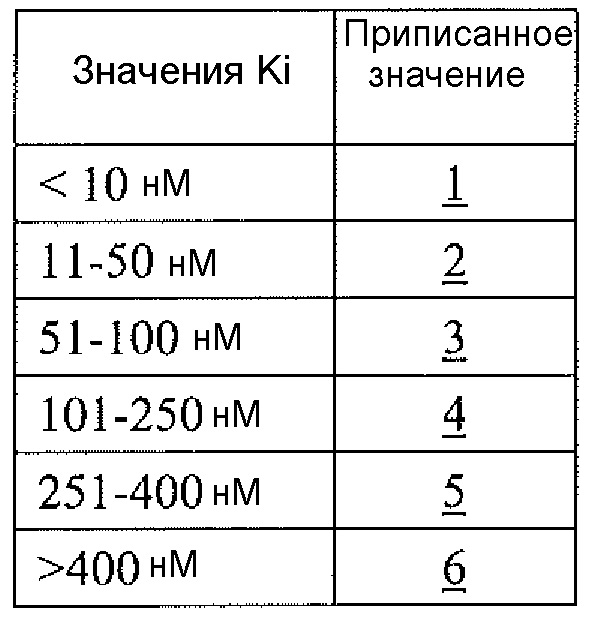
В таблице 3 продемонстрированы пиперазин-содержащие производные, которые сравнивают с прототипичным ингибитором аргиназы типа бороновой кислоты ABH. Представленные в ней данные иллюстрируют «эффект pH» в зависимости от длины боковой цепи. Например, пример 2 имеет линкер в виде 2-углеродной боковой цепи, пример 1 (таблица 2) имеет линкер в виде 3-углеродной боковой цепи, а пример 93 имеет линкер в виде 4-углеродной боковой цепи. Бензилпиперазин с линкером в виде 2-углеродной боковой цепи, пример 15, можно также сравнить с бензилпиперазином с линкером в виде 3-углеродной боковой цепи, пример 8 (таблица 2). Несмотря на то, что соединения демонстрируют «эффект pH», очевидно, что линкер в виде 3-углеродной боковой цепи является оптимальным.
В примерах 20 и 21 проксимальный азот заключен в пиперидиновое кольцо. Было установлено, что каждое из этих соединений является более активным против аргиназы I по сравнению с аргиназой II даже при pH 7,5, что означает, что оба примера демонстрируют некоторую селективность в отношении аргиназы I по сравнению с аргиназой II.
Примеры 7, 9 и 10, в случае всех из которых демонстрируется «эффект pH», имеют дифференциально замещенные бензильные группы при дистальном азоте. Каждый из примеров 16 и 41, в случае которых также демонстрируется «эффект pH», имеет ароматический заместитель при дистальном азоте. Кроме того, пример 14, в случае которого демонстрируется «эффект pH», имеет фенетильную группу при дистальном азоте, что говорит о том, что ароматические группы могут быть размещены на различных расстояниях от дистального азота без подавления «эффекта pH».
В таблице 3 также перечислены являющиеся пиперазиновыми производными соединения, в которых дистальный азот приведен в нейтральное состояние с помощью ацилирования или сульфонирования. Пример 129 содержит безамидогруппу, пример 142 является аналогом мочевины, а пример 151 содержит сульфамидогруппу. Каждое соединение продемонстрировало «эффект в зависимости от рН» в отношении аргиназы I человека, с селективностью в отношении аргиназы I по сравнению с аргиназой II при более низких значениях pH. Дальнейший анализ также показал, что не требуется, чтобы ацилированный дистальный азот находился в гетероциклическом кольце для обеспечения «эффекта pH». Примеры 31, 60, 112, 114 содержат ацилированный азот вне гетероциклического кольца, и эти соединения обладают «эффектом в зависимости от рН» с высокой активностью ингибирования одной или обеих из аргиназ человека - следует отметить, что проксимальный азот все еще является основным.
Как обсуждалось выше, пример 28 (таблица 2) говорит о том, что дистальный азот может быть заменен атомом углерода. Дополнительные аналоги (примеры 26, 27, 31, 32, 34, 36, 37, 39, 40, 44, 46, 101, 102, 103 и 106 в таблице 3), в которых проксимальный азот содержится в пиперидиновом кольце, говорят о том, что это пиперидиновое кольцо может быть замещено в различных положениях различными фрагментами, и при этом все еще имеется «эффект pH» в случае одной из двух аргиназ.
Пиперидиновое кольцо может быть замещено незамещенным или замещенным пирролидином или азетидином, и при этом все еще имеется «эффект pH», пока основный азот находится в проксимальном положении, как установлено по активности для примеров 50, 51, 54, 56, 57, 58, 60, 112 и 114.
Основный азот может содержаться в гетероциклических кольцах, таких как тетрагидроизохинолин (пример 45), морфолин (пример 64) и бензимидазол (пример 116), в которых проксимальный азот, согласно ожиданиям, является менее основным, чем когда он содержится в пиперазиновом или пиперидиновом кольце.
Кроме того, не требуется, чтобы проксимальный основный азот находился в гетероциклическом кольце, чтобы имелся этот «эффект pH». Примеры 67, 68, 77, 78, 85, 86, 88, 90, 117, 120 соответствуют различным замещенным вторичным или третичным аминам или с алкильными, или с арилалкильными фрагментами.
Этот проксимальный основный азот может также быть частью α-аминокислоты или α-аминоэфира, как продемонстрировано в примерах 98, 125 и 126. Активность этих примеров является уменьшенной при обоих значениях pH, но при их использовании все еще имеется «эффект pH» в случае по меньшей мере одной из аргиназ.
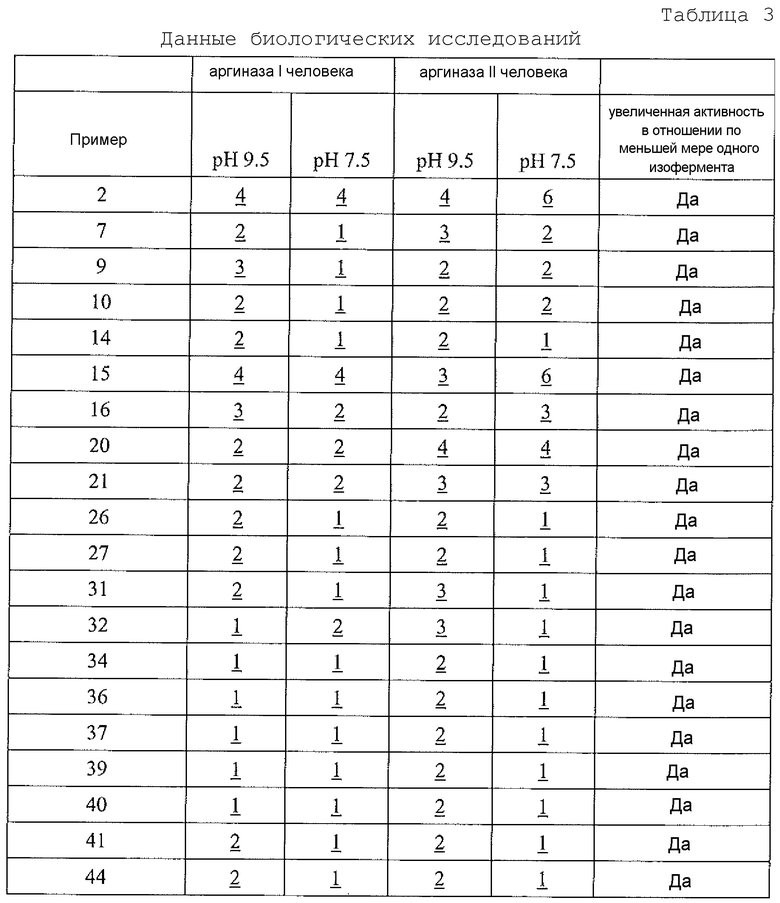
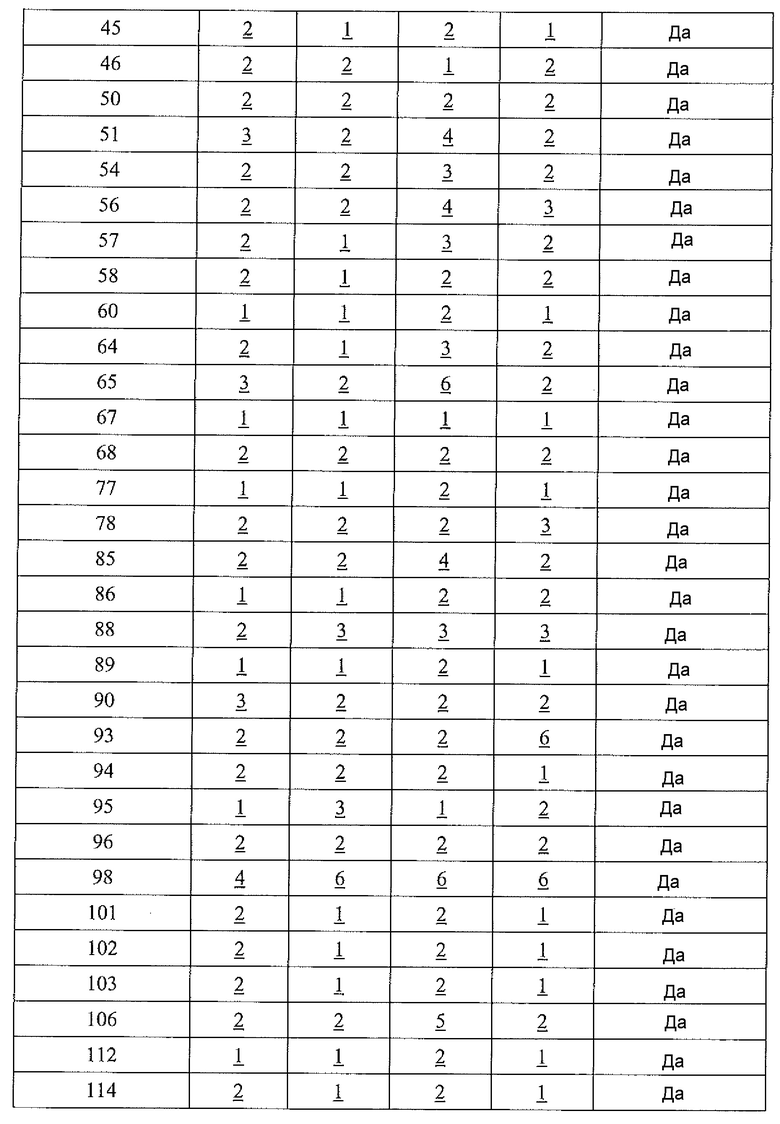
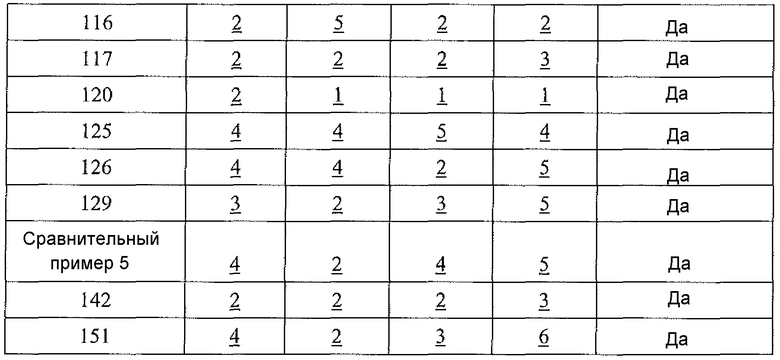
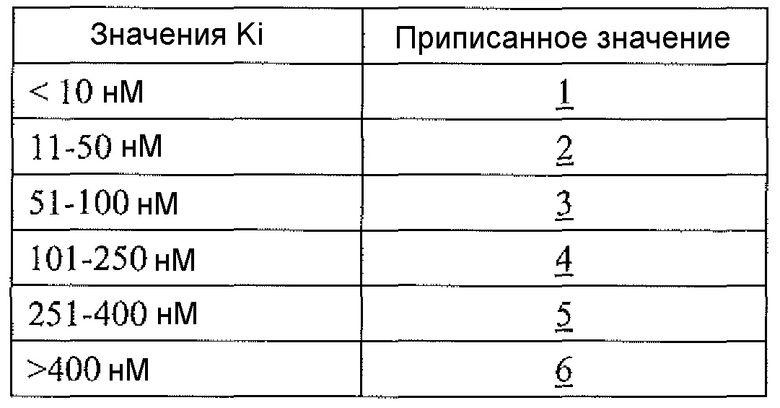
Без желания ограничиваться этой теорией, разработанное для этого класса ингибиторов аргиназы SAR говорит о том, что создается по меньшей мере одно дополнительное взаимодействие между боковой цепью R1 и ферментом. Были выяснены рентгеновские кристаллические структуры комплексов, образованных примером 8 и аргиназой I человека и аргиназой II человека. В этих структурах было подмечено, что часть «ABH» примера 8 связывается аналогичным образом, как описано для самой ABH. А именно, фрагменты в виде α-амина и α-карбоновой кислоты вставляются в соответствующие им карманы в двух ферментах в четком порядке образования водородных связей и ионных пар, отмеченном для этих фрагментов в ABH. Кроме того, бороновая кислота примера 8 была связана с каталитической молекулой воды аналогичным образом, который наблюдали для ABH. Было отмечено, что проксимальный основный азот, который был отделен тремя атомами углерода от α-углерода примера 8, образует ионную пару с боковыми цепями аспарагиновой кислоты вблизи активного центра. Что особенно важно, боковые цепи Asp181 и 183 в аргиназе I человека тесно контактировали с этим основным азотом. В соответствии с гипотезой авторов настоящего изобретения этот основный азот будет по большей части ионизированным с образованием положительно заряженных разновидностей при pH 7,5, тогда как при рН 9,5 этот атом будет по большей части неионизированным; поэтому образование ионной пары с отрицательно заряженными карбоновыми кислотами этих аспартаз будет минимизированным при последнем из двух названных значений pH. Некоторые исследованные лиганды, такие как примеры 98 и 116, как полагают, имеют меньшие pKa для проксимального N и не являются настолько активными, как примеры с проксимальными азотами с более высокими значениями pKa.
Как описано выше, примеры 2 и 93 (с этильным и бутильным спейсерами для проксимального азота) вызывают уменьшенное ингибирование по сравнению с примером 1. Взаимодействие в виде образования ионной пары, отмеченное в примере 8, в соответствии с ожиданием, является менее сильным в этих примерах, поскольку проксимальный основный азот размещен неоптимально.
В неограничивающем аспекте нечувствительность к pH, продемонстрированная этими аминокислотами, делает их особенно полезными в качестве ингибиторов аргиназы.
Фармацевтические композиции настоящего изобретения
В одном варианте осуществления настоящее изобретение относится к композиции, включающей по меньшей мере одно соединение настоящего изобретения или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
В другом варианте осуществления настоящее изобретение относится к фармацевтической композицию, включающей по меньшей мере одно соединение настоящего изобретения или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый носитель.
В одном варианте осуществления соединение или его фармацевтически приемлемая соль присутствуют в эффективном количестве в композиции. В другом варианте осуществления соединение или его фармацевтически приемлемая соль присутствуют на уровне от приблизительно 0,1% по массе до приблизительно 90% по массе, исходя из общей массы фармацевтической композиции. Предпочтительно, соединение или его фармацевтически приемлемая соль присутствуют на уровне, составляющем по меньшей мере приблизительно 1% по массе, исходя из общей массы фармацевтической композиции. Более предпочтительно, соединение или его фармацевтически приемлемая соль присутствуют на уровне, составляющем по меньшей мере приблизительно 5% по массе, исходя из общей массы фармацевтической композиции. Даже более предпочтительно, соединение или его фармацевтически приемлемая соль присутствуют на уровне, составляющем по меньшей мере приблизительно 10% по массе, исходя из общей массы фармацевтической композиции. Тем не менее, еще более предпочтительно, соединение или его фармацевтически приемлемая соль присутствуют на уровне, составляющем по меньшей мере приблизительно 25% по массе, исходя из общей массы фармацевтической композиции.
Настоящее изобретение включает комбинированные препараты, которые включают препараты, содержащие по меньшей мере одно соединение настоящего изобретения и второе терапевтическое средство, причем по меньшей мере одно соединение настоящего изобретения и второе терапевтическое средство являются комбинированными. Настоящее изобретение также включает комбинированные терапии, которые включают совместное введение ингибитора аргиназы этого изобретения с другим фармацевтически активным соединением или лекарственным средством. Более конкретно, термин «комбинированная терапия» относится к введению двух или более терапевтических средств или соединений для лечения по меньшей мере одного терапевтического состояния или нарушения, описанного в настоящем изобретении. Такое введение включает использование каждого типа терапевтического средства совместно или одновременно. Такое введение включает использование каждого типа терапевтического средства в одной и той же лекарственной форме с однократной дозой или в отдельных лекарственных формах с однократной дозой. В любом случае схема лечения обеспечивает выгодные эффекты комбинации лекарственных средств при лечении состояний или нарушений, описываемых в описании.
Соответственно, в некоторых вариантах осуществления настоящее изобретение относится к композициям, содержащим соединение настоящего изобретения или его фармацевтически приемлемую соль и ингибитор, выбираемый из группы, состоящей из ингибитора фосфодиэстеразы-1 (PDE1), ингибитора фосфодиэстеразы-2 (PDE2), ингибитора фосфодиэстеразы-3 (PDE3), ингибитора фосфодиэстеразы-4 (PDE4), ингибитора фосфодиэстеразы-5 (PDE5), неспецифического ингибитора PDE, который ингибирует по меньшей мере два фермента, выбираемых из группы, состоящей из PDE1, PDE2, PDE3, PDE4 и PDE5, и их комбинации, и необязательный фармацевтически приемлемый эксципиент.
В одном варианте осуществления соединения настоящего изобретения пригодны для лечения пациентов, которые не реагируют на ингибиторы PDE5. Без желания ограничиться этой теорией, ингибиторы аргиназы являются эффективными при лечении пациентов, которые не реагируют на ингибиторы PDE5, поскольку аргиназа действует на более ранней стадии в пути, приводящем к NO-зависимому расслаблению генитальной гладкомышечной ткани, необходимому для полового возбуждения.
Неограничивающие подходящие ингибиторы фосфодиэстеразы-1 (PDE1) включают SE3623 (доступный от Eisai), BAY 38304S (доступный от Bayer), HFV 1017 (этиловый эфир 7-бензолсульфониламино-3a-этил-1,2,3,3a,10,11b-гексагидро-11H-5a,11a-диаза-бензо[cd]флуорантен-S-карбоновой кислоты 2,3-дигидроксисукцинат, доступный от Oaiichi Fine Chemical), KF 19S14 (5-фенил-3-(3-пиридил)метил-3H-имидазо[4,5-c][1,8]нафтиридин-4(5H)-он, доступный от Kyowa Hakko) и SCH S1866 ((цис-5,6a,7,8,9,9a-гексагидро-2-[4-(трифторметил)фенилметил]-5-метил-циклопент[4,5]имидазо[2,1-b]пурин-4(3H)-он), доступный от Schering-Plough).
Неограничивающие подходящие ингибиторы фосфодиэстеразы-2 (PDE2) включают BAY 607550 (2-(3,4-диметоксибензил)-7-[1-(1-гидроксиэтил)-4-фенилбутил]-5-метил-3H-имидазо[5,1-f][1,2,4]триазин-4-он, доступный от Bayer).
Неограничивающие подходящие ингибиторы фосфодиэстеразы-5 (PDE5) включают силденафил (продаваемый под торговой маркой Viagra™), варденафил (продаваемый под торговой маркой Levitra™), тадалафил (продаваемый под торговой маркой Cialis™), мироденафил, уденафил, аванафил, дасантафил, NM 702 (4-бром-6-[3-(4-хлорфенил)пропокси]-5-[(пиридин-3-илметил)амино]-2H-пиридазин-3-она гидрохлорид, доступный от Nissan Chemical Industries), SLx-2101 (доступный от Surface Logix) и UK 369003 (доступный от Pfizer).
Неограничивающие подходящие неспецифические ингибиторы PDE, которые ингибируют по меньшей мере два фермента, выбираемых из группы, состоящей из PDE1, PDE2, PDE3, PDE4 и PDE5, или их комбинации, включают амлексанокс, цитрат кофеина, доксофиллин, левосимендан, мопидамол, пентоксифиллин, пемобендан, пропентофиллин, веснаринон и ибудиласт.
Такие композиции получают в соответствии с подходящими фармацевтическими процедурами, таким как описано в Remington's Pharmaceutical Sciences, 17th edition, ed. Alfonoso R. Gennaro, Mack Publishing Company, Easton, PA (1985). Фармацевтически приемлемыми носителями являются те, которые совместимы с другими ингредиентами в препарате и являются биологически приемлемыми.
В некоторых вариантах осуществления соединения настоящего изобретения могут быть введены перорально или парентерально, в чистом виде или в комбинации с традиционными фармацевтическими носителями. Подходящие твердые носители могут включать одно или более веществ, которые могут также действовать как корригенты, смазывающие вещества, солюбилизаторы, суспендирующие агенты, наполнители, вещества, способствующие скольжению, добавки для прессования, связующие вещества или агенты, вызывающие дезинтеграцию таблеток, или материалы для инкапсулирования. В порошках носитель представляет собой мелкозернистое твердое вещество в смеси с мелкозернистым активным ингредиентом. В таблетках активный ингредиент смешан с носителем, обладающим необходимыми способностями к прессованию, в подходящих пропорциях и спрессован в желаемую форму и до желаемого размера. Порошки и таблетки предпочтительно содержат вплоть до 99% активного ингредиента. Подходящие твердые носители включают, например, фосфат кальция, стеарат магния, тальк, сахара, лактозу, декстрин, крахмал, желатин, целлюлозу, метилцеллюлозу, натриевую соль карбоксиметилцеллюлозы, поливилпирролидин, легкоплавкие парафины и ионообменные смолы.
Жидкие носители могут быть использованы при получении растворов, суспензий, эмульсий, сиропов и эликсиров. Активный ингредиент этого изобретения может быть растворен или суспендирован в фармацевтически приемлемом жидком носителе, таком как вода, органический растворитель, смесь обоих или фармацевтически приемлемые масла или жир. Жидкий носитель может содержать другие подходящие фармацевтические добавки, такие как солюбилизаторы, эмульгаторы, буферы, консерванты, подсластители, корригенты, суспендирующие агенты, загустители, красители, регуляторы вязкости, стабилизаторы или осморегуляторы. Подходящие примеры жидких носителей для перорального и парентерального введения включают воду (в частности, содержащую вышеуказанные добавки, например, производные целлюлозы, предпочтительно раствор натриевой соли карбоксиметилцеллюлозы), спирты (включая одноатомные спирты и многоатомные спирты, например, глицерин) и их производные, и масла (например, фракционированное кокосовое масло и арахисовое масло). В случае парентерального введения носителем может также быть масляный эфир, такой как этилолеат и изопропилмиристат. Стерильные жидкие носители используются в стерильных композициях в жидкой форме для парентерального введения.
Жидкие фармацевтические композиции, которые являются стерильными растворами или суспензиями, могут быть введены, например, путем внутримышечной, внутрибрюшинной или подкожной инъекции. Стерильные растворы могут также быть введены внутривенно. В случае перорального введения может быть использована или жидкая, или твердая форма композиции.
Предпочтительно, фармацевтическая композиция находится в лекарственной форме с однократной дозой, например, в виде таблеток, капсул, порошков, растворов, суспензий, эмульсий, гранул или суппозиториев. В такой форме композиция подразделена на единичную дозу, содержащую соответствующие количества активного ингредиента; лекарственные формы с однократной дозой могут представлять собой упакованные композиции, например, упакованные порошки, флаконы, ампулы, предварительно наполненные шприцы или саше, содержащие жидкости. Лекарственной формой с однократной дозой может быть, например, сама капсула или таблетка, или она может представлять собой соответствующее число любых таких композиций в упакованной форме.
В другом варианте осуществления настоящего изобретения применимые в настоящем изобретении соединения можно вводить млекопитающему вместе с одним или более другими фармацевтическими активными агентами, такими как те агенты, которые используются для лечения любого другого заболевания, имеющегося у млекопитающего. Примеры таких фармацевтических активных агентов, применимых для таких комбинированных терапий, включают средства для купирования боли, антиангиогенные средства, антинеопластические средства, средства от диабета, противоинфекционные средства, желудочно-кишечные средства или их комбинации.
Один или более других фармацевтических активных агентов можно вводить в терапевтически эффективном количестве одновременно (например, по отдельности в одно и то же время или вместе в фармацевтической композиции) или последовательно с одним или более соединений настоящего изобретения.
Путь введения может быть любым путем, например, пероральным, назальным, легочным, чрескожным, таким как пассивная или ионтофоретическая доставка, или парентеральным, например, ректальным, инъекцией веществ замедленного всасывания, подкожным, внутривенным, интрауретральным, внутримышечным, интраназальным, раствором для глаз и мазью, в случае которого активное соединение настоящего изобретения эффективно переносится в соответствующее или желательное место действия. Кроме того, введение соединения настоящего изобретения с другими активными ингредиентами может быть совместным или одновременным.
