Область техники
[1] Настоящее изобретение относится к новому классу производных индола, в частности к соединению, представленному формулой (I), способу его получения, фармацевтической композиции и его применению в качестве понижающего регулятора рецепторов эстрогена для получения лекарственного средства для лечения эстроген-рецептор-положительного рака молочной железы.
Уровень техники
[2] Согласно статистике ВОЗ рак молочной железы стал раком со вторым самым высоким показателем заболеваемости в мире, а также раком с самым высоким показателем заболеваемости среди женщин. После многолетних исследований была подтверждена роль пути передачи сигнала посредством эстрогена-рецептора эстрогена в прогрессировании рака молочной железы; и рецептор эстрогена (ER) стал самым важным биомаркером рака молочной железы. В зависимости от экспрессии рецепторов эстрогена, рак молочной железы можно подразделить на эстроген-рецептор-положительный рак молочной железы и эстроген-рецептор-отрицательный рак молочной железы, при этом эстроген-рецептор-положительный рак молочной железы составляет более 70% от общего числа пациентов с раком молочной железы.
[3] Эндокринная терапия (ЭТ), которая нацелена на путь передачи сигнала посредством эстрогена-рецептора эстрогена в клетках рака молочной железы, была первым выбором для лечения эстроген-рецептор-положительного рака молочной железы вследствие ее минимального риска и значительной эффективности. Эндокринная терапия в основном включает три вида терапии, которые представляют собой терапию подавления функции яичников, применение ингибитора ароматазы (AI) и применение селективного модулятора рецепторов эстрогена (SERM). Терапию, включающую подавление функции яичников, применяют реже, чем два других вида терапии из-за низкой эффективности и низкой удовлетворенности пациентов. Ранние ингибиторы ароматазы (первое поколение, второе поколение) имеют низкую селективность по отношению к мишени и серьезные побочные эффекты. После многолетних исследований широко использовалось третье поколение ингибиторов ароматазы, которое значительно улучшило селективность по отношению к мишени и решило проблему более ранних ингибиторов ароматазы. В том числе летрозол применялся в качестве препарата первой линии для лечения эстроген-рецептор-положительного рака молочной железы. Селективный модулятор рецепторов эстрогена (SERM), который нацелен непосредственно на рецептор эстрогена для блокирования пути передачи сигнала, обладает значительной терапевтической эффективностью и длительной историей применения. В том числе тамоксифен является наиболее типичным селективным модулятором рецепторов эстрогена. В качестве препарата первой линии, рекомендованного для приоритетного применения, тамоксифен продемонстрировал значительную клиническую эффективность в профилактике и лечении эстроген-рецептор-положительного рака молочной железы.
[4] Хотя ингибитор ароматазы летрозол и селективный модулятор рецепторов эстрогена тамоксифен проявляют хорошую эффективность при лечении эстроген-рецептор-положительного рака молочной железы, устойчивость эстроген-рецептор-положительного рака молочной железы к ингибитору ароматазы и селективному модулятору рецепторов эстрогена становится все более значимой по мере их применения. В большом количестве исследований было показано, что механизм лекарственной устойчивости при раке молочной железы к указанным выше двум эндокринным терапиям не совсем одинаков. Что касается ингибитора ароматазы, рецептор эстрогена может соответствующим образом мутировать. Мутированный рецептор эстрогена сохраняет агонистическую конформацию в отсутствие эстрогена, поэтому продолжает функционировать как рецептор для стимулирования пролиферации клеток рака молочной железы. Механизм устойчивости к лекарственному средству при раке молочной железы в случае селективного модулятора рецепторов эстрогена тамоксифена более сложный. Во-первых, клетки рака молочной железы могут компенсировать потерю функции активатора функции рецептора эстрогена-2 ("activation of function-2"; AF-2), вызванную тамоксифеном, путем активации функции активатора функции рецептора эстрогена-1 ("activation of function-1"; AF-1). Между тем, клетки рака молочной железы могут адаптироваться к конформации связывания рецепторов эстрогена с тамоксифеном, регулируя структуру или концентрацию коактиватора рецепторов эстрогена для восстановления функции рецепторов эстрогена, тем самым вызывая лекарственную устойчивость.
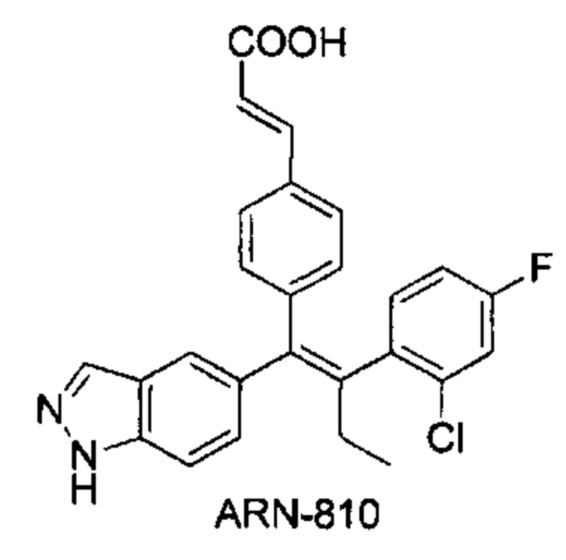
[5] Селективный понижающий регулятор рецепторов эстрогена (SERD) продемонстрировал уникальные преимущества при лечении рака молочной железы, устойчивого к двум эндокринным терапиям. Указанный механизм заключается в том, что селективная понижающая регуляция рецепторов эстрогена антагонизирует функцию рецепторов эстрогена, значительно ускоряет убиквитинирование и деградацию рецепторов эстрогена в клетках рака молочной железы (нормальных или мутантных) и полностью блокирует путь передачи сигналов через эстроген/рецептор эстрогена, что приводит к ингибированию роста и пролиферации нормальных или устойчивых к лекарственному средству клеток рака молочной железы. Исследования показали, что селективный понижающий регулятор рецепторов эстрогена может эффективно ингибировать пролиферацию устойчивых к эндокринным средствам клеток рака молочной железы. Фулвестрант, который является единственным селективным понижающим регулятором рецепторов эстрогена на рынке, показал хороший результат при лечении устойчивого к эндокринным средствам рака молочной железы, демонстрируя уникальное преимущество селективного понижающего регулятора рецепторов эстрогена. Однако у фулвестранта много недостатков. Во-первых, из-за плохих фармакокинетических свойств фулвестрант обладает нулевой пероральной биодоступностью и кроме того фулвестрант имеет более высокую скорость выведения (клиренс) из циркуляции. По указанным выше двум причинам данное лекарственное средство можно вводить только внутримышечно. Однако вследствие своей выраженной липофильной структуры фулвестрант, вводимый посредством внутримышечной инъекции, также имеет серьезные проблемы, связанные с распределением в тканях, его клинические проявления таковы, что только примерно 50% пациентов с раком молочной железы, получавших фулвестрант, демонстрируют клинический ответ. Таким образом, для лечения требуется понижающая регуляция рецепторов эстрогена с пероральной биодоступностью.
[6] В WO 2012037411 A2 описан пероральный селективный понижающий регулятор рецепторов эстрогена ARN-810, который находится на II фазе клинических исследований, для лечения ER-положительного рака молочной железы. Согласно докладу [J. Med. Chem. 2015, 58 (12), 4888-4904], важным фармакофором данной молекулы является индазольная структура на левой стороне молекулы и атом азота в указанной индазольной структуре действует как акцептор водородной связи при связывании с рецептором эстрогена.
Сущность настоящего изобретения
[7] В настоящем изобретении описано соединение, представленное формулой (I), его фармацевтически приемлемая соль, гидрат или пролекарство,
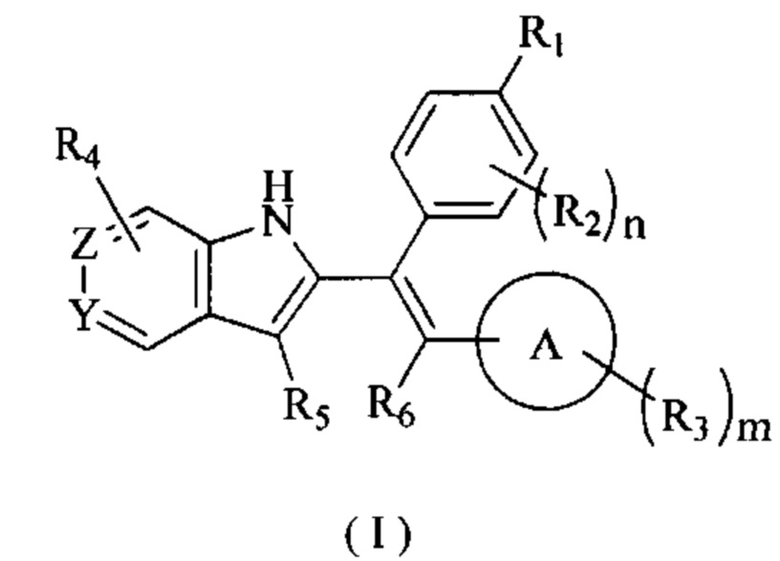
[8] где,
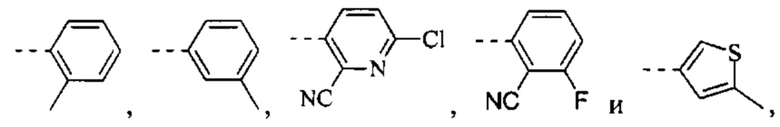
[9] R1 выбран из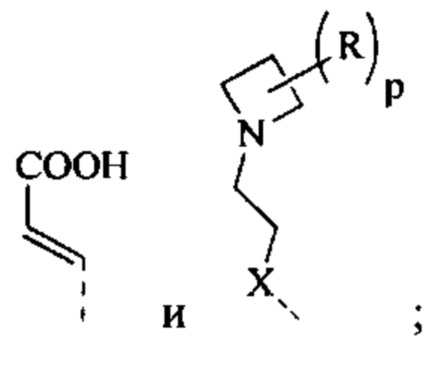
[10] Х выбран из одинарной связи, О и S;
[11] каждый Y и Z независимо выбран из N и СН;
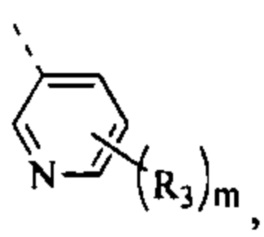
[12] кольцо А выбрано из 5-10-членного арила и 5-10-членного гетероарила;
[13] R2 выбран из Н, галогена, CN, NO2, ОН, СООН, NH2 или группы, состоящей из C1-6 алкила, C1-6 гетероалкила, С3-6 циклоалкила и 3-6-членного гетероциклоалкила, каждый из которых необязательно замещен 1, 2 или 3 R;
[14] R3 выбран из Н, галогена, CN, NO2, ОН, СООН, NH2 или группы, состоящей из C1-6 алкила, C1-6 гетероалкила, С3-6 циклоалкила и 3-6-членного гетероциклоалкила, каждый из которых необязательно замещен 1, 2 или 3 R;
[15] каждый R4 и R5 независимо выбран из Н, галогена, CN, NO2, ОН, СООН, NH2 или группы, состоящей из C1-6 алкила, C1-6 гетероалкила, С3-6 циклоалкила и 3-6-членного гетероциклоалкила, каждый из которых необязательно замещен 1, 2 или 3 R;
[16] R6 выбран из группы, состоящей из C1-6 алкила, C1-6 гетероалкила, С3-6 циклоалкила и 3-6-членного гетероциклоалкила, каждый из которых необязательно замещен 1, 2 или 3 R;
[17] R выбран из Н, F, Cl, Br, I, ОН, CN, NH2, СООН, C(=O)NH2 или группы, состоящей из С1-8 алкила, C1-8 гетероалкила, С3-6 циклоалкила, 3-6-членного гетероциклоалкила и С3-6 циклоалкил-С1-3 алкил-, каждый из которых необязательно замещен 1, 2 или 3 R';
[18] n выбран из 0, 1, 2, 3 и 4;
[19] m выбран из 0, 1, 2, 3 и 4;
[20] Р выбран из 0, 1, 2 и 3;
[21] или когда m представляет собой 2, R3 и R3 соединены вместе с образованием 5-6-членного кольца;
[22] R' выбран из группы, состоящей из F, Cl, Br, I, ОН, CN, NH2, СООН, Me, Et, CF3, CHF2, CH2F, NHCH3 и N(CH3)2;
[23] «гетеро» представляет собой гетероатом или гетероатомную группу, которая выбрана из группы, состоящей из -C(=O)N(R)-, -N(R)-, -S(=O)2N(R)-, -S(=O)N(R)-, -O-, -S-,=O,=S, -O-N=, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -N(R)C(=O)N(R)-;
[24] в любом из указанных выше случаев количество гетероатомов или гетероатомных групп независимо выбрано из 1, 2 и 3.
[25] В настоящем изобретении описано соединение, представленное формулой (I), его фармацевтически приемлемая соль, гидрат или пролекарство,
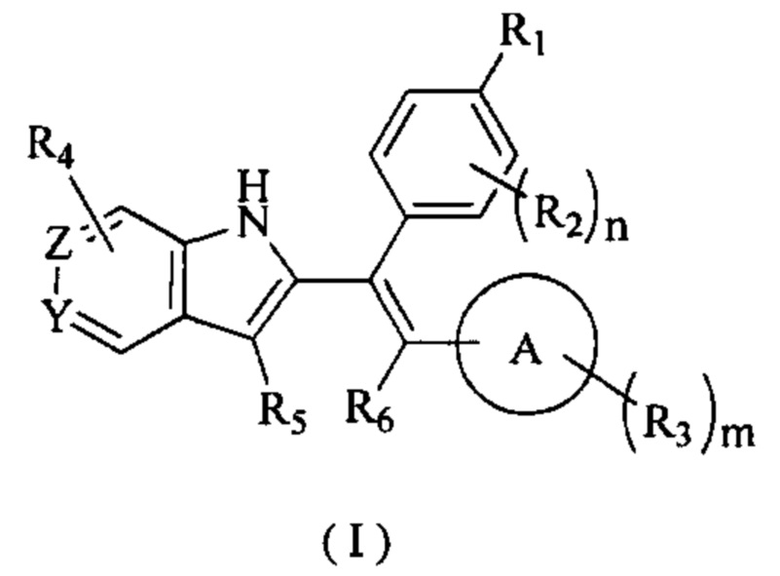
[26] где,
[27] R1 выбран из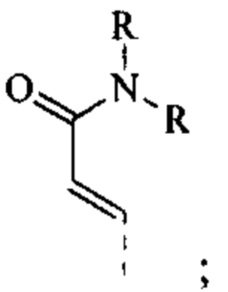
[28] каждый Y и Z независимо выбран из N и СН;
[29] кольцо А выбрано из 5-10-членного арила и 5-10-членного гетероарила;
[30] R2 выбран из Н, галогена, CN, NO2, ОН, СООН, NH2 или группы, состоящей из C1-6 алкила, C1-6 гетероалкила, С3-6 циклоалкила и 3-6-членного гетероциклоалкила, каждый из которых необязательно замещен 1, 2 или 3 R;
[31] R3 выбран из Н, галогена, CN, NO2, ОН, СООН, NH2 или группы, состоящей из C1-6 алкила, C1-6 гетероалкила, С3-6 циклоалкила и 3-6-членного гетероциклоалкила, каждый из которых необязательно замещен 1, 2 или 3 R;
[32] каждый R4 и R5 независимо выбран из Н, галогена, CN, NO2, ОН, СООН, NH2 или группы, состоящей из C1-6 алкила, C1-6 гетероалкила, С3-6 циклоалкила и 3-6-членного гетероциклоалкила, каждый из которых необязательно замещен 1, 2 или 3 R;
[33] R6 выбран из группы, состоящей из C1-6 алкила, C1-6 гетероалкила, С3-6 циклоалкила и 3-6-членного гетероциклоалкила, каждый из которых необязательно замещен 1, 2 или 3 R;
[34] R выбран из Н, F, Cl, Br, I, ОН, CN, NH2, СООН, C(=O)NH2 или группы, состоящей из C1-8 алкила, C1-8 гетероалкила, С3-6 циклоалкила, 3-6-членного гетероциклоалкила и С3-6 циклоалкил-C1-3 алкил-, каждый из которых необязательно замещен 1, 2 или 3 R';
[35] n выбран из 0, 1, 2, 3 и 4;
[36] m выбран из 0, 1, 2, 3 и 4;
[37] или когда m представляет собой 2, R3 и R3 соединены вместе с образованием 5-6-членного кольца;
[38] R' выбран из группы, состоящей из F, Cl, Br, I, ОН, CN, NH2, СООН, Me, Et, CF3, CHF2, CH2F, NHCH3 и N(CH3)2;
[39] «гетеро» представляет собой гетероатом или гетероатомную группу, которая выбрана из группы, состоящей из -C(=O)N(R)-, -N(R)-, -S(=O)2N(R)-, -S(=O)N(R)-, -O-, -S-, =O, =S, -O-N=, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -N(R)C(=O)N(R)-;
[40] в любом из указанных выше случаев количество гетероатомов или гетероатомных групп независимо выбрано из 1, 2 и 3.
[41] Согласно некоторым вариантам реализации R выбран из Н, F, Cl, Br, I, ОН, CN, NH2, СООН или группы, состоящей из С1-5 алкила, С1-5 гетероалкила, С3-6 циклоалкил-С1-3 алкил-, каждый из которых необязательно замещен 1, 2 или 3 R'.
[42] Согласно некоторым вариантам реализации R выбран из Н, F, Cl, Br, I, ОН, CN, NH2, СООН, C(=O)NH2 или группы, состоящей из СН3, CH3CH2, S(=O)CH3,
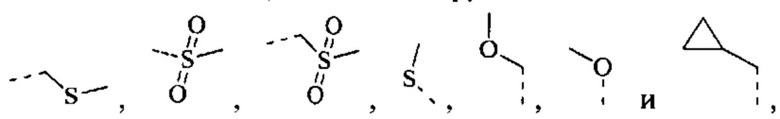 каждый из которых необязательно замещен 1, 2 или 3 R'.
каждый из которых необязательно замещен 1, 2 или 3 R'.
[43] Согласно некоторым вариантам реализации R выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, CN, NH2, СООН, C(=O)NH2, СН3, CH2Cl, CH2F, CHF2, CF3, OCF3, CH2OH, Et, S(=O)CH3, 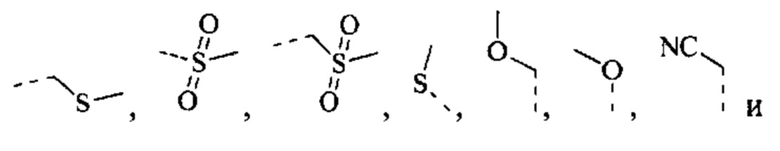
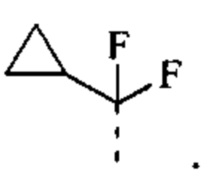
[44] Согласно некоторым вариантам реализации R выбран из Н, F, Cl, Br, I, ОН, CN, NH2, СООН или группы, состоящей из С1-5 алкила, С1-5 гетероалкила и С3-6 циклоалкил-С1-3 алкил-, каждый из которых необязательно замещен 1, 2 или 3 R'.
[45] Согласно некоторым вариантам реализации R выбран из Н, F, Cl, Br, I, ОН, CN, NH2, СООН, C(=O)NH2 или группы, состоящей из СН3, СН3СН2, S(=O)CH3,
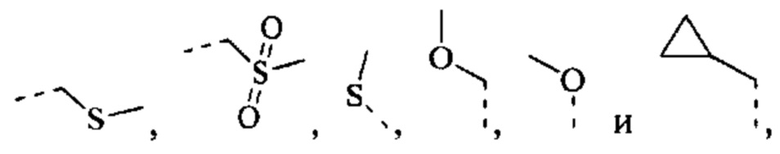
каждый из которых необязательно замещен 1, 2 или 3 R'.
[46] Согласно некоторым вариантам реализации R выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, CN, NH2, СООН, C(=O)NH2, СН3, CH2Cl, CH2F, CHF2, CF3, OCF3, СН2ОН, Et, S(=O)CH3,
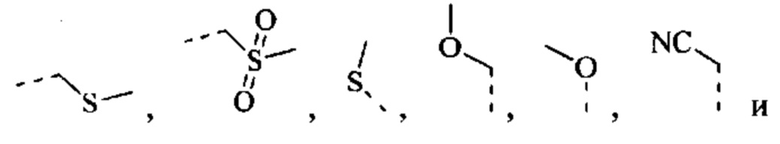
[47] Согласно некоторым вариантам реализации фрагмент  выбран из
выбран из
[48] Согласно некоторым вариантам реализации фрагмент 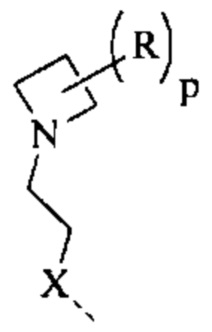 выбран из
выбран из
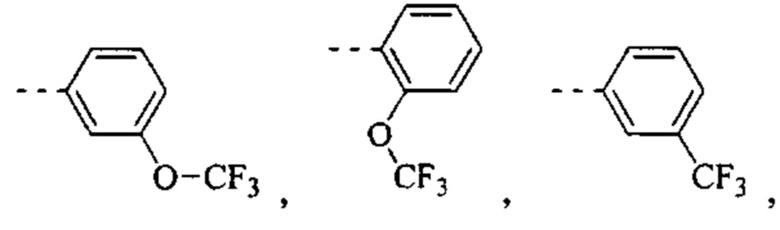
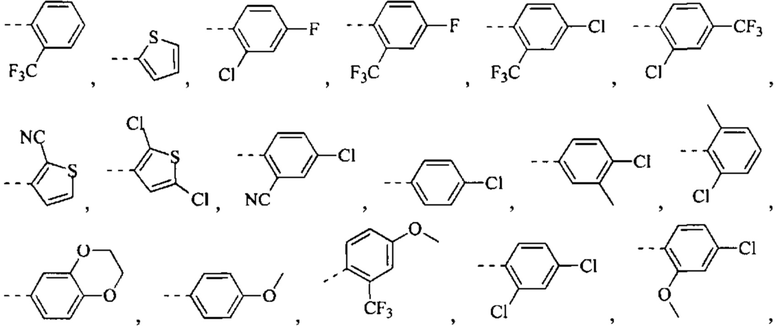
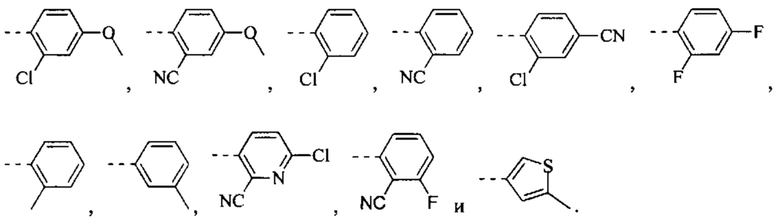
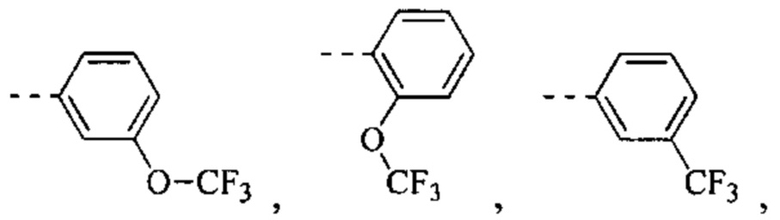
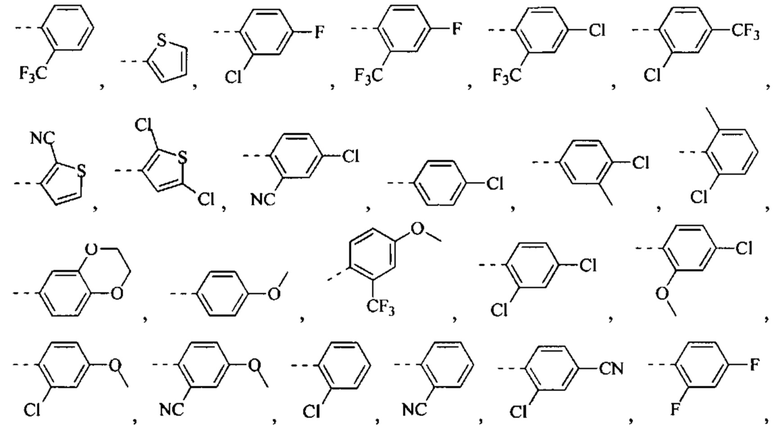
[49] Согласно некоторым вариантам реализации фрагмент 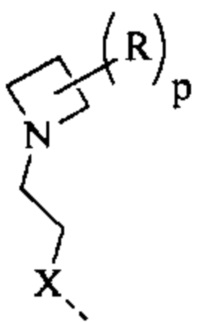 выбран из группы, состоящей из
выбран из группы, состоящей из
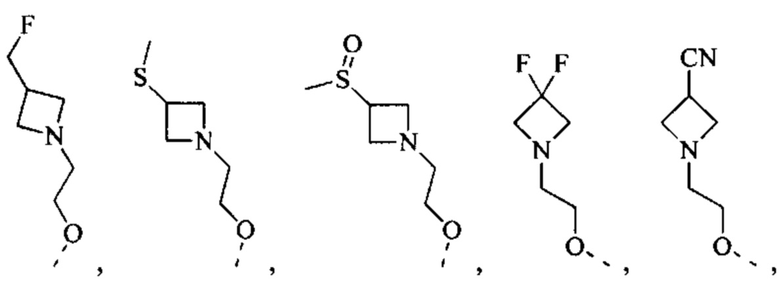
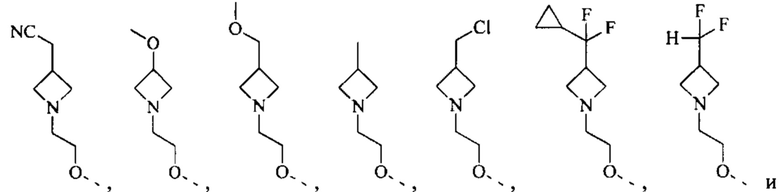
[50] Согласно некоторым вариантам реализации R2 выбран из Н, F, Cl, Br, I, CN, NO2, ОН, СООН, NH2 или группы, состоящей из С1-4 алкила, С1-4 алкокси, С1-4 алкилтио, NH(C1-4 алкил), N,N-ди(С1-3 алкил)амино и С3-6 циклоалкила, каждый из которых необязательно замещен 1, 2 или 3 R.
[51] Согласно некоторым вариантам реализации R3 выбран из Н, F, Cl, Br, I, CN, NO2, ОН, СООН, NH2 или группы, состоящей из С1-4 алкила, С1-4 алкокси, С1-4 алкилтио, NH(C1-4 алкил), N,N-ди(C1-3 алкил)амино, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, 5-6-членного арила и 5-6-членного гетероарила, каждый из которых необязательно замещен 1, 2 или 3 R.
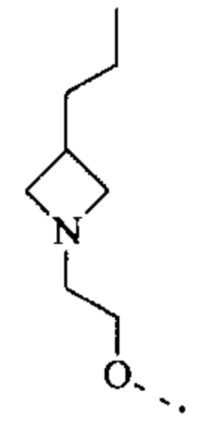
[52] Согласно некоторым вариантам реализации R3 выбран из Н, F, Cl, Br, I, CN, NO2, ОН, СООН, NH2 или группы, состоящей из СН3, СН3СН2, СН3О, СН3СН2О и  каждый из которых необязательно замещен 1, 2 или 3 R.
каждый из которых необязательно замещен 1, 2 или 3 R.
[53] Согласно некоторым вариантам реализации R3 выбран из группы, состоящей из Н, F, Cl, CN, СН3, CF3 и 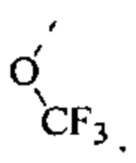
[54] Согласно некоторым вариантам реализации каждый R4 и R5 независимо выбран из Н, F, Cl, Br, I, CN, NO2, ОН, СООН, NH2 или группы, состоящей из C1-4 алкила, С1-4 алкокси, С1-4 алкилтио, NH(C1-4 алкил), N,N-ди(С1-3 алкил)амино и С3-6 циклоалкила, каждый из которых необязательно замещен 1, 2 или 3 R.
[55] Согласно некоторым вариантам реализации каждый R4 и R5 независимо выбран из группы, состоящей из Н, F, Cl, Br, CN и СН3.
[56] Согласно некоторым вариантам реализации кольцо А выбрано из группы, состоящей из фенила, пиридила, пиразинила, имидазолила, пиразолила, оксазолила, изоксазолила, тиазолила, изотиазолила, тиенила, бензофуранила, бензотиенила, индолила, бензимидазолила, бензотиазолила, пуринила, хинолинила и изохинолинила.
[57] Согласно некоторым вариантам реализации фрагмент  выбран из группы, состоящей из
выбран из группы, состоящей из
[58] Согласно некоторым вариантам реализации фрагмент 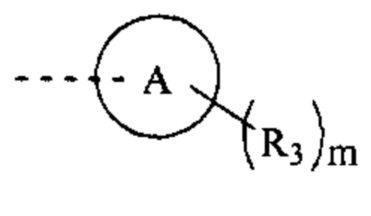 выбран из группы, состоящей из
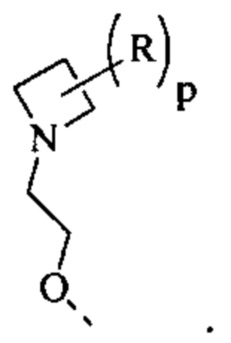
выбран из группы, состоящей из
[59] Согласно некоторым вариантам реализации R6 выбран из 
[60] Согласно некоторым вариантам реализации фрагмент 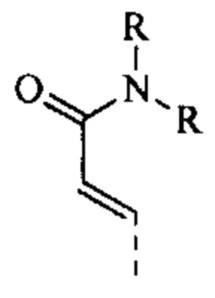 выбран из
выбран из 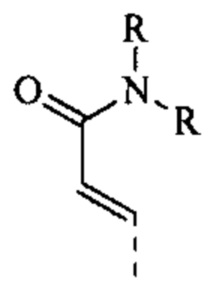
[61] Согласно некоторым вариантам реализации структурное звено выбрано из 
[62] Согласно некоторым вариантам реализации соединение выбрано из группы, состоящей из
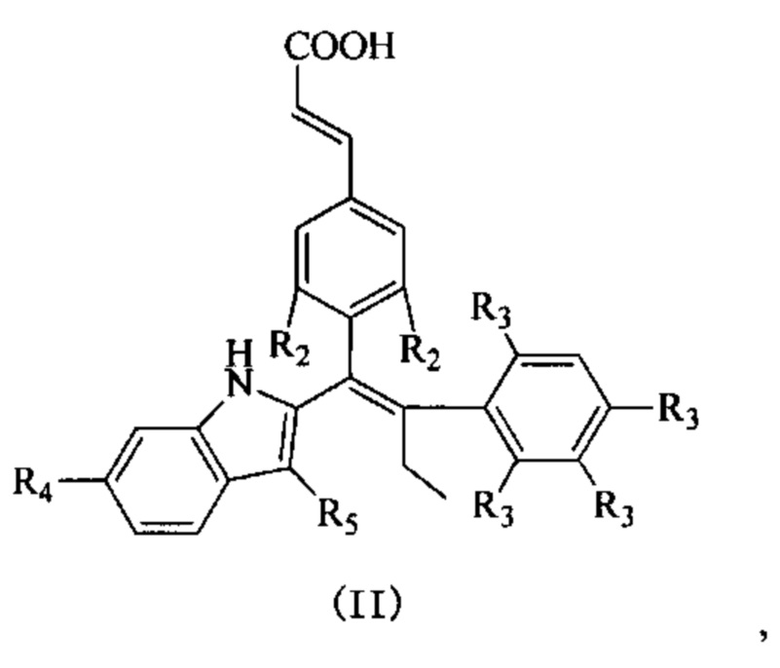
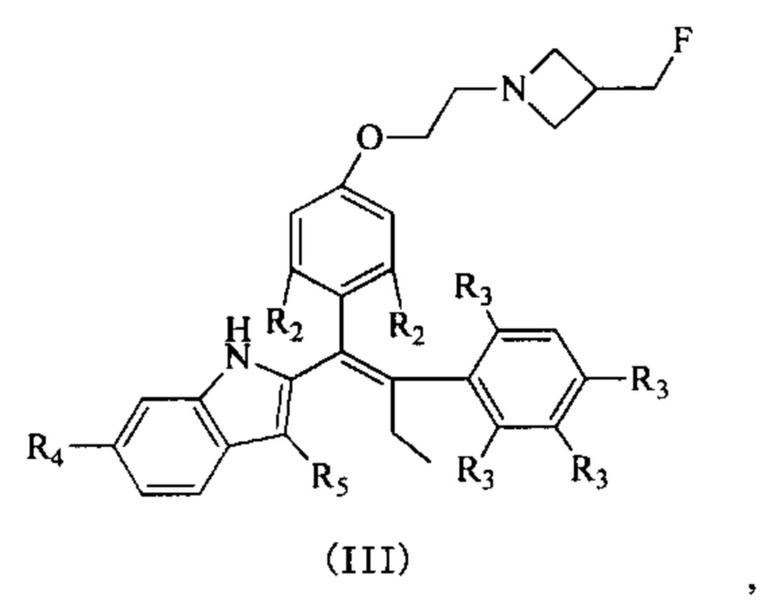
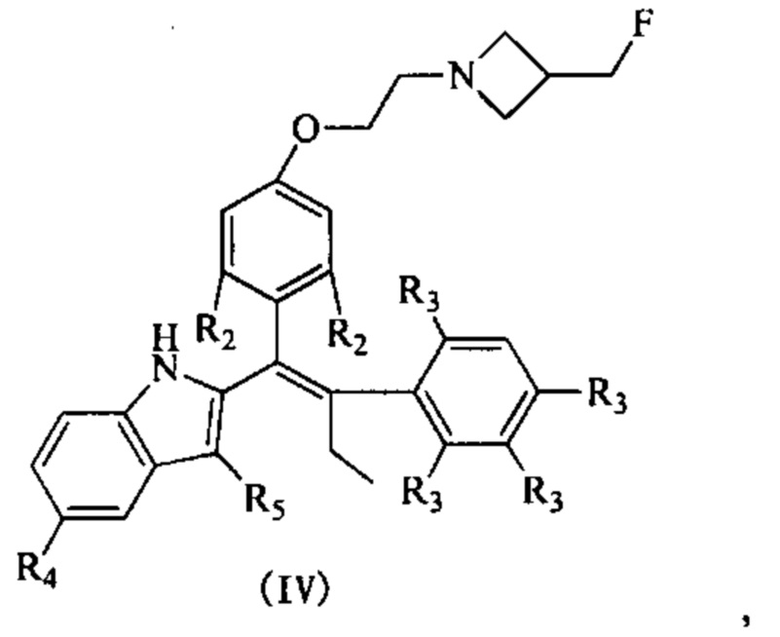
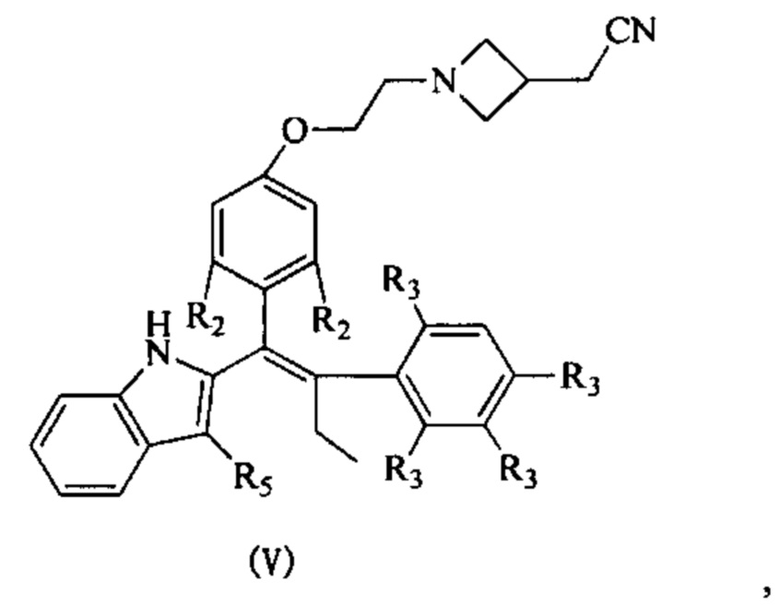
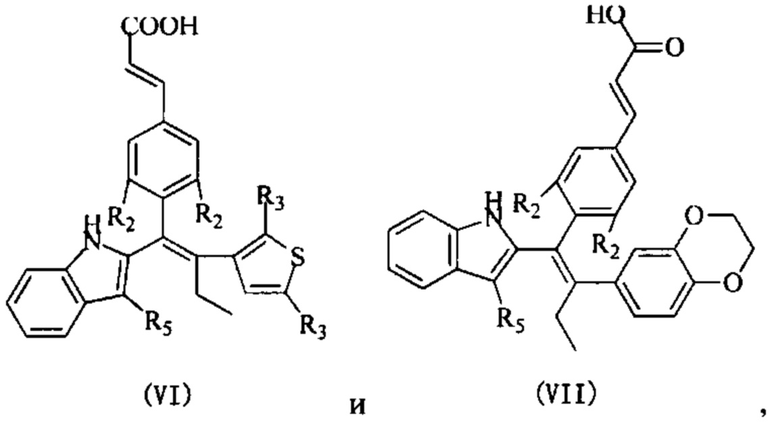
[63] где R2, R3, R4 и R5 являются такими, как определено выше.
[64] Согласно некоторым вариантам реализации соединение выбрано из
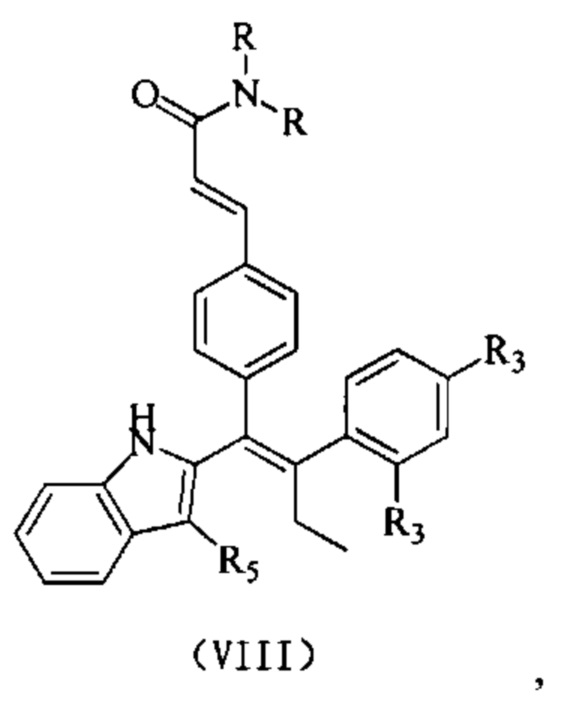
[65] где, R3, R5 и R являются такими, как определено выше.
[66] В настоящем изобретении также предложено соединение или его фармацевтически приемлемая соль, где соединение выбрано из группы, состоящей из
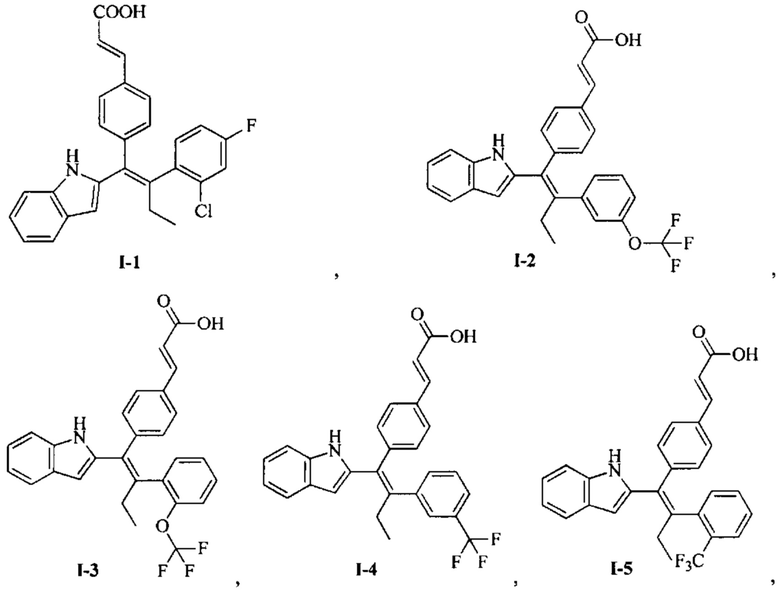
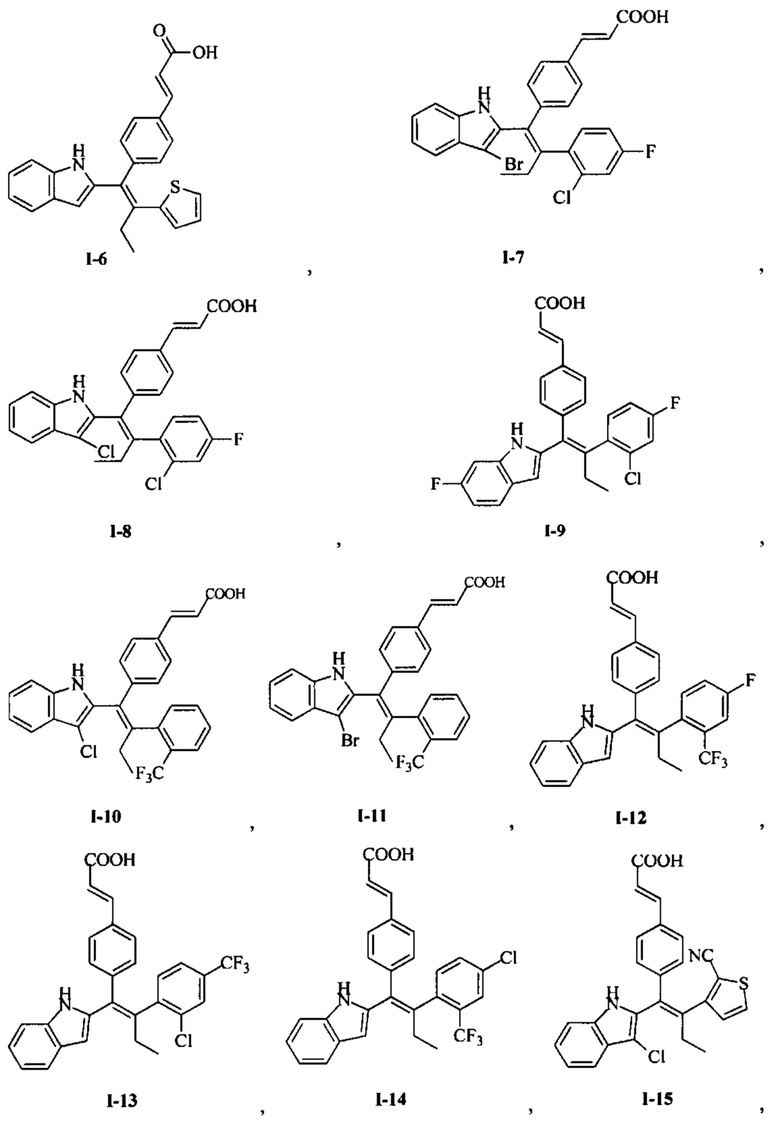
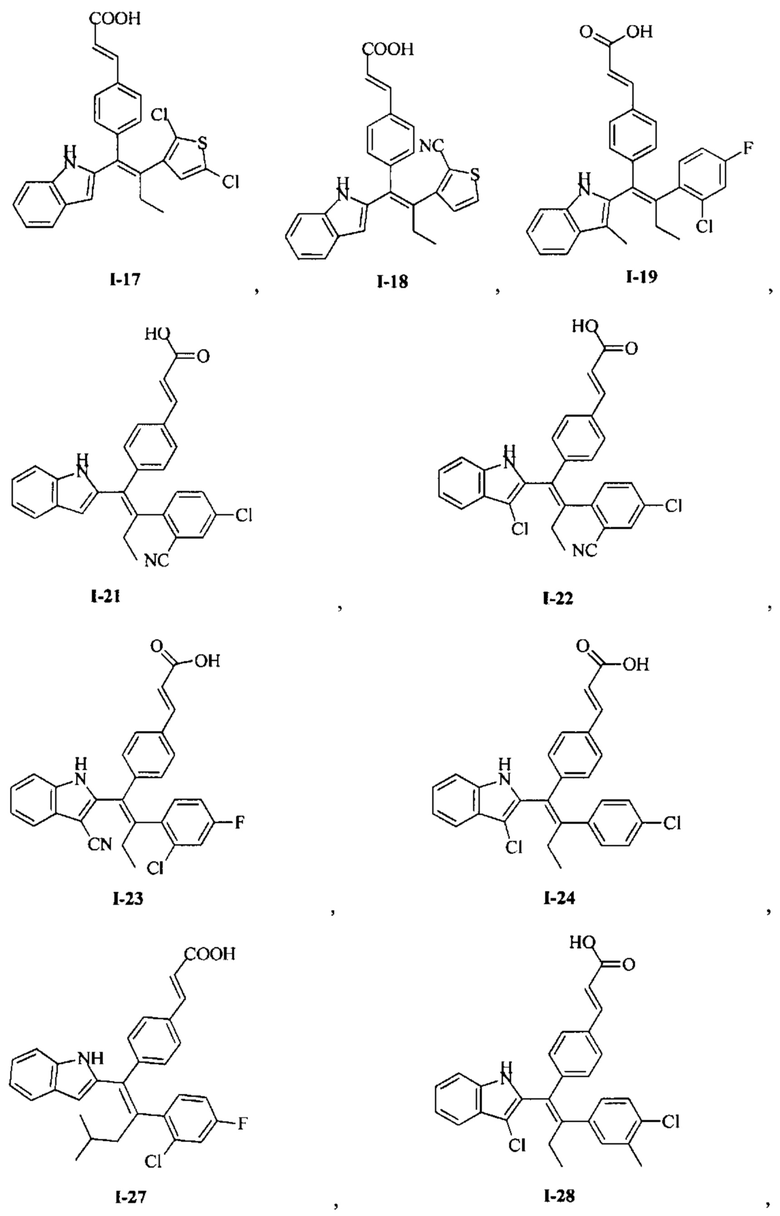
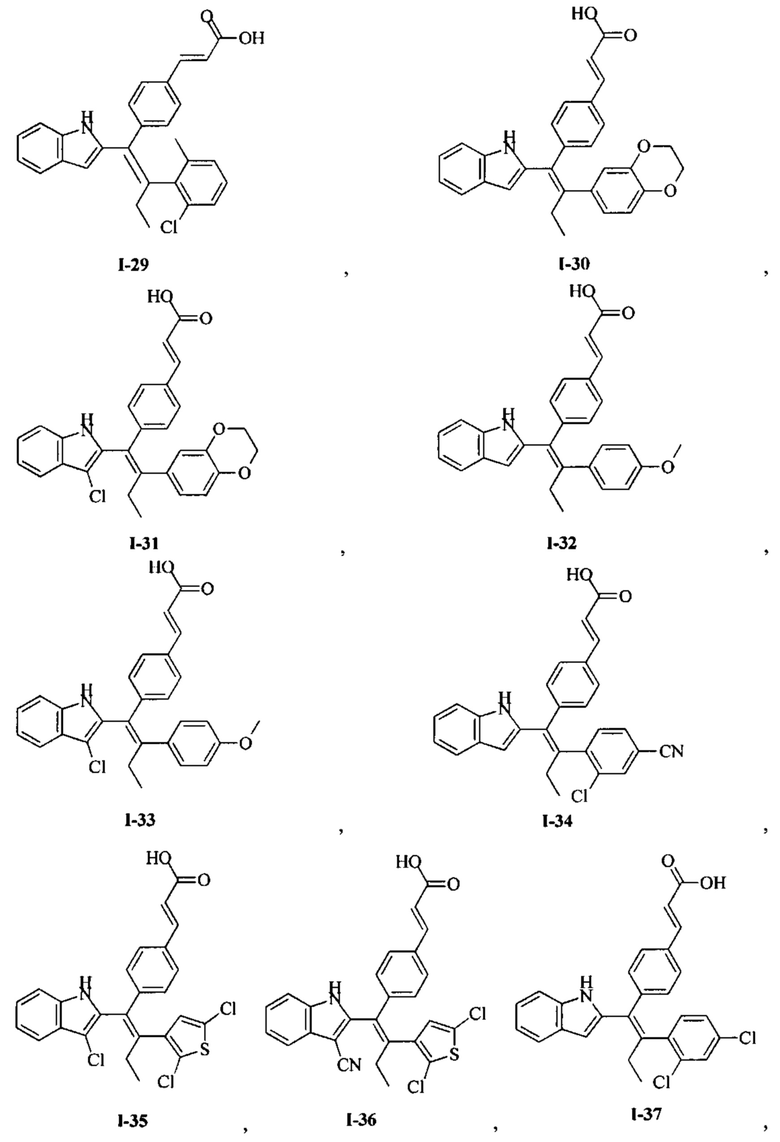
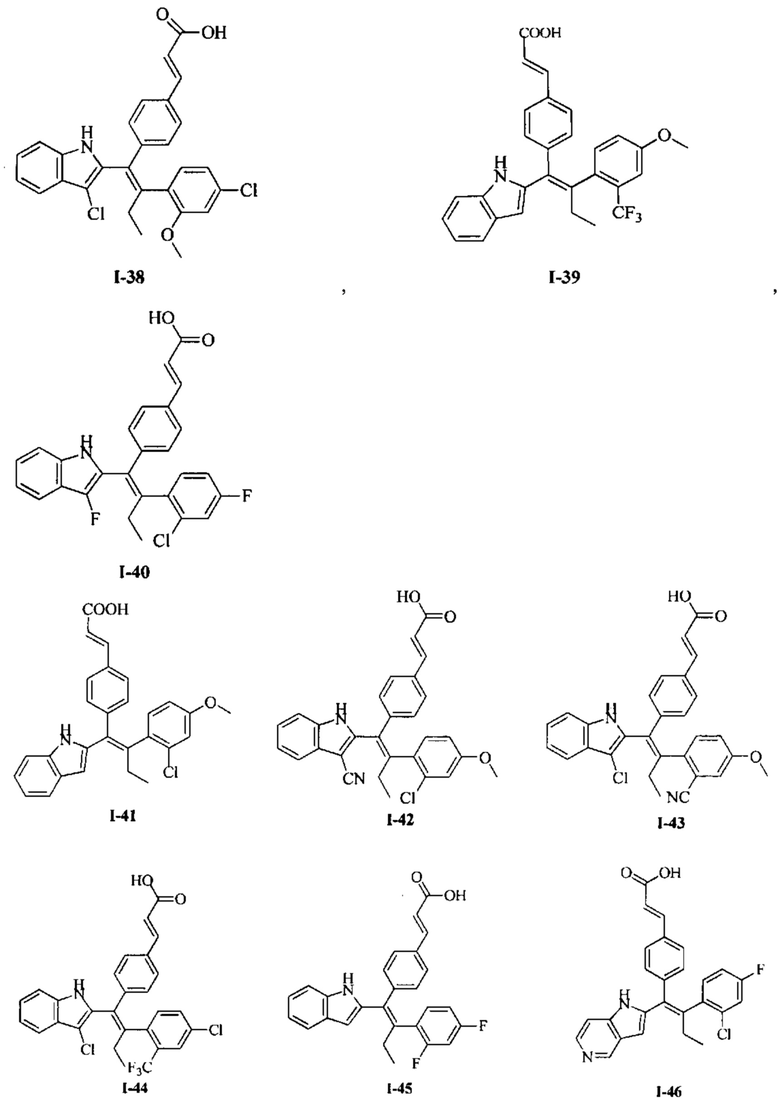
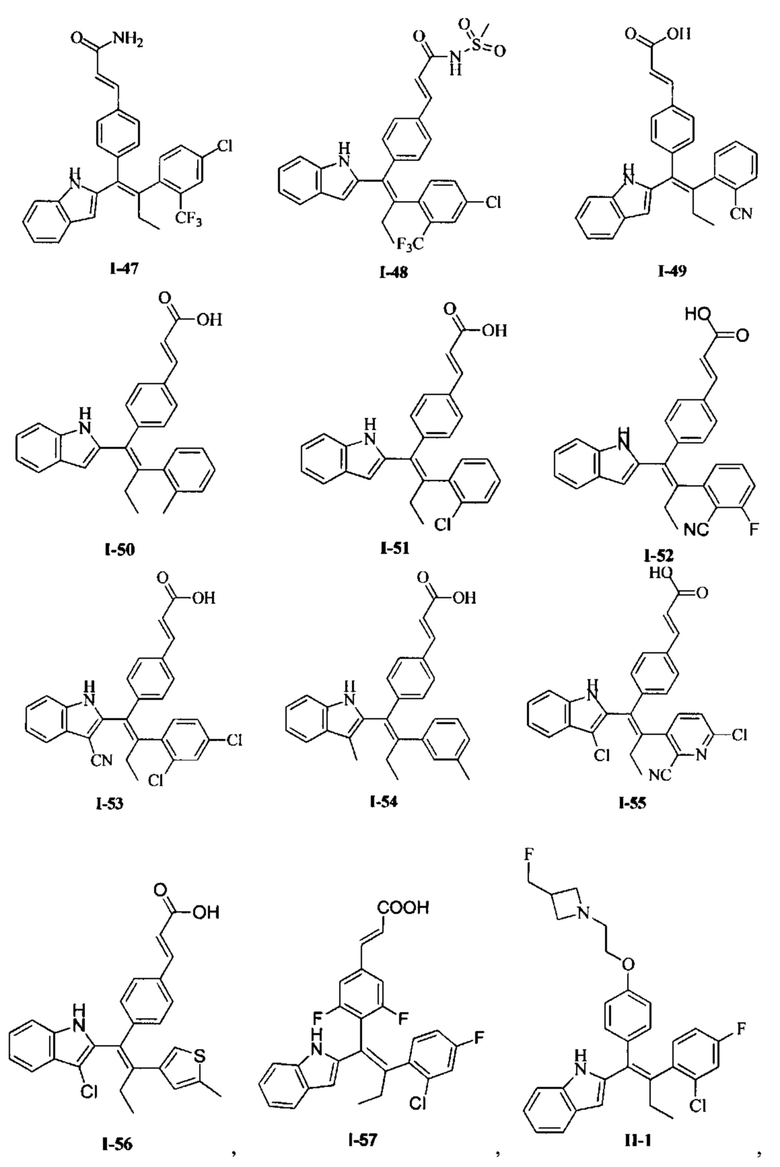
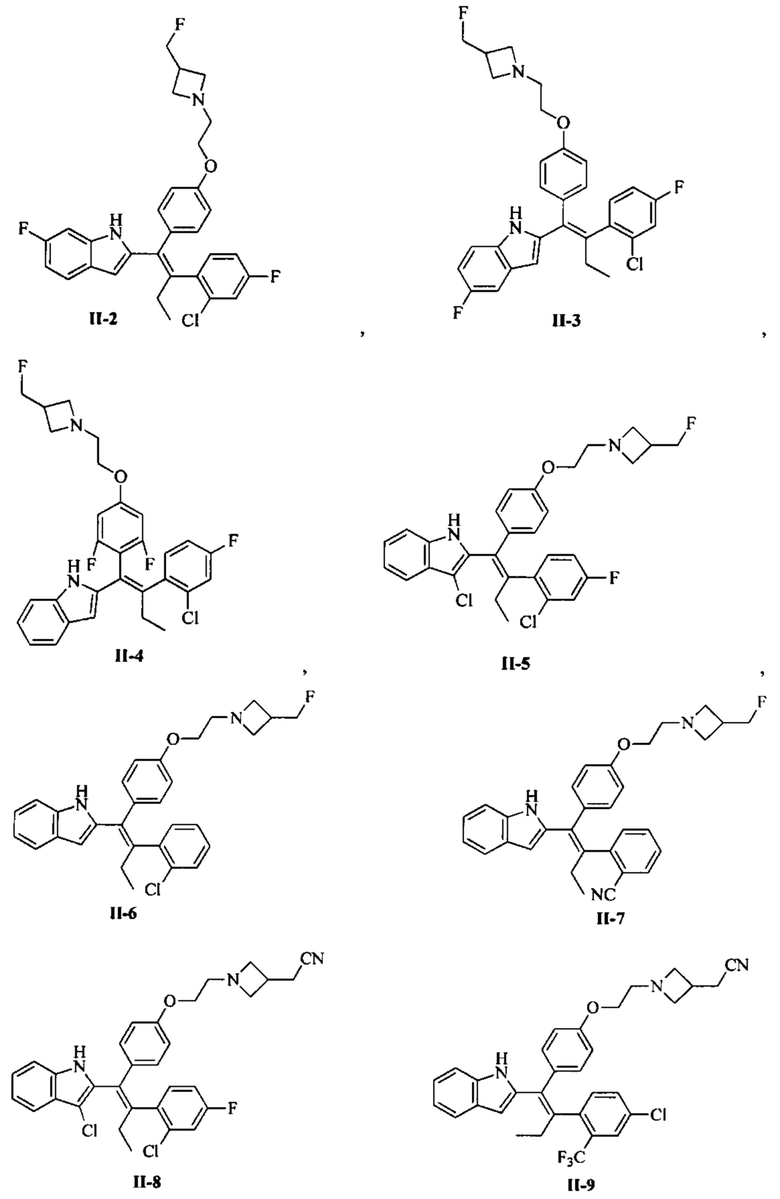
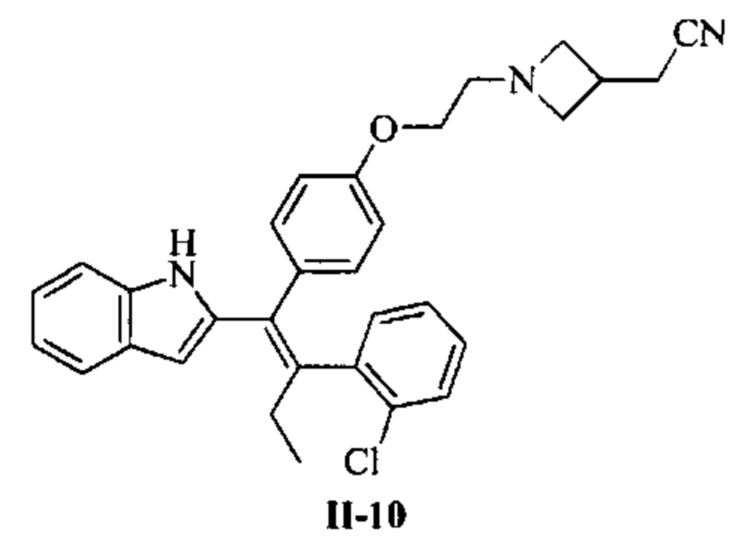
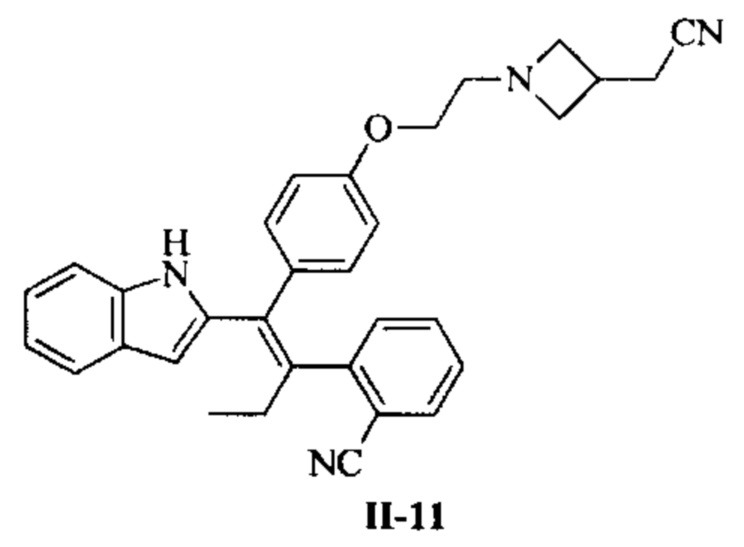
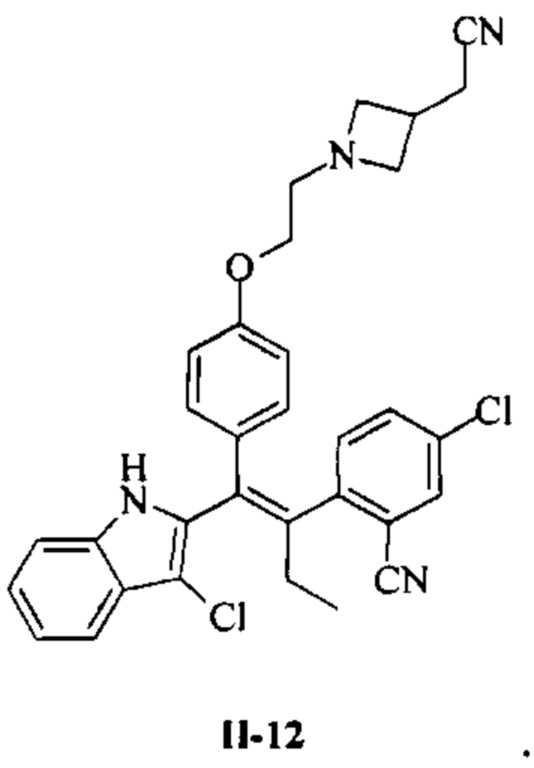
[67] В настоящем изобретении также предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли, как определено выше, и фармацевтически приемлемый носитель.
[68] В настоящем изобретении также предложено применение соединения или его фармацевтически приемлемой соли, как определено выше, или фармацевтической композиции, как определено выше, для получения лекарственного средства для лечения расстройства, связанного с рецептором эстрогена.
[69] Согласно некоторым вариантам реализации фрагмент 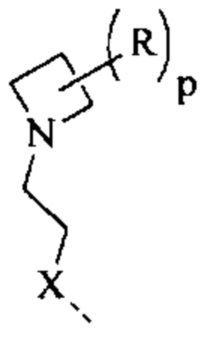 выбран из
выбран из 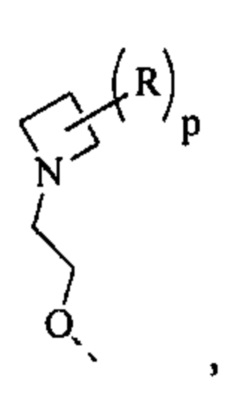 другие переменные являются такими, как определено выше.
другие переменные являются такими, как определено выше.
[70] Согласно некоторым вариантам реализации фрагмент  выбран из
выбран из  другие переменные являются такими, как определено выше.
другие переменные являются такими, как определено выше.
[71] Согласно некоторым вариантам реализации фрагмент  выбран из группы, состоящей из
выбран из группы, состоящей из 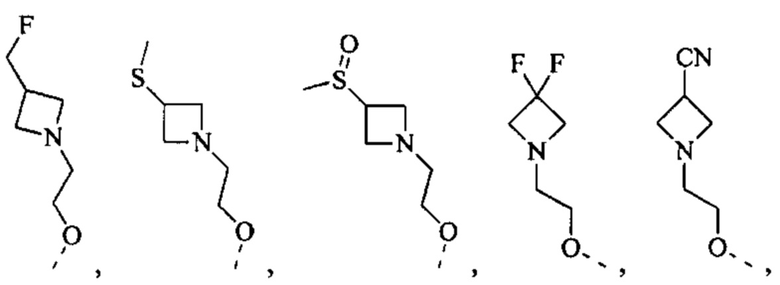
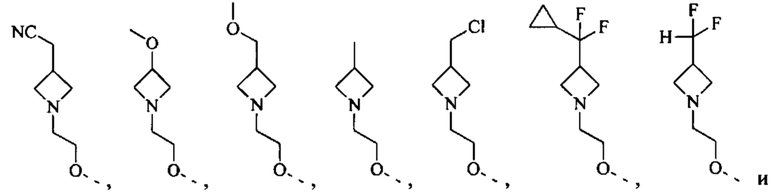
 другие переменные являются такими, как определено выше.
другие переменные являются такими, как определено выше.
[72] Согласно некоторым вариантам реализации R2 выбран из Н, F, Cl, Br, I, CN, NO2, ОН, СООН, NH2 или группы, состоящей из С1-4 алкила, С1-4 алкокси, С1-4 алкилтио, NH(C1-4 алкил), N,N-ди(C1-3 алкил)амино и С3-6 циклоалкила, каждый из которых необязательно замещен 1, 2 или 3 R, другие переменные являются такими, как определено выше.
[73] Согласно некоторым вариантам реализации R3 выбран из Н, F, Cl, Br, I, CN, NO2, ОН, СООН, NH2 или группы, состоящей из С1-4 алкила, С1-4 алкокси, С1-4 алкилтио, NH(C1-4 алкил), N,N-ди(C1-3 алкил)амино, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, 5-6-членного арила и 5-6-членного гетероарила, каждый из которых необязательно замещен 1, 2 или 3 R, другие переменные являются такими, как определено выше.
[74] Согласно некоторым вариантам реализации R3 выбран из Н, F, Cl, Br, I, CN, NO2, ОН, СООН, NH2 или группы, состоящей из СН3, СН3СН2, СН3О, СН3СН2О и 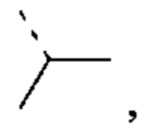 каждый из которых необязательно замещен 1, 2 или 3 R, другие переменные являются такими, как определено выше.
каждый из которых необязательно замещен 1, 2 или 3 R, другие переменные являются такими, как определено выше.
[75] Согласно некоторым вариантам реализации R3 выбран из Н, F, Cl, CN, СН3, CF3 и 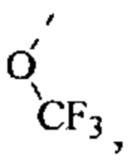 другие переменные являются такими, как определено выше.
другие переменные являются такими, как определено выше.
[76] Согласно некоторым вариантам реализации каждый R4 и R5 независимо выбран из Н, F, Cl, Br, I, CN, NO2, ОН, СООН, NH2 или группы, состоящей из C1-4 алкила, С1-4 алкокси, С1-4 алкилтио, NH(C1-4 алкил), N,N-ди(C1-3 алкил)амино и С3-6 циклоалкила, каждый из которых необязательно замещен 1, 2 или 3 R, другие переменные являются такими, как определено выше.
[77] Согласно некоторым вариантам реализации каждый R4 и R5 независимо выбран из группы, состоящей из Н, F, Cl, Br, CN и СН3, другие переменные являются такими, как определено выше.
[78] Согласно некоторым вариантам реализации кольцо А выбрано из группы, состоящей из фенила, пиридила, пиразинила, имидазолила, пиразолил,а оксазолила, изоксазолила, тиазолила, изотиазолила, тиенила, бензофуранила, бензотиенила, индолила, бензимидазолила, бензотиазолила, пуринила, хинолинила и изохинолинила, другие переменные являются такими, как определено выше.
[79] Согласно некоторым вариантам реализации фрагмент 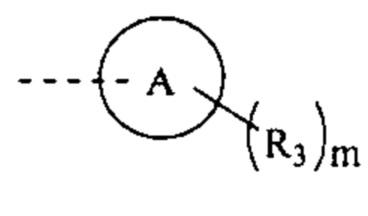 выбран из группы, состоящей из
выбран из группы, состоящей из 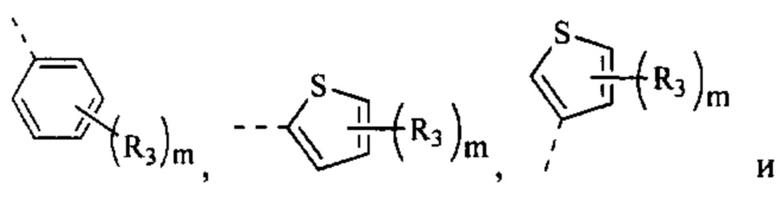
 другие переменные являются такими, как определено выше.
другие переменные являются такими, как определено выше.
[80] Согласно некоторым вариантам реализации фрагмент  выбран из группы, состоящей из
выбран из группы, состоящей из
другие переменные являются такими, как определено выше.
[81] Согласно некоторым вариантам реализации R6 выбран из  другие переменные являются такими, как определено выше.
другие переменные являются такими, как определено выше.
[82] Согласно некоторым вариантам реализации фрагмент  выбран из
выбран из  другие переменные являются такими, как определено выше.
другие переменные являются такими, как определено выше.
[83] Согласно некоторым вариантам реализации фрагмент  выбран из
выбран из  другие переменные являются такими, как определено выше.
другие переменные являются такими, как определено выше.
[84] Указанные выше переменные могут быть произвольно объединены, затем получены другие варианты реализации настоящего изобретения.
[85] Преимущественный эффект
[86] Соединение согласно настоящему изобретению в основном используют в качестве понижающего регулятора рецепторов эстрогена, который обеспечивает эффективную деградацию рецепторов эстрогена и уменьшение его внутриклеточной концентрации, тем самым блокируя путь передачи сигналов эстрогена и ингибируя прогрессирование связанных заболеваний. Фармакофором соединения по настоящему изобретению является 2-замещенный индол. По сравнению с уровнем техники соединение имеет новый фармакофор и демонстрирует новый механизм связывания с мишенью. На левой стороне структуры соединения отсутствует рецептор водородной связи, но соединение обладает превосходной внутриклеточной активностью по деградации рецепторов эстрогена. В то же время вследствие структурных изменений метаболический сайт соединения был уменьшен, в связи с чем соединение не так легко метаболизируется в организме и имеет длительный период полувыведения. Экспериментальные данные in vivo демонстрируют, что по сравнению с уровнем техники соединение по настоящему изобретению имеет более высокое системное содержание у животных, более высокую концентрацию лекарственного средства в опухолевом участке и более высокую концентрацию в плазме в тканях головного мозга, поскольку оно легко проходит гематоэнцефалический барьер. Полагают, что соединение окажет хорошее терапевтическое воздействие на метастатический рак в головном мозге. Соединение по настоящему изобретению может иметь меньшую клиническую дозировку и частоту дозирования, чем ARN-810, его легко вводить и оно имеет сниженные токсические побочные эффекты.
[87] Определения и описание
[88] Если не указано иное, следующие термины и фразы, используемые в настоящем документе, имеют следующие значения. Конкретный термин или фразу не следует считать неопределенным или неясным в отсутствие конкретного определения, но следует понимать в обычном смысле. Если в настоящем документе указывается торговое наименование, оно предназначено для обозначения соответствующего товара или его активного ингредиента. Термин «фармацевтически приемлемый» используется в настоящем документе в контексте соединений, материалов, композиций и/или лекарственных форм, которые подходят для применения с тканями человека и животных в рамках здравого медицинского суждения без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений, и соизмеримых с разумным соотношением польза/риск.
[89] Термин «фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, которую получают путем взаимодействия соединения, имеющего конкретный заместитель по настоящему изобретению, с относительно нетоксичной кислотной или основанием. Если соединение по настоящему изобретению содержит относительно кислотную функциональную группу, соль присоединения основания может быть получена путем приведения нейтральной формы соединения в контакт с достаточным количеством основания в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемая соль присоединения основания включает соль натрия, калия, кальция, аммония, органического амина или магния или подобные соли. Если соединение по настоящему изобретению содержит относительно основную функциональную группу, соль присоединения кислоты может быть получена путем приведения нейтральной формы соединения в контакт с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемой соли присоединения кислоты включают соль неорганической кислоты, где неорганическая кислота включает, например, хлористоводородную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, бикарбонат, фосфорную кислоту, моногидрофосфат, дигидрофосфат, серную кислоту, гидросульфат, йодистоводородную кислоту, фосфористую кислоту и тому подобное; и соль органической кислоты, где органическая кислота включает, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновая кислота, лимонная кислота, винная кислота и метансульфоновую кислоту и тому подобное; и соль аминокислоты (такой как аргинин и тому подобное), а также соль органической кислоты, такой как глюкуроновая кислота и тому подобное (см. Berge et al, "Pharmaceutical Salts", Journal of Pharmaceutical Science 66: 1-19 (1977)). Некоторые конкретные соединения по настоящему изобретению, которые содержат как основные, так и кислотные функциональные группы, могут быть превращены в любую соль присоединения основания или кислоты.
[90] Предпочтительно, нейтральную форму соединения восстанавливают путем приведения соли в контакт с основанием или кислотой традиционным способом, а затем отделения исходного соединения. Разница между исходной формой соединения и его различными солевыми формами заключается в конкретных физических свойствах, таких как разная растворимость в полярном растворителе.
[91] «Фармацевтически приемлемая соль», используемая в настоящем документе, относится к производному соединения по настоящему изобретению, где исходное соединение модифицируют путем образования соли с кислотой или основанием. Примеры фармацевтически приемлемой соли включают, но не ограничиваются ими, соль неорганической кислоты или органической кислоты и основного фрагмента, такого как амин, соль щелочного металла или органическую соль кислотного фрагмента, такого как карбоновая кислота и тому подобное. Фармацевтически приемлемая соль включает традиционную нетоксичную соль или четвертичную аммониевую соль исходного соединения, такую как соль, образованную нетоксичной неорганической кислотой или органической кислотой. Традиционная нетоксичная соль включает, но не ограничивается ей, соль, полученную из неорганической кислоты и органической кислоты, где неорганическая кислота или органическая кислота выбраны из группы, состоящей из 2-ацетоксибензойной кислоты, 2-гидроксиэтансульфоновой кислоты, уксусной кислоты, аскорбиновой кислоты, бензолсульфоновой кислоты, бензойной кислоты, бикарбоната, угольной кислоты, лимонной кислоты, этилендиаминтетрауксусной кислоты, этандисульфоновой кислоты, этансульфоновой кислоты, фумаровой кислоты, глюкогептозы, глюконовой кислоты, глутаминовой кислоты, гликолевой кислоты, бромистоводородной кислоты, хлористоводородной кислоты, гидроиодида, гидроксила, гидроксинафталина, изэтионовой кислоты, молочной кислоты, лактозы, додецилсульфоновой кислоты, малеиновой кислоты, яблочной кислоты, миндальной кислоты, метансульфоновой кислоты, азотная кислота, щавелевой кислоты, памовой кислоты, пантотеновой кислоты, фенилуксусной кислоты, фосфорной кислоты, полигалактановой кислоты, пропионовой кислоты, салициловой кислоты, стеариновой кислоты, слабой уксусной кислоты, янтарной кислоты, сульфаминовой кислоты, сульфаниловой кислоты, серной кислоты, танина, винной кислоты и п-толуолсульфоновой кислоты.
[92] Фармацевтически приемлемая соль по настоящему изобретению может быть получена из исходного соединения, которое содержит кислотный или основной фрагмент традиционным химическим способом. В целом, такую соль можно получить путем взаимодействия свободной кислотной или основной формы соединения со стехиометрическим количеством подходящего основания или кислоты в воде или органическим растворителем или их смеси. Обычно предпочтительными являются неводные среды, такие как простой эфир, этилацетат, этанол, изопропанол или ацетонитрил.
[93] В дополнение к солевой форме соединение, предложенное в настоящим изобретении, также существует в форме пролекарства. Пролекарством описанного в настоящем документе соединения является соединение, которое легко подвергается химическому изменению в физиологических условиях с превращением в соединение по настоящему изобретению. Кроме того, пролекарство может быть превращено в соединение по настоящему изобретению химическим или биохимическим способом в среде in vivo.
[94] Некоторые соединения по настоящему изобретению могут существовать в несольватированной форме или сольватированной форме, включая гидратированную форму. В целом, сольватированная форма эквивалентна несольватированной форме, и обе они включены в объем настоящего изобретения.
[95] Некоторые соединения по настоящему изобретению могут иметь асимметричный атом углерода (оптический центр) или двойную связь. Рацемат, диастереомер, геометрический изомер и индивидуальный изомер включены в объем настоящего изобретения.
[96] Если не указано иное, абсолютная конфигурация стереогенного центра представлена жирной клиновидной связью  и пунктирной клиновидной связью
и пунктирной клиновидной связью 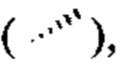 волнистая линия
волнистая линия  представляет собой жирную клиновидную связь или пунктирную клиновидную связь
представляет собой жирную клиновидную связь или пунктирную клиновидную связь  и относительная конфигурация стереогенного центра представлена жирной прямой связью
и относительная конфигурация стереогенного центра представлена жирной прямой связью 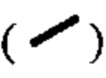 и пунктирной прямой связью
и пунктирной прямой связью  Если описанное в настоящем документе соединение содержит олефиновую двойную связь или другие геометрические асимметричные центры, то включены геометрические изомеры Е и Z, если не указано иное. Аналогично, все таутомерные формы включены в объем настоящего изобретения.
Если описанное в настоящем документе соединение содержит олефиновую двойную связь или другие геометрические асимметричные центры, то включены геометрические изомеры Е и Z, если не указано иное. Аналогично, все таутомерные формы включены в объем настоящего изобретения.
[97] Соединение по настоящему изобретению может иметь конкретную геометрическую или стереоизомерную форму. В настоящем изобретении предполагаются все такие соединения, включая цис и транс-изомер, (-)- и (+)-энантиомер, (R)- и (S)-энантиомер, диастереоизомер, (D)-изомер, (L)-изомер, и рацемическую смесь и другие смеси, например смесь, обогащенную энантиомером или диастереоизомером, все из которых включены в объем настоящего изобретения. Заместитель, такой как алкил, может иметь дополнительный асимметричный атом углерода. Все указанные изомеры и их смеси включены в объем настоящего изобретения.
[98] Оптически активный (R)- и (S)-изомер или D и L-изомер могут быть получены с использованием хирального синтеза или хиральных реагентов или других традиционных методов. Если необходимо получить один вид энантиомера определенного соединения по настоящему изобретению, чистый требуемый энантиомер может быть получен путем асимметричного синтеза или дериватизирующего действия хирального вспомогательного вещества с последующим разделением полученной диастереомерной смеси и отщеплением вспомогательной группы. Альтернативно, если молекула содержит основную функциональную группу (такую как амино) или кислотную функциональную группу (такую как карбоксил), соединение реагирует с соответствующей оптически активной кислотой или основанием с образованием соли диастереомерного изомера, которую затем подвергают диастереомерному разделению посредством традиционного способа в данной области с получением чистого энантиомера. Кроме того, энантиомер и диастереоизомер обычно выделяют хроматографией, в которой используют хиральную неподвижную фазу и которую необязательно объединяют со способом химической дериватизации (например, карбамат, полученный из амина).
[99] Соединение по настоящему изобретению может содержать неестественную долю изотопа атома одного или нескольких атомов, составляющих соединение. Например, соединение может быть помечено радиоактивным изотопом, таким как тритий (3Н), йод-125 (125I) или С-14 (14С). Все изотопные варианты соединения по настоящему изобретению, независимо от того, являются они радиоактивными или нет, включены в объем настоящего изобретения.
[100] Термин «фармацевтически приемлемый носитель» относится к любому агенту или носителю, который способен доставлять эффективное количество активного вещества по настоящему изобретению, не влияет на биологическую активность активного вещества и не проявляет токсических побочных эффектов на хозяина или пациента. Типичный носитель включает воду, масло, растительное и минеральное, кремовую основу, основу для лосьона, мазь и тому подобное. Основа включает суспендирующий агент, загуститель, усилитель проникновения и тому подобное. Их составы хорошо известны специалистам в области косметики или в области фармацевтических средств для местного введения. Дополнительная информация о носителях может быть найдена в Remington: The Science and Practice of Pharmacy, 21st Ed, Lippincott, Williams & Wilkins (2005), описание которой включено в настоящее описание посредством ссылки.
[101] Термин «вспомогательное вещество» в целом относится к носителю, разбавителю и/или среде, необходимому для получения эффективной фармацевтической композиции.
[102] Для лекарственного средства или фармакологически активного агента термин «эффективное количество» или «терапевтически эффективное количество» относится к нетоксичному, но достаточному количеству для достижения желаемого эффекта лекарственного средства или агента. Для пероральной лекарственной формы по настоящему изобретению «эффективное количество» активного вещества в композиции относится к количеству, требуемому для достижения желаемого эффекта при объединении с другим активным веществом в композиции. Эффективное количество варьируется от человека к человеку и определяется в зависимости от возраста и общего состояния реципиента, а также от конкретного активного вещества. Подходящее эффективное количество в отдельном случае может быть определено специалистом в данной области техники на основе рутинного эксперимента.
[103] Термин «активный ингредиент», «терапевтический агент», «активное вещество» или «активный агент» относится к химическому веществу, которое может эффективно лечить целевое нарушение, заболевание или состояние.
[104] «Необязательный» или «необязательно» означает, что последующее событие или условие могут возникать, но не необходимы, и данный термин включает случаи, в которых происходит событие или условие, и случаи, в которых событие или условие не происходит.
[105] Термин «замещенный» означает один или более атомов водорода на конкретном атоме, замещены заместителем, включая варианты с дейтерием и водородом, при условии, что валентность конкретного атома является нормальной и замещенное соединение является стабильным. Если заместитель представляет собой кетон (то есть=O), это означает, что замещены два атома водорода. Положения на ароматическом кольце не могут быть замещены кетоном. Термин «необязательно замещенный» означает, что атом может быть замещен заместителем или не замещен заместителем, если не указано иное, вид и количество заместителя могут быть произвольными, пока они являются химически осуществимыми.
[106] Если какая-либо переменная (например, R) встречается в составе или структуре соединения более одного раза, определение переменной в каждом случае является независимым. Так, например, если группа замещена 0-2 R, группа может необязательно содержать в качестве заместителей вплоть до двух R, где определение R в каждом случае является независимым. Кроме того, комбинация заместителя и/или его варианта допускается только тогда, когда комбинация приводит к стабильному соединению.
[107] Если количество связующих групп равно 0, например -(CRR)0-, это означает, что связующая группа представляет собой одинарную связь.
[108] Если одна из переменных выбрана из одинарной связи, это означает, что две группы, связанные одинарной связью, связаны напрямую. Например, когда L в A-L-Z представляет собой одинарную связь, структура A-L-Z фактически представляет собой A-Z.
[109] Если заместитель является свободным, это означает, что заместитель не существует. Например, если X является свободным в А-Х, то структура А-Х фактически представляет собой А. Если заместитель может быть связан с более чем одним атомом в кольце, такой заместитель может быть связан с любым атомом в кольце. Например, структурное звено 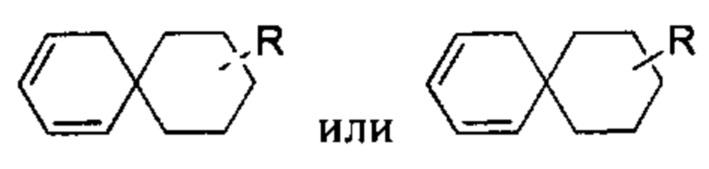 означает, что заместитель R может быть расположен в любом положении на циклогексиле или циклогексадиене. Если перечисленный заместитель не указывает, каким атомом он связан с замещаемой группой, такой заместитель может быть связан посредством любого его атома. Например, когда пиридил выступает в качестве заместителя, он может быть связан с замещаемой группой посредством любого атомом углерода на пиридиновом кольце. Если перечислительная связующая группа не указывает направление связывания, направление для связывания является произвольным, например, если связующая группа L, содержащаяся в
означает, что заместитель R может быть расположен в любом положении на циклогексиле или циклогексадиене. Если перечисленный заместитель не указывает, каким атомом он связан с замещаемой группой, такой заместитель может быть связан посредством любого его атома. Например, когда пиридил выступает в качестве заместителя, он может быть связан с замещаемой группой посредством любого атомом углерода на пиридиновом кольце. Если перечислительная связующая группа не указывает направление связывания, направление для связывания является произвольным, например, если связующая группа L, содержащаяся в 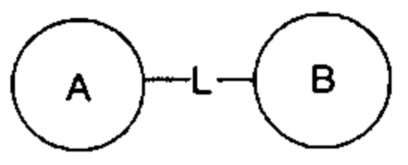 представляет собой -MW-, тогда - MW - может связывать кольцо А и кольцо В в виде
представляет собой -MW-, тогда - MW - может связывать кольцо А и кольцо В в виде 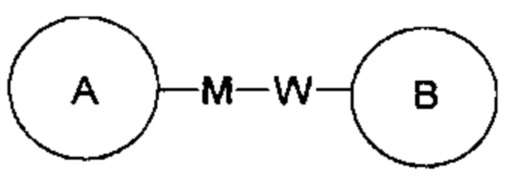 в том же порядке, что и порядок чтения слева направо, и образовывать
в том же порядке, что и порядок чтения слева направо, и образовывать 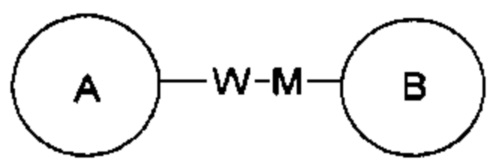 в направлении, противоположном порядку чтения слева направо. Объединение связующей группы, заместителя и/или их вариантов допускается только тогда, когда такое объединение может приводить к стабильному соединению.
в направлении, противоположном порядку чтения слева направо. Объединение связующей группы, заместителя и/или их вариантов допускается только тогда, когда такое объединение может приводить к стабильному соединению.
[110] Если не указано иное, термин «гетеро» представляет собой гетероатом или гетероатомную группу (например, атомную группу, содержащую гетероатом), включая атом, за исключением углерода (С) и водорода (Н), и атомную группу, содержащую указанный гетероатом, включающие, например, кислород (О), азот (N), серу (S), кремний (Si), германий (Ge), алюминий (Al), бор (В), -О-, -S-,=O,=S, -С(=O)O-, -С(=O)-, -C(=S)-, -S(=O), -S(=O)2-, и группу, состоящую из -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2N(H)- и -S(=O)N(H)-, каждый из которых необязательно замещен.
[111] Если не указано иное, термин «кольцо» относится к замещенному или незамещенному циклоалкилу, гетероциклоалкилу, циклоалкенилу, гетероциклоалкенилу, циклоалкинилу, гетероциклоалкинилу, арилу или гетероарилу. Так называемое кольцо включает одно кольцо, совокупность колец, спиральное кольцо, конденсированное кольцо или мостиковое кольцо. Количество атомов в кольце обычно определяется как количество членов в кольце, например «5-7-членное кольцо» означает, что в кольце содержится 5-7 атомов.
Если не указано иное, кольцо необязательно содержит от 1 до 3 гетероатомов. Следовательно, «5-7-членное кольцо» включает, например, фенил, пиридинил и пиперидинил; с другой стороны, термин «5-7-членное гетероциклоалкильное кольцо» включает пиридил и пиперидинил, но исключая фенил. Термин «кольцо» также включает кольцевую систему, содержащую по меньшей мере одно кольцо, причем каждое кольцо независимо соответствует указанному выше определению.
[112] Если не указано иное, термин «гетероцикл» или «гетероцикло» относится к стабильному моноциклическому, бициклическому или трициклическому кольцу, содержащему гетероатом или гетероатомную группу, которая может быть насыщенной, частично ненасыщенной или ненасыщенной (ароматической) и может содержать атомы углерода и 1, 2, 3 или 4 кольцевых гетероатома, независимо выбранных из N, О и S, где любой из указанных выше гетероциклов может быть конденсирован с бензольным кольцом с образованием бициклического кольца. Гетероатомы азота и серы могут быть необязательно окислены (то есть, NO и S(O)p, р равно 1 или 2). Атом азота может быть замещенным или незамещенным (то есть, N или NR, где R представляет собой Н или другие заместители, уже определенные в настоящем документе). Гетероцикл может быть присоединен к боковой группе любого гетероатома или атома углерода с образованием стабильной структуры. Если полученное соединение является стабильным, описанный в настоящем документе гетероцикл может иметь замещение по положению углерода или азота. Атом азота в гетероцикле необязательно кватернизирован. В предпочтительном варианте реализации, когда общее количество атомов S и О в гетероцикле больше 1, гетероатомы не являются соседними. В другом предпочтительном варианте реализации, общее количество атомов S и О в гетероцикле не превышает 1. Используемый в настоящем документе термин «ароматическая гетероциклическая группа» или «гетероарил» относится к стабильному 5-, 6- или 7-членному моноциклическому или бициклическому или 7-, 8-, 9- или 10-членному бициклическому гетероциклическому ароматическому кольцу, которое содержит атомы углерода и 1, 2, 3 или 4 кольцевых гетероатомов, независимо выбранных из N, О и S. Азот азота может быть замещенным или незамещенным (то есть, N или NR, где R представляет собой Н или другие заместители, уже определенные в настоящем документе). Гетероатомы азота и серы могут быть необязательно окислены (то есть, NO и S(O)p, р равно 1 или 2). Стоит отметить, что общее количество атомов S и О ароматического гетероцикла составляет не более одного. Мостиковое кольцо также включено в определение гетероцикла. Мостиковое кольцо образуется, когда один или несколько атомов (то есть, С, О, N или S) связывают два несмежных атома углерода или азота. Предпочтительное мостиковое кольцо включает (но не ограничивается ими) один атом углерода, два атома углерода, один атом азота, два атома азота и одну группу углерод-азот. Стоит отметить, что мостик всегда преобразует моноциклическое кольцо в трициклическое кольцо. В мостиковом кольце заместитель в кольце может также присутствовать в мостике.
[113] Примеры гетероциклического соединения включают, но не ограничиваются ими: акридинил, азоцинил, бензимидазолил, бензофуранил, бензомеркаптофуранил, бензомеркаптофенил, бензоксазолил, бензоксазолинил, бензотиазолил, бензотриазолил, бензотетразолил, бензоизоксазолил, бензоизотиазолил, бензоимидазолинил, карбазолил, 4aH-карбазолил, карбонил, хроманил, хромен, циннолинил декагидрохинолинил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индоленил, индолинил, индолизинил, индолил, 3H-индолил, изобензофуранил, изоиндолил, изоиндолинил, изохинолинил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, гидроксиндолил, пиримидинил, фенантридинил, фенантролинил, феназин, фенотиазин, бензоксантинил, фенолоксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонил, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазолил, пиридоимидазолил, пиридитиазолил, пиридинил, пирролидинил, пирролинил, 2Н-пирролил, пирролил, хиназолинил, хинолинил, 4Н-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, изотиазолилтиенил, тиенил, тиенооксазолил, тиенотиазолил, тиеноимидазолил, тиенил, триазинил, 1H-1,2,3-триазолил, 2H-1,2,3-триазолил, 1H-1,2,4-триазолил, 4H-1,2,4-триазолил и ксантенил. Также включены соединения с конденсированными кольцами и спиросоединения.
[114] Если не указано иное, термин «гидрокарбил» или его гипонимы (например, алкил, алкенил, алкинил и арил и т.д.) сам по себе или как часть другого заместителя относится к линейному, разветвленному или циклическому углеводородному радикалу или любой их комбинации. Они могут быть полностью насыщенными (например, алкил), моно- или полиненасыщенными (например, алкенил, алкинил и арил), могут быть моно-, ди- или полизамещенными, могут быть одновалентными (например, метил), двухвалентными (например, метилен) или поливалентными (например, метенил), могут также включать двухвалентную или поливалентную группу, имеют определенное количество атомов углерода (например, С1-С12 обозначает от 1 до 12 атомов углерода, С1-С12 выбран из С2, С3, С4, С5, С6, С7, C8, С9, С10, С11 и С12, С3-12 выбран из С3, С4, C5, С6, С7, C8, С9, С10, С11 и С12). Термин «гидрокарбил» включает, но не ограничивается ими, алифатический гидрокарбил и ароматический гидрокарбил. Алифатический гидрокарбил включает линейный и циклический гидрокарбил, в частности включает, но не ограничивается ими, алкил, алкенил и алкинил. Ароматический гидрокарбил включает, но не ограничивается ими, 6-12-членный ароматический гидрокарбил, такой как фенил, нафтил и тому подобное. Согласно некоторым вариантам реализации термин «гидрокарбил» относится к линейной или разветвленной группе или их комбинации, которая может быть полностью насыщенной, моно- или полиненасыщенной и может включать двухвалентную или поливалентную группу. Примеры насыщенной гидрокарбильной группы включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, циклогексил, (циклогексил)метил, циклопропилметил и гомолог или изомер н-амильной, н-гексильной, н-гептильной, н-октильной и других групп атомов. Ненасыщенный гидрокарбил содержит одну или несколько двойных или тройных связей. Примеры ненасыщенного алкила включают, но не ограничиваются ими, винил, 2-пропенил, бутенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и более высшие гомологи и изомеры.
[115] Если не указано иное, термин «гетерогидрокарбил» или его гипонимы (такие как гетероалкил, гетероалкенил, гетероалкинил и гетероарил и т.д.) сам по себе или как часть другого заместителя относится к стабильной линейной, разветвленной или циклической углеводородной группе или любой их комбинации, которая содержит определенное количество атомов углерода и по меньшей мере один гетероатом. Согласно некоторым вариантам реализации термин «гетероалкил» сам по себе или в сочетании с другим термином относится к стабильному линейному, разветвленному углеводородному радикалу или их комбинации, который содержит определенное количество атомов углерода и по меньшей мере один гетероатом. Согласно конкретному варианту реализации гетероатом выбирают из В, О, N и S, где атомы азота и серы необязательно окислены и атом азота необязательно кватернизирован. Гетероатом или гетероатомная группа может быть расположена в любом внутреннем положении гетерогидрокарбила, включая положение, по которому гидрокарбил присоединен к остальной части молекулы. Но термины «алкокси», «алкиламино» и «алкилтио» (или тиоалкил) используются в обычном значении и относятся к алкильной группе, связанной с остальной частью молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Примеры включают, но не ограничиваются ими, -СН2-СН2-О-СН3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-СН3, -CH2-S-CH2-CH3, -СН2-СН2, -S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-СН3, -CH2-CH=N-OCH3 и -CH=CH-N(CH3)-CH3. Могут присутствовать до двух последовательных гетероатомов, например, -CH2-NH-OCH3.
[116] Если не указано иное, термин «циклогидрокарбил», «гетероциклогидрокарбил» или его гипонимы (такие как арил, гетероарил, циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил и т.д.) сам по себе или в сочетании с другим термином относится к циклизованному «гидрокарбилу» или «гетерогидрокарбилу». Кроме того, для гетерогидрокарбила или гетероциклогидрокарбила (например, гетероалкила и гетероциклоалкила) один гетероатом может занимать положение, по которому гетероцикл присоединяется к остаточной части молекулы. Примеры циклоалкила включают, но не ограничиваются ими, циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и тому подобное. Неограничивающие примеры гетероциклоалкила включают 1-(1,2,5,6-тетрагидропиридил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, тетрагидротиофен-2-ил, тетрагидротиофен-3-ил, 1-пиперазинил и 2-пиперазинил.
[117] Если не указано иное, термин «алкил» относится к линейной или разветвленной насыщенной углеводородной группе, может быть монозамещен (например, -CH2F) или полизамещен (например, -CF3), может быть моновалентным (например, метил), двухвалентным (например, метилен) или поливалентным (например, метенил). Примеры алкила включают метил (Me), этил (Et), пропил (такой как н-пропил и изопропил), бутил (такой как н-бутил, изобутил, втор-бутил, трет-бутил), пентил (такой как н-пентил, изопентил, неопентил) и тому подобное.
[118] Если не указано иное, термин «алкенил» относится к алкильной группе, имеющей одну или несколько углерод-углеродных двойных связей в любом положении в цепи, может быть монозамещен или полизамещен и может быть моновалентным, двухвалентным или поливалентным. Примеры алкенила включают этенил, пропенил, бутенил, пентенил, гексенил, бутадиенил, пентадиенил, гексадиенил и тому подобное.
[119] Если не указано иное, термин «алкинил» относится к алкильной группе, имеющей одну или несколько углерод-углеродных тройных связей в любом положении цепи, может быть монозамещен или полизамещен и может быть моновалентным, двухвалентным или поливалентным. Примеры алкинила включают этинил, пропинил, бутинил, пентинил, гексинил и тому подобное.
[120] Если не указано иное, циклоалкил включает любой стабильный циклический или полициклический гидрокарбил и любой атом углерода насыщен, может быть монозамещенным или полизамещенным и может быть моновалентным, двухвалентным или поливалентным. Примеры циклоалкила включают, но не ограничиваются ими, циклопропил, норборнанил, [2.2.2]бициклооктан, [4.4.0]бициклодеканил и тому подобное.
[121] Если не указано иное, циклоалкенил включает любой стабильный циклический или полициклический гидрокарбил, имеющий одну или несколько ненасыщенных углерод-углеродных одинарных связей в любом положении в кольце, может быть монозамещенным или полизамещенным и может быть моновалентным, двухвалентным или поливалентный. Примеры циклоалкенила включают, но не ограничиваются ими, циклопентенил, циклогексенил и тому подобное.
[122] Если не указано иное, циклоалкинил включает любой стабильный циклический или полициклический гидрокарбил, имеющий одну или несколько тройных связей углерод-углерод в любом положении в кольце, может быть монозамещенным или полизамещенным и может быть моновалентным, двухвалентным или поливалентным.
[123] Если не указано иное, термин «галоген» или «гало» сам по себе или как часть другого заместителя относится к атому фтора, хлора, брома или йода. Кроме того, термин «галогеналкил» означает моногалогеналкил и полигалогеналкил. Например, термин «галоген(С1-С4) алкил» включает, но не ограничивается ими, трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и тому подобное. Примеры галогеналкила включают, но не ограничиваются ими, трифторметил, трихлорметил, пентафторэтил и пентахлорэтил.
[124] Термин «алкокси» представляет собой любой алкил, определенный выше, имеющий определенное количество атомов углерода, присоединенных посредством кислородного мостика. Если не указано иное, C1-6 алкокси включает C1, С2, С3, С4, C5 и С6алкокси. Примеры алкокси включают, но не ограничиваясь ими, метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и S-пентокси.
[125] Если не указано иное, термин «арил» относится к полиненасыщенному ароматическому заместителю, который может быть моно-, ди- или полизамещенным, может быть одновалентным, двухвалентным или поливалентным, может представлять собой одно кольцо или множество колец (например, от одного до трех колец, причем по меньшей мере одно кольцо является ароматическим), которые конденсированы или связаны ковалентно. Термин «гетероарил» относится к арилу (или кольцу), содержащему от одного до четырех гетероатомов. В иллюстративном примере гетероатом выбирают из В, О, N и S, где атомы азота и серы необязательно окислены и атом азота необязательно кватернизирован. Гетероарил может присоединяться к остальной части молекулы через гетероатом. Неограничивающие примеры арила или гетероарила включают фенил, нафтил, бифенил, пирролил, пиразолил, имидазолил, пиразинил, оксазолил, фенилоксазолил, изоксазолил, тиазолил, фуранил, тиенил, пиридил, пиримидинил, бензотиазолил, пуринил, бензимидазолил, индолил, изохинолил, хиноксалинил, хинолил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фурил, 3-фурил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидил, 4-пиримидил, 5-бензотиазолил, пуринил, 2-бензимидазолил, 5-индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалинил, 5-хиноксалинил, 3-хинолил и 6-хинолил. Заместитель любого из указанных выше арильных и гетероарильных кольцевых систем выбирают из приемлемого заместителя, описанного ниже.
[126] Если не указано иное, в сочетании с другими терминами (такими как арилокси, арилтио, арилалкил) арил включает арильное и гетероарильное кольцо, как определено выше. Таким образом, термин «аралкил» означает группу (например, бензил, фенэтил, пиридилметил и т.д.), где арил присоединен к алкилу, включая алкил, в котором атом углерода (например, метилен) заменен атомом, таким как кислород, например феноксиметил, 2-пиридилокси, 3-(1-нафтилокси)пропил и тому подобное.
[127] Термин «уходящая группа» относится к функциональной группе или атому, которая может быть заменена другой функциональной группой или атомом посредством реакции замещения (такой как реакция нуклеофильного замещения). Например, типичные уходящие группы включают трифлат; хлор, бром и йод; сульфонатную группу, такую как мезилат, тозилат, п-бромбензолсульфонат, п-толуолсульфонаты и тому подобное; ацилокси, такую как ацетокси, трифторацетокси и тому подобное.
[128] Термин «защитная группа» включает, но не ограничивается ими, «аминозащитную группу», «гидроксизащитную группу» или «тиозащитную группу». Термин «аминозащитная группа» относится к защитной группе, подходящей для блокирования боковой реакции по азоту аминогруппы. Типичные аминозащитные группы включают, но не ограничиваются ими: формил; ацил, такой как алканоил (например, ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такой как трет-бутоксикарбонил (Вос); арилметоксикарбонил, такой как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметил, такой как бензил (Bn), тритил (Tr), 1,1-бис-(4'-метоксифенил)метил; силил, такой как триметилсилил (ТМС) и трет-бутилдиметилсилил (TBS) и тому подобное. Термин «гидроксизащитная группа» относится к защитной группе, подходящей для блокирования боковой реакции по гидроксигруппе. Типичные гидроксизащитные группы включают, но не ограничиваются ими: алкил, такой как метил, этил и трет-бутил; ацил, такой как алканоил (например, ацетил); арилметил, такой как бензил (Bn), п-метоксибензил (РМВ), 9-флуоренилметил (Fm) и дифенилметил (бензгидрил, DPM); силил, такой как триметилсилил (ТМС) и трет-бутилдиметилсилил (TBS) и тому подобное.
[129] Соединение по настоящему изобретению может быть получено различными способами синтеза, хорошо известными специалистам в данной области техники, включая следующие перечислительные варианты реализации, варианты реализации, образованные следующими перечисленными вариантами реализации в сочетании с другими методами химического синтеза и эквивалентными заменами, хорошо известными специалистам в данной области техники. Предпочтительный вариант реализации включает, но не ограничивается этим, вариант реализации настоящего изобретения.
[130] Все растворители, используемые в настоящем изобретении, являются коммерчески доступными. Реакцию обычно проводили в безводном растворителе в инертной атмосфере азота. Данные протонного ядерного магнитного резонанса регистрировали на спектрометре Bruker Avance III 400 (400 МГц) и химический сдвиг был представлен как δ (ppm) в слабом поле тетраметилсилана. Масс-спектр получали на Agilent 1200 Series Plus 6110 (&1956А). ЖХ/МС или Shimadzu MS включали детектор DAD:SPD-M20A (LC) и Shimadzu Micromass 2020. Масс-спектрометр оснащен источником электрораспыления ионов (ESI), работающим в положительном или отрицательном режиме.
[131] В настоящем изобретении используются следующие сокращения: экв. представляет собой эквивалентный; ДХМ представляет собой дихлорметан; РЕ представляет собой петролейный эфир; ДМФА представляет собой N,N-диметилформамид; ДМСО представляет собой диметилсульфоксид; EtOAc или ЕА представляет собой этилацетат; EtOH представляет собой этанол; МеОН представляет собой метанол; Cbz представляет собой бензилоксикарбонил, который представляет собой аминозащитную группу; Boc представляет собой трет-бутилкарбонил, который представляет собой аминозащитную группу; ТГФ представляет собой тетрагидрофуран; Boc2O представляет собой ди-трет-бутилдикарбонат; DBU представляет 1,8-диазабицикло[5.4.0]ундец-7-ен; NBS представляет собой N-бромсукцинамид; NCS представляет собой N-хлорсукцинамид; НМРА представляет собой гексаметилфосфорамид; LDA представляет собой диизопропиламид лития; DPPF или dppf представляет бис(дифенилфосфино)ферроцен; MeCN представляет собой ацетонитрил; BBr3 представляет собой трибромид бора; Ad2nBuP представляет собой ди(адамантан-1 -ил)(бутил)фосфин.
[132] Анализ высокоэффективной жидкостной хроматографии проводили на системе Shimadzu LC20AB, оснащенной автоматическим пробоотборником Shimadzu SIL-20A и Shimadzu DAD детектора SPD-M20A. Использовали колонку Xtimate С18, 3 мкм, 2,1×300 мм. Способ 0-60АВ_6 мин: использовали линейный градиент, начиная с 100% А (А: 0,0655% ТФУ в воде) и заканчивая 60% В (В: 0,0625% ТФУ в MeCN) в течение 4,2 мин, с последующим элюированием с 60% В в течение 1 мин. Затем колонку повторно уравновешивали в течение 0,8 мин до 100:0 с общим временем хроматографирования 6 мин. Способ 10-80АВ_6 мин: использовали линейный градиент, начиная с 90% А (А: 0,0655% ТФУ в воде) и заканчивая 80% В (В: 0,0625% ТФУ в MeCN) в течение 4,2 минут, с последующим элюированием с 80% В в течение 1 мин. Затем колонку повторно уравновешивали в течение 0,8 мин до 90:10 с общим временем хроматографирования 6 мин. Температура колонки составляла 50°С, скорость потока составляла 0,8 мкл/мин. Детектор на диодной матрице сканировал от 200 до 400 нм.
[133] Тонкослойную хроматографию (ТСХ) проводили на пластинке силикагеля Sanpont-group GF254. Пятна обычно визуализировали методом УФ-облучения, а также использовали другие способы визуализации пятен на пластине ТСХ. В указанных методах I2(1 г I2 тщательно примешивали в 10 г силикагеля), ванилин (1 г ванилина, растворенного в 100 мл 10% H2SO4), нингидрин (поставляемый Aldrich) или специальный реагент (25 г (NH4)6Mo7O24⋅4H2O и 5 г (NH4)2Ce(IV)(NO3)6, растворенного в 450 мл H2O и 50 мл конц. H2SO4) использовали для визуализации пятен соединений. Фракционную колоночную хроматографию проводили на силикагеле Silicycle 40-63 мкм (230-400 меш) с помощью способов аналогичных, описанным в Still, W. С; Kahn, М; Mitra, М. J. Org. Chem. 1978, 43, 2923-2925. Элюент для колоночной флэш-хроматографии или ТСХ включал смешанные растворители дихлорметан/метанол, этилацетат/метанол или этилацетат/н-гексан.
[134] Препаративный хроматографический анализ проводили на системе Gilson-281 Prep LC 322 с детектором Gilson UV/VIS-156. Колонка представляла собой Agella Venusil ASB Prep С18, 5 мкм, 150×21,2 мм, Phenomenex Gemini C18, 5 мкм, 150×30 мм, Boston Symmetrix C18, 5 мкм, 150×30 мм или Phenomenex Synergi С18, 4 мкм, 150×30 мм. При скорости потока примерно 25 мл/мин соединение элюировали MeCN/H2O с низким градиентом, где вода содержала 0,05% HCl, 0,25% НСООН или 0,5% NH3.H2O, и общее время храматографирования составляло 8-15 мин.
[135] Анализ SFC проводили на системе Agilent 1260 Infinity SFC, оборудованной автосамплером Agilent 1260 и детектором Agilent DAD 1260. Использовали колонку Chiralcel OD-H 250×4,6 мм I.D., 5 мкм или Chiralpak AS-H 250×4,6 мм I.D., 5 мкм или Chiralpak AD-H 250×4,6 мм I.D., 5 мкм. В способе OD-Н_5_40_2.35 мл колонка была Chiralcel OD-H 250×4,6 мм I.D., 5 мкм, подвижная фаза составляла 40% этанол (0,05% DEA) в СО2, скорость потока составляла 2,35 мл/мин, длина волны составляла 220 нм. В способе AS-H_3_40_2.35 мл: колонка была Chiralpak AS-H 250×4,6 мм I.D., 5 мкм, подвижная фаза составляла 40% метанол (0,05% DEA) в СО2, скорость потока составляла 2,35 мл/мин, длина волны составляла 220 нм. В способе OD-H_3_40_2.35M: колонка была Chiralcel OD-H 250×4,6 мм I.D., 5 мкм, подвижная фаза составляла 40% метанол (0,05% DEA) в СО2, скорость потока составляла 2,35 мкл/мин, длина волны составляла 220 нм. В способе AD-H_2_50_2.35 мл: колонка была Chiralpak AD-H 250×4,6 мм I.D., 5 мкм, подвижная фаза составляла 50% метанол (0,1% МЕА) в СО2, скорость потока составляла 2,35 мл / мин, длина волны составляла 220 нм.
[136] Препаративный SFC-анализ проводили на системе Waters Thar 80 Pre-SFC с детектором Gilson UV. Использовали колонку Chiralcel OD-H 250×4,6 мм I.D., 5 мкм или Chiralpak AD-H 250×4,6 мм I.D., 5 мкм. Подвижную фазу этанола в СО2 или метанола в СО2 с низким градиентом использовали для элюирования соединения со скоростью потока 40-80 мкл/мин, где этанол или метанол содержал 0,05% NH3.H2O или 0,05% DEA или 0,1% МЕА, и общее время хроматографирования составляло 20-30 мин.
[137] Соединения называются вручную или программным обеспечением ChemDraw®, для коммерчески доступных соединений используются наименования поставщиков.
Подробное описание предпочтительного варианта реализации
[138] Настоящее изобретение будет конкретно описано ниже посредством вариантов реализации, однако объем настоящего изобретения ими не ограничивается.
[139] Специалисту в данной области техники будет понятно, что порядок стадий реакции в любой реакционной схеме может варьироваться для получения соединения по настоящему изобретению, и также включен в объем настоящего изобретения.