В одном варианте осуществления препарат настоящего изобретения вводят млекопитающему по меньшей мере одним путем, выбираемым из группы, состоящей из перорального, назального, легочного, чрескожного, интраназального, офтальмологического, ректального и парентерального введения, причем указанное парентеральное введение включает подкожное, внутривенное, интрауретральное или внутримышечное введение.
Особенно преимущественным является приготовление композиций в форме единицы дозирования для облегчения введения и единообразия дозы. Как используется в описании, форма единицы дозирования относится к физически дискретным единицам, подходящим в качестве однократных доз для пациента, подвергаемого лечению; при этом каждая единица содержит заранее заданное количество пептида, рассчитанное так, чтобы вызвать желаемый терапевтический эффект, вместе с необходимым фармацевтическим носителем. Спецификация для форм единиц дозирования обусловлена уникальными характеристиками активного соединения и конкретным терапевтическим эффектом, который должен быть достигнут, и ограничениями, связанными с областью составления смеси такого активного соединения для лечения пациентов, и напрямую зависит от них.
Фармацевтические композиции могут быть заключены в контейнер, пакет или дозатор вместе с инструкциями по введению.
Обычно дозы соединений настоящего изобретения, которые можно вводить животному, предпочтительно человеку, находятся в количественном диапазоне от 1 микрограмма до приблизительно 100 миллиграмм на килограмм массы тела животного. Точная вводимая доза варьирует в зависимости от любого ряда факторов, включающих, но не ограничиваясь ими, тип животного и тип нарушения, подвергаемого лечению, возраст животного и путь введения. Предпочтительно, доза соединения варьирует от приблизительно 10 микрограмм до приблизительно 10 миллиграмм на килограмм массы тела животного. Более предпочтительно, доза варьирует от приблизительно 100 микрограмм до приблизительно 5 миллиграмм на килограмм массы тела животного.
Обычно соединения настоящего изобретения можно вводить животному так часто, как несколько раз в день, или их можно вводить менее часто, например, один раз в день, один раз в неделю, один раз каждые две недели, один раз в месяц или даже менее часто, например, один раз в несколько месяцев или даже один раз в год или реже. Частота введения доз становится быстро очевидной квалифицированному специалисту и зависит от любого ряда факторов, таких как, но без ограничения, тип и тяжесть нарушения, подвергаемого лечению, тип и возраст животного.
Диагностические применения соединений настоящего изобретения
Медицинская диагностическая визуализация является важным элементом современного здравоохранения. Получение изображения эхографическим, радионуклидным, рентгеновским методом и методом магнитного резонанса облегчает диагностирование заболевания. Диагностические фармацевтические средства, часто называемые контрастными агентами, можно вводить пациенту вместо терапевтических ингибиторов аргиназы, или их можно вводить пациенту одновременно с терапевтическим средством для расширения применимости самого метода получения изображений. Такие формирующие изображение агенты действуют посредством изменения энергии или образа, которым энергия взаимодействует с тканями. Для медицинской диагностической визуализации часто используют нацеленные контрастные агенты, которые, при связывании или локализации в местах избирательно в организме, помогают получить представляющее диагностический интерес изображение.
Нацеленные контрастные агенты для диагностической визуализации, как правило, состоят из нацеливающего фрагмента, помеченного отслеживаемым, формирующим изображение фрагментом. Такие отслеживаемые, формирующие изображение фрагменты включают флуоресцентные метки; рентгеноконтрастные красители (например, йодированные ароматические соединения), радиоактивные элементы, такие как 3H, 18F, 125I, 129I; или диагностически применимые радиоактивные или парамагнитные металлы в хелатной форме, такие как Gd(III), Mn(II), Tc-99m, Re-186, Re-188, In-111 или Ga-67. Примеры применимых диагностических, формирующих изображение агентов настоящего изобретения включают соединения, в которых по меньшей мере один атом водорода первого и/или второго заместителей был замещен одним из вышеупомянутых формирующих изображение фрагментов.
Нацеливающий фрагмент переносит метку к представляющему диагностический интерес месту, в котором ее выявляют, например, с помощью ЯМР-томографической, эхографической, КТ или радионуклидной визуализации (включая SPECT и PET). В некоторых предпочтительных вариантах осуществления соединений настоящего изобретения соединение настоящего изобретения имеет остаток способного формировать изображение фрагмента, выбираемого из группы, состоящей из испускающего гамма-лучи радиоизотопа, испускающего позитроны радиоизотопа, контрастного агента для получения изображения методом магнитного резонанса, рентгеноконтрастного агента и контрастного агента для эхографии.
Посредством использования такого ингибитора аргиназы, соответственно конъюгированного со способным формировать изображение фрагментом, можно визуально наблюдать в организме пациента в режиме реального времени эндогенную аргиназную активность. Чтобы быть эффективной, способный формировать изображение фрагмент не должен вносить значительные помехи в связывание дериватизированного ингибитора аргиназы со своим субстратом. Например, конъюгат ингибитор аргиназы-способный формировать изображение фрагмент может иметь Ki, составляющую менее чем приблизительно 1000 нМ.
Настоящее изобретение включает дериватизированное соединение, помеченное флуоресцентной меткой. Настоящее изобретение также включает дериватизированный ингибитор аргиназы, помеченный флуоресцентной меткой. Например, в варианте осуществления спектроскопический зонд, такой как флуоресцентный фрагмент или чувствительный для ЯМР или ЯМР-томографии фрагмент или комплекс, ковалентно присоединен в качестве замещающей группы посредством гибкого линкера, достаточно длинного, так что зонд не создает отрицательные взаимодействия с поверхностью белка. Такой спектроскопический зонд является применимым диагностическим средством для неинвазивного определения сверхэкспрессии аргиназы, отмечаемой при некоторых заболеваниях, таких как, например, астма (сверхэкспрессия аргиназы дыхательных путей), рак (сверхэкспрессия аргиназы в некоторых типах рака молочной железы, злокачественные опухоли ободочной кишки и т.п.) или некоторые внутренние бактериальные инфекции (например, сверхэкспрессия аргиназы бактерии H. pylori во избежание иммунной реакции при язвах желудка у человека).
В одном аспекте настоящее изобретение включает способ диагностирования сверхэкспрессии аргиназы у пациента, включающий стадии введения пациенту диагностически эффективного количества соединения настоящего изобретения, или его фармацевтически приемлемой соли, которое включает второй заместитель, причем второй заместитель создает возможность для in vivo визуализации соединения; и получения изображения пациента.
В одном варианте осуществления сверхэкспрессия аргиназы связана с астмой, раком, бактериальными инфекциями или их комбинациями.
В другом аспекте настоящее изобретение включает способ диагностирования сверхэкспрессии аргиназы у пациента, включающий введение указанному пациенту диагностически эффективного количества соединения настоящего изобретения, или его фармацевтически приемлемой соли, которое включает способный формировать изображение фрагмент, и получение изображения указанного пациента.
В одном варианте осуществления сверхэкспрессия аргиназы связана с астмой, раком, бактериальными инфекциями или их комбинациями.
В еще одном аспекте настоящее изобретение относится к способу получения радиоизображения пациента, включающему введение пациенту эффективного количества соединения настоящего изобретения, которое имеет способный формировать радиоизображение фрагмент, и сканирование пациента с использованием устройства для получения радиоизображения.
В еще одном аспекте настоящее изобретение включает способ ингибирования аргиназы, включающий приведение аргиназы в контакт с соединением настоящего изобретения или его солью.
В одном варианте осуществления аргиназой является аргиназа дрожжей, бактерии, паразита или млекопитающего. В другом варианте осуществления аргиназой млекопитающего является аргиназа типа I человека или аргиназа типа II человека (например, аргиназа полового члена человека).
В еще одном аспекте настоящее изобретение включает диагностическую композицию, содержащую диагностически эффективное количество соединения настоящего изобретения или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель; причем соединение помечено диагностической меткой.
В еще одном аспекте настоящее изобретение включает диагностическую композицию, содержащую диагностически эффективное количество соединения настоящего изобретения или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель; причем соединение помечено способным формировать изображение фрагментом.
Терапевтические применения соединений настоящего изобретения
Настоящее изобретение включает соединения, которые ингибируют ферментативную активность аргиназы. Эти соединения, которые, как не было ранее известно, ингибируют этот фермент (и как не было ранее известно, имеют какое-либо применение), пригодны для ряда применений в медицине и исследовании.
Соединения, композиции и способы настоящего изобретения применимы для ингибирования активности аргиназы, в том числе, но без ограничения, аргиназы млекопитающего (например, человека), дрожжей и бактерий (таких как H. pylori). Соединения, композиции и способы, описываемые в описании, могут быть использованы для ингибирования аргиназной активности in vitro или in vivo, например, у человека. Эти композиции можно также использовать для лечения нарушения, характеризующегося либо необычно высокой аргиназной активностью в ткани млекопитающего, либо необычно низкой активностью синтазы оксида азота в ткани млекопитающего, предпочтительно человека. «Ингибирование» аргиназы с помощью ингибитора аргиназы, как используется в описании, означает снижение уровня аргиназной активности в присутствии ингибитора, по сравнению с уровнем аргиназной активности в отсутствие ингибитора.
Для ослабления связанного с аргиназой нарушения у млекопитающего ингибитор аргиназы, описываемый в описании, вводят млекопитающему, пораженному этим нарушением. Ингибитор предпочтительно вводят в комбинации с одним или более фармацевтически приемлемыми носителями, которые описаны в описании более подробно. Ингибитор (предпочтительно в комбинации с носителем) можно также вводить млекопитающему, пораженному нарушением, характеризующимся аберрантной активностью синтазы NO, или млекопитающему, который демонстрирует нормальные (т.е. не нездоровые) уровни аргиназной активности и активности синтазы NO, но у которого ингибирование аргиназной активности является желательным.
Существует несколько связанных с аргиназой заболеваний, некоторые из которых перечислены ниже. Они связаны с одним, двумя и всеми тремя явлениями, связанными с конститутивной и увеличенной аргиназной активностью, описываемыми в другом месте описания. Многие из этих заболеваний характеризуются двумя или даже тремя явлениями одновременно или последовательно, например, пролиферация клеток и накопление фиброзной ткани могут придать жесткость ткани дыхательных путей или сосудистой ткани в сокращенном состоянии, так что достижение NO-зависимого расслабления является более трудным.
Соединения настоящего изобретения могут быть использованы для лечения состояний или заболеваний у млекопитающего. В одном варианте осуществления заболевания или нарушения связаны с необычно высоким уровнем аргиназной активности или необычно низким уровнем активности синтазы NO. «Необычно высокий уровень аргиназной активности», как используется в описании, означает уровень аргиназной активности, который превышает уровень, обнаруживаемый в нормальной ткани, когда нормальная ткань не проявляет фенотип связанного с аргиназой нарушения. «Необычно низкий уровень NO», как используется в описании, означает уровень NO, который ниже уровня, обнаруживаемого в нормальной ткани.
В одном варианте осуществления описываемые в описании соединения могут быть использованы для лечения, предупреждения, устранения или диагностирования одного или более из следующих заболеваний, состояний или болезней: состояний, связанных с ишемически-реперфузионным повреждением (ишемией миокарда-реперфузионным повреждением, трансплантацией органа, острой почечной недостаточностью, вазоокклюзивными кризисами при серповидноклеточной анемии), идиопатическим фиброзом легких, легочной артериальной гипертензией, сильной коронарной вазодилатацией, астмой, острым респираторным дистресс-синдромом, хроническим обструктивным заболеванием легких (COPD), бронхолегочной дисплазией, гипоксической дыхательной недостаточностью, муковисцидозом, субарахноидальным кровоизлиянием, тромбозом, микробной инфекцией, раком, заживлением ран, консервацией крови, гипертрофией сердца, заболеванием желудочно-кишечного тракта, воспалительным заболеванием легких, нарушением полового возбуждения, сердечно-сосудистым нарушением, заболеванием, вызванным патогенным микроорганизмом, иммунологическим нарушением, раком, преждевременными родами, болезнью Рейно, псориазом, ревматоидным артритом и болезнью Пейрони.
В одном варианте осуществления описываемые в описании соединения могут быть использованы для лечения, предупреждения, устранения или диагностирования одного или более из следующих заболеваний, состояний или болезней, каждое из которых обсуждается по отдельности ниже: (1) заболевания желудочно-кишечного тракта, (2) воспалительные заболеваний легких, (3) нарушения полового возбуждения, (4) сердечно-сосудистые нарушения, (5) заболевания, вызванные патогенным микроорганизмом, (6) иммунологические нарушения, (7) рак, (8) преждевременные роды, (9) болезнь Рейно, (10) псориаз, (11) ревматоидный артрит и (12) болезнь Пейрони, среди прочих.
1. Заболевания желудочно-кишечного тракта
Увеличение аргиназной активности было связано с патофизиологией ряда состояний, включая ухудшение опосредуемого неадренергическими и нехолинергическими нервами (NANC) расслабления гладкой мышцы желудочно-кишечного тракта. Ингибитор аргиназы может быть использован для уменьшения такого ухудшения путем введения ингибитора млекопитающему, которое испытывает такое ухудшение, или млекопитающему, которое, как предвидят, будет испытывать такое ухудшение (например, человеку, пораженному нарушением моторики желудочно-кишечного тракта).
Соответственно, соединения настоящего изобретения могут быть применимыми при лечении или предупреждении нарушений моторики желудочно-кишечного тракта, которые основаны на данных наблюдений того, что аргиназа присутствует в мышце внутреннего анального сфинктера опоссума, и было установлено, что известный ингибитор аргиназы, ABH, расслабляет эту мышцу. См., например, Baggio et al., 1999, J. Pharm, Exp. Ther. 290, 1409-1416.
Соединения настоящего изобретения могут быть также применимыми при лечении или предупреждении воспалительного заболевания кишечника (IBD) (например, болезни Крона и неспецифического язвенного колита). В действительности, IBD, как было установлено, характеризуется увеличенной аргиназной активностью и эндотелиальной дисфункцией. См., например, Horowitz et al., 2007, Am. J. Physiol. Gastrointest. Liver Physiol. 292, G 1323-1336.
Также, соединения настоящего изобретения могут быть применимыми при лечении или предупреждении язв желудка, поскольку бактерия, которая вызывает язвы желудка, Helicobacter pylori, проявляет увеличенную аргиназную активность при колонизации во избежание иммунной реакции человека. См., например, Gobeli et al., 2001, Proc. Natl. Acad. Sci. (USA) 98, 13844-13849.
2. Воспалительные заболевания легких
Соединения настоящего изобретения могут быть применимыми при лечении или предупреждении астмы на основе данных наблюдений того, что экспрессия аргиназы повышена в дыхательных путях больных астмой. См., например, Zimmermann and Rothenberg, 2006, Eur. J. Pharmacol. 533:253-262. Кроме того, лечение с использованием аэрозольного аппарата морских свинок с помощью ABH в модели аллергической астмы предотвращает гиперчувствительность дыхательных путей. См., например, Maarsingh, "Arginase: A Novel Key Enzyme in the Pathophysiology of Allergic Asthma," Ph.D. dissertation, Chapter 9, University of Groningen, Netherlands (2006); Maarsingh et al., 2008, Am. J. Respir. Crit. Care Med. 178:565-573. Фенотип астмы характеризуется сужением дыхательных путей, гиперплазией гладких мышц дыхательных путей и постоянным накоплением фиброзной ткани; ингибитор аргиназы может расслабить гладкую мышцу дыхательный путей и уменьшить клеточную гиперплазию и фиброз.
Кроме того, соединения настоящего изобретения могут быть применимыми при лечении или предупреждении химически индуцированного фиброза легких, поскольку аргиназа I и II активируется при индуцированном блеомицином фиброзе легких для обеспечения большего количества L-орнитина для биосинтеза коллагена. См., например, Endo et al., 2003, Am. J. Physiol. Lung Cell Mol. Physiol. 285, L313-321.
Соединения настоящего изобретения могут быть также применимыми при лечении или предупреждении идиопатического фиброза легких на основе данных наблюдений того, что в модели на животных отмечается индуцированное вирусом увеличение экспрессии аргиназы I. См., например, Mora et al., 2006, Am. J. Respir. Cell Mol. Biol. 35:466-473.
Кроме того, соединения настоящего изобретения могут быть применимыми при лечении или предупреждении муковисцидоза. Увеличенная активность аргиназы в слюне вносит вклад в недостаток NO при заболевании легких в виде муковисцидоза; аргиназная активность также способствует развитию фиброза. См., например, Graseman et al., 2005, Am. J. Respir. Crit. Care Med. 172: 1523-1528.
3. Нарушения полового возбуждения
Эректильная дисфункция поражает половину мужской популяции старше сорока. Этот недуг часто является следствием дефектов сложных каскадов катализируемых ферментами реакций, контролирующих кровоток в пещеристое тело, полость из мышечной, губчатой ткани, и из него, которое наполняется кровью в состоянии эрекции. Дефекты, которые уменьшают кровоток в пещеристое тело, часто возникают как вторичные осложнения, связанные с другими состояниями здоровья, такими как болезнь сердца, гипертония, диабет, применение некоторых лекарственных средств и т.п.
В важном варианте осуществления настоящее изобретение относится к применению описываемого в описании ингибитора аргиназы для увеличения пенильной эректильной функции у млекопитающего (предпочтительно мужчины) или для ослабления эректильной дисфункции у млекопитающего. NO является важным регулятором эректильной функции и опосредует NANC-нейротрансмиссию в гладкой мышце пещеристого тела пениса, приводя к быстрому расслаблению, которое, в свою очередь, приводит к эрекции. По этой причине синтаза NO, которая катализирует окисление L-аргинина с образованием L-цитруллина и NO, является ключевым ферментом в физиологии пенильной гладкой мышцы. Аргиназа катализирует гидролиз L-аргинина с образованием L-орнитина и мочевины. Аргиназа регулирует активность синтазы NO посредством влияния на количество L-аргинина, доступного для окисления, катализируемого активностью синтазы NO. Таким образом, ингибирование аргиназной активности может увеличить активность синтазы NO, тем самым усиливая NO-зависимое расслабление гладкой мышцы в пещеристом теле и увеличивая пенальную эрекцию.
Аргиназа присутствует в пещеристом теле пениса кролика и человека, и ABH увеличивает NO-зависимое расслабление этой ткани. См., например, Cox et al., 1999, Nature Struct. Biol. 6:1043-1047. Ингибитор аргиназы, ABH, увеличивает эректильную реакцию у живых самцов кроликов. См., например, Cama et al., 2003, Biochemistry 42:8445-51. Экспрессия аргиназы II увеличена в пещеристом теле мужчины с диабетом, что приводит к уменьшению биосинтеза NO, которое, в свою очередь, приводит к эректильной дисфункции; введение ABH в ex vivo экспериментах восстанавливает биосинтез NO. См., например, Bivalacqua et al., 2001, Biochem. Biophys. Res. Commun. 283:923-927. Активность аргиназы I увеличена в пенисе старых мышей и ослабляет эректильную функцию. См., например, Bivalacqua et al., 2007, Am. J. Physiol. Heart Circ. Physiol. 292:H1340-51.
Соединения настоящего изобретения могут быть также применимыми при лечении или предупреждении нарушения полового возбуждения у женщин. Ингибитор аргиназы, ABH, усиливает гиперемическую реакцию в гениталиях самок кроликов. См., например, Cama et al., 2003, Biochemistry 42:8445-51.
4. Сердечно-сосудистые нарушения
Соединения настоящего изобретения могут быть применимыми при лечении или предупреждении эндотелиальной сосудистой дисфункции при атеросклерозе, гипертензии, гиперхолестеринемии и диабете. Аргиназа модулирует активность NOS посредством регулирования доступности L-аргинина, и вредные эффекты аргиназы могут быть блокированы с помощью ингибитора аргиназы. См., например, Berkowitz et al., 2003, Circulation 108:2000-2006 (2003); Yang and Ming, 2006, Clin. Med. Res. 4:53-65. Увеличенная аргиназная активность при диабете способствует развитию эндотелиальной сосудистой дисфункции в результате уменьшенной доступности L-аргинина для синтазы NO. См., например, Romero et al., 2008, Circ. Res 102:95-102. Ингибирование аргиназы уменьшает гипертензию у крыс со спонтанной гипертензией. См., например, Bagnost et al., 2010, Cardiovasc. Res. 87:569-577. Было установлено, что ингибирование аргиназы опосредует кардиопротекцию во время ишемии-реперфузии. См., например, Jung et al., 2010, Cardiovasc. Res. 85:147-154. Другие сосудистые заболевания включают заболевание периферических сосудов (PVD), заболевание периферических артерий (PAD) и субарахноидальное кровоизлияние. Аргиназа была идентифицирована как новая мишень для лекарственных средств для лечения атеросклероза. См., например, Yang and Ming, 2006, Curr. Hypertension Rep. 8:54-59.
Соединения настоящего изобретения могут быть применимыми при лечении или предупреждении легочной артериальной гипертензии. Увеличенная аргиназная активность способствует развитию эндотелиальной сосудистой дисфункции в результате уменьшения доступности L-аргинина для синтазы NO. См., например, Morris et al., 2007, Adv. Pulmonary Hypertension 5:31-36. Было также установлено, что экспрессия аргиназы II увеличена в артериях женщин с преэкламсией, состоянием с повышенной гипертензией. См. Sankaralingam et al., 2010, Cardiovasc. Res. 85:194-201.
5. Заболевания, вызванные патогенными микроорганизмами
Соединения настоящего изобретения могут быть применимыми при лечении или предупреждении африканского трипаносомоза, болезни Шагаса, лейшманиоза, малярии и других заболеваний, вызванных патогенными микроорганизмами. Участвующие в биосинтезе полиаминов ферменты важны для роста и выживания простейших. См., например, Heby et al., 2003, Biochem. Soc. Trans. 31:415-419. Аргиназа важна для жизнеспособности. См., например, Roberts et al., 2004, J. Biol. Chem. 279:23668-78. Поэтому ингибиторы аргиназ простейших могут уничтожить простейших.
Аргиназу можно ингибировать в дрожжах путем приведения дрожжей в контакт с композицией настоящего изобретения. Ингибирование аргиназы в дрожжах служит для минимизации продукции мочевины во время сбраживания алкогольных напитков.
6. Иммунологические нарушения
Соединения настоящего изобретения могут быть применимыми при лечении или предупреждении рассеянного склероза и, возможно, других аутоиммунных заболеваний на основе данных наблюдений того, что экспрессия аргиназы I увеличена в модели рассеянного склероза (экспериментального аутоиммунного энцефаломиелита) на животных, и введение ингибитора аргиназы ABH уменьшает оценку заболевания у животных. См., например, Xu et al., 2003, Immunology 110:141-148.
7. Рак
Индуцируемая опухолями толерантность уменьшает терапевтическую эффективность иммунотерапии; одним механизмом, приводящим к T-клеточной толерантности, является образование клеток-супрессоров миелоидного происхождения (MDSC), которые продуцируют аргиназу, тем самым истощая опухолевое микроокружение в отношении L-аргинина, что приводит к уменьшению передачи сигнала на T-клетки и их функционирование. Результатом является T-клеточная иммунологическая толерантность. Примечательно, что аргиназная активность является механизмом ускользания от иммунной системы, которым также обладают некоторые бактерии, например, Helicobacter pylori. MDSC рассматриваются в качестве «раковой защиты от иммунной атаки». См., например, Marx, 2008, Science 319:154-56.
Соответственно, экспрессия аргиназы увеличена в следующих типах рака, которые можно лечить ингибитором аргиназы, описываемым в описании: почечноклеточная карцинома (см., например, Zea et al., 2005, Cancer Res. 65:3044-48; Ochoa et al., 2007, Clin. Cancer Res. 13:721s-726s); рак предстательной железы (см., например, Bronte et al., 2005, J. Exp. Med. 201:1257-68) (ингибирование аргиназы N-гидрокси-L-аргинином способствует иммунотерапии опухолей); колоректальный рак (см., например, Leu and Wang, 1992, Cancer 70:733-36; Bronte and Zanovello, 2005, Nature Rev. Immunol. 5:641-54); рак молочной железы (см., например, Singh et al., 2000, Cancer Res. 60:3305-12; Bronte and Zanovello, 2005, Nature Rev. Immunol. 5:641-54) (ингибитор аргиназы N-гидрокси-L-аргинин ингибирует пролиферацию клеток и индуцирует апоптоз); рак кожи (плоскоклеточный рак и базально-клеточный рак) (см., например, Gokmen et al., 2001, J. Lab. Clin. Med. 137:340-44; Bronte and Zanovello, 2005, Nature Rev. Immunol. 5:641-54); рак легкого (см., например, Rodriguez et al., 2005, J. Exp. Med. 202:931-39; Bronte and Zanovello, 2005, Nature Rev. Immunol. 5:641-54); рак яичника (см., например, Melichar et al., J. Translational Med. 1, 1-5 (2003) (doi: 10.11861479-5876-1-5); и рак желудка (см., например, Wu et al., 1992, Life Sci. 51,1355-61); среди прочих.
8. Ведение преждевременных родов
Усиление расслабления маточной гладкой мышцы с помощью ингибитора аргиназы можно использовать для ведения преждевременных родов.