Вариант реализации 1
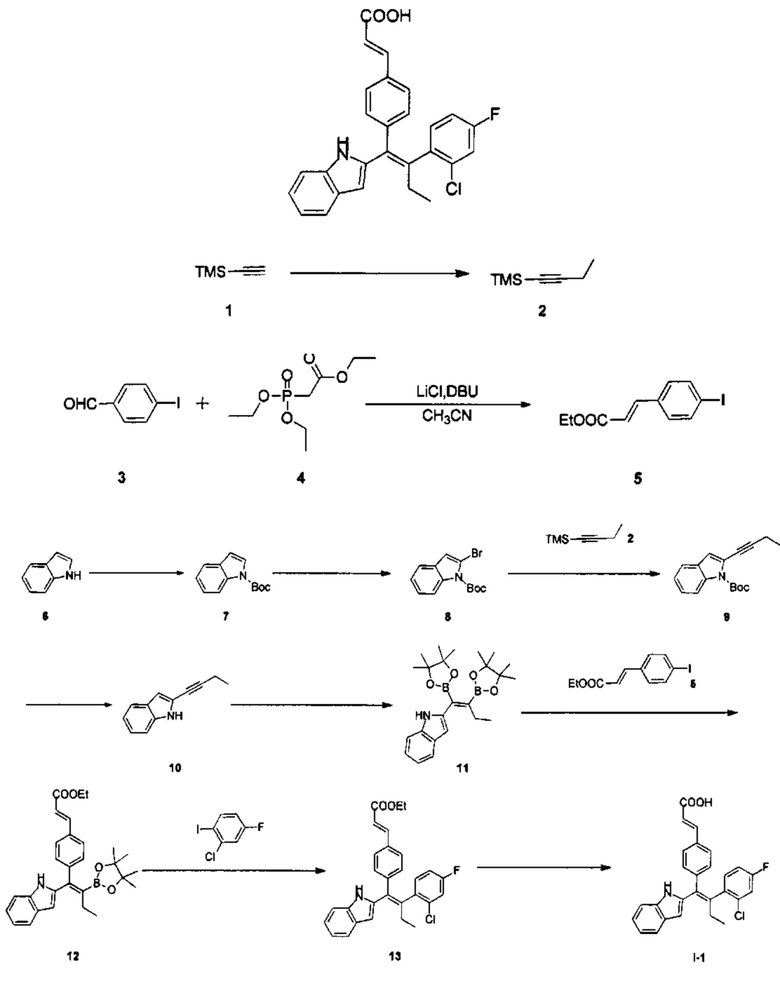
[140] Стадия А: N-бутиллитий (2,5М, 428,40 мл, 1,05 экв.) медленно добавляли по каплям к раствору 1 (100,00 г, 1,02 моль, 140,85 мл, 1,00 экв.) в тетрагидрофуране (500 мл) в течение 1 часа при -75°С в атмосфере азота. Реакционный раствор оставляли нагреваться до 0°С и перемешивали в течение 10 минут, затем охлаждали до -75°С и гексаметилтриамид фосфорной кислоты (201,06 г, 1,12 моль, 197,12 мл, 1,10 экв.) добавляли в течение 1 часа. После перемешивания реакционного раствора при -75°С в течение 1 часа, этилйодид (198,86 г, 1,27 моль, 101,98 мл, 1,25 экв.) добавляли в течение 1 часа.
Реакционный раствор оставляли нагреваться до 20°С и перемешивали в течение 10 часов. Затем добавляли 400 мл воды и смесь разделяли. Органическую фазу три раза промывали 400 мл воды, сушили над безводным сульфатом натрия, фильтровали и очищали путем перегонки с получением продукта 2 в виде бесцветной жидкости (65,00 г, 514,77 ммоль, выход 50,47%).
[141] Стадия В: Триэтилфосфоноацетат 4 (11,60 г, 51,72 ммоль, 10,26 мл, 1,20 экв.) и хлорид лития (3,65 г, 86,20 ммоль, 1,77 мл, 2,00 экв.) добавляли к раствору 3 (10,00 г, 43,10 ммоль, 1,00 экв.) в 100 мл ацетонитрила, затем раствор DBU (8,53 г, 56,03 ммоль, 8,45 мл, 1,30 экв.) в ацетонитриле добавляли по каплям в течение 30 мин при 0°С в атмосфере азота. После перемешивания реакционного раствора при 15°С в течение 1 часа добавляли 100 мл воды и смесь разделяли. Водную фазу экстрагировали дважды 70 мл дихлорметана. Органическую фазу объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (РЕ:ЕА=100:1-30:1) с получением продукта 5 в виде желтого твердого вещества (12,00 г, 39,72 ммоль, выход 92,16%).
[142] Стадия С: Диметиламинопиридин (3,65 г, 29,88 ммоль, 0,10 экв.) и Вос2О (68,46 г, 313,70 ммоль, 72,07 мл, 1,05 экв.) добавляли к раствору 6 (35,00 г, 298,76 ммоль, 1,00 экв.) в 400 мл дихлорметана. Реакционный раствор перемешивали при 20°С в течение 12 часов, затем дважды промывали 400 мл водного раствора хлорида аммония. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением продукта 7 в виде бесцветного масла (60,00 г, 276,17 ммоль, выход 92,44%).
[143] Стадия D: Диизопропиламид лития (2М, 75,95 мл, 1,10 экв.) медленно добавляли по каплям к раствору 7 (30,00 г, 138,08 ммоль, 1,00 экв.) в тетрагидрофуране (400 мл) при -75°С в атмосфере азота. Реакционный раствор перемешивали при -75°С в течение 30 минут, затем добавляли бромистый циан (55,40 г, 523,04 ммоль, 38,47 мл, 3,79экв.). Реакционный раствор оставляли нагреваться до 15°С и перемешивали в течение 12 часов, затем добавляли 400 мл воды. После разделения смеси органическую фазу три раза промывали 300 мл воды, сушили над безводным сульфатом натрия и фильтровали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (РЕ:ЕА=1:0-50:1), с получением продукта 8 в виде желтого масла (39,00 г, неочищенный продукт).
[144] Стадия Е: Карбонат цезия (85,81 г, 263,38 ммоль, 2,00 экв.), йодид меди (1,25 г, 6,58 ммоль, 0,05 экв.), ацетат палладия (1,48 г, 6,58 ммоль, 0,05 экв.) и 1,1'-бис(дифенилфосфино)ферроцен (3,65 г, 6,58 ммоль, 0,05 экв.) добавляли к раствору 8 (39,00 г, 131,69 ммоль, 1,00 экв.) в 300 мл N,N-диметилацетамиде, с последующим добавлением 2 (33,26 г, 263,38 ммоль, 2,00 экв.) в атмосфере азота. Реакционный раствор перемешивали при 80°С в течение 12 часов, затем добавляли 1 л этилацетат и 1 л воды. После разделения и фильтрования смеси органическую фазу три раза промывали 1 л воды, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (РЕ:ЕА=1:0-30:1), с получением продукта 9 в виде желтого масла (27,00 г, неочищенный продукт).
[145] Стадия F: Карбонат калия (69,27 г, 501,25 ммоль, 5,00 экв.) добавляли к раствору 9 (27,00 г, 100,25 ммоль, 1,00 экв.) в 300 мл метанола и 15 мл воды. Реакционный раствор перемешивали при 70°С в течение 12 часов, затем фильтровали и концентрировали. Добавляли 300 мл этилацетат и дважды промывали 300 мл воды. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (РЕ:ЕА=100:1-30:1) с получением продукта 10 в виде желтого твердого вещества (7,50 г, 44,32 ммоль, выход 44,21%). MS [ESI, M+1]: 170,1.
[146] Стадия G: Биспинаколатодибор (4,50 г, 17,73 ммоль, 1,00 экв.) и тетракис(трифенилфосфин)платину (1,10 г, 886,50 мкмоль, 0,05 экв.) добавляли к раствору 5 (3,00 г, 17,73 ммоль, 1,00 экв.) в 30 мл диметилтетрагидрофурана. Реакционный раствор перемешивали при 70°С в атмосфере азота в течение 5 часов и затем охлаждали до комнатной температуры. Продукт 6, содержащийся в реакционном растворе, использовали на следующей стадии без очистки.
[147] Стадия Н: Карбонат цезия (11,55 г, 35,44 ммоль, 2,00 экв.), соединение 5 (4,28 г, 14,18 ммоль, 0,80 экв.) и бис(трифенилфосфин)палладия дихлорид (622,02 мг, 886,00 мкмоль, 0,05 экв.) добавляли к раствору 10 (7,50 г, 17,72 ммоль, 1,00 экв.) в 70 мл диметилтетрагидрофурана и 3 мл воды. Реакционный раствор перемешивали при 15°С в атмосфере азота в течение 12 часов. Продукт 11, содержащийся в реакционном растворе, использовали на следующей стадии без очистки.
[148] Стадия I: 2-Хлор-4-фториодобензол (9,03 г, 35,22 ммоль, 2,00 экв.), раствор гидроксида калия (4М, 22,01 мл, 5,00 экв.) и бис(трифенилфосфин)палладия дихлорид (617,94 мг, 880,50 мкмоль, 0,05 экв.) добавляли к раствору 11 (8,30 г, 17,61 ммоль, 1,00 экв.) в 100 мл диметилтетрагидрофурана. Реакционный раствор перемешивали при 70°С в атмосфере азота в течение 12 часов, затем фильтровали через целит и фильтрат дважды промывали 100 мл солевого раствора. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (РЕ:ЕА=40:1-10:1) с получением продукта 13 в виде желтого твердого вещества (4,50 г, 5,57 ммоль, выход 31,65%, чистота 58,7%). MS [ESI, M+1]: 474,3.
[149] Стадия J: Гидроксид лития (1,33 г, 55,73 ммоль, 10,00 экв.) добавляли к раствору 13 (4,50 г, 5,57 ммоль, 1,00 экв.) в смешанном растворителе 30 мл метанола, 30 мл тетрагидрофурана и 10 мл воды. Реакционный раствор перемешивали при 35°С в течение 1 часа, затем добавляли 30 мл воды. Реакционный раствор доводили до рН 5 1М соляной кислотой, затем дважды экстрагировали 50 мл этилацетата. Органическую фазу объединяли, дважды промывали 60 мл воды, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ (система с муравьиной кислотой) с получением продукта I-1 (2,10 г, 4,69 ммоль, выход 84,13%, чистота 99,5%). MS [ESI, M+1]: 446,1.
[150] 1Н ЯМР EW3644-175-P1B (400 МГц, ДМСО-d6): δ 12,34 (s, 1H), 10,75 (s, 1Н), 7,56 (d, J=7,6 Гц, 1Н), 7,47-7,42 (m, 3Н), 7,36 (dd, J=8,8 Гц, J=1,6 Гц, 1Н), 7,30 (d, J=8,0 Гц, 1Н), 7,25 (dd, J=8,8 Гц, J=6,4 Гц, 1Н), 7,12 (dt, J=2,4 Гц, J=8,4 Гц, 1Н), 7,06 (t, J=7,6 Гц, 1Н), 7,01-6,96 (m, 3Н), 6,57 (s, 1H), 6,41 (d, J=16,0 Гц, 1 Н), 2,71-2,59 (m, 2 Н), 0,99 (t, J=7,2 Гц, 3 Н).
Вариант реализации 2
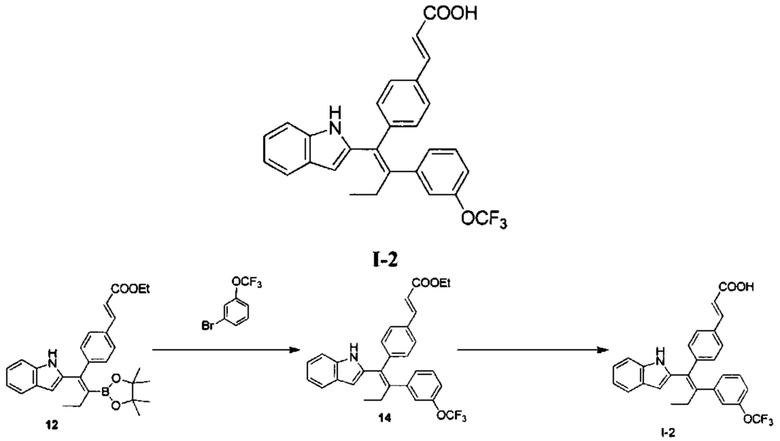
[151] Стадия А: Раствор соединения 12 (556,08 мг, 1,18 ммоль, 1,00 экв.), 3-трифторметилбромбензол (572,85 мг, 2,11 ммоль, 1,80 экв.), водный раствор гидроксида калия (4М, 1,64 мл, 5,60 экв.), дихлорбис(трифенилфосфин)палладий (24,64 мг, 35,10 мкмоль, 0,03экв.) в 20 мл 2-метилтетрагидрофурана три раза продували азотом. Реакционный раствор перемешивали при 80°С в атмосфере азота в течение 24 часов, затем разбавляли 20 мл воды, дважды экстрагировали 50 мл этилацетата каждый раз. Органическую фазу объединяли, промывали один раз 50 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии с получением соединения 14 в виде желтого масла (200,00 мг), которое непосредственно использовали на следующей стадии.
[152] Стадия В: Моногидрат гидроксида лития (83,00 мг, 1,98 ммоль, 5,00 экв.) добавляли к раствору соединения 14 (200,00 мг, 408,56 мкмоль, 1,00 экв.) в 20 мл метанола и 6 мл воды, реакционный раствор перемешивали при 30°С в течение 3 часов, затем выливали в 100 мл воды. Смесь доводили до рН 5-6 с помощью 3 моль/л соляной кислоты, затем дважды экстрагировали 100 мл дихлорметана каждый раз. Органическую фазу объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной хроматографии (муравьиная кислота) с получением I-2 (22,00 мг, 46,08 мкмоль, выход 11,65%). MS [ESI, M+1]: 478,1.
[153] 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,78 (br. s., 1H), 7,55 (d, J=7,2 Гц, 1Н), 7,50 - 7,27 (m, 5Н), 7,23 (d, J=7,6 Гц, 1Н), 7,14 (d, J=7,6 Гц, 1Н), 7,10 - 6,85 (m, 5Н), 6,54 (br. s., 1Н), 6,42 (d, J=16,0 Гц, 1Н), 2,65-2,78 (m, 2Н), 0,98 (t, J=6,8 Гц, 3Н).
Вариант реализации 3
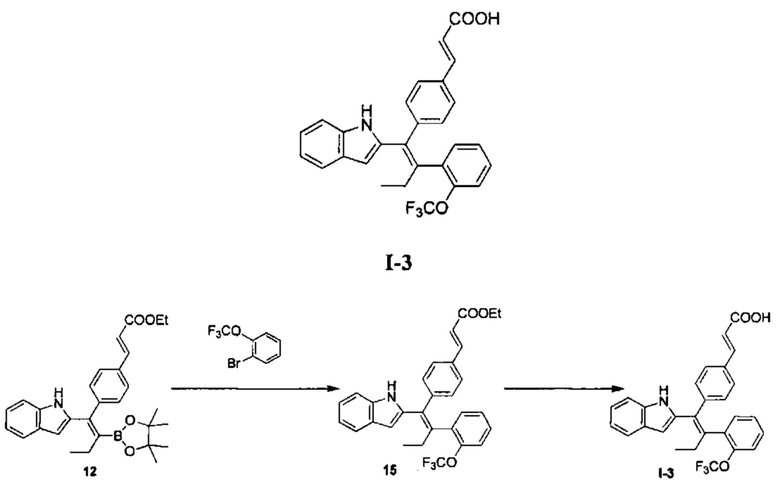
[154] Стадия А: Раствор соединения 12 (556,08 мг, 1,18 ммоль, 1.00 экв.). 2-трифторметилбромбензола (281,98 мг, 1,17 ммоль, 174,06 мкл, 1,00 экв.), водного раствора гидроксида калия (4М, 1,64 мл, 5,60 экв.) и дихлорбис(трифенилфосфин)палладия (24,64 мг, 35,10 мкмоль, 0,03экв.) в 20 мл 2-метилтетрагидрофурана три раза продували азотом. Реакционный раствор перемешивали при 80°С в атмосфере азота в течение 24 часов, затем разбавляли 20 мл воды. Смесь дважды экстрагировали 50 мл этилацетата каждый раз. Органическую фазу объединяли, промывали один раз 100 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии с получением соединения 15 в виде желтого масла (380,00 мг), которое непосредственно использовали на следующей стадии.
[155] Стадия В: Моногидрат гидроксида лития (83,00 мг, 1,98 ммоль, 5,00 экв.) добавляли к раствору соединения 15 (380,00 мг, 751,69 мкмоль, 1,00 экв.) в смешанном растворителе 6 мл тетрагидрофурана, 20 мл метанола и 6 мл воды, реакционный раствор перемешивали при 30°С в течение 7 часов. Реакционный раствор концентрировали до 1 мл, затем доводили до рН 5-6 с помощью 3 моль/л соляной кислоты, дважды экстрагировали 50 мл дихлорметана каждый раз. Органическую фазу объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной хроматографии (муравьиная кислота), с получением I-3 (45,00 мг, 94,25 мкмоль, выход 12,54%). MS [ESI, M+1]: 478,0.
[156] 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,77 (s, 1Н), 7,57 (d, J=7,6 Гц, 1H), 7,50 - 7,37 (m, 3Н), 7,36 - 7,15 (m, 5Н), 7,11 - 6,97 (m, 2Н), 6,92 (d, J=8,4 Гц, 2Н), 6,57 (s, 1Н), 6,42 (d, J=16,4 Гц, 1H), 2,80 - 2,60 (m, 2Н), 0,97 (t, J=7,6 Гц, 3Н).
Вариант реализации 4
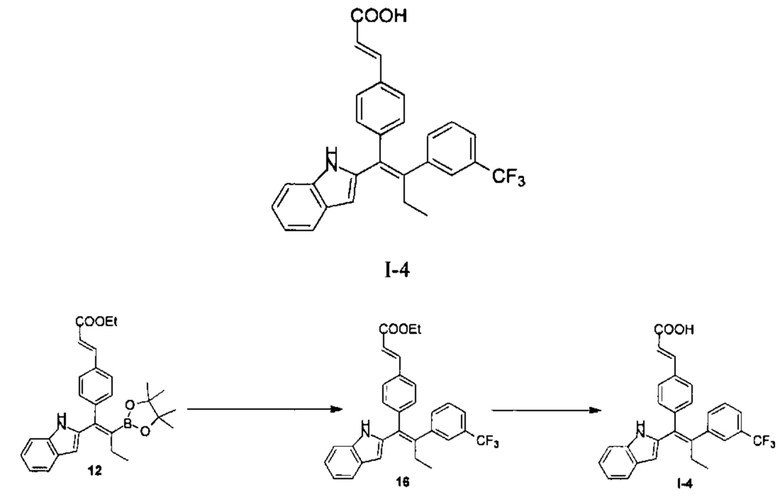
[157] Стадия А: 3-Трифторметилйодбензол (320,84 мг, 1,18 ммоль, 169,76 мкл, 2,00 экв.), водный раствор гидроксида калия (4М, 737,19 мкл, 5,00 экв.) и бис(трифенилфосфин)палладия дихлорид (20,70 мг, 29,49 мкмоль, 0,05 экв.) добавляли к раствору 12 (278,00 мг, 589,75 мкмоль, 1,00 экв.) в 10 мл диметилтетрагидрофурана. Реакционный раствор перемешивали при 70°С в атмосфере азота в течение 12 часов, затем добавляли 20 мл этилацетата. Смесь фильтровали через целит и фильтрат дважды промывали 30 мл насыщенного солевого раствора. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (РЕ:ЕА=20:1 - 10:1), с получением продукта 16 (200,00 мг, неочищенный продукт) в виде желтой слизи. MS (ESI, M+1): 490,2.
[158] Стадия В: Гидроксид лития (97,85 мг, 4,09 ммоль, 10,00 экв.) добавляли к раствору соединения 16 (200,00 мг, 408,56 мкмоль, 1,00 экв.) в смешанном растворителе 2 мл метанола, 2 мл тетрагидрофурана и 2 мл воды, реакционный раствор перемешивали при 30°С в течение 1 часа. Добавляли 10 мл воды, затем смесь доводили до рН 5 1М соляной кислотой и дважды экстрагировали 10 мл этилацетата. Органическую фазу объединяли, дважды промывали 10 мл воды, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ (система с муравьиной кислотой) с получением продукта I-4 (20,90 мг, 45,29 мкмоль, выход 11,09%, чистота 100%). MS (ESI, M+1): 462,2.
[159] 1Н ЯМР EW3644-177-P1B (400 МГц, ДМСО-d6): δ 10,77 (s, 1Н), 7,56 (d, J=8,0 Гц, 1Н), 7,51-7,46 (m, 3Н), 7,40 (d, J=8,8 Гц, 4Н), 7,30 (d, J=8,0 Гц, 1Н), 7,06 (t, J=8,0 Гц, 1Н), 6,99 (t, J=7,2 Гц, 1H), 6,90 (d, J=8,4 Гц, 2Н), 6,55 (d, J=1,6 Гц, 1Н), 6,40 (d, J=16,0 Гц, 1 Н), 2,75 (q, J=7,2 Гц 2 Н), 0,98 (t, J=7,2 Гц, 3 Н).
Вариант реализации 5
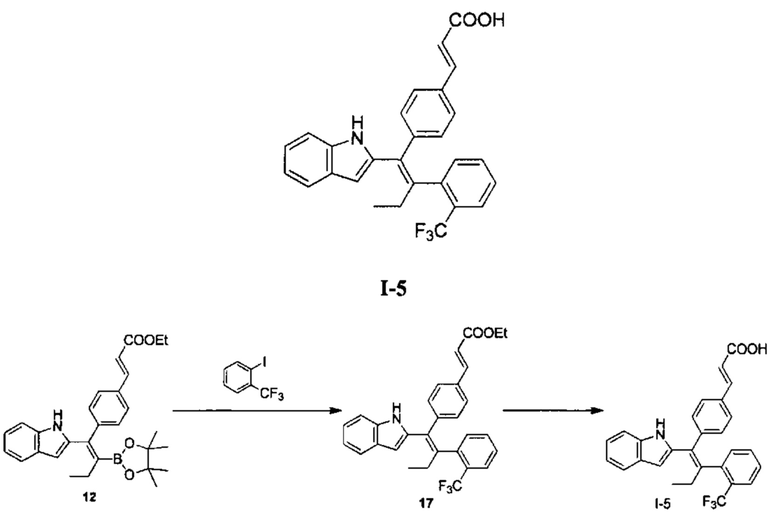
[160] Стадия А: Раствор соединения 12 (558,00 мг, 1,18 ммоль, 1,00 экв.), 2-трифторметилйодбензола (572,85 мг, 2,11 ммоль, 1,80 экв.), водного раствора гидроксида калия (4М, 1,64 мл, 5,60 экв.) и дихлорбис(трифенилфосфин)палладия (24,64 мг, 35,10 мкмоль, 0,03экв.) в 10 мл 2-метилтетрагидрофурана три раза продували азотом. Реакционный раствор перемешивали при 80°С в атмосфере азота в течение 24 часов, затем разбавляли 30 мл воды. Смесь дважды экстрагировали 50 мл этилацетата каждый раз. Органическую фазу объединяли, дважды промывали 50 мл насыщенного солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали, концентрировали, и очищали с помощью колоночной хроматографии с получением соединения 17 в виде желтого масла (200,00 мг), которое непосредственно использовали на следующей стадии.
[161] Стадия В: Моногидрат гидроксида лития (85,71 мг, 2,04 ммоль, 5,00 экв.) добавляли к раствору соединения 17 (200,00 мг, 408,56 мкмоль, 1,00 экв.) в смешанном растворителе 6 мл тетрагидрофурана, 20 мл метанола и 6 мл воды, реакционный раствор перемешивали при 30°С в течение 3 часов. Реакционный раствор концентрировали до 1 мл, затем доводили до рН 5-6 с помощью 3 моль/л соляной кислоты, дважды экстрагировали 50 мл дихлорметана каждый раз. Органическую фазу объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной хроматографии (муравьиная кислота) с получением продукта I-5 (44,90 мг, 97,30 мкмоль, выход 22,45%). MS [ESI, M+1]: 462,1.
[162] 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,79 (s, 1Н), 7,64 (d, J=8,0 Гц, 1Н), 7,60 - 7,50 (m, 2Н), 7,46 - 7,29 (m, 6Н), 7,12 - 6,90 (m, 4Н), 6,59 - 6,50 (m, 1Н), 6,39 (d, J=16,0 Гц, 1Н), 2,90 - 2,79 (m, 1Н), 2,48 - 2,41 (m, 1Н), 0,95 (t, J=7,6 Гц, 3Н).
Вариант реализации 6
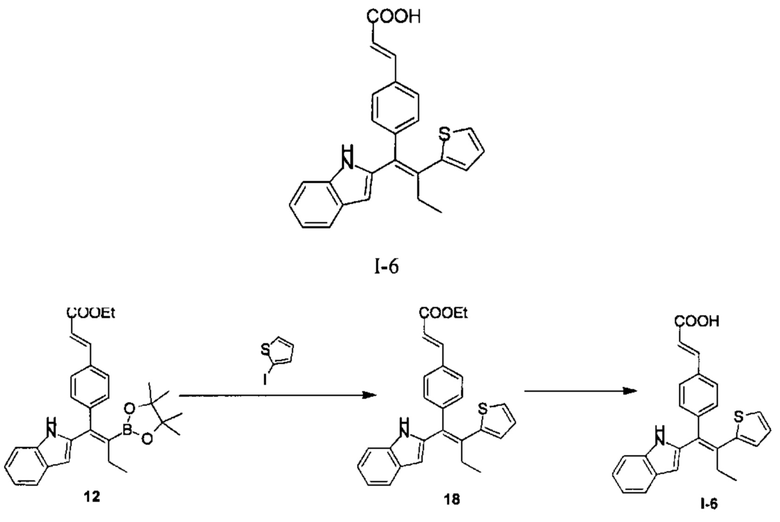
[163] Стадия А: Раствор соединения 12 (279,00 мг, 590,95 мкмоль, 1,00 экв.), 2-иодотиофена (247,74 мг, 1,18 ммоль, 120,26 мкл, 2,00 экв.), водного раствора гидроксида калия (4М, 737,19 мкл, 5,00 экв.), дихлорбис(трифенилфосфин)палладия (20,70 мг, 29,49 мкмоль, 0,05 экв.) в 10 мл 2-метилтетрагидрофурана три раза продували азотом и перемешивали при 70°С в течение 12 часов. Реакционный раствор разбавляли 20 мл этилацетата, фильтровали и фильтрат дважды промывали 30 мл насыщенного солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (РЕ:ЕА=20:1 - 10:1) с получением соединения 18 (200,00 мг) в виде желтого желе, которое непосредственно использовали на следующей стадии.
[164] Стадия В: Гидроксид лития (112,03 мг, 4,68 ммоль, 10,00 экв.) добавляли к раствору соединения 18 (200,00 мг, 467,77 мкмоль, 1,00 экв.) в смешанном растворителе 2 мл метанола, 2 мл тетрагидрофурана и 2 мл воды, реакционный раствор перемешивали при 30°С в течение 1 часа. Затем добавляли 10 мл воды, смесь доводили до рН 5 1М соляной кислотой, дважды экстрагировали 10 мл этилацетата каждый раз. Органическую фазу объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ (система с муравьиной кислотой) с получением продукта I-6 (12,40 мг, 30,83 мкмоль, выход 6,59%, чистота 99,34%). MS [ESI, M+1]: 400,1.
[165] 1Н ЯМР (400 МГц, ДМСО-d6): δ 10,72 (s, 1Н), 7,52 (d, J=8,0 Гц, 3Н), 7,48 (d, J=16,0 Гц, 1Н), 7,40 (dd, J=5,2 Гц, J=1,2 Гц, 1Н), 7,28 (d, J=8,4 Гц, 1Н), 7,09 (d, J=8,4 Гц, 2Н), 7,04 (t, J-6,0 Гц, 1Н), 6,97 (t, J=6,8 Гц, 1Н), 6,91 (dd, J=3,6 Гц, J=4,8 Гц, 1Н), 6,87 (dd, J=0,8 Гц, J=3,6 Гц, 1Н), 6,49 (d, J=1,6 Гц, 1Н), 6,46 (d, J=16,0 Гц, 1 Н), 2,71 (q, J=7,2 Гц 2 Н), 1,10 (t, J=7,2 Гц, 3 Н); MS(ESI, М+1): 400,1.
Вариант реализации 7
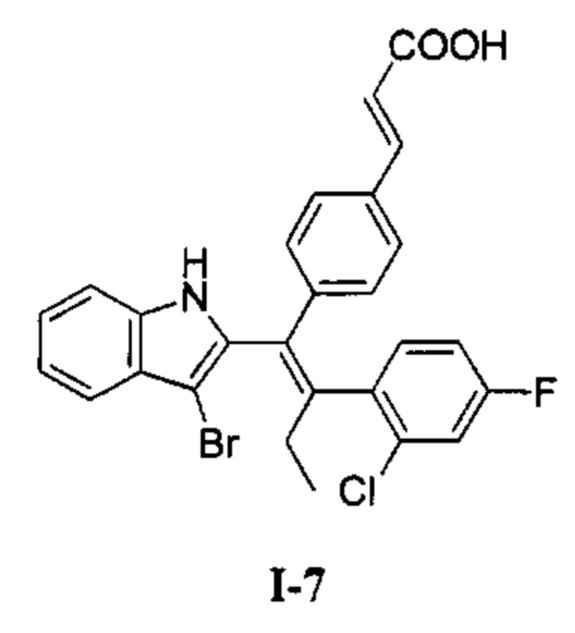
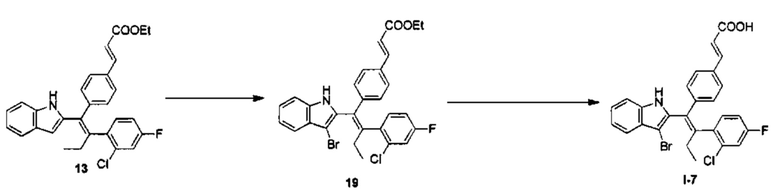
[166] Промежуточное соединение 13 получали способов в соответствии с процедурой реакции 1.
[167] Стадия А: Соединение 13 (200,00 мг, 409,32 мкмоль, 1,00 экв.) и N-бромсукцинимид (87,42 мг, 491,18 мкмоль, 1,20 экв.) растворяли в 5 мл дихлорметана. Реакционный раствор перемешивали при 15°С в атмосфере азота в течение 1 часа, затем концентрировали и очищали с помощью хроматографии на силикагеле с получением продукта 19 в виде желтого твердого вещества (150,00 мг, 271,32 мкмоль, выход 66,28%).
[168] Стадия С: Соединение 19 (80,00 мг, 144,70 мкмоль, 1,00 экв.), гидроксид лития (30,36 мг, 723,51 мкмоль, 5,00 экв.) растворяли в смешанном растворителе 1 мл метанола, 1 мл тетрагидрофурана и 0,5 мл воды. Реакционный раствор перемешивали при 30°С в атмосфере азота в течение 3 часов. Затем реакционный раствор доводили до рН 5-6 соляной кислотой (1 моль/л) и фильтровали с получением неочищенного продукта, который очищали с помощью препаративной хроматографии (система с муравьиной кислотой) с получением продукта I-7 (61,00 мг, 116,23 мкмоль, выход 80,32%, чистота 99,29%). MS [ESI, M+1]=526,0.
[169] 1Н ЯМР (400 МГц, ДМСО-d6) δ:11,66 (s, 1Н), 7,51 - 7,35 (m, 6Н), 7,30 -7,24 (m, 1Н), 7,22 - 7,09 (m, 3Н), 7,01 (d, J=8,4 Гц, 2Н), 6,39 (d, J=16,0 Гц, 1Н), 2,47 - 2,32 (m, 2Н), 0,88 (t, J=7,6 Гц, 3Н).
Вариант реализации 8
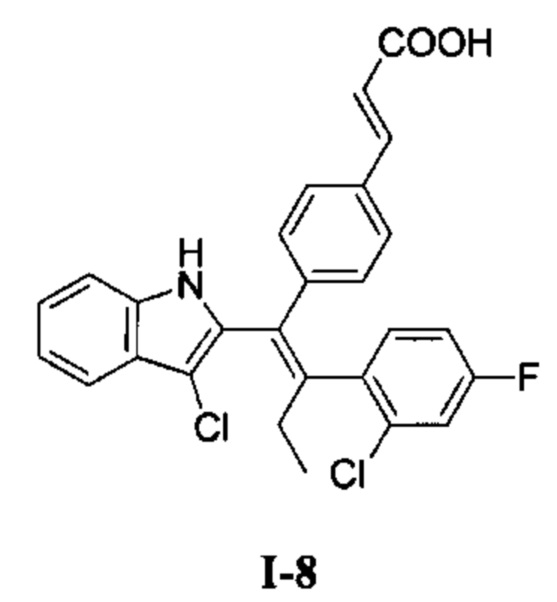
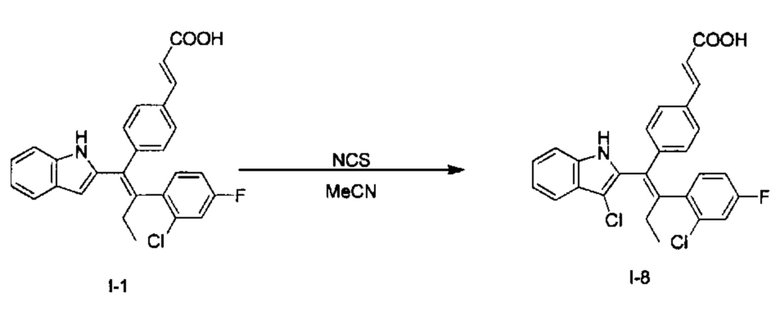
[170] Стадия А: Соединение I-1 (150,00 мг, 336,39 мкмоль, 1,00 экв.) и N-хлорсукцинимид (53,90 мг, 403,67 мкмоль, 1,20 экв.) растворяли в 5 мл ацетонитрила. Реакционный раствор перемешивали при 15°С в атмосфере азота в течение 6 часов. После завершения реакции реакционный раствор концентрировали и очищали с помощью препаративной хроматографии (система с муравьиной кислотой) с получением продукта I-8 (10,00 мг, 21,56 мкмоль, выход 11,78%, чистота 97,65%). MS [ESI, M+l]+=480,1.
[171] 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,51 (s, 1Н), 7,50 (d, J=8,0 Гц, 1Н), 7,47 -7,37 (m, 5Н), 7,29 (dd, J=6,4, 8,4 Гц, 1Н), 7,22 - 7,09 (m, 3Н), 7,01 (d, J=8,4 Гц, 2Н), 6,41 (d, J=16,4 Гц, 1Н), 2,49 - 2,36 (m, 2Н), 0,89 (t, J=7,6 Гц, 3Н).
Вариант реализации 9
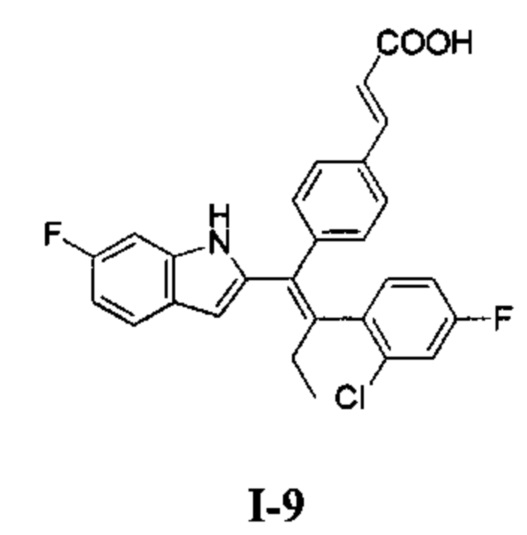
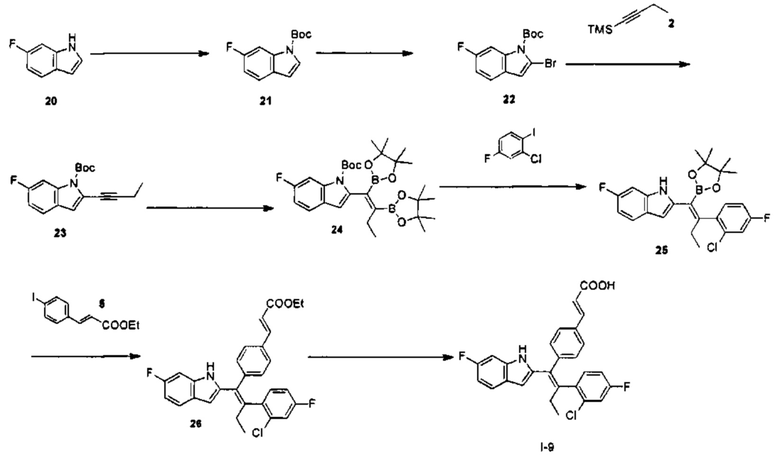
[172] Стадия А: N,N-диметилпиридин (406,83 мг, 3,33 ммоль, 0,05 экв.) и Вос2О (15,00 г, 68,73 ммоль, 15,79 мл, 1,03экв.) добавляли к раствору 20 (9,00 г, 66,60 ммоль, 1,00 экв.) в 180 мл дихлорметана. Реакционный раствор перемешивали при 15°С в течение 12 часов, затем гасили 100 мл воды при 0°С, экстрагировали три раза 210 мл этилацетата. Органическую фазу объединяли, промывали 200 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии с получением продукта 21 в виде бесцветного масла (15,40 г, 65,46 ммоль, выход 98,29%).
[173] Стадия В: Диизопропиламид лития (2М, 70,14 мл, 2,00 экв.) добавляли по каплям к раствору 21 (16,50 г, 70,14 ммоль, 1,00 экв.) в 300 мл тетрагидрофурана при -70°С в атмосфере азота. Реакционный раствор перемешивали при -70°С в течение 30 минут, затем бромистый циан (22,29 г, 210,42 ммоль, 15,48 мл, 3,00 экв.) добавляли к реакционному раствору. Реакционный раствор перемешивали при 15°С в течение 12 часов, затем гасили 50 мл насыщенного водного раствора хлорида аммония, разбавляли 50 мл воды и экстрагировали 300 мл этилацетата. Органическую фазу объединяли, дважды промывали 200 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии с получением неочищенного продукта 22 в виде желтого масла (14,10 г, 44,88 ммоль, выход 63,99%).
[174] Стадия С: Соединение 22 (12,00 г, 38,20 ммоль, 1,00 экв.), 1-триметилсилилбутин (9,65 г, 76,40 ммоль, 2,00 экв.), йодид меди (145,50 мг, 764,00 мкмоль, 0,02экв.), карбонат цезия (16,30 г, 50,04 ммоль, 1,31экв.), 1,1'-бис(дифенилфосфино)ферроцен (423,55 мг, 764,00 мкмоль, 0,02экв.) и ацетат палладия (171,53 мг, 764,00 мкмоль, 0,02экв.) растворяли в 100 мл N,N-диметилацетамида, реакционную систему три раза продували азотом и реакционный раствор перемешивали при 80°С в атмосфере азота в течение 6 часов. После завершения реакции реакционный раствор гасили 100 мл воды, разбавляли 100 мл этилацетата, фильтровали через целит, фильтрат экстрагировали три раза 300 мл этилацетата. Органическую фазу объединяли, промывали 300 мл солевого раствора три раза, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии с получением продукта 23 в виде желтого масла (6,00 г, 20,88 ммоль, выход 54,66%).
[175] Стадия D: Соединение 23 (6,00 г, 20,88 ммоль, 1,00 экв.), биспинаколатодибор (5,30 г, 20,88 ммоль, 1,00 экв.), тетракис(трифенилфосфин)платину (1,30 г, 1,04 ммоль, 0,05 экв.) растворяли в 100 мл 2-метилтетрагидрофурана, реакционную систему три раза продували азотом. Реакционный раствор перемешивали при 80°С в атмосфере азота в течение 4 часов. После завершения реакции реакционный раствор гасили 50 мл воды при 0°С, затем разбавляли 100 мл этилацетата, фильтровали через целит, и фильтрат экстрагировали три раза 300 мл этилацетата. Органическую фазу объединяли, промывали 100 мл солевого раствора, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии с получением продукта 24 в виде желтого масла (4,00 г, 7,17 ммоль, выход 34,33%, чистота 97%). MS [ESI, M+1]+=542,3.
[176] Стадия Е: Соединение 24 (515,46 мг, 923,75 мкмоль, 1,00 экв.), 2-хлор-4-фториодобензол (236,89 мг, 923,75 мкмоль, 1,00 экв.), карбонат цезия (601,95 мг, 1,85 ммоль, 2,00 экв.), дихлорбис(трифенилфосфин)палладия (64,84 мг, 92,38 мкмоль, 0,10 экв.) и 1,2 мл воды растворяли в 30 мл 2-метилтетрагидрофурана, и реакционную систему три раза продували азотом. Реакционный раствор перемешивали при 75°С в атмосфере азота в течение 12 часов. После завершения реакции реакционный раствор гасили 30 мл воды при 0°С, затем разбавляли 100 мл этилацетата, фильтровали через целит и фильтрат экстрагировали три раза 90 мл этилацетата. Органическую фазу объединяли, промывали 50 мл солевого раствора, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле с получением продукта 25 в виде желтого масла (500,00 мг), который непосредственно использовали на следующей стадии. MS [ESI, M+l]+=446,0.
[177] Стадия F: Соединение 25 (200,00 мг, 450,73 мкмоль, 1,00 экв.), соединение 5 (292,84 мг, 901,46 мкмоль, 2,00 экв.), дихлорбис(трифенилфосфин)палладия (63,27 мг, 90,15 мкмоль, 0,20 экв.) и водный раствор гидроксида калия (4М, 631,02 мкл, 5,60 экв.) растворяли в 5 мл 2-метилтетрагидрофурана, и реакционную систему три раза продували азотом. Реакционный раствор перемешивали при 80°С в атмосфере азота в течение 12 часов. После завершения реакции реакционный раствор гасили 30 мл воды при 0°С, затем разбавляли 30 мл этилацетата, фильтровали через целит и фильтрат экстрагировали три раза 90 мл этилацетата. Органическую фазу объединяли, промывали 50 мл солевого раствора, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле с получением неочищенного продукта 26 (90,00 мг), который непосредственно использовали на следующей стадии. MS [ESI, M+l]+-=492,2.
[178] Стадия G: Соединение 26 (90,00 мг, 182,94 мкмоль, 1,00 экв.) и гидроксид лития (38,38 мг, 914,70 мкмоль, 5,00 экв.) растворяли в смешанном растворителе 2 мл метанола, 1 мл тетрагидрофурана и 0,5 мл воды. Реакционный раствор перемешивали при 25°С в течение 12 часов, затем концентрировали до 3 мл, разбавляли 20 мл воды. Смесь доводили до рН 5-6 соляной кислотой (1 моль/л) и экстрагировали 200 мл дихлорметана. Органическую фазу объединяли, концентрировали и очищали с помощью препаративной хроматографии (система с муравьиной кислотой) с получением продукта I-9 (10,00 мг, 21,56 мкмоль, выход 11,78%). MS[ESI, М+1]+=464,2.
[179] 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,82 (s, 1H), 7,56 (dd, J=5,6, 8,8 Гц, 1H), 7,48 - 7,32 (m, 4Н), 7,25 (dd, J=6,4, 8,4 Гц, 1Н), 7,12 (dt, J=2,4, 8,4 Гц, 1H), 7,05 (dd, J=2,0, 10,0 Гц, 1Н), 6,96 (d, J=8,0 Гц, 2Н), 6,91 - 6,81 (m, 1Н), 6,60 (d, J=1,6 Гц, 1Н), 6,41 (d, J=14,4 Гц, 1Н), 2,70 - 2,59 (m, 2Н), 1,00 (t, J=7,2 Гц, 3Н).
Вариант реализации 10
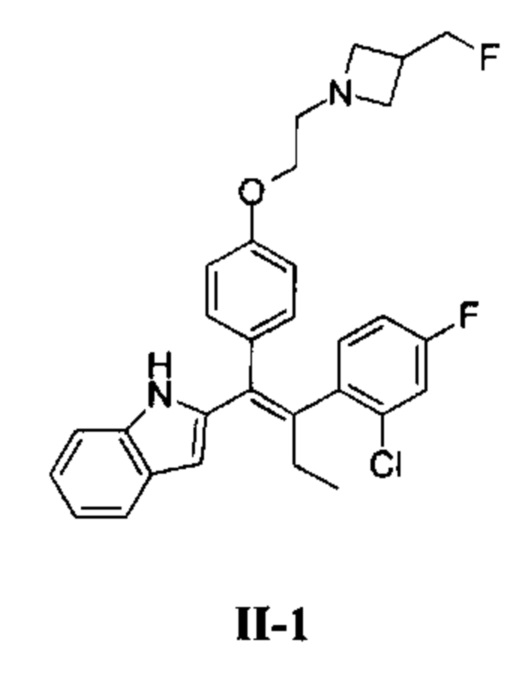
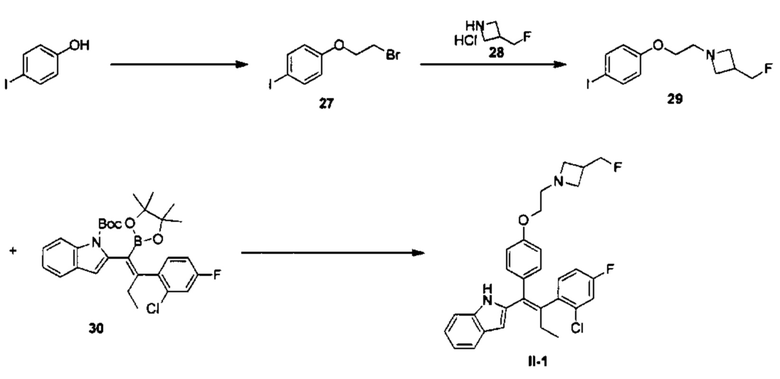
[180] Промежуточное соединение 30 получали используя процедуру, аналогичную процедуре, описанной в Варианте реализации 9, но с использованием индола в качестве исходного вещества.
[181] Стадия А: Карбонат цезия (6,52 г, 20,00 ммоль, 4,00 экв.) и 1,2-дибромэтан (4,70 г, 25,00 ммоль, 1,89 мл, 5,00 экв.) добавляли к раствору n-йодфенол (1,10 г, 5,00 ммоль, 1,00 экв.) в ацетонитриле (20,00 мл), реакционный раствор перемешивали при 85°С в течение 24 часов, затем концентрировали при пониженном давлении с удалением растворителя, разбавляли 20 мл воды и экстрагировали три раза 30 мл этилацетата каждый раз. Органическую фазу объединяли, промывали 30 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (РЕ/ЕА=100/1 до 20/1) с получением продукта 27 (1,40 г, 4,28 ммоль, выход 85,64%).
[182] Стадия В: Карбонат цезия (3,28 г, 10,08 ммоль, 3,00 экв.) и соединение 28 (506,30 мг, 4,03 ммоль, 1,20 экв.) добавляли к раствору соединения 27 (1,10 г, 3,36 ммоль, 1,00 экв.) в ацетонитриле (20,00 мл), реакционный раствор перемешивали при 85°С в течение 12 часов, затем разбавляли 50 мл воды и экстрагировали три раза 50 мл этилацетата каждый раз. Органическую фазу объединяли, дважды промывали 50 мл насыщенном солевым раствором каждый раз, сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (ДХМ/МеОН=100/1 до 10/1) с получением продукта 29 (700,00 мг, 2,09 ммоль, выход 62,16%).
[183] Стадия С: Водный раствор гидроксида калия (4М, 190,17 мкл, 4,00 экв.) добавляли к смеси соединения 30 (201,61 мг, 190,17 мкмоль, 1,00 экв.), соединения 29 (127,47 мг, 380,34 мкмоль, 2,00 экв.) и дихлорбис(трифенилфосфин)палладия (133,48 мг, 190,17 мкмоль, 1,00 экв.), реакционный раствор продували азотом и затем перемешивали при 80°С в атмосфере азота в течение 12 часов. Реакционный раствор концентрировали с получением неочищенного продукта, который очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением продукта II-1 (2,80 мг, 5,49 мкмоль, выход 2,89%, чистота 99,5%). MS(ESI, М+1): 507,1. 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,65 (s, 1Н), 8,32 (s, 1Н), 7,55 (d, J=7,6 Гц, 1Н), 7,36 (dd, J=2,4, 8,8 Гц, 1Н), 7,30 (d, J=8,0 Гц, 1Н), 7,24 - 7,18 (m, 1Н), 7,11 (dt, J=2,4, 8,4 Гц, 1Н), 7,08 - 7,02 (m, 1Н), 7,02 - 6,95 (m, 1Н), 6,84 (d, J=8,8 Гц, 2Н), 6,64 (d, J=8,8 Гц, 2Н), 6,53 (s, 1H), 4,54 (d, J=6,4 Гц, 1Н), 4,42 (d, J=6,4 Гц, 1Н), 3,79 (t, J=5,2 Гц, 2Н), 3,27 (t, J=7,2 Гц, 2Н), 2,96 (t, J=6,4 Гц, 2Н), 2,71 - 2,60 (m, 5Н), 0,99 (t, J=7,2 Гц, 3Н).
Вариант реализации 11
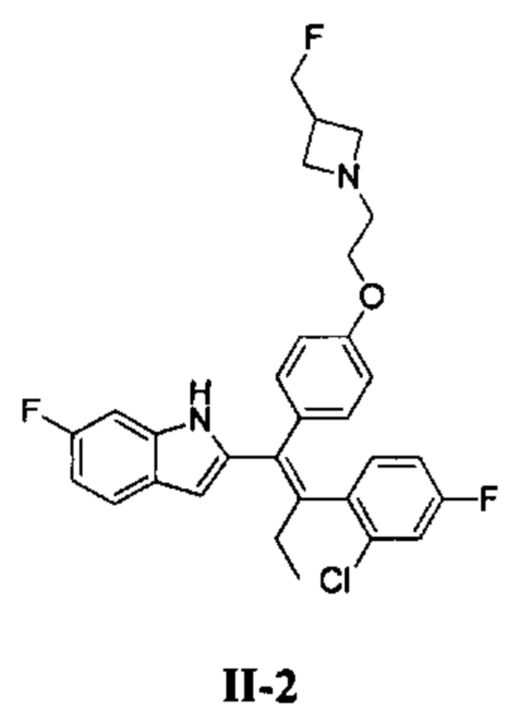
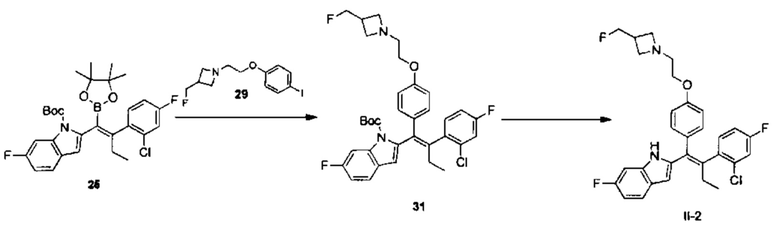
[184] Стадия А: Соединение 25 (340,00 мг, 625,18 мкмоль, 1,00 экв.) и соединение 29 (419,07 мг, 1,25 ммоль, 2,00 экв.), дихлорбис(трифенилфосфин)палладия (219,41 мг, 312,59 мкмоль, 0,50 экв.) и водный раствор гидроксида калия (4М, 875,26 мкл, 5,60 экв.) растворяли в 10 мл 2-метилтетрагидрофурана, реакционную систему три раза продували азотом. Реакционный раствор перемешивали при 80°С в атмосфере азота в течение 12 часов. После завершения реакции реакционный раствор охлаждали до 0°С, гасили 30 мл воды, разбавляли 30 мл этилацетата, фильтровали через целит и фильтрат экстрагировали три раза 20 мл этилацетата. Органическую фазу объединяли, промывали 20 мл солевого раствора, сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении и очищали с помощью препаративной тонкослойной хроматографии с получением неочищенного продукта 31 в виде желтого масла (110,00 мг, неочищенный), который непосредственно использовали на следующей стадии. MS [ESI, М-56]=625,2.
[185] Стадия В: Трифторуксусную кислоту (770,00 мг, 6,75 ммоль, 500,00 мкл, 42,22 экв.) добавляли к раствору соединения 31 (90,00 мг, 152,37 мкмоль, 1,00 экв.) в 33 мл дихлорметана и реакционный раствор перемешивали при 16°С в течение 3 часов. Реакционный раствор доводили до рН 7 - 8 с насыщенным водным раствором бикарбоната натрия, разбавляли 20 мл воды, дважды экстрагировали 100 мл дихлорметана каждый раз. Органическую фазу объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью препаративной хроматографии (система с муравьиной кислотой) с получением продукта II-2 (10,00 мг, 18,86 мкмоль, выход 12,38%, чистота 99,0%).
[186] MS [ESI,M+l]+=525,2.
[187] 1Н ЯМР (400 МГц, ДМСО-d6) δ=10,71 (s, 1Н), 8,29 (br. s., 1Н), 7,55 (dd, J=5,6, 8,4 Гц, 1Н), 7,35 (dd,.7=2,4, 8,8 Гц, 1Н), 7,21 (dd, 7=6,4, 8,4 Гц, 1Н), 7,13-7,02 (m, 2Н), 6,91 - 6,77 (m, 3Н), 6,65 (d, J=8,8 Гц, 2Н), 6,56 (d, J=1,2 Гц, 1Н), 4,54 (d, J=6,4 Гц, 1Н), 4,42 (d, J=6,4 Гц, 1Н), 3,79 (t, J=5,2 Гц, 2Н), 3,28 (t, J=6,8 Гц, 2Н), 2,97 (t, J=6,4 Гц, 2Н), 2,71 - 2,62 (m, 5Н), 0,99 (t, J=7,6 Гц, 3Н).
Вариант реализации 12
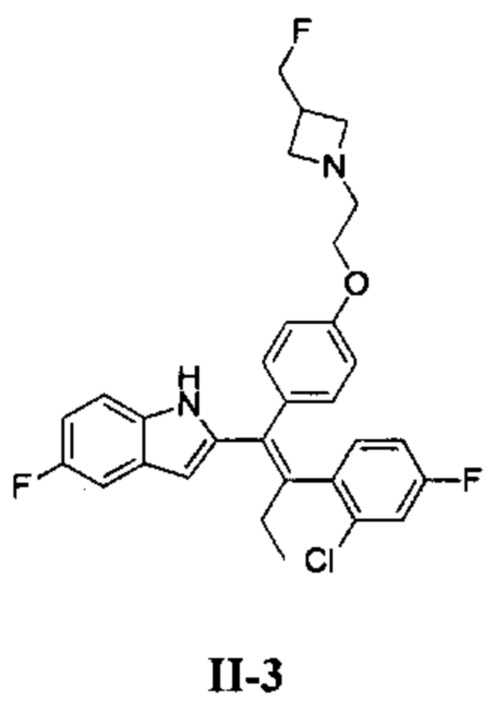
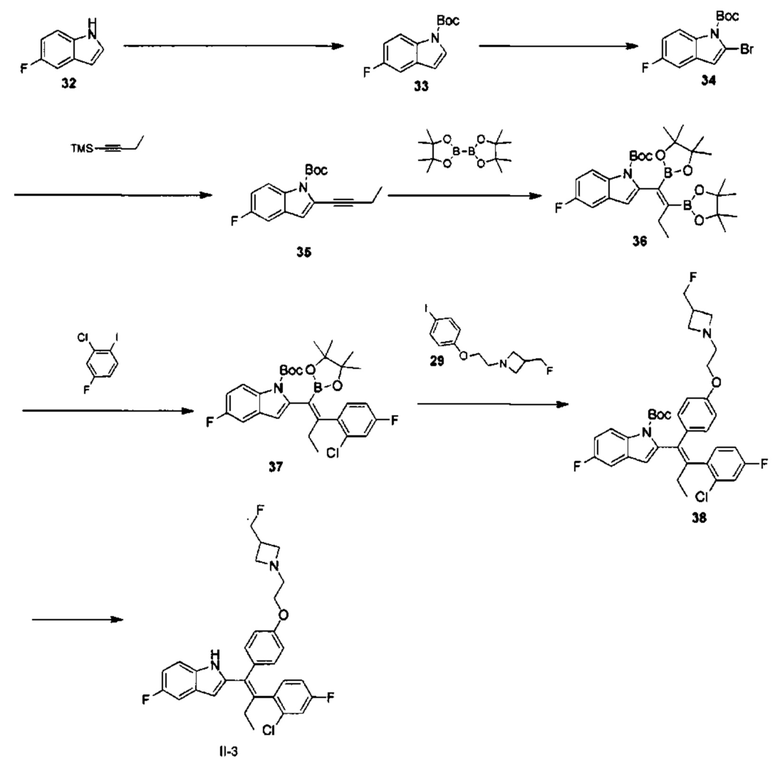
[188] Стадия А: N,N-диметилпиридин (135,61 мг, 1,11 ммоль, 0,05 экв.) и Вос2О (4,89 г, 22,42 ммоль, 1,01экв.) добавляли к раствору 32 (3,00 г, 22,20 ммоль, 1,00 экв.) в 30 мл дихлорметана. Реакционный раствор перемешивали при 10-15°С в течение 12 часов, затем гасили 50 мл водного раствора хлорида аммония при 0°С, дважды экстрагировали 25 мл дихлорметана каждый раз. Органическую фазу объединяли, промывали 50 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле с получением продукта 33 в виде белого масла (5,20 г, 22,10 ммоль, выход 99,57%).
[189] Стадия В: Диизопропиламид лития (2М, 14,96 мл, 1,10 экв.) добавляли по каплям к раствору 33 (6,40 г, 27,21 ммоль, 1,00 экв.) в 60 мл тетрагидрофурана при -70°С, после перемешивания при -70°С в течение 30 минут раствор бромистого циана (8,64 г, 81,62 ммоль, 6,00 мл, 3,00 экв.) в 20 мл тетрагидрофурана добавляли по каплям к указанному раствору. Реакционный раствор перемешивали при 15°С в течение 12 часов. Затем последовательно добавляли 40 мл воды и 40 мл этилацетата и смесь разделяли в делительной воронке. Органическую фазу три раза промывали 120 мл воды (40 мл каждый раз), сушили над безводным сульфатом натрия, концентрировали и фильтровали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии с получением продукта 34 в виде желтого масла (7,80 г, 24,83 ммоль, выход 91,25%).
[190] Стадия С: Соединение 34 (7,80 г, 24,83 ммоль, 1,00 экв.), 1-триметилсилилбутин (4,70 г, 37,24 ммоль, 1,50 экв.), йодид меди (236,43 мг, 1,24 ммоль, 0,05 экв.), карбонат цезия (10,52 г, 32,28 ммоль, 1,30 экв.), 1,1'-бис(дифенилфосфино)ферроцен (688,23 мг, 1,24 ммоль, 0,05 экв.) и ацетат палладия (278,72 мг, 1,24 ммоль, 0,05 экв.) растворяли в 80 мл N,N-диметилацетамида, реакционную систему три раза продували азотом и реакционный раствор перемешивали при 80-90°С в атмосфере азота в течение 48 часов. После завершения реакции реакционный раствор разбавляли 100 мл воды, дважды экстрагировали 100 мл этилацетата каждый раз. Органическую фазу объединяли, дважды промывали 100 мл солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии с получением продукта 35 в виде желтого масла (4,00 г, 13,92 ммоль, выход 56,07%).
[191] Стадия D: Соединение 35 (4,00 г, 13,92 ммоль, 1,00 экв.), биспинаколатодибор (3,57 г, 14,06 ммоль, 1,01экв.), тетракис(трифенилфосфин)платину (865,42 мг, 696,06 мкмоль, 0,05 экв.) растворяли в 50 мл 2-метилтетрагидрофурана, реакционную систему три раза продували азотом. Реакционный раствор перемешивали при 70-80°С в атмосфере азота в течение 5 часов, затем концентрировали при пониженном давлении с получением неочищенного продукта, который очищали с помощью колоночной хроматографии с получением продукта 36 в виде желтого твердого вещества (2,10 г, 3,88 ммоль, выход 27,87%).
[192] Стадия Е: Соединение 36 (2,10 г, 3,88 ммоль, 1,00 экв.), 2-хлор-4-фториодобензол (994,93 мг, 3,88 ммоль, 1,00 экв.), карбонат цезия (2,53 г, 7,76 ммоль, 2,00 экв.) и дихлорбис(трифенилфосфин)палладий (272,32 мг, 387,98 мкмоль, 0,10 экв.) растворяли в 10 мл 2-метилтетрагидрофурана и реакционную систему три раза продували азотом. Реакционный раствор перемешивали при 70-80°С в атмосфере азота в течение 12 часов, затем концентрировали при пониженном давлении, разбавляли 50 мл воды, дважды экстрагировали 50 мл этилацетата каждый раз. Органическую фазу объединяли, промывали 30 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле с получением продукта 37 в виде желтого масла (400,00 мг).
[193] Стадия F: Соединение 37 (200,00 мг, 367,76 мкмоль, 1,00 экв.), соединение 29 (246,52 мг, 735,52 мкмоль, 2,00 экв.), дихлорбис(трифенилфосфин)палладий (25,81 мг, 36,78 мкмоль, 0,10 экв.) и водный раствор гидроксида калия (4М, 514,86 мкл, 5,60 экв.) растворяли в 5 мл 2-метилтетрагидрофурана и реакционную систему три раза продували азотом. Реакционный раствор перемешивали при 80°С в атмосфере азота в течение 12 часов, затем концентрировали при пониженном давлении, и очищали с помощью препаративной тонкослойной хроматографии с получением неочищенного продукта 38 (100,00 мг), который непосредственно использовали на следующей стадии.
[194] Стадия G: Трифторуксусную кислоту (770,00 мг, 6,75 ммоль, 500,00 мкл, 42,22экв.) добавляли к раствору соединения 38 (100,00 мг, 159,97 мкмоль, 1,00 экв.) в 10 мл дихлорметана и реакционный раствор перемешивали при 10-16°С в течение 12 часов. Реакционный раствор доводили до рН 7-8 водным раствором бикарбоната натрия, разбавляли 20 мл воды, дважды экстрагировали 25 мл дихлорметан каждый раз. Органическую фазу объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью ВЭЖХ (система с муравьиной кислотой) с получением продукта II-3 (20,00 мг, 38,05 мкмоль, выход 23,79%, чистота 99,89%). MS (ESI, M+1): 525,2.
[195] 1Н ЯМР (400 МГц, ДМСО-d6) δ=10,75 (s, 1Н), 7,34 (dd, J=2,8, 9,2 Гц, 1H), 7,31 - 7,25 (m, 2Н), 7,20 (dd, J=6,4, 8,8 Гц, 1Н), 7,13 - 7,07 (m, 1Н), 6,88 (dt, J=2,6, 9,2 Гц, 1Н), 6,83 - 6,80 (m, 2Н), 6,65 - 6,62 (m, 2Н), 6,52 (d, J=l,6 Гц, 1Н), 4,53 (d, J=6,2 Гц, 1H), 4,41 (d, J=6,2 Гц, 1Н), 3,78 (t, J=5,6 Гц, 2Н), 3,28 - 3,23 (m, 3Н), 2,94 (t, J=6,4 Гц, 2Н), 2,68 - 2,59 (m, 4Н), 0,97 (t, J=7,2 Гц, 3Н).
Вариант реализации 13
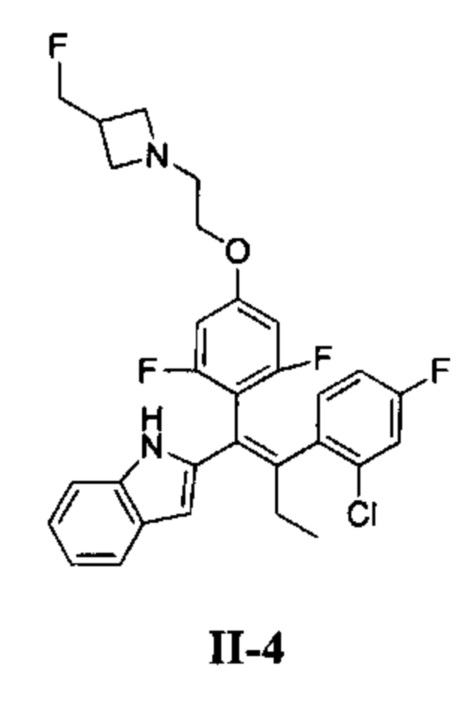
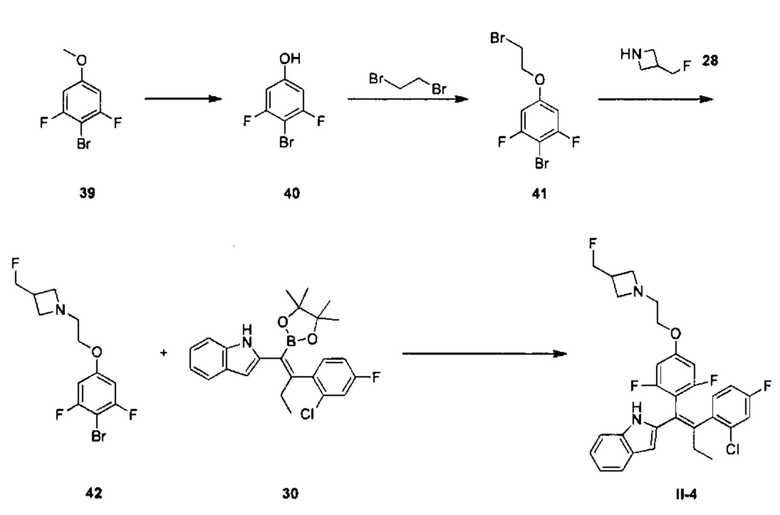
[196] Стадия А: Трибромид бора (6,74 г, 26,91 ммоль, 2,59 мл, 3,00 экв.) добавляли по каплям к раствору соединения 39 (2,00 г, 8,97 ммоль, 1,00 экв.) в дихлорметане (20,00 мл). После завершения добавления реакционный раствор перемешивали при 25°С в течение 12 часов, затем гасили путем добавления по каплям 40 мл метанола, концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле с получением продукта 40 в виде коричневого масла (2 г).
[197] Стадия В: Карбонат цезия (2,34 г, 7,18 ммоль, 1,00 экв.) и 1,2-дибромэтан (6,74 г, 35,90 ммоль, 2,71 мл, 5,00 экв.) добавляли к раствору соединения 40 (1,50 г, 7,18 ммоль, 1,00 экв.) в ацетонитриле (10,00 мл), реакционный раствор перемешивали при 80°С в течение 12 часов, затем выливали в 50 мл воды, дважды экстрагировали 50 мл этилацетата каждый раз. Органическую фазу объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (РЕ/ЕА=10/1) с получением продукта 41 в виде желтого масла (1,40 г, 4,43 ммоль, выход 61,72%).
[198] Стадия С: Карбонат цезия (2,06 г, 6,32 ммоль, 4,00 экв.) и соединение 28 (768,02 мг, 4,74 ммоль, 3,00 экв., 2HCl) добавляли к раствору соединения 41 (500,00 мг, 1,58 ммоль, 1,00 экв.) в ацетонитриле (20,00 мл), реакционный раствор перемешивали при 70°С в течение 1 часа, затем разбавляли 30 мл воды, экстрагировали 30 мл этилацетата, сушили над безводным сульфатом натрия, фильтровали, концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ТСХ (РЕ/ЕА=1/2) с получением продукта 42 в виде желтого твердого вещества (400,00 мг, 1,23 ммоль, выход 78,10%).
[199] Стадия D: Фосфат калия (2М, 1,64 мл, 4,00 экв.) и бифенил Ad2nBuP (54,62 мг, 81,77 мкмоль, 0,10 экв.) добавляли к раствору соединения 42 (430,00 мг, 817,72 мкмоль, 1,00 экв.) и соединения 30 (318,07 мг, 981,26 мкмоль, 1,20 экв.) в 8 мл тетрагидрофурана. Реакционный раствор перемешивали при 80°С в атмосфере азота, затем разбавляли 50 мл воды, дважды экстрагировали 50 мл этилацетата каждый раз в течение 36 часов, сушили над безводным сульфатом натрия, фильтровали, концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (РЕ/ЕА=15/1 до 0/1) и препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением продукта II-4 (530,00 мкг, 0,98 мкмоль, выход 0,12%). MS(ESI, М+1): 543,1.
[200] 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,70 (s, 1Н), 7,52 (d, J=8,0 Гц, 1Н), 7,35 -7,31 (m, 2Н), 7,26 - 7,20 (m, 1Н), 7,15 - 7,10 (m, 1Н), 7,10 - 7,03 (m, 1Н), 7,01 - 6,95 (m, 1Н), 6,50 (t, J=8,0 Гц, ЗН), 4,54 (d, J=6,4 Гц, 1H), 4,42 (d, J=6,4 Гц, 1Н), 3,84 (t, J=5,2 Гц, 2Н), 3,28 (t, J=7,2 Гц, 2Н), 2,95 (t, J=5,2 Гц, 2Н), 2,90 - 2,75 (m, 2Н), 2,74 - 2,60 (m, 3Н), 1,03 (t, J=7,2 Гц, 3Н).
Вариант реализации 14
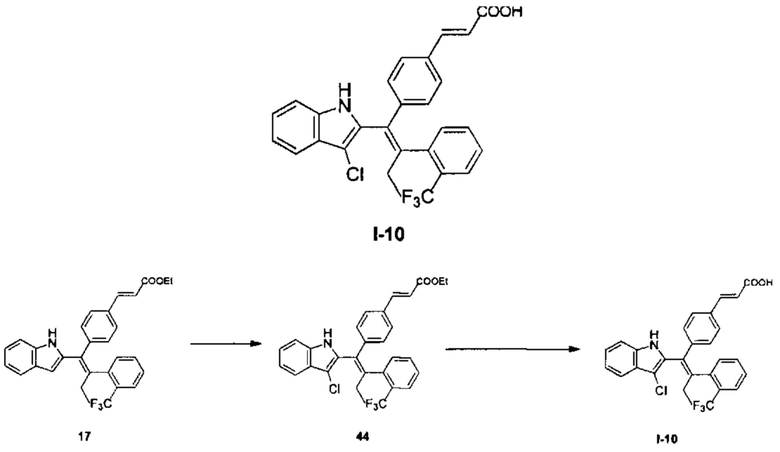
[201] Стадия А: N-хлорсукцинимид (390,71 мг, 2,93 ммоль, 1,10 экв.) добавляли к раствору неочищенного продукта 17 (1,30 г) в 10 мл ацетонитрила, реакционный раствор перемешивали при 15°С в атмосфере азота в течение 5 часов, затем гасили 40 мл воды, экстрагировали три раза 120 мл этилацетата (40 мл каждый раз). Органическую фазу объединяли, промывали 100 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта 44 (1,4 г, неочищенный).
[202] Стадия В: Гидроксид лития (319,96 мг, 13,35 ммоль, 5,00 экв.) добавляли к раствору неочищенного продукта 44 (1,40 г) в смешанном растворителе 10 мл тетрагидрофурана, 10 мл метанола и 2,5 мл воды, реакционный раствор перемешивали при 30°С в течение 1 часа, затем концентрировали до 1 мл при пониженном давлении, доводили до рН 5-6 3М соляной кислотой и фильтровали. Фильтрационный осадок сушили и очищали с помощью препаративной хроматографии (система с муравьиной кислотой) с получением продукта I-10 (691,00 мг, 1,38 ммоль, выход 51,66%, чистота 99%).
[203] 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,32 (brs, 1Н), 11,48 (s, 1Н), 7,71 (d,J=7,6 Гц, 1Н), 7,64 - 7,57 (m, 1Н), 7,53 - 7,45 (m, 2Н), 7,43 - 7,30 (m, 5Н), 7,22 - 7,10 (m, 2Н), 6,97 (d, J=8,4 Гц, 2Н), 6,37 (d, J=16,0 Гц, 1Н), 2,57 - 2,51 (m, 1Н), 2,38-2,29 (m, 1Н), 0,86 (t, J=7,6 Гц, 3Н). MS [ESI, М+1]: 496,1.
Вариант реализации 15
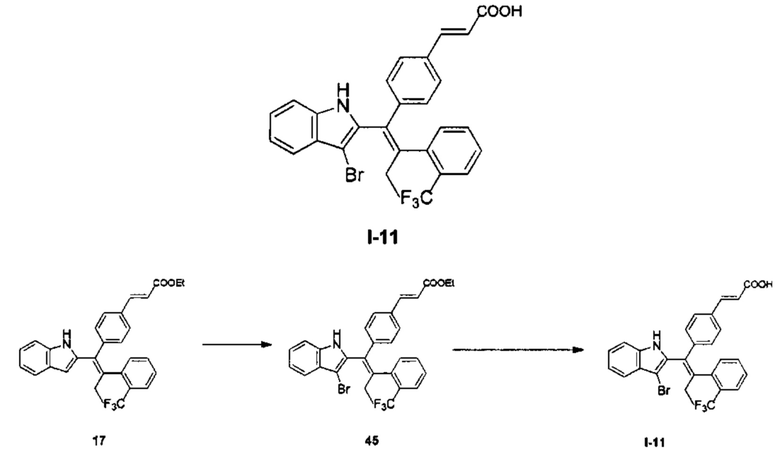
[204] Стадия А: N-бромсукцинимид (520,77 мг, 2,93 ммоль, 1,10 экв.) добавляли к раствору неочищенного продукта 17 (1,30 г, 2,66 ммоль, 1,00 экв.) в 10 мл ацетонитрила, реакционный раствор перемешивали при 15°С в атмосфере азота в течение 5 часов, затем гасили 40 мл воды, экстрагировали три раза 120 мл этилацетата (40 мл каждый раз). Органическую фазу объединяли, промывали 100 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта 45 (1,6 г, неочищенный), который непосредственно использовали на следующей стадии.
[205] Стадия В: Гидроксид лития (336,50 мг, 14,05 ммоль, 5,00 экв.) добавляли к раствору неочищенного продукта 45 (1,60 г, 2,81 ммоль, 1,00 экв.) в смешанном растворителе 10 мл тетрагидрофурана, 10 мл метанола и 2,5 мл воды, реакционный раствор перемешивали при 30°С в течение 1 часа. Реакционный раствор превратился из темно-желтого в коричневый, затем концентрировали его до 1 мл при пониженном давлении, доводили до рН 5-6 3М соляной кислотой и фильтровали. Фильтрационный осадок очищали с помощью препаративной хроматографии (система с муравьиной кислотой) и затем лиофилизировали с получением продукта I-11 в виде бледно-желтого твердого вещества (811,00 мг, 1,49 ммоль, выход 52,88%, чистота 99%).
Вариант реализации 16
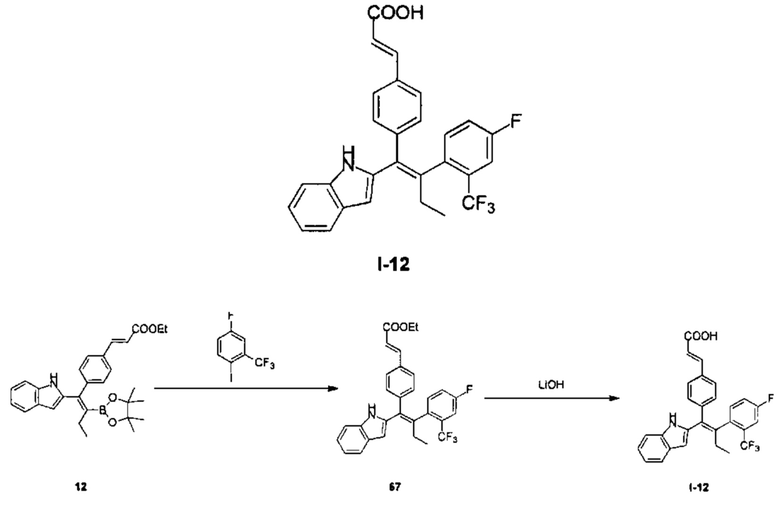
[206] Стадия А: Биспинаколатодибор (329,61 мг, 1,30 ммоль, 1,10 экв.) и тетракис(трифенилфосфин)платину (73,53 мг, 59,09 мкмоль, 0,05 экв.) добавляли к раствору соединения 10 (200,00 мг, 1,18 ммоль, 1,00 экв.) в 2-метилтетрагидрофуране (5,00 мл), реакционный раствор три раза продували азотом и перемешивали при 70°С в атмосфере азота в течение 5 часов. После охлаждения реакционного раствора до 0°С добавляли соединение 5 (285,58 мг, 945,27 мкмоль, 0,80 экв.), карбонат цезия (769,97 мг, 2,36 ммоль, 2,00 экв.), 0,4 мл воды, 5 мл 2-метилтетрагидрофурана и дихлорбис(трифенилфосфин)палладий (41,47 мг, 59,08 мкмоль, 0,05 экв.), затем реакционный раствор три раза продували азотом и перемешивали при 20°С в атмосфере азота в течение 12 часов. Затем 4-фтор-2-трифторметилйодбензол (678,60 мг, 2,34 ммоль, 2,00 экв.), водный раствор гидроксида калия (4М, 1,46 мл, 5,00 экв.), дихлорбис(трифенилфосфин)палладий (41,06 мг, 58,50 мкмоль, 0,05 экв.) и 10 мл 2-метилтетрагидрофурана добавляли к указанному раствору, который впоследствии три раза продували азотом. После перемешивания при 70°С в атмосфере азота в течение 12 часов реакционный раствор фильтровали и фильтрат непосредственно концентрировали и затем очищали с помощью колоночной хроматографии на силикагеле (РЕ:ЕА=40:1 до 15:1) с получением соединения 57 в виде желтого твердого вещества (350,00 мг, 628,94 мкмоль, выход 53,76%, чистота 91,2%).
[207] Стадия В: Моногидрат гидроксида лития (263,90 мг, 6,29 ммоль, 10,00 экв.) добавляли к раствору соединения 57 (350,00 мг, 628,94 мкмоль, 1,00 экв.) в смешанном растворителе 3 мл тетрагидрофурана, 3 мл метанола и 3 мл воды, реакционный раствор перемешивали при 35°С в течение 1 часа. 10 мл воды добавляли к реакционному раствору, затем смесь доводили до рН 5 с помощью 1 моль/л соляной кислоты и дважды экстрагировали 10 мл этилацетата каждый раз. Органическую фазу объединяли, дважды промывали 10 мл воды каждый раз, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением продукта I-12 (108,80 мг, 226,01 мкмоль, выход 35,94%, чистота 99,6%).
[208] 1Н ЯМР (400 МГц, ДМСО-d6): δ 10,78 (brs, 1Н), 9,57-9,54 (m, 2Н), 7,48-7,38 (m, 5Н), 7,31 (d, J=7,6 Гц, 1Н), 7,06 (dt, J=6,8 Гц, J=0,8 Гц, 1Н), 6,99 (dt, J=6,8 Гц, J=0,8 Гц, 1Н), 6,93 (d, J=8,0 Гц, 2Н), 6,53 (d, J=1,6 Гц, 1Н), 6,40 (d, J=16,0 Гц, 1H), 2,85-2,78 (m, 1 Н), 2,47-2,39 (m, 1 Н), 0,95 (t, J=7,6 Гц, 3 Н). MS(ESI, М+1): 480,2.
Вариант реализации 17
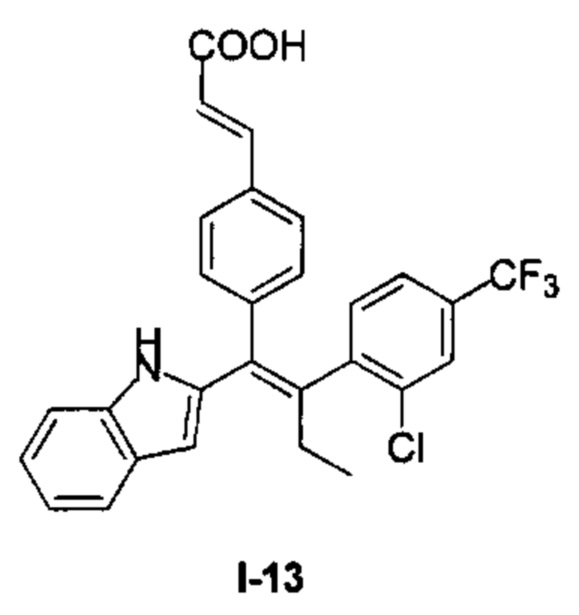
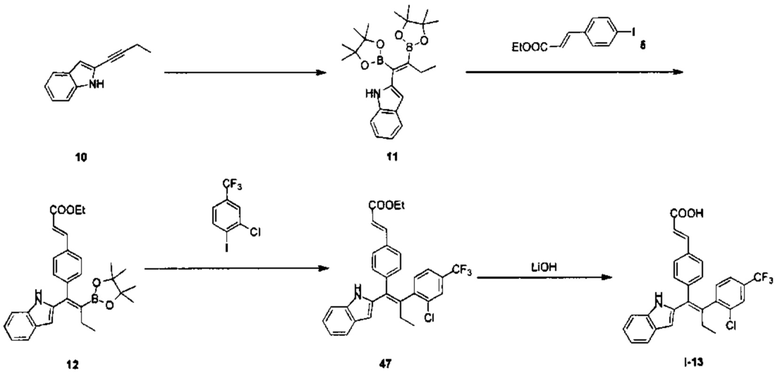
[209] Стадия А: Биспинаколатодибор (329,61 мг, 1,30 ммоль, 1,10 экв.) и тетракис(трифенилфосфин)платину (73,53 мг, 59,09 мкмоль, 0,05 экв.) добавляли к раствору соединения 10 (200,00 мг, 1,18 ммоль, 1,00 экв.) в 2-метилтетрагидрофуране (5,00 мл), реакционный раствор три раза продували азотом и перемешивали при 70°С в атмосфере азота в течение 5 часов. После охлаждения реакционного раствора до 0°С, добавляли соединение 5 (285,58 мг, 945,27 мкмоль, 0,80 экв.), карбонат цезия (769,97 мг, 2,36 ммоль, 2,00 экв.), 0,4 мл воды, 5 мл 2-метилтетрагидрофурана и дихлорбис(трифенилфосфин)палладий (41,47 мг, 59,08 мкмоль, 0,05 экв.), затем реакционный раствор три раза продували азотом и перемешивали при 20°С в атмосфере азота в течение 12 часов. Затем 2-хлор-4-трифторметилйодбензол (717,09 мг, 2,34 ммоль, 2,00 экв.), водный раствор гидроксида калия (4М, 1,46 мл, 5,00 экв.), дихлорбис(трифенилфосфин)палладий (41,06 мг, 58,50 мкмоль, 0,05 экв.) и 10 мл 2-метилтетрагидрофурана добавляли к реакционному раствору, который впоследствии три раза продували азотом. После перемешивания при 70°С в атмосфере азота в течение 12 часов реакционный раствор фильтровали через целит, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (РЕ:ЕА=40:1 до 15:1) с получением соединения 47 в виде желтого желе (738,00 мг), которое непосредственно использовали на следующей стадии.
[210] Стадия В: Моногидрат гидроксида лития (591,64 мг, 14,10 ммоль, 10,00 экв.) добавляли к раствору соединения 47 (738,80 мг, 1,41 ммоль, 1,00 экв.) в смешанном растворителе 3 мл метанола, 3 мл тетрагидрофурана и 3 мл воды, реакционный раствор перемешивали при 35°С в течение 1 часа. 10 мл воды добавляли к реакционному раствору, затем полученный раствор доводили до рН 5 с помощью 1 моль/л соляной кислоты, дважды экстрагировали 10 мл этилацетата каждый раз. Органическую фазу объединяли, дважды промывали 10 мл воды каждый раз, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением продукта I-13 (135,30 мг, 267,64 мкмоль, выход 18,98%, чистота 98,10%).
[211] 1Н ЯМР (400 МГц, ДМСО-d6): δ 10,77 (brs, 1Н), 7,81 (brs, 1Н), 7,62 (dd, J=8,0 Гц, J=1,2 Гц, 1Н), 7,57 (d, J=8,0 Гц, 1H), 7,47-7,40 (m, 4 Н), 7,31 (d, J=8,0 Гц, 1Н), 7,07 (dt, J=8,0 Гц, J=1,2 Гц, 1Н), 7,02-6,96 (m, 3Н), 6,61 (d, J=1,6 Гц, 1Н), 6,41 (d, J- 16,0 Гц, 1Н), 2,75-2,62 (m, 2 Н), 1,00 (t, J=7,6 Гц, 3 Н); MS(ESI, М+1): 496,2.
Вариант реализации 18
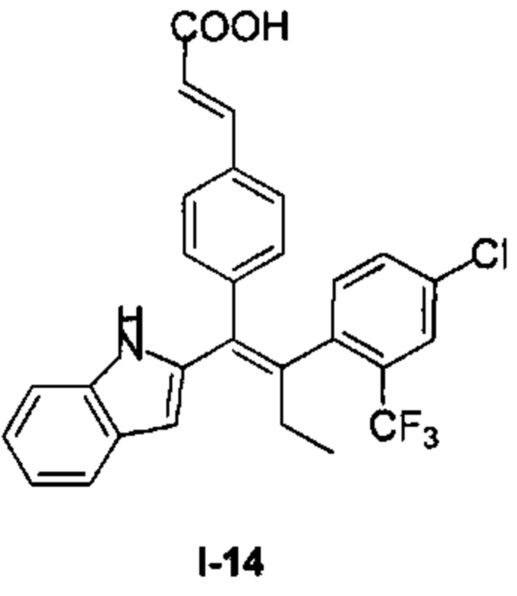
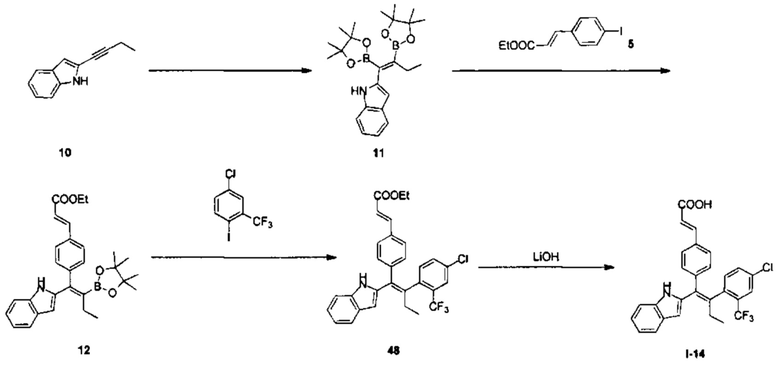
[212] Стадия А: Биспинаколатодибор (1,65 г, 6,50 ммоль, 1,10 экв.) и тетракис(трифенилфосфин)платину (367,64 мг, 295,50 мкмоль, 0,05 экв.) добавляли к раствору соединения 10 (1,09 г, 5,91 ммоль, 1,00 экв.) в 2-метилтетрагидрофуране (20,00 мл), реакционный раствор три раза продували азотом и перемешивали при 70°С в атмосфере азота в течение 5 часов. После охлаждения реакционного раствора до 0°С, добавляли соединение 5 (1,43 г, 4,73 ммоль, 0,80 экв.), карбонат цезия (3,85 г, 11,82 ммоль, 2,00 экв.), 1,2 мл воды, 10 мл 2-метилтетрагидрофурана и дихлорбис(трифенилфосфин)палладия (207,34 мг, 295,50 мкмоль, 0,05 экв.), затем реакционный раствор три раза продували азотом и перемешивали при 20°С в атмосфере азота в течение 12 часов. Затем 4-хлор-2-трифторметилйодбензол (3,63 г, 11,84 ммоль, 2,00 экв.), водный раствор гидроксида калия (4М, 7,40 мл, 5,00 экв.) и дихлорбис(трифенилфосфин)палладий (207,72 мг, 296,00 мкмоль, 0,05 экв.) добавляли к реакционному раствору, который впоследствии три раза продували азотом. После перемешивания при 70°С в атмосфере азота в течение 12 часов, реакционный раствор фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (РЕ:ЕА=50:1 до 5:1) с получением соединения 48 в виде желтого желе (2,60 г), которое непосредственно использовали на следующей стадии.
[213] Стадия В: Моногидрат гидроксида лития (2,08 г, 49,60 ммоль, 10,00 экв.) добавляли к раствору соединения 48 (2,60 г, 4,96 ммоль, 1,00 экв.) в смешанном растворителе 20 мл метанола, 20 мл тетрагидрофурана и 20 мл воды, реакционный раствор перемешивали при 35°С в течение 1 часа. 60 мл воды добавляли к реакционному раствору, затем смесь доводили до рН 5 с помощью 1 моль/л соляной кислоты, дважды экстрагировали 60 мл этилацетата каждый раз. Органическую фазу объединяли, дважды промывали 50 мл воды каждый раз, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением продукта I-14 (735,10 мг, 1,48 ммоль, выход 29,77%, чистота 99,6%).
[214] 1Н ЯМР (400 МГц, ДМСО-d6): δ 12,33 (brs, 1Н), 10,77 (brs, 1Н), 7,73 (d, J=2,0 Гц, 1Н), 7,65 (dd, J=8,4 Гц, J=2,0 Гц, 1H), 7,55 (d, J=8,0 Гц, 1Н), 7,45-7,37 (m, 4Н), 7,31 (d, J=8,0 Гц, 1Н), 7,06 (t, J=7,2 Гц, 1Н), 6,99 (t, J=7,2 Гц, 1Н), 6,94 (d, J=8,0 Гц, 2Н), 6,53 (d, J=1,2 Гц, 1Н), 6,40 (d, J=16,0 Гц, 1Н), 2,87-2,78 (m, 1 Н), 2,47-2,41 (m, 1 Н), 0,94 (t, J=7,6 Гц, 3 Н). MS (ESI,M+1): 496,0.
Вариант реализации 19
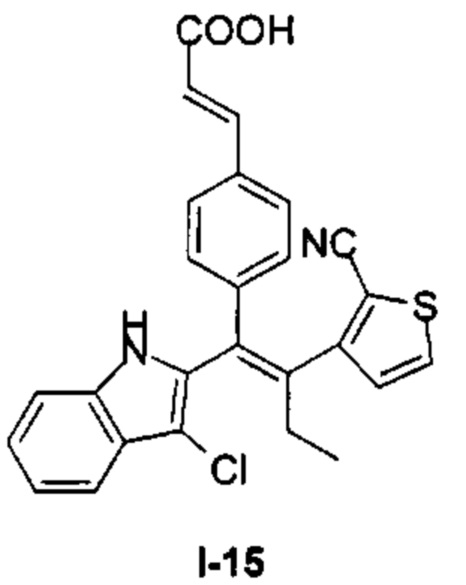
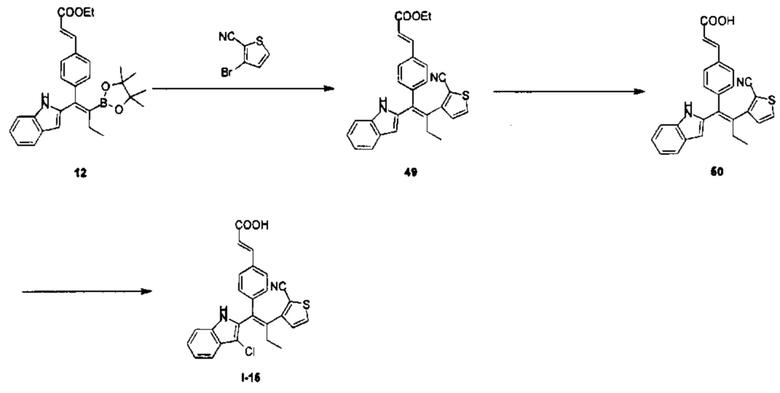
[215] Стадия А: 3-Бром-2-цианотиофен (195,55 мг, 1,04 ммоль, 2,00 экв.) и водный раствор гидроксида калия (4М, 520,00 мкл, 4,00 экв.) добавляли к раствору соединения 12 (300,00 мг, 519,95 мкмоль, 1,00 экв.) в 2-метилтетрагидрофуране (10,00 мл), после продувания реакционной системы азотом три раза добавляли дихлорбис(трифенилфосфин)палладия (18,25 мг, 26,00 мкмоль, 0,05 экв.). Реакционный раствор перемешивали при 70°С в атмосфере азота в течение 12 часов, затем разбавляли 20 мл этилацетата, фильтровали через целит.Фильтрат три раза промывали 20 мл солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением соединения 49 в виде коричневого желе (400,00 мг, неочищенный продукт).
[216] Стадия В: Моногидрат гидроксида лития (370,86 мг, 8,84 ммоль, 10,00 экв.) добавляли к раствору соединения 49 (400,00 мг, 883,84 мкмоль, 1,00 экв.) в смешанном растворителе 2 мл метанола, 2 мл тетрагидрофурана и 2 мл воды, реакционный раствор перемешивали при 45°С в течение 1 часа, затем доводили до рН 5 с помощью 1 моль/л соляной кислоты. Добавляли 20 мл этилацетата, затем дважды промывали 20 мл солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением промежуточного соединения 50 (75,00 мг, 163,07 мкмоль, выход 18,45%, чистота 92,3%).
[217] Стадия С: N-хлорсукцинимид (26,13 мг, 195,68 мкмоль, 1,20 экв.) добавляли к раствору соединения 50 (75,00 мг, 163,07 мкмоль, 1,00 экв.) в ацетонитриле (5,00 мл), реакционный раствор перемешивали при 30°С в течение 14 часов, затем концентрировали и очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением I-15 (7,40 мг, 16,12 мкмоль, выход 9,89%, чистота 100%).
[218] 1Н ЯМР (400 МГц, ДМСО-d6): δ 11,46 (brs, 1Н), 8,03 (d, J=5,2 Гц, 1H), 7,51 (d, J=7,6 Гц, 1Н), 7,44 (d, J=8,4 Гц, 2Н), 7,40 (d, J=16,4 Гц, 1Н), 7,37 (d, J=8,0 Гц, 1Н), 7,31 (d, J=5,2 Гц, 1Н), 7,19 (dt, J=7,2 Гц, J=1,2 Гц, 1Н), 7,13 (dt, J=8,0 Гц, J=1,2 Гц, 1Н), 6,92 (d, J=8,4 Гц, 2Н), 6,42 (d, J=16,0 Гц, 1Н), 2,50-2,46 (m, 2Н), 0,94 (t, J=7,6 Гц, 3 Н). MS (ESI, М+1, М+23): 459,0, 481,1.
Вариант реализации 20
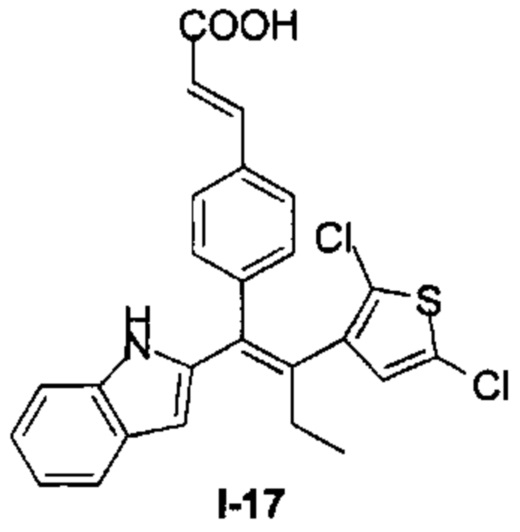
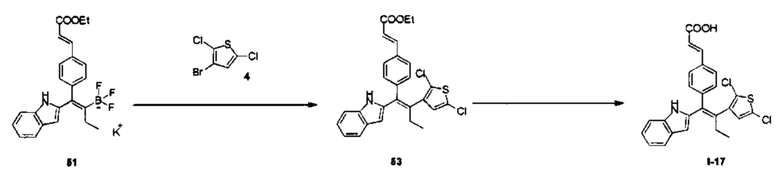
[219] Стадия А: Бис(дифенилфосфино)ферроцен дихлорпалладия (7,94 мг, 10,86 мкмоль, 0,05 экв.) добавляли к раствору 51 (100,00 мг, 217,14 мкмоль, 1,00 экв.), 2,5-дихлор-3-бромтиофена (70,50 мг, 304,00 мкмоль, 1,40 экв.), водного раствора гидроксида натрия (4М, 217,14 мкл, 4,00 экв.) в 2-метилтетрагидрофуране (10 мл) в 100 мл трехгорлой колбе. После продувания реакционной системы азотом при пониженном давлении реакционный раствор перемешивали при 70°С (внутренняя температура 60°С) в течение 12 часов, затем он поменял цвет с желтого на коричневый. 30 мл этилацетата добавляли к реакционному раствору и полученную смесь фильтровали через целит.Фильтрат три раза промывали 30 мл насыщенного солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением продукта 53 в виде коричневого твердого вещества (200,00 мг, неочищенный продукт), который непосредственно использовали на следующей стадии.
[220] Стадия В: Моногидрат гидроксида лития (169,04 мг, 4,03 ммоль, 10,00 экв.) добавляли к раствору соединения 53 (200,00 мг, 402,86 мкмоль, 1,00 экв.) в смешанном растворителе 2 мл метанола, 2 мл тетрагидрофурана и 2 мл воды в 100 мл одногорлой колбе, реакционный раствор перемешивали при 45°С в течение 1 часа и он поменял цвет с желтого на коричневый, затем доводили до рН 1 с помощью 1 моль/л соляной кислоты. Добавляли 30 мл этилацетата и 30 мл воды, затем полученную смесь разделяли. Органическую фазу дважды промывали 30 мл насыщенного солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением I-17 (36,00 мг, 76,71 мкмоль, выход 19,04%, чистота 99,8%)о 1Н ЯМР (400 МГц, ДМСО-d6): δ 10,73 (brs, 1Н), 7,70 (d, J = 3,6 Гц, 1Н), 7,53 (d, J=8,0 Гц, 3Н), 7,45 (d, J=16,0 Гц, 1Н), 7,29 (d, J=8,0 Гц, 1Н), 7,10-7,04 (m, 3Н), 6,98 (t, J=7,6 Гц, 1Н), 6,91 (d, J=3,6 Гц, 1Н), 6,54 (brs, 1Н), 6,47 (d, J=16,0 Гц, 1Н), 2,75 (q, J=7,2 Гц, 2Н), 2,43 (s, ЗН), 1,11 (t, J=7,6 Гц, 3 Н). MS (ESI, M+1): 468,0.
Вариант реализации 21
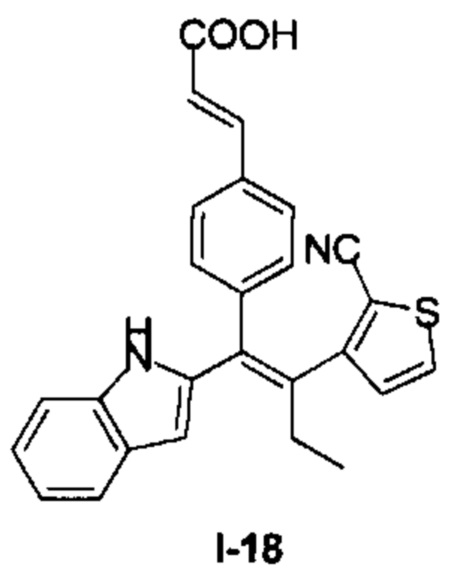
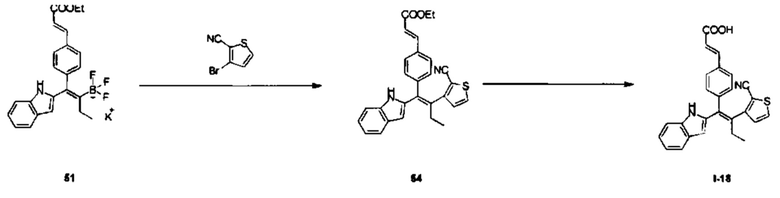
[221] Стадия А: Бис(дифенилфосфино)ферроцен дихлорпалладия (7,94 мг, 10,86 мкмоль, 0,05 экв.) добавляли к раствору 51 (100,00 мг, 217,14 мкмоль, 1,00 экв.), 3-бром-2-цианотиофена (57,17 мг, 304,00 мкмоль, 1,40 экв.), водного раствора гидроксида натрия (4М, 217,14 мкл, 4,00 экв.) в 2-метилтетрагидрофуране (10 мл) в 50 мл трехгорлой колбе. После продувания реакционной системы азотом при пониженном давлении, реакционный раствор перемешивали при 70°С в течение 12 часов, затем он поменял цвет с желтого на коричневый. 30 мл этилацетата добавляли к реакционному раствору, и смесь фильтровали через целит.Фильтрат три раза промывали 30 мл насыщенного солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением продукта 54 в виде коричневого твердого вещества (200,00 мг, неочищенный продукт), который непосредственно использовали на следующей стадии.
[222] Стадия В: Моногидрат гидроксида лития (185,43 мг, 4,42 ммоль, 10,00 экв.) добавляли к раствору соединения 54 (200,00 мг, 402,86 мкмоль, 1,00 экв.) в смешанном растворителе 2 мл метанола, 2 мл тетрагидрофурана и 2 мл воды в 100 мл одногорлой колбе, реакционный раствор перемешивали при 45°С в течение 1 часа и он поменял цвет с желтого на коричневый, затем доводили до рН 1 с помощью 1 моль/л соляной кислоты. Добавляли 30 мл этилацетата и 30 мл воды, затем смесь разделяли. Органическую фазу дважды промывали 30 мл насыщенного солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением I-18 (24,70 мг, 57,95 мкмоль, выход 13,11%, чистота 99,6%).
[223] 1Н ЯМР (400 МГц, ДМСО-d6): δ 10,78 (brs, 1Н), 7,99 (d, J=5,2 Гц, 1Н), 7,57 (d, J=8,0 Гц, 1Н), 7,49-7,45 (m, 3Н), 7,31-7,28 (m, 2Н), 7,07 (t, J=7,2 Гц, 1Н), 7,00 (t, J=7,2 Гц, 1H), 6,93 (d, J=8,4 Гц, 2H), 6,58 (d, J=1,2 Гц, 1H), 6,45 (d, J=16,0 Гц, 1H), 2,71 (q, J=7,6 Гц, 2H), 1,02 (t, J=7,6 Гц, 3 H). MS (ESI, M+1): 425,1.
Вариант реализации 22
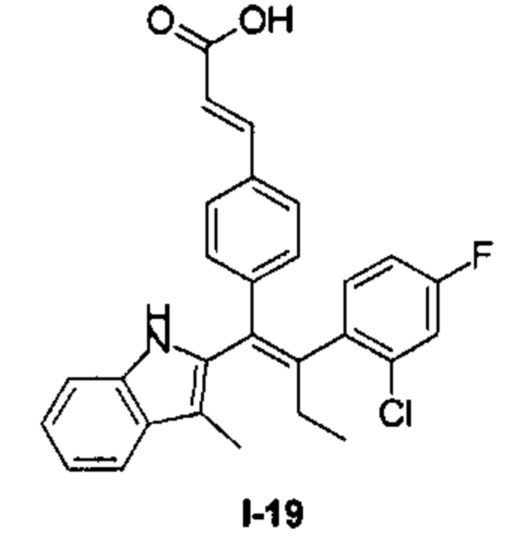
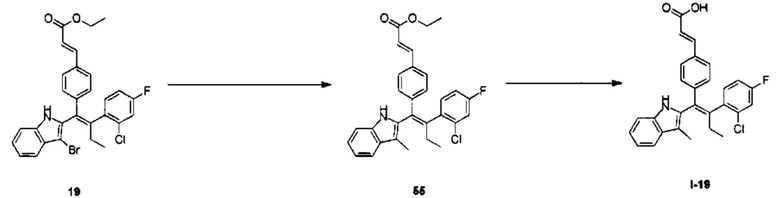
[224] Стадия A: 19 (50,00 мг, 90,44 мкмоль, 1,00 экв.), 2,4,6-триметилбороксин (34,06 мг, 271,32 мкмоль, 37,84 мкл, 3,00 экв.), карбонат цезия (88,40 мг, 271,32 мкмоль, 3,00 экв.) и 3 мл диоксан добавляли в одногорлую круглодонную колбу. После продувания реакционной системы азотом добавляли Pd(dppf)Cl2.ДХМ (7,39 мг, 9,04 мкмоль, 0,1 экв.) и реакционную систему продували азотом снова. Желтую эмульгируемую смесь перемешивали при 120°С в течение 1 часа и она поменяла цвет с желтого на темно желтый. Реакционный раствор фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ТСХ (РЕ:ЕА=5:1) с получением промежуточного соединения 55 (25,00 мг, 51,23 мкмоль, выход 56,65%). MS (ESI, M+1): 488,1.
[225] Стадия В: Моногидрат гидроксида лития (6,45 мг, 153,69 мкмоль, 3,00 экв.) добавляли к раствору соединения 55 (25,00 мг, 51,23 мкмоль, 1,00 экв.) в смешанном растворителе 2 мл тетрагидрофурана, 2 мл метанола и 2 мл воды, бесцветный прозрачный раствор перемешивали при 20°С в течение 72 часов, затем концентрировали при пониженном давлении, разбавляли 10 мл воды, доводили до рН 1-2 с помощью 1 моль/л соляной кислоты, экстрагировали три раза 20 мл этилацетата, промывали 20 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ТСХ (РЕ:ЕА=3:1) с получением продукта I-19 (20,00 мг, 43,44 мкмоль, выход 84,79%, чистота 99,9%).
[226] 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,80 (s, 1Н), 8,30 (s, 1Н), 7,49 (d, J=7,6 Гц, 1Н), 7,43 - 7,32 (m, 5H), 7,30 (d, J=8,0 Гц, 1H), 7,18 (dt, J=2,4, 8,5 Гц, 1H), 7,07 (dt, J=1,2, 7,6 Гц, 1H), 7,03 - 6,95 (m, 3H), 6,39 (d, J=16,0 Гц, 1H), 2,43 (q, J=7,6 Гц, 2H), 2,29 (s, 3H), 0,86 (t,.7=7,6 Гц, 3Н); MS (ESI,M+1): 460,1.
Вариант реализации 23
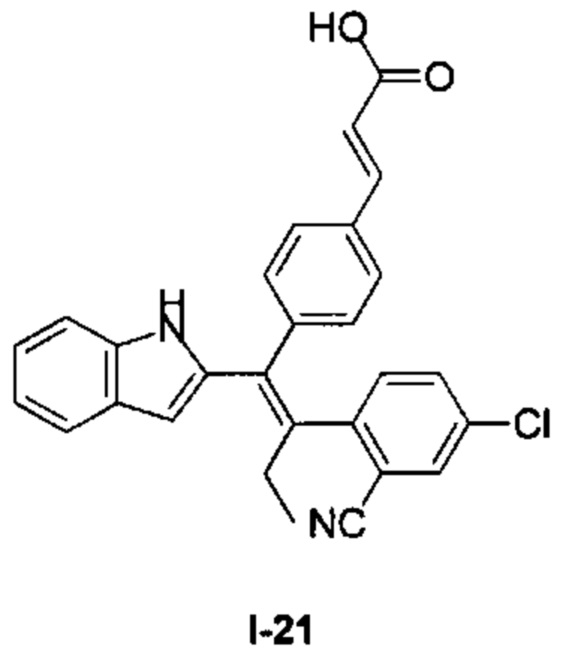
Вариант реализации 24
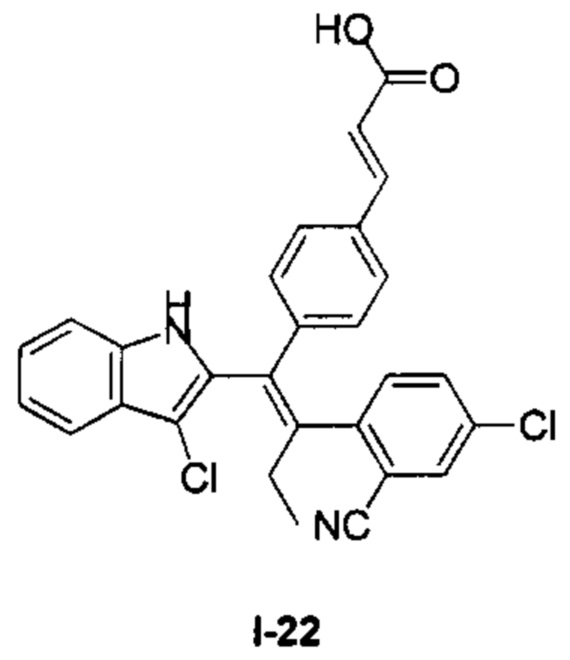
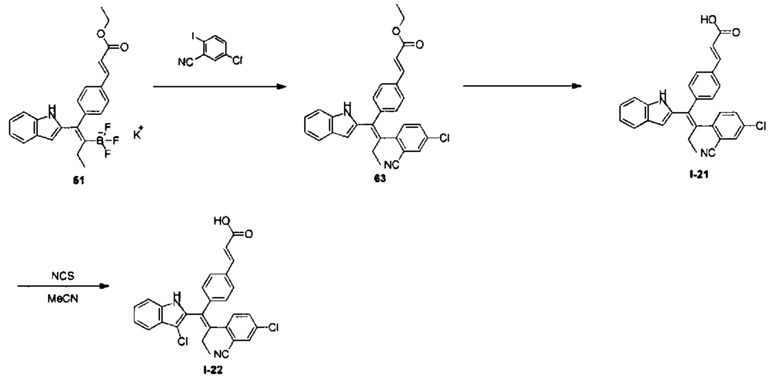
[227] Стадия А: 51 (101,68 мг, 221,57 мкмоль, 1,00 экв.), 5-хлор-2-йодбензонитрил (70,05 мг, 265,88 мкмоль, 1,20 экв.), Pd(dppf)Cl2 (8,11 мг, 11,08 мкмоль, 0,05 экв.) и 2-метилтетрагидрофуран (5,00 мл) добавляли в трехгорлую круглодонную колбу, после продувания реакционной системы азотом добавляли 4М гидроксид натрия (221,57 мкл, 4,00 экв.) и реакционную систему продували азотом снова. Реакционный раствор нагревали до 68°С и перемешивали в течение 10 часов, затем фильтровали. Фильтрационный осадок промывали 10 мл этилацетата и фильтрат концентрировали с получением промежуточного соединения 63 (125,00 мг), которое непосредственно использовали на следующей стадии. MS (ESI, М+1): 481,2.
[228] Стадия В: 63 (125,00 мг, 259,88 мкмоль, 1,00 экв.), тетрагидрофуран (2,00 мл) и метанол (2,00 мл) добавляли в одногорлую круглодонную колбу, с последующим добавлением моногидрата гидроксида лития (32,71 мг, 779,64 мкмоль, 3,00 экв.) и воды (2,00 мл). Реакционный раствор перемешивали при 30°С в течение 12 часов, затем концентрировали с удалением растворителя. 5 мл воды добавляли к реакционному раствору и полученную смесь доводили до рН 3 2М соляной кислотой, дважды экстрагировали 20 мл этилацетата каждый раз. Органическую фазу объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением продукта I-21 (32,00 мг, 70,51 мкмоль, выход 27,13%, чистота 99,8%). 1Н ЯМР (400 МГц, ДМСО-d6) δ=10,77 (s, 1Н), 7,88 (d, J=2,0 Гц, 1Н), 7,75 (dd, J=2,0, 8,4 Гц, 1Н), 7,61 (dd, 7=8,0, 12,0 Гц, 2Н), 7,49 - 7,37 (m, 3Н), 7,31 (br d, J=8,0 Гц, 1Н), 7,08 (br t, J=7,2 Гц, 1Н), 7,04 -6,99 (m, 1Н), 6,94 (br d, J=8,0 Гц, 2Н), 6,62 (s, 1Н), 6,44 (br d, J=16,0 Гц, 1Н), 2,95 - 2,57 (m, 2Н), 1,03 (br t, J=7,2 Гц, 3Н); MS (ESI, М+1): 453,1.