9. Болезнь Рейно
Болезнь Рейно является заболеванием микрососудистой сети. Поскольку подкожное введение ингибитора аргиназы BEC (который является аналогом ABH) людям расширяет кровеносные сосуды и увеличивает циркуляцию, ингибитор аргиназы может быть применимым при лечении болезни Рейно. См., например, Holowatz et al., 2006, J. Physiol, 574:573-81.
10. Псориаз
Аргиназа I является в высокой степени сверхэкспрессированной в гиперпролиферативном псориатическом эпидермисе в коже человека, и поэтому ингибиторы аргиназы могут быть применимыми при лечении псориаза. См., например, Bruch-Gerharz et al., 2003, Am. J. Pathology 162:203-11.
11. Ревматоидный артрит
Экспрессия аргиназы II увеличена в синовиальной жидкости являющихся людьми пациентов, и поэтому ингибиторы аргиназы могут быть применимыми при лечении артрита. См., например, Huang & Kaohsiung, 2001, J. Med. Sci. 17:358-63; Corraliza and Moncada, 2002, J. Rheumatol. 29:2261-65.
12. Болезнь Пейрони
Соединения настоящего изобретения могут быть применимыми при лечении или предупреждении болезни Пейрони. Экспрессия аргиназы II увеличена в пенисе крысы в модели этого заболевания на животных. См., например, Bivalacqua et al., 2001, J. Andrology 22:497-506. Хотя это нарушению может содействовать развитию эректильной дисфункции, оно в основном представляет собой воспалительное состояние, при котором в пенисе накапливается фиброзная ткань.
13. Общие сведения
Композиция настоящего изобретения может быть использована для лечения нарушения у млекопитающего, причем нарушение связано с экспрессией необычно высокого уровня аргиназной активности в ткани млекопитающего. Поскольку активность синтазы NO регулируется реципрокно относительно аргиназной активности у млекопитающих, более конкретно людей, соединения и композиции настоящего изобретения можно использовать для лечения нарушения у млекопитающего, которое связано с экспрессией необычно низкого уровня активности синтазы NO в ткани млекопитающего. Поскольку реципрокное взаимодействие аргиназы и синтазы NO вовлечено в функционирование гладкой мышцы, в настоящем изобретении также предусматривается применение описываемых в описании соединений для регуляции гладкомышечной активности. Соединение настоящего изобретения или композиция, содержащая соединение настоящего изобретения, которое включает описываемый в описании ингибитор аргиназы, также могут быть использованы для ингибирования аргиназы у млекопитающего, имеющего нормальные уровни аргиназной активности и активности синтазы NO, в частности, в том случае, когда физиология, на которую желательно оказать воздействие, является такой, на которую влияет активность аргиназы или синтазы NO, или в том случае, когда нарушение, причиной которого не являются аберрантные уровни аргиназы или NO, можно, тем не менее, ослабить или подавить посредством ингибирования аргиназной активности (например, некоторые формы эректильной дисфункции).
Настоящее изобретение включает способы ингибирования аргиназы у млекопитающего, включающие введение млекопитающему эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли.
Настоящее изобретение дополнительно включает способы лечения связанного с аргиназой нарушения у млекопитающего, включающие введение млекопитающему эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли. В другом варианте осуществления связанным с аргиназой нарушением является нарушение, связанное с необычно низким уровнем активности синтазы оксида азота в ткани человека, нарушение, связанное с необычно высоким уровнем аргиназной активности в ткани человека, или их комбинации, включая болезнь сердца, системную гипертензию, легочную гипертензию, эректильную дисфункцию, аутоиммунный энцефаломиелит, хроническую почечную недостаточность, нарушения моторики желудочно-кишечного тракта, различные типы рака желудка, уменьшенный печеночный кровоток, недостаточный печеночный кровоток, спазм церебральных сосудов или их комбинацию.
Настоящее изобретение также включает способы обеспечения освобождения от иммуносупрессии у млекопитающего, включающие введение млекопитающему эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли; причем указанное млекопитающее страдает заболеванием или состоянием, выбираемым из группы, состоящей из хронического инфекционного заболевания, бактериальной инфекции, паразитарной инфекции, травмы, лепры, туберкулеза, трансплантации печени, рака и их комбинации.
Настоящее изобретение дополнительно включает способы ингибирования продукции орнитина у млекопитающего, страдающего по меньшей мере одной опухолью, включающие введение млекопитающему эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли.
Настоящее изобретение также включает способы увеличения расслабления гладкой мышцы или расслабления гладкой мышцы у млекопитающего, включающие приведение гладкой мышцы в контакт с ингибитором аргиназы. Гладкая мышца предпочтительно находится в организме животного. Настоящее изобретение дополнительно включает способы расслабления гладкой мышцы у млекопитающего, включающие введение млекопитающему эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли. Тип гладкой мышцы, подвергаемой расслаблению, включает, но не ограничивается ими, гладкую мышцу желудочно-кишечного тракта, гладкую мышцу анального сфинктера, мышцу пищеводного сфинктера, сфинктер Одди, артериальную гладкую мышцу, сердечную гладкую мышцу, легочную гладкую мышцу, почечную гладкую мышцу, маточную гладкую мышцу, влагалищную гладкую мышцу, гладкую мышцу шейки матки, плацентарную гладкую мышцу и гладкую мышцу глаза. Когда гладкой мышцей является гладкая мышца желудочно-кишечного тракта, тип гладкой мышцы желудочно-кишечного тракта включает, но не ограничивается этим, мышцу внутреннего анального сфинктера.
Когда гладкая мышца находится в организме животного, настоящее изобретение включает способ ослабления (например, уменьшение степени или тяжести) или подавления (например, уменьшения вероятности развития, или предупреждения) связанного с аргиназой нарушения у животного. В предпочтительном варианте осуществления животным является человек.
В настоящем изобретении также предусматривается применение ингибитора аргиназы в in vitro функциональном исследовании ингибирования аргиназы/расслабления гладкой мышцы, с целью идентификации соединений, оказывающих влияние на функционирование гладкой мышцы. Идентифицированные подобным образом соединения, как считают, являются возможными антагонистами ингибитора аргиназы, поскольку эти соединения идентифицированы по их способности нейтрализовать ингибирование аргиназной активности. Например, эти соединения можно идентифицировать с помощью анализа на активность гладкой мышцы, используя мышцу внутреннего анального сфинктера и один из ингибиторов аргиназы настоящего изобретения. В этом анализе индуцируют расслабление полос мышцы внутреннего анального сфинктера, полученной от млекопитающего (например, взрослого опоссума) с помощью опосредуемого NANC нервами расслабления, используя стимуляцию электрическим полем (EFS); расслабление отменяют путем приведения мышечных полос в контакт с аргиназой; и возвращение расслабления осуществляют путем приведения мышцы в контакт с ингибитором аргиназы. Определяют эффект исследуемого соединения на последующее возвращение мышечного расслабления. Любое значительное возвращение состояния расслабления мышцы в присутствии исследуемого соединения, по сравнению с состоянием расслабления мышцы в отсутствие исследуемого соединения, является указанием на то, что исследуемое соединение является антагонистом ингибитора аргиназы.
Настоящее изобретение также включает способы лечения заболевания или состояния, связанного с увеличенной экспрессией аргиназы у млекопитающего, включающие введение млекопитающему эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли; причем заболеванием или состоянием является заболевание желудочно-кишечного тракта, воспалительное заболевание легких, нарушение полового возбуждения, сердечно-сосудистое заболевание, гемолитическое заболевание, аутоиммунное заболевание, заживление ран, заболевание, вызванное паразитическими простейшими, заболевание, вызванное бактериями, рак, преждевременные роды, псориаз или их комбинация.
Ингибирование аргиназы воздействует на рак двумя путями. Первый путь представляет собой освобождение от иммуносупрессии, которая приводит к толерантности опухоли, а второй путь имеет место в результате ограничения продукции орнитина и последующих полиаминов, которые играют роль в пролиферации.
В одном варианте осуществления заболеванием желудочно-кишечного тракта является нарушение моторики желудочно-кишечного тракта, воспалительное заболевание кишечника, болезнь Крона, неспецифический язвенный колит, язва желудка, аденотонзиллярная болезнь или их комбинация.
В одном варианте осуществления воспалительным заболевание легких является астма, химически индуцированный фиброз легких, идиопатический фиброз легких, муковисцидоз, хроническое обструктивное заболевание легких (COPD) или их комбинация.
В одном варианте осуществления нарушением полового возбуждения является эректильная дисфункция у мужчин, болезнь Пейрони или нарушение полового возбуждения у женщин.
В одном варианте осуществления сердечно-сосудистым заболеванием является эндотелиальная сосудистая дисфункция при атеросклерозе, гипертензии, ишемически-реперфузионном повреждении, заболевании периферических сосудов, заболевании периферических артерий, субарахноидальном кровоизлиянии, гиперхолестеринемии, диабете, сердечно-сосудистом заболевании при диабете, легочной артериальной гипертензии, болезни Рейно или их комбинации.
В одном варианте осуществления гемолитическим заболеванием является ночная пароксизмальная гемоглобинурия (PNH), серповидноклеточная анемия, талассемия, наследственный сфероцитоз и стоматоцитоз, микроангиопатическая гемолитическая анемия, недостаток пируваткиназы, реакция на переливание несовместимой по системе ABO крови, пароксизмальная холодовая гемоглобинурия, тяжелая идиопатическая аутоиммунная гемолитическая анемия, вызванная инфекцией анемия, малярия, сердечно-легочное шунтирование, индуцированная механическим клапаном сердца анемия, химически индуцированная анемия или их комбинация.
В одном варианте осуществления аутоиммунным заболеванием является энцефаломиелит, рассеянный склероз, антифосфолипидный синдром 1, аутоиммунная гемолитическая анемия, хроническая воспалительная демиелинизирующая полирадикулонейропатия, герпетиформный дерматит (глютеиновая болезнь), дерматомиозит, злокачественная миастения, пузырчатка, ревматоидный артрит, синдром скованного человека, диабет типа 1, анкилозирующий спондилоартрит или их комбинация.
В одном варианте осуществления состоянием является заживление раны.
В одном варианте осуществления заболеванием, вызванным паразитическими простейшими, является африканский трипаносомоз, болезнь Шагаса, лейшманиоз, малярия или их комбинация.
В одном варианте осуществления раком является почечноклеточный рак, рак предстательной железы, колоректальный рак, рак молочной железы, рак кожи, рак легкого, рак яичника, рак желудка или их комбинация. В другом варианте осуществления раком кожи является плоскоклеточный рак, базально-клеточный рак или их комбинация.
В одном варианте осуществления состояние представляет собой преждевременные роды.
В одном варианте осуществления состояние представляет собой болезнь Рейно.
Кроме того, соединения и композиции настоящего изобретения применимы в качестве антифунгицидов в экономически важной для сельского хозяйства или в ином отношении флоре. Соединения и композиции настоящего изобретения можно терапевтически наносить на растение путем опрыскивания или другим способом, хорошо известным в области биологии растений.
Квалифицированные в данной области техники специалисты различат, или будут способны определить, используя не более чем обычное экспериментирование, многочисленные эквиваленты конкретных процедур, вариантов осуществления, патентной формулы и примеров, описанных в описании. Такие эквиваленты, как считается, находятся в пределах объема этого изобретения и охватываются формулой изобретения, прилагаемой к нему. Например, следует понимать, что модификации условий реакций, включая, но не ограничиваясь ими, время реакций, масштаб/объем реакций и экспериментальные реагенты, такие как растворители, катализаторы, давления, атмосферные условия, например, атмосфера азота, и восстановители/окислители, с помощью принятых в данной области техники альтернатив и с использованием не более чем обычного экспериментирования, находятся в пределах объема настоящей заявки.
Должно быть понятно, что в любом месте, где в описании представлены величины и диапазоны, все значения и диапазоны, охватываемые этими значениями и диапазонами, как подразумевается, включены в объем настоящего изобретения. Кроме того, все значения, находящиеся в пределах этих диапазонов, а также верхние или нижние пределы диапазона значений, также предполагаются настоящим изобретением.
Следующие примеры дополнительно иллюстрируют аспекты настоящего изобретения. Однако они никоим образом не являются ограничением идей или описания настоящего изобретения, которые изложены в описании.
ПРИМЕРЫ
Настоящее изобретение теперь описывается со ссылкой на следующие примеры. Эти примеры представлены лишь с целью иллюстрации, и настоящее изобретение не ограничивается этими примерами, а наоборот охватывает все вариации, которые являются очевидными в результате представленных в описании идей.
Теперь описываются материалы и методы, использованные в экспериментах, и результаты экспериментов, представленных в этом примере.
Соединения настоящего изобретения могут быть получены с помощью одного или более из следующих общих методов. Все части и проценты являются массовыми, если не указано иное. При использовании в описании диапазонов для физических характеристик, таких как молекулярная масса, или химических характеристик, таких как химические формулы, все комбинации и субкомбинаций таких диапазонов внутри них, как предполагается, включены в качестве конкретных вариантов осуществления этого изобретения. Следует понимать, что эти примеры, хотя и указывают на предпочтительные варианты осуществления настоящего изобретения, представлены лишь в качестве иллюстрации. На основе вышеприведенного обсуждения и этих примеров квалифицированный в данной области техники специалист может определить существенные признаки этого изобретения и, не выходя за пределы его сущности и объема, может осуществить различные изменения и модификации настоящего изобретения для его адаптации к различным применениям и условиям. Чтобы эти способы синтеза можно было в более полной степени понять, также представлены некоторые примеры протоколов жидкофазного синтеза конкретных соединений. Все из исходных материалов коммерчески доступны или могут быть получены с помощью процедур, описанных на этих схемах, или с помощью процедур, которые будут хорошо известны любому специалисту в области органической химии.
Общая процедура A. Получение аминоэфира кетимина из α-бромацетата
Как продемонстрировано на фиг.1, бензофенонимин (6,68 мл, 40,0 ммоль) и трет-бутилбромацетат 3c (5,9 мл, 40,0 ммоль) растворяли в 40 мл ацетонитрила. Добавляли DIEA (6,95 мл, 40,0 ммоль) и реакционную смесь нагревали до кипения с обратным холодильником в течение 14 ч. Реакционную смесь охлаждали до комнатной температуры, нейтрализовали добавлением 50% водного раствора уксусной кислоты и охлаждали до 0°C. Твердую фазу собирали фильтрованием, а затем промывали холодным этанолом. Продукт 4c сушили в вакууме с получением 9,05 г (77%), который использовали без дальнейшей очистки. MS (масс-спектрометрия) (LC/MS (жидкостная хроматография с масс-спектрометрией, ESI (ионизация методом электрораспыления): 296 (M+H), 240 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, δ): 7,5-8,0 (м, 10Н), 4,5 (с, 2H), 1,4 (с, 9H). (см. O'Donnell, 2004, Acc. Chem. Res. 37, 506)
Общая процедура B. Синтез реагентов алкилйодидов
Как продемонстрировано на фиг.2, пиперазин 6 (3,0 ммоль) и 3-бром-1-пропанол (3,75 ммоль) растворяли в 15 мл 2-бутанона. Добавляли твердый K2CO3 (6,0 ммоль) и реакционную смесь перемешивали при кипении с обратным холодильником в течение 18-24 ч. Смеси давали охладиться до комнатной температуры и разбавляли EtOAc, промывали 3× водой и 1× насыщенным раствором соли. Органический раствор сушили над сульфатом магния и концентрировали в вакууме с получением 7. Полученное соединение 7 анализировали методом LC/MS и 1H ЯМР и использовали без дальнейшей очистки.
Как продемонстрировано на фиг.2, соединение 8 (1,03 г, 7,1 ммоль) смешивали с альдегидом 9 (7,1 ммоль) в 30 мл DCM/TMOF 1:1 в течение приблизительно 5 мин. К смеси четырьмя порциями добавляли триацетоксиборгидрид (4,15 г, 1,6 ммоль) и реакционную смесь перемешивали в течение 1,5 ч. Реакционную смесь разбавляли EtOAc и добавляли 1 н. NaOH. Слои разделяли и органический раствор промывали 2× 1 н. раствором NaOH и 1× насыщенным раствором соли. Органический раствор сушили над сульфатом магния и концентрировали в вакууме с получением 10. Полученное соединение 10 анализировали методом LC/MS и 1H ЯМР, и его использовали без дальнейшей очистки.
Как продемонстрировано на фиг.2, имидазол (960 мг, 14,2 ммоль) и смолу, с которой связан трифенилфосфин, (3,30 г, 3,0 ммоль/г, 9,9 ммоль) суспендироваали в 50 мл DCM в атмосфере аргона. К этой смеси добавляли йод (2,52 г, 10,0 ммоль) и перемешивали в течение 10-15 минут. Соединение 10 добавляли в 10 мл DCM, затем перемешивали в течение 12-18 ч. Реакционную смесь фильтровали и промывали 3 порциями DCM, каждый раз по 10 мл, для удаления смолы, а затем добавляли 25 мл насыщенного раствора тиосульфата натрия вместе с 10-15 мл воды и смесь перемешивали в течение 10-15 минут. Смесь разделяли и органический слой промывали 3× водой и 1× насыщенным раствором соли. Органический раствор сушили над сульфатом магния и концентрировали в вакууме с получением масла 11.
Общая процедура C. Алкилирование кетимин глицина с помощью неактивированных алкилгалогенидов
Как продемонстрировано на фиг.3 и 7, соединение 4c (1 ммоль) растворяли в 5 мл сухого ТГФ (тетрагидрофурана) в атмосфере аргона и охлаждали до -78°C. К реакционной смеси добавляли 1 M раствор основания, LiHMDS (гексаметилдисилазана лития), в ТГФ (1,05 мл) и перемешивали при -78°C в течение 45 минут, а затем добавляли алкилгалогенид, такой как соединение 11, (1,05 ммоль) в 3 мл сухого ТГФ. Реакционную смесь перемешивали при -78°C в течение 30 минут, а затем давали нагреться до комнатной температуры и перемешивали в течение 8-18 ч. Реакционную смесь разбавляли 20 мл EtOAc (этилацетата) и промывали водой, а затем насыщенным раствором соли. Органический раствор фильтровали через слой диатомовой земли марки Hydromatrix и концентрировали досуха. Этот остаток растворяли в небольшом количестве DCM, наносили на колонку с сухим силикагелем и элюировали, используя смесь EtOAc/гексан (1-5%), с получением соединения 12 или 24.
Общая процедура D. Алкилирование аминоэфира кетимина с помощью кротилбромида
Как продемонстрировано на фиг.3 и 7, соединение 12 или 24 (1 ммоль) растворяли в 5 мл сухого ТГФ в атмосфере аргона и охлаждали до -78°C. К реакционной смеси добавляли 0,5 M раствор KHMDS (гексаметилдисилазана калия) в толуоле (1,05 мл) и перемешивали при -78°C в течение 45 минут, а затем добавляли кротилбромид (1,05 экв.). Реакционную смесь перемешивали при температуре -78°C в течение 30 минут, а затем давали нагреться до комнатной температуры и перемешивали в течение 8-18 ч. Реакционную смесь разбавляли 20 мл EtOAc и промывали водой, а затем насыщенным раствором соли. Органический раствор фильтровали через слой диатомовой земли марки Hydromatrix и концентрировали досуха. Этот остаток растворяли в небольшом количестве DCM, наносили на колонку с сухим силикагелем и элюировали, используя смесь EtOAc/гексан (1-5%), с получением соединения 13 или 25.
Общая процедура E. Гидроборирование кротильной боковой цепи
Как продемонстрировано на фиг.3 и 7-8, в атмосфере аргона [Ir(cod)Cl]2 (34 мг, 0,05 ммоль, 5 мол.%) и DPPM (бис(дифенилфосфино)метан, 38 мг, 0,10 ммоль, 10 мол.%) растворяли в 5 мл сухого DCM. Пинаколборан (175 мкл, 1,20 ммоль) и соединение 13, 27 или 32 (1 ммоль) растворяли в 5 мл сухого DCM и добавляли. Реакционную смесь перемешивали при комнатной температуре в течение 24 ч. Добавляли 1 мл MeOH/H2O (1:1), чтобы погасить реакцию. Смесь концентрировали в вакууме, растворяли с помощью 20 мл EtOAc и промывали водой, а затем насыщенным раствором соли. Органический раствор фильтровали через слой диатомовой земли марки Hydromatrix и концентрировали досуха. Этот остаток растворяли в небольшом количестве DCM, наносили на колонку с сухим силикагелем и элюировали, используя смесь EtOAc/гексан (5-10%), с получением соединения 14, 28 или 33.
Общая процедура F. Избирательное удаление Boc
Как продемонстрировано на фиг.4 и 6, соединение 14 или 19 превращается в соединение 15 или 20 при обработке 25-50% ТФУК (трифторуксусной кислоты)/DCM в атмосфере аргона в течение 12-16 ч. Реакционную смесь концентрируют досуха в вакууме, добавляют несколько мл DCM и этот раствор вновь концентрируют досуха, затем остаток сушат в вакууме в течение нескольких часов, а затем помещают в атмосферу аргона.
Общая процедура G. Алкилирование пиперазиновой боковой цепи
Как продемонстрировано на фиг.4, соединение 15 растворяли в безводном ДМФА в атмосфере аргона и добавляли 2,5 экв. карбоната калия, а затем 1-2 экв. алкилгалогенида, такого как бензилбромид. Реакционную смесь перемешивали при комнатной температуре в течение 12-18 ч. Реакционную смесь разбавляли EtOAc и промывали 2× 1 н. NaOH, 1× водой и 1× насыщенным раствором соли. Органический раствор фильтровали через слой диатомовой земли марки Hydromatrix, концентрировали досуха и соединение 16 использовали как есть для последующей стадии снятия всех защитных групп.
Общая процедура H. Ацилирование пиперазиновой боковой цепи
Как продемонстрировано на фиг.4 и 6, соединение 15 или 20 растворяли в безводном ДМФА в атмосфере аргона и добавляли DIEA до тех пор, пока раствор не становился щелочным. Добавляли ацилирующий агент (сульфонилхлорид, хлорангидрид или изоцианат) (1,2 экв.) и реакционную смесь перемешивали в течение 12-18 ч. Реакционную смесь разбавляли EtOAc и промывали 2× 1 н. NaOH, 1× водой и 1× насыщенным раствором соли. Органический раствор фильтровали через слой диатомовой земли марки Hydromatrix и концентрировали досуха с получением соединения 17, 18, 21, 22 или 23, которое использовали как есть для последующей стадии снятия всех защитных групп.
Общая процедура I. Удаление кетимина и повторная защита α-амина группой Cbz
Как продемонстрировано на фиг.6, соединение 14 обрабатывали смесью 1 н. HCl/ТГФ (1:2) в течение от 30 мин до 1 ч при комнатной температуре. Реакционную смесь разбавляли EtOAc и 1 н. NaOH и слои разделяли. Органический слой промывали 1× 1 н. NaOH, 1× H2O и 1× насыщенным раствором соли. Органический раствор сушили над MgSO4, фильтровали и концентрировали досуха. Этот остаток растворяли в безводном ТГФ в атмосфере аргона, добавляли 1,2 экв. DIEA, а затем 1,2 экв. бензилхлорформиата, и эту смесь перемешивали при комнатной температуре в течение 12-18 ч. Смесь разбавляли EtOAc, промывали 3× 0,1 н. HCl и 1× насыщенным раствором соли. Органический раствор сушили над MgSO4, фильтровали и концентрировали досуха. Этот остаток растворяли в небольшом количестве DCM, наносили на колонку с сухим силикагелем и элюировали, используя смеси EtOAc/гексан (5-10%), с получением соединения 19.
Общая процедура J. Нейтральные условия для удаления кетимина и повторной защиты α-амина группой Cbz
Как продемонстрировано на фиг.7, 6,37 г (12,2 ммоль) соединения 25 растворяли в 60 мл сухого ТГФ в атмосфере аргона и к этому раствору добавляли 30 мл 1 M гидрохлорида гидроксиламина. Реакционную смесь быстро перемешивали при комнатной температуре до полного расхода исходного соединения. К реакционной смеси добавляли этилацетат (100 мл) и 1 н. NaOH (50 мл). Слои разделяли и водный раствор промывали 3× EtOAc. Органические растворы объединяли, промывали 1× насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме до масла. Этот остаток растворяли в небольшом количестве DCM, наносили на колонку с сухим силикагелем и элюировали, используя смеси EtOAc/гексан (5-10%), с получением чистого соединения 26. Соединение 26 растворяли в 40 мл сухого ТГФ в атмосфере аргона и добавляли 2,4 мл DIEA (13,8 ммоль), а затем 2,0 мл бензилхлорформиата (13,8 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли 100 мл EtOAc, промывали 3× 0,1 н. HCl и 1× насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме до получения масла, которое элюировали на колонке с силикагелем, используя смеси EtOAc/гексан (5-10%), с получением 3,28 г (59%) чистого соединения 27.
Общая процедура K. Защита с помощью Boc α-амина
Как продемонстрировано на фиг.8, соединение 26 растворяли в смеси ТГФ/вода (1:1) и к этому раствору добавляли 3,5 экв. NaHCO3 и 2 экв. ди-трет-бутилдикарбоната. Через 36 ч реакционную смесь разбавляли EtOAc и промывали 3× водой и 1× насыщенным раствором соли. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме до получения масла, которое очищали путем элюирования на колонке с силикагелем, используя смеси EtOAc/гексан (2-50%), с получением чистого соединения 32.
Общая процедура L. Удаление TBS эфира и превращение гидроксила в йодид
Как продемонстрировано на фиг.7 и 8, 0,8 ммоль соединения 28 или 33 растворяли в 5 мл сухого ТГФ в атмосфере аргона и к этому раствору добавляли 4 мл 0,2 н. HCl. Смесь быстро перемешивали при комнатной температуре в течение ночи. Смесь разбавляли 50 мл EtOAc, промывали 1× водой, 1× насыщенным раствором NaHCO3 и 1× насыщенным раствором соли, сушили над MgSOч, фильтровали и концентрировали в вакууме с получением 0,411 г масла коричневого цвета (29), которое можно использовать как есть в следующей реакции или которое можно элюировать на колонке с силикагелем, используя смеси EtOAc/гексаны (10-25%). Имидазол (0,11 г, 1,62 ммоль) и 0,375 г Ph3P-смолы (3,0 ммоль/г, 1,12 ммоль) помещали в атмосферу аргона и добавляли 5 мл сухого DCM. Добавляли йод (0,290 г, 1,15 ммоль) и смесь обрабатывали ультразвуком 2-3 мин для растворения йода. Затем соединение 29 в 2 мл сухого DCM добавляли к реакционной смеси, с последующим добавлением 2 мл DCM в качестве промывки, и эту смесь перемешивали в течение ночи при комнатной температуре. Смолу отфильтровывали и хорошо промывали DCM. Раствор DCM промывали 1× насыщенным раствором тиосульфата натрия, а затем 1× водой и 1× насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме с получением 459 мг (93%) масла, определенного как соединение 30 или 34.
Общая процедура M. Хиральная хроматография соединения 29
Как продемонстрировано на фиг.11 и 12, энантиомеры соединения 29 разделяли на хроматографической колонке CHIRALCEL®OZ 20мкм AD-H, используя смесь 10% изопропанол/гексан, с получением чистых соединений 36a и 36b. На аналитической колонке элюирование пика 1 происходило на 6,34 мин, а пика 2 на 13,52 мин.
Общая процедура N. Добавление нуклеофила к алкилйодидам
Как продемонстрировано на фиг.7 и 8, соединение 30 или 34 (0,149 ммоль) в 0,5 мл ДМФА добавляли к смеси нуклеофила, такого как, но без ограничения, замещенный гидрохлорид пиперидина (1,1 экв.), и сухого карбоната калия (0,080 г, 0,58 ммоль). Эту смесь быстро перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли 10 мл EtOAc, промывали 2× 1 н. NaOH, 1× водой и 1× насыщенным раствором соли. Раствор EtOAc фильтровали через колонку с Hydromatrix и раствор концентрировали в вакууме с получением неочищенного соединения 31 или 35. Эти соединения можно было использовать как есть, или их элюировали на колонке с силикагелем, используя смеси EtOAc/гексаны (20-50%).
Общая процедура O. Процедура снятия всех защитных групп (1)
Как продемонстрировано на фиг.5, соединение 14 растворяли в смеси 6 н. HCl/DCM 2:1 и перемешивали в течение ночи при 70°C. Эту реакционную смесь охлаждали и экстрагировали 3× диэтиловым эфиром. Водный слой лиофилизировали и остаток очищали методом препаративной ВЭЖХ, используя градиенты ацетонитрил/вода, содержащие 0,075% ТФУК, на препаративных колонках с диоксидом кремния C18.
Общая процедура P. Процедура снятия всех защитных групп (2)
Как продемонстрировано на фиг.5, 10 и 14, соединение 14, 35 или 47 растворяли в смеси 50-100% ТФУК/DCM в атмосфере аргона и перемешивали при комнатной температуре в течение 1-2 ч. Реакционную смесь концентрировали досуха в вакууме и остаток помещали в атмосферу аргона. Этот остаток растворяли в 3-5 мл 1 н. HCl плюс равное количество диэтилового эфира. Добавляли фенилбороновую кислоту (5 экв.) и реакционную смесь быстро перемешивали при комнатной температуре в течение 12-18 ч. Слои разделяли и водный слой промывали 5× диэтиловым эфиром. Водный слой лиофилизировали и остаток очищали методом препаративной ВЭЖХ, используя градиенты ацетонитрил/вода, содержащие 0,075% ТФУК, на препаративных колонках с диоксидом кремния C18.
Общая процедура O. Процедура снятия всех защитных групп (3)
Как продемонстрировано на фиг.10 и 14, соединение 31, 48 или 50 растворяли в ТГФ и добавляли 2-3 экв. 1 н. HCl. К этой смеси добавляли Pd/C (10%, 0,1 экв.) и реакционную смесь помещали в атмосферу водорода на 1-2 ч. Реакционную смесь фильтровали через целит и промывали ацетонитрилом 2-3 раза. Реакционную смесь концентрировали досуха в вакууме, остаток помещали в атмосферу аргона и для полного удаления защитных групп следовали общей процедуре P. Порядок реакции можно менять в зависимости от конкретного химического соответствия групп в R1 этих соединений.
Общая процедура R. Этерификация кислотными способами
Как продемонстрировано на схеме A на фиг.13, соединение 41 (75-100 мг) добавляли к 6 мл спирта (R2OH) и добавляли 0,5 мл тионилхлорида. Реакционную смесь кипятили с обратным холодильником в течение 16 ч, охлаждали и концентрировали в вакууме. Соединение 42 очищали методом препаративной ВЭЖХ, используя градиент ацетонитрил/вода.
Общая процедура S. Этерификация щелочными способами
Как продемонстрировано на схеме B на фиг.13, 100 мг соединения 43, 44, 45 или 46 растворяли в 5 мл ДМФА и добавляли 2,5 экв. K2CO3, а затем 1,2 экв. алкилгалогенида, такого как изопропилбромид. При использовании алкилхлорида к реакционной смеси также добавляли эквивалент NaI. Реакционную смесь перемешивали при 25-50°C в течение 4-16 ч. Продукт выделяли в результате разбавления с использованием EtOAc, промывали 3× водой и концентрировали в вакууме с получением соединения 47, 48, 49 или 50. Эти продукты элюировали на колонке с силикагелем, используя смеси EtOAc/гексаны (20-100%), или очищали методом ВЭЖХ с обращенной фазой, используя градиенты ацетонитрил/вода.
Общая процедура T. Этерификация с использованием DCC/DMAP
Как продемонстрировано на фиг.13, соединение 43, 44, 45 или 46 растворяли в DCM и добавляли триэтиламин (3 экв.), DCC (2 экв.), DMAP и 1,2 экв. спирта (R2OH). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, а затем ее концентрировали в вакууме. Остаток элюировали на колонке с силикагелем, используя смеси EtOAc/гексаны (20-50%), или очищали методом ВЭЖХ с обращенной фазой, используя градиенты ацетонитрил/вода, с получением соединения 47, 48, 49 или 50.
Общая процедура U. Этерификация с использованием заранее образованного хлорангидрида
Как продемонстрировано на схеме D на фиг.13, соединение 45 растворяли в DCM и добавляли 1-2 капли ДМФА, а затем 1,5 экв. оксалилхлорида при 0°C. Реакционной смеси давали нагреться до комнатной температуры, а затем перемешивали в течение 1-2 ч. Реакционную смесь концентрировали в вакууме и добавляли сухой DCM, а затем вновь концентрировали в вакууме. Остаток сушили в вакууме в течение 1-2 ч. Остаток растворяли в ДМФА, добавляли 1,5-3 экв. R2OH и реакционную смесь перемешивали в течение 16 ч, а затем концентрировали в вакууме. Остаток элюировали на колонке с силикагелем, используя смеси EtOAc/гексаны (20-50%), или очищали методом ВЭЖХ с обращенной фазой, используя градиенты ацетонитрил/вода, с получением соединения 49.
Общая процедура V. Снятие всех защитных групп с являющихся пролекарствами сложных эфиров
Как продемонстрировано на схеме A на фиг.14, снятие с соединения 47, 48 или 50 защитных групп при α-амине и бороновом эфире с получением соединений этого изобретения 42 осуществляли с помощью процедур, представленных на фиг.10, и общих процедур O, P или Q. Кроме того, защитную группу Cbz у соединения 48 удаляли путем кипячения с обратным холодильником в ТФУК в течение 16 ч.
Как продемонстрировано на схеме B на фиг.14, Fmoc-группа соединения 49 была отщеплена после обработки 5-20% пиперидином или пиперазином в ДМФА в течение от 30 мин до 4 ч при комнатной температуре.
Промежуточные продукты в виде пинаколборонатных эфиров растворяли в 3-5 мл 1 н. HCl плюс равное количество диэтилового эфира. Добавляли фенилбороновую кислоту (5 экв.) и реакционную смесь быстро перемешивали при комнатной температуре в течение 12-18 ч. Слои разделяли и водный раствор промывали 5× диэтиловым эфиром. Водный слой лиофилизировали и остаток очищали методом препаративной ВЭЖХ, используя градиенты ацетонитрил/вода, содержащие 0,075% ТФУК, на препаративных колонках с диоксидом кремния C18, получая соединение 42.
Сравнительный пример 4: Трифторацетатная соль 2-амино-6-бороно-2-(3-(пиперазин-1-ил)пропил)капроновой кислоты (1a)
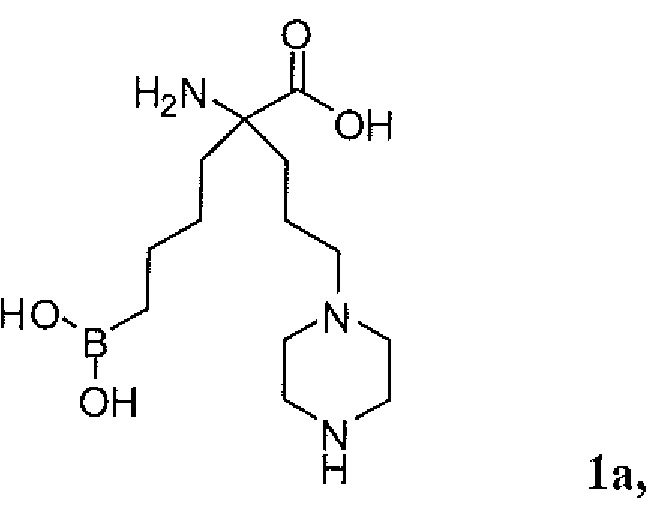
трет-Бутил 4-(5-трет-бутокси-4-(дифенилметиленамино)-5-оксопентилпиперазин-1-карбоксилат
Соединение 12a, 10,0 г (85%) получали после осуществления реакции 4c и 11 (где R=Boc), используя общую процедуру C, описанную выше. MS (LC/MS, ESI): 522 (M+H), 466 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, δ): 7,4-8,0 (м, 10Н), 4,9 (м, 1H), 3,2 (м, 4H), 2,4-2,5 (м, 6H), 2,0 (м, 2H), 1,4 (с, 18H), 1,3 (м, 2H).
трет-Бутил 4-(4-трет-бутоксикарбонил)-4-(дифенилметиленамино)окт-6-енил)пиперазин-1-карбоксилат
Соединение 13a, 10,4 г (94%), получали, используя общую процедуру D, описанную выше. MS (LC/MS, ESI): 576,5 (M+H), 520,5 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, δ): 7,4-8,0 (м, 10Н), 5,5 (м, 2H), 3,2 (м, 4H), 2,4-2,5 (м, 6Н), 2,05 (д, 3Н), 2,0 (м, 4H), 1,4 (с, 18H), 1,3 (м, 2H).
трет-Бутил 4-(4-трет-бутоксикарбонил)-4-(дифенилметиленамино)-8-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)октил)пиперазин-1-карбоксилат
Соединение 14a, 9,0 г (71%), получали, используя общую процедуру E, описанную выше. MS (LC/MS, ESI): 704,7 (M+H), 648,6 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, δ): 7,4-8,0 (м, 10Н), 3,2 (м, 4H), 2,4-2,5 (м, 6H), 2,0 (м, 4H), 1,4 (с, 18H), 1,3 (м, 4H), 1,25 (с, 12H), 0,8 (т, 2H).
Соединение 1a, 20 мг (45%), получали в виде соли ТФУК путем использования общей процедуры O, описанной выше, после очистки методом препаративной ВЭЖХ. MS (LC/MS, ESI): 284 (M-H2O+H). 1H ЯМР (300 МГц, D2O, δ): 2,8 (м, 4H), 2,4-2,5 (м, 6H), 2,0 (м, 4H), 1,3 (м, 4H), 0,8 (т, 211).
Пример 2
Следующее соединение, отмеченное в таблице 4, ниже, было синтезировано способом, аналогичным тому, который был описан выше для соединения 1a.
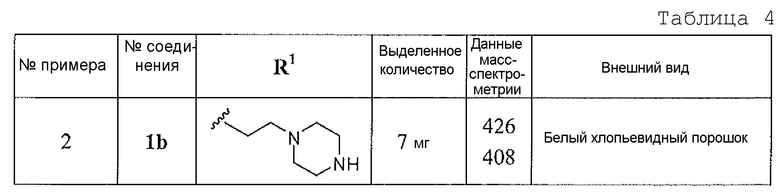
Пример 3: Трифторацетатная соль 2-амино-2-(3-(4-бензилпиперазин-1-ил)пропил)-6-боронокапроновой кислоты (1c)
Соединение 14a обрабатывали 20% TFA/DCM в атмосфере аргона, как описано выше в общей процедуре F, для получения соединения 15c, которое обрабатывали бензилбромидом, как описано выше в общей процедуре G, для получения соединения 16c. Это соединение использовали как есть в общей процедуре P с получением 13 мг 1c (19% в случае 3 стадий) после очистки методом препаративной ВЭЖХ в виде трифторацетатной соли. MS (LC/MS, ESI): 374 (M-H2O+H). 1H ЯМР (300 МГц, D2O, δ): 7,2-7,4 (м, 5H), 3,8 (с, 2H), 3,2 (м, 4H), 2,4-2,5 (м, 6H), 2,0 (м, 4H), 1,3 (м, 4H), 0,8 (т, 211).
Примеры 4-15
Следующие соединения, перечисленные в таблице 5, ниже, были синтезированы способом, аналогичным тому, который был описан выше для соединения 1c. В таблице 5 каждое соединение имеет следующую химическую структуру (каждый пример в таблице имеет другую группу R1):
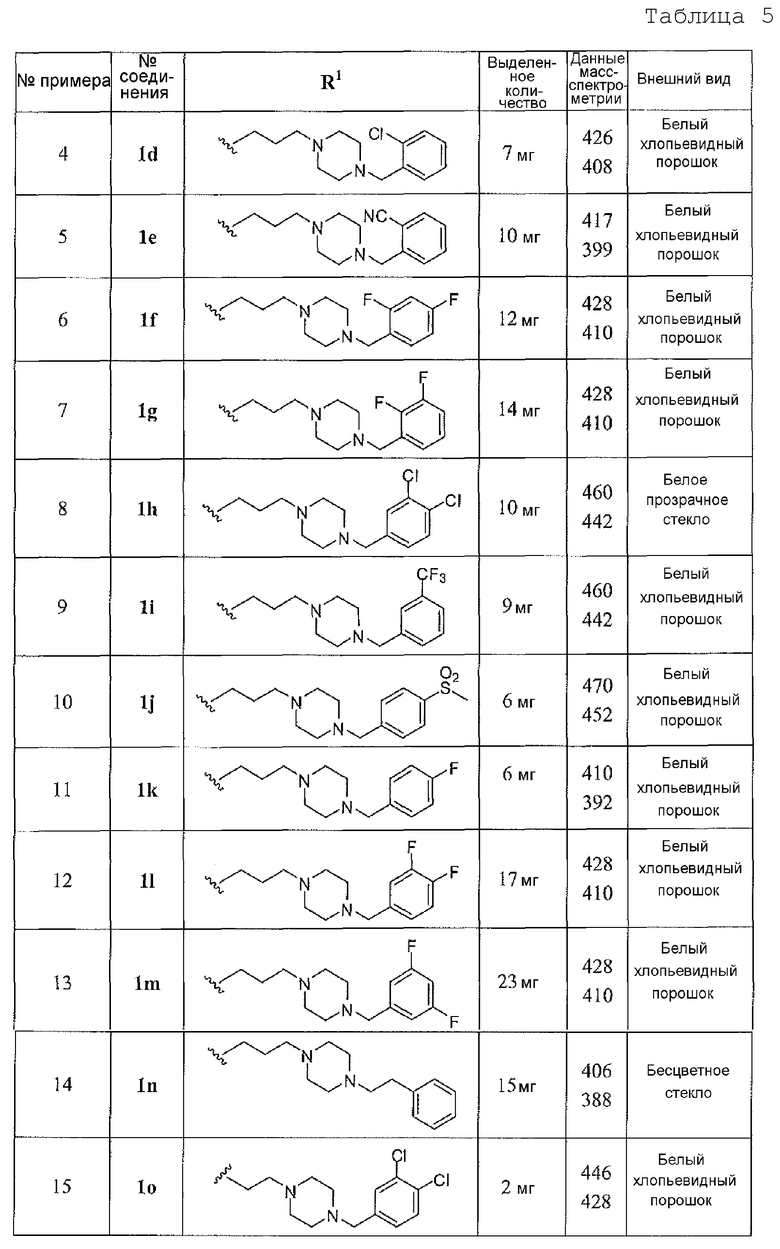
Пример 16: 2-амино-6-бороно-2-(3-(4-(3,4-дихлорфенил)пиперазин-1-ил)пропилкапроновая кислота (1p)
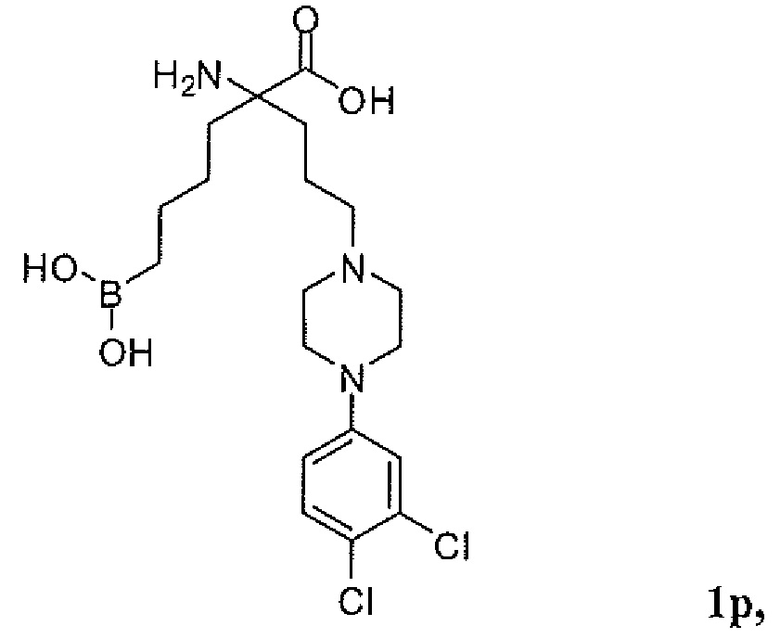
3-(4-(3,4-дихлорфенил)пиперазин-1-ил)пропан-1-ол (7p)
Соединение 5 (0,32 мл, 3,54 ммоль) смешивали с 4-(3,4-дихлорфенил)пиперазином (0,58 г, 2,5 ммоль) и карбонатом калия (0,78 г, 5,6 ммоль) в 15 мл 2-бутанона в течение ночи при 85°C, как описано в общей процедуре B, для получения соединения 7p и использовали как есть для следующей реакции. MS (LC/MS, ESI): 289 (M+H).
1-(3,4-дихлорфенил)-4-(3-йодпропил)пиперазин (11p)
Соединение 7p смешивали со смолой, с которой связан трифенилфосфин, имидазолом и йодом в DCM, как описано в общей процедуре B. Полученное соединение 11p использовали без дальнейшей очистки для следующей реакции. MS (LC/MS, ESI): 399 (M+H).
трет-Бутил 5-(4-(3-дихлорфенил)пиперазин-1-илl)-2-(дифенилметиленамино)пентаноат (12p)
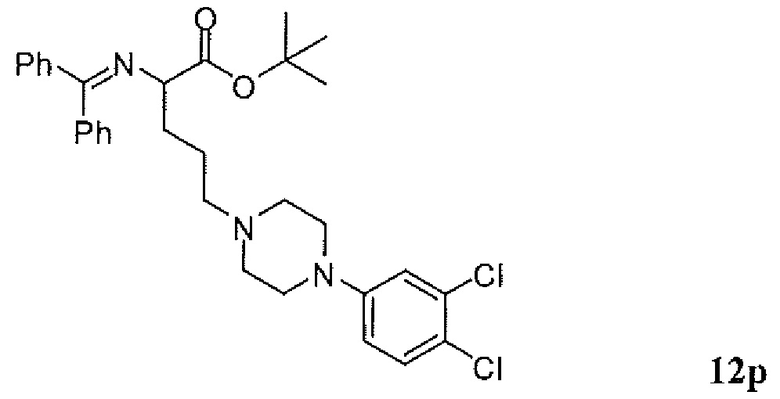
Соединение 12p, 0,497 г (37%), получали, используя общую процедуру C, описанную выше. MS (LC/MS, ESI): 566 (M+H), 510 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, δ): 7,4-8,0 (м, 10Н), 6,8-7,2 (м, 3H), 4,1 (т, 1H), 3,2-3,6 (м, 8Н), 2,4-2,5 (т, 2H), 1,7-2,0 (м, 4H), 1,4 (с, 9H).
трет-Бутил 2-(3-(4-(3,4-дихлорфенил)пиперазин-1-ил)пропил)-2-(дифенилметиленамино)гекс-4-еноат (13p)
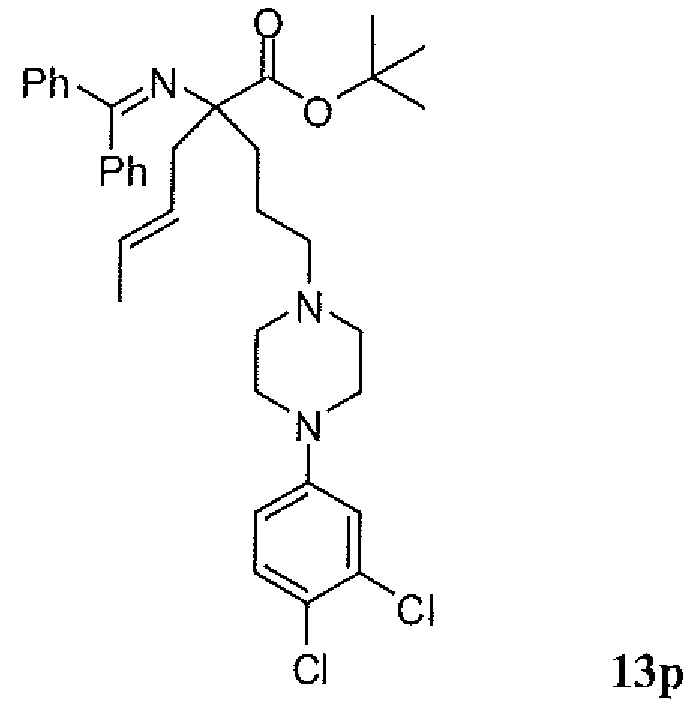
Соединение 13p, 0,359 г (94%), получали, используя общую процедуру D, описанную выше. MS (LC/MS, ESI): 620 (M+H), 564 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, δ): 7,4-8,0 (м, 10Н), 6,8-7,2 (м, 3H), 5,4-5,6 (м, 2H), 3,2-3,6 (м, 8H), 2,4-2,5 (м, 4H), 1,7-2,0 (м, 4H), 1,6 (д, 3H), 1,4 (с, 9H).
трет-Бутил 2-(3-(4-(3,4-дихлорфенил)пиперазин-1-ил)пропил)-2-(дифенилметиленамино)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексаноат (14p)
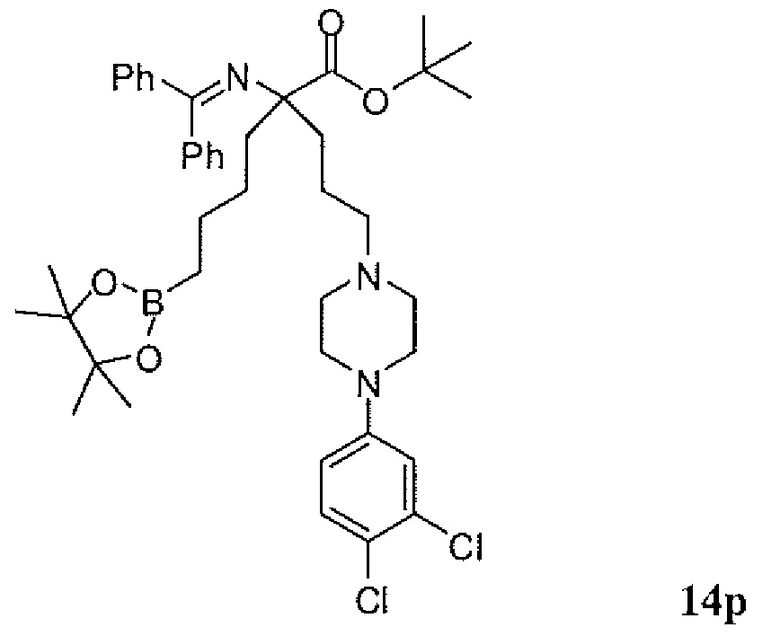
Соединение 14p, 0,302 г (70%), получали, используя общую процедуру E, описанную выше. MS (LC/MS, ESI): 748 (M+H), 692 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, δ): 7,4-8,0 (м, 10Н), 6,8-7,2 (м, 3H), 3,2-3,6 (м, 8H), 2,4-2,5 (м, 4H), 1,6-2,0 (м, 8H), 1,4 (с, 9H), 1,25 (с, 12H), 0,8 (т, 2H).