[229] Стадия С: I-21 (80,00 мг, 176,63 мкмоль, 1,00 экв.), 3 мл дихлорметан, 6 мл ацетонитрил и NCS (25,94 мг, 194,29 мкмоль, 1,10 экв.) добавляли в одногорлую круглодонную колбу в атмосфере азота. Реакционный раствор перемешивали при 25°С в течение 1 часа, затем гасили 10% тиосульфатом натрия (20 мл). Добавляли 20 мл дихлорметана и смесь разделяли, водную фазу экстрагировали три раза 10 мл дихлорметана каждый раз. Органическую фазу объединяли, промывали 20 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением продукта I-22 (25,00 мг, 50,99 мкмоль, выход 28,87%, чистота 99,4%). 1Н ЯМР (400 МГц, ДМСО-d6) δ=11,47 (br s, 1H), 7,94 (br s, 1H), 7,78 (br d, J=8,0 Гц, 1H), 7,60 - 7,49 (m, 2H), 7,48 - 7,36 (m, 4H), 7,25 - 7,09 (m, 2H), 6,96 (br d, J=7,2 Гц, 2H), 6,41 (br d, J=15,6 Гц, 1H), 2,61 - 2,53 (m, 2H), 0,93 (br t, J=6,8 Гц, 3Н); MS (ESI,M+1): 487,0.
Вариант реализации 25
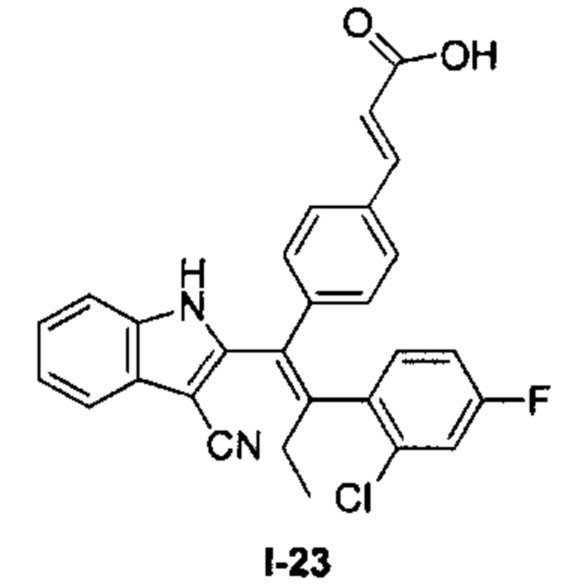
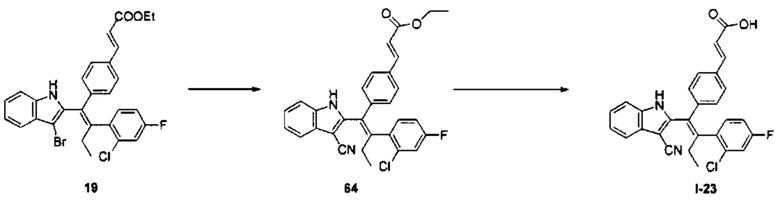
[230] Стадия А: Соединение 19 (50,00 мг, 85,92 мкмоль, 1,00 экв.) и CuCN (23,08 мг, 257,75 мкмоль, 3,00 экв.) растворяли в NMP (2,00 мл). Реакционный раствор продували азотом, перемешивали при 180°С в течение 2 часов в условиях микроволнового облучения и он поменял цвет с зеленого на черный. После завершения реакции добавляли воду (20 мл) и смесь доводили до рН 10 водным NaHCO3 (2 мл) и экстрагировали этилацетатом (20 мл*3). Органическую фазу объединяли, промывали насыщенным солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ТСХ (РЕ:ЕА=5:1) с получением продукта 64 в виде желтого масла (10,00 мг, 16,43 мкмоль, выход 19,13%, чистота 82%). MS (ESI,M+1): 499,1.
[231] Стадия В: Соединение 64 (80,00 мг, 160,33 мкмоль, 1,00 экв.) растворяли в смешанном растворителе тетрагидрофурана (1,50 мл), этанола (1,50 мл) и воды (1,50 мл), с последующим добавлением LiOH.Н2О (20,18 мг, 480,99 мкмоль, 3,00 экв.), затем реакционный раствор перемешивали при 30°С в течение 12 часов. Реакция была незавершенна, затем гидроксид натрия (9,62 мг, 240,50 мкмоль, 1,50 экв.) и этанол (1,50 мл) добавляли к реакционному раствору. Желтый реакционный раствор перемешивали при 45°С в течение 3 часов. После завершения реакции реакционный раствор концентрировали с получением неочищенного продукта, затем добавляли воду (1 мл) и 3 моль/л HCl к неочищенному продукту с доведением рН до 4. Затем выпадал твердый осадок, который выделяли фильтрованием. Твердое вещество очищали с помощью препаративной хроматографии (система с муравьиной кислотой) с получением продукта I-23 (25,90 мг, 54,45 мкмоль, выход 33,96%, чистота 99%). MS (ESI,M+1): 471,0.
[232] 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,36 (br s, 1Н), 7,65 (d, J=6,8 Гц, 1Н), 7,52 (d, J=8,0 Гц, 1Н), 7,48-7,41 (m, 3Н), 7,37 (br d, J=16,0 Гц, 1Н), 7,34-7,25 (m, 3Н), 7,25-7,17 (m, 1H), 7,01 (d, J=8,4 Гц, 2Н), 6,41 (br d, J=16,4 Гц, 1Н), 2,46-2,40 (m, 1Н), 2,46-2,40 (m, 1Н), 0,94 (t, J=7,6 Гц, 3Н).
Вариант реализации 26
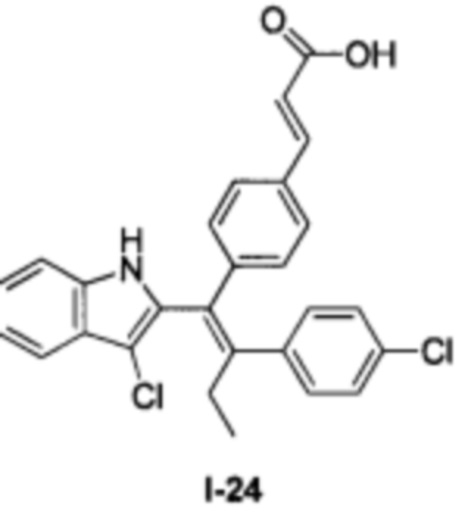
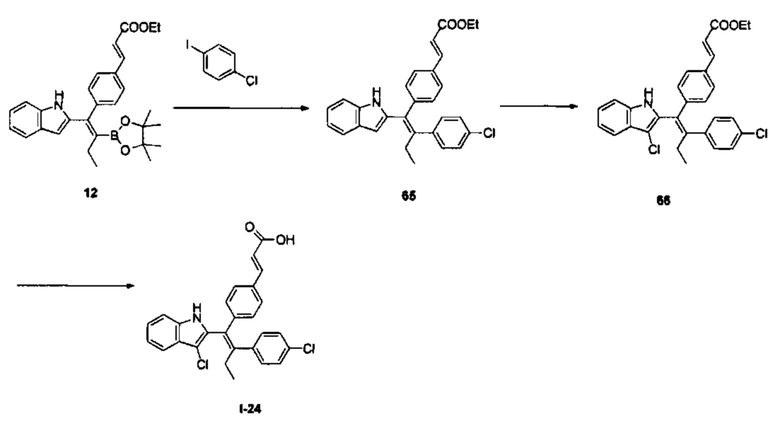
[233] Стадия А: Соединение 12 (340,91 мг, 636,42 мкмоль, 1,00 экв.) растворяли в 2-метилтетрагидрофуране (10,00 мл), затем добавляли гидроксид калия (4М, 890,99 мкл, 5,60 экв.) и 1-хлор-4-йодбензол (273,16 мг, 1,15 ммоль, 1,80 экв.). Реакционный раствор три раза продували азотом и перемешивали при 80°С в течение 12 часов. После завершения реакции воду (30 мл) добавляли к реакционному раствору и смесь экстрагировали этилацетатом (30 мл*3). Органическую фазу объединяли, промывали насыщенным солевым раствором (30 мл*2), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (РЕ:ЕА=1:0-20:1) с получением промежуточного соединения 65 (280,00 мг, 614,08 мкмоль, выход 96,49%). MS (ESI,M+1): 456,0.
[234] Стадия В: Соединение 65 (280,00 мг, 564,95 мкмоль, 1,00 экв.) растворяли в ацетонитриле (8,00 мл), добавляли NCS (82,98 мг, 621,44 мкмоль, 1,10 экв.) одной порцией. Реакционный раствор перемешивали при 20°С в течение 2 часов. После завершения реакции реакционный раствор концентрировали с получением неочищенного продукта. Затем воду (5 мл) добавляли к неочищенному продукту и смесь экстрагировали этилацетатом (5 мл*4). Органическую фазу объединяли, промывали насыщенным солевым раствором (10 мл*2), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта 66 (230,00 мг, 389,26 мкмоль, выход 68,90%, чистота 83%), который непосредственно использовали на следующей стадии без дополнительной очистки. MS (ESI, M+1): 490,0.
[235] Стадия С: Соединение 66 (230,00 мг, 389,26 мкмоль, 1,00 экв.) растворяли в смешанном растворителе воды (3,00 мл), метанола (6,00 мл) и тетрагидрофурана (6,00 мл), затем добавляли LiOH.H2O (81,67 мг, 1,95 ммоль, 5,00 экв.). Реакционный раствор перемешивали при 20°С в течение 12 часов. После завершения реакции реакционный раствор концентрировали, доводили до рН 4 с помощью 3 моль/л HCl (1 мл), фильтровали с получением твердого вещества, которое растворяли в 1 мл ДМФА и очищали с помощью препаративной хроматографии (система с муравьиной кислотой) с получением продукта I-24 в виде желтого твердого вещества (41,00 мг, 81,58 мкмоль, выход 20,96%, чистота 92%), но эта чистота была недостаточна. Продукт очищали с помощью щелочной препаративной хроматографии, но эта чистота также была недостаточна, затем очищали с помощью препаративной ТСХ (дихлорметан : этилацетат = 10:1) с получением продукта I-24 (9,70 мг, 20,56 мкмоль, выход 25,20%, чистота 98%).
[236] 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,47 (s, 1Н), 7,50 (d, J=7,6 Гц, 1Н), 7,47-7,30 (m, 6Н), 7,25-7,16 (m, 3Н), 7,16-7,10 (m, 1Н), 6,93 (d, J=8,4 Гц, 2Н), 6,40 (d, J=16,0 Гц, 1Н), 2,48-2,40 (m, 2Н), 0,90 (t, J=7,6 Гц, 3Н). MS (ESI,M+1): 462,0.
Вариант реализации 27
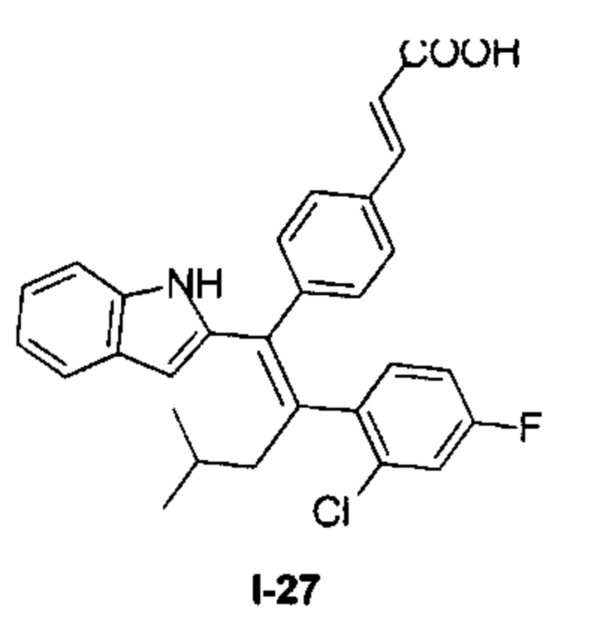
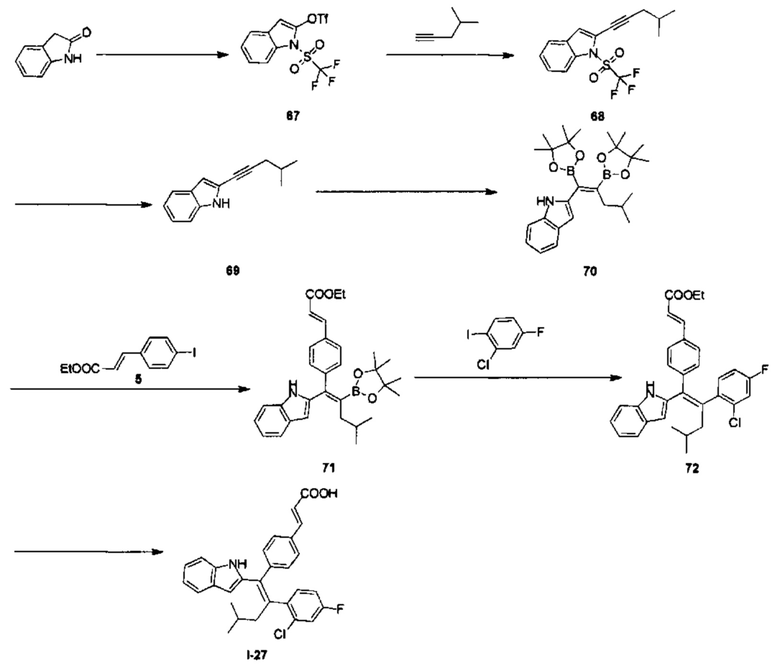
[237] Стадия А: Ангидрид трифторметансульфокислоты (19,07 г, 67,59 ммоль, 11,15 мл, 3,00 экв.) медленно добавляли по каплям к раствору оксиндола (3,00 г, 22,53 ммоль, 1,00 экв.) и триэтиламина (22,80 г, 225,30 ммоль, 31,23 мл, 10,00 экв.) в 100 мл 2-метилтетрагидрофурана при -78°С в атмосфере азота в реакционной колбе. Реакционный раствор перемешивали более 2 часов и он поменял цвет с бесцветного на оранжевый. Затем добавляли 4-метил-1-пентин (3,70 г, 45,06 ммоль, 5,29 мл, 2,00 экв.), триэтиламин (22,80 г, 225,30 ммоль, 31,23 мл, 10,00 экв.), тетракис(трифенилфосфин)платину (2,60 г, 2,25 ммоль, 0,10 экв.) и йодид меди (429,08 мг, 2,25 ммоль, 0,10 экв.). Реакционный раствор продували азотом, нагревали до 15°С и перемешивали в атмосфере азота в течение 12 часов. Серая суспензия стала черным раствором. Реакционный раствор гасили насыщенным водным раствором хлорида аммония (300 мл), экстрагировали три раза 100 мл этилацетата каждый раз. Органическую фазу промывали насыщенным солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле (ЕА:РЕ=0-5%) с получением соединения 68 (4,90 г, 14,88 ммоль, выход 66,04%).
[238] Стадия В: Карбонат калия (3,78 г, 27,32 ммоль, 2,00 экв.) добавляли к раствору соединения 68 (4,50 г, 13,66 ммоль, 1,00 экв.) а 50 мл тетрагидрофурана и 50 мл метанола. Реакционный раствор перемешивали при 25°С в течение 12 часов и бесцветная смесь поменяла цвет с желтого на коричневый. 200 мл этилацетата и 200 мл воды добавляли к реакционному раствору. Смесь разделяли в делительной воронке. Органическую фазу три раза промывали 100 мл воды каждый раз, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 69 (2,60 г, 13,18 ммоль, выход 96,48%).
[239] Стадия С: Тетракис(трифенилфосфин)платину (126,29 мг, 101,50 мкмоль, 0,05 экв.) добавляли к раствору соединения 69 (400,00 мг, 2,03 ммоль, 1,00 экв.) и биспинаколатодибора (515,50 мг, 2,03 ммоль, 1,00 экв.) в 10 мл 2-метилтетрагидрофурана. Реакционный раствор перемешивали при 70°С в течение 5 часов и он постепенно поменял цвет с желтого на коричневый. Реакционный раствор соединения 70 в 10 мл 2-метилтетрагидрофурана непосредственно использовали на следующей стадии.
[240] Стадия D: Соединение 5 (490,63 мг, 1,62 ммоль, 0,80 экв.), 10 мл 2-метилтетрагидрофурана и 3 мл воды добавляли к раствору соединения 70 (915,96 мг, 2,03 ммоль, 1,00 экв.) в 10 мл 2-метилтетрагидрофурана. Затем карбонат цезия (1,32 г, 4,06 ммоль, 2,00 экв.) и бис(трифенилфосфин)палладия дихлорид (142,49 мг, 203,00 мкмоль, 0,10 экв.) быстро добавляли к смеси при 0°С. Реакционный раствор перемешивали при 25°С в атмосфере азота в течение 12 часов и он постепенно поменял цвет с желтого на коричневый. Реакционный раствор соединения 71 в 20 мл 2-метилтетрагидрофурана непосредственно использовали на следующей стадии.
[241] Стадия Е: 2-Хлор-4-фтор-йодбензол (621,61 мг, 2,42 ммоль, 1,20 экв.) и 10 мл 2-метилтетрагидрофурана добавляли к раствору соединения 71 (1,01 г, 2,02 ммоль, 1,00 экв.) в 20 мл 2-метилтетрагидрофурана, затем водный раствор гидроксида калия (4М, 2,53 мл, 5,00 экв.) и бис(трифенилфосфин)палладия дихлорид (70,89 мг, 101,00 мкмоль, 0,05 экв.) добавляли при 25°С. Реакционный раствор перемешивали при 70°С в течение 12 часов, затем фильтровали и добавляли 30 мл этилацетата и 30 мл воды. Смесь разделяли в делительной воронке. Органическую фазу промывали 30 мл солевого раствора, сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (РЕ:ЕА=1:0 до 20:1) с получением промежуточного соединения 72 (500,00 мг, неочищенный продукт).
[242] Стадия F: Моногидрат гидроксида лития (417,91 мг, 9,96 ммоль, 10,00 экв.) добавляли к раствору соединения 72 (500,00 мг, 995,98 мкмоль, 1,00 экв.) в тетрагидрофуране (5,00 мл), метаноле (5,00 мл) и воде (5,00 мл) при 25°С, Смесь перемешивали при 40°С в течение 1 часа и она поменяла цвет с желтого на коричневый, затем доводили до рН 3 с помощью 1М HCl и добавляли 20 мл воды. Смесь дважды экстрагировали 20 мл этилацетата каждый раз. Органическую фазу объединяли, дважды промывали 20 мл солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали, концентрировали с получением неочищенного продукта, который очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением продукта I-27 (150,00 мг, 307,62 мкмоль, выход 30,89%, чистота 97,2%).
[243] 1Н ЯМР (400 МГц, ДМСО-d6): δ 10,78 (brs, 1Н), 7,56 (d, J=7,6 Гц, 1H), 7,48-7,38 (m, 3Н), 7,34 (dd, J=2,4 Гц, J=9,2 Гц, 1Н), 7,31-7,25 (m, 2Н), 7,11 (dt, J=2,4 Гц, J= 8,4 Гц, 1Н), 7,08-7,03 (m, 1Н), 7,01-6,96 (m, 3Н), 6,52 (d, J=1,2 Гц, 1Н), 6,40 (d, J=16,0 Гц, 1H), 2,62-2,53 (m, 2Н), 1,55-1,45 (m, 1Н), 0,87 (d, J=6,8 Гц, 3Н), 0,84 (d, J=6,8 Гц, 3Н). MS(ESI,M+1): 474,1.
Вариант реализации 28
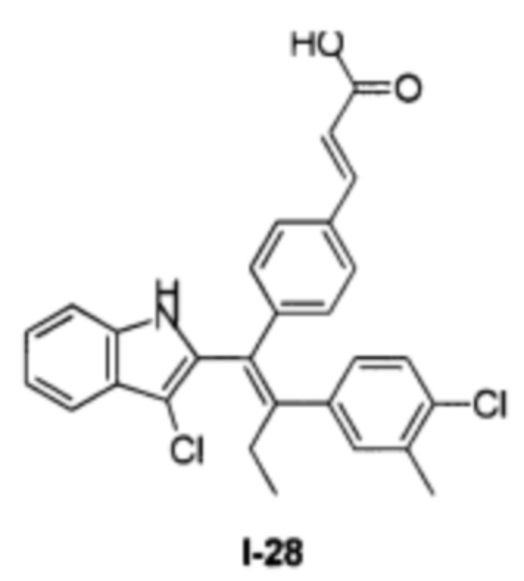
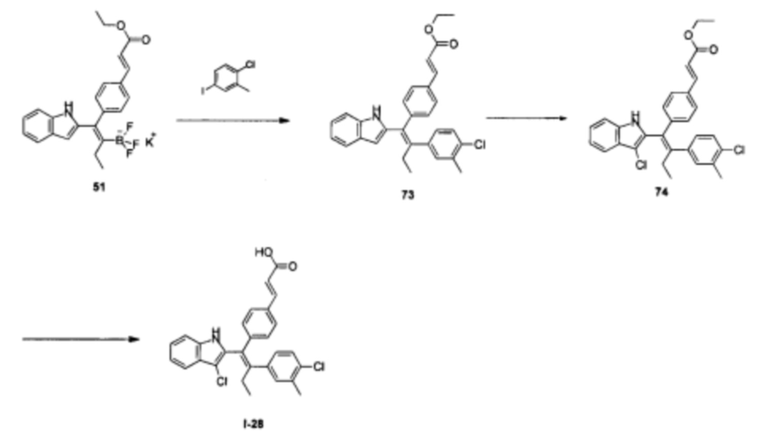
[244] Стадия A: NaOH (4М, 443,13 мкл, 4,00 экв.) добавляли к раствору соединения 51 (200,00 мг, 443,13 мкмоль, 1,00 экв.), 4-хлор-1-йод-2-толуола (156,63 мг, 620,38 мкмоль, 1,40 экв.), Pd(dppf)Cl2 (16,21 мг, 22,16 мкмоль, 0,05 экв.) в 12 мл 2-метилтетрагидрофурана, реакционный раствор три раза продували азотом, затем перемешивали при 65°С в течение 12 часов, он постепенно поменял цвет с желтого на черный. 20 мл воды добавляли к указанному раствору и смесь экстрагировали три раза этилацетатом (20 мл*3). Органическую фазу объединяли, промывали 20 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (400 меш, РЕ:ЕА=1:0 до 20:1) с получением соединения 73 (170,00 мг, 325,53 мкмоль, выход 73,46%, чистота 90%).
[245] Стадия В: NCS (38,35 мг, 287,23 мкмоль, 1,00 экв.) добавляли к раствору соединения 73 (150,00 мг, 287,23 мкмоль, 1,00 экв.) в ацетонитриле (6,00 мл) и дихлорметане (14,00 мл). Желтый раствор перемешивали при 30°С в течение 1,5 часов. Затем 30 мл воды добавляли к реакционному раствору и смесь дважды экстрагировали этилацетатом (30 мл*2). Органическую фазу объединяли, дважды промывали насыщенным солевым раствором (20 мл*2), сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (РЕ:ЕА=1:0 до 10:1) с получением промежуточного соединения 74 (170,00 мг, неочищенное).
[246] Стадия С: LiOH.H2O (70,70 мг, 1,69 ммоль, 5,00 экв.) добавляли к раствору соединения 74 (170,00 мг, 337,00 мкмоль, 1,00 экв.) в воде (2,00 мл), тетрагидрофуране (2,00 мл) и этаноле (2,00 мл). Желтый раствор перемешивали при 30°С в течение 1,5 часов, затем концентрировали при пониженном давлении с получением остатка. 1 мл воды добавляли к остатку, полученный раствор доводили до рН 4 3М соляной кислотой. Твердый осадок, выделяли фильтрованием и затем растворяли в 3 мл ДМФА и очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением продукта I-28 (46,50 мг, 96,63 мкмоль, выход 28,67%, чистота 99%).
[247] 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,46 (s, 1Н), 7,50 (d, J=7,6 Гц, 1Н), 7,46-7,37 (m, 4Н), 7,31-7,23 (m, 2Н), 7,22-7,09 (m, 2Н), 7,00-6,91 (m, 3Н), 6,40 (d, J=16,0 Гц, 1Н), 2,45 (q, J=7,6 Гц, 2Н), 2,27 (s, 3Н), 0,90 (t, 5=7,6Гц, 3Н). MS (ESI, M+1): 476,0.
Вариант реализации 29
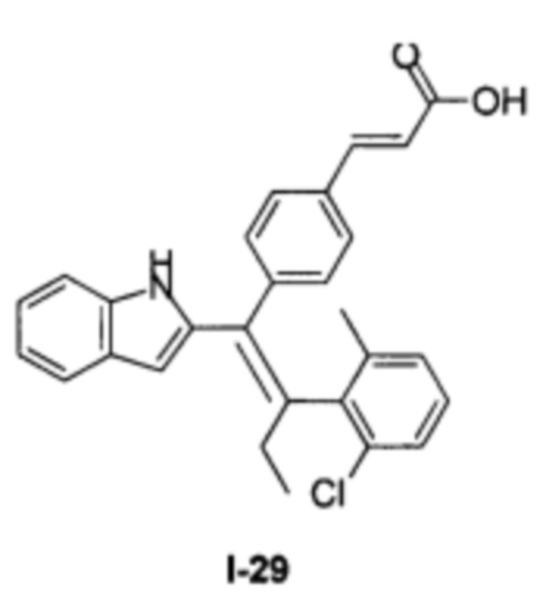
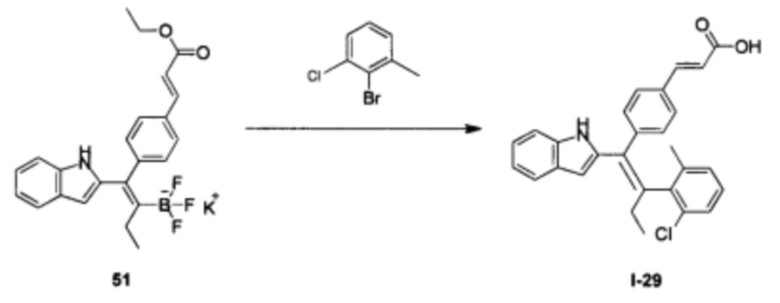
[248] Гидроксид натрия (34,87 мг, 871,64 мкмоль, 4,00 экв.) добавляли к смеси соединения 51 (100,00 мг, 217,91 мкмоль, 1,00 экв.), 2-бром-3-хлортолуола (62,69 мг, 305,07 мкмоль, 1,40 экв.) и 2-метилтетрагидрофурана (10,00 мл) в одногорлой колбе, с последующим добавлением 1,1'-бис(дифенилфосфино)ферроценпалладия хлорида (7,97 мг, 10,90 мкмоль, 0,05 экв.) в атмосфере азота. Реакционный раствор перемешивали при 70°С в течение 16 часов. После завершения реакции реакционный раствор доводили до рН 1 соляной кислотой (3М) при комнатной температуре, затем экстрагировали этилацетатом (20 мл). Органическую фазу концентрировали с получением неочищенного продукта, который очищали с помощью препаративной хроматографии с жидкой фазой (преп. ВЭЖХ: муравьиная кислота) с получением продукта I-29 (5,30 мг, 11,79 мкмоль, выход 5,41%, чистота 98,3%).
[249] MS (ESI, M+1): 442,0.
[250] 1Н ЯМР (400 МГц, ДМСО-d6) δ = 10,80 (s, 1Н), 8,40 (s, 1Н), 7,57 (d, J=7,3 Гц, 1Н), 7,39-7,27 (m, 5Н), 7,16-7,11 (m, 1H), 7,09-7,04 (m, 2Н), 7,03-6,97 (m, 3Н), 6,61 (d, J=1,4 Гц, 1Н), 6,38 (d, J=15,9 Гц, 1Н), 2,62-2,56 (m, 2Н), 2,16 (s, 3Н), 1,00 (t, J=7,5 Гц, 3Н).
Вариант реализации 30
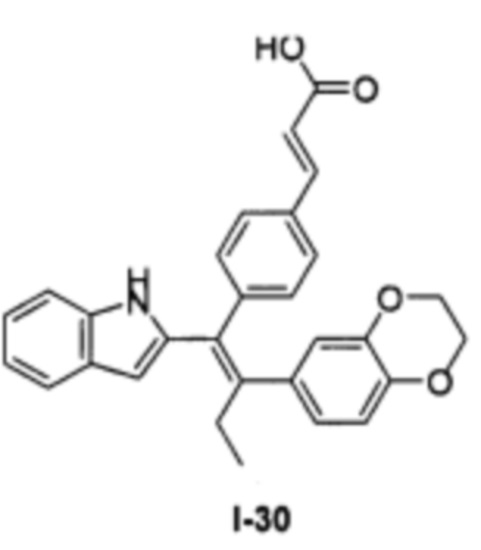
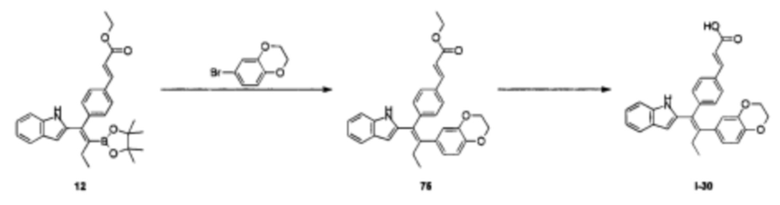
[251] Стадия А: Соединение 12 (100,00 мг, 212,13 мкмоль, 1,00 экв.) и 6-бром-1,4-бензодиоксан (68,42 мг, 318,19 мкмоль, 42,76 мкл, 1,50 экв.) взвешивали и растворяли в 2-метилтетрагидрофуране (4,00 мл) в атмосфере азота, с последующим добавлением гидроксида калия (66,65 мг, 1,19 ммоль, 5,60 экв.) (растворяли в 500,00 мкл воды) и дихлорбис(трифенилфосфин)палладия(II) (7,44 мг, 10,61 мкмоль, 0,05 экв.), реакционный раствор перемешивали при 75°С в течение 16 часов. После завершения реакции реакционный раствор фильтровали через силикагель (100-200 меш), силикагель промывали этилацетатом (30 мл), фильтрат объединяли и концентрировали с получением неочищенного продукта 75 в виде темно-коричневого масла (210,00 мг), который непосредственно использовали на следующей стадии.
[252] Стадия В: Моногидрат гидроксида лития (36,75 мг, 875,78 мкмоль, 2,00 экв.) добавляли к раствору неочищенного продукта 75 (210,00 мг) в тетрагидрофуране (4,00 мл), метаноле (1,00 мл) и воде (1,00 мл), реакционный раствор перемешивали при 25°С в течение 4 часов. После завершения реакции реакционный раствор доводили до рН 1 соляной кислотой (3М) при комнатной температуре, затем экстрагировали этилацетатом (30 мл). Органическую фазу промывали насыщенным солевым раствором (10 мл*2), концентрировали с получением неочищенного продукта, который очищали с помощью препаративной хроматографии с жидкой фазой (преп. ВЭЖХ: муравьиная кислота) с получением продукта I-30 (27,95 мг, 61,67 мкмоль, выход 14,08%, чистота 99,63%).
[253] MS (ESI, M+1): 452,0.
[254] 1Н ЯМР (400 МГц, ДМСО-d6) δ = 10,72 (s, 1Н), 7,53 (d, J=7,8 Гц, 1Н), 7,50-7,40 (m, 3Н), 7,28 (d, J=8,0 Гц, 1H), 7,09-6,90 (m, 4H), 6,72-6,64 (m, 2H), 6,55 (dd, J=1,9, 8,3 Гц, 1H), 6,47 (d, J=1,4 Гц, 1H), 6,42 (d, J=16,1 Гц, 1H), 4,19 (s, 4H), 2,60 (q, J=7,2 Гц, 2H), 0,96 (t, J=7,3 Гц, 3Н).
Вариант реализации 31
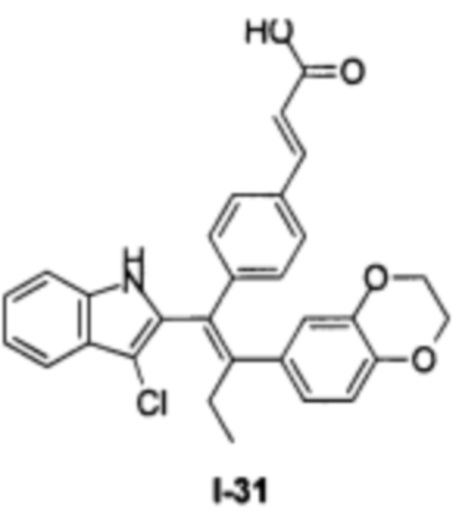
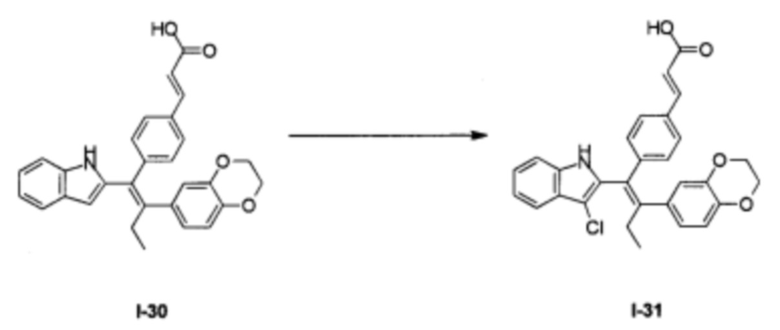
[255] Соединение I-30 (160,00 мг, 287,85 мкмоль, 1,00 экв.) взвешивали и растворяли в ацетонитриле (2,00 мл), N-хлорсукцинимид (38,44 мг, 287,85 мкмоль, 1,00 экв.) добавляли порциями, реакционный раствор перемешивали при 25°С в течение 2 часов. После завершения реакции насыщенный водный раствор тиосульфата натрия (3 мл) добавляли к реакционному раствору, смесь перемешивали в течение нескольких минут, затем экстрагировали дихлорметаном (30 мл*1). Органическую фазу концентрировали с получением неочищенного продукта, который очищали с помощью препаративной хроматографии с жидкой фазой (преп. ВЭЖХ: муравьиная кислота) с получением продукта I-31 в виде желтого твердого вещества (25,82 мг, 51,84 мкмоль, выход 18,01%, чистота 97,56%).
[256] 1Н ЯМР (400 МГц, ДМСО-d6) δ = 11,43 (s, 1Н), 8,40 (s, 1H), 7,48 (d, J=7,9 Гц, 1Н), 7,41-7,33 (m, 4Н), 7,20-7,10 (m, 2Н), 6,94 (d, J=8,3 Гц, 2Н), 6,77-6,68 (m, 2Н), 6,66-6,61 (m, 1Н), 6,38 (d, J=15,9 Гц, 1Н), 4,21 (s, 4Н), 2,39 (q, J=7,3 Гц, 2Н), 0,89 (t, J=7,4 Гц, 3Н); MS (ESI, M+1): 486,1.
Вариант реализации 32
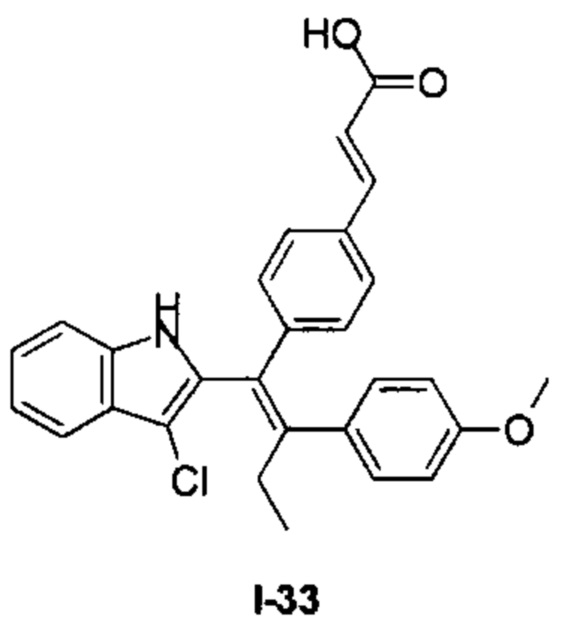
Вариант реализации 33
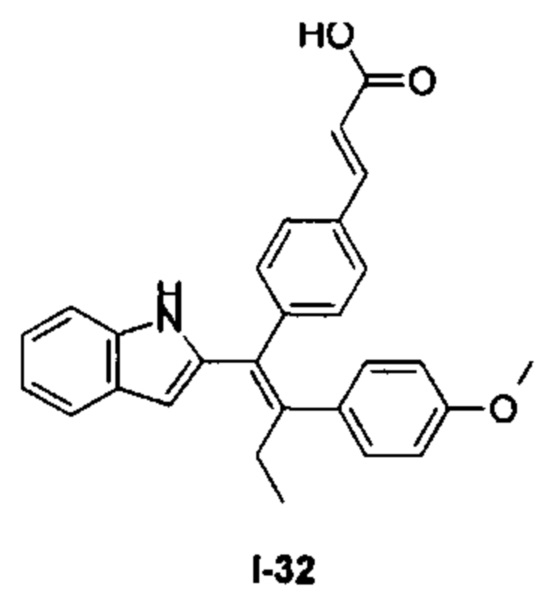
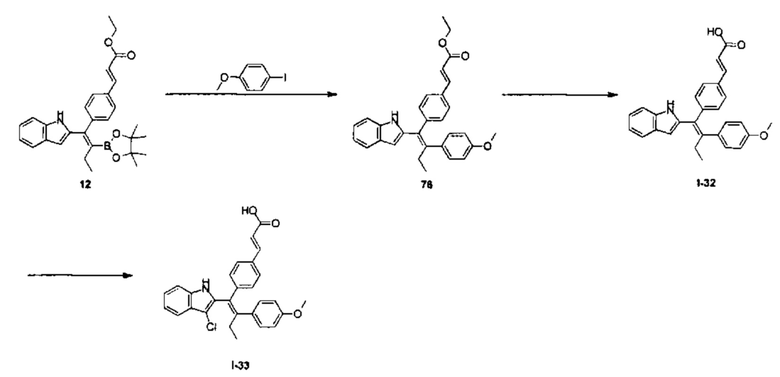
[257] Стадия А: Соединение 12 (100,00 мг, 212,13 мкмоль, 1,00 экв.) и 4-йоданизол (74,47 мг, 318,19 мкмоль, 1,50 экв.) взвешивали и растворяли в 2-метилтетрагидрофуране (4,00 мл) в атмосфере азота, с последующим добавлением гидроксида калия (66,65 мг, 1,19 ммоль, 5,60 экв.) (растворенного в 500,00 мкл воды) и дихлорбис(трифенилфосфин)палладия(II) (7,44 мг, 10,61 мкмоль, 0,05 экв.), реакционный раствор перемешивали при 75°С в течение 16 часов. После завершения реакции реакционный раствор фильтровали через силикагель (100-200 меш), силикагель промывали этилацетатом (30 мл*1), фильтрат объединяли и концентрировали с получением неочищенного продукта в виде темно-коричневого масла, который очищали с помощью пластинки с силикагелем (РЕ/ЕА=10/1) с получением промежуточного соединения 76 (50,00 мг).
[258] Стадия В: Моногидрат гидроксида лития (25,09 мг, 597,90 мкмоль, 15,00 экв.) добавляли к раствору неочищенного продукта 76 (50,00 мг) в смешанном растворителе тетрагидрофурана (2,00 мл), метанола (0,50 мл) и воды (0,50 мл), реакционный раствор перемешивали при 25°С в течение 4 часов. После завершения реакции реакционный раствор доводили до рН 1 соляной кислотой (3М) при комнатной температуре, затем экстрагировали этилацетатом (30 мл*1). Органическую фазу концентрировали с получением неочищенного продукта I-32 в виде желтого твердого вещества (68,00 мг).
[259] Стадия С: Соединение I-32 (68,00 мг) взвешивали и растворяли в ацетонитриле (2,00 мл) и дихлорметане (2,00 мл), N-хлорсукцинимид (10,72 мг, 80,28 мкмоль, 0,50 экв.) добавляли порциями, реакционный раствор перемешивали при 25°С в течение 2 часов. После завершения реакции насыщенный водный раствор тиосульфата натрия (3 мл) добавляли к реакционному раствору, полученную смесь перемешивали в течение нескольких минут, затем экстрагировали дихлорметаном (30 мл*1). Органическую фазу концентрировали с получением неочищенного продукта, который очищали с помощью препаративной хроматографии с жидкой фазой (преп. ВЭЖХ: муравьиная кислота) с получением продукта I-33 (8,71 мг, 18,39 мкмоль, выход 11,45%, чистота 96,67%).
[260] MS (ESI, M+1): 458,1.
[261] 1Н ЯМР (400 МГц, ДМСО-d6) δ = 11,44 (s, 1Н), 8,42 (s, 1Н), 7,48 (d, J=7,7 Гц, 1Н), 7,40-7,29 (m, 4Н), 7,20-7,16 (m, 1Н), 7,15-7,09 (m, 3Н), 6,92 (d, J=8,2 Гц, 2Н), 6,84 (d, J=8,7 Гц, 2Н), 6,36 (d, J=15,9 Гц, 1Н), 3,73 (s, 3Н), 2,43 (m, J=7,5 Гц, 2Н), 0,89 (t, J=7,3 Гц, 3Н).
Вариант реализации 34
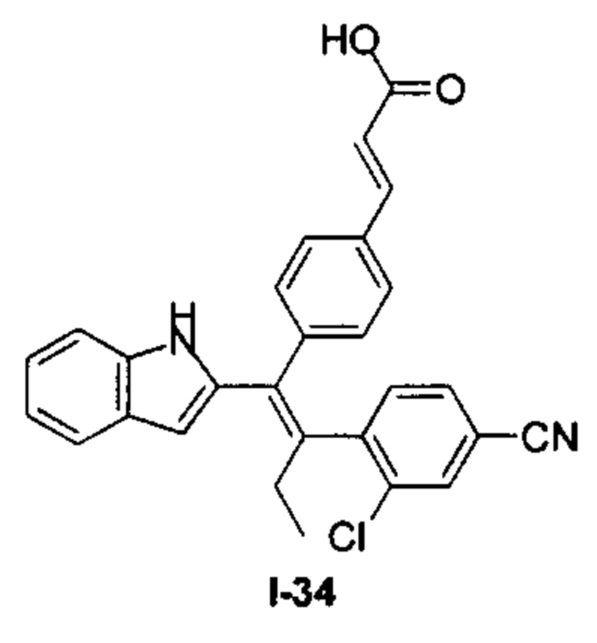
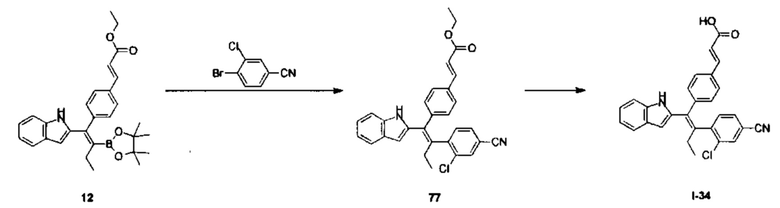
[262] Стадия А: Соединение 12 (200,00 мг, 339,42 мкмоль, 1,00 экв.) и 4-бром-3-хлорбензонитрил (110,21 мг, 509,13 мкмоль, 1,50 экв.) взвешивали и растворяли в 2-метилтетрагидрофуране (8,00 мл) в атмосфере азота, с последующим добавлением гидроксида калия (106,65 мг, 1,90 ммоль, 5,60 экв.) (растворяли в 1,00 мл воды) и дихлорбис(трифенилфосфин)палладия(II) (11,91 мг, 16,97 мкмоль, 0,05 экв.), реакционный раствор перемешивали при 75°С в течение 8 часов. После завершения реакции реакционный раствор концентрировали с удалением 2-метилтетрагидрофурана, экстрагировали этилацетатом (30 мл*1), сушили, концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ТСХ (РЕ/ЕА=5/1), дополнительно с помощью препаративной хроматографии с жидкой фазой (преп. ВЭЖХ: муравьиная кислота) с получением промежуточного соединения 77 (8,00 мг, 15,76 мкмоль, выход 4,64%, чистота 94,75%).
[263] Стадия В: Моногидрат гидроксида лития (5,00 мг, 119,16 мкмоль, 7,56 экв.) добавляли к раствору соединения 77 (8,00 мг) в смешанном растворителе тетрагидрофурана (2,00 мл), метанола (0,50 мл) и воды (0,50 мл), реакционный раствор перемешивали при 25°С в течение 4 часов. В тех же условиях, добавляли дополнительное количество моногидрата гидроксида лития (5,00 мг, 119,16 мкмоль, 7,56 экв.), и реакцию оставляли протекать дополнительные 2 часа. После завершения реакции реакционный раствор доводили до рН 1 соляной кислотой (4М) на ледяной бане, затем концентрировали, экстрагировали этилацетатом (15 мл*1). Органическую фазу концентрировали с получением неочищенного продукта, который очищали с помощью препаративной хроматографии с жидкой фазой (преп. ВЭЖХ: муравьиная кислота) с получением продукта I-34 (3,60 мг, 7,88 мкмоль, выход 50,02%, чистота 99,18%).
[264] MS (ESI, M+1): 453,0.
[265] 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,77 (s, 1Н), 8,38 (s, 1H), 7,99 (d, J=1,5 Гц, 1Н), 7,73 (dd, J=1,6, 7,9 Гц, 1Н), 7,57 (d, J=7,8 Гц, 1Н), 7,46 (d, J=8,1 Гц, 1Н), 7,43-7,29 (m, 4Н), 7,11-7,05 (m, 1Н), 7,04-6,94 (m, 3Н), 6,61 (d, J=1,5 Гц, 1Н), 6,40 (d, J=16,0 Гц, 1Н), 2,74-2,67 (m, 2Н), 1,00 (t, J=7,5 Гц, 3Н).
Вариант реализации 35
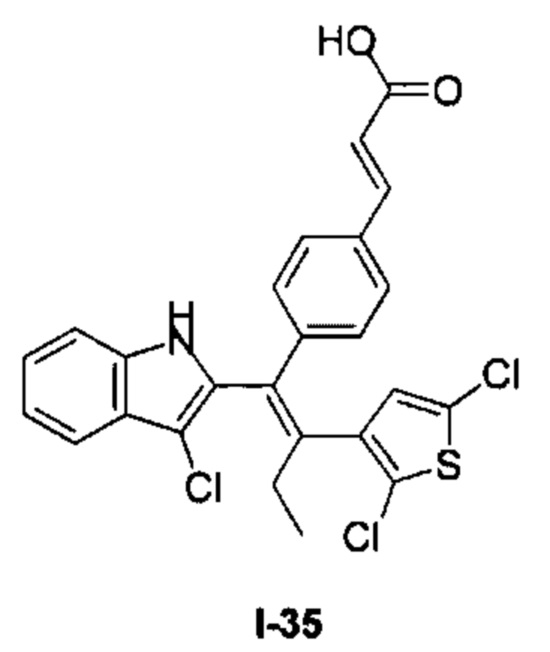
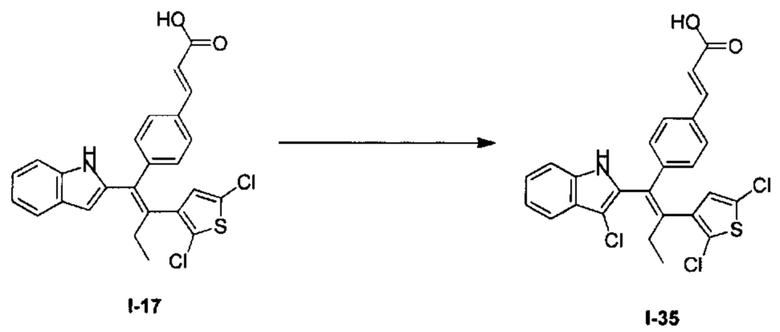
[266] Соединение I-17 (70,00 мг, 124,04 мкмоль, 1,00 экв.) взвешивали и растворяли в ацетонитриле (4,00 мл) и дихлорметане (4,00 мл), N-хлорсукцинимид (11,59 мг, 86,83 мкмоль, 0,70 экв.) добавляли порциями, реакционный раствор перемешивали при 25°С в течение 2 часов. После завершения реакции насыщенный водный раствор тиосульфата натрия (2 мл) добавляли к реакционному раствору, смесь перемешивали в течение нескольких минут, затем экстрагировали дихлорметаном (30 мл*1). Органическую фазу концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ТСХ пластины (дихлорметан/этилацетат=1/1, с небольшим количеством уксусной кислоты) с получением желтого твердого вещества 2 (20,00 мг, 35,25 мкмоль, выход 28,42%, чистота 88,63%). Твердое вещество 2 очищали с помощью препаративной ТСХ (100% этилацетат), дополнительно с помощью препаративной хроматографии с жидкой фазой (муравьиная кислота) с получением продукта I-35 в виде желтого твердого вещества (8,00 мг, 15,64 мкмоль, выход 47,38%, чистота 98,3%).
[267] 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ = 7,88 (s, 1Н), 7,72-7,64 (m, 2Н), 7,36-7,29 (m, 3Н), 7,28-7,21 (m, 2Н), 7,05 (d, J=8,4 Гц, 2Н), 6,72 (s, 1Н), 6,38 (d, J=16,0 Гц, 1Н), 2,53 (q, J=7,6 Гц, 2Н), 1,05 (t, J=7,6 Гц, 3Н); MS (ESI, M+1): 502,0.
Вариант реализации 36
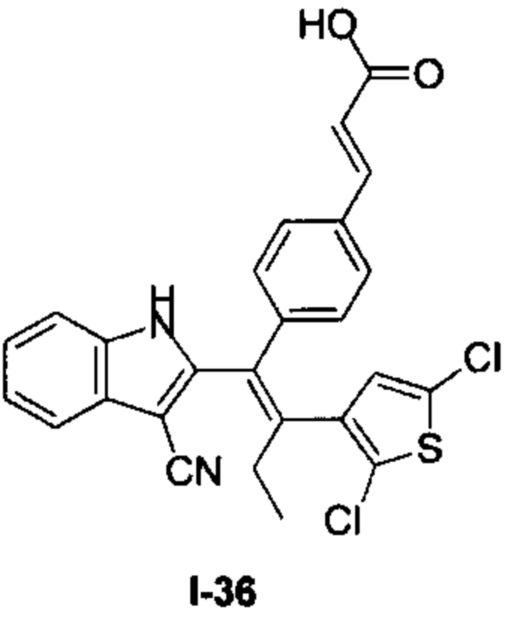
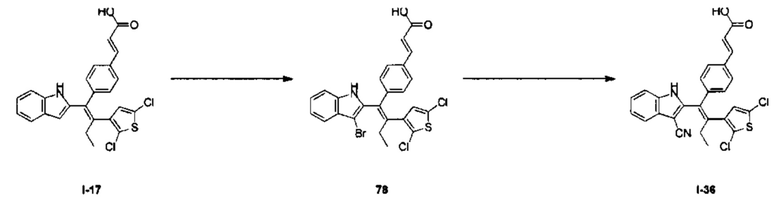
[268] Стадия А: Соединение I-17 (93,00 мг, 198,55 мкмоль, 1,00 экв.) взвешивали и растворяли в ацетонитриле (4,00 мл) и дихлорметане (4,00 мл), N-хлорсукцинимид (24,74 мг, 138,98 мкмоль, 0,70 экв.) добавляли порциями, реакционный раствор перемешивали при 25°С в течение 1 часа. После завершения реакции насыщенный водный раствор тиосульфата натрия (3 мл) добавляли к реакционному раствору, смесь перемешивали в течение нескольких минут, затем экстрагировали дихлорметаном (30 мл*1). Органическую фазу концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ТСХ (дихлорметан/этилацетат=1/1, с небольшим количеством уксусной кислоты) с получением промежуточного соединения 78 (95,00 мг, 151,02 мкмоль, выход 76,06%, чистота 87%).
[269] Стадия В: Соединение 78 (95,00 мг, 168,38 мкмоль, 1,00 экв.) растворяли в N-метилпирролидоне (2,00 мл) в микроволновой пробирке, с последующим добавлением цианида меди (31,06 мг, 346,86 мкмоль, 75,76 мкл, 2,06 экв.), реакционный раствор перемешивали при 180°С в течение 1,5 часов в условиях микроволнового облучения. В тех же условиях, добавляли дополнительное количество цианида меди (30,16 мг, 336,76 мкмоль, 73,56 мкл, 2,00 экв.) к реакционному раствору и реакцию оставляли протекать в течение 2 часов. В тех же условиях, добавляли дополнительное количество цианида меди (45,24 мг, 505,14 мкмоль, 110,34 мкл, 3,00 экв.) добавляли к реакционному раствору и реакцию оставляли протекать в течение 2 часов. После завершения реакции реакционный раствор выливали в ледяную воду (20 мл), экстрагировали этилацетатом (50 мл*2). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной жидкостной хроматографии (преп. ВЭЖХ: муравьиная кислота) с получением продукта I-36 в виде желтого твердого вещества (7,18 мг, 13,75 мкмоль, выход 8,16%, чистота 94,46%).
[270] 1Н ЯМР (400 МГц, ДМСО-d6) δ = 12,28 (s, 1H), 7,66 (d, J=7,1 Гц, 1H), 7,55 (d, J=8,3 Гц, 2Н), 7,52-7,46 (m, 2Н), 7,33-7,24 (m, 3Н), 7,00 (d, J=8,3 Гц, 2Н), 6,47 (d, J=16,0 Гц, 1Н), 2,44-2,39 (m, 2Н), 0,98 (t, J=7,4 Гц, 3Н); MS (ESI, M+1): 493,0.
Вариант реализации 37
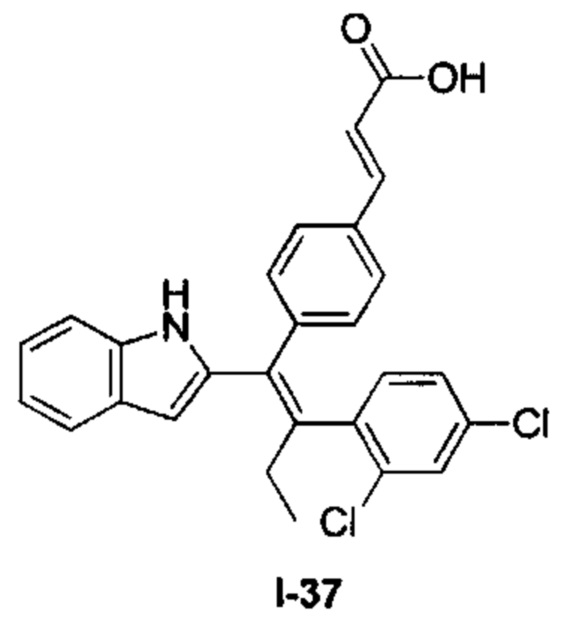
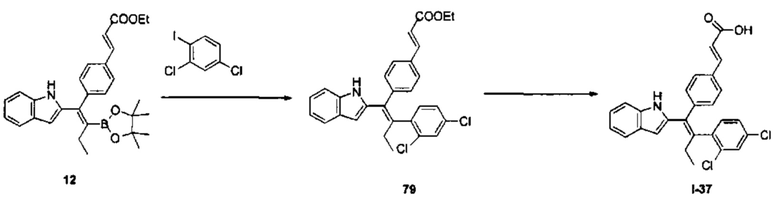
[271] Стадия А: Соединение 12 (300,00 мг, 560,05 мкмоль, 1,00 экв.) и 2,4-дихлор-1-йодбензол (275,11 мг, 1,01 ммоль, 1,80 экв.), 2-метилтетрагидрофуран (10,00 мл) и водный раствор гидроксида калия (4М, 784,06 мкл, 5,60 экв.) добавляли в 100 мл трехгорлую колбу с магнитной мешалкой. После продувания реакционной системы азотом три раза добавляли Pd(PPh3)2Cl2 (11,79 мг, 16,80 мкмоль, 0,03 экв.). Реакционную систему снова три раза продували азотом, затем реакционный раствор перемешивали при 80°С в течение 12 часов, и впоследствии он поменял цвет с желтого на черный. После завершения реакции воду (30 мл) добавляли к реакционному раствору и смесь экстрагировали дихлорметаном (30 мл*3). Органическую фазу объединяли, промывали насыщенным солевым раствором (30 мл*2), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (РЕ:ЕА=1:0 до 20:1) с получением соединения 79 (330,00 мг, неочищенного). MS (ESI,M+1): 490,0.
[272] Стадия В: Соединение 79 (330,00 мг, 672,89 мкмоль, 1,00 экв.) растворяли в смешанном растворителе воды (4,00 мл), метанола (8,00 мл) и тетрагидрофурана (8,00 мл). LiOH.Н2О (141,17 мг, 3,36 ммоль, 5,00 экв.) добавляли к указанному раствору, который впоследствии поменял цвет с желтого на светло-зеленый. Реакционный раствор перемешивали при 20°С в течение 12 часов. После завершения реакции реакционный раствор выпаривали с получением остатка. Затем 3 моль/л HCl (1,5 мл) добавляли к остатку для доведения рН полученного раствора до 5, раствор фильтровали с получением фильтрационного осадка, который очищали с помощью препаративной хроматографии (муравьиная кислота) с получением продукта I-37 (33,70 мг, 71,43 мкмоль, выход 10,62%, чистота 98%).
[273] 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,74 (s, 1Н), 8,31 (br s, 1Н), 7,62-7,49 (m, 2Н), 7,44-7,35 (m, 3Н), 7,34-7,27 (m, 2Н), 7,23 (d, J=8,0 Гц, 1Н), 7,10-7,03 (m, 1Н), 7,02-6,93 (m, 3Н), 6,57 (d, J=1,2 Гц, 1Н), 6,40 (d, J=16,0 Гц, 1Н), 2,75-2,58 (m, 2Н), 0,99 (t, 7=7,2Гц, 3Н); MS (ESI,M+1): 462,0.
Вариант реализации 38
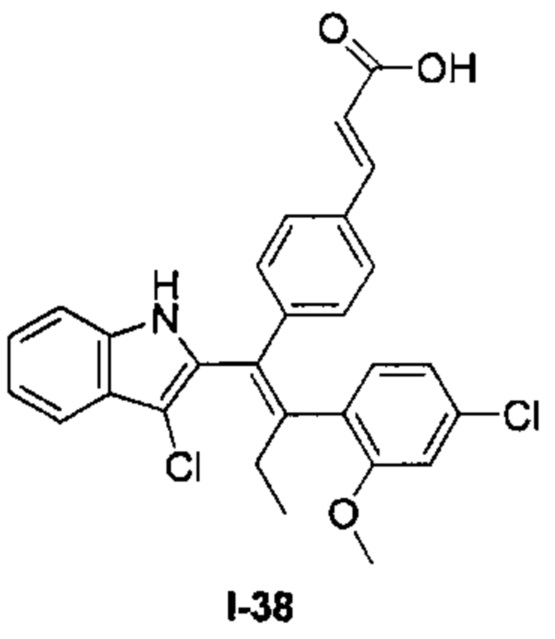
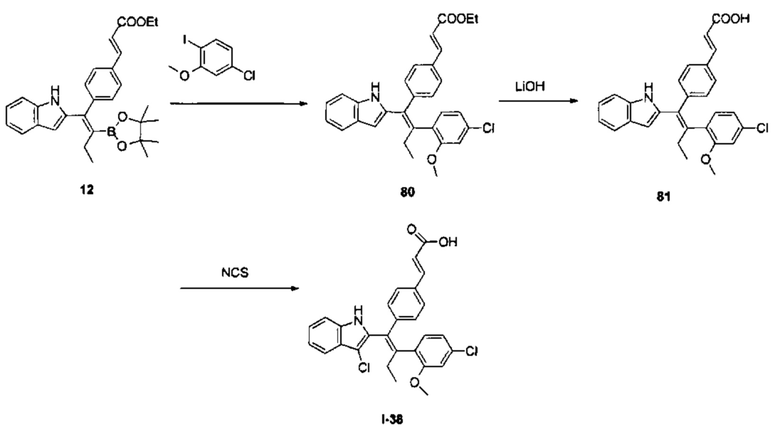
[274] Стадия А: Соединение 12 (150,00 мг, 318,20 мкмоль, 1,00 экв.), 2-mеТГФ (5,00 мл), 2-бром-5-хлоранизол (84,57 мг, 381,84 мкмоль, 1,20 экв.), воду (1,00 мл) и NaOH (50,91 мг, 1,27 ммоль, 4,00 экв.) последовательно добавляли в одногорлую колбу с магнитной мешалкой. Полученную смесь продували N2 три раза. Затем Pd(dppf)Cl2 (23,28 мг, 31,82 мкмоль, 0,10 экв.) добавляли к смеси в атмосфере азота. Реакционный раствор перемешивали при 75°С в течение 7 ч. 10 мл ЕА и 10 мл воды добавляли к реакционному раствору. Органическую фазу промывали 10 мл воды, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта (162,00 мг), который непосредственно использовали на следующей стадии на основании теоретического выхода.
[275] Стадия В: Соединение 80 (154,65 мг, 318,20 мкмоль, 1,00 экв.), ТГФ (3,00 мл), МеОН (1,00 мл), воду (1,00 мл) и LiOH (22,86 мг, 954,60 мкмоль, 3,00 экв.) последовательно добавляли в одногорлую колбу с магнитной мешалкой. Полученный черный раствор перемешивали при 45°С в течение 4 ч. 2 мл 1N HCl добавляли к реакционному раствору с последующим добавлением 10 мл воды, затем экстрагировали 30 мл EtOAc (10 мл*3). Органическую фазу объединяли, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта, который очищали с помощью препаративной хроматографии (муравьиная кислота) с получением соединения 81 (28,00 мг, 61,14 мкмоль, выход 19,21%).
[276] Соединение 81 (28,00 мг, 61,14 мкмоль, 1,00 экв.) и CH3CN (3,00 мл) последовательно добавляли в одногорлую колбу с магнитной мешалкой с получением желтого раствора, с последующим добавлением NCS (8,16 мг, 61,14 мкмоль, 1,00 экв.). Полученный раствор перемешивали при 30°С в течение 4 ч. Затем реакцию гасили 5 мл Na2S2O3 при 25°С и добавляли 20 мл воды. Смесь экстрагировали 60 мл EtOAc (20 мл*3). Органическую фазу объединяли, промывали 30 мл воды, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта, который очищали с помощью препаративной хроматографии (муравьиная кислота) с получением I-38 (18,10 мг, 36,43 мкмоль, выход 59,58%, чистота 99,1%), MS (ESI, M+1): 492,1.
[277] 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,44 (s, 1Н), 7,50 (d, J=8,0 Гц, 1H), 7,41-7,36 (m, 4Н), 7,20-7,10 (m, 3Н), 6,95-6,88(m, 4Н), 6,37 (d, J=16,0 Гц, 1Н), 3,82(s, 3Н), 2,01-1,98 (m, 2Н), 086-0,83 (m, 3Н).
Вариант реализации 39
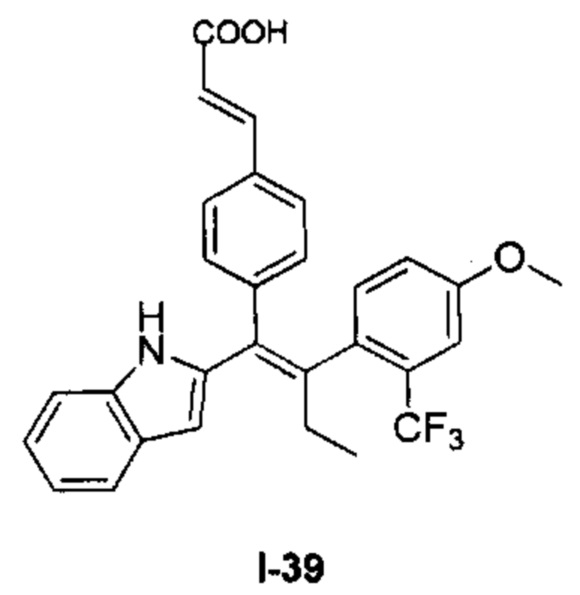
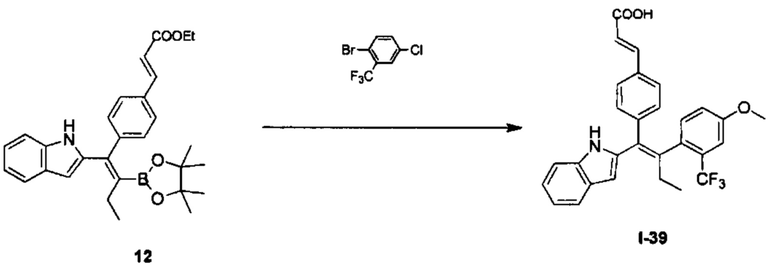
[278] Соединение 12 (110,00 мг, 190,65 мкмоль, 1,00 экв.) и 2-трифторметил-4-хлорбромбензол (53,7 мг, 210 мкмоль, 1,1 экв.) растворяли в 4 мл диметилтетрагидрофуране в 100 мл одногорлой колбе, с последующим добавлением раствора гидроксида натрия (4М, 142,99 мкл, 3,00 экв.) и Pd(PPh3)2Cl2 (6,69 мг, 9,53 мкмоль, 0,05 экв.). Реакционную систему три раза продували азотом и реакционный раствор перемешивали при 70°С в атмосфере азота в течение 16 часов. После завершения реакции реакционный раствор концентрировали с получением остатка, который очищали с помощью препаративной ТСХ (РЕ:EtOAc=0:1) с получением неочищенного продукта. Неочищенный продукт дополнительно очищали с помощью препаративной ВЭЖХ (FA) с получением I-39.
[279] 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ = 7,72 (s, 1Н), 7,67 (d, J=7,8 Гц, 1H), 7,60 (d, J=15,8 Гц, 1Н), 7,28-7,24 (m, 2Н), 7,21-7,12 (m, 2Н), 7,08 (d, J=2,6 Гц, 1Н), 7,05-6,99 (m, 3Н), 6,90 (dd, J=2,6, 8,7 Гц, 1Н), 6,72 (d, J=1,5 Гц, 1Н), 6,35 (d, J=15,9 Гц, 1Н), 3,81 (s, 3Н), 3,03 (m, J=7,5, 13,9 Гц, 1Н), 2,65-2,57 (m, 1Н), 1,08 (t, J=7,5 Гц, 3Н); MS (ESI, M+1): 492,5.
Вариант реализации 40
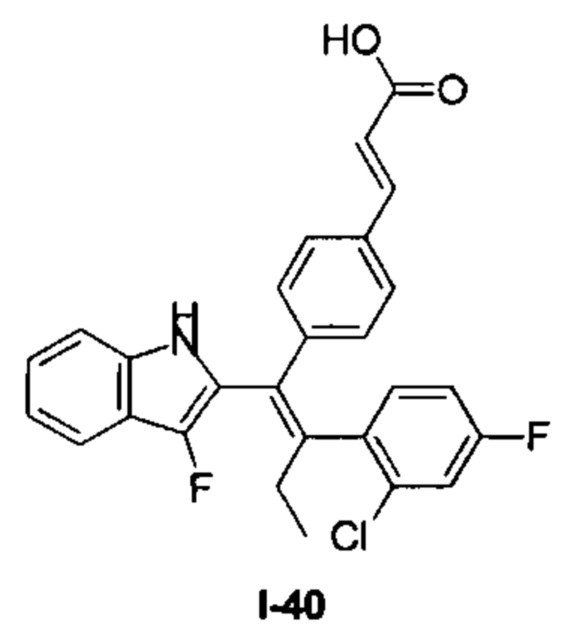
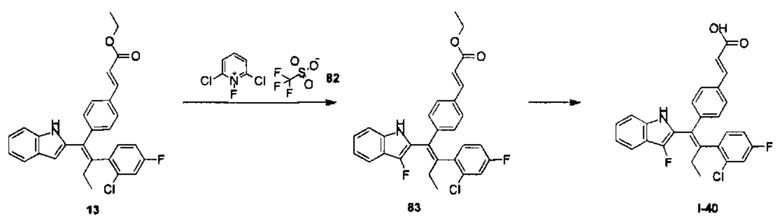
[280] Стадия А: Соединение 13 (1 г, 2,11 ммоль, 1экв.) и дихлорметан (20 мл) добавляли в 100 мл одногорлую колбу с магнитной мешалкой. Раствор продували азотом и охлаждали до 0°С с последующим добавлением соединения 82 (1,00 г, 3,16 ммоль, 1,5 экв.) при 0°С. Полученный темно-зеленый раствор перемешивали при 25°С в атмосфере азота в течение 16 часов. Добавляли 15 мл насыщенного водного раствора бикарбоната натрия, содержащего 10% тиосульфата натрия, и полученную смесь перемешивали в течение 10 минут. Водную фазу экстрагировали три раза 30 мл дихлорметана каждый раз. Органическую фазу объединяли, промывали 20 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (ЕА/РЕ=0%-20%) с получением продукта 83 в виде желтого твердого вещества.
[281] Стадия В: Соединение 3 (1 г, (0,15 г, 304,91 мкмоль, 1 экв.), 2 мл этанола и 3 мл тетрагидрофурана добавляли в 100 мл одногорлую колбу с магнитной мешалкой, с последующим добавлением раствора моногидрата гидроксида лития (38,38 мг, 914,72 мкмоль, 3 экв.) в 1 мл воды. Полученный желтый раствор перемешивали при 50°С в атмосфере азота в течение 2 часов, затем концентрировали и разбавляли 10 мл воды, доводили рН до 5-6 1М соляной кислотой, экстрагировали три раза 20 мл этилацетата каждый раз. Органическую фазу объединяли, промывали 20 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью препаративной высокоэффективной жидкостной хроматографии с получением I-40. 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,73 (s, 1Н), 7,55 (d, J=8,0 Гц, 1Н), 7,45-7,27 (m, 6Н), 7,19-7,11 (m, 2Н), 7,10-7,04 (m, 1Н), 6,95 (d, J=8,4 Гц, 2Н), 6,40 (d, J=16,0 Гц, 1Н), 2,64-2,56 (m, 2Н), 0,93 (t, J=7,6 Гц, 3Н).
Вариант реализации 41
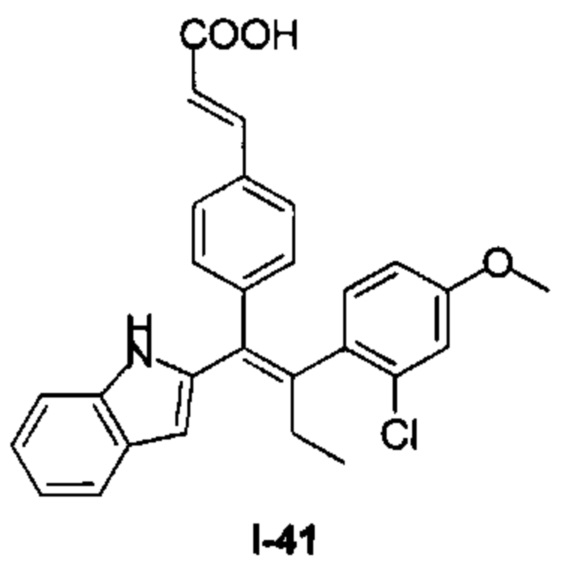
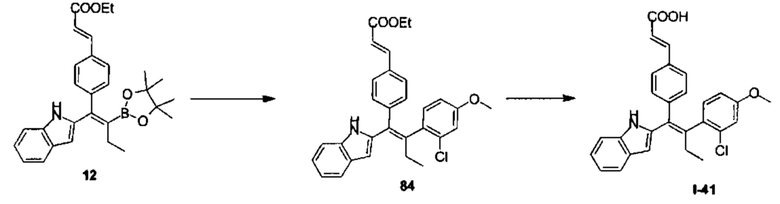
[282] Стадия А: Соединение 1 (400,00 мг, 693,27 мкмоль, 1,00 экв.), 2-хлор-4-метоксибромбензол (230,32 мг, 1,04 ммоль, 1,50 экв.) и 10 мл 2-метилтетрагидрофурана добавляли в 100 мл одногорлую колбу, с последующим последовательным добавлением водного раствора гидроксида калия (4М, 693,27 мкл, 4,00 экв.) и бис(трифенилфосфин)палладия дихлорида (24,33 мг, 34,66 мкмоль, 0,05 экв.). Реакционный раствор продували азотом, затем нагревали до 70°С и перемешивали в течение 12 часов. Реакционный раствор поменял цвет с желтого на коричневый. После завершения реакции 20 мл этилацетата добавляли к реакционному раствору и полученную смесь фильтровали через целит. Фильтрат три раза промывали 20 мл насыщенного солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта 84 в виде коричневого желе (570,00 мг, неочищенный продукт), которое непосредственно использовали на следующей стадии.
[283] Стадия В: Моногидрат гидроксида лития (490,93 мг, 11,70 ммоль, 10,00 экв.) добавляли к раствору соединения 84 (570,00 мг, 1,17 ммоль, 1,00 экв.) в смешанном растворителе 3 мл ТГФ, 3 мл метанола и 3 мл воды в колбе. Реакционный раствор перемешивали при 45°С в течение 1 часа и он оставался коричневым. После завершения реакции реакционный раствор доводили до рН 1 1М соляной кислотой с последующим добавлением 20 мл этилацетата. Органическую фазу дважды промывали 20 мл насыщенного солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта, который очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением соединения I-41. 1Н ЯМР (400 МГц, ДМСО-d6): δ 10,72 (brs, 1Н), 7,54 (d, J=7,6 Гц, 1Н), 7,43-7,39 (m, 3Н), 7,29 (d, J=8,0 Гц, 1H), 7,08-6,94 (m, 6H), 6,80 (dd, J=8,8 Гц, J=2,8 Гц, 1H), 6,54 (d, J=1,2 Гц, 1H), 6,39 (d, J=16,0 Гц, 1H), 3,71 (s, 3H), 2,66-2,55 (m, 2H), 0,98 (t, J=7,2 Гц, 3H). MS (ESI, M+1): 458,1.
Вариант реализации 42
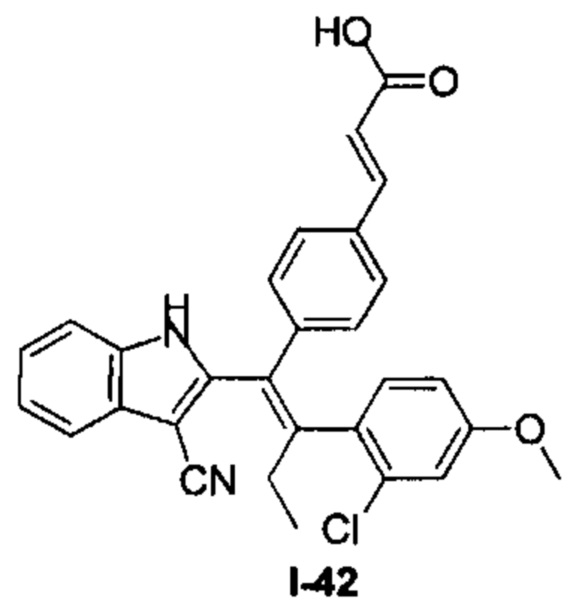
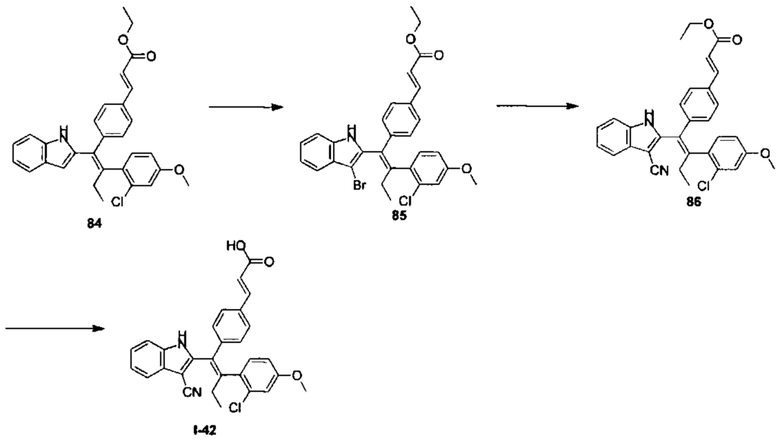
[284] Стадия А: Раствор NBS (42,85 мг, 240,74 мкмоль, 0,90 экв.) в дихлорметане (1 мл) добавляли к раствору соединения 84 (130,00 мг, 267,49 мкмоль, 1,00 экв.) в дихлорметане (4,00 мл) и ацетонитриле (4,00 мл). Реакционный раствор перемешивали в течение 1 часа, затем гасили водным раствором тиосульфата натрия и экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом натрия и концентрировали с получением промежуточного соединения 85 (180 мг, неочищенный продукт), которое непосредственно использовали на следующей стадии.
[285] Стадия В: Раствор промежуточного соединения 85 (180,00 мг, 318,64 мкмоль, 1,00 экв.) и цианида меди (171,22 мг, 1,91 ммоль, 417,61 мкл, 6,00 экв.) в N-метилпирролидоне (2,00 мл) нагревали до 180°С в условиях микроволнового облучения и перемешивали в течение 2 часов. Затем реакционный раствор охлаждали до комнатной температуры, и добавляли дополнительное количество цианида меди (57,07 мг, 637,28 мкмоль, 139,20 мкл, 2,00 экв.), и полученный реакционный раствор нагревали до 180°С в условиях микроволнового облучения и перемешивали в течение дополнительных 2 часов. Реакционный раствор охлаждали до комнатной температуры, выливали в 20 мл воды, экстрагировали этилацетатом и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ с получением промежуточного соединения 86 (30,00 мг, 54,60 мкмоль, выход 17,13%, чистота 93%).
[286] Стадия С: Гидроксид лития (12,32 мг, 293,61 мкмоль, 5,38 экв.) добавляли к раствору промежуточного соединения 86 (30,00 мг, 54,60 мкмоль, 1,00 экв.) в смешанном растворителе тетрагидрофурана (4,00 мл), метанола (1,00 мл) и воды (1,00 мл). Реакционный раствор перемешивали в течение 4 часов, затем доводили до рН 1 с помощью 4М HCl, концентрировали в вакууме и экстрагировали этилацетатом. Органическую фазу объединяли, сушили над сульфатом натрия и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ с получением I-42. 1Н ЯМР (400 МГц, ДМСО-d6): δ 12,32 (s, 1Н), 7,66 (d, J=8,8 Гц, 1Н), 7,50 (d, J=8,0 Гц, 1Н), 7,55-7,40 (m, 3Н), 7,33-7,23 (m, 2Н), 7,15 (d, J=8,8 Гц, 1Н), 7,10-7,00 (m, 3Н), 6,91 (dd, J=4,8 Гц, J=16 Гц, 1Н), 6,42 (d, J=16 Гц, 1Н), 3,75 st, 3Н), 2,53-2,31 (m, 2H), 0,93 (t, J=7,2 Гц, 3H). (ESI, M+1): 483,1.
Вариант реализации 43
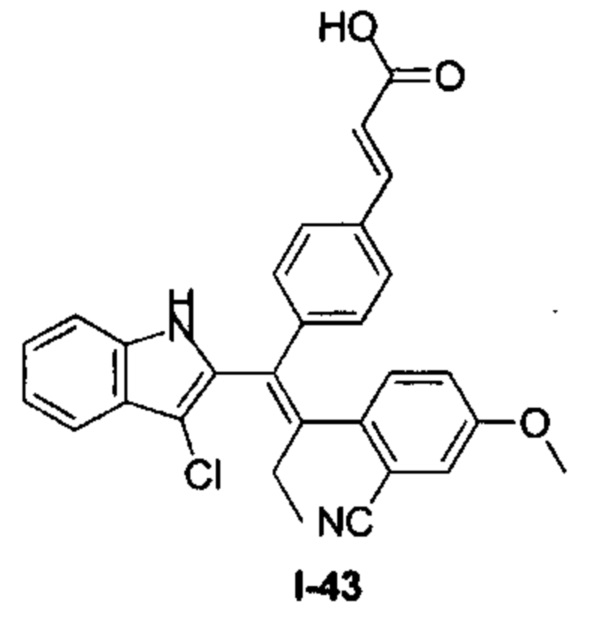
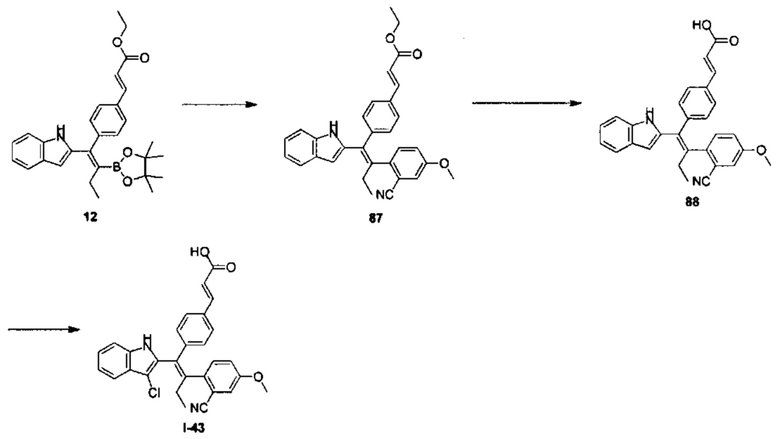
[287] Стадия А: KOH (4М, 259,66 мкл, 3,06 экв.) и Pd(Ph3P)2Cl2 (47,65 мг, 67,88 мкмоль, 0,20 экв.) добавляли к раствору промежуточного соединения 12 (200,00 мг, 339,42 мкмоль, 1,00 экв.) и 2-бром-5-метоксифенилцианида (108,00 мг, 509,13 мкмоль, 1,50 экв.) в 2-метилтетрагидрофуране. Реакционный раствор перемешивали при 65°С в атмосфере азота в течение 13 часов, затем охлаждали до комнатной температуры и добавляли дополнительное количество Pd(Ph3P)2Cl2 (20,00 мг, 33 мкмоль, 0,10 экв.). Реакционный раствор нагревали до 65°С и перемешивали в течение дополнительных 3 часов с получением 87, которое непосредственно использовали на следующей стадии. Реакционный раствор охлаждали до комнатной температуры, затем добавляли метанол (6,00 мл) и воду (3,00 мл) и полученную смесь перемешивали при 35°С в течение 2 часов. Добавляли гидроксид лития (15 мг), реакционный раствор нагревали до 45°С и перемешивали в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры, доводили рН до 5 с помощью 2М HCl, разбавляли водой, экстрагировали этилацетатом, сушили над сульфатом натрия, концентрировали и очищали с помощью колоночной хроматографии на силикагеле с получением промежуточного соединения 88.
[288] Стадия В: NCS (20,49 мг, 153,45 мкмоль, 1,00 экв.) добавляли к раствору промежуточного соединения 88 (70,00 мг, 153,45 мкмоль, 1,00 экв.) в ацетонитриле (3,00 мл). Реакционный раствор перемешивали при 20°С в течение 3 часов, затем гасили водным раствором тиосульфата натрия (10 мл), доводили рН до 5 с помощью 2М HCl, экстрагировали дихлорметаном. Органическую фазу объединяли, сушили над сульфатом натрия и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ с получением продукта I-43. 1Н ЯМР (400 МГц, ДМСО-d6): δ 11,43 (s, 1Н), 7,52 (d, J=8,4 Гц, 1Н), 7,42-7,39 (m, 5Н), 7,37-7,35 (m, 1Н), 7,31 (d, J=2,4 Гц, 1Н), 7,27-7,15 (m, 2Н), 6,94 (d, J=8,4 Гц, 2Н), 6,39 (d, J=16,0 Гц, 1Н), 3,78 (s, 3Н), 2,58-2,41 (m, 2Н), 0,91 (t, J=7,6 Гц, 3Н); MS (ESI, M+1): 483,1.
Вариант реализации 44
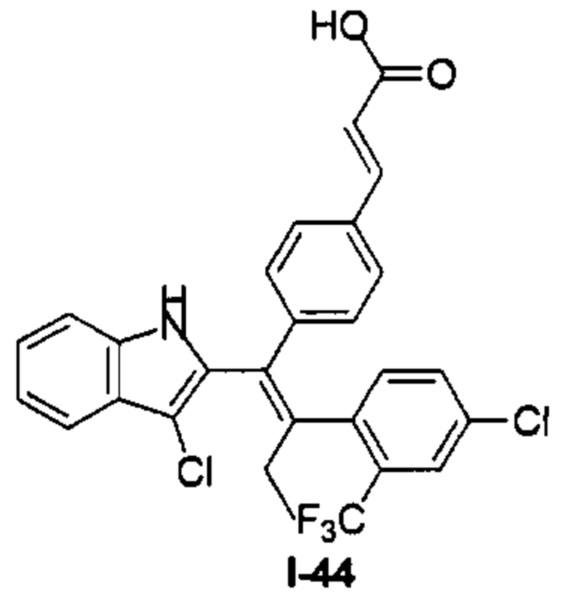
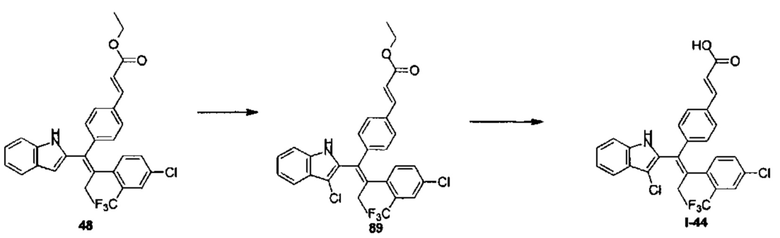
[289] Стадия A: NCS (15,80 мг, 118,33 мкмоль, 1,00 экв.) добавляли к раствору 48 (62,00 мг, 118,33 мкмоль, 1,00 экв.) в ацетонитриле (3,00 мл) и дихлорметане (5,00 мл). Реакционный раствор перемешивали при 15°С в течение 5 часов. Добавляли дополнительное количество NCS (4,00 мг, 29,96 мкмоль, 0,25 экв.) и реакционный раствор перемешивали в течение дополнительного 1 часа, затем гасили водным раствором тиосульфита натрия (20 мл), экстрагировали этилацетатом. Органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом натрия с получением промежуточного соединения 89 (66,00 мг, неочищенный продукт), которое непосредственно использовали на следующей стадии.
[290] Стадия В: LiOH (14,88 мг, 354,57 мкмоль, 3,00 экв.) добавляли к раствору промежуточного соединения 89 (66,00 мг, 118,19 мкмоль, 1,00 экв.) в смешанном растворителе метанола (3,00 мл), тетрагидрофурана (3,00 мл) и воды (3,00 мл). Реакционный раствор перемешивали при 15°С в течение 12 часов и концентрировали. Остаток разбавляли водой (10 мл), доводили рН до 5 с помощью 2М HCl, экстрагировали дихлорметаном. Органическую фазу объединяли, промывали насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ с получением I-44. 1Н ЯМР (400 МГц, ДМСО-d6): δ 11,51 (s, 1Н), 7,81 (d, J=2,0 Гц, 1Н), 7,73 (d, J=12 Гц, 1Н), 7,49 (d, J=9,6 Гц, 1Н), 7,46-7,35 (m, 5Н), 7,22-7,13 (m, 2Н), 6,97 (d, J=8,4 Гц, 2Н), 6,39 (s, J=16,0 Гц, 1H), 2,55-2,47 (m, 1H), 2,38-2,25 (m, 1H), 0,87 (t, J=7,2 Гц, 3Н); MS (ESI, M+1): 530,1.
Вариант реализации 45
[291] Стадия A: NCS (9,48 мг, 71,00 мкмоль, 0,9 экв.) добавляли к смеси соединения II-1 (0,04 г, 78,89 мкмоль, 1 экв.) и дихлорметана (4 мл) в одногорлой колбе с магнитной мешалкой. Желтый раствор перемешивали при 15°С в течение 2 часов, затем концентрировали с получением неочищенного продукта, который очищали с помощью препаративной высокоэффективной жидкостной хроматографии с получением II-5.
Вариант реализации 46
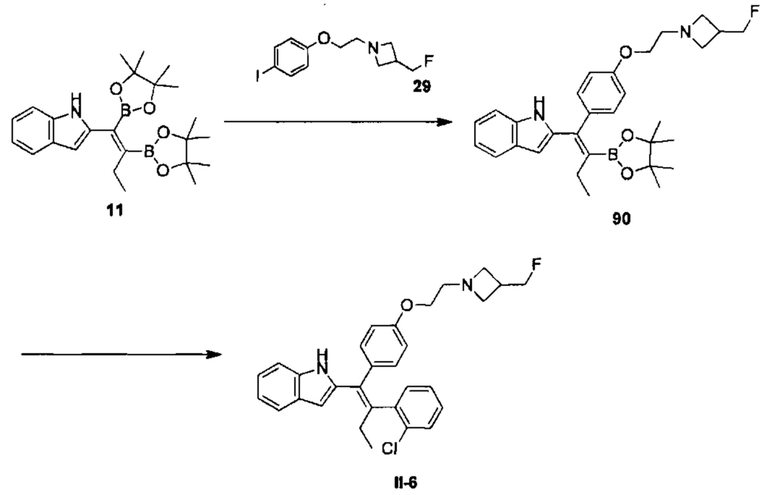
[292] Стадия А: Водный раствор гидроксида натрия (4М, 1,00 мл, 4,00 экв.) добавляли к смеси соединения 11 (423,16 мг, 1,00 ммоль, 1,00 экв.) (раствор в 10 мл 2-метилтетрагидрофурана) и соединения 29 (402,19 мг, 1,20 ммоль, 1,20 экв.) в трехгорлой колбе, с последующим добавлением бис(дифенилфосфино)ферроцен дихлорпалладия (36,59 мг, 50,00 мкмоль, 0,05 экв.) в атмосфере азота. Реакционный раствор перемешивали при 60°С в течение 4 часов. После завершения реакции реакционный раствор непосредственно использовали на следующей стадии без дополнительной доочистки на основе теоретического выхода для 90.
[293] Стадия В: Дихлорбис(трифенилфосфин)палладий(II) (42,12 мг, 60,00 мкмоль, 0,06 экв.) добавляли к смеси соединения 90 (504,44 мг, 1,00 ммоль, 1,00 экв.) (Стадия 1: раствор в 2-метилтетрагидрофуране и воде), гидроксида натрия (4М, 1,00 мл, 4,00 экв.) и 1-хлор-2-йодбензола (291,14 мг, 1,20 ммоль, 1,20 экв.) в атмосфере азота в трехгорлой колбе. Реакционный раствор добавляли при 70°С в атмосфере азота в течение 4 часов. После завершения реакции воду (40 мл) и этилацетат (30 мл) добавляли к реакционному раствору, затем экстрагировали этилацетатом (20 мл*3). Органическую фазу объединяли, промывали водой (30 мл), сушили над сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной жидкостной хроматографии (преп. ВЭЖХ: муравьиная кислота) с получением II-6. 1H ЯМР (400 МГц, ДМСО-d6) δ = 10,65 (s, 1Н), 8,35 (s, 1Н), 7,54-6,84 (m, 10Н), 6,62-6,54 (m, 3Н), 4,493 (dd, J=6,4 Гц, 48Гц, 2Н), 3,78 (d, J=5,2 Гц, 2Н), 2,95 (t, J=6,4 Гц, 2Н), 2,68-2,51 (m, 6Н), 0,99 (t, J=7,6 Гц, 3Н); MS (ESI, M+1): 489,1.
Вариант реализации 47
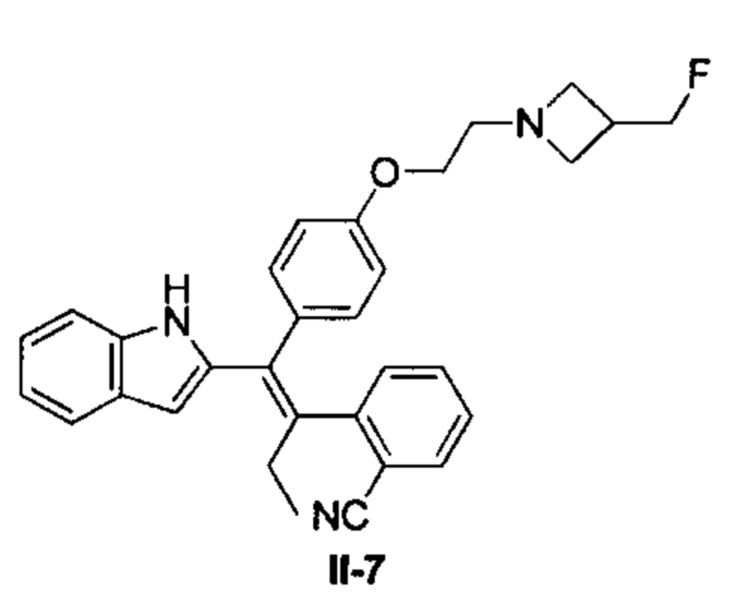
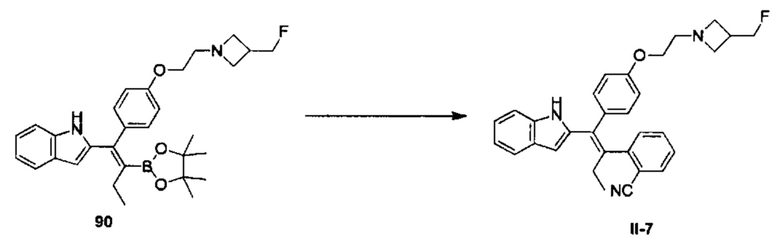
[294] Стадия А: 2-йодбензонитрил (329,79 мг, 1,44 ммоль, 1,20 экв.) добавляли к смеси соединения 90 (605,33 мг, 1,20 ммоль, 1,00 экв.) (раствор в 10 мл 2-метилтетрагидрофуране), воды (2,00 мл) и гидроксида натрия (192,00 мг, 4,80 ммоль, 4,00 экв.) в одногорлой колбе, которую затем три раза продували N2. Бис(трифенилфосфин)палладия хлорид (42,11 мг, 60,00 мкмоль, 0,05 экв.) добавляли в атмосфере азота. Реакционный раствор перемешивали при 70°С в течение 7 часов. После завершения реакции реакционный раствор разбавляли 40 мл воды, экстрагировали 45 мл этилацетата (15 мл*3). Органическую фазу объединяли, промывали 40 мл воды, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной хроматографии (система с муравьиной кислотой) с получением соединения II-7. 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,65 (s, 1Н), 8,27 (s, 1H), 7,66-7,51 (m, 4Н), 7,38-7,30 (m, 2Н), 7,06-7,00 (m, 2Н), 6,81 (d, J=8,8 Гц, 2Н), 6,62(d, J=8,8 Гц, 2Н), 6,57(d, 7=1,6 Гц, 1Н), 4,54 (d, J=6,0 Гц, 1H), 4,42(d, J=6,0 Гц, 1Н), 3,78 (t, J=5,6 Гц, 2Н), 2,96 (t, J=5,6 Гц, 2Н), 2,68-2,63 (m, 6Н), 2,03-1,96(m, 1H), 1,01 (t, J=7,6 Гц, 3Н); MS (ESI,M+1): 480,3.
Вариант реализации 48
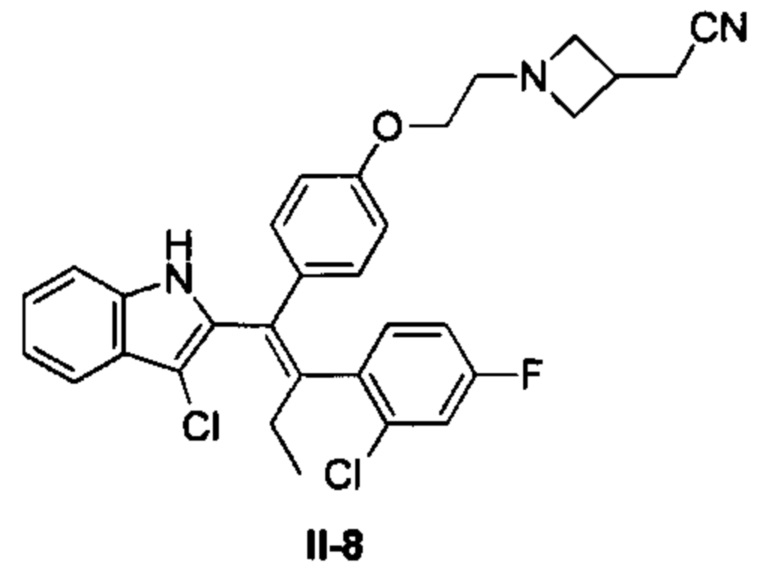
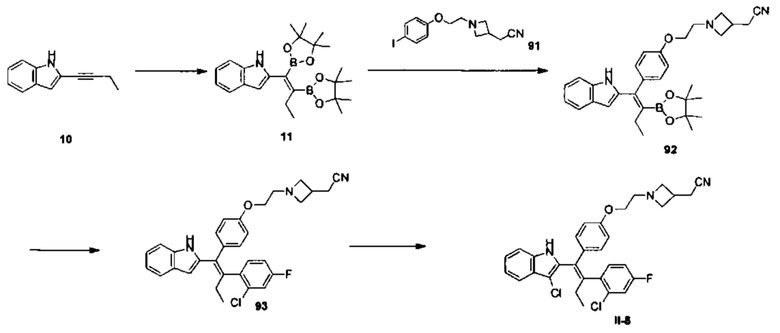
[295] Стадия А: Биспинаколатодибор (1,16 г, 4,57 ммоль, 1,05 экв.) и тетракис(трифенилфосфин)платину (271,04 мг, 217,84 мкмоль, 0,05 экв.) добавляли к раствору соединения 10 (750,00 мг, 4,36 ммоль, 1,00 экв.) в 2-метилтетрагидрофуране (20,00 мл), реакционный раствор три раза продували азотом, затем перемешивали при 70°С (внутренняя температура 60°С) в атмосфере азота в течение 5 часов, и полученное 11 не выделяли. После охлаждения реакционного раствора до 0°С добавляли соединение 91 (1,34 г, 3,91 ммоль, 0,90 экв.), карбонат калия (1,20 г, 8,70 ммоль, 2,00 экв.), 6 мл воды, 30 мл 2-метилтетрагидрофурана и дихлорбис(трифенилфосфин)палладия (152,60 мг, 217,41 мкмоль, 0,05 экв.), полученную смесь три раза продували азотом и перемешивали при 30°С в атмосфере азота в течение 12 часов, и полученное 92 не выделяли. Затем добавляли 4-фтор-2-хлорйодбензол (556,54 мг, 2,17 ммоль, 2,00 экв.), водный раствор гидроксида калия (4М, 1,09 мл, 4,00 экв.), дихлорбис(трифенилфосфин)палладий (76,17 мг, 108,51 мкмоль, 0,10 экв.) и 9 мл 2-метилтетрагидрофурана и полученную смесь три раза продували азотом и перемешивали при 60°С в атмосфере азота в течение 12 часов. Затем добавляли 20 мл этилацетата и смесь фильтровали. Фильтрат три раза промывали 20 мл насыщенного солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением соединения 93 в виде желтого твердого вещества (41,10 мг, 71,26 мкмоль, выход 6,54%, чистота 97,1%). MS (ESI, М+23): 536,1.
[296] Стадия В: N-хлорсукцинимид добавляли к раствору соединения 93 (41,10 мг, 77,64 мкмоль, 1,00 экв.) в 10 мл ацетонитрила и реакционный раствор перемешивали при 30°С в течение 10 часов. После завершения реакции реакционный раствор непосредственно концентрировали с получением неочищенного продукта, который очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением II-8. 1Н ЯМР (400 МГц, ДМСО-d6): δ 11,42 (s, 1H), 8,36 (s, 1Н), 7,49 (d, J=7,6 Гц, 1Н), 7,42-7,37 (m, 2Н), 7,26 (dd, J=8,8 Гц, J=6,4 Гц, 1H), 7,19-7,10 (m, 3Н), 6,92 (d, J=8,4 Гц, 2Н), 6,64 (d, J=8,8 Гц, 2Н), 3,77 (t, J=5,6 Гц, 2Н), 3,33 (t, J=7,2 Гц, 2Н), 2,91 (t, J=7,6 Гц, 2Н), 2,71 (d, J=6,8 Гц, 2Н), 2,65-2,59 (m, 3Н), 2,43-2,37 (m, 2Н), 0,92 (t, J=7,6 Гц, 3Н). MS (ESI,M+1): 548,1.
Вариант реализации 49
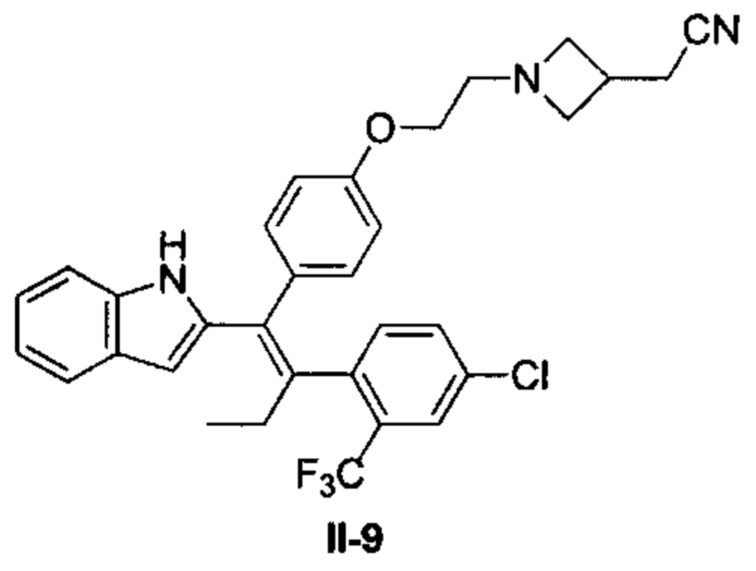
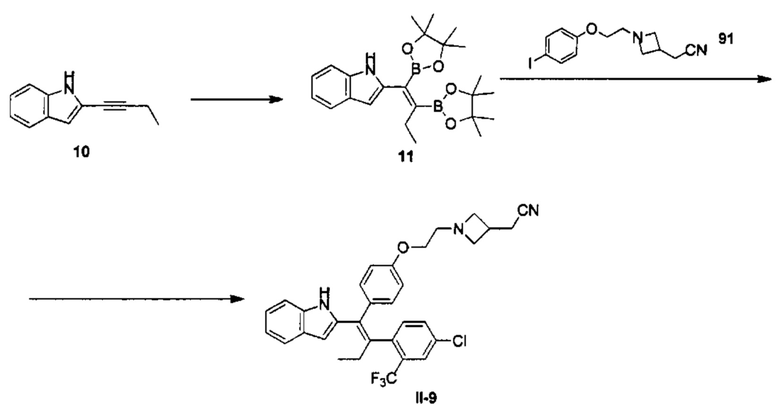
[297] Стадия А: Биспинаколатодибор (147,80 мг, 582,03 мкмоль, 1,05 экв.) и тетракис(трифенилфосфин)платину (34,48 мг, 27,72 мкмоль, 0,05 экв.) добавляли к раствору соединения 10 (100,00 мг, 554,31 мкмоль, 1,00 экв.) в 2-метилтетрагидрофуране (10,00 мл), реакционный раствор три раза продували азотом, затем перемешивали при 70°С в атмосфере азота в течение 5 часов, и полученное 11 не выделяли. После охлаждения реакционного раствора до 0°С добавляли соединение 91 (238,50 мг, 497,68 мкмоль, 0,90 экв.), карбонат калия K2CO3 (152,91 мг, 1,11 ммоль, 2,00 экв.), 2,5 мл воды, 10 мл 2-метилтетрагидрофурана и дихлорбис(трифенилфосфин)палладия (19,41 мг, 27,65 мкмоль, 0,05 экв.), полученную смесь три раза продували азотом и перемешивали при 30°С в атмосфере азота в течение 12 часов, и полученное 92 не выделяли. Затем добавляли 4-хлор-2-трифторметилйодбензол (338,89 мг, 1,11 ммоль, 2,00 экв.), водный раствор гидроксида калия (4М, 552,93 мкл, 4,00 экв.), дихлорбис(трифенилфосфин)палладия (19,40 мг, 27,65 мкмоль, 0,05 экв.) и 10 мл 2-метилтетрагидрофурана и полученную смесь три раза продували азотом и перемешивали при 70°С в атмосфере азота в течение 12 часов. Затем добавляли 20 мл этилацетата и смесь фильтровали. Фильтрат три раза промывали 20 мл насыщенного солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением соединения II-9. 1Н ЯМР (400 МГц, ДМСО-d6): δ 10,67 (s, 1Н), 8,38 (s, 1Н), 7,71 (d, J=1,6 Гц, 1Н), 7,63 (dd, J=8,4 Гц, J=1,6 Гц, 1H), 7,53 (d, J=8,0 Гц, 1Н), 7,32 (dd, J=8,0 Гц, J=12,0 Гц, 2Н), 7,04 (t, J=7,2 Гц, 1Н), 6,98 (t, J=7,2 Гц, 1Н), 6,80 (d, J=8,8 Гц, 2Н), 6,63 (d, J=8,4 Гц, 2Н), 6,48 (d, J=0,8 Гц, 1Н), 3,77 (t, J=5,6 Гц, 2Н), 3,33 (t, J=7,2 Гц, 2Н), 2,92 (t, J=7,6 Гц, 2Н), 2,84-2,77 (m, 1Н), 2,71 (d, J=6,8 Гц, 2Н), 2,66-2,61 (m, 3Н), 2,45-2,38 (m, 1Н), 0,93 (t, J=7,6 Гц, 3Н). MS (ESI, М+23): 564,2.
Вариант реализации 50
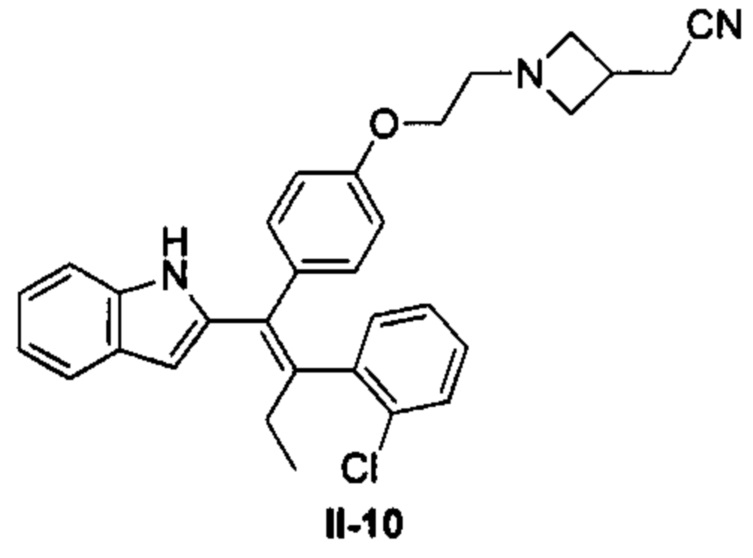
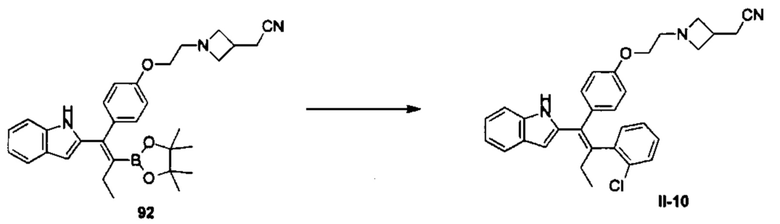
[298] Стадия А: 1-хлор-2-йодбензол (517,50 мг, 2,17 ммоль, 2,00 экв.), водный гидроксид калия (4М, 1,09 мл, 4,00 экв.) и 2-метилтетрагидрофуран (5,00 мл) добавляли к раствору соединения 92 (555,00 мг, 1,09 ммоль, 1,00 экв.) в 2-метилтетрагидрофуране (7,50 мл), реакционную систему три раза продували азотом, затем добавляли дихлорбис(трифенилфосфин)палладий (38,08 мг, 54,26 мкмоль, 0,05 экв.). Реакционный раствор перемешивали при 70°С в атмосфере азота в течение 12 часов, затем разбавляли 20 мл этилацетата и фильтровали. Фильтрат три раза промывали 20 мл солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью препаративной ВЭЖХ (система с муравьиной кислотой) с получением соединения II-10. 1Н ЯМР (400 МГц, ДМСО-d6): δ 10,64 (brs, 1Н), 8,29 (brs, 1Н), 7,54 (d, J=8,0 Гц, 1Н), 7,37-7,34 (m, 1Н), 7,29 (d, J=8,0 Гц, 1Н), 7,21-7,18 (m, 2Н), 7,14-7,12 (m, 1Н), 7,04 (dt, J=7,2 Гц, J=1,2 Гц, 1Н), 6,97 (dt, J=8,0 Гц, J=1,2 Гц, 1Н), 6,90-6,82 (m, 2Н), 6,61-6,59 (m, 2Н), 6,53 (d, J=1,2 Гц, 1Н), 3,76 (t, J=5,6 Гц, 2Н), 3,33 (t, J=7,2 Гц, 2Н), 2,92 (t, J=7,2 Гц, 2Н), 2,71 (d, J=6,8 Гц, 2Н), 2,67-2,55 (m, 5Н), 0,97 (t, J=7,6 Гц, 3Н). MS (ESI, M+1): 496,2.