Наконец, 128 мг (54%) соединения 1p, структура которого продемонстрирована выше, получали, используя общую процедуру O, описанную выше. MS (LC/MS, ESI): 427 (M-H2O+H). 1H ЯМР (300 МГц, D2O, δ): 7,4 (м, 1H), 6,9 (м, 2H), 3,2-3,6 (м, 8H), 2,4-2,5 (м, 4H), 2,0 (м, 4H), 1,3 (м, 4H), 0,8 (т, 2H).
Примеры 17-25
Следующие соединения, перечисленные в таблице 6, ниже, были синтезированы способом, аналогичным тому, который был описан выше для соединения 1p. В таблице 6 каждое соединение имеет следующую химическую структуру (каждый пример в таблице имеет другую группу R1):
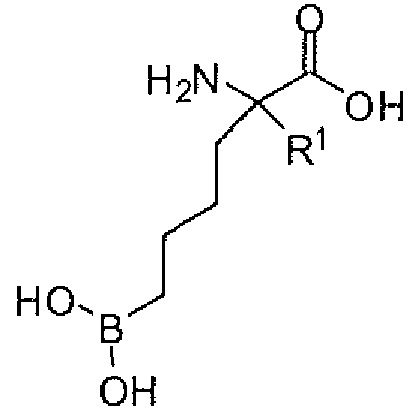
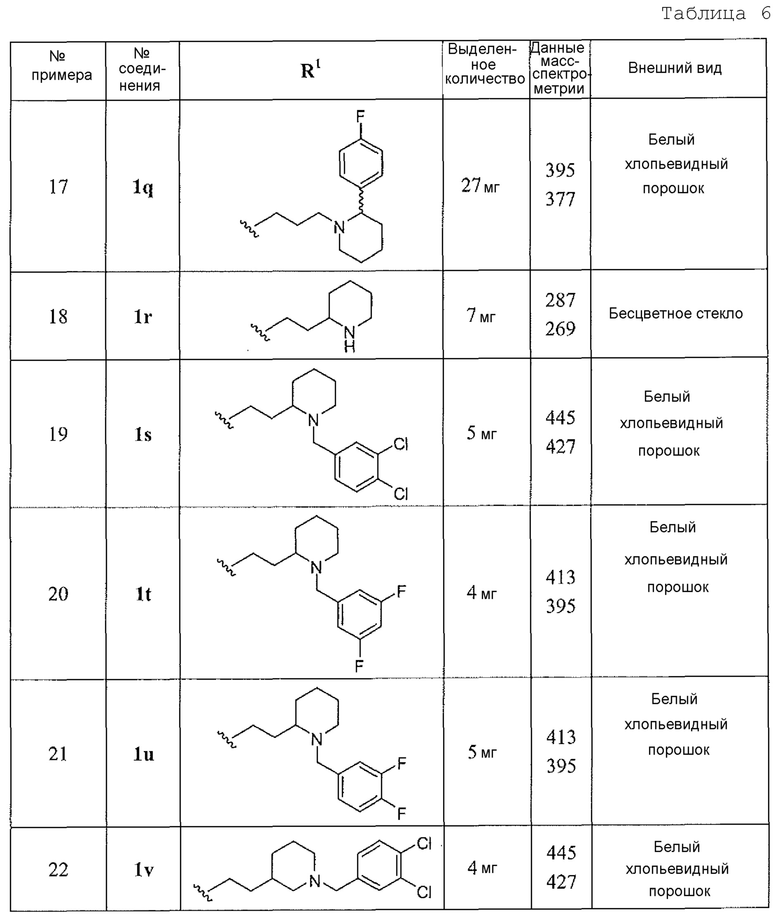
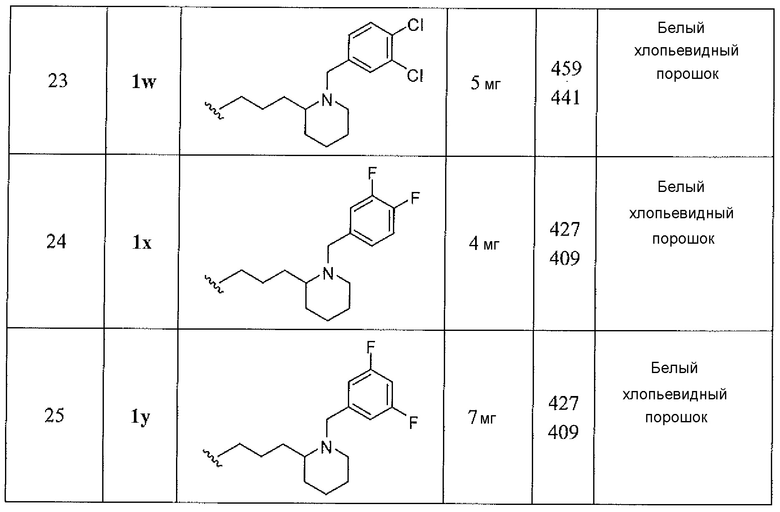
Пример 26: 2-амино-6-бороно-2-(3-(3-фенилпиперидин-1-ил)пропил)капроновая кислота (lz)
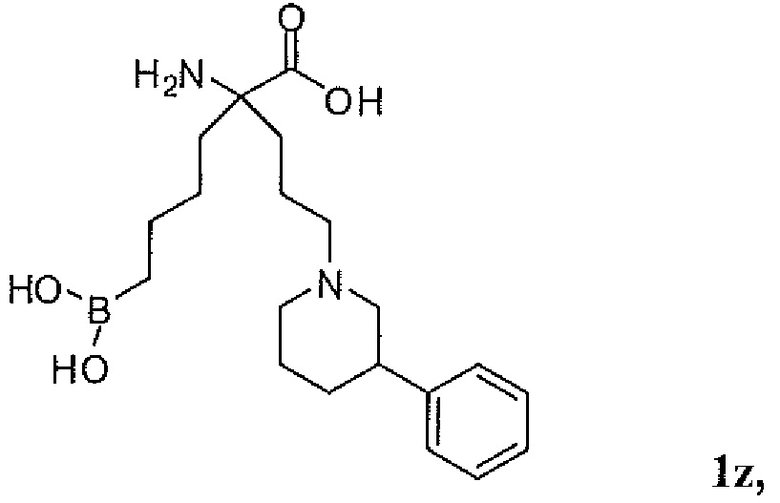
трет-Бутил 5-(трет-бутилдиметилсилилокси)-2-(дифенилметиленамино)пентаноат (24)
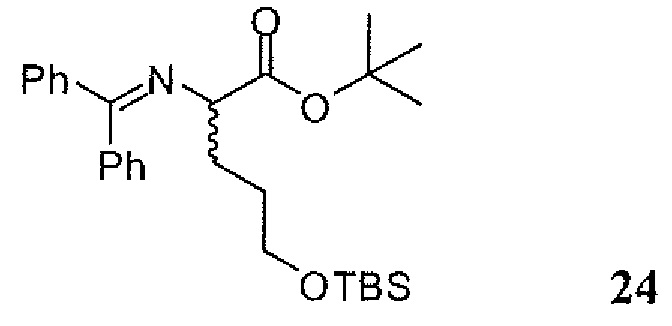
Соединение 24, 13,43 г (82%), получали, используя общую процедуру C, описанную выше. MS (LC/MS, ESI): 468,5 (M+H). 1H ЯМР (300 МГц, CDCl3, δ): 7,2-8,0 (м, 10Н), 3,9 (м, 1H), 3,6 (т, 2H), 2,0 (м, 2H), 1,4-1,6 (м, 2H), 1,4 (с, 9H), 0,9 (с, 9H), 0,05 (с, 6H).
трет-Бутил 2-(3-(трет-бутилдиметилсилилокси)пропил)-2-(дифенилметиленамино)гекс-4-еноат (25)
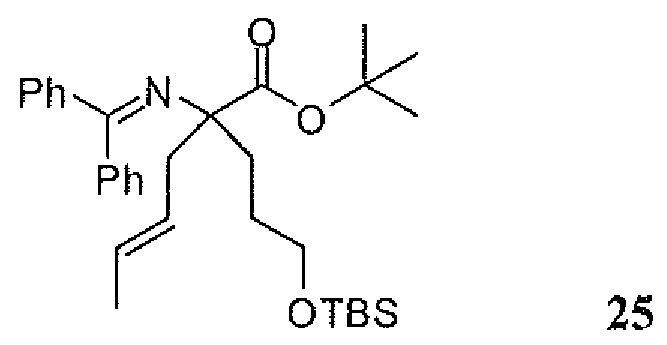
Соединение 25, 11,23 г (75%), получали, используя общую процедуру D, описанную выше. MS (LC/MS, ESI): 522,6 (M+H), 466,5 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, Δ): 7,3-7,8 (м, 10Н), 5,4-5,5 (м, 2H), 3,6 (т, 2H), 2,4-2,6 (м, 2H), 1,7-1,9 (м, 4H), 1,6 (д, 3H), 1,4 (с, 9H), 0,9 (с, 9H), 0,05 (с, 6H).
трет-Бутил 2-(бензилоксикарбониламино)-2-(3-(трет-бутилдиметилсилилокси)пропил)гекс-4-еноат (27)
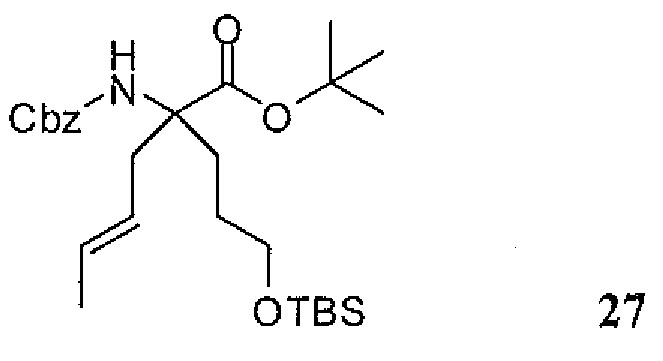
Соединение 27, 3,28 г (59%), получали, используя общую процедуру J, описанную выше. MS (LC/MS, ESI): 436,5 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, δ): 7,3 (с, 5Н), 5,8 (шир.с, 1H), 5,4-5,5 (м, 1Н), 5,2-5,3 (м, 1H), 5,0-5,2 (кв, 2H), 3,5-3,6 (м, 2H), 3,0-3,1 (м, 1H), 2,4-2,5 (м, 1Н), 2,2-2,3 (м, 1H), 1,8-1,9 (м, 1H), 1,6 (д, 3H), 1,4 (с, 9H), 0,9 (с, 9H), 0,05 (с, 6H).
трет-Бутил 2-(бензилоксикарбониламино)-2-(3-(трет-бутилдиметилсилилокси)пропил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексаноат (28)
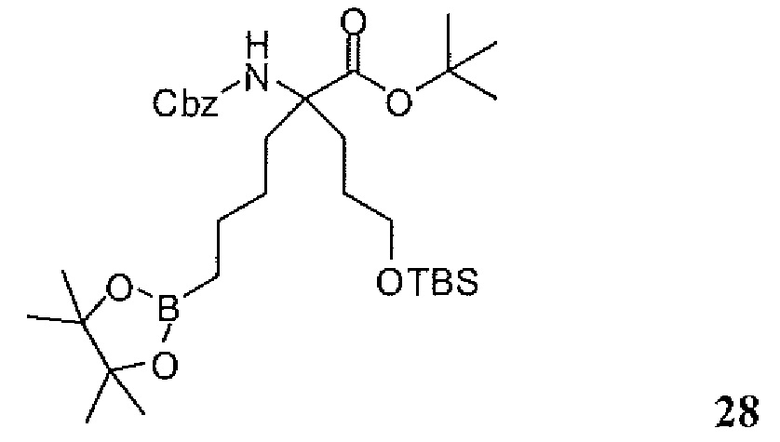
Соединение 28, 2,44 г (59%), получали, используя общую процедуру E, описанную выше. MS (LC/MS, ESI): 620,7 (M+H), 564,6 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, δ): 7,4 (с, 5H), 5,9 (шир.с, 1H), 5,1 (с, 2Н), 3,5-3,6 (м, 2H), 2,2-2,3 (м, 2H), 1,7-1,9 (м, 2H), 1,4 (с, 9H), 1,3-1,5 (м, 6H), 1,25 (с, 12H), 0,9 (с, 9H), 0,7 (т, 2H), 0,05 (с, 6H).
трет Бутил 2-(бензилоксикарбониламино)-2-(3-йодпропил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексаноат (30)
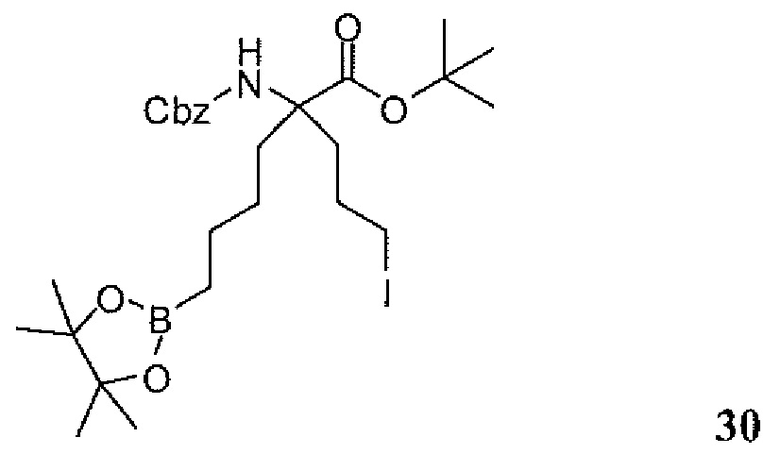
Соединение 30, 0,459 г (93%), получали, используя общую процедуру L, описанную выше. MS (LC/MS, ESI): 616 (M+H), 560 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, δ): 7,2 (с, 5H), 5,8 (шир.с, 1H), 5,0 (с, 2H), 2,9-3,2 (м, 2H), 2,1-2,3 (м, 2H), 1,7-1,9 (м, 3H), 1,4 (с, 9H), 1,3-1,5 (м, 5H), 1,25 (с, 12H), 0,6 (т, 2H).
трет Бутил-2-(бензилоксикарбониламино)-2-(3-(3-фенилпиперидин-1-ил)пропил)-6-(4,4,5,5-тетраметил-l,3,2-диоксаборолан-2-ил)гексаноат (31z)
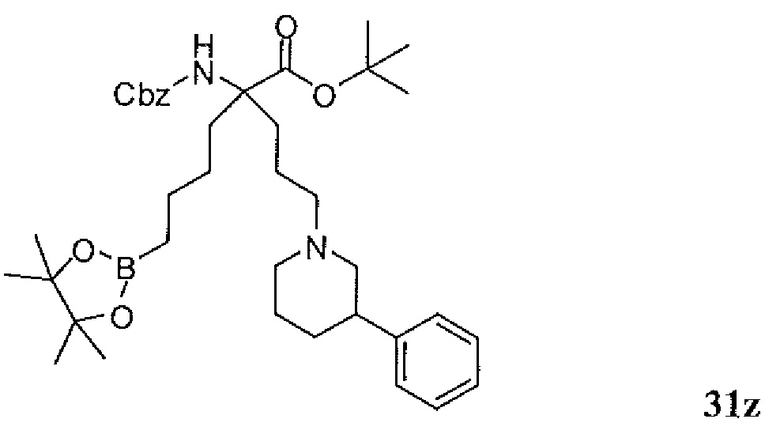
Соединение 31z, 0,134 г в виде неочищенной смеси получали, используя общую процедуру N, описанную выше. MS (LC/MS, ESI): 649 (M+H).
Наконец, 38 мг (42%) соединения 1z, структура которого продемонстрирована выше, получали, используя общую процедуру Q, описанную выше. MS (LC/MS, ESI): 359 (M-H2O+H). 1H ЯМР (300 МГц, D2O, δ): 7,2-7,4 (м, 5H), 2,8 (м, 1H), 2,4-2,5 (м, 6H), 2,0 (м, 4H), 1,4-1,8 (м, 6H), 1,3 (м, 4H), 0,8 (т, 2H).
Примеры 27-98
Следующие соединения, перечисленные в таблице 7, ниже, были синтезированы способом, аналогичным тому, который был описан выше для соединения 1z. Схемы снятия всех защитных групп могут быть различными для конкретных примеров вследствие химического соответствия группам R1 и защитным группам. В таблице 7 каждое соединение имеет следующую химическую структуру (каждый пример в таблице имеет другую группу R1):
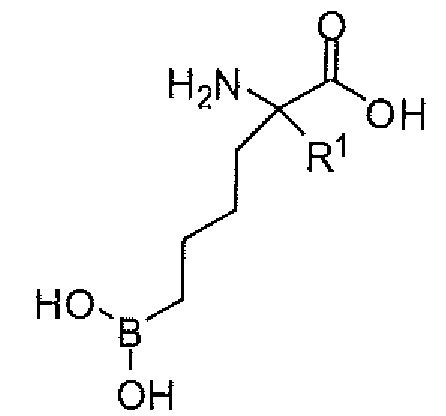
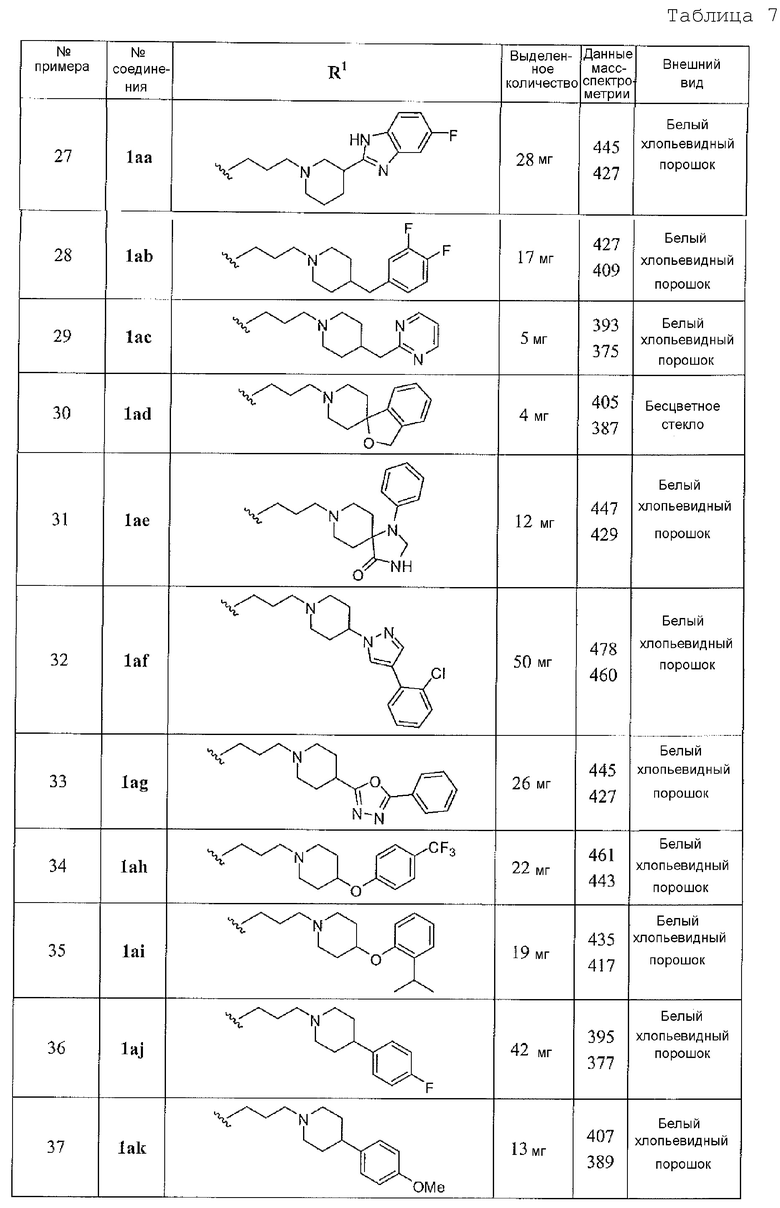
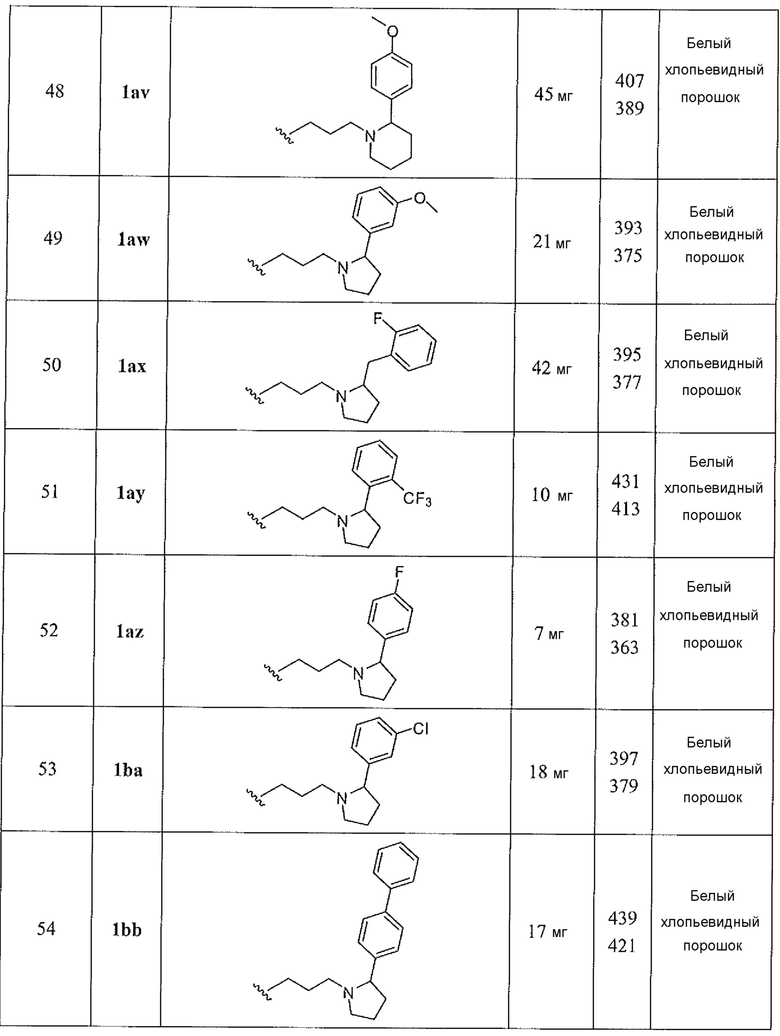
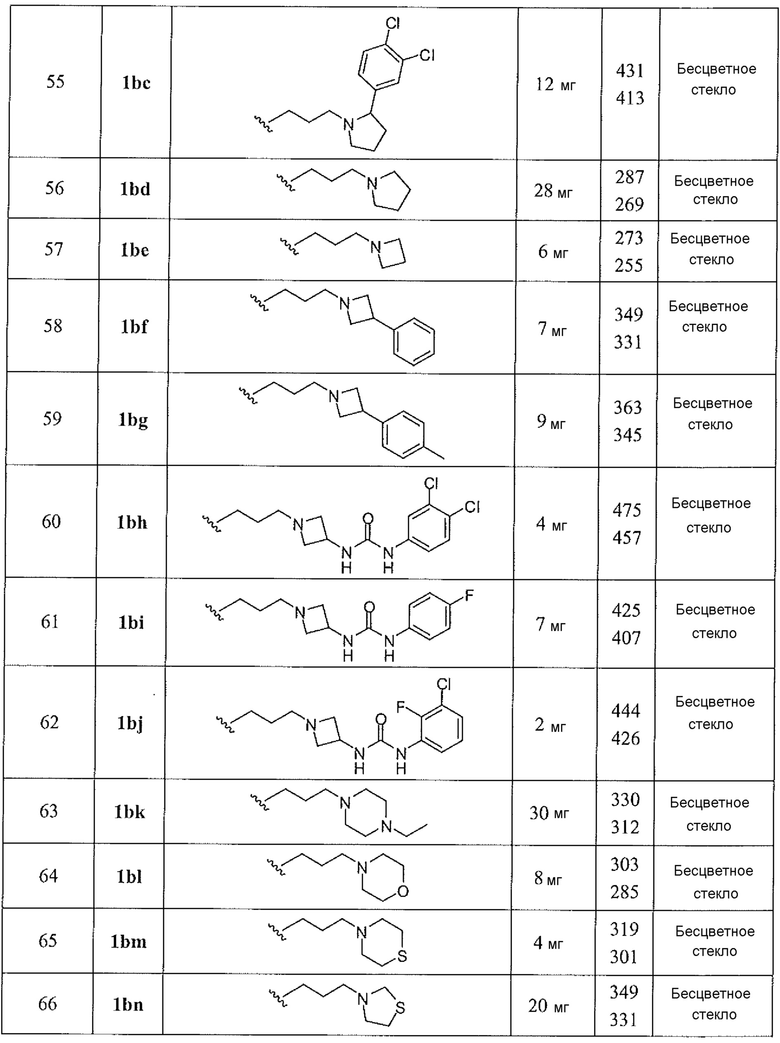
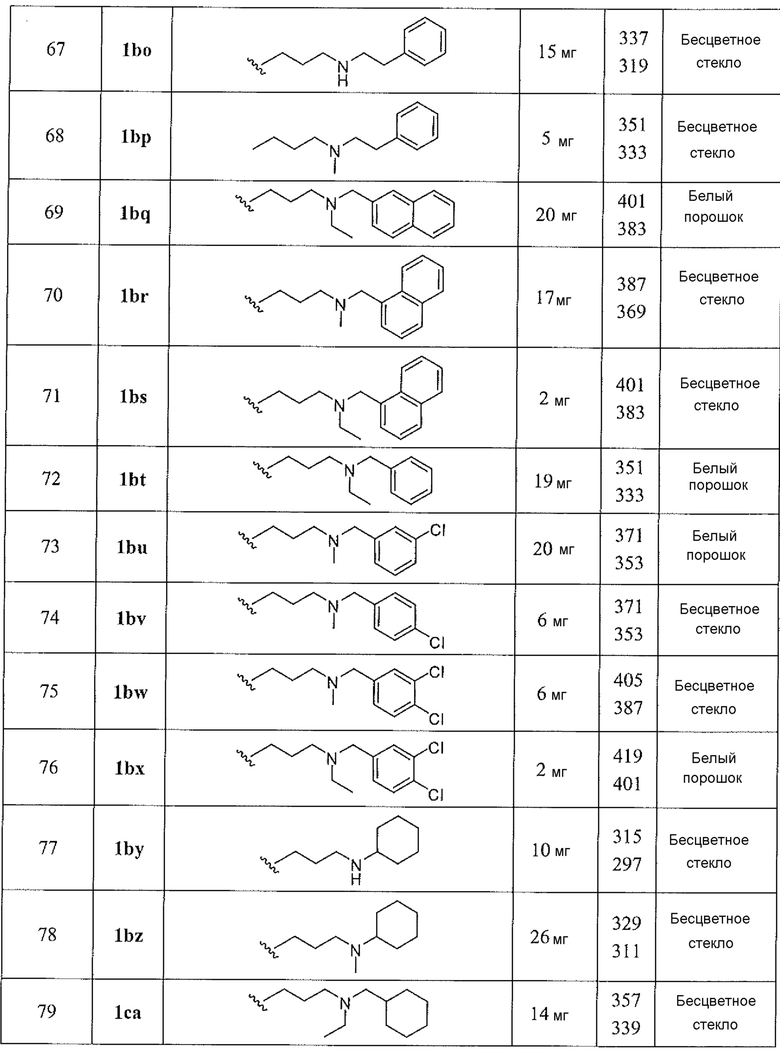
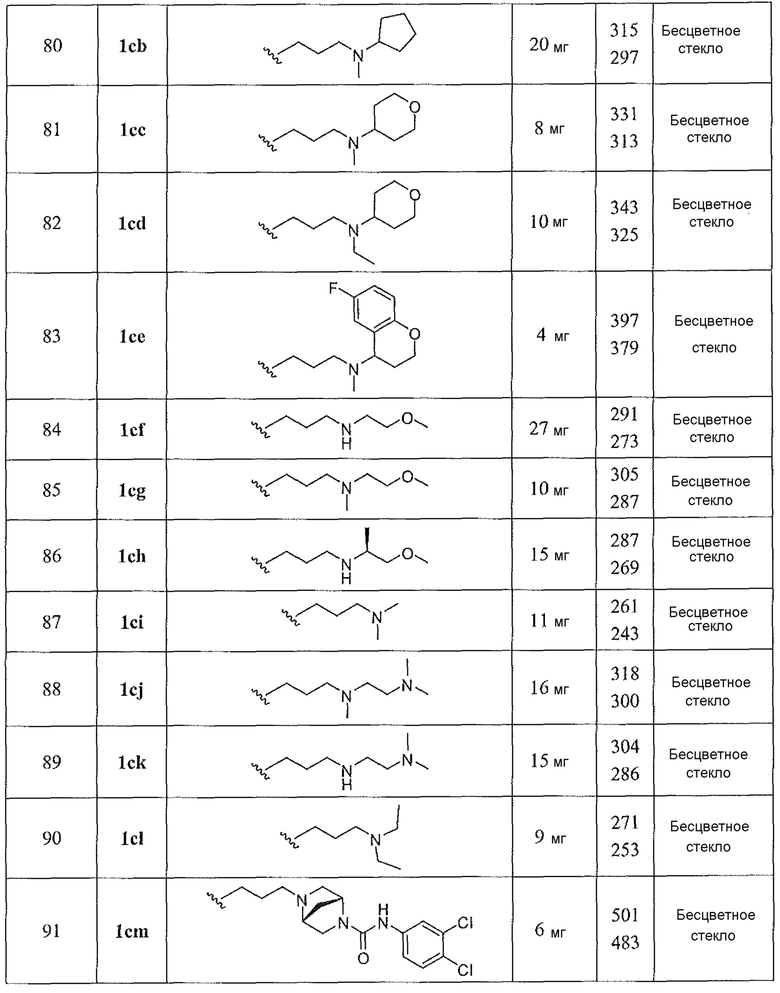
Пример 99: 2-амино-2-(3-(4-(бифенил-4-илметил)пиперазин-1-ил)пропил)-6-боронокапроновая кислота (1cu)
трет-Бутил 4-(бифенил-4-илметил)пиперазин-1-карбоксилат (51)
0,182 г (1,00 ммоль) бифенил-4-карбальдегида и 0,224 г (1,20 ммоль) трет-бутилпиперазин-1-карбоксилата растворяли в 10 мл сухого DCM. Затем 0,35 мл (3,0 ммоль) триметилортоформиата и 0,21 мл (1,50 ммоль) триэтиламина добавляли к реакционной смеси при комнатной температуре на 1 ч. Реакцию гасили раствором NaHCO3 и разбавляли EtOAc. Слои разделяли и водный раствор экстрагировали дважды EtOAc. Органический раствор сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением 0,35 г (100%) 51, которое использовали как есть для следующей реакции.
1-(Бифенил-4-илметил)пиперазин (52)
Растворяли 0,35 г (1,0 ммоль) соединения 51 в 5 мл метанола и 0,5 мл 12 н. HCl при комнатной температуре. Смесь перемешивали в течение 20 мин, добавляли к раствору NaHCO3 и экстрагировали 3× EtOAc. Органический раствор сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением 0,25 г (100%) 52.
(E)-трет-Бутил 2-амино-2-(3-(трет-бутилдиметилсилилокси)пропил)гекс-4-еноат (26)
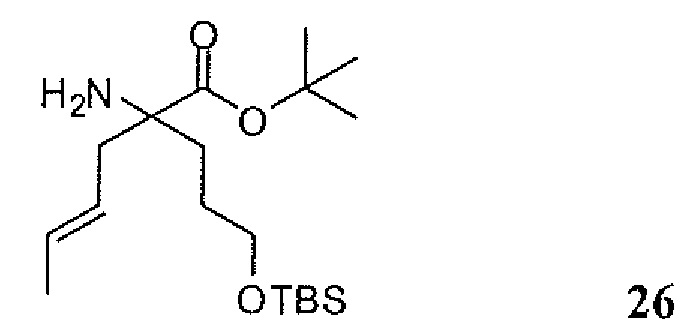
Получали 12,5 г (выход 81%) соединения 26 в виде масла из 40,0 г соединения 25, используя первую часть общей процедуры J, описанной выше. MS (LC/MS, ESI): 358 (M+H), 302 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, δ): 5,5 (м, 2H), 3,8 (т, 2H), 2,4-2,5 (дд, 2H), 2,05 (д, 3H), 1,6-1,8 (м, 4H), 1,38 (с, 9Н), 1,0 (с, 9H), 0,2 (с, 6H).
(E)-трет-Бутил 2-(трет-бутоксикарбониламино)-2-(3-(трет-бутилдиметилсилилокси)пропил)гекс-4-еноат (32)
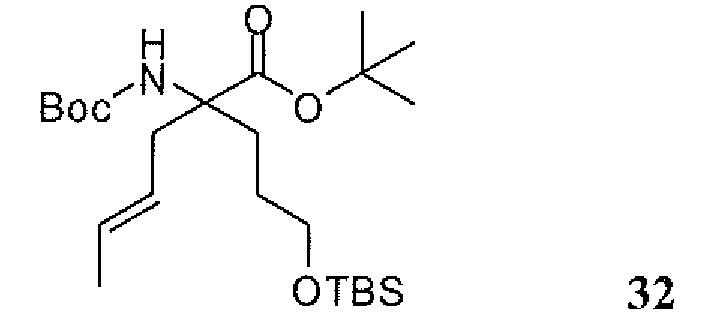
Получали 15,9 г (99%) соединения 32 из 12,5 г соединения 26, используя общую процедуру K, описанную выше. MS (LC/MS, ESI): 458 (M+H), 358 (M-Boc+H). 1H ЯМР (300 МГц, CDCl3, δ): 5,5 (м, 2H), 3,8 (т, 2H), 2,4-2,5 (дд, 2H), 2,05 (д, 3H), 1,6-1,8 (м, 4H), 1,4 (с, 9H), 1,38 (с, 9H), 1,0 (с, 9H), 0,2 (с, 6H).
трет-Бутил 2-(трет-бутоксикарбониламино)-2-(3-трет-бутилдиметилсилилокси)пропил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексаноат (33)
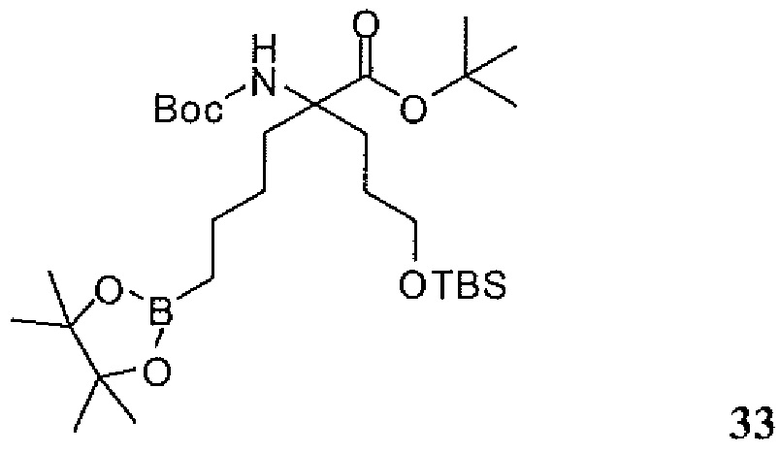
Получали 15,8 г (78%) соединения 33 из 15,9 г соединения 32, используя общую процедуру E, описанную выше. MS (LC/MS, ESI): 586 (M+H), 530 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, δ): 3,8 (т, 2H), 1,5-2,0 (м, 10Н), 1,4 (с, 9H), 1,38 (с, 9H), 1,25 (с, 12H), 1,0 (с, 9H), 0,8 (т, 2H), 0,2 (с, 6H).
трет-Бутил 2-(трет-бутокси)карбониламино)-2-(3-йодпропил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексаноат (34)
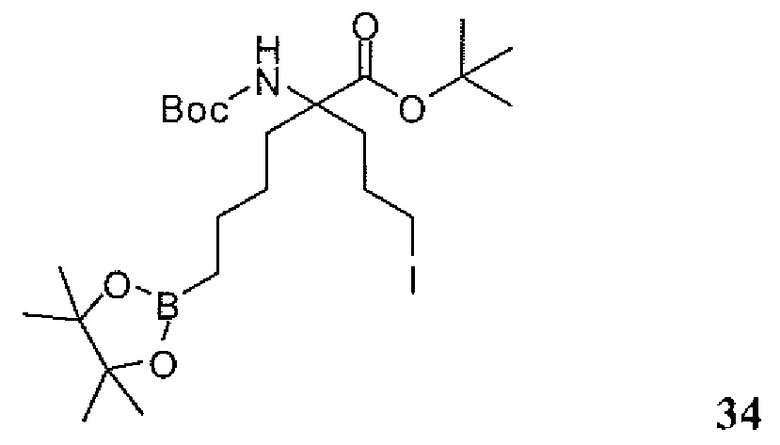
Получали 3,11 г (55%) соединения 34 из 5,77 г соединения 33, используя общую процедуру L, описанную выше. MS (LC/MS, ESI): 583 (M+H), 527 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, δ): 3,2 (т, 2H), 1,5-2,0 (м, 10Н), 1,4 (с, 9H), 1,38 (с, 9H), 1,25 (с, 12H), 1,0 (с, 9H), 0,8 (т, 2H), 0,2 (с, 6H).
трет-Бутил 2-(3-(4-(бифенил-4-илметил)пиперазин-1-ил)пропил)-2-(трет-бутоксикарбониламино)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексаноат (35cu)
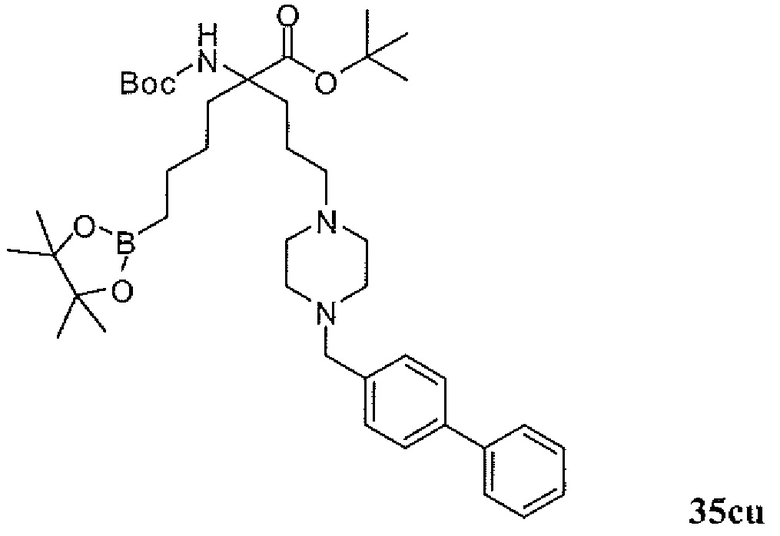
Получали 95 мг (67%) соединения 35cu из 0,116 г соединения 34 и 0,055 г соединения 52, описанных выше, используя общую процедуру N, описанную выше. MS (LC/MS, ESI): 706 (M+H).
Наконец, 30 мг соединения 1cu, структура которого продемонстрирована выше, получали, используя общую процедуру O, описанную выше. MS (LC/MS, ESI): 450 (M-H2O+H). 1H ЯМР (500 МГц, D2O, δ): 7,78 (д, 2H), 7,72 (д, 2H), 7,58 (д, 2H), 7,55 (д, 2H), 7,49 (т, 1H), 4,51 (с, 2Н), 3,70 (шир., 8H), 3,32 (т, 2H), 1,95-1,76 (м, 6H), 1,42 (м, 3H), 1,23 (м, 1H), 0,79 (т, 2H).
Примеры 100-128
Следующие соединения, перечисленные в таблице 8, ниже, были синтезированы способом, аналогичным тому, который был описан выше для соединения 1cu. В таблице 8 каждое соединение имеет следующую химическую структуру (каждый пример в таблице имеет другую группу R1):
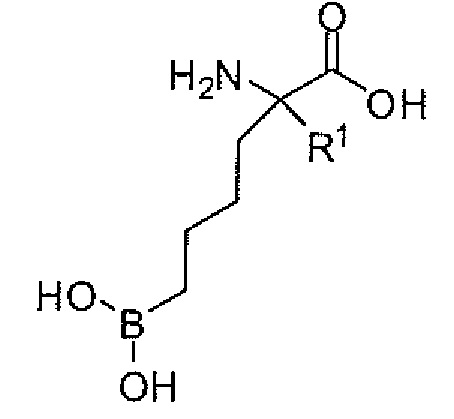
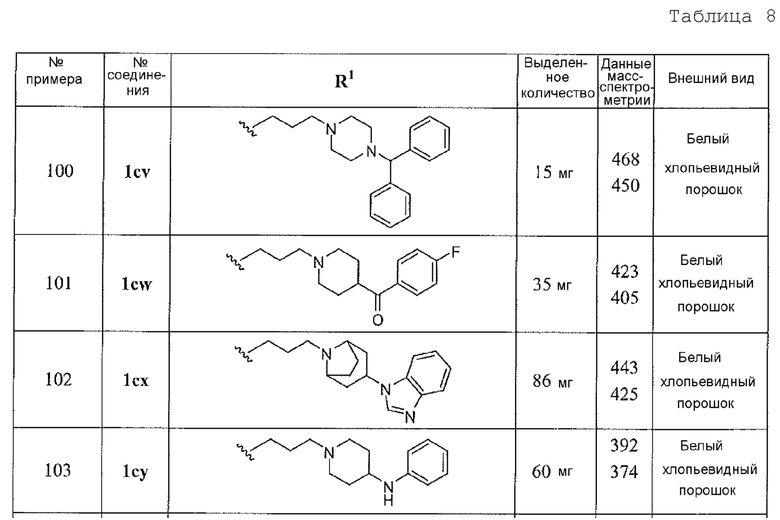
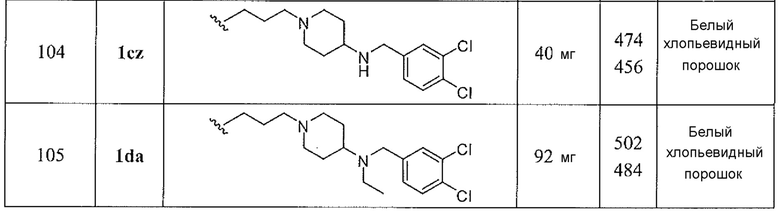
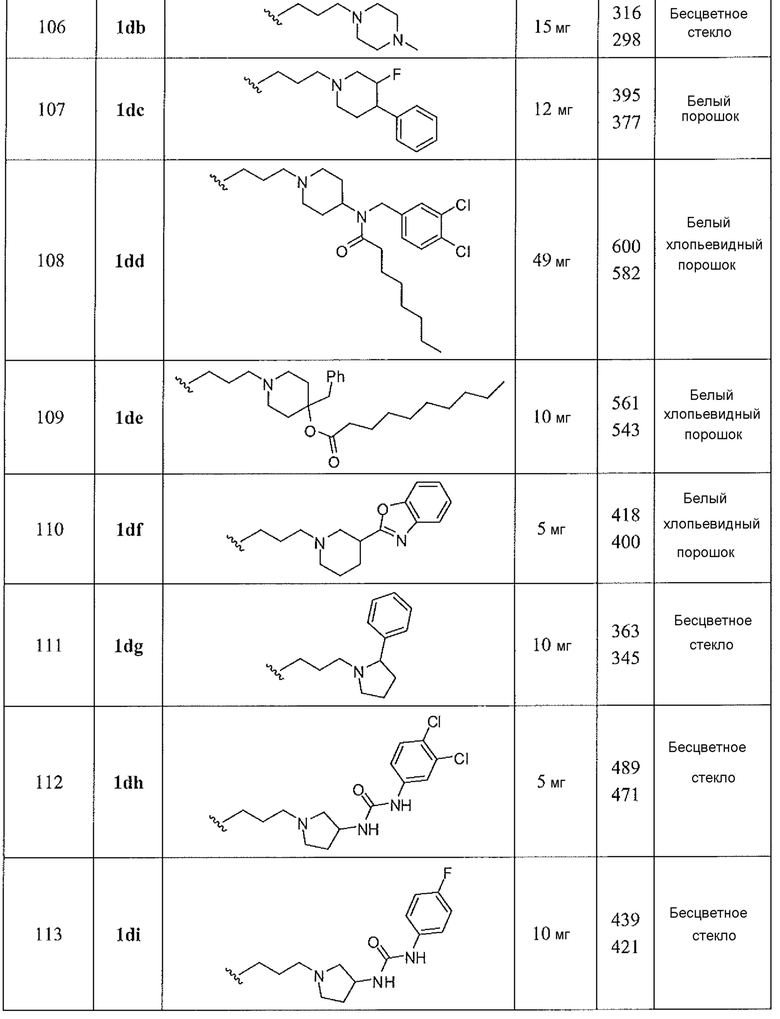
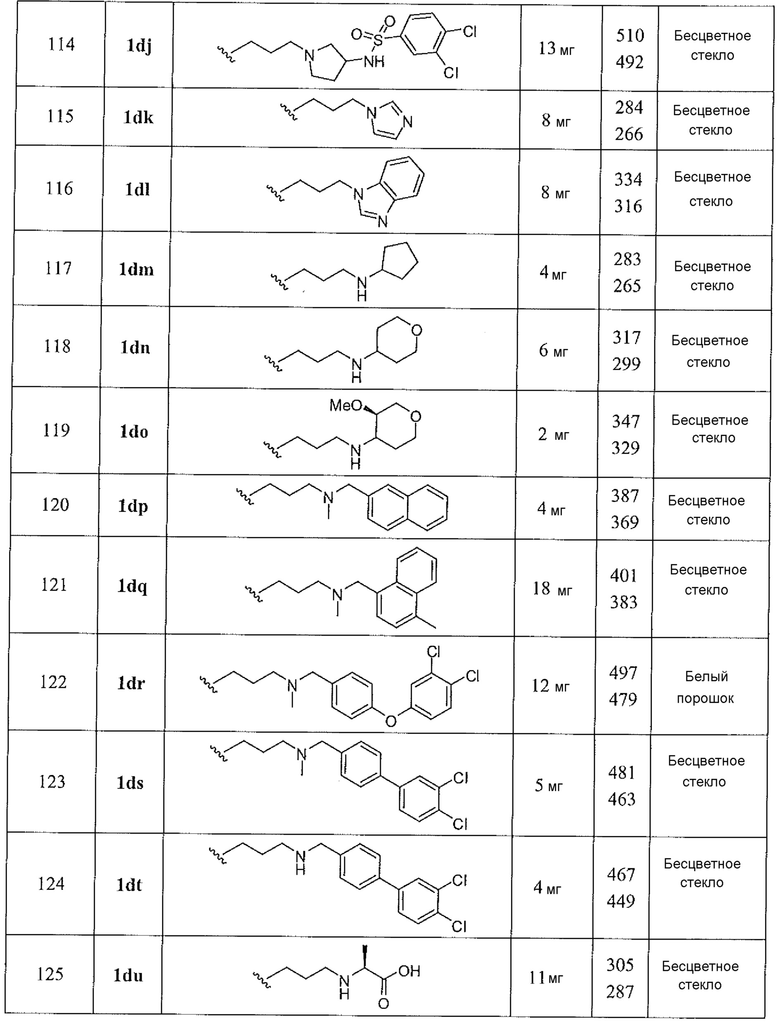
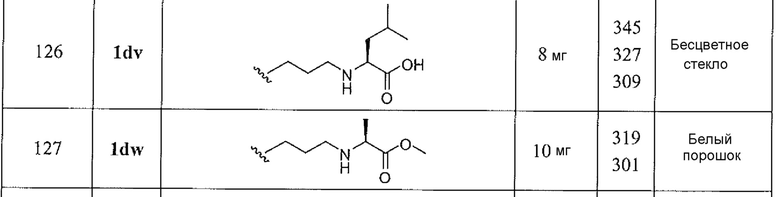
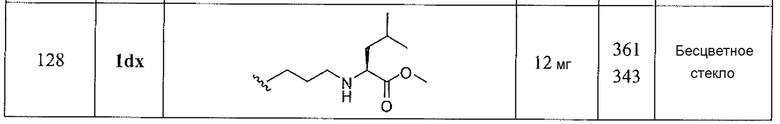
Пример 129: 2-амино-6-бороно-(3-(4-(3,4-дихлорбензоил)пиперазин-1-ил)пропил)капроновая кислота (1dy)
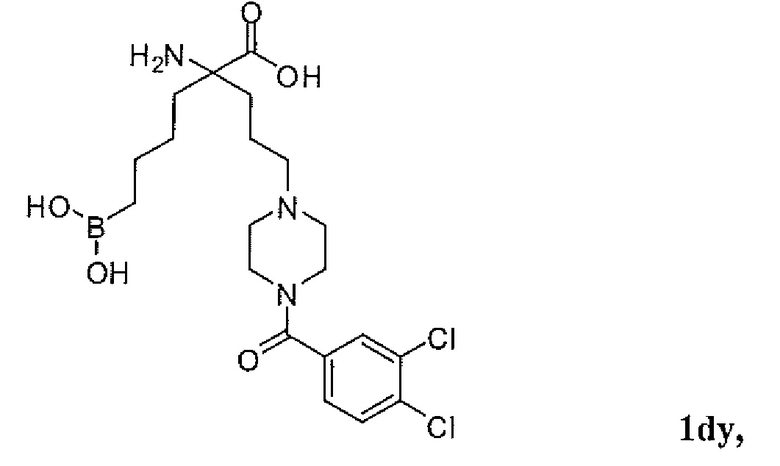
трет-Бутил 2-(3-(4-(3,4-дихлорбензоил)пиперазин-1-ил)пропил)-2-(дифенилметиленамино)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексаноат (17dy)
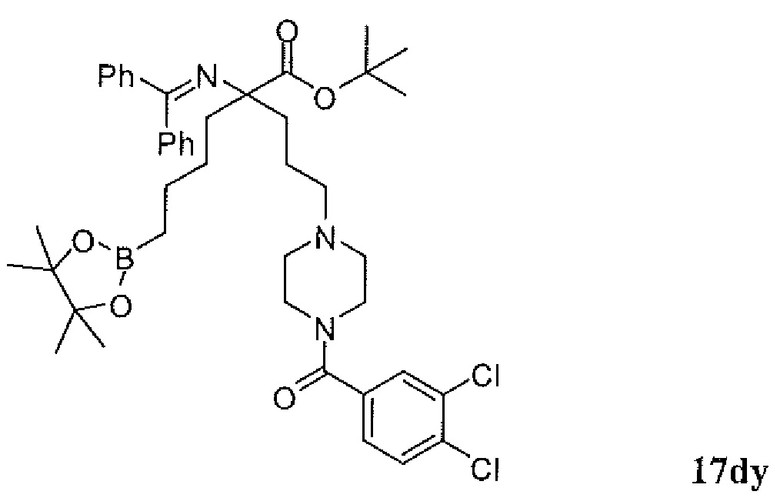
Соединение 17dy, 85 мг получали, используя общую процедуру H, описанную выше. MS (LC/MS, ESI): 776 (M+H).