Вариант реализации 51
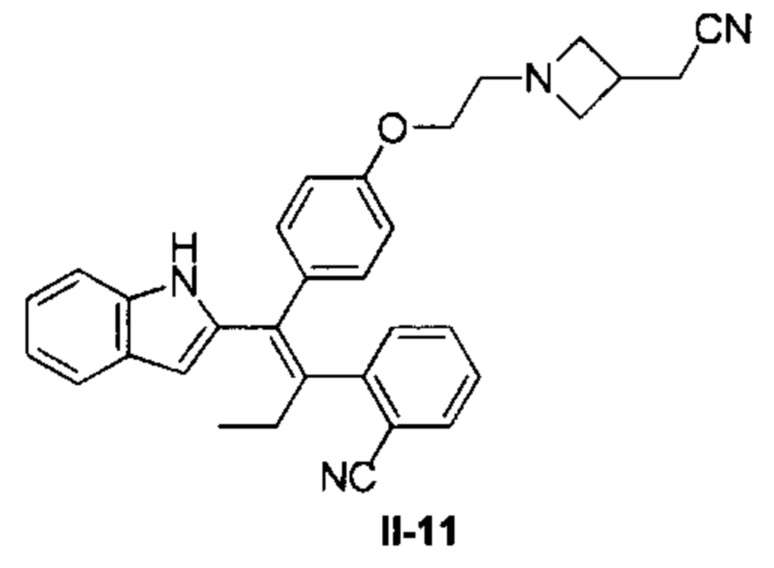
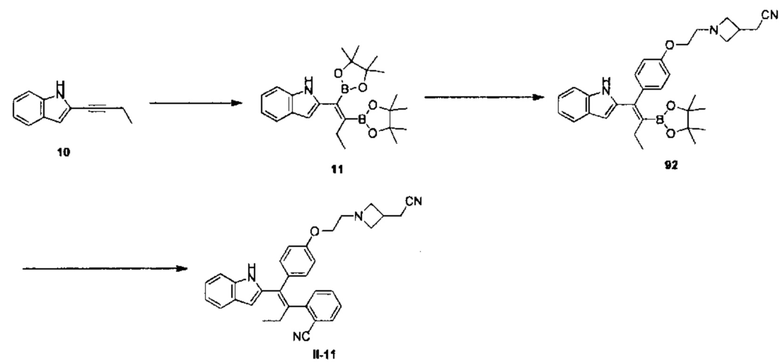
[299] Стадия А: Биспинаколатодибор (147,80 мг, 582,03 мкмоль, 1,05 экв.) и тетракис(трифенилфосфин)платину (34,48 мг, 27,72 мкмоль, 0,05 экв.) добавляли к раствору соединения 10 (100,00 мг, 554,31 мкмоль, 1,00 экв.) в 2-метилтетрагидрофуране (10,00 мл), реакционный раствор три раза продували азотом, затем перемешивали при 70°С в атмосфере азота в течение 5 часов и полученное 11 не выделяли. После охлаждения реакционного раствора до 0°С добавляли соединение 91 (238,50 мг, 497,68 мкмоль, 0,90 экв.), карбонат калия K2CO3 (152,91 мг, 1,11 ммоль, 2,00 экв.), 2,5 мл воды, 10 мл 2-метилтетрагидрофурана и дихлорбис(трифенилфосфин)палладий (19,41 мг, 27,65 мкмоль, 0,05 экв.), полученную смесь три раза продували азотом и перемешивали при 30°С в атмосфере азота в течение 12 часов, и полученное 92 не выделяли. Затем добавляли 2-йодбензонитрил (253,26 мг, 1,11 ммоль, 2,00 экв.), водный раствор гидроксида калия (4М, 552,93 мкл, 4,00 экв.), дихлорбис(трифенилфосфин)палладий (19,40 мг, 27,65 мкмоль, 0,05 экв.) и 5 мл 2-метилтетрагидрофурана и полученную смесь три раза продували азотом и перемешивали при 70°С в атмосфере азота в течение 12 часов. Затем добавляли 20 мл этилацетата и смесь фильтровали. Фильтрат три раза промывали 20 мл насыщенного солевого раствора каждый раз, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением соединения II-11. 1Н ЯМР (400 МГц, ДМСО-d6): δ 10,65 (s, 1Н), 8,28 (s, 1Н), 7,65-7,61 (m, 2Н), 7,56 (d, J=7,6 Гц, 1Н), 7,50 (d, J=7,6 Гц, 1Н), 7,35 (dt, J=7,6 Гц, J=0,8 Гц, 1Н), 7,30 (d, J=8,0 Гц, 1Н), 7,05 (dt, J=8,0 Гц, J=1,2 Гц, 1Н), 6,99 (dt, J=8,0 Гц, J=1,2 Гц, 1Н), 6,80 (d, J=8,8 Гц, 2Н), 6,61 (d, J=8,8 Гц, 2Н), 6,56 (d, J=1,6 Гц, 1Н), 3,77 (t, J=5,2 Гц, 2Н), 3,34 (t, J=7,2 Гц, 2Н), 2,92 (t, J=6,8 Гц, 2Н), 2,71 (d, J=7,2 Гц, 2Н), 2,67-2,57 (m, 3Н), 2,52-2,50 (m, 2Н), 1,00 (t, J=7,6 Гц, 3Н); MS (ESI, М+1, М+23): 492,2, 509,1.
Вариант реализации 52
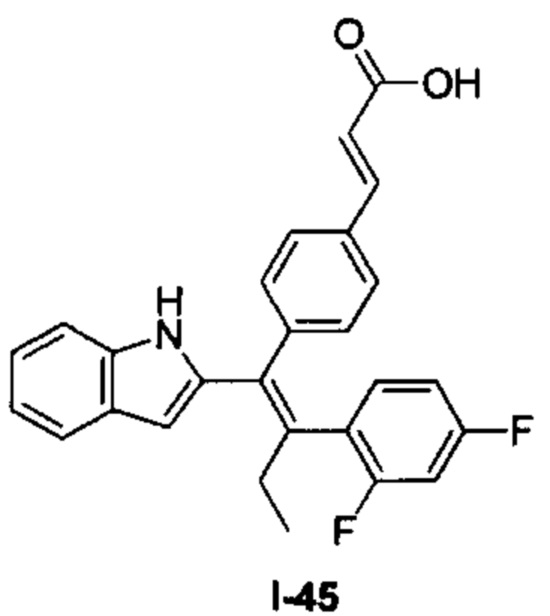
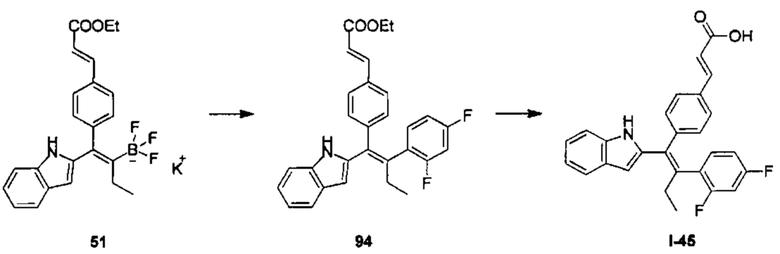
[300] Раствор 51 (100,00 мг, 218,58 мкмоль, 1,00 экв.), 2,4-дифторйодобензол (73,44 мг, 306,01 мкмоль, 1,40 экв.) и бис(дифенилфосфино)ферроцен дихлорпалладия (7,94 мг, 10,86 мкмоль, 0,05 экв.) в 2-метилтетрагидрофуране (15 мл) продували азотом, с последующим добавлением водного гидроксида натрия (4М, 218,58 мкл, 4,00 экв.). Реакционный раствор перемешивали при 65°С в течение 12 часов и он поменял цвет с желтого на черный. Полученный 15 мл черный раствор, содержащий соединение 94, теоретическое количество которого составило 150 мг, непосредственно использовали на следующей стадии. Указанный выше 15 мл черный раствор, содержащий соединение 94, теоретическое количество которого составило 150 мг, и метанол (8,00 мл) перемешивали при 30°С в течение 15 часов. 20 мл добавляли к реакционному раствору, затем смесь фильтровали. Фильтрат доводили до рН 4 3М соляной кислотой, затем экстрагировали три раза 20 мл этилацетата. Органическую фазу объединяли, промывали один раз 20 мл насыщенного солевого раствора, фильтровали, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, который растворяли в 3 мл ацетонитрила и очищали с помощью высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением I-45. 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,76 (s, 1Н), 7,55 (d, J=7,6 Гц, 1Н), 7,47-7,38 (m, 3Н), 7,33-7,19 (m, 2Н), 7,15-6,91 (m, 6Н), 6,54 (d, J=1,6 Гц, 1Н), 6,41 (d, J=16,4 Гц, 1Н), 2,63 (q, J=7,2 Гц, 2Н), 0,99 (t, J=7,6 Гц, 3Н). MS (ESI,M+1): 430,1.
Вариант реализации 53
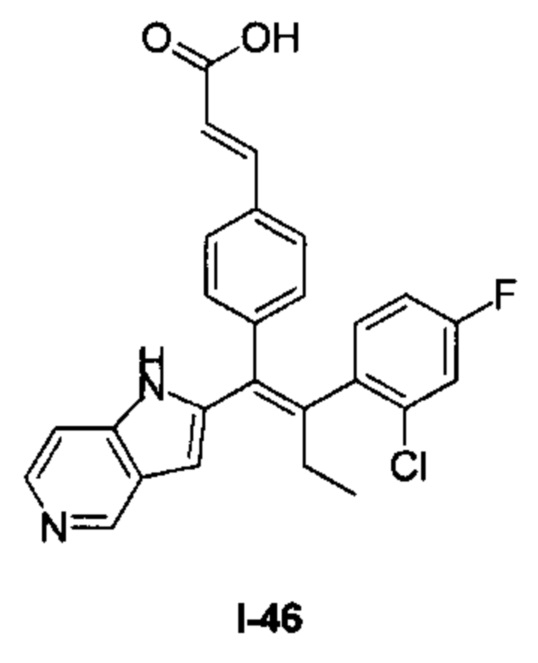
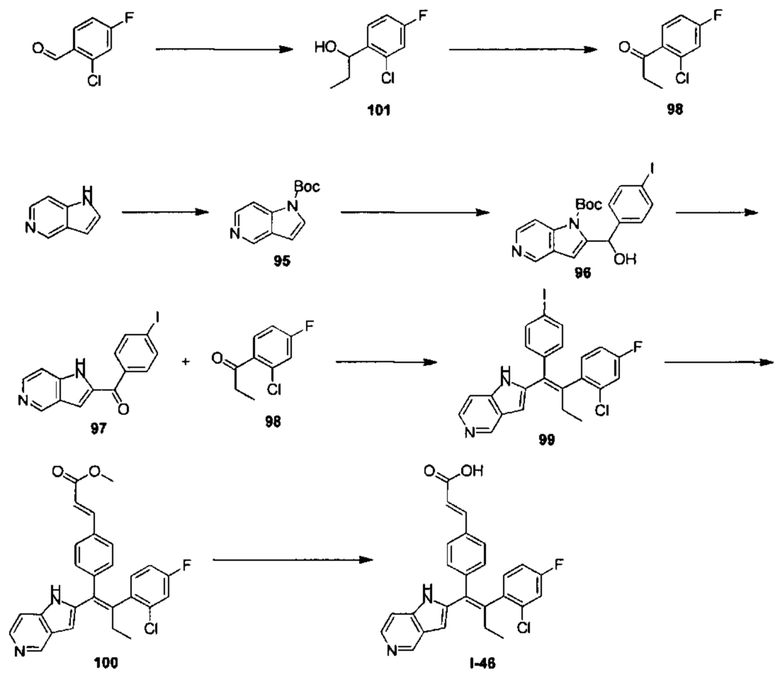
[301] Стадия А: Раствор 2-хлор-4-фторбензальдегида (5,00 г, 31,53 ммоль, 1,00 экв.) в 12 мл тетрагидрофуране охлаждали до -20°С, с последующим добавлением бромида этилмагния (2М, 17,34 мл, 1,10 экв.) в атмосфере азота. Реакционный раствор нагревали до 0°С и затем перемешивали при 20°С в течение 12 часов. Реакционный раствор гасили 15 мл насыщенного водного раствора хлорида аммония при 0°С, разбавляли 10 мл воды, разделяли, водную фазу три раза экстрагировали 50 мл этилацетата каждый раз. Органическую фазу объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (ЕА/РЕ=0% до 30%) с получением продукта 101 в виде желтого масла.
[302] Стадия В: 101 (1,70 г, 9,01 ммоль, 1,00 экв.) и 50 мл дихлорметана добавляли в одногорлую круглодонную колбу, затем охлаждали до 0°С и добавляли реагент Десса-Мартина (6,11 г, 14,42 ммоль, 4,46 мл, 1,60 экв.) в атмосфере азота. Реакционный раствор перемешивали при 0°С в течение 1 часа, затем гасили насыщенным водным раствором бикарбоната натрия, содержащим 15% тиосульфата натрия (20 мл), и фильтровали. Фильтрат разделяли и водную фазу три раза экстрагировали 50 мл дихлорметана каждый раз. Органическую фазу объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (ЕА/РЕ=0% до 10%) с получением 98.
[303] Стадия С: 5-Азаиндол (10,00 г, 84,65 ммоль, 1,00 экв.), Boc2O (19,40 г, 88,88 ммоль, 20,42 мл, 1,05 экв.) и 200 мл дихлорметана добавляли в одногорлую круглодонную колбу с последующим добавлением DMAP (1,03 г, 8,47 ммоль, 0,10 экв.) в атмосфере азота. Реакционный раствор перемешивали при 20°С в течение 12 часов, затем концентрировали и очищали с помощью колоночной хроматографии на силикагеле (ЕА/РЕ=0%-40%) с получением 95. 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 8,89 (s, 1Н), 8,47 (d, J=6,0 Гц, 1Н), 7,98 (d, J=5,6 Гц, 1Н), 7,61 (d, J=3,6 Гц, 1H), 6,65 (d, J=3,6 Гц, 1Н), 1,69 (s, 9Н).
[304] Стадия D: 95 (1,00 г, 4,58 ммоль, 1,00 экв.) растворяли в 10 мл тетрагидрофурана и охлаждали до -78°С. LDA (2М, 2,75 мл, 1,20 экв.) добавляли по каплям и температуру смеси поддерживали ниже -70°С в течение 30 минут, затем раствор 4-йодбензальдегида (1,06 г, 4,58 ммоль, 1,00 экв.) в 5 мл тетрагидрофурана добавляли по каплям при -70°С и реакционный раствор перемешивали при указанной температуре в течение 1 часа. Реакционный раствор медленно гасили 10 мл насыщенного водного раствора хлорида аммония, затем разбавляли 10 мл воды, разделяли и водную фазу три раза экстрагировали 30 мл этилацетата каждый раз. Органическую фазу объединяли, промывали 20 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (ЕА/РЕ=20%-100%) с получением продукта 96 в виде желтого твердого вещества. MS (ESI,M+1): 450,9.
[305] Стадия Е: 96 (6,10 г, 13,55 ммоль, 1,00 экв.) и 100 мл тетрагидрофурана добавляли в одногорлую круглодонную колбу с последующим добавлением диоксида марганца (11,78 г, 135,50 ммоль, 10,00 экв.). Реакционный раствор перемешивали при 70°С в течение 72 часов, затем фильтровали. Фильтрационный осадок промывали 30 мл дихлорметана. Фильтрат концентрировали и очищали с помощью колоночной хроматографии на силикагеле (метанол/дихлорметан = 0%-10%) и дополнительно очищали с помощью препаративной высокоэффективной жидкостной хроматографии (основной) с получением продукта 97 в виде желтого твердого вещества. MS (ESI,M+1): 349,0.
[306] Стадия D: Порошок цинка (657,39 мг, 10,05 ммоль, 14,00 экв.) добавляли к 5 мл тетрагидрофурана, с последующим добавлением по каплям тетрахлорида титана (817,26 мг, 4,31 ммоль, 472,40 мкл, 6,00 экв.) при 30°С. Затем добавляли раствор 97 (250,00 мг, 718,10 мкмоль, 1,00 экв.) и 98 (415,41 мг, 2,23 ммоль, 3,10 экв.) в тетрагидрофуране (20,00 мл). Реакционный раствор перемешивали при 70°С в течение 14 часов и он стал ярко-желтым. Реакционный раствор охлаждали до комнатной температуры, гасили 20 мл насыщенного водного раствора бикарбоната натрия, фильтровали с удалением желтого твердого вещества и разделяли. Водную фазу три раза экстрагировали 20 мл этилацетата каждый раз. Органическую фазу объединяли, промывали солевым раствором, сушили над безводным сульфатом натрия, фильтровали. Фильтрат фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (метанол/дихлорметан = 0%-5%) с получением продукта 99. MS (ESI, M+1): 503,0.
[307] Стадия F: 99 (230,00 мг, 457,48 мкмоль, 1,00 экв.), метилакрилат (196,92 мг, 2,29 ммоль, 205,13 мкл, 5,00 экв.), 1 мл триэтиламин, РОТ (69,62 мг, 228,74 мкмоль, 0,50 экв.) и 3 мл ДМФА добавляли в трехгорлую круглодонную колбу с последующим добавлением ацетата палладия (30,81 мг, 137,24 мкмоль, 0,30 экв.) в атмосфере азота. Реакционный раствор продували азотом и перемешивали при 110°С в течение 2 часов. Затем реакционный раствор фильтровали и фильтрационный осадок промывали 20 мл дихлорметана. Фильтрат концентрировали с получением 100 (250,00 мг, неочищенного). MS (ESI, M+1): 461,1.
[308] Стадия F: Моногидрат гидроксида лития (57,35 мг, 1,37 ммоль, 3,00 экв.) добавляли к раствору 100 (210,00 мг, 455,60 мкмоль, 1,00 экв.) в смешанном растворителе 5 мл тетрагидрофурана, 5 мл метанола и 5 мл воды. Желтую суспензию перемешивали при 20°С в течение 36 часов, затем концентрировали при пониженном давлении, разбавляли 10 мл воды и доводили до рН 4-5 лимонной кислотой. Твердый осадок собирали и очищали с помощью препаративной высокоэффективной жидкостной хроматографии (муравьиная кислота) с получением продукта I-46. 1Н ЯМР (400 МГц, ДМСО-d6) δ = 12,75 (s, 1Н), 9,31 (s, 1Н), 8,43 (d, J=6,8 Гц, 1Н), 7,86 (d, J=6,4 Гц, 1Н), 7,51-7,43 (m, 3Н), 7,39 (dd, J=2,4, 8,8 Гц, 1Н), 7,32 (dd, J=6,4, 8,4 Гц, 1H), 7,20-7,13 (m, 2Н), 7,01 (d, J=8,0 Гц, 2Н), 6,45 (d, J=16,0 Гц, 1Н), 2,61 (q, J=7,2 Гц, 2Н), 1,00 (br t, J=7,6 Гц, 3Н); MS(ESI,M+1): 447,0.
Вариант реализации 54
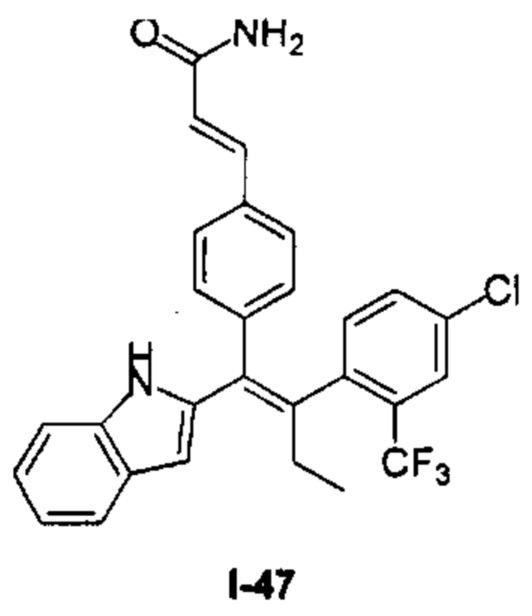
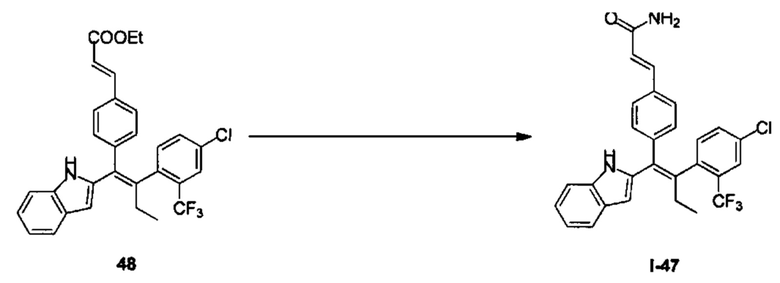
[309] Стадия А: ДМФА (5,00 мл) добавляли в 50 мл одногорлую колбу с последующим добавлением соединения 48 (100,00 мг, 190,85 мкмоль, 1,00 экв.) и формамида (51,58 мг, 1,15 ммоль, 45,64 мкл, 6,00 экв.). Раствор метилата натрия в метаноле (0,5М, 1,15 мл, 3,00 экв.) добавляли к раствору. Реакционный раствор перемешивали при 100°С в течение 2 часов и он поменял цвет с желтого на коричневый. После завершения реакции добавляли 30 мл этилацетата, затем три раза промывали 30 мл насыщенного солевого раствора каждый раз. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали и очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением продукта I-47. 1Н ЯМР (400 МГц, ДМСО-d6): δ 10,76 (brs, 1H), 7,72 (d, J=2,0 Гц, 1Н), 7,65 (dd, J=8,4 Гц, J=2,0 Гц, 1Н), 7,55 (d, J=7,6 Гц, 1H), 7,44 (brs, 1Н), 7,38 (d, J=8,4 Гц, 1Н), 7,32-7,27 (m, 3Н), 7,25 (d, J=15,6 Гц, 1Н), 7,08-6,97 (m, 3Н), 6,93 (d, J=8,0 Гц, 2Н), 6,52 (d, J=1,6 Гц, 1Н), 6,47 (d, J=16,0 Гц, 1Н), 2,85-2,80 (m, 1Н), 2,47-2,44 (m, 1Н), 0,94 (t, J=7,6 Гц, 3Н). MS (ESI, M+1): 495,2.
Вариант реализации 55
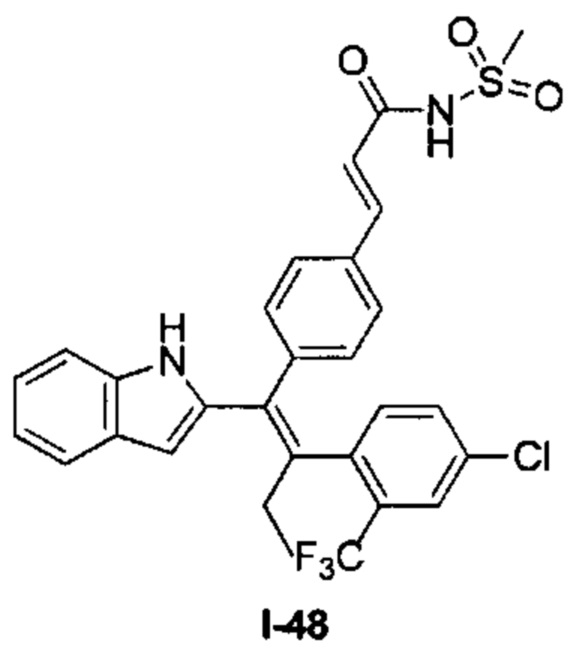
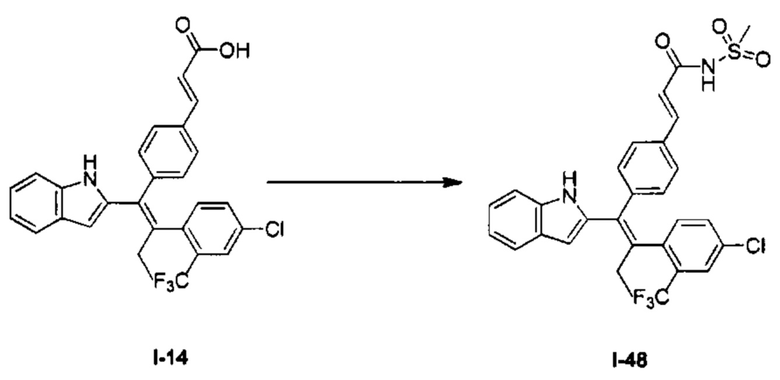
[310] Стадия А: Соединение I-14 (100,00 мг, 177,45 мкмоль, 1,00 экв.), EDCI (51,03 мг, 266,17 мкмоль, 1,50 экв.) и DMAP (49,86 мг, 408,13 мкмоль, 2,30 экв.) растворяли в дихлорметане (7,00 мл), с последующим добавлением метилсульфонамида (50,64 мг, 532,34 мкмоль, 3,00 экв.). Черный реакционный раствор перемешивали при 30°С в течение 12 часов. После завершения реакции добавляли воду (10 мл), затем экстрагировали этилацетатом (10 мл*3). Органическую фазу объединяли, промывали один раз 15 мл солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной хроматографии (муравьиная кислота) с получением продукта I-48. 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,80 (s, 1Н), 8,15 (s, 1Н), 7,75 (d, J=2,4 Гц, 1Н), 7,66 (dd, J=2,0, 8,4 Гц, 1Н), 7,57 (d, J=8,0 Гц, 1Н), 7,45-7,29 (m, 5Н), 7,11-6,91 (m, 4Н), 6,54 (d, J=2,0Гц, 1Н), 6,46 (d, J=15,4 Гц, 1H), 3,13 (s, 3Н), 2,89-2,78 (m, 1Н), 2,46 (br d, J=7,6 Гц, 1Н), 0,95 (t, J=7,6 Гц, 3Н); MS (ESI,M+1): 573,1.
Вариант реализации 56
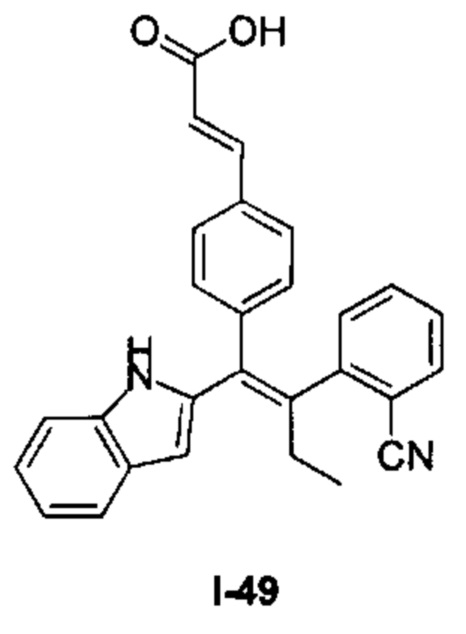
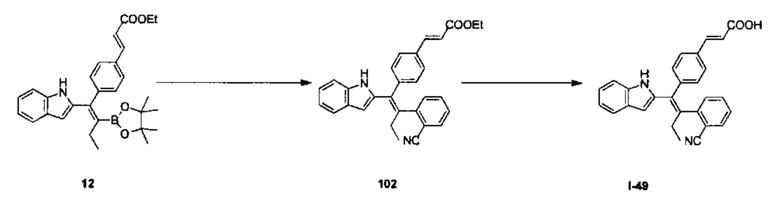
[311] Стадия А: Соединение 12 (272,73 мг, 509,13 мкмоль, 1,00 экв.), 2-циано бромбензол (139,01 мг, 763,70 мкмоль, 1,50 экв.), дихлорбис(трифенилфосфин)палладий (107,21 мг, 152,74 мкмоль, 0,30 экв.), водный раствор гидроксида калия (4М, 712,78 мкл, 5,60 экв.) растворяли в 20 мл 2-метилтетрагидрофурана, реакционный раствор три раза продували азотом, затем перемешивали при 75°С в атмосфере азота в течение 7 часов. Реакционный раствор поменял цвет с желтого на темно-коричневый и черное твердое вещество выпало в осадок. Реакционный раствор фильтровали через целит и фильтрат концентрировали и очищали с получением неочищенного продукта 102 в виде темно-коричневого масла (250,00 мг, неочищенный продукт), которое непосредственно использовали на следующей стадии без дополнительной очистки.
[312] Стадия В: Соединение 102 (250,00 мг, 559,86 мкмоль, 1,00 экв.), гидроксид лития (67,04 мг, 2,80 ммоль, 5,00 экв.) растворяли в смешанном растворителе 10 мл метанола, 5 мл тетрагидрофурана и 5 мл воды. Реакционный раствор перемешивали при 30°С в течение 1 часа, затем концентрировали до 1 мл, доводили рН до 5-6 соляной кислотой (3 моль/л), дважды экстрагировали 100 мл дихлорметана (50 мл каждый раз). Органическую фазу объединяли, сушили, фильтровали, концентрировали при пониженном давлении и очищали с помощью препаративной хроматографии (система с муравьиной кислотой) с получением продукта I-49 в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,76 (s, 1H), 7,69-7,62 (m, 2Н), 7,60-7,55 (m, 2Н), 7,44-7,39 (m, 3Н), 7,38-7,29 (m, 2Н), 7,07 (t, J=7,6 Гц, 1Н), 7,03-6,98 (m, 1Н), 6,94 (d, J=8,0 Гц, 2Н), 6,61 (s, 1Н), 6,41 (d, J=16,0 Гц, 1Н), 2,81-2,67 (m, 2Н), 1,02 (br t, J=7,2 Гц, 3Н); MS [ESI, M+1]:419,2.
Вариант реализации 57
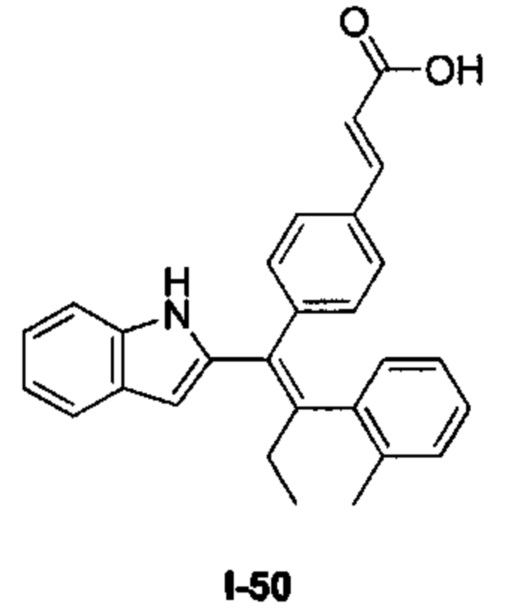
[313] I-50 получали с использованием промежуточного соединения 12 (227,27 мг, 424,27 мг, 1,00 экв.) и о-метилйодбензола (185,01 мг, 848,54 мкмоль, 108,19 мкл, 2,00 экв.) в качестве исходного вещества в соответствии с процедурой, аналогичной описанной для получения I-49.
[314] Неочищенный продукт очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением продукта I-50.
[315] 1Н ЯМР (400 МГц, ДМСО-d6): δ 10,81 (brs, 1H), 7,54 (d, J=8,0 Гц, 1Н), 7,39 (d, J=16,0 Гц, 1Н), 7,34 (d, J=8,4 Гц, 2Н), 7,30 (dd, J=8,0 Гц, J=0,8 Гц, 1Н), 7,18 (dt, J=7,6 Гц, J=1,6 Гц, 1Н), 7,14-7,03 (m, 4Н), 6,99 (dt, J=7,2 Гц, J=1,2 Гц, 1Н), 6,89 (d, J=8,4 Гц, 2Н), 6,53 (d, J=1,2 Гц, 1Н), 6,36 (d, J=16,0 Гц, 1Н), 2,59-2,54 (m, 2Н), 2,08 (s, 3Н), 0,95 (t, J=7,6 Гц, 3Н). MS (ESI, M+1): 408,1.
Вариант реализации 58
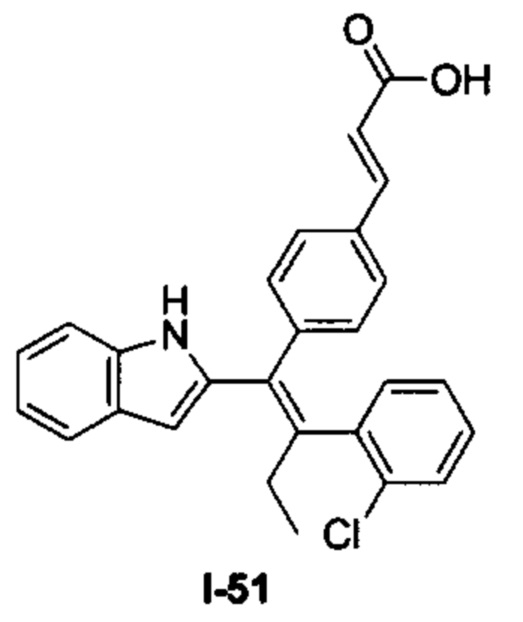
[316] I-51 получали с использованием промежуточного соединения 12 (327,27 мг, 610,96 мкмоль, 1,00 экв.) и о-хлорйодбензола (291,37 мг, 1,22 ммоль, 2,00 экв.) в качестве исходного вещества в соответствии с процедурой, аналогичной описанной для получения I-49.
[317] Неочищенный продукт очищали с помощью препаративной хроматографии (система с муравьиной кислотой) с получением продукта I-51. MS [ESI, M+1]+: 428,1, 1Н ЯМР (400 МГц, ДМСО-d6) δ = 10,74 (s, 1Н), 7,56 (d, J=8,0 Гц, 1Н), 7,42-7,35 (m, 4H), 7,30 (d, J=8,2 Гц, 1H), 7,24-7,15 (m, 3H), 7,09-6,94 (m, 4H), 6,57 (d, J=1,2 Гц, 1H), 6,38 (d, J=16,0 Гц, 1H), 2,75-2,59 (m, 1H), 2,75-2,56 (m, 1H), 1,00 (t, J=7,5 Гц, 3Н).
Вариант реализации 59
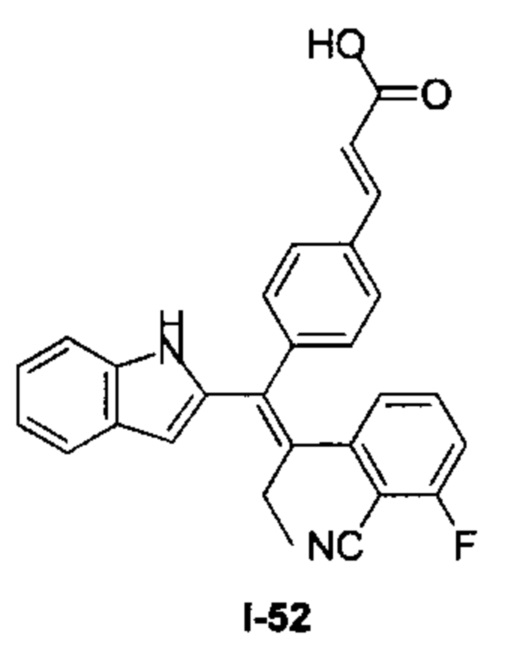
[318] I-52 получали с использованием промежуточного соединения 51 (101,68 мг, 221,57 мкмоль, 1,00 экв.) и 2-циано-3-фториодобензола (65,68 мг, 265,88 мкмоль, 1,20 экв.) в качестве исходного вещества в соответствии с процедурой, аналогичной описанной для получения I-28.
[319] Неочищенный продукт очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением продукта I-52. 1Н ЯМР (400 МГц, ДМСО-d6) δ = 10,79 (s, 1Н), 8,34 (br s, 1Н), 7,79-7,68 (m, 1Н), 7,60 (d, J=7,6 Гц, 1Н), 7,47-7,39 (m, 3Н), 7,34 (dd, J=8,0, 16,8 Гц, 3Н), 7,11-7,06 (m, 1Н), 7,04-6,99 (m, 1Н), 6,95 (d, J=8,0 Гц, 2Н), 6,62 (d, J=1,2 Гц, 1H), 6,43 (d, J=16,0 Гц, 1Н), 2,89-2,68 (m, 2Н), 1,05 (t, J=7,4 Гц, 3Н). MS (ESI, M+1): 437,1.
Вариант реализации 60
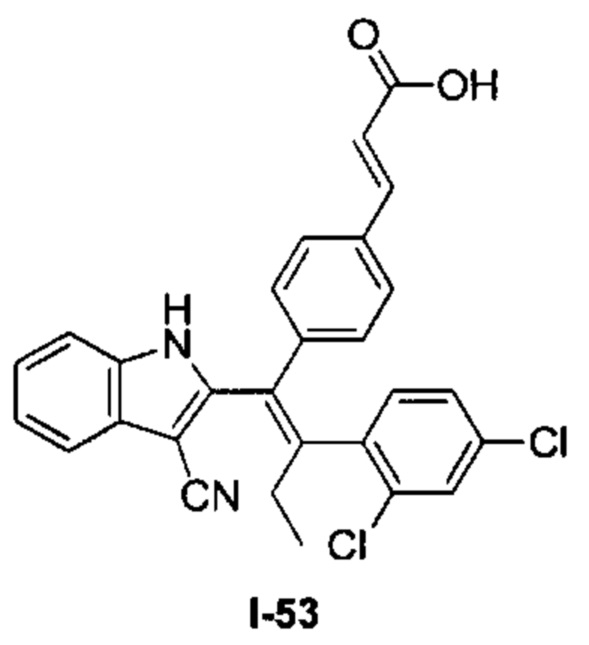
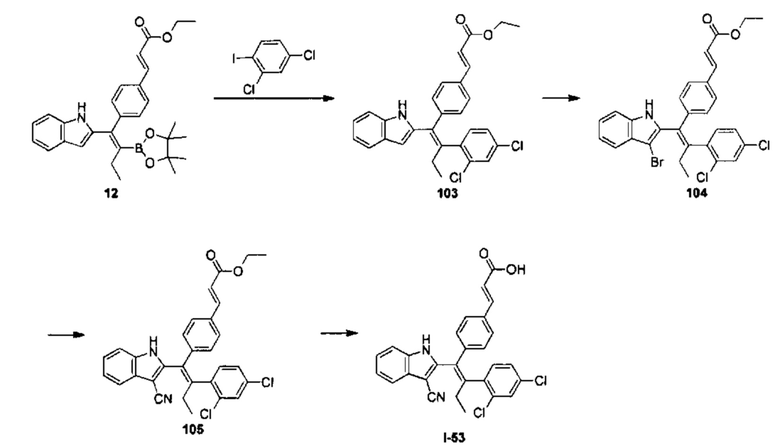
[320] Стадия А: Бис(трифенилфосфин)палладия дихлорид (30,00 мг, 42,74 мкмоль, 0,05 экв.) добавляли к раствору соединения 12 (500,00 мг, 848,55 мкмоль, 1,00 экв.), 2,4-дихлорйодбензола (250,00 мг, 916,43 мкмоль, 1,08 экв.), гидроксида калия (450,00 мг, 8,02 ммоль, 9,45 экв.) в 2-метилтетрагидрофуране (8,00 мл) при 25°С в атмосфере азота. Реакционный раствор перемешивали при 70°С в течение 18 часов, затем фильтровали, экстрагировали и очищали с помощью колоночной хроматографии на силикагеле с получением 103.
[321] Стадия В: NBS (115,00 мг, 646,14 мкмоль, 1,02 экв.) добавляли к раствору соединения 103 (330,00 мг, 632,52 мкмоль, 1,00 экв.) в 5 мл ацетонитрила. Реакционный раствор перемешивали при 25°С в течение 10 минут, затем добавляли 5 мл дихлорметана и реакционный раствор перемешивали при 25°С в течение дополнительных 50 минут. Реакционный раствор экстрагировали, разделяли, сушили над безводным сульфатом натрия и концентрировали с получением продукта 104 в виде желтого твердого вещества.
[322] Стадия С: Цианид меди (100,00 мг, 1,12 ммоль, 243,90 мкл, 21,46 экв.) добавляли к раствору соединения 104 (125,00 мг, 52,04 мкмоль, 1,00 экв.) в 5 мл NMP. Реакционный раствор перемешивали при 180°С в условиях микроволнового облучения в течение 3 часов. Затем реакционный раствор доводили рН до 9-10 с помощью 20 мл водного бикарбоната натрия, экстрагировали этилацетатом, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали с получением 105.
[323] Стадия D: Моногидрат гидроксида лития (75,00 мг, 1,79 ммоль, 10,24 экв.) добавляли к раствору соединения 105 (90,00 мг, 174,61 мкмоль, 1,00 экв.) в смешанном растворителе 2 мл ТГФ, 0,5 мл воды и 2 мл метанола. Реакционный раствор перемешивали при 25°С в течение 9 часов. 10 мл воды добавляли к реакционному раствору, затем полученный раствор доводили до рН 3-4 1М соляной кислотой, экстрагировали этилацетатом, концентрировали с получением неочищенного продукта, который очищали с помощью препаративной высокоэффективной жидкостной хроматографии с получением I-53. 1Н ЯМР (400 МГц, CDCl3): δ 8,50 (s, 1Н), 7,80-7,34 (m, 5Н), 7,26-7,75 (m, 1Н), 7,64 (d, 7=16,0 Гц, 1Н), 7,42-7,27 (m, 5Н), 7,16 (dd, J=2,0 Гц, J=9,6 Гц, 1Н), 7,08 (t, J=4,8 Гц, 3Н), 6,31 (d, J=16,0 Гц, 1 Н), 2,78-2,65 (m, 1Н), 7,26-7,75 (m, 1Н), 7,63-2,51 (m, 1Н), 1,05 (t, J=7,6 Гц, 3Н); MS (ESI, M+1): 487,2.
Вариант реализации 61
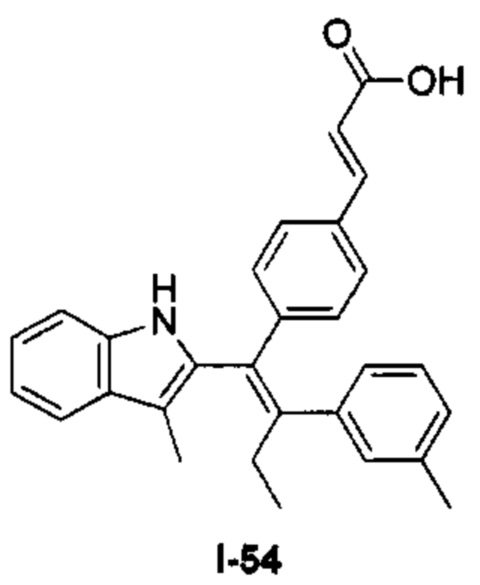
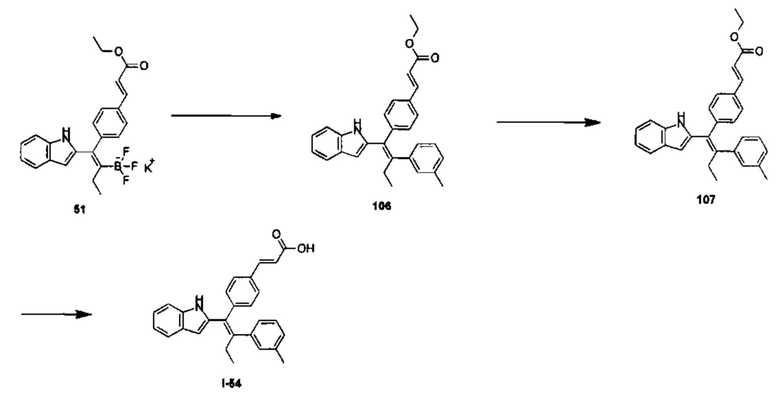
[324] Стадия А: Промежуточное соединение 106 получали с использованием промежуточного соединения 51 (200,00 мг, 443,13 мкмоль, 1,00 экв.) и m-метилйодбензола (135,26 мг, 620,38 мкмоль, 79,56 мкл, 1,40 экв.) в качестве исходного вещества в соответствии с процедурой, аналогичной описанной для получения промежуточного соединения 54.
[325] Стадия В: Промежуточное соединение 107 получали с использованием промежуточного соединения 106 (400,00 мг) и NBS (168,35 мг, 945,91 мкмоль, 1,03 экв.) в качестве исходного вещества в соответствии с процедурой, аналогичной описанной для получения промежуточного соединения 19.
[326] Стадия С: I-54 получали с использованием промежуточного соединения 107 (400,00 мг, 1 экв.) и триметилбороксина (300,13 мг, 2,39 ммоль, 333,48 мкл, 3,00 экв.) в качестве исходного вещества в соответствии с процедурой, аналогичной описанной для получения I-19. 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,78 (s, 1Н), 7,48 (d, J=7,6 Гц, 1Н), 7,37-7,27 (m, 4Н), 7,13-6,89 (m, 8Н), 6,37 (d, J=16 Гц, 1Н), 2,42 (t, J=7,6 Гц, 2H), 2,23 (s, 3Н), 2,19 (s, 3Н), 0,86 (t, J=7,6 Гц, 3Н); MS (ESI, M+1): 422,2.
Вариант реализации 62
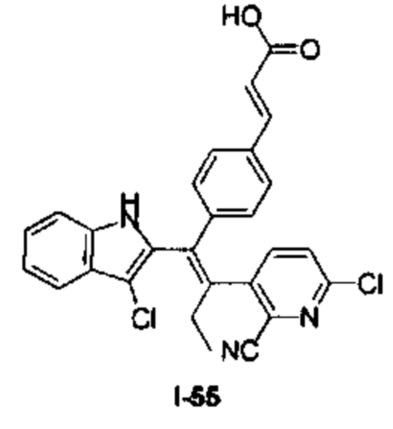
[327] I-55 получали с использованием промежуточного соединения 51 (300,00 мг, 0,656 ммоль, 1,00 экв.) и 2-хлор-5-бром-6-цианопиридина (214,21 мг, 985,10 мкмоль, 1,5 экв.) в качестве исходного вещества в соответствии с процедурой, аналогичной описанной для получения I-28.
[328] Неочищенный продукт очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением продукта I-55. 1Н ЯМР (400 МГц, ДМСО-d6): δ 11,46 (brs, 1Н), 8,25 (d, J=8,4 Гц, 1Н), 7,94 (d, J=8,4 Гц, 1Н), 7,61-7,34 (m, 5Н), 7,26-7,13 (m, 2Н), 6,96 (d, J=7,6 Гц, 1Н), 6,45 (brs, 1Н), 2,58 (q, J=7,2 Гц, 2Н), 0,98 (t, J=7,2 Гц, 3Н). MS (ESI, M+1): 488,0.
Вариант реализации 63
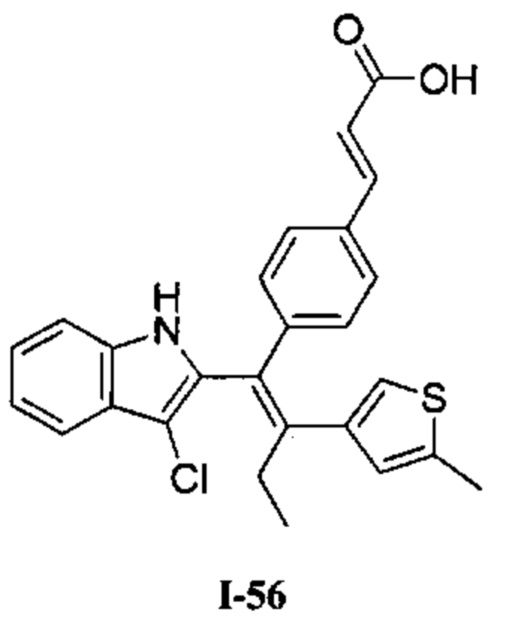
[329] I-56 получали с использованием промежуточного соединения 12 (1000 мг, 1,85 ммоль, 1,00 экв.) и 5-метил-3-бромтиофена (588,21 мг, 3,32m мкмоль, 1,8 экв.) в качестве исходного вещества в соответствии с процедурой, аналогичной описанной для получения I-24.
[330] Неочищенный продукт очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением продукта I-56. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,43 (s, 1Н), 7,53-7,43 (m, 4Н), 7,36 (d, J=8,0 Гц, 1Н), 7,22-7,09 (m, 2Н), 7,06-6,98 (m, 3Н), 6,53 (s, 1H), 6,44 (d, J=16 Гц, 1Н), 2,39 (q, J=7,2 Гц, 2Н), 2,34 (s, 3Н), 0,96 (t, J=7,2 Гц, 3Н); MS (ESI, M+1): 448,0.
Вариант реализации 64
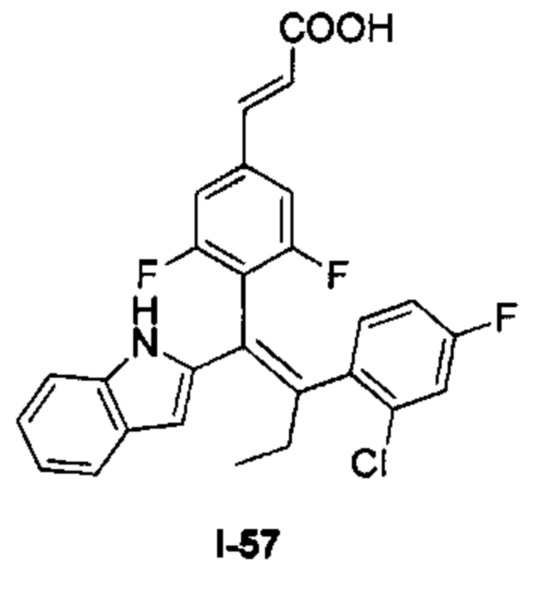
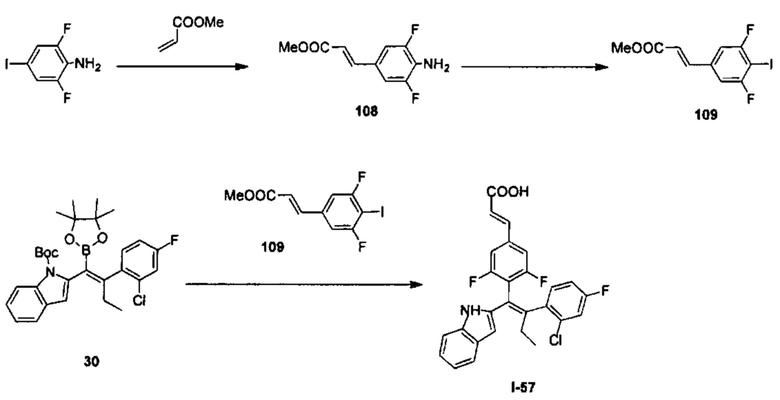
[331] Стадия А: Метилакрилат (25,32 г, 294,10 ммоль, 26,38 мл, 5,00 экв.), триэтиламин (11,90 г, 117,64 ммоль, 16,30 мл, 2,00 экв.) и 1,1'-бис(дифенилфосфино)ферроценпалладия хлорид (2,15 г, 2,94 ммоль, 0,05 экв.) добавляли к раствору 2,6-дифтор-4-йоданилина (15,00 г, 58,82 ммоль, 1,00 экв.) в 100 мл N,N-диметилформамида. Реакционный раствор перемешивали при 80°С в атмосфере азота в течение 12 часов. Затем добавляли 400 мл этилацетата и смесь фильтровали через целит. Фильтрат три раза промывали 400 мл воды. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле с получением промежуточного соединения 108.
[332] Стадия В: Йод (19,64 г, 77,40 ммоль, 15,59 мл, 3,00 экв.) добавляли к раствору 108 (5,50 г, 25,80 ммоль, 1,00 экв.) в 50 мл ацетонитрила, с последующим добавлением трет-бутилнитрита (3,99 г, 38,70 ммоль, 4,59 мл, 1,50 экв.) при 0°С в атмосфере азота. Реакционный раствор перемешивали при 15°С в атмосфере азота в течение 12 часов. Затем добавляли 150 мл этилацетата и смесь фильтровали через целит. Фильтрат дважды промывали 200 мл насыщенного водного сульфита натрия. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле с получением промежуточного соединения 109.
[333] Стадия С: Водный раствор гидроксида калия (4М, 3,33 мл, 5,00 экв.) и бис(трифенилфосфин)палладия дихлорид (93,44 мг, 133,00 мкмоль, 0,05 экв.) добавляли к раствору 30 (1,40 г, 2,66 ммоль, 1,00 экв.) в 30 мл диметилтетрагидрофурана. Реакционный раствор перемешивали при 70°С в атмосфере азота в течение 12 часов, затем фильтровали через целит. Фильтрат нейтрализовали до рН 5 соляной кислотой (1М). Добавляли 40 мл воды, затем дважды экстрагировали 30 мл этилацетата. Органическую фазу объединяли, дважды промывали 30 мл воды, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ (система с муравьиной кислотой) с получением продукта I-57. MS(ESI, М+1): 482,1 1Н ЯМР EW3644-119-Р1Е (400 МГц, ДМСО-d6): δ 12,52 (brs, 1Н), 10,81 (s, 1Н), 7,54 (d, J=8,0 Гц, 1Н), 7,45 (d, J=16,4 Гц, 1Н), 7,36-7,32 (m, 4Н), 7,23 (ddd, J=2,0 Гц, J=6,4 Гц, J=8,8 Гц, 1Н), 7,13 (dt, J=2,8 Гц, J=8,4 Гц, 1Н), 7,07 (dt, J=1,2 Гц, J=7,2 Гц, 1Н), 6,99 (dt, J=0,8 Гц, J=8,0 Гц, 1Н), 6,59 (d, J=16,0 Гц, 1Н), 6,55 (d, J=1,2 Гц, 1Н), 2,88-2,81 (m, 2Н), 1,05 (t, J=7,6 Гц, 3Н).
Вариант реализации 65
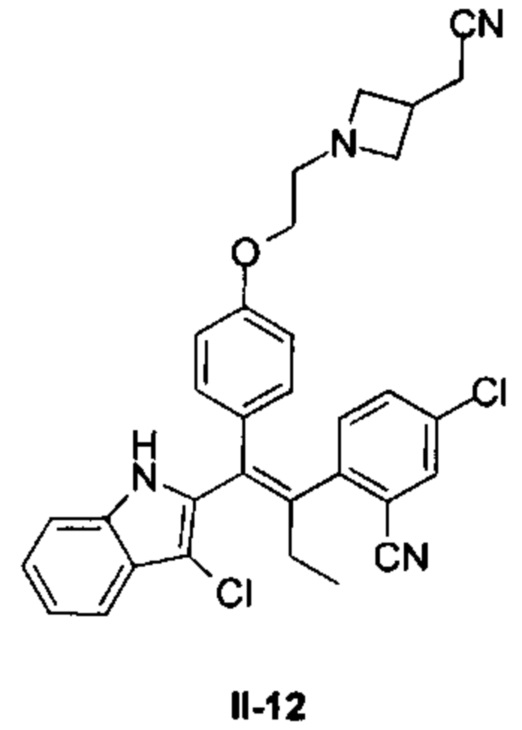
[334] II-12 получали с использованием промежуточного соединения 10 (500,00 мг, 2,92 ммоль, 1,00 экв.) и промежуточного соединения (920 мг, 2,56 ммоль, 0,9 экв.) и о-циано-n-хлорбромбензола (922 мг, 4,26 ммоль) в качестве исходного вещества в соответствии с процедурой, аналогичной описанной для получения II-8.
[335] Неочищенный продукт очищали с помощью препаративной высокоэффективной жидкостной хроматографии (система с муравьиной кислотой) с получением продукта I-56. 1Н ЯМР (400 МГц, ДМСО-d6) δ = 11,37 (s, 1Н), 7,94 (d, J=2,4 Гц, 1Н), 7,78-7,75 (m, 1H), 7,53-7,51 (m, 2Н), 7,39-7,37 (m, 1Н), 7,20-7,08 (m, 2Н), 6,82 (d, J=8,8 Гц, 2Н), 6,66 (d, J=8,8 Гц, 2Н), 3,78 (t, J=5,2 Гц, 2Н), 3,35-3,31 (m, 2Н), 2,29 (t, J=5,2 Гц, 2Н), 2,72-2,70 (m, 2Н), 2,56-2,55 (m, 5Н), 0,90 (t, J=7,6 Гц, 3Н); MS (ESI, M+1): 555,2.
[336] Эффект варианта реализации 1
[337] (1) Анализ ингибирования пролиферации клеток MCF-7
[338] Реагенты для эксперимента:
[339] Среда RPMI 1640, фетальная бычья сыворотка, реагент Promega CellTiter-Glo. Линия клеток MCF-7 была приобретена в Европейской коллекции клеточных культур (ЕСАСС). Многоканальный ридер Envision (PerkinElmer).