Наконец, 19 мг соединения 1dy, структура которого продемонстрирована выше, получали, используя общую процедуру O, описанную выше. MS (LC/MS, ESI): 456 (M-H2O+H). 1H ЯМР (300 МГц, D2O, δ): 8,1 (с, 1H), 7,9 (д, 1H), 7,6 (д, 1H), 3,4-3,5 (м, 4H), 2,4-2,5 (м, 6H), 2,0 (м, 6H), 1,3 (м, 4H), 0,8 (т, 2H).
Альтернативно, соединение 1dy можно получить с помощью следующей процедуры.
трет-Бутил 4-(4-(бензилоксикарбониламино)-4-(трет-бутоксикарбонил)-8-(4,4,5,5-5-тетраметил-1,3-2-диоксаборолан-2-ил)октил)пиперазин-1-карбоксилат (19)
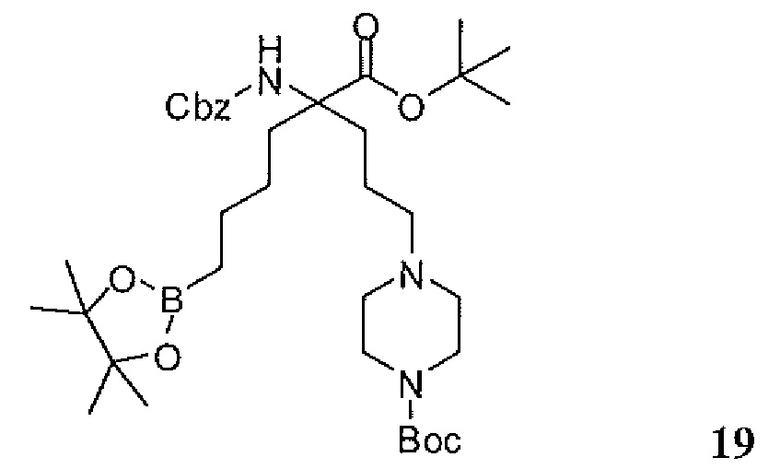
Соединение 19, 319 мг получали, используя общую процедуру I, описанную выше. MS (LC/MS, ESI): 674 (M+H). 1H ЯМР (300 МГц, CDCl3, δ): 7,4 (с, 5H), 5,9 (шир.с, 1H), 5,1 (с, 2H), 3,2-3,6 (м, 8H), 2,2-2,3 (м, 2H), 1,7-1,9 (м, 6H), 1,4 (с, 18H), 1,3-1,5 (м, 4H), 1,25 (с, 12H), 0,7 (т, 2H).
трет-Бутил 2-(бензилоксикарбониламино)-2-(3-пиперазин-1-ил)пропил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексаноат (20)
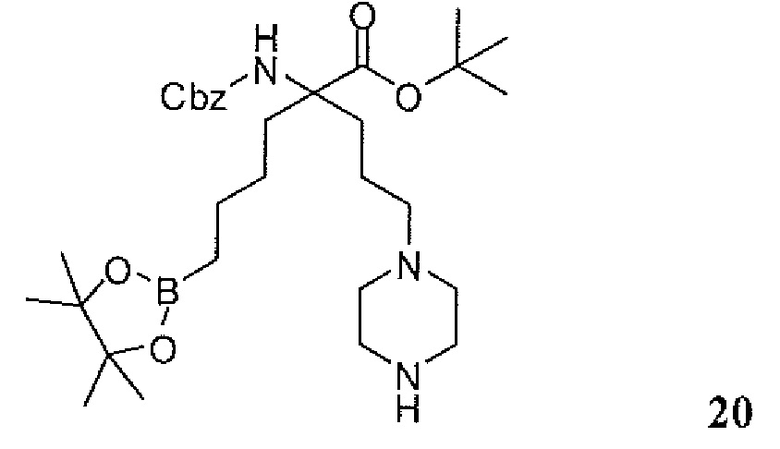
Соединение 20, 82 мг получали, используя общую процедуру F, описанную выше. MS (LC/MS, ESI): 574 (M+H).
трет-Бутил 2-(бензилоксикарбониламино)-2-(3-(4-(3,4-дихлорбензоил)пиперазин-1-ил)пропил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексаноат (22dy)
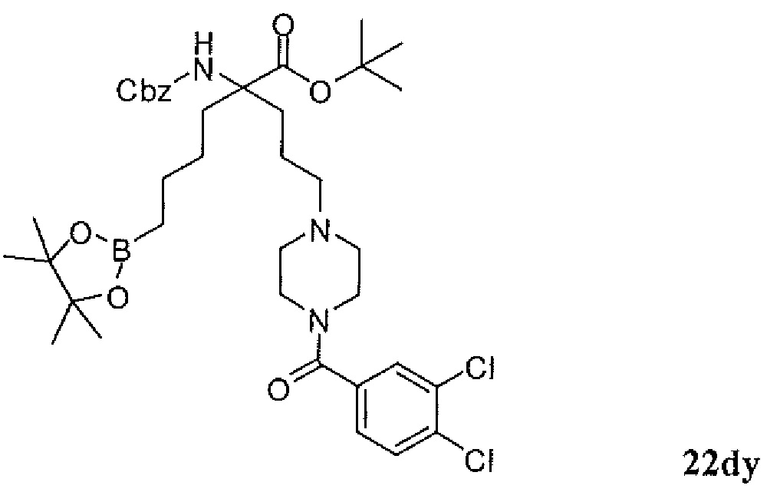
Соединение 22dy, 90 мг получали, используя общую процедуру H, описанную выше. MS (LC/MS, ESI): 746 (M+H).
Наконец, 9 мг соединения 1dy, структура которого продемонстрирована выше, получали, используя общую процедуру Q, описанную выше. MS (LC/MS, ESI): 456 (M-H2O+H). 1H ЯМР (300 МГц, D2O, δ): 8,1 (с, 1H), 7,9 (д, 1H), 7,6 (д, 1H), 3,4-3,5 (м, 4H), 2,4-2,5 (м, 6H), 2,0 (м, 6Н), 1,3 (м, 4H), 0,8 (т, 2H).
Примеры 131-154
Следующие соединения, перечисленные в таблице 9, ниже, были синтезированы способом, аналогичным тому, который был описан выше для соединения 1dy. В таблице 9 каждое соединение имеет следующую химическую структуру (каждый пример в таблице имеет другую группу R1):
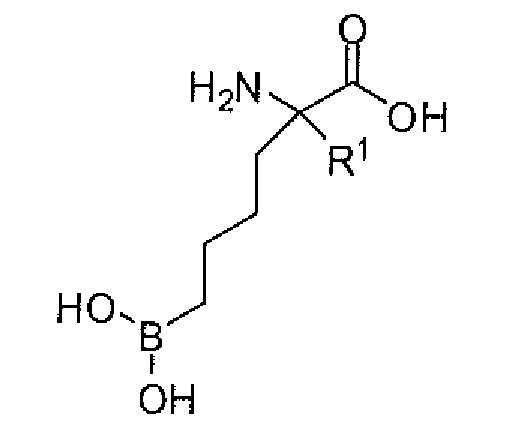
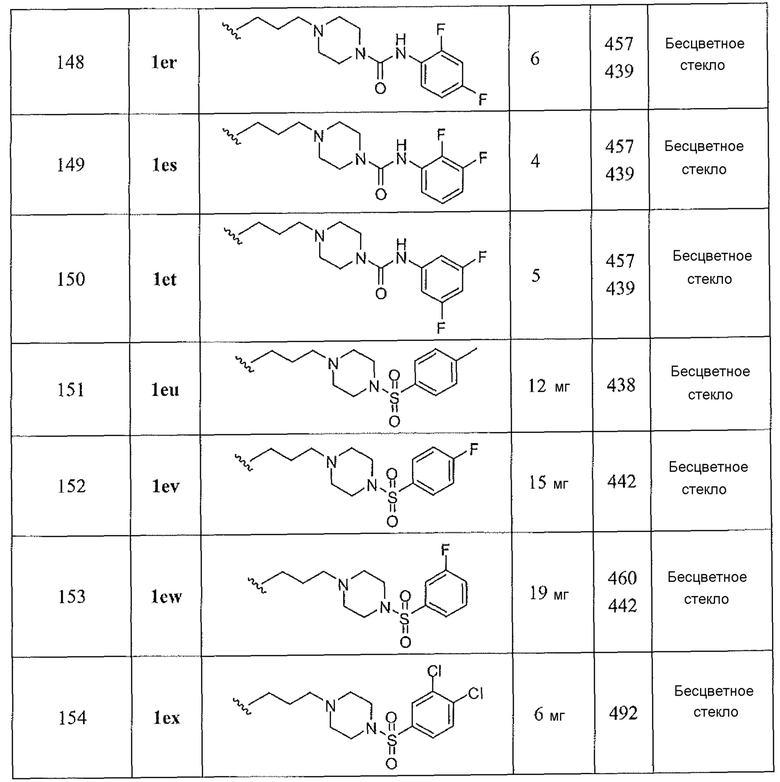
Следующие соединения, перечисленные в таблице 10, ниже, были синтезированы посредством общей процедуры R, S, T или U, описанной выше. Защитные группы, являющиеся интактными после этих реакций, удаляли посредством общей процедуры Q, описанной выше. В таблице 10 каждое соединение имеет следующую химическую структуру (каждый пример в таблице имеет другую группу R2):
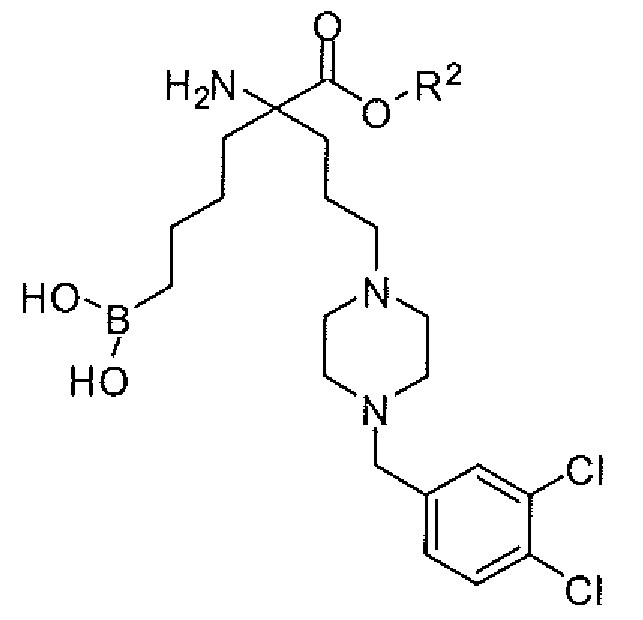
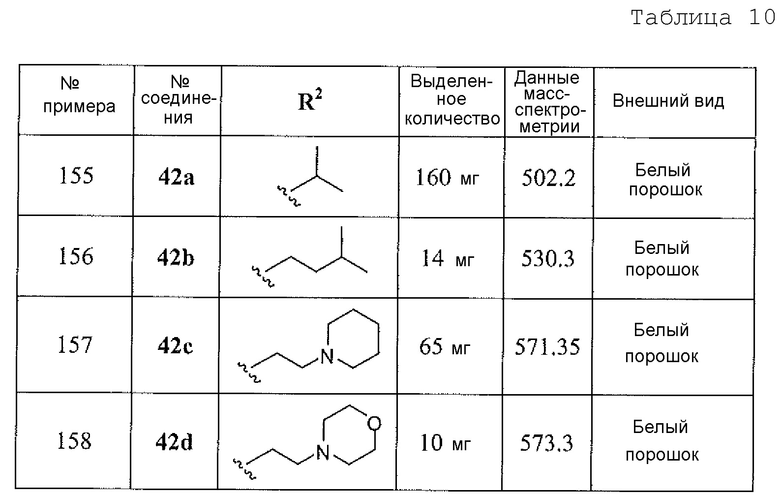
Следующие соединения, перечисленные в таблице 11, ниже, были синтезированы посредством общей процедуры R, S, T или U, описанной выше. Защитные группы, являющиеся интактными после этих реакций, удаляли посредством общей процедуры Q, описанной выше. В таблице 11 каждое соединение имеет следующую химическую структуру (каждый пример в таблице имеет другую группу R2):
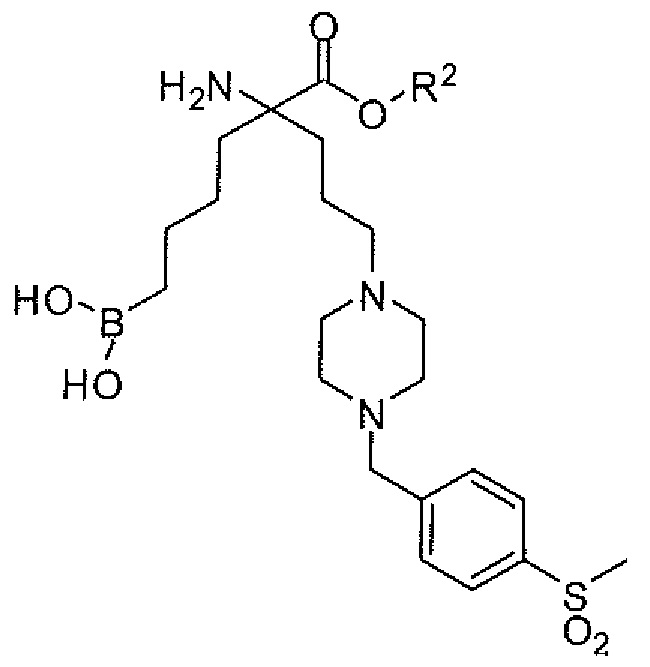
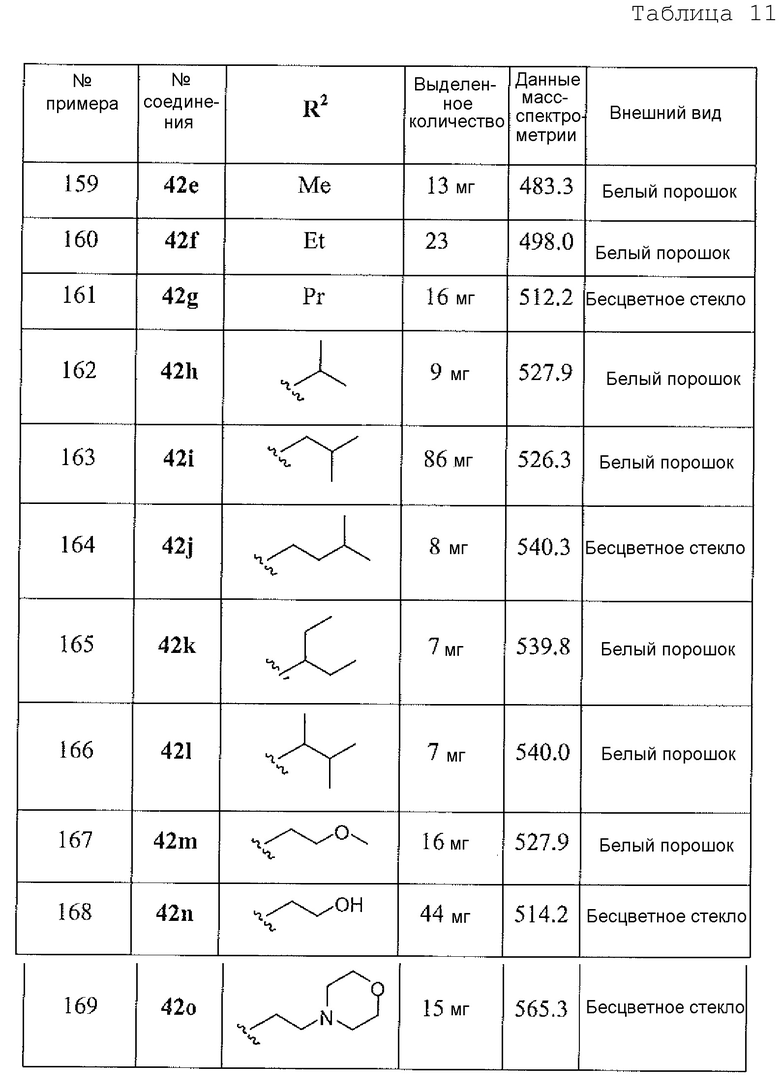
Пример 170. 5-амино-8-(4-(3,4-дихлорфенил)пиперазин-1-ил)-5-(метоксикарбонил)октилбороновая кислота (42p)
трет-Бутил 2-(бензилоксикарбониламино)-2-(3-(4-(3,4-дихлорфенил)пиперазин-1-ил)пропил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексаноат (53)
Соединение 53, 1,277 г (53%), получали, используя общую процедуру N, описанную выше. MS (LC/MS, ESI): 718 (M+H), 662 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, δ): 7,4 (с, 5H), 6,8-7,2 (м, 3H), 5,9 (шир.с, 1Н), 5,1 (с, 2H), 3,2-3,6 (м, 8H), 2,4-2,5 (м, 4H), 1,6-2,0 (м, 8H), 1,4 (с, 9H), 1,25 (с, 12H), 0,8 (т, 2H).
2-Бензилоксикарбониламино)-2-(3-(4-(3,4-дихлорфенил)пиперазин-1-ил)пропил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)капроновая кислота (54)
Соединение 51 обрабатывали 100% ТФУК при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали в вакууме, остаток промывали трижды DCM путем добавления нескольких мл DCM и удаления летучих компонентов на поверхности лужицы. Остаток, соединение 52, 0,125 г (100%), использовали как есть для следующей реакции. MS (LC/MS, ESI): 662 (M+H).
Метил 2-(бензилоксикарбонил)-2-(3-(4-(3,4-дихлорфенил)пиперазин-1-ил)пропил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексаноат (55)
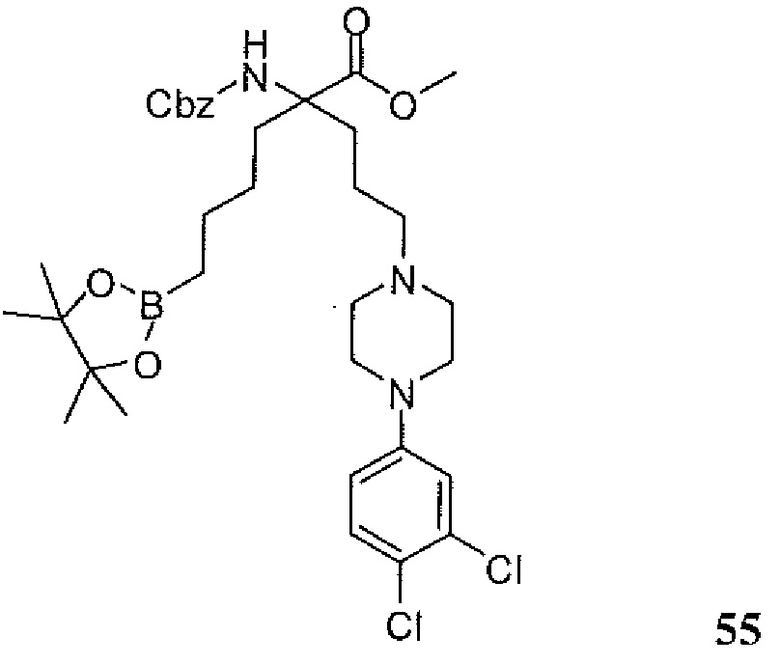
Соединение 54 (0,11 г, 0,163 ммоль) растворяли в 5 мл сухого толуола/метанола 1:1 в атмосфере аргона. TMS-диазометан в диэтиловом эфире добавляли по каплям при перемешивании при комнатной температуре до образования желтого раствора (всего 0,4 мл, 0,8 ммоль) и смесь перемешивали в течение еще 30 мин. Ледяную уксусную кислоту добавляли по каплям до исчезновения желтого цвета и реакционную смесь концентрировали в вакууме. Этот остаток элюировали на колонке с силикагелем, используя смеси EtOAc/гексаны (20-50%), с получением 90 мг (80%) соединения 55 в виде масла. MS (LC/MS, ESI): 718 (M+H), 662 (M-tBu+H). 1H ЯМР (300 МГц, CDCl3, δ): 7,4 (с, 5H), 6,8-7,2 (м, 3H), 5,9 (шир.с, 1H), 5,1 (с, 2H), 3,8 (с, 3H), 3,2-3,6 (м, 8H), 2,4-2,5 (м, 4H), 1,6-2,0 (м, 8H), 1,25 (с, 12H), 0,8 (т, 2H).
Наконец, 25 мг соединения 42p, структура которого продемонстрирована выше, получали, используя общую процедуру V. MS (LC/MS, ESI): 460 (M+H).
В таблице 12 демонстрируется неограничивающий набор соединений настоящего изобретения
Пример 171. Эффект рН в случае соединений настоящего изобретения
Ингибиторную активность выбранных соединений настоящего изобретения в отношении Arg I и/или Arg II определяли при значениях pH, равных 7,5 и 9,5. В одном варианте осуществления установили, что в случае соединений имеется «эффект рН», определенный в описании, если отношение активности при pH 7,5 по отношению к pH 9,5 составляет по меньшей мере 0,5. В случае некоторых соединений их ингибиторную активность при pH 7,5 сравнивали с ингибиторной активностью ABH при pH 7,5. Некоторые данные представлены в таблице 3.
Пример 172. Биологическое исследование ингибирования аргиназы
Количественное определение аргиназной активности выполняли в 96-луночном формате колориметрическим способом, используя набор для исследования аргиназы QuantiChrom™, доступный от BioAssay Systems (Hayward, California, каталожный № DARG-200), в соответствии с протоколом производителя. Были внесены изменения в количества реагентов для соответствия исследованию в высокой степени очищенного образца.
Кратко, в способе использовался хромоген, который образует окрашенный комплекс специфически с мочевиной, продуцируемой в аргиназной реакции (Mellerup, 1967, Clin. Chem. 13:900-08). Интенсивность окраски была прямо пропорциональна аргиназной активности в образце.
Степень продукции мочевины определяли в присутствии двенадцати различных концентраций каждого соединения - потенциального ингибитора. В типичном исследовании семь различных соединений проверяли относительно контрольного ингибитора (например, ABH) против одного фермента (или Arg I человека, или Arg II человека) при конкретном pH (или 9,5, или 7,5). Значение pH раствора регулировали, используя соответствующее pH-буферное соединение в концентрациях 60 мМ; например, 60 мМ глицин (pH 9,5) или 60 мМ HEPES (pH 7,5). Полумаксимальную ингибиторную концентрацию (IC50) определяли путем построения кривой зависимости эффекта от дозы. Значения IC50 зависят от условий измерения, значения IC50 преобразовывали в сродство связывания ингибитора (Ki), используя уравнение Ченга-Прусоффа и константу связывания по сродству (Km) для L-аргинина, которую определяли с помощью того же исследования активности (см., например, Cheng et al., 1973, Biochem. Pharmacol. 22:3099-108).
Сродства связывания ингибиторов для обеих аргиназ I и II («hArgI» и «hArgII», соответственно) представлены ниже в таблице 1 в описании в другом месте.