[340] Способ эксперимента:
[341] Клетки MCF-7 высевали в черные 384-луночные планшеты при плотности 600 клеток на лунку в 30 мл клеточной суспензии. Планшет для клеток помещали в инкубатор с диоксидом углерода в течение ночи.
[342] Тестируемое соединение разбавляли в 5 раз до 10-й концентрации с помощью Epmotion, разбавляя от 2,5 мМ до 1,28 нМ, на каждую концентрацию выделяли по две дублированные лунки. В промежуточный планшет добавляли 198 мкл среды, соединение с градиентными концентрациями переносили в промежуточный планшет при 2 мкл на лунку согласно соответствующему положению, затем смешивали и переносили в планшет для клеток при 20 мкл на лунку. Планшет для клеток инкубировали в инкубаторе с диоксидом углерода в течение 6 дней.
[343] Реагент Promega CellTiter-Glo добавляли в планшет для клеток при 25 мкл на лунку, клетки инкубировали в течение 10 минут при комнатной температуре для стабилизации сигнала люминесценции, который измеряли с использованием многоканального ридера PerkinElmer Envision.
[344] Анализ данных:
[345] Исходные данные были преобразованы в показатель ингибирования с использованием уравнения (Макс-Соотношение)/(Макс-Мин)*100%, и значение IC50 можно было получить путем подгонки кривой с четырьмя параметрами. (Модель 205 в XLFIT5, iDBS)
[346] (2) Анализ деградации ER в клетках MCF-7
[347] Реагенты эксперимента:
[348] среда RPMI 1640, фетальная бычья сыворотка, PBS, 16% параформальдегид, Triton, блокирующий раствор, антитело к рецептору эстрогена, вторичное козье антитело против кролика ближнего инфракрасного диапазона, краситель DRAQ5. Линия клеток MCF-7 была приобретена в Европейской коллекции клеточных культур (ЕСАСС). Система визуализации инфракрасного флуоресцентного сканирования Odyssey.
[349] Способ эксперимента:
[350] Клетки MCF-7 высевали в черные 384-луночные планшеты при плотности 3200 клеток на лунку в 30 мл суспензии клеток. Планшет для клеток помещали в инкубатор с диоксидом углерода на 4 дня.
[351] Тестируемое соединение разбавляли в 5 раз до 10-й концентрации с помощью Epmotion, фульвестрант разбавляли от 0,25 мМ до 0,28 нМ, другие соединения разбавляли от 2,5 мМ до 1,28 нМ, на каждую концентрацию выделяли по две дублированные лунки. В промежуточный планшет добавляли среду 198 мкл, соединение с градиентными концентрациями переносили в промежуточный планшет при 2 мкл на лунку согласно соответствующему положению, затем смешивали и переносили в планшет для клеток при 20 мкл на лунку. Планшет для клеток инкубировали в инкубаторе с диоксидом углерода в течение 20 часов.
[352] После добавления 8% параформальдегида в планшет для клеток при 50 мкл на лунку планшет для клеток инкубировали при комнатной температуре в течение 30 минут, затем дважды промывали PBS. После сушки добавляли 50 мкл PBS, содержащего 0,1% Triton, и планшет для клеток инкубировали при комнатной температуре в течение 15 минут, затем пять раз промывали PBS. После добавления 50 мкл блокирующего раствора планшет для клеток инкубировали при комнатной температуре в течение 1 часа. После сушки добавляли 50 г блокирующего раствора, содержащего 0,1% антитела к рецептору эстрогена, и планшет для клеток инкубировали при 4°С в течение ночи. На следующий день после того, как первичное антитело убрали, планшет пять раз промывали PBS, затем добавляли блокирующий раствор, содержащий 0,1% вторичного козьего антитела против кролика ближнего инфракрасного диапазона и 0,05% красителя DRAQ5. Планшет для клеток инкубировали при комнатной температуре в течение 1 часа, промывали PBS пять раз. После вытирания насухо планшет для клеток измеряли с использованием системы визуализации инфракрасного флуоресцентного сканирования Odyssey.
[353] Анализ данных:
[354] Исходные данные были преобразованы в показатель ингибирования с использованием уравнения (Макс-Соотношение)/(Макс-Мин)*100%, и значение IC50 можно было получить путем подгонки кривой с четырьмя параметрами. (Модель 205 в XLFIT5, iDBS)
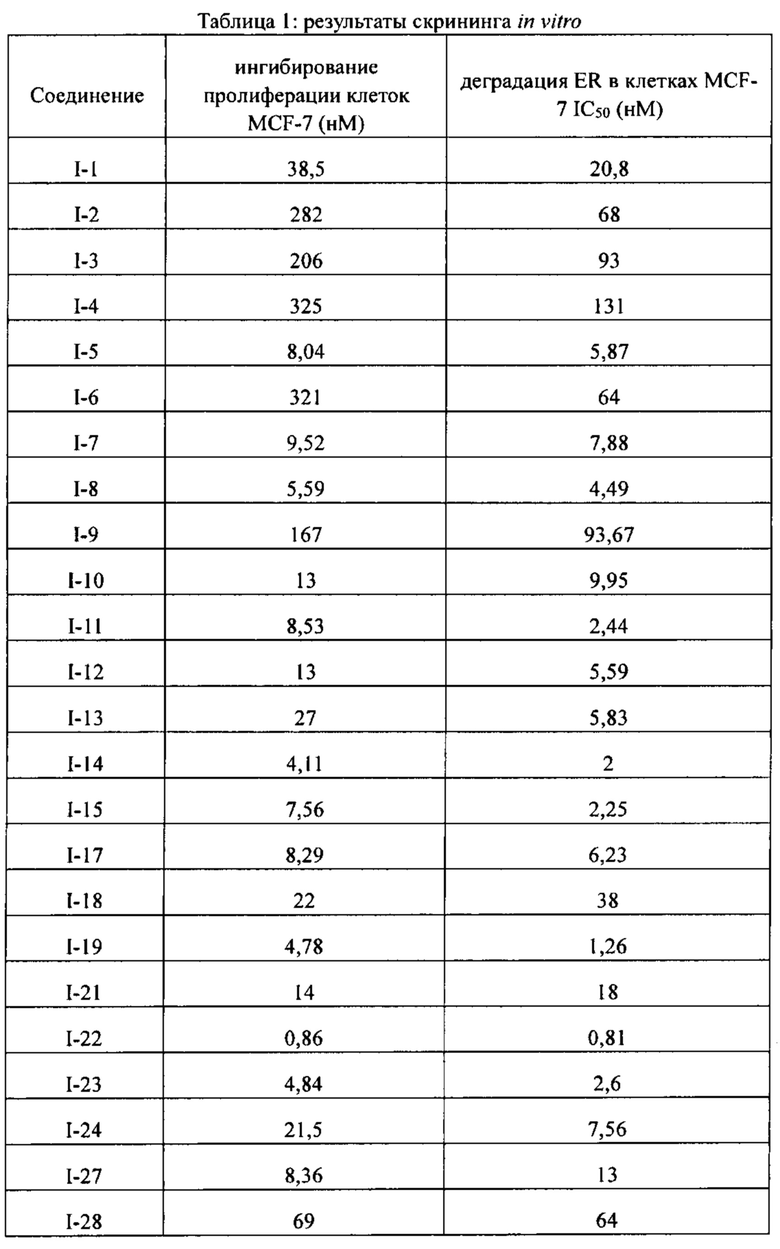
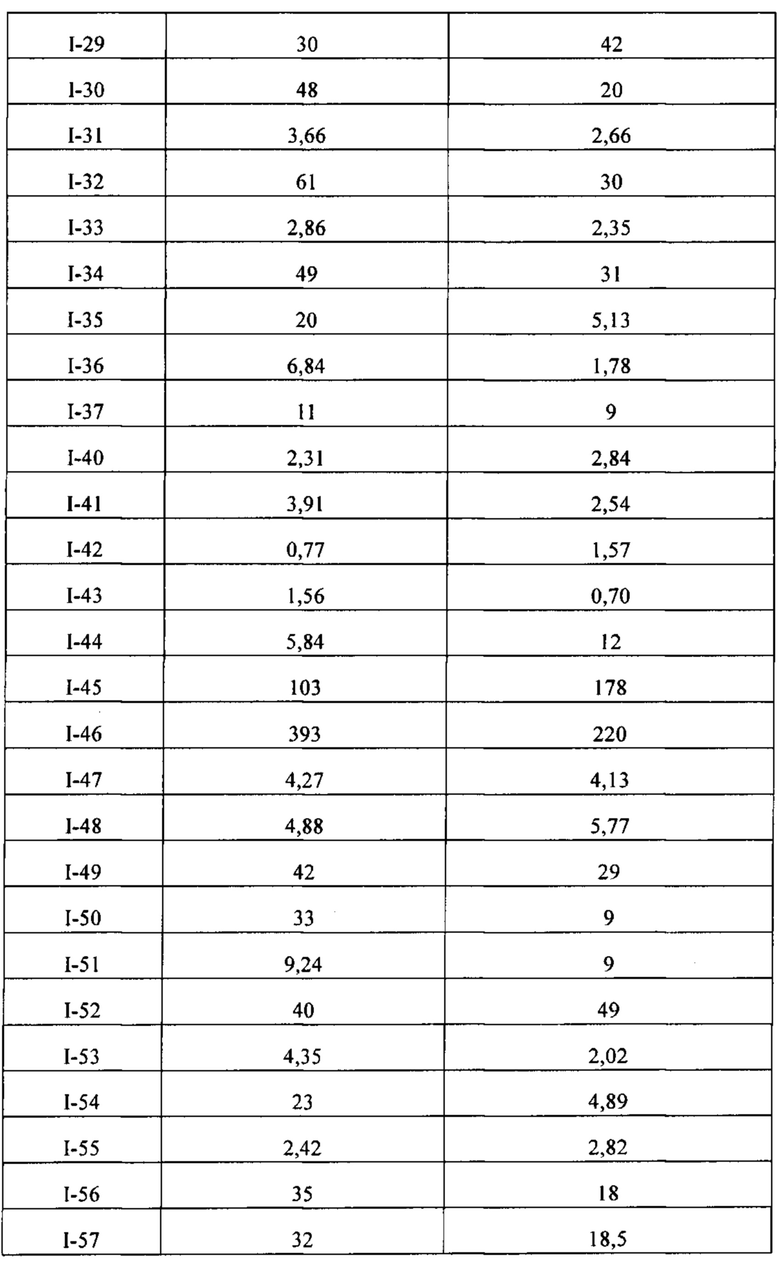
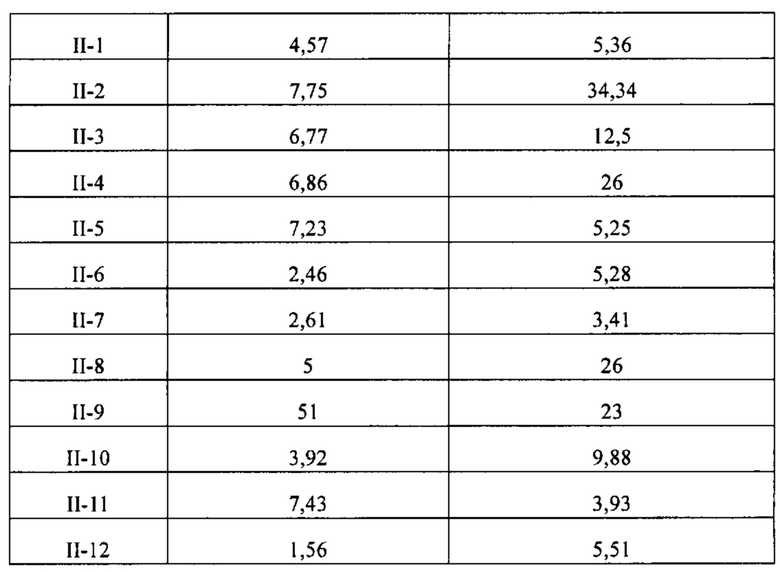
[355] Вывод:
[356] Соединение настоящего изобретения демонстрирует превосходную активность in vitro.
[357] Эффект варианта реализации 2: Оценка свойств DMPK in vitro
[358] (1) Результаты свойств DMPK in vitro показаны в таблице 2:

[359] Вывод:
[360] Соединение I-1 настоящего изобретения превосходит пероральный селективный понижающий регулятор рецепторов эстрогена ARN-810, находящийся во II фазе клинических исследований, с точки зрения пиковой концентрации в плазме Cmax после перорального введения, периода полувыведения, показателя клиренса (CL), ППК и пероральной биодоступности.
[361] (2) Результаты свойств ФК in vivo показаны в таблице 3:
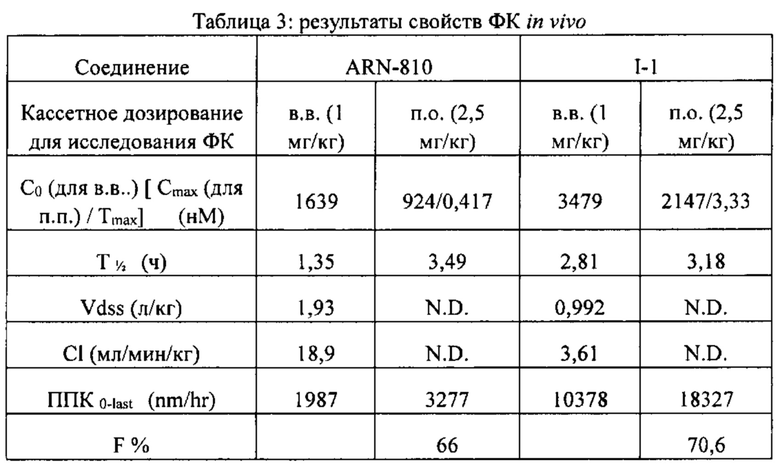
[362] Вывод:
[363] Соединение I-1 настоящего изобретения превосходит пероральный селективный понижающий регулятор рецептора эстрогена ARN-810, находящийся во II фазе клинических исследований, с точки зрения пиковой концентрации в плазме Cmax после перорального введения, периода полувыведения, показателя клиренса (CL), ППК и пероральной биодоступности.
[364] Эффект варианта реализации 3: оценка фармакодинамики in vivo
[365] Задача данного эксперимента состояла в оценке противоопухолевой активности перорального понижающего регулятора рецептора эстрогена ARN-810, находящегося во II фазе клинических исследований, и соединения I-1 на ксенотрансплантатный модели рака молочной железы MCF-7 у бестимусных мышей BALB/c (Beijing Vital River Laboratory Animal Technology Co., Ltd., 10 животных на экспериментальную группу). В данном эксперименте мышам, инокулированным клетками рака молочной железы MCF-7, перорально вводили ARN-810 с дозировкой 10/30 мг/кг, соединение I-1 с дозировкой 30 мг/кг и оценивали ингибирующую активность тестируемого лекарственного средства на рост опухоли.
[366] Мышам инокулировали подкожно 0,36 мг таблетку эстрогена с замедленным высвобождением (высвобождение в течение 60 дней) в левое плечо за три дня до инокуляции. Когда клетки находились в логарифмической фазе роста, клетки собирали, подсчитывали и доводили до концентрации 10×107 клеток/мл, затем добавляли равный объем Матригеля, перемешивали для инокуляции. 0,2 мкл суспензии опухолевых клеток MCF-7 (10×106) инокулировали подкожно в правое плечо каждой мыши. На 7-й день после инокуляции опухолевых клеток мышей распределяли по группам и вводили средство один раз в день, со средним объемом опухоли 169 мм3 и массой тела 23,33 г. Объем опухоли и массу тела измеряли два раза в неделю после распределения по группам, и показатель роста опухоли (Т/С) и показатель ингибирования роста опухоли (TGI) рассчитывали на основе последних данных измерений на 17-й день после распределения по группам. Результаты были следующими:
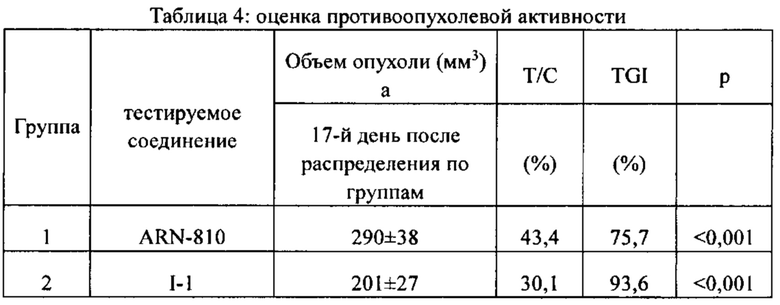
[367] а: Среднее ± SEM.
[368] Вывод:
[369] Противоопухолевая активность в результате перорального введения I-1 в дозировке 30 мг/кг превосходит противоопухолевую активность перорального понижающего регулятора рецептора эстрогена ARN-810, находящегося во II фазе клинических исследований, в дозировке 10/30 мг/кг.
[370] Эффект варианта реализации 4: оценка распределения лекарственного средства в тканях
[371] На 20-й день после распределения по группам образец собирали из плазмы, опухоли, молочной железы и мозга мышей в 4 временных точках (по 2-3 мыши на каждую временную точку), составляющих, соответственно, 0,5 часа, 1,5 часа, 6 часов и 24 часа после введения. Кровь (0,5 мл) собирали с использованием 1,5 мл антикоагуляционной пробирки ЭДТА-К2, немедленно центрифугировали при 4000 об/мин, 4°С в течение 10 минут для получения плазмы, образец плазмы хранили при -80°С для определения концентрации лекарственного средства. Опухоль собирали с использованием 2 мкл криопробирки и мозг собирали с использованием 5 мкл криопробирки, образцы немедленно помещали в жидкий азот для быстрого замораживания. Все образцы хранили при -80°С для определения концентрации лекарственного средства после отбора проб. Результат был преобразован на основе соответствующей ППК для сравнения распределения лекарственного средства.
[372] Результаты показаны в таблице 5.
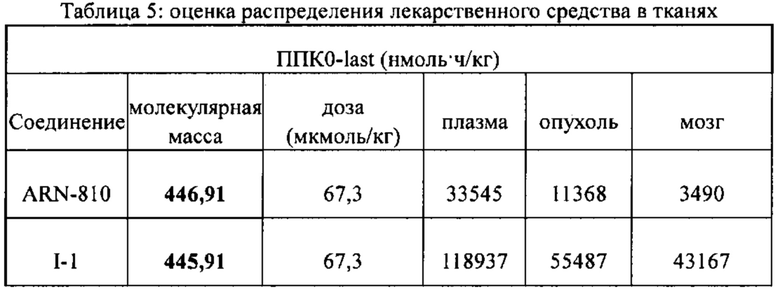
[373] Вывод:
[374] Распределение соединения I-1 в опухолевой ткани намного лучше, чем распределение ARN-810, что согласуется с его лучшей противоопухолевой активностью;
[375] Распределение соединения I-1 в ткани мозга намного лучше, чем распределение ARN-810, что указывает на то, что он будет иметь превосходную активность для лечения метастатического ER-положительного рака молочной железы в головном мозге.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| ПРОИЗВОДНОЕ НА ОСНОВЕ ДИГИДРОПИРИМИДО-КОЛЬЦА В КАЧЕСТВЕ ИНГИБИТОРА HBV | 2015 |
|
RU2693897C2 |
| НОВЫЙ АГОНИСТ β-РЕЦЕПТОРА ТИРЕОИДНЫХ ГОРМОНОВ | 2021 |
|
RU2839610C1 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| ИНГИБИТОР EGFR И ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2702631C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АГОНИСТОВ PPAR | 2017 |
|
RU2711991C1 |
| ТАКСАНОВЫЕ СОЕДИНЕНИЯ, А ТАКЖЕ СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2686675C1 |
| ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2809631C2 |
Изобретение относится к соединению индола формулы (I), где значения Y, Z, R1-R6, m, n определены в формуле изобретения, его фармацевтической композиции и применению в качестве понижающего регулятора рецепторов эстрогена при получении лекарств для лечения эстроген-рецептор-положительного рака молочной железы. 5 н. и 16 з.п. ф-лы, 5 табл., 65 пр.
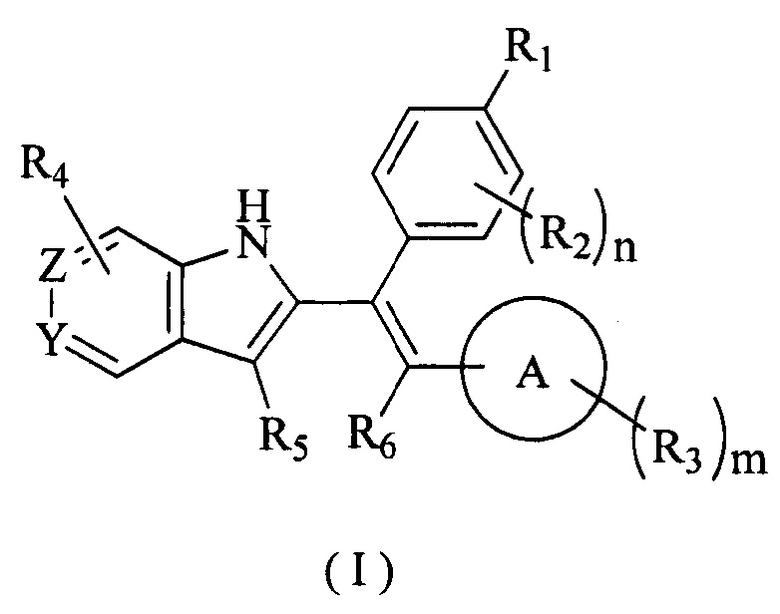
1. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль,
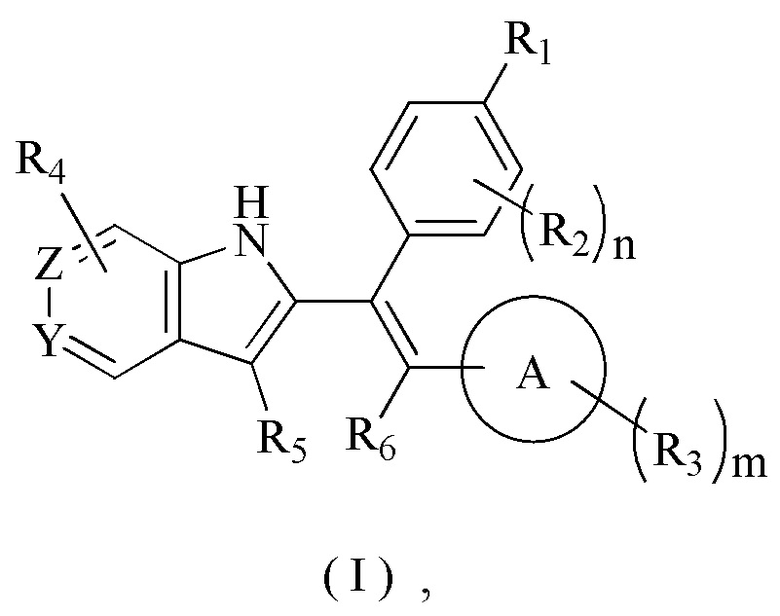
где R1 выбран из
 и
и  ;
;
X выбран из одинарной связи и O;
Y и Z представляют собой CH;
кольцо A выбрано из фенила, тиенила и пиридила;
R2 выбран из H и галогена;
R3 выбран из H, галогена, CN или группы, состоящей из C1-6 алкила и C1-6 алкокси, каждый из которых необязательно замещен 1, 2 или 3 R;
каждый R4 и R5 независимо выбран из H, галогена, CN и C1-6 алкила;
R6 представляет собой C1-6 алкил;
R выбран из H, F или C1-8 алкила, необязательно замещенного 1, 2 или 3 R’;
n выбран из 0, 1, 2, 3 и 4;
m выбран из 0, 1, 2, 3 и 4;
p выбран из 0, 1, 2 и 3;
или, когда m представляет собой 2, R3 и R3 соединены вместе с образованием 5-6-членного кольца;
R’ выбран из группы, состоящей из F и CN.
2. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль,
где R1 выбран из
 ;
;
Y и Z представляют собой CH;
кольцо A выбрано из фенила, тиенила и пиридила;
R2 выбран из H и галогена;
R3 выбран из H, галогена, CN или группы, состоящей из C1-6 алкила и C1-6 алкокси, каждый из которых необязательно замещен 1, 2 или 3 R;
каждый R4 и R5 независимо выбран из H, галогена, CN и C1-6 алкила;
R6 представляет собой C1-6 алкил;
R выбран из H, F и S(=O)CH3;
n выбран из 0, 1, 2, 3 и 4;
m выбран из 0, 1, 2, 3 и 4;
или, когда m представляет собой 2, R3 и R3 соединены вместе с образованием 5-6-членного кольца.
3. Соединение или его фармацевтически приемлемая соль по п. 1, отличающееся тем, что R выбран из H, F или C1-5 алкила, необязательно замещенного 1, 2 или 3 R’.
4. Соединение или его фармацевтически приемлемая соль по п. 3, отличающееся тем, что R выбран из H, F или группы, состоящей из CH3 и CH3CH2, каждый из которых необязательно замещен 1, 2 или 3 R’.
5. Соединение или его фармацевтически приемлемая соль по п. 4, отличающееся тем, что R выбран из группы, состоящей из H, F, CH3, CH2F, CHF2, CF3, Et и  .
.
6. Соединение или его фармацевтически приемлемая соль по п. 1, отличающееся тем, что структурное звено выбрано из 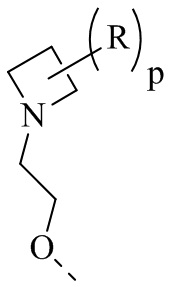 .
.
7. Соединение или его фармацевтически приемлемая соль по п. 6, отличающееся тем, что структурное звено выбрано из 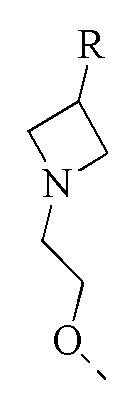 и
и 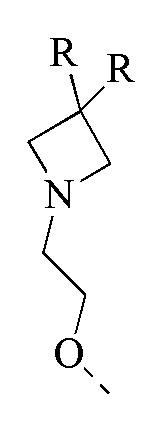 .
.
8. Соединение или его фармацевтически приемлемая соль по п. 7, отличающееся тем, что структурное звено выбрано из группы, состоящей из
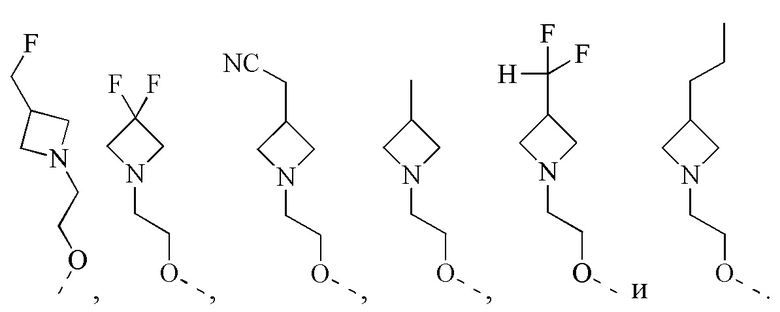
9. Соединение или его фармацевтически приемлемая соль по п. 1, отличающееся тем, что R2 выбран из H, F, Cl, Br или I, и/или
R3 выбран из H, F, Cl, Br, I, CN или группы, состоящей из C1-4 алкила и C1-4 алкокси, каждый из которых необязательно замещен 1, 2 или 3 R, и/или
каждый R4 и R5 независимо выбран из H, F, Cl, Br, I, CN или C1-4 алкила, и/или
кольцо A выбрано из группы, состоящей из фенила, пиридила и тиенила, и/или
R6 выбран из  и
и  .
.
10. Соединение или его фармацевтически приемлемая соль по п. 9, отличающееся тем, что R3 выбран из H, F, Cl, Br, I, CN или группы, состоящей из CH3, CH3CH2, CH3O, CH3CH2O и  , каждый из которых необязательно замещен 1, 2 или 3 R.
, каждый из которых необязательно замещен 1, 2 или 3 R.
11. Соединение или его фармацевтически приемлемая соль по п. 10, отличающееся тем, что R3 выбран из группы, состоящей из H, F, Cl, CN, CH3, CF3 и  .
.
12. Соединение или его фармацевтически приемлемая соль по п. 9, отличающееся тем, что каждый R4 и R5 независимо выбран из группы, состоящей из H, F, Cl, Br, CN и CH3.
13. Соединение или его фармацевтически приемлемая соль по п. 9, отличающееся тем, что структурное звено  выбрано из группы, состоящей из
выбрано из группы, состоящей из 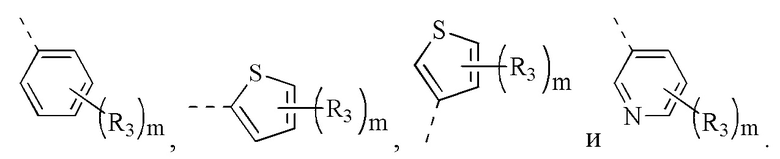
14. Соединение или его фармацевтически приемлемая соль по п. 13, отличающееся тем, что структурное звено выбрано из группы, состоящей из
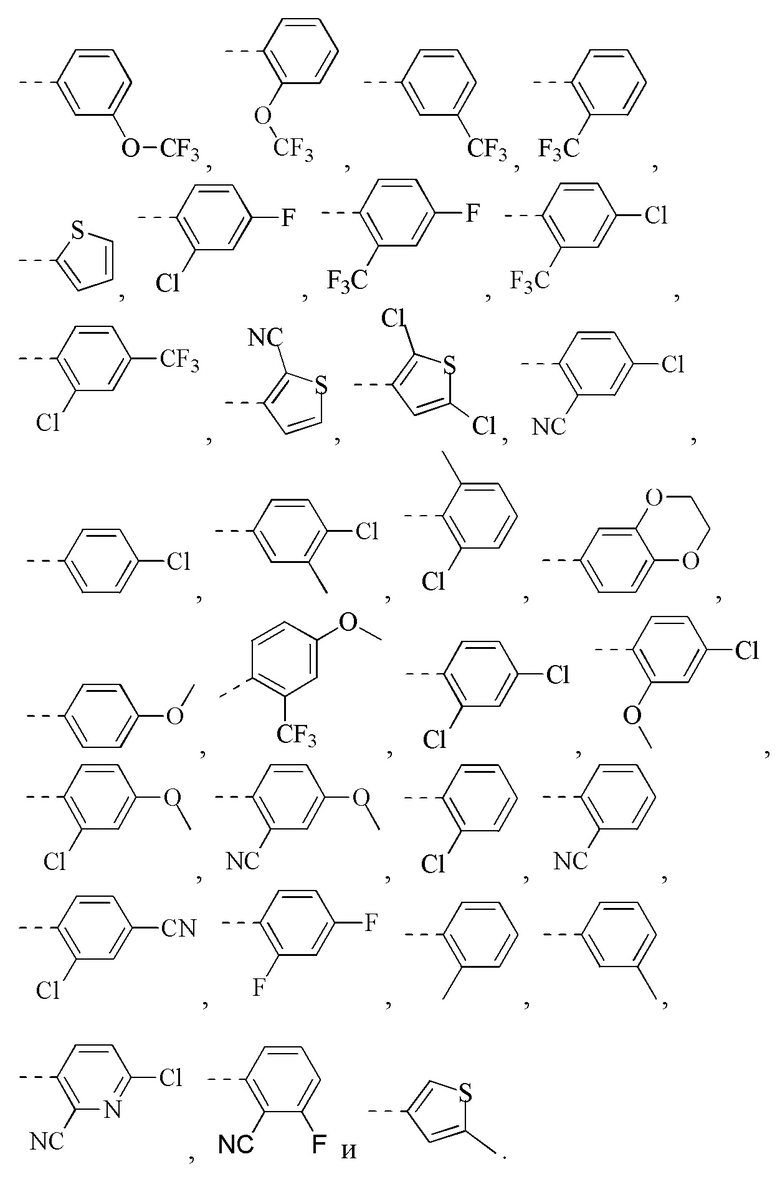
15. Соединение или его фармацевтически приемлемая соль по п. 2, отличающееся тем, что структурное звено  выбрано из
выбрано из  .
.
16. Соединение или его фармацевтически приемлемая соль по п. 15, отличающееся тем, что структурное звено выбрано из  и
и  .
.
17. Соединение или его фармацевтически приемлемая соль по п. 1, отличающееся тем, что соединение выбрано из группы, состоящей из
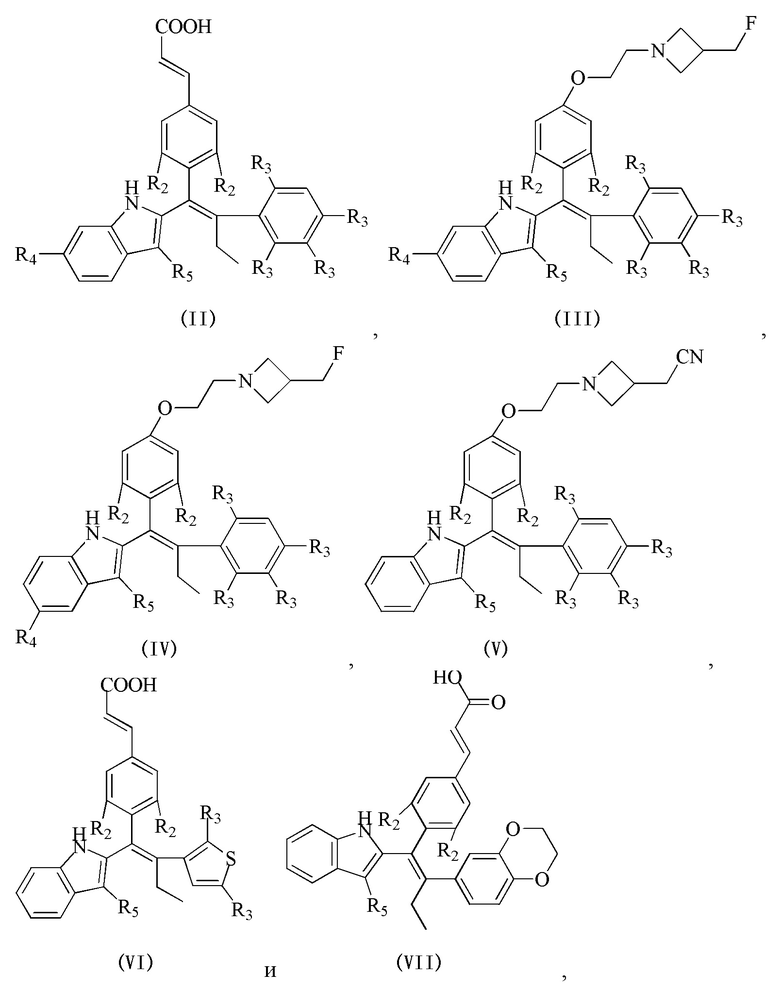
где R2, R3, R4 и R5 являются такими, как определено по п. 1.
18. Соединение или его фармацевтически приемлемая соль по п. 2, отличающееся тем, что соединение выбрано из
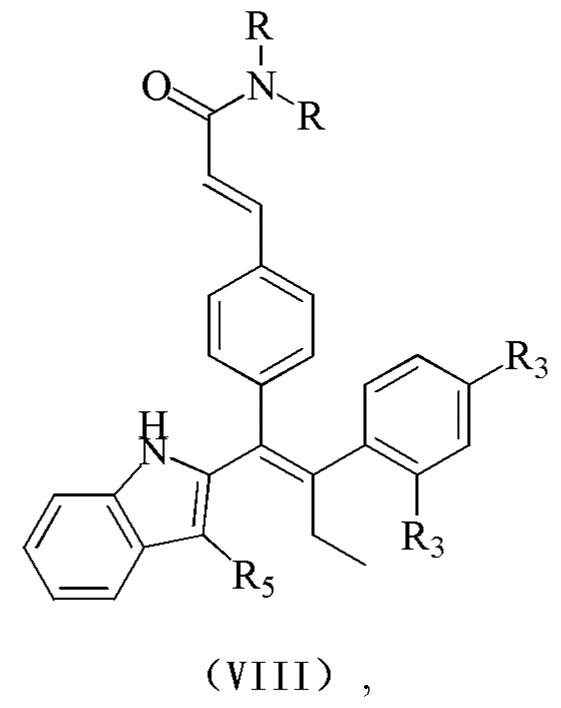
где R3, R5 и R являются такими, как определено по п. 2.
19. Соединение или его фармацевтически приемлемая соль, где соединение выбрано из группы, состоящей из
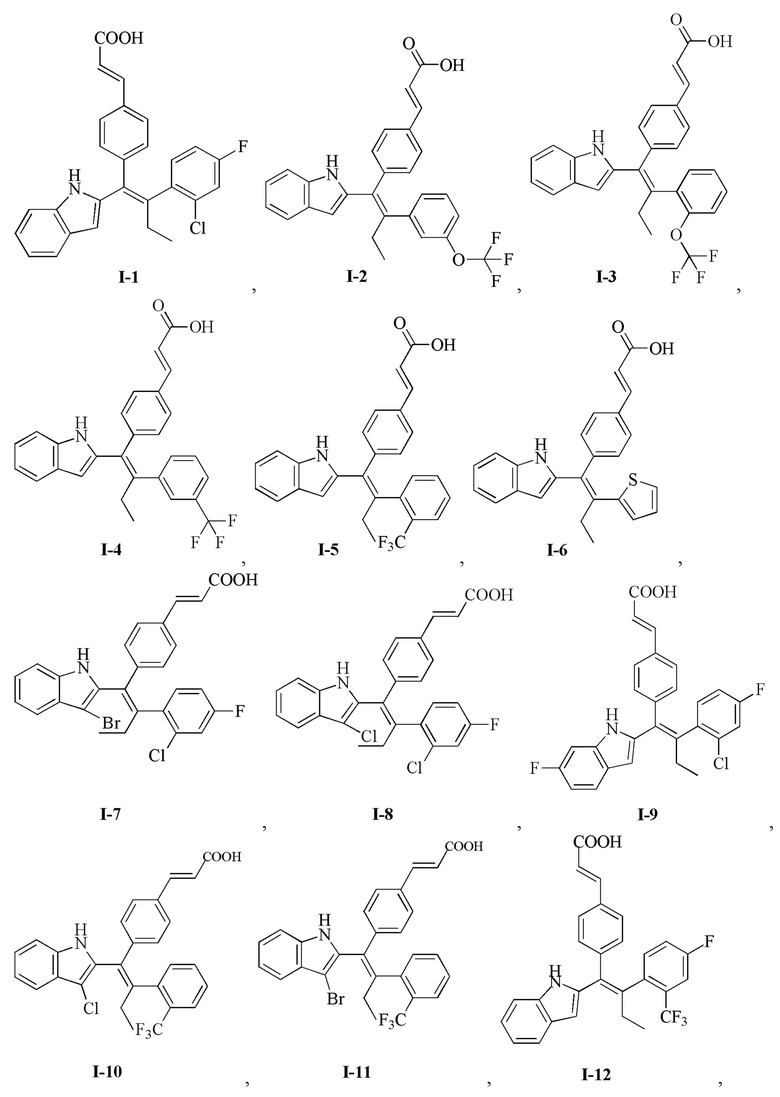
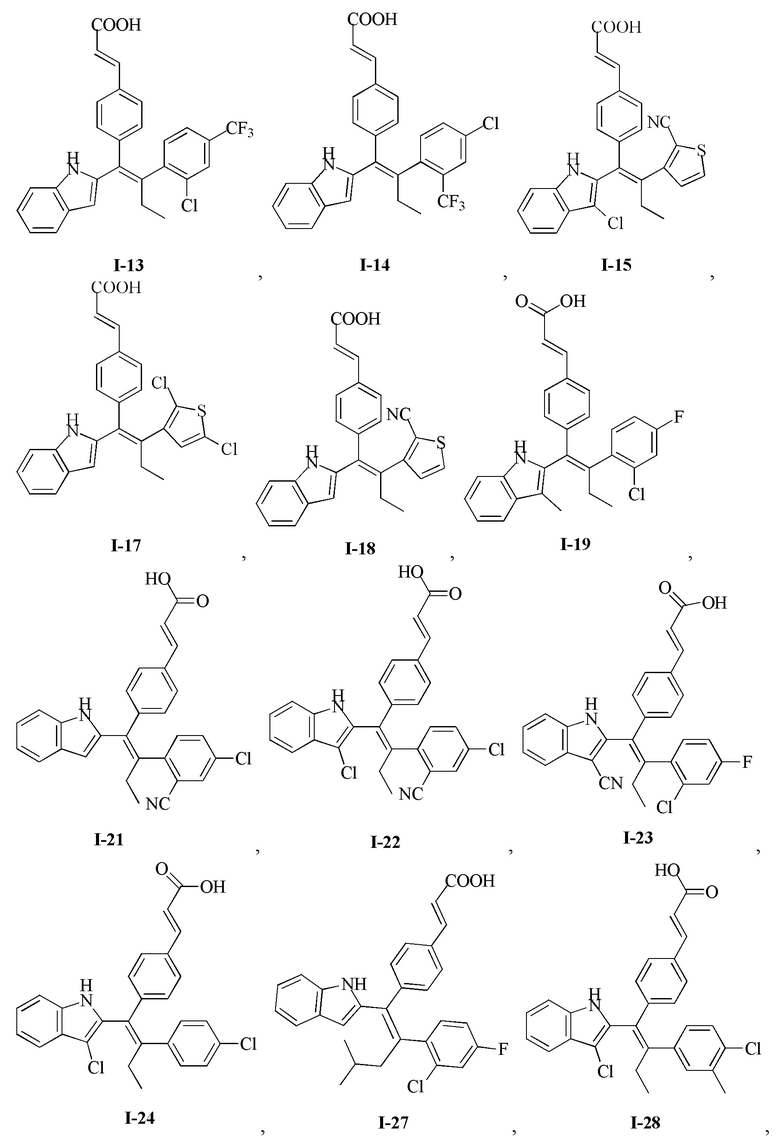
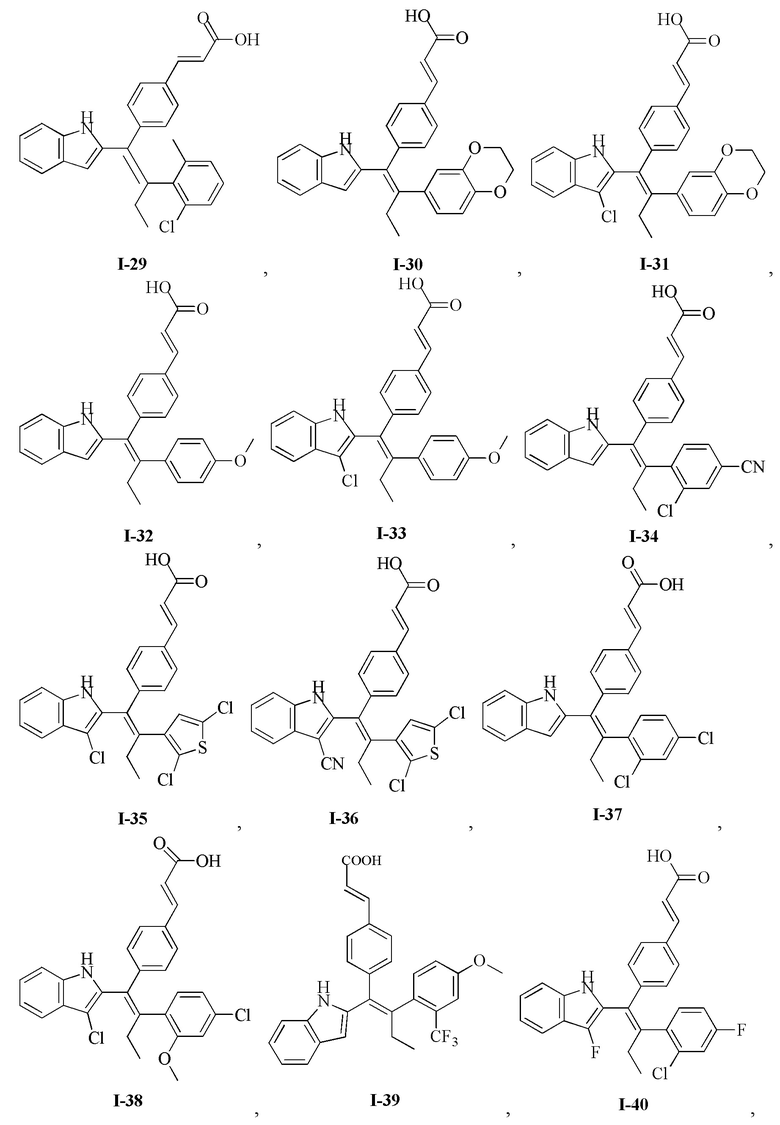
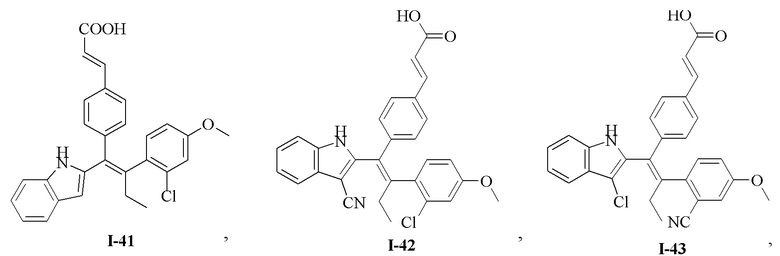
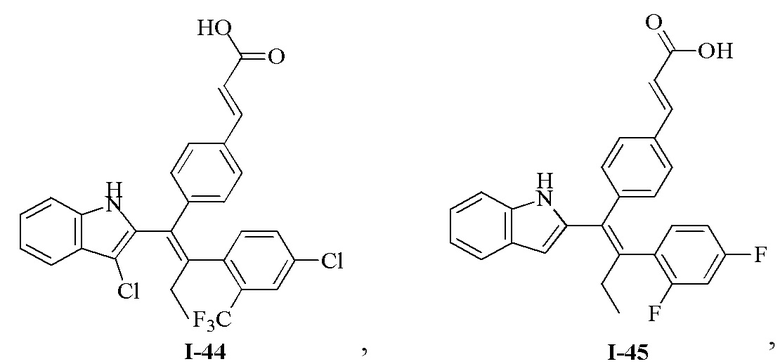
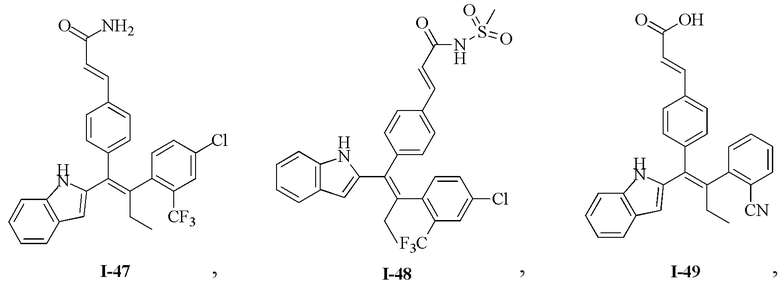
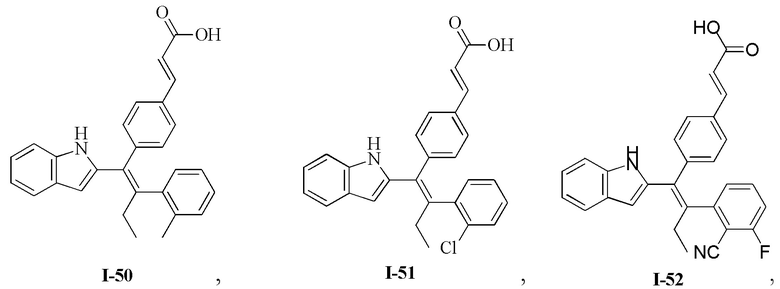
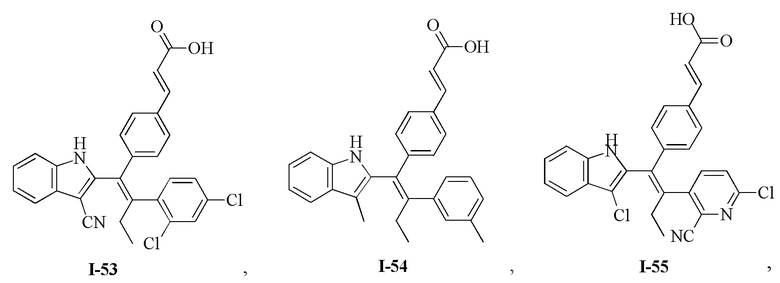
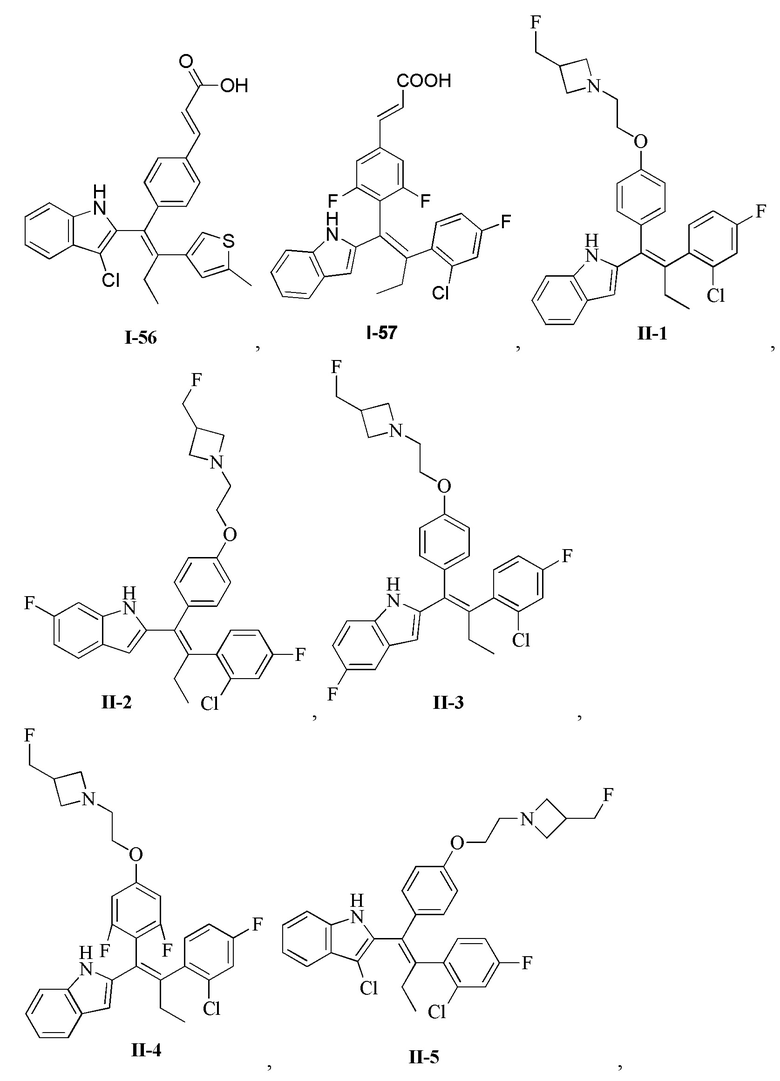
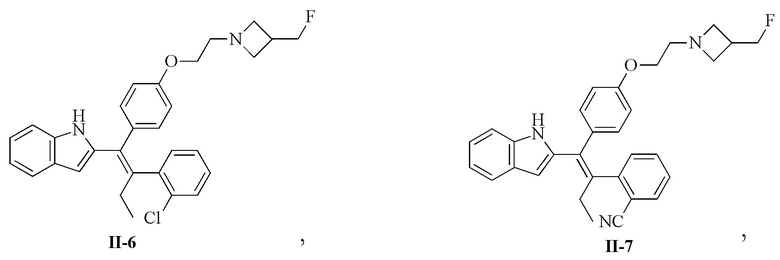
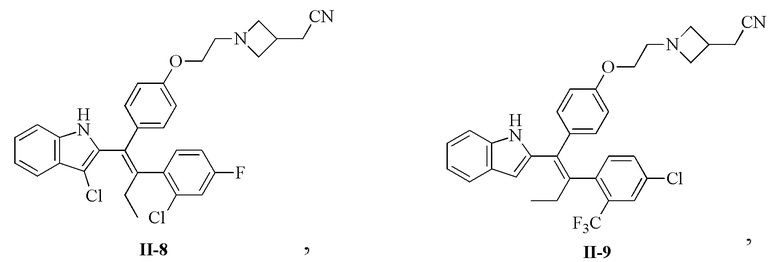
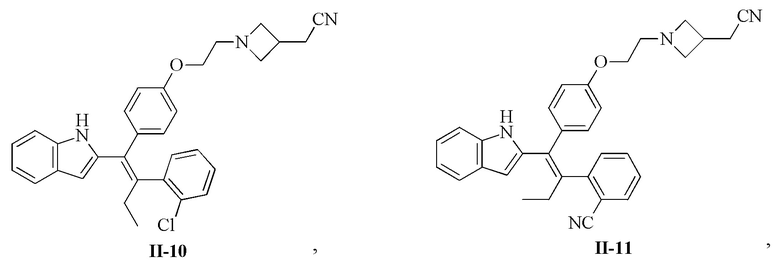
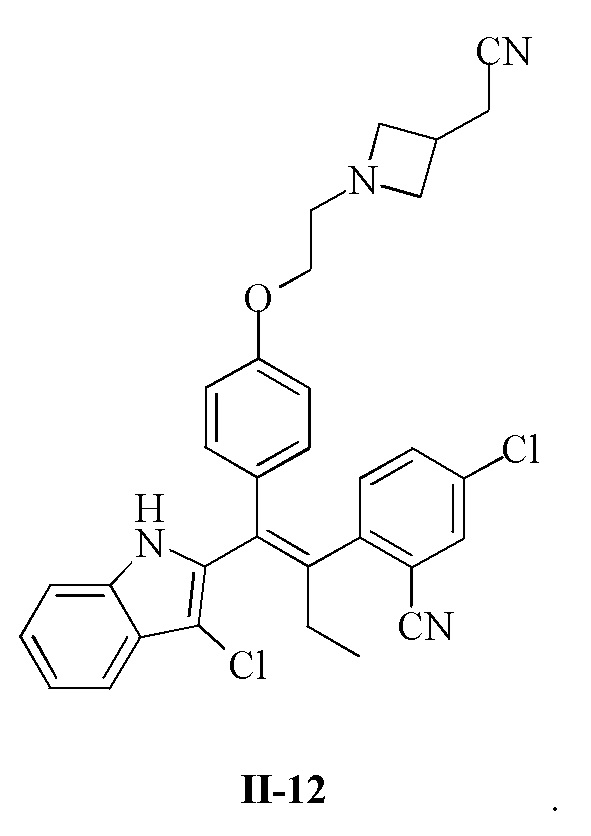
20. Фармацевтическая композиция для лечения эстроген-рецептор-положительного рака молочной железы, содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-19 и фармацевтически приемлемый носитель.
21. Способ лечения расстройства, связанного с рецептором эстрогена у субъекта, нуждающегося в этом, включающий введение эффективного количества соединения или его фармацевтически приемлемой соли по любому из пп. 1-19, где расстройство, связанное с рецептором эстрогена, представляет собой эстроген-рецептор-положительный рак молочной железы.
| Автомотриса для железных дорог | 1923 |
|
SU815A1 |
| WO 2012037411 A2, 22.03.2012 | |||
| WO 2015136017 A1, 17.09.2015 | |||
| WO 2015136016 A2, 17.09.2015. | |||