Пример 173. Ишемия миокарда-реперфузионное повреждение
Крыс внутрибрюшинно (IP) анестезировали с воздействием пентобарбитала (~50 мг/кг), брили и размещали в положении на спине с согнутыми ногами, интубировали и вентилировали (~90 вздохов/мин, ~2,5 мл дыхательный объем с использованием 95% О2/5% CO2), используя регулируемый небольшой вентилятор для животных (Harvard Apparatus). Анестезию поддерживали с помощью постоянной инфузии пентобарбитала натрия (для воздействия, ~3-5 мг/кг/ч, внутривенно) до завершения исследования через постоянный катетер, вставленный в периферическую вену (например, бедренную). Температуру тела постоянно контролировали на всем протяжении хирургических/экспериментальных процедур посредством ректального зонда и поддерживали в физиологическом диапазоне посредством нагреваемого, с контролем температуры (с обратной связью) небольшого хирургического столика для животных/блока регулирования (Vestavia Scientific).
После этого размещали трансторакальные игольчатые электроды для формирования ЭКГ в одном отведении (например, в отведении II). После достижения хирургического уровня анестезии бедренную артерию выделяли, отслаивали от окружающей ткани и канюлировали с использованием 2F высококачественного микроманометрического катетера (Millar Instruments). Для регистрации артериального/системного давления этот катетер продвигали к брюшной аорте. Постоянный катетер также вводили в вену (т.е. яремную) для введения исследуемого продукта/носителя, или красителей (см. ниже).
Наконец, животных размещали на правом боку и сердце обнажали с помощью левосторонней торакотомии/перикардиотомии. Вокруг проксимального отдела левой передней нисходящей артерии (LAD) накладывали неплотно лигатуры (используя колющие иглы). Затягивание этих хирургических петель (посредством небольших кусков полиэтиленовой трубки) приводило к временной ишемии части миокарда.
После хирургического приготовления животным давали возможность достичь гемодинамической стабильности (в течение приблизительно 15 мин) и получали данные на исходном уровне. Следует отметить, что для обеспечения однородности экспериментальных данных все животные должны были удовлетворять следующим критериям вхождения в исследование: частота сердцебиений >320 ударов в минуту и среднее артериальное давление >80 мм рт.ст. Схему анестезии можно регулировать для обеспечения надлежащей анестезии/обезболивания и для удовлетворения таким критериям включения.
После гемодинамической стабилизации и измерений на исходном уровне животных лечили или носителем, или исследуемым продуктом, доставляемым в виде внутривенной болюсной инъекции. Впоследствии (приблизительно через 15 мин после введения дозы) животных подвергали острому ишемическому инсульту в течение 30 мин посредством затягивания хирургической петли вокруг коронарной артерии LAD. Ишемию миокарда визуально подтверждали по появлению цианотичных изменений в дистальных локализациях LAD, а также по возникновению электрокардиографических изменений. Через приблизительно 30 мин вызванной ишемии хирургические петли вокруг коронарной артерии ослабляли и осуществляли реперфузию через ранее подвергнутый ишемии миокард в течение вплоть до 2 ч. Лечения не проводили ни в одном из периодов ишемии и/или реперфузии. Следует также отметить, что для минимизации какого-либо возможного смешивания эффектов на вызовы повреждения миокарда несамоустраняющиеся опасные аритмии/ритмы (например, вентрикулярную тахикардию/фибрилляцию), развивающиеся во время реперфузии, считали концом (т.е. эксперимент преждевременно прекращали).
На этапе завершения протокола оценивали необратимое повреждение миокарда (т.е. инфаркт), являющееся следствием I/R инсульта. Кратко, хирургические петли вокруг коронарных артерий вновь затягивали и внутривенно инъецировали синий краситель Эванса (1 мл/кг; Sigma, St. Louis, MO) для определения площади миокарда, подверженной риску (AR) во время ишемии. После этого сердце быстро извлекали, промывали холодным физиологически раствором, взвешивали, заворачивали в пищевую пленку и помещали в морозильную камеру приблизительно на 30 минут. Сердце извлекали из морозильной камеры, снова взвешивали и делали поперечные срезы; отбирали 2-3 коротких осевых сегментов (толщиной ~1,5 мм) от верхушки до основания. Срезы последовательно пронумеровывали, при этом «срез #1» был самым апикальным, и фотографировали/сканировали. После этого срезы инкубировали в течение приблизительно 15 минут в 1% хлориде трифенилтетразолия (TTC) приблизительно при 37°C и фиксировали в 10% нейтральном забуференном растворе формалина в течение приблизительно 60 минут.
После фиксации инфарктные и подверженные риску площади определяли и измеряли в цифровом формате. С такой целью толщину каждого среза измеряли с помощью микрометра и позже фотографировали/сканировали. Все фотографии импортировали в программу для анализа изображений (Image J; National Institutes of Health) и выполняли планиметрию с использованием вычислительной техники для определения общего размера инфаркта (IA) и подверженной риску площади (AR). Для каждого среза размер инфаркта (IA, не окрашенная ткань) представляли в виде процента от AR (IA/AR). Следует отметить, что во всех случаях количественную гистоморфометрию выполнял персонал, лишенный информации о назначении лечения/дизайне исследования. Результаты резюмированы в таблице 13.
Описания каждого и всякого патента, заявки на патент и публикации, приведенных в описании, тем самым включены в описание посредством ссылки в их полных объемах.
Хотя настоящее изобретение было раскрыто с учетом конкретных вариантов осуществления, очевидно, что другие варианты осуществления и вариации этого изобретения могут быть разработаны квалифицированными в данной области техники специалистами, не выходя за пределы истинной сущности и объема настоящего изобретения. Прилагаемая формула изобретения, как предполагается, рассматривается как включающая все такие варианты осуществления и эквивалентные вариации.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ АРГИНАЗЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2011 |
|
RU2586219C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНИЛ-АМИНОПИРИДИНОВЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ РАКА | 2010 |
|
RU2619463C2 |
| ТРИАЗОЛОПИРИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2561104C2 |
| АЦИЛИРОВАННЫЕ АРИЛЦИКЛОАКИЛАМИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИХ СРЕДСТВ | 2003 |
|
RU2337094C2 |
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ NAMPT | 2011 |
|
RU2617424C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ | 2002 |
|
RU2294326C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИМИДАЗОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2004 |
|
RU2330023C2 |
| КОМБИНАЦИЯ АНТАГОНИСТА С-МЕТ И АМИНОГЕТЕРОАРИЛА ДЛЯ ЛЕЧЕНИЯ РАКА | 2009 |
|
RU2526171C2 |
| ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ПРОТИВ ДИАБЕТА | 2006 |
|
RU2438671C2 |












Группа изобретений относится к области медицины, а именно к соединению, выбранному из:
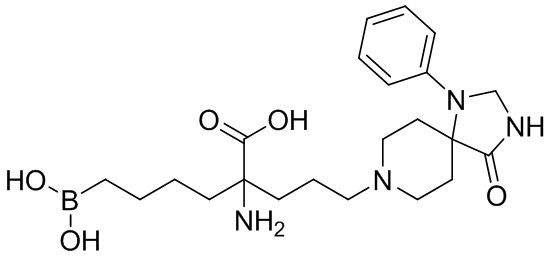 ,
, 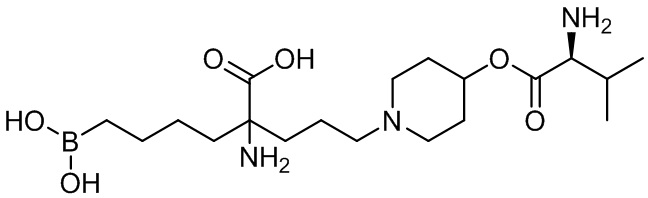 ,
, 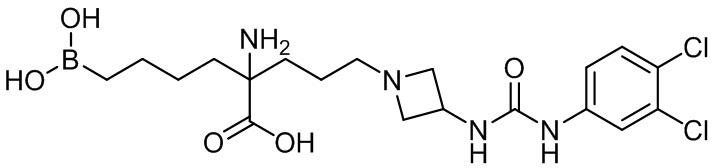 ,
,  ,
,  ,
, 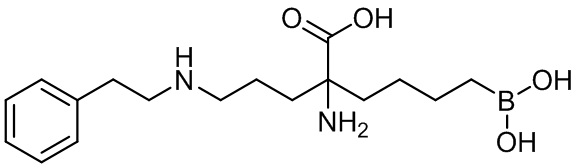 ,
, 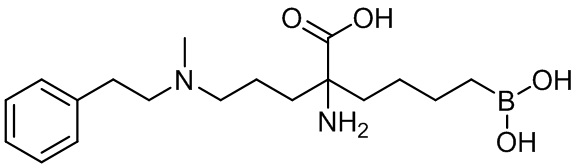 ,
, 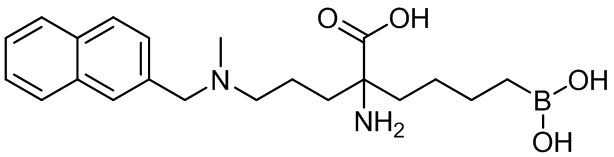 ,
, 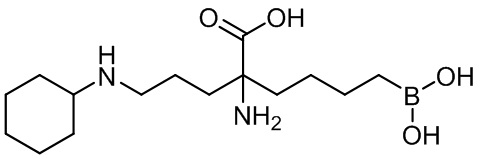 ,
, 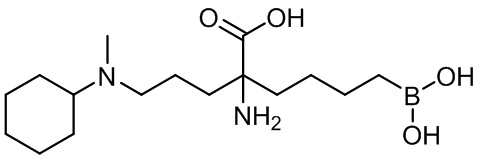 ,
, 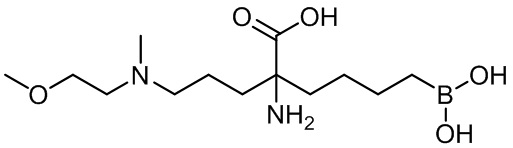 ,
, 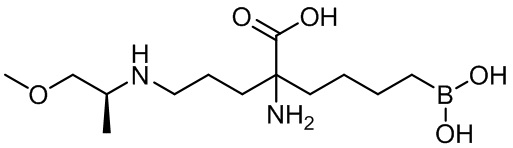 ,
, 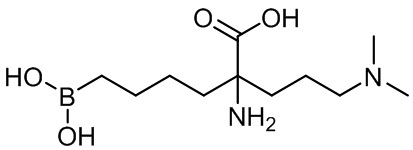 ,
, 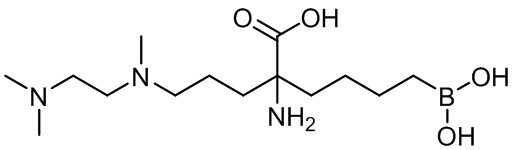 ,
, 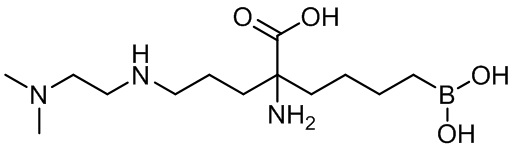 ,
, 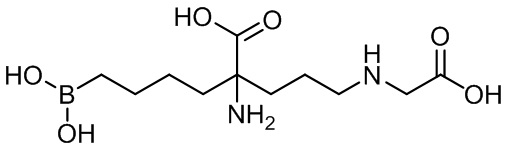 ,
, 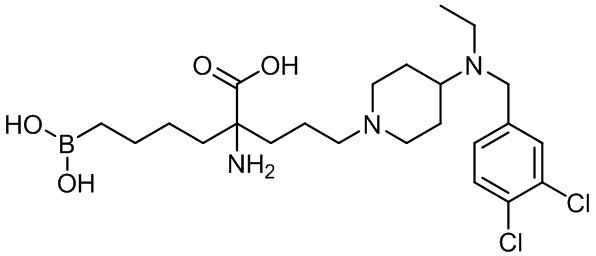 ,
, 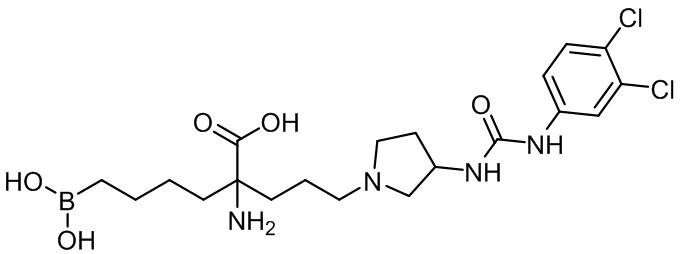 ,
, 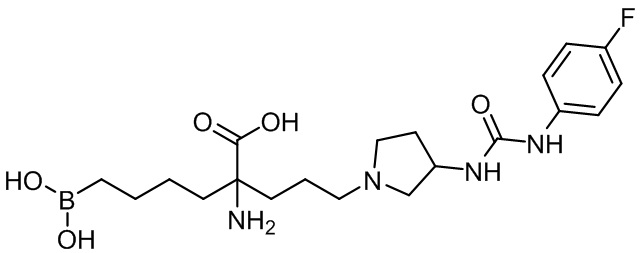 ,
, 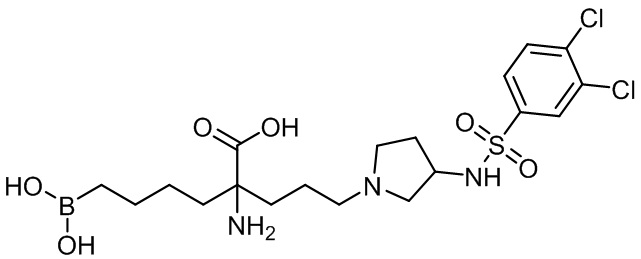 ,
,
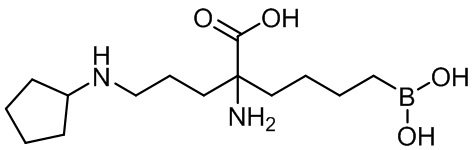 ,
, 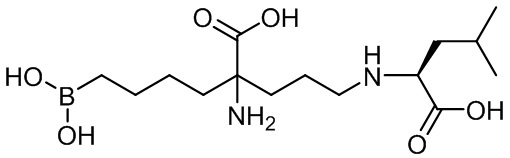 ,
,
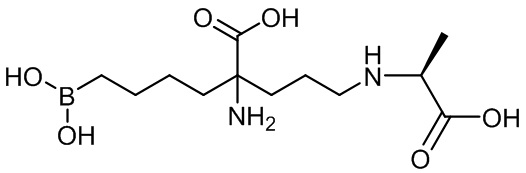 ,
, 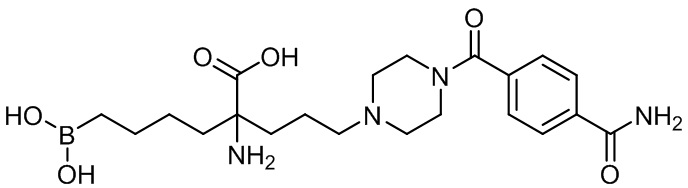
или его фармацевтически приемлемой соли, к применению вышеперечисленных соединений для лечения заболеваний, выбранных из группы, состоящей из хронического инфекционного заболевания, бактериальной инфекции, паразитарной инфекции, травмы, лепры, туберкулеза, трансплантации печени, рака, фиброзных заболеваний и их комбинации, и к фармацевтической композиции, включающей вышеперечисленные соединения. Группа изобретений обеспечивает получение ингибиторов аргиназной активности, которые могут использоваться для лечения заболевания у млекопитающего, причем заболевание характеризуется либо необычно высокой аргиназной активностью, либо необычно низкими уровнями оксида азота в ткани млекопитающего. 3 н.п. ф-лы, 14 ил., 13 табл., 173 пр.
1. Соединение, выбранное из:
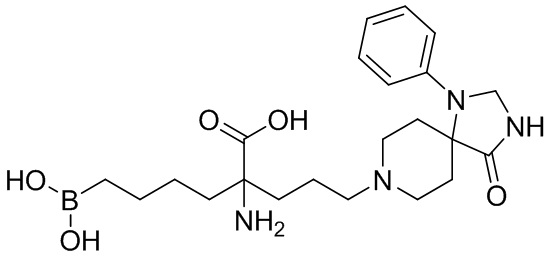 ,
,
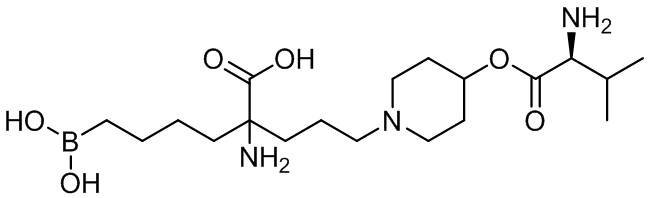 ,
, 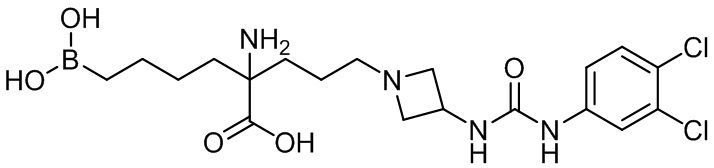 ,
, 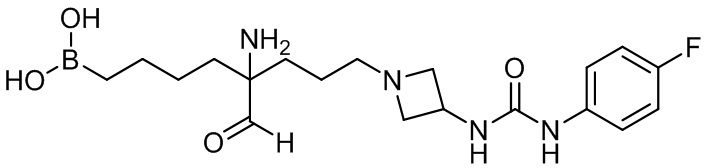 ,
, 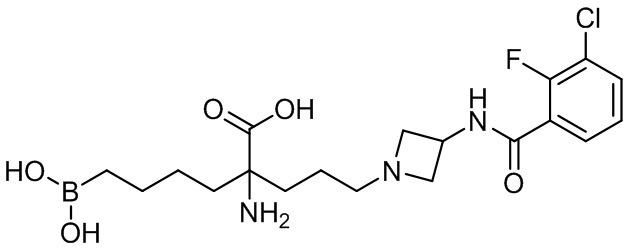 ,
, 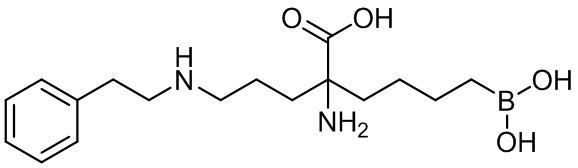 ,
, 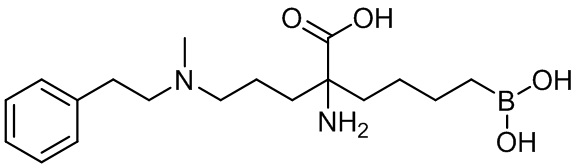 ,
, 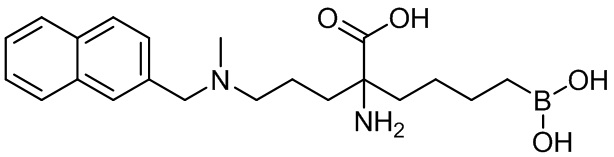 ,
, 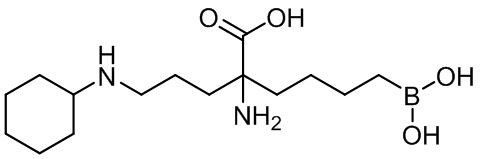 ,
, 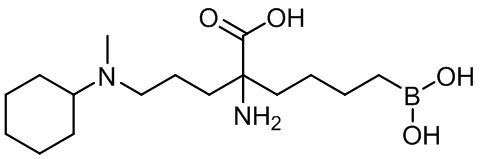 ,
, 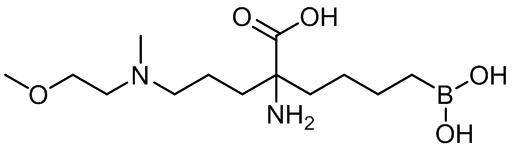 ,
, 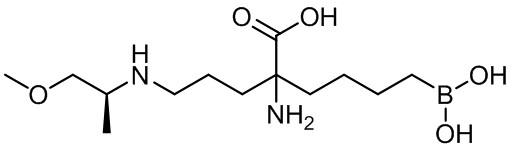 ,
, 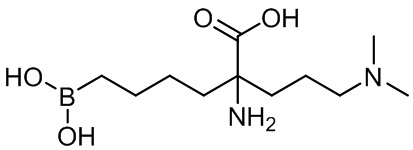 ,
, 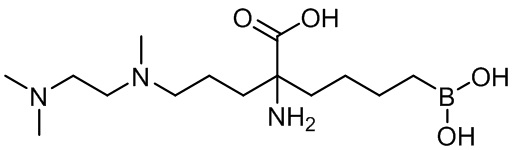 ,
, 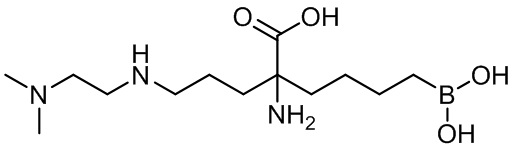 ,
, 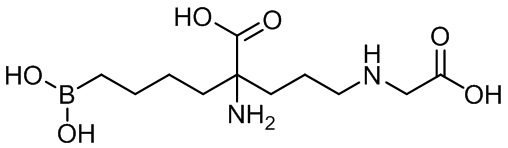 ,
, 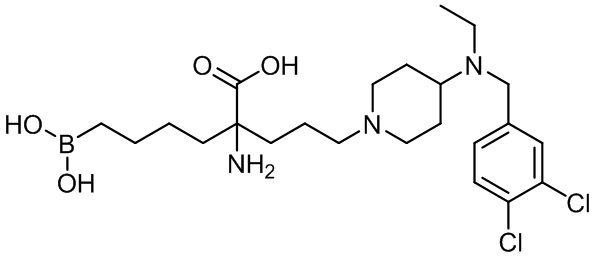 ,
, 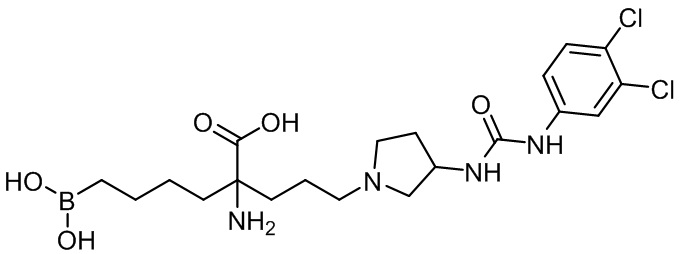 ,
, 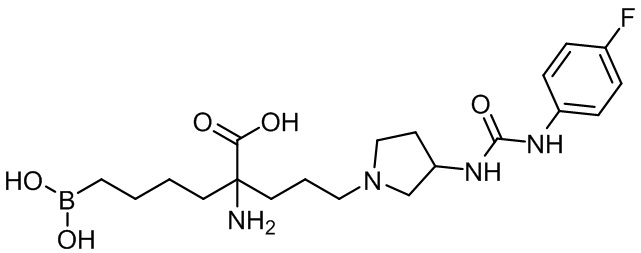 ,
, 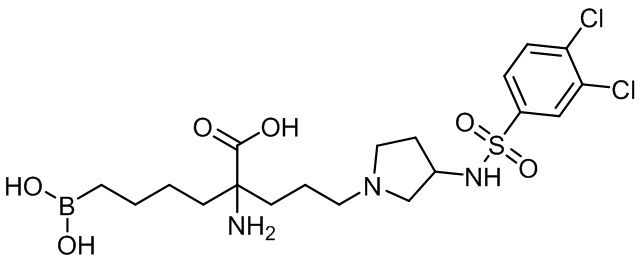 ,
,
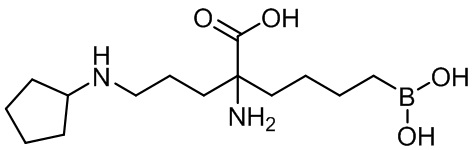 ,
, 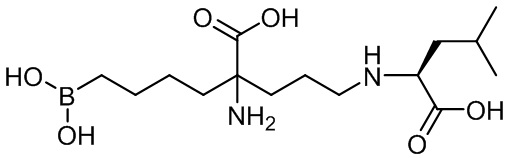 ,
,
.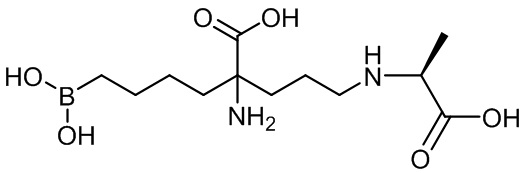 ,
, 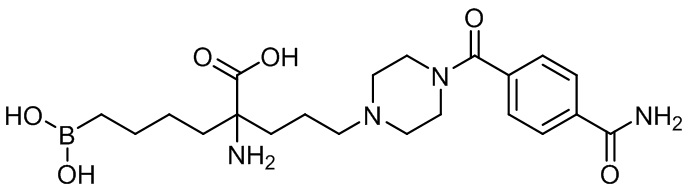
или его фармацевтически приемлемой соли.
2. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении аргиназы, включающая соединение по п.1 или его фармацевтически приемлемую соль в качестве активного ингредиента в терапевтически эффективном количестве и фармацевтически приемлемый носитель.
3. Применение соединения по п.1 для лечения заболевания, выбранного из группы, состоящей из хронического инфекционного заболевания, бактериальной инфекции, паразитарной инфекции, травмы, лепры, туберкулеза, трансплантации печени, рака, фиброзных заболеваний и их комбинации.
| WO 2010085797 A2, 29.07.2010 | |||
| WO 2010085797 A2, 29.07.2010. |

